
血球細胞の初期化法
【課題】体細胞から効率的に多能性幹細胞を製造する方法の提供。
【解決手段】体細胞から多能性幹細胞を製造する方法であって、(a)体細胞を、IL−6シグナル伝達因子刺激因子および少なくとも1種のサイトカインを含む細胞培養培地中で培養する工程、および(b)体細胞を脱分化させる工程を含んでなり、工程(a)の後に工程(b)が行われるか、または工程(a)と工程(b)が同時に行われる方法。
【解決手段】体細胞から多能性幹細胞を製造する方法であって、(a)体細胞を、IL−6シグナル伝達因子刺激因子および少なくとも1種のサイトカインを含む細胞培養培地中で培養する工程、および(b)体細胞を脱分化させる工程を含んでなり、工程(a)の後に工程(b)が行われるか、または工程(a)と工程(b)が同時に行われる方法。
【発明の詳細な説明】
【発明の背景】
【0001】
発明の分野
本発明は、分化した体細胞、特に血球系細胞を初期化または脱分化することにより多能性幹細胞を製造するための方法およびキットに関する。
【0002】
背景技術
胚性幹細胞(ES細胞)は、生体中の組織・臓器を構成するほぼ全ての種類の細胞に分化できる多能性を有したまま、長期間にわたる細胞増殖を無制限に維持できる細胞株である。この性質により、ES細胞は、パーキンソン病、I型糖尿病、脊髄損傷、心不全など、多くの疾患に対する移植療法のための細胞資源として、組織工学による人工組織・臓器作製のための多種類細胞材料の供給源として、さらには、基礎研究や創薬研究に用いられるヒト細胞の供給源としての利用が期待されている。しかしながら、ES細胞はヒトやマウスの初期胚を破壊して樹立された細胞株であることから、倫理的見地よりES細胞の利用に対する反対意見も多い。また、細胞移植療法については、他人の細胞を移植することになることから、臓器移植と同様に拒絶反応を惹起する危険性を伴っている。これらの問題を回避する方法として、患者自身の分化体細胞を初期化した、ES細胞に近い多能性と増殖能を有した誘導多能性幹細胞(iPS細胞)の利用が期待されている。
【0003】
分化体細胞の初期化法として、例えば、マウス胚性線維芽細胞(MEF細胞)にレトロウィルスベクターでOct4、Sox2、Klf4、およびc−mycの4遺伝子を同時感染させることによりiPS細胞を誘導する技術が報告されている(非特許文献1:K. Takahashi, S. Yamanaka, Cell, 126, pp.663-676, 2006;非特許文献2:K. Okita, T. Ichisaka, S. Yamanaka, Nature, 448, pp. 313-317, 2007;非特許文献3:M. Wernig et al., Nature, 448, pp.318-324, 2007)。マウス肝細胞および胃上皮細胞から同様の方法でiPS細胞を誘導する技術も報告されている(非特許文献4:T. Aoi et al., Science Express on 14 Feb. 2008)。これらの報告における初期化因子のスクリーニング方法については国際公開第2005/080598号パンフレット(特許文献1)に提案されており、また、当該初期化因子については、他のES細胞特異的遺伝子(初期化因子の候補遺伝子)と共に国際公開第2007/069666号パンフレット(特許文献2)に提案されている。これらの報告および提案においては、ヒト細胞について一部記載があるものの、この段階ではヒトES細胞と同等の性質を持ったヒトiPS細胞としての樹立は不完全なものである。また、肝細胞や胃上皮細胞からの誘導はヒトに適用する場合には侵襲性が多く、採取する患者の負担が大きくなることが予想される。
【0004】
ヒトiPS細胞を樹立する方法としては、成人皮膚由来線維芽細胞 (HDF細胞)にレトロウィルスを用いてOct4、Sox2、Klf4、およびc−mycの4因子を導入する方法が報告されている(非特許文献5:K. Takahashi, S. Yamanaka, Cell, 131, pp.861-872, 2007)。この方法においては、レトロウィルスを高効率に感染させるために、マウスエコトロピックウィルス受容体であるsolute carrier family 7(Slc7a1) をレンチウィルスにより発現させた細胞を用いている。その結果、レトロウィルスの感染後25日目頃よりヒトES細胞様のコロニーを認め、30日目以降にコロニーのピックアップを実施している。この方法では、新たにマウスエコトロピックウィルス受容体をレンチウィルスにより導入しているため、異種動物由来の遺伝子の発現および染色体へのDNA断片挿入機会の増加による、宿主細胞の癌化リスクの増大といった課題を抱えている。さらに、誘導日数が1ヶ月におよぶため、培養コストについても課題を抱えている。また、この報告では、iPS細胞の誘導に新生児包皮由来線維芽細胞 (BJ1細胞)も用いられており、その場合のヒトES細胞様コロニーの出現頻度はHDFを用いた場合よりも高いが、誘導日数については短縮されていない。
【0005】
他のグループでは、アデノウィルスによりマウスエコトロピックウィルス受容体であるmCAT1遺伝子を発現させたヒト新生児包皮由来線維芽細胞を用いて、同様の4因子をレトロウィルスにより導入し、iPS細胞を誘導した報告もある(非特許文献6:H.Masaki et al., Stem Cell Research, 1, pp.105-115, 2007)。この報告では、レトロウィルスの感染後17日目にiPS様のコロニーの出現が認められている。この方法では、アデノウィルスでウィルス受容体を発現させていることにより、染色体DNAへの異種遺伝子の挿入リスクは低減されているが、異種遺伝子が細胞内に導入されることに伴うリスクは以前として残る。同様の4因子をレトロウィルスによりヒト胎児肺線維芽細胞 (MRC5細胞)へ導入してiPS細胞を誘導した報告もある(非特許文献7:I. H. Park et al., Nature, 451, pp.141-146, 2008)。しかしながら、この報告では、BJ1細胞、成体間葉系幹細胞(MSC細胞)、およびHDF細胞からの4因子によるiPS細胞誘導は成功していない。また、初期化因子としてOct4、Sox2、NANOG、およびLin28の4因子を用いて、レンチウィルスで細胞に発現させることにより、fetal fibroblast cells(IMR90 cells)やnewborn foreskin fibroblastからES細胞様コロニーを取得している報告もある(非特許文献8:J. Yu et al., Science, 318, pp.1917-1920, 2007)。胎児や新生児の細胞を用いた場合、成人の表皮線維芽細胞を用いた場合に比べて、より短期間でiPS細胞が誘導できているが、治療や研究に用いる細胞源としては倫理的な課題が残る。この報告ではさらに、ヒトES細胞から分化誘導した血球細胞等を用いてiPS細胞が誘導可能であることが報告されている。しかし、ES細胞から分化誘導した細胞は正常な分化過程を経ておらず、成体由来の血球細胞と同一の細胞とはいえない。今のところ、成体から採取した血球細胞からのiPS細胞の誘導についての報告はない。
【0006】
Oct4、Sox2、Klf4、およびc−mycの4因子に加え、Nanogを共発現させることによりiPS細胞の誘導を効率化した報告もある(非特許文献9:W. E. Lowry et al., Proc. Natl. Acad. Sci. USA, 105, pp.2883-2888, 2008) 。Nanogの存在下でリプログラミング(初期化)効率が上がることは従来から報告されている(非特許文献10:J. Silva et al., Nature, 441, pp.997-1001, 2006) 。レトロウィルスを用いてHDF細胞に遺伝子導入を行い、ウィルス感染終了後21日目でES細胞様コロニーが出現する。Nanogを用いることによりiPS細胞の誘導効率が上がっているが、因子の種類を増やすことは、染色体に挿入される遺伝子の種類も増やすこととなり、初期化因子の種類を増やさない方法での効率改善が望まれる。
【0007】
また、マウスにおいては終末分化した成熟B細胞からiPS細胞を誘導した報告もある(非特許文献11:J. Hanna et al., Cell, 133, pp.250-264, 2008)。この報告においては、ドキシサイクリン依存的に4つの核初期化因子、Oct4、Sox2、Klf4、およびc−mycを発現するユニットをレンチウィルスベクターにより導入することによって、まずiPS細胞を誘導している。さらに、誘導されたiPS細胞を用いてトランスジェニックマウスを作製している。成熟B細胞からのiPS細胞誘導には、こうして得られたトランスジェニックマウスより調製したB細胞を用いており、同様の方法をヒトに適用することは不可能である。ヒトの終末分化した血液細胞からiPS細胞を誘導するためには本法を適用することはできず、ヒト初代細胞に適用できる初期化方法が望まれる。
【0008】
一方で、ES細胞のような多能性幹細胞の利用により治療効果が期待される医療領域では、ES細胞から分化誘導した細胞による治療法の検討と並行して、生体内に存在する体性幹細胞の利用が検討されてきた。そのような取組みの中、細胞移植療法への利用を目的として生体外で体性幹細胞を増幅する技術の報告がある。中でも造血幹細胞については、幹細胞因子(SCF)、Fltリガンド(FL)、トロンボポエチン(TPO)、インターロイキン−6(IL−6)と可溶化型IL−6受容体のα鎖(IL−6R)との融合タンパク質などのサイトカインを用いて、臍帯血由来の造血幹細胞や骨髄由来の造血幹細胞を増幅した報告がある(非特許文献12:T. Ueda et al., J. of Clinical Investigation, 105, pp-1013-1021, 2000;非特許文献13:L. Gammaitoni et al., Experimental Hematology, 31, pp. 261-270, 2003)。さらに、ストローマ細胞を用いて増幅率を向上させる技術もある(特許文献3:国際公開第2003/014336号パンフレット)。いずれも血球系の幹細胞または前駆細胞を増幅する技術であり、増幅の際に他の細胞系譜への分化能を付与することはできず、体細胞の初期化作用や脱分化作用はない。また、造血幹細胞の増幅には限界があり、ES細胞のように多能性を維持したまま無制限に増殖させることはできない。
【0009】
【特許文献1】国際公開第2005/080598号パンフレット
【特許文献2】国際公開第2007/069666号パンフレット
【特許文献3】国際公開第2003/014336号パンフレット
【非特許文献1】K. Takahashi, S. Yamanaka, Cell, 126, pp.663-676, 2006
【非特許文献2】K. Okita, T. Ichisaka, S. Yamanaka, Nature, 448, pp. 313-317, 2007
【非特許文献3】M. Wernig et al., Nature, 448, pp.318-324, 2007
【非特許文献4】T. Aoi et al., Science Express on 14 Feb. 2008
【非特許文献5】K. Takahashi, S. Yamanaka, Cell, 131, pp.861-872, 2007
【非特許文献6】H.Masaki et al., Stem Cell Research, 1, pp.105-115, 2007
【非特許文献7】I. H. Park et al., Nature, 451, pp.141-146, 2008
【非特許文献8】J. Yu et al., Science, 318, pp.1917-1920, 2007
【非特許文献9】W. E. Lowry et al., Proc. Natl. Acad. Sci. USA, 105, pp.2883-2888, 2008
【非特許文献10】J. Silva et al., Nature, 441, pp.997-1001, 2006
【非特許文献11】J. Hanna et al., Cell, 133, pp.250-264, 2008
【非特許文献12】T. Ueda et al., J. of Clinical Investigation, 105, pp-1013-1021, 2000
【非特許文献13】L. Gammaitoni et al., Experimental Hematology, 31, pp. 261-270, 2003
【発明の概要】
【0010】
本発明者らは、IL−6シグナル伝達因子(IL6ST)刺激因子とサイトカインカクテルとの存在下での培養と、細胞の脱分化とを組み合わせることにより、体細胞から多能性幹細胞を効率的に樹立できることを見出した。本発明はこの知見に基づくものである。
【0011】
従って、本発明の目的は、体細胞から効率的に多能性幹細胞を製造する方法を提供することにある。
【0012】
そして、本発明による多能性幹細胞の製造法は、体細胞から多能性幹細胞を製造する方法であって、(a)体細胞を、IL−6シグナル伝達因子刺激因子および少なくとも1種のサイトカインを含む細胞培養培地中で培養する工程、および(b)体細胞を脱分化させる工程を含んでなり、工程(a)の後に工程(b)が行われるか、または工程(a)と工程(b)が同時に行われる方法である。
【0013】
本発明によれば、人体より軽微な負担で採取できる体細胞、例えば末梢血液細胞などから、従来法よりも短期間で高効率に多能性幹細胞を樹立することができる。多能性幹細胞の樹立に要する期間は、例えば、従来法の約1/2にまで短縮することが可能である。よって、製造された多能性幹細胞を、脊髄損傷などの緊急性の高い疾患の治療に利用することができ、また、多能性幹細胞の製造にかかる費用も低減することが可能となる。
【発明の具体的説明】
【0014】
本発明において製造される「多能性幹細胞」とは、種々の分化系譜に分化可能な細胞を意味し、必ずしもES細胞と同等のすべての細胞系譜に分化可能な細胞に限られるものではなく、例えば、内胚葉系幹細胞、中胚葉系幹細胞、または外胚葉系幹細胞としてもよい。例えば、心不全の治療に使用する多能性幹細胞を製造するためには、心筋細胞に分化可能な細胞を製造できればよい。あるいは、I型糖尿病の治療に使用する多能性幹細胞を製造するためには、β細胞に分化可能な細胞を製造できればよい。使用目的に応じたレポーター遺伝子や特異的表面抗原に対する抗体を用いることで、目的の細胞を効率よく分離することが可能である。
【0015】
本発明の好ましい実施態様によれば、「多能性幹細胞」は、ES細胞と同等のすべての細胞系譜に分化可能な細胞とされる。このような細胞は、ヒトにおいては、SSEA−3、SSEA−4、TRA1−60、TRA1−81などのマーカー分子を発現する細胞である。マウスにおいては、SSEA−1などのマーカー分子を発現する細胞である。ヒトおよびマウスのそれらの細胞は、SCIDマウスやヌードマウスなどの免疫不全マウスの皮下に移植することにより、奇形腫を形成することが知られている。多能性幹細胞から形成された奇形腫には、内胚葉系、中胚葉系、外胚葉系、それぞれの系譜の分化細胞が含まれる。よって、ES細胞と同等のすべての細胞系譜に分化可能な多能性幹細胞は、このような奇形腫の形成により特徴づけられる。
【0016】
本発明による多能性幹細胞の製造法は、IL−6シグナル伝達因子(IL6ST)刺激因子および少なくとも1種のサイトカインを含む細胞培養培地中で培養する工程(以下「工程(a)」という)を含んでなる。
【0017】
本発明において「IL6シグナル伝達因子(IL6ST)」とは、interleukin 6 signal transducerを意味し、gp130とも呼ばれている。IL6STはシグナル領域、細胞外領域、膜貫通領域、および細胞内領域から構成される全長918アミノ酸残基のタンパク質を意味する。IL6STについては、シグナルペプチドであるN末端から22番目のグリシン残基までが欠失してもIL6STを介したシグナル伝達は維持されることが知られている。IL−6と結合したIL−6Rは2分子のIL6STと結合し、IL6STはホモ二量体を形成する。ホモ二量体を形成したIL6STは、細胞内領域のチロシン残基がリン酸化され、シグナル伝達が活性化されることが知られている。IL6STは、IL−11Rとも結合し、シグナルを伝達することが知られている。また、IL6STは白血球遊走阻止因子レセプター(LIFR)、オンコスタチンMレセプター(OSMR)、毛様体神経栄養因子レセプター(CNTFR)、カルジオトロピンレセプター(CT−1R)などとも結合し、ヘテロ二量体を形成することにより活性化されることが知られている。
【0018】
本発明において「IL6ST刺激因子」とは、ヒトIL6STを介してシグナルを伝達する分子を意味し、より具体的には、IL6STと会合することによりIL6STの活性化を引き起こす分子をいう。IL6ST刺激因子としては、IL−6またはその変異体とIL−6受容体(以下「IL−6R」という)またはその変異体との融合タンパク質、IL−6R(好ましくは、可溶性IL−6R(sIL−6R))またはその変異体とIL−6またはその変異体との組み合わせ(特表平10−509040号)、IL−6とIL−6受容体のα鎖との複合体、IL6STに対してアゴニストとして作用するIL6STに対する抗体(Z. J. Gu et al., Leukemia, 14, pp.188-197, 2000)、インターロイキン−11(IL−11)、白血球遊走阻止因子(LIF)、オンコスタチンM、およびカルジオトロピンが挙げられる。
【0019】
本発明において「IL−6」とは、IL−6として一般的に知られる、4つのへリックスから構成される全長212アミノ酸残基のタンパク質を意味する。また、本発明において「IL−6の変異体」とは、置換、欠失、挿入、および付加から選択される1以上の改変を有し、かつIL6ST刺激活性を有するIL−6の変異体を意味する。IL−6については、シグナルペプチドであるN末端から28番目のアラニン残基までが欠失してもIL6ST刺激活性を維持することが知られている(WO00/01731)。また、IL−6の37番目のリジン残基のC末端側がプロテアーゼの消化作用を受けること、および28番目のアラニン残基から37番目のリジン残基までの10アミノ酸残基が欠失してもIL−6活性に支障がないことが知られている(WO00/01731)。従って、本発明においては、N末端が欠失し、かつIL6ST刺激活性を有する改変IL−6をIL−6変異体として用いてもよい。このようなIL−6変異体としては、28番目までのN末端配列が欠失したIL−6、37番目までのN末端配列が欠失したIL−6が挙げられる。
【0020】
本発明において、「IL−6R」とは、IL−6受容体として一般的に知られる、シグナル領域、細胞外領域、膜貫通領域、および細胞内領域から構成される全長468アミノ酸残基のタンパク質を意味する。また、本発明において「IL−6Rの変異体」とは、置換、欠失、挿入、および付加から選択される1以上の改変を有し、かつIL6ST刺激活性を有するIL−6Rの変異体を意味する。IL−6Rについては、細胞外領域中に存在するサイトカインレセプター領域(112番目のバリン残基付近から323番目のアラニン残基付近までの領域)がIL−6との結合に必要十分であることが知られている。従って、本発明においては、IL−6Rの112番目のバリン残基付近から333番目のアラニン残基付近までの領域を含んでなる改変IL−6RをIL−6R変異体として用いてもよい。
【0021】
本発明の好ましい実施態様によれば、IL6ST刺激因子は、IL−6またはその変異体とIL−6Rまたはその変異体とが直接またはリンカーを介して連結された融合タンパク質とされる。このような融合タンパク質は、当技術分野において公知の方法、例えば、WO00/01731、WO99/02552、特表2000−506014号、またはFischer et al., Nature Biotech., 15, pp.142-145, 1997に記載の方法に従って製造することができる。
【0022】
本発明の特に好ましい実施態様によれば、前記融合タンパク質は、IL−6Rの112番目のバリン残基から333番目のアラニン残基までの断片のC末端と、IL−6の38番目のアスパラギン酸残基から212番目のメチオニン残基までの断片のN末端とが直接連結されたタンパク質(以下「FP6」という)とされる。この場合、この融合タンパク質をコードするDNA配列を宿主において発現可能なように連結したベクターを適当な宿主に導入し、形質転換された宿主を培養し、培養物からFP6を採取することにより融合タンパク質FP6を製造することができる(WO00/01731参照)。
【0023】
IL6ST刺激因子の細胞培養培地への添加量は特に制限されないが、好ましくは1ng/ml〜100μg/ml、より好ましくは10ng/ml〜20ng/mlとされる。
【0024】
本発明による多能性幹細胞の製造法において、IL−6ST刺激因子の使用は一つの主要な特徴である。特定の理論に拘束されるわけではないが、IL6STの活性化は、例えば、JAKキナーゼの活性化、STAT3のリン酸化、リン酸化によりホモ二量体を形成したSTAT3によるJunB、JABなどの遺伝子の転写促進を順次引き起こし、結果として細胞増殖、細胞分化調節等を生じることが考えられる。よって、本発明においては、IL6ST刺激因子に替えて、活性化型JAKキナーゼ、リン酸化型STAT3、ホモ二量体を形成させる変異を導入したSTAT3C(J.F.Bromberg et. al., Cell, 98, pp-295-303, 1999)、エストロゲン類縁体により誘導性にホモ二量体を形成させるSTAT3ER(T.Matsuda et al., EMBO Journal, 18, pp-4261-4269, 1999)、JAKキナーゼの阻害因子であるSOCS分子の阻害剤などを、遺伝子またはタンパク質の形で細胞内に導入することも考えられる。
【0025】
工程(a)において使用される「サイトカイン類」としては、トロンボポエチン(TPO)およびその変異体ならびにそれらの誘導体、c−mplリガンド(TPOを除く)およびその誘導体、幹細胞因子(SCF)、Flt−3リガンド(FL)、インターロイキン−3(IL−3)、インターロイキン−6(IL−6)、顆粒球コロニー刺激因子(G−CSF)、顆粒球マクロファージ・コロニー刺激因子(GM−CSF)、マクロファージ・コロニー刺激因子(M−CSF)、インターロイキン−7(IL−7)のような増殖因子;抗ヒトCD40抗体、抗ヒトCD3抗体、抗ヒトCD28抗体のような増殖刺激活性を有する抗体;リポ多糖(LPS)のような増殖刺激因子;およびこれらの組み合わせが挙げられる。
【0026】
本発明において「トロンボポエチン」(TPO)とは、全長332アミノ酸残基のアミノ酸配列からなる、血小板産生を特異的に刺激または増大させる活性を有するタンパク質をいう(WO95/21919)。また、本発明において「TPOの変異体」とは、置換、欠失、挿入、および付加から選択される1以上の改変を有し、かつ血小板産生を特異的に刺激または増大させるTPOの変異体を意味する。TPOの変異体としては、例えば、WO95/18858、特開平8−277296号公報、特開平8−228781号公報、およびWO95/21919等に記載されているものが挙げられ、好ましくは、アミノ酸残基1番目から163番目までのアミノ酸配列からなるTPOの変異体(断片)が挙げられる。
【0027】
本発明において「TPOおよびその変異体の誘導体」とは、水溶性ポリマーを連結してなるTPOおよびその変異体を意味する。水溶性ポリマーとしては、ポリエチレングリコール、好ましくは平均分子量が約5kDa〜約50kDaであるポリエチレングリコールが挙げられる。TPOおよびその変異体のポリエチレングリコール化(PEG化)は周知の方法に従って実施することができる(例えば、Focus on Growth Factors, 3, pp. 4-10, 1992参照)。水溶性ポリマー対「TPOおよびその変異体」タンパク質のモル比は1:1〜100:1とすることができ、ポリPEG化の場合には1:1〜20:1、モノPEG化の場合には1:1〜5:1とすることができる。
【0028】
本発明において「c−mplリガンド」とは、mplレセプターに結合可能なペプチド性リガンド、タンパク質性リガンド、および非ペプチド性リガンドを意味する。タンパク質性c−mplリガンド(TPOを除く)としては、巨核球の刺激剤としてのアゴニスト抗体(WO99/03495およびWO99/10494)や造血レセプターアゴニストなどの他のタンパク質( WO96/23888 、WO97/12978、WO97/12985、WO98/17810、WO96/34016およびWO00/24770)が挙げられる。ペプチド性c−mplリガンドとしては、WO96/40189、WO96/40750、WO98/25965、特開平10−72492号、WO99/42127、およびWO00/24770に記載されているものが挙げられる。非ペプチド性c−mplリガンドとしては、ベンゾジアゼピン誘導体(特開平11−1477号および特開平11−152276号)や他の低分子リガンド(WO99/11262、WO99/22733、WO99/22734、WO00/35446およびWO00/28987)が挙げられる。
【0029】
本発明において「c−mplリガンドの誘導体」とは、水溶性ポリマーを連結してなるc−mplリガンドおよびその変異体を意味する。水溶性ポリマーとしては、ポリエチレングリコール、好ましくは平均分子量が約5kDa〜約50kDaであるポリエチレングリコールが挙げられる。c−mplリガンドのポリエチレングリコール化(PEG化)は周知の方法に従って実施することができる(例えば、Focus on Growth Factors, 3, pp. 4-10, 1992参照)。水溶性ポリマー対c−mplリガンドタンパク質のモル比は1:1〜100:1とすることができ、ポリPEG化の場合には1:1〜20:1、モノPEG化の場合には1:1〜5:1とすることができる。
【0030】
本発明において「幹細胞因子」(SCF:stem cell factorまたはsteel factor)とは、造血および生殖発生に重要な機能を果たす、全長273アミノ酸残基のアミノ酸配列からなるタンパク質をいう。また、本発明において「SCFの変異体」とは、置換、欠失、挿入および付加から選択される1以上の改変を有し、かつSCFのレセプターであるc−kit分子を特異的に刺激して活性化させるSCFの変異体を意味する。SCFについては、シグナルペプチドであるN末端から25番目のスレオニン残基までが欠失しても活性を保持していることが知られている。SCFは、N末端から189番目のアラニン残基と190番目のアラニン残基との間で、あるいは190番目のアラニン残基と191番目のセリン残基との間でタンパク質分解酵素により消化されることにより可溶化型SCFとして存在することが知られている。また、複数のオルタナティブフォームが存在することが知られており、N末端から175番目のセリン残基から202番目のイソロイシン残基までを欠失した膜結合型のSCFが存在することが知られている。本発明においては、これらの変異体のいずれもが「SCFの変異体」として使用可能である。
【0031】
本発明において「IL−3」は、造血幹・前駆細胞に作用し、コロニー形成を支持する活性を持つタンパク質である。また、IL−3は、肥満細胞、好酸球、好塩基球、好中球、単球などの分化した細胞の増殖因子として重要な機能を有する。IL−3は、全長152アミノ酸残基のアミノ酸配列からなる。また、本発明において「IL−3の変異体」とは、置換、欠失、挿入および付加から選択される1以上の改変を有し、かつIL−3受容体を特異的に刺激して活性化させるIL−3の変異体を意味する。IL−3については、シグナルペプチドであるN末端から19番目のグルタミン残基までが欠失しても活性を保持していることが知られている。本発明においては、このような変異体を「IL−3の変異体」として使用することができる。
【0032】
本発明において「Flt−3リガンド」(FL)は、造血幹・前駆細胞の増殖因子として重要な機能を持つタンパク質である。FLは全長235アミノ酸残基のアミノ酸配列からなる膜結合蛋白質である。また、本発明において「FLの変異体」とは、置換、欠失、挿入および付加から選択される1以上の改変を有し、かつFlt−3チロシンキナーゼ受容体を特異的に刺激して活性化させるFLの変異体を意味し、好ましくはアミノ酸残基1番目から185番目のプロリン残基までのアミノ酸配列からなるFLの変異体(断片)が挙げられる。FLについては、シグナルペプチドであるN末端から26番目のグリシン残基までが欠失しても活性を保持していることが知られている。本発明においては、これらの変異体のいずれもが「FLの変異体」として使用可能である。
【0033】
本発明の好ましい実施態様によれば、サイトカインは、幹細胞因子(SCF)、トロンボポエチン(TPO)およびその変異体ならびにそれらの誘導体、Flt−3リガンド(FL)、インターロイキン−3(IL−3)およびその変異体、ならびにこれらの2以上の組合わせから選択され、より好ましくは、SCF、TPO、IL−3およびFlt−3リガンド、ならびにこれらの2以上の組合せから選択される。本発明の特に好ましい実施態様によれば、サイトカインは、SCF、TPOおよびIL−3の組合せ、またはSCF、TPO、IL−3およびFlt−3リガンドの組合せとされる。
【0034】
工程(a)において用いられる細胞培養培地は、例えば、下記の群:顆粒球コロニー刺激因子(G−CSF)、顆粒球マクロファージ・コロニー刺激因子(GM−CSF)、インターロイキン−7(IL−7)のような増殖因子;抗ヒトCD40抗体、抗ヒトCD3抗体、抗ヒトCD28抗体のような増殖刺激活性を有する抗体;リポ多糖(LPS)のような増殖刺激因子からなる群から選ばれる1種以上の増殖刺激因子をさらに含むものとしてもよい。これらの因子によるIL6STの発現亢進により、多能性幹細胞の誘導がより効率化する場合もある。
【0035】
これらサイトカインの細胞培養培地への添加量は特に制限されないが、好ましくは1ng/ml〜100μg/ml、より好ましくは10ng/ml〜100ng/mlとされる。
【0036】
培養に用いる基礎培地としては、血球細胞などの体細胞および多能性細胞の増殖および生存が害されない限り特に制限されないが、例えば、D−MEM培地、D−MEM/F12培地、MEM−α培地、Opti−MEM培地、IMDM培地、RPMI1640培地、StemPro培地(以上、Invitrogen社)、mTeSR 1培地(StemCell Technologies社)、HEScGRO培地(Millipore社)、N2B27培地、が好適に用いられる。培養温度は、通常25〜39℃、好ましくは33〜39℃である。また、培地に添加する物質としては、ウシ胎児血清、ヒト血清、ノックアウト血清リプレースメント(KnockOutSerumReplacement(KSR):Invitrogen社)、インシュリン、トランスフェリン、ラクトフェリン、エタノールアミン、亜セレン酸ナトリウム、モノチオグリセロール、2−メルカプトエタノール、ウシ血清アルブミン、ピルビン酸ナトリウム、ポリエチレングリコール、各種ビタミン、各種アミノ酸が挙げられる。CO2は、通常4〜15%であり、5〜10%が好ましい。O2は通常5〜25%である。また、培養にはトロンボポエチン(TPO)およびその変異体およびそれらの誘導体、c−mplリガンド(TPOを除く)およびその誘導体、幹細胞因子(SCF)、Flt−3リガンド(FL)、インターロイキン−3(IL−3)、インターロイキン−6(IL−6)、顆粒球コロニー刺激因子(G−CSF)、顆粒球マクロファージ・コロニー刺激因子(GM−CSF)、マクロファージ・コロニー刺激因子(M−CSF)、インターロイキン−7(IL−7)のような増殖因子;抗ヒトCD40抗体、抗ヒトCD3抗体、抗ヒトCD28抗体のような増殖刺激活性を有する抗体;リポ多糖(LPS)のような増殖刺激因子などのサイトカイン類の他に、トランスフォーミング成長因子−β(TGF−β)、骨形成因子(BMP)ファミリー、ヘッジホッグ因子(Hh)ファミリー、bFGFなどに代表される増殖因子、MIP−1α、SDF−1などのCCケモカインあるいはCXCケモカイン、エリスロポエチン(EPO)のような造血ホルモン、Wntファミリー遺伝子産物、Notchリガンドファミリー遺伝子産物、塩化リチウム、ピペコリン酸、γ―アミノ酪酸(GABA)などの化合物を10ng/mlから100μg/mlの範囲で添加することもできる。
【0037】
本発明における培養では、培養用のシャーレ、フラスコ、培養バックを用いた培養が可能であるが、培地組成、pHなどを機械的に制御し、高密度で培養が可能なバイオリアクターによって、その培養を改善することもできる。
【0038】
培養に用いるフィーダー細胞としては、マウス胚性線維芽細胞(MEF)、SNL細胞(A.P.McMahon and A.Bradley, Cell, 62, pp-1073-1085, 1990)、STO細胞などを用いることができる。フィーダー細胞の増殖を抑える方法としては、マイトマイシンC処理や放射線照射などの方法を用いることができる。また、フィーダー細胞に替えて、BDマトリゲル(BDバイオサイエンス社)などのマトリゲル、あるいはコラーゲン、ラミニン、フィブロネクチンなどの細胞外マトリックス蛋白質、あるいはゼラチンなどにより培養基材をコーティングする方法を用いることもできる。
【0039】
本発明による多能性幹細胞の製造法は、体細胞を脱分化させる工程(以下「工程(b)」という)をさらに含んでなる。この工程(b)は、工程(a)の後に行ってもよいし、または工程(a)と同時に行ってもよい。
【0040】
工程(a)と工程(b)が同時に行われる実施態様では、上述のIL6ST刺激因子およびサイトカインを含む上記細胞培養培地中で体細胞の脱分化が行われる。すなわち、工程(a)と工程(b)が同時に開始される。この実施態様では、脱分化の過程で培地を他の培地、例えばES細胞培養用の培地に変更することもできる。すなわち、この実施態様では、工程(a)のみを先に終了させ、工程(b)のみを継続してもよい。
【0041】
工程(a)の後に工程(b)が行われる実施態様では、上述のIL6ST刺激因子およびサイトカインを含む上記細胞培養培地中での体細胞の培養が行われ、その後、体細胞の脱分化が行われる。ここで、体細胞の脱分化は工程(a)で用いられる培地と同じ培地を用いて行うことができる。すなわち、この実施態様では、工程(a)は工程(b)よりも先に開始されるが、工程(b)を開始するときに、工程(a)は必ずしも終了していなくてもよい。さらに、この実施態様では、脱分化の過程で培地を他の培地、例えばES細胞培養用の培地に変更することもできる。すなわち、この実施態様では、工程(a)のみを先に終了させ、工程(b)のみを継続してもよい。
【0042】
本発明において「脱分化」とは、分化した組織や器官の細胞が、より未分化性の高い状態に戻ることを意味し、言い換えれば、分化とは逆のプロセスをいう。工程(b)における体細胞の脱分化は、当技術分野において公知の方法により行うことができる。
【0043】
本発明の好ましい実施態様によれば、工程(b)における体細胞の脱分化は、該体細胞の核初期化処理によって行われる。ここで、「核初期化」とは、体細胞の核が受精卵の状態に戻ることをいう。このような核初期化処理は、当技術分野において公知の方法、例えば、WO2007/069666に記載の方法に従って行うことができる。
【0044】
核初期化処理に用いられる核初期化因子としては、Octファミリー遺伝子(例えばOct3/4遺伝子)、Klfファミリー遺伝子(例えばKlf4遺伝子)、Soxファミリー遺伝子(例えばSox2遺伝子)およびMycファミリー遺伝子(例えばc−Myc遺伝子)の各遺伝子産物が知られている(WO2007/069666)。従って、核初期化処理は、体細胞をこれらの遺伝子産物に接触させることによって行うことができる。あるいは、核初期化処理は、体細胞において、これらの遺伝子を活性化または発現させることによって行うこともできる。本発明の好ましい実施態様によれば、体細胞の核初期化処理は、Octファミリー遺伝子(例えばOct3/4遺伝子)、Klfファミリー遺伝子(例えばKlf4遺伝子)、Soxファミリー遺伝子(例えばSox2遺伝子)およびMycファミリー遺伝子(例えばc−Myc遺伝子)を、発現可能な形で体細胞に導入することを含む。
【0045】
核初期化処理に用いられる培地は、例えば、下記の群:顆粒球コロニー刺激因子(G−CSF)、顆粒球マクロファージ・コロニー刺激因子(GM−CSF)、インターロイキン−7(IL−7)のような増殖因子;抗ヒトCD40抗体、抗ヒトCD3抗体、抗ヒトCD28抗体のような増殖刺激活性を有する抗体;リポ多糖(LPS)のような増殖刺激因子からなる群から選ばれる1種以上の増殖刺激因子を含むものとしてもよい。これらの因子による核初期化遺伝子ファミリーの発現亢進により、多能性幹細胞の誘導がより効率化する場合もある。
【0046】
核初期化処理に用いられる核初期化因子としては、上記の遺伝子に替えて、サイトカイン類、ケモカイン類、増殖因子類、低分子化合物、マイクロRNAなどのリボ核酸等を用いてもよい。このような核初期化因子としては、例えば、トランスフォーミング成長因子−β(TGF−β)、骨形成因子(BMP)ファミリー、ヘッジホッグ因子(Hh)ファミリー、bFGFなどに代表される増殖因子、MIP−1α、SDF−1などのCCケモカインあるいはCXCケモカイン、エリスロポエチン(EPO)のような造血ホルモン、Wntファミリー遺伝子産物、Notchリガンドファミリー遺伝子産物、塩化リチウム、ピペコリン酸、γ―アミノ酪酸(GABA)などの化合物、核初期化因子の発現を亢進させる低分子化合物、核初期化因子の転写調節に関わる因子のsiRNA等のマイクロRNA分子などが挙げられる。
【0047】
核初期化処理に用いられる核初期化因子の細胞導入法は特に制限されるものではなく、例えば、レトロウィルスベクター、レンチウィルスベクター、アデノ随伴ウィルスベクター、アデノウィルスベクター、単純ヘルペスウィルスベクター、ネコ内在ウィルスベクター、動物細胞用ベクター等を用いる方法が挙げられ、好ましくはレトロウィルスベクターまたはレンチウィルスベクターを用いる方法、より好ましくはレトロウィルスベクターを用いる方法が挙げられる。また、核初期化因子を細胞に注入する方法を用いてもよく、あるいは膜透過性の修飾を施すことにより各因子を細胞に添加する方法を用いてもよい。
【0048】
核初期化処理に用いられる遺伝子の細胞導入法は特に制限されるものではなく、動物細胞への遺伝子導入に用いられる一般的な方法、例えば、モロニーマウス白血病ウィルス等のレトロウィルスベクター、アデノウィルスベクター、アデノ随伴ウィルス(AAV)ベクター、単純ヘルペスウィルスベクター、レンチウィルスベクター等のウィルス由来の動物細胞用ベクターを用いる方法、リン酸カルシウム共沈法、DEAE−デキストラン法、エレクトロポレーション法、リポソーム法、リポフェクション法、マイクロインジェクション法、HVJリポソーム法等を用いることができる。本発明の好ましい実施態様によれば、核初期化処理に用いられる遺伝子の細胞導入法は、ウィルス由来の動物細胞用ベクター、より好ましくはレトロウィルスベクターを用いる方法とされる。
【0049】
本発明による多能性幹細胞の製造法において、出発材料として用いられる体細胞は特に制限されるものではなく、いかなる体細胞であってもよい。また、体細胞としては、採取された後に一旦凍結保存された細胞を用いてもよい。
【0050】
本発明の好ましい実施態様によれば、前記体細胞は血球細胞とされる。本発明において「血球細胞」とは、生体内の血流中に存在しうる血液細胞と同等の細胞群を意味し、必ずしも末梢血液細胞に限られるものではなく、例えば骨髄中や臍帯血中に存在するものであってもよい。より具体的には、「血球細胞」とは、CD45、CD11aなどの血液細胞のマーカー分子を発現する細胞をいう。また、本明細書において、骨髄中に存在することが確認されている血球細胞を用いることにより、他の組織細胞からよりも効率的に多能性幹細胞を樹立できることが見出されている。よって、本発明の好ましい実施態様によれば、前記血球細胞は骨髄由来単核球細胞とされる。さらに、本発明の他の好ましい実施態様によれば、前記血球細胞は分化血液細胞とされる。このような血球細胞は、採取された後に一旦凍結保存された細胞であってもよい。
【0051】
本発明の他の好ましい実施態様によれば、前記体細胞(特に血球細胞)は、IL6ST刺激因子と上記サイトカインとの存在下での培養により、水疱性口内炎ウィルスのGタンパク質を被覆タンパク質として持つレトロウィルスにより容易に感染する細胞とされる。このような細胞は、レトロウィルスベクターによって遺伝子を導入する上で有利である。
【0052】
本発明の他の好ましい実施態様によれば、前記体細胞(特に血球細胞)は、IL6STを高発現する細胞とされる。具体的には、IL6STはいずれの血球細胞においても発現しているが、中でも発現の高いT細胞、単球(Monocyte)、DC細胞、CD34陽性細胞などを用いることが考えられる。また、血球細胞以外でもIL6STを高発現する体細胞を用いることで、効率よく多能性幹細胞を製造することができる。具体的には、肝臓、腎臓、骨格筋、肺、すい臓、心臓、小腸、脊髄、唾液腺などの細胞も利用することが可能である。
【0053】
本発明の他の好ましい実施態様によれば、前記体細胞(特に血球細胞)は、工程(a)または(b)で用いられるサイトカインに対するレセプターを発現する細胞とされる。具体的には、c−kit(SCFレセプター)、c−mpl(TPOレセプター)、およびFlt3を発現する細胞として、CD34陽性細胞などを用いることが考えられる。また、IL−3Rα(IL−3レセプター)などを発現する細胞として、単球(Monocyte)などを用いることが考えられる。血球細胞以外でも、c−kitを発現する細胞などが挙げられる。
【0054】
本発明の他の好ましい実施態様によれば、前記体細胞(特に血球細胞)は、核初期化因子の少なくとも1種を発現する細胞とされる。例えば、核初期化因子の1つであるKlf4を発現する細胞としては、血球細胞の中では、単球(Monocyte)、DC細胞、CD34陽性細胞などが挙げられ、その他の組織では、肺、骨格筋、小腸などが挙げられる。Klf4以外についても、Klfファミリー遺伝子、Oct3/4ファミリー遺伝子、Mycファミリー遺伝子、またはSoxファミリー遺伝子を発現する体細胞を用いることができる。また、Oct3/4ファミリー遺伝子やNanog遺伝子などの初期化遺伝子については、それらの偽遺伝子を発現する細胞を体細胞として利用することもできる。
【0055】
本発明の他の好ましい実施態様によれば、前記体細胞(特に血球細胞)は、IL6ST刺激因子と上記のサイトカインとの存在下において核初期化因子の発現が上昇する細胞とされる。例えば、各サイトカインのレセプターであるc−kit(SCFレセプター)、c−mpl(TPOレセプター)、Flt3、IL−3Rα(IL−3レセプター)などを発現する細胞を用いることが考えられる。血球細胞の中では、CD34陽性細胞、DC細胞などが挙げられる。また、上記サイトカインによるこれらのレセプターへの刺激により、核初期化因子であるOct3/4ファミリー遺伝子、Klfファミリー遺伝子、Mycファミリー遺伝子、および/またはSoxファミリー遺伝子の発現が上昇する細胞を用いることにより、これらすべての初期化因子を用いずとも、多能性幹細胞の製造が可能となる。
【0056】
本発明の好ましい実施態様によれば、前記体細胞(特に血球細胞)はヒト細胞とされる。この実施態様では、本発明に用いられるIL6ST刺激因子、サイトカイン、核初期化因子またはその遺伝子は、全てヒト由来のものとすることが好ましい。これにより、ヒトの多能性幹細胞を効率よく製造することが可能となる。
【0057】
本発明に従って製造された細胞における脱分化または初期化の確認は、当技術分野において公知の方法によって行うことができる。このような方法としては、例えば、細胞表面に発現した未分化マーカーに対する抗体を用いて、蛍光励起細胞分離装置(FACS)を利用して解析する方法が挙げられる。具体的には、SSEA−3、SSEA−4、TRA1−60、TRA1−81などの未分化マーカーに対する抗体を利用することができる。また、NanogやOct3/4遺伝子といった未分化マーカーのレポーター遺伝子を細胞に導入して利用することもできる。
【0058】
本発明の工程(a)において用いられる、IL6ST刺激因子および少なくとも1種のサイトカインを含む細胞培養培地は、製造された多能性幹細胞の性質を維持するために利用することもできる。すなわち、IL6ST刺激因子と上記のサイトカイン類は、核初期化プロセスの促進のみならず、核初期化後の初期化状態の維持に利用することもできる。
【0059】
本発明による多能性幹細胞の製造法に用いられる試薬類をまとめて、キットとすることもできる。従って、本発明の他の態様によれば、体細胞から多能性幹細胞を製造するためのキットが提供され、該キットは、(a)IL−6シグナル伝達因子刺激因子および少なくとも1種のサイトカインを含む、体細胞を培養するための細胞培養培地、および(b)体細胞を脱分化させるための試薬を含んでなる。
【0060】
好適なIL−6シグナル伝達因子刺激因子および好適なサイトカインについては上述したとおりである。また、体細胞を脱分化させるための試薬についても上述したとおりであるが、好ましくは、Octファミリー遺伝子、klfファミリー遺伝子、Soxファミリー遺伝子、およびMycファミリー遺伝子を、発現可能な形で体細胞に導入するためのベクターを含む。本発明によるキットはさらに、多能性幹細胞の製造法に用いられる具体的方法に応じて、試薬類、反応容器、説明書等を含んでいてもよい。
【0061】
本発明に従って製造された多能性幹細胞は、各種分化細胞に誘導することが可能であるため、細胞再生医療に利用することができる。すなわち、この多能性幹細胞は、患者本人の体細胞を用いた自家の細胞移植および細胞バンク等より入手した細胞を利用する同種の細胞移植の両者の移植系への利用が可能である。既に存在する血球細胞の大型細胞バンクからも多能性幹細胞の細胞バンクを構築することができる。
【0062】
また、本発明によれば、各種疾病の患者からの血球細胞検体より、疾病情報に対応した多能性幹細胞バンクを構築することが可能である。さらに、健常人由来の血球細胞を初期化した多能性幹細胞を用いて各種分化細胞を誘導することにより、あるいは難治性疾患の患者より入手した血球細胞を用いて疾患のターゲット細胞を誘導することにより、創薬スクリーニングのツールとしての各種ヒトモデル細胞系の構築も可能である。
【実施例】
【0063】
以下、実施例を挙げて本発明をさらに具体的に説明するが、本発明はこれらに限定されるものではない。
【0064】
例1:ヒト骨髄由来単核球細胞からの多能性細胞の誘導(1)
A.ヒト骨髄由来単核球細胞の培養
凍結保存されたヒト骨髄由来単核球細胞(hBMMNCs)2.5×106細胞(Allcells社)を100mmペトリディッシュ2枚に播種し、37℃、5% CO2下、10%FBS入りDMEM培地で培養を開始した。一方のディッシュには、ヒトSCF(100ng/ml)、ヒトTPO(100ng/ml)、およびヒトIL−3(100ng/ml)を添加し、他方のディッシュにはSCF、TPOおよびIL−3に加えてFP6(20ng/ml)を添加した。ここで、SCF、TPOおよびIL−3からなるサイトカインカクテルを「ST3」と呼び、ST3にFP6を加えたカクテルを「ST3FP6」と呼ぶ(以下同様)。培養開始24時間後に、それぞれのディッシュにそれぞれのサイトカインカクテルを同濃度で再び添加した。培養開始48時間後にそれぞれの細胞を回収し、10%FBS入りDMEM培地500μlで懸濁した。ST3存在下で培養した細胞懸濁液にはST3を各300ng/mlの濃度で添加し、ST3FP6存在下で培養した細胞懸濁液には、ST3(各300ng/ml)に加えてFP6を60ng/mlの濃度で添加した。調製したhBMMNCsを次のウィルス感染実験に用いた。
【0065】
B.核初期化遺伝子を含む組換えレトロウィルスベクターの作製
コラーゲンタイプIコート100mmディッシュ12枚に、それぞれ4×106個のG3T−hi細胞(タカラバイオ社)を播種し、37℃、5% CO2下、10% FBS入りDMEM培地で培養した。培養24時間後に、核初期化遺伝子1種類についてディッシュ3枚を用いてウィルスベクターの遺伝子導入を行った。各ディッシュにつき、pGP vector(タカラバイオ社)6μg、pVSV−G vector(CLONTECH社)6μg、およびレトロウィルスベクターpMXs(T. Kitamura et al., Experimental Hematology, 31, pp.1007-1014, 2003;東京大学北村俊雄教授より供与)に核初期化遺伝子を挿入したプラスミド12μgを、遺伝子導入試薬FuGene6(ロシュダイグノスティック社)を用いて共導入した。用いた核初期化遺伝子は、ヒトOct3/4、ヒトSox2、ヒトKlf4、およびヒトc−mycの4種類であった。核初期化遺伝子発現レトロウィルスベクター:pMXshOct3/4、pMXshSox2、pMXshKlf4、およびpMXshc−mycの構築は、山中らの方法に従って行った(K. Takahashi, S. Yamanaka, Cell, 131, pp.861-872, 2007)。遺伝子導入の約48時間後に、核初期化遺伝子の種類ごとに培養液を回収し、培養液を0.45μmフィルターでろ過後、5800×g、4℃で4時間遠心し、培養上清を捨てることでウィルス沈殿を取得した。ウィルス沈殿は全て1つにまとめ、2mlの10%FBS入りDMEM培地で懸濁した。最終的に液量は2.4mlほどになった。
【0066】
C.ヒト骨髄由来単核球細胞のウィルス感染処理
予め50μg/mlのレトロネクチン(タカラバイオ社)でコートされた12ウェルプレートへ、懸濁したウィルス沈殿を1ウェルにつき1.2mlずつ添加し、1080×g、20℃で2時間遠心した。遠心後のプレートに前記のhBMMNCsの細胞懸濁液500μlを各ウェルに添加し、37℃、5% CO2にて培養を開始した。培養開始約24時間後に、それぞれにサイトカインを再び添加した。
【0067】
ウィルス感染開始から約48時間後にトリプシンEDTA液(GIBCO社)で各ウェルの細胞を回収し、予め5×105個/ディッシュのマウス胚性線維芽細胞をフィーダー細胞としたゼラチンコート100mmディッシュに10%FBS入りDMEM培地を用いて細胞を播き直した。それぞれのディッシュにはサイトカインを再び添加した。さらに、約24時間後および約72時間後に、サイトカイン入りの10%FBS入りDMEM培地で培地交換を実施した。その後、ウィルス感染開始から6日後からは、ST3またはST3FP6を含まないヒトES細胞培養用の培地に交換した。用いた培地の組成は、DMEM−F12培地(SIGMA社)500ml、非必須アミノ酸液(SIGMA社)5ml、200mM L−Glutamine(SIGMA社)6.25ml、KNOCKOUTTM Serum Replacement(KSR)(Invitorgen社)125ml、2−メルカプトエタノール(SIGMA社)5μl、5N NaOH 638μl、Human bFGF(UBI社)終濃度5ng/mlであった。
【0068】
その後、毎日培地交換を実施した。ウィルス感染開始から10日後には、両方のディッシュでいくつかのコロニーを確認した(図1B)。この時点でST3FP6存在下で培養したhBMMNCsの場合、比較的大きな扁平コロニーを含んでいたのに対し、ST3存在下で培養したhBMMNCsの場合は出現したコロニーはまだ小さく、マウスES細胞に似た形態のコロニーであった。15日後には各24個のコロニーをピックアップし、残りの細胞を予めフィーダー細胞を播いたゼラチンコート100mmディッシュに再播種した。20日後に再びコロニーをピックアップし、残りの細胞の半分を播種し、さらに残りの半分の細胞について、FACSによりヒトES細胞マーカーSSEA−4の発現確認を行った(図1A)。FACSによる解析では、回収した細胞をstaining medium(SM)(5% FBS、0.05% NaN3、および0.5mM EDTAを含むPBS)3〜5mlで洗浄後、フィコエリスリン結合抗SSEA4抗体(BDバイオサイエンス社)5μlを添加し、氷中で15分静置した。SM6mlで洗浄後、死細胞を検出するための試薬ヨウ化プロピジウム(PI)1μg/mlを含むSM液1mlで再懸濁し、FACS Calibur(BDバイオサイエンス社)またはFACS Aria SORP(BDバイオサイエンス社)を用いて測定を行った。その結果、ST3FP6存在下で培養したhBMMNCsを用いた場合、レトロウィルス感染20日後の時点で、幹細胞マーカーであるSSEA−4を発現する細胞が約40%に達し、FP6を添加しなかった場合の約15%に比して2.5倍以上に達していることが示された。また、ヒトES細胞においてはアルカリフォスファターゼが発現していることが知られている。そこで、27日後の時点で、常法に従ってアルカリフォスファターゼ活性染色を行った(図1C)。その結果、この時点でのコロニーのほとんどがアルカリフォスファターゼ陽性であることを確認した。以上より、ST3FP6存在下で培養したhBMMNCsを用いた場合、多能性幹細胞の出現時期、細胞数において、ST3存在下で培養したhBMMNCsを用いた場合を顕著に上回っていることが明らかとなった。
【0069】
例2:ヒト骨髄由来単核球細胞からの多能性細胞の誘導(2)
A.ヒト骨髄由来単核球細胞の培養
凍結保存されたヒト骨髄由来単核球細胞(hBMMNCs)2.5×106細胞(Allcells社)を100mmペトリディッシュ1枚に播種し、37℃、5% CO2下、10%FBS入りDMEM培地で培養を開始した。培地には、ヒトSCF(100ng/ml)、ヒトTPO(100ng/ml)、ヒトIL−3(100ng/ml)、ヒトFlt3リガンド(100ng/ml)、およびFP6(20ng/ml)を添加した。ここで、SCF、TPO、IL−3、Flt3リガンド、およびFP6からなるサイトカインカクテルを「ST3FLFP6」と呼ぶ(以下同様)。培養開始24時間後に、ST3FLFP6を同濃度で再び添加した。培養開始48時間後に細胞を回収し、総細胞数5×105個の細胞を10%FBS入りDMEM培地500μlで懸濁した。細胞懸濁液には、ST3FL(各300ng/ml)に加え、FP6を60ng/mlの濃度で添加した。調製したhBMMNCsを次のウィルス感染実験に用いた。
【0070】
B.核初期化遺伝子を含む組換えレトロウィルスベクターの作製
コラーゲンタイプIコート100mmディッシュ16枚に、それぞれ2.5×106個のG3T−hi細胞(タカラバイオ社)を播種し、37℃、5% CO2下、10% FBS入りDMEM培地で培養した。培養24時間後に、核初期化遺伝子1種類についてディッシュ4枚を用いてウィルスベクターの遺伝子導入を行った。各ディッシュにつき、pGP vector(タカラバイオ社)6μg、pVSV−G vector(CLONTECH社)6μg、およびレトロウィルスベクターpMXsに核初期化遺伝子を挿入したプラスミド12μgを、遺伝子導入試薬FuGene6(ロシュダイグノスティック社)を用いて共導入した。用いた核初期化遺伝子は、ヒトOct3/4、ヒトSox2、ヒトKlf4、ヒトc−mycの4種類であった。遺伝子導入の約48時間後に、核初期化遺伝子の種類ごとに培養液を回収し、培養液を0.75μmフィルターでろ過後、5000×g、4℃で4時間遠心し、培養上清を捨てることでウィルス沈殿を取得した。ウィルス沈殿は全て1つにまとめ、500μlの10%FBS入りDMEM培地を加えて再懸濁したところ、最終的に液量は1mlほどになった。
【0071】
C.ヒト骨髄由来単核球細胞のウィルス感染処理
予め50μg/mlのレトロネクチン(タカラバイオ社)でコートされた12ウェルプレート中の1つのウェルに、懸濁したウィルス沈殿を添加し、1080×g、20℃で2時間遠心した。遠心後のプレートに前記のhBMMNCsの細胞懸濁液500μlを添加し、37℃、5% CO2にて培養を開始した。培養開始約24時間後に、ST3FLFP6入りの培地で培地交換を実施した。
【0072】
ウィルス感染開始から約48時間後にトリプシンEDTA液(GIBCO社)で細胞を回収し、予め5×105個/ディッシュのマウス胚性線維芽細胞をフィーダー細胞としたゼラチンコート100mmディッシュに、10%FBSおよびST3FLFP6を補充したDMEM培地を用いて細胞を播き直した。さらに、約48時間後に10%FBSおよびST3FLFP6を補充したDMEM培地で培地交換を実施した。その後、ウィルス感染開始から6日後からは、ST3FLFP6を含まないヒトES細胞培養用の培地(例1Cに記載)に交換した。
【0073】
その後、毎日培地交換を実施した。ウィルス感染開始から11日後には、両方のディッシュでいくつかのコロニーを確認した。18日後には各24個のコロニーをピックアップし、残りの細胞を予めフィーダー細胞を播いたゼラチンコート100mmディッシュに再播種した。22日後の細胞を回収し、例1の方法に倣い、FACSによりヒトES細胞マーカーであるSSEA−4の発現を解析した。その結果を図2に示す。
【0074】
例3:レトロウィルス感染性ヒト骨髄由来単核球細胞の解析
A.ヒト骨髄由来単核球細胞のレトロウィルス感染性の分析
凍結保存されたhBMMNCs(Allcells社)を用いて、レトロウィルスの感染効率についての検討を行った。例1の方法に倣い、核初期化因子発現pMXsベクターの代わりに、緑色蛍光タンパク質EGFPを発現するpMXsIRES−EGFP(pMXsIG)ベクター(T. Kitamura et al., Experimental Hematology, 31, pp.1007-1014, 2003;東京大学北村俊雄教授より供与)を用いて実施した。
【0075】
hBMMNCsの培養条件は以下の3種類で実施した:(i)サイトカインを添加せずに、解凍後の細胞を用いてレトロウィルス感染を行った;(ii)例1と同様に、10%FBS入りDMEM培地にヒトSCF(100ng/ml)、ヒトTPO(100ng/ml)、およびヒトIL−3(100ng/ml)を加えて(ST3)、前培養およびレトロウィルス感染を実施した;(iii)例1と同様に、10%FBS入りDMEM培地にヒトSCF(100ng/ml)、ヒトTPO(100ng/ml)、ヒトIL−3(100ng/ml)、およびFP6(20ng/ml)を加えて(ST3FP6)、前培養およびレトロウィルス感染を実施した。
【0076】
レトロウィルスの調製および感染方法は例1と同様に実施した。感染2日後に細胞を回収し、細胞をSM液で洗浄後、PI(1μg/ml)を含むSM液1mlで再懸濁し、FACS Calibur(BDバイオサイエンス社)を用いてFACS解析を行った(図3)。その結果、サイトカインを添加した培養を行わない(i)の場合、レトロウィルスに感染する細胞は全く検出できなかったのに対して、ST3を添加((ii))することにより、生細胞中約4%の細胞がレトロウィルスに感染するようになり、さらにFP6を加えてST3FP6を添加((iii))することにより、レトロウィルスに感染する細胞は約8%に増加した。一方で、本例にて使用したレトロウィルスをコントロール細胞のNIH3T3細胞に感染させたところ、99%の効率で感染が可能であったことから、レトロウィルスそのものの感染能力は十分であった。
【0077】
B.感染細胞の表面抗原マーカーの分析
次に、例1および例2と同様にST3、ST3FP6またはST3FLFP6存在下で培養したhBMMNCsに対して、pMXsIGベクターより作製したレトロウィルスを感染させた場合の、EGFP陽性の感染細胞について、各種表面抗原マーカーに対する抗体を用いて免疫染色を行い、FACSを用いて解析を行った。
【0078】
回収した細胞をSM液3〜5 mlで洗浄後、各種抗体5μlを添加し、氷中で15分静置した。SM6mlで洗浄後、PI(1μg/ml)を含むSM液1mlで再懸濁し、FACS Calibur(BDバイオサイエンス社)にて測定を行った。用いた抗体は、アロフィコシアニン結合抗ヒトCD45抗体、アロフィコシアニン結合抗ヒトCD34抗体、フィコエリスリン結合抗ヒトCD38抗体、アロフィコシアニン結合抗ヒトCD11b抗体(以上、BDバイオサイエンス社)、フィコエリスリン結合抗ヒトCD3抗体、フィコエリスリン結合抗ヒトCD117抗体、およびフィコエリスリン結合抗ヒトCD19抗体(以上、IMMUNOTECH社)であった。CD45抗原は、すべてのヒト白血球、すなわちリンパ球、好酸球、単球、好塩基球、および好中球の表面に発現する。CD34抗原は、最も未分化な造血幹細胞と造血前駆細胞に発現する。CD38抗原は活性化したTおよびBリンパ球、NK細胞、単球、形質細胞および胸腺髄質細胞に発現する。CD11b抗原は、骨髄単球系細胞にみられ、NK細胞、顆粒球、単球/マクロファージに強く発現する。CD3抗原は、成熟T細胞および胸腺細胞に発現する。CD117抗原はごく少数の正常骨髄細胞にしか発現していないが、肥満細胞にも発現する。CD19抗原は初期B細胞を含むすべての正常B細胞に発現するが、形質細胞に成熟すると消失する。
【0079】
解析の結果、レトロウィルス感染細胞のほぼ全てにおいて白血球のマーカーであるCD45は陽性であり、ST3ではほぼすべてがCD34陽性細胞であり、ST3FP6またはST3FLFP6では約60%がCD34陽性細胞であった。一方で、CD11b、CD3、CD19といったマーカーはいずれも陰性であった(図4)。
【0080】
例4:レトロウィルス感染性ヒト末梢血由来単核球細胞の解析
凍結保存されたヒト末梢血由来単核球細胞(hPBMNCs)(Allcells社)に対して、例3と同様にpMXsIGベクターを用いてレトロウィルスの感染実験を行った。サイトカインカクテルを用いてhPBMNCsを培養した場合の感染効率とEGFP陽性の感染細胞について、各種表面抗原マーカーに対する抗体を用いて免疫染色を行い、FACSを用いて感染細胞の同定を行った。
【0081】
hPBMNCsの培養には、10%FBS入りDMEM培地にヒトSCF(100ng/ml)、ヒトTPO(100ng/ml)、ヒトIL−3(100ng/ml)、FP6(20ng/ml)、Flt3リガンド(100ng/ml)、ヒトGM−CSF(100ng/ml)、ヒトM−CSF(50ng/ml)を添加して培養したGM培養系と、ヒトSCF(100ng/ml)、ヒトTPO(100ng/ml)、ヒトIL−3(100ng/ml)、FP6(20ng/ml)、LPS(1μg/ml)、抗ヒトCD40抗体 #341(キリンファーマ社)(100ng/ml)、抗ヒトCD3抗体(OKT3 ヤンセンファーマ)(1μg/ml)、抗ヒトCD28抗体(eBioscience)(1μg/ml)を添加して前培養したTB培養系の2通りの培養系を用いて、レトロウィルス感染を実施した。レトロウィルスの調製および感染方法は例1と同様に実施した。感染2日後に細胞を回収し、回収した細胞をSM液3〜5mlで洗浄後、各種抗体5μlを添加し、氷中で15分静置した。SM6mlで洗浄後、PI(1μg/ml)を含むSM液1mlで再懸濁し、FACS Calibur(BDバイオサイエンス社)にて測定を行った。
【0082】
用いた抗体は、アロフィコシアニン結合抗ヒトCD45抗体、アロフィコシアニン結合抗ヒトCD34抗体、フィコエリスリン結合抗ヒトCD38抗体、アロフィコシアニン結合抗ヒトCD11b抗体、フィコエリスリン結合抗ヒトCD13抗体(以上、BDバイオサイエンス社)、フィコエリスリン結合抗ヒトCD3抗体、フィコエリスリン結合抗ヒトCD117抗体、およびフィコエリスリン結合抗ヒトCD19抗体(以上、IMMUNOTECH社)であった。CD13抗原は、正常末梢血の好中球、好酸球、好塩基球および単球など、ほとんどの骨髄系細胞に発現する。
【0083】
解析の結果、レトロウィルス感染細胞のほぼ全てにおいて白血球のマーカーであるCD45は陽性であり、GM培養系では骨髄細胞を用いた場合と同様に、約半数がCD34陽性であった。TB培養系においてはT細胞のマーカーであるCD3抗原陽性であった(図5)。本系を用いることにより、末梢血中に存在するT細胞などから多能性細胞を誘導できる可能性が考えられる。
【0084】
例5:ヒト誘導多能性細胞クローンの表面抗原解析
例1および例2の結果クローニングした細胞株について、コロニー形態の観察およびFACSによりヒトES細胞マーカーSSEA−4の発現確認を行った。FACS解析の方法は例1と同様に行った。
【0085】
ST3の培養条件を用いて取得したクローン#16および#30、ST3FPの培養条件を用いて取得したクローン#22および#33、ST3FLFPの培養条件を用いて取得したクローン#1および#4について解析した結果を図6に示す。いずれのクローンも幹細胞マーカーであるSSEA−4を有意に発現していることを確認した。
【0086】
例6:ヒト誘導多能性細胞クローンのRT−PCR解析
例5で解析したクローンを含め、例1および例2の結果取得した多能性幹細胞クローンについて、幹細胞特異的遺伝子のmRNAの発現をRT−PCR法を用いて解析した。
【0087】
各クローン細胞をマウス胚性線維芽細胞のフィーダー細胞上に播種し、ヒトES細胞用培地にて培養した細胞よりRNAを回収した。用いた培地の組成は、DMEM−F12培地(SIGMA社)500ml、非必須アミノ酸液(SIGMA社)5ml、200mM L−Glutamine(SIGMA社)6.25ml、KNOCKOUTTM Serum Replacement(KSR)(Invitorgen社)125ml、2−メルカプトエタノール(SIGMA社)5μl、5N NaOH 638μl、Human bFGF(UBI社)終濃度5ng/mlであった。回収した細胞はPBS緩衝液で洗浄後、細胞沈殿を液体窒素中にて凍結した。コントロール細胞として、凍結融解後のBMMNCsをフィーダー細胞上で数時間培養した細胞およびフィーダー細胞(MEF細胞)を用意した。凍結後の細胞サンプルより、RNeasy Plus Mini Kit(QIAGEN社)を用いて、添付プロトコルに従って全RNAを調製した。調製したRNAを用いて、SuperScriptIII First Strand Synthesis System(Invitrogen社)により添付のプロトコルに従ってcDNAを合成した。合成したcDNAを鋳型として、LA Taq DNA polymerase(タカラバイオ社)を用いて、96℃−20秒、55℃−30秒、および72℃−30秒からなる工程を1サイクルとして35サイクルのPCR反応を実施した。PCR反応に用いたプライマーは山中らと同様のものを用いており(K. Takahashi, S. Yamanaka, Cell, 131, pp.861-872, 2007)、各プライマーの塩基配列を表1に示す。各PCR反応は各遺伝子についてフォワード(Fw)プライマーとリバース(Rv)プライマーとの組合せを用いて実施した。RT反応およびPCR反応のコントロールとして、ハウスキーピング遺伝子であるglyceraldehyde−3−phosphate dehydrogenase(GAPDH)遺伝子を用いた。PCR反応物をエチジウムブロマイド入りのアガロースゲルにて電気泳動し、UVトランスイルミネーターにより観察した結果を図7に示す。解析の結果、いずれのクローンも幹細胞マーカー遺伝子を発現していることを確認した。
【0088】
【表1】

【0089】
例7:マウス骨髄由来単核球細胞からの多能性細胞の誘導(1)
A.マウス骨髄由来単核球細胞の取得
未分化な多能性幹細胞において緑色蛍光タンパク質EGFPを発現することが知られているOct4−EGFPトランスジェニックマウス(10〜20週齢)(K. Ohbo et al., Dev. Biol., 258, pp.209-225, 2003; 横浜市立大学 大保和之准教授より供与)の大腿骨内の骨髄を採取し、PBSに懸濁した。定法に従い(高津聖志、免疫研究の基礎技術、羊土社1995)、骨髄から比重遠心法により単核球細胞画分を濃縮し、これをマウス骨髄単核球細胞(mBMMNCs)として用いた。
【0090】
B.核初期化遺伝子を含む組換えレトロウィルスベクターの作製
コラーゲンタイプIコート60mmディッシュ10枚にそれぞれ1×106個、および6ウェルプレートの各ウェルに1×105個のレトロウィルス用のパッケージング用細胞:PLAT−E細胞(S.Morita et al., Gene Therapy, 7, pp.1063-1066, 2000;東京大学北村俊雄教授より供与)を播種し、37℃、5% CO2下、10% FBS入りDMEM培地で培養した。培養24時間後に、マウス核初期化遺伝子1種類について60mmディッシュ2枚を用いてウィルスベクターの遺伝子導入を行った。各ディッシュにつき、レトロウィルスベクターpMXsに核初期化遺伝子を挿入したプラスミド3μgを遺伝子導入試薬FuGene6(ロシュダイグノスティック社)を用いて導入した。用いた核初期化遺伝子は、マウスOct3/4、マウスSox2、マウスKlf4、およびマウスc−mycの4種類であった。核初期化遺伝子発現レトロウィルスベクター、pMXsmOct3/4、pMXsmSox2、pMXsmKlf4、およびpMXsmc−mycの構築は、山中らの方法に従って行った(K. Takahashi, S. Yamanaka, Cell, 126, pp.663-676, 2006)。その他に、60mmディッシュ1枚と6ウェルプレートの3ウェル分において、上記4種類の遺伝子各3μgのDNAを併せて導入した。また、コントロールとして緑色蛍光タンパク質EGFPを発現するpMXsIGベクター3μgを60mmディッシュ1枚に導入した。遺伝子導入の約48時間後に、各核初期化遺伝子について60mmディッシュ1枚ずつを集めて0.22μmフィルターでろ過後、5800×g、4℃で4時間遠心し、培養上清を捨てた後に3mlのマウスES細胞用培地で再懸濁した濃縮ウィルスサンプル(i)を取得した。マウスES細胞用培地としては、20%FBS入りDMEM培地に1/100容の2-メルカプトエタノール(SIGMA社)、1/1000容のESGRO(CHEMICON社)を添加したものを用いた。また、各核初期化遺伝子について60mmディッシュ1枚ずつを集めて0.22μmフィルターでろ過しただけのものを4ウィルスサンプル(ii)とした。4種類の遺伝子を同時に導入した60mmディッシュ1枚と6ウェルプレートの3ウェル分の培養上清に
ついては、0.22μmフィルターでろ過し、4遺伝子サンプル(iii)とした。コントロールのpMXsIGベクターを導入した培養上清については、0.22μmフィルターでろ過したものをコントロールサンプル(iv)とした。
【0091】
C.マウス骨髄由来単核球細胞のウィルス感染処理
予め33μg/mlのレトロネクチン(タカラバイオ社)でコートされた12ウェルプレートの2ウェルずつに上記(i)〜(iv)のサンプルを添加し、1080×g、25℃で2時間遠心した。遠心後のプレートからウィルスサンプルを除去し、新たなマウスES細胞用培地を1mlずつ添加した。さらに、(i)〜(iv)の各サンプルの一方のウェルには、マウスSCF(100ng/ml)、ヒトTPO(100ng/ml)、およびマウスIL−3(100ng/ml)(ST3)を添加した。その後、各ウェルに前記のOct4−EGFPマウスより調製したmBMMNCsを1×105個/ウェルになるよう添加し、37℃、5% CO2にて培養を開始した。
【0092】
また、ウィルス感染前日に、Oct4EGFPマウス由来胚性線維芽細胞(MEF)を12ウェルプレートに2×104個/ウェルで播種して培養していた各ウェルについても、上記(i)〜(iv)のサンプルを1ウェルずつに添加し、1080×g、32℃で40分間遠心した後、37℃、5% CO2にて培養を開始した。
【0093】
ウィルス感染2日後のコントロールサンプルにおけるEGFP発現細胞の割合は、MEF細胞では34%、サイトカインなしのmBMMNCsでは55%、ST3存在下のmBMMNCsでは60%であった。
【0094】
4ウィルスサンプル((ii))を感染させたmBMMNCsについては、サイトカインの添加、非添加に関わらず、ウィルス感染開始から5日後にはOct4−EGFPレポーター遺伝子によるEGFP遺伝子の発現が認められた(図8A・B)。一方で、MEFについてはこの時点ではEGFP遺伝子発現細胞の存在は認められなかった(図8C)。ウィルス感染7日後に、mBMMNCs由来各サンプルについて、Oct4−EGFPレポーター遺伝子の発現と血球細胞マーカーであるCD45抗原の発現のFACS解析を実施した。この解析のために、回収した細胞をSM液3〜5 mlで洗浄後、アロフィコシアニン結合抗マウスCD45抗体(BDバイオサイエンス社)5μlを添加し、氷中で15分静置した。SM6mlで洗浄後、PI(1μg/ml)を含むSM液1mlで再懸濁し、FACS Calibur(BDバイオサイエンス社)にて測定を行った。その結果を図9および表2に示す。FACS解析前の細胞の様子を図10に示す。感染前のmBMMNCsはそのほぼ全てがCD45抗原陽性であるのに対し、この時点でのOct4−EGFP陽性細胞の多くは既にCD45抗原陰性であった。また同時に、FACS sortingによりEGFP陽性細胞の分取を行い、それらの細胞からクローニングを実施し、マウス骨髄由来誘導多能性幹細胞(mBMiPS)クローン#7および#13を取得した。取得した各クローンについてもFACS解析を実施した(図11)。
【0095】
【表2】
【0096】
FACSによる解析では、回収した細胞をSM液3〜5 mlで洗浄後、各種抗体5μlを添加し、氷中で15分静置した。SM6mlで洗浄後、PI(1μg/ml)を含むSM液1mlで再懸濁し、FACS Calibur(BDバイオサイエンス社)またはFACS Aria SORP(BDバイオサイエンス社)を用いて測定を行った。抗体としては、アロフィコシアニン結合抗マウスCD45抗体(BDバイオサイエンス社)、およびAlexa Fluor647結合抗マウスSSEA−1抗体を用いた。Alexa Fluor647結合抗マウスSSEA−1抗体は、抗マウスSSEA−1抗体(CHEMICON社)100μgをAlexa Fluor 647 Protein Labeling Kit(Invitrogen社)を用いて標識することにより調製した。
【0097】
例8:マウス骨髄由来単核球細胞からの多能性細胞の誘導(2)
A.核初期化遺伝子を含む組換えレトロウィルスベクターの作製
コラーゲンタイプIコート60mmディッシュ12枚にそれぞれ7×105個のPLAT−E細胞を播種し、37℃、5% CO2下、10% FBS入りDMEM培地で培養した。培養24時間後に、各種遺伝子導入を行った。12枚のうち4枚については、マウス核初期化遺伝子1種類について60mmディッシュ1枚を用いてウィルスベクターの遺伝子導入を行った。各ディッシュにつき、レトロウィルスベクターpMYs(T. Kitamura et al., Experimental Hematology, 31, pp.1007-1014, 2003;東京大学北村俊雄教授より供与)に核初期化遺伝子を挿入したプラスミド4μgを遺伝子導入試薬FuGene6(ロシュダイグノスティック社)を用いて導入した。用いた核初期化遺伝子は、ヒトOct3/4、ヒトSox2、ヒトKlf4、およびヒトc−mycの4種類であった。60mmディッシュ4枚について、上記4種類の遺伝子各4μgのDNAを併せて導入した。また、コントロールとして緑色蛍光タンパク質EGFPを発現するpMYsIRES−EGFP(pMYsIG)ベクター16μgを60mmディッシュ4枚に導入した。
【0098】
遺伝子導入の約48時間後に、各核初期化遺伝子を個別に導入した60mmディッシュ計4枚を集めて4ウィルスサンプルとし、4種同時に導入した計4枚を集めて4遺伝子サンプルとし、pMYsIGベクターを導入した計4枚を集めてコントロールサンプルとした。いずれのサンプルについても、0.22μmフィルターでろ過後、5800×g、4℃で4時間遠心し、培養上清を捨てた後に2.5mlのマウスES細胞用培地で再懸濁し、再び0.22μmフィルターでろ過を行った。
【0099】
B.細胞のウィルス感染処理
レトロネクチン(タカラバイオ社)でコートされた12ウェルプレートの2ウェルずつに4ウィルスサンプル、4遺伝子サンプル、およびコントロールサンプルをそれぞれ添加し、1080×g、25℃で2時間遠心した。遠心後のプレートからウィルスサンプルを除去し、各サンプルのウェルに対して、マウスES細胞用培地で懸濁したOct4−EGFPマウスBMMNCsまたはOct4−EGFPマウスMEF細胞を1×105個/ウェルになるよう添加し、37℃、5% CO2にて培養を開始した。
【0100】
また、ウィルス感染前日に、Oct4−EGFPマウスMEF細胞をゼラチンコート12ウェルプレートに2×104個/ウェルで播種して培養していた各ウェルについても、上記3種のサンプルを1ウェルずつに添加し、1080×g、25℃で40分間遠心した後、37℃、5% CO2にて培養を開始した。
【0101】
ウィルス感染開始2日後のコントロールサンプルにおけるEGFP発現細胞の割合は、レトロネクチンコートプレートで感染させたMEF細胞では95%、ゼラチンコートプレートで予め培養していたMEF細胞では65%、レトロネクチンコートプレートで感染させたmBMMNCsでは26%であった(図12)。その後、ウィルス感染開始から7日目にFACS sortingを行い(図13)、12日目に出現コロニー数を計測した。その結果を表3にまとめた。
【0102】
【表3】
【0103】
例9:マウス骨髄由来多能性細胞の多分化能解析
mBMiPSクローン#7および#13について分化多能性を評価するため、ヌードマウス(Balb/cAJcl−nu)の皮下への移植を行った。未分化ES細胞は生体内に移植されると奇形腫を形成することが知られている。コントロール細胞としてマウスES細胞E14株を使用した。ヌードマウス1匹あたり各細胞1×106個を100μl血清非含有DMEM培地で懸濁して皮下投与した。その結果、マウスES細胞を移植した場合と同様の大きさの奇形腫瘍がmBMiPS細胞を移植した全例でも形成された。移植後2週間後および4週間後の腫瘍を摘出し、ホルマリン固定により組織標本を作製した。組織学的な解析の結果、いずれのクローン由来の奇形腫においても、神経組織、軟骨組織、筋組織、脂肪組織、食道粘膜様組織、気管上皮などが認められたことから、mBMMNCsより誘導された各クローンが多能性を有することが証明された(図14)。
【0104】
さらにmBMiPSクローン#7細胞をBalb/cマウスの受精卵から作製した胚盤胞に注入後、偽妊娠マウスに移植することによりキメラ個体を作出することを試みた。その結果、Balb/cマウスの産仔は白色の毛色を示すはずのところ、mBMiPS細胞に由来する黒色の毛色を有するキメラマウスが誕生した(図15および表4)。毛色は全身の細胞の一部を反映するものであり、他の組織・臓器においてもmBMiPS細胞に由来する細胞が個体を構成しているものと考えられた。
【0105】
【表4】
【0106】
例10:マウス骨髄由来多能性細胞の発現プロファイル解析
mBMiPSクローン#7および#13について、各5×105個の細胞からRNAeasy mini plus kit (QIAGEN社)を用いてRNAを調製し、Agilent Expression Array (Agilent社)を用いて発現プロファイル解析を実施した。コントロールとしてマウスES細胞E14株およびマウスBMMNC細胞を使用した。その結果、図16に示すような各遺伝子の発現パターンを示し、mBMiPSクローン#7および#13がES細胞と酷似した細胞であることが明らかとなった。
【図面の簡単な説明】
【0107】
【図1】ヒト骨髄由来単核球細胞(hBMMNCs)から誘導された多能性幹細胞の未分化マーカーの発現量を評価したFACS解析の結果、誘導された多能性幹細胞が形成するコロニーの形態および誘導された多能性幹細胞のアルカリフォスファターゼの活性染色の結果を示した。[A]は hBMMNCsに対して核初期化因子を発現するレトロウィルスを感染させた後20日目の未分化細胞マーカーSSEA−4の発現を示した。図の水平軸がSSEA−4の発現量を示し、垂直軸が示すPIの染色強度が低いものが生細胞を示した。図中の等高線は解析した細胞の存在頻度を示した。「ST3FP6」はレトロウィルス感染前2日間および核初期化プロセスにおいて、サイトカインカクテルとしてSCF、TPO、IL−3、FP6をhBMMNCsと共存させていることを示し、「ST3」はレトロウィルス感染前2日間および核初期化プロセスにおいて、サイトカインカクテルとしてSCF、TPO、IL−3をhBMMNCsと共存させていることを示した。[B]は核初期化プロセスにおいて形成された細胞コロニーの形態を示した。「Day10」、「Day20」はレトロウィルス感染開始からの日数を示した。[C]はhBMMNCsから誘導された多能性幹細胞のレトロウィルス感染開始から27日目のアルカリフォスファターゼ染色を示した。
【図2】ヒト骨髄由来単核球細胞(hBMMNCs)から誘導された多能性幹細胞の未分化マーカーの発現量を評価したFACS解析の結果および誘導された多能性幹細胞が形成するコロニーの形態を示した。レトロウィルス感染前2日間および核初期化プロセスにおいて、サイトカインカクテルとしてSCF、TPO、IL−3、FP6、FltリガンドをhBMMNCsと共存させた場合を示した。[A]は hBMMNCsに対して核初期化因子を発現するレトロウィルスを感染させた後22日目の未分化細胞マーカーSSEA−4の発現を示した。図の水平軸がSSEA−4の発現量を示し、垂直軸が細胞数を示した。 [B]は核初期化プロセスにおいて形成された細胞コロニーの形態を示した。
【図3】pMXsIGベクターを用いて調製されたレトロウィルスのヒト骨髄由来単核球細胞(hBMMNCs)に対する感染効率を評価するためのFACS解析結果を示した。「PI」はヨウ化プロピジウムによる染色強度を示し、図中の等高線は解析した細胞の存在頻度を示した。各図の水平軸は感染したレトロウィルスゲノムから発現する緑色蛍光タンパク質EGFPの発現強度を示した。「サイトカイン培養なし」はレトロウィルス感染前後でサイトカイン類を共存させない場合を示した。「ST3添加培養」はレトロウィルス感染2日前より、FACS解析を行ったレトロウィルス感染2日後までの間、hBMMNCsとSCF、TPO、IL−3のサイトカインを共存させた場合を示した。「ST3FP6添加培養」はレトロウィルス感染2日前より、FACS解析を行ったレトロウィルス感染2日後までの間、hBMMNCsとSCF、TPO、IL−3、FP6のサイトカインを共存させた場合を示した。
【図4】pMXsIGベクターを用いて調製されたレトロウィルスが感染したヒト骨髄由来単核球細胞(hBMMNCs)の細胞系譜を同定評価するためのFACS解析結果を示した。レトロウィルス感染2日前よりFACS解析を行ったレトロウィルス感染2日後までの間、hBMMNCsを、SCF、TPO、およびIL−3サイトカインと共存させた場合をST3として示し、SCF、TPO、IL−3、およびFP6サイトカインと共存させた場合をST3FP6として示し、SCF、TPO、IL−3、FP6、およびFlt3リガンドと共存させた場合をST3FLFP6として示した。各図の水平軸は感染したレトロウィルスゲノムから発現する緑色蛍光タンパク質EGFPの発現強度を示し、図中の等高線は解析した細胞の存在頻度を示した。「CD45APC」はアロフィコシアニン結合抗ヒトCD45抗体による染色強度を示した。「CD34APC」はアロフィコシアニン結合抗ヒトCD34抗体による染色強度を示した。「CD38PE」はフィコエリスリン結合抗ヒトCD38抗体による染色強度を示した。「CD117PE」はフィコエリスリン結合抗ヒトCD117抗体による染色強度を示した。「CD11bAPC」はアロフィコシアニン結合抗ヒトCD11b抗体による染色強度を示した。「CD3PE」はフィコエリスリン結合抗ヒトCD3抗体による染色強度を示した。「CD19PE」はフィコエリスリン結合抗ヒトCD19抗体による染色強度を示した。
【図5】pMXsIGベクターを用いて調製されたレトロウィルスが感染したヒト末梢血由来単核球細胞(hPBMNCs)の細胞系譜を同定評価するためのFACS解析結果を示した。レトロウィルス感染2日前よりFACS解析を行ったレトロウィルス感染2日後までの間、hPBMNCsをヒトSCF、ヒトTPO、ヒトIL−3、FP6、Flt3リガンド、ヒトGM−CSF、ヒトM−CSFと共存させた場合をGM培養として示し、ヒトSCF、ヒトTPO、ヒトIL−3、FP6、LPS、抗ヒトCD40抗体、抗ヒトCD3抗体、抗ヒトCD28抗体の因子と共存させた場合をTB培養として示した。各図の水平軸は感染したレトロウィルスゲノムから発現する緑色蛍光タンパク質EGFPの発現強度を示し、図中の等高線は解析した細胞の存在頻度を示した。「CD45APC」はアロフィコシアニン結合抗ヒトCD45抗体による染色強度を示した。「CD34APC」はアロフィコシアニン結合抗ヒトCD34抗体による染色強度を示した。「CD38PE」はフィコエリスリン結合抗ヒトCD38抗体による染色強度を示した。「CD117PE」はフィコエリスリン結合抗ヒトCD117抗体による染色強度を示した。「CD11bAPC」はアロフィコシアニン結合抗ヒトCD11b抗体による染色強度を示した。「CD3PE」はフィコエリスリン結合抗ヒトCD3抗体による染色強度を示した。「CD19PE」はフィコエリスリン結合抗ヒトCD19抗体による染色強度を示した。「CD13PE」はフィコエリスリン結合抗ヒトCD13抗体による染色強度を示した。
【図6】クローニングしたヒト誘導多能性細胞株について、コロニー形態およびFACSによるヒトES細胞マーカーSSEA−4の発現解析結果を示した。図の水平軸がSSEA−4の発現量を示し、垂直軸が細胞数を示した。
【図7】クローニングしたヒト誘導多能性細胞株についてのRT−PCR解析結果を示した。図の水平方向に各細胞株を配置し、ST3、ST3FP6、ST3FLFP6は各細胞の誘導条件を示し、その下の数字が各クローン番号を示した。図の垂直方向には多能性幹細胞マーカーとして知られる遺伝子を配置し、コントロール遺伝子としてハウスキーピング遺伝子であるGAPDHの結果を示した。図の黒色のバンドが各遺伝子についての発現を示している。
【図8】Oct4−EGFPトランスジェニックマウス骨髄由来単核球細胞(mBMMNCs)あるいはOct4−EGFPトランスジェニックマウス由来の胚性線維芽細胞(MEF)から誘導された多能性幹細胞が形成するコロニーの形態および未分化性のマーカーであるOct4−EGFPレポーター遺伝子からのEGFPタンパク質の発現を示した。[A]はOct4−EGFPトランスジェニックマウス由来のmBMMNCsに対して核初期化因子を発現するレトロウィルスを感染させた後5日目の細胞の形態および未分化性マーカーOct4−EGFPの発現を示した。「4遺伝子サンプル」はレトロウィルスを調製する際に、4つの核初期化遺伝子を同時にパッケージング細胞へ導入してレトロウィルスを作製した場合を示した。「4ウィルスサンプル」はレトロウィルスを調製する際に、4つの核初期化遺伝子をそれぞれ個別にパッケージング細胞へ導入してレトロウィルスを作製した後に、レトロウィルスを混合し細胞に感染させた場合を示した。[B]はOct4−EGFPトランスジェニックマウス由来のmBMMNCsにレトロウィルスを感染させる際に、SCF、TPO、IL−3のサイトカインを共存させた場合の、レトロウィルス感染後5日目の細胞の形態および未分化性マーカーOct4−EGFPの発現を示した。 [C]はOct4−EGFPトランスジェニックマウス由来のMEFに対して核初期化因子を発現するレトロウィルスを感染させた後5日目の細胞の形態および未分化性マーカーOct4−EGFPの発現を示した。
【図9】Oct4−EGFPトランスジェニックマウスの骨髄由来単核球細胞(mBMMNCs)から誘導された多能性幹細胞について、未分化性のマーカーであるOct4−EGFPレポーター遺伝子からのEGFPタンパク質の発現と血球細胞のマーカーであるCD45抗原の発現をFACSにて解析した結果を示した。Oct4−EGFPトランスジェニックマウス由来のmBMMNCsに対して核初期化因子を発現するレトロウィルスを感染させた後7日目の解析を示した。「4遺伝子」はレトロウィルスを調製する際に、4つの核初期化遺伝子を同時にパッケージング細胞へ導入してレトロウィルスを作製した場合を示した。「4ウィルス」はレトロウィルスを調製する際に、4つの核初期化遺伝子をそれぞれ個別にパッケージング細胞へ導入してレトロウィルスを作製した後に、レトロウィルスを混合し細胞に感染させた場合を示した。「濃縮ウィルス」は「4ウィルス」と同様に調製したレトロウィルスを濃縮して感染に用いた場合を示した。各図の水平軸は感染したレトロウィルスゲノムから発現する緑色蛍光タンパク質EGFPの発現強度を示した。「CD45APC」はアロフィコシアニン結合抗ヒトCD45抗体による染色強度を示し、図中の等高線は解析した細胞の存在頻度を示した。
【図10】Oct4−EGFPトランスジェニックマウス骨髄由来単核球細胞(mBMMNCs)あるいはOct4−EGFPトランスジェニックマウス由来の胚性線維芽細胞(MEF)から誘導された多能性幹細胞が形成するコロニーの形態および未分化性のマーカーであるOct4−EGFPレポーター遺伝子からのEGFPタンパク質の発現を示した。Oct4−EGFPトランスジェニックマウス由来のmBMMNCsあるいはMEFに対して核初期化因子を発現するレトロウィルスを感染させた後7日目の様子を示した。「mBMMNCs」はOct4−EGFPトランスジェニックマウス由来のmBMMNCsから誘導された細胞を示した。「MEF」はOct4−EGFPトランスジェニックマウス由来のMEFから誘導された細胞を示した。
【図11】Oct4−EGFPトランスジェニックマウスの骨髄由来単核球細胞(mBMMNCs)から誘導された多能性幹細胞クローンについて、未分化性のマーカーであるOct4−EGFPレポーター遺伝子からのEGFPタンパク質の発現とマウス未分化細胞のマーカーであるSSEA−1抗原の発現をFACSにて解析した結果および多能性幹細胞クローンが形成するコロニーの形態とコロニーにおける未分化性のマーカーであるOct4−EGFPレポーター遺伝子からのEGFPタンパク質の発現を示した。[A]はOct4−EGFPトランスジェニックマウス由来のmBMiPS細胞クローン#7およびクローン#13について未分化性マーカーOct4−EGFPの発現およびマウス未分化細胞のマーカーであるSSEA−1抗原の発現をFACSにて解析した結果を示した。「IgG APC」はアロフィコシアニン結合コントロールIgGによる染色強度を示した。「SSEA−1 Alexa647」はAlexa647結合抗マウスSSEA−1抗体による染色強度を示した。各図の水平軸は未分化性マーカーOct4−EGFPの発現強度を示し、図中の等高線は解析した細胞の存在頻度を示した。[B]はOct4−EGFPトランスジェニックマウス由来のmBMiPS細胞クローン#7およびクローン#13についてマウス未分化細胞のマーカーであるSSEA−1抗原の発現をFACSにて解析した結果を示した。水平軸はマウス未分化細胞のマーカーであるSSEA−1抗原の発現強度を示し、垂直軸は細胞数を示した。図の実線がAlexa647結合抗マウスSSEA−1抗体で染色した場合をしめし、破線はアロフィコシアニン結合コントロールIgGで染色した場合を示した。[C]はOct4−EGFPトランスジェニックマウス由来のmBMiPS細胞クローン#7およびクローン#13が形成するコロニーの形態および未分化性のマーカーであるOct4−EGFPレポーター遺伝子からのEGFPタンパク質の発現を示した。
【図12】pMYsIGベクターを用いて調製されたレトロウィルスのOct4−EGFPトランスジェニックマウス骨髄由来単核球細胞(mBMMNCs)あるいはOct4−EGFPトランスジェニックマウス由来の胚性線維芽細胞(MEF)に対する感染効率を評価するためのFACS解析結果を示した。図の水平軸は感染したレトロウィルスゲノムから発現する緑色蛍光タンパク質EGFPの発現強度を示し、垂直軸は細胞数を示した。レトロウィルス感染後2日目の解析結果を示した。「BMMNCs」はOct4−EGFPトランスジェニックマウス由来mBMMNCsを用いた場合を示し、「ゼラチン上に予め培養MEF」はウィルス感染前日からゼラチンコートプレート上で培養していたOct4−EGFPマウスMEFを用いた場合を示し、「レトロネクチン上MEF」はBMMNCsと同様にレトロネクチンコートプレートで感染させたMEFを用いた場合を示した。
【図13】Oct4−EGFPトランスジェニックマウスの骨髄由来単核球細胞(mBMMNCs)あるいはMEFからpMYs核初期化遺伝子ベクターを用いて調製されたレトロウィルスを用いて誘導された多能性幹細胞について、未分化性のマーカーであるOct4−EGFPレポーター遺伝子からのEGFPタンパク質の発現とマウス未分化細胞のマーカーであるSSEA−1抗原の発現をFACSにて解析した結果および多能性幹細胞クローンが形成するコロニーの形態とコロニーにおける未分化性のマーカーであるOct4−EGFPレポーター遺伝子からのEGFPタンパク質の発現を示した。[A]はレトロウィルス感染7日後のOct4−EGFPトランスジェニックマウス由来mBMMNCsおよびMEFの未分化性マーカーOct4−EGFPの発現およびマウス未分化細胞のマーカーであるSSEA−1抗原の発現をFACSにて解析した結果を示した。「SSEA−1 Alexa647」はAlexa647結合抗マウスSSEA−1抗体による染色強度を示した。各図の水平軸は未分化性マーカーOct4−EGFPの発現強度を示し、図中の等高線は解析した細胞の存在頻度を示した。[B]はレトロウィルス感染7日後にOct4−EGFPトランスジェニックマウス由来のmBMMNCsおよびMEFが形成するコロニーの形態および未分化性のマーカーであるOct4−EGFPレポーター遺伝子からのEGFPタンパク質の発現を示した。[C]はレトロウィルス感染12日後にOct4−EGFPトランスジェニックマウス由来のmBMMNCsおよびMEFが形成するコロニーの形態および未分化性のマーカーであるOct4−EGFPレポーター遺伝子からのEGFPタンパク質の発現を示した。「BMMNCs」はOct4−EGFPトランスジェニックマウス由来mBMMNCsを用いた場合を示し、「ゼラチン上に予め培養MEF」はウィルス感染前日からゼラチンコートプレート上で培養していたOct4−EGFPマウスMEFを用いた場合を示し、「レトロネクチン上MEF」はBMMNCsと同様にレトロネクチンコートプレートで感染させたMEFを用いた場合を示した。
【図14】ヌードマウスの皮下に移植されたマウス骨髄由来iPS#7細胞より形成された奇形腫を示す。[A]は皮下に移植されたマウス骨髄由来iPS#7細胞あるいはES細胞より奇形腫が形成されたヌードマウスを示す。[B]は形成された奇形腫より作製した病理標本を示し、標本中には、未分化神経組織、中枢神経、大脳皮質様組織、毛根組織、筋組織、軟骨組織、白色脂肪組織、気管粘液上皮細胞及び繊毛を有する気管上皮細胞、食道粘膜様組織、膵臓腺房細胞、唾液腺の漿液細胞および粘液細胞などが観察される。
【図15】マウス骨髄由来iPS#7細胞をBalb/cマウスの受精卵から作製した胚盤胞に注入後、偽妊娠マウスに移植することにより作出されたキメラ個体を示す。Balb/cマウスは本来白色の毛色を示し、骨髄由来iPS細胞は黒色の毛色を有するC57BL/6マウスに由来している。黒色の毛色を有する細胞は骨髄由来iPS細胞由来の細胞であることを示している。
【図16】マウス骨髄由来のiPS細胞クローンとマウスES細胞およびマウス骨髄由来単核球細胞について、各細胞で発現している遺伝子の発現強度の比較を示した図である。「iPS#7」はマウスiPS細胞クローンを示し、「BMMNC」はマウス骨髄由来単核球細胞を示す。各図の水平軸、垂直軸は各細胞で発現する遺伝子のメッセンジャーRNAの量を示し、図中の各点は一種類の遺伝子に対応している。比較する細胞間で発現する遺伝子のメッセンジャーRNAの量が同一である場合には、図中の各点は図の対角線上に位置することになる。
【発明の背景】
【0001】
発明の分野
本発明は、分化した体細胞、特に血球系細胞を初期化または脱分化することにより多能性幹細胞を製造するための方法およびキットに関する。
【0002】
背景技術
胚性幹細胞(ES細胞)は、生体中の組織・臓器を構成するほぼ全ての種類の細胞に分化できる多能性を有したまま、長期間にわたる細胞増殖を無制限に維持できる細胞株である。この性質により、ES細胞は、パーキンソン病、I型糖尿病、脊髄損傷、心不全など、多くの疾患に対する移植療法のための細胞資源として、組織工学による人工組織・臓器作製のための多種類細胞材料の供給源として、さらには、基礎研究や創薬研究に用いられるヒト細胞の供給源としての利用が期待されている。しかしながら、ES細胞はヒトやマウスの初期胚を破壊して樹立された細胞株であることから、倫理的見地よりES細胞の利用に対する反対意見も多い。また、細胞移植療法については、他人の細胞を移植することになることから、臓器移植と同様に拒絶反応を惹起する危険性を伴っている。これらの問題を回避する方法として、患者自身の分化体細胞を初期化した、ES細胞に近い多能性と増殖能を有した誘導多能性幹細胞(iPS細胞)の利用が期待されている。
【0003】
分化体細胞の初期化法として、例えば、マウス胚性線維芽細胞(MEF細胞)にレトロウィルスベクターでOct4、Sox2、Klf4、およびc−mycの4遺伝子を同時感染させることによりiPS細胞を誘導する技術が報告されている(非特許文献1:K. Takahashi, S. Yamanaka, Cell, 126, pp.663-676, 2006;非特許文献2:K. Okita, T. Ichisaka, S. Yamanaka, Nature, 448, pp. 313-317, 2007;非特許文献3:M. Wernig et al., Nature, 448, pp.318-324, 2007)。マウス肝細胞および胃上皮細胞から同様の方法でiPS細胞を誘導する技術も報告されている(非特許文献4:T. Aoi et al., Science Express on 14 Feb. 2008)。これらの報告における初期化因子のスクリーニング方法については国際公開第2005/080598号パンフレット(特許文献1)に提案されており、また、当該初期化因子については、他のES細胞特異的遺伝子(初期化因子の候補遺伝子)と共に国際公開第2007/069666号パンフレット(特許文献2)に提案されている。これらの報告および提案においては、ヒト細胞について一部記載があるものの、この段階ではヒトES細胞と同等の性質を持ったヒトiPS細胞としての樹立は不完全なものである。また、肝細胞や胃上皮細胞からの誘導はヒトに適用する場合には侵襲性が多く、採取する患者の負担が大きくなることが予想される。
【0004】
ヒトiPS細胞を樹立する方法としては、成人皮膚由来線維芽細胞 (HDF細胞)にレトロウィルスを用いてOct4、Sox2、Klf4、およびc−mycの4因子を導入する方法が報告されている(非特許文献5:K. Takahashi, S. Yamanaka, Cell, 131, pp.861-872, 2007)。この方法においては、レトロウィルスを高効率に感染させるために、マウスエコトロピックウィルス受容体であるsolute carrier family 7(Slc7a1) をレンチウィルスにより発現させた細胞を用いている。その結果、レトロウィルスの感染後25日目頃よりヒトES細胞様のコロニーを認め、30日目以降にコロニーのピックアップを実施している。この方法では、新たにマウスエコトロピックウィルス受容体をレンチウィルスにより導入しているため、異種動物由来の遺伝子の発現および染色体へのDNA断片挿入機会の増加による、宿主細胞の癌化リスクの増大といった課題を抱えている。さらに、誘導日数が1ヶ月におよぶため、培養コストについても課題を抱えている。また、この報告では、iPS細胞の誘導に新生児包皮由来線維芽細胞 (BJ1細胞)も用いられており、その場合のヒトES細胞様コロニーの出現頻度はHDFを用いた場合よりも高いが、誘導日数については短縮されていない。
【0005】
他のグループでは、アデノウィルスによりマウスエコトロピックウィルス受容体であるmCAT1遺伝子を発現させたヒト新生児包皮由来線維芽細胞を用いて、同様の4因子をレトロウィルスにより導入し、iPS細胞を誘導した報告もある(非特許文献6:H.Masaki et al., Stem Cell Research, 1, pp.105-115, 2007)。この報告では、レトロウィルスの感染後17日目にiPS様のコロニーの出現が認められている。この方法では、アデノウィルスでウィルス受容体を発現させていることにより、染色体DNAへの異種遺伝子の挿入リスクは低減されているが、異種遺伝子が細胞内に導入されることに伴うリスクは以前として残る。同様の4因子をレトロウィルスによりヒト胎児肺線維芽細胞 (MRC5細胞)へ導入してiPS細胞を誘導した報告もある(非特許文献7:I. H. Park et al., Nature, 451, pp.141-146, 2008)。しかしながら、この報告では、BJ1細胞、成体間葉系幹細胞(MSC細胞)、およびHDF細胞からの4因子によるiPS細胞誘導は成功していない。また、初期化因子としてOct4、Sox2、NANOG、およびLin28の4因子を用いて、レンチウィルスで細胞に発現させることにより、fetal fibroblast cells(IMR90 cells)やnewborn foreskin fibroblastからES細胞様コロニーを取得している報告もある(非特許文献8:J. Yu et al., Science, 318, pp.1917-1920, 2007)。胎児や新生児の細胞を用いた場合、成人の表皮線維芽細胞を用いた場合に比べて、より短期間でiPS細胞が誘導できているが、治療や研究に用いる細胞源としては倫理的な課題が残る。この報告ではさらに、ヒトES細胞から分化誘導した血球細胞等を用いてiPS細胞が誘導可能であることが報告されている。しかし、ES細胞から分化誘導した細胞は正常な分化過程を経ておらず、成体由来の血球細胞と同一の細胞とはいえない。今のところ、成体から採取した血球細胞からのiPS細胞の誘導についての報告はない。
【0006】
Oct4、Sox2、Klf4、およびc−mycの4因子に加え、Nanogを共発現させることによりiPS細胞の誘導を効率化した報告もある(非特許文献9:W. E. Lowry et al., Proc. Natl. Acad. Sci. USA, 105, pp.2883-2888, 2008) 。Nanogの存在下でリプログラミング(初期化)効率が上がることは従来から報告されている(非特許文献10:J. Silva et al., Nature, 441, pp.997-1001, 2006) 。レトロウィルスを用いてHDF細胞に遺伝子導入を行い、ウィルス感染終了後21日目でES細胞様コロニーが出現する。Nanogを用いることによりiPS細胞の誘導効率が上がっているが、因子の種類を増やすことは、染色体に挿入される遺伝子の種類も増やすこととなり、初期化因子の種類を増やさない方法での効率改善が望まれる。
【0007】
また、マウスにおいては終末分化した成熟B細胞からiPS細胞を誘導した報告もある(非特許文献11:J. Hanna et al., Cell, 133, pp.250-264, 2008)。この報告においては、ドキシサイクリン依存的に4つの核初期化因子、Oct4、Sox2、Klf4、およびc−mycを発現するユニットをレンチウィルスベクターにより導入することによって、まずiPS細胞を誘導している。さらに、誘導されたiPS細胞を用いてトランスジェニックマウスを作製している。成熟B細胞からのiPS細胞誘導には、こうして得られたトランスジェニックマウスより調製したB細胞を用いており、同様の方法をヒトに適用することは不可能である。ヒトの終末分化した血液細胞からiPS細胞を誘導するためには本法を適用することはできず、ヒト初代細胞に適用できる初期化方法が望まれる。
【0008】
一方で、ES細胞のような多能性幹細胞の利用により治療効果が期待される医療領域では、ES細胞から分化誘導した細胞による治療法の検討と並行して、生体内に存在する体性幹細胞の利用が検討されてきた。そのような取組みの中、細胞移植療法への利用を目的として生体外で体性幹細胞を増幅する技術の報告がある。中でも造血幹細胞については、幹細胞因子(SCF)、Fltリガンド(FL)、トロンボポエチン(TPO)、インターロイキン−6(IL−6)と可溶化型IL−6受容体のα鎖(IL−6R)との融合タンパク質などのサイトカインを用いて、臍帯血由来の造血幹細胞や骨髄由来の造血幹細胞を増幅した報告がある(非特許文献12:T. Ueda et al., J. of Clinical Investigation, 105, pp-1013-1021, 2000;非特許文献13:L. Gammaitoni et al., Experimental Hematology, 31, pp. 261-270, 2003)。さらに、ストローマ細胞を用いて増幅率を向上させる技術もある(特許文献3:国際公開第2003/014336号パンフレット)。いずれも血球系の幹細胞または前駆細胞を増幅する技術であり、増幅の際に他の細胞系譜への分化能を付与することはできず、体細胞の初期化作用や脱分化作用はない。また、造血幹細胞の増幅には限界があり、ES細胞のように多能性を維持したまま無制限に増殖させることはできない。
【0009】
【特許文献1】国際公開第2005/080598号パンフレット
【特許文献2】国際公開第2007/069666号パンフレット
【特許文献3】国際公開第2003/014336号パンフレット
【非特許文献1】K. Takahashi, S. Yamanaka, Cell, 126, pp.663-676, 2006
【非特許文献2】K. Okita, T. Ichisaka, S. Yamanaka, Nature, 448, pp. 313-317, 2007
【非特許文献3】M. Wernig et al., Nature, 448, pp.318-324, 2007
【非特許文献4】T. Aoi et al., Science Express on 14 Feb. 2008
【非特許文献5】K. Takahashi, S. Yamanaka, Cell, 131, pp.861-872, 2007
【非特許文献6】H.Masaki et al., Stem Cell Research, 1, pp.105-115, 2007
【非特許文献7】I. H. Park et al., Nature, 451, pp.141-146, 2008
【非特許文献8】J. Yu et al., Science, 318, pp.1917-1920, 2007
【非特許文献9】W. E. Lowry et al., Proc. Natl. Acad. Sci. USA, 105, pp.2883-2888, 2008
【非特許文献10】J. Silva et al., Nature, 441, pp.997-1001, 2006
【非特許文献11】J. Hanna et al., Cell, 133, pp.250-264, 2008
【非特許文献12】T. Ueda et al., J. of Clinical Investigation, 105, pp-1013-1021, 2000
【非特許文献13】L. Gammaitoni et al., Experimental Hematology, 31, pp. 261-270, 2003
【発明の概要】
【0010】
本発明者らは、IL−6シグナル伝達因子(IL6ST)刺激因子とサイトカインカクテルとの存在下での培養と、細胞の脱分化とを組み合わせることにより、体細胞から多能性幹細胞を効率的に樹立できることを見出した。本発明はこの知見に基づくものである。
【0011】
従って、本発明の目的は、体細胞から効率的に多能性幹細胞を製造する方法を提供することにある。
【0012】
そして、本発明による多能性幹細胞の製造法は、体細胞から多能性幹細胞を製造する方法であって、(a)体細胞を、IL−6シグナル伝達因子刺激因子および少なくとも1種のサイトカインを含む細胞培養培地中で培養する工程、および(b)体細胞を脱分化させる工程を含んでなり、工程(a)の後に工程(b)が行われるか、または工程(a)と工程(b)が同時に行われる方法である。
【0013】
本発明によれば、人体より軽微な負担で採取できる体細胞、例えば末梢血液細胞などから、従来法よりも短期間で高効率に多能性幹細胞を樹立することができる。多能性幹細胞の樹立に要する期間は、例えば、従来法の約1/2にまで短縮することが可能である。よって、製造された多能性幹細胞を、脊髄損傷などの緊急性の高い疾患の治療に利用することができ、また、多能性幹細胞の製造にかかる費用も低減することが可能となる。
【発明の具体的説明】
【0014】
本発明において製造される「多能性幹細胞」とは、種々の分化系譜に分化可能な細胞を意味し、必ずしもES細胞と同等のすべての細胞系譜に分化可能な細胞に限られるものではなく、例えば、内胚葉系幹細胞、中胚葉系幹細胞、または外胚葉系幹細胞としてもよい。例えば、心不全の治療に使用する多能性幹細胞を製造するためには、心筋細胞に分化可能な細胞を製造できればよい。あるいは、I型糖尿病の治療に使用する多能性幹細胞を製造するためには、β細胞に分化可能な細胞を製造できればよい。使用目的に応じたレポーター遺伝子や特異的表面抗原に対する抗体を用いることで、目的の細胞を効率よく分離することが可能である。
【0015】
本発明の好ましい実施態様によれば、「多能性幹細胞」は、ES細胞と同等のすべての細胞系譜に分化可能な細胞とされる。このような細胞は、ヒトにおいては、SSEA−3、SSEA−4、TRA1−60、TRA1−81などのマーカー分子を発現する細胞である。マウスにおいては、SSEA−1などのマーカー分子を発現する細胞である。ヒトおよびマウスのそれらの細胞は、SCIDマウスやヌードマウスなどの免疫不全マウスの皮下に移植することにより、奇形腫を形成することが知られている。多能性幹細胞から形成された奇形腫には、内胚葉系、中胚葉系、外胚葉系、それぞれの系譜の分化細胞が含まれる。よって、ES細胞と同等のすべての細胞系譜に分化可能な多能性幹細胞は、このような奇形腫の形成により特徴づけられる。
【0016】
本発明による多能性幹細胞の製造法は、IL−6シグナル伝達因子(IL6ST)刺激因子および少なくとも1種のサイトカインを含む細胞培養培地中で培養する工程(以下「工程(a)」という)を含んでなる。
【0017】
本発明において「IL6シグナル伝達因子(IL6ST)」とは、interleukin 6 signal transducerを意味し、gp130とも呼ばれている。IL6STはシグナル領域、細胞外領域、膜貫通領域、および細胞内領域から構成される全長918アミノ酸残基のタンパク質を意味する。IL6STについては、シグナルペプチドであるN末端から22番目のグリシン残基までが欠失してもIL6STを介したシグナル伝達は維持されることが知られている。IL−6と結合したIL−6Rは2分子のIL6STと結合し、IL6STはホモ二量体を形成する。ホモ二量体を形成したIL6STは、細胞内領域のチロシン残基がリン酸化され、シグナル伝達が活性化されることが知られている。IL6STは、IL−11Rとも結合し、シグナルを伝達することが知られている。また、IL6STは白血球遊走阻止因子レセプター(LIFR)、オンコスタチンMレセプター(OSMR)、毛様体神経栄養因子レセプター(CNTFR)、カルジオトロピンレセプター(CT−1R)などとも結合し、ヘテロ二量体を形成することにより活性化されることが知られている。
【0018】
本発明において「IL6ST刺激因子」とは、ヒトIL6STを介してシグナルを伝達する分子を意味し、より具体的には、IL6STと会合することによりIL6STの活性化を引き起こす分子をいう。IL6ST刺激因子としては、IL−6またはその変異体とIL−6受容体(以下「IL−6R」という)またはその変異体との融合タンパク質、IL−6R(好ましくは、可溶性IL−6R(sIL−6R))またはその変異体とIL−6またはその変異体との組み合わせ(特表平10−509040号)、IL−6とIL−6受容体のα鎖との複合体、IL6STに対してアゴニストとして作用するIL6STに対する抗体(Z. J. Gu et al., Leukemia, 14, pp.188-197, 2000)、インターロイキン−11(IL−11)、白血球遊走阻止因子(LIF)、オンコスタチンM、およびカルジオトロピンが挙げられる。
【0019】
本発明において「IL−6」とは、IL−6として一般的に知られる、4つのへリックスから構成される全長212アミノ酸残基のタンパク質を意味する。また、本発明において「IL−6の変異体」とは、置換、欠失、挿入、および付加から選択される1以上の改変を有し、かつIL6ST刺激活性を有するIL−6の変異体を意味する。IL−6については、シグナルペプチドであるN末端から28番目のアラニン残基までが欠失してもIL6ST刺激活性を維持することが知られている(WO00/01731)。また、IL−6の37番目のリジン残基のC末端側がプロテアーゼの消化作用を受けること、および28番目のアラニン残基から37番目のリジン残基までの10アミノ酸残基が欠失してもIL−6活性に支障がないことが知られている(WO00/01731)。従って、本発明においては、N末端が欠失し、かつIL6ST刺激活性を有する改変IL−6をIL−6変異体として用いてもよい。このようなIL−6変異体としては、28番目までのN末端配列が欠失したIL−6、37番目までのN末端配列が欠失したIL−6が挙げられる。
【0020】
本発明において、「IL−6R」とは、IL−6受容体として一般的に知られる、シグナル領域、細胞外領域、膜貫通領域、および細胞内領域から構成される全長468アミノ酸残基のタンパク質を意味する。また、本発明において「IL−6Rの変異体」とは、置換、欠失、挿入、および付加から選択される1以上の改変を有し、かつIL6ST刺激活性を有するIL−6Rの変異体を意味する。IL−6Rについては、細胞外領域中に存在するサイトカインレセプター領域(112番目のバリン残基付近から323番目のアラニン残基付近までの領域)がIL−6との結合に必要十分であることが知られている。従って、本発明においては、IL−6Rの112番目のバリン残基付近から333番目のアラニン残基付近までの領域を含んでなる改変IL−6RをIL−6R変異体として用いてもよい。
【0021】
本発明の好ましい実施態様によれば、IL6ST刺激因子は、IL−6またはその変異体とIL−6Rまたはその変異体とが直接またはリンカーを介して連結された融合タンパク質とされる。このような融合タンパク質は、当技術分野において公知の方法、例えば、WO00/01731、WO99/02552、特表2000−506014号、またはFischer et al., Nature Biotech., 15, pp.142-145, 1997に記載の方法に従って製造することができる。
【0022】
本発明の特に好ましい実施態様によれば、前記融合タンパク質は、IL−6Rの112番目のバリン残基から333番目のアラニン残基までの断片のC末端と、IL−6の38番目のアスパラギン酸残基から212番目のメチオニン残基までの断片のN末端とが直接連結されたタンパク質(以下「FP6」という)とされる。この場合、この融合タンパク質をコードするDNA配列を宿主において発現可能なように連結したベクターを適当な宿主に導入し、形質転換された宿主を培養し、培養物からFP6を採取することにより融合タンパク質FP6を製造することができる(WO00/01731参照)。
【0023】
IL6ST刺激因子の細胞培養培地への添加量は特に制限されないが、好ましくは1ng/ml〜100μg/ml、より好ましくは10ng/ml〜20ng/mlとされる。
【0024】
本発明による多能性幹細胞の製造法において、IL−6ST刺激因子の使用は一つの主要な特徴である。特定の理論に拘束されるわけではないが、IL6STの活性化は、例えば、JAKキナーゼの活性化、STAT3のリン酸化、リン酸化によりホモ二量体を形成したSTAT3によるJunB、JABなどの遺伝子の転写促進を順次引き起こし、結果として細胞増殖、細胞分化調節等を生じることが考えられる。よって、本発明においては、IL6ST刺激因子に替えて、活性化型JAKキナーゼ、リン酸化型STAT3、ホモ二量体を形成させる変異を導入したSTAT3C(J.F.Bromberg et. al., Cell, 98, pp-295-303, 1999)、エストロゲン類縁体により誘導性にホモ二量体を形成させるSTAT3ER(T.Matsuda et al., EMBO Journal, 18, pp-4261-4269, 1999)、JAKキナーゼの阻害因子であるSOCS分子の阻害剤などを、遺伝子またはタンパク質の形で細胞内に導入することも考えられる。
【0025】
工程(a)において使用される「サイトカイン類」としては、トロンボポエチン(TPO)およびその変異体ならびにそれらの誘導体、c−mplリガンド(TPOを除く)およびその誘導体、幹細胞因子(SCF)、Flt−3リガンド(FL)、インターロイキン−3(IL−3)、インターロイキン−6(IL−6)、顆粒球コロニー刺激因子(G−CSF)、顆粒球マクロファージ・コロニー刺激因子(GM−CSF)、マクロファージ・コロニー刺激因子(M−CSF)、インターロイキン−7(IL−7)のような増殖因子;抗ヒトCD40抗体、抗ヒトCD3抗体、抗ヒトCD28抗体のような増殖刺激活性を有する抗体;リポ多糖(LPS)のような増殖刺激因子;およびこれらの組み合わせが挙げられる。
【0026】
本発明において「トロンボポエチン」(TPO)とは、全長332アミノ酸残基のアミノ酸配列からなる、血小板産生を特異的に刺激または増大させる活性を有するタンパク質をいう(WO95/21919)。また、本発明において「TPOの変異体」とは、置換、欠失、挿入、および付加から選択される1以上の改変を有し、かつ血小板産生を特異的に刺激または増大させるTPOの変異体を意味する。TPOの変異体としては、例えば、WO95/18858、特開平8−277296号公報、特開平8−228781号公報、およびWO95/21919等に記載されているものが挙げられ、好ましくは、アミノ酸残基1番目から163番目までのアミノ酸配列からなるTPOの変異体(断片)が挙げられる。
【0027】
本発明において「TPOおよびその変異体の誘導体」とは、水溶性ポリマーを連結してなるTPOおよびその変異体を意味する。水溶性ポリマーとしては、ポリエチレングリコール、好ましくは平均分子量が約5kDa〜約50kDaであるポリエチレングリコールが挙げられる。TPOおよびその変異体のポリエチレングリコール化(PEG化)は周知の方法に従って実施することができる(例えば、Focus on Growth Factors, 3, pp. 4-10, 1992参照)。水溶性ポリマー対「TPOおよびその変異体」タンパク質のモル比は1:1〜100:1とすることができ、ポリPEG化の場合には1:1〜20:1、モノPEG化の場合には1:1〜5:1とすることができる。
【0028】
本発明において「c−mplリガンド」とは、mplレセプターに結合可能なペプチド性リガンド、タンパク質性リガンド、および非ペプチド性リガンドを意味する。タンパク質性c−mplリガンド(TPOを除く)としては、巨核球の刺激剤としてのアゴニスト抗体(WO99/03495およびWO99/10494)や造血レセプターアゴニストなどの他のタンパク質( WO96/23888 、WO97/12978、WO97/12985、WO98/17810、WO96/34016およびWO00/24770)が挙げられる。ペプチド性c−mplリガンドとしては、WO96/40189、WO96/40750、WO98/25965、特開平10−72492号、WO99/42127、およびWO00/24770に記載されているものが挙げられる。非ペプチド性c−mplリガンドとしては、ベンゾジアゼピン誘導体(特開平11−1477号および特開平11−152276号)や他の低分子リガンド(WO99/11262、WO99/22733、WO99/22734、WO00/35446およびWO00/28987)が挙げられる。
【0029】
本発明において「c−mplリガンドの誘導体」とは、水溶性ポリマーを連結してなるc−mplリガンドおよびその変異体を意味する。水溶性ポリマーとしては、ポリエチレングリコール、好ましくは平均分子量が約5kDa〜約50kDaであるポリエチレングリコールが挙げられる。c−mplリガンドのポリエチレングリコール化(PEG化)は周知の方法に従って実施することができる(例えば、Focus on Growth Factors, 3, pp. 4-10, 1992参照)。水溶性ポリマー対c−mplリガンドタンパク質のモル比は1:1〜100:1とすることができ、ポリPEG化の場合には1:1〜20:1、モノPEG化の場合には1:1〜5:1とすることができる。
【0030】
本発明において「幹細胞因子」(SCF:stem cell factorまたはsteel factor)とは、造血および生殖発生に重要な機能を果たす、全長273アミノ酸残基のアミノ酸配列からなるタンパク質をいう。また、本発明において「SCFの変異体」とは、置換、欠失、挿入および付加から選択される1以上の改変を有し、かつSCFのレセプターであるc−kit分子を特異的に刺激して活性化させるSCFの変異体を意味する。SCFについては、シグナルペプチドであるN末端から25番目のスレオニン残基までが欠失しても活性を保持していることが知られている。SCFは、N末端から189番目のアラニン残基と190番目のアラニン残基との間で、あるいは190番目のアラニン残基と191番目のセリン残基との間でタンパク質分解酵素により消化されることにより可溶化型SCFとして存在することが知られている。また、複数のオルタナティブフォームが存在することが知られており、N末端から175番目のセリン残基から202番目のイソロイシン残基までを欠失した膜結合型のSCFが存在することが知られている。本発明においては、これらの変異体のいずれもが「SCFの変異体」として使用可能である。
【0031】
本発明において「IL−3」は、造血幹・前駆細胞に作用し、コロニー形成を支持する活性を持つタンパク質である。また、IL−3は、肥満細胞、好酸球、好塩基球、好中球、単球などの分化した細胞の増殖因子として重要な機能を有する。IL−3は、全長152アミノ酸残基のアミノ酸配列からなる。また、本発明において「IL−3の変異体」とは、置換、欠失、挿入および付加から選択される1以上の改変を有し、かつIL−3受容体を特異的に刺激して活性化させるIL−3の変異体を意味する。IL−3については、シグナルペプチドであるN末端から19番目のグルタミン残基までが欠失しても活性を保持していることが知られている。本発明においては、このような変異体を「IL−3の変異体」として使用することができる。
【0032】
本発明において「Flt−3リガンド」(FL)は、造血幹・前駆細胞の増殖因子として重要な機能を持つタンパク質である。FLは全長235アミノ酸残基のアミノ酸配列からなる膜結合蛋白質である。また、本発明において「FLの変異体」とは、置換、欠失、挿入および付加から選択される1以上の改変を有し、かつFlt−3チロシンキナーゼ受容体を特異的に刺激して活性化させるFLの変異体を意味し、好ましくはアミノ酸残基1番目から185番目のプロリン残基までのアミノ酸配列からなるFLの変異体(断片)が挙げられる。FLについては、シグナルペプチドであるN末端から26番目のグリシン残基までが欠失しても活性を保持していることが知られている。本発明においては、これらの変異体のいずれもが「FLの変異体」として使用可能である。
【0033】
本発明の好ましい実施態様によれば、サイトカインは、幹細胞因子(SCF)、トロンボポエチン(TPO)およびその変異体ならびにそれらの誘導体、Flt−3リガンド(FL)、インターロイキン−3(IL−3)およびその変異体、ならびにこれらの2以上の組合わせから選択され、より好ましくは、SCF、TPO、IL−3およびFlt−3リガンド、ならびにこれらの2以上の組合せから選択される。本発明の特に好ましい実施態様によれば、サイトカインは、SCF、TPOおよびIL−3の組合せ、またはSCF、TPO、IL−3およびFlt−3リガンドの組合せとされる。
【0034】
工程(a)において用いられる細胞培養培地は、例えば、下記の群:顆粒球コロニー刺激因子(G−CSF)、顆粒球マクロファージ・コロニー刺激因子(GM−CSF)、インターロイキン−7(IL−7)のような増殖因子;抗ヒトCD40抗体、抗ヒトCD3抗体、抗ヒトCD28抗体のような増殖刺激活性を有する抗体;リポ多糖(LPS)のような増殖刺激因子からなる群から選ばれる1種以上の増殖刺激因子をさらに含むものとしてもよい。これらの因子によるIL6STの発現亢進により、多能性幹細胞の誘導がより効率化する場合もある。
【0035】
これらサイトカインの細胞培養培地への添加量は特に制限されないが、好ましくは1ng/ml〜100μg/ml、より好ましくは10ng/ml〜100ng/mlとされる。
【0036】
培養に用いる基礎培地としては、血球細胞などの体細胞および多能性細胞の増殖および生存が害されない限り特に制限されないが、例えば、D−MEM培地、D−MEM/F12培地、MEM−α培地、Opti−MEM培地、IMDM培地、RPMI1640培地、StemPro培地(以上、Invitrogen社)、mTeSR 1培地(StemCell Technologies社)、HEScGRO培地(Millipore社)、N2B27培地、が好適に用いられる。培養温度は、通常25〜39℃、好ましくは33〜39℃である。また、培地に添加する物質としては、ウシ胎児血清、ヒト血清、ノックアウト血清リプレースメント(KnockOutSerumReplacement(KSR):Invitrogen社)、インシュリン、トランスフェリン、ラクトフェリン、エタノールアミン、亜セレン酸ナトリウム、モノチオグリセロール、2−メルカプトエタノール、ウシ血清アルブミン、ピルビン酸ナトリウム、ポリエチレングリコール、各種ビタミン、各種アミノ酸が挙げられる。CO2は、通常4〜15%であり、5〜10%が好ましい。O2は通常5〜25%である。また、培養にはトロンボポエチン(TPO)およびその変異体およびそれらの誘導体、c−mplリガンド(TPOを除く)およびその誘導体、幹細胞因子(SCF)、Flt−3リガンド(FL)、インターロイキン−3(IL−3)、インターロイキン−6(IL−6)、顆粒球コロニー刺激因子(G−CSF)、顆粒球マクロファージ・コロニー刺激因子(GM−CSF)、マクロファージ・コロニー刺激因子(M−CSF)、インターロイキン−7(IL−7)のような増殖因子;抗ヒトCD40抗体、抗ヒトCD3抗体、抗ヒトCD28抗体のような増殖刺激活性を有する抗体;リポ多糖(LPS)のような増殖刺激因子などのサイトカイン類の他に、トランスフォーミング成長因子−β(TGF−β)、骨形成因子(BMP)ファミリー、ヘッジホッグ因子(Hh)ファミリー、bFGFなどに代表される増殖因子、MIP−1α、SDF−1などのCCケモカインあるいはCXCケモカイン、エリスロポエチン(EPO)のような造血ホルモン、Wntファミリー遺伝子産物、Notchリガンドファミリー遺伝子産物、塩化リチウム、ピペコリン酸、γ―アミノ酪酸(GABA)などの化合物を10ng/mlから100μg/mlの範囲で添加することもできる。
【0037】
本発明における培養では、培養用のシャーレ、フラスコ、培養バックを用いた培養が可能であるが、培地組成、pHなどを機械的に制御し、高密度で培養が可能なバイオリアクターによって、その培養を改善することもできる。
【0038】
培養に用いるフィーダー細胞としては、マウス胚性線維芽細胞(MEF)、SNL細胞(A.P.McMahon and A.Bradley, Cell, 62, pp-1073-1085, 1990)、STO細胞などを用いることができる。フィーダー細胞の増殖を抑える方法としては、マイトマイシンC処理や放射線照射などの方法を用いることができる。また、フィーダー細胞に替えて、BDマトリゲル(BDバイオサイエンス社)などのマトリゲル、あるいはコラーゲン、ラミニン、フィブロネクチンなどの細胞外マトリックス蛋白質、あるいはゼラチンなどにより培養基材をコーティングする方法を用いることもできる。
【0039】
本発明による多能性幹細胞の製造法は、体細胞を脱分化させる工程(以下「工程(b)」という)をさらに含んでなる。この工程(b)は、工程(a)の後に行ってもよいし、または工程(a)と同時に行ってもよい。
【0040】
工程(a)と工程(b)が同時に行われる実施態様では、上述のIL6ST刺激因子およびサイトカインを含む上記細胞培養培地中で体細胞の脱分化が行われる。すなわち、工程(a)と工程(b)が同時に開始される。この実施態様では、脱分化の過程で培地を他の培地、例えばES細胞培養用の培地に変更することもできる。すなわち、この実施態様では、工程(a)のみを先に終了させ、工程(b)のみを継続してもよい。
【0041】
工程(a)の後に工程(b)が行われる実施態様では、上述のIL6ST刺激因子およびサイトカインを含む上記細胞培養培地中での体細胞の培養が行われ、その後、体細胞の脱分化が行われる。ここで、体細胞の脱分化は工程(a)で用いられる培地と同じ培地を用いて行うことができる。すなわち、この実施態様では、工程(a)は工程(b)よりも先に開始されるが、工程(b)を開始するときに、工程(a)は必ずしも終了していなくてもよい。さらに、この実施態様では、脱分化の過程で培地を他の培地、例えばES細胞培養用の培地に変更することもできる。すなわち、この実施態様では、工程(a)のみを先に終了させ、工程(b)のみを継続してもよい。
【0042】
本発明において「脱分化」とは、分化した組織や器官の細胞が、より未分化性の高い状態に戻ることを意味し、言い換えれば、分化とは逆のプロセスをいう。工程(b)における体細胞の脱分化は、当技術分野において公知の方法により行うことができる。
【0043】
本発明の好ましい実施態様によれば、工程(b)における体細胞の脱分化は、該体細胞の核初期化処理によって行われる。ここで、「核初期化」とは、体細胞の核が受精卵の状態に戻ることをいう。このような核初期化処理は、当技術分野において公知の方法、例えば、WO2007/069666に記載の方法に従って行うことができる。
【0044】
核初期化処理に用いられる核初期化因子としては、Octファミリー遺伝子(例えばOct3/4遺伝子)、Klfファミリー遺伝子(例えばKlf4遺伝子)、Soxファミリー遺伝子(例えばSox2遺伝子)およびMycファミリー遺伝子(例えばc−Myc遺伝子)の各遺伝子産物が知られている(WO2007/069666)。従って、核初期化処理は、体細胞をこれらの遺伝子産物に接触させることによって行うことができる。あるいは、核初期化処理は、体細胞において、これらの遺伝子を活性化または発現させることによって行うこともできる。本発明の好ましい実施態様によれば、体細胞の核初期化処理は、Octファミリー遺伝子(例えばOct3/4遺伝子)、Klfファミリー遺伝子(例えばKlf4遺伝子)、Soxファミリー遺伝子(例えばSox2遺伝子)およびMycファミリー遺伝子(例えばc−Myc遺伝子)を、発現可能な形で体細胞に導入することを含む。
【0045】
核初期化処理に用いられる培地は、例えば、下記の群:顆粒球コロニー刺激因子(G−CSF)、顆粒球マクロファージ・コロニー刺激因子(GM−CSF)、インターロイキン−7(IL−7)のような増殖因子;抗ヒトCD40抗体、抗ヒトCD3抗体、抗ヒトCD28抗体のような増殖刺激活性を有する抗体;リポ多糖(LPS)のような増殖刺激因子からなる群から選ばれる1種以上の増殖刺激因子を含むものとしてもよい。これらの因子による核初期化遺伝子ファミリーの発現亢進により、多能性幹細胞の誘導がより効率化する場合もある。
【0046】
核初期化処理に用いられる核初期化因子としては、上記の遺伝子に替えて、サイトカイン類、ケモカイン類、増殖因子類、低分子化合物、マイクロRNAなどのリボ核酸等を用いてもよい。このような核初期化因子としては、例えば、トランスフォーミング成長因子−β(TGF−β)、骨形成因子(BMP)ファミリー、ヘッジホッグ因子(Hh)ファミリー、bFGFなどに代表される増殖因子、MIP−1α、SDF−1などのCCケモカインあるいはCXCケモカイン、エリスロポエチン(EPO)のような造血ホルモン、Wntファミリー遺伝子産物、Notchリガンドファミリー遺伝子産物、塩化リチウム、ピペコリン酸、γ―アミノ酪酸(GABA)などの化合物、核初期化因子の発現を亢進させる低分子化合物、核初期化因子の転写調節に関わる因子のsiRNA等のマイクロRNA分子などが挙げられる。
【0047】
核初期化処理に用いられる核初期化因子の細胞導入法は特に制限されるものではなく、例えば、レトロウィルスベクター、レンチウィルスベクター、アデノ随伴ウィルスベクター、アデノウィルスベクター、単純ヘルペスウィルスベクター、ネコ内在ウィルスベクター、動物細胞用ベクター等を用いる方法が挙げられ、好ましくはレトロウィルスベクターまたはレンチウィルスベクターを用いる方法、より好ましくはレトロウィルスベクターを用いる方法が挙げられる。また、核初期化因子を細胞に注入する方法を用いてもよく、あるいは膜透過性の修飾を施すことにより各因子を細胞に添加する方法を用いてもよい。
【0048】
核初期化処理に用いられる遺伝子の細胞導入法は特に制限されるものではなく、動物細胞への遺伝子導入に用いられる一般的な方法、例えば、モロニーマウス白血病ウィルス等のレトロウィルスベクター、アデノウィルスベクター、アデノ随伴ウィルス(AAV)ベクター、単純ヘルペスウィルスベクター、レンチウィルスベクター等のウィルス由来の動物細胞用ベクターを用いる方法、リン酸カルシウム共沈法、DEAE−デキストラン法、エレクトロポレーション法、リポソーム法、リポフェクション法、マイクロインジェクション法、HVJリポソーム法等を用いることができる。本発明の好ましい実施態様によれば、核初期化処理に用いられる遺伝子の細胞導入法は、ウィルス由来の動物細胞用ベクター、より好ましくはレトロウィルスベクターを用いる方法とされる。
【0049】
本発明による多能性幹細胞の製造法において、出発材料として用いられる体細胞は特に制限されるものではなく、いかなる体細胞であってもよい。また、体細胞としては、採取された後に一旦凍結保存された細胞を用いてもよい。
【0050】
本発明の好ましい実施態様によれば、前記体細胞は血球細胞とされる。本発明において「血球細胞」とは、生体内の血流中に存在しうる血液細胞と同等の細胞群を意味し、必ずしも末梢血液細胞に限られるものではなく、例えば骨髄中や臍帯血中に存在するものであってもよい。より具体的には、「血球細胞」とは、CD45、CD11aなどの血液細胞のマーカー分子を発現する細胞をいう。また、本明細書において、骨髄中に存在することが確認されている血球細胞を用いることにより、他の組織細胞からよりも効率的に多能性幹細胞を樹立できることが見出されている。よって、本発明の好ましい実施態様によれば、前記血球細胞は骨髄由来単核球細胞とされる。さらに、本発明の他の好ましい実施態様によれば、前記血球細胞は分化血液細胞とされる。このような血球細胞は、採取された後に一旦凍結保存された細胞であってもよい。
【0051】
本発明の他の好ましい実施態様によれば、前記体細胞(特に血球細胞)は、IL6ST刺激因子と上記サイトカインとの存在下での培養により、水疱性口内炎ウィルスのGタンパク質を被覆タンパク質として持つレトロウィルスにより容易に感染する細胞とされる。このような細胞は、レトロウィルスベクターによって遺伝子を導入する上で有利である。
【0052】
本発明の他の好ましい実施態様によれば、前記体細胞(特に血球細胞)は、IL6STを高発現する細胞とされる。具体的には、IL6STはいずれの血球細胞においても発現しているが、中でも発現の高いT細胞、単球(Monocyte)、DC細胞、CD34陽性細胞などを用いることが考えられる。また、血球細胞以外でもIL6STを高発現する体細胞を用いることで、効率よく多能性幹細胞を製造することができる。具体的には、肝臓、腎臓、骨格筋、肺、すい臓、心臓、小腸、脊髄、唾液腺などの細胞も利用することが可能である。
【0053】
本発明の他の好ましい実施態様によれば、前記体細胞(特に血球細胞)は、工程(a)または(b)で用いられるサイトカインに対するレセプターを発現する細胞とされる。具体的には、c−kit(SCFレセプター)、c−mpl(TPOレセプター)、およびFlt3を発現する細胞として、CD34陽性細胞などを用いることが考えられる。また、IL−3Rα(IL−3レセプター)などを発現する細胞として、単球(Monocyte)などを用いることが考えられる。血球細胞以外でも、c−kitを発現する細胞などが挙げられる。
【0054】
本発明の他の好ましい実施態様によれば、前記体細胞(特に血球細胞)は、核初期化因子の少なくとも1種を発現する細胞とされる。例えば、核初期化因子の1つであるKlf4を発現する細胞としては、血球細胞の中では、単球(Monocyte)、DC細胞、CD34陽性細胞などが挙げられ、その他の組織では、肺、骨格筋、小腸などが挙げられる。Klf4以外についても、Klfファミリー遺伝子、Oct3/4ファミリー遺伝子、Mycファミリー遺伝子、またはSoxファミリー遺伝子を発現する体細胞を用いることができる。また、Oct3/4ファミリー遺伝子やNanog遺伝子などの初期化遺伝子については、それらの偽遺伝子を発現する細胞を体細胞として利用することもできる。
【0055】
本発明の他の好ましい実施態様によれば、前記体細胞(特に血球細胞)は、IL6ST刺激因子と上記のサイトカインとの存在下において核初期化因子の発現が上昇する細胞とされる。例えば、各サイトカインのレセプターであるc−kit(SCFレセプター)、c−mpl(TPOレセプター)、Flt3、IL−3Rα(IL−3レセプター)などを発現する細胞を用いることが考えられる。血球細胞の中では、CD34陽性細胞、DC細胞などが挙げられる。また、上記サイトカインによるこれらのレセプターへの刺激により、核初期化因子であるOct3/4ファミリー遺伝子、Klfファミリー遺伝子、Mycファミリー遺伝子、および/またはSoxファミリー遺伝子の発現が上昇する細胞を用いることにより、これらすべての初期化因子を用いずとも、多能性幹細胞の製造が可能となる。
【0056】
本発明の好ましい実施態様によれば、前記体細胞(特に血球細胞)はヒト細胞とされる。この実施態様では、本発明に用いられるIL6ST刺激因子、サイトカイン、核初期化因子またはその遺伝子は、全てヒト由来のものとすることが好ましい。これにより、ヒトの多能性幹細胞を効率よく製造することが可能となる。
【0057】
本発明に従って製造された細胞における脱分化または初期化の確認は、当技術分野において公知の方法によって行うことができる。このような方法としては、例えば、細胞表面に発現した未分化マーカーに対する抗体を用いて、蛍光励起細胞分離装置(FACS)を利用して解析する方法が挙げられる。具体的には、SSEA−3、SSEA−4、TRA1−60、TRA1−81などの未分化マーカーに対する抗体を利用することができる。また、NanogやOct3/4遺伝子といった未分化マーカーのレポーター遺伝子を細胞に導入して利用することもできる。
【0058】
本発明の工程(a)において用いられる、IL6ST刺激因子および少なくとも1種のサイトカインを含む細胞培養培地は、製造された多能性幹細胞の性質を維持するために利用することもできる。すなわち、IL6ST刺激因子と上記のサイトカイン類は、核初期化プロセスの促進のみならず、核初期化後の初期化状態の維持に利用することもできる。
【0059】
本発明による多能性幹細胞の製造法に用いられる試薬類をまとめて、キットとすることもできる。従って、本発明の他の態様によれば、体細胞から多能性幹細胞を製造するためのキットが提供され、該キットは、(a)IL−6シグナル伝達因子刺激因子および少なくとも1種のサイトカインを含む、体細胞を培養するための細胞培養培地、および(b)体細胞を脱分化させるための試薬を含んでなる。
【0060】
好適なIL−6シグナル伝達因子刺激因子および好適なサイトカインについては上述したとおりである。また、体細胞を脱分化させるための試薬についても上述したとおりであるが、好ましくは、Octファミリー遺伝子、klfファミリー遺伝子、Soxファミリー遺伝子、およびMycファミリー遺伝子を、発現可能な形で体細胞に導入するためのベクターを含む。本発明によるキットはさらに、多能性幹細胞の製造法に用いられる具体的方法に応じて、試薬類、反応容器、説明書等を含んでいてもよい。
【0061】
本発明に従って製造された多能性幹細胞は、各種分化細胞に誘導することが可能であるため、細胞再生医療に利用することができる。すなわち、この多能性幹細胞は、患者本人の体細胞を用いた自家の細胞移植および細胞バンク等より入手した細胞を利用する同種の細胞移植の両者の移植系への利用が可能である。既に存在する血球細胞の大型細胞バンクからも多能性幹細胞の細胞バンクを構築することができる。
【0062】
また、本発明によれば、各種疾病の患者からの血球細胞検体より、疾病情報に対応した多能性幹細胞バンクを構築することが可能である。さらに、健常人由来の血球細胞を初期化した多能性幹細胞を用いて各種分化細胞を誘導することにより、あるいは難治性疾患の患者より入手した血球細胞を用いて疾患のターゲット細胞を誘導することにより、創薬スクリーニングのツールとしての各種ヒトモデル細胞系の構築も可能である。
【実施例】
【0063】
以下、実施例を挙げて本発明をさらに具体的に説明するが、本発明はこれらに限定されるものではない。
【0064】
例1:ヒト骨髄由来単核球細胞からの多能性細胞の誘導(1)
A.ヒト骨髄由来単核球細胞の培養
凍結保存されたヒト骨髄由来単核球細胞(hBMMNCs)2.5×106細胞(Allcells社)を100mmペトリディッシュ2枚に播種し、37℃、5% CO2下、10%FBS入りDMEM培地で培養を開始した。一方のディッシュには、ヒトSCF(100ng/ml)、ヒトTPO(100ng/ml)、およびヒトIL−3(100ng/ml)を添加し、他方のディッシュにはSCF、TPOおよびIL−3に加えてFP6(20ng/ml)を添加した。ここで、SCF、TPOおよびIL−3からなるサイトカインカクテルを「ST3」と呼び、ST3にFP6を加えたカクテルを「ST3FP6」と呼ぶ(以下同様)。培養開始24時間後に、それぞれのディッシュにそれぞれのサイトカインカクテルを同濃度で再び添加した。培養開始48時間後にそれぞれの細胞を回収し、10%FBS入りDMEM培地500μlで懸濁した。ST3存在下で培養した細胞懸濁液にはST3を各300ng/mlの濃度で添加し、ST3FP6存在下で培養した細胞懸濁液には、ST3(各300ng/ml)に加えてFP6を60ng/mlの濃度で添加した。調製したhBMMNCsを次のウィルス感染実験に用いた。
【0065】
B.核初期化遺伝子を含む組換えレトロウィルスベクターの作製
コラーゲンタイプIコート100mmディッシュ12枚に、それぞれ4×106個のG3T−hi細胞(タカラバイオ社)を播種し、37℃、5% CO2下、10% FBS入りDMEM培地で培養した。培養24時間後に、核初期化遺伝子1種類についてディッシュ3枚を用いてウィルスベクターの遺伝子導入を行った。各ディッシュにつき、pGP vector(タカラバイオ社)6μg、pVSV−G vector(CLONTECH社)6μg、およびレトロウィルスベクターpMXs(T. Kitamura et al., Experimental Hematology, 31, pp.1007-1014, 2003;東京大学北村俊雄教授より供与)に核初期化遺伝子を挿入したプラスミド12μgを、遺伝子導入試薬FuGene6(ロシュダイグノスティック社)を用いて共導入した。用いた核初期化遺伝子は、ヒトOct3/4、ヒトSox2、ヒトKlf4、およびヒトc−mycの4種類であった。核初期化遺伝子発現レトロウィルスベクター:pMXshOct3/4、pMXshSox2、pMXshKlf4、およびpMXshc−mycの構築は、山中らの方法に従って行った(K. Takahashi, S. Yamanaka, Cell, 131, pp.861-872, 2007)。遺伝子導入の約48時間後に、核初期化遺伝子の種類ごとに培養液を回収し、培養液を0.45μmフィルターでろ過後、5800×g、4℃で4時間遠心し、培養上清を捨てることでウィルス沈殿を取得した。ウィルス沈殿は全て1つにまとめ、2mlの10%FBS入りDMEM培地で懸濁した。最終的に液量は2.4mlほどになった。
【0066】
C.ヒト骨髄由来単核球細胞のウィルス感染処理
予め50μg/mlのレトロネクチン(タカラバイオ社)でコートされた12ウェルプレートへ、懸濁したウィルス沈殿を1ウェルにつき1.2mlずつ添加し、1080×g、20℃で2時間遠心した。遠心後のプレートに前記のhBMMNCsの細胞懸濁液500μlを各ウェルに添加し、37℃、5% CO2にて培養を開始した。培養開始約24時間後に、それぞれにサイトカインを再び添加した。
【0067】
ウィルス感染開始から約48時間後にトリプシンEDTA液(GIBCO社)で各ウェルの細胞を回収し、予め5×105個/ディッシュのマウス胚性線維芽細胞をフィーダー細胞としたゼラチンコート100mmディッシュに10%FBS入りDMEM培地を用いて細胞を播き直した。それぞれのディッシュにはサイトカインを再び添加した。さらに、約24時間後および約72時間後に、サイトカイン入りの10%FBS入りDMEM培地で培地交換を実施した。その後、ウィルス感染開始から6日後からは、ST3またはST3FP6を含まないヒトES細胞培養用の培地に交換した。用いた培地の組成は、DMEM−F12培地(SIGMA社)500ml、非必須アミノ酸液(SIGMA社)5ml、200mM L−Glutamine(SIGMA社)6.25ml、KNOCKOUTTM Serum Replacement(KSR)(Invitorgen社)125ml、2−メルカプトエタノール(SIGMA社)5μl、5N NaOH 638μl、Human bFGF(UBI社)終濃度5ng/mlであった。
【0068】
その後、毎日培地交換を実施した。ウィルス感染開始から10日後には、両方のディッシュでいくつかのコロニーを確認した(図1B)。この時点でST3FP6存在下で培養したhBMMNCsの場合、比較的大きな扁平コロニーを含んでいたのに対し、ST3存在下で培養したhBMMNCsの場合は出現したコロニーはまだ小さく、マウスES細胞に似た形態のコロニーであった。15日後には各24個のコロニーをピックアップし、残りの細胞を予めフィーダー細胞を播いたゼラチンコート100mmディッシュに再播種した。20日後に再びコロニーをピックアップし、残りの細胞の半分を播種し、さらに残りの半分の細胞について、FACSによりヒトES細胞マーカーSSEA−4の発現確認を行った(図1A)。FACSによる解析では、回収した細胞をstaining medium(SM)(5% FBS、0.05% NaN3、および0.5mM EDTAを含むPBS)3〜5mlで洗浄後、フィコエリスリン結合抗SSEA4抗体(BDバイオサイエンス社)5μlを添加し、氷中で15分静置した。SM6mlで洗浄後、死細胞を検出するための試薬ヨウ化プロピジウム(PI)1μg/mlを含むSM液1mlで再懸濁し、FACS Calibur(BDバイオサイエンス社)またはFACS Aria SORP(BDバイオサイエンス社)を用いて測定を行った。その結果、ST3FP6存在下で培養したhBMMNCsを用いた場合、レトロウィルス感染20日後の時点で、幹細胞マーカーであるSSEA−4を発現する細胞が約40%に達し、FP6を添加しなかった場合の約15%に比して2.5倍以上に達していることが示された。また、ヒトES細胞においてはアルカリフォスファターゼが発現していることが知られている。そこで、27日後の時点で、常法に従ってアルカリフォスファターゼ活性染色を行った(図1C)。その結果、この時点でのコロニーのほとんどがアルカリフォスファターゼ陽性であることを確認した。以上より、ST3FP6存在下で培養したhBMMNCsを用いた場合、多能性幹細胞の出現時期、細胞数において、ST3存在下で培養したhBMMNCsを用いた場合を顕著に上回っていることが明らかとなった。
【0069】
例2:ヒト骨髄由来単核球細胞からの多能性細胞の誘導(2)
A.ヒト骨髄由来単核球細胞の培養
凍結保存されたヒト骨髄由来単核球細胞(hBMMNCs)2.5×106細胞(Allcells社)を100mmペトリディッシュ1枚に播種し、37℃、5% CO2下、10%FBS入りDMEM培地で培養を開始した。培地には、ヒトSCF(100ng/ml)、ヒトTPO(100ng/ml)、ヒトIL−3(100ng/ml)、ヒトFlt3リガンド(100ng/ml)、およびFP6(20ng/ml)を添加した。ここで、SCF、TPO、IL−3、Flt3リガンド、およびFP6からなるサイトカインカクテルを「ST3FLFP6」と呼ぶ(以下同様)。培養開始24時間後に、ST3FLFP6を同濃度で再び添加した。培養開始48時間後に細胞を回収し、総細胞数5×105個の細胞を10%FBS入りDMEM培地500μlで懸濁した。細胞懸濁液には、ST3FL(各300ng/ml)に加え、FP6を60ng/mlの濃度で添加した。調製したhBMMNCsを次のウィルス感染実験に用いた。
【0070】
B.核初期化遺伝子を含む組換えレトロウィルスベクターの作製
コラーゲンタイプIコート100mmディッシュ16枚に、それぞれ2.5×106個のG3T−hi細胞(タカラバイオ社)を播種し、37℃、5% CO2下、10% FBS入りDMEM培地で培養した。培養24時間後に、核初期化遺伝子1種類についてディッシュ4枚を用いてウィルスベクターの遺伝子導入を行った。各ディッシュにつき、pGP vector(タカラバイオ社)6μg、pVSV−G vector(CLONTECH社)6μg、およびレトロウィルスベクターpMXsに核初期化遺伝子を挿入したプラスミド12μgを、遺伝子導入試薬FuGene6(ロシュダイグノスティック社)を用いて共導入した。用いた核初期化遺伝子は、ヒトOct3/4、ヒトSox2、ヒトKlf4、ヒトc−mycの4種類であった。遺伝子導入の約48時間後に、核初期化遺伝子の種類ごとに培養液を回収し、培養液を0.75μmフィルターでろ過後、5000×g、4℃で4時間遠心し、培養上清を捨てることでウィルス沈殿を取得した。ウィルス沈殿は全て1つにまとめ、500μlの10%FBS入りDMEM培地を加えて再懸濁したところ、最終的に液量は1mlほどになった。
【0071】
C.ヒト骨髄由来単核球細胞のウィルス感染処理
予め50μg/mlのレトロネクチン(タカラバイオ社)でコートされた12ウェルプレート中の1つのウェルに、懸濁したウィルス沈殿を添加し、1080×g、20℃で2時間遠心した。遠心後のプレートに前記のhBMMNCsの細胞懸濁液500μlを添加し、37℃、5% CO2にて培養を開始した。培養開始約24時間後に、ST3FLFP6入りの培地で培地交換を実施した。
【0072】
ウィルス感染開始から約48時間後にトリプシンEDTA液(GIBCO社)で細胞を回収し、予め5×105個/ディッシュのマウス胚性線維芽細胞をフィーダー細胞としたゼラチンコート100mmディッシュに、10%FBSおよびST3FLFP6を補充したDMEM培地を用いて細胞を播き直した。さらに、約48時間後に10%FBSおよびST3FLFP6を補充したDMEM培地で培地交換を実施した。その後、ウィルス感染開始から6日後からは、ST3FLFP6を含まないヒトES細胞培養用の培地(例1Cに記載)に交換した。
【0073】
その後、毎日培地交換を実施した。ウィルス感染開始から11日後には、両方のディッシュでいくつかのコロニーを確認した。18日後には各24個のコロニーをピックアップし、残りの細胞を予めフィーダー細胞を播いたゼラチンコート100mmディッシュに再播種した。22日後の細胞を回収し、例1の方法に倣い、FACSによりヒトES細胞マーカーであるSSEA−4の発現を解析した。その結果を図2に示す。
【0074】
例3:レトロウィルス感染性ヒト骨髄由来単核球細胞の解析
A.ヒト骨髄由来単核球細胞のレトロウィルス感染性の分析
凍結保存されたhBMMNCs(Allcells社)を用いて、レトロウィルスの感染効率についての検討を行った。例1の方法に倣い、核初期化因子発現pMXsベクターの代わりに、緑色蛍光タンパク質EGFPを発現するpMXsIRES−EGFP(pMXsIG)ベクター(T. Kitamura et al., Experimental Hematology, 31, pp.1007-1014, 2003;東京大学北村俊雄教授より供与)を用いて実施した。
【0075】
hBMMNCsの培養条件は以下の3種類で実施した:(i)サイトカインを添加せずに、解凍後の細胞を用いてレトロウィルス感染を行った;(ii)例1と同様に、10%FBS入りDMEM培地にヒトSCF(100ng/ml)、ヒトTPO(100ng/ml)、およびヒトIL−3(100ng/ml)を加えて(ST3)、前培養およびレトロウィルス感染を実施した;(iii)例1と同様に、10%FBS入りDMEM培地にヒトSCF(100ng/ml)、ヒトTPO(100ng/ml)、ヒトIL−3(100ng/ml)、およびFP6(20ng/ml)を加えて(ST3FP6)、前培養およびレトロウィルス感染を実施した。
【0076】
レトロウィルスの調製および感染方法は例1と同様に実施した。感染2日後に細胞を回収し、細胞をSM液で洗浄後、PI(1μg/ml)を含むSM液1mlで再懸濁し、FACS Calibur(BDバイオサイエンス社)を用いてFACS解析を行った(図3)。その結果、サイトカインを添加した培養を行わない(i)の場合、レトロウィルスに感染する細胞は全く検出できなかったのに対して、ST3を添加((ii))することにより、生細胞中約4%の細胞がレトロウィルスに感染するようになり、さらにFP6を加えてST3FP6を添加((iii))することにより、レトロウィルスに感染する細胞は約8%に増加した。一方で、本例にて使用したレトロウィルスをコントロール細胞のNIH3T3細胞に感染させたところ、99%の効率で感染が可能であったことから、レトロウィルスそのものの感染能力は十分であった。
【0077】
B.感染細胞の表面抗原マーカーの分析
次に、例1および例2と同様にST3、ST3FP6またはST3FLFP6存在下で培養したhBMMNCsに対して、pMXsIGベクターより作製したレトロウィルスを感染させた場合の、EGFP陽性の感染細胞について、各種表面抗原マーカーに対する抗体を用いて免疫染色を行い、FACSを用いて解析を行った。
【0078】
回収した細胞をSM液3〜5 mlで洗浄後、各種抗体5μlを添加し、氷中で15分静置した。SM6mlで洗浄後、PI(1μg/ml)を含むSM液1mlで再懸濁し、FACS Calibur(BDバイオサイエンス社)にて測定を行った。用いた抗体は、アロフィコシアニン結合抗ヒトCD45抗体、アロフィコシアニン結合抗ヒトCD34抗体、フィコエリスリン結合抗ヒトCD38抗体、アロフィコシアニン結合抗ヒトCD11b抗体(以上、BDバイオサイエンス社)、フィコエリスリン結合抗ヒトCD3抗体、フィコエリスリン結合抗ヒトCD117抗体、およびフィコエリスリン結合抗ヒトCD19抗体(以上、IMMUNOTECH社)であった。CD45抗原は、すべてのヒト白血球、すなわちリンパ球、好酸球、単球、好塩基球、および好中球の表面に発現する。CD34抗原は、最も未分化な造血幹細胞と造血前駆細胞に発現する。CD38抗原は活性化したTおよびBリンパ球、NK細胞、単球、形質細胞および胸腺髄質細胞に発現する。CD11b抗原は、骨髄単球系細胞にみられ、NK細胞、顆粒球、単球/マクロファージに強く発現する。CD3抗原は、成熟T細胞および胸腺細胞に発現する。CD117抗原はごく少数の正常骨髄細胞にしか発現していないが、肥満細胞にも発現する。CD19抗原は初期B細胞を含むすべての正常B細胞に発現するが、形質細胞に成熟すると消失する。
【0079】
解析の結果、レトロウィルス感染細胞のほぼ全てにおいて白血球のマーカーであるCD45は陽性であり、ST3ではほぼすべてがCD34陽性細胞であり、ST3FP6またはST3FLFP6では約60%がCD34陽性細胞であった。一方で、CD11b、CD3、CD19といったマーカーはいずれも陰性であった(図4)。
【0080】
例4:レトロウィルス感染性ヒト末梢血由来単核球細胞の解析
凍結保存されたヒト末梢血由来単核球細胞(hPBMNCs)(Allcells社)に対して、例3と同様にpMXsIGベクターを用いてレトロウィルスの感染実験を行った。サイトカインカクテルを用いてhPBMNCsを培養した場合の感染効率とEGFP陽性の感染細胞について、各種表面抗原マーカーに対する抗体を用いて免疫染色を行い、FACSを用いて感染細胞の同定を行った。
【0081】
hPBMNCsの培養には、10%FBS入りDMEM培地にヒトSCF(100ng/ml)、ヒトTPO(100ng/ml)、ヒトIL−3(100ng/ml)、FP6(20ng/ml)、Flt3リガンド(100ng/ml)、ヒトGM−CSF(100ng/ml)、ヒトM−CSF(50ng/ml)を添加して培養したGM培養系と、ヒトSCF(100ng/ml)、ヒトTPO(100ng/ml)、ヒトIL−3(100ng/ml)、FP6(20ng/ml)、LPS(1μg/ml)、抗ヒトCD40抗体 #341(キリンファーマ社)(100ng/ml)、抗ヒトCD3抗体(OKT3 ヤンセンファーマ)(1μg/ml)、抗ヒトCD28抗体(eBioscience)(1μg/ml)を添加して前培養したTB培養系の2通りの培養系を用いて、レトロウィルス感染を実施した。レトロウィルスの調製および感染方法は例1と同様に実施した。感染2日後に細胞を回収し、回収した細胞をSM液3〜5mlで洗浄後、各種抗体5μlを添加し、氷中で15分静置した。SM6mlで洗浄後、PI(1μg/ml)を含むSM液1mlで再懸濁し、FACS Calibur(BDバイオサイエンス社)にて測定を行った。
【0082】
用いた抗体は、アロフィコシアニン結合抗ヒトCD45抗体、アロフィコシアニン結合抗ヒトCD34抗体、フィコエリスリン結合抗ヒトCD38抗体、アロフィコシアニン結合抗ヒトCD11b抗体、フィコエリスリン結合抗ヒトCD13抗体(以上、BDバイオサイエンス社)、フィコエリスリン結合抗ヒトCD3抗体、フィコエリスリン結合抗ヒトCD117抗体、およびフィコエリスリン結合抗ヒトCD19抗体(以上、IMMUNOTECH社)であった。CD13抗原は、正常末梢血の好中球、好酸球、好塩基球および単球など、ほとんどの骨髄系細胞に発現する。
【0083】
解析の結果、レトロウィルス感染細胞のほぼ全てにおいて白血球のマーカーであるCD45は陽性であり、GM培養系では骨髄細胞を用いた場合と同様に、約半数がCD34陽性であった。TB培養系においてはT細胞のマーカーであるCD3抗原陽性であった(図5)。本系を用いることにより、末梢血中に存在するT細胞などから多能性細胞を誘導できる可能性が考えられる。
【0084】
例5:ヒト誘導多能性細胞クローンの表面抗原解析
例1および例2の結果クローニングした細胞株について、コロニー形態の観察およびFACSによりヒトES細胞マーカーSSEA−4の発現確認を行った。FACS解析の方法は例1と同様に行った。
【0085】
ST3の培養条件を用いて取得したクローン#16および#30、ST3FPの培養条件を用いて取得したクローン#22および#33、ST3FLFPの培養条件を用いて取得したクローン#1および#4について解析した結果を図6に示す。いずれのクローンも幹細胞マーカーであるSSEA−4を有意に発現していることを確認した。
【0086】
例6:ヒト誘導多能性細胞クローンのRT−PCR解析
例5で解析したクローンを含め、例1および例2の結果取得した多能性幹細胞クローンについて、幹細胞特異的遺伝子のmRNAの発現をRT−PCR法を用いて解析した。
【0087】
各クローン細胞をマウス胚性線維芽細胞のフィーダー細胞上に播種し、ヒトES細胞用培地にて培養した細胞よりRNAを回収した。用いた培地の組成は、DMEM−F12培地(SIGMA社)500ml、非必須アミノ酸液(SIGMA社)5ml、200mM L−Glutamine(SIGMA社)6.25ml、KNOCKOUTTM Serum Replacement(KSR)(Invitorgen社)125ml、2−メルカプトエタノール(SIGMA社)5μl、5N NaOH 638μl、Human bFGF(UBI社)終濃度5ng/mlであった。回収した細胞はPBS緩衝液で洗浄後、細胞沈殿を液体窒素中にて凍結した。コントロール細胞として、凍結融解後のBMMNCsをフィーダー細胞上で数時間培養した細胞およびフィーダー細胞(MEF細胞)を用意した。凍結後の細胞サンプルより、RNeasy Plus Mini Kit(QIAGEN社)を用いて、添付プロトコルに従って全RNAを調製した。調製したRNAを用いて、SuperScriptIII First Strand Synthesis System(Invitrogen社)により添付のプロトコルに従ってcDNAを合成した。合成したcDNAを鋳型として、LA Taq DNA polymerase(タカラバイオ社)を用いて、96℃−20秒、55℃−30秒、および72℃−30秒からなる工程を1サイクルとして35サイクルのPCR反応を実施した。PCR反応に用いたプライマーは山中らと同様のものを用いており(K. Takahashi, S. Yamanaka, Cell, 131, pp.861-872, 2007)、各プライマーの塩基配列を表1に示す。各PCR反応は各遺伝子についてフォワード(Fw)プライマーとリバース(Rv)プライマーとの組合せを用いて実施した。RT反応およびPCR反応のコントロールとして、ハウスキーピング遺伝子であるglyceraldehyde−3−phosphate dehydrogenase(GAPDH)遺伝子を用いた。PCR反応物をエチジウムブロマイド入りのアガロースゲルにて電気泳動し、UVトランスイルミネーターにより観察した結果を図7に示す。解析の結果、いずれのクローンも幹細胞マーカー遺伝子を発現していることを確認した。
【0088】
【表1】
【0089】
例7:マウス骨髄由来単核球細胞からの多能性細胞の誘導(1)
A.マウス骨髄由来単核球細胞の取得
未分化な多能性幹細胞において緑色蛍光タンパク質EGFPを発現することが知られているOct4−EGFPトランスジェニックマウス(10〜20週齢)(K. Ohbo et al., Dev. Biol., 258, pp.209-225, 2003; 横浜市立大学 大保和之准教授より供与)の大腿骨内の骨髄を採取し、PBSに懸濁した。定法に従い(高津聖志、免疫研究の基礎技術、羊土社1995)、骨髄から比重遠心法により単核球細胞画分を濃縮し、これをマウス骨髄単核球細胞(mBMMNCs)として用いた。
【0090】
B.核初期化遺伝子を含む組換えレトロウィルスベクターの作製
コラーゲンタイプIコート60mmディッシュ10枚にそれぞれ1×106個、および6ウェルプレートの各ウェルに1×105個のレトロウィルス用のパッケージング用細胞:PLAT−E細胞(S.Morita et al., Gene Therapy, 7, pp.1063-1066, 2000;東京大学北村俊雄教授より供与)を播種し、37℃、5% CO2下、10% FBS入りDMEM培地で培養した。培養24時間後に、マウス核初期化遺伝子1種類について60mmディッシュ2枚を用いてウィルスベクターの遺伝子導入を行った。各ディッシュにつき、レトロウィルスベクターpMXsに核初期化遺伝子を挿入したプラスミド3μgを遺伝子導入試薬FuGene6(ロシュダイグノスティック社)を用いて導入した。用いた核初期化遺伝子は、マウスOct3/4、マウスSox2、マウスKlf4、およびマウスc−mycの4種類であった。核初期化遺伝子発現レトロウィルスベクター、pMXsmOct3/4、pMXsmSox2、pMXsmKlf4、およびpMXsmc−mycの構築は、山中らの方法に従って行った(K. Takahashi, S. Yamanaka, Cell, 126, pp.663-676, 2006)。その他に、60mmディッシュ1枚と6ウェルプレートの3ウェル分において、上記4種類の遺伝子各3μgのDNAを併せて導入した。また、コントロールとして緑色蛍光タンパク質EGFPを発現するpMXsIGベクター3μgを60mmディッシュ1枚に導入した。遺伝子導入の約48時間後に、各核初期化遺伝子について60mmディッシュ1枚ずつを集めて0.22μmフィルターでろ過後、5800×g、4℃で4時間遠心し、培養上清を捨てた後に3mlのマウスES細胞用培地で再懸濁した濃縮ウィルスサンプル(i)を取得した。マウスES細胞用培地としては、20%FBS入りDMEM培地に1/100容の2-メルカプトエタノール(SIGMA社)、1/1000容のESGRO(CHEMICON社)を添加したものを用いた。また、各核初期化遺伝子について60mmディッシュ1枚ずつを集めて0.22μmフィルターでろ過しただけのものを4ウィルスサンプル(ii)とした。4種類の遺伝子を同時に導入した60mmディッシュ1枚と6ウェルプレートの3ウェル分の培養上清に
ついては、0.22μmフィルターでろ過し、4遺伝子サンプル(iii)とした。コントロールのpMXsIGベクターを導入した培養上清については、0.22μmフィルターでろ過したものをコントロールサンプル(iv)とした。
【0091】
C.マウス骨髄由来単核球細胞のウィルス感染処理
予め33μg/mlのレトロネクチン(タカラバイオ社)でコートされた12ウェルプレートの2ウェルずつに上記(i)〜(iv)のサンプルを添加し、1080×g、25℃で2時間遠心した。遠心後のプレートからウィルスサンプルを除去し、新たなマウスES細胞用培地を1mlずつ添加した。さらに、(i)〜(iv)の各サンプルの一方のウェルには、マウスSCF(100ng/ml)、ヒトTPO(100ng/ml)、およびマウスIL−3(100ng/ml)(ST3)を添加した。その後、各ウェルに前記のOct4−EGFPマウスより調製したmBMMNCsを1×105個/ウェルになるよう添加し、37℃、5% CO2にて培養を開始した。
【0092】
また、ウィルス感染前日に、Oct4EGFPマウス由来胚性線維芽細胞(MEF)を12ウェルプレートに2×104個/ウェルで播種して培養していた各ウェルについても、上記(i)〜(iv)のサンプルを1ウェルずつに添加し、1080×g、32℃で40分間遠心した後、37℃、5% CO2にて培養を開始した。
【0093】
ウィルス感染2日後のコントロールサンプルにおけるEGFP発現細胞の割合は、MEF細胞では34%、サイトカインなしのmBMMNCsでは55%、ST3存在下のmBMMNCsでは60%であった。
【0094】
4ウィルスサンプル((ii))を感染させたmBMMNCsについては、サイトカインの添加、非添加に関わらず、ウィルス感染開始から5日後にはOct4−EGFPレポーター遺伝子によるEGFP遺伝子の発現が認められた(図8A・B)。一方で、MEFについてはこの時点ではEGFP遺伝子発現細胞の存在は認められなかった(図8C)。ウィルス感染7日後に、mBMMNCs由来各サンプルについて、Oct4−EGFPレポーター遺伝子の発現と血球細胞マーカーであるCD45抗原の発現のFACS解析を実施した。この解析のために、回収した細胞をSM液3〜5 mlで洗浄後、アロフィコシアニン結合抗マウスCD45抗体(BDバイオサイエンス社)5μlを添加し、氷中で15分静置した。SM6mlで洗浄後、PI(1μg/ml)を含むSM液1mlで再懸濁し、FACS Calibur(BDバイオサイエンス社)にて測定を行った。その結果を図9および表2に示す。FACS解析前の細胞の様子を図10に示す。感染前のmBMMNCsはそのほぼ全てがCD45抗原陽性であるのに対し、この時点でのOct4−EGFP陽性細胞の多くは既にCD45抗原陰性であった。また同時に、FACS sortingによりEGFP陽性細胞の分取を行い、それらの細胞からクローニングを実施し、マウス骨髄由来誘導多能性幹細胞(mBMiPS)クローン#7および#13を取得した。取得した各クローンについてもFACS解析を実施した(図11)。
【0095】
【表2】
【0096】
FACSによる解析では、回収した細胞をSM液3〜5 mlで洗浄後、各種抗体5μlを添加し、氷中で15分静置した。SM6mlで洗浄後、PI(1μg/ml)を含むSM液1mlで再懸濁し、FACS Calibur(BDバイオサイエンス社)またはFACS Aria SORP(BDバイオサイエンス社)を用いて測定を行った。抗体としては、アロフィコシアニン結合抗マウスCD45抗体(BDバイオサイエンス社)、およびAlexa Fluor647結合抗マウスSSEA−1抗体を用いた。Alexa Fluor647結合抗マウスSSEA−1抗体は、抗マウスSSEA−1抗体(CHEMICON社)100μgをAlexa Fluor 647 Protein Labeling Kit(Invitrogen社)を用いて標識することにより調製した。
【0097】
例8:マウス骨髄由来単核球細胞からの多能性細胞の誘導(2)
A.核初期化遺伝子を含む組換えレトロウィルスベクターの作製
コラーゲンタイプIコート60mmディッシュ12枚にそれぞれ7×105個のPLAT−E細胞を播種し、37℃、5% CO2下、10% FBS入りDMEM培地で培養した。培養24時間後に、各種遺伝子導入を行った。12枚のうち4枚については、マウス核初期化遺伝子1種類について60mmディッシュ1枚を用いてウィルスベクターの遺伝子導入を行った。各ディッシュにつき、レトロウィルスベクターpMYs(T. Kitamura et al., Experimental Hematology, 31, pp.1007-1014, 2003;東京大学北村俊雄教授より供与)に核初期化遺伝子を挿入したプラスミド4μgを遺伝子導入試薬FuGene6(ロシュダイグノスティック社)を用いて導入した。用いた核初期化遺伝子は、ヒトOct3/4、ヒトSox2、ヒトKlf4、およびヒトc−mycの4種類であった。60mmディッシュ4枚について、上記4種類の遺伝子各4μgのDNAを併せて導入した。また、コントロールとして緑色蛍光タンパク質EGFPを発現するpMYsIRES−EGFP(pMYsIG)ベクター16μgを60mmディッシュ4枚に導入した。
【0098】
遺伝子導入の約48時間後に、各核初期化遺伝子を個別に導入した60mmディッシュ計4枚を集めて4ウィルスサンプルとし、4種同時に導入した計4枚を集めて4遺伝子サンプルとし、pMYsIGベクターを導入した計4枚を集めてコントロールサンプルとした。いずれのサンプルについても、0.22μmフィルターでろ過後、5800×g、4℃で4時間遠心し、培養上清を捨てた後に2.5mlのマウスES細胞用培地で再懸濁し、再び0.22μmフィルターでろ過を行った。
【0099】
B.細胞のウィルス感染処理
レトロネクチン(タカラバイオ社)でコートされた12ウェルプレートの2ウェルずつに4ウィルスサンプル、4遺伝子サンプル、およびコントロールサンプルをそれぞれ添加し、1080×g、25℃で2時間遠心した。遠心後のプレートからウィルスサンプルを除去し、各サンプルのウェルに対して、マウスES細胞用培地で懸濁したOct4−EGFPマウスBMMNCsまたはOct4−EGFPマウスMEF細胞を1×105個/ウェルになるよう添加し、37℃、5% CO2にて培養を開始した。
【0100】
また、ウィルス感染前日に、Oct4−EGFPマウスMEF細胞をゼラチンコート12ウェルプレートに2×104個/ウェルで播種して培養していた各ウェルについても、上記3種のサンプルを1ウェルずつに添加し、1080×g、25℃で40分間遠心した後、37℃、5% CO2にて培養を開始した。
【0101】
ウィルス感染開始2日後のコントロールサンプルにおけるEGFP発現細胞の割合は、レトロネクチンコートプレートで感染させたMEF細胞では95%、ゼラチンコートプレートで予め培養していたMEF細胞では65%、レトロネクチンコートプレートで感染させたmBMMNCsでは26%であった(図12)。その後、ウィルス感染開始から7日目にFACS sortingを行い(図13)、12日目に出現コロニー数を計測した。その結果を表3にまとめた。
【0102】
【表3】
【0103】
例9:マウス骨髄由来多能性細胞の多分化能解析
mBMiPSクローン#7および#13について分化多能性を評価するため、ヌードマウス(Balb/cAJcl−nu)の皮下への移植を行った。未分化ES細胞は生体内に移植されると奇形腫を形成することが知られている。コントロール細胞としてマウスES細胞E14株を使用した。ヌードマウス1匹あたり各細胞1×106個を100μl血清非含有DMEM培地で懸濁して皮下投与した。その結果、マウスES細胞を移植した場合と同様の大きさの奇形腫瘍がmBMiPS細胞を移植した全例でも形成された。移植後2週間後および4週間後の腫瘍を摘出し、ホルマリン固定により組織標本を作製した。組織学的な解析の結果、いずれのクローン由来の奇形腫においても、神経組織、軟骨組織、筋組織、脂肪組織、食道粘膜様組織、気管上皮などが認められたことから、mBMMNCsより誘導された各クローンが多能性を有することが証明された(図14)。
【0104】
さらにmBMiPSクローン#7細胞をBalb/cマウスの受精卵から作製した胚盤胞に注入後、偽妊娠マウスに移植することによりキメラ個体を作出することを試みた。その結果、Balb/cマウスの産仔は白色の毛色を示すはずのところ、mBMiPS細胞に由来する黒色の毛色を有するキメラマウスが誕生した(図15および表4)。毛色は全身の細胞の一部を反映するものであり、他の組織・臓器においてもmBMiPS細胞に由来する細胞が個体を構成しているものと考えられた。
【0105】
【表4】
【0106】
例10:マウス骨髄由来多能性細胞の発現プロファイル解析
mBMiPSクローン#7および#13について、各5×105個の細胞からRNAeasy mini plus kit (QIAGEN社)を用いてRNAを調製し、Agilent Expression Array (Agilent社)を用いて発現プロファイル解析を実施した。コントロールとしてマウスES細胞E14株およびマウスBMMNC細胞を使用した。その結果、図16に示すような各遺伝子の発現パターンを示し、mBMiPSクローン#7および#13がES細胞と酷似した細胞であることが明らかとなった。
【図面の簡単な説明】
【0107】
【図1】ヒト骨髄由来単核球細胞(hBMMNCs)から誘導された多能性幹細胞の未分化マーカーの発現量を評価したFACS解析の結果、誘導された多能性幹細胞が形成するコロニーの形態および誘導された多能性幹細胞のアルカリフォスファターゼの活性染色の結果を示した。[A]は hBMMNCsに対して核初期化因子を発現するレトロウィルスを感染させた後20日目の未分化細胞マーカーSSEA−4の発現を示した。図の水平軸がSSEA−4の発現量を示し、垂直軸が示すPIの染色強度が低いものが生細胞を示した。図中の等高線は解析した細胞の存在頻度を示した。「ST3FP6」はレトロウィルス感染前2日間および核初期化プロセスにおいて、サイトカインカクテルとしてSCF、TPO、IL−3、FP6をhBMMNCsと共存させていることを示し、「ST3」はレトロウィルス感染前2日間および核初期化プロセスにおいて、サイトカインカクテルとしてSCF、TPO、IL−3をhBMMNCsと共存させていることを示した。[B]は核初期化プロセスにおいて形成された細胞コロニーの形態を示した。「Day10」、「Day20」はレトロウィルス感染開始からの日数を示した。[C]はhBMMNCsから誘導された多能性幹細胞のレトロウィルス感染開始から27日目のアルカリフォスファターゼ染色を示した。
【図2】ヒト骨髄由来単核球細胞(hBMMNCs)から誘導された多能性幹細胞の未分化マーカーの発現量を評価したFACS解析の結果および誘導された多能性幹細胞が形成するコロニーの形態を示した。レトロウィルス感染前2日間および核初期化プロセスにおいて、サイトカインカクテルとしてSCF、TPO、IL−3、FP6、FltリガンドをhBMMNCsと共存させた場合を示した。[A]は hBMMNCsに対して核初期化因子を発現するレトロウィルスを感染させた後22日目の未分化細胞マーカーSSEA−4の発現を示した。図の水平軸がSSEA−4の発現量を示し、垂直軸が細胞数を示した。 [B]は核初期化プロセスにおいて形成された細胞コロニーの形態を示した。
【図3】pMXsIGベクターを用いて調製されたレトロウィルスのヒト骨髄由来単核球細胞(hBMMNCs)に対する感染効率を評価するためのFACS解析結果を示した。「PI」はヨウ化プロピジウムによる染色強度を示し、図中の等高線は解析した細胞の存在頻度を示した。各図の水平軸は感染したレトロウィルスゲノムから発現する緑色蛍光タンパク質EGFPの発現強度を示した。「サイトカイン培養なし」はレトロウィルス感染前後でサイトカイン類を共存させない場合を示した。「ST3添加培養」はレトロウィルス感染2日前より、FACS解析を行ったレトロウィルス感染2日後までの間、hBMMNCsとSCF、TPO、IL−3のサイトカインを共存させた場合を示した。「ST3FP6添加培養」はレトロウィルス感染2日前より、FACS解析を行ったレトロウィルス感染2日後までの間、hBMMNCsとSCF、TPO、IL−3、FP6のサイトカインを共存させた場合を示した。
【図4】pMXsIGベクターを用いて調製されたレトロウィルスが感染したヒト骨髄由来単核球細胞(hBMMNCs)の細胞系譜を同定評価するためのFACS解析結果を示した。レトロウィルス感染2日前よりFACS解析を行ったレトロウィルス感染2日後までの間、hBMMNCsを、SCF、TPO、およびIL−3サイトカインと共存させた場合をST3として示し、SCF、TPO、IL−3、およびFP6サイトカインと共存させた場合をST3FP6として示し、SCF、TPO、IL−3、FP6、およびFlt3リガンドと共存させた場合をST3FLFP6として示した。各図の水平軸は感染したレトロウィルスゲノムから発現する緑色蛍光タンパク質EGFPの発現強度を示し、図中の等高線は解析した細胞の存在頻度を示した。「CD45APC」はアロフィコシアニン結合抗ヒトCD45抗体による染色強度を示した。「CD34APC」はアロフィコシアニン結合抗ヒトCD34抗体による染色強度を示した。「CD38PE」はフィコエリスリン結合抗ヒトCD38抗体による染色強度を示した。「CD117PE」はフィコエリスリン結合抗ヒトCD117抗体による染色強度を示した。「CD11bAPC」はアロフィコシアニン結合抗ヒトCD11b抗体による染色強度を示した。「CD3PE」はフィコエリスリン結合抗ヒトCD3抗体による染色強度を示した。「CD19PE」はフィコエリスリン結合抗ヒトCD19抗体による染色強度を示した。
【図5】pMXsIGベクターを用いて調製されたレトロウィルスが感染したヒト末梢血由来単核球細胞(hPBMNCs)の細胞系譜を同定評価するためのFACS解析結果を示した。レトロウィルス感染2日前よりFACS解析を行ったレトロウィルス感染2日後までの間、hPBMNCsをヒトSCF、ヒトTPO、ヒトIL−3、FP6、Flt3リガンド、ヒトGM−CSF、ヒトM−CSFと共存させた場合をGM培養として示し、ヒトSCF、ヒトTPO、ヒトIL−3、FP6、LPS、抗ヒトCD40抗体、抗ヒトCD3抗体、抗ヒトCD28抗体の因子と共存させた場合をTB培養として示した。各図の水平軸は感染したレトロウィルスゲノムから発現する緑色蛍光タンパク質EGFPの発現強度を示し、図中の等高線は解析した細胞の存在頻度を示した。「CD45APC」はアロフィコシアニン結合抗ヒトCD45抗体による染色強度を示した。「CD34APC」はアロフィコシアニン結合抗ヒトCD34抗体による染色強度を示した。「CD38PE」はフィコエリスリン結合抗ヒトCD38抗体による染色強度を示した。「CD117PE」はフィコエリスリン結合抗ヒトCD117抗体による染色強度を示した。「CD11bAPC」はアロフィコシアニン結合抗ヒトCD11b抗体による染色強度を示した。「CD3PE」はフィコエリスリン結合抗ヒトCD3抗体による染色強度を示した。「CD19PE」はフィコエリスリン結合抗ヒトCD19抗体による染色強度を示した。「CD13PE」はフィコエリスリン結合抗ヒトCD13抗体による染色強度を示した。
【図6】クローニングしたヒト誘導多能性細胞株について、コロニー形態およびFACSによるヒトES細胞マーカーSSEA−4の発現解析結果を示した。図の水平軸がSSEA−4の発現量を示し、垂直軸が細胞数を示した。
【図7】クローニングしたヒト誘導多能性細胞株についてのRT−PCR解析結果を示した。図の水平方向に各細胞株を配置し、ST3、ST3FP6、ST3FLFP6は各細胞の誘導条件を示し、その下の数字が各クローン番号を示した。図の垂直方向には多能性幹細胞マーカーとして知られる遺伝子を配置し、コントロール遺伝子としてハウスキーピング遺伝子であるGAPDHの結果を示した。図の黒色のバンドが各遺伝子についての発現を示している。
【図8】Oct4−EGFPトランスジェニックマウス骨髄由来単核球細胞(mBMMNCs)あるいはOct4−EGFPトランスジェニックマウス由来の胚性線維芽細胞(MEF)から誘導された多能性幹細胞が形成するコロニーの形態および未分化性のマーカーであるOct4−EGFPレポーター遺伝子からのEGFPタンパク質の発現を示した。[A]はOct4−EGFPトランスジェニックマウス由来のmBMMNCsに対して核初期化因子を発現するレトロウィルスを感染させた後5日目の細胞の形態および未分化性マーカーOct4−EGFPの発現を示した。「4遺伝子サンプル」はレトロウィルスを調製する際に、4つの核初期化遺伝子を同時にパッケージング細胞へ導入してレトロウィルスを作製した場合を示した。「4ウィルスサンプル」はレトロウィルスを調製する際に、4つの核初期化遺伝子をそれぞれ個別にパッケージング細胞へ導入してレトロウィルスを作製した後に、レトロウィルスを混合し細胞に感染させた場合を示した。[B]はOct4−EGFPトランスジェニックマウス由来のmBMMNCsにレトロウィルスを感染させる際に、SCF、TPO、IL−3のサイトカインを共存させた場合の、レトロウィルス感染後5日目の細胞の形態および未分化性マーカーOct4−EGFPの発現を示した。 [C]はOct4−EGFPトランスジェニックマウス由来のMEFに対して核初期化因子を発現するレトロウィルスを感染させた後5日目の細胞の形態および未分化性マーカーOct4−EGFPの発現を示した。
【図9】Oct4−EGFPトランスジェニックマウスの骨髄由来単核球細胞(mBMMNCs)から誘導された多能性幹細胞について、未分化性のマーカーであるOct4−EGFPレポーター遺伝子からのEGFPタンパク質の発現と血球細胞のマーカーであるCD45抗原の発現をFACSにて解析した結果を示した。Oct4−EGFPトランスジェニックマウス由来のmBMMNCsに対して核初期化因子を発現するレトロウィルスを感染させた後7日目の解析を示した。「4遺伝子」はレトロウィルスを調製する際に、4つの核初期化遺伝子を同時にパッケージング細胞へ導入してレトロウィルスを作製した場合を示した。「4ウィルス」はレトロウィルスを調製する際に、4つの核初期化遺伝子をそれぞれ個別にパッケージング細胞へ導入してレトロウィルスを作製した後に、レトロウィルスを混合し細胞に感染させた場合を示した。「濃縮ウィルス」は「4ウィルス」と同様に調製したレトロウィルスを濃縮して感染に用いた場合を示した。各図の水平軸は感染したレトロウィルスゲノムから発現する緑色蛍光タンパク質EGFPの発現強度を示した。「CD45APC」はアロフィコシアニン結合抗ヒトCD45抗体による染色強度を示し、図中の等高線は解析した細胞の存在頻度を示した。
【図10】Oct4−EGFPトランスジェニックマウス骨髄由来単核球細胞(mBMMNCs)あるいはOct4−EGFPトランスジェニックマウス由来の胚性線維芽細胞(MEF)から誘導された多能性幹細胞が形成するコロニーの形態および未分化性のマーカーであるOct4−EGFPレポーター遺伝子からのEGFPタンパク質の発現を示した。Oct4−EGFPトランスジェニックマウス由来のmBMMNCsあるいはMEFに対して核初期化因子を発現するレトロウィルスを感染させた後7日目の様子を示した。「mBMMNCs」はOct4−EGFPトランスジェニックマウス由来のmBMMNCsから誘導された細胞を示した。「MEF」はOct4−EGFPトランスジェニックマウス由来のMEFから誘導された細胞を示した。
【図11】Oct4−EGFPトランスジェニックマウスの骨髄由来単核球細胞(mBMMNCs)から誘導された多能性幹細胞クローンについて、未分化性のマーカーであるOct4−EGFPレポーター遺伝子からのEGFPタンパク質の発現とマウス未分化細胞のマーカーであるSSEA−1抗原の発現をFACSにて解析した結果および多能性幹細胞クローンが形成するコロニーの形態とコロニーにおける未分化性のマーカーであるOct4−EGFPレポーター遺伝子からのEGFPタンパク質の発現を示した。[A]はOct4−EGFPトランスジェニックマウス由来のmBMiPS細胞クローン#7およびクローン#13について未分化性マーカーOct4−EGFPの発現およびマウス未分化細胞のマーカーであるSSEA−1抗原の発現をFACSにて解析した結果を示した。「IgG APC」はアロフィコシアニン結合コントロールIgGによる染色強度を示した。「SSEA−1 Alexa647」はAlexa647結合抗マウスSSEA−1抗体による染色強度を示した。各図の水平軸は未分化性マーカーOct4−EGFPの発現強度を示し、図中の等高線は解析した細胞の存在頻度を示した。[B]はOct4−EGFPトランスジェニックマウス由来のmBMiPS細胞クローン#7およびクローン#13についてマウス未分化細胞のマーカーであるSSEA−1抗原の発現をFACSにて解析した結果を示した。水平軸はマウス未分化細胞のマーカーであるSSEA−1抗原の発現強度を示し、垂直軸は細胞数を示した。図の実線がAlexa647結合抗マウスSSEA−1抗体で染色した場合をしめし、破線はアロフィコシアニン結合コントロールIgGで染色した場合を示した。[C]はOct4−EGFPトランスジェニックマウス由来のmBMiPS細胞クローン#7およびクローン#13が形成するコロニーの形態および未分化性のマーカーであるOct4−EGFPレポーター遺伝子からのEGFPタンパク質の発現を示した。
【図12】pMYsIGベクターを用いて調製されたレトロウィルスのOct4−EGFPトランスジェニックマウス骨髄由来単核球細胞(mBMMNCs)あるいはOct4−EGFPトランスジェニックマウス由来の胚性線維芽細胞(MEF)に対する感染効率を評価するためのFACS解析結果を示した。図の水平軸は感染したレトロウィルスゲノムから発現する緑色蛍光タンパク質EGFPの発現強度を示し、垂直軸は細胞数を示した。レトロウィルス感染後2日目の解析結果を示した。「BMMNCs」はOct4−EGFPトランスジェニックマウス由来mBMMNCsを用いた場合を示し、「ゼラチン上に予め培養MEF」はウィルス感染前日からゼラチンコートプレート上で培養していたOct4−EGFPマウスMEFを用いた場合を示し、「レトロネクチン上MEF」はBMMNCsと同様にレトロネクチンコートプレートで感染させたMEFを用いた場合を示した。
【図13】Oct4−EGFPトランスジェニックマウスの骨髄由来単核球細胞(mBMMNCs)あるいはMEFからpMYs核初期化遺伝子ベクターを用いて調製されたレトロウィルスを用いて誘導された多能性幹細胞について、未分化性のマーカーであるOct4−EGFPレポーター遺伝子からのEGFPタンパク質の発現とマウス未分化細胞のマーカーであるSSEA−1抗原の発現をFACSにて解析した結果および多能性幹細胞クローンが形成するコロニーの形態とコロニーにおける未分化性のマーカーであるOct4−EGFPレポーター遺伝子からのEGFPタンパク質の発現を示した。[A]はレトロウィルス感染7日後のOct4−EGFPトランスジェニックマウス由来mBMMNCsおよびMEFの未分化性マーカーOct4−EGFPの発現およびマウス未分化細胞のマーカーであるSSEA−1抗原の発現をFACSにて解析した結果を示した。「SSEA−1 Alexa647」はAlexa647結合抗マウスSSEA−1抗体による染色強度を示した。各図の水平軸は未分化性マーカーOct4−EGFPの発現強度を示し、図中の等高線は解析した細胞の存在頻度を示した。[B]はレトロウィルス感染7日後にOct4−EGFPトランスジェニックマウス由来のmBMMNCsおよびMEFが形成するコロニーの形態および未分化性のマーカーであるOct4−EGFPレポーター遺伝子からのEGFPタンパク質の発現を示した。[C]はレトロウィルス感染12日後にOct4−EGFPトランスジェニックマウス由来のmBMMNCsおよびMEFが形成するコロニーの形態および未分化性のマーカーであるOct4−EGFPレポーター遺伝子からのEGFPタンパク質の発現を示した。「BMMNCs」はOct4−EGFPトランスジェニックマウス由来mBMMNCsを用いた場合を示し、「ゼラチン上に予め培養MEF」はウィルス感染前日からゼラチンコートプレート上で培養していたOct4−EGFPマウスMEFを用いた場合を示し、「レトロネクチン上MEF」はBMMNCsと同様にレトロネクチンコートプレートで感染させたMEFを用いた場合を示した。
【図14】ヌードマウスの皮下に移植されたマウス骨髄由来iPS#7細胞より形成された奇形腫を示す。[A]は皮下に移植されたマウス骨髄由来iPS#7細胞あるいはES細胞より奇形腫が形成されたヌードマウスを示す。[B]は形成された奇形腫より作製した病理標本を示し、標本中には、未分化神経組織、中枢神経、大脳皮質様組織、毛根組織、筋組織、軟骨組織、白色脂肪組織、気管粘液上皮細胞及び繊毛を有する気管上皮細胞、食道粘膜様組織、膵臓腺房細胞、唾液腺の漿液細胞および粘液細胞などが観察される。
【図15】マウス骨髄由来iPS#7細胞をBalb/cマウスの受精卵から作製した胚盤胞に注入後、偽妊娠マウスに移植することにより作出されたキメラ個体を示す。Balb/cマウスは本来白色の毛色を示し、骨髄由来iPS細胞は黒色の毛色を有するC57BL/6マウスに由来している。黒色の毛色を有する細胞は骨髄由来iPS細胞由来の細胞であることを示している。
【図16】マウス骨髄由来のiPS細胞クローンとマウスES細胞およびマウス骨髄由来単核球細胞について、各細胞で発現している遺伝子の発現強度の比較を示した図である。「iPS#7」はマウスiPS細胞クローンを示し、「BMMNC」はマウス骨髄由来単核球細胞を示す。各図の水平軸、垂直軸は各細胞で発現する遺伝子のメッセンジャーRNAの量を示し、図中の各点は一種類の遺伝子に対応している。比較する細胞間で発現する遺伝子のメッセンジャーRNAの量が同一である場合には、図中の各点は図の対角線上に位置することになる。
【特許請求の範囲】
【請求項1】
体細胞から多能性幹細胞を製造する方法であって、
(a)体細胞を、IL−6シグナル伝達因子刺激因子および少なくとも1種のサイトカインを含む細胞培養培地中で培養する工程、および
(b)体細胞を脱分化させる工程
を含んでなり、工程(a)の後に工程(b)が行われるか、または工程(a)と工程(b)が同時に行われる、方法。
【請求項2】
前記IL−6シグナル伝達因子刺激因子が、IL−6もしくはその変異体とIL−6受容体もしくはその変異体とが連結された融合タンパク質である、請求項1に記載の方法。
【請求項3】
前記サイトカインが、SCF、TPO、IL−3およびFlt−3リガンド、ならびにこれらの2以上の組合せからなる群から選択されるものである、請求項1に記載の方法。
【請求項4】
工程(b)における体細胞の脱分化が、該体細胞の核初期化処理によって行われる、請求項1に記載の方法。
【請求項5】
体細胞の核初期化処理が、Octファミリー遺伝子、Klfファミリー遺伝子、Soxファミリー遺伝子、およびMycファミリー遺伝子を、発現可能な形で体細胞に導入することを含む、請求項4に記載の方法。
【請求項6】
体細胞が骨髄由来単核球細胞である、請求項1に記載の方法。
【請求項7】
体細胞が分化血液細胞である、請求項1に記載の方法。
【請求項8】
体細胞がヒト細胞である、請求項1に記載の方法。
【請求項9】
体細胞から多能性幹細胞を製造するためのキットであって、
(a)IL−6シグナル伝達因子刺激因子および少なくとも1種のサイトカインを含む、体細胞を培養するための細胞培養培地、および
(b)体細胞を脱分化させるための試薬
を含んでなる、キット。
【請求項10】
前記IL−6シグナル伝達因子刺激因子が、IL−6もしくはその変異体とIL−6受容体もしくはその変異体とが融合された融合タンパク質である、請求項9に記載のキット。
【請求項11】
前記サイトカインが、SCF、TPO、IL−3およびFlt−3リガンド、ならびにこれらの2以上の組合せからなる群から選択されるものである、請求項9に記載のキット。
【請求項12】
体細胞を脱分化させるための試薬が、Octファミリー遺伝子、klfファミリー遺伝子、Soxファミリー遺伝子、およびMycファミリー遺伝子を、発現可能な形で体細胞に導入するためのベクターを含むものである、請求項9に記載のキット。
【請求項13】
体細胞が骨髄由来単核球細胞である、請求項9に記載のキット。
【請求項14】
体細胞が分化血液細胞である、請求項9に記載のキット。
【請求項15】
体細胞がヒト細胞である、請求項9に記載のキット。
【請求項1】
体細胞から多能性幹細胞を製造する方法であって、
(a)体細胞を、IL−6シグナル伝達因子刺激因子および少なくとも1種のサイトカインを含む細胞培養培地中で培養する工程、および
(b)体細胞を脱分化させる工程
を含んでなり、工程(a)の後に工程(b)が行われるか、または工程(a)と工程(b)が同時に行われる、方法。
【請求項2】
前記IL−6シグナル伝達因子刺激因子が、IL−6もしくはその変異体とIL−6受容体もしくはその変異体とが連結された融合タンパク質である、請求項1に記載の方法。
【請求項3】
前記サイトカインが、SCF、TPO、IL−3およびFlt−3リガンド、ならびにこれらの2以上の組合せからなる群から選択されるものである、請求項1に記載の方法。
【請求項4】
工程(b)における体細胞の脱分化が、該体細胞の核初期化処理によって行われる、請求項1に記載の方法。
【請求項5】
体細胞の核初期化処理が、Octファミリー遺伝子、Klfファミリー遺伝子、Soxファミリー遺伝子、およびMycファミリー遺伝子を、発現可能な形で体細胞に導入することを含む、請求項4に記載の方法。
【請求項6】
体細胞が骨髄由来単核球細胞である、請求項1に記載の方法。
【請求項7】
体細胞が分化血液細胞である、請求項1に記載の方法。
【請求項8】
体細胞がヒト細胞である、請求項1に記載の方法。
【請求項9】
体細胞から多能性幹細胞を製造するためのキットであって、
(a)IL−6シグナル伝達因子刺激因子および少なくとも1種のサイトカインを含む、体細胞を培養するための細胞培養培地、および
(b)体細胞を脱分化させるための試薬
を含んでなる、キット。
【請求項10】
前記IL−6シグナル伝達因子刺激因子が、IL−6もしくはその変異体とIL−6受容体もしくはその変異体とが融合された融合タンパク質である、請求項9に記載のキット。
【請求項11】
前記サイトカインが、SCF、TPO、IL−3およびFlt−3リガンド、ならびにこれらの2以上の組合せからなる群から選択されるものである、請求項9に記載のキット。
【請求項12】
体細胞を脱分化させるための試薬が、Octファミリー遺伝子、klfファミリー遺伝子、Soxファミリー遺伝子、およびMycファミリー遺伝子を、発現可能な形で体細胞に導入するためのベクターを含むものである、請求項9に記載のキット。
【請求項13】
体細胞が骨髄由来単核球細胞である、請求項9に記載のキット。
【請求項14】
体細胞が分化血液細胞である、請求項9に記載のキット。
【請求項15】
体細胞がヒト細胞である、請求項9に記載のキット。
【図3】

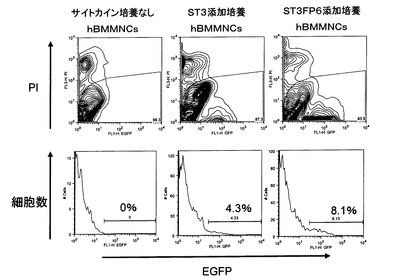
【図4】
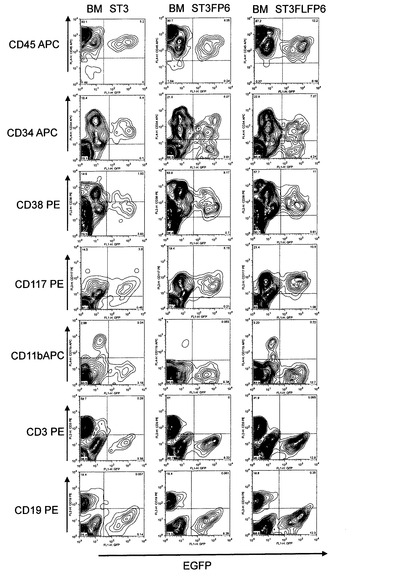
【図5】
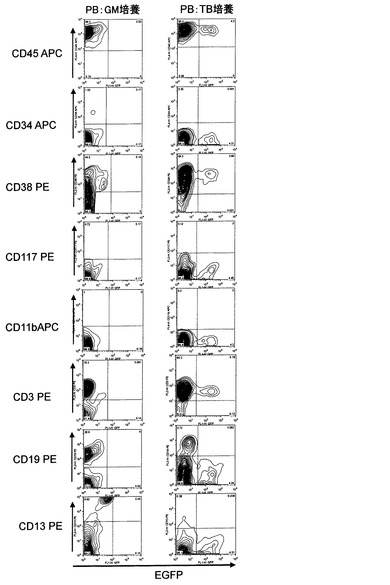
【図9】
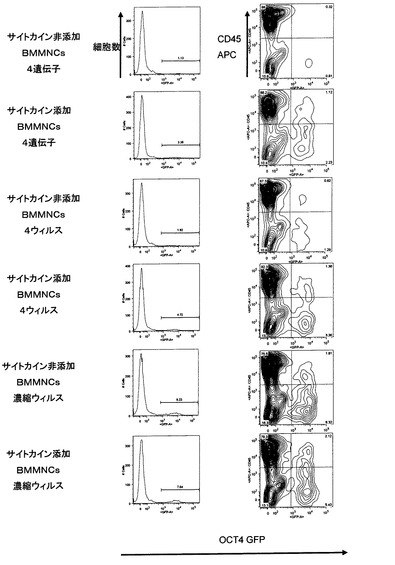
【図12】
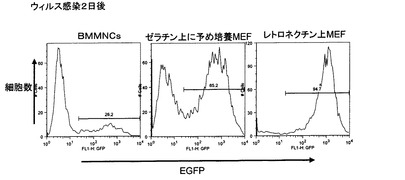
【図16】
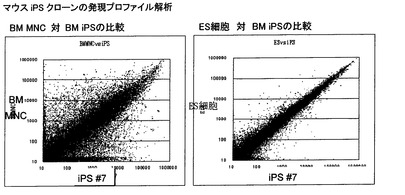
【図1】
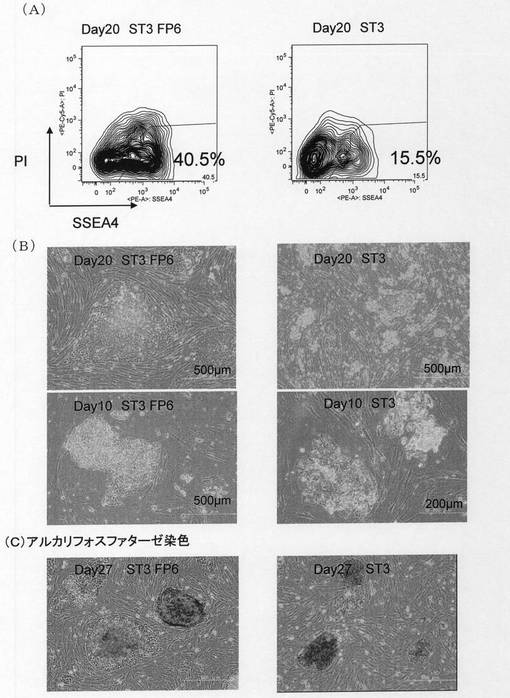
【図2】
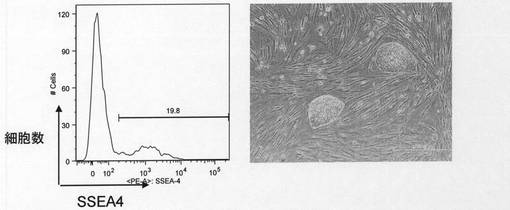
【図6】
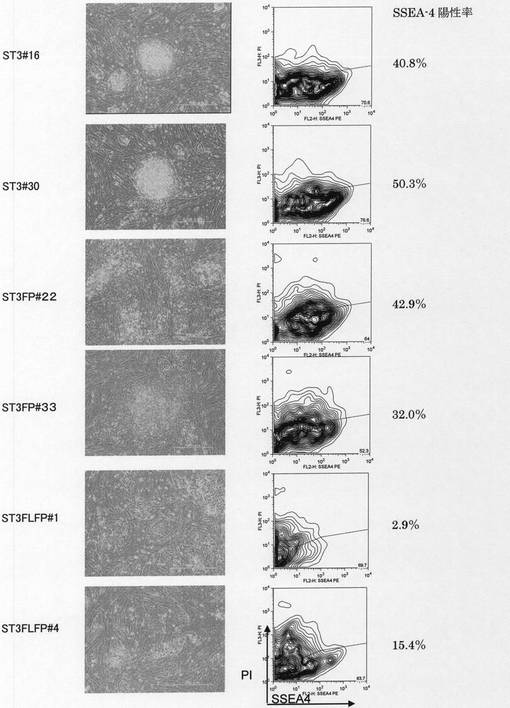
【図7】

【図8】
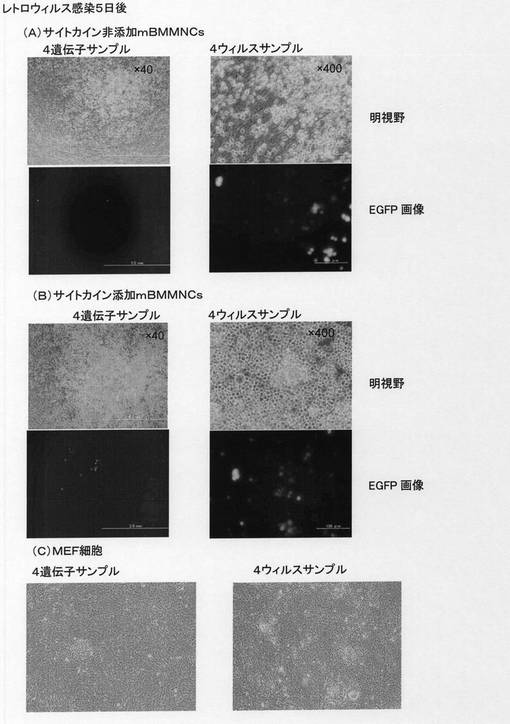
【図10】
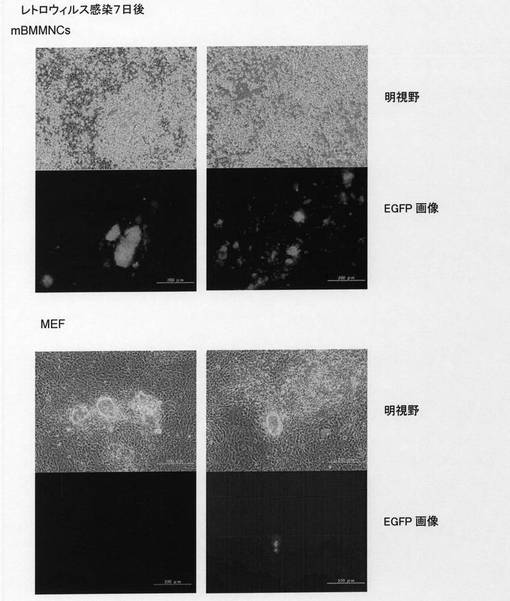
【図11】
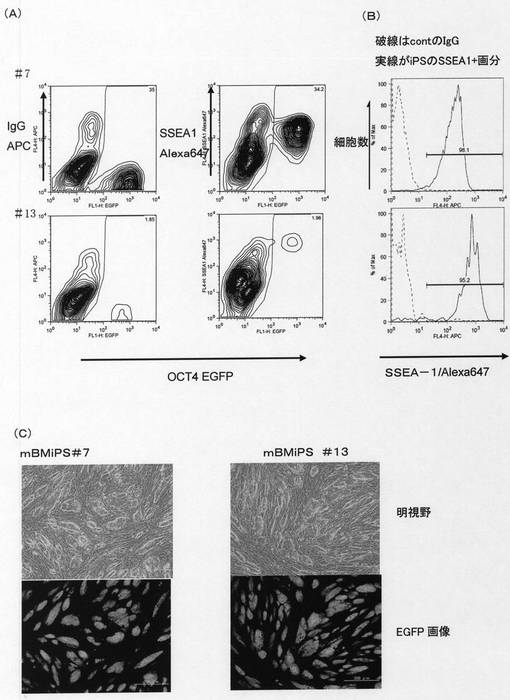
【図13】
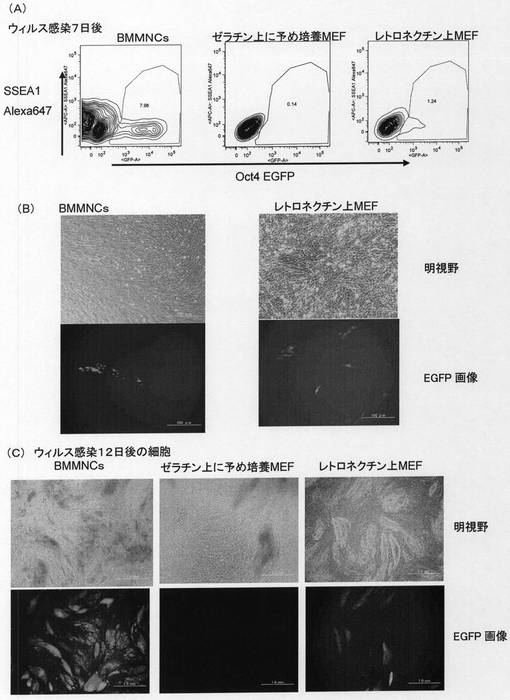
【図14】
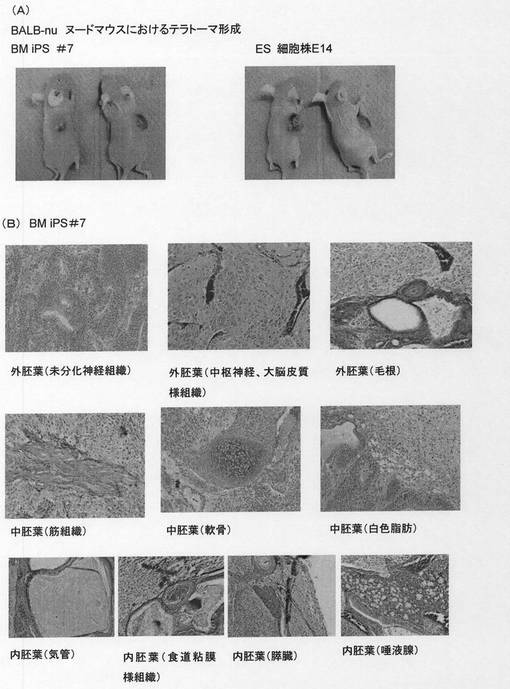
【図15】

【図4】
【図5】
【図9】
【図12】
【図16】
【図1】
【図2】
【図6】
【図7】
【図8】
【図10】
【図11】
【図13】
【図14】
【図15】
【公開番号】特開2011−160661(P2011−160661A)
【公開日】平成23年8月25日(2011.8.25)
【国際特許分類】
【出願番号】特願2008−145022(P2008−145022)
【出願日】平成20年6月2日(2008.6.2)
【出願人】(000001029)協和発酵キリン株式会社 (276)
【Fターム(参考)】
【公開日】平成23年8月25日(2011.8.25)
【国際特許分類】
【出願日】平成20年6月2日(2008.6.2)
【出願人】(000001029)協和発酵キリン株式会社 (276)
【Fターム(参考)】
[ Back to top ]