
血管構造及び/または機能のモジュレーションのための組成物及び方法
【課題】エンドセリン-1放出の刺激、血管収縮及び破裂した血管からの血液流出の減少よりなる群から選択される少なくとも1以上の一過性で局所性の生理学的応答を達成するための医薬の提供。
【解決手段】β-1→4コンフォメーションで共有結合した50〜150,000個のN-アセチルグルコサミン単糖を含んでなり、前記ポリマーが10,000ダルトン〜30 x 106ダルトンの分子量を有するポリ-β-1→4-N-アセチルグルコサミンポリマー。
【解決手段】β-1→4コンフォメーションで共有結合した50〜150,000個のN-アセチルグルコサミン単糖を含んでなり、前記ポリマーが10,000ダルトン〜30 x 106ダルトンの分子量を有するポリ-β-1→4-N-アセチルグルコサミンポリマー。
【発明の詳細な説明】
【技術分野】
【0001】
1.序
本発明は、半結晶ポリβ-1→4-N-アセチルグルコサミン(p-GlcNAc)多糖ポリマーを含む組成物、ならびにかかるポリマーを利用し、p-GlcNAc濃度依存的に、エンドセリン-1放出、血管収縮、及び/または破裂した血管からの血液流出の減少を一過性かつ局所的に刺激する方法に関する。これらの効果は、個々に及び/または共同して、出血の停止を助けるかまたは達成する。さらに特定すれば、本発明の方法は、N-アセチルグルコサミンの半結晶ポリマーを含む組成物及び物質の局所投与を含み、前記ポリマーがタンパク質を含まずかつ単一アミノ酸ならびにその他の有機及び無機不純物を実質的に含まず、そしてその成分である単糖類がβ-1→4コンフォメーションで結合されていることを特徴とする。
【背景技術】
【0002】
2.背景
血管ホメオスタシスは、部分的に、内皮細胞による生化学モジュレーターの調節された分泌に依存する。正常の生理学的条件下において、内皮細胞は一酸化窒素、プロスタサイクリン、PG12、アデノシン、過分極因子、組織因子経路インヒビター、及びscuプラスミノーゲンアクチベーターを合成しかつ分泌する。内皮細胞はまた、抗トロンビンIII及びプロテインCを活性化し、これらは共同して血管拡張を媒介して血小板接着、血小板活性化、トロンビン生成及びフィブリン沈着を抑制する。一酸化窒素は特に、血管ホメオスタシスに極めて重要な役割を果たす(Pearson, J. D. (2000) Lupus 9 (3):183-88;Beckerら (2000) Z Kardiol 89 (3):160-7;Schinin-Kerth, V. B. (1999) Transfus Clin Biol 6 (6):355-63)。
【0003】
強力な血管拡張薬ならびに血小板凝集及び活性化のインヒビターである一酸化窒素とプロスタサイクリンの産生が、内皮の抗血栓活性の基礎となっている(Yangら (1994) Circulation 89 (5): 2666-72)。一酸化窒素は構成的、基底レベルでアルギニンから一酸化窒素シンターゼにより合成され、かつこの合成は血管作用薬であるアセチルコリン及びブラジキニンにより刺激される。アルギニン類似体であるモノメチル-L-アルギニン(L-NMMA)及びニトロ-L-アルギニンメチルエステル(L-NAME)による一酸化窒素シンターゼの抑制は、一酸化窒素レベルを低下させ、血管内超音波イメージングにより測定すると、血管収縮をもたらすだけでなく血小板凝集も増加することが示されている(Yaoら (1992) Circulation 86 (4):1302-9;Emersonら (1999) Thromb Haemost 81 (6): 961-66)。
【0004】
アテローム性動脈硬化症、糖尿病、虚血後再灌流、炎症または高血圧症の結果としての内皮の混乱は、例えば前血栓症状態をもたらし、内皮はさらなる生化学モジュレーターのセットを作り出し、それらにはTNF-α、IL-8、ホンビルブラント(von Willebrand)因子、血小板活性化因子、組織プラスミノーゲンアクチベーター、及び1型プラスミノーゲンアクチベーターインヒビターが挙げられる(Pearson, J. D. (2000) Lupus 9 (3): 183-88;Beckerら (2000) Z Kardiol 89 (3): 160-7;Schinin-Kerth, V. B. (1999) Transfus Clin Biol 6 (6): 355-63)。さらに、血管内皮は、既知の最も強力な血管収縮薬であるエンドセリンを合成して作り出す。
【0005】
エンドセリンは、21-アミノ酸ペプチドのファミリー、すなわち、エンドセリン-1、エンドセリン-2、及びエンドセリン-3であり、元来その強力な血管収縮及び血管形成特性により特徴付けられる(例えば、Luscherら (1995), Agents Actions Suppl. (Switzerland) 45: 237-253;Yanagisawaら (1988) Nature 332: 411-415を参照)。エンドセリンファミリーの3つのイソペプチドであるエンドセリン-1、エンドセリン-2、及びエンドセリン-3は、3つの別の遺伝子がコードする高度に保存されたアミノ酸配列を有する(例えば、Inoueら (1989) Proc Natl Acad Sci USA 86: 2863-67;Saidaら (1989) J Biol Chem 264:14613-16を参照)。エンドセリン類は、平滑筋細胞を含む複数組織において合成されるが、エンドセリン-1は専ら血管内皮により合成される(Rosendorff, C. (1997) Cardiovasc Drugs 10 (6): 795-802)。エンドセリン類は203個のアミノ酸からなるプレプロエンドセリンとして合成される。エンドセリンシグナル配列が切断され、該タンパク質はさらなるタンパク分解によって、成熟した生物化学的に活性な21個のアミノ酸形態となる(例えば、Kashiwabaraら (1989) FEBS Lett 247: 337-40を参照)。エンドセリン合成は、エンドセリン及び非エンドセリン転化酵素を含む自己分泌機構を介してならびにキマーゼにより調節される(Batonら (1999) Curr Opin Nephrol Hypertens 8 (5): 549-56)。内皮からのエンドセリン-1の産生は、アンギオテンシンII、バソプレッシン、内毒素、及びとりわけシクロスポリンにより刺激され(例えば、Brooksら (1991) Eur J Pharm 194: 115-17を参照)、そして一酸化窒素により抑制される。
【0006】
エンドセリン活性は、2つの明確なGタンパク質結合受容体であるETA及びETBに対する優先的アフィニティによる結合を経由して自己分泌/パラ分泌的に媒介される(例えば、Hocherら (1997) Eur. J. Clin. Chem. Clin. Biochem. 35 (3): 175-189;Shichiriら (1991) J. Cardiovascular Pharmacol. 17: S76-S78を参照)。ETA受容体は血管収縮と連結した血管平滑筋上に見出され、心血管、腎、及び中枢神経系疾患と関連がある。ETB受容体はさらに複雑であり、アンタゴニスト作用を示す。内皮のETB受容体はクリアランスと血管拡張の二重の役割を有するのに対して、平滑筋細胞上のETB受容体は血管収縮も媒介する(Dupuis, J. (2000) Can J Cardiol 16 (1): 903-10)。内皮上のETB受容体は一酸化窒素及びプロスタサイクリンの放出と連結している(Rosendorff, C. (1997) Cardiovasc Drugs 10 (6): 795-802)。様々なエンドセリン受容体のアゴニスト及びアンタゴニストが存在し(Webbら (1997) Medicinal Research Reviews 17 (1): 17-67)、エンドセリンの作用機序を研究するのに利用されている。エンドセリンは強力な血管収縮活性を有することが知られているので、特に、エンドセリンアンタゴニスト(当技術分野では「エンドセリン受容体アンタゴニスト」とも呼ばれる)はヒトの疾患、最も著名なのは、心血管疾患、例えば高血圧、うっ血性心不全、アテローム性動脈硬化症、再狭窄、及び心筋梗塞を治療する上でのそれらの可能な役割について研究されている(Mateoら (1997) Pharmacological Res. 36 (5): 339-351)。
【0007】
さらに、エンドセリン-1は月経周期の正常な機能に関わることが示されている。月経は組織修復と置き換えの代表例であり、子宮を内張りする子宮内膜組織の新しい層の規則正しい再形成と再生を含む。この修復と再形成プロセスは瘢痕なしに達成され、一般的に身体の他の器官では見られない現象である点が注目される。この修復プロセスの欠陥が、月経過多と記録された女性ならびに避妊目的の皮下レボノルゲストレル・インプラント(NORPLANT)を装着した女性における過剰または異常な子宮内膜出血の原因であると考えられる。これらの両グループの女性においては、対照集団と比較して、非常に低レベルの子宮内膜エンドセリン-1が検出されるにすぎない。さらに、エンドセリン-1は月経性出血の停止ににおいて役割を果たすだけでなく、エンドセリン-1は月経後の子宮内膜組織の再生及び再形成に必要な分裂促進活性も有しうることが示されている。(例えば、Salamonsenら 1999, Balliere's Clinical Obstetrics and Gynaecology 13 (2): 161-79;Goldie 1999, Clinical and Experimental Pharmacology and Physiology 26: 145-48;Salamonsenら 1999, Clin. Exp. Phamaol. Physiol. 26 (2): 154-57を参照)。
【0008】
総括すると、血管ホメオスタシスは、血管内皮が媒介する2つの生理学的状態の間の動的バランスを反映する。抗血栓性と呼ばれる第1の状態は、とりわけ、一酸化窒素の産生、血管拡張、血小板接着及び活性化の抑制により、ならびにエンドセリン-1合成の抑圧により特徴付けられる。第2のまたはプロトロンビン生理学的状態は、とりわけ、エンドセリン-1の産生、血管収縮、血小板活性化、及び止血により特徴付けられる(Warner (1999), Clinical and Experimental Physiology 26: 347-52;Pearson, (2000), Lupus 9(3): 183-88)。
【先行技術文献】
【非特許文献】
【0009】
【非特許文献1】Pearson, J. D. (2000) Lupus 9 (3):183-88
【非特許文献2】Beckerら (2000) Z Kardiol 89 (3):160-7
【非特許文献3】Schinin-Kerth, V. B. (1999) Transfus Clin Biol 6 (6):355-63
【非特許文献4】Yaoら (1992) Circulation 86 (4):1302-9
【非特許文献5】Emersonら (1999) Thromb Haemost 81 (6): 961-66
【非特許文献6】Luscherら (1995), Agents Actions Suppl. (Switzerland) 45: 237-253
【非特許文献7】Yanagisawaら (1988) Nature 332: 411-415
【非特許文献8】Inoueら (1989) Proc Natl Acad Sci USA 86: 2863-67
【非特許文献9】Saidaら (1989) J Biol Chem 264:14613-16
【非特許文献10】Rosendorff, C. (1997) Cardiovasc Drugs 10 (6): 795-802
【非特許文献11】Kashiwabaraら (1989) FEBS Lett 247: 337-40
【非特許文献12】Batonら (1999) Curr Opin Nephrol Hypertens 8 (5): 549-56
【非特許文献13】Brooksら (1991) Eur J Pharm 194: 115-17
【非特許文献14】Hocherら (1997) Eur. J. Clin. Chem. Clin. Biochem. 35 (3): 175-189
【非特許文献15】Shichiriら (1991) J. Cardiovascular Pharmacol. 17: S76-S78
【非特許文献16】Webbら (1997) Medicinal Research Reviews 17 (1): 17-67
【非特許文献17】Mateoら (1997) Pharmacological Res. 36 (5): 339-351
【非特許文献18】Salamonsenら 1999, Balliere's Clinical Obstetrics and Gynaecology 13 (2): 161-79
【非特許文献19】Goldie 1999, Clinical and Experimental Pharmacology and Physiology 26: 145-48
【非特許文献20】Salamonsenら 1999, Clin. Exp. Phamaol. Physiol. 26 (2): 154-57
【非特許文献21】Warner (1999), Clinical and Experimental Physiology 26: 347-52
【発明の概要】
【発明が解決しようとする課題】
【0010】
血管ホメオスタシスの生理学的重要性に照らして考えると、上記プロセスの1以上の態様をモジュレートできる方法及び組成物に対するニーズがある。さらに特定すれば、エンドセリン放出、血管収縮、及び破裂した血管からの血液流出のモジュレーション、それによる出血の停止に有用でありうる組成物及び方法に対するニーズがある。すなわち、かかる組成物及び方法が物理的障害形成、凝血、または血餅形成に依存しない方法で作用しうるものであるにも関わらず、かかる組成物及び方法は、とりわけ、止血の達成に寄与しうる。従って、かかる方法及び組成物は、血管ホメオスタシスの混乱の結果として生じる疾患または症状の治療に対する治療上の用途を有すると期待しうる。さらに、例えば、患者への先に記載のエンドセリン-1アンタゴニストの投与から得られる全身効果を視野に入れると、限定されるものでないが、かかる患者におけるエンドセリン-1放出の刺激を含む、局所性で一過性の生理学的応答を産生する組成物及び方法に対するさらに大きなニーズがある。
【課題を解決するための手段】
【0011】
3.発明の概要
本発明は、出血障害を含む血管障害の治療または改善のための方法及び組成物に関する。さらに特定すれば、本発明は、半結晶ポリβ-1→4-N-アセチルグルコサミン(p-GlcNAc)多糖ポリマーを含む組成物、ならびに、例えばエンドセリン-1放出の刺激、血管収縮、及び/または破裂した血管からの血液流出の減少により、血管構造及び/または機能の一過性で局所性のモジュレーションを果たし、その結果、出血の停止を助けるかまたは達成する方法におけるかかるポリマーの使用に関する。
【0012】
本発明は、部分的には、半結晶ポリβ-1→4-N-アセチルグルコサミン(p-GlcNAc)多糖ポリマーを血管表面に局所適用すると、血管の収縮を誘導してそれにより血管の管腔を減少するだけでなく、適用した本明細書に開示した組成物及び物質に近接する組織においてエンドセリン-1放出の一過性で局所性の刺激も誘導するという本出願人の発見に基づく。
【0013】
本発明は、一態様においては、患者の血管構造及び/または機能の一過性で局所性の、モジュレーションを達成する方法であって、タンパク質を含まず、他の有機不純物を実質的に含まず、そして無機不純物を実質的に含まない半結晶ポリβ-1→4 N-アセチルグルコサミンポリマーを含む物質の局所投与を含んでなる前記方法に関する。これらの物質の投与は、限定されるものでないが、エンドセリン-1放出の刺激、血管収縮、及び破裂した血管からの血液流出の減少を含む一過性で局所性の生理学的応答を誘導する。
【0014】
本発明の一実施形態においては、血管内皮細胞からエンドセリン-1が放出される。この実施形態の他の態様においては、エンドセリン-1放出は他の内皮組織からまたは血小板から刺激される。
【0015】
一実施形態においては、ポリβ-1→4 N-アセチルグルコサミンポリマーは約50〜約4,000個のβ-1→4コンフォメーションで共有結合したN-アセチルグルコサミン単糖を含んでなり、約10,000ダルトン〜約800,000ダルトンの分子量を有する。他の実施形態においては、ポリβ-1→4 N-アセチルグルコサミンポリマーは約50〜約10,000個のβ-1→4コンフォメーションで共有結合したN-アセチルグルコサミン単糖を含んでなり、約10,000ダルトン〜約2 x 106ダルトンの分子量を有する。さらに他の実施形態においては、ポリβ-1→4 N-アセチルグルコサミンポリマーは約50〜約50,000個のβ-1→4コンフォメーションで共有結合したN-アセチルグルコサミン単糖を含んでなり、約10,000ダルトン〜約10 x 106ダルトンの分子量を有する。他の実施形態においては、ポリβ-1→4 N-アセチルグルコサミンポリマーは約50〜約150,000個のβ-1→4コンフォメーションで共有結合したN-アセチルグルコサミン単糖を含んでなり、約10,000ダルトン〜約30 x 106ダルトンの分子量を有する。
【0016】
本発明の好ましい実施形態においては、開示した方法を哺乳動物患者の治療に、かつさらに好ましい実施形態においては、かかる治療を必要とするヒトの治療に利用する。さらに特定すれば、血管構造及び/または機能のモジュレーションを利用して、特に凝血障害を患う患者における出血の停止を達成する。かかる障害は、血友病などのなどの遺伝的欠陥、または例えば、透析患者、心臓病患者もしくはその他の血管閉塞のリスクが高い患者へのクマジン(coumadin)などの全身抗凝血薬の投与を含む医学治療の結果でありうる。同様に、本発明の方法は、特に凝血障害症状を有する患者における、動脈瘤の外科修復または腫瘍もしくはポリープの切除中に、破裂した血管からの血液流出のの一過性で局所性の減少を達成するために利用し、それにより、かかる処置中の血液損失を最小化する。他の実施形態においては、本発明の方法は、子宮もしくは静脈瘤、特に食道静脈瘤の出血の治療に利用することができる。特定の理論もしくは機構に束縛されるのを欲しないが、本明細書に開示した方法によるかかる出血の停止は、凝血に依存しない方式で起こると考えられる。
【0017】
本発明の方法の他の実施形態においては、p-GlcNAcを含有する物質を患者の皮膚もしくはその他の器官の表面に局所投与するか、またはモジュレートすべき血管構造に直接適用してもよく、その血管構造は毛細管、血管または動脈であってもよい。
【0018】
本発明の方法のさらに他の実施形態においては、血管構造は破裂した血管であって、本発明のp-GlcNAcを含有する物質の局所適用を用いて出血の停止を達成する。
【0019】
本発明のさらなる実施形態においては、血管構造及び/または機能の一過性で局所性のモジュレーションの程度は、適用した半結晶ポリβ-1→4-N-アセチルグルコサミンの量と実質的に比例する。
【0020】
本発明はまた、タンパク質を含まず、その他の有機不純物を実質的に含まず、かつ無機不純物を実質的に含まない半結晶ポリβ-1→4 N-アセチルグルコサミンポリマーを含む生物分解性物質に関する。一実施形態においては、半結晶ポリβ-1→4 N-アセチルグルコサミンポリマーは約50〜約4,000個のβ-1→4コンフォメーションで共有結合したN-アセチルグルコサミン単糖を含みかつ約10,000ダルトン〜約800,000ダルトンの分子量を有する。他の実施形態においては、半結晶ポリβ-1→4 N-アセチルグルコサミンポリマーは約50〜約10,000個のβ-1→4コンフォメーションで共有結合したN-アセチルグルコサミン単糖を含みかつ約10,000ダルトン〜約2 x 106ダルトンの分子量を有する。さらに他の実施形態においては、ポリβ-1→4 N-アセチルグルコサミンポリマーは約50〜約50,000個のβ-1→4コンフォメーションで共有結合したN-アセチルグルコサミン単糖を含みかつ約10,000ダルトン〜約10 x 106ダルトンの分子量を有する。他の実施形態においては、ポリβ-14 N-アセチルグルコサミンポリマーは約50〜約150,000個のβ-1→4コンフォメーションで共有結合したN-アセチルグルコサミン単糖を含みかつ約10,000ダルトン〜約30 x 106ダルトンの分子量を有する。
【0021】
他の実施形態においては、半結晶ポリβ-1→4 N-アセチルグルコサミンポリマーを含む生物分解性物質は、バリヤーを形成しない物質である。
【0022】
さらに他の実施形態においては、半結晶ポリβ-1→4 N-アセチルグルコサミンポリマーは、少なくとも1つの脱アセチル化されたN-アセチルグルコサミン単糖を含む。この実施形態の他の態様においては、部分的に脱アセチル化されたポリβ-1→4 N-アセチルグルコサミンポリマーが、そのポリマーを下の実施例6に記載のIR吸収分光計により分析したときに鋭い明確なピークにより実証されるその半結晶構造を保持するのであれば、ポリβ-1→4 N-アセチルグルコサミンポリマーは、約10%、20%、30%、40%、50%または60%脱アセチル化された残基を含んでもよい。
【図面の簡単な説明】
【0023】
【図1】図1は、100% p-GlcNAcの化学構造である。「n」は約150,000までの範囲の整数を意味する。
【図2】図2は、p-GlcNAcの炭水化物分析、すなわちガスクロマトグラフィ-質量分析データである。黒正方形の点は、以下の第5.3.2節に記載の化学的/生物学的方法の酸処理/中和化変法を用いて精製したp-GlcNAcを表す。
【図3A】図3Aは、純粋なp-GlcNAc固体膜の円二色性スペクトルである。
【図3B】図3Bは、脱アセチル化p-GlcNAc固体膜の円二色性スペクトルである。純粋なp-GlcNAc(図3A)で観察された211nm最小値及び195nm最大値の消滅は、以下の第5.4節に記載の使用した条件下での完全な脱アセチル化を示す。
【図4A】図4Aは、機械力精製(上側)、及び化学的/生物学的精製法(下側)により調製した純粋な珪藻p-GlcNAcの薄膜の赤外分光分析である。
【図4B】図4Bは、以下の第5.5節に詳述した方法に従って、膜に成型した市販「キチン」の2つの調製物の赤外分光分析である。
【図4C】図4Cは、以下の第5.4節に詳述した方法に従って、熱変性により(上側)及び化学的脱アセチル化により(下側)改変した純粋p-GlcNAcの赤外分光分析である。
【図4D】図4Dは、以下の第5.3.2節に詳述した化学的/生物学的精製法を用いて珪藻タラシオシラ・フルビアチリス(Thalassiosira fluviatilis)から誘導したp-GlcNAc膜の赤外分光分析である。
【図4E】図4Eは、以下の第5.3.1節に記載の機械力精製法と次いでオートクレーブ処理により調製したp-GlcNAc膜の赤外分光分析である。
【図5A】図5Aは、以下の第5.3.2節に記載の化学的/生物学的精製法を用いて精製したp-GlcNAcのNMR分析である。表は、ピーク振幅、面積、及び基準対照に対する比、ならびにピークの全面積の比を示す。
【図5B】図5Bは、以下の第5.3.2節に記載の化学的/生物学的精製法を用いて精製したp-GlcNAcのNMR分析である。グラフはピークの全面積の比を表す。
【図6A】図6Aは、以下の5.3.1節に記載の機械力精製方法により調製したp-GlcNAc膜の透過型電子顕微鏡(TEM)写真である。拡大率:(図6A)、4190 x。
【図6B】図6Bは、以下の5.3.1節に記載の機械力精製方法により調製したp-GlcNAc膜の透過型電子顕微鏡(TEM)写真である。拡大率:(図6B)、16,250 x。
【図7A】図7Aは、以下の5.3.2節に記載の化学的/生物学的精製法の考察に記載のHF処理によるp-GlcNAc膜の透過型電子顕微鏡(TEM)写真である。拡大率:(図7A)、5270 x。
【図7B】図7Bは、以下の5.3.2節に記載の化学的/生物学的精製法の考察に記載のHF処理によるp-GlcNAc膜の透過型電子顕微鏡(TEM)写真である。拡大率:(図7B)、8150 x。
【図8A】図8Aは、以下の5.3.2節に記載の化学的/生物学的精製法の酸処理/中和化変法により調製したp-GlcNAc膜の透過型電子顕微鏡写真(TEM)である。拡大率:(図8A)、5270 x。
【図8B】図8Bは、以下の5.3.2節に記載の化学的/生物学的精製法の酸処理/中和化変法により調製したp-GlcNAc膜の透過型電子顕微鏡写真(TEM)である。拡大率:(図8B)、16,700 x。
【図9A】図9Aは、以下の5.3.2節に記載の化学的/生物学的精製法の酸処理/中和化変法により調製したp-GlcNAc膜の走査電子顕微鏡写真である。拡大率:200 x。
【図9B】図9Bは、以下の5.3.2節に記載の化学的/生物学的精製法の酸処理/中和化変法により調製したp-GlcNAc膜の走査電子顕微鏡写真である。拡大率:1000 x。
【図9C】図9Cは、以下の5.3.2節に記載の化学的/生物学的精製法の酸処理/中和化変法により調製したp-GlcNAc膜の走査電子顕微鏡写真である。拡大率:5000 x。
【図9D】図9Dは、以下の5.3.2節に記載の化学的/生物学的精製法の酸処理/中和化変法により調製したp-GlcNAc膜の走査電子顕微鏡写真である。拡大率:10,000 x。
【図9E】図9Eは、以下の5.3.2節に記載の化学的/生物学的精製法の酸処理/中和化変法により調製したp-GlcNAc膜の走査電子顕微鏡写真である。拡大率:20,000 x。
【図10A】図10Aは、純粋なp-GlcNAc膜の走査電子顕微鏡写真であり、この純粋なp-GlcNAc膜は、最初に以下の5.3節に記載の細胞溶解/中和化精製法を用いて生産し、ジメチルアセトアミド/塩化リチウムに溶解し、そして以下5.5節に記載のようにH2O中でマットに再沈降した物質から作った。拡大率:(図10A)、1000 x。
【図10B】図10Bは、純粋なp-GlcNAc膜の走査電子顕微鏡写真であり、この純粋なp-GlcNAc膜は、最初に以下の5.3節に記載の細胞溶解/中和化精製法を用いて生産し、ジメチルアセトアミド/塩化リチウムに溶解し、そして以下5.5節に記載のようにH2O中でマットに再沈降した物質から作った。拡大率:(図10B)、10,000 x。
【図11A】図11Aは、脱アセチル化p-GlcNAcマットの走査電子顕微鏡写真である。拡大率:(図11A)、1000 x。
【図11B】図11Bは、脱アセチル化p-GlcNAcマットの走査電子顕微鏡写真である。拡大率: (図11B)、10,000 x。
【図12A】図12Aは珪藻の写真である。珪藻細胞体から伸張するp-GlcNAc繊維に注目すること。
【図12B】図12Bは珪藻の写真である。珪藻細胞体から伸張するp-GlcNAc繊維に注目すること。
【図13】図13は、いくつかの可能なp-GlcNAc及びp-GlcNAcの脱アセチル化誘導体出発物質を示す図である(S. Hirano, 「日本におけるキチン及びキトサンの生産と応用("Production and Application of Chitin and Chitosan in Japan")」, in "Chitin and Chitosan," 1989, Skjak-Braek, Anthonsen, and Sanford, eds. Elsevier Science Publishing Co., pp. 37-43.から引用)。
【図14】図14は、それぞれの炭素原子に対する面積を得て、次いでCH3(面積)対C原子(面積)比を計算するために用いた、変換NMRデータ曲線である。
【図15】図15は、典型的なp-GlcNAc C13-NMRスペクトルである。個々のピークは、分子中のそれぞれのユニークな炭素原子のスペクトルへの寄与を表す。
【図16】図16は、CH3(面積)対C原子(面積)比を計算した値を表す変換NMRスペクトルデータである。上側:データのグラフ表示;下側:データ数値。
【図17A】p-GlcNAcマトリックスを蒸留水中で作製した。拡大率:200 x。これらの図のそれぞれに200ミクロンのスケールマーキングを示した。
【図17B】p-GlcNAcマトリックスを10%メタノールを含む蒸留水中で作製した。拡大率:200 x。これらの図のそれぞれに200ミクロンのスケールマーキングを示した。
【図17C】p-GlcNAcマトリックスを25%メタノールを含む蒸留水中で作製した。拡大率:200 x。これらの図のそれぞれに200ミクロンのスケールマーキングを示した。
【図17D】p-GlcNAcマトリックスを蒸留水中で作製した。拡大率:200 x。これらの図のそれぞれに200ミクロンのスケールマーキングを示した。
【図17E】p-GlcNAcマトリックスを10%エタノールを含む蒸留水中で作製した。拡大率:200 x。これらの図のそれぞれに200ミクロンのスケールマーキングを示した。
【図17F】p-GlcNAcマトリックスを25%エタノールを含む蒸留水中で作製した。拡大率:200 x。これらの図のそれぞれに200ミクロンのスケールマーキングを示した。
【図17G】p-GlcNAcマトリックスを40%エタノールを含む蒸留水中で作製した。拡大率:200 x。これらの図のそれぞれに200ミクロンのスケールマーキングを示した。
【図18】図18は、以下の第18.1節に記載の方法を用いて得た典型的な標準曲線である。このような標準曲線を、以下の第18.1節に同じく記載したリゾチーム-キチナーゼアッセイに用いた。
【図19】図19は、p-GlcNAcリゾチーム消化データである。ここに掲げたグラフは、p-GlcNAc膜がリゾチームにより消化される時間を通してのN-アセチルグルコサミンの蓄積を示す。グラフは、完全にアセチル化されたp-GlcNAcの分解速度を、部分的に(50%)脱アセチル化されたp-GlcNAcと比較し、部分的に脱アセチル化されたp-GlcNAcの分解速度が、完全にアセチル化されたp-GlcNAc物質のそれより有意に高いことを実証する。
【図20】図20は、p-GlcNAcリゾチーム消化データである。ここに掲げたグラフは、p-GlcNAc膜がリゾチームにより消化される時間を通してのN-アセチルグルコサミンの蓄積を示す。グラフは、2つの部分的に脱アセチル化されたp-GlcNAc膜(特に25%と50%脱アセチル化p-GlcNAc膜)を比較する。本データは、脱アセチル化のパーセントが増加すると分解速度が増加し、50%脱アセチル化p-GlcNAc膜の分解速度は25%脱アセチル化p-GlcNAc膜の分解速度より有意に高いことを実証する。
【図21A】p-GlcNAcのin vivo生分解性データである。第18.1節に記載のプロトタイプ1(完全にアセチル化されたp-GlcNAc)膜を腹部移植したラットを示す。図21Aは、移植0日のラットを示す。
【図21B】p-GlcNAcのin vivo生分解性データである。第18.1節に記載のプロトタイプ1(完全にアセチル化されたp-GlcNAc)膜を腹部移植したラットを示す。図21Bは、移植後14日のラットを示す。
【図21C】p-GlcNAcのin vivo生分解性データである。第18.1節に記載のプロトタイプ1(完全にアセチル化されたp-GlcNAc)膜を腹部移植したラットを示す。図21Cは、移植後21日のラットを示す。
【図21D】p-GlcNAcのin vivo生分解性データである。第18.1節に記載のプロトタイプ3A(凍結乾燥して部分的に脱アセチル化されたp-GlcNAc膜)を腹部移植したラットを示す。図21Dは、移植0日のラットを示す。
【図21E】p-GlcNAcのin vivo生分解性データである。第18.1節に記載のプロトタイプ3A(凍結乾燥して部分的に脱アセチル化されたp-GlcNAc膜)を腹部移植したラットを示す。図21Eは移植後14日のラットを示す。
【図22】図22A-22Bは、無傷内皮層の有る(図22A)、または内皮層除去後の(図22B)単離された大動脈環のp-GlcNAcによる用量依存的血管収縮である。無傷内皮層有りまたは無しで試験したp-GlcNAcの各濃度の報告値を得るために平均した収縮測定数を、図中の試験したp-GlcNAcの各濃度上に示した。
【図23】図23A-Eは、p-GlcNAcによる動脈血管収縮である。図23(A)は、動脈の片側にガーゼ包帯を適用した60分後に得たブタ動脈の断面図を示す。図23(B)は、動脈の片側にp-GlcNAc膜を適用した15分後に得たブタ動脈の断面図を示す。図23(C)は、動脈の片側にp-GlcNAc膜を適用した60分後に得たブタ動脈の断面図を示す。図23(D)は、動脈の片側にフィブリンコートしたコラーゲン包帯を適用した15分後に得たブタ動脈の断面図を示す。図23(E)は、動脈の片側にフィブリンコートしたコラーゲン包帯を適用した60分後に得たブタ動脈の断面図を示す。
【図24】図24は、p-GlcNAcによる動脈血管収縮である。図24は、示したように15または60分間、試験物質と直接接触した(1)、または接触しなかった(2)、ブタ動脈壁の厚さを記す。動脈の片側に適用した物質は、(A)ガーゼ包帯;(B)及び(C)p-GlcNAc膜;(D)及び(E)フィブリンコートしたコラーゲン包帯であった。
【発明を実施するための形態】
【0024】
5.発明の詳細な説明
本発明は、例えば、(1)エンドセリン-1放出の刺激、(2)血管収縮、及び(3)破裂した血管からの血液流出の減少により血管構造及び/または機能の一過性で局所性のモジュレーションを達成するのに有用であり、半結晶ポリβ-1→4-N-アセチルグルコサミン(p-GlcNAc)多糖ポリマーを含む組成物及び物質の局所投与を含んでなる、組成物ならびに方法に関する。標的組織におけるエンドセリン-1放出の刺激、血管収縮、及び破裂した血管からの血液流出の減少は、標的組織への本発明の物質の直接適用により、あるいは標的組織に隣接もしくは近接する皮膚またはその他の器官もしくは組織表面へのこれらの物質の適用により達成することができる。
【0025】
本発明は、従って、出血の停止を助けるかまたは直接達成する組成物及び方法にも関する。半結晶ポリβ-1→4 N-アセチルグルコサミンポリマーを含む本発明の物質の投与は、エンドセリン-1放出の刺激、血管収縮、及び破裂した血管における血液流出の減少をもたらす。
【0026】
これらの生理学的応答は、個々に及び/または共同して、毛細管、静脈、もしくは動脈であってもよい出血の停止を助けるかまたは直接達成する。特定の理論もしくは機構に束縛されることは欲しないが、かかる停止は凝血に依存しない方法で起こると考えられる。さらに、本発明の組成物及び方法を用いる出血の停止の達成はまた、血液凝固を促進する物理的バリヤーまたは力学的マトリックスの形成に依存するものでもない。すなわち、本発明によれば、本物質は、適用部位に接着して傷の境界を塞ぐ力学的マトリックスを与えるバリヤー形成物質である必要はない。対照的に、本発明の組成物及び方法は血管構造及び/または機能の一過性で局所性の変化を誘導し、そしてこの変化が血液凝固に依存しないで、それ自身、出血の停止を助けるかまたは直接達成するのである。
【0027】
さらに、本発明の組成物及び方法に係る好ましい物質は、完全にアセチル化された半結晶ポリβ-1→4 N-アセチルグルコサミンポリマーを含むものである。なぜなら、後掲の第16及び17節に記載の実施例ならびに図22に実証するように、70%脱アセチル化されたポリβ-1→4 N-アセチルグルコサミンポリマーを含む物質は決して血管収縮を誘導しないし、従って血管の管腔を減少しないし、それ故に、破裂した血管からの血液流出を減少しないからである。
【0028】
本発明は、部分的に、バリヤー形成物質である必要のない半結晶ポリβ-1→4-N-アセチルグルコサミン(p-GlcNAc)ポリマーを含む物質を局所に適用すると、単離したスプラグ・ドーリー(Sprague-Dawley)ラット動脈環の血管収縮が誘導されるという本出願人らの発見に基づく。この血液を含まない系において、完全にアセチル化されたポリβ-1→4-N-アセチルグルコサミンは、単離した動脈環の収縮を濃度依存的に誘導した。後掲の第17節に記載した実施例において、得られる血管収縮の程度は、単離した動脈環に適用したp-GlcNAcの濃度に実質的に比例した。対照的に、70%脱アセチル化されたポリβ-1→4-N-アセチルグルコサミンは、試験したいずれの濃度においても、単離した動脈環の血管収縮を誘導しなかった。
【0029】
本発明はまた、半結晶ポリβ-1→4 N-アセチルグルコサミンポリマーから作った膜を動脈の実験傷へin vivo適用すると、動脈組織と適用した膜の間の接触部位において即効性の血管収縮が刺激されたという本出願人らの発見にも部分的に基づいている。処置した組織を組織学的に分析すると、動脈収縮は、前記膜を適用した側の方が動脈の反対側より大きいことが示された。さらに、これらの組織サンプル免疫化学分析すると、エンドセリン-1放出に濃度勾配が存在すること、すなわち、エンドセリン-1放出の刺激が局所的な生理学的応答であることが示された。エンドセリン-1放出の刺激の程度は、半結晶ポリβ-1→4 N-アセチルグルコサミンポリマーを含有する膜が接触する表面において最大であり、隣接組織中に広がるものの、接触表面からの距離が増加するとその程度は減少した。類似の局在するエンドセリン-1放出の刺激は、半結晶ポリβ-1→4-N-アセチルグルコサミンを含む物質と接触する脾臓組織において観察された。
【0030】
本発明の方法は、一過性で局所性の(1)エンドセリン-1放出の亢進、(2)血管収縮、及び/または(3)破裂した血管からの血液流出の減少を達成する目的で、患者への治療上有効な形態のかつ治療上有効な量の半結晶ポリβ-1→4 N-アセチルグルコサミンポリマーを含む物質の局所投与を含む。
【0031】
以下に報じるのは、最初に、精製p-GlcNAc出発物質及びその再製剤(reformulation)の物理的特性の説明である。次に、肉微小藻類、好ましくは珪藻である出発供給源からp-GlcNAc出発物質を精製する方法を説明する。第3に、p-GlcNAcの再製剤、及びかかる再製剤を生産する方法を報じる。最後に、出発物質であるp-GlcNAc、p-GlcNAc誘導体及び/または出発物質のp-GlcNAc再製剤の用途を報じる。
【0032】
5.1. p-GlcNAc
p-GlcNAc出発物質は、本明細書に記載の技術、ならびにそのそれぞれが参照により本明細書にその全文が組み入れられる米国特許第5,686,115号、第5,624,679号、第5,623,064号、及び第5,622,834号に与えられた教示を利用して作ることができる。本明細書に使用されるp-GlcNAcポリマーは、約50〜約150,000個のN-アセチルグルコサミン単糖(図1)を含む。化学的及び物理的判定基準により確認すると、p-GlcNAc出発物質の純度は非常に高い。これらのなかには、化学組成物及び非多糖不純物がある。第1に、共に第5.3節に記載された2つの異なる精製方法を利用して生産した、p-GlcNAcの化学組成データを、以下の表Iに示した。見ればわかるように、両方の方法により生産したp-GlcNAcの化学組成は、実験誤差範囲内で、p-GlcNAcの化学式組成と同じである。第2に、表Iでもわかるように、生産されたp-GlcNAcは検出可能なタンパク質不純物を含まず、遊離アミノ酸などの他の有機不純物を実質的に含まず、そして灰及び金属イオンなどの無機不純物を実質的に含まない(p-GlcNAc出発物質は純粋なp-GlcNAcに対する炭素、水素、窒素及び酸素の理論値から2%以下の偏差がありうる)。従って、本明細書に使用される用語「有機不純物を実質的に含まない」及び「無機不純物を実質的に含まない」は、理論値から約2%以下の偏差の炭素、水素、窒素及び酸素のプロフィールを有するp-GlcNAcの組成物、そして好ましくはp-GlcNAc出発物質が表Iのp-GlcNAcマットの実験データに例示したプロフィール(該パーセント偏差を許容する)を有することを意味する。さらに、p-GlcNAc出発物質は束縛水のパーセントが低い。
【表1】

純粋なp-GlcNAc出発物質は、図2に示したのと実質的に同様な炭水化物分析プロフィールを示した。純粋なp-GlcNAc出発物質の一次単糖はN-アセチルグルコサミンである。さらに、純粋なp-GlcNAc出発物質は単糖グルコサミンを含有しない。
【0033】
p-GlcNAc出発物質の円二色性(CD)及び鋭い赤外スペクトル(IR)を、図3A、ならびに図4A、4D、及び4E、にそれぞれ示したが、これらは以下の第5.3節に記載の方法を用いて作った物質の分析である。かかる物理的データは、p-GlcNAc出発物質が高純度かつ半結晶であることを実証する。用語「半結晶」は、物質の高度に規則的な性質を意味する。当業者であれば、本発明のp-GlcNAcポリマーの赤外スペクトルに観察された、鋭く、よく分解されたピークは試験した物質の高度に規則的な結晶的性質(すなわち、「半結晶性」)を反映することを容易に理解するであろう。当業者はまた、例えば図4B及び4Cに挙げたようなIRスペクトルのブロードの、分解不十分なピークは、半結晶性の消失もしくは不足を示すことも理解するであろう。CD及びIRデータを得るために利用した方法は、以下の第6節に記載する実施例で説明する。
【0034】
純粋なp-GlcNAc出発物質のNMR分析は、図5A、14、15及び16に見られるのと実質的に類似したパターンを表す。かかるNMRパターンは、完全にアセチル化されたポリマーであるp-GlcNAc出発物質に一致するデータを示すだけでなく、p-GlcNAc種内に不純物の有機物質が存在しないことも実証する。以下の第5.3節に記載の方法を用いて生産しかつ以下の第8及び9節に記載する実施例で実証したp-GlcNAc出発物質の電子顕微鏡構造を、図6から図9Eにわたって示した。
【0035】
p-GlcNAc出発物質は高度の生体適合性を示す。生体適合性は、様々な技術により決定することができ、その技術は、限定されるものでないが、溶出試験、筋肉内移植、または動物被験者への皮内または全身注射などが挙げられる。簡単に説明すると、溶出試験(米国薬局方(U.S. Pharmacopeia) XXII, 1990, pp. 1415-1497;米国薬局方 XXII, 1991, 追補(Supplement)5, pp.2702-2703)は、試験材(test article)抽出物の生体適合性を評価するために設計されたものであり、抽出可能な細胞傷害性材に感受性がある哺乳動物細胞培養系(例えば、L929細胞系など)の試験材に応答する生物学的反応性をアッセイする。以下の第10節に記載する実施例はp-GlcNAc出発物質の高い生体適合性を実証する。
【0036】
5.2. p-GlcNAcの肉微小藻類供給源を生産する方法
5.2.1. p-GlcNAcの肉微小藻類供給源
p-GlcNAc出発物質は、肉微小藻類、好ましくは珪藻により生産し、かつそれから精製することができる。複数属の珪藻及びかかる属内の多数の種をp-GlcNAc出発供給源として利用することができる。これらの珪藻はそれぞれp-GlcNAcを生産する。かかる珪藻の写真は図12A-Bを参照すること。p-GlcNAc出発物質を生産する出発供給源として利用しうる珪藻は、限定されるものでないが、コアミケイソウ(Coscinodiscus)属、タイコケイソウ(Cyclotella)属、及びミカドケイソウ(Thalassiosira)属のメンバーが挙げられ、ミカドケイソウ(Thalassiosira)属が好ましい。
【0037】
コアミケイソウ(Coscinodiscus)属中で、p-GlcNAc出発物質を生産するために利用できる珪藻の種は、限定されるものでないが、concinnus及びradiatus種が挙げられる。タイコケイソウ(Cyclotella)属の利用しうる珪藻は、限定されるものでないが、caspia、cryptica、及びmeneghiniana種が挙げられる。p-GlcNAc出発物質を生産するために利用しうるミカドケイソウ(Thalassiosira)属珪藻は、限定されるものでないが、nitzschoides、aestivalis、antarctica、deciphens、eccentrica、floridana、fluviatilis、gravida、guillardii、hyalina、minima、nordenskioldii、oceanica、polychorda、pseudonana;rotula、tubifera、rumida、及びweissflogii種が挙げられ、fluviatilis及びweissflogii種が好ましい。
【0038】
上記の珪藻は、例えば、Bigelow海洋科学研究所の海洋植物プランクトンコレクションセンター(the Bigelow Laboratory for Ocean Sciences, Center for Collection of Marine Phytoplankton)(McKown Point, West Boothbay Harbor, Me., 04575)の培養コレクションから入手することができる。
【0039】
5.2.2. 珪藻類を増殖する方法
以上の第5.2.1節に記載のいずれの珪藻類も、例えば、本節に記載の方法を利用することにより、増殖させることができる。新しい珪藻培養は、無菌条件下で栄養培地(Nutrient Medium)に成熟珪藻培養物のアリコートを接種することにより開始する。栄養培地は全ての他の微生物を含んではならないので、栄養培地の調製に使用する水、有機成分、および無機成分を含む全ての物質は無菌でなければならない。さらに、この操作に関わる全ての工程、すなわち、全ての容器、ある容器から他の容器への物質の移動の全て、その他は、厳密な無菌条件のもとで実施することが必須である。一度に調製する栄養培地の量は、新しい培養を出発するために必要である量を超えてはならない。例えば、ほぼ1平方フィートの表面を占めるフェムバッハ(Fembach)フラスコを珪藻培養用の容器として使用してもよく、かかる容器は珪藻生物の最適な増殖のために1リットルの栄養培地を必要とする。
【0040】
栄養培地の調製は次の操作からなる:
a)海水の取得及び処理
b)脱イオン蒸留水の調製
c)一次栄養ストックの調製
d)栄養作業ストックの調製
e)最終栄養培地の調製
濾過海水は、例えば、海洋生物学研究所(Marine Biology Laboratory)(Woods Hole, Mass.)から入手できる。海水容器は5℃(±2℃)で保存しなければならない。必要があれば、所要容積の水を0.8ミクロン孔サイズのSupor-800ポリエーテルスルホンフィルター膜(Gelman, Inc.)を用いてブーフナー(Buchner)濾過ユニットを通して濾過してもよい。次いで、海水を例えば、121℃にて1リットル当たり少なくとも約15分間、オートクレーブ処理して無菌化する。無菌化プロセスが完了すると、栓をしたフラスコを、好ましくは、溶液をほぼ5℃(±2℃)の温度に到達させうる低温室へ移して即時冷却する。使用するときに、溶液を室温にあげる。
【0041】
水道水は、標準の設備と方法を用いて、蒸留しかつ脱イオンして回収し、そして清浄な密栓をした、好ましくはガラス製の容器に入れて保存する。
【0042】
以下に掲げるのは、栄養培地の調製に必要なストック溶液を調製するのに従いうる処方である。かかる処方は手引きとして利用されるが、かかる処方の日常的変法であって本明細書に記載のp-GlcNAc調製方法のために十分な肉微小藻類珪藻の増殖を持続しうる栄養培地の調製を助ける前記変法も、本発明の範囲内にあると意図することは理解されるべきである。
【0043】
I. 微量金属一次ストック(TMPS)
a. 39mM CuSO4.5H2O(硫酸銅[II]五水和物)(9.8g 硫酸銅[II]/L)
b. 7.5mM ZnSO4.7H2O(硫酸亜鉛七水和物)(22g 硫酸亜鉛/L)
c. 42mM CoCl2.6H2O(塩化コバルト[II]六水和物)(10g 塩化コバルト[II]/L)
d. 91mM MnCl2.4H2O(塩化マンガン[II]四水和物)(18g 塩化マンガン[II]/L)
e. 26mM NaMoO4.2H2O(モリブデン酸ナトリウム二水和物)(6.3g モリブデン酸ナトリウム/L)
f. 1mM H2SeO3(亜セレン酸)(0.129g 亜セレン酸/L)
0.2ミクロン孔サイズ以下のフィルターを用いて各栄養を滅菌濾過する。
【0044】
II. ビタミン一次ストック(VPS)
a. 1mg/ml ビタミンB12
b. 0.1mg/ml ビオチン
0.2ミクロン孔サイズ以下のフィルターを用いて両方のストックを滅菌濾過する。
【0045】
III. ナトリウム塩作業ストック(SSWS)
a. 硝酸ナトリウム作業ストック:0.88M(75g NaNO3/L)
b. 一塩基性リン酸ナトリウム一水和物作業ストック:36.2mM NaH2PO4・H2O(5g NaH2PO4・H2O/L)。メタケイ酸ナトリウム一水和物作業ストック:0.11M Na2SiO3・9H2O(30g Na2SiO3・9H2O/L)。
【0046】
0.2ミクロン孔サイズ以下のフィルターを用いてそれぞれのSSWSを滅菌濾過する。
【0047】
IV. 微量金属作業ストック(TMWS)
11.7mM Na2EDTA(エチレンジアミン四酢酸、二ナトリウム塩二水和物)(4.36 g/L)
11.7mM FeCl3・6H2O(塩化鉄[III]六水和物)(3.15 g/L)
先に掲げた6つの微量金属一次ストックのそれぞれの1ml/L。
【0048】
0.2ミクロン孔サイズ以下のフィルターを用いて滅菌濾過する。微量金属作業ストックは毎週新しく調製しなければならないことに注意する。
【0049】
V. ビタミン作業ストック(VWS)
1.0mμg/ml ビオチン(1.0ml 一次ビオチンストック/100ml)
1.0mμg/ml ビタミンB12(0.1ml ビタミンB12一次ストック/100ml)
0.20mg/ml チアミンHCl(20mg チアミン塩酸塩/100ml)。
【0050】
0.2ミクロン孔サイズ以下のフィルターを用いて滅菌濾過する。ビタミン作業ストックは毎週新しく調製しなければならないことに注意する。
【0051】
以下に記載したのは、栄養培地の調製及び珪藻培養のために従いうる技術である。これらの技術に加えて、本明細書に記載の調製方法として十分な珪藻増殖を持続できる栄養培地及び方法をもたらす本明細書に記載の処方及び/または操作のいずれの日常的変法も本発明の範囲内にあると意図することは理解されるべきである。
【0052】
栄養培地は、例えば、次のように調製することができる:濾過して無菌化した海水の各1リットルにNaNO3作業ストック1ml、NaH2PO4・H2O作業ストック1ml、微量金属作業ストック1ml、及びNa2SiO3・9H2O作業ストック1mlを加える。Na2SiO3・9H2Oの添加と同時に、1N HCl 2mlを加え、溶液を振とうして混合してもよい。次に、1N NaOH 1.5mlを加え、溶液を再び振とうして混合してもよい。最後に、ビタミン作業ストック0.5 mlを加えてもよい。
【0053】
新しい珪藻培養物を増殖する目的で、成熟培養物7ml(約1 x 105〜約1 x 106 細胞/mlの範囲内の細胞密度を有する)を、上記の方法に従って調製することができる無菌栄養培地100mlを含有する無菌容器に移してもよい。次いで、接種した培養物を8日間、次の条件のもとでインキュベートしてもよい:
温度:20℃、常時照明、
攪拌:1日1回、フラスコの緩やかな旋回。
【0054】
8日間のインキュベーション後に、このインキュベートした培養物80mlを、無菌条件下で、例えば、チーズ布で覆った綿ウール栓により保護した2.8Lフェルンバッハ(Fernbach)フラスコ中に含有される栄養培地1000mlに移してもよい。かかる培養物は、インキュベートして所望の細胞密度まで増殖させるか、あるいは、新しい珪藻培養物を接種するために利用することができる。培養物が所望の細胞密度に到達すると、培養物のp-GlcNAc繊維を収穫し、以下の第5.3節に記載の方法を利用して、p-GlcNAc出発物質を精製することができる。
【0055】
培養pHをほぼ7〜8、好ましくはほぼ7.4に維持するために、CO2を培養溶液に溶解してもよい。かかる中性pH環境を維持することにより、それぞれの珪藻培養物から得られるp-GlcNAc収率が大きく増加する。
【0056】
5.3. p-GlcNAc繊維を単離、精製、及び濃縮する方法
本節で記載するのは、以上の第5.2節で記載した珪藻培養物からp-GlcNAc繊維を調製するために利用しうる方法である。
【0057】
一方、以下に記載の肉微小藻類、好ましくは珪藻出発供給源からp-GlcNAcを精製する方法はそれぞれ、非常に純粋な、不純物を含まない半結晶p-GlcNAcを生産するものである。例えば、p-GlcNAc出発物質は、以下の第5.3.1節に記載する機械力方法を経由して精製することができる。第2の方法は化学的/生物学的方法と呼ばれ、第5.3.2節に記載され、機械力方法により生産される平均p-GlcNAc収率よりさらに高い平均収率を生産する。さらに、以下の第5.3.2節の化学的/生物学的方法の一部として記載した酸処理/中和化変法は、極めて長いp-GlcNAc繊維を生産し、ある繊維は100μmを超えかつ20-30 x 106ダルトンの非常に高分子量のp-GlcNAcポリマー分子を含有する。p-GlcNAcポリマー出発物質の分子量測定は、限定されるものでないが、固有粘度の測定を含む、当業者であれば周知のクロマトグラフィ及び物理化学的方法を利用して行う。
【0058】
5.3.1. 純粋なp-GlcNAcを調製するための機械力方法
p-GlcNAc繊維は、培養内容物を適当な機械力で処理することにより珪藻細胞体から分離することができる。かかる機械力は、限定されるものでないが、例えばコロイドミル、超音波装置、もしくは泡発生器により発生させる剪断力、または、例えばウエアリング(Waring)ブレンダーにより発生させる切断力が挙げられる。
【0059】
得られる珪藻細胞体及びp-GlcNAc繊維の懸濁液を、次いで分離する。例えば、懸濁液を一連の遠心分離工程で処理して細胞体からp-GlcNAc繊維を分離し、目視しうる凝集沈殿物質は、もし若干あっても、僅かでしかない透明な上清を得る。固定角の回転子及び約10℃の温度が遠心分離工程に好ましい。所要の速度、時間、及び遠心分離工程の総数は、例えば、使用される特定の遠心分離回転子に依って変化しうるが、かかるパラメーター値の決定は、当業者には明らかであろう。
【0060】
上清中のp-GlcNAc繊維は、次いで、当業者に周知の技術を利用して濃縮することができる。かかる技術は、限定されるものでないが、吸引及び濾過装置が挙げられる。
【0061】
最後に、濃縮したp-GlcNAc繊維は、例えば、脱イオン蒸留水、HCl及びエタノール、または他の適当な溶媒、好ましくは有機と無機物質の両方を溶解するアルコールなどの溶媒を用いて洗浄する。
【0062】
以下の第7節に記載する実施例は、p-GlcNAcを精製するためのこの方法の使用を示す。
【0063】
5.3.2. p-GlcNAcを精製するための化学的/生物学的方法
この方法においては、以下にさらに詳細に記載する化学的及び/または生物学的作薬剤で処理することにより、珪藻細胞体からp-GlcNAc繊維を分離する。
【0064】
珪藻培養物を、珪藻細胞壁を弱化しうる化学品を用いて処理し、p-GlcNAc繊維をその長さ及び構造を改変することなく放出させることができる。かかる化学品は、限定されるものでないが、フッ化水素酸(HF)が挙げられる。あるいは、成熟珪藻培養物を、生物学的プロセスを改変しうる生物学的薬剤により処理してp-GlcNAc繊維合成を抑制し、そして既に存在する繊維を放出させてもよい。例えば、かかる薬剤としては、限定されるものでないが、酵素N-アセチルグルコサミニル-P-トランスフェラーゼのインヒビターであるポリオキシン-Dが挙げられる。
【0065】
上記の化学的または生物学的薬剤を用いて処理した珪藻培養物中の細胞体及びp-GlcNAcを含有する繊維を、次に分離する。例えば、処理した珪藻培養の内容物を静置し、培養内容物から2つの明確な層を形成させてもよい。上層は主にp-GlcNAc繊維を含有するのに対して、下層は細胞体を含有する。上側のp-GlcNAc繊維を含有する層をサイフォンで吸い出し、底層の沈降した細胞物質を残してもよい。
【0066】
サイフォンで吸い出したp-GlcNAc繊維を含有する層を、次いでさらにp-GlcNAc繊維を損傷しないであろう界面活性剤を用いる処理により精製し、タンパク質及びその他の望ましくない物質を除去する。かかる界面活性剤としては、限定されるものでないが、ドデシル硫酸ナトリウム(SDS)が挙げられる。
【0067】
HF処理などの酸処理を用いて珪藻細胞体からp-GlcNAc繊維を分離するときに、繊維を分散させる工程を含んでもよい。かかる工程としては、限定されるものでないが、繊維をオービタルシェイカー(orbital shaker)の運動で処理する工程などの繊維分散のための機械力の使用が挙げられる。
【0068】
あるいは、酸処理した懸濁液を、場合により、中和した後に、界面活性剤処理によりさらに精製してもよい。かかる中和は、一般的に、懸濁液のpHをほぼ1.8からほぼ7.0に変えることであり、例えば、適当な容積の1M Tris(pH 8.0)の添加または適当な容積の水酸化ナトリウム(NaOH)の添加により達成しうる。中和すると、一般的に、本明細書で考察した他の精製方法より実質的に長い、純粋なp-GlcNAc繊維が得られる。
【0069】
精製したp-GlcNAc繊維は、次いで、吸引及び濾過装置を利用するなどの、当業者に周知の技術を利用して濃縮することができる。最後に、p-GlcNAc繊維を、脱イオン蒸留水、HCl及びエタノールまたは他の適当な溶媒、好ましくは、有機及び無機物質の両方を溶解するアルコールなどの溶媒を用いる一連の工程において洗浄する。
【0070】
以下の第8節に記載する実施例は、かかる精製法の成功を収めた利用を実証する。
【0071】
p-GlcNAc出発物質またはその部分的に脱アセチル化された誘導体を、制御した加水分解条件で処理すると、均一な、明確な分子量と他の物理的特性を有する分子のグループが得られる。かかる加水分解条件としては、例えば、酵素、リゾチームによる処理が挙げられる。p-GlcNAcを時間を変えてリゾチームに曝し、加水分解の程度を制御してもよい。かかる酵素による部分的消化反応はまた、基質の濃度、もしくは酵素の濃度、または基質と酵素の両方の濃度、ならびにpH及び温度を変えることにより制御することができる。さらに、加水分解速度は、リゾチーム処理されるp-GlcNAcが脱アセチル化されている程度の関数として制御することができる。脱アセチル化条件は、本節の初めに記載されている。約20〜90脱アセチル化パーセントの間で、p-GlcNAc分子がより完全に脱アセチル化されているほど、その分子は所与の時間により完全に加水分解されるであろう。加水分解及び/または脱アセチル化処理により、分子量の低下に加えて、物理特性の変化が誘発されるであろう。加水分解/脱アセチル化操作の結果は以下の第9節の実施例に記載する。
【0072】
5.4. p-GlcNAcの誘導体化
純粋な、完全にアセチル化されたp-GlcNAc出発物質は、様々な制御された条件と方法を利用することにより、広範囲の異なる化合物に誘導体化することができる。これらの化合物のいくつかを記載した図13を参照すること。かかる誘導体化した化合物としては、限定されるものでないが、以下にさらに詳しく記載する化学的及び/または酵素的手法を経由して改変された、部分的に脱アセチル化されたp-GlcNAcが挙げられる。さらに、p-GlcNAcまたはその部分的に脱アセチル化された誘導体は、硫酸化、リン酸化、及び/または硝酸化することにより誘導体化することができる。さらに、以下に詳述したように、O-スルホニル、N-アシル、O-アルキル、N-アルキル、及びN-アルキリデン及びN-アリーリデンならびにその他の誘導体を、p-GlcNAcまたは部分的に脱アセチル化されたp-GlcNAc出発物質から調製することができる。部分的に脱アセチル化されたp-GlcNAc出発物質を利用して、様々な有機塩及び/または金属キレートを調製することもできる。さらに、p-GlcNAc出発物質、またはその誘導体に、共有結合もしくは非共有結合により、任意の様々な分子が結合していてもよい。その上さらに、p-GlcNAc出発物質またはその誘導体を、制御された加水分解条件で処理し、均一かつ明確な分子量特性を有する分子のグループを得てもよい。かかる物質は、ポリマーをIR吸収分光計で分析したときに、p-GlcNAcポリマーが鋭く明確なピークにより示されるその半結晶構造を保持する限り、本発明に有用である。
【0073】
p-GlcNAc出発物質の1以上の単糖単位が脱アセチル化され、部分的に脱アセチル化されたポリβ-1→4-N-アセチルグルコサミン種を形成することができる。脱アセチル化モノマーは、一般的に、ポリマー全体にわたり本質的に無作為に分布してもよいし、またはポリβ-1→4 N-アセチルグルコサミンポリマー内の不連続なサブ領域として比較的かたまっていてもよい。ポリβ-1→4-N-アセチルグルコサミン種出発物質の単糖単位の一部分が脱アセチル化されているポリβ-1→4-N-グルコサミン種出発物質は、約30 x 106ダルトン以下の分子量を有し、β-1→4-Nコンフィギュレーションで共有結合した約150,000個のグルコサミン多糖を含む。一実施形態においては、ポリβ-1→4-N-グルコサミン種のグルコサミン単糖の少なくとも約90%はアセチル化されたまま残存し、一方、他の実施形態においては、ポリβ-1→4-N-グルコサミン種の単糖単位の少なくとも約80%、70%、60%、50%、または40%がアセチル化されたまま残存する。ただし、以下の実施例6に記載し、かつ図4B及び4Cに示した非結晶p-GlcNAcポリマーによって示されるIR吸収スペクトルと比較して図4A、4D、及び4Eに示したように、ポリマーをIR吸収分光計で分析したときに、部分的に脱アセチル化されたポリβ-1→4 N-アセチルグルコサミンポリマーは鋭く明確なピークにより示されるその半結晶構造を保持する。
【0074】
p-GlcNAc出発物質を塩基による処理で脱アセチル化し、遊離アミノ基をもつグルコサミンを得ることができる。この加水分解プロセスは、濃い水酸化ナトリウムまたは水酸化カリウム溶液を用いて高温で実施してもよい。しかし、脱アセチル化の程度を正確に制御するため及び多糖分子の炭水化物主鎖の分解を避けるため、キチンデアセチラーゼ酵素を使用する酵素手法を用いてp-GlcNAcを脱アセチル化するのが好ましい。かかるデアセチラーゼ酵素手法は当業者に周知であって、本明細書に参照によりその全文が組み入れられる(米国特許第5,219,749号)に従って実施することができる。
【0075】
p-GlcNAc出発物質の1以上の単糖単位は誘導体化されて、以下に記載のように、少なくとも1つの硫酸基を含有させるか、リン酸化もしくはニトロ化されていてもよい:
【化1】
[式中、Rおよび/またはR1は、水素の代わりに、及び/またはR2は、-COCH3の代わりに硫酸(-SHO3)、リン酸(-P(OH)2)、または硝酸(-NO2)基であってもよい]。
【0076】
以下に記載したのは、かかるp-GlcNAc誘導体を調製しうる方法である。本節に記載の方法を実施する前に、p-GlcNAc出発物質を最初に凍結乾燥し、液体窒素中で凍結し、そして粉末化することが有利でありうる。
【0077】
硫酸化p-GlcNAc誘導体は、例えば、2工程プロセスにより作製することができる。第1工程において、O-カルボキシメチルp-GlcNAcを、出発物質のp-GlcNAcおよび/またはp-GlcNAc誘導体から、例えば、Tokuraら(Tokura, S.ら, 1983, Polym. J. 15:485)が記載した技術を利用して調製することができる。第2に、硫酸化工程を、例えば、Schweiger(Schweiger, R. G., 1972, Carbohydrate Res. 21:219)が記載したような当業者に周知の技術に従い、N,N-ジメチル-ホルムアミド-三酸化硫黄を用いて実施することができる。得られた生成物は、ナトリウム塩として単離することができる。出発物質のリン酸化p-GlcNAc誘導体は、例えば、Nishiら(Nishi, N.ら, 1986, in "Chitin and Chitosan," Muzzarelliら, 編, Plenum Press, New York, pp. 297-299)が記載した当業者に周知の技術を利用することにより調製することができる。簡単に説明すると、p-GlcNAc/メタンスルホン酸混合物を、攪拌しながら約0℃〜5℃の温度にて五酸化リン(ほぼ0.5〜4.0モル当量)を用いて処理してもよい。処理は約2時間である。得られる生成物を次いで、当業者が周知する標準技術を用いて、沈殿させ洗浄してもよい。例えば、サンプルをエーテルなどの溶媒を用いて沈殿させ、遠心分離し、エーテル、アセトン、またはメタノールなどの溶媒を用いて洗浄し、そして乾燥してもよい。
【0078】
ニトロ化p-GlcNAc誘導体は、Schorigin及びHalt(Schorigin, R.及びHalt, E., 1934, Chem. Ber. 67:1712)が記載した当業者に周知の技術を利用して調製することができる。簡単に説明すると、p-GlcNAc及び/またはp-GlcNAc誘導体を濃硝酸を用いて処理して安定なニトロ化生成物を形成させる。
【0079】
p-GlcNAc出発物質の1以上の単糖単位は、以下に記載のように、スルホニル基を含有しうる:
【化2】
[式中、R3はアルキル、アリール、アルケニル、またはアルキニル部分であってもよい]。かかる誘導体は、Kuritaら(Kurita, K. ら, 1990, Polym. Prep (Am. Chem. Soc., Div. Polym. Chem.) 31:624-625)が記載した周知の方法により作製することができる。簡単に説明すれば、p-GlcNAcアルカリ水溶液を塩化トシルのクロロホルム溶液と反応させ、次いで低温で円滑に反応を進行させる。
【0080】
p-GlcNAc出発物質またはその脱アセチル化された誘導体の1以上の単糖は、1以上の以下に記載のO-アシル基を含有してもよい:
【化3】
[式中、R4及び/またはR5は、水素の代わりに、アルキル、アルケニル、またはアルキニル部分であってもよく、そしてR6はアルキル、アルケニル、またはアルキニル部分であってもよい]。かかる誘導体の例は、Komai(Komai, T.ら, 1986, in "Chitin and Chitosan,", Muzzarelliら, 編, Plenum Press, New York, pp. 497-506)が記載したような周知の方法により作製することができる。簡単に説明すると、p-GlcNAcを、メタンスルホン酸中で複数の適当な塩化アシルと反応させ、限定されるものでないが、カプロイル、カプリル、ラノイル、またはベンゾイル誘導体を含むp-GlcNAc誘導体を得ることができる。
【0081】
脱アセチル化p-GlcNAc出発物質の1以上の単糖は、以下に記載のN-アシル基を含有しうる:
【化4】
[式中、R7は、アルキル、アルケニル、またはアルキニル部分であってもよい]。かかる誘導体化は、Hiranoら(Hirano, S.ら, 1976, Carbohydrate Research 47: 315-320)が記載したような当業者に周知の技術を利用して得ることができる。
【0082】
脱アセチル化p-GlcNAcは、複数の有機酸水溶液に可溶である。選択したカルボン酸無水物を酢酸メタノール水溶液中のかかるp-GlcNAcを含有する溶液に加えると、N-アシルp-GlcNAc誘導体が形成される。
【0083】
脱アセチル化p-GlcNAc出発物質またはその脱アセチル化誘導体の1以上の単糖は、以下に記載のO-アルキル基を含有しうる:
【化5】
[式中、R8はアルキル、及びアルケニル、またはアルキニル部分であってもよい]。かかる誘導体化は、当業者が周知の技術を利用して得ることができる。例えば、Mareshら(Maresh, G.ら, in "Chitin and Chitosan," Skjak-Braek, G.ら, 編, 1989, Elsevier Publishing Co., pp. 389-395)が記載した方法が挙げられる。簡単に説明すると、脱アセチル化p-GlcNAcをジメトキシエタン(DME)中に分散させ、過剰のプロピレンオキシドと反応させる。反応時間は24時間であってもよく、反応はオートクレーブ中で40℃〜90℃にて行う。次いで混合物を水を用いて希釈し、濾過してもよい。DMEは蒸留により除去できる。最後に、最終生成物を凍結乾燥を経由して単離できる。
【0084】
p-GlcNAc出発物質の1以上の単糖単位は、以下に記載のアルカリ誘導体:
【化6】
であってもよい。かかる誘導体は、当業者に周知の技術により得ることができる。例えば、Noguchiら(Noguchi, J.ら, 1969, 工業化学雑誌 72:796-799)が記載したような方法を利用することができる。簡単に説明すると、p-GlcNAcを、真空下でNaOH(好ましくは43%)にほぼ2時間、0℃にて浸漬する。次いで過剰のNaOHを、例えば、かご型遠心分離機で遠心分離しかつ機械プレスにより除去してもよい。
【0085】
p-GlcNAc出発物質の脱アセチル化された誘導体の1以上の単糖単位は、以下に記載のN-アルキル基を含有してもよい:
【化7】
[式中、R9は、アルキル、アルケニル、またはアルキニル部分であってもよい]。かかる誘導体化は、例えば、O-アルキルp-GlcNAc誘導体の生成について上に記載した、Mareshら(Maresh, G.ら, in "Chitin and Chitosan," Skjak-Brack, G.ら, 編, 1989, Elsevier Publishing Co., pp.389-395)のような方法を利用して、得ることができる。
【0086】
p-GlcNAc出発物質の脱アセチル化された誘導体の1以上の単糖単位は、以下に記載の塩を形成してもよい:
【化8】
[式中、R11はアルキル、アルケニル、またはアルキニル部分であってもよい]。かかる誘導体化は、当業者が周知の技術を利用することにより得ることができる。例えば、Austin及びSennett(Austin, P. R.及びSennett, S., in "Chitin in Nature and Technology," 1986, Muzzarelli, R. A. A.ら, 編, Plenum Press, pp. 279-286)が記載したような方法を利用することができる。簡単に説明すると、脱アセチル化p-GlcNAcを例えば、酢酸エチルまたはイソプロパノールなどの有機媒質中に懸濁し、それに例えば、蟻酸、酢酸、グリコール酸、または乳酸などの適当な有機酸を加えてもよい。混合物をある時間(例えば、1〜3時間)静置してもよい。反応及び乾燥温度は約12℃〜約35℃で変化してもよく、20℃〜25℃が好ましい。次いで塩を濾過により分離し、新鮮な媒質を用いて洗浄し、そして残る媒質は蒸発させてもよい。
【0087】
p-GlcNAc出発物質の脱アセチル化された誘導体の1以上の単糖単位は、以下に記載の金属キレートを形成してもよい:
【化9】
[式中、R12は金属イオン、特に遷移金属の1つであってもよく、Xは脱アセチル化p-GlcNAc中に存在するアミノ及び置換アミノ基中に存在する窒素電子により確立された供与結合である]。
【0088】
p-GlcNAc出発物質の脱アセチル化された誘導体の1以上の単糖単位は、以下に記載のN-アルキリデンまたはN-アリーリデン基を含有してもよい:
【化10】
[式中、R13は、アルキル、アルケニル、アルキニル、またはアリール部分であってもよい]。かかる誘導体化は、当業者に周知の技術を利用して得ることができる。例えば、Hiranoら(Hirano, S.ら, 1981, J. Biomed. Mat. Res. 15:903-911)が記載した方法を利用してもよい。簡単に説明すると、脱アセチル化p-GlcNAcのN-置換反応を、カルボン酸無水物及び/またはアリールアルデヒドを用いて実施し、アシル-及び/またはアリーリデン誘導体を得てもよい。
【0089】
さらに、p-GlcNAc出発物質、またはその部分的に脱アセチル化された誘導体を、制御された加水分解条件で処理し、均一で明確な分子量及びその他の物理的特性を有する分子のグループを得ることができる。かかる加水分解条件は、例えば、酵素、リゾチームによる処理が挙げられる。p-GlcNAcを、加水分解の程度を制御するために、時間を変えて、リゾチームに曝してもよい。さらに、加水分解速度は、リゾチーム処理されるp-GlcNAcの脱アセチル化されている程度の関数として制御してもよい(例えば、第15節に提供し、かつ図18-20に示した実施例を参照)。かかる酵素による部分消化反応はまた、基質、酵素、または基質と酵素の両方の濃度、ならびにpH及び温度を変化させることにより制御することができる。他の実施形態においては、p-GlcNAcポリマーのサイズを超音波処理により小さくする。そのサイズは、使用する機器の動力だけでなくサンプルのpH、塩濃度、及び温度によって変化させることができる。p-GlcNAcもしくはその誘導体の可溶化は、第5.5節に記載した。従って、1以上のこれらの方法を単独でまたはお互いに組み合わせて利用することにより、より高分子量のp-GlcNAcポリマーを加水分解してより小さい断片とし、それを、例えばカラムクロマトグラフィを用いて、サイズに従ってクロマトグラフィ的に分離することができる。
【0090】
例えば、当業者であれば、p-GlcNAcの部分消化の程度を変えて所望の分子量範囲を有する反応生成物を提供するであろう。他の実施形態においては、リゾチームによる部分消化に使用する基質は、超音波処理されている及び/または部分的に脱アセチル化されているp-GlcNAcである。部分酵素消化を、カラムクロマトグラフィ、HPLC分離、または他の当技術分野で周知の技術及び方法などの分離技術と組み合わせることにより、当業者は狭い分子量分布範囲をもつ消化生成物を単離することができる。さらに、一連の部分消化反応の生成物を組み合わせることにより、当業者は、半結晶p-GlcNAc生成物の広範囲の分子量種を有するp-GlcNAcポリマーを含んでなる組成物をアセンブルすることができる。前記組成物は、例えば本明細書に開示された集団、すなわち一実施形態においては約50〜約150,000モノマー単位を含むポリマー、ならびに約50〜約50,000、約50〜約10,000、及び約50〜約4,000モノマー単位を含むポリマーを含むものである。。
【0091】
脱アセチル化条件は、本節の初めに記載されている。p-GlcNAc分子は約20〜90脱アセチル化パーセントの間でより完全に脱アセチル化されているほど、その分子は所与の時間により完全に加水分解されうる。分子量の低下に加えて、加水分解及び/または脱アセチル化処理により、物理的特性の変化が誘発されうる。
【0092】
さらに、様々な分子をp-GlcNAc出発物質の脱アセチル化された誘導体と、共有結合または非共有結合により機能的に結合させてもよい。かかる分子としては、限定されるものでないが、神経増殖因子などの増殖因子、ペプシンなどのプロテアーゼ、ホルモンのようなポリペプチド、またはRGD配列などのペプチド認識配列、フィブロネクチン認識配列、ラミニン、インテグリン、細胞接着分子、その他が挙げられる。例えば、以下の第5.6.1.1節に考察した化合物を参照すること。分子と曝された脱アセチル化p-GlcNAcの第一級アミンとの共有結合は、例えば、特定の長さの化学スペーサーとして作用する二官能性架橋試薬を利用する化学結合により実施することができる。かかる技術は当業者には周知であり、例えば、Davis及びPreston(Davis, M.及びPreston, J. F. 1981, Anal. Biochem. 116:404-407)ならびにStarosら(Staros, J. V.ら, 1986, Anal. Biochem. 156:220-222)の方法に類似する。簡単に説明すると、脱アセチル化されたまたは部分的に脱アセチル化されたp-GlcNAc出発物質と結合するペプチドのカルボン酸残基を活性化し、次いでp-GlcNAcと架橋することができる。活性化は、例えば、カルボジイミドEDC(1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド)などの溶液をリン酸バッファー中のペプチド溶液へ加えることにより実施できる。好ましくは、この溶液は、さらに、カップリングを増強するスルホ-NHS(N-ヒドロキシスルホスクシンイミド)などの試薬を含有しうる。活性化ペプチドは、炭酸バッファー(pH 9.0-9.2)などの高pHバッファー中で混合することにより脱アセチル化p-GlcNAcと架橋することができる。
【0093】
結合したペプチド(またはいずれかの共有結合した分子)の生物学的活性は、分子をp-GlcNAc出発物質と結合するために利用するリンカー分子(例えば、二官能性架橋化合物)の長さを変えることにより、維持することができる。結合分子の生物学的活性を変えることのない、所与の結合分子に対する適当なリンカー長さは、日常的に確かめることができる。例えば、所与の長さのリンカーを経由して結合されている分子の生物学的活性(例えば、治療上有効なレベルの生物学的活性)は、所与の結合される分子に特異的な周知のアッセイを利用して試験することができる。
【0094】
さらに、結合している分子の生物学的活性を維持するために、適当な天然の酵素により切断されてペプチド(またはいずれかの共有結合した分子)を放出しうるリンカーを利用することが必要な場合もある。
【0095】
上記のように、当業者が通常使用するアッセイを利用して、結合している特定分子の保持する生物学的活性を試験し、許容しうる活性レベル(例えば治療上有効な活性レベル)を保証することができる。
【0096】
あるいは、上記の分子を当業者が周知の技術を利用してp-GlcNAc及びその誘導体と非共有結合により結合することができる。例えば、選択した1以上の分子を、p-GlcNAc懸濁液もしくは部分的に脱アセチル化されたp-GlcNAc溶液と、p-GlcNAc-乳酸溶液と、脱アセチル化されたもしくは部分的に脱アセチル化されたp-GlcNAc塩溶液と、またはいずれかのp-GlcNAc誘導体溶液と混合してもよい。次いで混合物を凍結乾燥してもよい。凍結乾燥の後、分子は、恐らく疎水的、静電的及び他の非共有結合の相互作用を経由してp-GlcNAcマトリックスと結合する。従って、かかるp-GlcNAc製剤は製造が容易である。さらに、かかる製剤は、広いスペクトルの物理的特性及び最も疎水性から最も親水性までの水溶解特性を有する広範囲の分子を用いて、効果的に製造することができる。分子の結合にあたっては、当業者が特定の非共有結合した分子の活性を試験するために通常使用するアッセイを利用して、結合した分子により許容しうる活性のレベル(例えば、治療上有効な活性)が達成されたことを確認することができる。
【0097】
あるいは、p-GlcNAc及び/またはp-GlcNAc誘導体を含むハイブリッドを形成することができる。かかるハイブリッドは、p-GlcNAc及び/またはp-GlcNAc 誘導体に加えて、複数の天然の及び/または合成の物質を含有してもよい。例えば、ハイブリッドは、p-GlcNAc及び/またはp-GlcNAc誘導体に1以上の細胞外マトリックス(ECM)成分を加えて作製してもよい。かかるECM成分としては、限定されるものでないが、コラーゲン、フィブロネクチン、グリコサミノグリカン及び/またはペプチドグリカンが挙げられる。ハイブリッドはまた、p-GlcNAc及び/またはp-GlcNAc誘導体に1以上の合成物質、例えばポリエチレンなどを加えて作製してもよい。かかるp-GlcNAc/ポリエチレンまたはp-GlcNAc誘導体/ポリエチレンハイブリッドは、ハイブリッド成分を例えば、オートクレーブ処理を経由して熱的に結合して作ることができる。かかるハイブリッドポリマーは、ハイブリッドポリマーが、以下の実施例6に記載のIR吸収分光計により分析したとき、鋭い、明確なピークにより実証されるp-GlcNAc半結晶構造を保持する限り、本発明に有用である。
【0098】
コラーゲン/p-GlcNAcハイブリッドの場合、簡単に説明すると、p-GlcNAc懸濁液とコラーゲン懸濁液を混合し、凍結乾燥し、そして架橋、好ましくは脱水加熱して架橋することができる。かかるハイブリッドのコラーゲン種は天然でも合成でもよく、ヒトまたは例えばウシ起源などの非ヒト由来であってもよい。p-GlcNAc/コラーゲン及び/またはp-GlcNAc誘導体/コラーゲンハイブリッド物質は均一な物性を有しかつ多孔質マトリックスを形成する。以下の第13節に記載する実施例は、かかるp-GlcNAc/コラーゲンハイブリッドの作製、物性及び有用性を実証する。
【0099】
さらに、ヨード-p-GlcNAc誘導体を、例えば、スチレンと共重合して新規のプラスチック物質を製造することができる。ヨード-p-GlcNAcは、Kurita及びInoue(Kurita, K.及びInoue, S., 1989, in "Chitin and Chitosan," Skjak-Braekら, 編, Elsevier Science Publishing Co., Inc., p.365)が記載したのと同様な方法により、p-GlcNAcのトシル化とヨード化を経由して調製することができる。次いで、p-GlcNAcのヨード誘導体をニトロベンゼン中に分散させ、塩化スズ(IV)を触媒に使ってスチレンと反応させることができる。
【0100】
脱アセチル化p-GlcNAc及び、例えばアルギン酸ナトリウムなどの化合物の組合せ、ならびにカルボキシメチルp-GlcNAcを含んでなるハイブリッドを、本明細書に記載の技術を用いて製剤化することができる。かかる組合せを、例えば、膜及び繊維に成形または再成形してもよい。
【0101】
部分的に脱アセチル化されたp-GlcNAcと例えば、正及び負電荷の両方を有する、ポリアクリル酸もしくはペクチンなどのポリアニオンとの複合物を製剤化してもよい。かかる複合物の形成は、Mirelesら(Mireles, C.ら, 1992, in "Advances in Chitin and Chitosan," Brine, C. J.ら, 編, Elsevier Publishers, Ltd.)が記載したのと類似の方法に従って実施することができる。部分的に脱アセチル化されたp-GlcNAc及びポリアクリル酸、カラゲナンまたはペクチンを、例えば、HCl及びNaCl中にそれぞれ溶解し、等しいpHをもつ反応溶液と混合する。この操作は、正及び負の特性の両方を有する有効な分子を作り、例えば、酵素及び治療用化合物を固定するのに有用である。
【0102】
5.5. 再製剤化
以上の第5.4節に記載した、p-GlcNAc出発物質、ならびにその部分的に脱アセチル化された誘導体及び/またはそれらの誘導体を溶解し、次いで様々な形状と形態に再製剤化することができる。
【0103】
p-GlcNAc出発物質の溶解は、ジメチルアセトアミド(DMA)/塩化リチウムで処理することにより達成できる。p-GlcNAcは、5%LiCl(DMAの重量で)を含有するDMA溶液中で攪拌すると容易に溶解することができる。p-GlcNAc塩などの水溶性p-GlcNAc誘導体を水に溶解してもよい。部分的に脱アセチル化しておいたp-GlcNAcを1%酢酸などの弱い酸性溶液に溶解してもよい。水に不溶性のp-GlcNAc誘導体は、有機溶媒中の溶液としてもよい。
【0104】
DMA:LiCl中のp-GlcNAcのフェニルイソシアネートによる誘導体化を利用して、カルバニレートを作ることができる。さらに、DMA:LiCl中のp-GlcNAcをトルエン-p-スルホニルクロリドを用いて誘導体化して、トルエン-p-スルホネートを作ってもよい。
【0105】
溶液中のp-GlcNAc出発物質、その部分的に脱アセチル化された誘導体、及び/またはそれらの誘導体を、次いで沈降させ、限定されるものでないが、マット、ひも、ミクロスフェア(microsphere)、ミクロビーズ(microbead)、膜、繊維、ミクロファイバー(microfiber)、粉末、及びスポンジなどの形状に再製剤化することができる。さらに、超薄い(すなわち、約1ミクロンより薄い)均一な膜に製剤化してもよい。
【0106】
かかる製剤化は、例えば純粋なp-GlcNAcが、水及びアルコール、好ましくはエタノールなどの溶液に不溶であるという事実を利用して、達成することができる。例えばp-GlcNAcを含有するDMA/LiCl混合物を、注入などの通常の方法により、かかる水またはアルコール、好ましくはエタノール中に導入すると、溶液は溶存p-GlcNAcの再沈降、従って再製剤を起こしうる。かかる純粋なp-GlcNAc再製剤を、以下の第11節に記載する実施例において実証する。水溶性p-GlcNAc 誘導体の場合、再製剤化は、例えば、酢酸エチルまたはイソプロパノールなどの有機溶媒中で再沈降させることにより達成できる。部分的に脱アセチル化されているp-GlcNAcの再製剤化は、アルカリ溶液中で再沈降させることにより達成できる。水に不溶のp-GlcNAc誘導体は、例えば、水などの水溶液中で再沈降することにより再製剤化できる。
【0107】
p-GlcNAc膜及び三次元p-GlcNAcマトリックスは、膜またはマトリックス内に制御した平均孔サイズの形成を可能にする方法を経由して作ることができる。膜及びマトリックスにおける孔サイズは、使用するp-GlcNAc物質の量を変えることにより、ならびに、膜及び/またはマトリックスの形成前に、メタノールまたはエタノール、好ましくはエタノールなどの特定の溶媒を、約5%〜約40%の範囲の特定の量だけ加えることにより制御することができる。一般的に、溶媒のパーセントが高いほど、形成される平均孔サイズは小さくなる。以下の第15節に記載する実施例は、かかる多孔質p-GlcNAc構造の合成及び特性決定を実証する。
【0108】
他の実施形態においては、半結晶p-GlcNAcを、ミクロスフェア、ミクロビーズ、もしくはミクロフィブリル(microfibril)を含むゲル、フォーム、または溶液もしくは懸濁液として製剤化する。かかる製剤は、従って、さらに適当な量の製薬上許容されるビヒクルもしくは担体を含むものであって、半結晶p-GlcNAcの患者への投与に適当な形態を与える。
【0109】
特定の実施形態においては、用語「製薬上許容される」は、哺乳動物、特にヒトにおける使用について、連邦もしくは州政府の規制当局により認可されているかまたは米国薬局方またはその他の一般的に認識された薬局方に掲げられていることを意味する。用語「担体」は、治療剤と一緒に投与される希釈剤、アジュバント、添加剤、またはビヒクルを意味する。かかる製薬用担体は、水ならびにオイルなどの液体であってもよく、オイルとしては、落花生油、ダイズ油、鉱油、ゴマ油などの石油、動物、植物または合成起源のものが挙げられる。製薬用担体は、生理食塩水、アカシアゴム、ゼラチン、デンプンペースト、タルク、ケラチン、コロイド状シリカ、尿素、などである。さらに、補助剤、安定剤、粘稠剤、滑沢剤及び着色剤を使用してもよい。患者に投与するとき、p-GlcNAcと製薬上許容される担体は好ましくは無菌である。生理食塩水溶液ならびにデキストロース及びグリセロール水溶液を液状担体として利用することができる。適当な製薬用担体としてはまた、デンプン、グルコース、ラクトース、スクロース、ゼラチン、麦芽、コメ、小麦粉、チョーク、シリカゲル、ステアリン酸ナトリウム、モノステアリン酸グリセロール、タルク、塩化ナトリウム、乾燥スキムミルク、グリセロール、プロピレン、グリコール、水、エタノールなどが挙げられる。p-GlcNAc組成物はまた、所望により、最小量の湿潤もしくは乳化剤、またはpH緩衝剤を含有してもよい。
【0110】
p-GlcNAcを含有する組成物は、溶液、懸濁液、坐薬、エマルジョン、エーロゾル、スプレイ、または使用に適したいずれの他の形態をとってもよい。適当な製薬用担体のその他の例は、E. W. Martinによる「レミントンの製薬科学("Remington's Pharmaceutical Sciences")」に記載されている。
【0111】
p-GlcNAc製剤及び組成物はプレミックス投与形態として供給しうるが、他の実施形態においては、本明細書に開示された半結晶p-GlcNAcを、例えば、活性剤の量を示したアンプルまたは小袋(sachette)などの密封容器に入った乾燥した凍結乾燥粉末または無水濃縮物として別々に供給し、使用前に製薬上許容されるビヒクルまたは溶媒中に所望の濃度で懸濁または溶解してもよい。
【0112】
特定の障害または症状の治療に有効な半結晶p-GlcNAcの量は、障害または症状の性質に依存し、標準的な臨床技術により決定することができる。さらに、場合によってはin vitroまたはin vivoアッセイを実施して最適な用量範囲を決定する助けとしてもよい。組成物に使用される半結晶p-GlcNAcの正確な用量はまた、投与経路、及び疾患もしくは障害の重篤度にも依存し、担当医の判断とそれぞれの患者の状況に従って決定すべきである。しかし、半結晶p-GlcNAcは一般的に約1mg/cm2〜約500mg/cm2の範囲で局所投与される。他の実施形態においては、半結晶p-GlcNAcは一般的に約2mg/cm2〜約100mg/cm2、約5mg/cm2〜約50mg/cm2及び約10mg/cm2〜約20mg/cm2の範囲で局所投与される。有効用量は、in vitroまたは動物モデル試験系から誘導した用量-応答曲線から外挿することができる。第16及び17節に与えた後掲の実施例に記載するものの他に、当技術分野で周知のその他の動物モデル試験系としては、限定されるものでないが、次が挙げられる:(a)止血処置を評価するための、部分的肝切除のブタモデル、Davidsonら(Davidsonら 2000, Br. J. Surg. 87(6): 790-95)が記載;(b)止血の達成を意図する処置を評価するための、イヌ出血潰瘍モード、Pasrichaら(Pasrichaら 1999 Gastointest Endosc 49(5): 627-31)が記載;(c)肝切開の処置に基づく、ラットの外科出血モデル、Sirieixら(Sirieix ら 1998 Ann Vasc Surg 12(4): 311-16)が記載;(d)無傷内皮をもつ単離されたラット胸部大動脈環の血管収縮を評価する方法、Kimら(Kim ら 2000 J Lab Clin Med 135(2): 180-87;またGuo ら 1994 Methods Find Exp Clin Pharmaocl 15(5): 347-54も参照)が記載;(e)血管直径と血管を通過する血液流量の両方を測定することを意図する、ウサギの実験動物モデル、Caronら(Caron ら 1998 Artif Cells Blood Substit Immobil Biotechnol 26(3): 293-308)が記載;(f)ラットの子宮微小血管の直接観察を可能にし、適用した血管作用薬の量の関数として細動脈の外周径の評価を可能にする方法、Alsipら(Alsip ら 1996 Am J Obstet Bynecol 175(2):388-95)が記載;及び(g)自然発症高血圧ラットを用いたモデル形、とりわけ、ラジオイムノアッセイ手法を用いて血管中の免疫反応性エンドセリンのレベルを評価するモデル系、Schiffrinら(Schiffrin ら 1995 Br J Pharmacol 115(8): 1377-81)が記載。
【0113】
使用する半結晶p-GlcNAcの特定の製剤は、意図する用途に依存して異なる。例えば、接触可能な表面に直接適用するには、半結晶p-GlcNAcを膜または絆創膏などとして製剤化し製造することができる。かかる製剤においては、半結晶p-GlcNAcを、限定されるものでないが、天然または人工の繊維を含む1以上の他の物質と組み合わせてもよいし、及び/または、本明細書に記載のコポリマーとして再製剤化してもよい。かかる物質中に配合する1cm2当たりの半結晶p-GlcNAcの量は、意図する用途により、例えば、とりわけ、小さい切傷及び擦傷を治療する低用量範囲、及び重篤な傷害を治療する高用量p-GlcNAcレベルなどのように決定される。かかる物質のサイズ、形状、厚み、及びそれに配合する半結晶p-GlcNAcの総量を含む全組成は、同様に、意図する用途により決定される。
【0114】
半結晶p-GlcNAcを容易に接近し得ない表面、例えば、口内、鼻腔内、または身体の深い創傷に局所投与する場合、半結晶p-GlcNAcは、以上開示した製薬上許容される担体及びビヒクルを使用して、とりわけ、ゲル、フォーム、スプレイ、エマルジョン、懸濁液または溶液として製剤化される。かかる製剤は、通常、バリヤーを形成しない物質であり、一般的に、半結晶p-GlcNAcから形成されたミクロスフェア、ミクロビーズ、またはミクロフィブリルを含み、そして、限定されるものでないが、天然もしくは人工の繊維、及び/または本明細書に開示したコポリマーとして再製剤された半結晶p-GlcNAcを含む物質をさらに含んでいてもよい。再び、かかる製剤に含まれる半結晶p-GlcNAcの量及び/または濃度は、意図する用途に依存し、当業者には明らかでありかつin vitro及びin vivo試験を通して、特に当技術分野で周知の動物モデル系を用いて、容易に決定しうるであろう。
【0115】
半結晶p-GlcNAcの血管構造及び/または機能に対するモジュレーション効果は局所性でかつ一過性であるので、半結晶p-GlcNAcを含有する製剤の投与は、是正すべき症状が解消されるまで、間隔をおいて反復することができる。一般的に、かかる間隔は、約1時間であるが、治療する症状の性質及び適用する結晶p-GlcNAcの量に依存して、短いときも長いときもある。半結晶p-GlcNAc製剤を含む組成物を比較的接近しにくい表面に適用する事例においては、生物分解性組成物及び製剤が好ましい。
【0116】
5.6. 用途
p-GlcNAc出発物質は様々な用途を有し、例えば、エンドセリン-1放出の刺激、血管収縮、及び破裂した血管からの血液流出の減少を経由する血管構造及び/または機能のモジュレーション、ならびに出血の停止を助けるかまたは達成することが挙げられる。局所に適用された本発明のp-GlcNAcは、生体適合性、生分解性、無毒、及び非病原性である。本発明のp-GlcNAc物質はまた、免疫中性であるので、これらはヒトに免疫応答を誘発せず、従って、限定されるものでないが、フィルム、膜、ゲル、スポンジ、ミクロスフェア、ミクロビーズ、ミクロフィブリル、フォーム、及びスプレイを含む、本発明に開示したデバイスの製剤化に利用するのに特に有利である。また、該ポリマーが、以下の実施例6に記載のIR吸収分光計により分析したときに、ポリβ-1→4 N-アセチルグルコサミンポリマーが鋭く明確なピークにより実証されるその半結晶構造を保持する限りにおいて、天然のアルギネート及び場合によっては合成ポリマーのような特定のさらなる物質を、本明細書に記載のp-GlcNAcと組み合わせて、かかる物質及びデバイスの構築に利用することができる。一実施形態においては、p-GlcNAcは、本質的に完全にアセチル化されたβ-1→4-N-アセチルグルコサミンの半結晶ポリマーから成り、ここで前記ポリマーは、β-1→4コンフォメーションで共有結合した約50〜約150,000個のN-アセチルグルコサミン単糖を含んでなり、タンパク質を含まず、その他の有機不純物を実質的に含まず、無機不純物を実質的に含まずかつ約10,000ダルトン〜約30 x 106ダルトンの分子量を有する。他の実施形態においては、p-GlcNAcは、本質的に完全にアセチル化されたβ-1→4-N-アセチルグルコサミンの半結晶ポリマーからなり、ここで前記ポリマーは、β-1→4コンフォメーションで共有結合した約50〜約50,000個、約50〜約10,000個、約50〜約4,000個のN-アセチルグルコサミン単糖を含んでなり、タンパク質を含まず、その他の有機不純物を実質的に含まず、無機不純物を実質的に含まず、かつそれぞれ約10,000ダルトン〜約10 x 106ダルトン、約10,000ダルトン〜約2 x 106ダルトン、約10,000ダルトン〜約800,000ダルトンの分子量を有する。
【0117】
5.6.1. エンドセリン-1放出の刺激
本発明のp-GlcNAc物質は、以下の第16節に記載する実施例で成功裏に実証したように、例えばエンドセリン-1の放出を刺激するために利用される。エンドセリン-1放出の刺激は、とりわけ、子宮の子宮内膜組織によるエンドセリン-1の産生レベルの著しい低下に関連する月経過多の治療に利用される。
【0118】
エンドセリン-1放出の刺激は、ヒトまたは、限定されるものでないが、獣医学及び愛玩動物を含む非ヒト哺乳動物の標的組織へのp-GlcNAcを含む組成物及び物質の局所適用により達成される。かかる物質及び組成物は、本明細書に記載のp-GlcNAcと組み合わせて、天然のアルギネート及び、場合により、合成ポリマーなどのある特定のさらなる物質を含んでもよい。好ましい実施形態においては、かかる組成物及び物質のp-GlcNAcは、完全にアセチル化された、β-1→4-N-アセチルグルコサミンの半結晶ポリマーから本質的になり、タンパク質を含まず、その他の有機不純物を実質的に含まず、無機不純物を実質的に含まずかつ約30 x 106ダルトン以下の分子量を有する。
【0119】
p-GlcNAcを含む本発明の物質は、例えば、ゲル、フィルム、膜、及びスポンジとして製剤化しかつ適用する。かかる物質はまた、ミクロスフェア、ミクロビーズ、ミクロフィブリルの溶液もしくは懸濁液として、またはフォームもしくはスプレイとしても製剤化しかつ適用できる。従って、p-GlcNAcを含んでなる本発明の物質はバリヤー形成物質である必要はない。
【0120】
p-GlcNAcを含む本発明の組成物及び物質は、標的組織、すなわち、エンドセリン-1放出を刺激することを所望する組織に直接適用される。標的組織は、例えば月経過多を患う患者の子宮の子宮内膜組織でありうる。標的組織としては、一般的に、内皮組織、そして特に、動脈、静脈、または毛細血管であってもよい血管が挙げられる。半結晶p-GlcNAcを含む物質は、例えば、ゲル、フィルム、膜、スポンジ、スプレイもしくはフォーム、ならびにミクロスフェア、ミクロビーズもしくはミクロフィブリルの懸濁液、エマルジョンもしくは溶液として、局所に適用する。
【0121】
p-GlcNAcを含む本発明の組成物及び物質を局所適用すると、p-GlcNAc無処理の標的組織と比較して、標的組織において局所性かつ一過性で、投与p-GlcNAcの用量依存的に、エンドセリン-1の放出が刺激される。エンドセリン-1放出の刺激は局所性であり、p-GlcNAcを含む物質と直接接触する組織において最も顕著であり、さらに、隣接組織におけるエンドセリン-1放出の刺激の程度は、標的組織とp-GlcNAcを含む物質の接点からの距離が増加するとともに減少する(例えば、後掲の第16節に記載する実施例を参照)。
【0122】
エンドセリン-1放出の刺激は一過性であって、半結晶p-GlcNAcを含んでなる物質と接触する組織におけるエンドセリン-1のレベルはかかる物質の投与直後に最大となり、その後、刺激前に観察されたレベルへ減少する。すなわち、接触組織におけるエンドセリン-1濃度は一般的に半結晶p-GlcNAcの投与後15分までが最大であり、エンドセリン-1の濃度は半結晶p-GlcNAcの投与後約60分以内に接触直前に観察されたレベルに実質的に戻る(例えば、後掲の第16節に報じる実施例を参照)。従って、エンドセリン-1放出の長時間刺激を必要とする事例においては、半結晶p-GlcNAcを用いて製剤化した組成物及び/または物質のさらなるアリコートまたは用量を標的組織に逐次的に適用する。
【0123】
エンドセリン-1放出の刺激は用量に依存し、p-GlcNAcを含んでなる物質と接触した内皮組織により放出されるエンドセリン-1のレベルは、その物質中のp-GlcNAc量に実質的に比例する(かかる「実質的に比例する」効果の代表的実証については、例えば、後掲の第16節に記載する実施例を参照)。 従って、組成物及び物質は、必要なエンドセリン-1放出の刺激のレベルに要求されるp-GlcNAcのレベルを含有するように製剤化しかつ構築する。かかるレベルの決定は、日常のin vitro実験、及び動物モデル試験から容易に確認される。従って、より大きいエンドセリン-1放出の刺激を必要とする事例においては、組成物及び物質はp-GlcNAc濃度を増加して製剤化する。
【0124】
5.6.2. 血管収縮の誘導
本発明のp-GlcNAc物質は、例えば、後掲の第16及び17節に記載する実施例に成功裏に実証し、ならびに図22に示したように、血管収縮を誘導するために利用される。血管収縮は、半結晶p-GlcNAcを含んでなる組成物及び物質の、ヒトまたは、限定されるものでないが、獣医学及び愛玩動物を含む非ヒト哺乳動物の標的組織への局所適用により達成する。
【0125】
半結晶p-GlcNAcを含んでなる組成物の局所適用が有用である臨床応用としては、とりわけ、例えば、肝及び腎に生検傷を生じるかまたは血管に穿刺傷を生じる診断方法、例えば、心臓カテーテル法及びバルーン血管形成術における使用が挙げられる。それ故に、本発明の方法は、遺伝的欠陥からまたはクマヂンもしくはヘパリンなどの抗凝結薬の投与から生じうる凝血障害のいずれかの形態に悩む患者に特に有用である。特定の理論または機構に束縛されることを欲しないが、半結晶p-GlcNAcの局所適用により誘発される血管収縮は物理的に穿刺傷のサイズを縮小し、それにより、血液凝固に依存しない方法及び機構により出血の停止を容易にするかまたは達成すると考えられる。
【0126】
本発明に使用する物質及び組成物は、天然のアルギネート及び、いくつかの事例では、合成ポリマーなどのある特定の追加の物質を、本明細書に記載のp-GlcNAcと組み合わせて含んでもよい。好ましい実施形態においては、かかる組成物及び物質のp-GlcNAcは、本質的に完全にアセチル化されたβ-1→4-N-アセチルグルコサミンの半結晶ポリマーであって、タンパク質を含まず、その他の有機不純物を実質的に含まず、無機不純物を実質的に含まずかつ約30 x 106ダルトン以下の分子量を有する前記ポリマーから成る。
【0127】
p-GlcNAcを含んでなる本発明の物質は、例えば、ゲル、フィルム、膜、及びスポンジとして製剤化される。かかる物質はまた、ミクロスフェア、ミクロビーズ、ミクロフィブリルの溶液もしくは懸濁液またはスプレイもしくはフォームとして製剤化しかつ適用できる。従って、p-GlcNAcを含んでなる本発明の物質はバリヤー形成物質である必要はない。
【0128】
p-GlcNAcを含んでなる本発明の組成物及び物質は、標的組織、すなわち、血管収縮の誘導が所望される組織または血管に隣接したまたは近接した皮膚または他の組織に適用するか、あるいは標的組織に直接適用する。標的組織または血管としては、一般的に、動脈、静脈、または毛細血管が挙げられる。p-GlcNAcを含んでなる本発明の物質は、例えば、ゲル、フィルム、膜、またはスポンジ、スプレイもしくはフォームとして、またはミクロスフェア、ミクロビーズもしくはミクロフィブリルの懸濁液もしくは溶液として局所に適用する。
【0129】
p-GlcNAcを含んでなる本発明の組成物及び物質の局所適用は、局所性かつ一過性で、そして投与したp-GlcNAcの用量に依存して、血管収縮を刺激する。血管収縮の誘導は局所性であって、p-GlcNAcを含んでなる物質と直接接触する血管において最も顕著であり、さらに、血管収縮の刺激の程度は、p-GlcNAcを含んでなる物質と標的血管の接触点からの距離が増加するとともに減少する。
【0130】
血管収縮の刺激は一過性であって、血管における血管収縮の程度は、本発明のp-GlcNAc物質の投与直後に最大であり、その後、刺激前に観察されたレベルへ減少する。すなわち、血管収縮の程度は一般的にp-GlcNAcの投与後15分以内に最大となり、p-GlcNAcの投与後約60分以内に実質的に対照レベルに低下する。従って、長時間の血管収縮を必要とする事例においては、p-GlcNAcを用いて製剤化した組成物及び/または物質のさらなるアリコートまたは用量を標的組織に逐次的に適用する。
【0131】
血管収縮の誘導は用量に依存し、p-GlcNAcを含んでなる物質と接触した血管における血管収縮の程度は、その物質中のp-GlcNAc量に実質的に比例する。従って、組成物及び物質は、所望の血管収縮の程度に要求されるp-GlcNAcのレベルを含むように製剤化しかつ構築する。かかるp-GlcNAcのレベルの決定は、日常のin vitro実験、及び動物モデル試験から容易に確認される。従って、血管収縮のより大きい誘導を必要とする事例においては、組成物及び物質はp-GlcNAc濃度を増加して製剤化する。
【0132】
5.6.3. 破裂した血管からの血液流出の減少
p-GlcNAcを含む物質の局所投与を含んでなる本発明の方法はまた、例えば、標的組織の破裂した血管からの血液流出を減少するためにも利用する。破裂した血管からの血液流出の減少を達成するためのp-GlcNAcの局所適用のための臨床利用は、限定されるものでないが、腹部大動脈瘤の治療、腫瘍の塞栓形成治療、子宮類線維腫病変(uterine fibroid lesions)、及び脳性動脈瘤、例えば、脾臓、肝臓及び血管傷害を含む創傷、ならびに標準かつ最小限の侵入性外科的方法、例えば、子宮内膜症手術及び胆嚢の手術が挙げられる。これらの各例において、p-GlcNAcを含有する物質の局所適用の結果として、破裂した血管からの血液流出が減少し、手術中の血液損失が減少する。従って、本明細書に開示した血管収縮をもたらす組成物と方法の使用は、遺伝的欠陥からまたはクマジンもしくはヘパリンなどの抗凝血薬の投与から生じうる、いずれかの形の凝血障害に悩む患者のかかる症状を治療するのに特に有用でありうる。
【0133】
本発明の方法に用いる物質及び組成物は、天然のアルギネート及び、いくつかの事例では、合成ポリマーなどの特定の追加の物質を、本明細書に記載のp-GlcNAcと一緒に含んでもよい。好ましい実施形態においては、かかる組成物及び物質のp-GlcNAcは、完全にアセチル化されたβ-1→4-N-アセチルグルコサミンの半結晶ポリマーから本質的になり、ここで前記ポリマーは、タンパク質を含まず、その他の有機不純物を実質的に含まず、無機不純物を実質的に含まずかつ約30 x 106ダルトン以下の分子量を有する。
【0134】
p-GlcNAcを含んでなる本発明の物質は、例えば、ゲル、フィルム、膜、及びスポンジとして製剤化される。かかる物質はまた、ミクロスフェア、ミクロビーズ、もしくはミクロフィブリルの溶液または懸濁液として製剤化しかつ適用し、及び/またはフォームもしくはスプレイとして適用してもよい。従って、p-GlcNAcを含んでなる本発明の物質はバリヤー形成物質である必要はない。
【0135】
p-GlcNAcを含んでなる本発明の組成物及び物質は、標的組織(すなわち、破裂した血管からの血液流出を減少するのが所望される組織または血管)に隣接もしくは連続する皮膚もしくはその他の組織に適用されるか、または標的組織へ直接適用される。標的血管は動脈、静脈、または毛細血管でありうる。p-GlcNAcを含んでなる本発明の物質は、例えば、ゲル、フィルム、膜、スポンジ、スプレイもしくはフォームとして、またはミクロスフェア、ミクロビーズ、及び/もしくはミクロフィブリルの懸濁液もしくは溶液として局所に適用される。
【0136】
p-GlcNAcを含んでなる本発明の組成物及び物質の局所適用は、局所性かつ一過性で、投与したp-GlcNAcの用量に依存して、破裂した血管からの血液流出の減少を誘導する。破裂した血管からの血液流出の減少は局所性であって、p-GlcNAcを含んでなる物質と直接接触する血管において最も顕著であり、さらに、破裂した血管からの血液流出の減少の程度は、p-GlcNAcを含んでなる物質と標的血管の接触点からの距離が増加するとともに減少する。
【0137】
破裂した血管からの血液流出の減少は一過性であって、p-GlcNAcを含んでなる物質と接触したことによる血液流出の減少は、かかる物質の投与直後に最大であり、その後、破裂した血管からの血液流出は対照レベルへ戻る。すなわち、破裂した血管からの血液流出の減少の程度は一般的にp-GlcNAcの投与後15分以内に最大となり、その後、破裂した血管からの血液流出はp-GlcNAcの投与後約60分以内に対照レベルに戻る。従って、長時間の破裂した血管からの血液流出の減少を必要とする事例においては、p-GlcNAcを用いて製剤化した組成物及び/または物質のさらなるアリコートまたは用量を標的組織に逐次的に適用する。
【0138】
破裂した血管からの血液流出の減少は用量に依存し、p-GlcNAcを含む物質と接触した血管における破裂した血管からの血液流出の減少は、その物質中のp-GlcNAc量に実質的に比例する。従って、組成物及び物質は、所望の破裂した血管からの血液流出の減少に要求されるp-GlcNAcのレベルを含むように製剤化しかつ構築する。かかるp-GlcNAcのレベルの決定は、日常のin vitro実験、及び動物モデル試験から容易に確認される。従って、破裂した血管からの血液流出のより大きい程度の減少を必要とする事例においては、組成物及び物質はp-GlcNAc濃度を増加して製剤化する。
【0139】
5.6.4 開示した方法を利用する特定の適応症
エンドセリン-1放出の刺激、血管収縮、及び/または破裂した血管からの血液流出の減少、ならびに出血の停止が所望される特定の事例は、限定されるものでないが、例えば、肝及び腎に生検傷を生じる診断方法;限定されるものでないが、腹部大動脈瘤の血管内治療後の出血の防止、ならびに腫瘍、子宮類線維腫病変及び脳性動脈瘤の塞栓形成治療を含む塞栓形成方法;月経過多の治療に対する;例えば、脾臓、肝臓及び血管傷害を含む創傷;標準かつ最小限の侵入性外科的方法、例えば、子宮内膜症手術及び胆嚢の手術;軟及び硬組織創傷修復、例えば皮膚創傷及び火傷治癒;特に脾臓創傷に対する、外科的方法;ならびにカテーテル処置及びバルーン血管形成方法などの血管穿刺診断及び治療方法に対する利用が挙げられる。
【0140】
p-GlcNAcに基づく出発物質は、固体物質として、またはp-GlcNAcミクロビーズ、ミクロスフェア、またはミクロフィブリルを含んでなるゲル、フォーム、スプレイ、エマルジョン、懸濁液、または溶液として製剤化することができ、標準の外科方法を用いて適用することができ、かつ標準及び最小限侵入性外科的介入の両方によって利用することができる。本発明のゲルは、例えば、シリンジ形式のデバイスから押出しによりまたは膜もしくはフィルムと組み合わせて送達することができる。膜またはフィルムは、完全にアセチル化されたp-GlcNAcに基づく物質またはその他の天然もしくは合成の物質から製造することができる。
【0141】
上記の血管穿刺方法との関係では、エンドセリン-1分泌の刺激、血管収縮及び破裂した血管からの血液流出の減少のために利用される本発明の組成物及び物質は、カテーテル外筒を血管から除去した時点で、p-GlcNAcに基づく物質を人手で圧縮して皮膚に直接適用するか、またはカテーテル進路跡に導入することにより適用する。あるいは、血管からカテーテル外筒の除去を検出するデバイスを、血管内側と外側の組織の間の化学的、物理的もしくは他の相違をモニターする電子もしくは機械システムを用いて開発してもよい。例えば、プローブを血管から除去したときの、流体力学もしくは熱放散の変化を検出することができ;その時点でシグナルが送られ、p-GlcNAcを含んでなる組成物もしくは物質の適用が開始され、エンドセリン-1の放出を刺激し、血管収縮を誘導し、及び/または破裂した血管からの血液流出を減少しうる。
【0142】
p-GlcNAc、好ましくは完全にアセチル化されて、高度に規則正しいp-GlcNAcの半結晶ポリマーの局所投与を含んでなる、エンドセリン-1放出、血管収縮、及び破裂した血管からの血液流出の減少を誘導する本発明の方法は、止血を達成するために有用な方法及び組成物と一緒に使用してもよい。かかる他の方法及び組成物は、限定されるものでないが、(1)赤血球及び血小板を透過せずかつ血液凝固カスケードに必要な循環因子を濃縮しうるマトリックスを提供するバリヤー形成物質の適用、及び(2)例えば、トロンビン、フィブリノーゲン、及び第XIII因子を含む血液凝固カスケードの成分を含んでなる物質の創傷への適用が挙げられる。
【0143】
本発明の方法はまた、その必要が予想される場合に止血を達成する方法及び組成物の必要を最小限にするか、または効率を増加するために、予防的にも利用しうる。かかる必要のある例は、限定されるものでないが、胃腸病学的処置におけるポリープの除去、腫瘍組織の切除、及び抜歯である。かかる例においては、本発明の方法を利用し、標的組織に隣接または近接した組織及び血管において、一過性で局所性のエンドセリン-1放出、血管収縮、及び破裂した血管からの血液流出の減少を誘導し、それにより患者に実施した方法から生じるその後の出血を最小化する。
【実施例】
【0144】
6. 実施例:純粋なp-GlcNAc調製物の物理的特性の決定
本実施例に記載するのは、p-GlcNAc及び脱アセチル化p-GlcNAc膜の円二色性(CD)及び赤外分光(IR)分析である。
【0145】
6.1. 物質及び方法
p-GlcNAc及び市販「キチン(chitin)」調製物:
CD研究に用いたp-GlcNAcは、上の第5.3.1節に記載の機械力精製法を利用して調製した。
【0146】
市販「キチン」は、NovaChem, Ltd., PO Box 1030 Armdale, Halifax, Nova Scotia, Canada, B3L 4K9から購入した。
【0147】
IR研究に用いたp-GlcNAc膜は、上の第5.3.1節に記載の機械力精製法によるか、または上の第5.3.2節に記載の化学的/生物学的調製法により調製した。
【0148】
市販「p-GlcNAc」調製物を5%塩化リチウムを含有するジメチルアセトアミド溶液中に溶解し、脱イオン蒸留水上に重ねて膜を沈降させることにより、膜に成型した。
【0149】
p-GlcNAc誘導体及び処理:CD及びIR研究の両方に使用する脱アセチル化p-GlcNAcは、p-GlcNAcを50% NaOHを用いて60℃にて2時間処理することにより調製した。IR研究に使用した熱変性p-GlcNAc膜は0.2mM EDTA中で3分間煮沸して改変した。p-GlcNAcを30分間122℃にてオートクレーブ処理した。
【0150】
CD技術:固体CD技術は、本質的にDomard(Domard, A., 1986, Int. J. Macromol. 8:243-246)に従って実施した。
【0151】
6.2. 結果
6.2.1. CD分析
無処理p-GlcNAcから得たCDスペクトル(図3A)においては、p-GlcNAcのアセチル部分のカルボニル基の存在によって、予想したn-π*及びπ-π*光学活性電子遷移(220-185nM)が観察された。図3Bに示すように、かかるピークは、脱アセチル化p-GlcNAc生成物から得たCDスペクトルには全く存在しない。
【0152】
6.2.2. IR分光分析
本研究で得たIRスペクトルはp-GlcNAcの化学構造と一致する。さらに、各IRピークの鋭い鮮明度は、p-GlcNAc繊維における秩序正しくかつ規則的な(すなわち、半結晶)構造の存在を示す。機械力精製方法を経由して精製したp-GlcNAcのIRスペクトルである図4A、及び化学的/生物学的方法を経由して精製したp-GlcNAcのIRスペクトルである図4Dを参照すること。比較のため、市販「キチン」調製物のIRスペクトルを示す図4Bを参照すること。
【0153】
オートクレーブ処理したp-GlcNAc物質(図4E)から得たIRスペクトルは、図4Aで観察したIRスペクトルと可視的な相違はない。このデータは、p-GlcNAc物質はオートクレーブ処理によりポリマー構造の消失なしに無菌化しうることを示す。
【0154】
7. 実施例:機械力精製法を用いるp-GlcNAcの精製
本節においては、p-GlcNAcを、第5.3.1節に記載の機械力技術を用いて精製した。
【0155】
7.1. 物質及び方法/結果
珪藻培養条件:珪藻種タラシオシラ・フルビアチリス(Thalassiosira fluviatilis)を、以上の第5.1節及び第5.2節に記載の方法に従って、培養液中で増殖させた。
【0156】
SEM工程:本実施例で使用したSEM技術を、以下の第12.1節に記載する。
【0157】
p-GlcNAc精製手法:p-GlcNAcは、珪藻培養物から以上の第5.3.1節に記載の機械力技術を利用して精製した。特に、p-GlcNAc繊維は、培養内容物をウエアリング(Waring)ブレンダー中で、トップスピード混合運動の短いバースト3回で処理することにより珪藻細胞体から分離した。3回のバーストの全時間は約1秒であった。得られる懸濁液をソルヴォール(Sorvall)GS-4固定角ローター中で3500rpmで20分間、約10℃で遠心分離した。上清をデカントし、再び、今度は、ソルヴォールGS-4固定角ローター中で4000rpmで20分間、約10℃で遠心分離した。さらに再び、上清をデカントして4000rpmで、約10℃で遠心分離した。第3回目の遠心分離後の最終上清は透明であって、液中に浮遊する目視しうるフロックは、あっても僅かでしかなかった。透明な上清を、0.8μm孔サイズをもつSupor-800ポリエーテルスルホン・フィルター膜(Gelman, Inc.)を備えたブーフナー(Buchner)濾過ユニット中にデカントし、次いで吸引し、繊維懸濁液から液を濾過して膜上に繊維を回収した。回収した繊維を1リットルの脱イオン蒸留水を用いて70℃にて洗浄した。ほぼ全ての水が排水されたら、1リットルの1N HClを用いて70℃にて吸引して繊維を洗浄した。ほぼ全ての酸性溶液が排水されたら、1リットルの脱イオン蒸留水を用いて70℃にて吸引して繊維を洗浄した。ほぼ全ての洗浄水が排水されたら、1リットルの95%エタノールを用いて室温にて繊維を洗浄し、そして減圧した。次いで、白い繊維膜をその上に回収したフィルター膜を、濾過ユニットから除去し、膜とその膜支持体とを乾燥オーブン中で58℃にて20分間乾燥し、その後、膜とその支持体をデシケーター中に16時間置いた。
【0158】
この精製過程の後に、1000ml培養からのp-GlcNAcの収量は珪藻培養物1リットル当たり6.85ミリグラムであった。この技術で回収したp-GlcNAc繊維により形成される膜のSEM写真を図6に示す。
【0159】
8. 実施例:生物学的/化学的精製方法を利用するp-GlcNAcの精製
本節においては、上の第5.3.2節に記載の化学的/生物学的技術の2方法を利用して、p-GlcNAcを精製した。簡単に説明すると、第1の事例ではp-GlcNAcをHF処理を経由して、かつ第2の事例では酸処理/中和を経由して精製した。
【0160】
8.1. 物質及び方法/結果
珪藻培養条件:珪藻種タラシオシラ・フルビアチリス(Thalassiosira fluviatilis)を、上の第5.1節及び第5.2節に記載の方法に従って、培養液中で増殖させた。
【0161】
SEM工程:本実施例で使用した技術は、以下の第12.1節に記載した。
【0162】
精製方法:第1の事例においては、p-GlcNAcをHF処理により精製したが、その結果を図7に示す。具体的には、換気フードの下で、珪藻培養内容物に、元の細胞培養物の容積のそれぞれ1000mlに対して49%(29N)HF溶液2.42mlを室温で加え、0.07M HF溶液を得た。次いで混合物を約30秒間激しく振とうして、液を覆う持続性の泡を作った。容器を5〜6時間静置して重い微粒子を沈降させた。この時点の終わりには、泡の層が形成する一方、液自身は2層に分割された:第1層は容器の底に横たわる少量の非常に暗い緑色層であり、その上の第2層ははるかに明るい灰緑色の濁った相であって恐らく液の全容積の85〜90%を占めた。泡層を、毛細ガラス管及び真空吸引を用いて注意深くサイフォンで吸引除去した。次いで、灰色の曇った上清を、沈降した細胞体から主になる暗色の底層を乱さないように注意を払いながら、サイフォンで吸引除去し、別のプラスチック容器に移した。灰色の曇った上清を、さらに16時間静置した。液は最初、ほとんど無色、明灰色であるが、透明でなかった。16時間静置後、少量の泡が液本体の頂部に残りかつ少量の緑色物質が容器の底に沈降した。液は色がより明るくなったが、なお透明でなかった。液頂部の泡を上記のようにサイフォンで吸引除去した。次いで、液本体を注意深くサイフォンで吸引除去し、少量の沈降した緑色物質は容器の底に放置した。こうして単離した液は、p-GlcNAc繊維の大部分と若干の不純物を含有した。
【0163】
以上の工程中に珪藻から遊離されたタンパク質及び他の望ましくない物質を、繊維を含有する液から除去するために、繊維及び細胞膜残遺物の懸濁液をドデシル硫酸ナトリウム(SDS)を用いて洗浄した。具体的には、所要の容積の20%SDS溶液を加えて、最終濃度が容積基準で0.5%SDSの液を調製した。液を入れた容器をシールし、振とう機械上に水平位置にセットし、24時間、毎分100回で振とうして攪拌した。振とうを始めると直ぐに、大きな白色のp-GlcNAc繊維のフロックが懸濁液中に現れ、そしてかなりの量の泡が容器の上部空間に蓄積した。SDS洗浄が終わると、容器の内容物を、0.8μm孔サイズをもつSupor-800ポリエーテルスルホン・フィルター膜(Gelman, Inc.)を備えたブーフナー濾過設備へ移した。液を吸引濾過し、液中のp-GlcNAc繊維を濾過膜上に回収した。
【0164】
次いで、フィルター膜上に回収したp-GlcNAc繊維をさらに洗浄した。最初に、熱脱イオン蒸留水(70℃)を、元の懸濁液の容積の3倍用いて、繊維を洗浄した。脱イオン蒸留水を用いる水ジェットにより、ブーフナーフィルターのフィルター膜上に回収した白色繊維凝集塊をウエアリング(Waring)ブレンダーに移し、繊維凝集塊を約10回の短い混合バーストにより崩壊させた。崩壊した繊維の懸濁液を、上記のように、ポリエーテルスルホン膜を備えたブーフナー濾過漏斗へ移し、液を吸引下で除去した。回収した繊維を熱(70℃)1N HCl溶液1000mlを用いて洗浄し、続いてさらに熱(70℃)脱イオン蒸留水1000mlを用いて洗浄した。最後に、繊維を95%エタノール1000mlを用いて室温にて洗浄し、濾過して乾燥した。次いで、繊維膜及び繊維膜を支持するフィルター膜を、乾燥オーブンにて58℃で20分間乾燥した。次いで、膜と膜支持体をデシケーター内に16時間置いた。膜を注意してフィルター膜から剥がした。
【0165】
第2の事例においては、p-GlcNAcを以上の第5.3.2節で記載した酸処理/中和法を利用して精製した。具体的には、p-GlcNAcをSDS洗浄工程の前まで本節の始めに記載したように処理し、その時点で該溶液に2.9M Tris溶液を加えてほぼ7.0のpHに中和した。この特定の精製方法からのp-GlcNAc収率は珪藻培養物1リットル当たり20.20ミリグラムであったが、平均では、珪藻培養物1リットル当たりほぼ60ミリグラムが得られる。精製過程中に形成される膜のSEM顕微鏡写真を図8A〜B及び9A〜9Eに示す。
【0166】
9.実施例:p-GlcNAc脱アセチル化
p-GlcNAc膜を50%NaOH水溶液中に懸濁した。懸濁液を80℃にて2時間加熱した。得られる脱アセチル化膜を乾燥して走査電子顕微鏡により研究し、図11A-Bに示した。
【0167】
10.実施例:p-GlcNAc生体適合性
本実施例において、p-GlcNAc出発物質は、溶出試験、ウサギへの筋肉内移植、ウサギへの皮下注射、及びマウスへの全身注射によりアッセイしたところ検出可能な生物学的活性を表さないことが示された。
【0168】
10.1. 物質及び方法
10.1.1. 溶出試験
溶出試験の条件は、米国薬局方XXII、1990、1415-1497頁、及び米国薬局方XXII、追補5、1991、2702-2703頁に記載の規格に一致させた。
【0169】
細胞培養:マウス線維芽細胞L929細胞系統(American Type Culture Collection Rockville, Md.;ATCC No. CCL1;NCTCクローン929)を利用した。L929細胞の24時間コンフルエント単層を完全最小必須培地(MEM)において増殖させた。
【0170】
p-GlcNAc:以上の第5.3.1に記載した機械力精製法に従って調製しておいたp-GlcNAcの固体膜を、米国薬局方XXII(1990)要件に従って20ml血清補充MEM中で抽出した。
【0171】
対照:天然ゴムを陽性対照として使用し、かつシリコーンを陰性対照として使用した。対照を試験材、p-GlcNAcと同じ方法で試験した。
【0172】
抽出物:抽出物は、37℃にて、5%二酸化炭素を含有する加湿大気中、24時間で調製した。抽出物のpHの変化を測定し、pHを元の培地の±0.2のpH単位内に収めるように調節した。調節は、抽出pHの低下はHClにより、また抽出pHの上昇はNaHCO3により実施した。抽出物は、細胞単層に適用する前に、0.22ミクロンフィルターを通過させて滅菌濾過した。
【0173】
投薬:p-GlcNAcまたは対照抽出物の3mlを細胞培養の維持培地と置き換えた。全ての抽出物を繰り返して試験した。
【0174】
評価判定基準:細胞単層の応答を、目視または顕微鏡下のいずれかで評価した。生物学的反応、すなわち、細胞変性及び/または形成異常を以下に示したように0〜4の点数で評価した。試験システムは、もし陰性対照材に対して細胞反応性の徴候が認められず(評点0)かつ陽性対照材が中度(評点2)より大きい細胞反応性を示せば、適当である。試験材(すなわち、p-GlcNAc)は、もし試験材を用いて処理した培養であって、軽度より大きい反応性を示すものがなければ、生体適合性試験に合格する。
【表2】
10.1.2. 筋肉内移植
動物:健康な、ニュージーランド白ウサギ(New Zealand White Rabbit)、雄性及び雌性、(Eastern Rabbit Breeding Laboratory, Taunton, Mass.)を使用した。ウサギを個別に、懸架式ステンレス鋼製籠を用いて飼育した。動物を入手すると、8日間、実試験と同じ条件下で隔離した。ハードウッド(Hardwood)チップ(Sani-chips(TM), J. P. Murphy Forest Products, Montvale, N.J.)を籠下の無接触ろ床として使用した。動物施設は、華氏68±3度の温度、30-70%の相対湿度、1時間当たり少なくとも10-13回の完全空気交換、及びフルスペクトル蛍光灯を用いる12時間の明/暗サイクルに維持した。動物に、市販餌(Agway ProLab, Waverly, N.Y.)を制御した条件下で与え、かつ都市水道水を任意にとれるように補給した。餌、ろ床、または水の中に、試験結果を妨害すると思われる既知不純物は存在しなかった。研究用に選択した動物は、動物のより大きいプールから選んだ。ウサギは10g精度で体重測定し、耳の入れ墨により個々に同定した。
【0175】
p-GlcNAc:使用したp-GlcNAcは、上の第10.1.1節に記載したとおりである。
【0176】
移植試験:それぞれの移植試験に2匹のウサギを用いた。試験の日に、脊柱の両方の側の動物皮膚の毛を刈り込んだ。それぞれの動物を麻酔して筋肉運動を阻止した。無菌皮下針とスタイレットを用いて、試験p-GlcNAcの4つの細片(1mm x 1mm x 10mm)を、2匹のウサギのそれぞれに対して、脊椎の片側の脊椎傍筋中に移植した(正中線から2.5〜5cm、脊柱と平行に、そしてお互いから約2.5cmに)。同様な方式で、USP陰性対照プラスチックRS(1mm x 1mm x 10mm)の2つの細片をそれぞれの動物に対して、反対の筋肉中に移植した。動物を7日間維持した。観察期間の終わりに、動物の体重を測定し、注射用バルビツール酸塩、Euthanasia-5(Veterinary Laboratories, Inc., Lenexa, Kans.)により安楽死させた。組織が出血なしで切断されるために十分な時間が経過するのを待った。それぞれの移植片の中心部を囲む組織の領域を拡大レンズを用いて巨視的に試験した。止血、壊死、変色、及び感染を、次のスケールを用いてスコアを付けた:0=正常、1=軽度、2=中度、及び3=重度。もし嚢が存在すれば、最初に嚢の幅(すなわち、移植片の周縁から嚢の周縁までの距離)を0.1mmまで丸めて測定してスコアを付けた。嚢は次のとおりスコアを付けた:
【表3】
p-GlcNAcと陽性対照材の平均スコアの間の差を計算した。試験は、4つのp-GlcNAc細片の多くとも1つについて、もしそれぞれのウサギにおいて、p-GlcNAcに対する生物学的反応のそれぞれのカテゴリーに対する平均スコアと陽性対照プラスチック移植片部位に対するそれとの間の差が1.0を超えないか;または、それぞれのp-GlcNAc材に対する生物学的反応の全てのカテゴリーに対する平均スコアと全ての陽性対照プラスチック移植片部位に対する全てのカテゴリーに対する平均スコアとの間の差が、1.0を超えなければ、陰性と考えた。
【0177】
10.1.3. 皮内注射
動物:ニュージーランド白ウサギを使用し、以上の第10.1.2節に記載のように維持した。
【0178】
p-GlcNAc:以上の第5.3.1節に記載した精製の機械力方法に従って調製しておいたp-GlcNAcの固体膜を、抽出フラスコ内に置いて、これに適当な培地20mlを加えた。抽出は、70℃に24時間加熱して実施した。この過程の後に、抽出物を室温に冷却した。それぞれの抽出ボトルを激しく振とうした後に投与した。
【0179】
皮内試験:試験日に、動物の背中の毛を刈り込んだ。それぞれのp-GlcNAc抽出物の0.2ml容積を2匹のウサギのそれぞれの片側に5つの部位で皮内注射した。1匹のウサギ当たり1以上のp-GlcNAc抽出物を使用した。それぞれのウサギの他の側の5つの部位に、対応する対照0.2mlを注射した。注射部位を、紅斑、浮腫、及び壊死の徴候について、注射後24、48、及び72時間に観察した。観察結果を、皮膚反応スコアリング用ドレーズ(Draize)スケール(米国薬局方XXII, 1990, 1497-1500;米国薬局方XXII, 追補5, 1991, 2703-2705)に従ってスコアを付け、以下の表IIに示した:
【表4】
全ての紅斑及び浮腫のスコアを、24、48、及び72時間に個々に合計して12(すなわち、2動物 x 3スコアリング期間 x 2スコアリングカテゴリー)で除し、p-GlcNAcの全平均スコアを対応する対照に対して測定した。動物は、観察期間が終わると体重を測定し、バルビツール酸塩、Euthanasia-5(Veterinary Laboratories, Inc., Lenexa, Kans.)を注射して安楽死させた。試験結果は、もしp-GlcNAcと対照の平均反応スコア (紅斑/浮腫)の間の差が1.0以下であれば、合格とする。
【0180】
10.1.4. 全身注射
動物:アルビノ・スイス・マウス(Albino Swiss mice)(Mus musculus)、雌性、(Charles River Breeding Laboratories, Wilmington, Mass.)を使用した。5匹のマウスのグループをステンレス鋼蓋を取付けたポリプロピレン籠に入れて飼育した。ハードウッドチップ(SanichipsTM, J. P. Murphy Forest Products, Montvale, N.J.)を籠内の接触ろ床に使用した。動物施設は、限られたアクセス域として維持し、動物室は華氏68±3度の温度、相対湿度30-70%、1時間当たり少なくとも10-13回の完全空気交換、及び全スペクトル蛍光灯を用いる12時間の明/暗サイクルを保った。マウスに市販の餌及び都市水道水を任意でとれるようにで補給した。餌、ろ床、または水の中には、試験結果を妨害すると思われる既知不純物は存在しなかった。研究用に選択した動物は、より大きな動物のプールから選んだ。マウスは0.1g精度で体重測定し、個々に耳パンチにより同定した。
【0181】
p-GlcNAc:使用したサンプルは、以上の第10.1.1節に記載したとおりであった。抽出物は以上の第10.1.3節に記載の方法に従って調製した。
【0182】
全身注射試験:5匹のマウスのグループにp-GlcNAc抽出物または対応する対照材を、以下に掲げたのと同じ量をそして同じ経路により注射した。
【表5】
PEG400を用いて調製したp-GlcNAcの抽出物、及び対応する対照、を0.9%NaClにより希釈して1mlあたりPEG400 200mgとした。皮内試験用には、PEG400を0.9% NaClにより希釈して1mlあたりPEG400 120mgとした。
【0183】
動物を注射直後、注射後24、48、及び72時間に観察した。動物は、観察期間が終わると体重を測定し、二酸化炭素に暴露して安楽死させた。もしp-GlcNAcで処理した動物が対照材を用いて処理した動物より有意に大きい生物学的反応性を示さなければ、試験要件は合格とする。
【0184】
10.2. 結果
10.2.1. 溶出試験
p-GlcNAc試験材に対する細胞単層の応答は、目視及び顕微鏡下で評価した。評価に細胞化学染色は使わなかった。細胞の生物学的反応性の徴候なし(0級)は、陰性対照材もしくはp-GlcNAcに対する暴露後48時間に観察した。重度の反応性(4級)は以下の表IIIに示した通り、陽性対照材に対して認められた。
【表6】
従って、p-GlcNAc出発物質は、生体適合性に対する溶出試験の要件に合格し、かくして、非細胞傷害性である。
【0185】
10.2.2. 筋肉内移植
試験した両方のウサギ(A及びB)は、体重が増加し、毒性の徴候を示さなかった。データは表IVを参照。さらに、いずれの動物にも毒性の明確な徴候はなかった。試験及び対照材移植部位の目視評価では、炎症、嚢、出血、壊死または変色は認められなかった。結果は表IVを参照。従って、試験は、アッセイしたp-GlcNAcが生物学的反応性を示さず、それぞれのウサギにおいて、全てのp-GlcNAc移植部位に対する生物学的反応の全てのカテゴリーに対する平均スコアと、全ての対照移植部位に対する全てのカテゴリーに対する平均スコアとの差が1.0を超えないことを実証した。
【表7】
10.2.3 皮内試験
全動物の体重が増加した。データは表Vを参照。p-GlcNAcまたは対象材部位のいずれにおいても紅斑または浮腫の徴候は観察されなかった。毒性の明確な徴候はいずれの動物にも観察されなかった。p-GlcNAcと対照材平均反応スコア(紅斑/浮腫)との間の差はが1.0未満であったので、p-GlcNAcは皮下試験の要件に合格する。結果は表VIを参照。従って、この試験でアッセイされるとおり、p-GlcNAcは生物学的反応性を有しない。
【表8】
【表9】
10.2.4. 全身試験
p-GlcNAc抽出物または対照材を用いて処理した全てのマウスは体重が増加した。データは表VIIを参照。さらに、毒性の明確な徴候はいずれのp-GlcNAcまたは対照動物にも観察されなかった。結果は表VIを参照。従って、p-GlcNAc試験動物は対照材を用いて処理した動物と比較して有意に大きな生物学的反応性を示さなかったと結論される。
【表10】
11. 実施例:p-GlcNAc再製剤
本節に記載する実施例においては、p-GlcNAc膜(16.2 mg)を、5% LiClを含有するジメチルアセトアミド溶液1ml中に溶解した。p-GlcNAcを含有する溶液をシリンジ中に入れて純粋な水50ml中に押出し、繊維を沈降させた。得られた繊維を、図10A-Bに示す走査電子顕微鏡を用いて研究した。
【0186】
12. 実施例:p-GlcNAc/コラーゲン・ハイブリッド
本実施例で記載するのは、p-GlcNAc/コラーゲン・ハイブリッド物質の生成及び特性決定である。
【0187】
12.1. 物質及び方法
材料:ウシI型コラーゲンを、本研究に記載のハイブリッド調製に使用した。p-GlcNAcは以上の第5.3.2節に記載した機械力方法に従って調製した。
【0188】
ハイブリッド調製:コラーゲン(10ミリグラム/ml)とp-GlcNAc(0.25ミリグラム/ml)の水懸濁液を、様々な比で混合し、液体N2(-80℃)中で凍結し、-9℃にて4時間保って凍結乾燥した。材料を真空下(ほぼ0.030 Torr)で60℃にて3日間、脱水加熱して架橋した。
【0189】
細胞培養:マウス3T3線維芽細胞を、調製したコラーゲン/p-GlcNAc・ハイブリッド上で増殖した。標準培養方法に従い、培養の8日後にSEM顕微鏡写真を撮影した。
【0190】
12.2. 結果
コラーゲンとp-GlcNAc水懸濁液を、様々な比(すなわち、コラーゲン:p-GlcNAc懸濁液比=3:1、1:1、2:2、及びl:3)で混合し、凍結し、凍結乾燥し、そして架橋した。かかる方法によりコラーゲン/p-GlcNAcスラブを得た。得られた物質のSEM顕微鏡写真は、ハイブリッド物質の多孔質構造を示し、細胞の付着と増殖にとって効率的な三次元構造を提供した。
【0191】
13. 実施例:純粋なp-GlcNAc調製物のNMR特性決定
本実施例で記載するのは、純粋なp-GlcNAc調製物のNMR(核磁気共鳴)分析である。
【0192】
13.1. 物質及び方法
p-GlcNAc調製物:本節に記載のNMR研究に使用したp-GlcNAcは、以上の第5.3.2節に記載した化学精製法を利用し、フッ化水素酸を化学試薬に用いて調製した。
【0193】
NMR技術:固体NMRデータはブルーカー(Bruker)500MHz NMR分光計を用いて得た。コンピューター画像解析を利用して生のNMRスペクトルデータを変換し、バックグラウンドを排除しかつベースラインを正規化した。かかる変換データの一例を図14に示す。図14のような変換NMR曲線を利用して全ての炭素原子種に対する面積を求め、次いでCH3(面積)対C-原子(面積)の比を計算した。記載のようにして得たかかる値を図16に示す。
【0194】
13.2. 結果
固体NMRデータを、p-GlcNAcサンプル500mgの13C-NMRスペクトルを測定することにより取得した。典型的なNMRスペクトルを図15に示す。個々のピークは分子中のそれぞれのユニークな炭素原子のスペクトルへの寄与を表す。分子中のそれぞれの炭素原子種の相対的パーセントは、その炭素種が作ったピーク面積を、スペクトル中に観察される全てのNMRピーク下の面積の合計和により除して決定した。このようにして、基準原子により測定した分子におけるそれぞれの原子の比を計算することが可能であった。全てのp-GlcNAc分子は、定義により、C1、C2、C3、C4、C5及びC6原子を有するN-アセチルグルコサミン残基からなる。従って、もしポリマーの全てのグルコサミン残基がN-アセチル化されていれば、以上のいずれのグルコサミン残基炭素原子のピーク面積に対するN-アセチルCH3炭素原子のピーク面積の比も1.0となる。図14のようなデータを利用してCH3(面積)比の値を得た。
【0195】
図16で計算した比は、多くの場合、実験誤差内でほぼ1.0に等しく、例えば、CH3/C2=1.097、CH3/C6=0.984、CH3/C5=1.007、CH3/C1=0.886であった。これらの結果は、p-GlcNAc出発物質が不純物を含まず、完全にアセチル化されている(すなわち、グルコサミン残基の本質的に100%がN-アセチル化されている)という結論と一致する。
【0196】
14. 実施例:制御された孔サイズ三次元p-GlcNAcマトリックスの合成と生物学的特性の決定
以下に記載するのは、制御された平均孔サイズを有する多孔質マトリックスに基づく三次元p-GlcNAcを生産する方法である。かかるマトリックスは様々な重要な用途を有し、例えば、細胞の封入(encapsulation)が挙げられる。かかる細胞封入組成物は、移植しうる細胞に基づく治療薬として、及び軟骨再生などの他の細胞及び組織エンジニアリング用途に有用である。本明細書に実証したp-GlcNAc物質の形態と寸法を操作する能力は、p-GlcNAcポリマーを様々な形状に再製剤化する強力なツールを提供し、前記形状としては、限定されるものでないが、製薬上許容される担体、ビヒクル及び/または溶媒中のエマルジョン、懸濁液及び/または溶液として製剤化することができるミクロビーズ及びミクロスフェアが挙げられる。
【0197】
14.1. 物質及び方法
p-GlcNAc出発物質:p-GlcNAcを、以上の第5.3.2節に記載した化学的精製方法を利用し、化学試薬としてフッ化水素酸を用いて調製した。マトリックス製剤:凍結乾燥に先立って、p-GlcNAcサンプル20mgを含有する懸濁液(5ml)を以下の第14.2節に掲げた溶媒中で作った。次いでサンプルを組織培養ディッシュのウエルに注ぎ入れて-20℃にて凍結した。凍結したサンプルを次いで凍結乾燥し、得られた三次元マトリックスを除去した。
【0198】
操作電子顕微鏡技術:本明細書で利用した方法は、以上の第12.1節に記載したように実施した。図17A-Gに示す画像はマトリックス物質の200 x 拡大であり、これらの図のそれぞれに200ミクロンのスケールマーキングを示す。
【0199】
14.2. 結果
以上の第14.1節に記載したp-GlcNAc懸濁液は、次の各溶液を用いて得た:
A. 蒸留水
B. 10%メタノールを含む蒸留水
C. 25%メタノールを含む蒸留水
D. 蒸留水単独
E. 10%エタノールを含む蒸留水
F. 25%エタノールを含む蒸留水
G. 40%エタノールを含む蒸留水
各溶媒を用いて作ったマトリックスのサンプルを走査電子顕微鏡(SEM)分析にかけた。得られた結果を図17A-Gに示す。これらの図は、それぞれの懸濁液中のメタノールまたはエタノールのパーセントが増加すると、平均マトリックス孔サイズが減少することを示す。
【0200】
具体的には、2つの水懸濁液の孔サイズ(図17A及び17D)は平均して200ミクロンに近い。図17C及び17F(それぞれ25%メタノール及びエタノール)に示したサンプルの孔サイズは平均して30と50ミクロンの間にある。
【0201】
ここに示した結果は、エタノールとメタノールは両方ともp-GlcNAc孔サイズを制御するのに成功し、エタノールがメタノールより効率的でありうることを示唆する。
【0202】
15. 実施例:p-GlcNAc物質の生分解性
本節に記載する実施例は、制御可能なin vitro及びin vivo生物分解性及び再吸収速度を示すp-GlcNAc出発物質を調製しうることを実証する。
【0203】
15.1. 物質及び方法
p-GlcNAc物質:プロトタイプIを以上の第5.3.2節に記載した方法により、化学的方法を経由し、フッ化水素酸を化学試薬として用いて作った。プロトタイプIは100%アセチル化p-GlcNAcであることが示された。
【0204】
プロトタイプ3Aのp-GlcNAc出発物質を、以上の第5.3.2節に記載した方法により、化学的方法を経由し、フッ化水素酸を化学試薬として使うことによって作った。次いで、p-GlcNAc物質を、以上の第5.4節に記載した方法により脱アセチル化した。具体的には、p-GlcNAc物質を40% NaOH溶液を用いて60℃にて30分間処理した。得られるプロトタイプ3Aは、30%脱アセチル化されていることを確認した。
【0205】
プロトタイプ4のp-GlcNAc出発物質を、以上の第5.3.2節に記載した方法により、化学的方法を経由し、フッ化水素酸を化学試薬として使うことによって作った。次いでp-GlcNAc物質を40% NaOH溶液を用いて60℃にて、30分間処理することにより脱アセチル化した。次にその繊維を蒸留水中に懸濁し、-20℃にて凍結して凍結乾燥した。プロトタイプ4も、30%脱アセチル化されていることを確認した。
【0206】
腹部移植モデル:スプラグ・ドーリー・アルビノ(Sprague Dawley albino)ラットを使用して腹部移植モデル研究を行った。動物を麻酔して外科手術に備え、そして皮膚及び腹部筋肉を切開した。盲腸の位置を定めて外へ持ち上げた。1cm x 1cmのp-GlcNAc物質の膜を盲腸の上に置き、切開部をナイロンを用いて閉じた。対照動物は盲腸上に何も置かなかった。
【0207】
動物を、移植後14及び21日に開腹した。移植及び外植過程中に写真を撮影した(図23A-E)。外植過程後、組織病理学用の盲腸サンプルを調製した。
【0208】
p-GlcNAcのin vitro分解リゾチーム-キチナーゼアッセイ:本アッセイは、N-アセチルグルコサミンに対する比色アッセイであり、次のとおり実施した:反応サンプル150μlを13 x 100mmのガラス製使い捨て試験管中にピペットにより重複して採取し、0.25Mリン酸カリウムバッファー(pH 7.1)25μlをそれぞれの試験管に加え、次いで0.8Mホウ酸カリウム溶液(pH 9.8)35μlを加えた。試験管を直ちに氷浴に少なくとも2分間漬けた。次いでサンプルを氷浴から取り除き、新しく調製したDMAB試薬1mlを加え、そしてサンプルをボルテックス攪拌した。DMAB(ジメチルアミノベンズアルデヒド)試薬は、氷酢酸70ml及び11.6(濃)HCl 10mlをp-ジメチルアミノベンズアルデヒド8グラムに加えて作った。次いでサンプルを37℃にて20分間インキュベートした。
【0209】
標準曲線を調製するために、次の方法を利用した。GlcNAcストック溶液を0.010M酢酸ナトリウムバッファー(pH 4.5)を用いて0.1mg/mlに希釈し、希釈したGlcNAc溶液0μl、20μl、30μl、90μlまたは120μlを試験管のセットに加えた。次いで、0.010M酢酸ナトリウムバッファー(pH 4.5)150μl、130μl、60μlまたは30μlをそれぞれ試験管に加えた。次に、0.25Mリン酸カリウムバッファー(pH 7.1)25μl及び0.8Mホウ酸カリウムバッファー(pH 9.8)35μlをそれぞれの試験管に加えた。試験管の重複セットを同じ方法により調製した。
【0210】
試験管に栓をして100℃にて正確に3分間煮沸した。次いで試験管を氷浴に漬けた。試験管を氷浴から取出し、上記の方法に従って新しく調製したDMAB試薬1mlを各試験管に加えた。試験管を37℃にて20分間インキュベートした。各試験管の内容物の吸収を585nMにて読み取った。吸収はなるたけ速く読み取らねばならない。標準曲線をグラフ用紙上にプロットし、これを利用して反応サンプル中のN-アセチルグルコサミン濃度を決定した。典型的な標準曲線を図18に示す。
【0211】
15.2. 結果
p-GlcNAc物質のin-vitro生分解性を、p-GlcNAc膜物質のリゾチームによる分解に対する相対的感受性をアッセイする実験で研究した。p-GlcNAc膜を10mM酢酸バッファー中の過剰のリゾチームに曝し、その後のN-アセチルグルコサミンの放出を以上の第15.1節に記載したアッセイを用いて測定した。
【0212】
これらの実験の結果は、部分的に脱アセチル化された膜はリゾチームによる消化に対して、より感受性であること(図19を参照)及び、さらに、リゾチーム分解速度は脱アセチル化の程度と直接関係があること(図20を参照、50%脱アセチル化p-GlcNAc膜の分解速度を25%脱アセチル化p-GlcNAc膜に対して比較する)を示した。
【0213】
p-GlcNAcのin vivo分解
実験は、p-GlcNAc物質のin-vivo生分解性を目指して実施した。かかる実験は腹部移植モデルを利用した。次に掲げる3つのp-GlcNAc物質を試験した。
【0214】
試験したp-GlcNAc物質
1) 完全にアセチル化されたp-GlcNAc(プロトタイプ1と呼ぶ);
2) 部分的に脱アセチル化されたp-GlcNAc膜(プロトタイプ3Aと呼ぶ);及び
3) 凍結乾燥されかつ部分的に脱アセチル化されたp-GlcNAc膜(プロトタイプ4と呼ぶ)
結果
完全にアセチル化されたp-GlcNAc(プロトタイプ1)は、図21A-21Cに示すように21日内に再吸収された。部分的に脱アセチル化されたp-GlcNAc膜(プロトタイプ3A)は、図21D-21Eに示すように14日以内に完全に再吸収された。凍結乾燥しかつ部分的に脱アセチル化されたp-GlcNAc膜(プロトタイプ4)は、移植後21日の後にもなお完全には再吸収されなかった。
【0215】
組織病理学分析は、p-GlcNAc物質が再吸収されると、処理した動物から得た組織サンプルと対照動物からのそれとの間に検出しうる組織学的差異はないことを示した。
【0216】
16. 実施例:p-GIcNAcによるエンドセリン-1分泌の刺激と動脈血管収縮の誘導
本実施例は、本発明のp-GlcNAcを用いてin vivoでエンドセリン-1放出を刺激しかつ動脈血管収縮を誘導しうることを実証する。
【0217】
16.1. 大動脈切開の処置及び分析;材料及び方法
動物:本研究は、体重25〜30kg(平均27.5kg)の未熟雌性ヨークシャイア白ブタ(Yorkshire White swine)について実施した。次のプロトコルを全事例で使用した。
【0218】
プロトコル
1. 標準の前投薬の後に、動物を、100% O2及び1-2%ハロタンの吸入により麻酔する
2. CBC及び血小板計数のために対照血液サンプルを採取する
3. 腹部大動脈を露出する
4. タイでくくって固定し、大動脈に1cmの鉛直の傷を付ける
5. 試験材を適用する間、タイを解く
6. 1分間圧縮する
7. 圧縮を止めて、出血を観察する
8. もし出血すれば、ステップ4と5を繰り返す
9. もし15回の1分間圧縮で出血が停止しなければ、試験材は不合格である
10. 病理学用組織を回収する。
【0219】
16.2 脾臓切開の処置及び分析;材料及び方法
動物:本研究は、体重34〜37kgの未熟雌性ヨークシャイア白ブタ(Yorkshire White swine)について実施した。次のプロトコルを全事例で使用した。
【0220】
プロトコル
1. 標準の前投薬の後に、動物を、100% O2及び1-2%ハロタンの吸入により麻酔するCBC及び血小板計数のために対照血液サンプルを採取する
2. 絶対的止血を維持するため電気メスを用いて正中線腹部切開を行い、脾臓を摘出する
3. 脾臓をスポンジを用いて単離する
4. 脾臓表面上に深さ3mmの2cm x 2cm面積のカプセルストリップを作る
5. 傷から10秒間自由に出血させる
6. 蓄積した血液を外科スポンジを用いて除去する
7. 試験薬剤を適用する
8. 1分間、穏やかな圧力をかける
9. 圧力を除き、2分間出血を観察する
10. 傷が出血すれば、5及び6を繰り返す
11. 出血を制御するために必要な圧縮数及び止血までの時間を記録する
12. もし完全な出血の停止(出血の停止後2分間、再出血なしと定義する)が達成されれば記録する
13. 病理学研究用に組織を回収する。
【0221】
16.3 脾臓免疫染色のプロトコル
免疫染色は、わずかに改変を加えたPeninsula Laboratories社製ET-1染色キット(カタログ番号HIS-6901)を用いて実施した。
【0222】
スライド調製及び染色方法
1. 脾臓組織のサンプルを採取し、標準の方法を用いてスライド上にてサンプルをパラフィンに埋め込んで保存した。パラフィンは、その後、スライドを100%キシレン中で10分間インキュベートすることにより除去した。スライドを、100%エタノール、95%エタノール、及び次いで水道水のグレード別シリーズで各溶液に5回漬けることにより、再水和する。イム・エッジ(Imm Edge)耐水ペン(Vector Laboratories、カタログ番号H-4000)を用いて、組織サンプルの外周に線を引く。スライドをコプリンジャー(coplin jar)に入れたPBS pH7.4溶液中に保存する。
【0223】
2. 抗原アンマスキング溶液(Vector Laboratoriesカタログ番号H-3300)を100 Xに希釈し、30-45秒間、他のコプリンジャー内で加熱する。スライドをこの溶液へ移し20分間インキュベートする。乾燥し切るのを防止するために組織サンプルを覆うのに十分な溶液が存在することを確かめる。スライドをPBS pH7.4溶液を用いて2分間、よくリンス洗いする;2回繰り返す。過剰の溶液を除去するためにスライドを乾かすかまたは拭きとる。
【0224】
3. 正常のヤギ血清ブロッキング溶液の2滴もしくは100μLを各スライドに加える。室温にて20分間インキュベートする。スライドから過剰溶液を乾かすかまたは拭きとる。リンス洗いはしない。
【0225】
4. 凍結乾燥した一次抗体を、PBS pH7.4溶液32μlを用いて再構成する。このストック溶液から、一次抗体を400の希釈ファクターで希釈する。希釈した一次抗体の2滴もしくは100μLを各スライドに加える。スライドを、加湿室内の木製スティック上に水平に置き、一夜4℃にてインキュベートする。PBS pH7.4溶液を用いて2分間よくリンス洗いする;2回繰り返す。
【0226】
5. ビオチン化二次抗体の2滴もしくは100μLを各スライドに加える。30分間室温でインキュベートする。PBS pH7.4溶液を用いて2分間よくリンス洗いする;2回繰り返す。
【0227】
6. スライドを、コプリン・ジャーに入った3%証明付き過酸化水素(Fisherカタログ番号H 312-500)中に入れ、30分間室温にてクエンチする。PBS pH7.4溶液を用いて2分間、よくリンス洗いする;2回繰り返す。
【0228】
7. ストレプトアビジン-HRPコンジュゲート2滴または100μLを各スライドに加え、30分間室温でインキュベートする。PBS pH7.4溶液を用いて2分間、よくリンス洗いする;2回繰り返す。
【0229】
8. 蒸留水5.0mLをガラス製シンチレーション・バイアルに加えてDABクロマーゲン(Chromagen)溶液(Vector Laboratoriesカタログ番号sk-41067)を作る。バッファーストック溶液2滴を加えてよく混合する。次いで、DABストック溶液4滴を加えてよく混合する。最後に、過酸化水素溶液2滴を加えてよく混合する。DABクロマーゲン溶液200μLを各スライドに加える。3分間室温にてインキュベートする。蒸留水を用いてよくリンス洗いして拭きとる。
【0230】
9. スライドを、0.2%Working Light Green Solution(Sigmaカタログ番号L 5382)の希釈ファクター6のストック溶液を用いてカウンター染色する。スライドを3回Working Light Green Solutionに漬け、次いでスライドを5回それぞれ、脱水シリーズ、すなわち蒸留水、次いで95%エタノール、次いで100%エタノール、そして最後に100%キシレンに漬ける。スライドを乾かすかまたは拭きとって過剰キシレンを除去する。
【0231】
10. Cytoseal XYLマウンティング溶液(Stephens Scientificカタログ番号8312-4)の2滴を加え、スライドをマウントする。
【0232】
16.4 動脈の免疫染色プロトコル
動脈組織の免疫染色を、若干の改変を加えたPeninsula Laboratories社(カタログ番号HIS-6901)のET-1染色キットを用いて実施した。
【0233】
スライド調製
1. 肺動脈を、購入したシカから切除する
2. 動脈をRPMI培地100mL中に入れ、氷上に置く
3. メスを用いて動脈を切開する
4. 完全にアセチル化されたp-GlcNAc繊維からなる1cm X 1cm正方の膜で、切開部を覆って15分間置く
5. 膜適用部位において動脈の横断セクション薄片を作り、組織学研究用とする
6. そのセクションを9%ホルムアルデヒド中に置く。パラフィンを用いてスライドを作る。
【0234】
染色方法
1. スライドを10分間、100%キシレン中でインキュベートし、脱パラフィン化する。スライドを100%エタノール、95%エタノール、及び次いで水道水のグレード別シリーズを用い、それぞれの溶液に5回漬けて再水和する。イム・エッジ(Imm Edge)耐水ペン(Vector Laboratories、カタログ番号H-4000)を用いて、組織サンプルの外周に線を引く。スライドをコプリンジャー(coplin jar)に入れたPBS pH7.4溶液中に保存する。
【0235】
2. 抗原アンマスキング溶液(Vector Laboratoriesカタログ番号H-3300)を100倍希釈し、30-45秒間、他のコプリン・ジャー内で加熱する。スライドをこの溶液へ移し20分間インキュベートする。乾燥し切るのを防止するために組織サンプルを覆うのに十分な溶液をあることを確かめる。スライドをPBS pH7.4溶液を用いて2分間、よくリンス洗いする。2回繰り返す。過剰溶液を除去するためにスライドを乾かすかまたは拭きとる。
【0236】
3. 正常のヤギ血清ブロッキング溶液の2滴もしくは100μLを各スライドに加える。室温にて20分間インキュベートする。スライドから過剰溶液を乾かすかまたは拭きとる。リンス洗いはしない。
【0237】
4. 凍結乾燥した一次抗体をPBS pH7.4溶液32μlを用いて再構築する。このストック溶液から、一次抗体を希釈ファクター100で希釈する。希釈した一次抗体の2滴もしくは100μLを各スライドに加える。スライドを、加湿室内の木製スティック上に水平に置き、一夜4℃にてインキュベートする。PBS pH7.4溶液を用いて2分間よくリンス洗いする;2回繰り返す。
【0238】
5. ビオチン化二次抗体の2滴もしくは100μLを各スライドに加える。30分間室温でインキュベートする。PBS pH7.4溶液を用いて2分間よくリンス洗いする;2回繰り返す。
6. スライドを、コプリンジャーに入った3%証明付き過酸化水素(Fisherカタログ番号H 312-500)中に入れ、30分間室温にてクエンチする。PBS pH7.4を用いて2分間、よくリンス洗いする;2回繰り返す。
【0239】
7. ストレプトアビジン-HRPコンジュゲート2滴または100μLを各スライドに加え、30分間室温でインキュベートする。PBS pH7.4溶液を用いて2分間よくリンス洗いする;2回繰り返す。
【0240】
8. 蒸留水5.0 mLをガラス製シンチレーション・バイアルに加えることによりDABクロマーゲン-溶液(Vector Laboratoriesカタログ番号sk-41067)を作る。バッファーストック溶液2滴を加えてよく混合する。次いで、DABストック溶液4滴を加えてよく混合する。最後に、過酸化水素溶液2滴を加えてよく混合する。DABクロマーゲン-溶液200μLを各スライドに加える。3分間室温にてインキュベートする。蒸留水を用いてよくリンス洗いしかつ拭きとる。
【0241】
9. スライドを、0.2%Working Light Green Solution(Sigmaカタログ番号L 5382)の希釈ファクター6のストック溶液を用いてカウンター染色する。スライドを3回Working Light Green Solutionに漬け、次いでスライドを、脱水シリーズ、すなわち蒸留水、次いで95%エタノール、次いで100%エタノール、そして最後に100%キシレンにそれぞれ5回漬ける。スライドを乾かすかまたは拭きとって過剰キシレンを除去する。
【0242】
10. Cytoseal XYLマウンティング溶液(Stephens Scientificカタログ番号H 8312-4)の2滴を加え、スライドをマウントする。
【0243】
結果
完全にアセチル化されたp-GlcNAc繊維からなる膜を用いて処理した動脈組織の組織学的及び免疫学的試験は、傷害動脈組織とp-GlcNAcポリマーとの接触部位において即効性の血管収縮刺激を与えた。p-GlcNAc膜の適用により誘導される血管収縮は、組織学的により大きい実験動物においてより容易に見られた。動脈組織の収縮は、p-GlcNAc膜が適用された動脈サイドにおいてより顕著である。これらの分析結果を図23及び図24に示した。ガーゼ包帯のブタ動脈へを適用した60分後に(図23(A)、及び図24、サンプルA)、動脈壁とガーゼとの接触点(1)において、またはガーゼ包帯を適用したのと反対側の点において測定したところ、動脈壁厚さの値は比較しうるものあった。対照的に、半結晶p-GlcNAcで製剤化した膜のブタ動脈への適用(図23 (B)、図24、サンプルB)すると、膜を適用した15分後に、接触領域における壁厚さ(1)の著しい増加が誘導された。接触の60分後、p-GlcNAc膜との接触領域で測定した動脈壁の厚み(1)は、動脈の反対側の点で測定した厚み(2)と比較しうるレベルに戻った。
【0244】
エンドセリン-1に対する抗体を用いた免疫染色実験は、p-GlcNAc膜と生組織との間の接触部位におけるエンドセリン-1の分泌を示した。シカ肺動脈を用いたin vitro実験は、p-GlcNAc膜と動脈との接触表面上にのみエンドセリン-1が存在することを示した。In vivo実験は、処理した組織とp-GlcNAc膜との間の接触表面上においてだけでなく、組織のより深い層にも実質的に大きなエンドセリン-1放出を示した。p-GlcNAc膜の適用後15分以内に、処理組織とp-GlcNAc膜との間の接触のほんの60分後に実施した類似の分析と比較して、大きいエンドセリン-1の分泌が検出された。それにも関わらず、収縮効果は試験したその他のサンプルより強かった。
【0245】
他のサンプルのスライドについて同じエンドセリン-1免疫染色を観察したが、その結果はp-GlcNAcより遥かに低かった。p-GlcNAc膜と接触した脾臓組織の組織学的及び免疫学的分析は、エンドセリン-1放出の類似した亢進を示した。再び、実験膜の適用後15分以内に、エンドセリン-1はp-GlcNAc膜が適用されていたサンプルにおいてだけ観察された。実験膜と処理組織との間の接触の60分後に、全てのサンプルは比較しうるエンドセリン-1のレベルを示した。
【0246】
17. 実施例:血液産物不在のもとでの、p-GlcNAcによる血管収縮及びエンドセリン放出の誘導
本実施例は、本発明の完全にアセチル化された半結晶p-GlcNAcが、血液不在のもとで動脈血管収縮を誘導することを実証する。より具体的には、本実施例は、完全にアセチル化されたp-GlcNAcは、血液凝固カスケードのいずれの成分の不在のもとでも、内皮に依存する機構を経由して、部分的に内皮細胞からのエンドセリン-1放出により、単離されたラット大動脈環を有意に収縮することを実証する。
【0247】
17.1 物質及び方法
大動脈環を体重275-300gの雄性スプラグ・ドーリイ(Sprague-Dawley)ラットから得た。ラットを、腹腔内にペントバルビタールナトリウム(60 mg/kg)を注射して麻酔した。大動脈及びSMAをラットから急いで除去し、加温したクレープス-ヘンゼライト(Krebs-Henseleit)(KH)バッファー(単位mmol/lで、NaCl 118、KCl 4.75、CaCl2.2H2O 2.54、KH2PO4 1.19、MgSO4・7H2O 1.19、NaHCO3 12.5、及びグルコース 10.0からなる)中に懸濁した。単離した血管から注意深く結合組織を除去し、長さ2-3mmの環に切断した。次いで環をステンレス鋼フック上にマウントし、10-ml組織容器中に懸濁し、そしてFT-03力-変位変換器(Grass Instrument、Quincy、Mass.)と接続してGrass model 7 オシログラフレコーダー上で力の変化を記録した。容器をKHバッファーを用いて満たし、37℃にて95%O2+5%CO2を通気した。0.5gの静止力をSMA環に適用し、次いで環を90分間、平衡化した。この期間に、組織容器中のバッファーを15-20分毎に取り換え、血管環の静止力を調節して0.5gの前負荷を維持した。平衡化の90〜120分後に、環を100nM U-46619(9,11-ジデオキシ-9α-11α-メタンエポキシ-プロスタグランジンF2α;Biomol Research Laboratories、Plymouth Meeting、Pa.)、トロンボキサンA2模倣物に曝し、1.0gの発現力(developed force)を発生させた。安定した収縮を得ると、その容器に、典型的な内皮依存血管収縮薬であるアセチルコリンを0.1、1、10、及び100nMの累積濃度で加え、内皮の保全性を評価した。累積応答が安定化した後、環を洗浄し、再び、ベースラインに平衡化させた。
【0248】
この方法を、U-46619、次いでp-GlcNAcを用いて繰返した。p-GlcNAcは、図23に示すように14〜140g/mlで濃度依存的血管収縮を起こした。140g/mlの発生濃度において、p-GlcNAcは発現力218 ± 21mgで大動脈環を有意に収縮した(p<0.01)。内皮を剥いだ(すなわち、綿で覆われたひねりステンレス鋼ワイヤ上に大動脈環を静かに廻して内皮を除去した)大動脈環は、発現力33 ± 12mgでしか収縮しなかった。エンドセリンEtA受容体アンタゴニスト、JKC-301(シクロ[D-Asp-Pro-D-IIe-Leu-D-Trp]、Sigma Biochemicals and Reagents、St. Louis、Mo.)(0.5及び1M)による前処理は、p-GlcNAcが誘導する血管収縮を57〜61%だけ有意に減少させた。
【0249】
この方法を、U-46619を用い、次いで70%脱アセチル化p-GlcNAcにより繰返した。先に使用した完全にアセチル化した半結晶p-GlcNAcを70%脱アセチル化p-GlcNAcに置き換えると、この血液を含まないモデルシステムでは、全ての試験濃度において血管収縮が起こらなかった。
【0250】
本明細書で説明した本発明の多くの改変及び変法が、本発明の精神と範囲を逸脱することなしになされうることは明白である。上記の特定の実施形態は、例示としてだけ与えられたものであって、本発明は添付した請求項によってのみ制限されるものである。
【0251】
様々な出版物が本明細書に引用されたが、これらの開示は参照によりその全文が組み入れられる。
【技術分野】
【0001】
1.序
本発明は、半結晶ポリβ-1→4-N-アセチルグルコサミン(p-GlcNAc)多糖ポリマーを含む組成物、ならびにかかるポリマーを利用し、p-GlcNAc濃度依存的に、エンドセリン-1放出、血管収縮、及び/または破裂した血管からの血液流出の減少を一過性かつ局所的に刺激する方法に関する。これらの効果は、個々に及び/または共同して、出血の停止を助けるかまたは達成する。さらに特定すれば、本発明の方法は、N-アセチルグルコサミンの半結晶ポリマーを含む組成物及び物質の局所投与を含み、前記ポリマーがタンパク質を含まずかつ単一アミノ酸ならびにその他の有機及び無機不純物を実質的に含まず、そしてその成分である単糖類がβ-1→4コンフォメーションで結合されていることを特徴とする。
【背景技術】
【0002】
2.背景
血管ホメオスタシスは、部分的に、内皮細胞による生化学モジュレーターの調節された分泌に依存する。正常の生理学的条件下において、内皮細胞は一酸化窒素、プロスタサイクリン、PG12、アデノシン、過分極因子、組織因子経路インヒビター、及びscuプラスミノーゲンアクチベーターを合成しかつ分泌する。内皮細胞はまた、抗トロンビンIII及びプロテインCを活性化し、これらは共同して血管拡張を媒介して血小板接着、血小板活性化、トロンビン生成及びフィブリン沈着を抑制する。一酸化窒素は特に、血管ホメオスタシスに極めて重要な役割を果たす(Pearson, J. D. (2000) Lupus 9 (3):183-88;Beckerら (2000) Z Kardiol 89 (3):160-7;Schinin-Kerth, V. B. (1999) Transfus Clin Biol 6 (6):355-63)。
【0003】
強力な血管拡張薬ならびに血小板凝集及び活性化のインヒビターである一酸化窒素とプロスタサイクリンの産生が、内皮の抗血栓活性の基礎となっている(Yangら (1994) Circulation 89 (5): 2666-72)。一酸化窒素は構成的、基底レベルでアルギニンから一酸化窒素シンターゼにより合成され、かつこの合成は血管作用薬であるアセチルコリン及びブラジキニンにより刺激される。アルギニン類似体であるモノメチル-L-アルギニン(L-NMMA)及びニトロ-L-アルギニンメチルエステル(L-NAME)による一酸化窒素シンターゼの抑制は、一酸化窒素レベルを低下させ、血管内超音波イメージングにより測定すると、血管収縮をもたらすだけでなく血小板凝集も増加することが示されている(Yaoら (1992) Circulation 86 (4):1302-9;Emersonら (1999) Thromb Haemost 81 (6): 961-66)。
【0004】
アテローム性動脈硬化症、糖尿病、虚血後再灌流、炎症または高血圧症の結果としての内皮の混乱は、例えば前血栓症状態をもたらし、内皮はさらなる生化学モジュレーターのセットを作り出し、それらにはTNF-α、IL-8、ホンビルブラント(von Willebrand)因子、血小板活性化因子、組織プラスミノーゲンアクチベーター、及び1型プラスミノーゲンアクチベーターインヒビターが挙げられる(Pearson, J. D. (2000) Lupus 9 (3): 183-88;Beckerら (2000) Z Kardiol 89 (3): 160-7;Schinin-Kerth, V. B. (1999) Transfus Clin Biol 6 (6): 355-63)。さらに、血管内皮は、既知の最も強力な血管収縮薬であるエンドセリンを合成して作り出す。
【0005】
エンドセリンは、21-アミノ酸ペプチドのファミリー、すなわち、エンドセリン-1、エンドセリン-2、及びエンドセリン-3であり、元来その強力な血管収縮及び血管形成特性により特徴付けられる(例えば、Luscherら (1995), Agents Actions Suppl. (Switzerland) 45: 237-253;Yanagisawaら (1988) Nature 332: 411-415を参照)。エンドセリンファミリーの3つのイソペプチドであるエンドセリン-1、エンドセリン-2、及びエンドセリン-3は、3つの別の遺伝子がコードする高度に保存されたアミノ酸配列を有する(例えば、Inoueら (1989) Proc Natl Acad Sci USA 86: 2863-67;Saidaら (1989) J Biol Chem 264:14613-16を参照)。エンドセリン類は、平滑筋細胞を含む複数組織において合成されるが、エンドセリン-1は専ら血管内皮により合成される(Rosendorff, C. (1997) Cardiovasc Drugs 10 (6): 795-802)。エンドセリン類は203個のアミノ酸からなるプレプロエンドセリンとして合成される。エンドセリンシグナル配列が切断され、該タンパク質はさらなるタンパク分解によって、成熟した生物化学的に活性な21個のアミノ酸形態となる(例えば、Kashiwabaraら (1989) FEBS Lett 247: 337-40を参照)。エンドセリン合成は、エンドセリン及び非エンドセリン転化酵素を含む自己分泌機構を介してならびにキマーゼにより調節される(Batonら (1999) Curr Opin Nephrol Hypertens 8 (5): 549-56)。内皮からのエンドセリン-1の産生は、アンギオテンシンII、バソプレッシン、内毒素、及びとりわけシクロスポリンにより刺激され(例えば、Brooksら (1991) Eur J Pharm 194: 115-17を参照)、そして一酸化窒素により抑制される。
【0006】
エンドセリン活性は、2つの明確なGタンパク質結合受容体であるETA及びETBに対する優先的アフィニティによる結合を経由して自己分泌/パラ分泌的に媒介される(例えば、Hocherら (1997) Eur. J. Clin. Chem. Clin. Biochem. 35 (3): 175-189;Shichiriら (1991) J. Cardiovascular Pharmacol. 17: S76-S78を参照)。ETA受容体は血管収縮と連結した血管平滑筋上に見出され、心血管、腎、及び中枢神経系疾患と関連がある。ETB受容体はさらに複雑であり、アンタゴニスト作用を示す。内皮のETB受容体はクリアランスと血管拡張の二重の役割を有するのに対して、平滑筋細胞上のETB受容体は血管収縮も媒介する(Dupuis, J. (2000) Can J Cardiol 16 (1): 903-10)。内皮上のETB受容体は一酸化窒素及びプロスタサイクリンの放出と連結している(Rosendorff, C. (1997) Cardiovasc Drugs 10 (6): 795-802)。様々なエンドセリン受容体のアゴニスト及びアンタゴニストが存在し(Webbら (1997) Medicinal Research Reviews 17 (1): 17-67)、エンドセリンの作用機序を研究するのに利用されている。エンドセリンは強力な血管収縮活性を有することが知られているので、特に、エンドセリンアンタゴニスト(当技術分野では「エンドセリン受容体アンタゴニスト」とも呼ばれる)はヒトの疾患、最も著名なのは、心血管疾患、例えば高血圧、うっ血性心不全、アテローム性動脈硬化症、再狭窄、及び心筋梗塞を治療する上でのそれらの可能な役割について研究されている(Mateoら (1997) Pharmacological Res. 36 (5): 339-351)。
【0007】
さらに、エンドセリン-1は月経周期の正常な機能に関わることが示されている。月経は組織修復と置き換えの代表例であり、子宮を内張りする子宮内膜組織の新しい層の規則正しい再形成と再生を含む。この修復と再形成プロセスは瘢痕なしに達成され、一般的に身体の他の器官では見られない現象である点が注目される。この修復プロセスの欠陥が、月経過多と記録された女性ならびに避妊目的の皮下レボノルゲストレル・インプラント(NORPLANT)を装着した女性における過剰または異常な子宮内膜出血の原因であると考えられる。これらの両グループの女性においては、対照集団と比較して、非常に低レベルの子宮内膜エンドセリン-1が検出されるにすぎない。さらに、エンドセリン-1は月経性出血の停止ににおいて役割を果たすだけでなく、エンドセリン-1は月経後の子宮内膜組織の再生及び再形成に必要な分裂促進活性も有しうることが示されている。(例えば、Salamonsenら 1999, Balliere's Clinical Obstetrics and Gynaecology 13 (2): 161-79;Goldie 1999, Clinical and Experimental Pharmacology and Physiology 26: 145-48;Salamonsenら 1999, Clin. Exp. Phamaol. Physiol. 26 (2): 154-57を参照)。
【0008】
総括すると、血管ホメオスタシスは、血管内皮が媒介する2つの生理学的状態の間の動的バランスを反映する。抗血栓性と呼ばれる第1の状態は、とりわけ、一酸化窒素の産生、血管拡張、血小板接着及び活性化の抑制により、ならびにエンドセリン-1合成の抑圧により特徴付けられる。第2のまたはプロトロンビン生理学的状態は、とりわけ、エンドセリン-1の産生、血管収縮、血小板活性化、及び止血により特徴付けられる(Warner (1999), Clinical and Experimental Physiology 26: 347-52;Pearson, (2000), Lupus 9(3): 183-88)。
【先行技術文献】
【非特許文献】
【0009】
【非特許文献1】Pearson, J. D. (2000) Lupus 9 (3):183-88
【非特許文献2】Beckerら (2000) Z Kardiol 89 (3):160-7
【非特許文献3】Schinin-Kerth, V. B. (1999) Transfus Clin Biol 6 (6):355-63
【非特許文献4】Yaoら (1992) Circulation 86 (4):1302-9
【非特許文献5】Emersonら (1999) Thromb Haemost 81 (6): 961-66
【非特許文献6】Luscherら (1995), Agents Actions Suppl. (Switzerland) 45: 237-253
【非特許文献7】Yanagisawaら (1988) Nature 332: 411-415
【非特許文献8】Inoueら (1989) Proc Natl Acad Sci USA 86: 2863-67
【非特許文献9】Saidaら (1989) J Biol Chem 264:14613-16
【非特許文献10】Rosendorff, C. (1997) Cardiovasc Drugs 10 (6): 795-802
【非特許文献11】Kashiwabaraら (1989) FEBS Lett 247: 337-40
【非特許文献12】Batonら (1999) Curr Opin Nephrol Hypertens 8 (5): 549-56
【非特許文献13】Brooksら (1991) Eur J Pharm 194: 115-17
【非特許文献14】Hocherら (1997) Eur. J. Clin. Chem. Clin. Biochem. 35 (3): 175-189
【非特許文献15】Shichiriら (1991) J. Cardiovascular Pharmacol. 17: S76-S78
【非特許文献16】Webbら (1997) Medicinal Research Reviews 17 (1): 17-67
【非特許文献17】Mateoら (1997) Pharmacological Res. 36 (5): 339-351
【非特許文献18】Salamonsenら 1999, Balliere's Clinical Obstetrics and Gynaecology 13 (2): 161-79
【非特許文献19】Goldie 1999, Clinical and Experimental Pharmacology and Physiology 26: 145-48
【非特許文献20】Salamonsenら 1999, Clin. Exp. Phamaol. Physiol. 26 (2): 154-57
【非特許文献21】Warner (1999), Clinical and Experimental Physiology 26: 347-52
【発明の概要】
【発明が解決しようとする課題】
【0010】
血管ホメオスタシスの生理学的重要性に照らして考えると、上記プロセスの1以上の態様をモジュレートできる方法及び組成物に対するニーズがある。さらに特定すれば、エンドセリン放出、血管収縮、及び破裂した血管からの血液流出のモジュレーション、それによる出血の停止に有用でありうる組成物及び方法に対するニーズがある。すなわち、かかる組成物及び方法が物理的障害形成、凝血、または血餅形成に依存しない方法で作用しうるものであるにも関わらず、かかる組成物及び方法は、とりわけ、止血の達成に寄与しうる。従って、かかる方法及び組成物は、血管ホメオスタシスの混乱の結果として生じる疾患または症状の治療に対する治療上の用途を有すると期待しうる。さらに、例えば、患者への先に記載のエンドセリン-1アンタゴニストの投与から得られる全身効果を視野に入れると、限定されるものでないが、かかる患者におけるエンドセリン-1放出の刺激を含む、局所性で一過性の生理学的応答を産生する組成物及び方法に対するさらに大きなニーズがある。
【課題を解決するための手段】
【0011】
3.発明の概要
本発明は、出血障害を含む血管障害の治療または改善のための方法及び組成物に関する。さらに特定すれば、本発明は、半結晶ポリβ-1→4-N-アセチルグルコサミン(p-GlcNAc)多糖ポリマーを含む組成物、ならびに、例えばエンドセリン-1放出の刺激、血管収縮、及び/または破裂した血管からの血液流出の減少により、血管構造及び/または機能の一過性で局所性のモジュレーションを果たし、その結果、出血の停止を助けるかまたは達成する方法におけるかかるポリマーの使用に関する。
【0012】
本発明は、部分的には、半結晶ポリβ-1→4-N-アセチルグルコサミン(p-GlcNAc)多糖ポリマーを血管表面に局所適用すると、血管の収縮を誘導してそれにより血管の管腔を減少するだけでなく、適用した本明細書に開示した組成物及び物質に近接する組織においてエンドセリン-1放出の一過性で局所性の刺激も誘導するという本出願人の発見に基づく。
【0013】
本発明は、一態様においては、患者の血管構造及び/または機能の一過性で局所性の、モジュレーションを達成する方法であって、タンパク質を含まず、他の有機不純物を実質的に含まず、そして無機不純物を実質的に含まない半結晶ポリβ-1→4 N-アセチルグルコサミンポリマーを含む物質の局所投与を含んでなる前記方法に関する。これらの物質の投与は、限定されるものでないが、エンドセリン-1放出の刺激、血管収縮、及び破裂した血管からの血液流出の減少を含む一過性で局所性の生理学的応答を誘導する。
【0014】
本発明の一実施形態においては、血管内皮細胞からエンドセリン-1が放出される。この実施形態の他の態様においては、エンドセリン-1放出は他の内皮組織からまたは血小板から刺激される。
【0015】
一実施形態においては、ポリβ-1→4 N-アセチルグルコサミンポリマーは約50〜約4,000個のβ-1→4コンフォメーションで共有結合したN-アセチルグルコサミン単糖を含んでなり、約10,000ダルトン〜約800,000ダルトンの分子量を有する。他の実施形態においては、ポリβ-1→4 N-アセチルグルコサミンポリマーは約50〜約10,000個のβ-1→4コンフォメーションで共有結合したN-アセチルグルコサミン単糖を含んでなり、約10,000ダルトン〜約2 x 106ダルトンの分子量を有する。さらに他の実施形態においては、ポリβ-1→4 N-アセチルグルコサミンポリマーは約50〜約50,000個のβ-1→4コンフォメーションで共有結合したN-アセチルグルコサミン単糖を含んでなり、約10,000ダルトン〜約10 x 106ダルトンの分子量を有する。他の実施形態においては、ポリβ-1→4 N-アセチルグルコサミンポリマーは約50〜約150,000個のβ-1→4コンフォメーションで共有結合したN-アセチルグルコサミン単糖を含んでなり、約10,000ダルトン〜約30 x 106ダルトンの分子量を有する。
【0016】
本発明の好ましい実施形態においては、開示した方法を哺乳動物患者の治療に、かつさらに好ましい実施形態においては、かかる治療を必要とするヒトの治療に利用する。さらに特定すれば、血管構造及び/または機能のモジュレーションを利用して、特に凝血障害を患う患者における出血の停止を達成する。かかる障害は、血友病などのなどの遺伝的欠陥、または例えば、透析患者、心臓病患者もしくはその他の血管閉塞のリスクが高い患者へのクマジン(coumadin)などの全身抗凝血薬の投与を含む医学治療の結果でありうる。同様に、本発明の方法は、特に凝血障害症状を有する患者における、動脈瘤の外科修復または腫瘍もしくはポリープの切除中に、破裂した血管からの血液流出のの一過性で局所性の減少を達成するために利用し、それにより、かかる処置中の血液損失を最小化する。他の実施形態においては、本発明の方法は、子宮もしくは静脈瘤、特に食道静脈瘤の出血の治療に利用することができる。特定の理論もしくは機構に束縛されるのを欲しないが、本明細書に開示した方法によるかかる出血の停止は、凝血に依存しない方式で起こると考えられる。
【0017】
本発明の方法の他の実施形態においては、p-GlcNAcを含有する物質を患者の皮膚もしくはその他の器官の表面に局所投与するか、またはモジュレートすべき血管構造に直接適用してもよく、その血管構造は毛細管、血管または動脈であってもよい。
【0018】
本発明の方法のさらに他の実施形態においては、血管構造は破裂した血管であって、本発明のp-GlcNAcを含有する物質の局所適用を用いて出血の停止を達成する。
【0019】
本発明のさらなる実施形態においては、血管構造及び/または機能の一過性で局所性のモジュレーションの程度は、適用した半結晶ポリβ-1→4-N-アセチルグルコサミンの量と実質的に比例する。
【0020】
本発明はまた、タンパク質を含まず、その他の有機不純物を実質的に含まず、かつ無機不純物を実質的に含まない半結晶ポリβ-1→4 N-アセチルグルコサミンポリマーを含む生物分解性物質に関する。一実施形態においては、半結晶ポリβ-1→4 N-アセチルグルコサミンポリマーは約50〜約4,000個のβ-1→4コンフォメーションで共有結合したN-アセチルグルコサミン単糖を含みかつ約10,000ダルトン〜約800,000ダルトンの分子量を有する。他の実施形態においては、半結晶ポリβ-1→4 N-アセチルグルコサミンポリマーは約50〜約10,000個のβ-1→4コンフォメーションで共有結合したN-アセチルグルコサミン単糖を含みかつ約10,000ダルトン〜約2 x 106ダルトンの分子量を有する。さらに他の実施形態においては、ポリβ-1→4 N-アセチルグルコサミンポリマーは約50〜約50,000個のβ-1→4コンフォメーションで共有結合したN-アセチルグルコサミン単糖を含みかつ約10,000ダルトン〜約10 x 106ダルトンの分子量を有する。他の実施形態においては、ポリβ-14 N-アセチルグルコサミンポリマーは約50〜約150,000個のβ-1→4コンフォメーションで共有結合したN-アセチルグルコサミン単糖を含みかつ約10,000ダルトン〜約30 x 106ダルトンの分子量を有する。
【0021】
他の実施形態においては、半結晶ポリβ-1→4 N-アセチルグルコサミンポリマーを含む生物分解性物質は、バリヤーを形成しない物質である。
【0022】
さらに他の実施形態においては、半結晶ポリβ-1→4 N-アセチルグルコサミンポリマーは、少なくとも1つの脱アセチル化されたN-アセチルグルコサミン単糖を含む。この実施形態の他の態様においては、部分的に脱アセチル化されたポリβ-1→4 N-アセチルグルコサミンポリマーが、そのポリマーを下の実施例6に記載のIR吸収分光計により分析したときに鋭い明確なピークにより実証されるその半結晶構造を保持するのであれば、ポリβ-1→4 N-アセチルグルコサミンポリマーは、約10%、20%、30%、40%、50%または60%脱アセチル化された残基を含んでもよい。
【図面の簡単な説明】
【0023】
【図1】図1は、100% p-GlcNAcの化学構造である。「n」は約150,000までの範囲の整数を意味する。
【図2】図2は、p-GlcNAcの炭水化物分析、すなわちガスクロマトグラフィ-質量分析データである。黒正方形の点は、以下の第5.3.2節に記載の化学的/生物学的方法の酸処理/中和化変法を用いて精製したp-GlcNAcを表す。
【図3A】図3Aは、純粋なp-GlcNAc固体膜の円二色性スペクトルである。
【図3B】図3Bは、脱アセチル化p-GlcNAc固体膜の円二色性スペクトルである。純粋なp-GlcNAc(図3A)で観察された211nm最小値及び195nm最大値の消滅は、以下の第5.4節に記載の使用した条件下での完全な脱アセチル化を示す。
【図4A】図4Aは、機械力精製(上側)、及び化学的/生物学的精製法(下側)により調製した純粋な珪藻p-GlcNAcの薄膜の赤外分光分析である。
【図4B】図4Bは、以下の第5.5節に詳述した方法に従って、膜に成型した市販「キチン」の2つの調製物の赤外分光分析である。
【図4C】図4Cは、以下の第5.4節に詳述した方法に従って、熱変性により(上側)及び化学的脱アセチル化により(下側)改変した純粋p-GlcNAcの赤外分光分析である。
【図4D】図4Dは、以下の第5.3.2節に詳述した化学的/生物学的精製法を用いて珪藻タラシオシラ・フルビアチリス(Thalassiosira fluviatilis)から誘導したp-GlcNAc膜の赤外分光分析である。
【図4E】図4Eは、以下の第5.3.1節に記載の機械力精製法と次いでオートクレーブ処理により調製したp-GlcNAc膜の赤外分光分析である。
【図5A】図5Aは、以下の第5.3.2節に記載の化学的/生物学的精製法を用いて精製したp-GlcNAcのNMR分析である。表は、ピーク振幅、面積、及び基準対照に対する比、ならびにピークの全面積の比を示す。
【図5B】図5Bは、以下の第5.3.2節に記載の化学的/生物学的精製法を用いて精製したp-GlcNAcのNMR分析である。グラフはピークの全面積の比を表す。
【図6A】図6Aは、以下の5.3.1節に記載の機械力精製方法により調製したp-GlcNAc膜の透過型電子顕微鏡(TEM)写真である。拡大率:(図6A)、4190 x。
【図6B】図6Bは、以下の5.3.1節に記載の機械力精製方法により調製したp-GlcNAc膜の透過型電子顕微鏡(TEM)写真である。拡大率:(図6B)、16,250 x。
【図7A】図7Aは、以下の5.3.2節に記載の化学的/生物学的精製法の考察に記載のHF処理によるp-GlcNAc膜の透過型電子顕微鏡(TEM)写真である。拡大率:(図7A)、5270 x。
【図7B】図7Bは、以下の5.3.2節に記載の化学的/生物学的精製法の考察に記載のHF処理によるp-GlcNAc膜の透過型電子顕微鏡(TEM)写真である。拡大率:(図7B)、8150 x。
【図8A】図8Aは、以下の5.3.2節に記載の化学的/生物学的精製法の酸処理/中和化変法により調製したp-GlcNAc膜の透過型電子顕微鏡写真(TEM)である。拡大率:(図8A)、5270 x。
【図8B】図8Bは、以下の5.3.2節に記載の化学的/生物学的精製法の酸処理/中和化変法により調製したp-GlcNAc膜の透過型電子顕微鏡写真(TEM)である。拡大率:(図8B)、16,700 x。
【図9A】図9Aは、以下の5.3.2節に記載の化学的/生物学的精製法の酸処理/中和化変法により調製したp-GlcNAc膜の走査電子顕微鏡写真である。拡大率:200 x。
【図9B】図9Bは、以下の5.3.2節に記載の化学的/生物学的精製法の酸処理/中和化変法により調製したp-GlcNAc膜の走査電子顕微鏡写真である。拡大率:1000 x。
【図9C】図9Cは、以下の5.3.2節に記載の化学的/生物学的精製法の酸処理/中和化変法により調製したp-GlcNAc膜の走査電子顕微鏡写真である。拡大率:5000 x。
【図9D】図9Dは、以下の5.3.2節に記載の化学的/生物学的精製法の酸処理/中和化変法により調製したp-GlcNAc膜の走査電子顕微鏡写真である。拡大率:10,000 x。
【図9E】図9Eは、以下の5.3.2節に記載の化学的/生物学的精製法の酸処理/中和化変法により調製したp-GlcNAc膜の走査電子顕微鏡写真である。拡大率:20,000 x。
【図10A】図10Aは、純粋なp-GlcNAc膜の走査電子顕微鏡写真であり、この純粋なp-GlcNAc膜は、最初に以下の5.3節に記載の細胞溶解/中和化精製法を用いて生産し、ジメチルアセトアミド/塩化リチウムに溶解し、そして以下5.5節に記載のようにH2O中でマットに再沈降した物質から作った。拡大率:(図10A)、1000 x。
【図10B】図10Bは、純粋なp-GlcNAc膜の走査電子顕微鏡写真であり、この純粋なp-GlcNAc膜は、最初に以下の5.3節に記載の細胞溶解/中和化精製法を用いて生産し、ジメチルアセトアミド/塩化リチウムに溶解し、そして以下5.5節に記載のようにH2O中でマットに再沈降した物質から作った。拡大率:(図10B)、10,000 x。
【図11A】図11Aは、脱アセチル化p-GlcNAcマットの走査電子顕微鏡写真である。拡大率:(図11A)、1000 x。
【図11B】図11Bは、脱アセチル化p-GlcNAcマットの走査電子顕微鏡写真である。拡大率: (図11B)、10,000 x。
【図12A】図12Aは珪藻の写真である。珪藻細胞体から伸張するp-GlcNAc繊維に注目すること。
【図12B】図12Bは珪藻の写真である。珪藻細胞体から伸張するp-GlcNAc繊維に注目すること。
【図13】図13は、いくつかの可能なp-GlcNAc及びp-GlcNAcの脱アセチル化誘導体出発物質を示す図である(S. Hirano, 「日本におけるキチン及びキトサンの生産と応用("Production and Application of Chitin and Chitosan in Japan")」, in "Chitin and Chitosan," 1989, Skjak-Braek, Anthonsen, and Sanford, eds. Elsevier Science Publishing Co., pp. 37-43.から引用)。
【図14】図14は、それぞれの炭素原子に対する面積を得て、次いでCH3(面積)対C原子(面積)比を計算するために用いた、変換NMRデータ曲線である。
【図15】図15は、典型的なp-GlcNAc C13-NMRスペクトルである。個々のピークは、分子中のそれぞれのユニークな炭素原子のスペクトルへの寄与を表す。
【図16】図16は、CH3(面積)対C原子(面積)比を計算した値を表す変換NMRスペクトルデータである。上側:データのグラフ表示;下側:データ数値。
【図17A】p-GlcNAcマトリックスを蒸留水中で作製した。拡大率:200 x。これらの図のそれぞれに200ミクロンのスケールマーキングを示した。
【図17B】p-GlcNAcマトリックスを10%メタノールを含む蒸留水中で作製した。拡大率:200 x。これらの図のそれぞれに200ミクロンのスケールマーキングを示した。
【図17C】p-GlcNAcマトリックスを25%メタノールを含む蒸留水中で作製した。拡大率:200 x。これらの図のそれぞれに200ミクロンのスケールマーキングを示した。
【図17D】p-GlcNAcマトリックスを蒸留水中で作製した。拡大率:200 x。これらの図のそれぞれに200ミクロンのスケールマーキングを示した。
【図17E】p-GlcNAcマトリックスを10%エタノールを含む蒸留水中で作製した。拡大率:200 x。これらの図のそれぞれに200ミクロンのスケールマーキングを示した。
【図17F】p-GlcNAcマトリックスを25%エタノールを含む蒸留水中で作製した。拡大率:200 x。これらの図のそれぞれに200ミクロンのスケールマーキングを示した。
【図17G】p-GlcNAcマトリックスを40%エタノールを含む蒸留水中で作製した。拡大率:200 x。これらの図のそれぞれに200ミクロンのスケールマーキングを示した。
【図18】図18は、以下の第18.1節に記載の方法を用いて得た典型的な標準曲線である。このような標準曲線を、以下の第18.1節に同じく記載したリゾチーム-キチナーゼアッセイに用いた。
【図19】図19は、p-GlcNAcリゾチーム消化データである。ここに掲げたグラフは、p-GlcNAc膜がリゾチームにより消化される時間を通してのN-アセチルグルコサミンの蓄積を示す。グラフは、完全にアセチル化されたp-GlcNAcの分解速度を、部分的に(50%)脱アセチル化されたp-GlcNAcと比較し、部分的に脱アセチル化されたp-GlcNAcの分解速度が、完全にアセチル化されたp-GlcNAc物質のそれより有意に高いことを実証する。
【図20】図20は、p-GlcNAcリゾチーム消化データである。ここに掲げたグラフは、p-GlcNAc膜がリゾチームにより消化される時間を通してのN-アセチルグルコサミンの蓄積を示す。グラフは、2つの部分的に脱アセチル化されたp-GlcNAc膜(特に25%と50%脱アセチル化p-GlcNAc膜)を比較する。本データは、脱アセチル化のパーセントが増加すると分解速度が増加し、50%脱アセチル化p-GlcNAc膜の分解速度は25%脱アセチル化p-GlcNAc膜の分解速度より有意に高いことを実証する。
【図21A】p-GlcNAcのin vivo生分解性データである。第18.1節に記載のプロトタイプ1(完全にアセチル化されたp-GlcNAc)膜を腹部移植したラットを示す。図21Aは、移植0日のラットを示す。
【図21B】p-GlcNAcのin vivo生分解性データである。第18.1節に記載のプロトタイプ1(完全にアセチル化されたp-GlcNAc)膜を腹部移植したラットを示す。図21Bは、移植後14日のラットを示す。
【図21C】p-GlcNAcのin vivo生分解性データである。第18.1節に記載のプロトタイプ1(完全にアセチル化されたp-GlcNAc)膜を腹部移植したラットを示す。図21Cは、移植後21日のラットを示す。
【図21D】p-GlcNAcのin vivo生分解性データである。第18.1節に記載のプロトタイプ3A(凍結乾燥して部分的に脱アセチル化されたp-GlcNAc膜)を腹部移植したラットを示す。図21Dは、移植0日のラットを示す。
【図21E】p-GlcNAcのin vivo生分解性データである。第18.1節に記載のプロトタイプ3A(凍結乾燥して部分的に脱アセチル化されたp-GlcNAc膜)を腹部移植したラットを示す。図21Eは移植後14日のラットを示す。
【図22】図22A-22Bは、無傷内皮層の有る(図22A)、または内皮層除去後の(図22B)単離された大動脈環のp-GlcNAcによる用量依存的血管収縮である。無傷内皮層有りまたは無しで試験したp-GlcNAcの各濃度の報告値を得るために平均した収縮測定数を、図中の試験したp-GlcNAcの各濃度上に示した。
【図23】図23A-Eは、p-GlcNAcによる動脈血管収縮である。図23(A)は、動脈の片側にガーゼ包帯を適用した60分後に得たブタ動脈の断面図を示す。図23(B)は、動脈の片側にp-GlcNAc膜を適用した15分後に得たブタ動脈の断面図を示す。図23(C)は、動脈の片側にp-GlcNAc膜を適用した60分後に得たブタ動脈の断面図を示す。図23(D)は、動脈の片側にフィブリンコートしたコラーゲン包帯を適用した15分後に得たブタ動脈の断面図を示す。図23(E)は、動脈の片側にフィブリンコートしたコラーゲン包帯を適用した60分後に得たブタ動脈の断面図を示す。
【図24】図24は、p-GlcNAcによる動脈血管収縮である。図24は、示したように15または60分間、試験物質と直接接触した(1)、または接触しなかった(2)、ブタ動脈壁の厚さを記す。動脈の片側に適用した物質は、(A)ガーゼ包帯;(B)及び(C)p-GlcNAc膜;(D)及び(E)フィブリンコートしたコラーゲン包帯であった。
【発明を実施するための形態】
【0024】
5.発明の詳細な説明
本発明は、例えば、(1)エンドセリン-1放出の刺激、(2)血管収縮、及び(3)破裂した血管からの血液流出の減少により血管構造及び/または機能の一過性で局所性のモジュレーションを達成するのに有用であり、半結晶ポリβ-1→4-N-アセチルグルコサミン(p-GlcNAc)多糖ポリマーを含む組成物及び物質の局所投与を含んでなる、組成物ならびに方法に関する。標的組織におけるエンドセリン-1放出の刺激、血管収縮、及び破裂した血管からの血液流出の減少は、標的組織への本発明の物質の直接適用により、あるいは標的組織に隣接もしくは近接する皮膚またはその他の器官もしくは組織表面へのこれらの物質の適用により達成することができる。
【0025】
本発明は、従って、出血の停止を助けるかまたは直接達成する組成物及び方法にも関する。半結晶ポリβ-1→4 N-アセチルグルコサミンポリマーを含む本発明の物質の投与は、エンドセリン-1放出の刺激、血管収縮、及び破裂した血管における血液流出の減少をもたらす。
【0026】
これらの生理学的応答は、個々に及び/または共同して、毛細管、静脈、もしくは動脈であってもよい出血の停止を助けるかまたは直接達成する。特定の理論もしくは機構に束縛されることは欲しないが、かかる停止は凝血に依存しない方法で起こると考えられる。さらに、本発明の組成物及び方法を用いる出血の停止の達成はまた、血液凝固を促進する物理的バリヤーまたは力学的マトリックスの形成に依存するものでもない。すなわち、本発明によれば、本物質は、適用部位に接着して傷の境界を塞ぐ力学的マトリックスを与えるバリヤー形成物質である必要はない。対照的に、本発明の組成物及び方法は血管構造及び/または機能の一過性で局所性の変化を誘導し、そしてこの変化が血液凝固に依存しないで、それ自身、出血の停止を助けるかまたは直接達成するのである。
【0027】
さらに、本発明の組成物及び方法に係る好ましい物質は、完全にアセチル化された半結晶ポリβ-1→4 N-アセチルグルコサミンポリマーを含むものである。なぜなら、後掲の第16及び17節に記載の実施例ならびに図22に実証するように、70%脱アセチル化されたポリβ-1→4 N-アセチルグルコサミンポリマーを含む物質は決して血管収縮を誘導しないし、従って血管の管腔を減少しないし、それ故に、破裂した血管からの血液流出を減少しないからである。
【0028】
本発明は、部分的に、バリヤー形成物質である必要のない半結晶ポリβ-1→4-N-アセチルグルコサミン(p-GlcNAc)ポリマーを含む物質を局所に適用すると、単離したスプラグ・ドーリー(Sprague-Dawley)ラット動脈環の血管収縮が誘導されるという本出願人らの発見に基づく。この血液を含まない系において、完全にアセチル化されたポリβ-1→4-N-アセチルグルコサミンは、単離した動脈環の収縮を濃度依存的に誘導した。後掲の第17節に記載した実施例において、得られる血管収縮の程度は、単離した動脈環に適用したp-GlcNAcの濃度に実質的に比例した。対照的に、70%脱アセチル化されたポリβ-1→4-N-アセチルグルコサミンは、試験したいずれの濃度においても、単離した動脈環の血管収縮を誘導しなかった。
【0029】
本発明はまた、半結晶ポリβ-1→4 N-アセチルグルコサミンポリマーから作った膜を動脈の実験傷へin vivo適用すると、動脈組織と適用した膜の間の接触部位において即効性の血管収縮が刺激されたという本出願人らの発見にも部分的に基づいている。処置した組織を組織学的に分析すると、動脈収縮は、前記膜を適用した側の方が動脈の反対側より大きいことが示された。さらに、これらの組織サンプル免疫化学分析すると、エンドセリン-1放出に濃度勾配が存在すること、すなわち、エンドセリン-1放出の刺激が局所的な生理学的応答であることが示された。エンドセリン-1放出の刺激の程度は、半結晶ポリβ-1→4 N-アセチルグルコサミンポリマーを含有する膜が接触する表面において最大であり、隣接組織中に広がるものの、接触表面からの距離が増加するとその程度は減少した。類似の局在するエンドセリン-1放出の刺激は、半結晶ポリβ-1→4-N-アセチルグルコサミンを含む物質と接触する脾臓組織において観察された。
【0030】
本発明の方法は、一過性で局所性の(1)エンドセリン-1放出の亢進、(2)血管収縮、及び/または(3)破裂した血管からの血液流出の減少を達成する目的で、患者への治療上有効な形態のかつ治療上有効な量の半結晶ポリβ-1→4 N-アセチルグルコサミンポリマーを含む物質の局所投与を含む。
【0031】
以下に報じるのは、最初に、精製p-GlcNAc出発物質及びその再製剤(reformulation)の物理的特性の説明である。次に、肉微小藻類、好ましくは珪藻である出発供給源からp-GlcNAc出発物質を精製する方法を説明する。第3に、p-GlcNAcの再製剤、及びかかる再製剤を生産する方法を報じる。最後に、出発物質であるp-GlcNAc、p-GlcNAc誘導体及び/または出発物質のp-GlcNAc再製剤の用途を報じる。
【0032】
5.1. p-GlcNAc
p-GlcNAc出発物質は、本明細書に記載の技術、ならびにそのそれぞれが参照により本明細書にその全文が組み入れられる米国特許第5,686,115号、第5,624,679号、第5,623,064号、及び第5,622,834号に与えられた教示を利用して作ることができる。本明細書に使用されるp-GlcNAcポリマーは、約50〜約150,000個のN-アセチルグルコサミン単糖(図1)を含む。化学的及び物理的判定基準により確認すると、p-GlcNAc出発物質の純度は非常に高い。これらのなかには、化学組成物及び非多糖不純物がある。第1に、共に第5.3節に記載された2つの異なる精製方法を利用して生産した、p-GlcNAcの化学組成データを、以下の表Iに示した。見ればわかるように、両方の方法により生産したp-GlcNAcの化学組成は、実験誤差範囲内で、p-GlcNAcの化学式組成と同じである。第2に、表Iでもわかるように、生産されたp-GlcNAcは検出可能なタンパク質不純物を含まず、遊離アミノ酸などの他の有機不純物を実質的に含まず、そして灰及び金属イオンなどの無機不純物を実質的に含まない(p-GlcNAc出発物質は純粋なp-GlcNAcに対する炭素、水素、窒素及び酸素の理論値から2%以下の偏差がありうる)。従って、本明細書に使用される用語「有機不純物を実質的に含まない」及び「無機不純物を実質的に含まない」は、理論値から約2%以下の偏差の炭素、水素、窒素及び酸素のプロフィールを有するp-GlcNAcの組成物、そして好ましくはp-GlcNAc出発物質が表Iのp-GlcNAcマットの実験データに例示したプロフィール(該パーセント偏差を許容する)を有することを意味する。さらに、p-GlcNAc出発物質は束縛水のパーセントが低い。
【表1】
純粋なp-GlcNAc出発物質は、図2に示したのと実質的に同様な炭水化物分析プロフィールを示した。純粋なp-GlcNAc出発物質の一次単糖はN-アセチルグルコサミンである。さらに、純粋なp-GlcNAc出発物質は単糖グルコサミンを含有しない。
【0033】
p-GlcNAc出発物質の円二色性(CD)及び鋭い赤外スペクトル(IR)を、図3A、ならびに図4A、4D、及び4E、にそれぞれ示したが、これらは以下の第5.3節に記載の方法を用いて作った物質の分析である。かかる物理的データは、p-GlcNAc出発物質が高純度かつ半結晶であることを実証する。用語「半結晶」は、物質の高度に規則的な性質を意味する。当業者であれば、本発明のp-GlcNAcポリマーの赤外スペクトルに観察された、鋭く、よく分解されたピークは試験した物質の高度に規則的な結晶的性質(すなわち、「半結晶性」)を反映することを容易に理解するであろう。当業者はまた、例えば図4B及び4Cに挙げたようなIRスペクトルのブロードの、分解不十分なピークは、半結晶性の消失もしくは不足を示すことも理解するであろう。CD及びIRデータを得るために利用した方法は、以下の第6節に記載する実施例で説明する。
【0034】
純粋なp-GlcNAc出発物質のNMR分析は、図5A、14、15及び16に見られるのと実質的に類似したパターンを表す。かかるNMRパターンは、完全にアセチル化されたポリマーであるp-GlcNAc出発物質に一致するデータを示すだけでなく、p-GlcNAc種内に不純物の有機物質が存在しないことも実証する。以下の第5.3節に記載の方法を用いて生産しかつ以下の第8及び9節に記載する実施例で実証したp-GlcNAc出発物質の電子顕微鏡構造を、図6から図9Eにわたって示した。
【0035】
p-GlcNAc出発物質は高度の生体適合性を示す。生体適合性は、様々な技術により決定することができ、その技術は、限定されるものでないが、溶出試験、筋肉内移植、または動物被験者への皮内または全身注射などが挙げられる。簡単に説明すると、溶出試験(米国薬局方(U.S. Pharmacopeia) XXII, 1990, pp. 1415-1497;米国薬局方 XXII, 1991, 追補(Supplement)5, pp.2702-2703)は、試験材(test article)抽出物の生体適合性を評価するために設計されたものであり、抽出可能な細胞傷害性材に感受性がある哺乳動物細胞培養系(例えば、L929細胞系など)の試験材に応答する生物学的反応性をアッセイする。以下の第10節に記載する実施例はp-GlcNAc出発物質の高い生体適合性を実証する。
【0036】
5.2. p-GlcNAcの肉微小藻類供給源を生産する方法
5.2.1. p-GlcNAcの肉微小藻類供給源
p-GlcNAc出発物質は、肉微小藻類、好ましくは珪藻により生産し、かつそれから精製することができる。複数属の珪藻及びかかる属内の多数の種をp-GlcNAc出発供給源として利用することができる。これらの珪藻はそれぞれp-GlcNAcを生産する。かかる珪藻の写真は図12A-Bを参照すること。p-GlcNAc出発物質を生産する出発供給源として利用しうる珪藻は、限定されるものでないが、コアミケイソウ(Coscinodiscus)属、タイコケイソウ(Cyclotella)属、及びミカドケイソウ(Thalassiosira)属のメンバーが挙げられ、ミカドケイソウ(Thalassiosira)属が好ましい。
【0037】
コアミケイソウ(Coscinodiscus)属中で、p-GlcNAc出発物質を生産するために利用できる珪藻の種は、限定されるものでないが、concinnus及びradiatus種が挙げられる。タイコケイソウ(Cyclotella)属の利用しうる珪藻は、限定されるものでないが、caspia、cryptica、及びmeneghiniana種が挙げられる。p-GlcNAc出発物質を生産するために利用しうるミカドケイソウ(Thalassiosira)属珪藻は、限定されるものでないが、nitzschoides、aestivalis、antarctica、deciphens、eccentrica、floridana、fluviatilis、gravida、guillardii、hyalina、minima、nordenskioldii、oceanica、polychorda、pseudonana;rotula、tubifera、rumida、及びweissflogii種が挙げられ、fluviatilis及びweissflogii種が好ましい。
【0038】
上記の珪藻は、例えば、Bigelow海洋科学研究所の海洋植物プランクトンコレクションセンター(the Bigelow Laboratory for Ocean Sciences, Center for Collection of Marine Phytoplankton)(McKown Point, West Boothbay Harbor, Me., 04575)の培養コレクションから入手することができる。
【0039】
5.2.2. 珪藻類を増殖する方法
以上の第5.2.1節に記載のいずれの珪藻類も、例えば、本節に記載の方法を利用することにより、増殖させることができる。新しい珪藻培養は、無菌条件下で栄養培地(Nutrient Medium)に成熟珪藻培養物のアリコートを接種することにより開始する。栄養培地は全ての他の微生物を含んではならないので、栄養培地の調製に使用する水、有機成分、および無機成分を含む全ての物質は無菌でなければならない。さらに、この操作に関わる全ての工程、すなわち、全ての容器、ある容器から他の容器への物質の移動の全て、その他は、厳密な無菌条件のもとで実施することが必須である。一度に調製する栄養培地の量は、新しい培養を出発するために必要である量を超えてはならない。例えば、ほぼ1平方フィートの表面を占めるフェムバッハ(Fembach)フラスコを珪藻培養用の容器として使用してもよく、かかる容器は珪藻生物の最適な増殖のために1リットルの栄養培地を必要とする。
【0040】
栄養培地の調製は次の操作からなる:
a)海水の取得及び処理
b)脱イオン蒸留水の調製
c)一次栄養ストックの調製
d)栄養作業ストックの調製
e)最終栄養培地の調製
濾過海水は、例えば、海洋生物学研究所(Marine Biology Laboratory)(Woods Hole, Mass.)から入手できる。海水容器は5℃(±2℃)で保存しなければならない。必要があれば、所要容積の水を0.8ミクロン孔サイズのSupor-800ポリエーテルスルホンフィルター膜(Gelman, Inc.)を用いてブーフナー(Buchner)濾過ユニットを通して濾過してもよい。次いで、海水を例えば、121℃にて1リットル当たり少なくとも約15分間、オートクレーブ処理して無菌化する。無菌化プロセスが完了すると、栓をしたフラスコを、好ましくは、溶液をほぼ5℃(±2℃)の温度に到達させうる低温室へ移して即時冷却する。使用するときに、溶液を室温にあげる。
【0041】
水道水は、標準の設備と方法を用いて、蒸留しかつ脱イオンして回収し、そして清浄な密栓をした、好ましくはガラス製の容器に入れて保存する。
【0042】
以下に掲げるのは、栄養培地の調製に必要なストック溶液を調製するのに従いうる処方である。かかる処方は手引きとして利用されるが、かかる処方の日常的変法であって本明細書に記載のp-GlcNAc調製方法のために十分な肉微小藻類珪藻の増殖を持続しうる栄養培地の調製を助ける前記変法も、本発明の範囲内にあると意図することは理解されるべきである。
【0043】
I. 微量金属一次ストック(TMPS)
a. 39mM CuSO4.5H2O(硫酸銅[II]五水和物)(9.8g 硫酸銅[II]/L)
b. 7.5mM ZnSO4.7H2O(硫酸亜鉛七水和物)(22g 硫酸亜鉛/L)
c. 42mM CoCl2.6H2O(塩化コバルト[II]六水和物)(10g 塩化コバルト[II]/L)
d. 91mM MnCl2.4H2O(塩化マンガン[II]四水和物)(18g 塩化マンガン[II]/L)
e. 26mM NaMoO4.2H2O(モリブデン酸ナトリウム二水和物)(6.3g モリブデン酸ナトリウム/L)
f. 1mM H2SeO3(亜セレン酸)(0.129g 亜セレン酸/L)
0.2ミクロン孔サイズ以下のフィルターを用いて各栄養を滅菌濾過する。
【0044】
II. ビタミン一次ストック(VPS)
a. 1mg/ml ビタミンB12
b. 0.1mg/ml ビオチン
0.2ミクロン孔サイズ以下のフィルターを用いて両方のストックを滅菌濾過する。
【0045】
III. ナトリウム塩作業ストック(SSWS)
a. 硝酸ナトリウム作業ストック:0.88M(75g NaNO3/L)
b. 一塩基性リン酸ナトリウム一水和物作業ストック:36.2mM NaH2PO4・H2O(5g NaH2PO4・H2O/L)。メタケイ酸ナトリウム一水和物作業ストック:0.11M Na2SiO3・9H2O(30g Na2SiO3・9H2O/L)。
【0046】
0.2ミクロン孔サイズ以下のフィルターを用いてそれぞれのSSWSを滅菌濾過する。
【0047】
IV. 微量金属作業ストック(TMWS)
11.7mM Na2EDTA(エチレンジアミン四酢酸、二ナトリウム塩二水和物)(4.36 g/L)
11.7mM FeCl3・6H2O(塩化鉄[III]六水和物)(3.15 g/L)
先に掲げた6つの微量金属一次ストックのそれぞれの1ml/L。
【0048】
0.2ミクロン孔サイズ以下のフィルターを用いて滅菌濾過する。微量金属作業ストックは毎週新しく調製しなければならないことに注意する。
【0049】
V. ビタミン作業ストック(VWS)
1.0mμg/ml ビオチン(1.0ml 一次ビオチンストック/100ml)
1.0mμg/ml ビタミンB12(0.1ml ビタミンB12一次ストック/100ml)
0.20mg/ml チアミンHCl(20mg チアミン塩酸塩/100ml)。
【0050】
0.2ミクロン孔サイズ以下のフィルターを用いて滅菌濾過する。ビタミン作業ストックは毎週新しく調製しなければならないことに注意する。
【0051】
以下に記載したのは、栄養培地の調製及び珪藻培養のために従いうる技術である。これらの技術に加えて、本明細書に記載の調製方法として十分な珪藻増殖を持続できる栄養培地及び方法をもたらす本明細書に記載の処方及び/または操作のいずれの日常的変法も本発明の範囲内にあると意図することは理解されるべきである。
【0052】
栄養培地は、例えば、次のように調製することができる:濾過して無菌化した海水の各1リットルにNaNO3作業ストック1ml、NaH2PO4・H2O作業ストック1ml、微量金属作業ストック1ml、及びNa2SiO3・9H2O作業ストック1mlを加える。Na2SiO3・9H2Oの添加と同時に、1N HCl 2mlを加え、溶液を振とうして混合してもよい。次に、1N NaOH 1.5mlを加え、溶液を再び振とうして混合してもよい。最後に、ビタミン作業ストック0.5 mlを加えてもよい。
【0053】
新しい珪藻培養物を増殖する目的で、成熟培養物7ml(約1 x 105〜約1 x 106 細胞/mlの範囲内の細胞密度を有する)を、上記の方法に従って調製することができる無菌栄養培地100mlを含有する無菌容器に移してもよい。次いで、接種した培養物を8日間、次の条件のもとでインキュベートしてもよい:
温度:20℃、常時照明、
攪拌:1日1回、フラスコの緩やかな旋回。
【0054】
8日間のインキュベーション後に、このインキュベートした培養物80mlを、無菌条件下で、例えば、チーズ布で覆った綿ウール栓により保護した2.8Lフェルンバッハ(Fernbach)フラスコ中に含有される栄養培地1000mlに移してもよい。かかる培養物は、インキュベートして所望の細胞密度まで増殖させるか、あるいは、新しい珪藻培養物を接種するために利用することができる。培養物が所望の細胞密度に到達すると、培養物のp-GlcNAc繊維を収穫し、以下の第5.3節に記載の方法を利用して、p-GlcNAc出発物質を精製することができる。
【0055】
培養pHをほぼ7〜8、好ましくはほぼ7.4に維持するために、CO2を培養溶液に溶解してもよい。かかる中性pH環境を維持することにより、それぞれの珪藻培養物から得られるp-GlcNAc収率が大きく増加する。
【0056】
5.3. p-GlcNAc繊維を単離、精製、及び濃縮する方法
本節で記載するのは、以上の第5.2節で記載した珪藻培養物からp-GlcNAc繊維を調製するために利用しうる方法である。
【0057】
一方、以下に記載の肉微小藻類、好ましくは珪藻出発供給源からp-GlcNAcを精製する方法はそれぞれ、非常に純粋な、不純物を含まない半結晶p-GlcNAcを生産するものである。例えば、p-GlcNAc出発物質は、以下の第5.3.1節に記載する機械力方法を経由して精製することができる。第2の方法は化学的/生物学的方法と呼ばれ、第5.3.2節に記載され、機械力方法により生産される平均p-GlcNAc収率よりさらに高い平均収率を生産する。さらに、以下の第5.3.2節の化学的/生物学的方法の一部として記載した酸処理/中和化変法は、極めて長いp-GlcNAc繊維を生産し、ある繊維は100μmを超えかつ20-30 x 106ダルトンの非常に高分子量のp-GlcNAcポリマー分子を含有する。p-GlcNAcポリマー出発物質の分子量測定は、限定されるものでないが、固有粘度の測定を含む、当業者であれば周知のクロマトグラフィ及び物理化学的方法を利用して行う。
【0058】
5.3.1. 純粋なp-GlcNAcを調製するための機械力方法
p-GlcNAc繊維は、培養内容物を適当な機械力で処理することにより珪藻細胞体から分離することができる。かかる機械力は、限定されるものでないが、例えばコロイドミル、超音波装置、もしくは泡発生器により発生させる剪断力、または、例えばウエアリング(Waring)ブレンダーにより発生させる切断力が挙げられる。
【0059】
得られる珪藻細胞体及びp-GlcNAc繊維の懸濁液を、次いで分離する。例えば、懸濁液を一連の遠心分離工程で処理して細胞体からp-GlcNAc繊維を分離し、目視しうる凝集沈殿物質は、もし若干あっても、僅かでしかない透明な上清を得る。固定角の回転子及び約10℃の温度が遠心分離工程に好ましい。所要の速度、時間、及び遠心分離工程の総数は、例えば、使用される特定の遠心分離回転子に依って変化しうるが、かかるパラメーター値の決定は、当業者には明らかであろう。
【0060】
上清中のp-GlcNAc繊維は、次いで、当業者に周知の技術を利用して濃縮することができる。かかる技術は、限定されるものでないが、吸引及び濾過装置が挙げられる。
【0061】
最後に、濃縮したp-GlcNAc繊維は、例えば、脱イオン蒸留水、HCl及びエタノール、または他の適当な溶媒、好ましくは有機と無機物質の両方を溶解するアルコールなどの溶媒を用いて洗浄する。
【0062】
以下の第7節に記載する実施例は、p-GlcNAcを精製するためのこの方法の使用を示す。
【0063】
5.3.2. p-GlcNAcを精製するための化学的/生物学的方法
この方法においては、以下にさらに詳細に記載する化学的及び/または生物学的作薬剤で処理することにより、珪藻細胞体からp-GlcNAc繊維を分離する。
【0064】
珪藻培養物を、珪藻細胞壁を弱化しうる化学品を用いて処理し、p-GlcNAc繊維をその長さ及び構造を改変することなく放出させることができる。かかる化学品は、限定されるものでないが、フッ化水素酸(HF)が挙げられる。あるいは、成熟珪藻培養物を、生物学的プロセスを改変しうる生物学的薬剤により処理してp-GlcNAc繊維合成を抑制し、そして既に存在する繊維を放出させてもよい。例えば、かかる薬剤としては、限定されるものでないが、酵素N-アセチルグルコサミニル-P-トランスフェラーゼのインヒビターであるポリオキシン-Dが挙げられる。
【0065】
上記の化学的または生物学的薬剤を用いて処理した珪藻培養物中の細胞体及びp-GlcNAcを含有する繊維を、次に分離する。例えば、処理した珪藻培養の内容物を静置し、培養内容物から2つの明確な層を形成させてもよい。上層は主にp-GlcNAc繊維を含有するのに対して、下層は細胞体を含有する。上側のp-GlcNAc繊維を含有する層をサイフォンで吸い出し、底層の沈降した細胞物質を残してもよい。
【0066】
サイフォンで吸い出したp-GlcNAc繊維を含有する層を、次いでさらにp-GlcNAc繊維を損傷しないであろう界面活性剤を用いる処理により精製し、タンパク質及びその他の望ましくない物質を除去する。かかる界面活性剤としては、限定されるものでないが、ドデシル硫酸ナトリウム(SDS)が挙げられる。
【0067】
HF処理などの酸処理を用いて珪藻細胞体からp-GlcNAc繊維を分離するときに、繊維を分散させる工程を含んでもよい。かかる工程としては、限定されるものでないが、繊維をオービタルシェイカー(orbital shaker)の運動で処理する工程などの繊維分散のための機械力の使用が挙げられる。
【0068】
あるいは、酸処理した懸濁液を、場合により、中和した後に、界面活性剤処理によりさらに精製してもよい。かかる中和は、一般的に、懸濁液のpHをほぼ1.8からほぼ7.0に変えることであり、例えば、適当な容積の1M Tris(pH 8.0)の添加または適当な容積の水酸化ナトリウム(NaOH)の添加により達成しうる。中和すると、一般的に、本明細書で考察した他の精製方法より実質的に長い、純粋なp-GlcNAc繊維が得られる。
【0069】
精製したp-GlcNAc繊維は、次いで、吸引及び濾過装置を利用するなどの、当業者に周知の技術を利用して濃縮することができる。最後に、p-GlcNAc繊維を、脱イオン蒸留水、HCl及びエタノールまたは他の適当な溶媒、好ましくは、有機及び無機物質の両方を溶解するアルコールなどの溶媒を用いる一連の工程において洗浄する。
【0070】
以下の第8節に記載する実施例は、かかる精製法の成功を収めた利用を実証する。
【0071】
p-GlcNAc出発物質またはその部分的に脱アセチル化された誘導体を、制御した加水分解条件で処理すると、均一な、明確な分子量と他の物理的特性を有する分子のグループが得られる。かかる加水分解条件としては、例えば、酵素、リゾチームによる処理が挙げられる。p-GlcNAcを時間を変えてリゾチームに曝し、加水分解の程度を制御してもよい。かかる酵素による部分的消化反応はまた、基質の濃度、もしくは酵素の濃度、または基質と酵素の両方の濃度、ならびにpH及び温度を変えることにより制御することができる。さらに、加水分解速度は、リゾチーム処理されるp-GlcNAcが脱アセチル化されている程度の関数として制御することができる。脱アセチル化条件は、本節の初めに記載されている。約20〜90脱アセチル化パーセントの間で、p-GlcNAc分子がより完全に脱アセチル化されているほど、その分子は所与の時間により完全に加水分解されるであろう。加水分解及び/または脱アセチル化処理により、分子量の低下に加えて、物理特性の変化が誘発されるであろう。加水分解/脱アセチル化操作の結果は以下の第9節の実施例に記載する。
【0072】
5.4. p-GlcNAcの誘導体化
純粋な、完全にアセチル化されたp-GlcNAc出発物質は、様々な制御された条件と方法を利用することにより、広範囲の異なる化合物に誘導体化することができる。これらの化合物のいくつかを記載した図13を参照すること。かかる誘導体化した化合物としては、限定されるものでないが、以下にさらに詳しく記載する化学的及び/または酵素的手法を経由して改変された、部分的に脱アセチル化されたp-GlcNAcが挙げられる。さらに、p-GlcNAcまたはその部分的に脱アセチル化された誘導体は、硫酸化、リン酸化、及び/または硝酸化することにより誘導体化することができる。さらに、以下に詳述したように、O-スルホニル、N-アシル、O-アルキル、N-アルキル、及びN-アルキリデン及びN-アリーリデンならびにその他の誘導体を、p-GlcNAcまたは部分的に脱アセチル化されたp-GlcNAc出発物質から調製することができる。部分的に脱アセチル化されたp-GlcNAc出発物質を利用して、様々な有機塩及び/または金属キレートを調製することもできる。さらに、p-GlcNAc出発物質、またはその誘導体に、共有結合もしくは非共有結合により、任意の様々な分子が結合していてもよい。その上さらに、p-GlcNAc出発物質またはその誘導体を、制御された加水分解条件で処理し、均一かつ明確な分子量特性を有する分子のグループを得てもよい。かかる物質は、ポリマーをIR吸収分光計で分析したときに、p-GlcNAcポリマーが鋭く明確なピークにより示されるその半結晶構造を保持する限り、本発明に有用である。
【0073】
p-GlcNAc出発物質の1以上の単糖単位が脱アセチル化され、部分的に脱アセチル化されたポリβ-1→4-N-アセチルグルコサミン種を形成することができる。脱アセチル化モノマーは、一般的に、ポリマー全体にわたり本質的に無作為に分布してもよいし、またはポリβ-1→4 N-アセチルグルコサミンポリマー内の不連続なサブ領域として比較的かたまっていてもよい。ポリβ-1→4-N-アセチルグルコサミン種出発物質の単糖単位の一部分が脱アセチル化されているポリβ-1→4-N-グルコサミン種出発物質は、約30 x 106ダルトン以下の分子量を有し、β-1→4-Nコンフィギュレーションで共有結合した約150,000個のグルコサミン多糖を含む。一実施形態においては、ポリβ-1→4-N-グルコサミン種のグルコサミン単糖の少なくとも約90%はアセチル化されたまま残存し、一方、他の実施形態においては、ポリβ-1→4-N-グルコサミン種の単糖単位の少なくとも約80%、70%、60%、50%、または40%がアセチル化されたまま残存する。ただし、以下の実施例6に記載し、かつ図4B及び4Cに示した非結晶p-GlcNAcポリマーによって示されるIR吸収スペクトルと比較して図4A、4D、及び4Eに示したように、ポリマーをIR吸収分光計で分析したときに、部分的に脱アセチル化されたポリβ-1→4 N-アセチルグルコサミンポリマーは鋭く明確なピークにより示されるその半結晶構造を保持する。
【0074】
p-GlcNAc出発物質を塩基による処理で脱アセチル化し、遊離アミノ基をもつグルコサミンを得ることができる。この加水分解プロセスは、濃い水酸化ナトリウムまたは水酸化カリウム溶液を用いて高温で実施してもよい。しかし、脱アセチル化の程度を正確に制御するため及び多糖分子の炭水化物主鎖の分解を避けるため、キチンデアセチラーゼ酵素を使用する酵素手法を用いてp-GlcNAcを脱アセチル化するのが好ましい。かかるデアセチラーゼ酵素手法は当業者に周知であって、本明細書に参照によりその全文が組み入れられる(米国特許第5,219,749号)に従って実施することができる。
【0075】
p-GlcNAc出発物質の1以上の単糖単位は誘導体化されて、以下に記載のように、少なくとも1つの硫酸基を含有させるか、リン酸化もしくはニトロ化されていてもよい:
【化1】
[式中、Rおよび/またはR1は、水素の代わりに、及び/またはR2は、-COCH3の代わりに硫酸(-SHO3)、リン酸(-P(OH)2)、または硝酸(-NO2)基であってもよい]。
【0076】
以下に記載したのは、かかるp-GlcNAc誘導体を調製しうる方法である。本節に記載の方法を実施する前に、p-GlcNAc出発物質を最初に凍結乾燥し、液体窒素中で凍結し、そして粉末化することが有利でありうる。
【0077】
硫酸化p-GlcNAc誘導体は、例えば、2工程プロセスにより作製することができる。第1工程において、O-カルボキシメチルp-GlcNAcを、出発物質のp-GlcNAcおよび/またはp-GlcNAc誘導体から、例えば、Tokuraら(Tokura, S.ら, 1983, Polym. J. 15:485)が記載した技術を利用して調製することができる。第2に、硫酸化工程を、例えば、Schweiger(Schweiger, R. G., 1972, Carbohydrate Res. 21:219)が記載したような当業者に周知の技術に従い、N,N-ジメチル-ホルムアミド-三酸化硫黄を用いて実施することができる。得られた生成物は、ナトリウム塩として単離することができる。出発物質のリン酸化p-GlcNAc誘導体は、例えば、Nishiら(Nishi, N.ら, 1986, in "Chitin and Chitosan," Muzzarelliら, 編, Plenum Press, New York, pp. 297-299)が記載した当業者に周知の技術を利用することにより調製することができる。簡単に説明すると、p-GlcNAc/メタンスルホン酸混合物を、攪拌しながら約0℃〜5℃の温度にて五酸化リン(ほぼ0.5〜4.0モル当量)を用いて処理してもよい。処理は約2時間である。得られる生成物を次いで、当業者が周知する標準技術を用いて、沈殿させ洗浄してもよい。例えば、サンプルをエーテルなどの溶媒を用いて沈殿させ、遠心分離し、エーテル、アセトン、またはメタノールなどの溶媒を用いて洗浄し、そして乾燥してもよい。
【0078】
ニトロ化p-GlcNAc誘導体は、Schorigin及びHalt(Schorigin, R.及びHalt, E., 1934, Chem. Ber. 67:1712)が記載した当業者に周知の技術を利用して調製することができる。簡単に説明すると、p-GlcNAc及び/またはp-GlcNAc誘導体を濃硝酸を用いて処理して安定なニトロ化生成物を形成させる。
【0079】
p-GlcNAc出発物質の1以上の単糖単位は、以下に記載のように、スルホニル基を含有しうる:
【化2】
[式中、R3はアルキル、アリール、アルケニル、またはアルキニル部分であってもよい]。かかる誘導体は、Kuritaら(Kurita, K. ら, 1990, Polym. Prep (Am. Chem. Soc., Div. Polym. Chem.) 31:624-625)が記載した周知の方法により作製することができる。簡単に説明すれば、p-GlcNAcアルカリ水溶液を塩化トシルのクロロホルム溶液と反応させ、次いで低温で円滑に反応を進行させる。
【0080】
p-GlcNAc出発物質またはその脱アセチル化された誘導体の1以上の単糖は、1以上の以下に記載のO-アシル基を含有してもよい:
【化3】
[式中、R4及び/またはR5は、水素の代わりに、アルキル、アルケニル、またはアルキニル部分であってもよく、そしてR6はアルキル、アルケニル、またはアルキニル部分であってもよい]。かかる誘導体の例は、Komai(Komai, T.ら, 1986, in "Chitin and Chitosan,", Muzzarelliら, 編, Plenum Press, New York, pp. 497-506)が記載したような周知の方法により作製することができる。簡単に説明すると、p-GlcNAcを、メタンスルホン酸中で複数の適当な塩化アシルと反応させ、限定されるものでないが、カプロイル、カプリル、ラノイル、またはベンゾイル誘導体を含むp-GlcNAc誘導体を得ることができる。
【0081】
脱アセチル化p-GlcNAc出発物質の1以上の単糖は、以下に記載のN-アシル基を含有しうる:
【化4】
[式中、R7は、アルキル、アルケニル、またはアルキニル部分であってもよい]。かかる誘導体化は、Hiranoら(Hirano, S.ら, 1976, Carbohydrate Research 47: 315-320)が記載したような当業者に周知の技術を利用して得ることができる。
【0082】
脱アセチル化p-GlcNAcは、複数の有機酸水溶液に可溶である。選択したカルボン酸無水物を酢酸メタノール水溶液中のかかるp-GlcNAcを含有する溶液に加えると、N-アシルp-GlcNAc誘導体が形成される。
【0083】
脱アセチル化p-GlcNAc出発物質またはその脱アセチル化誘導体の1以上の単糖は、以下に記載のO-アルキル基を含有しうる:
【化5】
[式中、R8はアルキル、及びアルケニル、またはアルキニル部分であってもよい]。かかる誘導体化は、当業者が周知の技術を利用して得ることができる。例えば、Mareshら(Maresh, G.ら, in "Chitin and Chitosan," Skjak-Braek, G.ら, 編, 1989, Elsevier Publishing Co., pp. 389-395)が記載した方法が挙げられる。簡単に説明すると、脱アセチル化p-GlcNAcをジメトキシエタン(DME)中に分散させ、過剰のプロピレンオキシドと反応させる。反応時間は24時間であってもよく、反応はオートクレーブ中で40℃〜90℃にて行う。次いで混合物を水を用いて希釈し、濾過してもよい。DMEは蒸留により除去できる。最後に、最終生成物を凍結乾燥を経由して単離できる。
【0084】
p-GlcNAc出発物質の1以上の単糖単位は、以下に記載のアルカリ誘導体:
【化6】
であってもよい。かかる誘導体は、当業者に周知の技術により得ることができる。例えば、Noguchiら(Noguchi, J.ら, 1969, 工業化学雑誌 72:796-799)が記載したような方法を利用することができる。簡単に説明すると、p-GlcNAcを、真空下でNaOH(好ましくは43%)にほぼ2時間、0℃にて浸漬する。次いで過剰のNaOHを、例えば、かご型遠心分離機で遠心分離しかつ機械プレスにより除去してもよい。
【0085】
p-GlcNAc出発物質の脱アセチル化された誘導体の1以上の単糖単位は、以下に記載のN-アルキル基を含有してもよい:
【化7】
[式中、R9は、アルキル、アルケニル、またはアルキニル部分であってもよい]。かかる誘導体化は、例えば、O-アルキルp-GlcNAc誘導体の生成について上に記載した、Mareshら(Maresh, G.ら, in "Chitin and Chitosan," Skjak-Brack, G.ら, 編, 1989, Elsevier Publishing Co., pp.389-395)のような方法を利用して、得ることができる。
【0086】
p-GlcNAc出発物質の脱アセチル化された誘導体の1以上の単糖単位は、以下に記載の塩を形成してもよい:
【化8】
[式中、R11はアルキル、アルケニル、またはアルキニル部分であってもよい]。かかる誘導体化は、当業者が周知の技術を利用することにより得ることができる。例えば、Austin及びSennett(Austin, P. R.及びSennett, S., in "Chitin in Nature and Technology," 1986, Muzzarelli, R. A. A.ら, 編, Plenum Press, pp. 279-286)が記載したような方法を利用することができる。簡単に説明すると、脱アセチル化p-GlcNAcを例えば、酢酸エチルまたはイソプロパノールなどの有機媒質中に懸濁し、それに例えば、蟻酸、酢酸、グリコール酸、または乳酸などの適当な有機酸を加えてもよい。混合物をある時間(例えば、1〜3時間)静置してもよい。反応及び乾燥温度は約12℃〜約35℃で変化してもよく、20℃〜25℃が好ましい。次いで塩を濾過により分離し、新鮮な媒質を用いて洗浄し、そして残る媒質は蒸発させてもよい。
【0087】
p-GlcNAc出発物質の脱アセチル化された誘導体の1以上の単糖単位は、以下に記載の金属キレートを形成してもよい:
【化9】
[式中、R12は金属イオン、特に遷移金属の1つであってもよく、Xは脱アセチル化p-GlcNAc中に存在するアミノ及び置換アミノ基中に存在する窒素電子により確立された供与結合である]。
【0088】
p-GlcNAc出発物質の脱アセチル化された誘導体の1以上の単糖単位は、以下に記載のN-アルキリデンまたはN-アリーリデン基を含有してもよい:
【化10】
[式中、R13は、アルキル、アルケニル、アルキニル、またはアリール部分であってもよい]。かかる誘導体化は、当業者に周知の技術を利用して得ることができる。例えば、Hiranoら(Hirano, S.ら, 1981, J. Biomed. Mat. Res. 15:903-911)が記載した方法を利用してもよい。簡単に説明すると、脱アセチル化p-GlcNAcのN-置換反応を、カルボン酸無水物及び/またはアリールアルデヒドを用いて実施し、アシル-及び/またはアリーリデン誘導体を得てもよい。
【0089】
さらに、p-GlcNAc出発物質、またはその部分的に脱アセチル化された誘導体を、制御された加水分解条件で処理し、均一で明確な分子量及びその他の物理的特性を有する分子のグループを得ることができる。かかる加水分解条件は、例えば、酵素、リゾチームによる処理が挙げられる。p-GlcNAcを、加水分解の程度を制御するために、時間を変えて、リゾチームに曝してもよい。さらに、加水分解速度は、リゾチーム処理されるp-GlcNAcの脱アセチル化されている程度の関数として制御してもよい(例えば、第15節に提供し、かつ図18-20に示した実施例を参照)。かかる酵素による部分消化反応はまた、基質、酵素、または基質と酵素の両方の濃度、ならびにpH及び温度を変化させることにより制御することができる。他の実施形態においては、p-GlcNAcポリマーのサイズを超音波処理により小さくする。そのサイズは、使用する機器の動力だけでなくサンプルのpH、塩濃度、及び温度によって変化させることができる。p-GlcNAcもしくはその誘導体の可溶化は、第5.5節に記載した。従って、1以上のこれらの方法を単独でまたはお互いに組み合わせて利用することにより、より高分子量のp-GlcNAcポリマーを加水分解してより小さい断片とし、それを、例えばカラムクロマトグラフィを用いて、サイズに従ってクロマトグラフィ的に分離することができる。
【0090】
例えば、当業者であれば、p-GlcNAcの部分消化の程度を変えて所望の分子量範囲を有する反応生成物を提供するであろう。他の実施形態においては、リゾチームによる部分消化に使用する基質は、超音波処理されている及び/または部分的に脱アセチル化されているp-GlcNAcである。部分酵素消化を、カラムクロマトグラフィ、HPLC分離、または他の当技術分野で周知の技術及び方法などの分離技術と組み合わせることにより、当業者は狭い分子量分布範囲をもつ消化生成物を単離することができる。さらに、一連の部分消化反応の生成物を組み合わせることにより、当業者は、半結晶p-GlcNAc生成物の広範囲の分子量種を有するp-GlcNAcポリマーを含んでなる組成物をアセンブルすることができる。前記組成物は、例えば本明細書に開示された集団、すなわち一実施形態においては約50〜約150,000モノマー単位を含むポリマー、ならびに約50〜約50,000、約50〜約10,000、及び約50〜約4,000モノマー単位を含むポリマーを含むものである。。
【0091】
脱アセチル化条件は、本節の初めに記載されている。p-GlcNAc分子は約20〜90脱アセチル化パーセントの間でより完全に脱アセチル化されているほど、その分子は所与の時間により完全に加水分解されうる。分子量の低下に加えて、加水分解及び/または脱アセチル化処理により、物理的特性の変化が誘発されうる。
【0092】
さらに、様々な分子をp-GlcNAc出発物質の脱アセチル化された誘導体と、共有結合または非共有結合により機能的に結合させてもよい。かかる分子としては、限定されるものでないが、神経増殖因子などの増殖因子、ペプシンなどのプロテアーゼ、ホルモンのようなポリペプチド、またはRGD配列などのペプチド認識配列、フィブロネクチン認識配列、ラミニン、インテグリン、細胞接着分子、その他が挙げられる。例えば、以下の第5.6.1.1節に考察した化合物を参照すること。分子と曝された脱アセチル化p-GlcNAcの第一級アミンとの共有結合は、例えば、特定の長さの化学スペーサーとして作用する二官能性架橋試薬を利用する化学結合により実施することができる。かかる技術は当業者には周知であり、例えば、Davis及びPreston(Davis, M.及びPreston, J. F. 1981, Anal. Biochem. 116:404-407)ならびにStarosら(Staros, J. V.ら, 1986, Anal. Biochem. 156:220-222)の方法に類似する。簡単に説明すると、脱アセチル化されたまたは部分的に脱アセチル化されたp-GlcNAc出発物質と結合するペプチドのカルボン酸残基を活性化し、次いでp-GlcNAcと架橋することができる。活性化は、例えば、カルボジイミドEDC(1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド)などの溶液をリン酸バッファー中のペプチド溶液へ加えることにより実施できる。好ましくは、この溶液は、さらに、カップリングを増強するスルホ-NHS(N-ヒドロキシスルホスクシンイミド)などの試薬を含有しうる。活性化ペプチドは、炭酸バッファー(pH 9.0-9.2)などの高pHバッファー中で混合することにより脱アセチル化p-GlcNAcと架橋することができる。
【0093】
結合したペプチド(またはいずれかの共有結合した分子)の生物学的活性は、分子をp-GlcNAc出発物質と結合するために利用するリンカー分子(例えば、二官能性架橋化合物)の長さを変えることにより、維持することができる。結合分子の生物学的活性を変えることのない、所与の結合分子に対する適当なリンカー長さは、日常的に確かめることができる。例えば、所与の長さのリンカーを経由して結合されている分子の生物学的活性(例えば、治療上有効なレベルの生物学的活性)は、所与の結合される分子に特異的な周知のアッセイを利用して試験することができる。
【0094】
さらに、結合している分子の生物学的活性を維持するために、適当な天然の酵素により切断されてペプチド(またはいずれかの共有結合した分子)を放出しうるリンカーを利用することが必要な場合もある。
【0095】
上記のように、当業者が通常使用するアッセイを利用して、結合している特定分子の保持する生物学的活性を試験し、許容しうる活性レベル(例えば治療上有効な活性レベル)を保証することができる。
【0096】
あるいは、上記の分子を当業者が周知の技術を利用してp-GlcNAc及びその誘導体と非共有結合により結合することができる。例えば、選択した1以上の分子を、p-GlcNAc懸濁液もしくは部分的に脱アセチル化されたp-GlcNAc溶液と、p-GlcNAc-乳酸溶液と、脱アセチル化されたもしくは部分的に脱アセチル化されたp-GlcNAc塩溶液と、またはいずれかのp-GlcNAc誘導体溶液と混合してもよい。次いで混合物を凍結乾燥してもよい。凍結乾燥の後、分子は、恐らく疎水的、静電的及び他の非共有結合の相互作用を経由してp-GlcNAcマトリックスと結合する。従って、かかるp-GlcNAc製剤は製造が容易である。さらに、かかる製剤は、広いスペクトルの物理的特性及び最も疎水性から最も親水性までの水溶解特性を有する広範囲の分子を用いて、効果的に製造することができる。分子の結合にあたっては、当業者が特定の非共有結合した分子の活性を試験するために通常使用するアッセイを利用して、結合した分子により許容しうる活性のレベル(例えば、治療上有効な活性)が達成されたことを確認することができる。
【0097】
あるいは、p-GlcNAc及び/またはp-GlcNAc誘導体を含むハイブリッドを形成することができる。かかるハイブリッドは、p-GlcNAc及び/またはp-GlcNAc 誘導体に加えて、複数の天然の及び/または合成の物質を含有してもよい。例えば、ハイブリッドは、p-GlcNAc及び/またはp-GlcNAc誘導体に1以上の細胞外マトリックス(ECM)成分を加えて作製してもよい。かかるECM成分としては、限定されるものでないが、コラーゲン、フィブロネクチン、グリコサミノグリカン及び/またはペプチドグリカンが挙げられる。ハイブリッドはまた、p-GlcNAc及び/またはp-GlcNAc誘導体に1以上の合成物質、例えばポリエチレンなどを加えて作製してもよい。かかるp-GlcNAc/ポリエチレンまたはp-GlcNAc誘導体/ポリエチレンハイブリッドは、ハイブリッド成分を例えば、オートクレーブ処理を経由して熱的に結合して作ることができる。かかるハイブリッドポリマーは、ハイブリッドポリマーが、以下の実施例6に記載のIR吸収分光計により分析したとき、鋭い、明確なピークにより実証されるp-GlcNAc半結晶構造を保持する限り、本発明に有用である。
【0098】
コラーゲン/p-GlcNAcハイブリッドの場合、簡単に説明すると、p-GlcNAc懸濁液とコラーゲン懸濁液を混合し、凍結乾燥し、そして架橋、好ましくは脱水加熱して架橋することができる。かかるハイブリッドのコラーゲン種は天然でも合成でもよく、ヒトまたは例えばウシ起源などの非ヒト由来であってもよい。p-GlcNAc/コラーゲン及び/またはp-GlcNAc誘導体/コラーゲンハイブリッド物質は均一な物性を有しかつ多孔質マトリックスを形成する。以下の第13節に記載する実施例は、かかるp-GlcNAc/コラーゲンハイブリッドの作製、物性及び有用性を実証する。
【0099】
さらに、ヨード-p-GlcNAc誘導体を、例えば、スチレンと共重合して新規のプラスチック物質を製造することができる。ヨード-p-GlcNAcは、Kurita及びInoue(Kurita, K.及びInoue, S., 1989, in "Chitin and Chitosan," Skjak-Braekら, 編, Elsevier Science Publishing Co., Inc., p.365)が記載したのと同様な方法により、p-GlcNAcのトシル化とヨード化を経由して調製することができる。次いで、p-GlcNAcのヨード誘導体をニトロベンゼン中に分散させ、塩化スズ(IV)を触媒に使ってスチレンと反応させることができる。
【0100】
脱アセチル化p-GlcNAc及び、例えばアルギン酸ナトリウムなどの化合物の組合せ、ならびにカルボキシメチルp-GlcNAcを含んでなるハイブリッドを、本明細書に記載の技術を用いて製剤化することができる。かかる組合せを、例えば、膜及び繊維に成形または再成形してもよい。
【0101】
部分的に脱アセチル化されたp-GlcNAcと例えば、正及び負電荷の両方を有する、ポリアクリル酸もしくはペクチンなどのポリアニオンとの複合物を製剤化してもよい。かかる複合物の形成は、Mirelesら(Mireles, C.ら, 1992, in "Advances in Chitin and Chitosan," Brine, C. J.ら, 編, Elsevier Publishers, Ltd.)が記載したのと類似の方法に従って実施することができる。部分的に脱アセチル化されたp-GlcNAc及びポリアクリル酸、カラゲナンまたはペクチンを、例えば、HCl及びNaCl中にそれぞれ溶解し、等しいpHをもつ反応溶液と混合する。この操作は、正及び負の特性の両方を有する有効な分子を作り、例えば、酵素及び治療用化合物を固定するのに有用である。
【0102】
5.5. 再製剤化
以上の第5.4節に記載した、p-GlcNAc出発物質、ならびにその部分的に脱アセチル化された誘導体及び/またはそれらの誘導体を溶解し、次いで様々な形状と形態に再製剤化することができる。
【0103】
p-GlcNAc出発物質の溶解は、ジメチルアセトアミド(DMA)/塩化リチウムで処理することにより達成できる。p-GlcNAcは、5%LiCl(DMAの重量で)を含有するDMA溶液中で攪拌すると容易に溶解することができる。p-GlcNAc塩などの水溶性p-GlcNAc誘導体を水に溶解してもよい。部分的に脱アセチル化しておいたp-GlcNAcを1%酢酸などの弱い酸性溶液に溶解してもよい。水に不溶性のp-GlcNAc誘導体は、有機溶媒中の溶液としてもよい。
【0104】
DMA:LiCl中のp-GlcNAcのフェニルイソシアネートによる誘導体化を利用して、カルバニレートを作ることができる。さらに、DMA:LiCl中のp-GlcNAcをトルエン-p-スルホニルクロリドを用いて誘導体化して、トルエン-p-スルホネートを作ってもよい。
【0105】
溶液中のp-GlcNAc出発物質、その部分的に脱アセチル化された誘導体、及び/またはそれらの誘導体を、次いで沈降させ、限定されるものでないが、マット、ひも、ミクロスフェア(microsphere)、ミクロビーズ(microbead)、膜、繊維、ミクロファイバー(microfiber)、粉末、及びスポンジなどの形状に再製剤化することができる。さらに、超薄い(すなわち、約1ミクロンより薄い)均一な膜に製剤化してもよい。
【0106】
かかる製剤化は、例えば純粋なp-GlcNAcが、水及びアルコール、好ましくはエタノールなどの溶液に不溶であるという事実を利用して、達成することができる。例えばp-GlcNAcを含有するDMA/LiCl混合物を、注入などの通常の方法により、かかる水またはアルコール、好ましくはエタノール中に導入すると、溶液は溶存p-GlcNAcの再沈降、従って再製剤を起こしうる。かかる純粋なp-GlcNAc再製剤を、以下の第11節に記載する実施例において実証する。水溶性p-GlcNAc 誘導体の場合、再製剤化は、例えば、酢酸エチルまたはイソプロパノールなどの有機溶媒中で再沈降させることにより達成できる。部分的に脱アセチル化されているp-GlcNAcの再製剤化は、アルカリ溶液中で再沈降させることにより達成できる。水に不溶のp-GlcNAc誘導体は、例えば、水などの水溶液中で再沈降することにより再製剤化できる。
【0107】
p-GlcNAc膜及び三次元p-GlcNAcマトリックスは、膜またはマトリックス内に制御した平均孔サイズの形成を可能にする方法を経由して作ることができる。膜及びマトリックスにおける孔サイズは、使用するp-GlcNAc物質の量を変えることにより、ならびに、膜及び/またはマトリックスの形成前に、メタノールまたはエタノール、好ましくはエタノールなどの特定の溶媒を、約5%〜約40%の範囲の特定の量だけ加えることにより制御することができる。一般的に、溶媒のパーセントが高いほど、形成される平均孔サイズは小さくなる。以下の第15節に記載する実施例は、かかる多孔質p-GlcNAc構造の合成及び特性決定を実証する。
【0108】
他の実施形態においては、半結晶p-GlcNAcを、ミクロスフェア、ミクロビーズ、もしくはミクロフィブリル(microfibril)を含むゲル、フォーム、または溶液もしくは懸濁液として製剤化する。かかる製剤は、従って、さらに適当な量の製薬上許容されるビヒクルもしくは担体を含むものであって、半結晶p-GlcNAcの患者への投与に適当な形態を与える。
【0109】
特定の実施形態においては、用語「製薬上許容される」は、哺乳動物、特にヒトにおける使用について、連邦もしくは州政府の規制当局により認可されているかまたは米国薬局方またはその他の一般的に認識された薬局方に掲げられていることを意味する。用語「担体」は、治療剤と一緒に投与される希釈剤、アジュバント、添加剤、またはビヒクルを意味する。かかる製薬用担体は、水ならびにオイルなどの液体であってもよく、オイルとしては、落花生油、ダイズ油、鉱油、ゴマ油などの石油、動物、植物または合成起源のものが挙げられる。製薬用担体は、生理食塩水、アカシアゴム、ゼラチン、デンプンペースト、タルク、ケラチン、コロイド状シリカ、尿素、などである。さらに、補助剤、安定剤、粘稠剤、滑沢剤及び着色剤を使用してもよい。患者に投与するとき、p-GlcNAcと製薬上許容される担体は好ましくは無菌である。生理食塩水溶液ならびにデキストロース及びグリセロール水溶液を液状担体として利用することができる。適当な製薬用担体としてはまた、デンプン、グルコース、ラクトース、スクロース、ゼラチン、麦芽、コメ、小麦粉、チョーク、シリカゲル、ステアリン酸ナトリウム、モノステアリン酸グリセロール、タルク、塩化ナトリウム、乾燥スキムミルク、グリセロール、プロピレン、グリコール、水、エタノールなどが挙げられる。p-GlcNAc組成物はまた、所望により、最小量の湿潤もしくは乳化剤、またはpH緩衝剤を含有してもよい。
【0110】
p-GlcNAcを含有する組成物は、溶液、懸濁液、坐薬、エマルジョン、エーロゾル、スプレイ、または使用に適したいずれの他の形態をとってもよい。適当な製薬用担体のその他の例は、E. W. Martinによる「レミントンの製薬科学("Remington's Pharmaceutical Sciences")」に記載されている。
【0111】
p-GlcNAc製剤及び組成物はプレミックス投与形態として供給しうるが、他の実施形態においては、本明細書に開示された半結晶p-GlcNAcを、例えば、活性剤の量を示したアンプルまたは小袋(sachette)などの密封容器に入った乾燥した凍結乾燥粉末または無水濃縮物として別々に供給し、使用前に製薬上許容されるビヒクルまたは溶媒中に所望の濃度で懸濁または溶解してもよい。
【0112】
特定の障害または症状の治療に有効な半結晶p-GlcNAcの量は、障害または症状の性質に依存し、標準的な臨床技術により決定することができる。さらに、場合によってはin vitroまたはin vivoアッセイを実施して最適な用量範囲を決定する助けとしてもよい。組成物に使用される半結晶p-GlcNAcの正確な用量はまた、投与経路、及び疾患もしくは障害の重篤度にも依存し、担当医の判断とそれぞれの患者の状況に従って決定すべきである。しかし、半結晶p-GlcNAcは一般的に約1mg/cm2〜約500mg/cm2の範囲で局所投与される。他の実施形態においては、半結晶p-GlcNAcは一般的に約2mg/cm2〜約100mg/cm2、約5mg/cm2〜約50mg/cm2及び約10mg/cm2〜約20mg/cm2の範囲で局所投与される。有効用量は、in vitroまたは動物モデル試験系から誘導した用量-応答曲線から外挿することができる。第16及び17節に与えた後掲の実施例に記載するものの他に、当技術分野で周知のその他の動物モデル試験系としては、限定されるものでないが、次が挙げられる:(a)止血処置を評価するための、部分的肝切除のブタモデル、Davidsonら(Davidsonら 2000, Br. J. Surg. 87(6): 790-95)が記載;(b)止血の達成を意図する処置を評価するための、イヌ出血潰瘍モード、Pasrichaら(Pasrichaら 1999 Gastointest Endosc 49(5): 627-31)が記載;(c)肝切開の処置に基づく、ラットの外科出血モデル、Sirieixら(Sirieix ら 1998 Ann Vasc Surg 12(4): 311-16)が記載;(d)無傷内皮をもつ単離されたラット胸部大動脈環の血管収縮を評価する方法、Kimら(Kim ら 2000 J Lab Clin Med 135(2): 180-87;またGuo ら 1994 Methods Find Exp Clin Pharmaocl 15(5): 347-54も参照)が記載;(e)血管直径と血管を通過する血液流量の両方を測定することを意図する、ウサギの実験動物モデル、Caronら(Caron ら 1998 Artif Cells Blood Substit Immobil Biotechnol 26(3): 293-308)が記載;(f)ラットの子宮微小血管の直接観察を可能にし、適用した血管作用薬の量の関数として細動脈の外周径の評価を可能にする方法、Alsipら(Alsip ら 1996 Am J Obstet Bynecol 175(2):388-95)が記載;及び(g)自然発症高血圧ラットを用いたモデル形、とりわけ、ラジオイムノアッセイ手法を用いて血管中の免疫反応性エンドセリンのレベルを評価するモデル系、Schiffrinら(Schiffrin ら 1995 Br J Pharmacol 115(8): 1377-81)が記載。
【0113】
使用する半結晶p-GlcNAcの特定の製剤は、意図する用途に依存して異なる。例えば、接触可能な表面に直接適用するには、半結晶p-GlcNAcを膜または絆創膏などとして製剤化し製造することができる。かかる製剤においては、半結晶p-GlcNAcを、限定されるものでないが、天然または人工の繊維を含む1以上の他の物質と組み合わせてもよいし、及び/または、本明細書に記載のコポリマーとして再製剤化してもよい。かかる物質中に配合する1cm2当たりの半結晶p-GlcNAcの量は、意図する用途により、例えば、とりわけ、小さい切傷及び擦傷を治療する低用量範囲、及び重篤な傷害を治療する高用量p-GlcNAcレベルなどのように決定される。かかる物質のサイズ、形状、厚み、及びそれに配合する半結晶p-GlcNAcの総量を含む全組成は、同様に、意図する用途により決定される。
【0114】
半結晶p-GlcNAcを容易に接近し得ない表面、例えば、口内、鼻腔内、または身体の深い創傷に局所投与する場合、半結晶p-GlcNAcは、以上開示した製薬上許容される担体及びビヒクルを使用して、とりわけ、ゲル、フォーム、スプレイ、エマルジョン、懸濁液または溶液として製剤化される。かかる製剤は、通常、バリヤーを形成しない物質であり、一般的に、半結晶p-GlcNAcから形成されたミクロスフェア、ミクロビーズ、またはミクロフィブリルを含み、そして、限定されるものでないが、天然もしくは人工の繊維、及び/または本明細書に開示したコポリマーとして再製剤された半結晶p-GlcNAcを含む物質をさらに含んでいてもよい。再び、かかる製剤に含まれる半結晶p-GlcNAcの量及び/または濃度は、意図する用途に依存し、当業者には明らかでありかつin vitro及びin vivo試験を通して、特に当技術分野で周知の動物モデル系を用いて、容易に決定しうるであろう。
【0115】
半結晶p-GlcNAcの血管構造及び/または機能に対するモジュレーション効果は局所性でかつ一過性であるので、半結晶p-GlcNAcを含有する製剤の投与は、是正すべき症状が解消されるまで、間隔をおいて反復することができる。一般的に、かかる間隔は、約1時間であるが、治療する症状の性質及び適用する結晶p-GlcNAcの量に依存して、短いときも長いときもある。半結晶p-GlcNAc製剤を含む組成物を比較的接近しにくい表面に適用する事例においては、生物分解性組成物及び製剤が好ましい。
【0116】
5.6. 用途
p-GlcNAc出発物質は様々な用途を有し、例えば、エンドセリン-1放出の刺激、血管収縮、及び破裂した血管からの血液流出の減少を経由する血管構造及び/または機能のモジュレーション、ならびに出血の停止を助けるかまたは達成することが挙げられる。局所に適用された本発明のp-GlcNAcは、生体適合性、生分解性、無毒、及び非病原性である。本発明のp-GlcNAc物質はまた、免疫中性であるので、これらはヒトに免疫応答を誘発せず、従って、限定されるものでないが、フィルム、膜、ゲル、スポンジ、ミクロスフェア、ミクロビーズ、ミクロフィブリル、フォーム、及びスプレイを含む、本発明に開示したデバイスの製剤化に利用するのに特に有利である。また、該ポリマーが、以下の実施例6に記載のIR吸収分光計により分析したときに、ポリβ-1→4 N-アセチルグルコサミンポリマーが鋭く明確なピークにより実証されるその半結晶構造を保持する限りにおいて、天然のアルギネート及び場合によっては合成ポリマーのような特定のさらなる物質を、本明細書に記載のp-GlcNAcと組み合わせて、かかる物質及びデバイスの構築に利用することができる。一実施形態においては、p-GlcNAcは、本質的に完全にアセチル化されたβ-1→4-N-アセチルグルコサミンの半結晶ポリマーから成り、ここで前記ポリマーは、β-1→4コンフォメーションで共有結合した約50〜約150,000個のN-アセチルグルコサミン単糖を含んでなり、タンパク質を含まず、その他の有機不純物を実質的に含まず、無機不純物を実質的に含まずかつ約10,000ダルトン〜約30 x 106ダルトンの分子量を有する。他の実施形態においては、p-GlcNAcは、本質的に完全にアセチル化されたβ-1→4-N-アセチルグルコサミンの半結晶ポリマーからなり、ここで前記ポリマーは、β-1→4コンフォメーションで共有結合した約50〜約50,000個、約50〜約10,000個、約50〜約4,000個のN-アセチルグルコサミン単糖を含んでなり、タンパク質を含まず、その他の有機不純物を実質的に含まず、無機不純物を実質的に含まず、かつそれぞれ約10,000ダルトン〜約10 x 106ダルトン、約10,000ダルトン〜約2 x 106ダルトン、約10,000ダルトン〜約800,000ダルトンの分子量を有する。
【0117】
5.6.1. エンドセリン-1放出の刺激
本発明のp-GlcNAc物質は、以下の第16節に記載する実施例で成功裏に実証したように、例えばエンドセリン-1の放出を刺激するために利用される。エンドセリン-1放出の刺激は、とりわけ、子宮の子宮内膜組織によるエンドセリン-1の産生レベルの著しい低下に関連する月経過多の治療に利用される。
【0118】
エンドセリン-1放出の刺激は、ヒトまたは、限定されるものでないが、獣医学及び愛玩動物を含む非ヒト哺乳動物の標的組織へのp-GlcNAcを含む組成物及び物質の局所適用により達成される。かかる物質及び組成物は、本明細書に記載のp-GlcNAcと組み合わせて、天然のアルギネート及び、場合により、合成ポリマーなどのある特定のさらなる物質を含んでもよい。好ましい実施形態においては、かかる組成物及び物質のp-GlcNAcは、完全にアセチル化された、β-1→4-N-アセチルグルコサミンの半結晶ポリマーから本質的になり、タンパク質を含まず、その他の有機不純物を実質的に含まず、無機不純物を実質的に含まずかつ約30 x 106ダルトン以下の分子量を有する。
【0119】
p-GlcNAcを含む本発明の物質は、例えば、ゲル、フィルム、膜、及びスポンジとして製剤化しかつ適用する。かかる物質はまた、ミクロスフェア、ミクロビーズ、ミクロフィブリルの溶液もしくは懸濁液として、またはフォームもしくはスプレイとしても製剤化しかつ適用できる。従って、p-GlcNAcを含んでなる本発明の物質はバリヤー形成物質である必要はない。
【0120】
p-GlcNAcを含む本発明の組成物及び物質は、標的組織、すなわち、エンドセリン-1放出を刺激することを所望する組織に直接適用される。標的組織は、例えば月経過多を患う患者の子宮の子宮内膜組織でありうる。標的組織としては、一般的に、内皮組織、そして特に、動脈、静脈、または毛細血管であってもよい血管が挙げられる。半結晶p-GlcNAcを含む物質は、例えば、ゲル、フィルム、膜、スポンジ、スプレイもしくはフォーム、ならびにミクロスフェア、ミクロビーズもしくはミクロフィブリルの懸濁液、エマルジョンもしくは溶液として、局所に適用する。
【0121】
p-GlcNAcを含む本発明の組成物及び物質を局所適用すると、p-GlcNAc無処理の標的組織と比較して、標的組織において局所性かつ一過性で、投与p-GlcNAcの用量依存的に、エンドセリン-1の放出が刺激される。エンドセリン-1放出の刺激は局所性であり、p-GlcNAcを含む物質と直接接触する組織において最も顕著であり、さらに、隣接組織におけるエンドセリン-1放出の刺激の程度は、標的組織とp-GlcNAcを含む物質の接点からの距離が増加するとともに減少する(例えば、後掲の第16節に記載する実施例を参照)。
【0122】
エンドセリン-1放出の刺激は一過性であって、半結晶p-GlcNAcを含んでなる物質と接触する組織におけるエンドセリン-1のレベルはかかる物質の投与直後に最大となり、その後、刺激前に観察されたレベルへ減少する。すなわち、接触組織におけるエンドセリン-1濃度は一般的に半結晶p-GlcNAcの投与後15分までが最大であり、エンドセリン-1の濃度は半結晶p-GlcNAcの投与後約60分以内に接触直前に観察されたレベルに実質的に戻る(例えば、後掲の第16節に報じる実施例を参照)。従って、エンドセリン-1放出の長時間刺激を必要とする事例においては、半結晶p-GlcNAcを用いて製剤化した組成物及び/または物質のさらなるアリコートまたは用量を標的組織に逐次的に適用する。
【0123】
エンドセリン-1放出の刺激は用量に依存し、p-GlcNAcを含んでなる物質と接触した内皮組織により放出されるエンドセリン-1のレベルは、その物質中のp-GlcNAc量に実質的に比例する(かかる「実質的に比例する」効果の代表的実証については、例えば、後掲の第16節に記載する実施例を参照)。 従って、組成物及び物質は、必要なエンドセリン-1放出の刺激のレベルに要求されるp-GlcNAcのレベルを含有するように製剤化しかつ構築する。かかるレベルの決定は、日常のin vitro実験、及び動物モデル試験から容易に確認される。従って、より大きいエンドセリン-1放出の刺激を必要とする事例においては、組成物及び物質はp-GlcNAc濃度を増加して製剤化する。
【0124】
5.6.2. 血管収縮の誘導
本発明のp-GlcNAc物質は、例えば、後掲の第16及び17節に記載する実施例に成功裏に実証し、ならびに図22に示したように、血管収縮を誘導するために利用される。血管収縮は、半結晶p-GlcNAcを含んでなる組成物及び物質の、ヒトまたは、限定されるものでないが、獣医学及び愛玩動物を含む非ヒト哺乳動物の標的組織への局所適用により達成する。
【0125】
半結晶p-GlcNAcを含んでなる組成物の局所適用が有用である臨床応用としては、とりわけ、例えば、肝及び腎に生検傷を生じるかまたは血管に穿刺傷を生じる診断方法、例えば、心臓カテーテル法及びバルーン血管形成術における使用が挙げられる。それ故に、本発明の方法は、遺伝的欠陥からまたはクマヂンもしくはヘパリンなどの抗凝結薬の投与から生じうる凝血障害のいずれかの形態に悩む患者に特に有用である。特定の理論または機構に束縛されることを欲しないが、半結晶p-GlcNAcの局所適用により誘発される血管収縮は物理的に穿刺傷のサイズを縮小し、それにより、血液凝固に依存しない方法及び機構により出血の停止を容易にするかまたは達成すると考えられる。
【0126】
本発明に使用する物質及び組成物は、天然のアルギネート及び、いくつかの事例では、合成ポリマーなどのある特定の追加の物質を、本明細書に記載のp-GlcNAcと組み合わせて含んでもよい。好ましい実施形態においては、かかる組成物及び物質のp-GlcNAcは、本質的に完全にアセチル化されたβ-1→4-N-アセチルグルコサミンの半結晶ポリマーであって、タンパク質を含まず、その他の有機不純物を実質的に含まず、無機不純物を実質的に含まずかつ約30 x 106ダルトン以下の分子量を有する前記ポリマーから成る。
【0127】
p-GlcNAcを含んでなる本発明の物質は、例えば、ゲル、フィルム、膜、及びスポンジとして製剤化される。かかる物質はまた、ミクロスフェア、ミクロビーズ、ミクロフィブリルの溶液もしくは懸濁液またはスプレイもしくはフォームとして製剤化しかつ適用できる。従って、p-GlcNAcを含んでなる本発明の物質はバリヤー形成物質である必要はない。
【0128】
p-GlcNAcを含んでなる本発明の組成物及び物質は、標的組織、すなわち、血管収縮の誘導が所望される組織または血管に隣接したまたは近接した皮膚または他の組織に適用するか、あるいは標的組織に直接適用する。標的組織または血管としては、一般的に、動脈、静脈、または毛細血管が挙げられる。p-GlcNAcを含んでなる本発明の物質は、例えば、ゲル、フィルム、膜、またはスポンジ、スプレイもしくはフォームとして、またはミクロスフェア、ミクロビーズもしくはミクロフィブリルの懸濁液もしくは溶液として局所に適用する。
【0129】
p-GlcNAcを含んでなる本発明の組成物及び物質の局所適用は、局所性かつ一過性で、そして投与したp-GlcNAcの用量に依存して、血管収縮を刺激する。血管収縮の誘導は局所性であって、p-GlcNAcを含んでなる物質と直接接触する血管において最も顕著であり、さらに、血管収縮の刺激の程度は、p-GlcNAcを含んでなる物質と標的血管の接触点からの距離が増加するとともに減少する。
【0130】
血管収縮の刺激は一過性であって、血管における血管収縮の程度は、本発明のp-GlcNAc物質の投与直後に最大であり、その後、刺激前に観察されたレベルへ減少する。すなわち、血管収縮の程度は一般的にp-GlcNAcの投与後15分以内に最大となり、p-GlcNAcの投与後約60分以内に実質的に対照レベルに低下する。従って、長時間の血管収縮を必要とする事例においては、p-GlcNAcを用いて製剤化した組成物及び/または物質のさらなるアリコートまたは用量を標的組織に逐次的に適用する。
【0131】
血管収縮の誘導は用量に依存し、p-GlcNAcを含んでなる物質と接触した血管における血管収縮の程度は、その物質中のp-GlcNAc量に実質的に比例する。従って、組成物及び物質は、所望の血管収縮の程度に要求されるp-GlcNAcのレベルを含むように製剤化しかつ構築する。かかるp-GlcNAcのレベルの決定は、日常のin vitro実験、及び動物モデル試験から容易に確認される。従って、血管収縮のより大きい誘導を必要とする事例においては、組成物及び物質はp-GlcNAc濃度を増加して製剤化する。
【0132】
5.6.3. 破裂した血管からの血液流出の減少
p-GlcNAcを含む物質の局所投与を含んでなる本発明の方法はまた、例えば、標的組織の破裂した血管からの血液流出を減少するためにも利用する。破裂した血管からの血液流出の減少を達成するためのp-GlcNAcの局所適用のための臨床利用は、限定されるものでないが、腹部大動脈瘤の治療、腫瘍の塞栓形成治療、子宮類線維腫病変(uterine fibroid lesions)、及び脳性動脈瘤、例えば、脾臓、肝臓及び血管傷害を含む創傷、ならびに標準かつ最小限の侵入性外科的方法、例えば、子宮内膜症手術及び胆嚢の手術が挙げられる。これらの各例において、p-GlcNAcを含有する物質の局所適用の結果として、破裂した血管からの血液流出が減少し、手術中の血液損失が減少する。従って、本明細書に開示した血管収縮をもたらす組成物と方法の使用は、遺伝的欠陥からまたはクマジンもしくはヘパリンなどの抗凝血薬の投与から生じうる、いずれかの形の凝血障害に悩む患者のかかる症状を治療するのに特に有用でありうる。
【0133】
本発明の方法に用いる物質及び組成物は、天然のアルギネート及び、いくつかの事例では、合成ポリマーなどの特定の追加の物質を、本明細書に記載のp-GlcNAcと一緒に含んでもよい。好ましい実施形態においては、かかる組成物及び物質のp-GlcNAcは、完全にアセチル化されたβ-1→4-N-アセチルグルコサミンの半結晶ポリマーから本質的になり、ここで前記ポリマーは、タンパク質を含まず、その他の有機不純物を実質的に含まず、無機不純物を実質的に含まずかつ約30 x 106ダルトン以下の分子量を有する。
【0134】
p-GlcNAcを含んでなる本発明の物質は、例えば、ゲル、フィルム、膜、及びスポンジとして製剤化される。かかる物質はまた、ミクロスフェア、ミクロビーズ、もしくはミクロフィブリルの溶液または懸濁液として製剤化しかつ適用し、及び/またはフォームもしくはスプレイとして適用してもよい。従って、p-GlcNAcを含んでなる本発明の物質はバリヤー形成物質である必要はない。
【0135】
p-GlcNAcを含んでなる本発明の組成物及び物質は、標的組織(すなわち、破裂した血管からの血液流出を減少するのが所望される組織または血管)に隣接もしくは連続する皮膚もしくはその他の組織に適用されるか、または標的組織へ直接適用される。標的血管は動脈、静脈、または毛細血管でありうる。p-GlcNAcを含んでなる本発明の物質は、例えば、ゲル、フィルム、膜、スポンジ、スプレイもしくはフォームとして、またはミクロスフェア、ミクロビーズ、及び/もしくはミクロフィブリルの懸濁液もしくは溶液として局所に適用される。
【0136】
p-GlcNAcを含んでなる本発明の組成物及び物質の局所適用は、局所性かつ一過性で、投与したp-GlcNAcの用量に依存して、破裂した血管からの血液流出の減少を誘導する。破裂した血管からの血液流出の減少は局所性であって、p-GlcNAcを含んでなる物質と直接接触する血管において最も顕著であり、さらに、破裂した血管からの血液流出の減少の程度は、p-GlcNAcを含んでなる物質と標的血管の接触点からの距離が増加するとともに減少する。
【0137】
破裂した血管からの血液流出の減少は一過性であって、p-GlcNAcを含んでなる物質と接触したことによる血液流出の減少は、かかる物質の投与直後に最大であり、その後、破裂した血管からの血液流出は対照レベルへ戻る。すなわち、破裂した血管からの血液流出の減少の程度は一般的にp-GlcNAcの投与後15分以内に最大となり、その後、破裂した血管からの血液流出はp-GlcNAcの投与後約60分以内に対照レベルに戻る。従って、長時間の破裂した血管からの血液流出の減少を必要とする事例においては、p-GlcNAcを用いて製剤化した組成物及び/または物質のさらなるアリコートまたは用量を標的組織に逐次的に適用する。
【0138】
破裂した血管からの血液流出の減少は用量に依存し、p-GlcNAcを含む物質と接触した血管における破裂した血管からの血液流出の減少は、その物質中のp-GlcNAc量に実質的に比例する。従って、組成物及び物質は、所望の破裂した血管からの血液流出の減少に要求されるp-GlcNAcのレベルを含むように製剤化しかつ構築する。かかるp-GlcNAcのレベルの決定は、日常のin vitro実験、及び動物モデル試験から容易に確認される。従って、破裂した血管からの血液流出のより大きい程度の減少を必要とする事例においては、組成物及び物質はp-GlcNAc濃度を増加して製剤化する。
【0139】
5.6.4 開示した方法を利用する特定の適応症
エンドセリン-1放出の刺激、血管収縮、及び/または破裂した血管からの血液流出の減少、ならびに出血の停止が所望される特定の事例は、限定されるものでないが、例えば、肝及び腎に生検傷を生じる診断方法;限定されるものでないが、腹部大動脈瘤の血管内治療後の出血の防止、ならびに腫瘍、子宮類線維腫病変及び脳性動脈瘤の塞栓形成治療を含む塞栓形成方法;月経過多の治療に対する;例えば、脾臓、肝臓及び血管傷害を含む創傷;標準かつ最小限の侵入性外科的方法、例えば、子宮内膜症手術及び胆嚢の手術;軟及び硬組織創傷修復、例えば皮膚創傷及び火傷治癒;特に脾臓創傷に対する、外科的方法;ならびにカテーテル処置及びバルーン血管形成方法などの血管穿刺診断及び治療方法に対する利用が挙げられる。
【0140】
p-GlcNAcに基づく出発物質は、固体物質として、またはp-GlcNAcミクロビーズ、ミクロスフェア、またはミクロフィブリルを含んでなるゲル、フォーム、スプレイ、エマルジョン、懸濁液、または溶液として製剤化することができ、標準の外科方法を用いて適用することができ、かつ標準及び最小限侵入性外科的介入の両方によって利用することができる。本発明のゲルは、例えば、シリンジ形式のデバイスから押出しによりまたは膜もしくはフィルムと組み合わせて送達することができる。膜またはフィルムは、完全にアセチル化されたp-GlcNAcに基づく物質またはその他の天然もしくは合成の物質から製造することができる。
【0141】
上記の血管穿刺方法との関係では、エンドセリン-1分泌の刺激、血管収縮及び破裂した血管からの血液流出の減少のために利用される本発明の組成物及び物質は、カテーテル外筒を血管から除去した時点で、p-GlcNAcに基づく物質を人手で圧縮して皮膚に直接適用するか、またはカテーテル進路跡に導入することにより適用する。あるいは、血管からカテーテル外筒の除去を検出するデバイスを、血管内側と外側の組織の間の化学的、物理的もしくは他の相違をモニターする電子もしくは機械システムを用いて開発してもよい。例えば、プローブを血管から除去したときの、流体力学もしくは熱放散の変化を検出することができ;その時点でシグナルが送られ、p-GlcNAcを含んでなる組成物もしくは物質の適用が開始され、エンドセリン-1の放出を刺激し、血管収縮を誘導し、及び/または破裂した血管からの血液流出を減少しうる。
【0142】
p-GlcNAc、好ましくは完全にアセチル化されて、高度に規則正しいp-GlcNAcの半結晶ポリマーの局所投与を含んでなる、エンドセリン-1放出、血管収縮、及び破裂した血管からの血液流出の減少を誘導する本発明の方法は、止血を達成するために有用な方法及び組成物と一緒に使用してもよい。かかる他の方法及び組成物は、限定されるものでないが、(1)赤血球及び血小板を透過せずかつ血液凝固カスケードに必要な循環因子を濃縮しうるマトリックスを提供するバリヤー形成物質の適用、及び(2)例えば、トロンビン、フィブリノーゲン、及び第XIII因子を含む血液凝固カスケードの成分を含んでなる物質の創傷への適用が挙げられる。
【0143】
本発明の方法はまた、その必要が予想される場合に止血を達成する方法及び組成物の必要を最小限にするか、または効率を増加するために、予防的にも利用しうる。かかる必要のある例は、限定されるものでないが、胃腸病学的処置におけるポリープの除去、腫瘍組織の切除、及び抜歯である。かかる例においては、本発明の方法を利用し、標的組織に隣接または近接した組織及び血管において、一過性で局所性のエンドセリン-1放出、血管収縮、及び破裂した血管からの血液流出の減少を誘導し、それにより患者に実施した方法から生じるその後の出血を最小化する。
【実施例】
【0144】
6. 実施例:純粋なp-GlcNAc調製物の物理的特性の決定
本実施例に記載するのは、p-GlcNAc及び脱アセチル化p-GlcNAc膜の円二色性(CD)及び赤外分光(IR)分析である。
【0145】
6.1. 物質及び方法
p-GlcNAc及び市販「キチン(chitin)」調製物:
CD研究に用いたp-GlcNAcは、上の第5.3.1節に記載の機械力精製法を利用して調製した。
【0146】
市販「キチン」は、NovaChem, Ltd., PO Box 1030 Armdale, Halifax, Nova Scotia, Canada, B3L 4K9から購入した。
【0147】
IR研究に用いたp-GlcNAc膜は、上の第5.3.1節に記載の機械力精製法によるか、または上の第5.3.2節に記載の化学的/生物学的調製法により調製した。
【0148】
市販「p-GlcNAc」調製物を5%塩化リチウムを含有するジメチルアセトアミド溶液中に溶解し、脱イオン蒸留水上に重ねて膜を沈降させることにより、膜に成型した。
【0149】
p-GlcNAc誘導体及び処理:CD及びIR研究の両方に使用する脱アセチル化p-GlcNAcは、p-GlcNAcを50% NaOHを用いて60℃にて2時間処理することにより調製した。IR研究に使用した熱変性p-GlcNAc膜は0.2mM EDTA中で3分間煮沸して改変した。p-GlcNAcを30分間122℃にてオートクレーブ処理した。
【0150】
CD技術:固体CD技術は、本質的にDomard(Domard, A., 1986, Int. J. Macromol. 8:243-246)に従って実施した。
【0151】
6.2. 結果
6.2.1. CD分析
無処理p-GlcNAcから得たCDスペクトル(図3A)においては、p-GlcNAcのアセチル部分のカルボニル基の存在によって、予想したn-π*及びπ-π*光学活性電子遷移(220-185nM)が観察された。図3Bに示すように、かかるピークは、脱アセチル化p-GlcNAc生成物から得たCDスペクトルには全く存在しない。
【0152】
6.2.2. IR分光分析
本研究で得たIRスペクトルはp-GlcNAcの化学構造と一致する。さらに、各IRピークの鋭い鮮明度は、p-GlcNAc繊維における秩序正しくかつ規則的な(すなわち、半結晶)構造の存在を示す。機械力精製方法を経由して精製したp-GlcNAcのIRスペクトルである図4A、及び化学的/生物学的方法を経由して精製したp-GlcNAcのIRスペクトルである図4Dを参照すること。比較のため、市販「キチン」調製物のIRスペクトルを示す図4Bを参照すること。
【0153】
オートクレーブ処理したp-GlcNAc物質(図4E)から得たIRスペクトルは、図4Aで観察したIRスペクトルと可視的な相違はない。このデータは、p-GlcNAc物質はオートクレーブ処理によりポリマー構造の消失なしに無菌化しうることを示す。
【0154】
7. 実施例:機械力精製法を用いるp-GlcNAcの精製
本節においては、p-GlcNAcを、第5.3.1節に記載の機械力技術を用いて精製した。
【0155】
7.1. 物質及び方法/結果
珪藻培養条件:珪藻種タラシオシラ・フルビアチリス(Thalassiosira fluviatilis)を、以上の第5.1節及び第5.2節に記載の方法に従って、培養液中で増殖させた。
【0156】
SEM工程:本実施例で使用したSEM技術を、以下の第12.1節に記載する。
【0157】
p-GlcNAc精製手法:p-GlcNAcは、珪藻培養物から以上の第5.3.1節に記載の機械力技術を利用して精製した。特に、p-GlcNAc繊維は、培養内容物をウエアリング(Waring)ブレンダー中で、トップスピード混合運動の短いバースト3回で処理することにより珪藻細胞体から分離した。3回のバーストの全時間は約1秒であった。得られる懸濁液をソルヴォール(Sorvall)GS-4固定角ローター中で3500rpmで20分間、約10℃で遠心分離した。上清をデカントし、再び、今度は、ソルヴォールGS-4固定角ローター中で4000rpmで20分間、約10℃で遠心分離した。さらに再び、上清をデカントして4000rpmで、約10℃で遠心分離した。第3回目の遠心分離後の最終上清は透明であって、液中に浮遊する目視しうるフロックは、あっても僅かでしかなかった。透明な上清を、0.8μm孔サイズをもつSupor-800ポリエーテルスルホン・フィルター膜(Gelman, Inc.)を備えたブーフナー(Buchner)濾過ユニット中にデカントし、次いで吸引し、繊維懸濁液から液を濾過して膜上に繊維を回収した。回収した繊維を1リットルの脱イオン蒸留水を用いて70℃にて洗浄した。ほぼ全ての水が排水されたら、1リットルの1N HClを用いて70℃にて吸引して繊維を洗浄した。ほぼ全ての酸性溶液が排水されたら、1リットルの脱イオン蒸留水を用いて70℃にて吸引して繊維を洗浄した。ほぼ全ての洗浄水が排水されたら、1リットルの95%エタノールを用いて室温にて繊維を洗浄し、そして減圧した。次いで、白い繊維膜をその上に回収したフィルター膜を、濾過ユニットから除去し、膜とその膜支持体とを乾燥オーブン中で58℃にて20分間乾燥し、その後、膜とその支持体をデシケーター中に16時間置いた。
【0158】
この精製過程の後に、1000ml培養からのp-GlcNAcの収量は珪藻培養物1リットル当たり6.85ミリグラムであった。この技術で回収したp-GlcNAc繊維により形成される膜のSEM写真を図6に示す。
【0159】
8. 実施例:生物学的/化学的精製方法を利用するp-GlcNAcの精製
本節においては、上の第5.3.2節に記載の化学的/生物学的技術の2方法を利用して、p-GlcNAcを精製した。簡単に説明すると、第1の事例ではp-GlcNAcをHF処理を経由して、かつ第2の事例では酸処理/中和を経由して精製した。
【0160】
8.1. 物質及び方法/結果
珪藻培養条件:珪藻種タラシオシラ・フルビアチリス(Thalassiosira fluviatilis)を、上の第5.1節及び第5.2節に記載の方法に従って、培養液中で増殖させた。
【0161】
SEM工程:本実施例で使用した技術は、以下の第12.1節に記載した。
【0162】
精製方法:第1の事例においては、p-GlcNAcをHF処理により精製したが、その結果を図7に示す。具体的には、換気フードの下で、珪藻培養内容物に、元の細胞培養物の容積のそれぞれ1000mlに対して49%(29N)HF溶液2.42mlを室温で加え、0.07M HF溶液を得た。次いで混合物を約30秒間激しく振とうして、液を覆う持続性の泡を作った。容器を5〜6時間静置して重い微粒子を沈降させた。この時点の終わりには、泡の層が形成する一方、液自身は2層に分割された:第1層は容器の底に横たわる少量の非常に暗い緑色層であり、その上の第2層ははるかに明るい灰緑色の濁った相であって恐らく液の全容積の85〜90%を占めた。泡層を、毛細ガラス管及び真空吸引を用いて注意深くサイフォンで吸引除去した。次いで、灰色の曇った上清を、沈降した細胞体から主になる暗色の底層を乱さないように注意を払いながら、サイフォンで吸引除去し、別のプラスチック容器に移した。灰色の曇った上清を、さらに16時間静置した。液は最初、ほとんど無色、明灰色であるが、透明でなかった。16時間静置後、少量の泡が液本体の頂部に残りかつ少量の緑色物質が容器の底に沈降した。液は色がより明るくなったが、なお透明でなかった。液頂部の泡を上記のようにサイフォンで吸引除去した。次いで、液本体を注意深くサイフォンで吸引除去し、少量の沈降した緑色物質は容器の底に放置した。こうして単離した液は、p-GlcNAc繊維の大部分と若干の不純物を含有した。
【0163】
以上の工程中に珪藻から遊離されたタンパク質及び他の望ましくない物質を、繊維を含有する液から除去するために、繊維及び細胞膜残遺物の懸濁液をドデシル硫酸ナトリウム(SDS)を用いて洗浄した。具体的には、所要の容積の20%SDS溶液を加えて、最終濃度が容積基準で0.5%SDSの液を調製した。液を入れた容器をシールし、振とう機械上に水平位置にセットし、24時間、毎分100回で振とうして攪拌した。振とうを始めると直ぐに、大きな白色のp-GlcNAc繊維のフロックが懸濁液中に現れ、そしてかなりの量の泡が容器の上部空間に蓄積した。SDS洗浄が終わると、容器の内容物を、0.8μm孔サイズをもつSupor-800ポリエーテルスルホン・フィルター膜(Gelman, Inc.)を備えたブーフナー濾過設備へ移した。液を吸引濾過し、液中のp-GlcNAc繊維を濾過膜上に回収した。
【0164】
次いで、フィルター膜上に回収したp-GlcNAc繊維をさらに洗浄した。最初に、熱脱イオン蒸留水(70℃)を、元の懸濁液の容積の3倍用いて、繊維を洗浄した。脱イオン蒸留水を用いる水ジェットにより、ブーフナーフィルターのフィルター膜上に回収した白色繊維凝集塊をウエアリング(Waring)ブレンダーに移し、繊維凝集塊を約10回の短い混合バーストにより崩壊させた。崩壊した繊維の懸濁液を、上記のように、ポリエーテルスルホン膜を備えたブーフナー濾過漏斗へ移し、液を吸引下で除去した。回収した繊維を熱(70℃)1N HCl溶液1000mlを用いて洗浄し、続いてさらに熱(70℃)脱イオン蒸留水1000mlを用いて洗浄した。最後に、繊維を95%エタノール1000mlを用いて室温にて洗浄し、濾過して乾燥した。次いで、繊維膜及び繊維膜を支持するフィルター膜を、乾燥オーブンにて58℃で20分間乾燥した。次いで、膜と膜支持体をデシケーター内に16時間置いた。膜を注意してフィルター膜から剥がした。
【0165】
第2の事例においては、p-GlcNAcを以上の第5.3.2節で記載した酸処理/中和法を利用して精製した。具体的には、p-GlcNAcをSDS洗浄工程の前まで本節の始めに記載したように処理し、その時点で該溶液に2.9M Tris溶液を加えてほぼ7.0のpHに中和した。この特定の精製方法からのp-GlcNAc収率は珪藻培養物1リットル当たり20.20ミリグラムであったが、平均では、珪藻培養物1リットル当たりほぼ60ミリグラムが得られる。精製過程中に形成される膜のSEM顕微鏡写真を図8A〜B及び9A〜9Eに示す。
【0166】
9.実施例:p-GlcNAc脱アセチル化
p-GlcNAc膜を50%NaOH水溶液中に懸濁した。懸濁液を80℃にて2時間加熱した。得られる脱アセチル化膜を乾燥して走査電子顕微鏡により研究し、図11A-Bに示した。
【0167】
10.実施例:p-GlcNAc生体適合性
本実施例において、p-GlcNAc出発物質は、溶出試験、ウサギへの筋肉内移植、ウサギへの皮下注射、及びマウスへの全身注射によりアッセイしたところ検出可能な生物学的活性を表さないことが示された。
【0168】
10.1. 物質及び方法
10.1.1. 溶出試験
溶出試験の条件は、米国薬局方XXII、1990、1415-1497頁、及び米国薬局方XXII、追補5、1991、2702-2703頁に記載の規格に一致させた。
【0169】
細胞培養:マウス線維芽細胞L929細胞系統(American Type Culture Collection Rockville, Md.;ATCC No. CCL1;NCTCクローン929)を利用した。L929細胞の24時間コンフルエント単層を完全最小必須培地(MEM)において増殖させた。
【0170】
p-GlcNAc:以上の第5.3.1に記載した機械力精製法に従って調製しておいたp-GlcNAcの固体膜を、米国薬局方XXII(1990)要件に従って20ml血清補充MEM中で抽出した。
【0171】
対照:天然ゴムを陽性対照として使用し、かつシリコーンを陰性対照として使用した。対照を試験材、p-GlcNAcと同じ方法で試験した。
【0172】
抽出物:抽出物は、37℃にて、5%二酸化炭素を含有する加湿大気中、24時間で調製した。抽出物のpHの変化を測定し、pHを元の培地の±0.2のpH単位内に収めるように調節した。調節は、抽出pHの低下はHClにより、また抽出pHの上昇はNaHCO3により実施した。抽出物は、細胞単層に適用する前に、0.22ミクロンフィルターを通過させて滅菌濾過した。
【0173】
投薬:p-GlcNAcまたは対照抽出物の3mlを細胞培養の維持培地と置き換えた。全ての抽出物を繰り返して試験した。
【0174】
評価判定基準:細胞単層の応答を、目視または顕微鏡下のいずれかで評価した。生物学的反応、すなわち、細胞変性及び/または形成異常を以下に示したように0〜4の点数で評価した。試験システムは、もし陰性対照材に対して細胞反応性の徴候が認められず(評点0)かつ陽性対照材が中度(評点2)より大きい細胞反応性を示せば、適当である。試験材(すなわち、p-GlcNAc)は、もし試験材を用いて処理した培養であって、軽度より大きい反応性を示すものがなければ、生体適合性試験に合格する。
【表2】
10.1.2. 筋肉内移植
動物:健康な、ニュージーランド白ウサギ(New Zealand White Rabbit)、雄性及び雌性、(Eastern Rabbit Breeding Laboratory, Taunton, Mass.)を使用した。ウサギを個別に、懸架式ステンレス鋼製籠を用いて飼育した。動物を入手すると、8日間、実試験と同じ条件下で隔離した。ハードウッド(Hardwood)チップ(Sani-chips(TM), J. P. Murphy Forest Products, Montvale, N.J.)を籠下の無接触ろ床として使用した。動物施設は、華氏68±3度の温度、30-70%の相対湿度、1時間当たり少なくとも10-13回の完全空気交換、及びフルスペクトル蛍光灯を用いる12時間の明/暗サイクルに維持した。動物に、市販餌(Agway ProLab, Waverly, N.Y.)を制御した条件下で与え、かつ都市水道水を任意にとれるように補給した。餌、ろ床、または水の中に、試験結果を妨害すると思われる既知不純物は存在しなかった。研究用に選択した動物は、動物のより大きいプールから選んだ。ウサギは10g精度で体重測定し、耳の入れ墨により個々に同定した。
【0175】
p-GlcNAc:使用したp-GlcNAcは、上の第10.1.1節に記載したとおりである。
【0176】
移植試験:それぞれの移植試験に2匹のウサギを用いた。試験の日に、脊柱の両方の側の動物皮膚の毛を刈り込んだ。それぞれの動物を麻酔して筋肉運動を阻止した。無菌皮下針とスタイレットを用いて、試験p-GlcNAcの4つの細片(1mm x 1mm x 10mm)を、2匹のウサギのそれぞれに対して、脊椎の片側の脊椎傍筋中に移植した(正中線から2.5〜5cm、脊柱と平行に、そしてお互いから約2.5cmに)。同様な方式で、USP陰性対照プラスチックRS(1mm x 1mm x 10mm)の2つの細片をそれぞれの動物に対して、反対の筋肉中に移植した。動物を7日間維持した。観察期間の終わりに、動物の体重を測定し、注射用バルビツール酸塩、Euthanasia-5(Veterinary Laboratories, Inc., Lenexa, Kans.)により安楽死させた。組織が出血なしで切断されるために十分な時間が経過するのを待った。それぞれの移植片の中心部を囲む組織の領域を拡大レンズを用いて巨視的に試験した。止血、壊死、変色、及び感染を、次のスケールを用いてスコアを付けた:0=正常、1=軽度、2=中度、及び3=重度。もし嚢が存在すれば、最初に嚢の幅(すなわち、移植片の周縁から嚢の周縁までの距離)を0.1mmまで丸めて測定してスコアを付けた。嚢は次のとおりスコアを付けた:
【表3】
p-GlcNAcと陽性対照材の平均スコアの間の差を計算した。試験は、4つのp-GlcNAc細片の多くとも1つについて、もしそれぞれのウサギにおいて、p-GlcNAcに対する生物学的反応のそれぞれのカテゴリーに対する平均スコアと陽性対照プラスチック移植片部位に対するそれとの間の差が1.0を超えないか;または、それぞれのp-GlcNAc材に対する生物学的反応の全てのカテゴリーに対する平均スコアと全ての陽性対照プラスチック移植片部位に対する全てのカテゴリーに対する平均スコアとの間の差が、1.0を超えなければ、陰性と考えた。
【0177】
10.1.3. 皮内注射
動物:ニュージーランド白ウサギを使用し、以上の第10.1.2節に記載のように維持した。
【0178】
p-GlcNAc:以上の第5.3.1節に記載した精製の機械力方法に従って調製しておいたp-GlcNAcの固体膜を、抽出フラスコ内に置いて、これに適当な培地20mlを加えた。抽出は、70℃に24時間加熱して実施した。この過程の後に、抽出物を室温に冷却した。それぞれの抽出ボトルを激しく振とうした後に投与した。
【0179】
皮内試験:試験日に、動物の背中の毛を刈り込んだ。それぞれのp-GlcNAc抽出物の0.2ml容積を2匹のウサギのそれぞれの片側に5つの部位で皮内注射した。1匹のウサギ当たり1以上のp-GlcNAc抽出物を使用した。それぞれのウサギの他の側の5つの部位に、対応する対照0.2mlを注射した。注射部位を、紅斑、浮腫、及び壊死の徴候について、注射後24、48、及び72時間に観察した。観察結果を、皮膚反応スコアリング用ドレーズ(Draize)スケール(米国薬局方XXII, 1990, 1497-1500;米国薬局方XXII, 追補5, 1991, 2703-2705)に従ってスコアを付け、以下の表IIに示した:
【表4】
全ての紅斑及び浮腫のスコアを、24、48、及び72時間に個々に合計して12(すなわち、2動物 x 3スコアリング期間 x 2スコアリングカテゴリー)で除し、p-GlcNAcの全平均スコアを対応する対照に対して測定した。動物は、観察期間が終わると体重を測定し、バルビツール酸塩、Euthanasia-5(Veterinary Laboratories, Inc., Lenexa, Kans.)を注射して安楽死させた。試験結果は、もしp-GlcNAcと対照の平均反応スコア (紅斑/浮腫)の間の差が1.0以下であれば、合格とする。
【0180】
10.1.4. 全身注射
動物:アルビノ・スイス・マウス(Albino Swiss mice)(Mus musculus)、雌性、(Charles River Breeding Laboratories, Wilmington, Mass.)を使用した。5匹のマウスのグループをステンレス鋼蓋を取付けたポリプロピレン籠に入れて飼育した。ハードウッドチップ(SanichipsTM, J. P. Murphy Forest Products, Montvale, N.J.)を籠内の接触ろ床に使用した。動物施設は、限られたアクセス域として維持し、動物室は華氏68±3度の温度、相対湿度30-70%、1時間当たり少なくとも10-13回の完全空気交換、及び全スペクトル蛍光灯を用いる12時間の明/暗サイクルを保った。マウスに市販の餌及び都市水道水を任意でとれるようにで補給した。餌、ろ床、または水の中には、試験結果を妨害すると思われる既知不純物は存在しなかった。研究用に選択した動物は、より大きな動物のプールから選んだ。マウスは0.1g精度で体重測定し、個々に耳パンチにより同定した。
【0181】
p-GlcNAc:使用したサンプルは、以上の第10.1.1節に記載したとおりであった。抽出物は以上の第10.1.3節に記載の方法に従って調製した。
【0182】
全身注射試験:5匹のマウスのグループにp-GlcNAc抽出物または対応する対照材を、以下に掲げたのと同じ量をそして同じ経路により注射した。
【表5】
PEG400を用いて調製したp-GlcNAcの抽出物、及び対応する対照、を0.9%NaClにより希釈して1mlあたりPEG400 200mgとした。皮内試験用には、PEG400を0.9% NaClにより希釈して1mlあたりPEG400 120mgとした。
【0183】
動物を注射直後、注射後24、48、及び72時間に観察した。動物は、観察期間が終わると体重を測定し、二酸化炭素に暴露して安楽死させた。もしp-GlcNAcで処理した動物が対照材を用いて処理した動物より有意に大きい生物学的反応性を示さなければ、試験要件は合格とする。
【0184】
10.2. 結果
10.2.1. 溶出試験
p-GlcNAc試験材に対する細胞単層の応答は、目視及び顕微鏡下で評価した。評価に細胞化学染色は使わなかった。細胞の生物学的反応性の徴候なし(0級)は、陰性対照材もしくはp-GlcNAcに対する暴露後48時間に観察した。重度の反応性(4級)は以下の表IIIに示した通り、陽性対照材に対して認められた。
【表6】
従って、p-GlcNAc出発物質は、生体適合性に対する溶出試験の要件に合格し、かくして、非細胞傷害性である。
【0185】
10.2.2. 筋肉内移植
試験した両方のウサギ(A及びB)は、体重が増加し、毒性の徴候を示さなかった。データは表IVを参照。さらに、いずれの動物にも毒性の明確な徴候はなかった。試験及び対照材移植部位の目視評価では、炎症、嚢、出血、壊死または変色は認められなかった。結果は表IVを参照。従って、試験は、アッセイしたp-GlcNAcが生物学的反応性を示さず、それぞれのウサギにおいて、全てのp-GlcNAc移植部位に対する生物学的反応の全てのカテゴリーに対する平均スコアと、全ての対照移植部位に対する全てのカテゴリーに対する平均スコアとの差が1.0を超えないことを実証した。
【表7】
10.2.3 皮内試験
全動物の体重が増加した。データは表Vを参照。p-GlcNAcまたは対象材部位のいずれにおいても紅斑または浮腫の徴候は観察されなかった。毒性の明確な徴候はいずれの動物にも観察されなかった。p-GlcNAcと対照材平均反応スコア(紅斑/浮腫)との間の差はが1.0未満であったので、p-GlcNAcは皮下試験の要件に合格する。結果は表VIを参照。従って、この試験でアッセイされるとおり、p-GlcNAcは生物学的反応性を有しない。
【表8】
【表9】
10.2.4. 全身試験
p-GlcNAc抽出物または対照材を用いて処理した全てのマウスは体重が増加した。データは表VIIを参照。さらに、毒性の明確な徴候はいずれのp-GlcNAcまたは対照動物にも観察されなかった。結果は表VIを参照。従って、p-GlcNAc試験動物は対照材を用いて処理した動物と比較して有意に大きな生物学的反応性を示さなかったと結論される。
【表10】
11. 実施例:p-GlcNAc再製剤
本節に記載する実施例においては、p-GlcNAc膜(16.2 mg)を、5% LiClを含有するジメチルアセトアミド溶液1ml中に溶解した。p-GlcNAcを含有する溶液をシリンジ中に入れて純粋な水50ml中に押出し、繊維を沈降させた。得られた繊維を、図10A-Bに示す走査電子顕微鏡を用いて研究した。
【0186】
12. 実施例:p-GlcNAc/コラーゲン・ハイブリッド
本実施例で記載するのは、p-GlcNAc/コラーゲン・ハイブリッド物質の生成及び特性決定である。
【0187】
12.1. 物質及び方法
材料:ウシI型コラーゲンを、本研究に記載のハイブリッド調製に使用した。p-GlcNAcは以上の第5.3.2節に記載した機械力方法に従って調製した。
【0188】
ハイブリッド調製:コラーゲン(10ミリグラム/ml)とp-GlcNAc(0.25ミリグラム/ml)の水懸濁液を、様々な比で混合し、液体N2(-80℃)中で凍結し、-9℃にて4時間保って凍結乾燥した。材料を真空下(ほぼ0.030 Torr)で60℃にて3日間、脱水加熱して架橋した。
【0189】
細胞培養:マウス3T3線維芽細胞を、調製したコラーゲン/p-GlcNAc・ハイブリッド上で増殖した。標準培養方法に従い、培養の8日後にSEM顕微鏡写真を撮影した。
【0190】
12.2. 結果
コラーゲンとp-GlcNAc水懸濁液を、様々な比(すなわち、コラーゲン:p-GlcNAc懸濁液比=3:1、1:1、2:2、及びl:3)で混合し、凍結し、凍結乾燥し、そして架橋した。かかる方法によりコラーゲン/p-GlcNAcスラブを得た。得られた物質のSEM顕微鏡写真は、ハイブリッド物質の多孔質構造を示し、細胞の付着と増殖にとって効率的な三次元構造を提供した。
【0191】
13. 実施例:純粋なp-GlcNAc調製物のNMR特性決定
本実施例で記載するのは、純粋なp-GlcNAc調製物のNMR(核磁気共鳴)分析である。
【0192】
13.1. 物質及び方法
p-GlcNAc調製物:本節に記載のNMR研究に使用したp-GlcNAcは、以上の第5.3.2節に記載した化学精製法を利用し、フッ化水素酸を化学試薬に用いて調製した。
【0193】
NMR技術:固体NMRデータはブルーカー(Bruker)500MHz NMR分光計を用いて得た。コンピューター画像解析を利用して生のNMRスペクトルデータを変換し、バックグラウンドを排除しかつベースラインを正規化した。かかる変換データの一例を図14に示す。図14のような変換NMR曲線を利用して全ての炭素原子種に対する面積を求め、次いでCH3(面積)対C-原子(面積)の比を計算した。記載のようにして得たかかる値を図16に示す。
【0194】
13.2. 結果
固体NMRデータを、p-GlcNAcサンプル500mgの13C-NMRスペクトルを測定することにより取得した。典型的なNMRスペクトルを図15に示す。個々のピークは分子中のそれぞれのユニークな炭素原子のスペクトルへの寄与を表す。分子中のそれぞれの炭素原子種の相対的パーセントは、その炭素種が作ったピーク面積を、スペクトル中に観察される全てのNMRピーク下の面積の合計和により除して決定した。このようにして、基準原子により測定した分子におけるそれぞれの原子の比を計算することが可能であった。全てのp-GlcNAc分子は、定義により、C1、C2、C3、C4、C5及びC6原子を有するN-アセチルグルコサミン残基からなる。従って、もしポリマーの全てのグルコサミン残基がN-アセチル化されていれば、以上のいずれのグルコサミン残基炭素原子のピーク面積に対するN-アセチルCH3炭素原子のピーク面積の比も1.0となる。図14のようなデータを利用してCH3(面積)比の値を得た。
【0195】
図16で計算した比は、多くの場合、実験誤差内でほぼ1.0に等しく、例えば、CH3/C2=1.097、CH3/C6=0.984、CH3/C5=1.007、CH3/C1=0.886であった。これらの結果は、p-GlcNAc出発物質が不純物を含まず、完全にアセチル化されている(すなわち、グルコサミン残基の本質的に100%がN-アセチル化されている)という結論と一致する。
【0196】
14. 実施例:制御された孔サイズ三次元p-GlcNAcマトリックスの合成と生物学的特性の決定
以下に記載するのは、制御された平均孔サイズを有する多孔質マトリックスに基づく三次元p-GlcNAcを生産する方法である。かかるマトリックスは様々な重要な用途を有し、例えば、細胞の封入(encapsulation)が挙げられる。かかる細胞封入組成物は、移植しうる細胞に基づく治療薬として、及び軟骨再生などの他の細胞及び組織エンジニアリング用途に有用である。本明細書に実証したp-GlcNAc物質の形態と寸法を操作する能力は、p-GlcNAcポリマーを様々な形状に再製剤化する強力なツールを提供し、前記形状としては、限定されるものでないが、製薬上許容される担体、ビヒクル及び/または溶媒中のエマルジョン、懸濁液及び/または溶液として製剤化することができるミクロビーズ及びミクロスフェアが挙げられる。
【0197】
14.1. 物質及び方法
p-GlcNAc出発物質:p-GlcNAcを、以上の第5.3.2節に記載した化学的精製方法を利用し、化学試薬としてフッ化水素酸を用いて調製した。マトリックス製剤:凍結乾燥に先立って、p-GlcNAcサンプル20mgを含有する懸濁液(5ml)を以下の第14.2節に掲げた溶媒中で作った。次いでサンプルを組織培養ディッシュのウエルに注ぎ入れて-20℃にて凍結した。凍結したサンプルを次いで凍結乾燥し、得られた三次元マトリックスを除去した。
【0198】
操作電子顕微鏡技術:本明細書で利用した方法は、以上の第12.1節に記載したように実施した。図17A-Gに示す画像はマトリックス物質の200 x 拡大であり、これらの図のそれぞれに200ミクロンのスケールマーキングを示す。
【0199】
14.2. 結果
以上の第14.1節に記載したp-GlcNAc懸濁液は、次の各溶液を用いて得た:
A. 蒸留水
B. 10%メタノールを含む蒸留水
C. 25%メタノールを含む蒸留水
D. 蒸留水単独
E. 10%エタノールを含む蒸留水
F. 25%エタノールを含む蒸留水
G. 40%エタノールを含む蒸留水
各溶媒を用いて作ったマトリックスのサンプルを走査電子顕微鏡(SEM)分析にかけた。得られた結果を図17A-Gに示す。これらの図は、それぞれの懸濁液中のメタノールまたはエタノールのパーセントが増加すると、平均マトリックス孔サイズが減少することを示す。
【0200】
具体的には、2つの水懸濁液の孔サイズ(図17A及び17D)は平均して200ミクロンに近い。図17C及び17F(それぞれ25%メタノール及びエタノール)に示したサンプルの孔サイズは平均して30と50ミクロンの間にある。
【0201】
ここに示した結果は、エタノールとメタノールは両方ともp-GlcNAc孔サイズを制御するのに成功し、エタノールがメタノールより効率的でありうることを示唆する。
【0202】
15. 実施例:p-GlcNAc物質の生分解性
本節に記載する実施例は、制御可能なin vitro及びin vivo生物分解性及び再吸収速度を示すp-GlcNAc出発物質を調製しうることを実証する。
【0203】
15.1. 物質及び方法
p-GlcNAc物質:プロトタイプIを以上の第5.3.2節に記載した方法により、化学的方法を経由し、フッ化水素酸を化学試薬として用いて作った。プロトタイプIは100%アセチル化p-GlcNAcであることが示された。
【0204】
プロトタイプ3Aのp-GlcNAc出発物質を、以上の第5.3.2節に記載した方法により、化学的方法を経由し、フッ化水素酸を化学試薬として使うことによって作った。次いで、p-GlcNAc物質を、以上の第5.4節に記載した方法により脱アセチル化した。具体的には、p-GlcNAc物質を40% NaOH溶液を用いて60℃にて30分間処理した。得られるプロトタイプ3Aは、30%脱アセチル化されていることを確認した。
【0205】
プロトタイプ4のp-GlcNAc出発物質を、以上の第5.3.2節に記載した方法により、化学的方法を経由し、フッ化水素酸を化学試薬として使うことによって作った。次いでp-GlcNAc物質を40% NaOH溶液を用いて60℃にて、30分間処理することにより脱アセチル化した。次にその繊維を蒸留水中に懸濁し、-20℃にて凍結して凍結乾燥した。プロトタイプ4も、30%脱アセチル化されていることを確認した。
【0206】
腹部移植モデル:スプラグ・ドーリー・アルビノ(Sprague Dawley albino)ラットを使用して腹部移植モデル研究を行った。動物を麻酔して外科手術に備え、そして皮膚及び腹部筋肉を切開した。盲腸の位置を定めて外へ持ち上げた。1cm x 1cmのp-GlcNAc物質の膜を盲腸の上に置き、切開部をナイロンを用いて閉じた。対照動物は盲腸上に何も置かなかった。
【0207】
動物を、移植後14及び21日に開腹した。移植及び外植過程中に写真を撮影した(図23A-E)。外植過程後、組織病理学用の盲腸サンプルを調製した。
【0208】
p-GlcNAcのin vitro分解リゾチーム-キチナーゼアッセイ:本アッセイは、N-アセチルグルコサミンに対する比色アッセイであり、次のとおり実施した:反応サンプル150μlを13 x 100mmのガラス製使い捨て試験管中にピペットにより重複して採取し、0.25Mリン酸カリウムバッファー(pH 7.1)25μlをそれぞれの試験管に加え、次いで0.8Mホウ酸カリウム溶液(pH 9.8)35μlを加えた。試験管を直ちに氷浴に少なくとも2分間漬けた。次いでサンプルを氷浴から取り除き、新しく調製したDMAB試薬1mlを加え、そしてサンプルをボルテックス攪拌した。DMAB(ジメチルアミノベンズアルデヒド)試薬は、氷酢酸70ml及び11.6(濃)HCl 10mlをp-ジメチルアミノベンズアルデヒド8グラムに加えて作った。次いでサンプルを37℃にて20分間インキュベートした。
【0209】
標準曲線を調製するために、次の方法を利用した。GlcNAcストック溶液を0.010M酢酸ナトリウムバッファー(pH 4.5)を用いて0.1mg/mlに希釈し、希釈したGlcNAc溶液0μl、20μl、30μl、90μlまたは120μlを試験管のセットに加えた。次いで、0.010M酢酸ナトリウムバッファー(pH 4.5)150μl、130μl、60μlまたは30μlをそれぞれ試験管に加えた。次に、0.25Mリン酸カリウムバッファー(pH 7.1)25μl及び0.8Mホウ酸カリウムバッファー(pH 9.8)35μlをそれぞれの試験管に加えた。試験管の重複セットを同じ方法により調製した。
【0210】
試験管に栓をして100℃にて正確に3分間煮沸した。次いで試験管を氷浴に漬けた。試験管を氷浴から取出し、上記の方法に従って新しく調製したDMAB試薬1mlを各試験管に加えた。試験管を37℃にて20分間インキュベートした。各試験管の内容物の吸収を585nMにて読み取った。吸収はなるたけ速く読み取らねばならない。標準曲線をグラフ用紙上にプロットし、これを利用して反応サンプル中のN-アセチルグルコサミン濃度を決定した。典型的な標準曲線を図18に示す。
【0211】
15.2. 結果
p-GlcNAc物質のin-vitro生分解性を、p-GlcNAc膜物質のリゾチームによる分解に対する相対的感受性をアッセイする実験で研究した。p-GlcNAc膜を10mM酢酸バッファー中の過剰のリゾチームに曝し、その後のN-アセチルグルコサミンの放出を以上の第15.1節に記載したアッセイを用いて測定した。
【0212】
これらの実験の結果は、部分的に脱アセチル化された膜はリゾチームによる消化に対して、より感受性であること(図19を参照)及び、さらに、リゾチーム分解速度は脱アセチル化の程度と直接関係があること(図20を参照、50%脱アセチル化p-GlcNAc膜の分解速度を25%脱アセチル化p-GlcNAc膜に対して比較する)を示した。
【0213】
p-GlcNAcのin vivo分解
実験は、p-GlcNAc物質のin-vivo生分解性を目指して実施した。かかる実験は腹部移植モデルを利用した。次に掲げる3つのp-GlcNAc物質を試験した。
【0214】
試験したp-GlcNAc物質
1) 完全にアセチル化されたp-GlcNAc(プロトタイプ1と呼ぶ);
2) 部分的に脱アセチル化されたp-GlcNAc膜(プロトタイプ3Aと呼ぶ);及び
3) 凍結乾燥されかつ部分的に脱アセチル化されたp-GlcNAc膜(プロトタイプ4と呼ぶ)
結果
完全にアセチル化されたp-GlcNAc(プロトタイプ1)は、図21A-21Cに示すように21日内に再吸収された。部分的に脱アセチル化されたp-GlcNAc膜(プロトタイプ3A)は、図21D-21Eに示すように14日以内に完全に再吸収された。凍結乾燥しかつ部分的に脱アセチル化されたp-GlcNAc膜(プロトタイプ4)は、移植後21日の後にもなお完全には再吸収されなかった。
【0215】
組織病理学分析は、p-GlcNAc物質が再吸収されると、処理した動物から得た組織サンプルと対照動物からのそれとの間に検出しうる組織学的差異はないことを示した。
【0216】
16. 実施例:p-GIcNAcによるエンドセリン-1分泌の刺激と動脈血管収縮の誘導
本実施例は、本発明のp-GlcNAcを用いてin vivoでエンドセリン-1放出を刺激しかつ動脈血管収縮を誘導しうることを実証する。
【0217】
16.1. 大動脈切開の処置及び分析;材料及び方法
動物:本研究は、体重25〜30kg(平均27.5kg)の未熟雌性ヨークシャイア白ブタ(Yorkshire White swine)について実施した。次のプロトコルを全事例で使用した。
【0218】
プロトコル
1. 標準の前投薬の後に、動物を、100% O2及び1-2%ハロタンの吸入により麻酔する
2. CBC及び血小板計数のために対照血液サンプルを採取する
3. 腹部大動脈を露出する
4. タイでくくって固定し、大動脈に1cmの鉛直の傷を付ける
5. 試験材を適用する間、タイを解く
6. 1分間圧縮する
7. 圧縮を止めて、出血を観察する
8. もし出血すれば、ステップ4と5を繰り返す
9. もし15回の1分間圧縮で出血が停止しなければ、試験材は不合格である
10. 病理学用組織を回収する。
【0219】
16.2 脾臓切開の処置及び分析;材料及び方法
動物:本研究は、体重34〜37kgの未熟雌性ヨークシャイア白ブタ(Yorkshire White swine)について実施した。次のプロトコルを全事例で使用した。
【0220】
プロトコル
1. 標準の前投薬の後に、動物を、100% O2及び1-2%ハロタンの吸入により麻酔するCBC及び血小板計数のために対照血液サンプルを採取する
2. 絶対的止血を維持するため電気メスを用いて正中線腹部切開を行い、脾臓を摘出する
3. 脾臓をスポンジを用いて単離する
4. 脾臓表面上に深さ3mmの2cm x 2cm面積のカプセルストリップを作る
5. 傷から10秒間自由に出血させる
6. 蓄積した血液を外科スポンジを用いて除去する
7. 試験薬剤を適用する
8. 1分間、穏やかな圧力をかける
9. 圧力を除き、2分間出血を観察する
10. 傷が出血すれば、5及び6を繰り返す
11. 出血を制御するために必要な圧縮数及び止血までの時間を記録する
12. もし完全な出血の停止(出血の停止後2分間、再出血なしと定義する)が達成されれば記録する
13. 病理学研究用に組織を回収する。
【0221】
16.3 脾臓免疫染色のプロトコル
免疫染色は、わずかに改変を加えたPeninsula Laboratories社製ET-1染色キット(カタログ番号HIS-6901)を用いて実施した。
【0222】
スライド調製及び染色方法
1. 脾臓組織のサンプルを採取し、標準の方法を用いてスライド上にてサンプルをパラフィンに埋め込んで保存した。パラフィンは、その後、スライドを100%キシレン中で10分間インキュベートすることにより除去した。スライドを、100%エタノール、95%エタノール、及び次いで水道水のグレード別シリーズで各溶液に5回漬けることにより、再水和する。イム・エッジ(Imm Edge)耐水ペン(Vector Laboratories、カタログ番号H-4000)を用いて、組織サンプルの外周に線を引く。スライドをコプリンジャー(coplin jar)に入れたPBS pH7.4溶液中に保存する。
【0223】
2. 抗原アンマスキング溶液(Vector Laboratoriesカタログ番号H-3300)を100 Xに希釈し、30-45秒間、他のコプリンジャー内で加熱する。スライドをこの溶液へ移し20分間インキュベートする。乾燥し切るのを防止するために組織サンプルを覆うのに十分な溶液が存在することを確かめる。スライドをPBS pH7.4溶液を用いて2分間、よくリンス洗いする;2回繰り返す。過剰の溶液を除去するためにスライドを乾かすかまたは拭きとる。
【0224】
3. 正常のヤギ血清ブロッキング溶液の2滴もしくは100μLを各スライドに加える。室温にて20分間インキュベートする。スライドから過剰溶液を乾かすかまたは拭きとる。リンス洗いはしない。
【0225】
4. 凍結乾燥した一次抗体を、PBS pH7.4溶液32μlを用いて再構成する。このストック溶液から、一次抗体を400の希釈ファクターで希釈する。希釈した一次抗体の2滴もしくは100μLを各スライドに加える。スライドを、加湿室内の木製スティック上に水平に置き、一夜4℃にてインキュベートする。PBS pH7.4溶液を用いて2分間よくリンス洗いする;2回繰り返す。
【0226】
5. ビオチン化二次抗体の2滴もしくは100μLを各スライドに加える。30分間室温でインキュベートする。PBS pH7.4溶液を用いて2分間よくリンス洗いする;2回繰り返す。
【0227】
6. スライドを、コプリン・ジャーに入った3%証明付き過酸化水素(Fisherカタログ番号H 312-500)中に入れ、30分間室温にてクエンチする。PBS pH7.4溶液を用いて2分間、よくリンス洗いする;2回繰り返す。
【0228】
7. ストレプトアビジン-HRPコンジュゲート2滴または100μLを各スライドに加え、30分間室温でインキュベートする。PBS pH7.4溶液を用いて2分間、よくリンス洗いする;2回繰り返す。
【0229】
8. 蒸留水5.0mLをガラス製シンチレーション・バイアルに加えてDABクロマーゲン(Chromagen)溶液(Vector Laboratoriesカタログ番号sk-41067)を作る。バッファーストック溶液2滴を加えてよく混合する。次いで、DABストック溶液4滴を加えてよく混合する。最後に、過酸化水素溶液2滴を加えてよく混合する。DABクロマーゲン溶液200μLを各スライドに加える。3分間室温にてインキュベートする。蒸留水を用いてよくリンス洗いして拭きとる。
【0230】
9. スライドを、0.2%Working Light Green Solution(Sigmaカタログ番号L 5382)の希釈ファクター6のストック溶液を用いてカウンター染色する。スライドを3回Working Light Green Solutionに漬け、次いでスライドを5回それぞれ、脱水シリーズ、すなわち蒸留水、次いで95%エタノール、次いで100%エタノール、そして最後に100%キシレンに漬ける。スライドを乾かすかまたは拭きとって過剰キシレンを除去する。
【0231】
10. Cytoseal XYLマウンティング溶液(Stephens Scientificカタログ番号8312-4)の2滴を加え、スライドをマウントする。
【0232】
16.4 動脈の免疫染色プロトコル
動脈組織の免疫染色を、若干の改変を加えたPeninsula Laboratories社(カタログ番号HIS-6901)のET-1染色キットを用いて実施した。
【0233】
スライド調製
1. 肺動脈を、購入したシカから切除する
2. 動脈をRPMI培地100mL中に入れ、氷上に置く
3. メスを用いて動脈を切開する
4. 完全にアセチル化されたp-GlcNAc繊維からなる1cm X 1cm正方の膜で、切開部を覆って15分間置く
5. 膜適用部位において動脈の横断セクション薄片を作り、組織学研究用とする
6. そのセクションを9%ホルムアルデヒド中に置く。パラフィンを用いてスライドを作る。
【0234】
染色方法
1. スライドを10分間、100%キシレン中でインキュベートし、脱パラフィン化する。スライドを100%エタノール、95%エタノール、及び次いで水道水のグレード別シリーズを用い、それぞれの溶液に5回漬けて再水和する。イム・エッジ(Imm Edge)耐水ペン(Vector Laboratories、カタログ番号H-4000)を用いて、組織サンプルの外周に線を引く。スライドをコプリンジャー(coplin jar)に入れたPBS pH7.4溶液中に保存する。
【0235】
2. 抗原アンマスキング溶液(Vector Laboratoriesカタログ番号H-3300)を100倍希釈し、30-45秒間、他のコプリン・ジャー内で加熱する。スライドをこの溶液へ移し20分間インキュベートする。乾燥し切るのを防止するために組織サンプルを覆うのに十分な溶液をあることを確かめる。スライドをPBS pH7.4溶液を用いて2分間、よくリンス洗いする。2回繰り返す。過剰溶液を除去するためにスライドを乾かすかまたは拭きとる。
【0236】
3. 正常のヤギ血清ブロッキング溶液の2滴もしくは100μLを各スライドに加える。室温にて20分間インキュベートする。スライドから過剰溶液を乾かすかまたは拭きとる。リンス洗いはしない。
【0237】
4. 凍結乾燥した一次抗体をPBS pH7.4溶液32μlを用いて再構築する。このストック溶液から、一次抗体を希釈ファクター100で希釈する。希釈した一次抗体の2滴もしくは100μLを各スライドに加える。スライドを、加湿室内の木製スティック上に水平に置き、一夜4℃にてインキュベートする。PBS pH7.4溶液を用いて2分間よくリンス洗いする;2回繰り返す。
【0238】
5. ビオチン化二次抗体の2滴もしくは100μLを各スライドに加える。30分間室温でインキュベートする。PBS pH7.4溶液を用いて2分間よくリンス洗いする;2回繰り返す。
6. スライドを、コプリンジャーに入った3%証明付き過酸化水素(Fisherカタログ番号H 312-500)中に入れ、30分間室温にてクエンチする。PBS pH7.4を用いて2分間、よくリンス洗いする;2回繰り返す。
【0239】
7. ストレプトアビジン-HRPコンジュゲート2滴または100μLを各スライドに加え、30分間室温でインキュベートする。PBS pH7.4溶液を用いて2分間よくリンス洗いする;2回繰り返す。
【0240】
8. 蒸留水5.0 mLをガラス製シンチレーション・バイアルに加えることによりDABクロマーゲン-溶液(Vector Laboratoriesカタログ番号sk-41067)を作る。バッファーストック溶液2滴を加えてよく混合する。次いで、DABストック溶液4滴を加えてよく混合する。最後に、過酸化水素溶液2滴を加えてよく混合する。DABクロマーゲン-溶液200μLを各スライドに加える。3分間室温にてインキュベートする。蒸留水を用いてよくリンス洗いしかつ拭きとる。
【0241】
9. スライドを、0.2%Working Light Green Solution(Sigmaカタログ番号L 5382)の希釈ファクター6のストック溶液を用いてカウンター染色する。スライドを3回Working Light Green Solutionに漬け、次いでスライドを、脱水シリーズ、すなわち蒸留水、次いで95%エタノール、次いで100%エタノール、そして最後に100%キシレンにそれぞれ5回漬ける。スライドを乾かすかまたは拭きとって過剰キシレンを除去する。
【0242】
10. Cytoseal XYLマウンティング溶液(Stephens Scientificカタログ番号H 8312-4)の2滴を加え、スライドをマウントする。
【0243】
結果
完全にアセチル化されたp-GlcNAc繊維からなる膜を用いて処理した動脈組織の組織学的及び免疫学的試験は、傷害動脈組織とp-GlcNAcポリマーとの接触部位において即効性の血管収縮刺激を与えた。p-GlcNAc膜の適用により誘導される血管収縮は、組織学的により大きい実験動物においてより容易に見られた。動脈組織の収縮は、p-GlcNAc膜が適用された動脈サイドにおいてより顕著である。これらの分析結果を図23及び図24に示した。ガーゼ包帯のブタ動脈へを適用した60分後に(図23(A)、及び図24、サンプルA)、動脈壁とガーゼとの接触点(1)において、またはガーゼ包帯を適用したのと反対側の点において測定したところ、動脈壁厚さの値は比較しうるものあった。対照的に、半結晶p-GlcNAcで製剤化した膜のブタ動脈への適用(図23 (B)、図24、サンプルB)すると、膜を適用した15分後に、接触領域における壁厚さ(1)の著しい増加が誘導された。接触の60分後、p-GlcNAc膜との接触領域で測定した動脈壁の厚み(1)は、動脈の反対側の点で測定した厚み(2)と比較しうるレベルに戻った。
【0244】
エンドセリン-1に対する抗体を用いた免疫染色実験は、p-GlcNAc膜と生組織との間の接触部位におけるエンドセリン-1の分泌を示した。シカ肺動脈を用いたin vitro実験は、p-GlcNAc膜と動脈との接触表面上にのみエンドセリン-1が存在することを示した。In vivo実験は、処理した組織とp-GlcNAc膜との間の接触表面上においてだけでなく、組織のより深い層にも実質的に大きなエンドセリン-1放出を示した。p-GlcNAc膜の適用後15分以内に、処理組織とp-GlcNAc膜との間の接触のほんの60分後に実施した類似の分析と比較して、大きいエンドセリン-1の分泌が検出された。それにも関わらず、収縮効果は試験したその他のサンプルより強かった。
【0245】
他のサンプルのスライドについて同じエンドセリン-1免疫染色を観察したが、その結果はp-GlcNAcより遥かに低かった。p-GlcNAc膜と接触した脾臓組織の組織学的及び免疫学的分析は、エンドセリン-1放出の類似した亢進を示した。再び、実験膜の適用後15分以内に、エンドセリン-1はp-GlcNAc膜が適用されていたサンプルにおいてだけ観察された。実験膜と処理組織との間の接触の60分後に、全てのサンプルは比較しうるエンドセリン-1のレベルを示した。
【0246】
17. 実施例:血液産物不在のもとでの、p-GlcNAcによる血管収縮及びエンドセリン放出の誘導
本実施例は、本発明の完全にアセチル化された半結晶p-GlcNAcが、血液不在のもとで動脈血管収縮を誘導することを実証する。より具体的には、本実施例は、完全にアセチル化されたp-GlcNAcは、血液凝固カスケードのいずれの成分の不在のもとでも、内皮に依存する機構を経由して、部分的に内皮細胞からのエンドセリン-1放出により、単離されたラット大動脈環を有意に収縮することを実証する。
【0247】
17.1 物質及び方法
大動脈環を体重275-300gの雄性スプラグ・ドーリイ(Sprague-Dawley)ラットから得た。ラットを、腹腔内にペントバルビタールナトリウム(60 mg/kg)を注射して麻酔した。大動脈及びSMAをラットから急いで除去し、加温したクレープス-ヘンゼライト(Krebs-Henseleit)(KH)バッファー(単位mmol/lで、NaCl 118、KCl 4.75、CaCl2.2H2O 2.54、KH2PO4 1.19、MgSO4・7H2O 1.19、NaHCO3 12.5、及びグルコース 10.0からなる)中に懸濁した。単離した血管から注意深く結合組織を除去し、長さ2-3mmの環に切断した。次いで環をステンレス鋼フック上にマウントし、10-ml組織容器中に懸濁し、そしてFT-03力-変位変換器(Grass Instrument、Quincy、Mass.)と接続してGrass model 7 オシログラフレコーダー上で力の変化を記録した。容器をKHバッファーを用いて満たし、37℃にて95%O2+5%CO2を通気した。0.5gの静止力をSMA環に適用し、次いで環を90分間、平衡化した。この期間に、組織容器中のバッファーを15-20分毎に取り換え、血管環の静止力を調節して0.5gの前負荷を維持した。平衡化の90〜120分後に、環を100nM U-46619(9,11-ジデオキシ-9α-11α-メタンエポキシ-プロスタグランジンF2α;Biomol Research Laboratories、Plymouth Meeting、Pa.)、トロンボキサンA2模倣物に曝し、1.0gの発現力(developed force)を発生させた。安定した収縮を得ると、その容器に、典型的な内皮依存血管収縮薬であるアセチルコリンを0.1、1、10、及び100nMの累積濃度で加え、内皮の保全性を評価した。累積応答が安定化した後、環を洗浄し、再び、ベースラインに平衡化させた。
【0248】
この方法を、U-46619、次いでp-GlcNAcを用いて繰返した。p-GlcNAcは、図23に示すように14〜140g/mlで濃度依存的血管収縮を起こした。140g/mlの発生濃度において、p-GlcNAcは発現力218 ± 21mgで大動脈環を有意に収縮した(p<0.01)。内皮を剥いだ(すなわち、綿で覆われたひねりステンレス鋼ワイヤ上に大動脈環を静かに廻して内皮を除去した)大動脈環は、発現力33 ± 12mgでしか収縮しなかった。エンドセリンEtA受容体アンタゴニスト、JKC-301(シクロ[D-Asp-Pro-D-IIe-Leu-D-Trp]、Sigma Biochemicals and Reagents、St. Louis、Mo.)(0.5及び1M)による前処理は、p-GlcNAcが誘導する血管収縮を57〜61%だけ有意に減少させた。
【0249】
この方法を、U-46619を用い、次いで70%脱アセチル化p-GlcNAcにより繰返した。先に使用した完全にアセチル化した半結晶p-GlcNAcを70%脱アセチル化p-GlcNAcに置き換えると、この血液を含まないモデルシステムでは、全ての試験濃度において血管収縮が起こらなかった。
【0250】
本明細書で説明した本発明の多くの改変及び変法が、本発明の精神と範囲を逸脱することなしになされうることは明白である。上記の特定の実施形態は、例示としてだけ与えられたものであって、本発明は添付した請求項によってのみ制限されるものである。
【0251】
様々な出版物が本明細書に引用されたが、これらの開示は参照によりその全文が組み入れられる。
【特許請求の範囲】
【請求項1】
エンドセリン-1放出の刺激、血管収縮及び破裂した血管からの血液流出の減少よりなる群から選択される少なくとも1以上の一過性で局所性の生理学的応答を達成するための医薬の製造のためのポリ-β-1→4-N-アセチルグルコサミンポリマーを含むバリヤー非形成物質の使用であって、前記医薬が前記応答を必要とする患者に局所投与されるものである、前記使用。
【請求項2】
生理学的応答がエンドセリン-1放出の刺激を含む、請求項1に記載の使用。
【請求項3】
エンドセリン-1が血管内皮細胞から放出される、請求項2に記載の使用。
【請求項4】
生理学的応答が血管収縮を含む、請求項1に記載の使用
【請求項5】
生理学的応答が破裂した血管からの血液流出の減少を含む、請求項1に記載の使用。
【請求項6】
破裂した血管からの血液流出の減少が破裂した血管からの血液流出の停止を含む、請求項5に記載の使用。
【請求項7】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがβ-1→4コンフォメーションで共有結合した50〜150,000個のN-アセチルグルコサミン単糖を含んでなり、前記ポリマーが10,000ダルトン〜30 x 106ダルトンの分子量を有する、請求項1に記載の使用。
【請求項8】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがβ-1→4コンフォメーションで共有結合した50〜50,000個のN-アセチルグルコサミン単糖を含んでなり、前記ポリマーが10,000ダルトン〜10 x 106ダルトンの分子量を有する、請求項7に記載の使用。
【請求項9】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがβ-1→4コンフォメーションで共有結合した50〜10,000個のN-アセチルグルコサミン単糖を含んでなり、前記ポリマーが10,000ダルトン〜2 x 106ダルトンの分子量を有する、請求項8に記載の使用。
【請求項10】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがβ-1→4コンフォメーションで共有結合した50〜4,000個のN-アセチルグルコサミン単糖を含んでなり、前記ポリマーが10,000ダルトン〜800,000ダルトンの分子量を有する、請求項9に記載の使用。
【請求項11】
ポリ-β-1→4-N-アセチルグルコサミンポリマーが少なくとも1個のアセチル化されていないグルコサミン単糖単位を含んでなり、かつグルコサミン単糖単位の少なくとも40%がN-アセチル化されている、請求項7に記載の使用。
【請求項12】
患者がヒトである、請求項1に記載の使用。
【請求項13】
物質がフォーム、スプレイ、エマルジョン、懸濁液または溶液の形態である、請求項1に記載の使用。
【請求項14】
物質が血管に直接適用するためのものである、請求項1に記載の使用。
【請求項15】
生理学的応答が毛細管、静脈、及び動脈よりなる群から選択される血管に影響を及ぼす、請求項1に記載の使用。
【請求項16】
血管が破裂した血管である、請求項15に記載の使用。
【請求項17】
生理学的応答が出血の停止を含む、請求項16に記載の使用。
【請求項18】
生理学的応答の程度が、投与されたポリ-β-1→4-N-アセチルグルコサミンポリマーの量に比例する、請求項1に記載の使用。
【請求項19】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがタンパク質を含んでいない、請求項1に記載の使用。
【請求項20】
ポリ-β-1→4-N-アセチルグルコサミンポリマーが半結晶である、請求項1に記載の使用。
【請求項21】
β-1→4コンフォメーションで共有結合した50〜150,000個のN-アセチルグルコサミン単糖を含むポリ-β-1→4-N-アセチルグルコサミンポリマーを含む、生分解性でバリヤーを形成しない物質であって、前記ポリマーが10,000ダルトン〜30 x 106ダルトンの分子量を有する前記物質。
【請求項22】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがβ-1→4コンフォメーションで共有結合した50〜50,000個のN-アセチルグルコサミン単糖を含んでなり、そして前記ポリマーが10,000ダルトン〜10 x 106ダルトンの分子量を有する、請求項21に記載の物質。
【請求項23】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがβ-1→4コンフォメーションで共有結合した50〜10,000個のN-アセチルグルコサミン単糖を含んでなり、そして前記ポリマーが10,000ダルトン〜2 x 106ダルトンの分子量を有する、請求項22に記載の物質。
【請求項24】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがβ-1→4コンフォメーションで共有結合した50〜4,000個のN-アセチルグルコサミン単糖を含んでなり、そして前記ポリマーが10,000ダルトン〜800,000ダルトンの分子量を有する、請求項23に記載の物質。
【請求項25】
ポリ-β-1→4-N-アセチルグルコサミンポリマーが少なくとも1個のアセチル化されていないグルコサミン単糖単位を含んでなり、かつグルコサミン単糖単位の少なくとも40%がN-アセチル化されている、請求項21に記載の物質。
【請求項26】
物質がフォーム、スプレイ、エマルジョン、懸濁液または溶液である、請求項21に記載の物質。
【請求項27】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがタンパク質を含んでいない、請求項21に記載の物質。
【請求項28】
ポリ-β-1→4-N-アセチルグルコサミンポリマーが半結晶である、請求項21に記載の物質。
【請求項29】
血管障害を有する患者を治療するための医薬の製造のためのポリ-β-1→4-N-アセチルグルコサミンポリマーを含むバリヤー非形成物質の使用であって、前記医薬が血管障害を有する患者に局所投与されるものである前記使用。
【請求項30】
血管障害が、月経過多、脳性動脈瘤、腹部動脈瘤、子宮類線維腫病変及び血管破裂よりなる群から選択される、請求項29に記載の使用。
【請求項31】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがβ-1→4コンフォメーションで共有結合した50〜150,000個のN-アセチルグルコサミン単糖を含んでなり、そして前記ポリマーが10,000ダルトン〜30 x 106ダルトンの分子量を有する、請求項29に記載の使用。
【請求項32】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがβ-1→4コンフォメーションで共有結合した50〜50,000個のN-アセチルグルコサミン単糖を含んでなり、そして前記ポリマーが10,000ダルトン〜10 x 106ダルトンの分子量を有する、請求項31に記載の使用。
【請求項33】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがβ-1→4コンフォメーションで共有結合した50〜10,000個のN-アセチルグルコサミン単糖を含んでなり、そして前記ポリマーが10,000ダルトン〜2 x 106ダルトンの分子量を有する、請求項32に記載の使用。
【請求項34】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがβ-1→4コンフォメーションで共有結合した50〜4,000個のN-アセチルグルコサミン単糖を含んでなり、そして前記ポリマーが10,000ダルトン〜800,000ダルトンの分子量を有する、請求項33に記載の使用。
【請求項35】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがタンパク質を含んでいない、請求項29に記載の使用。
【請求項36】
ポリ-β-1→4-N-アセチルグルコサミンポリマーが半結晶である、請求項29に記載の使用。
【請求項37】
局所投与が患者における生理学的応答を達成し、生理学的応答がエンドセリン-1放出の刺激を含む、請求項29に記載の使用。
【請求項38】
局所投与が患者における生理学的応答を達成し、生理学的応答が血管収縮を含む、請求項29に記載の使用。
【請求項39】
局所投与が患者における生理学的応答を達成し、生理学的応答が破裂した血管からの血液流出の減少を含む、請求項29に記載の使用。
【請求項40】
物質がフォーム、スプレイ、エマルジョン、懸濁液または溶液の形態である、請求項29に記載の使用。
【請求項41】
患者の血管における破裂又は穿刺傷を治療するための医薬の製造のためのポリ-β-1→4-N-アセチルグルコサミンポリマーを含むフィルム又は膜の使用であって、破裂又は穿刺傷が心臓カテーテル処置又はバルーン血管形成処置によって引き起こされ、前記医薬が血管破裂又は穿刺傷の部位で圧迫して患者の皮膚に直接適用されるものであり、前記ポリ-β-1→4-N-アセチルグルコサミンポリマーの量が破裂又は穿刺傷からの血液流出を停止又は減少させるのに有効であり、それにより前記破裂又は穿刺傷を治療する、前記使用。
【請求項42】
患者の破裂又は穿刺した血管における止血を達成するための時間を短縮するための医薬の製造のためのポリ-β-1→4-N-アセチルグルコサミンポリマーの量を含むフィルム又は膜の使用であって、破裂又は穿刺傷が心臓カテーテル処置又はバルーン血管形成処置によって引き起こされ、前記医薬が、単独で圧迫するのと比較して止血を達成するための時間を短縮するために、血管破裂又は穿刺傷の部位で圧迫して患者の皮膚に直接適用されるものであり、それにより前記破裂又は穿刺した血管における止血を達成するための時間を短縮する、前記使用。
【請求項43】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがβ-1→4コンフォメーションで共有結合した50〜150,000個のN-アセチルグルコサミン単糖を含んでなり、そして前記ポリマーが10,000ダルトン〜30 x 106ダルトンの分子量を有する、請求項41又は42に記載の使用。
【請求項44】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがβ-1→4コンフォメーションで共有結合した50〜50,000個のN-アセチルグルコサミン単糖を含んでなり、そして前記ポリマーが10,000ダルトン〜10 x 106ダルトンの分子量を有する、請求項43に記載の使用。
【請求項45】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがβ-1→4コンフォメーションで共有結合した50〜10,000個のN-アセチルグルコサミン単糖を含んでなり、そして前記ポリマーが10,000ダルトン〜2 x 106ダルトンの分子量を有する、請求項44に記載の使用。
【請求項46】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがβ-1→4コンフォメーションで共有結合した50〜4,000個のN-アセチルグルコサミン単糖を含んでなり、そして前記ポリマーが10,000ダルトン〜800,000ダルトンの分子量を有する、請求項45に記載の使用。
【請求項47】
ポリ-β-1→4-N-アセチルグルコサミンポリマーが少なくとも1個のアセチル化されていないグルコサミン単糖単位を含んでなり、かつグルコサミン単糖単位の少なくとも40%がN-アセチル化されている、請求項43に記載の使用。
【請求項48】
ポリ-β-1→4-N-アセチルグルコサミンポリマーが半結晶である、請求項41又は42に記載の使用。
【請求項49】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがタンパク質を含んでいない、請求項41又は42に記載の使用。
【請求項50】
患者がヒトである、請求項41又は42に記載の使用。
【請求項51】
フィルム又は膜がバリヤー形成物質を含む、請求項41又は42に記載の使用。
【請求項52】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがマット、ひも、ミクロビーズ、ミクロスフィア、又はミクロフィブリルとして製剤化される、請求項41又は42に記載の使用。
【請求項53】
医薬がコラーゲンをさらに含む、請求項41又は42に記載の使用。
【請求項54】
医薬が1ミクロン未満の厚さの超薄型の均一な膜をさらに含む、請求項41又は42に記載の使用。
【請求項55】
医薬が医薬担体をさらに含む、請求項41又は42に記載の使用。
【請求項56】
医薬が血液凝固カスケードに必要とされる少なくとも1種の因子さらに含む、請求項41又は42に記載の使用。
【請求項57】
因子がトロンビン、フィブリノーゲン及び第XIII因子よりなる群から選択される、請求項56に記載の使用。
【請求項58】
医薬が血管収縮剤をさらに含む、請求項41又は42に記載の使用。
【請求項59】
血管収縮剤がU-46619である、請求項58に記載の使用。
【請求項60】
医薬がクマヂン又はヘパリンをさらに含む、請求項41又は42に記載の使用。
【請求項1】
エンドセリン-1放出の刺激、血管収縮及び破裂した血管からの血液流出の減少よりなる群から選択される少なくとも1以上の一過性で局所性の生理学的応答を達成するための医薬の製造のためのポリ-β-1→4-N-アセチルグルコサミンポリマーを含むバリヤー非形成物質の使用であって、前記医薬が前記応答を必要とする患者に局所投与されるものである、前記使用。
【請求項2】
生理学的応答がエンドセリン-1放出の刺激を含む、請求項1に記載の使用。
【請求項3】
エンドセリン-1が血管内皮細胞から放出される、請求項2に記載の使用。
【請求項4】
生理学的応答が血管収縮を含む、請求項1に記載の使用
【請求項5】
生理学的応答が破裂した血管からの血液流出の減少を含む、請求項1に記載の使用。
【請求項6】
破裂した血管からの血液流出の減少が破裂した血管からの血液流出の停止を含む、請求項5に記載の使用。
【請求項7】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがβ-1→4コンフォメーションで共有結合した50〜150,000個のN-アセチルグルコサミン単糖を含んでなり、前記ポリマーが10,000ダルトン〜30 x 106ダルトンの分子量を有する、請求項1に記載の使用。
【請求項8】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがβ-1→4コンフォメーションで共有結合した50〜50,000個のN-アセチルグルコサミン単糖を含んでなり、前記ポリマーが10,000ダルトン〜10 x 106ダルトンの分子量を有する、請求項7に記載の使用。
【請求項9】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがβ-1→4コンフォメーションで共有結合した50〜10,000個のN-アセチルグルコサミン単糖を含んでなり、前記ポリマーが10,000ダルトン〜2 x 106ダルトンの分子量を有する、請求項8に記載の使用。
【請求項10】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがβ-1→4コンフォメーションで共有結合した50〜4,000個のN-アセチルグルコサミン単糖を含んでなり、前記ポリマーが10,000ダルトン〜800,000ダルトンの分子量を有する、請求項9に記載の使用。
【請求項11】
ポリ-β-1→4-N-アセチルグルコサミンポリマーが少なくとも1個のアセチル化されていないグルコサミン単糖単位を含んでなり、かつグルコサミン単糖単位の少なくとも40%がN-アセチル化されている、請求項7に記載の使用。
【請求項12】
患者がヒトである、請求項1に記載の使用。
【請求項13】
物質がフォーム、スプレイ、エマルジョン、懸濁液または溶液の形態である、請求項1に記載の使用。
【請求項14】
物質が血管に直接適用するためのものである、請求項1に記載の使用。
【請求項15】
生理学的応答が毛細管、静脈、及び動脈よりなる群から選択される血管に影響を及ぼす、請求項1に記載の使用。
【請求項16】
血管が破裂した血管である、請求項15に記載の使用。
【請求項17】
生理学的応答が出血の停止を含む、請求項16に記載の使用。
【請求項18】
生理学的応答の程度が、投与されたポリ-β-1→4-N-アセチルグルコサミンポリマーの量に比例する、請求項1に記載の使用。
【請求項19】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがタンパク質を含んでいない、請求項1に記載の使用。
【請求項20】
ポリ-β-1→4-N-アセチルグルコサミンポリマーが半結晶である、請求項1に記載の使用。
【請求項21】
β-1→4コンフォメーションで共有結合した50〜150,000個のN-アセチルグルコサミン単糖を含むポリ-β-1→4-N-アセチルグルコサミンポリマーを含む、生分解性でバリヤーを形成しない物質であって、前記ポリマーが10,000ダルトン〜30 x 106ダルトンの分子量を有する前記物質。
【請求項22】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがβ-1→4コンフォメーションで共有結合した50〜50,000個のN-アセチルグルコサミン単糖を含んでなり、そして前記ポリマーが10,000ダルトン〜10 x 106ダルトンの分子量を有する、請求項21に記載の物質。
【請求項23】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがβ-1→4コンフォメーションで共有結合した50〜10,000個のN-アセチルグルコサミン単糖を含んでなり、そして前記ポリマーが10,000ダルトン〜2 x 106ダルトンの分子量を有する、請求項22に記載の物質。
【請求項24】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがβ-1→4コンフォメーションで共有結合した50〜4,000個のN-アセチルグルコサミン単糖を含んでなり、そして前記ポリマーが10,000ダルトン〜800,000ダルトンの分子量を有する、請求項23に記載の物質。
【請求項25】
ポリ-β-1→4-N-アセチルグルコサミンポリマーが少なくとも1個のアセチル化されていないグルコサミン単糖単位を含んでなり、かつグルコサミン単糖単位の少なくとも40%がN-アセチル化されている、請求項21に記載の物質。
【請求項26】
物質がフォーム、スプレイ、エマルジョン、懸濁液または溶液である、請求項21に記載の物質。
【請求項27】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがタンパク質を含んでいない、請求項21に記載の物質。
【請求項28】
ポリ-β-1→4-N-アセチルグルコサミンポリマーが半結晶である、請求項21に記載の物質。
【請求項29】
血管障害を有する患者を治療するための医薬の製造のためのポリ-β-1→4-N-アセチルグルコサミンポリマーを含むバリヤー非形成物質の使用であって、前記医薬が血管障害を有する患者に局所投与されるものである前記使用。
【請求項30】
血管障害が、月経過多、脳性動脈瘤、腹部動脈瘤、子宮類線維腫病変及び血管破裂よりなる群から選択される、請求項29に記載の使用。
【請求項31】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがβ-1→4コンフォメーションで共有結合した50〜150,000個のN-アセチルグルコサミン単糖を含んでなり、そして前記ポリマーが10,000ダルトン〜30 x 106ダルトンの分子量を有する、請求項29に記載の使用。
【請求項32】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがβ-1→4コンフォメーションで共有結合した50〜50,000個のN-アセチルグルコサミン単糖を含んでなり、そして前記ポリマーが10,000ダルトン〜10 x 106ダルトンの分子量を有する、請求項31に記載の使用。
【請求項33】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがβ-1→4コンフォメーションで共有結合した50〜10,000個のN-アセチルグルコサミン単糖を含んでなり、そして前記ポリマーが10,000ダルトン〜2 x 106ダルトンの分子量を有する、請求項32に記載の使用。
【請求項34】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがβ-1→4コンフォメーションで共有結合した50〜4,000個のN-アセチルグルコサミン単糖を含んでなり、そして前記ポリマーが10,000ダルトン〜800,000ダルトンの分子量を有する、請求項33に記載の使用。
【請求項35】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがタンパク質を含んでいない、請求項29に記載の使用。
【請求項36】
ポリ-β-1→4-N-アセチルグルコサミンポリマーが半結晶である、請求項29に記載の使用。
【請求項37】
局所投与が患者における生理学的応答を達成し、生理学的応答がエンドセリン-1放出の刺激を含む、請求項29に記載の使用。
【請求項38】
局所投与が患者における生理学的応答を達成し、生理学的応答が血管収縮を含む、請求項29に記載の使用。
【請求項39】
局所投与が患者における生理学的応答を達成し、生理学的応答が破裂した血管からの血液流出の減少を含む、請求項29に記載の使用。
【請求項40】
物質がフォーム、スプレイ、エマルジョン、懸濁液または溶液の形態である、請求項29に記載の使用。
【請求項41】
患者の血管における破裂又は穿刺傷を治療するための医薬の製造のためのポリ-β-1→4-N-アセチルグルコサミンポリマーを含むフィルム又は膜の使用であって、破裂又は穿刺傷が心臓カテーテル処置又はバルーン血管形成処置によって引き起こされ、前記医薬が血管破裂又は穿刺傷の部位で圧迫して患者の皮膚に直接適用されるものであり、前記ポリ-β-1→4-N-アセチルグルコサミンポリマーの量が破裂又は穿刺傷からの血液流出を停止又は減少させるのに有効であり、それにより前記破裂又は穿刺傷を治療する、前記使用。
【請求項42】
患者の破裂又は穿刺した血管における止血を達成するための時間を短縮するための医薬の製造のためのポリ-β-1→4-N-アセチルグルコサミンポリマーの量を含むフィルム又は膜の使用であって、破裂又は穿刺傷が心臓カテーテル処置又はバルーン血管形成処置によって引き起こされ、前記医薬が、単独で圧迫するのと比較して止血を達成するための時間を短縮するために、血管破裂又は穿刺傷の部位で圧迫して患者の皮膚に直接適用されるものであり、それにより前記破裂又は穿刺した血管における止血を達成するための時間を短縮する、前記使用。
【請求項43】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがβ-1→4コンフォメーションで共有結合した50〜150,000個のN-アセチルグルコサミン単糖を含んでなり、そして前記ポリマーが10,000ダルトン〜30 x 106ダルトンの分子量を有する、請求項41又は42に記載の使用。
【請求項44】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがβ-1→4コンフォメーションで共有結合した50〜50,000個のN-アセチルグルコサミン単糖を含んでなり、そして前記ポリマーが10,000ダルトン〜10 x 106ダルトンの分子量を有する、請求項43に記載の使用。
【請求項45】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがβ-1→4コンフォメーションで共有結合した50〜10,000個のN-アセチルグルコサミン単糖を含んでなり、そして前記ポリマーが10,000ダルトン〜2 x 106ダルトンの分子量を有する、請求項44に記載の使用。
【請求項46】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがβ-1→4コンフォメーションで共有結合した50〜4,000個のN-アセチルグルコサミン単糖を含んでなり、そして前記ポリマーが10,000ダルトン〜800,000ダルトンの分子量を有する、請求項45に記載の使用。
【請求項47】
ポリ-β-1→4-N-アセチルグルコサミンポリマーが少なくとも1個のアセチル化されていないグルコサミン単糖単位を含んでなり、かつグルコサミン単糖単位の少なくとも40%がN-アセチル化されている、請求項43に記載の使用。
【請求項48】
ポリ-β-1→4-N-アセチルグルコサミンポリマーが半結晶である、請求項41又は42に記載の使用。
【請求項49】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがタンパク質を含んでいない、請求項41又は42に記載の使用。
【請求項50】
患者がヒトである、請求項41又は42に記載の使用。
【請求項51】
フィルム又は膜がバリヤー形成物質を含む、請求項41又は42に記載の使用。
【請求項52】
ポリ-β-1→4-N-アセチルグルコサミンポリマーがマット、ひも、ミクロビーズ、ミクロスフィア、又はミクロフィブリルとして製剤化される、請求項41又は42に記載の使用。
【請求項53】
医薬がコラーゲンをさらに含む、請求項41又は42に記載の使用。
【請求項54】
医薬が1ミクロン未満の厚さの超薄型の均一な膜をさらに含む、請求項41又は42に記載の使用。
【請求項55】
医薬が医薬担体をさらに含む、請求項41又は42に記載の使用。
【請求項56】
医薬が血液凝固カスケードに必要とされる少なくとも1種の因子さらに含む、請求項41又は42に記載の使用。
【請求項57】
因子がトロンビン、フィブリノーゲン及び第XIII因子よりなる群から選択される、請求項56に記載の使用。
【請求項58】
医薬が血管収縮剤をさらに含む、請求項41又は42に記載の使用。
【請求項59】
血管収縮剤がU-46619である、請求項58に記載の使用。
【請求項60】
医薬がクマヂン又はヘパリンをさらに含む、請求項41又は42に記載の使用。
【図1】

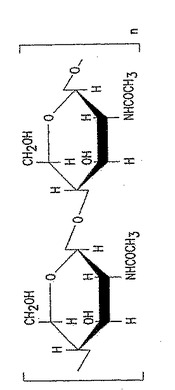
【図2】
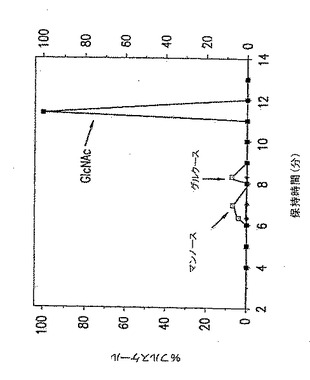
【図3A】
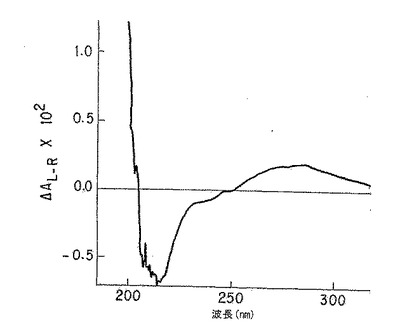
【図3B】
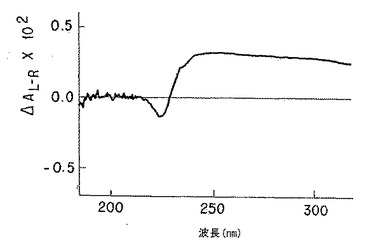
【図4A】
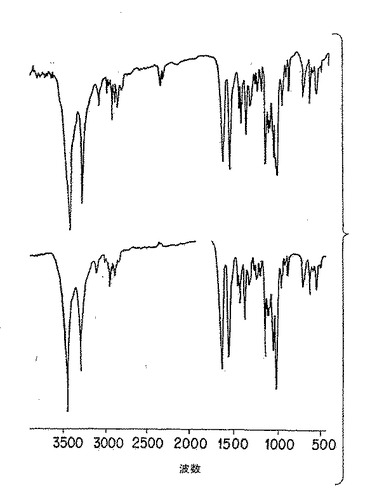
【図4B】
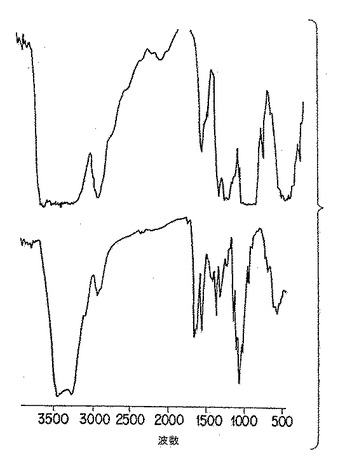
【図4C】
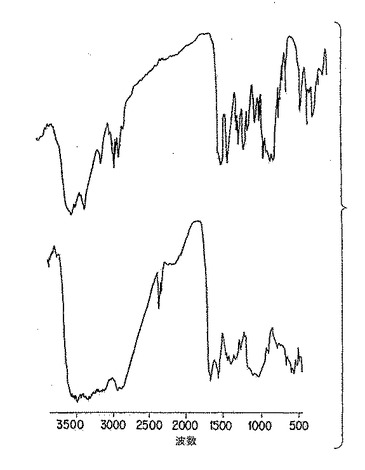
【図4D】
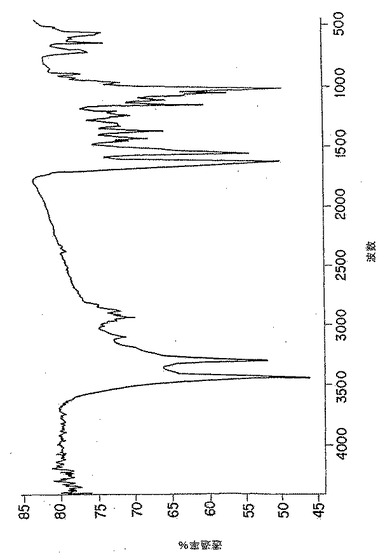
【図4E】
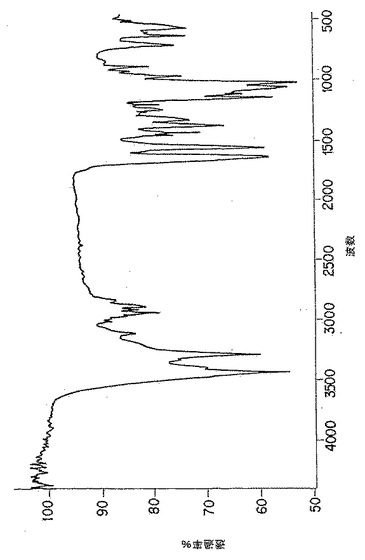
【図5A】
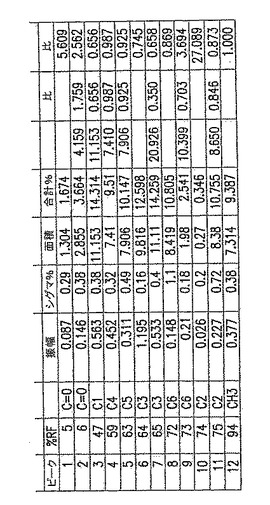
【図5B】
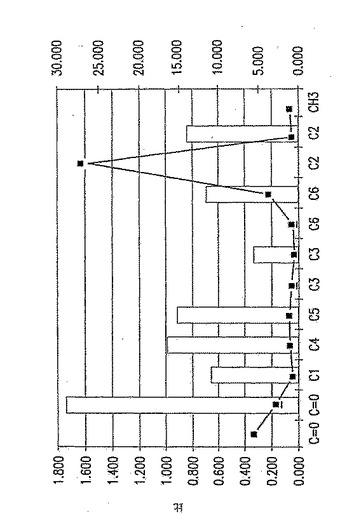
【図6A】

【図6B】

【図7A】

【図7B】
【図8A】

【図8B】

【図9A】

【図9B】

【図9C】

【図9D】
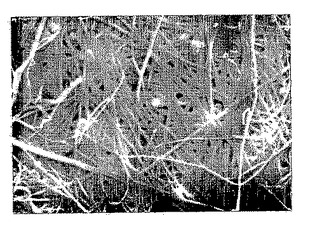
【図9E】

【図10A】

【図10B】
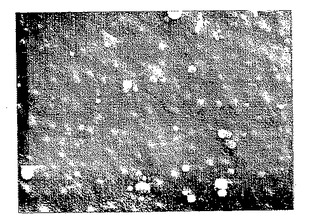
【図11A】

【図11B】

【図12A】

【図12B】
【図13】
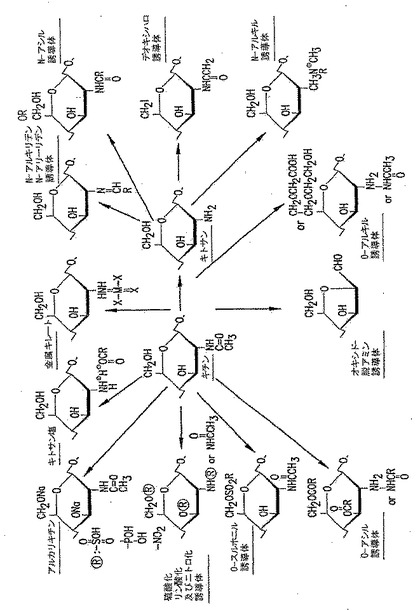
【図14】
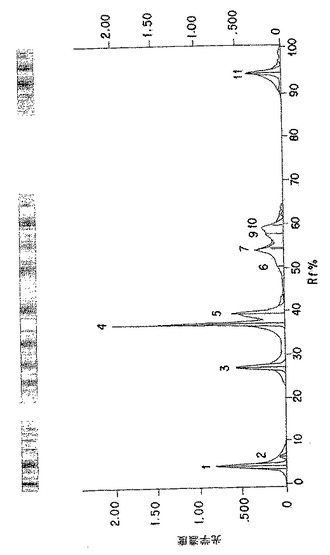
【図15】
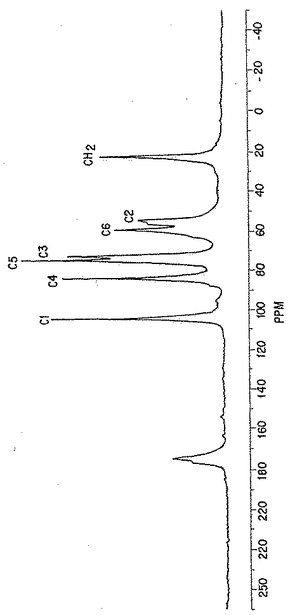
【図16】
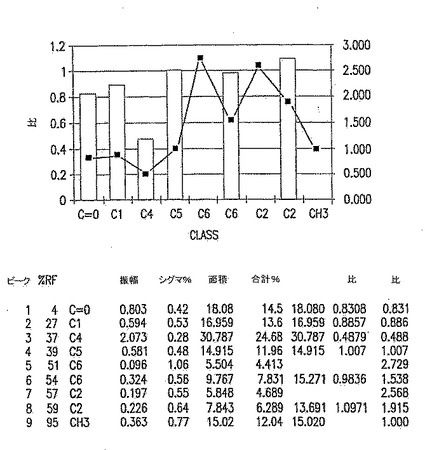
【図17A】

【図17B】

【図17C】
【図17D】

【図17E】
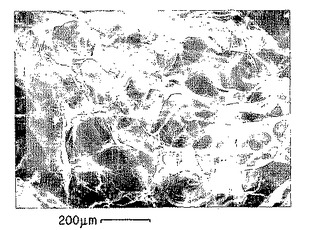
【図17F】
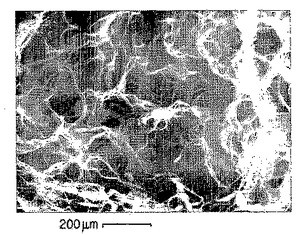
【図17G】

【図18】
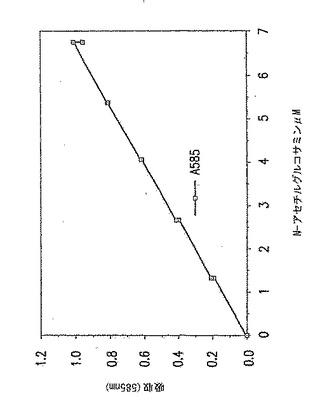
【図19】
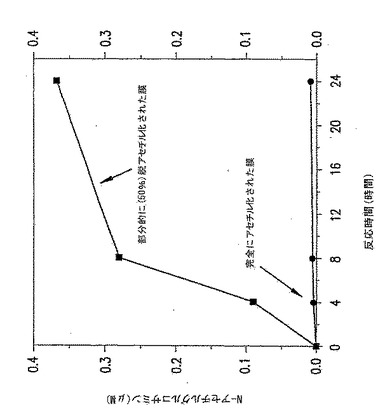
【図20】
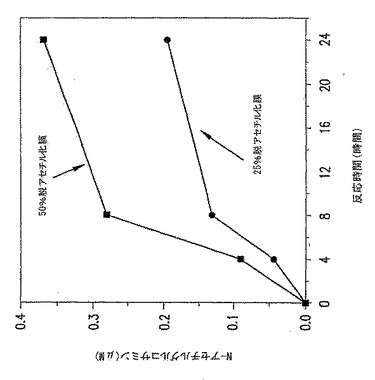
【図21A】
【図21B】

【図21C】

【図21D】

【図21E】

【図22】
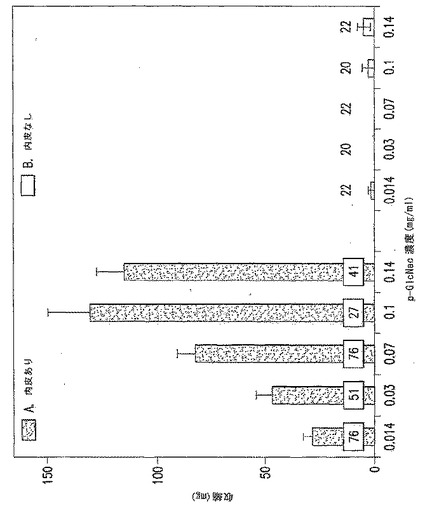
【図23】
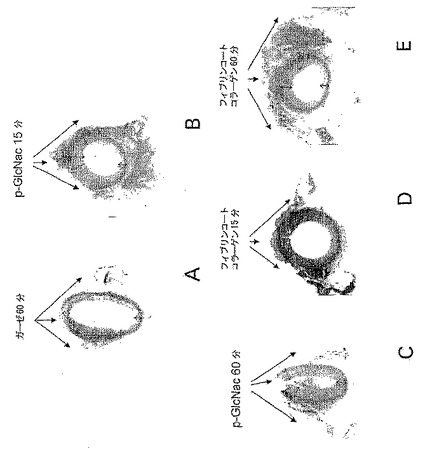
【図24】
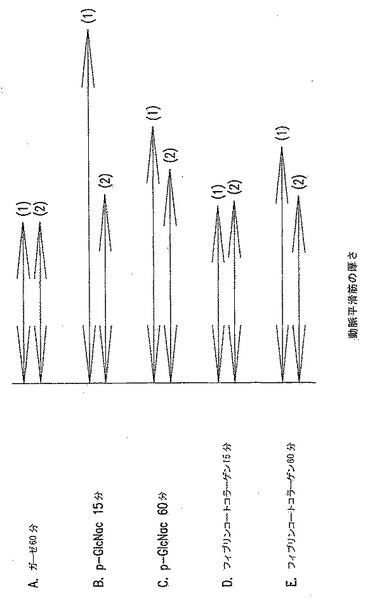
【図2】
【図3A】
【図3B】
【図4A】
【図4B】
【図4C】
【図4D】
【図4E】
【図5A】
【図5B】
【図6A】
【図6B】
【図7A】
【図7B】
【図8A】
【図8B】
【図9A】
【図9B】
【図9C】
【図9D】
【図9E】
【図10A】
【図10B】
【図11A】
【図11B】
【図12A】
【図12B】
【図13】
【図14】
【図15】
【図16】
【図17A】
【図17B】
【図17C】
【図17D】
【図17E】
【図17F】
【図17G】
【図18】
【図19】
【図20】
【図21A】
【図21B】
【図21C】
【図21D】
【図21E】
【図22】
【図23】
【図24】
【公開番号】特開2010−265285(P2010−265285A)
【公開日】平成22年11月25日(2010.11.25)
【国際特許分類】
【出願番号】特願2010−151827(P2010−151827)
【出願日】平成22年7月2日(2010.7.2)
【分割の表示】特願2002−563772(P2002−563772)の分割
【原出願日】平成14年2月8日(2002.2.8)
【出願人】(501250337)マリン ポリマー テクノロジーズ,インコーポレーテッド (8)
【Fターム(参考)】
【公開日】平成22年11月25日(2010.11.25)
【国際特許分類】
【出願日】平成22年7月2日(2010.7.2)
【分割の表示】特願2002−563772(P2002−563772)の分割
【原出願日】平成14年2月8日(2002.2.8)
【出願人】(501250337)マリン ポリマー テクノロジーズ,インコーポレーテッド (8)
【Fターム(参考)】
[ Back to top ]