
血糖値を降下させるペプチド
【課題】 哺乳動物の血糖レベルを低下させる、安定かつ効果的な化合物が必要とされている。
【解決手段】 本発明は、安定性が高く、過度に高い血糖値の治療に有用な新規ペプチド結合体に関する。
【解決手段】 本発明は、安定性が高く、過度に高い血糖値の治療に有用な新規ペプチド結合体に関する。
【発明の詳細な説明】
【技術分野】
【0001】
発明の属する技術分野
本発明は、GLP-1活性に対するアゴニストである新規ペプチドアゴニストに関する。より具体的には、本発明は、エキセンジン-4ポリペプチド配列の変異体を含む血糖レベルを低下させる新規ペプチド、およびGLP-4またはエキセンジン-4ポリペプチド配列の変異体を含むペプチド結合体であって、薬理学的活性を有しかつ安定で、GLP-1活性のアゴニストとして過剰な血糖レベルの調節および/または胃内容排出の調節などが有効である疾患(糖尿病および摂食障害など)の治療に有用である上記ペプチドおよびペプチド結合体に関する。本発明はまた、上記新規ペプチドの製造方法、本発明のペプチドと生理学的に許容されうる担体とを含む組成物(例えば、医薬組成物など)、治療に使用するための該ペプチド、障害の治療方法、および治療に使用するための医薬組成物を製造するための該ペプチドの使用に関する。
【背景技術】
【0002】
発明の背景
血糖レベルを低下させる多くのホルモンは、腸内の栄養分の存在および吸収に応答して胃腸管粘膜から放出される。これらのホルモンには、ガストリン、セクレチン、グルコース依存性インスリン分泌刺激(insulinotropic)ポリペプチド(GIP)およびグルカゴン様ペプチド-1(GLP-1)が含まれる。公知の最も強力なものは、GLP-1である(Orskov, 1992, Diabetologia 35: 701-711)。グルカゴン様ペプチド-1(GLP-1)は、180アミノ酸からなるペプチドであるプログルカゴンの産物である(Drucker、1998、Diabetes 47: 159-169)。プログルカゴンの全配列には、グルカゴンの29アミノ酸の配列、GLP-1の36または37アミノ酸配列および腸作用性(intestinotrophic)ペプチドであるグルカゴン様ペプチド-2(GLP-2)の34アミノ酸配列が含まれる。GLP-1は多くの機能を有している。これは、正常なヒトにおいてインスリン分泌に対する作用を増強する生理学的ホルモンであり、従って内分泌性ホルモンである。さらに、GLP-1はまた、グルカゴン濃度を低下させ、胃内容排泄を遅延させ、(プロ)インスリンの生合成を刺激し、かつ、インスリン感受性を増進させる(Nauck, 1997, Horm. Metab. Res. 47: 1253-1258)。該ペプチドはまた、グルコース耐性に障害のある被験体においてはβ細胞のグルコースに対する感知能および応答能を増進させる(Byrne, 1998, Eur. J. Clin. Invest. 28: 72-78)。ヒトのGLP-1のインスリン分泌刺激作用は、グルコースの消失速度を増大させる。これは、一部には、インスリンレベルが増大するからであり、また、一部には、インスリン感受性が増大するからである(D'Alessio, 1994, Eur. J. Clin. Invest. 28: 72-78)。このことにより、GLP-1はII型糖尿病の治療に有望な物質として位置づけられてきた。GLP-1の活性断片はGLP-1(7〜36)およびGLP-1(7〜37)であることがわかっている。しかし、天然型GLP-1の薬理学的に主要とされる問題は、半減期が短いことである。ヒトおよびラットにおいては、GLP-1はジペプチジルペプチダーゼ-IV(DPP-IV)により、GLP-1(9-36)アミドに迅速に分解され、これは内因性GLP-1受容体アンタゴニストとして作用する(Deacon, 1998, Diabetologia 41: 271-278)。この問題を回避する種々のストラテジーが提唱されてきたが、その中のいくつかはDPP-IVの阻害剤および他のDPP-IV耐性のGLP-1(7-36)アミドの類似体を用いるものである(Deacon, 1998, Diabetologia 41: 271-278; Deaconら、1998, Diabetes 47: 764-769; Ritzel, 1998, J. Endocrinol. 159: 93-102;米国特許第5,545,618号; Pederson, 1998, Diabetes 47: 1253-1258)。
【0003】
血糖レベルを低下させるもう一つのグループのペプチドであるエキセンジンは、GLP-1[7-36]NH2にある程度の配列類似性を有している(53%)(Goke ら、1993 J. Biol. Chem. 268: 19650-55)。エキセンジンは、毒トカゲ(Helodermatidae)またはアメリカドクトカゲ(Raufman, 1996, Reg. Peptides 61:1-18)の毒に認められる。エキセンジン-3は、メキシコアメリカドクトカゲ Heloderma horridumの毒に存在し、エキセンジン-4は、ヒーラモンスター Heloderma suspectumの毒に存在する。エキセンジン-4は、2および3箇所だけがエキセンジン-3とは異なっている。エキセンジン-4のアミノ末端に連結した47アミノ酸のペプチドであるエキセンジン-4前駆体タンパク質をコードするcDNAが、クローニングされ配列決定された(Pohl ら、1998, J. Biol. Chem. 273:9778-9784およびWO98/35033)。エキセンジン-3およびエキセンジン-4の両方とも、モルモット膵腺房細胞において、エキセンジン受容体と相互作用することにより細胞のcAMP産生の増加を刺激する(Raufman, 1996, Reg. Peptides 61: 1-18)。エキセンジン-3が誘導する細胞のcAMP産生の増加は二相性であるが、エキセンジン-3が誘導する膵腺房細胞のアミラーゼ放出の増加は単相性のである。対照的に、エキセンジン-4が誘導するcAMP産生の増加は単相性であるが、エキセンジン-4はアミラーゼ放出には変化を及ぼさない。
【0004】
エキセンジン-4は、単離されたラットインスリノーマ細胞におけるGLP-1受容体の強力なアゴニストである(Gokeら、1993, J. Biol. Chem. 268: 19650-55)。DPP-IVにより認識されるGLP-1の(His Ala)ドメインがエキセンジン-4に存在しないことから、エキセンジン-4は有望視されている(Gokeら、1993, J. Biol. Chem. 268: 19650-55)。[125I]GLP-1の孤束核への結合は、非標識GLP-1および[Tyr39]エキセンジン-4により濃度依存的に阻害され、この際のKi値はそれぞれ、3.5 nMおよび9.4 nMであり、これらと同様の値が細胞系でも認められている(Gokeら、1995, Eur. J. Neurosci. 7: 2294-2300およびGokeら、1993, J. Biol. Chem. 268: 19650-55)。さらに、糖尿病db/dbマウスでは、エキセンジン-4の全身投与により血糖レベルが40%低下する(WO99/07404)。最近、Griegらにより、糖尿病ob/obマウスで、1日1回のエキセンジン-4の腹腔内注射による血糖低下効果が長時間持続すること示された(Griegら、1999, Diabetologia 42: 45-50)。米国特許第5,424,286号には、インスリン分泌刺激活性を維持するためにはN末端配列の大部分(エキセンジン-4(1-31)およびY31-エキセンジン-4(1-31))が必須であり、N末端が切断されたエキセンジン(エキセンジン-4(9-39))は阻害特性を有することが開示されている。
【0005】
真性糖尿病の治療、胃の運動性の低減、胃内容排出の遅延および高血糖の予防(米国特許第5,424,286号、WO98/05351)、ならびに食物摂取の低減(WO98/30231)のために、エキセンジン-3、エキセンジン-4、およびエキセンジンアゴニストの使用が提案されてきた。天然のエキセンジン配列を改変することによる新規化合物を得る方法が提唱されている。その1つは、例えば、WO98/43708の記載に開示されている、C末端アミノ酸残基に結合した親油性置換基を1つだけ有するエキセンジンの誘導体のように、分子に親油性置換基を結合させるものである。
【0006】
主要な手法は、天然のエキセンジン-4の配列のアミノ酸置換および/またはC末端の切断により特性解析されたエキセンジン類似体を考案することであった。この手法は、WO99/07404、WO99/25727およびWO99/25728に記載の化合物により示されている。WO99/07404は、一般式Iのエキセンジンアゴニストを開示する。該式は、4〜5位はGly Thrであり、11〜13位はSer Lys Gln であり、15〜21位はGlu Glu Glu Ala Val Arg Leu であり、26〜30位はLeu Lys Asn Gly Gly であり、32〜35位はSer Ser Gly Ala であり、残りの位置には野生型エキセンジンのアミノ酸残基か、または特定のアミノ酸置換を有しうる、39アミノ酸残基からなるペプチド配列を示す。該式Iは、新規化合物であるdesPro36-エキセンジン-4(1-39)、エキセンジン-4(1-39)-K6またはdesPro36-エキセンジン-4(1-39)-K6などの本明細書に記載するような、特定のアミノ酸の欠失を有し、および/または結合体であるエキセンジンアゴニストまたは類似体は全く包含していない。
【0007】
WO99/25727は、4位にGlyおよび18位にAlaを有し、残りの位置には野生型エキセンジンのアミノ酸残基か、または特定のアミノ酸置換を有しうる、28〜38アミノ酸残基からなるペプチド配列を示す一般式Iを有するエキセンジンアゴニストを開示している。式Iは、C末端アミノ酸としてSerを有するペプチド配列、ならびに新規化合物であるdesPro36-エキセンジン-4(1-39)、エキセンジン-4(1-39)-K6またはdesPro36-エキセンジン-4(1-39)-K6などの本明細書に記載するような、特定のアミノ酸の欠失を有し、および/または結合体であるエキセンジンアゴニストまたは類似体は全く包含していない。さらにWO99/25727に記載の式IIは、式Iに類似したペプチド配列を示しているが、27位または28位にあるリジンに、C(1〜10)アルカノイルまたはシクロアルキルアルカノイル置換基を有するエキセンジン誘導体を含む。異常な食後血糖レベルの治療の際に、該化合物を頻繁に(例えば1日に1回、2回、または3回)投与する。
【0008】
WO99/25728は、18位に固定のAlaを有し、残りの位置には野生型エキセンジンのアミノ酸残基であるか、または特定のアミノ酸置換を有しうる、28〜39アミノ酸残基からなるペプチド配列を示す一般式Iを有するエキセンジンアゴニストを開示している。該エキセンジンアゴニストはすべて、アミノ酸置換の程度が異なる末端切断型エキセンジン類似体に相当する。34〜38のアミノ酸残基からなるペプチド配列は、C末端にSerを有していない。39アミノ酸残基からなるペプチド配列は、C末端にSerまたはTyrのいずれかを有することができ、ほかの残基は有していない。本明細書中に記載するような、特定のアミノ酸欠失を有し、および/または結合体である本発明のエキセンジンアゴニストまたは類似体は、式Iには含まれない。さらに、式IIは式Iに類似しているが27または28位のリジンに、C(1〜10)アルカノイルまたはシクロアルキルアルカノイル置換基を有するエキセンジン誘導体を含むペプチド配列を示す。
【0009】
WO99/46283(1999年9月16日公開)は、薬理学的活性のあるペプチドXおよび該Xに共有結合している4〜20アミノ酸残基からなる安定化ペプチド配列Zを含むペプチド結合体を開示しており、該結合体は、半減期がXの半減期に比べて増大しているという特徴を有する。Xはエキセンジン-4またはエキセンジン-3であってよい。
【発明の開示】
【発明が解決しようとする課題】
【0010】
本発明の目的
哺乳動物の血糖レベルを低下させる、安定かつ効果的な化合物が必要とされている。従って、本発明の目的は、哺乳動物の血糖レベルを低下させる新規化合物を提供することである。理想的には、経口投与である場合に効果的なものである。さらなる本発明の目的は、GLP-1活性および/またはエキセンジン-4活性の新規ペプチドアゴニストを提供することである。またさらに、半減期が増大し、および/もしくはクリアランスが低減された、GLP-1活性ならびに/またはエキセンジン-4活性についてのペプチドアゴニストを提供することも、さらなる本発明の目的である。
【課題を解決するための手段】
【0011】
発明の概要
本発明は、
(a) エキセンジン(exendin)-4に対して少なくとも90%の相同性を有するエキセンジン、
(b) 34〜39位の1〜5個の欠失からなる群より選択される改変を有し、かつ40位に親油性置換基を有するLysを有する、該エキセンジンの変異体、または
(c) (i) 8位のアラニンの、D-アラニン、グリシンもしくはα-アミノイソ酪酸への置換、および
(ii) 親油性置換基
からなる群より選択される少なくとも1個の改変を有するGLP-1(7-36)またはGLP-1(7-37)、
からなる群より選択されるペプチドX(ただし、Xはエキセンジン-4またはエキセンジン-3ではない)と、
4〜20個のアミノ酸単位からなり、各アミノ酸単位が、Ala、Leu、Ser、Thr、Tyr、Asn、Gln、Asp、Glu、Lys、Arg、His、Met、Ornおよび一般式I
-NH-C(R1)(R2)-C(=O)- (I)
(式中、R1およびR2は、水素、C1-6-アルキル、フェニルおよびフェニル-メチルからなる群より選択され、C1-6-アルキルは、ハロゲン、ヒドロキシ、アミノ、シアノ、ニトロ、スルホノおよびカルボキシからなる群より選択される1〜3個の置換基で置換されていてもよく、フェニルおよびフェニル-メチルは、C1-6-アルキル、C2-6-アルケニル、ハロゲン、ヒドロキシ、アミノ、シアノ、ニトロ、スルホノおよびカルボキシからなる群より選択される1〜3個の置換基で置換されていてもよく、あるいはR1とR2は、それらが結合している炭素原子と一緒になって、シクロペンチル、シクロヘキシルまたはシクロヘプチル環を形成している。)
で表されるアミノ酸単位(例えば2,4-ジアミノブタン酸および2,3-ジアミノプロパン酸)からなる群より選択される、前記変異体に共有結合したペプチド配列Zと、
を含むペプチド結合体に関する。
【0012】
ペプチドXは、糖尿病の哺乳動物のグルコース耐性の改善に有効であることをさらに特徴とする。
【0013】
さらに、本発明は、親エキセンジンの新規変異体であって、該親エキセンジンは、エキセンジン-4に対して少なくとも90%の相同性を有するアミノ酸配列を有し、該変異体は、哺乳動物において血糖値を降下させ、かつGLP-1受容体に結合し、かつ
(a)34〜38位の1〜5個の欠失、および
(b)εアミノ基に親油性置換基が結合している40位のLys、
からなる群より選択される少なくとも1個の改変を有する、前記変異体に関する。
【発明を実施するための最良の形態】
【0014】
詳細な説明
本発明の化合物は、親エキセンジンの今まで知られていない欠失変異体を含む。エキセンジン-4(1-39)の既知の置換および/または末端切断変異体とは対照的に、本発明の新規化合物は、安定性に優れ、結合特性が減じられないかまたは高められた恒常的なαヘリックス構造を示すものと考えられる。さらに、新規変異体、すなわち改変型GLP-1(7-36)-NH2、および改変型GLP-1(7-37)を、特定の短いペプチド配列(Z)に結合させると、薬理学的特性を損なうことなくこれらの化合物が安定になる。これらの結合により、ペプチド分子にin vivoでの安定性および親水性が付与される。Zは、アミノ酸残基から構成され、単独ではαヘリックス構造の面でなんら構造的特徴をもたない。しかし、円偏光二色性と核磁気共鳴(NMR)分光法の両方を用いた研究から、Zの付加によって、ペプチドのαヘリックス構造の量の増大により証明されたように、いくつかのペプチドの構造的特徴が変化する。例えば、円偏光二色性により、Z-改変された(Gly8)-GLP-1は、(Gly8)-GLP-1よりもはるかに多量のαヘリックス構造を有することが示された。薬理学的結果と一緒になって、その構造分析により、Zがペプチドのコンホメーションをその能力を失うことなく改変し、より高い酵素安定性をもたらしていることが示唆される。ペプチドの物理的および化学的特性も、Z改変によってかなり変更されうるものであり、薬理学的な処方戦略に影響を及ぼすものである。
エキセンジン変異体
本発明のエキセンジン変異体は、エキセンジン-4に対して少なくとも約90%、最も好ましくは少なくとも約95%の相同性を有する、親エキセンジンペプチドの変異体であり、エキセンジン活性をもち、哺乳動物において血糖値を降下させ、GLP-1受容体に結合する。好ましい実施形態では、親エキセンジンペプチドはエキセンジン-4(1-39)のアミノ酸配列と、5個のアミノ酸残基、好ましくは4個のアミノ酸残基、より好ましくは3個のアミノ酸残基、さらにより好ましくは2個のアミノ酸残基、その上より好ましくは1個のアミノ酸残基が異なるアミノ酸配列を有する。
【0015】
1つの実施形態では、エキセンジン変異体は、34〜38位に1〜5個の欠失を含む。好ましくは、変異体は34〜38位に1〜4個の欠失を含み、より好ましくは36〜38位に1〜3個の欠失を含む。好ましくは、親エキセンジンはエキセンジン-4であり、ペプチド結合体にペプチドXとして含まれている好ましい変異体は、36位、37位および38位のPro残基の1個、2個または3個がエキセンジン-4のアミノ酸配列から、好ましくはエキセンジン-4(1-39)のアミノ酸配列から欠失しているアミノ酸配列を有する。
【0016】
本発明において、XペプチドへのZ配列の結合がこれらの化合物の安定性を高めるものと考えられる。プロリンは、Xペプチドの構造を安定化させるZの作用を妨害する可能性がある剛性(rigid)アミノ酸である。したがって、本発明の親エキセンジンの変異体を含むペプチド結合体においては、例えば糖尿病のdb/dbマウスにおける経口グルコース負荷試験(OGTT)で測定された該結合体の有効性が悪影響を受けないかぎり、エキセンジン骨格の36位、37位および38位にあるプロリンアミノ酸のうちの1個、2個または全部が欠失していることが好ましい。
【0017】
別の実施形態では、変異体は、40位に追加の残基、すなわち、アミド結合を介してリジンのεアミノ基に結合している親油性置換基を含むリジン残基を含む。親油性置換基は、直鎖状もしくは分岐状脂肪酸または直鎖状もしくは分枝状アルカンα,ω-ジカルボン酸のアシル基であり得る。アシル基は、式CH3(CH2)nCO-(式中、nは4〜38の整数であり、好ましくは4〜24の整数である)で表され得る。特定の実施形態では、アシル基は、CH3(CH2)6CO-、CH3(CH2)8CO-、CH3(CH2)10CO-、CH3(CH2)12CO-、CH3(CH2)14CO-、CH3(CH2)16CO-、CH3(CH2)18CO-、CH3(CH2)20CO-およびCH3(CH2)22CO-からなる群より選択される。アシル基は、式HOOC(CH2)mCO- (式中、mは4〜38の整数であり、好ましくは4〜24の整数である)で表され得る。特定の実施形態では、アシル基は、HOOC(CH2)14CO-、HOOC(CH2)16CO-、HOOC(CH2)18CO-、HOOC(CH2)20CO-およびHOOC(CH2)22CO-からなる群より選択される。より特定的な実施形態では、親油性置換基は、テトラデカノイル、ω-カルボキシノナデカノイル、7-デオキシコロイル、コロイル、パルミトイルおよびリトコリルからなる群より選択される。最も特定的な実施形態では、親油性置換基はパルミトイルである。
【0018】
あるいは、親油性置換基はNH基を有していてもよい。特定の実施形態としては、限定するものではないが、式CH3(CH2)a((CH2)bCOOH)CHNHCO(CH2)2CO- (式中、aおよびbは整数であり、a+bは8〜33の整数、好ましくは12〜28の整数である);CH3(CH2)cCONHCH(COOH)(CH2)2CO- (式中、cは10〜24の整数である);CH3(CH2)dCONHCH(CH2)2(COOH)CO- (式中、dは8〜24の整数である);COOH(CH2)eCO- (式中、eは8〜24の整数である);-NHCH(COOH)(CH2)4NHCO(CH2)fCH3(式中、fは8〜18の整数である);-NHCH(COOH)(CH2)4NHCOCH(CH2)2COOH)NHCO(CH2)gCH3(式中、gは10〜16の整数である);および-NHCH(COOH)(CH2)4NHCO(CH2)2CH(COOH)NHCO(CH2)hCH3(式中、hは0または1〜22の整数、好ましくは10〜16の整数である)が挙げられる。
【0019】
40位に親油性置換基を有するリジン残基を含むエキセンジン変異体は、任意に、34〜39位、好ましくは34〜38位に、1〜5個の欠失、好ましくは1〜3個の欠失をさらに含み、例えば、[desSer39,Lys40(パルミトイル)]エキセンジン-4(1-39)、[desPro36,Lys40(パルミトイル)]エキセンジン-4(1-39)および[desPro36,Lys40(パルミトイル)]エキセンジン-4(1-40)が挙げられる。
【0020】
最も特定的な実施形態では、変異体は、
化合物1:desPro36-エキセンジン-4(1-39)-NH2(配列番号101)、
desPro36-エキセンジン-4(1-40)-NH2、
化合物14:desPro36,Pro37,Pro38-エキセンジン-4(1-39)-NH2 、
desPro36,Pro37,Pro38-エキセンジン-4(1-40)-NH2 、
desPro36,Pro37-エキセンジン-4(1-39)-NH2 、
desAla35-エキセンジン-4(1-39)-NH2(配列番号105)、
desGly34-エキセンジン-4(1-39)-NH2(配列番号106)、
desSer39-(Lys40(パルミトイル))エキセンジン-4(1-39)-NH2(配列番号107)、
desGly34-(Lys40(パルミトイル))エキセンジン-4(1-39)-NH2(配列番号108)、
desAla35-(Lys40(パルミトイル))エキセンジン-4(1-39)-NH2(配列番号109)、
desPro36-(Lys40(パルミトイル))エキセンジン-4(1-39)-NH2(配列番号110)、ならびにその遊離酸およびその薬学的に許容される塩からなる群より選択され得る。
改変型GLP-1
本発明のペプチド結合体中にペプチドXとして含まれる好ましい改変型GLP-1は、8位のアラニンがグリシンに置換されているGLP-1(7-36)-NH2またはGLP-1(7-37)のアミノ酸配列を有する。あるいは、好ましい改変型GLP-1は、8位のアラニンがグリシンに置換されており、26位、34位または37位の1個のリジン残基に親油性置換基、好ましくはパルミトイル基を有するGLP-1(7-36)またはGLP-1(7-37)のアミノ酸配列を有する。親油性置換基は、好ましくは、該リジンのεアミノ基に結合しており、エキセンジン変異体について上記した特定の実施形態を含む。本発明の結合体中でXとして用いられる改変型GLP-1(7-36)またはGLP-1(7-37)は、親油性置換基を含むWO99/43707およびWO98/08871に引用されたもの、またはより好ましくは8位にグリシン置換基を有するそのGLP-1類似体であってもよい。好ましくはペプチドXは、
Gly8-GLP-1(7-36)、
Gly8-GLP-1(7-37)、および
Gly8-GLP-1(7-36)-Lys37(パルミトイル)
である。
【0021】
親油性置換基を有する本発明の化合物は、WO98/08871においてGLP-1誘導体について示されたように、親ペプチドよりも遅延性の作用プロファイルを示すだろう。
ペプチド結合体
ペプチド配列Zは、ペプチド配列XのC末端またはN末端に結合していてもよく、あるいは2つのペプチド配列がXのC末端とN末端の両方にそれぞれ結合していてもよい。天然の(native)ペプチドXが遊離のC末端カルボン酸を有する場合、ペプチド配列Zは、ペプチドXのC末端またはペプチドXのN末端のいずれかに結合することができ、あるいはXのC末端およびN末端が両方ともそれぞれ個々のペプチド配列Zに結合していてもよい。あるいは、Zは、ペプチド配列X内のどこかにある、リジン、ヒスチジンもしくはアルギニンの側鎖上の窒素原子またはグルタミン酸もしくはアスパラギン酸の側鎖上のカルボニル基に結合していてもよい。1つの実施形態では、Zは、Xの配列内部に結合していてもよく、XのNおよび/またはC末端に結合していてもよい。配列がペプチド配列XのC末端に、N末端に、もしくはその両方に、またはその内部に結合すべきかどうかは、その特定のペプチドXによって異なり、当業者により容易に決定され得る。好ましくは、Xは、ペプチド結合を介して、好ましくはXのC末端によりZに結合する。
【0022】
本発明の1つの態様は、(a) エキセンジン-4に対して少なくとも90%の相同性を有するエキセンジン、
(b) 34〜39位の1〜5個の欠失からなる群より選択される改変を有し、かつ40位に親油性置換基を有するLysを有する、該エキセンジンの変異体、または
(c) (i) 8位のアラニンの、D-アラニン、グリシンもしくはα-アミノイソ酪酸(Aib)への置換、および
(ii) 親油性置換基
からなる群より選択される少なくとも1個の改変を有するGLP-1(7-36)またはGLP-1(7-37)、
である哺乳動物の血糖値を降下させるペプチドXと、
4〜20個のアミノ酸単位からなり、各アミノ酸単位が、Ala、Leu、Ser、Thr、Tyr、Asn、Gln、Asp、Glu、Lys、Arg、His、Met、Ornおよび一般式I
-NH-C(R1)(R2)-C(=O)- (I)
(式中、R1およびR2は、水素、C1-6-アルキル、フェニルおよびフェニル-メチルからなる群より選択され、C1-6-アルキルは、ハロゲン、ヒドロキシ、アミノ、シアノ、ニトロ、スルホノおよびカルボキシからなる群より選択される1〜3個の置換基で置換されていてもよく、フェニルおよびフェニル-メチルは、C1-6-アルキル、C2-6-アルケニル、ハロゲン、ヒドロキシ、アミノ、シアノ、ニトロ、スルホノおよびカルボキシからなる群より選択される1〜3個の置換基で置換されていてもよく、あるいはR1とR2は、それらが結合している炭素原子と一緒になって、シクロペンチル、シクロヘキシルまたはシクロヘプチル環を形成している。)
で表されるアミノ酸単位(例えば2,4-ジアミノブタン酸および2,3-ジアミノプロパン酸)からなる群より選択される、Xに共有結合したペプチド配列Zと、
を含むペプチド結合体に関する。好ましくは、Xは、GLP-1受容体に結合し、エキセンジン-4またはエキセンジン-3を含まない。
【0023】
Zは、典型的には、4〜20個のアミノ酸残基、例えば4〜15個、より好ましくは4〜10個、特に4〜7個のアミノ酸残基、例えば4個、5個、6個、7個、8個または10個のアミノ酸残基(6個のアミノ酸残基が好ましい)からなるペプチド配列である。好ましくは、Zは、少なくとも1個のLys残基を含む。本発明の好ましい実施形態では、ペプチド配列Z中の各アミノ酸残基は、互いに独立に、Ala、Leu、Ser、Thr、Tyr、Asn、Gln、Asp、Glu、Lys、Arg、His、Met、Ornならびにジアミノブタン酸およびジアミノプロパン酸からなる群より選択される。好ましくは、アミノ酸残基は、Glu、LysおよびMetから選択され、特に、Lysであり、あるいはアミノ酸残基は、Asn、GluおよびLysからなる群より選択される。上記のアミノ酸は、D-またはL-配置のいずれかであってよいが、好ましくは上記のアミノ酸はL-配置である。本発明の好ましい実施形態では、Zは少なくとも1個のリジン残基を含むか、またはZがペプチド結合を介してペプチドXのN末端に結合する場合には、ZはAsn-(Glu)n(式中、nは3〜7の整数である)からなる群より選択されるアミノ酸配列を有する。
【0024】
したがって、ペプチド配列Zの実例は、

【0025】
(ここで、各Xaaは、互いに独立に、Ala、Leu、Ser、Thr、Tyr、Asn、Gln、Asp、Glu、Arg、His、Met、Orn、および上記で定義した式Iで表されるアミノ酸、例えばDbuまたはDprからなる群より選択される)である。
【0026】
上述したように、Zのアミノ酸残基はもちろん全て異なっていても全て同一であってもよい。しかし、本発明の興味深い実施形態では、Z中のアミノ酸残基は、2個または3個の異なるアミノ酸から選択されるか、または同一のアミノ酸である。Z中のアミノ酸残基が同一である適当なペプチド配列の例は、例えば(Lys)n (式中、nは4〜15、好ましくは4〜10、例えば4〜8など、例えば4〜7の範囲の整数である)であり、例えば、Lys4(配列番号1)、Lys5(配列番号2)、Lys6(配列番号8)、Lys7(配列番号30)である。好ましくは、XのC末端にペプチド結合を介して結合した(Lys)6である。
【0027】
Z中のアミノ酸残基が約2個の異なるアミノ酸から選択される適当なペプチド配列の例は、(Lys-Xaa)mまたは(Xaa-Lys)m(式中、mは約2〜7、好ましくは2〜5、例えば2〜4などの整数、例えば3であり、Xaaは、互いに独立に、Ser、Thr、Tyr、Asn、Gln、Asp、Glu、Arg、His、Orn、2,4-ジアミノブタン酸、2,3-ジアミノプロパン酸およびMetからなる群より選択される)である。より好ましくは、そのようなペプチド配列は、例えば、(Lys-Xaa)3または(Xaa-Lys)3(式中、Xaaは前記のとおりである)であり、例えば、(Lys-Glu)3(配列番号83)または(Glu-Lys)3(配列番号84)である。Z中のアミノ酸残基が約2個のアミノ酸残基から選択される適当なペプチド配列の他の例は、例えば、Lysp-XaaqまたはXaap-Lysq(式中、pおよびqは1〜14の整数であり、ただしp+qは4〜15の範囲、好ましくは4〜10の範囲、例えば4〜8の範囲など、例えば4〜6の範囲、例えば4、5または6であり、Xaaは、互いに独立に、Ser、Thr、Tyr、Asn、Gln、Asp、Glu、Arg、HisおよびMetからなる群より選択される)である。より好ましくは、そのようなペプチド配列は、例えば、Lys3-Xaa3またはXaa3-Lys3(式中、Xaaは前記のとおりである)であり、例えば、Lys3-Glu3(配列番号85)またはGlu3-Lys3(配列番号86)である。より好ましいZ配列は、AsnおよびGlnから選択されるアミノ酸残基と、GluおよびAspから選択される4〜7個のアミノ酸残基とを有する配列からなり、例えば、Asn-(Glu)5、Asn-(Glu)6、Gln-(Glu)5、Asn-(Asp)5、およびGln-(Asp)5、であり、本発明のペプチド結合体のN末端部分である。
【0028】
Z中のアミノ酸残基が3個の異なるアミノ酸から選択される適当なペプチド配列の例は、例えば、
【0029】
(式中、xおよびyは約1〜5の範囲の整数であるが、ただしx+yは最大6であり、Xaa1およびXaa2は、互いに独立に、Ala、Leu、Ser、Thr、Tyr、Asn、Gln、Asp、Glu、Arg、His、Met、Orn、2,3-ジアミノプロパン酸、2,4-ジアミノブタン酸、および上記で定義した式Iで表されるアミノ酸からなる群より選択される)などである。
【0030】
本発明の好ましい実施形態では、ペプチド結合体の経口最小有効量とペプチドXの最小有効量の比は少なくとも1:5である。
【0031】
本発明の最も好ましい実施形態は、
desPro36-エキセンジン-4(1-39)-NH2(配列番号101)、
desPro36-エキセンジン-4(1-40)-NH2、
desPro36-desPro37-エキセンジン-4(1-39)-NH2 、
desPro36-desPro37-desPro38-エキセンジン-4(1-39)-NH2 、
desPro36-desPro37-desPro38-エキセンジン-4(1-40)-NH2 、
desAla35-エキセンジン-4(1-39)-NH2(配列番号105)、
desGly34-エキセンジン-4(1-39)-NH2(配列番号106)、
desGly34-(Lys40(パルミトイル))エキセンジン-4(1-39)-NH2(配列番号108)、
desAla35-(Lys40(パルミトイル))エキセンジン-4(1-39)-NH2(配列番号109)、
desPro36-(Lys40(パルミトイル))エキセンジン-4(1-39)-NH2(配列番号110)、
化合物(iii) Gly8-GLP-1(7-36)-NH2 、Gly8-GLP-1(7-37)、および
Gly8-GLP-1(7-36)-Lys37(パルミトイル)-NH2、
からなる群より選択され、ペプチド結合を介して(Lys)n(式中、nは4〜8の整数であり、好ましくはnは6である)からなる群より選択されるペプチド配列ZにC末端により結合している、GLP-1および/またはエキセンジン-4活性のアゴニストであるペプチドXを含む新規ペプチド結合体に関する。
【0032】
本発明のペプチド結合体は、好ましくはアミド(NH2)もしくは遊離酸(OH)形態、またはその塩形態であってもよいことは理解されるべきである。本発明のペプチド結合体の例は、
Gly8-GLP-1(7-36)-Lys6-NH2(配列番号88)、
(Gly8,Lys37(パルミトイル)-GLP-1(7-36)(ヒト)-Lys7-NH2(配列番号89)、
desSer39-エキセンジン-4(1-39)-Lys6-NH2(配列番号91)、
エキセンジン-4(1-39)-Lys6-NH2(配列番号92)、
desPro36-エキセンジン-4(1-39)-Lys6-NH2(配列番号93)、
desAla35-エキセンジン-4(1-39)-Lys6-NH2(配列番号94)、
desGly34-エキセンジン-4(1-39)-Lys6-NH2(配列番号95)、
desSer39-(Lys40(パルミトイル))エキセンジン-4(1-39)-Lys7-NH2(配列番号96)、
desGly34-(Lys40(パルミトイル))エキセンジン-4(1-39)-Lys7-NH2(配列番号97)、
desAla35-(Lys40(パルミトイル))エキセンジン-4(1-39)-Lys7-NH2(配列番号98)、
desPro36-(Lys40(パルミトイル))エキセンジン-4(1-39)-Lys7-NH2(配列番号99)、
Lys40(パルミトイル)エキセンジン-4(1-39)-Lys7-NH2(配列番号100)、
desPro36,Pro37-エキセンジン-4(1-39)-Lys6-NH2、
Lys6-desPro36,Pro37,Pro38-エキセンジン-4(1-39)-NH2、
Asn(Glu)5-desPro36,Pro37,Pro38-エキセンジン-4(1-39)-NH2、
Lys6-desPro36,Pro37,Pro38-エキセンジン-4(1-39)-Lys6-NH2、
Asn(Glu)5-desPro36,Pro37,Pro38-エキセンジン-4(1-39)-Lys6-NH2、
desPro36,Pro37,Pro38-エキセンジン-4(1-39)-Lys6-NH2、
Ser8-GLP-1(7-36)-Lys6-NH2、
Aib8-GLP-1(7-36)-Lys6-NH2、
Lys6-Gly8-GLP-1(7-36)-Lys6-NH2、
Lys6-Gly8-GLP-1(7-36)-NH2、
(Gly8,Lys26(パルミトイル)-GLP-1(7-36)(ヒト)-Lys6-NH2 、
(Gly8,Lys34(パルミトイル)-GLP-1(7-36)(ヒト)-Lys6-NH2 、
Gly8-GLP-1(7-36)-Lys8-NH2 、
Gly8-GLP-1(7-36)-Lys10-NH2 、
Gly8-GLP-1(7-37)-Lys6-NH2 、ならびにその遊離酸およびその薬学的に許容される塩、である。
【0033】
好ましいペプチド結合体は、
desPro36-エキセンジン-4(1-39)-Lys6-NH2(配列番号93)、
Gly8-GLP-1(7-36)-Lys6-NH2(配列番号88)、
desPro36,Pro37,Pro38-エキセンジン-4(1-39)-Lys6-NH2、および
その上記で定義した塩である。
【0034】
最も特定的な実施形態では、結合体は、Gly8-GLP-1-(7-36)(ヒト)-NH2、Gly8-GLP-1-(7-36)(ヒト)-Lys6-NH2、Gly8Lys37(パルミトイル)-GLP-1-(7-36)(ヒト)-Lys7-NH2、Gly8Lys34(パルミトイル)-GLP-1-(7-36)(ヒト)-Lys6-NH2、desSer39-エキセンジン-4(1-39)-Lys6-NH2、エキセンジン-4(1-39)-Lys6-NH2、desPro36-エキセンジン-4(1-39)-Lys6-NH2、desAla35-エキセンジン-4(1-39)-Lys6-NH2、desGly34-エキセンジン-4(1-39)-Lys6-NH2、desSer39-(Lys40(パルミトイル))エキセンジン-4(1-39)-Lys7-NH2、desGly34-(Lys40(パルミトイル))エキセンジン-4(1-39)-Lys7-NH2、desAla35-(Lys40(パルミトイル))エキセンジン-4(1-39)-Lys7-NH2、desPro36-(Lys40(パルミトイル)エキセンジン-4(1-39)-Lys7-NH2およびLys40(パルミトイル)エキセンジン-4(1-39)-Lys7-NH2からなる群より選択される。
【0035】
本発明のペプチド結合体の提供により、GLP-1およびエキセンジンならびにこれらの活性類似体などの血糖低下ペプチドの経口投与が可能になる。本明細書中に示す好ましい末端ペプチド断片であるZは、Xの所望の活性に顕著な影響を与えることなく、ペプチドXをα-らせん構造に誘導するよう選択される。結合していないペプチドと比較すると結合したペプチドの半減期は2〜3倍増加している(下記表5参照)ということによって実証されているように、該らせん構造は、例えば分解などに対して、このペプチド鎖を安定化する。ペプチド配列Zは、最小有効量が少なくも5倍低減されるように特定の構造を分子に導入するの関与するペプチド結合体の一部である。最小有効量は、好ましくは少なくとも10倍、より好ましくは少なくとも25倍、さらに好ましくは40倍、最も好ましくは50倍低減される。従って、本発明はまた、上記に定義したペプチド配列Zの上記に定義したペプチド結合体の調製への使用に関する。
【0036】
従って、本発明はまた、本明細書中に定義するペプチドXを含む新規ペプチド結合体に関する。この場合のXは、哺乳動物において血糖レベルを低下させ、該ペプチド結合体の経口最小有効量と、ペプチドXの経口最小有効量の比は少なくとも1:5である。
【0037】
詳細には、本発明により、インスリン分泌刺激に有効な量の本発明のペプチド結合体を投与することを含む哺乳動物のインスリン放出を刺激する方法、哺乳動物の血糖レベルを低下させるのに有効な量の本発明のペプチド結合体を投与することを含む哺乳動物の血糖レベルを低下させる方法、胃の運動性を低減させるのに有効な量の本発明のペプチド結合体により哺乳動物の胃の運動性を低減させる方法、胃内容排出を遅延させるのに有効な量の本発明のペプチド結合体により哺乳動物の胃内容排出を遅延させる方法、食物摂取を抑制するのに有効な量の本発明のペプチド結合体により哺乳動物の食物摂取を抑制する方法、および哺乳動物において血漿脂肪レベルを低減するのに有効な量の本発明のペプチド結合体を哺乳動物に投与することを含む哺乳動物の血漿脂肪レベルを低減させる方法が導かれる。特に、本発明のペプチド結合体を、I型またはII型糖尿病、肥満、摂食障害、高血糖症、代謝異常、胃の疾患およびインスリン抵抗性症候群の治療に用いることができる。
【0038】
本発明はまた、組換えDNA技術により上記ペプチド結合体を調製する方法であって、(a)上記ペプチド結合体をコードする核酸配列を宿主細胞に導入するステップ、(b)該宿主細胞を培養するステップ、および(c)該培養物から上記ペプチド結合体を単離するステップを含む方法、または、(a)上記ペプチド結合体をコードする核酸配列を含む組換え宿主細胞を、該ペプチド結合体の産生が可能な条件下で培養するステップ、および(b)該培養物から上記ペプチド結合体を単離するステップを含む方法に関する。
【0039】
該方法はまた、ペプチドXを、組換えDNA法を用い、ペプチドを単離することにより得る、ペプチド結合体を調製する方法にも関する。その後、固相支持体に結合しているか、または固相合成法により調製しておいたZにXを結合させる。さらに、本発明は、ペプチド合成法による本発明のペプチド結合体の調製に関する。さらに、本発明は、ペプチド合成法による、本発明のペプチド結合体の調製に関する。
【0040】
受容体への結合(インスリン分泌刺激活性)に必須であると考えられている天然のエキセンジン-4 N末端配列に対して実質的に相同性を有する33〜39のアミノ酸残基、好ましくは36〜38アミノ酸残基のN末端配列、およびC末端配列Zを含む本発明の結合体は、天然型エキセンジンおよびエキセンジンのC末端側末端切断型に比べて安定性が向上しているという利点をさらに有している。同様に、GLP-1ペプチド結合体である化合物4は、結合していない化合物(iii)に比べて安定性の向上を示す。
組成物
本発明はまた、本発明のエキセンジン変異体またはペプチド結合体を生理学的に許容されうる担体と組み合わせて含有する組成物に関する。かかる組成物は、経口投与、非経口投与(皮下投与(s.c.)、静脈内投与(i.v.)、筋肉内投与(i.m.)、硬膜外投与、直接脳内投与および腹腔内投与(i.p.)を含む)、直腸投与、気管内投与、鼻腔内投与、皮膚投与、腟内投与、口腔内投与、眼内投与、または肺投与に適した形態とすることができ、皮下投与または経口投与に適した形態とすることが好ましい。そして、かかる組成物は、例えば「Remington's Pharmaceutical Sciences」(第17版、Alfonso R. Gennaro (編)、Mark Publishing Company, Easton PA, U.S.A., 1985)およびさらに新しい版、ならびに「Drugs and the Pharmaceutical Sciences」シリーズ(Marcel Dekker)中の研究論文などの一般的な記載にあるような当業者に公知の方法で調製できる。該組成物は、例えば、カプセル、錠剤、エアロゾル、局所適用形態、液状または半液状形態(溶液、懸濁液、分散液、エマルジョン、ミセルまたはリポソーム等)などの通常の形態とすることができる。皮下投与に適切な液状組成物が好ましい。好ましい実施形態では、本発明の組成物は、皮下投与される。また別の好ましい実施形態では、本発明の組成物は、経口投与され、この場合は錠剤またはカプセルが好ましい投与形態の1つとして挙げられる。
【0041】
用いる薬学的担体または希釈剤は、従来からの固体または液体の担体でよい。固体担体の例としては、ラクトース、白土、スクロース、シクロデキストリン、タルク、ゼラチン、寒天、ペクチン、アラビアゴム、ステアリン酸マグネシウム、ステアリン酸またはセルロースの低級アルキルエーテル等が挙げられる。液体担体の例としては、シロップ、ピーナッツ油、オリーブ油、リン脂質、ステロール、脂肪酸、脂肪酸アミン、ポリオキシエチレン、等張バッファー溶液および水が挙げられる。同様に、担体または希釈剤は、当業者に公知のあらゆる徐放性物質(グリセリルモノステアレートまたはグリセリルジステアレートなど)を単独でもしくは蜜鑞と混合した形で含有してよい。固体担体を経口投与に用いる場合は、錠剤または硬質ゼラチンカプセル内に入った粉末もしくはペレット形態に調剤するか、あるいはトローチまたはロゼンジの形態に調剤してもよい。固体担体の量は、広い範囲内で様々であるが、通常は約25mg〜約1gであろう。
【0042】
従来の錠剤化技法により調製される典型的錠剤は、
- コア:活性化合物(本発明の化合物の遊離型または塩)100 mg;コロイド状シリコン二酸化物(Aerosil)1.5 mg;セルロース、ミクロクリスト(microcryst; Avicel)70 mg;修飾セルロースガム(Ac-Di-Sol)7.5 mg;ステアリン酸マグネシウム
- コーティング:HPMC 約9 mg;*Mywacett 9-40T 約0.9 mg;フィルムコーティングの可塑剤として用いる*アセチル化モノグリセリド
を含んでよい。
【0043】
液体の担体を用いる場合、調剤は、シロップ、エマルジョン、軟質ゼラチンカプセルまたは注射可能な滅菌済みの液体(水性または非水性の液体懸濁液または溶液など)の形態とすることができる。
【0044】
鼻内投与用には、調剤は、エアロゾルを適用するための液体担体(特に水性担体)に溶解または懸濁した本発明の化合物、好ましくは本発明の結合体を含んでよい。担体は可溶化剤(例えば、プロピレングリコール、胆汁酸塩またはポリオキシエチレン高級アルコールエーテルなどの界面活性剤、吸収増強剤(レシチン(ホスファチジルコリン)またはシクロデキストリンなど)または保存剤(パラビンなど)等を含んでよい。
【0045】
上記組成物はまた、局所的もしくは全身的注射または注入に適する形態であってよく、すなわち、滅菌済みの水または等張生理的食塩水もしくはグルコース溶液を用いて製剤化することができる。該組成物は、当技術分野に公知の従来からの滅菌技法により滅菌してもよい。得られた水性溶液は用途によって包装をしてもよく、または衛生的な条件下で濾過して凍結乾燥してもよい。凍結乾燥した製剤は、投与前に滅菌済み水性溶液と混合する。静脈投与、皮下投与または経口投与に使用する製剤は、活性化合物をバッファーに溶かした溶液が好ましい。該製剤は、活性薬物と滅菌済みバッファー溶液から使用の直前に調製する。好ましい滅菌方法の1つは、使用の直前に調製した溶液を滅菌濾過することでありうる。組成物は、適切な生理学的条件に合致するように薬学的に許容される助剤(例えば、緩衝剤、等張調整剤など)を含んでよい。その例としては、酢酸ナトリウム、乳酸ナトリウム、塩化ナトリウム、塩化カリウム、塩化カルシウムなどがある。
【0046】
本発明の化合物は、有効な薬理学的特性(例えば、タンパク質分解酵素に対する安定性など)を有する。選択されたタンパク質分解酵素の存在下での本発明のペプチドおよびペプチド結合体のin vitro安定性試験では、該新規ペプチドは従来技術におけるペプチドに比べて半減期が増大していることが判明した。従って、本発明の化合物は、GLP-1および他のGLP-1のアゴニストに比べてin vitroで作用する時間がかなり延長されている。さらに、本発明の化合物はcAMPの生成を刺激する。この作用は、cAMPアッセイ(例えば、WO98/08871の記載など参照)において実証することができる。
【0047】
本発明のペプチド化合物は、GLP-1活性および/またはエキセンジン-4活性についてのアゴニストであり、特定のペプチドに対する当技術分野で公知のアッセイにより測定したところ糖尿病哺乳動物において血中のグルコース耐性を向上させる。このようなアッセイの例は本明細書中に記載している。従って、本発明はまた、治療に使用するための上述のエキセンジン変異体およびペプチド結合体、ならびに治療(例えば、I型またはII型糖尿病、肥満、摂食障害およびインシュリン耐性症候群の治療)に使用するための医薬組成物の製造における上述のペプチド結合体の使用にも関する。
【0048】
特定の実施形態では、脊椎動物または哺乳動物のインスリン放出の刺激、血糖レベルの低下、胃の運動性の低減、胃内容排出の遅延化、食物摂取の阻害(例えば、食欲の抑制、または血漿脂質レベルの低減などによる)のために本発明のエキセンジン変異体およびペプチド結合体を用いることができる。本発明の新規化合物はまた、高血糖症の危険を伴う真性糖尿病(即ち、インスリン感受性が、ストレス、心筋梗塞(myocardia infection)、発作および梗塞などにより低下しているか、または妊娠中のインスリン抵抗性が起こっているような場合)の治療に一般に使用することも可能である。該新規化合物はまた、末端肥大症、クッシング症候群、褐色細胞腫、グルカゴノーマ、ソマトスタチノーマ、原発性アルドステロン症などの他の内分泌性疾患の二次的作用である場合、または高血糖症をもたらす特定のホルモンの投与の二次的作用もしくは特定の薬剤(抗高血圧剤、サイアザイド利尿薬、エストロゲンを含む製剤、向精神薬、交感神経作用剤、など)の二次的作用の場合が考えられる他の型の糖尿病の治療に用いることもできる。さらに、本発明の新規化合物を用いて高血糖症の危険を伴う疾患および症状(即ち、アルコール摂取などによる内因性グルコース産生が低下している場合、あるいは下垂体機能不全もしくは原発性副腎皮質不全の患者などにおけるインスリン感受性が増大している場合または進行性腎不全によりインスリンクリアランスが低下している場合など)の治療に一般的に用いることができる。
【0049】
他の特別の治療用途は、WO99/40788(エキセンジンおよびGLP-1の筋収縮性および利尿作用に関する)、WO98/39022(エキセンジンもしくはGLP-1またはエキセンジンもしくはGLP-1のアゴニストを被験体に投与して、被験体に鎮静効果または不安解消作用をもたらすことを含む、中枢または抹消神経系の活性化の増大した被験体の哺乳動物を鎮静させる方法に関する)、WO93/18786(GLP-1(7-37)またはGLP-1(7-36)アミドを用いる治療法に加えて、スルホニル尿素などの経口血糖降下剤による治療をさらに含み、強い相乗効果を生み出す糖尿病の治療に関する)、WO98/19698(肥満の調節におけるGLP-1類似体の使用に関する)、WO98/08531(心筋梗塞後の死亡率および罹患率を低減させる方法におけるGLP-1またはその類似体の使用に関する)、WO98/08873(外科手術後の異化作用の変化およびストレスに対するホルモン応答を緩和する方法におけるGLP-1またはその類似体の使用に関する)に記載されている。また、本発明の化合物は、他の抗糖尿病薬(インスリン、メトホルミン、スルホニル尿素およびサイアゾリジンジオンなど)と組み合わせた治療にも適切であり、または、他の抗肥満薬(レプチン、デキスフェンフルラミン、アンフェタミンなど)と組み合わせた治療にも適切である。
定義
本明細書中で用いる「ペプチド」という用語は、1つのアミノ酸のカルボキシル基ともう1つのアミノ酸のアミノ基とでアミドを形成することのより生成するあらゆる化合物をいう。ペプチド内のアミド結合は、ペプチド結合と呼ぶこともできる。ペプチドという用語は、通常、化合物内のアミド結合が、1つのアミノ酸のC-1ともう1つのアミノ酸のN-2との間で形成されている(ユーペプチド結合(eupeptide bond)と呼ぶこともある)化合物に用いられるが、他のアミド結合(イソペプチド結合と呼ぶこともある)により結合した残基を有する化合物を含む。約10〜20残基より少数の残基からなるペプチドは、オリゴペプチドと呼ぶ場合もあり、それ以上の残基からなるペプチドは、ポリペプチドと呼ぶ場合もある。約50残基より大きい特定の配列のポリペプチドは通常、タンパク質として知られている。本明細書中で用いる「天然のポリペプチド配列」という用語は、天然のL−アミノ酸残基からなり、かつ組換え宿主細胞により発現可能なポリペプチド配列をいう。本明細書中の化合物Xは全て40アミノ酸残基以下のペプチド配列である。
【0050】
本明細書中で用いる「GLP-1」という用語は、GLP-1(7-37)-OH、GLP-1(7-37)-NH2、GLP-1(7-36)-OH、およびGLP-1(7-36)-NH2を含む。
【0051】
「アゴニスト」という用語は、受容体と相互作用でき、該受容体に特徴的な生理学的または薬理学的応答特性(収縮、弛緩、分泌、酵素の活性化など)を誘起し得る内因性の物質または薬剤を指す。
【0052】
「アンタゴニスト」という用語は、他のものの生理学的作用に拮抗する薬剤または化合物を指す。受容体レベルでは、通常他の生物学的に活性な物質により誘導される受容体関連応答に拮抗する化学物質である。
【0053】
「部分的アゴニスト」という用語は、適用する薬剤の量に関係なく、受容体集団の最大限の活性化を誘導することはできないアゴニストを指す。「部分的アゴニスト」という用語は、所定の組織内における「中間的な本質的効力を有するアゴニスト」ということもできる。さらに、部分的アゴニストは、同一の受容体に作用する完全なアゴニストの効果に拮抗し得る。
【0054】
「受容体」という用語は、細胞内もしくは細胞上にあり、分子性伝達物質(神経伝達物質、ホルモン、リンホカイン、レクチン、薬剤など)として作用する化合物を特異的に認識してこれに結合する分子または多量体構造物を指す。
【0055】
本発明の「エキセンジン変異体」という用語は、エキセンジン-4に対して少なくとも約90%の相同性を有し、最も好ましくはエキセンジン-4(1-39)に対して少なくとも約95%の相同性を有し、エキセンジン活性(例えば、哺乳動物において血糖レベルを低下させ、かつGLP-1受容体に結合する)を有する、親エキセンジンペプチドの変異体であると解釈されるべきである。本明細書中に用いる「エキセンジン-4」という用語は、アミノ酸配列が米国特許第5,424,286号中の配列番号2に開示されているエキセンジン-4(1-39)、およびThe Journal of Biological Chemistry, Vol. 272, No7, pp.4108-15中にChenおよびDruckerによって開示されているエキセンジン-4(1-40)(これは、C末端のアミノ酸残基として40位にグリシンを有するという点のみ相違する)を指す。親エキセンジンの相同性は、2つのタンパク質の配列間での同一性の程度(第2の配列からの第1の配列の派生を示す)として決定される。相同性は、GCGプログラムパッケージ (Program Manual for the Wisconsin Package, Version 8, August 1994, Genetics Computer Group. 575 Science Drive, Madison, Wisconsin, USA 53711)(Needleman, S.B.およびWunsch, C.D., (1970), J. Mol. Biol. 48: 443-453)中に提供されるGAPなどの当技術分野で公知のコンピュータプログラムにより適切に決定することができる。ポリペプチド配列比較のための以下の設定;GAPクリエーションペナルティ3.0、およびGAPエクステンションペナルティ0.1を用いることができる。
【0056】
「塩」という用語は、薬学的に許容され得る塩、即ち、酸付加塩および塩基性塩などを含む。酸付加塩の例としては、塩酸塩、ナトリウム塩、臭化水素酸塩などが挙げられる。塩基性塩の例としては、ナトリウムおよびカリウムなどのアルカリ金属カルシウムなどのアルカリ土類金属、ならびにアンモニウムイオンである+N(R3)3(R4)(この場合、R3およびR4は独立して、任意に置換されたC1-6アルキル、任意に置換されたC2-6アルケニル、任意に置換されたアリールまたは任意に置換されたヘテロアリールを指す)から選択されるカチオンを含む塩がある。薬学的に許容され得る塩の他の例としては、例えば、「Remington's Pharmaceutical Sciences」第17版、Alfonso R. Gennaro(編)、Mark Publishing Company, Easton, PA, U.S.A., 1985 およびさらに新しい版、ならびにEncyclopedia of Pharmaceutical Technologyに記載されているものなどが挙げられる。
変異体および結合体の調製
本発明のエキセンジン変異体およびペプチド結合体は、当技術分野に公知の方法により調製することができる。即ち、変異体ならびにペプチド配列XおよびZは、溶液合成またはMerrifield型固相合成などの標準的なペプチド調製技法により調製できる。Boc(tert.ブチルオキシカルボニル)およびFmoc(9-フルオレニルメチルオキシカルボニル)のストラテジーが適用可能であると考えられる。
【0057】
1つの可能な合成ストラテジーでは、本発明のペプチド結合体は、まず公知の標準的な保護法、カップリング法および脱保護法を用いてペプチド配列Zを構築した後、続いてZの構築と同様の方法でペプチド配列XをZにカップリングさせ、最後に担体からペプチド結合体全体を切り離すという固相合成により調製できる。このストラテジーで、ペプチド配列ZがペプチドXのC末端のカルボニル基に共有結合しているペプチド結合体が得られる。しかしながら、所望のペプチド結合体が、2つの安定化配列Zが独立して、ペプチドXのC末端およびN末端の両方に共有結合により結合しているペプチド結合体である場合、上記のストラテジーも適用できるが、C末端結合ペプチド結合体を固相支持体から切り離す前に、2つめのペプチド配列ZをXのN末端に上記と同様の方法でさらにカップリングさせる必要があることは、当業者には理解されるであろう。このストラテジーを用いて、GluまたはAspの側鎖のカルボニル基にZを結合させることもできる。ペプチド配列ZがXのN末端の窒素原子に共有結合しているか、またはXのLys、ArgまたはHisの側鎖の窒素原子に共有結合しているペプチド結合体を調製するのに可能なストラテジーは、上記の方法に類似した方法である。即ち、ペプチド結合体を、まず公知の標準的な保護法、カップリング法および脱保護法によりペプチド配列Xを構築した後、続いてXの構築と同様の方法でペプチド配列ZをXにカップリングさせて、最後にペプチド結合体全体を担体から切り離すという固相合成により調製できる。他の可能なストラテジーは、溶液合成、固相合成、組換え技法、または酵素的合成により、XとZの2つの配列(またはその一部)のうちの1つまたは両方を別々に調製し、続いて溶液中でもしくは固相技法またはその組み合わせによる公知の断片濃縮法により2つの配列をカップリングすることである。1つの実施形態では、Xを組換えDNA法により調製し、Zを固相合成により調製するということもできる。XとZの結合は化学的連結反応により行うことができる。この技術によると、特異性の高い方法により、全く保護されていないペプチド断片の構築が可能である(Liuら、1996、J. Am. Chem. Soc. 118: 307-312およびDawsonら、1996、226: 776)。この結合は、ペプチド結合により全く保護されていないペプチド断片をペプチド結合を介して結合させる特異性の高い技法であるプロテアーゼ触媒性ペプチド結合形成により実施することも可能である(W. Kullmann, 1987, Enzymatic Peptide Synthesis, CRC Press. Boca Raton. Florida. pp.41-59)。
【0058】
ペプチド配列ZにあるLys、Arg、His、Trp、Ser、Thr、Cys、Tyr、AspおよびGluの側鎖の誘導体化は、当技術分野で公知の適切な直交的(orthogonal)な保護スキームを用いて従来からの収束性ペプチド合成により、または、同じく公知の、適切な鎖の直交的(orthogonal)で除去可能な保護を用いる一般的な固相法により実施することができる。
【0059】
さらに、例えば、還元型ペプチド結合などの等電子的結合(isosteric bond)を含むペプチドXなどから得られる改変ペプチド配列をペプチド配列Zにカップリングさせるような場合は、上記のストラテジーの組合せが特に適用可能である場合があると考えられる。このような場合、アミノ酸の連続的カップリングによりZの固定化断片を調製し、その後完全なペプチド配列X(溶液中で調製するか、または全てもしくは部分的に固相技法を用いるか、あるいは組換え技術により調製する)を該断片にカップリングさせることが有利であり得る。
【0060】
適切な固相支持体材料(SSM)の例としては、例えば、官能基化された樹脂(ポリスチレン、ポリアクリルアミド、ポリジメチルアクリルアミド、ポリエチレングリコール、セルロース、ポリエチレン、ポリスチレンにグラフト化されたポリエチレングリコール、ラテックス、ダイナビーズなど)がある。ペプチド配列ZのC末端のアミノ酸またはペプチドXのC末端のアミノ酸を、一般的なリンカー(2,4-ジメトキシ-4'-ヒドロキシ-ベンゾフェノン、4-(4-ヒドロキシ-メチル-3-メトキシフェノキシ)-酪酸、4-ヒドロキシ-メチル安息香酸、4-ヒドロキシメチル-フェノキシ酢酸、3-(4-ヒドロキシメチルフェノキシ)プロピオン酸、およびp-[(R,S)-a[1-(9H-フルオレン-9-イル)メトキシホルムアミド]-2,4-ジメトキシベンジル]-フェノキシ-酢酸など)により固相支持体材料に結合させることが必要または望ましいことは理解されよう。
【0061】
本発明の変異体およびペプチド結合体を、トリフルオロ酢酸、トリフルオロメタンスルホン酸、臭化水素、塩化水素、フッ化水素などの酸を用いて、また場合によっては、酸を、目的に適合する1種以上の「スカベンジャー」(例えば、エタンジチオール、トリイソプロピルシラン、フェノール、チオアニソールなど)と組み合わせて用いることにより、固相支持体材料から切断することができる。あるいは、塩基(アンモニア、ヒドラジンなど)、アルコキシド(ナトリウムエトキシドなど)、水酸化物(水酸化ナトリウムなど)などを用いて、本発明のペプチド結合体を固相支持体から切断することができる。
【0062】
従って、本発明はまた、Zが、好ましくはペプチド結合を介してXに共有結合している、薬理学的活性のあるペプチド結合体を調製する方法に関する。式I(X-Z)で表されるペプチド結合体の調製方法は、以下のステップ:
a) N-α-保護基を含む適切な保護基を有する、活性化型であるアミノ酸またはジペプチドを固定化ペプチド配列H-Z-SSMにカップリングされることにより、N-α-保護ペプチド断片を形成させ、
b) N-α-保護基を除去して、N末端が保護されていない固定化ペプチド断片を形成させ、
c) N-α-保護基を含む適切な保護基を有する、カルボキシルが活性化型であるさらなるアミノ酸またはジペプチドを固定化ペプチド断片のN末端にカップリングさせ、所望のペプチド配列Xが得られるまで、ステップb)とステップc)の除去/カップリングのステップを繰り返し、そして、
d) 固相支持体材料からペプチド結合体を切り離すこと、
を含む。
【0063】
式II(Z-X)で表されるペプチド結合体の調製方法は、以下のステップ:
a) N-α-保護基を含む適切な保護基を有する、活性化型であるアミノ酸またはジペプチドを固相支持体材料(SSM)にカップリングされることにより、固定化保護アミノ酸または固定化保護ジペプチドを形成させ、
b) N-α-保護基を除去して、N末端が保護されていない固定化アミノ酸または固定化ペプチド断片を形成させ、
c) N-α-保護基を含む適切な保護基を有する、カルボキシルが活性化型であるさらなるアミノ酸またはジペプチドを固定化アミノ酸または固定化ペプチド断片のN末端にカップリングさせ、所望のペプチド配列Xが得られるまで、ステップb)とステップc)の除去/カップリングのステップを繰り返し、そして、
d) N-α-保護基を含む適切な保護基を有する、カルボキシルが活性化型であるさらなるアミノ酸またはジペプチドを固定化ペプチド断片のN末端にカップリングさせ、所望のペプチド配列Zが得られるまで、ステップb)とステップd)の除去/カップリングのステップを繰り返し、そして、
e) 固相支持体材料からペプチド結合体を切り離すこと、
を含む。
【0064】
さらに、式III(Z-X-Z)で表されるペプチド結合体の調製方法は、以下のステップ:
a) N-α-保護基を含む適切な保護基を有する、カルボキシルが活性化型であるアミノ酸またはジペプチドを固定化ペプチド配列H-Z-SSMにカップリングすることにより、固定化N-α-保護ペプチド断片を形成させ、
b) 該N-α-保護基を除去して、N末端が保護されていない固定化ペプチド断片を形成させ、
c) N-α-保護基を含む適切な保護基を有する、カルボキシルが活性化型であるさらなるアミノ酸またはジペプチドを固定化ペプチド断片のN末端にカップリングさせ、所望のペプチド配列Xが得られるまで、ステップb)とステップc)の除去/カップリングのステップを繰り返し、そして、
d) N-α-保護基を含む適切な保護基を有する、カルボキシルが活性化型であるさらなるアミノ酸またはジペプチドを固定化ペプチド断片のN末端にカップリングさせ、所望のペプチド配列Zが得られるまで、ステップb)とステップd)の除去/カップリングのステップを繰り返し、そして、
e) 固相支持体材料からペプチド結合体を切り離すこと、
を含む。
【0065】
カップリング、除去および切断のステップは、保護ストラテジーおよび選択した固相材料を考慮して、当業者に公知の方法により実施する。しかし、一般にはBoc(tert.ブチルオキシカルボニル)およびFmoc(9-フルオレニルメチルオキシカルボニル)の保護ストラテジーが適用可能であると考えられ、さらに、当業者に公知の様々な活性化手法を用いて、例えば、ペプチド化学の分野の当業者には公知のように、適切なアミノ酸またはペプチドのC末端の活性化誘導体(酸ハロゲン化物、酸無水物、HObt-エステルなどの活性化エステルなど)を、適切なアミノ酸またはペプチドのアミノ基と反応させるなどにより、ペプチド結合を形成することができると考えられる。さらに、一般的な条件下で反応性のある官能基を担持するアミノ酸残基を用いる場合は、側鎖保護基を含有させることが必要または望ましい場合がある。必要な保護スキームは、当業者には公知であろう(例えば、M. BodanszkyおよびA. Bodanszky,「The Practice of Peptide Synthesis」第2版、Springer-Verlag、1994、J.Jones、「The Chemical Synthesis of Peptides」、Clarendon Press, 1991、およびDrylandら、1986、J. Chem.Soc., Perkin Trans. 1: 125-137など参照)。
【0066】
また本発明のペプチドおよびペプチド結合体は、当業者に公知の一般的方法および原則を用いた組換えDNA技術によって調製されうる。ペプチドおよびペプチド結合体をコードする核酸配列は、確立された標準的方法、例えばS.L. BeaucageおよびM.H.Caruthers. Tetrahedron Letters 22. 1981, pp.1859-1869に記載のホスホアミダイト法、またはMatthesら, EMBO Journal 3, 1984. pp.801-805に記載の方法によって合成的に調製されうる。ホスホアミダイト法に従えば、オリゴヌクレオチドは、例えば自動DNA合成器において合成され、精製、アニーリング、連結後、好適なベクター中にクローニングされる。ペプチドXをコードする核酸配列を単離もしくはクローニングするために使用される技法は当技術分野において公知であり、これらの技法には、ゲノムDNAからの単離、cDNAからの調製、またはそれらの組合せが含まれる。かかるゲノムDNAから本発明の核酸配列をクローニングすることは、例えば周知のポリメラーゼ連鎖反応(PCR)または共有の構造的特徴を有するクローニングされたDNA断片を検出するための発現ライブラリーの抗体スクリーニングを用いることによって達成することができる。例えば、Innisら, 1990. A Guide to Methods and Application. Academic Press. New Yorkを参照されたい。リガーゼ連鎖反応(LCR)、連結活性化転写(ligated activated transcription:LAT)および核酸配列に基づく増幅(NASBA)のような他の核酸増幅法を用いることができる。次いで、それをZをコードする核酸配列に連結することができる。
【0067】
次いで、ペプチドおよびペプチド結合体をコードする核酸配列を、組換え発現ベクター(これは、組換えDNA法に都合よく利用することができる任意のベクターであってよい)に挿入する。ベクターの選択は、多くの場合、それを導入しようとする宿主細胞に依存するであろう。従って、ベクターは、自律複製ベクター、すなわち染色体外の実在として存在し、その複製が染色体複製から独立しているベクター、例えばプラスミドであってよい。あるいは、ベクターは、宿主細胞中に導入する場合、宿主細胞ゲノムに組込まれ、かつ組み込まれた染色体と一緒に複製するベクターのうちの1つであってよい。
【0068】
ベクターにおいて、本発明のペプチドおよびペプチド結合体をコードする核酸配列は、好適なプロモーター配列に機能的に連結されるべきである。プロモーターは、選択した宿主細胞において転写活性を示す任意の核酸配列であってよく、かつ宿主細胞に対して同種の、もしくは異種のタンパク質をコードする遺伝子に由来していてもよい。哺乳動物細胞において該ペプチドおよびペプチド結合体をコードする核酸配列の転写を指令する好適なプロモーターの例としては、SV40プロモーター(Subramaniら, Mol. Cell Biol. 1. 1981. pp.854-864)、MT-1(メタロチオネイン遺伝子)プロモーター(Palmiterら, Science 222. 1983 pp.809-814)またはアデノウイルス2主要後期プロモーター、ラウス肉腫ウイルス(RSV)プロモーター、サイトメガロウイルス(CMV)プロモーター(Boshartら, 1981. Cell 41: 521-530)およびウシパピローマウイルスプロモーター(BPV)が挙げられる。昆虫細胞において使用するのに好適なプロモーターは、ポリへドリンプロモーター(Vasuvedanら, FEBS Lett. 311. 1992. pp.7-11)である。
【0069】
特に細菌宿主細胞において、該ペプチドおよびペプチド結合体をコードする核酸配列の転写を指令するのに好適なプロモーターの例としては、大腸菌(E.coli)lacオペロン、ストレプトミセス・コエリカラー(Streptomyces coelicolor)アガラーゼ遺伝子(dagA)、枯草菌(Bacillus subtilis)レバンスクラーゼ遺伝子(sacB)、バシラス・リへニホルミス(Bacillus licheniformis)α-アミラーゼ遺伝子(amyL)、バシラス・ステアロサルモフィルス(Bacillus stearothermophilus)マルトース生成性(maltogenic)アミラーゼ遺伝子(amyM)、バシラス・アミロリクエファシエンス(Bacillus amyloliquefaciens)αアミラーゼ遺伝子(amyQ)、バシラス・リへニホルミス(Bacillus licheniformis)ぺニシリナーゼ遺伝子(penP)、枯草菌(Bacillus subtilis)xylAおよびxylB遺伝子、および原核生物性β-ラクタマーゼ遺伝子(Villa-Kamaroffら, 1978. Proceedings of the National Academy of Sciences USA 75: 3727-3731)から得られるプロモーター、ならびにtacプロモーター(DeBoerら, 1983. Proceedings of the National Academy of Sciences USA 80: 21-25)が挙げられる。さらにプロモーターは、Scientific American. 1980. 242: 74-94中の「組換え細菌に由来する有用なタンパク質(Useful ptoteins from recombinant bacteria)」;Sambrookら, 1989. 前掲に記載されている。糸状菌宿主細胞において、ペプチドおよびペプチド結合体をコードする核酸配列の転写を指令するのに好適なプロモーターの例としては、コウジカビ(Aspergillus oryzae)TAKAアミラーゼ、リゾムコール・ミエへイ(Rhizomucor miehei)アスパラギン酸プロテイナーゼ、黒コウジカビ(Aspergillus niger)中性α-アミラーゼ、黒コウジカビ(Aspergillus niger)酸安定性α-アミラーゼ、黒コウジカビ(Aspergillus niger)もしくはアスペルギルス・アワモリ(Aspergillus awamori)グルコアミラーゼ(glaA)、リゾムコール・ミエへイ(Rhizomucor miehei)リパーゼ、コウジカビ(Aspergillus oryzae)アルカリ性プロテアーゼ、コウジカビ(Aspergillus oryzae)トリオースリン酸イソメラーゼ、アスペルギルス・ニダランス(Aspergillus nidulans)アセトアミダーゼ、サトイモ乾腐病菌(Fusarium oxysporum)トリプシン様プロテアーゼ(米国特許第4,288,627号に記載されており、これは参照により本明細書に組み入れる)をコードする遺伝子から得られるプロモーター、およびこれらのハイブリッドである。糸状菌宿主細胞において使用するのに特に好ましいプロモーターは、TAKAアミラーゼ、NA2-tpi(黒コウジカビ中性アミラーゼおよびコウジカビトリオースリン酸イソメラーゼをコードする遺伝子に由来するプロモーターのハイブリッド)、およびglaAプロモーターである。酵母宿主において有用なプロモーターは、サッカロミセス・セレビシエ(Saccharomyces cerevisiae)エノラーゼ(ENO-1)遺伝子、サッカロミセス・セレビシエ(Saccharomyces cerevisiae)ガラクトキナーゼ遺伝子(GAL1)、サッカロミセス・セレビシエ(Saccharomyces cerevisiae)アルコールデヒドロゲナーゼ/グリセルアルデヒド-3-リン酸デヒドロゲナーゼ遺伝子(ADH2/GAP)、およびサッカロミセス・セレビシエ(Saccharomyces cerevisiae)3-ホスホグリセリン酸キナーゼ遺伝子から得られる。酵母宿主細胞に有用な他のプロモーターは、Romanosら, 1992, Yeast 8: 423-488によって記載されている。
【0070】
また該ペプチドおよびペプチド結合体をコードする核酸配列は、好適なターミネーター、例えば、ヒト成長ホルモンターミネーター(Palmiterら,前掲書中)に機能的に連結し得る。糸状菌宿主細胞において好ましいターミネーターは、コウジカビTAKAアミラーゼ、黒コウジカビグルコアミラーゼ、アスペルギルス・ニダランスアントラニル酸シンターゼ、黒コウジカビα-グルコシダーゼ、およびサトイモ乾腐病菌トリプシン様プロテアーゼをコードする遺伝子から得られる。酵母宿主細胞にとって好ましいターミネーターは、サッカロミセス・セレビシエ エノラーゼ、サッカロミセス・セレビシエ チトクロームC(CYC1)、またはサッカロミセス・セレビシエ グリセルアルデヒド-3-リン酸デヒドロゲナーゼをコードする遺伝子から得られる。酵母宿主細胞にとって有用な他のターミネーターは、Romanosら, 1992, 前掲によって記載されている。
【0071】
さらにベクターは、ポリアデニル化シグナル(例えば、SV40またはアデノウイルス5Elb領域に由来)、転写エンハンサー配列(例えば、SV40エンハンサー)および翻訳エンハンサー配列(例えば、アデノウイルスVA RNAsをコードするもの)のようなエレメントを含んでいてもよい。さらに、糸状菌宿主細胞に好ましいポリアデニル化配列は、コウジカビTAKAアミラーゼ、黒コウジカビグルコアミラーゼ、アスペルギルス・ニダランス アントラニル酸シンターゼ、および黒コウジカビα-グルコシダーゼをコードする遺伝子から得られる。酵母宿主細胞にとって有用なポリアデニル化配列は、GuoおよびSherman, 1995, Molecular Cellular Biology 15: 5983-5990によって記載されている。
【0072】
さらに組換え発現ベクターは、ベクターが当該の宿主細胞で複製することを可能にするDNA配列を含んでいてもよい。かかる配列(宿主細胞が哺乳動物細胞である場合)の例としては、SV40またはポリオーマの複製開始点が挙げられる。細菌の複製開始点の例としては、プラスミドpBR322、pUC19、pACYC177、pACYC184、pUB110、pE194、pTA1060、およびpAMβ1の複製開始点が挙げられる。酵母宿主細胞において使用するための複製開始点の例としては、2ミクロンの複製開始点、CEN6とARS4との組合せ、およびCEN3とARS1との組合せが挙げられる。複製開始点は、宿主細胞においてその機能を温度感受性にする突然変異を有するものであってよい(例えば、Ehrlich, 1978, Proc. Natl. Acad. Sci. USA 75: 1433を参照されたい)。
【0073】
またベクターは、選択マーカー、例えば、ジヒドロ葉酸還元酵素(DHFR)または薬物(例えば、ネオマイシン、ジェネティシン、アンピシリン、またはハイグロマイシン)に対する耐性を付与するものをコードする遺伝子などの宿主細胞の欠損を補う遺伝子の産物を含んでいてもよい。酵母宿主細胞に好適なマーカーは、ADE2、HIS3、LEU2、LYS2、MET3、TRP1およびURA3である。糸状菌宿主細胞において使用するための選択マーカーは、限定されるものではないが、amdS(アセトアミダーゼ)、argB(オルニチンカルバモイルトランスフェラーゼ)、bar(ホスフィノトリシン(phosphinothricin))アセチルトランスフェラーゼ)、hygB(ハイグロマイシンホスホトランスフェラーゼ)、niaD(硝酸還元酵素)、pyrG(オロチジン-5'-リン酸デカルボキシラーゼ)、sC(硫酸アデニルトランスフェラーゼ)、trpC(アントラニル酸シンターゼ)、およびグルホシネート耐性マーカー、ならびに他の種に由来する等価物を含む群から選択され得る。コウジカビ細胞において使用するのに好ましいものは、アスペルギルス・ニダランスもしくはコウジカビのamdSおよびpyrGマーカー、ならびにストレプトミセス・ハイグロスコピカスのbarマーカーである。さらに、選択マーカーが別々のベクター上にある場合には、選択は、例えばWO91/17243に記載のように同時形質転換によって成し遂げることができる。
【0074】
ペプチドおよびペプチド結合体、プロモーター、ならびにターミネーターのそれぞれをコードする核酸配列を連結するために使用する方法、およびそれらを複製に必要な情報を含有する好適なベクターに挿入するための方法は、当業者に周知である(例えば、Sambrookら, 前掲書中を参照されたい)。
【0075】
発現ベクターを導入する宿主細胞は、ペプチドおよびペプチド結合体を産生することが可能である細胞のいずれであってもよく、真核細胞、例えば無セキツイ動物(昆虫)細胞、またはセキツイ細胞(例えば、アフリカツメガエル(Xenopus laevis)卵母細胞もしくは哺乳動物細胞)、特に昆虫細胞および哺乳動物細胞であってよい。好適な哺乳動物細胞系の例としては、COS(例えば、ATCC CRL 1650)、BHK(例えば、ATCC CRL 1632、ATCC CCL 10)、またはCHO(例えば、ATCC CCL 61)細胞系が挙げられる。
【0076】
哺乳動物細胞をトランスフェクトする方法および細胞に導入したDNA配列を発現する方法は、例えばKaufmanおよびSharp, 1982, J. Mol. Biol. 159: 601-621; SouthernおよびBerg, 1982, J. Mol. Appl. Genet. 1: 327-341; Loyterら, 1982, Proc. Natl. Acad. Sci. USA 79: 422-426; Wiglerら, 1978, Cell 14: 725; CorsaroおよびPearson, 1981, Somatic Cell Genetics 7: 603; Grahamおよびvan der Eb. 1973, Virology 52: 456; Fraleyら, 1980, JBC 225: 10431; Capecchi, 1980, Cell 22: 479; Wibergら, 1983, NAR 11: 7287;およびNeumannら, 1982, EMBO J. 1: 841-845に記載されている。また宿主細胞は、単細胞病原体(例えば原核生物)または非単細胞病原体(例えば、真核生物)であってよい。有用な単細胞は、細菌細胞であり、例えば、限定されるものではないが、バシラス細胞(例えば、バシラス・アルカノフィルス(Bacillus alkalophilus)、バシラス・アミロリクエファシエンス、バシラス・ブレビス(Bacillus brevis)、バシラス・サークランス(Bacillus circulans)、バシラス・コアグランス(Bacillus coagulans)、バシラス・ロータス(Bacillus lautus)、バシラス・レンタス(Bacillus lentus)、バシラス・リへニホルミス、巨大菌(Bacillus megaterium)、バシラス・ステアロサルモフィルス、枯草菌、およびバシラス・スリンジエンシス(Bacillus thuringiensis));またはストレプトミセス細胞(例えば、ストレプトミセス・リビダンス(Streptomyces lividans)もしくはストレプトミセス・ムリヌス(Streptomyces murinus))を含めたグラム陽性細菌、または大腸菌およびシュードモナス(Pseudomonas)種のようなグラム陰性細菌である。好ましい実施形態において、細菌宿主細胞はバシラス・レンタス、バシラス・リへニホルミス、バシラス・ステアロサルモフィルスまたは枯草菌細胞である。細菌宿主細胞の形質転換は、例えばプロトプラスト形質転換法(例えば、ChangおよびCohen, 1979, Molecular General Genetics 168:111-115を参照されたい)、コンピテント細胞の使用(例えば、YoungおよびSpizizin, 1961, Journal of Bacteriology 81: 823-829、またはDubnarおよびDavidoff Abelson, 1971, Journal of Molecular Biology 56: 209-221を参照されたい)、エレクトロポレーション法(例えば、ShigekawaおよびDower, 1988, Biotechniques 6: 742-751を参照されたい)、または接合(例えば、KoehlerおよびThorne, 1987, Journal of Bacteriology 169: 5771-5278を参照されたい)によって達成されうる。宿主細胞は、真菌細胞であってよい。また真菌宿主細胞は、酵母細胞であってよい。本明細書において使用される「酵母」という用語には、子嚢胞子形成酵母(ascosporogenous yeast)(エンドミケス目(Endomyoetales))、担子胞子形成酵母(basidiosporogenous yeast)および不完全菌類(Fungi Imperfecti)(ブラストミセテス(Blastomycetes))に属する酵母が含まれる。
【0077】
細胞の培養に用いる培地は、哺乳動物細胞を生育するのに好適な通常の培地、例えば適当な補充物を含有する血清含有培地もしくは無血清培地、または昆虫細胞、酵母細胞、もしくは真菌細胞を生育するのに好適な培地であってよい。好適な培地は、製造業者から入手可能であり、または公表されている配合表(例えば、American Type Culture Collectionのカタログ中の配合表)に従って調製できる。
【0078】
従って、また本発明は、本発明の天然のポリペプチド配列を有するエキセンジン変異体およびペプチド結合体を製造する方法であって、以下の手順:
a) 本発明のエキセンジン変異体またはペプチド結合体のペプチド配列を含んでなるポリペプチド配列をコードする核酸配列および選択マーカーを含有する核酸構築物またはベクターを宿主細胞中に導入することによって、組換え宿主細胞を得て;
b) 該組換え宿主細胞を選別し;
c) 該組換え宿主細胞を該ポリペプチド配列の産生を可能にする条件下で培養し;
d) 該ポリペプチド配列をこの培養物から単離し;さらに
e) 場合によって、適当なプロテアーゼを用いて該ポリペプチド配列を切断することによって該ペプチド結合体を得ること、
を含む方法に関する。
【0079】
次いで、このように細胞によって産生された天然のポリペプチド配列を有する本発明の変異体およびペプチド結合体を、遠心分離もしくは濾過によって培地から宿主細胞を分離し、塩(例えば、硫酸アンモニウム)によって上清もしくは濾液のタンパク質成分を沈澱させ、種々のクロマトグラフィー法(例えば、イオン交換クロマトグラフィー、アフィニティクロマトグラフィーなど)によって精製することを含めた通常の方法によって培養培地から回収することができる。親油性置換基を、当技術分野において公知の方法を用いて、本発明のペプチドに結合させることができる。一実施形態において、親油性置換基は、標準的な合成方法において、すでに親油性置換基が結合したアミノ酸を組込むことによって結合されうる(例えば、実施例の節における化合物7の合成を参照されたい)。あるいは、この置換基は、例えばWO98/08871に記載のように、ペプチドを合成し、単離した後に結合させることができる。
【0080】
本発明を以下の実施例でさらに説明する。
【実施例】
【0081】
実施例
ペプチド合成、一般的手順
・装置および合成方法
ペプチドを、濾過用のポリプロピレンフィルターを備えたポリエチレン容器で、N-α-アミノ保護基として9-フルオレニルメチルオキシカルボニル(Fmoc)を、および側鎖官能基については好適な慣用の保護基を用いて、バッチ式で合成する(Drylandら, 1986, J. Chem. Soc., Perkin Trans. 1: 125-137)。
・溶媒
溶媒DMF(N,N-ジメチルホルムアミド、Riedel de-Haen, Germany)を、強陽イオン交換樹脂(Lewatit S 100 MB/H 強酸、Bayer AG Leverkusen, Germany)を充填したカラムに通して精製し、使用前に3,4-ジヒドロ-3-ヒドロキシ-4-オキソ-1,2,3-ベンゾトリアジン(Dhbt-OH)を加えて遊離アミンについて分析する(遊離アミンが存在する場合には黄色(Dhbt-O-陰イオン)となる)。溶媒DCM(ジクロロメタン、分析用グレード、Riedel de-Haen, Germany)は精製せずにそのまま使用する。THF(テトラヒドロフラン、分析用グレード、Riedel de-Haen, Germany)は精製せずにそのまま使用する。
・アミノ酸
Fmoc保護アミノ酸は好適に側鎖が保護されたものをMilliGen(UK)およびPerSeptive Biosystems GmbH Hamburg, Germanyから購入する。FmocLys(パルミトイル)-OHはBachem(Switzerland)から購入する。
・リンカー
(4-ヒドロキシメチルフェノキシ)酢酸(HMPA), Novabiochem, SwitzerlandをDICによって、プレフォーム型(preformed)またはin situ生成による1-ヒドロキシベンゾトリアゾール(HObt)エステルのいずれかで樹脂と結合させる。
・カップリング試薬
カップリング試薬ジイソプロピルカルボジイミド(DIC)はRiedel de-Haen, Germanyから購入し、使用前に蒸留する。ジシクロヘキシルカルボジイミド(DCC)はMerck-Schuchardt, Munchen, Germanyから購入し、蒸留精製する。
・固相支持体
Fmoc法により合成されるペプチドをDMF中0.05M以上の濃度のFmoc保護活性化アミノ酸を用いて次の種類の固相支持体上で合成する。TentaGel S樹脂0.22〜0.31mmol/g(TentaGel S-Ram、TentaGel S RAM-Lys(Boc)Fmoc:Rapp polymere, Germany)。
・触媒および他の試薬
ジイソプロピルエチルアミン(DIEA)はAldrich, Germanyから、エチレンジアミンはFlukaから、ピペリジンおよびピリジンはRiedel de-Haen, Frankfurt, Germanyから購入する。4-(N,N-ジ-メチルアミノ)ピリジン(DMAP)はFluka, Switzerlandから購入し、対称無水物(symmetrical anhydrides)を伴うカップリング反応の触媒として用いる。エタンジチオールはRiedel de-Haen, Frankfurt, Germanyから購入する。3,4-ジヒドロ-3-ヒドロキシ-4-オキソ-1,2,3-ベンゾトリアジン(Dhbt-OH)および1-ヒドロキシベンゾトリアゾール(HObt)はFluka, Switzerlandから入手する。
・カップリング手順
第1のアミノ酸をDICまたはDCCによって、好適なN-α-保護アミノ酸から生成されるDMF中の対称無水物として結合させる。次のアミノ酸はDMF中でDICによって、好適なN-α-保護アミノ酸およびHObtから作製される、プレフォームHObtエステルとして結合させる。ニンヒドリン試験を80℃で行ってアシル化を確認し、試験中のFmoc脱保護を防ぐ(Larsen. B. D.およびHolm. A., 1994. Int. J. Peptide Protein Res. 43: 1-9)。
・HObtエステルとしてのカップリング
方法a. 3当量のN-α-アミノ保護アミノ酸を3当量のHObtおよび3当量のDICとともにDMFに溶解する。この溶液を室温で10分間置いた後、予め活性化したアミノ酸を加える前にDMF中0.2%Dhbt-OHの溶液で洗浄した樹脂に加える。
【0082】
方法b. 3当量のN-α-アミノ保護アミノ酸を3当量のHObtとともにDMFに溶解する。使用直前に3当量のDICを加える。最終溶液を樹脂に加える。
・プレフォーム対称無水物
6当量のN-α-アミノ保護アミノ酸をDCMに溶解し、0℃まで冷却する。DDCまたはDIC(3当量)を加え、反応を10分間続ける。溶媒を真空で留去し、残渣をDMFに溶解する。DCCを用いた場合にはDMF溶液を濾過し、すぐに樹脂、続いて0.1当量のDMAPを加える。
・N-α-アミノFmoc保護基の脱保護
Fmoc基の脱保護は、DMF中20%ピペリジン溶液で処理(1×5および1×10分)し、続いてDhbt-OHを排液のDMFに加えて黄色(Dhbt-O-)が検出されなくなるまでDMFで洗浄して行う。
・酸による樹脂からのペプチドの切断
方法a. ペプチドは、95%トリフルオロ酢酸(TFA, Riedel de-Haen, Frankfurt, Germany)-水v/vまたは95%TFAおよび5%エタンジチオールv/vで室温で2時間処理して樹脂から切断する。濾過した樹脂を95%TFA-水v/vで洗浄し、濾液および洗液を10%酢酸を加えて希釈する。得られた混合物をエーテルで3回抽出し、最後に凍結乾燥する。凍結乾燥粗生成物を高速液体クロマトグラフィー(HPLC)で分析し、質量分析法(MS)により同定する。
・TentaGel S-RAM上でのバッチ式ペプチド合成
TentaGel S-RAM樹脂(100〜1000mg、0.22〜0.31mmol/g)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れる。樹脂をDMF(5〜10ml)で膨潤させ、Fmoc基を上記の手順に従って除去する。その配列に従う次のアミノ酸を、上記のようにDICによって、in situ生成されるFmoc保護HObtエステル(3当量)として結合させる。特に断りのない限り、カップリングは3時間続ける。樹脂から溶媒を抜き(drained)、DMF(4×5〜10ml、各2分)で洗浄し、過剰の試薬を除去する。ニンヒドリン試験を80℃で行ってすべてのアシル化を確認する。合成完了後、ペプチド-樹脂をDMF(3×5〜10ml、各5分)、DCM(3×5〜10ml、各1分)、さらに最後にジエチルエーテル(3×5〜10ml、各1分)で洗浄し、真空乾燥する。
・HPLC条件
定組成(isocratic)HPLC分析は、LC-6Aポンプ、215nmで作動させるMERCK HITACHI L-4000 UV検出器および20μlループのRheodyne 7125 注入バルブからなるShimadzu systemで行う。定組成分析に用いるカラムは、Spherisorb ODS-2(100×3mm: 5-μm粒子)(MicroLab, Aarhus, Denmark)である。グラジェントを用いるHPLC分析は、MERCK HITACHI L-6200 インテリジェントポンプ、215nmで作動させるMERCK HITACHI L-4000 UV検出器および20μlループのRheodyne 7125 注入バルブで、またはWater 996 フォトダイオードアレイ検出器を備えたWater 600 E装置で行う。用いるカラムはRescorce(商標)RPC 1ml(Waters)またはLiChroCART 125-4、LiChrospher 100 RP-18(5μm)(Merck)である。バッファーAは水中0.1容量%TFA、バッファーBは90容量%アセトニトリル、9.9容量%水および0.1容量%TFAである。バッファーは、次のペプチド分析用のグラジェント、1)0%〜100%Bの直線グラジェント(30分)または2)0%B(2分)、0%〜50%B(23分)、50%〜100%B(5分)の直線グラジェントのいずれかを用いて流速1.3〜1.5ml/分でカラムを通して供給する。分取HPLCについては、Water 996 フォトダイオードアレイ検出器を備えたWater 600 E装置で精製を行う。用いるカラムはWaters Delta-Pak C-18 15μm, 100Å, 25×100mmである。グラジェント”2)”を流速9ml/分で用いる。
・質量分析法
質量スペクトルは、エレクトロスプレー(ESI)プローブ(ES-MS)を備えたFinnigan Mat LCQ装置およびマトリックスとしてβ-シアノ-p-ヒドロキシ桂皮酸を用いるTofSpec E. Fisons Instrument(MALDI-TOF)で得る。また、Micromass LCT装置によりスペクトルを得てもよい。
先行技術ペプチドのペプチド合成
(i)化合物(i)のペプチド合成
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Pro-Ser-NH2
TentaGel S-RAM上での(エキセンジン-4(1-39)-NH2)(配列番号102)
乾燥TentaGel S-RAM樹脂(0.25mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。Fmoc基を上記の手順に従って除去し、その配列に従うペプチドを、「TentaGel S-RAM樹脂上でのバッチ式ペプチド合成」に記載したように構築(assembled)する。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。粗ペプチドを上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が90%を超えていることがわかる。ペプチドはES-MSにより同定する。収率17%。
(ii)化合物(ii)のペプチド合成
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Pro-NH2
TentaGel S-RAM上での(des Ser39 エキセンジン-4(1-39)-NH2)
乾燥TentaGel S-RAM樹脂(0.25mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。Fmoc基を上記の手順に従って除去し、その配列に従うペプチドを、「TentaGel S-RAM樹脂上でのバッチ式ペプチド合成」に記載したように構成する。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。粗ペプチドを上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が97%を超えていることがわかる。ペプチドはES-MSにより同定する。収率22%。
(iii)化合物(iii)のペプチド合成
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Gly-Gln-Ala-Ala-Lys-Glu-Phe-Ile-Ala-Trp-Leu-Val-Lys-Gly-Arg-NH2
TentaGel S-RAM上での(Gly8-GLP1-(7-36)(ヒト)-NH2)(配列番号87)
乾燥TentaGel S-RAM樹脂(0.25mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。Fmoc基を上記の手順に従って除去し、その配列に従うペプチドを、「TentaGel S-RAM樹脂上でのバッチ式ペプチド合成」に記載したように構成する。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。粗ペプチドを上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が95%を超えていることがわかる。ペプチドはES-MSにより同定する。収率9%。
本発明のペプチド配列の合成
1.化合物1のペプチド合成
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Ser-NH2
TentaGel S-RAM上での(des-Pro36-エキセンジン-4(1-39)-NH2)(配列番号101)
乾燥TentaGel S-RAM樹脂(0.25mmol/g、1500mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。Fmoc基を上記の手順に従って除去し、その配列に従うペプチドを、「TentaGel S-RAM樹脂上でのバッチ式ペプチド合成」に記載したように構築する。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。粗ペプチドを上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が95%を超えていることがわかる。ペプチドはES-MSにより同定する。収率18.3%。
2.化合物2のペプチド合成
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Ser-(Lys)6-NH2
TentaGel S-RAM-Lys(Boc)Fmoc上での(des-Pro36-エキセンジン-4(1-39)-Lys6-NH2)(配列番号93)
乾燥TentaGel S-RAM-Lys(Boc)Fmoc樹脂(0.22mmol/g、1500mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。第1のリジンのFmoc基を上記のように除去し、「TentaGel S-Ram-Lys(Boc)Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が95%を超えていることがわかる。ペプチドはES-MSにより同定する。収率22.1%。
3.化合物3のペプチド合成
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Pro-Ser-(Lys)6-NH2
TentaGel S-RAM-Lys(Boc)Fmoc上での(エキセンジン-4(1-39)-Lys6-NH2)(配列番号92)
乾燥TentaGel S-RAM-Lys(Boc)Fmoc樹脂(0.22mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。第1のリジンのFmoc基を上記のように除去し、「TentaGel S-Ram-Lys(Boc)Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が90%を超えていることがわかる。ペプチドはES-MSにより同定する。収率20.5%。
4.化合物4のペプチド合成
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Gly-Gln-Ala-Ala-Lys-Glu-Phe-lle-Ala-Trp-Leu-Val-Lys-Gly-Arg-(Lys)6-NH2
TentaGel S-RAM-Lys(Boc)Fmoc上での(Gly8-GLP1-(7-36)(ヒト)-Lys6-NH2)(配列番号88)
乾燥TentaGel S-RAM-Lys(Boc)Fmoc樹脂(0.22mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。第1のリジンのFmoc基を上記のように除去し、「TentaGel S-Ram-Lys(Boc)Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が95%を超えていることがわかる。ペプチドはES-MSにより同定する。収率11.7%。
4a.化合物4のペプチド合成
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Gly-Gln-Ala-Ala-Lys-Glu-Phe-lle-Ala-Trp-Leu-Val-Lys-Gly-Arg-Lys-Lys-Lys-Lys-Lys-Lys-NH2
TentaGel S-RAM-Lys(Boc)Fmoc上での([Gly8]hGLP-1(7-36)-(Lys)6-NH2)(配列番号88)
乾燥TentaGel S-RAM-Lys(Boc)Fmoc樹脂(0.22mmol/g、2013mg)を濾過用のポリプロピレンフィルターを備えたガラス容器に入れ、DMF(5ml)で2時間膨潤させる。第1のリジンのFmoc基を上記のように除去し、「TentaGel S-Ram-Lys(Boc)Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が90%を超えていることがわかる。ペプチドはES-MSにより同定する。収率13%。
5.化合物5のペプチド合成
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Gly-Gln-Ala-Ala-Lys-Glu-Phe-lle-Ala-Trp-Leu-Val-Lys-Gly-Arg-Lys(パルミトイル)-(Lys)6-NH2
TentaGel S-RAM-Lys(Boc)Fmoc上での([Gly8,Lys37(パルミトイル)]GLP1-(7-36)(ヒト)-(Lys)7-NH2)(配列番号89)
乾燥TentaGel S-RAM-Lys(Boc)Fmoc樹脂(0.22mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。第1のリジンのFmoc基を上記のように除去し、「TentaGel S-Ram-Lys(Boc)Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。試薬Fmoc-Lys(パルミトイル)-OHはDMFにわずかしか溶解しないため、若干方法を改変して結合させる。Fmoc-Lys(パルミトイル)-OH約400mgをDMFではなくTHF約6mlに溶解させる。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法bに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が95%を超えていることがわかる。ペプチドはES-MSにより同定する。収率9.3%。
6.化合物6のペプチド合成
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Gly-Gln-Ala-Ala-Lys-Glu-Phe-lle-Ala-Trp-Leu-Val-Lys(パルミトイル)-Gly-Arg-(Lys)6-NH2
TentaGel S-RAM-Lys(Boc)Fmoc上での([Gly8,Lys34(パルミトイル)]GLP1-(7-36)(ヒト)-(Lys)6-NH2)(配列番号90)
乾燥TentaGel S-RAM-Lys(Boc)Fmoc樹脂(0.22mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。第1のリジンのFmoc基を上記のように除去し、「TentaGel S-Ram-Lys(Boc)Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。試薬Fmoc-Lys(パルミトイル)-OHはDMFにわずかしか溶解しないため、若干方法を改変して結合させる。Fmoc-Lys(パルミトイル)-OH約400mgをDMFではなくTHF約6mlに溶解させる。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が90%を超えていることがわかる。ペプチドはES-MSにより同定する。収率4.2%。
7.化合物7のペプチド合成
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Gly-Gln-Ala-Ala-Lys(パルミトイル)-Glu-Phe-Ile-Ala-Trp-Leu-Val-Lys-Gly-Arg-(Lys)6-NH2
TentaGel S-RAM-Lys(Boc)Fmoc上での([Gly8,Lys26(パルミトイル)]GLP1-(7-36)(ヒト)-(Lys)6-NH2)(配列番号103)
乾燥TentaGel S-RAM-Lys(Boc)Fmoc樹脂(0.22mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。第1のリジンのFmoc基を上記のように除去し、「TentaGel S-Ram-Lys(Boc)Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。試薬Fmoc-Lys(パルミトイル)-OHはDMFにわずかしか溶解しないため、若干方法を改変して結合させる。Fmoc-Lys(パルミトイル)-OH約400mgをDMFではなくTHF約6mlに溶解させる。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が90%を超えていることがわかる。ペプチドはES-MSにより同定する。収率2.2%。
8.化合物8のペプチド合成
H-Lys-Lys-Lys-Lys-Lys-Lys-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Ser-NH2
TentaGel S-RAM-Fmoc上での(H-(Lys)6-des Pro36 エキセンジン-4(1-39)-NH2)
乾燥TentaGel S-RAM-Fmoc樹脂(0.23mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。樹脂のFmoc基を上記のように除去し、「TentaGel S-Ram-Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が95%を超えていることがわかる。ペプチドはES-MSにより同定する。収率26%。
9.化合物9のペプチド合成
H-Lys6-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Ser-(Lys)6-NH2
TentaGel S-RAM-Lys(Boc)Fmoc上での(H-Lys6-des Pro36 エキセンジン-4(1-39)-Lys6-NH2)
乾燥TentaGel S-RAM-Lys(Boc)Fmoc樹脂(0.22mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。第1のリジンのFmoc基を上記のように除去し、「TentaGel S-Ram-Lys(Boc)Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が90%を超えていることがわかる。ペプチドはES-MSにより同定する。収率32%。
10.化合物10のペプチド合成
H-Lys-Lys-Lys-Lys-Lys-Lys-His-Gly-Glu-Gly-Thr-PheThr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Gly-Gln-Ala-Ala-Lys-Glu-Phe-lle-Ala-Trp-Leu-Val-LysGly-Arg-Lys-Lys-Lys-Lys-Lys-Lys-NH2
TentaGel S-RAM-Lys(Boc)Fmoc上での(H-(Lys)6-([Gly8]hGLP-1(7-36)-(Lys)6-NH2)
乾燥TentaGel S-RAM-Lys(Boc)Fmoc樹脂(0.22mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。第1のリジンのFmoc基を上記のように除去し、「TentaGel S-Ram-Lys(Boc)Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が90%を超えていることがわかる。ペプチドはES-MSにより同定する。収率18%。
11.化合物11のペプチド合成
H-Lys-Lys-Lys-Lys-Lys-Lys-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Gly-Gln-Ala-Ala-Lys-Glu-Phe-Ile-Ala-Trp-Leu-Val-Lys-Gly-Arg-NH2
TentaGel S-RAM-Lys(Boc)Fmoc上での(H-(Lys)6-[Gly8]hGLP-1(7-36)-NH2)
乾燥TentaGel S-RAM-Fmoc樹脂(0.23mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。樹脂のFmoc基を上記のように除去し、「TentaGel S-Ram-Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が98%を超えていることがわかる。ペプチドはES-MSにより同定する。収率15%。
12.化合物12のペプチド合成
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Gly-Gln-Ala-Ala-Lys-Glu-Phe-Ile-Ala-Trp-Leu-Val-Lys-Gly-Arg-Lys-Lys-Lys-Lys-Lys-Lys-Lys-Lys-NH2
TentaGel S-RAM-Lys(Boc)Fmoc上での([Gly8]hGLP-1(7-36)-(Lys)8-NH2)
乾燥TentaGel S-RAM-Lys(Boc)Fmoc樹脂(0.22mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。第1のリジンのFmoc基を上記のように除去し、「TentaGel S-Ram-Lys(Boc)Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が98%を超えていることがわかる。ペプチドはES-MSにより同定する。収率4.2%。
13.化合物13のペプチド合成
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Gly-Gln-Ala-Ala-Lys-Glu-Phe-Ile-Ala-Trp-Leu-Val-Lys-Gly-Arg-Lys-Lys-Lys-Lys-Lys-Lys-Lys-Lys-Lys-Lys-NH2
TentaGel S-RAM-Lys(Boc)Fmoc上での([Gly8]hGLP-1(7-36)-(Lys)10-NH2)
乾燥TentaGel S-RAM-Lys(Boc)Fmoc樹脂(0.22mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。第1のリジンのFmoc基を上記のように除去し、「TentaGel S-Ram-Lys(Boc)Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が95%を超えていることがわかる。ペプチドはES-MSにより同定する。収率2%。
14.化合物14のペプチド合成
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Ser-NH2
TentaGel S-RAM-Fmoc上での(H-des Pro36,Pro37,Pro38エキセンジン-4(1-39)-NH2)
乾燥TentaGel S-RAM-Fmoc樹脂(0.23mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。樹脂のFmoc基を上記のように除去し、「TentaGel S-Ram-Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が95%を超えていることがわかる。ペプチドはES-MSにより同定する。収率11%。
15.化合物15のペプチド合成
H-Lys-Lys-Lys-Lys-Lys-Lys-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Ser-NH2
TentaGel S-RAM-Fmoc上での(H-(Lys)6-des Pro36,Pro37,Pro38 エキセンジン-4(1-39)-NH2)
乾燥TentaGel S-RAM-Fmoc樹脂(0.23mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。樹脂のFmoc基を上記のように除去し、「TentaGel S-Ram-Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が94%を超えていることがわかる。ペプチドはES-MSにより同定する。収率17%。
16.化合物16のペプチド合成
H-Asn-Glu-Glu-Glu-Glu-Glu-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-lle-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Ser-NH2
TentaGel S-RAM-Fmoc上での(H-Asn-(Glu)5-des Pro36,Pro37,Pro38 エキセンジン-4(1-39)-NH2)
乾燥TentaGel S-RAM-Fmoc樹脂(0.23mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。樹脂のFmoc基を上記のように除去し、「TentaGel S-Ram-Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が90%を超えていることがわかる。ペプチドはES-MSにより同定する。収率9%。
17.化合物17のペプチド合成.化合物3.
H-(Lys)6-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Ser-(Lys)6-NH2
TentaGel S-RAM-Lys(Boc)Fmoc上での(H-(Lys)6-des Pro36,Pro37,Pro38 エキセンジン-4(1-39)-(Lys)6-NH2)
乾燥TentaGel S-RAM-Lys(Boc)Fmoc樹脂(0.22mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。第1のリジンのFmoc基を上記のように除去し、「TentaGel S-Ram-Lys(Boc)Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が90%を超えていることがわかる。ペプチドはES-MSにより同定する。収率10%。
18.化合物18のペプチド合成
H-Asn-Glu-Glu-Glu-Glu-Glu-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-lle-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Ser-(Lys)6-NH2
TentaGel S-RAM-Lys(Boc)Fmoc上での(H-Asn-(Glu)5-des Pro36,Pro37,Pro38 エキセンジン-4(1-39)-(Lys)6-NH2)
乾燥TentaGel S-RAM-Lys(Boc)Fmoc樹脂(0.22mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。第1のリジンのFmoc基を上記のように除去し、「TentaGel S-Ram-Lys(Boc)Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が92%を超えていることがわかる。ペプチドはES-MSにより同定する。収率14%。
19.化合物19のペプチド合成
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Ser-(Lys)6-NH2
TentaGel S-RAM-Lys(Boc)Fmoc上での(des Pro36,Pro37,Pro38エキセンジン-4(1-39)-(Lys)6-NH2)
乾燥TentaGel S-RAM-Lys(Boc)Fmoc樹脂(0.22mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。第1のリジンのFmoc基を上記のように除去し、「TentaGel S-Ram-Lys(Boc)Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が97%を超えていることがわかる。ペプチドはES-MSにより同定する。収率19%。
20.化合物2の組換え調製物
pYES0010発現ベクターの構築
酵母での異種構造発現のため、合成cDNAを構築した。化合物2をコードするタンパク質配列をサッカロミセス・セレビシエ(Saccharomyces cerevisiae)コドン利用表((Saccharomyces Genomeデータベース)を用いて逆翻訳した。合成cDNAの翻訳を可能にするため、付加的ATG開始コドンを5’末端に付加し、TAA停止コドンを3’末端に付加した。構築物をアンピシリン耐性遺伝子を含むpYES2シャトルベクターのHindIIIおよびEcoRI部位に挿入し、新たな構築物をpYES0010と名付けた(図6参照)。続いてpYES0010を大腸菌(E.coli)へ形質転換し、アンピシリン選択圧下に置いた。陽性クローンを選択して配列決定した。
酵母への形質転換
pYES0010を酵母ハプロイドINVSc1:MaTa his3deltal leu2 trp1-289 ura3-52へと形質転換させる目的で酵母をYPD培地(1%酵母抽出物、2%ペプトン、2%グルコースおよび0.004%硫酸アデニン)で30℃で飽和状態まで増殖させた。1mlの培養物を形質転換用に回収した。2μlの10mg/ml担体DNAを加え、1μgのpYES0010を加えて混合した。0.5ml(45%PEG 4000、1M LiOAc、0.5M EDTAおよび1M Tris-HCl(pH7.5)を加えて混合した。最後に20μl 1M DTTを加え、混合物を室温で16時間インキュベートした。インキュベーション後、細胞を42℃で10分間熱ショックを与え、選択プレート(6.7%酵母窒素ベース、2%グルコース、20μg/mlアデニン、20μg/mlアルギニン、29μg/mlイソロイシン、20μg/mlヒスチジン、60μg/mlロイシン、20μg/mlリジン、20μg/mlトリプトファン、20μg/mlメチオニン、50μg/mlフェニルアラニン、150μg/mlバリン、30μg/mlチロシンおよび2.5%寒天)に播種した。形質転換細胞が現れるまで、プレートを30℃で3〜5日間インキュベートした。
化合物2の発現および精製
形質転換細胞を選択培地(6.7%酵母窒素ベース、2%グルコース、20μg/mlアデニン、20μg/mlアルギニン、29μg/mlイソロイシン、20μg/mlヒスチジン、60μg/mlロイシン、20μg/mlリジン、20μg/mlトリプトファン、20μg/mlメチオニン、50μg/mlフェニルアラニン、150μg/mlバリン、30μg/mlチロシン)で1.5日間培養した。細胞を回収し、ガラクトース誘導培地(6.7%酵母窒素ベース、4%ガラクトース、20μg/mlアデニン、20μg/mlアルギニン、29μg/mlイソロイシン、20μg/mlヒスチジン、60μg/mlロイシン、20μg/mlリジン、20μg/mlトリプトファン、20μg/mlメチオニン、50μg/mlフェニルアラニン、150μg/mlバリン、30μg/mlチロシン)に1日間再懸濁した。細胞を回収し、プロテアーゼ阻害剤(Roche)を含む10mM Tris-HCl pH7.5でホモジナイズした。溶解物を20,000×gで30分間遠心分離して清澄化した。上清を10mM Tris-HCl(pH7.5)で平衡状態にしたSuperdex 12 HR 10/30カラム(Amersham Pharmacia Biotech)に入れた。カラムを50mM 重炭酸アンモニウムバッファーpH8.0で溶出した。組換え化合物2を含むサンプルをプールした。N末端メチオニンをメチオニンアミノペプチダーゼにより除去し、サンプルをHPLCカラムでさらに精製した。
化合物2精製のためのHPLC設定
HPLCカラム:Kromasil RP C8; K 100-10-C8 nr. CER 2230.compound
温度: 22℃
流速: 35ml/分
HPLC溶媒:
A:水中0.10%トリフルオロ酢酸
B:アセトニトリル:水 90:10中0.10%トリフルオロ酢酸
化合物2をHPLCカラムから20%〜80%アセトニトリル中0.10%トリフルオロ酢酸で40分間溶出した。
21.ペプチド注射製剤
ペプチドの静脈注射用定用量製剤は、ペプチドを無菌、等張生理食塩水に溶解し、得られた溶液を滅菌条件下で不活性ガスを充填したガラス製アンプルに入れて調製する。各用量のペプチドを、不活性ガスを充填した乾燥状態のアンプルまたはキャップ付バイアルに入れる。ペプチドの静脈注射用の複数用量製剤は、ペプチドを無菌、等張生理食塩水に溶解し、得られた溶液をキャップ付バイアルに入れて、必要に応じて保存剤(例えば、0.1%パラヒドロキシベンゾエート、1%ベンジルアルコールまたは0.1%クロロクレゾール)を加えて、調製する。
複数用量ペプチド製剤の例
化合物2 12.25mg
リン酸二水素ナトリウム 1.380g
パラヒドロキシベンゾエート 0.1g
注射水溶液 100ml
【0083】
22.安定性試験
in vivoにおけるタンパク質分解からの上記ペプチドの保護について調べる手段として、選択したタンパク質分解酵素の存在下でのペプチドおよびペプチド複合体のin vitroにおける安定性試験を用いる。実施する試験のねらいは、1種以上の酵素、ロイシンアミノペプチダーゼ、カルボキシペプチダーゼAおよびジペプチジルアミノペプチダーゼIVの溶液中、37℃における化合物4、5、6および7のin vitroにおける安定性と先行技術の化合物、化合物(iii)H-(Gly8)-hGLP-1(7-36)-NH2)およびhGLP-1(7-36)-NH2のものを測定して比較することであった。
in vitro安定性についての材料および装置
用いた水はMilli-Q 水処理システム(Millipore, Bedford, MA, USA)から得られる最高品質のものであった。アセトニトリル(ACN)はLabscan Ltd.(Dublin, Ireland)から入手されるスーパーグラジェント級のものであった。トリフルオロ酢酸(TFA)99.9%、リン酸二水素ナトリウム(NaH2PO4)、水酸化ナトリウム(NaOH)および用いた他のすべての化学薬品は分析用のものであった。ロイシンアミノペプチダーゼ(EC 3.4.11.1)、カルボキシペプチダーゼA(EC 3.4.17.1)およびジペプチジルペプチダーゼ(ジペプチジルアミノペプチダーゼIV、EC 3.4.14.5)はすべてSigma(St. Louis, MO, USA)から入手した。HP 1100バイナリーポンプ、HP 1100オートサンプラー、HP 1100カラムサーモスタットおよびHP 1100可変波長検出器からなるHewlett Packard HP 1100 HPLCシステムを用いてHPLC分析を行った。LCソフトウェア(Rev. A. 06. 01)用のHewlett Packard Chemstationを装置の制御およびデータ取得用に用いた。5μm、C18、300Å粒子を充填したVydac 238TP54(150×4.6mm I.D.)カラムを装置に取り付けて用いた。Stuart ScientificのSHT200Dブロックヒーターを安定性試験中のペプチド/酵素溶液の加熱に用いた。ロイシンアミノペプチダーゼ(25U/ml)もしくはカルボキシペプチダーゼA(1U/ml)を含むpH7.4の50mMリン酸バッファー溶液またはジペプチジルアミノペプチダーゼIV(0.5U/ml)を含むpH8.0の100mM重炭酸アンモニウムバッファーでの試験化合物の分解を37℃で調べた。1アリコート(100μl)の水中ペプチドストック溶液(1mg/ml)をエッペンドルフマイクロバイアル中の予め熱した酵素溶液900μlに加えて試験を開始した。その際、ペプチドの開始濃度を0.1mg/ml(約1.7・10-5〜1.8・10-5M)とした。ペプチド/酵素溶液を37℃に保ち、適当な時間間隔をおいて100μlのサンプルをペプチド/酵素溶液から回収し、アセトニトリル中の25%TFA20μlと十分に混合して酵素分解の進行を止めた。不活性化したサンプルをオートサンプラーバイアルに移し、インタクトな試験化合物の含量を以下に記載するHPLCにより調べた。酵素溶液中での試験化合物の半減期(t1/2)を、自然対数のプロットないし時間に対する残留濃度(すなわち、HPLCピーク高さ)から以下の式:
t1/2=1/kobs・ln(2)
(なお、kobsは観測した分解の見かけの一次速度定数)
を用いて算出した。
HPLC分析
上記のように実施した安定性試験のサンプルを上記の装置および次の試験条件を用いてグラジェントHPLC分析により分析した。
カラム温度:30℃
注入量:10μl
移動相A:水中0.1%TFA
移動相B:アセトニトリル(ACN)中0.085%TFA
グラジェント:32〜52% B、21分
検出:215nmのUV
各安定性試験から得られた結果を以下の表1に示している。この表から、調べたすべての酵素を含む溶液中で本発明の化合物の半減期がかなり延びていることがわかる。
【0084】
【表1】
【0085】
LAP:ロイシンアミノペプチダーゼ、CPA:カルボキシペプチダーゼA、DPPIV:ジペプチジルアミノペプチダーゼIV
【0086】
23.化合物(iii)および化合物4のラット血漿におけるin vitro安定性試験
ヘパリンにより安定させたラット(Sprague-Dawley)血漿中で2種類の試験化合物の分解を行った後、固相抽出およびLC-MSを組み合わせて行った。分解は720分間血漿中で行った。化合物(iii)の半減期がラット血漿中では238分であることがわかった。この試験結果とラット血漿中で466分であるとわかった化合物4の半減期とを比較した。
材料および方法
ヘパリンナトリウム中のブランクラット血漿(5000単位/mL)はHarlan Sera Lab Ltd.(Loughborough, UK)から入手した。試験に用いた試験物質については以下の表に記載している。in vitro試験では100μg/ml Milli-Q水の原液を用いた(26.0μM化合物(iii)H-(Gly8)-GLP-1-NH2または17.8μM 化合物4について記載)。
【0087】
LC-MS分析は、オンライン脱ガス装置、クォーターナリーグラジェントポンプ、オートサンプラー、カラムオーブンからなるHP 1100装置、Hewlett Packard(Wilmington, DE. USA)とMicromass(Altrincham, UK)のQuattro Ultima質量分析計とを組み合わせて行った。LCおよびMSはともにMassLynx3.3ソフトウェアで制御した。MS検出の前にVydac 218MS52(2.1×250mm)カラム(Hesperia, CA, USA)でLC分離を行った。開始血漿量は1000μl(37℃)であった。開始血漿量から100μlを0.75ml HPLCバイアル(ブランクとして用いる)へ移し、560μlの抽出溶液(MeCN:0.18M 炭酸アンモニウム pH9.5(6:94v/v) 4C)と混合し、ASPEC XL4 Robotを用いて固相抽出法で抽出した。100μlのストック溶液を残る900μlの血漿に加えて十分に混合し、37℃でインキュベートした(開始濃度は試験化合物10μg/mlとなる)。各時点(各々0.2、60、120、180、240、360、480、662および720分)において血漿を含む薬剤100μlを回収し、560μlの氷冷抽出溶液と混合し、直ちに上記の固相抽出法により抽出した。抽出した血漿サンプルをLC-MSにより分析した。LC-MS分析はQuattro Ultima II 三連四重極MS装置とを組み合わせたHP 1100系LCで行った。サンプルをオートサンプラートレイで18℃に保ち、その後10μlを注入した。分離はVydac 218MS52(2.1×250mm)LCカラムで30℃で行い、14分間で15〜50%Bの直線濃度勾配、流速250μl/分を用いた。水中0.1%蟻酸を移動相A、MeCN中0.1%蟻酸を移動相Bとして用いた。化合物4および化合物(iii)を、それぞれ6H+(m/z=676.7)および4H+(m/z=822.1)イオン種を用い、単一イオン記録方式(SIR)により検出した。化合物4および化合物(iii)の分析用のconc電圧はそれぞれ100および70Vに設定した。化合物4および化合物(iii)のin vitro安定性をラット血漿中、LC-MSにより調べた。2種類の化合物の分解は720分間行い、結果を、時間に対するピーク面積の自然対数でプロットした。化合物の分解速度(kobs)が線形回帰による傾きとして、半減期(t1/2)がln2/kobsとして示されることがわかった。試験の結果を以下に示す。
【0088】
よって、試験の結果から、(Gly8)hGLP-1(7-36)配列にC末端Lys6ペプチドが結合していることで、ラット血漿における安定性が2倍増加することとなる。
24.化合物5の単回用量の経口および非経口投与の糖尿病ob/obマウスの血中グルコースレベルへの影響
本発明の化合物は血中グルコースを低減させる特性を有している。このことについて、化合物5を用いて腹膜内(i.p.)および経口(p.o.)投与後のob/ob変異体マウスの血中グルコース(BG)レベルへの影響を試験し、考察した。化合物5の腹膜内投与では110μg/マウスの用量で糖尿病マウスのBGレベルが低下した。同様に、化合物5の経口投与では、より低い用量ではないが、1100μg/マウスの用量で同様のBGレベルの低下が認められた。
試験
優性変異レプチン(Tomita, T., Doull. V., Pollock. H. G., およびKrizsan, D. 1992. Pancreatic islets of obese hyperglycemic mice(ob/ob). Pancreas 7: 367-75)が原因で肥満である、40匹の雌糖尿病ob/obマウス(Umea系統、Bomholtgaard)を12:12時間明暗周期による制御された周囲条件下で収容し(3マウス/ケージ)、水を得るための自由な通路を設けた標準Altromin no 1324 dietで餌を与えた。到着時、動物は8週齢であった。マウスを2週間、新環境に順応させた後、試験を開始した。試験時点で動物は13週齢、体重41.8±3.2g(平均±SD; n=42)であった。ストレスにより誘導されるBG変動を軽減するため、試験1日前および3日前にマウスに処置を施した。試験当日、照明をつけて2〜3時間後に尾の先から採血した。分析のため、1滴の血液(<5μl)をグルコースストリップに落とし、Elite Autoanalyser、Bayer, Denmarkにより測定した。全血中グルコース(BG)濃度を固定化グルコースオキシダーゼ法により調べた。血中グルコースレベルは正常血糖から重症高血糖症との間で(範囲: 3.6〜15.6mM; 平均±SD; 9.4±3.3mM; n=42)変動した。BG<5.8mMであった6動物を試験から除外した(全体n=36)。残りの動物をBGレベルに基づいて等級別に分類し、各群間で平均BGに差がないようにした。最初の対照血液サンプリングから1時間後に薬剤を投与し、BGをt=60分、t=120分、t=240分、t=480分に測定した。
ペプチドおよび他の材料
化合物5(バッチ番号ZP 3.12画分1-2、精製)は、Department of Chemistry, Zealand Pharmaceuticalsによって合成された。投与の直前にペプチドを滅菌等張NaClに溶解して容量を0.2mlとした。同じ溶液をp.o.およびi.p.投与の両方に使用した。各血液サンプリング時に、データ記録用紙に各動物について記入した。
薬剤投与
動物に化合物5を投与した。最大用量は1100μg/マウス、最小用量は1.1μg/マウスであった。陰性対照として生理食塩水をp.o.により投与し、陽性対照として試験化合物をi.p.により110μg/マウスの用量を与えた。
【0089】
制御条件下での非絶食ob/obマウスのBGレベルはすべての群で同等であった(各群のデータは示していない)が、群内のBGレベルは大きく分散していた(すべての動物のBG範囲: 5.8〜15.6mM)。そのため、高血糖症の変動程度を補正するために結果を基線との相対差(%対照)として示した。
【0090】
化合物5 110μgの腹膜内投与によりBGの持続的な低下がもたらされ、化合物の投与後1〜2時間後には最下点に達した。生理食塩水で処置した動物には変化が見られなかった。薬剤投与から4〜8時間後の間に、ほとんどの群(5/6)でBGが増加した。化合物5は、糖尿病ob/obマウスにi.p.投与した場合、110μg/マウスの用量でBGレベルが低下した(データは示していない)。60分後には抗糖尿病性効果が認められ、化合物投与から2〜4時間後に最大となった。さらに長時間にわたる効果(>8時間)から、化合物5が、よく知られてはいるが、短時間しか作用しない天然のGLP-1(Bailey, C. J. & Flatt, P. R. 1987, Glucagon-like peptide-1 and the entero-insular axis in obese hyperglycaemic (ob/ob)mice. Life Sci. 40. 521-5)よりも長時間の作用を有していることがわかる。用量1100μg/マウス p.o.では、110μg i.p.で処置した動物で認められたものと同様なBGの低下が示された。
【0091】
発明者らは化合物5が、化合物110μg/マウスのi.p.投与により、糖尿病ob/obマウスのBGレベルを効果的に低下させることを示した。また、経口経路により与えた場合でも、化合物5 1100μg/マウスで同様な効果が認められた。このことから、化合物が胃腸管から吸収されていることがわかる。
25.in vivo試験
化合物1(des Pro36-エキセンジン-4(1-39)-NH2(配列番号101))、
化合物2(des Pro36-エキセンジン-4(1-39)-Lys6-NH2(配列番号93))、
化合物(ii)(Gly8-GLP1-(7-36)(ヒト)-NH2(配列番号87))、
化合物4(Gly8-GLP1-(7-36)(ヒト)-Lys6-NH2(配列番号88))および
化合物5(Gly8 Lys37(パルミトイル)-GLP1-(7-36)(ヒト)-Lys7-NH2(配列番号89))
について
ob/obマウスに、各ペプチドを種々の濃度で、経口および腹腔内投与し、これらの化合物が血糖値に影響を与えるか否かを調べた。用いた試験条件は実施例24に記載したものと同じである。
ペプチドおよび他の材料
des Pro36-エキセンジン-4(1-39)-NH2(化合物1. 配列番号101)およびC末端に結合した付加的配列Lys6以外は同じペプチド、des Pro36-エキセンジン-4(1-39)-Lys6-NH2(化合物2. 配列番号93)、Gly8-GLP1-(7-36)(ヒト)-NH2(化合物(ii). 配列番号87)およびC末端に結合した付加的配列Lys6以外は同じペプチド、Gly8-GLP1-(7-36)(ヒト)-Lys6-NH2(化合物4. 配列番号88)ならびにGly8 Lys37(パルミトイル)-GLP1-(7-36)(ヒト)-Lys7-NH2(化合物5. 配列番号89)は、上記の方法で合成する。溶液については、投与当日の朝、動物に投与する直前に調製する。同じ溶液を経口および腹腔内投与の両方に使用する。すべてのペプチドを滅菌等張NaClに溶解して容量を0.2mlとする。すべての試験を同じマウスで行い、表2に示すペプチドの有効量を比較する。上記のように血液サンプリングを行い、表3に示す用量を動物に投与する。陰性対照として生理食塩水を経口投与する。この結果を表4に示している。
【0092】
【表2】
【0093】
【表3】
【0094】
群のデータには、各処置群の個々の結果の平均±SEMを記載している。化合物の効果を調べるため、ベースラインとの絶対および相対(t=0の%)差を各時点について算出した。
【0095】
【表4】
【0096】
得られた結果を表4および図1〜3に示している。
【0097】
これらの結果により、試験したすべての化合物が血糖値の低下作用を有することがわかる。化合物1を腹腔(ip)内に与えると、この効果が最大に現れ、また100μgの化合物2の経口投与(po)による効果と1000μgの化合物2の腹腔内投与(ip)による効果とが同等である。腹腔内から与える場合の化合物1(des Pro36-エキセンジン-4(1-39)-NH2. 配列番号101)および化合物2(des Pro36-エキセンジン-4(1-39)-Lys6-NH2. 配列番号93)の有効性が、エキセンジン-4(1-39)-NH2(化合物(i))自体を同じ方法で投与する場合(データは示していない)と極めて近いことがわかる。
【0098】
化合物1. des Pro36-エキセンジン-4(1-39)-NH2(配列番号101)では、化合物を経口投与する場合、400μg/マウスまでの用量では血糖値の低下には効果がないが、Lys断片を付加した同じ化合物では、10μg/マウスの用量でも効果があることがわかる。このことから、des Pro36-エキセンジン-4(1-39)-Lys6-NH2(配列番号93)の最小有効経口用量が、des Pro36-エキセンジン-4(1-39)-NH2(配列番号101)の少なくとも40倍低いことが示される。
【0099】
これらの結果から、腹膜内投与する場合では配列Zの結合による種々のペプチドの有効性への有意な効果はないが、経口投与する場合には化合物の有効性がかなり高まっていることが示される。
26.覚醒ラット十二指腸における胃腸送達後の化合物4および化合物(iii)の生物学的利用性
種々のペプチドに基づくGLP-1類似体が非経口使用のために開発されてきたが、これらのうちで経口投与後に薬理学的効果を有する物質はなかった[Holst, J. J. Enteroglucagon. Annu Rev Physiol. 59: 257-271. 1997]。これについては、覚醒ラット十二指腸からの試験化合物の吸収を調べることとした。化合物(iii)(Gly8)hGLP-1(7-36)-NH2を対照として用いた。
化学物質および試薬
ヘパリンナトリウム中のブランクラット血漿(5000単位/mL)はHarlan Sera Lab Ltd.(Loughborough, UK)から入手した。OASIS(商標)HLB固相抽出カラム、1cc、30mg吸着剤はWaters(Milford, MA, USA)から入手し、ISOLUTE C18(EC)、1cc、SPEカラムはIST(Mid Glamorgan, U.K.)から入手した。LC/MS分析は、オンライン脱ガス装置、バイナリーグラジェントポンプ、オートサンプラー、カラムオーブンからなるHP 1100装置、Hewlett Packard(Wilmington, DE. USA)とMicromass(Altrincham, UK)のQuattro Ultima質量分析計とを組み合わせて行った。LCおよびMSはともにMassLynx3.3ソフトウェアで制御した。MS検出の前にVydac 218MS52(2.1×250mm)カラム(Hesperia, CA, USA)でLC分離を行った。
薬剤および用量レベル
化合物4(バッチ番号ZP 7.97-5-F、4053g/mol)および化合物(iii)(バッチ番号ZP 7.73-2-G、3854g/mol)はFmoc法により自所で合成した。質量分析法により同定を行い、RP-HPLCにより試験化合物の両方のバッチの純度をそれぞれ97および99.7%と決定した。バッチのペプチド含量はZP 7.97-5-FおよびZP 7.73-2-Gそれぞれ72%および80%であった。ペプチドをパイロジェンを含んでいない等張生理食塩水に溶解し、1,000または10,000nmol/kgの用量を100μlの容量で十二指腸内カテーテルにより投与した。
動物
体重250〜350gの14匹のSprague-Dawleyラットを試験に用いた。動脈血サンプリングのため、皮下注射(s.c.)でHypnorm(登録商標)-Dormicum(登録商標)を用いてラットに麻酔をかけ、大腿動脈にカテーテルを挿入した。さらに十二指腸にも胃を切開することによりカテーテルを挿入した。手術後1週間ラットを回復させてから試験を開始した。試験当日、手術したラットは覚醒していた。十二指腸内のカテーテルが十二指腸にあるどうかを確認するため、試験直後にラットの解剖を行った。
サンプル処理
血液サンプルをt=-5、5、10、15、20、40および60分に採取した。血液を氷冷チューブを含むEDTAに採取し、直ちに4℃で5分間遠心分離(4,000×g)した。血漿(250μl)を250μlの抽出溶液(MeCN:0.18M 炭酸アンモニウム pH9.5、10:90v/v)の入った氷冷0.75ml PLCバイアルへ移した。血漿サンプルはSPEおよびLC/MS分析まで-20℃で保存した。
固相抽出
血漿サンプル(400μl)を含有する薬剤を950μl MeCN、次に950μl水で予め調整した固相抽出カラムに載せた。カラムを950μl水中2%TFA、次に同容量のMeCN:水(20:78 v/v)中2%TFAで洗浄した。分析物を500μl MeCN:水(60:38 v/v)中2%TFAで溶出し、LC/MSにより分析した。
LC/MS
サンプルをオートサンプラートレイ中で18℃に保ち、その後LCカラム(Vydac 218MS52(2.1×250mm))に20〜50μlを注入した。分離は30℃で行い、流速250μl/分および表1のグラジェントを用いた。試験化合物および対照薬剤の両方を、それぞれm/z=676.7およびm/z=1095.2および821.8(イオン種)を用い、単一イオン記録方式(SIR)により検出した。すべての装置の条件はMassLynxソフトウェアバージョン3.3で制御した。
【0100】
血漿サンプルを材料および方法に記載したように分析した。化合物4の生物学的利用性については、1,000(n=4)および10,000(n=5)nmol/kgの用量で調べたが、化合物(iii)の生物学的利用性については、10,000(n=5)nmol/kgの用量でのみ試験した。
【0101】
化合物(iii)の濃度は、調査したすべての時点で検出限界(約0.5nM)以下であったので、正確な生物学的利用性が評価できなかった。これに対し、化合物4は1,000nmol/kgの十二指腸内投与後では4匹のラットのうち2匹、および10,000nmol/kgの投与後では5匹のラットのうち4匹の血漿サンプルから検出された。
27.ウサギおよびブタへのi.v.投与後の化合物1、化合物2、化合物4および化合物(iii)のin vivo薬物動態
本発明者らはラット血漿でGLP-1アゴニスト化合物4のin vitro安定性が対照薬剤である化合物(iii)に比べて向上していたことを示した。この効果がin vivoにおいても維持されるかどうかを確かめるため、2種類の化合物の薬物動態パラメーターをウサギで調べることとした。同じ試験条件を用いて、化合物1および2についてのこれらのパラメーターをウサギで測定した。同じ試験条件を用いてブタでも測定した。
化学薬品および試薬
ヘパリンナトリウム中のブランクウサギ血漿(5000単位/mL)はHarlan Sera Lab Ltd.(Loughborough, UK)から入手した。OASIS(商標)HLB固相抽出カラム、1cc、30mg吸着剤はWaters(Milford, MA, USA)から入手し、ISOLUTE C18(EC)、1cc、SPEカラムはIST(Mid Glamorgan, U.K.)から入手した。LC/MS分析は、オンライン脱ガス装置、バイナリーグラジェントポンプ、オートサンプラー、カラムオーブンからなるHP 1100装置、Hewlett Packard(Wilmington, DE. USA)とMicromass(Altrincham, UK)のQuattro Ultima質量分析計とを組み合わせて行った。LCおよびMSはともにMassLynx3.3ソフトウェアで制御した。MS検出の前にVydac 218MS52(2.1×250mm)カラム(Hesperia, CA, USA)でLC分離を行った。
薬剤および用量レベル
化合物4(バッチ番号ZP 7.97-5-F、4053g/mol)および化合物(iii)(バッチ番号ZP 7.73-2-G、3854g/mol)はFmoc法により所内で合成した。質量分析法により同定を行い、RP-HPLCにより試験化合物の両方のバッチの純度をそれぞれ97および99.7%と決定した。バッチのペプチド含量はZP 7.97-5-FおよびZP 7.73-2-Gそれぞれ72%および80%であった。ペプチドをパイロジェンを含んでいない等張生理食塩水に溶解し、両方のペプチドを1,000nmol/kgの用量でウサギおよびラットにi.v.投与した。
ウサギ
体重2.5〜3.0kgの15羽のニュージーランドシロウサギ(New Zealand White rabbit)を試験に用いた。試験当日、薬剤のi.v.投与および動脈血サンプリングのため、i.m.でHypnorm(登録商標)を、次にi.v.でDormicum(登録商標)を用いてウサギに麻酔をかけ、大腿静脈および動脈にカテーテルを挿入した。試験中、ウサギは覚醒していない状態にあった。
サンプル処理
血液サンプルをt=1、3、5、10、15、20、30、40、60、90、120、150、180および240分の時点で採取した。血液をEDTAを含む氷冷チューブに採取し、直ちに4℃で5分間遠心分離(20,000×g)した。血漿(250μl)を250μlの抽出溶液(MeCN:0.18M 炭酸アンモニウム pH9.5、10:90 v/v)の入った氷冷0.75ml PLCバイアルへ移した。血漿サンプルはSPEおよびLC/MS分析まで-20℃で保存した。
固相抽出
血漿サンプル(400μl)を含有する薬剤を950μl MeCN、次に950μl水で予め調整したOASIS(商標)HLB(化合物4)またはISOLUTE(商標)(化合物(iii))固相抽出カラムに入れた。カラムを950μl水中2%TFA、次に同容量のMeCN:水(20:78 v/v)中2%TFAで洗浄した。分析物を500μl MeCN:水(60:38 v/v)中2%TFAで溶出し、LC/MSにより分析した。
LC/MS
サンプルをオートサンプラートレイで18℃に保ち、その後LCカラム(Vydac 218MS52(2.1×250mm))に20ないし50μlを注入した。分離は30℃で行い、流速250μl/分および以下の表に示す通りのグラジェントを用いた。試験化合物および対照薬剤の両方を、それぞれm/z=676.7およびm/z=1095.2および821.8(イオン種)を用い、単一イオン記録方式(SIR)により検出した。すべての装置の条件はMassLynxソフトウェアバージョン3.3で制御した。
【0102】
血漿サンプルを材料および方法に記載したように分析し、血漿濃度(Cpl)を時間に対して片対数グラフにプロットした。ウサギは血漿濃度について3時間追跡した。ラットでは血液量が限られているため、血液サンプリングは1時間までに制限した。各ウサギのCpl対時間曲線は、WinNonlin 3.1(Pharsight Corp.(Mountain View. CA))の1/y2 重み付き最小二乗法を用い、ツー・コンパートメント・オープン・モデル(図は示していない)にフィットさせた。分析データから得られる薬物動態定数を表5に記載し、化合物4および化合物(iii)それぞれの1μmol/kgのi.v.注射後のウサギにおける分解動態を図4に示している。
【0103】
【表5】
【0104】
28.化合物2、14〜16、18および19の化合物(i)と比較したグルコース負荷試験
雄の糖尿病db/dbマウス(M&B. Bomholtgaard, LI, Skensved, Denmark)を用いる。この十分に解明されているモデルマウスはレプチン受容体の変異により遺伝的にグルコース代謝不全を有している。ホモ接合体db/dbマウスは制御できないインスリン非依存型糖尿病(NIDDM)のヒト患者のように煩渇多飲症、多尿症および糖尿、ならびに高血糖症の程度にかかわらず、生涯の最初の3週間での体重の増加を経験する。しかしながら、このモデルでの高血糖症は進行性ランゲルハンス島萎縮症と関連しており、ケトン症の可能性もあって、6〜8月齢で死亡することもある。そのため、それらの病状の進行およびその程度には注意を払わねばならない。それゆえ、GLP-1類似体の薬剤試験には16週齢未満のdb/dbマウスだけを用いることが好ましい。
【0105】
すべてのマウスを少なくとも1週間、新しい環境に順応させ、2日間毎日処置を施した後、最初の経口グルコース負荷試験(OGTT)を行う。さらに、ストレスにより誘導されるグルコースの混乱を軽減するため、試験前に下記の化合物を加えないで少なくとも1回OGTTに付しておくべきである。糖尿病マウス間ではグルコース負荷が大きく分散しているため、最初に使用する前にOGTTにより動物を等級別に分類する。
ペプチド
ペプチドを0.1%ウシアルブミンを含有する0.1Mリン酸緩衝溶液(PBS)(5M NaOHを加えてpHを7.4に調整)に溶解する。すべての溶液については、試験直前の朝新たに調整する。動物を処置するビヒクルは、0.1%アルブミンを含有するPBSのみとする。
グルコース負荷試験および適用
経口グルコース負荷試験の前、動物に17時間(午後4時から翌日の午前9時まで)絶食させる。午前9時から、尾の先から採血し(t=-15分)、血中グルコースを測定する。全血中グルコース(mM)濃度を、製造業者のマニュアルに従って1滴の血液(<5μl、Elite Autoanalyser、Bayer, Denmark)を用いる固定化グルコースオキシダーゼ法により調べる。重症糖尿病の動物(>10mM)は排除する。動物には、最初の血液サンプリング直後にビヒクルまたは1用量の抗糖尿病性化合物の腹腔内(i.p.)注射を施す。すべての群において注入量は200μg/体重50gマウスとする。物質のi.p.投与から15分後、水に溶解した1g/kgグルコース(Sigma, St. Louis)の経口用量を与えて(200μg/体重50g)動物をホームケージに戻す(t=0)。血中グルコースレベルをt=30分、t=60分、t=120分およびt=240分に測定する。観察期間中、動物に絶食をさせる。各血液サンプリング時には、データ記録用紙に各動物について記入した。
算出および統計値
化合物の効果を調べるため、ベースライン(t=0)との絶対および相対差を各時点について算出する。全試験の曲線下の面積(AUC 0〜240分)を台形法を用いて求める。分類化当日に、すべての群でグルコース負荷が同じようになるようにマウスを振り分ける。しかしながら、時間内での糖尿病の進行を補正するため、試験中、ビヒクルで処置した対照群を毎日試験して、薬剤に対する応答をビヒクルで処置した時間制御動物で観察される応答と比較して示す。
【0106】
各物質の用量応答曲線をプロットし(図5参照)、薬剤の効果を、ビヒクルで処置して得られる応答と比較して、ANCOVA分析(共分散分析)を用いて調べる。処置(薬剤またはビヒクル)は独立変数と考えられ、ビヒクル処置した時間制御マウスの応答率%で示されるAUC 0〜240分は従属変数であり、薬剤用量は共変量として定義される。Post-hoc分析はフィッシャーの最小有意差検定を用いて行う。0.05レベルでは有意差があると考えられる。Windows(登録商標) NT版Statistica バージョン5.5、StatSoft, Tulsa, Oklahoma, U.S.A.を用いて統計解析を行った。図5に示される用量応答曲線では、試験したすべての化合物が対照薬剤のものと同等のグルコース低下効果を示していることがはっきりとわかる。
29.db/dbマウスにおける化合物2および化合物(i)のOGGTへの効果
図7は実施例28に記載した同じ試験条件を用いて行ったOGTTでの化合物2および化合物(i)のAUCのプロットである。この図から、化合物2の血糖の低下効果と先行技術の化合物(iii)の効果とが同等であることがわかる。
30.化合物2、100nmol/kgをOGTTの24時間前までにi.p.投与した場合の経口グルコース負荷試験(OGTT)における化合物の長期効果
この試験ではdb/dbマウス、その他に最大用量100nmol/kg i.p.を用い、実施例28に記載した同じ試験条件を用いる。結果を図8に示している。この試験から、化合物2の作用期間はdb/dbマウスでは18時間までであるという結論になる。
【0107】
本明細書に記載されかつ特許請求される本発明は、本明細書に開示される特定の実施形態に限定されるものではなく、これらの実施形態は本発明のいくつかの態様を例示するものである。同等の実施形態はいずれも本発明の範囲内とされる。実際に、当業者ならば、本明細書に示され、記載されたものに加え、本発明の様々な改変も先行技術から理解できよう。かかる改変もまた添付の請求項の範囲に入る。本明細書において様々な参考文献が引用されているが、これらの明細は参照によりすべて本明細書に組み入れるものとする。
【図面の簡単な説明】
【0108】
【図1】マウスの血糖値における化合物1(配列番号101)(desPro36-エキセンジン-4(1-39)-NH2)の作用を示す(実施例25参照)。
【図2】マウスの血糖値における化合物2(配列番号93)(desPro36-エキセンジン-4(1-39)-Lys6-NH2)の作用を示す(実施例25参照)。
【図3】マウスの血糖値における化合物5(配列番号89)(Gly8,Lys37(パルミトイル)-GLP-1(7-36)(ヒト)-(Lys)7-NH2)の作用を示す(実施例25参照)。
【図4】ウサギにおいて1μmol/kgの化合物4および化合物(iii)を静脈内(i.v.)注射した後のin vivoでの分解速度論を示す(実施例27参照)。
【図5】経口グルコース負荷試験(OGTT)における化合物2、14〜16、18および19についてのAUC(曲線より下の面積、area under the curve)値(平均±SEM)のグラフ(実施例28参照)。
【図6】酵母における化合物2の異種発現のために構築した合成cDNAを示す図である。この新規構築物をpYES0010と命名した(実施例20参照)。
【図7】化合物2および化合物(i)についての相対AUC0-240min値(平均±SEM)に基づいたdb/dbマウスでのGTTにおける用量応答のグラフ(実施例29参照)。
【図8】OGTTの24時間前までに投与した場合の経口グルコース負荷試験(OGTT)における化合物2の最大量、すなわち100nmol/kg i.p.(腹腔内)の効果を示す。
【技術分野】
【0001】
発明の属する技術分野
本発明は、GLP-1活性に対するアゴニストである新規ペプチドアゴニストに関する。より具体的には、本発明は、エキセンジン-4ポリペプチド配列の変異体を含む血糖レベルを低下させる新規ペプチド、およびGLP-4またはエキセンジン-4ポリペプチド配列の変異体を含むペプチド結合体であって、薬理学的活性を有しかつ安定で、GLP-1活性のアゴニストとして過剰な血糖レベルの調節および/または胃内容排出の調節などが有効である疾患(糖尿病および摂食障害など)の治療に有用である上記ペプチドおよびペプチド結合体に関する。本発明はまた、上記新規ペプチドの製造方法、本発明のペプチドと生理学的に許容されうる担体とを含む組成物(例えば、医薬組成物など)、治療に使用するための該ペプチド、障害の治療方法、および治療に使用するための医薬組成物を製造するための該ペプチドの使用に関する。
【背景技術】
【0002】
発明の背景
血糖レベルを低下させる多くのホルモンは、腸内の栄養分の存在および吸収に応答して胃腸管粘膜から放出される。これらのホルモンには、ガストリン、セクレチン、グルコース依存性インスリン分泌刺激(insulinotropic)ポリペプチド(GIP)およびグルカゴン様ペプチド-1(GLP-1)が含まれる。公知の最も強力なものは、GLP-1である(Orskov, 1992, Diabetologia 35: 701-711)。グルカゴン様ペプチド-1(GLP-1)は、180アミノ酸からなるペプチドであるプログルカゴンの産物である(Drucker、1998、Diabetes 47: 159-169)。プログルカゴンの全配列には、グルカゴンの29アミノ酸の配列、GLP-1の36または37アミノ酸配列および腸作用性(intestinotrophic)ペプチドであるグルカゴン様ペプチド-2(GLP-2)の34アミノ酸配列が含まれる。GLP-1は多くの機能を有している。これは、正常なヒトにおいてインスリン分泌に対する作用を増強する生理学的ホルモンであり、従って内分泌性ホルモンである。さらに、GLP-1はまた、グルカゴン濃度を低下させ、胃内容排泄を遅延させ、(プロ)インスリンの生合成を刺激し、かつ、インスリン感受性を増進させる(Nauck, 1997, Horm. Metab. Res. 47: 1253-1258)。該ペプチドはまた、グルコース耐性に障害のある被験体においてはβ細胞のグルコースに対する感知能および応答能を増進させる(Byrne, 1998, Eur. J. Clin. Invest. 28: 72-78)。ヒトのGLP-1のインスリン分泌刺激作用は、グルコースの消失速度を増大させる。これは、一部には、インスリンレベルが増大するからであり、また、一部には、インスリン感受性が増大するからである(D'Alessio, 1994, Eur. J. Clin. Invest. 28: 72-78)。このことにより、GLP-1はII型糖尿病の治療に有望な物質として位置づけられてきた。GLP-1の活性断片はGLP-1(7〜36)およびGLP-1(7〜37)であることがわかっている。しかし、天然型GLP-1の薬理学的に主要とされる問題は、半減期が短いことである。ヒトおよびラットにおいては、GLP-1はジペプチジルペプチダーゼ-IV(DPP-IV)により、GLP-1(9-36)アミドに迅速に分解され、これは内因性GLP-1受容体アンタゴニストとして作用する(Deacon, 1998, Diabetologia 41: 271-278)。この問題を回避する種々のストラテジーが提唱されてきたが、その中のいくつかはDPP-IVの阻害剤および他のDPP-IV耐性のGLP-1(7-36)アミドの類似体を用いるものである(Deacon, 1998, Diabetologia 41: 271-278; Deaconら、1998, Diabetes 47: 764-769; Ritzel, 1998, J. Endocrinol. 159: 93-102;米国特許第5,545,618号; Pederson, 1998, Diabetes 47: 1253-1258)。
【0003】
血糖レベルを低下させるもう一つのグループのペプチドであるエキセンジンは、GLP-1[7-36]NH2にある程度の配列類似性を有している(53%)(Goke ら、1993 J. Biol. Chem. 268: 19650-55)。エキセンジンは、毒トカゲ(Helodermatidae)またはアメリカドクトカゲ(Raufman, 1996, Reg. Peptides 61:1-18)の毒に認められる。エキセンジン-3は、メキシコアメリカドクトカゲ Heloderma horridumの毒に存在し、エキセンジン-4は、ヒーラモンスター Heloderma suspectumの毒に存在する。エキセンジン-4は、2および3箇所だけがエキセンジン-3とは異なっている。エキセンジン-4のアミノ末端に連結した47アミノ酸のペプチドであるエキセンジン-4前駆体タンパク質をコードするcDNAが、クローニングされ配列決定された(Pohl ら、1998, J. Biol. Chem. 273:9778-9784およびWO98/35033)。エキセンジン-3およびエキセンジン-4の両方とも、モルモット膵腺房細胞において、エキセンジン受容体と相互作用することにより細胞のcAMP産生の増加を刺激する(Raufman, 1996, Reg. Peptides 61: 1-18)。エキセンジン-3が誘導する細胞のcAMP産生の増加は二相性であるが、エキセンジン-3が誘導する膵腺房細胞のアミラーゼ放出の増加は単相性のである。対照的に、エキセンジン-4が誘導するcAMP産生の増加は単相性であるが、エキセンジン-4はアミラーゼ放出には変化を及ぼさない。
【0004】
エキセンジン-4は、単離されたラットインスリノーマ細胞におけるGLP-1受容体の強力なアゴニストである(Gokeら、1993, J. Biol. Chem. 268: 19650-55)。DPP-IVにより認識されるGLP-1の(His Ala)ドメインがエキセンジン-4に存在しないことから、エキセンジン-4は有望視されている(Gokeら、1993, J. Biol. Chem. 268: 19650-55)。[125I]GLP-1の孤束核への結合は、非標識GLP-1および[Tyr39]エキセンジン-4により濃度依存的に阻害され、この際のKi値はそれぞれ、3.5 nMおよび9.4 nMであり、これらと同様の値が細胞系でも認められている(Gokeら、1995, Eur. J. Neurosci. 7: 2294-2300およびGokeら、1993, J. Biol. Chem. 268: 19650-55)。さらに、糖尿病db/dbマウスでは、エキセンジン-4の全身投与により血糖レベルが40%低下する(WO99/07404)。最近、Griegらにより、糖尿病ob/obマウスで、1日1回のエキセンジン-4の腹腔内注射による血糖低下効果が長時間持続すること示された(Griegら、1999, Diabetologia 42: 45-50)。米国特許第5,424,286号には、インスリン分泌刺激活性を維持するためにはN末端配列の大部分(エキセンジン-4(1-31)およびY31-エキセンジン-4(1-31))が必須であり、N末端が切断されたエキセンジン(エキセンジン-4(9-39))は阻害特性を有することが開示されている。
【0005】
真性糖尿病の治療、胃の運動性の低減、胃内容排出の遅延および高血糖の予防(米国特許第5,424,286号、WO98/05351)、ならびに食物摂取の低減(WO98/30231)のために、エキセンジン-3、エキセンジン-4、およびエキセンジンアゴニストの使用が提案されてきた。天然のエキセンジン配列を改変することによる新規化合物を得る方法が提唱されている。その1つは、例えば、WO98/43708の記載に開示されている、C末端アミノ酸残基に結合した親油性置換基を1つだけ有するエキセンジンの誘導体のように、分子に親油性置換基を結合させるものである。
【0006】
主要な手法は、天然のエキセンジン-4の配列のアミノ酸置換および/またはC末端の切断により特性解析されたエキセンジン類似体を考案することであった。この手法は、WO99/07404、WO99/25727およびWO99/25728に記載の化合物により示されている。WO99/07404は、一般式Iのエキセンジンアゴニストを開示する。該式は、4〜5位はGly Thrであり、11〜13位はSer Lys Gln であり、15〜21位はGlu Glu Glu Ala Val Arg Leu であり、26〜30位はLeu Lys Asn Gly Gly であり、32〜35位はSer Ser Gly Ala であり、残りの位置には野生型エキセンジンのアミノ酸残基か、または特定のアミノ酸置換を有しうる、39アミノ酸残基からなるペプチド配列を示す。該式Iは、新規化合物であるdesPro36-エキセンジン-4(1-39)、エキセンジン-4(1-39)-K6またはdesPro36-エキセンジン-4(1-39)-K6などの本明細書に記載するような、特定のアミノ酸の欠失を有し、および/または結合体であるエキセンジンアゴニストまたは類似体は全く包含していない。
【0007】
WO99/25727は、4位にGlyおよび18位にAlaを有し、残りの位置には野生型エキセンジンのアミノ酸残基か、または特定のアミノ酸置換を有しうる、28〜38アミノ酸残基からなるペプチド配列を示す一般式Iを有するエキセンジンアゴニストを開示している。式Iは、C末端アミノ酸としてSerを有するペプチド配列、ならびに新規化合物であるdesPro36-エキセンジン-4(1-39)、エキセンジン-4(1-39)-K6またはdesPro36-エキセンジン-4(1-39)-K6などの本明細書に記載するような、特定のアミノ酸の欠失を有し、および/または結合体であるエキセンジンアゴニストまたは類似体は全く包含していない。さらにWO99/25727に記載の式IIは、式Iに類似したペプチド配列を示しているが、27位または28位にあるリジンに、C(1〜10)アルカノイルまたはシクロアルキルアルカノイル置換基を有するエキセンジン誘導体を含む。異常な食後血糖レベルの治療の際に、該化合物を頻繁に(例えば1日に1回、2回、または3回)投与する。
【0008】
WO99/25728は、18位に固定のAlaを有し、残りの位置には野生型エキセンジンのアミノ酸残基であるか、または特定のアミノ酸置換を有しうる、28〜39アミノ酸残基からなるペプチド配列を示す一般式Iを有するエキセンジンアゴニストを開示している。該エキセンジンアゴニストはすべて、アミノ酸置換の程度が異なる末端切断型エキセンジン類似体に相当する。34〜38のアミノ酸残基からなるペプチド配列は、C末端にSerを有していない。39アミノ酸残基からなるペプチド配列は、C末端にSerまたはTyrのいずれかを有することができ、ほかの残基は有していない。本明細書中に記載するような、特定のアミノ酸欠失を有し、および/または結合体である本発明のエキセンジンアゴニストまたは類似体は、式Iには含まれない。さらに、式IIは式Iに類似しているが27または28位のリジンに、C(1〜10)アルカノイルまたはシクロアルキルアルカノイル置換基を有するエキセンジン誘導体を含むペプチド配列を示す。
【0009】
WO99/46283(1999年9月16日公開)は、薬理学的活性のあるペプチドXおよび該Xに共有結合している4〜20アミノ酸残基からなる安定化ペプチド配列Zを含むペプチド結合体を開示しており、該結合体は、半減期がXの半減期に比べて増大しているという特徴を有する。Xはエキセンジン-4またはエキセンジン-3であってよい。
【発明の開示】
【発明が解決しようとする課題】
【0010】
本発明の目的
哺乳動物の血糖レベルを低下させる、安定かつ効果的な化合物が必要とされている。従って、本発明の目的は、哺乳動物の血糖レベルを低下させる新規化合物を提供することである。理想的には、経口投与である場合に効果的なものである。さらなる本発明の目的は、GLP-1活性および/またはエキセンジン-4活性の新規ペプチドアゴニストを提供することである。またさらに、半減期が増大し、および/もしくはクリアランスが低減された、GLP-1活性ならびに/またはエキセンジン-4活性についてのペプチドアゴニストを提供することも、さらなる本発明の目的である。
【課題を解決するための手段】
【0011】
発明の概要
本発明は、
(a) エキセンジン(exendin)-4に対して少なくとも90%の相同性を有するエキセンジン、
(b) 34〜39位の1〜5個の欠失からなる群より選択される改変を有し、かつ40位に親油性置換基を有するLysを有する、該エキセンジンの変異体、または
(c) (i) 8位のアラニンの、D-アラニン、グリシンもしくはα-アミノイソ酪酸への置換、および
(ii) 親油性置換基
からなる群より選択される少なくとも1個の改変を有するGLP-1(7-36)またはGLP-1(7-37)、
からなる群より選択されるペプチドX(ただし、Xはエキセンジン-4またはエキセンジン-3ではない)と、
4〜20個のアミノ酸単位からなり、各アミノ酸単位が、Ala、Leu、Ser、Thr、Tyr、Asn、Gln、Asp、Glu、Lys、Arg、His、Met、Ornおよび一般式I
-NH-C(R1)(R2)-C(=O)- (I)
(式中、R1およびR2は、水素、C1-6-アルキル、フェニルおよびフェニル-メチルからなる群より選択され、C1-6-アルキルは、ハロゲン、ヒドロキシ、アミノ、シアノ、ニトロ、スルホノおよびカルボキシからなる群より選択される1〜3個の置換基で置換されていてもよく、フェニルおよびフェニル-メチルは、C1-6-アルキル、C2-6-アルケニル、ハロゲン、ヒドロキシ、アミノ、シアノ、ニトロ、スルホノおよびカルボキシからなる群より選択される1〜3個の置換基で置換されていてもよく、あるいはR1とR2は、それらが結合している炭素原子と一緒になって、シクロペンチル、シクロヘキシルまたはシクロヘプチル環を形成している。)
で表されるアミノ酸単位(例えば2,4-ジアミノブタン酸および2,3-ジアミノプロパン酸)からなる群より選択される、前記変異体に共有結合したペプチド配列Zと、
を含むペプチド結合体に関する。
【0012】
ペプチドXは、糖尿病の哺乳動物のグルコース耐性の改善に有効であることをさらに特徴とする。
【0013】
さらに、本発明は、親エキセンジンの新規変異体であって、該親エキセンジンは、エキセンジン-4に対して少なくとも90%の相同性を有するアミノ酸配列を有し、該変異体は、哺乳動物において血糖値を降下させ、かつGLP-1受容体に結合し、かつ
(a)34〜38位の1〜5個の欠失、および
(b)εアミノ基に親油性置換基が結合している40位のLys、
からなる群より選択される少なくとも1個の改変を有する、前記変異体に関する。
【発明を実施するための最良の形態】
【0014】
詳細な説明
本発明の化合物は、親エキセンジンの今まで知られていない欠失変異体を含む。エキセンジン-4(1-39)の既知の置換および/または末端切断変異体とは対照的に、本発明の新規化合物は、安定性に優れ、結合特性が減じられないかまたは高められた恒常的なαヘリックス構造を示すものと考えられる。さらに、新規変異体、すなわち改変型GLP-1(7-36)-NH2、および改変型GLP-1(7-37)を、特定の短いペプチド配列(Z)に結合させると、薬理学的特性を損なうことなくこれらの化合物が安定になる。これらの結合により、ペプチド分子にin vivoでの安定性および親水性が付与される。Zは、アミノ酸残基から構成され、単独ではαヘリックス構造の面でなんら構造的特徴をもたない。しかし、円偏光二色性と核磁気共鳴(NMR)分光法の両方を用いた研究から、Zの付加によって、ペプチドのαヘリックス構造の量の増大により証明されたように、いくつかのペプチドの構造的特徴が変化する。例えば、円偏光二色性により、Z-改変された(Gly8)-GLP-1は、(Gly8)-GLP-1よりもはるかに多量のαヘリックス構造を有することが示された。薬理学的結果と一緒になって、その構造分析により、Zがペプチドのコンホメーションをその能力を失うことなく改変し、より高い酵素安定性をもたらしていることが示唆される。ペプチドの物理的および化学的特性も、Z改変によってかなり変更されうるものであり、薬理学的な処方戦略に影響を及ぼすものである。
エキセンジン変異体
本発明のエキセンジン変異体は、エキセンジン-4に対して少なくとも約90%、最も好ましくは少なくとも約95%の相同性を有する、親エキセンジンペプチドの変異体であり、エキセンジン活性をもち、哺乳動物において血糖値を降下させ、GLP-1受容体に結合する。好ましい実施形態では、親エキセンジンペプチドはエキセンジン-4(1-39)のアミノ酸配列と、5個のアミノ酸残基、好ましくは4個のアミノ酸残基、より好ましくは3個のアミノ酸残基、さらにより好ましくは2個のアミノ酸残基、その上より好ましくは1個のアミノ酸残基が異なるアミノ酸配列を有する。
【0015】
1つの実施形態では、エキセンジン変異体は、34〜38位に1〜5個の欠失を含む。好ましくは、変異体は34〜38位に1〜4個の欠失を含み、より好ましくは36〜38位に1〜3個の欠失を含む。好ましくは、親エキセンジンはエキセンジン-4であり、ペプチド結合体にペプチドXとして含まれている好ましい変異体は、36位、37位および38位のPro残基の1個、2個または3個がエキセンジン-4のアミノ酸配列から、好ましくはエキセンジン-4(1-39)のアミノ酸配列から欠失しているアミノ酸配列を有する。
【0016】
本発明において、XペプチドへのZ配列の結合がこれらの化合物の安定性を高めるものと考えられる。プロリンは、Xペプチドの構造を安定化させるZの作用を妨害する可能性がある剛性(rigid)アミノ酸である。したがって、本発明の親エキセンジンの変異体を含むペプチド結合体においては、例えば糖尿病のdb/dbマウスにおける経口グルコース負荷試験(OGTT)で測定された該結合体の有効性が悪影響を受けないかぎり、エキセンジン骨格の36位、37位および38位にあるプロリンアミノ酸のうちの1個、2個または全部が欠失していることが好ましい。
【0017】
別の実施形態では、変異体は、40位に追加の残基、すなわち、アミド結合を介してリジンのεアミノ基に結合している親油性置換基を含むリジン残基を含む。親油性置換基は、直鎖状もしくは分岐状脂肪酸または直鎖状もしくは分枝状アルカンα,ω-ジカルボン酸のアシル基であり得る。アシル基は、式CH3(CH2)nCO-(式中、nは4〜38の整数であり、好ましくは4〜24の整数である)で表され得る。特定の実施形態では、アシル基は、CH3(CH2)6CO-、CH3(CH2)8CO-、CH3(CH2)10CO-、CH3(CH2)12CO-、CH3(CH2)14CO-、CH3(CH2)16CO-、CH3(CH2)18CO-、CH3(CH2)20CO-およびCH3(CH2)22CO-からなる群より選択される。アシル基は、式HOOC(CH2)mCO- (式中、mは4〜38の整数であり、好ましくは4〜24の整数である)で表され得る。特定の実施形態では、アシル基は、HOOC(CH2)14CO-、HOOC(CH2)16CO-、HOOC(CH2)18CO-、HOOC(CH2)20CO-およびHOOC(CH2)22CO-からなる群より選択される。より特定的な実施形態では、親油性置換基は、テトラデカノイル、ω-カルボキシノナデカノイル、7-デオキシコロイル、コロイル、パルミトイルおよびリトコリルからなる群より選択される。最も特定的な実施形態では、親油性置換基はパルミトイルである。
【0018】
あるいは、親油性置換基はNH基を有していてもよい。特定の実施形態としては、限定するものではないが、式CH3(CH2)a((CH2)bCOOH)CHNHCO(CH2)2CO- (式中、aおよびbは整数であり、a+bは8〜33の整数、好ましくは12〜28の整数である);CH3(CH2)cCONHCH(COOH)(CH2)2CO- (式中、cは10〜24の整数である);CH3(CH2)dCONHCH(CH2)2(COOH)CO- (式中、dは8〜24の整数である);COOH(CH2)eCO- (式中、eは8〜24の整数である);-NHCH(COOH)(CH2)4NHCO(CH2)fCH3(式中、fは8〜18の整数である);-NHCH(COOH)(CH2)4NHCOCH(CH2)2COOH)NHCO(CH2)gCH3(式中、gは10〜16の整数である);および-NHCH(COOH)(CH2)4NHCO(CH2)2CH(COOH)NHCO(CH2)hCH3(式中、hは0または1〜22の整数、好ましくは10〜16の整数である)が挙げられる。
【0019】
40位に親油性置換基を有するリジン残基を含むエキセンジン変異体は、任意に、34〜39位、好ましくは34〜38位に、1〜5個の欠失、好ましくは1〜3個の欠失をさらに含み、例えば、[desSer39,Lys40(パルミトイル)]エキセンジン-4(1-39)、[desPro36,Lys40(パルミトイル)]エキセンジン-4(1-39)および[desPro36,Lys40(パルミトイル)]エキセンジン-4(1-40)が挙げられる。
【0020】
最も特定的な実施形態では、変異体は、
化合物1:desPro36-エキセンジン-4(1-39)-NH2(配列番号101)、
desPro36-エキセンジン-4(1-40)-NH2、
化合物14:desPro36,Pro37,Pro38-エキセンジン-4(1-39)-NH2 、
desPro36,Pro37,Pro38-エキセンジン-4(1-40)-NH2 、
desPro36,Pro37-エキセンジン-4(1-39)-NH2 、
desAla35-エキセンジン-4(1-39)-NH2(配列番号105)、
desGly34-エキセンジン-4(1-39)-NH2(配列番号106)、
desSer39-(Lys40(パルミトイル))エキセンジン-4(1-39)-NH2(配列番号107)、
desGly34-(Lys40(パルミトイル))エキセンジン-4(1-39)-NH2(配列番号108)、
desAla35-(Lys40(パルミトイル))エキセンジン-4(1-39)-NH2(配列番号109)、
desPro36-(Lys40(パルミトイル))エキセンジン-4(1-39)-NH2(配列番号110)、ならびにその遊離酸およびその薬学的に許容される塩からなる群より選択され得る。
改変型GLP-1
本発明のペプチド結合体中にペプチドXとして含まれる好ましい改変型GLP-1は、8位のアラニンがグリシンに置換されているGLP-1(7-36)-NH2またはGLP-1(7-37)のアミノ酸配列を有する。あるいは、好ましい改変型GLP-1は、8位のアラニンがグリシンに置換されており、26位、34位または37位の1個のリジン残基に親油性置換基、好ましくはパルミトイル基を有するGLP-1(7-36)またはGLP-1(7-37)のアミノ酸配列を有する。親油性置換基は、好ましくは、該リジンのεアミノ基に結合しており、エキセンジン変異体について上記した特定の実施形態を含む。本発明の結合体中でXとして用いられる改変型GLP-1(7-36)またはGLP-1(7-37)は、親油性置換基を含むWO99/43707およびWO98/08871に引用されたもの、またはより好ましくは8位にグリシン置換基を有するそのGLP-1類似体であってもよい。好ましくはペプチドXは、
Gly8-GLP-1(7-36)、
Gly8-GLP-1(7-37)、および
Gly8-GLP-1(7-36)-Lys37(パルミトイル)
である。
【0021】
親油性置換基を有する本発明の化合物は、WO98/08871においてGLP-1誘導体について示されたように、親ペプチドよりも遅延性の作用プロファイルを示すだろう。
ペプチド結合体
ペプチド配列Zは、ペプチド配列XのC末端またはN末端に結合していてもよく、あるいは2つのペプチド配列がXのC末端とN末端の両方にそれぞれ結合していてもよい。天然の(native)ペプチドXが遊離のC末端カルボン酸を有する場合、ペプチド配列Zは、ペプチドXのC末端またはペプチドXのN末端のいずれかに結合することができ、あるいはXのC末端およびN末端が両方ともそれぞれ個々のペプチド配列Zに結合していてもよい。あるいは、Zは、ペプチド配列X内のどこかにある、リジン、ヒスチジンもしくはアルギニンの側鎖上の窒素原子またはグルタミン酸もしくはアスパラギン酸の側鎖上のカルボニル基に結合していてもよい。1つの実施形態では、Zは、Xの配列内部に結合していてもよく、XのNおよび/またはC末端に結合していてもよい。配列がペプチド配列XのC末端に、N末端に、もしくはその両方に、またはその内部に結合すべきかどうかは、その特定のペプチドXによって異なり、当業者により容易に決定され得る。好ましくは、Xは、ペプチド結合を介して、好ましくはXのC末端によりZに結合する。
【0022】
本発明の1つの態様は、(a) エキセンジン-4に対して少なくとも90%の相同性を有するエキセンジン、
(b) 34〜39位の1〜5個の欠失からなる群より選択される改変を有し、かつ40位に親油性置換基を有するLysを有する、該エキセンジンの変異体、または
(c) (i) 8位のアラニンの、D-アラニン、グリシンもしくはα-アミノイソ酪酸(Aib)への置換、および
(ii) 親油性置換基
からなる群より選択される少なくとも1個の改変を有するGLP-1(7-36)またはGLP-1(7-37)、
である哺乳動物の血糖値を降下させるペプチドXと、
4〜20個のアミノ酸単位からなり、各アミノ酸単位が、Ala、Leu、Ser、Thr、Tyr、Asn、Gln、Asp、Glu、Lys、Arg、His、Met、Ornおよび一般式I
-NH-C(R1)(R2)-C(=O)- (I)
(式中、R1およびR2は、水素、C1-6-アルキル、フェニルおよびフェニル-メチルからなる群より選択され、C1-6-アルキルは、ハロゲン、ヒドロキシ、アミノ、シアノ、ニトロ、スルホノおよびカルボキシからなる群より選択される1〜3個の置換基で置換されていてもよく、フェニルおよびフェニル-メチルは、C1-6-アルキル、C2-6-アルケニル、ハロゲン、ヒドロキシ、アミノ、シアノ、ニトロ、スルホノおよびカルボキシからなる群より選択される1〜3個の置換基で置換されていてもよく、あるいはR1とR2は、それらが結合している炭素原子と一緒になって、シクロペンチル、シクロヘキシルまたはシクロヘプチル環を形成している。)
で表されるアミノ酸単位(例えば2,4-ジアミノブタン酸および2,3-ジアミノプロパン酸)からなる群より選択される、Xに共有結合したペプチド配列Zと、
を含むペプチド結合体に関する。好ましくは、Xは、GLP-1受容体に結合し、エキセンジン-4またはエキセンジン-3を含まない。
【0023】
Zは、典型的には、4〜20個のアミノ酸残基、例えば4〜15個、より好ましくは4〜10個、特に4〜7個のアミノ酸残基、例えば4個、5個、6個、7個、8個または10個のアミノ酸残基(6個のアミノ酸残基が好ましい)からなるペプチド配列である。好ましくは、Zは、少なくとも1個のLys残基を含む。本発明の好ましい実施形態では、ペプチド配列Z中の各アミノ酸残基は、互いに独立に、Ala、Leu、Ser、Thr、Tyr、Asn、Gln、Asp、Glu、Lys、Arg、His、Met、Ornならびにジアミノブタン酸およびジアミノプロパン酸からなる群より選択される。好ましくは、アミノ酸残基は、Glu、LysおよびMetから選択され、特に、Lysであり、あるいはアミノ酸残基は、Asn、GluおよびLysからなる群より選択される。上記のアミノ酸は、D-またはL-配置のいずれかであってよいが、好ましくは上記のアミノ酸はL-配置である。本発明の好ましい実施形態では、Zは少なくとも1個のリジン残基を含むか、またはZがペプチド結合を介してペプチドXのN末端に結合する場合には、ZはAsn-(Glu)n(式中、nは3〜7の整数である)からなる群より選択されるアミノ酸配列を有する。
【0024】
したがって、ペプチド配列Zの実例は、
【0025】
(ここで、各Xaaは、互いに独立に、Ala、Leu、Ser、Thr、Tyr、Asn、Gln、Asp、Glu、Arg、His、Met、Orn、および上記で定義した式Iで表されるアミノ酸、例えばDbuまたはDprからなる群より選択される)である。
【0026】
上述したように、Zのアミノ酸残基はもちろん全て異なっていても全て同一であってもよい。しかし、本発明の興味深い実施形態では、Z中のアミノ酸残基は、2個または3個の異なるアミノ酸から選択されるか、または同一のアミノ酸である。Z中のアミノ酸残基が同一である適当なペプチド配列の例は、例えば(Lys)n (式中、nは4〜15、好ましくは4〜10、例えば4〜8など、例えば4〜7の範囲の整数である)であり、例えば、Lys4(配列番号1)、Lys5(配列番号2)、Lys6(配列番号8)、Lys7(配列番号30)である。好ましくは、XのC末端にペプチド結合を介して結合した(Lys)6である。
【0027】
Z中のアミノ酸残基が約2個の異なるアミノ酸から選択される適当なペプチド配列の例は、(Lys-Xaa)mまたは(Xaa-Lys)m(式中、mは約2〜7、好ましくは2〜5、例えば2〜4などの整数、例えば3であり、Xaaは、互いに独立に、Ser、Thr、Tyr、Asn、Gln、Asp、Glu、Arg、His、Orn、2,4-ジアミノブタン酸、2,3-ジアミノプロパン酸およびMetからなる群より選択される)である。より好ましくは、そのようなペプチド配列は、例えば、(Lys-Xaa)3または(Xaa-Lys)3(式中、Xaaは前記のとおりである)であり、例えば、(Lys-Glu)3(配列番号83)または(Glu-Lys)3(配列番号84)である。Z中のアミノ酸残基が約2個のアミノ酸残基から選択される適当なペプチド配列の他の例は、例えば、Lysp-XaaqまたはXaap-Lysq(式中、pおよびqは1〜14の整数であり、ただしp+qは4〜15の範囲、好ましくは4〜10の範囲、例えば4〜8の範囲など、例えば4〜6の範囲、例えば4、5または6であり、Xaaは、互いに独立に、Ser、Thr、Tyr、Asn、Gln、Asp、Glu、Arg、HisおよびMetからなる群より選択される)である。より好ましくは、そのようなペプチド配列は、例えば、Lys3-Xaa3またはXaa3-Lys3(式中、Xaaは前記のとおりである)であり、例えば、Lys3-Glu3(配列番号85)またはGlu3-Lys3(配列番号86)である。より好ましいZ配列は、AsnおよびGlnから選択されるアミノ酸残基と、GluおよびAspから選択される4〜7個のアミノ酸残基とを有する配列からなり、例えば、Asn-(Glu)5、Asn-(Glu)6、Gln-(Glu)5、Asn-(Asp)5、およびGln-(Asp)5、であり、本発明のペプチド結合体のN末端部分である。
【0028】
Z中のアミノ酸残基が3個の異なるアミノ酸から選択される適当なペプチド配列の例は、例えば、
【0029】
(式中、xおよびyは約1〜5の範囲の整数であるが、ただしx+yは最大6であり、Xaa1およびXaa2は、互いに独立に、Ala、Leu、Ser、Thr、Tyr、Asn、Gln、Asp、Glu、Arg、His、Met、Orn、2,3-ジアミノプロパン酸、2,4-ジアミノブタン酸、および上記で定義した式Iで表されるアミノ酸からなる群より選択される)などである。
【0030】
本発明の好ましい実施形態では、ペプチド結合体の経口最小有効量とペプチドXの最小有効量の比は少なくとも1:5である。
【0031】
本発明の最も好ましい実施形態は、
desPro36-エキセンジン-4(1-39)-NH2(配列番号101)、
desPro36-エキセンジン-4(1-40)-NH2、
desPro36-desPro37-エキセンジン-4(1-39)-NH2 、
desPro36-desPro37-desPro38-エキセンジン-4(1-39)-NH2 、
desPro36-desPro37-desPro38-エキセンジン-4(1-40)-NH2 、
desAla35-エキセンジン-4(1-39)-NH2(配列番号105)、
desGly34-エキセンジン-4(1-39)-NH2(配列番号106)、
desGly34-(Lys40(パルミトイル))エキセンジン-4(1-39)-NH2(配列番号108)、
desAla35-(Lys40(パルミトイル))エキセンジン-4(1-39)-NH2(配列番号109)、
desPro36-(Lys40(パルミトイル))エキセンジン-4(1-39)-NH2(配列番号110)、
化合物(iii) Gly8-GLP-1(7-36)-NH2 、Gly8-GLP-1(7-37)、および
Gly8-GLP-1(7-36)-Lys37(パルミトイル)-NH2、
からなる群より選択され、ペプチド結合を介して(Lys)n(式中、nは4〜8の整数であり、好ましくはnは6である)からなる群より選択されるペプチド配列ZにC末端により結合している、GLP-1および/またはエキセンジン-4活性のアゴニストであるペプチドXを含む新規ペプチド結合体に関する。
【0032】
本発明のペプチド結合体は、好ましくはアミド(NH2)もしくは遊離酸(OH)形態、またはその塩形態であってもよいことは理解されるべきである。本発明のペプチド結合体の例は、
Gly8-GLP-1(7-36)-Lys6-NH2(配列番号88)、
(Gly8,Lys37(パルミトイル)-GLP-1(7-36)(ヒト)-Lys7-NH2(配列番号89)、
desSer39-エキセンジン-4(1-39)-Lys6-NH2(配列番号91)、
エキセンジン-4(1-39)-Lys6-NH2(配列番号92)、
desPro36-エキセンジン-4(1-39)-Lys6-NH2(配列番号93)、
desAla35-エキセンジン-4(1-39)-Lys6-NH2(配列番号94)、
desGly34-エキセンジン-4(1-39)-Lys6-NH2(配列番号95)、
desSer39-(Lys40(パルミトイル))エキセンジン-4(1-39)-Lys7-NH2(配列番号96)、
desGly34-(Lys40(パルミトイル))エキセンジン-4(1-39)-Lys7-NH2(配列番号97)、
desAla35-(Lys40(パルミトイル))エキセンジン-4(1-39)-Lys7-NH2(配列番号98)、
desPro36-(Lys40(パルミトイル))エキセンジン-4(1-39)-Lys7-NH2(配列番号99)、
Lys40(パルミトイル)エキセンジン-4(1-39)-Lys7-NH2(配列番号100)、
desPro36,Pro37-エキセンジン-4(1-39)-Lys6-NH2、
Lys6-desPro36,Pro37,Pro38-エキセンジン-4(1-39)-NH2、
Asn(Glu)5-desPro36,Pro37,Pro38-エキセンジン-4(1-39)-NH2、
Lys6-desPro36,Pro37,Pro38-エキセンジン-4(1-39)-Lys6-NH2、
Asn(Glu)5-desPro36,Pro37,Pro38-エキセンジン-4(1-39)-Lys6-NH2、
desPro36,Pro37,Pro38-エキセンジン-4(1-39)-Lys6-NH2、
Ser8-GLP-1(7-36)-Lys6-NH2、
Aib8-GLP-1(7-36)-Lys6-NH2、
Lys6-Gly8-GLP-1(7-36)-Lys6-NH2、
Lys6-Gly8-GLP-1(7-36)-NH2、
(Gly8,Lys26(パルミトイル)-GLP-1(7-36)(ヒト)-Lys6-NH2 、
(Gly8,Lys34(パルミトイル)-GLP-1(7-36)(ヒト)-Lys6-NH2 、
Gly8-GLP-1(7-36)-Lys8-NH2 、
Gly8-GLP-1(7-36)-Lys10-NH2 、
Gly8-GLP-1(7-37)-Lys6-NH2 、ならびにその遊離酸およびその薬学的に許容される塩、である。
【0033】
好ましいペプチド結合体は、
desPro36-エキセンジン-4(1-39)-Lys6-NH2(配列番号93)、
Gly8-GLP-1(7-36)-Lys6-NH2(配列番号88)、
desPro36,Pro37,Pro38-エキセンジン-4(1-39)-Lys6-NH2、および
その上記で定義した塩である。
【0034】
最も特定的な実施形態では、結合体は、Gly8-GLP-1-(7-36)(ヒト)-NH2、Gly8-GLP-1-(7-36)(ヒト)-Lys6-NH2、Gly8Lys37(パルミトイル)-GLP-1-(7-36)(ヒト)-Lys7-NH2、Gly8Lys34(パルミトイル)-GLP-1-(7-36)(ヒト)-Lys6-NH2、desSer39-エキセンジン-4(1-39)-Lys6-NH2、エキセンジン-4(1-39)-Lys6-NH2、desPro36-エキセンジン-4(1-39)-Lys6-NH2、desAla35-エキセンジン-4(1-39)-Lys6-NH2、desGly34-エキセンジン-4(1-39)-Lys6-NH2、desSer39-(Lys40(パルミトイル))エキセンジン-4(1-39)-Lys7-NH2、desGly34-(Lys40(パルミトイル))エキセンジン-4(1-39)-Lys7-NH2、desAla35-(Lys40(パルミトイル))エキセンジン-4(1-39)-Lys7-NH2、desPro36-(Lys40(パルミトイル)エキセンジン-4(1-39)-Lys7-NH2およびLys40(パルミトイル)エキセンジン-4(1-39)-Lys7-NH2からなる群より選択される。
【0035】
本発明のペプチド結合体の提供により、GLP-1およびエキセンジンならびにこれらの活性類似体などの血糖低下ペプチドの経口投与が可能になる。本明細書中に示す好ましい末端ペプチド断片であるZは、Xの所望の活性に顕著な影響を与えることなく、ペプチドXをα-らせん構造に誘導するよう選択される。結合していないペプチドと比較すると結合したペプチドの半減期は2〜3倍増加している(下記表5参照)ということによって実証されているように、該らせん構造は、例えば分解などに対して、このペプチド鎖を安定化する。ペプチド配列Zは、最小有効量が少なくも5倍低減されるように特定の構造を分子に導入するの関与するペプチド結合体の一部である。最小有効量は、好ましくは少なくとも10倍、より好ましくは少なくとも25倍、さらに好ましくは40倍、最も好ましくは50倍低減される。従って、本発明はまた、上記に定義したペプチド配列Zの上記に定義したペプチド結合体の調製への使用に関する。
【0036】
従って、本発明はまた、本明細書中に定義するペプチドXを含む新規ペプチド結合体に関する。この場合のXは、哺乳動物において血糖レベルを低下させ、該ペプチド結合体の経口最小有効量と、ペプチドXの経口最小有効量の比は少なくとも1:5である。
【0037】
詳細には、本発明により、インスリン分泌刺激に有効な量の本発明のペプチド結合体を投与することを含む哺乳動物のインスリン放出を刺激する方法、哺乳動物の血糖レベルを低下させるのに有効な量の本発明のペプチド結合体を投与することを含む哺乳動物の血糖レベルを低下させる方法、胃の運動性を低減させるのに有効な量の本発明のペプチド結合体により哺乳動物の胃の運動性を低減させる方法、胃内容排出を遅延させるのに有効な量の本発明のペプチド結合体により哺乳動物の胃内容排出を遅延させる方法、食物摂取を抑制するのに有効な量の本発明のペプチド結合体により哺乳動物の食物摂取を抑制する方法、および哺乳動物において血漿脂肪レベルを低減するのに有効な量の本発明のペプチド結合体を哺乳動物に投与することを含む哺乳動物の血漿脂肪レベルを低減させる方法が導かれる。特に、本発明のペプチド結合体を、I型またはII型糖尿病、肥満、摂食障害、高血糖症、代謝異常、胃の疾患およびインスリン抵抗性症候群の治療に用いることができる。
【0038】
本発明はまた、組換えDNA技術により上記ペプチド結合体を調製する方法であって、(a)上記ペプチド結合体をコードする核酸配列を宿主細胞に導入するステップ、(b)該宿主細胞を培養するステップ、および(c)該培養物から上記ペプチド結合体を単離するステップを含む方法、または、(a)上記ペプチド結合体をコードする核酸配列を含む組換え宿主細胞を、該ペプチド結合体の産生が可能な条件下で培養するステップ、および(b)該培養物から上記ペプチド結合体を単離するステップを含む方法に関する。
【0039】
該方法はまた、ペプチドXを、組換えDNA法を用い、ペプチドを単離することにより得る、ペプチド結合体を調製する方法にも関する。その後、固相支持体に結合しているか、または固相合成法により調製しておいたZにXを結合させる。さらに、本発明は、ペプチド合成法による本発明のペプチド結合体の調製に関する。さらに、本発明は、ペプチド合成法による、本発明のペプチド結合体の調製に関する。
【0040】
受容体への結合(インスリン分泌刺激活性)に必須であると考えられている天然のエキセンジン-4 N末端配列に対して実質的に相同性を有する33〜39のアミノ酸残基、好ましくは36〜38アミノ酸残基のN末端配列、およびC末端配列Zを含む本発明の結合体は、天然型エキセンジンおよびエキセンジンのC末端側末端切断型に比べて安定性が向上しているという利点をさらに有している。同様に、GLP-1ペプチド結合体である化合物4は、結合していない化合物(iii)に比べて安定性の向上を示す。
組成物
本発明はまた、本発明のエキセンジン変異体またはペプチド結合体を生理学的に許容されうる担体と組み合わせて含有する組成物に関する。かかる組成物は、経口投与、非経口投与(皮下投与(s.c.)、静脈内投与(i.v.)、筋肉内投与(i.m.)、硬膜外投与、直接脳内投与および腹腔内投与(i.p.)を含む)、直腸投与、気管内投与、鼻腔内投与、皮膚投与、腟内投与、口腔内投与、眼内投与、または肺投与に適した形態とすることができ、皮下投与または経口投与に適した形態とすることが好ましい。そして、かかる組成物は、例えば「Remington's Pharmaceutical Sciences」(第17版、Alfonso R. Gennaro (編)、Mark Publishing Company, Easton PA, U.S.A., 1985)およびさらに新しい版、ならびに「Drugs and the Pharmaceutical Sciences」シリーズ(Marcel Dekker)中の研究論文などの一般的な記載にあるような当業者に公知の方法で調製できる。該組成物は、例えば、カプセル、錠剤、エアロゾル、局所適用形態、液状または半液状形態(溶液、懸濁液、分散液、エマルジョン、ミセルまたはリポソーム等)などの通常の形態とすることができる。皮下投与に適切な液状組成物が好ましい。好ましい実施形態では、本発明の組成物は、皮下投与される。また別の好ましい実施形態では、本発明の組成物は、経口投与され、この場合は錠剤またはカプセルが好ましい投与形態の1つとして挙げられる。
【0041】
用いる薬学的担体または希釈剤は、従来からの固体または液体の担体でよい。固体担体の例としては、ラクトース、白土、スクロース、シクロデキストリン、タルク、ゼラチン、寒天、ペクチン、アラビアゴム、ステアリン酸マグネシウム、ステアリン酸またはセルロースの低級アルキルエーテル等が挙げられる。液体担体の例としては、シロップ、ピーナッツ油、オリーブ油、リン脂質、ステロール、脂肪酸、脂肪酸アミン、ポリオキシエチレン、等張バッファー溶液および水が挙げられる。同様に、担体または希釈剤は、当業者に公知のあらゆる徐放性物質(グリセリルモノステアレートまたはグリセリルジステアレートなど)を単独でもしくは蜜鑞と混合した形で含有してよい。固体担体を経口投与に用いる場合は、錠剤または硬質ゼラチンカプセル内に入った粉末もしくはペレット形態に調剤するか、あるいはトローチまたはロゼンジの形態に調剤してもよい。固体担体の量は、広い範囲内で様々であるが、通常は約25mg〜約1gであろう。
【0042】
従来の錠剤化技法により調製される典型的錠剤は、
- コア:活性化合物(本発明の化合物の遊離型または塩)100 mg;コロイド状シリコン二酸化物(Aerosil)1.5 mg;セルロース、ミクロクリスト(microcryst; Avicel)70 mg;修飾セルロースガム(Ac-Di-Sol)7.5 mg;ステアリン酸マグネシウム
- コーティング:HPMC 約9 mg;*Mywacett 9-40T 約0.9 mg;フィルムコーティングの可塑剤として用いる*アセチル化モノグリセリド
を含んでよい。
【0043】
液体の担体を用いる場合、調剤は、シロップ、エマルジョン、軟質ゼラチンカプセルまたは注射可能な滅菌済みの液体(水性または非水性の液体懸濁液または溶液など)の形態とすることができる。
【0044】
鼻内投与用には、調剤は、エアロゾルを適用するための液体担体(特に水性担体)に溶解または懸濁した本発明の化合物、好ましくは本発明の結合体を含んでよい。担体は可溶化剤(例えば、プロピレングリコール、胆汁酸塩またはポリオキシエチレン高級アルコールエーテルなどの界面活性剤、吸収増強剤(レシチン(ホスファチジルコリン)またはシクロデキストリンなど)または保存剤(パラビンなど)等を含んでよい。
【0045】
上記組成物はまた、局所的もしくは全身的注射または注入に適する形態であってよく、すなわち、滅菌済みの水または等張生理的食塩水もしくはグルコース溶液を用いて製剤化することができる。該組成物は、当技術分野に公知の従来からの滅菌技法により滅菌してもよい。得られた水性溶液は用途によって包装をしてもよく、または衛生的な条件下で濾過して凍結乾燥してもよい。凍結乾燥した製剤は、投与前に滅菌済み水性溶液と混合する。静脈投与、皮下投与または経口投与に使用する製剤は、活性化合物をバッファーに溶かした溶液が好ましい。該製剤は、活性薬物と滅菌済みバッファー溶液から使用の直前に調製する。好ましい滅菌方法の1つは、使用の直前に調製した溶液を滅菌濾過することでありうる。組成物は、適切な生理学的条件に合致するように薬学的に許容される助剤(例えば、緩衝剤、等張調整剤など)を含んでよい。その例としては、酢酸ナトリウム、乳酸ナトリウム、塩化ナトリウム、塩化カリウム、塩化カルシウムなどがある。
【0046】
本発明の化合物は、有効な薬理学的特性(例えば、タンパク質分解酵素に対する安定性など)を有する。選択されたタンパク質分解酵素の存在下での本発明のペプチドおよびペプチド結合体のin vitro安定性試験では、該新規ペプチドは従来技術におけるペプチドに比べて半減期が増大していることが判明した。従って、本発明の化合物は、GLP-1および他のGLP-1のアゴニストに比べてin vitroで作用する時間がかなり延長されている。さらに、本発明の化合物はcAMPの生成を刺激する。この作用は、cAMPアッセイ(例えば、WO98/08871の記載など参照)において実証することができる。
【0047】
本発明のペプチド化合物は、GLP-1活性および/またはエキセンジン-4活性についてのアゴニストであり、特定のペプチドに対する当技術分野で公知のアッセイにより測定したところ糖尿病哺乳動物において血中のグルコース耐性を向上させる。このようなアッセイの例は本明細書中に記載している。従って、本発明はまた、治療に使用するための上述のエキセンジン変異体およびペプチド結合体、ならびに治療(例えば、I型またはII型糖尿病、肥満、摂食障害およびインシュリン耐性症候群の治療)に使用するための医薬組成物の製造における上述のペプチド結合体の使用にも関する。
【0048】
特定の実施形態では、脊椎動物または哺乳動物のインスリン放出の刺激、血糖レベルの低下、胃の運動性の低減、胃内容排出の遅延化、食物摂取の阻害(例えば、食欲の抑制、または血漿脂質レベルの低減などによる)のために本発明のエキセンジン変異体およびペプチド結合体を用いることができる。本発明の新規化合物はまた、高血糖症の危険を伴う真性糖尿病(即ち、インスリン感受性が、ストレス、心筋梗塞(myocardia infection)、発作および梗塞などにより低下しているか、または妊娠中のインスリン抵抗性が起こっているような場合)の治療に一般に使用することも可能である。該新規化合物はまた、末端肥大症、クッシング症候群、褐色細胞腫、グルカゴノーマ、ソマトスタチノーマ、原発性アルドステロン症などの他の内分泌性疾患の二次的作用である場合、または高血糖症をもたらす特定のホルモンの投与の二次的作用もしくは特定の薬剤(抗高血圧剤、サイアザイド利尿薬、エストロゲンを含む製剤、向精神薬、交感神経作用剤、など)の二次的作用の場合が考えられる他の型の糖尿病の治療に用いることもできる。さらに、本発明の新規化合物を用いて高血糖症の危険を伴う疾患および症状(即ち、アルコール摂取などによる内因性グルコース産生が低下している場合、あるいは下垂体機能不全もしくは原発性副腎皮質不全の患者などにおけるインスリン感受性が増大している場合または進行性腎不全によりインスリンクリアランスが低下している場合など)の治療に一般的に用いることができる。
【0049】
他の特別の治療用途は、WO99/40788(エキセンジンおよびGLP-1の筋収縮性および利尿作用に関する)、WO98/39022(エキセンジンもしくはGLP-1またはエキセンジンもしくはGLP-1のアゴニストを被験体に投与して、被験体に鎮静効果または不安解消作用をもたらすことを含む、中枢または抹消神経系の活性化の増大した被験体の哺乳動物を鎮静させる方法に関する)、WO93/18786(GLP-1(7-37)またはGLP-1(7-36)アミドを用いる治療法に加えて、スルホニル尿素などの経口血糖降下剤による治療をさらに含み、強い相乗効果を生み出す糖尿病の治療に関する)、WO98/19698(肥満の調節におけるGLP-1類似体の使用に関する)、WO98/08531(心筋梗塞後の死亡率および罹患率を低減させる方法におけるGLP-1またはその類似体の使用に関する)、WO98/08873(外科手術後の異化作用の変化およびストレスに対するホルモン応答を緩和する方法におけるGLP-1またはその類似体の使用に関する)に記載されている。また、本発明の化合物は、他の抗糖尿病薬(インスリン、メトホルミン、スルホニル尿素およびサイアゾリジンジオンなど)と組み合わせた治療にも適切であり、または、他の抗肥満薬(レプチン、デキスフェンフルラミン、アンフェタミンなど)と組み合わせた治療にも適切である。
定義
本明細書中で用いる「ペプチド」という用語は、1つのアミノ酸のカルボキシル基ともう1つのアミノ酸のアミノ基とでアミドを形成することのより生成するあらゆる化合物をいう。ペプチド内のアミド結合は、ペプチド結合と呼ぶこともできる。ペプチドという用語は、通常、化合物内のアミド結合が、1つのアミノ酸のC-1ともう1つのアミノ酸のN-2との間で形成されている(ユーペプチド結合(eupeptide bond)と呼ぶこともある)化合物に用いられるが、他のアミド結合(イソペプチド結合と呼ぶこともある)により結合した残基を有する化合物を含む。約10〜20残基より少数の残基からなるペプチドは、オリゴペプチドと呼ぶ場合もあり、それ以上の残基からなるペプチドは、ポリペプチドと呼ぶ場合もある。約50残基より大きい特定の配列のポリペプチドは通常、タンパク質として知られている。本明細書中で用いる「天然のポリペプチド配列」という用語は、天然のL−アミノ酸残基からなり、かつ組換え宿主細胞により発現可能なポリペプチド配列をいう。本明細書中の化合物Xは全て40アミノ酸残基以下のペプチド配列である。
【0050】
本明細書中で用いる「GLP-1」という用語は、GLP-1(7-37)-OH、GLP-1(7-37)-NH2、GLP-1(7-36)-OH、およびGLP-1(7-36)-NH2を含む。
【0051】
「アゴニスト」という用語は、受容体と相互作用でき、該受容体に特徴的な生理学的または薬理学的応答特性(収縮、弛緩、分泌、酵素の活性化など)を誘起し得る内因性の物質または薬剤を指す。
【0052】
「アンタゴニスト」という用語は、他のものの生理学的作用に拮抗する薬剤または化合物を指す。受容体レベルでは、通常他の生物学的に活性な物質により誘導される受容体関連応答に拮抗する化学物質である。
【0053】
「部分的アゴニスト」という用語は、適用する薬剤の量に関係なく、受容体集団の最大限の活性化を誘導することはできないアゴニストを指す。「部分的アゴニスト」という用語は、所定の組織内における「中間的な本質的効力を有するアゴニスト」ということもできる。さらに、部分的アゴニストは、同一の受容体に作用する完全なアゴニストの効果に拮抗し得る。
【0054】
「受容体」という用語は、細胞内もしくは細胞上にあり、分子性伝達物質(神経伝達物質、ホルモン、リンホカイン、レクチン、薬剤など)として作用する化合物を特異的に認識してこれに結合する分子または多量体構造物を指す。
【0055】
本発明の「エキセンジン変異体」という用語は、エキセンジン-4に対して少なくとも約90%の相同性を有し、最も好ましくはエキセンジン-4(1-39)に対して少なくとも約95%の相同性を有し、エキセンジン活性(例えば、哺乳動物において血糖レベルを低下させ、かつGLP-1受容体に結合する)を有する、親エキセンジンペプチドの変異体であると解釈されるべきである。本明細書中に用いる「エキセンジン-4」という用語は、アミノ酸配列が米国特許第5,424,286号中の配列番号2に開示されているエキセンジン-4(1-39)、およびThe Journal of Biological Chemistry, Vol. 272, No7, pp.4108-15中にChenおよびDruckerによって開示されているエキセンジン-4(1-40)(これは、C末端のアミノ酸残基として40位にグリシンを有するという点のみ相違する)を指す。親エキセンジンの相同性は、2つのタンパク質の配列間での同一性の程度(第2の配列からの第1の配列の派生を示す)として決定される。相同性は、GCGプログラムパッケージ (Program Manual for the Wisconsin Package, Version 8, August 1994, Genetics Computer Group. 575 Science Drive, Madison, Wisconsin, USA 53711)(Needleman, S.B.およびWunsch, C.D., (1970), J. Mol. Biol. 48: 443-453)中に提供されるGAPなどの当技術分野で公知のコンピュータプログラムにより適切に決定することができる。ポリペプチド配列比較のための以下の設定;GAPクリエーションペナルティ3.0、およびGAPエクステンションペナルティ0.1を用いることができる。
【0056】
「塩」という用語は、薬学的に許容され得る塩、即ち、酸付加塩および塩基性塩などを含む。酸付加塩の例としては、塩酸塩、ナトリウム塩、臭化水素酸塩などが挙げられる。塩基性塩の例としては、ナトリウムおよびカリウムなどのアルカリ金属カルシウムなどのアルカリ土類金属、ならびにアンモニウムイオンである+N(R3)3(R4)(この場合、R3およびR4は独立して、任意に置換されたC1-6アルキル、任意に置換されたC2-6アルケニル、任意に置換されたアリールまたは任意に置換されたヘテロアリールを指す)から選択されるカチオンを含む塩がある。薬学的に許容され得る塩の他の例としては、例えば、「Remington's Pharmaceutical Sciences」第17版、Alfonso R. Gennaro(編)、Mark Publishing Company, Easton, PA, U.S.A., 1985 およびさらに新しい版、ならびにEncyclopedia of Pharmaceutical Technologyに記載されているものなどが挙げられる。
変異体および結合体の調製
本発明のエキセンジン変異体およびペプチド結合体は、当技術分野に公知の方法により調製することができる。即ち、変異体ならびにペプチド配列XおよびZは、溶液合成またはMerrifield型固相合成などの標準的なペプチド調製技法により調製できる。Boc(tert.ブチルオキシカルボニル)およびFmoc(9-フルオレニルメチルオキシカルボニル)のストラテジーが適用可能であると考えられる。
【0057】
1つの可能な合成ストラテジーでは、本発明のペプチド結合体は、まず公知の標準的な保護法、カップリング法および脱保護法を用いてペプチド配列Zを構築した後、続いてZの構築と同様の方法でペプチド配列XをZにカップリングさせ、最後に担体からペプチド結合体全体を切り離すという固相合成により調製できる。このストラテジーで、ペプチド配列ZがペプチドXのC末端のカルボニル基に共有結合しているペプチド結合体が得られる。しかしながら、所望のペプチド結合体が、2つの安定化配列Zが独立して、ペプチドXのC末端およびN末端の両方に共有結合により結合しているペプチド結合体である場合、上記のストラテジーも適用できるが、C末端結合ペプチド結合体を固相支持体から切り離す前に、2つめのペプチド配列ZをXのN末端に上記と同様の方法でさらにカップリングさせる必要があることは、当業者には理解されるであろう。このストラテジーを用いて、GluまたはAspの側鎖のカルボニル基にZを結合させることもできる。ペプチド配列ZがXのN末端の窒素原子に共有結合しているか、またはXのLys、ArgまたはHisの側鎖の窒素原子に共有結合しているペプチド結合体を調製するのに可能なストラテジーは、上記の方法に類似した方法である。即ち、ペプチド結合体を、まず公知の標準的な保護法、カップリング法および脱保護法によりペプチド配列Xを構築した後、続いてXの構築と同様の方法でペプチド配列ZをXにカップリングさせて、最後にペプチド結合体全体を担体から切り離すという固相合成により調製できる。他の可能なストラテジーは、溶液合成、固相合成、組換え技法、または酵素的合成により、XとZの2つの配列(またはその一部)のうちの1つまたは両方を別々に調製し、続いて溶液中でもしくは固相技法またはその組み合わせによる公知の断片濃縮法により2つの配列をカップリングすることである。1つの実施形態では、Xを組換えDNA法により調製し、Zを固相合成により調製するということもできる。XとZの結合は化学的連結反応により行うことができる。この技術によると、特異性の高い方法により、全く保護されていないペプチド断片の構築が可能である(Liuら、1996、J. Am. Chem. Soc. 118: 307-312およびDawsonら、1996、226: 776)。この結合は、ペプチド結合により全く保護されていないペプチド断片をペプチド結合を介して結合させる特異性の高い技法であるプロテアーゼ触媒性ペプチド結合形成により実施することも可能である(W. Kullmann, 1987, Enzymatic Peptide Synthesis, CRC Press. Boca Raton. Florida. pp.41-59)。
【0058】
ペプチド配列ZにあるLys、Arg、His、Trp、Ser、Thr、Cys、Tyr、AspおよびGluの側鎖の誘導体化は、当技術分野で公知の適切な直交的(orthogonal)な保護スキームを用いて従来からの収束性ペプチド合成により、または、同じく公知の、適切な鎖の直交的(orthogonal)で除去可能な保護を用いる一般的な固相法により実施することができる。
【0059】
さらに、例えば、還元型ペプチド結合などの等電子的結合(isosteric bond)を含むペプチドXなどから得られる改変ペプチド配列をペプチド配列Zにカップリングさせるような場合は、上記のストラテジーの組合せが特に適用可能である場合があると考えられる。このような場合、アミノ酸の連続的カップリングによりZの固定化断片を調製し、その後完全なペプチド配列X(溶液中で調製するか、または全てもしくは部分的に固相技法を用いるか、あるいは組換え技術により調製する)を該断片にカップリングさせることが有利であり得る。
【0060】
適切な固相支持体材料(SSM)の例としては、例えば、官能基化された樹脂(ポリスチレン、ポリアクリルアミド、ポリジメチルアクリルアミド、ポリエチレングリコール、セルロース、ポリエチレン、ポリスチレンにグラフト化されたポリエチレングリコール、ラテックス、ダイナビーズなど)がある。ペプチド配列ZのC末端のアミノ酸またはペプチドXのC末端のアミノ酸を、一般的なリンカー(2,4-ジメトキシ-4'-ヒドロキシ-ベンゾフェノン、4-(4-ヒドロキシ-メチル-3-メトキシフェノキシ)-酪酸、4-ヒドロキシ-メチル安息香酸、4-ヒドロキシメチル-フェノキシ酢酸、3-(4-ヒドロキシメチルフェノキシ)プロピオン酸、およびp-[(R,S)-a[1-(9H-フルオレン-9-イル)メトキシホルムアミド]-2,4-ジメトキシベンジル]-フェノキシ-酢酸など)により固相支持体材料に結合させることが必要または望ましいことは理解されよう。
【0061】
本発明の変異体およびペプチド結合体を、トリフルオロ酢酸、トリフルオロメタンスルホン酸、臭化水素、塩化水素、フッ化水素などの酸を用いて、また場合によっては、酸を、目的に適合する1種以上の「スカベンジャー」(例えば、エタンジチオール、トリイソプロピルシラン、フェノール、チオアニソールなど)と組み合わせて用いることにより、固相支持体材料から切断することができる。あるいは、塩基(アンモニア、ヒドラジンなど)、アルコキシド(ナトリウムエトキシドなど)、水酸化物(水酸化ナトリウムなど)などを用いて、本発明のペプチド結合体を固相支持体から切断することができる。
【0062】
従って、本発明はまた、Zが、好ましくはペプチド結合を介してXに共有結合している、薬理学的活性のあるペプチド結合体を調製する方法に関する。式I(X-Z)で表されるペプチド結合体の調製方法は、以下のステップ:
a) N-α-保護基を含む適切な保護基を有する、活性化型であるアミノ酸またはジペプチドを固定化ペプチド配列H-Z-SSMにカップリングされることにより、N-α-保護ペプチド断片を形成させ、
b) N-α-保護基を除去して、N末端が保護されていない固定化ペプチド断片を形成させ、
c) N-α-保護基を含む適切な保護基を有する、カルボキシルが活性化型であるさらなるアミノ酸またはジペプチドを固定化ペプチド断片のN末端にカップリングさせ、所望のペプチド配列Xが得られるまで、ステップb)とステップc)の除去/カップリングのステップを繰り返し、そして、
d) 固相支持体材料からペプチド結合体を切り離すこと、
を含む。
【0063】
式II(Z-X)で表されるペプチド結合体の調製方法は、以下のステップ:
a) N-α-保護基を含む適切な保護基を有する、活性化型であるアミノ酸またはジペプチドを固相支持体材料(SSM)にカップリングされることにより、固定化保護アミノ酸または固定化保護ジペプチドを形成させ、
b) N-α-保護基を除去して、N末端が保護されていない固定化アミノ酸または固定化ペプチド断片を形成させ、
c) N-α-保護基を含む適切な保護基を有する、カルボキシルが活性化型であるさらなるアミノ酸またはジペプチドを固定化アミノ酸または固定化ペプチド断片のN末端にカップリングさせ、所望のペプチド配列Xが得られるまで、ステップb)とステップc)の除去/カップリングのステップを繰り返し、そして、
d) N-α-保護基を含む適切な保護基を有する、カルボキシルが活性化型であるさらなるアミノ酸またはジペプチドを固定化ペプチド断片のN末端にカップリングさせ、所望のペプチド配列Zが得られるまで、ステップb)とステップd)の除去/カップリングのステップを繰り返し、そして、
e) 固相支持体材料からペプチド結合体を切り離すこと、
を含む。
【0064】
さらに、式III(Z-X-Z)で表されるペプチド結合体の調製方法は、以下のステップ:
a) N-α-保護基を含む適切な保護基を有する、カルボキシルが活性化型であるアミノ酸またはジペプチドを固定化ペプチド配列H-Z-SSMにカップリングすることにより、固定化N-α-保護ペプチド断片を形成させ、
b) 該N-α-保護基を除去して、N末端が保護されていない固定化ペプチド断片を形成させ、
c) N-α-保護基を含む適切な保護基を有する、カルボキシルが活性化型であるさらなるアミノ酸またはジペプチドを固定化ペプチド断片のN末端にカップリングさせ、所望のペプチド配列Xが得られるまで、ステップb)とステップc)の除去/カップリングのステップを繰り返し、そして、
d) N-α-保護基を含む適切な保護基を有する、カルボキシルが活性化型であるさらなるアミノ酸またはジペプチドを固定化ペプチド断片のN末端にカップリングさせ、所望のペプチド配列Zが得られるまで、ステップb)とステップd)の除去/カップリングのステップを繰り返し、そして、
e) 固相支持体材料からペプチド結合体を切り離すこと、
を含む。
【0065】
カップリング、除去および切断のステップは、保護ストラテジーおよび選択した固相材料を考慮して、当業者に公知の方法により実施する。しかし、一般にはBoc(tert.ブチルオキシカルボニル)およびFmoc(9-フルオレニルメチルオキシカルボニル)の保護ストラテジーが適用可能であると考えられ、さらに、当業者に公知の様々な活性化手法を用いて、例えば、ペプチド化学の分野の当業者には公知のように、適切なアミノ酸またはペプチドのC末端の活性化誘導体(酸ハロゲン化物、酸無水物、HObt-エステルなどの活性化エステルなど)を、適切なアミノ酸またはペプチドのアミノ基と反応させるなどにより、ペプチド結合を形成することができると考えられる。さらに、一般的な条件下で反応性のある官能基を担持するアミノ酸残基を用いる場合は、側鎖保護基を含有させることが必要または望ましい場合がある。必要な保護スキームは、当業者には公知であろう(例えば、M. BodanszkyおよびA. Bodanszky,「The Practice of Peptide Synthesis」第2版、Springer-Verlag、1994、J.Jones、「The Chemical Synthesis of Peptides」、Clarendon Press, 1991、およびDrylandら、1986、J. Chem.Soc., Perkin Trans. 1: 125-137など参照)。
【0066】
また本発明のペプチドおよびペプチド結合体は、当業者に公知の一般的方法および原則を用いた組換えDNA技術によって調製されうる。ペプチドおよびペプチド結合体をコードする核酸配列は、確立された標準的方法、例えばS.L. BeaucageおよびM.H.Caruthers. Tetrahedron Letters 22. 1981, pp.1859-1869に記載のホスホアミダイト法、またはMatthesら, EMBO Journal 3, 1984. pp.801-805に記載の方法によって合成的に調製されうる。ホスホアミダイト法に従えば、オリゴヌクレオチドは、例えば自動DNA合成器において合成され、精製、アニーリング、連結後、好適なベクター中にクローニングされる。ペプチドXをコードする核酸配列を単離もしくはクローニングするために使用される技法は当技術分野において公知であり、これらの技法には、ゲノムDNAからの単離、cDNAからの調製、またはそれらの組合せが含まれる。かかるゲノムDNAから本発明の核酸配列をクローニングすることは、例えば周知のポリメラーゼ連鎖反応(PCR)または共有の構造的特徴を有するクローニングされたDNA断片を検出するための発現ライブラリーの抗体スクリーニングを用いることによって達成することができる。例えば、Innisら, 1990. A Guide to Methods and Application. Academic Press. New Yorkを参照されたい。リガーゼ連鎖反応(LCR)、連結活性化転写(ligated activated transcription:LAT)および核酸配列に基づく増幅(NASBA)のような他の核酸増幅法を用いることができる。次いで、それをZをコードする核酸配列に連結することができる。
【0067】
次いで、ペプチドおよびペプチド結合体をコードする核酸配列を、組換え発現ベクター(これは、組換えDNA法に都合よく利用することができる任意のベクターであってよい)に挿入する。ベクターの選択は、多くの場合、それを導入しようとする宿主細胞に依存するであろう。従って、ベクターは、自律複製ベクター、すなわち染色体外の実在として存在し、その複製が染色体複製から独立しているベクター、例えばプラスミドであってよい。あるいは、ベクターは、宿主細胞中に導入する場合、宿主細胞ゲノムに組込まれ、かつ組み込まれた染色体と一緒に複製するベクターのうちの1つであってよい。
【0068】
ベクターにおいて、本発明のペプチドおよびペプチド結合体をコードする核酸配列は、好適なプロモーター配列に機能的に連結されるべきである。プロモーターは、選択した宿主細胞において転写活性を示す任意の核酸配列であってよく、かつ宿主細胞に対して同種の、もしくは異種のタンパク質をコードする遺伝子に由来していてもよい。哺乳動物細胞において該ペプチドおよびペプチド結合体をコードする核酸配列の転写を指令する好適なプロモーターの例としては、SV40プロモーター(Subramaniら, Mol. Cell Biol. 1. 1981. pp.854-864)、MT-1(メタロチオネイン遺伝子)プロモーター(Palmiterら, Science 222. 1983 pp.809-814)またはアデノウイルス2主要後期プロモーター、ラウス肉腫ウイルス(RSV)プロモーター、サイトメガロウイルス(CMV)プロモーター(Boshartら, 1981. Cell 41: 521-530)およびウシパピローマウイルスプロモーター(BPV)が挙げられる。昆虫細胞において使用するのに好適なプロモーターは、ポリへドリンプロモーター(Vasuvedanら, FEBS Lett. 311. 1992. pp.7-11)である。
【0069】
特に細菌宿主細胞において、該ペプチドおよびペプチド結合体をコードする核酸配列の転写を指令するのに好適なプロモーターの例としては、大腸菌(E.coli)lacオペロン、ストレプトミセス・コエリカラー(Streptomyces coelicolor)アガラーゼ遺伝子(dagA)、枯草菌(Bacillus subtilis)レバンスクラーゼ遺伝子(sacB)、バシラス・リへニホルミス(Bacillus licheniformis)α-アミラーゼ遺伝子(amyL)、バシラス・ステアロサルモフィルス(Bacillus stearothermophilus)マルトース生成性(maltogenic)アミラーゼ遺伝子(amyM)、バシラス・アミロリクエファシエンス(Bacillus amyloliquefaciens)αアミラーゼ遺伝子(amyQ)、バシラス・リへニホルミス(Bacillus licheniformis)ぺニシリナーゼ遺伝子(penP)、枯草菌(Bacillus subtilis)xylAおよびxylB遺伝子、および原核生物性β-ラクタマーゼ遺伝子(Villa-Kamaroffら, 1978. Proceedings of the National Academy of Sciences USA 75: 3727-3731)から得られるプロモーター、ならびにtacプロモーター(DeBoerら, 1983. Proceedings of the National Academy of Sciences USA 80: 21-25)が挙げられる。さらにプロモーターは、Scientific American. 1980. 242: 74-94中の「組換え細菌に由来する有用なタンパク質(Useful ptoteins from recombinant bacteria)」;Sambrookら, 1989. 前掲に記載されている。糸状菌宿主細胞において、ペプチドおよびペプチド結合体をコードする核酸配列の転写を指令するのに好適なプロモーターの例としては、コウジカビ(Aspergillus oryzae)TAKAアミラーゼ、リゾムコール・ミエへイ(Rhizomucor miehei)アスパラギン酸プロテイナーゼ、黒コウジカビ(Aspergillus niger)中性α-アミラーゼ、黒コウジカビ(Aspergillus niger)酸安定性α-アミラーゼ、黒コウジカビ(Aspergillus niger)もしくはアスペルギルス・アワモリ(Aspergillus awamori)グルコアミラーゼ(glaA)、リゾムコール・ミエへイ(Rhizomucor miehei)リパーゼ、コウジカビ(Aspergillus oryzae)アルカリ性プロテアーゼ、コウジカビ(Aspergillus oryzae)トリオースリン酸イソメラーゼ、アスペルギルス・ニダランス(Aspergillus nidulans)アセトアミダーゼ、サトイモ乾腐病菌(Fusarium oxysporum)トリプシン様プロテアーゼ(米国特許第4,288,627号に記載されており、これは参照により本明細書に組み入れる)をコードする遺伝子から得られるプロモーター、およびこれらのハイブリッドである。糸状菌宿主細胞において使用するのに特に好ましいプロモーターは、TAKAアミラーゼ、NA2-tpi(黒コウジカビ中性アミラーゼおよびコウジカビトリオースリン酸イソメラーゼをコードする遺伝子に由来するプロモーターのハイブリッド)、およびglaAプロモーターである。酵母宿主において有用なプロモーターは、サッカロミセス・セレビシエ(Saccharomyces cerevisiae)エノラーゼ(ENO-1)遺伝子、サッカロミセス・セレビシエ(Saccharomyces cerevisiae)ガラクトキナーゼ遺伝子(GAL1)、サッカロミセス・セレビシエ(Saccharomyces cerevisiae)アルコールデヒドロゲナーゼ/グリセルアルデヒド-3-リン酸デヒドロゲナーゼ遺伝子(ADH2/GAP)、およびサッカロミセス・セレビシエ(Saccharomyces cerevisiae)3-ホスホグリセリン酸キナーゼ遺伝子から得られる。酵母宿主細胞に有用な他のプロモーターは、Romanosら, 1992, Yeast 8: 423-488によって記載されている。
【0070】
また該ペプチドおよびペプチド結合体をコードする核酸配列は、好適なターミネーター、例えば、ヒト成長ホルモンターミネーター(Palmiterら,前掲書中)に機能的に連結し得る。糸状菌宿主細胞において好ましいターミネーターは、コウジカビTAKAアミラーゼ、黒コウジカビグルコアミラーゼ、アスペルギルス・ニダランスアントラニル酸シンターゼ、黒コウジカビα-グルコシダーゼ、およびサトイモ乾腐病菌トリプシン様プロテアーゼをコードする遺伝子から得られる。酵母宿主細胞にとって好ましいターミネーターは、サッカロミセス・セレビシエ エノラーゼ、サッカロミセス・セレビシエ チトクロームC(CYC1)、またはサッカロミセス・セレビシエ グリセルアルデヒド-3-リン酸デヒドロゲナーゼをコードする遺伝子から得られる。酵母宿主細胞にとって有用な他のターミネーターは、Romanosら, 1992, 前掲によって記載されている。
【0071】
さらにベクターは、ポリアデニル化シグナル(例えば、SV40またはアデノウイルス5Elb領域に由来)、転写エンハンサー配列(例えば、SV40エンハンサー)および翻訳エンハンサー配列(例えば、アデノウイルスVA RNAsをコードするもの)のようなエレメントを含んでいてもよい。さらに、糸状菌宿主細胞に好ましいポリアデニル化配列は、コウジカビTAKAアミラーゼ、黒コウジカビグルコアミラーゼ、アスペルギルス・ニダランス アントラニル酸シンターゼ、および黒コウジカビα-グルコシダーゼをコードする遺伝子から得られる。酵母宿主細胞にとって有用なポリアデニル化配列は、GuoおよびSherman, 1995, Molecular Cellular Biology 15: 5983-5990によって記載されている。
【0072】
さらに組換え発現ベクターは、ベクターが当該の宿主細胞で複製することを可能にするDNA配列を含んでいてもよい。かかる配列(宿主細胞が哺乳動物細胞である場合)の例としては、SV40またはポリオーマの複製開始点が挙げられる。細菌の複製開始点の例としては、プラスミドpBR322、pUC19、pACYC177、pACYC184、pUB110、pE194、pTA1060、およびpAMβ1の複製開始点が挙げられる。酵母宿主細胞において使用するための複製開始点の例としては、2ミクロンの複製開始点、CEN6とARS4との組合せ、およびCEN3とARS1との組合せが挙げられる。複製開始点は、宿主細胞においてその機能を温度感受性にする突然変異を有するものであってよい(例えば、Ehrlich, 1978, Proc. Natl. Acad. Sci. USA 75: 1433を参照されたい)。
【0073】
またベクターは、選択マーカー、例えば、ジヒドロ葉酸還元酵素(DHFR)または薬物(例えば、ネオマイシン、ジェネティシン、アンピシリン、またはハイグロマイシン)に対する耐性を付与するものをコードする遺伝子などの宿主細胞の欠損を補う遺伝子の産物を含んでいてもよい。酵母宿主細胞に好適なマーカーは、ADE2、HIS3、LEU2、LYS2、MET3、TRP1およびURA3である。糸状菌宿主細胞において使用するための選択マーカーは、限定されるものではないが、amdS(アセトアミダーゼ)、argB(オルニチンカルバモイルトランスフェラーゼ)、bar(ホスフィノトリシン(phosphinothricin))アセチルトランスフェラーゼ)、hygB(ハイグロマイシンホスホトランスフェラーゼ)、niaD(硝酸還元酵素)、pyrG(オロチジン-5'-リン酸デカルボキシラーゼ)、sC(硫酸アデニルトランスフェラーゼ)、trpC(アントラニル酸シンターゼ)、およびグルホシネート耐性マーカー、ならびに他の種に由来する等価物を含む群から選択され得る。コウジカビ細胞において使用するのに好ましいものは、アスペルギルス・ニダランスもしくはコウジカビのamdSおよびpyrGマーカー、ならびにストレプトミセス・ハイグロスコピカスのbarマーカーである。さらに、選択マーカーが別々のベクター上にある場合には、選択は、例えばWO91/17243に記載のように同時形質転換によって成し遂げることができる。
【0074】
ペプチドおよびペプチド結合体、プロモーター、ならびにターミネーターのそれぞれをコードする核酸配列を連結するために使用する方法、およびそれらを複製に必要な情報を含有する好適なベクターに挿入するための方法は、当業者に周知である(例えば、Sambrookら, 前掲書中を参照されたい)。
【0075】
発現ベクターを導入する宿主細胞は、ペプチドおよびペプチド結合体を産生することが可能である細胞のいずれであってもよく、真核細胞、例えば無セキツイ動物(昆虫)細胞、またはセキツイ細胞(例えば、アフリカツメガエル(Xenopus laevis)卵母細胞もしくは哺乳動物細胞)、特に昆虫細胞および哺乳動物細胞であってよい。好適な哺乳動物細胞系の例としては、COS(例えば、ATCC CRL 1650)、BHK(例えば、ATCC CRL 1632、ATCC CCL 10)、またはCHO(例えば、ATCC CCL 61)細胞系が挙げられる。
【0076】
哺乳動物細胞をトランスフェクトする方法および細胞に導入したDNA配列を発現する方法は、例えばKaufmanおよびSharp, 1982, J. Mol. Biol. 159: 601-621; SouthernおよびBerg, 1982, J. Mol. Appl. Genet. 1: 327-341; Loyterら, 1982, Proc. Natl. Acad. Sci. USA 79: 422-426; Wiglerら, 1978, Cell 14: 725; CorsaroおよびPearson, 1981, Somatic Cell Genetics 7: 603; Grahamおよびvan der Eb. 1973, Virology 52: 456; Fraleyら, 1980, JBC 225: 10431; Capecchi, 1980, Cell 22: 479; Wibergら, 1983, NAR 11: 7287;およびNeumannら, 1982, EMBO J. 1: 841-845に記載されている。また宿主細胞は、単細胞病原体(例えば原核生物)または非単細胞病原体(例えば、真核生物)であってよい。有用な単細胞は、細菌細胞であり、例えば、限定されるものではないが、バシラス細胞(例えば、バシラス・アルカノフィルス(Bacillus alkalophilus)、バシラス・アミロリクエファシエンス、バシラス・ブレビス(Bacillus brevis)、バシラス・サークランス(Bacillus circulans)、バシラス・コアグランス(Bacillus coagulans)、バシラス・ロータス(Bacillus lautus)、バシラス・レンタス(Bacillus lentus)、バシラス・リへニホルミス、巨大菌(Bacillus megaterium)、バシラス・ステアロサルモフィルス、枯草菌、およびバシラス・スリンジエンシス(Bacillus thuringiensis));またはストレプトミセス細胞(例えば、ストレプトミセス・リビダンス(Streptomyces lividans)もしくはストレプトミセス・ムリヌス(Streptomyces murinus))を含めたグラム陽性細菌、または大腸菌およびシュードモナス(Pseudomonas)種のようなグラム陰性細菌である。好ましい実施形態において、細菌宿主細胞はバシラス・レンタス、バシラス・リへニホルミス、バシラス・ステアロサルモフィルスまたは枯草菌細胞である。細菌宿主細胞の形質転換は、例えばプロトプラスト形質転換法(例えば、ChangおよびCohen, 1979, Molecular General Genetics 168:111-115を参照されたい)、コンピテント細胞の使用(例えば、YoungおよびSpizizin, 1961, Journal of Bacteriology 81: 823-829、またはDubnarおよびDavidoff Abelson, 1971, Journal of Molecular Biology 56: 209-221を参照されたい)、エレクトロポレーション法(例えば、ShigekawaおよびDower, 1988, Biotechniques 6: 742-751を参照されたい)、または接合(例えば、KoehlerおよびThorne, 1987, Journal of Bacteriology 169: 5771-5278を参照されたい)によって達成されうる。宿主細胞は、真菌細胞であってよい。また真菌宿主細胞は、酵母細胞であってよい。本明細書において使用される「酵母」という用語には、子嚢胞子形成酵母(ascosporogenous yeast)(エンドミケス目(Endomyoetales))、担子胞子形成酵母(basidiosporogenous yeast)および不完全菌類(Fungi Imperfecti)(ブラストミセテス(Blastomycetes))に属する酵母が含まれる。
【0077】
細胞の培養に用いる培地は、哺乳動物細胞を生育するのに好適な通常の培地、例えば適当な補充物を含有する血清含有培地もしくは無血清培地、または昆虫細胞、酵母細胞、もしくは真菌細胞を生育するのに好適な培地であってよい。好適な培地は、製造業者から入手可能であり、または公表されている配合表(例えば、American Type Culture Collectionのカタログ中の配合表)に従って調製できる。
【0078】
従って、また本発明は、本発明の天然のポリペプチド配列を有するエキセンジン変異体およびペプチド結合体を製造する方法であって、以下の手順:
a) 本発明のエキセンジン変異体またはペプチド結合体のペプチド配列を含んでなるポリペプチド配列をコードする核酸配列および選択マーカーを含有する核酸構築物またはベクターを宿主細胞中に導入することによって、組換え宿主細胞を得て;
b) 該組換え宿主細胞を選別し;
c) 該組換え宿主細胞を該ポリペプチド配列の産生を可能にする条件下で培養し;
d) 該ポリペプチド配列をこの培養物から単離し;さらに
e) 場合によって、適当なプロテアーゼを用いて該ポリペプチド配列を切断することによって該ペプチド結合体を得ること、
を含む方法に関する。
【0079】
次いで、このように細胞によって産生された天然のポリペプチド配列を有する本発明の変異体およびペプチド結合体を、遠心分離もしくは濾過によって培地から宿主細胞を分離し、塩(例えば、硫酸アンモニウム)によって上清もしくは濾液のタンパク質成分を沈澱させ、種々のクロマトグラフィー法(例えば、イオン交換クロマトグラフィー、アフィニティクロマトグラフィーなど)によって精製することを含めた通常の方法によって培養培地から回収することができる。親油性置換基を、当技術分野において公知の方法を用いて、本発明のペプチドに結合させることができる。一実施形態において、親油性置換基は、標準的な合成方法において、すでに親油性置換基が結合したアミノ酸を組込むことによって結合されうる(例えば、実施例の節における化合物7の合成を参照されたい)。あるいは、この置換基は、例えばWO98/08871に記載のように、ペプチドを合成し、単離した後に結合させることができる。
【0080】
本発明を以下の実施例でさらに説明する。
【実施例】
【0081】
実施例
ペプチド合成、一般的手順
・装置および合成方法
ペプチドを、濾過用のポリプロピレンフィルターを備えたポリエチレン容器で、N-α-アミノ保護基として9-フルオレニルメチルオキシカルボニル(Fmoc)を、および側鎖官能基については好適な慣用の保護基を用いて、バッチ式で合成する(Drylandら, 1986, J. Chem. Soc., Perkin Trans. 1: 125-137)。
・溶媒
溶媒DMF(N,N-ジメチルホルムアミド、Riedel de-Haen, Germany)を、強陽イオン交換樹脂(Lewatit S 100 MB/H 強酸、Bayer AG Leverkusen, Germany)を充填したカラムに通して精製し、使用前に3,4-ジヒドロ-3-ヒドロキシ-4-オキソ-1,2,3-ベンゾトリアジン(Dhbt-OH)を加えて遊離アミンについて分析する(遊離アミンが存在する場合には黄色(Dhbt-O-陰イオン)となる)。溶媒DCM(ジクロロメタン、分析用グレード、Riedel de-Haen, Germany)は精製せずにそのまま使用する。THF(テトラヒドロフラン、分析用グレード、Riedel de-Haen, Germany)は精製せずにそのまま使用する。
・アミノ酸
Fmoc保護アミノ酸は好適に側鎖が保護されたものをMilliGen(UK)およびPerSeptive Biosystems GmbH Hamburg, Germanyから購入する。FmocLys(パルミトイル)-OHはBachem(Switzerland)から購入する。
・リンカー
(4-ヒドロキシメチルフェノキシ)酢酸(HMPA), Novabiochem, SwitzerlandをDICによって、プレフォーム型(preformed)またはin situ生成による1-ヒドロキシベンゾトリアゾール(HObt)エステルのいずれかで樹脂と結合させる。
・カップリング試薬
カップリング試薬ジイソプロピルカルボジイミド(DIC)はRiedel de-Haen, Germanyから購入し、使用前に蒸留する。ジシクロヘキシルカルボジイミド(DCC)はMerck-Schuchardt, Munchen, Germanyから購入し、蒸留精製する。
・固相支持体
Fmoc法により合成されるペプチドをDMF中0.05M以上の濃度のFmoc保護活性化アミノ酸を用いて次の種類の固相支持体上で合成する。TentaGel S樹脂0.22〜0.31mmol/g(TentaGel S-Ram、TentaGel S RAM-Lys(Boc)Fmoc:Rapp polymere, Germany)。
・触媒および他の試薬
ジイソプロピルエチルアミン(DIEA)はAldrich, Germanyから、エチレンジアミンはFlukaから、ピペリジンおよびピリジンはRiedel de-Haen, Frankfurt, Germanyから購入する。4-(N,N-ジ-メチルアミノ)ピリジン(DMAP)はFluka, Switzerlandから購入し、対称無水物(symmetrical anhydrides)を伴うカップリング反応の触媒として用いる。エタンジチオールはRiedel de-Haen, Frankfurt, Germanyから購入する。3,4-ジヒドロ-3-ヒドロキシ-4-オキソ-1,2,3-ベンゾトリアジン(Dhbt-OH)および1-ヒドロキシベンゾトリアゾール(HObt)はFluka, Switzerlandから入手する。
・カップリング手順
第1のアミノ酸をDICまたはDCCによって、好適なN-α-保護アミノ酸から生成されるDMF中の対称無水物として結合させる。次のアミノ酸はDMF中でDICによって、好適なN-α-保護アミノ酸およびHObtから作製される、プレフォームHObtエステルとして結合させる。ニンヒドリン試験を80℃で行ってアシル化を確認し、試験中のFmoc脱保護を防ぐ(Larsen. B. D.およびHolm. A., 1994. Int. J. Peptide Protein Res. 43: 1-9)。
・HObtエステルとしてのカップリング
方法a. 3当量のN-α-アミノ保護アミノ酸を3当量のHObtおよび3当量のDICとともにDMFに溶解する。この溶液を室温で10分間置いた後、予め活性化したアミノ酸を加える前にDMF中0.2%Dhbt-OHの溶液で洗浄した樹脂に加える。
【0082】
方法b. 3当量のN-α-アミノ保護アミノ酸を3当量のHObtとともにDMFに溶解する。使用直前に3当量のDICを加える。最終溶液を樹脂に加える。
・プレフォーム対称無水物
6当量のN-α-アミノ保護アミノ酸をDCMに溶解し、0℃まで冷却する。DDCまたはDIC(3当量)を加え、反応を10分間続ける。溶媒を真空で留去し、残渣をDMFに溶解する。DCCを用いた場合にはDMF溶液を濾過し、すぐに樹脂、続いて0.1当量のDMAPを加える。
・N-α-アミノFmoc保護基の脱保護
Fmoc基の脱保護は、DMF中20%ピペリジン溶液で処理(1×5および1×10分)し、続いてDhbt-OHを排液のDMFに加えて黄色(Dhbt-O-)が検出されなくなるまでDMFで洗浄して行う。
・酸による樹脂からのペプチドの切断
方法a. ペプチドは、95%トリフルオロ酢酸(TFA, Riedel de-Haen, Frankfurt, Germany)-水v/vまたは95%TFAおよび5%エタンジチオールv/vで室温で2時間処理して樹脂から切断する。濾過した樹脂を95%TFA-水v/vで洗浄し、濾液および洗液を10%酢酸を加えて希釈する。得られた混合物をエーテルで3回抽出し、最後に凍結乾燥する。凍結乾燥粗生成物を高速液体クロマトグラフィー(HPLC)で分析し、質量分析法(MS)により同定する。
・TentaGel S-RAM上でのバッチ式ペプチド合成
TentaGel S-RAM樹脂(100〜1000mg、0.22〜0.31mmol/g)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れる。樹脂をDMF(5〜10ml)で膨潤させ、Fmoc基を上記の手順に従って除去する。その配列に従う次のアミノ酸を、上記のようにDICによって、in situ生成されるFmoc保護HObtエステル(3当量)として結合させる。特に断りのない限り、カップリングは3時間続ける。樹脂から溶媒を抜き(drained)、DMF(4×5〜10ml、各2分)で洗浄し、過剰の試薬を除去する。ニンヒドリン試験を80℃で行ってすべてのアシル化を確認する。合成完了後、ペプチド-樹脂をDMF(3×5〜10ml、各5分)、DCM(3×5〜10ml、各1分)、さらに最後にジエチルエーテル(3×5〜10ml、各1分)で洗浄し、真空乾燥する。
・HPLC条件
定組成(isocratic)HPLC分析は、LC-6Aポンプ、215nmで作動させるMERCK HITACHI L-4000 UV検出器および20μlループのRheodyne 7125 注入バルブからなるShimadzu systemで行う。定組成分析に用いるカラムは、Spherisorb ODS-2(100×3mm: 5-μm粒子)(MicroLab, Aarhus, Denmark)である。グラジェントを用いるHPLC分析は、MERCK HITACHI L-6200 インテリジェントポンプ、215nmで作動させるMERCK HITACHI L-4000 UV検出器および20μlループのRheodyne 7125 注入バルブで、またはWater 996 フォトダイオードアレイ検出器を備えたWater 600 E装置で行う。用いるカラムはRescorce(商標)RPC 1ml(Waters)またはLiChroCART 125-4、LiChrospher 100 RP-18(5μm)(Merck)である。バッファーAは水中0.1容量%TFA、バッファーBは90容量%アセトニトリル、9.9容量%水および0.1容量%TFAである。バッファーは、次のペプチド分析用のグラジェント、1)0%〜100%Bの直線グラジェント(30分)または2)0%B(2分)、0%〜50%B(23分)、50%〜100%B(5分)の直線グラジェントのいずれかを用いて流速1.3〜1.5ml/分でカラムを通して供給する。分取HPLCについては、Water 996 フォトダイオードアレイ検出器を備えたWater 600 E装置で精製を行う。用いるカラムはWaters Delta-Pak C-18 15μm, 100Å, 25×100mmである。グラジェント”2)”を流速9ml/分で用いる。
・質量分析法
質量スペクトルは、エレクトロスプレー(ESI)プローブ(ES-MS)を備えたFinnigan Mat LCQ装置およびマトリックスとしてβ-シアノ-p-ヒドロキシ桂皮酸を用いるTofSpec E. Fisons Instrument(MALDI-TOF)で得る。また、Micromass LCT装置によりスペクトルを得てもよい。
先行技術ペプチドのペプチド合成
(i)化合物(i)のペプチド合成
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Pro-Ser-NH2
TentaGel S-RAM上での(エキセンジン-4(1-39)-NH2)(配列番号102)
乾燥TentaGel S-RAM樹脂(0.25mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。Fmoc基を上記の手順に従って除去し、その配列に従うペプチドを、「TentaGel S-RAM樹脂上でのバッチ式ペプチド合成」に記載したように構築(assembled)する。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。粗ペプチドを上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が90%を超えていることがわかる。ペプチドはES-MSにより同定する。収率17%。
(ii)化合物(ii)のペプチド合成
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Pro-NH2
TentaGel S-RAM上での(des Ser39 エキセンジン-4(1-39)-NH2)
乾燥TentaGel S-RAM樹脂(0.25mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。Fmoc基を上記の手順に従って除去し、その配列に従うペプチドを、「TentaGel S-RAM樹脂上でのバッチ式ペプチド合成」に記載したように構成する。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。粗ペプチドを上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が97%を超えていることがわかる。ペプチドはES-MSにより同定する。収率22%。
(iii)化合物(iii)のペプチド合成
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Gly-Gln-Ala-Ala-Lys-Glu-Phe-Ile-Ala-Trp-Leu-Val-Lys-Gly-Arg-NH2
TentaGel S-RAM上での(Gly8-GLP1-(7-36)(ヒト)-NH2)(配列番号87)
乾燥TentaGel S-RAM樹脂(0.25mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。Fmoc基を上記の手順に従って除去し、その配列に従うペプチドを、「TentaGel S-RAM樹脂上でのバッチ式ペプチド合成」に記載したように構成する。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。粗ペプチドを上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が95%を超えていることがわかる。ペプチドはES-MSにより同定する。収率9%。
本発明のペプチド配列の合成
1.化合物1のペプチド合成
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Ser-NH2
TentaGel S-RAM上での(des-Pro36-エキセンジン-4(1-39)-NH2)(配列番号101)
乾燥TentaGel S-RAM樹脂(0.25mmol/g、1500mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。Fmoc基を上記の手順に従って除去し、その配列に従うペプチドを、「TentaGel S-RAM樹脂上でのバッチ式ペプチド合成」に記載したように構築する。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。粗ペプチドを上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が95%を超えていることがわかる。ペプチドはES-MSにより同定する。収率18.3%。
2.化合物2のペプチド合成
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Ser-(Lys)6-NH2
TentaGel S-RAM-Lys(Boc)Fmoc上での(des-Pro36-エキセンジン-4(1-39)-Lys6-NH2)(配列番号93)
乾燥TentaGel S-RAM-Lys(Boc)Fmoc樹脂(0.22mmol/g、1500mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。第1のリジンのFmoc基を上記のように除去し、「TentaGel S-Ram-Lys(Boc)Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が95%を超えていることがわかる。ペプチドはES-MSにより同定する。収率22.1%。
3.化合物3のペプチド合成
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Pro-Ser-(Lys)6-NH2
TentaGel S-RAM-Lys(Boc)Fmoc上での(エキセンジン-4(1-39)-Lys6-NH2)(配列番号92)
乾燥TentaGel S-RAM-Lys(Boc)Fmoc樹脂(0.22mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。第1のリジンのFmoc基を上記のように除去し、「TentaGel S-Ram-Lys(Boc)Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が90%を超えていることがわかる。ペプチドはES-MSにより同定する。収率20.5%。
4.化合物4のペプチド合成
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Gly-Gln-Ala-Ala-Lys-Glu-Phe-lle-Ala-Trp-Leu-Val-Lys-Gly-Arg-(Lys)6-NH2
TentaGel S-RAM-Lys(Boc)Fmoc上での(Gly8-GLP1-(7-36)(ヒト)-Lys6-NH2)(配列番号88)
乾燥TentaGel S-RAM-Lys(Boc)Fmoc樹脂(0.22mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。第1のリジンのFmoc基を上記のように除去し、「TentaGel S-Ram-Lys(Boc)Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が95%を超えていることがわかる。ペプチドはES-MSにより同定する。収率11.7%。
4a.化合物4のペプチド合成
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Gly-Gln-Ala-Ala-Lys-Glu-Phe-lle-Ala-Trp-Leu-Val-Lys-Gly-Arg-Lys-Lys-Lys-Lys-Lys-Lys-NH2
TentaGel S-RAM-Lys(Boc)Fmoc上での([Gly8]hGLP-1(7-36)-(Lys)6-NH2)(配列番号88)
乾燥TentaGel S-RAM-Lys(Boc)Fmoc樹脂(0.22mmol/g、2013mg)を濾過用のポリプロピレンフィルターを備えたガラス容器に入れ、DMF(5ml)で2時間膨潤させる。第1のリジンのFmoc基を上記のように除去し、「TentaGel S-Ram-Lys(Boc)Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が90%を超えていることがわかる。ペプチドはES-MSにより同定する。収率13%。
5.化合物5のペプチド合成
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Gly-Gln-Ala-Ala-Lys-Glu-Phe-lle-Ala-Trp-Leu-Val-Lys-Gly-Arg-Lys(パルミトイル)-(Lys)6-NH2
TentaGel S-RAM-Lys(Boc)Fmoc上での([Gly8,Lys37(パルミトイル)]GLP1-(7-36)(ヒト)-(Lys)7-NH2)(配列番号89)
乾燥TentaGel S-RAM-Lys(Boc)Fmoc樹脂(0.22mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。第1のリジンのFmoc基を上記のように除去し、「TentaGel S-Ram-Lys(Boc)Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。試薬Fmoc-Lys(パルミトイル)-OHはDMFにわずかしか溶解しないため、若干方法を改変して結合させる。Fmoc-Lys(パルミトイル)-OH約400mgをDMFではなくTHF約6mlに溶解させる。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法bに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が95%を超えていることがわかる。ペプチドはES-MSにより同定する。収率9.3%。
6.化合物6のペプチド合成
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Gly-Gln-Ala-Ala-Lys-Glu-Phe-lle-Ala-Trp-Leu-Val-Lys(パルミトイル)-Gly-Arg-(Lys)6-NH2
TentaGel S-RAM-Lys(Boc)Fmoc上での([Gly8,Lys34(パルミトイル)]GLP1-(7-36)(ヒト)-(Lys)6-NH2)(配列番号90)
乾燥TentaGel S-RAM-Lys(Boc)Fmoc樹脂(0.22mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。第1のリジンのFmoc基を上記のように除去し、「TentaGel S-Ram-Lys(Boc)Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。試薬Fmoc-Lys(パルミトイル)-OHはDMFにわずかしか溶解しないため、若干方法を改変して結合させる。Fmoc-Lys(パルミトイル)-OH約400mgをDMFではなくTHF約6mlに溶解させる。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が90%を超えていることがわかる。ペプチドはES-MSにより同定する。収率4.2%。
7.化合物7のペプチド合成
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Gly-Gln-Ala-Ala-Lys(パルミトイル)-Glu-Phe-Ile-Ala-Trp-Leu-Val-Lys-Gly-Arg-(Lys)6-NH2
TentaGel S-RAM-Lys(Boc)Fmoc上での([Gly8,Lys26(パルミトイル)]GLP1-(7-36)(ヒト)-(Lys)6-NH2)(配列番号103)
乾燥TentaGel S-RAM-Lys(Boc)Fmoc樹脂(0.22mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。第1のリジンのFmoc基を上記のように除去し、「TentaGel S-Ram-Lys(Boc)Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。試薬Fmoc-Lys(パルミトイル)-OHはDMFにわずかしか溶解しないため、若干方法を改変して結合させる。Fmoc-Lys(パルミトイル)-OH約400mgをDMFではなくTHF約6mlに溶解させる。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が90%を超えていることがわかる。ペプチドはES-MSにより同定する。収率2.2%。
8.化合物8のペプチド合成
H-Lys-Lys-Lys-Lys-Lys-Lys-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Ser-NH2
TentaGel S-RAM-Fmoc上での(H-(Lys)6-des Pro36 エキセンジン-4(1-39)-NH2)
乾燥TentaGel S-RAM-Fmoc樹脂(0.23mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。樹脂のFmoc基を上記のように除去し、「TentaGel S-Ram-Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が95%を超えていることがわかる。ペプチドはES-MSにより同定する。収率26%。
9.化合物9のペプチド合成
H-Lys6-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Ser-(Lys)6-NH2
TentaGel S-RAM-Lys(Boc)Fmoc上での(H-Lys6-des Pro36 エキセンジン-4(1-39)-Lys6-NH2)
乾燥TentaGel S-RAM-Lys(Boc)Fmoc樹脂(0.22mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。第1のリジンのFmoc基を上記のように除去し、「TentaGel S-Ram-Lys(Boc)Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が90%を超えていることがわかる。ペプチドはES-MSにより同定する。収率32%。
10.化合物10のペプチド合成
H-Lys-Lys-Lys-Lys-Lys-Lys-His-Gly-Glu-Gly-Thr-PheThr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Gly-Gln-Ala-Ala-Lys-Glu-Phe-lle-Ala-Trp-Leu-Val-LysGly-Arg-Lys-Lys-Lys-Lys-Lys-Lys-NH2
TentaGel S-RAM-Lys(Boc)Fmoc上での(H-(Lys)6-([Gly8]hGLP-1(7-36)-(Lys)6-NH2)
乾燥TentaGel S-RAM-Lys(Boc)Fmoc樹脂(0.22mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。第1のリジンのFmoc基を上記のように除去し、「TentaGel S-Ram-Lys(Boc)Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が90%を超えていることがわかる。ペプチドはES-MSにより同定する。収率18%。
11.化合物11のペプチド合成
H-Lys-Lys-Lys-Lys-Lys-Lys-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Gly-Gln-Ala-Ala-Lys-Glu-Phe-Ile-Ala-Trp-Leu-Val-Lys-Gly-Arg-NH2
TentaGel S-RAM-Lys(Boc)Fmoc上での(H-(Lys)6-[Gly8]hGLP-1(7-36)-NH2)
乾燥TentaGel S-RAM-Fmoc樹脂(0.23mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。樹脂のFmoc基を上記のように除去し、「TentaGel S-Ram-Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が98%を超えていることがわかる。ペプチドはES-MSにより同定する。収率15%。
12.化合物12のペプチド合成
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Gly-Gln-Ala-Ala-Lys-Glu-Phe-Ile-Ala-Trp-Leu-Val-Lys-Gly-Arg-Lys-Lys-Lys-Lys-Lys-Lys-Lys-Lys-NH2
TentaGel S-RAM-Lys(Boc)Fmoc上での([Gly8]hGLP-1(7-36)-(Lys)8-NH2)
乾燥TentaGel S-RAM-Lys(Boc)Fmoc樹脂(0.22mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。第1のリジンのFmoc基を上記のように除去し、「TentaGel S-Ram-Lys(Boc)Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が98%を超えていることがわかる。ペプチドはES-MSにより同定する。収率4.2%。
13.化合物13のペプチド合成
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Gly-Gln-Ala-Ala-Lys-Glu-Phe-Ile-Ala-Trp-Leu-Val-Lys-Gly-Arg-Lys-Lys-Lys-Lys-Lys-Lys-Lys-Lys-Lys-Lys-NH2
TentaGel S-RAM-Lys(Boc)Fmoc上での([Gly8]hGLP-1(7-36)-(Lys)10-NH2)
乾燥TentaGel S-RAM-Lys(Boc)Fmoc樹脂(0.22mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。第1のリジンのFmoc基を上記のように除去し、「TentaGel S-Ram-Lys(Boc)Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が95%を超えていることがわかる。ペプチドはES-MSにより同定する。収率2%。
14.化合物14のペプチド合成
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Ser-NH2
TentaGel S-RAM-Fmoc上での(H-des Pro36,Pro37,Pro38エキセンジン-4(1-39)-NH2)
乾燥TentaGel S-RAM-Fmoc樹脂(0.23mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。樹脂のFmoc基を上記のように除去し、「TentaGel S-Ram-Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が95%を超えていることがわかる。ペプチドはES-MSにより同定する。収率11%。
15.化合物15のペプチド合成
H-Lys-Lys-Lys-Lys-Lys-Lys-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Ser-NH2
TentaGel S-RAM-Fmoc上での(H-(Lys)6-des Pro36,Pro37,Pro38 エキセンジン-4(1-39)-NH2)
乾燥TentaGel S-RAM-Fmoc樹脂(0.23mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。樹脂のFmoc基を上記のように除去し、「TentaGel S-Ram-Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が94%を超えていることがわかる。ペプチドはES-MSにより同定する。収率17%。
16.化合物16のペプチド合成
H-Asn-Glu-Glu-Glu-Glu-Glu-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-lle-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Ser-NH2
TentaGel S-RAM-Fmoc上での(H-Asn-(Glu)5-des Pro36,Pro37,Pro38 エキセンジン-4(1-39)-NH2)
乾燥TentaGel S-RAM-Fmoc樹脂(0.23mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。樹脂のFmoc基を上記のように除去し、「TentaGel S-Ram-Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が90%を超えていることがわかる。ペプチドはES-MSにより同定する。収率9%。
17.化合物17のペプチド合成.化合物3.
H-(Lys)6-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Ser-(Lys)6-NH2
TentaGel S-RAM-Lys(Boc)Fmoc上での(H-(Lys)6-des Pro36,Pro37,Pro38 エキセンジン-4(1-39)-(Lys)6-NH2)
乾燥TentaGel S-RAM-Lys(Boc)Fmoc樹脂(0.22mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。第1のリジンのFmoc基を上記のように除去し、「TentaGel S-Ram-Lys(Boc)Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が90%を超えていることがわかる。ペプチドはES-MSにより同定する。収率10%。
18.化合物18のペプチド合成
H-Asn-Glu-Glu-Glu-Glu-Glu-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-lle-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Ser-(Lys)6-NH2
TentaGel S-RAM-Lys(Boc)Fmoc上での(H-Asn-(Glu)5-des Pro36,Pro37,Pro38 エキセンジン-4(1-39)-(Lys)6-NH2)
乾燥TentaGel S-RAM-Lys(Boc)Fmoc樹脂(0.22mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。第1のリジンのFmoc基を上記のように除去し、「TentaGel S-Ram-Lys(Boc)Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が92%を超えていることがわかる。ペプチドはES-MSにより同定する。収率14%。
19.化合物19のペプチド合成
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Ser-(Lys)6-NH2
TentaGel S-RAM-Lys(Boc)Fmoc上での(des Pro36,Pro37,Pro38エキセンジン-4(1-39)-(Lys)6-NH2)
乾燥TentaGel S-RAM-Lys(Boc)Fmoc樹脂(0.22mmol/g、1000mg)を濾過用のポリプロピレンフィルターを備えたポリエチレン容器に入れ、DMF(5ml)で2時間膨潤させる。第1のリジンのFmoc基を上記のように除去し、「TentaGel S-Ram-Lys(Boc)Fmoc上でのバッチ式ペプチド合成」に記載したようにペプチド配列が完成するまで合成を続ける。合成完了後、ペプチド-樹脂をDMF(3×5ml、各1分)、DCM(3×5ml、各1分)、ジエチルエーテル(3×5ml、各1分)で洗浄し、真空乾燥する。上記の方法aに従ってペプチドを樹脂から切断し、酢酸から凍結乾燥する。凍結乾燥粗生成物を上記の手順を用いる分取HPLCにより精製する。精製した生成物が均質であり、かつ純度が97%を超えていることがわかる。ペプチドはES-MSにより同定する。収率19%。
20.化合物2の組換え調製物
pYES0010発現ベクターの構築
酵母での異種構造発現のため、合成cDNAを構築した。化合物2をコードするタンパク質配列をサッカロミセス・セレビシエ(Saccharomyces cerevisiae)コドン利用表((Saccharomyces Genomeデータベース)を用いて逆翻訳した。合成cDNAの翻訳を可能にするため、付加的ATG開始コドンを5’末端に付加し、TAA停止コドンを3’末端に付加した。構築物をアンピシリン耐性遺伝子を含むpYES2シャトルベクターのHindIIIおよびEcoRI部位に挿入し、新たな構築物をpYES0010と名付けた(図6参照)。続いてpYES0010を大腸菌(E.coli)へ形質転換し、アンピシリン選択圧下に置いた。陽性クローンを選択して配列決定した。
酵母への形質転換
pYES0010を酵母ハプロイドINVSc1:MaTa his3deltal leu2 trp1-289 ura3-52へと形質転換させる目的で酵母をYPD培地(1%酵母抽出物、2%ペプトン、2%グルコースおよび0.004%硫酸アデニン)で30℃で飽和状態まで増殖させた。1mlの培養物を形質転換用に回収した。2μlの10mg/ml担体DNAを加え、1μgのpYES0010を加えて混合した。0.5ml(45%PEG 4000、1M LiOAc、0.5M EDTAおよび1M Tris-HCl(pH7.5)を加えて混合した。最後に20μl 1M DTTを加え、混合物を室温で16時間インキュベートした。インキュベーション後、細胞を42℃で10分間熱ショックを与え、選択プレート(6.7%酵母窒素ベース、2%グルコース、20μg/mlアデニン、20μg/mlアルギニン、29μg/mlイソロイシン、20μg/mlヒスチジン、60μg/mlロイシン、20μg/mlリジン、20μg/mlトリプトファン、20μg/mlメチオニン、50μg/mlフェニルアラニン、150μg/mlバリン、30μg/mlチロシンおよび2.5%寒天)に播種した。形質転換細胞が現れるまで、プレートを30℃で3〜5日間インキュベートした。
化合物2の発現および精製
形質転換細胞を選択培地(6.7%酵母窒素ベース、2%グルコース、20μg/mlアデニン、20μg/mlアルギニン、29μg/mlイソロイシン、20μg/mlヒスチジン、60μg/mlロイシン、20μg/mlリジン、20μg/mlトリプトファン、20μg/mlメチオニン、50μg/mlフェニルアラニン、150μg/mlバリン、30μg/mlチロシン)で1.5日間培養した。細胞を回収し、ガラクトース誘導培地(6.7%酵母窒素ベース、4%ガラクトース、20μg/mlアデニン、20μg/mlアルギニン、29μg/mlイソロイシン、20μg/mlヒスチジン、60μg/mlロイシン、20μg/mlリジン、20μg/mlトリプトファン、20μg/mlメチオニン、50μg/mlフェニルアラニン、150μg/mlバリン、30μg/mlチロシン)に1日間再懸濁した。細胞を回収し、プロテアーゼ阻害剤(Roche)を含む10mM Tris-HCl pH7.5でホモジナイズした。溶解物を20,000×gで30分間遠心分離して清澄化した。上清を10mM Tris-HCl(pH7.5)で平衡状態にしたSuperdex 12 HR 10/30カラム(Amersham Pharmacia Biotech)に入れた。カラムを50mM 重炭酸アンモニウムバッファーpH8.0で溶出した。組換え化合物2を含むサンプルをプールした。N末端メチオニンをメチオニンアミノペプチダーゼにより除去し、サンプルをHPLCカラムでさらに精製した。
化合物2精製のためのHPLC設定
HPLCカラム:Kromasil RP C8; K 100-10-C8 nr. CER 2230.compound
温度: 22℃
流速: 35ml/分
HPLC溶媒:
A:水中0.10%トリフルオロ酢酸
B:アセトニトリル:水 90:10中0.10%トリフルオロ酢酸
化合物2をHPLCカラムから20%〜80%アセトニトリル中0.10%トリフルオロ酢酸で40分間溶出した。
21.ペプチド注射製剤
ペプチドの静脈注射用定用量製剤は、ペプチドを無菌、等張生理食塩水に溶解し、得られた溶液を滅菌条件下で不活性ガスを充填したガラス製アンプルに入れて調製する。各用量のペプチドを、不活性ガスを充填した乾燥状態のアンプルまたはキャップ付バイアルに入れる。ペプチドの静脈注射用の複数用量製剤は、ペプチドを無菌、等張生理食塩水に溶解し、得られた溶液をキャップ付バイアルに入れて、必要に応じて保存剤(例えば、0.1%パラヒドロキシベンゾエート、1%ベンジルアルコールまたは0.1%クロロクレゾール)を加えて、調製する。
複数用量ペプチド製剤の例
化合物2 12.25mg
リン酸二水素ナトリウム 1.380g
パラヒドロキシベンゾエート 0.1g
注射水溶液 100ml
【0083】
22.安定性試験
in vivoにおけるタンパク質分解からの上記ペプチドの保護について調べる手段として、選択したタンパク質分解酵素の存在下でのペプチドおよびペプチド複合体のin vitroにおける安定性試験を用いる。実施する試験のねらいは、1種以上の酵素、ロイシンアミノペプチダーゼ、カルボキシペプチダーゼAおよびジペプチジルアミノペプチダーゼIVの溶液中、37℃における化合物4、5、6および7のin vitroにおける安定性と先行技術の化合物、化合物(iii)H-(Gly8)-hGLP-1(7-36)-NH2)およびhGLP-1(7-36)-NH2のものを測定して比較することであった。
in vitro安定性についての材料および装置
用いた水はMilli-Q 水処理システム(Millipore, Bedford, MA, USA)から得られる最高品質のものであった。アセトニトリル(ACN)はLabscan Ltd.(Dublin, Ireland)から入手されるスーパーグラジェント級のものであった。トリフルオロ酢酸(TFA)99.9%、リン酸二水素ナトリウム(NaH2PO4)、水酸化ナトリウム(NaOH)および用いた他のすべての化学薬品は分析用のものであった。ロイシンアミノペプチダーゼ(EC 3.4.11.1)、カルボキシペプチダーゼA(EC 3.4.17.1)およびジペプチジルペプチダーゼ(ジペプチジルアミノペプチダーゼIV、EC 3.4.14.5)はすべてSigma(St. Louis, MO, USA)から入手した。HP 1100バイナリーポンプ、HP 1100オートサンプラー、HP 1100カラムサーモスタットおよびHP 1100可変波長検出器からなるHewlett Packard HP 1100 HPLCシステムを用いてHPLC分析を行った。LCソフトウェア(Rev. A. 06. 01)用のHewlett Packard Chemstationを装置の制御およびデータ取得用に用いた。5μm、C18、300Å粒子を充填したVydac 238TP54(150×4.6mm I.D.)カラムを装置に取り付けて用いた。Stuart ScientificのSHT200Dブロックヒーターを安定性試験中のペプチド/酵素溶液の加熱に用いた。ロイシンアミノペプチダーゼ(25U/ml)もしくはカルボキシペプチダーゼA(1U/ml)を含むpH7.4の50mMリン酸バッファー溶液またはジペプチジルアミノペプチダーゼIV(0.5U/ml)を含むpH8.0の100mM重炭酸アンモニウムバッファーでの試験化合物の分解を37℃で調べた。1アリコート(100μl)の水中ペプチドストック溶液(1mg/ml)をエッペンドルフマイクロバイアル中の予め熱した酵素溶液900μlに加えて試験を開始した。その際、ペプチドの開始濃度を0.1mg/ml(約1.7・10-5〜1.8・10-5M)とした。ペプチド/酵素溶液を37℃に保ち、適当な時間間隔をおいて100μlのサンプルをペプチド/酵素溶液から回収し、アセトニトリル中の25%TFA20μlと十分に混合して酵素分解の進行を止めた。不活性化したサンプルをオートサンプラーバイアルに移し、インタクトな試験化合物の含量を以下に記載するHPLCにより調べた。酵素溶液中での試験化合物の半減期(t1/2)を、自然対数のプロットないし時間に対する残留濃度(すなわち、HPLCピーク高さ)から以下の式:
t1/2=1/kobs・ln(2)
(なお、kobsは観測した分解の見かけの一次速度定数)
を用いて算出した。
HPLC分析
上記のように実施した安定性試験のサンプルを上記の装置および次の試験条件を用いてグラジェントHPLC分析により分析した。
カラム温度:30℃
注入量:10μl
移動相A:水中0.1%TFA
移動相B:アセトニトリル(ACN)中0.085%TFA
グラジェント:32〜52% B、21分
検出:215nmのUV
各安定性試験から得られた結果を以下の表1に示している。この表から、調べたすべての酵素を含む溶液中で本発明の化合物の半減期がかなり延びていることがわかる。
【0084】
【表1】
【0085】
LAP:ロイシンアミノペプチダーゼ、CPA:カルボキシペプチダーゼA、DPPIV:ジペプチジルアミノペプチダーゼIV
【0086】
23.化合物(iii)および化合物4のラット血漿におけるin vitro安定性試験
ヘパリンにより安定させたラット(Sprague-Dawley)血漿中で2種類の試験化合物の分解を行った後、固相抽出およびLC-MSを組み合わせて行った。分解は720分間血漿中で行った。化合物(iii)の半減期がラット血漿中では238分であることがわかった。この試験結果とラット血漿中で466分であるとわかった化合物4の半減期とを比較した。
材料および方法
ヘパリンナトリウム中のブランクラット血漿(5000単位/mL)はHarlan Sera Lab Ltd.(Loughborough, UK)から入手した。試験に用いた試験物質については以下の表に記載している。in vitro試験では100μg/ml Milli-Q水の原液を用いた(26.0μM化合物(iii)H-(Gly8)-GLP-1-NH2または17.8μM 化合物4について記載)。
【0087】
LC-MS分析は、オンライン脱ガス装置、クォーターナリーグラジェントポンプ、オートサンプラー、カラムオーブンからなるHP 1100装置、Hewlett Packard(Wilmington, DE. USA)とMicromass(Altrincham, UK)のQuattro Ultima質量分析計とを組み合わせて行った。LCおよびMSはともにMassLynx3.3ソフトウェアで制御した。MS検出の前にVydac 218MS52(2.1×250mm)カラム(Hesperia, CA, USA)でLC分離を行った。開始血漿量は1000μl(37℃)であった。開始血漿量から100μlを0.75ml HPLCバイアル(ブランクとして用いる)へ移し、560μlの抽出溶液(MeCN:0.18M 炭酸アンモニウム pH9.5(6:94v/v) 4C)と混合し、ASPEC XL4 Robotを用いて固相抽出法で抽出した。100μlのストック溶液を残る900μlの血漿に加えて十分に混合し、37℃でインキュベートした(開始濃度は試験化合物10μg/mlとなる)。各時点(各々0.2、60、120、180、240、360、480、662および720分)において血漿を含む薬剤100μlを回収し、560μlの氷冷抽出溶液と混合し、直ちに上記の固相抽出法により抽出した。抽出した血漿サンプルをLC-MSにより分析した。LC-MS分析はQuattro Ultima II 三連四重極MS装置とを組み合わせたHP 1100系LCで行った。サンプルをオートサンプラートレイで18℃に保ち、その後10μlを注入した。分離はVydac 218MS52(2.1×250mm)LCカラムで30℃で行い、14分間で15〜50%Bの直線濃度勾配、流速250μl/分を用いた。水中0.1%蟻酸を移動相A、MeCN中0.1%蟻酸を移動相Bとして用いた。化合物4および化合物(iii)を、それぞれ6H+(m/z=676.7)および4H+(m/z=822.1)イオン種を用い、単一イオン記録方式(SIR)により検出した。化合物4および化合物(iii)の分析用のconc電圧はそれぞれ100および70Vに設定した。化合物4および化合物(iii)のin vitro安定性をラット血漿中、LC-MSにより調べた。2種類の化合物の分解は720分間行い、結果を、時間に対するピーク面積の自然対数でプロットした。化合物の分解速度(kobs)が線形回帰による傾きとして、半減期(t1/2)がln2/kobsとして示されることがわかった。試験の結果を以下に示す。
【0088】
よって、試験の結果から、(Gly8)hGLP-1(7-36)配列にC末端Lys6ペプチドが結合していることで、ラット血漿における安定性が2倍増加することとなる。
24.化合物5の単回用量の経口および非経口投与の糖尿病ob/obマウスの血中グルコースレベルへの影響
本発明の化合物は血中グルコースを低減させる特性を有している。このことについて、化合物5を用いて腹膜内(i.p.)および経口(p.o.)投与後のob/ob変異体マウスの血中グルコース(BG)レベルへの影響を試験し、考察した。化合物5の腹膜内投与では110μg/マウスの用量で糖尿病マウスのBGレベルが低下した。同様に、化合物5の経口投与では、より低い用量ではないが、1100μg/マウスの用量で同様のBGレベルの低下が認められた。
試験
優性変異レプチン(Tomita, T., Doull. V., Pollock. H. G., およびKrizsan, D. 1992. Pancreatic islets of obese hyperglycemic mice(ob/ob). Pancreas 7: 367-75)が原因で肥満である、40匹の雌糖尿病ob/obマウス(Umea系統、Bomholtgaard)を12:12時間明暗周期による制御された周囲条件下で収容し(3マウス/ケージ)、水を得るための自由な通路を設けた標準Altromin no 1324 dietで餌を与えた。到着時、動物は8週齢であった。マウスを2週間、新環境に順応させた後、試験を開始した。試験時点で動物は13週齢、体重41.8±3.2g(平均±SD; n=42)であった。ストレスにより誘導されるBG変動を軽減するため、試験1日前および3日前にマウスに処置を施した。試験当日、照明をつけて2〜3時間後に尾の先から採血した。分析のため、1滴の血液(<5μl)をグルコースストリップに落とし、Elite Autoanalyser、Bayer, Denmarkにより測定した。全血中グルコース(BG)濃度を固定化グルコースオキシダーゼ法により調べた。血中グルコースレベルは正常血糖から重症高血糖症との間で(範囲: 3.6〜15.6mM; 平均±SD; 9.4±3.3mM; n=42)変動した。BG<5.8mMであった6動物を試験から除外した(全体n=36)。残りの動物をBGレベルに基づいて等級別に分類し、各群間で平均BGに差がないようにした。最初の対照血液サンプリングから1時間後に薬剤を投与し、BGをt=60分、t=120分、t=240分、t=480分に測定した。
ペプチドおよび他の材料
化合物5(バッチ番号ZP 3.12画分1-2、精製)は、Department of Chemistry, Zealand Pharmaceuticalsによって合成された。投与の直前にペプチドを滅菌等張NaClに溶解して容量を0.2mlとした。同じ溶液をp.o.およびi.p.投与の両方に使用した。各血液サンプリング時に、データ記録用紙に各動物について記入した。
薬剤投与
動物に化合物5を投与した。最大用量は1100μg/マウス、最小用量は1.1μg/マウスであった。陰性対照として生理食塩水をp.o.により投与し、陽性対照として試験化合物をi.p.により110μg/マウスの用量を与えた。
【0089】
制御条件下での非絶食ob/obマウスのBGレベルはすべての群で同等であった(各群のデータは示していない)が、群内のBGレベルは大きく分散していた(すべての動物のBG範囲: 5.8〜15.6mM)。そのため、高血糖症の変動程度を補正するために結果を基線との相対差(%対照)として示した。
【0090】
化合物5 110μgの腹膜内投与によりBGの持続的な低下がもたらされ、化合物の投与後1〜2時間後には最下点に達した。生理食塩水で処置した動物には変化が見られなかった。薬剤投与から4〜8時間後の間に、ほとんどの群(5/6)でBGが増加した。化合物5は、糖尿病ob/obマウスにi.p.投与した場合、110μg/マウスの用量でBGレベルが低下した(データは示していない)。60分後には抗糖尿病性効果が認められ、化合物投与から2〜4時間後に最大となった。さらに長時間にわたる効果(>8時間)から、化合物5が、よく知られてはいるが、短時間しか作用しない天然のGLP-1(Bailey, C. J. & Flatt, P. R. 1987, Glucagon-like peptide-1 and the entero-insular axis in obese hyperglycaemic (ob/ob)mice. Life Sci. 40. 521-5)よりも長時間の作用を有していることがわかる。用量1100μg/マウス p.o.では、110μg i.p.で処置した動物で認められたものと同様なBGの低下が示された。
【0091】
発明者らは化合物5が、化合物110μg/マウスのi.p.投与により、糖尿病ob/obマウスのBGレベルを効果的に低下させることを示した。また、経口経路により与えた場合でも、化合物5 1100μg/マウスで同様な効果が認められた。このことから、化合物が胃腸管から吸収されていることがわかる。
25.in vivo試験
化合物1(des Pro36-エキセンジン-4(1-39)-NH2(配列番号101))、
化合物2(des Pro36-エキセンジン-4(1-39)-Lys6-NH2(配列番号93))、
化合物(ii)(Gly8-GLP1-(7-36)(ヒト)-NH2(配列番号87))、
化合物4(Gly8-GLP1-(7-36)(ヒト)-Lys6-NH2(配列番号88))および
化合物5(Gly8 Lys37(パルミトイル)-GLP1-(7-36)(ヒト)-Lys7-NH2(配列番号89))
について
ob/obマウスに、各ペプチドを種々の濃度で、経口および腹腔内投与し、これらの化合物が血糖値に影響を与えるか否かを調べた。用いた試験条件は実施例24に記載したものと同じである。
ペプチドおよび他の材料
des Pro36-エキセンジン-4(1-39)-NH2(化合物1. 配列番号101)およびC末端に結合した付加的配列Lys6以外は同じペプチド、des Pro36-エキセンジン-4(1-39)-Lys6-NH2(化合物2. 配列番号93)、Gly8-GLP1-(7-36)(ヒト)-NH2(化合物(ii). 配列番号87)およびC末端に結合した付加的配列Lys6以外は同じペプチド、Gly8-GLP1-(7-36)(ヒト)-Lys6-NH2(化合物4. 配列番号88)ならびにGly8 Lys37(パルミトイル)-GLP1-(7-36)(ヒト)-Lys7-NH2(化合物5. 配列番号89)は、上記の方法で合成する。溶液については、投与当日の朝、動物に投与する直前に調製する。同じ溶液を経口および腹腔内投与の両方に使用する。すべてのペプチドを滅菌等張NaClに溶解して容量を0.2mlとする。すべての試験を同じマウスで行い、表2に示すペプチドの有効量を比較する。上記のように血液サンプリングを行い、表3に示す用量を動物に投与する。陰性対照として生理食塩水を経口投与する。この結果を表4に示している。
【0092】
【表2】
【0093】
【表3】
【0094】
群のデータには、各処置群の個々の結果の平均±SEMを記載している。化合物の効果を調べるため、ベースラインとの絶対および相対(t=0の%)差を各時点について算出した。
【0095】
【表4】
【0096】
得られた結果を表4および図1〜3に示している。
【0097】
これらの結果により、試験したすべての化合物が血糖値の低下作用を有することがわかる。化合物1を腹腔(ip)内に与えると、この効果が最大に現れ、また100μgの化合物2の経口投与(po)による効果と1000μgの化合物2の腹腔内投与(ip)による効果とが同等である。腹腔内から与える場合の化合物1(des Pro36-エキセンジン-4(1-39)-NH2. 配列番号101)および化合物2(des Pro36-エキセンジン-4(1-39)-Lys6-NH2. 配列番号93)の有効性が、エキセンジン-4(1-39)-NH2(化合物(i))自体を同じ方法で投与する場合(データは示していない)と極めて近いことがわかる。
【0098】
化合物1. des Pro36-エキセンジン-4(1-39)-NH2(配列番号101)では、化合物を経口投与する場合、400μg/マウスまでの用量では血糖値の低下には効果がないが、Lys断片を付加した同じ化合物では、10μg/マウスの用量でも効果があることがわかる。このことから、des Pro36-エキセンジン-4(1-39)-Lys6-NH2(配列番号93)の最小有効経口用量が、des Pro36-エキセンジン-4(1-39)-NH2(配列番号101)の少なくとも40倍低いことが示される。
【0099】
これらの結果から、腹膜内投与する場合では配列Zの結合による種々のペプチドの有効性への有意な効果はないが、経口投与する場合には化合物の有効性がかなり高まっていることが示される。
26.覚醒ラット十二指腸における胃腸送達後の化合物4および化合物(iii)の生物学的利用性
種々のペプチドに基づくGLP-1類似体が非経口使用のために開発されてきたが、これらのうちで経口投与後に薬理学的効果を有する物質はなかった[Holst, J. J. Enteroglucagon. Annu Rev Physiol. 59: 257-271. 1997]。これについては、覚醒ラット十二指腸からの試験化合物の吸収を調べることとした。化合物(iii)(Gly8)hGLP-1(7-36)-NH2を対照として用いた。
化学物質および試薬
ヘパリンナトリウム中のブランクラット血漿(5000単位/mL)はHarlan Sera Lab Ltd.(Loughborough, UK)から入手した。OASIS(商標)HLB固相抽出カラム、1cc、30mg吸着剤はWaters(Milford, MA, USA)から入手し、ISOLUTE C18(EC)、1cc、SPEカラムはIST(Mid Glamorgan, U.K.)から入手した。LC/MS分析は、オンライン脱ガス装置、バイナリーグラジェントポンプ、オートサンプラー、カラムオーブンからなるHP 1100装置、Hewlett Packard(Wilmington, DE. USA)とMicromass(Altrincham, UK)のQuattro Ultima質量分析計とを組み合わせて行った。LCおよびMSはともにMassLynx3.3ソフトウェアで制御した。MS検出の前にVydac 218MS52(2.1×250mm)カラム(Hesperia, CA, USA)でLC分離を行った。
薬剤および用量レベル
化合物4(バッチ番号ZP 7.97-5-F、4053g/mol)および化合物(iii)(バッチ番号ZP 7.73-2-G、3854g/mol)はFmoc法により自所で合成した。質量分析法により同定を行い、RP-HPLCにより試験化合物の両方のバッチの純度をそれぞれ97および99.7%と決定した。バッチのペプチド含量はZP 7.97-5-FおよびZP 7.73-2-Gそれぞれ72%および80%であった。ペプチドをパイロジェンを含んでいない等張生理食塩水に溶解し、1,000または10,000nmol/kgの用量を100μlの容量で十二指腸内カテーテルにより投与した。
動物
体重250〜350gの14匹のSprague-Dawleyラットを試験に用いた。動脈血サンプリングのため、皮下注射(s.c.)でHypnorm(登録商標)-Dormicum(登録商標)を用いてラットに麻酔をかけ、大腿動脈にカテーテルを挿入した。さらに十二指腸にも胃を切開することによりカテーテルを挿入した。手術後1週間ラットを回復させてから試験を開始した。試験当日、手術したラットは覚醒していた。十二指腸内のカテーテルが十二指腸にあるどうかを確認するため、試験直後にラットの解剖を行った。
サンプル処理
血液サンプルをt=-5、5、10、15、20、40および60分に採取した。血液を氷冷チューブを含むEDTAに採取し、直ちに4℃で5分間遠心分離(4,000×g)した。血漿(250μl)を250μlの抽出溶液(MeCN:0.18M 炭酸アンモニウム pH9.5、10:90v/v)の入った氷冷0.75ml PLCバイアルへ移した。血漿サンプルはSPEおよびLC/MS分析まで-20℃で保存した。
固相抽出
血漿サンプル(400μl)を含有する薬剤を950μl MeCN、次に950μl水で予め調整した固相抽出カラムに載せた。カラムを950μl水中2%TFA、次に同容量のMeCN:水(20:78 v/v)中2%TFAで洗浄した。分析物を500μl MeCN:水(60:38 v/v)中2%TFAで溶出し、LC/MSにより分析した。
LC/MS
サンプルをオートサンプラートレイ中で18℃に保ち、その後LCカラム(Vydac 218MS52(2.1×250mm))に20〜50μlを注入した。分離は30℃で行い、流速250μl/分および表1のグラジェントを用いた。試験化合物および対照薬剤の両方を、それぞれm/z=676.7およびm/z=1095.2および821.8(イオン種)を用い、単一イオン記録方式(SIR)により検出した。すべての装置の条件はMassLynxソフトウェアバージョン3.3で制御した。
【0100】
血漿サンプルを材料および方法に記載したように分析した。化合物4の生物学的利用性については、1,000(n=4)および10,000(n=5)nmol/kgの用量で調べたが、化合物(iii)の生物学的利用性については、10,000(n=5)nmol/kgの用量でのみ試験した。
【0101】
化合物(iii)の濃度は、調査したすべての時点で検出限界(約0.5nM)以下であったので、正確な生物学的利用性が評価できなかった。これに対し、化合物4は1,000nmol/kgの十二指腸内投与後では4匹のラットのうち2匹、および10,000nmol/kgの投与後では5匹のラットのうち4匹の血漿サンプルから検出された。
27.ウサギおよびブタへのi.v.投与後の化合物1、化合物2、化合物4および化合物(iii)のin vivo薬物動態
本発明者らはラット血漿でGLP-1アゴニスト化合物4のin vitro安定性が対照薬剤である化合物(iii)に比べて向上していたことを示した。この効果がin vivoにおいても維持されるかどうかを確かめるため、2種類の化合物の薬物動態パラメーターをウサギで調べることとした。同じ試験条件を用いて、化合物1および2についてのこれらのパラメーターをウサギで測定した。同じ試験条件を用いてブタでも測定した。
化学薬品および試薬
ヘパリンナトリウム中のブランクウサギ血漿(5000単位/mL)はHarlan Sera Lab Ltd.(Loughborough, UK)から入手した。OASIS(商標)HLB固相抽出カラム、1cc、30mg吸着剤はWaters(Milford, MA, USA)から入手し、ISOLUTE C18(EC)、1cc、SPEカラムはIST(Mid Glamorgan, U.K.)から入手した。LC/MS分析は、オンライン脱ガス装置、バイナリーグラジェントポンプ、オートサンプラー、カラムオーブンからなるHP 1100装置、Hewlett Packard(Wilmington, DE. USA)とMicromass(Altrincham, UK)のQuattro Ultima質量分析計とを組み合わせて行った。LCおよびMSはともにMassLynx3.3ソフトウェアで制御した。MS検出の前にVydac 218MS52(2.1×250mm)カラム(Hesperia, CA, USA)でLC分離を行った。
薬剤および用量レベル
化合物4(バッチ番号ZP 7.97-5-F、4053g/mol)および化合物(iii)(バッチ番号ZP 7.73-2-G、3854g/mol)はFmoc法により所内で合成した。質量分析法により同定を行い、RP-HPLCにより試験化合物の両方のバッチの純度をそれぞれ97および99.7%と決定した。バッチのペプチド含量はZP 7.97-5-FおよびZP 7.73-2-Gそれぞれ72%および80%であった。ペプチドをパイロジェンを含んでいない等張生理食塩水に溶解し、両方のペプチドを1,000nmol/kgの用量でウサギおよびラットにi.v.投与した。
ウサギ
体重2.5〜3.0kgの15羽のニュージーランドシロウサギ(New Zealand White rabbit)を試験に用いた。試験当日、薬剤のi.v.投与および動脈血サンプリングのため、i.m.でHypnorm(登録商標)を、次にi.v.でDormicum(登録商標)を用いてウサギに麻酔をかけ、大腿静脈および動脈にカテーテルを挿入した。試験中、ウサギは覚醒していない状態にあった。
サンプル処理
血液サンプルをt=1、3、5、10、15、20、30、40、60、90、120、150、180および240分の時点で採取した。血液をEDTAを含む氷冷チューブに採取し、直ちに4℃で5分間遠心分離(20,000×g)した。血漿(250μl)を250μlの抽出溶液(MeCN:0.18M 炭酸アンモニウム pH9.5、10:90 v/v)の入った氷冷0.75ml PLCバイアルへ移した。血漿サンプルはSPEおよびLC/MS分析まで-20℃で保存した。
固相抽出
血漿サンプル(400μl)を含有する薬剤を950μl MeCN、次に950μl水で予め調整したOASIS(商標)HLB(化合物4)またはISOLUTE(商標)(化合物(iii))固相抽出カラムに入れた。カラムを950μl水中2%TFA、次に同容量のMeCN:水(20:78 v/v)中2%TFAで洗浄した。分析物を500μl MeCN:水(60:38 v/v)中2%TFAで溶出し、LC/MSにより分析した。
LC/MS
サンプルをオートサンプラートレイで18℃に保ち、その後LCカラム(Vydac 218MS52(2.1×250mm))に20ないし50μlを注入した。分離は30℃で行い、流速250μl/分および以下の表に示す通りのグラジェントを用いた。試験化合物および対照薬剤の両方を、それぞれm/z=676.7およびm/z=1095.2および821.8(イオン種)を用い、単一イオン記録方式(SIR)により検出した。すべての装置の条件はMassLynxソフトウェアバージョン3.3で制御した。
【0102】
血漿サンプルを材料および方法に記載したように分析し、血漿濃度(Cpl)を時間に対して片対数グラフにプロットした。ウサギは血漿濃度について3時間追跡した。ラットでは血液量が限られているため、血液サンプリングは1時間までに制限した。各ウサギのCpl対時間曲線は、WinNonlin 3.1(Pharsight Corp.(Mountain View. CA))の1/y2 重み付き最小二乗法を用い、ツー・コンパートメント・オープン・モデル(図は示していない)にフィットさせた。分析データから得られる薬物動態定数を表5に記載し、化合物4および化合物(iii)それぞれの1μmol/kgのi.v.注射後のウサギにおける分解動態を図4に示している。
【0103】
【表5】
【0104】
28.化合物2、14〜16、18および19の化合物(i)と比較したグルコース負荷試験
雄の糖尿病db/dbマウス(M&B. Bomholtgaard, LI, Skensved, Denmark)を用いる。この十分に解明されているモデルマウスはレプチン受容体の変異により遺伝的にグルコース代謝不全を有している。ホモ接合体db/dbマウスは制御できないインスリン非依存型糖尿病(NIDDM)のヒト患者のように煩渇多飲症、多尿症および糖尿、ならびに高血糖症の程度にかかわらず、生涯の最初の3週間での体重の増加を経験する。しかしながら、このモデルでの高血糖症は進行性ランゲルハンス島萎縮症と関連しており、ケトン症の可能性もあって、6〜8月齢で死亡することもある。そのため、それらの病状の進行およびその程度には注意を払わねばならない。それゆえ、GLP-1類似体の薬剤試験には16週齢未満のdb/dbマウスだけを用いることが好ましい。
【0105】
すべてのマウスを少なくとも1週間、新しい環境に順応させ、2日間毎日処置を施した後、最初の経口グルコース負荷試験(OGTT)を行う。さらに、ストレスにより誘導されるグルコースの混乱を軽減するため、試験前に下記の化合物を加えないで少なくとも1回OGTTに付しておくべきである。糖尿病マウス間ではグルコース負荷が大きく分散しているため、最初に使用する前にOGTTにより動物を等級別に分類する。
ペプチド
ペプチドを0.1%ウシアルブミンを含有する0.1Mリン酸緩衝溶液(PBS)(5M NaOHを加えてpHを7.4に調整)に溶解する。すべての溶液については、試験直前の朝新たに調整する。動物を処置するビヒクルは、0.1%アルブミンを含有するPBSのみとする。
グルコース負荷試験および適用
経口グルコース負荷試験の前、動物に17時間(午後4時から翌日の午前9時まで)絶食させる。午前9時から、尾の先から採血し(t=-15分)、血中グルコースを測定する。全血中グルコース(mM)濃度を、製造業者のマニュアルに従って1滴の血液(<5μl、Elite Autoanalyser、Bayer, Denmark)を用いる固定化グルコースオキシダーゼ法により調べる。重症糖尿病の動物(>10mM)は排除する。動物には、最初の血液サンプリング直後にビヒクルまたは1用量の抗糖尿病性化合物の腹腔内(i.p.)注射を施す。すべての群において注入量は200μg/体重50gマウスとする。物質のi.p.投与から15分後、水に溶解した1g/kgグルコース(Sigma, St. Louis)の経口用量を与えて(200μg/体重50g)動物をホームケージに戻す(t=0)。血中グルコースレベルをt=30分、t=60分、t=120分およびt=240分に測定する。観察期間中、動物に絶食をさせる。各血液サンプリング時には、データ記録用紙に各動物について記入した。
算出および統計値
化合物の効果を調べるため、ベースライン(t=0)との絶対および相対差を各時点について算出する。全試験の曲線下の面積(AUC 0〜240分)を台形法を用いて求める。分類化当日に、すべての群でグルコース負荷が同じようになるようにマウスを振り分ける。しかしながら、時間内での糖尿病の進行を補正するため、試験中、ビヒクルで処置した対照群を毎日試験して、薬剤に対する応答をビヒクルで処置した時間制御動物で観察される応答と比較して示す。
【0106】
各物質の用量応答曲線をプロットし(図5参照)、薬剤の効果を、ビヒクルで処置して得られる応答と比較して、ANCOVA分析(共分散分析)を用いて調べる。処置(薬剤またはビヒクル)は独立変数と考えられ、ビヒクル処置した時間制御マウスの応答率%で示されるAUC 0〜240分は従属変数であり、薬剤用量は共変量として定義される。Post-hoc分析はフィッシャーの最小有意差検定を用いて行う。0.05レベルでは有意差があると考えられる。Windows(登録商標) NT版Statistica バージョン5.5、StatSoft, Tulsa, Oklahoma, U.S.A.を用いて統計解析を行った。図5に示される用量応答曲線では、試験したすべての化合物が対照薬剤のものと同等のグルコース低下効果を示していることがはっきりとわかる。
29.db/dbマウスにおける化合物2および化合物(i)のOGGTへの効果
図7は実施例28に記載した同じ試験条件を用いて行ったOGTTでの化合物2および化合物(i)のAUCのプロットである。この図から、化合物2の血糖の低下効果と先行技術の化合物(iii)の効果とが同等であることがわかる。
30.化合物2、100nmol/kgをOGTTの24時間前までにi.p.投与した場合の経口グルコース負荷試験(OGTT)における化合物の長期効果
この試験ではdb/dbマウス、その他に最大用量100nmol/kg i.p.を用い、実施例28に記載した同じ試験条件を用いる。結果を図8に示している。この試験から、化合物2の作用期間はdb/dbマウスでは18時間までであるという結論になる。
【0107】
本明細書に記載されかつ特許請求される本発明は、本明細書に開示される特定の実施形態に限定されるものではなく、これらの実施形態は本発明のいくつかの態様を例示するものである。同等の実施形態はいずれも本発明の範囲内とされる。実際に、当業者ならば、本明細書に示され、記載されたものに加え、本発明の様々な改変も先行技術から理解できよう。かかる改変もまた添付の請求項の範囲に入る。本明細書において様々な参考文献が引用されているが、これらの明細は参照によりすべて本明細書に組み入れるものとする。
【図面の簡単な説明】
【0108】
【図1】マウスの血糖値における化合物1(配列番号101)(desPro36-エキセンジン-4(1-39)-NH2)の作用を示す(実施例25参照)。
【図2】マウスの血糖値における化合物2(配列番号93)(desPro36-エキセンジン-4(1-39)-Lys6-NH2)の作用を示す(実施例25参照)。
【図3】マウスの血糖値における化合物5(配列番号89)(Gly8,Lys37(パルミトイル)-GLP-1(7-36)(ヒト)-(Lys)7-NH2)の作用を示す(実施例25参照)。
【図4】ウサギにおいて1μmol/kgの化合物4および化合物(iii)を静脈内(i.v.)注射した後のin vivoでの分解速度論を示す(実施例27参照)。
【図5】経口グルコース負荷試験(OGTT)における化合物2、14〜16、18および19についてのAUC(曲線より下の面積、area under the curve)値(平均±SEM)のグラフ(実施例28参照)。
【図6】酵母における化合物2の異種発現のために構築した合成cDNAを示す図である。この新規構築物をpYES0010と命名した(実施例20参照)。
【図7】化合物2および化合物(i)についての相対AUC0-240min値(平均±SEM)に基づいたdb/dbマウスでのGTTにおける用量応答のグラフ(実施例29参照)。
【図8】OGTTの24時間前までに投与した場合の経口グルコース負荷試験(OGTT)における化合物2の最大量、すなわち100nmol/kg i.p.(腹腔内)の効果を示す。
【特許請求の範囲】
【請求項1】
(a) エキセンジン-4に対して少なくとも90%の相同性を有するエキセンジン、
(b) 34〜39位の1〜5個の欠失からなる群より選択される改変を有し、かつ40位に親油性置換基を有するLysを有する、該エキセンジンの変異体、または
(c) (i) 8位のアラニンの、D-アラニン、グリシンもしくはα-アミノイソ酪酸への置換、および
(ii) 親油性置換基
からなる群より選択される少なくとも1個の改変を有するGLP-1(7-36)またはGLP-1(7-37)、
からなる群より選択されるペプチドX(ただし、Xはエキセンジン-4またはエキセンジン-3ではない)と、
4〜20個のアミノ酸単位からなり、各アミノ酸単位が、Ala、Leu、Ser、Thr、Tyr、Asn、Gln、Asp、Glu、Lys、Arg、His、Met、Ornおよび一般式I
-NH-C(R1)(R2)-C(=O)- (I)
(式中、R1およびR2は、水素、C1-6-アルキル、フェニルおよびフェニル-メチルからなる群より選択され、C1-6-アルキルは、ハロゲン、ヒドロキシ、アミノ、シアノ、ニトロ、スルホノおよびカルボキシからなる群より選択される1〜3個の置換基で置換されていてもよく、フェニルおよびフェニル-メチルは、C1-6-アルキル、C2-6-アルケニル、ハロゲン、ヒドロキシ、アミノ、シアノ、ニトロ、スルホノおよびカルボキシからなる群より選択される1〜3個の置換基で置換されていてもよく、あるいはR1とR2は、それらが結合している炭素原子と一緒になって、シクロペンチル、シクロヘキシルまたはシクロヘプチル環を形成している。)
で表されるアミノ酸単位(例えば2,4-ジアミノブタン酸および2,3-ジアミノプロパン酸)からなる群より選択される、前記変異体に共有結合したペプチド配列Zと、
を含むペプチド結合体、ならびに該ペプチド結合体の薬学的に許容される塩またはC末端アミド。
【請求項2】
ペプチドXが、糖尿病の哺乳動物におけるグルコース耐性の改善に有効であることをさらに特徴とする、請求項1に記載のペプチド結合体。
【請求項3】
Zが、少なくとも1個のLysアミノ酸単位、好ましくは少なくとも2個のLysアミノ酸単位、例えば少なくとも3個のLysアミノ酸単位など、例えば少なくとも4個のLysアミノ酸単位、より好ましくは少なくとも5個のLysアミノ酸単位、例えば6個または7個のLysアミノ酸単位など、を含む請求項1または2に記載のペプチド結合体。
【請求項4】
ペプチド結合体の最小有効量と、ペプチドXの経口最小有効量の比が少なくとも1:5であることを特徴とする、請求項1〜3のいずれか1項に記載のペプチド結合体。
【請求項5】
親油性基が、ペプチドXのLysのεアミノ基に結合したパルミトイルである、請求項1〜4のいずれか1項に記載のペプチド結合体。
【請求項6】
Xが、Gly8-GLP-1-(7-36)(ヒト)-NH2、Gly8-GLP-1-(7-36)(ヒト)、Gly8Lys37(パルミトイル)-GLP-1-(7-36)(ヒト)、Gly8Lys34(パルミトイル)-GLP-1-(7-36)(ヒト)、desSer39-エキセンジン-4(1-39)、エキセンジン-4(1-39)、desPro36-エキセンジン-4(1-39)、desAla35-エキセンジン-4(1-39)、desGly34-エキセンジン-4(1-39)、desSer39-(Lys40(パルミトイル))エキセンジン-4(1-39)、desGly34-(Lys40(パルミトイル))エキセンジン-4(1-39)、desAla35-(Lys40(パルミトイル))エキセンジン-4(1-39)、desPro36-(Lys40(パルミトイル))エキセンジン-4(1-39)およびLys40(パルミトイル)エキセンジン-4(1-39)、ならびにその遊離酸またはC末端第1級アミドおよび薬学的に許容される塩からなる群より選択される、請求項1〜5のいずれか1項に記載のペプチド結合体。
【請求項7】
ペプチドXが、8位のアラニンのグリシンへの置換および26位、34位または37位のリジンへの親油性パルミトイル基の結合からなる群より選択される少なくとも1個の改変を有するGLP-1(7-36)-NH2、ならびにその遊離酸および薬学的に許容される塩からなる群より選択される、請求項1〜5のいずれか1項に記載のペプチド結合体。
【請求項8】
親油性置換基が、GLP-1のLysのεアミノ基に結合している、請求項7に記載のペプチド結合体。
【請求項9】
ペプチドXが、エキセンジン-4のアミノ酸配列から36〜38位のPro残基の1個、2個または3個が欠失したアミノ酸配列を有するエキセンジン変異体からなる群より選択され、好ましくはXが、エキセンジン-4(1-39)のアミノ酸配列から36〜38位のPro残基の1個、2個または3個が欠失したアミノ酸配列を有するエキセンジン変異体、その遊離酸またはC末端第1級アミドおよび薬学的に許容される塩からなる群より選択される、請求項1〜5のいずれか1項に記載のペプチド結合体。
【請求項10】
Xが、
desPro36-エキセンジン-4(1-39)-NH2、
desPro36,Pro37-エキセンジン-4(1-39)-NH2、および
desPro36,Pro37,Pro38-エキセンジン-4(1-39)-NH2、
からなる群より選択される、請求項9に記載のペプチド結合体。
【請求項11】
Xがペプチド結合を介してZに結合していることを特徴とする、請求項1〜10のいずれか1項に記載のペプチド結合体。
【請求項12】
ZがXのC末端カルボニル基に共有結合しているか、またはZが該変異体のN末端窒素原子に共有結合しているか、または第1配列(Z)が該変異体のC末端カルボニル基に共有結合し、かつ第2配列(Z)がXのN末端窒素原子に共有結合しているか、またはZが該変異体のリジン、アルギニンもしくはヒスチジン残基の側鎖にある窒素原子もしくはグルタミン酸もしくはアスパラギン酸の側鎖にあるカルボニル基に共有結合している、請求項1に記載のペプチド結合体。
【請求項13】
Zが、4〜15個、好ましくは4〜10個、より好ましくは4〜7個、最も好ましくは6〜7個のアミノ酸単位からなる、請求項1〜12のいずれか1項に記載のペプチド結合体。
【請求項14】
ペプチド配列(Z)の全体の電荷が、pH7で0〜+15の範囲、好ましくは0〜+10の範囲、例えば0〜+8の範囲など、より好ましくは0〜+7または0〜+6の範囲である、請求項1〜13のいずれか1項に記載のペプチド結合体。
【請求項15】
Z中の各アミノ酸単位が、互いに独立に、Ser、Thr、Tyr、Asn、Gln、Asp、Glu、Lys、Arg、His、Orn、2,4-ジアミノブタン酸、2,3-ジアミノプロパン酸およびMetからなる群より選択される、請求項1〜14のいずれか1項に記載のペプチド結合体。
【請求項16】
Zが(Lys)n(式中、nは4〜15の整数、好ましくは4〜10の整数、例えば4〜8の整数など、例えば4〜7の整数である)である、請求項1〜15のいずれか1項に記載のペプチド結合体。
【請求項17】
ZがLys6またはLys7である、請求項1〜16のいずれか1項に記載のペプチド結合体。
【請求項18】
ZがXのC末端に結合したLys6である、請求項1に記載のペプチド結合体。
【請求項19】
Zがペプチド結合を介してXのN末端に結合しており、アミノ酸残基が、Asn、Asp、GluおよびGlnからなる群、好ましくはAsnおよびGluからなる群より選択され、好ましくはZがAsn-(Glu)n(式中、nは3〜7の整数、好ましくは5である)からなる群より選択されるアミノ酸配列を有する、請求項1に記載のペプチド結合体。
【請求項20】
Zが、
〔ここで、各Xaaは、互いに独立に、Ala、Leu、Ser、Thr、Tyr、Asn、Gln、Asp、Glu、Arg、His、Met、Orn、および式I
-NH-C(R1)(R2)-C(=O)- (I)
(式中、R1およびR2は、水素、C1-6-アルキル、フェニルおよびフェニル-メチルからなる群より選択され、C1-6-アルキルは、ハロゲン、ヒドロキシ、アミノ、シアノ、ニトロ、スルホノおよびカルボキシからなる群より選択される1〜3個の置換基で置換されていてもよく、フェニルおよびフェニル-メチルは、C1-6-アルキル、C2-6-アルケニル、ハロゲン、ヒドロキシ、アミノ、シアノ、ニトロ、スルホノおよびカルボキシからなる群より選択される1〜3個の置換基で置換されていてもよく、あるいはR1とR2は、それらが結合している炭素原子と一緒になって、シクロペンチル、シクロヘキシルまたはシクロヘプチル環を形成している。)
で表されるアミノ酸(例えば2,4-ジアミノブタン酸(Dbu)および2,3-ジアミノプロパン酸(Dpr))からなる群より選択されるものである。〕
である、請求項13に記載のペプチド結合体。
【請求項21】
アミノ酸残基がL-配置である、請求項1〜20のいずれか1項に記載のペプチド結合体。
【請求項22】
ペプチド結合体が、
desSer39-エキセンジン-4(1-39)-Lys6-NH2(配列番号91)、
desPro36-エキセンジン-4(1-39)-Lys6-NH2(配列番号93)、
desAla35-エキセンジン-4(1-39)-Lys6-NH2(配列番号94)、
desGly34-エキセンジン-4(1-39)-Lys6-NH2(配列番号95)、
desSer39-(Lys40(パルミトイル))エキセンジン-4(1-39)-Lys7-NH2(配列番号96)、
desGly34-(Lys40(パルミトイル))エキセンジン-4(1-39)-Lys7-NH2(配列番号97)、
desAla35-(Lys40(パルミトイル))エキセンジン-4(1-39)-Lys7-NH2(配列番号98)、
desPro36-(Lys40(パルミトイル))エキセンジン-4(1-39)-Lys7-NH2(配列番号99)、
Lys40(パルミトイル)エキセンジン-4(1-39)-Lys7-NH2(配列番号100)、
desPro36,Pro37-エキセンジン-4(1-39)-Lys6-NH2、
Lys6-desPro36,Pro37,Pro38-エキセンジン-4(1-39)-NH2、
Asn(Glu)5-desPro36,Pro37,Pro38-エキセンジン-4(1-39)-NH2、
Lys6-desPro36,Pro37,Pro38-エキセンジン-4(1-39)-Lys6-NH2、
Asn(Glu)5-desPro36,Pro37,Pro38-エキセンジン-4(1-39)-Lys6-NH2、
desPro36,Pro37,Pro38-エキセンジン-4(1-39)-Lys6-NH2、
Gly8-GLP-1(7-36)-Lys6-NH2(配列番号88)、
Lys6-Gly8-GLP-1(7-36)-Lys6-NH2 、
Lys6-Gly8-GLP-1(7-36)-NH2 、
(Gly8,Lys37(パルミトイル)-GLP-1(7-36)(ヒト)-Lys7-NH2(配列番号89)、
(Gly8,Lys26(パルミトイル)-GLP-1(7-36)(ヒト)-Lys6-NH2 、
Gly8,Lys34(パルミトイル)-GLP-1(7-36)(ヒト)-Lys6-NH2 、
Gly8-GLP-1(7-36)-Lys8-NH2 、
Gly8-GLP-1(7-36)-Lys10-NH2 、
Gly8-GLP-1(7-37)-Lys6-NH2 、ならびにその遊離酸およびその薬学的に許容される塩、
からなる群より選択される、請求項1に記載のペプチド結合体。
【請求項23】
GLP-1活性および/またはエキセンジン-4活性のペプチドアゴニストであるXを含む新規ペプチド結合体であって、Xが、
desPro36-エキセンジン-4(1-39)-NH2(配列番号101)、
desPro36-desPro37-エキセンジン-4(1-39)-NH2 、
desPro36-desPro37-desPro38-エキセンジン-4(1-39)-NH2 、
desAla35-エキセンジン-4(1-39)-NH2(配列番号105)、
desGly34-エキセンジン-4(1-39)-NH2(配列番号106)、
desGly34-(Lys40(パルミトイル))エキセンジン-4(1-39)-NH2(配列番号108)、
desAla35-(Lys40(パルミトイル))エキセンジン-4(1-39)-NH2(配列番号109)、
desPro36-(Lys40(パルミトイル))エキセンジン-4(1-39)-NH2(配列番号110)、
Gly8-Glp-1(7-36)-NH2 、およびGly8-Glp-1(7-36)、
からなる群より選択され、かつXがペプチド結合を介して(Lys)n(式中、nは4〜8の整数であり、好ましくはnは6である)からなる群より選択されるペプチド配列ZにC末端により結合している、前記ペプチド結合体、ならびにその遊離酸およびその薬学的に許容される塩。
【請求項24】
エキセンジン-4(1-39)-Lys6-NH2(配列番号92)ならびにその遊離酸およびその薬学的に許容される塩である、ペプチド結合体。
【請求項25】
治療に使用するための、請求項1〜24のいずれか1項に記載の薬学的に活性な結合体。
【請求項26】
請求項1〜24のいずれか1項に記載の薬学的に活性なペプチド結合体および製薬上許容される担体を含む、医薬組成物。
【請求項27】
活性ペプチド結合体が、
desPro36-エキセンジン-4(1-39)-Lys6-NH2(配列番号93)、
Lys6-desPro36,Pro37,Pro38-エキセンジン-4(1-39)-NH2 、
Asn(Glu)5-desPro36,Pro37,Pro38-エキセンジン-4(1-39)-NH2、
Asn(Glu)5-desPro36,Pro37,Pro38-エキセンジン-4(1-39)-Lys6-NH2、
desPro36,Pro37,Pro38-エキセンジン-4(1-39)-Lys6-NH2、ならびにその遊離酸および薬学的に許容される塩、
からなる群より選択される、請求項25または26に記載の医薬組成物。
【請求項28】
活性ペプチド結合体が、Gly8-GLP-1(7-36)-Lys6-NH2(配列番号88)、Gly8-GLP-1(7-36)-Lys7-NH2 、ならびにその遊離酸および薬学的に許容される塩からなる群より選択される、請求項25または26に記載の医薬組成物。
【請求項29】
親エキセンジンの変異体であって、該親エキセンジンは、エキセンジン-4に対して少なくとも90%の相同性を有するアミノ酸配列を有し、該変異体は、血糖値を降下させ、かつ哺乳動物のGLP-1受容体に結合し、かつ
(a)34〜38位の1〜5個の欠失、および
(b)εアミノ基にアミド結合を介して親油性置換基が結合している40位のLys、
からなる群より選択される少なくとも1個の改変を有する、前記変異体。
【請求項30】
親エキセンジンがエキセンジン-4である、請求項29に記載の変異体。
【請求項31】
エキセンジン-4がエキセンジン-4(1-39)である、請求項30に記載の変異体。
【請求項32】
該変異体が、εアミノ基にアミド結合を介してパルミトイル基が結合している40位のLysを含む、請求項29〜31のいずれか1項に記載の変異体。
【請求項33】
変異体が、desPro36-エキセンジン-4(1-39)-NH2、desPro36,Pro37,Pro38-エキセンジン-4(1-39)-NH2 、desPro36,Pro37-エキセンジン-4(1-39)-NH2 、desAla35-エキセンジン-4(1-39)-NH2 、desGly34-エキセンジン-4(1-39)-NH2 、desSer39-(Lys40(パルミトイル)エキセンジン-4(1-39)-NH2 、desGly34-(Lys40(パルミトイル))エキセンジン-4(1-39)-NH2 、desAla35-(Lys40(パルミトイル)エキセンジン-4(1-39)-NH2 、ならびにその遊離酸およびその薬学的に許容される塩からなる群より選択される、請求項29〜32のいずれか1項に記載の変異体。
【請求項34】
請求項29〜33のいずれか1項に記載の親エキセンジンの変異体および生理学的に許容される担体を含む、医薬組成物。
【請求項35】
変異体が、desPro36-エキセンジン-4(1-39)-NH2、ならびにその遊離酸およびその薬学的に許容される塩からなる群より選択される、請求項34に記載の医薬組成物。
【請求項36】
治療に使用するための請求項35に記載の組成物。
【請求項37】
請求項1〜24のいずれか1項に記載のペプチド結合体または天然ポリペプチド配列を有する請求項29〜33のいずれか1項に記載のエキセンジン変異体を製造する方法であって、
f) 該エキセンジン変異体または該ペプチド結合体のペプチド配列を含むポリペプチド配列をコードする核酸配列および選択マーカーを含有する核酸構築物またはベクターを、宿主細胞に導入することによって組換え宿主細胞を得て、
g) 該組換え宿主細胞を選別し、
h) 該組換え宿主細胞を、該ポリペプチド配列の産生を可能にする条件下で培養し、
i) 該ポリペプチド配列を培養物から分離し、そして
j) 場合によって、適当なプロテアーゼを用いて該ポリペプチド配列を切断することによって該ペプチド結合体を得ること、
を含む前記方法。
【請求項38】
天然ポリペプチド配列を有する、請求項1〜37のいずれか1項に記載のペプチド結合体をコードするポリペプチド配列。
【請求項39】
医薬組成物の製造のための、請求項1〜24のいずれか1項に記載のペプチド結合体または請求項29〜33のいずれか1項に記載のエキセンジン変異体の使用。
【請求項40】
I型糖尿病またはII型糖尿病、インスリン抵抗性症候群、肥満、摂食障害、高血糖症、代謝異常および胃の疾患の治療に使用する医薬組成物を製造するための、請求項1〜24のいずれか1項に記載の薬学的に活性なペプチド結合体または請求項29〜33のいずれか1項に記載のエキセンジン変異体の使用。
【請求項41】
血糖値を上昇させることが知られているホルモン、例えばアドレナリン、グルココルチコイド、成長ホルモンおよびグルカゴンを含むカテコールアミンによって誘発された血糖値の上昇に関連した疾患状態の治療に使用する医薬組成物を製造するための、請求項1〜24のいずれか1項に記載の薬学的に活性なペプチド結合体または請求項29〜33のいずれか1項に記載のエキセンジン変異体の使用。
【請求項42】
血糖値の調節に使用する医薬組成物を製造するための、請求項1〜24のいずれか1項に記載の薬学的に活性なペプチド結合体または請求項29〜33のいずれか1項に記載のエキセンジン変異体の使用。
【請求項43】
胃内容排出の調節に使用する医薬組成物を製造するための、請求項1〜24のいずれか1項に記載の薬学的に活性なペプチド結合体または請求項29〜33のいずれか1項に記載のエキセンジン変異体の使用。
【請求項44】
哺乳動物においてインスリン放出を刺激する方法であって、インスリン分泌刺激に有効な量の請求項1〜24のいずれか1項に記載のペプチド結合体または請求項29〜33のいずれか1項に記載のエキセンジン変異体を投与することを含む、前記方法。
【請求項45】
哺乳動物において血糖値を降下させる方法であって、該哺乳動物の血糖値を降下させるのに有効な量の請求項1〜24のいずれか1項に記載のペプチド結合体または請求項29〜33のいずれか1項に記載のエキセンジン変異体を投与することを含む、前記方法。
【請求項46】
哺乳動物において血漿脂質レベルを降下させる方法であって、該哺乳動物の血漿脂質レベルを降下させるのに有効な量の請求項1〜24のいずれか1項に記載のペプチド結合体または請求項29〜33のいずれか1項に記載のエキセンジン変異体を投与することを含む、前記方法。
【請求項47】
哺乳動物において心筋梗塞後の死亡率および罹患率を低下させる方法であって、心筋梗塞後の死亡率および罹患率を低下させるのに有効な量の請求項1〜24のいずれか1項に記載のペプチド結合体または請求項29〜33のいずれか1項に記載のエキセンジン変異体を投与することを含む、前記方法。
【請求項48】
エキセンジン4およびエキセンジン4変異体の血漿内半減期を増大させる方法であって、好ましくは6個のリジン残基からなるポリペプチド配列を、ペプチド結合を介して該エキセンジン4またはエキセンジン4変異体のC末端に連結させることを含む、前記方法。
【請求項1】
(a) エキセンジン-4に対して少なくとも90%の相同性を有するエキセンジン、
(b) 34〜39位の1〜5個の欠失からなる群より選択される改変を有し、かつ40位に親油性置換基を有するLysを有する、該エキセンジンの変異体、または
(c) (i) 8位のアラニンの、D-アラニン、グリシンもしくはα-アミノイソ酪酸への置換、および
(ii) 親油性置換基
からなる群より選択される少なくとも1個の改変を有するGLP-1(7-36)またはGLP-1(7-37)、
からなる群より選択されるペプチドX(ただし、Xはエキセンジン-4またはエキセンジン-3ではない)と、
4〜20個のアミノ酸単位からなり、各アミノ酸単位が、Ala、Leu、Ser、Thr、Tyr、Asn、Gln、Asp、Glu、Lys、Arg、His、Met、Ornおよび一般式I
-NH-C(R1)(R2)-C(=O)- (I)
(式中、R1およびR2は、水素、C1-6-アルキル、フェニルおよびフェニル-メチルからなる群より選択され、C1-6-アルキルは、ハロゲン、ヒドロキシ、アミノ、シアノ、ニトロ、スルホノおよびカルボキシからなる群より選択される1〜3個の置換基で置換されていてもよく、フェニルおよびフェニル-メチルは、C1-6-アルキル、C2-6-アルケニル、ハロゲン、ヒドロキシ、アミノ、シアノ、ニトロ、スルホノおよびカルボキシからなる群より選択される1〜3個の置換基で置換されていてもよく、あるいはR1とR2は、それらが結合している炭素原子と一緒になって、シクロペンチル、シクロヘキシルまたはシクロヘプチル環を形成している。)
で表されるアミノ酸単位(例えば2,4-ジアミノブタン酸および2,3-ジアミノプロパン酸)からなる群より選択される、前記変異体に共有結合したペプチド配列Zと、
を含むペプチド結合体、ならびに該ペプチド結合体の薬学的に許容される塩またはC末端アミド。
【請求項2】
ペプチドXが、糖尿病の哺乳動物におけるグルコース耐性の改善に有効であることをさらに特徴とする、請求項1に記載のペプチド結合体。
【請求項3】
Zが、少なくとも1個のLysアミノ酸単位、好ましくは少なくとも2個のLysアミノ酸単位、例えば少なくとも3個のLysアミノ酸単位など、例えば少なくとも4個のLysアミノ酸単位、より好ましくは少なくとも5個のLysアミノ酸単位、例えば6個または7個のLysアミノ酸単位など、を含む請求項1または2に記載のペプチド結合体。
【請求項4】
ペプチド結合体の最小有効量と、ペプチドXの経口最小有効量の比が少なくとも1:5であることを特徴とする、請求項1〜3のいずれか1項に記載のペプチド結合体。
【請求項5】
親油性基が、ペプチドXのLysのεアミノ基に結合したパルミトイルである、請求項1〜4のいずれか1項に記載のペプチド結合体。
【請求項6】
Xが、Gly8-GLP-1-(7-36)(ヒト)-NH2、Gly8-GLP-1-(7-36)(ヒト)、Gly8Lys37(パルミトイル)-GLP-1-(7-36)(ヒト)、Gly8Lys34(パルミトイル)-GLP-1-(7-36)(ヒト)、desSer39-エキセンジン-4(1-39)、エキセンジン-4(1-39)、desPro36-エキセンジン-4(1-39)、desAla35-エキセンジン-4(1-39)、desGly34-エキセンジン-4(1-39)、desSer39-(Lys40(パルミトイル))エキセンジン-4(1-39)、desGly34-(Lys40(パルミトイル))エキセンジン-4(1-39)、desAla35-(Lys40(パルミトイル))エキセンジン-4(1-39)、desPro36-(Lys40(パルミトイル))エキセンジン-4(1-39)およびLys40(パルミトイル)エキセンジン-4(1-39)、ならびにその遊離酸またはC末端第1級アミドおよび薬学的に許容される塩からなる群より選択される、請求項1〜5のいずれか1項に記載のペプチド結合体。
【請求項7】
ペプチドXが、8位のアラニンのグリシンへの置換および26位、34位または37位のリジンへの親油性パルミトイル基の結合からなる群より選択される少なくとも1個の改変を有するGLP-1(7-36)-NH2、ならびにその遊離酸および薬学的に許容される塩からなる群より選択される、請求項1〜5のいずれか1項に記載のペプチド結合体。
【請求項8】
親油性置換基が、GLP-1のLysのεアミノ基に結合している、請求項7に記載のペプチド結合体。
【請求項9】
ペプチドXが、エキセンジン-4のアミノ酸配列から36〜38位のPro残基の1個、2個または3個が欠失したアミノ酸配列を有するエキセンジン変異体からなる群より選択され、好ましくはXが、エキセンジン-4(1-39)のアミノ酸配列から36〜38位のPro残基の1個、2個または3個が欠失したアミノ酸配列を有するエキセンジン変異体、その遊離酸またはC末端第1級アミドおよび薬学的に許容される塩からなる群より選択される、請求項1〜5のいずれか1項に記載のペプチド結合体。
【請求項10】
Xが、
desPro36-エキセンジン-4(1-39)-NH2、
desPro36,Pro37-エキセンジン-4(1-39)-NH2、および
desPro36,Pro37,Pro38-エキセンジン-4(1-39)-NH2、
からなる群より選択される、請求項9に記載のペプチド結合体。
【請求項11】
Xがペプチド結合を介してZに結合していることを特徴とする、請求項1〜10のいずれか1項に記載のペプチド結合体。
【請求項12】
ZがXのC末端カルボニル基に共有結合しているか、またはZが該変異体のN末端窒素原子に共有結合しているか、または第1配列(Z)が該変異体のC末端カルボニル基に共有結合し、かつ第2配列(Z)がXのN末端窒素原子に共有結合しているか、またはZが該変異体のリジン、アルギニンもしくはヒスチジン残基の側鎖にある窒素原子もしくはグルタミン酸もしくはアスパラギン酸の側鎖にあるカルボニル基に共有結合している、請求項1に記載のペプチド結合体。
【請求項13】
Zが、4〜15個、好ましくは4〜10個、より好ましくは4〜7個、最も好ましくは6〜7個のアミノ酸単位からなる、請求項1〜12のいずれか1項に記載のペプチド結合体。
【請求項14】
ペプチド配列(Z)の全体の電荷が、pH7で0〜+15の範囲、好ましくは0〜+10の範囲、例えば0〜+8の範囲など、より好ましくは0〜+7または0〜+6の範囲である、請求項1〜13のいずれか1項に記載のペプチド結合体。
【請求項15】
Z中の各アミノ酸単位が、互いに独立に、Ser、Thr、Tyr、Asn、Gln、Asp、Glu、Lys、Arg、His、Orn、2,4-ジアミノブタン酸、2,3-ジアミノプロパン酸およびMetからなる群より選択される、請求項1〜14のいずれか1項に記載のペプチド結合体。
【請求項16】
Zが(Lys)n(式中、nは4〜15の整数、好ましくは4〜10の整数、例えば4〜8の整数など、例えば4〜7の整数である)である、請求項1〜15のいずれか1項に記載のペプチド結合体。
【請求項17】
ZがLys6またはLys7である、請求項1〜16のいずれか1項に記載のペプチド結合体。
【請求項18】
ZがXのC末端に結合したLys6である、請求項1に記載のペプチド結合体。
【請求項19】
Zがペプチド結合を介してXのN末端に結合しており、アミノ酸残基が、Asn、Asp、GluおよびGlnからなる群、好ましくはAsnおよびGluからなる群より選択され、好ましくはZがAsn-(Glu)n(式中、nは3〜7の整数、好ましくは5である)からなる群より選択されるアミノ酸配列を有する、請求項1に記載のペプチド結合体。
【請求項20】
Zが、
〔ここで、各Xaaは、互いに独立に、Ala、Leu、Ser、Thr、Tyr、Asn、Gln、Asp、Glu、Arg、His、Met、Orn、および式I
-NH-C(R1)(R2)-C(=O)- (I)
(式中、R1およびR2は、水素、C1-6-アルキル、フェニルおよびフェニル-メチルからなる群より選択され、C1-6-アルキルは、ハロゲン、ヒドロキシ、アミノ、シアノ、ニトロ、スルホノおよびカルボキシからなる群より選択される1〜3個の置換基で置換されていてもよく、フェニルおよびフェニル-メチルは、C1-6-アルキル、C2-6-アルケニル、ハロゲン、ヒドロキシ、アミノ、シアノ、ニトロ、スルホノおよびカルボキシからなる群より選択される1〜3個の置換基で置換されていてもよく、あるいはR1とR2は、それらが結合している炭素原子と一緒になって、シクロペンチル、シクロヘキシルまたはシクロヘプチル環を形成している。)
で表されるアミノ酸(例えば2,4-ジアミノブタン酸(Dbu)および2,3-ジアミノプロパン酸(Dpr))からなる群より選択されるものである。〕
である、請求項13に記載のペプチド結合体。
【請求項21】
アミノ酸残基がL-配置である、請求項1〜20のいずれか1項に記載のペプチド結合体。
【請求項22】
ペプチド結合体が、
desSer39-エキセンジン-4(1-39)-Lys6-NH2(配列番号91)、
desPro36-エキセンジン-4(1-39)-Lys6-NH2(配列番号93)、
desAla35-エキセンジン-4(1-39)-Lys6-NH2(配列番号94)、
desGly34-エキセンジン-4(1-39)-Lys6-NH2(配列番号95)、
desSer39-(Lys40(パルミトイル))エキセンジン-4(1-39)-Lys7-NH2(配列番号96)、
desGly34-(Lys40(パルミトイル))エキセンジン-4(1-39)-Lys7-NH2(配列番号97)、
desAla35-(Lys40(パルミトイル))エキセンジン-4(1-39)-Lys7-NH2(配列番号98)、
desPro36-(Lys40(パルミトイル))エキセンジン-4(1-39)-Lys7-NH2(配列番号99)、
Lys40(パルミトイル)エキセンジン-4(1-39)-Lys7-NH2(配列番号100)、
desPro36,Pro37-エキセンジン-4(1-39)-Lys6-NH2、
Lys6-desPro36,Pro37,Pro38-エキセンジン-4(1-39)-NH2、
Asn(Glu)5-desPro36,Pro37,Pro38-エキセンジン-4(1-39)-NH2、
Lys6-desPro36,Pro37,Pro38-エキセンジン-4(1-39)-Lys6-NH2、
Asn(Glu)5-desPro36,Pro37,Pro38-エキセンジン-4(1-39)-Lys6-NH2、
desPro36,Pro37,Pro38-エキセンジン-4(1-39)-Lys6-NH2、
Gly8-GLP-1(7-36)-Lys6-NH2(配列番号88)、
Lys6-Gly8-GLP-1(7-36)-Lys6-NH2 、
Lys6-Gly8-GLP-1(7-36)-NH2 、
(Gly8,Lys37(パルミトイル)-GLP-1(7-36)(ヒト)-Lys7-NH2(配列番号89)、
(Gly8,Lys26(パルミトイル)-GLP-1(7-36)(ヒト)-Lys6-NH2 、
Gly8,Lys34(パルミトイル)-GLP-1(7-36)(ヒト)-Lys6-NH2 、
Gly8-GLP-1(7-36)-Lys8-NH2 、
Gly8-GLP-1(7-36)-Lys10-NH2 、
Gly8-GLP-1(7-37)-Lys6-NH2 、ならびにその遊離酸およびその薬学的に許容される塩、
からなる群より選択される、請求項1に記載のペプチド結合体。
【請求項23】
GLP-1活性および/またはエキセンジン-4活性のペプチドアゴニストであるXを含む新規ペプチド結合体であって、Xが、
desPro36-エキセンジン-4(1-39)-NH2(配列番号101)、
desPro36-desPro37-エキセンジン-4(1-39)-NH2 、
desPro36-desPro37-desPro38-エキセンジン-4(1-39)-NH2 、
desAla35-エキセンジン-4(1-39)-NH2(配列番号105)、
desGly34-エキセンジン-4(1-39)-NH2(配列番号106)、
desGly34-(Lys40(パルミトイル))エキセンジン-4(1-39)-NH2(配列番号108)、
desAla35-(Lys40(パルミトイル))エキセンジン-4(1-39)-NH2(配列番号109)、
desPro36-(Lys40(パルミトイル))エキセンジン-4(1-39)-NH2(配列番号110)、
Gly8-Glp-1(7-36)-NH2 、およびGly8-Glp-1(7-36)、
からなる群より選択され、かつXがペプチド結合を介して(Lys)n(式中、nは4〜8の整数であり、好ましくはnは6である)からなる群より選択されるペプチド配列ZにC末端により結合している、前記ペプチド結合体、ならびにその遊離酸およびその薬学的に許容される塩。
【請求項24】
エキセンジン-4(1-39)-Lys6-NH2(配列番号92)ならびにその遊離酸およびその薬学的に許容される塩である、ペプチド結合体。
【請求項25】
治療に使用するための、請求項1〜24のいずれか1項に記載の薬学的に活性な結合体。
【請求項26】
請求項1〜24のいずれか1項に記載の薬学的に活性なペプチド結合体および製薬上許容される担体を含む、医薬組成物。
【請求項27】
活性ペプチド結合体が、
desPro36-エキセンジン-4(1-39)-Lys6-NH2(配列番号93)、
Lys6-desPro36,Pro37,Pro38-エキセンジン-4(1-39)-NH2 、
Asn(Glu)5-desPro36,Pro37,Pro38-エキセンジン-4(1-39)-NH2、
Asn(Glu)5-desPro36,Pro37,Pro38-エキセンジン-4(1-39)-Lys6-NH2、
desPro36,Pro37,Pro38-エキセンジン-4(1-39)-Lys6-NH2、ならびにその遊離酸および薬学的に許容される塩、
からなる群より選択される、請求項25または26に記載の医薬組成物。
【請求項28】
活性ペプチド結合体が、Gly8-GLP-1(7-36)-Lys6-NH2(配列番号88)、Gly8-GLP-1(7-36)-Lys7-NH2 、ならびにその遊離酸および薬学的に許容される塩からなる群より選択される、請求項25または26に記載の医薬組成物。
【請求項29】
親エキセンジンの変異体であって、該親エキセンジンは、エキセンジン-4に対して少なくとも90%の相同性を有するアミノ酸配列を有し、該変異体は、血糖値を降下させ、かつ哺乳動物のGLP-1受容体に結合し、かつ
(a)34〜38位の1〜5個の欠失、および
(b)εアミノ基にアミド結合を介して親油性置換基が結合している40位のLys、
からなる群より選択される少なくとも1個の改変を有する、前記変異体。
【請求項30】
親エキセンジンがエキセンジン-4である、請求項29に記載の変異体。
【請求項31】
エキセンジン-4がエキセンジン-4(1-39)である、請求項30に記載の変異体。
【請求項32】
該変異体が、εアミノ基にアミド結合を介してパルミトイル基が結合している40位のLysを含む、請求項29〜31のいずれか1項に記載の変異体。
【請求項33】
変異体が、desPro36-エキセンジン-4(1-39)-NH2、desPro36,Pro37,Pro38-エキセンジン-4(1-39)-NH2 、desPro36,Pro37-エキセンジン-4(1-39)-NH2 、desAla35-エキセンジン-4(1-39)-NH2 、desGly34-エキセンジン-4(1-39)-NH2 、desSer39-(Lys40(パルミトイル)エキセンジン-4(1-39)-NH2 、desGly34-(Lys40(パルミトイル))エキセンジン-4(1-39)-NH2 、desAla35-(Lys40(パルミトイル)エキセンジン-4(1-39)-NH2 、ならびにその遊離酸およびその薬学的に許容される塩からなる群より選択される、請求項29〜32のいずれか1項に記載の変異体。
【請求項34】
請求項29〜33のいずれか1項に記載の親エキセンジンの変異体および生理学的に許容される担体を含む、医薬組成物。
【請求項35】
変異体が、desPro36-エキセンジン-4(1-39)-NH2、ならびにその遊離酸およびその薬学的に許容される塩からなる群より選択される、請求項34に記載の医薬組成物。
【請求項36】
治療に使用するための請求項35に記載の組成物。
【請求項37】
請求項1〜24のいずれか1項に記載のペプチド結合体または天然ポリペプチド配列を有する請求項29〜33のいずれか1項に記載のエキセンジン変異体を製造する方法であって、
f) 該エキセンジン変異体または該ペプチド結合体のペプチド配列を含むポリペプチド配列をコードする核酸配列および選択マーカーを含有する核酸構築物またはベクターを、宿主細胞に導入することによって組換え宿主細胞を得て、
g) 該組換え宿主細胞を選別し、
h) 該組換え宿主細胞を、該ポリペプチド配列の産生を可能にする条件下で培養し、
i) 該ポリペプチド配列を培養物から分離し、そして
j) 場合によって、適当なプロテアーゼを用いて該ポリペプチド配列を切断することによって該ペプチド結合体を得ること、
を含む前記方法。
【請求項38】
天然ポリペプチド配列を有する、請求項1〜37のいずれか1項に記載のペプチド結合体をコードするポリペプチド配列。
【請求項39】
医薬組成物の製造のための、請求項1〜24のいずれか1項に記載のペプチド結合体または請求項29〜33のいずれか1項に記載のエキセンジン変異体の使用。
【請求項40】
I型糖尿病またはII型糖尿病、インスリン抵抗性症候群、肥満、摂食障害、高血糖症、代謝異常および胃の疾患の治療に使用する医薬組成物を製造するための、請求項1〜24のいずれか1項に記載の薬学的に活性なペプチド結合体または請求項29〜33のいずれか1項に記載のエキセンジン変異体の使用。
【請求項41】
血糖値を上昇させることが知られているホルモン、例えばアドレナリン、グルココルチコイド、成長ホルモンおよびグルカゴンを含むカテコールアミンによって誘発された血糖値の上昇に関連した疾患状態の治療に使用する医薬組成物を製造するための、請求項1〜24のいずれか1項に記載の薬学的に活性なペプチド結合体または請求項29〜33のいずれか1項に記載のエキセンジン変異体の使用。
【請求項42】
血糖値の調節に使用する医薬組成物を製造するための、請求項1〜24のいずれか1項に記載の薬学的に活性なペプチド結合体または請求項29〜33のいずれか1項に記載のエキセンジン変異体の使用。
【請求項43】
胃内容排出の調節に使用する医薬組成物を製造するための、請求項1〜24のいずれか1項に記載の薬学的に活性なペプチド結合体または請求項29〜33のいずれか1項に記載のエキセンジン変異体の使用。
【請求項44】
哺乳動物においてインスリン放出を刺激する方法であって、インスリン分泌刺激に有効な量の請求項1〜24のいずれか1項に記載のペプチド結合体または請求項29〜33のいずれか1項に記載のエキセンジン変異体を投与することを含む、前記方法。
【請求項45】
哺乳動物において血糖値を降下させる方法であって、該哺乳動物の血糖値を降下させるのに有効な量の請求項1〜24のいずれか1項に記載のペプチド結合体または請求項29〜33のいずれか1項に記載のエキセンジン変異体を投与することを含む、前記方法。
【請求項46】
哺乳動物において血漿脂質レベルを降下させる方法であって、該哺乳動物の血漿脂質レベルを降下させるのに有効な量の請求項1〜24のいずれか1項に記載のペプチド結合体または請求項29〜33のいずれか1項に記載のエキセンジン変異体を投与することを含む、前記方法。
【請求項47】
哺乳動物において心筋梗塞後の死亡率および罹患率を低下させる方法であって、心筋梗塞後の死亡率および罹患率を低下させるのに有効な量の請求項1〜24のいずれか1項に記載のペプチド結合体または請求項29〜33のいずれか1項に記載のエキセンジン変異体を投与することを含む、前記方法。
【請求項48】
エキセンジン4およびエキセンジン4変異体の血漿内半減期を増大させる方法であって、好ましくは6個のリジン残基からなるポリペプチド配列を、ペプチド結合を介して該エキセンジン4またはエキセンジン4変異体のC末端に連結させることを含む、前記方法。
【図1】

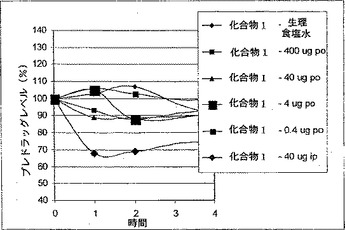
【図2】
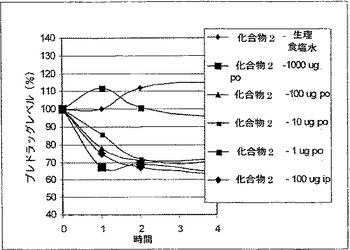
【図3】
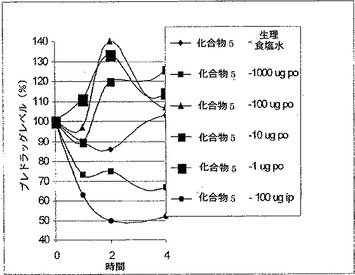
【図4】
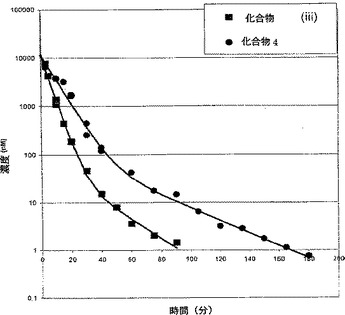
【図5】
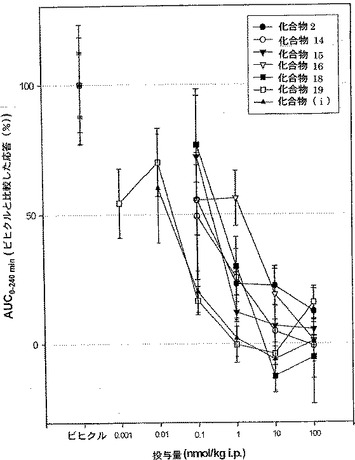
【図6】
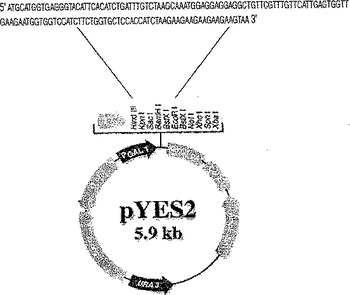
【図7】
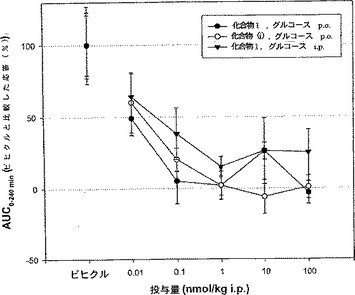
【図8】
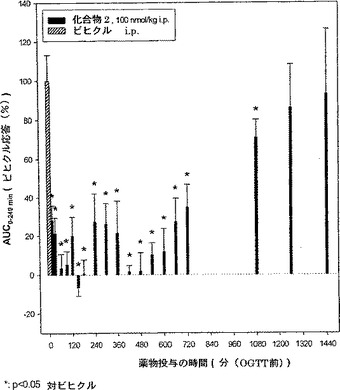
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公開番号】特開2007−1987(P2007−1987A)
【公開日】平成19年1月11日(2007.1.11)
【国際特許分類】
【出願番号】特願2006−243389(P2006−243389)
【出願日】平成18年9月7日(2006.9.7)
【分割の表示】特願2001−509765(P2001−509765)の分割
【原出願日】平成12年7月12日(2000.7.12)
【出願人】(502017216)ジーランド ファーマ エー/エス (2)
【Fターム(参考)】
【公開日】平成19年1月11日(2007.1.11)
【国際特許分類】
【出願日】平成18年9月7日(2006.9.7)
【分割の表示】特願2001−509765(P2001−509765)の分割
【原出願日】平成12年7月12日(2000.7.12)
【出願人】(502017216)ジーランド ファーマ エー/エス (2)
【Fターム(参考)】
[ Back to top ]