
表面分析装置及び表面分析方法
【課題】多価イオンを用いて、試料表面の分析を高精度で短時間に行うことができる表面分析装置を提供する。
【解決手段】表面分析装置1は、試料5を搭載する試料台6と、試料台6に搭載した試料5に価数が15以上の多価イオンビーム4を照射する多価イオン発生源3と、試料5に多価イオンビーム4を照射することにより生じる二次イオン7を検出する質量分析部8と、試料5に多価イオンビーム4を照射することにより生じる二次電子9を検出する二次電子検出部10と、二次電子検出部10からの二次電子検出信号を受け分析開始信号を生成し質量分析部へ送信する質量分析制御部12を、備えて構成される。
【解決手段】表面分析装置1は、試料5を搭載する試料台6と、試料台6に搭載した試料5に価数が15以上の多価イオンビーム4を照射する多価イオン発生源3と、試料5に多価イオンビーム4を照射することにより生じる二次イオン7を検出する質量分析部8と、試料5に多価イオンビーム4を照射することにより生じる二次電子9を検出する二次電子検出部10と、二次電子検出部10からの二次電子検出信号を受け分析開始信号を生成し質量分析部へ送信する質量分析制御部12を、備えて構成される。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、多価イオンを試料へのイオン源として用いた表面分析装置及び表面分析方法に関する。
【背景技術】
【0002】
原子から電子を取り除くと正のイオンができるが、例えばXe44+イオンのように、電子を2個以上取り去ったものを正の多価イオンという。多価イオンは極めて大きな内部エネルギーを持っており、多価イオンが固体表面と衝突すると、多数の二次電子が放出されたり(非特許文献1参照)、多価イオンの入射点まわりにナノメートルの大きさの構造変化が誘起されたりする(非特許文献2参照)など、数多くの特異的な現象が起こる。
この多価イオンと物質の特異な相互作用は、単一イオンインプランテーションや量子ドット作製などのナノメートル領域のプロセス技術に応用できる可能性があり、注目を集めている(非特許文献3参照)。
【0003】
このような多価イオンを生成するイオン源としては、電子サイクロトロン共鳴(ECR)型イオン源(ECRIS)と電子ビーム型イオン源(EBIS)とが知られている。後者は前者に比べて得られるイオンの電離度が高いという特徴がある。
【0004】
EBISとしては、原子物理学の研究用に開発された核融合研究所の装置が知られている(例えば、非特許文献4参照)。この装置は、電子源(カソード)、ドリフトチューブ、コレクタ、ソレノイド磁石、イオン引き出し用レンズなどから構成されている。カソードから出射した電子は、磁場中に配置されたドリフトチューブを通り、コレクタに捕集される。電子は、ドリフトチューブに形成される強磁場で圧縮され大電流密度の電子ビームとなっている。一方、カソード付近から導入された気体は、イオンに対して障壁となる、ドリフトチューブ内に形成される井戸型ポテンシャルによって、電子による衝突電離が進み、多価イオンとなる。
【0005】
さらに、1988年には、EBISを改良したEBIT(電子ビームイオントラップ)が開発された(非特許文献5参照)。このEBITの多価イオン発生原理はEBISと同じであるが、超伝導ヘルムホルツ型コイルを用い、ドリフトチューブを従来よりも短くして、ドリフトチューブ内のプラズマの不安定性を回避することにより、イオンの閉じ込め時間を改善し、高価数の多価イオンを安定に保持できる。このため、EBITでは、ドリフトチューブでの電子ビームの絞り込みを究極まで行い、高電離イオンを生成することを可能にした。
【0006】
また、EBITとして、ウラン(U)まで完全電離できるように、電子の加速電圧を最大300kVとした装置が本発明者らにより開発されている(非特許文献6参照。)。このEBITは、原子物理学の研究のために開発されたもので、生成可能な多価イオンの内部エネルギーとしては世界最高の性能を持っている。
【0007】
ところで、イオンを用いた分析方法としては、従来から1価のイオンをスパッタ源とする二次イオン質量分析装置(SIMS)が知られている。
【0008】
さらに、スパッタ源として低速の多価イオンを用いた表面分析の試みがなされている(非特許文献6参照。)。非特許文献6では、2keV〜5keVの低速で、価数が4〜12の多価イオンをSi表面に照射した場合のH+イオン(プロトン)の検出が報告されている。
【0009】
【非特許文献1】J. W. McDonald, D. Schneider, M. W. ClarkandD. DeWitt, Phys. Rev. Lett., Vol. 68, (1992), p.2297
【非特許文献2】T. Meguro et al., Appl. Phys. Lett., Vol.79, (2001), p.3866
【非特許文献3】T. Schenkel et al., Appl. Phys., Vol.94, (2003), p.7017
【非特許文献4】小林 信夫、大谷 俊介 他7 名、名古屋大学プラズマ研究所資料・技術報告、IPPJ−DT−84、1981年
【非特許文献5】MA. Levin他7名, Physica Scr., T22, (1988), p.157
【非特許文献6】大谷俊介、桜井誠、プラズマ・核融合学会誌、73,(1997),p.1063
【非特許文献7】山崎泰規、第52回応用物理学関係連合講演会、講演予稿集、29p−C−3(2005年春)
【発明の開示】
【発明が解決しようとする課題】
【0010】
従来の技術では、12価以下の多価イオンを用いた軽元素の分析手法は知られていたが、検出感度が低いものであり、固体等の表面に吸着した水素原子のような軽元素を分析することは困難であった。このため、軽元素の分析には、例えば12時間程の非常に長い時間を要していた。従って、例えば被測定試料を超高真空下に保持して測定を行った場合であっても、真空槽の残留ガスや真空容器の内壁からの脱出ガスが被測定試料へ再付着するので、正しい分析を行うことが困難であった。
【0011】
また、従来の1価のイオンをスパッタ源とする二次イオン質量分析装置により化合物からなる試料を分析する場合、試料の表面からスパッタされた粒子、すなわち二次イオンのイオン化率が違うので、化合物の化学両論的な組成が反映された二次イオン強度を得ることは困難であった。
【0012】
本発明は、上記課題に鑑み、高い多価数を有する多価イオンを用いて被測定物の表面または表面近傍に存在する原子や分子を効率良く検出することができる表面分析装置と表面分析方法を提供することを目的としている。
【課題を解決するための手段】
【0013】
上記目的を達成するため、本発明の表面分析装置は、試料を搭載する試料台と、試料台に搭載された試料に価数が15以上の多価イオンビームを照射する多価イオン発生源と、試料に多価イオンビームが照射されることにより生じる二次イオンを検出する質量分析部と、試料に多価イオンビームが照射されることにより生じる二次電子を検出する二次電子検出部と、二次電子検出部からの二次電子検出信号を受けて分析開始信号を生成し質量分析部へ送信する質量分析制御部を、備えている。
上記構成において、二次電子検出部は、好ましくは、電気的に独立した複数の二次電子検出器を有する。
二次電子検出器は、好ましくは、複数のチャンネルトロン又は複数のアノード電極を持つマイクロチャンネルプレートからなる。
多価イオン発生源は、好ましくは、一端に多価イオン発生源からの多価イオンビームが入射され他端から多価イオンビームが出射される多価イオンガイドを備えており、多価イオンガイドの他端部はキャピラリー構造である。
【0014】
上記構成によれば、試料の表面に15価以上の多価イオンビームを照射することで、他手法では分析が困難であった所謂質量の小さいH+等の元素を、高感度でかつ短時間で測定することができる。
【0015】
本発明の表面分析方法は、多価イオンビームを15価以上の価数で変化させて試料へ照射し、試料表面の特定の元素から生じる二次イオンのカウント数と価数との関係を算出して、特定の元素に対する多価イオンビームの価数を決定する。
上記構成において、好ましくは、試料が2以上の構成元素からなり、各構成元素から生じる二次イオンのカウント数と価数との関係を算出して、各構成元素の組成比を与える多価イオンの価数を決定する。
【0016】
上記構成によれば、試料の表面に検出対象となる元素に対して15価以上の最適な多価イオンビームを照射することで、他手法では分析が困難であった所謂、質量の小さいH+等の元素を、高感度でかつ短時間で測定することができる。
【発明の効果】
【0017】
本発明によれば、15価以上の多価イオンビームを試料に照射することにより試料から発生する二次イオンの信号は、価数の4乗等のべき数に比例して増大するために、試料表面の分析を短時間で分析することができる。このため、表面清浄化を施した試料の分析においても残留ガス等の再付着が起こらず高精度な分析を行うことができる。
【発明を実施するための最良の形態】
【0018】
以下、図面に示した実施形態に基づいて本発明を詳細に説明する。
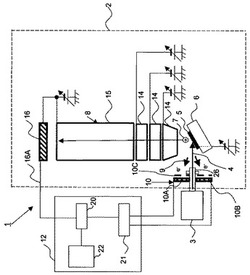
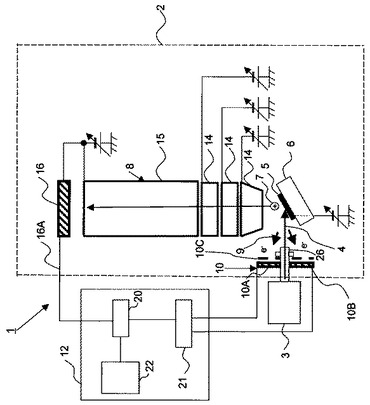
図1は、本発明による多価イオンを用いた表面分析装置1の構成を模式的に示す断面図である。
図1において、本発明の多価イオンを用いた表面分析装置1は、試料5を搭載する試料台6と、試料台6に搭載された試料5に価数が15以上の多価イオンビーム4を照射する多価イオン発生源3と、試料5に多価イオンビーム4が照射されることで生じる二次イオン7を検出する質量分析部8と、試料5に多価イオンビーム4が照射されることで生じる二次電子9を検出する二次電子検出部10と、二次電子検出部10からの二次電子検出信号を受け分析開始信号を生成し質量分析部8へ送信する質量分析制御部12を、を含んで構成されている。
ここで、真空容器2に多価イオン発生源3が接続され、この真空容器2内には試料台6と質量分析部8と二次電子検出部10とが配設され、質量分析制御部12は、真空容器2の外部に配設されている。真空容器2は、図示しない真空排気装置により後述する多価イオン発生源3の真空排気装置とは独立に真空排気されてもよい。
【0019】
本実施形態では、質量分析部8はTOF(飛行時間:Time of Flight)型の質量分析部8から構成されており、レンズ14と飛行管15と二次イオン7を検出する二次イオン検出器16と、を備えているが、質量分析部8は各種の方式を用いることができる。
【0020】
質量分析制御部12は、二次イオン検出器16からの信号を飛行時間毎に検出する多チャンネルアナライザ20と、多価イオンビーム4が試料5に入射した時間を検出し、分析開始信号を多チャンネルアナライザ20に送出する分析開始信号生成手段21と、多チャンネルアナライザ20からの信号を処理するコンピュータ22と、から構成されている。
【0021】
本発明による多価イオンを用いた表面分析装置1が、従来の質量分析装置と異なる点は下記の通りである。
(1)従来の質量分析装置装置が1価のイオンを試料に照射するのに対して15価以上の多価イオンを用いる点。
(2)多価イオンが照射された試料5から発生する二次イオン7を、質量分析部8及び質量分析制御部12で検出する点。
【0022】
最初に、上記(1)の点に関し、多価イオン発生源3について説明する。本発明に用いる多価イオン発生源3は、15価以上の多価イオンを発生できればその発生方法を問わない。
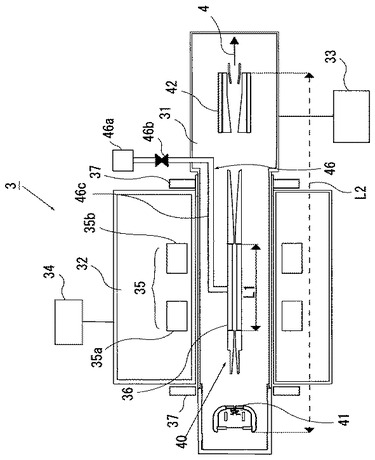
図2は多価イオン発生源3の構成例を模式的に示す断面図である。多価イオン発生源3において、第1の真空容器31及び第2の真空容器32は、それぞれ、真空排気装置33,34により独立に排気されるようになっている。第1の真空容器31内は、多価イオン発生のためには、1×10−5Pa(パスカル)以下、特に1×10−9Pa以下の極高真空とする。例えば、U92+の生成には10−10Pa(10−12Torr)の真空が必要である。このため、第1の真空容器31は脱ガスのために、図示しないベーキング用ヒータなどを用いたベーキング手段を備えている。このベーキング手段を用いることで、第1の真空容器31は250〜300℃程度にまでベーキングすることができる。
ここで、第1及び第2の真空容器31,32は、少なくともそれらの対向面が、超伝導磁石35からの磁場をドリフトチューブ36に印加するように、非磁性材料から形成されている。非磁性材料としては、オーステナイト系ステンレス鋼(例えば、SUS−304,SUS−316)やアルミニウムを用いることができる。
【0023】
第2の真空容器32には、外部と熱的に遮断するために超伝導磁石35が収容され、この超伝導磁石35は、例えば10K(−263℃)の極低温に冷却される。また、第2の真空容器32の真空度は、10−4Pa程度の高真空にされる。このような高真空を得るためのベーキング手段は不要である。そして、第2の真空容器32の第1の真空容器31側の外周部には、磁気シールド部37が設けられている。この磁気シールド部37は、超伝導磁石35からの磁場が、イオン源電極40の電子源41及びコレクタ42側のドリフトチューブ36に漏れないようにするために配設するものである。この磁気シールド部37には、軟鉄などの強磁性材料を用いればよい。
【0024】
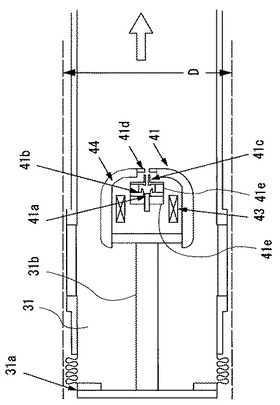
図3は図2に示す電子源41の構成を模式的に示す拡大断面図である。図示するように、電子源41は、第1の真空容器31の左側面の超高真空用フランジ31aに支持部31bを介して、第1の真空容器31の水平方向の中心軸上に配設されている。第2の真空容器32内に収容した超伝導磁石35の空隙(ボア)の直径はDで示している。そして、図中の矢印は、排気経路を示している。電子源41は、左から右方向に、フィラメント(カソード)電極41a、フォーカス電極41b、アノード電極41c、スノート電極41dの順に配置され、それぞれが、絶縁碍子41e,41eにより絶縁されている。さらに、電子源41は、カソード近傍の磁場を0(零)とするためのバッキングコイル(Bucking coil)43を備え、磁気シールド部44に収容されている。
【0025】
ここで、図2に示すように電子源41から発生される電子ビームを、ドリフトチューブ36に当てることなくコレクタ42に導く必要がある。このため、上記のカソード電極41a、アノード電極41c及びスノート電極41dの各電極と絶縁碍子41e,41eとは、嵌め合いにより組み立てることで、相互の軸を高い精度で一致させている。
【0026】
一例として、300mAの電流をカソード電極41aから引き出す際は、フィラメント電位を基準にして、アノード電極41cには+10kV、スノート電極41dには+15kVの電位を与える。このように、各電極とバッキングコイル43及び磁気シールド部44には最大+15kVの高電圧が印加されるが、各部品を小型化することで相互の絶縁を確保しながら、磁気シールド部44の直径を100mmに抑えることができる。この場合には、外径が162mmで内径が102mmの超高真空用フランジ31aに電子源41を収容することができ、第2の真空容器32の電子源41側の直径を152mmに抑えることができる。このため、超高真空用フランジ31aの内径は、磁気シールド部44及びドリフトチューブ36の直径よりも十分大きいので、電子源41領域からの放出ガスを効率良く排気できる。このように、電子源41の寸法を小さくすることで、超伝導磁石35を収容する第2の真空容器32の環状の空隙、即ち超伝導磁石35のボア径を小さくすることができる。
【0027】
ドリフトチューブ36は、幾つかに分割された円筒形電極から構成されていて、その両端が、イオンに対して障壁(井戸型ポテンシャル)を形成するような電場配置となっている。
【0028】
イオンとなる気体は、気体イオン導入手段46によりドリフトチューブ36の側面からその内部に導入される。気体イオン導入手段46は、気体源46a,流量調整部46b,第1の真空容器31内への配管部46cなどから構成されている。
【0029】
一方、固体をイオンとする場合には、ソースとして固体イオン源を用いればよい。
図4は、固体のイオン源を有する多価イオン発生源3の構成を模式的に示す断面図である。図4に示す多価イオン発生源3が、図2に示す多価イオン発生源3と異なるのは、気体イオン導入手段46の代わりに、コレクタ42の右側に固体イオン導入手段48を備えていることである。このような固体イオン導入手段48は、所望のイオンを発生させる針状の固体材料を真空アーク放電させ、1価のイオンにする真空アーク型イオン源などを用いることができる。固体イオン導入手段48から発生したイオンは、第1の真空容器31中でコレクタ42を通過してドリフトチューブ36に注入される。
【0030】
上記した気体イオン導入手段46又は固体イオン導入手段48からなるイオン導入手段によりドリフトチューブ36に注入されるイオンは、一定時間、井戸型ポテンシャルに閉じ込められ、電子による衝突電離が進み多価イオンとなる。この多価イオンのうち、電子の衝突により運動エネルギーが増加して障壁を越えたものが、多価イオン発生源3の外部に取り出される。この際、イオンの電離度が究極、即ち、これ以上電子を当て続けても電離が進まない状態に達したときに、電場配置を変えて多価イオンが外に飛び出すようにして多価イオンビーム4を生成させる。
【0031】
コレクタ42は、ドリフトチューブ36を通過する電子を捕集するための電極であり、例えば、サプレッサ電極、コレクタ電極、エクストラクター電極などの電極から構成されている。カソード及びコレクタ42には、アースに対して最大−数十kVから−300kV程度の電圧が印加されている。この加速電圧は、所望の多価イオンが得られるための電圧であればよい。電子は、ドリフトチューブ36の入射直前に最大加速電圧が印加され、ドリフトチューブ36を通過した電子は、コレクタ42の手前で2〜3kV程度に減速されてコレクタ42に捕集される。このため、コレクタ42には、この電圧と電子ビーム電流との積の電力が捕集される。この電力は、上記コレクタ42手前の電圧が3kVで電子ビーム電流が300mAの場合には900Wとなり、絞られた状態でコレクタ42に当たると、当たった場所が溶ける。したがって、コレクタ42は、冷媒で冷却した電極構造として電子ビームを広げながら電子を捕集する。
【0032】
次に、超伝導磁石35について説明する。
図2及び図4に示すように、超伝導磁石35は第1のコイル35a及び第2のコイル35bからなる、所謂ヘルムホルツ型コイルを用いている。第1のコイル35a及び第2のコイル35bの半径をaとし、これらのコイルの間隔を半径aと同じaとしたときに、対向するコイルの中央部分の磁場を一様にすることができる。ここで、超伝導磁石35は、図示しないHeを冷媒とするクライオスタットや閉サイクル冷凍機により冷却されて、超伝導状態に保持される。
【0033】
上記多価イオン発生源3の特徴は、第2の真空容器32内にイオン閉じ込めのための超伝導磁石35が配置され、第2の真空容器32の環状の空隙に第1の真空容器31が挿入されていることである。つまり、超伝導磁石35の空隙(ボア)内に第1の真空容器31が挿入されていることである。上記のように、この第1の真空容器31内には、電子源41、ドリフトチューブ36及びコレクタ42からなるイオン源電極40が配置されている。また、超伝導磁石35を収容する第2の真空容器32は、第1の真空容器31から両者の真空を破らずに脱着が可能である。このため、イオン源電極40を第1の真空容器31に収納したものを作ればよく、超伝導磁石35は市販品を用いることができるので製作が容易である。したがって、本発明に用いる多価イオン発生源3においては、従来の多価イオン発生源のように超伝導磁石35とイオン源電極40とを一体化した構造を精度良く製作する必要がない。
【0034】
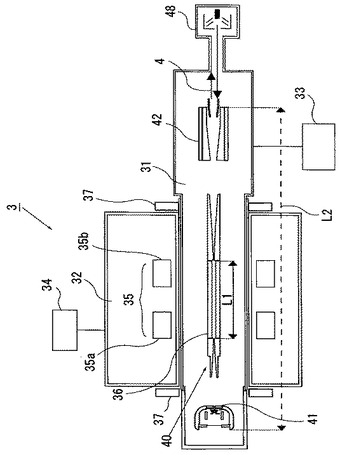
本発明に用いることができる多価イオン発生源3は以上のように構成されており、次にその動作について説明する。
図5は、多価イオン発生源3に印加される電源を説明するための模式的なブロック図である。電子の加速電圧が40kVの一例を示している。図示するように、電子源41のフィラメント電極41aには15V,2A、フォーカス電極41bには50V,1mA、アノード電極41cには10kV、スノート電極41dには15kV,1mA、の電源が−30kVの電源に上乗せされて供給されている。また、ドリフトチューブ36の各電極には、10kVの電源上に、第1電極500V、第2電極500V、第3電極500Vがそれぞれ上乗せされて供給されている。
さらに、コレクタ42のサプレッサ電極、コレクタ電極、エクストラクター電極には、それぞれ、2kV、3kV,500mA、−3kVの電源が、−30kVの電源に上乗せされて供給されている。
【0035】
上述のように、電子源41から発生された電子は、超伝導磁石35の磁場中に配置されたドリフトチューブ36を通る。図5に示す電源を用いた場合には、電子源41とドリフトチューブ36の間で電子が最大40kVまで加速される。この加速されてドリフトチューブ36に入射した電子は、超伝導磁石35によりドリフトチューブ36に形成される強磁場で圧縮され大電流密度(〜1000A/cm2)の電子ビームとなり、コレクタ42に捕集される。図5に示す電源を用いた場合には、コレクタ42には2〜3kV位まで減速されて捕集される。この際、電子は、ドリフトチューブ36の直前で最大加速電圧が印加される。原子を電離するためには、束縛電子の電離エネルギー以上のエネルギーを持った電子と衝突させなければならないので、重元素(例えば、キセノン(Xe)など)を完全に電離させるには1s軌道の電離エネルギー(Xeでは35keV)より高いエネルギーが必要で、加速電圧35kV以上が必要である。
【0036】
一方、気体イオン導入手段46から導入されるイオンとなる気体は、ドリフトチューブ36内でイオン化される。ドリフトチューブ36の電極は幾つかに分割されており、両端がイオンに対して障壁(井戸型ポテンシャル)が形成されるように電位が与えられている。イオンは一定時間、この井戸型ポテンシャルに閉じ込められ、気体の原子を電子ビームに繰り返し衝突させることにより電離が進む。電子による衝突電離が進み多価イオンビーム4となる。この多価イオンのうち、電子の衝突により運動エネルギーが増加し障壁を越えたものがイオン源外部に取り出される。このように、イオンの電離度が究極、即ち、これ以上電子を当て続けても電離が進まない状態に達したときに、電場配置を変えて多価イオンが外に飛び出すようにして多価イオンビーム4を生成させる。
このドリフトチューブ36で生成した多価イオンが、ドリフトチューブ36から引き出される際には、ドリフトチューブ36とコレクタ42との間において40kVで一且加速され、第1の真空容器31を出る際は、この第1の真空容器31はアース電位であるので10kVまで減速されて出射する。
【0037】
得られる多価イオンビーム4の強度は、イオンがトラップされているドリフトチューブ36の長さL1(図2参照)及びエミッション電流に比例し、電離度が究極に至るまでの時間は電子の電流密度に比例する。なお、ドリフトチューブ36の長さが長すぎる場合にはプラズマの不安定性が出現し、高電離の多価イオンができなくなる。
また、電子ビーム電流密度は、超伝導磁石35によりドリフトチューブ36に印加される磁場強度が高いほど高くなる。但し、比例関係ではない。このため、単位時間当たりに得られる多価イオンビーム4の強度は、電子ビーム電流密度が高くトラップ領域が長いほど高くすることができる。
【0038】
多価イオン発生源3は、以上の機構において従来の多価イオン発生源と同様である。
本実施形態の多価イオン発生源3の特徴は、イオン源電極40を収納する第1の真空容器31のための真空排気装置33と、超伝導磁石35を収容する第2の真空容器32のための真空排気装置34とを着脱して分離することができる点にある。このため、超伝導磁石35を収容する第2の真空容器32を、第1の真空容器31から分離した状態で、第1の真空容器31及び収容されているイオン源電極40を十分に加熱して脱ガスをすることができる。したがって、多価イオン発生のための第1の真空容器31を短時間で極高真空とすることができる。例えば、誤って電子ビームをドリフトチューブ36に当てたときには、多価イオン発生源3では、イオン源電極40を収納した第1の真空容器31のみ修理すればよい。
これにより、多価イオン発生源3においては、イオン源電極40の真空立ち上げが短時間で済むので、操作性、保守性に優れた多価イオン発生源3となる。
【0039】
試料台6は、その位置が制御できるように可動な試料台とすればよい。例えば、試料台6は、XYZステージなどを用いることができる。このXYZステージ6は、電歪素子やステップモーターを用いることができる。
【0040】
次に、上記(2)の点に関し、多価イオンビーム4が照射された試料5から発生する二次イオン7の検出を行う質量分析部8及び質量分析制御部12について詳しく説明する。
質量分析部8において、試料5へパルス状の多価イオンビーム4が照射されると試料5の表面に存在する元素から二次イオン7及び二次電子9が発生する。発生した二次イオン7は、レンズ14で集められ飛行管15へ入射し、二次イオン7の質量等で決まる飛行時間を経て、二次イオン検出器16に入射する。
【0041】
一方、試料5に多価イオンビーム4が照射したときに発生する二次電子9は、二次電子検出部10によって検出される。この場合、1個の多価イオンビーム4の照射により数十個から数百個の二次電子9が発生するので、照射された多価イオンビーム4の数を正確に計測できる。この二次電子検出部10は、試料5へ1個の多価イオンビーム4が照射したときに発生する二次電子9を高感度で検出できる二次電子検出器10A等を用いることが好ましい。
【0042】
図1に示す二次電子検出部10は、入射する多価イオンビーム4の試料5へのビーム進行方向に対して垂直面内に配置され、上下方向の1個ずつ、つまり2個の二次電子検出器10A,10Bが配設されている。図示するように、二次電子検出部10と試料5との間には、二次電子9を集めるためのレンズ作用を有するグリッド10Cなどのレンズを備えてもよい。さらに、二次電子増倍のために二次電子検出器10A等に印加する電圧を外部に漏らさないようにシールド部をさらに設けてもよい。このシールド部は金属からなり、二次電子検出器10A,10Bを取り囲む形状とすればよい。
【0043】
分析開始信号を多チャンネルアナライザ20に送出する分析開始信号生成手段21は、二次電子検出部10からの出力が入力され、その測定データの処理を行う。一般に、二次電子信号は雑音が多いため誤計測をする可能性がある。二次電子検出部10は、生じる雑音による計測誤差を無くすために、上記のように電気的に独立した複数の二次電子検出器10A,10Bを備えて構成することができる。
【0044】
複数の二次電子検出器10A,10Bを備えた場合には、分析開始信号生成手段21には、試料5から発生する複数の二次電子9の計測データが一致するか否かを判定する判定回路を設けることが好ましい。そして、複数の二次電子検出器10A,10Bの計測データが一致した場合に、正しい計測データであると判定すればよい。このように、多価イオンビーム4の照射では多数の二次電子9が放出されるため、少なくとも2個の二次電子検出器10A,10Bから得られた信号が一致したときのみ真の信号と見做すことで、正確なイオン数の計測が可能になる。
【0045】
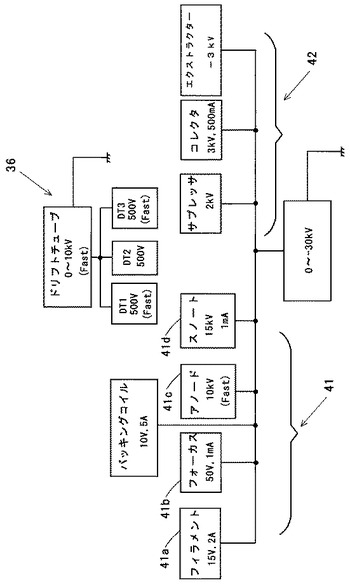
図6は、(A)が分析開始信号生成手段21の構成を示す模式図であり、(B)が分析開始信号生成手段21で用いるAND回路24の真理値表を示す図である。
図6(A)に示すように、分析開始信号生成手段21は、判定回路として論理積演算を行うAND回路24等を備えている。その入力24A,24Bには、2つの二次電子検出器10A,10Bの出力が接続されている。二次電子検出器10A,10Bの出力は図示しない増幅器によりAND回路24のハイレベル及びローレベルの信号を出力するように信号処理がされている。上記の二次電子検出器10Aとしては、二次電子9も検出できる所謂光電子増倍管を使用できる。このような電子増倍効果を有する二次電子検出器10Aとしては、複数のチャンネルトロンや複数のアノード電極を持つマイクロチャンネルプレートを用いることができる。
【0046】
図6に示す二次電子検出器10A,10Bにおいては、多価イオン1個による二次電子9を検出したときには、ハイレベル、即ち1の信号をAND回路24に送出する。逆に、二次電子9を検出しない場合にはローレベル即ち0の信号をAND回路24に送出する。したがって、図6(B)のAND回路24の真理値表に示すように、AND回路24においては、その2入力24A,24Bが共に1の場合に出力24Cが1となる。これ以外の入力の場合の出力24Cは何れも0である。つまり、2つの二次電子検出器10A,10Bが共に、多価イオン1個による二次電子を検出したときには、AND回路24は多価イオン1個が発生したと判断する。AND回路24を用いた判定回路によれば、特に、試料5へ入射する多価イオンの強度が著しく弱い場合、例えば多価イオンのパルス周波数が1Hz又は1Hz以下の場合であっても多価イオンの発生を検出することができる。
【0047】
なお、2入力AND回路24を用いた判定回路について説明したが、精度を上げるために3入力以上の二次電子検出器を用いる場合には、二次電子検出器の数だけの入力を備えたAND回路24で行えばよい。これにより、複数の二次電子検出器の計測データが一致した場合に、多価イオンビーム4が照射されたという判定をすることができるので、雑音を除去して精度を高めることができる。
【0048】
上記判定回路24は、多価イオンのパルス周波数が100Hz以上の場合には、二次電子検出部10からの信号を、高速プリアンプで増幅し、この信号を時間弁別器(Timing Discriminator)を介して、多チャンネルアナライザ20に入力する構成としてもよい。
【0049】
本実施形態の表面分析装置1は以上のように構成されており、次にその動作について説明する。
多価イオン発生源3から発生した多価イオンビーム4が、試料5に照射される。この場合、試料5への多価イオンビーム4の照射位置は、可動な試料台6により位置制御がされている。
試料5の位置が決定された後、多価イオンビーム4が試料5に照射されると、試料5の所定位置に存在する原子に応じた二次イオン7が発生する。この場合に発生する二次電子9が二次電子検出器10A,10Bにより検出される。その有無が分析開始信号生成手段21により判定される。したがって、分析開始信号生成手段21のAND回路24の出力がハイレベルの場合が、試料5に多価イオンビーム4が照射された時間に対応する。
【0050】
質量分析部8がTOF型の質量分析装置である場合には、質量分析制御部12に入力されるAND回路の出力24Cがハイレベルとなった時刻が測定開始の時間となり、この信号を質量分析測定の開始信号と呼ぶことにする。
これにより、開始信号以降に得られるTOF型の質量分析部8における二次イオン検出器16からの信号が、試料5から発生した二次イオン7の信号である。
次に、二次イオン7の信号検出が終了した時点で、二次イオン検出器16から質量分析制御部12へ、測定停止信号16Aが送出される。
したがって、上記開始信号から測定停止信号16Aまでの信号を解析することにより、試料5の所定位置に存在していた原子の質量分析を行うことができる。
【0051】
本発明の表面分析装置1によれば、特に、15価以上の多価イオンビーム4を用いることで、短時間に試料5から二次イオン7を発生させたり、スパッタリング効率を顕著に増加させたりすることができる。これにより、表面分析装置1によれば、15価以上の多価イオンビーム4を試料5に照射することにより試料5から発生する二次イオン7の信号は、価数の4乗等のべき数に比例して増大するため、試料5の表面の分析を短時間で分析することができる。このため、表面清浄化を施した試料5の分析においても残留ガス等の再付着が起こらず高精度な分析を行うことができる。
【0052】
本発明の表面分析装置1によれば、試料5の表面に15価以上の多価イオンビーム4を照射することで、他手法では分析が困難であった所謂質量の小さいH+等の元素を、高感度でかつ短時間で測定することができる。
【0053】
また、試料5の位置決めを、nmオーダーの分解能を有するXYZステージ6により制御する場合には空間分解能を著しく高めることができる。したがって、表面分析装置1によれば、空間分解能が高く、微量な元素を高感度で、かつ、短時間で分析することができる。
【0054】
上記の本発明の表面分析装置1においては、さらに、試料5の表面の観察するための観察装置を備えていてもよい。このような観察装置としては、電子線回折装置、走査型電子顕微鏡(SEM)などが挙げられる。
【0055】
本発明の表面分析装置1によれば、多チャンネルアナライザ20の検出値からコンピュータ22を用いた解析により種々の表面分析を行うことができる。
次に、表面分析方法の解析例について説明する。
(イ)15価以上の多価イオンビーム4をパルスとして試料5に照射し、パルス毎の測定を所定の回数繰り返し行う。
(ロ)測定誤差を除去するために、上記(イ)の測定で取得した複数のデータをコンピュータ22を用いて積算して、平均値が一定の値に収束した段階で多価イオンビーム4の照射を停止する。
上記の表面分析方法によれば、単元素からなる試料5の表面分析を短時間で行うことができる。
【0056】
試料5が2つの元素A,Bからなり、その原子比がx:yである場合の表面分析方法について説明する。この場合、試料5の化学式は、AxByである。
(イ)15価の多価イオンビーム4をパルスとして試料5に照射し、パルス毎の測定を所定の回数繰り返し行う。
(ロ)測定誤差を除去するために、上記(イ)の測定で取得した複数のデータをコンピュータ22を用いて積算して、平均値が一定の値に収束した段階で多価イオンビーム4の照射を停止する。
(ハ)15価以上の多価イオンビーム4(価数=15+n)をパルスとして試料5に照射し、1回パルス毎の測定を所定の回数繰り返し行う。
(ニ)測定誤差を除去するために、上記繰り返し測定したデータをコンピュータ22を用いて積算して、平均値が一定の値に収束した段階で多価イオンビーム4の照射を停止する。
(ホ)上記(ハ)及び(ニ)の測定を繰り返し行い、所定の多価数、例えば価数が50までのデータを得る。
(へ)上記測定のデータから、構成元素A及びBの二次イオン7のカウント数と価数との関係を算出する。構成元素A及びBのカウント数の比(x:y)が得られる価数を求める。
(ト)上記(へ)のステップで構成元素A及びBのカウント数の比(x:y)が得られる価数を得た場合には、同じ組成の試料5を次回の測定の際には、この価数の多価イオンビーム4を照射すれば、化学量的組成が反映された表面分析を行うことができる。
なお、試料5が3つ以上の元素からなる場合にも、上記(イ)〜(ト)の測定において、さらに第3以上の元素C等の二次イオン7のカウント数と価数との関係を算出することにより表面分析ができる。
【0057】
試料5が2つの元素A,Bからなり、その組成が不明である場合の表面分析方法について説明する。
上記の原子比(x:y)が判明している試料5を標準試料とする。2つの元素A,Bからなり、その組成が不明である試料5に対して、標準試料の化学量的組成が反映された分析ができる価数を有する多価イオンビーム4を照射し、上記(イ)及び(ロ)の測定を行い、元素A,Bのカウント数を得る。得られた元素A,Bのカウント数を、それぞれ、na、nbとする。この元素A,Bのカウント数na,nbを、標準試料のカウント数を基準として比較することで、組成の不明な試料5の組成を求めることができる。
ここで、試料5が3つ以上の元素からなる場合にも、上記測定において、さらに第3以上の元素C等の二次イオン7のカウント数と標準試料のカウント数との関係を算出することにより表面分析ができる。
【0058】
表面分析装置1においては、多価イオン発生源3と試料5との間に、さらに減速レンズを設けてもよい。減速レンズを設けることで、多価イオン発生源3から入射する多価イオンビーム4の加速電圧を制御することができる。この多価イオンビーム4が種々の多価イオンを含む場合には、所望の多価イオンを選択するために、さらに、イオン分離器を設けてもよい。イオン分離器としては、分析磁石(偏向磁石)や磁場及び電場によるウィーンフィルターなどを用いることができる。
【0059】
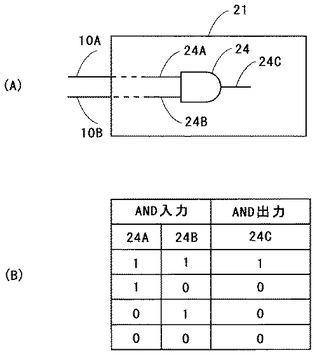
表面分析装置1においては、多価イオン発生源3と試料5との間に、さらに多価イオンガイド26を設けてもよい。
図7は、図1に示した多価イオンガイド26の先端部の構成を模式的に示す断面図である。多価イオンガイド26においては、その一端26Aに多価イオンビーム4を入射し、他端26Bから多価イオンビーム4を出射する。つまり、多価イオンビーム4が、試料5を載置する試料台6に導くためのガイドとなっている。そして、多価イオンガイドの他端側26Bの先端部26Cが、直径がおおよそ100nm以下程度の開口部を有する、所謂キャピラリー構造となっている。この多価イオンガイド26は、好ましくは絶縁体からなり、例えばセラミックや石英ガラスを用いることができる。このようなキャピラリーは、例えば石英の細管を加熱溶融し、溶融状態にて引き伸ばし加工を行うことで製作することができる。
【0060】
多価イオンビーム4が多価イオンガイド26に入射すると、多価イオンガイド26の内壁が帯電する。このために、多価イオンビーム4は多価イオンガイド26の内壁から反発されて、多価イオンガイド26の中心部を通過し、キャピラリー構造となっている先端部26Cから試料5に入射する。これにより、多価イオンビーム4は、多価イオンガイドの先端部26Cでそのビーム径が絞られ、即ち、収束して試料5に入射する。
【0061】
(分析例1)
以下、本発明の表面分析装置1による分析例について説明する。
試料5となるSi(シリコン)の(100)表面を1200℃で5秒間の加熱を3回行った後、表面に50価のXeイオン(Xe50+)を、加速電圧を3keVで照射した。二次電子検出器10A,10Bとしてはチャンネルトロンを用いた。さらに、試料5に照射される多価イオンビーム4を計測するために、試料5の裏面側に別のチャンネルトロンを配置した。
図8は、分析例1のTOF−SIMSによるプロファイルを示す図である。図8において、横軸は飛行時間(ns)を、縦軸は検出した二次イオン7のカウント数を示している。図8から明らかなように、Si表面からはH+,H2+,O+,O2+,Si+,Si2+,Si3+,Si4+,Si2+,SiO+の各イオンが検出されていることが分かる。
【0062】
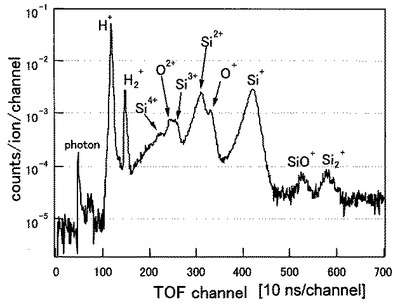
(分析例2)
Si(シリコン)の(100)表面を、所謂白木法によりウエット処理をし、900℃で3分間のアニールを施した。この試料5の表面に50価のXeイオン(Xe50+)を、分析例1と同じ条件で照射した。
図9に分析例2のTOF−SIMSによるプロファイルを示す。図9の横軸及び縦軸は図8と同じである。図9から明らかなように、Si表面からはH+,H2+,O+,O2+,Si+,Si2+,Si3+,Si4+,Si2+,SiO+の各イオンが検出されていることが分かる。
【0063】
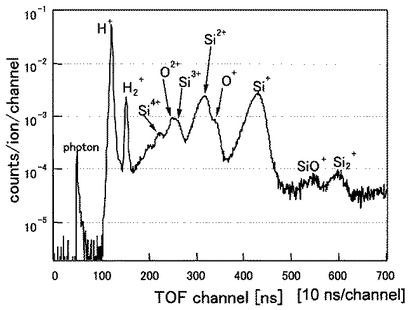
(分析例3)
試料5となるSi(シリコン)の(100)表面を1200℃で12時間加熱して表面清浄化を行った後、表面に50価のXeイオン(Xe50+)を、分析例1と同じ条件で照射した。
図10に分析例3のTOF−SIMSによるプロファイルを示す。図10の横軸及び縦軸は図8と同じである。図10から明らかなように、Si表面からはH+,H2+,Si+,Si2+,Si3+,Si4+,Si2+の各イオンが検出されていることが分かる。
【0064】
(分析例1〜3の比較)
分析例1及び分析例2のSi表面から検出される二次イオン7は、その種類が同じであることが分かる。また、検出されたH+イオンのカウント数もほぼ同じ位である。
一方、分析例3の試料5からは、分析例1及び分析例2で検出されたO+,O2+,SiO+が検出されないことが分かる。また、検出されたH+イオンのカウント数は、分析例1及び分析例2よりもほぼ1桁小さいことが分かる。これらの結果から、分析例3の場合には、Siの表面における水素の量が非常に少ないことが分かる。
【0065】
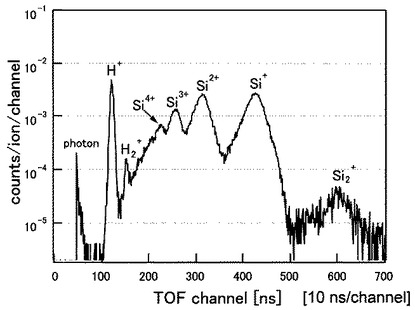
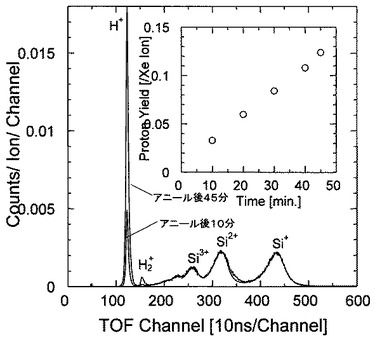
(分析例4)
試料5となるSiの(100)表面を1200℃で12時間加熱して表面清浄化を行った後、表面に50価のXeイオン(Xe50+)を、分析例1と同じ条件で照射した。
図11に分析例4のTOF−SIMSによるプロファイルを示す。図11は、Si(100)表面をアニールした後10分及び45分経過したときのプロファイルを示しており、横軸及び縦軸は図8と同じである。
図11から明らかなように、Si表面からはH+,H2+,Si+,Si2+,Si3+の各イオンが検出され、特に45分経過後では、H+のカウント数が著しく増大していることが分かる。
【0066】
図11の挿入図は、アニール後のH+イオンのイールドの時間依存性を示す図である。図の横軸はアニール後の経過時間(分)を示し、縦軸はH+イオンのイールドを示している。この挿入図から、アニール直後ではH+イオンは殆ど検出されないことが分かり、10分,20分,30分,40分,45分経過後にはH+イオンが検出され、しかも、イールドは経過時間と共に増加することが分かる。
分析例4の結果によれば、Si表面の清浄化処理直後からの経過時間と共に水素が増大していく様子を観察することができた。
【0067】
分析例4に示すように、長時間の測定時間を要する従来技術では、分析が困難であった試料5となるSi表面に形成した清浄表面を正確に評価できることが判明した。
【0068】
(分析例5)
試料5となるSi(シリコン)の(111)表面を1200℃で5秒間の加熱を3回行った後、表面へ29価,34価,38価,44価,50価のXeイオン(Xe50+)を、加速電圧175keVで照射した。
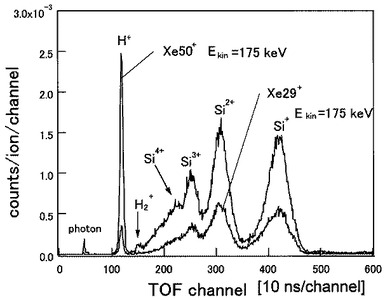
図12は、分析例5において、29価のXeイオン(Xe29+)及び50価のXeイオン(Xe50+)を照射したときのTOF−SIMSによるプロファイルを示す図である。図12において、横軸は飛行時間(ns)を、縦軸は検出した二次イオン7のカウント数を示している。
図12から明らかなように、29価のXeイオン(Xe29+)の照射によりSi表面からH+,Si3+,Si2+,Si+の各イオンが検出されていることが分かる。50価のXeイオン(Xe50+)の照射によりSi表面から検出されるイオンは、29価のXeイオン(Xe29+)の照射により検出されたイオンの他に、さらに、H2+,Si4+が検出され、しかも検出されたイオンのカウント数が多いことが分かる。
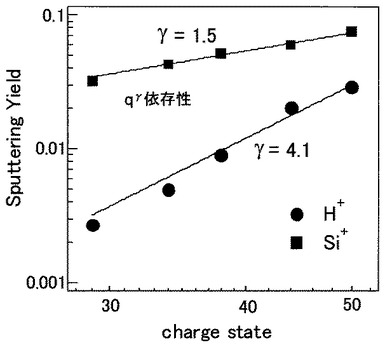
【0069】
図13は、分析例5において検出したH+イオン及びSi+イオンのスパッタリングイールドの価数依存性を示す図である。図13において、横軸は照射したXeイオンの価数を示し、縦軸はH+イオン及びSi+イオンのスパッタリングイールドを示している。
図13から明らかなように、H+イオンのスパッタリングイールドは、価数qに対して、4.1乗、つまり、q4.1で増加していることが分かる。同様に、Si+イオンのスパッタリングイールドは、価数qに対して、1.5乗、つまりq1.5で増加することが分かった。
【0070】
(分析例6)
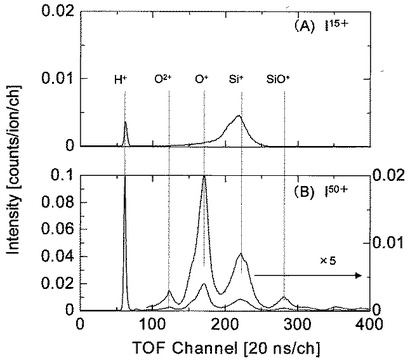
試料5となるSiO2の表面へ15価から50価まで5価ずつ増加させた種々の価数のIイオン(I15+,I20+,I25+,I30+,I35+,I40+,I45+,I50+)を加速電圧175keVで照射した。
図14は、分析例6において、15価のIイオン(I15+)及び50価のIイオン(I50+)を照射したときのTOF−SIMSによるプロファイルを示す図である。図14において、横軸は飛行時間(ns)を、縦軸は検出した二次イオン7のカウント数を示している。
図14から明らかなように、15価のIイオン(I15+)照射ではSi表面からH+,Si+の各イオンが検出されていることが分かる。50価のIイオン(I50+)の照射によりSi表面から検出されるイオンは、15価のIイオン(I29+)の照射により検出されたイオン(H+,Si+)の他に、さらに、O+,O2+,SiO+が検出され、しかも検出された各イオンのカウント数が多いことが分かる。
【0071】
分析例6における多価イオンによるSiO2膜の表面分析の場合には、酸素が陽イオンとして検出されている点が多価イオンによる表面分析の特徴である。これに対して、1価イオンを照射源とする従来のSIMS分析の場合には、酸素が陰イオンとして検出されていた。
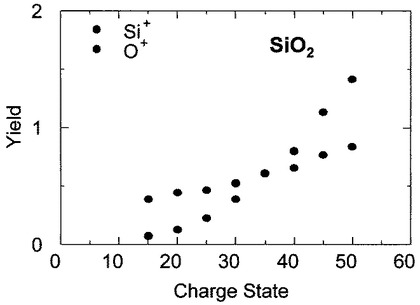
【0072】
図15は、分析例6において検出したSi+イオン及びO+イオンのスパッタリングイールドの価数依存性を示す図である。図15において、横軸は照射したIイオンの価数を、縦軸はSi+イオン及びO+イオンのスパッタリングイールドを示している。
図15から明らかなように、Si+イオンのスパッタリングイールドは、価数qに対してほぼ比例して増加していることが分かる。
一方、O+イオンのスパッタリングイールドは、価数qに対して2.5乗、つまりq2.5で増加することが分かった。価数が35価よりも大きい場合(q>35)では、O+イオンのイールドがSi+イオンのイールドよりも大きくなることが判明した。従って、価数が50価(I50+)の場合には、O+のスパッタリングイールドは、Si+のスパッタリングイールドの約2倍であり、SiO2の化学量論的組成を反映した二次イオン7の収率となっていることが判明した。
【0073】
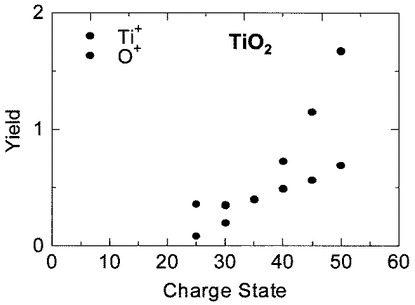
(分析例7)
試料5をSiO2からTiO2とした以外は、分析例6と同様に15価から50価まで5価ずつに増加させた種々の価数のIイオン(I15+,I20+,I25+,I30+,I35+,I40+,I45+,I50+)を、加速電圧175keVで照射した。試料5がTiO2の場合には、価数が25価以上でTi+やO+のイオン信号が得られた。
図16は、分析例7において検出したTi+イオン及びO+イオンのスパッタリングイールドの価数依存性を示す図である。図16において、横軸は照射したIイオンの価数を、縦軸はTi+イオン及びO+イオンのスパッタリングイールドを示している。
図16から明らかなように、Ti+イオンのスパッタリングイールドは、価数qが30以上ではほぼ比例して増加していることが分かる。
一方、O+イオンのスパッタリングイールドは、価数qに対して4乗、つまりq4で増加することが分かった。価数が35価よりも大きい場合(q>35)では、O+イオンのイールドがTi+イオンのイールドよりも大きくなることが判明した。従って、価数が50価(I50+)の場合には、O+のスパッタリングイールドは、Ti+のスパッタリングイールドの約2倍であり、TiO2の化学量論的組成を反映した二次イオン7の収率となっていること判明した。
【0074】
本発明は上記実施例に限定されることなく、特許請求の範囲に記載した発明の範囲内で種々の変形が可能であり、それらも本発明の範囲内に含まれることはいうまでもない。例えば、上記実施の形態において、多価イオンガイド26の形状や真空容器2の構造や付属させる観察機器の構成などは、試料5に応じて適宜変更できる。
【図面の簡単な説明】
【0075】
【図1】本発明による多価イオンを用いた表面分析装置の構成を模式的に示す断面図である。
【図2】多価イオン発生源の構成例を模式的に示す断面図である。
【図3】図2に示す電子源の構成を模式的に示す拡大断面図である。
【図4】固体のイオン源を有する多価イオン発生源の構成を模式的に示す断面図である。
【図5】多価イオン発生源に印加される電源を説明するための模式的なブロック図である。
【図6】(A)は分析開始信号生成手段の構成を示す模式図、(B)は分析開始信号生成手段で用いるAND回路の真理値表を示す図である。
【図7】図1に示した多価イオンガイドの先端部の構成の模式的断面図である。
【図8】分析例1のTOF−SIMSによるプロファイルを示す図である。
【図9】分析例2のTOF−SIMSによるプロファイルを示す図である。
【図10】分析例3のTOF−SIMSによるプロファイルを示す図である。
【図11】分析例4のTOF−SIMSによるプロファイルを示す図である。
【図12】分析例5において、29価のXeイオン(Xe29+)及び50価のXeイオン(Xe50+)を照射したときのTOF−SIMSによるプロファイルを示す図である。
【図13】分析例5において検出したH+イオン及びSi+イオンのスパッタリングイールドの価数依存性を示す図である。
【図14】分析例6において、15価のIイオン(I15+)及び50価のIイオン(I50+)を照射したときのTOF−SIMSによるプロファイルを示す図である。
【図15】分析例6において検出したSi+イオン及びO+イオンのスパッタリングイールドの価数依存性を示す図である。
【図16】分析例7において検出したTi+イオン及びO+イオンのスパッタリングイールドの価数依存性を示す図である。
【符号の説明】
【0076】
1:表面分析装置
2:真空容器
3:多価イオン発生源
4:多価イオンビーム
5:試料
6:試料台
7:二次イオン
8:質量分析部
9:二次電子
10:二次電子検出部
10A,10B:二次電子検出器
10C:グリッド
12:質量分析制御部
14:レンズ
15:飛行管
16:二次イオン検出器
16A:測定停止信号
20:多チャンネルアナライザ
21:分析開始信号生成手段
22:コンピュータ
24:AND回路
24A,24B:入力
24C:出力
26:多価イオンガイド
26C:先端部
31:第1の真空容器
31a:超高真空用フランジ
31b:支持部
32:第2の真空容器
33:第1の真空容器の真空排気装置
34:第2の真空容器の真空排気装置
35:超伝導磁石
35a:第1のコイル
35b:第2のコイル
36:ドリフトチューブ
37,44:磁気シールド部
40:イオン源電極
41:電子源
41a:フィラメント(カソード)電極
41b:フォーカス電極
41c:アノード電極
41d:スノート電極
41e:絶縁碍子
42:コレクタ
43:バッキングコイル
46:気体イオン導入手段
46a:気体源
46b:流量調整部
46c;配管部
48:固体イオン導入手段
【技術分野】
【0001】
本発明は、多価イオンを試料へのイオン源として用いた表面分析装置及び表面分析方法に関する。
【背景技術】
【0002】
原子から電子を取り除くと正のイオンができるが、例えばXe44+イオンのように、電子を2個以上取り去ったものを正の多価イオンという。多価イオンは極めて大きな内部エネルギーを持っており、多価イオンが固体表面と衝突すると、多数の二次電子が放出されたり(非特許文献1参照)、多価イオンの入射点まわりにナノメートルの大きさの構造変化が誘起されたりする(非特許文献2参照)など、数多くの特異的な現象が起こる。
この多価イオンと物質の特異な相互作用は、単一イオンインプランテーションや量子ドット作製などのナノメートル領域のプロセス技術に応用できる可能性があり、注目を集めている(非特許文献3参照)。
【0003】
このような多価イオンを生成するイオン源としては、電子サイクロトロン共鳴(ECR)型イオン源(ECRIS)と電子ビーム型イオン源(EBIS)とが知られている。後者は前者に比べて得られるイオンの電離度が高いという特徴がある。
【0004】
EBISとしては、原子物理学の研究用に開発された核融合研究所の装置が知られている(例えば、非特許文献4参照)。この装置は、電子源(カソード)、ドリフトチューブ、コレクタ、ソレノイド磁石、イオン引き出し用レンズなどから構成されている。カソードから出射した電子は、磁場中に配置されたドリフトチューブを通り、コレクタに捕集される。電子は、ドリフトチューブに形成される強磁場で圧縮され大電流密度の電子ビームとなっている。一方、カソード付近から導入された気体は、イオンに対して障壁となる、ドリフトチューブ内に形成される井戸型ポテンシャルによって、電子による衝突電離が進み、多価イオンとなる。
【0005】
さらに、1988年には、EBISを改良したEBIT(電子ビームイオントラップ)が開発された(非特許文献5参照)。このEBITの多価イオン発生原理はEBISと同じであるが、超伝導ヘルムホルツ型コイルを用い、ドリフトチューブを従来よりも短くして、ドリフトチューブ内のプラズマの不安定性を回避することにより、イオンの閉じ込め時間を改善し、高価数の多価イオンを安定に保持できる。このため、EBITでは、ドリフトチューブでの電子ビームの絞り込みを究極まで行い、高電離イオンを生成することを可能にした。
【0006】
また、EBITとして、ウラン(U)まで完全電離できるように、電子の加速電圧を最大300kVとした装置が本発明者らにより開発されている(非特許文献6参照。)。このEBITは、原子物理学の研究のために開発されたもので、生成可能な多価イオンの内部エネルギーとしては世界最高の性能を持っている。
【0007】
ところで、イオンを用いた分析方法としては、従来から1価のイオンをスパッタ源とする二次イオン質量分析装置(SIMS)が知られている。
【0008】
さらに、スパッタ源として低速の多価イオンを用いた表面分析の試みがなされている(非特許文献6参照。)。非特許文献6では、2keV〜5keVの低速で、価数が4〜12の多価イオンをSi表面に照射した場合のH+イオン(プロトン)の検出が報告されている。
【0009】
【非特許文献1】J. W. McDonald, D. Schneider, M. W. ClarkandD. DeWitt, Phys. Rev. Lett., Vol. 68, (1992), p.2297
【非特許文献2】T. Meguro et al., Appl. Phys. Lett., Vol.79, (2001), p.3866
【非特許文献3】T. Schenkel et al., Appl. Phys., Vol.94, (2003), p.7017
【非特許文献4】小林 信夫、大谷 俊介 他7 名、名古屋大学プラズマ研究所資料・技術報告、IPPJ−DT−84、1981年
【非特許文献5】MA. Levin他7名, Physica Scr., T22, (1988), p.157
【非特許文献6】大谷俊介、桜井誠、プラズマ・核融合学会誌、73,(1997),p.1063
【非特許文献7】山崎泰規、第52回応用物理学関係連合講演会、講演予稿集、29p−C−3(2005年春)
【発明の開示】
【発明が解決しようとする課題】
【0010】
従来の技術では、12価以下の多価イオンを用いた軽元素の分析手法は知られていたが、検出感度が低いものであり、固体等の表面に吸着した水素原子のような軽元素を分析することは困難であった。このため、軽元素の分析には、例えば12時間程の非常に長い時間を要していた。従って、例えば被測定試料を超高真空下に保持して測定を行った場合であっても、真空槽の残留ガスや真空容器の内壁からの脱出ガスが被測定試料へ再付着するので、正しい分析を行うことが困難であった。
【0011】
また、従来の1価のイオンをスパッタ源とする二次イオン質量分析装置により化合物からなる試料を分析する場合、試料の表面からスパッタされた粒子、すなわち二次イオンのイオン化率が違うので、化合物の化学両論的な組成が反映された二次イオン強度を得ることは困難であった。
【0012】
本発明は、上記課題に鑑み、高い多価数を有する多価イオンを用いて被測定物の表面または表面近傍に存在する原子や分子を効率良く検出することができる表面分析装置と表面分析方法を提供することを目的としている。
【課題を解決するための手段】
【0013】
上記目的を達成するため、本発明の表面分析装置は、試料を搭載する試料台と、試料台に搭載された試料に価数が15以上の多価イオンビームを照射する多価イオン発生源と、試料に多価イオンビームが照射されることにより生じる二次イオンを検出する質量分析部と、試料に多価イオンビームが照射されることにより生じる二次電子を検出する二次電子検出部と、二次電子検出部からの二次電子検出信号を受けて分析開始信号を生成し質量分析部へ送信する質量分析制御部を、備えている。
上記構成において、二次電子検出部は、好ましくは、電気的に独立した複数の二次電子検出器を有する。
二次電子検出器は、好ましくは、複数のチャンネルトロン又は複数のアノード電極を持つマイクロチャンネルプレートからなる。
多価イオン発生源は、好ましくは、一端に多価イオン発生源からの多価イオンビームが入射され他端から多価イオンビームが出射される多価イオンガイドを備えており、多価イオンガイドの他端部はキャピラリー構造である。
【0014】
上記構成によれば、試料の表面に15価以上の多価イオンビームを照射することで、他手法では分析が困難であった所謂質量の小さいH+等の元素を、高感度でかつ短時間で測定することができる。
【0015】
本発明の表面分析方法は、多価イオンビームを15価以上の価数で変化させて試料へ照射し、試料表面の特定の元素から生じる二次イオンのカウント数と価数との関係を算出して、特定の元素に対する多価イオンビームの価数を決定する。
上記構成において、好ましくは、試料が2以上の構成元素からなり、各構成元素から生じる二次イオンのカウント数と価数との関係を算出して、各構成元素の組成比を与える多価イオンの価数を決定する。
【0016】
上記構成によれば、試料の表面に検出対象となる元素に対して15価以上の最適な多価イオンビームを照射することで、他手法では分析が困難であった所謂、質量の小さいH+等の元素を、高感度でかつ短時間で測定することができる。
【発明の効果】
【0017】
本発明によれば、15価以上の多価イオンビームを試料に照射することにより試料から発生する二次イオンの信号は、価数の4乗等のべき数に比例して増大するために、試料表面の分析を短時間で分析することができる。このため、表面清浄化を施した試料の分析においても残留ガス等の再付着が起こらず高精度な分析を行うことができる。
【発明を実施するための最良の形態】
【0018】
以下、図面に示した実施形態に基づいて本発明を詳細に説明する。
図1は、本発明による多価イオンを用いた表面分析装置1の構成を模式的に示す断面図である。
図1において、本発明の多価イオンを用いた表面分析装置1は、試料5を搭載する試料台6と、試料台6に搭載された試料5に価数が15以上の多価イオンビーム4を照射する多価イオン発生源3と、試料5に多価イオンビーム4が照射されることで生じる二次イオン7を検出する質量分析部8と、試料5に多価イオンビーム4が照射されることで生じる二次電子9を検出する二次電子検出部10と、二次電子検出部10からの二次電子検出信号を受け分析開始信号を生成し質量分析部8へ送信する質量分析制御部12を、を含んで構成されている。
ここで、真空容器2に多価イオン発生源3が接続され、この真空容器2内には試料台6と質量分析部8と二次電子検出部10とが配設され、質量分析制御部12は、真空容器2の外部に配設されている。真空容器2は、図示しない真空排気装置により後述する多価イオン発生源3の真空排気装置とは独立に真空排気されてもよい。
【0019】
本実施形態では、質量分析部8はTOF(飛行時間:Time of Flight)型の質量分析部8から構成されており、レンズ14と飛行管15と二次イオン7を検出する二次イオン検出器16と、を備えているが、質量分析部8は各種の方式を用いることができる。
【0020】
質量分析制御部12は、二次イオン検出器16からの信号を飛行時間毎に検出する多チャンネルアナライザ20と、多価イオンビーム4が試料5に入射した時間を検出し、分析開始信号を多チャンネルアナライザ20に送出する分析開始信号生成手段21と、多チャンネルアナライザ20からの信号を処理するコンピュータ22と、から構成されている。
【0021】
本発明による多価イオンを用いた表面分析装置1が、従来の質量分析装置と異なる点は下記の通りである。
(1)従来の質量分析装置装置が1価のイオンを試料に照射するのに対して15価以上の多価イオンを用いる点。
(2)多価イオンが照射された試料5から発生する二次イオン7を、質量分析部8及び質量分析制御部12で検出する点。
【0022】
最初に、上記(1)の点に関し、多価イオン発生源3について説明する。本発明に用いる多価イオン発生源3は、15価以上の多価イオンを発生できればその発生方法を問わない。
図2は多価イオン発生源3の構成例を模式的に示す断面図である。多価イオン発生源3において、第1の真空容器31及び第2の真空容器32は、それぞれ、真空排気装置33,34により独立に排気されるようになっている。第1の真空容器31内は、多価イオン発生のためには、1×10−5Pa(パスカル)以下、特に1×10−9Pa以下の極高真空とする。例えば、U92+の生成には10−10Pa(10−12Torr)の真空が必要である。このため、第1の真空容器31は脱ガスのために、図示しないベーキング用ヒータなどを用いたベーキング手段を備えている。このベーキング手段を用いることで、第1の真空容器31は250〜300℃程度にまでベーキングすることができる。
ここで、第1及び第2の真空容器31,32は、少なくともそれらの対向面が、超伝導磁石35からの磁場をドリフトチューブ36に印加するように、非磁性材料から形成されている。非磁性材料としては、オーステナイト系ステンレス鋼(例えば、SUS−304,SUS−316)やアルミニウムを用いることができる。
【0023】
第2の真空容器32には、外部と熱的に遮断するために超伝導磁石35が収容され、この超伝導磁石35は、例えば10K(−263℃)の極低温に冷却される。また、第2の真空容器32の真空度は、10−4Pa程度の高真空にされる。このような高真空を得るためのベーキング手段は不要である。そして、第2の真空容器32の第1の真空容器31側の外周部には、磁気シールド部37が設けられている。この磁気シールド部37は、超伝導磁石35からの磁場が、イオン源電極40の電子源41及びコレクタ42側のドリフトチューブ36に漏れないようにするために配設するものである。この磁気シールド部37には、軟鉄などの強磁性材料を用いればよい。
【0024】
図3は図2に示す電子源41の構成を模式的に示す拡大断面図である。図示するように、電子源41は、第1の真空容器31の左側面の超高真空用フランジ31aに支持部31bを介して、第1の真空容器31の水平方向の中心軸上に配設されている。第2の真空容器32内に収容した超伝導磁石35の空隙(ボア)の直径はDで示している。そして、図中の矢印は、排気経路を示している。電子源41は、左から右方向に、フィラメント(カソード)電極41a、フォーカス電極41b、アノード電極41c、スノート電極41dの順に配置され、それぞれが、絶縁碍子41e,41eにより絶縁されている。さらに、電子源41は、カソード近傍の磁場を0(零)とするためのバッキングコイル(Bucking coil)43を備え、磁気シールド部44に収容されている。
【0025】
ここで、図2に示すように電子源41から発生される電子ビームを、ドリフトチューブ36に当てることなくコレクタ42に導く必要がある。このため、上記のカソード電極41a、アノード電極41c及びスノート電極41dの各電極と絶縁碍子41e,41eとは、嵌め合いにより組み立てることで、相互の軸を高い精度で一致させている。
【0026】
一例として、300mAの電流をカソード電極41aから引き出す際は、フィラメント電位を基準にして、アノード電極41cには+10kV、スノート電極41dには+15kVの電位を与える。このように、各電極とバッキングコイル43及び磁気シールド部44には最大+15kVの高電圧が印加されるが、各部品を小型化することで相互の絶縁を確保しながら、磁気シールド部44の直径を100mmに抑えることができる。この場合には、外径が162mmで内径が102mmの超高真空用フランジ31aに電子源41を収容することができ、第2の真空容器32の電子源41側の直径を152mmに抑えることができる。このため、超高真空用フランジ31aの内径は、磁気シールド部44及びドリフトチューブ36の直径よりも十分大きいので、電子源41領域からの放出ガスを効率良く排気できる。このように、電子源41の寸法を小さくすることで、超伝導磁石35を収容する第2の真空容器32の環状の空隙、即ち超伝導磁石35のボア径を小さくすることができる。
【0027】
ドリフトチューブ36は、幾つかに分割された円筒形電極から構成されていて、その両端が、イオンに対して障壁(井戸型ポテンシャル)を形成するような電場配置となっている。
【0028】
イオンとなる気体は、気体イオン導入手段46によりドリフトチューブ36の側面からその内部に導入される。気体イオン導入手段46は、気体源46a,流量調整部46b,第1の真空容器31内への配管部46cなどから構成されている。
【0029】
一方、固体をイオンとする場合には、ソースとして固体イオン源を用いればよい。
図4は、固体のイオン源を有する多価イオン発生源3の構成を模式的に示す断面図である。図4に示す多価イオン発生源3が、図2に示す多価イオン発生源3と異なるのは、気体イオン導入手段46の代わりに、コレクタ42の右側に固体イオン導入手段48を備えていることである。このような固体イオン導入手段48は、所望のイオンを発生させる針状の固体材料を真空アーク放電させ、1価のイオンにする真空アーク型イオン源などを用いることができる。固体イオン導入手段48から発生したイオンは、第1の真空容器31中でコレクタ42を通過してドリフトチューブ36に注入される。
【0030】
上記した気体イオン導入手段46又は固体イオン導入手段48からなるイオン導入手段によりドリフトチューブ36に注入されるイオンは、一定時間、井戸型ポテンシャルに閉じ込められ、電子による衝突電離が進み多価イオンとなる。この多価イオンのうち、電子の衝突により運動エネルギーが増加して障壁を越えたものが、多価イオン発生源3の外部に取り出される。この際、イオンの電離度が究極、即ち、これ以上電子を当て続けても電離が進まない状態に達したときに、電場配置を変えて多価イオンが外に飛び出すようにして多価イオンビーム4を生成させる。
【0031】
コレクタ42は、ドリフトチューブ36を通過する電子を捕集するための電極であり、例えば、サプレッサ電極、コレクタ電極、エクストラクター電極などの電極から構成されている。カソード及びコレクタ42には、アースに対して最大−数十kVから−300kV程度の電圧が印加されている。この加速電圧は、所望の多価イオンが得られるための電圧であればよい。電子は、ドリフトチューブ36の入射直前に最大加速電圧が印加され、ドリフトチューブ36を通過した電子は、コレクタ42の手前で2〜3kV程度に減速されてコレクタ42に捕集される。このため、コレクタ42には、この電圧と電子ビーム電流との積の電力が捕集される。この電力は、上記コレクタ42手前の電圧が3kVで電子ビーム電流が300mAの場合には900Wとなり、絞られた状態でコレクタ42に当たると、当たった場所が溶ける。したがって、コレクタ42は、冷媒で冷却した電極構造として電子ビームを広げながら電子を捕集する。
【0032】
次に、超伝導磁石35について説明する。
図2及び図4に示すように、超伝導磁石35は第1のコイル35a及び第2のコイル35bからなる、所謂ヘルムホルツ型コイルを用いている。第1のコイル35a及び第2のコイル35bの半径をaとし、これらのコイルの間隔を半径aと同じaとしたときに、対向するコイルの中央部分の磁場を一様にすることができる。ここで、超伝導磁石35は、図示しないHeを冷媒とするクライオスタットや閉サイクル冷凍機により冷却されて、超伝導状態に保持される。
【0033】
上記多価イオン発生源3の特徴は、第2の真空容器32内にイオン閉じ込めのための超伝導磁石35が配置され、第2の真空容器32の環状の空隙に第1の真空容器31が挿入されていることである。つまり、超伝導磁石35の空隙(ボア)内に第1の真空容器31が挿入されていることである。上記のように、この第1の真空容器31内には、電子源41、ドリフトチューブ36及びコレクタ42からなるイオン源電極40が配置されている。また、超伝導磁石35を収容する第2の真空容器32は、第1の真空容器31から両者の真空を破らずに脱着が可能である。このため、イオン源電極40を第1の真空容器31に収納したものを作ればよく、超伝導磁石35は市販品を用いることができるので製作が容易である。したがって、本発明に用いる多価イオン発生源3においては、従来の多価イオン発生源のように超伝導磁石35とイオン源電極40とを一体化した構造を精度良く製作する必要がない。
【0034】
本発明に用いることができる多価イオン発生源3は以上のように構成されており、次にその動作について説明する。
図5は、多価イオン発生源3に印加される電源を説明するための模式的なブロック図である。電子の加速電圧が40kVの一例を示している。図示するように、電子源41のフィラメント電極41aには15V,2A、フォーカス電極41bには50V,1mA、アノード電極41cには10kV、スノート電極41dには15kV,1mA、の電源が−30kVの電源に上乗せされて供給されている。また、ドリフトチューブ36の各電極には、10kVの電源上に、第1電極500V、第2電極500V、第3電極500Vがそれぞれ上乗せされて供給されている。
さらに、コレクタ42のサプレッサ電極、コレクタ電極、エクストラクター電極には、それぞれ、2kV、3kV,500mA、−3kVの電源が、−30kVの電源に上乗せされて供給されている。
【0035】
上述のように、電子源41から発生された電子は、超伝導磁石35の磁場中に配置されたドリフトチューブ36を通る。図5に示す電源を用いた場合には、電子源41とドリフトチューブ36の間で電子が最大40kVまで加速される。この加速されてドリフトチューブ36に入射した電子は、超伝導磁石35によりドリフトチューブ36に形成される強磁場で圧縮され大電流密度(〜1000A/cm2)の電子ビームとなり、コレクタ42に捕集される。図5に示す電源を用いた場合には、コレクタ42には2〜3kV位まで減速されて捕集される。この際、電子は、ドリフトチューブ36の直前で最大加速電圧が印加される。原子を電離するためには、束縛電子の電離エネルギー以上のエネルギーを持った電子と衝突させなければならないので、重元素(例えば、キセノン(Xe)など)を完全に電離させるには1s軌道の電離エネルギー(Xeでは35keV)より高いエネルギーが必要で、加速電圧35kV以上が必要である。
【0036】
一方、気体イオン導入手段46から導入されるイオンとなる気体は、ドリフトチューブ36内でイオン化される。ドリフトチューブ36の電極は幾つかに分割されており、両端がイオンに対して障壁(井戸型ポテンシャル)が形成されるように電位が与えられている。イオンは一定時間、この井戸型ポテンシャルに閉じ込められ、気体の原子を電子ビームに繰り返し衝突させることにより電離が進む。電子による衝突電離が進み多価イオンビーム4となる。この多価イオンのうち、電子の衝突により運動エネルギーが増加し障壁を越えたものがイオン源外部に取り出される。このように、イオンの電離度が究極、即ち、これ以上電子を当て続けても電離が進まない状態に達したときに、電場配置を変えて多価イオンが外に飛び出すようにして多価イオンビーム4を生成させる。
このドリフトチューブ36で生成した多価イオンが、ドリフトチューブ36から引き出される際には、ドリフトチューブ36とコレクタ42との間において40kVで一且加速され、第1の真空容器31を出る際は、この第1の真空容器31はアース電位であるので10kVまで減速されて出射する。
【0037】
得られる多価イオンビーム4の強度は、イオンがトラップされているドリフトチューブ36の長さL1(図2参照)及びエミッション電流に比例し、電離度が究極に至るまでの時間は電子の電流密度に比例する。なお、ドリフトチューブ36の長さが長すぎる場合にはプラズマの不安定性が出現し、高電離の多価イオンができなくなる。
また、電子ビーム電流密度は、超伝導磁石35によりドリフトチューブ36に印加される磁場強度が高いほど高くなる。但し、比例関係ではない。このため、単位時間当たりに得られる多価イオンビーム4の強度は、電子ビーム電流密度が高くトラップ領域が長いほど高くすることができる。
【0038】
多価イオン発生源3は、以上の機構において従来の多価イオン発生源と同様である。
本実施形態の多価イオン発生源3の特徴は、イオン源電極40を収納する第1の真空容器31のための真空排気装置33と、超伝導磁石35を収容する第2の真空容器32のための真空排気装置34とを着脱して分離することができる点にある。このため、超伝導磁石35を収容する第2の真空容器32を、第1の真空容器31から分離した状態で、第1の真空容器31及び収容されているイオン源電極40を十分に加熱して脱ガスをすることができる。したがって、多価イオン発生のための第1の真空容器31を短時間で極高真空とすることができる。例えば、誤って電子ビームをドリフトチューブ36に当てたときには、多価イオン発生源3では、イオン源電極40を収納した第1の真空容器31のみ修理すればよい。
これにより、多価イオン発生源3においては、イオン源電極40の真空立ち上げが短時間で済むので、操作性、保守性に優れた多価イオン発生源3となる。
【0039】
試料台6は、その位置が制御できるように可動な試料台とすればよい。例えば、試料台6は、XYZステージなどを用いることができる。このXYZステージ6は、電歪素子やステップモーターを用いることができる。
【0040】
次に、上記(2)の点に関し、多価イオンビーム4が照射された試料5から発生する二次イオン7の検出を行う質量分析部8及び質量分析制御部12について詳しく説明する。
質量分析部8において、試料5へパルス状の多価イオンビーム4が照射されると試料5の表面に存在する元素から二次イオン7及び二次電子9が発生する。発生した二次イオン7は、レンズ14で集められ飛行管15へ入射し、二次イオン7の質量等で決まる飛行時間を経て、二次イオン検出器16に入射する。
【0041】
一方、試料5に多価イオンビーム4が照射したときに発生する二次電子9は、二次電子検出部10によって検出される。この場合、1個の多価イオンビーム4の照射により数十個から数百個の二次電子9が発生するので、照射された多価イオンビーム4の数を正確に計測できる。この二次電子検出部10は、試料5へ1個の多価イオンビーム4が照射したときに発生する二次電子9を高感度で検出できる二次電子検出器10A等を用いることが好ましい。
【0042】
図1に示す二次電子検出部10は、入射する多価イオンビーム4の試料5へのビーム進行方向に対して垂直面内に配置され、上下方向の1個ずつ、つまり2個の二次電子検出器10A,10Bが配設されている。図示するように、二次電子検出部10と試料5との間には、二次電子9を集めるためのレンズ作用を有するグリッド10Cなどのレンズを備えてもよい。さらに、二次電子増倍のために二次電子検出器10A等に印加する電圧を外部に漏らさないようにシールド部をさらに設けてもよい。このシールド部は金属からなり、二次電子検出器10A,10Bを取り囲む形状とすればよい。
【0043】
分析開始信号を多チャンネルアナライザ20に送出する分析開始信号生成手段21は、二次電子検出部10からの出力が入力され、その測定データの処理を行う。一般に、二次電子信号は雑音が多いため誤計測をする可能性がある。二次電子検出部10は、生じる雑音による計測誤差を無くすために、上記のように電気的に独立した複数の二次電子検出器10A,10Bを備えて構成することができる。
【0044】
複数の二次電子検出器10A,10Bを備えた場合には、分析開始信号生成手段21には、試料5から発生する複数の二次電子9の計測データが一致するか否かを判定する判定回路を設けることが好ましい。そして、複数の二次電子検出器10A,10Bの計測データが一致した場合に、正しい計測データであると判定すればよい。このように、多価イオンビーム4の照射では多数の二次電子9が放出されるため、少なくとも2個の二次電子検出器10A,10Bから得られた信号が一致したときのみ真の信号と見做すことで、正確なイオン数の計測が可能になる。
【0045】
図6は、(A)が分析開始信号生成手段21の構成を示す模式図であり、(B)が分析開始信号生成手段21で用いるAND回路24の真理値表を示す図である。
図6(A)に示すように、分析開始信号生成手段21は、判定回路として論理積演算を行うAND回路24等を備えている。その入力24A,24Bには、2つの二次電子検出器10A,10Bの出力が接続されている。二次電子検出器10A,10Bの出力は図示しない増幅器によりAND回路24のハイレベル及びローレベルの信号を出力するように信号処理がされている。上記の二次電子検出器10Aとしては、二次電子9も検出できる所謂光電子増倍管を使用できる。このような電子増倍効果を有する二次電子検出器10Aとしては、複数のチャンネルトロンや複数のアノード電極を持つマイクロチャンネルプレートを用いることができる。
【0046】
図6に示す二次電子検出器10A,10Bにおいては、多価イオン1個による二次電子9を検出したときには、ハイレベル、即ち1の信号をAND回路24に送出する。逆に、二次電子9を検出しない場合にはローレベル即ち0の信号をAND回路24に送出する。したがって、図6(B)のAND回路24の真理値表に示すように、AND回路24においては、その2入力24A,24Bが共に1の場合に出力24Cが1となる。これ以外の入力の場合の出力24Cは何れも0である。つまり、2つの二次電子検出器10A,10Bが共に、多価イオン1個による二次電子を検出したときには、AND回路24は多価イオン1個が発生したと判断する。AND回路24を用いた判定回路によれば、特に、試料5へ入射する多価イオンの強度が著しく弱い場合、例えば多価イオンのパルス周波数が1Hz又は1Hz以下の場合であっても多価イオンの発生を検出することができる。
【0047】
なお、2入力AND回路24を用いた判定回路について説明したが、精度を上げるために3入力以上の二次電子検出器を用いる場合には、二次電子検出器の数だけの入力を備えたAND回路24で行えばよい。これにより、複数の二次電子検出器の計測データが一致した場合に、多価イオンビーム4が照射されたという判定をすることができるので、雑音を除去して精度を高めることができる。
【0048】
上記判定回路24は、多価イオンのパルス周波数が100Hz以上の場合には、二次電子検出部10からの信号を、高速プリアンプで増幅し、この信号を時間弁別器(Timing Discriminator)を介して、多チャンネルアナライザ20に入力する構成としてもよい。
【0049】
本実施形態の表面分析装置1は以上のように構成されており、次にその動作について説明する。
多価イオン発生源3から発生した多価イオンビーム4が、試料5に照射される。この場合、試料5への多価イオンビーム4の照射位置は、可動な試料台6により位置制御がされている。
試料5の位置が決定された後、多価イオンビーム4が試料5に照射されると、試料5の所定位置に存在する原子に応じた二次イオン7が発生する。この場合に発生する二次電子9が二次電子検出器10A,10Bにより検出される。その有無が分析開始信号生成手段21により判定される。したがって、分析開始信号生成手段21のAND回路24の出力がハイレベルの場合が、試料5に多価イオンビーム4が照射された時間に対応する。
【0050】
質量分析部8がTOF型の質量分析装置である場合には、質量分析制御部12に入力されるAND回路の出力24Cがハイレベルとなった時刻が測定開始の時間となり、この信号を質量分析測定の開始信号と呼ぶことにする。
これにより、開始信号以降に得られるTOF型の質量分析部8における二次イオン検出器16からの信号が、試料5から発生した二次イオン7の信号である。
次に、二次イオン7の信号検出が終了した時点で、二次イオン検出器16から質量分析制御部12へ、測定停止信号16Aが送出される。
したがって、上記開始信号から測定停止信号16Aまでの信号を解析することにより、試料5の所定位置に存在していた原子の質量分析を行うことができる。
【0051】
本発明の表面分析装置1によれば、特に、15価以上の多価イオンビーム4を用いることで、短時間に試料5から二次イオン7を発生させたり、スパッタリング効率を顕著に増加させたりすることができる。これにより、表面分析装置1によれば、15価以上の多価イオンビーム4を試料5に照射することにより試料5から発生する二次イオン7の信号は、価数の4乗等のべき数に比例して増大するため、試料5の表面の分析を短時間で分析することができる。このため、表面清浄化を施した試料5の分析においても残留ガス等の再付着が起こらず高精度な分析を行うことができる。
【0052】
本発明の表面分析装置1によれば、試料5の表面に15価以上の多価イオンビーム4を照射することで、他手法では分析が困難であった所謂質量の小さいH+等の元素を、高感度でかつ短時間で測定することができる。
【0053】
また、試料5の位置決めを、nmオーダーの分解能を有するXYZステージ6により制御する場合には空間分解能を著しく高めることができる。したがって、表面分析装置1によれば、空間分解能が高く、微量な元素を高感度で、かつ、短時間で分析することができる。
【0054】
上記の本発明の表面分析装置1においては、さらに、試料5の表面の観察するための観察装置を備えていてもよい。このような観察装置としては、電子線回折装置、走査型電子顕微鏡(SEM)などが挙げられる。
【0055】
本発明の表面分析装置1によれば、多チャンネルアナライザ20の検出値からコンピュータ22を用いた解析により種々の表面分析を行うことができる。
次に、表面分析方法の解析例について説明する。
(イ)15価以上の多価イオンビーム4をパルスとして試料5に照射し、パルス毎の測定を所定の回数繰り返し行う。
(ロ)測定誤差を除去するために、上記(イ)の測定で取得した複数のデータをコンピュータ22を用いて積算して、平均値が一定の値に収束した段階で多価イオンビーム4の照射を停止する。
上記の表面分析方法によれば、単元素からなる試料5の表面分析を短時間で行うことができる。
【0056】
試料5が2つの元素A,Bからなり、その原子比がx:yである場合の表面分析方法について説明する。この場合、試料5の化学式は、AxByである。
(イ)15価の多価イオンビーム4をパルスとして試料5に照射し、パルス毎の測定を所定の回数繰り返し行う。
(ロ)測定誤差を除去するために、上記(イ)の測定で取得した複数のデータをコンピュータ22を用いて積算して、平均値が一定の値に収束した段階で多価イオンビーム4の照射を停止する。
(ハ)15価以上の多価イオンビーム4(価数=15+n)をパルスとして試料5に照射し、1回パルス毎の測定を所定の回数繰り返し行う。
(ニ)測定誤差を除去するために、上記繰り返し測定したデータをコンピュータ22を用いて積算して、平均値が一定の値に収束した段階で多価イオンビーム4の照射を停止する。
(ホ)上記(ハ)及び(ニ)の測定を繰り返し行い、所定の多価数、例えば価数が50までのデータを得る。
(へ)上記測定のデータから、構成元素A及びBの二次イオン7のカウント数と価数との関係を算出する。構成元素A及びBのカウント数の比(x:y)が得られる価数を求める。
(ト)上記(へ)のステップで構成元素A及びBのカウント数の比(x:y)が得られる価数を得た場合には、同じ組成の試料5を次回の測定の際には、この価数の多価イオンビーム4を照射すれば、化学量的組成が反映された表面分析を行うことができる。
なお、試料5が3つ以上の元素からなる場合にも、上記(イ)〜(ト)の測定において、さらに第3以上の元素C等の二次イオン7のカウント数と価数との関係を算出することにより表面分析ができる。
【0057】
試料5が2つの元素A,Bからなり、その組成が不明である場合の表面分析方法について説明する。
上記の原子比(x:y)が判明している試料5を標準試料とする。2つの元素A,Bからなり、その組成が不明である試料5に対して、標準試料の化学量的組成が反映された分析ができる価数を有する多価イオンビーム4を照射し、上記(イ)及び(ロ)の測定を行い、元素A,Bのカウント数を得る。得られた元素A,Bのカウント数を、それぞれ、na、nbとする。この元素A,Bのカウント数na,nbを、標準試料のカウント数を基準として比較することで、組成の不明な試料5の組成を求めることができる。
ここで、試料5が3つ以上の元素からなる場合にも、上記測定において、さらに第3以上の元素C等の二次イオン7のカウント数と標準試料のカウント数との関係を算出することにより表面分析ができる。
【0058】
表面分析装置1においては、多価イオン発生源3と試料5との間に、さらに減速レンズを設けてもよい。減速レンズを設けることで、多価イオン発生源3から入射する多価イオンビーム4の加速電圧を制御することができる。この多価イオンビーム4が種々の多価イオンを含む場合には、所望の多価イオンを選択するために、さらに、イオン分離器を設けてもよい。イオン分離器としては、分析磁石(偏向磁石)や磁場及び電場によるウィーンフィルターなどを用いることができる。
【0059】
表面分析装置1においては、多価イオン発生源3と試料5との間に、さらに多価イオンガイド26を設けてもよい。
図7は、図1に示した多価イオンガイド26の先端部の構成を模式的に示す断面図である。多価イオンガイド26においては、その一端26Aに多価イオンビーム4を入射し、他端26Bから多価イオンビーム4を出射する。つまり、多価イオンビーム4が、試料5を載置する試料台6に導くためのガイドとなっている。そして、多価イオンガイドの他端側26Bの先端部26Cが、直径がおおよそ100nm以下程度の開口部を有する、所謂キャピラリー構造となっている。この多価イオンガイド26は、好ましくは絶縁体からなり、例えばセラミックや石英ガラスを用いることができる。このようなキャピラリーは、例えば石英の細管を加熱溶融し、溶融状態にて引き伸ばし加工を行うことで製作することができる。
【0060】
多価イオンビーム4が多価イオンガイド26に入射すると、多価イオンガイド26の内壁が帯電する。このために、多価イオンビーム4は多価イオンガイド26の内壁から反発されて、多価イオンガイド26の中心部を通過し、キャピラリー構造となっている先端部26Cから試料5に入射する。これにより、多価イオンビーム4は、多価イオンガイドの先端部26Cでそのビーム径が絞られ、即ち、収束して試料5に入射する。
【0061】
(分析例1)
以下、本発明の表面分析装置1による分析例について説明する。
試料5となるSi(シリコン)の(100)表面を1200℃で5秒間の加熱を3回行った後、表面に50価のXeイオン(Xe50+)を、加速電圧を3keVで照射した。二次電子検出器10A,10Bとしてはチャンネルトロンを用いた。さらに、試料5に照射される多価イオンビーム4を計測するために、試料5の裏面側に別のチャンネルトロンを配置した。
図8は、分析例1のTOF−SIMSによるプロファイルを示す図である。図8において、横軸は飛行時間(ns)を、縦軸は検出した二次イオン7のカウント数を示している。図8から明らかなように、Si表面からはH+,H2+,O+,O2+,Si+,Si2+,Si3+,Si4+,Si2+,SiO+の各イオンが検出されていることが分かる。
【0062】
(分析例2)
Si(シリコン)の(100)表面を、所謂白木法によりウエット処理をし、900℃で3分間のアニールを施した。この試料5の表面に50価のXeイオン(Xe50+)を、分析例1と同じ条件で照射した。
図9に分析例2のTOF−SIMSによるプロファイルを示す。図9の横軸及び縦軸は図8と同じである。図9から明らかなように、Si表面からはH+,H2+,O+,O2+,Si+,Si2+,Si3+,Si4+,Si2+,SiO+の各イオンが検出されていることが分かる。
【0063】
(分析例3)
試料5となるSi(シリコン)の(100)表面を1200℃で12時間加熱して表面清浄化を行った後、表面に50価のXeイオン(Xe50+)を、分析例1と同じ条件で照射した。
図10に分析例3のTOF−SIMSによるプロファイルを示す。図10の横軸及び縦軸は図8と同じである。図10から明らかなように、Si表面からはH+,H2+,Si+,Si2+,Si3+,Si4+,Si2+の各イオンが検出されていることが分かる。
【0064】
(分析例1〜3の比較)
分析例1及び分析例2のSi表面から検出される二次イオン7は、その種類が同じであることが分かる。また、検出されたH+イオンのカウント数もほぼ同じ位である。
一方、分析例3の試料5からは、分析例1及び分析例2で検出されたO+,O2+,SiO+が検出されないことが分かる。また、検出されたH+イオンのカウント数は、分析例1及び分析例2よりもほぼ1桁小さいことが分かる。これらの結果から、分析例3の場合には、Siの表面における水素の量が非常に少ないことが分かる。
【0065】
(分析例4)
試料5となるSiの(100)表面を1200℃で12時間加熱して表面清浄化を行った後、表面に50価のXeイオン(Xe50+)を、分析例1と同じ条件で照射した。
図11に分析例4のTOF−SIMSによるプロファイルを示す。図11は、Si(100)表面をアニールした後10分及び45分経過したときのプロファイルを示しており、横軸及び縦軸は図8と同じである。
図11から明らかなように、Si表面からはH+,H2+,Si+,Si2+,Si3+の各イオンが検出され、特に45分経過後では、H+のカウント数が著しく増大していることが分かる。
【0066】
図11の挿入図は、アニール後のH+イオンのイールドの時間依存性を示す図である。図の横軸はアニール後の経過時間(分)を示し、縦軸はH+イオンのイールドを示している。この挿入図から、アニール直後ではH+イオンは殆ど検出されないことが分かり、10分,20分,30分,40分,45分経過後にはH+イオンが検出され、しかも、イールドは経過時間と共に増加することが分かる。
分析例4の結果によれば、Si表面の清浄化処理直後からの経過時間と共に水素が増大していく様子を観察することができた。
【0067】
分析例4に示すように、長時間の測定時間を要する従来技術では、分析が困難であった試料5となるSi表面に形成した清浄表面を正確に評価できることが判明した。
【0068】
(分析例5)
試料5となるSi(シリコン)の(111)表面を1200℃で5秒間の加熱を3回行った後、表面へ29価,34価,38価,44価,50価のXeイオン(Xe50+)を、加速電圧175keVで照射した。
図12は、分析例5において、29価のXeイオン(Xe29+)及び50価のXeイオン(Xe50+)を照射したときのTOF−SIMSによるプロファイルを示す図である。図12において、横軸は飛行時間(ns)を、縦軸は検出した二次イオン7のカウント数を示している。
図12から明らかなように、29価のXeイオン(Xe29+)の照射によりSi表面からH+,Si3+,Si2+,Si+の各イオンが検出されていることが分かる。50価のXeイオン(Xe50+)の照射によりSi表面から検出されるイオンは、29価のXeイオン(Xe29+)の照射により検出されたイオンの他に、さらに、H2+,Si4+が検出され、しかも検出されたイオンのカウント数が多いことが分かる。
【0069】
図13は、分析例5において検出したH+イオン及びSi+イオンのスパッタリングイールドの価数依存性を示す図である。図13において、横軸は照射したXeイオンの価数を示し、縦軸はH+イオン及びSi+イオンのスパッタリングイールドを示している。
図13から明らかなように、H+イオンのスパッタリングイールドは、価数qに対して、4.1乗、つまり、q4.1で増加していることが分かる。同様に、Si+イオンのスパッタリングイールドは、価数qに対して、1.5乗、つまりq1.5で増加することが分かった。
【0070】
(分析例6)
試料5となるSiO2の表面へ15価から50価まで5価ずつ増加させた種々の価数のIイオン(I15+,I20+,I25+,I30+,I35+,I40+,I45+,I50+)を加速電圧175keVで照射した。
図14は、分析例6において、15価のIイオン(I15+)及び50価のIイオン(I50+)を照射したときのTOF−SIMSによるプロファイルを示す図である。図14において、横軸は飛行時間(ns)を、縦軸は検出した二次イオン7のカウント数を示している。
図14から明らかなように、15価のIイオン(I15+)照射ではSi表面からH+,Si+の各イオンが検出されていることが分かる。50価のIイオン(I50+)の照射によりSi表面から検出されるイオンは、15価のIイオン(I29+)の照射により検出されたイオン(H+,Si+)の他に、さらに、O+,O2+,SiO+が検出され、しかも検出された各イオンのカウント数が多いことが分かる。
【0071】
分析例6における多価イオンによるSiO2膜の表面分析の場合には、酸素が陽イオンとして検出されている点が多価イオンによる表面分析の特徴である。これに対して、1価イオンを照射源とする従来のSIMS分析の場合には、酸素が陰イオンとして検出されていた。
【0072】
図15は、分析例6において検出したSi+イオン及びO+イオンのスパッタリングイールドの価数依存性を示す図である。図15において、横軸は照射したIイオンの価数を、縦軸はSi+イオン及びO+イオンのスパッタリングイールドを示している。
図15から明らかなように、Si+イオンのスパッタリングイールドは、価数qに対してほぼ比例して増加していることが分かる。
一方、O+イオンのスパッタリングイールドは、価数qに対して2.5乗、つまりq2.5で増加することが分かった。価数が35価よりも大きい場合(q>35)では、O+イオンのイールドがSi+イオンのイールドよりも大きくなることが判明した。従って、価数が50価(I50+)の場合には、O+のスパッタリングイールドは、Si+のスパッタリングイールドの約2倍であり、SiO2の化学量論的組成を反映した二次イオン7の収率となっていることが判明した。
【0073】
(分析例7)
試料5をSiO2からTiO2とした以外は、分析例6と同様に15価から50価まで5価ずつに増加させた種々の価数のIイオン(I15+,I20+,I25+,I30+,I35+,I40+,I45+,I50+)を、加速電圧175keVで照射した。試料5がTiO2の場合には、価数が25価以上でTi+やO+のイオン信号が得られた。
図16は、分析例7において検出したTi+イオン及びO+イオンのスパッタリングイールドの価数依存性を示す図である。図16において、横軸は照射したIイオンの価数を、縦軸はTi+イオン及びO+イオンのスパッタリングイールドを示している。
図16から明らかなように、Ti+イオンのスパッタリングイールドは、価数qが30以上ではほぼ比例して増加していることが分かる。
一方、O+イオンのスパッタリングイールドは、価数qに対して4乗、つまりq4で増加することが分かった。価数が35価よりも大きい場合(q>35)では、O+イオンのイールドがTi+イオンのイールドよりも大きくなることが判明した。従って、価数が50価(I50+)の場合には、O+のスパッタリングイールドは、Ti+のスパッタリングイールドの約2倍であり、TiO2の化学量論的組成を反映した二次イオン7の収率となっていること判明した。
【0074】
本発明は上記実施例に限定されることなく、特許請求の範囲に記載した発明の範囲内で種々の変形が可能であり、それらも本発明の範囲内に含まれることはいうまでもない。例えば、上記実施の形態において、多価イオンガイド26の形状や真空容器2の構造や付属させる観察機器の構成などは、試料5に応じて適宜変更できる。
【図面の簡単な説明】
【0075】
【図1】本発明による多価イオンを用いた表面分析装置の構成を模式的に示す断面図である。
【図2】多価イオン発生源の構成例を模式的に示す断面図である。
【図3】図2に示す電子源の構成を模式的に示す拡大断面図である。
【図4】固体のイオン源を有する多価イオン発生源の構成を模式的に示す断面図である。
【図5】多価イオン発生源に印加される電源を説明するための模式的なブロック図である。
【図6】(A)は分析開始信号生成手段の構成を示す模式図、(B)は分析開始信号生成手段で用いるAND回路の真理値表を示す図である。
【図7】図1に示した多価イオンガイドの先端部の構成の模式的断面図である。
【図8】分析例1のTOF−SIMSによるプロファイルを示す図である。
【図9】分析例2のTOF−SIMSによるプロファイルを示す図である。
【図10】分析例3のTOF−SIMSによるプロファイルを示す図である。
【図11】分析例4のTOF−SIMSによるプロファイルを示す図である。
【図12】分析例5において、29価のXeイオン(Xe29+)及び50価のXeイオン(Xe50+)を照射したときのTOF−SIMSによるプロファイルを示す図である。
【図13】分析例5において検出したH+イオン及びSi+イオンのスパッタリングイールドの価数依存性を示す図である。
【図14】分析例6において、15価のIイオン(I15+)及び50価のIイオン(I50+)を照射したときのTOF−SIMSによるプロファイルを示す図である。
【図15】分析例6において検出したSi+イオン及びO+イオンのスパッタリングイールドの価数依存性を示す図である。
【図16】分析例7において検出したTi+イオン及びO+イオンのスパッタリングイールドの価数依存性を示す図である。
【符号の説明】
【0076】
1:表面分析装置
2:真空容器
3:多価イオン発生源
4:多価イオンビーム
5:試料
6:試料台
7:二次イオン
8:質量分析部
9:二次電子
10:二次電子検出部
10A,10B:二次電子検出器
10C:グリッド
12:質量分析制御部
14:レンズ
15:飛行管
16:二次イオン検出器
16A:測定停止信号
20:多チャンネルアナライザ
21:分析開始信号生成手段
22:コンピュータ
24:AND回路
24A,24B:入力
24C:出力
26:多価イオンガイド
26C:先端部
31:第1の真空容器
31a:超高真空用フランジ
31b:支持部
32:第2の真空容器
33:第1の真空容器の真空排気装置
34:第2の真空容器の真空排気装置
35:超伝導磁石
35a:第1のコイル
35b:第2のコイル
36:ドリフトチューブ
37,44:磁気シールド部
40:イオン源電極
41:電子源
41a:フィラメント(カソード)電極
41b:フォーカス電極
41c:アノード電極
41d:スノート電極
41e:絶縁碍子
42:コレクタ
43:バッキングコイル
46:気体イオン導入手段
46a:気体源
46b:流量調整部
46c;配管部
48:固体イオン導入手段
【特許請求の範囲】
【請求項1】
試料を搭載する試料台と、
上記試料台に搭載された試料に価数が15以上の多価イオンビームを照射する多価イオン発生源と、
上記試料に多価イオンビームが照射されることにより生じる二次イオンを検出する質量分析部と、
上記試料に多価イオンビームが照射されることにより生じる二次電子を検出する二次電子検出部と、
上記二次電子検出部からの二次電子検出信号を受けて分析開始信号を生成し上記質量分析部へ送信する質量分析制御部を、備えた、表面分析装置。
【請求項2】
前記二次電子検出部は、電気的に独立した複数の二次電子検出器を有する、請求項1に記載の表面分析装置。
【請求項3】
前記二次電子検出器は、複数のチャンネルトロン又は複数のアノード電極を持つマイクロチャンネルプレートからなる、請求項2に記載の表面分析装置。
【請求項4】
前記多価イオン発生源は、一端に前記多価イオン発生源からの多価イオンビームが入射され他端から前記多価イオンビームが出射される多価イオンガイドを備えており、該多価イオンガイドの他端部はキャピラリー構造である、請求項1に記載の表面分析装置。
【請求項5】
多価イオンビームを15価以上の価数で変化させて試料へ照射し、
上記試料表面の特定の元素から生じる二次イオンのカウント数と価数との関係を算出して、上記特定の元素に対する多価イオンビームの価数を決定する、表面分析方法。
【請求項6】
前記試料が2以上の構成元素からなり、各構成元素から生じる二次イオンのカウント数と価数との関係を算出し、各構成元素の組成比を与える多価イオンの価数を決定する、請求項5に記載の表面分析方法。
【請求項1】
試料を搭載する試料台と、
上記試料台に搭載された試料に価数が15以上の多価イオンビームを照射する多価イオン発生源と、
上記試料に多価イオンビームが照射されることにより生じる二次イオンを検出する質量分析部と、
上記試料に多価イオンビームが照射されることにより生じる二次電子を検出する二次電子検出部と、
上記二次電子検出部からの二次電子検出信号を受けて分析開始信号を生成し上記質量分析部へ送信する質量分析制御部を、備えた、表面分析装置。
【請求項2】
前記二次電子検出部は、電気的に独立した複数の二次電子検出器を有する、請求項1に記載の表面分析装置。
【請求項3】
前記二次電子検出器は、複数のチャンネルトロン又は複数のアノード電極を持つマイクロチャンネルプレートからなる、請求項2に記載の表面分析装置。
【請求項4】
前記多価イオン発生源は、一端に前記多価イオン発生源からの多価イオンビームが入射され他端から前記多価イオンビームが出射される多価イオンガイドを備えており、該多価イオンガイドの他端部はキャピラリー構造である、請求項1に記載の表面分析装置。
【請求項5】
多価イオンビームを15価以上の価数で変化させて試料へ照射し、
上記試料表面の特定の元素から生じる二次イオンのカウント数と価数との関係を算出して、上記特定の元素に対する多価イオンビームの価数を決定する、表面分析方法。
【請求項6】
前記試料が2以上の構成元素からなり、各構成元素から生じる二次イオンのカウント数と価数との関係を算出し、各構成元素の組成比を与える多価イオンの価数を決定する、請求項5に記載の表面分析方法。
【図1】


【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【公開番号】特開2009−250903(P2009−250903A)
【公開日】平成21年10月29日(2009.10.29)
【国際特許分類】
【出願番号】特願2008−101984(P2008−101984)
【出願日】平成20年4月9日(2008.4.9)
【出願人】(503360115)独立行政法人科学技術振興機構 (1,734)
【出願人】(504133110)国立大学法人電気通信大学 (383)
【出願人】(504150450)国立大学法人神戸大学 (421)
【Fターム(参考)】
【公開日】平成21年10月29日(2009.10.29)
【国際特許分類】
【出願日】平成20年4月9日(2008.4.9)
【出願人】(503360115)独立行政法人科学技術振興機構 (1,734)
【出願人】(504133110)国立大学法人電気通信大学 (383)
【出願人】(504150450)国立大学法人神戸大学 (421)
【Fターム(参考)】
[ Back to top ]