
表面機能性部材の製造方法、表面機能性部材及び電気泳動素子

【課題】優れた使用耐久性を有し、かつ長時間に亘って機能を維持し得る表面機能性部材の製造方法、及び表面機能性部材並びに電気泳動素子を提供する。
【解決手段】表面にカルボキシル基を有する基材上に、アミノ基を有する機能性素材を、縮合剤の存在下、直接結合させて得られる表面機能性部材の製造方法であって、水系連続相中で反応させることを特徴とする表面機能性部材の製造方法を提供する。
【解決手段】表面にカルボキシル基を有する基材上に、アミノ基を有する機能性素材を、縮合剤の存在下、直接結合させて得られる表面機能性部材の製造方法であって、水系連続相中で反応させることを特徴とする表面機能性部材の製造方法を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、表面機能性部材の製造方法、及び表面機能性部材及び電気泳動素子に関するものであり、詳細には、アミノ基を有する各種機能性素材を、縮合剤の存在下で、直接結合して得られる表面機能性部材の製造方法、及び当該方法により作製された表面機能性部材、並びに該表面機能性部材を用いた電気泳動素子に関するものであり、更に詳細には、PLD(ペーパーライクディスプレー)、電子写真現像剤(トナー)、インクジェット(インク)等各種の用途に用いられる機能性表面層を有する応用範囲の広い、簡便な表面機能性部材の製造方法、及び表面機能性部材及び電気泳動素子に関する。
【背景技術】
【0002】
従来、任意の基材に、高分子機能性素材、又は低分子機能性素材を、吸着させ、あるいは反応させることにより、各種機能を有する表面層が形成された表面機能性部材について、種々の技術提案がなされている。
特に、機材との密着力が強く、機械的耐磨耗性や有機溶剤耐性に優れ、かつ優れた耐久性を有している表面機能性部材が得られることを目的として、基材と共有結合により、直接結合している方式の表面機能性部材が注目されている。
【0003】
例えば、(1)クロム硫酸水溶液などの薬品処理、(2)カップリング剤による処理、(3)蒸気処理、(4)グラフト化処理、(5)電気化学的な方法等が知られている(例えば、下記非特許文献1参照。)。このうち、(1)及び(5)は、酸化反応、あるいは還元反応により新たに反応性の置換基を形成するもので機能性素材の導入には不適である。
また、上記(3)の、蒸気処理による機能性素材の導入においては、機能性素材の沸点がある程度低いことが条件となり、装置が大掛かりとなりコスト上においても課題を有している。
【0004】
一方、上記(2)のカップリング剤による処理は、基材と直接共有結合により結合しているため、基材との密着性が強く、機械的な耐磨耗性や有機溶剤耐性が高く、耐久性にも優れた表面機能性部材が得られていた。
しかしながら、カップリング剤による処理は、基本的に加熱反応であり、通常200℃以上、触媒を使用しても150℃以上の温度が必要となる。機能性素材の熱劣化を防止するためには、温度は低く抑える方が好ましい。
また、機能性素材のカップリング剤化(機能性素材中間体)を行う工程が必要であるため、製造コストの点からは不利である。
(4)のグラフト化処理は、光、プラズマ、放射線等の反応エネルギーを付与しながら、気相、あるいは液相処理を行う方法であるが、これにおいては、大きな光(反応)エネルギー下に晒されることになるため、機能性素材の光劣化が大きな課題となっていた。
特に基材として粒子を選定した場合には、最も一般的な表面機能化方法として、シード重合法があり、現在、活発な研究開発が進められている。シード重合法は、重合体粒子(シード粒子)に単量体及び必要に応じて種々の機能性化合物を吸収・膨潤させて、このシード粒子に吸収された単量体を粒子表面で重合することにより製造する方法である。
例えば、架橋性単量体を使用し、吸収・膨潤が困難であるため、非常に複雑で、精度の高い重合法が開示されており(例えば、下記特許文献1参照。)、また、単量体と油溶性染料によるシード重合を膨潤剤(添加剤)を含有しない水系媒体中での重合が開示されている(例えば、下記特許文献2参照。)。
シード重合法による表面機能化は、吸収・膨潤が非常に困難で、多段階吸収法、膨潤剤の使用等、多数提案されているが、吸収・膨潤は基本的にはシード粒子と水系媒体との分配によるため、攪拌方法、温度制御など非常に精密な反応制御が必要となる。また、吸収・膨潤したシード粒子は、非常に不安定(粒子表面の粘着力が増加)で、たびたび、歪な粒子の生成、或いはシード粒子間の合一が発生し、粒子径、及び粒径分布の制御が困難となる。
以上のように、表面機能性部材における機能性素材の安定、且つ保持特性及びその持続性の向上、加えて応用範囲の広い、簡便な表面機能性部材の製造方法については、さらなる改良が望まれているのが現状である。
【0005】
【特許文献1】特許第3434036号公報
【特許文献2】特開2001−89510号公報
【非特許文献1】「高分子の表面改質と応用」(監修:角田光雄、発行:(株)シーエムシー)、第13頁の第2章、化学的表面改質技術
【発明の開示】
【発明が解決しようとする課題】
【0006】
そこで本発明においては、上述した従来の技術が包含していた各種問題点に鑑みて、機能性素材と基材とを共有結合により直接結合している方法を選定し、使用環境下においても優れた機能発現、及び耐久性を実現し得ること、及び各種の用途に用いられる応用範囲の広い、簡便な表面機能性部材を製造する方法、及び当該方法により得られた表面機能性部材、並びに該表面機能性部材を用いた電気泳動素子を提供することとした。
【課題を解決するための手段】
【0007】
本発明者は、表面にカルボキシル基を有する基材上に、アミノ基を有する機能性素材を縮合剤の存在下に、直接結合させて成る表面機能性部材において、水系連続相中で、反応させることにより、上記従来技術における課題が解決されることを見出し、本発明に想到するに至った。
【0008】
請求項1の発明においては、表面にカルボキシル基を有する基材上に、アミノ基を有する機能性素材を、縮合剤の存在下、直接結合させて得られる表面機能性部材の製造方法であって、水系連続相中で反応させることを特徴とする表面機能性部材の製造方法を提供する。
【0009】
請求項2の発明においては、前記表面にカルボキシル基を有する基材が、乳化重合、懸濁重合、シード重合、あるいはソープフリー重合のいずれかの重合方式により得られた粒子であり、かつ平均粒子径が0.1〜10μmであることを特徴とする請求項1の表面機能性部材の製造方法を提供する。
【0010】
請求項3の発明においては、前記縮合剤が、下記一般式(1)で表わされるトリアジン化合物であることを特徴とする請求項1又は2の表面機能性部材の製造方法を提供する。
【0011】
【化1】

【0012】
但し、R1、R2は、置換もしくは無置換のアルキル基を表す。
【0013】
請求項4の発明においては、表面にカルボキシル基を有する基材上に、アミノ基を有する機能性素材を、縮合剤の存在下、直接結合させて得られる表面機能性部材の製造方法であって、水系連続相中に、更に非イオン性界面活性剤を添加する工程を有することを特徴とする請求項1乃至3のいずれかの表面機能性部材の製造方法を提供する。
【0014】
請求項5の発明においては、前記表面にカルボキシル基を有する基材上に、アミノ基を有する機能性素材を、縮合剤の存在下、水を主体とする連続相中で直接結合させて得られる表面機能性部材の製造方法であって、前記、水系連続相中に非イオン性界面活性剤を添加したことを特徴とする表面機能性部材の製造方法を提供する。
【0015】
請求項6の発明においては、前記アミノ基を有する機能性素材が、シロキサン骨格を有する置換基を含有する機能性素材であることを特徴とする請求項1乃至5のいずれか一項に記載の表面機能性部材の製造方法を提供する。
【0016】
請求項7の発明においては、前記アミノ基を有する機能性素材が、下記一般式(I)及び一般式(II)で表されるシリコーン化合物より選ばれる少なくとも一種のシリコーン化合物であることを特徴とする請求項1乃至5のいずれか一項に記載の表面機能性部材の製造方法を提供する。
H2N−B11−X ・・・(I)
H2N−B12−NH−B13−X ・・・(II)
(式中、B11,B12及びB13は、炭素数2から6のアルキレン基、Xはシロキサン骨格を示す。)
【0017】
請求項8の発明においては、表面にカルボキシル基を有する基材上に、アミノ基を有する機能性素材を、縮合剤の存在下で直接結合させて得られる表面機能性部材であって、前記結合の反応は、水系連続相中で行われたものであることを特徴とする表面機能性部材を提供する。
【0018】
請求項9の発明においては、前記表面にカルボキシル基を有する基材が着色剤を含有していることを特徴とする請求項8の表面機能性部材を提供する。
【0019】
請求項10の発明においては、前記基材が球状粒子であり、かつ平均粒子径が0.1〜10μmであることを特徴とする請求項8又は9の表面機能性部材を提供する。
【0020】
請求項11の発明においては、前記アミノ基を有する機能性素材が、シロキサン骨格を有する置換基を含有する機能性素材であることを特徴とする請求項8乃至10のいずれか一項に記載の表面機能性部材を提供する。
【0021】
請求項12の発明においては、前記アミノ基を有する機能性素材が、下記一般式(I)及び一般式(II)で表されるシリコーン化合物より選ばれる少なくとも一種のシリコーン化合物であることを特徴とする請求項8乃至10のいずれか一項に記載の表面機能性部材を提供する。
H2N−B11−X ・・・(I)
H2N−B12−NH−B13−X ・・・(II)
(式中、B11,B12及びB13は、炭素数2から6のアルキレン基、Xはシロキサン骨格を示す。)
【0022】
請求項13の発明においては、絶縁性液体中に、請求項8乃至12のいずれか一項に記載の表面機能性部材を分散してなることを特徴とする電気泳動素子を提供する。
【0023】
請求項14においては、シリコーンオイル中に、請求項11又は12に記載の表面機能性部材を分散してなることを特徴とする電気泳動素子を提供する。
【0024】
本発明方法によれば、水系連続相中で、表面にカルボキシル基を有する基材上にアミノ基を有する機能性素材を、縮合剤の存在下で反応させるものであるので、非常に安易かつ環境面にも配慮された工程により所望の表面機能性部材が得られる。
また、基材と機能性素材との結合力が極めて強く、耐久性に優れた表面機能性部材が得られる。
【発明の効果】
【0025】
本発明によれば、基材とアミド基を介して直接機能性素材を導入することにより耐久性に優れ、かつ長時間に亘って機能を維持し得る表面機能性部材の適応範囲の広い製造方法、及びこの製造方法により得られた表面機能性部材を提供することができた。さらに、該表面機能性部材を用いた電気泳動素子を提供することができた。
【発明を実施するための最良の形態】
【0026】
以下、本発明について、具体的に説明する。
本発明においては、水系連続相中で、基材上のカルボキシル基と、機能性素材のアミノ基を縮合剤の存在下に直接結合させて表面機能性部材を得る。
【0027】
先ず、カルボキシル基を有する基材について説明する。
基材としては、板状物、あるいは球状物等、いかなる形状のものも使用でき、最終的に目的とする表面機能性部材の使用目的に応じて高分子樹脂からなる表面を有する基材を適宜選択する。例えば、樹脂フィルム、表面に樹脂が被覆されているガラス等の無機基材、表面が樹脂層からなる複合材等が挙げられる。
特に形状の観点からは、表面積が大きく表面機能性部材の効果が期待できる粉体や粒子が好適である。更に、粒径の均一な粒子が作製できること、及び水系連続相中で造粒出来るという観点からは、乳化重合、懸濁重合、シード重合、及びソープフリー重合より得られた粒子が好適である。
材料としては、寸度的に安定なものが好適であり、最終的に目的とする表面機能性部材の所望の可撓性、強度、耐久性、物性等を有していればよい。
光透過性を必要とする材料を選択する場合は、例えば、ガラス、プラスチックフィルム(例えば、二酢酸セルロース、三酢酸セルロース、プロピオン酸セルロース、酪酸セルロース、酢酸酪酸セルロース、硝酸セルロース、ポリエチレンテレフタレート、ポリエチレン、ポリスチレン、ポリプロピレン、ポリカーボネート、ポリビニルアセタール等)等が挙げられる。
特に光透過性に優れた材料としては、例えば、ポリメチルメタクリレート、ポリカーボネート、ポリスチレン、ポリジエチレングリコールビスアリルカーボネート等の樹脂材料が好適である。
光透過性を有さない材料を選択する場合は、例えば、紙、プラスチックがラミネートされた紙、金属板(例えば、アルミニウム、亜鉛、銅等)、金属がラミネート若しくは蒸着された紙若しくはプラスチックフィルム等を挙げることができる。
基材の材料としては、最終的に目的とする表面機能性部材の用途や、反応される機能性素材との関係に応じて適宜材料選択するが、加工性、透明性の観点からは、高分子樹脂からなる表面を有する材料が好ましく、具体的には、樹脂フィルム、表面に樹脂が被覆されているガラス等の透明無機基材、表面層が樹脂層からなる複合材が好適である。
【0028】
基材の材料として、表面に樹脂が被覆されているものを選択するときには、例えば、表面に樹脂フィルムが貼着された積層板、プライマー処理された基材、ハードコート処理された基材等が挙げられる(例えば、特開2006−56949号公報に開示されている。)。
表面層が樹脂層からなる基材とは、具体的には、裏面に接着剤層が設けられた樹脂シール材、ガラスと樹脂との積層体である合わせガラス等が挙げられる。
また、基材は、最終的に目的とする表面機能性部材の使用目的に応じて、粗面化処理を施してもよい。次行程である機能性素材の単位面積当たりの反応量をより向上、あるいは反応性を向上させるために表面積を増加させる目的で、基材表面を予め粗面化しておくことが有効である。
粗面化方法としては、公知の方法を選択することができる。例えば、基材が樹脂フィルムの場合には、グロー放電処理、スパッタリング、サンドブラスト研磨法、バフ研磨法、粒子付着法、粒子塗布法等が挙げられる。また、基材がガラス板等の無機材料の場合には、機械的に粗面化する方法が適用できる。機械的方法としては、ボール研磨法、ブラシ研磨法、ブラスト研磨法、バフ研磨法等を適用できる。
【0029】
次に、基材の表面にカルボキシル基を導入する方法について説明する。
カルボキシル基の導入方法としては、従来公知の方法を適宜使用できる。例えば、予め表面にカルボキシル基を有している基材を使用する方法、基材上にカルボキシル基を有する樹脂層(中間層)を形成する方法が挙げられる。
【0030】
基材が板状物である場合は、カルボキシル基を有する樹脂を所定の溶剤に溶解、分散、あるいは熱溶融して、これをスピンコーティング、ブレードコーティング、スプレーコーティング、ディップコーティング等公知の塗工方法により塗布して樹脂層を形成することができる。
【0031】
また、上記のように層状の樹脂層を形成するのみならず、カルボキシル基を有する樹脂により球状物を作製しこれを基材としてもよい。この場合、カルボキシル基を有する樹脂に対する溶解度差を利用して造粒する方法が好適であり、具体的には、カルボキシル基を有する樹脂を溶剤に溶解し、カルボキシル基を有する樹脂に対して貧溶媒中に滴下する方法、カルボキシル基を有する樹脂を溶剤に溶解し、溶媒を徐々に留去する方法等が挙げられる。特に、カルボキシル基を有する樹脂を溶剤に溶解し、分散安定剤等を添加した水系連続相中で造粒する方法が次工程への移行が容易である点、及び極性基であるカルボキシル基が特異的に粒子表面に形成される点で好適である(コアセルベーション法)。
なお、樹脂中には必要に応じて、着色剤、分散剤、熱安定剤、酸化防止剤、紫外線吸収剤等の各種添加剤を均一に溶解、分散するようにしてもよい。
上記樹脂としては、例えば、アクリル樹脂、スチレン系樹脂、エポキシ樹脂、ウレタン樹脂、ビニル樹脂、フェノール樹脂、ポリエステル樹脂、ポリアミド樹脂、メラミン系樹脂等の合成樹脂、ゼラチン、カゼイン、セルロースデンプンなどの天然樹脂、あるいはこれらの共重合樹脂等がいずれも適用できる。また、これら樹脂は使用目的に応じて架橋構造を形成したものとしてもよい。
【0032】
その他、紫外線、エキシマレーザー等の光処理、電子線処理、イオンビーム処理、あるいはプラズマ処理等の化学作用により、新たにカルボキシル基を生成する方法が挙げられる。この方法における樹脂としては、上記に掲げたものを適宜使用できる。
【0033】
また、基材面へのカルボキシル基の導入方法として、グラフト化による生成方法も適用できる。
この場合、基材は、グラフトポリマーが化学的に結合できるような表面を有しているものを適用する。なお基材自体の表面がこのような特性を有していてもよく、このような特性を有する樹脂層(中間層)を基材表面に形成してもよい。樹脂層(中間層)としては、光グラフト重合法、放射線照射グラフト重合法によりグラフトポリマーを合成する場合には、上記に掲げた樹脂を使用できる。また、光グラフト重合法、プラズマ照射グラフト重合法、放射線照射グラフと重合法等は、グラフト重合の開始が樹脂層(中間層)の水素引き抜きから反応が進行するため、水素が引き抜かれやすい樹脂、例えばアクリル樹脂、ウレタン樹脂、スチレン系樹脂、ビニル系樹脂、ポリアミド系樹脂、エポキシ樹脂等が製造適性の観点から好適である。この場合、基材表面、あるいは樹脂層(中間層)表面に、重合性化合物と重合開始剤を添加し、所定のエネルギーを付与することにより重合開始能を発現することができる。
重合性化合物は、活性光線照射等のエネルギー付与により、カルボキシル基含有モノマー、あるいはカルボキシル基含有ポリマーが付与し得るものであれば制限はない。特に、分子内にカルボキシル基を有する重合性基含有ポリマーが好適である。
重合開始剤は、所定のエネルギー、例えば、活性光線の照射、加熱、電子線の照射等により、重合開始能を発現しうる公知の熱重合開始剤、光重合開始剤等を、目的に応じて、適宜選択して適用できる。特に、熱重合よりも反応速度(重合速度)が高い光重合を利用することが製造適性の観点から好適であるため、光重合開始剤の使用がより好ましい。
光重合開始剤は、照射される活性光線に対して活性であり、重合性化合物を重合させることができれば、特に制限はなく、例えば、ラジカル重合開始剤、アニオン重合開始剤、カチオン重合開始剤等が適用できる。
【0034】
カルボキシル基を有する基材が微粒子である場合には、上述のコアセルベーション法と重合法がある。重合法は、重合性単量体に、重合開始剤と、必要に応じて着色剤、及び架橋剤等の各種添加剤とともに均一に溶解、あるいは分散させた重合単量体組成物を調整し、昇温して重合することにより、所望の粒径を有する微粒子を得ることができる。また、必要に応じて、連鎖移動剤、ワックス、及び帯電制御剤等の各種添加剤等を添加してもよい。
【0035】
上記粒子の粒径分布は、製造される本発明の表面機能性部材の粒径分布に反映されることから、単分散性の高いものか好ましく、相対標準偏差(CV値)として30%以下であることが好ましい。相対標準偏差(CV値)が30%を超えると上記粒子から製造される表面機能性部材の粒径分布が広がり、粒径の不均一性より生じる粒子の特性が著しく損なわれ、例えば、電気泳動移動度に差が生じ問題となる。
上記相対標準偏差(CV値)は下記数式(1)により算定される。
相対標準偏差(CV値(%))=(sd/m)×100……(1)
(式中、sdは粒子径の標準偏差を、mは平均径である。)
なお、上記sd、及びmは、粒径アナライザー(FPAR−1000:大塚電子社製)による動的光散乱法により得られる数値である。
【0036】
上記カルボキシル基を有する微粒子の作製に用いる重合性単量体について説明する。
重合性単量体としては、ラジカル重合性単量体を必須の構成成分とする。
上記所望の微粒子の作製に適用する必須成分であるカルボキシル基を有する単量体としては、例えば、4−ビニル安息香酸、3−ビニル安息香酸などのスチレン誘導体、(メタ)アクリル酸、イタコン酸、イタコン酸モノブチルエステル、マレイン酸、マレイン酸モノメチルエステル、マレイン酸モノブチルエステル、2−カルボキシルエチルアクリレート等のアクリル酸系誘導体が挙げられる。
上記カルボキシル基を有する単量体の配合量は、以下に示す通常単量体と併せた総単量体100重量部に対して1〜50重量部とすることが好適である。
通常単量体は、特に限定されるものではない。例えば、スチレン、α−メチルスチレン、p−メチルスチレン、p−クロロスチレン、クロロメチルスチレン等のスチレン誘導体、塩化ビニル、酢酸ビニル、プロピロン酸ビニル等のビニルエステル類、アクリロニトリル等の不飽和ニトリル類、(メタ)アクリル酸メチル、(メタ)アクリル酸エチル、(メタ)アクリル酸ブチル、(メタ)アクリル酸−2−エチルへキシル、(メタ)アクリル酸ステアリル、エチレングリコール(メタ)アクリレート、トリフルオロエチル(メタ)アクリレート、ペンタフルオロプロピル(メタ)アクリレート、シクロヘキシル(メタ)アクリレート等の(メタ)アクリル酸エステル誘導体が挙げられる。これらは単独で用いてもよく、2種以上併用しても良い。また、必要に応じて、通常単量体中ビニル基を2個以上含有する架橋性単量体を含有してもよい。ビニル基を2個以上含有する架橋性単量体としては特に限定されず、例えば、ジビニルベンゼン、ジビニルビフェニル、ジビニルナフタレン、エチレングリコールジ(メタ)アクリレート、ブタジエングリコールジ(メタ)アクリレート、ポリエチレングリコールジ(メタ)アクリレート等が挙げられる。上記架橋性単量体は単独で用いても良く、二種以上を併用しても良い。
また、本発明では、マクロモノマーをカルボキシル基を有する単量体、及び通常単量体と共に重合性単量体として使用することができる。マクロモノマーは、分子鎖の末端にビニル重合性官能基を有するもので、数平均分子量が、通常、1000〜30000のオリゴマー又はポリマーである。
マクロモノマー分子鎖の末端に有するビニル重合性官能基としては、アクロイル基、メタクロイル基等を挙げることができる。マクロモノマーの量は、重合性単量体100重量部に対して、通常、0.01〜10重量部とすることが好適である。
重合組成物には樹脂の重合度を制御し、軟化点や分子量等の物性を調節するために連鎖移動剤を添加することができる。連鎖移動剤としては、ラジカル重合反応で一般的に用いられる連鎖移動剤を用いることが可能であり、特に限定されるものではない。例えば、オクチルメルカプタン、ドデシルメルカプタン、tert−ドデシルメルカプタン等のメルカプタン、及びスチレンダイマー等が挙げられる。
【0037】
上記カルボキシル基を有する微粒子の作製に用いるラジカル重合開始剤について説明する。
ラジカル重合開始剤は、特に限定されず、従来公知のものを適用できる。
例えば、過硫酸カリウム、過硫酸アンモニウム等の過硫酸塩、過酸化ベンゾイル、過酸化ラウロイル、オキソクロロ過酸化ベンゾイル、t−ブチルパーオキシ−2−エチルヘキサニエート、ジ−t−ブチルパ−オキサイド等の過酸化物、2,2’−アゾビスイソブチロニトリル、1,1’−アゾビス(シクロヘキサン−1−カルボニトリル)、2,2’−アゾビス(2,4−ジメチルバレロニトリル)、2,2’−アゾビス[2−メチル−N−(2−ヒドリキシエチル)プロピオンアミド]、2,2’アゾビス(2−メチルプロピオンアミジン)塩、2,2’−アゾビス[N−(2−カルビキシエチル)−2−メチルプロピオンアミジン]、4,4’−アゾビス(4−シアノ吉草酸)等のアゾ系化合物が挙げられる。中でも、2,2’−アゾビス[N−(2−カルボキシエチル)−2−メチルプロピオンアミジン]、4,4’−アゾビス(4−シアノ吉草酸)などの水溶性アゾ系化合物が好適である。上記ラジカル重合開始剤は、必要に応じて還元剤と組み合わせたレドックス系開始剤として使用してもよい。レドックス開始剤を用いることで、重合活性が上昇し重合温度の低下が図れ、更に重合時間の短縮が期待できる。また、上記ラジカル重合開始剤の配合量は、重合性単量体100重量部に対して0.1〜10重量部とすることが好適である。
【0038】
次に、上記カルボキシル基を有する微粒子の作製に用いる添加剤について説明する。
カルボキシル基を含有する微粒子を作製する際には、必要に応じて、界面活性剤を添加してもよい。界面活性剤としては特に限定されるものでは無いが、下記のイオン性及びノニオン性界面活性剤が好適に用いられる。
イオン性界面活性剤としては、例えば、ドデシルベンゼンスルホン酸ナトリウム、アリールアルキルポリエーテルスルホン酸ナトリウム、3,3−ジスルホンジフェニル尿素−4,4−ジアゾビス−アミノ−8−ナフトール−6−スルホン酸ナトリウム、等のスルホン酸塩、ドデシル硫酸ナトリウム、テトラデシル硫酸ナトリウム、ペンタデシル硫酸ナトリウム、ジアルキルスルホコハク酸エステルナトリウム、等の硫酸エステル、オレイン酸ナトリウム、ラウリン酸ナトリウム、カプリン酸ナトリウム、カプロン酸ナトリウム、ステアリン酸カリウム等の脂肪酸塩などが挙げられる。
非イオン性界面活性剤としては、ポリエチレンオキシド、ポリポロピレンオキシド、ポリエチレンオキシドとポリプロピレンオキシドの組み合わせ、ポリエチレングリコールの高級脂肪酸エステル、ポリプロピレンオキシドの高級脂肪酸エステル、アルキルフェノールポリエチレンオキシド、アルキルフェノールポリプロピレンオキシド、ポリエチレンオキシドアルキルエーテル、ポリプロピレンオキシドアルキルエーテルグリコール、ソルビタンエステルなどが挙げられる。
界面活性剤の配合量は、総単量体100重量部に対して、0.1〜5重量部とすることが好適である。
更に、必要に応じて、分散安定剤を添加してもよい。分散安定剤としては、例えば、部分鹸化されたポリビニルアルコール、ポリビニルエーテル、ゼラチン、メチルセルロース、エチルセルロース、ヒドロキシエチルセルロース、カルボキシメチルセルロースのナトリウム塩等の高分子分散安定剤、リン酸カルシウム、リン酸マグネシウム、リン酸アルミニウム、炭酸カルシウム、炭酸マグネシウム、硫酸バリウム、水酸化マグネシウム、ベントナイト等の無機分散安定剤が挙げられる。分散安定剤の配合量は、総単量体100重量部に対して0.1〜5重量部とすることが好適である。
【0039】
なお、上記の他、着色剤を添加してもよい。着色剤粒子分散液は、着色剤を水系媒体中、あるいは溶剤系媒体中に分散することにより調整することができる。着色剤の分散処理は、水中の場合は、界面活性剤を臨界ミセル濃度(CMC)以上にした状態で行われ、用いられる界面活性剤はイオン性界面活性剤やノニオン性界面活性剤が使用でき、これらを単独、或いは適当な組成で混合して使用すればよい。溶剤系では着色剤と充分に湿潤する溶剤を選択し、必要に応じ界面活性剤も使用される。用いられる溶剤としては、メタノール、エタノール、イソプロパノール、アセトン、メチルエチルケトン、メチルイソブチルケトン、テトラヒドロフランなどが挙げられる。着色剤の分散処理に使用する分散機は特に限定されないが、好ましくは超音波分散機、機械的ホモジナイザーや圧力式ホモジナイザー等の加圧分散機、サンドグラインダー、ダイヤモンドファインミル、ビーズミル等の媒体型分散機が挙げられる。
着色剤としては、公知の顔料が使用可能で、例えば、カーボンブラック、アニリンブルー、カルコイルブルー、クロムイエロー、ウルトラマリンブルー、デュポンオイルレッド、キノリンイエロー、メチレンブルークロリド、銅フタロシアニン、マラカイトグリーンオキサレート、ランプブラック、ローズベンガル、C.I.ピグメント・レッド48:1,C.I.ピグメント・レッド122,C.I.ピグメント・レッド57:1,C.I.ピグメント・レッド184,C.I.ピグメント・イエロー97,C.I.ピグメント・イエロー12,C.I.ピグメント・イエロー17,C.I.ソルベント・イエロー162,C.I.ピグメント・イエロー180,C.I.ピグメント・イエロー185,C.I.ピグメント・ブルー15:1、C.I.ピグメント・ブルー15:3等を挙げることができる。
着色剤の使用量は、粒子中において樹脂成分100重量部に対する含有量が2〜30重量部、好ましくは5〜25重量部となるような量が好適である。
本発明の「表面にカルボキシル基を有する基材」は、界面重合法、乳化重合法、ソープフリー乳化重合法、非水分散重合法等の湿式重合法、気相法、コアセルベーション法、表面改質法等種々の方法で作成した高分子化合物を用いることができる。高分子化合物としては、スチレン系、(メタ)アクリル系、ポリエステル系、ベンゾグアナミン、メラミン、テフロン(登録商標)、シリコン、ポリエチレン、ポリプロピレン、或いはこれらの共重合化合物等を用いることができる。
本発明に係わる樹脂粒子の体積平均粒径は、0.1〜10μmで粒径分布の揃った樹脂粒子であることが好ましく、より好ましくは0.1〜5μmの範囲で粒径分布の揃った粒子である。また本発明に係わる樹脂粒子のガラス転移点(Tg)は特に制限されないが、好ましくは−10〜120℃の範囲である。また本発明に係わる樹脂粒子の分子量は特に制限されないが、好ましくは質量平均分子量で2000〜1000000である。
【0040】
次に、表面機能性部材の作製方法について説明する。
上述したような、各種表面にカルボキシル基を有する基材上に、アミノ基を有する機能性素材を、縮合剤の存在下で直接結合させて表面機能性部材を作製する。なお、反応工程において、水系連続相中で行う。
基材のカルボキシル基と、機能性素材の極性基とを結合させる反応は、公知の活性アミド化法である。なお、縮合剤として、下記式(2)中の、一般式(3)で表わされるトリアジン化合物を適用した反応については、Tetrahedron55(1999)13159−13170に記載されている。
【0041】
【化2】
【0042】
但し、R1、R2は、置換もしくは無置換のアルキル基を表す。
R1、R2で用いられる置換基として、置換もしくは無置換のアルキル基としては以下のものが挙げられる。
例えば、炭素数1〜18の直鎖、分岐鎖又は環状のアルキル基であり、これらのアルキル基は更に炭素数1〜18のアルコキシ基、フェニル基、又は炭素数1〜6の直鎖、分岐鎖又は環状のアルキル基で置換されたフェニル基を含有しても良い。具体的には、メチル基、エチル基、n−プロピル基、i−プロピル基、n−ブチル基、i−ブチル基、s−ブチル基、t−ブチル基、n−ヘキシル基、n−オクチル基、n−デシル基、n−ドデシル基、n−オクタデシル基、トリフルオロメチル基、2−シアノエチル基、メトキシメチル基、メトキシエチル基、ベンジル基、4−クロロベンジル基、シクロペンチル基、シクロヘキシル基などが挙げられる。これらの中で、メチル基、エチル基プロピル基など低級アルキル基は、水に対する溶解性が優れるため、好適に使用できる。一方、炭素数10〜18の直鎖アルキル基も好適に使用できる。これは、R1、R2に長鎖アルキル基を導入することにより、界面活性剤的性質により分散性の改良に寄与したものと考えている。
【0043】
上記式(2)は、カルボキシル基を有する化合物(4)と、アミノ基を有する化合物(5)とを、式(3)で表されるトリアジン化合物縮合剤下で、中間体(6)を経て、アミド化合物(7)と、副生成物ヒドロキシトリアジン化合物(8)が生成する反応である。
式(3)のトリアジン化合物は、水中でも安定であると記載されているが、通常は反応溶媒としてTHF(テトラヒドロフラン)が適用されている。
なお、本発明においては、上記活性アミド化反応を、水系連続相中で行うことに特徴を有している。ただし、Tetrahedron57(2001)1551−1558には、反応溶媒として、水、或いは水/メタノール(1:1)を使用する反応の記載がある。この場合、カルボキシル基を有する化合物は水溶性であるか、難水溶性の場合は反応溶媒として水/メタノール(1:1)を使用し、少なくともカルボキシル基を有する化合物は溶解した反応系で使用している。
それに対して、本発明の特徴は、上記の水系連続相中での活性アミド化反応を、完全に不溶なカルボキシル基を有する化合物を使用している点、すなわち、固体/液体間の不均一状態で行う点である。一般的に、固体/液体間の反応は反応試薬の衝突回数の問題で、極度に反応性が低下、あるいは反応しないことが知られている。特に、カルボキシル基を有する化合物に粒子を適用する場合、粒径が小さくなるほど(凝集力が増加し)、反応性が低下し、上記の水系連続相中での活性アミド化反応は、更に困難となる。
【0044】
本発明においては、表面にカルボキシル基を有する基材上に、アミノ基を有する機能性素材を、縮合剤の存在下、水系連続相中で直接結合させて得られる表面機能性部材であって、前記、水系連続相中に非イオン性界面活性剤を添加することにより、従来問題となっていた上記課題が解決されることを見いだした(請求項4)。
特に、非イオン性界面活性剤が優れている理由は、上記式(2)の反応で使用する酸性及び塩基性の試薬に対して、不活性であることに起因していると考えられる。実際、イオン性界面活性剤を使用すると、非常に反応性が悪いか、或いは全く反応しない場合がある。これは、一般式(2)の反応で使用する試薬とイオン性界面活性剤の間に何らかのイオン的相互作用があり、反応を阻害しているものと考えている。
非イオン性界面活性剤としては、前述の化合物が使用でき、必要に応じて2種以上混合して使用しても良い。これらの非イオン性界面活性剤中、HLB(Hydrophile-Lipophile Balance)が12〜18の非イオン性界面活性剤が好適である。また、非イオン性界面活性剤を2種以上使用する場合のHLBは、その加重平均になる。HLBが12以下では、親水性部位が不安定化し、HLBが18以上では、疎水性部位が不安定化する。従って、HLBが12〜18の非イオン性界面活性剤、更にHLBが12.5〜17の非イオン性界面活性剤が好適に使用できる。
【0045】
ここで、HLBは、以下に示すグリフィンの式より求められる。
HLB=(親水性部分の分子量)/(全体の分子量)×100÷5
本発明で使用される縮合剤として、上記式(2)中の、式(3)で表わされるトリアジン化合物以外に、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩等のカルボジイミド化合物、及びフェニルスルホン酸ビス(2−ニトロフェニルエステル)等のスルホン酸化合物は相間移動触媒とともに水溶液中で使用されるが、反応性、及び取り扱い容易性により、式(3)で表わされるトリアジン化合物を好適に使用することができる。
これら表面機能性部材を作製する場合、表面にカルボキシル基を有する基材、アミノ基を有する機能性素材、及び必要に応じて界面活性剤等の添加剤を水系連続相中に溶解、或いは分散させた機能性部材組成物を調整し、縮合剤を含む水を添加することにより、所望の機能性素材が直接結合した基材を得ることができる。表面にカルボキシル基を有する基材、アミノ基を有する機能性素材、及び必要に応じて界面活性剤等の添加剤を水系連続相中に分散する場合は、前述の分散機を使用することができる。また、カルボキシル基を有する基材の分散安定性を改善する目的で、水系連続相を中性〜弱アルカリ性(pH9程度)とすることが好ましく、更に、アミノ基を有する機能性素材量をカボキシル基の当量より多く使用し水系連続相を弱アルカリ性とすることが好ましい。この場合のアミノ基を有する機能性素材量は、カボキシル基の1.2〜2.0倍当量が好適である。
【0046】
本発明のアミノ基を有する機能性素材中、アミノ基としては、カルボキシル基と反応してアミド結合を形成できるものであれば特に制限はないが、立体障害が大きい場合、反応性の点で二級アミノ基より一級アミノ基の方が好ましい。機能性素材としては、特に制限はなく、発生する機能に応じて、従来公知の原子、置換基、及び官能基等が選ばれる。例えば、表面硬度に関しては架橋構造が形成される官能基、表面滑性に関してはフッ素原子、或いはジメチルシロキサン骨格を有する置換基、及び、分散安定性、表面の帯電性、導電性、着色剤等の機能性基を挙げることができる。
反応は、縮合剤を含む水溶液を添加することにより始まり、通常、0℃から室温(25℃)で6時間程度終わる。ただし、電子的、立体的に反応性が悪い場合は、昇温、或いは長時間の反応が必要となる。昇温する場合は、安定性の問題で70℃程度までとした方がよい。また、化学的安定性のため、反応系内をアルゴンガス、窒素ガス等の不活性ガス雰囲気下で行うこともできる。
【0047】
本発明の表面機能性部材は、非常に簡便な方法で、基材と機能性素材を直接アミド結合して得られるため、機能性素材を高安定に、しかも高耐久性・高持続性を有する。しかも、本発明の製造方法は、アミノ基を有する多くの機能性素材に広範囲に応用可能であり、基材上に非常に均一に単分子層を形成可能な優れた方法である。また、本発明で得られる粒子は、平均粒子径が0.1〜10μm、の範囲内であり、CV値は30%以下で非常に単分散性の高いほぼ真球状粒子である。また本発明の製造方法によれば、非常に容易に、しかも平均粒子径、相対標準偏差(CV値)等の粒子特性を損なうことなく着色粒子を提供することができる。
【0048】
本発明方法を利用して作製された表面機能性部材を構成する着色粒子に関しては、特に、帯電性着色粒子への応用が考えられ、PLD(ペーパーライクディスプレー)、電子写真用液体現像剤、静電インクジェットプリンター用インク等に用いられる。
具体的には、本発明の着色微粒子のシイルコーンオイル、高純度石油等の絶縁性液体分散液内で、電気的に帯電して、泳動させる装置(電気泳動素子)に応用できる。なお、前記絶縁性液体の具体的な市販品としては、前記高純度石油では、例えば、アイソパーG,H,L,M(エクソン化学社製)やノルパー12(エクソン化学社製)等が挙げられ、前記シリコーンオイルでは、例えばSH−200シリーズ(東レ・ダウコーニング社製)、KF−96シリーズ(信越化学社製)、L−45シリーズ(日本ユニカー社製)、及びAKシリーズ(旭化成ワッカーシリコーン社製)等が挙げられる。
このような帯電性着色粒子は、対向電極セル内の電気絶縁性液体中での帯電性、電気泳動特性が極めて重要である。その帯電性着色粒子に求められる電気泳動特性とは、所定の電解(又は電場)下にある粒子表面が、電気的に応答し易い帯電性を呈する着色粒子である。しかも、帯電性着色粒子を所定の電解下にスムーズに泳動させるためには、その粒子が泳動セル中で凝集して粗大化粒子になり難い着色粒子であることも重要である。
これらの電気泳動特性は、主に、着色粒子の表面状態、及び表面の置換基に左右される。
すなわち、上記式(2)中の機能性素材(5)を構成するB1、B2で表される置換基の種類によって左右されるものであり、その帯電性としては、それぞれ(−)帯電性と(+)帯電性を示す場合があり、少なくとも二種以上の複数種の置換基を使用するときには、その(−)及び(+)帯電性を示すもののうち、同種帯電性の置換基同士を複数組み合わせて使用することが好ましい。
【0049】
(−)帯電性の傾向にある置換基(B1、B2)としては、例えば、フェニル基、ベンジル基、2−フェニルエチル基等の芳香族基、2−クロロエチル基等の塩素原子含有アルキル基、p−クロロベンジル基などの塩素原子含有芳香族基、2−シアノエチル基等のシアノ基含有アルキル基、2−ヒドロキシエチル基、2−ヒドロキシプロピル基等のヒドロキシ基含有アルキル基、2−フルオロエチル基、2,2,2−トリフルオロエチル基等のフッ素原子含有アルキル基、パーフルオロベンゼン等のフッ素原子含有芳香族基等が挙げられる。
一方、(+)帯電性の傾向がある置換基(B1、B2)として、例えば、2−ジメチルアミノエチル基、2−ジブチルアミノエチル基、2−(1−ピペリジノ)エチル基、2−プロピリアミノエチル基、2−フェニルアミノエチル基等のアミノ基含有アルキル基、4−ジエチルアミノフェニル基、4−ジメチルアミノベンジル基等のアミノ基含有芳香族基等が挙げられる。
【0050】
また、このように泳動セル中で優れた非凝集性を有しているためには、その形状が定形で、その定形粒子が球状粒子であることが好ましく、更には、その粒径分布がより狭いことが望ましい。
更に、着色粒子の表面状態、及び表面の置換基にも左右されるものであり、アミノ基を有する機能性素材である上記式(5)中のB1、B2で表わされる置換基の種類によって左右される。
具体的に、非凝集性粒子を設計するためには、絶縁性液体がシリコーンオイルの場合は置換基(B1、B2)がシロキサン骨格を有する置換基としたり、高純度石油の場合は置換基(B1、B2)が長鎖アルキル基としたりする等、絶縁性液体と類似の骨格を有する置換値を導入することが好ましい。これは着色粒子と絶縁性液体間の相溶性が増加し、分散、及び分散安定性が増加するためである。
【0051】
絶縁性液体がシリコーンオイルの場合は、上記式(2)中の機能性素材(5)として一般的なアミノ変性シリコーンを使用することができるが、この中でも下記一般式(I)及び一般式(II)で表されるシリコーン化合物より選ばれた1種又は2種以上のシリコーン化合物が、特に好ましい。
H2N−B11−X ・・・(I)
H2N−B12−NH−B13−X ・・・(II)
(式中、B11,B12及びB13は、炭素数2から6のアルキレン基、Xはシロキサン骨格を示す。)
【0052】
ここで、式(I),(II)中、B11,B12及びB13である炭素数2から6のアルキレン基としては、直鎖或いは分岐のアルキレン基で、例えばエチレン、プロピレン、1−メチルエチレン、ブチレン、1,2−ジメチルエチレン、ペンチレン、へキシレン等が挙げられ、Xのシロキサン骨格は、例えば下記一般式(III)で示す片末端型、或いは下記一般式(IV)で示す側鎖型を挙げることができる。なお、式中l、m、nは、0以上、30以下の整数を表す。
【0053】
【化3】
【0054】
【化4】
【0055】
本発明方法により得られる表面機能性部材を適用した帯電性着色粒子を、PLD、電子写真画像形成装置の静電着色トナー、静電インクジェットプリンター用インク等として使用する場合に、所望の帯電特性、電気泳動性を発揮させるためには、その粒子形状も重要である。この点については、少なくとも定形粒子であって、かつ粒子表面により均質に帯電させるために略球状であることが好適である。また、平均粒径が0.1〜10μmの範囲であることが、上記用途として好適である。
【0056】
また、電気泳動及び転写時に、印可電解による粒子間のクーロン反発により粒子を凝集粗大化させ難くさせる効果が期待できることから、平均粒径のバラツキが著しく低いことが好ましい。
この粒子の均斉度を相対標準偏差(CV値)で表すこととし、これが30%以下の略単分散粒子であると、帯電性、電気泳動特性に優れた帯電性着色粒子を提供できることが確かめられた。
また、本発明の表面機能性部材を利用した帯電性着色粒子には、必要に応じて公知の添加剤を加えてもよい。例えば、分散剤、電荷制御剤、熱安定剤、防腐剤、表面張力調整剤、酸化防止剤、近赤外線吸収剤、紫外線吸収剤、蛍光剤、蛍光増白剤等が挙げられる。
【実施例】
【0057】
以下、本発明について具体的な実施例を挙げて説明するが、本発明はこれらの例に限定されるものではない。
【0058】
先ず、表面機能性部材の評価に用いる方法と装置を下記に示す。
<粒径、及び相対標準偏差(CV値)>
粒径アナライザー(FPAR-1000:大塚電子製)−試料−1.0重量%水溶液で測定した。
<赤外線吸収スペクトル>
顕微FT-IR(サーモエレクトロン製)、顕微透過法で測定した。
<XPS(X-ray photoelectron spectroscopy)>
・測定装置:AXIS−ULTRA(Kratos社製)
X線源 :Alモノクロメータ使用
X線パワー :225W
測定領域 :900μm×600μm
パスエネルギー:wide scan(ワイドスキャン)=80eV
:narrow scan(ナロースキャン)=20eV<SEM観察>日本電子製 電界放射型走査電子顕微鏡 JSM-7401F
―観察条件―導電処理:Osコート、加速電圧:5.0kV、検出器:SEI
<ζ(Zeta)電位測定>
・測定装置:ESA9800(Matec Applied Science社製)
【0059】
次に、下記の〔合成例1〜6〕のそれぞれにおいて、表面にカルボキシル基を有する粒子を作製する。
【0060】
〔合成例1〕
温度計と窒素導入管とを装着した500mlの4つ口フラスコに、カルボキシル基を有する単量体であるメタクリル酸1.30重量部、通常単量体であるメチルメタクリレート28.70重量部、リン酸二水素カリウム0.147重量部、1N−水酸化カリウム水溶液16.17重量部、及びイオン交換水254重量部を投入し、強攪拌下で30分間、混合した。
次に、窒素気流下で攪拌しながら65℃に昇温した。この反応液を65℃の温度に保持しながら、過硫酸ナトリウム0.163重量部、及びイオン交換水20重量部より成る重合開始剤水溶液を一時間要して滴下し、更に6時間反応を行い、その後、放冷し、濾過し、水洗処理を3回行い、乾燥処理を行うことにより、表面にカルボキシル基を有する粒子(A)が25.88重量部得られた。
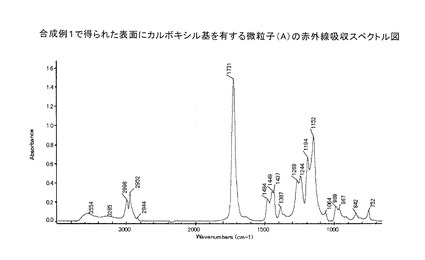
この粒子(A)の粒度分布測定を行ったところ、平均粒径:0.64μmであり、その相対標準偏差(CV値)は22.0%であった。赤外線吸収スペクトルを図1に示す。
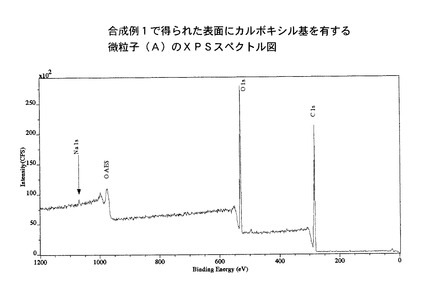
また、XPSスペクトルを図2に示す。
【0061】
〔合成例2〕
温度計と窒素導入管とを装着した500mlの4つ口フラスコに、カルボキシル基を有する単量体であるアクリル酸2.19重量部、通常単量体であるメチルメタクリレート27.38重量部、リン酸二水素カリウム0.147重量部、1N−水酸化カリウム水溶液16.17重量部、及びイオン交換水254重量部を投入し、強攪拌下で30分間、混合した。
次に、窒素気流下で攪拌を行いながら65℃に昇温した。この反応液を65℃の温度に保持しながら、過硫酸ナトリウム0.163重量部、及びイオン交換水20重量部より成る重合開始剤水溶液を一時間要して滴下し、更に6時間反応を行い、その後、放冷し、濾過し、水洗処理を3回行い、乾燥処理を行うことにより、表面にカルボキシル基を有する粒子(B)が24.20重量部得られた。
この粒子の粒度分布測定を行ったところ、平均粒径:0.39μmであり、その相対標準偏差(CV値)は22.5%であった。
【0062】
〔合成例3〕
温度計と窒素導入管とを装着した500mlの4つ口フラスコに、カルボキシル基を有する単量体であるメタクリル酸2.62重量部、通常単量体であるメチルメタクリレート27.38重量部、リン酸二水素カリウム0.147重量部、1N−水酸化カリウム水溶液31.47重量部、及びイオン交換水238重量部を投入し、強攪拌下で30分間、混合した。
次に、窒素気流下で攪拌を行いながら、68℃に昇温した。この反応液を68℃の温度に保持しながら、水溶性重合開始剤(和光純薬社製、商品名「VA−057」=2,2’−アゾビス[N−(2−カルボキシエチル)−2−メチルプロピオンアミジン])0.504重量部及びイオン交換水30重量部より成る重合開始剤水溶液を一時間要して滴下し、更に6時間反応を行い、その後、放冷し、濾過し、水洗処理を3回行い、乾燥処理を行うことにより、表面にカルボキシル基を有する粒子(C)が23.60重量部得られた。
この粒子の粒度分布測定を行ったところ、平均粒径:0.59μmであり、その相対標準偏差(CV値)は23.6%であった。
【0063】
〔合成例4〕
温度計と窒素導入管とを装着した500mlの4つ口フラスコに、カルボキシル基を有する単量体であるメタクリル酸2.62重量部、通常単量体であるメチルメタクリレート27.38重量部、リン酸二水素カリウム0.147重量部、1N−水酸化カリウム水溶液31.47重量部、及びイオン交換水225重量部を投入し、強攪拌下でメタノール13.50重量部を10分間要して滴下し、更に30分間、強攪拌下混合した。
次に、窒素気流下で攪拌を行いながら、68℃に昇温した。この反応液を68℃の温度に保持しながら、水溶性重合開始剤(和光純薬社製、商品名「VA−057」=2,2’−アゾビス[N−(2−カルボキシエチル)−2−メチルプロピオンアミジン])0.504重量部、及びイオン交換水30重量部より成る重合開始剤水溶液を一時間要して滴下し、更に6時間反応を行い、その後、放冷し、濾過し、水洗処理を3回行い、乾燥処理を行うことにより、表面にカルボキシル基を有する粒子(D)が23.52重量部得られた。
この粒子の粒度分布測定を行ったところ、平均粒径:0.82μmであり、その相対標準偏差(CV値)は26.5%であった。
【0064】
〔合成例5〕
カーボンブラック(三菱化学社製、商品名「MA−100」)16重量部、ドデシルベンゼンスルホン酸ナトリウム0.64重量部、及びイオン交換水64重量部をメディア式湿式粉砕器を用い、20重量%のカーボンブラック分散液を得た。また、水酸化マグネシウム(和光純薬製、0.07μm)20重量部、及びイオン交換水180重量部をメディア式湿式粉砕器を用い、10重量%の水酸化マグネシウム分散液(無機分散安定剤)を得た。
他方、温度計と窒素導入管とを装着した500mlの4つ口フラスコに、カルボキシル基を有する単量体であるメタクリル酸2.35重量部、通常単量体であるメチルメタクリレート24.65重量部、リン酸二水素カリウム0.147重量部、1N−水酸化カリウム水溶液31.47重量部、及びイオン交換水200重量部を投入し、強攪拌下で10重量%の水酸化マグネシウム分散液18.5重量部を加え、更に20重量%のカーボンブラック分散液15重量部を10分間要して滴下し、更に30分間、強攪拌下混合した。
次に、窒素気流下で攪拌を行いながら、68℃に昇温した。この反応液を68℃の温度に保持しながら、水溶性重合開始剤(和光純薬社製、商品名「VA−057」=2,2’−アゾビス[N−(2−カルボキシエチル)−2−メチルプロピオンアミジン])0.453重量部及びイオン交換水30重量部より成る重合開始剤水溶液を一時間要して滴下し、更に6時間反応を行い、その後、放冷し、濾過し、水洗処理を3回行い、乾燥処理を行うことにより、表面にカルボキシル基を有する粒子(E)が24.32重量部得られた。
この粒子の粒度分布測定を行ったところ、平均粒径:0.77μmであり、その相対標準偏差(CV値)は25.2%であった。
【0065】
〔合成例6〕
温度計と窒素導入管とを装着した500mlの4つ口フラスコに、カルボキシル基を有する単量体であるメタクリル酸4.25重量部、通常単量体であるメチルメタクリレート19.75重量部、リン酸二水素カリウム0.147重量部、1N−水酸化カリウム水溶液50.41重量部、ポリメタクリル酸エステルマクロマー(東亞合成化学工業社製、商品名「AA6」)0.94重量部、及びイオン交換水220重量部を投入し、強攪拌下で20重量%のカーボンブラック分散液30重量部を10分間要して滴下し、更に30分間、強攪拌下混合した。
次に、窒素気流下で攪拌を行いながら、68℃に昇温した。この反応液を68℃の温度に保持しながら、水溶性重合開始剤(和光純薬社製、商品名「VA−057」=2,2’−アゾビス[N−(2−カルボキシエチル)−2−メチルプロピオンアミジン])0.409重量部及びイオン交換水30重量部より成る重合開始剤水溶液を一時間要して滴下し、更に6時間反応を行い、その後、放冷し、濾過し、水洗処理を3回行い、乾燥処理を行うことにより、表面にカルボキシル基を有する粒子(F)が25.61重量部得られた。
この粒子の粒度分布測定を行ったところ、平均粒径:0.93μmであり、その相対標準偏差(CV値)は28.7%であった。
【0066】
〔合成例7〕
フタロシアニンブルー(C.I.ピグメントブルー15:3)20重量部、ドデシルベンゼンスルホン酸ナトリウム0.32重量部、及びイオン交換水80重量部をメディア式湿式粉砕器を用い、20重量%のフタロシアニンブルー分散液を得た。
他方、リン酸二水素カリウム0.147重量部、1N−水酸化カリウム水溶液20.59重量部、及びイオン交換水200重量部をホモジナイザー(IKA社製:ウルトラタックスT25)で高速攪拌下(8000rpm)で、10重量%のポリビニルアルコール(和光純薬製、重合度約500)水溶液1.2重量部を加え、更にカルボキシル基を有する単量体であるアクリル酸1.41重量部、通常単量体であるメチルメタクリレート7.03重量部、及びベンジルメタクリレート18.56重量部を投入、更に前記20重量%のフタロシアニンブルー分散液15重量部を、10分間要して滴下し、更に30分間、高速攪拌下混合し反応液を得た。
続いて、予め用意しておいた温度計と窒素導入管とを装着した500mlの4つ口セパラブルフラスコに上記反応液を投入し、更に、窒素気流下で攪拌を行いながら、70℃に昇温した。この反応液を70℃の温度に保持しながら、水溶性重合開始剤(和光純薬社製、商品名「V−501」=2,2’−アゾビス[4−シアノ吉草酸])0.219重量部、1N−水酸化カルシウム水溶液1.56重量部、及びイオン交換水20重量部より成る重合開始剤水溶液を一時間要して滴下し、更に6時間反応を行い、その後、放冷し、濾過し、水洗処理を3回行い、乾燥処理を行うことにより、表面にカルボキシル基を有する粒子(G)が25.74重量部得られた。
この粒子の粒度分布測定を行ったところ、平均粒径:0.68μmであり、その相対標準偏差(CV値)は22.9%であった。
【0067】
次に、上記〔合成例1〜7〕で得られた表面にカルボキシル基を有する粒子に対してアミド化反応を行った。
【0068】
〔実施例1〕
<非イオン界面活性剤のHLB=14.31>
ノニオン系乳化剤EA−177(第一工業製薬(株)製)0.143重量部、XL−60(第一工業製薬(株)製)0.143重量部、及びイオン交換水285重量部をホモジナイザー(IKA社製:ウルトラタックスT25)で高速攪拌下(8000rpm)、シクロヘキシルアミン1.50重量部を、10分間かけて滴下した。
次に、上記〔合成例1〕で作製した表面にカルボキシル基を有する粒子(A)15.00重量部を加え、更に20分間、高速攪拌し、粒子乳化液を得た。
続いて、予め用意しておいた、温度計と窒素導入管とを装着した500mlの4つ口フラスコに、上記粒子乳化液を投入し、更に、上記一般式(1)に示したトリアジン化合物(但し、R1=R2=CH3)4.18重量部を、イオン交換水10重量部の溶液を加え、室温下、窒素気流下で攪拌を6時間行った。
反応終了後、1Nの硫酸を用いて酸性にし、濾過、水洗を3回行った。これを乾燥処理後、表面機能性部材が14.30重量部(収率91.6%)が得られた。
この表面機能性部材粒子の粒度分布測定を行ったところ、平均粒径:0.75μmであり、その相対標準偏差(CV値)は27.0%であった。
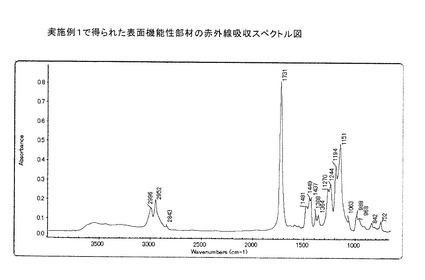
この例において得られた表面機能性部材粒子の赤外線吸収スペクトル図を図3に示す。
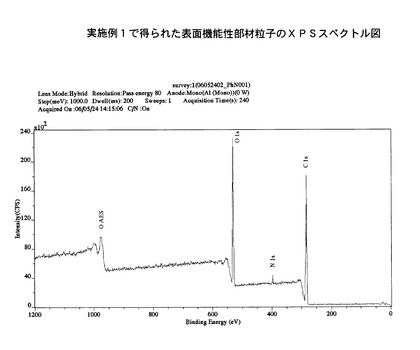
また、XPSスペクトル図を図4に示す。
図2と図4を比較すると、実施例1により作製された表面機能性部材粒子(図4)に、アミド結合に基づくN1sピークが現れていることが判り、表面機能化の実施が確認された。
更に、XPS(400eV付近のナロースキャン)による測定を行い、合成例1の粒子(A)と実施例1の表面機能性部材粒子を併せて図5に示した。
また、実施例1の表面機能性部材粒子をSEM観察(×10,000)したときの状態を図6に示す。これによると凝集が効果的に回避できており均斉度に優れていることが確かめられた。
【0069】
〔実施例2〕
<非イオン界面活性剤のHLB=17.00>
ノニオン系乳化剤NL-250(第一工業製薬(株)製)0.143重量部、XL−60(第一工業製薬(株)製)0.143重量部、及びイオン交換水285重量部をホモジナイザー(IKA社製:ウルトラタックスT25)で高速攪拌下(8000rpm)、アニリン2.87重量部を、10分間かけて滴下した。
次に、上記〔合成例2〕で作製した表面にカルボキシル基を有する粒子(B)15.00重量部を加え、更に20分間、高速攪拌し、粒子乳化液を得た。
続いて予め用意しておいた、温度計と窒素導入管とを装着した500mlの4つ口フラスコに、上記において得た粒子乳化液を投入し、更に、一般式(1)に示したトリアジン化合物(但し、R1=R2=CH3)8.53重量部を、イオン交換水10重量部の溶液を加え、室温下、窒素気流下で攪拌を6時間行った。
反応終了後、1Nの硫酸を用いて酸性にし、濾過、水洗を3回行った。これを乾燥処理後、表面機能性部材が13.51重量部(収率83.6%)が得られた。
この表面機能性部材粒子の、粒度分布測定を行ったところ、平均粒径:0.45μmであり、その相対標準偏差(CV値)は25.5%であった。
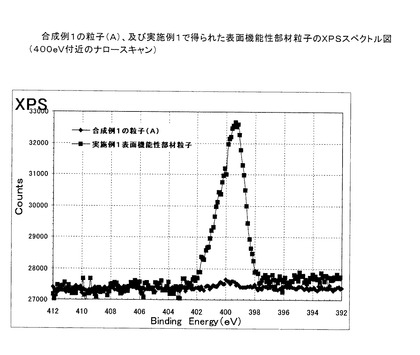
この例において得られた表面機能性部材粒子の赤外線吸収スペクトル図を図7に示す。
【0070】
〔実施例3〕
<非イオン界面活性剤のHLB=13.78>
ノニオン系乳化剤EA−177(第一工業製薬(株)製)0.143重量部、XL−60(第一工業製薬(株)製)0.285重量部、及びイオン交換水285重量部をホモジナイザー(IKA社製:ウルトラタックスT25)で高速攪拌下(8000rpm)、2−フェネチルアミン3.68重量部を、10分間かけて滴下した。
次に、上記〔合成例3〕で作製した表面にカルボキシル基を有する粒子(C)15.00重量部を加え、更に20分間高速攪拌し、粒子乳化液を得た。
続いて、予め用意しておいた、温度計と窒素導入管とを装着した500mlの4つ口フラスコに、上記において得た粒子乳化液を投入し、更に一般式(1)に示したトリアジン化合物(但し、R1=R2=CH3)8.41重量部を、イオン交換水10重量部の溶液を加え、室温下、窒素気流下で攪拌を6時間行った。
反応終了後、1Nの硫酸を用いて酸性にし、濾過、水洗を3回行った。これを乾燥処理後、表面機能性部材が13.79重量部(収率82.8%)が得られた。
この表面機能性部材粒子の、粒度分布測定を行ったところ、平均粒径:0.65μmであり、その相対標準偏差(CV値)は21.0%であった。
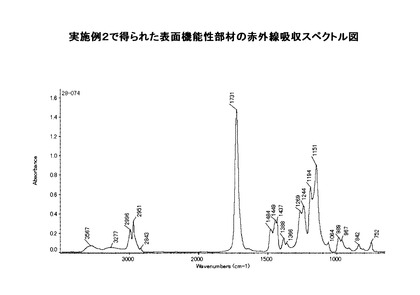
この例において得られた表面機能性部材粒子の赤外線吸収スペクトル図を図8に示す。
【0071】
〔実施例4〕
<非イオン界面活性剤のHLB=14.31>
ノニオン系乳化剤EA−177(第一工業製薬(株)製)0.143重量部、XL−60(第一工業製薬(株)製)0.143重量部、及びイオン交換水285重量部をホモジナイザー(IKA社製:ウルトラタックスT25)で高速攪拌下(8000rpm)、ベンジルアミン3.26重量部を、10分間かけて滴下した。
次に、上記〔合成例4〕で作製した表面にカルボキシル基を有する粒子(D)15.00重量部を加え、更に20分間、高速攪拌し、粒子乳化液を得た。
続いて、予め用意しておいた、温度計と窒素導入管とを装着した500mlの4つ口フラスコに、上記において得た粒子乳化液を投入し、更に、上記一般式(1)に示したトリアジン化合物(但し、R1=R2=CH3)8.41重量部を、イオン交換水10重量部の溶液を加え、室温下、窒素気流下で攪拌を6時間行った。
反応終了後、1Nの硫酸を用いて酸性にし、濾過、水洗を3回行った。これを乾燥処理後、表面機能性部材が14.89重量部(収率90.5%)が得られた。
この表面機能性部材粒子の粒度分布測定を行ったところ、平均粒径:0.90μmであり、その相対標準偏差(CV値)は22.9%であった。
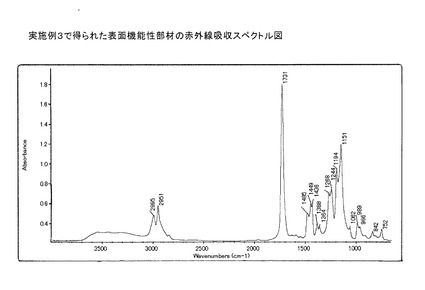
この例において得られた表面機能性部材粒子の赤外線吸収スペクトル図を図9に示す。
【0072】
〔実施例5〕
<非イオン界面活性剤のHLB=14.31>
ノニオン系乳化剤EA−177(第一工業製薬(株)製)0.143重量部、XL−60(第一工業製薬(株)製)0.143重量部、及びイオン交換水285重量部をホモジナイザー(IKA社製:ウルトラタックスT25)で高速攪拌下(8000rpm)、1−(2−アミノエチル)ピペリジン3.90重量部を10分間かけて滴下した。
次に、上記〔合成例5〕で作製した表面にカルボキシル基を有する粒子(E)15.00重量部を加え、更に20分間、高速攪拌し、粒子乳化液を得た。
続いて予め用意しておいた、温度計と窒素導入管とを装着した500mlの4つ口フラスコに、上記において得た粒子乳化液を投入し、更に、上記一般式(1)に示したトリアジン化合物(但し、R1=R2=CH3)8.41重量部を、イオン交換水10重量部の溶液を加え、室温下、窒素気流下で攪拌を6時間行った。
反応終了後、1Nの硫酸を用いて酸性にし、濾過、水洗を3回行った。これを乾燥処理後、表面機能性部材が14.77重量部(収率91.0%)が得られた。
この表面機能性部材粒子の粒度分布測定を行ったところ、平均粒径:0.89μmであり、その相対標準偏差(CV値)は20.7%であった。
【0073】
〔実施例6〕
<非イオン界面活性剤のHLB=14.31>
ノニオン系乳化剤EA−177(第一工業製薬(株)製)0.143重量部、XL−60(第一工業製薬(株)製)0.143重量部、及びイオン交換水285重量部をホモジナイザー(IKA社製:ウルトラタックスT25)で高速攪拌下(8000rpm)、N,N−ジ−n−ブチルエチレンジアミン10.62重量部を、10分間かけて滴下した。
次に、上記〔合成例5〕で作製した表面にカルボキシル基を有する粒子(E)15.00重量部を加え、更に20分間、高速攪拌し、粒子乳化液を得た。
続いて、予め用意しておいた、温度計と窒素導入管とを装着した500mlの4つ口フラスコに、上記において得た粒子乳化液を投入し、更に、上記一般式(1)に示したトリアジン化合物(但し、R1=R2=CH3)17.06重量部を、イオン交換水10重量部の溶液を加え、室温下、窒素気流下で攪拌を6時間行った。
反応終了後、1Nの硫酸を用いて酸性にし、濾過、水洗を3回行った。これを乾燥処理後、表面機能性部材が15.82重量部(収率90.4%)が得られた。
この表面機能性部材粒子の粒度分布測定を行ったところ、平均粒径:0.98μmであり、その相対標準偏差(CV値)は25.3%であった。
【0074】
〔実施例7〕
<非イオン界面活性剤のHLB=13.78>
ノニオン系乳化剤EA−177(第一工業製薬(株)製)0.146重量部、XL−60(第一工業製薬(株)製)0.291重量部、及びイオン交換水291重量部をホモジナイザー(IKA社製:ウルトラタックスT25)で高速攪拌下(8000rpm)、N,N−ジメチルエチレンジアミン1.63重量部を、10分間かけて滴下した。
次に、上記〔合成例7〕で作製した表面にカルボキシル基を有する粒子(G)9.00重量部を加え、更に20分間高速攪拌し、粒子乳化液を得た。
続いて、予め用意しておいた、温度計と窒素導入管とを装着した500mlの4つ口フラスコに、上記において得た粒子乳化液を投入し、更に一般式(1)に示したトリアジン化合物(但し、R1=R2=CH3)5.12重量部を、イオン交換水20重量部の溶液を加え、室温下、窒素気流下で攪拌を6時間行った。
反応終了後、1Nの硫酸を用いて酸性にし、濾過、水洗を3回行った。これを乾燥処理後、表面機能性部材が8.76重量部(収率90.8%)が得られた。
この表面機能性部材粒子の、粒度分布測定を行ったところ、平均粒径:0.81μmであり、その相対標準偏差(CV値)は21.0%であった。
【0075】
〔実施例8〕
<非イオン界面活性剤のHLB=13.78>
ノニオン系乳化剤EA−177(第一工業製薬(株)製)0.147重量部、XL−60(第一工業製薬(株)製)0.294重量部、及びイオン交換水284重量部をホモジナイザー(IKA社製:ウルトラタックスT25)で高速攪拌下(8000rpm)、アミノ基を有するシリコーン化合物として3−アミノプロピルメチル−ビス(トリメチルシロキシ)シラン(アズマックス社製)3.45重量部を、10分間かけて滴下した。
次に、上記〔合成例7〕で作製した表面にカルボキシル基を有する粒子(G)6.00重量部を加え、更に20分間高速攪拌し、粒子乳化液を得た。
続いて、予め用意しておいた、温度計と窒素導入管とを装着した500mlの4つ口フラスコに、上記において得た粒子乳化液を投入し、更に一般式(1)に示したトリアジン化合物(但し、R1=R2=CH3)3.41重量部を、イオン交換水20重量部の溶液を加え、室温下、窒素気流下で攪拌を6時間行った。
反応終了後、1Nの硫酸を用いて酸性にし、濾過、水洗を3回行った。これを乾燥処理後、表面機能性部材が13.79重量部(収率82.8%)が得られた。
この表面機能性部材粒子の、粒度分布測定を行ったところ、平均粒径:0.65μmであり、その相対標準偏差(CV値)は19.4%であった。
【0076】
〔比較例〕
<シード重合:非イオン界面活性剤のHLB=14.31>
ノニオン系乳化剤EA−177(第一工業製薬(株)製)0.143重量部、XL−60(第一工業製薬(株)製)0.143重量部、及びイオン交換水276重量部を強攪拌下、メチルメタクリレート9.60重量部を10分間かけて滴下した。
次に、上記〔合成例1〕で作製した粒子(A)14.40重量部を加え、更に20分間、高速攪拌し、粒子乳化液を得た。
続いて、予め用意しておいた、温度計と窒素導入管とを装着した500mlの4つ口フラスコに、上記において得た粒子乳化液を投入し、更に、水溶性重合開始剤(和光純薬社製、商品名「VA−057」=2,2’−アゾビス[N−(2−カルボキシエチル)−2−メチルプロピオンアミジン])0.487重量部のイオン交換水30重量部の水溶液を加え、窒素気流下で、静かに攪拌しながら室温より40℃まで0.5℃/分の速度で昇温し、更に40℃で2時間吸収・膨潤し、その後、68℃まで0.2℃/分の速度で昇温し、更に68℃で4時間反応した。
反応終了後、1Nの硫酸を用いて酸性にし、濾過、水洗を3回行った。
これを乾燥処理後、シード重合による比較例粒子が19.32重量部(収率80.5%)が得られた。
この比較例粒子の、粒度分布測定を行ったところ、平均粒径:1.03μmであり、その相対標準偏差(CV値)は45.3%であった。
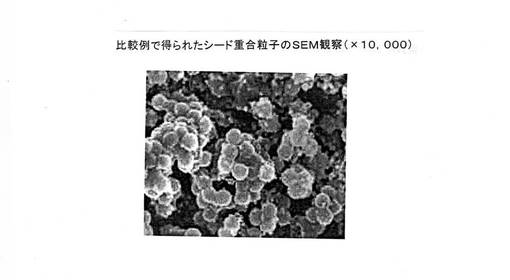
この例において得られた粒子のSEM観察(×10,000)を図10に示す。
図6の実施例1で得られた表面機能性部材粒子と、図10の比較例で得られたシード重合粒子とを比較する明らかなように、シード重合粒子は微小粒子、及び粒子間の合一と思われる凝集物が観察された。
その結果、相対標準偏差(CV値)はシード粒子(合成例1で作製した粒子(A))と比較して大幅に増加した。
【0077】
上述したように、本発明方法によれば、相対標準偏差(CV値)を損なうことなく、非常に容易な工程で、単分散性の高い、略真球状の表面機能性部材粒子が作製できた。
【0078】
〔応用例〕
実施例4、実施例6及び実施例8で得られた表面機能性部材を絶縁性液体であるシリコーンオイルSH−200(東レ・ダウコーニング社製)に2重量%の濃度に分散し、ζ電位を測定した。次に、図11に示す装置により、電気泳動を確認した。
【0079】
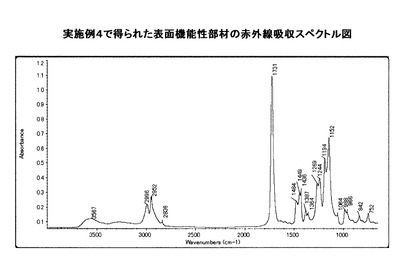
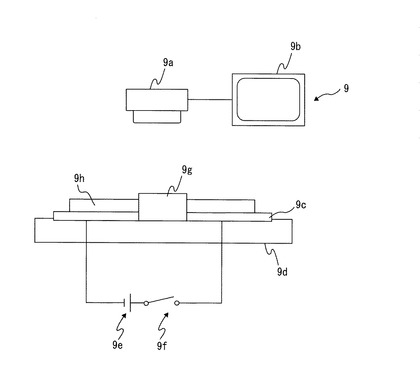
ここで、図11の電気泳動装置について説明する。この装置は、ITO電極9c間の表面機能性部材の分散液9gの電気泳動の評価を、高速度カメラ撮影装置9を用いて行うものである。すなわち、まずガラス基板9d上にITO電極9cを100μmの間隔をあけて厚さ100nm蒸着し、その上に25μm厚のガラス板9hを100μm間隔でITO電極9c上に接着し液溜を設ける。次に、液溜に上記表面機能性部材の分散液9gを注入し、ITO電極9c間に電源9eから1000Vの電圧を印加した時に液滴が泳動される様子を上方の高速度カメラ9aで撮影してモニター9bで観察し、電気泳動の評価を行った。
【0080】
〔応用比較例〕
応用比較例として、合成例1で得られた表面にカルボキシル基を有する粒子(A)を、上記応用例と同じ条件で分散し、ζ電位の測定及び電気泳動の確認を行った。
以上の応用例、応用比較例の結果を併せて、表1に示す。
【0081】
【表1】
【図面の簡単な説明】
【0082】
【図1】合成例1で得られた微粒子(A)の赤外線吸収スペクトル図を示す。
【図2】合成例1で得られた微粒子(A)のXPSスペクトル図を示す。
【図3】実施例1で得られた表面機能性部材の赤外線吸収スペクトル図を示す。
【図4】実施例1で得られた表面機能性部材のXPSスペクトル図を示す。
【図5】合成例1の粒子(A)、及び実施例1の表面機能性部材微粒子のXPSスペクトル図を示す。
【図6】実施例1で得られた表面機能性部材粒子のSEM観察状態図を示す。
【図7】実施例2で得られた表面機能性部材の赤外線吸収スペクトル図を示す。
【図8】実施例3で得られた表面機能性部材の赤外線吸収スペクトル図を示す。
【図9】実施例4で得られた表面機能性部材の赤外線吸収スペクトル図を示す。
【図10】比較例で得られた表面機能性部材粒子のSEM観察状態図を示す。
【図11】電気泳動装置の構成図を示す。
【符号の説明】
【0083】
9 高速度カメラ撮影装置
9a 高速度カメラ
9b モニター
9c ITO電極
9d ガラス基板
9e 電源
9f スイッチ
9g 表面機能性部材の分散液
9h ガラス板
【技術分野】
【0001】
本発明は、表面機能性部材の製造方法、及び表面機能性部材及び電気泳動素子に関するものであり、詳細には、アミノ基を有する各種機能性素材を、縮合剤の存在下で、直接結合して得られる表面機能性部材の製造方法、及び当該方法により作製された表面機能性部材、並びに該表面機能性部材を用いた電気泳動素子に関するものであり、更に詳細には、PLD(ペーパーライクディスプレー)、電子写真現像剤(トナー)、インクジェット(インク)等各種の用途に用いられる機能性表面層を有する応用範囲の広い、簡便な表面機能性部材の製造方法、及び表面機能性部材及び電気泳動素子に関する。
【背景技術】
【0002】
従来、任意の基材に、高分子機能性素材、又は低分子機能性素材を、吸着させ、あるいは反応させることにより、各種機能を有する表面層が形成された表面機能性部材について、種々の技術提案がなされている。
特に、機材との密着力が強く、機械的耐磨耗性や有機溶剤耐性に優れ、かつ優れた耐久性を有している表面機能性部材が得られることを目的として、基材と共有結合により、直接結合している方式の表面機能性部材が注目されている。
【0003】
例えば、(1)クロム硫酸水溶液などの薬品処理、(2)カップリング剤による処理、(3)蒸気処理、(4)グラフト化処理、(5)電気化学的な方法等が知られている(例えば、下記非特許文献1参照。)。このうち、(1)及び(5)は、酸化反応、あるいは還元反応により新たに反応性の置換基を形成するもので機能性素材の導入には不適である。
また、上記(3)の、蒸気処理による機能性素材の導入においては、機能性素材の沸点がある程度低いことが条件となり、装置が大掛かりとなりコスト上においても課題を有している。
【0004】
一方、上記(2)のカップリング剤による処理は、基材と直接共有結合により結合しているため、基材との密着性が強く、機械的な耐磨耗性や有機溶剤耐性が高く、耐久性にも優れた表面機能性部材が得られていた。
しかしながら、カップリング剤による処理は、基本的に加熱反応であり、通常200℃以上、触媒を使用しても150℃以上の温度が必要となる。機能性素材の熱劣化を防止するためには、温度は低く抑える方が好ましい。
また、機能性素材のカップリング剤化(機能性素材中間体)を行う工程が必要であるため、製造コストの点からは不利である。
(4)のグラフト化処理は、光、プラズマ、放射線等の反応エネルギーを付与しながら、気相、あるいは液相処理を行う方法であるが、これにおいては、大きな光(反応)エネルギー下に晒されることになるため、機能性素材の光劣化が大きな課題となっていた。
特に基材として粒子を選定した場合には、最も一般的な表面機能化方法として、シード重合法があり、現在、活発な研究開発が進められている。シード重合法は、重合体粒子(シード粒子)に単量体及び必要に応じて種々の機能性化合物を吸収・膨潤させて、このシード粒子に吸収された単量体を粒子表面で重合することにより製造する方法である。
例えば、架橋性単量体を使用し、吸収・膨潤が困難であるため、非常に複雑で、精度の高い重合法が開示されており(例えば、下記特許文献1参照。)、また、単量体と油溶性染料によるシード重合を膨潤剤(添加剤)を含有しない水系媒体中での重合が開示されている(例えば、下記特許文献2参照。)。
シード重合法による表面機能化は、吸収・膨潤が非常に困難で、多段階吸収法、膨潤剤の使用等、多数提案されているが、吸収・膨潤は基本的にはシード粒子と水系媒体との分配によるため、攪拌方法、温度制御など非常に精密な反応制御が必要となる。また、吸収・膨潤したシード粒子は、非常に不安定(粒子表面の粘着力が増加)で、たびたび、歪な粒子の生成、或いはシード粒子間の合一が発生し、粒子径、及び粒径分布の制御が困難となる。
以上のように、表面機能性部材における機能性素材の安定、且つ保持特性及びその持続性の向上、加えて応用範囲の広い、簡便な表面機能性部材の製造方法については、さらなる改良が望まれているのが現状である。
【0005】
【特許文献1】特許第3434036号公報
【特許文献2】特開2001−89510号公報
【非特許文献1】「高分子の表面改質と応用」(監修:角田光雄、発行:(株)シーエムシー)、第13頁の第2章、化学的表面改質技術
【発明の開示】
【発明が解決しようとする課題】
【0006】
そこで本発明においては、上述した従来の技術が包含していた各種問題点に鑑みて、機能性素材と基材とを共有結合により直接結合している方法を選定し、使用環境下においても優れた機能発現、及び耐久性を実現し得ること、及び各種の用途に用いられる応用範囲の広い、簡便な表面機能性部材を製造する方法、及び当該方法により得られた表面機能性部材、並びに該表面機能性部材を用いた電気泳動素子を提供することとした。
【課題を解決するための手段】
【0007】
本発明者は、表面にカルボキシル基を有する基材上に、アミノ基を有する機能性素材を縮合剤の存在下に、直接結合させて成る表面機能性部材において、水系連続相中で、反応させることにより、上記従来技術における課題が解決されることを見出し、本発明に想到するに至った。
【0008】
請求項1の発明においては、表面にカルボキシル基を有する基材上に、アミノ基を有する機能性素材を、縮合剤の存在下、直接結合させて得られる表面機能性部材の製造方法であって、水系連続相中で反応させることを特徴とする表面機能性部材の製造方法を提供する。
【0009】
請求項2の発明においては、前記表面にカルボキシル基を有する基材が、乳化重合、懸濁重合、シード重合、あるいはソープフリー重合のいずれかの重合方式により得られた粒子であり、かつ平均粒子径が0.1〜10μmであることを特徴とする請求項1の表面機能性部材の製造方法を提供する。
【0010】
請求項3の発明においては、前記縮合剤が、下記一般式(1)で表わされるトリアジン化合物であることを特徴とする請求項1又は2の表面機能性部材の製造方法を提供する。
【0011】
【化1】
【0012】
但し、R1、R2は、置換もしくは無置換のアルキル基を表す。
【0013】
請求項4の発明においては、表面にカルボキシル基を有する基材上に、アミノ基を有する機能性素材を、縮合剤の存在下、直接結合させて得られる表面機能性部材の製造方法であって、水系連続相中に、更に非イオン性界面活性剤を添加する工程を有することを特徴とする請求項1乃至3のいずれかの表面機能性部材の製造方法を提供する。
【0014】
請求項5の発明においては、前記表面にカルボキシル基を有する基材上に、アミノ基を有する機能性素材を、縮合剤の存在下、水を主体とする連続相中で直接結合させて得られる表面機能性部材の製造方法であって、前記、水系連続相中に非イオン性界面活性剤を添加したことを特徴とする表面機能性部材の製造方法を提供する。
【0015】
請求項6の発明においては、前記アミノ基を有する機能性素材が、シロキサン骨格を有する置換基を含有する機能性素材であることを特徴とする請求項1乃至5のいずれか一項に記載の表面機能性部材の製造方法を提供する。
【0016】
請求項7の発明においては、前記アミノ基を有する機能性素材が、下記一般式(I)及び一般式(II)で表されるシリコーン化合物より選ばれる少なくとも一種のシリコーン化合物であることを特徴とする請求項1乃至5のいずれか一項に記載の表面機能性部材の製造方法を提供する。
H2N−B11−X ・・・(I)
H2N−B12−NH−B13−X ・・・(II)
(式中、B11,B12及びB13は、炭素数2から6のアルキレン基、Xはシロキサン骨格を示す。)
【0017】
請求項8の発明においては、表面にカルボキシル基を有する基材上に、アミノ基を有する機能性素材を、縮合剤の存在下で直接結合させて得られる表面機能性部材であって、前記結合の反応は、水系連続相中で行われたものであることを特徴とする表面機能性部材を提供する。
【0018】
請求項9の発明においては、前記表面にカルボキシル基を有する基材が着色剤を含有していることを特徴とする請求項8の表面機能性部材を提供する。
【0019】
請求項10の発明においては、前記基材が球状粒子であり、かつ平均粒子径が0.1〜10μmであることを特徴とする請求項8又は9の表面機能性部材を提供する。
【0020】
請求項11の発明においては、前記アミノ基を有する機能性素材が、シロキサン骨格を有する置換基を含有する機能性素材であることを特徴とする請求項8乃至10のいずれか一項に記載の表面機能性部材を提供する。
【0021】
請求項12の発明においては、前記アミノ基を有する機能性素材が、下記一般式(I)及び一般式(II)で表されるシリコーン化合物より選ばれる少なくとも一種のシリコーン化合物であることを特徴とする請求項8乃至10のいずれか一項に記載の表面機能性部材を提供する。
H2N−B11−X ・・・(I)
H2N−B12−NH−B13−X ・・・(II)
(式中、B11,B12及びB13は、炭素数2から6のアルキレン基、Xはシロキサン骨格を示す。)
【0022】
請求項13の発明においては、絶縁性液体中に、請求項8乃至12のいずれか一項に記載の表面機能性部材を分散してなることを特徴とする電気泳動素子を提供する。
【0023】
請求項14においては、シリコーンオイル中に、請求項11又は12に記載の表面機能性部材を分散してなることを特徴とする電気泳動素子を提供する。
【0024】
本発明方法によれば、水系連続相中で、表面にカルボキシル基を有する基材上にアミノ基を有する機能性素材を、縮合剤の存在下で反応させるものであるので、非常に安易かつ環境面にも配慮された工程により所望の表面機能性部材が得られる。
また、基材と機能性素材との結合力が極めて強く、耐久性に優れた表面機能性部材が得られる。
【発明の効果】
【0025】
本発明によれば、基材とアミド基を介して直接機能性素材を導入することにより耐久性に優れ、かつ長時間に亘って機能を維持し得る表面機能性部材の適応範囲の広い製造方法、及びこの製造方法により得られた表面機能性部材を提供することができた。さらに、該表面機能性部材を用いた電気泳動素子を提供することができた。
【発明を実施するための最良の形態】
【0026】
以下、本発明について、具体的に説明する。
本発明においては、水系連続相中で、基材上のカルボキシル基と、機能性素材のアミノ基を縮合剤の存在下に直接結合させて表面機能性部材を得る。
【0027】
先ず、カルボキシル基を有する基材について説明する。
基材としては、板状物、あるいは球状物等、いかなる形状のものも使用でき、最終的に目的とする表面機能性部材の使用目的に応じて高分子樹脂からなる表面を有する基材を適宜選択する。例えば、樹脂フィルム、表面に樹脂が被覆されているガラス等の無機基材、表面が樹脂層からなる複合材等が挙げられる。
特に形状の観点からは、表面積が大きく表面機能性部材の効果が期待できる粉体や粒子が好適である。更に、粒径の均一な粒子が作製できること、及び水系連続相中で造粒出来るという観点からは、乳化重合、懸濁重合、シード重合、及びソープフリー重合より得られた粒子が好適である。
材料としては、寸度的に安定なものが好適であり、最終的に目的とする表面機能性部材の所望の可撓性、強度、耐久性、物性等を有していればよい。
光透過性を必要とする材料を選択する場合は、例えば、ガラス、プラスチックフィルム(例えば、二酢酸セルロース、三酢酸セルロース、プロピオン酸セルロース、酪酸セルロース、酢酸酪酸セルロース、硝酸セルロース、ポリエチレンテレフタレート、ポリエチレン、ポリスチレン、ポリプロピレン、ポリカーボネート、ポリビニルアセタール等)等が挙げられる。
特に光透過性に優れた材料としては、例えば、ポリメチルメタクリレート、ポリカーボネート、ポリスチレン、ポリジエチレングリコールビスアリルカーボネート等の樹脂材料が好適である。
光透過性を有さない材料を選択する場合は、例えば、紙、プラスチックがラミネートされた紙、金属板(例えば、アルミニウム、亜鉛、銅等)、金属がラミネート若しくは蒸着された紙若しくはプラスチックフィルム等を挙げることができる。
基材の材料としては、最終的に目的とする表面機能性部材の用途や、反応される機能性素材との関係に応じて適宜材料選択するが、加工性、透明性の観点からは、高分子樹脂からなる表面を有する材料が好ましく、具体的には、樹脂フィルム、表面に樹脂が被覆されているガラス等の透明無機基材、表面層が樹脂層からなる複合材が好適である。
【0028】
基材の材料として、表面に樹脂が被覆されているものを選択するときには、例えば、表面に樹脂フィルムが貼着された積層板、プライマー処理された基材、ハードコート処理された基材等が挙げられる(例えば、特開2006−56949号公報に開示されている。)。
表面層が樹脂層からなる基材とは、具体的には、裏面に接着剤層が設けられた樹脂シール材、ガラスと樹脂との積層体である合わせガラス等が挙げられる。
また、基材は、最終的に目的とする表面機能性部材の使用目的に応じて、粗面化処理を施してもよい。次行程である機能性素材の単位面積当たりの反応量をより向上、あるいは反応性を向上させるために表面積を増加させる目的で、基材表面を予め粗面化しておくことが有効である。
粗面化方法としては、公知の方法を選択することができる。例えば、基材が樹脂フィルムの場合には、グロー放電処理、スパッタリング、サンドブラスト研磨法、バフ研磨法、粒子付着法、粒子塗布法等が挙げられる。また、基材がガラス板等の無機材料の場合には、機械的に粗面化する方法が適用できる。機械的方法としては、ボール研磨法、ブラシ研磨法、ブラスト研磨法、バフ研磨法等を適用できる。
【0029】
次に、基材の表面にカルボキシル基を導入する方法について説明する。
カルボキシル基の導入方法としては、従来公知の方法を適宜使用できる。例えば、予め表面にカルボキシル基を有している基材を使用する方法、基材上にカルボキシル基を有する樹脂層(中間層)を形成する方法が挙げられる。
【0030】
基材が板状物である場合は、カルボキシル基を有する樹脂を所定の溶剤に溶解、分散、あるいは熱溶融して、これをスピンコーティング、ブレードコーティング、スプレーコーティング、ディップコーティング等公知の塗工方法により塗布して樹脂層を形成することができる。
【0031】
また、上記のように層状の樹脂層を形成するのみならず、カルボキシル基を有する樹脂により球状物を作製しこれを基材としてもよい。この場合、カルボキシル基を有する樹脂に対する溶解度差を利用して造粒する方法が好適であり、具体的には、カルボキシル基を有する樹脂を溶剤に溶解し、カルボキシル基を有する樹脂に対して貧溶媒中に滴下する方法、カルボキシル基を有する樹脂を溶剤に溶解し、溶媒を徐々に留去する方法等が挙げられる。特に、カルボキシル基を有する樹脂を溶剤に溶解し、分散安定剤等を添加した水系連続相中で造粒する方法が次工程への移行が容易である点、及び極性基であるカルボキシル基が特異的に粒子表面に形成される点で好適である(コアセルベーション法)。
なお、樹脂中には必要に応じて、着色剤、分散剤、熱安定剤、酸化防止剤、紫外線吸収剤等の各種添加剤を均一に溶解、分散するようにしてもよい。
上記樹脂としては、例えば、アクリル樹脂、スチレン系樹脂、エポキシ樹脂、ウレタン樹脂、ビニル樹脂、フェノール樹脂、ポリエステル樹脂、ポリアミド樹脂、メラミン系樹脂等の合成樹脂、ゼラチン、カゼイン、セルロースデンプンなどの天然樹脂、あるいはこれらの共重合樹脂等がいずれも適用できる。また、これら樹脂は使用目的に応じて架橋構造を形成したものとしてもよい。
【0032】
その他、紫外線、エキシマレーザー等の光処理、電子線処理、イオンビーム処理、あるいはプラズマ処理等の化学作用により、新たにカルボキシル基を生成する方法が挙げられる。この方法における樹脂としては、上記に掲げたものを適宜使用できる。
【0033】
また、基材面へのカルボキシル基の導入方法として、グラフト化による生成方法も適用できる。
この場合、基材は、グラフトポリマーが化学的に結合できるような表面を有しているものを適用する。なお基材自体の表面がこのような特性を有していてもよく、このような特性を有する樹脂層(中間層)を基材表面に形成してもよい。樹脂層(中間層)としては、光グラフト重合法、放射線照射グラフト重合法によりグラフトポリマーを合成する場合には、上記に掲げた樹脂を使用できる。また、光グラフト重合法、プラズマ照射グラフト重合法、放射線照射グラフと重合法等は、グラフト重合の開始が樹脂層(中間層)の水素引き抜きから反応が進行するため、水素が引き抜かれやすい樹脂、例えばアクリル樹脂、ウレタン樹脂、スチレン系樹脂、ビニル系樹脂、ポリアミド系樹脂、エポキシ樹脂等が製造適性の観点から好適である。この場合、基材表面、あるいは樹脂層(中間層)表面に、重合性化合物と重合開始剤を添加し、所定のエネルギーを付与することにより重合開始能を発現することができる。
重合性化合物は、活性光線照射等のエネルギー付与により、カルボキシル基含有モノマー、あるいはカルボキシル基含有ポリマーが付与し得るものであれば制限はない。特に、分子内にカルボキシル基を有する重合性基含有ポリマーが好適である。
重合開始剤は、所定のエネルギー、例えば、活性光線の照射、加熱、電子線の照射等により、重合開始能を発現しうる公知の熱重合開始剤、光重合開始剤等を、目的に応じて、適宜選択して適用できる。特に、熱重合よりも反応速度(重合速度)が高い光重合を利用することが製造適性の観点から好適であるため、光重合開始剤の使用がより好ましい。
光重合開始剤は、照射される活性光線に対して活性であり、重合性化合物を重合させることができれば、特に制限はなく、例えば、ラジカル重合開始剤、アニオン重合開始剤、カチオン重合開始剤等が適用できる。
【0034】
カルボキシル基を有する基材が微粒子である場合には、上述のコアセルベーション法と重合法がある。重合法は、重合性単量体に、重合開始剤と、必要に応じて着色剤、及び架橋剤等の各種添加剤とともに均一に溶解、あるいは分散させた重合単量体組成物を調整し、昇温して重合することにより、所望の粒径を有する微粒子を得ることができる。また、必要に応じて、連鎖移動剤、ワックス、及び帯電制御剤等の各種添加剤等を添加してもよい。
【0035】
上記粒子の粒径分布は、製造される本発明の表面機能性部材の粒径分布に反映されることから、単分散性の高いものか好ましく、相対標準偏差(CV値)として30%以下であることが好ましい。相対標準偏差(CV値)が30%を超えると上記粒子から製造される表面機能性部材の粒径分布が広がり、粒径の不均一性より生じる粒子の特性が著しく損なわれ、例えば、電気泳動移動度に差が生じ問題となる。
上記相対標準偏差(CV値)は下記数式(1)により算定される。
相対標準偏差(CV値(%))=(sd/m)×100……(1)
(式中、sdは粒子径の標準偏差を、mは平均径である。)
なお、上記sd、及びmは、粒径アナライザー(FPAR−1000:大塚電子社製)による動的光散乱法により得られる数値である。
【0036】
上記カルボキシル基を有する微粒子の作製に用いる重合性単量体について説明する。
重合性単量体としては、ラジカル重合性単量体を必須の構成成分とする。
上記所望の微粒子の作製に適用する必須成分であるカルボキシル基を有する単量体としては、例えば、4−ビニル安息香酸、3−ビニル安息香酸などのスチレン誘導体、(メタ)アクリル酸、イタコン酸、イタコン酸モノブチルエステル、マレイン酸、マレイン酸モノメチルエステル、マレイン酸モノブチルエステル、2−カルボキシルエチルアクリレート等のアクリル酸系誘導体が挙げられる。
上記カルボキシル基を有する単量体の配合量は、以下に示す通常単量体と併せた総単量体100重量部に対して1〜50重量部とすることが好適である。
通常単量体は、特に限定されるものではない。例えば、スチレン、α−メチルスチレン、p−メチルスチレン、p−クロロスチレン、クロロメチルスチレン等のスチレン誘導体、塩化ビニル、酢酸ビニル、プロピロン酸ビニル等のビニルエステル類、アクリロニトリル等の不飽和ニトリル類、(メタ)アクリル酸メチル、(メタ)アクリル酸エチル、(メタ)アクリル酸ブチル、(メタ)アクリル酸−2−エチルへキシル、(メタ)アクリル酸ステアリル、エチレングリコール(メタ)アクリレート、トリフルオロエチル(メタ)アクリレート、ペンタフルオロプロピル(メタ)アクリレート、シクロヘキシル(メタ)アクリレート等の(メタ)アクリル酸エステル誘導体が挙げられる。これらは単独で用いてもよく、2種以上併用しても良い。また、必要に応じて、通常単量体中ビニル基を2個以上含有する架橋性単量体を含有してもよい。ビニル基を2個以上含有する架橋性単量体としては特に限定されず、例えば、ジビニルベンゼン、ジビニルビフェニル、ジビニルナフタレン、エチレングリコールジ(メタ)アクリレート、ブタジエングリコールジ(メタ)アクリレート、ポリエチレングリコールジ(メタ)アクリレート等が挙げられる。上記架橋性単量体は単独で用いても良く、二種以上を併用しても良い。
また、本発明では、マクロモノマーをカルボキシル基を有する単量体、及び通常単量体と共に重合性単量体として使用することができる。マクロモノマーは、分子鎖の末端にビニル重合性官能基を有するもので、数平均分子量が、通常、1000〜30000のオリゴマー又はポリマーである。
マクロモノマー分子鎖の末端に有するビニル重合性官能基としては、アクロイル基、メタクロイル基等を挙げることができる。マクロモノマーの量は、重合性単量体100重量部に対して、通常、0.01〜10重量部とすることが好適である。
重合組成物には樹脂の重合度を制御し、軟化点や分子量等の物性を調節するために連鎖移動剤を添加することができる。連鎖移動剤としては、ラジカル重合反応で一般的に用いられる連鎖移動剤を用いることが可能であり、特に限定されるものではない。例えば、オクチルメルカプタン、ドデシルメルカプタン、tert−ドデシルメルカプタン等のメルカプタン、及びスチレンダイマー等が挙げられる。
【0037】
上記カルボキシル基を有する微粒子の作製に用いるラジカル重合開始剤について説明する。
ラジカル重合開始剤は、特に限定されず、従来公知のものを適用できる。
例えば、過硫酸カリウム、過硫酸アンモニウム等の過硫酸塩、過酸化ベンゾイル、過酸化ラウロイル、オキソクロロ過酸化ベンゾイル、t−ブチルパーオキシ−2−エチルヘキサニエート、ジ−t−ブチルパ−オキサイド等の過酸化物、2,2’−アゾビスイソブチロニトリル、1,1’−アゾビス(シクロヘキサン−1−カルボニトリル)、2,2’−アゾビス(2,4−ジメチルバレロニトリル)、2,2’−アゾビス[2−メチル−N−(2−ヒドリキシエチル)プロピオンアミド]、2,2’アゾビス(2−メチルプロピオンアミジン)塩、2,2’−アゾビス[N−(2−カルビキシエチル)−2−メチルプロピオンアミジン]、4,4’−アゾビス(4−シアノ吉草酸)等のアゾ系化合物が挙げられる。中でも、2,2’−アゾビス[N−(2−カルボキシエチル)−2−メチルプロピオンアミジン]、4,4’−アゾビス(4−シアノ吉草酸)などの水溶性アゾ系化合物が好適である。上記ラジカル重合開始剤は、必要に応じて還元剤と組み合わせたレドックス系開始剤として使用してもよい。レドックス開始剤を用いることで、重合活性が上昇し重合温度の低下が図れ、更に重合時間の短縮が期待できる。また、上記ラジカル重合開始剤の配合量は、重合性単量体100重量部に対して0.1〜10重量部とすることが好適である。
【0038】
次に、上記カルボキシル基を有する微粒子の作製に用いる添加剤について説明する。
カルボキシル基を含有する微粒子を作製する際には、必要に応じて、界面活性剤を添加してもよい。界面活性剤としては特に限定されるものでは無いが、下記のイオン性及びノニオン性界面活性剤が好適に用いられる。
イオン性界面活性剤としては、例えば、ドデシルベンゼンスルホン酸ナトリウム、アリールアルキルポリエーテルスルホン酸ナトリウム、3,3−ジスルホンジフェニル尿素−4,4−ジアゾビス−アミノ−8−ナフトール−6−スルホン酸ナトリウム、等のスルホン酸塩、ドデシル硫酸ナトリウム、テトラデシル硫酸ナトリウム、ペンタデシル硫酸ナトリウム、ジアルキルスルホコハク酸エステルナトリウム、等の硫酸エステル、オレイン酸ナトリウム、ラウリン酸ナトリウム、カプリン酸ナトリウム、カプロン酸ナトリウム、ステアリン酸カリウム等の脂肪酸塩などが挙げられる。
非イオン性界面活性剤としては、ポリエチレンオキシド、ポリポロピレンオキシド、ポリエチレンオキシドとポリプロピレンオキシドの組み合わせ、ポリエチレングリコールの高級脂肪酸エステル、ポリプロピレンオキシドの高級脂肪酸エステル、アルキルフェノールポリエチレンオキシド、アルキルフェノールポリプロピレンオキシド、ポリエチレンオキシドアルキルエーテル、ポリプロピレンオキシドアルキルエーテルグリコール、ソルビタンエステルなどが挙げられる。
界面活性剤の配合量は、総単量体100重量部に対して、0.1〜5重量部とすることが好適である。
更に、必要に応じて、分散安定剤を添加してもよい。分散安定剤としては、例えば、部分鹸化されたポリビニルアルコール、ポリビニルエーテル、ゼラチン、メチルセルロース、エチルセルロース、ヒドロキシエチルセルロース、カルボキシメチルセルロースのナトリウム塩等の高分子分散安定剤、リン酸カルシウム、リン酸マグネシウム、リン酸アルミニウム、炭酸カルシウム、炭酸マグネシウム、硫酸バリウム、水酸化マグネシウム、ベントナイト等の無機分散安定剤が挙げられる。分散安定剤の配合量は、総単量体100重量部に対して0.1〜5重量部とすることが好適である。
【0039】
なお、上記の他、着色剤を添加してもよい。着色剤粒子分散液は、着色剤を水系媒体中、あるいは溶剤系媒体中に分散することにより調整することができる。着色剤の分散処理は、水中の場合は、界面活性剤を臨界ミセル濃度(CMC)以上にした状態で行われ、用いられる界面活性剤はイオン性界面活性剤やノニオン性界面活性剤が使用でき、これらを単独、或いは適当な組成で混合して使用すればよい。溶剤系では着色剤と充分に湿潤する溶剤を選択し、必要に応じ界面活性剤も使用される。用いられる溶剤としては、メタノール、エタノール、イソプロパノール、アセトン、メチルエチルケトン、メチルイソブチルケトン、テトラヒドロフランなどが挙げられる。着色剤の分散処理に使用する分散機は特に限定されないが、好ましくは超音波分散機、機械的ホモジナイザーや圧力式ホモジナイザー等の加圧分散機、サンドグラインダー、ダイヤモンドファインミル、ビーズミル等の媒体型分散機が挙げられる。
着色剤としては、公知の顔料が使用可能で、例えば、カーボンブラック、アニリンブルー、カルコイルブルー、クロムイエロー、ウルトラマリンブルー、デュポンオイルレッド、キノリンイエロー、メチレンブルークロリド、銅フタロシアニン、マラカイトグリーンオキサレート、ランプブラック、ローズベンガル、C.I.ピグメント・レッド48:1,C.I.ピグメント・レッド122,C.I.ピグメント・レッド57:1,C.I.ピグメント・レッド184,C.I.ピグメント・イエロー97,C.I.ピグメント・イエロー12,C.I.ピグメント・イエロー17,C.I.ソルベント・イエロー162,C.I.ピグメント・イエロー180,C.I.ピグメント・イエロー185,C.I.ピグメント・ブルー15:1、C.I.ピグメント・ブルー15:3等を挙げることができる。
着色剤の使用量は、粒子中において樹脂成分100重量部に対する含有量が2〜30重量部、好ましくは5〜25重量部となるような量が好適である。
本発明の「表面にカルボキシル基を有する基材」は、界面重合法、乳化重合法、ソープフリー乳化重合法、非水分散重合法等の湿式重合法、気相法、コアセルベーション法、表面改質法等種々の方法で作成した高分子化合物を用いることができる。高分子化合物としては、スチレン系、(メタ)アクリル系、ポリエステル系、ベンゾグアナミン、メラミン、テフロン(登録商標)、シリコン、ポリエチレン、ポリプロピレン、或いはこれらの共重合化合物等を用いることができる。
本発明に係わる樹脂粒子の体積平均粒径は、0.1〜10μmで粒径分布の揃った樹脂粒子であることが好ましく、より好ましくは0.1〜5μmの範囲で粒径分布の揃った粒子である。また本発明に係わる樹脂粒子のガラス転移点(Tg)は特に制限されないが、好ましくは−10〜120℃の範囲である。また本発明に係わる樹脂粒子の分子量は特に制限されないが、好ましくは質量平均分子量で2000〜1000000である。
【0040】
次に、表面機能性部材の作製方法について説明する。
上述したような、各種表面にカルボキシル基を有する基材上に、アミノ基を有する機能性素材を、縮合剤の存在下で直接結合させて表面機能性部材を作製する。なお、反応工程において、水系連続相中で行う。
基材のカルボキシル基と、機能性素材の極性基とを結合させる反応は、公知の活性アミド化法である。なお、縮合剤として、下記式(2)中の、一般式(3)で表わされるトリアジン化合物を適用した反応については、Tetrahedron55(1999)13159−13170に記載されている。
【0041】
【化2】
【0042】
但し、R1、R2は、置換もしくは無置換のアルキル基を表す。
R1、R2で用いられる置換基として、置換もしくは無置換のアルキル基としては以下のものが挙げられる。
例えば、炭素数1〜18の直鎖、分岐鎖又は環状のアルキル基であり、これらのアルキル基は更に炭素数1〜18のアルコキシ基、フェニル基、又は炭素数1〜6の直鎖、分岐鎖又は環状のアルキル基で置換されたフェニル基を含有しても良い。具体的には、メチル基、エチル基、n−プロピル基、i−プロピル基、n−ブチル基、i−ブチル基、s−ブチル基、t−ブチル基、n−ヘキシル基、n−オクチル基、n−デシル基、n−ドデシル基、n−オクタデシル基、トリフルオロメチル基、2−シアノエチル基、メトキシメチル基、メトキシエチル基、ベンジル基、4−クロロベンジル基、シクロペンチル基、シクロヘキシル基などが挙げられる。これらの中で、メチル基、エチル基プロピル基など低級アルキル基は、水に対する溶解性が優れるため、好適に使用できる。一方、炭素数10〜18の直鎖アルキル基も好適に使用できる。これは、R1、R2に長鎖アルキル基を導入することにより、界面活性剤的性質により分散性の改良に寄与したものと考えている。
【0043】
上記式(2)は、カルボキシル基を有する化合物(4)と、アミノ基を有する化合物(5)とを、式(3)で表されるトリアジン化合物縮合剤下で、中間体(6)を経て、アミド化合物(7)と、副生成物ヒドロキシトリアジン化合物(8)が生成する反応である。
式(3)のトリアジン化合物は、水中でも安定であると記載されているが、通常は反応溶媒としてTHF(テトラヒドロフラン)が適用されている。
なお、本発明においては、上記活性アミド化反応を、水系連続相中で行うことに特徴を有している。ただし、Tetrahedron57(2001)1551−1558には、反応溶媒として、水、或いは水/メタノール(1:1)を使用する反応の記載がある。この場合、カルボキシル基を有する化合物は水溶性であるか、難水溶性の場合は反応溶媒として水/メタノール(1:1)を使用し、少なくともカルボキシル基を有する化合物は溶解した反応系で使用している。
それに対して、本発明の特徴は、上記の水系連続相中での活性アミド化反応を、完全に不溶なカルボキシル基を有する化合物を使用している点、すなわち、固体/液体間の不均一状態で行う点である。一般的に、固体/液体間の反応は反応試薬の衝突回数の問題で、極度に反応性が低下、あるいは反応しないことが知られている。特に、カルボキシル基を有する化合物に粒子を適用する場合、粒径が小さくなるほど(凝集力が増加し)、反応性が低下し、上記の水系連続相中での活性アミド化反応は、更に困難となる。
【0044】
本発明においては、表面にカルボキシル基を有する基材上に、アミノ基を有する機能性素材を、縮合剤の存在下、水系連続相中で直接結合させて得られる表面機能性部材であって、前記、水系連続相中に非イオン性界面活性剤を添加することにより、従来問題となっていた上記課題が解決されることを見いだした(請求項4)。
特に、非イオン性界面活性剤が優れている理由は、上記式(2)の反応で使用する酸性及び塩基性の試薬に対して、不活性であることに起因していると考えられる。実際、イオン性界面活性剤を使用すると、非常に反応性が悪いか、或いは全く反応しない場合がある。これは、一般式(2)の反応で使用する試薬とイオン性界面活性剤の間に何らかのイオン的相互作用があり、反応を阻害しているものと考えている。
非イオン性界面活性剤としては、前述の化合物が使用でき、必要に応じて2種以上混合して使用しても良い。これらの非イオン性界面活性剤中、HLB(Hydrophile-Lipophile Balance)が12〜18の非イオン性界面活性剤が好適である。また、非イオン性界面活性剤を2種以上使用する場合のHLBは、その加重平均になる。HLBが12以下では、親水性部位が不安定化し、HLBが18以上では、疎水性部位が不安定化する。従って、HLBが12〜18の非イオン性界面活性剤、更にHLBが12.5〜17の非イオン性界面活性剤が好適に使用できる。
【0045】
ここで、HLBは、以下に示すグリフィンの式より求められる。
HLB=(親水性部分の分子量)/(全体の分子量)×100÷5
本発明で使用される縮合剤として、上記式(2)中の、式(3)で表わされるトリアジン化合物以外に、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩等のカルボジイミド化合物、及びフェニルスルホン酸ビス(2−ニトロフェニルエステル)等のスルホン酸化合物は相間移動触媒とともに水溶液中で使用されるが、反応性、及び取り扱い容易性により、式(3)で表わされるトリアジン化合物を好適に使用することができる。
これら表面機能性部材を作製する場合、表面にカルボキシル基を有する基材、アミノ基を有する機能性素材、及び必要に応じて界面活性剤等の添加剤を水系連続相中に溶解、或いは分散させた機能性部材組成物を調整し、縮合剤を含む水を添加することにより、所望の機能性素材が直接結合した基材を得ることができる。表面にカルボキシル基を有する基材、アミノ基を有する機能性素材、及び必要に応じて界面活性剤等の添加剤を水系連続相中に分散する場合は、前述の分散機を使用することができる。また、カルボキシル基を有する基材の分散安定性を改善する目的で、水系連続相を中性〜弱アルカリ性(pH9程度)とすることが好ましく、更に、アミノ基を有する機能性素材量をカボキシル基の当量より多く使用し水系連続相を弱アルカリ性とすることが好ましい。この場合のアミノ基を有する機能性素材量は、カボキシル基の1.2〜2.0倍当量が好適である。
【0046】
本発明のアミノ基を有する機能性素材中、アミノ基としては、カルボキシル基と反応してアミド結合を形成できるものであれば特に制限はないが、立体障害が大きい場合、反応性の点で二級アミノ基より一級アミノ基の方が好ましい。機能性素材としては、特に制限はなく、発生する機能に応じて、従来公知の原子、置換基、及び官能基等が選ばれる。例えば、表面硬度に関しては架橋構造が形成される官能基、表面滑性に関してはフッ素原子、或いはジメチルシロキサン骨格を有する置換基、及び、分散安定性、表面の帯電性、導電性、着色剤等の機能性基を挙げることができる。
反応は、縮合剤を含む水溶液を添加することにより始まり、通常、0℃から室温(25℃)で6時間程度終わる。ただし、電子的、立体的に反応性が悪い場合は、昇温、或いは長時間の反応が必要となる。昇温する場合は、安定性の問題で70℃程度までとした方がよい。また、化学的安定性のため、反応系内をアルゴンガス、窒素ガス等の不活性ガス雰囲気下で行うこともできる。
【0047】
本発明の表面機能性部材は、非常に簡便な方法で、基材と機能性素材を直接アミド結合して得られるため、機能性素材を高安定に、しかも高耐久性・高持続性を有する。しかも、本発明の製造方法は、アミノ基を有する多くの機能性素材に広範囲に応用可能であり、基材上に非常に均一に単分子層を形成可能な優れた方法である。また、本発明で得られる粒子は、平均粒子径が0.1〜10μm、の範囲内であり、CV値は30%以下で非常に単分散性の高いほぼ真球状粒子である。また本発明の製造方法によれば、非常に容易に、しかも平均粒子径、相対標準偏差(CV値)等の粒子特性を損なうことなく着色粒子を提供することができる。
【0048】
本発明方法を利用して作製された表面機能性部材を構成する着色粒子に関しては、特に、帯電性着色粒子への応用が考えられ、PLD(ペーパーライクディスプレー)、電子写真用液体現像剤、静電インクジェットプリンター用インク等に用いられる。
具体的には、本発明の着色微粒子のシイルコーンオイル、高純度石油等の絶縁性液体分散液内で、電気的に帯電して、泳動させる装置(電気泳動素子)に応用できる。なお、前記絶縁性液体の具体的な市販品としては、前記高純度石油では、例えば、アイソパーG,H,L,M(エクソン化学社製)やノルパー12(エクソン化学社製)等が挙げられ、前記シリコーンオイルでは、例えばSH−200シリーズ(東レ・ダウコーニング社製)、KF−96シリーズ(信越化学社製)、L−45シリーズ(日本ユニカー社製)、及びAKシリーズ(旭化成ワッカーシリコーン社製)等が挙げられる。
このような帯電性着色粒子は、対向電極セル内の電気絶縁性液体中での帯電性、電気泳動特性が極めて重要である。その帯電性着色粒子に求められる電気泳動特性とは、所定の電解(又は電場)下にある粒子表面が、電気的に応答し易い帯電性を呈する着色粒子である。しかも、帯電性着色粒子を所定の電解下にスムーズに泳動させるためには、その粒子が泳動セル中で凝集して粗大化粒子になり難い着色粒子であることも重要である。
これらの電気泳動特性は、主に、着色粒子の表面状態、及び表面の置換基に左右される。
すなわち、上記式(2)中の機能性素材(5)を構成するB1、B2で表される置換基の種類によって左右されるものであり、その帯電性としては、それぞれ(−)帯電性と(+)帯電性を示す場合があり、少なくとも二種以上の複数種の置換基を使用するときには、その(−)及び(+)帯電性を示すもののうち、同種帯電性の置換基同士を複数組み合わせて使用することが好ましい。
【0049】
(−)帯電性の傾向にある置換基(B1、B2)としては、例えば、フェニル基、ベンジル基、2−フェニルエチル基等の芳香族基、2−クロロエチル基等の塩素原子含有アルキル基、p−クロロベンジル基などの塩素原子含有芳香族基、2−シアノエチル基等のシアノ基含有アルキル基、2−ヒドロキシエチル基、2−ヒドロキシプロピル基等のヒドロキシ基含有アルキル基、2−フルオロエチル基、2,2,2−トリフルオロエチル基等のフッ素原子含有アルキル基、パーフルオロベンゼン等のフッ素原子含有芳香族基等が挙げられる。
一方、(+)帯電性の傾向がある置換基(B1、B2)として、例えば、2−ジメチルアミノエチル基、2−ジブチルアミノエチル基、2−(1−ピペリジノ)エチル基、2−プロピリアミノエチル基、2−フェニルアミノエチル基等のアミノ基含有アルキル基、4−ジエチルアミノフェニル基、4−ジメチルアミノベンジル基等のアミノ基含有芳香族基等が挙げられる。
【0050】
また、このように泳動セル中で優れた非凝集性を有しているためには、その形状が定形で、その定形粒子が球状粒子であることが好ましく、更には、その粒径分布がより狭いことが望ましい。
更に、着色粒子の表面状態、及び表面の置換基にも左右されるものであり、アミノ基を有する機能性素材である上記式(5)中のB1、B2で表わされる置換基の種類によって左右される。
具体的に、非凝集性粒子を設計するためには、絶縁性液体がシリコーンオイルの場合は置換基(B1、B2)がシロキサン骨格を有する置換基としたり、高純度石油の場合は置換基(B1、B2)が長鎖アルキル基としたりする等、絶縁性液体と類似の骨格を有する置換値を導入することが好ましい。これは着色粒子と絶縁性液体間の相溶性が増加し、分散、及び分散安定性が増加するためである。
【0051】
絶縁性液体がシリコーンオイルの場合は、上記式(2)中の機能性素材(5)として一般的なアミノ変性シリコーンを使用することができるが、この中でも下記一般式(I)及び一般式(II)で表されるシリコーン化合物より選ばれた1種又は2種以上のシリコーン化合物が、特に好ましい。
H2N−B11−X ・・・(I)
H2N−B12−NH−B13−X ・・・(II)
(式中、B11,B12及びB13は、炭素数2から6のアルキレン基、Xはシロキサン骨格を示す。)
【0052】
ここで、式(I),(II)中、B11,B12及びB13である炭素数2から6のアルキレン基としては、直鎖或いは分岐のアルキレン基で、例えばエチレン、プロピレン、1−メチルエチレン、ブチレン、1,2−ジメチルエチレン、ペンチレン、へキシレン等が挙げられ、Xのシロキサン骨格は、例えば下記一般式(III)で示す片末端型、或いは下記一般式(IV)で示す側鎖型を挙げることができる。なお、式中l、m、nは、0以上、30以下の整数を表す。
【0053】
【化3】
【0054】
【化4】
【0055】
本発明方法により得られる表面機能性部材を適用した帯電性着色粒子を、PLD、電子写真画像形成装置の静電着色トナー、静電インクジェットプリンター用インク等として使用する場合に、所望の帯電特性、電気泳動性を発揮させるためには、その粒子形状も重要である。この点については、少なくとも定形粒子であって、かつ粒子表面により均質に帯電させるために略球状であることが好適である。また、平均粒径が0.1〜10μmの範囲であることが、上記用途として好適である。
【0056】
また、電気泳動及び転写時に、印可電解による粒子間のクーロン反発により粒子を凝集粗大化させ難くさせる効果が期待できることから、平均粒径のバラツキが著しく低いことが好ましい。
この粒子の均斉度を相対標準偏差(CV値)で表すこととし、これが30%以下の略単分散粒子であると、帯電性、電気泳動特性に優れた帯電性着色粒子を提供できることが確かめられた。
また、本発明の表面機能性部材を利用した帯電性着色粒子には、必要に応じて公知の添加剤を加えてもよい。例えば、分散剤、電荷制御剤、熱安定剤、防腐剤、表面張力調整剤、酸化防止剤、近赤外線吸収剤、紫外線吸収剤、蛍光剤、蛍光増白剤等が挙げられる。
【実施例】
【0057】
以下、本発明について具体的な実施例を挙げて説明するが、本発明はこれらの例に限定されるものではない。
【0058】
先ず、表面機能性部材の評価に用いる方法と装置を下記に示す。
<粒径、及び相対標準偏差(CV値)>
粒径アナライザー(FPAR-1000:大塚電子製)−試料−1.0重量%水溶液で測定した。
<赤外線吸収スペクトル>
顕微FT-IR(サーモエレクトロン製)、顕微透過法で測定した。
<XPS(X-ray photoelectron spectroscopy)>
・測定装置:AXIS−ULTRA(Kratos社製)
X線源 :Alモノクロメータ使用
X線パワー :225W
測定領域 :900μm×600μm
パスエネルギー:wide scan(ワイドスキャン)=80eV
:narrow scan(ナロースキャン)=20eV<SEM観察>日本電子製 電界放射型走査電子顕微鏡 JSM-7401F
―観察条件―導電処理:Osコート、加速電圧:5.0kV、検出器:SEI
<ζ(Zeta)電位測定>
・測定装置:ESA9800(Matec Applied Science社製)
【0059】
次に、下記の〔合成例1〜6〕のそれぞれにおいて、表面にカルボキシル基を有する粒子を作製する。
【0060】
〔合成例1〕
温度計と窒素導入管とを装着した500mlの4つ口フラスコに、カルボキシル基を有する単量体であるメタクリル酸1.30重量部、通常単量体であるメチルメタクリレート28.70重量部、リン酸二水素カリウム0.147重量部、1N−水酸化カリウム水溶液16.17重量部、及びイオン交換水254重量部を投入し、強攪拌下で30分間、混合した。
次に、窒素気流下で攪拌しながら65℃に昇温した。この反応液を65℃の温度に保持しながら、過硫酸ナトリウム0.163重量部、及びイオン交換水20重量部より成る重合開始剤水溶液を一時間要して滴下し、更に6時間反応を行い、その後、放冷し、濾過し、水洗処理を3回行い、乾燥処理を行うことにより、表面にカルボキシル基を有する粒子(A)が25.88重量部得られた。
この粒子(A)の粒度分布測定を行ったところ、平均粒径:0.64μmであり、その相対標準偏差(CV値)は22.0%であった。赤外線吸収スペクトルを図1に示す。
また、XPSスペクトルを図2に示す。
【0061】
〔合成例2〕
温度計と窒素導入管とを装着した500mlの4つ口フラスコに、カルボキシル基を有する単量体であるアクリル酸2.19重量部、通常単量体であるメチルメタクリレート27.38重量部、リン酸二水素カリウム0.147重量部、1N−水酸化カリウム水溶液16.17重量部、及びイオン交換水254重量部を投入し、強攪拌下で30分間、混合した。
次に、窒素気流下で攪拌を行いながら65℃に昇温した。この反応液を65℃の温度に保持しながら、過硫酸ナトリウム0.163重量部、及びイオン交換水20重量部より成る重合開始剤水溶液を一時間要して滴下し、更に6時間反応を行い、その後、放冷し、濾過し、水洗処理を3回行い、乾燥処理を行うことにより、表面にカルボキシル基を有する粒子(B)が24.20重量部得られた。
この粒子の粒度分布測定を行ったところ、平均粒径:0.39μmであり、その相対標準偏差(CV値)は22.5%であった。
【0062】
〔合成例3〕
温度計と窒素導入管とを装着した500mlの4つ口フラスコに、カルボキシル基を有する単量体であるメタクリル酸2.62重量部、通常単量体であるメチルメタクリレート27.38重量部、リン酸二水素カリウム0.147重量部、1N−水酸化カリウム水溶液31.47重量部、及びイオン交換水238重量部を投入し、強攪拌下で30分間、混合した。
次に、窒素気流下で攪拌を行いながら、68℃に昇温した。この反応液を68℃の温度に保持しながら、水溶性重合開始剤(和光純薬社製、商品名「VA−057」=2,2’−アゾビス[N−(2−カルボキシエチル)−2−メチルプロピオンアミジン])0.504重量部及びイオン交換水30重量部より成る重合開始剤水溶液を一時間要して滴下し、更に6時間反応を行い、その後、放冷し、濾過し、水洗処理を3回行い、乾燥処理を行うことにより、表面にカルボキシル基を有する粒子(C)が23.60重量部得られた。
この粒子の粒度分布測定を行ったところ、平均粒径:0.59μmであり、その相対標準偏差(CV値)は23.6%であった。
【0063】
〔合成例4〕
温度計と窒素導入管とを装着した500mlの4つ口フラスコに、カルボキシル基を有する単量体であるメタクリル酸2.62重量部、通常単量体であるメチルメタクリレート27.38重量部、リン酸二水素カリウム0.147重量部、1N−水酸化カリウム水溶液31.47重量部、及びイオン交換水225重量部を投入し、強攪拌下でメタノール13.50重量部を10分間要して滴下し、更に30分間、強攪拌下混合した。
次に、窒素気流下で攪拌を行いながら、68℃に昇温した。この反応液を68℃の温度に保持しながら、水溶性重合開始剤(和光純薬社製、商品名「VA−057」=2,2’−アゾビス[N−(2−カルボキシエチル)−2−メチルプロピオンアミジン])0.504重量部、及びイオン交換水30重量部より成る重合開始剤水溶液を一時間要して滴下し、更に6時間反応を行い、その後、放冷し、濾過し、水洗処理を3回行い、乾燥処理を行うことにより、表面にカルボキシル基を有する粒子(D)が23.52重量部得られた。
この粒子の粒度分布測定を行ったところ、平均粒径:0.82μmであり、その相対標準偏差(CV値)は26.5%であった。
【0064】
〔合成例5〕
カーボンブラック(三菱化学社製、商品名「MA−100」)16重量部、ドデシルベンゼンスルホン酸ナトリウム0.64重量部、及びイオン交換水64重量部をメディア式湿式粉砕器を用い、20重量%のカーボンブラック分散液を得た。また、水酸化マグネシウム(和光純薬製、0.07μm)20重量部、及びイオン交換水180重量部をメディア式湿式粉砕器を用い、10重量%の水酸化マグネシウム分散液(無機分散安定剤)を得た。
他方、温度計と窒素導入管とを装着した500mlの4つ口フラスコに、カルボキシル基を有する単量体であるメタクリル酸2.35重量部、通常単量体であるメチルメタクリレート24.65重量部、リン酸二水素カリウム0.147重量部、1N−水酸化カリウム水溶液31.47重量部、及びイオン交換水200重量部を投入し、強攪拌下で10重量%の水酸化マグネシウム分散液18.5重量部を加え、更に20重量%のカーボンブラック分散液15重量部を10分間要して滴下し、更に30分間、強攪拌下混合した。
次に、窒素気流下で攪拌を行いながら、68℃に昇温した。この反応液を68℃の温度に保持しながら、水溶性重合開始剤(和光純薬社製、商品名「VA−057」=2,2’−アゾビス[N−(2−カルボキシエチル)−2−メチルプロピオンアミジン])0.453重量部及びイオン交換水30重量部より成る重合開始剤水溶液を一時間要して滴下し、更に6時間反応を行い、その後、放冷し、濾過し、水洗処理を3回行い、乾燥処理を行うことにより、表面にカルボキシル基を有する粒子(E)が24.32重量部得られた。
この粒子の粒度分布測定を行ったところ、平均粒径:0.77μmであり、その相対標準偏差(CV値)は25.2%であった。
【0065】
〔合成例6〕
温度計と窒素導入管とを装着した500mlの4つ口フラスコに、カルボキシル基を有する単量体であるメタクリル酸4.25重量部、通常単量体であるメチルメタクリレート19.75重量部、リン酸二水素カリウム0.147重量部、1N−水酸化カリウム水溶液50.41重量部、ポリメタクリル酸エステルマクロマー(東亞合成化学工業社製、商品名「AA6」)0.94重量部、及びイオン交換水220重量部を投入し、強攪拌下で20重量%のカーボンブラック分散液30重量部を10分間要して滴下し、更に30分間、強攪拌下混合した。
次に、窒素気流下で攪拌を行いながら、68℃に昇温した。この反応液を68℃の温度に保持しながら、水溶性重合開始剤(和光純薬社製、商品名「VA−057」=2,2’−アゾビス[N−(2−カルボキシエチル)−2−メチルプロピオンアミジン])0.409重量部及びイオン交換水30重量部より成る重合開始剤水溶液を一時間要して滴下し、更に6時間反応を行い、その後、放冷し、濾過し、水洗処理を3回行い、乾燥処理を行うことにより、表面にカルボキシル基を有する粒子(F)が25.61重量部得られた。
この粒子の粒度分布測定を行ったところ、平均粒径:0.93μmであり、その相対標準偏差(CV値)は28.7%であった。
【0066】
〔合成例7〕
フタロシアニンブルー(C.I.ピグメントブルー15:3)20重量部、ドデシルベンゼンスルホン酸ナトリウム0.32重量部、及びイオン交換水80重量部をメディア式湿式粉砕器を用い、20重量%のフタロシアニンブルー分散液を得た。
他方、リン酸二水素カリウム0.147重量部、1N−水酸化カリウム水溶液20.59重量部、及びイオン交換水200重量部をホモジナイザー(IKA社製:ウルトラタックスT25)で高速攪拌下(8000rpm)で、10重量%のポリビニルアルコール(和光純薬製、重合度約500)水溶液1.2重量部を加え、更にカルボキシル基を有する単量体であるアクリル酸1.41重量部、通常単量体であるメチルメタクリレート7.03重量部、及びベンジルメタクリレート18.56重量部を投入、更に前記20重量%のフタロシアニンブルー分散液15重量部を、10分間要して滴下し、更に30分間、高速攪拌下混合し反応液を得た。
続いて、予め用意しておいた温度計と窒素導入管とを装着した500mlの4つ口セパラブルフラスコに上記反応液を投入し、更に、窒素気流下で攪拌を行いながら、70℃に昇温した。この反応液を70℃の温度に保持しながら、水溶性重合開始剤(和光純薬社製、商品名「V−501」=2,2’−アゾビス[4−シアノ吉草酸])0.219重量部、1N−水酸化カルシウム水溶液1.56重量部、及びイオン交換水20重量部より成る重合開始剤水溶液を一時間要して滴下し、更に6時間反応を行い、その後、放冷し、濾過し、水洗処理を3回行い、乾燥処理を行うことにより、表面にカルボキシル基を有する粒子(G)が25.74重量部得られた。
この粒子の粒度分布測定を行ったところ、平均粒径:0.68μmであり、その相対標準偏差(CV値)は22.9%であった。
【0067】
次に、上記〔合成例1〜7〕で得られた表面にカルボキシル基を有する粒子に対してアミド化反応を行った。
【0068】
〔実施例1〕
<非イオン界面活性剤のHLB=14.31>
ノニオン系乳化剤EA−177(第一工業製薬(株)製)0.143重量部、XL−60(第一工業製薬(株)製)0.143重量部、及びイオン交換水285重量部をホモジナイザー(IKA社製:ウルトラタックスT25)で高速攪拌下(8000rpm)、シクロヘキシルアミン1.50重量部を、10分間かけて滴下した。
次に、上記〔合成例1〕で作製した表面にカルボキシル基を有する粒子(A)15.00重量部を加え、更に20分間、高速攪拌し、粒子乳化液を得た。
続いて、予め用意しておいた、温度計と窒素導入管とを装着した500mlの4つ口フラスコに、上記粒子乳化液を投入し、更に、上記一般式(1)に示したトリアジン化合物(但し、R1=R2=CH3)4.18重量部を、イオン交換水10重量部の溶液を加え、室温下、窒素気流下で攪拌を6時間行った。
反応終了後、1Nの硫酸を用いて酸性にし、濾過、水洗を3回行った。これを乾燥処理後、表面機能性部材が14.30重量部(収率91.6%)が得られた。
この表面機能性部材粒子の粒度分布測定を行ったところ、平均粒径:0.75μmであり、その相対標準偏差(CV値)は27.0%であった。
この例において得られた表面機能性部材粒子の赤外線吸収スペクトル図を図3に示す。
また、XPSスペクトル図を図4に示す。
図2と図4を比較すると、実施例1により作製された表面機能性部材粒子(図4)に、アミド結合に基づくN1sピークが現れていることが判り、表面機能化の実施が確認された。
更に、XPS(400eV付近のナロースキャン)による測定を行い、合成例1の粒子(A)と実施例1の表面機能性部材粒子を併せて図5に示した。
また、実施例1の表面機能性部材粒子をSEM観察(×10,000)したときの状態を図6に示す。これによると凝集が効果的に回避できており均斉度に優れていることが確かめられた。
【0069】
〔実施例2〕
<非イオン界面活性剤のHLB=17.00>
ノニオン系乳化剤NL-250(第一工業製薬(株)製)0.143重量部、XL−60(第一工業製薬(株)製)0.143重量部、及びイオン交換水285重量部をホモジナイザー(IKA社製:ウルトラタックスT25)で高速攪拌下(8000rpm)、アニリン2.87重量部を、10分間かけて滴下した。
次に、上記〔合成例2〕で作製した表面にカルボキシル基を有する粒子(B)15.00重量部を加え、更に20分間、高速攪拌し、粒子乳化液を得た。
続いて予め用意しておいた、温度計と窒素導入管とを装着した500mlの4つ口フラスコに、上記において得た粒子乳化液を投入し、更に、一般式(1)に示したトリアジン化合物(但し、R1=R2=CH3)8.53重量部を、イオン交換水10重量部の溶液を加え、室温下、窒素気流下で攪拌を6時間行った。
反応終了後、1Nの硫酸を用いて酸性にし、濾過、水洗を3回行った。これを乾燥処理後、表面機能性部材が13.51重量部(収率83.6%)が得られた。
この表面機能性部材粒子の、粒度分布測定を行ったところ、平均粒径:0.45μmであり、その相対標準偏差(CV値)は25.5%であった。
この例において得られた表面機能性部材粒子の赤外線吸収スペクトル図を図7に示す。
【0070】
〔実施例3〕
<非イオン界面活性剤のHLB=13.78>
ノニオン系乳化剤EA−177(第一工業製薬(株)製)0.143重量部、XL−60(第一工業製薬(株)製)0.285重量部、及びイオン交換水285重量部をホモジナイザー(IKA社製:ウルトラタックスT25)で高速攪拌下(8000rpm)、2−フェネチルアミン3.68重量部を、10分間かけて滴下した。
次に、上記〔合成例3〕で作製した表面にカルボキシル基を有する粒子(C)15.00重量部を加え、更に20分間高速攪拌し、粒子乳化液を得た。
続いて、予め用意しておいた、温度計と窒素導入管とを装着した500mlの4つ口フラスコに、上記において得た粒子乳化液を投入し、更に一般式(1)に示したトリアジン化合物(但し、R1=R2=CH3)8.41重量部を、イオン交換水10重量部の溶液を加え、室温下、窒素気流下で攪拌を6時間行った。
反応終了後、1Nの硫酸を用いて酸性にし、濾過、水洗を3回行った。これを乾燥処理後、表面機能性部材が13.79重量部(収率82.8%)が得られた。
この表面機能性部材粒子の、粒度分布測定を行ったところ、平均粒径:0.65μmであり、その相対標準偏差(CV値)は21.0%であった。
この例において得られた表面機能性部材粒子の赤外線吸収スペクトル図を図8に示す。
【0071】
〔実施例4〕
<非イオン界面活性剤のHLB=14.31>
ノニオン系乳化剤EA−177(第一工業製薬(株)製)0.143重量部、XL−60(第一工業製薬(株)製)0.143重量部、及びイオン交換水285重量部をホモジナイザー(IKA社製:ウルトラタックスT25)で高速攪拌下(8000rpm)、ベンジルアミン3.26重量部を、10分間かけて滴下した。
次に、上記〔合成例4〕で作製した表面にカルボキシル基を有する粒子(D)15.00重量部を加え、更に20分間、高速攪拌し、粒子乳化液を得た。
続いて、予め用意しておいた、温度計と窒素導入管とを装着した500mlの4つ口フラスコに、上記において得た粒子乳化液を投入し、更に、上記一般式(1)に示したトリアジン化合物(但し、R1=R2=CH3)8.41重量部を、イオン交換水10重量部の溶液を加え、室温下、窒素気流下で攪拌を6時間行った。
反応終了後、1Nの硫酸を用いて酸性にし、濾過、水洗を3回行った。これを乾燥処理後、表面機能性部材が14.89重量部(収率90.5%)が得られた。
この表面機能性部材粒子の粒度分布測定を行ったところ、平均粒径:0.90μmであり、その相対標準偏差(CV値)は22.9%であった。
この例において得られた表面機能性部材粒子の赤外線吸収スペクトル図を図9に示す。
【0072】
〔実施例5〕
<非イオン界面活性剤のHLB=14.31>
ノニオン系乳化剤EA−177(第一工業製薬(株)製)0.143重量部、XL−60(第一工業製薬(株)製)0.143重量部、及びイオン交換水285重量部をホモジナイザー(IKA社製:ウルトラタックスT25)で高速攪拌下(8000rpm)、1−(2−アミノエチル)ピペリジン3.90重量部を10分間かけて滴下した。
次に、上記〔合成例5〕で作製した表面にカルボキシル基を有する粒子(E)15.00重量部を加え、更に20分間、高速攪拌し、粒子乳化液を得た。
続いて予め用意しておいた、温度計と窒素導入管とを装着した500mlの4つ口フラスコに、上記において得た粒子乳化液を投入し、更に、上記一般式(1)に示したトリアジン化合物(但し、R1=R2=CH3)8.41重量部を、イオン交換水10重量部の溶液を加え、室温下、窒素気流下で攪拌を6時間行った。
反応終了後、1Nの硫酸を用いて酸性にし、濾過、水洗を3回行った。これを乾燥処理後、表面機能性部材が14.77重量部(収率91.0%)が得られた。
この表面機能性部材粒子の粒度分布測定を行ったところ、平均粒径:0.89μmであり、その相対標準偏差(CV値)は20.7%であった。
【0073】
〔実施例6〕
<非イオン界面活性剤のHLB=14.31>
ノニオン系乳化剤EA−177(第一工業製薬(株)製)0.143重量部、XL−60(第一工業製薬(株)製)0.143重量部、及びイオン交換水285重量部をホモジナイザー(IKA社製:ウルトラタックスT25)で高速攪拌下(8000rpm)、N,N−ジ−n−ブチルエチレンジアミン10.62重量部を、10分間かけて滴下した。
次に、上記〔合成例5〕で作製した表面にカルボキシル基を有する粒子(E)15.00重量部を加え、更に20分間、高速攪拌し、粒子乳化液を得た。
続いて、予め用意しておいた、温度計と窒素導入管とを装着した500mlの4つ口フラスコに、上記において得た粒子乳化液を投入し、更に、上記一般式(1)に示したトリアジン化合物(但し、R1=R2=CH3)17.06重量部を、イオン交換水10重量部の溶液を加え、室温下、窒素気流下で攪拌を6時間行った。
反応終了後、1Nの硫酸を用いて酸性にし、濾過、水洗を3回行った。これを乾燥処理後、表面機能性部材が15.82重量部(収率90.4%)が得られた。
この表面機能性部材粒子の粒度分布測定を行ったところ、平均粒径:0.98μmであり、その相対標準偏差(CV値)は25.3%であった。
【0074】
〔実施例7〕
<非イオン界面活性剤のHLB=13.78>
ノニオン系乳化剤EA−177(第一工業製薬(株)製)0.146重量部、XL−60(第一工業製薬(株)製)0.291重量部、及びイオン交換水291重量部をホモジナイザー(IKA社製:ウルトラタックスT25)で高速攪拌下(8000rpm)、N,N−ジメチルエチレンジアミン1.63重量部を、10分間かけて滴下した。
次に、上記〔合成例7〕で作製した表面にカルボキシル基を有する粒子(G)9.00重量部を加え、更に20分間高速攪拌し、粒子乳化液を得た。
続いて、予め用意しておいた、温度計と窒素導入管とを装着した500mlの4つ口フラスコに、上記において得た粒子乳化液を投入し、更に一般式(1)に示したトリアジン化合物(但し、R1=R2=CH3)5.12重量部を、イオン交換水20重量部の溶液を加え、室温下、窒素気流下で攪拌を6時間行った。
反応終了後、1Nの硫酸を用いて酸性にし、濾過、水洗を3回行った。これを乾燥処理後、表面機能性部材が8.76重量部(収率90.8%)が得られた。
この表面機能性部材粒子の、粒度分布測定を行ったところ、平均粒径:0.81μmであり、その相対標準偏差(CV値)は21.0%であった。
【0075】
〔実施例8〕
<非イオン界面活性剤のHLB=13.78>
ノニオン系乳化剤EA−177(第一工業製薬(株)製)0.147重量部、XL−60(第一工業製薬(株)製)0.294重量部、及びイオン交換水284重量部をホモジナイザー(IKA社製:ウルトラタックスT25)で高速攪拌下(8000rpm)、アミノ基を有するシリコーン化合物として3−アミノプロピルメチル−ビス(トリメチルシロキシ)シラン(アズマックス社製)3.45重量部を、10分間かけて滴下した。
次に、上記〔合成例7〕で作製した表面にカルボキシル基を有する粒子(G)6.00重量部を加え、更に20分間高速攪拌し、粒子乳化液を得た。
続いて、予め用意しておいた、温度計と窒素導入管とを装着した500mlの4つ口フラスコに、上記において得た粒子乳化液を投入し、更に一般式(1)に示したトリアジン化合物(但し、R1=R2=CH3)3.41重量部を、イオン交換水20重量部の溶液を加え、室温下、窒素気流下で攪拌を6時間行った。
反応終了後、1Nの硫酸を用いて酸性にし、濾過、水洗を3回行った。これを乾燥処理後、表面機能性部材が13.79重量部(収率82.8%)が得られた。
この表面機能性部材粒子の、粒度分布測定を行ったところ、平均粒径:0.65μmであり、その相対標準偏差(CV値)は19.4%であった。
【0076】
〔比較例〕
<シード重合:非イオン界面活性剤のHLB=14.31>
ノニオン系乳化剤EA−177(第一工業製薬(株)製)0.143重量部、XL−60(第一工業製薬(株)製)0.143重量部、及びイオン交換水276重量部を強攪拌下、メチルメタクリレート9.60重量部を10分間かけて滴下した。
次に、上記〔合成例1〕で作製した粒子(A)14.40重量部を加え、更に20分間、高速攪拌し、粒子乳化液を得た。
続いて、予め用意しておいた、温度計と窒素導入管とを装着した500mlの4つ口フラスコに、上記において得た粒子乳化液を投入し、更に、水溶性重合開始剤(和光純薬社製、商品名「VA−057」=2,2’−アゾビス[N−(2−カルボキシエチル)−2−メチルプロピオンアミジン])0.487重量部のイオン交換水30重量部の水溶液を加え、窒素気流下で、静かに攪拌しながら室温より40℃まで0.5℃/分の速度で昇温し、更に40℃で2時間吸収・膨潤し、その後、68℃まで0.2℃/分の速度で昇温し、更に68℃で4時間反応した。
反応終了後、1Nの硫酸を用いて酸性にし、濾過、水洗を3回行った。
これを乾燥処理後、シード重合による比較例粒子が19.32重量部(収率80.5%)が得られた。
この比較例粒子の、粒度分布測定を行ったところ、平均粒径:1.03μmであり、その相対標準偏差(CV値)は45.3%であった。
この例において得られた粒子のSEM観察(×10,000)を図10に示す。
図6の実施例1で得られた表面機能性部材粒子と、図10の比較例で得られたシード重合粒子とを比較する明らかなように、シード重合粒子は微小粒子、及び粒子間の合一と思われる凝集物が観察された。
その結果、相対標準偏差(CV値)はシード粒子(合成例1で作製した粒子(A))と比較して大幅に増加した。
【0077】
上述したように、本発明方法によれば、相対標準偏差(CV値)を損なうことなく、非常に容易な工程で、単分散性の高い、略真球状の表面機能性部材粒子が作製できた。
【0078】
〔応用例〕
実施例4、実施例6及び実施例8で得られた表面機能性部材を絶縁性液体であるシリコーンオイルSH−200(東レ・ダウコーニング社製)に2重量%の濃度に分散し、ζ電位を測定した。次に、図11に示す装置により、電気泳動を確認した。
【0079】
ここで、図11の電気泳動装置について説明する。この装置は、ITO電極9c間の表面機能性部材の分散液9gの電気泳動の評価を、高速度カメラ撮影装置9を用いて行うものである。すなわち、まずガラス基板9d上にITO電極9cを100μmの間隔をあけて厚さ100nm蒸着し、その上に25μm厚のガラス板9hを100μm間隔でITO電極9c上に接着し液溜を設ける。次に、液溜に上記表面機能性部材の分散液9gを注入し、ITO電極9c間に電源9eから1000Vの電圧を印加した時に液滴が泳動される様子を上方の高速度カメラ9aで撮影してモニター9bで観察し、電気泳動の評価を行った。
【0080】
〔応用比較例〕
応用比較例として、合成例1で得られた表面にカルボキシル基を有する粒子(A)を、上記応用例と同じ条件で分散し、ζ電位の測定及び電気泳動の確認を行った。
以上の応用例、応用比較例の結果を併せて、表1に示す。
【0081】
【表1】
【図面の簡単な説明】
【0082】
【図1】合成例1で得られた微粒子(A)の赤外線吸収スペクトル図を示す。
【図2】合成例1で得られた微粒子(A)のXPSスペクトル図を示す。
【図3】実施例1で得られた表面機能性部材の赤外線吸収スペクトル図を示す。
【図4】実施例1で得られた表面機能性部材のXPSスペクトル図を示す。
【図5】合成例1の粒子(A)、及び実施例1の表面機能性部材微粒子のXPSスペクトル図を示す。
【図6】実施例1で得られた表面機能性部材粒子のSEM観察状態図を示す。
【図7】実施例2で得られた表面機能性部材の赤外線吸収スペクトル図を示す。
【図8】実施例3で得られた表面機能性部材の赤外線吸収スペクトル図を示す。
【図9】実施例4で得られた表面機能性部材の赤外線吸収スペクトル図を示す。
【図10】比較例で得られた表面機能性部材粒子のSEM観察状態図を示す。
【図11】電気泳動装置の構成図を示す。
【符号の説明】
【0083】
9 高速度カメラ撮影装置
9a 高速度カメラ
9b モニター
9c ITO電極
9d ガラス基板
9e 電源
9f スイッチ
9g 表面機能性部材の分散液
9h ガラス板
【特許請求の範囲】
【請求項1】
表面にカルボキシル基を有する基材上に、アミノ基を有する機能性素材を、縮合剤の存在下、直接結合させて得られる表面機能性部材の製造方法であって、
水系連続相中で反応させることを特徴とする表面機能性部材の製造方法。
【請求項2】
前記表面にカルボキシル基を有する基材が、乳化重合、懸濁重合、シード重合、あるいはソープフリー重合のいずれかの重合方式により得られた粒子であり、かつ平均粒子径が0.1〜10μmであることを特徴とする請求項1に記載の表面機能性部材の製造方法。
【請求項3】
前記縮合剤が、下記一般式(1)で表わされるトリアジン化合物であることを特徴とする請求項1又は2に記載の表面機能性部材の製造方法。
【化1】
但し、R1、R2は、置換もしくは無置換のアルキル基を表す。
【請求項4】
表面にカルボキシル基を有する基材上に、アミノ基を有する機能性素材を、縮合剤の存在下、直接結合させて得られる表面機能性部材の製造方法であって、
水系連続相中に、更に非イオン性界面活性剤を添加する工程を有することを特徴とする請求項1乃至3のいずれか一項に記載の表面機能性部材の製造方法。
【請求項5】
前記表面にカルボキシル基を有する基材上に、アミノ基を有する機能性素材を、縮合剤の存在下、水を主体とする連続相中で直接結合させて得られる表面機能性部材の製造方法であって、
前記、水系連続相中に非イオン性界面活性剤を添加することを特徴とする表面機能性部材の製造方法。
【請求項6】
前記アミノ基を有する機能性素材が、シロキサン骨格を有する置換基を含有する機能性素材であることを特徴とする請求項1乃至5のいずれか一項に記載の表面機能性部材の製造方法。
【請求項7】
前記アミノ基を有する機能性素材が、下記一般式(I)及び一般式(II)で表されるシリコーン化合物より選ばれる少なくとも一種のシリコーン化合物であることを特徴とする請求項1乃至5のいずれか一項に記載の表面機能性部材の製造方法。
H2N−B11−X ・・・(I)
H2N−B12−NH−B13−X ・・・(II)
(式中、B11,B12及びB13は、炭素数2から6のアルキレン基、Xはシロキサン骨格を示す。)
【請求項8】
表面にカルボキシル基を有する基材上に、アミノ基を有する機能性素材を、縮合剤の存在下で直接結合させて得られる表面機能性部材であって、
前記結合の反応は、水系連続相中で行われたものであることを特徴とする表面機能性部材。
【請求項9】
前記表面にカルボキシル基を有する基材が着色剤を含有していることを特徴とする請求項8に記載の表面機能性部材。
【請求項10】
前記基材が球状粒子であり、かつ平均粒子径が0.1〜10μmであることを特徴とする請求項8又は9に記載の表面機能性部材。
【請求項11】
前記アミノ基を有する機能性素材が、シロキサン骨格を有する置換基を含有する機能性素材であることを特徴とする請求項8乃至10のいずれか一項に記載の表面機能性部材。
【請求項12】
前記アミノ基を有する機能性素材が、下記一般式(I)及び一般式(II)で表されるシリコーン化合物より選ばれる少なくとも一種のシリコーン化合物であることを特徴とする請求項8乃至10のいずれか一項に記載の表面機能性部材。
H2N−B11−X ・・・(I)
H2N−B12−NH−B13−X ・・・(II)
(式中、B11,B12及びB13は、炭素数2から6のアルキレン基、Xはシロキサン骨格を示す。)
【請求項13】
絶縁性液体中に、請求項8乃至12のいずれか一項に記載の表面機能性部材を分散してなることを特徴とする電気泳動素子。
【請求項14】
シリコーンオイル中に、請求項11又は12に記載の表面機能性部材を分散してなることを特徴とする電気泳動素子。
【請求項1】
表面にカルボキシル基を有する基材上に、アミノ基を有する機能性素材を、縮合剤の存在下、直接結合させて得られる表面機能性部材の製造方法であって、
水系連続相中で反応させることを特徴とする表面機能性部材の製造方法。
【請求項2】
前記表面にカルボキシル基を有する基材が、乳化重合、懸濁重合、シード重合、あるいはソープフリー重合のいずれかの重合方式により得られた粒子であり、かつ平均粒子径が0.1〜10μmであることを特徴とする請求項1に記載の表面機能性部材の製造方法。
【請求項3】
前記縮合剤が、下記一般式(1)で表わされるトリアジン化合物であることを特徴とする請求項1又は2に記載の表面機能性部材の製造方法。
【化1】
但し、R1、R2は、置換もしくは無置換のアルキル基を表す。
【請求項4】
表面にカルボキシル基を有する基材上に、アミノ基を有する機能性素材を、縮合剤の存在下、直接結合させて得られる表面機能性部材の製造方法であって、
水系連続相中に、更に非イオン性界面活性剤を添加する工程を有することを特徴とする請求項1乃至3のいずれか一項に記載の表面機能性部材の製造方法。
【請求項5】
前記表面にカルボキシル基を有する基材上に、アミノ基を有する機能性素材を、縮合剤の存在下、水を主体とする連続相中で直接結合させて得られる表面機能性部材の製造方法であって、
前記、水系連続相中に非イオン性界面活性剤を添加することを特徴とする表面機能性部材の製造方法。
【請求項6】
前記アミノ基を有する機能性素材が、シロキサン骨格を有する置換基を含有する機能性素材であることを特徴とする請求項1乃至5のいずれか一項に記載の表面機能性部材の製造方法。
【請求項7】
前記アミノ基を有する機能性素材が、下記一般式(I)及び一般式(II)で表されるシリコーン化合物より選ばれる少なくとも一種のシリコーン化合物であることを特徴とする請求項1乃至5のいずれか一項に記載の表面機能性部材の製造方法。
H2N−B11−X ・・・(I)
H2N−B12−NH−B13−X ・・・(II)
(式中、B11,B12及びB13は、炭素数2から6のアルキレン基、Xはシロキサン骨格を示す。)
【請求項8】
表面にカルボキシル基を有する基材上に、アミノ基を有する機能性素材を、縮合剤の存在下で直接結合させて得られる表面機能性部材であって、
前記結合の反応は、水系連続相中で行われたものであることを特徴とする表面機能性部材。
【請求項9】
前記表面にカルボキシル基を有する基材が着色剤を含有していることを特徴とする請求項8に記載の表面機能性部材。
【請求項10】
前記基材が球状粒子であり、かつ平均粒子径が0.1〜10μmであることを特徴とする請求項8又は9に記載の表面機能性部材。
【請求項11】
前記アミノ基を有する機能性素材が、シロキサン骨格を有する置換基を含有する機能性素材であることを特徴とする請求項8乃至10のいずれか一項に記載の表面機能性部材。
【請求項12】
前記アミノ基を有する機能性素材が、下記一般式(I)及び一般式(II)で表されるシリコーン化合物より選ばれる少なくとも一種のシリコーン化合物であることを特徴とする請求項8乃至10のいずれか一項に記載の表面機能性部材。
H2N−B11−X ・・・(I)
H2N−B12−NH−B13−X ・・・(II)
(式中、B11,B12及びB13は、炭素数2から6のアルキレン基、Xはシロキサン骨格を示す。)
【請求項13】
絶縁性液体中に、請求項8乃至12のいずれか一項に記載の表面機能性部材を分散してなることを特徴とする電気泳動素子。
【請求項14】
シリコーンオイル中に、請求項11又は12に記載の表面機能性部材を分散してなることを特徴とする電気泳動素子。
【図1】

【図2】
【図3】
【図4】
【図5】
【図7】
【図8】
【図9】
【図11】
【図6】

【図10】
【図2】
【図3】
【図4】
【図5】
【図7】
【図8】
【図9】
【図11】
【図6】
【図10】
【公開番号】特開2008−138162(P2008−138162A)
【公開日】平成20年6月19日(2008.6.19)
【国際特許分類】
【出願番号】特願2007−115082(P2007−115082)
【出願日】平成19年4月25日(2007.4.25)
【出願人】(000006747)株式会社リコー (37,907)
【Fターム(参考)】
【公開日】平成20年6月19日(2008.6.19)
【国際特許分類】
【出願日】平成19年4月25日(2007.4.25)
【出願人】(000006747)株式会社リコー (37,907)
【Fターム(参考)】
[ Back to top ]