
表面被覆微粒子の医薬組成物
【課題】低分子量薬物及びペプチドやタンパク質などの高分子化合物を注射以外の方法で効率良く投与するために使用され得る医薬組成物、並びにかかる組成物を製造するための方法を提供すること。
【解決手段】(a)所定のpHで正若しくは負の電荷を有する薬物、(b)医薬上許容される微粒子及び(c)該pHで帯電しうる医薬上許容される表面被覆ポリマーを含む経粘膜投与用医薬組成物であって、該表面被覆ポリマーは該微粒子の表面を被覆しており、該薬物は該表面被覆ポリマーを介して該微粒子の表面に固定化されており、且つ、該微粒子と該表面被覆ポリマーとが非共有結合的に相互作用し、同時に該表面被覆ポリマーと該薬物とが互いに静電相互作用することにより複合体を形成することを特徴とする、組成物。
【解決手段】(a)所定のpHで正若しくは負の電荷を有する薬物、(b)医薬上許容される微粒子及び(c)該pHで帯電しうる医薬上許容される表面被覆ポリマーを含む経粘膜投与用医薬組成物であって、該表面被覆ポリマーは該微粒子の表面を被覆しており、該薬物は該表面被覆ポリマーを介して該微粒子の表面に固定化されており、且つ、該微粒子と該表面被覆ポリマーとが非共有結合的に相互作用し、同時に該表面被覆ポリマーと該薬物とが互いに静電相互作用することにより複合体を形成することを特徴とする、組成物。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、経粘膜投与用医薬組成物及びその製造方法に関する。より詳細には、本発明は、薬物、微粒子及び表面被覆ポリマーからなる複合体を含み、ここで該表面被覆ポリマーは該微粒子の表面を被覆しており、該薬物は該表面被覆ポリマーを介して該微粒子の表面に固定化されており、且つ、該微粒子と該表面被覆ポリマーとが非共有結合的に相互作用し、同時に該表面被覆ポリマーと該薬物とが静電相互作用することにより該複合体を形成することを特徴とする新規経粘膜投与用医薬組成物;並びにその製造方法に関する。
【背景技術】
【0002】
バイオテクノロジーの発達は、ペプチド、タンパク質、多糖、ポリ核酸、siRNA、RNA、抗体、抗原、低分子量薬物などの多くの治療化合物の発見をもたらした。しかしながら、これらの化合物の多くは、その物理化学的特性(例えば、大きな分子量、親水性、不安定性など)のために、注射以外の方法による体内への投与が難しい。これらの化合物のなかには注射により毎日複数回投与する必要があるものがあるが、若年患者は必ずしも、そのような化合物についてのこの服用計画を順守するわけではないことが特に問題となっている(非特許文献1及び2)。
【0003】
経口経路、或いは肺、口腔、膣及び鼻などの粘膜を通じたその他の粘膜送達経路による投与において、そのような薬物は、その物理的大きさや親水性によって吸収されにくい。更に、それらの薬物は、ペプチダーゼやプロテアーゼなどの酵素による分解を受けやすく、特に消化管においてそのことが問題となる。それらの薬物の粘膜表面を通じた輸送を改善するために、吸収促進剤を含有する製剤が使用されてきたが、そのような製剤は特に経鼻経路及び肺経路による送達においてある程度の成功を収めてきた。しかしながら、より大きな分子量を持つ化合物の粘膜表面を通じた輸送を達成するための効果的な方法及び組成物の開発が望まれている。
【0004】
ペプチドやタンパク質などの高分子薬物の粘膜表面を通じた輸送のための手段として、ナノ粒子などの微粒子を使用するシステムが広く検討されてきた(非特許文献3−5)。ペプチド及びタンパク質薬物の場合、ナノ粒子のマトリックス内に薬物を封入しなければ安定性が低いことが示されてきたが、それらの化合物のサイズが大きいことやナノ粒子のマトリックス内が通常疎水的環境であることから、それらの化合物のナノ粒子への封入は困難であり、その結果、通常非常に低い担持能となっており、従って粘膜表面に多量のナノ粒子を投与する必要がある。更には、粘膜を通じたナノ粒子の輸送は容易に達成されるものではないことが、文献において明らかにされている(非特許文献6)。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Drug Discovery Today, Vol.7, pp.1184-1189 (2002)
【非特許文献2】J. Control. Rel., Vol.87, pp.187-198 (2003)
【非特許文献3】J. Pharm. Sci., Vol.96, pp.473-483 (2007)
【非特許文献4】Biomaterials, Vol.23, pp.3193-3201 (2002)
【非特許文献5】Int. J. Pharm., Vol.342, pp.240-249 (2007)
【非特許文献6】J. Pharm. Sci., Vol.96, pp.473-483 (2007)
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明の目的は、低分子量薬物又はペプチドやタンパク質などの高分子化合物を注射以外の方法で効率良く投与するために使用され得る医薬組成物を提供することにある。より詳細には、本発明の目的は、低分子量薬物又はペプチドやタンパク質などの高分子薬物を鼻などの粘膜を通じた経路で効率良く投与するための微粒子を含む医薬組成物であって、これまでの経粘膜投与用微粒子製剤と比較して優れた薬物の担持率及び担持能を有すると同時に薬物の安定性向上も達成した微粒子含有医薬組成物を提供することにある。本発明はまた、かかる医薬組成物を製造するための方法を提供することもその目的とする。
【課題を解決するための手段】
【0007】
本発明者らは、薬物(例えば、ペプチド、タンパク質、DNA、RNA、siRNA、多糖、抗体、抗原、低分子量化合物など)を注射以外の方法で効率良く投与するための方法として、ナノ粒子などの微粒子システムを使用した経粘膜投与に着目し鋭意検討を重ねた。その結果、薬物と表面被覆ポリマー(即ち、微粒子の表面に付着するポリマー)との間の静電相互作用によって形成されている薬物−表面被覆ポリマー複合体が、微粒子と表面被覆ポリマーとの間の非共有結合的な相互作用によって微粒子表面に固定化されている複合体を含む組成物を作製することにより、同じ薬物の溶液製剤と比較して著しく薬物の安定性が向上することを見出した。更に、該組成物は、薬物を封入するタイプの微粒子製剤と比較して、優れた薬物担持能を有することも見出した。これらの知見から、該組成物を使用することにより、従来法より優れた薬物送達システムを達成することが可能であることを本発明者らは見出し、本発明を完成するに至った。
即ち、本発明は以下の通りである。
【0008】
[1](a)所定のpHで正若しくは負の電荷を有する薬物、(b)医薬上許容される微粒子及び(c)該pHで帯電しうる医薬上許容される表面被覆ポリマーを含む経粘膜投与用医薬組成物であって、該表面被覆ポリマーは該微粒子の表面を被覆しており、該薬物は該表面被覆ポリマーを介して該微粒子の表面に固定化されており、且つ、該微粒子と該表面被覆ポリマーとが非共有結合的に相互作用し、同時に該表面被覆ポリマーと該薬物とが静電相互作用することにより複合体を形成することを特徴とする、組成物。
[2]前記微粒子と前記表面被覆ポリマーとの非共有結合的な相互作用が静電相互作用であることを特徴とする、上記[1]記載の組成物。
[3]所定のpHが投与部位の生理的pHである、上記[1]又は[2]記載の組成物。
[4]前記薬物がペプチド、タンパク質、DNA、RNA、siRNA、多糖、抗原及び低分子量薬物からなる群より選択される、上記[1]〜[3]のいずれか1つに記載の組成物。
[5]前記薬物が薬効或いはワクチン効果をもたらしうる薬物である、上記[1]〜[4]のいずれか1つに記載の組成物。
[6]前記薬物がインスリンである、上記[4]記載の組成物。
[7]前記薬物がブロムヘキシン、ゾルミトリプタン及びそれらの塩からなる群から選択される少なくとも1種類の薬物である、上記[4]記載の組成物。
[8]前記表面被覆ポリマーが前記所定のpHにおいて単独では難水溶性である、上記[1]〜[7]のいずれか1つに記載の組成物。
[9]前記表面被覆ポリマーがキトサン、ポリアルギニン、ポリアクリル酸、ポリガンマグルタミン酸及びそれらの塩からなる群から選択される少なくとも1種類のポリマーである、上記[1]〜[8]のいずれか1つに記載の組成物。
[10]前記表面被覆ポリマーが粘膜付着性であり、且つ/又は経粘膜吸収促進因子として機能する、上記[1]〜[9]のいずれか1つに記載の組成物。
[11]前記微粒子がカルボキシル基又はアミノ基を含むポリマーを含む、上記[1]〜[10]のいずれか1つに記載の組成物。
[12]前記微粒子がポリ乳酸グリコール酸共重合体からなる、上記[1]〜[11]のいずれか1つに記載の組成物。
[13]前記所定のpHにおける前記複合体の平均粒径が10nm以上50μm以下である、上記[1]〜[12]のいずれか1つに記載の組成物。
[14]上記[8]記載の組成物の製造方法であって、
(a)前記表面被覆ポリマーが易水溶性であるpHで、前記薬物、前記微粒子及び前記表面被覆ポリマーを混合し、
(b)該混合液のpHを前記所定のpHに調整することを特徴とする、方法。
[15]上記[1]〜[13]のいずれか1つに記載の組成物の製造方法であって、
(a)前記薬物と前記表面被覆ポリマーとが同符号の電荷を持つようなpH条件下で、前記薬物、前記表面被覆ポリマー及び前記微粒子を混合した後、
(b)該混合物のpHを前記薬物の電荷が反対符号に変化するようなpHに調整すること
を含み、ここで前記薬物は両性薬物である、方法。
[16]上記[15]記載の製造方法であって、
前記工程(a)のpH条件において、前記微粒子が、前記薬物及び前記表面被覆ポリマーの電荷と反対符号に帯電していることを特徴とする方法。
[17]上記[1]〜[13]のいずれか1つに記載の組成物の製造方法であって、
(a)前記微粒子材料の有機溶媒溶液を前記表面被覆ポリマーの水溶液中へ滴下し、
(b)該有機溶媒を揮散させた後、
(c)前記薬物を添加して混合し、
(d)該混合液のpHを前記所定のpHに調整することを特徴とする、方法。
【発明の効果】
【0009】
本発明の組成物を使用することにより、これまで注射以外の方法で投与することが難しかった低分子量薬物及びペプチドやタンパク質などの高分子薬物を効率良く経粘膜投与することが可能となる。本発明の組成物中に含有された薬物は、反対電荷の表面被覆ポリマー、及び微粒子と複合体を形成することにより、溶液製剤に含有された場合よりも高い安定性(例えば、対酵素安定性、保存安定性)を有し、尚且つ、微粒子のマトリックス中に薬物を封入した微粒子製剤と比較して、高い薬物担持能を有する。更に、微粒子表面で薬物と複合体を形成する表面被覆ポリマーの種類によって、薬物の徐放性及び速放性を達成すること、並びに経粘膜吸収性の調節をすることが可能となる。
【図面の簡単な説明】
【0010】
【図1】インスリン−キトサン複合体で表面被覆されたPLGAコア粒子からなる表面被覆微粒子(表面キャリアシステム)の作製のメカニズムを説明する図である。
【図2】PLGA微粒子表面でのインスリン−キトサン複合体の形成について試験した結果を示す図である。左のヒストグラムはpH 6.0の緩衝液中のインスリンの放出、右のヒストグラムはpH 4.5の緩衝液中のインスリンの放出を示しており、縦軸は放出されたインスリン量(μg)を表す。
【図3】インスリン−キトサン複合体で表面被覆されたPLGAコア粒子の、飛行時間型2次イオン質量分析(Time of Flight Secondary Ion Mass Spectrometry; ToF-SIMS)での分析結果を示す図である。上段は各試料(PLGA、キトサン(Chitosan)、インスリン(Insulin)及び表面被覆微粒子(Carrier System))中のm/z=33ピークの検出結果を示し(横軸:m/z;縦軸:検出強度)、下段は、観察視野における表面被覆微粒子のm/z=33ピークの分布画像(左図)と全イオンの分布画像(右図)をそれぞれ示す。
【図4】培養後にキモトリプシン溶液中に残った分解されなかったインスリンの割合(%)を示す図である。上の棒は遊離インスリン溶液の場合、下の棒はPLGA/キトサン/インスリン表面被覆微粒子の場合に対応する。
【図5】ブタ鼻粘膜に対するインスリン透過試験に用いた方法を説明する模式図である。
【図6】ブタ鼻粘膜に対するインスリン透過試験の結果を示す図である。横軸は透過試験開始後の時間(分)を示し、縦軸は所定の時間までにブタ鼻粘膜を透過したインスリン量(ng/cm2)を示す。
【図7】表面被覆微粒子或いはキトサン/インスリン混合物の粒子径を測定した結果を示す図である。(a)は表面被覆微粒子(コア粒子を有する;PLGA100/キトサン/インスリン=1250/900/200 μg/ml)の粒子径分布、(b)はキトサン/インスリン混合物(コア粒子を持たない;キトサン/インスリン=900/200 μg/ml)の粒子径分布を示す。横軸は粒子の半径(nm)を示し、縦軸は強度を示す。
【図8】表面被覆微粒子(PLGA100/キトサン/インスリン=250/180/40 μg/ml)の凍結乾燥前及び凍結乾燥再懸濁後の粒子径を測定した結果を示す図である。(a)は凍結乾燥前の表面被覆微粒子の粒子径分布、(b)は凍結乾燥再懸濁後の表面被覆微粒子の粒子径分布を示す。横軸は粒子の半径(nm)を示し、縦軸は強度を示す。
【図9】実施例7の試料及び比較対照の試料をラットに経鼻投与した後の血中グルコースレベルを示す。結果は試料投与前グルコースレベルを100%として、相対的なグルコースレベル低下を%表示した。平均値±標準誤差で示した。
【図10】実施例8の試料を用いたインビトロ放出試験(N=3)(5mM MES等張バッファ(pH6)、37℃)の結果を示す。
【発明を実施するための形態】
【0011】
本発明は、(a)所定のpHで正若しくは負の電荷を有する薬物、(b)医薬上許容される微粒子及び(c)該pHで帯電しうる医薬上許容される表面被覆ポリマーを含む経粘膜投与用医薬組成物を提供する。該組成物において、該表面被覆ポリマーは該微粒子の表面を被覆しており、該薬物は該表面被覆ポリマーを介して該微粒子の表面に固定化されており、且つ、該微粒子と該表面被覆ポリマーとが非共有結合的に相互作用し、同時に該表面被覆ポリマーと該薬物とが静電相互作用することにより複合体(該複合体を以下、「表面被覆微粒子」ともいう)を形成する。
【0012】
「経粘膜投与用医薬組成物」とは、処置(treatment)及び/又は治療(therapy)を必要とする対象の粘膜に対して、塗布、スプレー、噴霧、貼付など適切な方法で投与され、薬物が粘膜組織へと、又は粘膜組織を通して循環器系或いは免疫系へと送達されることにより薬効或いはワクチン効果をもたらし得る医薬組成物を意味する。経粘膜投与用医薬組成物は、例えば、全身送達のために、粘膜を通して吸収され得る。粘膜の例としては、肺、口腔、眼、膣、胃腸管及び鼻などの粘膜が挙げられる。投与の簡便さの観点から、前記粘膜としては鼻粘膜が好ましい。
【0013】
表面被覆微粒子を構成する上記(c)の医薬上許容される表面被覆ポリマーは、上記の通り、微粒子の表面を被覆する。ここでいう「被覆」は、該表面被覆ポリマーが、該微粒子の内部ではなく、該微粒子の表面に、該微粒子と該表面被覆ポリマーとの間の非共有結合的な相互作用によって付着していることを単に意味しており、該微粒子の表面全体が該表面被覆ポリマーによって覆われることを必ずしも意味しない。
表面被覆ポリマーは、生体適合性であることが望ましい。本明細書において、用語「生体適合性」とは、ある物質及びその分解産物が、生体組織又は生体システム(例、血液循環系、神経系、免疫系など)に対して中毒的又は損傷的な影響を与えないことを意味する。生体適合性のポリマーは、ヒト又は他の動物への投与に適したものである。該表面被覆ポリマーはまた、生分解性であることがより好ましい。本明細書において、用語「生分解性」とは、ある物質が、酵素的、化学的若しくは物理的プロセスなどにより生体内で許容される時間内に分解されて、より小さな化学種を形成することを意味する。ある物質の生体適合性及び生分解性を検査する方法は、当該技術分野において周知である。
【0014】
表面被覆ポリマーは、天然ポリマーであっても、或いは合成ポリマーであっても良い。表面被覆ポリマーは、所定のpHにおいて、微粒子の表面で薬物とイオン結合するため、該所定のpHで該薬物と反対電荷を有するポリマーでなければならない。該所定のpHで同じ符号に帯電しうるポリマー同士であれば、必要に応じて2種類以上のポリマーを表面被覆ポリマーのために併用することもできる。
【0015】
本発明の医薬組成物は、例えば後述する製造法により製造され得るが、当該製造法により製造する場合、前記所定のpHにおいて単独では難水溶性であっても、表面被覆ポリマーが易水溶性であるpHで薬物及び微粒子と混合した後、該混合液のpHを前記所定のpHに調整することによって製造が可能である。従って、表面被覆ポリマーのために、前記所定のpHにおいて易水溶性であるポリマーだけでなく、前記所定のpHにおいて難水溶性であるポリマーも使用され得る。
【0016】
表面被覆ポリマーに使用され得るポリマーは、以下に限定されないが、ポリアニオン性又はポリカチオン性の多糖、ポリアミノ酸及びその他の帯電したポリマーから選択され得る。該ポリマーは、使用される薬物の種類、表面被覆ポリマーの電荷、および薬物の電荷などに応じて適宜選択される。
【0017】
本発明に使用され得るポリアニオン性の多糖とは、カルボキシル基、硫酸基又はリン酸基などの1つ以上の酸性極性基を構成単位中に有する多糖類である。そのようなポリアニオン性の多糖の例としては、以下に限定されないが、コンドロイチン硫酸、デキストラン硫酸、カルボキシメチルセルロース、アルギン酸、ペクチン、ヒアルロン酸、これらの誘導体や塩などが挙げられる。
【0018】
本発明に使用され得るポリカチオン性の多糖とは、アミノ基などの1つ以上の塩基性極性基を構成単位中に有する多糖類である。そのようなポリカチオン性の多糖の例としては、以下に限定されないが、キチン、キトサン、これらの誘導体や塩などが挙げられる。キトサンおよびキトサン誘導体は、様々な分子量及び脱アセチル化の程度のものから選択することができ、また、キトサン誘導体については、様々な置換の程度のものから選択することができる。
【0019】
本発明に使用され得るポリアニオン性のポリアミノ酸とは、等電点が生理的pHよりも酸性側にあるポリアミノ酸であり、その例としては、以下に限定されないが、ポリグルタミン酸、ポリアスパラギン酸、これらの誘導体や塩などが挙げられる。
【0020】
本発明に使用され得るポリカチオン性のポリアミノ酸とは、等電点が生理的pHよりも塩基性側にあるポリアミノ酸であり、その例としては、以下に限定されないが、ポリリジン、ポリアルギニン、これらの誘導体や塩などが挙げられる。
【0021】
表面被覆ポリマーに使用され得るポリマーとして、上記の多糖及びポリアミノ酸以外に、ポリエチレンイミンやポリアクリル酸、これらの誘導体や塩なども挙げられる。
【0022】
表面被覆ポリマーは、ポリエチレングリコール化(PEG化)及び/又はグリコシル化されていても良い。
【0023】
表面被覆ポリマーは、更に、粘膜付着性であってもよく、且つ/又は経粘膜吸収促進因子として機能しても良い。粘膜付着性ポリマーとしては、例えば、キトサン、ポリアクリル酸、アルギン酸ナトリウム、カルボキシメチルセルロースなどの他に、それらのPEG化されたポリマー類などが挙げられる。また、経粘膜吸収促進因子として機能するポリマーとしては、例えば、キトサン、ポリアクリル酸、ポリアルギニン、これらの塩や誘導体などが挙げられる。
【0024】
表面被覆ポリマーの分子量は、分解速度、機械強度、溶解度、表面被覆ポリマーと共に複合体を形成することになる薬物の種類などの要素を考慮して当業者は決定することができる。典型的には、ゲル浸透クロマトグラフィー法により測定した表面被覆ポリマーの重量平均分子量は、好ましくは1,000Da以上、より好ましくは2,000Da以上であるべきであり、また、好ましくは1,000,000Da以下、より好ましくは500,000Da以下であるべきである。従って、典型的には、表面被覆ポリマーの重量平均分子量は、好ましくは1,000−1,000,000Daであり、より好ましくは2,000−500,000Daである。例えば、キチン又はキトサンの重量平均分子量は、1,000−1,000,000Daであり得、キチン又はキトサンの脱アセチル化の程度は、20−100%であり得る。
【0025】
本発明の組成物は、表面被覆ポリマーの選択により、徐放性又は速放性の組成物として薬物の放出速度の制御、及び薬物の経粘膜吸収性の調節が可能となる。当業者は、該組成物が所望の薬動力学的特性を持つよう、表面被覆ポリマーを適切に選択することができる。
【0026】
本発明の組成物に使用される薬物は、その使用目的に応じて選択される。本発明の組成物において、薬物は、微粒子の表面で適切な表面被覆ポリマーとイオン結合するため、該組成物において使用され得る薬物は、所定のpHで正若しくは負に帯電する(即ち、表面被覆ポリマーと反対の電荷を有する)薬物でなければならない。かかる条件を満足する限り、任意の薬物が本発明の組成物で使用され得る。なお該所定のpHで同じ符号に帯電しうる薬物同士であれば、必要に応じて2種類以上の薬物を併用することもできる。
【0027】
該薬物は、以下に限定されないが、ペプチド、タンパク質、DNA、RNA、siRNA、多糖、抗体、抗原、低分子量化合物などであり得る。本発明の医薬組成物は、例えば後述する製造法により製造され得るが、当該製造法により製造する場合、薬物が表面被覆ポリマーと静電相互作用しない帯電条件でそれらをよく混合した後にpH変化により薬物と表面被覆ポリマーとの間にイオン結合を生じさせることが可能であるため、pHの変化により調製過程で帯電(符号又は強さ)を変化させることができる薬物が特に好適であり得る。そのような薬物としては、pHによって正にも負にも帯電するペプチドやタンパク質などの両性薬物、調製過程と組成物中に存在する場合とで帯電の強さが大きく変化するような酸解離定数(pKa)又は塩基解離定数(pKb)を有する薬物、及び水に溶解して、pHにあまり依存しない電荷を有し得る塩酸塩、硫酸塩、酢酸塩など塩形態の低分子量薬物が挙げられる。
【0028】
薬物としては、以下に限定されないが、例えば、抗高血圧剤、抗低血圧剤、鎮痛剤、抗精神病剤、抗鬱剤、抗躁剤、抗不安剤、鎮静剤、催眠剤、抗癲癇剤、オピオイドアゴニスト、喘息治療剤、麻酔剤、抗不整脈剤、関節炎治療剤、鎮痙剤、ACEインヒビター、鬱血除去剤、抗生物質、抗狭心症剤、利尿剤、抗パーキンソン病剤、気管支拡張剤、分娩促進剤、抗利尿剤、抗高脂血症剤、免疫抑制剤、免疫調節剤、制吐剤、抗感染症剤、抗新生物剤、抗真菌剤、抗ウイルス剤、抗糖尿病剤、抗アレルギー剤、解熱剤、抗腫瘍剤、抗痛風剤、抗ヒスタミン剤、止痒剤、骨調節剤、心血管剤、コレステロール低下剤、抗マラリア剤、喫煙を中止するための薬剤、鎮咳剤、去痰剤、粘液溶解剤、鼻詰り用薬剤、ドパミン作動剤、消化管用薬剤、筋弛緩剤、神経筋遮断剤、副交感神経作動剤、プロスタグランジン、興奮薬、食欲抑制剤、甲状腺剤又は抗甲状腺剤、ホルモン、抗偏頭痛剤、抗肥満剤、及び抗炎症剤などが挙げられる。薬物は予防的ワクチン、免疫療法、抗体治療、遺伝子治療、遺伝子発現抑制などを目的とした様々なペプチド、タンパク質、多糖、抗原、抗体、DNA、RNA、siRNA、低分子量薬物などから選択しても良い。
【0029】
薬物の具体例としては、以下に限定されないが、インスリン、グルカゴン、ロイプロリド、成長ホルモン、副甲状腺ホルモン、カルシトニン、血管内皮成長因子、エリスロポエチン、ヘパリン、シクロスポリン、オキシトシン、チロシン、エンケファリン、チロトロピン放出ホルモン、卵胞刺激ホルモン、黄体形成ホルモン、バソプレシン、バソプレシン類似体、カタラーゼ、スーパーオキシドジスムターゼ、インターロイキンII、インターフェロン、コロニー刺激因子、腫瘍壊死因子、メラニン細胞刺激ホルモン、グルカゴン様ペプチド−1、グルカゴン様ペプチド−2、カタカルシン、コレシステキニン−12、コレシステキニン−8、エキセンディン、ゴナドリベリン関連ペプチド、インスリン様タンパク質、ロイシン−エンケファリン、メチオニン−エンケファリン、ロイモルフィン、ニューロフィジン、コペプチン、ニューロペプチドY、ニューロペプチドAF、PACAP関連ペプチド、膵臓ホルモン、ペプチドYY、ウロテンシン、腸ペプチド、副腎皮質刺激ペプチド、上皮成長因子、プロラクチン、黄体形成ホルモン放出ホルモン(LHRH)、LHRHアゴニスト、成長ホルモン放出因子、ソマトスタチン、ガストリン、テトラガストリン、ペンタガストリン、エンドルフィン、アンジオテンシン、顆粒球コロニー刺激因子、顆粒球−マクロファージコロニー刺激因子、ヘパリナーゼ、インフルエンザワクチン用抗原、破傷風毒素、癌ワクチン用ペプチド、β−アミロイド、免疫グロブリン、肝硬変治療用siRNA、癌治療用siRNA、ブロムヘキシン、グラニセトロン、ゾルミトリプタン、スマトリプタンなどの低分子量薬物やそれらの薬学的に許容される塩などが挙げられる。
【0030】
上述したように、薬物及び表面被覆ポリマーは、微粒子表面において静電相互作用により結合して薬物−表面被覆ポリマー複合体を形成する。該複合体を形成する薬物と表面被覆ポリマーとの組み合わせについて、所定のpHにおいて正電荷を有する薬物と負電荷を有する表面被覆ポリマーとの組み合わせであっても良く、或いは所定のpHにおいて負電荷を有する薬物と正電荷を有する表面被覆ポリマーとの組み合わせであっても良い。
【0031】
前記医薬上許容される微粒子(本明細書において、用語「微粒子」は、上記(b)の微粒子を意味しているが、「表面被覆微粒子」との区別を特に明確にするために、該微粒子を「コア微粒子」と呼ぶこともある)は、生体適合性のポリマーからなることが望ましい。該ポリマーは、生分解性であっても非生分解性であっても良いが、生体に対する安全性の観点から、生分解性のものが好ましい。また、該ポリマーは、天然ポリマーであっても、或いは合成ポリマーであっても良い。
【0032】
微粒子のために使用され得る生体適合性且つ生分解性のポリマーとしては、以下に限定されないが、例えば、ポリエチレングリコール(PEG)、ポリ乳酸(PLA)、ポリグリコール酸(PGA)、ポリ乳酸グリコール酸共重合体(PLGA)、PEGとPLGAとのブロック共重合体(PEG−PLGA)、ポリ無水化合物、ポリ(ε−カプロラクトン)、ポリヒドロキシブチラート、ポリアミノ酸、ポリオルトエステル、ポリホスホエステル、ポリジアキサノン、ポリエステルアミド、ポリホスファゼン、ポリシアノアクリラート、キトサン、キトサン誘導体、スターチ、スターチ誘導体、アルブミン、フィブリン、フィブリノゲン、セルロース、コラーゲン、ヒアルロン酸、これらの物質の混合物及びコポリマー類などが挙げられる。
【0033】
微粒子のために使用され得る生体適合性且つ非生分解性のポリマーとしては、以下に限定されないが、例えば、ポリアクリラート、ポリアクリラートエステル、ポロキサマー、テトロニクス、ポリエチレン、ポリメチルメタクリラート、ポリメチルメタクリラートエステル、ポリスチレン、エチレンビニルアセタート、アシル化酢酸セルロース、ポリウレタン、塩化ポリビニル、これらの物質の混合物及びコポリマー類などが挙げられる。
【0034】
上記微粒子は、親水性であっても疎水性であっても良い。水系での微粒子の調製過程及び表面被覆微粒子の調製過程において、微粒子の形状及び大きさを維持するのが容易という観点から、疎水性の微粒子が好ましい。好適な例として、カルボキシル基又は第1級、第2級若しくは第3級アミノ基を有する疎水性ポリマーが微粒子に使用され得る。
【0035】
該微粒子のためのポリマーの分子量は、分解速度、機械強度、溶解度などの要素を考慮して当業者によって決定され得る。典型的には、ゲル浸透クロマトグラフィー法により測定した該ポリマーの重量平均分子量として、1,000Da(ダルトン)以上が好ましく、2,000Da以上がより好ましく、また、1,000,000Da以下が好ましく、500,000Da以下がより好ましい。従って、典型的には、該ポリマーの重量平均分子量として、1,000−1,000,000Daが好ましく、2,000−500,000Daがより好ましい。
【0036】
コア微粒子の大きさとしては、1nm以上、好ましくは5nm以上、より好ましくは10nm以上の平均粒径を持ち、尚且つ50μm以下、好ましくは20μm以下、より好ましくは10μm以下の平均粒径を持つ粒子が挙げられる。
なお、ここでいう粒径は、微粒子を前記“所定のpH”の水溶液中に分散させて測定した値である。粒径は、粒子径測定装置により測定し、粒子の形状を球であると仮定して算出した直径のことである。粒子径測定装置及び平均粒径の算出方法は、粒子サイズに応じて使い分ける。即ち、動的光散乱測定装置において測定可能な粒子サイズの場合(通常7μm以下)は、動的光散乱測定装置において測定し、散乱強度分布から求めた流体力学的直径の平均値を平均粒径として採用する。動的光散乱測定装置により測定できない大きい粒子サイズの場合(通常7μmよりも大きい場合)は、レーザー回折式粒度分布測定装置を用いて測定し、頻度分布を算術平均して求めた平均径を平均粒径として採用する。
ここで、コア微粒子の平均粒径が、例えば、10nm以上であるとは、上記の動的光散乱測定装置の散乱強度分布の各粒子径ピークの割合(全ピーク累積散乱強度に対する各粒子径ピークの累積散乱強度の割合)、又は上記のレーザー回折式粒度分布測定装置における頻度分布の各粒子径ピークの割合(全ピーク累積頻度に対する各粒子径ピークの累積頻度の割合)において、粒子径ピークの平均粒径のうちの10%以上、好ましくは20%以上、より好ましくは30%以上、更に好ましくは40%以上、特に好ましくは50%以上が10nm以上であることをいう。
【0037】
本発明の組成物を粘膜に投与したとき、表面被覆微粒子は、粘膜表面に達するか又は粘膜組織に取り込まれ得、そこで薬物を放出する。薬物はその後、血流に輸送される。微粒子が十分小さい場合(例えば、微粒子の粒径が20nm以下)、細胞間隙を通過して血流に到達する可能性がある。或いは、微粒子は、鼻又は腸などの一部の粘膜においてM細胞又はM様細胞に取り込まれて免疫系或いはリンパ系に輸送され得る。
【0038】
上記微粒子の製造は、文献に記載された様々な方法により行うことができる。該文献としては、例えば、Champion JA. et al., Proc. Natl. Acad. Sci. USA, Vol.104, pp.11901-4 (2007); Chattopadhyay P. et al., Adv. Drug Deliv. Rev., Vol.59, pp.443-53 (2007); Zhou WY et al., J. Mater. Sci. Mater. Med., Vol.19, pp.103-110 (2008); Schaffazick SR et al., Pharmazie, Vol.62, pp.354-60 (2007); Almeida AJ et al., Adv. Drug Deliv. Rev., Vol.59, pp.478-90 (2007); Muller, R.H., “Colloidal Carriers for Controlled Drug Delivery and Targeting: Modification, Characterization and In vivo Distribution”, CRC Press (1991); Jorg Kreuter (ed.), Colloidal Drug Delivery Systems, Marcel Dekker (1994)などが挙げられる。該微粒子の製造に使用され得る方法としては、例えば、ナノ沈殿(nanoprecipitation)、相分離、乳化、自己集合、高圧均質化、複合体形成(complexation)、イオンゲル化などが挙げられる。
【0039】
上述した通り、微粒子は、表面被覆ポリマーと非共有結合的に相互作用して表面被覆微粒子を生成するものである必要がある。ここでいう非共有結合的な相互作用とは、静電相互作用、疎水的相互作用、ファンデルワールス相互作用、水素結合など、共有結合によらない相互作用のことである。このうち例えば静電相互作用を利用する場合には、該微粒子は、静電相互作用するため、所定のpHで表面被覆ポリマーと逆の符号の電荷を有するポリマーでなければならない。従って、組成物に使用する薬物、表面被覆ポリマー及び微粒子のためのポリマーの選択に関して、所定のpHにおいて正の電荷を有する薬物、負の電荷を有する表面被覆ポリマー、及び正の電荷を有する微粒子のためのポリマーの組み合わせ、或いは所定のpHにおいて負の電荷を有する薬物、正の電荷を有する表面被覆ポリマー、及び負の電荷を有する微粒子のためのポリマーの組み合わせとなる。そのような組み合わせとなるために薬物、表面被覆ポリマー及び微粒子のためのポリマーが満たすべき等電点(pI)又は酸解離定数(pKa)若しくは塩基解離定数(pKb)に関する条件はそれぞれ以下の通りである:薬物及び微粒子のためのポリマーのpI又はpKa若しくは(14−pKb)の値が製造後の組成物のpHよりも高く、且つ表面被覆ポリマーのpI又はpKa若しくは(14−pKb)の値が製造後の組成物のpHよりも低い;或いは薬物及び微粒子のためのポリマーのpI又はpKa若しくは(14−pKb)の値が製造後の組成物のpHよりも低く、且つ表面被覆ポリマーのpI又はpKa若しくは(14−pKb)の値が製造後の組成物のpHよりも高い。各化合物について、等電点及び/又は酸解離定数を決定することは当業者の通常の技術的範囲内である。或いは、水中で溶解して帯電しうる塩酸塩、硫酸塩、酢酸塩など塩形態の低分子量薬物の場合は、該組成物は、水中での薬物の帯電とは逆の符号の電荷を有する表面被覆ポリマー、水中での薬物の帯電と同じ符号の電荷を有する微粒子とを、帯電した薬物と組み合わせて調製してもよい。
【0040】
前記所定のpH、即ち製造後の組成物のpHは、局所刺激を回避するために、投与部位の生理的pHに設定することが望ましい。上述したように、本発明の組成物は、肺、口腔、眼、膣、腸及び鼻などの粘膜に投与され得るが、これらの様々の粘膜の生理的pHは様々である。例えば、胃腸管の生理的pHは、その長さに沿って、胃における約pH1から、結腸におけるpH8まで上昇する;口腔は、6.8付近のpHを有する;鼻の流体のpHは、約pH5.5〜6.5の範囲にわたる;膣のpHは、4.5付近である。例えば、本発明の組成物が鼻粘膜投与用である場合、その好ましいpH値として約6.0が挙げられる。
【0041】
上述した通り、本発明の組成物における薬物として、インスリンが使用され得る。該組成物のpHが6.0である場合、インスリンの等電点は約pH5.3であるため、インスリンは組成物中で負に帯電している。従って、表面被覆ポリマーとしてはpH6.0で正電荷を有するポリマーである必要がある。非共有結合性の相互作用として静電相互作用を利用して表面被覆ポリマーと微粒子を相互作用させる場合は、微粒子としてはpH6.0で負電荷を有するポリマーからなる微粒子を好適な一例として用いることができる。かかる表面被覆ポリマーとしてはキトサンが挙げられ、微粒子のためのポリマーとしてはポリ乳酸グリコール酸共重合体(PLGA)が挙げられる。
上述した通り、本発明の組成物における薬物として、ブロムヘキシン、ゾルミトリプタン及びそれらの塩が使用され得る。該組成物のpHが6.0〜7.0である場合、該薬物は水中で正符号に帯電するため、表面被覆ポリマーとしては該pHで負電荷を有するポリマーである必要がある。非共有結合性の相互作用として静電相互作用を利用して表面被覆ポリマーと微粒子を相互作用させる場合は、微粒子としては該pHで正電荷を有するポリマーからなる微粒子を好適な一例として用いることができる。かかる表面被覆ポリマーとしてはポリアクリル酸及び又はポリガンマグルタミン酸又はそれらの塩が例として挙げられ、微粒子としてはキトサン微粒子やアミノ修飾したポリスチレン粒子が一例として挙げられる。
【0042】
表面被覆微粒子の大きさとしては、10nm以上、好ましくは20nm以上、より好ましくは40nm以上の平均粒径を持ち、尚且つ50μm以下、好ましくは20μm、より好ましくは10μm以下の平均粒径を持つ粒子が挙げられる。
なお、ここでいう粒径は、表面被覆微粒子を前記“所定のpH”の水溶液中に分散させて測定した値である。具体的には、表面被覆微粒子が懸濁液の場合は、懸濁液と同じpH(前記所定のpH)の水溶液で測定に適した濃度に希釈して測定する。表面被覆微粒子の剤型が、ドライパウダー、シート剤など、懸濁液以外の剤型に加工されていて、そのままでは粒径を測定できない場合は、水、又は適当なpH緩衝液を加えて、前記“所定のpH”の懸濁液に調製した後に粒径を測定する。粒径は、粒子径測定装置により測定し、粒子の形状を球であると仮定して算出した直径のことである。粒子径測定装置及び平均粒径の算出方法は、粒子サイズに応じて使い分ける。即ち、動的光散乱測定装置において測定可能な粒子サイズの場合(通常7μm以下)は、動的光散乱測定装置において測定し、散乱強度分布から求めた流体力学的直径の平均値を平均粒径として採用する。動的光散乱測定装置により測定できない大きい粒子サイズの場合(通常7μmよりも大きい場合)は、レーザー回折式粒度分布測定装置を用いて測定し、頻度分布を算術平均して求めた平均径を平均粒径として採用する。
ここで、表面被覆微粒子の平均粒径が、例えば、10nm以上であるとは、上記の動的光散乱測定装置の散乱強度分布の各粒子径ピークの割合(全ピーク累積散乱強度に対する各粒子径ピークの累積散乱強度の割合)、又は上記のレーザー回折式粒度分布測定装置における頻度分布の各粒子径ピークの割合(全ピーク累積頻度に対する各粒子径ピークの累積頻度の割合)において、粒子径ピークの平均粒子径のうちの10%以上、好ましくは20%以上、より好ましくは30%以上、更に好ましくは40%以上、特に好ましくは50%以上が10nm以上であることをいう。
【0043】
本発明の組成物における表面被覆微粒子は、コアとしての微粒子が存在することにより、単純に表面被覆ポリマーと薬物とを混合して複合体を形成させた場合と比べて、粒子径が単分散である。従って、本発明のような表面被覆微粒子の構成をとることによって、特性の揃った製剤の調製が容易となる。かかる特徴も本発明の利点である。
【0044】
本発明の組成物は、表面被覆微粒子が、標的粘膜部位に直接到達できる製剤として送達される必要がある。該製剤の例としては、経肺投与剤、経口投与剤、口腔内投与剤、眼内投与剤、膣内投与剤、鼻腔内投与剤、坐剤などが挙げられる。
【0045】
経肺投与剤としては、肺用吸入器により肺胞に送達される吸入剤が好ましい。
【0046】
経口投与剤としては、通常の経口投与製剤、例えば錠剤、顆粒剤、細粒剤、カプセル剤などであるが、小腸内で薬物が放出されるように工夫された剤型、例えば腸溶性錠剤、腸溶性顆粒剤、腸溶性カプセル剤、腸溶性細粒剤が好ましい。
【0047】
口腔内投与剤、眼内投与剤及び鼻腔内投与剤としては、口腔錠剤、口腔スプレー、点眼剤、点鼻剤、エアゾール、軟膏剤、ゲル剤、クリーム剤、液剤、懸濁液剤、ローション剤、ドライパウダー剤、シート剤、貼付剤などが挙げられる。
【0048】
膣内投与剤及び坐剤としては、軟膏剤、ゲル剤、クリーム剤、液剤、懸濁液剤、ローション剤、ドライパウダー剤、シート剤、カプセル剤などが挙げられる。
【0049】
上記の剤形に製する方法としては、当該分野で一般的に用いられている公知の製造方法を適用することができる。また、上記の剤形に製する場合には、必要に応じて、特定の剤形に製する際に通常用いられる賦形剤、結合剤、崩壊剤、滑沢剤などの担体、甘味剤、界面活性剤、懸濁化剤、乳化剤、着色剤、保存剤、安定剤などの各種製剤添加物などを適宜、適量含有させて製造することができる。また、本発明の組成物は、懸濁液を凍結乾燥するなどしてドライパウダー化した状態で保存し、使用時に該ドライパウダーに水を加えて再懸濁することもできる。かかる方法を採ることにより、組成物の保存安定性を向上させるために薬物、微粒子のためのポリマー、及び/又は表面被覆ポリマーの加水分解を回避することが可能となる。
【0050】
本発明の組成物において、微粒子のためのポリマー、表面被覆ポリマー及び薬物の好適な組成比は、使用する微粒子、表面被覆ポリマー及び薬物によって変動するため一概には言えないが、例えば、微粒子のためのポリマーとしてポリ乳酸グリコール酸共重合体(PLGA)、表面被覆ポリマーとしてキトサン、薬物としてインスリンを使用する場合、組成物中の重量比として、PLGA:キトサン:インスリン=1:0.1〜100:0.01〜100であり得る。
【0051】
本発明の医薬組成物は、安定かつ低毒性で安全に使用することができる。その投与頻度及び1回の投与量は、使用する薬物、患者の状態や体重、投与経路、治療方針などによって異なり一概には言えないが、例えば、糖尿病などの患者に薬物としてインスリンを使用した本発明の組成物を経鼻投与する場合には、一つの治療方針として、成人(体重約60kg)に対して有効成分(インスリン)として約2mgから約6mgを毎食の前に投与することができる。
【0052】
本発明はまた、上述した医薬組成物を製造する方法を提供する。本発明の方法は、適当なpHの溶液中で薬物と表面被覆ポリマーと微粒子とを混合し、任意でpHを変化させて、薬物と表面被覆ポリマーとの間で静電相互作用させ、表面被覆ポリマーと微粒子との間で非共有結合的に相互作用させることを含む。該方法は、加熱処理などが必要なく、簡便である。
【0053】
該方法においては、事前に、薬物、表面被覆ポリマー及び微粒子のためのポリマーの組み合わせ並びに本発明の組成物のpH(所定のpH)を決定しておく。これらの要素の決定は、本発明の組成物の説明において上述した通りに行うことができる。また、通常、薬物、表面被覆ポリマー及び微粒子を混合する前に、上述した方法により微粒子を作製しておく。次いで、薬物と表面被覆ポリマーと微粒子との混合、及び任意でpHの調整を行い、本発明の表面被覆微粒子を製造する。混合及び任意でのpH調整は、以下のa)〜c)からなる群から選択されるいずれか1つ:
a)薬物と表面被覆ポリマーとを、それらの複合体が形成されないpHを持つ溶液中で混合し、次いで微粒子を該溶液中に加えた後、溶液のpHを変化させて薬物と表面被覆ポリマーとの複合体形成及び微粒子表面への薬物−表面被覆ポリマー複合体の固定化を促進すること;
b)薬物と表面被覆ポリマーとを、それらの複合体が形成されるpHを持つ溶液中で混合した後、微粒子を該溶液中に加えて薬物−表面被覆ポリマー複合体を微粒子表面に固定化させること;
c)微粒子と表面被覆ポリマーとを、溶液中で混合して表面被覆ポリマーを微粒子の表面に固定化させ、次いで薬物を該溶液に加えた後、溶液のpHを調整して微粒子表面に固定化された表面被覆ポリマーと、薬物との複合体形成を促進すること、
を含む。
【0054】
特に、表面被覆ポリマーが所定のpHにおいて単独では難水溶性である場合、まず、(a)前記表面被覆ポリマーが易水溶性であるpHで、前記薬物、前記微粒子及び前記表面被覆ポリマーを混合し、次いで、(b)該混合液のpHを前記所定のpHに調整することが望ましい。
【0055】
また、本発明の医薬組成物に用いる薬物として、pHに応じて帯電の符号が変わる、いわゆる両性薬物を用いる場合には、本発明の医薬組成物の製造方法として、次の方法を用いることもできる。即ち、第一段階として、薬物の電荷と表面被覆ポリマーの電荷とが同符号となるようなpH条件下で、薬物、表面被覆ポリマー及び微粒子を混合することで、薬物及び表面被覆ポリマーの両方を微粒子の表面に静電相互作用などの非共有結合性相互作用により引き寄せることができる。その上で、第二段階として、該混合物のpHを薬物の電荷が反対符号に変化するようなpHに調整すれば、微粒子表面において、そこに集められた薬物と表面被覆ポリマーとの間で効率よく静電相互作用による結合を形成させ、本発明の医薬組成物を効率よく製造することができる。この製造方法は、副生成物としての遊離の薬物−表面被覆ポリマー複合体(微粒子表面に固定化されていない)の生成を抑制できるので有用である。
この方法をインスリン(薬物;等電点:約5.3)、キトサン(表面被覆ポリマー)、PLGA微粒子(微粒子)を例にして説明すると、インスリンが正に帯電する“等電点未満のpH”(例えば、pH4.5など)でインスリン、キトサン、PLGA微粒子を混合した後に、インスリンが負に帯電する“等電点より高いpH”(pH6.0など)にpHを調整するという方法である。pH4.5及びpH6.0のいずれにおいてもキトサンは正帯電、PLGA粒子は負帯電であるが、インスリンの電荷はpH4.5で正帯電、pH6.0で負帯電になるので、pH6.0において、互いにイオン結合したキトサン及びインスリンがPLGA微粒子の表面に固定化された本発明の組成物の一つを製造することもできる。なお、図1は、この実施態様を説明している。
即ち、本発明は、本発明の組成物の製造方法であって、
(a)薬物と表面被覆ポリマーとが同符号の電荷を持つようなpH条件下で、薬物、表面被覆ポリマー及び微粒子を混合した後、
(b)該混合物のpHを薬物の電荷が反対符号に変化するようなpHに調整すること
を含み、ここで該薬物は両性薬物である方法を提供する。
【0056】
或いは、また別の製造方法として、特にコア粒子の粒子径を小さくすることで産物としての表面被覆微粒子の粒子径を小さく制御したい場合に有用な方法として、次の方法も利用できる。この方法では、予め微粒子を調製しておくのではなく、微粒子の調製と、微粒子及び表面被覆ポリマーとの静電相互作用とを同時に開始させる。具体的には、この方法では、上記したような微粒子の材料(例、ポリマー)を含む適当な有機溶媒溶液(例、アセトン溶液など)を表面被覆ポリマー水溶液中へ滴下し、攪拌などにより溶液中から有機溶媒を揮散させることで、コア粒子としての微粒子生成と、表面被覆ポリマーによる微粒子被覆とを開始させる。そこに薬物を添加し、混合した上で、任意でpHを変化させて、薬物と表面被覆ポリマーとの間並びに表面被覆ポリマーと微粒子の間で静電相互作用を促進させることで、表面被覆微粒子を製造する。
即ち、本発明は、本発明の組成物の製造方法であって、
(a)前記微粒子材料の有機溶媒溶液を前記表面被覆ポリマーの水溶液中へ滴下し、
(b)該有機溶媒を揮散させた後、
(c)前記薬物を添加して混合し、
(d)該混合液のpHを前記所定のpHに調整することを特徴とする方法を提供する。
【0057】
以下、実施例及び試験例を挙げて本発明を更に詳細に説明するが、本発明は以下の実施例などによって制限されない。
【実施例】
【0058】
作製例1:様々な粒子径のポリ乳酸グリコール酸共重合体(PLGA)微粒子の作製
ラクチド:グリコリド比が50:50であるPLGA(RESOMER RG 502H,Bohringer Ingelheim)を使用して、PLGA微粒子を作製した。HPLCグレードのアセトンにPLGAを必要濃度で溶解した。PLGA/アセトン溶液を純水に1:3の割合となるまで、常時撹拌しながら滴下で加えた。アセトンが完全に蒸発するまで(約4時間)混合液を撹拌した。
その結果得られた微粒子の粒子径の分布を動的光散乱測定装置(DLS 802,Viscotek)により測定した。PLGAの濃度と得られた粒子の直径との関係を表1に示す。最初の有機溶媒溶液中のポリマー濃度を低くすることで、より微小な粒子が容易且つ再現性良く得られることが明らかとなった。
【0059】
【表1】

【0060】
実施例1:2種類の緩衝系における薬物−表面被覆ポリマー複合体で表面被覆された微粒子システムの作製
タンパク質薬物としてインスリン(pI 約5.3)を使用し、正に帯電した表面被覆ポリマーとしてキトサンを使用した。
ウシインスリン(Sigma,160μg/ml)の0.5mMクエン酸溶液(pH 4.5)1.5mlをキトサン(Bioneer 143kDa,0.72mg/ml)の0.5mMクエン酸溶液(pH 4.5)1.5mlに加え、最低30分間室温で放置した。作製例1に記載したようにして作製したPLGA微粒子(直径 約100nm;以下、「PLGA 100」ともいう)の0.5mMクエン酸溶液(pH 4.5;PLGA微粒子濃度:500μg/ml)の懸濁液3mlをキトサン/インスリン溶液に加え、最低1時間室温で放置した。NaOH(0.1−2.5N)を加えてpHを6.0に上昇させ、塩及び添加物を加えて、懸濁液の溶媒組成を表2又は表3に記載の緩衝液と同じ組成とした。
【0061】
【表2】
【0062】
【表3】
【0063】
表面被覆微粒子の粒子径及びゼータ電位をそれぞれDLS 802(Viscotek)及びZeta sizer 2000(Malvern)により測定した(表4)。
表4から分かるように、2種類の緩衝系について同様且つ好適な粒子径及びゼータ電位が得られた。また、2種類の表面被覆微粒子は、被覆していない微粒子の約2倍の粒子径で、ゼータ電位は大きな正の電位であった。両方の表面被覆微粒子はコロイド懸濁液として安定であることが分かった。
【0064】
【表4】
【0065】
実施例2:2種類のインスリン濃度での20mM MES緩衝系における薬物−表面被覆ポリマー複合体で表面被覆された微粒子システムの作製
タンパク質薬物としてインスリン(pI 約5.3)を使用し、正に帯電した表面被覆ポリマーとしてキトサンを使用した。
ウシインスリン(Sigma,160μg/ml又は800μg/ml)の0.5mMクエン酸溶液(pH 4.5)2mlをキトサン(Bioneer 143kDa,0.72mg/ml又は3.6mg/ml)の0.5mMクエン酸溶液(pH 4.5)2mlに加え、最低30分間室温で放置した。作製例1に記載したようにして作製したPLGA微粒子(直径 約100nm)の0.5mMクエン酸溶液(pH 4.5;PLGA微粒子濃度:0.5mg/ml又は2.5mg/ml)の懸濁液4mlをキトサン/インスリン溶液に加え、最低1時間室温で放置した。NaOH(0.1〜2.5N)を加えてpHを6.0に上昇させ、塩及び添加物を加えて、懸濁液の溶媒組成を表5に記載の緩衝液と同じ組成とした。
【0066】
【表5】
【0067】
表面被覆微粒子の粒子径及びゼータ電位をそれぞれDLS 802(Viscotek)及びZeta sizer 2000(Malvern)により測定した。両方の表面被覆微粒子サンプル共に好適な粒子径及びゼータ電位(表6)を示し、粒子径は被覆していない粒子(表4)の約2倍、ゼータ電位は大きな正の電位であった。また、両方の表面被覆微粒子はコロイド懸濁液として安定であった。
【0068】
【表6】
【0069】
実施例3:表面被覆ポリマーとしてポリ−L−アルギニンを使用した表面被覆PLGA微粒子システムの作製
タンパク質薬物としてインスリン(pI 約5.3)を使用し、正に帯電した表面被覆ポリマーとしてポリ−L−アルギニンを使用した。
ウシインスリン(Sigma,40μg/ml)の0.5mMクエン酸溶液(pH 6.0)3mlをポリ−L−アルギニン(MW 125kDa,Sigma;2.88mg/ml)の0.5mMクエン酸溶液(pH 6.0)3mlに加え、最低30分間室温で放置した。作製例1に記載したようにして作製したPLGA微粒子(直径 約100nm)の0.5mMクエン酸溶液(pH 6.0;PLGA微粒子濃度:250μg/ml)の懸濁液6mlをポリ−L−アルギニン/インスリン溶液に加え、最低1時間室温で放置した。塩及び添加物を加えて、懸濁液の溶媒組成を表5に記載の緩衝液と同じ組成とした。表面被覆微粒子の粒子径及びゼータ電位をそれぞれDLS 802(Viscotek)及びZeta sizer 2000(Malvern)により測定した。該粒子の平均径は285.9±90.6nm、ゼータ電位は+48.3±0.9mVであった。
【0070】
実施例4:表面被覆ポリマーとしてキトサン及びポリ−L−アルギニンを使用した表面被覆PLGA微粒子システムの作製
タンパク質薬物としてインスリン(pI 約5.3)を使用し、正に帯電した表面被覆ポリマーとしてキトサン及びポリ−L−アルギニンを使用した。
ウシインスリン(Sigma,800μg/ml)の0.5mMクエン酸溶液(pH 4.5)2mlをキトサン(MW 143kDa,0.36mg/ml)及びポリ−L−アルギニン(MW 125kDa,Sigma;1.8mg/ml)の0.5mMクエン酸溶液(pH 4.5)の混合液2mlに加え、最低30分間室温で放置した。作製例1に記載したようにして作製したPLGA微粒子(直径 約100nm)の0.5mMクエン酸溶液(pH 4.5;PLGA微粒子濃度:2.5mg/ml)の懸濁液4mlをキトサン/ポリ−L−アルギニン/インスリン溶液に加え、最低1時間室温で放置した。塩及び添加物を加えて、懸濁液の溶媒組成を表7に記載の緩衝液と同じ組成とした。表面被覆微粒子の粒子径及びゼータ電位をそれぞれDLS 802(Viscotek)及びZeta sizer 2000(Malvern)により測定した。該粒子の平均径は336.1±20.8nm、ゼータ電位は+40.3±3.4mVであった。
【0071】
【表7】
【0072】
実施例5:他の作製法を用いたインスリン/キトサン表面被覆PLGA微粒子システムの作製
0.1% w/vポリスチレン微粒子(MolecularProbe,カルボキシル化FluoSpheres)の0.5mMクエン酸溶液(pH 4.5)3mlをキトサン(Bioneer 143kDa,180μg/ml)の0.5mMクエン酸溶液(pH 4.5)3mlに加え、最低1時間室温で放置した。ウシインスリン(Sigma,20μg/ml)の0.5mMクエン酸溶液(pH 4.5)6mlをポリスチレンとキトサンとの上記混合液に加え、最低1時間室温で放置した。NaOH(0.1−2.5N)を加えてpHを6.0に上昇させ、塩及び添加物を加えて、懸濁液の溶媒組成を表5に記載の緩衝液と同じ組成とした。
表面被覆微粒子の粒子径及びゼータ電位をそれぞれDLS 802(Viscotek)及びZeta sizer 2000(Malvern)により測定した。該粒子の平均径は302.0±68.6nm、ゼータ電位は+27.9±1.7mVであった。また、ポリスチレンコア粒子の直径は196.7±27.5nmであったが、これはポリスチレンコア粒子の周囲に厚さ約50nmの層があることを示している。
【0073】
実施例6:他の作製法を用いたインスリン/キトサン表面被覆PLGA微粒子システムの作製
ラクチド:グリコリド比が50:50であるPLGA(RESOMER RG 502H,Bohringer Ingelheim)のアセトン溶液(PLGA0.01%(w/v):約20nmのPLGA粒子を調製することができる濃度)1.8mlをキトサン(Bioneer 73kDa,0.25mg/ml)の0.5mMクエン酸溶液(pH 4.5)6mlに加え、アセトンが完全に蒸発するまで室温で放置した。このPLGA/キトサン懸濁液3mlにウシインスリン(Sigma,160μg/ml)の0.5mMクエン酸溶液(pH 4.5)3mlを加え、最低1時間室温で放置した。NaOH(0.1−2.5N)を加えてpHを6.0に上昇させ、塩及び添加物を加えて、懸濁液の溶媒組成を表2に記載の緩衝液と同じ組成とした。表面被覆微粒子の粒子径をDLS 802(Viscotek)により測定した。該粒子の平均径は146.1±35.8nmであった。該粒子はコロイド懸濁液として安定であった。
【0074】
実施例7:動物実験用のインスリン/キトサン表面被覆PLGA微粒子システムの作製
タンパク質薬物としてインスリン(pI 約5.3)を使用し、正に帯電した表面被覆ポリマーとしてキトサンを使用した。動物実験用に高濃度試料(インスリン濃度6mg/mL)として調製した。
キトサン水溶液(甲陽ケミカル製、コーヨーキトサンFL−80、1.44mg/mL、pH4.5)15mlにウシインスリン水溶液(Sigma,320μg/ml、pH 4.5)15mlと50mMクエン酸水溶液0.02mlを加えて、pHを4.5±0.1に調整し約1時間静置した。その後PLGA微粒子懸濁液(粒子径約100nm、PLGA微粒子濃度1mg/mL、pH4.5)30ml、50mMクエン酸水溶液0.02mlを加え、pHを4.5に調整した。1時間静置した後にpHを6.0に調整し、マルトース(0.421g)を加えて溶解、pH6であることを確認した。この調製液を液体窒素で凍結後、凍結乾燥を行った。凍結乾燥品は凍結乾燥前の溶液の1/15ボリュームの蒸留水で再分散させた。再懸濁液を遠心分離し(19400×G、3時間、4℃)4/5容量の上清を除くことで粒子分画を濃縮して、動物実験用のサンプル(インスリン濃度6mg/mL)を得た。
表面被覆微粒子の粒子径及びゼータ電位をZeta sizer Nano(Malvern)により測定した。該粒子の平均径は252nm、ゼータ電位は+10.6mVであった。該粒子はコロイド懸濁液として安定であった。また、本実施例の表面被覆微粒子に結合しているインスリンの割合を後述する方法にて測定したところ担持率は93%であった。
【0075】
実施例8:放出試験用のインスリン/キトサン表面被覆PLGA微粒子システムの作製
タンパク質薬物としてインスリン(pI 約5.3)を使用し、正に帯電した表面被覆ポリマーとしてキトサンを使用した。
キトサン水溶液(甲陽ケミカル製、コーヨーキトサンFL−80、1.44mg/mL、pH4.5)15mlにウシインスリン水溶液(Sigma,320μg/ml、pH 4.5)15mlと50mMクエン酸水溶液0.02mlを加えて、pHを4.5±0.1に調整し約1時間静置した。その後PLGA微粒子懸濁液(粒子径約100nm、PLGA微粒子濃度1mg/mL、pH4.5)30ml、50mMクエン酸水溶液0.02mlを加え、pHを4.5に調整した。1時間静置した後にpHを6.0に調整し、マルトース(0.421g)を加えて溶解、pH6であることを確認した。
【0076】
実施例9:表面被覆ポリマーとしてカチオン系キトサン誘導体を使用した表面被覆PLGA微粒子システム(pH8)の作製
タンパク質薬物としてインスリン(pI 約5.3)を使用し、正に帯電した表面被覆ポリマーとしてカチオン系キトサン誘導体を使用した。
ウシインスリン(Sigma,0.32mg/ml)の0.5mMクエン酸溶液(pH 4.5)2mlをカチオン系キトサン誘導体(大日精化製、カチオン系キトサン誘導体水溶液,1.44mg/ml)の0.5mMクエン酸溶液(pH 4.5)2mlに加え、最低30分間室温で放置した。作製例1に記載したようにして作製したPLGA微粒子(直径 約100nm)の0.5mMクエン酸溶液(pH 4.5;PLGA微粒子濃度:1.0mg/ml)の懸濁液4mlをカチオン系キトサン誘導体/インスリン溶液に加え、最低1時間室温で放置した。10%w/vの濃度となるようにマルトースを添加し、NaOHを添加してpHを8に調整した。
表面被覆微粒子の粒子径をZeta sizer Nano(Malvern)により測定した。該粒子の平均径は230nmであった。該粒子はコロイド懸濁液として安定であった。また、本実施例の表面被覆微粒子に結合しているインスリンの割合を後述する方法にて測定したところ担持率は74%w/wであった。
【0077】
実施例10:表面被覆ポリマーとしてカチオン系キトサン誘導体を使用した表面被覆PLGA微粒子システム(pH7)の作製
タンパク質薬物としてインスリン(pI 約5.3)を使用し、正に帯電した表面被覆ポリマーとしてカチオン系キトサン誘導体を使用した。
ウシインスリン(Sigma,0.32mg/ml)の0.5mMクエン酸溶液(pH 7.0)2mlをカチオン系キトサン誘導体(大日精化製、カチオン系キトサン誘導体水溶液,1.44mg/ml)の0.5mMクエン酸溶液(pH 7.0)2mlに加え、最低30分間室温で放置した。作製例1に記載したようにして作製したPLGA微粒子(直径 約100nm)の0.5mMクエン酸溶液(pH 7.0;PLGA微粒子濃度:1.0mg/ml)の懸濁液4mlをカチオン系キトサン誘導体/インスリン溶液に加え、最低1時間室温で放置した。10%w/vの濃度となるようにマルトースを添加し、pH7であることを確認した。
表面被覆微粒子の粒子径及びゼータ電位をZeta sizer Nano(Malvern)により測定した。該粒子の平均径は234nm、ゼータ電位は+11.3mVであった。該粒子はコロイド懸濁液として安定であった。また、本実施例の表面被覆微粒子に結合しているインスリンの量を後述する方法にて測定したところ担持率は65%w/wであった。
【0078】
実施例11:正帯電薬物と負帯電表面被覆ポリマーを使用した表面被覆微粒子システムの作製
正帯電の低分子量薬物としてゾルミトリプタン(pKa=9.5)を使用し、負に帯電した表面被覆ポリマーとしてポリアクリル酸を用いた。
ポリアクリル酸水溶液(和光純薬製、平均分子量25万、1.44mg/ml)2mlにトリメチルアミン修飾ポリスチレン粒子(micromer NR3+ 100nm、コアフロント株式会社)の水懸濁液(1mg/ml)4mlを加え軽く混ぜ、約1時間後にゾルミトリプタン水溶液(640μg/ml)2mlを加え、軽く混ぜた後にpHを6.0に調整した。
表面被覆微粒子の粒子径及びゼータ電位をZeta sizer Nano(Malvern)により測定した。該粒子の平均径は330nm、ゼータ電位は−75mVであった。該粒子はコロイド懸濁液として安定であった。また、本実施例の表面被覆微粒子に結合している薬物の量を後述する方法に準じて(HPLC条件は別条件)測定したところ担持率は14%w/wであった。
【0079】
実施例12:正帯電薬物と負帯電表面被覆ポリマーを使用した表面被覆微粒子システムの作製
正帯電の低分子量薬物としてブロムヘキシン塩酸塩を使用し、負に帯電した表面被覆ポリマーとしてポリアクリル酸ナトリウムを用いた。
ポリアクリル酸ナトリウム水溶液(重合度2,700〜7,500,和光純薬製、1.44mg/ml)2mlにブロムヘキシン塩酸塩水溶液(640μg/ml)1mlと蒸留水1mlを加えて軽く混ぜ、約1時間後にトリメチルアミン修飾ポリスチレン粒子(micromer NR3+ 100nm、コアフロント株式会社)の水懸濁液(1mg/ml)4mlを加え、軽く混ぜた後にpHを6に調整した。
表面被覆微粒子の粒子径及びゼータ電位をZeta sizer Nano(Malvern)により測定した。該粒子の平均径は160nm、ゼータ電位は−57mVであった。該粒子はコロイド懸濁液として安定であった。また、本実施例の表面被覆微粒子に結合している薬物の量を後述する方法に準じて(HPLC条件は別条件)測定したところ担持率は88%w/wであった。
【0080】
実施例13:正帯電薬物と負帯電表面被覆ポリマーを使用した表面被覆微粒子システムの作製
正帯電の低分子量薬物としてブロムヘキシン塩酸塩を使用し、負に帯電した表面被覆ポリマーとしてポリガンマグルタミン酸ナトリウムを用いた。
ポリガンマグルタミン酸ナトリウム水溶液(平均分子量20万〜50万,和光純薬製、1.44mg/ml)2mlにブロムヘキシン塩酸塩水溶液(640μg/ml)1mlと蒸留水1mlを加えて軽く混ぜ、約1時間後にトリメチルアミン修飾ポリスチレン粒子(micromer NR3+ 100nm、コアフロント株式会社)の水懸濁液(1mg/ml)4mlを加え、軽く混ぜた後にpHを6に調整した。
表面被覆微粒子の粒子径及びゼータ電位をZeta sizer Nano(Malvern)により測定した。該粒子の平均径は203nm、ゼータ電位は−63mVであった。該粒子はコロイド懸濁液として安定であった。また、本実施例の表面被覆微粒子に結合している薬物の量を後述する方法に準じて(HPLC条件は別条件)測定したところ担持率は32%w/wであった。
【0081】
試験例1:微粒子表面での薬物と表面被覆ポリマーとの相互作用の評価
表5に記載の緩衝液中のインスリン/キトサン表面被覆PLGA微粒子の懸濁液(PLGA 100/キトサン/インスリン=500/360/80μg/ml)1mlをマイクロチューブに入れ、18000rpm(23900×g)で60分間遠心分離した。沈殿物を表5に記載の緩衝液で洗った。更に、沈殿物にpH 4.5又はpH6.0の解離用緩衝液を加え、それぞれのチューブを2時間室温で振盪した。懸濁液を同条件で15分間再び遠心分離し、上清を回収した。その後、各上清緩衝液中のインスリン濃度をELISAで定量した。
キトサン及びインスリンが共に正に帯電するため静電力による両者の引力相互作用が弱いpH 4.5の緩衝液の場合、両者が逆電荷を持つため静電引力が強くなるpH6.0の場合と比べてインスリン量が顕著に多かった(図2)。なお、遊離のインスリン及びキトサンは、解離の実験をする前の上記洗浄操作により除去されている。これらの結果は、微粒子表面にキトサン/インスリン複合体が存在することを示している。
【0082】
実施例1に記載の方法にて調製した、表2に記載の緩衝液中のインスリン/キトサン表面被覆PLGA微粒子の懸濁液(PLGA 100/キトサン/インスリン=250/180/40μg/ml)1mlをマイクロチューブに入れ、18000rpm(23900×g)で60分間遠心分離した。沈殿物を表2に記載の緩衝液で洗った。洗浄後の沈殿物をバッファーにて再懸濁したものを、ガラス板上に塗り広げ、約48時間自然乾燥させて、TOF−SIMS用試料とした。比較対照として、インスリン、キトサン、又は作製例1の方法で調製したPLGA 100をガラス板上に塗り広げた試料も用意した。
これらの各試料について、TOF−SIMS IV装置(ION−TOF GmbH)にて、インスリンのシステイン残基のSHに相当するピーク(m/z=33)の有無を測定した。その結果、PLGAとキトサンからはm/z=33のピークは有意なレベルで検出されなかったが、上記の洗浄済み表面被覆微粒子の試料とインスリンの試料とでは強いピークが検出された。各試料についての測定結果を図3上段に示す。
また、上記の洗浄済み表面被覆微粒子については、観察視野中のm/z=33の分布(図3下段左)とトータルイオンの分布(図3下段右)の様子も確認し、両者の分布パターンが一致すること、すなわち、粒子からm/z=33が検出されていることも確認した。
以上の特異性に関する測定結果と、粒子分布画像の結果は、洗浄済み表面被覆微粒子の表面にインスリンが存在することを示している。
【0083】
試験例2:薬物担持率及び担持能の評価
試験に供したサンプルは以下のようにして作製した。
【0084】
[PLGA/キトサン/インスリン(1000/720/160μg/ml)表面被覆微粒子(表8に記載の緩衝液中)]
タンパク質薬物としてインスリン(pI 約5.3)を使用し、正に帯電した表面被覆ポリマーとしてキトサンを使用した。
ウシインスリン(Sigma,0.64mg/ml)の0.5mMクエン酸溶液(pH 4.5)4.5mlをキトサン(Bioneer 143kDa,2.88mg/ml)の0.5mMクエン酸溶液(pH 4.5)4.5mlに加え、最低30分間室温で放置した。作製例1に記載したようにして作製したPLGA微粒子(直径 約100nm)の0.5mMクエン酸溶液(pH 4.5;PLGA微粒子濃度:2.0mg/ml)の懸濁液9mlをキトサン/インスリン溶液に加え、最低1時間室温で放置した。NaOH(0.1−2.5N)を加えてpHを6.0に上昇させ、グルコースを加えて、懸濁液の溶媒組成を表8に記載の緩衝液と同じ組成とした。
【0085】
【表8】
【0086】
表面被覆微粒子の粒子径及びゼータ電位をそれぞれDLS 802(Viscotek)及びZeta sizer 2000(Malvern)により測定した。該粒子の平均径は248.8±94.2nm、ゼータ電位は+8.7±0.5mVであった。
【0087】
[PLGA/キトサン/インスリン(250/90/40μg/ml)表面被覆微粒子(表8に記載の緩衝液中)]
タンパク質薬物としてインスリン(pI 約5.3)を使用し、正に帯電した表面被覆ポリマーとしてキトサンを使用した。
ウシインスリン(Sigma,160μg/ml)の0.5mMクエン酸溶液(pH 4.5)3mlをキトサン(Bioneer 143kDa,0.36mg/ml)の0.5mMクエン酸溶液(pH 4.5)3mlに加え、最低30分間室温で放置した。作製例1に記載したようにして作製したPLGA微粒子(直径 約100nm)の0.5mMクエン酸溶液(pH 4.5;PLGA微粒子濃度:500μg/ml)の懸濁液6mlをキトサン/インスリン溶液に加え、最低1時間室温で放置した。NaOH(0.1−2.5N)を加えてpHを6.0に上昇させ、グルコースを加えて、懸濁液の溶媒組成を表8に記載の緩衝液と同じ組成とした。
【0088】
表面被覆微粒子の粒子径及びゼータ電位をそれぞれDLS 802(Viscotek)及びZeta sizer 2000(Malvern)により測定した。該粒子の平均径は253.7±31.3nm、ゼータ電位は+6.4±1.8mVであった。
【0089】
[PLGA/ポリ−L−アルギニン/インスリン(125/720/40μg/ml)表面被覆微粒子(表5に記載の緩衝液中)]
タンパク質薬物としてインスリン(pI 約5.3)を使用し、正に帯電した表面被覆ポリマーとしてポリ−L−アルギニンを使用した。
ウシインスリン(Sigma,160μg/ml)の0.5mMクエン酸溶液(pH 6.0)3mlをポリ−L−アルギニン(MW 125kDa,Sigma;2.88mg/ml)の0.5mMクエン酸溶液(pH 6.0)3mlに加え、最低30分間室温で放置した。作製例1に記載したようにして作製したPLGA微粒子(直径 約100nm)の0.5mMクエン酸溶液(pH 6.0;PLGA微粒子濃度:250μg/ml)の懸濁液6mlをポリ−L−アルギニン/インスリン溶液に加え、最低1時間室温で放置した。塩及び添加物を加えて、懸濁液の溶媒組成を表5に記載の緩衝液と同じ組成とした。表面被覆微粒子の粒子径及びゼータ電位をそれぞれDLS 802(Viscotek)及びZeta sizer 2000(Malvern)により測定した。該粒子の平均径は497.4±141.9nm、ゼータ電位は+44.3±2.1mVであった。
【0090】
インスリンの担持率及び担持能を以下に記載する方法により測定した。
[結合したインスリンの解析法]
上記各サンプル0.5ml又は1mlを1.5mlマイクロチューブに入れ、遠心分離した(15000rpm(21900×g),180分,4℃)。上清を回収し、上清中のインスリン濃度をインスリンELISAキット(Mercodia Bovine insulin ELISA)及び希釈緩衝液(Mercodia Diabetes sample buffer)又はHPLCを使用して測定した(当該インスリン濃度をAとする)。コントロールとして、1.5mlマイクロチューブ中の上記各サンプル0.5ml又は1mlを4℃で180分間静置し、全サンプル中のインスリン濃度を同様にして測定した(当該インスリン濃度をBとする)。
担持率及び担持能は以下のようにして計算される:
担持率(%)=100×((Bの平均値)−A)/(Bの平均値));
担持能(%)=コア粒子総質量に対する結合したインスリンの質量
=100×((Bの平均値)−A)/コア粒子総質量
(コア粒子総質量=粒子数×4/3×3.14×(コア粒子の半径)3×密度)
【0091】
上記方法によるインスリン担持の解析結果を表9に示す。
【0092】
【表9】
【0093】
試験例3:微粒子表面で複合体化したインスリンの安定性評価
試験例2で使用した、表8に記載の緩衝液中のPLGA/キトサン/インスリン(1000/720/160μg/ml)表面被覆微粒子を本試験のサンプルとして使用した。また、コントロールとして、表8に記載の緩衝液と同じ溶媒組成のインスリン溶液(pH 6;インスリン濃度:160μg/ml)を使用した。
【0094】
酵素反応させるサンプルの調製は以下のようにして行った。
ウシの膵臓由来のα−キモトリプシン(Fluka Biochemika;code No.27270)を濃度が40μg/mlになるまで5mM MES緩衝液(pH 6.0)に溶解した。サンプル1mlを37℃で15分間温めた後、同様にして温めておいた5mM MES緩衝液(pH 6.0)0.6ml及び同様にして温めておいたα−キモトリプシンの5mM MES緩衝溶液(pH 6.0;α−キモトリプシン濃度:40μg/ml)0.4mlを加えた。混合物を37℃で30分間振盪培養した後、氷で冷却した氷酢酸1mlを加えて酵素反応を停止させた。次いでミキサーで1分間混合し、1時間以上室温で放置した後、フィルター径0.1μmのフィルターでろ過して、HPLC分析用試料を得た。
【0095】
一方、酵素を含有しない比較対照サンプルの調製は以下のようにして行った。
サンプル1mlを5mM MES緩衝液(pH 6.0)1ml及び氷酢酸1mlと混合した。次いで、混合物をミキサーで1分間混合し、1時間以上室温で放置した後、フィルター径0.1μmのフィルターでろ過して、HPLC分析用試料を得た。
【0096】
また、インスリン定量の検量線用標準サンプルの調製は以下のようにして行った。
インスリン溶液(40−160μg/ml)1mlを5mM MES緩衝液(pH 6.0)1ml及び氷酢酸1mlと混合し、ミキサーで混合した。該混合物をHPLC分析の検量線用の標準サンプルとして使用した。
【0097】
サンプルのHPLC分析は以下の条件で行った:
C18カラム(Inertsil ODS−2,5μm,250mm×4.6mm);
移動相A:0.1% TFA水溶液,移動相B:0.1% TFA CH3CN溶液;
グラジエント条件(移動相B濃度):0分時:30%,10分時:40%,11分時:30%,16分時:30%;
カラムオーブン温度:40℃,流速:1.0ml/分,注入量:20μl,検出:UV275nm
【0098】
上記条件による分析の結果、酵素反応後に残存していたインスリン量の割合は、PLGA/キトサン/インスリンについて83.9%±2.3%、インスリン溶液について61.8%±2.0%であった。即ち、遊離のインスリン溶液での38.2%に対して、表面被覆複合体化したインスリンの16.1%のみが、本実験の時間中に分解された(図4)。
この結果は、酵素反応に対するインスリン安定性が大きく向上したことを示している。
【0099】
試験例4:表面被覆微粒子を使用したブタ鼻粘膜に対するインスリン透過試験
タンパク質薬物としてインスリン(pI 約5.3)を使用し、正に帯電した表面被覆ポリマーとしてキトサン及びポリ−L−アルギニンを使用した。表面被覆微粒子の作製は、実施例4に記載したのと同様にして行った。また、コントロールとして、表7に記載の緩衝液と同じ溶媒組成のインスリン溶液(pH 6;インスリン濃度:200μg/ml)を使用した。鼻の呼吸部粘膜組織をブタの鼻腔(呼吸部)から摘出した。摘出した組織は、水平拡散チャンバに配置するまで、実施例4で使用したものと同じ組成の緩衝液(酸素処理して冷却したもの)の中で保存した。当該組織を適当な大きさに切断し、図5に示すように水平拡散チャンバ中のドナーセルとレセプターセルとの間に配置した(粘膜の有効面積:0.79cm2)。新鮮な(上記の)緩衝液(酸素処理して冷却したもの)をドナーセル及びレセプターセルに注入し、両セルを29±1℃に加温した循環水で30分間温めて、組織を平衡化させた。透過試験の間も、ドナーセル及びレセプターセルに注入した緩衝液を酸素処理し、また、29±1℃に加温した循環水で両セルを温めた。透過試験の前後で、組織が生存していること、及び損傷を受けていないことをAlamar Blue Assay及び組織のTEER値の測定により確かめた。
ドナーセル中の全ての緩衝液を同量の各サンプルに置換することにより透過試験を開始した。選択した時点において、200μlのサンプルをレセプターセルから採取し、同量の新鮮な緩衝液(酸素処理後、29±1℃に加温しておいたもの)で置換した。レセプターセルから採取したサンプルをインスリンELISAキット及び希釈緩衝液を使用して分析した。分析結果を図6に示す。図6は、コントロールのインスリン溶液を投与した場合と比較して、キトサン/ポリ−L−アルギニン/インスリン表面被覆PLGA微粒子を投与した場合により多くのインスリンが摘出鼻呼吸部粘膜組織を透過したことを示している。
【0100】
試験例5:表面被覆微粒子の粒子径分布の測定
タンパク質薬物としてインスリン(pI 約5.3)を使用し、正に帯電した表面被覆ポリマーとしてキトサンを使用した。
ウシインスリン(Sigma,0.8mg/ml)の0.5mMクエン酸溶液(pH 4.5)1.5mlをキトサン(Bioneer 143kDa,3.6mg/ml)の0.5mMクエン酸溶液(pH 4.5)1.5mlに加え、最低30分間室温で放置した。作製例1に記載したようにして作製したPLGA微粒子(直径 約100nm;以下、「PLGA 100」ともいう)の0.5mMクエン酸溶液(pH 4.5;PLGA微粒子濃度:2.5mg/ml)の懸濁液3ml、或いは単なる0.5mMクエン酸溶液(pH 4.5)をキトサン/インスリン溶液に加え、最低1時間室温で放置した。NaOH(0.1−2.5N)を加えてpHを6.0に上昇させ、塩及び添加物を加えて、懸濁液の溶媒組成を表2に記載の緩衝液と同じ組成とした。表面被覆微粒子或いはキトサン/インスリン混合物の粒子径をDLS 802(Viscotek)により測定した。
その結果を図7に示す。図7(a)は表面被覆微粒子、図7(b)はキトサン/インスリン混合物についての粒子径分布を示しており、また、そのそれぞれについて、ピーク領域における強度のパーセンテージ、粒子径及び標準偏差を表10及び11に示す。
【0101】
【表10】
【0102】
【表11】
【0103】
PLGA粒子を添加した場合は、単分散の粒子径が得られたのに対して、PLGA粒子を添加しなかった場合は、粒子径の大きなものも含む多分散の粒子径分布となった。この結果より、コア粒子の存在により、粒子径の揃った表面被覆微粒子が得られることがわかる。
【0104】
試験例6:表面被覆微粒子懸濁液をドライパウダーとして製剤化する可能性の確認
実施例1の方法にて、[PLGA100/キトサン/インスリン(250/180/40μg/ml)表面被覆微粒子(表2に記載の緩衝液中)]を調製した。この懸濁液を凍結乾燥し、凍結乾燥前と同量の水で再懸濁させた。凍結乾燥前の粒子径、及び凍結乾燥再懸濁後の粒子径をDLS 802(Viscotek)を用いて測定した。粒子径の測定結果を図8に示す。図8(a)は凍結乾燥前、図8(b)は凍結乾燥再懸濁後の粒子径分布を示しており、また、そのそれぞれについて、ピーク領域における強度のパーセンテージ、粒子径及び標準偏差を表12及び13に示す。
【0105】
【表12】
【0106】
【表13】
【0107】
凍結乾燥前の粒子径は183.0±21.2nm、凍結乾燥再懸濁後の粒子径は229.4±47.9nmであり、凍結乾燥と再懸濁の操作により主成分の粒子径は有意に変化せず、目だった凝集物も生成していないことがわかる。この結果より、本発明の表面被覆微粒子は懸濁液としてだけでなく、ドライパウダーなど他の剤型としても使用可能であることがわかる。
【0108】
試験例7:表面被覆微粒子を使用したラット鼻腔に対するインビボ投与試験
試験にはラット(系統:SDラット、7週齢、オス、ブリーダー:日本エスエルシー)を用いた。ラットは試験前日の夕方から絶食させた。試験は筋肉注射及び腹腔点滴による麻酔の下で行った。血中グルコース濃度が安定化してから、鼻腔へのサンプル投与を行い、投与してから10、20、30、60、90、120分後に尾静脈からの採血を行い、血中グルコース検出キット(テルモ製、メディセーフミニ)を用いて血中グルコースレベルを測定した。
試料として実施例7に記載の表面被覆微粒子を20μl/体重300g(400μgインスリン/kg体重)の量だけラット鼻腔に投与した。また、コントロールとして、いずれも実施例7と同じ溶媒組成である、クエン酸0.5mMマルトース10%w/v溶液(pH6)(バッファー液)、インスリン溶液(pH6;インスリン濃度:6mg/ml、クエン酸0.5mMマルトース10%w/v)、キトサン/インスリン混液(pH6;キトサン濃度:27mg/ml、インスリン濃度:6mg/ml、クエン酸0.5mMマルトース10%w/v)も同様にラット鼻腔に投与した。
試験結果を図9に示す。図9は、コントロールのバッファー液やインスリン溶液を投与した場合と比較して、大きく血中グルコースレベルが低下していること、また、キトサン/インスリン混液と比べても初期の血中グルコースレベルの低下が早いことを示しており、いずれも本発明の実施例がペプチド薬物の経粘膜デリバリーにおいて優れた促進効果を有することを示している。
【0109】
試験例8:表面被覆微粒子を使用したインビトロ放出試験
実施例8の試料0.5mLをエッペンドルフチューブ1.5mLに添加し遠心分離(13600rpm(19400xG)、3hr、4℃)して沈殿物を得た。放出試験液として5mM MES 152mM NaCl水溶液(pH 6.0、生理的等張イオン強度154mM)を準備した。
該沈殿物に放出試験液0.5mLを添加し、ピペッティングとミキサーにより再分散させたものを10mL容ガラスバイアルに移した。エッペンドルフチューブを新たな放出試験液0.5mLで洗い、洗い液も上記ガラスバイアルに移した(懸濁液合計1mL)。懸濁液入りガラスバイアルを37℃、75rpmにて振盪することで放出試験を行った。所定時間(0、0.5、3時間)後、懸濁液を0.1μmフィルター(Sartorius)にてろ過し、ろ液を得た。ろ液とその1/2容量の5%リン酸をミキサーを用いて混合し、放出インスリン量分析用のHPLC試料とした。
また、放出前のインスリン含量を定量する目的で、該沈殿物に蒸留水1mLと5%リン酸0.5mLを加えてミキサー混合し、4℃にて一夜放置した。再度ミキサー混合した後に0.1μmフィルター(Sartorius)でろ過して、該沈殿物中インスリン量定量用のHPLC試料とした。
前記のインスリンHPLC分析条件を用いてインスリン定量を行った。
放出率(%)=100×(放出液中のインスリン量/放出前沈殿中のインスリン量)
放出試験の結果を図10に示す。該沈殿物に含まれるほぼ全てのインスリンが放出試験の3時間以内に放出された。
【産業上の利用可能性】
【0110】
本発明の組成物を使用することにより、これまで注射以外の方法で投与することが難しかった低分子量薬物及びペプチドやタンパク質などの高分子薬物を効率良く経粘膜投与することが可能となる。本発明の組成物中に含有された薬物は、表面被覆ポリマー及び微粒子と複合体を形成することにより、溶液製剤に含有された場合よりも高い安定性(例えば、対酵素安定性、保存安定性)を有し、尚且つ、微粒子のマトリックス中に薬物を封入した微粒子製剤と比較して、高い薬物担持能を有する。更に、微粒子表面で薬物と複合体を形成する表面被覆ポリマーの種類によって、薬物が徐放性及び速放性を示す表面被覆微粒子を作製すること、並びに経粘膜吸収性の調節をすることが可能となる。
【技術分野】
【0001】
本発明は、経粘膜投与用医薬組成物及びその製造方法に関する。より詳細には、本発明は、薬物、微粒子及び表面被覆ポリマーからなる複合体を含み、ここで該表面被覆ポリマーは該微粒子の表面を被覆しており、該薬物は該表面被覆ポリマーを介して該微粒子の表面に固定化されており、且つ、該微粒子と該表面被覆ポリマーとが非共有結合的に相互作用し、同時に該表面被覆ポリマーと該薬物とが静電相互作用することにより該複合体を形成することを特徴とする新規経粘膜投与用医薬組成物;並びにその製造方法に関する。
【背景技術】
【0002】
バイオテクノロジーの発達は、ペプチド、タンパク質、多糖、ポリ核酸、siRNA、RNA、抗体、抗原、低分子量薬物などの多くの治療化合物の発見をもたらした。しかしながら、これらの化合物の多くは、その物理化学的特性(例えば、大きな分子量、親水性、不安定性など)のために、注射以外の方法による体内への投与が難しい。これらの化合物のなかには注射により毎日複数回投与する必要があるものがあるが、若年患者は必ずしも、そのような化合物についてのこの服用計画を順守するわけではないことが特に問題となっている(非特許文献1及び2)。
【0003】
経口経路、或いは肺、口腔、膣及び鼻などの粘膜を通じたその他の粘膜送達経路による投与において、そのような薬物は、その物理的大きさや親水性によって吸収されにくい。更に、それらの薬物は、ペプチダーゼやプロテアーゼなどの酵素による分解を受けやすく、特に消化管においてそのことが問題となる。それらの薬物の粘膜表面を通じた輸送を改善するために、吸収促進剤を含有する製剤が使用されてきたが、そのような製剤は特に経鼻経路及び肺経路による送達においてある程度の成功を収めてきた。しかしながら、より大きな分子量を持つ化合物の粘膜表面を通じた輸送を達成するための効果的な方法及び組成物の開発が望まれている。
【0004】
ペプチドやタンパク質などの高分子薬物の粘膜表面を通じた輸送のための手段として、ナノ粒子などの微粒子を使用するシステムが広く検討されてきた(非特許文献3−5)。ペプチド及びタンパク質薬物の場合、ナノ粒子のマトリックス内に薬物を封入しなければ安定性が低いことが示されてきたが、それらの化合物のサイズが大きいことやナノ粒子のマトリックス内が通常疎水的環境であることから、それらの化合物のナノ粒子への封入は困難であり、その結果、通常非常に低い担持能となっており、従って粘膜表面に多量のナノ粒子を投与する必要がある。更には、粘膜を通じたナノ粒子の輸送は容易に達成されるものではないことが、文献において明らかにされている(非特許文献6)。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Drug Discovery Today, Vol.7, pp.1184-1189 (2002)
【非特許文献2】J. Control. Rel., Vol.87, pp.187-198 (2003)
【非特許文献3】J. Pharm. Sci., Vol.96, pp.473-483 (2007)
【非特許文献4】Biomaterials, Vol.23, pp.3193-3201 (2002)
【非特許文献5】Int. J. Pharm., Vol.342, pp.240-249 (2007)
【非特許文献6】J. Pharm. Sci., Vol.96, pp.473-483 (2007)
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明の目的は、低分子量薬物又はペプチドやタンパク質などの高分子化合物を注射以外の方法で効率良く投与するために使用され得る医薬組成物を提供することにある。より詳細には、本発明の目的は、低分子量薬物又はペプチドやタンパク質などの高分子薬物を鼻などの粘膜を通じた経路で効率良く投与するための微粒子を含む医薬組成物であって、これまでの経粘膜投与用微粒子製剤と比較して優れた薬物の担持率及び担持能を有すると同時に薬物の安定性向上も達成した微粒子含有医薬組成物を提供することにある。本発明はまた、かかる医薬組成物を製造するための方法を提供することもその目的とする。
【課題を解決するための手段】
【0007】
本発明者らは、薬物(例えば、ペプチド、タンパク質、DNA、RNA、siRNA、多糖、抗体、抗原、低分子量化合物など)を注射以外の方法で効率良く投与するための方法として、ナノ粒子などの微粒子システムを使用した経粘膜投与に着目し鋭意検討を重ねた。その結果、薬物と表面被覆ポリマー(即ち、微粒子の表面に付着するポリマー)との間の静電相互作用によって形成されている薬物−表面被覆ポリマー複合体が、微粒子と表面被覆ポリマーとの間の非共有結合的な相互作用によって微粒子表面に固定化されている複合体を含む組成物を作製することにより、同じ薬物の溶液製剤と比較して著しく薬物の安定性が向上することを見出した。更に、該組成物は、薬物を封入するタイプの微粒子製剤と比較して、優れた薬物担持能を有することも見出した。これらの知見から、該組成物を使用することにより、従来法より優れた薬物送達システムを達成することが可能であることを本発明者らは見出し、本発明を完成するに至った。
即ち、本発明は以下の通りである。
【0008】
[1](a)所定のpHで正若しくは負の電荷を有する薬物、(b)医薬上許容される微粒子及び(c)該pHで帯電しうる医薬上許容される表面被覆ポリマーを含む経粘膜投与用医薬組成物であって、該表面被覆ポリマーは該微粒子の表面を被覆しており、該薬物は該表面被覆ポリマーを介して該微粒子の表面に固定化されており、且つ、該微粒子と該表面被覆ポリマーとが非共有結合的に相互作用し、同時に該表面被覆ポリマーと該薬物とが静電相互作用することにより複合体を形成することを特徴とする、組成物。
[2]前記微粒子と前記表面被覆ポリマーとの非共有結合的な相互作用が静電相互作用であることを特徴とする、上記[1]記載の組成物。
[3]所定のpHが投与部位の生理的pHである、上記[1]又は[2]記載の組成物。
[4]前記薬物がペプチド、タンパク質、DNA、RNA、siRNA、多糖、抗原及び低分子量薬物からなる群より選択される、上記[1]〜[3]のいずれか1つに記載の組成物。
[5]前記薬物が薬効或いはワクチン効果をもたらしうる薬物である、上記[1]〜[4]のいずれか1つに記載の組成物。
[6]前記薬物がインスリンである、上記[4]記載の組成物。
[7]前記薬物がブロムヘキシン、ゾルミトリプタン及びそれらの塩からなる群から選択される少なくとも1種類の薬物である、上記[4]記載の組成物。
[8]前記表面被覆ポリマーが前記所定のpHにおいて単独では難水溶性である、上記[1]〜[7]のいずれか1つに記載の組成物。
[9]前記表面被覆ポリマーがキトサン、ポリアルギニン、ポリアクリル酸、ポリガンマグルタミン酸及びそれらの塩からなる群から選択される少なくとも1種類のポリマーである、上記[1]〜[8]のいずれか1つに記載の組成物。
[10]前記表面被覆ポリマーが粘膜付着性であり、且つ/又は経粘膜吸収促進因子として機能する、上記[1]〜[9]のいずれか1つに記載の組成物。
[11]前記微粒子がカルボキシル基又はアミノ基を含むポリマーを含む、上記[1]〜[10]のいずれか1つに記載の組成物。
[12]前記微粒子がポリ乳酸グリコール酸共重合体からなる、上記[1]〜[11]のいずれか1つに記載の組成物。
[13]前記所定のpHにおける前記複合体の平均粒径が10nm以上50μm以下である、上記[1]〜[12]のいずれか1つに記載の組成物。
[14]上記[8]記載の組成物の製造方法であって、
(a)前記表面被覆ポリマーが易水溶性であるpHで、前記薬物、前記微粒子及び前記表面被覆ポリマーを混合し、
(b)該混合液のpHを前記所定のpHに調整することを特徴とする、方法。
[15]上記[1]〜[13]のいずれか1つに記載の組成物の製造方法であって、
(a)前記薬物と前記表面被覆ポリマーとが同符号の電荷を持つようなpH条件下で、前記薬物、前記表面被覆ポリマー及び前記微粒子を混合した後、
(b)該混合物のpHを前記薬物の電荷が反対符号に変化するようなpHに調整すること
を含み、ここで前記薬物は両性薬物である、方法。
[16]上記[15]記載の製造方法であって、
前記工程(a)のpH条件において、前記微粒子が、前記薬物及び前記表面被覆ポリマーの電荷と反対符号に帯電していることを特徴とする方法。
[17]上記[1]〜[13]のいずれか1つに記載の組成物の製造方法であって、
(a)前記微粒子材料の有機溶媒溶液を前記表面被覆ポリマーの水溶液中へ滴下し、
(b)該有機溶媒を揮散させた後、
(c)前記薬物を添加して混合し、
(d)該混合液のpHを前記所定のpHに調整することを特徴とする、方法。
【発明の効果】
【0009】
本発明の組成物を使用することにより、これまで注射以外の方法で投与することが難しかった低分子量薬物及びペプチドやタンパク質などの高分子薬物を効率良く経粘膜投与することが可能となる。本発明の組成物中に含有された薬物は、反対電荷の表面被覆ポリマー、及び微粒子と複合体を形成することにより、溶液製剤に含有された場合よりも高い安定性(例えば、対酵素安定性、保存安定性)を有し、尚且つ、微粒子のマトリックス中に薬物を封入した微粒子製剤と比較して、高い薬物担持能を有する。更に、微粒子表面で薬物と複合体を形成する表面被覆ポリマーの種類によって、薬物の徐放性及び速放性を達成すること、並びに経粘膜吸収性の調節をすることが可能となる。
【図面の簡単な説明】
【0010】
【図1】インスリン−キトサン複合体で表面被覆されたPLGAコア粒子からなる表面被覆微粒子(表面キャリアシステム)の作製のメカニズムを説明する図である。
【図2】PLGA微粒子表面でのインスリン−キトサン複合体の形成について試験した結果を示す図である。左のヒストグラムはpH 6.0の緩衝液中のインスリンの放出、右のヒストグラムはpH 4.5の緩衝液中のインスリンの放出を示しており、縦軸は放出されたインスリン量(μg)を表す。
【図3】インスリン−キトサン複合体で表面被覆されたPLGAコア粒子の、飛行時間型2次イオン質量分析(Time of Flight Secondary Ion Mass Spectrometry; ToF-SIMS)での分析結果を示す図である。上段は各試料(PLGA、キトサン(Chitosan)、インスリン(Insulin)及び表面被覆微粒子(Carrier System))中のm/z=33ピークの検出結果を示し(横軸:m/z;縦軸:検出強度)、下段は、観察視野における表面被覆微粒子のm/z=33ピークの分布画像(左図)と全イオンの分布画像(右図)をそれぞれ示す。
【図4】培養後にキモトリプシン溶液中に残った分解されなかったインスリンの割合(%)を示す図である。上の棒は遊離インスリン溶液の場合、下の棒はPLGA/キトサン/インスリン表面被覆微粒子の場合に対応する。
【図5】ブタ鼻粘膜に対するインスリン透過試験に用いた方法を説明する模式図である。
【図6】ブタ鼻粘膜に対するインスリン透過試験の結果を示す図である。横軸は透過試験開始後の時間(分)を示し、縦軸は所定の時間までにブタ鼻粘膜を透過したインスリン量(ng/cm2)を示す。
【図7】表面被覆微粒子或いはキトサン/インスリン混合物の粒子径を測定した結果を示す図である。(a)は表面被覆微粒子(コア粒子を有する;PLGA100/キトサン/インスリン=1250/900/200 μg/ml)の粒子径分布、(b)はキトサン/インスリン混合物(コア粒子を持たない;キトサン/インスリン=900/200 μg/ml)の粒子径分布を示す。横軸は粒子の半径(nm)を示し、縦軸は強度を示す。
【図8】表面被覆微粒子(PLGA100/キトサン/インスリン=250/180/40 μg/ml)の凍結乾燥前及び凍結乾燥再懸濁後の粒子径を測定した結果を示す図である。(a)は凍結乾燥前の表面被覆微粒子の粒子径分布、(b)は凍結乾燥再懸濁後の表面被覆微粒子の粒子径分布を示す。横軸は粒子の半径(nm)を示し、縦軸は強度を示す。
【図9】実施例7の試料及び比較対照の試料をラットに経鼻投与した後の血中グルコースレベルを示す。結果は試料投与前グルコースレベルを100%として、相対的なグルコースレベル低下を%表示した。平均値±標準誤差で示した。
【図10】実施例8の試料を用いたインビトロ放出試験(N=3)(5mM MES等張バッファ(pH6)、37℃)の結果を示す。
【発明を実施するための形態】
【0011】
本発明は、(a)所定のpHで正若しくは負の電荷を有する薬物、(b)医薬上許容される微粒子及び(c)該pHで帯電しうる医薬上許容される表面被覆ポリマーを含む経粘膜投与用医薬組成物を提供する。該組成物において、該表面被覆ポリマーは該微粒子の表面を被覆しており、該薬物は該表面被覆ポリマーを介して該微粒子の表面に固定化されており、且つ、該微粒子と該表面被覆ポリマーとが非共有結合的に相互作用し、同時に該表面被覆ポリマーと該薬物とが静電相互作用することにより複合体(該複合体を以下、「表面被覆微粒子」ともいう)を形成する。
【0012】
「経粘膜投与用医薬組成物」とは、処置(treatment)及び/又は治療(therapy)を必要とする対象の粘膜に対して、塗布、スプレー、噴霧、貼付など適切な方法で投与され、薬物が粘膜組織へと、又は粘膜組織を通して循環器系或いは免疫系へと送達されることにより薬効或いはワクチン効果をもたらし得る医薬組成物を意味する。経粘膜投与用医薬組成物は、例えば、全身送達のために、粘膜を通して吸収され得る。粘膜の例としては、肺、口腔、眼、膣、胃腸管及び鼻などの粘膜が挙げられる。投与の簡便さの観点から、前記粘膜としては鼻粘膜が好ましい。
【0013】
表面被覆微粒子を構成する上記(c)の医薬上許容される表面被覆ポリマーは、上記の通り、微粒子の表面を被覆する。ここでいう「被覆」は、該表面被覆ポリマーが、該微粒子の内部ではなく、該微粒子の表面に、該微粒子と該表面被覆ポリマーとの間の非共有結合的な相互作用によって付着していることを単に意味しており、該微粒子の表面全体が該表面被覆ポリマーによって覆われることを必ずしも意味しない。
表面被覆ポリマーは、生体適合性であることが望ましい。本明細書において、用語「生体適合性」とは、ある物質及びその分解産物が、生体組織又は生体システム(例、血液循環系、神経系、免疫系など)に対して中毒的又は損傷的な影響を与えないことを意味する。生体適合性のポリマーは、ヒト又は他の動物への投与に適したものである。該表面被覆ポリマーはまた、生分解性であることがより好ましい。本明細書において、用語「生分解性」とは、ある物質が、酵素的、化学的若しくは物理的プロセスなどにより生体内で許容される時間内に分解されて、より小さな化学種を形成することを意味する。ある物質の生体適合性及び生分解性を検査する方法は、当該技術分野において周知である。
【0014】
表面被覆ポリマーは、天然ポリマーであっても、或いは合成ポリマーであっても良い。表面被覆ポリマーは、所定のpHにおいて、微粒子の表面で薬物とイオン結合するため、該所定のpHで該薬物と反対電荷を有するポリマーでなければならない。該所定のpHで同じ符号に帯電しうるポリマー同士であれば、必要に応じて2種類以上のポリマーを表面被覆ポリマーのために併用することもできる。
【0015】
本発明の医薬組成物は、例えば後述する製造法により製造され得るが、当該製造法により製造する場合、前記所定のpHにおいて単独では難水溶性であっても、表面被覆ポリマーが易水溶性であるpHで薬物及び微粒子と混合した後、該混合液のpHを前記所定のpHに調整することによって製造が可能である。従って、表面被覆ポリマーのために、前記所定のpHにおいて易水溶性であるポリマーだけでなく、前記所定のpHにおいて難水溶性であるポリマーも使用され得る。
【0016】
表面被覆ポリマーに使用され得るポリマーは、以下に限定されないが、ポリアニオン性又はポリカチオン性の多糖、ポリアミノ酸及びその他の帯電したポリマーから選択され得る。該ポリマーは、使用される薬物の種類、表面被覆ポリマーの電荷、および薬物の電荷などに応じて適宜選択される。
【0017】
本発明に使用され得るポリアニオン性の多糖とは、カルボキシル基、硫酸基又はリン酸基などの1つ以上の酸性極性基を構成単位中に有する多糖類である。そのようなポリアニオン性の多糖の例としては、以下に限定されないが、コンドロイチン硫酸、デキストラン硫酸、カルボキシメチルセルロース、アルギン酸、ペクチン、ヒアルロン酸、これらの誘導体や塩などが挙げられる。
【0018】
本発明に使用され得るポリカチオン性の多糖とは、アミノ基などの1つ以上の塩基性極性基を構成単位中に有する多糖類である。そのようなポリカチオン性の多糖の例としては、以下に限定されないが、キチン、キトサン、これらの誘導体や塩などが挙げられる。キトサンおよびキトサン誘導体は、様々な分子量及び脱アセチル化の程度のものから選択することができ、また、キトサン誘導体については、様々な置換の程度のものから選択することができる。
【0019】
本発明に使用され得るポリアニオン性のポリアミノ酸とは、等電点が生理的pHよりも酸性側にあるポリアミノ酸であり、その例としては、以下に限定されないが、ポリグルタミン酸、ポリアスパラギン酸、これらの誘導体や塩などが挙げられる。
【0020】
本発明に使用され得るポリカチオン性のポリアミノ酸とは、等電点が生理的pHよりも塩基性側にあるポリアミノ酸であり、その例としては、以下に限定されないが、ポリリジン、ポリアルギニン、これらの誘導体や塩などが挙げられる。
【0021】
表面被覆ポリマーに使用され得るポリマーとして、上記の多糖及びポリアミノ酸以外に、ポリエチレンイミンやポリアクリル酸、これらの誘導体や塩なども挙げられる。
【0022】
表面被覆ポリマーは、ポリエチレングリコール化(PEG化)及び/又はグリコシル化されていても良い。
【0023】
表面被覆ポリマーは、更に、粘膜付着性であってもよく、且つ/又は経粘膜吸収促進因子として機能しても良い。粘膜付着性ポリマーとしては、例えば、キトサン、ポリアクリル酸、アルギン酸ナトリウム、カルボキシメチルセルロースなどの他に、それらのPEG化されたポリマー類などが挙げられる。また、経粘膜吸収促進因子として機能するポリマーとしては、例えば、キトサン、ポリアクリル酸、ポリアルギニン、これらの塩や誘導体などが挙げられる。
【0024】
表面被覆ポリマーの分子量は、分解速度、機械強度、溶解度、表面被覆ポリマーと共に複合体を形成することになる薬物の種類などの要素を考慮して当業者は決定することができる。典型的には、ゲル浸透クロマトグラフィー法により測定した表面被覆ポリマーの重量平均分子量は、好ましくは1,000Da以上、より好ましくは2,000Da以上であるべきであり、また、好ましくは1,000,000Da以下、より好ましくは500,000Da以下であるべきである。従って、典型的には、表面被覆ポリマーの重量平均分子量は、好ましくは1,000−1,000,000Daであり、より好ましくは2,000−500,000Daである。例えば、キチン又はキトサンの重量平均分子量は、1,000−1,000,000Daであり得、キチン又はキトサンの脱アセチル化の程度は、20−100%であり得る。
【0025】
本発明の組成物は、表面被覆ポリマーの選択により、徐放性又は速放性の組成物として薬物の放出速度の制御、及び薬物の経粘膜吸収性の調節が可能となる。当業者は、該組成物が所望の薬動力学的特性を持つよう、表面被覆ポリマーを適切に選択することができる。
【0026】
本発明の組成物に使用される薬物は、その使用目的に応じて選択される。本発明の組成物において、薬物は、微粒子の表面で適切な表面被覆ポリマーとイオン結合するため、該組成物において使用され得る薬物は、所定のpHで正若しくは負に帯電する(即ち、表面被覆ポリマーと反対の電荷を有する)薬物でなければならない。かかる条件を満足する限り、任意の薬物が本発明の組成物で使用され得る。なお該所定のpHで同じ符号に帯電しうる薬物同士であれば、必要に応じて2種類以上の薬物を併用することもできる。
【0027】
該薬物は、以下に限定されないが、ペプチド、タンパク質、DNA、RNA、siRNA、多糖、抗体、抗原、低分子量化合物などであり得る。本発明の医薬組成物は、例えば後述する製造法により製造され得るが、当該製造法により製造する場合、薬物が表面被覆ポリマーと静電相互作用しない帯電条件でそれらをよく混合した後にpH変化により薬物と表面被覆ポリマーとの間にイオン結合を生じさせることが可能であるため、pHの変化により調製過程で帯電(符号又は強さ)を変化させることができる薬物が特に好適であり得る。そのような薬物としては、pHによって正にも負にも帯電するペプチドやタンパク質などの両性薬物、調製過程と組成物中に存在する場合とで帯電の強さが大きく変化するような酸解離定数(pKa)又は塩基解離定数(pKb)を有する薬物、及び水に溶解して、pHにあまり依存しない電荷を有し得る塩酸塩、硫酸塩、酢酸塩など塩形態の低分子量薬物が挙げられる。
【0028】
薬物としては、以下に限定されないが、例えば、抗高血圧剤、抗低血圧剤、鎮痛剤、抗精神病剤、抗鬱剤、抗躁剤、抗不安剤、鎮静剤、催眠剤、抗癲癇剤、オピオイドアゴニスト、喘息治療剤、麻酔剤、抗不整脈剤、関節炎治療剤、鎮痙剤、ACEインヒビター、鬱血除去剤、抗生物質、抗狭心症剤、利尿剤、抗パーキンソン病剤、気管支拡張剤、分娩促進剤、抗利尿剤、抗高脂血症剤、免疫抑制剤、免疫調節剤、制吐剤、抗感染症剤、抗新生物剤、抗真菌剤、抗ウイルス剤、抗糖尿病剤、抗アレルギー剤、解熱剤、抗腫瘍剤、抗痛風剤、抗ヒスタミン剤、止痒剤、骨調節剤、心血管剤、コレステロール低下剤、抗マラリア剤、喫煙を中止するための薬剤、鎮咳剤、去痰剤、粘液溶解剤、鼻詰り用薬剤、ドパミン作動剤、消化管用薬剤、筋弛緩剤、神経筋遮断剤、副交感神経作動剤、プロスタグランジン、興奮薬、食欲抑制剤、甲状腺剤又は抗甲状腺剤、ホルモン、抗偏頭痛剤、抗肥満剤、及び抗炎症剤などが挙げられる。薬物は予防的ワクチン、免疫療法、抗体治療、遺伝子治療、遺伝子発現抑制などを目的とした様々なペプチド、タンパク質、多糖、抗原、抗体、DNA、RNA、siRNA、低分子量薬物などから選択しても良い。
【0029】
薬物の具体例としては、以下に限定されないが、インスリン、グルカゴン、ロイプロリド、成長ホルモン、副甲状腺ホルモン、カルシトニン、血管内皮成長因子、エリスロポエチン、ヘパリン、シクロスポリン、オキシトシン、チロシン、エンケファリン、チロトロピン放出ホルモン、卵胞刺激ホルモン、黄体形成ホルモン、バソプレシン、バソプレシン類似体、カタラーゼ、スーパーオキシドジスムターゼ、インターロイキンII、インターフェロン、コロニー刺激因子、腫瘍壊死因子、メラニン細胞刺激ホルモン、グルカゴン様ペプチド−1、グルカゴン様ペプチド−2、カタカルシン、コレシステキニン−12、コレシステキニン−8、エキセンディン、ゴナドリベリン関連ペプチド、インスリン様タンパク質、ロイシン−エンケファリン、メチオニン−エンケファリン、ロイモルフィン、ニューロフィジン、コペプチン、ニューロペプチドY、ニューロペプチドAF、PACAP関連ペプチド、膵臓ホルモン、ペプチドYY、ウロテンシン、腸ペプチド、副腎皮質刺激ペプチド、上皮成長因子、プロラクチン、黄体形成ホルモン放出ホルモン(LHRH)、LHRHアゴニスト、成長ホルモン放出因子、ソマトスタチン、ガストリン、テトラガストリン、ペンタガストリン、エンドルフィン、アンジオテンシン、顆粒球コロニー刺激因子、顆粒球−マクロファージコロニー刺激因子、ヘパリナーゼ、インフルエンザワクチン用抗原、破傷風毒素、癌ワクチン用ペプチド、β−アミロイド、免疫グロブリン、肝硬変治療用siRNA、癌治療用siRNA、ブロムヘキシン、グラニセトロン、ゾルミトリプタン、スマトリプタンなどの低分子量薬物やそれらの薬学的に許容される塩などが挙げられる。
【0030】
上述したように、薬物及び表面被覆ポリマーは、微粒子表面において静電相互作用により結合して薬物−表面被覆ポリマー複合体を形成する。該複合体を形成する薬物と表面被覆ポリマーとの組み合わせについて、所定のpHにおいて正電荷を有する薬物と負電荷を有する表面被覆ポリマーとの組み合わせであっても良く、或いは所定のpHにおいて負電荷を有する薬物と正電荷を有する表面被覆ポリマーとの組み合わせであっても良い。
【0031】
前記医薬上許容される微粒子(本明細書において、用語「微粒子」は、上記(b)の微粒子を意味しているが、「表面被覆微粒子」との区別を特に明確にするために、該微粒子を「コア微粒子」と呼ぶこともある)は、生体適合性のポリマーからなることが望ましい。該ポリマーは、生分解性であっても非生分解性であっても良いが、生体に対する安全性の観点から、生分解性のものが好ましい。また、該ポリマーは、天然ポリマーであっても、或いは合成ポリマーであっても良い。
【0032】
微粒子のために使用され得る生体適合性且つ生分解性のポリマーとしては、以下に限定されないが、例えば、ポリエチレングリコール(PEG)、ポリ乳酸(PLA)、ポリグリコール酸(PGA)、ポリ乳酸グリコール酸共重合体(PLGA)、PEGとPLGAとのブロック共重合体(PEG−PLGA)、ポリ無水化合物、ポリ(ε−カプロラクトン)、ポリヒドロキシブチラート、ポリアミノ酸、ポリオルトエステル、ポリホスホエステル、ポリジアキサノン、ポリエステルアミド、ポリホスファゼン、ポリシアノアクリラート、キトサン、キトサン誘導体、スターチ、スターチ誘導体、アルブミン、フィブリン、フィブリノゲン、セルロース、コラーゲン、ヒアルロン酸、これらの物質の混合物及びコポリマー類などが挙げられる。
【0033】
微粒子のために使用され得る生体適合性且つ非生分解性のポリマーとしては、以下に限定されないが、例えば、ポリアクリラート、ポリアクリラートエステル、ポロキサマー、テトロニクス、ポリエチレン、ポリメチルメタクリラート、ポリメチルメタクリラートエステル、ポリスチレン、エチレンビニルアセタート、アシル化酢酸セルロース、ポリウレタン、塩化ポリビニル、これらの物質の混合物及びコポリマー類などが挙げられる。
【0034】
上記微粒子は、親水性であっても疎水性であっても良い。水系での微粒子の調製過程及び表面被覆微粒子の調製過程において、微粒子の形状及び大きさを維持するのが容易という観点から、疎水性の微粒子が好ましい。好適な例として、カルボキシル基又は第1級、第2級若しくは第3級アミノ基を有する疎水性ポリマーが微粒子に使用され得る。
【0035】
該微粒子のためのポリマーの分子量は、分解速度、機械強度、溶解度などの要素を考慮して当業者によって決定され得る。典型的には、ゲル浸透クロマトグラフィー法により測定した該ポリマーの重量平均分子量として、1,000Da(ダルトン)以上が好ましく、2,000Da以上がより好ましく、また、1,000,000Da以下が好ましく、500,000Da以下がより好ましい。従って、典型的には、該ポリマーの重量平均分子量として、1,000−1,000,000Daが好ましく、2,000−500,000Daがより好ましい。
【0036】
コア微粒子の大きさとしては、1nm以上、好ましくは5nm以上、より好ましくは10nm以上の平均粒径を持ち、尚且つ50μm以下、好ましくは20μm以下、より好ましくは10μm以下の平均粒径を持つ粒子が挙げられる。
なお、ここでいう粒径は、微粒子を前記“所定のpH”の水溶液中に分散させて測定した値である。粒径は、粒子径測定装置により測定し、粒子の形状を球であると仮定して算出した直径のことである。粒子径測定装置及び平均粒径の算出方法は、粒子サイズに応じて使い分ける。即ち、動的光散乱測定装置において測定可能な粒子サイズの場合(通常7μm以下)は、動的光散乱測定装置において測定し、散乱強度分布から求めた流体力学的直径の平均値を平均粒径として採用する。動的光散乱測定装置により測定できない大きい粒子サイズの場合(通常7μmよりも大きい場合)は、レーザー回折式粒度分布測定装置を用いて測定し、頻度分布を算術平均して求めた平均径を平均粒径として採用する。
ここで、コア微粒子の平均粒径が、例えば、10nm以上であるとは、上記の動的光散乱測定装置の散乱強度分布の各粒子径ピークの割合(全ピーク累積散乱強度に対する各粒子径ピークの累積散乱強度の割合)、又は上記のレーザー回折式粒度分布測定装置における頻度分布の各粒子径ピークの割合(全ピーク累積頻度に対する各粒子径ピークの累積頻度の割合)において、粒子径ピークの平均粒径のうちの10%以上、好ましくは20%以上、より好ましくは30%以上、更に好ましくは40%以上、特に好ましくは50%以上が10nm以上であることをいう。
【0037】
本発明の組成物を粘膜に投与したとき、表面被覆微粒子は、粘膜表面に達するか又は粘膜組織に取り込まれ得、そこで薬物を放出する。薬物はその後、血流に輸送される。微粒子が十分小さい場合(例えば、微粒子の粒径が20nm以下)、細胞間隙を通過して血流に到達する可能性がある。或いは、微粒子は、鼻又は腸などの一部の粘膜においてM細胞又はM様細胞に取り込まれて免疫系或いはリンパ系に輸送され得る。
【0038】
上記微粒子の製造は、文献に記載された様々な方法により行うことができる。該文献としては、例えば、Champion JA. et al., Proc. Natl. Acad. Sci. USA, Vol.104, pp.11901-4 (2007); Chattopadhyay P. et al., Adv. Drug Deliv. Rev., Vol.59, pp.443-53 (2007); Zhou WY et al., J. Mater. Sci. Mater. Med., Vol.19, pp.103-110 (2008); Schaffazick SR et al., Pharmazie, Vol.62, pp.354-60 (2007); Almeida AJ et al., Adv. Drug Deliv. Rev., Vol.59, pp.478-90 (2007); Muller, R.H., “Colloidal Carriers for Controlled Drug Delivery and Targeting: Modification, Characterization and In vivo Distribution”, CRC Press (1991); Jorg Kreuter (ed.), Colloidal Drug Delivery Systems, Marcel Dekker (1994)などが挙げられる。該微粒子の製造に使用され得る方法としては、例えば、ナノ沈殿(nanoprecipitation)、相分離、乳化、自己集合、高圧均質化、複合体形成(complexation)、イオンゲル化などが挙げられる。
【0039】
上述した通り、微粒子は、表面被覆ポリマーと非共有結合的に相互作用して表面被覆微粒子を生成するものである必要がある。ここでいう非共有結合的な相互作用とは、静電相互作用、疎水的相互作用、ファンデルワールス相互作用、水素結合など、共有結合によらない相互作用のことである。このうち例えば静電相互作用を利用する場合には、該微粒子は、静電相互作用するため、所定のpHで表面被覆ポリマーと逆の符号の電荷を有するポリマーでなければならない。従って、組成物に使用する薬物、表面被覆ポリマー及び微粒子のためのポリマーの選択に関して、所定のpHにおいて正の電荷を有する薬物、負の電荷を有する表面被覆ポリマー、及び正の電荷を有する微粒子のためのポリマーの組み合わせ、或いは所定のpHにおいて負の電荷を有する薬物、正の電荷を有する表面被覆ポリマー、及び負の電荷を有する微粒子のためのポリマーの組み合わせとなる。そのような組み合わせとなるために薬物、表面被覆ポリマー及び微粒子のためのポリマーが満たすべき等電点(pI)又は酸解離定数(pKa)若しくは塩基解離定数(pKb)に関する条件はそれぞれ以下の通りである:薬物及び微粒子のためのポリマーのpI又はpKa若しくは(14−pKb)の値が製造後の組成物のpHよりも高く、且つ表面被覆ポリマーのpI又はpKa若しくは(14−pKb)の値が製造後の組成物のpHよりも低い;或いは薬物及び微粒子のためのポリマーのpI又はpKa若しくは(14−pKb)の値が製造後の組成物のpHよりも低く、且つ表面被覆ポリマーのpI又はpKa若しくは(14−pKb)の値が製造後の組成物のpHよりも高い。各化合物について、等電点及び/又は酸解離定数を決定することは当業者の通常の技術的範囲内である。或いは、水中で溶解して帯電しうる塩酸塩、硫酸塩、酢酸塩など塩形態の低分子量薬物の場合は、該組成物は、水中での薬物の帯電とは逆の符号の電荷を有する表面被覆ポリマー、水中での薬物の帯電と同じ符号の電荷を有する微粒子とを、帯電した薬物と組み合わせて調製してもよい。
【0040】
前記所定のpH、即ち製造後の組成物のpHは、局所刺激を回避するために、投与部位の生理的pHに設定することが望ましい。上述したように、本発明の組成物は、肺、口腔、眼、膣、腸及び鼻などの粘膜に投与され得るが、これらの様々の粘膜の生理的pHは様々である。例えば、胃腸管の生理的pHは、その長さに沿って、胃における約pH1から、結腸におけるpH8まで上昇する;口腔は、6.8付近のpHを有する;鼻の流体のpHは、約pH5.5〜6.5の範囲にわたる;膣のpHは、4.5付近である。例えば、本発明の組成物が鼻粘膜投与用である場合、その好ましいpH値として約6.0が挙げられる。
【0041】
上述した通り、本発明の組成物における薬物として、インスリンが使用され得る。該組成物のpHが6.0である場合、インスリンの等電点は約pH5.3であるため、インスリンは組成物中で負に帯電している。従って、表面被覆ポリマーとしてはpH6.0で正電荷を有するポリマーである必要がある。非共有結合性の相互作用として静電相互作用を利用して表面被覆ポリマーと微粒子を相互作用させる場合は、微粒子としてはpH6.0で負電荷を有するポリマーからなる微粒子を好適な一例として用いることができる。かかる表面被覆ポリマーとしてはキトサンが挙げられ、微粒子のためのポリマーとしてはポリ乳酸グリコール酸共重合体(PLGA)が挙げられる。
上述した通り、本発明の組成物における薬物として、ブロムヘキシン、ゾルミトリプタン及びそれらの塩が使用され得る。該組成物のpHが6.0〜7.0である場合、該薬物は水中で正符号に帯電するため、表面被覆ポリマーとしては該pHで負電荷を有するポリマーである必要がある。非共有結合性の相互作用として静電相互作用を利用して表面被覆ポリマーと微粒子を相互作用させる場合は、微粒子としては該pHで正電荷を有するポリマーからなる微粒子を好適な一例として用いることができる。かかる表面被覆ポリマーとしてはポリアクリル酸及び又はポリガンマグルタミン酸又はそれらの塩が例として挙げられ、微粒子としてはキトサン微粒子やアミノ修飾したポリスチレン粒子が一例として挙げられる。
【0042】
表面被覆微粒子の大きさとしては、10nm以上、好ましくは20nm以上、より好ましくは40nm以上の平均粒径を持ち、尚且つ50μm以下、好ましくは20μm、より好ましくは10μm以下の平均粒径を持つ粒子が挙げられる。
なお、ここでいう粒径は、表面被覆微粒子を前記“所定のpH”の水溶液中に分散させて測定した値である。具体的には、表面被覆微粒子が懸濁液の場合は、懸濁液と同じpH(前記所定のpH)の水溶液で測定に適した濃度に希釈して測定する。表面被覆微粒子の剤型が、ドライパウダー、シート剤など、懸濁液以外の剤型に加工されていて、そのままでは粒径を測定できない場合は、水、又は適当なpH緩衝液を加えて、前記“所定のpH”の懸濁液に調製した後に粒径を測定する。粒径は、粒子径測定装置により測定し、粒子の形状を球であると仮定して算出した直径のことである。粒子径測定装置及び平均粒径の算出方法は、粒子サイズに応じて使い分ける。即ち、動的光散乱測定装置において測定可能な粒子サイズの場合(通常7μm以下)は、動的光散乱測定装置において測定し、散乱強度分布から求めた流体力学的直径の平均値を平均粒径として採用する。動的光散乱測定装置により測定できない大きい粒子サイズの場合(通常7μmよりも大きい場合)は、レーザー回折式粒度分布測定装置を用いて測定し、頻度分布を算術平均して求めた平均径を平均粒径として採用する。
ここで、表面被覆微粒子の平均粒径が、例えば、10nm以上であるとは、上記の動的光散乱測定装置の散乱強度分布の各粒子径ピークの割合(全ピーク累積散乱強度に対する各粒子径ピークの累積散乱強度の割合)、又は上記のレーザー回折式粒度分布測定装置における頻度分布の各粒子径ピークの割合(全ピーク累積頻度に対する各粒子径ピークの累積頻度の割合)において、粒子径ピークの平均粒子径のうちの10%以上、好ましくは20%以上、より好ましくは30%以上、更に好ましくは40%以上、特に好ましくは50%以上が10nm以上であることをいう。
【0043】
本発明の組成物における表面被覆微粒子は、コアとしての微粒子が存在することにより、単純に表面被覆ポリマーと薬物とを混合して複合体を形成させた場合と比べて、粒子径が単分散である。従って、本発明のような表面被覆微粒子の構成をとることによって、特性の揃った製剤の調製が容易となる。かかる特徴も本発明の利点である。
【0044】
本発明の組成物は、表面被覆微粒子が、標的粘膜部位に直接到達できる製剤として送達される必要がある。該製剤の例としては、経肺投与剤、経口投与剤、口腔内投与剤、眼内投与剤、膣内投与剤、鼻腔内投与剤、坐剤などが挙げられる。
【0045】
経肺投与剤としては、肺用吸入器により肺胞に送達される吸入剤が好ましい。
【0046】
経口投与剤としては、通常の経口投与製剤、例えば錠剤、顆粒剤、細粒剤、カプセル剤などであるが、小腸内で薬物が放出されるように工夫された剤型、例えば腸溶性錠剤、腸溶性顆粒剤、腸溶性カプセル剤、腸溶性細粒剤が好ましい。
【0047】
口腔内投与剤、眼内投与剤及び鼻腔内投与剤としては、口腔錠剤、口腔スプレー、点眼剤、点鼻剤、エアゾール、軟膏剤、ゲル剤、クリーム剤、液剤、懸濁液剤、ローション剤、ドライパウダー剤、シート剤、貼付剤などが挙げられる。
【0048】
膣内投与剤及び坐剤としては、軟膏剤、ゲル剤、クリーム剤、液剤、懸濁液剤、ローション剤、ドライパウダー剤、シート剤、カプセル剤などが挙げられる。
【0049】
上記の剤形に製する方法としては、当該分野で一般的に用いられている公知の製造方法を適用することができる。また、上記の剤形に製する場合には、必要に応じて、特定の剤形に製する際に通常用いられる賦形剤、結合剤、崩壊剤、滑沢剤などの担体、甘味剤、界面活性剤、懸濁化剤、乳化剤、着色剤、保存剤、安定剤などの各種製剤添加物などを適宜、適量含有させて製造することができる。また、本発明の組成物は、懸濁液を凍結乾燥するなどしてドライパウダー化した状態で保存し、使用時に該ドライパウダーに水を加えて再懸濁することもできる。かかる方法を採ることにより、組成物の保存安定性を向上させるために薬物、微粒子のためのポリマー、及び/又は表面被覆ポリマーの加水分解を回避することが可能となる。
【0050】
本発明の組成物において、微粒子のためのポリマー、表面被覆ポリマー及び薬物の好適な組成比は、使用する微粒子、表面被覆ポリマー及び薬物によって変動するため一概には言えないが、例えば、微粒子のためのポリマーとしてポリ乳酸グリコール酸共重合体(PLGA)、表面被覆ポリマーとしてキトサン、薬物としてインスリンを使用する場合、組成物中の重量比として、PLGA:キトサン:インスリン=1:0.1〜100:0.01〜100であり得る。
【0051】
本発明の医薬組成物は、安定かつ低毒性で安全に使用することができる。その投与頻度及び1回の投与量は、使用する薬物、患者の状態や体重、投与経路、治療方針などによって異なり一概には言えないが、例えば、糖尿病などの患者に薬物としてインスリンを使用した本発明の組成物を経鼻投与する場合には、一つの治療方針として、成人(体重約60kg)に対して有効成分(インスリン)として約2mgから約6mgを毎食の前に投与することができる。
【0052】
本発明はまた、上述した医薬組成物を製造する方法を提供する。本発明の方法は、適当なpHの溶液中で薬物と表面被覆ポリマーと微粒子とを混合し、任意でpHを変化させて、薬物と表面被覆ポリマーとの間で静電相互作用させ、表面被覆ポリマーと微粒子との間で非共有結合的に相互作用させることを含む。該方法は、加熱処理などが必要なく、簡便である。
【0053】
該方法においては、事前に、薬物、表面被覆ポリマー及び微粒子のためのポリマーの組み合わせ並びに本発明の組成物のpH(所定のpH)を決定しておく。これらの要素の決定は、本発明の組成物の説明において上述した通りに行うことができる。また、通常、薬物、表面被覆ポリマー及び微粒子を混合する前に、上述した方法により微粒子を作製しておく。次いで、薬物と表面被覆ポリマーと微粒子との混合、及び任意でpHの調整を行い、本発明の表面被覆微粒子を製造する。混合及び任意でのpH調整は、以下のa)〜c)からなる群から選択されるいずれか1つ:
a)薬物と表面被覆ポリマーとを、それらの複合体が形成されないpHを持つ溶液中で混合し、次いで微粒子を該溶液中に加えた後、溶液のpHを変化させて薬物と表面被覆ポリマーとの複合体形成及び微粒子表面への薬物−表面被覆ポリマー複合体の固定化を促進すること;
b)薬物と表面被覆ポリマーとを、それらの複合体が形成されるpHを持つ溶液中で混合した後、微粒子を該溶液中に加えて薬物−表面被覆ポリマー複合体を微粒子表面に固定化させること;
c)微粒子と表面被覆ポリマーとを、溶液中で混合して表面被覆ポリマーを微粒子の表面に固定化させ、次いで薬物を該溶液に加えた後、溶液のpHを調整して微粒子表面に固定化された表面被覆ポリマーと、薬物との複合体形成を促進すること、
を含む。
【0054】
特に、表面被覆ポリマーが所定のpHにおいて単独では難水溶性である場合、まず、(a)前記表面被覆ポリマーが易水溶性であるpHで、前記薬物、前記微粒子及び前記表面被覆ポリマーを混合し、次いで、(b)該混合液のpHを前記所定のpHに調整することが望ましい。
【0055】
また、本発明の医薬組成物に用いる薬物として、pHに応じて帯電の符号が変わる、いわゆる両性薬物を用いる場合には、本発明の医薬組成物の製造方法として、次の方法を用いることもできる。即ち、第一段階として、薬物の電荷と表面被覆ポリマーの電荷とが同符号となるようなpH条件下で、薬物、表面被覆ポリマー及び微粒子を混合することで、薬物及び表面被覆ポリマーの両方を微粒子の表面に静電相互作用などの非共有結合性相互作用により引き寄せることができる。その上で、第二段階として、該混合物のpHを薬物の電荷が反対符号に変化するようなpHに調整すれば、微粒子表面において、そこに集められた薬物と表面被覆ポリマーとの間で効率よく静電相互作用による結合を形成させ、本発明の医薬組成物を効率よく製造することができる。この製造方法は、副生成物としての遊離の薬物−表面被覆ポリマー複合体(微粒子表面に固定化されていない)の生成を抑制できるので有用である。
この方法をインスリン(薬物;等電点:約5.3)、キトサン(表面被覆ポリマー)、PLGA微粒子(微粒子)を例にして説明すると、インスリンが正に帯電する“等電点未満のpH”(例えば、pH4.5など)でインスリン、キトサン、PLGA微粒子を混合した後に、インスリンが負に帯電する“等電点より高いpH”(pH6.0など)にpHを調整するという方法である。pH4.5及びpH6.0のいずれにおいてもキトサンは正帯電、PLGA粒子は負帯電であるが、インスリンの電荷はpH4.5で正帯電、pH6.0で負帯電になるので、pH6.0において、互いにイオン結合したキトサン及びインスリンがPLGA微粒子の表面に固定化された本発明の組成物の一つを製造することもできる。なお、図1は、この実施態様を説明している。
即ち、本発明は、本発明の組成物の製造方法であって、
(a)薬物と表面被覆ポリマーとが同符号の電荷を持つようなpH条件下で、薬物、表面被覆ポリマー及び微粒子を混合した後、
(b)該混合物のpHを薬物の電荷が反対符号に変化するようなpHに調整すること
を含み、ここで該薬物は両性薬物である方法を提供する。
【0056】
或いは、また別の製造方法として、特にコア粒子の粒子径を小さくすることで産物としての表面被覆微粒子の粒子径を小さく制御したい場合に有用な方法として、次の方法も利用できる。この方法では、予め微粒子を調製しておくのではなく、微粒子の調製と、微粒子及び表面被覆ポリマーとの静電相互作用とを同時に開始させる。具体的には、この方法では、上記したような微粒子の材料(例、ポリマー)を含む適当な有機溶媒溶液(例、アセトン溶液など)を表面被覆ポリマー水溶液中へ滴下し、攪拌などにより溶液中から有機溶媒を揮散させることで、コア粒子としての微粒子生成と、表面被覆ポリマーによる微粒子被覆とを開始させる。そこに薬物を添加し、混合した上で、任意でpHを変化させて、薬物と表面被覆ポリマーとの間並びに表面被覆ポリマーと微粒子の間で静電相互作用を促進させることで、表面被覆微粒子を製造する。
即ち、本発明は、本発明の組成物の製造方法であって、
(a)前記微粒子材料の有機溶媒溶液を前記表面被覆ポリマーの水溶液中へ滴下し、
(b)該有機溶媒を揮散させた後、
(c)前記薬物を添加して混合し、
(d)該混合液のpHを前記所定のpHに調整することを特徴とする方法を提供する。
【0057】
以下、実施例及び試験例を挙げて本発明を更に詳細に説明するが、本発明は以下の実施例などによって制限されない。
【実施例】
【0058】
作製例1:様々な粒子径のポリ乳酸グリコール酸共重合体(PLGA)微粒子の作製
ラクチド:グリコリド比が50:50であるPLGA(RESOMER RG 502H,Bohringer Ingelheim)を使用して、PLGA微粒子を作製した。HPLCグレードのアセトンにPLGAを必要濃度で溶解した。PLGA/アセトン溶液を純水に1:3の割合となるまで、常時撹拌しながら滴下で加えた。アセトンが完全に蒸発するまで(約4時間)混合液を撹拌した。
その結果得られた微粒子の粒子径の分布を動的光散乱測定装置(DLS 802,Viscotek)により測定した。PLGAの濃度と得られた粒子の直径との関係を表1に示す。最初の有機溶媒溶液中のポリマー濃度を低くすることで、より微小な粒子が容易且つ再現性良く得られることが明らかとなった。
【0059】
【表1】
【0060】
実施例1:2種類の緩衝系における薬物−表面被覆ポリマー複合体で表面被覆された微粒子システムの作製
タンパク質薬物としてインスリン(pI 約5.3)を使用し、正に帯電した表面被覆ポリマーとしてキトサンを使用した。
ウシインスリン(Sigma,160μg/ml)の0.5mMクエン酸溶液(pH 4.5)1.5mlをキトサン(Bioneer 143kDa,0.72mg/ml)の0.5mMクエン酸溶液(pH 4.5)1.5mlに加え、最低30分間室温で放置した。作製例1に記載したようにして作製したPLGA微粒子(直径 約100nm;以下、「PLGA 100」ともいう)の0.5mMクエン酸溶液(pH 4.5;PLGA微粒子濃度:500μg/ml)の懸濁液3mlをキトサン/インスリン溶液に加え、最低1時間室温で放置した。NaOH(0.1−2.5N)を加えてpHを6.0に上昇させ、塩及び添加物を加えて、懸濁液の溶媒組成を表2又は表3に記載の緩衝液と同じ組成とした。
【0061】
【表2】
【0062】
【表3】
【0063】
表面被覆微粒子の粒子径及びゼータ電位をそれぞれDLS 802(Viscotek)及びZeta sizer 2000(Malvern)により測定した(表4)。
表4から分かるように、2種類の緩衝系について同様且つ好適な粒子径及びゼータ電位が得られた。また、2種類の表面被覆微粒子は、被覆していない微粒子の約2倍の粒子径で、ゼータ電位は大きな正の電位であった。両方の表面被覆微粒子はコロイド懸濁液として安定であることが分かった。
【0064】
【表4】
【0065】
実施例2:2種類のインスリン濃度での20mM MES緩衝系における薬物−表面被覆ポリマー複合体で表面被覆された微粒子システムの作製
タンパク質薬物としてインスリン(pI 約5.3)を使用し、正に帯電した表面被覆ポリマーとしてキトサンを使用した。
ウシインスリン(Sigma,160μg/ml又は800μg/ml)の0.5mMクエン酸溶液(pH 4.5)2mlをキトサン(Bioneer 143kDa,0.72mg/ml又は3.6mg/ml)の0.5mMクエン酸溶液(pH 4.5)2mlに加え、最低30分間室温で放置した。作製例1に記載したようにして作製したPLGA微粒子(直径 約100nm)の0.5mMクエン酸溶液(pH 4.5;PLGA微粒子濃度:0.5mg/ml又は2.5mg/ml)の懸濁液4mlをキトサン/インスリン溶液に加え、最低1時間室温で放置した。NaOH(0.1〜2.5N)を加えてpHを6.0に上昇させ、塩及び添加物を加えて、懸濁液の溶媒組成を表5に記載の緩衝液と同じ組成とした。
【0066】
【表5】
【0067】
表面被覆微粒子の粒子径及びゼータ電位をそれぞれDLS 802(Viscotek)及びZeta sizer 2000(Malvern)により測定した。両方の表面被覆微粒子サンプル共に好適な粒子径及びゼータ電位(表6)を示し、粒子径は被覆していない粒子(表4)の約2倍、ゼータ電位は大きな正の電位であった。また、両方の表面被覆微粒子はコロイド懸濁液として安定であった。
【0068】
【表6】
【0069】
実施例3:表面被覆ポリマーとしてポリ−L−アルギニンを使用した表面被覆PLGA微粒子システムの作製
タンパク質薬物としてインスリン(pI 約5.3)を使用し、正に帯電した表面被覆ポリマーとしてポリ−L−アルギニンを使用した。
ウシインスリン(Sigma,40μg/ml)の0.5mMクエン酸溶液(pH 6.0)3mlをポリ−L−アルギニン(MW 125kDa,Sigma;2.88mg/ml)の0.5mMクエン酸溶液(pH 6.0)3mlに加え、最低30分間室温で放置した。作製例1に記載したようにして作製したPLGA微粒子(直径 約100nm)の0.5mMクエン酸溶液(pH 6.0;PLGA微粒子濃度:250μg/ml)の懸濁液6mlをポリ−L−アルギニン/インスリン溶液に加え、最低1時間室温で放置した。塩及び添加物を加えて、懸濁液の溶媒組成を表5に記載の緩衝液と同じ組成とした。表面被覆微粒子の粒子径及びゼータ電位をそれぞれDLS 802(Viscotek)及びZeta sizer 2000(Malvern)により測定した。該粒子の平均径は285.9±90.6nm、ゼータ電位は+48.3±0.9mVであった。
【0070】
実施例4:表面被覆ポリマーとしてキトサン及びポリ−L−アルギニンを使用した表面被覆PLGA微粒子システムの作製
タンパク質薬物としてインスリン(pI 約5.3)を使用し、正に帯電した表面被覆ポリマーとしてキトサン及びポリ−L−アルギニンを使用した。
ウシインスリン(Sigma,800μg/ml)の0.5mMクエン酸溶液(pH 4.5)2mlをキトサン(MW 143kDa,0.36mg/ml)及びポリ−L−アルギニン(MW 125kDa,Sigma;1.8mg/ml)の0.5mMクエン酸溶液(pH 4.5)の混合液2mlに加え、最低30分間室温で放置した。作製例1に記載したようにして作製したPLGA微粒子(直径 約100nm)の0.5mMクエン酸溶液(pH 4.5;PLGA微粒子濃度:2.5mg/ml)の懸濁液4mlをキトサン/ポリ−L−アルギニン/インスリン溶液に加え、最低1時間室温で放置した。塩及び添加物を加えて、懸濁液の溶媒組成を表7に記載の緩衝液と同じ組成とした。表面被覆微粒子の粒子径及びゼータ電位をそれぞれDLS 802(Viscotek)及びZeta sizer 2000(Malvern)により測定した。該粒子の平均径は336.1±20.8nm、ゼータ電位は+40.3±3.4mVであった。
【0071】
【表7】
【0072】
実施例5:他の作製法を用いたインスリン/キトサン表面被覆PLGA微粒子システムの作製
0.1% w/vポリスチレン微粒子(MolecularProbe,カルボキシル化FluoSpheres)の0.5mMクエン酸溶液(pH 4.5)3mlをキトサン(Bioneer 143kDa,180μg/ml)の0.5mMクエン酸溶液(pH 4.5)3mlに加え、最低1時間室温で放置した。ウシインスリン(Sigma,20μg/ml)の0.5mMクエン酸溶液(pH 4.5)6mlをポリスチレンとキトサンとの上記混合液に加え、最低1時間室温で放置した。NaOH(0.1−2.5N)を加えてpHを6.0に上昇させ、塩及び添加物を加えて、懸濁液の溶媒組成を表5に記載の緩衝液と同じ組成とした。
表面被覆微粒子の粒子径及びゼータ電位をそれぞれDLS 802(Viscotek)及びZeta sizer 2000(Malvern)により測定した。該粒子の平均径は302.0±68.6nm、ゼータ電位は+27.9±1.7mVであった。また、ポリスチレンコア粒子の直径は196.7±27.5nmであったが、これはポリスチレンコア粒子の周囲に厚さ約50nmの層があることを示している。
【0073】
実施例6:他の作製法を用いたインスリン/キトサン表面被覆PLGA微粒子システムの作製
ラクチド:グリコリド比が50:50であるPLGA(RESOMER RG 502H,Bohringer Ingelheim)のアセトン溶液(PLGA0.01%(w/v):約20nmのPLGA粒子を調製することができる濃度)1.8mlをキトサン(Bioneer 73kDa,0.25mg/ml)の0.5mMクエン酸溶液(pH 4.5)6mlに加え、アセトンが完全に蒸発するまで室温で放置した。このPLGA/キトサン懸濁液3mlにウシインスリン(Sigma,160μg/ml)の0.5mMクエン酸溶液(pH 4.5)3mlを加え、最低1時間室温で放置した。NaOH(0.1−2.5N)を加えてpHを6.0に上昇させ、塩及び添加物を加えて、懸濁液の溶媒組成を表2に記載の緩衝液と同じ組成とした。表面被覆微粒子の粒子径をDLS 802(Viscotek)により測定した。該粒子の平均径は146.1±35.8nmであった。該粒子はコロイド懸濁液として安定であった。
【0074】
実施例7:動物実験用のインスリン/キトサン表面被覆PLGA微粒子システムの作製
タンパク質薬物としてインスリン(pI 約5.3)を使用し、正に帯電した表面被覆ポリマーとしてキトサンを使用した。動物実験用に高濃度試料(インスリン濃度6mg/mL)として調製した。
キトサン水溶液(甲陽ケミカル製、コーヨーキトサンFL−80、1.44mg/mL、pH4.5)15mlにウシインスリン水溶液(Sigma,320μg/ml、pH 4.5)15mlと50mMクエン酸水溶液0.02mlを加えて、pHを4.5±0.1に調整し約1時間静置した。その後PLGA微粒子懸濁液(粒子径約100nm、PLGA微粒子濃度1mg/mL、pH4.5)30ml、50mMクエン酸水溶液0.02mlを加え、pHを4.5に調整した。1時間静置した後にpHを6.0に調整し、マルトース(0.421g)を加えて溶解、pH6であることを確認した。この調製液を液体窒素で凍結後、凍結乾燥を行った。凍結乾燥品は凍結乾燥前の溶液の1/15ボリュームの蒸留水で再分散させた。再懸濁液を遠心分離し(19400×G、3時間、4℃)4/5容量の上清を除くことで粒子分画を濃縮して、動物実験用のサンプル(インスリン濃度6mg/mL)を得た。
表面被覆微粒子の粒子径及びゼータ電位をZeta sizer Nano(Malvern)により測定した。該粒子の平均径は252nm、ゼータ電位は+10.6mVであった。該粒子はコロイド懸濁液として安定であった。また、本実施例の表面被覆微粒子に結合しているインスリンの割合を後述する方法にて測定したところ担持率は93%であった。
【0075】
実施例8:放出試験用のインスリン/キトサン表面被覆PLGA微粒子システムの作製
タンパク質薬物としてインスリン(pI 約5.3)を使用し、正に帯電した表面被覆ポリマーとしてキトサンを使用した。
キトサン水溶液(甲陽ケミカル製、コーヨーキトサンFL−80、1.44mg/mL、pH4.5)15mlにウシインスリン水溶液(Sigma,320μg/ml、pH 4.5)15mlと50mMクエン酸水溶液0.02mlを加えて、pHを4.5±0.1に調整し約1時間静置した。その後PLGA微粒子懸濁液(粒子径約100nm、PLGA微粒子濃度1mg/mL、pH4.5)30ml、50mMクエン酸水溶液0.02mlを加え、pHを4.5に調整した。1時間静置した後にpHを6.0に調整し、マルトース(0.421g)を加えて溶解、pH6であることを確認した。
【0076】
実施例9:表面被覆ポリマーとしてカチオン系キトサン誘導体を使用した表面被覆PLGA微粒子システム(pH8)の作製
タンパク質薬物としてインスリン(pI 約5.3)を使用し、正に帯電した表面被覆ポリマーとしてカチオン系キトサン誘導体を使用した。
ウシインスリン(Sigma,0.32mg/ml)の0.5mMクエン酸溶液(pH 4.5)2mlをカチオン系キトサン誘導体(大日精化製、カチオン系キトサン誘導体水溶液,1.44mg/ml)の0.5mMクエン酸溶液(pH 4.5)2mlに加え、最低30分間室温で放置した。作製例1に記載したようにして作製したPLGA微粒子(直径 約100nm)の0.5mMクエン酸溶液(pH 4.5;PLGA微粒子濃度:1.0mg/ml)の懸濁液4mlをカチオン系キトサン誘導体/インスリン溶液に加え、最低1時間室温で放置した。10%w/vの濃度となるようにマルトースを添加し、NaOHを添加してpHを8に調整した。
表面被覆微粒子の粒子径をZeta sizer Nano(Malvern)により測定した。該粒子の平均径は230nmであった。該粒子はコロイド懸濁液として安定であった。また、本実施例の表面被覆微粒子に結合しているインスリンの割合を後述する方法にて測定したところ担持率は74%w/wであった。
【0077】
実施例10:表面被覆ポリマーとしてカチオン系キトサン誘導体を使用した表面被覆PLGA微粒子システム(pH7)の作製
タンパク質薬物としてインスリン(pI 約5.3)を使用し、正に帯電した表面被覆ポリマーとしてカチオン系キトサン誘導体を使用した。
ウシインスリン(Sigma,0.32mg/ml)の0.5mMクエン酸溶液(pH 7.0)2mlをカチオン系キトサン誘導体(大日精化製、カチオン系キトサン誘導体水溶液,1.44mg/ml)の0.5mMクエン酸溶液(pH 7.0)2mlに加え、最低30分間室温で放置した。作製例1に記載したようにして作製したPLGA微粒子(直径 約100nm)の0.5mMクエン酸溶液(pH 7.0;PLGA微粒子濃度:1.0mg/ml)の懸濁液4mlをカチオン系キトサン誘導体/インスリン溶液に加え、最低1時間室温で放置した。10%w/vの濃度となるようにマルトースを添加し、pH7であることを確認した。
表面被覆微粒子の粒子径及びゼータ電位をZeta sizer Nano(Malvern)により測定した。該粒子の平均径は234nm、ゼータ電位は+11.3mVであった。該粒子はコロイド懸濁液として安定であった。また、本実施例の表面被覆微粒子に結合しているインスリンの量を後述する方法にて測定したところ担持率は65%w/wであった。
【0078】
実施例11:正帯電薬物と負帯電表面被覆ポリマーを使用した表面被覆微粒子システムの作製
正帯電の低分子量薬物としてゾルミトリプタン(pKa=9.5)を使用し、負に帯電した表面被覆ポリマーとしてポリアクリル酸を用いた。
ポリアクリル酸水溶液(和光純薬製、平均分子量25万、1.44mg/ml)2mlにトリメチルアミン修飾ポリスチレン粒子(micromer NR3+ 100nm、コアフロント株式会社)の水懸濁液(1mg/ml)4mlを加え軽く混ぜ、約1時間後にゾルミトリプタン水溶液(640μg/ml)2mlを加え、軽く混ぜた後にpHを6.0に調整した。
表面被覆微粒子の粒子径及びゼータ電位をZeta sizer Nano(Malvern)により測定した。該粒子の平均径は330nm、ゼータ電位は−75mVであった。該粒子はコロイド懸濁液として安定であった。また、本実施例の表面被覆微粒子に結合している薬物の量を後述する方法に準じて(HPLC条件は別条件)測定したところ担持率は14%w/wであった。
【0079】
実施例12:正帯電薬物と負帯電表面被覆ポリマーを使用した表面被覆微粒子システムの作製
正帯電の低分子量薬物としてブロムヘキシン塩酸塩を使用し、負に帯電した表面被覆ポリマーとしてポリアクリル酸ナトリウムを用いた。
ポリアクリル酸ナトリウム水溶液(重合度2,700〜7,500,和光純薬製、1.44mg/ml)2mlにブロムヘキシン塩酸塩水溶液(640μg/ml)1mlと蒸留水1mlを加えて軽く混ぜ、約1時間後にトリメチルアミン修飾ポリスチレン粒子(micromer NR3+ 100nm、コアフロント株式会社)の水懸濁液(1mg/ml)4mlを加え、軽く混ぜた後にpHを6に調整した。
表面被覆微粒子の粒子径及びゼータ電位をZeta sizer Nano(Malvern)により測定した。該粒子の平均径は160nm、ゼータ電位は−57mVであった。該粒子はコロイド懸濁液として安定であった。また、本実施例の表面被覆微粒子に結合している薬物の量を後述する方法に準じて(HPLC条件は別条件)測定したところ担持率は88%w/wであった。
【0080】
実施例13:正帯電薬物と負帯電表面被覆ポリマーを使用した表面被覆微粒子システムの作製
正帯電の低分子量薬物としてブロムヘキシン塩酸塩を使用し、負に帯電した表面被覆ポリマーとしてポリガンマグルタミン酸ナトリウムを用いた。
ポリガンマグルタミン酸ナトリウム水溶液(平均分子量20万〜50万,和光純薬製、1.44mg/ml)2mlにブロムヘキシン塩酸塩水溶液(640μg/ml)1mlと蒸留水1mlを加えて軽く混ぜ、約1時間後にトリメチルアミン修飾ポリスチレン粒子(micromer NR3+ 100nm、コアフロント株式会社)の水懸濁液(1mg/ml)4mlを加え、軽く混ぜた後にpHを6に調整した。
表面被覆微粒子の粒子径及びゼータ電位をZeta sizer Nano(Malvern)により測定した。該粒子の平均径は203nm、ゼータ電位は−63mVであった。該粒子はコロイド懸濁液として安定であった。また、本実施例の表面被覆微粒子に結合している薬物の量を後述する方法に準じて(HPLC条件は別条件)測定したところ担持率は32%w/wであった。
【0081】
試験例1:微粒子表面での薬物と表面被覆ポリマーとの相互作用の評価
表5に記載の緩衝液中のインスリン/キトサン表面被覆PLGA微粒子の懸濁液(PLGA 100/キトサン/インスリン=500/360/80μg/ml)1mlをマイクロチューブに入れ、18000rpm(23900×g)で60分間遠心分離した。沈殿物を表5に記載の緩衝液で洗った。更に、沈殿物にpH 4.5又はpH6.0の解離用緩衝液を加え、それぞれのチューブを2時間室温で振盪した。懸濁液を同条件で15分間再び遠心分離し、上清を回収した。その後、各上清緩衝液中のインスリン濃度をELISAで定量した。
キトサン及びインスリンが共に正に帯電するため静電力による両者の引力相互作用が弱いpH 4.5の緩衝液の場合、両者が逆電荷を持つため静電引力が強くなるpH6.0の場合と比べてインスリン量が顕著に多かった(図2)。なお、遊離のインスリン及びキトサンは、解離の実験をする前の上記洗浄操作により除去されている。これらの結果は、微粒子表面にキトサン/インスリン複合体が存在することを示している。
【0082】
実施例1に記載の方法にて調製した、表2に記載の緩衝液中のインスリン/キトサン表面被覆PLGA微粒子の懸濁液(PLGA 100/キトサン/インスリン=250/180/40μg/ml)1mlをマイクロチューブに入れ、18000rpm(23900×g)で60分間遠心分離した。沈殿物を表2に記載の緩衝液で洗った。洗浄後の沈殿物をバッファーにて再懸濁したものを、ガラス板上に塗り広げ、約48時間自然乾燥させて、TOF−SIMS用試料とした。比較対照として、インスリン、キトサン、又は作製例1の方法で調製したPLGA 100をガラス板上に塗り広げた試料も用意した。
これらの各試料について、TOF−SIMS IV装置(ION−TOF GmbH)にて、インスリンのシステイン残基のSHに相当するピーク(m/z=33)の有無を測定した。その結果、PLGAとキトサンからはm/z=33のピークは有意なレベルで検出されなかったが、上記の洗浄済み表面被覆微粒子の試料とインスリンの試料とでは強いピークが検出された。各試料についての測定結果を図3上段に示す。
また、上記の洗浄済み表面被覆微粒子については、観察視野中のm/z=33の分布(図3下段左)とトータルイオンの分布(図3下段右)の様子も確認し、両者の分布パターンが一致すること、すなわち、粒子からm/z=33が検出されていることも確認した。
以上の特異性に関する測定結果と、粒子分布画像の結果は、洗浄済み表面被覆微粒子の表面にインスリンが存在することを示している。
【0083】
試験例2:薬物担持率及び担持能の評価
試験に供したサンプルは以下のようにして作製した。
【0084】
[PLGA/キトサン/インスリン(1000/720/160μg/ml)表面被覆微粒子(表8に記載の緩衝液中)]
タンパク質薬物としてインスリン(pI 約5.3)を使用し、正に帯電した表面被覆ポリマーとしてキトサンを使用した。
ウシインスリン(Sigma,0.64mg/ml)の0.5mMクエン酸溶液(pH 4.5)4.5mlをキトサン(Bioneer 143kDa,2.88mg/ml)の0.5mMクエン酸溶液(pH 4.5)4.5mlに加え、最低30分間室温で放置した。作製例1に記載したようにして作製したPLGA微粒子(直径 約100nm)の0.5mMクエン酸溶液(pH 4.5;PLGA微粒子濃度:2.0mg/ml)の懸濁液9mlをキトサン/インスリン溶液に加え、最低1時間室温で放置した。NaOH(0.1−2.5N)を加えてpHを6.0に上昇させ、グルコースを加えて、懸濁液の溶媒組成を表8に記載の緩衝液と同じ組成とした。
【0085】
【表8】
【0086】
表面被覆微粒子の粒子径及びゼータ電位をそれぞれDLS 802(Viscotek)及びZeta sizer 2000(Malvern)により測定した。該粒子の平均径は248.8±94.2nm、ゼータ電位は+8.7±0.5mVであった。
【0087】
[PLGA/キトサン/インスリン(250/90/40μg/ml)表面被覆微粒子(表8に記載の緩衝液中)]
タンパク質薬物としてインスリン(pI 約5.3)を使用し、正に帯電した表面被覆ポリマーとしてキトサンを使用した。
ウシインスリン(Sigma,160μg/ml)の0.5mMクエン酸溶液(pH 4.5)3mlをキトサン(Bioneer 143kDa,0.36mg/ml)の0.5mMクエン酸溶液(pH 4.5)3mlに加え、最低30分間室温で放置した。作製例1に記載したようにして作製したPLGA微粒子(直径 約100nm)の0.5mMクエン酸溶液(pH 4.5;PLGA微粒子濃度:500μg/ml)の懸濁液6mlをキトサン/インスリン溶液に加え、最低1時間室温で放置した。NaOH(0.1−2.5N)を加えてpHを6.0に上昇させ、グルコースを加えて、懸濁液の溶媒組成を表8に記載の緩衝液と同じ組成とした。
【0088】
表面被覆微粒子の粒子径及びゼータ電位をそれぞれDLS 802(Viscotek)及びZeta sizer 2000(Malvern)により測定した。該粒子の平均径は253.7±31.3nm、ゼータ電位は+6.4±1.8mVであった。
【0089】
[PLGA/ポリ−L−アルギニン/インスリン(125/720/40μg/ml)表面被覆微粒子(表5に記載の緩衝液中)]
タンパク質薬物としてインスリン(pI 約5.3)を使用し、正に帯電した表面被覆ポリマーとしてポリ−L−アルギニンを使用した。
ウシインスリン(Sigma,160μg/ml)の0.5mMクエン酸溶液(pH 6.0)3mlをポリ−L−アルギニン(MW 125kDa,Sigma;2.88mg/ml)の0.5mMクエン酸溶液(pH 6.0)3mlに加え、最低30分間室温で放置した。作製例1に記載したようにして作製したPLGA微粒子(直径 約100nm)の0.5mMクエン酸溶液(pH 6.0;PLGA微粒子濃度:250μg/ml)の懸濁液6mlをポリ−L−アルギニン/インスリン溶液に加え、最低1時間室温で放置した。塩及び添加物を加えて、懸濁液の溶媒組成を表5に記載の緩衝液と同じ組成とした。表面被覆微粒子の粒子径及びゼータ電位をそれぞれDLS 802(Viscotek)及びZeta sizer 2000(Malvern)により測定した。該粒子の平均径は497.4±141.9nm、ゼータ電位は+44.3±2.1mVであった。
【0090】
インスリンの担持率及び担持能を以下に記載する方法により測定した。
[結合したインスリンの解析法]
上記各サンプル0.5ml又は1mlを1.5mlマイクロチューブに入れ、遠心分離した(15000rpm(21900×g),180分,4℃)。上清を回収し、上清中のインスリン濃度をインスリンELISAキット(Mercodia Bovine insulin ELISA)及び希釈緩衝液(Mercodia Diabetes sample buffer)又はHPLCを使用して測定した(当該インスリン濃度をAとする)。コントロールとして、1.5mlマイクロチューブ中の上記各サンプル0.5ml又は1mlを4℃で180分間静置し、全サンプル中のインスリン濃度を同様にして測定した(当該インスリン濃度をBとする)。
担持率及び担持能は以下のようにして計算される:
担持率(%)=100×((Bの平均値)−A)/(Bの平均値));
担持能(%)=コア粒子総質量に対する結合したインスリンの質量
=100×((Bの平均値)−A)/コア粒子総質量
(コア粒子総質量=粒子数×4/3×3.14×(コア粒子の半径)3×密度)
【0091】
上記方法によるインスリン担持の解析結果を表9に示す。
【0092】
【表9】
【0093】
試験例3:微粒子表面で複合体化したインスリンの安定性評価
試験例2で使用した、表8に記載の緩衝液中のPLGA/キトサン/インスリン(1000/720/160μg/ml)表面被覆微粒子を本試験のサンプルとして使用した。また、コントロールとして、表8に記載の緩衝液と同じ溶媒組成のインスリン溶液(pH 6;インスリン濃度:160μg/ml)を使用した。
【0094】
酵素反応させるサンプルの調製は以下のようにして行った。
ウシの膵臓由来のα−キモトリプシン(Fluka Biochemika;code No.27270)を濃度が40μg/mlになるまで5mM MES緩衝液(pH 6.0)に溶解した。サンプル1mlを37℃で15分間温めた後、同様にして温めておいた5mM MES緩衝液(pH 6.0)0.6ml及び同様にして温めておいたα−キモトリプシンの5mM MES緩衝溶液(pH 6.0;α−キモトリプシン濃度:40μg/ml)0.4mlを加えた。混合物を37℃で30分間振盪培養した後、氷で冷却した氷酢酸1mlを加えて酵素反応を停止させた。次いでミキサーで1分間混合し、1時間以上室温で放置した後、フィルター径0.1μmのフィルターでろ過して、HPLC分析用試料を得た。
【0095】
一方、酵素を含有しない比較対照サンプルの調製は以下のようにして行った。
サンプル1mlを5mM MES緩衝液(pH 6.0)1ml及び氷酢酸1mlと混合した。次いで、混合物をミキサーで1分間混合し、1時間以上室温で放置した後、フィルター径0.1μmのフィルターでろ過して、HPLC分析用試料を得た。
【0096】
また、インスリン定量の検量線用標準サンプルの調製は以下のようにして行った。
インスリン溶液(40−160μg/ml)1mlを5mM MES緩衝液(pH 6.0)1ml及び氷酢酸1mlと混合し、ミキサーで混合した。該混合物をHPLC分析の検量線用の標準サンプルとして使用した。
【0097】
サンプルのHPLC分析は以下の条件で行った:
C18カラム(Inertsil ODS−2,5μm,250mm×4.6mm);
移動相A:0.1% TFA水溶液,移動相B:0.1% TFA CH3CN溶液;
グラジエント条件(移動相B濃度):0分時:30%,10分時:40%,11分時:30%,16分時:30%;
カラムオーブン温度:40℃,流速:1.0ml/分,注入量:20μl,検出:UV275nm
【0098】
上記条件による分析の結果、酵素反応後に残存していたインスリン量の割合は、PLGA/キトサン/インスリンについて83.9%±2.3%、インスリン溶液について61.8%±2.0%であった。即ち、遊離のインスリン溶液での38.2%に対して、表面被覆複合体化したインスリンの16.1%のみが、本実験の時間中に分解された(図4)。
この結果は、酵素反応に対するインスリン安定性が大きく向上したことを示している。
【0099】
試験例4:表面被覆微粒子を使用したブタ鼻粘膜に対するインスリン透過試験
タンパク質薬物としてインスリン(pI 約5.3)を使用し、正に帯電した表面被覆ポリマーとしてキトサン及びポリ−L−アルギニンを使用した。表面被覆微粒子の作製は、実施例4に記載したのと同様にして行った。また、コントロールとして、表7に記載の緩衝液と同じ溶媒組成のインスリン溶液(pH 6;インスリン濃度:200μg/ml)を使用した。鼻の呼吸部粘膜組織をブタの鼻腔(呼吸部)から摘出した。摘出した組織は、水平拡散チャンバに配置するまで、実施例4で使用したものと同じ組成の緩衝液(酸素処理して冷却したもの)の中で保存した。当該組織を適当な大きさに切断し、図5に示すように水平拡散チャンバ中のドナーセルとレセプターセルとの間に配置した(粘膜の有効面積:0.79cm2)。新鮮な(上記の)緩衝液(酸素処理して冷却したもの)をドナーセル及びレセプターセルに注入し、両セルを29±1℃に加温した循環水で30分間温めて、組織を平衡化させた。透過試験の間も、ドナーセル及びレセプターセルに注入した緩衝液を酸素処理し、また、29±1℃に加温した循環水で両セルを温めた。透過試験の前後で、組織が生存していること、及び損傷を受けていないことをAlamar Blue Assay及び組織のTEER値の測定により確かめた。
ドナーセル中の全ての緩衝液を同量の各サンプルに置換することにより透過試験を開始した。選択した時点において、200μlのサンプルをレセプターセルから採取し、同量の新鮮な緩衝液(酸素処理後、29±1℃に加温しておいたもの)で置換した。レセプターセルから採取したサンプルをインスリンELISAキット及び希釈緩衝液を使用して分析した。分析結果を図6に示す。図6は、コントロールのインスリン溶液を投与した場合と比較して、キトサン/ポリ−L−アルギニン/インスリン表面被覆PLGA微粒子を投与した場合により多くのインスリンが摘出鼻呼吸部粘膜組織を透過したことを示している。
【0100】
試験例5:表面被覆微粒子の粒子径分布の測定
タンパク質薬物としてインスリン(pI 約5.3)を使用し、正に帯電した表面被覆ポリマーとしてキトサンを使用した。
ウシインスリン(Sigma,0.8mg/ml)の0.5mMクエン酸溶液(pH 4.5)1.5mlをキトサン(Bioneer 143kDa,3.6mg/ml)の0.5mMクエン酸溶液(pH 4.5)1.5mlに加え、最低30分間室温で放置した。作製例1に記載したようにして作製したPLGA微粒子(直径 約100nm;以下、「PLGA 100」ともいう)の0.5mMクエン酸溶液(pH 4.5;PLGA微粒子濃度:2.5mg/ml)の懸濁液3ml、或いは単なる0.5mMクエン酸溶液(pH 4.5)をキトサン/インスリン溶液に加え、最低1時間室温で放置した。NaOH(0.1−2.5N)を加えてpHを6.0に上昇させ、塩及び添加物を加えて、懸濁液の溶媒組成を表2に記載の緩衝液と同じ組成とした。表面被覆微粒子或いはキトサン/インスリン混合物の粒子径をDLS 802(Viscotek)により測定した。
その結果を図7に示す。図7(a)は表面被覆微粒子、図7(b)はキトサン/インスリン混合物についての粒子径分布を示しており、また、そのそれぞれについて、ピーク領域における強度のパーセンテージ、粒子径及び標準偏差を表10及び11に示す。
【0101】
【表10】
【0102】
【表11】
【0103】
PLGA粒子を添加した場合は、単分散の粒子径が得られたのに対して、PLGA粒子を添加しなかった場合は、粒子径の大きなものも含む多分散の粒子径分布となった。この結果より、コア粒子の存在により、粒子径の揃った表面被覆微粒子が得られることがわかる。
【0104】
試験例6:表面被覆微粒子懸濁液をドライパウダーとして製剤化する可能性の確認
実施例1の方法にて、[PLGA100/キトサン/インスリン(250/180/40μg/ml)表面被覆微粒子(表2に記載の緩衝液中)]を調製した。この懸濁液を凍結乾燥し、凍結乾燥前と同量の水で再懸濁させた。凍結乾燥前の粒子径、及び凍結乾燥再懸濁後の粒子径をDLS 802(Viscotek)を用いて測定した。粒子径の測定結果を図8に示す。図8(a)は凍結乾燥前、図8(b)は凍結乾燥再懸濁後の粒子径分布を示しており、また、そのそれぞれについて、ピーク領域における強度のパーセンテージ、粒子径及び標準偏差を表12及び13に示す。
【0105】
【表12】
【0106】
【表13】
【0107】
凍結乾燥前の粒子径は183.0±21.2nm、凍結乾燥再懸濁後の粒子径は229.4±47.9nmであり、凍結乾燥と再懸濁の操作により主成分の粒子径は有意に変化せず、目だった凝集物も生成していないことがわかる。この結果より、本発明の表面被覆微粒子は懸濁液としてだけでなく、ドライパウダーなど他の剤型としても使用可能であることがわかる。
【0108】
試験例7:表面被覆微粒子を使用したラット鼻腔に対するインビボ投与試験
試験にはラット(系統:SDラット、7週齢、オス、ブリーダー:日本エスエルシー)を用いた。ラットは試験前日の夕方から絶食させた。試験は筋肉注射及び腹腔点滴による麻酔の下で行った。血中グルコース濃度が安定化してから、鼻腔へのサンプル投与を行い、投与してから10、20、30、60、90、120分後に尾静脈からの採血を行い、血中グルコース検出キット(テルモ製、メディセーフミニ)を用いて血中グルコースレベルを測定した。
試料として実施例7に記載の表面被覆微粒子を20μl/体重300g(400μgインスリン/kg体重)の量だけラット鼻腔に投与した。また、コントロールとして、いずれも実施例7と同じ溶媒組成である、クエン酸0.5mMマルトース10%w/v溶液(pH6)(バッファー液)、インスリン溶液(pH6;インスリン濃度:6mg/ml、クエン酸0.5mMマルトース10%w/v)、キトサン/インスリン混液(pH6;キトサン濃度:27mg/ml、インスリン濃度:6mg/ml、クエン酸0.5mMマルトース10%w/v)も同様にラット鼻腔に投与した。
試験結果を図9に示す。図9は、コントロールのバッファー液やインスリン溶液を投与した場合と比較して、大きく血中グルコースレベルが低下していること、また、キトサン/インスリン混液と比べても初期の血中グルコースレベルの低下が早いことを示しており、いずれも本発明の実施例がペプチド薬物の経粘膜デリバリーにおいて優れた促進効果を有することを示している。
【0109】
試験例8:表面被覆微粒子を使用したインビトロ放出試験
実施例8の試料0.5mLをエッペンドルフチューブ1.5mLに添加し遠心分離(13600rpm(19400xG)、3hr、4℃)して沈殿物を得た。放出試験液として5mM MES 152mM NaCl水溶液(pH 6.0、生理的等張イオン強度154mM)を準備した。
該沈殿物に放出試験液0.5mLを添加し、ピペッティングとミキサーにより再分散させたものを10mL容ガラスバイアルに移した。エッペンドルフチューブを新たな放出試験液0.5mLで洗い、洗い液も上記ガラスバイアルに移した(懸濁液合計1mL)。懸濁液入りガラスバイアルを37℃、75rpmにて振盪することで放出試験を行った。所定時間(0、0.5、3時間)後、懸濁液を0.1μmフィルター(Sartorius)にてろ過し、ろ液を得た。ろ液とその1/2容量の5%リン酸をミキサーを用いて混合し、放出インスリン量分析用のHPLC試料とした。
また、放出前のインスリン含量を定量する目的で、該沈殿物に蒸留水1mLと5%リン酸0.5mLを加えてミキサー混合し、4℃にて一夜放置した。再度ミキサー混合した後に0.1μmフィルター(Sartorius)でろ過して、該沈殿物中インスリン量定量用のHPLC試料とした。
前記のインスリンHPLC分析条件を用いてインスリン定量を行った。
放出率(%)=100×(放出液中のインスリン量/放出前沈殿中のインスリン量)
放出試験の結果を図10に示す。該沈殿物に含まれるほぼ全てのインスリンが放出試験の3時間以内に放出された。
【産業上の利用可能性】
【0110】
本発明の組成物を使用することにより、これまで注射以外の方法で投与することが難しかった低分子量薬物及びペプチドやタンパク質などの高分子薬物を効率良く経粘膜投与することが可能となる。本発明の組成物中に含有された薬物は、表面被覆ポリマー及び微粒子と複合体を形成することにより、溶液製剤に含有された場合よりも高い安定性(例えば、対酵素安定性、保存安定性)を有し、尚且つ、微粒子のマトリックス中に薬物を封入した微粒子製剤と比較して、高い薬物担持能を有する。更に、微粒子表面で薬物と複合体を形成する表面被覆ポリマーの種類によって、薬物が徐放性及び速放性を示す表面被覆微粒子を作製すること、並びに経粘膜吸収性の調節をすることが可能となる。
【特許請求の範囲】
【請求項1】
(a)所定のpHで正若しくは負の電荷を有する薬物、(b)医薬上許容される微粒子及び(c)該pHで帯電しうる医薬上許容される表面被覆ポリマーを含む経粘膜投与用医薬組成物であって、該表面被覆ポリマーは該微粒子の表面を被覆しており、該薬物は該表面被覆ポリマーを介して該微粒子の表面に固定化されており、且つ、該微粒子と該表面被覆ポリマーとが非共有結合的に相互作用し、同時に該表面被覆ポリマーと該薬物とが静電相互作用することにより複合体を形成することを特徴とする、組成物。
【請求項2】
前記微粒子と前記表面被覆ポリマーとの非共有結合的な相互作用が静電相互作用であることを特徴とする、請求項1記載の組成物。
【請求項3】
所定のpHが投与部位の生理的pHである、請求項1又は2記載の組成物。
【請求項4】
前記薬物がペプチド、タンパク質、DNA、RNA、siRNA、多糖、抗原及び低分子量薬物からなる群より選択される、請求項1〜3のいずれか1項記載の組成物。
【請求項5】
前記薬物が薬効或いはワクチン効果をもたらしうる薬物である、請求項1〜4のいずれか1項記載の組成物。
【請求項6】
前記薬物がインスリンである、請求項4記載の組成物。
【請求項7】
前記薬物がブロムヘキシン、ゾルミトリプタン及びそれらの塩からなる群から選択される少なくとも1種類の薬物である、請求項4記載の組成物。
【請求項8】
前記表面被覆ポリマーが前記所定のpHにおいて単独では難水溶性である、請求項1〜7のいずれか1項記載の組成物。
【請求項9】
前記表面被覆ポリマーがキトサン、ポリアルギニン、ポリアクリル酸、ポリガンマグルタミン酸、及びそれらの塩からなる群から選択される少なくとも1種類のポリマーである、請求項1〜8のいずれか1項記載の組成物。
【請求項10】
前記表面被覆ポリマーが粘膜付着性であり、且つ/又は経粘膜吸収促進因子として機能する、請求項1〜9のいずれか1項記載の組成物。
【請求項11】
前記微粒子がカルボキシル基又はアミノ基を含むポリマーを含む、請求項1〜10のいずれか1項記載の組成物。
【請求項12】
前記微粒子がポリ乳酸グリコール酸共重合体からなる、請求項1〜11のいずれか1項記載の組成物。
【請求項13】
前記所定のpHにおける前記複合体の平均粒径が10nm以上50μm以下である、請求項1〜12のいずれか1項記載の組成物。
【請求項14】
請求項8記載の組成物の製造方法であって、
(a)前記表面被覆ポリマーが易水溶性であるpHで、前記薬物、前記微粒子及び前記表面被覆ポリマーを混合し、
(b)該混合液のpHを前記所定のpHに調整することを特徴とする、方法。
【請求項15】
請求項1〜13のいずれか1項記載の組成物の製造方法であって、
(a)前記薬物と前記表面被覆ポリマーとが同符号の電荷を持つようなpH条件下で、前記薬物、前記表面被覆ポリマー及び前記微粒子を混合した後、
(b)該混合物のpHを前記薬物の電荷が反対符号に変化するようなpHに調整すること
を含み、ここで前記薬物は両性薬物である、方法。
【請求項16】
請求項15記載の製造方法であって、
前記工程(a)のpH条件において、前記微粒子が、前記薬物及び前記表面被覆ポリマーの電荷と反対符号に帯電していることを特徴とする方法。
【請求項17】
請求項1〜13のいずれか1項記載の組成物の製造方法であって、
(a)前記微粒子材料の有機溶媒溶液を前記表面被覆ポリマーの水溶液中へ滴下し、
(b)該有機溶媒を揮散させた後、
(c)前記薬物を添加して混合し、
(d)該混合液のpHを前記所定のpHに調整することを特徴とする、方法。
【請求項1】
(a)所定のpHで正若しくは負の電荷を有する薬物、(b)医薬上許容される微粒子及び(c)該pHで帯電しうる医薬上許容される表面被覆ポリマーを含む経粘膜投与用医薬組成物であって、該表面被覆ポリマーは該微粒子の表面を被覆しており、該薬物は該表面被覆ポリマーを介して該微粒子の表面に固定化されており、且つ、該微粒子と該表面被覆ポリマーとが非共有結合的に相互作用し、同時に該表面被覆ポリマーと該薬物とが静電相互作用することにより複合体を形成することを特徴とする、組成物。
【請求項2】
前記微粒子と前記表面被覆ポリマーとの非共有結合的な相互作用が静電相互作用であることを特徴とする、請求項1記載の組成物。
【請求項3】
所定のpHが投与部位の生理的pHである、請求項1又は2記載の組成物。
【請求項4】
前記薬物がペプチド、タンパク質、DNA、RNA、siRNA、多糖、抗原及び低分子量薬物からなる群より選択される、請求項1〜3のいずれか1項記載の組成物。
【請求項5】
前記薬物が薬効或いはワクチン効果をもたらしうる薬物である、請求項1〜4のいずれか1項記載の組成物。
【請求項6】
前記薬物がインスリンである、請求項4記載の組成物。
【請求項7】
前記薬物がブロムヘキシン、ゾルミトリプタン及びそれらの塩からなる群から選択される少なくとも1種類の薬物である、請求項4記載の組成物。
【請求項8】
前記表面被覆ポリマーが前記所定のpHにおいて単独では難水溶性である、請求項1〜7のいずれか1項記載の組成物。
【請求項9】
前記表面被覆ポリマーがキトサン、ポリアルギニン、ポリアクリル酸、ポリガンマグルタミン酸、及びそれらの塩からなる群から選択される少なくとも1種類のポリマーである、請求項1〜8のいずれか1項記載の組成物。
【請求項10】
前記表面被覆ポリマーが粘膜付着性であり、且つ/又は経粘膜吸収促進因子として機能する、請求項1〜9のいずれか1項記載の組成物。
【請求項11】
前記微粒子がカルボキシル基又はアミノ基を含むポリマーを含む、請求項1〜10のいずれか1項記載の組成物。
【請求項12】
前記微粒子がポリ乳酸グリコール酸共重合体からなる、請求項1〜11のいずれか1項記載の組成物。
【請求項13】
前記所定のpHにおける前記複合体の平均粒径が10nm以上50μm以下である、請求項1〜12のいずれか1項記載の組成物。
【請求項14】
請求項8記載の組成物の製造方法であって、
(a)前記表面被覆ポリマーが易水溶性であるpHで、前記薬物、前記微粒子及び前記表面被覆ポリマーを混合し、
(b)該混合液のpHを前記所定のpHに調整することを特徴とする、方法。
【請求項15】
請求項1〜13のいずれか1項記載の組成物の製造方法であって、
(a)前記薬物と前記表面被覆ポリマーとが同符号の電荷を持つようなpH条件下で、前記薬物、前記表面被覆ポリマー及び前記微粒子を混合した後、
(b)該混合物のpHを前記薬物の電荷が反対符号に変化するようなpHに調整すること
を含み、ここで前記薬物は両性薬物である、方法。
【請求項16】
請求項15記載の製造方法であって、
前記工程(a)のpH条件において、前記微粒子が、前記薬物及び前記表面被覆ポリマーの電荷と反対符号に帯電していることを特徴とする方法。
【請求項17】
請求項1〜13のいずれか1項記載の組成物の製造方法であって、
(a)前記微粒子材料の有機溶媒溶液を前記表面被覆ポリマーの水溶液中へ滴下し、
(b)該有機溶媒を揮散させた後、
(c)前記薬物を添加して混合し、
(d)該混合液のpHを前記所定のpHに調整することを特徴とする、方法。
【図1】

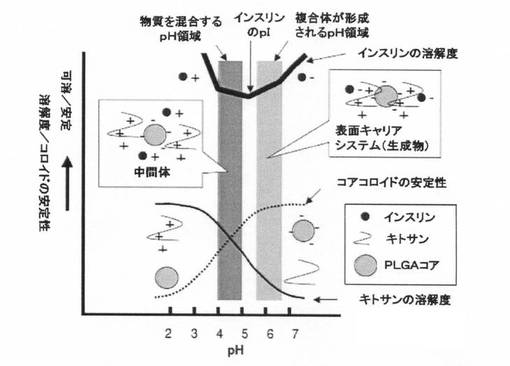
【図2】
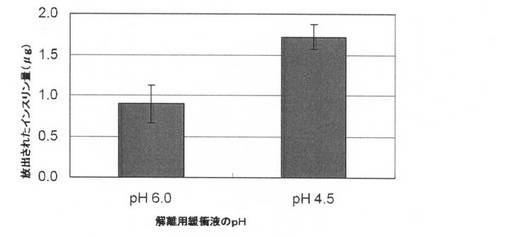
【図3】
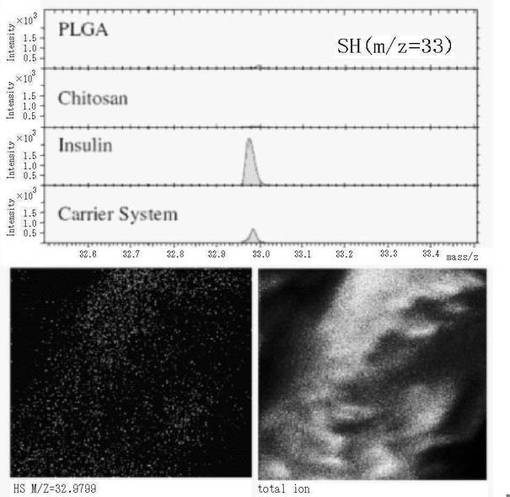
【図4】
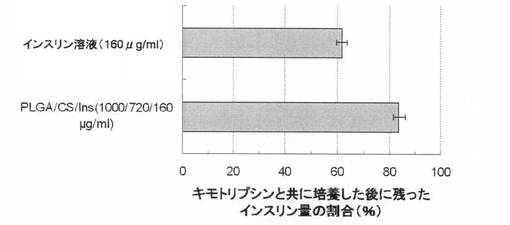
【図5】
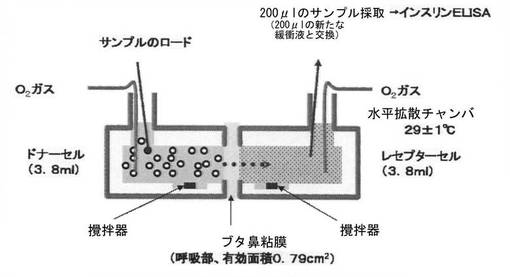
【図6】
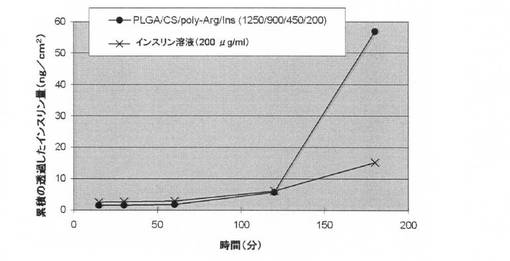
【図7】
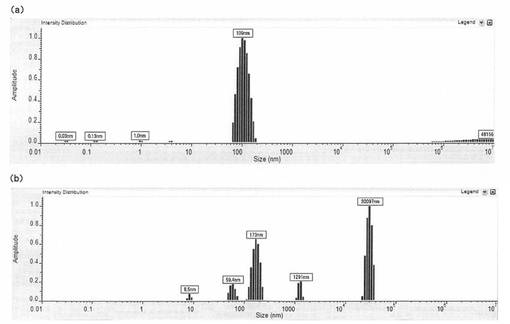
【図8】
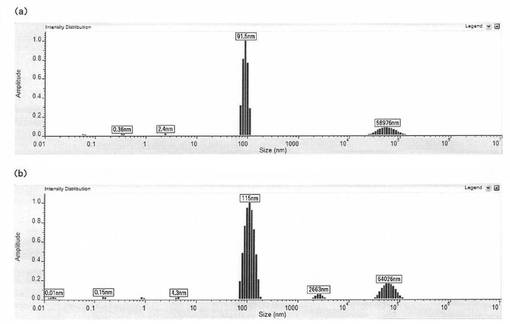
【図9】
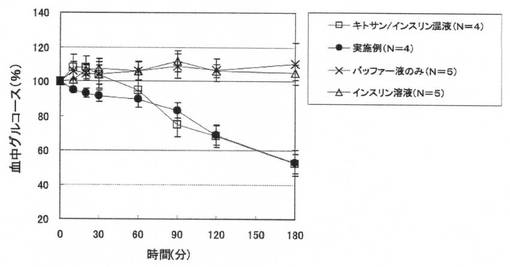
【図10】
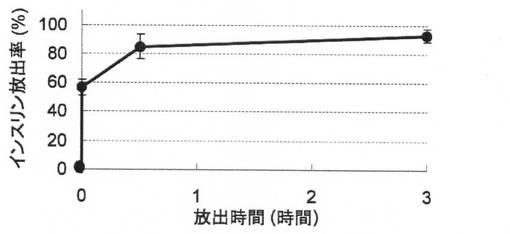
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【公開番号】特開2010−31003(P2010−31003A)
【公開日】平成22年2月12日(2010.2.12)
【国際特許分類】
【出願番号】特願2009−157253(P2009−157253)
【出願日】平成21年7月1日(2009.7.1)
【出願人】(000003964)日東電工株式会社 (5,557)
【Fターム(参考)】
【公開日】平成22年2月12日(2010.2.12)
【国際特許分類】
【出願日】平成21年7月1日(2009.7.1)
【出願人】(000003964)日東電工株式会社 (5,557)
【Fターム(参考)】
[ Back to top ]