
被験物質の有害性を評価する遺伝子改変動物
【課題】 被験物質の有害性または影響を評価するための遺伝子改変動物、および当該動物またはその一部における化学物質の影響を評価する方法を提供する。
【解決手段】 チロシン水酸化酵素遺伝子の5’上流に存在するアリルハイドロカーボン受容体結合エンハンサーと、任意のプロモーターと、レポーター遺伝子と、ポリ(A)付加シグナルとを機能的に連結したDNAを導入された遺伝子改変動物。
【解決手段】 チロシン水酸化酵素遺伝子の5’上流に存在するアリルハイドロカーボン受容体結合エンハンサーと、任意のプロモーターと、レポーター遺伝子と、ポリ(A)付加シグナルとを機能的に連結したDNAを導入された遺伝子改変動物。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、被験物質の有害性を評価するための遺伝子改変動物、および当該動物またはその一部における被験物質の作用を評価する方法に関する。
【背景技術】
【0002】
ダイオキシン類、PCB類、臭素系難燃剤および一部の農薬など、環境中の汚染化学物質がヒトの健康に及ぼす影響が懸念されている。また、産業活動や日々の生活の中で使用されている多種多様な化学物質の中にも、ヒトの健康に対する影響が未知の化学物質も多い。これらの化学物質は、肝機能、生殖機能、免疫機能および脳神経機能などの生体機能に作用して、ヒトの健康に有害な影響を及ぼすとされる。
【0003】
従って、ある特定の化学物質が作用しうる生体機能を検出および/または評価する技術は、化学物質の取り扱いや規制を判断する、あるいは化学物質に暴露されたヒトの治療をおこなう上で非常に重要となる。特に近年では、胎児および幼児期の化学物質暴露が児童に自閉症や多動性障害等の高次脳機能障害を引き起こす可能性が指摘されており、脳神経機能に対する化学物質の影響を評価するための技術開発が望まれている。
【0004】
現在、化学物質の生体機能への影響を評価する技術は、大きく分類すると、単一細胞を用いたin vitro評価技術と動物個体を用いたin vivo評価技術に分類される。
【0005】
in vitro評価技術には、マウス肝臓由来細胞Hepa1c1c7を用いて被験物質の肝機能への作用を評価する技術(特許文献1)、ヒト乳腺由来細胞MGF-7を用いて被験物質の生殖機能への作用を評価する技術(特許文献2)等がある。また、脳神経機能に限れば、マウス神経芽細胞腫Neuro2aを用いたin vitro評価方法を本出願人は既に報告している。当該方法では、当該細胞に、チロシン水酸化酵素遺伝子(ここでは、「TH遺伝子」と記す)のアリルハイドロカーボン受容体結合エンハンサー配列とTH遺伝子コアプロモーター配列で構成されるプロモーターの制御下にあるレポーター遺伝子を導入して得られた遺伝子改変細胞において、被験物質の脳神経系への影響が評価される。in vivo評価技術では、Mvh遺伝子あるいはc-mos遺伝子のプロモーターの制御下にあるレポーター遺伝子を導入された遺伝子改変マウスを用いた被験物質の生殖機能への影響を評価する方法が報告されている(特許文献3、特許文献4)。
【特許文献1】US Patent 5,854,010
【特許文献2】特開2002−253231
【特許文献3】特開2001−8577
【特許文献4】特開2001−8578
【発明の開示】
【発明が解決しようとする課題】
【0006】
上述のような状況に鑑み本発明の目的は、被験物質の生体に対する有害性または影響を評価するための遺伝子改変動物、および当該動物またはその一部における被験物質の影響を評価する方法を提供することである。詳しくは、被験物質の脳神経機能への作用を評価するために使用される遺伝子変異動物、および該遺伝子変異動物またはその一部を使用して被験物質の脳神経機能への影響を評価する方法を提供することである。
【課題を解決するための手段】
【0007】
上記目的を達成するための本発明は、
(1)チロシン水酸化酵素遺伝子の5’上流に存在するアリルハイドロカーボン受容体結合エンハンサーと、任意のプロモーターと、レポーター遺伝子と、ポリ(A)付加シグナルとを機能的に連結したDNAを導入された遺伝子改変動物;および
(2)前記(1)に記載の遺伝子改変動物またはその一部と、被験物質とを接触させることと、当該レポーター遺伝子の発現を検出することと、を具備する被験物質の有害性を判定する方法;
である。
【発明の効果】
【0008】
本発明により、被験物質の生体に対する有害性または影響を評価するための遺伝子改変動物、および当該動物またはその一部における被験物質の影響を評価する方法が提供される。詳しくは、被験物質の脳神経機能への作用を評価するために使用される遺伝子変異動物、および該遺伝子変異動物またはその一部を使用して被験物質の脳神経機能への影響を評価する方法が提供される。
【発明を実施するための最良の形態】
【0009】
現在、被験物質の脳神経機能に対する作用を評価するin vitro評価技術として、マウス神経芽細胞腫Neuro2aを用いた方法が既に報告されている。しかしながら、このような単一細胞を使用した評価技術では、動物個体レベルでの被験物質の作用を正確に評価することは難しい。即ち、これは、例えば、単一細胞では、被験物質の取り込みや分布、あるいは被験物質の代謝等が必ずしも動物個体と同等に再現されない可能性が大きいと考えられるからである。従って、被験物質の生体機能に対する作用評価には、in vitro評価技術とin vivo評価技術の併用が望ましいと考えられる。そこで、本発明者らは、脳神経機能を評価する場合に有効なin vivo評価技術が未だに確立されていないことに着目し、被験物質の脳神経機能に対する影響を評価するためのin vivo評価技術の開発を行った。
【0010】
TH遺伝子はドーパミン合成酵素をコードする遺伝子であり、化学物質暴露による該遺伝子の発現変化は脳神経系の機能に影響することが知られている。本発明者等は、TH遺伝子の発現を指標とした、脳神経系への化学物質作用のin vivo評価技術を鋭意検討した結果、本発明に従う、TH遺伝子のアリルハイドロカーボン受容体(ここでは「AhR」とも記す)結合エンハンサーと任意のプロモーターとレポーター遺伝子とポリ(A)付加シグナルを連結したDNAを導入した遺伝子改変マウスを作製するに至った。
【0011】
本発明の遺伝子改変マウスは、脳神経系でレポーター遺伝子を発現し、その発現はTH遺伝子の発現と相関する。したがって、該遺伝子改変マウスを使用すれば、被験物質がTH遺伝子の発現に及ぼす作用、すなわち被験物質の脳神経系に対する作用を、レポーター遺伝子の発現を指標としたin vivoの生化学的測定により、迅速かつ容易に評価することが可能となる。
【0012】
[遺伝子改変マウス]
以下、本発明を詳細に説明する。本発明の遺伝子改変マウスは、TH遺伝子のAhR結合エンハンサーと任意のプロモーターとレポーター遺伝子とポリ(A)付加シグナルとを機能的に連結したDNAを、受精卵または初期胚に導入して得られる遺伝子改変マウスである。
【0013】
ここで、「機能的に連結した」とは、連結された領域が、その領域の機能を発揮するように連結されていることを意味する。例えば、プロモーターまたはレポーター遺伝子が「機能的に連結」されたとは、本発明に従う遺伝子改変動物において、プロモーター活性を発揮し、レポーター遺伝子の発現を増強させるように連結されていることをいう。
【0014】
本発明において、マウスに導入するDNAは、AhR結合エンハンサーと任意のプロモーターとレポーター遺伝子とにより構成される。
【0015】
[エンハンサー]
本発明でいうエンハンサーとは、基質により活性化された転写因子が結合するゲノム上の特異的な塩基配列であり、転写因子との結合によって下流遺伝子の転写を活性化する機能を持つ塩基配列である。AhR結合エンハンサーとは、ダイオキシンとの結合で活性化されたAhR(受容体型の転写因子)が結合する塩基配列であり、この結合により下流遺伝子の転写を活性化する機能を持つ塩基配列である。
【0016】
本発明において、AhR結合エンハンサーとして最低限含まれるべき塩基配列は、配列番号1で表される塩基配列である。
【化1】

【0017】
当該塩基配列は、マウスのTH遺伝子5’上流域(-214bp 〜 -237bp)に由来する。本発明では、配列番号1で表される塩基配列が含まれれば、AhR結合エンハンサーとして、例えば、配列番号2で表される塩基配列のようにTH遺伝子5’上流域のより広範囲の塩基配列(-175bp 〜 -237bp)を使用してもよい。
【化2】
【0018】
また、AhR結合エンハンサーは、複数個連結して使用してもよいし、マウスのゲノム上での向きと同じ向き(順)で使用しても、或いは異なる向き(逆)で使用してもよい。複数個のAhR結合エンハンサーを連結して使用する場合には、該エンハンサーの機能が維持される限りにおいて、連結部に、1〜50塩基程度のリンカー配列等の塩基配列を含んでもよい。本発明のAhR結合エンハンサーの例として、配列番号3で表される塩基配列が挙げられる。
【化3】
【0019】
当該エンハンサーは、配列番号2で表される塩基配列を5’側から順に「逆・逆・順・順・順・順」の向きで6個連結したものである。このようなエンハンサーは、ポリメラーゼ連鎖反応(PCR)などの既知の遺伝子操作法を組み合わせることにより作製することができる。
【0020】
また、当該エンハンサーは、配列番号1または配列番号2で表される塩基配列を1つ以上、例えば、1、2、3、4、5、6、7、8、9または10以上で含めばよく、好ましくは2〜8で含めばよく、最も好ましくは3〜6で含めばよい。上述したように、エンハンサーに含まれる配列番号1または配列番号2の向きは、マウスTH遺伝子の5’上流におけるエンハンサー配列のTH構造遺伝子に対する向きと同じ向き(即ち、順向き)であっても、異なる向き(逆向き)であってもよく、またその組合せであってもよい。
【0021】
本発明において使用されるエンハンサー領域は上述したような配列番号1、2および3で示される配列以外にも、TH遺伝子の転写制御領域内の被験物質に応答して下流遺伝子の転写活性を増強する領域であればよい。TH遺伝子の転写制御領域は、例えば、配列番号17に記載の領域(2.5kb)である。TH遺伝子の転写制御領域は、任意の生物のTH遺伝子の5’上流領域(2500bp)であり得、例えば、ヒト、マウス、またはラットのTH遺伝子の5’上流領域(2500bp)であってもよい。
【0022】
また、被験物質に応答して下流遺伝子の転写活性を増強するエンハンサー領域は、プロモーター活性を増強する領域を含んでもよい。
【0023】
[プロモーター]
プロモーターとは、RNAポリメラーゼの結合配列を含む、遺伝子の転写開始に不可欠な塩基配列である。本発明におけるプロモーターは、配列番号4に記載したTH遺伝子のプロモーターであることが好ましい。
【化4】
【0024】
ただし、これに限定されるものではなく、例えば、シミアンウイルス40(SV40)の初期、或いは後期プロモーター、ヒトヘルペスウイルス1チミジンキナーゼ・プロモーター、サイトメガロウイルス・プロモーター等の哺乳動物で活性を持つプロモーターであればそれ自身公知の何れのプロモーターが使用されてもよい。
【0025】
このようなプロモーターは、ポリメラーゼ連鎖反応(PCR)などの既知の遺伝子操作法を組み合わせることにより取得することができる。取得されたプロモーターは、前記エンハンサーの3’末端側に、DNA連結酵素などを使用して連結する。エンハンサーとプロモーターの連結では、該エンハンサーと該プロモーターの機能が維持される限りにおいて、連結部に、1〜50塩基程度のリンカー配列等の塩基配列が含まれてもよい。
【0026】
本発明におけるエンハンサーとプロモーターを機能的に連結した塩基配列の例としては、配列番号5に記載した塩基配列が挙げられる。
【化5】
【0027】
この配列番号5で表される塩基配列は、配列番号3に記載のエンハンサーに、リンカー配列を介して、配列番号4に記載のTH遺伝子プロモーターを連結したものである。
【0028】
[レポーター遺伝子]
本発明におけるレポーター遺伝子は、酵素反応の速度や反応の定量性などから、ホタルの発光反応触媒酵素をコードするルシフェラーゼ遺伝子が好ましい。ただし、これに限定されるものではなく、例えば、緑色蛍光タンパク質遺伝子、βガラクトシダーゼ遺伝子、クロラムフェニコールアセチルトランスフェラーゼ遺伝子等のそれ自身公知のレポーター遺伝子を使用することができる。
【0029】
レポーター遺伝子は、天然から得られた遺伝子の塩基配列のまま使用してもよいし、部位特異的突然変異によって改変された遺伝子を用いてもよい。或いは、レポーター遺伝子がコードするタンパク質の機能が失われない限りにおいて、他のタンパク質やポリペプチドとの融合タンパク質として発現するように塩基配列を改変した遺伝子を使用してもよい。
【0030】
このようなレポーター遺伝子は、市販されている遺伝子を購入して使用されてもよいし、ポリメラーゼ連鎖反応(PCR)などの既知の遺伝子操作法を組み合わせて作製されてもよい。ルシフェラーゼ遺伝子の場合は、例えば、PGVベクター・シリーズ(TOYO B-Net)に組み込まれている、配列番号7に記載のホタルのルシフェラーゼ遺伝子が使用されてもよい。
【化6】
【0031】
当該ベクターのルシフェラーゼ遺伝子を融合遺伝子として用いる場合は、ルシフェラーゼ遺伝子の開始コドン上に組み込まれたNco Iサイトに、融合したいタンパク質、或いはペプチドをコードするDNAをin frameで挿入して作製すればよい。N末端にc-mycタグ配列が融合されたルシフェラーゼタンパク質をコードする融合遺伝子を作製する場合は、例えば、配列番号8と配列番号9で表される塩基配列をアニールさせた後に、PGV-P2ベクターのHin dIIIとNco Iサイトに挿入すればよい。これにより、配列番号10に記載のc-mycタグ-ルシフェラーゼ融合遺伝子を作製できる。
【化7】
【0032】
【化8】
【0033】
【化9】
【0034】
このような融合遺伝子から産生されるレポータータンパク質は、タグ特異的な抗体で検出できるため、有効な抗体の存在しないレポータータンパク質の検出に有効である。このように作製したレポーター遺伝子は、エンハンサーとプロモーターを機能的に連結した塩基配列、例えば、配列番号5で表される塩基配列の3’末端側に、DNA連結酵素などを使用して連結すればよい。プロモーターとレポーターの連結では、該プロモーターと該レポーター遺伝子の機能が維持される限りにおいて、連結部に、リンカー配列等の1〜50塩基程度の塩基配列が含まれてもよい。
【0035】
[ポリ(A)付加シグナル]
本発明におけるポリ(A)付加シグナルは、哺乳動物の遺伝子の転写終結に機能する転写終結シグナル配列であればそれ自身公知の何れの配列を使用してもよい。例としては、SV40ウイルスの後期ポリ(A)付加シグナルや、牛成長ホルモン遺伝子のポリ(A)付加シグナルなどが挙げられる。配列番号11に、SV40ウイルス後期ポリ(A)付加シグナルの塩基配列の例を示す。
【化10】
【0036】
ただし、本発明の実施において使用可能なSV40ウイルス後期ポリ(A)付加シグナルは、これに限定されるものではなく、ポリ(A)付加シグナルとしての機能を損なわない限りにおいては、遺伝子配列を改変したものを用いてもよい。
【0037】
ポリ(A)付加シグナルは、レポーター遺伝子の3’末端側に連結して使用する。レポーター遺伝子とポリ(A)付加シグナルは、DNA連結酵素などを使用して連結してもよいし、市販のベクター等で既にレポーター遺伝子とポリ(A)付加シグナルが連結済みのものをそのまま利用してもよい。レポーター遺伝子とポリ(A)付加シグナルの連結部には、該レポーター遺伝子と該ポリ(A)付加シグナルの機能が維持される限りにおいて、リンカー配列等の1〜50塩基程度の塩基配列が含まれてもよい。
【0038】
[導入DNA]
本発明の1態様では、前記のエンハンサーとプロモーターとレポーター遺伝子とポリ(A)付加シグナルを機能的に連結したDNAをマウスに導入して遺伝子改変マウスを作製する。マウスに導入するDNAの例としては、配列番号6に記載のDNAが挙げられる。
【化11−1】
【0039】
【化11−2】
この配列番号6で表されるDNAは、配列番号5で表されるエンハンサーとプロモーターを連結した塩基配列と、配列番号10で表されるN末端にc-mycタグを融合したルシフェラーゼ遺伝子と、配列番号11で表されるSV40ウイルスの後期ポリ(A)付加シグナルを連結したものである。該DNAにおいては、配列番号5と配列番号10、及び配列番号10と配列番号11の連結部にリンカー配列が含まれる。
【0040】
[遺伝子改変動物]
遺伝子改変マウスは、前記のDNAをマウスに導入して、該DNAをゲノムに持つマウスを選択することにより得ることができる。本発明の実施において、マウスに導入するDNAは、配列番号6と80%以上の相同性を有するDNAであることが望ましいが、これに限定されるものではない。エンハンサーとプロモーターとレポーター遺伝子が機能的に連結されたDNAであり、かつエンハンサーに1つ以上のAhR結合エンハンサーが含まれれば、必ずしも配列番号6と80%以上の相同性を持つ必要はない。当該DNAの導入に用いるマウスは、C57BL/6、DBA2、Balb/cなどの近交系の系統が好ましい。
【0041】
当該DNAを導入するべき動物は、マウスのみならず、ラット、ハムスター、およびウサギなどを含んでよい。その場合、当該DNAを導入されるべき動物種に応じて、チロシン水酸化酵素遺伝子の5’上流に存在するアリルハイドロカーボン受容体結合エンハンサーと、任意のプロモーターと、レポーター遺伝子と、ポリ(A)付加シグナルとを選択し、それらを機能的に連結したDNAを用いればよい。
【0042】
[DNAの導入]
動物へのDNA導入は、マイクロインジェクション法によるのが最も一般的である。具体的には、エンハンサーとプロモーターとレポーター遺伝子と転写終結シグナル配列とを機能的に連結したDNAを、マイクロマニュピュレーターを用いて、1細胞期の受精卵の雄性前核中へ顕微鏡下微量注入する。
【0043】
生き残った受精卵を仮親の卵管に移植した後、移植したマウスを飼育して、生まれた仔マウスから、サザンブロット解析、PCR などの方法で、導入DNAを有する個体を選択する。例えば、得られたマウスの尾から得たゲノムDNAをサザンブロットで解析して、導入DNAを受け継いだマウス個体を選択する。これは導入DNAが次世代に受け継がれることを示すものであり、動物モデルとして有用な遺伝子改変マウスの作製を示すものである。
【0044】
その他の方法としては、DNAは含有するウイルスベクターによる方法、或いはES細胞による方法などがある。ウイルスベクター法では、透明帯を除去した4〜8細胞期胚にウイルスベクターを感染後、胚盤胞まで発生させて偽妊娠させた代理母の子宮に戻して発生させることにより、遺伝子改変マウス個体を得ることができる。
【0045】
ES細胞(embryonic stem cell、胚性幹細胞)を用いる方法では、DNAを導入したES細胞からキメラマウスを経て、遺伝子改変マウス個体を得ることができる。このような遺伝子改変マウスは、導入遺伝子を相同染色体の両方に持つホモ接合体のマウスを取得し、その雌雄のマウスを交配することによって、すべての子孫が該遺伝子を安定に保持する状態で繁殖継代することができる。遺伝子改変マウスの遺伝形質を維持するために使用される受精卵は、雄マウスと雌マウスを交配させることによって得られる。受精卵は、自然交配によっても得られるが、雌マウスの性周期を人工的に調節後、雄マウスを交配させるのが好ましい。
【0046】
本発明の態様により、汎用の酵素活性検出試薬キットと測定機器を利用できる指標形質(レポーター)を脳神経系で発現する遺伝子改変マウスが得られる。該遺伝子改変マウスを用いれば、被験物質の脳神経系への作用をin vivoで迅速かつ容易に評価することができる。すなわち、本発明に従う遺伝子改変マウスは、レポーター遺伝子を脳神経系で発現する特性を持ち、このレポーター遺伝子の発現を指標にした生化学的定量による被験物質の毒性検査で、当該被験物質の脳神経系への作用を評価するための生体試験材料として使用できる。
【0047】
[被験物質の作用を評価する方法]
本発明の一つの実施態様は、本発明の遺伝子改変マウスに被験物質を投与した後に、該遺伝子改変マウスの脳神経系におけるレポーター遺伝子の発現を測定することにより、被験物質の脳神経系に対する作用を評価する方法である。
【0048】
レポーター遺伝子がホタルのルシフェラーゼ遺伝子である場合は、被験物質を遺伝子改変マウスに投与してから、6-72時間後に、該遺伝子改変マウスの腹腔内にルシフェラーゼの基質であるルシフェリンを投与してIVISシステム(In Vivo Imaging System、Xenogen社)等の発光測定装置で個体、或いは脳のルシフェラーゼ酵素活性由来の発光を測定すればよい。
【0049】
或いは、遺伝子改変マウスの脳を摘出してから、適当なタンパク質抽出試薬を用いてタンパク質を抽出し、これにルシフェリンを添加後、市販のルミノメーター等でルシフェラーゼ酵素活性由来の発光量を測定してもよい。このような測定で、被験物質を投与した遺伝子改変マウスにおいて、被験物質を投与していない対照区の遺伝子改変マウスに比して、有意に発光量が変化している場合に、被験物質が脳神経系に対して作用すると評価する。このとき、被験物質の投与による発光量変化の大きさが、被験物質が有する脳神経系への作用の大きさを判断するための指標となる。
【0050】
本発明の別の一つの実施態様は、遺伝子改変マウスから分離した初代神経細胞を被験物質に暴露した後に、該細胞におけるレポーター遺伝子の発現を測定することにより、被験物質の脳神経系に対する作用を評価する方法である。レポーター遺伝子がホタルのルシフェラーゼ遺伝子である場合は、遺伝子改変マウスから分離した初代神経細胞を被験物質に暴露し、2〜72時間後に、細胞を回収してタンパク質を抽出し、抽出液にルシフェラーゼの基質であるルシフェリンを投与してルミノメーターでルシフェラーゼ酵素活性由来の発光を測定すればよい。
【0051】
このような評価では、被験物質を投与した遺伝子改変マウスの初代神経細胞が、被験物質を投与していない対照区の遺伝子改変マウスの初代神経細胞に比して、有意に発光量が変化している場合に、被験物質が脳神経系に対して作用すると評価する。このとき、被験物質の投与による発光量変化の大きさが、被験物質が有する脳神経系への作用の大きさを判断するための指標となる。
【0052】
ここで「被験物質」とは、ダイオキシン様活性を有する物質であり得る。本発明者は、ダイオキシン様活性を有する物質が、上述のエンハンサー領域に作用して下流遺伝子の活性を増強できることを見出し、既に報告している。ダイオキシン様活性を有する物質は、アリルハイドロカーボン受容体と複合体を形成し、かかる複合体がエンハンサー領域に結合し、これにより下流遺伝子の転写活性を増強すると推定される。
【0053】
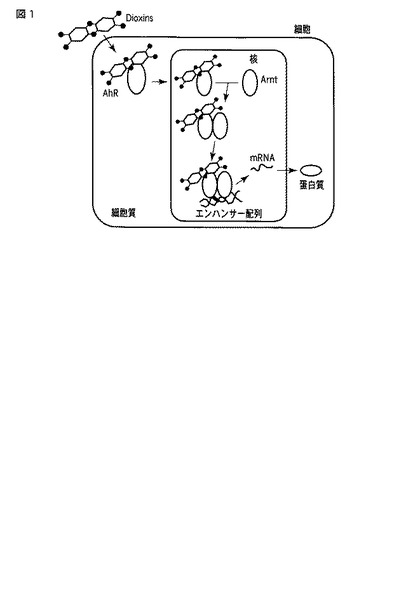
図1にアリルハイドロカーボン受容体(AhR)による遺伝子の転写活性メカニズムの1つを示す。TCDDなどのダイオキシン類は、細胞内のAhRに結合して複合体を形成し、遺伝子の特定の配列に結合することにより、下流遺伝子の発現を活性化することが知られている。
【0054】
図1において、Arntはアリルハイドロカーボン受容体核内トランスロケーターを示す。ダイオキシン様活性を有する化学物質と結合したAhRは核内でArntと複合体を形成することによってエンハンサーと結合し、下流の遺伝子の発現を活性化することが知られている。
【0055】
本発明に従うと、遺伝子改変動物を使用した被験物質の有害性を判定する方法は、当該遺伝子改変動物と被験物質を接触させることと、当該レポーター遺伝子の発現を検出することを具備すればよい。
【0056】
例えば、そのような接触は、被験物質の経口投与であってもよく、例えば、腹腔内注射、静脈内注射、筋肉内注射、皮下注射、皮内注射、経膣投与、経腸投与、経皮投与、経鼻投与、経肺投与および点眼などの非経口投与などであってよい。また、被験物質への暴露も当該接触と同様の手段により行ってよい。
【0057】
そのような接触の後に、発現される当該レポーター遺伝子に適した手段により、当該遺伝子の発現の有無または強弱を検出又は測定すればよい。その場合、動物全体で観察および/または測定してもよく、または摘出した脳を用いて、所望に応じてホモジナイズ、切断、ミンスおよび/またはタンパク質の抽出などの処理を行った後に観察および/または測定してもよい。
【0058】
また、本発明の遺伝子改変動物を使用した被験物質の有害性を判定する方法は、当該遺伝子改変動物の一部と被験物質を接触させることにより行ってもよい。その場合、遺伝子改変動物の一部の器官または臓器などと、被験物質を接触させればよい。その後、発現されるレポーター遺伝子に適した手段により、当該遺伝子の発現の有無または強弱を検出または測定すればよい。
【0059】
以上、遺伝子改変動物の代表的な例として遺伝子改変マウスについて主に説明してきたが、マウス以外の動物についても同様にそれ自身公知の方法により当該DNAを導入し、遺伝子改変動物を得ることが可能であり、且つ上述した何れの項目についてもマウスと同様に適用することが可能である。また更に、何れの動物の場合においても、上述のマウスと同様に被験物質の有害性を判定する方法に使用することが可能である。
【0060】
[例]
以下に実施例を挙げて、本発明をより具体的に説明する。ただし、これらの実施例は説明のためのものであって、本範囲の技術範囲を限定するものではない。
【0061】
1)マウス導入用ベクターの調整
配列番号1に記載の配列を含むTH遺伝子の5’上流-175bp 〜 -237bpの領域を2回繰り返した1本鎖DNA(即ち、TH遺伝子のAhR結合エンハンサー(配列番号1)を2つ含む)を合成した。このDNAを鋳型として、特異的なプライマーでPCRをおこない、一本鎖DNAを2本鎖に変換した。一本鎖DNA(配列番号12)、及び特異的なプライマーの塩基配列(配列番号13および14)を以下に示した。
【表A】
【0062】
【化12】
【0063】
【化13】
【0064】
【化14】
【0065】
PCRで得た2本鎖DNAを、T4ポリヌクレオチドキナーゼでリン酸化後にT4 DNAリガーゼで連結し、アガロースゲル電気泳動で連結個数が3つのDNA断片を分離してから、QIA quick ゲル抽出キット(キアゲン)で精製した。精製したDNA断片は、AhR結合エンハンサーの6回繰り返しからなる。このDNAをルシフェラーゼ発現ベクターであるPGV-P2ベクター(TOYO B-NET)に組み込んだ。まず、PGV-P2上のSV40プロモーターを制限酵素Hin dIIIとXho Iで切り出して、TH遺伝子のプロモーター[転写開始点(0 bp)から5’上流 -100 bpまでの領域(配列番号4)]と組み換えたベクター(PGV-THp)を作製した。次に、PGV-THpのHin dIIIとNco Iサイトに、下記のc-mycタグ配列を挿入した。
【表B】
c-mycタグ配列は、上記の相補的な1本鎖DNAをアニーリングさせて得た。c-mycタグの末端には、ベクターへの組み込みを容易にするために、Hin dIII認識配列(5’末端)とNco I認識配列(3’末端)を付加した。2本鎖にしたc-mycタグ配列は、PGV-THpのHin dIIIとNco I部位に組み込んだ。この操作で得られたベクター、すなわちPGV-THp上のルシフェラーゼ遺伝子N末端にin-frameでc-mycタグ配列が付加されたベクター:PGV-THp-MLucをSma Iで切断し、このサイトにAhR結合エンハンサーの6回繰り返しとTH遺伝子のプロモーターからなるDNA配列(配列番号5)を組み込んで、マウス導入用のベクター:pTHEn-MLucを作製した。
【0066】
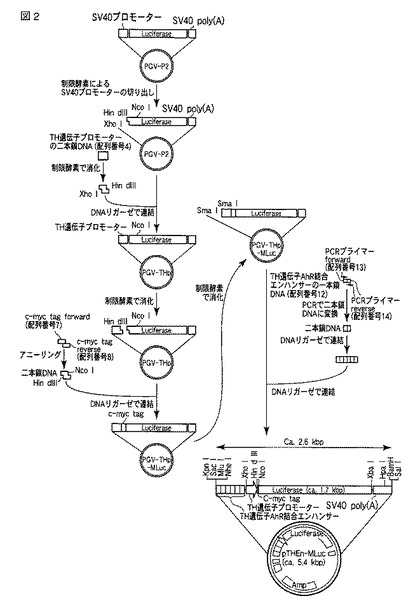
pTHEn-MLuc上のAhR結合エンハンサーからc-mycタグ配列の塩基配列は、シーケンシングで確認した。ベクター作製のフローを図2に示した。
【0067】
2) 遺伝子改変候補マウスの作出
C57BL6系統のマウスを使用した。遺伝子導入操作の3日前に、4-6週齢の雌マウスに対して、血清性性腺刺激ホルモン(PMSG)を腹腔内に注射し、前日には胎盤性性腺刺激ホルモン(hCG)を腹腔内注射した。注射後、このマウスを雄マウスと交配させて、翌日、膣栓があった雌マウスから卵管を摘出した。ヒアルロニダーゼを含む培養液中で卵管膨大部を破いて取り出した卵塊を暫く放置して卵丘細胞と受精卵を解離させてから、受精卵を採卵用ピペットで採取した。採取した受精卵は、培養液で数回洗浄してから、CO2インキュベーター内で培養した。
【0068】
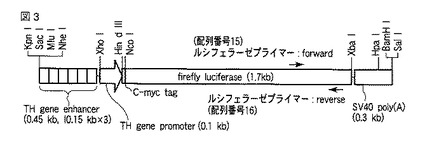
受精卵に導入する遺伝子には、pTHEn-MLucから制限酵素Kpn IとBam HIで配列番号6で表されるDNA(図3を参照されたい、TH遺伝子エンハンサーとプロモーター::c-mycタグ-ルシフェラーゼ遺伝子::SV40ポリAシグナル)を切り出した後に精製し、TEバッファー[10mM Tris-HCl (pH7.5), 0.1mM EDTA]に1〜2 ng/μlに溶解したものを用いた。
【0069】
受精卵をCO2インキュベーターから取り出して、顕微鏡にセットしたチャンバーの培地に入れて、低倍率下、ホールディングピペットで保持してから、高倍率の微分干渉レンズ下で、受精卵に、先に述べた方法で調整した配列番号6で表されるDNAをインジェクションピペットで導入した。インジェクション後の受精卵は、培地を交換後にCO2インキュベーター内で24時間培養した。この受精卵を偽妊娠雌マウスに移植して発生させ、遺伝子改変候補マウスを作出した。
【0070】
3)ジェノタイピングによる遺伝子改変マウスの選択
遺伝子改変マウスは、2)の候補マウスの中からサザンブロット並びにPCRで、ゲノム上の導入DNA(配列番号6、受精卵に導入したDNA)の一部を検出して選択した。2 cm程度に切断したマウス(4-6週齢)の尾を遠心チューブに入れ、50℃で8から10時間プロテアーゼK処理をおこなった後、溶液に等量のフェノール/クロロホルム/イソアミルアルコール(25:24:1)を加えて混合してから、15,000 rpm,3分間の遠心で水層とフェノール層に分離した。新しいチューブに水層を取り分けて1/4量の10 M 酢酸アンモニウムと2.5 倍量のエタノールを加えてよく混ぜた後、15,000 rpm,3分間の遠心によって、溶液中のゲノムDNAを沈殿させて上清を除いた。沈殿を70% エタノールで洗浄してから風乾し、TEバッファーに再溶解してマウスのゲノムDNA溶液を得た。
【0071】
サザンブロッティングによるジェノタイピングでは、このゲノムDNA溶液に制限酵素Xho Iを加えて完全消化して、アガロースゲル電気泳動をおこなった後、キャピラリートランスファー法で、ゲル中のゲノムDNAナイロンメンブレンHybond N+(GEヘルスケア・バイオサイエンス)に転写した。メンブレンへのDNA固定は、紫外線照射でおこなった。このメンブレンをハイブリダイゼーションバッファー(1/20量 Denhardt’s溶液, 50 μg/mlサケ精子DNA)に浸して、プレ・ハイブリダイゼーションを65℃で1時間おこない、次に放射性同位体(32P)ラベルしたプローブDNAを含んだハイブリダイゼーションバッファー中で65℃、1晩のハイブリダイゼーションをおこなった。非特異的なプローブ吸着は、メンブレンを洗浄液A(2 x SSC, 0.1% SDS)中で65℃、5分間×2回、続いて洗浄液B(0.5 x SSC, 0.1% SDS)中で65℃,15分間×2回洗浄して除去した。ハイブリダイゼーション・プローブには、pTHEN-MLucのHin dIIIとSph I断片を放射線同位体(32P)ラベルしたものを用いた。ハイブリダイゼーションのシグナルはX線フィルムで検出した。この結果、#176、#177、#184の3匹の遺伝子改変候補マウスで導入DNAに由来するシグナルが検出された(図4)。
【0072】
PCRによるジェノタイピングは、マウスの尾から抽出したゲノムDNAを鋳型として、特異的なプライマーを使用して、pTHEn-MLucのルシフェラーゼ構造遺伝子の一部が増幅されるかどうかで判定した。以下に使用したプライマーの塩基配列を示した(配列番号15および配列番号16)。
【表C】
【0073】
【化15】
【0074】
【化16】
【0075】
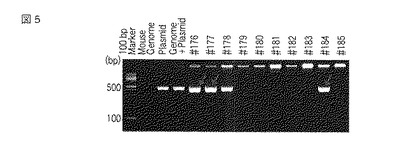
増幅の確認は、アガロースゲル電気泳動でおこなった。PCRによるジェノタイピングでは、図5に示したように、#176、#177、#178、#184の4匹の遺伝子改変候補マウスで導入DNAに由来するシグナルが検出された。
【0076】
これら2種類のジェノタイピング、すなわちサザンハイブリダイゼーションとPCRの両方法で導入DNAのシグナルが検出された3匹のマウス(#176、#177、#184)を遺伝子改変マウスとして選択し、以降の実験に使用した。#176のマウスについては、受精卵と精子を凍結保存し、特許生物寄託センターへの寄託をおこなった(寄託番号FERM BP-10993)。
【0077】
3)遺伝子改変マウス・ラインの維持
2)で選択した3匹の遺伝子改変マウス(遺伝子改変マウスの0世代:T0世代目)をC57BL/6系統の野生型マウスと交配(戻し交配=バッククロス)して子孫を得た。この遺伝子改変マウスの子孫とC57BL/6系統の野生型マウス、或いは遺伝子改変マウスの子孫と交配することにより、遺伝子改変マウスを維持した。
【0078】
4)β遺伝子改変マウスにおけるNF暴露時のルシフェラーゼ発光の測定
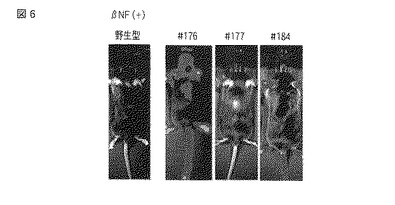
被験物質にはダイオキシンのアナログであるβナフトフラボン(βNF)を用いた。#176(4週齢)、#177(6週齢)、#184(6週齢)のT1世代の遺伝子改変マウスにコーンオイルに溶解したβNFを40 mg/kgとなるように腹腔内に注射した。対照区のマウス(T1世代の遺伝子改変マウス)は、腹腔内にβNF投与区と等量のコーンオイルを投与した。投与から72時間後、これらのマウスの腹腔内にルシフェリン溶液を150 mg/kg BWとなるように投与してから、5分後にIVISシステム(in vivo imaging system 100 series、Xenogen社製)でマウスのルシフェラーゼ発光を検出したところ、全ての遺伝子改変マウスでルシフェラーゼ発光が観察された。(図6)。更に、#176の遺伝子改変マウスに関しては、脳は中脳付近で切断して、IVISシステムで切断面の発光を測定した。この結果、βNF投与マウスの中脳・海馬付近において発光量の増加が検出された(図7)。以上の結果から、遺伝子改変マウス(#176、#177、#184)を用いて、レポーター遺伝子の発現変化を指標に、βNFの脳神経系に対する作用を評価できることが示された。
【0079】
5)遺伝子改変マウスから分離した初代神経細胞におけるダイオキシン(TCDD)暴露時のルシフェラーゼ発光の測定
#176の遺伝子改変マウス(ヘテロ、♂)と野生型マウス(♀)を交配し、プラグの確認から12日後に、胎児(E12 = 胎齢12日)の脳を無菌環境下で摘出した。摘出した胎児の脳を全てまとめてから、0.25% トリプシンを加え、37℃で15分間インキュベートした。トリプシンを除いて、ハンクス溶液で3回洗浄後、細胞を血清入りDF1:1培地(ダルベッコ-F12等比混合培地+10%牛胎児血清)に縣濁してから、培養プレート(オルニチン-フィブロネクチンでコート)に播種し、37℃/5% CO2雰囲気下で培養した。一晩培養後、神経細胞を優先的に増殖させるために、培地を無血清培地[ダルベッコMEM/ハムF12等比混合培地+TIPS(トランスフェリン/インスリン/ペニシリン/ストレプトマイシン)]に交換して培養した(図8)。
【0080】
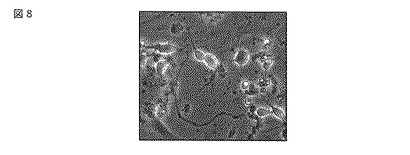
4日後、培地に終濃度が0,0.1,10 ng/mlとなるようにTCDD(2,3,7,8-テトラクロロジベンゾ-p-ジオキシン、関東化学)を添加し、24時間後に細胞を回収した。回収した細胞は、リン酸緩衝液で2回洗浄した後、200 μlのタンパク質抽出溶液(TOYO B-NET)を加えて15分間室温で攪拌後、細胞を凍結(-80℃)-融解(室温)によって破砕した。この細胞破砕液を1.5 mlチューブに回収後、遠心をおこない、上清をタンパク質抽出溶液として回収した。このようにして得たタンパク質抽出溶液をルシフェラーゼ発光の測定に使用した。ルシフェラーゼ発光は、タンパク質回収液10 μlにアッセイ溶液(ピッカジーン・アッセイ・キット、TOYO B-NET)を100 μlを加えて攪拌後、直ぐにルミノメーター LB-940(ベルトルード)で2分間当たりの発光量を測定した。得られた発光量は、タンパク質溶液の総タンパク量で除算して、相対発光強度(RLU / min / μg protein)に換算した。溶液の総タンパク量はプロテインアッセイ・キット(バイオラッド)で測定した。この結果、遺伝子改変マウスから分離した初代神経細胞は、0.1 nM TCDD暴露で未暴露時の1.3倍、10 nM TCDD暴露時には3.1倍と、TCDD濃度に依存した発光量の増加を示した(図9)。この結果より、遺伝子改変マウス(#176)から分離した初代神経細胞を用いて、レポーター遺伝子の発現変化を指標に、TCDDの脳神経系に対する作用を評価できることが示された。
【図面の簡単な説明】
【0081】
【図1】遺伝子転写活性メカニズムを示す図。
【図2】ベクター作製のフローを示す図。
【図3】受精卵に導入するDNAを示す図。
【図4】導入DNAに由来するシグナルを示すサザンハイブリダイゼーションの結果を示す図。
【図5】導入DNAに由来するシグナルを示す電気泳動の結果を示す図。
【図6】遺伝子改変マウスにおけるルシフェラーゼ発光を示す図。
【図7】マウス中脳・海馬付近における発光の増加を示す図。
【図8】培養神経細胞を示す図。
【図9】TCDD濃度に依存した発光量の増加を示す図。
【技術分野】
【0001】
本発明は、被験物質の有害性を評価するための遺伝子改変動物、および当該動物またはその一部における被験物質の作用を評価する方法に関する。
【背景技術】
【0002】
ダイオキシン類、PCB類、臭素系難燃剤および一部の農薬など、環境中の汚染化学物質がヒトの健康に及ぼす影響が懸念されている。また、産業活動や日々の生活の中で使用されている多種多様な化学物質の中にも、ヒトの健康に対する影響が未知の化学物質も多い。これらの化学物質は、肝機能、生殖機能、免疫機能および脳神経機能などの生体機能に作用して、ヒトの健康に有害な影響を及ぼすとされる。
【0003】
従って、ある特定の化学物質が作用しうる生体機能を検出および/または評価する技術は、化学物質の取り扱いや規制を判断する、あるいは化学物質に暴露されたヒトの治療をおこなう上で非常に重要となる。特に近年では、胎児および幼児期の化学物質暴露が児童に自閉症や多動性障害等の高次脳機能障害を引き起こす可能性が指摘されており、脳神経機能に対する化学物質の影響を評価するための技術開発が望まれている。
【0004】
現在、化学物質の生体機能への影響を評価する技術は、大きく分類すると、単一細胞を用いたin vitro評価技術と動物個体を用いたin vivo評価技術に分類される。
【0005】
in vitro評価技術には、マウス肝臓由来細胞Hepa1c1c7を用いて被験物質の肝機能への作用を評価する技術(特許文献1)、ヒト乳腺由来細胞MGF-7を用いて被験物質の生殖機能への作用を評価する技術(特許文献2)等がある。また、脳神経機能に限れば、マウス神経芽細胞腫Neuro2aを用いたin vitro評価方法を本出願人は既に報告している。当該方法では、当該細胞に、チロシン水酸化酵素遺伝子(ここでは、「TH遺伝子」と記す)のアリルハイドロカーボン受容体結合エンハンサー配列とTH遺伝子コアプロモーター配列で構成されるプロモーターの制御下にあるレポーター遺伝子を導入して得られた遺伝子改変細胞において、被験物質の脳神経系への影響が評価される。in vivo評価技術では、Mvh遺伝子あるいはc-mos遺伝子のプロモーターの制御下にあるレポーター遺伝子を導入された遺伝子改変マウスを用いた被験物質の生殖機能への影響を評価する方法が報告されている(特許文献3、特許文献4)。
【特許文献1】US Patent 5,854,010
【特許文献2】特開2002−253231
【特許文献3】特開2001−8577
【特許文献4】特開2001−8578
【発明の開示】
【発明が解決しようとする課題】
【0006】
上述のような状況に鑑み本発明の目的は、被験物質の生体に対する有害性または影響を評価するための遺伝子改変動物、および当該動物またはその一部における被験物質の影響を評価する方法を提供することである。詳しくは、被験物質の脳神経機能への作用を評価するために使用される遺伝子変異動物、および該遺伝子変異動物またはその一部を使用して被験物質の脳神経機能への影響を評価する方法を提供することである。
【課題を解決するための手段】
【0007】
上記目的を達成するための本発明は、
(1)チロシン水酸化酵素遺伝子の5’上流に存在するアリルハイドロカーボン受容体結合エンハンサーと、任意のプロモーターと、レポーター遺伝子と、ポリ(A)付加シグナルとを機能的に連結したDNAを導入された遺伝子改変動物;および
(2)前記(1)に記載の遺伝子改変動物またはその一部と、被験物質とを接触させることと、当該レポーター遺伝子の発現を検出することと、を具備する被験物質の有害性を判定する方法;
である。
【発明の効果】
【0008】
本発明により、被験物質の生体に対する有害性または影響を評価するための遺伝子改変動物、および当該動物またはその一部における被験物質の影響を評価する方法が提供される。詳しくは、被験物質の脳神経機能への作用を評価するために使用される遺伝子変異動物、および該遺伝子変異動物またはその一部を使用して被験物質の脳神経機能への影響を評価する方法が提供される。
【発明を実施するための最良の形態】
【0009】
現在、被験物質の脳神経機能に対する作用を評価するin vitro評価技術として、マウス神経芽細胞腫Neuro2aを用いた方法が既に報告されている。しかしながら、このような単一細胞を使用した評価技術では、動物個体レベルでの被験物質の作用を正確に評価することは難しい。即ち、これは、例えば、単一細胞では、被験物質の取り込みや分布、あるいは被験物質の代謝等が必ずしも動物個体と同等に再現されない可能性が大きいと考えられるからである。従って、被験物質の生体機能に対する作用評価には、in vitro評価技術とin vivo評価技術の併用が望ましいと考えられる。そこで、本発明者らは、脳神経機能を評価する場合に有効なin vivo評価技術が未だに確立されていないことに着目し、被験物質の脳神経機能に対する影響を評価するためのin vivo評価技術の開発を行った。
【0010】
TH遺伝子はドーパミン合成酵素をコードする遺伝子であり、化学物質暴露による該遺伝子の発現変化は脳神経系の機能に影響することが知られている。本発明者等は、TH遺伝子の発現を指標とした、脳神経系への化学物質作用のin vivo評価技術を鋭意検討した結果、本発明に従う、TH遺伝子のアリルハイドロカーボン受容体(ここでは「AhR」とも記す)結合エンハンサーと任意のプロモーターとレポーター遺伝子とポリ(A)付加シグナルを連結したDNAを導入した遺伝子改変マウスを作製するに至った。
【0011】
本発明の遺伝子改変マウスは、脳神経系でレポーター遺伝子を発現し、その発現はTH遺伝子の発現と相関する。したがって、該遺伝子改変マウスを使用すれば、被験物質がTH遺伝子の発現に及ぼす作用、すなわち被験物質の脳神経系に対する作用を、レポーター遺伝子の発現を指標としたin vivoの生化学的測定により、迅速かつ容易に評価することが可能となる。
【0012】
[遺伝子改変マウス]
以下、本発明を詳細に説明する。本発明の遺伝子改変マウスは、TH遺伝子のAhR結合エンハンサーと任意のプロモーターとレポーター遺伝子とポリ(A)付加シグナルとを機能的に連結したDNAを、受精卵または初期胚に導入して得られる遺伝子改変マウスである。
【0013】
ここで、「機能的に連結した」とは、連結された領域が、その領域の機能を発揮するように連結されていることを意味する。例えば、プロモーターまたはレポーター遺伝子が「機能的に連結」されたとは、本発明に従う遺伝子改変動物において、プロモーター活性を発揮し、レポーター遺伝子の発現を増強させるように連結されていることをいう。
【0014】
本発明において、マウスに導入するDNAは、AhR結合エンハンサーと任意のプロモーターとレポーター遺伝子とにより構成される。
【0015】
[エンハンサー]
本発明でいうエンハンサーとは、基質により活性化された転写因子が結合するゲノム上の特異的な塩基配列であり、転写因子との結合によって下流遺伝子の転写を活性化する機能を持つ塩基配列である。AhR結合エンハンサーとは、ダイオキシンとの結合で活性化されたAhR(受容体型の転写因子)が結合する塩基配列であり、この結合により下流遺伝子の転写を活性化する機能を持つ塩基配列である。
【0016】
本発明において、AhR結合エンハンサーとして最低限含まれるべき塩基配列は、配列番号1で表される塩基配列である。
【化1】
【0017】
当該塩基配列は、マウスのTH遺伝子5’上流域(-214bp 〜 -237bp)に由来する。本発明では、配列番号1で表される塩基配列が含まれれば、AhR結合エンハンサーとして、例えば、配列番号2で表される塩基配列のようにTH遺伝子5’上流域のより広範囲の塩基配列(-175bp 〜 -237bp)を使用してもよい。
【化2】
【0018】
また、AhR結合エンハンサーは、複数個連結して使用してもよいし、マウスのゲノム上での向きと同じ向き(順)で使用しても、或いは異なる向き(逆)で使用してもよい。複数個のAhR結合エンハンサーを連結して使用する場合には、該エンハンサーの機能が維持される限りにおいて、連結部に、1〜50塩基程度のリンカー配列等の塩基配列を含んでもよい。本発明のAhR結合エンハンサーの例として、配列番号3で表される塩基配列が挙げられる。
【化3】
【0019】
当該エンハンサーは、配列番号2で表される塩基配列を5’側から順に「逆・逆・順・順・順・順」の向きで6個連結したものである。このようなエンハンサーは、ポリメラーゼ連鎖反応(PCR)などの既知の遺伝子操作法を組み合わせることにより作製することができる。
【0020】
また、当該エンハンサーは、配列番号1または配列番号2で表される塩基配列を1つ以上、例えば、1、2、3、4、5、6、7、8、9または10以上で含めばよく、好ましくは2〜8で含めばよく、最も好ましくは3〜6で含めばよい。上述したように、エンハンサーに含まれる配列番号1または配列番号2の向きは、マウスTH遺伝子の5’上流におけるエンハンサー配列のTH構造遺伝子に対する向きと同じ向き(即ち、順向き)であっても、異なる向き(逆向き)であってもよく、またその組合せであってもよい。
【0021】
本発明において使用されるエンハンサー領域は上述したような配列番号1、2および3で示される配列以外にも、TH遺伝子の転写制御領域内の被験物質に応答して下流遺伝子の転写活性を増強する領域であればよい。TH遺伝子の転写制御領域は、例えば、配列番号17に記載の領域(2.5kb)である。TH遺伝子の転写制御領域は、任意の生物のTH遺伝子の5’上流領域(2500bp)であり得、例えば、ヒト、マウス、またはラットのTH遺伝子の5’上流領域(2500bp)であってもよい。
【0022】
また、被験物質に応答して下流遺伝子の転写活性を増強するエンハンサー領域は、プロモーター活性を増強する領域を含んでもよい。
【0023】
[プロモーター]
プロモーターとは、RNAポリメラーゼの結合配列を含む、遺伝子の転写開始に不可欠な塩基配列である。本発明におけるプロモーターは、配列番号4に記載したTH遺伝子のプロモーターであることが好ましい。
【化4】
【0024】
ただし、これに限定されるものではなく、例えば、シミアンウイルス40(SV40)の初期、或いは後期プロモーター、ヒトヘルペスウイルス1チミジンキナーゼ・プロモーター、サイトメガロウイルス・プロモーター等の哺乳動物で活性を持つプロモーターであればそれ自身公知の何れのプロモーターが使用されてもよい。
【0025】
このようなプロモーターは、ポリメラーゼ連鎖反応(PCR)などの既知の遺伝子操作法を組み合わせることにより取得することができる。取得されたプロモーターは、前記エンハンサーの3’末端側に、DNA連結酵素などを使用して連結する。エンハンサーとプロモーターの連結では、該エンハンサーと該プロモーターの機能が維持される限りにおいて、連結部に、1〜50塩基程度のリンカー配列等の塩基配列が含まれてもよい。
【0026】
本発明におけるエンハンサーとプロモーターを機能的に連結した塩基配列の例としては、配列番号5に記載した塩基配列が挙げられる。
【化5】
【0027】
この配列番号5で表される塩基配列は、配列番号3に記載のエンハンサーに、リンカー配列を介して、配列番号4に記載のTH遺伝子プロモーターを連結したものである。
【0028】
[レポーター遺伝子]
本発明におけるレポーター遺伝子は、酵素反応の速度や反応の定量性などから、ホタルの発光反応触媒酵素をコードするルシフェラーゼ遺伝子が好ましい。ただし、これに限定されるものではなく、例えば、緑色蛍光タンパク質遺伝子、βガラクトシダーゼ遺伝子、クロラムフェニコールアセチルトランスフェラーゼ遺伝子等のそれ自身公知のレポーター遺伝子を使用することができる。
【0029】
レポーター遺伝子は、天然から得られた遺伝子の塩基配列のまま使用してもよいし、部位特異的突然変異によって改変された遺伝子を用いてもよい。或いは、レポーター遺伝子がコードするタンパク質の機能が失われない限りにおいて、他のタンパク質やポリペプチドとの融合タンパク質として発現するように塩基配列を改変した遺伝子を使用してもよい。
【0030】
このようなレポーター遺伝子は、市販されている遺伝子を購入して使用されてもよいし、ポリメラーゼ連鎖反応(PCR)などの既知の遺伝子操作法を組み合わせて作製されてもよい。ルシフェラーゼ遺伝子の場合は、例えば、PGVベクター・シリーズ(TOYO B-Net)に組み込まれている、配列番号7に記載のホタルのルシフェラーゼ遺伝子が使用されてもよい。
【化6】
【0031】
当該ベクターのルシフェラーゼ遺伝子を融合遺伝子として用いる場合は、ルシフェラーゼ遺伝子の開始コドン上に組み込まれたNco Iサイトに、融合したいタンパク質、或いはペプチドをコードするDNAをin frameで挿入して作製すればよい。N末端にc-mycタグ配列が融合されたルシフェラーゼタンパク質をコードする融合遺伝子を作製する場合は、例えば、配列番号8と配列番号9で表される塩基配列をアニールさせた後に、PGV-P2ベクターのHin dIIIとNco Iサイトに挿入すればよい。これにより、配列番号10に記載のc-mycタグ-ルシフェラーゼ融合遺伝子を作製できる。
【化7】
【0032】
【化8】
【0033】
【化9】
【0034】
このような融合遺伝子から産生されるレポータータンパク質は、タグ特異的な抗体で検出できるため、有効な抗体の存在しないレポータータンパク質の検出に有効である。このように作製したレポーター遺伝子は、エンハンサーとプロモーターを機能的に連結した塩基配列、例えば、配列番号5で表される塩基配列の3’末端側に、DNA連結酵素などを使用して連結すればよい。プロモーターとレポーターの連結では、該プロモーターと該レポーター遺伝子の機能が維持される限りにおいて、連結部に、リンカー配列等の1〜50塩基程度の塩基配列が含まれてもよい。
【0035】
[ポリ(A)付加シグナル]
本発明におけるポリ(A)付加シグナルは、哺乳動物の遺伝子の転写終結に機能する転写終結シグナル配列であればそれ自身公知の何れの配列を使用してもよい。例としては、SV40ウイルスの後期ポリ(A)付加シグナルや、牛成長ホルモン遺伝子のポリ(A)付加シグナルなどが挙げられる。配列番号11に、SV40ウイルス後期ポリ(A)付加シグナルの塩基配列の例を示す。
【化10】
【0036】
ただし、本発明の実施において使用可能なSV40ウイルス後期ポリ(A)付加シグナルは、これに限定されるものではなく、ポリ(A)付加シグナルとしての機能を損なわない限りにおいては、遺伝子配列を改変したものを用いてもよい。
【0037】
ポリ(A)付加シグナルは、レポーター遺伝子の3’末端側に連結して使用する。レポーター遺伝子とポリ(A)付加シグナルは、DNA連結酵素などを使用して連結してもよいし、市販のベクター等で既にレポーター遺伝子とポリ(A)付加シグナルが連結済みのものをそのまま利用してもよい。レポーター遺伝子とポリ(A)付加シグナルの連結部には、該レポーター遺伝子と該ポリ(A)付加シグナルの機能が維持される限りにおいて、リンカー配列等の1〜50塩基程度の塩基配列が含まれてもよい。
【0038】
[導入DNA]
本発明の1態様では、前記のエンハンサーとプロモーターとレポーター遺伝子とポリ(A)付加シグナルを機能的に連結したDNAをマウスに導入して遺伝子改変マウスを作製する。マウスに導入するDNAの例としては、配列番号6に記載のDNAが挙げられる。
【化11−1】
【0039】
【化11−2】
この配列番号6で表されるDNAは、配列番号5で表されるエンハンサーとプロモーターを連結した塩基配列と、配列番号10で表されるN末端にc-mycタグを融合したルシフェラーゼ遺伝子と、配列番号11で表されるSV40ウイルスの後期ポリ(A)付加シグナルを連結したものである。該DNAにおいては、配列番号5と配列番号10、及び配列番号10と配列番号11の連結部にリンカー配列が含まれる。
【0040】
[遺伝子改変動物]
遺伝子改変マウスは、前記のDNAをマウスに導入して、該DNAをゲノムに持つマウスを選択することにより得ることができる。本発明の実施において、マウスに導入するDNAは、配列番号6と80%以上の相同性を有するDNAであることが望ましいが、これに限定されるものではない。エンハンサーとプロモーターとレポーター遺伝子が機能的に連結されたDNAであり、かつエンハンサーに1つ以上のAhR結合エンハンサーが含まれれば、必ずしも配列番号6と80%以上の相同性を持つ必要はない。当該DNAの導入に用いるマウスは、C57BL/6、DBA2、Balb/cなどの近交系の系統が好ましい。
【0041】
当該DNAを導入するべき動物は、マウスのみならず、ラット、ハムスター、およびウサギなどを含んでよい。その場合、当該DNAを導入されるべき動物種に応じて、チロシン水酸化酵素遺伝子の5’上流に存在するアリルハイドロカーボン受容体結合エンハンサーと、任意のプロモーターと、レポーター遺伝子と、ポリ(A)付加シグナルとを選択し、それらを機能的に連結したDNAを用いればよい。
【0042】
[DNAの導入]
動物へのDNA導入は、マイクロインジェクション法によるのが最も一般的である。具体的には、エンハンサーとプロモーターとレポーター遺伝子と転写終結シグナル配列とを機能的に連結したDNAを、マイクロマニュピュレーターを用いて、1細胞期の受精卵の雄性前核中へ顕微鏡下微量注入する。
【0043】
生き残った受精卵を仮親の卵管に移植した後、移植したマウスを飼育して、生まれた仔マウスから、サザンブロット解析、PCR などの方法で、導入DNAを有する個体を選択する。例えば、得られたマウスの尾から得たゲノムDNAをサザンブロットで解析して、導入DNAを受け継いだマウス個体を選択する。これは導入DNAが次世代に受け継がれることを示すものであり、動物モデルとして有用な遺伝子改変マウスの作製を示すものである。
【0044】
その他の方法としては、DNAは含有するウイルスベクターによる方法、或いはES細胞による方法などがある。ウイルスベクター法では、透明帯を除去した4〜8細胞期胚にウイルスベクターを感染後、胚盤胞まで発生させて偽妊娠させた代理母の子宮に戻して発生させることにより、遺伝子改変マウス個体を得ることができる。
【0045】
ES細胞(embryonic stem cell、胚性幹細胞)を用いる方法では、DNAを導入したES細胞からキメラマウスを経て、遺伝子改変マウス個体を得ることができる。このような遺伝子改変マウスは、導入遺伝子を相同染色体の両方に持つホモ接合体のマウスを取得し、その雌雄のマウスを交配することによって、すべての子孫が該遺伝子を安定に保持する状態で繁殖継代することができる。遺伝子改変マウスの遺伝形質を維持するために使用される受精卵は、雄マウスと雌マウスを交配させることによって得られる。受精卵は、自然交配によっても得られるが、雌マウスの性周期を人工的に調節後、雄マウスを交配させるのが好ましい。
【0046】
本発明の態様により、汎用の酵素活性検出試薬キットと測定機器を利用できる指標形質(レポーター)を脳神経系で発現する遺伝子改変マウスが得られる。該遺伝子改変マウスを用いれば、被験物質の脳神経系への作用をin vivoで迅速かつ容易に評価することができる。すなわち、本発明に従う遺伝子改変マウスは、レポーター遺伝子を脳神経系で発現する特性を持ち、このレポーター遺伝子の発現を指標にした生化学的定量による被験物質の毒性検査で、当該被験物質の脳神経系への作用を評価するための生体試験材料として使用できる。
【0047】
[被験物質の作用を評価する方法]
本発明の一つの実施態様は、本発明の遺伝子改変マウスに被験物質を投与した後に、該遺伝子改変マウスの脳神経系におけるレポーター遺伝子の発現を測定することにより、被験物質の脳神経系に対する作用を評価する方法である。
【0048】
レポーター遺伝子がホタルのルシフェラーゼ遺伝子である場合は、被験物質を遺伝子改変マウスに投与してから、6-72時間後に、該遺伝子改変マウスの腹腔内にルシフェラーゼの基質であるルシフェリンを投与してIVISシステム(In Vivo Imaging System、Xenogen社)等の発光測定装置で個体、或いは脳のルシフェラーゼ酵素活性由来の発光を測定すればよい。
【0049】
或いは、遺伝子改変マウスの脳を摘出してから、適当なタンパク質抽出試薬を用いてタンパク質を抽出し、これにルシフェリンを添加後、市販のルミノメーター等でルシフェラーゼ酵素活性由来の発光量を測定してもよい。このような測定で、被験物質を投与した遺伝子改変マウスにおいて、被験物質を投与していない対照区の遺伝子改変マウスに比して、有意に発光量が変化している場合に、被験物質が脳神経系に対して作用すると評価する。このとき、被験物質の投与による発光量変化の大きさが、被験物質が有する脳神経系への作用の大きさを判断するための指標となる。
【0050】
本発明の別の一つの実施態様は、遺伝子改変マウスから分離した初代神経細胞を被験物質に暴露した後に、該細胞におけるレポーター遺伝子の発現を測定することにより、被験物質の脳神経系に対する作用を評価する方法である。レポーター遺伝子がホタルのルシフェラーゼ遺伝子である場合は、遺伝子改変マウスから分離した初代神経細胞を被験物質に暴露し、2〜72時間後に、細胞を回収してタンパク質を抽出し、抽出液にルシフェラーゼの基質であるルシフェリンを投与してルミノメーターでルシフェラーゼ酵素活性由来の発光を測定すればよい。
【0051】
このような評価では、被験物質を投与した遺伝子改変マウスの初代神経細胞が、被験物質を投与していない対照区の遺伝子改変マウスの初代神経細胞に比して、有意に発光量が変化している場合に、被験物質が脳神経系に対して作用すると評価する。このとき、被験物質の投与による発光量変化の大きさが、被験物質が有する脳神経系への作用の大きさを判断するための指標となる。
【0052】
ここで「被験物質」とは、ダイオキシン様活性を有する物質であり得る。本発明者は、ダイオキシン様活性を有する物質が、上述のエンハンサー領域に作用して下流遺伝子の活性を増強できることを見出し、既に報告している。ダイオキシン様活性を有する物質は、アリルハイドロカーボン受容体と複合体を形成し、かかる複合体がエンハンサー領域に結合し、これにより下流遺伝子の転写活性を増強すると推定される。
【0053】
図1にアリルハイドロカーボン受容体(AhR)による遺伝子の転写活性メカニズムの1つを示す。TCDDなどのダイオキシン類は、細胞内のAhRに結合して複合体を形成し、遺伝子の特定の配列に結合することにより、下流遺伝子の発現を活性化することが知られている。
【0054】
図1において、Arntはアリルハイドロカーボン受容体核内トランスロケーターを示す。ダイオキシン様活性を有する化学物質と結合したAhRは核内でArntと複合体を形成することによってエンハンサーと結合し、下流の遺伝子の発現を活性化することが知られている。
【0055】
本発明に従うと、遺伝子改変動物を使用した被験物質の有害性を判定する方法は、当該遺伝子改変動物と被験物質を接触させることと、当該レポーター遺伝子の発現を検出することを具備すればよい。
【0056】
例えば、そのような接触は、被験物質の経口投与であってもよく、例えば、腹腔内注射、静脈内注射、筋肉内注射、皮下注射、皮内注射、経膣投与、経腸投与、経皮投与、経鼻投与、経肺投与および点眼などの非経口投与などであってよい。また、被験物質への暴露も当該接触と同様の手段により行ってよい。
【0057】
そのような接触の後に、発現される当該レポーター遺伝子に適した手段により、当該遺伝子の発現の有無または強弱を検出又は測定すればよい。その場合、動物全体で観察および/または測定してもよく、または摘出した脳を用いて、所望に応じてホモジナイズ、切断、ミンスおよび/またはタンパク質の抽出などの処理を行った後に観察および/または測定してもよい。
【0058】
また、本発明の遺伝子改変動物を使用した被験物質の有害性を判定する方法は、当該遺伝子改変動物の一部と被験物質を接触させることにより行ってもよい。その場合、遺伝子改変動物の一部の器官または臓器などと、被験物質を接触させればよい。その後、発現されるレポーター遺伝子に適した手段により、当該遺伝子の発現の有無または強弱を検出または測定すればよい。
【0059】
以上、遺伝子改変動物の代表的な例として遺伝子改変マウスについて主に説明してきたが、マウス以外の動物についても同様にそれ自身公知の方法により当該DNAを導入し、遺伝子改変動物を得ることが可能であり、且つ上述した何れの項目についてもマウスと同様に適用することが可能である。また更に、何れの動物の場合においても、上述のマウスと同様に被験物質の有害性を判定する方法に使用することが可能である。
【0060】
[例]
以下に実施例を挙げて、本発明をより具体的に説明する。ただし、これらの実施例は説明のためのものであって、本範囲の技術範囲を限定するものではない。
【0061】
1)マウス導入用ベクターの調整
配列番号1に記載の配列を含むTH遺伝子の5’上流-175bp 〜 -237bpの領域を2回繰り返した1本鎖DNA(即ち、TH遺伝子のAhR結合エンハンサー(配列番号1)を2つ含む)を合成した。このDNAを鋳型として、特異的なプライマーでPCRをおこない、一本鎖DNAを2本鎖に変換した。一本鎖DNA(配列番号12)、及び特異的なプライマーの塩基配列(配列番号13および14)を以下に示した。
【表A】
【0062】
【化12】
【0063】
【化13】
【0064】
【化14】
【0065】
PCRで得た2本鎖DNAを、T4ポリヌクレオチドキナーゼでリン酸化後にT4 DNAリガーゼで連結し、アガロースゲル電気泳動で連結個数が3つのDNA断片を分離してから、QIA quick ゲル抽出キット(キアゲン)で精製した。精製したDNA断片は、AhR結合エンハンサーの6回繰り返しからなる。このDNAをルシフェラーゼ発現ベクターであるPGV-P2ベクター(TOYO B-NET)に組み込んだ。まず、PGV-P2上のSV40プロモーターを制限酵素Hin dIIIとXho Iで切り出して、TH遺伝子のプロモーター[転写開始点(0 bp)から5’上流 -100 bpまでの領域(配列番号4)]と組み換えたベクター(PGV-THp)を作製した。次に、PGV-THpのHin dIIIとNco Iサイトに、下記のc-mycタグ配列を挿入した。
【表B】
c-mycタグ配列は、上記の相補的な1本鎖DNAをアニーリングさせて得た。c-mycタグの末端には、ベクターへの組み込みを容易にするために、Hin dIII認識配列(5’末端)とNco I認識配列(3’末端)を付加した。2本鎖にしたc-mycタグ配列は、PGV-THpのHin dIIIとNco I部位に組み込んだ。この操作で得られたベクター、すなわちPGV-THp上のルシフェラーゼ遺伝子N末端にin-frameでc-mycタグ配列が付加されたベクター:PGV-THp-MLucをSma Iで切断し、このサイトにAhR結合エンハンサーの6回繰り返しとTH遺伝子のプロモーターからなるDNA配列(配列番号5)を組み込んで、マウス導入用のベクター:pTHEn-MLucを作製した。
【0066】
pTHEn-MLuc上のAhR結合エンハンサーからc-mycタグ配列の塩基配列は、シーケンシングで確認した。ベクター作製のフローを図2に示した。
【0067】
2) 遺伝子改変候補マウスの作出
C57BL6系統のマウスを使用した。遺伝子導入操作の3日前に、4-6週齢の雌マウスに対して、血清性性腺刺激ホルモン(PMSG)を腹腔内に注射し、前日には胎盤性性腺刺激ホルモン(hCG)を腹腔内注射した。注射後、このマウスを雄マウスと交配させて、翌日、膣栓があった雌マウスから卵管を摘出した。ヒアルロニダーゼを含む培養液中で卵管膨大部を破いて取り出した卵塊を暫く放置して卵丘細胞と受精卵を解離させてから、受精卵を採卵用ピペットで採取した。採取した受精卵は、培養液で数回洗浄してから、CO2インキュベーター内で培養した。
【0068】
受精卵に導入する遺伝子には、pTHEn-MLucから制限酵素Kpn IとBam HIで配列番号6で表されるDNA(図3を参照されたい、TH遺伝子エンハンサーとプロモーター::c-mycタグ-ルシフェラーゼ遺伝子::SV40ポリAシグナル)を切り出した後に精製し、TEバッファー[10mM Tris-HCl (pH7.5), 0.1mM EDTA]に1〜2 ng/μlに溶解したものを用いた。
【0069】
受精卵をCO2インキュベーターから取り出して、顕微鏡にセットしたチャンバーの培地に入れて、低倍率下、ホールディングピペットで保持してから、高倍率の微分干渉レンズ下で、受精卵に、先に述べた方法で調整した配列番号6で表されるDNAをインジェクションピペットで導入した。インジェクション後の受精卵は、培地を交換後にCO2インキュベーター内で24時間培養した。この受精卵を偽妊娠雌マウスに移植して発生させ、遺伝子改変候補マウスを作出した。
【0070】
3)ジェノタイピングによる遺伝子改変マウスの選択
遺伝子改変マウスは、2)の候補マウスの中からサザンブロット並びにPCRで、ゲノム上の導入DNA(配列番号6、受精卵に導入したDNA)の一部を検出して選択した。2 cm程度に切断したマウス(4-6週齢)の尾を遠心チューブに入れ、50℃で8から10時間プロテアーゼK処理をおこなった後、溶液に等量のフェノール/クロロホルム/イソアミルアルコール(25:24:1)を加えて混合してから、15,000 rpm,3分間の遠心で水層とフェノール層に分離した。新しいチューブに水層を取り分けて1/4量の10 M 酢酸アンモニウムと2.5 倍量のエタノールを加えてよく混ぜた後、15,000 rpm,3分間の遠心によって、溶液中のゲノムDNAを沈殿させて上清を除いた。沈殿を70% エタノールで洗浄してから風乾し、TEバッファーに再溶解してマウスのゲノムDNA溶液を得た。
【0071】
サザンブロッティングによるジェノタイピングでは、このゲノムDNA溶液に制限酵素Xho Iを加えて完全消化して、アガロースゲル電気泳動をおこなった後、キャピラリートランスファー法で、ゲル中のゲノムDNAナイロンメンブレンHybond N+(GEヘルスケア・バイオサイエンス)に転写した。メンブレンへのDNA固定は、紫外線照射でおこなった。このメンブレンをハイブリダイゼーションバッファー(1/20量 Denhardt’s溶液, 50 μg/mlサケ精子DNA)に浸して、プレ・ハイブリダイゼーションを65℃で1時間おこない、次に放射性同位体(32P)ラベルしたプローブDNAを含んだハイブリダイゼーションバッファー中で65℃、1晩のハイブリダイゼーションをおこなった。非特異的なプローブ吸着は、メンブレンを洗浄液A(2 x SSC, 0.1% SDS)中で65℃、5分間×2回、続いて洗浄液B(0.5 x SSC, 0.1% SDS)中で65℃,15分間×2回洗浄して除去した。ハイブリダイゼーション・プローブには、pTHEN-MLucのHin dIIIとSph I断片を放射線同位体(32P)ラベルしたものを用いた。ハイブリダイゼーションのシグナルはX線フィルムで検出した。この結果、#176、#177、#184の3匹の遺伝子改変候補マウスで導入DNAに由来するシグナルが検出された(図4)。
【0072】
PCRによるジェノタイピングは、マウスの尾から抽出したゲノムDNAを鋳型として、特異的なプライマーを使用して、pTHEn-MLucのルシフェラーゼ構造遺伝子の一部が増幅されるかどうかで判定した。以下に使用したプライマーの塩基配列を示した(配列番号15および配列番号16)。
【表C】
【0073】
【化15】
【0074】
【化16】
【0075】
増幅の確認は、アガロースゲル電気泳動でおこなった。PCRによるジェノタイピングでは、図5に示したように、#176、#177、#178、#184の4匹の遺伝子改変候補マウスで導入DNAに由来するシグナルが検出された。
【0076】
これら2種類のジェノタイピング、すなわちサザンハイブリダイゼーションとPCRの両方法で導入DNAのシグナルが検出された3匹のマウス(#176、#177、#184)を遺伝子改変マウスとして選択し、以降の実験に使用した。#176のマウスについては、受精卵と精子を凍結保存し、特許生物寄託センターへの寄託をおこなった(寄託番号FERM BP-10993)。
【0077】
3)遺伝子改変マウス・ラインの維持
2)で選択した3匹の遺伝子改変マウス(遺伝子改変マウスの0世代:T0世代目)をC57BL/6系統の野生型マウスと交配(戻し交配=バッククロス)して子孫を得た。この遺伝子改変マウスの子孫とC57BL/6系統の野生型マウス、或いは遺伝子改変マウスの子孫と交配することにより、遺伝子改変マウスを維持した。
【0078】
4)β遺伝子改変マウスにおけるNF暴露時のルシフェラーゼ発光の測定
被験物質にはダイオキシンのアナログであるβナフトフラボン(βNF)を用いた。#176(4週齢)、#177(6週齢)、#184(6週齢)のT1世代の遺伝子改変マウスにコーンオイルに溶解したβNFを40 mg/kgとなるように腹腔内に注射した。対照区のマウス(T1世代の遺伝子改変マウス)は、腹腔内にβNF投与区と等量のコーンオイルを投与した。投与から72時間後、これらのマウスの腹腔内にルシフェリン溶液を150 mg/kg BWとなるように投与してから、5分後にIVISシステム(in vivo imaging system 100 series、Xenogen社製)でマウスのルシフェラーゼ発光を検出したところ、全ての遺伝子改変マウスでルシフェラーゼ発光が観察された。(図6)。更に、#176の遺伝子改変マウスに関しては、脳は中脳付近で切断して、IVISシステムで切断面の発光を測定した。この結果、βNF投与マウスの中脳・海馬付近において発光量の増加が検出された(図7)。以上の結果から、遺伝子改変マウス(#176、#177、#184)を用いて、レポーター遺伝子の発現変化を指標に、βNFの脳神経系に対する作用を評価できることが示された。
【0079】
5)遺伝子改変マウスから分離した初代神経細胞におけるダイオキシン(TCDD)暴露時のルシフェラーゼ発光の測定
#176の遺伝子改変マウス(ヘテロ、♂)と野生型マウス(♀)を交配し、プラグの確認から12日後に、胎児(E12 = 胎齢12日)の脳を無菌環境下で摘出した。摘出した胎児の脳を全てまとめてから、0.25% トリプシンを加え、37℃で15分間インキュベートした。トリプシンを除いて、ハンクス溶液で3回洗浄後、細胞を血清入りDF1:1培地(ダルベッコ-F12等比混合培地+10%牛胎児血清)に縣濁してから、培養プレート(オルニチン-フィブロネクチンでコート)に播種し、37℃/5% CO2雰囲気下で培養した。一晩培養後、神経細胞を優先的に増殖させるために、培地を無血清培地[ダルベッコMEM/ハムF12等比混合培地+TIPS(トランスフェリン/インスリン/ペニシリン/ストレプトマイシン)]に交換して培養した(図8)。
【0080】
4日後、培地に終濃度が0,0.1,10 ng/mlとなるようにTCDD(2,3,7,8-テトラクロロジベンゾ-p-ジオキシン、関東化学)を添加し、24時間後に細胞を回収した。回収した細胞は、リン酸緩衝液で2回洗浄した後、200 μlのタンパク質抽出溶液(TOYO B-NET)を加えて15分間室温で攪拌後、細胞を凍結(-80℃)-融解(室温)によって破砕した。この細胞破砕液を1.5 mlチューブに回収後、遠心をおこない、上清をタンパク質抽出溶液として回収した。このようにして得たタンパク質抽出溶液をルシフェラーゼ発光の測定に使用した。ルシフェラーゼ発光は、タンパク質回収液10 μlにアッセイ溶液(ピッカジーン・アッセイ・キット、TOYO B-NET)を100 μlを加えて攪拌後、直ぐにルミノメーター LB-940(ベルトルード)で2分間当たりの発光量を測定した。得られた発光量は、タンパク質溶液の総タンパク量で除算して、相対発光強度(RLU / min / μg protein)に換算した。溶液の総タンパク量はプロテインアッセイ・キット(バイオラッド)で測定した。この結果、遺伝子改変マウスから分離した初代神経細胞は、0.1 nM TCDD暴露で未暴露時の1.3倍、10 nM TCDD暴露時には3.1倍と、TCDD濃度に依存した発光量の増加を示した(図9)。この結果より、遺伝子改変マウス(#176)から分離した初代神経細胞を用いて、レポーター遺伝子の発現変化を指標に、TCDDの脳神経系に対する作用を評価できることが示された。
【図面の簡単な説明】
【0081】
【図1】遺伝子転写活性メカニズムを示す図。
【図2】ベクター作製のフローを示す図。
【図3】受精卵に導入するDNAを示す図。
【図4】導入DNAに由来するシグナルを示すサザンハイブリダイゼーションの結果を示す図。
【図5】導入DNAに由来するシグナルを示す電気泳動の結果を示す図。
【図6】遺伝子改変マウスにおけるルシフェラーゼ発光を示す図。
【図7】マウス中脳・海馬付近における発光の増加を示す図。
【図8】培養神経細胞を示す図。
【図9】TCDD濃度に依存した発光量の増加を示す図。
【特許請求の範囲】
【請求項1】
チロシン水酸化酵素遺伝子の5’上流に存在するアリルハイドロカーボン受容体結合エンハンサーと、任意のプロモーターと、レポーター遺伝子と、ポリ(A)付加シグナルとを機能的に連結したDNAを導入された遺伝子改変動物。
【請求項2】
前記動物がマウスである遺伝子改変動物。
【請求項3】
前記エンハンサーが、配列番号1で表される塩基配列を含むことを特徴とする、請求項2に記載の遺伝子改変動物。
【請求項4】
前記エンハンサーが、配列番号2で表される塩基配列を含むことを特徴とする、請求項2に記載の遺伝子改変動物。
【請求項5】
前記エンハンサーが、配列番号3で表される塩基配列を含むことを特徴とする、請求項2に記載の遺伝子改変動物。
【請求項6】
前記プロモーターが、配列番号4で表される塩基配列からなることを特徴とする、請求項2に記載の遺伝子改変動物。
【請求項7】
前記エンハンサーとプロモーターを連結した塩基配列が、配列番号5で表される塩基配列からなることを特徴とする、請求項2に記載の遺伝子改変動物。
【請求項8】
前記レポーター遺伝子が、ルシフェラーゼ遺伝子、緑色蛍光タンパク質遺伝子、βガラクトシダーゼ遺伝子およびクロラムフェニコールアセチルトランスフェラーゼ遺伝子からなる群より選択されることを特徴とする、請求項1から7の何れか1項に記載の遺伝子改変動物。
【請求項9】
請求項8に記載の遺伝子改変動物であって、前記レポーター遺伝子が、ルシフェラーゼ遺伝子であることを特徴とする遺伝子改変動物。
【請求項10】
前記DNAが、配列番号6で表される塩基配列からなることを特徴とする、請求項2に記載の遺伝子改変動物。
【請求項11】
受託番号FERM BP-10993である請求項10に記載の遺伝子改変動物。
【請求項12】
請求項1から11の何れか1項に記載の遺伝子改変動物であって、前記レポーター遺伝子の発現が脳神経系において誘導されることを特徴とする遺伝子改変動物。
【請求項13】
請求項1から11の何れか1項に記載の遺伝子改変動物またはその一部と、被験物質とを接触させることと、当該レポーター遺伝子の発現を検出することと、を具備する被験物質の有害性を判定する方法。
【請求項14】
請求項9、10または11に記載の遺伝子改変動物またはその一部と、被験物質とを接触させることと、当該ルシフェラーゼ遺伝子の発現を検出することとを具備する被験物質の有害性を判定する方法。
【請求項15】
請求項1から11の何れか1項に記載の遺伝子改変動物またはその一部と、被験物質とを接触させることと、被験物質のドーパミン合成への影響を検出する方法。
【請求項16】
請求項9、10または11の何れか1項に記載の遺伝子改変動物またはその一部と、被験物質とを接触させることと、被験物質により誘導されるルシフェラーゼ遺伝子の発現を検出することによって、被験物質のドーパミン合成への影響を判定する方法。
【請求項1】
チロシン水酸化酵素遺伝子の5’上流に存在するアリルハイドロカーボン受容体結合エンハンサーと、任意のプロモーターと、レポーター遺伝子と、ポリ(A)付加シグナルとを機能的に連結したDNAを導入された遺伝子改変動物。
【請求項2】
前記動物がマウスである遺伝子改変動物。
【請求項3】
前記エンハンサーが、配列番号1で表される塩基配列を含むことを特徴とする、請求項2に記載の遺伝子改変動物。
【請求項4】
前記エンハンサーが、配列番号2で表される塩基配列を含むことを特徴とする、請求項2に記載の遺伝子改変動物。
【請求項5】
前記エンハンサーが、配列番号3で表される塩基配列を含むことを特徴とする、請求項2に記載の遺伝子改変動物。
【請求項6】
前記プロモーターが、配列番号4で表される塩基配列からなることを特徴とする、請求項2に記載の遺伝子改変動物。
【請求項7】
前記エンハンサーとプロモーターを連結した塩基配列が、配列番号5で表される塩基配列からなることを特徴とする、請求項2に記載の遺伝子改変動物。
【請求項8】
前記レポーター遺伝子が、ルシフェラーゼ遺伝子、緑色蛍光タンパク質遺伝子、βガラクトシダーゼ遺伝子およびクロラムフェニコールアセチルトランスフェラーゼ遺伝子からなる群より選択されることを特徴とする、請求項1から7の何れか1項に記載の遺伝子改変動物。
【請求項9】
請求項8に記載の遺伝子改変動物であって、前記レポーター遺伝子が、ルシフェラーゼ遺伝子であることを特徴とする遺伝子改変動物。
【請求項10】
前記DNAが、配列番号6で表される塩基配列からなることを特徴とする、請求項2に記載の遺伝子改変動物。
【請求項11】
受託番号FERM BP-10993である請求項10に記載の遺伝子改変動物。
【請求項12】
請求項1から11の何れか1項に記載の遺伝子改変動物であって、前記レポーター遺伝子の発現が脳神経系において誘導されることを特徴とする遺伝子改変動物。
【請求項13】
請求項1から11の何れか1項に記載の遺伝子改変動物またはその一部と、被験物質とを接触させることと、当該レポーター遺伝子の発現を検出することと、を具備する被験物質の有害性を判定する方法。
【請求項14】
請求項9、10または11に記載の遺伝子改変動物またはその一部と、被験物質とを接触させることと、当該ルシフェラーゼ遺伝子の発現を検出することとを具備する被験物質の有害性を判定する方法。
【請求項15】
請求項1から11の何れか1項に記載の遺伝子改変動物またはその一部と、被験物質とを接触させることと、被験物質のドーパミン合成への影響を検出する方法。
【請求項16】
請求項9、10または11の何れか1項に記載の遺伝子改変動物またはその一部と、被験物質とを接触させることと、被験物質により誘導されるルシフェラーゼ遺伝子の発現を検出することによって、被験物質のドーパミン合成への影響を判定する方法。
【図1】

【図2】
【図3】
【図4】

【図5】
【図6】
【図7】
【図8】
【図9】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公開番号】特開2010−75065(P2010−75065A)
【公開日】平成22年4月8日(2010.4.8)
【国際特許分類】
【出願番号】特願2008−244392(P2008−244392)
【出願日】平成20年9月24日(2008.9.24)
【出願人】(000003078)株式会社東芝 (54,554)
【Fターム(参考)】
【公開日】平成22年4月8日(2010.4.8)
【国際特許分類】
【出願日】平成20年9月24日(2008.9.24)
【出願人】(000003078)株式会社東芝 (54,554)
【Fターム(参考)】
[ Back to top ]