
複合材料
【課題】グラフェンライク炭素材料の樹脂からなる基材に対する密着性に優れた複合材料を提供する。
【解決手段】樹脂からなる基材と、基材表面の少なくとも一部を覆うように設けられたグラフェンライク炭素材料層とを備え、基材の表面にグラフェンライク炭素が密着している複合材料。
【解決手段】樹脂からなる基材と、基材表面の少なくとも一部を覆うように設けられたグラフェンライク炭素材料層とを備え、基材の表面にグラフェンライク炭素が密着している複合材料。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、樹脂と、グラフェンもしくは薄片化黒鉛のようなグラフェンライク炭素材料との複合材料に関し、より詳細には、樹脂からなる基材に対するグラフェンライク炭素材料の密着性が高められている複合材料及びその製造方法に関する。
【背景技術】
【0002】
従来、黒鉛、カーボンナノチューブ、カーボンファイバーまたはカーボン粒子などの炭素材料が、樹脂への補強剤や充填剤として広く用いられている。また、近年、黒鉛を剥離して得られる、グラフェン積層数がより少ない薄片化黒鉛がより注目されている。
【0003】
上記のような炭素材料と樹脂との複合材料として、例えば下記の特許文献1に示すように、エポキシ樹脂中に炭素繊維などの炭素材料を分散させてなる複合材料が知られている。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特開2003−277471号公報
【発明の概要】
【発明が解決しようとする課題】
【0005】
しかしながら、従来の樹脂と炭素材料との複合材料では、樹脂と炭素材料との密着強度が充分でないという問題があった。例えば、特許文献1に記載のように樹脂中に炭素材料を分散させてなる複合材料では、添加されている炭素材料により物性の改善が図られているものの、炭素材料との密着強度は充分ではなかった。
【0006】
本発明の目的は、上述した従来技術の欠点を解消し、樹脂とグラフェンライク炭素材料との密着性に優れた複合材料及びその製造方法を提供することにある。
【課題を解決するための手段】
【0007】
本発明に係る複合材料は、樹脂からなる基材と、前記基材表面の少なくとも一部を覆うように設けられたグラフェンライク炭素材料層とを備え、前記基材の表面にグラフェンライク炭素が密着している。
【0008】
本発明に係る複合材料のある特定の局面では、グラフェンライク炭素の一部が基材の表面から内部に入り込んでいる。従って、両者の密着性がより一層高められる。
【0009】
本発明に係る複合材料の他の特定の局面では、前記樹脂からなる基材が、樹脂微粒子であり、前記樹脂微粒子の外表面を覆うように前記グラフェンライク炭素材料層が形成されている。この場合には、樹脂微粒子の外表面が、グラフェンライク炭素材料層で覆われており、かつグラフェンライク炭素の一部が樹脂微粒子表面に入り込んでいるため、樹脂微粒子とグラフェンライク炭素材料層との密着性が高められている。しかも、グラフェンライク炭素材料層表面に有する微粒子状の複合材料は、凝集し難い。従って、いわゆる自由流動性粉体(free flowing powder)として取り扱うことができる。
【0010】
本発明に係る複合材料の他の特定の局面では、前記樹脂からなる基材がシート状基材であって、該シート状基材の少なくとも一方面に前記グラフェンライク炭素材料層が設けられている。この場合には、本発明に従って、炭素材料層のシート状基材との密着性に優れたシート状の複合材料を提供することができる。
【0011】
本発明に係る複合材料では、好ましくは、グラフェンライク炭素材料がグラフェンまたは薄片化黒鉛からなる。グラフェンまたは薄片化黒鉛はアスペクト比が大きく、かつ、グラフェン積層数が少ないため、少量の添加で複合材料の物性を高めることができる。
【0012】
本発明に係る複合材料の製造方法は、本発明に従って構成された複合材料の製造方法であって、樹脂からなる基材と、グラフェンライク炭素材料とを用意する工程と、前記樹脂からなる基材の表面の少なくとも一部に前記グラフェンライク炭素材料を接触させ、超臨界または亜臨界状態の流体を作用させつつ加熱する工程とを備える。
【0013】
本発明に係る製造方法では、好ましくは、前記超臨界または亜臨界の流体として、超臨界または亜臨界状態のCO2を用いる。CO2は、31.1℃程度の温度かつ7.52MPa程度の圧力下で超臨界状態となる。従って、H2Oなどに比べて、穏やかな条件で樹脂からなる基材の表面を膨潤させることができる。そのため、ガラス転移温度が低い樹脂を用いた場合においても、本発明の複合材料を確実に得ることができる。
【発明の効果】
【0014】
本発明に係る複合材料では、樹脂からなる基材にグラフェンライク炭素が密着しているため、グラフェンライク炭素材料層と基材との密着性に優れた複合材料を得ることが可能となる。
【0015】
また、本発明に係る製造方法によれば、超臨界または亜臨界状態の流体を樹脂に作用させつつ加熱することにより、樹脂からなる基材の表面にグラフェンライク炭素が密着するようにして、樹脂表面にグラフェンライク炭素材料層が形成される。よって、樹脂からなる基材に対して密着性に優れたグラフェンライク炭素材料層を有する本発明の複合材料を得ることができる。また、本発明の製造方法では、上記のようにして、グラフェンライク炭素材料層を基材表面に形成するため、基材の形状も特に限定されない。よって、樹脂微粒子のような微粒子状、シート状基材などの形状に限らず、複雑な形状の樹脂からなる基材の表面にも、本発明に従ってグラフェンライク炭素材料層を容易に形成することができる。
【0016】
また、被分散体である炭素材料微粒子は凝集性が高く、均一に分散させ、付着させる事は容易ではなかったところ、発明の製造方法では、容易に炭素材料微粒子を基材表面に密着させることができる。
【図面の簡単な説明】
【0017】
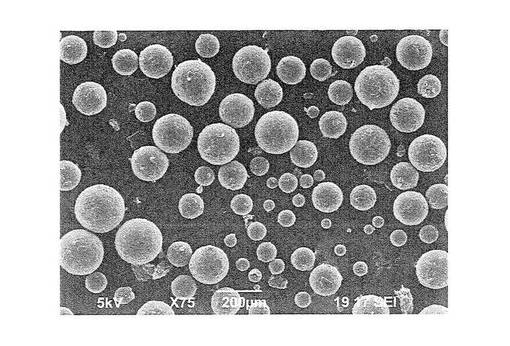
【図1】図1は、ポリメチルメタクリレート微粒子1gと薄片化黒鉛0.01gを混合し、超臨界二酸化炭素(65℃、35MPa)で5時間作用させた後の微粒子の走査型電子顕微鏡写真(75倍)である。
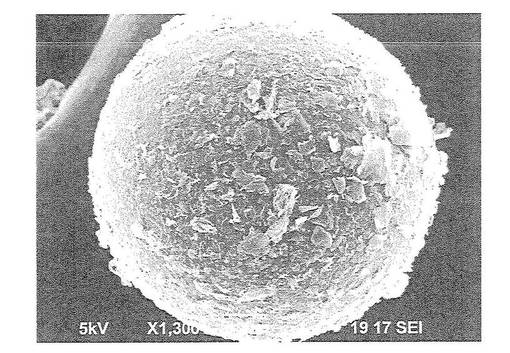
【図2】図2は、ポリメチルメタクリレート微粒子1gと薄片化黒鉛0.01gを混合し、超臨界二酸化炭素(65℃、35MPa)で5時間作用させた後の微粒子の走査型電子顕微鏡写真(1300倍)である。
【図3】図3は、ポリメチルメタクリレート微粒子1gと薄片化黒鉛0.01gを混合し、超臨界二酸化炭素(65℃、35MPa)で5時間作用させた後の微粒子の断面の走査型電子顕微鏡写真(550倍)である。
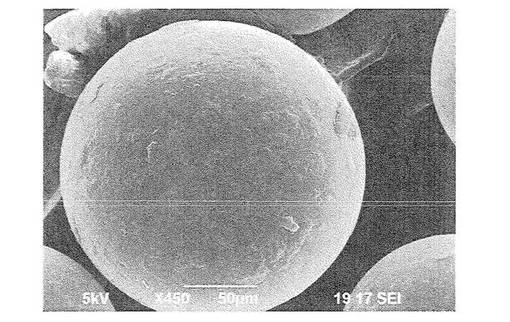
【図4】図4は、ポリメチルメタクリレート微粒子1gと薄片化黒鉛0.005gを混合し、超臨界二酸化炭素(65℃、35MPa)で5時間作用させた後の微粒子の走査型電子顕微鏡写真(450倍)である。
【図5】図5は、ポリメチルメタクリレート微粒子を超臨界二酸化炭素(65℃、35MPa)で5時間作用させた後の微粒子の走査型電子顕微鏡写真(120倍)である。
【図6】図6は、ポリスチレン微粒子(積水化成品工業株式会社製の品番S−20)1gと薄片化黒鉛0.01gを混合し、超臨界二酸化炭素(55℃、28MPa)で12時間作用させた後の微粒子の走査型電子顕微鏡写真(200倍)である。
【図7】図7は、ポリスチレン微粒子(積水化成品工業株式会社製の品番S−20)1gと薄片化黒鉛0.01gを混合し、超臨界二酸化炭素(55℃、28MPa)で12時間作用させた後の微粒子の走査型電子顕微鏡写真(500倍)である。
【図8】図8は、ポリスチレン微粒子(積水化成品工業株式会社製の品番S−20)を超臨界二酸化炭素(55℃、28MPa)で5時間作用させた後の微粒子の走査型電子顕微鏡写真(200倍)である。
【図9】図9は、ポリスチレン微粒子(積水化成品工業株式会社製の品番S−30)1gと薄片化黒鉛0.001gを混合し、超臨界二酸化炭素(60℃、28MPa)で4.5時間作用させた後の微粒子の走査型電子顕微鏡写真(100倍)である。
【図10】図10は、ポリスチレン微粒子(積水化成品工業株式会社製の品番S−30)1gと薄片化黒鉛0.001gを混合し、超臨界二酸化炭素(60℃、28MPa)で4.5時間作用させた後の微粒子の走査型電子顕微鏡写真(500倍)である。
【図11】図11は、ポリスチレン微粒子(積水化成品工業株式会社製の品番S−30)を超臨界二酸化炭素(60℃、28MPa)で4.5時間作用させた後の微粒子の走査型電子顕微鏡写真(100倍)である。
【図12】図12は、ポリスチレン微粒子(積水化成品工業株式会社製の品番S−30)を超臨界二酸化炭素(60℃、28MPa)で4.5時間作用させた後の微粒子の走査型電子顕微鏡写真(500倍)である。
【図13】図13は、ポリスチレン微粒子(積水化成品工業株式会社製の品番S−40)1gと薄片化黒鉛0.005gを混合し、超臨界二酸化炭素(60℃、28MPa)で4.5時間作用させた後の微粒子の走査型電子顕微鏡写真(100倍)である。
【図14】図14は、ポリスチレン微粒子(積水化成品工業株式会社製の品番S−40)を超臨界二酸化炭素(60℃、28MPa)で4.5時間作用させた後の微粒子の走査型電子顕微鏡写真(100倍)である。
【図15】図15は、ポリスチレン及びメタクリル酸2−ヒドロキシエチルの共重合体からなる微粒子(積水化成品工業株式会社製の品番CS−10)1gと薄片化黒鉛0.3gを混合し、超臨界二酸化炭素(35℃、21MPa)で12時間作用させた後の微粒子の走査型電子顕微鏡写真(500倍)である。
【図16】図16は、ポリスチレン及びメタクリル酸2−ヒドロキシエチルの共重合体からなる微粒子(積水化成品工業株式会社製の品番CS−10)1gと薄片化黒鉛0.3gを混合し、超臨界二酸化炭素(35℃、21MPa)で12時間作用させた後の微粒子の走査型電子顕微鏡写真(5000倍)である。
【図17】図17は、ポリスチレン及びメタクリル酸2−ヒドロキシエチルの共重合体からなる微粒子(積水化成品工業株式会社製の品番CS−10)を超臨界二酸化炭素(35℃、21MPa)で12時間作用させた後の微粒子の走査型電子顕微鏡写真(450倍)である。
【図18】図18は、ポリスチレン及びメタクリル酸2−ヒドロキシエチルの共重合体からなる微粒子(積水化成品工業株式会社製の品番CS−50)1gと薄片化黒鉛0.001gを混合し、超臨界二酸化炭素(室温、28MPa)で12時間作用させた後の微粒子の走査型電子顕微鏡写真(70倍)である。
【図19】図19は、ポリスチレン及びメタクリル酸2−ヒドロキシエチルの共重合体からなる微粒子(積水化成品工業株式会社製の品番CS−50)を超臨界二酸化炭素(室温、28MPa)で12時間作用させた後の微粒子の走査型電子顕微鏡写真(80倍)である。
【発明を実施するための形態】
【0018】
以下、本発明の具体的な実施形態を説明することにより、本発明を明らかにする。
【0019】
(樹脂からなる基材)
本発明に係る複合材料及びその製造方法では、樹脂からなる基材を用いる。基材を構成する樹脂については、超臨界または亜臨界状態の流体を作用させつつ加熱することにより、表面を柔らかくし得る適宜の樹脂を用いることができる。このような樹脂は、合成樹脂であってもよく、天然樹脂であってもよい。
【0020】
上記樹脂としては、超臨界または亜臨界状態の流体が作用する温度において軟化する程度のガラス転移温度Tgを有するものが好ましい。後述するように、CO2が超臨界または亜臨界状態で作用させる流体として好適に用いられる。従って、樹脂としては、ポリスチレン、ポリプロピレン、ポリメチルメタクリレート(PMMA)、セルロースなどを好適に用いることができる。また、樹脂は、これらのポリマーを構成するモノマーの共重合体であってもよい。もっとも、本発明において用いる樹脂材料としては、PMMA以外、様々な(メタ)アクリル系樹脂、ポリプロピレン以外の様々なポリオレフィン等も用いることができる。
【0021】
上記樹脂からなる基材の形状についても特に限定されない。基材は、樹脂微粒子であってもよい。すなわち、微粒子状基材であってもよい。なお、微粒子の直径は特に限定されないが、平均200μm以下の微粒子が好適に用いられ、これよりも大きい粒子状の樹脂からなる基材を用いてもよい。樹脂からなる基材が粒子状である場合、本発明に従って得られた複合材料では、後述するように、凝集が生じ難い。従って、自由流動性粉末(free flowing powder)として取り扱うことができる。
【0022】
また、樹脂からなる基材は、シート状であってもよい。シート状基材の場合には、シート状基材の一面及び/または反対側の面の少なくとも一部に本発明に従って、グラフェンライク炭素材料層を形成することができる。
【0023】
さらに、本発明で用いられる樹脂からなる基材は、粒子状やシート状である必要は必ずしもない。すなわち、グラフェンライク炭素材料を樹脂からなる基材表面の少なくとも一部に接触させ、その状態で超臨界または亜臨界状態の流体を作用させ得る限り、樹脂からなる基材の形状は複雑な立体的形状を有するものであってもよい。その場合においても、本発明に従って、表面にグラフェンライク炭素材料層を有する複雑な立体的形状を有する複合材料を得ることができる。
【0024】
また、複雑な表面の基材の一部に、グラフェンライク炭素材料を選択的に形成することもできる。
【0025】
(グラフェンライク炭素材料層)
本発明に係る複合材料では、上記樹脂からなる基材の表面の少なくとも一部を覆うように、グラフェンライク炭素材料層が設けられている。このグラフェンライク炭素材料層を構成するグラフェンライク炭素材料としては、グラフェンまたは薄片化黒鉛を好適に用いることができる。周知のように、黒鉛は、グラフェンの積層体である。薄片化黒鉛は、黒鉛を剥離することにより得られ、薄片化黒鉛の積層体におけるグラフェンの積層数は数層〜200層程度である。また、薄片化黒鉛の比表面積は黒鉛よりもはるかに大きく、600m2/g以上である。本発明において、上記薄片化黒鉛とは、黒鉛を剥離することにより得られ、上記のようなグラフェン積層数のグラフェン積層体をいうものとする。
【0026】
上記薄片化黒鉛としては、市販されている薄片化黒鉛を用いてもよい。また、黒鉛を剥離する様々な処理により薄片化黒鉛を得てもよい。
【0027】
上記のように薄片化黒鉛を得る方法としては特に限定されず、黒鉛を膨張して行われた膨張化黒鉛を剥離することにより得ることができる。黒鉛を膨潤し膨張化黒鉛とする工程は、1)電解質溶液中に層状黒鉛を浸漬し加熱する方法、及び2)電気分解法などを用いることができる。
【0028】
1)の方法では、硝酸や硫酸中に層状黒鉛を浸漬し、加熱し、硝酸イオンや硫酸イオンを層間にインターカレートする方法である。この場合、硝酸濃度及び硫酸濃度は、40重量%〜70重量%程度であることが望ましい。この範囲内であれば、硝酸イオンや硫酸イオンを確実に層間にインターカレートすることができる。また、加熱温度については、20℃以上、50℃以下であることが好ましい。この範囲内の温度であれば、上記硝酸イオンや硫酸イオンを確実に層間にインターカレートすることができる。
【0029】
2)の電気分解法では、層状黒鉛を作用極とし、該作用極をPtなどからなる対照極と共に硝酸や硫酸中に浸漬し、電気分解する。それによって、層状黒鉛の層間すなわちグラフェン間に硝酸イオンや硫酸イオン等の電解質イオンをインターカレートすることができ、層間を広げることができる。
【0030】
次に、上記のようにして得られた膨張化黒鉛からなるシートを水等により洗浄し、乾燥し、硝酸イオンや硫酸イオン等を除去する。このようにして、乾燥した膨張化黒鉛からなるシートを得ることができる。膨張化黒鉛を剥離して薄片化黒鉛を得るには、加熱、超音波を加える方法などを用いることができる。
【0031】
なお、本発明におけるグラフェンライク炭素材料としては、グラフェンまたは薄片化黒鉛に限らず、カーボンナノチューブなどの表面にグラフェンシート構造を有する様々なグラフェンライク炭素材料を用いてもよい。
【0032】
本発明においては、上記グラフェンライク炭素材料層の厚みは特に限定されず、用途に応じて適宜選択すればよい。もっとも、樹脂微粒子からなる基材表面にグラフェンライク炭素材料層を設ける場合には、グラフェンライク炭素材料層の厚みは0.5nm〜500nm程度である。また、シート状基材の少なくとも一方面にグラフェンライク炭素材料層を設ける場合においても、0.5nm〜500nm程度の厚みとすればよい。
【0033】
グラフェンライク炭素材料層の厚みが厚すぎると、樹脂物性の効果が発現しなくなることがある。逆に、グラフェンライク炭素材料層の厚みが薄くなりすぎると、グラフェンライク炭素材料層を設けたことによる物性改善効果が充分に得られないことがある。
【0034】
本発明においては、上記グラフェンライク炭素材料層を構成しているグラフェンライク炭素の一部が、基材の表面に密着している。好ましくは、グラフェンライク炭素の一部が基材の表面から内部に向かって入り込んでいる。そのため、アンカー効果により、グラフェンライク炭素材料層と樹脂からなる基材との密着性が効果的に高められる。
【0035】
グラフェンライク炭素の基材表面への密着性に優れた本発明の複合材料、より好ましくは、グラフェンライク炭素の一部が基材の表面に内部に入り込んでいる本発明の複合材料は、本発明の製造方法に従って得ることができる。
【0036】
(製造方法)
本発明の製造方法では、先ず、樹脂からなる上記基材と、上記グラフェンライク炭素材料とを用意する。次に、樹脂からなる基材の表面の少なくとも一部に、上記グラフェンライク炭素材料を接触させ、その状態で、超臨界または亜臨界状態の流体を作用させつつ加熱する。超臨界または亜臨界状態の流体としては、CO2、H2O、などを用いることができる。
【0037】
CO2は、31.1℃の温度及び7.52Mpa程度の気圧で超臨界状態となる。また、−56.6℃〜31.1℃及び0.528Mpa〜7.52MPa程度の気圧の範囲で亜臨界状態となる。超臨界または亜臨界状態の流体を作用させつつ加熱することにより、樹脂からなる基材表面が軟化する。従って、グラフェンライク炭素が軟化した基材表面と接触する。また、好ましくはグラフェンライク炭素の一部が基材表面に入り込む。そのため、加熱後冷却すれば、グラフェンライク炭素を基材表面と密着するようにして、基材表面の少なくとも一部を覆うようにグラフェンライク炭素材料層が形成される。すなわち、本発明の複合材料を得ることができる。
【0038】
よって、上記樹脂からなる基材を構成する樹脂のガラス転移温度Tgが、上記超臨界または亜臨界状態の流体を作用させつつ加熱する工程における温度雰囲気にあることが望ましい。より具体的には、樹脂のガラス転移温度Tgは、上記加熱温度−100℃〜+100℃の範囲にあることが望ましい。この範囲内にあれば、樹脂からなる基材表面にグラフェンライク炭素の一部を確実に入り込ませることができる。
【0039】
上記樹脂からなる基材の表面にグラフェンライク炭素材料を接触させるにあたっては、上記の通り、基材の表面の少なくとも一部に接触させればよい。もっとも、基材の全表面にグラフェンライク炭素材料を接触させてもよい。
【0040】
さらに、上記のように、基材表面の少なくとも一部にグラフェンライク炭素材料を接触させた状態で超臨界または亜臨界状態の流体を作用させつつ加熱するものであるため、基材表面の一部に選択的にグラフェンライク炭素材料を接触させ、基材表面の一部に選択的にグラフェンライク炭素材料層が設けられている複合材料も容易に得ることができる。加えて、前述したように、複雑な立体的な形状を有する基材を用いた場合においても、その表面に本発明に従ってグラフェンライク炭素材料層を容易にかつ確実に形成することができる。
【0041】
(複合材料の物性)
本発明に係る複合材料では、上記のように、グラフェンライク炭素材料の基材表面への密着性が高められ、好ましくは、グラフェンライク炭素材料の一部が樹脂からなる基材表面に入り込むようにして、グラフェンライク炭素材料層が形成されている。従って、グラフェンライク炭素材料層と基材との密着性を効果的に高め得る。よって、熱履歴を伴う環境に晒されたとしても、グラフェンライク炭素材料層の基材からの剥離等が生じ難い。また、密着性に優れているため、炭素材料による機械的強度の改善等も効果的に図ることができる。
【0042】
加えて、本願発明者の実験によれば、例えば、PMMAに上記グラフェンライク炭素材料層を本発明に従って形成してなる複合材料では、複合材料のガラス転移温度Tgも高められることがわかった。従って、耐熱性に優れた複合材料を提供することができる。このように、複合材料のTgが高められるのは、グラフェンライク炭素材料とPMMAの表面との密着性が高められるため、グラフェンライク炭素と樹脂との相互作用が強くなることによると考えられる。
【0043】
なお、PMMAに限らず、他の樹脂を用いた場合においても、本発明に従ってグラフェンライク炭素材料層を形成することにより、複合材料のTgを元の樹脂のTgに比べて効果的に高めることができる。従って、耐熱性に優れた複合材料を提供することが可能となる。
【0044】
以下、本発明の実施例及び比較例を説明する。なお、本発明は以下の実施例に限定されるものではない。
【0045】
(実施例1)
1)薄片化黒鉛の調製
原料の黒鉛シートとして東洋炭素社製、品番:PF100−UHPを用意した。この黒鉛シートと同じ製法で、圧延処理時の圧延倍率を下げて密度0.7、厚み1mmの低密度黒鉛シートを用意した。
【0046】
上記のようにして得られた密度0.7の黒鉛シートを3cm×3cmの大きさに切断し、電極材料としての黒鉛シートを得た。この黒鉛シートに、2本のスリットを、スリットの長さが1cm、幅が1cmとなるようにカッターナイフにより切削し、形成した。上記2本のスリットが形成された黒鉛シートに、Ptからなる電極を挿入した。このようにして用意した黒鉛シートを作用極(陽極)として、Ptからなる対照極(陰極)及び、Ag/AgClからなる参照極とともに60重量%濃度の硝酸水溶液中に浸漬し、直流電圧を印加し電気化学処理を行った。このようにして、陽極に作用極として用いた黒鉛を膨張化黒鉛とした。
【0047】
次に、得られた膨張化黒鉛を乾燥し、1cm角に切断し、その1つをカーボンるつぼに入れて電磁誘導加熱処理を行った。誘導加熱装置はSKメディカル社製MU1700Dを用い、アルゴンガス雰囲気下で最高到達温度550℃となるように10Aの電流量で行った。電磁誘導加熱により膨張化黒鉛は薄片化され、得られた薄片化黒鉛の粉末を島津製作所製の比表面積測定装置ASAP−2000で窒素ガスを用いて測定したところ、1回測定で850m2/gの比表面積を示した。
【0048】
2)複合材料の製造
樹脂からなる基材として、ポリメチルメタクリレートからなる微粒子(Aldrich社製、品番:445746−500G、Mw:35万、Tg:122℃)を用意した。このポリメチルメタクリレートからなる微粒子1.0gと、上記のようにして得られた薄片化黒鉛0.01gを圧力容器内に配置し、室温(23℃)及び10MPaの圧力で超臨界状態とされているCO2を10mL加えた後、いったんCO2を除去した(水分を除去し、乾燥させるため)。その後、再度室温(23℃)及び10MPaの圧力で超臨界状態とされているCO2を10mL加えた。その後、温度を65℃まで上昇させ5時間かき混ぜながら加熱した。このときの圧力は約35MPaまで上昇した。しかる後、室温まで冷却し、複合材料を得た。得られた複合材料は粒子状であるその平均粒径は120μmであった。また、このようにして得られた複合材料としての粒子の表面を走査型電子顕微鏡(日本電子社製 JCM−5700)により観察した。
【0049】
図1は、実施例1で得た複合材料としての粒子の75倍の電子顕微鏡写真を示し、図2はその表面を1300倍に拡大して示す電子顕微鏡写真である。
【0050】
上記粒子を乳鉢を用いて部分的に切断し、部分的に切断されている粒子を同じく走査型電子顕微鏡(日本電子社製 JCM−5700)により観察した。図3は、このようにして得られた550倍の倍率の走査型電子顕微鏡写真を示す。図1〜図3から明らかなように、得られた複合材料では、微粒子表面にグラフェンライク炭素材料が形成されており、特に図3の写真から、グラフェンライク炭素は基材粒子表面に集積していることがわかる。
【0051】
(実施例2)
実施例1と同様の薄片化黒鉛0.005gとポリメチルメタクリレートからなる微粒子(Aldrich社製、品番:445746−500G、Mw:35万、Tg:122℃)1.0gを混合し、実施例1と同様に圧力容器内に配置し、室温(23℃)及び10MPaの圧力で超臨界状態とされているCO2を10mL加えた後、いったんCO2を除去した(水分を除去し、乾燥させるため)。その後、再度室温(23℃)及び10MPaの圧力で超臨界状態とされているCO2を10mL加えた。その後、温度を65℃まで上昇させ5時間かき混ぜながら加熱した。このときの圧力は約35MPaまで上昇した。しかる後、室温まで冷却し、複合材料を得た。得られた複合材料は粒子状であるその平均粒径は120μmであった。また、このようにして得られた複合材料としての粒子の表面を走査型電子顕微鏡(日本電子社製 JCM−5700)により観察した。
【0052】
図4は、実施例2で得た複合材料としての粒子の450倍の走査型電子顕微鏡写真である。図4の写真から、グラフェンライク炭素の一部が樹脂からなる元の基材粒子表面の内側に入り込んでいることがわかる。
【0053】
(比較例1)
比較例として、実施例で用意した材料としてのポリメチルメタクリレート微粒子を用意した。このポリメチルメタクリレート微粒子の120倍の走査型電子顕微鏡写真を図5に示す。図5から明らかなように、このポリメチルメタクリレート微粒子では、表面にグラフェンライク炭素が全く存在しないため、平滑な表面を呈していることがわかる。
【0054】
(実施例3)
実施例1と同様の薄片化黒鉛0.01gとポリスチレンからなる微粒子(積水化成品工業株式会社製、品番:S−20、平均粒子径:300μm、Tg:106℃)1.0gを混合し、実施例1と同様に圧力容器内に配置し、室温(23℃)及び10MPaの圧力で超臨界状態とされているCO2を10mL加えた後、いったんCO2を除去した(水分を除去し、乾燥させるため)。その後、再度室温(23℃)及び10MPaの圧力で超臨界状態とされているCO2を10mL加えた。その後、温度を55℃まで上昇させ12時間かき混ぜながら加熱した。このときの圧力は約28MPaまで上昇した。しかる後、室温まで冷却し、複合材料を得た。得られた複合材料としての粒子の表面を走査型電子顕微鏡(日本電子社製 JCM−5700)により観察した。
【0055】
図6及び図7は、ぞれぞれ、実施例3で得た複合材料としての粒子の200倍及び500倍の走査型電子顕微鏡写真である。図6及び図7の写真から、グラフェンライク炭素の一部が樹脂からなる元の基材粒子表面の内側に入り込んでいることがわかる。
【0056】
(比較例2)
実施例3で用意した材料としてのポリスチレン微粒子を用意した。このポリスチレン微粒子の200倍の走査型電子顕微鏡写真を図8に示す。図8から明らかなように、このポリスチレン微粒子では、表面にグラフェンライク炭素が全く存在しないため、平滑な表面を呈していることがわかる。
【0057】
(実施例4)
実施例1と同様の薄片化黒鉛0.001gとポリスチレンからなる微粒子(積水化成品工業株式会社製、品番:S−30、平均粒子径:800μm、Tg:105℃)1.0gを混合したこと、及び温度を60℃まで上昇させ4.5時間かき混ぜながら加熱したこと以外は、実施例3と同様にして、複合材料を得た。なお、混合時の圧力は28MPaまで上昇した。得られた複合材料としての粒子の表面を走査型電子顕微鏡(日本電子社製 JCM−5700)により観察した。
【0058】
図9及び図10は、それぞれ、実施例4で得た複合材料としての粒子の100倍及び500倍の走査型電子顕微鏡写真である。図5の写真から、グラフェンライク炭素の一部が樹脂からなる元の基材粒子表面の内側に入り込んでいることがわかる。
【0059】
(比較例3)
実施例4で用意した材料としてのポリスチレン微粒子を用意した。このポリスチレン微粒子の100倍及び500倍の走査型電子顕微鏡写真を、それぞれ図11及び図12に示す。図11及び図12から明らかなように、このポリメチルメタクリレート微粒子では、表面にグラフェンライク炭素が全く存在しないため、平滑な表面を呈していることがわかる。
【0060】
(実施例5)
実施例1と同様の薄片化黒鉛0.005gとポリスチレンからなる微粒子(積水化成品工業株式会社製、品番:S−40、平均粒子径:600μm、Tg:105℃)1.0gを混合したこと以外は、実施例4と同様にして、複合材料を得た。なお、混合時の圧力は約28MPaまで上昇した。得られた複合材料としての粒子の表面を走査型電子顕微鏡(日本電子社製 JCM−5700)により観察した。
【0061】
図13は、実施例5で得た複合材料としての粒子の100倍の走査型電子顕微鏡写真である。図13の写真から、グラフェンライク炭素の一部が樹脂からなる元の基材粒子表面の内側に入り込んでいることがわかる。
【0062】
(比較例4)
実施例5で用意した材料としてのポリスチレン微粒子を用意した。このポリスチレン微粒子の100倍の走査型電子顕微鏡写真を図14に示す。図14から明らかなように、この微粒子では、表面にグラフェンライク炭素が全く存在しないため、平滑な表面を呈していることがわかる。
【0063】
(実施例6)
実施例1と同様の薄片化黒鉛0.3gと、ポリスチレン及びメタクリル酸2−ヒドロキシエチルの共重合体からなる微粒子(積水化成品工業株式会社製、品番:CS−10、平均粒子径:100μm、Tg:98℃)1.0gを混合したこと、及び温度を35℃まで上昇させ12時間かき混ぜたこと以外は、実施例3と同様にして、複合材料を得た。なお、混合時の圧力は約21MPaまで上昇した。得られた複合材料としての粒子の表面を走査型電子顕微鏡(日本電子社製 JCM−5700)により観察した。
【0064】
図15及び図16は、それぞれ、実施例6で得た複合材料としての粒子の500倍及び5000倍の走査型電子顕微鏡写真である。図15及び図16の写真から、グラフェンライク炭素の一部が樹脂からなる元の基材粒子表面の内側に入り込んでいることがわかる。
【0065】
(比較例5)
実施例6で用意した材料としてのポリスチレン及びメタクリル酸2−ヒドロキシエチルの共重合体微粒子を用意した。この微粒子の450倍の走査型電子顕微鏡写真を図17に示す。図17から明らかなように、この微粒子では、表面にグラフェンライク炭素が全く存在しないため、平滑な表面を呈していることがわかる。
【0066】
(実施例7)
実施例1と同様の薄片化黒鉛0.001gと、ポリスチレン及びアクリル酸ブチルの共重合体からなる微粒子(積水化成品工業株式会社製、品番:CS−50、平均粒子径:1100μm、Tg:46℃)1.0gを混合したこと、及び温度を室温のまま12時間かき混ぜたこと以外は、実施例3と同様にして、複合材料を得た。なお、混合時の圧力は約28MPaまで上昇した。得られた複合材料としての粒子の表面を走査型電子顕微鏡(日本電子社製 JCM−5700)により観察した。
【0067】
図18は、実施例7で得た複合材料としての粒子の70倍の走査型電子顕微鏡写真である。図18の写真から、グラフェンライク炭素の一部が樹脂からなる元の基材粒子表面の内側に入り込んでいることがわかる。
【0068】
(比較例6)
実施例7で用意した材料としてのポリスチレン及びアクリル酸ブチルの共重合体微粒子を用意した。この微粒子の80倍の走査型電子顕微鏡写真を図19に示す。図19から明らかなように、この微粒子では、表面にグラフェンライク炭素が全く存在しないため、平滑な表面を呈していることがわかる。
【技術分野】
【0001】
本発明は、樹脂と、グラフェンもしくは薄片化黒鉛のようなグラフェンライク炭素材料との複合材料に関し、より詳細には、樹脂からなる基材に対するグラフェンライク炭素材料の密着性が高められている複合材料及びその製造方法に関する。
【背景技術】
【0002】
従来、黒鉛、カーボンナノチューブ、カーボンファイバーまたはカーボン粒子などの炭素材料が、樹脂への補強剤や充填剤として広く用いられている。また、近年、黒鉛を剥離して得られる、グラフェン積層数がより少ない薄片化黒鉛がより注目されている。
【0003】
上記のような炭素材料と樹脂との複合材料として、例えば下記の特許文献1に示すように、エポキシ樹脂中に炭素繊維などの炭素材料を分散させてなる複合材料が知られている。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特開2003−277471号公報
【発明の概要】
【発明が解決しようとする課題】
【0005】
しかしながら、従来の樹脂と炭素材料との複合材料では、樹脂と炭素材料との密着強度が充分でないという問題があった。例えば、特許文献1に記載のように樹脂中に炭素材料を分散させてなる複合材料では、添加されている炭素材料により物性の改善が図られているものの、炭素材料との密着強度は充分ではなかった。
【0006】
本発明の目的は、上述した従来技術の欠点を解消し、樹脂とグラフェンライク炭素材料との密着性に優れた複合材料及びその製造方法を提供することにある。
【課題を解決するための手段】
【0007】
本発明に係る複合材料は、樹脂からなる基材と、前記基材表面の少なくとも一部を覆うように設けられたグラフェンライク炭素材料層とを備え、前記基材の表面にグラフェンライク炭素が密着している。
【0008】
本発明に係る複合材料のある特定の局面では、グラフェンライク炭素の一部が基材の表面から内部に入り込んでいる。従って、両者の密着性がより一層高められる。
【0009】
本発明に係る複合材料の他の特定の局面では、前記樹脂からなる基材が、樹脂微粒子であり、前記樹脂微粒子の外表面を覆うように前記グラフェンライク炭素材料層が形成されている。この場合には、樹脂微粒子の外表面が、グラフェンライク炭素材料層で覆われており、かつグラフェンライク炭素の一部が樹脂微粒子表面に入り込んでいるため、樹脂微粒子とグラフェンライク炭素材料層との密着性が高められている。しかも、グラフェンライク炭素材料層表面に有する微粒子状の複合材料は、凝集し難い。従って、いわゆる自由流動性粉体(free flowing powder)として取り扱うことができる。
【0010】
本発明に係る複合材料の他の特定の局面では、前記樹脂からなる基材がシート状基材であって、該シート状基材の少なくとも一方面に前記グラフェンライク炭素材料層が設けられている。この場合には、本発明に従って、炭素材料層のシート状基材との密着性に優れたシート状の複合材料を提供することができる。
【0011】
本発明に係る複合材料では、好ましくは、グラフェンライク炭素材料がグラフェンまたは薄片化黒鉛からなる。グラフェンまたは薄片化黒鉛はアスペクト比が大きく、かつ、グラフェン積層数が少ないため、少量の添加で複合材料の物性を高めることができる。
【0012】
本発明に係る複合材料の製造方法は、本発明に従って構成された複合材料の製造方法であって、樹脂からなる基材と、グラフェンライク炭素材料とを用意する工程と、前記樹脂からなる基材の表面の少なくとも一部に前記グラフェンライク炭素材料を接触させ、超臨界または亜臨界状態の流体を作用させつつ加熱する工程とを備える。
【0013】
本発明に係る製造方法では、好ましくは、前記超臨界または亜臨界の流体として、超臨界または亜臨界状態のCO2を用いる。CO2は、31.1℃程度の温度かつ7.52MPa程度の圧力下で超臨界状態となる。従って、H2Oなどに比べて、穏やかな条件で樹脂からなる基材の表面を膨潤させることができる。そのため、ガラス転移温度が低い樹脂を用いた場合においても、本発明の複合材料を確実に得ることができる。
【発明の効果】
【0014】
本発明に係る複合材料では、樹脂からなる基材にグラフェンライク炭素が密着しているため、グラフェンライク炭素材料層と基材との密着性に優れた複合材料を得ることが可能となる。
【0015】
また、本発明に係る製造方法によれば、超臨界または亜臨界状態の流体を樹脂に作用させつつ加熱することにより、樹脂からなる基材の表面にグラフェンライク炭素が密着するようにして、樹脂表面にグラフェンライク炭素材料層が形成される。よって、樹脂からなる基材に対して密着性に優れたグラフェンライク炭素材料層を有する本発明の複合材料を得ることができる。また、本発明の製造方法では、上記のようにして、グラフェンライク炭素材料層を基材表面に形成するため、基材の形状も特に限定されない。よって、樹脂微粒子のような微粒子状、シート状基材などの形状に限らず、複雑な形状の樹脂からなる基材の表面にも、本発明に従ってグラフェンライク炭素材料層を容易に形成することができる。
【0016】
また、被分散体である炭素材料微粒子は凝集性が高く、均一に分散させ、付着させる事は容易ではなかったところ、発明の製造方法では、容易に炭素材料微粒子を基材表面に密着させることができる。
【図面の簡単な説明】
【0017】
【図1】図1は、ポリメチルメタクリレート微粒子1gと薄片化黒鉛0.01gを混合し、超臨界二酸化炭素(65℃、35MPa)で5時間作用させた後の微粒子の走査型電子顕微鏡写真(75倍)である。
【図2】図2は、ポリメチルメタクリレート微粒子1gと薄片化黒鉛0.01gを混合し、超臨界二酸化炭素(65℃、35MPa)で5時間作用させた後の微粒子の走査型電子顕微鏡写真(1300倍)である。
【図3】図3は、ポリメチルメタクリレート微粒子1gと薄片化黒鉛0.01gを混合し、超臨界二酸化炭素(65℃、35MPa)で5時間作用させた後の微粒子の断面の走査型電子顕微鏡写真(550倍)である。
【図4】図4は、ポリメチルメタクリレート微粒子1gと薄片化黒鉛0.005gを混合し、超臨界二酸化炭素(65℃、35MPa)で5時間作用させた後の微粒子の走査型電子顕微鏡写真(450倍)である。
【図5】図5は、ポリメチルメタクリレート微粒子を超臨界二酸化炭素(65℃、35MPa)で5時間作用させた後の微粒子の走査型電子顕微鏡写真(120倍)である。
【図6】図6は、ポリスチレン微粒子(積水化成品工業株式会社製の品番S−20)1gと薄片化黒鉛0.01gを混合し、超臨界二酸化炭素(55℃、28MPa)で12時間作用させた後の微粒子の走査型電子顕微鏡写真(200倍)である。
【図7】図7は、ポリスチレン微粒子(積水化成品工業株式会社製の品番S−20)1gと薄片化黒鉛0.01gを混合し、超臨界二酸化炭素(55℃、28MPa)で12時間作用させた後の微粒子の走査型電子顕微鏡写真(500倍)である。
【図8】図8は、ポリスチレン微粒子(積水化成品工業株式会社製の品番S−20)を超臨界二酸化炭素(55℃、28MPa)で5時間作用させた後の微粒子の走査型電子顕微鏡写真(200倍)である。
【図9】図9は、ポリスチレン微粒子(積水化成品工業株式会社製の品番S−30)1gと薄片化黒鉛0.001gを混合し、超臨界二酸化炭素(60℃、28MPa)で4.5時間作用させた後の微粒子の走査型電子顕微鏡写真(100倍)である。
【図10】図10は、ポリスチレン微粒子(積水化成品工業株式会社製の品番S−30)1gと薄片化黒鉛0.001gを混合し、超臨界二酸化炭素(60℃、28MPa)で4.5時間作用させた後の微粒子の走査型電子顕微鏡写真(500倍)である。
【図11】図11は、ポリスチレン微粒子(積水化成品工業株式会社製の品番S−30)を超臨界二酸化炭素(60℃、28MPa)で4.5時間作用させた後の微粒子の走査型電子顕微鏡写真(100倍)である。
【図12】図12は、ポリスチレン微粒子(積水化成品工業株式会社製の品番S−30)を超臨界二酸化炭素(60℃、28MPa)で4.5時間作用させた後の微粒子の走査型電子顕微鏡写真(500倍)である。
【図13】図13は、ポリスチレン微粒子(積水化成品工業株式会社製の品番S−40)1gと薄片化黒鉛0.005gを混合し、超臨界二酸化炭素(60℃、28MPa)で4.5時間作用させた後の微粒子の走査型電子顕微鏡写真(100倍)である。
【図14】図14は、ポリスチレン微粒子(積水化成品工業株式会社製の品番S−40)を超臨界二酸化炭素(60℃、28MPa)で4.5時間作用させた後の微粒子の走査型電子顕微鏡写真(100倍)である。
【図15】図15は、ポリスチレン及びメタクリル酸2−ヒドロキシエチルの共重合体からなる微粒子(積水化成品工業株式会社製の品番CS−10)1gと薄片化黒鉛0.3gを混合し、超臨界二酸化炭素(35℃、21MPa)で12時間作用させた後の微粒子の走査型電子顕微鏡写真(500倍)である。
【図16】図16は、ポリスチレン及びメタクリル酸2−ヒドロキシエチルの共重合体からなる微粒子(積水化成品工業株式会社製の品番CS−10)1gと薄片化黒鉛0.3gを混合し、超臨界二酸化炭素(35℃、21MPa)で12時間作用させた後の微粒子の走査型電子顕微鏡写真(5000倍)である。
【図17】図17は、ポリスチレン及びメタクリル酸2−ヒドロキシエチルの共重合体からなる微粒子(積水化成品工業株式会社製の品番CS−10)を超臨界二酸化炭素(35℃、21MPa)で12時間作用させた後の微粒子の走査型電子顕微鏡写真(450倍)である。
【図18】図18は、ポリスチレン及びメタクリル酸2−ヒドロキシエチルの共重合体からなる微粒子(積水化成品工業株式会社製の品番CS−50)1gと薄片化黒鉛0.001gを混合し、超臨界二酸化炭素(室温、28MPa)で12時間作用させた後の微粒子の走査型電子顕微鏡写真(70倍)である。
【図19】図19は、ポリスチレン及びメタクリル酸2−ヒドロキシエチルの共重合体からなる微粒子(積水化成品工業株式会社製の品番CS−50)を超臨界二酸化炭素(室温、28MPa)で12時間作用させた後の微粒子の走査型電子顕微鏡写真(80倍)である。
【発明を実施するための形態】
【0018】
以下、本発明の具体的な実施形態を説明することにより、本発明を明らかにする。
【0019】
(樹脂からなる基材)
本発明に係る複合材料及びその製造方法では、樹脂からなる基材を用いる。基材を構成する樹脂については、超臨界または亜臨界状態の流体を作用させつつ加熱することにより、表面を柔らかくし得る適宜の樹脂を用いることができる。このような樹脂は、合成樹脂であってもよく、天然樹脂であってもよい。
【0020】
上記樹脂としては、超臨界または亜臨界状態の流体が作用する温度において軟化する程度のガラス転移温度Tgを有するものが好ましい。後述するように、CO2が超臨界または亜臨界状態で作用させる流体として好適に用いられる。従って、樹脂としては、ポリスチレン、ポリプロピレン、ポリメチルメタクリレート(PMMA)、セルロースなどを好適に用いることができる。また、樹脂は、これらのポリマーを構成するモノマーの共重合体であってもよい。もっとも、本発明において用いる樹脂材料としては、PMMA以外、様々な(メタ)アクリル系樹脂、ポリプロピレン以外の様々なポリオレフィン等も用いることができる。
【0021】
上記樹脂からなる基材の形状についても特に限定されない。基材は、樹脂微粒子であってもよい。すなわち、微粒子状基材であってもよい。なお、微粒子の直径は特に限定されないが、平均200μm以下の微粒子が好適に用いられ、これよりも大きい粒子状の樹脂からなる基材を用いてもよい。樹脂からなる基材が粒子状である場合、本発明に従って得られた複合材料では、後述するように、凝集が生じ難い。従って、自由流動性粉末(free flowing powder)として取り扱うことができる。
【0022】
また、樹脂からなる基材は、シート状であってもよい。シート状基材の場合には、シート状基材の一面及び/または反対側の面の少なくとも一部に本発明に従って、グラフェンライク炭素材料層を形成することができる。
【0023】
さらに、本発明で用いられる樹脂からなる基材は、粒子状やシート状である必要は必ずしもない。すなわち、グラフェンライク炭素材料を樹脂からなる基材表面の少なくとも一部に接触させ、その状態で超臨界または亜臨界状態の流体を作用させ得る限り、樹脂からなる基材の形状は複雑な立体的形状を有するものであってもよい。その場合においても、本発明に従って、表面にグラフェンライク炭素材料層を有する複雑な立体的形状を有する複合材料を得ることができる。
【0024】
また、複雑な表面の基材の一部に、グラフェンライク炭素材料を選択的に形成することもできる。
【0025】
(グラフェンライク炭素材料層)
本発明に係る複合材料では、上記樹脂からなる基材の表面の少なくとも一部を覆うように、グラフェンライク炭素材料層が設けられている。このグラフェンライク炭素材料層を構成するグラフェンライク炭素材料としては、グラフェンまたは薄片化黒鉛を好適に用いることができる。周知のように、黒鉛は、グラフェンの積層体である。薄片化黒鉛は、黒鉛を剥離することにより得られ、薄片化黒鉛の積層体におけるグラフェンの積層数は数層〜200層程度である。また、薄片化黒鉛の比表面積は黒鉛よりもはるかに大きく、600m2/g以上である。本発明において、上記薄片化黒鉛とは、黒鉛を剥離することにより得られ、上記のようなグラフェン積層数のグラフェン積層体をいうものとする。
【0026】
上記薄片化黒鉛としては、市販されている薄片化黒鉛を用いてもよい。また、黒鉛を剥離する様々な処理により薄片化黒鉛を得てもよい。
【0027】
上記のように薄片化黒鉛を得る方法としては特に限定されず、黒鉛を膨張して行われた膨張化黒鉛を剥離することにより得ることができる。黒鉛を膨潤し膨張化黒鉛とする工程は、1)電解質溶液中に層状黒鉛を浸漬し加熱する方法、及び2)電気分解法などを用いることができる。
【0028】
1)の方法では、硝酸や硫酸中に層状黒鉛を浸漬し、加熱し、硝酸イオンや硫酸イオンを層間にインターカレートする方法である。この場合、硝酸濃度及び硫酸濃度は、40重量%〜70重量%程度であることが望ましい。この範囲内であれば、硝酸イオンや硫酸イオンを確実に層間にインターカレートすることができる。また、加熱温度については、20℃以上、50℃以下であることが好ましい。この範囲内の温度であれば、上記硝酸イオンや硫酸イオンを確実に層間にインターカレートすることができる。
【0029】
2)の電気分解法では、層状黒鉛を作用極とし、該作用極をPtなどからなる対照極と共に硝酸や硫酸中に浸漬し、電気分解する。それによって、層状黒鉛の層間すなわちグラフェン間に硝酸イオンや硫酸イオン等の電解質イオンをインターカレートすることができ、層間を広げることができる。
【0030】
次に、上記のようにして得られた膨張化黒鉛からなるシートを水等により洗浄し、乾燥し、硝酸イオンや硫酸イオン等を除去する。このようにして、乾燥した膨張化黒鉛からなるシートを得ることができる。膨張化黒鉛を剥離して薄片化黒鉛を得るには、加熱、超音波を加える方法などを用いることができる。
【0031】
なお、本発明におけるグラフェンライク炭素材料としては、グラフェンまたは薄片化黒鉛に限らず、カーボンナノチューブなどの表面にグラフェンシート構造を有する様々なグラフェンライク炭素材料を用いてもよい。
【0032】
本発明においては、上記グラフェンライク炭素材料層の厚みは特に限定されず、用途に応じて適宜選択すればよい。もっとも、樹脂微粒子からなる基材表面にグラフェンライク炭素材料層を設ける場合には、グラフェンライク炭素材料層の厚みは0.5nm〜500nm程度である。また、シート状基材の少なくとも一方面にグラフェンライク炭素材料層を設ける場合においても、0.5nm〜500nm程度の厚みとすればよい。
【0033】
グラフェンライク炭素材料層の厚みが厚すぎると、樹脂物性の効果が発現しなくなることがある。逆に、グラフェンライク炭素材料層の厚みが薄くなりすぎると、グラフェンライク炭素材料層を設けたことによる物性改善効果が充分に得られないことがある。
【0034】
本発明においては、上記グラフェンライク炭素材料層を構成しているグラフェンライク炭素の一部が、基材の表面に密着している。好ましくは、グラフェンライク炭素の一部が基材の表面から内部に向かって入り込んでいる。そのため、アンカー効果により、グラフェンライク炭素材料層と樹脂からなる基材との密着性が効果的に高められる。
【0035】
グラフェンライク炭素の基材表面への密着性に優れた本発明の複合材料、より好ましくは、グラフェンライク炭素の一部が基材の表面に内部に入り込んでいる本発明の複合材料は、本発明の製造方法に従って得ることができる。
【0036】
(製造方法)
本発明の製造方法では、先ず、樹脂からなる上記基材と、上記グラフェンライク炭素材料とを用意する。次に、樹脂からなる基材の表面の少なくとも一部に、上記グラフェンライク炭素材料を接触させ、その状態で、超臨界または亜臨界状態の流体を作用させつつ加熱する。超臨界または亜臨界状態の流体としては、CO2、H2O、などを用いることができる。
【0037】
CO2は、31.1℃の温度及び7.52Mpa程度の気圧で超臨界状態となる。また、−56.6℃〜31.1℃及び0.528Mpa〜7.52MPa程度の気圧の範囲で亜臨界状態となる。超臨界または亜臨界状態の流体を作用させつつ加熱することにより、樹脂からなる基材表面が軟化する。従って、グラフェンライク炭素が軟化した基材表面と接触する。また、好ましくはグラフェンライク炭素の一部が基材表面に入り込む。そのため、加熱後冷却すれば、グラフェンライク炭素を基材表面と密着するようにして、基材表面の少なくとも一部を覆うようにグラフェンライク炭素材料層が形成される。すなわち、本発明の複合材料を得ることができる。
【0038】
よって、上記樹脂からなる基材を構成する樹脂のガラス転移温度Tgが、上記超臨界または亜臨界状態の流体を作用させつつ加熱する工程における温度雰囲気にあることが望ましい。より具体的には、樹脂のガラス転移温度Tgは、上記加熱温度−100℃〜+100℃の範囲にあることが望ましい。この範囲内にあれば、樹脂からなる基材表面にグラフェンライク炭素の一部を確実に入り込ませることができる。
【0039】
上記樹脂からなる基材の表面にグラフェンライク炭素材料を接触させるにあたっては、上記の通り、基材の表面の少なくとも一部に接触させればよい。もっとも、基材の全表面にグラフェンライク炭素材料を接触させてもよい。
【0040】
さらに、上記のように、基材表面の少なくとも一部にグラフェンライク炭素材料を接触させた状態で超臨界または亜臨界状態の流体を作用させつつ加熱するものであるため、基材表面の一部に選択的にグラフェンライク炭素材料を接触させ、基材表面の一部に選択的にグラフェンライク炭素材料層が設けられている複合材料も容易に得ることができる。加えて、前述したように、複雑な立体的な形状を有する基材を用いた場合においても、その表面に本発明に従ってグラフェンライク炭素材料層を容易にかつ確実に形成することができる。
【0041】
(複合材料の物性)
本発明に係る複合材料では、上記のように、グラフェンライク炭素材料の基材表面への密着性が高められ、好ましくは、グラフェンライク炭素材料の一部が樹脂からなる基材表面に入り込むようにして、グラフェンライク炭素材料層が形成されている。従って、グラフェンライク炭素材料層と基材との密着性を効果的に高め得る。よって、熱履歴を伴う環境に晒されたとしても、グラフェンライク炭素材料層の基材からの剥離等が生じ難い。また、密着性に優れているため、炭素材料による機械的強度の改善等も効果的に図ることができる。
【0042】
加えて、本願発明者の実験によれば、例えば、PMMAに上記グラフェンライク炭素材料層を本発明に従って形成してなる複合材料では、複合材料のガラス転移温度Tgも高められることがわかった。従って、耐熱性に優れた複合材料を提供することができる。このように、複合材料のTgが高められるのは、グラフェンライク炭素材料とPMMAの表面との密着性が高められるため、グラフェンライク炭素と樹脂との相互作用が強くなることによると考えられる。
【0043】
なお、PMMAに限らず、他の樹脂を用いた場合においても、本発明に従ってグラフェンライク炭素材料層を形成することにより、複合材料のTgを元の樹脂のTgに比べて効果的に高めることができる。従って、耐熱性に優れた複合材料を提供することが可能となる。
【0044】
以下、本発明の実施例及び比較例を説明する。なお、本発明は以下の実施例に限定されるものではない。
【0045】
(実施例1)
1)薄片化黒鉛の調製
原料の黒鉛シートとして東洋炭素社製、品番:PF100−UHPを用意した。この黒鉛シートと同じ製法で、圧延処理時の圧延倍率を下げて密度0.7、厚み1mmの低密度黒鉛シートを用意した。
【0046】
上記のようにして得られた密度0.7の黒鉛シートを3cm×3cmの大きさに切断し、電極材料としての黒鉛シートを得た。この黒鉛シートに、2本のスリットを、スリットの長さが1cm、幅が1cmとなるようにカッターナイフにより切削し、形成した。上記2本のスリットが形成された黒鉛シートに、Ptからなる電極を挿入した。このようにして用意した黒鉛シートを作用極(陽極)として、Ptからなる対照極(陰極)及び、Ag/AgClからなる参照極とともに60重量%濃度の硝酸水溶液中に浸漬し、直流電圧を印加し電気化学処理を行った。このようにして、陽極に作用極として用いた黒鉛を膨張化黒鉛とした。
【0047】
次に、得られた膨張化黒鉛を乾燥し、1cm角に切断し、その1つをカーボンるつぼに入れて電磁誘導加熱処理を行った。誘導加熱装置はSKメディカル社製MU1700Dを用い、アルゴンガス雰囲気下で最高到達温度550℃となるように10Aの電流量で行った。電磁誘導加熱により膨張化黒鉛は薄片化され、得られた薄片化黒鉛の粉末を島津製作所製の比表面積測定装置ASAP−2000で窒素ガスを用いて測定したところ、1回測定で850m2/gの比表面積を示した。
【0048】
2)複合材料の製造
樹脂からなる基材として、ポリメチルメタクリレートからなる微粒子(Aldrich社製、品番:445746−500G、Mw:35万、Tg:122℃)を用意した。このポリメチルメタクリレートからなる微粒子1.0gと、上記のようにして得られた薄片化黒鉛0.01gを圧力容器内に配置し、室温(23℃)及び10MPaの圧力で超臨界状態とされているCO2を10mL加えた後、いったんCO2を除去した(水分を除去し、乾燥させるため)。その後、再度室温(23℃)及び10MPaの圧力で超臨界状態とされているCO2を10mL加えた。その後、温度を65℃まで上昇させ5時間かき混ぜながら加熱した。このときの圧力は約35MPaまで上昇した。しかる後、室温まで冷却し、複合材料を得た。得られた複合材料は粒子状であるその平均粒径は120μmであった。また、このようにして得られた複合材料としての粒子の表面を走査型電子顕微鏡(日本電子社製 JCM−5700)により観察した。
【0049】
図1は、実施例1で得た複合材料としての粒子の75倍の電子顕微鏡写真を示し、図2はその表面を1300倍に拡大して示す電子顕微鏡写真である。
【0050】
上記粒子を乳鉢を用いて部分的に切断し、部分的に切断されている粒子を同じく走査型電子顕微鏡(日本電子社製 JCM−5700)により観察した。図3は、このようにして得られた550倍の倍率の走査型電子顕微鏡写真を示す。図1〜図3から明らかなように、得られた複合材料では、微粒子表面にグラフェンライク炭素材料が形成されており、特に図3の写真から、グラフェンライク炭素は基材粒子表面に集積していることがわかる。
【0051】
(実施例2)
実施例1と同様の薄片化黒鉛0.005gとポリメチルメタクリレートからなる微粒子(Aldrich社製、品番:445746−500G、Mw:35万、Tg:122℃)1.0gを混合し、実施例1と同様に圧力容器内に配置し、室温(23℃)及び10MPaの圧力で超臨界状態とされているCO2を10mL加えた後、いったんCO2を除去した(水分を除去し、乾燥させるため)。その後、再度室温(23℃)及び10MPaの圧力で超臨界状態とされているCO2を10mL加えた。その後、温度を65℃まで上昇させ5時間かき混ぜながら加熱した。このときの圧力は約35MPaまで上昇した。しかる後、室温まで冷却し、複合材料を得た。得られた複合材料は粒子状であるその平均粒径は120μmであった。また、このようにして得られた複合材料としての粒子の表面を走査型電子顕微鏡(日本電子社製 JCM−5700)により観察した。
【0052】
図4は、実施例2で得た複合材料としての粒子の450倍の走査型電子顕微鏡写真である。図4の写真から、グラフェンライク炭素の一部が樹脂からなる元の基材粒子表面の内側に入り込んでいることがわかる。
【0053】
(比較例1)
比較例として、実施例で用意した材料としてのポリメチルメタクリレート微粒子を用意した。このポリメチルメタクリレート微粒子の120倍の走査型電子顕微鏡写真を図5に示す。図5から明らかなように、このポリメチルメタクリレート微粒子では、表面にグラフェンライク炭素が全く存在しないため、平滑な表面を呈していることがわかる。
【0054】
(実施例3)
実施例1と同様の薄片化黒鉛0.01gとポリスチレンからなる微粒子(積水化成品工業株式会社製、品番:S−20、平均粒子径:300μm、Tg:106℃)1.0gを混合し、実施例1と同様に圧力容器内に配置し、室温(23℃)及び10MPaの圧力で超臨界状態とされているCO2を10mL加えた後、いったんCO2を除去した(水分を除去し、乾燥させるため)。その後、再度室温(23℃)及び10MPaの圧力で超臨界状態とされているCO2を10mL加えた。その後、温度を55℃まで上昇させ12時間かき混ぜながら加熱した。このときの圧力は約28MPaまで上昇した。しかる後、室温まで冷却し、複合材料を得た。得られた複合材料としての粒子の表面を走査型電子顕微鏡(日本電子社製 JCM−5700)により観察した。
【0055】
図6及び図7は、ぞれぞれ、実施例3で得た複合材料としての粒子の200倍及び500倍の走査型電子顕微鏡写真である。図6及び図7の写真から、グラフェンライク炭素の一部が樹脂からなる元の基材粒子表面の内側に入り込んでいることがわかる。
【0056】
(比較例2)
実施例3で用意した材料としてのポリスチレン微粒子を用意した。このポリスチレン微粒子の200倍の走査型電子顕微鏡写真を図8に示す。図8から明らかなように、このポリスチレン微粒子では、表面にグラフェンライク炭素が全く存在しないため、平滑な表面を呈していることがわかる。
【0057】
(実施例4)
実施例1と同様の薄片化黒鉛0.001gとポリスチレンからなる微粒子(積水化成品工業株式会社製、品番:S−30、平均粒子径:800μm、Tg:105℃)1.0gを混合したこと、及び温度を60℃まで上昇させ4.5時間かき混ぜながら加熱したこと以外は、実施例3と同様にして、複合材料を得た。なお、混合時の圧力は28MPaまで上昇した。得られた複合材料としての粒子の表面を走査型電子顕微鏡(日本電子社製 JCM−5700)により観察した。
【0058】
図9及び図10は、それぞれ、実施例4で得た複合材料としての粒子の100倍及び500倍の走査型電子顕微鏡写真である。図5の写真から、グラフェンライク炭素の一部が樹脂からなる元の基材粒子表面の内側に入り込んでいることがわかる。
【0059】
(比較例3)
実施例4で用意した材料としてのポリスチレン微粒子を用意した。このポリスチレン微粒子の100倍及び500倍の走査型電子顕微鏡写真を、それぞれ図11及び図12に示す。図11及び図12から明らかなように、このポリメチルメタクリレート微粒子では、表面にグラフェンライク炭素が全く存在しないため、平滑な表面を呈していることがわかる。
【0060】
(実施例5)
実施例1と同様の薄片化黒鉛0.005gとポリスチレンからなる微粒子(積水化成品工業株式会社製、品番:S−40、平均粒子径:600μm、Tg:105℃)1.0gを混合したこと以外は、実施例4と同様にして、複合材料を得た。なお、混合時の圧力は約28MPaまで上昇した。得られた複合材料としての粒子の表面を走査型電子顕微鏡(日本電子社製 JCM−5700)により観察した。
【0061】
図13は、実施例5で得た複合材料としての粒子の100倍の走査型電子顕微鏡写真である。図13の写真から、グラフェンライク炭素の一部が樹脂からなる元の基材粒子表面の内側に入り込んでいることがわかる。
【0062】
(比較例4)
実施例5で用意した材料としてのポリスチレン微粒子を用意した。このポリスチレン微粒子の100倍の走査型電子顕微鏡写真を図14に示す。図14から明らかなように、この微粒子では、表面にグラフェンライク炭素が全く存在しないため、平滑な表面を呈していることがわかる。
【0063】
(実施例6)
実施例1と同様の薄片化黒鉛0.3gと、ポリスチレン及びメタクリル酸2−ヒドロキシエチルの共重合体からなる微粒子(積水化成品工業株式会社製、品番:CS−10、平均粒子径:100μm、Tg:98℃)1.0gを混合したこと、及び温度を35℃まで上昇させ12時間かき混ぜたこと以外は、実施例3と同様にして、複合材料を得た。なお、混合時の圧力は約21MPaまで上昇した。得られた複合材料としての粒子の表面を走査型電子顕微鏡(日本電子社製 JCM−5700)により観察した。
【0064】
図15及び図16は、それぞれ、実施例6で得た複合材料としての粒子の500倍及び5000倍の走査型電子顕微鏡写真である。図15及び図16の写真から、グラフェンライク炭素の一部が樹脂からなる元の基材粒子表面の内側に入り込んでいることがわかる。
【0065】
(比較例5)
実施例6で用意した材料としてのポリスチレン及びメタクリル酸2−ヒドロキシエチルの共重合体微粒子を用意した。この微粒子の450倍の走査型電子顕微鏡写真を図17に示す。図17から明らかなように、この微粒子では、表面にグラフェンライク炭素が全く存在しないため、平滑な表面を呈していることがわかる。
【0066】
(実施例7)
実施例1と同様の薄片化黒鉛0.001gと、ポリスチレン及びアクリル酸ブチルの共重合体からなる微粒子(積水化成品工業株式会社製、品番:CS−50、平均粒子径:1100μm、Tg:46℃)1.0gを混合したこと、及び温度を室温のまま12時間かき混ぜたこと以外は、実施例3と同様にして、複合材料を得た。なお、混合時の圧力は約28MPaまで上昇した。得られた複合材料としての粒子の表面を走査型電子顕微鏡(日本電子社製 JCM−5700)により観察した。
【0067】
図18は、実施例7で得た複合材料としての粒子の70倍の走査型電子顕微鏡写真である。図18の写真から、グラフェンライク炭素の一部が樹脂からなる元の基材粒子表面の内側に入り込んでいることがわかる。
【0068】
(比較例6)
実施例7で用意した材料としてのポリスチレン及びアクリル酸ブチルの共重合体微粒子を用意した。この微粒子の80倍の走査型電子顕微鏡写真を図19に示す。図19から明らかなように、この微粒子では、表面にグラフェンライク炭素が全く存在しないため、平滑な表面を呈していることがわかる。
【特許請求の範囲】
【請求項1】
樹脂からなる基材と、前記基材表面の少なくとも一部を覆うように設けられたグラフェンライク炭素材料層とを備え、前記基材の表面にグラフェンライク炭素が密着している、複合材料。
【請求項2】
前記グラフェンライク炭素の一部が前記基材の表面から内部に入り込んでいる、請求項1に記載の複合材料。
【請求項3】
前記樹脂からなる基材が、樹脂微粒子であり、前記樹脂微粒子の外表面を覆うように前記グラフェンライク炭素材料層が形成されている、請求項1または2に記載の複合材料。
【請求項4】
前記樹脂からなる基材がシート状基材であって、該シート状基材の少なくとも一方面に前記グラフェンライク炭素材料層が設けられている、請求項1または2に記載の複合材料。
【請求項5】
前記グラフェンライク炭素材料がグラフェンまたは薄片化黒鉛からなる、請求項1〜4のいずれか1項に記載の複合材料。
【請求項1】
樹脂からなる基材と、前記基材表面の少なくとも一部を覆うように設けられたグラフェンライク炭素材料層とを備え、前記基材の表面にグラフェンライク炭素が密着している、複合材料。
【請求項2】
前記グラフェンライク炭素の一部が前記基材の表面から内部に入り込んでいる、請求項1に記載の複合材料。
【請求項3】
前記樹脂からなる基材が、樹脂微粒子であり、前記樹脂微粒子の外表面を覆うように前記グラフェンライク炭素材料層が形成されている、請求項1または2に記載の複合材料。
【請求項4】
前記樹脂からなる基材がシート状基材であって、該シート状基材の少なくとも一方面に前記グラフェンライク炭素材料層が設けられている、請求項1または2に記載の複合材料。
【請求項5】
前記グラフェンライク炭素材料がグラフェンまたは薄片化黒鉛からなる、請求項1〜4のいずれか1項に記載の複合材料。
【図1】


【図2】
【図3】

【図4】
【図5】

【図6】

【図7】

【図8】

【図9】

【図10】

【図11】

【図12】

【図13】

【図14】

【図15】

【図16】

【図17】

【図18】

【図19】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【公開番号】特開2013−60364(P2013−60364A)
【公開日】平成25年4月4日(2013.4.4)
【国際特許分類】
【出願番号】特願2012−242353(P2012−242353)
【出願日】平成24年11月2日(2012.11.2)
【分割の表示】特願2012−526560(P2012−526560)の分割
【原出願日】平成24年5月28日(2012.5.28)
【出願人】(000002174)積水化学工業株式会社 (5,781)
【出願人】(504159235)国立大学法人 熊本大学 (314)
【Fターム(参考)】
【公開日】平成25年4月4日(2013.4.4)
【国際特許分類】
【出願日】平成24年11月2日(2012.11.2)
【分割の表示】特願2012−526560(P2012−526560)の分割
【原出願日】平成24年5月28日(2012.5.28)
【出願人】(000002174)積水化学工業株式会社 (5,781)
【出願人】(504159235)国立大学法人 熊本大学 (314)
【Fターム(参考)】
[ Back to top ]