
複素環化合物及びその医薬用途
【課題】11β−HSD1阻害活性を有する化合物を提供するとともに、副作用が軽減された新たなメカニズムの糖尿病の治療薬及びインスリン抵抗性の改善薬を提供すること。
【解決手段】本発明は、下記の一般式で表される複素環化合物又はその薬学的に許容される塩を提供する。

【解決手段】本発明は、下記の一般式で表される複素環化合物又はその薬学的に許容される塩を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、複素環化合物及びその医薬用途に関する。
【背景技術】
【0002】
糖尿病とは、インスリンの作用の不足による慢性の高血糖状態を主徴とする代謝疾患群であり、1型と2型に大別される。1型糖尿病は、インスリンを合成及び分泌する膵ランゲルハンス島β細胞の破壊や消失によるインスリン分泌量の低下を主要な原因として発症するのに対し、2型糖尿病は、インスリン分泌量の低下やインスリン抵抗性をきたす素因を含む複数の遺伝因子に、過食(特に高脂肪食)、運動不足、肥満、ストレス等の環境因子及び加齢が加わって発症する。
【0003】
また、糖尿病による長期間の高血糖状態の持続は、糖尿病三大合併症(糖尿病性網膜症、糖尿病性腎症及び糖尿病性神経障害)の他にも失明や心血管障害等を引き起こす危険性が高いことが知られている。このため、糖尿病の発症時に、血糖をコントロールして正常に近い状態へと導く治療を行い、耐糖能異常、食後高血糖及び空腹時高血糖を改善していくことが、患者のQOL(quality of life)の観点からも重要であると考えられている(非特許文献1)。
【0004】
現在使用されている糖尿病治療薬としては、インスリン製剤、スルホニルウレア剤(例えば、グリベンクラミド)、ビグアナイド剤(例えば、メトホルミン)、α−グルコシダーゼ阻害剤(例えば、ボグリボース)、チアゾリジンジオン系薬剤(例えば、ピオグリタゾン)等が挙げられる(非特許文献1〜5)。
【0005】
インスリン製剤は、強力な血糖低下作用を有するが、しばしば重篤な低血糖又は体重増加を引き起こすことが知られ、さらに注射による投与が必要であるため、患者への負担が大きいというデメリットがある。スルホニルウレア剤は、血糖低下作用を有するが、同様に重篤な低血糖や体重増加を引き起こし、長期間投与の場合には、2次無効と呼ばれる血糖値コントロールの悪化が起こることが知られている。ビクアナイド剤は、血糖低下作用及びインスリン抵抗性改善作用等を有するが、下痢又は膨満等の胃腸管障害を引き起こすことがあり、さらには重篤な乳酸アシドーシスを引き起こすリスクもあるため、腎障害又は肝機能障害のある患者には禁忌とされている。α−グルコシダーゼ阻害剤は、食後の血糖上昇(食後過血糖)を抑制する作用を有するが、腹痛、下痢又は膨満等の胃腸障害を引き起こすことが知られている。チアゾリジンジオン系薬剤は、インスリン抵抗性の改善を介した血糖降下作用を有するが、しばしば体重増加、浮腫、肝障害を引き起こし、心不全患者や心不全の既往者には不適とされ、治療期間中には定期的な肝機能検査が必要であるとされている。
【0006】
一方、インスリン抵抗性とは、血中のインスリン濃度に見合ったインスリン作用が得られない状態をいい、インスリン抵抗性の症状は、糖尿病を患っていない境界型のヒトにも認められることが知られている。また、インスリン抵抗性は、内臓脂肪型肥満とともに、メタボリックシンドロームの基盤病態であり、動脈硬化、肥満、高血圧及び脂質代謝異常の病態形成に密接に関わることが明らかにされている。ここでメタボリックシンドロームとは、肥満を基盤に、主として脂質代謝異常(高中性脂肪及び低HDLコレステロール)、高血圧及び耐糖能異常の持続が虚血性の心血管疾患の発症リスクを高めることを想定した概念である。
【0007】
このため、インスリン抵抗性の改善は、糖代謝異常の改善のみならず、メタボリックシンドローム、心血管障害さらには脳血管障害の治療にも重要であると考えられている(非特許文献1)。
【0008】
また、11β−ヒドロキシステロイドデヒドロゲナーゼ1(11β−hydroxysteroid dehydrogenase type 1;以下、11β−HSD1)は、不活性型グルココルチコイド(ヒトではコルチゾン、げっ歯類では11−デヒドロコルチコステロン)を活性型グルココルチコイド(ヒトではコルチゾール、げっ歯類ではコルチコステロン)に細胞内で変換する酵素であり、肝臓、脂肪組織及び骨格筋において高発現している(非特許文献6)。活性型グルココルチコイドは、肝臓での糖新生亢進及び解糖系抑制又は脂肪組織及び筋肉での糖の取り込み抑制により、高血糖等の代謝異常を惹起することが報告されている(非特許文献7)。11β−HSD1ノックアウトマウスは、ストレス負荷や高脂肪食負荷に対する肝臓の糖新生酵素(PEPCKやG6Pase等)が誘導されず、糖尿病の発症に対して明らかな抵抗性を示し、高脂肪食負荷による内臓脂肪の蓄積が選択的に抑制されるために、メタボリックシンドロームの発症や進展にも抵抗性を示すことが報告されている(非特許文献8)。
【0009】
なお、特許文献1〜5には、11β−HSD1阻害作用を有する化合物が開示されている。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】国際公開第08/099145号
【特許文献2】国際公開第09/060232号
【特許文献3】国際公開第09/098501号
【特許文献4】国際公開第10/087770号
【特許文献5】国際公開第09/010416号
【非特許文献】
【0011】
【非特許文献1】日本糖尿病学会、「糖尿病治療ガイド2008−2009」、2008年
【非特許文献2】Moller、Nature、2001年、第414巻、p.821−827
【非特許文献3】Skyler、Journal of Medicinal Chemistry、2004年、第47巻、p.4113−4117
【非特許文献4】Rossら、Chemical Review、2004年、第104巻、p.1255−1282
【非特許文献5】Stumvollら、Lancet、2005年、第365巻、p.1333−1346
【非特許文献6】Secklら、Endocrinology、2001年、第142巻、p.1371
【非特許文献7】土田ら、日本臨床、2002年、第60巻、増刊号7、p.280
【非特許文献8】Kotelevtsevら、Proceedings of the National Academy of Sciences of the United States of America、1997年、第94巻、p.14924−14929
【発明の概要】
【発明が解決しようとする課題】
【0012】
しかしながら、上記のように、糖尿病治療薬の既存薬は、そのいずれもが薬効は示すが、種々の欠点や副作用を有しているのが現状であるため、慢性疾患や複合疾患を持つ患者に対する投与や長期的な投与が可能な使い勝手の良い糖尿病治療薬の創出が望まれている。
【0013】
また、11β−HSD1阻害作用を有する化合物は、不活性型グルココルチコイドの変換を阻害し、活性型グルココルチコイドの産生を抑制することが可能であるため、既存の医薬品に比べて新たなメカニズムで高血糖や肝糖新生を抑制し、インスリン抵抗性及びメタボリックシンドロームの改善にも薬効を示し得ることが期待されるが、未だに、11β−HSD1阻害作用を有する糖尿病治療薬やインスリン抵抗性の改善薬は医薬品として承認されていない。
【0014】
そこで本発明は、11β−HSD1阻害活性を有する化合物を提供するとともに、副作用が軽減された新たなメカニズムの糖尿病の治療薬及びインスリン抵抗性の改善薬を提供することを目的とする。
【課題を解決するための手段】
【0015】
本発明者らは上記の目的を達成するため鋭意検討した結果、新規な複素環化合物又はその薬学的に許容される塩が、強力な11β−HSD1阻害活性を有し、糖尿病の治療作用及びインスリン抵抗性の改善作用を有することを見出し、本発明を完成させるに至った。
【0016】
すなわち、本発明は、以下の一般式(I)で表される複素環化合物又はその薬学的に許容される塩を提供する。
【化1】
[式中、R1は、炭素数1〜6のアルキル、炭素数3〜10のシクロアルキル、環構成炭素数6〜10のアリール又は環構成原子数5〜10のヘテロアリール(ここで、該アリール及び該ヘテロアリールは、それぞれ独立して、1以上のハロゲンによって置換されていてもよい炭素数1〜6のアルキル、又は、炭素数1〜6のアルキルオキシ若しくはハロゲン、によって置換されていてもよい)を示し、R2〜R5は、それぞれ独立して、炭素数1〜6のアルキル又は水素原子を示し、R6は、ヒドロキシ又は水素原子を示す。]
【0017】
R1は、炭素数1〜6のアルキル、環構成炭素数6〜10のアリール又は環構成原子数5〜10のヘテロアリール(ここで、該アリール及び該ヘテロアリールは、それぞれ独立して、1以上のハロゲンによって置換されていてもよい炭素数1〜6のアルキル、炭素数1〜6のアルキルオキシ又はハロゲンによって置換されていてもよい)を示すことが好ましく、フェニル又は3−クロロフェニルを示すことがより好ましい。
【0018】
この場合、R1を限定することで、より強い11β−HSD1阻害活性が得られる。
【0019】
また、R2〜R5は、それぞれ独立してメチル基又は水素原子を示すことが好ましく、R2及びR3はメチル基を、R4及びR5は水素原子を示すことがより好ましい。
【0020】
この場合、R2〜R5を限定することで、さらに強い11β−HSD1阻害活性が得られる。
【0021】
さらにR6は、ヒドロキシを示すことが好ましい。
【0022】
この場合、R6を限定することで、より高い代謝安定性が得られる。
【0023】
また本発明は、上記の複素環化合物又はその薬学的に許容される塩を有効成分として含有する、11β−HSD1阻害剤、糖尿病の治療薬又は予防薬を提供する。上記の糖尿病は、2型糖尿病であることが好ましい。
【0024】
さらに本発明は、上記の複素環化合物又はその薬学的に許容される塩を有効成分として含有する、インスリン抵抗性の改善薬を提供する。
【発明の効果】
【0025】
本発明の複素環化合物又はその薬学的に許容される塩は、in vivoにおいて顕著な11β−HSD1阻害活性を示し、糖尿病疾患、インスリン抵抗性を伴う疾患に対し顕著な治療効果を有している。また本発明の糖尿病の治療薬及びインスリン抵抗性の改善薬は、既存の糖尿病治療薬等で認められる体重増加等の副作用が軽減されており、慢性疾患や複合疾患を持つ患者に対する投与や長期的な投与が可能となる。
【図面の簡単な説明】
【0026】
【図1】実施例10の化合物の空腹時血糖低下作用を示す図である。
【図2】実施例22の化合物の空腹時血糖低下作用を示す図である。
【図3】実施例10の化合物のインスリン抵抗性改善作用を示す図である。
【図4】実施例22の化合物のインスリン抵抗性改善作用を示す図である。
【図5】実施例10の化合物の体重に対する影響を示す図である。
【図6】実施例22の化合物の体重に対する影響を示す図である。
【発明を実施するための形態】
【0027】
本明細書で使用する次の用語は、特に断りがない限り、下記の定義のとおりである。
【0028】
「アルキル」とは、直鎖又は分岐の飽和炭化水素基を意味し、例えば、メチル、エチル、1−プロピル、2−プロピル、1−ブチル、2−ブチル、2−メチル−2−プロピル、1,1−ジメチルエチル、1−ペンチル、2−ペンチル、3−ペンチル又は1−ヘキシルが挙げられる。
【0029】
「シクロアルキル」とは、部分的に不飽和結合を有していてもよく、あるいは架橋環やスピロ環を形成していてもよい、飽和脂環式炭化水素基を意味し、例えば、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロオクチル、ビシクロ[2,2,1]ヘキサン−1−イル、ビシクロ[2,2,1]ヘプタン−2−イル、ビシクロ[3,1,1]ヘプタン−3−イル、ビシクロ[2,2,2]オクタン−1−イル、アダマンチル(アダマンタン−1−イル又はアダマンタン−2−イル)又はノルボルニルが挙げられる。
【0030】
「アリール」とは、芳香族基を意味し、例えば、フェニル又はナフチルが挙げられる。
【0031】
「ヘテロアリール」とは、硫黄原子、窒素原子及び酸素原子からなる群から任意に選択されるヘテロ原子1〜4個を含む(窒素原子は酸化されていてもよい)複素環式芳香族基を意味し、例えば、チエニル、ピロリル、フリル、チアゾリル、イミダゾリル、オキサゾリル、ピラゾリル、イソキサゾリル、1H−1,2,3−トリアゾリル、1H−1,2,4−トリアゾリル、2H−1,2,3−トリアゾリル、1,3,4−オキサジアゾール−2−イル、1H−テトラゾリル、2H−テトラゾリル、ピリジル(ピリジン−2−イル、ピリジン−3−イル又はピリジン−4−イル)、ピリジン−1−オキシド−2−イル、ピリジン−1−オキシド−3−イル、ピリジン−1−オキシド−4−イル、ピリミジニル、ピラジニル、インドリル、ベンゾイミダゾリル、ベンゾチアゾリル、キノリル又はイソキノリルが挙げられる。
【0032】
「アルキルオキシ」とは、アルキルが酸素原子に結合した官能基を意味し、例えば、メトキシ、エトキシ、1−プロポキシ、2−プロポキシ又は1−ブトキシが挙げられる。
【0033】
「ハロゲン」とは、フッ素、塩素、臭素又はヨウ素を意味する。
【0034】
「糖尿病」とは、世界保健機構(WHO)、日本糖尿病学会、米国糖尿病協会又は欧州糖尿病協会等の診断基準に該当する病態を意味し、例えば、1型糖尿病、2型糖尿病又は妊娠糖尿病が挙げられる。
【0035】
「インスリン抵抗性」とは、インスリン抵抗性指数(空腹時血糖値(mg/dL)×空腹時インスリン(μU/mL)÷405)、グルコースクランプ法等を診断基準とする、血中のインスリン濃度に見合ったインスリン作用が得られない病態を意味し、シンドロームXもこれに包含される。
【0036】
「インスリン抵抗性の改善薬」とは、インスリン感受性を増加させる効果を示す薬剤のことを意味し、糖尿病における高インスリン血症の改善作用や空腹時血糖の改善作用等の効果も含まれる。
【0037】
「治療薬又は予防薬」には、治療又は予防の一方に用いられるもののみならず、治療及び予防の双方を目的として同時に用いられるものも包含される。
【0038】
本発明の複素環化合物は、以下の一般式(I)で表されることを特徴としている。
【化2】
[式中、R1は、炭素数1〜6のアルキル、炭素数3〜10のシクロアルキル、環構成炭素数6〜10のアリール又は環構成原子数5〜10のヘテロアリール(ここで、該アリール及び該ヘテロアリールは、それぞれ独立して、1以上のハロゲンによって置換されていてもよい炭素数1〜6のアルキル、又は、炭素数1〜6のアルキルオキシ若しくはハロゲン、によって置換されていてもよい)を示し、R2〜R5は、それぞれ独立して、炭素数1〜6のアルキル又は水素原子を示し、R6は、ヒドロキシ又は水素原子を示す。]
【0039】
R1は、炭素数1〜6のアルキル、環構成炭素数6〜10のアリール又は環構成原子数5〜10のヘテロアリール(ここで、該アリール及び該ヘテロアリールは、それぞれ独立して、1以上のハロゲンによって置換されていてもよい炭素数1〜6のアルキル、炭素数1〜6のアルキルオキシ又はハロゲンによって置換されていてもよい)を示すことが好ましく、フェニル又は3−クロロフェニルを示すことがより好ましい。
【0040】
また、R2〜R5は、それぞれ独立してメチル基又は水素原子を示すことが好ましく、R2及びR3はメチル基を、R4及びR5は水素原子を示すことがより好ましい。
【0041】
さらにR6は、ヒドロキシを示すことが好ましい。
【0042】
なお、一般式(I)で表される複素環化合物は、場合によってはエナンチオマー、ラセミ体又はジアステレオマー等の立体異性体が存在し得るが、本発明の複素環化合物には、全ての立体異性体及びそれぞれの異性体の混合物が包含される。
【0043】
一般式(I)で表される複素環化合物における好ましい置換基の組合せの例を、表1〜8に示す。
【化3】
【0044】
【表1】
【0045】
【表2】
【0046】
【表3】
【0047】
【表4】
【0048】
【表5】
【0049】
【表6】
【0050】
【表7】
【0051】
【表8】
【0052】
一般式(I)で表される複素環化合物の「薬学的に許容される塩」としては、該複素環化合物が塩基性置換基を有する場合には、例えば、塩酸塩、硫酸塩、硝酸塩、臭化水素酸塩、ヨウ化水素酸塩若しくはリン酸塩等の無機酸塩、酢酸塩、トリフルオロ酢酸塩、乳酸塩、クエン酸塩、シュウ酸塩、グルタル酸塩、リンゴ酸塩、酒石酸塩、フマル酸塩、マンデル酸塩、マレイン酸塩、安息香酸塩若しくはフタル酸塩等の有機カルボン酸塩、メタンスルホン酸塩、エタンスルホン酸塩、ベンゼンスルホン酸塩、p−トルエンスルホン酸塩若しくはカンファースルホン酸塩等の有機スルホン酸塩又はアスパラギン酸塩若しくはグルタミン酸塩等の酸性アミノ酸塩が挙げられるが、塩酸塩、臭化水素酸塩、リン酸塩、トリフルオロ酢酸塩、クエン酸、フマル酸、酒石酸塩又はメタンスルホン酸塩が好ましい。
【0053】
また、該複素環化合物が酸性置換基を有する場合には、例えば、ナトリウム塩若しくはカリウム塩等のアルカリ金属塩、カルシウム塩若しくはマグネシウム塩等のアルカリ土類金属塩、アルミニウム塩、アンモニウム塩、トリメチルアミン塩、トリエチルアミン塩、ピリジン塩、ピコリン塩、エタノールアミン塩、ジエタノールアミン塩、トリエタノールアミン塩、ジシクロヘキシルアミン塩若しくはN,N−ジベンジルエチレンジアミン塩等の有機塩基塩又はアルギニン塩、リジン塩若しくはオルニチン塩等の塩基性アミノ酸塩が挙げられるが、ナトリウム塩、カリウム塩又はエタノールアミン塩が好ましい。
【0054】
一般式(I)で表される複素環化合物又はその薬学的に許容される塩は、文献既知の合成法を適宜組み合わせることによって製造することができる。スキーム1に代表的な製造法を例示する。各反応式中の記号の意味は、特に断りのない限り、上記の定義のとおりである。
【0055】
下記の製造法により得られた一般式(I)で表される複素環化合物は、公知の手段によって単離精製することができる。単離精製のための公知の手段としては、例えば、溶媒抽出、再結晶又はクロマトグラフィーが挙げられる。
【0056】
一般式(I)で表される複素環化合物が、光学異性体又は立体異性体を含有する場合には、公知の方法により、それぞれの異性体を単一化合物として得ることができる。
【化4】
【0057】
スキーム1中、Xは、炭素数1〜6のアルキルを示す。
【0058】
〔第1工程〕
一般式(III)で表されるカルボン酸は、一般式(II)で表されるエステルの酸性又は塩基性条件での加水分解反応によって合成することができる。
【0059】
上記の加水分解反応は、Greenの方法(Protective Groups in Organic Synthesis、第5版、John Wieley & Sons.、1999年)又は実験化学講座(第4版、22巻、p.271−309)等に記載の方法に準じて実施可能である。
【0060】
上記の加水分解反応に用いる酸又は塩基の量は、一般式(II)で表されるエステルに対して0.5〜100当量が好ましく、1〜30当量がより好ましい。
【0061】
上記の加水分解反応に用いる反応溶媒は、通常反応を阻害しない溶媒から適宜選択されるが、例えば、テトラヒドロフラン(以下、「THF」)、1,4−ジオキサン、エチレングリコールジメチルエーテル若しくはジメトキシエタン等のエーテル系溶媒、ジクロロメタン、クロロホルム若しくは1,2−ジクロロエタン等のハロゲン系溶媒、ベンゼン若しくはトルエン等の芳香族炭化水素系溶媒、N,N−ジメチルホルムアミド(以下、「DMF」)若しくはジメチルスルホキシド(以下、「DMSO」)等の非プロトン性極性溶媒、アセトン若しくはメチルエチルケトン等のケトン系溶媒、アセトニトリル若しくはプロピオニトリル等のニトリル系溶媒、メタノール、エタノール若しくは2−プロパノール等のプロトン性アルコール溶媒又は水あるいはそれらの混合溶媒が挙げられ、中でもエーテル系溶媒、非プロトン性極性溶媒、プロトン性アルコール溶媒若しくは水又はそれらの混合溶媒が好ましい。
【0062】
上記の加水分解反応開始時点における、一般式(II)で表されるエステルの反応液中の濃度は、通常0.01mM〜1Mの範囲であることが好ましい。
【0063】
上記の加水分解反応温度は、−78〜200℃が好ましく、−20〜100℃がより好ましい。
【0064】
上記の加水分解反応時間は、反応温度等の条件に応じて適宜選択されるが、通常1〜30時間程度で満足すべき結果が得られる。
【0065】
〔第2工程〕
一般式(I)で表される複素環化合物は、一般式(III)で表されるカルボン酸の分子内縮合反応によって合成することができる。
【0066】
上記の縮合反応は、一般的なペプチド合成法(「実験化学講座(第16巻)有機化合物の合成IV」、第5版、p.258−270等)に準じて実施可能である。
【0067】
上記の縮合反応に用いる試薬(以下、「縮合剤」)としては、例えば、シクロヘキシルカルボジイミド、N−エチル−N’−3−ジメチルアミノプロピルカルボジイミド塩酸塩、N,N−カルボジイミダゾール、O−(7−アザベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウム ヘキサフルオロホスファート又はオルト−(ベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウムヘキサフルオロホスファート(以下、「HBTU」)が挙げられるが、HBTUが好ましい。
【0068】
上記の縮合剤の量は、一般式(III)で表されるカルボン酸に対して1〜10当量が好ましく、1〜3当量がより好ましい。
【0069】
上記の縮合反応に適する反応溶媒は、上記の縮合剤の種類により異なるが、例えば、THF、1,4−ジオキサン、エチレングリコールジメチルエーテル若しくはジメトキシエタン等のエーテル系溶媒、ジクロロメタン、クロロホルム若しくは1,2−ジクロロエタン等のハロゲン系溶媒、DMF若しくはDMSO等の非プロトン性極性溶媒又はアセトニトリル若しくはプロピオニトリル等のニトリル系溶媒が挙げられ、中でもハロゲン系溶媒若しくは非プロトン性極性溶媒が好ましく、ハロゲン系溶媒がより好ましい。
【0070】
上記の縮合反応開始時点における、一般式(III)で表されるカルボン酸の反応液中の濃度は、通常0.01mM〜1Mの範囲であることが好ましい。
【0071】
上記の縮合反応温度は、−78〜200℃が好ましく、0〜100℃がより好ましい。
【0072】
上記の縮合反応時間は、反応温度等の条件に応じて適宜選択されるが、通常6〜48時間程度で満足すべき結果が得られる。
【0073】
スキーム1に例示された製造法の出発原料である、一般式(II)で表されるエステルは、例えば、スキーム2に示す方法で合成することができる。各反応式中の記号の意味は、特に断りのない限り、上記の定義のとおりである。
【0074】
【化5】
【0075】
〔第1工程〕
一般式(VI)で表されるアルデヒドは、一般式(V)で表されるアルコールの酸化反応によって合成することができる。
【0076】
上記の酸化反応は、一般的な酸化反応(「実験化学講座(第15巻)有機化合物の合成III」、第5版、p.9−44等)に準じて実施可能である。
【0077】
上記の酸化反応に用いる試薬(以下、「酸化剤」)としては、例えば、Dess−Martin試薬又はDMSO−塩化オキサリルが挙げられるが、Dess−Martin試薬が好ましい。
【0078】
上記の酸化剤の量は、一般式(V)で表されるアルコールに対して1〜10当量が好ましく、1〜3当量がより好ましい。
【0079】
上記の酸化反応に適する反応溶媒は、上記の酸化剤の種類により異なるが、例えば、THF、1,4−ジオキサン、エチレングリコールジメチルエーテル若しくはジメトキシエタン等のエーテル系溶媒、ジクロロメタン、クロロホルム若しくは1,2−ジクロロエタン等のハロゲン系溶媒、DMF若しくはDMSO等の非プロトン性極性溶媒又はアセトニトリル若しくはプロピオニトリル等のニトリル系溶媒が挙げられ、中でもハロゲン系溶媒が好ましい。
【0080】
上記の酸化反応開始時点における、一般式(V)で表されるアルコールの反応液中の濃度は、通常0.01mM〜1Mの範囲であることが好ましい。
【0081】
上記の酸化反応温度は、−78〜200℃が好ましく、0〜100℃がより好ましい。
【0082】
上記の酸化反応時間は、反応温度等の条件に応じて適宜選択されるが、通常1〜48時間程度で満足すべき結果が得られる。
【0083】
〔第2工程〕
一般式(II)で表されるアミンは、一般式(VI)で表されるアルデヒドと一般式(IV)で表されるアミンとの還元的アミノ化反応によって合成することができる。
【0084】
上記の還元的アミノ化反応は、一般的な還元的アミノ化反応(「実験化学講座(第14巻)有機化合物の合成II」、第5版、p.371等)に準じて実施可能である。
【0085】
上記の還元的アミノ化反応に用いる試薬(以下、「還元剤」)としては、例えば、水素化リチウムアルミニウム、水素化ホウ素ナトリウム、水素化シアノホウ素ナトリウム、水素化トリアセトキシホウ素ナトリウム又はパラジウム/炭素が挙げられるが、水素化トリアセトキシホウ素ナトリウムが好ましい。
【0086】
上記の還元剤の量は、一般式(VI)で表されるアルデヒドに対して1〜10当量が好ましく、1〜3当量がより好ましい。
【0087】
上記の還元的アミノ化反応に適する反応溶媒は、上記の還元剤の種類により異なるが、例えば、THF、1,4−ジオキサン、エチレングリコールジメチルエーテル若しくはジメトキシエタン等のエーテル系溶媒、ジクロロメタン、クロロホルム若しくは1,2−ジクロロエタン等のハロゲン系溶媒、メタノール若しくはエタノール等のプロトン性極性溶媒、DMF若しくはDMSO等の非プロトン性極性溶媒又はアセトニトリル若しくはプロピオニトリル等のニトリル系溶媒が挙げられ、中でもプロトン性極性溶媒が好ましい。
【0088】
上記の還元的アミノ化反応開始時点における、一般式(VI)で表されるアルデヒドの反応液中の濃度は、通常0.01mM〜1Mの範囲であることが好ましい。
【0089】
上記の還元的アミノ化反応温度は、−78〜200℃が好ましく、0〜100℃がより好ましい。
【0090】
上記の還元的アミノ化反応時間は、反応温度等の条件に応じて適宜選択されるが、通常6〜60時間程度で満足すべき結果が得られる。
【0091】
スキーム2に例示された製造法の出発原料である、一般式(V)で表されるアルコールは、例えば、スキーム3に示す方法で合成することができる。各反応式中の記号の意味は、特に断りのない限り、上記の定義のとおりである。
【化6】
【0092】
スキーム3中、Yは、置換されていてもよいベンジル又はトリアルキルシリルを示す。
【0093】
一般式(V)で表されるアルコールは、一般式(VII)で表されるベンジルエーテル又はトリアルキルシリルエーテルの脱保護反応によって合成することができる。
【0094】
上記の脱保護反応は、一般的なアルコール保護基の脱保護法(「実験化学講座(第14巻)有機化合物の合成と反応IV」、第3版、p.2497−2505等)準じて実施可能である。
【0095】
上記の脱保護反応に用いる試薬(以下、「脱保護剤」)としては、ベンジルエーテルに対しては、例えば、水素雰囲気下におけるパラジウム/炭素、水酸化パラジウム/炭素又は白金/炭素が挙げられるが、水素雰囲気下におけるパラジウム/炭素が好ましい。
【0096】
また、トリアルキルシリルエーテルに対しては、例えば、塩酸又はテトラブチルアンモニウムフルオリド(以下、「TBAF」)が好ましく、TBAFがより好ましい。
【0097】
上記の脱保護剤の量は、一般式(VII)で表されるベンジルエーテル又はトリアルキルシリルエーテルに対して0.1〜10当量が好ましく、0.1〜3当量がより好ましい。
【0098】
上記の脱保護反応に適する反応溶媒は、上記の脱保護剤の種類により異なるが、例えば、THF、1,4−ジオキサン、エチレングリコールジメチルエーテル若しくはジメトキシエタン等のエーテル系溶媒、ジクロロメタン、クロロホルム若しくは1,2−ジクロロエタン等のハロゲン系溶媒、DMF若しくはDMSO等の非プロトン性極性溶媒、メタノール等のプロトン性極性溶媒又はアセトニトリル若しくはプロピオニトリル等のニトリル系溶媒が挙げられ、中でもエーテル系溶媒又はプロトン性極性溶媒が好ましい。
【0099】
上記の脱保護反応開始時点における、一般式(VII)で表されるベンジルエーテル又はトリアルキルシリルエーテルの反応液中の濃度は、通常0.01mM〜1Mの範囲であることが好ましい。
【0100】
上記の脱保護反応温度は、−78〜200℃が好ましく、0〜100℃がより好ましい。
【0101】
上記の脱保護反応時間は、反応温度等の条件に応じて適宜選択されるが、通常6〜48時間程度で満足すべき結果が得られる。
【0102】
スキーム3に例示された製造法の出発原料である、一般式(VII)で表されるベンジルエーテル又はトリアルキルシリルエーテルは、例えば、スキーム4に示す方法で合成することができる。各反応式中の記号の意味は、特に断りのない限り、上記の定義のとおりである。
【0103】
スキーム4における、一般式(X)で表されるヒドラジンとしては、塩酸塩等の塩を用いても構わない。
【化7】
【0104】
〔第1工程〕
一般式(VIII)で表される化合物は、一般式(IX)で表される化合物とジメチルホルムアミドジメチルアセタールとのアミノメチル化反応によって合成することができる。
【0105】
上記のアミノメチル化反応に用いるジメチルホルムアミドジメチルアセタールの量は、一般式(IX)で表されるアセト酢酸エステルに対して1〜20当量が好ましく、3〜10当量がより好ましい。
【0106】
上記のアミノメチル化反応は、反応溶媒を用いず、無溶媒で行うことが好ましい。一方で、上記のアミノメチル化反応に反応溶媒を用いる場合にあっては、該反応溶媒は通常反応を阻害しない溶媒から適宜選択される。そのような溶媒としては、例えば、THF、1,4−ジオキサン若しくはエチレングリコールジメチルエーテル等のエーテル系溶媒、ジクロロメタン、クロロホルム若しくは1,2−ジクロロエタン等のハロゲン系溶媒、ベンゼン若しくはトルエン等の芳香族炭化水素系溶媒、DMF若しくはDMSO等の非プロトン性極性溶媒、メタノール、エタノール若しくは2−プロパノール等のプロトン性アルコール溶媒若しくは水又は混合溶媒が挙げられるが、ハロゲン系溶媒、芳香族炭化水素系溶媒、非プロトン性極性溶媒又はプロトン性アルコール溶媒が好ましい。
【0107】
上記のアミノメチル化反応開始時点における、一般式(IX)で表される化合物の反応液中の濃度は、通常0.1mM〜10Mの範囲であることが好ましい。
【0108】
上記のアミノメチル化反応温度は、0〜200℃が好ましく、20〜120℃がより好ましい。
【0109】
上記のアミノメチル化反応時間は、反応温度等の条件に応じて適宜選択されるが、通常0.5〜12時間程度で満足すべき結果が得られる。
【0110】
〔第2工程〕
一般式(VII)で表されるベンジルエーテル又はトリアルキルシリルエーテルは、一般式(VIII)で表される化合物に、一般式(X)で表されるヒドラジン化合物を作用させる環化反応によって合成することができる。
【0111】
上記の環化反応に用いるヒドラジン化合物の量は、一般式(VIII)で表されるアセト酢酸エステルに対して1〜10当量が好ましく、1〜5当量がより好ましい。
【0112】
上記の環化反応に用いる反応溶媒は、通常反応を阻害しない溶媒から適宜選択されるが、例えば、DMF若しくはDMSO等の非プロトン性極性溶媒、メタノール、エタノール若しくは2−プロパノール等のプロトン性アルコール溶媒、アセトニトリル若しくはプロピオニトリル等のニトリル系溶媒、ギ酸、酢酸若しくはプロピオン酸等の有機カルボン酸又は水あるいはそれらの混合溶媒が挙げられ、中でもプロトン性アルコール溶媒又はニトリル系溶媒が好ましく、プロトン性アルコール溶媒がより好ましい。
【0113】
上記の環化反応開始時点における、一般式(VIII)で表される化合物の濃度は、通常0.01mM〜10Mの範囲であることが好ましい。
【0114】
上記の環化反応温度は、0〜200℃が好ましく、20〜120℃がより好ましい。
【0115】
上記の環化反応時間は、反応温度等の条件に応じて適宜選択されるが、通常1〜12時間程度で満足すべき結果が得られる。
【0116】
一般式(IV)
【化8】
で表されるアミンは、例えば、文献記載の方法(Tetrahedron Letters、2006年、第47巻、第46号、p.8063−8067;Journal of Medicinal Chemistry、2007年、第50巻、第1号、p.149−164等)により合成することができる。
【0117】
また、本発明の11β−HSD1阻害剤、糖尿病の治療薬及び予防薬並びにインスリン抵抗性の改善薬は、上記の一般式(I)で表される複素環化合物又はその薬学的に許容される塩を有効成分として含有することを特徴としている。
【0118】
上記の11β−HSD1阻害剤、糖尿病の治療薬及び予防薬並びにインスリン抵抗性の改善薬の薬効は、血中グリコヘモグロビン値(HbA1c)、空腹時血糖値、経口糖負荷試験(OGTT;oral glucose tolerance test)における血糖の2時間値、血中c−ペプチド値、血中グリコアルブミン値、1,5−AG(1,5−アンヒドログルシトール)、血中空腹時インスリン値、HOMA−IR(homeostasis model assessment of insulin resistance)、グルコースクランプ(euglycemic−hyperinsulinemic clamp)試験における糖注入率(GIR; glucose infusion rate)、糖産生率(HGP;Hepatic glucose production)、インスリン負荷試験(ITT;insulin tolerance test)又はピルビン酸負荷試験(PTT;Pyruvate tolerance test)等で評価できる。
【0119】
一般式(I)で表される複素環化合物又はその薬学的に許容される塩は、糖尿病、特に2型糖尿病の改善に効果的であり、具体的には、HbA1c、グリコアルブミン、フルクトサミン、1,5−AG、随時高血糖、空腹時高血糖、耐糖能異常、肝糖新生の亢進、HOMA−IR及びグルコースクランプ試験における糖注入率(GIR)や糖産生率(HGP)の改善が期待できる。さらに、一般式(I)で表される複素環化合物又はその薬学的に許容される塩は、インスリン抵抗性の改善に用いることができるため、メタボリックシンドロームの治療薬又は予防薬として適している。
【0120】
一般式(I)で表される複素環化合物を、糖尿病の治療薬若しくは予防薬又はインスリン抵抗性の改善薬として臨床で投与する場合にあっては、一般式(I)で表される複素環化合物のフリー体又はその薬学的に許容される塩をそのまま投与もよいし、さらに賦形剤、安定化剤、保存剤、緩衝剤、溶解補助剤、乳化剤、希釈剤又は等張化剤等の添加剤が適宜混合されたものを投与しても構わない。その投与形態としては、例えば、錠剤、カプセル剤、顆粒剤、散剤若しくはシロップ剤等の経口剤による経口投与、注射剤、坐剤若しくは液剤等による非経口投与又は軟膏剤、クリーム剤若しくは貼付剤等による局所投与が挙げられる。
【0121】
上記の各投与形態の製剤は、公知の製法によって作ることができるが、一般式(I)で表される複素環化合物又はその薬学的に許容される塩を0.00001〜90重量%含有することが好ましく、0.0001〜70重量%含有することがより好ましい。
【0122】
一般式(I)で表される複素環化合物又はその薬学的に許容される塩の投与量は、症状、年齢、体重及び投与方法等に応じて適宜選択されるが、成人に対して経口剤として投与する場合、有効成分量として1日1μg〜10gが好ましく、それぞれ1回又は数回に分けて投与することができる。
【0123】
一般式(I)で表される複素環化合物又はその薬学的に許容される塩を有効成分として含有する医薬、すなわち糖尿病の治療薬若しくは予防薬又はインスリン抵抗性の改善薬は、その他の糖尿病治療薬、脂質異常症治療薬、肥満症治療薬又はメタボリックシンドローム治療薬(以下、「併用薬」)と組み合わせて用いても構わない。
【0124】
この際、一般式(I)で表される複素環化合物又はその薬学的に許容される塩を有効成分とする医薬と、併用薬との投与時期は限定されず、これらを併用又は配合により同時に投与してもよいし、時間差をおいて投与してもよい。併用薬の投与量は、臨床上用いられている用量を基準として適宜選択することができる。また、一般式(I)で表される複素環化合物又はその薬学的に許容される塩を有効成分とする医薬と、併用薬との併用容量比あるいは配合比は、投与対象、投与形態、症状又は各薬剤の組み合わせ等により適宜選択することができる。
【0125】
上記の併用薬としては、例えば、インスリン製剤(超速効型インスリン製剤、速効型インスリン製剤、混合型インスリン製剤、中間型インスリン製剤、持続型インスリン製剤、持効型溶解インスリン製剤、経肺インスリン製剤、経口インスリン製剤等)、インスリン抵抗性改善薬(ピオグリタゾン(pioglitazone)、ロシグリタゾン(rosiglitazone)、ネトグリタゾン(netoglitazone)、ファルグリタザール(farglitazar)、リボグリタゾン(rivoglitazone)、バラグリタゾン(balaglitazone)等)、α−グルコシダーゼ阻害薬(アカルボース(acarbose)、ボグリボース(voglibose)、ミグリトール(miglitol)、エミグリテート(emiglitate)等)、ビグアナイド剤(メトホルミン(metformin)、ブホルミン(buformin)等)、スルホニルウレア剤(トルブタミド(tolbutamide)、アセトヘキサミド(acetohexamide)、クロルプロパミド(chlorpropamide)、トラザミド(tolazamide)、グリクロピラミド(glyclopyramide)、グリブゾール(glybuzole)、グリベンクラミド(glibenclamide)、グリクラジド(gliclazide)、グリメピリド(glimepiride)、グリピジド(glipizide)、グリキドン(gliquidone)等)、速効型インスリン分泌促進薬(ナテグリニド(nateglinide)、レパグリニド(repaglinide)、ミチグリニド(mitiglinide)等)、GLP−1作動薬(エクセナチド(exenatide)、リラグルチド(liraglutide)等)、アミリン作動薬(プラムリンチド(pramlintide)等)、DPP−IV阻害薬(ビルダグリプチン(vildagliptin)、シタグリプチン(sitagliptin)、サクサグリプチン(saxagliptin)、アログリプチン(alogliptin)、デナグリプチン(denagliptin)、ジェミグリプチン(Gemigliptin)、ドゥトグリプチン(Dutogliptin)等)、β3作動薬(ソラベグロン(solabegron)、ミラベグロン(mirabegron)、KRP−204等)、フルクトース−1,6−ビスホスファターゼ阻害薬(MB−6322、MB−07803等)、SGLT(sodium−dependent renal glucose transporter)阻害薬(セルグリフロジン(sergliflozin)、レモグリフロジン(remogliflozin)、AVE−2268、TS−033、KGA−2727、SAR−7226等)、11β−HSD1阻害薬(BVT−3498、AMG−221、INCB−13739、INCB−20817、JTT−654、PF−915275等)、PTP−1B(protein tyrosine phosphatase−1B)阻害薬(ISIS−113715、JTT−551等)、GSK3β(glycogen synthase kinase 3β)阻害薬(SAR−502250等)、グルカゴン拮抗薬(BAY−27−9955、NN−2501等)、グリコーゲンホスホリラーゼ阻害薬(イソファゴミン(Isofagomine)、PSN−357等)、CPT1(カルニチン O−パルミトイルトランスフェラーゼ1)阻害薬(テグリカール(teglicar)等)、グルココルチコイド拮抗薬(ミフェプリストン(mifepristone)、KB−3305等)、HMG−CoA還元酵素阻害薬(プラバスタチン(pravastatin)、シンバスタチン(simvastatin)、フルバスタチン(fluvastatin)、アトルバスタチン(atorvastatin)、ピタバスタチン(pitavastatin)等)、陰イオン交換樹脂(コレスチラミン(colestyramine)、コレスチミド(colestimide)等)、フィブラート系薬(クロフィブラート(clofibrate)、クリノフィブラート(clinofibrate)、ベザフィブラート(bezafibrate)、フェノフィブラート(fenofibrate)等)、ニコチン酸系薬(ニコチン酸トコフェロール(tocopherol nicotinate)、CB1(カンナビノイド1)拮抗薬(リモナバン(rimonabant)、スリナバン(surinabant)、タラナバン(taranabant)、ドリナバン(drinabant)等)、リパーゼ阻害剤(オルリスタット(orlistat)等)又は中枢性食欲抑制剤(マジンドール(mazindol)、フェンフルラミン(fenfluramine)、デクスフェンフルラミン(dexfenfluramine)又はシブトラミン(sibutramine)、フェンターミン(phentermine)等)が挙げられる。
【実施例】
【0126】
以下、実施例を示して本発明を具体的に詳述するが、本発明はこれらに限定されるものではない。
【0127】
一般式(I)で表される複素環化合物の原料及び中間体は、以下の参考例に記載する方法で合成した。また、参考例化合物の合成に使用される化合物で合成法の記載のないものについては、市販の化合物を使用した。
【0128】
(参考例1) trans−5−ヒドロキシ−2−アダマンチルアミン(以下、「参考例1の化合物」)の合成:
【化9】
5−ヒドロキシ−2−アダマンタノン(3.0g,18mmol)をトルエン(60mL)に溶解し、(−)−1−フェニルエチルアミン(2.4mL,19mmol)、アルミニウムイソプロポキシド(3.7g,18mmol)、水酸化パラジウム/炭素(10%wet,0.25g,1.2mmol)を加え水素雰囲気下、50℃で撹拌した。16時間後、反応液を室温まで冷却し、セライト濾過し、濾液を濃縮した。得られた粗生成物をメタノール(130mL)に溶解し、水酸化パラジウム/炭素(10%wet,0.15g,0.72mmol)を加え水素雰囲気下、室温で撹拌した。14時間後、反応液をセライト濾過し、濾液を濃縮した。得られた粗生成物をヘキサン/酢酸エチルを溶媒として再結晶を行い、表題化合物0.98g(55%)を白色固体として得た。
1H−NMR(400MHz,CDCl3)
δ:3.79(3H,s),6.92−6.94(2H,m),7.05(1H,dd,J=4.8,7.6Hz),7.09−7.11(2H,m),7.97(1H,dd,J=2.0,7.6Hz),8.28(1H,dd,J=2.0,4.8Hz).
MS(ESI)[M+H]+
227(M+H)+
【0129】
(参考例2) エチル 6−ベンジルオキシ−3−オキソヘキサノエート(以下、「参考例2の化合物」)の合成:
【化10】
4−ベンジルオキシ酪酸(5.1g,26mmol)をアセトニトリル(50mL)に溶解し、カルボニルジイミダゾール(4.5g,28mmol)を加えて室温で撹拌した(反応液A)。別途、マグネシウムクロリド(5.5g,58mmol)をアセトニトリル(50mL)に溶解し、マロン酸エチルカリウム塩(9.4g,55mmol)、トリエチルアミン(17mL,120mmol)を加え室温で撹拌した(反応液B)。2.5時間後、反応液Aを反応液Bに加え90℃で撹拌した。2時間後、反応液を室温まで冷却し、1N 塩酸を加えpH=5とした後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=100/0〜98/2)で精製し、表題化合物4.7g(95%)を無色液体として得た。1H−NMR(400MHz,CDCl3)
δ:1.27(3H,t,J=7.2Hz),1.82−1.96(2H,m),2.66(2H,t,J=7.2Hz),3.43(2H,s),3.49(2H,t,J=6.0Hz),4.19(2H,q,J=7.2Hz),4.48(2H,s),7.27−7.37(5H,m).
MS(ESI)[M+H]+
265
【0130】
(参考例3) エチル 6−ベンジルオキシ−4,4−ジメチル−3−オキソヘキサノエート(以下、「参考例3の化合物」)の合成:
〔第1工程〕
メチル 4−ベンジルオキシブタノエートの合成:
【化11】
4−ベンジルオキシ酪酸(5.0g,26mmol)をメタノール(52mL)に溶解し、濃硫酸(1.0mL,19mmol)を加え室温で撹拌した。3時間後、炭酸水素ナトリウムを加え中和した後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=100/0〜76/24)で精製し、表題化合物5.4g(99%)を無色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:1.90−1.98(2H,m),2.45(2H,t,J=7.6Hz),3.51(2H,t,J=6.0Hz),3.65(3H,m),4.49(2H,m),7.26−7.35(5H,s).
MS(ESI)[M+H]+
209
【0131】
〔第2工程〕
メチル 4−ベンジルオキシ−2−メチルブタノエートの合成:
【化12】
ジイソプロピルアミン(5.1mL,36mmol)をTHF(90mL)に溶解し、n−ブチルリチウム(2.6N,14mL,36mmol)を加え−78℃で撹拌した。20分後、メチル 4−ベンジルオキシブタノエート(0.74g,2.0mmol)のTHF溶液(10mL)を加え−78℃で撹拌した。20分後、ヨードメタン(3.0mL,48mmol)を加え0℃で撹拌した。3時間後、水、1N 塩酸を加えた後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=98/2〜80/20)で精製し、表題化合物5.0g(94%)を無色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:1.17(3H,d,J=6.8Hz),1.65−1.75(1H,m),1.98−2.06(1H,m),2.61−2.71(1H,m),3.47−3.52(2H,m),3.64(3H,s),4.48(2H,s),7.26−7.35(5H,m).
MS(ESI)[M+H]+
223
【0132】
〔第3工程〕
メチル 4−ベンジルオキシ−2,2−ジメチルブタノエートの合成:
【化13】
ジイソプロピルアミン(2.4mL,17mmol)をTHF(45mL)に溶解し、n−ブチルリチウム(2.6N,6.5mL,17mmol)を加え−78℃で撹拌した。20分後、メチル 4−ベンジルオキシ−2−メチルブタノエート(0.74g,2.0mmol)のTHF溶液(5.0mL)を加え−78℃で撹拌した。20分後、ヨードメタン(1.4mL,22mmol)を加え0℃で撹拌した。3時間後、水、1N 塩酸を加えた後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=100/0〜86/14)で精製し、表題化合物2.5g(94%)を無色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:1.20(6H,s),1.90(2H,t,J=6.8Hz),3.50(2H,t,J=6.8Hz),3.59(3H,s),4.45(2H,s),7.26−7.36(5H,m).
MS(ESI)[M+H]+
237
【0133】
〔第4工程〕
参考例3の化合物の合成:
【化14】
メチル 4−ベンジルオキシ−2,2−ジメチルブタノエート(4.0g,17mmol)をメタノール(25mL)に溶解し、1N 水酸化ナトリウム(25mL,25mmol)を加え60℃で撹拌した。3時間後、反応液を室温まで冷却し、濃縮しクロロホルムで逆抽出を行い、1N 塩酸を加え中和した後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をアセトニトリル(34mL)に溶解し、カルボニルジイミダゾール(2.9g,18mmol)を加え室温で撹拌した(反応液A)。別途、マグネシウムクロリド(3.5g,37mmol)をアセトニトリル(34mL)に溶解し、マロン酸エチルカリウム塩(6.1g,36mmol)、トリエチルアミン(11mL,76mmol)を加え室温で撹拌した(反応液B)。14時間後、反応液Aを反応液Bに加え90℃で撹拌した。2時間後、反応液を室温まで冷却し、1N 塩酸を加えpH=5とした後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=100/0〜86/14)で精製し、表題化合物4.7g(95%)を無色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:1.26(6H,s),1.38(3H,t,J=6.8Hz),1.54−1.55(2H,m),4.30(2H,q,J=6.8Hz),7.43−7.48(5H,m),8.06(1H,s),9.58−9.62(1H,m).
MS(ESI)[M+H]+
293
【0134】
(比較例1) 5−アダマンタン−2−イル−1−フェニル−6,7−ジヒドロ−1H−ピラゾロ[4,3−C]ピリジン−4(5H)−オン(以下、「比較例1の化合物」)の合成:
〔第1工程〕
(1−アダマンタン−2−イル)ピペリジン−2,4−ジオンの合成:
【化15】
アダマンタン−2−アミン(2.5g,17mmol)をTHF(32mL)に溶解し、エチルアクリレート(2.3mL,21mmol)を加え20℃で撹拌した。16時間後、エチル 3−クロロ−オキソプロパノエート(2.4mL,19mmol)、トリエチルアミン(3.0mL,21mmol)を加え20℃で撹拌した。2時間後、水を加えた後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をTHF(14mL)に溶解し、ナトリウムエトキサイド(0.23g,3.4mmol)を加え90℃で撹拌した。2時間後、塩酸水溶液(6.0N,8.0mL,48mmol)を加えた後、100℃で撹拌した。4時間後、反応液を室温まで冷却し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=70/30〜40/60)で精製し、表題化合物0.41g(10%)を白色固体として得た。
1H−NMR(400MHz,CDCl3)
δ:1.71−1.77(5H,m),1.83−1.93(7H,m),2.27(2H,s),2.60(2H,s),3.40(2H,s),3.84(2H,s),4.27(1H,s)
MS(ESI)[M+H]+
248
【0135】
〔第2工程〕
比較例1の化合物の合成:
【化16】
1−アダマンタン−2−イルピペリジン−2,4−ジオン(0.41g,1.7mmol)に1,1−ジメトキシ−N,N−ジメチルメタンアミン(1.1mL,8.3mmol)を加え90℃で撹拌した。2時間後、反応液を室温まで冷却し、水を加えた後、クロロホルムで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗成生物をエタノール(7.0mL)に溶解し、フェニルヒドラジン(0.27mL,2.7mmol)を加え90℃で撹拌した。5時間後、反応液を室温まで冷却し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=80/20〜40/60)で精製し、表題化合物0.41g(86%)を白色固体として得た。
1H−NMR(400MHz,CDCl3)
δ:1.66−1.77(5H,m),1.87−2.00(7H,m),2.32(2H,s),3.10(2H,t,J=6.4Hz),3.77(2H,t,J=6.4Hz),4.27(1H,s),7.39−7.41(1H,m),7.50−7.51(4H,m),8.05(1H,s)
MS(ESI)[M+H]+
348
【0136】
(実施例1) 1−フェニル−5−ヒドロキシアダマンタン−2−イル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例1の化合物」)の合成:
〔第1工程〕
エチル 5−(2−シアノエチル)−1−フェニル−1H−ピラゾール−4−カルボキシレートの合成:
【化17】
エチル 5−シアノ−3−オキソペンタノエート(1.5g,8.8mmol)にジメチルホルムアミドジメチルアセタール(5.9mL,44mmol)を加え80℃で撹拌した。3時間後、反応液を室温まで冷却し、水を加えた後、ジエチルエーテルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をエタノール(40mL)に溶解し、フェニルヒドラジン(2.0g,18mmol)を加え80℃で撹拌した。5時間後、室温まで冷却し、水を加えた後、濃縮後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=70/30〜12/88)で精製し、表題化合物0.46g(20%)を黄色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:1.40(3H,t,J=7.2Hz),2.75(2H,t,J=8.0Hz),3.27(2H,t,J=8.0Hz),4.35(2H,q,J=7.2Hz),7.40−7.44(2H,m),7.52−7.58(3H,m),8.06(1H,s).
MS(ESI)[M+H]+
270
【0137】
〔第2工程〕
エチル 5−(3−アミノプロピル)−1−フェニル−1H−ピラゾール−4−カルボキシレートの合成:
【化18】
エチル 5−(2−シアノエチル)−1−フェニル−1H−ピラゾール−4−カルボキシレート(0.45g,1.7mmol)をエタノール(17mL)に溶解し、パラジウム/炭素(10%wet,0.18g,0.17mmol)、濃塩酸(1.0mL)を加え水素雰囲気下、室温で撹拌した。25時間後、反応液をセライト濾過し、濾液を濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=100/0〜76/24)で精製し、表題化合物0.16g(35%)を無色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:1.38(3H,t,J=7.2Hz),1.83(2H,t,J=6.8Hz),2.58−2.78(4H,m),3.05(2H,t,J=7.2Hz),4.33(2H,q,J=7.2Hz),7.38−7.41(2H,m),7.43−7.48(3H,m),8.00(1H,s),
MS(ESI)[M+H]+
274
【0138】
〔第3工程〕
実施例1の化合物の合成:
【化19】
エチル 5−(3−アミノプロピル)−1−フェニル−1H−ピラゾール−4−カルボキシレート(81mg,0.30mmol)をエタノール(3.0mL)に溶解し、アダマンタン−2−オン(49mg,0.33mmol)、チタニウム(IV)テトライソプロポキシド(180mL,0.59mmol)を加え室温で撹拌した。20時間後、水素化トリアセトキシホウ素ナトリウム(0.13g,0.59mmol)を加え室温で撹拌した。2時間後、1N 塩酸を加えた後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をクロロホルム(5.0mL)に溶解し、ジイソプロピルエチルアミン(0.14mL,0.26mmol)、HBTU(61mg,0.26mmol)を加え室温で撹拌した。1時間後、水を加えた後、クロロホルムで抽出した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=90/10〜47/53)で精製し、表題化合物82mg(77%)を白色固体として得た。
1H−NMR(400MHz,CDCl3)
δ:1.66−1.77(3H,m),1.85−2.01(8H,m),2.12−2.20(2H,m),2.21−2.29(2H,m),2.90(2H,t,J=7.2Hz),3.56−3.62(2H,m),4.37−4.41(1H,m),7.39−7.51(5H,m),8.11(1H,s).
MS(ESI)[M+H]+
362
【0139】
(実施例2) 1−メチル−5−(trans−5−ヒドロキシアダマンタン−2−イル)5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例2の化合物」)の合成:
【化20】
エチル 6−ベンジルオキシ−3−オキソヘキサノエート(2.0g,7.6mmol)にジメチルホルムアミドジメチルアセタール(5.1mL,38mmol)を加え90℃で撹拌した。2時間後、反応液を室温まで冷却し、水を加えた後、ジエチルエーテルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をエタノール(35mL)に溶解し、メチルヒドラジン(0.50g,11mmol)を加え80℃で撹拌した。4時間後、反応液を室温まで冷却し、濃縮し水を加えた後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=100/0〜81/19)で精製した。得られた粗精製物をエタノール(27mL)に溶解し、水酸化パラジウム/炭素(10%wet,0.075g,0.54mmol)を加え水素雰囲気下、室温で撹拌した。5時間後、反応液をセライト濾過し、濾液を濃縮した。得られた粗生成物をジクロロメタン(27mL)に溶解し、Dess−Martin試薬(1.1g,2.7mmol)を加え室温で撹拌した。4時間後、反応液をセライト濾過し、濾液を濃縮した。得られた粗生成物をクロロホルム(28mL)に溶解し、trans−5−ヒドロキシ−2−アダマンチルアミン(450mg,2.7mmol)、酢酸(0.47mL,8.1mmol)、水素化トリアセトキシホウ素ナトリウム(1.5g,8.1mmol)を加え室温で撹拌した。16時間後、水を加えた後、クロロホルムで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=100/0〜81/19)で精製した。得られた粗精製物をメタノール(1.0mL)に溶解し、1N 水酸化ナトリウム(1.0mL,1.0mmol)を加え室温で撹拌した。16時間後、濃縮した。得られた粗生成物をクロロホルム(14mL)に溶解し、ジイソプロピルエチルアミン(0.37mL,2.1mmol)、HBTU(160mg,0.70mmol)を加え室温で撹拌した。16時間後、水を加えた後、クロロホルムで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=100/0〜91/9)で精製し、表題化合物45mg(3%)を白色固体として得た。
1H−NMR(400MHz,CDCl3)
δ:1.75−2.84(7H,m),1.95−2.00(2H,m),2.15−2.25(3H,m),2.44−2.48(2H,m),2.86(2H,t,J=7.2Hz),3.53−3.57(2H,m),3.80(3H,s),4.28−4.31(1H,m),7.95(1H,s).
MS(ESI)[M+H]+
316
【0140】
(実施例3) 1−エチル−5−(trans−5−ヒドロキシアダマンタン−2−イル)5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例3の化合物」)の合成:
参考例1及び2並びに実施例2と同様の手順により、以下の実施例3の化合物を合成した。
【化21】
1H−NMR(400MHz,CDCl3)
δ:1.43(3H,t,J=7.2Hz),1.50−1.60(2H,m),1.75−1.85(6H,m),1.95−2.01(2H,m),2.15−2.24(2H,m),2.43−2.49(2H,m),2.88(2H,t,J=7.2Hz),3.52−3.57(2H,m),4.08(2H,q,J=7.2Hz),4.08−4.11(1H,m),7.97(1H,s).
MS(ESI)[M+H]+
330
【0141】
(実施例4) 1−(tert−ブチル)−5−(trans−5−ヒドロキシアダマンタン−2−イル)5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例4の化合物」)の合成:
参考例1及び2並びに実施例2と同様の手順により、以下の実施例4の化合物を合成した。
【化22】
1H−NMR(400MHz,CDCl3)
δ:1.49−1.58(2H,m),1.66(9H,s),1.76−1.86(6H,m),1.95−2.02(2H,m),2.10−2.24(3H,m),2.41−2.47(2H,m),3.12(2H,t,J=7.2Hz),3.44−3.49(2H,m),7.82(1H,s).
MS(ESI)[M+H]+
358
【0142】
(実施例5) 1−シクロヘキシル−5−(trans−5−ヒドロキシアダマンタン−2−イル)5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例5の化合物」)の合成:
参考例1及び2並びに実施例2と同様の手順により、以下の実施例5の化合物を合成した。
【化23】
1H−NMR(400MHz,CDCl3)
δ:1.31−1.40(4H,m),1.48−1.59(5H,m),1.78−2.00(11H,m),2.15−2.25(2H,m),2.43−2.49(2H,m),2.83−2.85(2H,m),3.53−3.57(2H,m),3.85−3.94(1H,m),4.26−4.32(1H,m),7.99(1H,s).
MS(ESI)[M+H]+
384
【0143】
(実施例6) 1−シクロヘプチル−5−(trans−5−ヒドロキシアダマンタン−2−イル)5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例6の化合物」)の合成:
参考例1及び2並びに実施例2と同様の手順により、以下の実施例6の化合物を合成した。
【化24】
1H−NMR(400MHz,CDCl3)
δ:1.54−1.59(7H,m),1.62−1.68(3H,m),1.75−2.24(15H,m),2.44−2.48(2H,m),2.85−2.90(2H,m),3.52−3.56(2H,m),4.06−4.15(1H,m),4.28−4.32(1H,m),7.98(1H,s).
MS(ESI)[M+H]+
398
【0144】
(実施例7) 1−フェニル−5−(trans−5−ヒドロキシアダマンタン−2−イル)5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例7の化合物」)の合成:
参考例1及び2並びに実施例2と同様の手順により、以下の実施例7の化合物を合成した。
【化25】
1H−NMR(400MHz,CDCl3)
δ:1.66−2.00(10H,m),2.12−2.20(2H,m),2.24−2.30(2H,m),2.90(2H,t,J=7.2Hz),3.56−3.64(2H,m),4.38−4.42(1H,m),7.40−7.52(5H,m),8.11(1H,s).
MS(ESI)[M+H]+
378
【0145】
(実施例8) 1−(ピリジン−2−イル)−5−(trans−5−ヒドロキシアダマンタン−2−イル)5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例8の化合物」)の合成:
参考例1及び2並びに実施例2と同様の手順により、以下の実施例8の化合物を合成した。
【化26】
1H−NMR(400MHz,CDCl3)
δ:1.38−1.42(1H,m),1.55−1.61(1H,m),1.77−1.88(6H,m),1.97−2.02(2H,m),2.19−2.24(2H,m),2.47−2.52(2H,m),3.47−3.52(2H,m),3.59−3.63(2H,m),4.32−4.34(1H,m),7.23−7.26(1H,m),7.85(1H,ddd,J=1.6,7.2,8.0Hz),7.91(1H,d,J=8.0Hz),8.16(1H,s),8.43−8.46(1H,m).
MS(ESI)[M+H]+
379
【0146】
(実施例9) 1−フェニル−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8−メチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例9の化合物」)の合成:
【化27】
メチル 4−ベンジルオキシ−2−メチルブタノエート(1.0g,4.2mmol)をメタノール(7.0mL)に溶解し、1N 水酸化ナトリウム(6.8mL,6.8mmol)を加え室温で撹拌した。16時間後、濃縮した後1N 塩酸を加え中和した後、酢酸エチルで抽出した。得られた粗生成物をアセトニトリル(9.0mL)に溶解し、カルボニルジイミダゾール(0.74g,4.5mmol)を加え室温で撹拌した(反応液A)。別途、マグネシウムクロリド(0.91g,9.5mmol)をアセトニトリル(9.0mL)に溶解し、マロン酸エチルカリウム塩(1.5g,9.1mmol)、トリエチルアミン(2.7mL,19mmol)を加え室温で撹拌した(反応液B)。14時間後、反応液Aを反応液Bに加え90℃で撹拌した。2時間後、反応液を室温まで冷却し、1N 塩酸を加えpH=5とした後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=100/0〜79/21)で精製した。得られた粗精製物にジメチルホルムアミドジメチルアセタール(2.9mL,22mmol)を加え80℃で撹拌した。4時間後、反応液を室温まで冷却し、濃縮した。得られた粗生成物をエタノール(30mL)に溶解し、フェニルヒドラジン(0.93g,8.6mmol)を加え80℃で撹拌した。5時間後、反応液を室温まで冷却し、濃縮し水を加えた後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=94/6〜76/24)で精製した。
【0147】
得られた粗精製物をエタノール(32mL)に溶解し、水酸化パラジウム/炭素(10%wet,0.45g,0.32mmol)を加え水素雰囲気下、室温で撹拌した。21時間後、反応液をセライト濾過し、濾液を濃縮した。得られた粗生成物をジクロロメタン(32mL)に溶解し、Dess−Martin試薬(1.4g,3.2mmol)を加え室温で撹拌した。2時間後、反応液をセライト濾過し、濾液を濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=100/0〜66/34)で精製した。得られた粗精製物をクロロホルム(23mL)に溶解し、trans−5−ヒドロキシ−2−アダマンチルアミン(390mg,2.3mmol)、酢酸(0.34mL,6.9mmol)、水素化トリアセトキシホウ素ナトリウム(1.3g,6.9mmol)を加え室温で撹拌した。16時間後、飽和炭酸水素ナトリウム水溶液を加え中和した後、クロロホルムで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をメタノール(4.0mL)に溶解し、1N 水酸化ナトリウム(3.5mL,3.5mmol)を加え室温で撹拌した。16時間後、濃縮した。得られた粗生成物をクロロホルム(23mL)に溶解し、ジイソプロピルエチルアミン(1.2mL,6.9mmol)、HBTU(540mg,2.3mmol)を加え室温で撹拌した。16時間後、水を加えた後、クロロホルムで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=100/0〜96/4)で精製した。この際得られた固形物を酢酸エチル/ヘキサン=1/1溶液によりスラリー洗浄し、表題化合物110mg(6%)を白色固体として得た。
1H−NMR(400MHz,CDCl3)
δ:1.03(3H,d,J=6.8Hz),1.50−1.64(3H,m),1.74−2.04(7H,m),2.20−2.25(1H,m),2.31−2.36(1H,m),2.38−2.49(1H,m),2.55−2.65(2H,m),2.92−3.00(1H,m),3.14−3.22(1H,m),3.50−3.57(1H,m),4.32−4.36(1H,m),7.39−7.52(5H,m),8.10(1H,s).
MS(ESI)[M+H]+
392
【0148】
(実施例10) 1−フェニル−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8,8−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例10の化合物」)の合成:
参考例1及び3並びに実施例2と同様の手順により、以下の実施例10の化合物を合成した。
【化28】
1H−NMR(400MHz,CDCl3)
δ:1.14(6H,s),1.40−1.47(1H,m),1.56−1.62(1H,m),1.76−1.85(1H,m),1.76−1.85(6H,m),1.96−2.05(4H,m),2.20−2.25(1H,m),3.59−3.64(2H,m),4.30−4.33(1H,m),7.36−7.40(2H,m),7.44−7.52(3H,m),8.12(1H,s).
MS(ESI)[M+H]+
406
【0149】
(実施例11) 1−フェニル−5−(trans−5−ヒドロキシアダマンタン−2−イル)−7−メチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例11の化合物」)の合成:
〔第1工程〕
4−(ベンジルオキシ)−2−メチルブトキシ−tert−ブチルジメチルシランの合成:
【化29】
メチル 4−(ベンジルオキシ)−2−メチルブタノエート(1.5g,6.8mmol)をTHF(34mL)に溶解し、水素化リチウムアルミニウム(0.14g,20mmol)を加え室温で撹拌した。2時間後、硫酸ナトリウム十水和物を加え室温で撹拌した。1時間後、反応液をセライト濾過し、濾液を濃縮した。得られた粗生成物をDMF(14mL)に溶解し、イミダゾール(0.92g,14mmol)、tert−ブチルジメチルシリルクロリド(1.5g,10mmol)を加え室温で撹拌した。2時間後、水を加えた後、ジエチルエーテルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=100/0〜81/19)で精製し、表題化合物2.0g(94%)を無色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:0.02(6H,s),0.86−0.90(12H,m),1.35−1.45(1H,m),1.70−1.80(2H,m),3.38−3.55(4H,m),4.50(2H,s),7.26−7.35(5H,m).
MS(ESI)[M+H]+
309
【0150】
〔第2工程〕
4−(tert−ブチルジメチルシリルオキシ)−3−メチルブタナールの合成:
【化30】
4−(ベンジルオキシ)−2−メチルブトキシ−tert−ブチルジメチルシラン(1.8g,5.8mmol)を酢酸エチル(58mL)に溶解し、パラジウム/炭素(10%wet,0.62g,5.8mmol)を加え水素雰囲気下、室温で撹拌した。4時間後、反応液をセライト濾過し、濾液を濃縮した。得られた粗生成物をジクロロメタン(55mL)に溶解し、Dess−Martin試薬(2.4g,5.5mmol)を加え室温で撹拌した。2時間後、反応液をセライト濾過し、濾液を濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=100/0〜81/19)で精製し、表題化合物0.75g(60%)を無色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:0.03(6H,s),0.88(9H,s),0.94(3H,d,J=6.4Hz),2.19−2.32(2H,m),2.46−2.54(1H,m),3.34−3.39(1H,m),3.56(1H,dd,J=5.2,10.0Hz),9.77(1H,t,J=2.0Hz).
MS(ESI)[M+H]+
217
【0151】
〔第3工程〕
エチル 1−フェニル−5−(2−メチル−3−(tert−ブチルジメチルシリルオキシ)プロピル)−1H−ピラゾール−4−カルボキシレートの合成:
【化31】
4−(tert−ブチルジメチルシリルオキシ)−3−メチルブタナール(750mg,3.5mmol)をtert−ブチルアルコール(10mL)に溶解し、水(20mL)、リン酸水素二ナトリウム(1.5g,10mmol)を加え室温で撹拌した。1時間後、亜塩素酸ナトリウム(1.9g,21mmol)を加え室温で撹拌した。4時間後、水、亜硫酸水素ナトリウム(6.6g,52mmol)を加えた後、酢酸エチルで抽出した。得られた粗生成物をアセトニトリル(9.0mL)に溶解し、カルボニルジイミダゾール(0.67g,4.2mmol)を加え室温で撹拌した(反応液A)。別途、マグネシウムクロリド(0.83g,8.7mmol)をアセトニトリル(9.0mL)に溶解し、マロン酸エチルカリウム塩(1.4g,8.3mmol)、トリエチルアミン(2.5mL,18mmol)を加え撹拌した。2時間後、反応液を室温まで冷却し、1N 塩酸を加えpH=5とした後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物にジメチルホルムアミドジメチルアセタール(2.7mL,20mmol)を加え90℃で撹拌した。2時間後、反応液を室温まで冷却し、濃縮した。得られた粗生成物をエタノール(28mL)に溶解し、フェニルヒドラジン(0.86g,7.9mmol)を加え80℃で撹拌した。5時間後、反応液を室温まで冷却し、濃縮し水を加えた後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=93/7〜79/21)で精製し、表題化合物0.76g(54%)を黄色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:0.00(6H,s),0.68(3H,d,J=6.8Hz),0.80(9H,s),1.37(3H,t,J=7.2Hz),1.80−1.99(1H,m),2.82−2.96(1H,m),3.05−3.11(1H,m),3.26−3.35(2H,m),4.32(2H,q,J=7.2Hz),7.38−7.51(5H,m),8.01(1H,s).
MS(ESI)[M+H]+
403
【0152】
〔第4工程〕
エチル 1−フェニル−5−(2−メチル−3−ヒドロキシプロピル)−1H−ピラゾール−4−カルボキシレートの合成:
【化32】
エチル 1−フェニル−5−(2−メチル−3−(tert−ブチルジメチルシリルオキシ)プロピル)−1H−ピラゾール−4−カルボキシレート(0.76g,1.9mmol)をTHF(10mL)に溶解し、TBAF(1.0N,3.8mL,3.8mmol)を加え室温で撹拌した。16時間後、水を加えた後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=93/7〜66/34)で精製し、表題化合物0.48g(88%)を黄色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:0.78(3H,d,J=7.2Hz),1.39(3H,t,J=7.2Hz),1.68−1.78(1H,m),2.48−2.54(1H,m),2.74−2.81(1H,m),3.33−3.42(3H,m),4.34(2H,q,J=7.2Hz),7.37−7.42(2H,m),7.47−7.55(3H,m),8.03(1H,s).
MS(ESI)[M+H]+
289
【0153】
〔第5工程〕
エチル 1−フェニル−5−(2−メチル−3−オキソプロピル)−1H−ピラゾール−4−カルボキシレートの合成:
【化33】
エチル 1−フェニル−5−(2−メチル−3−ヒドロキシプロピル)−1H−ピラゾール−4−カルボキシレート(480mg,1.7mmol)をジクロロメタン(17mL)に溶解し、Dess−Martin試薬(710mg,1.7mmol)を加え室温で撹拌した。2時間後、反応液をセライト濾過し、濾液を濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=96/4〜69/31)で精製し、表題化合物0.48g(76%)を無色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:0.93(3H,q,J=7.2Hz),1.37(3H,t,J=7.2Hz),2.70−2.79(1H,m),2.97−3.05(1H,m),3.41−3.48(1H,m)、4.32(2H,q,J=7.2Hz),7.36−7.40(2H,m),7.48−7.54(3H,m),8.06(1H,s),9.50−9.52(1H,m).
MS(ESI)[M+H]+
287
【0154】
〔第6工程〕
実施例11の化合物の合成:
【化34】
エチル 1−フェニル−5−(2−メチル−3−オキソプロピル)−1H−ピラゾール−4−カルボキシレート(360mg,1.3mmol)をクロロホルム(13mL)に溶解し、trans−5−ヒドロキシ−2−アダマンチルアミン(210mg,1.3mmol)、酢酸(0.22mL,3.8mmol)、水素化トリアセトキシホウ素ナトリウム(710mg,3.8mmol)を加え室温で撹拌した。16時間後、飽和炭酸水素ナトリウム水溶液を加え中和した後、クロロホルムで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をメタノール(2.0mL)に溶解し、1N 水酸化ナトリウム(1.9mL,1.9mmol)を加え室温で撹拌した。16時間後、濃縮した。得られた粗生成物をクロロホルム(13mL)に溶解し、ジイソプロピルエチルアミン(0.66mL,3.8mmol)、HBTU(300mg,1.3mmol)を加え室温で撹拌した。16時間後、水を加えた後、クロロホルムで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。この際得られた固形物を酢酸エチル/ヘキサン=1/1溶液によりスラリー洗浄し、表題化合物51mg(10%)を白色固体として得た。
1H−NMR(400MHz,CDCl3)
δ:1.03(3H,d,J=7.2Hz),1.58−1.65(3H,m),1.74−2.05(7H,m),2.20−2.22(1H,m),2.34−2.37(1H,m),2.37−2.48(1H,m),2.57−2.65(2H,m),3.20(1H,dd,J=9.6,15.6Hz),3.54(1H,dd,J=3.2,15.6Hz),4.32−4.35(1H,m),7.40−7.53(5H,m),8.11(1H,s).
MS(ESI)[M+H]+
392
【0155】
(実施例12) 1−フェニル−5−(trans−5−ヒドロキシアダマンタン−2−イル)−7,7−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例12の化合物」)の合成:
〔第1工程〕
4−(ベンジルオキシ)−2,2−ジメチルブトキシ−tert−ブチルジメチルシランの合成:
【化35】
メチル 4−(ベンジルオキシ)−2,2−ジメチルブタノエート(1.5g,6.4mmol)をTHF(32mL)に溶解し、水素化リチウムアルミニウム(0.13g,19mmol)を加え室温で撹拌した。2時間後、硫酸ナトリウム十水和物を加え室温で撹拌した。1時間後、反応液をセライト濾過し、濾液を濃縮した。得られた粗生成物をDMF(13mL)に溶解し、イミダゾール(0.86g,13mmol)、tert−ブチルジメチルシリルクロリド(1.4g,9.5mmol)を加え室温で撹拌した。2時間後、水を加えた後、ジエチルエーテルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=100/0〜81/19)で精製し、表題化合物1.8g(86%)を無色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:0.00(6H,s),0.86(6H,s),0.88(9H,s),1.61(2H,t,J=7.6Hz),3.24(2H,s),3.53(2H,t,J=7.6Hz),4.49(2H,s),7.26−7.34(5H,m).
MS(ESI)[M+H]+
323
【0156】
〔第2工程〕
4−(tert−ブチルジメチルシリルオキシ)−3,3−ジメチルブタナールエチルの合成:
【化36】
4−(ベンジルオキシ)−2,2−ジメチルブトキシ−tert−ブチルジメチルシラン(1.7g,5.3mmol)を酢酸エチル(53mL)に溶解し、パラジウム/炭素(10%wet,0.56g,5.3mmol)を加え水素雰囲気下、室温で撹拌した。4時間後、反応液をセライト濾過し、濾液を濃縮した。得られた粗生成物をジクロロメタン(52mL)に溶解し、Dess−Martin試薬(2.2g,5.2mmol)を加え室温で撹拌した。2時間後、反応液をセライト濾過し、濾液を濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=100/0〜81/19)で精製し、表題化合物0.96g(79%)を無色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:0.03(6H,s),0.89(9H,s),1.02(6H,s),2.28(2H,d,J=3.2Hz),3.35(2H,s),9.84(1H,d,J=3.2Hz).
MS(ESI)[M+H]+
231
【0157】
〔第3工程〕
エチル 1−フェニル−5−(2,2−ジメチル−3−(tert−ブチルジメチルシリルオキシ)プロピル)−1H−ピラゾール−4−カルボキシレートの合成:
【化37】
4−(tert−ブチルジメチルシリルオキシ)−3,3−ジメチルブタナール(960mg,4.2mmol)をtert−ブチルアルコール(13mL)に溶解し、水(26mL)、リン酸水素二ナトリウム(1.8g,13mmol)を加え室温で撹拌した。1時間後、亜塩素酸ナトリウム(2.3g,25mmol)を加え室温で撹拌した。4時間後、水、亜硫酸水素ナトリウム(7.9g,63mmol)を加えた後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をアセトニトリル(9.0mL)に溶解し、カルボニルジイミダゾール(0.71g,4.4mmol)を加え室温で撹拌した(反応液A)。別途、マグネシウムクロリド(0.87g,9.1mmol)をアセトニトリル(9.0mL)に溶解し、マロン酸エチルカリウム塩(1.5g,8.7mmol)、トリエチルアミン(2.6mL,19mmol)を加え室温で撹拌した(反応液B)。2時間後、反応液Aを反応液Bに加え90℃で撹拌した。2時間後、反応液を室温まで冷却し、1N 塩酸を加えpH=5とした後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物にジメチルホルムアミドジメチルアセタール(2.8mL,21mmol)を加え90℃で撹拌した。2時間後、反応液を室温まで冷却し、濃縮した。得られた粗生成物をエタノール(28mL)に溶解し、フェニルヒドラジン(0.87g,8.1mmol)を加え80℃で撹拌した。5時間後、反応液を室温まで冷却し、濃縮し水を加えた後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=93/7〜79/21)で精製し、表題化合物0.80g(46%)を黄色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:0.00(6H,s),0.57(6H,s),0.81(9H,s),1.38(3H,t,J=7.2Hz),3.22(1H,s),3.22(2H,s),4.31(2H,q,J=7.2Hz),7.38−7.50(5H,m),8.06(1H,s).
MS(ESI)[M+H]+
417
【0158】
〔第4工程〕
エチル 1−フェニル−5−(2,2−ジメチル−3−ヒドロキシプロピル)−1H−ピラゾール−4−カルボキシレートの合成:
【化38】
エチル 1−フェニル−5−(2,2−ジメチル−3−(tert−ブチルジメチルシリルオキシ)プロピル)−1H−ピラゾール−4−カルボキシレート(0.80g,1.9mmol)をTHF(10mL)に溶解し、TBAF(1.0N,3.8mL,3.8mmol)を加え室温で撹拌した。16時間後、水を加えた後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=93/7〜66/34)で精製し、表題化合物0.48g(83%)を黄色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:0.60(6H,s),1.39(3H,t,J=7.2Hz),3.03(2H,d,J=7.2Hz),3.12−3.19(2H,m),4.35(2H,q,J=7.2Hz),7.37−7.41(2H,m),7.44−7.54(3H,m),8.07(1H,s).
MS(ESI)[M+H]+
303
【0159】
〔第5工程〕
エチル 1−フェニル−5−(2,2−ジメチル−3−オキソプロピル)−1H−ピラゾール−4−カルボキシレートの合成:
【化39】
エチル 1−フェニル−5−(2,2−ジメチル−3−ヒドロキシプロピル)−1H−ピラゾール−4−カルボキシレート(480mg,1.6mmol)をジクロロメタン(16mL)に溶解し、Dess−Martin試薬(680mg,1.6mmol)を加え室温で撹拌した。2時間後、反応液をセライト濾過し、濾液を濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=96/4〜69/31)で精製し、表題化合物0.34g(72%)を無色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:0.83(6H,s),1.37(3H,t,J=7.2Hz),3.42(2H,s),4.31(2H,q,J=7.2Hz),7.36−7.40(2H,m),7.46−7.54(3H,m),8.05(1H,m),9.22(1H,s).
MS(ESI)[M+H]+
301
【0160】
〔第6工程〕
実施例12の化合物の合成:
【化40】
エチル 1−フェニル−5−(2,2−ジメチル−3−オキソプロピル)−1H−ピラゾール−4−カルボキシレート(340mg,1.1mmol)をクロロホルム(11mL)に溶解し、trans−5−ヒドロキシ−2−アダマンチルアミン(190mg,1.1mmol)、酢酸(0.19mL,3.4mmol)、水素化トリアセトキシホウ素ナトリウム(640mg,3.4mmol)を加え室温で撹拌した。16時間後、飽和炭酸水素ナトリウム水溶液を加え中和した後、クロロホルムで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をメタノール(2.0mL)に溶解し、1N 水酸化ナトリウム(1.7mL,1.7mmol)を加え室温で撹拌した。16時間後、濃縮した。得られた粗生成物をクロロホルム(11mL)に溶解し、ジイソプロピルエチルアミン(0.59mL,3.4mmol)、HBTU(270mg,1.1mmol)を加え室温で撹拌した。16時間後、水を加えた後、クロロホルムで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。この際得られた固形物を酢酸エチル/ヘキサン=1/1溶液によりスラリー洗浄し、表題化合物100mg(22%)を白色固体として得た。
1H−NMR(400MHz,CDCl3)
δ:1.08(6H,s),1.56−1.62(2H,m),1.76−1.83(4H,m),1.88−1.98(2H,m),2.00−2.08(2H,m),2.20−2.27(1H,m),2.43−2.49(2H,m),2 64(2H,s),3.49(2H,s),4.42−4.46(1H,m),7.40−7.53(5H,m),8.16(1H,s).
MS(ESI)[M+H]+
406
【0161】
(実施例13) 1−エチル−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8,8−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例13の化合物」)の合成:
参考例1及び3並びに実施例2と同様の手順により、以下の実施例13の化合物を合成した。
【化41】
1H−NMR(400MHz,CDCl3)
δ:1.56(6H,s),1.57−1.59(2H,m),1.76−1.82(6H,m),1.95−2.03(4H,m),2.18−2.24(1H,m),2.44−2.50(2H,m),3.63−3.68(2H,m),4.20−4.29(3H,m),8.02(1H,s).
MS(ESI)[M+H]+
358
【0162】
(実施例14) 1−シクロヘキシル−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8,8−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例14の化合物」)の合成:
参考例1及び3並びに実施例2と同様の手順により、以下の実施例14の化合物を合成した。
【化42】
1H−NMR(400MHz,CDCl3)
δ:1.32−1.37(2H,m),1.40(6H,s),1.70−2.12(19H,m),2.18−2.23(1H,m),2.44−2.50(2H,m),3.53−3.58(2H,m),4.16−4.28(2H,m),8.02(1H,s).
MS(ESI)[M+H]+
412
【0163】
(実施例15) 1−(ピリジン−2−イル)−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8,8−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例15の化合物」)の合成:
参考例1及び3並びに実施例2と同様の手順により、以下の実施例15の化合物を合成した。
【化43】
1H−NMR(400MHz,CDCl3)
δ:1.24(6H,s),1.56−1.62(2H,m),1.77−1.86(6H,m),1.98−2.04(4H,m),2.20−2.25(1H,m),2.48−2.54(2H,m),3.62−3.67(3H,m),4.30−4.33(1H,m),7.40−7.46(2H,m),7.89(1H,ddd,J=2.0,7.6,8.0Hz),8.18(1H,s),8.57−8.60(1H,m).
MS(ESI)[M+H]+
407
【0164】
(実施例16) 1−(2−メチルフェニル)−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8,8−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例16の化合物」)の合成:
参考例1及び3並びに実施例2と同様の手順により、以下の実施例16の化合物を合成した。
【化44】
1H−NMR(400MHz,CDCl3)
δ:0.98(3H,s),1.25(3H,s),1.55−1.63(2H,m),1.75−1.86(6H,m),1.96−2.03(7H,m),2.19−2.25(1H,m),2.45−2.55(2H,m),3.58−3.65(2H,m),4.30−4.35(1H,m),7.26−7.42(4H,m),8.16(1H,s).
MS(ESI)[M+H]+
420
【0165】
(実施例17) 1−(3−メチルフェニル)−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8,8−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例17の化合物」)の合成:
参考例1及び3並びに実施例2と同様の手順により、以下の実施例17の化合物を合成した。
【化45】
1H−NMR(400MHz,CDCl3)
δ:1.15(6H,s),1.55−1.63(2H,m),1.78−1.85(6H,m),1.99−2.05(4H,m),2.19−2.25(1H,m),2.40(3H,s),2.48−2.52(2H,m),3.58−3.65(2H,m),4.30−4.34(1H,m),7.15−7.20(2H,m),7.27−7.37(2H,m),8.10(1H,s).
MS(ESI)[M+H]+
420
【0166】
(実施例18) 1−(4−メチルフェニル)−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8,8−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例18の化合物」)の合成:
参考例1及び3並びに実施例2と同様の手順により、以下の実施例18の化合物を合成した。
【化46】
1H−NMR(400MHz,CDCl3)
δ:1.14(6H,s),1.40−1.47(1H,m),1.56−1.62(1H,m),1.76−1.85(1H,m),1.76−1.85(6H,m),1.96−2.05(4H,m),2.20−2.25(1H,m),3.59−3.64(2H,m),4.30−4.33(1H,m),7.36−7.40(2H,m),7.44−7.52(3H,m),8.12(1H,s).
MS(ESI)[M+H]+
420
【0167】
(実施例19) 1−(3−フルオロフェニル)−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8,8−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例19の化合物」)の合成:
参考例1及び3並びに実施例2と同様の手順により、以下の実施例19の化合物を合成した。
【化47】
1H−NMR(400MHz,CDCl3)
δ:1.17(6H,s),1.56−1.60(2H,m),1.78−1.82(6H,m),1.99−2.01(4H,m),2.20−2・24(1H,m),2.48−2.52(2H,brs),3.60−3.62(2H,m),4.31(1H,s),7.13−7.24(3H,m),7.42−7.48(1H,m),8.12(1H,s).
MS(ESI)[M+H]+
424
【0168】
実施例20 1−(4−フルオロフェニル)−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8,8−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例20の化合物」)の合成:
参考例1及び3並びに実施例2と同様の手順により、以下の実施例20の化合物を合成した。
【化48】
1H−NMR(400MHz,CDCl3)
δ:1.14(6H,s),1.55−1.60(2H,m),1.78−1.82(6H,m),1.98−2.03(4H,m),2.20−2・24(1H,m),2.47−2.51(2H,m),3.60−3.62(2H,m),4.32(1H,s),7.14−7.19(2H,m),7.35−7.39(2H,m),8.11(1H,s).
MS(ESI)[M+H]+
424
【0169】
(実施例21) 1−(3−クロロフェニル)−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8,8−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例21の化合物」)の合成:
参考例1及び3並びに実施例2と同様の手順により、以下の実施例21の化合物を合成した。
【化49】
1H−NMR(400MHz,CDCl3)
δ:1.14(6H,s),1.56−1.63(2H,m),1.78−1.82(6H,m),1.98−2.01(4H,m),2.20−2.23(1H,m),2.49(2H,brs),3.60−3.62(2H,m),4.31(1H,s),7.27−7.31(1H,m),7.40−7.51(3H,m),8.12(1H,s).
MS(ESI)[M+H]+
440
【0170】
(実施例22) 1−(4−クロロフェニル)−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8,8−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例22の化合物」)の合成:
参考例1及び3並びに実施例2と同様の手順により、以下の実施例22の化合物を合成した。
【化50】
1H−NMR(400MHz,CDCl3)
δ:1.14(6H,s),1.55−1.60(2H,m),1.78−1.82(6H,m),1.98−2.01(4H,m),2.20−2・24(1H,m),2.47−2.51(2H,m),3.60−3.62(2H,m),4.29−4.33(1H,m),7.34(2H,d,J=8.8Hz),7.47(2H,d,J=8.8Hz),8.12(1H,s).
MS(ESI)[M+H]+
440
【0171】
(実施例23) 1−(3−トリフルオロメチルフェニル)−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8,8−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例23の化合物」)の合成:
参考例1及び3並びに実施例2と同様の手順により、以下の実施例23の化合物を合成した。
【化51】
1H−NMR(400MHz,CDCl3)
δ:1.14(6H,s),1.54−2.05(12H,m),2.21−2.25(1H,m),2.50−2.53(2H,m),3.60−3.64(2H,m),4.30−4.34(1H,m),7.60−7.68(3H,m),7.78−7.80(1H,m),8.14(1H,s).
MS(ESI)[M+H]+
474
【0172】
(実施例24) 1−(4−トリフルオロメチルフェニル)−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8,8−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例24の化合物」)の合成:
参考例1及び3並びに実施例2と同様の手順により、以下の実施例24の化合物を合成した。
【化52】
1H−NMR(400MHz,CDCl3)
δ:1.15(6H,s),1.59−1.62(2H,m),1.77−1.85(6H,m),1.99−2.02(4H,m),2.20−2.25(1H,m),2.48−2.53(2H,m),3.60−3.65(2H,m),4.30−4.33(1H,m),7.53(2H,d,J=8.4Hz),7.77(2H,d,J=8.4Hz),8.14(1H,s).
MS(ESI)[M+H]+
474
【0173】
(実施例25) 1−(2−メトキシフェニルフェニル)−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8,8−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例25の化合物」)の合成:
参考例1及び3並びに実施例2と同様の手順により、以下の実施例25の化合物を合成した。
【化53】
1H−NMR(400MHz,CDCl3)
δ:1.03(3H,s),1.18(3H,s),1.52−1.60(2H,m),1.76−2.04(10H,m),2.20−2.24(1H,m),2.46−2.54(2H,m),3.59−3.63(2H,m),3.77(3H,s),4.30−4.33(1H,m),6.98−7.06(2H,m),7.35(1H,dd,J=1.7,7.6Hz),7.44−7.49(1H,m),8.16(1H,s).
MS(ESI)[M+H]+
436
【0174】
(実施例26) 1−(4−メトキシフェニル)−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8,8−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例26の化合物」)の合成:
参考例1及び3並びに実施例2と同様の手順により、以下の実施例26の化合物を合成した。
【化54】
1H−NMR(400MHz,CDCl3)
δ:1.15(6H,s),1.40−2.02(13H,m),2.18−2.22(1H,m),2.49−2.52(2H,m),3.59−3.63(2H,m),3.87(3H,s),4.19−4.22(1H,m),6.95(2H,d,J=8.8Hz),7.28(2H,d,J=8.8Hz),8.10(1H,s).
MS(ESI)[M+H]+
436
【0175】
(実施例27) 11β−HSD1阻害実験:無細胞評価系:
ヒト11β−HSD1遺伝子発現ベクターをHEK293H細胞(インビトロジェン社)に一過性導入して調製した細胞抽出液(以下、「細胞ライセート」)を用いて、本発明の複素環化合物又はその薬学的に許容される塩(以下、「実施例の化合物群」)の11β−HSD1阻害作用を評価した。
【0176】
1.試験方法
a.酵素源
I.M.A.G.Eクローン(クローンID:5193867、インビトロジェン社)より調製したヒト11β−HSD1のcDNA配列を含むプラスミドDNA(pCMV−SPORT6ベクターに挿入されている)を鋳型にして、ヒト11β−HSD1のcDNA配列を含むDNA断片をPCRで増幅し、pTA2(東洋紡社)にサブクローニングした。塩基配列を解析して変異の無いことを確認したポジティブクローンを制限酵素XhoI及びXbaIで切断し、ヒト11β−HSD1のcDNA配列を含むDNA断片をpCI−neo(プロメガ社)のXhoI、XbaI開裂部位に挿入し、ヒト11β−HSD1遺伝子発現ベクターh11β−HSD1/pCI−neoを得た。
【0177】
細胞ライセートの調製は以下の方法で行った。HEK293H細胞の細胞数をカウントし、2×106cells/5mL/6cm dishとなるように播種し、37℃、5%CO2インキュベーターで一晩培養した。細胞の培養液を交換(2mL培養液/dish)した後、h11β−HSD1/pCI−neoをFuGENE HD Transfection Reagent(ロシュ・アプライド・サイエンス社)を用いて細胞にトランスフェクションした。トランスフェクションは試薬添付の調製方法に従って行った。細胞をPBSで縣濁後、遠心して得た細胞塊を液体窒素で凍結した。細胞塊を6cm dish1枚当たり200μLの溶解バッファー(0.25mol/L sucrose、1mmol/L EDTA・2Na、10mmol/L Tris−HCl(pH7.4)、1% protease inhibitor cocktail)に縣濁し、凍結融解を3回繰り返し行った。このようにして得られたヒト11β−HSD1発現細胞ライセートは、protein assay(バイオ・ラッド社)を用いてBradford法によりタンパク質定量を行い、10%グリセロールとなるようにグリセロールを添加して、−80℃にて使用時まで凍結保存した。
【0178】
b.酵素阻害実験
被験化合物は、DMSOにて溶解、種々の濃度に希釈後、さらにアッセイバッファー(20mmol/L HEPES(pH6.0)、5mmol/L EDTA、0.1%BSA)にて希釈した(DMSO最終濃度:1%)。アッセイバッファーにて最終濃度の2.67倍に希釈したヒト11β−HSD1発現細胞ライセート(最終タンパク質濃度 100〜200μg/mL)12μLを96ウェルV底プレートに添加し、そこにアッセイバッファー又は希釈した被験化合物溶液4μLを添加して、37℃で10分間プレインキュベーションした。384ウェルブラックプレート(コーニング社)に、アッセイバッファーにて希釈した補酵素NADPH(シグマ社)、基質コルチゾン(シグマ社)の等量混合液5μLを添加し、さらに、プレインキュベーションした細胞ライセート希釈液5μLを添加し、37℃にて2時間インキュベーションした。反応により生成されたコルチゾールは、HTRFコルチゾールキット(シスバイオ社)を用いて測定した。すなわち、384ウェルブラックプレートの各ウェルにコルチゾールと競合するコルチゾール−d2、及びクリプテート標識抗コルチゾールモノクローナル抗体を各5μL添加し、遮光下、室温にて2時間インキュベーションした。その後、反応液の蛍光(励起波長340nm、蛍光波長615nm及び665nm)をマルチラベルカウンター(Arvo SX;パーキンエルマー社)を用いて測定した。コルチゾール検量線を作成し、反応液中の基質コルチゾンから変換されたコルチゾール濃度を算出した。
【0179】
測定結果より被験化合物の各濃度について阻害率(% of inhibition)を求めた。以下に阻害率の求め方を示す。
Ratio=(A665nm/B615nm)×104
A665nm:665nmにおける測定値
B615nm:615nmにおける測定値
阻害率(% of inhibition)=((被験化合物のRatio−被験化合物非添加のRatio)/(細胞ライセート非添加かつ被験化合物非添加のRatio−被験化合物非添加のRatio))×100
【0180】
2.結果
11β−HSD1活性を50%阻害する化合物濃度をIC50値として算出した。結果を表9に示す。表9に示した実施例の化合物群は、強力な11β−HSD1阻害活性を示した。一方、比較対照化合物として用いた比較例1の化合物は僅かな11β−HSD1阻害活性しか示さなかった。
【0181】
【表9】
【0182】
(実施例28) 病態モデルでのインスリン抵抗性改善作用及び体重への影響:
マウスDIO(Diet induced−obesity)モデル(以下、「DIOマウス」)を用いて、実施例の化合物群のインスリン抵抗性改善作用を評価した(Reuter、Drug Discovery Today:Disease Models、2007年、4巻、p.3−8)。
【0183】
1.試験方法
C57BL/6Nマウス(雄性;日本チャールス・リバー社)を4週齢で入荷し、1週間通常飼料(CRF−1)で飼育した後、60%脂肪を含む固形飼料(D12492;リサーチダイエット社)に切り替え、12〜13週間給餌・飼育したものをDIOマウスとして実験に用いた。入荷以降、通常飼料を継続給餌したマウスを正常動物(正常群)とした。
【0184】
実施例の化合物群及びピオグリタゾン塩酸塩は、瑪瑙乳鉢を用いて0.5%メチルセルロース(以下、「MC」)にて3mg/mLとなるよう懸濁した。懸濁調製した投与薬液をマウスに、ディスポーザブルシリンジ及び経口投与用ゾンデを用いて、14日間(投与開始日を0日目として、0日目〜13日目の間)、1日1回、10mL/kgの容量で無麻酔下強制経口投与した。対照となる、DIOマウスの溶媒投与群(溶媒群)及び正常群には0.5%MCを投与した。なお、DIOマウスの群分けは投与開始前日(−1日目)の時点で各群の体重が均等になるように実施した。
【0185】
14日目に、予め絶食用5連ケージ内で一晩絶食させたマウスの尾静脈血を採取し、その血糖値(空腹時血糖値)を簡易型血糖測定装置(メディセンス・プレシジョンエクシード;アボットジャパン社)を用いて測定した。
【0186】
空腹時血糖値測定後、マウスのインスリン抵抗性評価のため、インスリン負荷試験(ITT)を実施した。100unit/mLの市販インスリン溶液(ヒューマリンR注;イーライ・リリー社)を0.1%BSA含有生理食塩水で0.06unit/mLに希釈して調製したインスリン溶液を、マウスに腹腔内投与(0.3unit/kg)した。インスリン投与前(0分)及びインスリン投与後30、60、120分に、上記と同様の方法で血糖値を測定した。
【0187】
実施例の化合物群及びピオグリタゾン塩酸塩の、体重への影響は、投与最終日(13日目)時点での、投与開始日からの体重変化量(g)によって評価した。
【0188】
2.結果
実施例の化合物群を代表する、実施例10の化合物及び実施例22の化合物の評価結果を図1〜6、表10及び11に示す。空腹時血糖値に対する作用を図1及び2に、インスリン負荷試験における作用を図3及び4、表10及び11に、体重に対する影響を図5及び6に示す。図表中の「溶媒」はDIOマウスに0.5%MCを投与した群(溶媒群)を示し、「実施例10の化合物」はDIOマウスに実施例10の化合物を30mg/kg/day投与した群を示し、「実施例22の化合物」はDIOマウスに実施例22の化合物を30mg/kg/day投与した群を示し、「ピオグリタゾン」はDIOマウスにピオグリタゾン塩酸塩を30mg/kg/day投与した群を示し、「正常」は正常動物に0.5%MCを投与した群(正常群)を示す。図表の各値は5〜8匹/群の平均値±標準誤差で示す。
【0189】
【表10】
【0190】
【表11】
【0191】
図1及び2に示すように、空腹時血糖値は、実施例10の化合物及び実施例22の化合物を投与した群において、溶媒群より低値で、且つピオグリタゾン投与群と同等の値を示した。この結果から、実施例の化合物群は、空腹時血糖低下作用を有することが明らかになった。
【0192】
図3及び4に示すように、インスリン投与後の血糖値は、実施例10の化合物及び実施例22の化合物を投与した群において、溶媒群より低値で、且つピオグリタゾン投与群と同等の推移を示した。また、表10及び11に示すように、血糖値AUCは、実施例10及び22の化合物投与群とピオグリタゾン投与群で同等の値であった。これらの結果から、実施例の化合物群は、インスリン抵抗性改善作用を有することが明らかになった。
【0193】
図5及び6に、投与開始後13日目時点での投与開始日(0日目)からの体重変化量を示す。実施例10の化合物及び実施例22の化合物を投与した群の体重変化量は、溶媒群より低値であり、溶媒群より高値を示したピオグリタゾン投与群とは対照的であった。この結果から、実施例の化合物群には、体重増加の副作用がないことが明らかになった。
【産業上の利用可能性】
【0194】
本発明の複素環化合物又はその薬学的に許容される塩は、顕著な11β−HSD1阻害活性を示すとともに副作用が軽減されているため、医薬の分野において、糖尿病の治療薬及び予防薬並びにインスリン抵抗性に対する改善薬として用いることができる。
【技術分野】
【0001】
本発明は、複素環化合物及びその医薬用途に関する。
【背景技術】
【0002】
糖尿病とは、インスリンの作用の不足による慢性の高血糖状態を主徴とする代謝疾患群であり、1型と2型に大別される。1型糖尿病は、インスリンを合成及び分泌する膵ランゲルハンス島β細胞の破壊や消失によるインスリン分泌量の低下を主要な原因として発症するのに対し、2型糖尿病は、インスリン分泌量の低下やインスリン抵抗性をきたす素因を含む複数の遺伝因子に、過食(特に高脂肪食)、運動不足、肥満、ストレス等の環境因子及び加齢が加わって発症する。
【0003】
また、糖尿病による長期間の高血糖状態の持続は、糖尿病三大合併症(糖尿病性網膜症、糖尿病性腎症及び糖尿病性神経障害)の他にも失明や心血管障害等を引き起こす危険性が高いことが知られている。このため、糖尿病の発症時に、血糖をコントロールして正常に近い状態へと導く治療を行い、耐糖能異常、食後高血糖及び空腹時高血糖を改善していくことが、患者のQOL(quality of life)の観点からも重要であると考えられている(非特許文献1)。
【0004】
現在使用されている糖尿病治療薬としては、インスリン製剤、スルホニルウレア剤(例えば、グリベンクラミド)、ビグアナイド剤(例えば、メトホルミン)、α−グルコシダーゼ阻害剤(例えば、ボグリボース)、チアゾリジンジオン系薬剤(例えば、ピオグリタゾン)等が挙げられる(非特許文献1〜5)。
【0005】
インスリン製剤は、強力な血糖低下作用を有するが、しばしば重篤な低血糖又は体重増加を引き起こすことが知られ、さらに注射による投与が必要であるため、患者への負担が大きいというデメリットがある。スルホニルウレア剤は、血糖低下作用を有するが、同様に重篤な低血糖や体重増加を引き起こし、長期間投与の場合には、2次無効と呼ばれる血糖値コントロールの悪化が起こることが知られている。ビクアナイド剤は、血糖低下作用及びインスリン抵抗性改善作用等を有するが、下痢又は膨満等の胃腸管障害を引き起こすことがあり、さらには重篤な乳酸アシドーシスを引き起こすリスクもあるため、腎障害又は肝機能障害のある患者には禁忌とされている。α−グルコシダーゼ阻害剤は、食後の血糖上昇(食後過血糖)を抑制する作用を有するが、腹痛、下痢又は膨満等の胃腸障害を引き起こすことが知られている。チアゾリジンジオン系薬剤は、インスリン抵抗性の改善を介した血糖降下作用を有するが、しばしば体重増加、浮腫、肝障害を引き起こし、心不全患者や心不全の既往者には不適とされ、治療期間中には定期的な肝機能検査が必要であるとされている。
【0006】
一方、インスリン抵抗性とは、血中のインスリン濃度に見合ったインスリン作用が得られない状態をいい、インスリン抵抗性の症状は、糖尿病を患っていない境界型のヒトにも認められることが知られている。また、インスリン抵抗性は、内臓脂肪型肥満とともに、メタボリックシンドロームの基盤病態であり、動脈硬化、肥満、高血圧及び脂質代謝異常の病態形成に密接に関わることが明らかにされている。ここでメタボリックシンドロームとは、肥満を基盤に、主として脂質代謝異常(高中性脂肪及び低HDLコレステロール)、高血圧及び耐糖能異常の持続が虚血性の心血管疾患の発症リスクを高めることを想定した概念である。
【0007】
このため、インスリン抵抗性の改善は、糖代謝異常の改善のみならず、メタボリックシンドローム、心血管障害さらには脳血管障害の治療にも重要であると考えられている(非特許文献1)。
【0008】
また、11β−ヒドロキシステロイドデヒドロゲナーゼ1(11β−hydroxysteroid dehydrogenase type 1;以下、11β−HSD1)は、不活性型グルココルチコイド(ヒトではコルチゾン、げっ歯類では11−デヒドロコルチコステロン)を活性型グルココルチコイド(ヒトではコルチゾール、げっ歯類ではコルチコステロン)に細胞内で変換する酵素であり、肝臓、脂肪組織及び骨格筋において高発現している(非特許文献6)。活性型グルココルチコイドは、肝臓での糖新生亢進及び解糖系抑制又は脂肪組織及び筋肉での糖の取り込み抑制により、高血糖等の代謝異常を惹起することが報告されている(非特許文献7)。11β−HSD1ノックアウトマウスは、ストレス負荷や高脂肪食負荷に対する肝臓の糖新生酵素(PEPCKやG6Pase等)が誘導されず、糖尿病の発症に対して明らかな抵抗性を示し、高脂肪食負荷による内臓脂肪の蓄積が選択的に抑制されるために、メタボリックシンドロームの発症や進展にも抵抗性を示すことが報告されている(非特許文献8)。
【0009】
なお、特許文献1〜5には、11β−HSD1阻害作用を有する化合物が開示されている。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】国際公開第08/099145号
【特許文献2】国際公開第09/060232号
【特許文献3】国際公開第09/098501号
【特許文献4】国際公開第10/087770号
【特許文献5】国際公開第09/010416号
【非特許文献】
【0011】
【非特許文献1】日本糖尿病学会、「糖尿病治療ガイド2008−2009」、2008年
【非特許文献2】Moller、Nature、2001年、第414巻、p.821−827
【非特許文献3】Skyler、Journal of Medicinal Chemistry、2004年、第47巻、p.4113−4117
【非特許文献4】Rossら、Chemical Review、2004年、第104巻、p.1255−1282
【非特許文献5】Stumvollら、Lancet、2005年、第365巻、p.1333−1346
【非特許文献6】Secklら、Endocrinology、2001年、第142巻、p.1371
【非特許文献7】土田ら、日本臨床、2002年、第60巻、増刊号7、p.280
【非特許文献8】Kotelevtsevら、Proceedings of the National Academy of Sciences of the United States of America、1997年、第94巻、p.14924−14929
【発明の概要】
【発明が解決しようとする課題】
【0012】
しかしながら、上記のように、糖尿病治療薬の既存薬は、そのいずれもが薬効は示すが、種々の欠点や副作用を有しているのが現状であるため、慢性疾患や複合疾患を持つ患者に対する投与や長期的な投与が可能な使い勝手の良い糖尿病治療薬の創出が望まれている。
【0013】
また、11β−HSD1阻害作用を有する化合物は、不活性型グルココルチコイドの変換を阻害し、活性型グルココルチコイドの産生を抑制することが可能であるため、既存の医薬品に比べて新たなメカニズムで高血糖や肝糖新生を抑制し、インスリン抵抗性及びメタボリックシンドロームの改善にも薬効を示し得ることが期待されるが、未だに、11β−HSD1阻害作用を有する糖尿病治療薬やインスリン抵抗性の改善薬は医薬品として承認されていない。
【0014】
そこで本発明は、11β−HSD1阻害活性を有する化合物を提供するとともに、副作用が軽減された新たなメカニズムの糖尿病の治療薬及びインスリン抵抗性の改善薬を提供することを目的とする。
【課題を解決するための手段】
【0015】
本発明者らは上記の目的を達成するため鋭意検討した結果、新規な複素環化合物又はその薬学的に許容される塩が、強力な11β−HSD1阻害活性を有し、糖尿病の治療作用及びインスリン抵抗性の改善作用を有することを見出し、本発明を完成させるに至った。
【0016】
すなわち、本発明は、以下の一般式(I)で表される複素環化合物又はその薬学的に許容される塩を提供する。
【化1】
[式中、R1は、炭素数1〜6のアルキル、炭素数3〜10のシクロアルキル、環構成炭素数6〜10のアリール又は環構成原子数5〜10のヘテロアリール(ここで、該アリール及び該ヘテロアリールは、それぞれ独立して、1以上のハロゲンによって置換されていてもよい炭素数1〜6のアルキル、又は、炭素数1〜6のアルキルオキシ若しくはハロゲン、によって置換されていてもよい)を示し、R2〜R5は、それぞれ独立して、炭素数1〜6のアルキル又は水素原子を示し、R6は、ヒドロキシ又は水素原子を示す。]
【0017】
R1は、炭素数1〜6のアルキル、環構成炭素数6〜10のアリール又は環構成原子数5〜10のヘテロアリール(ここで、該アリール及び該ヘテロアリールは、それぞれ独立して、1以上のハロゲンによって置換されていてもよい炭素数1〜6のアルキル、炭素数1〜6のアルキルオキシ又はハロゲンによって置換されていてもよい)を示すことが好ましく、フェニル又は3−クロロフェニルを示すことがより好ましい。
【0018】
この場合、R1を限定することで、より強い11β−HSD1阻害活性が得られる。
【0019】
また、R2〜R5は、それぞれ独立してメチル基又は水素原子を示すことが好ましく、R2及びR3はメチル基を、R4及びR5は水素原子を示すことがより好ましい。
【0020】
この場合、R2〜R5を限定することで、さらに強い11β−HSD1阻害活性が得られる。
【0021】
さらにR6は、ヒドロキシを示すことが好ましい。
【0022】
この場合、R6を限定することで、より高い代謝安定性が得られる。
【0023】
また本発明は、上記の複素環化合物又はその薬学的に許容される塩を有効成分として含有する、11β−HSD1阻害剤、糖尿病の治療薬又は予防薬を提供する。上記の糖尿病は、2型糖尿病であることが好ましい。
【0024】
さらに本発明は、上記の複素環化合物又はその薬学的に許容される塩を有効成分として含有する、インスリン抵抗性の改善薬を提供する。
【発明の効果】
【0025】
本発明の複素環化合物又はその薬学的に許容される塩は、in vivoにおいて顕著な11β−HSD1阻害活性を示し、糖尿病疾患、インスリン抵抗性を伴う疾患に対し顕著な治療効果を有している。また本発明の糖尿病の治療薬及びインスリン抵抗性の改善薬は、既存の糖尿病治療薬等で認められる体重増加等の副作用が軽減されており、慢性疾患や複合疾患を持つ患者に対する投与や長期的な投与が可能となる。
【図面の簡単な説明】
【0026】
【図1】実施例10の化合物の空腹時血糖低下作用を示す図である。
【図2】実施例22の化合物の空腹時血糖低下作用を示す図である。
【図3】実施例10の化合物のインスリン抵抗性改善作用を示す図である。
【図4】実施例22の化合物のインスリン抵抗性改善作用を示す図である。
【図5】実施例10の化合物の体重に対する影響を示す図である。
【図6】実施例22の化合物の体重に対する影響を示す図である。
【発明を実施するための形態】
【0027】
本明細書で使用する次の用語は、特に断りがない限り、下記の定義のとおりである。
【0028】
「アルキル」とは、直鎖又は分岐の飽和炭化水素基を意味し、例えば、メチル、エチル、1−プロピル、2−プロピル、1−ブチル、2−ブチル、2−メチル−2−プロピル、1,1−ジメチルエチル、1−ペンチル、2−ペンチル、3−ペンチル又は1−ヘキシルが挙げられる。
【0029】
「シクロアルキル」とは、部分的に不飽和結合を有していてもよく、あるいは架橋環やスピロ環を形成していてもよい、飽和脂環式炭化水素基を意味し、例えば、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロオクチル、ビシクロ[2,2,1]ヘキサン−1−イル、ビシクロ[2,2,1]ヘプタン−2−イル、ビシクロ[3,1,1]ヘプタン−3−イル、ビシクロ[2,2,2]オクタン−1−イル、アダマンチル(アダマンタン−1−イル又はアダマンタン−2−イル)又はノルボルニルが挙げられる。
【0030】
「アリール」とは、芳香族基を意味し、例えば、フェニル又はナフチルが挙げられる。
【0031】
「ヘテロアリール」とは、硫黄原子、窒素原子及び酸素原子からなる群から任意に選択されるヘテロ原子1〜4個を含む(窒素原子は酸化されていてもよい)複素環式芳香族基を意味し、例えば、チエニル、ピロリル、フリル、チアゾリル、イミダゾリル、オキサゾリル、ピラゾリル、イソキサゾリル、1H−1,2,3−トリアゾリル、1H−1,2,4−トリアゾリル、2H−1,2,3−トリアゾリル、1,3,4−オキサジアゾール−2−イル、1H−テトラゾリル、2H−テトラゾリル、ピリジル(ピリジン−2−イル、ピリジン−3−イル又はピリジン−4−イル)、ピリジン−1−オキシド−2−イル、ピリジン−1−オキシド−3−イル、ピリジン−1−オキシド−4−イル、ピリミジニル、ピラジニル、インドリル、ベンゾイミダゾリル、ベンゾチアゾリル、キノリル又はイソキノリルが挙げられる。
【0032】
「アルキルオキシ」とは、アルキルが酸素原子に結合した官能基を意味し、例えば、メトキシ、エトキシ、1−プロポキシ、2−プロポキシ又は1−ブトキシが挙げられる。
【0033】
「ハロゲン」とは、フッ素、塩素、臭素又はヨウ素を意味する。
【0034】
「糖尿病」とは、世界保健機構(WHO)、日本糖尿病学会、米国糖尿病協会又は欧州糖尿病協会等の診断基準に該当する病態を意味し、例えば、1型糖尿病、2型糖尿病又は妊娠糖尿病が挙げられる。
【0035】
「インスリン抵抗性」とは、インスリン抵抗性指数(空腹時血糖値(mg/dL)×空腹時インスリン(μU/mL)÷405)、グルコースクランプ法等を診断基準とする、血中のインスリン濃度に見合ったインスリン作用が得られない病態を意味し、シンドロームXもこれに包含される。
【0036】
「インスリン抵抗性の改善薬」とは、インスリン感受性を増加させる効果を示す薬剤のことを意味し、糖尿病における高インスリン血症の改善作用や空腹時血糖の改善作用等の効果も含まれる。
【0037】
「治療薬又は予防薬」には、治療又は予防の一方に用いられるもののみならず、治療及び予防の双方を目的として同時に用いられるものも包含される。
【0038】
本発明の複素環化合物は、以下の一般式(I)で表されることを特徴としている。
【化2】
[式中、R1は、炭素数1〜6のアルキル、炭素数3〜10のシクロアルキル、環構成炭素数6〜10のアリール又は環構成原子数5〜10のヘテロアリール(ここで、該アリール及び該ヘテロアリールは、それぞれ独立して、1以上のハロゲンによって置換されていてもよい炭素数1〜6のアルキル、又は、炭素数1〜6のアルキルオキシ若しくはハロゲン、によって置換されていてもよい)を示し、R2〜R5は、それぞれ独立して、炭素数1〜6のアルキル又は水素原子を示し、R6は、ヒドロキシ又は水素原子を示す。]
【0039】
R1は、炭素数1〜6のアルキル、環構成炭素数6〜10のアリール又は環構成原子数5〜10のヘテロアリール(ここで、該アリール及び該ヘテロアリールは、それぞれ独立して、1以上のハロゲンによって置換されていてもよい炭素数1〜6のアルキル、炭素数1〜6のアルキルオキシ又はハロゲンによって置換されていてもよい)を示すことが好ましく、フェニル又は3−クロロフェニルを示すことがより好ましい。
【0040】
また、R2〜R5は、それぞれ独立してメチル基又は水素原子を示すことが好ましく、R2及びR3はメチル基を、R4及びR5は水素原子を示すことがより好ましい。
【0041】
さらにR6は、ヒドロキシを示すことが好ましい。
【0042】
なお、一般式(I)で表される複素環化合物は、場合によってはエナンチオマー、ラセミ体又はジアステレオマー等の立体異性体が存在し得るが、本発明の複素環化合物には、全ての立体異性体及びそれぞれの異性体の混合物が包含される。
【0043】
一般式(I)で表される複素環化合物における好ましい置換基の組合せの例を、表1〜8に示す。
【化3】
【0044】
【表1】
【0045】
【表2】
【0046】
【表3】
【0047】
【表4】
【0048】
【表5】
【0049】
【表6】
【0050】
【表7】
【0051】
【表8】
【0052】
一般式(I)で表される複素環化合物の「薬学的に許容される塩」としては、該複素環化合物が塩基性置換基を有する場合には、例えば、塩酸塩、硫酸塩、硝酸塩、臭化水素酸塩、ヨウ化水素酸塩若しくはリン酸塩等の無機酸塩、酢酸塩、トリフルオロ酢酸塩、乳酸塩、クエン酸塩、シュウ酸塩、グルタル酸塩、リンゴ酸塩、酒石酸塩、フマル酸塩、マンデル酸塩、マレイン酸塩、安息香酸塩若しくはフタル酸塩等の有機カルボン酸塩、メタンスルホン酸塩、エタンスルホン酸塩、ベンゼンスルホン酸塩、p−トルエンスルホン酸塩若しくはカンファースルホン酸塩等の有機スルホン酸塩又はアスパラギン酸塩若しくはグルタミン酸塩等の酸性アミノ酸塩が挙げられるが、塩酸塩、臭化水素酸塩、リン酸塩、トリフルオロ酢酸塩、クエン酸、フマル酸、酒石酸塩又はメタンスルホン酸塩が好ましい。
【0053】
また、該複素環化合物が酸性置換基を有する場合には、例えば、ナトリウム塩若しくはカリウム塩等のアルカリ金属塩、カルシウム塩若しくはマグネシウム塩等のアルカリ土類金属塩、アルミニウム塩、アンモニウム塩、トリメチルアミン塩、トリエチルアミン塩、ピリジン塩、ピコリン塩、エタノールアミン塩、ジエタノールアミン塩、トリエタノールアミン塩、ジシクロヘキシルアミン塩若しくはN,N−ジベンジルエチレンジアミン塩等の有機塩基塩又はアルギニン塩、リジン塩若しくはオルニチン塩等の塩基性アミノ酸塩が挙げられるが、ナトリウム塩、カリウム塩又はエタノールアミン塩が好ましい。
【0054】
一般式(I)で表される複素環化合物又はその薬学的に許容される塩は、文献既知の合成法を適宜組み合わせることによって製造することができる。スキーム1に代表的な製造法を例示する。各反応式中の記号の意味は、特に断りのない限り、上記の定義のとおりである。
【0055】
下記の製造法により得られた一般式(I)で表される複素環化合物は、公知の手段によって単離精製することができる。単離精製のための公知の手段としては、例えば、溶媒抽出、再結晶又はクロマトグラフィーが挙げられる。
【0056】
一般式(I)で表される複素環化合物が、光学異性体又は立体異性体を含有する場合には、公知の方法により、それぞれの異性体を単一化合物として得ることができる。
【化4】
【0057】
スキーム1中、Xは、炭素数1〜6のアルキルを示す。
【0058】
〔第1工程〕
一般式(III)で表されるカルボン酸は、一般式(II)で表されるエステルの酸性又は塩基性条件での加水分解反応によって合成することができる。
【0059】
上記の加水分解反応は、Greenの方法(Protective Groups in Organic Synthesis、第5版、John Wieley & Sons.、1999年)又は実験化学講座(第4版、22巻、p.271−309)等に記載の方法に準じて実施可能である。
【0060】
上記の加水分解反応に用いる酸又は塩基の量は、一般式(II)で表されるエステルに対して0.5〜100当量が好ましく、1〜30当量がより好ましい。
【0061】
上記の加水分解反応に用いる反応溶媒は、通常反応を阻害しない溶媒から適宜選択されるが、例えば、テトラヒドロフラン(以下、「THF」)、1,4−ジオキサン、エチレングリコールジメチルエーテル若しくはジメトキシエタン等のエーテル系溶媒、ジクロロメタン、クロロホルム若しくは1,2−ジクロロエタン等のハロゲン系溶媒、ベンゼン若しくはトルエン等の芳香族炭化水素系溶媒、N,N−ジメチルホルムアミド(以下、「DMF」)若しくはジメチルスルホキシド(以下、「DMSO」)等の非プロトン性極性溶媒、アセトン若しくはメチルエチルケトン等のケトン系溶媒、アセトニトリル若しくはプロピオニトリル等のニトリル系溶媒、メタノール、エタノール若しくは2−プロパノール等のプロトン性アルコール溶媒又は水あるいはそれらの混合溶媒が挙げられ、中でもエーテル系溶媒、非プロトン性極性溶媒、プロトン性アルコール溶媒若しくは水又はそれらの混合溶媒が好ましい。
【0062】
上記の加水分解反応開始時点における、一般式(II)で表されるエステルの反応液中の濃度は、通常0.01mM〜1Mの範囲であることが好ましい。
【0063】
上記の加水分解反応温度は、−78〜200℃が好ましく、−20〜100℃がより好ましい。
【0064】
上記の加水分解反応時間は、反応温度等の条件に応じて適宜選択されるが、通常1〜30時間程度で満足すべき結果が得られる。
【0065】
〔第2工程〕
一般式(I)で表される複素環化合物は、一般式(III)で表されるカルボン酸の分子内縮合反応によって合成することができる。
【0066】
上記の縮合反応は、一般的なペプチド合成法(「実験化学講座(第16巻)有機化合物の合成IV」、第5版、p.258−270等)に準じて実施可能である。
【0067】
上記の縮合反応に用いる試薬(以下、「縮合剤」)としては、例えば、シクロヘキシルカルボジイミド、N−エチル−N’−3−ジメチルアミノプロピルカルボジイミド塩酸塩、N,N−カルボジイミダゾール、O−(7−アザベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウム ヘキサフルオロホスファート又はオルト−(ベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウムヘキサフルオロホスファート(以下、「HBTU」)が挙げられるが、HBTUが好ましい。
【0068】
上記の縮合剤の量は、一般式(III)で表されるカルボン酸に対して1〜10当量が好ましく、1〜3当量がより好ましい。
【0069】
上記の縮合反応に適する反応溶媒は、上記の縮合剤の種類により異なるが、例えば、THF、1,4−ジオキサン、エチレングリコールジメチルエーテル若しくはジメトキシエタン等のエーテル系溶媒、ジクロロメタン、クロロホルム若しくは1,2−ジクロロエタン等のハロゲン系溶媒、DMF若しくはDMSO等の非プロトン性極性溶媒又はアセトニトリル若しくはプロピオニトリル等のニトリル系溶媒が挙げられ、中でもハロゲン系溶媒若しくは非プロトン性極性溶媒が好ましく、ハロゲン系溶媒がより好ましい。
【0070】
上記の縮合反応開始時点における、一般式(III)で表されるカルボン酸の反応液中の濃度は、通常0.01mM〜1Mの範囲であることが好ましい。
【0071】
上記の縮合反応温度は、−78〜200℃が好ましく、0〜100℃がより好ましい。
【0072】
上記の縮合反応時間は、反応温度等の条件に応じて適宜選択されるが、通常6〜48時間程度で満足すべき結果が得られる。
【0073】
スキーム1に例示された製造法の出発原料である、一般式(II)で表されるエステルは、例えば、スキーム2に示す方法で合成することができる。各反応式中の記号の意味は、特に断りのない限り、上記の定義のとおりである。
【0074】
【化5】
【0075】
〔第1工程〕
一般式(VI)で表されるアルデヒドは、一般式(V)で表されるアルコールの酸化反応によって合成することができる。
【0076】
上記の酸化反応は、一般的な酸化反応(「実験化学講座(第15巻)有機化合物の合成III」、第5版、p.9−44等)に準じて実施可能である。
【0077】
上記の酸化反応に用いる試薬(以下、「酸化剤」)としては、例えば、Dess−Martin試薬又はDMSO−塩化オキサリルが挙げられるが、Dess−Martin試薬が好ましい。
【0078】
上記の酸化剤の量は、一般式(V)で表されるアルコールに対して1〜10当量が好ましく、1〜3当量がより好ましい。
【0079】
上記の酸化反応に適する反応溶媒は、上記の酸化剤の種類により異なるが、例えば、THF、1,4−ジオキサン、エチレングリコールジメチルエーテル若しくはジメトキシエタン等のエーテル系溶媒、ジクロロメタン、クロロホルム若しくは1,2−ジクロロエタン等のハロゲン系溶媒、DMF若しくはDMSO等の非プロトン性極性溶媒又はアセトニトリル若しくはプロピオニトリル等のニトリル系溶媒が挙げられ、中でもハロゲン系溶媒が好ましい。
【0080】
上記の酸化反応開始時点における、一般式(V)で表されるアルコールの反応液中の濃度は、通常0.01mM〜1Mの範囲であることが好ましい。
【0081】
上記の酸化反応温度は、−78〜200℃が好ましく、0〜100℃がより好ましい。
【0082】
上記の酸化反応時間は、反応温度等の条件に応じて適宜選択されるが、通常1〜48時間程度で満足すべき結果が得られる。
【0083】
〔第2工程〕
一般式(II)で表されるアミンは、一般式(VI)で表されるアルデヒドと一般式(IV)で表されるアミンとの還元的アミノ化反応によって合成することができる。
【0084】
上記の還元的アミノ化反応は、一般的な還元的アミノ化反応(「実験化学講座(第14巻)有機化合物の合成II」、第5版、p.371等)に準じて実施可能である。
【0085】
上記の還元的アミノ化反応に用いる試薬(以下、「還元剤」)としては、例えば、水素化リチウムアルミニウム、水素化ホウ素ナトリウム、水素化シアノホウ素ナトリウム、水素化トリアセトキシホウ素ナトリウム又はパラジウム/炭素が挙げられるが、水素化トリアセトキシホウ素ナトリウムが好ましい。
【0086】
上記の還元剤の量は、一般式(VI)で表されるアルデヒドに対して1〜10当量が好ましく、1〜3当量がより好ましい。
【0087】
上記の還元的アミノ化反応に適する反応溶媒は、上記の還元剤の種類により異なるが、例えば、THF、1,4−ジオキサン、エチレングリコールジメチルエーテル若しくはジメトキシエタン等のエーテル系溶媒、ジクロロメタン、クロロホルム若しくは1,2−ジクロロエタン等のハロゲン系溶媒、メタノール若しくはエタノール等のプロトン性極性溶媒、DMF若しくはDMSO等の非プロトン性極性溶媒又はアセトニトリル若しくはプロピオニトリル等のニトリル系溶媒が挙げられ、中でもプロトン性極性溶媒が好ましい。
【0088】
上記の還元的アミノ化反応開始時点における、一般式(VI)で表されるアルデヒドの反応液中の濃度は、通常0.01mM〜1Mの範囲であることが好ましい。
【0089】
上記の還元的アミノ化反応温度は、−78〜200℃が好ましく、0〜100℃がより好ましい。
【0090】
上記の還元的アミノ化反応時間は、反応温度等の条件に応じて適宜選択されるが、通常6〜60時間程度で満足すべき結果が得られる。
【0091】
スキーム2に例示された製造法の出発原料である、一般式(V)で表されるアルコールは、例えば、スキーム3に示す方法で合成することができる。各反応式中の記号の意味は、特に断りのない限り、上記の定義のとおりである。
【化6】
【0092】
スキーム3中、Yは、置換されていてもよいベンジル又はトリアルキルシリルを示す。
【0093】
一般式(V)で表されるアルコールは、一般式(VII)で表されるベンジルエーテル又はトリアルキルシリルエーテルの脱保護反応によって合成することができる。
【0094】
上記の脱保護反応は、一般的なアルコール保護基の脱保護法(「実験化学講座(第14巻)有機化合物の合成と反応IV」、第3版、p.2497−2505等)準じて実施可能である。
【0095】
上記の脱保護反応に用いる試薬(以下、「脱保護剤」)としては、ベンジルエーテルに対しては、例えば、水素雰囲気下におけるパラジウム/炭素、水酸化パラジウム/炭素又は白金/炭素が挙げられるが、水素雰囲気下におけるパラジウム/炭素が好ましい。
【0096】
また、トリアルキルシリルエーテルに対しては、例えば、塩酸又はテトラブチルアンモニウムフルオリド(以下、「TBAF」)が好ましく、TBAFがより好ましい。
【0097】
上記の脱保護剤の量は、一般式(VII)で表されるベンジルエーテル又はトリアルキルシリルエーテルに対して0.1〜10当量が好ましく、0.1〜3当量がより好ましい。
【0098】
上記の脱保護反応に適する反応溶媒は、上記の脱保護剤の種類により異なるが、例えば、THF、1,4−ジオキサン、エチレングリコールジメチルエーテル若しくはジメトキシエタン等のエーテル系溶媒、ジクロロメタン、クロロホルム若しくは1,2−ジクロロエタン等のハロゲン系溶媒、DMF若しくはDMSO等の非プロトン性極性溶媒、メタノール等のプロトン性極性溶媒又はアセトニトリル若しくはプロピオニトリル等のニトリル系溶媒が挙げられ、中でもエーテル系溶媒又はプロトン性極性溶媒が好ましい。
【0099】
上記の脱保護反応開始時点における、一般式(VII)で表されるベンジルエーテル又はトリアルキルシリルエーテルの反応液中の濃度は、通常0.01mM〜1Mの範囲であることが好ましい。
【0100】
上記の脱保護反応温度は、−78〜200℃が好ましく、0〜100℃がより好ましい。
【0101】
上記の脱保護反応時間は、反応温度等の条件に応じて適宜選択されるが、通常6〜48時間程度で満足すべき結果が得られる。
【0102】
スキーム3に例示された製造法の出発原料である、一般式(VII)で表されるベンジルエーテル又はトリアルキルシリルエーテルは、例えば、スキーム4に示す方法で合成することができる。各反応式中の記号の意味は、特に断りのない限り、上記の定義のとおりである。
【0103】
スキーム4における、一般式(X)で表されるヒドラジンとしては、塩酸塩等の塩を用いても構わない。
【化7】
【0104】
〔第1工程〕
一般式(VIII)で表される化合物は、一般式(IX)で表される化合物とジメチルホルムアミドジメチルアセタールとのアミノメチル化反応によって合成することができる。
【0105】
上記のアミノメチル化反応に用いるジメチルホルムアミドジメチルアセタールの量は、一般式(IX)で表されるアセト酢酸エステルに対して1〜20当量が好ましく、3〜10当量がより好ましい。
【0106】
上記のアミノメチル化反応は、反応溶媒を用いず、無溶媒で行うことが好ましい。一方で、上記のアミノメチル化反応に反応溶媒を用いる場合にあっては、該反応溶媒は通常反応を阻害しない溶媒から適宜選択される。そのような溶媒としては、例えば、THF、1,4−ジオキサン若しくはエチレングリコールジメチルエーテル等のエーテル系溶媒、ジクロロメタン、クロロホルム若しくは1,2−ジクロロエタン等のハロゲン系溶媒、ベンゼン若しくはトルエン等の芳香族炭化水素系溶媒、DMF若しくはDMSO等の非プロトン性極性溶媒、メタノール、エタノール若しくは2−プロパノール等のプロトン性アルコール溶媒若しくは水又は混合溶媒が挙げられるが、ハロゲン系溶媒、芳香族炭化水素系溶媒、非プロトン性極性溶媒又はプロトン性アルコール溶媒が好ましい。
【0107】
上記のアミノメチル化反応開始時点における、一般式(IX)で表される化合物の反応液中の濃度は、通常0.1mM〜10Mの範囲であることが好ましい。
【0108】
上記のアミノメチル化反応温度は、0〜200℃が好ましく、20〜120℃がより好ましい。
【0109】
上記のアミノメチル化反応時間は、反応温度等の条件に応じて適宜選択されるが、通常0.5〜12時間程度で満足すべき結果が得られる。
【0110】
〔第2工程〕
一般式(VII)で表されるベンジルエーテル又はトリアルキルシリルエーテルは、一般式(VIII)で表される化合物に、一般式(X)で表されるヒドラジン化合物を作用させる環化反応によって合成することができる。
【0111】
上記の環化反応に用いるヒドラジン化合物の量は、一般式(VIII)で表されるアセト酢酸エステルに対して1〜10当量が好ましく、1〜5当量がより好ましい。
【0112】
上記の環化反応に用いる反応溶媒は、通常反応を阻害しない溶媒から適宜選択されるが、例えば、DMF若しくはDMSO等の非プロトン性極性溶媒、メタノール、エタノール若しくは2−プロパノール等のプロトン性アルコール溶媒、アセトニトリル若しくはプロピオニトリル等のニトリル系溶媒、ギ酸、酢酸若しくはプロピオン酸等の有機カルボン酸又は水あるいはそれらの混合溶媒が挙げられ、中でもプロトン性アルコール溶媒又はニトリル系溶媒が好ましく、プロトン性アルコール溶媒がより好ましい。
【0113】
上記の環化反応開始時点における、一般式(VIII)で表される化合物の濃度は、通常0.01mM〜10Mの範囲であることが好ましい。
【0114】
上記の環化反応温度は、0〜200℃が好ましく、20〜120℃がより好ましい。
【0115】
上記の環化反応時間は、反応温度等の条件に応じて適宜選択されるが、通常1〜12時間程度で満足すべき結果が得られる。
【0116】
一般式(IV)
【化8】
で表されるアミンは、例えば、文献記載の方法(Tetrahedron Letters、2006年、第47巻、第46号、p.8063−8067;Journal of Medicinal Chemistry、2007年、第50巻、第1号、p.149−164等)により合成することができる。
【0117】
また、本発明の11β−HSD1阻害剤、糖尿病の治療薬及び予防薬並びにインスリン抵抗性の改善薬は、上記の一般式(I)で表される複素環化合物又はその薬学的に許容される塩を有効成分として含有することを特徴としている。
【0118】
上記の11β−HSD1阻害剤、糖尿病の治療薬及び予防薬並びにインスリン抵抗性の改善薬の薬効は、血中グリコヘモグロビン値(HbA1c)、空腹時血糖値、経口糖負荷試験(OGTT;oral glucose tolerance test)における血糖の2時間値、血中c−ペプチド値、血中グリコアルブミン値、1,5−AG(1,5−アンヒドログルシトール)、血中空腹時インスリン値、HOMA−IR(homeostasis model assessment of insulin resistance)、グルコースクランプ(euglycemic−hyperinsulinemic clamp)試験における糖注入率(GIR; glucose infusion rate)、糖産生率(HGP;Hepatic glucose production)、インスリン負荷試験(ITT;insulin tolerance test)又はピルビン酸負荷試験(PTT;Pyruvate tolerance test)等で評価できる。
【0119】
一般式(I)で表される複素環化合物又はその薬学的に許容される塩は、糖尿病、特に2型糖尿病の改善に効果的であり、具体的には、HbA1c、グリコアルブミン、フルクトサミン、1,5−AG、随時高血糖、空腹時高血糖、耐糖能異常、肝糖新生の亢進、HOMA−IR及びグルコースクランプ試験における糖注入率(GIR)や糖産生率(HGP)の改善が期待できる。さらに、一般式(I)で表される複素環化合物又はその薬学的に許容される塩は、インスリン抵抗性の改善に用いることができるため、メタボリックシンドロームの治療薬又は予防薬として適している。
【0120】
一般式(I)で表される複素環化合物を、糖尿病の治療薬若しくは予防薬又はインスリン抵抗性の改善薬として臨床で投与する場合にあっては、一般式(I)で表される複素環化合物のフリー体又はその薬学的に許容される塩をそのまま投与もよいし、さらに賦形剤、安定化剤、保存剤、緩衝剤、溶解補助剤、乳化剤、希釈剤又は等張化剤等の添加剤が適宜混合されたものを投与しても構わない。その投与形態としては、例えば、錠剤、カプセル剤、顆粒剤、散剤若しくはシロップ剤等の経口剤による経口投与、注射剤、坐剤若しくは液剤等による非経口投与又は軟膏剤、クリーム剤若しくは貼付剤等による局所投与が挙げられる。
【0121】
上記の各投与形態の製剤は、公知の製法によって作ることができるが、一般式(I)で表される複素環化合物又はその薬学的に許容される塩を0.00001〜90重量%含有することが好ましく、0.0001〜70重量%含有することがより好ましい。
【0122】
一般式(I)で表される複素環化合物又はその薬学的に許容される塩の投与量は、症状、年齢、体重及び投与方法等に応じて適宜選択されるが、成人に対して経口剤として投与する場合、有効成分量として1日1μg〜10gが好ましく、それぞれ1回又は数回に分けて投与することができる。
【0123】
一般式(I)で表される複素環化合物又はその薬学的に許容される塩を有効成分として含有する医薬、すなわち糖尿病の治療薬若しくは予防薬又はインスリン抵抗性の改善薬は、その他の糖尿病治療薬、脂質異常症治療薬、肥満症治療薬又はメタボリックシンドローム治療薬(以下、「併用薬」)と組み合わせて用いても構わない。
【0124】
この際、一般式(I)で表される複素環化合物又はその薬学的に許容される塩を有効成分とする医薬と、併用薬との投与時期は限定されず、これらを併用又は配合により同時に投与してもよいし、時間差をおいて投与してもよい。併用薬の投与量は、臨床上用いられている用量を基準として適宜選択することができる。また、一般式(I)で表される複素環化合物又はその薬学的に許容される塩を有効成分とする医薬と、併用薬との併用容量比あるいは配合比は、投与対象、投与形態、症状又は各薬剤の組み合わせ等により適宜選択することができる。
【0125】
上記の併用薬としては、例えば、インスリン製剤(超速効型インスリン製剤、速効型インスリン製剤、混合型インスリン製剤、中間型インスリン製剤、持続型インスリン製剤、持効型溶解インスリン製剤、経肺インスリン製剤、経口インスリン製剤等)、インスリン抵抗性改善薬(ピオグリタゾン(pioglitazone)、ロシグリタゾン(rosiglitazone)、ネトグリタゾン(netoglitazone)、ファルグリタザール(farglitazar)、リボグリタゾン(rivoglitazone)、バラグリタゾン(balaglitazone)等)、α−グルコシダーゼ阻害薬(アカルボース(acarbose)、ボグリボース(voglibose)、ミグリトール(miglitol)、エミグリテート(emiglitate)等)、ビグアナイド剤(メトホルミン(metformin)、ブホルミン(buformin)等)、スルホニルウレア剤(トルブタミド(tolbutamide)、アセトヘキサミド(acetohexamide)、クロルプロパミド(chlorpropamide)、トラザミド(tolazamide)、グリクロピラミド(glyclopyramide)、グリブゾール(glybuzole)、グリベンクラミド(glibenclamide)、グリクラジド(gliclazide)、グリメピリド(glimepiride)、グリピジド(glipizide)、グリキドン(gliquidone)等)、速効型インスリン分泌促進薬(ナテグリニド(nateglinide)、レパグリニド(repaglinide)、ミチグリニド(mitiglinide)等)、GLP−1作動薬(エクセナチド(exenatide)、リラグルチド(liraglutide)等)、アミリン作動薬(プラムリンチド(pramlintide)等)、DPP−IV阻害薬(ビルダグリプチン(vildagliptin)、シタグリプチン(sitagliptin)、サクサグリプチン(saxagliptin)、アログリプチン(alogliptin)、デナグリプチン(denagliptin)、ジェミグリプチン(Gemigliptin)、ドゥトグリプチン(Dutogliptin)等)、β3作動薬(ソラベグロン(solabegron)、ミラベグロン(mirabegron)、KRP−204等)、フルクトース−1,6−ビスホスファターゼ阻害薬(MB−6322、MB−07803等)、SGLT(sodium−dependent renal glucose transporter)阻害薬(セルグリフロジン(sergliflozin)、レモグリフロジン(remogliflozin)、AVE−2268、TS−033、KGA−2727、SAR−7226等)、11β−HSD1阻害薬(BVT−3498、AMG−221、INCB−13739、INCB−20817、JTT−654、PF−915275等)、PTP−1B(protein tyrosine phosphatase−1B)阻害薬(ISIS−113715、JTT−551等)、GSK3β(glycogen synthase kinase 3β)阻害薬(SAR−502250等)、グルカゴン拮抗薬(BAY−27−9955、NN−2501等)、グリコーゲンホスホリラーゼ阻害薬(イソファゴミン(Isofagomine)、PSN−357等)、CPT1(カルニチン O−パルミトイルトランスフェラーゼ1)阻害薬(テグリカール(teglicar)等)、グルココルチコイド拮抗薬(ミフェプリストン(mifepristone)、KB−3305等)、HMG−CoA還元酵素阻害薬(プラバスタチン(pravastatin)、シンバスタチン(simvastatin)、フルバスタチン(fluvastatin)、アトルバスタチン(atorvastatin)、ピタバスタチン(pitavastatin)等)、陰イオン交換樹脂(コレスチラミン(colestyramine)、コレスチミド(colestimide)等)、フィブラート系薬(クロフィブラート(clofibrate)、クリノフィブラート(clinofibrate)、ベザフィブラート(bezafibrate)、フェノフィブラート(fenofibrate)等)、ニコチン酸系薬(ニコチン酸トコフェロール(tocopherol nicotinate)、CB1(カンナビノイド1)拮抗薬(リモナバン(rimonabant)、スリナバン(surinabant)、タラナバン(taranabant)、ドリナバン(drinabant)等)、リパーゼ阻害剤(オルリスタット(orlistat)等)又は中枢性食欲抑制剤(マジンドール(mazindol)、フェンフルラミン(fenfluramine)、デクスフェンフルラミン(dexfenfluramine)又はシブトラミン(sibutramine)、フェンターミン(phentermine)等)が挙げられる。
【実施例】
【0126】
以下、実施例を示して本発明を具体的に詳述するが、本発明はこれらに限定されるものではない。
【0127】
一般式(I)で表される複素環化合物の原料及び中間体は、以下の参考例に記載する方法で合成した。また、参考例化合物の合成に使用される化合物で合成法の記載のないものについては、市販の化合物を使用した。
【0128】
(参考例1) trans−5−ヒドロキシ−2−アダマンチルアミン(以下、「参考例1の化合物」)の合成:
【化9】
5−ヒドロキシ−2−アダマンタノン(3.0g,18mmol)をトルエン(60mL)に溶解し、(−)−1−フェニルエチルアミン(2.4mL,19mmol)、アルミニウムイソプロポキシド(3.7g,18mmol)、水酸化パラジウム/炭素(10%wet,0.25g,1.2mmol)を加え水素雰囲気下、50℃で撹拌した。16時間後、反応液を室温まで冷却し、セライト濾過し、濾液を濃縮した。得られた粗生成物をメタノール(130mL)に溶解し、水酸化パラジウム/炭素(10%wet,0.15g,0.72mmol)を加え水素雰囲気下、室温で撹拌した。14時間後、反応液をセライト濾過し、濾液を濃縮した。得られた粗生成物をヘキサン/酢酸エチルを溶媒として再結晶を行い、表題化合物0.98g(55%)を白色固体として得た。
1H−NMR(400MHz,CDCl3)
δ:3.79(3H,s),6.92−6.94(2H,m),7.05(1H,dd,J=4.8,7.6Hz),7.09−7.11(2H,m),7.97(1H,dd,J=2.0,7.6Hz),8.28(1H,dd,J=2.0,4.8Hz).
MS(ESI)[M+H]+
227(M+H)+
【0129】
(参考例2) エチル 6−ベンジルオキシ−3−オキソヘキサノエート(以下、「参考例2の化合物」)の合成:
【化10】
4−ベンジルオキシ酪酸(5.1g,26mmol)をアセトニトリル(50mL)に溶解し、カルボニルジイミダゾール(4.5g,28mmol)を加えて室温で撹拌した(反応液A)。別途、マグネシウムクロリド(5.5g,58mmol)をアセトニトリル(50mL)に溶解し、マロン酸エチルカリウム塩(9.4g,55mmol)、トリエチルアミン(17mL,120mmol)を加え室温で撹拌した(反応液B)。2.5時間後、反応液Aを反応液Bに加え90℃で撹拌した。2時間後、反応液を室温まで冷却し、1N 塩酸を加えpH=5とした後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=100/0〜98/2)で精製し、表題化合物4.7g(95%)を無色液体として得た。1H−NMR(400MHz,CDCl3)
δ:1.27(3H,t,J=7.2Hz),1.82−1.96(2H,m),2.66(2H,t,J=7.2Hz),3.43(2H,s),3.49(2H,t,J=6.0Hz),4.19(2H,q,J=7.2Hz),4.48(2H,s),7.27−7.37(5H,m).
MS(ESI)[M+H]+
265
【0130】
(参考例3) エチル 6−ベンジルオキシ−4,4−ジメチル−3−オキソヘキサノエート(以下、「参考例3の化合物」)の合成:
〔第1工程〕
メチル 4−ベンジルオキシブタノエートの合成:
【化11】
4−ベンジルオキシ酪酸(5.0g,26mmol)をメタノール(52mL)に溶解し、濃硫酸(1.0mL,19mmol)を加え室温で撹拌した。3時間後、炭酸水素ナトリウムを加え中和した後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=100/0〜76/24)で精製し、表題化合物5.4g(99%)を無色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:1.90−1.98(2H,m),2.45(2H,t,J=7.6Hz),3.51(2H,t,J=6.0Hz),3.65(3H,m),4.49(2H,m),7.26−7.35(5H,s).
MS(ESI)[M+H]+
209
【0131】
〔第2工程〕
メチル 4−ベンジルオキシ−2−メチルブタノエートの合成:
【化12】
ジイソプロピルアミン(5.1mL,36mmol)をTHF(90mL)に溶解し、n−ブチルリチウム(2.6N,14mL,36mmol)を加え−78℃で撹拌した。20分後、メチル 4−ベンジルオキシブタノエート(0.74g,2.0mmol)のTHF溶液(10mL)を加え−78℃で撹拌した。20分後、ヨードメタン(3.0mL,48mmol)を加え0℃で撹拌した。3時間後、水、1N 塩酸を加えた後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=98/2〜80/20)で精製し、表題化合物5.0g(94%)を無色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:1.17(3H,d,J=6.8Hz),1.65−1.75(1H,m),1.98−2.06(1H,m),2.61−2.71(1H,m),3.47−3.52(2H,m),3.64(3H,s),4.48(2H,s),7.26−7.35(5H,m).
MS(ESI)[M+H]+
223
【0132】
〔第3工程〕
メチル 4−ベンジルオキシ−2,2−ジメチルブタノエートの合成:
【化13】
ジイソプロピルアミン(2.4mL,17mmol)をTHF(45mL)に溶解し、n−ブチルリチウム(2.6N,6.5mL,17mmol)を加え−78℃で撹拌した。20分後、メチル 4−ベンジルオキシ−2−メチルブタノエート(0.74g,2.0mmol)のTHF溶液(5.0mL)を加え−78℃で撹拌した。20分後、ヨードメタン(1.4mL,22mmol)を加え0℃で撹拌した。3時間後、水、1N 塩酸を加えた後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=100/0〜86/14)で精製し、表題化合物2.5g(94%)を無色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:1.20(6H,s),1.90(2H,t,J=6.8Hz),3.50(2H,t,J=6.8Hz),3.59(3H,s),4.45(2H,s),7.26−7.36(5H,m).
MS(ESI)[M+H]+
237
【0133】
〔第4工程〕
参考例3の化合物の合成:
【化14】
メチル 4−ベンジルオキシ−2,2−ジメチルブタノエート(4.0g,17mmol)をメタノール(25mL)に溶解し、1N 水酸化ナトリウム(25mL,25mmol)を加え60℃で撹拌した。3時間後、反応液を室温まで冷却し、濃縮しクロロホルムで逆抽出を行い、1N 塩酸を加え中和した後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をアセトニトリル(34mL)に溶解し、カルボニルジイミダゾール(2.9g,18mmol)を加え室温で撹拌した(反応液A)。別途、マグネシウムクロリド(3.5g,37mmol)をアセトニトリル(34mL)に溶解し、マロン酸エチルカリウム塩(6.1g,36mmol)、トリエチルアミン(11mL,76mmol)を加え室温で撹拌した(反応液B)。14時間後、反応液Aを反応液Bに加え90℃で撹拌した。2時間後、反応液を室温まで冷却し、1N 塩酸を加えpH=5とした後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=100/0〜86/14)で精製し、表題化合物4.7g(95%)を無色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:1.26(6H,s),1.38(3H,t,J=6.8Hz),1.54−1.55(2H,m),4.30(2H,q,J=6.8Hz),7.43−7.48(5H,m),8.06(1H,s),9.58−9.62(1H,m).
MS(ESI)[M+H]+
293
【0134】
(比較例1) 5−アダマンタン−2−イル−1−フェニル−6,7−ジヒドロ−1H−ピラゾロ[4,3−C]ピリジン−4(5H)−オン(以下、「比較例1の化合物」)の合成:
〔第1工程〕
(1−アダマンタン−2−イル)ピペリジン−2,4−ジオンの合成:
【化15】
アダマンタン−2−アミン(2.5g,17mmol)をTHF(32mL)に溶解し、エチルアクリレート(2.3mL,21mmol)を加え20℃で撹拌した。16時間後、エチル 3−クロロ−オキソプロパノエート(2.4mL,19mmol)、トリエチルアミン(3.0mL,21mmol)を加え20℃で撹拌した。2時間後、水を加えた後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をTHF(14mL)に溶解し、ナトリウムエトキサイド(0.23g,3.4mmol)を加え90℃で撹拌した。2時間後、塩酸水溶液(6.0N,8.0mL,48mmol)を加えた後、100℃で撹拌した。4時間後、反応液を室温まで冷却し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=70/30〜40/60)で精製し、表題化合物0.41g(10%)を白色固体として得た。
1H−NMR(400MHz,CDCl3)
δ:1.71−1.77(5H,m),1.83−1.93(7H,m),2.27(2H,s),2.60(2H,s),3.40(2H,s),3.84(2H,s),4.27(1H,s)
MS(ESI)[M+H]+
248
【0135】
〔第2工程〕
比較例1の化合物の合成:
【化16】
1−アダマンタン−2−イルピペリジン−2,4−ジオン(0.41g,1.7mmol)に1,1−ジメトキシ−N,N−ジメチルメタンアミン(1.1mL,8.3mmol)を加え90℃で撹拌した。2時間後、反応液を室温まで冷却し、水を加えた後、クロロホルムで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗成生物をエタノール(7.0mL)に溶解し、フェニルヒドラジン(0.27mL,2.7mmol)を加え90℃で撹拌した。5時間後、反応液を室温まで冷却し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=80/20〜40/60)で精製し、表題化合物0.41g(86%)を白色固体として得た。
1H−NMR(400MHz,CDCl3)
δ:1.66−1.77(5H,m),1.87−2.00(7H,m),2.32(2H,s),3.10(2H,t,J=6.4Hz),3.77(2H,t,J=6.4Hz),4.27(1H,s),7.39−7.41(1H,m),7.50−7.51(4H,m),8.05(1H,s)
MS(ESI)[M+H]+
348
【0136】
(実施例1) 1−フェニル−5−ヒドロキシアダマンタン−2−イル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例1の化合物」)の合成:
〔第1工程〕
エチル 5−(2−シアノエチル)−1−フェニル−1H−ピラゾール−4−カルボキシレートの合成:
【化17】
エチル 5−シアノ−3−オキソペンタノエート(1.5g,8.8mmol)にジメチルホルムアミドジメチルアセタール(5.9mL,44mmol)を加え80℃で撹拌した。3時間後、反応液を室温まで冷却し、水を加えた後、ジエチルエーテルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をエタノール(40mL)に溶解し、フェニルヒドラジン(2.0g,18mmol)を加え80℃で撹拌した。5時間後、室温まで冷却し、水を加えた後、濃縮後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=70/30〜12/88)で精製し、表題化合物0.46g(20%)を黄色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:1.40(3H,t,J=7.2Hz),2.75(2H,t,J=8.0Hz),3.27(2H,t,J=8.0Hz),4.35(2H,q,J=7.2Hz),7.40−7.44(2H,m),7.52−7.58(3H,m),8.06(1H,s).
MS(ESI)[M+H]+
270
【0137】
〔第2工程〕
エチル 5−(3−アミノプロピル)−1−フェニル−1H−ピラゾール−4−カルボキシレートの合成:
【化18】
エチル 5−(2−シアノエチル)−1−フェニル−1H−ピラゾール−4−カルボキシレート(0.45g,1.7mmol)をエタノール(17mL)に溶解し、パラジウム/炭素(10%wet,0.18g,0.17mmol)、濃塩酸(1.0mL)を加え水素雰囲気下、室温で撹拌した。25時間後、反応液をセライト濾過し、濾液を濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=100/0〜76/24)で精製し、表題化合物0.16g(35%)を無色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:1.38(3H,t,J=7.2Hz),1.83(2H,t,J=6.8Hz),2.58−2.78(4H,m),3.05(2H,t,J=7.2Hz),4.33(2H,q,J=7.2Hz),7.38−7.41(2H,m),7.43−7.48(3H,m),8.00(1H,s),
MS(ESI)[M+H]+
274
【0138】
〔第3工程〕
実施例1の化合物の合成:
【化19】
エチル 5−(3−アミノプロピル)−1−フェニル−1H−ピラゾール−4−カルボキシレート(81mg,0.30mmol)をエタノール(3.0mL)に溶解し、アダマンタン−2−オン(49mg,0.33mmol)、チタニウム(IV)テトライソプロポキシド(180mL,0.59mmol)を加え室温で撹拌した。20時間後、水素化トリアセトキシホウ素ナトリウム(0.13g,0.59mmol)を加え室温で撹拌した。2時間後、1N 塩酸を加えた後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をクロロホルム(5.0mL)に溶解し、ジイソプロピルエチルアミン(0.14mL,0.26mmol)、HBTU(61mg,0.26mmol)を加え室温で撹拌した。1時間後、水を加えた後、クロロホルムで抽出した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=90/10〜47/53)で精製し、表題化合物82mg(77%)を白色固体として得た。
1H−NMR(400MHz,CDCl3)
δ:1.66−1.77(3H,m),1.85−2.01(8H,m),2.12−2.20(2H,m),2.21−2.29(2H,m),2.90(2H,t,J=7.2Hz),3.56−3.62(2H,m),4.37−4.41(1H,m),7.39−7.51(5H,m),8.11(1H,s).
MS(ESI)[M+H]+
362
【0139】
(実施例2) 1−メチル−5−(trans−5−ヒドロキシアダマンタン−2−イル)5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例2の化合物」)の合成:
【化20】
エチル 6−ベンジルオキシ−3−オキソヘキサノエート(2.0g,7.6mmol)にジメチルホルムアミドジメチルアセタール(5.1mL,38mmol)を加え90℃で撹拌した。2時間後、反応液を室温まで冷却し、水を加えた後、ジエチルエーテルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をエタノール(35mL)に溶解し、メチルヒドラジン(0.50g,11mmol)を加え80℃で撹拌した。4時間後、反応液を室温まで冷却し、濃縮し水を加えた後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=100/0〜81/19)で精製した。得られた粗精製物をエタノール(27mL)に溶解し、水酸化パラジウム/炭素(10%wet,0.075g,0.54mmol)を加え水素雰囲気下、室温で撹拌した。5時間後、反応液をセライト濾過し、濾液を濃縮した。得られた粗生成物をジクロロメタン(27mL)に溶解し、Dess−Martin試薬(1.1g,2.7mmol)を加え室温で撹拌した。4時間後、反応液をセライト濾過し、濾液を濃縮した。得られた粗生成物をクロロホルム(28mL)に溶解し、trans−5−ヒドロキシ−2−アダマンチルアミン(450mg,2.7mmol)、酢酸(0.47mL,8.1mmol)、水素化トリアセトキシホウ素ナトリウム(1.5g,8.1mmol)を加え室温で撹拌した。16時間後、水を加えた後、クロロホルムで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=100/0〜81/19)で精製した。得られた粗精製物をメタノール(1.0mL)に溶解し、1N 水酸化ナトリウム(1.0mL,1.0mmol)を加え室温で撹拌した。16時間後、濃縮した。得られた粗生成物をクロロホルム(14mL)に溶解し、ジイソプロピルエチルアミン(0.37mL,2.1mmol)、HBTU(160mg,0.70mmol)を加え室温で撹拌した。16時間後、水を加えた後、クロロホルムで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=100/0〜91/9)で精製し、表題化合物45mg(3%)を白色固体として得た。
1H−NMR(400MHz,CDCl3)
δ:1.75−2.84(7H,m),1.95−2.00(2H,m),2.15−2.25(3H,m),2.44−2.48(2H,m),2.86(2H,t,J=7.2Hz),3.53−3.57(2H,m),3.80(3H,s),4.28−4.31(1H,m),7.95(1H,s).
MS(ESI)[M+H]+
316
【0140】
(実施例3) 1−エチル−5−(trans−5−ヒドロキシアダマンタン−2−イル)5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例3の化合物」)の合成:
参考例1及び2並びに実施例2と同様の手順により、以下の実施例3の化合物を合成した。
【化21】
1H−NMR(400MHz,CDCl3)
δ:1.43(3H,t,J=7.2Hz),1.50−1.60(2H,m),1.75−1.85(6H,m),1.95−2.01(2H,m),2.15−2.24(2H,m),2.43−2.49(2H,m),2.88(2H,t,J=7.2Hz),3.52−3.57(2H,m),4.08(2H,q,J=7.2Hz),4.08−4.11(1H,m),7.97(1H,s).
MS(ESI)[M+H]+
330
【0141】
(実施例4) 1−(tert−ブチル)−5−(trans−5−ヒドロキシアダマンタン−2−イル)5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例4の化合物」)の合成:
参考例1及び2並びに実施例2と同様の手順により、以下の実施例4の化合物を合成した。
【化22】
1H−NMR(400MHz,CDCl3)
δ:1.49−1.58(2H,m),1.66(9H,s),1.76−1.86(6H,m),1.95−2.02(2H,m),2.10−2.24(3H,m),2.41−2.47(2H,m),3.12(2H,t,J=7.2Hz),3.44−3.49(2H,m),7.82(1H,s).
MS(ESI)[M+H]+
358
【0142】
(実施例5) 1−シクロヘキシル−5−(trans−5−ヒドロキシアダマンタン−2−イル)5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例5の化合物」)の合成:
参考例1及び2並びに実施例2と同様の手順により、以下の実施例5の化合物を合成した。
【化23】
1H−NMR(400MHz,CDCl3)
δ:1.31−1.40(4H,m),1.48−1.59(5H,m),1.78−2.00(11H,m),2.15−2.25(2H,m),2.43−2.49(2H,m),2.83−2.85(2H,m),3.53−3.57(2H,m),3.85−3.94(1H,m),4.26−4.32(1H,m),7.99(1H,s).
MS(ESI)[M+H]+
384
【0143】
(実施例6) 1−シクロヘプチル−5−(trans−5−ヒドロキシアダマンタン−2−イル)5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例6の化合物」)の合成:
参考例1及び2並びに実施例2と同様の手順により、以下の実施例6の化合物を合成した。
【化24】
1H−NMR(400MHz,CDCl3)
δ:1.54−1.59(7H,m),1.62−1.68(3H,m),1.75−2.24(15H,m),2.44−2.48(2H,m),2.85−2.90(2H,m),3.52−3.56(2H,m),4.06−4.15(1H,m),4.28−4.32(1H,m),7.98(1H,s).
MS(ESI)[M+H]+
398
【0144】
(実施例7) 1−フェニル−5−(trans−5−ヒドロキシアダマンタン−2−イル)5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例7の化合物」)の合成:
参考例1及び2並びに実施例2と同様の手順により、以下の実施例7の化合物を合成した。
【化25】
1H−NMR(400MHz,CDCl3)
δ:1.66−2.00(10H,m),2.12−2.20(2H,m),2.24−2.30(2H,m),2.90(2H,t,J=7.2Hz),3.56−3.64(2H,m),4.38−4.42(1H,m),7.40−7.52(5H,m),8.11(1H,s).
MS(ESI)[M+H]+
378
【0145】
(実施例8) 1−(ピリジン−2−イル)−5−(trans−5−ヒドロキシアダマンタン−2−イル)5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例8の化合物」)の合成:
参考例1及び2並びに実施例2と同様の手順により、以下の実施例8の化合物を合成した。
【化26】
1H−NMR(400MHz,CDCl3)
δ:1.38−1.42(1H,m),1.55−1.61(1H,m),1.77−1.88(6H,m),1.97−2.02(2H,m),2.19−2.24(2H,m),2.47−2.52(2H,m),3.47−3.52(2H,m),3.59−3.63(2H,m),4.32−4.34(1H,m),7.23−7.26(1H,m),7.85(1H,ddd,J=1.6,7.2,8.0Hz),7.91(1H,d,J=8.0Hz),8.16(1H,s),8.43−8.46(1H,m).
MS(ESI)[M+H]+
379
【0146】
(実施例9) 1−フェニル−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8−メチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例9の化合物」)の合成:
【化27】
メチル 4−ベンジルオキシ−2−メチルブタノエート(1.0g,4.2mmol)をメタノール(7.0mL)に溶解し、1N 水酸化ナトリウム(6.8mL,6.8mmol)を加え室温で撹拌した。16時間後、濃縮した後1N 塩酸を加え中和した後、酢酸エチルで抽出した。得られた粗生成物をアセトニトリル(9.0mL)に溶解し、カルボニルジイミダゾール(0.74g,4.5mmol)を加え室温で撹拌した(反応液A)。別途、マグネシウムクロリド(0.91g,9.5mmol)をアセトニトリル(9.0mL)に溶解し、マロン酸エチルカリウム塩(1.5g,9.1mmol)、トリエチルアミン(2.7mL,19mmol)を加え室温で撹拌した(反応液B)。14時間後、反応液Aを反応液Bに加え90℃で撹拌した。2時間後、反応液を室温まで冷却し、1N 塩酸を加えpH=5とした後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=100/0〜79/21)で精製した。得られた粗精製物にジメチルホルムアミドジメチルアセタール(2.9mL,22mmol)を加え80℃で撹拌した。4時間後、反応液を室温まで冷却し、濃縮した。得られた粗生成物をエタノール(30mL)に溶解し、フェニルヒドラジン(0.93g,8.6mmol)を加え80℃で撹拌した。5時間後、反応液を室温まで冷却し、濃縮し水を加えた後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=94/6〜76/24)で精製した。
【0147】
得られた粗精製物をエタノール(32mL)に溶解し、水酸化パラジウム/炭素(10%wet,0.45g,0.32mmol)を加え水素雰囲気下、室温で撹拌した。21時間後、反応液をセライト濾過し、濾液を濃縮した。得られた粗生成物をジクロロメタン(32mL)に溶解し、Dess−Martin試薬(1.4g,3.2mmol)を加え室温で撹拌した。2時間後、反応液をセライト濾過し、濾液を濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=100/0〜66/34)で精製した。得られた粗精製物をクロロホルム(23mL)に溶解し、trans−5−ヒドロキシ−2−アダマンチルアミン(390mg,2.3mmol)、酢酸(0.34mL,6.9mmol)、水素化トリアセトキシホウ素ナトリウム(1.3g,6.9mmol)を加え室温で撹拌した。16時間後、飽和炭酸水素ナトリウム水溶液を加え中和した後、クロロホルムで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をメタノール(4.0mL)に溶解し、1N 水酸化ナトリウム(3.5mL,3.5mmol)を加え室温で撹拌した。16時間後、濃縮した。得られた粗生成物をクロロホルム(23mL)に溶解し、ジイソプロピルエチルアミン(1.2mL,6.9mmol)、HBTU(540mg,2.3mmol)を加え室温で撹拌した。16時間後、水を加えた後、クロロホルムで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=100/0〜96/4)で精製した。この際得られた固形物を酢酸エチル/ヘキサン=1/1溶液によりスラリー洗浄し、表題化合物110mg(6%)を白色固体として得た。
1H−NMR(400MHz,CDCl3)
δ:1.03(3H,d,J=6.8Hz),1.50−1.64(3H,m),1.74−2.04(7H,m),2.20−2.25(1H,m),2.31−2.36(1H,m),2.38−2.49(1H,m),2.55−2.65(2H,m),2.92−3.00(1H,m),3.14−3.22(1H,m),3.50−3.57(1H,m),4.32−4.36(1H,m),7.39−7.52(5H,m),8.10(1H,s).
MS(ESI)[M+H]+
392
【0148】
(実施例10) 1−フェニル−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8,8−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例10の化合物」)の合成:
参考例1及び3並びに実施例2と同様の手順により、以下の実施例10の化合物を合成した。
【化28】
1H−NMR(400MHz,CDCl3)
δ:1.14(6H,s),1.40−1.47(1H,m),1.56−1.62(1H,m),1.76−1.85(1H,m),1.76−1.85(6H,m),1.96−2.05(4H,m),2.20−2.25(1H,m),3.59−3.64(2H,m),4.30−4.33(1H,m),7.36−7.40(2H,m),7.44−7.52(3H,m),8.12(1H,s).
MS(ESI)[M+H]+
406
【0149】
(実施例11) 1−フェニル−5−(trans−5−ヒドロキシアダマンタン−2−イル)−7−メチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例11の化合物」)の合成:
〔第1工程〕
4−(ベンジルオキシ)−2−メチルブトキシ−tert−ブチルジメチルシランの合成:
【化29】
メチル 4−(ベンジルオキシ)−2−メチルブタノエート(1.5g,6.8mmol)をTHF(34mL)に溶解し、水素化リチウムアルミニウム(0.14g,20mmol)を加え室温で撹拌した。2時間後、硫酸ナトリウム十水和物を加え室温で撹拌した。1時間後、反応液をセライト濾過し、濾液を濃縮した。得られた粗生成物をDMF(14mL)に溶解し、イミダゾール(0.92g,14mmol)、tert−ブチルジメチルシリルクロリド(1.5g,10mmol)を加え室温で撹拌した。2時間後、水を加えた後、ジエチルエーテルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=100/0〜81/19)で精製し、表題化合物2.0g(94%)を無色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:0.02(6H,s),0.86−0.90(12H,m),1.35−1.45(1H,m),1.70−1.80(2H,m),3.38−3.55(4H,m),4.50(2H,s),7.26−7.35(5H,m).
MS(ESI)[M+H]+
309
【0150】
〔第2工程〕
4−(tert−ブチルジメチルシリルオキシ)−3−メチルブタナールの合成:
【化30】
4−(ベンジルオキシ)−2−メチルブトキシ−tert−ブチルジメチルシラン(1.8g,5.8mmol)を酢酸エチル(58mL)に溶解し、パラジウム/炭素(10%wet,0.62g,5.8mmol)を加え水素雰囲気下、室温で撹拌した。4時間後、反応液をセライト濾過し、濾液を濃縮した。得られた粗生成物をジクロロメタン(55mL)に溶解し、Dess−Martin試薬(2.4g,5.5mmol)を加え室温で撹拌した。2時間後、反応液をセライト濾過し、濾液を濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=100/0〜81/19)で精製し、表題化合物0.75g(60%)を無色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:0.03(6H,s),0.88(9H,s),0.94(3H,d,J=6.4Hz),2.19−2.32(2H,m),2.46−2.54(1H,m),3.34−3.39(1H,m),3.56(1H,dd,J=5.2,10.0Hz),9.77(1H,t,J=2.0Hz).
MS(ESI)[M+H]+
217
【0151】
〔第3工程〕
エチル 1−フェニル−5−(2−メチル−3−(tert−ブチルジメチルシリルオキシ)プロピル)−1H−ピラゾール−4−カルボキシレートの合成:
【化31】
4−(tert−ブチルジメチルシリルオキシ)−3−メチルブタナール(750mg,3.5mmol)をtert−ブチルアルコール(10mL)に溶解し、水(20mL)、リン酸水素二ナトリウム(1.5g,10mmol)を加え室温で撹拌した。1時間後、亜塩素酸ナトリウム(1.9g,21mmol)を加え室温で撹拌した。4時間後、水、亜硫酸水素ナトリウム(6.6g,52mmol)を加えた後、酢酸エチルで抽出した。得られた粗生成物をアセトニトリル(9.0mL)に溶解し、カルボニルジイミダゾール(0.67g,4.2mmol)を加え室温で撹拌した(反応液A)。別途、マグネシウムクロリド(0.83g,8.7mmol)をアセトニトリル(9.0mL)に溶解し、マロン酸エチルカリウム塩(1.4g,8.3mmol)、トリエチルアミン(2.5mL,18mmol)を加え撹拌した。2時間後、反応液を室温まで冷却し、1N 塩酸を加えpH=5とした後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物にジメチルホルムアミドジメチルアセタール(2.7mL,20mmol)を加え90℃で撹拌した。2時間後、反応液を室温まで冷却し、濃縮した。得られた粗生成物をエタノール(28mL)に溶解し、フェニルヒドラジン(0.86g,7.9mmol)を加え80℃で撹拌した。5時間後、反応液を室温まで冷却し、濃縮し水を加えた後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=93/7〜79/21)で精製し、表題化合物0.76g(54%)を黄色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:0.00(6H,s),0.68(3H,d,J=6.8Hz),0.80(9H,s),1.37(3H,t,J=7.2Hz),1.80−1.99(1H,m),2.82−2.96(1H,m),3.05−3.11(1H,m),3.26−3.35(2H,m),4.32(2H,q,J=7.2Hz),7.38−7.51(5H,m),8.01(1H,s).
MS(ESI)[M+H]+
403
【0152】
〔第4工程〕
エチル 1−フェニル−5−(2−メチル−3−ヒドロキシプロピル)−1H−ピラゾール−4−カルボキシレートの合成:
【化32】
エチル 1−フェニル−5−(2−メチル−3−(tert−ブチルジメチルシリルオキシ)プロピル)−1H−ピラゾール−4−カルボキシレート(0.76g,1.9mmol)をTHF(10mL)に溶解し、TBAF(1.0N,3.8mL,3.8mmol)を加え室温で撹拌した。16時間後、水を加えた後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=93/7〜66/34)で精製し、表題化合物0.48g(88%)を黄色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:0.78(3H,d,J=7.2Hz),1.39(3H,t,J=7.2Hz),1.68−1.78(1H,m),2.48−2.54(1H,m),2.74−2.81(1H,m),3.33−3.42(3H,m),4.34(2H,q,J=7.2Hz),7.37−7.42(2H,m),7.47−7.55(3H,m),8.03(1H,s).
MS(ESI)[M+H]+
289
【0153】
〔第5工程〕
エチル 1−フェニル−5−(2−メチル−3−オキソプロピル)−1H−ピラゾール−4−カルボキシレートの合成:
【化33】
エチル 1−フェニル−5−(2−メチル−3−ヒドロキシプロピル)−1H−ピラゾール−4−カルボキシレート(480mg,1.7mmol)をジクロロメタン(17mL)に溶解し、Dess−Martin試薬(710mg,1.7mmol)を加え室温で撹拌した。2時間後、反応液をセライト濾過し、濾液を濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=96/4〜69/31)で精製し、表題化合物0.48g(76%)を無色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:0.93(3H,q,J=7.2Hz),1.37(3H,t,J=7.2Hz),2.70−2.79(1H,m),2.97−3.05(1H,m),3.41−3.48(1H,m)、4.32(2H,q,J=7.2Hz),7.36−7.40(2H,m),7.48−7.54(3H,m),8.06(1H,s),9.50−9.52(1H,m).
MS(ESI)[M+H]+
287
【0154】
〔第6工程〕
実施例11の化合物の合成:
【化34】
エチル 1−フェニル−5−(2−メチル−3−オキソプロピル)−1H−ピラゾール−4−カルボキシレート(360mg,1.3mmol)をクロロホルム(13mL)に溶解し、trans−5−ヒドロキシ−2−アダマンチルアミン(210mg,1.3mmol)、酢酸(0.22mL,3.8mmol)、水素化トリアセトキシホウ素ナトリウム(710mg,3.8mmol)を加え室温で撹拌した。16時間後、飽和炭酸水素ナトリウム水溶液を加え中和した後、クロロホルムで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をメタノール(2.0mL)に溶解し、1N 水酸化ナトリウム(1.9mL,1.9mmol)を加え室温で撹拌した。16時間後、濃縮した。得られた粗生成物をクロロホルム(13mL)に溶解し、ジイソプロピルエチルアミン(0.66mL,3.8mmol)、HBTU(300mg,1.3mmol)を加え室温で撹拌した。16時間後、水を加えた後、クロロホルムで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。この際得られた固形物を酢酸エチル/ヘキサン=1/1溶液によりスラリー洗浄し、表題化合物51mg(10%)を白色固体として得た。
1H−NMR(400MHz,CDCl3)
δ:1.03(3H,d,J=7.2Hz),1.58−1.65(3H,m),1.74−2.05(7H,m),2.20−2.22(1H,m),2.34−2.37(1H,m),2.37−2.48(1H,m),2.57−2.65(2H,m),3.20(1H,dd,J=9.6,15.6Hz),3.54(1H,dd,J=3.2,15.6Hz),4.32−4.35(1H,m),7.40−7.53(5H,m),8.11(1H,s).
MS(ESI)[M+H]+
392
【0155】
(実施例12) 1−フェニル−5−(trans−5−ヒドロキシアダマンタン−2−イル)−7,7−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例12の化合物」)の合成:
〔第1工程〕
4−(ベンジルオキシ)−2,2−ジメチルブトキシ−tert−ブチルジメチルシランの合成:
【化35】
メチル 4−(ベンジルオキシ)−2,2−ジメチルブタノエート(1.5g,6.4mmol)をTHF(32mL)に溶解し、水素化リチウムアルミニウム(0.13g,19mmol)を加え室温で撹拌した。2時間後、硫酸ナトリウム十水和物を加え室温で撹拌した。1時間後、反応液をセライト濾過し、濾液を濃縮した。得られた粗生成物をDMF(13mL)に溶解し、イミダゾール(0.86g,13mmol)、tert−ブチルジメチルシリルクロリド(1.4g,9.5mmol)を加え室温で撹拌した。2時間後、水を加えた後、ジエチルエーテルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=100/0〜81/19)で精製し、表題化合物1.8g(86%)を無色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:0.00(6H,s),0.86(6H,s),0.88(9H,s),1.61(2H,t,J=7.6Hz),3.24(2H,s),3.53(2H,t,J=7.6Hz),4.49(2H,s),7.26−7.34(5H,m).
MS(ESI)[M+H]+
323
【0156】
〔第2工程〕
4−(tert−ブチルジメチルシリルオキシ)−3,3−ジメチルブタナールエチルの合成:
【化36】
4−(ベンジルオキシ)−2,2−ジメチルブトキシ−tert−ブチルジメチルシラン(1.7g,5.3mmol)を酢酸エチル(53mL)に溶解し、パラジウム/炭素(10%wet,0.56g,5.3mmol)を加え水素雰囲気下、室温で撹拌した。4時間後、反応液をセライト濾過し、濾液を濃縮した。得られた粗生成物をジクロロメタン(52mL)に溶解し、Dess−Martin試薬(2.2g,5.2mmol)を加え室温で撹拌した。2時間後、反応液をセライト濾過し、濾液を濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=100/0〜81/19)で精製し、表題化合物0.96g(79%)を無色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:0.03(6H,s),0.89(9H,s),1.02(6H,s),2.28(2H,d,J=3.2Hz),3.35(2H,s),9.84(1H,d,J=3.2Hz).
MS(ESI)[M+H]+
231
【0157】
〔第3工程〕
エチル 1−フェニル−5−(2,2−ジメチル−3−(tert−ブチルジメチルシリルオキシ)プロピル)−1H−ピラゾール−4−カルボキシレートの合成:
【化37】
4−(tert−ブチルジメチルシリルオキシ)−3,3−ジメチルブタナール(960mg,4.2mmol)をtert−ブチルアルコール(13mL)に溶解し、水(26mL)、リン酸水素二ナトリウム(1.8g,13mmol)を加え室温で撹拌した。1時間後、亜塩素酸ナトリウム(2.3g,25mmol)を加え室温で撹拌した。4時間後、水、亜硫酸水素ナトリウム(7.9g,63mmol)を加えた後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をアセトニトリル(9.0mL)に溶解し、カルボニルジイミダゾール(0.71g,4.4mmol)を加え室温で撹拌した(反応液A)。別途、マグネシウムクロリド(0.87g,9.1mmol)をアセトニトリル(9.0mL)に溶解し、マロン酸エチルカリウム塩(1.5g,8.7mmol)、トリエチルアミン(2.6mL,19mmol)を加え室温で撹拌した(反応液B)。2時間後、反応液Aを反応液Bに加え90℃で撹拌した。2時間後、反応液を室温まで冷却し、1N 塩酸を加えpH=5とした後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物にジメチルホルムアミドジメチルアセタール(2.8mL,21mmol)を加え90℃で撹拌した。2時間後、反応液を室温まで冷却し、濃縮した。得られた粗生成物をエタノール(28mL)に溶解し、フェニルヒドラジン(0.87g,8.1mmol)を加え80℃で撹拌した。5時間後、反応液を室温まで冷却し、濃縮し水を加えた後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=93/7〜79/21)で精製し、表題化合物0.80g(46%)を黄色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:0.00(6H,s),0.57(6H,s),0.81(9H,s),1.38(3H,t,J=7.2Hz),3.22(1H,s),3.22(2H,s),4.31(2H,q,J=7.2Hz),7.38−7.50(5H,m),8.06(1H,s).
MS(ESI)[M+H]+
417
【0158】
〔第4工程〕
エチル 1−フェニル−5−(2,2−ジメチル−3−ヒドロキシプロピル)−1H−ピラゾール−4−カルボキシレートの合成:
【化38】
エチル 1−フェニル−5−(2,2−ジメチル−3−(tert−ブチルジメチルシリルオキシ)プロピル)−1H−ピラゾール−4−カルボキシレート(0.80g,1.9mmol)をTHF(10mL)に溶解し、TBAF(1.0N,3.8mL,3.8mmol)を加え室温で撹拌した。16時間後、水を加えた後、酢酸エチルで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=93/7〜66/34)で精製し、表題化合物0.48g(83%)を黄色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:0.60(6H,s),1.39(3H,t,J=7.2Hz),3.03(2H,d,J=7.2Hz),3.12−3.19(2H,m),4.35(2H,q,J=7.2Hz),7.37−7.41(2H,m),7.44−7.54(3H,m),8.07(1H,s).
MS(ESI)[M+H]+
303
【0159】
〔第5工程〕
エチル 1−フェニル−5−(2,2−ジメチル−3−オキソプロピル)−1H−ピラゾール−4−カルボキシレートの合成:
【化39】
エチル 1−フェニル−5−(2,2−ジメチル−3−ヒドロキシプロピル)−1H−ピラゾール−4−カルボキシレート(480mg,1.6mmol)をジクロロメタン(16mL)に溶解し、Dess−Martin試薬(680mg,1.6mmol)を加え室温で撹拌した。2時間後、反応液をセライト濾過し、濾液を濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=96/4〜69/31)で精製し、表題化合物0.34g(72%)を無色液体として得た。
1H−NMR(400MHz,CDCl3)
δ:0.83(6H,s),1.37(3H,t,J=7.2Hz),3.42(2H,s),4.31(2H,q,J=7.2Hz),7.36−7.40(2H,m),7.46−7.54(3H,m),8.05(1H,m),9.22(1H,s).
MS(ESI)[M+H]+
301
【0160】
〔第6工程〕
実施例12の化合物の合成:
【化40】
エチル 1−フェニル−5−(2,2−ジメチル−3−オキソプロピル)−1H−ピラゾール−4−カルボキシレート(340mg,1.1mmol)をクロロホルム(11mL)に溶解し、trans−5−ヒドロキシ−2−アダマンチルアミン(190mg,1.1mmol)、酢酸(0.19mL,3.4mmol)、水素化トリアセトキシホウ素ナトリウム(640mg,3.4mmol)を加え室温で撹拌した。16時間後、飽和炭酸水素ナトリウム水溶液を加え中和した後、クロロホルムで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。得られた粗生成物をメタノール(2.0mL)に溶解し、1N 水酸化ナトリウム(1.7mL,1.7mmol)を加え室温で撹拌した。16時間後、濃縮した。得られた粗生成物をクロロホルム(11mL)に溶解し、ジイソプロピルエチルアミン(0.59mL,3.4mmol)、HBTU(270mg,1.1mmol)を加え室温で撹拌した。16時間後、水を加えた後、クロロホルムで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムにより乾燥し、濃縮した。この際得られた固形物を酢酸エチル/ヘキサン=1/1溶液によりスラリー洗浄し、表題化合物100mg(22%)を白色固体として得た。
1H−NMR(400MHz,CDCl3)
δ:1.08(6H,s),1.56−1.62(2H,m),1.76−1.83(4H,m),1.88−1.98(2H,m),2.00−2.08(2H,m),2.20−2.27(1H,m),2.43−2.49(2H,m),2 64(2H,s),3.49(2H,s),4.42−4.46(1H,m),7.40−7.53(5H,m),8.16(1H,s).
MS(ESI)[M+H]+
406
【0161】
(実施例13) 1−エチル−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8,8−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例13の化合物」)の合成:
参考例1及び3並びに実施例2と同様の手順により、以下の実施例13の化合物を合成した。
【化41】
1H−NMR(400MHz,CDCl3)
δ:1.56(6H,s),1.57−1.59(2H,m),1.76−1.82(6H,m),1.95−2.03(4H,m),2.18−2.24(1H,m),2.44−2.50(2H,m),3.63−3.68(2H,m),4.20−4.29(3H,m),8.02(1H,s).
MS(ESI)[M+H]+
358
【0162】
(実施例14) 1−シクロヘキシル−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8,8−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例14の化合物」)の合成:
参考例1及び3並びに実施例2と同様の手順により、以下の実施例14の化合物を合成した。
【化42】
1H−NMR(400MHz,CDCl3)
δ:1.32−1.37(2H,m),1.40(6H,s),1.70−2.12(19H,m),2.18−2.23(1H,m),2.44−2.50(2H,m),3.53−3.58(2H,m),4.16−4.28(2H,m),8.02(1H,s).
MS(ESI)[M+H]+
412
【0163】
(実施例15) 1−(ピリジン−2−イル)−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8,8−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例15の化合物」)の合成:
参考例1及び3並びに実施例2と同様の手順により、以下の実施例15の化合物を合成した。
【化43】
1H−NMR(400MHz,CDCl3)
δ:1.24(6H,s),1.56−1.62(2H,m),1.77−1.86(6H,m),1.98−2.04(4H,m),2.20−2.25(1H,m),2.48−2.54(2H,m),3.62−3.67(3H,m),4.30−4.33(1H,m),7.40−7.46(2H,m),7.89(1H,ddd,J=2.0,7.6,8.0Hz),8.18(1H,s),8.57−8.60(1H,m).
MS(ESI)[M+H]+
407
【0164】
(実施例16) 1−(2−メチルフェニル)−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8,8−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例16の化合物」)の合成:
参考例1及び3並びに実施例2と同様の手順により、以下の実施例16の化合物を合成した。
【化44】
1H−NMR(400MHz,CDCl3)
δ:0.98(3H,s),1.25(3H,s),1.55−1.63(2H,m),1.75−1.86(6H,m),1.96−2.03(7H,m),2.19−2.25(1H,m),2.45−2.55(2H,m),3.58−3.65(2H,m),4.30−4.35(1H,m),7.26−7.42(4H,m),8.16(1H,s).
MS(ESI)[M+H]+
420
【0165】
(実施例17) 1−(3−メチルフェニル)−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8,8−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例17の化合物」)の合成:
参考例1及び3並びに実施例2と同様の手順により、以下の実施例17の化合物を合成した。
【化45】
1H−NMR(400MHz,CDCl3)
δ:1.15(6H,s),1.55−1.63(2H,m),1.78−1.85(6H,m),1.99−2.05(4H,m),2.19−2.25(1H,m),2.40(3H,s),2.48−2.52(2H,m),3.58−3.65(2H,m),4.30−4.34(1H,m),7.15−7.20(2H,m),7.27−7.37(2H,m),8.10(1H,s).
MS(ESI)[M+H]+
420
【0166】
(実施例18) 1−(4−メチルフェニル)−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8,8−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例18の化合物」)の合成:
参考例1及び3並びに実施例2と同様の手順により、以下の実施例18の化合物を合成した。
【化46】
1H−NMR(400MHz,CDCl3)
δ:1.14(6H,s),1.40−1.47(1H,m),1.56−1.62(1H,m),1.76−1.85(1H,m),1.76−1.85(6H,m),1.96−2.05(4H,m),2.20−2.25(1H,m),3.59−3.64(2H,m),4.30−4.33(1H,m),7.36−7.40(2H,m),7.44−7.52(3H,m),8.12(1H,s).
MS(ESI)[M+H]+
420
【0167】
(実施例19) 1−(3−フルオロフェニル)−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8,8−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例19の化合物」)の合成:
参考例1及び3並びに実施例2と同様の手順により、以下の実施例19の化合物を合成した。
【化47】
1H−NMR(400MHz,CDCl3)
δ:1.17(6H,s),1.56−1.60(2H,m),1.78−1.82(6H,m),1.99−2.01(4H,m),2.20−2・24(1H,m),2.48−2.52(2H,brs),3.60−3.62(2H,m),4.31(1H,s),7.13−7.24(3H,m),7.42−7.48(1H,m),8.12(1H,s).
MS(ESI)[M+H]+
424
【0168】
実施例20 1−(4−フルオロフェニル)−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8,8−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例20の化合物」)の合成:
参考例1及び3並びに実施例2と同様の手順により、以下の実施例20の化合物を合成した。
【化48】
1H−NMR(400MHz,CDCl3)
δ:1.14(6H,s),1.55−1.60(2H,m),1.78−1.82(6H,m),1.98−2.03(4H,m),2.20−2・24(1H,m),2.47−2.51(2H,m),3.60−3.62(2H,m),4.32(1H,s),7.14−7.19(2H,m),7.35−7.39(2H,m),8.11(1H,s).
MS(ESI)[M+H]+
424
【0169】
(実施例21) 1−(3−クロロフェニル)−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8,8−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例21の化合物」)の合成:
参考例1及び3並びに実施例2と同様の手順により、以下の実施例21の化合物を合成した。
【化49】
1H−NMR(400MHz,CDCl3)
δ:1.14(6H,s),1.56−1.63(2H,m),1.78−1.82(6H,m),1.98−2.01(4H,m),2.20−2.23(1H,m),2.49(2H,brs),3.60−3.62(2H,m),4.31(1H,s),7.27−7.31(1H,m),7.40−7.51(3H,m),8.12(1H,s).
MS(ESI)[M+H]+
440
【0170】
(実施例22) 1−(4−クロロフェニル)−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8,8−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例22の化合物」)の合成:
参考例1及び3並びに実施例2と同様の手順により、以下の実施例22の化合物を合成した。
【化50】
1H−NMR(400MHz,CDCl3)
δ:1.14(6H,s),1.55−1.60(2H,m),1.78−1.82(6H,m),1.98−2.01(4H,m),2.20−2・24(1H,m),2.47−2.51(2H,m),3.60−3.62(2H,m),4.29−4.33(1H,m),7.34(2H,d,J=8.8Hz),7.47(2H,d,J=8.8Hz),8.12(1H,s).
MS(ESI)[M+H]+
440
【0171】
(実施例23) 1−(3−トリフルオロメチルフェニル)−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8,8−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例23の化合物」)の合成:
参考例1及び3並びに実施例2と同様の手順により、以下の実施例23の化合物を合成した。
【化51】
1H−NMR(400MHz,CDCl3)
δ:1.14(6H,s),1.54−2.05(12H,m),2.21−2.25(1H,m),2.50−2.53(2H,m),3.60−3.64(2H,m),4.30−4.34(1H,m),7.60−7.68(3H,m),7.78−7.80(1H,m),8.14(1H,s).
MS(ESI)[M+H]+
474
【0172】
(実施例24) 1−(4−トリフルオロメチルフェニル)−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8,8−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例24の化合物」)の合成:
参考例1及び3並びに実施例2と同様の手順により、以下の実施例24の化合物を合成した。
【化52】
1H−NMR(400MHz,CDCl3)
δ:1.15(6H,s),1.59−1.62(2H,m),1.77−1.85(6H,m),1.99−2.02(4H,m),2.20−2.25(1H,m),2.48−2.53(2H,m),3.60−3.65(2H,m),4.30−4.33(1H,m),7.53(2H,d,J=8.4Hz),7.77(2H,d,J=8.4Hz),8.14(1H,s).
MS(ESI)[M+H]+
474
【0173】
(実施例25) 1−(2−メトキシフェニルフェニル)−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8,8−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例25の化合物」)の合成:
参考例1及び3並びに実施例2と同様の手順により、以下の実施例25の化合物を合成した。
【化53】
1H−NMR(400MHz,CDCl3)
δ:1.03(3H,s),1.18(3H,s),1.52−1.60(2H,m),1.76−2.04(10H,m),2.20−2.24(1H,m),2.46−2.54(2H,m),3.59−3.63(2H,m),3.77(3H,s),4.30−4.33(1H,m),6.98−7.06(2H,m),7.35(1H,dd,J=1.7,7.6Hz),7.44−7.49(1H,m),8.16(1H,s).
MS(ESI)[M+H]+
436
【0174】
(実施例26) 1−(4−メトキシフェニル)−5−(trans−5−ヒドロキシアダマンタン−2−イル)−8,8−ジメチル−5,6,7,8−テトラヒドロピラゾロ[4,3−c]アゼピン−4(1H)−オン(以下、「実施例26の化合物」)の合成:
参考例1及び3並びに実施例2と同様の手順により、以下の実施例26の化合物を合成した。
【化54】
1H−NMR(400MHz,CDCl3)
δ:1.15(6H,s),1.40−2.02(13H,m),2.18−2.22(1H,m),2.49−2.52(2H,m),3.59−3.63(2H,m),3.87(3H,s),4.19−4.22(1H,m),6.95(2H,d,J=8.8Hz),7.28(2H,d,J=8.8Hz),8.10(1H,s).
MS(ESI)[M+H]+
436
【0175】
(実施例27) 11β−HSD1阻害実験:無細胞評価系:
ヒト11β−HSD1遺伝子発現ベクターをHEK293H細胞(インビトロジェン社)に一過性導入して調製した細胞抽出液(以下、「細胞ライセート」)を用いて、本発明の複素環化合物又はその薬学的に許容される塩(以下、「実施例の化合物群」)の11β−HSD1阻害作用を評価した。
【0176】
1.試験方法
a.酵素源
I.M.A.G.Eクローン(クローンID:5193867、インビトロジェン社)より調製したヒト11β−HSD1のcDNA配列を含むプラスミドDNA(pCMV−SPORT6ベクターに挿入されている)を鋳型にして、ヒト11β−HSD1のcDNA配列を含むDNA断片をPCRで増幅し、pTA2(東洋紡社)にサブクローニングした。塩基配列を解析して変異の無いことを確認したポジティブクローンを制限酵素XhoI及びXbaIで切断し、ヒト11β−HSD1のcDNA配列を含むDNA断片をpCI−neo(プロメガ社)のXhoI、XbaI開裂部位に挿入し、ヒト11β−HSD1遺伝子発現ベクターh11β−HSD1/pCI−neoを得た。
【0177】
細胞ライセートの調製は以下の方法で行った。HEK293H細胞の細胞数をカウントし、2×106cells/5mL/6cm dishとなるように播種し、37℃、5%CO2インキュベーターで一晩培養した。細胞の培養液を交換(2mL培養液/dish)した後、h11β−HSD1/pCI−neoをFuGENE HD Transfection Reagent(ロシュ・アプライド・サイエンス社)を用いて細胞にトランスフェクションした。トランスフェクションは試薬添付の調製方法に従って行った。細胞をPBSで縣濁後、遠心して得た細胞塊を液体窒素で凍結した。細胞塊を6cm dish1枚当たり200μLの溶解バッファー(0.25mol/L sucrose、1mmol/L EDTA・2Na、10mmol/L Tris−HCl(pH7.4)、1% protease inhibitor cocktail)に縣濁し、凍結融解を3回繰り返し行った。このようにして得られたヒト11β−HSD1発現細胞ライセートは、protein assay(バイオ・ラッド社)を用いてBradford法によりタンパク質定量を行い、10%グリセロールとなるようにグリセロールを添加して、−80℃にて使用時まで凍結保存した。
【0178】
b.酵素阻害実験
被験化合物は、DMSOにて溶解、種々の濃度に希釈後、さらにアッセイバッファー(20mmol/L HEPES(pH6.0)、5mmol/L EDTA、0.1%BSA)にて希釈した(DMSO最終濃度:1%)。アッセイバッファーにて最終濃度の2.67倍に希釈したヒト11β−HSD1発現細胞ライセート(最終タンパク質濃度 100〜200μg/mL)12μLを96ウェルV底プレートに添加し、そこにアッセイバッファー又は希釈した被験化合物溶液4μLを添加して、37℃で10分間プレインキュベーションした。384ウェルブラックプレート(コーニング社)に、アッセイバッファーにて希釈した補酵素NADPH(シグマ社)、基質コルチゾン(シグマ社)の等量混合液5μLを添加し、さらに、プレインキュベーションした細胞ライセート希釈液5μLを添加し、37℃にて2時間インキュベーションした。反応により生成されたコルチゾールは、HTRFコルチゾールキット(シスバイオ社)を用いて測定した。すなわち、384ウェルブラックプレートの各ウェルにコルチゾールと競合するコルチゾール−d2、及びクリプテート標識抗コルチゾールモノクローナル抗体を各5μL添加し、遮光下、室温にて2時間インキュベーションした。その後、反応液の蛍光(励起波長340nm、蛍光波長615nm及び665nm)をマルチラベルカウンター(Arvo SX;パーキンエルマー社)を用いて測定した。コルチゾール検量線を作成し、反応液中の基質コルチゾンから変換されたコルチゾール濃度を算出した。
【0179】
測定結果より被験化合物の各濃度について阻害率(% of inhibition)を求めた。以下に阻害率の求め方を示す。
Ratio=(A665nm/B615nm)×104
A665nm:665nmにおける測定値
B615nm:615nmにおける測定値
阻害率(% of inhibition)=((被験化合物のRatio−被験化合物非添加のRatio)/(細胞ライセート非添加かつ被験化合物非添加のRatio−被験化合物非添加のRatio))×100
【0180】
2.結果
11β−HSD1活性を50%阻害する化合物濃度をIC50値として算出した。結果を表9に示す。表9に示した実施例の化合物群は、強力な11β−HSD1阻害活性を示した。一方、比較対照化合物として用いた比較例1の化合物は僅かな11β−HSD1阻害活性しか示さなかった。
【0181】
【表9】
【0182】
(実施例28) 病態モデルでのインスリン抵抗性改善作用及び体重への影響:
マウスDIO(Diet induced−obesity)モデル(以下、「DIOマウス」)を用いて、実施例の化合物群のインスリン抵抗性改善作用を評価した(Reuter、Drug Discovery Today:Disease Models、2007年、4巻、p.3−8)。
【0183】
1.試験方法
C57BL/6Nマウス(雄性;日本チャールス・リバー社)を4週齢で入荷し、1週間通常飼料(CRF−1)で飼育した後、60%脂肪を含む固形飼料(D12492;リサーチダイエット社)に切り替え、12〜13週間給餌・飼育したものをDIOマウスとして実験に用いた。入荷以降、通常飼料を継続給餌したマウスを正常動物(正常群)とした。
【0184】
実施例の化合物群及びピオグリタゾン塩酸塩は、瑪瑙乳鉢を用いて0.5%メチルセルロース(以下、「MC」)にて3mg/mLとなるよう懸濁した。懸濁調製した投与薬液をマウスに、ディスポーザブルシリンジ及び経口投与用ゾンデを用いて、14日間(投与開始日を0日目として、0日目〜13日目の間)、1日1回、10mL/kgの容量で無麻酔下強制経口投与した。対照となる、DIOマウスの溶媒投与群(溶媒群)及び正常群には0.5%MCを投与した。なお、DIOマウスの群分けは投与開始前日(−1日目)の時点で各群の体重が均等になるように実施した。
【0185】
14日目に、予め絶食用5連ケージ内で一晩絶食させたマウスの尾静脈血を採取し、その血糖値(空腹時血糖値)を簡易型血糖測定装置(メディセンス・プレシジョンエクシード;アボットジャパン社)を用いて測定した。
【0186】
空腹時血糖値測定後、マウスのインスリン抵抗性評価のため、インスリン負荷試験(ITT)を実施した。100unit/mLの市販インスリン溶液(ヒューマリンR注;イーライ・リリー社)を0.1%BSA含有生理食塩水で0.06unit/mLに希釈して調製したインスリン溶液を、マウスに腹腔内投与(0.3unit/kg)した。インスリン投与前(0分)及びインスリン投与後30、60、120分に、上記と同様の方法で血糖値を測定した。
【0187】
実施例の化合物群及びピオグリタゾン塩酸塩の、体重への影響は、投与最終日(13日目)時点での、投与開始日からの体重変化量(g)によって評価した。
【0188】
2.結果
実施例の化合物群を代表する、実施例10の化合物及び実施例22の化合物の評価結果を図1〜6、表10及び11に示す。空腹時血糖値に対する作用を図1及び2に、インスリン負荷試験における作用を図3及び4、表10及び11に、体重に対する影響を図5及び6に示す。図表中の「溶媒」はDIOマウスに0.5%MCを投与した群(溶媒群)を示し、「実施例10の化合物」はDIOマウスに実施例10の化合物を30mg/kg/day投与した群を示し、「実施例22の化合物」はDIOマウスに実施例22の化合物を30mg/kg/day投与した群を示し、「ピオグリタゾン」はDIOマウスにピオグリタゾン塩酸塩を30mg/kg/day投与した群を示し、「正常」は正常動物に0.5%MCを投与した群(正常群)を示す。図表の各値は5〜8匹/群の平均値±標準誤差で示す。
【0189】
【表10】
【0190】
【表11】
【0191】
図1及び2に示すように、空腹時血糖値は、実施例10の化合物及び実施例22の化合物を投与した群において、溶媒群より低値で、且つピオグリタゾン投与群と同等の値を示した。この結果から、実施例の化合物群は、空腹時血糖低下作用を有することが明らかになった。
【0192】
図3及び4に示すように、インスリン投与後の血糖値は、実施例10の化合物及び実施例22の化合物を投与した群において、溶媒群より低値で、且つピオグリタゾン投与群と同等の推移を示した。また、表10及び11に示すように、血糖値AUCは、実施例10及び22の化合物投与群とピオグリタゾン投与群で同等の値であった。これらの結果から、実施例の化合物群は、インスリン抵抗性改善作用を有することが明らかになった。
【0193】
図5及び6に、投与開始後13日目時点での投与開始日(0日目)からの体重変化量を示す。実施例10の化合物及び実施例22の化合物を投与した群の体重変化量は、溶媒群より低値であり、溶媒群より高値を示したピオグリタゾン投与群とは対照的であった。この結果から、実施例の化合物群には、体重増加の副作用がないことが明らかになった。
【産業上の利用可能性】
【0194】
本発明の複素環化合物又はその薬学的に許容される塩は、顕著な11β−HSD1阻害活性を示すとともに副作用が軽減されているため、医薬の分野において、糖尿病の治療薬及び予防薬並びにインスリン抵抗性に対する改善薬として用いることができる。
【特許請求の範囲】
【請求項1】
以下の一般式(I)
【化1】
[式中、R1は、炭素数1〜6のアルキル、炭素数3〜10のシクロアルキル、環構成炭素数6〜10のアリール又は環構成原子数5〜10のヘテロアリール(ここで、該アリール及び該ヘテロアリールは、それぞれ独立して、1以上のハロゲンによって置換されていてもよい炭素数1〜6のアルキル、又は、炭素数1〜6のアルキルオキシ若しくはハロゲン、によって置換されていてもよい)を示し、R2〜R5は、それぞれ独立して、炭素数1〜6のアルキル又は水素原子を示し、R6は、ヒドロキシ又は水素原子を示す。]
で表される、複素環化合物又はその薬学的に許容される塩。
【請求項2】
R1は、炭素数1〜6のアルキル、環構成炭素数6〜10のアリール又は環構成原子数5〜10のヘテロアリール(ここで、該アリール及び該ヘテロアリールは、それぞれ独立して、1以上のハロゲンによって置換されていてもよい炭素数1〜6のアルキル、炭素数1〜6のアルキルオキシ又はハロゲンによって置換されていてもよい)を示す、請求項1記載の複素環化合物又はその薬学的に許容される塩。
【請求項3】
R1は、フェニル又は3−クロロフェニルを示す、請求項2記載の複素環化合物又はその薬学的に許容される塩。
【請求項4】
R2〜R5は、それぞれ独立して、メチル又は水素原子を示す、請求項1〜3のいずれか一項記載の複素環化合物又はその薬学的に許容される塩。
【請求項5】
R2及びR3は、メチル基、R4及びR5は、水素原子を示す、請求項1〜4のいずれか一項記載の複素環化合物又はその薬学的に許容される塩。
【請求項6】
R6は、ヒドロキシを示す、請求項1〜5のいずれか一項記載の複素環化合物又はその薬学的に許容される塩。
【請求項7】
請求項1〜6のいずれか一項記載の複素環化合物又はその薬学的に許容される塩を有効成分として含有する、11β−ヒドロキシステロイドデヒドロゲナーゼ1阻害剤。
【請求項8】
請求項1〜6のいずれか一項記載の複素環化合物又はその薬学的に許容される塩を有効成分として含有する、糖尿病の治療薬又は予防薬。
【請求項9】
前記糖尿病は、2型糖尿病である、請求項8記載の治療薬又は予防薬。
【請求項10】
請求項1〜6のいずれか一項記載の複素環化合物又はその薬学的に許容される塩を有効成分として含有する、インスリン抵抗性の改善薬。
【請求項1】
以下の一般式(I)
【化1】
[式中、R1は、炭素数1〜6のアルキル、炭素数3〜10のシクロアルキル、環構成炭素数6〜10のアリール又は環構成原子数5〜10のヘテロアリール(ここで、該アリール及び該ヘテロアリールは、それぞれ独立して、1以上のハロゲンによって置換されていてもよい炭素数1〜6のアルキル、又は、炭素数1〜6のアルキルオキシ若しくはハロゲン、によって置換されていてもよい)を示し、R2〜R5は、それぞれ独立して、炭素数1〜6のアルキル又は水素原子を示し、R6は、ヒドロキシ又は水素原子を示す。]
で表される、複素環化合物又はその薬学的に許容される塩。
【請求項2】
R1は、炭素数1〜6のアルキル、環構成炭素数6〜10のアリール又は環構成原子数5〜10のヘテロアリール(ここで、該アリール及び該ヘテロアリールは、それぞれ独立して、1以上のハロゲンによって置換されていてもよい炭素数1〜6のアルキル、炭素数1〜6のアルキルオキシ又はハロゲンによって置換されていてもよい)を示す、請求項1記載の複素環化合物又はその薬学的に許容される塩。
【請求項3】
R1は、フェニル又は3−クロロフェニルを示す、請求項2記載の複素環化合物又はその薬学的に許容される塩。
【請求項4】
R2〜R5は、それぞれ独立して、メチル又は水素原子を示す、請求項1〜3のいずれか一項記載の複素環化合物又はその薬学的に許容される塩。
【請求項5】
R2及びR3は、メチル基、R4及びR5は、水素原子を示す、請求項1〜4のいずれか一項記載の複素環化合物又はその薬学的に許容される塩。
【請求項6】
R6は、ヒドロキシを示す、請求項1〜5のいずれか一項記載の複素環化合物又はその薬学的に許容される塩。
【請求項7】
請求項1〜6のいずれか一項記載の複素環化合物又はその薬学的に許容される塩を有効成分として含有する、11β−ヒドロキシステロイドデヒドロゲナーゼ1阻害剤。
【請求項8】
請求項1〜6のいずれか一項記載の複素環化合物又はその薬学的に許容される塩を有効成分として含有する、糖尿病の治療薬又は予防薬。
【請求項9】
前記糖尿病は、2型糖尿病である、請求項8記載の治療薬又は予防薬。
【請求項10】
請求項1〜6のいずれか一項記載の複素環化合物又はその薬学的に許容される塩を有効成分として含有する、インスリン抵抗性の改善薬。
【図1】

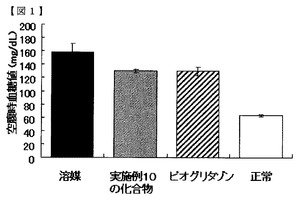
【図2】
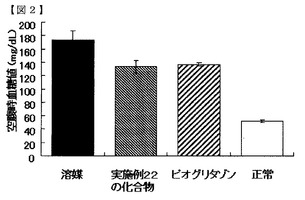
【図3】
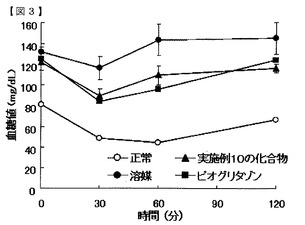
【図4】
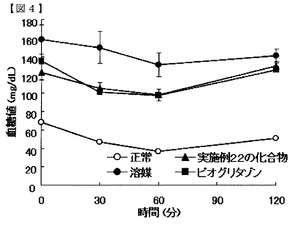
【図5】
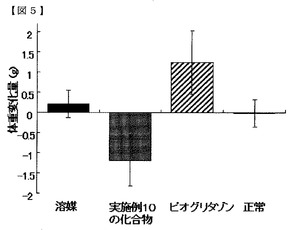
【図6】
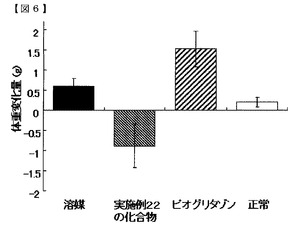
【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2013−28576(P2013−28576A)
【公開日】平成25年2月7日(2013.2.7)
【国際特許分類】
【出願番号】特願2011−167539(P2011−167539)
【出願日】平成23年7月29日(2011.7.29)
【出願人】(000003159)東レ株式会社 (7,677)
【Fターム(参考)】
【公開日】平成25年2月7日(2013.2.7)
【国際特許分類】
【出願日】平成23年7月29日(2011.7.29)
【出願人】(000003159)東レ株式会社 (7,677)
【Fターム(参考)】
[ Back to top ]