
複製欠損アデノウイルスベクターの製造のためのアデノウイルスアンプリコンおよびプロデューサー細胞、その製造方法および用途
本発明は、非構造および構造遺伝子の複数の欠失を含有するヘルパー非依存性アデノウイルスベクターの製造のための効率的プロデューサー細胞系の開発に使用しうるプラスミドに関する。より詳しくは、本発明は、多欠失アデノウイルスベクターを相補し高力価調製物を得るために使用しうる新規アデノウイルスアンプリコンを含むプロデューサー細胞を提供する。該アンプリコンは、左および右ITRの共有結合の形態の、Ad5 E2ウイルス遺伝子(すなわち、ポリメラーゼ、プレ末端タンパク質およびDNA結合タンパク質)およびE4 orf6、EBV潜在性複製起点(OriP)ならびにアデノウイルス複製起点を発現するエピソームプラスミドである。このプラスミドはAd5 E2遺伝子発現の誘導に際して自己複製可能である。本発明は更に、開示されているプロデューサー細胞の製造方法、および治療用途に十分な規模でウイルスベクターを製造するための、該細胞の使用を含む。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、分子生物学の分野に関し、特に、多欠失または完全欠失ヘルパー非依存性アデノウイルスベクターの高クローニング容量プロデューサー細胞系を製造するための、誘導可能な自己複製能を有するエピソームプラスミドの開発および使用に関する。
【背景技術】
【0002】
アデノウイルス(Ad)は、多種多様な組織の休止細胞および増殖細胞の両方に感染しうる広範な指向性により特徴づけられる。一般に、野生型ヒトAd5ウイルスによる許容細胞の感染は約104〜105個のウイルス粒子の産生を引き起こす。ウイルスゲノムの扱い易さと共に、高力価増殖能は、Adベクターを、ワクチン接種および遺伝子治療のための並びに細胞培養内での遺伝子発現のための遺伝子導入ベクターとしての使用に魅力的なものにする。
【0003】
安全性プロファイルの改善(例えば、ウイルス遺伝子発現から生じる毒性の最小化)および前世代のベクターのクローニング容量の増加を目的として、ヒトAd5およびAd2に基づく幾つかのベクター系が開発されている。代替ベクター系の開発のための方法は、典型的には、ベクターバックボーンからアデノウイルス遺伝子を欠失させることを含む。アデノウイルスゲノムは機能的には、非構造産物および構造産物をコードする遺伝子を含む、初期領域および後期領域に細分される。第1の領域は、ウイルスDNA複製の前に発現されるポリペプチドをコードする初期(E)遺伝子を含む。第2の領域は、ウイルス複製の後続段階で必要なポリペプチドをコードする後期(L)遺伝子を含む。アデノウイルスゲノムのL領域は本質的には、ウイルス粒子の集合に必要な構造タンパク質をコードする。
【0004】
コンピテント細胞の感染後転写される第1の領域は、E遺伝子およびL遺伝子の両方のトランス活性化に関与するタンパク質をコードするE1a領域である。続いて転写されるE1b領域は、RNA合成を調節し、E1aによるアポトーシス作用から宿主細胞を防御するポリペプチドをコードする。したがって、E1a/E1bの遺伝子/機能はウイルス複製に必須である。第1世代(FG)アデノウイルスベクターは、典型的に、アデノウイルスE1遺伝子内の欠失を含む。これらの欠失は、修飾転写単位のタンパク質産物がトランスで与えられない限り、アデノウイルスを複製欠損にする。一般に、FGアデノウイルスベクターの最高容量は8kbを超えない。FG Ad5ベクターはE1領域の欠失または修飾により弱毒化されるが、一般に、いくつかの腫瘍細胞系内での複製能の維持および漏出遺伝子発現の両方の結果として、インビトロで細胞毒性が見られる。典型的に、FG Adベクターでのインビボ形質導入は比較的短いトランスジーン発現をもたらす。
【0005】
追加的なウイルス遺伝子の欠失に基づく第2および第3世代ベクター系はアデノウイルス遺伝子発現の更なる弱毒化およびベクター容量の増加をもたらす。より詳しくは、より新しい世代のベクターはウイルスE2、E3および/またはE4遺伝子内の追加的な欠失を含む。ΔE1/E3/E4ベクターのクローニング容量は約11kbに近い。E2領域は、ウイルスDNAポリメラーゼ、プレ末端タンパク質およびウイルスDNAに結合するタンパク質を含むウイルス複製に直接関与するタンパク質をコードする。E3領域は、ウイルス複製には要求されないが宿主免疫応答を制御するようインビボで機能するタンパク質をコードすることが公知である。E4領域遺伝子は、宿主細胞の遺伝子発現を低下させアデノウイルスゲノムのE2およびL領域の転写を増強するよう機能するポリペプチドをコードする。種々の組合せのE1、E2a/b、E3および/またはE4欠失を有する多欠失ベクターの使用は、古典的なFG(2−4,23,24,33,45,52)ベクターほどはインビトロで細胞毒性ではなく、マウス肝臓において、より安定であることが認められている。しかし、より新しい世代のアデノウイルスベクターが有意に長い持続能を有するという決定的な証拠は存在しない。さらに、追加的な欠失の導入は、生じる力価を有意に低下させ、臨床応用のための該ベクターの大規模製造をより困難にする(33,18)。実際、ほとんど全ての場合に、パッケージング/プロデューサー細胞系内に安定に導入された相補遺伝子の発現は、複数の欠失を相補しなければならない場合非効率的である(5,54)。
【0006】
現在のところ、ヘルパー依存性(HD)完全欠失アデノウイルスベクター遺伝子が、インビボ遺伝子導入のための最も効率的で安全なベクターの1つとみなされている(5,15,28,36,39−41,43,54)。完全欠失Adベクターは、複製およびパッケージング(すなわち、包膜)に必要なシス要素のみを含有し、すべてのアデノウイルス遺伝子を欠く。伝統的には、必要なアデノウイルス遺伝子はヘルパーウイルスによりトランスで供与される。しかし、HDベクターはいくつかの欠点により特徴づけられる。なかでも、該系は、トランスジーンを含有するHDベクターと必要なウイルスタンパク質をトランスで供与するヘルパーウイルスとによるパッケージング細胞系の共感染を要するため、3つの独立した成分の制御を要することが挙げられる。実際、医薬規模でのヘルパー依存性アデノウイルスベクターの生産は、克服するのが困難な問題を伴い、高すぎる生産コストを伴う。また、ヘルパーウイルスの使用はほとんど常に、HDベクター製剤を汚染する。
【0007】
E2遺伝子および/またはE4領域のいくつかを欠失させ異なる初期遺伝子の欠失を組合せることにより、多欠失ヘルパー非依存性Adベクターも構築されている(2−4,23,24,33,45,52)。典型的に、必要な相補遺伝子を並列で相補パッケージング細胞系内に安定的に導入する。しかし、この方法は低コピー数のウイルス遺伝子の染色体組み込みを要し、多数の欠失を相補しなければならない場合非効率的となりうる。Andrews J.L.ら(5)は、E1、E2a、E3およびE4領域が欠失したベクターは高い力価までは増殖し得ないことを示した。Zhou H.ら(54)は、第1世代アデノウイルスベクターにより通常達する力価に近い力価でE1/E2a欠失ベクターを効率的に増殖させるためにDBP遺伝子の複数の組込みコピーが必要であることを示した。
【発明の開示】
【発明が解決しようとする課題】
【0008】
効率的なパッケージング/プロデューサー細胞系の開発は、ヘルパー非依存性アデノウイルスベクターの開発に関連した最も困難な課題の1つである。したがって、アデノウイルス由来ベクターの絶えざる開発および使用のための重要な要件は、多または完全欠失アデノウイルスベクターの高力価調製物の製造を促進するヘルパー非依存性プロデューサー細胞系の設計である。理想的な解決策は、完全欠失ヘルパー非依存性アデノウイルスベクターの高力価増殖に適したヘルパーまたはプロデューサー細胞系を使用するアデノウイルスベクター系の開発であろう。
【課題を解決するための手段】
【0009】
発明の概要
本発明は、哺乳類細胞の核内での誘導性自己複製能を有する、本明細書においてはアデノウイルスアンプリコンまたはレプリコンと称されるエピソームプラスミドを提供する。開示するアデノウイルスアンプリコンは以下の特性により特徴づけられる。(i)これはEBV潜伏性複製起点(oriP)およびヒトAd5逆末端反復(ITR)結合部を含有する。(ii)これは、全3個のアデノウイルス5型初期領域2(E2)遺伝子および初期領域4(E4)ORF6を、Tet依存性プロモーターの制御下、誘導可能に発現する。本明細書中に示すとおり、Tet転写サイレンサー(tTS)および逆Tetトランスアクチベーター(rtTA2)を発現する293EBNA細胞を形質転換するために、開示するアンプリコンを使用した場合、得られた安定な細胞系(2E2)は、ドキシサイクリンの存在下、第1世代Adベクターに感染した293細胞より高いレベルのポリメラーゼ、前駆体末端タンパク質(pTP)およびDNA結合タンパク質(DBP)を産生した。本明細書に記載のデータは更に、本明細書中に開示するプロデューサー細胞系(すなわち、2E2)が、多欠失ΔE1,E2,E3,E4 Adベクターの増殖のために使用しうることを証明している。したがって、開示するAd/EBVアンプリコンは、多または完全欠失アデノウイルスベクターの高力価増殖に適した効率的なヘルパー細胞系の産生に対する重要な寄与をもたらす。
【0010】
本発明の第1の態様は、(a)EBNA−1タンパク質を発現する分裂細胞の核内のアンプリコンの維持を促すEBV由来複製起点(Ori−P)、(b)Adに基づく様態での増幅を可能にするAd5ウイルスITR結合部の形態のAd5複製起点、(c)Ad5由来ポリメラーゼおよびプレ末端タンパク質をコードする核酸配列よりなる第1転写単位、(d)Ad5 DNA結合タンパク質およびE4 ORF6をコードする核酸配列よりなる第2転写単位、ならびに(e)選択マーカーを含んでなり、第1転写単位および第2転写単位が二方向性テトラサイクリン依存性プロモーターに融合している、アデノウイルスアンプリコンを提供する。特定の実施形態においては、本発明はAd5 E2/E4 ORF6アンプリコン,pE2を提供する。
【0011】
もう1つの態様においては、本発明は更に、ブダペスト条約に基づき原寄託物としてBelgian Coordinated Collections of Microorganisms Laboratory of Molecular Biology(BCCM/LMBP,Ghent University,Technologiepark 927,B−9052 Gent−Zwijnaarde,Belgium)Plasmid Collectionに2004年10月15日付けで寄託されたプラスミドのヌクレオチド配列を含んでなるエピソームプラスミドを提供する。該寄託物には受託番号LMBP 4972が付与された。この寄託物は、Budapest Treaty on the International Recognition of the Deposit of Microorganisms for the Purposes of Patent Procedureに基づいて維持される。この寄託は単に当業者にとっての便宜としてなされたものであり、寄託が35 U.S.C.§112に基づいて要求されることを自認するものではない。寄託された物質の公への入手可能性に対する全ての制限は、37 C.F.R.§1.808(b)に規定された要件の場合を除き、特許の付与に際して、変更不能に除かれる。
【0012】
OriP潜伏性複製起点と組合されたEBV核抗原−1(EBNA−1)の存在は、細胞周期当たり1回だけ複製する安定なコピー数での核内保持および自律的エピソーム性複製の機能をもたらす(48)。E4orf6ならびにアデノウイルスDNA複製をもたらすAd5ポリメラーゼ、pTPおよびDBPのコード配列はTetプロモーターの制御下で2つのビ−シストロン転写単位内に配置されるため、Ad/EBVエピソームが転写的にサイレントな場合には。それは潜伏性ウイルス要素として維持される。本明細書に示すとおり、開示されているアンプリコンはE2遺伝子発現の誘導に際して複製されて、コピー数の増加をもたらす。
【0013】
もう1つの実施形態においては、本発明は、プロモーターに融合した関心のあるトランスジーンをコードする発現カセットを更に含むAd5 E2/E4ORF6アンプリコンを含む。関心のあるトランスジーンには、限定的なものではないが例えば免疫グロブリンまたは免疫グロブリンのフラグメント、一本鎖抗体、二重特異性抗体、エリスロポエチン、成長ホルモン、サイトカイン、例えばIL−2およびIL−10関連サイトカイン(例えば、IL−19、IL−20、IL−22、IL−24,IL−26、IL−28およびIL−29遺伝子)のようなタンパク質をコードするヒト遺伝子;HCVのコア、E1、E2または非構造領域、HIVのHIV−1 gp41、GP120、gag、pol、nef、HSV−2糖タンパク質D、HPV Ll、L2、E6およびE7タンパク質、SARS−CoVのスパイク(S)糖タンパク質、細胞膜タンパク質、例えばウイルス受容体、例えばSARS−CoV ACE2受容体、HIV−1受容体CD4およびケモカインコレセプター、HCV受容体CD81、SRB1、L−SIGNおよび硫酸ヘパリンシンデカン、Gタンパク質共役受容体(GPCR)、チロシンキナーゼ細胞表面受容体をコードするウイルス遺伝子が含まれる。
【0014】
本発明の第2の態様は、本発明のアデノウイルスアンプリコンを含んでなるプロデューサー/ヘルパー細胞系を提供する。より詳しくは、本発明は、(a)Ad5 E1タンパク質、(b)EBV由来EBNAタンパク質、(c)Tet転写サイレンサー、(d)Tet逆トランスアクチベーター、(e)アデノウイルスアンプリコン[該アデノウイルスアンプリコンは、第2転写単位(第2転写単位はEBV由来OriP、アデノウイルスITR結合部ならびにAd5 E4 ORF6およびDNA結合タンパク質をコードする核酸配列よりなる。)と組合された第1転写単位(第1転写単位はAd5由来ポリメラーゼおよびプレ末端タンパク質をコードする核酸配列よりなる。)よりなり、第1転写単位および第2転写単位は二方向性テトラサイクリン依存性プロモーターに融合している。]、ならびに(f)選択マーカーを発現するアデノウイルスパッケージング細胞系を提供する。
【0015】
特定の実施形態において、本発明のこの態様は、本明細書においては、293EBNAtet細胞(Tet転写サイレンサーtTSkidおよびtet逆トランスアクチベーターrtTA2を発現する293EBNA細胞として定義される。)をpE2で形質転換して、ΔE1,E2,E3,E4 Adベクターの増殖のためのプロデューサー細胞系としての使用に適した細胞系を得ることにより例示される。本明細書中に例示されているパッケージング細胞系は2E2と称される。開示されている系のAd5 ΔE1,E2,E3,E4 Adベクターは、12.4Kbまでのクローニング容量により、およびウイルス遺伝子発現の漏出の減少により特徴づけられる。本発明のプロデューサー細胞は、とりわけ、遺伝子治療およびワクチン接種のために設計された組換えアデノウイルスの製造に有用である。
【0016】
本発明のもう1つの態様は、治療用の複製欠損アデノウイルスベクターの製造方法を提供する。例えば、特定の実施形態において、本発明は、感染因子/病原体により発現される抗原に対する免疫原性応答を誘発するためのワクチンとしての使用のための免疫原性組成物を提供する。もう1つの実施形態において、本発明は、腫瘍抗原に対する免疫応答を誘導するのに適したワクチンを提供する。本発明のこの態様は、本明細書中において、全HCVポリタンパク質を発現するΔE1−E4発現ベクターの構築、および免疫実験における該ベクターの利用により例示される。
【0017】
1つの実施形態においては、本発明は、パッケージング細胞内に多欠失アデノウイルス発現ベクターを導入すること{前記パッケージング細胞は、EBV由来EBNAタンパク質;Tet転写サイレンサー;Tet逆トランスアクチベーター;アデノウイルス発現ベクター[前記アデノウイルス発現ベクターはEBV由来oriP、アデノウイルスITR結合部および第1転写単位(前記第1転写単位はAd5 DNA結合タンパク質およびE4 ORF6をコードする核酸配列よりなる第2転写単位と組合されたAd5 E2由来ポリメラーゼ、プレ末端タンパク質をコードする核酸配列よりなる。)よりなり、第1転写単位および第2転写単位は二方向性テトラサイクリン依存性プロモーターに融合している。]ならびにプロモーターに融合した関心のあるトランスジーンをコードする発現カセットを発現する。}、E2およびE4ORF6コード配列の発現を誘導すること、および産生した複製欠損アデノウイルスを回収することを含んでなる、関心のあるトランスジーンを含む複製欠損アデノウイルスの製造方法を提供する。特定の実施形態においては、E2およびE4ORF6コード配列の過剰発現により特徴づけられるpE2アンプリコンの複製を誘発するドキシサイクリンと該パッケージング細胞とを接触させることにより、E2およびE4ORF6コード配列の発現を誘導する。
【0018】
代替的実施形態において、本発明の方法は、E1、E2、E3およびE4遺伝子を欠く多欠失ヒトAd5アデノウイルスベクターのためのパッケージング細胞としての、tTSkidおよびrtTA2を発現する293EBNA細胞の使用を含む。本発明は更に、本明細書に開示されており特許請求されている製造方法により回収され精製された組換え複製欠損アデノウイルス粒子を提供する。
【0019】
本発明の他の特徴および利点は本明細書に記載の開示から明らかである。実施例(具体例)は、特許請求されている本発明の実施に有用な種々の成分および方法を例示する。該実施例は本発明を限定するものではないと解釈されると理解されるべきである。本開示に基づき、当業者は、本発明の実施のための他の成分および方法を特定し使用することが可能である。
【0020】
図面の簡単な説明
図1A〜1Bは、安定な293EBNATetクローンを得るために使用したプラスミドの概要図を示す。パネルAはプラスミド成分の直線的表示を示す。以下の略語が使用されている:逆Tetトランスアクチベーター(rtTA);Tetサイレンサー(tTS);ECMV内部リボソーム進入部位(IRES);イントロン配列(intS);およびピューロマイシン耐性(PuroR)。
【0021】
パネルBはpE2プラスミドの概要図を示す。pFG140由来のAd5逆末端反復配列の頭−尾結合体を該プラスミド内にクローニングした(ITR、灰色矢じり)。Ad5初期遺伝子が黒色矢印で示されている。ポリメラーゼ(Pol)、プレ末端タンパク質(pTP)、DNA結合タンパク質(DBP)およびE4orf6を、Tet応答性要素(TRE、白色太棒線)により駆動される2つのビシストロン発現カセット内に挿入した。ニワトリβグロビン絶縁(insulator)配列(HS4)に隣接するEBV潜在性複製起点(OriP)が付点太棒線および灰色太棒線で示されている。
【0022】
図2はAdTetLuc感染クローンにおけるルシフェラーゼ発現のグラフ表示を示す。293EBNA細胞および種々の293EBNA/Tetクローンに、1μg/ml ドキシサイクリンの存在下(黒色棒線)または非存在下(白色棒線)、AdTetLucを感染させた(m.o.i 10)。細胞溶解物におけるルシフェラーゼ活性を感染の48時間後に評価した。
【0023】
図3は環状および直線形態のpE2の概要図を示す。pE2の環状形態と直線形態との識別を可能にするNotI消化により得たDNA断片も示されている。
【0024】
図4A〜4B。パネルAは、ドキシサイクリンによるAd5 E2遺伝子発現の活性化の際のpE2の複製を示すサザンブロット分析を示す。NotIで消化されたpE2の108個のコピーを第1レーンにローディングした。ドキシサイクリンの非存在下/存在下のpE2での形質移入の48時間後に293 EBNA Tet細胞から抽出されNotIおよびDpnIで消化されたエピソームDNAをレーン2および3にローディングした。環状および直線状単量体形態を示す12.6および4.4Kbのバンドが黒色矢印で示されている。DNAマーカーのサイズが図の右側に示されている(Kb)。
【0025】
パネルBは、E2タンパク質のtet誘導性発現を示すウエスタンブロット分析を示す。ドキシサイクリンの存在下(+)(レーン3、6および9)または非存在下(−)(レーン2、5および8)でpE2アンプリコンで形質移入された293EBNATet細胞におけるDBP、pTPおよびポリメラーゼタンパク質のウエスタンブロット分析。陰性(非形質移入)対照がレーン1、4および7に示されている。特異的ウサギ抗血清(ポリメラーゼ、pTP)またはマウスモノクローナル抗体(DBP)でE2タンパク質を検出した。
【0026】
図5A〜5B。2E2クローンから抽出されたpE2の構造およびE2タンパク質の発現。パネルAは、クローン293EBNATetおよび2E2から抽出されたpE2の構造を示すサザンブロット分析である。293EBNATetおよび2E2クローンから抽出されたDNAのサザンブロット分析。Hirt法により抽出されたDNAをBamHIで消化し、1%アガロースゲル上で分離し、ナイロンメンブレン上に移し、32Pで標識されたpE2 DNAでハイブリド形成させた。参照体として第1レーンにpE2ベクターをローディングした。293EBNATet細胞から(陰性対照)および2E2クローンから抽出されたDNAをそれぞれ第2および第3レーンにローディングした。
【0027】
パネルBは、Ad5ΔE1ベクターに感染した細胞におけるE2発現と比較した場合の2E2安定細胞系におけるE2タンパク質のtet誘導性発現を示すウエスタンブロット分析である。2E2クローンによるE2aおよびE2bタンパク質発現を、ドキシサイクリン(1μg/ml)(レーン、2、3;5、6;8、9)の存在下(+)(レーン3)および非存在下(−)(レーン2)、ウエスタンブロットにより評価し、500のm.o.i.のFG Ad5ΔE1ベクターに感染した非誘導2E2細胞からのE2タンパク質の発現レベル(レーン1)と比較した。分子量マーカーの移動度(kDa)が図の左に示されている。
【0028】
図6は、EGFPを発現するAd ΔE1−4ベクターをレスキューし増殖させるための2E2細胞の使用を示す一連の写真を示す。サイレンシングされた(‐doxy)または活性化された(+doxy)E2/orf6遺伝子発現を伴う2E2クローンにおけるAd5ΔE1−4EGFPウイルス増殖。P0=形質移入、P1およびP2は、前感染経過からの全粗溶解物の1/10を細胞に感染させることにより得た。
【0029】
図7A〜7B。パネルAはAd5ウイルスの概要地図である。この図にはすべての欠失領域が示されている。E1、E2a、E3、および7個のE4 orfのうちのorf3を除く6個を、ベクターバックボーンから完全に欠失させた。ポリメラーゼおよびプレ末端タンパク質の欠失は部分的であるに過ぎない。E1領域は、MCMVプロモーターにより駆動されるHCVポリタンパク質発現カセットで置換される。ベクターゲノムの制限分析において使用するHindIII制限部位が示されている(┃)。
【0030】
パネルBは、該多欠失ベクターのE1領域内に導入されたHCV(BK株)ポリタンパク質発現カセットの概要図を示す。HCV 5’および3’UTR配列が除去されており、最適化コザック配列が該ポリタンパク質の5’側に融合されている。発現はマウスCMVプロモーター(mCMV)およびウシ成長ホルモンポリA(BGHポリA)により調節される。
【0031】
図8はAd ΔE1−4orf3+HCVの制限分析を示す。プラスミドDNAおよびCsCl精製ウイルス粒子から抽出されたウイルスDNAをHindIIIで消化し、クレノウ酵素でのフィルイン反応により(33P)dATPで末端標識した。精製されたAd ΔE1−4orf3+HCVベクター(レーン4)のウイルスDNA制限パターンを元のプラスミド(レーン3)と比較した。HindIIIで制限処理されたFG(ΔE1−E3)Ad5(レーン1)およびAd5ΔE1−4orf3+空ベクター(レーン2)バックボーンを該ゲル内に加えた。a、b、c、dは、欠失を含有する多欠失ベクターDNAバンド、およびFG(ΔE1−E3)Ad5パターンにおける対応バンドを示す。
【0032】
図9は、Ad5ΔE1−4HCV感染細胞におけるHCVタンパク質の発現を示すウエスタンブロット分析を示す。HeLa細胞にAd5ΔE1−4HCVを10のm.o.i.で感染させた。HCV特異的抗体でのウエスタンブロット分析により細胞抽出物においてHCVタンパク質を検出した。感染の48時間後に調製した、HeLa細胞からの溶解物をレーン3にローディングし、未感染対照細胞からの溶解物(レーン1)、およびmCMV−HCVベクターDNAで形質移入したHeLa細胞からの溶解物(レーン2)と比較した。特定のバンドが矢印で示されている。
【0033】
図10は、マウスにおけるAd5ΔE1−4HCVウイルス免疫化に対するインビボCD8+ T細胞応答を特徴づけるFACSデータのグラフ表示を示す。A2.1(a)およびCB6F1(b)。1010vpで筋肉内に免疫化されたマウスの新鮮に単離された脾細胞を、3週間後のIFN−γに関する細胞内染色により、HCV−ペプチドのプールに対するCD8+ T細胞応答に関して試験した。x軸 抗INF−γ、y軸 抗CD8。プールC(コア)、プールF−G(NS3)、プールH(NS4)、プールI−L−M(NS5a/b)。
【0034】
図11は、本開示に記載されているポリヌクレオチドおよびポリペプチド配列のヌクレオチドおよび/またはアミノ酸配列(すなわち、配列番号1〜21)を示す。
【0035】
図12A〜12Cは、NS3ヘリカーゼ内のエピトープのマッピングに用いた方法を示す。Ad5ΔE1−E4HCVで初回抗原刺激されpSh−Ad5−HCVで追加抗原刺激された2匹のマウス(マウス4,黒塗り棒線、マウス5,付点棒線)から精製した脾細胞を、全NS3ヘリカーゼ領域に及ぶペプチド(I〜XVIII)の二次元サブセット上でγ−IFN−Elispotにおいて試験した(図12A)。図12Bに示されているデータは、免疫応答を特徴づけするために使用したパネルAにおいて特定された陽性閾値を超える応答を惹起したペプチドプール間の交差点を要約したものである。パネル12Cは、該応答をもたらすNS3エピトープを特定するために行ったγ−IFN−Elispotアッセイの結果を要約したものである。
【0036】
図13は、VV−NSチャレンジに対する防御の誘導における免疫化の効力を示す。灰色のドットは相乗平均力価(N=5)を表す。アステリスクは対照に対するp<0.05を示す(Mann−Whitneyランク)。
【0037】
図14A〜Bは、Ad5ΔE1−E4HCV免疫化に応答してアカゲザルにおいて惹起された免疫応答を要約したものである。パネルAは、Ad5ΔE1−E4HCVの1回の投与に際してサル4061において惹起されγ−IFN−Elispotにより分析された経時的免疫応答を表す。結果は106PBMC当たりのγ−IFNスポット形成細胞数(SFC)として表されている。各棒グラフは、別々のペプチドプールに対する応答を表す。パネルBは、Ad5ΔE1−E4HCVの1回の投与により3頭の個々のサルにおいて誘導され注射の6週間後にγ−IFN−Elispotにより分析された免疫応答を示す。結果は、106PBMC当たりのγ−IFNスポット形成細胞数(SFC)として表されている。各棒グラフは、別々のペプチドプールに対する応答を表す。
【0038】
発明の詳細な説明
個々の文の終わりに示されている数字の列挙は、本明細書の終わりに番号付きで示されている参考文献の一覧に対応する。本明細書中に引用されている参考文献は本発明の先行技術であると自認されるものではない。
【0039】
本明細書中で用いるすべての科学技術用語は、特に定義されない限り、当業者に一般に理解されているものと同じ意義を有することに留意することが、本発明の理解に重要である。以下に幾つかの用語を説明する。あるいは、ある種の用語は、初めて用いられる際に本明細書中で定義されうる。
【0040】
本明細書中で用いる「アンプリコン」なる語は、必須遺伝子機能が付与されると複製されうるエピソームまたは染色体外DNA要素を意味する。一般に、アデノウイルスアンプリコンは、ウイルスDNAの複製を支持する各末端反復配列の少なくとも一部分を含むと理解される。真核ウイルスアンプリコンは、好ましくは、完全なITR配列の少なくとも約90%を含む。したがって、「アデノウイルスアンプリコン」はITR結合部および適切な複製起点のいずれかを含む。
【0041】
本明細書中で用いる「形質移入」なる語は、細胞が生物学的に生存可能なままでDNAを細胞の外部から細胞の内部へ導入する適切な方法のいずれかを意味する。本明細書中で用いるこの用語は、限定的なものではないがエピソームおよび他の環状または直鎖状DNA形態の形質移入を含む任意の手段による宿主細胞内へのDNAの導入を含む。この用語は、例えば本明細書に記載されているような遺伝子治療法を含む。本発明を実施するためには、限定的なものではないがリン酸カルシウム共沈法、エレクトロポレーション、遺伝子銃形質移入、リポフェクションまたは他のカチオン脂質に基づく形質移入を含む適切な形質移入法のいずれも用いることが可能である。これらの技術は当業者によく知られている。
【0042】
本明細書中で用いる用語は本発明を限定するものではない。例えば、「遺伝子」なる語は、遺伝子産物をコードするcDNA、RNAまたは他のポリヌクレオチドを含む。「核酸」、「RNA」、「DNA」などの用語を用いる際には、特定の段階で用いられうる化学構造を限定するものではない。例えば、DNAの代わりにRNAが一般に用いられうることは当業者によく知られており、したがって、「DNA」なる語の使用はこの代用を含むと解釈されるべきである。また、種々の核酸類似体および誘導体も本発明の範囲内であることが公知である。遺伝子または核酸の「発現」は、細胞遺伝子発現だけでなく、クローニング系および任意の他のコンテクストにおける核酸の転写および翻訳をも含む。
【0043】
自然感染からのアデノウイルスの大規模生産はウイルスDNA複製と主要後期プロモーター活性との協同作用の結果であると理解される。実際、この方法は、ビリオンをパッケージングするのに要する大きなプールを生成する効果を有する、高コピー数の転写活性鋳型の蓄積をもたらす。先行技術は、1以上のウイルスタンパク質が欠損したアデノウイルスベクターの産生のための1以上のウイルスタンパク質を発現するいくつかのヘルパー細胞系を含むが、自然感染を模倣した一連の協同事象(例えば、ウイルスDNA複製および必要な構造タンパク質の発現)により特徴づけられる生産系は未だ記載されていない。宿主細胞染色体内への相補遺伝子の組込みに基づく相補ヘルパー細胞系の製造を要する方法を用いる代わりに、宿主細胞内で自己複製可能なアデノウイルス複製に必要な非構造E2遺伝子のすべてを含有するエピソームプラスミドが得られた。
【0044】
ヘルパーウイルス依存性生産系の使用に伴う、先行技術で認められている問題点に対処して、本発明は、多または完全欠失アデノウイルスベクターを相補しうるプロデューサー細胞系を作製するために使用されうる新規アデノウイルスアンプリコンを提供する。該アンプリコンは、ヘルパーウイルス非依存性ベクター産生の効率が最大となるよう自然アデノウイルス感染の段階を模倣する態様で機能するように設計されている。本明細書中に示すとおり、これは、ウイルス転写カスケードの開始に要するアデノウイルス初期遺伝子の発現を能動的に抑制し/遅延させることにより開示のエピソーム(すなわち、アデノウイルスアンプリコン)がパッケージング細胞系の核内の潜伏相において維持されるパッケージング細胞を操作することにより達成される。
【0045】
潜伏性は、エプスタインバーウイルス(EBV)由来DNA複製要素の核保持特性の利用および誘導性発現系(本明細書中ではテトラサイクリン調節発現系により例示される。)の使用により達成される。誘導により、複製に必要なアデノウイルスE2遺伝子(すなわち、ポリメラーゼ、プレ末端タンパク質およびDNA結合タンパク質)の発現を招くエピソーム配列の転写を引き起こす複製相の活性化がもたらされる。実際、潜伏相から複製相への移行により、トランスジーンを含む多または完全欠失アデノウイルスベクターを効率的にパッケージングするのに必要な大量の相補ウイルスタンパク質の蓄積が促される。したがって、典型的に自然感染の後期に高力価のビリオン産生をもたらす一連の事象を模倣する態様で該パッケージング細胞が機能するのを可能にするよう、該アンプリコンが設計される。したがって、開示されているアンプリコンおよびパッケージング細胞系は、ヘルパーウイルス非依存性医薬等級ベクターの効率的高力価製造方法を可能にする。
【0046】
エピソームプラスミド(pE2)は以下の特徴により特徴づけられる:(i)これは、該エピソームを自律複製可能にし核内保持の促進によりエピソームを多コピーで維持するEBVプラスミド複製起点のような要素を含む、(ii)これは、直鎖形態でのDNA複製を可能にするAd5逆末端反復(ITR)結合部を含む、(iii)これは、アデノウイルスの複製に必要なE2アデノウイルス遺伝子、例えばポリメラーゼ、プレ末端タンパク質およびDNA結合タンパク質ならびに初期領域4(E4)ORF6の誘導性発現をもたらす。
【0047】
ウイルスDNA複製のためには、Ad5ウイルスDNA(GenBank BK000408に開示されている。)の僅か2つの領域のみがシスで要求されることが公知であることを、当業者は認識する。これらは左逆転末端反復配列またはITR(Ad5のbp1〜約103)および右ITR(Ad5のbp35833〜35935)である。EBV由来の複製起点系の存在は、アンプリコンが多コピーで核内に保持され染色体DNAと同時に複製されるのを可能にし、一方、アデノウイルスITR結合部の存在は、E2領域によりコードされるタンパク質の存在下、アンプリコンが多コピー数で複製されるのを可能にする。該アンプリコン上での誘導性プロモーターの使用は、エピソームの複製に及びトランスジーンを含む多または完全欠失アデノウイルスベクターの増殖に要求されるアデノウイルス遺伝子を、厳密に調節応答性である誘導性プロモーターの制御下に配置する。一般には、誘導性プロモーターは、アクチベーターにより誘導されるプロモーターである。誘導性プロモーターに作用するインデューサーの非存在下では、エピソーム上に含有されるアデノウイルス遺伝子は発現されず、ウイルスタンパク質の産生は伴わず、ウイルスタンパク質誘導性細胞毒性のリスクは最小である。
【0048】
実際、開示されているアンプリコン(例えば、エピソーム)は、前記特性の1以上を同時に達成するよう、宿主細胞内に存在する他の要素(例えば、活性化因子)と協同して働きうる要素を含みうる。例えば、同一要素(DNA配列)が自己増殖能を付与し核内保持を促進しうる。代替的ウイルス複製系に由来するDNA配列も本発明の実施に使用されうると理解されるべきである。例えば、ウシパピローマウイルス(BPV)由来の複製起点および活性化因子(60)またはSV40由来T抗原系に基づくベクターに由来する配列が、適切な代替物となる。
【0049】
本明細書中の実施例は、EBNA−1を発現する宿主細胞の使用を記載しているが、所望により、アデノウイルス遺伝子を含有する同一エピソーム単位(アンプリコン)上もしくは複製可能な第2の遺伝単位上にコード配列を含めることにより又は宿主細胞ゲノム内への安定組込みにより他の活性化因子を導入することが可能であることは容易に理解される。例えば、本明細書に記載のとおりにEBNA1抗原およびEBV複製起点を使用する代わりに、BPV複製起点と共にウシパピローマウイルス(BPV)E1およびE2抗原を使用することが可能である。E1抗原は、複製の開始および伸長に必要なヘリカーゼであり、E2抗原は、複製起点へのE1抗原の結合を補助する転写因子である(61)。総合すると、これらのウイルスタンパク質はまた、適当な形質移入に関してコンピテントである細胞におけるエピソームの核内保持を促進することが公知である。
【0050】
霊長類細胞内において非組込み型自律的複製性エピソームベクターを安定に維持し及びプラスミドの安定な複製を支持する、EBVゲノムの確定された遺伝的要素が公知である。必要な遺伝的要素には、シス作用性プラスミド複製起点(oriP)およびトランス作用性エプスタインバー核抗原(EBNA−1)タンパク質が含まれる。より詳しくは、EBV由来要素、すなわちoriPおよびEBNA−1は、これらのベクターにより形質移入された哺乳類細胞内において1〜90個の範囲の数で非組込み型染色体外分子として専ら存在する組換えエピソームの安定な複製を支持するために使用されている。
【0051】
EBVゲノムの複製起点oriPを含有しEBNA1ウイルスタンパク質(641アミノ酸)の発現を可能にするプラスミドは、形質移入されたヒト細胞内で安定なエピソーム様態で維持され、それらの複製は細胞分裂と同時に生じる。本明細書に示すとおり、活性化因子EBNA−1の存在下で使用するEBV複製起点(OriP)は自己複製能を付与し、核内保持を促進する。理論により束縛されるものではないが、EBNA1タンパク質は複製開始のレベルで30bpの反復配列に結合し、S相の時点での細胞性因子のリクルートメント、およびoriP配列をシスで有するプラスミドの、細胞分裂との同期的複製を可能にすると考えられている。さらに、EBNA1は、おそらく染色体構造体および反復単位のレベルでの同時結合により、細胞分裂時のエピソームの分離および核内維持を可能にするのであろう。これらの要素は、それ自身で、複製時に、細胞当たり複数のコピーのプラスミドベクターのエピソーム維持および分離を可能にする。
【0052】
本発明で使用するエピソームにおいて、適切なEBV複製起点DNA配列のいずれも使用することができる。適切なEBV複製起点配列(oriP)の一例はGenBank V01555に開示されている。oriP領域はこのGenBank配列のヌクレオチド7333からヌクレオチド9312までの配列に及んでいる。本明細書に記載のエピソームにおいて使用されるoriP配列は、65bpの逆転復配列単位により形成され30bp単位の4個の不完全なコピーを含む複製起点から960bp離れた、30bpの20単位の反復から構成されている。
【0053】
エプスタインバーウイルス(EBV)由来oriPは、EBNA−1結合配列の2つのクラスター(反復配列のファミリーおよび二回対称配列)から構成されている。どちらの要素もEBNA−1への複数の結合部位を有し、oriPを含有するプラスミドの複製および核内保持に必須である。宿主細胞因子は、本明細書に開示するエピソームの複製および核内保持を補助すると考えられている。一般に、適当なoriP配列は、oriP機能に必要なことが知られている反復配列のファミリーおよび二回対称の領域を含む。本発明において使用されうるEBV oriP配列には、天然に存在する配列からの修飾を含有するもの、例えば、天然配列の欠失、挿入、置換および重複を含有するものが含まれる。そのような誘導体配列は、例えば、oriP機能に必要な前記公知領域を維持させることにより入手できる。また、保存的置換はよく知られており、当業者に入手可能である。使用するoriP配列は、十分に多い量のEBNA1タンパク質の存在下でoriP配列が存在するエピソームの複製を導くよう宿主細胞内で有効に機能するものである。
【0054】
適切なEBNA1タンパク質のいずれかをコードするDNAが本発明のプロデューサー細胞により発現されうる。EBNA1をコードするDNAはInvitrogenから商業的に入手可能であり、そのEBV系列プラスミドのいくつかの中に含有される。さらに、EBNAタンパク質をコードするDNAは、発現されるタンパク質が宿主細胞におけるEBV oriP含有エピソームの複製を支持する、天然に存在するEBNA1アミノ酸配列の変異体、例えば、欠失、付加、挿入または置換を含有するものをコードすることが可能である。この変異体は、本明細書に記載の他の配列の場合と同様に、アミノ酸配列保存的変異体をコードする機能的に保存的な核酸配列、BLASTまたはFASTAアルゴリズムによる測定で90%を超える、好ましくは95%を超える同一性または相同性を有する配列、および高いストリンジェンシーのハイブリダイゼーション条件下でハイブリダイズする配列を含む。さらに、同一EBNA1タンパク質をコードする縮重DNA配列を使用することが可能である。同一アミノ酸配列を発現しうる縮重DNA配列は、そのようなDNA配列の構築および発現方法と同様、当技術分野でよく知られている。
【0055】
EBNA1は、よく知られた技術を用いて任意の霊長類またはイヌ細胞内に安定に形質移入されることができ、組込み遺伝子コピーからEBNA1を発現する得られた細胞系は、適当な生産細胞系を作製するために使用されうる。あるいは、EBNA1が発現される限り、感染性または欠損性EBVを既に含有する細胞系を使用することが可能である。これは、ATCCから入手可能な多数のEBV形質転換リンパ芽球を含む。EBNA1を既に発現する細胞系の形質移入が非常に好都合である。なぜなら、そのような細胞はエピソーム構築物を安定に維持する能力は数桁増強されることが可能であり、僅か2〜3週間のうちに安定細胞系が作製されうるからである(62)。しかし、これらの方法は、組込み遺伝子からEBNA1を構成的に発現する細胞系を製造する追加的な工程を要する。
【0056】
本明細書中に示すとおり、テトラサイクリンプロモーターは、テトラサイクリンまたはその一般的類似体の1つ(例えば、ドキシサイクリン(Dox))に応答性であり、開示されているエピソーム単位(例えば、アンプリコンまたはレプリコン)における使用に適している。テトラサイクリンの類似体であるドキシサイクリンは、ヒトにおけるその安全な使用、細菌テトラサイクリンリプレッサー(TetT)に対するその特異性のため、広く受け入れられている。簡潔に説明すると、テトラサイクリン依存性調節系(tet系)は、単純ヘルペスウイルスVP16タンパク質のアクチベータードメインに融合した原核性TetRよりなるテトラサイクリントランスアクチベーター(tTA)と、最小プロモーターに融合した原核性テトラサイクリンオペレーター部位(tetO)のいくつかのコピーよりなるテトラサイクリン応答性要素(TRE)との間の相互作用に基づく(68)。テトラサイクリン(tet)の存在下、tTAは、TREに結合するその能力を失い、発現が遮断される。逆トランスアクチベーター(rtTA)は突然変異誘発によりtTAから誘導されている。tTAとは対照的に、rtTAはtetの存在下でTREに結合するに過ぎない。
【0057】
転写の基底レベルを減少させることにより遺伝子発現の緊縮調節を得るために、本発明者らは、最近記載された逆tetトランスアクチベーターの新規改良形態(29)とTet転写サイレンサーtTSkid(16)との組合せを利用するTet調節系を使用した。tTSkidは、転写のリプレッサーとして機能することが公知の腎タンパク質Kid−1のKRABドメインを含有する。tTSkidは、エフェクター薬の非存在下でTetプロモーターに結合して、転写の基底レベルを減少させる。逆Tetトランスアクチベーターとの組合せは、ドキシサイクリン添加により調節される活性化/抑制系の構築を可能にする。この目的のために、図1Aに記載のとおりにEMCV IRESを使用することによりそれらの2つの遺伝子をビシストロン転写単位において組合せた。
【0058】
真核細胞における誘導性遺伝子発現を導きうるいくつかのプロモーター系が入手可能である。これらには、重金属イオン(63)、(64)、イソプロピル−β−D−チオガラクトシド(65)、ホルモン、例えばコルチコステロイド(66)、プロゲステロンアンタゴニスト(67)またはテトラサイクリン(68)に応答して修飾される活性を有するプロモーターが含まれる。しかし、エクジソン、ラパマイシン、RU486、デキサメタゾンおよび重金属(すなわち、ZnまたはCd)のようなアクチベーターに対して応答性である他のよく知られた誘導性調節要素も適している。代替的な調節要素を本発明での使用に応用することは当業者の能力の範囲内に十分に含まれる。本発明の目的には、十分なレベルまたは調節制御を保証するものであり、医薬用途に許容されるアクチベーターにより誘導可能なものである限り、調節要素のいずれも使用することが可能である。さらに、遺伝子発現の厳密な調節を促進するために、他の調節要素、例えばテトラサイクリン応答性トランスアクチベーターおよび/またはサイレンサー(rtTAおよびtTs)に誘導性プロモーターを機能的に連結しうると理解されるべきである。
【0059】
発現カセット(関心のあるトランスジーンと、本明細書中に開示されている哺乳類細胞内での発現を導くための必要な調節配列とを含むものとして定義される。)のすべては、腸菌(E.coli)BJ5183内での相同組換えによるpAd5ΔE1−4orf3+のE1領域における挿入が可能となるよう、CMVプロモーターおよびトランスジーン発現用BGHポリAシグナルに加えてAd5配列[nt]1−450(左)(配列番号1)および[nt]3511−5792(右)(配列番号2)を含有するAd−シャトルベクターのコンテクストにおいて構築した。EGFP cDNAはpEGFPプラスミド(Clontech)から得、ついでAd−シャトルプラスミド内にクローニングしてpShAd5 EGFPを得た。
【0060】
本明細書に記載の方法において、所望の配列をコードするエピソームにより成功裏に形質移入された細胞を選択するために、通常の選択マーカーを使用する。そのような選択は、通常、形質移入された細胞を抗生物質または関連選択過程を開始させる他の物質にさらすことを含む。本発明で使用するエピソームにおいて使用する選択マーカー遺伝子は、これらの遺伝子を含有する細胞が対応抗生物質または因子の存在下で成長するのを可能にする、特定の抗生物質および/または因子に対する耐性を付与するタンパク質をコードする遺伝子である。真核性選択マーカーの非限定的な例には、ヒグロマイシン(Life Technologies,Inc.;Gaithesboro,Md.から商業的に入手可能なhygまたはhph)、ネオマイシン(Life Technologies,Inc.Gaithesboro,Md.から商業的に入手可能なneo)、ゼオシン(Pharmingen,San Diego Calif.から商業的に入手可能なSh Ble)、ピューロマイシン(Clontech,Palo Alto Calif.から入手可能なpac、ピューロマイシン−N−アセチル−トランスフェラーゼ)、ウアバイン(Pharmingenから入手可能なoua)およびブラスチシジン(Invitrogenから入手可能)に対する耐性を付与する抗生物質耐性遺伝子が含まれる。
【0061】
多欠失ヒトAd5ベクターバックボーンの概要図を図7Aに示す。E1およびE3領域の古典的欠失(11に概説されている)のほかに、本発明者らは、L4イントロンを含め、r鎖においてコードされる他の機能に何ら影響を及ぼすことなくDNA結合タンパク質の全コード配列([nt]22245−24029;(配列番号3)1784bp欠失)を除去した。三成分リーダー配列のイントロンおよび主要後期単位に対応するプレ末端タンパク質(Ad5[nt]8919−9462(配列番号5)543bp欠失)遺伝子およびポリメラーゼ([nt]7274−7883;(配列番号4)609bp欠失)の部分を欠失させてE2b遺伝子発現をノックアウトした。さらに、ポリメラーゼのトランケート化非活性形態の産生を妨げるために、ATG開始コドンをCTGに突然変異させた。E4プロモーターに直接的に融合したorf3を除き、E4領域を完全に欠失させた([nt]32830−34316(配列番号6)および34895−35443;(配列番号7))。
【0062】
全初期遺伝子の欠失を組合せることによりAd5バックボーンにおいて生じた理論的空間は約12.4Kbである。該新規ベクター系の大きな容量を利用して、マウスサイトメガロウイルス(MCMV)プロモーターに融合した全HCVポリタンパク質遺伝子用の発現カセットを挿入した。該HCVポリタンパク質発現カセットは、5’および3’非翻訳領域を除去し、コアATGの上流に最適コザック配列を挿入し、NS5Bレプリカーゼの触媒ドメインを変異させて酵素活性を除去することにより構築した(32)。トランスジーンの発現効率を増加させるために、ヒトCMVプロモーターを、FGアデノウイルスベクターにおいて4〜30倍強力であると報告されているマウスCMVプロモーター(1)で置換した。
【0063】
図7Bは、多欠失ベクターのE1領域内に導入されたHCV(株BK)ポリタンパク質発現カセットの概要図を示す。HCV 5’および3’UTR配列を除去し、最適化コザック配列を該ポリタンパク質の5’側に融合させた。発現はマウスCMVプロモーター(mCMV)およびウシ成長ホルモンポリA(BGHポリA)により調節される。
【0064】
オープンリーディングフレーム3の維持は、内部CMVプロモーターにより調節されるトランスジーンのインビボおよびインビトロでの持続的発現に必要であることが公知である(18,34)。したがって、ΔE1E3FGベクターの5700bpの欠失、およびそれによる、wt(6)の105%のゲノムサイズのパケーンジング容量に加えて、該新規Ad5ΔE1−4orf3+ウイルスベクターは12.4Kbまでのトランスジーンを収容しうる。この目的には、本発明のアデノウイルスアンプリコンおよびプロデューサー細胞ならびに本発明の方法を用いて、関心のある多数のトランスジーンを含む欠損アデノウイルスベクターを得ることが可能であると予想される。
【0065】
本明細書中に開示されている多欠失Ad5ウイルスバックボーンにおける使用のための適当なトランスジーンには、特定の哺乳類種における発現に関してコドンが最適化された、免疫原をコードする核酸配列(すなわち、トランスジーン)が含まれるが、これに限定されるものではない。1つの実施形態においては、本発明は、感染因子により発現される抗原に対する免疫応答を誘導するための免疫原性組成物(例えば、ワクチン)を提供する。例えば、ヒトおよび/または非ヒト動物種に感染するウイルスに対する免疫応答を惹起することが望ましい。
【0066】
多欠失Ad5ベクターは、細菌、真菌、寄生生物を含む病原体により発現されるタンパク質に対するヒトまたは動物における免疫応答を刺激するのにも適していよう。スタフィロコッカス・アウレスス(Staphylococcus aureus)、ストレプトコッカス・ピロゲネス(streptococcus pyogenes)、ストレプトコッカス・ニューモニエ(streptococcus pneumoniae)、ビブリオ・コレラ(vibrio cholerae)、クロストリジウム・テタニ(clostridium tetani)、ナイゼリア・メテンジチス(neisseria meningitis)、コリネバクテリウム・ジフテリアエ(corynebacterium diphteriae)、ミコバクテリウム・ツバクロシス(mycobacteria tuberculosis)およびレプラエ(leprae)、リステリア・モノサイトジェネス(listeria monocytogenes)、レジオネラ・ニューモフィラ(legionella pneumofila)は、望ましいであろう、免疫応答を惹起する対象となる細菌の具体例であるが、これらに限定されるものではない。真菌および寄生生物の具体例としては、カンジタ・アルビカンス(Candida albicans)、アスペルギルス・フミガツス(aspergillus fumigatus)、ヒストプラズマ・カプスラツム(histoplasma capsulatum)、プラスモジウム・マラリアエ(Plasmodium malariae)、レイシュマニア・マジョール(Leishmania major)、トリパノソマ・クルジ(trypanosome cruzi)およびブルセイ(brucei)、シストロソマ・ヘマトビウム(Schistosoma haematobium)、マナソニ(mansoni)およびジャポニカム(japonicum);エンタモエバ・ヒストリチカ(Entamoeba histolytica)、ヒトフィラリア症を引き起こすフィラリア(Filaria)の種々の種が挙げられうる。
【0067】
予防的および/または治療的免疫応答が望ましいであろうウイルス科の具体例には、アフトウイルス属、カルヂオウイルス属、エンテロウイルス属、ヘパトウイルス属、パレコウイルス属、ライノウイルス属のような6つの異なる属を含むピコルナウイルス科が含まれる。これらのすべては、脊椎動物に感染するウイルスを含有する。免疫応答が望ましいであろうピコルナウイルスの具体例としては、口蹄疫ウイルス、脳心筋炎ウイルス、ポリオウイルス、コクサッキーウイルス、ヒトA型肝炎ウイルス、ヒトパレコウイルス(parechoviruses)、ライノウイルスが挙げられる。カリシウイルス科は、ノーウォーク群のウイルスにより引き起こされるヒトにおける流行性胃腸炎、およびウサギ出血性疾患ウイルスに関連したウサギにおける出血性疾患またはネコカリシウイルスにより引き起こされるネコにおける呼吸疾患のような動物における他の症候群に関連した種々の属を含む。もう1つの科は、ヒトおよび多数の異なる動物種から分離されたウイルスを含むアストロウイルス科である。ヒトアストロウイルスは胃腸炎および乳幼児の下痢に関連している。トガウイルス科はアルファウイルス属およびルビウイルス属の2つの属を含む。アルファウイルス属は、ヒトおよび獣医学的疾患、例えば関節炎(すなわち、チクングニヤウイルス、シンドビスウイルス)または脳炎(すなわち、東部ウマ脳炎ウイルス、西部ウマ脳炎ウイルス)に関連している。風疹ウイルスは、発熱およびリンオアデノパシーに関連した軽度の発疹性疾患の突発を引き起こすルビウイルス属の唯一のメンバーである。風疹ウイルス感染は、妊娠初期に母親により獲得された場合には、胎児異常にも関連する。フラビウイルス科は、重要なヒトおよび動物病原体を含むフラビウイルス属、ペスチウイルス属およびヘパシウイルス属の3つの属よりなるもう1つのウイルス科である。フラビウイルス属のメンバーの多くは、発熱、脳炎および出血熱を含む種々の疾患を引き起こす節足動物媒介ヒト病原体である。デング熱ウイルス、黄熱ウイルス、日本脳炎ウイルス、西ナイル熱ウイルス、ダニ媒介脳炎ウイルスは、世界的または地域的(地域流行性)に懸念される主要な病原体である。ペスチウイルス属は、経済的に非常に重要な動物病原体、例えばウシウイルス性下痢ウイルス、古典的ブタコレラウイルス、ボーダー病ウイルスを含む。C型関連ウイルスは、急性および慢性肝炎を引き起こすヘパシウイルス属の唯一のメンバーである。組換えアデノウイルスにより発現されるHCVタンパク質は、世界中で1億7000万人を冒すウイルス感染の結果を抑制する防御用および治療用免疫応答を惹起しうる。
【0068】
コロナウイルス科のメンバーに由来する抗原を組換えアデノウイルスベクターにより発現させて、感染に対する防御を得ることが可能である。重篤な急性呼吸症候群コロナウイルス(SARS−Coウイルス)に対する防御は、限定的なものではないがヌクレオカプシド(N)タンパク質、ポリメラーゼ(P)タンパク質、膜(M)糖タンパク質、スパイク(S)糖タンパク質、小エンベロープ(E)タンパク質または該ウイルスにより発現される他のポリペプチドのいずれかを含むSARS−CoVタンパク質の組合せを発現する多欠失Ad5ベクターで免疫することにより得られうる。狂犬病ウイルスを含むラブドウイルス科のメンバーは、ウイルスタンパク質を発現する組換えワクチンの標的となりうる。他の考えられうる標的には、いくつかの出血熱の突発を引き起こすエボラ様ウイルスおよびマールブルグ様ウイルス属を含むフィロウイルス科;麻疹、呼吸器合胞体、パラインフルエンザウイルスのようなヒトにおいて公知の最も一般的なウイルスおよびニューカッスル病および牛疫ウイルスのような獣医学的に関心の持たれるウイルスのいくつかを含むパラミクソウイルス科;インフルエンザA、B、Cウイルスを含むオルトミクソウイルス科;のようなリフトバレー熱、シン・ノンブレ(Sin Nombre)、ハンタ、プーマラウイルスのような重要なヒト病原体を含む、主として節足動物により脊椎動物宿主に伝染するブニヤウイルス科;リンパ球性脈絡髄膜炎、ラッサ熱、アルゼンチン出血熱、ボリビア出血熱ウイルスを含むアレナウイルス科;主としてウマおよびヒツジにおける中枢神経系疾患を引き起こすウイルスを含むボルナウイルス(Bornaviridae)科;世界的な乳児および幼児における重篤な下痢疾患の最も重要な原因であるロタウイルス、ヒトおよび他の哺乳動物の両方を冒しうるオルビウイルス(ブルータング、流行性出血熱ウイルス)を含むレオウイルス科が含まれる。
【0069】
ウイルス抗原をコードする適切なトランスジーンは、重要なヒト病原体を含む大きなウイルス群であるレトロウイルス科のメンバー、例えばヒト免疫不全ウイルス1および2(HIV−1およびHIV−2)ならびにヒトt細胞白血病ウイルス1および2型(HTLV1および2)ならびに非ヒトレンチウイルス、例えばヒツジおよびヤギを冒すマエディウイルス/ビスナウイルス、ウマを冒すウマ伝染性貧血ウイルス、ウシを冒すウシ免疫不全ウイルス、ネコを冒すネコ免疫不全ウイルス;ポリオーマウイルス科群小DNA腫瘍原性ウイルス[原型ウイルスは、それぞれマウスおよびアカゲザルを冒すポリオーマウイルスおよびSV40である(SV40に密接に関連しているBKおよびJCウイルスはヒト患者から分離された。)。]からも得ることが可能である。
【0070】
パピローマウイルス科は、疣贅およびコンジロームを生成する、ヒトを含む高等脊椎動物を冒すDNAウイルス群からなる。パピローマウイルスの感染はヒトおよび動物の両方における癌の発生に関連づけられた。ヒトパピローマウイルスは子宮頚癌、膣癌および皮膚癌に関連している。ヘルペスウイルス科は、ヒトおよび他の哺乳動物に対する多数の重要な病原体が分類される亜科を含む。他の抗原源には、単純ヘルペスウイルス1および2、水痘・帯状疱疹ウイルス、エプスタインバーウイルス、サイトメガロウイルス、ヒトヘルペスウイルス6A。6Bおよび7、カポジ肉腫関連ヘルペスウイルスが含まれるが、これらに限定されるものではない。さらに適した抗原源としては、ポックスウイルス科のメンバー、例えばサル痘ウイルス、伝染性軟属腫ウイルス、痘瘡ウイルス;ヘパドナウイルス科の原型メンバーであるB型肝炎ウイルスならびに急性および/慢性肝炎を引き起こす他のウイルス、例えばデルタ型肝炎ウイルス、E型肝炎ウイルスが挙げられる。
【0071】
第2の実施形態において、本発明は、腫瘍抗原に対する免疫応答を誘導するための免疫原性組成物(例えば、ワクチン)を提供する。適切な組成物は、腫瘍抗原をコードする最適化核酸配列を含む組換えチンパンジーアデノウイルスと生理的に許容される担体とを含有するであろう。特定の実施形態において、該カセットのコード配列要素は、単一の免疫原、例えば病原因子由来の抗原、または自己抗原、例えば腫瘍関連抗原をコードしうる。他の実施形態において、該コード配列は2以上の免疫原をコードしうる。例えば、これは、Her2 Neu、CEA、Hepcam、PSA、PSMA、テロメラーゼ(Telomerase)、gp100、メラン(Melan)−A/MART−1、Muc−1、NY−ESO−1、スルビビン(Survivin)、ストロメリシン(Stromelysin)3、チロシナーゼ(Tyrosinase)、MAGE3、CML68、CML66、OY−TES−1、 SSX−2、SART−1、SART−2、SART−3、NY−CO−58、NY−BR−62、hKLP2、VEGF、5T4のような自己抗原の組合せをコードしうる。
【0072】
トランスジーンの発現を導くために使用する転写プロモーターは、好ましくは、真核性RNAポリメラーゼにより認識される。好ましい実施形態においては、該プロモーターは「強力」または「効率的」プロモーター、例えば、本明細書に記載の実施例において使用するマウスCMVプロモーター(mCMV)である。もう1つの強力プロモーターの一例は、好ましくはイントロン配列を伴わない、最初期ヒトサイトメガロウイルスプロモーター(参照により本明細書に組み入れるChapmanら,1991 Nucl.Acids Res 19:3979−3986)である。したがって、本明細書中に開示されており特許請求されているエピソームにおける使用に適した1つの代替的プロモーターには、ヒトCMVプロモーターが含まれる。当業者は、任意の多数の他の公知プロモーター、例えば強力な免疫グロブリンまたは他の初期もしくは後期ウイルスプロモーター、例えばSV40初期もしくは後期プロモーター、ラウス肉腫ウイルス(RSV)初期プロモーター;真核細胞プロモーター、例えばβアクチンプロモーター(Ng,S.Y.,Nuc. Acid Res.17:601−615,1989,Quitscheら,J.Biol.Chem.264:9539−9545,1989)、GADPHプロモーター(Alexanderら,Proc.Nat.Acad.Sci.USA 85:5092−5096,1988,Ercolaniら,J.Biol.Chem.263:15335−15341,1988)、メタロチオネインプロモーター(Karinら Cell 36:371−379,1989;Richardsら,Cell 37:263−272,1984)ならびにコンカテネーション化応答要素プロモーター、例えばサイクリックAMP応答要素プロモーター(cre)、血清応答要素プロモーター(sre)、ホルボールエステルプロモーター(TPA)および応答要素プロモーター(tre)(最小TATAボックス近傍のもの)を使用しうると理解する。
【0073】
遺伝子発現カセット内に存在する好ましい転写終結配列は、ウシ成長ホルモンターミネーター/ポリアデニル化シグナル(bGHpA)である。代替的転写終結/ポリアデニル化配列には、チミジンキナーゼ(tk)遺伝子に由来するもの又はSV40由来配列、例えばpCEP4ベクター(Invitrogen)において見出されるものが含まれるが、これらに限定されるものではない。
【0074】
本発明の目的、利点、用途および方法について全般的に説明してきたが、本発明の種々の実施形態を詳細に説明するために、以下に非限定的な実施例を記載する。しかし、本明細書に記載の発明は、以下の実施例の詳細には限定されず、これらは、本発明の実施を望む者のための単なる指針として記載されているに過ぎないと理解されるべきである。本発明の範囲は、完全な開示および本明細書に添付されている特許請求の範囲に関して評価されるべきである。
【0075】
材料および方法
本発明の実施は、特に示さない限り、分子生物学(組換え技術を含む)、微生物学、細胞生物学および生化学の通常の技術を用い、これらは当技術分野の通常の技術の範囲内である。このような技術は、例えば“Molecular Cloning:A Laboratory Manual”,2nd edition(Sambrookら,1989);“Oligonucleotide Synthesis”(M.J.Gait編,1984);“Animal Cell Culture”(R.I.Freshney編,1987);“Methods in Enzymology”(Academic Press,Inc.);“Handbook of Experimental Immunology”,4th edition(D.M.Weir & C.C.Blackwell編,Blackwell Science Inc.,1987);“Gene Transfer Vectors for Mammalian Cells”(J.M.Miller & M.P.Calos編,1987);“Current Protocols in Molecular Biology”(F.M.Ausubelら編,1987);および“PCR:The Polymerase Chain Reaction”(Mullisら編,1994)のような参考文献に十分に説明されている。
【0076】
配列決定は、標識プライマーもしくはターミネーターを使用する商業的に入手可能な自動シークエンサーまたは配列決定用のゲルに基づく方法を用いて行うことが可能である。また、配列分析は、標的DNAまたはRNA分子上でお互いに直に隣接してアニールするオリゴヌクレオチド配列の連結に基づく方法により行う(WuおよびWallace,Genomics 4:560−569(1989);Landrenら,Proc.Natl.Acad.Sci.87:8923−8927(1990);Barany,F.,Proc.Natl.Acad.Sci.88:189−193(1991))。図11は、本開示に記載されているポリヌクレオチドおよびポリペプチド配列のヌクレオチドおよび/またはアミノ酸配列(すなわち、配列番号1〜21)を示す。
【0077】
本開示の全体にわたって記載されている具体的な塩基対の番号に関する基準として、野生型アデノウイルス血清型5を使用する。野生型アデノウイルス血清型5の配列は公知であり、当技術分野において記載されている。参照により本明細書に組み入れるChroboczekら,1992 J.Virology 186:280を参照されたい。当業者は、アデノウイルス血清型5に関する塩基対により定められる領域に対する配列相同性により、他のアデノウイルス血清型(例えば、血清型2、4、6、12、16、17、24、31、33および42)における前記領域を容易に特定することが可能である。したがって、ヒトアデノウイルス血清型5を使用する以下の実施例は限定的なものではないと理解されるべきである。当業者は、異なるアデノウイルス血清型(例えば、Ad2)に関して、類似したプラスミド、ウイルスおよび技術を利用しうると認識するであろう。同様に、異なるヒトアデノウイルス、特にチンパンジーアデノウイルスに関して、類似したプラスミド、ウイルスおよび技術を利用しうるため、ヒトAdの使用は限定的なものではない。
【0078】
プラスミドの構築
Tetサイレンサーおよび逆Tetトランスアクチベーター発現カセットを含有するpIRESTの構築を図1Aに記載する。該Tet系は、単一の発現ベクターにおいて、以下のとおりに構築した。Tetサイレンサー(tTS)を含有するEcoRI−ClaI DNA断片をプラスミドpUHS6−1(E.Bujardにより快く提供されたもの)から単離し、ベクターpIRES−Neo(Clontech)内のIRES配列の下流に挿入してNeo遺伝子を置換した。その新たなベクターpIRES−tTSを、ヒトサイトメガロウイルスIEプロモーターの下流の唯一のEcoRV制限部位内へのrtTA2遺伝子(E.Bujardにより快く提供されたpUHD 52−1に由来する。)の挿入により修飾して、pIREStTS/rtTAを得た。図2は、AdTetLuc感染クローンにおけるルシフェラーゼ発現のグラフ表示を示す。簡潔に説明すると、293EBNA細胞および種々の293EBNA/Tetクローンに、1μg/ml ドキシサイクリンの存在下(黒色棒線)または非存在下(白色棒線)、AdTetLucを感染させた(m.o.i 10)。細胞溶解物におけるルシフェラーゼ活性を感染の48時間後に評価した。
【0079】
Tetタンパク質を安定に発現する細胞クローンを単離するための選択ーマーカーを導入するために、pPURベクター(Clontech)から得たピューロマイシン耐性発現カセットをpIREStTS/rtTAのXhoI部位内に挿入してpIREStTS/rtTApuroを得た。pE2の構造を図1Bに記載する。
【0080】
Ad5ポリメラーゼcDNAを含むプラスミドpVacPolから得たClaI/SphI断片、およびAd5−pTP cDNAを含有するpVACpTPからのAcc65/EcoRV断片を誘導性Tetプロモーターの制御下のベクターpBI(Clontech)内に挿入することにより、Ad5ポリメラーゼおよびプレ末端タンパク質を発現するビシストロン発現ベクターを構築した(pVacPolおよびpVACpTPはP.C.van der Vlietにより快く提供されたものである。)。オリゴヌクレオチド:5’−TTATACGCGTGCCACCATGACTACGTCCGG−3’(配列番号9)および5’−TTATGCTAGCGCGAAGGAGAAGTCCACG−3’(配列番号10)ならびにpFG140(19)から得たAd5 DBP遺伝子(Ad5[nt]22443−24032)(配列番号11)を使用するPCRにより得たAd5 E4 orf6(Ad5[nt]33193−34077)(配列番号8)を同じベクターpBI内に挿入することにより、第2の二方向性誘導性カセットを構築した。
【0081】
HS4絶縁体(insulator)に隣接するpCEP4に由来するEBV−OriP(EBV[nt]7333〜ヌクレオチド9312;GenBank V01555)(配列番号12)領域をpJC13−1(9)のBamHI部位内への直接的クローニングにより得た。オリゴヌクレオチド:5’−AACTACAATTCCCAACACATAC−3’(配列番号13)および5’−CACATCCGTCGCTTACATG−3’(配列番号14)を使用するpFG140からのPCRにより、Ad5 ITR結合部を増幅した。
【0082】
最後に、tk−ヒグロマイシン−Bホスホトランスフェラーゼ(HPH)カセットをpCEP4(Invitrogen)から誘導した。pE2を構成するすべての要素をpBI−pol/pTPベクター内に順次導入して、最終的にpE2を得た。
【0083】
pAd5ΔE1−4の構築
pAdCMV/LacZ/ΔPolベクター(A.Amalfitanoから快く提供されたもの(4))およびAd5dl308ΔpTPβ−gal(J.Schaackから快く提供されたもの)(45)からのそれぞれAd5ポリメラーゼ(Ad5[nt]7274−7883)(配列番号4)およびプレ末端タンパク質(Ad5[nt]8915−9462)(配列番号5)の部分欠失をMRKpAd5E3(52)内に導入することにより、E2b遺伝子が欠失したAd5ΔE1−E3バックボーンを得た。また、ATGからCTGへのポリメラーゼ開始コドンの部位特異的突然変異誘発も行って、最終的にpAd5 ΔE1,E3,E2Bベクターを得た。DBP遺伝子を含むDraI−MscI断片(Ad5[nt]22445−24029)が欠失したAd5のBamHI/XhoI断片([nt]21563−24797)を含有するpBluescriptKSII+(Stratagene)はRocco Savinoにより快く提供された。
【0084】
***pAd−ΔE1−2ベクターは、ΔDBP断片およびAdΔE1,E3,E2Bベクターを大腸菌(E.coli)Bj5183内に同時形質転換する相同組換えにより得た。orf3を除く完全なE4単位([nt]32830−34316および[nt]34895−35443)の欠失を、以下に記載するとおりに行った。AvrIIおよびMfeI制限部位を末端に有するorf3領域をPCR(ΔE4orf fw AvrII:5’− GCCTAGGGATGCGTGTCATAATCAGTGTGGGTTC−3’(配列番号15);ΔE4orf3 rev MfeI:5’−CAATTGAAAAGTGAGCGGGAAGAGCTGGAAGAACCATG−3’(配列番号16))により増幅し、同じ酵素で消化されたE4−シャトルベクター内にクローニングした。E4orf3はE4プロモーターおよびポリAシグナルを維持する。pAd5ΔE1−4orf3+ベクターを、そのようなDNAをpAd5ΔE1−2ベクターと共に大腸菌(E.coli)BJ5183内に同時形質転換することにより得た。
【0085】
すべての発現カセットは、トランスジーン発現用のCMVプロモーターおよびBGHポリAシグナルに加えて、記載されている(53)とおりに大腸菌(E.coli)BJ5183における相同組換えによりpAd5ΔE1−4orf3+のE1領域内の挿入を可能にするAd5配列[nt]1−450(左)および[nt]3511−5792(右)を含有するAd5−シャトルベクターのコンテクストにおいて構築した。EGFP cDNAをpEGFPプラスミド(Clontech,BD Bioscience,San Jose,CA,USA)から得、ついでAd−シャトルプラスミド内にクローニングしてpShAd5 EGFPを得た。5’および3’非翻訳末端反復配列(UTR)が欠失したHCV−BK cDNA(HCV BK[nt]342−9374(配列番号17))をプラスミドpCMV(1−9.4)(14)から誘導した。
【0086】
NS5B ORFは、ウイルスRNA依存性RNA−ポリメラーゼの触媒三つ組み残基に対応する3つのアミノ酸位置において変異させて(G−2737をAへ、D−2738をAへ、およびD−2739をGへ)、酵素活性を喪失させた(Nicosiaら,未公開データ)。最適化コザック配列に融合したHCV cDNAを、HCMVプロモーターをMCMVプロモーターで置換することにより得たpAd5−シャトルの修飾形態内にクローニングして、最終的にpShAd5HCVを構築した。pAd5ΔE1−4orf3+のE1領域内へのすべての発現カセットの挿入を、記載されているとおりに(43)、大腸菌(E.coli)内での相同組換えにより達成した。
【0087】
細胞
293EBNA細胞系(Invitrogen)をダルベッコ変法イーグル培地(DMEM)+10%ウシ胎児血清(FBS)、ペニシリン(100U/ml)、ストレプトマイシン(100μg/ml)、2mM グルタミンおよび250μg/ml G−418(GIBCO BRL)内で培養した。0.5μg/ml ピューロマイシンを含有する同じ培地を使用することにより、293EBNATet細胞を選択した。2E2細胞を選択することにより、90μg/mlのヒグロマイシンBを、既に記載されている培地に加えた。Lipofectamine2000(Invitrogen)を該製造業者の説明に従い使用してプラスミドDNA形質移入を行った。逆TetトランスアクチベーターおよびTetサイレンサータンパク質を発現する293EBNAクローンを得るために、形質移入の1日前に、1×106個の293EBNA細胞を6cmプレート内に播き、SapIで線状化された5μgのpIREStTS/rtTApuroで形質移入した。形質移入の48時間後、細胞をトリプシン処理し、15cmプレート内のピューロマイシン含有DMEM内に播いた。耐性クローンを単離し、ついで、Tet−ルシフェラーゼカセットを含有する組換えAd5でスクリーニングした。各クローンの5×105個の細胞を24ウェルプレート内に三重に播き、該細胞にAd5 Tet−lucをドキシサイクリンの存在下および非存在下で10の感染多重度(moi)で感染させた。感染の24時間後、細胞を回収し、細胞溶解物においてルシフェラーゼ活性を測定した(ルシフェラーゼアッセイ系;Promega)。遺伝子発現の誘導およびサイレンシングの両方を各クローンに関して、親293EBNA細胞において行った対照実験において得た相対光単位(rlu)値に対する比として評価した。
【0088】
2E2パッケージング細胞系を得るために、前記プロトコールに従い293EBNA Tet細胞をpE2ベクターで形質移入した。90μg/mlのヒグロマイシンBを含有するDMEMを使用して、安定なトランスフェクタントを選択した。耐性クローンを増殖させ、Ad5ΔE1−2EGFP DNAの形質移入によりスクリーニングした。陽性クローンをCPEの出現により特定し、Ad5ΔE1−2EGFPベクターの連続継代により確認した。1×106 2E2細胞(n=3)からのエピソームコピー数を染色体外DNA上で直接的に定量的リアルタイムPCRにより評価した。プローブ(5’−FAM−TGGCATGACACTACGACCAACACGATCT−3’−TAMRA)(配列番号18)ならびにプライマーE4−fw(5’−ACTACGTCCGGCGTTCCAT−3’)(配列番号19)およびE4−rw(5’−GGAGTGCGCCGAGACAAC−3’)(配列番号20)。
【0089】
ウイルスの増幅および力価測定
多欠失ウイルスの産生を2E2パッケージング細胞系において行った。アデノウイルスゲノムをPacI消化によりそれぞれのプラスミドから遊離させ、1μg/ml ドキシサイクリンの存在下で2E2細胞内に形質移入した。形質移入の4〜6日後、細胞を3サイクルの凍結/解凍により細胞溶解し、該溶解物の5分の1を使用して、該ウイルスを連続継代により増幅した。2層細胞ファクトリー(factory)(NUNC)内に播いた2E2細胞に感染させることにより、大規模増幅を行った。アデノウイルスベクターをCsCl勾配により精製し、透析し、リアルタイムPCRにより定量した。該CsCl精製ベクターの感染度を組織培養感染量50%(TICD50)として2E2上で評価した(43)。
【0090】
サザンブロット分析
pE2複製をサザンブロット分析により評価した。293EBNATet細胞を6cmディッシュ内に播き、ドキシサイクリン(1μg/ml)の存在下または非存在下、Lipofectamine2000(Invitrogen)により5μgのpE2ベクターで形質移入した。48時間後、Hirt法(22)により染色体外DNAを単離した。ついでDNAをNotIおよびDpnIで消化し、32P DNAプローブを使用する標準的な方法によるサザン分析に付した。Phosphor Imager(商標)系(Molecular Dynamics)を使用するオートラジオグラフィーによりシグナルを検出した。
【0091】
安定なpE2クローンからのエピソームDNAをHirtプロトコールに従い抽出し、BamHIで消化し、32P標識pE2 DNAをプローブとして使用するサザンブロット法により分析した。
【0092】
ウエスタンブロット分析
形質移入の48時間後、以下のとおりにタンパク質発現の分析を行った。2E2歳棒をリン酸緩衝食塩水(PBS)で2回洗浄し、6cmプレート当たり0.5mlのRIPAバッファー(1×PBS、1% NP−40、0.5% デオキシコール酸ナトリウム、0.1% SDS、0.05mM PMSF)を加えることにより細胞溶解した。プレートを氷上で1時間インキュベートし、ついで10,000×g、4℃での遠心分離後、可溶性タンパク質を細胞溶解物から集めた。30μgのタンパク質上でウエスタンブロット分析を行った。サンプルを10% SDS PAGE上で分離し、Protanニトロセルロースメンブレン(SchleicherおよびSchuell)上にブロッティングした。該メンブレンをポリメラーゼまたはpTPに対するウサギ抗血清と共に、および抗DBP mab(F.Graham,Mc Master University,Hamilton,Canadaにより快く提供されたクローンH2−19)と共にインキュベートした。ホースラディッシュペルオキシダーゼ結合二次抗体の存在下でのインキュベーション後、タンパク質をSupersignal West Pico化学発光基質(PIERCE)により検出した。以下の試薬を使用することによりHCVタンパク質発現を検出した:抗コアモノクローナル抗体(mab)B12.F8(M.Mondelli,University of Paviaにより快く提供された);抗E2、mab 185.C7;抗NS3 mab 10E5/24;抗NS5a、ウサギポリクローナル抗血清;抗NS5b mab 20B6/13。
【0093】
免疫化プロトコールおよび免疫応答の分析
6〜8週齢の雌C57BL/6、A2.1およびCB6F1マウス(Charles River Breeding Laboratories)を、該ウイルスを両方の大腿四頭筋内に注射することにより免疫した。注射の3週間後に免疫応答を分析した。
【0094】
該ウイルスを大腿四頭筋内に注射することにより、アカゲザル(Rhesus macaques)を免疫した。注射の4、6、8および12週間(W)後に免疫応答を分析した。
【0095】
E2タンパク質に対する抗体価を、Zucchelli,S.ら(55)に記載されているとおりにELISAにより測定した。細胞性免疫応答を以下に記載のとおりに評価した。HCVコア、NS3、NS4、NS5aおよびNS5bタンパク質の全配列を含む15マーの重複ペプチドのプールを使用して、HCV特異的IFNγ分泌細胞を明らかにした。いくつかの実験においては、CD8+エピトープを含有する9マーペプチドを使用して免疫応答を評価した(HCV NS3タンパク質からのpepl480,GAVQNEVTL(配列番号21))。以下のとおりにIFNγ酵素結合イムノスポットアッセイ(ELIspot)によりIFNγ分泌細胞を定量した。マルチスクリーン96ウェル濾過プレート(Millipore)を100μlの抗マウスIFNγ Mab(PharMingen)でコートし、4℃で一晩インキュベートし、ついで1×PBSで洗浄し、ウェル当たり200μlのR10培地で2時間ブロッキングした。脾細胞を免疫化マウスから調製し、R10培地(10% ウシ胎児血清、2mM L−グルタミン、50U/ml ペニシリン、50μg/ml ストレプトマイシン、10mM HEPES、50μM 2−メルカプトエタノールで補足されたRPMI 1640)内に再懸濁させ、ついで、2.5×105または5×105/ウェルの密度で抗IFNγ mabでコートされたマルチスクリーン96ウェルプレート上でプレーティングした。ついで脾細胞を200ng/ウェルのペプチドプールの存在下で24時間インキュベートした。十分な洗浄(1×PBS、0.005% Tween)の後、ビオチン化ラット抗マウスIFNγ抗体(PharMingen,San Diego,Calif.)を加え、室温で3時間インキュベートした。最後に、ストレプトアビジン−アルカリホスファターゼ(PharMigen)および1−Step NBT−BClP現像液(Pierce,Rockford,Ill.)を該ウェルに加えた。ELIspotリーダー(Bioline)を使用してスポットを計数した。
【0096】
IFNγ細胞内染色およびFACS分析を以下のとおりに行った。Elispotアッセイに関して記載されているとおりに調製した脾細胞を、タンパク質輸送を抑制するブレフェルジン(GolgiPlug,PharMingen)を含有するR10培地内でペプチドプールと共に一晩インキュベートした。FACSバッファー(PBS w/o CaおよびMg、1% FCS、0.01% NaN3)中、飽和量の精製抗マウスCD16/CD32と共に細胞をインキュベートすることにより、細胞ブロッキングを行った。FACSバッファーでの洗浄の後、APC結合抗マウスCD3e、PE結合抗マウスCD4およびPerCP結合抗マウスCD8a抗体を該細胞に加え、室温で30分間インキュベートした。ついで、Cytofix/Cytoperm Plus(GolgiPlugを伴う)キットを使用して、細胞を4℃で20分間にわたり透過性亢進させた。Perm/Washでの洗浄の後、FITC結合抗マウスIFNγを加え、該細胞を室温で30分間インキュベートした。該最終染色工程後、該細胞を洗浄し、1% ホルムアルデヒド中で固定した。データを、FACSCalibur(Beckton & Dickinson)を使用して集め、Cell−Questソフトウェア(BD Biosciences)を使用して分析した。すべての抗体および二次試薬はBD PharMingen(San Diego,CA)から購入した。
【0097】
Accuspin Istopaqueチューブ(Sigma Aldrich cat A0561)を該製造業者の説明に従い使用して、サルPBMCをEDTA処理末梢血から単離した。簡潔に説明すると、血液を、等容量のHBSS(ハンクス平衡溶液Gibco cat 14175−053)を含有するAccuspinチューブ内に移し、800g、室温で15分間遠心分離した。PBMCバンドを集め、HBSSで1回、R10で1回洗浄し、最後にR10に再懸濁させた。各ウェル内でプレーティングする細胞の量が異なる以外は前記のとおりに(2×105および4×105/ウェル)、g−IFN−Elispotを行った。
【0098】
ワクシニアウイルスチャレンジ
免疫化マウスに5×106pfuのVV−NSをi.p.注射した。5日後に摘出した個々のマウスからの対(ペア)になった卵巣を均質化し、3回、凍結−解凍し、Hul43TK−細胞の単層上で10倍希釈物をプレーティングすることにより力価測定した。48時間後、0.5% クリスタルバイオレットでの染色により力価を読取った。
【0099】
実施例
本発明の種々の特徴を更に例示するために、以下に実施例を記載する。該実施例は、特許請求されている発明の実施のための有用な方法をも例示するものである。該実施例は本発明を限定するものではない。
【実施例1】
【0100】
Tet−S/rtTA2を共発現する細胞系の開発
293EBNA細胞系におけるpIREStTS/rtTApuro形質移入およびそれに続くピューロマイシン選択(「材料および方法」を参照されたい)により得た安定なクローンを、Tet誘導性ルシフェラーゼ発現カセットを含有する第1世代Adベクター(Ad Tet−luc)を使用することによりスクリーニングした。ピューロマイシン耐性クローンを24ウェルプレート内に三重に播き、10のmoiで細胞にAd Tet−lucを感染させ、該細胞をドキシサイクリン(1μg/ml)の存在下/非存在下で維持した。感染の24時間後、該粗細胞溶解物においてルシフェラーゼの発現を測定した。20倍を超えるルシフェラーゼ活性の誘導を示すクローンを選択し、増殖させた。図2に示すとおり、選択されたすべてのクローンにおいて、Tetサイレンサーの共発現はルシフェラーゼの基底レベルを減少させた。ドキシサイクリンの存在下で基底発現に対して25倍の減少を示し最良の誘導比(100倍超)を伴うクローン1.1(293EBNATetと称される)を選択し、増殖させた。
【実施例2】
【0101】
E2/E4 orf6アデノウイルスアンプリコン(pE2)の構築
すべての初期遺伝子が欠失したAdベクターを機能的に相補するために、以下の要素を含有する、Ad5に基づくアンプリコンを構築した:i)EBNA−1タンパク質を発現する分裂細胞の核における安定な維持のためのEBVの潜在性複製起点(Ori−P)(48);ii)tk−ヒグロマイシンB選択マーカー;iii)Adに基づく様態でのプラスミド複製を可能にするpFG140(19)由来のAd5ウイルスITR結合部;iv)二方向性テトラサイクリン誘導性プロモーターの制御下の2つの分岐転写単位において配置されたAd5 E2(ポリメラーゼ、プレ末端タンパク質およびDNA結合タンパク質)およびE4−orf6遺伝子。E2およびE4 orf6遠位プロモーター(17)に対するOriPのエンハンサー効果を減少させるために、OriP要素に隣接する2つのニワトリβグロビンHS4絶縁体(insulator)二量体(9)をも導入した。得られたプラスミド(pE2)の構造を図1Bに示す。
【0102】
pE2で形質移入された293EBNATet細胞の培地におけるドキシサイクリンの添加の際のE2遺伝子発現の誘導をウエスタンブロットにより測定した。図4B(レーン2、5および8)に示すとおり、エフェクター薬の非存在下では形質移入の48時間後にタンパク質発現は検出されなかったが、1μg/mlのドキシサイクリンを加えることにより転写を誘導した場合には、すべてのE2タンパク質の強力な発現が明らかであった(レーン3、6および9)。形質移入の4および6日後にも同様の結果が得られた(データ非表示)。
【0103】
Ad複製に必要なシスおよびトランス作用性要素の両方が前記系内に存在するため、E2遺伝子発現の誘導が293EBNATetにより形質移入された細胞におけるpE2 DNA複製をも誘発するのかどうかを試験した。形質移入の48時間後、全DNA上のサザンブロットによりプラスミド複製を検出した。サンプルを、まず、DpnIで消化して投入プラスミドDNAを除去し、ついでNotIで消化して、Ad ITRを介して複製された直鎖状形態と天然環状プラスミド形態とを区別化した(図3)。図3に示すとおりにpE2から誘導されたDNAプローブとブロットとをハイブリド形成させたところ、該ブロットはpE2の環状形態に関する12.6Kbのプラスミド由来のバンドを示した(図4A、レーン1)。pE2で形質移入された細胞からの全DNAを分析したところ、複製されたアンプリコンの予想サイズのバンドは、ドキシサイクリンで細胞を誘導した場合にのみ、視認可能であった(図4A、レーン2および3)。ITR結合部が欠失したpE2プラスミド誘導体の形質移入からはDNA複製の証拠は検出されなかった(データ非表示)。これらのデータは、tTS/rtTAサイレンシング/活性化系が、該プラスミドが一過性形質移入後に高コピー数で存在する場合であっても、pE2機能の完全な遮断を可能にするが、初期遺伝子発現のデオキシサイクリン誘導に際してはアデノウイルス特異的に該アンプリコンの効率的な複製を支持しうることを示した。それらは、初期遺伝子が欠失したアデノウイルスベクターのレスキューおよび増殖のための適当な系としての293EBNATet細胞と組合されたpE2アンプリコンを支持する確固たる証拠を提供した。
【実施例3】
【0104】
E1、E2、E4相補細胞系の作製
E2遺伝子発現の誘導は、アデノウイルス複製装置の活性化を介して、直鎖状DNAとしてのpE2の複製をもたらすことが観察された。pE2を使用して、Tet転写サイレンサー(tTS)および逆Tetトランスアクチベーター2(rtTA2)を発現する293EBNA細胞を形質転換して、2E2安定細胞系を得た。2E2細胞は、ドキシサイクリンを培地に加えた場合には、Ad5第1世代(FG)ベクターに感染した293細胞より高いレベルのポリメラーゼ、前駆体末端タンパク質(pTP)およびDNA結合タンパク質(DBP)を産生した。E2およびE4ORF6遺伝子の発現は、誘導されると、第1世代(FG)アデノウイルスベクターと比較しうるレベルまでの、E1、E2、E3およびE4遺伝子を欠く多欠失Ad5ベクターの増幅を効率的に支持した。
【0105】
簡潔に説明すると、pE2で形質移入された293EBNATet細胞を、「材料および方法」に記載されているとおりに、ヒグロマイシンBの存在下で選択した。個々のクローンを増殖させ、E2遺伝子欠失を含有するAd5ベクターのレスキューおよび増殖を調べることによりスクリーニングした。6ウェルプレート内に播いた細胞をdoxy存在下のAd5ΔE1−2ベクターでの形質移入に付した。形質移入の7日後、細胞を凍結−解凍により細胞溶解し、500μlの細胞溶解物を使用して各対応クローンの新鮮単層に感染させた。継代1におけるCPEの直接観察により陽性クローンの評価を行った。ついで該ベクターを該選択クローンにおいて連続継代し、リアルタイムPCRにより増殖を評価した。2回の連続継代の後、ウイルスゲノムは細胞溶解物1ml当たり約1×1010 ゲノムのプラトー付近に達し、ついでこれを、感染moiの上昇にもかかわらず維持された(データ非表示)。サザンブロット分析で染色体外DNAを評価することにより、未修飾無傷エピソーム要素としてpE2を含有するいくつかのクローンを特定した。
【0106】
細胞系の特徴づけ及びΔE1,E2,E3,E4ベクターの増幅の後続工程のために、2E2と称されるクローン11を選択した。まず、2E2細胞におけるpE2のコピー数を、染色体外DNA上のリアルタイムPCRにより、16コピー/細胞の平均として決定した(n=3)。継代にわたる2E2細胞におけるpE2エピソームプラスミドの安定性を評価するために、15継代後にHirt法(22)によりエピソームDNAを抽出し、ついでBamHIで消化し、プラスミド全体をプローブとして使用するサザンブロットにより分析した。図5Aには、元のプラスミドと比較した、細胞系から抽出されたエピソームの制限パターンが示されている。それらのパターンは同一であった。このことは、該エピソームが細胞核内で経時的に安定に維持されること、およびプラスミド構造内の再構成が生じなかったことを示している。エピソームDNAの安定性は、ドキシサイクリンの非存在下で検出可能な発現を伴わない一過性形質移入実験で既に観察されているものに類似した細胞系の15継代後にウエスタンブロットによりE2タンパク質の発現を分析することによっても証明された(図5B)。E2タンパク質発現レベルを比較するために、FGウイルスに感染した未誘導2E2細胞から得た細胞抽出物を含めた。
【実施例4】
【0107】
ΔE1−4orf3+ Ad5バックボーンの構築
該ベクター構築を促進するために、左ITRおよびパッケージングシグナル(Ad5[nt]1−450)(配列番号1)ならびに発現カセットに隣接するpIX遺伝子(Ad5[nt]3511−5792)(配列番号2)を含むAd5断片を含有するシャトルベクターを構築した。大腸菌(E.coli)株BJ5183における相同組換えにより発現カセットをAd5 E1領域内に組換えた。その新たなベクター系を評価するために、pAd5ΔE1−4orf3+EGFPを構築した。pAd5ΔE1−4orf3+EGFPベクターをPacIで線状化して感染性ウイルスDNAをプラスミド配列から遊離させ、2E2細胞内に形質移入し、それをドキシサイクリンの存在下または非存在下でインキュベートした。2連続継代後に得た結果を図6に示す。EGFP形質導入ウイルス粒子およびCPEは、培地へのドキシサイクリン添加によりE2およびorf6遺伝子が誘導された場合にのみ生じた。orf3およびorf6タンパク質の共発現は、E4単位が欠失したAdベクターの高力価増幅に寄与する(25)。相補遺伝子誘導の非存在下ではウイルス粒子は生じなかった。ベクター産生のプラトーは、細胞の100%がEGFP陽性である場合に、2連続継代の直後に観察された(図6)。高力価への多欠失ベクターの増殖の支持における該新規細胞系の効率を評価するために、5つの2層細胞ファクトリー(Nunc Inc.)内に播かれた約5×108個の2E2細胞を感染させることにより、大規模製造を試みた。該ウイルスをCsCl勾配の2回通過により精製し、リアルタイムPCRにより定量して、最終的に、約5000vp/細胞の生産で2×1012 vp/mlの力価を得た。EGFPおよびHCVを発現する多欠失ベクターとFGベクターとの間の比較を表1に示す。
【0108】
【表1】
【0109】
すべての初期遺伝子の欠失を組合せることによりAd5バックボーン内に生じた理論的空間は約12.4Kbである。該新規ベクター系の大きな容量を利用して、マウスサイトメガロウイルス(MCMV)プロモーターに融合した全HCVポリタンパク質遺伝子用の発現カセットを挿入した。該HCVポリタンパク質発現カセットは、5’および3’非翻訳領域を除去し、コアATGの上流に最適コザック配列を挿入し、NS5Bレプリカーゼの触媒ドメインを突然変異させて酵素活性を除去することにより構築された(44)。トランスジーンの発現効率を増加させるために、ヒトCMVプロモーターを、FGアデノウイルスベクターにおいて4〜30倍強力であると報告されているマウスCMVプロモーター(1)で置換した。
【0110】
Ad5ΔE1−4orf3+HCVベクターは2E2細胞内の形質移入により成功裏にレスキューされた。培地にドキシサイクリンを1μg/mlの最終濃度で加えることにより、形質移入の直後にE2遺伝子発現を誘導した。2E2細胞内での連続継代によりAd5ΔE1−4orf3+HCVベクターを増幅した。「材料および方法」に記載されているとおりに、粗細胞溶解物内のウイルスゲノム濃度をリアルタイムPCRにより評価した。大規模調製物を得るために、4回の連続増幅継代の後に得た粗溶解物を使用して、2.8×109個の2E2細胞に約100ゲノム/細胞のmoiで感染させた。感染の48時間後、完全なCPEが明らかに認められた時点で細胞を回収した。精製ウイルスの最終収量を表1に示す。293細胞内で増殖させたEGFPを発現するΔE1E3FGベクターと異ならない約5000粒子/細胞の生産を得た。
【0111】
ポリメラーゼおよびプレ末端タンパク質の欠失は、これらの2つの遺伝子の一部分のみを含むものであったため、AdベクターとpE2エピソームとの間の相同性の領域は、野生型遺伝子をウイルスゲノム内にレスキューして戻すのに理論的に十分なものである。連続継代に際してのAd5ΔE1−4orf3+HCVベクターの構造的完全性を評価するために、および野生型初期遺伝子を含有するウイルスの再構成が2E2細胞内でのベクター増幅中に生じうるかどうかを試験するために、CsCl精製ベクターのDNA構造を決定した。5継代後にウイルス粒子から精製したDNAから得たパターンとプレ−アデノプラスミドの放射能標識制限パターンとの比較を図8に示す。
【0112】
該野生型遺伝子を含有する断片のサイズを比較するために、FGプラスミドベクターをゲル(レーン1)内に含めた。Ad5ΔE1−4orf3+HCVベクターの制限パターンは親プラスミドのものと同一であるようであり、再構成または野生型E2−E4遺伝子を含有するベクター種の出現の証拠は認められなかった。
【0113】
種々のmoiのベクターを使用する293およびHeLa細胞系のインビトロ感染により、HCVタンパク質の発現の効率を評価した。HCVコア、E1、E2、NS3、NS4、NS5aおよびNS5Bに対する特異的モノクローナル抗体またはポリクローナル抗血清でのウエスタンブロット分析は該感染細胞におけるHCVタンパク質の存在を示したが、このことはHCVポリタンパク質の正しいプロセシングを示している(図9)。未プロセシング産物は検出されなかった。
【0114】
2E2細胞系は、FGベクターに感染した293細胞より高いレベルのE2aおよびE2bを発現することを、データが示していることに注目すべきである。理論により束縛されるものではないが、比較的高いレベルのE2aおよびE2bの産生が細胞当たりの多欠失ベクター粒子の高収量につながったと考えられる。細胞当たりの多欠失粒子の収量は、FGベクターから得られた収量と一貫して比較しうるものであった。さらに、PolおよびpTP遺伝子がpE2との相同組換えによりベクターバックボーンにおいてレスキューされうる理論的可能性にもかかわらず、Ad5ΔE1−4orf3+ベクターは連続継代にわたり安定であることが示された。相補タンパク質の発現の、観察された高いレベルは、おそらく、多欠失ベクターに対するE2b野生型ウイルスの選択的利点を低下させるであろう。
【実施例5】
【0115】
AdΔE1−4orf3+HCVベクターでの免疫化はマウスにおける強力なCMI応答を誘導する
ヒトおよびチンパンジーにおいて観察されたHCV感染の消散は、典型的には、複数のエイトープに対する強力なT細胞応答を伴っている(56,57)。感染対象において特定されたHLAクラスI限定エピトープは、クラスタリングの証拠を伴うことなく、ゲノム全体に広がっている(58に概説されている)。細胞性免疫応答を惹起するHCVポリタンパク質を発現する多欠失Adベクターの効力をマウス免疫実験において評価した。該ベクターは、感染細胞のウエスタンブロット分析により示されるとおり成熟産物へと正しくプロセシングされるポリタンパク質前駆体全体の合成を導く。また、種々の系統のマウスにおいてマッピングされたHCV−BK CD8+エピトープを含有するオリゴペプチドを使用して、該免疫化を観察した。HCVエピトープに特異的なCD4+およびCD8+ T細胞を、コア、E2およびNSタンパク質の配列全体に及ぶ重複15マーペプチドのプールを使用することにより、IFN−γ Elispotおよび細胞内染色(ICS)により決定した。
【0116】
CB6F1およびHLA A2.1マウス系統を免疫することにより、複数のウイルス決定基に対する強力なT細胞免疫応答(全CD8+細胞の0.2〜2.25%)を測定した。免疫化マウスの脾細胞の分析は、低レベルのCD4+抗原特異的T細胞を伴うCD8+偏向免疫応答を示した。この免疫応答特性は、送達された抗原に伴われているというよりもワクチン接種方法に伴われたものである。Casimiroらにより、Ad5gagワクチンで免疫されたアカゲザルにおいて、低い比のCD4+/CD8+が観察され、CD8+表現型の応答細胞の大部分は強力なCTL活性伴っていた(59)。これに対して、筋肉内DNA注射によるHCV抗原での遺伝的免疫化は、マウスおよび非ヒト霊長類の両方において、より釣りあったCD4+/CD8+応答をもたらした(Nicosia A.未公開の結果)。
【0117】
種々の量のベクターにより誘導された細胞性免疫を、漸増量の筋肉内注射Ad5ΔE1−4orf3+HCV(1×107〜1×1011 vp/マウス)でC57Bl6マウスを免疫することにより測定した。免疫化の3週間後、NS3タンパク質のヘリカーゼドメイン内にマッピングされたCD8+T細胞エピトープ(GAVQNEVTL(配列番号21)aa 1629−1637 HCV 1b)に対するT細胞応答に関してマウスを試験した。新鮮に単離された脾細胞を9マーペプチドと共に一晩インキュベートし、ついでIFNγELISPOTアッセイにより分析した。
【0118】
表2に示すとおり、誘導されたT細胞応答の大きさはAd5ΔE1−4HCVの用量に依存し、最初の陽性結果は1×108vp/用量で観察された。この用量のベクターで、5匹中4匹のマウスがNS3に対する免疫応答を引き起こし、特異的T細胞の度数は1×106脾細胞当たり100〜180 CD8+細胞であった。
【0119】
表2におけるデータは、5匹の免疫化マウスから得た100万個の脾細胞当たりのIFNγスポット形成細胞(SFC)の数を要約している。C57Bl6マウスにおけるBK NS3ヘリカーゼドメイン内にマッピングされたCD8+エピトープを含有する1480ノナマー(GAVQNEVTL)(配列番号21)と共に脾細胞をインキュベートした。1匹の動物から得た値および免疫化マウスの各群から計算した相乗平均が該表に示されている。抗原特異的CD8+ T細胞の度数は、1011 vp/動物を注射することにより百万個の脾細胞当たり400〜1000のSFCの範囲で568の相乗平均値まで用量と共に増加した。
【0120】
【表2】
【実施例6】
【0121】
ヒトHLA−A2.1を発現するトランスジェニックマウスにおける多特異的CMI応答の誘導
防御性HCVワクチンは、ヒトMHC対立遺伝子の及びウイルスの遺伝的多様性のため、一般的集団における広範な細胞性免疫応答を誘導する必要がある。Ad5ΔE1−4HCVベクターでのマウスの免疫化は、HCVポリタンパク質の複数のエピトープに対する強力なCD8+T細胞応答を誘導した。より詳しくは、複数のHCVエピトープに対する細胞性免疫応答を惹起するAd5ΔE1−4orf3+HCVベクターの能力を測定した。免疫応答の拘束のため、CB6F1およびHLA A2.1トランスジェニックマウスにおいて、ベクター免疫化により惹起されるHCV特異的T細胞応答を測定した。
【0122】
5匹/群の動物の大腿四頭筋にAd5ΔE1−4orf3+HCVの1×1010ウイルス粒子を注射した。注射の3週間後、ワクチンにより誘導された抗HCV免疫の強度および質をICS法により評価することにより、マウスを分析した。脾細胞からの抗原特異的IFNγ分泌を、コア(aa 1−190)およびNS3〜NS5bタンパク質の非構造領域(aa 1026−3009)に及ぶ10残基重複する15マーから構成される7個のペプチドプールで刺激した。5匹のマウスのプールに関して脾細胞の分析を行った。
【0123】
図10に示す結果は、Ad5ΔE1−E4−HCVベクターが、ヒトHLA−A2.1を発現するトランスジェニックマウスにおいて、強力かつ多特異的なT細胞応答を誘導することを示している。より詳しくは、該免疫化は該ペプチドプールのすべてに対するT細胞応答を引き起こした。CD4+およびCD8+の両方の抗原特異的Tリンパ球が観察されたが、応答細胞の大部分はCD8+であった。抗原特異的CD8+リンパ球の度数は、該コアに対するCD8+ T細胞の2.2%から、NS5BのC末端部分に応答するCD8+ Tの0.2%まで、抗原によって様々である。
【実施例7】
【0124】
Ad5ΔE1−E4−HCV免疫化はA2.1マウスにおいてCMI応答を惹起する
トランスジェニックA.21マウスをW=0およびW=2に、1010pp/マウス/注射の用量のAd5ΔE1−E4HCVまたは50μg/マウス/注射の用量の対応Ad5シャトルベクター(pShAd5HCV)で免疫した。1)Ad5ΔE1−E4−HCVで初回抗原刺激および追加抗原刺激し、ならびに2)pSh−Ad5−HCVで初回抗原刺激しAd5ΔE1−E4−HCVで追加抗原刺激し、第4週に動物から得た精製脾細胞の免疫応答を、HCVポリタンパク質全体に及ぶペプチドプールを使用するγIFN−ElispotおよびγIFN細胞内染色により分析した。
【0125】
NS3ヘリカーゼ領域全体に及ぶペプチド(I〜XVIII)のサブセットを使用する実験において、該応答の特異性を測定した。簡潔に説明すると、Ad5DE1−E4HCVで初回抗原刺激しpSh−Ad5−HCVで追加抗原刺激した2匹のマウスから脾細胞を単離した。図12AにおけるElispotの結果は、該マウスが、NS3領域に及ぶペプチドに対して強反応性である広範な免疫応答で応答したことを示している。該データは更に、Ad5ΔE1−E4−HCVまたはpSh−Ad5−HCVのいずれかで免疫されたマウスにおけるHCV特異的免疫応答の初回抗原刺激および追加抗原刺激の両方にAd5ΔE1−E4−HCVを使用しうることを証明した。
【0126】
陽性閾値を超える応答を惹起するプール間の交差点は、考えられうる刺激性ペプチドを表す(ハイライト表示)(図12B)。ついでこれらのペプチドをγIFN−Elispotアッセイにおいて個々に試験した。図12Cに要約した結果は、ペプチド95(LAAKLSGLGINAVAY)(NS3 aal403−1417)(配列番号22)エピトープに対する特異性/反応性を示している。したがって、これらのマウスにおいて惹起された免疫応答は、HCV患者において既に特定されているNS3ヘリカーゼ領域におけるCD8+エピトープに対する特異性を含む特異性により特徴づけられる。
【実施例8】
【0127】
Ad5ΔE1−E4−HCVでの免疫化はVV−NSでのチャレンジに対する防御をもたらす
この実験は、惹起された免疫応答が、HCV非構造タンパク質を発現する組換えワクシニアウイルスでのチャレンジに対する防御をもたらしうることを証明している。Ad5ΔE1−E4HCVでの免疫化により惹起された免疫応答が後続のウイルスチャレンジに対する防御をもたらすのかどうかを判定するために、実施例7に記載のプロトコールに従い免疫したマウスを第4週に、HCV非構造領域(VV−NS)を発現する組換えワクチンウイルス(5×106 pfuの用量)でチャレンジした。実験対照として、W=0およびW=2にAd5ΔE1−E4EGFPの2回の注射で免疫したマウスを使用した。5日後、対(ペア)になった卵巣を摘出し、VVを力価測定した。図13に示す結果は、対照マウスと比較した場合の免疫化マウスのVV力価における有意な減少を示している。灰色のドットは相乗平均力価(N=5)を表す。* = 対照に対するp<0.05(Mann−Whitneyランク)。
【実施例9】
【0128】
Ad5ΔE1−E4HCVでの免疫化はアカゲザルにおいてHCV特異的免疫応答を初回刺激する
アカゲザルの大腿四頭筋に1010ppのAd5ΔE1−E4−HCVを注射した。該免疫化の効力を注射後の種々の時点(W=4、W=6、W=8、W=12)で末梢血単核細胞(PBMC)に関するγIFN−Elispotアッセイにより評価した。該注射により惹起された免疫応答は注射の6週間後にピークとなり、複数のHCVエピトープに対するものであった。これらの3頭のサルのうちの1頭は、注射の12週間後までの、長期にわたる応答を示した。これらのデータは、霊長類動物モデルにおいて免疫応答を惹起するためにAd5ΔE1−E4−HCVを使用しうることを示している。
【0129】
図14Aは、Ad5DE1−E4−HCVの1回の投与の後にサル4061において惹起された経時的応答を表す。結果は注射の4、6、8および12週間後の106PBMC当たりのg−IFNスポット形成細胞数(SFC)として表されている。各棒グラフは、別々のペプチドプールに対する応答を表す。
【0130】
図14Bは、γIFN−Elispot6アッセイにおいて評価された、Ad5DE1−E4−HCVの注射の6週間後の3頭のサル(番号4061、9003、7023)の免疫応答を示す。結果は、106PBMC当たりのg−IFNスポット形成細胞数(SFC)として表されている。各棒グラフは、別々のペプチドプールに対する応答を表す。
【0131】
本発明は、特定の実施形態とみなされるものに関して説明されているが、本発明はそのような実施形態には限定されないと理解されるべきである。逆に、本発明は、添付の特許請求の範囲の精神および範囲内に含まれる種々の修飾および均等物を含むと意図される。
【0132】
本開示の全体にわたって引用されているすべての参考文献を、参照により明示的に本明細書に組み入れることとする。
【0133】
【表3】
【図面の簡単な説明】
【0134】
【図1A】安定な293EBNATetクローンを得るために使用したプラスミドの概要図を示し、プラスミド成分の直線的表示を示す。
【図1B】安定な293EBNATetクローンを得るために使用したプラスミドの概要図をし、pE2プラスミドの概要図を示す。
【図2】図2はAdTetLuc感染クローンにおけるルシフェラーゼ発現のグラフ表示を示す。
【図3】図3は環状および直線形態のpE2の概要図を示す。pE2の環状形態と直線形態との識別を可能にするNotI消化により得たDNA断片も示されている。
【図4A】ドキシサイクリンによるAd5 E2遺伝子発現の活性化の際のpE2の複製を示すサザンブロット分析を示す。
【図4B】E2タンパク質のtet誘導性発現を示すウエスタンブロット分析を示す。
【図5A】2E2クローンから抽出されたpE2の構造およびE2タンパク質の発現を示し、クローン293EBNATetおよび2E2から抽出されたpE2の構造を示すサザンブロット分析である。
【図5B】Ad5ΔE1ベクターに感染した細胞におけるE2発現と比較した場合の2E2安定細胞系におけるE2タンパク質のtet誘導性発現を示すウエスタンブロット分析である。
【図6】図6は、EGFPを発現するAd ΔE1−4ベクターをレスキューし増殖させるための2E2細胞の使用を示す一連の写真を示す。
【図7A】Ad5ウイルスの概要地図である。この図にはすべての欠失領域が示されている。
【図7B】多欠失ベクターのE1領域内に導入されたHCV(BK株)ポリタンパク質発現カセットの概要図を示す。
【図8】図8はAd ΔE1−4orf3+HCVの制限分析を示す。
【図9】図9は、Ad5ΔE1−4HCV感染細胞におけるHCVタンパク質の発現を示すウエスタンブロット分析を示す。
【図10A】マウスにおけるAd5ΔE1−4HCVウイルス免疫化に対するインビボCD8+ T細胞応答を特徴づけるFACSデータのグラフ表示を示す。
【図10B】マウスにおけるAd5ΔE1−4HCVウイルス免疫化に対するインビボCD8+ T細胞応答を特徴づけるFACSデータのグラフ表示を示す。
【図11A】図11は、本開示に記載されているポリヌクレオチドおよびポリペプチド配列のヌクレオチドおよび/またはアミノ酸配列(すなわち、配列番号1〜21)を示す。
【図11B】図11は、本開示に記載されているポリヌクレオチドおよびポリペプチド配列のヌクレオチドおよび/またはアミノ酸配列(すなわち、配列番号1〜21)を示す。
【図11C】図11は、本開示に記載されているポリヌクレオチドおよびポリペプチド配列のヌクレオチドおよび/またはアミノ酸配列(すなわち、配列番号1〜21)を示す。
【図11D】図11は、本開示に記載されているポリヌクレオチドおよびポリペプチド配列のヌクレオチドおよび/またはアミノ酸配列(すなわち、配列番号1〜21)を示す。
【図11E】図11は、本開示に記載されているポリヌクレオチドおよびポリペプチド配列のヌクレオチドおよび/またはアミノ酸配列(すなわち、配列番号1〜21)を示す。
【図11F】図11は、本開示に記載されているポリヌクレオチドおよびポリペプチド配列のヌクレオチドおよび/またはアミノ酸配列(すなわち、配列番号1〜21)を示す。
【図11G】図11は、本開示に記載されているポリヌクレオチドおよびポリペプチド配列のヌクレオチドおよび/またはアミノ酸配列(すなわち、配列番号1〜21)を示す。
【図11H】図11は、本開示に記載されているポリヌクレオチドおよびポリペプチド配列のヌクレオチドおよび/またはアミノ酸配列(すなわち、配列番号1〜21)を示す。
【図12A】NS3ヘリカーゼ内のエピトープのマッピングに用いた方法を示す。Ad5ΔE1−E4HCVで初回抗原刺激されpSh−Ad5−HCVで追加抗原刺激された2匹のマウス(マウス4,黒塗り棒線、マウス5,付点棒線)から精製した脾細胞を、全NS3ヘリカーゼ領域に及ぶペプチド(I〜XVIII)の二次元サブセット上でγ−IFN−Elispotにおいて試験した。
【図12B】NS3ヘリカーゼ内のエピトープのマッピングに用いた方法を示す。図12Bに示されているデータは、免疫応答を特徴づけするために使用したパネルAにおいて特定された陽性閾値を超える応答を惹起したペプチドプール間の交差点を要約したものである。
【図12C】NS3ヘリカーゼ内のエピトープのマッピングに用いた方法を示す。パネル12Cは、該応答をもたらすNS3エピトープを特定するために行ったγ−IFN−Elispotアッセイの結果を要約したものである。
【図13】図13は、VV−NSチャレンジに対する防御の誘導における免疫化の効力を示す。灰色のドットは相乗平均力価(N=5)を表す。アステリスクは対照に対するp<0.05を示す(Mann−Whitneyランク)。
【図14A】Ad5ΔE1−E4HCV免疫化に応答してアカゲザルにおいて惹起された免疫応答を要約したものであり、Ad5ΔE1−E4HCVの1回の投与に際してサル4061において惹起されγ−IFN−Elispotにより分析された経時的免疫応答を表す。
【図14B】Ad5ΔE1−E4HCV免疫化に応答してアカゲザルにおいて惹起された免疫応答を要約したものであり、Ad5ΔE1−E4HCVの1回の投与により3頭の個々のサルにおいて誘導され注射の6週間後にγ−IFN−Elispotにより分析された免疫応答を示す。
【技術分野】
【0001】
本発明は、分子生物学の分野に関し、特に、多欠失または完全欠失ヘルパー非依存性アデノウイルスベクターの高クローニング容量プロデューサー細胞系を製造するための、誘導可能な自己複製能を有するエピソームプラスミドの開発および使用に関する。
【背景技術】
【0002】
アデノウイルス(Ad)は、多種多様な組織の休止細胞および増殖細胞の両方に感染しうる広範な指向性により特徴づけられる。一般に、野生型ヒトAd5ウイルスによる許容細胞の感染は約104〜105個のウイルス粒子の産生を引き起こす。ウイルスゲノムの扱い易さと共に、高力価増殖能は、Adベクターを、ワクチン接種および遺伝子治療のための並びに細胞培養内での遺伝子発現のための遺伝子導入ベクターとしての使用に魅力的なものにする。
【0003】
安全性プロファイルの改善(例えば、ウイルス遺伝子発現から生じる毒性の最小化)および前世代のベクターのクローニング容量の増加を目的として、ヒトAd5およびAd2に基づく幾つかのベクター系が開発されている。代替ベクター系の開発のための方法は、典型的には、ベクターバックボーンからアデノウイルス遺伝子を欠失させることを含む。アデノウイルスゲノムは機能的には、非構造産物および構造産物をコードする遺伝子を含む、初期領域および後期領域に細分される。第1の領域は、ウイルスDNA複製の前に発現されるポリペプチドをコードする初期(E)遺伝子を含む。第2の領域は、ウイルス複製の後続段階で必要なポリペプチドをコードする後期(L)遺伝子を含む。アデノウイルスゲノムのL領域は本質的には、ウイルス粒子の集合に必要な構造タンパク質をコードする。
【0004】
コンピテント細胞の感染後転写される第1の領域は、E遺伝子およびL遺伝子の両方のトランス活性化に関与するタンパク質をコードするE1a領域である。続いて転写されるE1b領域は、RNA合成を調節し、E1aによるアポトーシス作用から宿主細胞を防御するポリペプチドをコードする。したがって、E1a/E1bの遺伝子/機能はウイルス複製に必須である。第1世代(FG)アデノウイルスベクターは、典型的に、アデノウイルスE1遺伝子内の欠失を含む。これらの欠失は、修飾転写単位のタンパク質産物がトランスで与えられない限り、アデノウイルスを複製欠損にする。一般に、FGアデノウイルスベクターの最高容量は8kbを超えない。FG Ad5ベクターはE1領域の欠失または修飾により弱毒化されるが、一般に、いくつかの腫瘍細胞系内での複製能の維持および漏出遺伝子発現の両方の結果として、インビトロで細胞毒性が見られる。典型的に、FG Adベクターでのインビボ形質導入は比較的短いトランスジーン発現をもたらす。
【0005】
追加的なウイルス遺伝子の欠失に基づく第2および第3世代ベクター系はアデノウイルス遺伝子発現の更なる弱毒化およびベクター容量の増加をもたらす。より詳しくは、より新しい世代のベクターはウイルスE2、E3および/またはE4遺伝子内の追加的な欠失を含む。ΔE1/E3/E4ベクターのクローニング容量は約11kbに近い。E2領域は、ウイルスDNAポリメラーゼ、プレ末端タンパク質およびウイルスDNAに結合するタンパク質を含むウイルス複製に直接関与するタンパク質をコードする。E3領域は、ウイルス複製には要求されないが宿主免疫応答を制御するようインビボで機能するタンパク質をコードすることが公知である。E4領域遺伝子は、宿主細胞の遺伝子発現を低下させアデノウイルスゲノムのE2およびL領域の転写を増強するよう機能するポリペプチドをコードする。種々の組合せのE1、E2a/b、E3および/またはE4欠失を有する多欠失ベクターの使用は、古典的なFG(2−4,23,24,33,45,52)ベクターほどはインビトロで細胞毒性ではなく、マウス肝臓において、より安定であることが認められている。しかし、より新しい世代のアデノウイルスベクターが有意に長い持続能を有するという決定的な証拠は存在しない。さらに、追加的な欠失の導入は、生じる力価を有意に低下させ、臨床応用のための該ベクターの大規模製造をより困難にする(33,18)。実際、ほとんど全ての場合に、パッケージング/プロデューサー細胞系内に安定に導入された相補遺伝子の発現は、複数の欠失を相補しなければならない場合非効率的である(5,54)。
【0006】
現在のところ、ヘルパー依存性(HD)完全欠失アデノウイルスベクター遺伝子が、インビボ遺伝子導入のための最も効率的で安全なベクターの1つとみなされている(5,15,28,36,39−41,43,54)。完全欠失Adベクターは、複製およびパッケージング(すなわち、包膜)に必要なシス要素のみを含有し、すべてのアデノウイルス遺伝子を欠く。伝統的には、必要なアデノウイルス遺伝子はヘルパーウイルスによりトランスで供与される。しかし、HDベクターはいくつかの欠点により特徴づけられる。なかでも、該系は、トランスジーンを含有するHDベクターと必要なウイルスタンパク質をトランスで供与するヘルパーウイルスとによるパッケージング細胞系の共感染を要するため、3つの独立した成分の制御を要することが挙げられる。実際、医薬規模でのヘルパー依存性アデノウイルスベクターの生産は、克服するのが困難な問題を伴い、高すぎる生産コストを伴う。また、ヘルパーウイルスの使用はほとんど常に、HDベクター製剤を汚染する。
【0007】
E2遺伝子および/またはE4領域のいくつかを欠失させ異なる初期遺伝子の欠失を組合せることにより、多欠失ヘルパー非依存性Adベクターも構築されている(2−4,23,24,33,45,52)。典型的に、必要な相補遺伝子を並列で相補パッケージング細胞系内に安定的に導入する。しかし、この方法は低コピー数のウイルス遺伝子の染色体組み込みを要し、多数の欠失を相補しなければならない場合非効率的となりうる。Andrews J.L.ら(5)は、E1、E2a、E3およびE4領域が欠失したベクターは高い力価までは増殖し得ないことを示した。Zhou H.ら(54)は、第1世代アデノウイルスベクターにより通常達する力価に近い力価でE1/E2a欠失ベクターを効率的に増殖させるためにDBP遺伝子の複数の組込みコピーが必要であることを示した。
【発明の開示】
【発明が解決しようとする課題】
【0008】
効率的なパッケージング/プロデューサー細胞系の開発は、ヘルパー非依存性アデノウイルスベクターの開発に関連した最も困難な課題の1つである。したがって、アデノウイルス由来ベクターの絶えざる開発および使用のための重要な要件は、多または完全欠失アデノウイルスベクターの高力価調製物の製造を促進するヘルパー非依存性プロデューサー細胞系の設計である。理想的な解決策は、完全欠失ヘルパー非依存性アデノウイルスベクターの高力価増殖に適したヘルパーまたはプロデューサー細胞系を使用するアデノウイルスベクター系の開発であろう。
【課題を解決するための手段】
【0009】
発明の概要
本発明は、哺乳類細胞の核内での誘導性自己複製能を有する、本明細書においてはアデノウイルスアンプリコンまたはレプリコンと称されるエピソームプラスミドを提供する。開示するアデノウイルスアンプリコンは以下の特性により特徴づけられる。(i)これはEBV潜伏性複製起点(oriP)およびヒトAd5逆末端反復(ITR)結合部を含有する。(ii)これは、全3個のアデノウイルス5型初期領域2(E2)遺伝子および初期領域4(E4)ORF6を、Tet依存性プロモーターの制御下、誘導可能に発現する。本明細書中に示すとおり、Tet転写サイレンサー(tTS)および逆Tetトランスアクチベーター(rtTA2)を発現する293EBNA細胞を形質転換するために、開示するアンプリコンを使用した場合、得られた安定な細胞系(2E2)は、ドキシサイクリンの存在下、第1世代Adベクターに感染した293細胞より高いレベルのポリメラーゼ、前駆体末端タンパク質(pTP)およびDNA結合タンパク質(DBP)を産生した。本明細書に記載のデータは更に、本明細書中に開示するプロデューサー細胞系(すなわち、2E2)が、多欠失ΔE1,E2,E3,E4 Adベクターの増殖のために使用しうることを証明している。したがって、開示するAd/EBVアンプリコンは、多または完全欠失アデノウイルスベクターの高力価増殖に適した効率的なヘルパー細胞系の産生に対する重要な寄与をもたらす。
【0010】
本発明の第1の態様は、(a)EBNA−1タンパク質を発現する分裂細胞の核内のアンプリコンの維持を促すEBV由来複製起点(Ori−P)、(b)Adに基づく様態での増幅を可能にするAd5ウイルスITR結合部の形態のAd5複製起点、(c)Ad5由来ポリメラーゼおよびプレ末端タンパク質をコードする核酸配列よりなる第1転写単位、(d)Ad5 DNA結合タンパク質およびE4 ORF6をコードする核酸配列よりなる第2転写単位、ならびに(e)選択マーカーを含んでなり、第1転写単位および第2転写単位が二方向性テトラサイクリン依存性プロモーターに融合している、アデノウイルスアンプリコンを提供する。特定の実施形態においては、本発明はAd5 E2/E4 ORF6アンプリコン,pE2を提供する。
【0011】
もう1つの態様においては、本発明は更に、ブダペスト条約に基づき原寄託物としてBelgian Coordinated Collections of Microorganisms Laboratory of Molecular Biology(BCCM/LMBP,Ghent University,Technologiepark 927,B−9052 Gent−Zwijnaarde,Belgium)Plasmid Collectionに2004年10月15日付けで寄託されたプラスミドのヌクレオチド配列を含んでなるエピソームプラスミドを提供する。該寄託物には受託番号LMBP 4972が付与された。この寄託物は、Budapest Treaty on the International Recognition of the Deposit of Microorganisms for the Purposes of Patent Procedureに基づいて維持される。この寄託は単に当業者にとっての便宜としてなされたものであり、寄託が35 U.S.C.§112に基づいて要求されることを自認するものではない。寄託された物質の公への入手可能性に対する全ての制限は、37 C.F.R.§1.808(b)に規定された要件の場合を除き、特許の付与に際して、変更不能に除かれる。
【0012】
OriP潜伏性複製起点と組合されたEBV核抗原−1(EBNA−1)の存在は、細胞周期当たり1回だけ複製する安定なコピー数での核内保持および自律的エピソーム性複製の機能をもたらす(48)。E4orf6ならびにアデノウイルスDNA複製をもたらすAd5ポリメラーゼ、pTPおよびDBPのコード配列はTetプロモーターの制御下で2つのビ−シストロン転写単位内に配置されるため、Ad/EBVエピソームが転写的にサイレントな場合には。それは潜伏性ウイルス要素として維持される。本明細書に示すとおり、開示されているアンプリコンはE2遺伝子発現の誘導に際して複製されて、コピー数の増加をもたらす。
【0013】
もう1つの実施形態においては、本発明は、プロモーターに融合した関心のあるトランスジーンをコードする発現カセットを更に含むAd5 E2/E4ORF6アンプリコンを含む。関心のあるトランスジーンには、限定的なものではないが例えば免疫グロブリンまたは免疫グロブリンのフラグメント、一本鎖抗体、二重特異性抗体、エリスロポエチン、成長ホルモン、サイトカイン、例えばIL−2およびIL−10関連サイトカイン(例えば、IL−19、IL−20、IL−22、IL−24,IL−26、IL−28およびIL−29遺伝子)のようなタンパク質をコードするヒト遺伝子;HCVのコア、E1、E2または非構造領域、HIVのHIV−1 gp41、GP120、gag、pol、nef、HSV−2糖タンパク質D、HPV Ll、L2、E6およびE7タンパク質、SARS−CoVのスパイク(S)糖タンパク質、細胞膜タンパク質、例えばウイルス受容体、例えばSARS−CoV ACE2受容体、HIV−1受容体CD4およびケモカインコレセプター、HCV受容体CD81、SRB1、L−SIGNおよび硫酸ヘパリンシンデカン、Gタンパク質共役受容体(GPCR)、チロシンキナーゼ細胞表面受容体をコードするウイルス遺伝子が含まれる。
【0014】
本発明の第2の態様は、本発明のアデノウイルスアンプリコンを含んでなるプロデューサー/ヘルパー細胞系を提供する。より詳しくは、本発明は、(a)Ad5 E1タンパク質、(b)EBV由来EBNAタンパク質、(c)Tet転写サイレンサー、(d)Tet逆トランスアクチベーター、(e)アデノウイルスアンプリコン[該アデノウイルスアンプリコンは、第2転写単位(第2転写単位はEBV由来OriP、アデノウイルスITR結合部ならびにAd5 E4 ORF6およびDNA結合タンパク質をコードする核酸配列よりなる。)と組合された第1転写単位(第1転写単位はAd5由来ポリメラーゼおよびプレ末端タンパク質をコードする核酸配列よりなる。)よりなり、第1転写単位および第2転写単位は二方向性テトラサイクリン依存性プロモーターに融合している。]、ならびに(f)選択マーカーを発現するアデノウイルスパッケージング細胞系を提供する。
【0015】
特定の実施形態において、本発明のこの態様は、本明細書においては、293EBNAtet細胞(Tet転写サイレンサーtTSkidおよびtet逆トランスアクチベーターrtTA2を発現する293EBNA細胞として定義される。)をpE2で形質転換して、ΔE1,E2,E3,E4 Adベクターの増殖のためのプロデューサー細胞系としての使用に適した細胞系を得ることにより例示される。本明細書中に例示されているパッケージング細胞系は2E2と称される。開示されている系のAd5 ΔE1,E2,E3,E4 Adベクターは、12.4Kbまでのクローニング容量により、およびウイルス遺伝子発現の漏出の減少により特徴づけられる。本発明のプロデューサー細胞は、とりわけ、遺伝子治療およびワクチン接種のために設計された組換えアデノウイルスの製造に有用である。
【0016】
本発明のもう1つの態様は、治療用の複製欠損アデノウイルスベクターの製造方法を提供する。例えば、特定の実施形態において、本発明は、感染因子/病原体により発現される抗原に対する免疫原性応答を誘発するためのワクチンとしての使用のための免疫原性組成物を提供する。もう1つの実施形態において、本発明は、腫瘍抗原に対する免疫応答を誘導するのに適したワクチンを提供する。本発明のこの態様は、本明細書中において、全HCVポリタンパク質を発現するΔE1−E4発現ベクターの構築、および免疫実験における該ベクターの利用により例示される。
【0017】
1つの実施形態においては、本発明は、パッケージング細胞内に多欠失アデノウイルス発現ベクターを導入すること{前記パッケージング細胞は、EBV由来EBNAタンパク質;Tet転写サイレンサー;Tet逆トランスアクチベーター;アデノウイルス発現ベクター[前記アデノウイルス発現ベクターはEBV由来oriP、アデノウイルスITR結合部および第1転写単位(前記第1転写単位はAd5 DNA結合タンパク質およびE4 ORF6をコードする核酸配列よりなる第2転写単位と組合されたAd5 E2由来ポリメラーゼ、プレ末端タンパク質をコードする核酸配列よりなる。)よりなり、第1転写単位および第2転写単位は二方向性テトラサイクリン依存性プロモーターに融合している。]ならびにプロモーターに融合した関心のあるトランスジーンをコードする発現カセットを発現する。}、E2およびE4ORF6コード配列の発現を誘導すること、および産生した複製欠損アデノウイルスを回収することを含んでなる、関心のあるトランスジーンを含む複製欠損アデノウイルスの製造方法を提供する。特定の実施形態においては、E2およびE4ORF6コード配列の過剰発現により特徴づけられるpE2アンプリコンの複製を誘発するドキシサイクリンと該パッケージング細胞とを接触させることにより、E2およびE4ORF6コード配列の発現を誘導する。
【0018】
代替的実施形態において、本発明の方法は、E1、E2、E3およびE4遺伝子を欠く多欠失ヒトAd5アデノウイルスベクターのためのパッケージング細胞としての、tTSkidおよびrtTA2を発現する293EBNA細胞の使用を含む。本発明は更に、本明細書に開示されており特許請求されている製造方法により回収され精製された組換え複製欠損アデノウイルス粒子を提供する。
【0019】
本発明の他の特徴および利点は本明細書に記載の開示から明らかである。実施例(具体例)は、特許請求されている本発明の実施に有用な種々の成分および方法を例示する。該実施例は本発明を限定するものではないと解釈されると理解されるべきである。本開示に基づき、当業者は、本発明の実施のための他の成分および方法を特定し使用することが可能である。
【0020】
図面の簡単な説明
図1A〜1Bは、安定な293EBNATetクローンを得るために使用したプラスミドの概要図を示す。パネルAはプラスミド成分の直線的表示を示す。以下の略語が使用されている:逆Tetトランスアクチベーター(rtTA);Tetサイレンサー(tTS);ECMV内部リボソーム進入部位(IRES);イントロン配列(intS);およびピューロマイシン耐性(PuroR)。
【0021】
パネルBはpE2プラスミドの概要図を示す。pFG140由来のAd5逆末端反復配列の頭−尾結合体を該プラスミド内にクローニングした(ITR、灰色矢じり)。Ad5初期遺伝子が黒色矢印で示されている。ポリメラーゼ(Pol)、プレ末端タンパク質(pTP)、DNA結合タンパク質(DBP)およびE4orf6を、Tet応答性要素(TRE、白色太棒線)により駆動される2つのビシストロン発現カセット内に挿入した。ニワトリβグロビン絶縁(insulator)配列(HS4)に隣接するEBV潜在性複製起点(OriP)が付点太棒線および灰色太棒線で示されている。
【0022】
図2はAdTetLuc感染クローンにおけるルシフェラーゼ発現のグラフ表示を示す。293EBNA細胞および種々の293EBNA/Tetクローンに、1μg/ml ドキシサイクリンの存在下(黒色棒線)または非存在下(白色棒線)、AdTetLucを感染させた(m.o.i 10)。細胞溶解物におけるルシフェラーゼ活性を感染の48時間後に評価した。
【0023】
図3は環状および直線形態のpE2の概要図を示す。pE2の環状形態と直線形態との識別を可能にするNotI消化により得たDNA断片も示されている。
【0024】
図4A〜4B。パネルAは、ドキシサイクリンによるAd5 E2遺伝子発現の活性化の際のpE2の複製を示すサザンブロット分析を示す。NotIで消化されたpE2の108個のコピーを第1レーンにローディングした。ドキシサイクリンの非存在下/存在下のpE2での形質移入の48時間後に293 EBNA Tet細胞から抽出されNotIおよびDpnIで消化されたエピソームDNAをレーン2および3にローディングした。環状および直線状単量体形態を示す12.6および4.4Kbのバンドが黒色矢印で示されている。DNAマーカーのサイズが図の右側に示されている(Kb)。
【0025】
パネルBは、E2タンパク質のtet誘導性発現を示すウエスタンブロット分析を示す。ドキシサイクリンの存在下(+)(レーン3、6および9)または非存在下(−)(レーン2、5および8)でpE2アンプリコンで形質移入された293EBNATet細胞におけるDBP、pTPおよびポリメラーゼタンパク質のウエスタンブロット分析。陰性(非形質移入)対照がレーン1、4および7に示されている。特異的ウサギ抗血清(ポリメラーゼ、pTP)またはマウスモノクローナル抗体(DBP)でE2タンパク質を検出した。
【0026】
図5A〜5B。2E2クローンから抽出されたpE2の構造およびE2タンパク質の発現。パネルAは、クローン293EBNATetおよび2E2から抽出されたpE2の構造を示すサザンブロット分析である。293EBNATetおよび2E2クローンから抽出されたDNAのサザンブロット分析。Hirt法により抽出されたDNAをBamHIで消化し、1%アガロースゲル上で分離し、ナイロンメンブレン上に移し、32Pで標識されたpE2 DNAでハイブリド形成させた。参照体として第1レーンにpE2ベクターをローディングした。293EBNATet細胞から(陰性対照)および2E2クローンから抽出されたDNAをそれぞれ第2および第3レーンにローディングした。
【0027】
パネルBは、Ad5ΔE1ベクターに感染した細胞におけるE2発現と比較した場合の2E2安定細胞系におけるE2タンパク質のtet誘導性発現を示すウエスタンブロット分析である。2E2クローンによるE2aおよびE2bタンパク質発現を、ドキシサイクリン(1μg/ml)(レーン、2、3;5、6;8、9)の存在下(+)(レーン3)および非存在下(−)(レーン2)、ウエスタンブロットにより評価し、500のm.o.i.のFG Ad5ΔE1ベクターに感染した非誘導2E2細胞からのE2タンパク質の発現レベル(レーン1)と比較した。分子量マーカーの移動度(kDa)が図の左に示されている。
【0028】
図6は、EGFPを発現するAd ΔE1−4ベクターをレスキューし増殖させるための2E2細胞の使用を示す一連の写真を示す。サイレンシングされた(‐doxy)または活性化された(+doxy)E2/orf6遺伝子発現を伴う2E2クローンにおけるAd5ΔE1−4EGFPウイルス増殖。P0=形質移入、P1およびP2は、前感染経過からの全粗溶解物の1/10を細胞に感染させることにより得た。
【0029】
図7A〜7B。パネルAはAd5ウイルスの概要地図である。この図にはすべての欠失領域が示されている。E1、E2a、E3、および7個のE4 orfのうちのorf3を除く6個を、ベクターバックボーンから完全に欠失させた。ポリメラーゼおよびプレ末端タンパク質の欠失は部分的であるに過ぎない。E1領域は、MCMVプロモーターにより駆動されるHCVポリタンパク質発現カセットで置換される。ベクターゲノムの制限分析において使用するHindIII制限部位が示されている(┃)。
【0030】
パネルBは、該多欠失ベクターのE1領域内に導入されたHCV(BK株)ポリタンパク質発現カセットの概要図を示す。HCV 5’および3’UTR配列が除去されており、最適化コザック配列が該ポリタンパク質の5’側に融合されている。発現はマウスCMVプロモーター(mCMV)およびウシ成長ホルモンポリA(BGHポリA)により調節される。
【0031】
図8はAd ΔE1−4orf3+HCVの制限分析を示す。プラスミドDNAおよびCsCl精製ウイルス粒子から抽出されたウイルスDNAをHindIIIで消化し、クレノウ酵素でのフィルイン反応により(33P)dATPで末端標識した。精製されたAd ΔE1−4orf3+HCVベクター(レーン4)のウイルスDNA制限パターンを元のプラスミド(レーン3)と比較した。HindIIIで制限処理されたFG(ΔE1−E3)Ad5(レーン1)およびAd5ΔE1−4orf3+空ベクター(レーン2)バックボーンを該ゲル内に加えた。a、b、c、dは、欠失を含有する多欠失ベクターDNAバンド、およびFG(ΔE1−E3)Ad5パターンにおける対応バンドを示す。
【0032】
図9は、Ad5ΔE1−4HCV感染細胞におけるHCVタンパク質の発現を示すウエスタンブロット分析を示す。HeLa細胞にAd5ΔE1−4HCVを10のm.o.i.で感染させた。HCV特異的抗体でのウエスタンブロット分析により細胞抽出物においてHCVタンパク質を検出した。感染の48時間後に調製した、HeLa細胞からの溶解物をレーン3にローディングし、未感染対照細胞からの溶解物(レーン1)、およびmCMV−HCVベクターDNAで形質移入したHeLa細胞からの溶解物(レーン2)と比較した。特定のバンドが矢印で示されている。
【0033】
図10は、マウスにおけるAd5ΔE1−4HCVウイルス免疫化に対するインビボCD8+ T細胞応答を特徴づけるFACSデータのグラフ表示を示す。A2.1(a)およびCB6F1(b)。1010vpで筋肉内に免疫化されたマウスの新鮮に単離された脾細胞を、3週間後のIFN−γに関する細胞内染色により、HCV−ペプチドのプールに対するCD8+ T細胞応答に関して試験した。x軸 抗INF−γ、y軸 抗CD8。プールC(コア)、プールF−G(NS3)、プールH(NS4)、プールI−L−M(NS5a/b)。
【0034】
図11は、本開示に記載されているポリヌクレオチドおよびポリペプチド配列のヌクレオチドおよび/またはアミノ酸配列(すなわち、配列番号1〜21)を示す。
【0035】
図12A〜12Cは、NS3ヘリカーゼ内のエピトープのマッピングに用いた方法を示す。Ad5ΔE1−E4HCVで初回抗原刺激されpSh−Ad5−HCVで追加抗原刺激された2匹のマウス(マウス4,黒塗り棒線、マウス5,付点棒線)から精製した脾細胞を、全NS3ヘリカーゼ領域に及ぶペプチド(I〜XVIII)の二次元サブセット上でγ−IFN−Elispotにおいて試験した(図12A)。図12Bに示されているデータは、免疫応答を特徴づけするために使用したパネルAにおいて特定された陽性閾値を超える応答を惹起したペプチドプール間の交差点を要約したものである。パネル12Cは、該応答をもたらすNS3エピトープを特定するために行ったγ−IFN−Elispotアッセイの結果を要約したものである。
【0036】
図13は、VV−NSチャレンジに対する防御の誘導における免疫化の効力を示す。灰色のドットは相乗平均力価(N=5)を表す。アステリスクは対照に対するp<0.05を示す(Mann−Whitneyランク)。
【0037】
図14A〜Bは、Ad5ΔE1−E4HCV免疫化に応答してアカゲザルにおいて惹起された免疫応答を要約したものである。パネルAは、Ad5ΔE1−E4HCVの1回の投与に際してサル4061において惹起されγ−IFN−Elispotにより分析された経時的免疫応答を表す。結果は106PBMC当たりのγ−IFNスポット形成細胞数(SFC)として表されている。各棒グラフは、別々のペプチドプールに対する応答を表す。パネルBは、Ad5ΔE1−E4HCVの1回の投与により3頭の個々のサルにおいて誘導され注射の6週間後にγ−IFN−Elispotにより分析された免疫応答を示す。結果は、106PBMC当たりのγ−IFNスポット形成細胞数(SFC)として表されている。各棒グラフは、別々のペプチドプールに対する応答を表す。
【0038】
発明の詳細な説明
個々の文の終わりに示されている数字の列挙は、本明細書の終わりに番号付きで示されている参考文献の一覧に対応する。本明細書中に引用されている参考文献は本発明の先行技術であると自認されるものではない。
【0039】
本明細書中で用いるすべての科学技術用語は、特に定義されない限り、当業者に一般に理解されているものと同じ意義を有することに留意することが、本発明の理解に重要である。以下に幾つかの用語を説明する。あるいは、ある種の用語は、初めて用いられる際に本明細書中で定義されうる。
【0040】
本明細書中で用いる「アンプリコン」なる語は、必須遺伝子機能が付与されると複製されうるエピソームまたは染色体外DNA要素を意味する。一般に、アデノウイルスアンプリコンは、ウイルスDNAの複製を支持する各末端反復配列の少なくとも一部分を含むと理解される。真核ウイルスアンプリコンは、好ましくは、完全なITR配列の少なくとも約90%を含む。したがって、「アデノウイルスアンプリコン」はITR結合部および適切な複製起点のいずれかを含む。
【0041】
本明細書中で用いる「形質移入」なる語は、細胞が生物学的に生存可能なままでDNAを細胞の外部から細胞の内部へ導入する適切な方法のいずれかを意味する。本明細書中で用いるこの用語は、限定的なものではないがエピソームおよび他の環状または直鎖状DNA形態の形質移入を含む任意の手段による宿主細胞内へのDNAの導入を含む。この用語は、例えば本明細書に記載されているような遺伝子治療法を含む。本発明を実施するためには、限定的なものではないがリン酸カルシウム共沈法、エレクトロポレーション、遺伝子銃形質移入、リポフェクションまたは他のカチオン脂質に基づく形質移入を含む適切な形質移入法のいずれも用いることが可能である。これらの技術は当業者によく知られている。
【0042】
本明細書中で用いる用語は本発明を限定するものではない。例えば、「遺伝子」なる語は、遺伝子産物をコードするcDNA、RNAまたは他のポリヌクレオチドを含む。「核酸」、「RNA」、「DNA」などの用語を用いる際には、特定の段階で用いられうる化学構造を限定するものではない。例えば、DNAの代わりにRNAが一般に用いられうることは当業者によく知られており、したがって、「DNA」なる語の使用はこの代用を含むと解釈されるべきである。また、種々の核酸類似体および誘導体も本発明の範囲内であることが公知である。遺伝子または核酸の「発現」は、細胞遺伝子発現だけでなく、クローニング系および任意の他のコンテクストにおける核酸の転写および翻訳をも含む。
【0043】
自然感染からのアデノウイルスの大規模生産はウイルスDNA複製と主要後期プロモーター活性との協同作用の結果であると理解される。実際、この方法は、ビリオンをパッケージングするのに要する大きなプールを生成する効果を有する、高コピー数の転写活性鋳型の蓄積をもたらす。先行技術は、1以上のウイルスタンパク質が欠損したアデノウイルスベクターの産生のための1以上のウイルスタンパク質を発現するいくつかのヘルパー細胞系を含むが、自然感染を模倣した一連の協同事象(例えば、ウイルスDNA複製および必要な構造タンパク質の発現)により特徴づけられる生産系は未だ記載されていない。宿主細胞染色体内への相補遺伝子の組込みに基づく相補ヘルパー細胞系の製造を要する方法を用いる代わりに、宿主細胞内で自己複製可能なアデノウイルス複製に必要な非構造E2遺伝子のすべてを含有するエピソームプラスミドが得られた。
【0044】
ヘルパーウイルス依存性生産系の使用に伴う、先行技術で認められている問題点に対処して、本発明は、多または完全欠失アデノウイルスベクターを相補しうるプロデューサー細胞系を作製するために使用されうる新規アデノウイルスアンプリコンを提供する。該アンプリコンは、ヘルパーウイルス非依存性ベクター産生の効率が最大となるよう自然アデノウイルス感染の段階を模倣する態様で機能するように設計されている。本明細書中に示すとおり、これは、ウイルス転写カスケードの開始に要するアデノウイルス初期遺伝子の発現を能動的に抑制し/遅延させることにより開示のエピソーム(すなわち、アデノウイルスアンプリコン)がパッケージング細胞系の核内の潜伏相において維持されるパッケージング細胞を操作することにより達成される。
【0045】
潜伏性は、エプスタインバーウイルス(EBV)由来DNA複製要素の核保持特性の利用および誘導性発現系(本明細書中ではテトラサイクリン調節発現系により例示される。)の使用により達成される。誘導により、複製に必要なアデノウイルスE2遺伝子(すなわち、ポリメラーゼ、プレ末端タンパク質およびDNA結合タンパク質)の発現を招くエピソーム配列の転写を引き起こす複製相の活性化がもたらされる。実際、潜伏相から複製相への移行により、トランスジーンを含む多または完全欠失アデノウイルスベクターを効率的にパッケージングするのに必要な大量の相補ウイルスタンパク質の蓄積が促される。したがって、典型的に自然感染の後期に高力価のビリオン産生をもたらす一連の事象を模倣する態様で該パッケージング細胞が機能するのを可能にするよう、該アンプリコンが設計される。したがって、開示されているアンプリコンおよびパッケージング細胞系は、ヘルパーウイルス非依存性医薬等級ベクターの効率的高力価製造方法を可能にする。
【0046】
エピソームプラスミド(pE2)は以下の特徴により特徴づけられる:(i)これは、該エピソームを自律複製可能にし核内保持の促進によりエピソームを多コピーで維持するEBVプラスミド複製起点のような要素を含む、(ii)これは、直鎖形態でのDNA複製を可能にするAd5逆末端反復(ITR)結合部を含む、(iii)これは、アデノウイルスの複製に必要なE2アデノウイルス遺伝子、例えばポリメラーゼ、プレ末端タンパク質およびDNA結合タンパク質ならびに初期領域4(E4)ORF6の誘導性発現をもたらす。
【0047】
ウイルスDNA複製のためには、Ad5ウイルスDNA(GenBank BK000408に開示されている。)の僅か2つの領域のみがシスで要求されることが公知であることを、当業者は認識する。これらは左逆転末端反復配列またはITR(Ad5のbp1〜約103)および右ITR(Ad5のbp35833〜35935)である。EBV由来の複製起点系の存在は、アンプリコンが多コピーで核内に保持され染色体DNAと同時に複製されるのを可能にし、一方、アデノウイルスITR結合部の存在は、E2領域によりコードされるタンパク質の存在下、アンプリコンが多コピー数で複製されるのを可能にする。該アンプリコン上での誘導性プロモーターの使用は、エピソームの複製に及びトランスジーンを含む多または完全欠失アデノウイルスベクターの増殖に要求されるアデノウイルス遺伝子を、厳密に調節応答性である誘導性プロモーターの制御下に配置する。一般には、誘導性プロモーターは、アクチベーターにより誘導されるプロモーターである。誘導性プロモーターに作用するインデューサーの非存在下では、エピソーム上に含有されるアデノウイルス遺伝子は発現されず、ウイルスタンパク質の産生は伴わず、ウイルスタンパク質誘導性細胞毒性のリスクは最小である。
【0048】
実際、開示されているアンプリコン(例えば、エピソーム)は、前記特性の1以上を同時に達成するよう、宿主細胞内に存在する他の要素(例えば、活性化因子)と協同して働きうる要素を含みうる。例えば、同一要素(DNA配列)が自己増殖能を付与し核内保持を促進しうる。代替的ウイルス複製系に由来するDNA配列も本発明の実施に使用されうると理解されるべきである。例えば、ウシパピローマウイルス(BPV)由来の複製起点および活性化因子(60)またはSV40由来T抗原系に基づくベクターに由来する配列が、適切な代替物となる。
【0049】
本明細書中の実施例は、EBNA−1を発現する宿主細胞の使用を記載しているが、所望により、アデノウイルス遺伝子を含有する同一エピソーム単位(アンプリコン)上もしくは複製可能な第2の遺伝単位上にコード配列を含めることにより又は宿主細胞ゲノム内への安定組込みにより他の活性化因子を導入することが可能であることは容易に理解される。例えば、本明細書に記載のとおりにEBNA1抗原およびEBV複製起点を使用する代わりに、BPV複製起点と共にウシパピローマウイルス(BPV)E1およびE2抗原を使用することが可能である。E1抗原は、複製の開始および伸長に必要なヘリカーゼであり、E2抗原は、複製起点へのE1抗原の結合を補助する転写因子である(61)。総合すると、これらのウイルスタンパク質はまた、適当な形質移入に関してコンピテントである細胞におけるエピソームの核内保持を促進することが公知である。
【0050】
霊長類細胞内において非組込み型自律的複製性エピソームベクターを安定に維持し及びプラスミドの安定な複製を支持する、EBVゲノムの確定された遺伝的要素が公知である。必要な遺伝的要素には、シス作用性プラスミド複製起点(oriP)およびトランス作用性エプスタインバー核抗原(EBNA−1)タンパク質が含まれる。より詳しくは、EBV由来要素、すなわちoriPおよびEBNA−1は、これらのベクターにより形質移入された哺乳類細胞内において1〜90個の範囲の数で非組込み型染色体外分子として専ら存在する組換えエピソームの安定な複製を支持するために使用されている。
【0051】
EBVゲノムの複製起点oriPを含有しEBNA1ウイルスタンパク質(641アミノ酸)の発現を可能にするプラスミドは、形質移入されたヒト細胞内で安定なエピソーム様態で維持され、それらの複製は細胞分裂と同時に生じる。本明細書に示すとおり、活性化因子EBNA−1の存在下で使用するEBV複製起点(OriP)は自己複製能を付与し、核内保持を促進する。理論により束縛されるものではないが、EBNA1タンパク質は複製開始のレベルで30bpの反復配列に結合し、S相の時点での細胞性因子のリクルートメント、およびoriP配列をシスで有するプラスミドの、細胞分裂との同期的複製を可能にすると考えられている。さらに、EBNA1は、おそらく染色体構造体および反復単位のレベルでの同時結合により、細胞分裂時のエピソームの分離および核内維持を可能にするのであろう。これらの要素は、それ自身で、複製時に、細胞当たり複数のコピーのプラスミドベクターのエピソーム維持および分離を可能にする。
【0052】
本発明で使用するエピソームにおいて、適切なEBV複製起点DNA配列のいずれも使用することができる。適切なEBV複製起点配列(oriP)の一例はGenBank V01555に開示されている。oriP領域はこのGenBank配列のヌクレオチド7333からヌクレオチド9312までの配列に及んでいる。本明細書に記載のエピソームにおいて使用されるoriP配列は、65bpの逆転復配列単位により形成され30bp単位の4個の不完全なコピーを含む複製起点から960bp離れた、30bpの20単位の反復から構成されている。
【0053】
エプスタインバーウイルス(EBV)由来oriPは、EBNA−1結合配列の2つのクラスター(反復配列のファミリーおよび二回対称配列)から構成されている。どちらの要素もEBNA−1への複数の結合部位を有し、oriPを含有するプラスミドの複製および核内保持に必須である。宿主細胞因子は、本明細書に開示するエピソームの複製および核内保持を補助すると考えられている。一般に、適当なoriP配列は、oriP機能に必要なことが知られている反復配列のファミリーおよび二回対称の領域を含む。本発明において使用されうるEBV oriP配列には、天然に存在する配列からの修飾を含有するもの、例えば、天然配列の欠失、挿入、置換および重複を含有するものが含まれる。そのような誘導体配列は、例えば、oriP機能に必要な前記公知領域を維持させることにより入手できる。また、保存的置換はよく知られており、当業者に入手可能である。使用するoriP配列は、十分に多い量のEBNA1タンパク質の存在下でoriP配列が存在するエピソームの複製を導くよう宿主細胞内で有効に機能するものである。
【0054】
適切なEBNA1タンパク質のいずれかをコードするDNAが本発明のプロデューサー細胞により発現されうる。EBNA1をコードするDNAはInvitrogenから商業的に入手可能であり、そのEBV系列プラスミドのいくつかの中に含有される。さらに、EBNAタンパク質をコードするDNAは、発現されるタンパク質が宿主細胞におけるEBV oriP含有エピソームの複製を支持する、天然に存在するEBNA1アミノ酸配列の変異体、例えば、欠失、付加、挿入または置換を含有するものをコードすることが可能である。この変異体は、本明細書に記載の他の配列の場合と同様に、アミノ酸配列保存的変異体をコードする機能的に保存的な核酸配列、BLASTまたはFASTAアルゴリズムによる測定で90%を超える、好ましくは95%を超える同一性または相同性を有する配列、および高いストリンジェンシーのハイブリダイゼーション条件下でハイブリダイズする配列を含む。さらに、同一EBNA1タンパク質をコードする縮重DNA配列を使用することが可能である。同一アミノ酸配列を発現しうる縮重DNA配列は、そのようなDNA配列の構築および発現方法と同様、当技術分野でよく知られている。
【0055】
EBNA1は、よく知られた技術を用いて任意の霊長類またはイヌ細胞内に安定に形質移入されることができ、組込み遺伝子コピーからEBNA1を発現する得られた細胞系は、適当な生産細胞系を作製するために使用されうる。あるいは、EBNA1が発現される限り、感染性または欠損性EBVを既に含有する細胞系を使用することが可能である。これは、ATCCから入手可能な多数のEBV形質転換リンパ芽球を含む。EBNA1を既に発現する細胞系の形質移入が非常に好都合である。なぜなら、そのような細胞はエピソーム構築物を安定に維持する能力は数桁増強されることが可能であり、僅か2〜3週間のうちに安定細胞系が作製されうるからである(62)。しかし、これらの方法は、組込み遺伝子からEBNA1を構成的に発現する細胞系を製造する追加的な工程を要する。
【0056】
本明細書中に示すとおり、テトラサイクリンプロモーターは、テトラサイクリンまたはその一般的類似体の1つ(例えば、ドキシサイクリン(Dox))に応答性であり、開示されているエピソーム単位(例えば、アンプリコンまたはレプリコン)における使用に適している。テトラサイクリンの類似体であるドキシサイクリンは、ヒトにおけるその安全な使用、細菌テトラサイクリンリプレッサー(TetT)に対するその特異性のため、広く受け入れられている。簡潔に説明すると、テトラサイクリン依存性調節系(tet系)は、単純ヘルペスウイルスVP16タンパク質のアクチベータードメインに融合した原核性TetRよりなるテトラサイクリントランスアクチベーター(tTA)と、最小プロモーターに融合した原核性テトラサイクリンオペレーター部位(tetO)のいくつかのコピーよりなるテトラサイクリン応答性要素(TRE)との間の相互作用に基づく(68)。テトラサイクリン(tet)の存在下、tTAは、TREに結合するその能力を失い、発現が遮断される。逆トランスアクチベーター(rtTA)は突然変異誘発によりtTAから誘導されている。tTAとは対照的に、rtTAはtetの存在下でTREに結合するに過ぎない。
【0057】
転写の基底レベルを減少させることにより遺伝子発現の緊縮調節を得るために、本発明者らは、最近記載された逆tetトランスアクチベーターの新規改良形態(29)とTet転写サイレンサーtTSkid(16)との組合せを利用するTet調節系を使用した。tTSkidは、転写のリプレッサーとして機能することが公知の腎タンパク質Kid−1のKRABドメインを含有する。tTSkidは、エフェクター薬の非存在下でTetプロモーターに結合して、転写の基底レベルを減少させる。逆Tetトランスアクチベーターとの組合せは、ドキシサイクリン添加により調節される活性化/抑制系の構築を可能にする。この目的のために、図1Aに記載のとおりにEMCV IRESを使用することによりそれらの2つの遺伝子をビシストロン転写単位において組合せた。
【0058】
真核細胞における誘導性遺伝子発現を導きうるいくつかのプロモーター系が入手可能である。これらには、重金属イオン(63)、(64)、イソプロピル−β−D−チオガラクトシド(65)、ホルモン、例えばコルチコステロイド(66)、プロゲステロンアンタゴニスト(67)またはテトラサイクリン(68)に応答して修飾される活性を有するプロモーターが含まれる。しかし、エクジソン、ラパマイシン、RU486、デキサメタゾンおよび重金属(すなわち、ZnまたはCd)のようなアクチベーターに対して応答性である他のよく知られた誘導性調節要素も適している。代替的な調節要素を本発明での使用に応用することは当業者の能力の範囲内に十分に含まれる。本発明の目的には、十分なレベルまたは調節制御を保証するものであり、医薬用途に許容されるアクチベーターにより誘導可能なものである限り、調節要素のいずれも使用することが可能である。さらに、遺伝子発現の厳密な調節を促進するために、他の調節要素、例えばテトラサイクリン応答性トランスアクチベーターおよび/またはサイレンサー(rtTAおよびtTs)に誘導性プロモーターを機能的に連結しうると理解されるべきである。
【0059】
発現カセット(関心のあるトランスジーンと、本明細書中に開示されている哺乳類細胞内での発現を導くための必要な調節配列とを含むものとして定義される。)のすべては、腸菌(E.coli)BJ5183内での相同組換えによるpAd5ΔE1−4orf3+のE1領域における挿入が可能となるよう、CMVプロモーターおよびトランスジーン発現用BGHポリAシグナルに加えてAd5配列[nt]1−450(左)(配列番号1)および[nt]3511−5792(右)(配列番号2)を含有するAd−シャトルベクターのコンテクストにおいて構築した。EGFP cDNAはpEGFPプラスミド(Clontech)から得、ついでAd−シャトルプラスミド内にクローニングしてpShAd5 EGFPを得た。
【0060】
本明細書に記載の方法において、所望の配列をコードするエピソームにより成功裏に形質移入された細胞を選択するために、通常の選択マーカーを使用する。そのような選択は、通常、形質移入された細胞を抗生物質または関連選択過程を開始させる他の物質にさらすことを含む。本発明で使用するエピソームにおいて使用する選択マーカー遺伝子は、これらの遺伝子を含有する細胞が対応抗生物質または因子の存在下で成長するのを可能にする、特定の抗生物質および/または因子に対する耐性を付与するタンパク質をコードする遺伝子である。真核性選択マーカーの非限定的な例には、ヒグロマイシン(Life Technologies,Inc.;Gaithesboro,Md.から商業的に入手可能なhygまたはhph)、ネオマイシン(Life Technologies,Inc.Gaithesboro,Md.から商業的に入手可能なneo)、ゼオシン(Pharmingen,San Diego Calif.から商業的に入手可能なSh Ble)、ピューロマイシン(Clontech,Palo Alto Calif.から入手可能なpac、ピューロマイシン−N−アセチル−トランスフェラーゼ)、ウアバイン(Pharmingenから入手可能なoua)およびブラスチシジン(Invitrogenから入手可能)に対する耐性を付与する抗生物質耐性遺伝子が含まれる。
【0061】
多欠失ヒトAd5ベクターバックボーンの概要図を図7Aに示す。E1およびE3領域の古典的欠失(11に概説されている)のほかに、本発明者らは、L4イントロンを含め、r鎖においてコードされる他の機能に何ら影響を及ぼすことなくDNA結合タンパク質の全コード配列([nt]22245−24029;(配列番号3)1784bp欠失)を除去した。三成分リーダー配列のイントロンおよび主要後期単位に対応するプレ末端タンパク質(Ad5[nt]8919−9462(配列番号5)543bp欠失)遺伝子およびポリメラーゼ([nt]7274−7883;(配列番号4)609bp欠失)の部分を欠失させてE2b遺伝子発現をノックアウトした。さらに、ポリメラーゼのトランケート化非活性形態の産生を妨げるために、ATG開始コドンをCTGに突然変異させた。E4プロモーターに直接的に融合したorf3を除き、E4領域を完全に欠失させた([nt]32830−34316(配列番号6)および34895−35443;(配列番号7))。
【0062】
全初期遺伝子の欠失を組合せることによりAd5バックボーンにおいて生じた理論的空間は約12.4Kbである。該新規ベクター系の大きな容量を利用して、マウスサイトメガロウイルス(MCMV)プロモーターに融合した全HCVポリタンパク質遺伝子用の発現カセットを挿入した。該HCVポリタンパク質発現カセットは、5’および3’非翻訳領域を除去し、コアATGの上流に最適コザック配列を挿入し、NS5Bレプリカーゼの触媒ドメインを変異させて酵素活性を除去することにより構築した(32)。トランスジーンの発現効率を増加させるために、ヒトCMVプロモーターを、FGアデノウイルスベクターにおいて4〜30倍強力であると報告されているマウスCMVプロモーター(1)で置換した。
【0063】
図7Bは、多欠失ベクターのE1領域内に導入されたHCV(株BK)ポリタンパク質発現カセットの概要図を示す。HCV 5’および3’UTR配列を除去し、最適化コザック配列を該ポリタンパク質の5’側に融合させた。発現はマウスCMVプロモーター(mCMV)およびウシ成長ホルモンポリA(BGHポリA)により調節される。
【0064】
オープンリーディングフレーム3の維持は、内部CMVプロモーターにより調節されるトランスジーンのインビボおよびインビトロでの持続的発現に必要であることが公知である(18,34)。したがって、ΔE1E3FGベクターの5700bpの欠失、およびそれによる、wt(6)の105%のゲノムサイズのパケーンジング容量に加えて、該新規Ad5ΔE1−4orf3+ウイルスベクターは12.4Kbまでのトランスジーンを収容しうる。この目的には、本発明のアデノウイルスアンプリコンおよびプロデューサー細胞ならびに本発明の方法を用いて、関心のある多数のトランスジーンを含む欠損アデノウイルスベクターを得ることが可能であると予想される。
【0065】
本明細書中に開示されている多欠失Ad5ウイルスバックボーンにおける使用のための適当なトランスジーンには、特定の哺乳類種における発現に関してコドンが最適化された、免疫原をコードする核酸配列(すなわち、トランスジーン)が含まれるが、これに限定されるものではない。1つの実施形態においては、本発明は、感染因子により発現される抗原に対する免疫応答を誘導するための免疫原性組成物(例えば、ワクチン)を提供する。例えば、ヒトおよび/または非ヒト動物種に感染するウイルスに対する免疫応答を惹起することが望ましい。
【0066】
多欠失Ad5ベクターは、細菌、真菌、寄生生物を含む病原体により発現されるタンパク質に対するヒトまたは動物における免疫応答を刺激するのにも適していよう。スタフィロコッカス・アウレスス(Staphylococcus aureus)、ストレプトコッカス・ピロゲネス(streptococcus pyogenes)、ストレプトコッカス・ニューモニエ(streptococcus pneumoniae)、ビブリオ・コレラ(vibrio cholerae)、クロストリジウム・テタニ(clostridium tetani)、ナイゼリア・メテンジチス(neisseria meningitis)、コリネバクテリウム・ジフテリアエ(corynebacterium diphteriae)、ミコバクテリウム・ツバクロシス(mycobacteria tuberculosis)およびレプラエ(leprae)、リステリア・モノサイトジェネス(listeria monocytogenes)、レジオネラ・ニューモフィラ(legionella pneumofila)は、望ましいであろう、免疫応答を惹起する対象となる細菌の具体例であるが、これらに限定されるものではない。真菌および寄生生物の具体例としては、カンジタ・アルビカンス(Candida albicans)、アスペルギルス・フミガツス(aspergillus fumigatus)、ヒストプラズマ・カプスラツム(histoplasma capsulatum)、プラスモジウム・マラリアエ(Plasmodium malariae)、レイシュマニア・マジョール(Leishmania major)、トリパノソマ・クルジ(trypanosome cruzi)およびブルセイ(brucei)、シストロソマ・ヘマトビウム(Schistosoma haematobium)、マナソニ(mansoni)およびジャポニカム(japonicum);エンタモエバ・ヒストリチカ(Entamoeba histolytica)、ヒトフィラリア症を引き起こすフィラリア(Filaria)の種々の種が挙げられうる。
【0067】
予防的および/または治療的免疫応答が望ましいであろうウイルス科の具体例には、アフトウイルス属、カルヂオウイルス属、エンテロウイルス属、ヘパトウイルス属、パレコウイルス属、ライノウイルス属のような6つの異なる属を含むピコルナウイルス科が含まれる。これらのすべては、脊椎動物に感染するウイルスを含有する。免疫応答が望ましいであろうピコルナウイルスの具体例としては、口蹄疫ウイルス、脳心筋炎ウイルス、ポリオウイルス、コクサッキーウイルス、ヒトA型肝炎ウイルス、ヒトパレコウイルス(parechoviruses)、ライノウイルスが挙げられる。カリシウイルス科は、ノーウォーク群のウイルスにより引き起こされるヒトにおける流行性胃腸炎、およびウサギ出血性疾患ウイルスに関連したウサギにおける出血性疾患またはネコカリシウイルスにより引き起こされるネコにおける呼吸疾患のような動物における他の症候群に関連した種々の属を含む。もう1つの科は、ヒトおよび多数の異なる動物種から分離されたウイルスを含むアストロウイルス科である。ヒトアストロウイルスは胃腸炎および乳幼児の下痢に関連している。トガウイルス科はアルファウイルス属およびルビウイルス属の2つの属を含む。アルファウイルス属は、ヒトおよび獣医学的疾患、例えば関節炎(すなわち、チクングニヤウイルス、シンドビスウイルス)または脳炎(すなわち、東部ウマ脳炎ウイルス、西部ウマ脳炎ウイルス)に関連している。風疹ウイルスは、発熱およびリンオアデノパシーに関連した軽度の発疹性疾患の突発を引き起こすルビウイルス属の唯一のメンバーである。風疹ウイルス感染は、妊娠初期に母親により獲得された場合には、胎児異常にも関連する。フラビウイルス科は、重要なヒトおよび動物病原体を含むフラビウイルス属、ペスチウイルス属およびヘパシウイルス属の3つの属よりなるもう1つのウイルス科である。フラビウイルス属のメンバーの多くは、発熱、脳炎および出血熱を含む種々の疾患を引き起こす節足動物媒介ヒト病原体である。デング熱ウイルス、黄熱ウイルス、日本脳炎ウイルス、西ナイル熱ウイルス、ダニ媒介脳炎ウイルスは、世界的または地域的(地域流行性)に懸念される主要な病原体である。ペスチウイルス属は、経済的に非常に重要な動物病原体、例えばウシウイルス性下痢ウイルス、古典的ブタコレラウイルス、ボーダー病ウイルスを含む。C型関連ウイルスは、急性および慢性肝炎を引き起こすヘパシウイルス属の唯一のメンバーである。組換えアデノウイルスにより発現されるHCVタンパク質は、世界中で1億7000万人を冒すウイルス感染の結果を抑制する防御用および治療用免疫応答を惹起しうる。
【0068】
コロナウイルス科のメンバーに由来する抗原を組換えアデノウイルスベクターにより発現させて、感染に対する防御を得ることが可能である。重篤な急性呼吸症候群コロナウイルス(SARS−Coウイルス)に対する防御は、限定的なものではないがヌクレオカプシド(N)タンパク質、ポリメラーゼ(P)タンパク質、膜(M)糖タンパク質、スパイク(S)糖タンパク質、小エンベロープ(E)タンパク質または該ウイルスにより発現される他のポリペプチドのいずれかを含むSARS−CoVタンパク質の組合せを発現する多欠失Ad5ベクターで免疫することにより得られうる。狂犬病ウイルスを含むラブドウイルス科のメンバーは、ウイルスタンパク質を発現する組換えワクチンの標的となりうる。他の考えられうる標的には、いくつかの出血熱の突発を引き起こすエボラ様ウイルスおよびマールブルグ様ウイルス属を含むフィロウイルス科;麻疹、呼吸器合胞体、パラインフルエンザウイルスのようなヒトにおいて公知の最も一般的なウイルスおよびニューカッスル病および牛疫ウイルスのような獣医学的に関心の持たれるウイルスのいくつかを含むパラミクソウイルス科;インフルエンザA、B、Cウイルスを含むオルトミクソウイルス科;のようなリフトバレー熱、シン・ノンブレ(Sin Nombre)、ハンタ、プーマラウイルスのような重要なヒト病原体を含む、主として節足動物により脊椎動物宿主に伝染するブニヤウイルス科;リンパ球性脈絡髄膜炎、ラッサ熱、アルゼンチン出血熱、ボリビア出血熱ウイルスを含むアレナウイルス科;主としてウマおよびヒツジにおける中枢神経系疾患を引き起こすウイルスを含むボルナウイルス(Bornaviridae)科;世界的な乳児および幼児における重篤な下痢疾患の最も重要な原因であるロタウイルス、ヒトおよび他の哺乳動物の両方を冒しうるオルビウイルス(ブルータング、流行性出血熱ウイルス)を含むレオウイルス科が含まれる。
【0069】
ウイルス抗原をコードする適切なトランスジーンは、重要なヒト病原体を含む大きなウイルス群であるレトロウイルス科のメンバー、例えばヒト免疫不全ウイルス1および2(HIV−1およびHIV−2)ならびにヒトt細胞白血病ウイルス1および2型(HTLV1および2)ならびに非ヒトレンチウイルス、例えばヒツジおよびヤギを冒すマエディウイルス/ビスナウイルス、ウマを冒すウマ伝染性貧血ウイルス、ウシを冒すウシ免疫不全ウイルス、ネコを冒すネコ免疫不全ウイルス;ポリオーマウイルス科群小DNA腫瘍原性ウイルス[原型ウイルスは、それぞれマウスおよびアカゲザルを冒すポリオーマウイルスおよびSV40である(SV40に密接に関連しているBKおよびJCウイルスはヒト患者から分離された。)。]からも得ることが可能である。
【0070】
パピローマウイルス科は、疣贅およびコンジロームを生成する、ヒトを含む高等脊椎動物を冒すDNAウイルス群からなる。パピローマウイルスの感染はヒトおよび動物の両方における癌の発生に関連づけられた。ヒトパピローマウイルスは子宮頚癌、膣癌および皮膚癌に関連している。ヘルペスウイルス科は、ヒトおよび他の哺乳動物に対する多数の重要な病原体が分類される亜科を含む。他の抗原源には、単純ヘルペスウイルス1および2、水痘・帯状疱疹ウイルス、エプスタインバーウイルス、サイトメガロウイルス、ヒトヘルペスウイルス6A。6Bおよび7、カポジ肉腫関連ヘルペスウイルスが含まれるが、これらに限定されるものではない。さらに適した抗原源としては、ポックスウイルス科のメンバー、例えばサル痘ウイルス、伝染性軟属腫ウイルス、痘瘡ウイルス;ヘパドナウイルス科の原型メンバーであるB型肝炎ウイルスならびに急性および/慢性肝炎を引き起こす他のウイルス、例えばデルタ型肝炎ウイルス、E型肝炎ウイルスが挙げられる。
【0071】
第2の実施形態において、本発明は、腫瘍抗原に対する免疫応答を誘導するための免疫原性組成物(例えば、ワクチン)を提供する。適切な組成物は、腫瘍抗原をコードする最適化核酸配列を含む組換えチンパンジーアデノウイルスと生理的に許容される担体とを含有するであろう。特定の実施形態において、該カセットのコード配列要素は、単一の免疫原、例えば病原因子由来の抗原、または自己抗原、例えば腫瘍関連抗原をコードしうる。他の実施形態において、該コード配列は2以上の免疫原をコードしうる。例えば、これは、Her2 Neu、CEA、Hepcam、PSA、PSMA、テロメラーゼ(Telomerase)、gp100、メラン(Melan)−A/MART−1、Muc−1、NY−ESO−1、スルビビン(Survivin)、ストロメリシン(Stromelysin)3、チロシナーゼ(Tyrosinase)、MAGE3、CML68、CML66、OY−TES−1、 SSX−2、SART−1、SART−2、SART−3、NY−CO−58、NY−BR−62、hKLP2、VEGF、5T4のような自己抗原の組合せをコードしうる。
【0072】
トランスジーンの発現を導くために使用する転写プロモーターは、好ましくは、真核性RNAポリメラーゼにより認識される。好ましい実施形態においては、該プロモーターは「強力」または「効率的」プロモーター、例えば、本明細書に記載の実施例において使用するマウスCMVプロモーター(mCMV)である。もう1つの強力プロモーターの一例は、好ましくはイントロン配列を伴わない、最初期ヒトサイトメガロウイルスプロモーター(参照により本明細書に組み入れるChapmanら,1991 Nucl.Acids Res 19:3979−3986)である。したがって、本明細書中に開示されており特許請求されているエピソームにおける使用に適した1つの代替的プロモーターには、ヒトCMVプロモーターが含まれる。当業者は、任意の多数の他の公知プロモーター、例えば強力な免疫グロブリンまたは他の初期もしくは後期ウイルスプロモーター、例えばSV40初期もしくは後期プロモーター、ラウス肉腫ウイルス(RSV)初期プロモーター;真核細胞プロモーター、例えばβアクチンプロモーター(Ng,S.Y.,Nuc. Acid Res.17:601−615,1989,Quitscheら,J.Biol.Chem.264:9539−9545,1989)、GADPHプロモーター(Alexanderら,Proc.Nat.Acad.Sci.USA 85:5092−5096,1988,Ercolaniら,J.Biol.Chem.263:15335−15341,1988)、メタロチオネインプロモーター(Karinら Cell 36:371−379,1989;Richardsら,Cell 37:263−272,1984)ならびにコンカテネーション化応答要素プロモーター、例えばサイクリックAMP応答要素プロモーター(cre)、血清応答要素プロモーター(sre)、ホルボールエステルプロモーター(TPA)および応答要素プロモーター(tre)(最小TATAボックス近傍のもの)を使用しうると理解する。
【0073】
遺伝子発現カセット内に存在する好ましい転写終結配列は、ウシ成長ホルモンターミネーター/ポリアデニル化シグナル(bGHpA)である。代替的転写終結/ポリアデニル化配列には、チミジンキナーゼ(tk)遺伝子に由来するもの又はSV40由来配列、例えばpCEP4ベクター(Invitrogen)において見出されるものが含まれるが、これらに限定されるものではない。
【0074】
本発明の目的、利点、用途および方法について全般的に説明してきたが、本発明の種々の実施形態を詳細に説明するために、以下に非限定的な実施例を記載する。しかし、本明細書に記載の発明は、以下の実施例の詳細には限定されず、これらは、本発明の実施を望む者のための単なる指針として記載されているに過ぎないと理解されるべきである。本発明の範囲は、完全な開示および本明細書に添付されている特許請求の範囲に関して評価されるべきである。
【0075】
材料および方法
本発明の実施は、特に示さない限り、分子生物学(組換え技術を含む)、微生物学、細胞生物学および生化学の通常の技術を用い、これらは当技術分野の通常の技術の範囲内である。このような技術は、例えば“Molecular Cloning:A Laboratory Manual”,2nd edition(Sambrookら,1989);“Oligonucleotide Synthesis”(M.J.Gait編,1984);“Animal Cell Culture”(R.I.Freshney編,1987);“Methods in Enzymology”(Academic Press,Inc.);“Handbook of Experimental Immunology”,4th edition(D.M.Weir & C.C.Blackwell編,Blackwell Science Inc.,1987);“Gene Transfer Vectors for Mammalian Cells”(J.M.Miller & M.P.Calos編,1987);“Current Protocols in Molecular Biology”(F.M.Ausubelら編,1987);および“PCR:The Polymerase Chain Reaction”(Mullisら編,1994)のような参考文献に十分に説明されている。
【0076】
配列決定は、標識プライマーもしくはターミネーターを使用する商業的に入手可能な自動シークエンサーまたは配列決定用のゲルに基づく方法を用いて行うことが可能である。また、配列分析は、標的DNAまたはRNA分子上でお互いに直に隣接してアニールするオリゴヌクレオチド配列の連結に基づく方法により行う(WuおよびWallace,Genomics 4:560−569(1989);Landrenら,Proc.Natl.Acad.Sci.87:8923−8927(1990);Barany,F.,Proc.Natl.Acad.Sci.88:189−193(1991))。図11は、本開示に記載されているポリヌクレオチドおよびポリペプチド配列のヌクレオチドおよび/またはアミノ酸配列(すなわち、配列番号1〜21)を示す。
【0077】
本開示の全体にわたって記載されている具体的な塩基対の番号に関する基準として、野生型アデノウイルス血清型5を使用する。野生型アデノウイルス血清型5の配列は公知であり、当技術分野において記載されている。参照により本明細書に組み入れるChroboczekら,1992 J.Virology 186:280を参照されたい。当業者は、アデノウイルス血清型5に関する塩基対により定められる領域に対する配列相同性により、他のアデノウイルス血清型(例えば、血清型2、4、6、12、16、17、24、31、33および42)における前記領域を容易に特定することが可能である。したがって、ヒトアデノウイルス血清型5を使用する以下の実施例は限定的なものではないと理解されるべきである。当業者は、異なるアデノウイルス血清型(例えば、Ad2)に関して、類似したプラスミド、ウイルスおよび技術を利用しうると認識するであろう。同様に、異なるヒトアデノウイルス、特にチンパンジーアデノウイルスに関して、類似したプラスミド、ウイルスおよび技術を利用しうるため、ヒトAdの使用は限定的なものではない。
【0078】
プラスミドの構築
Tetサイレンサーおよび逆Tetトランスアクチベーター発現カセットを含有するpIRESTの構築を図1Aに記載する。該Tet系は、単一の発現ベクターにおいて、以下のとおりに構築した。Tetサイレンサー(tTS)を含有するEcoRI−ClaI DNA断片をプラスミドpUHS6−1(E.Bujardにより快く提供されたもの)から単離し、ベクターpIRES−Neo(Clontech)内のIRES配列の下流に挿入してNeo遺伝子を置換した。その新たなベクターpIRES−tTSを、ヒトサイトメガロウイルスIEプロモーターの下流の唯一のEcoRV制限部位内へのrtTA2遺伝子(E.Bujardにより快く提供されたpUHD 52−1に由来する。)の挿入により修飾して、pIREStTS/rtTAを得た。図2は、AdTetLuc感染クローンにおけるルシフェラーゼ発現のグラフ表示を示す。簡潔に説明すると、293EBNA細胞および種々の293EBNA/Tetクローンに、1μg/ml ドキシサイクリンの存在下(黒色棒線)または非存在下(白色棒線)、AdTetLucを感染させた(m.o.i 10)。細胞溶解物におけるルシフェラーゼ活性を感染の48時間後に評価した。
【0079】
Tetタンパク質を安定に発現する細胞クローンを単離するための選択ーマーカーを導入するために、pPURベクター(Clontech)から得たピューロマイシン耐性発現カセットをpIREStTS/rtTAのXhoI部位内に挿入してpIREStTS/rtTApuroを得た。pE2の構造を図1Bに記載する。
【0080】
Ad5ポリメラーゼcDNAを含むプラスミドpVacPolから得たClaI/SphI断片、およびAd5−pTP cDNAを含有するpVACpTPからのAcc65/EcoRV断片を誘導性Tetプロモーターの制御下のベクターpBI(Clontech)内に挿入することにより、Ad5ポリメラーゼおよびプレ末端タンパク質を発現するビシストロン発現ベクターを構築した(pVacPolおよびpVACpTPはP.C.van der Vlietにより快く提供されたものである。)。オリゴヌクレオチド:5’−TTATACGCGTGCCACCATGACTACGTCCGG−3’(配列番号9)および5’−TTATGCTAGCGCGAAGGAGAAGTCCACG−3’(配列番号10)ならびにpFG140(19)から得たAd5 DBP遺伝子(Ad5[nt]22443−24032)(配列番号11)を使用するPCRにより得たAd5 E4 orf6(Ad5[nt]33193−34077)(配列番号8)を同じベクターpBI内に挿入することにより、第2の二方向性誘導性カセットを構築した。
【0081】
HS4絶縁体(insulator)に隣接するpCEP4に由来するEBV−OriP(EBV[nt]7333〜ヌクレオチド9312;GenBank V01555)(配列番号12)領域をpJC13−1(9)のBamHI部位内への直接的クローニングにより得た。オリゴヌクレオチド:5’−AACTACAATTCCCAACACATAC−3’(配列番号13)および5’−CACATCCGTCGCTTACATG−3’(配列番号14)を使用するpFG140からのPCRにより、Ad5 ITR結合部を増幅した。
【0082】
最後に、tk−ヒグロマイシン−Bホスホトランスフェラーゼ(HPH)カセットをpCEP4(Invitrogen)から誘導した。pE2を構成するすべての要素をpBI−pol/pTPベクター内に順次導入して、最終的にpE2を得た。
【0083】
pAd5ΔE1−4の構築
pAdCMV/LacZ/ΔPolベクター(A.Amalfitanoから快く提供されたもの(4))およびAd5dl308ΔpTPβ−gal(J.Schaackから快く提供されたもの)(45)からのそれぞれAd5ポリメラーゼ(Ad5[nt]7274−7883)(配列番号4)およびプレ末端タンパク質(Ad5[nt]8915−9462)(配列番号5)の部分欠失をMRKpAd5E3(52)内に導入することにより、E2b遺伝子が欠失したAd5ΔE1−E3バックボーンを得た。また、ATGからCTGへのポリメラーゼ開始コドンの部位特異的突然変異誘発も行って、最終的にpAd5 ΔE1,E3,E2Bベクターを得た。DBP遺伝子を含むDraI−MscI断片(Ad5[nt]22445−24029)が欠失したAd5のBamHI/XhoI断片([nt]21563−24797)を含有するpBluescriptKSII+(Stratagene)はRocco Savinoにより快く提供された。
【0084】
***pAd−ΔE1−2ベクターは、ΔDBP断片およびAdΔE1,E3,E2Bベクターを大腸菌(E.coli)Bj5183内に同時形質転換する相同組換えにより得た。orf3を除く完全なE4単位([nt]32830−34316および[nt]34895−35443)の欠失を、以下に記載するとおりに行った。AvrIIおよびMfeI制限部位を末端に有するorf3領域をPCR(ΔE4orf fw AvrII:5’− GCCTAGGGATGCGTGTCATAATCAGTGTGGGTTC−3’(配列番号15);ΔE4orf3 rev MfeI:5’−CAATTGAAAAGTGAGCGGGAAGAGCTGGAAGAACCATG−3’(配列番号16))により増幅し、同じ酵素で消化されたE4−シャトルベクター内にクローニングした。E4orf3はE4プロモーターおよびポリAシグナルを維持する。pAd5ΔE1−4orf3+ベクターを、そのようなDNAをpAd5ΔE1−2ベクターと共に大腸菌(E.coli)BJ5183内に同時形質転換することにより得た。
【0085】
すべての発現カセットは、トランスジーン発現用のCMVプロモーターおよびBGHポリAシグナルに加えて、記載されている(53)とおりに大腸菌(E.coli)BJ5183における相同組換えによりpAd5ΔE1−4orf3+のE1領域内の挿入を可能にするAd5配列[nt]1−450(左)および[nt]3511−5792(右)を含有するAd5−シャトルベクターのコンテクストにおいて構築した。EGFP cDNAをpEGFPプラスミド(Clontech,BD Bioscience,San Jose,CA,USA)から得、ついでAd−シャトルプラスミド内にクローニングしてpShAd5 EGFPを得た。5’および3’非翻訳末端反復配列(UTR)が欠失したHCV−BK cDNA(HCV BK[nt]342−9374(配列番号17))をプラスミドpCMV(1−9.4)(14)から誘導した。
【0086】
NS5B ORFは、ウイルスRNA依存性RNA−ポリメラーゼの触媒三つ組み残基に対応する3つのアミノ酸位置において変異させて(G−2737をAへ、D−2738をAへ、およびD−2739をGへ)、酵素活性を喪失させた(Nicosiaら,未公開データ)。最適化コザック配列に融合したHCV cDNAを、HCMVプロモーターをMCMVプロモーターで置換することにより得たpAd5−シャトルの修飾形態内にクローニングして、最終的にpShAd5HCVを構築した。pAd5ΔE1−4orf3+のE1領域内へのすべての発現カセットの挿入を、記載されているとおりに(43)、大腸菌(E.coli)内での相同組換えにより達成した。
【0087】
細胞
293EBNA細胞系(Invitrogen)をダルベッコ変法イーグル培地(DMEM)+10%ウシ胎児血清(FBS)、ペニシリン(100U/ml)、ストレプトマイシン(100μg/ml)、2mM グルタミンおよび250μg/ml G−418(GIBCO BRL)内で培養した。0.5μg/ml ピューロマイシンを含有する同じ培地を使用することにより、293EBNATet細胞を選択した。2E2細胞を選択することにより、90μg/mlのヒグロマイシンBを、既に記載されている培地に加えた。Lipofectamine2000(Invitrogen)を該製造業者の説明に従い使用してプラスミドDNA形質移入を行った。逆TetトランスアクチベーターおよびTetサイレンサータンパク質を発現する293EBNAクローンを得るために、形質移入の1日前に、1×106個の293EBNA細胞を6cmプレート内に播き、SapIで線状化された5μgのpIREStTS/rtTApuroで形質移入した。形質移入の48時間後、細胞をトリプシン処理し、15cmプレート内のピューロマイシン含有DMEM内に播いた。耐性クローンを単離し、ついで、Tet−ルシフェラーゼカセットを含有する組換えAd5でスクリーニングした。各クローンの5×105個の細胞を24ウェルプレート内に三重に播き、該細胞にAd5 Tet−lucをドキシサイクリンの存在下および非存在下で10の感染多重度(moi)で感染させた。感染の24時間後、細胞を回収し、細胞溶解物においてルシフェラーゼ活性を測定した(ルシフェラーゼアッセイ系;Promega)。遺伝子発現の誘導およびサイレンシングの両方を各クローンに関して、親293EBNA細胞において行った対照実験において得た相対光単位(rlu)値に対する比として評価した。
【0088】
2E2パッケージング細胞系を得るために、前記プロトコールに従い293EBNA Tet細胞をpE2ベクターで形質移入した。90μg/mlのヒグロマイシンBを含有するDMEMを使用して、安定なトランスフェクタントを選択した。耐性クローンを増殖させ、Ad5ΔE1−2EGFP DNAの形質移入によりスクリーニングした。陽性クローンをCPEの出現により特定し、Ad5ΔE1−2EGFPベクターの連続継代により確認した。1×106 2E2細胞(n=3)からのエピソームコピー数を染色体外DNA上で直接的に定量的リアルタイムPCRにより評価した。プローブ(5’−FAM−TGGCATGACACTACGACCAACACGATCT−3’−TAMRA)(配列番号18)ならびにプライマーE4−fw(5’−ACTACGTCCGGCGTTCCAT−3’)(配列番号19)およびE4−rw(5’−GGAGTGCGCCGAGACAAC−3’)(配列番号20)。
【0089】
ウイルスの増幅および力価測定
多欠失ウイルスの産生を2E2パッケージング細胞系において行った。アデノウイルスゲノムをPacI消化によりそれぞれのプラスミドから遊離させ、1μg/ml ドキシサイクリンの存在下で2E2細胞内に形質移入した。形質移入の4〜6日後、細胞を3サイクルの凍結/解凍により細胞溶解し、該溶解物の5分の1を使用して、該ウイルスを連続継代により増幅した。2層細胞ファクトリー(factory)(NUNC)内に播いた2E2細胞に感染させることにより、大規模増幅を行った。アデノウイルスベクターをCsCl勾配により精製し、透析し、リアルタイムPCRにより定量した。該CsCl精製ベクターの感染度を組織培養感染量50%(TICD50)として2E2上で評価した(43)。
【0090】
サザンブロット分析
pE2複製をサザンブロット分析により評価した。293EBNATet細胞を6cmディッシュ内に播き、ドキシサイクリン(1μg/ml)の存在下または非存在下、Lipofectamine2000(Invitrogen)により5μgのpE2ベクターで形質移入した。48時間後、Hirt法(22)により染色体外DNAを単離した。ついでDNAをNotIおよびDpnIで消化し、32P DNAプローブを使用する標準的な方法によるサザン分析に付した。Phosphor Imager(商標)系(Molecular Dynamics)を使用するオートラジオグラフィーによりシグナルを検出した。
【0091】
安定なpE2クローンからのエピソームDNAをHirtプロトコールに従い抽出し、BamHIで消化し、32P標識pE2 DNAをプローブとして使用するサザンブロット法により分析した。
【0092】
ウエスタンブロット分析
形質移入の48時間後、以下のとおりにタンパク質発現の分析を行った。2E2歳棒をリン酸緩衝食塩水(PBS)で2回洗浄し、6cmプレート当たり0.5mlのRIPAバッファー(1×PBS、1% NP−40、0.5% デオキシコール酸ナトリウム、0.1% SDS、0.05mM PMSF)を加えることにより細胞溶解した。プレートを氷上で1時間インキュベートし、ついで10,000×g、4℃での遠心分離後、可溶性タンパク質を細胞溶解物から集めた。30μgのタンパク質上でウエスタンブロット分析を行った。サンプルを10% SDS PAGE上で分離し、Protanニトロセルロースメンブレン(SchleicherおよびSchuell)上にブロッティングした。該メンブレンをポリメラーゼまたはpTPに対するウサギ抗血清と共に、および抗DBP mab(F.Graham,Mc Master University,Hamilton,Canadaにより快く提供されたクローンH2−19)と共にインキュベートした。ホースラディッシュペルオキシダーゼ結合二次抗体の存在下でのインキュベーション後、タンパク質をSupersignal West Pico化学発光基質(PIERCE)により検出した。以下の試薬を使用することによりHCVタンパク質発現を検出した:抗コアモノクローナル抗体(mab)B12.F8(M.Mondelli,University of Paviaにより快く提供された);抗E2、mab 185.C7;抗NS3 mab 10E5/24;抗NS5a、ウサギポリクローナル抗血清;抗NS5b mab 20B6/13。
【0093】
免疫化プロトコールおよび免疫応答の分析
6〜8週齢の雌C57BL/6、A2.1およびCB6F1マウス(Charles River Breeding Laboratories)を、該ウイルスを両方の大腿四頭筋内に注射することにより免疫した。注射の3週間後に免疫応答を分析した。
【0094】
該ウイルスを大腿四頭筋内に注射することにより、アカゲザル(Rhesus macaques)を免疫した。注射の4、6、8および12週間(W)後に免疫応答を分析した。
【0095】
E2タンパク質に対する抗体価を、Zucchelli,S.ら(55)に記載されているとおりにELISAにより測定した。細胞性免疫応答を以下に記載のとおりに評価した。HCVコア、NS3、NS4、NS5aおよびNS5bタンパク質の全配列を含む15マーの重複ペプチドのプールを使用して、HCV特異的IFNγ分泌細胞を明らかにした。いくつかの実験においては、CD8+エピトープを含有する9マーペプチドを使用して免疫応答を評価した(HCV NS3タンパク質からのpepl480,GAVQNEVTL(配列番号21))。以下のとおりにIFNγ酵素結合イムノスポットアッセイ(ELIspot)によりIFNγ分泌細胞を定量した。マルチスクリーン96ウェル濾過プレート(Millipore)を100μlの抗マウスIFNγ Mab(PharMingen)でコートし、4℃で一晩インキュベートし、ついで1×PBSで洗浄し、ウェル当たり200μlのR10培地で2時間ブロッキングした。脾細胞を免疫化マウスから調製し、R10培地(10% ウシ胎児血清、2mM L−グルタミン、50U/ml ペニシリン、50μg/ml ストレプトマイシン、10mM HEPES、50μM 2−メルカプトエタノールで補足されたRPMI 1640)内に再懸濁させ、ついで、2.5×105または5×105/ウェルの密度で抗IFNγ mabでコートされたマルチスクリーン96ウェルプレート上でプレーティングした。ついで脾細胞を200ng/ウェルのペプチドプールの存在下で24時間インキュベートした。十分な洗浄(1×PBS、0.005% Tween)の後、ビオチン化ラット抗マウスIFNγ抗体(PharMingen,San Diego,Calif.)を加え、室温で3時間インキュベートした。最後に、ストレプトアビジン−アルカリホスファターゼ(PharMigen)および1−Step NBT−BClP現像液(Pierce,Rockford,Ill.)を該ウェルに加えた。ELIspotリーダー(Bioline)を使用してスポットを計数した。
【0096】
IFNγ細胞内染色およびFACS分析を以下のとおりに行った。Elispotアッセイに関して記載されているとおりに調製した脾細胞を、タンパク質輸送を抑制するブレフェルジン(GolgiPlug,PharMingen)を含有するR10培地内でペプチドプールと共に一晩インキュベートした。FACSバッファー(PBS w/o CaおよびMg、1% FCS、0.01% NaN3)中、飽和量の精製抗マウスCD16/CD32と共に細胞をインキュベートすることにより、細胞ブロッキングを行った。FACSバッファーでの洗浄の後、APC結合抗マウスCD3e、PE結合抗マウスCD4およびPerCP結合抗マウスCD8a抗体を該細胞に加え、室温で30分間インキュベートした。ついで、Cytofix/Cytoperm Plus(GolgiPlugを伴う)キットを使用して、細胞を4℃で20分間にわたり透過性亢進させた。Perm/Washでの洗浄の後、FITC結合抗マウスIFNγを加え、該細胞を室温で30分間インキュベートした。該最終染色工程後、該細胞を洗浄し、1% ホルムアルデヒド中で固定した。データを、FACSCalibur(Beckton & Dickinson)を使用して集め、Cell−Questソフトウェア(BD Biosciences)を使用して分析した。すべての抗体および二次試薬はBD PharMingen(San Diego,CA)から購入した。
【0097】
Accuspin Istopaqueチューブ(Sigma Aldrich cat A0561)を該製造業者の説明に従い使用して、サルPBMCをEDTA処理末梢血から単離した。簡潔に説明すると、血液を、等容量のHBSS(ハンクス平衡溶液Gibco cat 14175−053)を含有するAccuspinチューブ内に移し、800g、室温で15分間遠心分離した。PBMCバンドを集め、HBSSで1回、R10で1回洗浄し、最後にR10に再懸濁させた。各ウェル内でプレーティングする細胞の量が異なる以外は前記のとおりに(2×105および4×105/ウェル)、g−IFN−Elispotを行った。
【0098】
ワクシニアウイルスチャレンジ
免疫化マウスに5×106pfuのVV−NSをi.p.注射した。5日後に摘出した個々のマウスからの対(ペア)になった卵巣を均質化し、3回、凍結−解凍し、Hul43TK−細胞の単層上で10倍希釈物をプレーティングすることにより力価測定した。48時間後、0.5% クリスタルバイオレットでの染色により力価を読取った。
【0099】
実施例
本発明の種々の特徴を更に例示するために、以下に実施例を記載する。該実施例は、特許請求されている発明の実施のための有用な方法をも例示するものである。該実施例は本発明を限定するものではない。
【実施例1】
【0100】
Tet−S/rtTA2を共発現する細胞系の開発
293EBNA細胞系におけるpIREStTS/rtTApuro形質移入およびそれに続くピューロマイシン選択(「材料および方法」を参照されたい)により得た安定なクローンを、Tet誘導性ルシフェラーゼ発現カセットを含有する第1世代Adベクター(Ad Tet−luc)を使用することによりスクリーニングした。ピューロマイシン耐性クローンを24ウェルプレート内に三重に播き、10のmoiで細胞にAd Tet−lucを感染させ、該細胞をドキシサイクリン(1μg/ml)の存在下/非存在下で維持した。感染の24時間後、該粗細胞溶解物においてルシフェラーゼの発現を測定した。20倍を超えるルシフェラーゼ活性の誘導を示すクローンを選択し、増殖させた。図2に示すとおり、選択されたすべてのクローンにおいて、Tetサイレンサーの共発現はルシフェラーゼの基底レベルを減少させた。ドキシサイクリンの存在下で基底発現に対して25倍の減少を示し最良の誘導比(100倍超)を伴うクローン1.1(293EBNATetと称される)を選択し、増殖させた。
【実施例2】
【0101】
E2/E4 orf6アデノウイルスアンプリコン(pE2)の構築
すべての初期遺伝子が欠失したAdベクターを機能的に相補するために、以下の要素を含有する、Ad5に基づくアンプリコンを構築した:i)EBNA−1タンパク質を発現する分裂細胞の核における安定な維持のためのEBVの潜在性複製起点(Ori−P)(48);ii)tk−ヒグロマイシンB選択マーカー;iii)Adに基づく様態でのプラスミド複製を可能にするpFG140(19)由来のAd5ウイルスITR結合部;iv)二方向性テトラサイクリン誘導性プロモーターの制御下の2つの分岐転写単位において配置されたAd5 E2(ポリメラーゼ、プレ末端タンパク質およびDNA結合タンパク質)およびE4−orf6遺伝子。E2およびE4 orf6遠位プロモーター(17)に対するOriPのエンハンサー効果を減少させるために、OriP要素に隣接する2つのニワトリβグロビンHS4絶縁体(insulator)二量体(9)をも導入した。得られたプラスミド(pE2)の構造を図1Bに示す。
【0102】
pE2で形質移入された293EBNATet細胞の培地におけるドキシサイクリンの添加の際のE2遺伝子発現の誘導をウエスタンブロットにより測定した。図4B(レーン2、5および8)に示すとおり、エフェクター薬の非存在下では形質移入の48時間後にタンパク質発現は検出されなかったが、1μg/mlのドキシサイクリンを加えることにより転写を誘導した場合には、すべてのE2タンパク質の強力な発現が明らかであった(レーン3、6および9)。形質移入の4および6日後にも同様の結果が得られた(データ非表示)。
【0103】
Ad複製に必要なシスおよびトランス作用性要素の両方が前記系内に存在するため、E2遺伝子発現の誘導が293EBNATetにより形質移入された細胞におけるpE2 DNA複製をも誘発するのかどうかを試験した。形質移入の48時間後、全DNA上のサザンブロットによりプラスミド複製を検出した。サンプルを、まず、DpnIで消化して投入プラスミドDNAを除去し、ついでNotIで消化して、Ad ITRを介して複製された直鎖状形態と天然環状プラスミド形態とを区別化した(図3)。図3に示すとおりにpE2から誘導されたDNAプローブとブロットとをハイブリド形成させたところ、該ブロットはpE2の環状形態に関する12.6Kbのプラスミド由来のバンドを示した(図4A、レーン1)。pE2で形質移入された細胞からの全DNAを分析したところ、複製されたアンプリコンの予想サイズのバンドは、ドキシサイクリンで細胞を誘導した場合にのみ、視認可能であった(図4A、レーン2および3)。ITR結合部が欠失したpE2プラスミド誘導体の形質移入からはDNA複製の証拠は検出されなかった(データ非表示)。これらのデータは、tTS/rtTAサイレンシング/活性化系が、該プラスミドが一過性形質移入後に高コピー数で存在する場合であっても、pE2機能の完全な遮断を可能にするが、初期遺伝子発現のデオキシサイクリン誘導に際してはアデノウイルス特異的に該アンプリコンの効率的な複製を支持しうることを示した。それらは、初期遺伝子が欠失したアデノウイルスベクターのレスキューおよび増殖のための適当な系としての293EBNATet細胞と組合されたpE2アンプリコンを支持する確固たる証拠を提供した。
【実施例3】
【0104】
E1、E2、E4相補細胞系の作製
E2遺伝子発現の誘導は、アデノウイルス複製装置の活性化を介して、直鎖状DNAとしてのpE2の複製をもたらすことが観察された。pE2を使用して、Tet転写サイレンサー(tTS)および逆Tetトランスアクチベーター2(rtTA2)を発現する293EBNA細胞を形質転換して、2E2安定細胞系を得た。2E2細胞は、ドキシサイクリンを培地に加えた場合には、Ad5第1世代(FG)ベクターに感染した293細胞より高いレベルのポリメラーゼ、前駆体末端タンパク質(pTP)およびDNA結合タンパク質(DBP)を産生した。E2およびE4ORF6遺伝子の発現は、誘導されると、第1世代(FG)アデノウイルスベクターと比較しうるレベルまでの、E1、E2、E3およびE4遺伝子を欠く多欠失Ad5ベクターの増幅を効率的に支持した。
【0105】
簡潔に説明すると、pE2で形質移入された293EBNATet細胞を、「材料および方法」に記載されているとおりに、ヒグロマイシンBの存在下で選択した。個々のクローンを増殖させ、E2遺伝子欠失を含有するAd5ベクターのレスキューおよび増殖を調べることによりスクリーニングした。6ウェルプレート内に播いた細胞をdoxy存在下のAd5ΔE1−2ベクターでの形質移入に付した。形質移入の7日後、細胞を凍結−解凍により細胞溶解し、500μlの細胞溶解物を使用して各対応クローンの新鮮単層に感染させた。継代1におけるCPEの直接観察により陽性クローンの評価を行った。ついで該ベクターを該選択クローンにおいて連続継代し、リアルタイムPCRにより増殖を評価した。2回の連続継代の後、ウイルスゲノムは細胞溶解物1ml当たり約1×1010 ゲノムのプラトー付近に達し、ついでこれを、感染moiの上昇にもかかわらず維持された(データ非表示)。サザンブロット分析で染色体外DNAを評価することにより、未修飾無傷エピソーム要素としてpE2を含有するいくつかのクローンを特定した。
【0106】
細胞系の特徴づけ及びΔE1,E2,E3,E4ベクターの増幅の後続工程のために、2E2と称されるクローン11を選択した。まず、2E2細胞におけるpE2のコピー数を、染色体外DNA上のリアルタイムPCRにより、16コピー/細胞の平均として決定した(n=3)。継代にわたる2E2細胞におけるpE2エピソームプラスミドの安定性を評価するために、15継代後にHirt法(22)によりエピソームDNAを抽出し、ついでBamHIで消化し、プラスミド全体をプローブとして使用するサザンブロットにより分析した。図5Aには、元のプラスミドと比較した、細胞系から抽出されたエピソームの制限パターンが示されている。それらのパターンは同一であった。このことは、該エピソームが細胞核内で経時的に安定に維持されること、およびプラスミド構造内の再構成が生じなかったことを示している。エピソームDNAの安定性は、ドキシサイクリンの非存在下で検出可能な発現を伴わない一過性形質移入実験で既に観察されているものに類似した細胞系の15継代後にウエスタンブロットによりE2タンパク質の発現を分析することによっても証明された(図5B)。E2タンパク質発現レベルを比較するために、FGウイルスに感染した未誘導2E2細胞から得た細胞抽出物を含めた。
【実施例4】
【0107】
ΔE1−4orf3+ Ad5バックボーンの構築
該ベクター構築を促進するために、左ITRおよびパッケージングシグナル(Ad5[nt]1−450)(配列番号1)ならびに発現カセットに隣接するpIX遺伝子(Ad5[nt]3511−5792)(配列番号2)を含むAd5断片を含有するシャトルベクターを構築した。大腸菌(E.coli)株BJ5183における相同組換えにより発現カセットをAd5 E1領域内に組換えた。その新たなベクター系を評価するために、pAd5ΔE1−4orf3+EGFPを構築した。pAd5ΔE1−4orf3+EGFPベクターをPacIで線状化して感染性ウイルスDNAをプラスミド配列から遊離させ、2E2細胞内に形質移入し、それをドキシサイクリンの存在下または非存在下でインキュベートした。2連続継代後に得た結果を図6に示す。EGFP形質導入ウイルス粒子およびCPEは、培地へのドキシサイクリン添加によりE2およびorf6遺伝子が誘導された場合にのみ生じた。orf3およびorf6タンパク質の共発現は、E4単位が欠失したAdベクターの高力価増幅に寄与する(25)。相補遺伝子誘導の非存在下ではウイルス粒子は生じなかった。ベクター産生のプラトーは、細胞の100%がEGFP陽性である場合に、2連続継代の直後に観察された(図6)。高力価への多欠失ベクターの増殖の支持における該新規細胞系の効率を評価するために、5つの2層細胞ファクトリー(Nunc Inc.)内に播かれた約5×108個の2E2細胞を感染させることにより、大規模製造を試みた。該ウイルスをCsCl勾配の2回通過により精製し、リアルタイムPCRにより定量して、最終的に、約5000vp/細胞の生産で2×1012 vp/mlの力価を得た。EGFPおよびHCVを発現する多欠失ベクターとFGベクターとの間の比較を表1に示す。
【0108】
【表1】
【0109】
すべての初期遺伝子の欠失を組合せることによりAd5バックボーン内に生じた理論的空間は約12.4Kbである。該新規ベクター系の大きな容量を利用して、マウスサイトメガロウイルス(MCMV)プロモーターに融合した全HCVポリタンパク質遺伝子用の発現カセットを挿入した。該HCVポリタンパク質発現カセットは、5’および3’非翻訳領域を除去し、コアATGの上流に最適コザック配列を挿入し、NS5Bレプリカーゼの触媒ドメインを突然変異させて酵素活性を除去することにより構築された(44)。トランスジーンの発現効率を増加させるために、ヒトCMVプロモーターを、FGアデノウイルスベクターにおいて4〜30倍強力であると報告されているマウスCMVプロモーター(1)で置換した。
【0110】
Ad5ΔE1−4orf3+HCVベクターは2E2細胞内の形質移入により成功裏にレスキューされた。培地にドキシサイクリンを1μg/mlの最終濃度で加えることにより、形質移入の直後にE2遺伝子発現を誘導した。2E2細胞内での連続継代によりAd5ΔE1−4orf3+HCVベクターを増幅した。「材料および方法」に記載されているとおりに、粗細胞溶解物内のウイルスゲノム濃度をリアルタイムPCRにより評価した。大規模調製物を得るために、4回の連続増幅継代の後に得た粗溶解物を使用して、2.8×109個の2E2細胞に約100ゲノム/細胞のmoiで感染させた。感染の48時間後、完全なCPEが明らかに認められた時点で細胞を回収した。精製ウイルスの最終収量を表1に示す。293細胞内で増殖させたEGFPを発現するΔE1E3FGベクターと異ならない約5000粒子/細胞の生産を得た。
【0111】
ポリメラーゼおよびプレ末端タンパク質の欠失は、これらの2つの遺伝子の一部分のみを含むものであったため、AdベクターとpE2エピソームとの間の相同性の領域は、野生型遺伝子をウイルスゲノム内にレスキューして戻すのに理論的に十分なものである。連続継代に際してのAd5ΔE1−4orf3+HCVベクターの構造的完全性を評価するために、および野生型初期遺伝子を含有するウイルスの再構成が2E2細胞内でのベクター増幅中に生じうるかどうかを試験するために、CsCl精製ベクターのDNA構造を決定した。5継代後にウイルス粒子から精製したDNAから得たパターンとプレ−アデノプラスミドの放射能標識制限パターンとの比較を図8に示す。
【0112】
該野生型遺伝子を含有する断片のサイズを比較するために、FGプラスミドベクターをゲル(レーン1)内に含めた。Ad5ΔE1−4orf3+HCVベクターの制限パターンは親プラスミドのものと同一であるようであり、再構成または野生型E2−E4遺伝子を含有するベクター種の出現の証拠は認められなかった。
【0113】
種々のmoiのベクターを使用する293およびHeLa細胞系のインビトロ感染により、HCVタンパク質の発現の効率を評価した。HCVコア、E1、E2、NS3、NS4、NS5aおよびNS5Bに対する特異的モノクローナル抗体またはポリクローナル抗血清でのウエスタンブロット分析は該感染細胞におけるHCVタンパク質の存在を示したが、このことはHCVポリタンパク質の正しいプロセシングを示している(図9)。未プロセシング産物は検出されなかった。
【0114】
2E2細胞系は、FGベクターに感染した293細胞より高いレベルのE2aおよびE2bを発現することを、データが示していることに注目すべきである。理論により束縛されるものではないが、比較的高いレベルのE2aおよびE2bの産生が細胞当たりの多欠失ベクター粒子の高収量につながったと考えられる。細胞当たりの多欠失粒子の収量は、FGベクターから得られた収量と一貫して比較しうるものであった。さらに、PolおよびpTP遺伝子がpE2との相同組換えによりベクターバックボーンにおいてレスキューされうる理論的可能性にもかかわらず、Ad5ΔE1−4orf3+ベクターは連続継代にわたり安定であることが示された。相補タンパク質の発現の、観察された高いレベルは、おそらく、多欠失ベクターに対するE2b野生型ウイルスの選択的利点を低下させるであろう。
【実施例5】
【0115】
AdΔE1−4orf3+HCVベクターでの免疫化はマウスにおける強力なCMI応答を誘導する
ヒトおよびチンパンジーにおいて観察されたHCV感染の消散は、典型的には、複数のエイトープに対する強力なT細胞応答を伴っている(56,57)。感染対象において特定されたHLAクラスI限定エピトープは、クラスタリングの証拠を伴うことなく、ゲノム全体に広がっている(58に概説されている)。細胞性免疫応答を惹起するHCVポリタンパク質を発現する多欠失Adベクターの効力をマウス免疫実験において評価した。該ベクターは、感染細胞のウエスタンブロット分析により示されるとおり成熟産物へと正しくプロセシングされるポリタンパク質前駆体全体の合成を導く。また、種々の系統のマウスにおいてマッピングされたHCV−BK CD8+エピトープを含有するオリゴペプチドを使用して、該免疫化を観察した。HCVエピトープに特異的なCD4+およびCD8+ T細胞を、コア、E2およびNSタンパク質の配列全体に及ぶ重複15マーペプチドのプールを使用することにより、IFN−γ Elispotおよび細胞内染色(ICS)により決定した。
【0116】
CB6F1およびHLA A2.1マウス系統を免疫することにより、複数のウイルス決定基に対する強力なT細胞免疫応答(全CD8+細胞の0.2〜2.25%)を測定した。免疫化マウスの脾細胞の分析は、低レベルのCD4+抗原特異的T細胞を伴うCD8+偏向免疫応答を示した。この免疫応答特性は、送達された抗原に伴われているというよりもワクチン接種方法に伴われたものである。Casimiroらにより、Ad5gagワクチンで免疫されたアカゲザルにおいて、低い比のCD4+/CD8+が観察され、CD8+表現型の応答細胞の大部分は強力なCTL活性伴っていた(59)。これに対して、筋肉内DNA注射によるHCV抗原での遺伝的免疫化は、マウスおよび非ヒト霊長類の両方において、より釣りあったCD4+/CD8+応答をもたらした(Nicosia A.未公開の結果)。
【0117】
種々の量のベクターにより誘導された細胞性免疫を、漸増量の筋肉内注射Ad5ΔE1−4orf3+HCV(1×107〜1×1011 vp/マウス)でC57Bl6マウスを免疫することにより測定した。免疫化の3週間後、NS3タンパク質のヘリカーゼドメイン内にマッピングされたCD8+T細胞エピトープ(GAVQNEVTL(配列番号21)aa 1629−1637 HCV 1b)に対するT細胞応答に関してマウスを試験した。新鮮に単離された脾細胞を9マーペプチドと共に一晩インキュベートし、ついでIFNγELISPOTアッセイにより分析した。
【0118】
表2に示すとおり、誘導されたT細胞応答の大きさはAd5ΔE1−4HCVの用量に依存し、最初の陽性結果は1×108vp/用量で観察された。この用量のベクターで、5匹中4匹のマウスがNS3に対する免疫応答を引き起こし、特異的T細胞の度数は1×106脾細胞当たり100〜180 CD8+細胞であった。
【0119】
表2におけるデータは、5匹の免疫化マウスから得た100万個の脾細胞当たりのIFNγスポット形成細胞(SFC)の数を要約している。C57Bl6マウスにおけるBK NS3ヘリカーゼドメイン内にマッピングされたCD8+エピトープを含有する1480ノナマー(GAVQNEVTL)(配列番号21)と共に脾細胞をインキュベートした。1匹の動物から得た値および免疫化マウスの各群から計算した相乗平均が該表に示されている。抗原特異的CD8+ T細胞の度数は、1011 vp/動物を注射することにより百万個の脾細胞当たり400〜1000のSFCの範囲で568の相乗平均値まで用量と共に増加した。
【0120】
【表2】
【実施例6】
【0121】
ヒトHLA−A2.1を発現するトランスジェニックマウスにおける多特異的CMI応答の誘導
防御性HCVワクチンは、ヒトMHC対立遺伝子の及びウイルスの遺伝的多様性のため、一般的集団における広範な細胞性免疫応答を誘導する必要がある。Ad5ΔE1−4HCVベクターでのマウスの免疫化は、HCVポリタンパク質の複数のエピトープに対する強力なCD8+T細胞応答を誘導した。より詳しくは、複数のHCVエピトープに対する細胞性免疫応答を惹起するAd5ΔE1−4orf3+HCVベクターの能力を測定した。免疫応答の拘束のため、CB6F1およびHLA A2.1トランスジェニックマウスにおいて、ベクター免疫化により惹起されるHCV特異的T細胞応答を測定した。
【0122】
5匹/群の動物の大腿四頭筋にAd5ΔE1−4orf3+HCVの1×1010ウイルス粒子を注射した。注射の3週間後、ワクチンにより誘導された抗HCV免疫の強度および質をICS法により評価することにより、マウスを分析した。脾細胞からの抗原特異的IFNγ分泌を、コア(aa 1−190)およびNS3〜NS5bタンパク質の非構造領域(aa 1026−3009)に及ぶ10残基重複する15マーから構成される7個のペプチドプールで刺激した。5匹のマウスのプールに関して脾細胞の分析を行った。
【0123】
図10に示す結果は、Ad5ΔE1−E4−HCVベクターが、ヒトHLA−A2.1を発現するトランスジェニックマウスにおいて、強力かつ多特異的なT細胞応答を誘導することを示している。より詳しくは、該免疫化は該ペプチドプールのすべてに対するT細胞応答を引き起こした。CD4+およびCD8+の両方の抗原特異的Tリンパ球が観察されたが、応答細胞の大部分はCD8+であった。抗原特異的CD8+リンパ球の度数は、該コアに対するCD8+ T細胞の2.2%から、NS5BのC末端部分に応答するCD8+ Tの0.2%まで、抗原によって様々である。
【実施例7】
【0124】
Ad5ΔE1−E4−HCV免疫化はA2.1マウスにおいてCMI応答を惹起する
トランスジェニックA.21マウスをW=0およびW=2に、1010pp/マウス/注射の用量のAd5ΔE1−E4HCVまたは50μg/マウス/注射の用量の対応Ad5シャトルベクター(pShAd5HCV)で免疫した。1)Ad5ΔE1−E4−HCVで初回抗原刺激および追加抗原刺激し、ならびに2)pSh−Ad5−HCVで初回抗原刺激しAd5ΔE1−E4−HCVで追加抗原刺激し、第4週に動物から得た精製脾細胞の免疫応答を、HCVポリタンパク質全体に及ぶペプチドプールを使用するγIFN−ElispotおよびγIFN細胞内染色により分析した。
【0125】
NS3ヘリカーゼ領域全体に及ぶペプチド(I〜XVIII)のサブセットを使用する実験において、該応答の特異性を測定した。簡潔に説明すると、Ad5DE1−E4HCVで初回抗原刺激しpSh−Ad5−HCVで追加抗原刺激した2匹のマウスから脾細胞を単離した。図12AにおけるElispotの結果は、該マウスが、NS3領域に及ぶペプチドに対して強反応性である広範な免疫応答で応答したことを示している。該データは更に、Ad5ΔE1−E4−HCVまたはpSh−Ad5−HCVのいずれかで免疫されたマウスにおけるHCV特異的免疫応答の初回抗原刺激および追加抗原刺激の両方にAd5ΔE1−E4−HCVを使用しうることを証明した。
【0126】
陽性閾値を超える応答を惹起するプール間の交差点は、考えられうる刺激性ペプチドを表す(ハイライト表示)(図12B)。ついでこれらのペプチドをγIFN−Elispotアッセイにおいて個々に試験した。図12Cに要約した結果は、ペプチド95(LAAKLSGLGINAVAY)(NS3 aal403−1417)(配列番号22)エピトープに対する特異性/反応性を示している。したがって、これらのマウスにおいて惹起された免疫応答は、HCV患者において既に特定されているNS3ヘリカーゼ領域におけるCD8+エピトープに対する特異性を含む特異性により特徴づけられる。
【実施例8】
【0127】
Ad5ΔE1−E4−HCVでの免疫化はVV−NSでのチャレンジに対する防御をもたらす
この実験は、惹起された免疫応答が、HCV非構造タンパク質を発現する組換えワクシニアウイルスでのチャレンジに対する防御をもたらしうることを証明している。Ad5ΔE1−E4HCVでの免疫化により惹起された免疫応答が後続のウイルスチャレンジに対する防御をもたらすのかどうかを判定するために、実施例7に記載のプロトコールに従い免疫したマウスを第4週に、HCV非構造領域(VV−NS)を発現する組換えワクチンウイルス(5×106 pfuの用量)でチャレンジした。実験対照として、W=0およびW=2にAd5ΔE1−E4EGFPの2回の注射で免疫したマウスを使用した。5日後、対(ペア)になった卵巣を摘出し、VVを力価測定した。図13に示す結果は、対照マウスと比較した場合の免疫化マウスのVV力価における有意な減少を示している。灰色のドットは相乗平均力価(N=5)を表す。* = 対照に対するp<0.05(Mann−Whitneyランク)。
【実施例9】
【0128】
Ad5ΔE1−E4HCVでの免疫化はアカゲザルにおいてHCV特異的免疫応答を初回刺激する
アカゲザルの大腿四頭筋に1010ppのAd5ΔE1−E4−HCVを注射した。該免疫化の効力を注射後の種々の時点(W=4、W=6、W=8、W=12)で末梢血単核細胞(PBMC)に関するγIFN−Elispotアッセイにより評価した。該注射により惹起された免疫応答は注射の6週間後にピークとなり、複数のHCVエピトープに対するものであった。これらの3頭のサルのうちの1頭は、注射の12週間後までの、長期にわたる応答を示した。これらのデータは、霊長類動物モデルにおいて免疫応答を惹起するためにAd5ΔE1−E4−HCVを使用しうることを示している。
【0129】
図14Aは、Ad5DE1−E4−HCVの1回の投与の後にサル4061において惹起された経時的応答を表す。結果は注射の4、6、8および12週間後の106PBMC当たりのg−IFNスポット形成細胞数(SFC)として表されている。各棒グラフは、別々のペプチドプールに対する応答を表す。
【0130】
図14Bは、γIFN−Elispot6アッセイにおいて評価された、Ad5DE1−E4−HCVの注射の6週間後の3頭のサル(番号4061、9003、7023)の免疫応答を示す。結果は、106PBMC当たりのg−IFNスポット形成細胞数(SFC)として表されている。各棒グラフは、別々のペプチドプールに対する応答を表す。
【0131】
本発明は、特定の実施形態とみなされるものに関して説明されているが、本発明はそのような実施形態には限定されないと理解されるべきである。逆に、本発明は、添付の特許請求の範囲の精神および範囲内に含まれる種々の修飾および均等物を含むと意図される。
【0132】
本開示の全体にわたって引用されているすべての参考文献を、参照により明示的に本明細書に組み入れることとする。
【0133】
【表3】
【図面の簡単な説明】
【0134】
【図1A】安定な293EBNATetクローンを得るために使用したプラスミドの概要図を示し、プラスミド成分の直線的表示を示す。
【図1B】安定な293EBNATetクローンを得るために使用したプラスミドの概要図をし、pE2プラスミドの概要図を示す。
【図2】図2はAdTetLuc感染クローンにおけるルシフェラーゼ発現のグラフ表示を示す。
【図3】図3は環状および直線形態のpE2の概要図を示す。pE2の環状形態と直線形態との識別を可能にするNotI消化により得たDNA断片も示されている。
【図4A】ドキシサイクリンによるAd5 E2遺伝子発現の活性化の際のpE2の複製を示すサザンブロット分析を示す。
【図4B】E2タンパク質のtet誘導性発現を示すウエスタンブロット分析を示す。
【図5A】2E2クローンから抽出されたpE2の構造およびE2タンパク質の発現を示し、クローン293EBNATetおよび2E2から抽出されたpE2の構造を示すサザンブロット分析である。
【図5B】Ad5ΔE1ベクターに感染した細胞におけるE2発現と比較した場合の2E2安定細胞系におけるE2タンパク質のtet誘導性発現を示すウエスタンブロット分析である。
【図6】図6は、EGFPを発現するAd ΔE1−4ベクターをレスキューし増殖させるための2E2細胞の使用を示す一連の写真を示す。
【図7A】Ad5ウイルスの概要地図である。この図にはすべての欠失領域が示されている。
【図7B】多欠失ベクターのE1領域内に導入されたHCV(BK株)ポリタンパク質発現カセットの概要図を示す。
【図8】図8はAd ΔE1−4orf3+HCVの制限分析を示す。
【図9】図9は、Ad5ΔE1−4HCV感染細胞におけるHCVタンパク質の発現を示すウエスタンブロット分析を示す。
【図10A】マウスにおけるAd5ΔE1−4HCVウイルス免疫化に対するインビボCD8+ T細胞応答を特徴づけるFACSデータのグラフ表示を示す。
【図10B】マウスにおけるAd5ΔE1−4HCVウイルス免疫化に対するインビボCD8+ T細胞応答を特徴づけるFACSデータのグラフ表示を示す。
【図11A】図11は、本開示に記載されているポリヌクレオチドおよびポリペプチド配列のヌクレオチドおよび/またはアミノ酸配列(すなわち、配列番号1〜21)を示す。
【図11B】図11は、本開示に記載されているポリヌクレオチドおよびポリペプチド配列のヌクレオチドおよび/またはアミノ酸配列(すなわち、配列番号1〜21)を示す。
【図11C】図11は、本開示に記載されているポリヌクレオチドおよびポリペプチド配列のヌクレオチドおよび/またはアミノ酸配列(すなわち、配列番号1〜21)を示す。
【図11D】図11は、本開示に記載されているポリヌクレオチドおよびポリペプチド配列のヌクレオチドおよび/またはアミノ酸配列(すなわち、配列番号1〜21)を示す。
【図11E】図11は、本開示に記載されているポリヌクレオチドおよびポリペプチド配列のヌクレオチドおよび/またはアミノ酸配列(すなわち、配列番号1〜21)を示す。
【図11F】図11は、本開示に記載されているポリヌクレオチドおよびポリペプチド配列のヌクレオチドおよび/またはアミノ酸配列(すなわち、配列番号1〜21)を示す。
【図11G】図11は、本開示に記載されているポリヌクレオチドおよびポリペプチド配列のヌクレオチドおよび/またはアミノ酸配列(すなわち、配列番号1〜21)を示す。
【図11H】図11は、本開示に記載されているポリヌクレオチドおよびポリペプチド配列のヌクレオチドおよび/またはアミノ酸配列(すなわち、配列番号1〜21)を示す。
【図12A】NS3ヘリカーゼ内のエピトープのマッピングに用いた方法を示す。Ad5ΔE1−E4HCVで初回抗原刺激されpSh−Ad5−HCVで追加抗原刺激された2匹のマウス(マウス4,黒塗り棒線、マウス5,付点棒線)から精製した脾細胞を、全NS3ヘリカーゼ領域に及ぶペプチド(I〜XVIII)の二次元サブセット上でγ−IFN−Elispotにおいて試験した。
【図12B】NS3ヘリカーゼ内のエピトープのマッピングに用いた方法を示す。図12Bに示されているデータは、免疫応答を特徴づけするために使用したパネルAにおいて特定された陽性閾値を超える応答を惹起したペプチドプール間の交差点を要約したものである。
【図12C】NS3ヘリカーゼ内のエピトープのマッピングに用いた方法を示す。パネル12Cは、該応答をもたらすNS3エピトープを特定するために行ったγ−IFN−Elispotアッセイの結果を要約したものである。
【図13】図13は、VV−NSチャレンジに対する防御の誘導における免疫化の効力を示す。灰色のドットは相乗平均力価(N=5)を表す。アステリスクは対照に対するp<0.05を示す(Mann−Whitneyランク)。
【図14A】Ad5ΔE1−E4HCV免疫化に応答してアカゲザルにおいて惹起された免疫応答を要約したものであり、Ad5ΔE1−E4HCVの1回の投与に際してサル4061において惹起されγ−IFN−Elispotにより分析された経時的免疫応答を表す。
【図14B】Ad5ΔE1−E4HCV免疫化に応答してアカゲザルにおいて惹起された免疫応答を要約したものであり、Ad5ΔE1−E4HCVの1回の投与により3頭の個々のサルにおいて誘導され注射の6週間後にγ−IFN−Elispotにより分析された免疫応答を示す。
【特許請求の範囲】
【請求項1】
a.EBV由来複製起点(Ori−P)、
b.Ad5(ITR結合部)、
c.Ad5由来ポリメラーゼおよびプレ末端タンパク質をコードする核酸配列よりなる第1転写単位、
d.Ad5 E4 ORF6およびDNA結合タンパク質をコードする核酸配列よりなる第2転写単位、ならびに
e.選択マーカー
を含んでなり、第1転写単位および第2転写単位が二方向性テトラサイクリン依存性プロモーターに融合している、アデノウイルスアンプリコン。
【請求項2】
該アンプリコンがpE2のヌクレオチド配列を含む、請求項1記載のアデノウイルスアンプリコン。
【請求項3】
プラスミドLMBP4972のヌクレオチド配列よりなるエピソームプラスミド。
【請求項4】
プロモーターに融合したトランスジーンをコードする発現カセットを更に含む、請求項1記載のアデノウイルスアンプリコン。
【請求項5】
a.EBV由来EBNA1タンパク質、
b.Tet転写サイレンサー、
c.Tet逆トランスアクチベーター、
d.アデノウイルスアンプリコン[該アデノウイルスアンプリコンは、第2転写単位(第2転写単位はEBV由来OriP、アデノウイルスITR結合部ならびにAd5 E4 ORF6およびDNA結合タンパク質をコードする核酸配列よりなる。)と組合された第1転写単位(第1転写単位はAd5由来ポリメラーゼおよびプレ末端タンパク質をコードする核酸配列よりなる。)よりなり、第1転写単位および第2転写単位は二方向性テトラサイクリン依存性プロモーターに融合している。]、および
e.選択マーカー
を発現するアデノウイルスプロデューサー細胞。
【請求項6】
該細胞が、EBNA1タンパク質を発現する霊長類由来細胞系である、請求項5記載のプロデューサー細胞。
【請求項7】
該細胞が293EBNAである、請求項6記載のプロデューサー細胞系。
【請求項8】
Tet転写サイレンサーがtTSkidである、請求項5記載のプロデューサー細胞。
【請求項9】
Tet逆トランスアクチベーターがrtTA2である、請求項8記載のプロデューサー細胞系。
【請求項10】
2E2(pE2で形質転換された293EBNA細胞)プロデューサー細胞。
【請求項11】
a.プロデューサー細胞内に多欠失アデノウイルス発現ベクターを導入すること{該プロデューサー細胞は、
i.EBV由来EBNAタンパク質、
ii.Tet転写サイレンサー、
iii.Tet逆トランスアクチベーター、
iv.アデノウイルスアンプリコン[該アデノウイルスアンプリコンは、第2転写単位(第2転写単位はEBV由来ori−P、アデノウイルスITR結合部ならびにAd5 E4 ORF6およびDNA結合タンパク質をコードする核酸配列よりなる。)と組合された第1転写単位(第1転写単位はAd E2由来ポリメラーゼおよびプレ末端タンパク質をコードする核酸配列よりなる。)よりなり、第1転写単位および第2転写単位は二方向性テトラサイクリン依存性プロモーターに融合している。]、
を発現する。}、
b.E2およびE4ORF6コード配列の発現を誘導すること、および
c.産生された複製欠損アデノウイルスを回収すること
を含んでなる、関心のある遺伝子を含む複製欠損アデノウイルスの製造方法。
【請求項12】
該プロデューサー細胞系が、アデノウイルスE1タンパク質、EBNA1および転写調節系を発現するヒト細胞系である、請求項11記載の製造方法。
【請求項13】
該プロデューサー細胞が、tTskidとrETA2とを発現する293EBNA細胞である、請求項12記載の製造方法。
【請求項14】
多欠失アデノウイルスベクターがアデノウイルスE1、E2、E3およびE4遺伝子を欠く、請求項13記載の製造方法。
【請求項15】
多欠失アデノウイルスベクターがヒトAd5バックボーンよりなる、請求項14記載の製造方法。
【請求項16】
E2およびE4ORF6コード配列の発現が、該プロデューサー細胞をドキシサイクリンと接触させることにより誘導される、請求項11記載の製造方法。
【請求項17】
EBNA1、Tet転写サイレンサーおよびTet逆トランスアクチベーターを発現する哺乳類細胞内に請求項4記載のアデノウイルスアンプリコンを導入し、E2およびE4ORF6コード配列の発現を誘導し、産生された複製欠損アデノウイルスを回収することを含んでなる、複製欠損アデノウイルス粒子の製造方法。
【請求項18】
該プロデューサー細胞が、tTSkidとrtTA2とを発現する293EBNA細胞である、請求項17記載の製造方法。
【請求項19】
E2およびE4ORF6コード配列の発現が、該パッケージング細胞をドキシサイクリンと接触させることにより誘導される、請求項17記載の製造方法。
【請求項20】
請求項17記載の製造方法により回収され精製された組換え複製欠損アデノウイルス粒子。
【請求項1】
a.EBV由来複製起点(Ori−P)、
b.Ad5(ITR結合部)、
c.Ad5由来ポリメラーゼおよびプレ末端タンパク質をコードする核酸配列よりなる第1転写単位、
d.Ad5 E4 ORF6およびDNA結合タンパク質をコードする核酸配列よりなる第2転写単位、ならびに
e.選択マーカー
を含んでなり、第1転写単位および第2転写単位が二方向性テトラサイクリン依存性プロモーターに融合している、アデノウイルスアンプリコン。
【請求項2】
該アンプリコンがpE2のヌクレオチド配列を含む、請求項1記載のアデノウイルスアンプリコン。
【請求項3】
プラスミドLMBP4972のヌクレオチド配列よりなるエピソームプラスミド。
【請求項4】
プロモーターに融合したトランスジーンをコードする発現カセットを更に含む、請求項1記載のアデノウイルスアンプリコン。
【請求項5】
a.EBV由来EBNA1タンパク質、
b.Tet転写サイレンサー、
c.Tet逆トランスアクチベーター、
d.アデノウイルスアンプリコン[該アデノウイルスアンプリコンは、第2転写単位(第2転写単位はEBV由来OriP、アデノウイルスITR結合部ならびにAd5 E4 ORF6およびDNA結合タンパク質をコードする核酸配列よりなる。)と組合された第1転写単位(第1転写単位はAd5由来ポリメラーゼおよびプレ末端タンパク質をコードする核酸配列よりなる。)よりなり、第1転写単位および第2転写単位は二方向性テトラサイクリン依存性プロモーターに融合している。]、および
e.選択マーカー
を発現するアデノウイルスプロデューサー細胞。
【請求項6】
該細胞が、EBNA1タンパク質を発現する霊長類由来細胞系である、請求項5記載のプロデューサー細胞。
【請求項7】
該細胞が293EBNAである、請求項6記載のプロデューサー細胞系。
【請求項8】
Tet転写サイレンサーがtTSkidである、請求項5記載のプロデューサー細胞。
【請求項9】
Tet逆トランスアクチベーターがrtTA2である、請求項8記載のプロデューサー細胞系。
【請求項10】
2E2(pE2で形質転換された293EBNA細胞)プロデューサー細胞。
【請求項11】
a.プロデューサー細胞内に多欠失アデノウイルス発現ベクターを導入すること{該プロデューサー細胞は、
i.EBV由来EBNAタンパク質、
ii.Tet転写サイレンサー、
iii.Tet逆トランスアクチベーター、
iv.アデノウイルスアンプリコン[該アデノウイルスアンプリコンは、第2転写単位(第2転写単位はEBV由来ori−P、アデノウイルスITR結合部ならびにAd5 E4 ORF6およびDNA結合タンパク質をコードする核酸配列よりなる。)と組合された第1転写単位(第1転写単位はAd E2由来ポリメラーゼおよびプレ末端タンパク質をコードする核酸配列よりなる。)よりなり、第1転写単位および第2転写単位は二方向性テトラサイクリン依存性プロモーターに融合している。]、
を発現する。}、
b.E2およびE4ORF6コード配列の発現を誘導すること、および
c.産生された複製欠損アデノウイルスを回収すること
を含んでなる、関心のある遺伝子を含む複製欠損アデノウイルスの製造方法。
【請求項12】
該プロデューサー細胞系が、アデノウイルスE1タンパク質、EBNA1および転写調節系を発現するヒト細胞系である、請求項11記載の製造方法。
【請求項13】
該プロデューサー細胞が、tTskidとrETA2とを発現する293EBNA細胞である、請求項12記載の製造方法。
【請求項14】
多欠失アデノウイルスベクターがアデノウイルスE1、E2、E3およびE4遺伝子を欠く、請求項13記載の製造方法。
【請求項15】
多欠失アデノウイルスベクターがヒトAd5バックボーンよりなる、請求項14記載の製造方法。
【請求項16】
E2およびE4ORF6コード配列の発現が、該プロデューサー細胞をドキシサイクリンと接触させることにより誘導される、請求項11記載の製造方法。
【請求項17】
EBNA1、Tet転写サイレンサーおよびTet逆トランスアクチベーターを発現する哺乳類細胞内に請求項4記載のアデノウイルスアンプリコンを導入し、E2およびE4ORF6コード配列の発現を誘導し、産生された複製欠損アデノウイルスを回収することを含んでなる、複製欠損アデノウイルス粒子の製造方法。
【請求項18】
該プロデューサー細胞が、tTSkidとrtTA2とを発現する293EBNA細胞である、請求項17記載の製造方法。
【請求項19】
E2およびE4ORF6コード配列の発現が、該パッケージング細胞をドキシサイクリンと接触させることにより誘導される、請求項17記載の製造方法。
【請求項20】
請求項17記載の製造方法により回収され精製された組換え複製欠損アデノウイルス粒子。
【図1A】

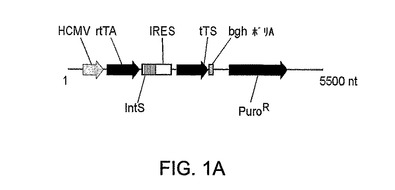
【図1B】
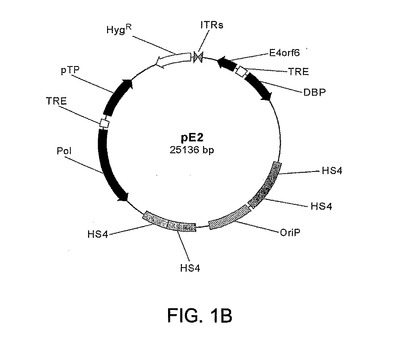
【図2】
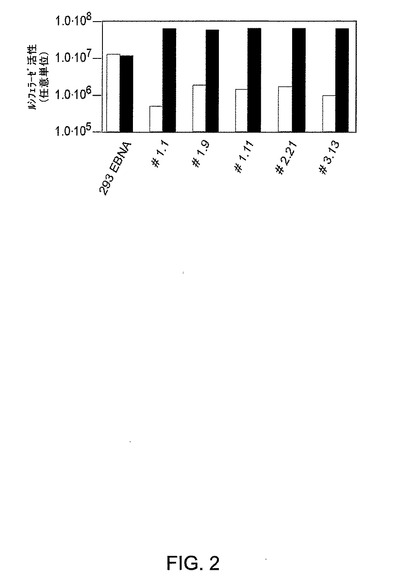
【図3】
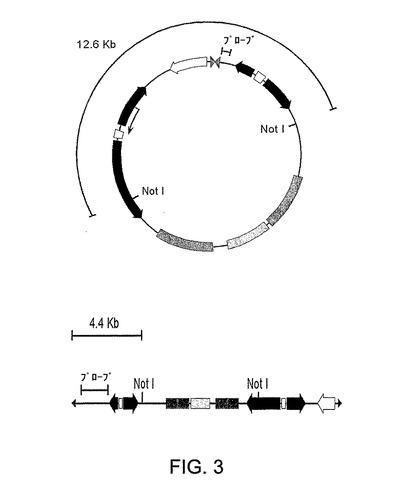
【図4A】
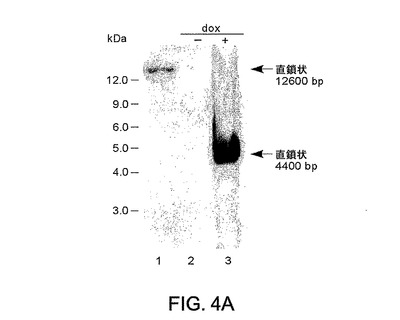
【図4B】

【図5A】
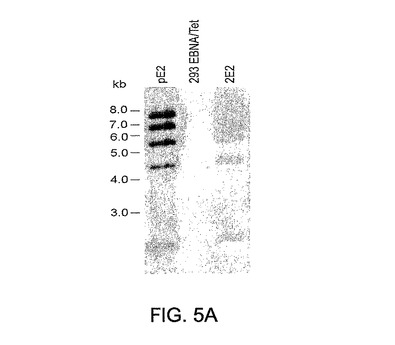
【図5B】
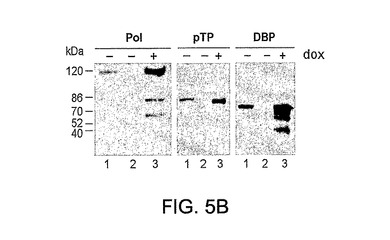
【図6】
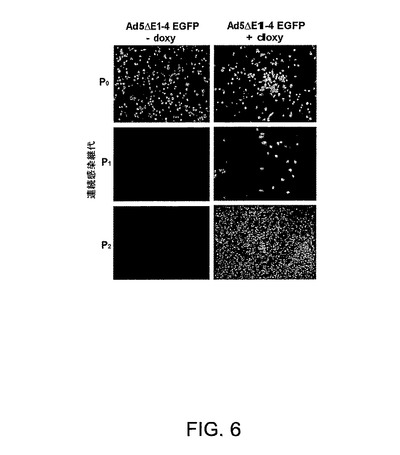
【図7A】
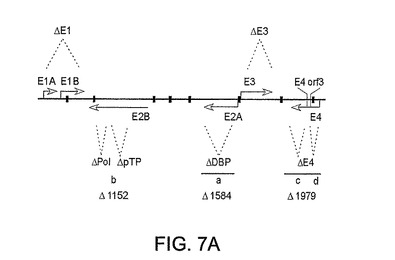
【図7B】
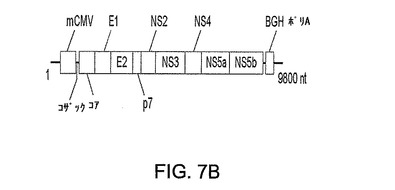
【図8】
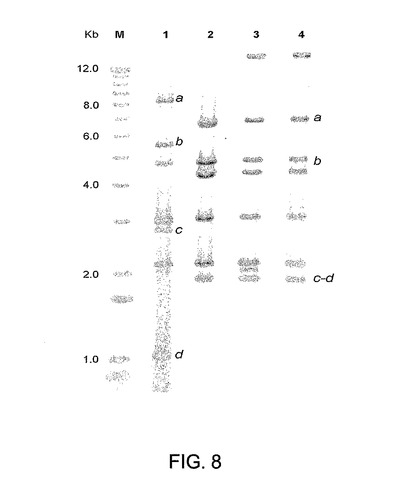
【図9】
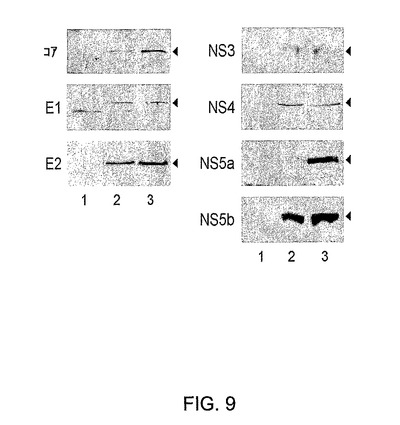
【図10A】
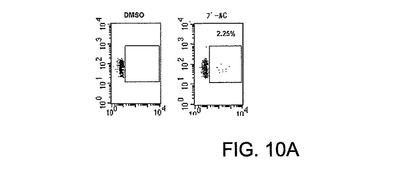
【図10B】
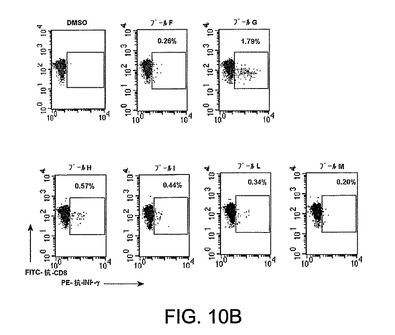
【図11A】

【図11B】

【図11C】

【図11D】

【図11E】

【図11F】

【図11G】

【図11H】

【図12A】
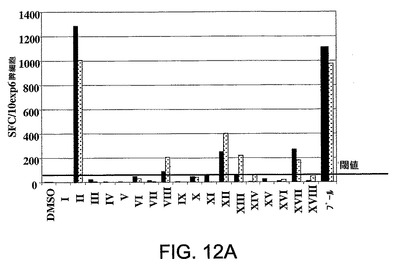
【図12B】
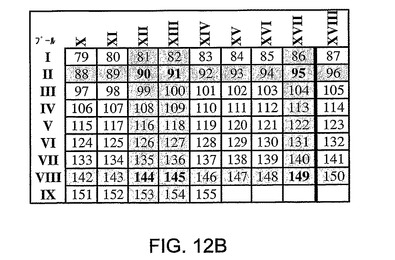
【図12C】
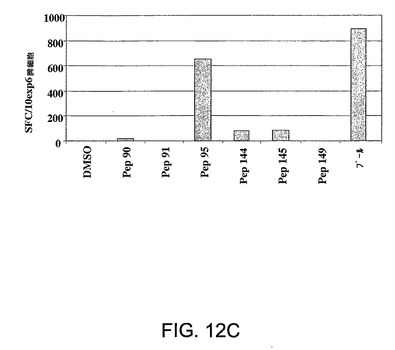
【図13】
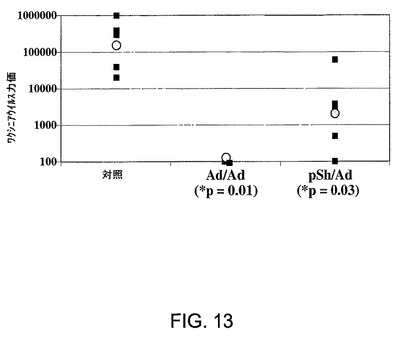
【図14A】
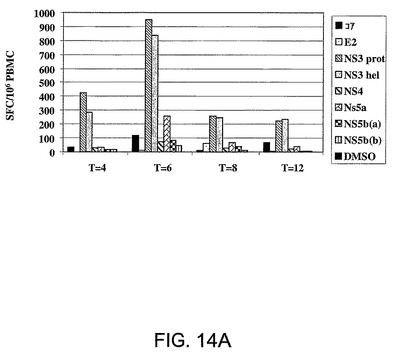
【図14B】
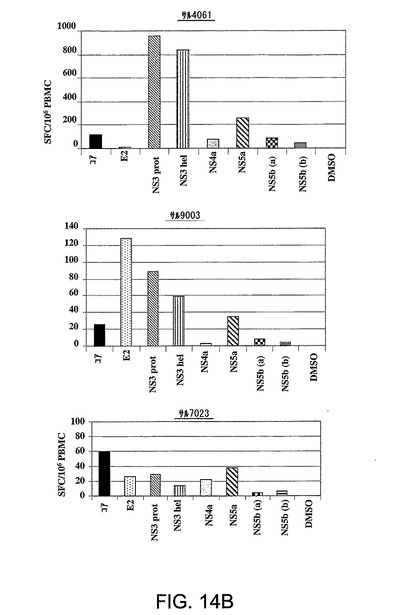
【図1B】
【図2】
【図3】
【図4A】
【図4B】
【図5A】
【図5B】
【図6】
【図7A】
【図7B】
【図8】
【図9】
【図10A】
【図10B】
【図11A】
【図11B】
【図11C】
【図11D】
【図11E】
【図11F】
【図11G】
【図11H】
【図12A】
【図12B】
【図12C】
【図13】
【図14A】
【図14B】
【公表番号】特表2008−518591(P2008−518591A)
【公表日】平成20年6月5日(2008.6.5)
【国際特許分類】
【出願番号】特願2007−538349(P2007−538349)
【出願日】平成17年10月27日(2005.10.27)
【国際出願番号】PCT/EP2005/011629
【国際公開番号】WO2006/048215
【国際公開日】平成18年5月11日(2006.5.11)
【出願人】(501209427)イステイチユート・デイ・リチエルケ・デイ・ビオロジア・モレコラーレ・ピ・アンジエレツテイ・エツセ・ピー・アー (90)
【Fターム(参考)】
【公表日】平成20年6月5日(2008.6.5)
【国際特許分類】
【出願日】平成17年10月27日(2005.10.27)
【国際出願番号】PCT/EP2005/011629
【国際公開番号】WO2006/048215
【国際公開日】平成18年5月11日(2006.5.11)
【出願人】(501209427)イステイチユート・デイ・リチエルケ・デイ・ビオロジア・モレコラーレ・ピ・アンジエレツテイ・エツセ・ピー・アー (90)
【Fターム(参考)】
[ Back to top ]