
試料調製法および装置
【課題】試料中の複数種の核酸(cDNA、mRNA)を、同時、個別に増幅し、取り出して、解析する方法および装置の提供。
【解決手段】分析対象となるDNA分子1〜3をゲル状の微小液滴4,5,7内に1個以下含む試料溶液を、DNA分子の総数Nよりも多いM個の微小液滴に分画し、該液滴を含むエマルジョン11中で、各液的内のDNAをPCRにより増幅し、その後に各微小液滴中に得られた増幅産物の有無(量)を、インターカレーター等を用いた蛍光検出により検出する方法、およびそのための装置。
【解決手段】分析対象となるDNA分子1〜3をゲル状の微小液滴4,5,7内に1個以下含む試料溶液を、DNA分子の総数Nよりも多いM個の微小液滴に分画し、該液滴を含むエマルジョン11中で、各液的内のDNAをPCRにより増幅し、その後に各微小液滴中に得られた増幅産物の有無(量)を、インターカレーター等を用いた蛍光検出により検出する方法、およびそのための装置。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は遺伝子解析技術のための試料調製法に関する。より詳細には、1つの細胞に含まれるmRNAのデジタル解析あるいは多数の標的分子を同時に個別解析する方法のための試料調製法に関する。
【背景技術】
【0002】
ヒトゲノム配列解読の完了に伴い、種々ゲノムの情報が精力的に調べられそれらを活用する時代になってきた。ゲノム情報はmRNAに転写され、蛋白質へと翻訳される。こうした遺伝子の発現プロフィール解析は、生命活動の詳細を調べるために不可欠である。これまで主流となっている解析方法は多くの細胞からmRNAを取りだし、蛍光標識をした後にDNAプローブアレー(DNAチップ)に作用させ、mRNAと相補配列を持ちプローブに標識mRNAを捕獲して検出する方法であった。これに対し、多くの細胞からmRNAを取りだし、そのcDNAを作製し、それを電気泳動的に分離して計測する方法もある。この方法は種々のmRNAの量をアナログ的に計測する方法であるが、計測感度の問題から、多くの細胞を用いてmRNAを取りだし、計測しなければならない。
【0003】
一方、生命活動は多くの細胞が協調して一つのシステムを構成することで成り立っており、組織の中の個々の細胞にはそれぞれ異なる役割があると考えられている。本当の生命を理解するには、このような個々の細胞の働きをモニターすることが重要であり、1つの細胞に含まれるmRNAあるいは蛋白質の計測が重要視され始めている。それには1つの細胞に微量含まれるmRNAの種類と量を正確に定量分析することが必要であるがこのような方法は未だ確立されていない。
【0004】
発明者らはこの課題を克服するために1細胞に含まれる全てのmRNAあるいは計測が必要と考えられる複数種類のmRNAをデジタルカウントすることにより、定量分析することを目指している。デジタルカウントとは、それぞれのmRNA(あるいはcDNA断片)の配列を決定して種類を決め、その配列を有するmRNAが何個含まれるかをカウントして定量分析する方法である。
【0005】
すなわち、細胞のような小さな領域に含まれる複数のmRNAあるいはDNA断片のそれぞれについて配列解析することによりデジタルカウントするが、そのためには個々のmRNA(あるいはcDNA断片)を個別に増幅して配列解析できるようにする必要がある。ここで重要なことは全てのmRNA(あるいはcDNA断片)をもれなく独立に増幅することである。
【0006】
上記の方法では、一分子のDNAまたはmRNAを出発材料に並列して多くのPCR増幅を行うが、試料は溶液状でその中には数十から百万オーダーのmRNAあるいはcDNA断片が含まれている。これらを一括してPCR増幅すると複数の増幅産物の混合物が得られるだけで目的の計測試料は得られない。個々のmRNAを独立して、漏れがないように増幅してそれらを別々に取り出すことが必要である。個々のmRNAを独立に増幅するために、それぞれが分離された状態で個別にPCR反応を行うが、そのためには一反応体積あたりの反応開始時のDNAまたはRNA分子数の期待値が1以下になるまで試料溶液を希釈、分画を行った上で各々の分画について独立してPCR増幅反応を行う必要がある。例えば、ある試料中の増幅対象分子数が10万分子と見込まれる場合には、試料溶液を希釈して数十万以上に分画し、各々個別にPCR(ポリメラーゼ連鎖反応)等の増幅反応を行うことで試料中の全ての分子を独立して増幅すること、すなわちクローン増幅することができる。
【0007】
このような方法で複数のDNAを個別に増幅しようとする試みは近年いくつか試みられている。たとえば非常に多くの微小反応セルを平面プレート上に設け、ターゲットDNA断片と増幅に必要な酵素や反応基質を含む溶液をプレート上に流して微小反応セルに分画する。分画されたPCR溶液は相互に分離されているので独立に増幅することができる。1つの分画に入るDNA試料の量が平均1個以下になるように調整することで個別増幅が可能となる。この方法の一例が、アナリティカルケミストリーに開示されている(非特許文献1)。本例では、シリコン基板上に1万個のウエルを構築し高集積化している。しかし、100万個のDNA断片を増幅するためには更に多くの反応セルが必要である。また、対象となる試料溶液を全て余すことなく微小反応セルに注入することは不可能であって、溶液が余ったり、反応セルの内壁等に吸着されたりしてPCR増幅反応に使われないDNAが生じてしまう。
【0008】
また、1分子からの増幅を目指した試みではないが、微小容量のタイタープレートではなく平面に配置されたゲルドットマトリックスを用いてPCRする例もある(特許文献1)。従来より、低温でゲル化する材料を用いてPCR産物試料溶液をゲル化して試料の操作性を向上させる方法は公知だが(特許文献2)、この例では、ゲル化した試料をマトリクス的に配置した遺伝子検査用チップを利用する。しかし、この方法は空間的に固定された反応セルを用いるため、反応セルの中には目的の増幅産物が含まれるものも存在するが、全く含まれないものも存在してしまう。そのため、増幅されなかったターゲット試料が出てしまい、どのように目的の増幅産物を選別するかが問題となる。
【0009】
さらにもう一つの有力な方法として、エマルジョンPCRと呼ばれる方法がある。この方法では、試料毎に独立した反応容器を用いる代わりに、オイル中に多数形成した微小液滴内で反応を行う。この方法では、液滴の微小化が攪拌等により容易に行えるため、数十万個以上の反応容器相当の微小液滴を100マイクロリットル程度の一つの容器内に形成可能である。
【0010】
しかしエマルジョンを用いる方法では、個々の微小液滴から試料を個別に回収することは容易でないため、微小液滴中にDNAまたはRNAを固定化するため、プローブ付きのビーズを入れて、反応物が生成した後に反応物を捕獲したビーズを溶液から分離することによって各微小液滴中のDNAまたはRNAを回収している。こうしたビーズ固相を用いた試料の回収時には、酵素反応等によって得られたDNAまたはRNAを回収するために、生成物が得られている固相を得られていない固相と分けて回収する必要がある。そのため、PCR反応によって得られたDNAの一部と相補的な配列を持つプローブを固定化した磁気ビーズを用意し、プローブと増幅されたDNA断片をハイブリダイズさせた後、磁石を用いて選別回収する方法が用いられている。この方法を活用して多くのDNA断片を増幅してゲノム配列決定に活用した例が、ネイチャー等に掲載されている(特許文献3および非特許文献2)。しかし、この技術を全てのmRNA増幅とその配列測定に活用しようとするとやはり重大な問題がある。この系ではビーズとターゲットDNA1コピーがエマルジョン中の1反応液滴に含まれる必要があり、生成した液滴中にビーズが2個以上含まれる、1つのmRNAが重複してカウントされてしまい、デジタルカウントはできない。この問題を解消するため、ビーズの量をDNAと同様に少なくするとDNAは含まれるがビーズが含まれない液滴が多数生じてしまいやはり不都合である。固体ビーズを用いて生成したDNAを回収することは良い方法で、重複したDNA試料を用いてゲノム配列を決定するなどの目的には十分使い得る方法であるが、デジタルカウントには適さない。
【0011】
【特許文献1】特開2004−337064号公報
【特許文献2】特開平10−004963号公報
【特許文献3】WO2005/10145(PCT/US2004/015587)
【非特許文献1】Anal.Chem.2001,73,p1043−1047
【非特許文献2】Nature.2005,437,p376−380,Supplementary Information)
【発明の開示】
【発明が解決しようとする課題】
【0012】
前述のとおり、従来の方法にはいずれも問題点があった。まず、タイタープレートを用いた技術は多数試料を同時処理して増幅産物を取り出す時の液体ハンドリングについて配慮がされておらず、増幅された反応産物を見分けて多数試料を液体状態のまま個別に回収するため、試料数に応じた大量の試料容器や煩雑な取り扱い作業が必要となるという問題があった。
【0013】
ビーズ表面に増幅産物を捕獲して回収する方法では、ビーズに反応に必要なプライマー等をあらかじめ固定化しておく必要があるが、固相に固定したプライマーを増幅用のプライマーとして用いて固相表面に増幅産物を得ようとすると増幅効率が低下するという問題があった。これは、酵素反応の基質となるDNAあるいはRNA等の分子が固定化されていることにより分子運動の自由度が下がるため、溶液系に比べて反応効率が大きく低下するためである。さらに、固相表面へのDNAまたはRNAの非特異的吸着という問題もあった。すなわち、最初の増幅の鋳型となるDNA断片が固相に吸着されていると鋳型として巧く働かず、エマルジョン中にDNA鋳型が1コピー含まれているものの増幅産物が得られない。特にクローン増幅の目的のために1反応液あたり1分子という極低濃度のDNAまたはRNA試料を出発材料に用いる場合には、非特異的吸着による影響が相対的に大きくなるため深刻である。さらに前述のとおり、個々の微小エマルジョン反応液にビーズを均一に入れることは困難である。特に、エマルジョンを攪拌操作によって調製する場合には全ての液滴に同数のビーズ等の固相を入れることは不可能であり、一つの液滴に複数のビーズが入ったり、一個も入らなかったりする。液滴あたりのビーズ等の固相の数をコントロールできないと試料中の全分子を対象とする一分子計測を精度よく行うことが困難になる。
【0014】
このように従来の方法は、いずれも試料となるDNA断片プール(1細胞から得たmRNAあるいはcDNA断片の集合)を構成する全ての成分を同時に増幅して回収するという目的には適さなかった。
【0015】
本発明は、このような従来技術の問題点を克服するためになされたもので、1細胞に含まれるmRNAを取りだし、これをcDNAに転写し、1分子毎に増幅して回収し、DNA配列決定試料を調製することを課題とする。すなわち、簡便な方法でDNA断片プールに含まれる全ての成分を1分子毎に増幅してそれらを個別に回収する技術を提供することを目的とする。
【課題を解決するための手段】
【0016】
上記課題を解決するために発明者らは鋭意検討し、1分子毎の増幅を確実に実現し、増幅された反応産物だけを取り出す工夫をすることで、1細胞に含まれる全てのmRNA(cDNA)を1分子毎に増幅してそれらを個別に回収することに成功した。
【0017】
すなわち、本発明は、試料中の複数種の核酸を個別に増幅し取り出す方法であって、1つの微小液滴内に含まれる核酸が1を超えないように希釈された試料に対し、疎水性溶媒中の微小液滴内でPCR反応を行い、PCR反応終了後に反応液を固体またはゲルの状態で分離することを特徴とする方法に関する。
【0018】
前記方法は、PCR反応液中に増幅産物に結合あるいは取り込まれる蛍光試薬をあらかじめ添加することにより、増幅産物の含まれる液滴だけを選別分取する工程を含んでいてもよい。そのような蛍光試薬としては、インターカレーターや、蛍光ラベルされたモレキュラービーコン等を挙げることができる。
【0019】
試料中の複数種の核酸には、あらかじめ単一のPCRプライマーで増幅可能なようにアダプタ配列を導入しておくことが望ましい。
【0020】
本発明では液滴毎に独立して増幅を行なうため、PCR反応は、疎水性溶媒中に分散された微小液滴エマルジョンに対して行なうか、あるいは、相互に分離された微小反応セルの並んだプレートの微小反応セル内で行うことが望ましい。
【0021】
PCR反応液には、反応後の液を固体またはゲルの状態で分離するために、あらかじめヒドロゲルを形成するための、アガロース、ゼラチン、でんぷん、カラギーナン、ペクチン、アガロペクチン、ポリアクリルアミド、ポリアクリル酸、ポリビニルアルコール、ポリビニルピロリドン等の水溶性合成ポリマー等のゲル化剤を添加しておく。
【0022】
本発明で用いられる疎水性溶媒としては、シリコーン系オイルまたはパラフィン系オイルを主剤とするものが好ましい。
【0023】
また、PCR反応液には、疎水性媒体中での微小液滴の安定性を向上させるため、界面活性剤(両親媒性物質等)および/または被膜形成剤をあらかじめ添加しておくことが好ましい。
【0024】
本発明はまた、前記した方法によって個別に増幅し取り出された複数種の核酸を検出あるいは定量する工程を含む、核酸解析方法も提供する。
【0025】
さらに本発明は、前記した方法に用いるための装置であって、1)ゲル化剤を溶液状態で保存するための温調機構、ゲル化剤を反応液と混合するための液体ハンドリング機構、攪拌機構から構成される試料分注混合装置、2)振動または回転式のミキサー、インクジェットおよび微細流路のいずれかで構成される微小液滴調製装置、3)PCR反応のためのサーマルサイクル機能を持つ温調装置、および、4)画像検出方式またはフローセル方式の検出器を具備する蛍光検出装置を含む装置を提供する。
【0026】
前記装置では、4)のフローセル方式において、フローセルが流路切替えによる分取機能を具備していることが望ましい。
【0027】
さらに本発明は、前記装置と、DNAシーケンサーおよび/またはフローサイトメトリーを含む、核酸解析システムも提供する。
【発明の効果】
【0028】
本発明によれば、1細胞に含まれる全てのmRNAのような、多数の微量試料を同時に個別にPCRで増幅し、得られた増幅産物を蛍光で確認し、ゲルの微小液滴として回収が可能である。回収のために、反応液中に固相を設ける必要がなく、このためのコストと手間が省ける。また、固相を用いることによる試料のロスや反応効率の低下を防止できる。
【発明を実施するための最良の形態】
【0029】
本発明では、増幅の阻害要因となる固体ビーズをPCR反応に加えたり、あるいは固体からなる微小反応セルを活用したりすることなく、エマルジョン反応液を用いて微小液滴内で数多くのPCR反応を同時に行い、DNA相補鎖合成が行われた反応溶液だけを回収する。
【0030】
本発明において「微小液滴」とは、1つの液滴が1つの核酸を含みうるような微細な液滴を意味し、その大きさは特に限定されないが、直径1μm〜150μm程度であることが好ましい。また、「微小セル」とは、前記した微小液滴1つを格納するセルであって、その大きさは特に限定されないが、直径3μm〜250μm程度で、10万個以上設けられることが好ましい。
【0031】
なお、試料は前記した微小液滴内に含まれる核酸が1を超えないよう十分に希釈して用いる。また、試料中の核酸には単一プライマーで増幅可能なように、アダプタ配列をあらかじめ導入しておく。アダプタ配列の導入は、たとえば、mRNAからcDNAを合成する際に、アダプタ配列を含むプライマーを用いて行なうなど、公知の方法により実施できる。
【0032】
PCR反応はビーズなどの固体相は共存させず溶液状態で行い、効率よくPCR反応を進める。つぎに、増幅産物を含むエマルジョンは、温度を下げて固体あるいはゲル状態で取り出す。PCRは通常50〜96℃の高温で行われるので室温あるいはそれ以下の温度でエマルジョンを固体あるいはゲルとして取り出すことは可能である。すなわち、高温では液体、低温では固体あるいはゲルとなる物質を共存させてこれを実現する。
【0033】
相補鎖合成が行われたか否かを見分ける方法は種々存在するが、本発明の実施例では2本鎖DNAの間に入り込み蛍光を発するインターカレーターを用いた蛍光検出の例を示した。インターカレーターとしてはサイバーグリーンI、ピコグリーン、エチジウムブロマイドなどを挙げることができる。検出は、インターカレーターに限定されず、モレキュラービーコンのように相補鎖合成が行われたときに蛍光を発するプローブを利用してもよい。
【0034】
取りだしたゲルあるいは反応溶液ビーズ(固体状態あるいはゲル状態なのでこのように呼ぶ)にレーザーを照射して蛍光を発するものを選別捕獲する。これにはフローサイトメトリーなど既存の装置を使うことができるほか、マイクロ流路を用いたビーズセレクターなどが利用できる。
【0035】
上記の方法において、反応物を固化あるいはゲル化させて取り出すための素材としては、アガロース、ゼラチン、でんぷん(アミロース)、ポリアクリルアミドなどの親水性ゲル化剤を用いることができる。これらのゲル化剤は熱溶解性であるため、反応効率の良い溶液系での反応を実現できる。すなわち、これらのゲル化剤の水溶液は一般的な耐熱酵素の反応温度である50℃以上の条件では溶液状態であり、反応産物の取り出し時の室温の条件ではゲル化する。37℃程度で溶液状態にする必要がある場合には低融点アガロースを用いることも可能である。もちろんこのほかにDNA相補鎖合成に伴い固体状になる物質を加えても良い。
【実施例】
【0036】
以下、本発明を実施例により詳細に説明するが、本発明はこれらの実施例に限定されるものではない。
【0037】
実施例1:
本実施例ではアガロースを含んだエマルジョンをオイル中で攪拌して調製した例を示す。
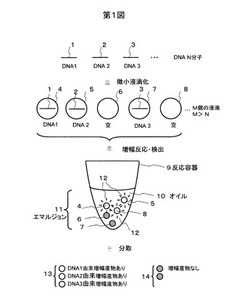
本方法の基本概念を図1に示した。分析対象となるDNA分子1−3を含む試料溶液をDNA分子の総数Nよりも多いM個の微小液滴4−8に分画することでDNAが入った微小液滴4,5,7,DNAの入らなかった微小液滴6,8が形成される。微小液滴4−8は反応容器9のオイル10中に分散させてエマルジョン11を形成する。この微小液滴を含むエマルジョンをPCR等の増幅反応を行った後に、各微小液滴中に得られた増幅産物の有無(量)をインターカレーター等を用いた蛍光検出により検出し、蛍光12の検出される各DNA1−3由来の増幅産物がある微小液滴13と蛍光の検出されない増幅産物がない微小液滴14に分ける。微小液滴にはあらかじめ常温でゲルまたは固体となるゲル化剤を含有させることで個々の微小液滴の分取を可能にする。すなわち、レーザーを照射して発光する液滴(ゲル)だけを回収したり、発光しない液滴(ゲル)を溶かして除去したりすることにより目的の増幅産物を得ることができる。
【0038】
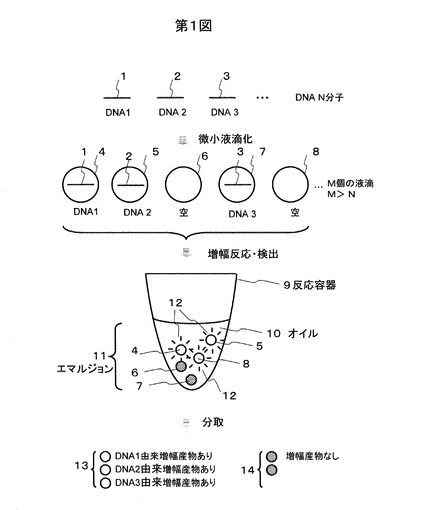
つぎに、1細胞由来のcDNAを対象とする場合を図2を用い説明する。1個の細胞21から得たmRNA22を磁気ビーズ23などに固定されたポリTオリゴマー24をプローブとして捕獲し、相補鎖DNA25を逆転写酵素で合成する(1stストランド合成)。mRNA22をRNaseHで分解した後、ランダムプライマーを用いてcDNA2本鎖26を形成する(2ndストランド合成)。次いでMboIなどの制限酵素により配列特異的にDNA2本鎖を切断する。切断箇所27に配列既知のアダプタ配列28をライゲーションにより結合させ、PCRプライミングサイトとする。このようして得たビーズ固定の2本鎖DNA断片29を含む溶液を昇温して2本鎖を乖離させ、ビーズ23に固定化された一本鎖31から遊離した一本鎖DNA30を得る。この一本鎖DNAの両端の配列は5’末端がアダプタ配列28の既知配列で3’末端がポリAなので、アダプタ配列28とポリTプライマーを共通に用いてPCR増幅が可能である。遊離した一本鎖DNA30と二つのプライマー32,33および相補鎖合成基質・酵素を加えて図1に示す微小液滴中でPCR増幅する。このときに上述したように低温でゲル化するアガロースとインターカレーターを加えておく。PCR反応の詳細については後述するが、50−96℃程度の熱サイクルで行われるので、この温度ではアガロースは液体状である。PCRが終了したら室温にして、アガロースが含まれる反応液をゲルビーズとして回収する。一方、特定の配列を持つ複数のmRNAをカウントする場合にはそれら配列に特異的な配列を持つプライマー34を用いるが、特異的でないプライミング配列35を持つ部分をアンカーしておいて、この部分をPCR増幅プライマーに用いてもよい。
【0039】
本実施例では加えたDNA鋳型の数が分かるようにモデルサンプルを用いて実験を行ったが、実際のcDNA計測でも同様のプライマーを用いて個別増幅を行うことができる。
【0040】
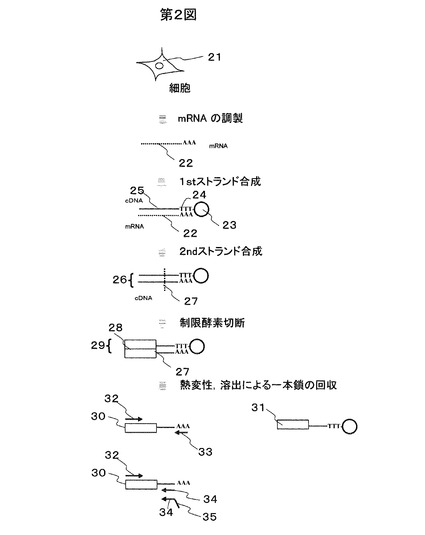
以下増幅工程について図3により説明する。本増幅工程は(1)同一反応容器内の疎水性溶媒中にゲル化剤および蛍光色素を含む増幅用反応液の微小液滴を増幅対象の鋳型分子数よりも多数調製する工程41、(2)増幅反応を行う工程42、(3)増幅産物を含むゲル化した微小液滴を確認する工程43、(4)増幅産物を含むゲル化した微小液滴を分取する工程44からなる。以下、4つの工程について詳述する。
【0041】
(1)同一反応容器内の疎水性溶媒中にゲル化剤および蛍光色素を含む増幅用反応液の微小液滴を増幅対象の鋳型分子数よりも多数調製する工程:
以下の組成のPCR反応液を準備する:120mM Tris-SO4(pH8.9),36mM Ammonium Sulfate,4mM MgSO4,0.4mM dNTPs,Fプライマー0.4μM(GTTTTCCCAGTCACGACGTTG:配列番号1),Rプライマー0.4μM(ATGACCATGATTACGCCAAGC:配列番号2),増幅用酵素Platinum Taq DNA ポリメラーゼ High Fidelity(インビトロジェン社)0.04unit/μL(一反応あたりの体積は50μl)。
【0042】
上記反応液には、鋳型DNAとしてコピー数を見積もることのできるように市販のpUC19プラスミドDNA(2686塩基,タカラバイオ)を用いた。実際にはこの鋳型が一反応あたり104〜108分子含まれるように調製して増幅効率などを確認した。プラスミドDNAの分子数は製品の添付資料の原液の濃度(0.5μg/μl,1.7x1011分子/μl)より求めた。また、反応液にはPCR産物の蛍光検出用色素として、サイバーグリーン(SYBR Green)I溶液(インビトロジェン社,S7563)を原液の2500倍の希釈度になるように加えた。なお、本品のモル濃度は開示されていないため希釈度は絶対的な数値ではない。
【0043】
蛍光色素としてはサイバーグリーンIの他、ピコグリーン、エチジウムブロマイドなどの2本鎖DNAと結合することによって蛍光強度が増強するインターカレータが適用可能である。そのほか、モレキュラービーコンのように相補鎖合成が行われたときに蛍光を発するプローブを用いてもよい。
【0044】
ゲル化剤としてはアガロースを用いた。アガロースにはゲル強度が1800g/平方センチ(1%(w/v)ゲル)以上の高ゲル強度であるSeakem Gold Agarose(タカラバイオ社)を用いた。
【0045】
ゲル濃度は反応セットアップ時の液体の取り扱いやすさと取り出し時のゲル状態でのハンドリングに必要な硬さの双方を考慮すると、アガロースの場合1−1.5%(w/v)が好適である。ただし、ゲル強度は同じゲル材料でも製品によって大きく異なるため、材料毎に最適な濃度は異なる。取り出し後のゲルの硬さ、あるいは、ゲルの水分を除去したあとの乾燥状態でのサイズを確保したい場合には、さらに高濃度のゲルを用いてもよい。アガロースについては2.5%(w/v)、ゼラチンについては5.0%(w/v)まではPCR反応に大きな支障はない。
【0046】
アガロースは粉末からは溶解しにくいのであらかじめオートクレーブを用いて121℃まで加熱して2.5%(w/v)の均一な水溶液を調製し、ピペッティングが容易な粘度になる50℃以上の溶液で準備する。この2.5%アガロース水溶液を、50℃程度の上述のPCR反応液と等体積(一反応あたり50μl)ずつ手早く混合し、最終アガロース濃度が1.25%(w/v)の反応液(一反応あたり計100μl)を調製する。混合時の温度は耐熱性酵素に影響を与えない90℃以下で行う。
【0047】
エマルジョン調製用のオイルにはシリコーン系混合オイルを用いた。組成は上述の文献(Nature, 2005, 437, p376-380, (Supplementary Information))中の記述を参考に、以下の組成とした:(1)Polyphenylmethylsiloxane(フルカ社,商品名AR20)を25%(v/v),(2)10%(v/v) PEG/PPG-18/18 Dimethiconeポリマー, Decamethylpentacyclosiloxane溶液(東レ・ダウコーニング社,商品名 DC5225C) を50%(v/v),(3) 50%(v/v)Trimethylsiloxysilicate, Decamethylpentacyclosiloxane溶液(東レ・ダウコーニング社,商品名BY11-018)を25%(v/v)。
【0048】
各成分はPolyphenylmethylsiloxaneはベースオイル、Decamethylpentacyclosiloxaneは溶剤、PEG/PPG-18/18 Dimethiconeポリマーは界面活性作用および増粘性のあるポリマー、Trimethylsiloxysilicateは水との界面にケイ酸の被膜を形成する成分である。
【0049】
本混合オイルを、上記のゲル化剤入りの反応液と同量(一反応あたり100μl)混合し、エマルジョンを調製する(混合後、一反応あたり200μl)。混合液を2mlのサンプルチューブに入れて、ボルテックスミキサ(タイテック社,2500rpm)で2-5秒程度攪拌することで直径50-100μm程度の微小液滴が得られる。
【0050】
液滴のサイズは、目的とする増幅倍率および、増幅対象の分子数によって変化させてよいが、直径20-200μmが好適であり、特に一細胞中に存在している遺伝子数に相当する10万分子程度を対象とする場合には直径50-100μm程度が増幅に必要な十分な試薬成分量を確保した上で、総反応液量が操作の容易な1ml以下となるため好適である。
【0051】
反応液の微小液滴エマルジョンを形成する方法は特に制限するものでなく、上記のミキサーによる攪拌の他、インクジェット法、微細流路を用いた方法(Angew. Chem. Int. Ed. 2005, 44, p724-728)などを用いることが可能である。
【0052】
得られたエマルジョンは一般的なプラスチック製反応容器中で増幅反応を行えばよい。一般的な反応容器以外にも、反応後の観察を容易にする目的で、相互に分離された微小反応セルの並んだプレートの微小反応セル内で増幅反応を行ってもよい。
【0053】
オイルの各成分の混合比の変化は反応液の微小液滴自体の形成に関しては大きな影響を与えないが、エマルジョンとしての安定性には多少の影響がある。オイルがベースオイル成分のPolyphenylmethylsiloxane 100%の場合にはエマルジョン形成後もオイル部分が白濁せずに透明なので、光学的検出には特に好適であるが、反応液の微小液滴同士がくっつきやすくなる。ただし、反応液にゲル化剤が入っているため、微小液滴同士がひとつに融合することはない。ベースオイル成分のPolyphenylmethylsiloxane に、Trimethylsiloxysilicateを成分量で5%程度(50%溶液で1/10容)以上加えると、微小液滴同士のくっつきは解消される。同成分を成分量で25%(50%溶液で1/2容)まで増やしても効果は同様である。
【0054】
また、ベースオイル成分のPolyphenylmethylsiloxane にPEG/PPG-18/18を成分量で1%(v/v)(10%溶液で1/10容)以上加えると、エマルジョン形成後に全体が白濁するが、エマルジョン中の微小液滴のオイルとの分離は抑制されエマルジョンの安定性が向上する。PEG/PPG-18/18を成分量で7%(v/v)(10%溶液で7/10容)まで増やしても効果に大きな変化はない。
【0055】
上記で用いた面活性剤成分、増粘成分、被膜形成成分は類似の物質でも代用可能である。
【0056】
疎水性溶媒としては、上記のシリコーン(有機ケイ素)系オイルの他、ミネラルオイル等のパラフィン系オイルなどが使用可能である。シリコーン系オイルは比重が0.98程度で反応液の溶媒である水の比重1に近いことと、温度による粘度の変化が少ないことにより、反応液と安定なエマルジョン形成が可能で特に好適である。
【0057】
(2)増幅反応を行う工程
調製した上記のエマルジョン状態の反応溶液は、0.2mlチューブに50μlずつ分注し、94℃15秒,55℃30秒,70℃1分の熱サイクル条件でPCRによる増幅反応を行う。サイクル数は40サイクルである。熱サイクル用装置としてサーマルサイクラー9700(アプライドバイオシステムズ社)を使うことができる。
熱サイクル装置には、PCRの熱サイクルの機能に加え、反応セットアップ時にゲル化剤水溶液、反応液、混合オイルを高温状態で保つための50℃以上の恒温槽機能が付加されていることが望ましい。
【0058】
(3)増幅産物を含むゲル化した微小液滴を確認する工程
反応後に、エマルジョンに対して5倍容のイソプロパノールを加えてエマルジョンを1液化し、スピンダウンしてゲル化した微小液滴のビーズを回収する。
【0059】
回収した増幅産物を含むゲル化した微小液滴ビーズは、ゲル電気泳動(アジレント社バイオアナライザ,DNA500キット,または2%アガロースゲル)を用いて電気泳動を行い、増幅産物のサイズおよび量を確認することができる。図4は鋳型を1x106分子加えた場合の増幅産物に対するアジレント社バイオアナライザを用いた電気泳動分析の結果である。同じ反応液組成で、ゲル化成分を加えていない試料(試料1)、微小液滴のエマルジョン状態にしなかった試料(試料2)も並行して調製し比較した。
【0060】
上記の回収した試料(試料3)のバンド47は、ゲル化成分が入っていない試料(試料1)のバンド45、ゲル化成分ありでエマルジョン状態にしなかった試料(試料2)のバンド46と同じ111塩基の位置に泳動されており、比較対象と同一サイズの産物の生成が確認できた。
【0061】
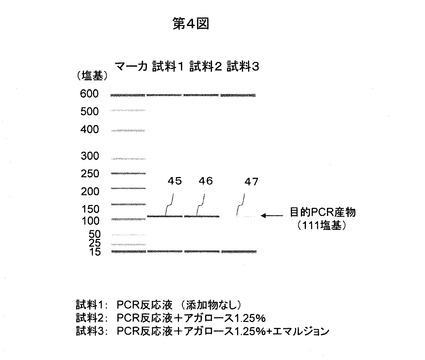
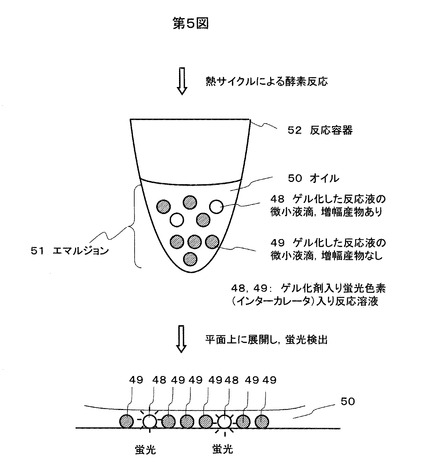
反応後のエマルジョン状態の反応溶液は蛍光顕微鏡(構成例:オリンパスBX51,UlS-2光学系,対物レンズUplanSApo,ミラーユニットWIB-UMWIB3)で上記のような精製を行うことなく、直接観察が可能である。観察の状態を模式的に図5に示す。鋳型を1x105分子加えた場合の蛍光で観察した結果の一例を図6に示す。ゲル化した反応液の微小液滴48,49のうち、増幅産物がある微小液滴48はサイバーグリーンIの蛍光で明るく観測され、増幅産物がない微小液滴49は暗く観測される。
【0062】
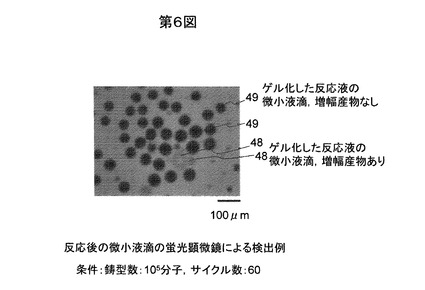
一反応あたりの鋳型数を変化させて上記図5および図6と同じ要領で観察を行い、増幅産物による蛍光が観測された微小液滴48の数の割合を、サーマルサイクル数が40サイクルの場合71、サーマルサイクル数が60サイクルの場合72について、それぞれプロットしたグラフを図7に示す。
【0063】
図7に示すようにサーマルサイクル数が40サイクルの場合と60サイクルの場合の変化は小さく、効率よく増幅されて40サイクルでほぼプラトーに達していることが示唆された。
【0064】
微小液滴の平均直径を50μmと仮定すると、一個あたりの平均体積は65plで、1反応(100μl)あたりの微小液滴の個数は1.5x106個であり、反応開始時に鋳型を105個加えた場合には、1割弱の微小液滴に一分子の鋳型が含まれ、鋳型を107個加えた場合には、ほぼ全ての微小液滴に一分子以上の鋳型が含まれることが期待されるが、図7に示される増幅産物が検出された微小液滴の割合の実測の結果は、鋳型を105個加えた場合には数%、106個加えた場合には10数%、107個加えた場合にはほぼ100%と上記の期待値に近い値になっており、本実施例における増幅がうまく進んでいることを示している。
【0065】
また、増幅産物量についても考察を行った。上記図4に示す回収した増幅産物(試料3)の電気泳動分析結果中の111塩基の産物のバンド47の濃度は約1ng/μlと定量(アジレントバイオアナライザ2100による定量値)されたので、1反応100μlあたり約100ngの増幅産物が回収されたことになる。111塩基の2本鎖DNAの100ngは1.4pM、8x1011分子に相当する。
【0066】
増幅率についても考察した。図7に示す結果より、反応開始時に鋳型を106個加えた場合には、約10%の微小液滴から増幅産物が認められるので、1反応100μlあたりの微小液滴の個数を上記の仮定より1.5x106個とすると、増幅産物が得られている微小液滴はその10%の1.5x105個であり、増幅産物が得られている微小液滴1個あたりのPCR産物は約5x106分子となり、これは増幅率が5x106倍と良好であることを示す。増幅産物の観察は、上記のほか、後述するフローサイトメトリーを用いてもよい。
【0067】
(4)増幅産物を含むゲル化した微小液滴を分取する工程
本実施例では、増幅産物を含むゲル化した微小液滴は顕微鏡観察下でキャピラリ管を装着したピペット(ドラモンド社製シーケンシングピペットなど)を用いて回収を行った。
回収した上記の微小液滴は、その中に含まれている増幅産物量をリアルタイムPCRで定量することが可能であった。また、増幅産物に対して、再度増幅の過程を加えてサンガー法、あるいは、パイロシーケンシング法による塩基配列決定に供することも可能である。
【0068】
回収の方法には上記の他に後述するフローサイトメトリーも適用可能である。本実施例によれば、106個の多数の微量試料を同時に個別に5x106倍までPCRで増幅し、得られた増幅産物を蛍光で確認し、ゲルの微小液滴として回収が可能である。個別回収のために、反応液中に固相を設ける必要がなく、このためのコストと手間が省ける。また、固相を用いることによる反応効率の低下を防止できる。
【0069】
実施例2:反応容器の形状
本実施例は反応容器の形状を相互に分離された微小反応セルの並んだプレートで構成するものである。
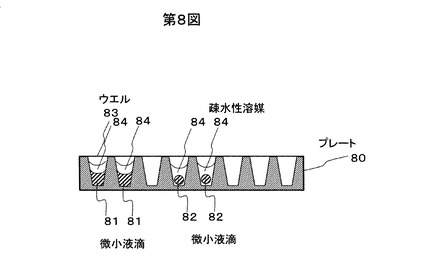
本実施例を図8−10を用いて説明する。図8のように、プレート80には個々の微小液滴81,82が収まるウエル83が多数設けられている。ウエル83は図9に示すように、2次元的に配列させてプレート80を構成する。微小液滴は直接ウエル83に入っていて疎水性溶媒84等でふたをされていてもよいし、ウエル83中の疎水性溶媒84の中に入っていてもよい。
【0070】
この場合の疎水性溶媒84は、エマルジョン形成の目的以外に、反応液中の水分蒸発の防止の機能、微小液滴の形状を球状に保つ機能、ゲルの取り出し時にゲルと容器表面との固着を防ぐ機能も果たす。
【0071】
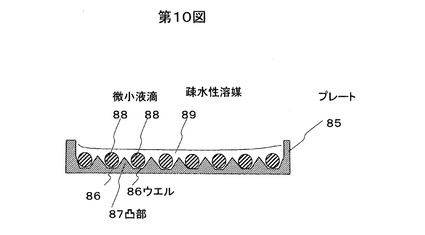
微小液滴81または82は相互に分離されている必要があるが、必ずしもウエル83そのものが相互に分離されている必要はなく、図10のプレート85のように、ウエル86間にある凸部87により、微小液滴88の移動を制限し、各ウエルを満たす疎水性溶媒89により複数の微小液滴88間を分離してもよい。
【0072】
各ウエルの直径は5μmから150μmが多数試料の同時増幅のためには好ましい。ウエルの数には特に制限はないが、一細胞由来の全発現遺伝子の増幅を目的とする場合には10万以上あることが望ましい。
【0073】
プレートの材質はポリカーボネート等の耐熱性の透明なプラスチックあるいはガラスが熱サイクルおよび光学的測定に好適である。
【0074】
本実施例によれば、反応後の微小液滴がプレートの平面上に展開されるために、増幅反応後の観察が容易である。また、平面上の各微小液滴の位置が固定されるために、その位置による各微小液滴の識別が可能である。
【0075】
実施例3:
本実施例は微小液滴の製法の別の例を示すものである。
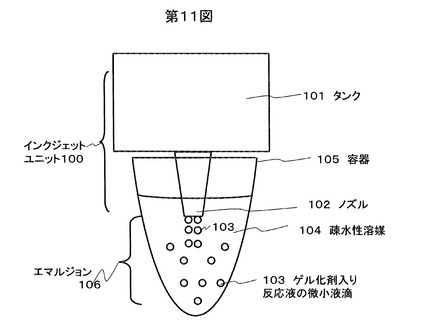
本実施例について図11を用いて説明する。本実施例では微小液滴の形成にインクジェットユニット100を用いる。インクジェットユニット100は,微小液滴103を調製するための溶液を格納するためのタンク101と、微小液滴化して噴出させるノズル102から構成される。ノズル内では反応液が瞬間的に加熱されることにより一定量の反応液が噴出する。微小液滴103は疎水性溶媒中104に直接噴出あるいは落下させられるように容器105に対して配置する。微小液滴103は疎水性溶媒中104に噴出または落下させられることにより、エマルジョン106が調製される。
【0076】
本実施例は微小液滴のサイズ、および数量のコントロールに適しており、特に、0.5plから10pl程度(直径10μm-30μm程度)の液滴を調製するのに適している。疎水性溶媒中に直接微小液滴を噴出する場合には、試料の相互コンタミネーション防止に有効である。
【0077】
実施例4:
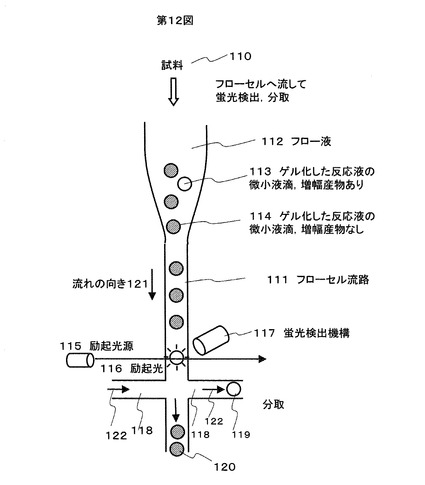
本実施例は増幅産物が得られている微小液滴の検出・分取にフローセルを用いた構成に関するものである。
本実施例について図12を用いて説明する。増幅反応後の微小液滴113,114を含む試料110を、光学セルを形成しているフローセル流路111にフロー液112の流れの向き121に従って流す。流れは自然落下で流すほか、ポンプを用いてもよい。励起光源115からの励起光116を微小液滴に照射し、得られる蛍光を光検出器、レンズ、フィルタなどから構成される蛍光検出機構117で検出する。得られた蛍光強度により増幅産物の量(または有無)を判定する。流路に流すフロー液112は上記実施例1のエマルジョン組成の場合にはシリコーン系のオイル(例:Polyphenylmethylsiloxane)が好ましい。
【0078】
蛍光強度が一定レベル以上の微小液滴119を、別の流路118に流れ122を生じさせることで、蛍光強度が一定レベル以下の微小液滴120と分離し、回収する。蛍光強度が一定レベル以下の微小液滴に対して同様の分離回収を行ってもよい。別の流路118に回収する際には、微小液滴119をレーザー等で局部的に加熱してゲルを溶解して回収してもよい。
【0079】
本実施例によれば、増幅反応後の微小液滴を、含まれる増幅産物の量によって分離回収する作業を連続的、自動的に行うことができる。
【0080】
実施例5:
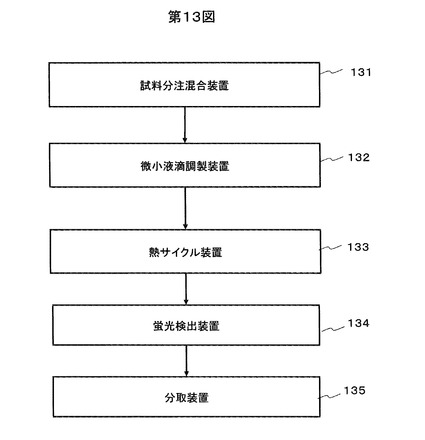
本実施例では、本発明の方法を行うための装置について説明する。
装置のブロック図を図13に示す。本実施例の装置は、試料分注混合装置131、微小液滴調製装置132、熱サイクル装置133、蛍光検出装置134、分取装置135から構成される。
【0081】
試料分注混合装置131は、ゲル化剤を溶液状態で保存するための温調機構、ゲル化剤を反応液と混合するための液体ハンドリング機構および攪拌機構を具備する。温調機構の温調範囲は0-120℃で、ゲル化剤の迅速な溶解に必要な温度に対応する。
【0082】
微小液滴調製装置132はいずれかの方式の攪拌機構から構成される。すなわち、振動または回転式のミキサー、実施例3に記述のインクジェット、微細流路を用いた方法のいずれかで構成する。
【0083】
熱サイクル装置133は一般のPCR用サーマルサイクラーと同様の温調機構を具備する装置である。上記131の温調機構と兼用してもよい。
【0084】
蛍光検出装置134は、蛍光顕微鏡の画像検出方式、またはフローセル方式の検出器から構成する。
【0085】
分取装置135は、実施例4に記述のようにフローセルに付随して設ける流路切替え機構を具備する。
【0086】
本実施例によれば多数の微量試料を同時に個別にPCRで増幅し、得られた増幅産物を蛍光で確認し、ゲルの微小液滴として回収する工程を省力化できる。
【産業上の利用可能性】
【0087】
本発明は、1細胞に含まれる全てのmRNAあるいは計測が必要と考えられる複数種類のmRNAをデジタルカウントする定量分析に必要な要素技術である。したがって、生物分野、医療分野、化学分野をはじめ単分子解析を必要とするあらゆる分野において有用である。
【図面の簡単な説明】
【0088】
【図1】図1は、本発明の方法の概念図である。
【図2】図2は、本発明の方法をcDNAに適用した場合の概念図である。
【図3】図3は、本発明の方法の工程図である。
【図4】図4は、本発明の実施例1のデータである。
【図5】図5は、本発明の実施例1の説明図である。
【図6】図6は、本発明の実施例1のデータである。
【図7】図7は、本発明の実施例1のデータである。
【図8】図8は、本発明の実施例2の説明図である。
【図9】図9は、本発明の実施例2の説明図である。
【図10】図10は、本発明の実施例2の説明図である。
【図11】図11は、本発明の実施例3の説明図である。
【図12】図12は、本発明の実施例4の説明図である。
【図13】図13は、本発明の実施例5の説明図である。
【符号の説明】
【0089】
1-3:DNA分子、4,5,7:DNAの入った微小液滴、6,8:DNAの入らなかった微小液滴、9:反応容器、10:オイル、11:エマルジョン、12:蛍光、13:DNA1−3由来の増幅産物がある微小液滴、14:増幅産物がない微小液滴、21:1個の細胞、22:1個の細胞から得たmRNA、23:磁気ビーズ、24:ポリTオリゴマー、25:相補鎖DNA、26:cDNA、27:切断箇所、28:アダプタ配列、29:DNA断片、30:遊離した一本鎖DNA、31:ビーズに固定化された一本鎖DNA、32,33,34:プライマー、35:プライミング配列、41:同一反応容器内の疎水性溶媒中にゲル化剤および蛍光色素を含む増幅用反応液の微小液滴を増幅対象の鋳型分子数よりも多数調製する工程、42:増幅反応を行う工程、43:増幅産物を含むゲル化した微小液滴を確認する工程、44:増幅産物を含むゲル化した微小液滴を分取する工程、45,46,47:目的PCR産物のバンド、48,49:ゲル化した反応液の微小液滴、50:オイル、51:エマルジョン、71:鋳型分子数と蛍光検出された液滴数の関係(40サイクル)、72:鋳型分子数と蛍光検出された液滴数の関係(60サイクル)、80,85:プレート、81,82,88:微小液滴、83,86:ウエル、84,89:疎水性溶媒、87:凸部、100:インクジェットユニット、101:タンク、102:ノズル、103:微小液滴、104:疎水性溶媒、105:容器、106:エマルジョン、110:試料、111:フローセル流路、112:フロー液、113,114:微小液滴、115:励起光源、116:励起光、117:蛍光検出機構、118:流路、119,120:微小液滴、121:流れの向き、122:流れ、131:試料分注混合装置、132:微小液滴調製装置、133:熱サイクル装置、134:蛍光検出装置、135:分取装置
【配列表フリーテキスト】
【0090】
配列番号1:プライマー
配列番号2:プライマー
【技術分野】
【0001】
本発明は遺伝子解析技術のための試料調製法に関する。より詳細には、1つの細胞に含まれるmRNAのデジタル解析あるいは多数の標的分子を同時に個別解析する方法のための試料調製法に関する。
【背景技術】
【0002】
ヒトゲノム配列解読の完了に伴い、種々ゲノムの情報が精力的に調べられそれらを活用する時代になってきた。ゲノム情報はmRNAに転写され、蛋白質へと翻訳される。こうした遺伝子の発現プロフィール解析は、生命活動の詳細を調べるために不可欠である。これまで主流となっている解析方法は多くの細胞からmRNAを取りだし、蛍光標識をした後にDNAプローブアレー(DNAチップ)に作用させ、mRNAと相補配列を持ちプローブに標識mRNAを捕獲して検出する方法であった。これに対し、多くの細胞からmRNAを取りだし、そのcDNAを作製し、それを電気泳動的に分離して計測する方法もある。この方法は種々のmRNAの量をアナログ的に計測する方法であるが、計測感度の問題から、多くの細胞を用いてmRNAを取りだし、計測しなければならない。
【0003】
一方、生命活動は多くの細胞が協調して一つのシステムを構成することで成り立っており、組織の中の個々の細胞にはそれぞれ異なる役割があると考えられている。本当の生命を理解するには、このような個々の細胞の働きをモニターすることが重要であり、1つの細胞に含まれるmRNAあるいは蛋白質の計測が重要視され始めている。それには1つの細胞に微量含まれるmRNAの種類と量を正確に定量分析することが必要であるがこのような方法は未だ確立されていない。
【0004】
発明者らはこの課題を克服するために1細胞に含まれる全てのmRNAあるいは計測が必要と考えられる複数種類のmRNAをデジタルカウントすることにより、定量分析することを目指している。デジタルカウントとは、それぞれのmRNA(あるいはcDNA断片)の配列を決定して種類を決め、その配列を有するmRNAが何個含まれるかをカウントして定量分析する方法である。
【0005】
すなわち、細胞のような小さな領域に含まれる複数のmRNAあるいはDNA断片のそれぞれについて配列解析することによりデジタルカウントするが、そのためには個々のmRNA(あるいはcDNA断片)を個別に増幅して配列解析できるようにする必要がある。ここで重要なことは全てのmRNA(あるいはcDNA断片)をもれなく独立に増幅することである。
【0006】
上記の方法では、一分子のDNAまたはmRNAを出発材料に並列して多くのPCR増幅を行うが、試料は溶液状でその中には数十から百万オーダーのmRNAあるいはcDNA断片が含まれている。これらを一括してPCR増幅すると複数の増幅産物の混合物が得られるだけで目的の計測試料は得られない。個々のmRNAを独立して、漏れがないように増幅してそれらを別々に取り出すことが必要である。個々のmRNAを独立に増幅するために、それぞれが分離された状態で個別にPCR反応を行うが、そのためには一反応体積あたりの反応開始時のDNAまたはRNA分子数の期待値が1以下になるまで試料溶液を希釈、分画を行った上で各々の分画について独立してPCR増幅反応を行う必要がある。例えば、ある試料中の増幅対象分子数が10万分子と見込まれる場合には、試料溶液を希釈して数十万以上に分画し、各々個別にPCR(ポリメラーゼ連鎖反応)等の増幅反応を行うことで試料中の全ての分子を独立して増幅すること、すなわちクローン増幅することができる。
【0007】
このような方法で複数のDNAを個別に増幅しようとする試みは近年いくつか試みられている。たとえば非常に多くの微小反応セルを平面プレート上に設け、ターゲットDNA断片と増幅に必要な酵素や反応基質を含む溶液をプレート上に流して微小反応セルに分画する。分画されたPCR溶液は相互に分離されているので独立に増幅することができる。1つの分画に入るDNA試料の量が平均1個以下になるように調整することで個別増幅が可能となる。この方法の一例が、アナリティカルケミストリーに開示されている(非特許文献1)。本例では、シリコン基板上に1万個のウエルを構築し高集積化している。しかし、100万個のDNA断片を増幅するためには更に多くの反応セルが必要である。また、対象となる試料溶液を全て余すことなく微小反応セルに注入することは不可能であって、溶液が余ったり、反応セルの内壁等に吸着されたりしてPCR増幅反応に使われないDNAが生じてしまう。
【0008】
また、1分子からの増幅を目指した試みではないが、微小容量のタイタープレートではなく平面に配置されたゲルドットマトリックスを用いてPCRする例もある(特許文献1)。従来より、低温でゲル化する材料を用いてPCR産物試料溶液をゲル化して試料の操作性を向上させる方法は公知だが(特許文献2)、この例では、ゲル化した試料をマトリクス的に配置した遺伝子検査用チップを利用する。しかし、この方法は空間的に固定された反応セルを用いるため、反応セルの中には目的の増幅産物が含まれるものも存在するが、全く含まれないものも存在してしまう。そのため、増幅されなかったターゲット試料が出てしまい、どのように目的の増幅産物を選別するかが問題となる。
【0009】
さらにもう一つの有力な方法として、エマルジョンPCRと呼ばれる方法がある。この方法では、試料毎に独立した反応容器を用いる代わりに、オイル中に多数形成した微小液滴内で反応を行う。この方法では、液滴の微小化が攪拌等により容易に行えるため、数十万個以上の反応容器相当の微小液滴を100マイクロリットル程度の一つの容器内に形成可能である。
【0010】
しかしエマルジョンを用いる方法では、個々の微小液滴から試料を個別に回収することは容易でないため、微小液滴中にDNAまたはRNAを固定化するため、プローブ付きのビーズを入れて、反応物が生成した後に反応物を捕獲したビーズを溶液から分離することによって各微小液滴中のDNAまたはRNAを回収している。こうしたビーズ固相を用いた試料の回収時には、酵素反応等によって得られたDNAまたはRNAを回収するために、生成物が得られている固相を得られていない固相と分けて回収する必要がある。そのため、PCR反応によって得られたDNAの一部と相補的な配列を持つプローブを固定化した磁気ビーズを用意し、プローブと増幅されたDNA断片をハイブリダイズさせた後、磁石を用いて選別回収する方法が用いられている。この方法を活用して多くのDNA断片を増幅してゲノム配列決定に活用した例が、ネイチャー等に掲載されている(特許文献3および非特許文献2)。しかし、この技術を全てのmRNA増幅とその配列測定に活用しようとするとやはり重大な問題がある。この系ではビーズとターゲットDNA1コピーがエマルジョン中の1反応液滴に含まれる必要があり、生成した液滴中にビーズが2個以上含まれる、1つのmRNAが重複してカウントされてしまい、デジタルカウントはできない。この問題を解消するため、ビーズの量をDNAと同様に少なくするとDNAは含まれるがビーズが含まれない液滴が多数生じてしまいやはり不都合である。固体ビーズを用いて生成したDNAを回収することは良い方法で、重複したDNA試料を用いてゲノム配列を決定するなどの目的には十分使い得る方法であるが、デジタルカウントには適さない。
【0011】
【特許文献1】特開2004−337064号公報
【特許文献2】特開平10−004963号公報
【特許文献3】WO2005/10145(PCT/US2004/015587)
【非特許文献1】Anal.Chem.2001,73,p1043−1047
【非特許文献2】Nature.2005,437,p376−380,Supplementary Information)
【発明の開示】
【発明が解決しようとする課題】
【0012】
前述のとおり、従来の方法にはいずれも問題点があった。まず、タイタープレートを用いた技術は多数試料を同時処理して増幅産物を取り出す時の液体ハンドリングについて配慮がされておらず、増幅された反応産物を見分けて多数試料を液体状態のまま個別に回収するため、試料数に応じた大量の試料容器や煩雑な取り扱い作業が必要となるという問題があった。
【0013】
ビーズ表面に増幅産物を捕獲して回収する方法では、ビーズに反応に必要なプライマー等をあらかじめ固定化しておく必要があるが、固相に固定したプライマーを増幅用のプライマーとして用いて固相表面に増幅産物を得ようとすると増幅効率が低下するという問題があった。これは、酵素反応の基質となるDNAあるいはRNA等の分子が固定化されていることにより分子運動の自由度が下がるため、溶液系に比べて反応効率が大きく低下するためである。さらに、固相表面へのDNAまたはRNAの非特異的吸着という問題もあった。すなわち、最初の増幅の鋳型となるDNA断片が固相に吸着されていると鋳型として巧く働かず、エマルジョン中にDNA鋳型が1コピー含まれているものの増幅産物が得られない。特にクローン増幅の目的のために1反応液あたり1分子という極低濃度のDNAまたはRNA試料を出発材料に用いる場合には、非特異的吸着による影響が相対的に大きくなるため深刻である。さらに前述のとおり、個々の微小エマルジョン反応液にビーズを均一に入れることは困難である。特に、エマルジョンを攪拌操作によって調製する場合には全ての液滴に同数のビーズ等の固相を入れることは不可能であり、一つの液滴に複数のビーズが入ったり、一個も入らなかったりする。液滴あたりのビーズ等の固相の数をコントロールできないと試料中の全分子を対象とする一分子計測を精度よく行うことが困難になる。
【0014】
このように従来の方法は、いずれも試料となるDNA断片プール(1細胞から得たmRNAあるいはcDNA断片の集合)を構成する全ての成分を同時に増幅して回収するという目的には適さなかった。
【0015】
本発明は、このような従来技術の問題点を克服するためになされたもので、1細胞に含まれるmRNAを取りだし、これをcDNAに転写し、1分子毎に増幅して回収し、DNA配列決定試料を調製することを課題とする。すなわち、簡便な方法でDNA断片プールに含まれる全ての成分を1分子毎に増幅してそれらを個別に回収する技術を提供することを目的とする。
【課題を解決するための手段】
【0016】
上記課題を解決するために発明者らは鋭意検討し、1分子毎の増幅を確実に実現し、増幅された反応産物だけを取り出す工夫をすることで、1細胞に含まれる全てのmRNA(cDNA)を1分子毎に増幅してそれらを個別に回収することに成功した。
【0017】
すなわち、本発明は、試料中の複数種の核酸を個別に増幅し取り出す方法であって、1つの微小液滴内に含まれる核酸が1を超えないように希釈された試料に対し、疎水性溶媒中の微小液滴内でPCR反応を行い、PCR反応終了後に反応液を固体またはゲルの状態で分離することを特徴とする方法に関する。
【0018】
前記方法は、PCR反応液中に増幅産物に結合あるいは取り込まれる蛍光試薬をあらかじめ添加することにより、増幅産物の含まれる液滴だけを選別分取する工程を含んでいてもよい。そのような蛍光試薬としては、インターカレーターや、蛍光ラベルされたモレキュラービーコン等を挙げることができる。
【0019】
試料中の複数種の核酸には、あらかじめ単一のPCRプライマーで増幅可能なようにアダプタ配列を導入しておくことが望ましい。
【0020】
本発明では液滴毎に独立して増幅を行なうため、PCR反応は、疎水性溶媒中に分散された微小液滴エマルジョンに対して行なうか、あるいは、相互に分離された微小反応セルの並んだプレートの微小反応セル内で行うことが望ましい。
【0021】
PCR反応液には、反応後の液を固体またはゲルの状態で分離するために、あらかじめヒドロゲルを形成するための、アガロース、ゼラチン、でんぷん、カラギーナン、ペクチン、アガロペクチン、ポリアクリルアミド、ポリアクリル酸、ポリビニルアルコール、ポリビニルピロリドン等の水溶性合成ポリマー等のゲル化剤を添加しておく。
【0022】
本発明で用いられる疎水性溶媒としては、シリコーン系オイルまたはパラフィン系オイルを主剤とするものが好ましい。
【0023】
また、PCR反応液には、疎水性媒体中での微小液滴の安定性を向上させるため、界面活性剤(両親媒性物質等)および/または被膜形成剤をあらかじめ添加しておくことが好ましい。
【0024】
本発明はまた、前記した方法によって個別に増幅し取り出された複数種の核酸を検出あるいは定量する工程を含む、核酸解析方法も提供する。
【0025】
さらに本発明は、前記した方法に用いるための装置であって、1)ゲル化剤を溶液状態で保存するための温調機構、ゲル化剤を反応液と混合するための液体ハンドリング機構、攪拌機構から構成される試料分注混合装置、2)振動または回転式のミキサー、インクジェットおよび微細流路のいずれかで構成される微小液滴調製装置、3)PCR反応のためのサーマルサイクル機能を持つ温調装置、および、4)画像検出方式またはフローセル方式の検出器を具備する蛍光検出装置を含む装置を提供する。
【0026】
前記装置では、4)のフローセル方式において、フローセルが流路切替えによる分取機能を具備していることが望ましい。
【0027】
さらに本発明は、前記装置と、DNAシーケンサーおよび/またはフローサイトメトリーを含む、核酸解析システムも提供する。
【発明の効果】
【0028】
本発明によれば、1細胞に含まれる全てのmRNAのような、多数の微量試料を同時に個別にPCRで増幅し、得られた増幅産物を蛍光で確認し、ゲルの微小液滴として回収が可能である。回収のために、反応液中に固相を設ける必要がなく、このためのコストと手間が省ける。また、固相を用いることによる試料のロスや反応効率の低下を防止できる。
【発明を実施するための最良の形態】
【0029】
本発明では、増幅の阻害要因となる固体ビーズをPCR反応に加えたり、あるいは固体からなる微小反応セルを活用したりすることなく、エマルジョン反応液を用いて微小液滴内で数多くのPCR反応を同時に行い、DNA相補鎖合成が行われた反応溶液だけを回収する。
【0030】
本発明において「微小液滴」とは、1つの液滴が1つの核酸を含みうるような微細な液滴を意味し、その大きさは特に限定されないが、直径1μm〜150μm程度であることが好ましい。また、「微小セル」とは、前記した微小液滴1つを格納するセルであって、その大きさは特に限定されないが、直径3μm〜250μm程度で、10万個以上設けられることが好ましい。
【0031】
なお、試料は前記した微小液滴内に含まれる核酸が1を超えないよう十分に希釈して用いる。また、試料中の核酸には単一プライマーで増幅可能なように、アダプタ配列をあらかじめ導入しておく。アダプタ配列の導入は、たとえば、mRNAからcDNAを合成する際に、アダプタ配列を含むプライマーを用いて行なうなど、公知の方法により実施できる。
【0032】
PCR反応はビーズなどの固体相は共存させず溶液状態で行い、効率よくPCR反応を進める。つぎに、増幅産物を含むエマルジョンは、温度を下げて固体あるいはゲル状態で取り出す。PCRは通常50〜96℃の高温で行われるので室温あるいはそれ以下の温度でエマルジョンを固体あるいはゲルとして取り出すことは可能である。すなわち、高温では液体、低温では固体あるいはゲルとなる物質を共存させてこれを実現する。
【0033】
相補鎖合成が行われたか否かを見分ける方法は種々存在するが、本発明の実施例では2本鎖DNAの間に入り込み蛍光を発するインターカレーターを用いた蛍光検出の例を示した。インターカレーターとしてはサイバーグリーンI、ピコグリーン、エチジウムブロマイドなどを挙げることができる。検出は、インターカレーターに限定されず、モレキュラービーコンのように相補鎖合成が行われたときに蛍光を発するプローブを利用してもよい。
【0034】
取りだしたゲルあるいは反応溶液ビーズ(固体状態あるいはゲル状態なのでこのように呼ぶ)にレーザーを照射して蛍光を発するものを選別捕獲する。これにはフローサイトメトリーなど既存の装置を使うことができるほか、マイクロ流路を用いたビーズセレクターなどが利用できる。
【0035】
上記の方法において、反応物を固化あるいはゲル化させて取り出すための素材としては、アガロース、ゼラチン、でんぷん(アミロース)、ポリアクリルアミドなどの親水性ゲル化剤を用いることができる。これらのゲル化剤は熱溶解性であるため、反応効率の良い溶液系での反応を実現できる。すなわち、これらのゲル化剤の水溶液は一般的な耐熱酵素の反応温度である50℃以上の条件では溶液状態であり、反応産物の取り出し時の室温の条件ではゲル化する。37℃程度で溶液状態にする必要がある場合には低融点アガロースを用いることも可能である。もちろんこのほかにDNA相補鎖合成に伴い固体状になる物質を加えても良い。
【実施例】
【0036】
以下、本発明を実施例により詳細に説明するが、本発明はこれらの実施例に限定されるものではない。
【0037】
実施例1:
本実施例ではアガロースを含んだエマルジョンをオイル中で攪拌して調製した例を示す。
本方法の基本概念を図1に示した。分析対象となるDNA分子1−3を含む試料溶液をDNA分子の総数Nよりも多いM個の微小液滴4−8に分画することでDNAが入った微小液滴4,5,7,DNAの入らなかった微小液滴6,8が形成される。微小液滴4−8は反応容器9のオイル10中に分散させてエマルジョン11を形成する。この微小液滴を含むエマルジョンをPCR等の増幅反応を行った後に、各微小液滴中に得られた増幅産物の有無(量)をインターカレーター等を用いた蛍光検出により検出し、蛍光12の検出される各DNA1−3由来の増幅産物がある微小液滴13と蛍光の検出されない増幅産物がない微小液滴14に分ける。微小液滴にはあらかじめ常温でゲルまたは固体となるゲル化剤を含有させることで個々の微小液滴の分取を可能にする。すなわち、レーザーを照射して発光する液滴(ゲル)だけを回収したり、発光しない液滴(ゲル)を溶かして除去したりすることにより目的の増幅産物を得ることができる。
【0038】
つぎに、1細胞由来のcDNAを対象とする場合を図2を用い説明する。1個の細胞21から得たmRNA22を磁気ビーズ23などに固定されたポリTオリゴマー24をプローブとして捕獲し、相補鎖DNA25を逆転写酵素で合成する(1stストランド合成)。mRNA22をRNaseHで分解した後、ランダムプライマーを用いてcDNA2本鎖26を形成する(2ndストランド合成)。次いでMboIなどの制限酵素により配列特異的にDNA2本鎖を切断する。切断箇所27に配列既知のアダプタ配列28をライゲーションにより結合させ、PCRプライミングサイトとする。このようして得たビーズ固定の2本鎖DNA断片29を含む溶液を昇温して2本鎖を乖離させ、ビーズ23に固定化された一本鎖31から遊離した一本鎖DNA30を得る。この一本鎖DNAの両端の配列は5’末端がアダプタ配列28の既知配列で3’末端がポリAなので、アダプタ配列28とポリTプライマーを共通に用いてPCR増幅が可能である。遊離した一本鎖DNA30と二つのプライマー32,33および相補鎖合成基質・酵素を加えて図1に示す微小液滴中でPCR増幅する。このときに上述したように低温でゲル化するアガロースとインターカレーターを加えておく。PCR反応の詳細については後述するが、50−96℃程度の熱サイクルで行われるので、この温度ではアガロースは液体状である。PCRが終了したら室温にして、アガロースが含まれる反応液をゲルビーズとして回収する。一方、特定の配列を持つ複数のmRNAをカウントする場合にはそれら配列に特異的な配列を持つプライマー34を用いるが、特異的でないプライミング配列35を持つ部分をアンカーしておいて、この部分をPCR増幅プライマーに用いてもよい。
【0039】
本実施例では加えたDNA鋳型の数が分かるようにモデルサンプルを用いて実験を行ったが、実際のcDNA計測でも同様のプライマーを用いて個別増幅を行うことができる。
【0040】
以下増幅工程について図3により説明する。本増幅工程は(1)同一反応容器内の疎水性溶媒中にゲル化剤および蛍光色素を含む増幅用反応液の微小液滴を増幅対象の鋳型分子数よりも多数調製する工程41、(2)増幅反応を行う工程42、(3)増幅産物を含むゲル化した微小液滴を確認する工程43、(4)増幅産物を含むゲル化した微小液滴を分取する工程44からなる。以下、4つの工程について詳述する。
【0041】
(1)同一反応容器内の疎水性溶媒中にゲル化剤および蛍光色素を含む増幅用反応液の微小液滴を増幅対象の鋳型分子数よりも多数調製する工程:
以下の組成のPCR反応液を準備する:120mM Tris-SO4(pH8.9),36mM Ammonium Sulfate,4mM MgSO4,0.4mM dNTPs,Fプライマー0.4μM(GTTTTCCCAGTCACGACGTTG:配列番号1),Rプライマー0.4μM(ATGACCATGATTACGCCAAGC:配列番号2),増幅用酵素Platinum Taq DNA ポリメラーゼ High Fidelity(インビトロジェン社)0.04unit/μL(一反応あたりの体積は50μl)。
【0042】
上記反応液には、鋳型DNAとしてコピー数を見積もることのできるように市販のpUC19プラスミドDNA(2686塩基,タカラバイオ)を用いた。実際にはこの鋳型が一反応あたり104〜108分子含まれるように調製して増幅効率などを確認した。プラスミドDNAの分子数は製品の添付資料の原液の濃度(0.5μg/μl,1.7x1011分子/μl)より求めた。また、反応液にはPCR産物の蛍光検出用色素として、サイバーグリーン(SYBR Green)I溶液(インビトロジェン社,S7563)を原液の2500倍の希釈度になるように加えた。なお、本品のモル濃度は開示されていないため希釈度は絶対的な数値ではない。
【0043】
蛍光色素としてはサイバーグリーンIの他、ピコグリーン、エチジウムブロマイドなどの2本鎖DNAと結合することによって蛍光強度が増強するインターカレータが適用可能である。そのほか、モレキュラービーコンのように相補鎖合成が行われたときに蛍光を発するプローブを用いてもよい。
【0044】
ゲル化剤としてはアガロースを用いた。アガロースにはゲル強度が1800g/平方センチ(1%(w/v)ゲル)以上の高ゲル強度であるSeakem Gold Agarose(タカラバイオ社)を用いた。
【0045】
ゲル濃度は反応セットアップ時の液体の取り扱いやすさと取り出し時のゲル状態でのハンドリングに必要な硬さの双方を考慮すると、アガロースの場合1−1.5%(w/v)が好適である。ただし、ゲル強度は同じゲル材料でも製品によって大きく異なるため、材料毎に最適な濃度は異なる。取り出し後のゲルの硬さ、あるいは、ゲルの水分を除去したあとの乾燥状態でのサイズを確保したい場合には、さらに高濃度のゲルを用いてもよい。アガロースについては2.5%(w/v)、ゼラチンについては5.0%(w/v)まではPCR反応に大きな支障はない。
【0046】
アガロースは粉末からは溶解しにくいのであらかじめオートクレーブを用いて121℃まで加熱して2.5%(w/v)の均一な水溶液を調製し、ピペッティングが容易な粘度になる50℃以上の溶液で準備する。この2.5%アガロース水溶液を、50℃程度の上述のPCR反応液と等体積(一反応あたり50μl)ずつ手早く混合し、最終アガロース濃度が1.25%(w/v)の反応液(一反応あたり計100μl)を調製する。混合時の温度は耐熱性酵素に影響を与えない90℃以下で行う。
【0047】
エマルジョン調製用のオイルにはシリコーン系混合オイルを用いた。組成は上述の文献(Nature, 2005, 437, p376-380, (Supplementary Information))中の記述を参考に、以下の組成とした:(1)Polyphenylmethylsiloxane(フルカ社,商品名AR20)を25%(v/v),(2)10%(v/v) PEG/PPG-18/18 Dimethiconeポリマー, Decamethylpentacyclosiloxane溶液(東レ・ダウコーニング社,商品名 DC5225C) を50%(v/v),(3) 50%(v/v)Trimethylsiloxysilicate, Decamethylpentacyclosiloxane溶液(東レ・ダウコーニング社,商品名BY11-018)を25%(v/v)。
【0048】
各成分はPolyphenylmethylsiloxaneはベースオイル、Decamethylpentacyclosiloxaneは溶剤、PEG/PPG-18/18 Dimethiconeポリマーは界面活性作用および増粘性のあるポリマー、Trimethylsiloxysilicateは水との界面にケイ酸の被膜を形成する成分である。
【0049】
本混合オイルを、上記のゲル化剤入りの反応液と同量(一反応あたり100μl)混合し、エマルジョンを調製する(混合後、一反応あたり200μl)。混合液を2mlのサンプルチューブに入れて、ボルテックスミキサ(タイテック社,2500rpm)で2-5秒程度攪拌することで直径50-100μm程度の微小液滴が得られる。
【0050】
液滴のサイズは、目的とする増幅倍率および、増幅対象の分子数によって変化させてよいが、直径20-200μmが好適であり、特に一細胞中に存在している遺伝子数に相当する10万分子程度を対象とする場合には直径50-100μm程度が増幅に必要な十分な試薬成分量を確保した上で、総反応液量が操作の容易な1ml以下となるため好適である。
【0051】
反応液の微小液滴エマルジョンを形成する方法は特に制限するものでなく、上記のミキサーによる攪拌の他、インクジェット法、微細流路を用いた方法(Angew. Chem. Int. Ed. 2005, 44, p724-728)などを用いることが可能である。
【0052】
得られたエマルジョンは一般的なプラスチック製反応容器中で増幅反応を行えばよい。一般的な反応容器以外にも、反応後の観察を容易にする目的で、相互に分離された微小反応セルの並んだプレートの微小反応セル内で増幅反応を行ってもよい。
【0053】
オイルの各成分の混合比の変化は反応液の微小液滴自体の形成に関しては大きな影響を与えないが、エマルジョンとしての安定性には多少の影響がある。オイルがベースオイル成分のPolyphenylmethylsiloxane 100%の場合にはエマルジョン形成後もオイル部分が白濁せずに透明なので、光学的検出には特に好適であるが、反応液の微小液滴同士がくっつきやすくなる。ただし、反応液にゲル化剤が入っているため、微小液滴同士がひとつに融合することはない。ベースオイル成分のPolyphenylmethylsiloxane に、Trimethylsiloxysilicateを成分量で5%程度(50%溶液で1/10容)以上加えると、微小液滴同士のくっつきは解消される。同成分を成分量で25%(50%溶液で1/2容)まで増やしても効果は同様である。
【0054】
また、ベースオイル成分のPolyphenylmethylsiloxane にPEG/PPG-18/18を成分量で1%(v/v)(10%溶液で1/10容)以上加えると、エマルジョン形成後に全体が白濁するが、エマルジョン中の微小液滴のオイルとの分離は抑制されエマルジョンの安定性が向上する。PEG/PPG-18/18を成分量で7%(v/v)(10%溶液で7/10容)まで増やしても効果に大きな変化はない。
【0055】
上記で用いた面活性剤成分、増粘成分、被膜形成成分は類似の物質でも代用可能である。
【0056】
疎水性溶媒としては、上記のシリコーン(有機ケイ素)系オイルの他、ミネラルオイル等のパラフィン系オイルなどが使用可能である。シリコーン系オイルは比重が0.98程度で反応液の溶媒である水の比重1に近いことと、温度による粘度の変化が少ないことにより、反応液と安定なエマルジョン形成が可能で特に好適である。
【0057】
(2)増幅反応を行う工程
調製した上記のエマルジョン状態の反応溶液は、0.2mlチューブに50μlずつ分注し、94℃15秒,55℃30秒,70℃1分の熱サイクル条件でPCRによる増幅反応を行う。サイクル数は40サイクルである。熱サイクル用装置としてサーマルサイクラー9700(アプライドバイオシステムズ社)を使うことができる。
熱サイクル装置には、PCRの熱サイクルの機能に加え、反応セットアップ時にゲル化剤水溶液、反応液、混合オイルを高温状態で保つための50℃以上の恒温槽機能が付加されていることが望ましい。
【0058】
(3)増幅産物を含むゲル化した微小液滴を確認する工程
反応後に、エマルジョンに対して5倍容のイソプロパノールを加えてエマルジョンを1液化し、スピンダウンしてゲル化した微小液滴のビーズを回収する。
【0059】
回収した増幅産物を含むゲル化した微小液滴ビーズは、ゲル電気泳動(アジレント社バイオアナライザ,DNA500キット,または2%アガロースゲル)を用いて電気泳動を行い、増幅産物のサイズおよび量を確認することができる。図4は鋳型を1x106分子加えた場合の増幅産物に対するアジレント社バイオアナライザを用いた電気泳動分析の結果である。同じ反応液組成で、ゲル化成分を加えていない試料(試料1)、微小液滴のエマルジョン状態にしなかった試料(試料2)も並行して調製し比較した。
【0060】
上記の回収した試料(試料3)のバンド47は、ゲル化成分が入っていない試料(試料1)のバンド45、ゲル化成分ありでエマルジョン状態にしなかった試料(試料2)のバンド46と同じ111塩基の位置に泳動されており、比較対象と同一サイズの産物の生成が確認できた。
【0061】
反応後のエマルジョン状態の反応溶液は蛍光顕微鏡(構成例:オリンパスBX51,UlS-2光学系,対物レンズUplanSApo,ミラーユニットWIB-UMWIB3)で上記のような精製を行うことなく、直接観察が可能である。観察の状態を模式的に図5に示す。鋳型を1x105分子加えた場合の蛍光で観察した結果の一例を図6に示す。ゲル化した反応液の微小液滴48,49のうち、増幅産物がある微小液滴48はサイバーグリーンIの蛍光で明るく観測され、増幅産物がない微小液滴49は暗く観測される。
【0062】
一反応あたりの鋳型数を変化させて上記図5および図6と同じ要領で観察を行い、増幅産物による蛍光が観測された微小液滴48の数の割合を、サーマルサイクル数が40サイクルの場合71、サーマルサイクル数が60サイクルの場合72について、それぞれプロットしたグラフを図7に示す。
【0063】
図7に示すようにサーマルサイクル数が40サイクルの場合と60サイクルの場合の変化は小さく、効率よく増幅されて40サイクルでほぼプラトーに達していることが示唆された。
【0064】
微小液滴の平均直径を50μmと仮定すると、一個あたりの平均体積は65plで、1反応(100μl)あたりの微小液滴の個数は1.5x106個であり、反応開始時に鋳型を105個加えた場合には、1割弱の微小液滴に一分子の鋳型が含まれ、鋳型を107個加えた場合には、ほぼ全ての微小液滴に一分子以上の鋳型が含まれることが期待されるが、図7に示される増幅産物が検出された微小液滴の割合の実測の結果は、鋳型を105個加えた場合には数%、106個加えた場合には10数%、107個加えた場合にはほぼ100%と上記の期待値に近い値になっており、本実施例における増幅がうまく進んでいることを示している。
【0065】
また、増幅産物量についても考察を行った。上記図4に示す回収した増幅産物(試料3)の電気泳動分析結果中の111塩基の産物のバンド47の濃度は約1ng/μlと定量(アジレントバイオアナライザ2100による定量値)されたので、1反応100μlあたり約100ngの増幅産物が回収されたことになる。111塩基の2本鎖DNAの100ngは1.4pM、8x1011分子に相当する。
【0066】
増幅率についても考察した。図7に示す結果より、反応開始時に鋳型を106個加えた場合には、約10%の微小液滴から増幅産物が認められるので、1反応100μlあたりの微小液滴の個数を上記の仮定より1.5x106個とすると、増幅産物が得られている微小液滴はその10%の1.5x105個であり、増幅産物が得られている微小液滴1個あたりのPCR産物は約5x106分子となり、これは増幅率が5x106倍と良好であることを示す。増幅産物の観察は、上記のほか、後述するフローサイトメトリーを用いてもよい。
【0067】
(4)増幅産物を含むゲル化した微小液滴を分取する工程
本実施例では、増幅産物を含むゲル化した微小液滴は顕微鏡観察下でキャピラリ管を装着したピペット(ドラモンド社製シーケンシングピペットなど)を用いて回収を行った。
回収した上記の微小液滴は、その中に含まれている増幅産物量をリアルタイムPCRで定量することが可能であった。また、増幅産物に対して、再度増幅の過程を加えてサンガー法、あるいは、パイロシーケンシング法による塩基配列決定に供することも可能である。
【0068】
回収の方法には上記の他に後述するフローサイトメトリーも適用可能である。本実施例によれば、106個の多数の微量試料を同時に個別に5x106倍までPCRで増幅し、得られた増幅産物を蛍光で確認し、ゲルの微小液滴として回収が可能である。個別回収のために、反応液中に固相を設ける必要がなく、このためのコストと手間が省ける。また、固相を用いることによる反応効率の低下を防止できる。
【0069】
実施例2:反応容器の形状
本実施例は反応容器の形状を相互に分離された微小反応セルの並んだプレートで構成するものである。
本実施例を図8−10を用いて説明する。図8のように、プレート80には個々の微小液滴81,82が収まるウエル83が多数設けられている。ウエル83は図9に示すように、2次元的に配列させてプレート80を構成する。微小液滴は直接ウエル83に入っていて疎水性溶媒84等でふたをされていてもよいし、ウエル83中の疎水性溶媒84の中に入っていてもよい。
【0070】
この場合の疎水性溶媒84は、エマルジョン形成の目的以外に、反応液中の水分蒸発の防止の機能、微小液滴の形状を球状に保つ機能、ゲルの取り出し時にゲルと容器表面との固着を防ぐ機能も果たす。
【0071】
微小液滴81または82は相互に分離されている必要があるが、必ずしもウエル83そのものが相互に分離されている必要はなく、図10のプレート85のように、ウエル86間にある凸部87により、微小液滴88の移動を制限し、各ウエルを満たす疎水性溶媒89により複数の微小液滴88間を分離してもよい。
【0072】
各ウエルの直径は5μmから150μmが多数試料の同時増幅のためには好ましい。ウエルの数には特に制限はないが、一細胞由来の全発現遺伝子の増幅を目的とする場合には10万以上あることが望ましい。
【0073】
プレートの材質はポリカーボネート等の耐熱性の透明なプラスチックあるいはガラスが熱サイクルおよび光学的測定に好適である。
【0074】
本実施例によれば、反応後の微小液滴がプレートの平面上に展開されるために、増幅反応後の観察が容易である。また、平面上の各微小液滴の位置が固定されるために、その位置による各微小液滴の識別が可能である。
【0075】
実施例3:
本実施例は微小液滴の製法の別の例を示すものである。
本実施例について図11を用いて説明する。本実施例では微小液滴の形成にインクジェットユニット100を用いる。インクジェットユニット100は,微小液滴103を調製するための溶液を格納するためのタンク101と、微小液滴化して噴出させるノズル102から構成される。ノズル内では反応液が瞬間的に加熱されることにより一定量の反応液が噴出する。微小液滴103は疎水性溶媒中104に直接噴出あるいは落下させられるように容器105に対して配置する。微小液滴103は疎水性溶媒中104に噴出または落下させられることにより、エマルジョン106が調製される。
【0076】
本実施例は微小液滴のサイズ、および数量のコントロールに適しており、特に、0.5plから10pl程度(直径10μm-30μm程度)の液滴を調製するのに適している。疎水性溶媒中に直接微小液滴を噴出する場合には、試料の相互コンタミネーション防止に有効である。
【0077】
実施例4:
本実施例は増幅産物が得られている微小液滴の検出・分取にフローセルを用いた構成に関するものである。
本実施例について図12を用いて説明する。増幅反応後の微小液滴113,114を含む試料110を、光学セルを形成しているフローセル流路111にフロー液112の流れの向き121に従って流す。流れは自然落下で流すほか、ポンプを用いてもよい。励起光源115からの励起光116を微小液滴に照射し、得られる蛍光を光検出器、レンズ、フィルタなどから構成される蛍光検出機構117で検出する。得られた蛍光強度により増幅産物の量(または有無)を判定する。流路に流すフロー液112は上記実施例1のエマルジョン組成の場合にはシリコーン系のオイル(例:Polyphenylmethylsiloxane)が好ましい。
【0078】
蛍光強度が一定レベル以上の微小液滴119を、別の流路118に流れ122を生じさせることで、蛍光強度が一定レベル以下の微小液滴120と分離し、回収する。蛍光強度が一定レベル以下の微小液滴に対して同様の分離回収を行ってもよい。別の流路118に回収する際には、微小液滴119をレーザー等で局部的に加熱してゲルを溶解して回収してもよい。
【0079】
本実施例によれば、増幅反応後の微小液滴を、含まれる増幅産物の量によって分離回収する作業を連続的、自動的に行うことができる。
【0080】
実施例5:
本実施例では、本発明の方法を行うための装置について説明する。
装置のブロック図を図13に示す。本実施例の装置は、試料分注混合装置131、微小液滴調製装置132、熱サイクル装置133、蛍光検出装置134、分取装置135から構成される。
【0081】
試料分注混合装置131は、ゲル化剤を溶液状態で保存するための温調機構、ゲル化剤を反応液と混合するための液体ハンドリング機構および攪拌機構を具備する。温調機構の温調範囲は0-120℃で、ゲル化剤の迅速な溶解に必要な温度に対応する。
【0082】
微小液滴調製装置132はいずれかの方式の攪拌機構から構成される。すなわち、振動または回転式のミキサー、実施例3に記述のインクジェット、微細流路を用いた方法のいずれかで構成する。
【0083】
熱サイクル装置133は一般のPCR用サーマルサイクラーと同様の温調機構を具備する装置である。上記131の温調機構と兼用してもよい。
【0084】
蛍光検出装置134は、蛍光顕微鏡の画像検出方式、またはフローセル方式の検出器から構成する。
【0085】
分取装置135は、実施例4に記述のようにフローセルに付随して設ける流路切替え機構を具備する。
【0086】
本実施例によれば多数の微量試料を同時に個別にPCRで増幅し、得られた増幅産物を蛍光で確認し、ゲルの微小液滴として回収する工程を省力化できる。
【産業上の利用可能性】
【0087】
本発明は、1細胞に含まれる全てのmRNAあるいは計測が必要と考えられる複数種類のmRNAをデジタルカウントする定量分析に必要な要素技術である。したがって、生物分野、医療分野、化学分野をはじめ単分子解析を必要とするあらゆる分野において有用である。
【図面の簡単な説明】
【0088】
【図1】図1は、本発明の方法の概念図である。
【図2】図2は、本発明の方法をcDNAに適用した場合の概念図である。
【図3】図3は、本発明の方法の工程図である。
【図4】図4は、本発明の実施例1のデータである。
【図5】図5は、本発明の実施例1の説明図である。
【図6】図6は、本発明の実施例1のデータである。
【図7】図7は、本発明の実施例1のデータである。
【図8】図8は、本発明の実施例2の説明図である。
【図9】図9は、本発明の実施例2の説明図である。
【図10】図10は、本発明の実施例2の説明図である。
【図11】図11は、本発明の実施例3の説明図である。
【図12】図12は、本発明の実施例4の説明図である。
【図13】図13は、本発明の実施例5の説明図である。
【符号の説明】
【0089】
1-3:DNA分子、4,5,7:DNAの入った微小液滴、6,8:DNAの入らなかった微小液滴、9:反応容器、10:オイル、11:エマルジョン、12:蛍光、13:DNA1−3由来の増幅産物がある微小液滴、14:増幅産物がない微小液滴、21:1個の細胞、22:1個の細胞から得たmRNA、23:磁気ビーズ、24:ポリTオリゴマー、25:相補鎖DNA、26:cDNA、27:切断箇所、28:アダプタ配列、29:DNA断片、30:遊離した一本鎖DNA、31:ビーズに固定化された一本鎖DNA、32,33,34:プライマー、35:プライミング配列、41:同一反応容器内の疎水性溶媒中にゲル化剤および蛍光色素を含む増幅用反応液の微小液滴を増幅対象の鋳型分子数よりも多数調製する工程、42:増幅反応を行う工程、43:増幅産物を含むゲル化した微小液滴を確認する工程、44:増幅産物を含むゲル化した微小液滴を分取する工程、45,46,47:目的PCR産物のバンド、48,49:ゲル化した反応液の微小液滴、50:オイル、51:エマルジョン、71:鋳型分子数と蛍光検出された液滴数の関係(40サイクル)、72:鋳型分子数と蛍光検出された液滴数の関係(60サイクル)、80,85:プレート、81,82,88:微小液滴、83,86:ウエル、84,89:疎水性溶媒、87:凸部、100:インクジェットユニット、101:タンク、102:ノズル、103:微小液滴、104:疎水性溶媒、105:容器、106:エマルジョン、110:試料、111:フローセル流路、112:フロー液、113,114:微小液滴、115:励起光源、116:励起光、117:蛍光検出機構、118:流路、119,120:微小液滴、121:流れの向き、122:流れ、131:試料分注混合装置、132:微小液滴調製装置、133:熱サイクル装置、134:蛍光検出装置、135:分取装置
【配列表フリーテキスト】
【0090】
配列番号1:プライマー
配列番号2:プライマー
【特許請求の範囲】
【請求項1】
試料中の複数種の核酸を個別に増幅し取り出す方法であって、1つの微小液滴内に含まれる核酸が1を超えないように希釈された試料に対し、疎水性溶媒中の微小液滴内でPCR反応を行い、PCR反応終了後に反応液を固体またはゲルの状態で分離することを特徴とする方法。
【請求項2】
PCR反応液中に増幅産物に結合あるいは取り込まれる蛍光試薬をあらかじめ添加することにより、増幅産物の含まれる液滴だけを選別分取する工程を含むことを特徴とする、請求項1に記載の方法。
【請求項3】
PCR反応が疎水性溶媒中に分散された微小液滴エマルジョン中で行われることを特徴とする、請求項1または2に記載の方法。
【請求項4】
PCR反応が、相互に分離された微小反応セルの並んだプレートの微小反応セル内で行われることを特徴とする、請求項1または2に記載の方法。
【請求項5】
試料中の複数種の核酸が、あらかじめ単一のPCRプライマーで増幅可能なようにアダプタ配列を導入されていることを特徴とする、請求項1〜4のいずれか1項に記載の方法。
【請求項6】
反応液を固体またはゲルの状態で分離するために、PCR反応液中にアガロース、ゼラチン、でんぷん、カラギーナン、ペクチン、アガロペクチン、ポリアクリルアミド、ポリアクリル酸、ポリビニルアルコール、およびポリビニルピロリドンから選ばれるいずれか1つのゲル化剤をあらかじめを添加しておくことを特徴とする、請求項1〜5のいずれか1項に記載の方法。
【請求項7】
前記疎水性溶媒がシリコーン系オイルまたはパラフィン系オイルを主剤とするものである、請求項1〜6のいずれか1項に記載の方法。
【請求項8】
さらに、PCR反応液中に界面活性剤および/または被膜形成剤をあらかじめ添加しておくことを特徴とする、請求項1〜7のいずれか1項に記載の方法。
【請求項9】
請求項1〜8の方法によって個別に増幅し取り出された複数種の核酸を検出あるいは定量する工程を含む、核酸解析方法。
【請求項10】
1)ゲル化剤を溶液状態で保存するための温調機構、ゲル化剤を反応液と混合するための液体ハンドリング機構、攪拌機構から構成される試料分注混合装置、2)振動または回転式のミキサー、インクジェットおよび微細流路のいずれかで構成される微小液滴調製装置、3)PCR反応のためのサーマルサイクル機能を持つ温調装置、および、4)画像検出方式またはフローセル方式の検出器を具備する蛍光検出装置を含む、複数種の核酸を個別に増幅し取り出すための装置。
【請求項11】
前記4)のフローセル方式において、フローセルが流路切替えによる分取機能を具備していることを特徴とする、請求項10に記載の装置。
【請求項12】
請求項10または11に記載の装置と、DNAシーケンサーおよび/またはフローサイトメトリーを含む、核酸解析システム。
【請求項1】
試料中の複数種の核酸を個別に増幅し取り出す方法であって、1つの微小液滴内に含まれる核酸が1を超えないように希釈された試料に対し、疎水性溶媒中の微小液滴内でPCR反応を行い、PCR反応終了後に反応液を固体またはゲルの状態で分離することを特徴とする方法。
【請求項2】
PCR反応液中に増幅産物に結合あるいは取り込まれる蛍光試薬をあらかじめ添加することにより、増幅産物の含まれる液滴だけを選別分取する工程を含むことを特徴とする、請求項1に記載の方法。
【請求項3】
PCR反応が疎水性溶媒中に分散された微小液滴エマルジョン中で行われることを特徴とする、請求項1または2に記載の方法。
【請求項4】
PCR反応が、相互に分離された微小反応セルの並んだプレートの微小反応セル内で行われることを特徴とする、請求項1または2に記載の方法。
【請求項5】
試料中の複数種の核酸が、あらかじめ単一のPCRプライマーで増幅可能なようにアダプタ配列を導入されていることを特徴とする、請求項1〜4のいずれか1項に記載の方法。
【請求項6】
反応液を固体またはゲルの状態で分離するために、PCR反応液中にアガロース、ゼラチン、でんぷん、カラギーナン、ペクチン、アガロペクチン、ポリアクリルアミド、ポリアクリル酸、ポリビニルアルコール、およびポリビニルピロリドンから選ばれるいずれか1つのゲル化剤をあらかじめを添加しておくことを特徴とする、請求項1〜5のいずれか1項に記載の方法。
【請求項7】
前記疎水性溶媒がシリコーン系オイルまたはパラフィン系オイルを主剤とするものである、請求項1〜6のいずれか1項に記載の方法。
【請求項8】
さらに、PCR反応液中に界面活性剤および/または被膜形成剤をあらかじめ添加しておくことを特徴とする、請求項1〜7のいずれか1項に記載の方法。
【請求項9】
請求項1〜8の方法によって個別に増幅し取り出された複数種の核酸を検出あるいは定量する工程を含む、核酸解析方法。
【請求項10】
1)ゲル化剤を溶液状態で保存するための温調機構、ゲル化剤を反応液と混合するための液体ハンドリング機構、攪拌機構から構成される試料分注混合装置、2)振動または回転式のミキサー、インクジェットおよび微細流路のいずれかで構成される微小液滴調製装置、3)PCR反応のためのサーマルサイクル機能を持つ温調装置、および、4)画像検出方式またはフローセル方式の検出器を具備する蛍光検出装置を含む、複数種の核酸を個別に増幅し取り出すための装置。
【請求項11】
前記4)のフローセル方式において、フローセルが流路切替えによる分取機能を具備していることを特徴とする、請求項10に記載の装置。
【請求項12】
請求項10または11に記載の装置と、DNAシーケンサーおよび/またはフローサイトメトリーを含む、核酸解析システム。
【図1】


【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【公開番号】特開2008−245612(P2008−245612A)
【公開日】平成20年10月16日(2008.10.16)
【国際特許分類】
【出願番号】特願2007−93618(P2007−93618)
【出願日】平成19年3月30日(2007.3.30)
【出願人】(000005108)株式会社日立製作所 (27,607)
【Fターム(参考)】
【公開日】平成20年10月16日(2008.10.16)
【国際特許分類】
【出願日】平成19年3月30日(2007.3.30)
【出願人】(000005108)株式会社日立製作所 (27,607)
【Fターム(参考)】
[ Back to top ]