
認識機能障害(cognitiveimpairment)を治療するための、組み合わされたセロトニン再取り込み、5−HT3および5−HT1A活性を有する化合物としての1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピペラジン
【課題】
うつ病患者における認識機能障害の治療に格別に有用な化合物を提供すること。
【解決手段】
1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピペラジンは、S
ERT、5−HT3および5−HT1Aに及ぼす強力な活性を呈し、そのようなものとし
て、認識機能障害の治療、特にはうつ病患者における認識機能障害の治療に有用であり得
る。
うつ病患者における認識機能障害の治療に格別に有用な化合物を提供すること。
【解決手段】
1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピペラジンは、S
ERT、5−HT3および5−HT1Aに及ぼす強力な活性を呈し、そのようなものとし
て、認識機能障害の治療、特にはうつ病患者における認識機能障害の治療に有用であり得
る。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、セロトニン受容体1A(5−HT1A)およびセロトニン受容体3(5−H
T3)に及ぼす活性と組み合わされたセロトニン再取り込み阻害活性を呈する化合物に関
し、これらの化合物はCNS関連疾患の治療に有用である。
【背景技術】
【0002】
選択的セロトニン再取り込み阻害薬(SSRI)は、それ以前に使用されていた化合物
、即ち、古典的な三環式化合物に比べ、より効果的であり、耐容性に優れ、好ましい安全
性プロフィールを有しているため、長年にわたり、特定のCNS関連疾患の治療、特には
うつ病、不安および社会恐怖症の治療に用いられる第一選択薬であった。
【0003】
それにもかかわらず、SSRIを用いる治療上の処置は、有意な割合の無応答者、即ち
、SSRI治療に応答しない患者または限られた程度にしか応答しない患者により阻まれ
ている。その上、典型的には、SSRI治療は、治療の数週間後まで効果を示し始めない
。
【0004】
SSRI治療のこれらの欠点のうちのいくつかを回避するため、精神科医は、時々、増
強(augmentation)戦略を利用する。抗うつ剤の増強は、例えば気分安定剤、例えばリチ
ウムカルボナートもしくはトリヨードチロニンなどと組み合わせることにより、または電
気ショック療法と併用することにより達成することができる。
【0005】
セロトニントランスポーター(SERT)の阻害と1種またはそれ以上のセロトニン受
容体に及ぼす活性との組み合わせが有益であり得ることは公知である。これまでに、セロ
トニン再取り込み阻害剤と5−HT2Cアンタゴニスト作用または逆アゴニスト作用を有
する化合物(5−HT2C受容体において負の効能を有する化合物)との組み合わせは、
微小透析実験で測定したときに、抹消(terminal)領域における5−HT(セロトニン)
レベルのかなりの増大をもたらすことが見出されている(特許文献1)。これは、臨床に
おける抗うつ効果の比較的短期間での発現およびセロトニン再取り込み阻害剤(SRI)
の治療効果の増強または相乗作用を示唆していたのであろう。
【0006】
同様に、5−HT1Aの部分的アゴニストであるピンドロールとセロトニン再取り込み
阻害剤との組み合わせが効果の速やかな発現をもたらすことも報告されている(非特許文
献1)。
【0007】
CNS関連疾患、例えばうつ病、不安および統合失調症などは、しばしば、他の障害ま
たは機能不全、例えば認知障害または認識機能障害などと併存する(非特許文献2、非特
許文献3)。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】国際公開第01/41701号パンフレット
【非特許文献】
【0009】
【非特許文献1】Psych.Res.、125、81−86、2004
【非特許文献2】Scand.J.Psych.、43、239−251、2002
【非特許文献3】Am.J.Psych.、158、1722−1725、2001
【発明の概要】
【発明が解決しようとする課題】
【0010】
幾つかの神経伝達物質がこの認識力を調節する神経学的事象にかかわっているものと推
測されている。特に、コリン作動系は認識に関して重要な役割を果たしており、従って、
コリン作動系に影響を及ぼす化合物は認識機能障害の治療に潜在的に有用である。5−H
T1A受容体および/または5−HT3受容体に影響を及ぼす化合物はコリン作動系に影
響を及ぼすことが知られており、それらの化合物は、そのようなものとして、認識機能障
害の治療に有用であり得る。
【0011】
このような理由から、5−HT1Aおよび/または5−HT3受容体活性を機能させる
化合物は、認識機能障害の治療に有用であると期待することができよう。その上にSER
T活性も機能させる化合物は、そのような化合物がうつ病の治療において速やかな治療効
果の発現をももたらすものと考えられるため、うつ病患者における認識機能障害の治療に
格別に有用であろう。
【0012】
国際公開第03/029232号パンフレットは、例えばSERT活性を有する化合物
として化合物1−[2−(2,4−ジメチルフェニルスルファニル)フェニル]ピペラジ
ン(実施例1e)を開示している。
【課題を解決するための手段】
【0013】
驚くべきことに、本発明者らは、1−[2−(2,4−ジメチルフェニルスルファニル
)フェニル]ピペラジンがSERT阻害、5−HT3アンタゴニズムおよび5−HT1A
部分的アゴニズムの組み合わせを発揮することを見出した。従って、1つの実施形態にお
いては、本発明は、1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]
ピペラジンである化合物Iおよびその化合物の薬学的に許容可能な塩を提供し、但し、前
述の化合物は非結晶質形態における遊離塩基ではないものとする。
【0014】
1つの実施形態においては、本発明は、治療における化合物Iの使用を提供する。
【0015】
1つの実施形態においては、本発明は、化合物Iを含む医薬組成物を提供する。
【0016】
1つの実施形態においては、本発明は治療方法を提供し、その治療方法は、治療を必要
としている患者に有効量の化合物Iを投与することを含む。
【0017】
1つの実施形態においては、本発明は、薬剤の製造における化合物Iの使用を提供する
。
【図面の簡単な説明】
【0018】
【図1】結晶質塩基のXRPD。
【図2】アルファ型の臭化水素酸塩のXRPD。
【図3】ベータ型の臭化水素酸塩のXRPD。
【図4】ガンマ型の臭化水素酸塩のXRPD。
【図5】半水和物の形態の臭化水素酸塩のXRPD。
【図6】エチルアセタート溶媒和物およびアルファ型の臭化水素酸塩の混合物のXRPD。
【図7】塩酸塩のXRPD。
【図8】一水和物の形態の塩酸塩のXRPD。
【図9】メシル酸塩のXRPD。
【図10】フマル酸塩のXRPD。
【図11】マレイン酸塩のXRPD。
【図12】メソ−酒石酸塩のXRPD。
【図13】L−(+)−酒石酸塩のXRPD。
【図14】D−(−)−酒石酸塩のXRPD。
【図15】硫酸塩のXRPD。
【図16】リン酸塩のXRPD。
【図17】硝酸塩のXRPD。
【図18a】皮内ホルマリン試験における本発明の化合物の効果。X−軸は投与された化合物の量を示している;Y−軸は足をなめるのに費やした時間的な量(秒)を示している。0−5分の時間的期間内における応答。
【図18b】皮内ホルマリン試験における本発明の化合物の効果。X−軸は投与された化合物の量を示している;Y−軸は足をなめるのに費やした時間的な量(秒)を示している。20−30分の時間的期間内における応答。
【図19a】1−[2−(2,4−ジメチルフェニルスルファニル)フェニル]ピペラジンHBr塩の投与時における自由行動ラットの前前頭皮質における細胞外アセチルコリンレベル。
【図19b】1−[2−(2,4−ジメチルフェニルスルファニル)フェニル]ピペラジンHBr塩の投与時における自由行動ラットの腹側海馬における細胞外アセチルコリンレベル。
【図20】獲得(acquisition)の60分前に与えられたときの、Sprague−Dawleyラットにおける文脈的恐怖条件付けに及ぼす1−[2−(2,4−ジメチルフェニルスルファニル)フェニル]ピペラジンHBr塩の効果。フットショックUSに先立つ58秒間の馴化期間の間、すくみ行動(freezing behaviour)のスコアが付けられた(ショック前獲得)(白色のバー)。すくみ行動はトレーニングの24時間後に測定された(保持(retention)テスト)(黒色のバー)。
【図21】保持テストの1時間前に与えられたときの、Sprague−Dawleyラットにおける文脈的恐怖条件付けに及ぼす1−[2−(2,4−ジメチルフェニルスルファニル)フェニル]ピペラジンHBr塩の効果。フットショックUSに先立って、58秒間の間、すくみ行動のスコアが付けられた(獲得)(白色のバー)。すくみ行動はトレーニングの24時間後に測定された(保持テスト)(黒色のバー)。
【図22】獲得の直後に与えられたときの、Sprague−Dawleyラットにおける文脈的恐怖条件付けに及ぼすAA21004の効果。フットショックUSに先立って、58秒間の間、すくみ行動のスコアが付けられた(ショック前獲得)(白色のバー)。すくみ行動はトレーニングの24時間後に測定された(記憶保持テスト)(黒色のバー)。
【発明を実施するための形態】
【0019】
本発明は、その構造が
【0020】
【化1】

【0021】
である化合物I、1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピ
ペラジンおよびその化合物Iの薬学的に許容可能な塩に関し、但し、化合物Iは非結晶質
形態における遊離塩基ではないものとする。
【0022】
1つの実施形態においては、上述の薬学的に許容可能な塩は、無毒な酸の酸付加塩であ
る。そのような塩は、有機酸から生成される塩、例えばマレイン酸、フマル酸、安息香酸
、アスコルビン酸、コハク酸、シュウ酸、ビス−メチレンサリチル酸、メタンスルホン酸
、エタンジスルホン酸、酢酸、プロピオン酸、酒石酸、サリチル酸、クエン酸、グルコン
酸、乳酸、リンゴ酸、マンデル酸、ケイ皮酸、シトラコン酸、アスパラギン酸、ステアリ
ン酸、パルミチン酸、イタコン酸、グリコール酸、p−アミノ安息香酸、グルタミン酸、
ベンゼンスルホン酸、テオフィリン酢酸、ならびに8−ハロテオフィリン、例えば8−ブ
ロモテオフィリンなどから生成される塩を含む。また、そのような塩は、無機塩から生成
されてもよく、例えば塩酸、臭化水素酸、硫酸、スルファミン酸、リン酸および硝酸など
から生成されてもよい。特別なものとして、メタンスルホン酸、マレイン酸、フマル酸、
メソ−酒石酸、(+)−酒石酸、(−)−酒石酸、塩酸、臭化水素酸、硫酸、亜リン酸お
よび硝酸から生成される塩を挙げることができる。格別なものとして臭化水素酸塩を挙げ
ることができる。
【0023】
経口用剤形、特に錠剤は、投与しやすく、その結果、良好なコンプライアンスが得られ
るため、患者および医療実践者により好まれることが多い。錠剤の場合、活性成分は結晶
質であることが好適である。1つの実施形態においては、本発明の化合物は結晶質である
。
【0024】
1つの実施形態においては、本発明の結晶は溶媒和物、即ち、溶媒分子がその結晶構造
の一部を形成する結晶である。この溶媒和物は水から形成されてよく、その場合には、そ
れらの溶媒和物は水和物と呼ばれることが多い。代替的に、これらの溶媒和物は、他の溶
媒、例えばエタノール、アセトンまたはエチルアセタートなどから形成されてもよい。そ
の溶媒和の正確な量はそれらの条件に依存することが多い。例えば、水和物は、典型的に
は、温度が高められた場合または相対湿度が下げられた場合には、水分を失うであろう。
【0025】
1つの実施形態においては、本発明の化合物は非溶媒和型の結晶である。
【0026】
幾つかの化合物は吸湿性であり、即ち、それらの化合物は湿気に晒されたときに水分を
吸収する。吸湿性は、一般的に、薬剤配合物中、特には乾性配合物中、例えば錠剤中など
に存在させるべき化合物にとっては望ましくない特性であると見なされている。1つの実
施形態においては、本発明は、吸湿性の低い結晶を提供する。結晶質の活性成分を用いる
経口用剤形の場合には、それらの結晶が明確に定義されたものであることも有益である。
この文脈において、「明確に定義された」という用語は、具体的には、その化学量論が明
確に定義されたものであることを意味し、即ち、その塩を形成しているイオン間の比が小
さな整数間の比、例えば1:1、1:2、2:1、1:1:1などであることを意味する
。1つの実施形態においては、本発明の化合物は明確に定義された結晶である。
【0027】
本発明の結晶質化合物は1つより多くの形態で存在していてよく、即ち、それらの化合
物は多形性形態で存在していてよい。多形性形態は、化合物が1つより多くの形態で結晶
化し得る場合に存在する。本発明は、純粋な化合物またはそれらの化合物の混合物のどち
らの場合にも、すべてのそのような多形性形態を包含すべく意図されている。
【0028】
1つの実施形態においては、本発明の化合物は精製された形態である。「精製された形
態」という用語は、化合物が、場合によって、他の化合物または他の形態のそれと同一の
化合物を本質的に含んでいないことを表すべく意図されている。
【0029】
1つの実施形態においては、本発明は、図1〜17、特には図2、3、4および5に示
されているようなXRDPを有する本発明の化合物の結晶質の塩を提供する。
【0030】
以下の表は、本発明の化合物に対する主要なXRDP反射(XRDP reflections)を示し
ている。
【0031】
選択されたX線ピーク位置(°2θ)、すべての値+−0.1°
【0032】
【表1】
【0033】
例えば図2〜5により証拠だてられているように、本発明の化合物、この場合には臭化
水素酸塩は、幾つかの形態で存在することができ、即ち、多形性であり得る。それらの多
形性形態は、実施例4dで示されているように、異なる特性を有している。ベータ型の臭
化水素酸塩は、より高いDSC融点およびより低い溶解度により示されているように、高
い安定性を有している。その上、前述のベータ型は低い吸湿性と溶解度との魅力的な複合
特性を有しており、その複合特性により、この化合物は錠剤を製造するのに特に適したも
のと成っている。従って、1つの実施形態においては、本発明は、約6.89、9.73
、13.78および14.62(°2θ)におけるXRDP反射、特には図3に示されて
いるようなXRPDを有する、1−[2−(2,4−ジメチルフェニルスルファニル)−
フェニル]ピペラジンの臭化水素酸塩を提供する。
【0034】
また、活性成分の溶解度も、バイオアベイラビリティーに直接的な影響を及ぼし得るた
め、剤形を選択する上で重要である。経口用剤形の場合には、活性成分のより高い溶解度
は、バイオアベイラビリティーを高めるため、一般的には、有益であると考えられている
。
【0035】
皮質および海馬のコリン作動性神経伝達は認識力にとって非常に重要であり、また、数
多くの前臨床観察がこの系にとってのセロトニン受容体1A(5−HT1A)の重要性を
指摘している。T.Koyamaは、Neurosci.Lett.、265、33〜3
6、1999において、5−HT1AアゴニストであるBAYX3702がラットの皮質
および海馬からのアセチルコリン流出を増大させることを報じている。興味深いことに、
5−HT1AアンタゴニストであるWAY−100635はBAYX3702の効果を排
除することができ、これは、BAYX3702の効果が5−HT1A媒介性であることを
示している。
【0036】
数多くの研究が、認識機能障害に及ぼす5−HT1Aのモジュレーターの効果について
報じている。A.Menesesは、Neurobiol.Learn.Memory、
71、207〜218、1999において、部分的5−HT1Aアゴニストである(±)
−8−ヒドロキシ−2−(ジ−n−プロピルアミノ)−テトラリン、HCl(8−OH−
DPAT)が、正常なラットにおける学習の統合(consolidation)を促進し、また、認
識機能障害を負ったラットにおける認識機能を正常化することを報じている。
【0037】
これらの前臨床観察結果は臨床においても反映されているようである。T.Sumiy
oshiは、Am.J.Psych.、158、1722〜1725、2001において
ある研究について報じており、その研究では、患者に、プラシーボまたは5−HT1Aア
ゴニストであるタンドスピロンと組み合わせて、定型抗精神病薬、例えばハロペリドール
、スルピリドおよびピモジドなど(すべてのものが5−HT1A活性を欠く)が投与され
た。抗精神病薬に加えてタンドスピロンが投与された患者は認識能力の改善を示したが、
一方、プラシーボが投与された患者は改善を示さなかった。同様に、同じく5−HT1A
アゴニストである非定型抗精神病薬、例えばクロザピンなども統合失調症患者における認
識力を増強するが、一方、5−HT1A活性を有さない定型抗精神病薬、例えばハロペリ
ドールなどは認識力を増強しない(Y.Chung、Brain Res.、1023、
54〜63、2004)。
【0038】
上で述べられているように、コリン作動系は認識力を調節する神経学的事象にかかわっ
ているものと考えられており、また、コリン作動系は、セロトニン受容体3(5−HT3
)による抑制制御の影響を受けやすいものと考えられる[Giovanniniら、J.
Pharmacol.Exp.Ther.、1998、285:1219〜1225;C
ostallおよびNaylor、Current Drug Targets−CNS
&Neurobiol.Disord.2004、3:27〜37]。
【0039】
マウスにおける馴化試験において、ラットにおけるT字型迷路強制交替試験(T-maze r
einforced alternation task)において、ならびにマーモセットにおける物体弁別(obje
ct discrimination)および逆転学習課題において、オンダンセトロンは、ムスカリン性
アンタゴニストであるスコポラミンにより引き起こされる機能障害または基底核から現れ
るコリン作動性経路の病変を低減した(Barnesら、Pharmacol.Bioc
hem.Behav.1990、35:955〜962;Careyら、Pharmac
ol.Biochem.Behav.1992、42:75〜83)。Boastら(N
eurobiol.Learn.Mem.1999、71:259〜271)は、NMD
A受容体の非競合的アンタゴニストであるMK−801を使用して、遅延標本非照合放射
状迷路課題で訓練されたラットの認識能力を混乱させた。オンダンセトロンは認識機能障
害をブロックすることが示された。その上、マウスでの受動的回避課題におけるエタノー
ルの記憶喪失作用についての研究において、エタノールのこの記憶喪失作用はオンダンセ
トロンにより正常状態へ向けて部分的に回復された(Napiorkowska−Paw
lakら、Fundam.Clin.pharmacol.2000、14:125〜1
31)。従って、前臨床モデルにおけるコリン作動系の機能障害後の5−HT3アンタゴ
ニズムによるコリン作動性伝達の促進(Diez−Arizaら、Psychophar
macology、2003、169:35〜41;Gil−Beaら、Neuroph
armcol.2004、47:225〜232)は、認知障害の治療においてこの処置
を用いることに対する根拠を示唆している。
【0040】
健常な男性の被検者におけるランダム化二重盲検クロスオーバー試験において行われた
言葉および空間の記憶ならびに注意の持続についての評価は、5−HT3アンタゴニスト
であるアロセトロンが言葉および空間の記憶におけるスコポラミン誘発性の障害を軽減す
ることを示した(Preston、Recent Advances in the t
reatment of Neurodegenerative disorders
and cognitive function、1994、(eds.)Racagn
iおよびLanger、Basel Karger、p.89〜93)。
【0041】
結論として、5−HT3アンタゴニスト活性と組み合わせて5−HT1A部分的アゴニ
スト活性を発揮する化合物は、認識機能障害の治療に特別に有用であると考えられる。さ
らに、セロトニン再取り込み阻害を発揮する化合物は、5−HT1A部分的アゴニズムと
組み合わされたセロトニン再取り込み阻害がより迅速なうつ病の治療効果の発現をもたら
すため、うつ病に伴う認識機能障害の治療に格別に有用であろう。
【0042】
実施例1で示されているように、本発明の化合物はヒトセロトニントランスポーターの
強力な阻害剤であり、即ち、それらの化合物はセロトニン再取り込みを阻害する。その上
、それらの化合物は、マウス、ラット、モルモットおよびイヌ5−HT3受容体における
強力なアンタゴニストである。卵母細胞にクローニングされたヒト5−HT3受容体にお
いて、それらの化合物は低濃度ではアンタゴニストであり(IC50=約30nM)、そ
の一方で、より高い濃度では、それらの化合物はアゴニスト特性を示す(ED50=2.
1μM)ことが判明した。高濃度における本発明の化合物のその後の適用は何らアゴニス
ト応答を示さなかったが、これは、インビトロにおける急速な脱感作(desenitisation)
または直接的なアンタゴニズムによるものであったとすることができよう。従って、低濃
度において、本発明の化合物は、他の種からの5−HT3受容体で観測されたのと同様に
、ヒト5−HT3受容体においても著しいアンタゴニズムを示す。
【0043】
本発明の化合物は、ラットおよびマウスの両者の脳ホモジネートにおける5−HT1A
受容体に低い親和性で結合する。しかし、本発明の化合物は、40nMのKiでヒト5−
HT1A受容体に結合する。その上、機能データ(functional data)は、本発明の化合
物がヒト5−HT1A受容体における部分的アゴニストであり、85%の有効性をもたら
すことを示している。
【0044】
SERT、5−HT3受容体および5−HT1A受容体における本発明の活性は、ヒト
における本化合物のインビボプロフィールに寄与するものと予想される。
【0045】
実施例26に示されているように、本発明の化合物は、ラットにおいて、前前頭皮質お
よび腹側海馬における細胞外アセチルコリンレベルの増大をもたらす。これらの前臨床知
見は、認識機能障害の治療におけるアセチルコリンエステラーゼ阻害剤の使用と比較して
、認識機能障害の治療、例えばアルツハイマー病の治療における臨床的な効果に変わるも
のと期待される。この見解に対する更なる支持を実施例27に見出すことができ、そこで
のデータは、本発明の化合物がラットにおける文脈的(contextual)記憶を増強すること
を示している。結局のところ、ラットにおけるアセチルコリンレベルに及ぼす効果および
記憶に及ぼす効果と組み合わせた本発明の化合物の薬理学的プロフィールは、本発明の化
合物が認識機能障害の治療に有用であることを強く示唆している。
【0046】
1つの実施形態においては、本発明は認知障害または認識機能障害を治療するための方
法に関し、その方法は、治療を必要としている患者に治療的有効量の本発明の化合物を投
与することを含む。
【0047】
認知障害または認識機能障害は、認知機能または認知領域の低下を含み、例えば作業記
憶、注意および覚醒、言語的学習および記憶、視覚的な学習および記憶、論理的思考およ
び問題の解決、例えば実行機能、処理速度および/または社会的認知の低下を含む。特に
、認知障害または認識機能障害は、注意の欠如、解体した思考、思考の減退(slow think
ing)、理解困難(difficulty in understanding)、集中力不足(poor concentration)
、問題解決能力の低下(impairment of problem solving)、記憶力貧困(poor memory)
、思考の表現困難(difficulties in expressing thoughts)および/または思考、感情
および行動の統合困難(difficulties in integrating thoughts, feelings and behavio
ur)、または見当違いな思考の消去困難(difficulties in extinction of irrelevant t
houghts)を表し得る。「認知障害」および「認識機能障害」という用語は同じものを指
示すべく意図されており、互換可能に使用される。
【0048】
1つの実施形態においては、その患者は、別のCNS障害、例えば情動障害、例えばう
つ病;全般性(generalised)うつ病;大うつ病性障害;一般的(general)不安障害およ
びパニック障害を含めた不安障害;強迫性障害;統合失調症;パーキンソン病;認知症;
エイズによる認知症;ADHD;加齢による記憶障害;またはアルツハイマー病などの診
断も同時に下されている。
【0049】
認識機能障害は、例えば大うつ病性障害などのうつ病の典型的な特徴の一つである。認
知障害は、うつ状態の改善が認識機能障害の改善をももたらすという意味で、ある程度、
うつ病に対して続発性であり得る。しかし、認知障害が実際にはうつ病から独立している
ことを示す明らかな証拠も存在する。例えば、何件かの研究が、うつ病からの回復時に存
続する認識機能障害を示している[J.Nervous Mental Disease
、185、748〜754、197]。その上、うつ病および認識機能障害に及ぼす抗う
つ薬の示差的な効果が、たとえしばしば併存する状態であるとしても、うつ病と認識機能
障害とは独立しているという考えに対する更なる支持を与えている。セロトニンおよびノ
ルアドレナリン医薬は抑うつ症状の同程度の改善をもたらすが、幾つかの研究は、ノルア
ドレナリン作動系の調節がセロトニン調節程には認識機能を改善しなかったことを示して
いる[Brain Res.Bull.、58、345〜350、2002;Hum P
sychpharmacol.、8、41〜47、1993]。
【0050】
本発明の化合物を投与することによるうつ病患者における認識機能障害の治療は格別に
有利であると考えられる。本発明の化合物の多面的な薬効薬理、特にSERT、5−HT
3および5−HT1A活性は、抑うつ状態の迅速な治療効果の発現と組み合わされた認識
機能の改善をもたらすことが期待される。
【0051】
認識機能障害は、高齢者における特に重要な考慮すべき事項である。認識機能障害は加
齢とともに普通に進行するが、更に、うつ病でも進行する。従って、1つの実施形態にお
いては、認識機能障害の治療が為される患者は高齢者であり、特にうつ病を伴った高齢者
である。
【0052】
認識機能は、上で述べられているように、統合失調症患者で損なわれることが多い。ま
た、何件かの研究は、認識機能が統合失調症における職業上の機能(vocational functio
ning)と関連していると結論付けてもいる[Scizophrenia Res.、45
、175〜184、2000]。1つの実施形態においては、認識機能障害の治療が為さ
れる患者は統合失調症患者である。
【0053】
5−HT3受容体アンタゴニストは、更に、種々の疾患の治療用、例えば嘔吐、化学療
法起因性嘔吐、渇望、物質乱用、疼痛、過敏性腸症候群(IBS)、統合失調症および摂
食障害などの治療用としても示唆されている[Eur.J.Pharmacol.、56
0、1〜8、2007;Pharmacol.Therapeut.、111、855〜
876、2006;Alimentary Pharmacol.Ther.、24、1
83〜205、2006]。
【0054】
ある臨床研究は、臨床応答が不充分なうつ病患者(治療抵抗性うつ病(TRD)または
難治性うつ病)を治療するには、ミルタザピン(mirtazipine)とSSRIとを組み合わ
せた場合の方がSSRIを単独で使用する場合よりも優れていることを示している[Ps
ychother.Psychosom.、75、139〜153、2006]。ミルタ
ザピンは5−HT2および5−HT3アンタゴニストであり、これは、本発明の化合物が
TRDの治療に有用であるという概念を支持するものである。
【0055】
顔面紅潮は閉経過渡期に関連する症状である。女性によっては、この顔面紅潮が、睡眠
または一般的な活動を妨害し、治療が必要な程度にまで成り得る。数十年間にわたり、エ
ストロゲンを用いるホルモン代償療法が既定の治療法であったが、最近では、乳癌および
心事象などの副作用に関する懸念が表明されている。SSRIを用いる臨床試験は、エス
トロゲンの場合程ではないにしても、これらの化合物が顔面紅潮に有効であることを示し
ている[J.Am.Med.Ass.、295、2057〜2071、2006]。しか
し、セロトニン再取り込みを阻害する化合物、例えば本発明の化合物を用いる顔面紅潮の
治療法は、エストロゲンを受け入れることができない女性または受け入れない女性に対す
る代替的治療法である。
【0056】
睡眠時無呼吸もしくは閉塞型睡眠時無呼吸−低呼吸症候群または閉塞型睡眠呼吸障害は
、効果的な薬物療法が特定されるべき状態のまま留まっている障害である。しかし、動物
における幾つかの研究は、5−HT3アンタゴニスト、例えば本発明の化合物がこれらの
疾患の治療に効果的であり得ることを示唆している[Sleep、21、131〜136
、1998;Sleep、8、871、878、2001]。
【0057】
1つの実施形態においては、本発明は、情動障害;うつ病;大うつ病性障害;産後うつ
病;双極性障害、アルツハイマー病、精神病、癌、加齢(age)もしくはパーキンソン病
に伴ううつ病;不安;全般性不安障害;社会不安障害;強迫性障害;パニック障害;パニ
ック発作;恐怖症;社会恐怖症;広場恐怖症;ストレス性尿失禁;嘔吐;IBS;摂食障
害;慢性疼痛;部分的応答者(partial responders);治療抵抗性うつ病;アルツハイマ
ー病;認識機能障害;ADHD;メランコリー;PTSD;顔面紅潮;睡眠時無呼吸;ア
ルコール、ニコチンもしくは炭水化物渇望;物質乱用;およびアルコールもしくは薬物乱
用から選択される疾患を治療する方法に関係し、その方法は、治療を必要としている患者
に治療的有効量の本発明の化合物を投与することを含む。1つの実施形態においては、上
で列挙されている疾患のうちのいずれかに関して治療される前述の患者は、最初にその疾
患の診断が下されている。
【0058】
一般的には抗うつ薬での治療、特にはSSRIでの治療は、性的機能不全を伴い、しば
しば治療の中止をもたらすことが周知である。SSRIで治療を受ける患者のうちの30
〜70%もの多くの患者が性機能の障害を訴えており[J.Clin.Psych.、6
6、844〜848、2005]、それらの障害は、リビドーの減退、オルガスムの遅延
、低減または欠落、性的興奮の減少および勃起機能不全を含む。臨床試験において、合計
で114人の被検者が本発明の化合物を投与された;これら114人の被検者のうち、僅
か1人の被検者のみが性的機能不全を訴えた。これらのデータは、本発明の化合物を用い
る臨床的介入が驚くべき程僅かな数の性的機能障害しか伴わないことを示唆している。
【0059】
上で述べられているように、本発明の化合物は、特に、慢性疼痛の治療に非常に適して
いる。慢性疼痛は、幻肢痛、神経障害性疼痛、糖尿病性ニューロパシー、ヘルペス後神経
痛(PHN)、手根管症候群(CTS)、HIVニューロパシー、複合性局所疼痛症候群
(CPRS)、三叉神経の神経痛/三叉神経痛/疼痛性チック、外科的介入(例えば術後
鎮痛薬)、糖尿病性血管症、膵島炎を伴った毛細血管抵抗もしくは糖尿病症状、アンギナ
に関連した疼痛、月経に関連した疼痛、癌に関連した疼痛、歯痛、頭痛、片頭痛、緊張性
疼痛、三叉神経痛、顎関節症候群、筋筋膜疼痛筋肉損傷(myofascial pain muscular inj
ury)、線維筋痛症候群、骨関節疼痛(変形性関節症)、慢性関節リウマチ、やけどを伴
う外傷に由来する慢性関節リウマチおよび浮腫、変形性関節症、骨粗鬆症、骨への転移も
しくは不明の理由による捻挫もしくは骨折の骨痛、痛風、結合組織炎、筋筋膜疼痛、胸郭
出口症候群、上背部痛もしくは下背部痛(ここで、この背部痛は系統的、局所的もしくは
主要な脊椎疾患(神経根障害)に由来する)、骨盤痛、心臓性胸痛、非心臓性胸痛、脊髄
損傷(SCI)関連疼痛、中枢性脳卒中後疼痛、癌ニューロパシー、エイズによる疼痛、
鎌状赤血球による疼痛、または老人性疼痛(geriatric pain)などの適応症を含む。
【0060】
実施例16で提示されているデータは、本発明の化合物が疼痛の治療に有用であり、ま
た、それらの化合物が鎮痛効果さえ有し得ることを示しており、更に、神経障害性疼痛に
関する動物モデルにおける研究がこの見解を確証している。
【0061】
本明細書で使用する場合、「治療的有効量」の化合物という用語は、上述の化合物を投
与することを含む治療的介入において、所与の疾患およびそれの合併症の臨床的徴候を治
し、緩和し、または部分的に停止させるのに充分な量を意味する。これを果たすのに適切
な量が「治療的有効量」として定義される。それぞれの目的での有効量は、その疾患また
は損傷の重症度、更にはその被検者の体重および全身状態に依存するであろう。適切な用
量の決定は、数値のマトリックスを構築し、そのマトリックスの種々の異なるポイントで
試験すること(これらはすべて、訓練を積んだ医師の通常の技術能力の範囲内である)に
より、ルーチン的な実験を用いて果たし得ることが理解されよう。
【0062】
本明細書で使用する場合、「治療」および「治療する」という用語は、ある状態、例え
ば疾患または障害などと闘うことを目的とした患者の管理およびケアを意味する。この用
語は、患者が患っている、ある与えられた状態に対する治療の全スペクトル、例えばそれ
らの症状もしくは合併症を緩和するため、その疾患、障害もしくは状態の進行を遅らせる
ため、それらの症状および合併症を緩和もしくは軽減するため、および/またはその疾患
、障害もしくは状態を治癒もしくは排除するため、更には、その状態を予防するための、
活性化合物の投与などを含めるべく意図されており、ここで、予防は、疾患、状態もしく
は障害と闘うことを目的とする患者の管理およびケアを意味するものと理解すべきであり
、それらの症状もしくは合併症の発現を防ぐための活性化合物の投与を含む。とはいえ、
予防的治療(防止を目的とした治療)および治療的処置(治すことを目的とした処置)は
本発明の2つの別個の態様である。治療されるべき患者は、好適には哺乳動物であり、特
には人間である。
【0063】
典型的には、本発明の治療は本発明の化合物を毎日投与することを含むであろう。これ
は、毎日1回の投与、または1日に2回の投与もしくはもっと頻繁な回数の投与さえをも
含み得る。
【0064】
1つの実施形態においては、本発明は、情動障害;うつ病;大うつ病性障害;産後うつ
病;双極性障害、アルツハイマー病、精神病、癌、加齢もしくはパーキンソン病に伴うう
つ病;不安;全般性不安障害;社会不安障害;強迫性障害;パニック障害;パニック発作
;恐怖症;社会恐怖症;広場恐怖症;ストレス性尿失禁;嘔吐;IBS;摂食障害;慢性
疼痛;部分的応答者;治療抵抗性うつ病;アルツハイマー病;認識機能障害;ADHD;
メランコリー;PTSD;顔面紅潮;睡眠時無呼吸;アルコール、ニコチンもしくは炭水
化物渇望;物質乱用;またはアルコールもしくは薬物乱用を治療するための薬剤を製造す
るための本発明の化合物の使用に関する。
【0065】
1つの実施形態においては、本発明は、情動障害、うつ病、大うつ病性障害、産後うつ
病、双極性障害、アルツハイマー病、精神病、癌、加齢もしくはパーキンソン病に伴うう
つ病、不安、全般性不安障害、社会不安障害、強迫性障害、パニック障害、パニック発作
、恐怖症、社会恐怖症、広場恐怖症、ストレス性尿失禁、嘔吐、IBS、摂食障害、慢性
疼痛、部分的応答者、治療抵抗性うつ病、アルツハイマー病、認識機能障害、ADHD、
メランコリー、PTSD、顔面紅潮、睡眠時無呼吸、アルコール、ニコチンもしくは炭水
化物渇望、物質乱用、またはアルコールもしくは薬物乱用から選択される疾患の治療にお
いて使用するための本発明の化合物に関する。
【0066】
ヒトの認識に及ぼす本発明の化合物の効果は数多くの仕方で評価することができる。そ
の効果は、健常なボランティアに本化合物を投与し、その後、認められているテスト、例
えばAuditory Verbal Learning Test(AVLT)、Wi
sconsin Card Sorting Test(WCST)などで認識能力を測
定する試験、または持続的注意[Psycopharmacol.、163、106〜1
10、2002;Psychiatry Clin.Neurosci.、60、70〜
76、2006]を測定する試験で評価することができる。その効果は、勿論、同じ種類
の試験を用いて、認識機能障害を患っている患者で評価されてもよい。代替的に、認知モ
デルが用いられてもよく、そこでは、健常なボランティアにおいて認識機能障害が誘発さ
れ、本発明の化合物の回復効果が測定される。認識機能障害は、例えばスコポラミン、睡
眠剥奪、アルコール、およびトリプトファン枯渇により誘発することができる。
【0067】
本発明の薬剤配合物は当分野における従来の方法で製造することができる。特に錠剤に
ついて言及すれば、錠剤は、活性成分を通常の佐剤および/または希釈剤と混合し、その
後、その混合物を通常の打錠機で圧縮することにより製造することができる。佐剤または
希釈剤の例は:無水カルシウム水素ホスファート、PVP、PVP−VAコポリマー、微
結晶性セルロース、ナトリウムデンプングリコラート、トウモロコシデンプン、マンニト
ール、ジャガイモデンプン、タルカム、マグネシウムステアラート、ゼラチン、ラクトー
ス、ゴムなどを含む。そのような目的で通常使用されるあらゆる他の佐剤または添加剤、
例えば着色剤、矯味矯臭剤、保存剤なども、それらが活性成分と適合することを条件とし
て、使用することができる。
【0068】
注射用液剤は、活性成分および可能な添加剤を注射用溶剤、好適には蒸留水の一部に溶
解し、その溶液を所望の体積に調節し、得られた溶液を滅菌して適切なアンプルまたはバ
イアル中に充填することにより製造することができる。当分野において普通に使用されて
いるあらゆる適切な添加剤、例えば等張化剤、保存剤、酸化防止剤などが加えられてよい
。
【0069】
本発明の医薬組成物またはこの発明に従って製造される医薬組成物はあらゆる適切な経
路で投与することができ、例えば、錠剤、カプセル剤、散剤、シロップ剤などの形態で経
口的に投与されてもよいし、または注射用溶液の形態で非経口的に投与されてもよい。そ
のような組成物を製造するために、当分野において周知の種々の方法が用いられてよく、
また、薬学的に許容可能なあらゆる担体、希釈剤、賦形剤または当分野において普通に使
用されている他の添加剤を使用することができる。
【0070】
都合よくは、本発明の化合物は、その化合物を約1mgから50mgまでの量で含有す
る単位投薬形態(unit dosage form)で投与される。上限は、5−HT3活性の濃度依存
性によって設定されるものと考えられる。合計1日量は、通常、約1〜20mgの範囲の
本発明の化合物、例えば約1mg〜10mgまで、約5〜10mg、約10〜20mgま
たは約10〜15mgなどの範囲の本発明の化合物である。特別な1日量として、5mg
、10mg、15mgまたは20mgを挙げることができる。
【0071】
本発明の化合物を含有する錠剤は、都合よくは、湿式造粒法により製造することができ
る。この方法を用いる場合、乾燥固体(活性成分、増量剤、結合剤など)がブレンドされ
、水または他の湿潤剤(例えばアルコール)で湿らされ、それらの湿気を帯びた固体で凝
集塊または顆粒が構築される。望ましい均一な粒径が達成されるまで湿式集塊形成が続け
られ、望ましい粒径になった後、その顆粒化された生成物が乾燥される。本発明の化合物
は、典型的には、高剪断ミキサー内において水と共にラクトース一水和物、トウモロコシ
デンプンおよびコポビドンと混合される。顆粒の形成後、これらの顆粒は、適切なシーブ
サイズを有する篩でふるいにかけられ、乾燥されてよい。次いで、結果として得られたこ
れらの乾燥した顆粒が微結晶性セルロース、クロスカルメロースナトリウムおよびマグネ
シウムステアラートと混合され、その後、錠剤が圧縮される。代替的に、本発明の化合物
の湿式造粒は、マンニトール、トウモロコシデンプンおよびコポビドンを用い、錠剤を圧
縮する前に、それらの顆粒を微結晶性セルロース、ナトリウムデンプングリコラートおよ
びマグネシウムステアラートと混合することにより達成されてもよい。代替的に、本発明
の化合物の湿式造粒は、無水カルシウム水素ホスファート、トウモロコシデンプンおよび
コポビドンを用い、錠剤を圧縮する前に、それらの顆粒を微結晶性セルロース、ナトリウ
ムデンプングリコラート(タイプA)、タルクおよびマグネシウムステアラートと混合す
ることにより達成されてもよい。コポビドンはPVP−VAコポリマーである。
【0072】
1つの実施形態においては、本発明の化合物は例えばベータ型の臭化水素酸塩であり、
適切な錠剤は以下のとおりのものから成っていてよい(示されている百分率はw/w%で
ある):
HBr塩:2〜20%
ラクトース一水和物:30〜50%
デンプン:15〜30%
コポビドン:3〜5%
微結晶性セルロース:15〜25%
クロスカルメロースナトリウム:2〜5%
Mgステアラート:0.5〜5%。
【0073】
特に、錠剤は以下のとおりのものから成っていてよい:
HBr塩:3〜4%
ラクトース一水和物:44〜46%
デンプン:22〜23%
コポビドン:3〜4%
微結晶性セルロース:20〜22%
クロスカルメロースナトリウム:3〜3.5%
Mgステアラート:0.5〜1%
または
HBr塩:15〜16%
ラクトース一水和物:35〜38%
デンプン:18〜20%
コポビドン:3〜4%
微結晶性セルロース:20〜22%
クロスカルメロースナトリウム:3〜3.5%
Mgステアラート:0.5〜1%
または
HBr塩:1〜2%
ラクトース一水和物:44〜46%
デンプン:20〜24%
コポビドン:3〜4%
微結晶性セルロース:22〜24%
クロスカルメロースナトリウム:3〜4%
Mgステアラート:0.5〜1%。
【0074】
1つの実施形態においては、本発明の化合物は例えばベータ型の臭化水素酸塩であり、
適切な錠剤は以下のとおりのものから成っていてよい:
HBr塩:2〜30%
マンニトール:25〜45%
トウモロコシデンプン:10〜20%
コポビドン:2〜4%
微結晶性セルロース:22〜27%
ナトリウムデンプングリコラート:4〜5%
Mgステアラート:0.25〜5%、例えば0.25〜2%など。
【0075】
特に、錠剤は以下のとおりのものから成っていてよい:
HBr塩:20〜22%
マンニトール:35〜36%
トウモロコシデンプン:10〜12%
コポビドン:2.5〜3%
微結晶性セルロース:24〜25%
ナトリウムデンプングリコラート:3〜4%
Mgステアラート:0.25〜1%
または
HBr塩:12〜13%
マンニトール:36〜37%
トウモロコシデンプン:18〜19%
コポビドン:3〜4%
微結晶性セルロース:24〜25%
ナトリウムデンプングリコラート:3〜4%
Mgステアラート:0.25〜1%
または
HBr塩:25〜27%
マンニトール:27〜29%
トウモロコシデンプン:13〜15%
コポビドン:3〜4%
微結晶性セルロース:24〜25%
ナトリウムデンプングリコラート:3〜5%
Mgステアラート:0.25〜1%
または
HBr塩:3〜4%
マンニトール:40〜42%
トウモロコシデンプン:20〜22%
コポビドン:3〜4%
微結晶性セルロース:26〜28%
ナトリウムデンプングリコラート:3〜5%
Mgステアラート:0.5%。
【0076】
1つの実施形態においては、本発明の化合物は臭化水素酸塩であり、適切な錠剤は以下
のとおりのものから成っていてよい:
HBr塩:3〜8%
無水カルシウム水素ホスファート:35〜45%
トウモロコシデンプン:15〜25%
コポビドン:2〜6%
微結晶性セルロース:20〜30%
ナトリウムデンプングリコラート:1〜3%
タルク:2〜6%
マグネシウムステアラート:0.5〜2%。
【0077】
特に、錠剤は以下のとおりのものから成っていてよい:
HBr塩:約5%
無水カルシウム水素ホスファート:約39%
トウモロコシデンプン:約20%
コポビドン:約3%
微結晶性セルロース:約25%
ナトリウムデンプングリコラート:約3%
タルク:約4%
マグネシウムステアラート:約1%。
【0078】
種々の異なる量の活性化合物を有する錠剤、例えば2.5mg、5mg、10mg、2
0mg、25mg、30mg、40mg、50mg、60mgまたは80mgの遊離塩基
に該当した錠剤などは、正しい量の本発明の化合物を適切なサイズの錠剤と組み合わせて
選ぶことにより得ることができる。
【0079】
本発明の化合物を含有した錠剤を製造するために使用される結晶のサイズは重要である
。もし結晶が小さすぎた場合には、それらの結晶が錠剤機のプランジャーにくっついてし
まう可能性がある。その一方で、それらの結晶は大きすぎてもいけない。腸内における溶
解速度は、結晶のサイズが大きくなると減少する。従って、もし結晶が大きすぎた場合に
は、本化合物のバイオアベイラビリティーが損なわれ得る。分位値(quantiles)、例え
ばD5%、D10%、D50%、D90%、D95%およびD98%などを用いて粒径分
布を表現することができる。本明細書で使用する場合、「粒径分布」という用語は、Sy
mpatec Helos装置において1バールの分散圧におけるレーザー回折により決
定したときの、等価球形粒子の直径の累積体積サイズ分布を意味する。
【0080】
1つの実施形態においては、本発明の化合物の結晶、特にベータ型の臭化水素酸塩の結
晶は、D98%:650〜680μm;D50%:230〜250μm;およびD5%:
40〜60μmに該当する粒径分布を有している。1つの更なる実施形態においては、そ
の粒径分布は、D98%:370〜390μm;D50%:100〜120μm;および
D5%:5〜15μmに該当する。尚も更なる1つの実施形態においては、その粒径分布
は、D98%:100〜125μm;D50%:15〜25μm;およびD5%:1〜3
μmに該当する。尚も更なる1つの実施形態においては、その粒径分布は、D98%:5
0〜70μm;D50%:3〜7μm;およびD5%:0.5〜2μmに該当する。
【0081】
本発明の遊離塩基は、国際公開第2003/029232号パンフレットに開示されて
いるようにして製造することができる。本発明の塩は、その遊離塩基を適切な溶剤中に溶
解し、関連する酸を加え、続いて沈殿させることにより製造することができる。沈殿は、
第二溶剤の添加、および/または蒸発、および/または冷却のいずれかにより果たすこと
ができる。代替的に、本発明の遊離塩基および最終的には本発明の化合物は、以下で説明
されているようにパラジウム触媒反応において合成することもできる。
【0082】
芳香族炭素−ヘテロ原子結合の形成は、芳香族求核置換または銅媒介Ullman反応
により達成することができる。より最近になって、パラジウムはそのような結合の形成、
特にはC−NおよびC−S結合の形成での強力な触媒であることが示されている(例えば
、米国特許第5,573,460号を参照のこと)。
【0083】
1つの実施形態においては、本発明は
【0084】
【化2】
【0085】
を製造するための方法を提供し、その方法は、化合物II
【0086】
【化3】
【0087】
[式中、R’は水素または一価の金属イオンを表す]を、溶剤、塩基、ならびにパラジウ
ムのソースおよびホスフィン配位子からなるパラジウム触媒の存在下において、60℃か
ら130℃までの間の温度で、式III
【0088】
【化4】
【0089】
[式中、X1およびX2は独立してハロゲンを表す]の化合物、および式IV
【0090】
【化5】
【0091】
[式中、Rは水素または保護基を表す]の化合物と反応させることを含む。
【0092】
1つの実施形態においては、この方法はサブプロセスに分けられ、そのサブプロセスで
は、化合物IIおよび化合物IIIが第一の反応において反応させられ、以下の式
【0093】
【化6】
【0094】
の化合物をもたらす。次いで、この化合物が、場合によっては適切な程度にまで精製され
た後、4−[2−(2,4−ジメチル−フェニルスルファニル)−フェニル]−ピペラジ
ンをもたらすべく化合物IVと反応させられる。
【0095】
ワンポット合成、即ち、すべての反応物が反応または方法の開始時に一緒に混合される
合成は、それらが有する固有の簡易性のため、特に有用である。その一方で、起こり得る
望ましくない副反応の数が劇的に増大し、これは、再度、望ましくない副次生成物の数お
よび/または量が増大し、それに対応して、望ましい生成物の収率が低下し得ることを意
味している。具体的には、本願方法の場合、ピペラジンは2個の窒素を有しており、潜在
的にそれぞれの窒素がC−N結合の形成に関与するということが観測され得る。驚くべき
ことに、本願方法は、純粋な化合物の高収率を維持しながら、ワンポット合成、即ち、最
初から化合物II、化合物IIIおよび化合物IVが混合される方法として実行できるこ
とが判明した。
【0096】
化合物IIはチオールまたは対応するチオラートである。労働衛生上の観点から判断す
ると、チオールに付随する臭気の問題を回避するため、チオラート、例えばLi+、Na
+またはK+チオラートなどを使用することが有益であり得る。とはいえ、1つの実施形
態においては、R’は水素である。
【0097】
化合物IIIは1,2−ジハロゲン活性化ベンゼンであり、それらのハロゲンはCl、
BrおよびIのいずれであってもよい。特には、化合物IIは1−ブロモ−2−ヨード−
ベンゼンまたは1,2−ジブロモ−ベンゼンである。
【0098】
本発明の方法において使用される溶剤は、反応温度範囲内、即ち、60〜130℃の沸
点を有する非プロトン性有機溶剤またはそのような溶剤の混合物から選択されてよい。典
型的には、その溶剤は、トルエン、キシレン、トリエチルアミン、トリブチルアミン、ジ
オキサン、N−メチルピロリドンから選択され、またはそれらの任意の混合物から選択さ
れる。特には、溶剤としてトルエンの名を挙げることができる。
【0099】
前記方法の中核を成すものは、それ無しでは反応が起こらないパラジウム触媒の使用で
ある。このパラジウム触媒は、パラジウム源とホスフィン配位子とからなっている。有用
なパラジウム源は、種々の異なる酸化状態、例えば0およびIIなどのパラジウムを含む
。本発明の方法において使用され得るパラジウム源の例は、Pd2dba3、Pddba
2およびPd(OAc)2である。dbaはジベンジリデンアセトンの略号である。特に
、Pddba2およびPd2dba3の名を挙げることができる。このパラジウム源は、
典型的には、0.1〜10モル%、例えば1〜10モル%など、例えば1〜5モル%など
の量で適用される。この出願全体を通じ、モル%は限定反応物(limiting reactant)に
関して算出される。
【0100】
単座および二座のどちらも、数多くのホスフィン配位子が公知である。有用なホスフィ
ン配位子は、ラセミ2,2’−ビス−ジフェニルホスファニル−[1,1’]ビナフタレ
ニル(rac−BINAP)、1,1’−ビス(ジフェニルホスフィノ)フェロセン(D
PPF)、ビス−(2−ジフェニルホスフィノフェニル)エーテル(DPEphos)、
トリ−t−ブチル−ホスフィン(Fu’s塩)、ビフェニル−2−イル−ジ−t−ブチル
−ホスフィン、ビフェニル−2−イル−ジシクロヘキシル−ホスフィン、(2’−ジシク
ロヘキシルホスファニル−ビフェニル−2−イル)−ジメチル−アミン、[2’−(ジ−
t−ブチル−ホスファニル)−ビフェニル−2−イル]−ジメチル−アミン、およびジシ
クロヘキシル−(2’,4’,6’−トリ−プロピル−ビフェニル−2−イル)−ホスフ
ァンを含む。更に、カルベン配位子、例えば1,3−ビス−(2,6−ジ−イソプロピル
−フェニル)−3H−イミダゾル−1−イウムなど;クロリドをホスフィン配位子の代わ
りに使用することもできる。1つの実施形態においては、そのホスフィン配位子はrac
−BINAP、DPPFまたはDPEphosであり、特にはrac−BINAPである
。このホスフィン配位子は、通常、0.1モル%から10モル%までの間、例えば1モル
%から5モル%までの間などで適用され、典型的には約1〜2モル%の量で適用される。
【0101】
pHを高めるために、塩基が反応混合物に加えられる。特には、NaOt−Bu、KO
t−BuおよびCs2CO3から選択された塩基が有用である。有機塩基、例えば1,8
−ジアザビシクロ[5.4.0]ウンデカ−7−エン(DBU)および1,4−ジアザビ
シクロ[2.2.2]オクタン(DABCO)なども同様に適用することができる。特に
、NaO(t−Bu)およびKO(t−Bu)の名を挙げることができる。典型的には、
この塩基は約1〜5当量の量で加えられ、例えば1〜3当量など、例えば2〜3当量など
の量で加えられる。
【0102】
化合物IVはピペラジン化合物である。ピペラジンは2個の窒素を有しており、そのう
ちの1個のみがC−N結合の形成に関与する。1つの実施形態においては、第二の窒素に
対する結合の形成は、モノ−保護ピペラジンを用いることにより、即ち、式中のRが保護
基である実施形態を用いることにより回避されている。多くの保護基が当分野において公
知であり、有用な例はboc、Bn、Cbz、C(=O)OおよびMeを含み、特にはb
ocである。Bnはベンジルの略号であり;bocはt−ブチルオキシカルボニルの略号
であり;cbzはベンジルオキシカルボニルの略号である。もし保護型のピペラジンがそ
れらの反応で使用される場合には、その保護基は以降のステップで取り除かれなければな
らず、典型的には酸水溶液を加えることにより除去される。メチルが保護基として使用さ
れる場合には、そのメチルは、カルバマートとの反応およびそれ以降のこの基の除去によ
り取り除かれてよい。
【0103】
驚くべきことに、第二の窒素への望ましくない結合の形成を伴うことなく、非保護型ピ
ペラジンも同様に使用され得ることが判明した。保護型のピペラジンおよび非保護型のピ
ペラジンは、種々の異なる溶剤中において異なる溶解度を有している;一例を挙げれば、
ピペラジンはトルエン中に事実上溶けないが、その一方で、bocで保護されたピペラジ
ンはトルエンに高度に可溶性である。通常は、適用された溶剤中にすべての反応物が容易
に溶けることが反応の成功にとっての必要条件であると期待されよう。それにもかかわら
ず、本発明の方法は、溶剤としてトルエンを用い、且つ、非保護型のピペラジン、即ち、
式中のRが水素である実施形態のピペラジンを用いて、高い収率で実施されることが判明
した。従って、1つの実施形態においては、溶剤はトルエンであり、化合物IVはピペラ
ジンである。1つの更なる実施形態においては、この条件の組み合わせがワンポット合成
で用いられる。
【0104】
1つの実施形態においては、方法を実施するための温度は、約80℃〜約120℃であ
る。
【0105】
1つの実施形態においては、1−[2−(2,4−ジメチル−フェニルスルファニル)
フェニル]−ピペラジンが以下のステップを含む方法で製造される:
a.1〜1.5当量の化合物II、IIIおよびIVをトルエン中に溶解または分散さ
せて、混合物Aを得るステップ;
b.場合によってはトルエン中に分散もしくは溶解または分散された、1〜2モル%の
Pddba2および1〜2モル%のrac−BINAPを2〜3当量のNaOt−Buと
共に混合物Aに加えて混合物Bを得、化合物IIおよびIIIが完全に変換されるまで、
典型的には5〜10時間、この混合物Bを約100℃に加熱するステップ;
c.化合物IVが完全に変換されるまで、典型的には16〜32時間、ステップbで得
られた混合物の温度を約120℃に高めるステップ;および
d.場合によって、化合物IIIが保護型のピペラジンであるときに、酸水溶液を加え
ることによりその保護基を取り除くステップ。
【0106】
場合によっては、上の一連の反応ステップに精製ステップが含められてよい。
【0107】
1つの実施形態においては、1〜1.5当量の2,4−ジメチル−チオール、1−ブロ
モ−2−ヨードベンゼン(または1,2−ジブロモ−ベンゼン)およびピペラジンがトル
エン中に分散され、続いて、トルエン中に分散された2〜5当量、例えば3当量などのN
aOt−Buならびに1〜2モル%のPd2dba3およびrac−BINAPを加えて
混合物を得、この混合物を2〜10時間、典型的には3〜5時間還流させて1−[2−(
2,4−ジメチル−フェニルスルファニル)−フェニル]−ピペラジンを得る。場合によ
っては、対応する臭化水素酸付加塩を得るため、この生成物を更にHBr水溶液と反応さ
せることもできる。
【0108】
1つの実施形態においては、2〜5当量のNaOt−Bu、2〜5当量のピペラジン、
0.2〜0.6モル%のPddba2および0.6〜1モル%のrac−BINAPをト
ルエン中に分散させて混合物A’を得、その混合物に約1当量の2−ブロモ−ヨードベン
ゼンを加えて混合物B’を得、その混合物に1当量の2,4−ジメチルチオフェノールを
加え、結果として生じた混合物を3〜7時間、例えば4〜6時間加熱して還流させ、1−
[2−(2,4−ジメチル−フェニルスルファニル)−フェニル]−ピペラジンを得る。
場合によっては、対応する臭化水素酸付加塩を得るため、この生成物を更にHBr水溶液
と反応させることもできる。
【0109】
ある状況においては、遊離塩基としてよりもむしろ、1−[2−(2,4−ジメチル−
フェニルスルファニル)−フェニル]−ピペラジンの酸付加塩を得る方が望ましい場合が
あり得る。酸付加塩は更なるプロセスステップにおいて得られてよく、そのステップでは
、得られた遊離塩基が適切な酸、例えばフマル酸、硫酸、塩酸または臭化水素酸などと反
応させられる。その酸は、反応混合物に直接的に加えられてもよいし、または、代替的に
、遊離塩基がそのようなステップの前に最初に何らかの適切な程度にまで精製されてもよ
い。もしも遊離塩基が乾燥化合物として単離されている場合には、酸との反応に先立って
その遊離塩基を溶液にするため、溶剤を使用することが必要になるであろう。1つの実施
形態においては、何らその遊離塩基の初期精製を伴うことなく、臭化水素酸水溶液が反応
混合物に直接的に加えられる。
【0110】
保護型のピペラジンが使用されている方法においては、上で説明されているように、酸
水溶液を加えることによって保護基を取り除かなければならない。1つの実施形態におい
ては、上述の酸水溶液は、2つの変換、即ち、保護型ピペラジンの脱保護および酸付加塩
の形成を達成することができるように選択されてよい。特に、臭化水素酸水溶液を用いて
、1つのプロセスステップで、保護型ピペラジンを脱保護し、且つ、臭化水素酸付加塩を
得ることができる。
【0111】
ここで述べられているすべての反応および反応混合物に対して、それらを不活性ガスで
パージすることまたはそれらを不活性ガスのブランケットの下で実行することが有利であ
り得るという考えが当てはまる。窒素は、安価で容易に入手可能な不活性ガスの例である
。
【0112】
出版物、特許出願および特許を含め、ここで引用されているすべての参考文献は、これ
をもって、参照により、それらの内容全体が、また、本明細書のどこか別の個所で為され
ている何らかの別個に与えられた特定の書類の組み込みに関係なく、それぞれの参考文献
が参照により組み込まれるべく個別的および具体的に指示されているのと同じ程度にまで
、および、その内容全体が本明細書において説明されているのと同じ程度にまで(法律に
より許容される最大限の程度にまで)本明細書に組み込まれる。
【0113】
本発明を説明する文脈における「a」、「an」および「the」という用語ならびに
同様な指示対象の使用は、本明細書で別な具合に指示されていない限り、または文脈によ
って明らかに否定されていない限り、単数形と複数形との両方をカバーするものと解釈す
べきである。例えば、「the compound(本化合物)」という語句は、別な具
合に指示されていない限り、本発明または特定の開述されている態様の様々な化合物を表
すものと理解すべきである。
【0114】
別な具合に指示されていない限り、本明細書で与えられているすべての正確な値は、対
応する大凡の値の代表である(例えば、特定のファクターまたは測量に関して与えられて
いるすべての正確な例証的値は、適切な個所では「約」に変更し、対応する大凡の測量も
与えているものと見なすことができる)。
【0115】
1つの構成要素または複数の構成要素に関する「含む(comprising)」、「有する(ha
ving)」、「含有する(including)」または「含む(containing)」などの用語を用い
た何らかの態様または本発明の態様についての本明細書での説明は、別な具合に述べられ
ていない限り、または文脈によって明らかに否定されていない限り、その特定の構成要素
または複数の特定の構成要素に関する「からなる」、「本質的に(そのような構成要素)
からなる」、または「(そのような構成要素)を実質的に含む」同様な態様または本発明
の態様に対する支持を与えるべく意図されている(例えば、特定の構成要素を含むものと
して本明細書で説明されている組成物は、別な具合に述べられていない限り、または文脈
によって明らかに否定されていない限り、その構成要素からなる組成物をも説明している
ものとして理解されるべきである)。
【実施例】
【0116】
分析方法
1H NMRスペクトルが500.13MHzにおいてBruker Avance
DRX500装置に記録される。溶剤としてジメチルスルホキシド(99.8%D)が使
用され、内部参照標準としてテトラメチルシラン(TMS)が使用される。
【0117】
融点が示差走査熱量測定法(DSC)を用いて測定される。その機器は、開始値として
融点を与えるべく5°/分で校正されたTA−InstrumentsのDSC−Q10
00である。約2mgのサンプルが窒素フロー下におけるゆるく閉じられたパン内におい
て5°/分で加熱される。
【0118】
乾燥された材料の溶剤/水含有量を見積もるために使用される熱重量分析(TGA)は
、TA−InstrumentsのTGA−Q500を用いて実施される。1〜10mg
のサンプルが窒素フロー下における開放されたパン内において10°/分で加熱される。
【0119】
粉末X線回折図形が、CuKα1放射線を用いるPANalytical X’Per
t PRO X−Ray Diffractometerで測定された。サンプルは、X
’celerator検出器を用いて2θ−レンジ5〜40°における反射モードで測定
された。
【0120】
実施例1:インビトロ受容体薬理学
ラットセロトニントランスポーター:IC50 5.3nM(5−HT取り込みの遮断
)
ヒトセロトニントランスポーター:IC50 40nM(5−HT取り込みの遮断)
ヒト5−HT1A受容体:部分的アゴニズムを有し、Ki 40nM(効力85%)
ラット5−HT3受容体:IC50 0.2nM(機能分析におけるアンタゴニズム)
ヒト5−HT3A受容体:IC50約20nM(機能分析におけるアンタゴニズム)。
より高い濃度では、前記化合物は、2.1μMのED50を有するアゴニスト活性を示す
。また、本発明の化合物は、インビトロ結合アッセイにおいて、ヒト5HT3受容体に対
して高い親和性も示した(Ki 4.5nM)。
【0121】
実施例2:認識作用
上で検討されているように、本発明の化合物はコリン作動系と相互作用し、また、イン
ビボモデルにおいて、以下の項目のうちの1つまたはそれ以上で効果をみることが期待さ
れよう。
【0122】
・5選択反応時間試験(5−CSRT)(連続的な注意に及ぼす効果を実証するのに有
用)
・空間Y字型迷路試験(短期記憶、長期記憶および作業記憶に及ぼす効果を実証するの
に有用)
・注意セットの移行モデル(attentional set shifting model)(実行機能、即ち、論
理的思考および問題解決に及ぼす効果を実証するのに有用)。
【0123】
実施例3a:遊離塩基の化合物Iの製造
10グラムの1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピペ
ラジンヒドロブロミドを100mlの3MのNaOHおよび100mlのエチルアセター
トの攪拌混合物で10分間処理した。その有機相を分離し、100mlの15%wtのN
aCl(aq)で洗浄し、MgSO4上で乾燥させ、濾過し、真空下で濃縮することによ
り、透明な無色の油状物として、7.7グラム(98%)の化合物Iの塩基を得た。
NMRは構造に適合する。
【0124】
実施例3b:結晶質塩基の化合物Iの製造
3.0グラムの1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピ
ペラジンの無色の油状物を70mlのアセトニトリルで処理し、加熱して還流させた。そ
のほとんど透明な溶液を濾過し、得られた透明な濾液を自然発生的に冷却すると、濾過後
間もなく沈殿が始まった。その混合物を室温(22℃)で2時間撹拌し、得られた生成物
を濾過により単離し、真空下(40℃)で一晩乾燥させた。2.7グラム(90%)の白
色の固体として結晶質塩基が単離された。NMRは構造に適合する。元素分析:72.4
0%のC、9.28%のN、7.58%のH(理論値:72.26%のC、9.36%の
N、7.42%のH)。
【0125】
実施例3c:結晶質塩基の化合物Iの特性描写
実施例3bのようにして製造されるこの塩基は結晶質である(XRPD)−図1参照。
この結晶質塩基は約117℃の融点を有している。また、この塩基は吸湿性ではなく、且
つ、水中における0.1mg/mlの溶解度を有している。
【0126】
実施例4a:化合物Iのアルファ型の臭化水素酸塩の製造
2.0グラムの1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピ
ペラジンを熱い30mlのエチルアセタート中に溶解し、0.73mlの48%wtのH
Br(aq)を加えた。この添加は濃厚なスラリーの形成をもたらし、適切に撹拌できる
ようにするため、更に10mlのエチルアセタートを加えた。そのスラリーを室温で1時
間撹拌した。濾過し、真空下(20℃)で一晩乾燥させることにより、白色の固体として
2.0グラムの生成物が得られた(80%)。NMRは構造に適合する。元素分析:57
.05%のC、7.18%のN、6.16%のH(1:1の塩に対する理論値:56.9
9%のC、7.39%のN、6.11%のH)。
【0127】
実施例4b:化合物Iのアルファ型のヒドロブロミドの特性描写
実施例4aのようにして製造されるこのアルファ型のヒドロブロミドは結晶質である(
XRPD)−図2参照。この結晶質ヒドロブロミドは約226℃の融点を有している。ま
た、このヒドロブロミドは高い相対湿度に晒されたときに約0.3%の水分を吸収し、且
つ、水中における2mg/mlの溶解度を有している。
【0128】
実施例4c:化合物Iのベータ型の臭化水素酸塩の製造
49.5グラムの1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]
ピペラジンの無色の油状物を500mlのエチルアセタート中に溶解し、18.5mlの
48%wtのHBr(aq)を加えた。この添加は濃厚なスラリーの形成をもたらし、そ
のスラリーを室温で一晩撹拌した。濾過し、真空下(50℃)で一晩乾燥させることによ
り、白色の固体として29.6グラムの生成物が得られた(47%)。
NMRは構造に適合する。元素分析:56.86%のC、7.35%のN、6.24%の
H(1:1の塩に対する理論値:56.99%のC、7.39%のN、6.11%のH)
。
【0129】
実施例4d:化合物Iのベータ型のヒドロブロミドの特性描写
実施例4cのようにして製造されるこのベータ型のヒドロブロミドは結晶質である(X
RPD)−図3参照。この結晶質ヒドロブロミドは約231℃の融点を有している。また
、このヒドロブロミドは高い相対湿度に晒されたときに約0.6%の水分を吸収し、且つ
、水中における1.2mg/mlの溶解度を有している。
【0130】
実施例4e:化合物Iのガンマ型の臭化水素酸塩の製造
実施例4aのようにして製造された1gの1−[2−(2,4−ジメチルフェニルスル
ファニル)−フェニル]ピペラジンヒドロブロミドを20mlの水に加え、85℃に加熱
した。その溶液はほとんど透明であった。1滴のHBrを加えることによりその溶液が透
明になった。曇り点が観測されるまでHBrを加えた。その溶液を室温にまで冷却し、乾
燥させた。NMRは構造に適合する。元素分析:56.63%のC、7.18%のN、6
.21%のH(1:1の塩に対する理論値:56.99%のC、7.39%のN、6.1
1%のH)。
【0131】
実施例4f:化合物Iのガンマ型のヒドロブロミドの特性描写
実施例6eのようにして製造されるこのヒドロブロミドは結晶質である(XRPD)−
図4参照。このDSC曲線は約100℃における幾つかの熱的事象:恐らくは結晶形の変
化;を示している。その後、このヒドロブロミドは約220℃で溶融する。この結晶質ヒ
ドロブロミドは高い相対湿度に晒されたときに約4.5%の水分を吸収し、また、室温で
30%のRHのときには約2%の水分が吸収される。
【0132】
実施例4g:化合物Iのヒドロブロミド水和物の製造
1.4グラムの1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピ
ペラジンの油状物を20mlの水に加え、60℃に加熱した。48%のHBrを用いてp
Hを1に調節した。その溶液を室温にまで冷却し、乾燥させた。NMRは構造に適合する
。元素分析:55.21%のC、7.16%のN、6.34%のH(1:1の塩半水和物
に対する理論値:55.68%のC、7.21%のN、6.23%のH)。
【0133】
実施例4h:化合物Iのヒドロブロミドの半水和物の特性描写
実施例4gのようにして製造されるこの水和物は結晶質である(XRPD)−図5参照
。水分の含有量は相対湿度に強く依存する。室温で95%のRHのときには、含水率は約
3.7%である。約100℃に加熱することにより脱水が起こる。
【0134】
実施例4i:化合物Iの臭化水素酸塩のエチルアセタート溶媒和物の製造
0.9グラムの1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピ
ペラジンの油状物を35mlのエチルアセタート中に溶解し、0.5mlの48%wtの
HBr(aq)を加えた。この添加は濃厚なスラリーの形成をもたらし、そのスラリーを
室温で一晩撹拌した。濾過し、30mlのジエチルエーテルで洗った後、真空下(50℃
)で一晩乾燥させることにより、1.0グラム(65%)の1−[2−(2,4−ジメチ
ルフェニルスルファニル)−フェニル]ピペラジンHBrEtOAc溶媒和物が得られた
。NMRは構造に適合する。元素分析:56.19%のC、6.60%のN、6.56%
のH(TGAおよびKFにより決定した際の8%のエチルアセタートおよび0.5%の水
に対して補正されたときの1:1の塩に対する理論値:56.51%のC、6.76%の
N、6.38%のH)。
【0135】
実施例4j:化合物Iのヒドロブロミドのエチルアセタート溶媒和物の特性描写
実施例4iのようにして製造されるこのエチルアセタート溶媒和物は結晶質である(X
RPD)−図6参照。そのバッチは、恐らくは乾燥が部分的に脱溶媒和を引き起こしたた
め、化合物Iの溶媒和物とアルファ型との混合物を含んでいる。この脱溶媒和は、10°
/分で加熱した場合、約75℃で始まる。脱溶媒和後、アルファ型が形成される。高い相
対湿度に晒されると、エチルアセタートが水で置換され、その後に湿度が下げられたとき
にその水分が放出される。結果として得られる固体は吸湿性であり、高い相対湿度のとき
には3.2%の水分を吸収する。
【0136】
実施例5a:化合物Iの塩酸塩の製造
1.0グラムの1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピ
ペラジンの油状物を、穏やかに加熱(30℃)しながら、20mlのエチルアセタート中
に溶解した。透明な溶液が得られたときに、ジエチルエーテル中における2MのHClの
溶液を、pHが約1〜2になるまでゆっくりと加えた。この添加中に、自然発生的な沈殿
が観測された。最後の添加後、その懸濁液を1時間撹拌し、その後、濾過し、真空下(4
0℃)で一晩乾燥させることにより、白色の沈殿物が単離された。1.1グラム(99%
)の1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピペラジンヒド
ロクロリドが単離された。
NMRは構造に適合する。元素分析:64.18%のC、8.25%のN、6.96%の
H(TGAにより決定した際の0.66%の水に対して補正されたときの1:1の塩に対
する理論値:64.13%のC、8.31%のN、6.95%のH)。
【0137】
実施例5b:化合物Iのヒドロクロリドの特性描写
実施例5aのようにして製造されるこのヒドロクロリドは結晶質である(XRPD)−
図7参照。この結晶質ヒドロクロリドは約236℃の融点を有している。また、このヒド
ロクロリドは高い相対湿度に晒されたときに約1.5%の水分を吸収し、且つ、水中にお
ける3mg/mlの溶解度を有している。
【0138】
実施例5c:化合物Iのヒドロクロリド一水和物の製造
11.9グラムの1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]
ピペラジンの油状物を、加熱しながら、100mlのエタノール中に溶解した。均質な溶
液が得られたときに行われた3.5mlの濃HCl(aq)の添加は、白色の固体の即座
の沈殿を引き起こした。得られた懸濁液を最初に5分間攪拌し、次いで、氷浴上で更に1
時間攪拌した後、濾過した。その白色の固体を100mlの新鮮な冷たいエタノール(−
18℃で2時間冷凍庫に置いておいたもの)、50mlのアセトンおよび最後に50ml
のジエチルエーテルを用いて洗った後、真空下(50℃)において一晩乾燥させた。5.
1グラム(38%)の1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル
]ピペラジンHClが単離された。
NMRは構造に適合する。元素分析:61.23%のC、7.91%のN、7.16%の
H(1:1の塩一水和物に対する理論値:61.26%のC、7.94%のN、7.14
%のH)。
【0139】
実施例5d:化合物Iのヒドロクロリド一水和物の特性描写
実施例5cのようにして製造されるこのヒドロクロリド一水和物は結晶質である(XR
PD)−図8参照。この結晶質の一水和物は約50℃で水分を失い始める。更なる加熱に
より幾つかの熱的事象、恐らくは転位が起こり、この一水和物は約230℃で溶融し、続
いて、約236℃で再結晶化および融解する。この結晶質一水和物は高い相対湿度に晒さ
れたときに更なる量の水分を吸収せず、また、この水和物に結合された水は、相対湿度が
室温で10%RH以下に下がるまで放出されない。この結晶質一水和物は、水中における
約2mg/mlの溶解度を有している。
【0140】
実施例6a:化合物Iのメシル酸塩の製造
1.0グラムの1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピ
ペラジンの油状物を、加熱(70℃)により、20mlのエチルアセタート中に溶解した
。透明な溶液が得られたときに、0.35グラムのメタンスルホン酸(1.1当量)をゆ
っくりと加えた。最後の添加後、その溶液を氷上で冷却し、ジエチルエーテルをゆっくり
と加えることにより、生成物の沈殿がもたらされた。その懸濁液を氷上で2時間撹拌した
後、生じた白色の沈殿物を濾過により単離し、真空下(40℃)において一晩乾燥させた
。1.1グラム(85%)の1−[2−(2,4−ジメチルフェニルスルファニル)−フ
ェニル]ピペラジンメシラートが単離された。NMRは構造に適合する。元素分析:57
.81%のC、6.81%のN、6.68%のH(1:1の塩に対する理論値:57.8
1%のC、7.10%のN、6.64%のH)。
【0141】
実施例6b:化合物Iのメシラートの特性描写
実施例7aのようにして製造されるこのメシラートは結晶質である(XRPD)−図9
参照。この結晶質メシラートは約163℃の融点を有している。また、このメシラートは
吸湿性である(80%の相対湿度に晒されたときに約8%の水分を吸収し、これにより、
水和された形態に変換される。吸収された水のうちの最後の6%は相対湿度が10%RH
以下になるまで放出されない。この結晶質メシラートは、水中における非常に高い溶解度
(>45mg/ml)を有している。
【0142】
実施例7a:化合物Iのフマラートの製造
50mlのメタノールおよび50mlのエチルアセタートの混合物中において、5.5
グラムの1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピペラジン
の油状物を加熱し、還流させた。その溶液を放置して冷却した僅か後に2.1グラムのフ
マル酸を加えることにより、発熱反応が起こり、白色の固体の沈殿がもたらされた。得ら
れた懸濁液を撹拌し、その間、室温にまで冷却させ、続いて、−18℃において冷凍庫内
で2時間冷却した。その白色の固体を濾過により収集し、20mlの冷たいエチルアセタ
ートで洗った後、真空下(50℃)において一晩乾燥させた。3.1グラム(44%)の
生成物が単離された。
【0143】
NMRは構造に適合する。元素分析:63.42%のC、6.64%のN、6.42%
のH(1:1の塩に対する理論値:63.74%のC、6.76%のN、6.32%のH
)。
【0144】
実施例7b:化合物Iのフマラートの特性描写
実施例7aのようにして製造されるこのフマラートは結晶質である(XRPD)−図1
0参照。この結晶質フマラートは約194℃の融点を有している。水中における溶解度は
0.4mg/mlである。
【0145】
実施例8a:化合物Iのマレアートの製造
2.5グラムの1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピ
ペラジンの油状物を50mlのエチルアセタート中に溶解し、60度に加熱した後、1.
1グラムのマレイン酸を加えた。その混合物を再び加熱して5分間還流させ、撹拌しなが
ら放置して室温にまで冷却した。冷却している間に沈殿が始まり、冷凍庫(−18℃)内
に4時間入れることにより終結された。その白色の固体を濾過により収集し、50mlの
ジエチルエーテルで洗った後、真空下(50℃)において一晩乾燥させた。これにより1
.3グラムの1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピペラ
ジンマレアート(38%)が得られ、このマレアートを還流時に40mlのエチルアセタ
ートおよび5mlのメタノールで処理することにより再結晶化した。その透明な溶液を室
温にまで冷却し、続いて、冷凍庫(−18℃)内で2時間冷却した後、濾過し、10ml
の冷たいエチルアセタートで2回洗浄し、その後、真空下(50℃)において2日間乾燥
させた。0.9グラム(69%)の1−[2−(2,4−ジメチルフェニルスルファニル
)−フェニル]ピペラジンマレアートが単離された。NMRは構造に適合する。元素分析
:63.57%のC、6.79%のN、6.39%のH(1:1の塩に対する理論値:6
3.74%のC、6.76%のN、6.32%のH)。
【0146】
実施例8b:化合物Iのマレアートの特性描写
実施例8aのようにして製造されるこのマレアートは結晶質である(XRPD)−図1
1参照。この結晶質フマラートは約152℃の融点を有している。水中における溶解度は
約1mg/mlである。
【0147】
実施例9a:化合物Iのメソ−タルトラートの製造
アセトン中における1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル
]ピペラジンの11.1mlの0.30M溶液を5mlのアセトン中に溶解された0.5
グラムのメソ−酒石酸で処理した。得られた混合物を室温で30分間撹拌し、その間に沈
殿が起った。濾過し、最初に5mlのアセトン次いで3mlのジエチルエーテルで洗うこ
とにより、白色の固体として生成物が得られ、その生成物を真空下(50℃)において一
晩乾燥させた。1.4グラム(93%)の1−[2−(2,4−ジメチルフェニルスルフ
ァニル)−フェニル]ピペラジンメソ−酒石酸が単離された。NMRは構造に適合する。
元素分析:58.58%のC、6.29%のN、6.40%のH(1:1の塩に対する理
論値:58.91%のC、6.25%のN、6.29%のH)。
【0148】
実施例9b:化合物Iのメソ−タルトラートの特性描写
実施例9aのようにして製造されるこのメソ−タルトラートは結晶質である(XRPD
)−図12参照。この結晶質のメソ−タルトラートは約164℃の融点を有している。水
中における溶解度は約0.7mg/mlである。
【0149】
実施例10a:化合物IのL−(+)−タルトラートの製造
アセトン中における1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル
]ピペラジンの11.1mlの0.30M溶液を5mlのアセトン中に溶解された0.5
グラムのL−(+)−酒石酸で処理した。得られた混合物を室温で30分間撹拌し、その
間に沈殿が起った。濾過し、最初に5mlのアセトン次いで3mlのジエチルエーテルで
洗うことにより、白色の固体として生成物が得られ、その生成物を真空下(50℃)にお
いて一晩乾燥させた。1.2グラム(81%)の1−[2−(2,4−ジメチルフェニル
スルファニル)−フェニル]ピペラジン(+)−酒石酸が単離された。NMRは構造に適
合する。元素分析:58.86%のC、6.30%のN、6.38%のH(1:1の塩に
対する理論値:58.91%のC、6.25%のN、6.29%のH)。
【0150】
実施例10b:化合物IのL−(+)−タルトラートの特性描写
実施例10aのようにして製造されるこのL−(+)−タルトラートは結晶質である(
XRPD)−図13参照。この結晶質のL−(+)−タルトラートは約171℃の融点を
有している。水中における溶解度は約0.4mg/mlである。
【0151】
実施例11a:化合物IのD−(−)−タルトラートの製造
アセトン中における1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル
]ピペラジンの11.1mlの0.30M溶液を5mlのアセトン中に溶解された0.5
グラムのD−(−)−酒石酸で処理した。得られた混合物を室温で30分間撹拌し、その
間に沈殿が起った。濾過し、最初に5mlのアセトン次いで3mlのジエチルエーテルで
洗うことにより、白色の固体として生成物が得られ、その生成物を真空下(50℃)にお
いて一晩乾燥させた。1.0グラム(68%)の1−[2−(2,4−ジメチルフェニル
スルファニル)−フェニル]ピペラジンD−(−)−酒石酸が単離された。NMRは構造
に適合する。元素分析:58.90%のC、6.26%のN、6.35%のH(1:1の
塩に対する理論値:58.91%のC、6.25%のN、6.29%のH)。
【0152】
実施例11b:化合物IのD−(−)−タルトラートの特性描写
実施例11aのようにして製造されるこのD−(+)−タルトラートは結晶質である(
XRPD)−図14参照。この結晶質のD−(−)−タルトラートは約175℃の融点を
有している。水中における溶解度は約0.4mg/mlである。
【0153】
実施例12a:化合物Iのスルファートの製造
アセトン中における1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル
]ピペラジンの11.1mlの0.30M溶液をH2SO4(aq)の2.2mlの3M
溶液で処理した。得られた混合物を室温で30分間撹拌し、次いで、氷浴上で更に4時間
撹拌すると沈殿が起こり、その沈殿が終結された。濾過し、最初に5mlのアセトン次い
で3mlのジエチルエーテルで洗うことにより、白色の固体として生成物が得られ、その
生成物を真空下(50℃)において一晩乾燥させた。0.51グラム(39%)の1−[
2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピペラジンスルファートが
単離された。NMRは構造に適合する。元素分析:54.53%のC、7.22%のN、
6.28%のH(1:1の塩に対する理論値:54.52%のC、7.07%のN、6.
10%のH)。
【0154】
実施例12b:化合物Iのスルファートの特性描写
実施例12aのようにして製造されるこのスルファートは結晶質である(XRPD)−
図15参照。この結晶質スルファートは約166℃の融点を有している。水中における溶
解度は約0.1mg/mlである。
【0155】
実施例13a:化合物Iのホスファートの製造
アセトン中における1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル
]ピペラジンの11.1mlの0.30M溶液を0.2mlの65%のH3PO4(aq
)で処理した。得られた混合物を室温で30分間撹拌し、その間に沈殿が起った。濾過し
、最初に5mlのアセトン次いで3mlのジエチルエーテルで洗うことにより、白色の固
体として生成物が得られ、その生成物を真空下(50℃)において一晩乾燥させた。1.
23グラム(94%)の1−[2−(2,4−ジメチルフェニルスルファニル)−フェニ
ル]ピペラジンホスファートが単離された。NMRは構造に適合する。元素分析:54.
21%のC、7.15%のN、6.43%のH(1:1の塩に対する理論値:54.53
%のC、7.07%のN、6.36%のH)。
【0156】
実施例13b:化合物Iのホスファートの特性描写
実施例13aのようにして製造されるこのホスファートは結晶質である(XRPD)−
図16参照。この結晶質ホスファートは約224℃の融点を有している。水中における溶
解度は約1mg/mlである。
【0157】
実施例14a:化合物Iのニトラートの製造
アセトン中における1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル
]ピペラジンの11.1mlの0.30M溶液を0.2mlの16.5MのHNO3(a
q)で処理した。得られた混合物を室温で30分間撹拌し、その間に沈殿が起った。濾過
し、最初に5mlのアセトン次いで3mlのジエチルエーテルで洗うことにより、白色の
固体として生成物が得られ、その生成物を真空下(50℃)において一晩乾燥させた。0
.87グラム(73%)の1−[2−(2,4−ジメチルフェニルスルファニル)−フェ
ニル]ピペラジンニトラートが単離された。NMRは構造に適合する。元素分析:59.
80%のC、11.67%のN、6.51%のH(1:1の塩に対する理論値:59.8
1%のC、11.63%のN、6.41%のH)。
【0158】
実施例14b:化合物Iのニトラートの特性描写
実施例14aのようにして製造されるこのニトラートは結晶質である(XRPD)−図
17参照。この結晶質ニトラートは溶融しないが、約160℃における発熱反応下におい
て分解する。水中における溶解度は約0.8mg/mlである。
【0159】
実施例15:錠剤
以下の実施例は、本発明の化合物を含む錠剤を製造し得る方法の代表的な例を示してい
る。すべての実施例でベータ型の臭化水素酸塩が使用された。
【0160】
実施例15a
63.55gの本臭化水素酸塩、923.65gのLactosum 350M、46
1.8gのトウモロコシデンプンおよび76.0gのKollidon VA64をDi
osna PP1高剪断混合機内において1000rpmのインペラー速度で2分間混合
した。次いで、インペラーの速度を800rpmに下げ、短時間の間に220gの水を加
えた。集塊形成(massing)を7分間実行し、結果として得られた顆粒を4000μmの
サイズの篩に通した。それらの顆粒を乾燥させ、710μmのサイズの篩に通した。結果
として得られた1383.5gの顆粒を400gのAvicel PH200および60
gのAc−Di−Solと混合した。15gのマグネシウムステアラートと混合すること
によるそのブレンドの潤滑化の後、得られた粉末ブレンドをタブレット成形機へ移した。
5mgの本遊離塩基に相当する目標含有量を有する錠剤を得るため、200mgの目標コ
ア重量および8mmの直径を有する錠剤が製造された。
【0161】
実施例15b
317.75gの本臭化水素酸塩、754.15gのLactosum 350M、3
77.1gのトウモロコシデンプンおよび76.0gのKollidon VA64をD
iosna PP1高剪断混合機内において1000rpmのインペラー速度で2分間混
合した。次いで、インペラーの速度を800rpmに下げ、短時間の間に210gの水を
加えた。集塊形成を7分間実行し、結果として得られた顆粒を4000μmのサイズの篩
に通した。それらの顆粒を乾燥させ、710μmのサイズの篩に通した。結果として得ら
れた1386.2gの顆粒を400gのAvicel PH200および60gのAc−
Di−Solと混合した。15gのマグネシウムステアラートと混合することによるその
ブレンドの潤滑化の後、得られた粉末ブレンドをタブレット成形機へ移した。25mgの
本遊離塩基に相当する目標含有量を有する錠剤を得るため、200mgの目標コア重量お
よび8mmの直径を有する錠剤が製造された。
【0162】
実施例15c
32.2gの本臭化水素酸塩、944.82gのLactosum 350M、472
.4gのトウモロコシデンプンおよび76.0gのKollidon VA64をDio
sna PP1高剪断混合機内において1000rpmのインペラー速度で2分間混合し
た。次いで、インペラーの速度を800rpmに下げ、短時間の間に220gの水を加え
た。集塊形成を7分間実行し、結果として得られた顆粒を4000μmのサイズの篩に通
した。それらの顆粒を乾燥させ、710μmの篩に通した。結果として得られた1317
gの顆粒を400gのAvicel PH200および60gのAc−Di−Solと混
合した。15gのマグネシウムステアラートと混合することによるそのブレンドの潤滑化
の後、得られた粉末ブレンドをタブレット成形機へ移した。2.5mgの本遊離塩基に相
当する目標含有量を有する錠剤を得るため、208mgの目標コア重量および8mmの直
径を有する錠剤が製造された。
【0163】
実施例15d
540.85gの本臭化水素酸塩、953.00gのPearlitol 50C、2
96.22gのトウモロコシデンプンおよび70.5gのKollidon VA64を
Aeromatic−Fielder PMA1高剪断混合機内において1000rpm
のインペラー速度で2分間混合した。次いで、そのインペラー速度を800rpmに下げ
、短時間の間に241.87gの水を加えた。集塊形成を7分間実行し、結果として得ら
れた顆粒を4000μmのサイズの篩に通した。それらの顆粒を乾燥させ、710μmの
サイズの篩に通した。結果として得られた1500gの顆粒を531.91gのAvic
el PH200および85.11gのPrimojelと混合した。10.64gのマ
グネシウムステアラートと混合することによるそのブレンドの潤滑化の後、得られた粉末
ブレンドをタブレット成形機へ移した。25mgの本遊離塩基に相当する目標含有量を有
する錠剤を得るため、125mgの目標コア重量および6mmの直径を有する錠剤が製造
された。
【0164】
実施例15e
270.45gの本臭化水素酸塩、772.0gのPearlitol 50C、38
6.41gのトウモロコシデンプンおよび70.5gのKollidon VA64をA
eromatic−Fielder PMA1高剪断混合機内において1000rpmの
インペラー速度で2分間混合した。次いで、そのインペラー速度を800rpmに下げ、
短時間の間に195gの水を加えた。集塊形成を5.5分間実行し、結果として得られた
顆粒を4000μmのサイズの篩に通した。それらの顆粒を乾燥させ、710μmのサイ
ズの篩に通した。結果として得られた1200.3gの顆粒を425.5gのAvice
l PH200および68.09gのPrimojelと混合した。8.8gのマグネシ
ウムステアラートと混合することによるそのブレンドの潤滑化の後、得られた粉末ブレン
ドをタブレット成形機へ移した。10mgの本遊離塩基に相当する目標含有量を有する錠
剤を得るため、100の目標コア重量および6mmの直径を有する錠剤が製造された。
【0165】
実施例15f
504.85gの本遊離塩基、552.95gのPearlitol 50C、276
.53gのトウモロコシデンプンおよび65.7gのKollidon VA64をAe
romatic−Fielder PMA1高剪断混合機内において1000rpmのイ
ンペラー速度で2分間混合した。次いで、そのインペラー速度を800rpmに下げ、短
時間の間に182gの水を加えた。集塊形成を5.5分間実行し、結果として得られた顆
粒を4000μmのサイズの篩に通した。それらの顆粒を乾燥させ、710μmのサイズ
の篩に通した。結果として得られた1250.7gの顆粒を443.31gのAvice
l PH200および70.8gのPrimojelと混合した。8.92gのマグネシ
ウムステアラートと混合することによるそのブレンドの潤滑化の後、得られた粉末ブレン
ドをタブレット成形機へ移した。50mgの本遊離塩基に相当する目標含有量を有する錠
剤を得るため、250mgの目標コア重量および8mmの直径を有する錠剤が製造された
。
【0166】
実施例15g
135.23gの本臭化水素酸塩、863.2gのPearlitol 50C、43
2.69gのトウモロコシデンプンおよび70.66gのKollidon VA64を
Aeromatic−Fielder PMA1高剪断混合機内において1000rpm
のインペラー速度で2分間混合した。次いで、そのインペラー速度を800rpmに下げ
、短時間の間に195gの水を加えた。集塊形成を5.5分間実行し、結果として得られ
た顆粒を4000μmのサイズの篩に通した。それらの顆粒を乾燥させ、710μmのサ
イズの篩に通した。結果として得られた1200gの顆粒を425.28gのAvice
l PH200および68.2gのPrimojelと混合した。8.58gのマグネシ
ウムステアラートと混合することによるそのブレンドの潤滑化の後、得られた粉末ブレン
ドをタブレット成形機へ移した。5mgの本遊離塩基に相当する目標含有量を有する錠剤
を得るため、100mgの目標コア重量および6mmの直径を有する錠剤が製造された。
【0167】
実施例15h
67.6gの本臭化水素酸塩、908.0gのPearlitol 50C、453.
9gのトウモロコシデンプンおよび70.51gのKollidon VA64をDio
sna PP1高剪断混合機内において1000rpmのインペラー速度で2分間混合し
た。次いで、そのインペラー速度を800rpmに下げ、短時間の間に195gの水を加
えた。集塊形成を5.5分間実行し、結果として得られた顆粒を4000μmのサイズの
篩に通した。それらの顆粒を乾燥させ、710μmのサイズの篩に通した。結果として得
られた1325gの顆粒を531.91gのAvicel PH200および85.11
gのPrimojelと混合した。10.64gのマグネシウムステアラートと混合する
ことによるそのブレンドの潤滑化の後、得られた粉末ブレンドをタブレット成形機へ移し
た。5mgの本遊離塩基に相当する目標含有量を有する錠剤を得るため、207.8mg
の目標コア重量および7mmの直径を有する錠剤が製造された。
【0168】
実施例15i
2290.1gの本臭化水素酸塩、17568gの無水カルシウム水素ホスファートお
よび8783gのトウモロコシデンプンならびに1510gのコポビドンをAeroma
tic−Fielder PMA100高剪断混合機内において200rpmのインペラ
ー速度で3分間混合した。次いで、150rpmのインペラー速度における2分間の間に
5130gの水を加えた。集塊形成を15分間実行し、結果として得られた顆粒を、9.
525mmのサイズのスクリーンを備えた、約2700rpmで作動する円錐型ミルに通
した。それらの顆粒を乾燥させ、2.388mmのサイズのスクリーンを備えた、約15
00rpmで作動する円錐型ミルに通した。結果として得られた28747gの顆粒を1
1250gの微結晶性セルロース、1350gのナトリウムデンプングリコラート(タイ
プA)および1800gのタルクと混合した。450gのマグネシウムステアラートと混
合することによるそのブレンドの潤滑化の後、得られた粉末ブレンドをタブレット成形機
へ移した。5mgの本遊離塩基に相当する本臭化水素酸塩の目標含有量を有する錠剤を得
るため、125mgの目標コア重量および6mmの直径を有する錠剤が製造された。更に
、10mgの本遊離塩基に相当する本臭化水素酸塩の目標含有量を有する錠剤を得るため
、250mgの目標コア重量および8mmの直径を有する錠剤が製造された。
【0169】
実施例16:マウスでの皮内ホルマリン試験における疼痛効果(pain effects)
このモデルにおいては、マウスが左後足にホルマリン(4.5%、20μl)の注入を
受ける。このホルマリン注射により引き起こされた刺激は特徴的な二相性の行動応答を誘
発し、これは損傷を負った足をなめるのに費やした時間の量により定量化される。第一相
(約0〜10分)は直接的な化学刺激および侵害受容を表し、一方、第二相(約20〜3
0分)は神経障害性起源の痛みを表すものと考えられる。これら2つの相は鎮静期によっ
て分離されており、この鎮静期には行動が正常に戻る。疼痛性刺激を低減するための試験
化合物の有効性は、これら2つの相での損傷を負った足をなめるのに費やした時間の量を
計時することにより評価される。
【0170】
本発明の化合物は第二相における疼痛スコアの有意な低減を示し(図18b)、これは
、神経障害性起源の疼痛に対して効力を有することを示したものである。その上、本発明
の化合物は第一相におけるスコアの有意な低減も示し(図18a)、これは、最高の用量
において、より高い鎮痛作用を有することを示したものである。要約すると、これらの結
果は、本発明の化合物が疼痛性障害の治療に効果的である可能性が高いことを指示してい
る。
【0171】
実施例17
【0172】
【化7】
【0173】
20gの2−ブロモヨードベンゼン(71mmol)および9.8gの2,4−ジメチ
ルチオフェノール(71mmol)を100mlのトルエン中に溶解した。324mgの
Pd2dba3(0.35mmol;1mol%)および381mgのDPEPhos(
0.71mmol;1mol%)の前に、その溶液が窒素でパージされた。その反応混合
物を5分間攪拌し、その間に、色が暗赤色からオレンジ色に変わった。8.7gのKOB
ut(78mmol)が加えられ、直ちに不均一な混合物が形成された。その懸濁液を窒
素下において100℃に加熱した。1時間後、その混合物を0℃に冷却し、2時間撹拌し
た後、その混合物をセライトのパッドを通じて濾過した。得られた濾過ケーキを2×50
mlのトルエンで洗浄し、それらを合わせた濾液を蒸発させることにより、21gのオレ
ンジ色−赤色調の油状物が得られ(99%の収率)、この油状物は、HPLCおよびGC
−MSにより>96%の純度であることが判明した。
【0174】
実施例18
【0175】
【化8】
【0176】
機械式のスターラーを伴う1L用の三つ口丸形ボトルに500mlのトルエンを入れ、
203mgのPddba2(0.35mmol;0.1mol%)および760mgのD
PEPhos(1.5mmol;0.4mol%)を加えた。その暗赤色の溶液を窒素で
5分間パージした後、100gの2−ブロモヨードベンゼン(353mmol)および4
8.9gの2,4−ジメチルチオフェノール(353mmol)の添加が行われた。43
.6gのKOBut(389mmol)の添加は発熱反応を引き起こし、温度を20℃か
ら36℃に高め、それと同時に、不均一な混合物の形成をもたらした。その懸濁液を窒素
下において100℃に加熱した。7時間後、その混合物を0℃に冷却し、2時間撹拌した
後、セライトのパッドを通じてその混合物を濾過した。得られた濾過ケーキを2×200
mlのトルエンで洗浄し、それらを合わせた濾液を蒸発させることにより、104gのオ
レンジ色の油状物が得られ(105%の収率)、この油状物はHPLCにより97%の純
度であることが判明し、また、NMRにより望ましい構造であることが確認された。この
油状物は室温で静置している間に固化した。
【0177】
実施例19
【0178】
【化9】
【0179】
50mlの乾燥トルエン中における10グラムの1−(2−ブロモ−フェニルスルファ
ニル)−2,4−ジメチル−ベンゼン(34mmol)の溶液に7グラムのboc−ピペ
ラジン(38mmol)を加え、窒素で5分間脱気し、312mgのPd2dba3(2
mol%)および637mgのrac−BINAP(3mol%)を加え、更に5分間脱
気した後、3.9グラムのButONa(41mmol)を加え、80℃に15時間加熱
した。その反応混合物を室温にまで冷却し、20mlの15%のブラインで2回抽出し、
Na2SO4上で乾燥させ、活性炭を加え、15分間還流させ、セライトを通じて濾過し
、蒸発させることにより、NMRで決定した際に95%の純度を有する、14.2グラム
の褐色がかった油状物(4−[2−(2,4−ジメチル−フェニルスルファニル)−フェ
ニル]−BOC−ピペラジン)が得られた。この粗製油状物を200mlのMeOHおよ
び20mlの6MのHCl(aq.)中に溶解し、1時間還流させた後、HPLCは完全
な脱保護を示した。室温に冷却した後、メタノールを回転蒸発器での真空により除去し、
20mlの濃NaOH(pHは13〜14であった)を加え、その後、得られた混合物を
100mlのEtOAcとともに15分間攪拌した。その有機相を収集し、30mlの1
5%のブラインで2回抽出し、Na2SO4上で乾燥させ、30mlのMeOH中におけ
る5.2gのフマル酸(44mmol)を加えた。加熱して還流させている間に均一な溶
液が形成され、更なる加熱または冷却のどちらかの間に、その溶液から急速な沈殿が生じ
る。その沈殿物を収集し、20mlのEtOAcおよび20mlのアセトンで洗浄し、真
空下で乾燥させることにより、66%の全収率で、白色の粉末として9.3グラムの1−
[2−(2,4−ジメチル−フェニルスルファニル)−フェニル]−ピペラジンフマラー
ト(22mmol)が得られ、このフマラートは、LC−MSにより99.5%の純度を
有していた。
【0180】
実施例20
【0181】
【化10】
【0182】
100gの1,2−ジブロモベンゼン(424mmol)および58.6gの2,4−
ジメチルチオフェノール(424mmol)を800mlのトルエン中に溶解する。4.
6gのPddba2(8mmol;2mol%)および13.1gのrac−BINAP
(21mmol;5mol%)の前に、その溶液が窒素でパージされる。反応混合物を5
分間攪拌し、その間に、色が暗赤色からオレンジ色に変わる。61gのNaOBut(6
36mmol)および200mlのトルエンが加えられ、直ちに不均一な混合物が形成さ
れた。その懸濁液を窒素下において80℃に加熱した。10時間後、その混合物を60℃
に冷却した後、500mlのトルエン中における102.6gのboc−ピペラジン(5
51mmol)および別の61gのNaOBut(636mmol)を加える。その反応
混合物を窒素でパージした後、別な部の4.6gのPddba2(8mmol;2mol
%)および13.1gのrac−BINAP(21mmol;5mol%)を加えた。今
度は、更に6時間またはHPLCが完全な変換を示すまで、その混合物を加熱して還流さ
せた(110℃)。得られた反応混合物を氷上で2時間冷却した後、その混合物をセライ
トのパッドを通じて濾過した。得られた濾過ケーキを2×200mlのトルエンで洗浄し
、それらを合わせた濾液を蒸発させることにより、242gの赤色の油状物が得られる。
この油状物を1000mlのMeOH中に溶解し、115mlの48wt%のHBr(a
q.)をゆっくりと加え、続いて、加熱して2時間還流させた後、HPLCにより完全な
脱保護が検出された。その混合物を冷却し、1000mlのEtOAcを加え、蒸発によ
りMeOHを除去した。1000mlのEt2Oの添加は沈殿を引き起こした。室温で2
時間撹拌し続けた後、そのスラリーを冷凍庫内で一晩放置した(−18℃)。濾過し、2
00mlのEt2Oで2回洗浄し、40℃において真空下で乾燥させることにより、17
2gの褐色がかった固体が得られた。その褐色がかった固体を1500mlの沸騰したH
2Oで1時間処理した後、更に2時間かけて室温にまで冷却した。濾過し、40℃におい
て真空下で一晩乾燥させることにより、98gの4−[2−(2,4−ジメチル−フェニ
ルスルファニル)−フェニル]−ピペラジンヒドロブロミド(61%)が得られた。
【0183】
実施例21
【0184】
【化11】
【0185】
102gの2−ブロモ−ヨードベンゼン(362mmol)および50gの2,4−ジ
メチルチオフェノール(362mmol)を1000mlのトルエン中に溶解する。この
溶液に81gのBOC−ピペラジン(434mmol)を加え、続いて、2.08gのP
ddba2(1mol%)および4.51gのrac−BINAP(2mol%)を加え
た。この混合物を窒素で5分間パージした後、300mlのトルエン中における87gの
NaOBut(905mmol)のスラリーを加えた。得られた懸濁液を窒素下において
一晩100℃に加熱した。GCMS分析は中間生成物(1−(2−ブロモ−フェニルスル
ファニル)−2,4−ジメチル−ベンゼン)への完全な変換を示し、その温度を上昇させ
て更に24時間還流させた(120℃)。HPLC分析は中間体(1−BOC−4−[2
−(2,4−ジメチル−フェニルスルファニル)−フェニル]−ピペラジン)への完全な
変換を示した。その反応混合物を氷上で1時間冷却した後、その混合物を濾過した。得ら
れた濾過ケーキを2×200mlのトルエンで洗浄し、それらを合わせた濾液に80ml
の48wt%のHBr(aq.)を加え、続いて、加熱して18時間還流させた後、HP
LCにより完全な脱保護が検出された。得られた混合物を氷上で2時間冷却し、濾過した
。その褐色がかった固体を、活性炭(25g)とともに、1000mlの沸騰したH2O
中に1時間溶解し、熱いうちに濾過し、放置して冷却した。生じた沈殿物を濾過により収
集し、40℃において真空下で一晩乾燥させることにより、白色の固体として49gの4
−[2−(2,4−ジメチル−フェニルスルファニル)−フェニル]−ピペラジンヒドロ
ブロミド(36%)が得られた。
【0186】
実施例22
【0187】
【化12】
【0188】
機械式のスターラーを伴う1L用の三つ口丸形ボトルに500mlのトルエンを入れ、
809mgのPd2dba3(0.88mmol;0.5mol%)および952mgの
DPEPhos(1.77mmol;0.5mol%)を加えた。その暗赤色の溶液を窒
素で5分間パージした後、100gの2−ブロモヨードベンゼン(353mmol)およ
び48.9gの2,4−ジメチルチオフェノール(353mmol)の添加が行われた。
43.6gのKOBut(389mmol)の添加は発熱反応を引き起こし、温度を20
℃から42℃に高め、それと同時に、不均一な混合物の形成がもたらされ、また、その色
が暗赤色からオレンジ色/褐色がかった色に変わった。その懸濁液を窒素下において10
0℃に加熱した。僅か20分後、HPLCは1−(2−ブロモ−フェニルスルファニル)
−2,4−ジメチル−ベンゼンへの完全な変換を示した。その混合物を40℃に冷却し、
600mlの15wt%のNaClを加え、5分間攪拌した。その有機相を分離し、水性
相を2×100mlのトルエンで洗った。それらを合わせた有機相を100mlの2Mの
HCl(aq.)、100mlの15wt%のNaClで洗浄し、Na2SO4上で乾燥
させ、活性炭(10g)とともに15分間還流させ、2回濾過し、蒸発させることにより
、107.3gのオレンジ色−赤色の油状物が得られ(103%)、この油状物はHPL
Cにより98%の純度であることが判明した。
【0189】
500mlの乾燥トルエン中における90グラムの上述のオレンジ色−赤色の油状物(
307mmol)の溶液に57グラムのboc−ピペラジン(307mmol)を加え、
窒素で5分間脱気し、1.4gのPd2dba3(1.53mmol;0.5mol%)
および2.9gのrac−BINAP(4.6mmol;1.5mol%)を加え、更に
2分間脱気した後、35.4グラムのButONa(368mmol)を加え、80℃に
18時間加熱した。HPLCは完全な変換を示し、その反応混合物を室温にまで冷却した
後、濾過し、得られた濾過ケーキを2×100mlのトルエンで洗った。それらを合わせ
た濾液を2×150mlの15wt%のNaClで2回抽出し、Na2SO4上で乾燥さ
せ、活性炭を加え、30分間還流させ、2回濾過し、蒸発させることにより、140.7
グラムの褐色がかった油状物(4−[2−(2,4−ジメチル−フェニルスルファニル)
−フェニル]−BOC−ピペラジン)が得られた。この粗製油状物を300mlのMeO
Hおよび200mlの6MのHCl(aq.)中に溶解し、1時間還流させた後、HPL
Cは完全な脱保護を示した。室温に冷却した後、メタノールを回転蒸発器での真空により
除去し、200mlの濃NaOH(pHは13〜14であった)を加え、その後、得られ
た混合物を1000mlのEtOAcとともに15分間攪拌した。その有機相を収集し、
300mlの15wt%のブラインで抽出し、Na2SO4上で乾燥させ、300mlの
MeOH中における46.3gのフマル酸(399mmol)の溶液に加えた。その混合
物を加熱して還流させ、室温にまで冷却し、その後、冷凍庫内で一晩放置した(−18℃
)。生じた沈殿物を収集し、100mlのEtOAcおよび100mlのアセトンで洗浄
し、真空下(50℃)で乾燥させることにより、81%の全収率で、白色の粉末として1
03.2gの1−[2−(2,4−ジメチル−フェニルスルファニル)−フェニル]−ピ
ペラジンフマラート(249mmol)が得られ、このフマラートは、LC−MSにより
99%の純度を有していた。このフマラートを、EtOAc/H2O/濃NaOHを用い
て、遊離塩基(1−[2−(2,4−ジメチル−フェニルスルファニル)−フェニル]−
ピペラジン)に変換し、その有機相をブラインで洗浄し、Na2SO4上で乾燥させ、濾
過し、その濾液に34mlの48wt%のHBr(aq.)を加えることにより、白色の
固体の沈殿が引き起こされた。その固体を収集し、1000mlの沸騰したH2Oで処理
し、これを室温にまで冷却するとスラリーが形成された。この最終生成物(1−[2−(
2,4−ジメチル−フェニルスルファニル)−フェニル]−ピペラジンヒドロブロミド)
を濾過により収集し、真空下(50℃)において乾燥させることにより、83gの白色の
粉末(71%の全収率)が得られた[CHN(teo.)56.99;6.11;7.3
9;CHN(実測値)57.11;6.15;7.35]。
【0190】
実施例23
【0191】
【化13】
【0192】
815gのNaOBut(8.48mol)、844gのピペラジン(9.8mol)
、6.6gのPd(dba)2(11.48mmol)および13.6gのrac−BI
NAP(21.84mmol)を4Lのトルエンとともに50分間攪拌した。その後、8
40gの2−ブロモ−ヨードベンゼン(2.97mol)を1.5Lのトルエンとともに
加え、30分間攪拌し続け、最後に、390.8gの2,4−ジメチルチオフェノール(
2.83mmol)を1.5Lのトルエンとともに加えた。この懸濁液を加熱して還流さ
せ、5時間還流し続けた。その反応混合物を一晩冷ました。2Lの水を加え、1時間撹拌
した後、濾過助剤を通じてその混合物を濾過した。その後、得られた濾液を3×1Lのブ
ラインで洗った。この後、それらを合わせた水性相を600mlのトルエンで抽出した。
この後、それらを合わせたトルエン相を70℃に加熱し、続いて、329.2mlの48
wt%のHBr(aq.)および164.6mlの水を加えた。得られた混合物を室温に
まで一晩冷却した。その最終生成物(1−[2−(2,4−ジメチル−フェニルスルファ
ニル)−フェニル]−ピペラジンヒドロブロミド)を濾過により収集し、真空下(60℃
)において乾燥させることにより、895g(84%の収率)の生成物が得られた。
【0193】
実施例24
【0194】
【化14】
【0195】
40.76gのNaOBut(424.1mol)、0.33gのPddba2(0.
57mmol)および0.68gのrac−BINAP(1.09mmol)を200m
lのトルエンとともに攪拌した。その後、42gの2−ブロモ−ヨードベンゼン(362
mmol)および19.54gの2,4−ジメチルチオフェノール(362mmol)を
50mlのトルエンとともに加えた。この懸濁液を加熱して還流させ、一晩還流し続けた
。HPLC分析は、中間生成物(1−(2−ブロモ−フェニルスルファニル)−2,4−
ジメチル−ベンゼン)への完全な変換を示した。その反応混合物を室温にまで冷却し、濾
過助剤を通じて濾過した。その濾液を40.76gのNaOBut(424.1mmol
)、42.2gのピペラジン(489.9mmol)、0.33gのPddba2(0.
57mmol)および0.68gのrac−BINAP(1.09mmol)の混合物に
加え、加熱して2時間還流させた。その反応混合物を一晩冷ました。100mlの水を加
え、その水性相を分離して取り除いた。濾過助剤を通じてその有機相を濾過し、その後、
得られた濾液を3×80mlのブラインで洗った。この後、それらを合わせた水性相を5
0mlのトルエンで抽出した。その後、それらを合わせたトルエン相を70℃に加熱し、
続いて、16.5mlの48wt%のHBr(aq.)および8.25mlの水を加えた
。得られた混合物を室温にまで一晩冷却した。その最終生成物(1−[2−(2,4−ジ
メチル−フェニルスルファニル)−フェニル]−ピペラジンヒドロブロミド)を濾過によ
り収集し、真空下(60℃)において乾燥させることにより、40.18gのオフ・ホワ
イトの粉末が得られた(75%の収率)。
【0196】
実施例25
【0197】
【化15】
【0198】
40.76gのNaOBut(424.1mmol)、0.33gのPddba2(0
.57mmol)および0.68gのrac−BINAP(1.09mmol)を200
mlのトルエンとともに攪拌した。その後、42gの2−ブロモ−ヨードベンゼン(14
8.5mmol)および19.54gの2,4−ジメチルチオフェノール(141.4m
mol)を50mlのトルエンとともに加えた。この懸濁液を加熱して還流させ、一晩還
流し続けた。HPLC分析は、中間生成物(1−(2−ブロモ−フェニルスルファニル)
−2,4−ジメチル−ベンゼン)への完全な変換を示した。反応を50℃に冷却し、42
.2gのピペラジン(489.9mmol)を100mlのトルエンとともに加えた。得
られた混合物を加熱して4時間還流させた。その反応混合物を室温にまで一晩冷却した。
100mlの水を加え、濾過助剤を通じてその反応混合物を濾過した。その後、得られた
濾過ケーキを50mlのトルエンで洗った。
【0199】
その水性相を分離して取り除いた後、その有機相を3×25mlのブラインおよび25
mlの水で洗った。この後、それらを合わせた水性相を30mlのトルエンで抽出した。
その後、それらを合わせたトルエン相を70℃に加熱し、続いて、16.46mlの48
wt%のHBr(aq.)および8.23mlの水を加えた。得られた混合物を室温にま
で一晩冷却した。その最終生成物(1−[2−(2,4−ジメチル−フェニルスルファニ
ル)−フェニル]−ピペラジンヒドロブロミド)を濾過により収集し、真空下(60℃)
において乾燥させることにより、46.8g(87%の収率)の生成物が得られた。
【0200】
実施例26:自由行動ラットの脳におけるアセチルコリンの細胞外レベルに及ぼす効果
方法
動物に1−[2−(2,4−ジメチルフェニルスルファニル)フェニル]ピペラジン、
HBr塩を投与した。
【0201】
動物
最初に計量したときの体重が275〜300gの雄のSprague−Dawleyラ
ットを使用した。これらの動物は、食物および水道水を自由に摂取することができる状態
で、規則的な室内温度(21±2℃)および湿度(55±5%)が得られるように制御さ
れた条件下において、12時間の明/暗サイクルの下で飼育された。
【0202】
手術および微小透析実験
ラットにヒプノルム/ドルミカム(2ml/kg)で麻酔をかけ、腹側海馬(座標:ブ
レグマの5.6mm後方、側方−5.0mm、硬膜の7.0mm腹側)または前前頭皮質
(座標:ブレグマの3.2mm前方;側方、0.8mm;硬膜の4.0mm腹側)に透析
プローブチップを位置付けることを目的として、脳内ガイドカニューレ(CMA/12)
を脳内に定位的に埋め込んだ。アンカースクリューおよびアクリルセメントを用いてその
ガイドカニューレを固定した。動物の体温が直腸プローブによりモニタリングされ、37
℃に維持された。それらのラットは、2日間、手術からの回復が許され、一匹ずつケージ
内で飼育された。実験の当日、上述のガイドカニューレを通じて微小透析プロープ(CM
A/12、直径0.5mm、長さ3mm)が挿入された。
【0203】
それらのプローブは、複式チャンネルスイベルを介してマイクロインジェクションポン
プに接続された。濾過されたリンゲル溶液(0.5μMのネオスチグミンを含有する、1
45mmのNaCl、3mMのKCl、1mMのMgCl2、1.2mMのCaCl2)
での微小透析プローブの潅流は、脳内にプローブを挿入する少し前に始められ、1μl/
分の一定の流量で実験の期間中続けられた。安定化の180分後、実験が開始された。透
析液が20分毎に収集された。実験後、これらの動物を犠牲にし、それらの脳を取り除い
て凍結し、プローブの配置を確認するために薄片化した。
【0204】
化合物を10%のHPbetaCD中に溶解し、皮下に注射した(2.5〜10mg/
kg)。用量は塩のmg/kg体重として表現されている。化合物は2.5ml/kgの
量で投与された。
【0205】
透析液アセチルコリンの分析
透析液中におけるアセチルコリン(ACh)の濃度が、100mMの二ナトリウム水素
ホスファート、2.0mMのオクタンスルホン酸、0.5mMのテトラメチルアンモニウ
ムクロリドおよび0.005%のMB(ESA)からなるpH8.0の移動相を用いる電
気化学検出でのHPLCにより分析された。固定化されたコリンオキシダーゼを含有する
プレカラム酵素反応器(ESA)が、分析カラム(ESA ACH−250);流量0.
35ml/分、温度:35℃でのAChの分離に先立ち、注入されたサンプル(10μl
)からコリンを排除した。分析カラムの後、サンプルは、固定化されたアセチルコリンエ
ステラーゼおよびコリンオキシダーゼを含有するポストカラム固相反応器(ESA)に通
された。後者の反応器はAChをコリンに変換し、その後、コリンをベタインおよびH2
O2に変換した。後者のH2O2が白金電極(分析セル:ESA、モデル5040)を用
いることにより電気化学的に検出された。
【0206】
データの表現
単回注入実験において、化合物投与の直前の先行する3つの連続したAChサンプルの
平均値が各実験での基礎レベルとして用いられ、データが基礎(100%に正規化された
平均基礎注入前値)の百分率に変換された。
【0207】
結果
本化合物は、ラットの前前頭皮質および腹側海馬におけるAChの細胞外レベルを有意
に上昇させた−図19aおよび19b参照。
【0208】
実施例27:ラットにおける文脈的恐怖条件付け
この実験で投与された化合物は1−[2−(2,4−ジメチルフェニルスルファニル)
フェニル]ピペラジンHBr塩であった。
【0209】
我々は、ラットにおいて、文脈的恐怖条件付けの獲得、固定および想起に及ぼす本化合
物の効果について調べた。この恐怖条件付けパラダイムにおいては、動物は、中立的環境
(状況、トレーニングチャンバー、CS)を嫌悪経験(電気的フットショック、US)と
関連付けることを学ぶ。そのトレーニングチャンバーに再び暴露されると、それらの動物
はすくみ行動を示し、このすくみ行動はその恐怖関連記憶の直接的な尺度であると考えら
れている[Pavlov J.Biol.Sci.、15、177〜182、1980]
。この文脈的恐怖条件付けに関する神経解剖学は充分に研究されており、幾つかの研究が
、この記憶の形成には海馬および扁桃体が必要であることを示している[Hippoca
mpus、11、8〜17、2001;J.Neurosci.、19、1106〜11
14、1999;Behav.Neurosci.、106、274〜285、1992
]。
【0210】
動物および薬剤
12時間の明/暗サイクルの下で1つのケージ当たり2匹が飼育された、Charle
s River Laboratoriesから入手した、成体の雄のSprague−
Dawleyラット(トレーニング時の体重、250〜300g)を使用した。食物およ
び水は自由に摂取することができた。それらのラットは到着してから1週間後に使用され
た。化合物は10%のHPbetaCD中に溶解され、皮下に注入された。薬剤は、2.
5ml/kgの量で投与された。
【0211】
機器
トレーニングおよび試験は、隔離された部屋に収容され、且つ、換気システムに接続さ
れた防音チャンバー(30×20×40cm)内で実施された。照明は白色灯(60ワッ
ト)により与えられた。チャンバーの床は、電気ショック発生装置に取り付けられた金属
製の格子から成っていた。トレーニングおよび試験をする前に、そのチャンバーを70%
のエタノール溶液で清浄にした。ビデオカメラにより、オフライン分析でのトレーニング
セッションの行動の観察および記録が可能化された。
【0212】
獲得および保持試験
獲得の間、動物は、1分間の馴化期間の間、新しい環境を自由に探索することを許され
、その1分間の馴化期間は、帯電可能な格子製の床を通じて与えられる1回の避けられな
いフットショック(無条件刺激、US)と同時に終結した。そのフットショックは、持続
時間が2秒であり、0.75mAの強度であった。動物は、US後、更に60秒間、その
条件付けチャンバー内に留められた。この状況に対するベースライン−すくみ行動応答を
決定するため、最初の58秒間(ショック前獲得;実験者にはグループ情報が知らされて
いない)中にすくみ行動のスコアが付けられた。獲得が終了すると、動物は優しく取り出
され、元の自分のケージに入れられた。24時間後、同じ動物が上述のトレーニング状況
(恐怖条件付けチャンバー)に再度入れられ、2分間の保持試験が実施された。この期間
中、フットショックは加えられなかった。グループに関する情報が知らされていない実験
者によって、全試験期間中におけるすくみ行動のスコアが付けられ、合計試験期間の百分
率として提示された。
【0213】
結果および検討
ラットにおける文脈的恐怖条件付けに及ぼす化合物の効果
ラットにおける文脈的恐怖条件付けに及ぼす化合物の効果が、(i)獲得(獲得の前に
薬剤適用、図20)、(ii)記憶の想起(試験の前に薬剤適用、図21)および(ii
i)固定(獲得の直後に薬剤適用、図22)に関して調べられた。この第1セットの実験
においては、化合物(1mg/kg、5mg/kgおよび10mg/kg)が獲得セッシ
ョンの1時間前に投与された。図20は、トレーニング期間(フードショックの前の58
秒間)中および24時間後の保持試験におけるすくみ行動の獲得を描いている。以下の知
見が認められた:
・本化合物は、試験されたどの用量においても、フットショックを与える前のベースラ
インすくみ行動に影響を及ぼさない。
【0214】
・5mg/kgの用量における本化合物は、獲得の24時間後に行われる記憶保持試験
中のすくみ行動に費やす時間を増大させる傾向を有している(39.24±13.76%
、n=6、対、賦形剤で治療された動物における24.30±4.40%、n=16)。
【0215】
・10mg/kgの用量における本化合物は、獲得の24時間後に行われる保持試験中
のすくみ行動に費やす時間を有意に増大させる(52.15±5.68%、n=10、対
、賦形剤で治療された動物における24.30±4.40%、n=16、p<0.01)
。
【0216】
図20に記載されているようにこの恐怖条件付けモデルは、学習および記憶の研究用に
、文献で説明されている標準的な手順である。記憶想起に及ぼすこの薬剤の急性効果を更
に解明するため、化合物(5mg/kg、10mg/kgおよび20mg/kg)を保持
試験の1時間前に適用した。本化合物は5mg/kgの用量で記憶試験中のすくみ行動の
発現を抑制することが観測された(12.86±3.57%、n=9、対、賦形剤で治療
された動物における33.61±4.29%、n=13、p<0.05)(図21)。
【0217】
上述されているように、本化合物は、それ自体では、USを開始する前のベースライン
すくみ行動に影響を及ぼさず(図20)、従って、一見して最ももっともらしい仮説は、
図21で観測された効果が抗不安作用によるものであるという仮説である。条件付けされ
た記憶はすくみ行動、潜在的な抗不安作用を有する化合物により低減される応答により評
価される。この実験は、記憶想起の直前に与えられた本化合物が抗不安効果を有している
ことを示しており、従って、図20に示されているすくみ行動の増大が本化合物の不安惹
起作用によるものであるとは考えにくい。
【0218】
本化合物が、不安惹起性ではないが、認識力を増強させる潜在的能力(pro-cognitive
potential)を備えていることを補強的に示すため、獲得セッションの後に5mg/kg
、10mg/kgおよび20mg/kgの用量で本化合物を投与した。その結果、このセ
ットの実験において、本化合物は、獲得の間にも保持試験全体を通じてもオンボード(on
board)でなかった。ここで、5mg/kgの用量における本化合物は、獲得セッション
の24時間後に行われた保持試験中のすくみ行動に費やす時間を有意に増大させることが
観測された(45.58±4.50%、n=8、対、賦形剤で治療された動物における2
5.26±3.57%、n=19、p<0.05)。状況への再暴露中のすくみ行動に費
やす時間のパーセンテージが恐怖関連記憶の尺度として記述されており[Pavlov
J.Biol.Sci.、15、177〜182、1980]、賦形剤で治療された動物
と比べたときに、化合物で治療されたラットでは、これが増大されている(図20および
21)。ひとまとめにして考えると、これらのデータは、本化合物が文脈的記憶を高める
ことを示している。
【技術分野】
【0001】
本発明は、セロトニン受容体1A(5−HT1A)およびセロトニン受容体3(5−H
T3)に及ぼす活性と組み合わされたセロトニン再取り込み阻害活性を呈する化合物に関
し、これらの化合物はCNS関連疾患の治療に有用である。
【背景技術】
【0002】
選択的セロトニン再取り込み阻害薬(SSRI)は、それ以前に使用されていた化合物
、即ち、古典的な三環式化合物に比べ、より効果的であり、耐容性に優れ、好ましい安全
性プロフィールを有しているため、長年にわたり、特定のCNS関連疾患の治療、特には
うつ病、不安および社会恐怖症の治療に用いられる第一選択薬であった。
【0003】
それにもかかわらず、SSRIを用いる治療上の処置は、有意な割合の無応答者、即ち
、SSRI治療に応答しない患者または限られた程度にしか応答しない患者により阻まれ
ている。その上、典型的には、SSRI治療は、治療の数週間後まで効果を示し始めない
。
【0004】
SSRI治療のこれらの欠点のうちのいくつかを回避するため、精神科医は、時々、増
強(augmentation)戦略を利用する。抗うつ剤の増強は、例えば気分安定剤、例えばリチ
ウムカルボナートもしくはトリヨードチロニンなどと組み合わせることにより、または電
気ショック療法と併用することにより達成することができる。
【0005】
セロトニントランスポーター(SERT)の阻害と1種またはそれ以上のセロトニン受
容体に及ぼす活性との組み合わせが有益であり得ることは公知である。これまでに、セロ
トニン再取り込み阻害剤と5−HT2Cアンタゴニスト作用または逆アゴニスト作用を有
する化合物(5−HT2C受容体において負の効能を有する化合物)との組み合わせは、
微小透析実験で測定したときに、抹消(terminal)領域における5−HT(セロトニン)
レベルのかなりの増大をもたらすことが見出されている(特許文献1)。これは、臨床に
おける抗うつ効果の比較的短期間での発現およびセロトニン再取り込み阻害剤(SRI)
の治療効果の増強または相乗作用を示唆していたのであろう。
【0006】
同様に、5−HT1Aの部分的アゴニストであるピンドロールとセロトニン再取り込み
阻害剤との組み合わせが効果の速やかな発現をもたらすことも報告されている(非特許文
献1)。
【0007】
CNS関連疾患、例えばうつ病、不安および統合失調症などは、しばしば、他の障害ま
たは機能不全、例えば認知障害または認識機能障害などと併存する(非特許文献2、非特
許文献3)。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】国際公開第01/41701号パンフレット
【非特許文献】
【0009】
【非特許文献1】Psych.Res.、125、81−86、2004
【非特許文献2】Scand.J.Psych.、43、239−251、2002
【非特許文献3】Am.J.Psych.、158、1722−1725、2001
【発明の概要】
【発明が解決しようとする課題】
【0010】
幾つかの神経伝達物質がこの認識力を調節する神経学的事象にかかわっているものと推
測されている。特に、コリン作動系は認識に関して重要な役割を果たしており、従って、
コリン作動系に影響を及ぼす化合物は認識機能障害の治療に潜在的に有用である。5−H
T1A受容体および/または5−HT3受容体に影響を及ぼす化合物はコリン作動系に影
響を及ぼすことが知られており、それらの化合物は、そのようなものとして、認識機能障
害の治療に有用であり得る。
【0011】
このような理由から、5−HT1Aおよび/または5−HT3受容体活性を機能させる
化合物は、認識機能障害の治療に有用であると期待することができよう。その上にSER
T活性も機能させる化合物は、そのような化合物がうつ病の治療において速やかな治療効
果の発現をももたらすものと考えられるため、うつ病患者における認識機能障害の治療に
格別に有用であろう。
【0012】
国際公開第03/029232号パンフレットは、例えばSERT活性を有する化合物
として化合物1−[2−(2,4−ジメチルフェニルスルファニル)フェニル]ピペラジ
ン(実施例1e)を開示している。
【課題を解決するための手段】
【0013】
驚くべきことに、本発明者らは、1−[2−(2,4−ジメチルフェニルスルファニル
)フェニル]ピペラジンがSERT阻害、5−HT3アンタゴニズムおよび5−HT1A
部分的アゴニズムの組み合わせを発揮することを見出した。従って、1つの実施形態にお
いては、本発明は、1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]
ピペラジンである化合物Iおよびその化合物の薬学的に許容可能な塩を提供し、但し、前
述の化合物は非結晶質形態における遊離塩基ではないものとする。
【0014】
1つの実施形態においては、本発明は、治療における化合物Iの使用を提供する。
【0015】
1つの実施形態においては、本発明は、化合物Iを含む医薬組成物を提供する。
【0016】
1つの実施形態においては、本発明は治療方法を提供し、その治療方法は、治療を必要
としている患者に有効量の化合物Iを投与することを含む。
【0017】
1つの実施形態においては、本発明は、薬剤の製造における化合物Iの使用を提供する
。
【図面の簡単な説明】
【0018】
【図1】結晶質塩基のXRPD。
【図2】アルファ型の臭化水素酸塩のXRPD。
【図3】ベータ型の臭化水素酸塩のXRPD。
【図4】ガンマ型の臭化水素酸塩のXRPD。
【図5】半水和物の形態の臭化水素酸塩のXRPD。
【図6】エチルアセタート溶媒和物およびアルファ型の臭化水素酸塩の混合物のXRPD。
【図7】塩酸塩のXRPD。
【図8】一水和物の形態の塩酸塩のXRPD。
【図9】メシル酸塩のXRPD。
【図10】フマル酸塩のXRPD。
【図11】マレイン酸塩のXRPD。
【図12】メソ−酒石酸塩のXRPD。
【図13】L−(+)−酒石酸塩のXRPD。
【図14】D−(−)−酒石酸塩のXRPD。
【図15】硫酸塩のXRPD。
【図16】リン酸塩のXRPD。
【図17】硝酸塩のXRPD。
【図18a】皮内ホルマリン試験における本発明の化合物の効果。X−軸は投与された化合物の量を示している;Y−軸は足をなめるのに費やした時間的な量(秒)を示している。0−5分の時間的期間内における応答。
【図18b】皮内ホルマリン試験における本発明の化合物の効果。X−軸は投与された化合物の量を示している;Y−軸は足をなめるのに費やした時間的な量(秒)を示している。20−30分の時間的期間内における応答。
【図19a】1−[2−(2,4−ジメチルフェニルスルファニル)フェニル]ピペラジンHBr塩の投与時における自由行動ラットの前前頭皮質における細胞外アセチルコリンレベル。
【図19b】1−[2−(2,4−ジメチルフェニルスルファニル)フェニル]ピペラジンHBr塩の投与時における自由行動ラットの腹側海馬における細胞外アセチルコリンレベル。
【図20】獲得(acquisition)の60分前に与えられたときの、Sprague−Dawleyラットにおける文脈的恐怖条件付けに及ぼす1−[2−(2,4−ジメチルフェニルスルファニル)フェニル]ピペラジンHBr塩の効果。フットショックUSに先立つ58秒間の馴化期間の間、すくみ行動(freezing behaviour)のスコアが付けられた(ショック前獲得)(白色のバー)。すくみ行動はトレーニングの24時間後に測定された(保持(retention)テスト)(黒色のバー)。
【図21】保持テストの1時間前に与えられたときの、Sprague−Dawleyラットにおける文脈的恐怖条件付けに及ぼす1−[2−(2,4−ジメチルフェニルスルファニル)フェニル]ピペラジンHBr塩の効果。フットショックUSに先立って、58秒間の間、すくみ行動のスコアが付けられた(獲得)(白色のバー)。すくみ行動はトレーニングの24時間後に測定された(保持テスト)(黒色のバー)。
【図22】獲得の直後に与えられたときの、Sprague−Dawleyラットにおける文脈的恐怖条件付けに及ぼすAA21004の効果。フットショックUSに先立って、58秒間の間、すくみ行動のスコアが付けられた(ショック前獲得)(白色のバー)。すくみ行動はトレーニングの24時間後に測定された(記憶保持テスト)(黒色のバー)。
【発明を実施するための形態】
【0019】
本発明は、その構造が
【0020】
【化1】
【0021】
である化合物I、1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピ
ペラジンおよびその化合物Iの薬学的に許容可能な塩に関し、但し、化合物Iは非結晶質
形態における遊離塩基ではないものとする。
【0022】
1つの実施形態においては、上述の薬学的に許容可能な塩は、無毒な酸の酸付加塩であ
る。そのような塩は、有機酸から生成される塩、例えばマレイン酸、フマル酸、安息香酸
、アスコルビン酸、コハク酸、シュウ酸、ビス−メチレンサリチル酸、メタンスルホン酸
、エタンジスルホン酸、酢酸、プロピオン酸、酒石酸、サリチル酸、クエン酸、グルコン
酸、乳酸、リンゴ酸、マンデル酸、ケイ皮酸、シトラコン酸、アスパラギン酸、ステアリ
ン酸、パルミチン酸、イタコン酸、グリコール酸、p−アミノ安息香酸、グルタミン酸、
ベンゼンスルホン酸、テオフィリン酢酸、ならびに8−ハロテオフィリン、例えば8−ブ
ロモテオフィリンなどから生成される塩を含む。また、そのような塩は、無機塩から生成
されてもよく、例えば塩酸、臭化水素酸、硫酸、スルファミン酸、リン酸および硝酸など
から生成されてもよい。特別なものとして、メタンスルホン酸、マレイン酸、フマル酸、
メソ−酒石酸、(+)−酒石酸、(−)−酒石酸、塩酸、臭化水素酸、硫酸、亜リン酸お
よび硝酸から生成される塩を挙げることができる。格別なものとして臭化水素酸塩を挙げ
ることができる。
【0023】
経口用剤形、特に錠剤は、投与しやすく、その結果、良好なコンプライアンスが得られ
るため、患者および医療実践者により好まれることが多い。錠剤の場合、活性成分は結晶
質であることが好適である。1つの実施形態においては、本発明の化合物は結晶質である
。
【0024】
1つの実施形態においては、本発明の結晶は溶媒和物、即ち、溶媒分子がその結晶構造
の一部を形成する結晶である。この溶媒和物は水から形成されてよく、その場合には、そ
れらの溶媒和物は水和物と呼ばれることが多い。代替的に、これらの溶媒和物は、他の溶
媒、例えばエタノール、アセトンまたはエチルアセタートなどから形成されてもよい。そ
の溶媒和の正確な量はそれらの条件に依存することが多い。例えば、水和物は、典型的に
は、温度が高められた場合または相対湿度が下げられた場合には、水分を失うであろう。
【0025】
1つの実施形態においては、本発明の化合物は非溶媒和型の結晶である。
【0026】
幾つかの化合物は吸湿性であり、即ち、それらの化合物は湿気に晒されたときに水分を
吸収する。吸湿性は、一般的に、薬剤配合物中、特には乾性配合物中、例えば錠剤中など
に存在させるべき化合物にとっては望ましくない特性であると見なされている。1つの実
施形態においては、本発明は、吸湿性の低い結晶を提供する。結晶質の活性成分を用いる
経口用剤形の場合には、それらの結晶が明確に定義されたものであることも有益である。
この文脈において、「明確に定義された」という用語は、具体的には、その化学量論が明
確に定義されたものであることを意味し、即ち、その塩を形成しているイオン間の比が小
さな整数間の比、例えば1:1、1:2、2:1、1:1:1などであることを意味する
。1つの実施形態においては、本発明の化合物は明確に定義された結晶である。
【0027】
本発明の結晶質化合物は1つより多くの形態で存在していてよく、即ち、それらの化合
物は多形性形態で存在していてよい。多形性形態は、化合物が1つより多くの形態で結晶
化し得る場合に存在する。本発明は、純粋な化合物またはそれらの化合物の混合物のどち
らの場合にも、すべてのそのような多形性形態を包含すべく意図されている。
【0028】
1つの実施形態においては、本発明の化合物は精製された形態である。「精製された形
態」という用語は、化合物が、場合によって、他の化合物または他の形態のそれと同一の
化合物を本質的に含んでいないことを表すべく意図されている。
【0029】
1つの実施形態においては、本発明は、図1〜17、特には図2、3、4および5に示
されているようなXRDPを有する本発明の化合物の結晶質の塩を提供する。
【0030】
以下の表は、本発明の化合物に対する主要なXRDP反射(XRDP reflections)を示し
ている。
【0031】
選択されたX線ピーク位置(°2θ)、すべての値+−0.1°
【0032】
【表1】
【0033】
例えば図2〜5により証拠だてられているように、本発明の化合物、この場合には臭化
水素酸塩は、幾つかの形態で存在することができ、即ち、多形性であり得る。それらの多
形性形態は、実施例4dで示されているように、異なる特性を有している。ベータ型の臭
化水素酸塩は、より高いDSC融点およびより低い溶解度により示されているように、高
い安定性を有している。その上、前述のベータ型は低い吸湿性と溶解度との魅力的な複合
特性を有しており、その複合特性により、この化合物は錠剤を製造するのに特に適したも
のと成っている。従って、1つの実施形態においては、本発明は、約6.89、9.73
、13.78および14.62(°2θ)におけるXRDP反射、特には図3に示されて
いるようなXRPDを有する、1−[2−(2,4−ジメチルフェニルスルファニル)−
フェニル]ピペラジンの臭化水素酸塩を提供する。
【0034】
また、活性成分の溶解度も、バイオアベイラビリティーに直接的な影響を及ぼし得るた
め、剤形を選択する上で重要である。経口用剤形の場合には、活性成分のより高い溶解度
は、バイオアベイラビリティーを高めるため、一般的には、有益であると考えられている
。
【0035】
皮質および海馬のコリン作動性神経伝達は認識力にとって非常に重要であり、また、数
多くの前臨床観察がこの系にとってのセロトニン受容体1A(5−HT1A)の重要性を
指摘している。T.Koyamaは、Neurosci.Lett.、265、33〜3
6、1999において、5−HT1AアゴニストであるBAYX3702がラットの皮質
および海馬からのアセチルコリン流出を増大させることを報じている。興味深いことに、
5−HT1AアンタゴニストであるWAY−100635はBAYX3702の効果を排
除することができ、これは、BAYX3702の効果が5−HT1A媒介性であることを
示している。
【0036】
数多くの研究が、認識機能障害に及ぼす5−HT1Aのモジュレーターの効果について
報じている。A.Menesesは、Neurobiol.Learn.Memory、
71、207〜218、1999において、部分的5−HT1Aアゴニストである(±)
−8−ヒドロキシ−2−(ジ−n−プロピルアミノ)−テトラリン、HCl(8−OH−
DPAT)が、正常なラットにおける学習の統合(consolidation)を促進し、また、認
識機能障害を負ったラットにおける認識機能を正常化することを報じている。
【0037】
これらの前臨床観察結果は臨床においても反映されているようである。T.Sumiy
oshiは、Am.J.Psych.、158、1722〜1725、2001において
ある研究について報じており、その研究では、患者に、プラシーボまたは5−HT1Aア
ゴニストであるタンドスピロンと組み合わせて、定型抗精神病薬、例えばハロペリドール
、スルピリドおよびピモジドなど(すべてのものが5−HT1A活性を欠く)が投与され
た。抗精神病薬に加えてタンドスピロンが投与された患者は認識能力の改善を示したが、
一方、プラシーボが投与された患者は改善を示さなかった。同様に、同じく5−HT1A
アゴニストである非定型抗精神病薬、例えばクロザピンなども統合失調症患者における認
識力を増強するが、一方、5−HT1A活性を有さない定型抗精神病薬、例えばハロペリ
ドールなどは認識力を増強しない(Y.Chung、Brain Res.、1023、
54〜63、2004)。
【0038】
上で述べられているように、コリン作動系は認識力を調節する神経学的事象にかかわっ
ているものと考えられており、また、コリン作動系は、セロトニン受容体3(5−HT3
)による抑制制御の影響を受けやすいものと考えられる[Giovanniniら、J.
Pharmacol.Exp.Ther.、1998、285:1219〜1225;C
ostallおよびNaylor、Current Drug Targets−CNS
&Neurobiol.Disord.2004、3:27〜37]。
【0039】
マウスにおける馴化試験において、ラットにおけるT字型迷路強制交替試験(T-maze r
einforced alternation task)において、ならびにマーモセットにおける物体弁別(obje
ct discrimination)および逆転学習課題において、オンダンセトロンは、ムスカリン性
アンタゴニストであるスコポラミンにより引き起こされる機能障害または基底核から現れ
るコリン作動性経路の病変を低減した(Barnesら、Pharmacol.Bioc
hem.Behav.1990、35:955〜962;Careyら、Pharmac
ol.Biochem.Behav.1992、42:75〜83)。Boastら(N
eurobiol.Learn.Mem.1999、71:259〜271)は、NMD
A受容体の非競合的アンタゴニストであるMK−801を使用して、遅延標本非照合放射
状迷路課題で訓練されたラットの認識能力を混乱させた。オンダンセトロンは認識機能障
害をブロックすることが示された。その上、マウスでの受動的回避課題におけるエタノー
ルの記憶喪失作用についての研究において、エタノールのこの記憶喪失作用はオンダンセ
トロンにより正常状態へ向けて部分的に回復された(Napiorkowska−Paw
lakら、Fundam.Clin.pharmacol.2000、14:125〜1
31)。従って、前臨床モデルにおけるコリン作動系の機能障害後の5−HT3アンタゴ
ニズムによるコリン作動性伝達の促進(Diez−Arizaら、Psychophar
macology、2003、169:35〜41;Gil−Beaら、Neuroph
armcol.2004、47:225〜232)は、認知障害の治療においてこの処置
を用いることに対する根拠を示唆している。
【0040】
健常な男性の被検者におけるランダム化二重盲検クロスオーバー試験において行われた
言葉および空間の記憶ならびに注意の持続についての評価は、5−HT3アンタゴニスト
であるアロセトロンが言葉および空間の記憶におけるスコポラミン誘発性の障害を軽減す
ることを示した(Preston、Recent Advances in the t
reatment of Neurodegenerative disorders
and cognitive function、1994、(eds.)Racagn
iおよびLanger、Basel Karger、p.89〜93)。
【0041】
結論として、5−HT3アンタゴニスト活性と組み合わせて5−HT1A部分的アゴニ
スト活性を発揮する化合物は、認識機能障害の治療に特別に有用であると考えられる。さ
らに、セロトニン再取り込み阻害を発揮する化合物は、5−HT1A部分的アゴニズムと
組み合わされたセロトニン再取り込み阻害がより迅速なうつ病の治療効果の発現をもたら
すため、うつ病に伴う認識機能障害の治療に格別に有用であろう。
【0042】
実施例1で示されているように、本発明の化合物はヒトセロトニントランスポーターの
強力な阻害剤であり、即ち、それらの化合物はセロトニン再取り込みを阻害する。その上
、それらの化合物は、マウス、ラット、モルモットおよびイヌ5−HT3受容体における
強力なアンタゴニストである。卵母細胞にクローニングされたヒト5−HT3受容体にお
いて、それらの化合物は低濃度ではアンタゴニストであり(IC50=約30nM)、そ
の一方で、より高い濃度では、それらの化合物はアゴニスト特性を示す(ED50=2.
1μM)ことが判明した。高濃度における本発明の化合物のその後の適用は何らアゴニス
ト応答を示さなかったが、これは、インビトロにおける急速な脱感作(desenitisation)
または直接的なアンタゴニズムによるものであったとすることができよう。従って、低濃
度において、本発明の化合物は、他の種からの5−HT3受容体で観測されたのと同様に
、ヒト5−HT3受容体においても著しいアンタゴニズムを示す。
【0043】
本発明の化合物は、ラットおよびマウスの両者の脳ホモジネートにおける5−HT1A
受容体に低い親和性で結合する。しかし、本発明の化合物は、40nMのKiでヒト5−
HT1A受容体に結合する。その上、機能データ(functional data)は、本発明の化合
物がヒト5−HT1A受容体における部分的アゴニストであり、85%の有効性をもたら
すことを示している。
【0044】
SERT、5−HT3受容体および5−HT1A受容体における本発明の活性は、ヒト
における本化合物のインビボプロフィールに寄与するものと予想される。
【0045】
実施例26に示されているように、本発明の化合物は、ラットにおいて、前前頭皮質お
よび腹側海馬における細胞外アセチルコリンレベルの増大をもたらす。これらの前臨床知
見は、認識機能障害の治療におけるアセチルコリンエステラーゼ阻害剤の使用と比較して
、認識機能障害の治療、例えばアルツハイマー病の治療における臨床的な効果に変わるも
のと期待される。この見解に対する更なる支持を実施例27に見出すことができ、そこで
のデータは、本発明の化合物がラットにおける文脈的(contextual)記憶を増強すること
を示している。結局のところ、ラットにおけるアセチルコリンレベルに及ぼす効果および
記憶に及ぼす効果と組み合わせた本発明の化合物の薬理学的プロフィールは、本発明の化
合物が認識機能障害の治療に有用であることを強く示唆している。
【0046】
1つの実施形態においては、本発明は認知障害または認識機能障害を治療するための方
法に関し、その方法は、治療を必要としている患者に治療的有効量の本発明の化合物を投
与することを含む。
【0047】
認知障害または認識機能障害は、認知機能または認知領域の低下を含み、例えば作業記
憶、注意および覚醒、言語的学習および記憶、視覚的な学習および記憶、論理的思考およ
び問題の解決、例えば実行機能、処理速度および/または社会的認知の低下を含む。特に
、認知障害または認識機能障害は、注意の欠如、解体した思考、思考の減退(slow think
ing)、理解困難(difficulty in understanding)、集中力不足(poor concentration)
、問題解決能力の低下(impairment of problem solving)、記憶力貧困(poor memory)
、思考の表現困難(difficulties in expressing thoughts)および/または思考、感情
および行動の統合困難(difficulties in integrating thoughts, feelings and behavio
ur)、または見当違いな思考の消去困難(difficulties in extinction of irrelevant t
houghts)を表し得る。「認知障害」および「認識機能障害」という用語は同じものを指
示すべく意図されており、互換可能に使用される。
【0048】
1つの実施形態においては、その患者は、別のCNS障害、例えば情動障害、例えばう
つ病;全般性(generalised)うつ病;大うつ病性障害;一般的(general)不安障害およ
びパニック障害を含めた不安障害;強迫性障害;統合失調症;パーキンソン病;認知症;
エイズによる認知症;ADHD;加齢による記憶障害;またはアルツハイマー病などの診
断も同時に下されている。
【0049】
認識機能障害は、例えば大うつ病性障害などのうつ病の典型的な特徴の一つである。認
知障害は、うつ状態の改善が認識機能障害の改善をももたらすという意味で、ある程度、
うつ病に対して続発性であり得る。しかし、認知障害が実際にはうつ病から独立している
ことを示す明らかな証拠も存在する。例えば、何件かの研究が、うつ病からの回復時に存
続する認識機能障害を示している[J.Nervous Mental Disease
、185、748〜754、197]。その上、うつ病および認識機能障害に及ぼす抗う
つ薬の示差的な効果が、たとえしばしば併存する状態であるとしても、うつ病と認識機能
障害とは独立しているという考えに対する更なる支持を与えている。セロトニンおよびノ
ルアドレナリン医薬は抑うつ症状の同程度の改善をもたらすが、幾つかの研究は、ノルア
ドレナリン作動系の調節がセロトニン調節程には認識機能を改善しなかったことを示して
いる[Brain Res.Bull.、58、345〜350、2002;Hum P
sychpharmacol.、8、41〜47、1993]。
【0050】
本発明の化合物を投与することによるうつ病患者における認識機能障害の治療は格別に
有利であると考えられる。本発明の化合物の多面的な薬効薬理、特にSERT、5−HT
3および5−HT1A活性は、抑うつ状態の迅速な治療効果の発現と組み合わされた認識
機能の改善をもたらすことが期待される。
【0051】
認識機能障害は、高齢者における特に重要な考慮すべき事項である。認識機能障害は加
齢とともに普通に進行するが、更に、うつ病でも進行する。従って、1つの実施形態にお
いては、認識機能障害の治療が為される患者は高齢者であり、特にうつ病を伴った高齢者
である。
【0052】
認識機能は、上で述べられているように、統合失調症患者で損なわれることが多い。ま
た、何件かの研究は、認識機能が統合失調症における職業上の機能(vocational functio
ning)と関連していると結論付けてもいる[Scizophrenia Res.、45
、175〜184、2000]。1つの実施形態においては、認識機能障害の治療が為さ
れる患者は統合失調症患者である。
【0053】
5−HT3受容体アンタゴニストは、更に、種々の疾患の治療用、例えば嘔吐、化学療
法起因性嘔吐、渇望、物質乱用、疼痛、過敏性腸症候群(IBS)、統合失調症および摂
食障害などの治療用としても示唆されている[Eur.J.Pharmacol.、56
0、1〜8、2007;Pharmacol.Therapeut.、111、855〜
876、2006;Alimentary Pharmacol.Ther.、24、1
83〜205、2006]。
【0054】
ある臨床研究は、臨床応答が不充分なうつ病患者(治療抵抗性うつ病(TRD)または
難治性うつ病)を治療するには、ミルタザピン(mirtazipine)とSSRIとを組み合わ
せた場合の方がSSRIを単独で使用する場合よりも優れていることを示している[Ps
ychother.Psychosom.、75、139〜153、2006]。ミルタ
ザピンは5−HT2および5−HT3アンタゴニストであり、これは、本発明の化合物が
TRDの治療に有用であるという概念を支持するものである。
【0055】
顔面紅潮は閉経過渡期に関連する症状である。女性によっては、この顔面紅潮が、睡眠
または一般的な活動を妨害し、治療が必要な程度にまで成り得る。数十年間にわたり、エ
ストロゲンを用いるホルモン代償療法が既定の治療法であったが、最近では、乳癌および
心事象などの副作用に関する懸念が表明されている。SSRIを用いる臨床試験は、エス
トロゲンの場合程ではないにしても、これらの化合物が顔面紅潮に有効であることを示し
ている[J.Am.Med.Ass.、295、2057〜2071、2006]。しか
し、セロトニン再取り込みを阻害する化合物、例えば本発明の化合物を用いる顔面紅潮の
治療法は、エストロゲンを受け入れることができない女性または受け入れない女性に対す
る代替的治療法である。
【0056】
睡眠時無呼吸もしくは閉塞型睡眠時無呼吸−低呼吸症候群または閉塞型睡眠呼吸障害は
、効果的な薬物療法が特定されるべき状態のまま留まっている障害である。しかし、動物
における幾つかの研究は、5−HT3アンタゴニスト、例えば本発明の化合物がこれらの
疾患の治療に効果的であり得ることを示唆している[Sleep、21、131〜136
、1998;Sleep、8、871、878、2001]。
【0057】
1つの実施形態においては、本発明は、情動障害;うつ病;大うつ病性障害;産後うつ
病;双極性障害、アルツハイマー病、精神病、癌、加齢(age)もしくはパーキンソン病
に伴ううつ病;不安;全般性不安障害;社会不安障害;強迫性障害;パニック障害;パニ
ック発作;恐怖症;社会恐怖症;広場恐怖症;ストレス性尿失禁;嘔吐;IBS;摂食障
害;慢性疼痛;部分的応答者(partial responders);治療抵抗性うつ病;アルツハイマ
ー病;認識機能障害;ADHD;メランコリー;PTSD;顔面紅潮;睡眠時無呼吸;ア
ルコール、ニコチンもしくは炭水化物渇望;物質乱用;およびアルコールもしくは薬物乱
用から選択される疾患を治療する方法に関係し、その方法は、治療を必要としている患者
に治療的有効量の本発明の化合物を投与することを含む。1つの実施形態においては、上
で列挙されている疾患のうちのいずれかに関して治療される前述の患者は、最初にその疾
患の診断が下されている。
【0058】
一般的には抗うつ薬での治療、特にはSSRIでの治療は、性的機能不全を伴い、しば
しば治療の中止をもたらすことが周知である。SSRIで治療を受ける患者のうちの30
〜70%もの多くの患者が性機能の障害を訴えており[J.Clin.Psych.、6
6、844〜848、2005]、それらの障害は、リビドーの減退、オルガスムの遅延
、低減または欠落、性的興奮の減少および勃起機能不全を含む。臨床試験において、合計
で114人の被検者が本発明の化合物を投与された;これら114人の被検者のうち、僅
か1人の被検者のみが性的機能不全を訴えた。これらのデータは、本発明の化合物を用い
る臨床的介入が驚くべき程僅かな数の性的機能障害しか伴わないことを示唆している。
【0059】
上で述べられているように、本発明の化合物は、特に、慢性疼痛の治療に非常に適して
いる。慢性疼痛は、幻肢痛、神経障害性疼痛、糖尿病性ニューロパシー、ヘルペス後神経
痛(PHN)、手根管症候群(CTS)、HIVニューロパシー、複合性局所疼痛症候群
(CPRS)、三叉神経の神経痛/三叉神経痛/疼痛性チック、外科的介入(例えば術後
鎮痛薬)、糖尿病性血管症、膵島炎を伴った毛細血管抵抗もしくは糖尿病症状、アンギナ
に関連した疼痛、月経に関連した疼痛、癌に関連した疼痛、歯痛、頭痛、片頭痛、緊張性
疼痛、三叉神経痛、顎関節症候群、筋筋膜疼痛筋肉損傷(myofascial pain muscular inj
ury)、線維筋痛症候群、骨関節疼痛(変形性関節症)、慢性関節リウマチ、やけどを伴
う外傷に由来する慢性関節リウマチおよび浮腫、変形性関節症、骨粗鬆症、骨への転移も
しくは不明の理由による捻挫もしくは骨折の骨痛、痛風、結合組織炎、筋筋膜疼痛、胸郭
出口症候群、上背部痛もしくは下背部痛(ここで、この背部痛は系統的、局所的もしくは
主要な脊椎疾患(神経根障害)に由来する)、骨盤痛、心臓性胸痛、非心臓性胸痛、脊髄
損傷(SCI)関連疼痛、中枢性脳卒中後疼痛、癌ニューロパシー、エイズによる疼痛、
鎌状赤血球による疼痛、または老人性疼痛(geriatric pain)などの適応症を含む。
【0060】
実施例16で提示されているデータは、本発明の化合物が疼痛の治療に有用であり、ま
た、それらの化合物が鎮痛効果さえ有し得ることを示しており、更に、神経障害性疼痛に
関する動物モデルにおける研究がこの見解を確証している。
【0061】
本明細書で使用する場合、「治療的有効量」の化合物という用語は、上述の化合物を投
与することを含む治療的介入において、所与の疾患およびそれの合併症の臨床的徴候を治
し、緩和し、または部分的に停止させるのに充分な量を意味する。これを果たすのに適切
な量が「治療的有効量」として定義される。それぞれの目的での有効量は、その疾患また
は損傷の重症度、更にはその被検者の体重および全身状態に依存するであろう。適切な用
量の決定は、数値のマトリックスを構築し、そのマトリックスの種々の異なるポイントで
試験すること(これらはすべて、訓練を積んだ医師の通常の技術能力の範囲内である)に
より、ルーチン的な実験を用いて果たし得ることが理解されよう。
【0062】
本明細書で使用する場合、「治療」および「治療する」という用語は、ある状態、例え
ば疾患または障害などと闘うことを目的とした患者の管理およびケアを意味する。この用
語は、患者が患っている、ある与えられた状態に対する治療の全スペクトル、例えばそれ
らの症状もしくは合併症を緩和するため、その疾患、障害もしくは状態の進行を遅らせる
ため、それらの症状および合併症を緩和もしくは軽減するため、および/またはその疾患
、障害もしくは状態を治癒もしくは排除するため、更には、その状態を予防するための、
活性化合物の投与などを含めるべく意図されており、ここで、予防は、疾患、状態もしく
は障害と闘うことを目的とする患者の管理およびケアを意味するものと理解すべきであり
、それらの症状もしくは合併症の発現を防ぐための活性化合物の投与を含む。とはいえ、
予防的治療(防止を目的とした治療)および治療的処置(治すことを目的とした処置)は
本発明の2つの別個の態様である。治療されるべき患者は、好適には哺乳動物であり、特
には人間である。
【0063】
典型的には、本発明の治療は本発明の化合物を毎日投与することを含むであろう。これ
は、毎日1回の投与、または1日に2回の投与もしくはもっと頻繁な回数の投与さえをも
含み得る。
【0064】
1つの実施形態においては、本発明は、情動障害;うつ病;大うつ病性障害;産後うつ
病;双極性障害、アルツハイマー病、精神病、癌、加齢もしくはパーキンソン病に伴うう
つ病;不安;全般性不安障害;社会不安障害;強迫性障害;パニック障害;パニック発作
;恐怖症;社会恐怖症;広場恐怖症;ストレス性尿失禁;嘔吐;IBS;摂食障害;慢性
疼痛;部分的応答者;治療抵抗性うつ病;アルツハイマー病;認識機能障害;ADHD;
メランコリー;PTSD;顔面紅潮;睡眠時無呼吸;アルコール、ニコチンもしくは炭水
化物渇望;物質乱用;またはアルコールもしくは薬物乱用を治療するための薬剤を製造す
るための本発明の化合物の使用に関する。
【0065】
1つの実施形態においては、本発明は、情動障害、うつ病、大うつ病性障害、産後うつ
病、双極性障害、アルツハイマー病、精神病、癌、加齢もしくはパーキンソン病に伴うう
つ病、不安、全般性不安障害、社会不安障害、強迫性障害、パニック障害、パニック発作
、恐怖症、社会恐怖症、広場恐怖症、ストレス性尿失禁、嘔吐、IBS、摂食障害、慢性
疼痛、部分的応答者、治療抵抗性うつ病、アルツハイマー病、認識機能障害、ADHD、
メランコリー、PTSD、顔面紅潮、睡眠時無呼吸、アルコール、ニコチンもしくは炭水
化物渇望、物質乱用、またはアルコールもしくは薬物乱用から選択される疾患の治療にお
いて使用するための本発明の化合物に関する。
【0066】
ヒトの認識に及ぼす本発明の化合物の効果は数多くの仕方で評価することができる。そ
の効果は、健常なボランティアに本化合物を投与し、その後、認められているテスト、例
えばAuditory Verbal Learning Test(AVLT)、Wi
sconsin Card Sorting Test(WCST)などで認識能力を測
定する試験、または持続的注意[Psycopharmacol.、163、106〜1
10、2002;Psychiatry Clin.Neurosci.、60、70〜
76、2006]を測定する試験で評価することができる。その効果は、勿論、同じ種類
の試験を用いて、認識機能障害を患っている患者で評価されてもよい。代替的に、認知モ
デルが用いられてもよく、そこでは、健常なボランティアにおいて認識機能障害が誘発さ
れ、本発明の化合物の回復効果が測定される。認識機能障害は、例えばスコポラミン、睡
眠剥奪、アルコール、およびトリプトファン枯渇により誘発することができる。
【0067】
本発明の薬剤配合物は当分野における従来の方法で製造することができる。特に錠剤に
ついて言及すれば、錠剤は、活性成分を通常の佐剤および/または希釈剤と混合し、その
後、その混合物を通常の打錠機で圧縮することにより製造することができる。佐剤または
希釈剤の例は:無水カルシウム水素ホスファート、PVP、PVP−VAコポリマー、微
結晶性セルロース、ナトリウムデンプングリコラート、トウモロコシデンプン、マンニト
ール、ジャガイモデンプン、タルカム、マグネシウムステアラート、ゼラチン、ラクトー
ス、ゴムなどを含む。そのような目的で通常使用されるあらゆる他の佐剤または添加剤、
例えば着色剤、矯味矯臭剤、保存剤なども、それらが活性成分と適合することを条件とし
て、使用することができる。
【0068】
注射用液剤は、活性成分および可能な添加剤を注射用溶剤、好適には蒸留水の一部に溶
解し、その溶液を所望の体積に調節し、得られた溶液を滅菌して適切なアンプルまたはバ
イアル中に充填することにより製造することができる。当分野において普通に使用されて
いるあらゆる適切な添加剤、例えば等張化剤、保存剤、酸化防止剤などが加えられてよい
。
【0069】
本発明の医薬組成物またはこの発明に従って製造される医薬組成物はあらゆる適切な経
路で投与することができ、例えば、錠剤、カプセル剤、散剤、シロップ剤などの形態で経
口的に投与されてもよいし、または注射用溶液の形態で非経口的に投与されてもよい。そ
のような組成物を製造するために、当分野において周知の種々の方法が用いられてよく、
また、薬学的に許容可能なあらゆる担体、希釈剤、賦形剤または当分野において普通に使
用されている他の添加剤を使用することができる。
【0070】
都合よくは、本発明の化合物は、その化合物を約1mgから50mgまでの量で含有す
る単位投薬形態(unit dosage form)で投与される。上限は、5−HT3活性の濃度依存
性によって設定されるものと考えられる。合計1日量は、通常、約1〜20mgの範囲の
本発明の化合物、例えば約1mg〜10mgまで、約5〜10mg、約10〜20mgま
たは約10〜15mgなどの範囲の本発明の化合物である。特別な1日量として、5mg
、10mg、15mgまたは20mgを挙げることができる。
【0071】
本発明の化合物を含有する錠剤は、都合よくは、湿式造粒法により製造することができ
る。この方法を用いる場合、乾燥固体(活性成分、増量剤、結合剤など)がブレンドされ
、水または他の湿潤剤(例えばアルコール)で湿らされ、それらの湿気を帯びた固体で凝
集塊または顆粒が構築される。望ましい均一な粒径が達成されるまで湿式集塊形成が続け
られ、望ましい粒径になった後、その顆粒化された生成物が乾燥される。本発明の化合物
は、典型的には、高剪断ミキサー内において水と共にラクトース一水和物、トウモロコシ
デンプンおよびコポビドンと混合される。顆粒の形成後、これらの顆粒は、適切なシーブ
サイズを有する篩でふるいにかけられ、乾燥されてよい。次いで、結果として得られたこ
れらの乾燥した顆粒が微結晶性セルロース、クロスカルメロースナトリウムおよびマグネ
シウムステアラートと混合され、その後、錠剤が圧縮される。代替的に、本発明の化合物
の湿式造粒は、マンニトール、トウモロコシデンプンおよびコポビドンを用い、錠剤を圧
縮する前に、それらの顆粒を微結晶性セルロース、ナトリウムデンプングリコラートおよ
びマグネシウムステアラートと混合することにより達成されてもよい。代替的に、本発明
の化合物の湿式造粒は、無水カルシウム水素ホスファート、トウモロコシデンプンおよび
コポビドンを用い、錠剤を圧縮する前に、それらの顆粒を微結晶性セルロース、ナトリウ
ムデンプングリコラート(タイプA)、タルクおよびマグネシウムステアラートと混合す
ることにより達成されてもよい。コポビドンはPVP−VAコポリマーである。
【0072】
1つの実施形態においては、本発明の化合物は例えばベータ型の臭化水素酸塩であり、
適切な錠剤は以下のとおりのものから成っていてよい(示されている百分率はw/w%で
ある):
HBr塩:2〜20%
ラクトース一水和物:30〜50%
デンプン:15〜30%
コポビドン:3〜5%
微結晶性セルロース:15〜25%
クロスカルメロースナトリウム:2〜5%
Mgステアラート:0.5〜5%。
【0073】
特に、錠剤は以下のとおりのものから成っていてよい:
HBr塩:3〜4%
ラクトース一水和物:44〜46%
デンプン:22〜23%
コポビドン:3〜4%
微結晶性セルロース:20〜22%
クロスカルメロースナトリウム:3〜3.5%
Mgステアラート:0.5〜1%
または
HBr塩:15〜16%
ラクトース一水和物:35〜38%
デンプン:18〜20%
コポビドン:3〜4%
微結晶性セルロース:20〜22%
クロスカルメロースナトリウム:3〜3.5%
Mgステアラート:0.5〜1%
または
HBr塩:1〜2%
ラクトース一水和物:44〜46%
デンプン:20〜24%
コポビドン:3〜4%
微結晶性セルロース:22〜24%
クロスカルメロースナトリウム:3〜4%
Mgステアラート:0.5〜1%。
【0074】
1つの実施形態においては、本発明の化合物は例えばベータ型の臭化水素酸塩であり、
適切な錠剤は以下のとおりのものから成っていてよい:
HBr塩:2〜30%
マンニトール:25〜45%
トウモロコシデンプン:10〜20%
コポビドン:2〜4%
微結晶性セルロース:22〜27%
ナトリウムデンプングリコラート:4〜5%
Mgステアラート:0.25〜5%、例えば0.25〜2%など。
【0075】
特に、錠剤は以下のとおりのものから成っていてよい:
HBr塩:20〜22%
マンニトール:35〜36%
トウモロコシデンプン:10〜12%
コポビドン:2.5〜3%
微結晶性セルロース:24〜25%
ナトリウムデンプングリコラート:3〜4%
Mgステアラート:0.25〜1%
または
HBr塩:12〜13%
マンニトール:36〜37%
トウモロコシデンプン:18〜19%
コポビドン:3〜4%
微結晶性セルロース:24〜25%
ナトリウムデンプングリコラート:3〜4%
Mgステアラート:0.25〜1%
または
HBr塩:25〜27%
マンニトール:27〜29%
トウモロコシデンプン:13〜15%
コポビドン:3〜4%
微結晶性セルロース:24〜25%
ナトリウムデンプングリコラート:3〜5%
Mgステアラート:0.25〜1%
または
HBr塩:3〜4%
マンニトール:40〜42%
トウモロコシデンプン:20〜22%
コポビドン:3〜4%
微結晶性セルロース:26〜28%
ナトリウムデンプングリコラート:3〜5%
Mgステアラート:0.5%。
【0076】
1つの実施形態においては、本発明の化合物は臭化水素酸塩であり、適切な錠剤は以下
のとおりのものから成っていてよい:
HBr塩:3〜8%
無水カルシウム水素ホスファート:35〜45%
トウモロコシデンプン:15〜25%
コポビドン:2〜6%
微結晶性セルロース:20〜30%
ナトリウムデンプングリコラート:1〜3%
タルク:2〜6%
マグネシウムステアラート:0.5〜2%。
【0077】
特に、錠剤は以下のとおりのものから成っていてよい:
HBr塩:約5%
無水カルシウム水素ホスファート:約39%
トウモロコシデンプン:約20%
コポビドン:約3%
微結晶性セルロース:約25%
ナトリウムデンプングリコラート:約3%
タルク:約4%
マグネシウムステアラート:約1%。
【0078】
種々の異なる量の活性化合物を有する錠剤、例えば2.5mg、5mg、10mg、2
0mg、25mg、30mg、40mg、50mg、60mgまたは80mgの遊離塩基
に該当した錠剤などは、正しい量の本発明の化合物を適切なサイズの錠剤と組み合わせて
選ぶことにより得ることができる。
【0079】
本発明の化合物を含有した錠剤を製造するために使用される結晶のサイズは重要である
。もし結晶が小さすぎた場合には、それらの結晶が錠剤機のプランジャーにくっついてし
まう可能性がある。その一方で、それらの結晶は大きすぎてもいけない。腸内における溶
解速度は、結晶のサイズが大きくなると減少する。従って、もし結晶が大きすぎた場合に
は、本化合物のバイオアベイラビリティーが損なわれ得る。分位値(quantiles)、例え
ばD5%、D10%、D50%、D90%、D95%およびD98%などを用いて粒径分
布を表現することができる。本明細書で使用する場合、「粒径分布」という用語は、Sy
mpatec Helos装置において1バールの分散圧におけるレーザー回折により決
定したときの、等価球形粒子の直径の累積体積サイズ分布を意味する。
【0080】
1つの実施形態においては、本発明の化合物の結晶、特にベータ型の臭化水素酸塩の結
晶は、D98%:650〜680μm;D50%:230〜250μm;およびD5%:
40〜60μmに該当する粒径分布を有している。1つの更なる実施形態においては、そ
の粒径分布は、D98%:370〜390μm;D50%:100〜120μm;および
D5%:5〜15μmに該当する。尚も更なる1つの実施形態においては、その粒径分布
は、D98%:100〜125μm;D50%:15〜25μm;およびD5%:1〜3
μmに該当する。尚も更なる1つの実施形態においては、その粒径分布は、D98%:5
0〜70μm;D50%:3〜7μm;およびD5%:0.5〜2μmに該当する。
【0081】
本発明の遊離塩基は、国際公開第2003/029232号パンフレットに開示されて
いるようにして製造することができる。本発明の塩は、その遊離塩基を適切な溶剤中に溶
解し、関連する酸を加え、続いて沈殿させることにより製造することができる。沈殿は、
第二溶剤の添加、および/または蒸発、および/または冷却のいずれかにより果たすこと
ができる。代替的に、本発明の遊離塩基および最終的には本発明の化合物は、以下で説明
されているようにパラジウム触媒反応において合成することもできる。
【0082】
芳香族炭素−ヘテロ原子結合の形成は、芳香族求核置換または銅媒介Ullman反応
により達成することができる。より最近になって、パラジウムはそのような結合の形成、
特にはC−NおよびC−S結合の形成での強力な触媒であることが示されている(例えば
、米国特許第5,573,460号を参照のこと)。
【0083】
1つの実施形態においては、本発明は
【0084】
【化2】
【0085】
を製造するための方法を提供し、その方法は、化合物II
【0086】
【化3】
【0087】
[式中、R’は水素または一価の金属イオンを表す]を、溶剤、塩基、ならびにパラジウ
ムのソースおよびホスフィン配位子からなるパラジウム触媒の存在下において、60℃か
ら130℃までの間の温度で、式III
【0088】
【化4】
【0089】
[式中、X1およびX2は独立してハロゲンを表す]の化合物、および式IV
【0090】
【化5】
【0091】
[式中、Rは水素または保護基を表す]の化合物と反応させることを含む。
【0092】
1つの実施形態においては、この方法はサブプロセスに分けられ、そのサブプロセスで
は、化合物IIおよび化合物IIIが第一の反応において反応させられ、以下の式
【0093】
【化6】
【0094】
の化合物をもたらす。次いで、この化合物が、場合によっては適切な程度にまで精製され
た後、4−[2−(2,4−ジメチル−フェニルスルファニル)−フェニル]−ピペラジ
ンをもたらすべく化合物IVと反応させられる。
【0095】
ワンポット合成、即ち、すべての反応物が反応または方法の開始時に一緒に混合される
合成は、それらが有する固有の簡易性のため、特に有用である。その一方で、起こり得る
望ましくない副反応の数が劇的に増大し、これは、再度、望ましくない副次生成物の数お
よび/または量が増大し、それに対応して、望ましい生成物の収率が低下し得ることを意
味している。具体的には、本願方法の場合、ピペラジンは2個の窒素を有しており、潜在
的にそれぞれの窒素がC−N結合の形成に関与するということが観測され得る。驚くべき
ことに、本願方法は、純粋な化合物の高収率を維持しながら、ワンポット合成、即ち、最
初から化合物II、化合物IIIおよび化合物IVが混合される方法として実行できるこ
とが判明した。
【0096】
化合物IIはチオールまたは対応するチオラートである。労働衛生上の観点から判断す
ると、チオールに付随する臭気の問題を回避するため、チオラート、例えばLi+、Na
+またはK+チオラートなどを使用することが有益であり得る。とはいえ、1つの実施形
態においては、R’は水素である。
【0097】
化合物IIIは1,2−ジハロゲン活性化ベンゼンであり、それらのハロゲンはCl、
BrおよびIのいずれであってもよい。特には、化合物IIは1−ブロモ−2−ヨード−
ベンゼンまたは1,2−ジブロモ−ベンゼンである。
【0098】
本発明の方法において使用される溶剤は、反応温度範囲内、即ち、60〜130℃の沸
点を有する非プロトン性有機溶剤またはそのような溶剤の混合物から選択されてよい。典
型的には、その溶剤は、トルエン、キシレン、トリエチルアミン、トリブチルアミン、ジ
オキサン、N−メチルピロリドンから選択され、またはそれらの任意の混合物から選択さ
れる。特には、溶剤としてトルエンの名を挙げることができる。
【0099】
前記方法の中核を成すものは、それ無しでは反応が起こらないパラジウム触媒の使用で
ある。このパラジウム触媒は、パラジウム源とホスフィン配位子とからなっている。有用
なパラジウム源は、種々の異なる酸化状態、例えば0およびIIなどのパラジウムを含む
。本発明の方法において使用され得るパラジウム源の例は、Pd2dba3、Pddba
2およびPd(OAc)2である。dbaはジベンジリデンアセトンの略号である。特に
、Pddba2およびPd2dba3の名を挙げることができる。このパラジウム源は、
典型的には、0.1〜10モル%、例えば1〜10モル%など、例えば1〜5モル%など
の量で適用される。この出願全体を通じ、モル%は限定反応物(limiting reactant)に
関して算出される。
【0100】
単座および二座のどちらも、数多くのホスフィン配位子が公知である。有用なホスフィ
ン配位子は、ラセミ2,2’−ビス−ジフェニルホスファニル−[1,1’]ビナフタレ
ニル(rac−BINAP)、1,1’−ビス(ジフェニルホスフィノ)フェロセン(D
PPF)、ビス−(2−ジフェニルホスフィノフェニル)エーテル(DPEphos)、
トリ−t−ブチル−ホスフィン(Fu’s塩)、ビフェニル−2−イル−ジ−t−ブチル
−ホスフィン、ビフェニル−2−イル−ジシクロヘキシル−ホスフィン、(2’−ジシク
ロヘキシルホスファニル−ビフェニル−2−イル)−ジメチル−アミン、[2’−(ジ−
t−ブチル−ホスファニル)−ビフェニル−2−イル]−ジメチル−アミン、およびジシ
クロヘキシル−(2’,4’,6’−トリ−プロピル−ビフェニル−2−イル)−ホスフ
ァンを含む。更に、カルベン配位子、例えば1,3−ビス−(2,6−ジ−イソプロピル
−フェニル)−3H−イミダゾル−1−イウムなど;クロリドをホスフィン配位子の代わ
りに使用することもできる。1つの実施形態においては、そのホスフィン配位子はrac
−BINAP、DPPFまたはDPEphosであり、特にはrac−BINAPである
。このホスフィン配位子は、通常、0.1モル%から10モル%までの間、例えば1モル
%から5モル%までの間などで適用され、典型的には約1〜2モル%の量で適用される。
【0101】
pHを高めるために、塩基が反応混合物に加えられる。特には、NaOt−Bu、KO
t−BuおよびCs2CO3から選択された塩基が有用である。有機塩基、例えば1,8
−ジアザビシクロ[5.4.0]ウンデカ−7−エン(DBU)および1,4−ジアザビ
シクロ[2.2.2]オクタン(DABCO)なども同様に適用することができる。特に
、NaO(t−Bu)およびKO(t−Bu)の名を挙げることができる。典型的には、
この塩基は約1〜5当量の量で加えられ、例えば1〜3当量など、例えば2〜3当量など
の量で加えられる。
【0102】
化合物IVはピペラジン化合物である。ピペラジンは2個の窒素を有しており、そのう
ちの1個のみがC−N結合の形成に関与する。1つの実施形態においては、第二の窒素に
対する結合の形成は、モノ−保護ピペラジンを用いることにより、即ち、式中のRが保護
基である実施形態を用いることにより回避されている。多くの保護基が当分野において公
知であり、有用な例はboc、Bn、Cbz、C(=O)OおよびMeを含み、特にはb
ocである。Bnはベンジルの略号であり;bocはt−ブチルオキシカルボニルの略号
であり;cbzはベンジルオキシカルボニルの略号である。もし保護型のピペラジンがそ
れらの反応で使用される場合には、その保護基は以降のステップで取り除かれなければな
らず、典型的には酸水溶液を加えることにより除去される。メチルが保護基として使用さ
れる場合には、そのメチルは、カルバマートとの反応およびそれ以降のこの基の除去によ
り取り除かれてよい。
【0103】
驚くべきことに、第二の窒素への望ましくない結合の形成を伴うことなく、非保護型ピ
ペラジンも同様に使用され得ることが判明した。保護型のピペラジンおよび非保護型のピ
ペラジンは、種々の異なる溶剤中において異なる溶解度を有している;一例を挙げれば、
ピペラジンはトルエン中に事実上溶けないが、その一方で、bocで保護されたピペラジ
ンはトルエンに高度に可溶性である。通常は、適用された溶剤中にすべての反応物が容易
に溶けることが反応の成功にとっての必要条件であると期待されよう。それにもかかわら
ず、本発明の方法は、溶剤としてトルエンを用い、且つ、非保護型のピペラジン、即ち、
式中のRが水素である実施形態のピペラジンを用いて、高い収率で実施されることが判明
した。従って、1つの実施形態においては、溶剤はトルエンであり、化合物IVはピペラ
ジンである。1つの更なる実施形態においては、この条件の組み合わせがワンポット合成
で用いられる。
【0104】
1つの実施形態においては、方法を実施するための温度は、約80℃〜約120℃であ
る。
【0105】
1つの実施形態においては、1−[2−(2,4−ジメチル−フェニルスルファニル)
フェニル]−ピペラジンが以下のステップを含む方法で製造される:
a.1〜1.5当量の化合物II、IIIおよびIVをトルエン中に溶解または分散さ
せて、混合物Aを得るステップ;
b.場合によってはトルエン中に分散もしくは溶解または分散された、1〜2モル%の
Pddba2および1〜2モル%のrac−BINAPを2〜3当量のNaOt−Buと
共に混合物Aに加えて混合物Bを得、化合物IIおよびIIIが完全に変換されるまで、
典型的には5〜10時間、この混合物Bを約100℃に加熱するステップ;
c.化合物IVが完全に変換されるまで、典型的には16〜32時間、ステップbで得
られた混合物の温度を約120℃に高めるステップ;および
d.場合によって、化合物IIIが保護型のピペラジンであるときに、酸水溶液を加え
ることによりその保護基を取り除くステップ。
【0106】
場合によっては、上の一連の反応ステップに精製ステップが含められてよい。
【0107】
1つの実施形態においては、1〜1.5当量の2,4−ジメチル−チオール、1−ブロ
モ−2−ヨードベンゼン(または1,2−ジブロモ−ベンゼン)およびピペラジンがトル
エン中に分散され、続いて、トルエン中に分散された2〜5当量、例えば3当量などのN
aOt−Buならびに1〜2モル%のPd2dba3およびrac−BINAPを加えて
混合物を得、この混合物を2〜10時間、典型的には3〜5時間還流させて1−[2−(
2,4−ジメチル−フェニルスルファニル)−フェニル]−ピペラジンを得る。場合によ
っては、対応する臭化水素酸付加塩を得るため、この生成物を更にHBr水溶液と反応さ
せることもできる。
【0108】
1つの実施形態においては、2〜5当量のNaOt−Bu、2〜5当量のピペラジン、
0.2〜0.6モル%のPddba2および0.6〜1モル%のrac−BINAPをト
ルエン中に分散させて混合物A’を得、その混合物に約1当量の2−ブロモ−ヨードベン
ゼンを加えて混合物B’を得、その混合物に1当量の2,4−ジメチルチオフェノールを
加え、結果として生じた混合物を3〜7時間、例えば4〜6時間加熱して還流させ、1−
[2−(2,4−ジメチル−フェニルスルファニル)−フェニル]−ピペラジンを得る。
場合によっては、対応する臭化水素酸付加塩を得るため、この生成物を更にHBr水溶液
と反応させることもできる。
【0109】
ある状況においては、遊離塩基としてよりもむしろ、1−[2−(2,4−ジメチル−
フェニルスルファニル)−フェニル]−ピペラジンの酸付加塩を得る方が望ましい場合が
あり得る。酸付加塩は更なるプロセスステップにおいて得られてよく、そのステップでは
、得られた遊離塩基が適切な酸、例えばフマル酸、硫酸、塩酸または臭化水素酸などと反
応させられる。その酸は、反応混合物に直接的に加えられてもよいし、または、代替的に
、遊離塩基がそのようなステップの前に最初に何らかの適切な程度にまで精製されてもよ
い。もしも遊離塩基が乾燥化合物として単離されている場合には、酸との反応に先立って
その遊離塩基を溶液にするため、溶剤を使用することが必要になるであろう。1つの実施
形態においては、何らその遊離塩基の初期精製を伴うことなく、臭化水素酸水溶液が反応
混合物に直接的に加えられる。
【0110】
保護型のピペラジンが使用されている方法においては、上で説明されているように、酸
水溶液を加えることによって保護基を取り除かなければならない。1つの実施形態におい
ては、上述の酸水溶液は、2つの変換、即ち、保護型ピペラジンの脱保護および酸付加塩
の形成を達成することができるように選択されてよい。特に、臭化水素酸水溶液を用いて
、1つのプロセスステップで、保護型ピペラジンを脱保護し、且つ、臭化水素酸付加塩を
得ることができる。
【0111】
ここで述べられているすべての反応および反応混合物に対して、それらを不活性ガスで
パージすることまたはそれらを不活性ガスのブランケットの下で実行することが有利であ
り得るという考えが当てはまる。窒素は、安価で容易に入手可能な不活性ガスの例である
。
【0112】
出版物、特許出願および特許を含め、ここで引用されているすべての参考文献は、これ
をもって、参照により、それらの内容全体が、また、本明細書のどこか別の個所で為され
ている何らかの別個に与えられた特定の書類の組み込みに関係なく、それぞれの参考文献
が参照により組み込まれるべく個別的および具体的に指示されているのと同じ程度にまで
、および、その内容全体が本明細書において説明されているのと同じ程度にまで(法律に
より許容される最大限の程度にまで)本明細書に組み込まれる。
【0113】
本発明を説明する文脈における「a」、「an」および「the」という用語ならびに
同様な指示対象の使用は、本明細書で別な具合に指示されていない限り、または文脈によ
って明らかに否定されていない限り、単数形と複数形との両方をカバーするものと解釈す
べきである。例えば、「the compound(本化合物)」という語句は、別な具
合に指示されていない限り、本発明または特定の開述されている態様の様々な化合物を表
すものと理解すべきである。
【0114】
別な具合に指示されていない限り、本明細書で与えられているすべての正確な値は、対
応する大凡の値の代表である(例えば、特定のファクターまたは測量に関して与えられて
いるすべての正確な例証的値は、適切な個所では「約」に変更し、対応する大凡の測量も
与えているものと見なすことができる)。
【0115】
1つの構成要素または複数の構成要素に関する「含む(comprising)」、「有する(ha
ving)」、「含有する(including)」または「含む(containing)」などの用語を用い
た何らかの態様または本発明の態様についての本明細書での説明は、別な具合に述べられ
ていない限り、または文脈によって明らかに否定されていない限り、その特定の構成要素
または複数の特定の構成要素に関する「からなる」、「本質的に(そのような構成要素)
からなる」、または「(そのような構成要素)を実質的に含む」同様な態様または本発明
の態様に対する支持を与えるべく意図されている(例えば、特定の構成要素を含むものと
して本明細書で説明されている組成物は、別な具合に述べられていない限り、または文脈
によって明らかに否定されていない限り、その構成要素からなる組成物をも説明している
ものとして理解されるべきである)。
【実施例】
【0116】
分析方法
1H NMRスペクトルが500.13MHzにおいてBruker Avance
DRX500装置に記録される。溶剤としてジメチルスルホキシド(99.8%D)が使
用され、内部参照標準としてテトラメチルシラン(TMS)が使用される。
【0117】
融点が示差走査熱量測定法(DSC)を用いて測定される。その機器は、開始値として
融点を与えるべく5°/分で校正されたTA−InstrumentsのDSC−Q10
00である。約2mgのサンプルが窒素フロー下におけるゆるく閉じられたパン内におい
て5°/分で加熱される。
【0118】
乾燥された材料の溶剤/水含有量を見積もるために使用される熱重量分析(TGA)は
、TA−InstrumentsのTGA−Q500を用いて実施される。1〜10mg
のサンプルが窒素フロー下における開放されたパン内において10°/分で加熱される。
【0119】
粉末X線回折図形が、CuKα1放射線を用いるPANalytical X’Per
t PRO X−Ray Diffractometerで測定された。サンプルは、X
’celerator検出器を用いて2θ−レンジ5〜40°における反射モードで測定
された。
【0120】
実施例1:インビトロ受容体薬理学
ラットセロトニントランスポーター:IC50 5.3nM(5−HT取り込みの遮断
)
ヒトセロトニントランスポーター:IC50 40nM(5−HT取り込みの遮断)
ヒト5−HT1A受容体:部分的アゴニズムを有し、Ki 40nM(効力85%)
ラット5−HT3受容体:IC50 0.2nM(機能分析におけるアンタゴニズム)
ヒト5−HT3A受容体:IC50約20nM(機能分析におけるアンタゴニズム)。
より高い濃度では、前記化合物は、2.1μMのED50を有するアゴニスト活性を示す
。また、本発明の化合物は、インビトロ結合アッセイにおいて、ヒト5HT3受容体に対
して高い親和性も示した(Ki 4.5nM)。
【0121】
実施例2:認識作用
上で検討されているように、本発明の化合物はコリン作動系と相互作用し、また、イン
ビボモデルにおいて、以下の項目のうちの1つまたはそれ以上で効果をみることが期待さ
れよう。
【0122】
・5選択反応時間試験(5−CSRT)(連続的な注意に及ぼす効果を実証するのに有
用)
・空間Y字型迷路試験(短期記憶、長期記憶および作業記憶に及ぼす効果を実証するの
に有用)
・注意セットの移行モデル(attentional set shifting model)(実行機能、即ち、論
理的思考および問題解決に及ぼす効果を実証するのに有用)。
【0123】
実施例3a:遊離塩基の化合物Iの製造
10グラムの1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピペ
ラジンヒドロブロミドを100mlの3MのNaOHおよび100mlのエチルアセター
トの攪拌混合物で10分間処理した。その有機相を分離し、100mlの15%wtのN
aCl(aq)で洗浄し、MgSO4上で乾燥させ、濾過し、真空下で濃縮することによ
り、透明な無色の油状物として、7.7グラム(98%)の化合物Iの塩基を得た。
NMRは構造に適合する。
【0124】
実施例3b:結晶質塩基の化合物Iの製造
3.0グラムの1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピ
ペラジンの無色の油状物を70mlのアセトニトリルで処理し、加熱して還流させた。そ
のほとんど透明な溶液を濾過し、得られた透明な濾液を自然発生的に冷却すると、濾過後
間もなく沈殿が始まった。その混合物を室温(22℃)で2時間撹拌し、得られた生成物
を濾過により単離し、真空下(40℃)で一晩乾燥させた。2.7グラム(90%)の白
色の固体として結晶質塩基が単離された。NMRは構造に適合する。元素分析:72.4
0%のC、9.28%のN、7.58%のH(理論値:72.26%のC、9.36%の
N、7.42%のH)。
【0125】
実施例3c:結晶質塩基の化合物Iの特性描写
実施例3bのようにして製造されるこの塩基は結晶質である(XRPD)−図1参照。
この結晶質塩基は約117℃の融点を有している。また、この塩基は吸湿性ではなく、且
つ、水中における0.1mg/mlの溶解度を有している。
【0126】
実施例4a:化合物Iのアルファ型の臭化水素酸塩の製造
2.0グラムの1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピ
ペラジンを熱い30mlのエチルアセタート中に溶解し、0.73mlの48%wtのH
Br(aq)を加えた。この添加は濃厚なスラリーの形成をもたらし、適切に撹拌できる
ようにするため、更に10mlのエチルアセタートを加えた。そのスラリーを室温で1時
間撹拌した。濾過し、真空下(20℃)で一晩乾燥させることにより、白色の固体として
2.0グラムの生成物が得られた(80%)。NMRは構造に適合する。元素分析:57
.05%のC、7.18%のN、6.16%のH(1:1の塩に対する理論値:56.9
9%のC、7.39%のN、6.11%のH)。
【0127】
実施例4b:化合物Iのアルファ型のヒドロブロミドの特性描写
実施例4aのようにして製造されるこのアルファ型のヒドロブロミドは結晶質である(
XRPD)−図2参照。この結晶質ヒドロブロミドは約226℃の融点を有している。ま
た、このヒドロブロミドは高い相対湿度に晒されたときに約0.3%の水分を吸収し、且
つ、水中における2mg/mlの溶解度を有している。
【0128】
実施例4c:化合物Iのベータ型の臭化水素酸塩の製造
49.5グラムの1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]
ピペラジンの無色の油状物を500mlのエチルアセタート中に溶解し、18.5mlの
48%wtのHBr(aq)を加えた。この添加は濃厚なスラリーの形成をもたらし、そ
のスラリーを室温で一晩撹拌した。濾過し、真空下(50℃)で一晩乾燥させることによ
り、白色の固体として29.6グラムの生成物が得られた(47%)。
NMRは構造に適合する。元素分析:56.86%のC、7.35%のN、6.24%の
H(1:1の塩に対する理論値:56.99%のC、7.39%のN、6.11%のH)
。
【0129】
実施例4d:化合物Iのベータ型のヒドロブロミドの特性描写
実施例4cのようにして製造されるこのベータ型のヒドロブロミドは結晶質である(X
RPD)−図3参照。この結晶質ヒドロブロミドは約231℃の融点を有している。また
、このヒドロブロミドは高い相対湿度に晒されたときに約0.6%の水分を吸収し、且つ
、水中における1.2mg/mlの溶解度を有している。
【0130】
実施例4e:化合物Iのガンマ型の臭化水素酸塩の製造
実施例4aのようにして製造された1gの1−[2−(2,4−ジメチルフェニルスル
ファニル)−フェニル]ピペラジンヒドロブロミドを20mlの水に加え、85℃に加熱
した。その溶液はほとんど透明であった。1滴のHBrを加えることによりその溶液が透
明になった。曇り点が観測されるまでHBrを加えた。その溶液を室温にまで冷却し、乾
燥させた。NMRは構造に適合する。元素分析:56.63%のC、7.18%のN、6
.21%のH(1:1の塩に対する理論値:56.99%のC、7.39%のN、6.1
1%のH)。
【0131】
実施例4f:化合物Iのガンマ型のヒドロブロミドの特性描写
実施例6eのようにして製造されるこのヒドロブロミドは結晶質である(XRPD)−
図4参照。このDSC曲線は約100℃における幾つかの熱的事象:恐らくは結晶形の変
化;を示している。その後、このヒドロブロミドは約220℃で溶融する。この結晶質ヒ
ドロブロミドは高い相対湿度に晒されたときに約4.5%の水分を吸収し、また、室温で
30%のRHのときには約2%の水分が吸収される。
【0132】
実施例4g:化合物Iのヒドロブロミド水和物の製造
1.4グラムの1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピ
ペラジンの油状物を20mlの水に加え、60℃に加熱した。48%のHBrを用いてp
Hを1に調節した。その溶液を室温にまで冷却し、乾燥させた。NMRは構造に適合する
。元素分析:55.21%のC、7.16%のN、6.34%のH(1:1の塩半水和物
に対する理論値:55.68%のC、7.21%のN、6.23%のH)。
【0133】
実施例4h:化合物Iのヒドロブロミドの半水和物の特性描写
実施例4gのようにして製造されるこの水和物は結晶質である(XRPD)−図5参照
。水分の含有量は相対湿度に強く依存する。室温で95%のRHのときには、含水率は約
3.7%である。約100℃に加熱することにより脱水が起こる。
【0134】
実施例4i:化合物Iの臭化水素酸塩のエチルアセタート溶媒和物の製造
0.9グラムの1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピ
ペラジンの油状物を35mlのエチルアセタート中に溶解し、0.5mlの48%wtの
HBr(aq)を加えた。この添加は濃厚なスラリーの形成をもたらし、そのスラリーを
室温で一晩撹拌した。濾過し、30mlのジエチルエーテルで洗った後、真空下(50℃
)で一晩乾燥させることにより、1.0グラム(65%)の1−[2−(2,4−ジメチ
ルフェニルスルファニル)−フェニル]ピペラジンHBrEtOAc溶媒和物が得られた
。NMRは構造に適合する。元素分析:56.19%のC、6.60%のN、6.56%
のH(TGAおよびKFにより決定した際の8%のエチルアセタートおよび0.5%の水
に対して補正されたときの1:1の塩に対する理論値:56.51%のC、6.76%の
N、6.38%のH)。
【0135】
実施例4j:化合物Iのヒドロブロミドのエチルアセタート溶媒和物の特性描写
実施例4iのようにして製造されるこのエチルアセタート溶媒和物は結晶質である(X
RPD)−図6参照。そのバッチは、恐らくは乾燥が部分的に脱溶媒和を引き起こしたた
め、化合物Iの溶媒和物とアルファ型との混合物を含んでいる。この脱溶媒和は、10°
/分で加熱した場合、約75℃で始まる。脱溶媒和後、アルファ型が形成される。高い相
対湿度に晒されると、エチルアセタートが水で置換され、その後に湿度が下げられたとき
にその水分が放出される。結果として得られる固体は吸湿性であり、高い相対湿度のとき
には3.2%の水分を吸収する。
【0136】
実施例5a:化合物Iの塩酸塩の製造
1.0グラムの1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピ
ペラジンの油状物を、穏やかに加熱(30℃)しながら、20mlのエチルアセタート中
に溶解した。透明な溶液が得られたときに、ジエチルエーテル中における2MのHClの
溶液を、pHが約1〜2になるまでゆっくりと加えた。この添加中に、自然発生的な沈殿
が観測された。最後の添加後、その懸濁液を1時間撹拌し、その後、濾過し、真空下(4
0℃)で一晩乾燥させることにより、白色の沈殿物が単離された。1.1グラム(99%
)の1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピペラジンヒド
ロクロリドが単離された。
NMRは構造に適合する。元素分析:64.18%のC、8.25%のN、6.96%の
H(TGAにより決定した際の0.66%の水に対して補正されたときの1:1の塩に対
する理論値:64.13%のC、8.31%のN、6.95%のH)。
【0137】
実施例5b:化合物Iのヒドロクロリドの特性描写
実施例5aのようにして製造されるこのヒドロクロリドは結晶質である(XRPD)−
図7参照。この結晶質ヒドロクロリドは約236℃の融点を有している。また、このヒド
ロクロリドは高い相対湿度に晒されたときに約1.5%の水分を吸収し、且つ、水中にお
ける3mg/mlの溶解度を有している。
【0138】
実施例5c:化合物Iのヒドロクロリド一水和物の製造
11.9グラムの1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]
ピペラジンの油状物を、加熱しながら、100mlのエタノール中に溶解した。均質な溶
液が得られたときに行われた3.5mlの濃HCl(aq)の添加は、白色の固体の即座
の沈殿を引き起こした。得られた懸濁液を最初に5分間攪拌し、次いで、氷浴上で更に1
時間攪拌した後、濾過した。その白色の固体を100mlの新鮮な冷たいエタノール(−
18℃で2時間冷凍庫に置いておいたもの)、50mlのアセトンおよび最後に50ml
のジエチルエーテルを用いて洗った後、真空下(50℃)において一晩乾燥させた。5.
1グラム(38%)の1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル
]ピペラジンHClが単離された。
NMRは構造に適合する。元素分析:61.23%のC、7.91%のN、7.16%の
H(1:1の塩一水和物に対する理論値:61.26%のC、7.94%のN、7.14
%のH)。
【0139】
実施例5d:化合物Iのヒドロクロリド一水和物の特性描写
実施例5cのようにして製造されるこのヒドロクロリド一水和物は結晶質である(XR
PD)−図8参照。この結晶質の一水和物は約50℃で水分を失い始める。更なる加熱に
より幾つかの熱的事象、恐らくは転位が起こり、この一水和物は約230℃で溶融し、続
いて、約236℃で再結晶化および融解する。この結晶質一水和物は高い相対湿度に晒さ
れたときに更なる量の水分を吸収せず、また、この水和物に結合された水は、相対湿度が
室温で10%RH以下に下がるまで放出されない。この結晶質一水和物は、水中における
約2mg/mlの溶解度を有している。
【0140】
実施例6a:化合物Iのメシル酸塩の製造
1.0グラムの1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピ
ペラジンの油状物を、加熱(70℃)により、20mlのエチルアセタート中に溶解した
。透明な溶液が得られたときに、0.35グラムのメタンスルホン酸(1.1当量)をゆ
っくりと加えた。最後の添加後、その溶液を氷上で冷却し、ジエチルエーテルをゆっくり
と加えることにより、生成物の沈殿がもたらされた。その懸濁液を氷上で2時間撹拌した
後、生じた白色の沈殿物を濾過により単離し、真空下(40℃)において一晩乾燥させた
。1.1グラム(85%)の1−[2−(2,4−ジメチルフェニルスルファニル)−フ
ェニル]ピペラジンメシラートが単離された。NMRは構造に適合する。元素分析:57
.81%のC、6.81%のN、6.68%のH(1:1の塩に対する理論値:57.8
1%のC、7.10%のN、6.64%のH)。
【0141】
実施例6b:化合物Iのメシラートの特性描写
実施例7aのようにして製造されるこのメシラートは結晶質である(XRPD)−図9
参照。この結晶質メシラートは約163℃の融点を有している。また、このメシラートは
吸湿性である(80%の相対湿度に晒されたときに約8%の水分を吸収し、これにより、
水和された形態に変換される。吸収された水のうちの最後の6%は相対湿度が10%RH
以下になるまで放出されない。この結晶質メシラートは、水中における非常に高い溶解度
(>45mg/ml)を有している。
【0142】
実施例7a:化合物Iのフマラートの製造
50mlのメタノールおよび50mlのエチルアセタートの混合物中において、5.5
グラムの1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピペラジン
の油状物を加熱し、還流させた。その溶液を放置して冷却した僅か後に2.1グラムのフ
マル酸を加えることにより、発熱反応が起こり、白色の固体の沈殿がもたらされた。得ら
れた懸濁液を撹拌し、その間、室温にまで冷却させ、続いて、−18℃において冷凍庫内
で2時間冷却した。その白色の固体を濾過により収集し、20mlの冷たいエチルアセタ
ートで洗った後、真空下(50℃)において一晩乾燥させた。3.1グラム(44%)の
生成物が単離された。
【0143】
NMRは構造に適合する。元素分析:63.42%のC、6.64%のN、6.42%
のH(1:1の塩に対する理論値:63.74%のC、6.76%のN、6.32%のH
)。
【0144】
実施例7b:化合物Iのフマラートの特性描写
実施例7aのようにして製造されるこのフマラートは結晶質である(XRPD)−図1
0参照。この結晶質フマラートは約194℃の融点を有している。水中における溶解度は
0.4mg/mlである。
【0145】
実施例8a:化合物Iのマレアートの製造
2.5グラムの1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピ
ペラジンの油状物を50mlのエチルアセタート中に溶解し、60度に加熱した後、1.
1グラムのマレイン酸を加えた。その混合物を再び加熱して5分間還流させ、撹拌しなが
ら放置して室温にまで冷却した。冷却している間に沈殿が始まり、冷凍庫(−18℃)内
に4時間入れることにより終結された。その白色の固体を濾過により収集し、50mlの
ジエチルエーテルで洗った後、真空下(50℃)において一晩乾燥させた。これにより1
.3グラムの1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピペラ
ジンマレアート(38%)が得られ、このマレアートを還流時に40mlのエチルアセタ
ートおよび5mlのメタノールで処理することにより再結晶化した。その透明な溶液を室
温にまで冷却し、続いて、冷凍庫(−18℃)内で2時間冷却した後、濾過し、10ml
の冷たいエチルアセタートで2回洗浄し、その後、真空下(50℃)において2日間乾燥
させた。0.9グラム(69%)の1−[2−(2,4−ジメチルフェニルスルファニル
)−フェニル]ピペラジンマレアートが単離された。NMRは構造に適合する。元素分析
:63.57%のC、6.79%のN、6.39%のH(1:1の塩に対する理論値:6
3.74%のC、6.76%のN、6.32%のH)。
【0146】
実施例8b:化合物Iのマレアートの特性描写
実施例8aのようにして製造されるこのマレアートは結晶質である(XRPD)−図1
1参照。この結晶質フマラートは約152℃の融点を有している。水中における溶解度は
約1mg/mlである。
【0147】
実施例9a:化合物Iのメソ−タルトラートの製造
アセトン中における1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル
]ピペラジンの11.1mlの0.30M溶液を5mlのアセトン中に溶解された0.5
グラムのメソ−酒石酸で処理した。得られた混合物を室温で30分間撹拌し、その間に沈
殿が起った。濾過し、最初に5mlのアセトン次いで3mlのジエチルエーテルで洗うこ
とにより、白色の固体として生成物が得られ、その生成物を真空下(50℃)において一
晩乾燥させた。1.4グラム(93%)の1−[2−(2,4−ジメチルフェニルスルフ
ァニル)−フェニル]ピペラジンメソ−酒石酸が単離された。NMRは構造に適合する。
元素分析:58.58%のC、6.29%のN、6.40%のH(1:1の塩に対する理
論値:58.91%のC、6.25%のN、6.29%のH)。
【0148】
実施例9b:化合物Iのメソ−タルトラートの特性描写
実施例9aのようにして製造されるこのメソ−タルトラートは結晶質である(XRPD
)−図12参照。この結晶質のメソ−タルトラートは約164℃の融点を有している。水
中における溶解度は約0.7mg/mlである。
【0149】
実施例10a:化合物IのL−(+)−タルトラートの製造
アセトン中における1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル
]ピペラジンの11.1mlの0.30M溶液を5mlのアセトン中に溶解された0.5
グラムのL−(+)−酒石酸で処理した。得られた混合物を室温で30分間撹拌し、その
間に沈殿が起った。濾過し、最初に5mlのアセトン次いで3mlのジエチルエーテルで
洗うことにより、白色の固体として生成物が得られ、その生成物を真空下(50℃)にお
いて一晩乾燥させた。1.2グラム(81%)の1−[2−(2,4−ジメチルフェニル
スルファニル)−フェニル]ピペラジン(+)−酒石酸が単離された。NMRは構造に適
合する。元素分析:58.86%のC、6.30%のN、6.38%のH(1:1の塩に
対する理論値:58.91%のC、6.25%のN、6.29%のH)。
【0150】
実施例10b:化合物IのL−(+)−タルトラートの特性描写
実施例10aのようにして製造されるこのL−(+)−タルトラートは結晶質である(
XRPD)−図13参照。この結晶質のL−(+)−タルトラートは約171℃の融点を
有している。水中における溶解度は約0.4mg/mlである。
【0151】
実施例11a:化合物IのD−(−)−タルトラートの製造
アセトン中における1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル
]ピペラジンの11.1mlの0.30M溶液を5mlのアセトン中に溶解された0.5
グラムのD−(−)−酒石酸で処理した。得られた混合物を室温で30分間撹拌し、その
間に沈殿が起った。濾過し、最初に5mlのアセトン次いで3mlのジエチルエーテルで
洗うことにより、白色の固体として生成物が得られ、その生成物を真空下(50℃)にお
いて一晩乾燥させた。1.0グラム(68%)の1−[2−(2,4−ジメチルフェニル
スルファニル)−フェニル]ピペラジンD−(−)−酒石酸が単離された。NMRは構造
に適合する。元素分析:58.90%のC、6.26%のN、6.35%のH(1:1の
塩に対する理論値:58.91%のC、6.25%のN、6.29%のH)。
【0152】
実施例11b:化合物IのD−(−)−タルトラートの特性描写
実施例11aのようにして製造されるこのD−(+)−タルトラートは結晶質である(
XRPD)−図14参照。この結晶質のD−(−)−タルトラートは約175℃の融点を
有している。水中における溶解度は約0.4mg/mlである。
【0153】
実施例12a:化合物Iのスルファートの製造
アセトン中における1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル
]ピペラジンの11.1mlの0.30M溶液をH2SO4(aq)の2.2mlの3M
溶液で処理した。得られた混合物を室温で30分間撹拌し、次いで、氷浴上で更に4時間
撹拌すると沈殿が起こり、その沈殿が終結された。濾過し、最初に5mlのアセトン次い
で3mlのジエチルエーテルで洗うことにより、白色の固体として生成物が得られ、その
生成物を真空下(50℃)において一晩乾燥させた。0.51グラム(39%)の1−[
2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピペラジンスルファートが
単離された。NMRは構造に適合する。元素分析:54.53%のC、7.22%のN、
6.28%のH(1:1の塩に対する理論値:54.52%のC、7.07%のN、6.
10%のH)。
【0154】
実施例12b:化合物Iのスルファートの特性描写
実施例12aのようにして製造されるこのスルファートは結晶質である(XRPD)−
図15参照。この結晶質スルファートは約166℃の融点を有している。水中における溶
解度は約0.1mg/mlである。
【0155】
実施例13a:化合物Iのホスファートの製造
アセトン中における1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル
]ピペラジンの11.1mlの0.30M溶液を0.2mlの65%のH3PO4(aq
)で処理した。得られた混合物を室温で30分間撹拌し、その間に沈殿が起った。濾過し
、最初に5mlのアセトン次いで3mlのジエチルエーテルで洗うことにより、白色の固
体として生成物が得られ、その生成物を真空下(50℃)において一晩乾燥させた。1.
23グラム(94%)の1−[2−(2,4−ジメチルフェニルスルファニル)−フェニ
ル]ピペラジンホスファートが単離された。NMRは構造に適合する。元素分析:54.
21%のC、7.15%のN、6.43%のH(1:1の塩に対する理論値:54.53
%のC、7.07%のN、6.36%のH)。
【0156】
実施例13b:化合物Iのホスファートの特性描写
実施例13aのようにして製造されるこのホスファートは結晶質である(XRPD)−
図16参照。この結晶質ホスファートは約224℃の融点を有している。水中における溶
解度は約1mg/mlである。
【0157】
実施例14a:化合物Iのニトラートの製造
アセトン中における1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル
]ピペラジンの11.1mlの0.30M溶液を0.2mlの16.5MのHNO3(a
q)で処理した。得られた混合物を室温で30分間撹拌し、その間に沈殿が起った。濾過
し、最初に5mlのアセトン次いで3mlのジエチルエーテルで洗うことにより、白色の
固体として生成物が得られ、その生成物を真空下(50℃)において一晩乾燥させた。0
.87グラム(73%)の1−[2−(2,4−ジメチルフェニルスルファニル)−フェ
ニル]ピペラジンニトラートが単離された。NMRは構造に適合する。元素分析:59.
80%のC、11.67%のN、6.51%のH(1:1の塩に対する理論値:59.8
1%のC、11.63%のN、6.41%のH)。
【0158】
実施例14b:化合物Iのニトラートの特性描写
実施例14aのようにして製造されるこのニトラートは結晶質である(XRPD)−図
17参照。この結晶質ニトラートは溶融しないが、約160℃における発熱反応下におい
て分解する。水中における溶解度は約0.8mg/mlである。
【0159】
実施例15:錠剤
以下の実施例は、本発明の化合物を含む錠剤を製造し得る方法の代表的な例を示してい
る。すべての実施例でベータ型の臭化水素酸塩が使用された。
【0160】
実施例15a
63.55gの本臭化水素酸塩、923.65gのLactosum 350M、46
1.8gのトウモロコシデンプンおよび76.0gのKollidon VA64をDi
osna PP1高剪断混合機内において1000rpmのインペラー速度で2分間混合
した。次いで、インペラーの速度を800rpmに下げ、短時間の間に220gの水を加
えた。集塊形成(massing)を7分間実行し、結果として得られた顆粒を4000μmの
サイズの篩に通した。それらの顆粒を乾燥させ、710μmのサイズの篩に通した。結果
として得られた1383.5gの顆粒を400gのAvicel PH200および60
gのAc−Di−Solと混合した。15gのマグネシウムステアラートと混合すること
によるそのブレンドの潤滑化の後、得られた粉末ブレンドをタブレット成形機へ移した。
5mgの本遊離塩基に相当する目標含有量を有する錠剤を得るため、200mgの目標コ
ア重量および8mmの直径を有する錠剤が製造された。
【0161】
実施例15b
317.75gの本臭化水素酸塩、754.15gのLactosum 350M、3
77.1gのトウモロコシデンプンおよび76.0gのKollidon VA64をD
iosna PP1高剪断混合機内において1000rpmのインペラー速度で2分間混
合した。次いで、インペラーの速度を800rpmに下げ、短時間の間に210gの水を
加えた。集塊形成を7分間実行し、結果として得られた顆粒を4000μmのサイズの篩
に通した。それらの顆粒を乾燥させ、710μmのサイズの篩に通した。結果として得ら
れた1386.2gの顆粒を400gのAvicel PH200および60gのAc−
Di−Solと混合した。15gのマグネシウムステアラートと混合することによるその
ブレンドの潤滑化の後、得られた粉末ブレンドをタブレット成形機へ移した。25mgの
本遊離塩基に相当する目標含有量を有する錠剤を得るため、200mgの目標コア重量お
よび8mmの直径を有する錠剤が製造された。
【0162】
実施例15c
32.2gの本臭化水素酸塩、944.82gのLactosum 350M、472
.4gのトウモロコシデンプンおよび76.0gのKollidon VA64をDio
sna PP1高剪断混合機内において1000rpmのインペラー速度で2分間混合し
た。次いで、インペラーの速度を800rpmに下げ、短時間の間に220gの水を加え
た。集塊形成を7分間実行し、結果として得られた顆粒を4000μmのサイズの篩に通
した。それらの顆粒を乾燥させ、710μmの篩に通した。結果として得られた1317
gの顆粒を400gのAvicel PH200および60gのAc−Di−Solと混
合した。15gのマグネシウムステアラートと混合することによるそのブレンドの潤滑化
の後、得られた粉末ブレンドをタブレット成形機へ移した。2.5mgの本遊離塩基に相
当する目標含有量を有する錠剤を得るため、208mgの目標コア重量および8mmの直
径を有する錠剤が製造された。
【0163】
実施例15d
540.85gの本臭化水素酸塩、953.00gのPearlitol 50C、2
96.22gのトウモロコシデンプンおよび70.5gのKollidon VA64を
Aeromatic−Fielder PMA1高剪断混合機内において1000rpm
のインペラー速度で2分間混合した。次いで、そのインペラー速度を800rpmに下げ
、短時間の間に241.87gの水を加えた。集塊形成を7分間実行し、結果として得ら
れた顆粒を4000μmのサイズの篩に通した。それらの顆粒を乾燥させ、710μmの
サイズの篩に通した。結果として得られた1500gの顆粒を531.91gのAvic
el PH200および85.11gのPrimojelと混合した。10.64gのマ
グネシウムステアラートと混合することによるそのブレンドの潤滑化の後、得られた粉末
ブレンドをタブレット成形機へ移した。25mgの本遊離塩基に相当する目標含有量を有
する錠剤を得るため、125mgの目標コア重量および6mmの直径を有する錠剤が製造
された。
【0164】
実施例15e
270.45gの本臭化水素酸塩、772.0gのPearlitol 50C、38
6.41gのトウモロコシデンプンおよび70.5gのKollidon VA64をA
eromatic−Fielder PMA1高剪断混合機内において1000rpmの
インペラー速度で2分間混合した。次いで、そのインペラー速度を800rpmに下げ、
短時間の間に195gの水を加えた。集塊形成を5.5分間実行し、結果として得られた
顆粒を4000μmのサイズの篩に通した。それらの顆粒を乾燥させ、710μmのサイ
ズの篩に通した。結果として得られた1200.3gの顆粒を425.5gのAvice
l PH200および68.09gのPrimojelと混合した。8.8gのマグネシ
ウムステアラートと混合することによるそのブレンドの潤滑化の後、得られた粉末ブレン
ドをタブレット成形機へ移した。10mgの本遊離塩基に相当する目標含有量を有する錠
剤を得るため、100の目標コア重量および6mmの直径を有する錠剤が製造された。
【0165】
実施例15f
504.85gの本遊離塩基、552.95gのPearlitol 50C、276
.53gのトウモロコシデンプンおよび65.7gのKollidon VA64をAe
romatic−Fielder PMA1高剪断混合機内において1000rpmのイ
ンペラー速度で2分間混合した。次いで、そのインペラー速度を800rpmに下げ、短
時間の間に182gの水を加えた。集塊形成を5.5分間実行し、結果として得られた顆
粒を4000μmのサイズの篩に通した。それらの顆粒を乾燥させ、710μmのサイズ
の篩に通した。結果として得られた1250.7gの顆粒を443.31gのAvice
l PH200および70.8gのPrimojelと混合した。8.92gのマグネシ
ウムステアラートと混合することによるそのブレンドの潤滑化の後、得られた粉末ブレン
ドをタブレット成形機へ移した。50mgの本遊離塩基に相当する目標含有量を有する錠
剤を得るため、250mgの目標コア重量および8mmの直径を有する錠剤が製造された
。
【0166】
実施例15g
135.23gの本臭化水素酸塩、863.2gのPearlitol 50C、43
2.69gのトウモロコシデンプンおよび70.66gのKollidon VA64を
Aeromatic−Fielder PMA1高剪断混合機内において1000rpm
のインペラー速度で2分間混合した。次いで、そのインペラー速度を800rpmに下げ
、短時間の間に195gの水を加えた。集塊形成を5.5分間実行し、結果として得られ
た顆粒を4000μmのサイズの篩に通した。それらの顆粒を乾燥させ、710μmのサ
イズの篩に通した。結果として得られた1200gの顆粒を425.28gのAvice
l PH200および68.2gのPrimojelと混合した。8.58gのマグネシ
ウムステアラートと混合することによるそのブレンドの潤滑化の後、得られた粉末ブレン
ドをタブレット成形機へ移した。5mgの本遊離塩基に相当する目標含有量を有する錠剤
を得るため、100mgの目標コア重量および6mmの直径を有する錠剤が製造された。
【0167】
実施例15h
67.6gの本臭化水素酸塩、908.0gのPearlitol 50C、453.
9gのトウモロコシデンプンおよび70.51gのKollidon VA64をDio
sna PP1高剪断混合機内において1000rpmのインペラー速度で2分間混合し
た。次いで、そのインペラー速度を800rpmに下げ、短時間の間に195gの水を加
えた。集塊形成を5.5分間実行し、結果として得られた顆粒を4000μmのサイズの
篩に通した。それらの顆粒を乾燥させ、710μmのサイズの篩に通した。結果として得
られた1325gの顆粒を531.91gのAvicel PH200および85.11
gのPrimojelと混合した。10.64gのマグネシウムステアラートと混合する
ことによるそのブレンドの潤滑化の後、得られた粉末ブレンドをタブレット成形機へ移し
た。5mgの本遊離塩基に相当する目標含有量を有する錠剤を得るため、207.8mg
の目標コア重量および7mmの直径を有する錠剤が製造された。
【0168】
実施例15i
2290.1gの本臭化水素酸塩、17568gの無水カルシウム水素ホスファートお
よび8783gのトウモロコシデンプンならびに1510gのコポビドンをAeroma
tic−Fielder PMA100高剪断混合機内において200rpmのインペラ
ー速度で3分間混合した。次いで、150rpmのインペラー速度における2分間の間に
5130gの水を加えた。集塊形成を15分間実行し、結果として得られた顆粒を、9.
525mmのサイズのスクリーンを備えた、約2700rpmで作動する円錐型ミルに通
した。それらの顆粒を乾燥させ、2.388mmのサイズのスクリーンを備えた、約15
00rpmで作動する円錐型ミルに通した。結果として得られた28747gの顆粒を1
1250gの微結晶性セルロース、1350gのナトリウムデンプングリコラート(タイ
プA)および1800gのタルクと混合した。450gのマグネシウムステアラートと混
合することによるそのブレンドの潤滑化の後、得られた粉末ブレンドをタブレット成形機
へ移した。5mgの本遊離塩基に相当する本臭化水素酸塩の目標含有量を有する錠剤を得
るため、125mgの目標コア重量および6mmの直径を有する錠剤が製造された。更に
、10mgの本遊離塩基に相当する本臭化水素酸塩の目標含有量を有する錠剤を得るため
、250mgの目標コア重量および8mmの直径を有する錠剤が製造された。
【0169】
実施例16:マウスでの皮内ホルマリン試験における疼痛効果(pain effects)
このモデルにおいては、マウスが左後足にホルマリン(4.5%、20μl)の注入を
受ける。このホルマリン注射により引き起こされた刺激は特徴的な二相性の行動応答を誘
発し、これは損傷を負った足をなめるのに費やした時間の量により定量化される。第一相
(約0〜10分)は直接的な化学刺激および侵害受容を表し、一方、第二相(約20〜3
0分)は神経障害性起源の痛みを表すものと考えられる。これら2つの相は鎮静期によっ
て分離されており、この鎮静期には行動が正常に戻る。疼痛性刺激を低減するための試験
化合物の有効性は、これら2つの相での損傷を負った足をなめるのに費やした時間の量を
計時することにより評価される。
【0170】
本発明の化合物は第二相における疼痛スコアの有意な低減を示し(図18b)、これは
、神経障害性起源の疼痛に対して効力を有することを示したものである。その上、本発明
の化合物は第一相におけるスコアの有意な低減も示し(図18a)、これは、最高の用量
において、より高い鎮痛作用を有することを示したものである。要約すると、これらの結
果は、本発明の化合物が疼痛性障害の治療に効果的である可能性が高いことを指示してい
る。
【0171】
実施例17
【0172】
【化7】
【0173】
20gの2−ブロモヨードベンゼン(71mmol)および9.8gの2,4−ジメチ
ルチオフェノール(71mmol)を100mlのトルエン中に溶解した。324mgの
Pd2dba3(0.35mmol;1mol%)および381mgのDPEPhos(
0.71mmol;1mol%)の前に、その溶液が窒素でパージされた。その反応混合
物を5分間攪拌し、その間に、色が暗赤色からオレンジ色に変わった。8.7gのKOB
ut(78mmol)が加えられ、直ちに不均一な混合物が形成された。その懸濁液を窒
素下において100℃に加熱した。1時間後、その混合物を0℃に冷却し、2時間撹拌し
た後、その混合物をセライトのパッドを通じて濾過した。得られた濾過ケーキを2×50
mlのトルエンで洗浄し、それらを合わせた濾液を蒸発させることにより、21gのオレ
ンジ色−赤色調の油状物が得られ(99%の収率)、この油状物は、HPLCおよびGC
−MSにより>96%の純度であることが判明した。
【0174】
実施例18
【0175】
【化8】
【0176】
機械式のスターラーを伴う1L用の三つ口丸形ボトルに500mlのトルエンを入れ、
203mgのPddba2(0.35mmol;0.1mol%)および760mgのD
PEPhos(1.5mmol;0.4mol%)を加えた。その暗赤色の溶液を窒素で
5分間パージした後、100gの2−ブロモヨードベンゼン(353mmol)および4
8.9gの2,4−ジメチルチオフェノール(353mmol)の添加が行われた。43
.6gのKOBut(389mmol)の添加は発熱反応を引き起こし、温度を20℃か
ら36℃に高め、それと同時に、不均一な混合物の形成をもたらした。その懸濁液を窒素
下において100℃に加熱した。7時間後、その混合物を0℃に冷却し、2時間撹拌した
後、セライトのパッドを通じてその混合物を濾過した。得られた濾過ケーキを2×200
mlのトルエンで洗浄し、それらを合わせた濾液を蒸発させることにより、104gのオ
レンジ色の油状物が得られ(105%の収率)、この油状物はHPLCにより97%の純
度であることが判明し、また、NMRにより望ましい構造であることが確認された。この
油状物は室温で静置している間に固化した。
【0177】
実施例19
【0178】
【化9】
【0179】
50mlの乾燥トルエン中における10グラムの1−(2−ブロモ−フェニルスルファ
ニル)−2,4−ジメチル−ベンゼン(34mmol)の溶液に7グラムのboc−ピペ
ラジン(38mmol)を加え、窒素で5分間脱気し、312mgのPd2dba3(2
mol%)および637mgのrac−BINAP(3mol%)を加え、更に5分間脱
気した後、3.9グラムのButONa(41mmol)を加え、80℃に15時間加熱
した。その反応混合物を室温にまで冷却し、20mlの15%のブラインで2回抽出し、
Na2SO4上で乾燥させ、活性炭を加え、15分間還流させ、セライトを通じて濾過し
、蒸発させることにより、NMRで決定した際に95%の純度を有する、14.2グラム
の褐色がかった油状物(4−[2−(2,4−ジメチル−フェニルスルファニル)−フェ
ニル]−BOC−ピペラジン)が得られた。この粗製油状物を200mlのMeOHおよ
び20mlの6MのHCl(aq.)中に溶解し、1時間還流させた後、HPLCは完全
な脱保護を示した。室温に冷却した後、メタノールを回転蒸発器での真空により除去し、
20mlの濃NaOH(pHは13〜14であった)を加え、その後、得られた混合物を
100mlのEtOAcとともに15分間攪拌した。その有機相を収集し、30mlの1
5%のブラインで2回抽出し、Na2SO4上で乾燥させ、30mlのMeOH中におけ
る5.2gのフマル酸(44mmol)を加えた。加熱して還流させている間に均一な溶
液が形成され、更なる加熱または冷却のどちらかの間に、その溶液から急速な沈殿が生じ
る。その沈殿物を収集し、20mlのEtOAcおよび20mlのアセトンで洗浄し、真
空下で乾燥させることにより、66%の全収率で、白色の粉末として9.3グラムの1−
[2−(2,4−ジメチル−フェニルスルファニル)−フェニル]−ピペラジンフマラー
ト(22mmol)が得られ、このフマラートは、LC−MSにより99.5%の純度を
有していた。
【0180】
実施例20
【0181】
【化10】
【0182】
100gの1,2−ジブロモベンゼン(424mmol)および58.6gの2,4−
ジメチルチオフェノール(424mmol)を800mlのトルエン中に溶解する。4.
6gのPddba2(8mmol;2mol%)および13.1gのrac−BINAP
(21mmol;5mol%)の前に、その溶液が窒素でパージされる。反応混合物を5
分間攪拌し、その間に、色が暗赤色からオレンジ色に変わる。61gのNaOBut(6
36mmol)および200mlのトルエンが加えられ、直ちに不均一な混合物が形成さ
れた。その懸濁液を窒素下において80℃に加熱した。10時間後、その混合物を60℃
に冷却した後、500mlのトルエン中における102.6gのboc−ピペラジン(5
51mmol)および別の61gのNaOBut(636mmol)を加える。その反応
混合物を窒素でパージした後、別な部の4.6gのPddba2(8mmol;2mol
%)および13.1gのrac−BINAP(21mmol;5mol%)を加えた。今
度は、更に6時間またはHPLCが完全な変換を示すまで、その混合物を加熱して還流さ
せた(110℃)。得られた反応混合物を氷上で2時間冷却した後、その混合物をセライ
トのパッドを通じて濾過した。得られた濾過ケーキを2×200mlのトルエンで洗浄し
、それらを合わせた濾液を蒸発させることにより、242gの赤色の油状物が得られる。
この油状物を1000mlのMeOH中に溶解し、115mlの48wt%のHBr(a
q.)をゆっくりと加え、続いて、加熱して2時間還流させた後、HPLCにより完全な
脱保護が検出された。その混合物を冷却し、1000mlのEtOAcを加え、蒸発によ
りMeOHを除去した。1000mlのEt2Oの添加は沈殿を引き起こした。室温で2
時間撹拌し続けた後、そのスラリーを冷凍庫内で一晩放置した(−18℃)。濾過し、2
00mlのEt2Oで2回洗浄し、40℃において真空下で乾燥させることにより、17
2gの褐色がかった固体が得られた。その褐色がかった固体を1500mlの沸騰したH
2Oで1時間処理した後、更に2時間かけて室温にまで冷却した。濾過し、40℃におい
て真空下で一晩乾燥させることにより、98gの4−[2−(2,4−ジメチル−フェニ
ルスルファニル)−フェニル]−ピペラジンヒドロブロミド(61%)が得られた。
【0183】
実施例21
【0184】
【化11】
【0185】
102gの2−ブロモ−ヨードベンゼン(362mmol)および50gの2,4−ジ
メチルチオフェノール(362mmol)を1000mlのトルエン中に溶解する。この
溶液に81gのBOC−ピペラジン(434mmol)を加え、続いて、2.08gのP
ddba2(1mol%)および4.51gのrac−BINAP(2mol%)を加え
た。この混合物を窒素で5分間パージした後、300mlのトルエン中における87gの
NaOBut(905mmol)のスラリーを加えた。得られた懸濁液を窒素下において
一晩100℃に加熱した。GCMS分析は中間生成物(1−(2−ブロモ−フェニルスル
ファニル)−2,4−ジメチル−ベンゼン)への完全な変換を示し、その温度を上昇させ
て更に24時間還流させた(120℃)。HPLC分析は中間体(1−BOC−4−[2
−(2,4−ジメチル−フェニルスルファニル)−フェニル]−ピペラジン)への完全な
変換を示した。その反応混合物を氷上で1時間冷却した後、その混合物を濾過した。得ら
れた濾過ケーキを2×200mlのトルエンで洗浄し、それらを合わせた濾液に80ml
の48wt%のHBr(aq.)を加え、続いて、加熱して18時間還流させた後、HP
LCにより完全な脱保護が検出された。得られた混合物を氷上で2時間冷却し、濾過した
。その褐色がかった固体を、活性炭(25g)とともに、1000mlの沸騰したH2O
中に1時間溶解し、熱いうちに濾過し、放置して冷却した。生じた沈殿物を濾過により収
集し、40℃において真空下で一晩乾燥させることにより、白色の固体として49gの4
−[2−(2,4−ジメチル−フェニルスルファニル)−フェニル]−ピペラジンヒドロ
ブロミド(36%)が得られた。
【0186】
実施例22
【0187】
【化12】
【0188】
機械式のスターラーを伴う1L用の三つ口丸形ボトルに500mlのトルエンを入れ、
809mgのPd2dba3(0.88mmol;0.5mol%)および952mgの
DPEPhos(1.77mmol;0.5mol%)を加えた。その暗赤色の溶液を窒
素で5分間パージした後、100gの2−ブロモヨードベンゼン(353mmol)およ
び48.9gの2,4−ジメチルチオフェノール(353mmol)の添加が行われた。
43.6gのKOBut(389mmol)の添加は発熱反応を引き起こし、温度を20
℃から42℃に高め、それと同時に、不均一な混合物の形成がもたらされ、また、その色
が暗赤色からオレンジ色/褐色がかった色に変わった。その懸濁液を窒素下において10
0℃に加熱した。僅か20分後、HPLCは1−(2−ブロモ−フェニルスルファニル)
−2,4−ジメチル−ベンゼンへの完全な変換を示した。その混合物を40℃に冷却し、
600mlの15wt%のNaClを加え、5分間攪拌した。その有機相を分離し、水性
相を2×100mlのトルエンで洗った。それらを合わせた有機相を100mlの2Mの
HCl(aq.)、100mlの15wt%のNaClで洗浄し、Na2SO4上で乾燥
させ、活性炭(10g)とともに15分間還流させ、2回濾過し、蒸発させることにより
、107.3gのオレンジ色−赤色の油状物が得られ(103%)、この油状物はHPL
Cにより98%の純度であることが判明した。
【0189】
500mlの乾燥トルエン中における90グラムの上述のオレンジ色−赤色の油状物(
307mmol)の溶液に57グラムのboc−ピペラジン(307mmol)を加え、
窒素で5分間脱気し、1.4gのPd2dba3(1.53mmol;0.5mol%)
および2.9gのrac−BINAP(4.6mmol;1.5mol%)を加え、更に
2分間脱気した後、35.4グラムのButONa(368mmol)を加え、80℃に
18時間加熱した。HPLCは完全な変換を示し、その反応混合物を室温にまで冷却した
後、濾過し、得られた濾過ケーキを2×100mlのトルエンで洗った。それらを合わせ
た濾液を2×150mlの15wt%のNaClで2回抽出し、Na2SO4上で乾燥さ
せ、活性炭を加え、30分間還流させ、2回濾過し、蒸発させることにより、140.7
グラムの褐色がかった油状物(4−[2−(2,4−ジメチル−フェニルスルファニル)
−フェニル]−BOC−ピペラジン)が得られた。この粗製油状物を300mlのMeO
Hおよび200mlの6MのHCl(aq.)中に溶解し、1時間還流させた後、HPL
Cは完全な脱保護を示した。室温に冷却した後、メタノールを回転蒸発器での真空により
除去し、200mlの濃NaOH(pHは13〜14であった)を加え、その後、得られ
た混合物を1000mlのEtOAcとともに15分間攪拌した。その有機相を収集し、
300mlの15wt%のブラインで抽出し、Na2SO4上で乾燥させ、300mlの
MeOH中における46.3gのフマル酸(399mmol)の溶液に加えた。その混合
物を加熱して還流させ、室温にまで冷却し、その後、冷凍庫内で一晩放置した(−18℃
)。生じた沈殿物を収集し、100mlのEtOAcおよび100mlのアセトンで洗浄
し、真空下(50℃)で乾燥させることにより、81%の全収率で、白色の粉末として1
03.2gの1−[2−(2,4−ジメチル−フェニルスルファニル)−フェニル]−ピ
ペラジンフマラート(249mmol)が得られ、このフマラートは、LC−MSにより
99%の純度を有していた。このフマラートを、EtOAc/H2O/濃NaOHを用い
て、遊離塩基(1−[2−(2,4−ジメチル−フェニルスルファニル)−フェニル]−
ピペラジン)に変換し、その有機相をブラインで洗浄し、Na2SO4上で乾燥させ、濾
過し、その濾液に34mlの48wt%のHBr(aq.)を加えることにより、白色の
固体の沈殿が引き起こされた。その固体を収集し、1000mlの沸騰したH2Oで処理
し、これを室温にまで冷却するとスラリーが形成された。この最終生成物(1−[2−(
2,4−ジメチル−フェニルスルファニル)−フェニル]−ピペラジンヒドロブロミド)
を濾過により収集し、真空下(50℃)において乾燥させることにより、83gの白色の
粉末(71%の全収率)が得られた[CHN(teo.)56.99;6.11;7.3
9;CHN(実測値)57.11;6.15;7.35]。
【0190】
実施例23
【0191】
【化13】
【0192】
815gのNaOBut(8.48mol)、844gのピペラジン(9.8mol)
、6.6gのPd(dba)2(11.48mmol)および13.6gのrac−BI
NAP(21.84mmol)を4Lのトルエンとともに50分間攪拌した。その後、8
40gの2−ブロモ−ヨードベンゼン(2.97mol)を1.5Lのトルエンとともに
加え、30分間攪拌し続け、最後に、390.8gの2,4−ジメチルチオフェノール(
2.83mmol)を1.5Lのトルエンとともに加えた。この懸濁液を加熱して還流さ
せ、5時間還流し続けた。その反応混合物を一晩冷ました。2Lの水を加え、1時間撹拌
した後、濾過助剤を通じてその混合物を濾過した。その後、得られた濾液を3×1Lのブ
ラインで洗った。この後、それらを合わせた水性相を600mlのトルエンで抽出した。
この後、それらを合わせたトルエン相を70℃に加熱し、続いて、329.2mlの48
wt%のHBr(aq.)および164.6mlの水を加えた。得られた混合物を室温に
まで一晩冷却した。その最終生成物(1−[2−(2,4−ジメチル−フェニルスルファ
ニル)−フェニル]−ピペラジンヒドロブロミド)を濾過により収集し、真空下(60℃
)において乾燥させることにより、895g(84%の収率)の生成物が得られた。
【0193】
実施例24
【0194】
【化14】
【0195】
40.76gのNaOBut(424.1mol)、0.33gのPddba2(0.
57mmol)および0.68gのrac−BINAP(1.09mmol)を200m
lのトルエンとともに攪拌した。その後、42gの2−ブロモ−ヨードベンゼン(362
mmol)および19.54gの2,4−ジメチルチオフェノール(362mmol)を
50mlのトルエンとともに加えた。この懸濁液を加熱して還流させ、一晩還流し続けた
。HPLC分析は、中間生成物(1−(2−ブロモ−フェニルスルファニル)−2,4−
ジメチル−ベンゼン)への完全な変換を示した。その反応混合物を室温にまで冷却し、濾
過助剤を通じて濾過した。その濾液を40.76gのNaOBut(424.1mmol
)、42.2gのピペラジン(489.9mmol)、0.33gのPddba2(0.
57mmol)および0.68gのrac−BINAP(1.09mmol)の混合物に
加え、加熱して2時間還流させた。その反応混合物を一晩冷ました。100mlの水を加
え、その水性相を分離して取り除いた。濾過助剤を通じてその有機相を濾過し、その後、
得られた濾液を3×80mlのブラインで洗った。この後、それらを合わせた水性相を5
0mlのトルエンで抽出した。その後、それらを合わせたトルエン相を70℃に加熱し、
続いて、16.5mlの48wt%のHBr(aq.)および8.25mlの水を加えた
。得られた混合物を室温にまで一晩冷却した。その最終生成物(1−[2−(2,4−ジ
メチル−フェニルスルファニル)−フェニル]−ピペラジンヒドロブロミド)を濾過によ
り収集し、真空下(60℃)において乾燥させることにより、40.18gのオフ・ホワ
イトの粉末が得られた(75%の収率)。
【0196】
実施例25
【0197】
【化15】
【0198】
40.76gのNaOBut(424.1mmol)、0.33gのPddba2(0
.57mmol)および0.68gのrac−BINAP(1.09mmol)を200
mlのトルエンとともに攪拌した。その後、42gの2−ブロモ−ヨードベンゼン(14
8.5mmol)および19.54gの2,4−ジメチルチオフェノール(141.4m
mol)を50mlのトルエンとともに加えた。この懸濁液を加熱して還流させ、一晩還
流し続けた。HPLC分析は、中間生成物(1−(2−ブロモ−フェニルスルファニル)
−2,4−ジメチル−ベンゼン)への完全な変換を示した。反応を50℃に冷却し、42
.2gのピペラジン(489.9mmol)を100mlのトルエンとともに加えた。得
られた混合物を加熱して4時間還流させた。その反応混合物を室温にまで一晩冷却した。
100mlの水を加え、濾過助剤を通じてその反応混合物を濾過した。その後、得られた
濾過ケーキを50mlのトルエンで洗った。
【0199】
その水性相を分離して取り除いた後、その有機相を3×25mlのブラインおよび25
mlの水で洗った。この後、それらを合わせた水性相を30mlのトルエンで抽出した。
その後、それらを合わせたトルエン相を70℃に加熱し、続いて、16.46mlの48
wt%のHBr(aq.)および8.23mlの水を加えた。得られた混合物を室温にま
で一晩冷却した。その最終生成物(1−[2−(2,4−ジメチル−フェニルスルファニ
ル)−フェニル]−ピペラジンヒドロブロミド)を濾過により収集し、真空下(60℃)
において乾燥させることにより、46.8g(87%の収率)の生成物が得られた。
【0200】
実施例26:自由行動ラットの脳におけるアセチルコリンの細胞外レベルに及ぼす効果
方法
動物に1−[2−(2,4−ジメチルフェニルスルファニル)フェニル]ピペラジン、
HBr塩を投与した。
【0201】
動物
最初に計量したときの体重が275〜300gの雄のSprague−Dawleyラ
ットを使用した。これらの動物は、食物および水道水を自由に摂取することができる状態
で、規則的な室内温度(21±2℃)および湿度(55±5%)が得られるように制御さ
れた条件下において、12時間の明/暗サイクルの下で飼育された。
【0202】
手術および微小透析実験
ラットにヒプノルム/ドルミカム(2ml/kg)で麻酔をかけ、腹側海馬(座標:ブ
レグマの5.6mm後方、側方−5.0mm、硬膜の7.0mm腹側)または前前頭皮質
(座標:ブレグマの3.2mm前方;側方、0.8mm;硬膜の4.0mm腹側)に透析
プローブチップを位置付けることを目的として、脳内ガイドカニューレ(CMA/12)
を脳内に定位的に埋め込んだ。アンカースクリューおよびアクリルセメントを用いてその
ガイドカニューレを固定した。動物の体温が直腸プローブによりモニタリングされ、37
℃に維持された。それらのラットは、2日間、手術からの回復が許され、一匹ずつケージ
内で飼育された。実験の当日、上述のガイドカニューレを通じて微小透析プロープ(CM
A/12、直径0.5mm、長さ3mm)が挿入された。
【0203】
それらのプローブは、複式チャンネルスイベルを介してマイクロインジェクションポン
プに接続された。濾過されたリンゲル溶液(0.5μMのネオスチグミンを含有する、1
45mmのNaCl、3mMのKCl、1mMのMgCl2、1.2mMのCaCl2)
での微小透析プローブの潅流は、脳内にプローブを挿入する少し前に始められ、1μl/
分の一定の流量で実験の期間中続けられた。安定化の180分後、実験が開始された。透
析液が20分毎に収集された。実験後、これらの動物を犠牲にし、それらの脳を取り除い
て凍結し、プローブの配置を確認するために薄片化した。
【0204】
化合物を10%のHPbetaCD中に溶解し、皮下に注射した(2.5〜10mg/
kg)。用量は塩のmg/kg体重として表現されている。化合物は2.5ml/kgの
量で投与された。
【0205】
透析液アセチルコリンの分析
透析液中におけるアセチルコリン(ACh)の濃度が、100mMの二ナトリウム水素
ホスファート、2.0mMのオクタンスルホン酸、0.5mMのテトラメチルアンモニウ
ムクロリドおよび0.005%のMB(ESA)からなるpH8.0の移動相を用いる電
気化学検出でのHPLCにより分析された。固定化されたコリンオキシダーゼを含有する
プレカラム酵素反応器(ESA)が、分析カラム(ESA ACH−250);流量0.
35ml/分、温度:35℃でのAChの分離に先立ち、注入されたサンプル(10μl
)からコリンを排除した。分析カラムの後、サンプルは、固定化されたアセチルコリンエ
ステラーゼおよびコリンオキシダーゼを含有するポストカラム固相反応器(ESA)に通
された。後者の反応器はAChをコリンに変換し、その後、コリンをベタインおよびH2
O2に変換した。後者のH2O2が白金電極(分析セル:ESA、モデル5040)を用
いることにより電気化学的に検出された。
【0206】
データの表現
単回注入実験において、化合物投与の直前の先行する3つの連続したAChサンプルの
平均値が各実験での基礎レベルとして用いられ、データが基礎(100%に正規化された
平均基礎注入前値)の百分率に変換された。
【0207】
結果
本化合物は、ラットの前前頭皮質および腹側海馬におけるAChの細胞外レベルを有意
に上昇させた−図19aおよび19b参照。
【0208】
実施例27:ラットにおける文脈的恐怖条件付け
この実験で投与された化合物は1−[2−(2,4−ジメチルフェニルスルファニル)
フェニル]ピペラジンHBr塩であった。
【0209】
我々は、ラットにおいて、文脈的恐怖条件付けの獲得、固定および想起に及ぼす本化合
物の効果について調べた。この恐怖条件付けパラダイムにおいては、動物は、中立的環境
(状況、トレーニングチャンバー、CS)を嫌悪経験(電気的フットショック、US)と
関連付けることを学ぶ。そのトレーニングチャンバーに再び暴露されると、それらの動物
はすくみ行動を示し、このすくみ行動はその恐怖関連記憶の直接的な尺度であると考えら
れている[Pavlov J.Biol.Sci.、15、177〜182、1980]
。この文脈的恐怖条件付けに関する神経解剖学は充分に研究されており、幾つかの研究が
、この記憶の形成には海馬および扁桃体が必要であることを示している[Hippoca
mpus、11、8〜17、2001;J.Neurosci.、19、1106〜11
14、1999;Behav.Neurosci.、106、274〜285、1992
]。
【0210】
動物および薬剤
12時間の明/暗サイクルの下で1つのケージ当たり2匹が飼育された、Charle
s River Laboratoriesから入手した、成体の雄のSprague−
Dawleyラット(トレーニング時の体重、250〜300g)を使用した。食物およ
び水は自由に摂取することができた。それらのラットは到着してから1週間後に使用され
た。化合物は10%のHPbetaCD中に溶解され、皮下に注入された。薬剤は、2.
5ml/kgの量で投与された。
【0211】
機器
トレーニングおよび試験は、隔離された部屋に収容され、且つ、換気システムに接続さ
れた防音チャンバー(30×20×40cm)内で実施された。照明は白色灯(60ワッ
ト)により与えられた。チャンバーの床は、電気ショック発生装置に取り付けられた金属
製の格子から成っていた。トレーニングおよび試験をする前に、そのチャンバーを70%
のエタノール溶液で清浄にした。ビデオカメラにより、オフライン分析でのトレーニング
セッションの行動の観察および記録が可能化された。
【0212】
獲得および保持試験
獲得の間、動物は、1分間の馴化期間の間、新しい環境を自由に探索することを許され
、その1分間の馴化期間は、帯電可能な格子製の床を通じて与えられる1回の避けられな
いフットショック(無条件刺激、US)と同時に終結した。そのフットショックは、持続
時間が2秒であり、0.75mAの強度であった。動物は、US後、更に60秒間、その
条件付けチャンバー内に留められた。この状況に対するベースライン−すくみ行動応答を
決定するため、最初の58秒間(ショック前獲得;実験者にはグループ情報が知らされて
いない)中にすくみ行動のスコアが付けられた。獲得が終了すると、動物は優しく取り出
され、元の自分のケージに入れられた。24時間後、同じ動物が上述のトレーニング状況
(恐怖条件付けチャンバー)に再度入れられ、2分間の保持試験が実施された。この期間
中、フットショックは加えられなかった。グループに関する情報が知らされていない実験
者によって、全試験期間中におけるすくみ行動のスコアが付けられ、合計試験期間の百分
率として提示された。
【0213】
結果および検討
ラットにおける文脈的恐怖条件付けに及ぼす化合物の効果
ラットにおける文脈的恐怖条件付けに及ぼす化合物の効果が、(i)獲得(獲得の前に
薬剤適用、図20)、(ii)記憶の想起(試験の前に薬剤適用、図21)および(ii
i)固定(獲得の直後に薬剤適用、図22)に関して調べられた。この第1セットの実験
においては、化合物(1mg/kg、5mg/kgおよび10mg/kg)が獲得セッシ
ョンの1時間前に投与された。図20は、トレーニング期間(フードショックの前の58
秒間)中および24時間後の保持試験におけるすくみ行動の獲得を描いている。以下の知
見が認められた:
・本化合物は、試験されたどの用量においても、フットショックを与える前のベースラ
インすくみ行動に影響を及ぼさない。
【0214】
・5mg/kgの用量における本化合物は、獲得の24時間後に行われる記憶保持試験
中のすくみ行動に費やす時間を増大させる傾向を有している(39.24±13.76%
、n=6、対、賦形剤で治療された動物における24.30±4.40%、n=16)。
【0215】
・10mg/kgの用量における本化合物は、獲得の24時間後に行われる保持試験中
のすくみ行動に費やす時間を有意に増大させる(52.15±5.68%、n=10、対
、賦形剤で治療された動物における24.30±4.40%、n=16、p<0.01)
。
【0216】
図20に記載されているようにこの恐怖条件付けモデルは、学習および記憶の研究用に
、文献で説明されている標準的な手順である。記憶想起に及ぼすこの薬剤の急性効果を更
に解明するため、化合物(5mg/kg、10mg/kgおよび20mg/kg)を保持
試験の1時間前に適用した。本化合物は5mg/kgの用量で記憶試験中のすくみ行動の
発現を抑制することが観測された(12.86±3.57%、n=9、対、賦形剤で治療
された動物における33.61±4.29%、n=13、p<0.05)(図21)。
【0217】
上述されているように、本化合物は、それ自体では、USを開始する前のベースライン
すくみ行動に影響を及ぼさず(図20)、従って、一見して最ももっともらしい仮説は、
図21で観測された効果が抗不安作用によるものであるという仮説である。条件付けされ
た記憶はすくみ行動、潜在的な抗不安作用を有する化合物により低減される応答により評
価される。この実験は、記憶想起の直前に与えられた本化合物が抗不安効果を有している
ことを示しており、従って、図20に示されているすくみ行動の増大が本化合物の不安惹
起作用によるものであるとは考えにくい。
【0218】
本化合物が、不安惹起性ではないが、認識力を増強させる潜在的能力(pro-cognitive
potential)を備えていることを補強的に示すため、獲得セッションの後に5mg/kg
、10mg/kgおよび20mg/kgの用量で本化合物を投与した。その結果、このセ
ットの実験において、本化合物は、獲得の間にも保持試験全体を通じてもオンボード(on
board)でなかった。ここで、5mg/kgの用量における本化合物は、獲得セッション
の24時間後に行われた保持試験中のすくみ行動に費やす時間を有意に増大させることが
観測された(45.58±4.50%、n=8、対、賦形剤で治療された動物における2
5.26±3.57%、n=19、p<0.05)。状況への再暴露中のすくみ行動に費
やす時間のパーセンテージが恐怖関連記憶の尺度として記述されており[Pavlov
J.Biol.Sci.、15、177〜182、1980]、賦形剤で治療された動物
と比べたときに、化合物で治療されたラットでは、これが増大されている(図20および
21)。ひとまとめにして考えると、これらのデータは、本化合物が文脈的記憶を高める
ことを示している。
【特許請求の範囲】
【請求項1】
化合物が非結晶質形態の遊離塩基ではないことを条件とする、1−[2−(2,4−ジ
メチルフェニルスルファニル)−フェニル]ピペラジンおよびその薬学的に許容可能な塩
。
【請求項2】
化合物が臭化水素酸塩、塩酸塩、メシル酸塩、フマル酸塩、マレイン酸塩、メソ−酒石
酸塩、L−(+)−酒石酸塩、D−(−)−酒石酸塩、硫酸塩、リン酸塩または硝酸塩で
ある、請求項1に記載の化合物。
【請求項3】
化合物が結晶質である、請求項1または2に記載の化合物。
【請求項4】
化合物が図1〜17のいずれかに示されているようなXRDPを有している、請求項3
に記載の化合物。
【請求項5】
結晶質形態の1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピペ
ラジン臭化水素酸塩である、請求項1に記載の化合物。
【請求項6】
化合物が約6.89、9.73、13.78および14.64(°2θ)におけるXR
PD反射を有している、請求項5に記載の化合物。
【請求項7】
化合物が図3に示されているようなXRDPを有している、請求項5に記載の化合物。
【請求項8】
化合物が:
D98%:650〜680μm;D50%:230〜250μm;およびD5%:40
〜60μm;
D98%:370〜390;d50%:100〜120μm;D5%:5〜15μm;
D98%:100〜125μm;D50%:15〜25μm;およびD5%:1〜3μ
m;または
D98%:50〜70μm;D50%:3〜7μm;およびD5%:0.5〜2、
に対応する粒径分布を有している、請求項6に記載の化合物。
【請求項9】
治療において使用するための請求項1〜8のいずれかに記載の化合物。
【請求項10】
薬学的に許容可能な賦形剤とともに請求項1〜8のいずれかに記載の化合物を含む医薬
組成物。
【請求項11】
請求項5または6に記載の化合物を含む、請求項10に記載の組成物。
【請求項12】
湿式造粒法により製造される錠剤であり、そして、無水カルシウム水素ホスファート、
トウモロコシデンプン、PVP−VAコポリマー、微結晶性セルロース、ナトリウムデン
プングリコラート、タルクおよびマグネシウムステアラートを含む、請求項10に記載の
組成物。
【請求項13】
HBr塩:3〜8%
無水カルシウム水素ホスファート:35〜45%
トウモロコシデンプン:15〜25%
PVP−VAコポリマー:2〜6%
微結晶性セルロース:20〜30%
ナトリウムデンプングリコラート:1〜3%
タルク:2〜6%
マグネシウムステアラート:0.5〜2%
を含む、請求項12に記載の組成物。
【請求項14】
HBr塩:約5%
無水カルシウム水素ホスファート:約39%
トウモロコシデンプン:約20%
PVP−VAコポリマー:約3%
微結晶性セルロース:約25%
ナトリウムデンプングリコラート:約3%
タルク:約4%
マグネシウムステアラート:約1%
を含む、請求項12に記載の組成物。
【請求項15】
情動障害、うつ病、大うつ病性障害、産後うつ病、双極性障害、アルツハイマー病、精
神病、癌、加齢もしくはパーキンソン病に伴ううつ病、不安、全般性不安障害、社会不安
障害、強迫性障害、パニック障害、パニック発作、恐怖症、社会恐怖症、広場恐怖症、ス
トレス性尿失禁、嘔吐、IBS、摂食障害、慢性疼痛、部分的応答者、治療抵抗性うつ病
、アルツハイマー病、認識機能障害、ADHD、メランコリー、PTSD、顔面紅潮、睡
眠時無呼吸、アルコール、ニコチンもしくは炭水化物渇望、物質乱用、およびアルコール
もしくは薬物乱用から選択される疾患を治療する方法であって、治療を必要としている患
者に請求項1〜8のいずれかに記載の治療的有効量の化合物を投与することを含む、治療
方法。
【請求項16】
情動障害、うつ病、大うつ病性障害、産後うつ病、双極性障害、アルツハイマー病、精
神病、癌、加齢もしくはパーキンソン病に伴ううつ病、不安、全般性不安障害、社会不安
障害、強迫性障害、パニック障害、パニック発作、恐怖症、社会恐怖症、広場恐怖症、ス
トレス性尿失禁、嘔吐、IBS、摂食障害、慢性疼痛、部分的応答者、治療抵抗性うつ病
、アルツハイマー病、認識機能障害、ADHD、メランコリー、PTSD、顔面紅潮、睡
眠時無呼吸、アルコール、ニコチンもしくは炭水化物渇望、物質乱用、またはアルコール
もしくは薬物乱用を治療するための薬剤を製造するための請求項1〜8のいずれかに記載
の化合物の使用。
【請求項17】
情動障害、うつ病、大うつ病性障害、産後うつ病、双極性障害、アルツハイマー病、精
神病、癌、加齢もしくはパーキンソン病に伴ううつ病、不安、全般性不安障害、社会不安
障害、強迫性障害、パニック障害、パニック発作、恐怖症、社会恐怖症、広場恐怖症、ス
トレス性尿失禁、嘔吐、IBS、摂食障害、慢性疼痛、部分的応答者、治療抵抗性うつ病
、アルツハイマー病、認識機能障害、ADHD、メランコリー、PTSD、顔面紅潮、睡
眠時無呼吸、アルコール、ニコチンもしくは炭水化物渇望、物質乱用、およびアルコール
もしくは薬物乱用から選択される疾患の治療において使用するための請求項1〜8のいず
れかに記載の化合物。
【請求項18】
以下:
【化1】
を製造するための方法であって、溶剤、塩基、ならびにパラジウム源およびホスフィン配
位子からなるパラジウム触媒の存在下において、60℃から130℃までの間の温度で、
化合物II
【化2】
[式中、R’は水素または一価の金属イオンを表す]を、式III
【化3】
[式中、X1およびX2は独立してハロゲンを表す]の化合物および式IV
【化4】
[式中、Rは水素または保護基を表す]の化合物と反応させることを含む、前記の方法。
【請求項19】
化合物IIおよび化合物IIIが第一の反応で反応させられ、場合により前記第一反応
での反応生成物
【化5】
が単離および精製され、続いて、前記化合物IVとのその後の反応が行われる、請求項1
8に記載の方法。
【請求項20】
化合物II、化合物IIIおよび化合物IVが前記方法の開始時に一緒に混合される、
請求項18に記載の方法。
【請求項21】
X1およびX2が独立してBrまたはIを表す、請求項18〜20のいずれかに記載の
方法。
【請求項22】
X1がBrを表し、X2がIを表す、請求項21に記載の方法。
【請求項23】
前記溶剤が非プロトン性溶剤である、請求項18〜22のいずれかに記載の方法。
【請求項24】
前記溶剤がトルエン、キシレン、トリエチルアミン、トリブチルアミン、ジオキシンま
たはN−メチルピロリドンから選択される、請求項18〜23のいずれかに記載の方法。
【請求項25】
前記溶剤がトルエンである、請求項24に記載の方法。
【請求項26】
前記パラジウム源がPddba2、Pd(OAc)2またはPd2dba3から選択さ
れる、請求項18〜25のいずれかに記載の方法。
【請求項27】
前記パラジウム源がPddba2またはPd2dba3である、請求項26に記載の方
法。
【請求項28】
前記ホスフィン配位子が
2,2’−ビス−ジフェニルホスファニル−[1,1’]ビナフタレニル(rac−B
INAP);
1,1’−ビス(ジフェニルホスフィノ)フェロセン(DPPF);
ビス−(2−ジフェニルホスフィノフェニル)エーテル(DPEphos);
トリ−t−ブチルホスフィン(Fu’s塩);
ビフェニル−2−イル−ジ−t−ブチル−ホスフィン;
ビフェニル−2−イル−ジシクロヘキシル−ホスフィン;
(2’−ジシクロヘキシルホスファニル−ビフェニル−2−イル)−ジメチル−アミン
;
[2’−(ジ−t−ブチル−ホスファニル)−ビフェニル−2−イル]−ジメチル−ア
ミン;および
ジシクロヘキシル−(2’,4’,6’−トリ−プロピル−ビフェニル−2−イル)−
ホスファン;
から選択される、請求項18〜27のいずれかに記載の方法。
【請求項29】
前記ホスフィン配位子がrac−BINAPである、請求項28に記載の方法。
【請求項30】
前記塩基がNaO(t−Bu)、KO(t−Bu)、Cs2CO3、DBUまたはDA
BCOから選択される、請求項18〜28のいずれかに記載の方法。
【請求項31】
前記塩基がNaO(t−Bu)である、請求項30に記載の方法。
【請求項32】
Rが水素を表す、請求項18〜31のいずれかに記載の方法。
【請求項33】
RがBoc、Bn、Cbz、C(=O)OetまたはMeから選択される保護基を表す
、請求項18〜31のいずれかに記載の方法。
【請求項34】
R’が水素である、請求項18〜33のいずれかに記載の方法。
【請求項35】
前記温度が約80℃から約120℃までの間である、請求項18〜34のいずれかに記
載の方法。
【請求項36】
以下のステップ:
a.1〜1.5当量の化合物I、IIおよびIIIをトルエン中に溶解または分散させ
て混合物Aを得るステップ;
b.1〜2モル%のPddba2および1〜2モル%のrac−BINAPを2〜3当
量のNaO(t−Bu)とともに混合物Aに加えて混合物Bを得るステップであって、こ
こで、化合物IIおよびIIIが完全に変換されるまで前記混合物Bが約100℃に加熱
されるステップ;
c.ステップbで得られた混合物の温度を、化合物IVが完全に変換されるまで、約1
20℃に高めるステップ;および
d.場合によって、化合物IVが保護型ピペラジンであるときに、酸水溶液を加えるこ
とによって前記保護基を取り除くステップ;
を含む、請求項20に記載の方法。
【請求項37】
1〜1.5当量の2,4−ジメチル−チオール、1−ブロモ−1−ヨード−ベンゼン(
または1,2−ジブロモ−ベンゼン)およびピペラジンをトルエン中に分散し、続いて、
トルエン中に分散された2〜5当量のNaO(t−Bu)および1〜2モル%のPd2d
ba3およびrac−BINAPを加えて混合物を得、前記混合物を100〜130℃に
2〜10時間加熱して生成物1−[2−(2,4−ジメチル−フェニルスルファニル)−
フェニル]−ピペラジンを得る、請求項20に記載の方法。
【請求項38】
前記混合物が3〜5時間加熱還流され、そして、後続のステップであって、得られた生
成物が対応する臭化水素酸付加塩を得るべく、更にHBr水溶液と反応させられるステッ
プがそれに続く、請求項37に記載の方法。
【請求項39】
2〜5当量のNaO(t−Bu)、2〜5当量のピペラジン、0.2〜0.6モル%の
Pddba2および0.6〜1モル%のrac−BINAPをトルエン中に分散して混合
物A’を得、前記混合物A’に約1当量の2−ブロモ−ヨードベンゼンを加えて混合物B
’を得、前記混合物B’に1当量の2,4−ジメチルチオフェノールを加え、得られた混
合物を3〜7時間加熱還流して1−[2−(2,4−ジメチル−フェニルスルファニル)
−フェニル]−ピペラジンを得る、請求項20に記載の方法。
【請求項40】
前記の得られた混合物が4〜6時間加熱還流され、そして、後続のステップであって、
得られた生成物が対応する臭化水素酸付加塩を得るべく、更にHBr水溶液と反応させら
れるステップがそれに続く、請求項39に記載の方法。
【請求項41】
対応する臭化水素酸塩を得るために、HBr水溶液を生成物
【化6】
に加える付加的なステップを伴い、前記生成物が場合によっては精製された形態である、
請求項18〜37のいずれかに記載の方法。
【請求項42】
1−[2−(2,4−ジメチル−フェニルスルファニル)−フェニル]−ピペラジン臭
化水素酸付加塩を製造するための方法であって、1〜1.5当量の2,4−ジメチル−チ
オール、1−ブロモ−1−ヨード−ベンゼン(または1,2−ジブロモ−ベンゼン)およ
びピペラジンをトルエン中に分散し、続いて、トルエン中に分散された2〜5当量のNa
O(t−Bu)および1〜2モル%のPd2dba3およびrac−BINAPを加えて
混合物を得、前記混合物を3〜5時間加熱還流して生成物1−[2−(2,4−ジメチル
−フェニルスルファニル)−フェニル]−ピペラジンを得、前記ピペラジンを更に臭化水
素酸水溶液と反応させる、前記の方法。
【請求項43】
1−[2−(2,4−ジメチル−フェニルスルファニル)−フェニル]−ピペラジン臭
化水素酸付加塩を製造するための方法であって、2〜5当量のNaO(t−Bu)、2〜
5当量のピペラジン、0.2〜0.6モル%のPddba2および0.6〜1モル%のr
ac−BINAPをトルエン中に分散して混合物A’を得、前記混合物A’に約1当量の
2−ブロモ−ヨードベンゼンを加えて混合物B’を得、前記混合物B’に1当量の2,4
−ジメチルチオフェノールを加え、結果として得られた混合物を4〜6時間加熱還流して
1−[2−(2,4−ジメチル−フェニルスルファニル)−フェニル]−ピペラジンを得
、前記ピペラジンを更に臭化水素酸水溶液と反応させる、前記の方法。
【請求項1】
化合物が非結晶質形態の遊離塩基ではないことを条件とする、1−[2−(2,4−ジ
メチルフェニルスルファニル)−フェニル]ピペラジンおよびその薬学的に許容可能な塩
。
【請求項2】
化合物が臭化水素酸塩、塩酸塩、メシル酸塩、フマル酸塩、マレイン酸塩、メソ−酒石
酸塩、L−(+)−酒石酸塩、D−(−)−酒石酸塩、硫酸塩、リン酸塩または硝酸塩で
ある、請求項1に記載の化合物。
【請求項3】
化合物が結晶質である、請求項1または2に記載の化合物。
【請求項4】
化合物が図1〜17のいずれかに示されているようなXRDPを有している、請求項3
に記載の化合物。
【請求項5】
結晶質形態の1−[2−(2,4−ジメチルフェニルスルファニル)−フェニル]ピペ
ラジン臭化水素酸塩である、請求項1に記載の化合物。
【請求項6】
化合物が約6.89、9.73、13.78および14.64(°2θ)におけるXR
PD反射を有している、請求項5に記載の化合物。
【請求項7】
化合物が図3に示されているようなXRDPを有している、請求項5に記載の化合物。
【請求項8】
化合物が:
D98%:650〜680μm;D50%:230〜250μm;およびD5%:40
〜60μm;
D98%:370〜390;d50%:100〜120μm;D5%:5〜15μm;
D98%:100〜125μm;D50%:15〜25μm;およびD5%:1〜3μ
m;または
D98%:50〜70μm;D50%:3〜7μm;およびD5%:0.5〜2、
に対応する粒径分布を有している、請求項6に記載の化合物。
【請求項9】
治療において使用するための請求項1〜8のいずれかに記載の化合物。
【請求項10】
薬学的に許容可能な賦形剤とともに請求項1〜8のいずれかに記載の化合物を含む医薬
組成物。
【請求項11】
請求項5または6に記載の化合物を含む、請求項10に記載の組成物。
【請求項12】
湿式造粒法により製造される錠剤であり、そして、無水カルシウム水素ホスファート、
トウモロコシデンプン、PVP−VAコポリマー、微結晶性セルロース、ナトリウムデン
プングリコラート、タルクおよびマグネシウムステアラートを含む、請求項10に記載の
組成物。
【請求項13】
HBr塩:3〜8%
無水カルシウム水素ホスファート:35〜45%
トウモロコシデンプン:15〜25%
PVP−VAコポリマー:2〜6%
微結晶性セルロース:20〜30%
ナトリウムデンプングリコラート:1〜3%
タルク:2〜6%
マグネシウムステアラート:0.5〜2%
を含む、請求項12に記載の組成物。
【請求項14】
HBr塩:約5%
無水カルシウム水素ホスファート:約39%
トウモロコシデンプン:約20%
PVP−VAコポリマー:約3%
微結晶性セルロース:約25%
ナトリウムデンプングリコラート:約3%
タルク:約4%
マグネシウムステアラート:約1%
を含む、請求項12に記載の組成物。
【請求項15】
情動障害、うつ病、大うつ病性障害、産後うつ病、双極性障害、アルツハイマー病、精
神病、癌、加齢もしくはパーキンソン病に伴ううつ病、不安、全般性不安障害、社会不安
障害、強迫性障害、パニック障害、パニック発作、恐怖症、社会恐怖症、広場恐怖症、ス
トレス性尿失禁、嘔吐、IBS、摂食障害、慢性疼痛、部分的応答者、治療抵抗性うつ病
、アルツハイマー病、認識機能障害、ADHD、メランコリー、PTSD、顔面紅潮、睡
眠時無呼吸、アルコール、ニコチンもしくは炭水化物渇望、物質乱用、およびアルコール
もしくは薬物乱用から選択される疾患を治療する方法であって、治療を必要としている患
者に請求項1〜8のいずれかに記載の治療的有効量の化合物を投与することを含む、治療
方法。
【請求項16】
情動障害、うつ病、大うつ病性障害、産後うつ病、双極性障害、アルツハイマー病、精
神病、癌、加齢もしくはパーキンソン病に伴ううつ病、不安、全般性不安障害、社会不安
障害、強迫性障害、パニック障害、パニック発作、恐怖症、社会恐怖症、広場恐怖症、ス
トレス性尿失禁、嘔吐、IBS、摂食障害、慢性疼痛、部分的応答者、治療抵抗性うつ病
、アルツハイマー病、認識機能障害、ADHD、メランコリー、PTSD、顔面紅潮、睡
眠時無呼吸、アルコール、ニコチンもしくは炭水化物渇望、物質乱用、またはアルコール
もしくは薬物乱用を治療するための薬剤を製造するための請求項1〜8のいずれかに記載
の化合物の使用。
【請求項17】
情動障害、うつ病、大うつ病性障害、産後うつ病、双極性障害、アルツハイマー病、精
神病、癌、加齢もしくはパーキンソン病に伴ううつ病、不安、全般性不安障害、社会不安
障害、強迫性障害、パニック障害、パニック発作、恐怖症、社会恐怖症、広場恐怖症、ス
トレス性尿失禁、嘔吐、IBS、摂食障害、慢性疼痛、部分的応答者、治療抵抗性うつ病
、アルツハイマー病、認識機能障害、ADHD、メランコリー、PTSD、顔面紅潮、睡
眠時無呼吸、アルコール、ニコチンもしくは炭水化物渇望、物質乱用、およびアルコール
もしくは薬物乱用から選択される疾患の治療において使用するための請求項1〜8のいず
れかに記載の化合物。
【請求項18】
以下:
【化1】
を製造するための方法であって、溶剤、塩基、ならびにパラジウム源およびホスフィン配
位子からなるパラジウム触媒の存在下において、60℃から130℃までの間の温度で、
化合物II
【化2】
[式中、R’は水素または一価の金属イオンを表す]を、式III
【化3】
[式中、X1およびX2は独立してハロゲンを表す]の化合物および式IV
【化4】
[式中、Rは水素または保護基を表す]の化合物と反応させることを含む、前記の方法。
【請求項19】
化合物IIおよび化合物IIIが第一の反応で反応させられ、場合により前記第一反応
での反応生成物
【化5】
が単離および精製され、続いて、前記化合物IVとのその後の反応が行われる、請求項1
8に記載の方法。
【請求項20】
化合物II、化合物IIIおよび化合物IVが前記方法の開始時に一緒に混合される、
請求項18に記載の方法。
【請求項21】
X1およびX2が独立してBrまたはIを表す、請求項18〜20のいずれかに記載の
方法。
【請求項22】
X1がBrを表し、X2がIを表す、請求項21に記載の方法。
【請求項23】
前記溶剤が非プロトン性溶剤である、請求項18〜22のいずれかに記載の方法。
【請求項24】
前記溶剤がトルエン、キシレン、トリエチルアミン、トリブチルアミン、ジオキシンま
たはN−メチルピロリドンから選択される、請求項18〜23のいずれかに記載の方法。
【請求項25】
前記溶剤がトルエンである、請求項24に記載の方法。
【請求項26】
前記パラジウム源がPddba2、Pd(OAc)2またはPd2dba3から選択さ
れる、請求項18〜25のいずれかに記載の方法。
【請求項27】
前記パラジウム源がPddba2またはPd2dba3である、請求項26に記載の方
法。
【請求項28】
前記ホスフィン配位子が
2,2’−ビス−ジフェニルホスファニル−[1,1’]ビナフタレニル(rac−B
INAP);
1,1’−ビス(ジフェニルホスフィノ)フェロセン(DPPF);
ビス−(2−ジフェニルホスフィノフェニル)エーテル(DPEphos);
トリ−t−ブチルホスフィン(Fu’s塩);
ビフェニル−2−イル−ジ−t−ブチル−ホスフィン;
ビフェニル−2−イル−ジシクロヘキシル−ホスフィン;
(2’−ジシクロヘキシルホスファニル−ビフェニル−2−イル)−ジメチル−アミン
;
[2’−(ジ−t−ブチル−ホスファニル)−ビフェニル−2−イル]−ジメチル−ア
ミン;および
ジシクロヘキシル−(2’,4’,6’−トリ−プロピル−ビフェニル−2−イル)−
ホスファン;
から選択される、請求項18〜27のいずれかに記載の方法。
【請求項29】
前記ホスフィン配位子がrac−BINAPである、請求項28に記載の方法。
【請求項30】
前記塩基がNaO(t−Bu)、KO(t−Bu)、Cs2CO3、DBUまたはDA
BCOから選択される、請求項18〜28のいずれかに記載の方法。
【請求項31】
前記塩基がNaO(t−Bu)である、請求項30に記載の方法。
【請求項32】
Rが水素を表す、請求項18〜31のいずれかに記載の方法。
【請求項33】
RがBoc、Bn、Cbz、C(=O)OetまたはMeから選択される保護基を表す
、請求項18〜31のいずれかに記載の方法。
【請求項34】
R’が水素である、請求項18〜33のいずれかに記載の方法。
【請求項35】
前記温度が約80℃から約120℃までの間である、請求項18〜34のいずれかに記
載の方法。
【請求項36】
以下のステップ:
a.1〜1.5当量の化合物I、IIおよびIIIをトルエン中に溶解または分散させ
て混合物Aを得るステップ;
b.1〜2モル%のPddba2および1〜2モル%のrac−BINAPを2〜3当
量のNaO(t−Bu)とともに混合物Aに加えて混合物Bを得るステップであって、こ
こで、化合物IIおよびIIIが完全に変換されるまで前記混合物Bが約100℃に加熱
されるステップ;
c.ステップbで得られた混合物の温度を、化合物IVが完全に変換されるまで、約1
20℃に高めるステップ;および
d.場合によって、化合物IVが保護型ピペラジンであるときに、酸水溶液を加えるこ
とによって前記保護基を取り除くステップ;
を含む、請求項20に記載の方法。
【請求項37】
1〜1.5当量の2,4−ジメチル−チオール、1−ブロモ−1−ヨード−ベンゼン(
または1,2−ジブロモ−ベンゼン)およびピペラジンをトルエン中に分散し、続いて、
トルエン中に分散された2〜5当量のNaO(t−Bu)および1〜2モル%のPd2d
ba3およびrac−BINAPを加えて混合物を得、前記混合物を100〜130℃に
2〜10時間加熱して生成物1−[2−(2,4−ジメチル−フェニルスルファニル)−
フェニル]−ピペラジンを得る、請求項20に記載の方法。
【請求項38】
前記混合物が3〜5時間加熱還流され、そして、後続のステップであって、得られた生
成物が対応する臭化水素酸付加塩を得るべく、更にHBr水溶液と反応させられるステッ
プがそれに続く、請求項37に記載の方法。
【請求項39】
2〜5当量のNaO(t−Bu)、2〜5当量のピペラジン、0.2〜0.6モル%の
Pddba2および0.6〜1モル%のrac−BINAPをトルエン中に分散して混合
物A’を得、前記混合物A’に約1当量の2−ブロモ−ヨードベンゼンを加えて混合物B
’を得、前記混合物B’に1当量の2,4−ジメチルチオフェノールを加え、得られた混
合物を3〜7時間加熱還流して1−[2−(2,4−ジメチル−フェニルスルファニル)
−フェニル]−ピペラジンを得る、請求項20に記載の方法。
【請求項40】
前記の得られた混合物が4〜6時間加熱還流され、そして、後続のステップであって、
得られた生成物が対応する臭化水素酸付加塩を得るべく、更にHBr水溶液と反応させら
れるステップがそれに続く、請求項39に記載の方法。
【請求項41】
対応する臭化水素酸塩を得るために、HBr水溶液を生成物
【化6】
に加える付加的なステップを伴い、前記生成物が場合によっては精製された形態である、
請求項18〜37のいずれかに記載の方法。
【請求項42】
1−[2−(2,4−ジメチル−フェニルスルファニル)−フェニル]−ピペラジン臭
化水素酸付加塩を製造するための方法であって、1〜1.5当量の2,4−ジメチル−チ
オール、1−ブロモ−1−ヨード−ベンゼン(または1,2−ジブロモ−ベンゼン)およ
びピペラジンをトルエン中に分散し、続いて、トルエン中に分散された2〜5当量のNa
O(t−Bu)および1〜2モル%のPd2dba3およびrac−BINAPを加えて
混合物を得、前記混合物を3〜5時間加熱還流して生成物1−[2−(2,4−ジメチル
−フェニルスルファニル)−フェニル]−ピペラジンを得、前記ピペラジンを更に臭化水
素酸水溶液と反応させる、前記の方法。
【請求項43】
1−[2−(2,4−ジメチル−フェニルスルファニル)−フェニル]−ピペラジン臭
化水素酸付加塩を製造するための方法であって、2〜5当量のNaO(t−Bu)、2〜
5当量のピペラジン、0.2〜0.6モル%のPddba2および0.6〜1モル%のr
ac−BINAPをトルエン中に分散して混合物A’を得、前記混合物A’に約1当量の
2−ブロモ−ヨードベンゼンを加えて混合物B’を得、前記混合物B’に1当量の2,4
−ジメチルチオフェノールを加え、結果として得られた混合物を4〜6時間加熱還流して
1−[2−(2,4−ジメチル−フェニルスルファニル)−フェニル]−ピペラジンを得
、前記ピペラジンを更に臭化水素酸水溶液と反応させる、前記の方法。
【図1】

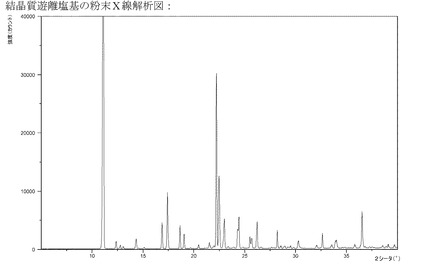
【図2】
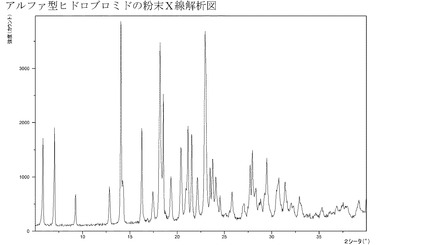
【図3】
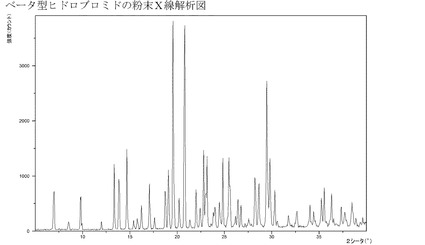
【図4】
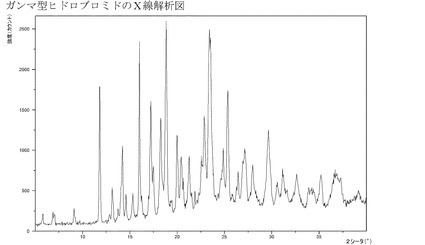
【図5】
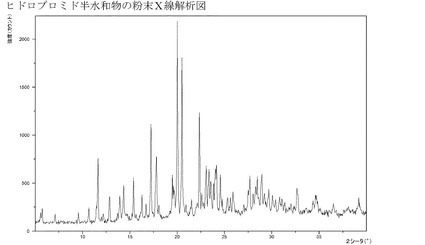
【図6】
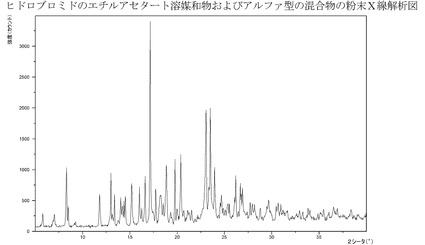
【図7】
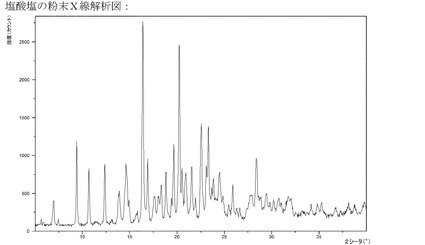
【図8】
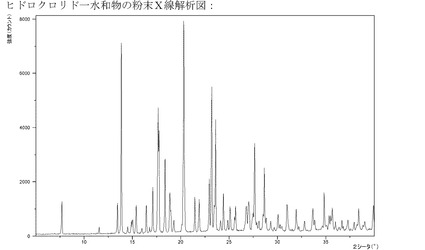
【図9】
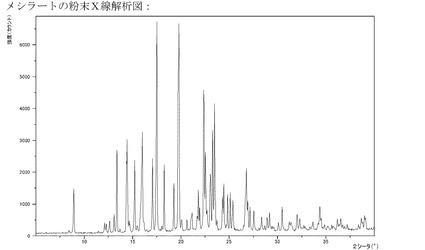
【図10】
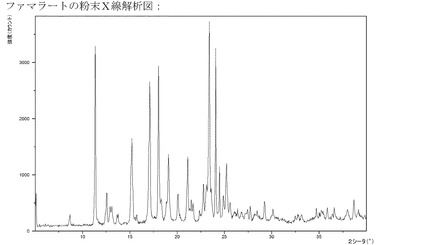
【図11】
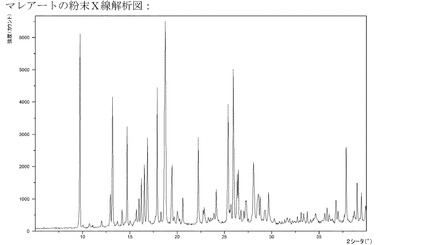
【図12】
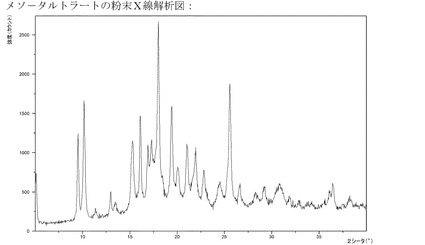
【図13】
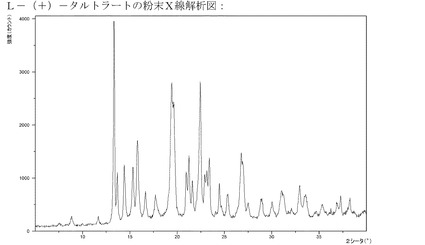
【図14】
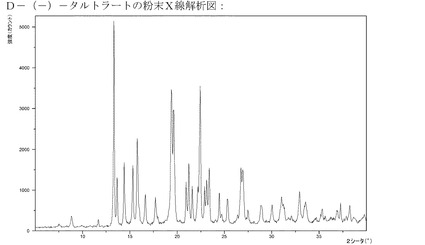
【図15】
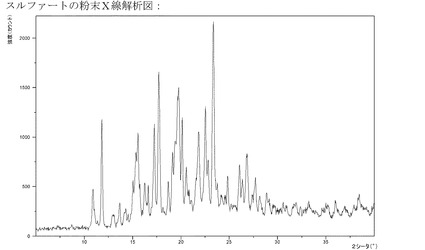
【図16】
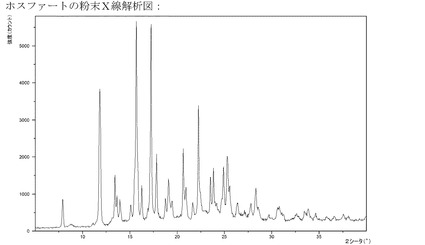
【図17】
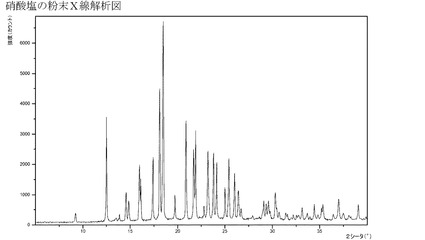
【図18a】
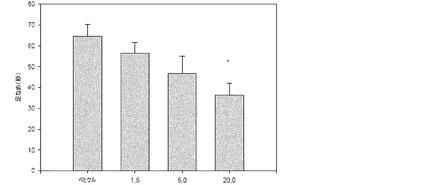
【図18b】
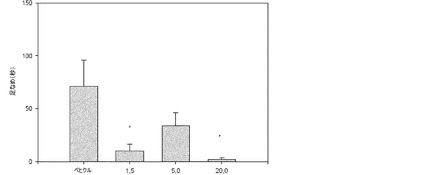
【図19a】
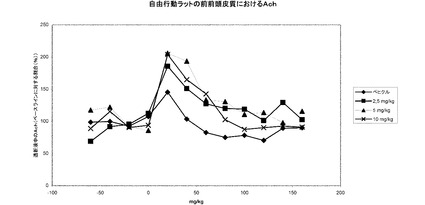
【図19b】
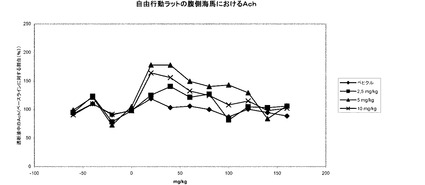
【図20】
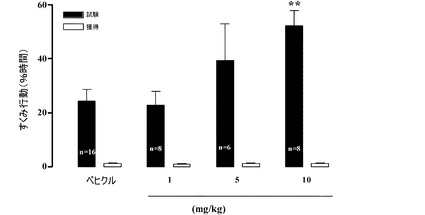
【図21】
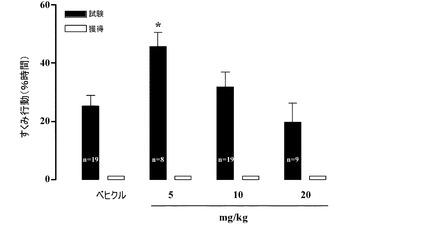
【図22】
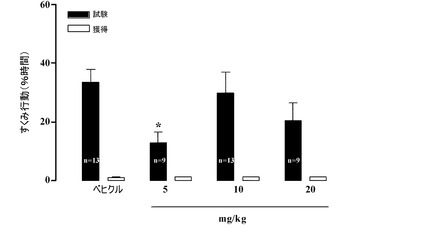
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18a】
【図18b】
【図19a】
【図19b】
【図20】
【図21】
【図22】
【公開番号】特開2013−56933(P2013−56933A)
【公開日】平成25年3月28日(2013.3.28)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−259835(P2012−259835)
【出願日】平成24年11月28日(2012.11.28)
【分割の表示】特願2009−295187(P2009−295187)の分割
【原出願日】平成19年6月15日(2007.6.15)
【出願人】(591143065)ハー・ルンドベック・アクチエゼルスカベット (129)
【Fターム(参考)】
【公開日】平成25年3月28日(2013.3.28)
【国際特許分類】
【出願番号】特願2012−259835(P2012−259835)
【出願日】平成24年11月28日(2012.11.28)
【分割の表示】特願2009−295187(P2009−295187)の分割
【原出願日】平成19年6月15日(2007.6.15)
【出願人】(591143065)ハー・ルンドベック・アクチエゼルスカベット (129)
【Fターム(参考)】
[ Back to top ]