
誘導体化炭素
本発明は、アミノ酸もしくはその誘導体が炭素に結合している誘導体化炭素を提供する。本発明の誘導体化炭素は、液体媒体中の金属イオンの検出および液体媒体からの金属イオンの除去に対して有用である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、誘導体化炭素(derivatised carbon)に関し、具体的には、所望の特性を付与するよう化学的に変性された表面を有するグラファイトおよび他の形態の炭素に関する。
【背景技術】
【0002】
有毒物質(特に有毒重金属)の環境への蓄積と放出が、過去数十年にわたって大幅に増大した。採掘作業と重工業が環境に及ぼす影響により、高濃度の有毒重金属イオン(例えば、CuIIイオン、CdIIイオン、PbIIイオン、およびHgIIイオン)が湖や川に蓄積されてきており、これらの汚染物は、ほとんどが非分解性であって、自然界で再循環している。水性媒体や飲用水中に重金属が存在すると、曝露レベルと重金属の化学的形態によっては、ヒトと水生生物の健康にとって危険となる可能性がある。重金属汚染が引き起こした人道面での悲惨な結果の1つの例が、アルゼンチン、中国、メキシコ、台湾、インド、および特にバングラデシュ等の国々における、何百万という人々の広範囲にわたる被毒である。バングラデシュの地下水の最大で60%が、世界保健機構(WHO)のガイドラインである10ppbをはるかに超える自然発生的な濃度のヒ素を含有している。これら重金属イオンの塩の多くが水溶性であるので、通常の物理的な分離方法は役に立たない。飲用水濾過および/または環境浄化において使用するための水性媒体から有毒重金属イオンを除去できる、簡単で、迅速で、かつ低コストの方法を開発することが急務である。
【0003】
ポリ-L-ヒスチジン、ポリ-L-アスパラギン酸、ポリ-L-グルタミン酸、および特にポリ-L-システイン等のポリペプチドは、金属イオン(例えば、CdIIイオン、PbIIイオン、NiIIイオン、およびCuIIイオン)とキレート環を形成することが知られており、種々の担体に結びつき、これら金属の痕跡分析に使用されている(Malachowski et al,Anal.Chim.Acta.2003, 495, 151;Malachowski et al,Anal.Chim.Acta.2004, 517, 187;Malachowski et al,Pure Appl.Chem.2004, 76, 777;Johnson et al,Anal.Chim.Acta.2005, 77, 30;Howard et al,Anal.At.Spectrom.1999, 14, 1209;およびJurbergs et al,Anal.Chem.1997, 69, 1893)。バイオホモポリマー(biohomopolymers)および他のペプチドは、金属の抽出や再生に対し、従来の方法〔例えば、単純な濾過や沈殿(後者は、環境機関の厳しい規制に適合するよう、ターゲット金属の濃度を減少させることができない場合が多い)〕を凌ぐ大きな利点を有する。
【0004】
グラファイトの表面は、種々の比較的簡単な方法(例えば、所定の化学的または生物学的成分の、物理吸着および化学的もしくは電気化学的に開始される化学吸着)を使用して化学的に変性させることができる。誘導体化表面(derivatised surfaces)を有するグラファイトは、様々な用途にて(例えば、バッテリー技術における電極物質として、およびセンサーとして)使用することができる。グラファイト物質の表面にヒドロキシル部分やカルボキシル部分等の反応性基が存在していることは知られているけれども、化学的に誘導体化されたグラファイトを合成化学用途のための固体担体として使用することはまだ限定されている。
【発明の開示】
【0005】
本発明は、段階的な合成を行う上での炭素ベースの固体担体を提供する。このような物質の表面を“ビルディングブロック(building-block)”様式にて誘導体化することで、所望の特性(例えば、ターゲット検体に対する感受性)を付与することが可能となる。このように、アミノ酸、ペプチド、低分子タンパク質、および核酸等の化学種を、比較的簡単な仕方で炭素(例えばグラファイト)粒子に連結することができる。炭素の表面を最初に誘導体化する化学種の化学を変えることによって、ビルディングブロック分子を炭素表面に連結する種々の方法が可能となる。本発明は特に、アミノ酸もしくはその誘導体が結合する誘導体化炭素(特にグラファイト)を提供する。アミノ酸は、モノマー(例えばシステイン)であっても、あるいは金属イオンと結合することができるポリペプチド(例えばポリ-L-システイン)であってもよい。したがって本発明は特に、有毒重金属の検出、および水や他の水性媒体からの有毒重金属の除去に関連している。
【0006】
本発明の第1の態様によれば、アミノ酸もしくはその誘導体が炭素に結合している誘導体化炭素が提供される。この結合は、直接的な結合であっても、あるいは間接的な結合(例えばフェニルアミン基を介して)であってもよい。
【0007】
本発明はさらに、ニトロフェニル誘導体化炭素が得られるような条件下にて、炭素とニトロベンゼンジアゾニウム化合物とを接触させる、という誘導体化炭素の製造方法を提供する。
【0008】
本発明はさらに、炭素を、炭素の表面のカルボキシル基を介してアミノ酸もしくはその誘導体に直接結合させる、という誘導体化炭素の製造方法を提供する。この製造方法は、炭素の表面のカルボキシル基をハロゲン化アシル基に転化させること、次いで得られる生成物とアミノ酸もしくはその誘導体とを接触させることを含む。
【0009】
本発明はさらに、本発明の誘導体化炭素を含んだ炭素電極を提供する。
本発明はさらに、本発明の電極を含んだ電気化学的デバイスを提供する。本発明の電気化学的デバイスは、電気化学的センサーの形態であっても、あるいは電気化学的リアクターであってもよい。
【0010】
本発明はさらに、本発明の誘導体化炭素と水性媒体とを接触させることを含む、液体媒体から金属イオンを除去する方法を提供する。
本発明はさらに、本発明の電気化学的デバイスを使用して、液体媒体をボルタンメトリー分析にかけることを含む、液体媒体中における金属イオンの存在を検出する方法を提供する。
【0011】
本発明の誘導体化炭素は、液体媒体(水および他の水性媒体を含む)中の金属イオンの検出、液体媒体からの金属イオンの除去、および液体媒体中の金属イオンの滴定に対して有用である。このような金属イオンとしては、例えば、Cd(II)イオン、Pb(II)イオン、Zn(II)イオン、Cu(II)イオン、およびAs(III)イオンなどがある。誘導体化炭素は粒状形態(例えば粉末形態)であってよい。表面積が大きいことから、グラファイト粉末やガラス質炭素粉末等の粒状物質が望ましい(表面積が大きいと、比較的多量のアミノ酸もしくはそれらの誘導体を連結させることができる)。したがって本発明の誘導体化炭素は、公知の変性固体物質よりはるかに多い量の金属イオンを結びつけることができる。
【0012】
(種々の実施態様の説明)
本発明は、アミノ酸もしくはその誘導体が炭素に結合している誘導体化炭素を提供する。アミノ酸もしくはその誘導体は、炭素に直接結合していても、あるいは間接的に(すなわち、リンカーを介して)結合していてもよい。特に挙げておきたいのは、アミノ酸もしくはその誘導体が、炭素上に存在するカルボキシル基やフェニルアミン基を介して結合している状態の炭素である。
【0013】
1つの実施態様においては、アミノ酸はイオウ含有アミノ酸(例えば、システイン、グルタチオン、チロシン、またはこれらの誘導体)である。イオウ含有アミノ酸は、ペンダントのチオール基またはチオール様の基を有してよい。アミノ酸は、エステル〔例えば、メチルエステルやエチルエステル(特定の例としてはL-システインメチルエステルがある)〕の形態をとってよい。アミノ酸の誘導体は、アミノ酸のオリゴマーとポリマーを含む。例えば、システイン誘導体はポリシステインまたはシステアミンであってよく、またグルタチオン誘導体はポリグルタチオンであってよい。代表的なポリマーアミノ酸は、ポリマー鎖1つ当たり50〜100のシステイン残基を含有する、S-ベンジル保護されたホモポリマーである。アミノ酸もしくはその誘導体は、保護されていても、あるいは保護されていなくてもよい。1つの例は、ポリ-S-ベンジル-L-システイン等のポリシステインである。
【0014】
炭素は、粒状物の形態(例えば粉末形態)をとってよい。粒状炭素は、1〜100μm(例えば2〜50μm)の直径を有する粒子を含んでよい。特に挙げておきたいのは、グラファイト粉末、ガラス質炭素球形粉末、および熱分解グラファイトの形態物である。これとは別に、炭素は、カーボンナノチューブの形態〔例えば、多層カーボンナノチューブ(MWCNT)〕をとってもよい。
【0015】
本発明の誘導体化炭素の例としては、システインで変性されたガラス質炭素;グルタチオンもしくはシステアミン、またはこれらの誘導体;およびポリシステインもしくはポリグルタチオンで変性された炭素粉末;などがある。言うまでもないが、本発明は他のアミノ酸ポリマーやそれらの誘導体に対して、そしてさらにアミノ酸モノマーやそれらのチオール含有誘導体(例えばシステイン)に対して適用可能であり、これらがガラス質炭素に連結される。特定の例としては、システインもしくはその誘導体〔例えば、システインのエステル(システインメチルエステル等)、またはシステインのポリマー(ポリシステインやポリ-S-ベンジル-L-システイン等)〕を使用して誘導体化された炭素粉末(例えば、グラファイト粉末やガラス質炭素球形粉末)がある。
【0016】
誘導体化炭素は、炭素とニトロベンゼンジアゾニウム化合物とを、ニトロフェニル誘導体化炭素が生成されるような条件下にて接触させることによって得ることができる。この反応は、適切な試剤(次亜リン酸等)の存在下にて行うことができる。ニトロフェニル誘導体化炭素を還元して、アニリン誘導体化炭素を形成させることができる。この生成物をさらに反応させて、置換アニリン誘導体化炭素を得ることができる。特に、アニリン誘導体化炭素をアミノ酸もしくはその誘導体(例えば、ポリ-S-ベンジル-L-システイン等のポリシステイン)と反応させることができる。
【0017】
誘導体化炭素はさらに、炭素の表面に存在しているカルボキシル基をハロゲン化アシル基に転化させ、次いで得られた生成物とアミノ酸もしくはその誘導体とを接触させることによって得ることができる。ハロゲン化アシルは、例えば塩化アシルであってよい。アミノ酸もしくはその誘導体に存在している任意のカルボキシル基を保護することができる。
【0018】
本発明の誘導体化炭素は、金属イオンの検出(例えば電気化学的検出)、金属イオンの滴定、または液体媒体からの金属イオンの除去に対して使用することができる。金属イオンは、例えば、Cd(II)、Pb(II)、Zn(II)、Cu(II)、およびAs(III)の1種以上であってよい。液体媒体は、例えば水性媒体であってよい。
【0019】
本発明の誘導体化炭素(特に、システイン誘導体化炭素やポリシステイン誘導体化炭素)は、ヒ素の検出に対して有用である。例えば、本発明の誘導体化炭素は、比較的高価な飲用水濾過装置中に組み込むことができる。これに反して、本発明の誘導体化炭素がAs(III)以外の金属イオンに対して選択性がある限りにおいて、As(III)の検出を妨害するイオン〔Cu(II)イオン等〕を除去すべく、本発明の誘導体化炭素をヒ素センサー中に組み込むこともできる。したがって本発明は、天然の供給物がしばしば有毒重金属(例えば、ヒ素やカドミウム)で汚染されている場合に、水の浄化、工場廃液からの金属の回収もしくは抽出、および飲用水の濾過において使用するための安価で且つ魅力的な物質を提供する。
【0020】
本発明はさらに、金属イオン封鎖において有用な物質を提供する。例えば、炭素上に固定されたポリシステインは一般に、公知の基質(例えば、ガラスやポリマービーズ等)の場合より金属取り込み量(物質1グラム当たり)がはるかに多い。単位面積当たりの金属イオン封鎖ユニットの密度も、ナノスケールの変性が使用されている場合(例えば、ナノチューブの場合)は、活性表面積が増大するので、従来技術の基質の場合よりはるかに大きい。したがって、金属イオン取り込みの熱力学と反応速度(kinetic rate)の両方を高めることができる。
【0021】
本発明は特に、バイオホモポリマー(特に、ポリ-L-ヒスチジン、ポリ-L-アスパラギン酸、ポリ-L-グルタミン酸、および特にポリ-L-システインから選択されるポリペプチド)をグラファイト粉末に連結することによってもたらされる固体担体物質を提供する。前述したように、このようなポリマーは、アルカリ金属やアルカリ土類金属(例えば、ナトリウムやカルシウム)に対して殆ど親和性をもたない有毒重金属(例えば、カドミウム、鉛、ニッケル、および銅)とキレート環を形成することが知られている。本発明のシステイン誘導体化グラファイト粉末またはポリ-L-システイン誘導体化グラファイト粉末を使用して、水性媒体から金属イオン〔例えばCd(II)イオン〕を定量的に滴定することができる。グラファイト粉末の表面積が大きく、グラファイト粉末に多量のアミノ酸を連結できることから、システイン変性炭素またはポリシステイン変性炭素は、他のいかなる担体物質に結合したポリ-L-システインよりはるかに多くの量のCd(II)イオンとキレート環を形成する。したがって本発明の誘導体化炭素は、工場廃液からの有毒重金属の回収、環境浄化、および飲用水の濾過に使用するのに特に適している。
【0022】
以下に実施例を挙げて本発明を説明する。
【実施例】
【0023】
実施例1:ポリ-L-システインによるグラファイト粉末の誘導体化
試剤と化学物質
塩化カリウム(Riedel de Haenから購入)を除いて、試剤は全てアルドリッチ社から購入し、入手しうる最高グレードであり、さらなる精製を行わずに使用した。使用した合成グラファイト粉末は、直径が2〜20μmの異形粒子からなり、アルドリッチ社から購入した。水溶液は全て、18.2MΩcm以上の抵抗率を有する、エルガスタット(Elgastat)UHQグレードシステム(Elga)からの脱イオン水を使用して調製した。
【0024】
脱イオン水中pHが1.0〜12.0の範囲の溶液を以下のように調製した:pH1.0,0.10MのHCl;pH1.7,0.1Mのテトラシュウ酸カリウム(potassium tetraoxalate);pH4.6,[0.10Mの酢酸]+[0.10Mの酢酸ナトリウム];pH5.04,0.5Mの酢酸ナトリウム;pH6.8,[0.025MのNa2HPO4]+[0.025MのKH2PO4];pH9.2,0.05Mのテトラホウ酸二ナトリウム;pH10.5,0.1Mのテトラホウ酸二ナトリウム;およびpH12.0,0.01Mの水酸化ナトリウム。これらの溶液はさらに、0.10MのKClを支持電解質として含有した。pHの測定は、ハンナ(Hanna)pH213pHメーターを使用して行った。
【0025】
計測
電気化学的測定値は、標準的な3電極配置構成を有するμAutolabコンピュータ制御ポテンシオスタット〔エコケミー社(Ecochemie)〕を使用して記録した。電気化学的実験は、25cm3の体積のガラスセル中にて行った。熱分解グラファイト底面電極(BPPG,直径5mm,ル・カルボネ社)またはホウ素ドープダイヤモンド電極(BDD,直径3mm,ウィンザー・サイエンティフィック社)が作用電極として作動した。白金コイル(純度99.99%,グッドフェロー社)がカウンター電極として作動した。特に明記しない限り、飽和カロメル電極(SCE,ラジオメーター社)を参照電極として使用して、セル集成体を仕上げた。電気化学的実験は全て、高純度のN2ガス(BOCガス)を使用して溶液を30分脱気した後に、20±2℃にて行った。
【0026】
4-ニトロフェニル誘導体化炭素をSn/HClで還元し、そして4-ニトロ安息香酸と連結させた後のX線光電子スペクトロスコピー(XPS)は、アルミニウムKαバンド(hν=1486.7eV)からのX線照射を使用して(供給源を14hνおよび200mAに設定)、サイエンタ(Scienta)ESCA300機器により行った。スペクトルは全て、150evのパスエネルギーと90°のテイクオフ角度を使用して記録した。特に明記しない限り、1.9mmのスリット幅を使用した。分析チャンバー中のベース圧を2.0×10-9ミリバール以下に保持した。
【0027】
S-ベンジル保護ポリ-L-システインと脱保護ポリ-L-システインのXPSは、アルミニウムKαバンド(hν=1486.7eV)からのX線照射を使用して、VG Clam4MCDアナライザシステムにより行った。XPS実験は全て、90°のテイクオフ角度を有する100eVのアナライザエネルギー(analyzer energy)を使用して記録した。分析チャンバー中のベース圧を2.0×10-9ミリバール以下に保持した。検討した誘導体化炭素サンプルはいずれも、両面接着テープを使用してスタブ上に貼り付け、次いでスペクトロメーターの超高真空分析チャンバー中に配置した。X線照射を受けたときに、光電子を放出して正に帯電するのを防ぐために、スペクトロメーター分析チャンバー内の“フラッドガン”からの電子ビーム(10eV)で、サンプルの表面に衝撃を与えた。得られたスペクトルの分析は、マイクロカル・オリジン(Microcal Origin)6.0を使用して行った。
【0028】
一般的な反応スキーム
スキーム1は、グラファイト粉末を誘導体化するための合成経路を示しており、“ビルディングブロック”の化学、およびグラファイト粉末へのポリ-L-システインの連結の背景となっている原理を示している。
【0029】
【化1】

【0030】
4-ニトロフェニルでグラファイト粉末を誘導体化してNP炭素を作製する
ファストレッドGG(4-ニトロベンゼンジアゾニウムテトラフルオロボレート)の5mM溶液10cm3中に0.5gのグラファイト粉末を混合して攪拌し、これに50cm3の次亜リン酸(H3PO2、50重量%水溶液)を加えた。次いでこの溶液を、穏やかに攪拌しながら5℃で30分放置してから水吸引によって濾過し、脱イオン水で洗浄して過剰の酸を除去し、そして最後にアセトニトリルで洗浄して未反応のジアゾニウム塩を除去した。4-ニトロフェニル誘導体化グラファイト粉末(“NP炭素”)を、ドラフト中に12時間置くことによって風乾してから、使用するまでは気密容器中に保存した(Pandurangappa et al,Analyst,2002,127,1568;およびPandurangappa et al,Analyst,2003,128,473)。
【0031】
NP炭素を還元してAN炭素を作製する
NP炭素粉末(1.02g)とスズ(1.63g,13.7ミリモル)を水(12ml)中に懸濁させた。濃塩酸(4.5ml,53.8ミリモル)を加え、本混合物を加熱還流した。反応混合物を、アルゴン雰囲気下にて100℃で攪拌した。18時間後、混合物を濾過し、得られた固体を、塩酸(1M水溶液100ml)、メタノール(100ml)、水酸化カリウム(1M水溶液50ml)、およびメタノール(50ml)で洗浄した。固体を減圧乾燥して、グラファイト表面に共有結合的に誘導体化されたp-アニリン部分からなる、還元形態のNP炭素である黒色粉末(180.4mg)(“AN炭素”)を得た。
【0032】
4-ニトロ安息香酸をAN炭素に連結させる
AN炭素(500mg)、1-ヒドロキシベンゾトリアゾール水和物(HOBt,670mg,5.0ミリモル)、ベンゾトリアゾール-1-イル-オキシトリピロリジノホスホニウムヘキサフルオロホスフェート(PyBop,2.6g,5ミリモル)、およびp-ニトロ安息香酸(840mg,5ミリモル)をフラスコ中に仕込み、DMF(8ml)を加えた。エチルジイソプロピルアミン(1.7ml,10ミリモル)を加えた。反応混合物を、アルゴン雰囲気下にて室温で攪拌した。18時間後、混合物を濾過し、固体をメタノール(50ml)、アセトニトリル(50ml)、およびDCM(50ml)で洗浄した。固体を減圧乾燥して、アミド結合を介してAN炭素表面に連結した4-ニトロ安息香酸からなる黒色粉末(“NBAN炭素”)を得た。
【0033】
NP炭素、AN炭素、およびNBAN炭素のボルタンメトリー特性決定とXPS特性決定
誘導体化されたNP炭素、AN炭素、およびNBAN炭素のボルタンメトリー特性決定は、先ずそれぞれの誘導体化炭素を、「Leventis et al,Talanta,2004,63,1039」に記載のように、bppg電極の表面上に別々に(separately)そして研磨的に(abrasively)不動態化した後に、pH1.0〜pH12.0の範囲にわたって行った。
【0034】
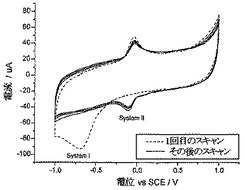
図1aは、pH6.8でのNP炭素のボルタンメトリーを示している。還元方向への1回目のスキャンでは、SCEに対して約−0.685Vにて大きな還元ウェーブ(reduction wave)が観察された(図1aにおいて“システムI”として表示されている)。SCEに対して−1.0Vにてスキャン方向を反転させると、システムIに対する対応した酸化ウェーブ(oxidation wave)は観察されなかった。このことは、プロセスが電気化学的に不可逆性であることを示している。しかしながら、SCEに対して約+0.025Vにて酸化ウェーブが観察された。引き続きスキャンを行うと、SCEに対して約−0.095Vにて対応する還元ウェーブ観察され、これは、電気化学的にほぼ可逆性のプロセス(“システムII”と呼ぶ)に対応している。電気化学的に不可逆性のシステムIは、その後のスキャンにおいては存在せず、このことは、全ての4-ニトロフェニル部分が還元されていることを示している。
【0035】
観察されたボルタンメトリー挙動とウェーブ形状は、NP炭素についての従来の研究(Pandurangappa et al,Analyst,2002,127,1586)と矛盾せず、水性媒体中における表面結合ニトロ基の電気化学的還元に対応している。スキーム2は、ニトロベンゼン自体の一般的な例に対するこうした挙動を示している(「Pandurangappa et al,Analyst,2002,127,1586」および「Rubinstein,J.Electroanal.Chem.,1971,29,309」)。
【0036】
【化2】
【0037】
このメカニズムにおいては、システムIは、ニトロ基を4電子-4プロトンプロセスにて化学的・電気化学的に不可逆性還元して、アリールヒドロキシルアミンを形成することに相当している。次いで、電気化学的にほぼ可逆性の2電子-2プロトン酸化を受けて(システムII)アリールニトロソ化学種を形成する。このボルタンメトリー挙動は、検討した全てのpHにおいて観察されたけれども、同時に起こるプロトン移動により、システムIとIIに対するピーク電位はpHに依存し、pH1.0〜pH12.0の範囲にわたって、線形ネルンスト式(a linear, Nernstian fashion)にて、それぞれ55.4mV/pHユニットおよび54.4mV/pHユニットだけ変化した(従来の研究と一致する)。
【0038】
十分に確立したボルタンメトリー特性決定プロトコル(「Leventis et al,Talanta 2004,63,1039」および「Wildgoose et al,Talanta,2003,60,887」)を、ほぼ可逆性のシステムIIに対して、検討したそれぞれのpHにおいて実施し、4-ニトロフェニル部分が、実際にグラファイト粒子の表面にとどめられていることが確認された。
【0039】
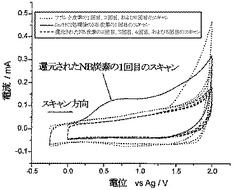
AN炭素のボルタンメトリー特性決定により、システムIまたはシステムIIに対応するボルタンメトリーウェーブ(voltammetric waves)は観察されない、ということが明らかになった。したがって全ての4-ニトロフェニル基が対応するアニリン様部分に還元された、と結論付けることができる。0.1Mの過塩素酸テトラブチルアンモニウム(TBAP)を含有するアセトニトリル中でのAN炭素のボルタンメトリーは、1回目のスキャンにおいては、アニリンからそのラジカルカチオンへの1電子酸化に対応する電位において、銀疑似参照電極に対して約+0.700Vにて酸化ウェーブを示した(図1b)。
【0040】
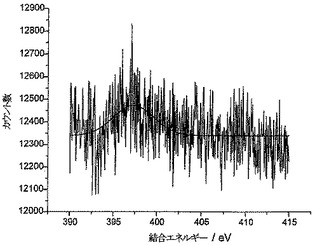
XPSを使用して、AN炭素のさらなる特性決定を行った。図2aによれば、スペクトルのN1s領域に単一のピークが観察されており、400.1eVの結合エネルギーは芳香族アミン部分と合致している。ニトロ部分内のN1sレベルまたはO1sレベルから放出される光電子に対応した結合エネルギーにおけるシグナルは観察されなかった。
【0041】
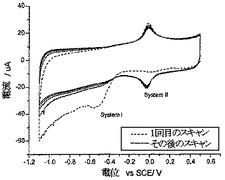
NBAN炭素のボルタンメトリー特性決定により、ニトロ基の予想される特徴的還元がこの場合も観察されること、およびボルタンメトリーが表面結合化学した種に対応していることが明らかになった(図1C)。図2bは、NBAN炭素のXPSスペクトルのN1s領域を示している。400.6eVと405.4eVの結合エネルギーを有する2つのピークが観察され、ピーク高さの比はほぼ1:1である。XPSのデータベースと比較することにより、これらのピークは、それぞれアミド基およびニトロ基中の窒素原子に対応していることが確認された。さらに、スペクトルのO1s領域(図示せず)のガウス解析は、それぞれ、アミド基と芳香族ニトロ基内の酸素原子に合致した530.7eVと533.6eVの結合エネルギーを有するピークを明らかにしている。これらの結果を考慮すると、連結はもっぱら、4-ニトロ安息香酸分子と、AN炭素の表面上のアニリン様部分との間で起こる、と結論づけることができる。
【0042】
ポリ-S-ベンジル-L-システインをAN炭素に連結してPSB炭素を作製する
ポリ-S-ベンジル-L-システイン(PSBC,170mg,0.02ミリモル)を1,4-ジオキサン(3ml)中に溶解した。塩化トリメチルシリル(5.6μl,0.04ミリモル)をDMF(3ml)中に溶解して得た溶液を加えて、ペプチドホモポリマーの溶解性を高めた。反応混合物を、アルゴン雰囲気下にて50℃で攪拌した。1時間後、反応混合物を室温に冷却した。エチルジイソプロピルアミン(6.5μl,0.04ミリモル)を加え、本混合物を0℃に冷却してから塩化9-フルオロエニルメトキシカルボキシル(Fmoc,5.7mg,0.02ミリモル)を加えた。本混合物を室温に自然加温した。1時間半後、減圧にて溶媒を除去して白色固体を得た。得られた残留物に、1-ヒドロキシベンゾトリアゾール水和物(HOBt,4.2mg,0.2ミリモル)、ベンゾトリアゾール-1-イル-オキシトリピロリジノホスホニウムヘキサフルオロホスフェート(PyBop,11.6g,0.02ミリモル)、AN炭素(104mg)、およびDMF(10ml)を加えた。エチルジイソプロピルアミン(17.7μl,0.04ミリモル)を加えた。反応混合物を、アルゴン雰囲気下にて室温で攪拌した。19時間後、混合物を濾過し、得られた固体を、DMF(10ml)、メタノール(10ml)、アセトニトリル(50ml)、およびDCM(50ml)で洗浄した。固体を減圧乾燥して、アミド結合を介してAN炭素に連結したS-ベンジル保護ポリ-L-システインからなる黒色粉末(200mg)(“PSBC炭素”)を得た。
【0043】
PSBC炭素の脱保護
ポリ-L-システイン中のチオール基の脱保護は、バーチ還元法を使用して達成された。PSBC炭素(124mg)とナトリウム(120mg,5.2ミリモル)を収容するフラスコ中に、液体アンモニア(約10ml)を濃縮した。本溶液を、アルゴン雰囲気下にて−78℃で攪拌した。20分後、1-ブタノール(0.3ml)を加え、反応混合物をさらに5分攪拌してから室温に自然加温した。アンモニアが蒸発した後、塩化アンモニウム(約4mlの飽和水溶液)を加えて反応混合物をクエンチした。懸濁液を濾過し、得られた固体を、水(20ml)、メタノール(20ml)、およびDCM(20ml)で洗浄した。固体を減圧乾燥して、アミド結合を介してAN炭素に連結したポリ-L-システインからなる黒色粉末(106mg)(“PC炭素”)を得た。
【0044】
PSBC炭素とPC炭素のXPS特性決定
図3aと3bはそれぞれ、PSBC炭素とPC炭素に対して得られたXPSスペクトルを示している。PSBC炭素においては、S2p3/2レベルとS2sレベルから放出される光電子に対応した162.5eVと226.5eVの結合エネルギーを有する2つのピークが観察された(S-ベンジル保護ポリシステインに対する文献値とよく一致している)。脱保護されたPC炭素においては、S2p3/2光電子とS2s光電子の結合エネルギーが163.5eVと227.5eVにわずかにシフトした(この場合も、ポリシステイン中の遊離チオールに対する文献値とよく一致している)。PSBC炭素とPC炭素の両方に対し、O1sピークとN1sピークはそれぞれ531.5eVと400.5eVに位置しており、ポリシステイン中のアミド結合からの寄与によって特色づけられている(文献値とよく一致している)。PSBC炭素サンプルとPC炭素サンプルの元素分析から、グラファイト表面に連結したポリ-L-システインの相対量(relative amounts)は、バーチ還元を使用したチオール基の脱保護後でも変化していないことがわかった(全イオウシグナル、全酸素シグナル、および全窒素シグナルは、表面元素組成のそれぞれに対して約7±1.4%であった)。このことは、比較的多量のポリシステインが表面に連結していることを示している。したがって、ポリ-L-システインがAN炭素に連結していて、ポリ-L-システイン中のチオール基を脱保護すべくバーチ還元を行った後でも連結したままである、と結論づけることができる。
【0045】
実施例2:PC炭素の使用による、水性媒体中のカドミウムの定量分析
水溶液からのCd2+の取り込みを、線形掃引ストリッピングボルタンメトリー(LSV)ストリッピングプロトコルを使用して、ホウ素ドープダイヤモンド(BDD)電極(「Banks et al,Talanta,2004,62,279」によって開発)にて電気化学的にモニターした。
【0046】
Cd2+検出のための最適化pHはpH5であり、したがってPC炭素によるCd2+のキレート化およびキレート化したCd2+の量のLSV検出に対して、0.05Mの酢酸ナトリウム緩衝液(pH5.04)を使用した。カドミウム検出のためのLSVプロトコルは、溶液を攪拌しながら、電位をSCEに対して−1.5Vに60秒保持することによって、Cd2+をBDD電極上にCd0として堆積させることを含んだ。次いで100mVs-1にて−1.1Vから−0.3Vまで電位をスキャンすることによってLSVを実施し、SCEに対して約−0.8Vにてカドミウムストリッピングピークを観察した。このプロトコルの精度を確認するために、標準的な5nM Cd2+の添加量によってCd(NO3)2の“ブラインド”溶液を分析し、[ピーク高さvs.Cd2+濃度]の標準添加量プロットを作成した。
【0047】
図4は、このようにして得られる、Cd2+の量を増大させるためのオーバーレイドLSVボルタンモグラムと標準添加量プロットを示している。Cd2+の濃度は、LSVプロトコルによって20.5nM±0.1nMであると測定された(検出限界(3σ)は0.2nM)。実際のCd2+濃度は20nM±0.1nMであり、LSVプロトコルが、1〜100nMの濃度範囲にわたって微量のCd2+を測定するための正確な方法であることがわかる。
【0048】
PC炭素によってキレート形成されたCd2+の量を測定するために、pH5の酢酸ナトリウム緩衝液中1mMのCd(NO3)2溶液を調製した。この溶液の10μlサンプルを採り、初期のCd2+濃度がLSVプロトコルによって測定できるよう105倍に希釈した。次いで、1mM、2mM、および3mMのCd(NO3)2の10cm3に、それぞれ5mg、10mg、および20mgのPC炭素を加え、10分攪拌した。PC炭素を濾別し、再び濾液の10μlサンプルを採って希釈してから、サンプル中に残留しているCd2+の量を、LSVプロトコルを使用して測定した。PC炭素の各添加量に対して、この手順を3回繰り返した。
【0049】
表1は、量をいろいろ変えたときのPC炭素に対する、キレート化されたCd2+の量を示している。ある時間にわたって実験を繰り返し、10分から12時間まで時間を種々変えて、PC炭素をCd2+と共に攪拌した。PC炭素に対するCd2+の曝露時間を増大させても、キレート化Cd2+の量は増大しなかった。比較のため、ブランクのグラファイト粉末を使用して、同様の実験を行った。ブランクのグラファイト粉末によるCd2+の取り込み量は測定不可であった。表1に記載の結果から、PC炭素が、PC炭素1g当たり1218μモル±200μモルのCd2+をキレート化する、ということを算出することができた。
【0050】
【表1】
【0051】
PC炭素によるCd2+の取り込みは、ポリシステインを他の基質に連結させている従来の研究(「Jurbergs et al,Anal.Chem.,1997,69,1893」;「Malachowski et al,Pure Appl.Chem.,2004,76,777」;「Johnson et al,Anal.Chem.2005,77,30」;および「Howard et al,J.Anal.At.Spectrom.,1999,14,1209」)と比較して、1g当たり最大で100倍大きいことがわかった。特定の理論で拘束されるつもりはないが、これは、グラファイト粉末の表面積が大きいこと、および炭素表面上の多くのエッジ・プレイン様(edge-plane-like)欠陥部位に連結させることができる4-ニトロフェニル基の割合が大きいこと(これにより、他の固体担体に対するよりもはるかに多くのポリシステインをグラファイト粉末に連結させることができる)によるものであると考えられる。さらに、PC炭素をカドミウム(II)溶液に曝露することで、PC炭素によるCd2+イオンの定量的な滴定を迅速に行える。
【0052】
従来の研究により、チオール基との単純なプロトン交換よりむしろ第三立体構造変化(tertiary conformational changes)の結果として、硝酸を使用してポリシステインからCd2+を定量的に回収することができる、ということが実証されている(「Howard et al,J.Anal.At.Spectrom.,1999,14,1209」;および「Miller et al,Anal.Chem.,2001,73,4087」)。カドミウムイオンは、濾過したPC炭素サンプルを1MのHNO3中にて攪拌することによって、PC炭素から回収された。PC炭素の各サンプルを、1.0MのHNO310cm3中にて30分または5時間攪拌した後に、懸濁液を濾過した。濾液の10μlサンプルを取り出し、pH5の緩衝液中に希釈してから、サンプル中に残留しているCd2+の量を、LSVプロトコルを使用して測定した。いずれの場合も、サンプルを30分処理したか、あるいは5時間処理したかに関係なく、キレート化Cd2+の40%±10%が回収された。このことは、Howardらの研究(ポリシステインが、Cd2+に対して弱い結合部位と強い結合部位の両方を示す)と一致している。
【0053】
実施例3:チロシンによるグラファイト粉末とMWCNTの誘導体化
実施例1に記載のジアゾニウム塩化学により、グラファイトとMWCNTに4-ニトロフェニル基を連結した。ニトロ基をSn/HClで還元して、アニリン変性炭素とアニリン変性MWCNTを得た。次いでアニリン基をジアゾ化し、チロシンに連結して、金属をキレート化できる物質を、そしてさらに、アミノ酸またはチオール含有分子をチロシン変性炭素とチロシン変性MWCNTに連結させるための経路を得た。さらに、誘導体化炭素と誘導体化MWCNTの表面上のアニリン部分のアミン基を、金属キレート化/回収に使用すべくチオール基に転化させた。
【0054】
実施例4:L-システインメチルエステルによるガラス質炭素粉末の誘導体化
2gのガラス質炭素球形粉末(GC,直径10〜20μm,タイプI,Alfa Aesar)を10cm3のSOCl2と共に1時間攪拌してから、乾燥CH3Clで洗浄した。これにより、カルボキシル表面基が塩化アシル類縁体に転化された。次いで、この物質と0.5gのL-システイン-メチルエステル塩酸塩(シグマ-アルドリッチ社)とを、10cm3の乾燥CH2Cl2中にて、攪拌しながら、そして0.27cm3のEt3Nを徐々に加えながら反応させた。反応混合物を12時間(一晩)攪拌して、L-システインメチルエステル誘導体化GC球形粉末(“CysMeO-GC”)を得た。このプロセスをスキーム3に示す。
【0055】
【化3】
【0056】
同様の手順にて、ガラス質炭素球形粉末と、グルタチオン(還元形,<99%,アルドリッチ社)およびシステアミン塩酸塩(アクロス・オーガニックス社)とを連結した。
【0057】
実施例5:CysMeO-GC粉末の使用による水からのカドミウムの除去
カドミウムの検出
使用した線形掃引ボルタンメトリー(LSV)ストリッピングプロトコルは、従来の検出プロトコル(Kruusman et al,Electroanalysis,2004,16,399)に基づいた。ホウ素ドープダイヤモンド電極(BDD,直径3mm,ウインザー・サイエンティフィック社)を作用電極として使用し、白金コイルと飽和カロメル電極が、それぞれカウンター電極および参照電極として作用した。電気化学的実験は、pH5.04の0.05M酢酸ナトリウム緩衝液中にて0.1MのKClを支持電解質として加えて、コンピュータ制御のポテンシオスタット(μオートラブ社)を使用して行った。
【0058】
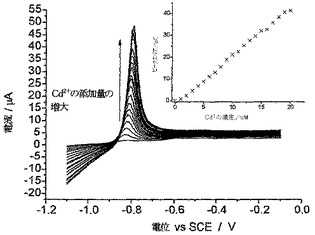
Cd(II)のLSV検出は、下記のパラメーターを使用して行った。酢酸ナトリウム緩衝液10cm3に、試験しようとするサンプルの10μlアリコートを加えた。攪拌しながら、SCEに対して−1.5Vの電位にて60秒間、BDD電極上にカドミウムを堆積させた。電位を、100mVs-1にてSCEに対して−1.1V〜−0.6Vまで掃引し、SCEに対して約−0.780Vにてカドミウムのストリッピングピークが観察された。0.1μMのCd(II)の標準添加量を0.1〜1.0μMの範囲にわたって加え、対応する添加プロットを作成し、これを使用して、最初のサンプル中におけるバックグラウンドCd(II)濃度を算出した。
【0059】
川水からのカドミウムの除去
オックスフォードのチャーウェル川から川水のサンプルを採取した(未処理)。この川水の10cm3サンプルをスパイクして、有毒カドミウム廃棄物の環境的にひどい流出をシミュレートするよう、約1.5mMのカドミウム(II)濃度を生成させた。このコネクションは、ネバ川(ロシアのサンクト・ペテルスブルグを流れており、極めて汚染されていることがよく知られている)における算出された平均Cd(II)濃度である。この“実際の(real)”マトリックスサンプルに10mgのCysMeO-GC粉末を加え、攪拌した。サンプルを濾過し、分析のために10μlアリコートを、上記のLSV Cd(II)ストリッピングプロトコルを使用して5分後に、次いで10分間隔ごとに1時間にわたって取り出した。
【0060】
図5は、こうして得られたCd(II)濃度プロフィールを示している。10mgのCysMeO-GC粉末によって、サンプルからCd(II)の約87%が除去されたことがわかる。残留Cd(II)の濃度は、サンクト・ペテルスブルグにおける蛇口からの供給上水中のCd(II)の算出飲用水濃度の約半分であった(それでもまだ、WHO、EU、およびEPAのガイドラインより高い)。CysMeO-GC粉末は、環境浄化および/または金属イオン封鎖に使用するための、安価で極めて有効な物質として使用することができる。
【0061】
ミネラルウォーターからのカドミウムの除去
50ppb(飲用水に対するEPA勧告最大限度の10倍)のCd(II)濃度が得られるよう、エヴィアン・ミネラルウォーターの10cm3サンプルにCd(II)をスパイクすることによって、飲用上水の汚染をシミュレートした。次いで、この“実際の”マトリックスを10mgのCysMeO-GC粉末と共に攪拌し、前述のように分析した。Cd(II)の除去結果を図6に示す。
【0062】
CysMeO-GC粉末に曝露してから10分以内に、ミネラルウォーター中のCd(II)濃度は、EPA勧告最大限度(5ppb)未満になった。したがってCys-GCは、有毒重金属〔Cd(II)など〕を除去すべく飲用水の濾過に使用するための優れた物質である。
【0063】
実施例6:CysMeO-GC粉末の使用による、水からの銅の除去
使用したスクエアウェーブボルタンメトリー(square wave voltammetry)(SWV)ストリッピングプロトコルは、従来の検出プロトコル(Banks et al,Phys.Chem.Chem.Phys.,2003,5,1652)に基づいた。直径50μmの金ディスク電極(<99.99%,グッドフェロー社)を作用電極として使用し、白金コイルと飽和カロメル電極(SCE,ラジオメーター社)が、それぞれカウンター電極および参照電極として作用した。電気化学的実験は、pH2.00の0.1Mリン酸(H3PO4)緩衝液中にて0.1MのKClを支持電解質として加えて、コンピュータ制御のポテンシオスタット(μオートラブ社)を使用して行った。
【0064】
Cu(II)のSWV検出は、下記のパラメーターを使用して行った:周波数50Hz,ステップ電位2mV,振幅25mV。リン酸緩衝液9.5cm3に、試験しようとするサンプルの0.5cm3アリコートを加えた。攪拌しながら、SCEに対して−1.5Vの電位にて15秒間、作用電極上に銅を堆積させた。電位を、SCEに対して−1.0V〜+0.6Vまで掃引し、SCEに対して約−0.05Vにて銅のストリッピングピークが観察された。1.0μMのCu(II)の標準添加量を1.0〜10.0μMの範囲にわたって加え、対応する添加プロットを作成し、これを使用して、最初のサンプル中におけるバックグラウンドCu(II)濃度を算出した。
【0065】
川水からの銅の除去
チャーウェル川から川水のサンプル10cm3(未処理)を、上記のSWV銅ストリッピングプロトコルを使用して分析した。約30μMのCu(II)濃度(1.3mgL-1または20.1μMというEPA限度より少し上)を有することが見出され、したがってCu(II)濃度をスパイクせずに使用した。この場合も、サンプルを10mgのCysMeO-GCに曝露し、種々の間隔で1時間にわたって分析して、残留しているCu(II)濃度を測定した。
図7は、サンプルからのCu(II)の除去結果を示している。
【0066】
実施例7:PC炭素粉末およびCysMeO-GC粉末の使用による、水からのヒ素の除去
試剤と化学物質
使用した化学物質は全て分析用グレードであり、受け入れたままの状態にて、さらなる精製を施さずに使用した。これらの化学物質は、(メタ)亜ヒ酸ナトリウム(フルカ社、+99.0%)と硝酸(アルドリッチ社、二重蒸留したPPB/テフロン(登録商標)グレード、ICP-MSによる測定によれば金属不純物はpptレベル)であった。溶液は全て、18.2MΩcm以上の抵抗率を有する脱イオン水(ビベンジウォーターシステムズ社)を使用して調製した。飲用水のサンプルはバングラデシュから入手した。
【0067】
計測
ボルタンメトリー測定は、μオートラブIII(ECO-ケミー社)ポテンシオスタットを使用して行った。測定は全て、3電極セルを使用して行った。作用電極は、金マイクロディスク電極(直径1mm)であり、テフロン(登録商標)ハウジング中に金ワイヤをシールすることによって社内で作製した。カウンター電極は、光沢のある白金ワイヤであり、飽和カロメル電極(ラジオメーター社)が参照電極として使用されている。金電極は、軟らかなラップパッド上にて0.1μmのアルミナスラリーを使用して研磨した。
【0068】
直径3mmのチタン合金マイクロチップ(ジェンコンズ社)を取り付けた、20kHzの周波数で作動する超音波ホーン〔モデルCV26(ソニックス&マテリアルズ社)〕を使用してソノボルタンメトリーによる検討(sonovoltammetric studies)を行った。超音波の強さを熱量測定法により測定し(「Banks et al,Phys.Chem.Chem.Phys.2004,6,3147」;「Magulis et al,Russ.J.Phys.Chem.1969,43,592」;および「Magulis et al,Ultrasonic.Sonochem.,2003,10,343」)、10%にて57Wcm-2であることがわかった。作用電極を超音波ホーンに対して正面向き配列で配置し、超音波ホーンを段付きチップ(stepped tip)のショルダーの向こう側に沈めて、超音波が溶液に確実に効率的に加えられるようにした。ヒ素の検出に対しては、オートラブソフトウェア(三次多項式補正を使用する)を使用してボルタンメトリー曲線をベースライン補正した。
【0069】
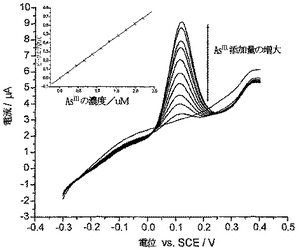
PC炭素の使用によるヒ素の除去
ポリシステイン誘導体化炭素粉末を、純水中においてAs(III)と錯形成する能力に関して試験した。堆積プロセス時に超音波によってアシストされる金電極にて、陽極ストリッピングボルタンメトリー(ASV)を使用してAs(III)の濃度を測定した。超音波を強めることで、ASVを金電極にて使用するヒ素検出の感度が大幅に高まった。「Simm et al,Electroanalysis 2005,17,335」に記載の最適化された条件を使用した。各サンプルを錯形成リガンドに曝露する前に対照実験を行って、標準添加量法(standard additions method)によって測定されるAs(III)の濃度が、本方法の検出限度内に入るよう確実に正確になるようにした。
【0070】
(メタ)亜ヒ酸ナトリウムを超純水中に溶解してAs(III)の1.1mM溶液(pH5.4)を調製し、本溶液25mlをフラスコ中に入れて攪拌し、これに10mgのポリシステイン炭素粉末(PC炭素)を加えた。10分、30分、および60分の間隔にて溶液から50μlのサンプルを採取し、次いでこれを0.1Mの硝酸中に希釈し、レベルを追跡して分析を行った。この分析は、金電極をSCEに対して−0.6Vに60秒間保持することによって行い、この時間中、超音波をホーンにて使用して、20mmの距離と5%の振幅をチップした。次いで電位を、堆積電位から1V(SCEに対して)まで100mV/秒のスキャン速度でポジテイブに掃引し、約0.1V(SCEに対して)でのヒ素のストリッピングシグナルを明らかにした。各分析に対し、この初期値を3回測定し、平均値を算出した。それぞれの測定に対して2.4×10-7MのAs(III)を加え、標準添加量法によって存在するAs(III)の最初の濃度を測定するために、これを3回繰り返した。
【0071】
図8は、時間の経過に対するAs(III)濃度の減少を示しており、攪拌60分後にAs(III)の濃度は1.1mMから0.7mMまで減少(36%の減少)し、ポイントを通して一次指数関数的減衰ラインが適合した。本溶液を、さらなる攪拌を行うことなく20日間にわたって静置した後、濃度が0.55mMに減少したことがわかった。
【0072】
CysMeO-GC粉末の使用によるヒ素の除去
(メタ)亜ヒ酸ナトリウムを超純水中に溶解してAs(III)の0.98mM溶液(pH5.4)を調製し、本溶液25mlをフラスコ中に入れて攪拌し、これに10mgのCys-GC粉末を加えた。10分、20分、および60分の間隔にて溶液から50μlのサンプルを採取し、次いでこれを0.1Mの硝酸中に希釈し、レベルを追跡して分析を行った。
【0073】
図9は、時間の経過に対するAs(III)濃度の減少を示しており、攪拌60分後にAs(III)の濃度は0.98mMから0.7mMまで減少(28.6%の減少)した。本溶液を、さらなる攪拌を行うことなく3日間にわたって静置したが、ヒ素濃度のさらなる減少は見られなかった。
【0074】
次いで、バングラデシュ等の地域からの飲用水中に見られると思われる程度のトレースレベルにて実験を行った(Anawar et al,Environment International 2002,27,597)。サンプルは、50ppbというバングラデシュの限度より4倍大きい200ppb(2.66μM)のAs(III)レベルに調製した。200mgのCysMeO-GC粉末を25mlのサンプル中に混合し、次いでこれを所定時間攪拌してから、システインによるAs(III)の錯形成を停止させるために、濾紙を使用してCysMeO-GC粉末を濾過した。次いでサンプルを0.1Mの硝酸溶液中に1:1に希釈して分析に供した。
【0075】
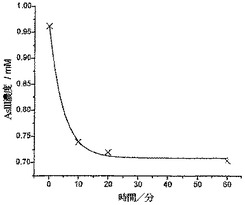
図10は、わずか10分後にヒ素濃度が200ppbから77ppbに大幅に減少し、そして30分後にレベルが55ppbに減少した、ということを示している。60分での分析結果は、ヒ素の濃度がこのレベルで一定となり(73%の減少)、As(III)が、バングラデシュの飲用安全限度よりやや高い濃度にて存在している、ということを示している。
【0076】
次いで実際のサンプルを使用して、CysMeO-GC粉末が、真正のバングラデシュ井戸水サンプル中のヒ素と錯形成する能力を調べた。サンプルを最初にASV法によって試験して、存在するAs(III)の濃度を測定した。しかしながら、As(III)の濃度が検出可能限界(1×10-8M)未満であることがわかり、したがって実験に使用すべく、水サンプルを120ppbの値にスパイクした。上記実験のように、25mlの水サンプルに200mgのCysMeO-GC粉末を加え、これを所定時間(5分、10分、30分、および45分)攪拌してから濾過して、溶液から粉末を除去した。再び、サンプルを0.1Mの硝酸中に1:1に希釈して分析実験に供した。
【0077】
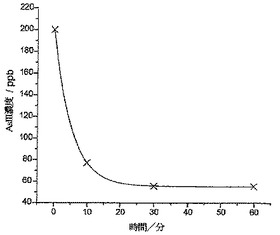
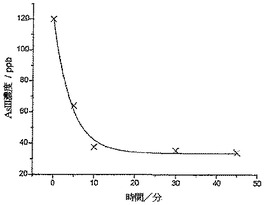
図11は、一次指数関数的減衰に適合した分析結果を示している。わずか5分の攪拌後に、存在するヒ素の濃度が47%ほど減少し(64ppb)、10分の攪拌後では、濃度がさらに69%ほど減少している(38ppb;すなわち、バングラデシュ飲用安全限度より12ppb少ない)ことがわかる。45分後、濃度が34ppb(すなわち、初期値の28%)で横ばい状態になった。純水ではなく実際のサンプルで分析を行ったので、実験では、バングラデシュの上水中に一般的に見られる多くの微量金属に曝露された(銅、鉛、および水銀など;Anawar et al,Environment International 2002,27,597)。図12は、30分サンプルの分析からのASVプロットを示しており、SCEに対して約0.4Vにて、これら汚染物質のうちの1種による大きなストリッピングウェーブを見ることができる。
【0078】
実施例8:L-システインメチルエステルによる炭素粉末の誘導体化
試剤と装置
ガラス質炭素ミクロ球形粉末(Alfa Aesar,タイプI,直径10〜20μm)と塩化カリウム(Riedel de Haen)を除いて、試剤は全てアルドリッチ社から購入し、市販の最高グレードであり、さらなる精製を行わずに使用した。水溶液は全て、18.2MΩcm以上の抵抗率を有する脱イオン水(ビベンジウォーターシステムズ社)を使用して調製した。pHの測定は、ハンナ・インスツルメントpH213pHメーターを使用して行った。
【0079】
X線光電子スペクトロスコピー(XPS)は、VGクラム4MCDアナライザシステムを使用し、アルミニウムKαバンド(hν=1486.7eV)からのX線照射を使用して行った。XPS実験は全て、90°のテイクオフ角度を有する100eVのアナライザエネルギーを使用して記録した。分析チャンバー中のベース圧を2.0×10-9ミリバール以下に保持した。各炭素粉末サンプルを、両面接着テープを使用してスタブ上に貼り付け、次いでスペクトロメーターの超高真空分析チャンバー中に配置した。X線照射を受けたときに、光電子の放出によって正に帯電するのを防ぐために、スペクトロメーター分析チャンバー内の“フラッドガン”からの電子ビーム(10eV)で、サンプルの表面に衝撃を与えた。フラッドガンがスペクトル線のピーク位置に及ぼす影響を明らかにする上で、記載のピーク位置が、286.6eVというC1s文献値に関して補正されていない点に留意しておかねばならない。得られたスペクトルの分析は、マイクロカル・オリジン6.0を使用して行った。スペクトルピークの帰属は、UKSAFデータベースとNISTデータベースを使用して行った。
【0080】
CysOMe炭素のサンプルに対する燃焼分析は、標準的な方法と装置を使用してC、N、およびSの元素含量(%)を測定することによって行った。
【0081】
炭素粉末へのL-システインメチルエステルの連結
酸素含有表面基(例えば、ヒドロキシル部分やキノニル部分)(グラファイト表面上のエッジ・プレイン欠陥部位をデコレートすることが知られている)を酸化することによって、およびグラファイト粉末を濃硝酸(HNO3)中で18時間攪拌することによって、グラファイト表面にカルボキシル部分を導入した。粉末サンプルから硝酸を除去するために、酸化されたグラファイト粉末を、洗浄液が中性になるまで多量の純水で洗浄した。
【0082】
次いでグラファイト粉末の変性を下記のように達成した。表面カルボキシル基を対応する塩化アシル部分に転化させるために、2gの酸化グラファイト粉末を10cm3の塩化チオニル(SO2Cl2)中にて90分攪拌し、その後、得られた物質を乾燥クロロホルムで洗浄して、未反応の塩化チオニル不純物を除去した。次いで、0.5gのシステインメチルエステル塩酸塩を含有する10cm3の乾燥クロロホルム中に粉末を懸濁させた。この懸濁液に0.27cm3の乾燥トリエチルアミンを滴下し、反応混合物を、不活性アルゴン雰囲気下にて室温で12時間攪拌した。最後に、得られた変性グラファイト粉末(“CysOMe-炭素”)を、多量のクロロホルム、アセトニトリル、アセトン、および純水で洗浄して未反応の化学種を除去した。
【0083】
CysOMe-炭素粉末の特性決定
XPSを使用して、どの程度のCysOMeがグラファイの表面に結合しているかを調べた。CysOMe-炭素粉末のサンプルをXPSスペクトロメーター中に据え付け、図13に示すように、スキャンを0eVから1200eVまで行った。UKSAFデータベースとNISTデータベースを使用して、ピークの帰属を行った。
【0084】
各元素の個別のX線断面積によって調整される広域スペクトルにおける各ピークの下の面積から表面の元素組成(%)を算出した。種々の元素に対する関連した原子感度ファクターを考慮にいれると、CysOMeが表面元素の約10%を構成しており、異なったサンプル作製間の変動は±3%である、ということが見出された。この表面被覆率は、燃焼分析を使用して得られる表面被覆率とよく一致している(燃焼分析は、10〜14%のCysOMeの表面被覆率をもたらし、この値は、CysOMe-GC粉末に対する値の約2倍である)。
【0085】
CuII溶液、CdII溶液、またはAsIII溶液に、金属イオンの取り込みが完全となるよう十分な時間にわたって曝露した後のCysOMe-炭素粉末のサンプルに対して、さらにXPS分析を行った(下記のセクションを参照)。図14は、As3sスペクトル、As3p3/2スペクトル、S2sスペクトル、およびS2p3/2スペクトルが観察される区域に対してAsIIIに曝露した後のCysOMe-炭素のXPSスペクトルを示している。AsIII対CysOMeの比(イオウのスペクトル線エリアによって測定)は、相対原子感度ファクターを考慮に入れると約1:1であることがわかった。検討した他の金属に対するXPSの結果は、同様の化学量論関係を示す。
【0086】
実施例9:CysOMe-炭素粉末の使用による、種々の金属イオンの検出と除去
試剤と装置
μAutolabコンピュータ制御ポテンシオスタット(エコケミー社)を使用して電気化学的測定を行った。全体を通して、10cm3の溶液を含んだ3電極セルを使用した。作用電極は、ガラス質炭素電極(GC,直径3mm,BAS)、正方形のホウ素ドープダイヤモンド電極(BDD,3mm×3mm,ウィンザー・サイエンティフィック社)、または金マクロディスク電極(直径1mm,グッドフェロー社)で構成された。光沢のある白金ワイヤ(99.99%,グッドフェロー社)をカウンター電極として作動させ、銀ワイヤの偽電極(99.99%,グッドフェロー社)または飽和カロメル電極(SCE,ラジオメーター社)を参照電極として作動させて3電極集成体を仕上げた。溶液は全て、高純度N2(BOCガス)を使用して20分にわたって脱気し、その後に電気化学的実験を行った。
【0087】
溶液中のAsIII濃度に対する誘導結合プラズマ原子発光分光法(ICPAES)による分析を、パーキンエルマーオプティマ5300DVエミッションICP機器を使用して行った。推奨発光波長は188.979nmであり、最良の検出を得るためにはアキシアル・ビュー(axial view)が推奨される。これは200nmの限界点未満であるので、光学機器をアルゴンの高速流れでパージして、水と空気による光の吸収を最小限に抑えた。
【0088】
5ポイント(0ppb,50ppb,100ppb,150ppb,および200ppb)を使用するAsIIIキャリブレーションは0.9993の相関係数をもたらし、検出限度(ブランクの標準偏差の3倍として定義され、それぞれ3回の繰り返しにて測定された4ブランクチェックからの平均)は9.78ppbすなわち0.0098ppmであることが見出された。パーキンエルマーによる期待値は、この波長に対して1〜10ppbであり、したがって感度は受け入れ可能である。ブランクチェック(blank check)の溶液は、4チェックに対して2.0〜4.5ppbをもたらした。
【0089】
CysOMe-炭素粉末の使用による、CuII除去とCdII除去の熱力学と反応速度論
重金属イオン(CuIIイオン、CdIIイオン、およびAsIIIイオンなど)の除去に対するCysOMe-炭素粉末の有効性を調べた。25mgの変性炭素粉末を、種々の濃度の溶液25cm3中にて種々の時間にわたって攪拌することによって、pH2.0溶液からのCuIIの除去、またはpH5.0溶液からのCdIIの除去に対する濃度-時間プロフィールを作成した。使用した濃度範囲を5μM〜500μMの範囲で変え、このとき正確な濃度(exact solution)は、グラファイト粉末による実験を始める前にLSASV分析を使用して測定した。攪拌時間は、連続状態にて2〜30分であった。
【0090】
CysOMe-炭素とCysOMe-GCとの、CuII取り込み又はCdII取り込みに関する濃度-時間プロフィールの比較から、いずれの場合も変性炭素粉末が、図15に示すように、より多くの量の金属イオンをより速やかな仕方にて除去した、ということがわかる。これは、ガラス質炭素より、CysOMeを含んだグラファイト粉末のほうが、表面積がより大きいことによるものであると考えることができる。
【0091】
ラングミュア等温線モデルとフロイントリッヒ等温線モデルを使用して、実験データを解析した。熱力学的パラメーター、K’とn(CuIIとCdIIの取り込みに関して、CysOMe-炭素とCysOMe-GCの両方に対して得た)を表2に示す。K’とnは、最大吸着容量に関係したフロイントリッヒ定数であり、K’の値が大きくなるほど、そしてnの値が小さくなるほど、被吸着体(adsorbens)に対する吸着剤(adsorbent)の親和性が高くなる。
【0092】
【表2】
【0093】
CysOMe-炭素による金属イオン吸着速度を、検討した各濃度において、対応する濃度-時間プロフィールからの初期金属イオン吸着速度を使用して調べた。比較のため、CysOMe-GCとCysOMe-炭素によるCuIIとCdIIの平均吸着速度定数(kads)を表3に示す。
【0094】
【表3】
【0095】
CysOMe-GC粉末と比較してCysOMe-炭素粉末の吸着速度がより速い(GCミクロ球体の吸着速度の約2倍)のは、グラファイト粒子に対するCysOMeの表面被覆率が増大していることを示している。
【0096】
CysOMe-炭素粉末によるAsIIIイオンの吸着
CysOMe-炭素粉末によるAsIIIイオンの取り込みを下記のように測定した。種々の濃度(10〜150μM)のヒ素を含有する20cm3溶液に40mgの変性炭素粉末を加え、数分〜数時間の範囲にわたって攪拌した。粉末を濾別し、LSASVを使用して溶液を分析して、残留しているAsIIIの濃度を調べた。LSASV法によって分析した一組のサンプルを、次いでICP-AESを使用してAsIII濃度に関して分析した。ICP-AES分析の結果は、LSASVによって得られる結果とよく一致している(5%以内)ことが見出され、このことは、電気分析プロトコルによって正確で信頼できる結果が得られたことを示している。
【0097】
CysMeO-炭素粉末の使用による、微量のAsIIIの除去
初期のAsIII濃度が約70ppbであることがわかっている20cm3溶液中に40mgのCysOMe-炭素粉末を加え、最大30分までの種々の時間にわたって攪拌し、溶液中に残留しているAsIIIの濃度を、前述の微量分析プロトコルを使用してモニターした。
【0098】
図16は、濃度-時間プロフィールの結果を示している。AsIIIの初期濃度は、少量のCysOMe-炭素に曝露してから10分以内にWHO限度(10ppb)未満にまで減少し、20分の曝露後に、本方法の検出限界未満にまで減少した。
【0099】
CysMeO-炭素粉末によるCdII取り込み量の測定
CysOMe-炭素粉末への曝露後にサンプル中に残留しているCdIIの濃度を、pH5.0の酢酸ナトリウム緩衝液中において、Bankら(Talanta 2004,62,279)によって開発されたホウ素ドープダイヤモンジ電極(BDD)にてLSASVプロトコルを使用して測定した。LSASV分析は、下記のパラメーターを使用して行った。BDD電極は、攪拌しながら、SCEに対して−1.5Vの堆積電位に60秒間保持した。次いで、0.1Vs-1のスキャン速度にて、SCEに対して−1.2Vから−0.1Vまで電位を掃引した。カドミウムストリッピングピークは、SCEに対して−0.8Vにて観察された。
【0100】
未知のCdII濃度を有するサンプルを分析する前に、ブランクの酢酸塩緩衝液からなるサンプルに対して標準添加量法を使用して線形範囲を測定した。得られた結果から、LSASV分析プロトコルにより、1〜20μMの線形検出範囲(3σに基づいた検出限界は0.96μM)が得られたことがわかる。必要に応じて、CdIIの濃度が線形範囲内に入るよう、分析の前にサンプルを希釈した。
【0101】
分析しようとするサンプルに標準的な1μM CdII添加量を加え、図17に示すように、標準添加量プロットを作成することによって未知のCdII濃度を決定した。この分析を3回繰り返し、サンプル中に残留しているCdIIの濃度を、3つの結果の平均として算出した。
【0102】
CysMeO-炭素粉末によるCdII取り込み量の測定
サンプル中のCuII濃度を、下記のプロトコルにしたがって、上記の標準添加量法とLSASVプロトコルを使用して測定した。CuIIの分析は、電着プロセス時に塩化銅(I)沈殿物が形成されるのを避けるために(SCE参照電極が使用され、LSASV分析に対して問題がある場合は、沈殿物が形成されることがある)、GC作用電極とAg偽参照電極を使用して0.1MのH3PO4(pH2.0)中にて行った。銅ストリッピングピークは、Agに対して約−0.1Vにおいて観察することができた。1μM CuIIの標準添加量を使用する線形分析濃度範囲は2〜20μMであることがわかった。したがってサンプルはいずれも、この範囲内にはいるよう必要に応じて希釈した。LSASVは、Agに対して−1.5Vの堆積電位、30秒の堆積時間、100mVs-1のスキャン速度、およびAgに対して−1.5Vから+0.8Vまでのスキャン、を使用して行った。
【0103】
CysMeO-炭素粉末によるAsIII取り込み量の測定
LSASVは、参照電極として作用するSCEを含んだ金作用電極(直径1mm)を使用して、0.1M HClの10cm3溶液(pH1.0)中にて行った。LSASV分析は、比較的高い濃度のサンプルに対して、下記のパラメーターを使用して行った:SCEに対して−0.3Vの堆積電位、最初の5秒攪拌して60秒の堆積時間。次いでLSASVボルタンメトリーを、100mVs-1のスキャン速度にて、5mVのステップ電位にて、SCEに対して−0.3Vから+0.4Vまで行った。標準的な2.2μM溶液添加量(5μlの4.4mM標準溶液)を加え、測定した未知サンプル濃度を使用して、標準添加量プロットを作成した。AsIII検出に対する線形範囲は、2〜20μMであることがわかった(3σ値に基づいた検出限界は1.25μM)。濃度がこの範囲内に入るよう、分析の前に、必要に応じて溶液を希釈した。
【0104】
微量分析実験のため、プロトコルを若干変えた。他のパラメーターを上記したものと同一にして、60秒の堆積時間全体にわたって溶液を攪拌した。分析サンプルに加える5μlアリコートが0.22μM標準添加量に相当するよう、標準AsIII溶液を希釈した。得られたボルタンメトリーを図18に示す。線形範囲は0〜2.2μMであることが決定され(検出限界は0.03μM)、したがって分析の前にサンプルを希釈する必要はなかった。
【図面の簡単な説明】
【0105】
【図1a】pH6.8の緩衝液中における4-ニトロフェニル誘導体化炭素(“NP炭素”)のレスポンスを示している連続的なボルタンモグラムである。
【図1b】0.1Mの過塩素酸テトラブチルアンモニウム(TBAP)を担持用電解質として含有するアセトニトリル中における、ブランクのグラファイト粉末とアニリン誘導体化炭素(“AN炭素”)のオーバーレイドのボルタンモグラムである。
【図1c】pH6.8の緩衝液中における4-ニトロ安息香酸誘導体化炭素(“NBAN炭素”)のレスポンスを示している連続的なボルタンモグラムである。
【図2a】AN炭素のX線光電子分光法(XPS)スペクトルのN1s領域を示している。
【図2b】NBAN炭素のXPSスペクトルのN1s領域を示している。
【図3a】ポリ-S-ベンジル-L-システイン誘導体化炭素(“PSBC炭素”)のXPSワイドスペクトルである。
【図3b】ポリ-L-システイン誘導体化炭素(“PC炭素”)のXPSワイドスペクトルである。
【図4】標準的なCd2+添加量の場合の、Cd2+検出に対する線形掃引ストリッピングボルタンモグラムである。差し込み図は、対応する標準添加量プロットを示している。
【図5】10mgのシステインメチルエステル誘導体化ガラス質炭素(“CysMeO-GC”)に曝露した後の、川の水の10cm3サンプル〔当初のCd(II)濃度は約1.5mM〕中に残留しているカドミウムの濃度プロフィールである。
【図6】10mgのCysMeO-GCに曝露した後の、ミネラルウォーターの10cm3サンプル〔当初のCd(II)濃度は50ppb〕中に残留しているカドミウムの濃度プロフィールである。
【図7】10mgのCysMeO-GCに種々の時間にわたって曝露した後の、川の水の10cm3サンプル中に残留している銅の濃度プロフィールである。
【図8】10mgのPC炭素粉末に曝露して、所定時間にわたって攪拌した後に残留しているAs(III)の濃度を示している。この曲線は、一次指数関数的減衰がデータに適合していることを示している。
【図9】10mgのCysMeo-GC粉末に曝露して、所定時間にわたって攪拌した後に残留しているAs(III)の濃度を示している。この曲線は、一次指数関数的減衰がデータに適合していることを示している。
【図10】2000ppbのAs(III)溶液に対し、200mgのCysMeo-GC粉末に曝露して、所定時間にわたって攪拌した後に残留しているAs(III)の濃度を示している。この曲線は、一次指数関数的減衰がデータに適合していることを示している。
【図11】バングラデシュの水サンプルである120ppbのAs(III)溶液に対し、200mgのCysMeo-GC粉末に曝露して、所定時間にわたって攪拌した後に残留しているAs(III)の濃度を示している。この曲線は、一次指数関数的減衰がデータに適合していることを示している。
【図12】200mgのCysMeo-GC球形粉末に曝露して、30分攪拌した後の、120ppbのAs(III)バングラデシュ水サンプルの陽極ストリッピングボルタンモグラムである。線形掃引ボルタンメトリー(LSV)を100mV/sにて行い、2.4×10-7Mの標準添加量を使用した。
【図13】L-システインメチルエステル変性炭素粉末(“CysOMe-炭素”)のXPSスペクトルである。
【図14】AsIIIに曝露した後のCysOMe-炭素粉末の、ベースライン補正したXPSスペクトルである。重要な区域が120〜260eVであることがわかる。点線は、マイクロカル・オリジン・ソトウェア・パッケージ(MicroCal Origin software package)を使用して行われたガウス分布ピークの適合性を示している。
【図15】Cd(NO3)2をpH5.0の酢酸塩緩衝液中に溶解して得られる約55μM溶液からのCdIIの除去に対する、CysOMe-GC粉末吸着剤とCysOMe-炭素粉末吸着剤とを比較しているオーバーレイドの濃度-時間プロフィールである。
【図16】痕跡量のAsIIIのWHO勧告限度未満(10ppb)までの除去に対する濃度-時間プロフィールである。
【図17】標準的なCd2+添加量を1μMずつ増大(0〜20μM)させた場合の、CdII線形掃引陽極ストリッピングボルタンメトリー(LSASV)によるオーバーレイドのボルタンモグラムである。差し込み図は、対応する標準添加量プロットを示している。
【図18】標準的なAsIII添加量を0.22μMずつ増大(0〜2.2μM)させた場合の、オーバーレイドのAsIIILSASVボルタンモグラムである。差し込み図は、対応する標準添加量プロットを示している。
【技術分野】
【0001】
本発明は、誘導体化炭素(derivatised carbon)に関し、具体的には、所望の特性を付与するよう化学的に変性された表面を有するグラファイトおよび他の形態の炭素に関する。
【背景技術】
【0002】
有毒物質(特に有毒重金属)の環境への蓄積と放出が、過去数十年にわたって大幅に増大した。採掘作業と重工業が環境に及ぼす影響により、高濃度の有毒重金属イオン(例えば、CuIIイオン、CdIIイオン、PbIIイオン、およびHgIIイオン)が湖や川に蓄積されてきており、これらの汚染物は、ほとんどが非分解性であって、自然界で再循環している。水性媒体や飲用水中に重金属が存在すると、曝露レベルと重金属の化学的形態によっては、ヒトと水生生物の健康にとって危険となる可能性がある。重金属汚染が引き起こした人道面での悲惨な結果の1つの例が、アルゼンチン、中国、メキシコ、台湾、インド、および特にバングラデシュ等の国々における、何百万という人々の広範囲にわたる被毒である。バングラデシュの地下水の最大で60%が、世界保健機構(WHO)のガイドラインである10ppbをはるかに超える自然発生的な濃度のヒ素を含有している。これら重金属イオンの塩の多くが水溶性であるので、通常の物理的な分離方法は役に立たない。飲用水濾過および/または環境浄化において使用するための水性媒体から有毒重金属イオンを除去できる、簡単で、迅速で、かつ低コストの方法を開発することが急務である。
【0003】
ポリ-L-ヒスチジン、ポリ-L-アスパラギン酸、ポリ-L-グルタミン酸、および特にポリ-L-システイン等のポリペプチドは、金属イオン(例えば、CdIIイオン、PbIIイオン、NiIIイオン、およびCuIIイオン)とキレート環を形成することが知られており、種々の担体に結びつき、これら金属の痕跡分析に使用されている(Malachowski et al,Anal.Chim.Acta.2003, 495, 151;Malachowski et al,Anal.Chim.Acta.2004, 517, 187;Malachowski et al,Pure Appl.Chem.2004, 76, 777;Johnson et al,Anal.Chim.Acta.2005, 77, 30;Howard et al,Anal.At.Spectrom.1999, 14, 1209;およびJurbergs et al,Anal.Chem.1997, 69, 1893)。バイオホモポリマー(biohomopolymers)および他のペプチドは、金属の抽出や再生に対し、従来の方法〔例えば、単純な濾過や沈殿(後者は、環境機関の厳しい規制に適合するよう、ターゲット金属の濃度を減少させることができない場合が多い)〕を凌ぐ大きな利点を有する。
【0004】
グラファイトの表面は、種々の比較的簡単な方法(例えば、所定の化学的または生物学的成分の、物理吸着および化学的もしくは電気化学的に開始される化学吸着)を使用して化学的に変性させることができる。誘導体化表面(derivatised surfaces)を有するグラファイトは、様々な用途にて(例えば、バッテリー技術における電極物質として、およびセンサーとして)使用することができる。グラファイト物質の表面にヒドロキシル部分やカルボキシル部分等の反応性基が存在していることは知られているけれども、化学的に誘導体化されたグラファイトを合成化学用途のための固体担体として使用することはまだ限定されている。
【発明の開示】
【0005】
本発明は、段階的な合成を行う上での炭素ベースの固体担体を提供する。このような物質の表面を“ビルディングブロック(building-block)”様式にて誘導体化することで、所望の特性(例えば、ターゲット検体に対する感受性)を付与することが可能となる。このように、アミノ酸、ペプチド、低分子タンパク質、および核酸等の化学種を、比較的簡単な仕方で炭素(例えばグラファイト)粒子に連結することができる。炭素の表面を最初に誘導体化する化学種の化学を変えることによって、ビルディングブロック分子を炭素表面に連結する種々の方法が可能となる。本発明は特に、アミノ酸もしくはその誘導体が結合する誘導体化炭素(特にグラファイト)を提供する。アミノ酸は、モノマー(例えばシステイン)であっても、あるいは金属イオンと結合することができるポリペプチド(例えばポリ-L-システイン)であってもよい。したがって本発明は特に、有毒重金属の検出、および水や他の水性媒体からの有毒重金属の除去に関連している。
【0006】
本発明の第1の態様によれば、アミノ酸もしくはその誘導体が炭素に結合している誘導体化炭素が提供される。この結合は、直接的な結合であっても、あるいは間接的な結合(例えばフェニルアミン基を介して)であってもよい。
【0007】
本発明はさらに、ニトロフェニル誘導体化炭素が得られるような条件下にて、炭素とニトロベンゼンジアゾニウム化合物とを接触させる、という誘導体化炭素の製造方法を提供する。
【0008】
本発明はさらに、炭素を、炭素の表面のカルボキシル基を介してアミノ酸もしくはその誘導体に直接結合させる、という誘導体化炭素の製造方法を提供する。この製造方法は、炭素の表面のカルボキシル基をハロゲン化アシル基に転化させること、次いで得られる生成物とアミノ酸もしくはその誘導体とを接触させることを含む。
【0009】
本発明はさらに、本発明の誘導体化炭素を含んだ炭素電極を提供する。
本発明はさらに、本発明の電極を含んだ電気化学的デバイスを提供する。本発明の電気化学的デバイスは、電気化学的センサーの形態であっても、あるいは電気化学的リアクターであってもよい。
【0010】
本発明はさらに、本発明の誘導体化炭素と水性媒体とを接触させることを含む、液体媒体から金属イオンを除去する方法を提供する。
本発明はさらに、本発明の電気化学的デバイスを使用して、液体媒体をボルタンメトリー分析にかけることを含む、液体媒体中における金属イオンの存在を検出する方法を提供する。
【0011】
本発明の誘導体化炭素は、液体媒体(水および他の水性媒体を含む)中の金属イオンの検出、液体媒体からの金属イオンの除去、および液体媒体中の金属イオンの滴定に対して有用である。このような金属イオンとしては、例えば、Cd(II)イオン、Pb(II)イオン、Zn(II)イオン、Cu(II)イオン、およびAs(III)イオンなどがある。誘導体化炭素は粒状形態(例えば粉末形態)であってよい。表面積が大きいことから、グラファイト粉末やガラス質炭素粉末等の粒状物質が望ましい(表面積が大きいと、比較的多量のアミノ酸もしくはそれらの誘導体を連結させることができる)。したがって本発明の誘導体化炭素は、公知の変性固体物質よりはるかに多い量の金属イオンを結びつけることができる。
【0012】
(種々の実施態様の説明)
本発明は、アミノ酸もしくはその誘導体が炭素に結合している誘導体化炭素を提供する。アミノ酸もしくはその誘導体は、炭素に直接結合していても、あるいは間接的に(すなわち、リンカーを介して)結合していてもよい。特に挙げておきたいのは、アミノ酸もしくはその誘導体が、炭素上に存在するカルボキシル基やフェニルアミン基を介して結合している状態の炭素である。
【0013】
1つの実施態様においては、アミノ酸はイオウ含有アミノ酸(例えば、システイン、グルタチオン、チロシン、またはこれらの誘導体)である。イオウ含有アミノ酸は、ペンダントのチオール基またはチオール様の基を有してよい。アミノ酸は、エステル〔例えば、メチルエステルやエチルエステル(特定の例としてはL-システインメチルエステルがある)〕の形態をとってよい。アミノ酸の誘導体は、アミノ酸のオリゴマーとポリマーを含む。例えば、システイン誘導体はポリシステインまたはシステアミンであってよく、またグルタチオン誘導体はポリグルタチオンであってよい。代表的なポリマーアミノ酸は、ポリマー鎖1つ当たり50〜100のシステイン残基を含有する、S-ベンジル保護されたホモポリマーである。アミノ酸もしくはその誘導体は、保護されていても、あるいは保護されていなくてもよい。1つの例は、ポリ-S-ベンジル-L-システイン等のポリシステインである。
【0014】
炭素は、粒状物の形態(例えば粉末形態)をとってよい。粒状炭素は、1〜100μm(例えば2〜50μm)の直径を有する粒子を含んでよい。特に挙げておきたいのは、グラファイト粉末、ガラス質炭素球形粉末、および熱分解グラファイトの形態物である。これとは別に、炭素は、カーボンナノチューブの形態〔例えば、多層カーボンナノチューブ(MWCNT)〕をとってもよい。
【0015】
本発明の誘導体化炭素の例としては、システインで変性されたガラス質炭素;グルタチオンもしくはシステアミン、またはこれらの誘導体;およびポリシステインもしくはポリグルタチオンで変性された炭素粉末;などがある。言うまでもないが、本発明は他のアミノ酸ポリマーやそれらの誘導体に対して、そしてさらにアミノ酸モノマーやそれらのチオール含有誘導体(例えばシステイン)に対して適用可能であり、これらがガラス質炭素に連結される。特定の例としては、システインもしくはその誘導体〔例えば、システインのエステル(システインメチルエステル等)、またはシステインのポリマー(ポリシステインやポリ-S-ベンジル-L-システイン等)〕を使用して誘導体化された炭素粉末(例えば、グラファイト粉末やガラス質炭素球形粉末)がある。
【0016】
誘導体化炭素は、炭素とニトロベンゼンジアゾニウム化合物とを、ニトロフェニル誘導体化炭素が生成されるような条件下にて接触させることによって得ることができる。この反応は、適切な試剤(次亜リン酸等)の存在下にて行うことができる。ニトロフェニル誘導体化炭素を還元して、アニリン誘導体化炭素を形成させることができる。この生成物をさらに反応させて、置換アニリン誘導体化炭素を得ることができる。特に、アニリン誘導体化炭素をアミノ酸もしくはその誘導体(例えば、ポリ-S-ベンジル-L-システイン等のポリシステイン)と反応させることができる。
【0017】
誘導体化炭素はさらに、炭素の表面に存在しているカルボキシル基をハロゲン化アシル基に転化させ、次いで得られた生成物とアミノ酸もしくはその誘導体とを接触させることによって得ることができる。ハロゲン化アシルは、例えば塩化アシルであってよい。アミノ酸もしくはその誘導体に存在している任意のカルボキシル基を保護することができる。
【0018】
本発明の誘導体化炭素は、金属イオンの検出(例えば電気化学的検出)、金属イオンの滴定、または液体媒体からの金属イオンの除去に対して使用することができる。金属イオンは、例えば、Cd(II)、Pb(II)、Zn(II)、Cu(II)、およびAs(III)の1種以上であってよい。液体媒体は、例えば水性媒体であってよい。
【0019】
本発明の誘導体化炭素(特に、システイン誘導体化炭素やポリシステイン誘導体化炭素)は、ヒ素の検出に対して有用である。例えば、本発明の誘導体化炭素は、比較的高価な飲用水濾過装置中に組み込むことができる。これに反して、本発明の誘導体化炭素がAs(III)以外の金属イオンに対して選択性がある限りにおいて、As(III)の検出を妨害するイオン〔Cu(II)イオン等〕を除去すべく、本発明の誘導体化炭素をヒ素センサー中に組み込むこともできる。したがって本発明は、天然の供給物がしばしば有毒重金属(例えば、ヒ素やカドミウム)で汚染されている場合に、水の浄化、工場廃液からの金属の回収もしくは抽出、および飲用水の濾過において使用するための安価で且つ魅力的な物質を提供する。
【0020】
本発明はさらに、金属イオン封鎖において有用な物質を提供する。例えば、炭素上に固定されたポリシステインは一般に、公知の基質(例えば、ガラスやポリマービーズ等)の場合より金属取り込み量(物質1グラム当たり)がはるかに多い。単位面積当たりの金属イオン封鎖ユニットの密度も、ナノスケールの変性が使用されている場合(例えば、ナノチューブの場合)は、活性表面積が増大するので、従来技術の基質の場合よりはるかに大きい。したがって、金属イオン取り込みの熱力学と反応速度(kinetic rate)の両方を高めることができる。
【0021】
本発明は特に、バイオホモポリマー(特に、ポリ-L-ヒスチジン、ポリ-L-アスパラギン酸、ポリ-L-グルタミン酸、および特にポリ-L-システインから選択されるポリペプチド)をグラファイト粉末に連結することによってもたらされる固体担体物質を提供する。前述したように、このようなポリマーは、アルカリ金属やアルカリ土類金属(例えば、ナトリウムやカルシウム)に対して殆ど親和性をもたない有毒重金属(例えば、カドミウム、鉛、ニッケル、および銅)とキレート環を形成することが知られている。本発明のシステイン誘導体化グラファイト粉末またはポリ-L-システイン誘導体化グラファイト粉末を使用して、水性媒体から金属イオン〔例えばCd(II)イオン〕を定量的に滴定することができる。グラファイト粉末の表面積が大きく、グラファイト粉末に多量のアミノ酸を連結できることから、システイン変性炭素またはポリシステイン変性炭素は、他のいかなる担体物質に結合したポリ-L-システインよりはるかに多くの量のCd(II)イオンとキレート環を形成する。したがって本発明の誘導体化炭素は、工場廃液からの有毒重金属の回収、環境浄化、および飲用水の濾過に使用するのに特に適している。
【0022】
以下に実施例を挙げて本発明を説明する。
【実施例】
【0023】
実施例1:ポリ-L-システインによるグラファイト粉末の誘導体化
試剤と化学物質
塩化カリウム(Riedel de Haenから購入)を除いて、試剤は全てアルドリッチ社から購入し、入手しうる最高グレードであり、さらなる精製を行わずに使用した。使用した合成グラファイト粉末は、直径が2〜20μmの異形粒子からなり、アルドリッチ社から購入した。水溶液は全て、18.2MΩcm以上の抵抗率を有する、エルガスタット(Elgastat)UHQグレードシステム(Elga)からの脱イオン水を使用して調製した。
【0024】
脱イオン水中pHが1.0〜12.0の範囲の溶液を以下のように調製した:pH1.0,0.10MのHCl;pH1.7,0.1Mのテトラシュウ酸カリウム(potassium tetraoxalate);pH4.6,[0.10Mの酢酸]+[0.10Mの酢酸ナトリウム];pH5.04,0.5Mの酢酸ナトリウム;pH6.8,[0.025MのNa2HPO4]+[0.025MのKH2PO4];pH9.2,0.05Mのテトラホウ酸二ナトリウム;pH10.5,0.1Mのテトラホウ酸二ナトリウム;およびpH12.0,0.01Mの水酸化ナトリウム。これらの溶液はさらに、0.10MのKClを支持電解質として含有した。pHの測定は、ハンナ(Hanna)pH213pHメーターを使用して行った。
【0025】
計測
電気化学的測定値は、標準的な3電極配置構成を有するμAutolabコンピュータ制御ポテンシオスタット〔エコケミー社(Ecochemie)〕を使用して記録した。電気化学的実験は、25cm3の体積のガラスセル中にて行った。熱分解グラファイト底面電極(BPPG,直径5mm,ル・カルボネ社)またはホウ素ドープダイヤモンド電極(BDD,直径3mm,ウィンザー・サイエンティフィック社)が作用電極として作動した。白金コイル(純度99.99%,グッドフェロー社)がカウンター電極として作動した。特に明記しない限り、飽和カロメル電極(SCE,ラジオメーター社)を参照電極として使用して、セル集成体を仕上げた。電気化学的実験は全て、高純度のN2ガス(BOCガス)を使用して溶液を30分脱気した後に、20±2℃にて行った。
【0026】
4-ニトロフェニル誘導体化炭素をSn/HClで還元し、そして4-ニトロ安息香酸と連結させた後のX線光電子スペクトロスコピー(XPS)は、アルミニウムKαバンド(hν=1486.7eV)からのX線照射を使用して(供給源を14hνおよび200mAに設定)、サイエンタ(Scienta)ESCA300機器により行った。スペクトルは全て、150evのパスエネルギーと90°のテイクオフ角度を使用して記録した。特に明記しない限り、1.9mmのスリット幅を使用した。分析チャンバー中のベース圧を2.0×10-9ミリバール以下に保持した。
【0027】
S-ベンジル保護ポリ-L-システインと脱保護ポリ-L-システインのXPSは、アルミニウムKαバンド(hν=1486.7eV)からのX線照射を使用して、VG Clam4MCDアナライザシステムにより行った。XPS実験は全て、90°のテイクオフ角度を有する100eVのアナライザエネルギー(analyzer energy)を使用して記録した。分析チャンバー中のベース圧を2.0×10-9ミリバール以下に保持した。検討した誘導体化炭素サンプルはいずれも、両面接着テープを使用してスタブ上に貼り付け、次いでスペクトロメーターの超高真空分析チャンバー中に配置した。X線照射を受けたときに、光電子を放出して正に帯電するのを防ぐために、スペクトロメーター分析チャンバー内の“フラッドガン”からの電子ビーム(10eV)で、サンプルの表面に衝撃を与えた。得られたスペクトルの分析は、マイクロカル・オリジン(Microcal Origin)6.0を使用して行った。
【0028】
一般的な反応スキーム
スキーム1は、グラファイト粉末を誘導体化するための合成経路を示しており、“ビルディングブロック”の化学、およびグラファイト粉末へのポリ-L-システインの連結の背景となっている原理を示している。
【0029】
【化1】
【0030】
4-ニトロフェニルでグラファイト粉末を誘導体化してNP炭素を作製する
ファストレッドGG(4-ニトロベンゼンジアゾニウムテトラフルオロボレート)の5mM溶液10cm3中に0.5gのグラファイト粉末を混合して攪拌し、これに50cm3の次亜リン酸(H3PO2、50重量%水溶液)を加えた。次いでこの溶液を、穏やかに攪拌しながら5℃で30分放置してから水吸引によって濾過し、脱イオン水で洗浄して過剰の酸を除去し、そして最後にアセトニトリルで洗浄して未反応のジアゾニウム塩を除去した。4-ニトロフェニル誘導体化グラファイト粉末(“NP炭素”)を、ドラフト中に12時間置くことによって風乾してから、使用するまでは気密容器中に保存した(Pandurangappa et al,Analyst,2002,127,1568;およびPandurangappa et al,Analyst,2003,128,473)。
【0031】
NP炭素を還元してAN炭素を作製する
NP炭素粉末(1.02g)とスズ(1.63g,13.7ミリモル)を水(12ml)中に懸濁させた。濃塩酸(4.5ml,53.8ミリモル)を加え、本混合物を加熱還流した。反応混合物を、アルゴン雰囲気下にて100℃で攪拌した。18時間後、混合物を濾過し、得られた固体を、塩酸(1M水溶液100ml)、メタノール(100ml)、水酸化カリウム(1M水溶液50ml)、およびメタノール(50ml)で洗浄した。固体を減圧乾燥して、グラファイト表面に共有結合的に誘導体化されたp-アニリン部分からなる、還元形態のNP炭素である黒色粉末(180.4mg)(“AN炭素”)を得た。
【0032】
4-ニトロ安息香酸をAN炭素に連結させる
AN炭素(500mg)、1-ヒドロキシベンゾトリアゾール水和物(HOBt,670mg,5.0ミリモル)、ベンゾトリアゾール-1-イル-オキシトリピロリジノホスホニウムヘキサフルオロホスフェート(PyBop,2.6g,5ミリモル)、およびp-ニトロ安息香酸(840mg,5ミリモル)をフラスコ中に仕込み、DMF(8ml)を加えた。エチルジイソプロピルアミン(1.7ml,10ミリモル)を加えた。反応混合物を、アルゴン雰囲気下にて室温で攪拌した。18時間後、混合物を濾過し、固体をメタノール(50ml)、アセトニトリル(50ml)、およびDCM(50ml)で洗浄した。固体を減圧乾燥して、アミド結合を介してAN炭素表面に連結した4-ニトロ安息香酸からなる黒色粉末(“NBAN炭素”)を得た。
【0033】
NP炭素、AN炭素、およびNBAN炭素のボルタンメトリー特性決定とXPS特性決定
誘導体化されたNP炭素、AN炭素、およびNBAN炭素のボルタンメトリー特性決定は、先ずそれぞれの誘導体化炭素を、「Leventis et al,Talanta,2004,63,1039」に記載のように、bppg電極の表面上に別々に(separately)そして研磨的に(abrasively)不動態化した後に、pH1.0〜pH12.0の範囲にわたって行った。
【0034】
図1aは、pH6.8でのNP炭素のボルタンメトリーを示している。還元方向への1回目のスキャンでは、SCEに対して約−0.685Vにて大きな還元ウェーブ(reduction wave)が観察された(図1aにおいて“システムI”として表示されている)。SCEに対して−1.0Vにてスキャン方向を反転させると、システムIに対する対応した酸化ウェーブ(oxidation wave)は観察されなかった。このことは、プロセスが電気化学的に不可逆性であることを示している。しかしながら、SCEに対して約+0.025Vにて酸化ウェーブが観察された。引き続きスキャンを行うと、SCEに対して約−0.095Vにて対応する還元ウェーブ観察され、これは、電気化学的にほぼ可逆性のプロセス(“システムII”と呼ぶ)に対応している。電気化学的に不可逆性のシステムIは、その後のスキャンにおいては存在せず、このことは、全ての4-ニトロフェニル部分が還元されていることを示している。
【0035】
観察されたボルタンメトリー挙動とウェーブ形状は、NP炭素についての従来の研究(Pandurangappa et al,Analyst,2002,127,1586)と矛盾せず、水性媒体中における表面結合ニトロ基の電気化学的還元に対応している。スキーム2は、ニトロベンゼン自体の一般的な例に対するこうした挙動を示している(「Pandurangappa et al,Analyst,2002,127,1586」および「Rubinstein,J.Electroanal.Chem.,1971,29,309」)。
【0036】
【化2】
【0037】
このメカニズムにおいては、システムIは、ニトロ基を4電子-4プロトンプロセスにて化学的・電気化学的に不可逆性還元して、アリールヒドロキシルアミンを形成することに相当している。次いで、電気化学的にほぼ可逆性の2電子-2プロトン酸化を受けて(システムII)アリールニトロソ化学種を形成する。このボルタンメトリー挙動は、検討した全てのpHにおいて観察されたけれども、同時に起こるプロトン移動により、システムIとIIに対するピーク電位はpHに依存し、pH1.0〜pH12.0の範囲にわたって、線形ネルンスト式(a linear, Nernstian fashion)にて、それぞれ55.4mV/pHユニットおよび54.4mV/pHユニットだけ変化した(従来の研究と一致する)。
【0038】
十分に確立したボルタンメトリー特性決定プロトコル(「Leventis et al,Talanta 2004,63,1039」および「Wildgoose et al,Talanta,2003,60,887」)を、ほぼ可逆性のシステムIIに対して、検討したそれぞれのpHにおいて実施し、4-ニトロフェニル部分が、実際にグラファイト粒子の表面にとどめられていることが確認された。
【0039】
AN炭素のボルタンメトリー特性決定により、システムIまたはシステムIIに対応するボルタンメトリーウェーブ(voltammetric waves)は観察されない、ということが明らかになった。したがって全ての4-ニトロフェニル基が対応するアニリン様部分に還元された、と結論付けることができる。0.1Mの過塩素酸テトラブチルアンモニウム(TBAP)を含有するアセトニトリル中でのAN炭素のボルタンメトリーは、1回目のスキャンにおいては、アニリンからそのラジカルカチオンへの1電子酸化に対応する電位において、銀疑似参照電極に対して約+0.700Vにて酸化ウェーブを示した(図1b)。
【0040】
XPSを使用して、AN炭素のさらなる特性決定を行った。図2aによれば、スペクトルのN1s領域に単一のピークが観察されており、400.1eVの結合エネルギーは芳香族アミン部分と合致している。ニトロ部分内のN1sレベルまたはO1sレベルから放出される光電子に対応した結合エネルギーにおけるシグナルは観察されなかった。
【0041】
NBAN炭素のボルタンメトリー特性決定により、ニトロ基の予想される特徴的還元がこの場合も観察されること、およびボルタンメトリーが表面結合化学した種に対応していることが明らかになった(図1C)。図2bは、NBAN炭素のXPSスペクトルのN1s領域を示している。400.6eVと405.4eVの結合エネルギーを有する2つのピークが観察され、ピーク高さの比はほぼ1:1である。XPSのデータベースと比較することにより、これらのピークは、それぞれアミド基およびニトロ基中の窒素原子に対応していることが確認された。さらに、スペクトルのO1s領域(図示せず)のガウス解析は、それぞれ、アミド基と芳香族ニトロ基内の酸素原子に合致した530.7eVと533.6eVの結合エネルギーを有するピークを明らかにしている。これらの結果を考慮すると、連結はもっぱら、4-ニトロ安息香酸分子と、AN炭素の表面上のアニリン様部分との間で起こる、と結論づけることができる。
【0042】
ポリ-S-ベンジル-L-システインをAN炭素に連結してPSB炭素を作製する
ポリ-S-ベンジル-L-システイン(PSBC,170mg,0.02ミリモル)を1,4-ジオキサン(3ml)中に溶解した。塩化トリメチルシリル(5.6μl,0.04ミリモル)をDMF(3ml)中に溶解して得た溶液を加えて、ペプチドホモポリマーの溶解性を高めた。反応混合物を、アルゴン雰囲気下にて50℃で攪拌した。1時間後、反応混合物を室温に冷却した。エチルジイソプロピルアミン(6.5μl,0.04ミリモル)を加え、本混合物を0℃に冷却してから塩化9-フルオロエニルメトキシカルボキシル(Fmoc,5.7mg,0.02ミリモル)を加えた。本混合物を室温に自然加温した。1時間半後、減圧にて溶媒を除去して白色固体を得た。得られた残留物に、1-ヒドロキシベンゾトリアゾール水和物(HOBt,4.2mg,0.2ミリモル)、ベンゾトリアゾール-1-イル-オキシトリピロリジノホスホニウムヘキサフルオロホスフェート(PyBop,11.6g,0.02ミリモル)、AN炭素(104mg)、およびDMF(10ml)を加えた。エチルジイソプロピルアミン(17.7μl,0.04ミリモル)を加えた。反応混合物を、アルゴン雰囲気下にて室温で攪拌した。19時間後、混合物を濾過し、得られた固体を、DMF(10ml)、メタノール(10ml)、アセトニトリル(50ml)、およびDCM(50ml)で洗浄した。固体を減圧乾燥して、アミド結合を介してAN炭素に連結したS-ベンジル保護ポリ-L-システインからなる黒色粉末(200mg)(“PSBC炭素”)を得た。
【0043】
PSBC炭素の脱保護
ポリ-L-システイン中のチオール基の脱保護は、バーチ還元法を使用して達成された。PSBC炭素(124mg)とナトリウム(120mg,5.2ミリモル)を収容するフラスコ中に、液体アンモニア(約10ml)を濃縮した。本溶液を、アルゴン雰囲気下にて−78℃で攪拌した。20分後、1-ブタノール(0.3ml)を加え、反応混合物をさらに5分攪拌してから室温に自然加温した。アンモニアが蒸発した後、塩化アンモニウム(約4mlの飽和水溶液)を加えて反応混合物をクエンチした。懸濁液を濾過し、得られた固体を、水(20ml)、メタノール(20ml)、およびDCM(20ml)で洗浄した。固体を減圧乾燥して、アミド結合を介してAN炭素に連結したポリ-L-システインからなる黒色粉末(106mg)(“PC炭素”)を得た。
【0044】
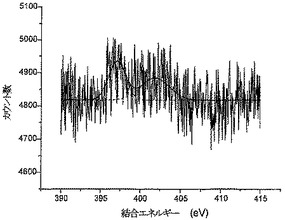
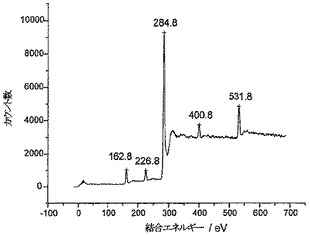
PSBC炭素とPC炭素のXPS特性決定
図3aと3bはそれぞれ、PSBC炭素とPC炭素に対して得られたXPSスペクトルを示している。PSBC炭素においては、S2p3/2レベルとS2sレベルから放出される光電子に対応した162.5eVと226.5eVの結合エネルギーを有する2つのピークが観察された(S-ベンジル保護ポリシステインに対する文献値とよく一致している)。脱保護されたPC炭素においては、S2p3/2光電子とS2s光電子の結合エネルギーが163.5eVと227.5eVにわずかにシフトした(この場合も、ポリシステイン中の遊離チオールに対する文献値とよく一致している)。PSBC炭素とPC炭素の両方に対し、O1sピークとN1sピークはそれぞれ531.5eVと400.5eVに位置しており、ポリシステイン中のアミド結合からの寄与によって特色づけられている(文献値とよく一致している)。PSBC炭素サンプルとPC炭素サンプルの元素分析から、グラファイト表面に連結したポリ-L-システインの相対量(relative amounts)は、バーチ還元を使用したチオール基の脱保護後でも変化していないことがわかった(全イオウシグナル、全酸素シグナル、および全窒素シグナルは、表面元素組成のそれぞれに対して約7±1.4%であった)。このことは、比較的多量のポリシステインが表面に連結していることを示している。したがって、ポリ-L-システインがAN炭素に連結していて、ポリ-L-システイン中のチオール基を脱保護すべくバーチ還元を行った後でも連結したままである、と結論づけることができる。
【0045】
実施例2:PC炭素の使用による、水性媒体中のカドミウムの定量分析
水溶液からのCd2+の取り込みを、線形掃引ストリッピングボルタンメトリー(LSV)ストリッピングプロトコルを使用して、ホウ素ドープダイヤモンド(BDD)電極(「Banks et al,Talanta,2004,62,279」によって開発)にて電気化学的にモニターした。
【0046】
Cd2+検出のための最適化pHはpH5であり、したがってPC炭素によるCd2+のキレート化およびキレート化したCd2+の量のLSV検出に対して、0.05Mの酢酸ナトリウム緩衝液(pH5.04)を使用した。カドミウム検出のためのLSVプロトコルは、溶液を攪拌しながら、電位をSCEに対して−1.5Vに60秒保持することによって、Cd2+をBDD電極上にCd0として堆積させることを含んだ。次いで100mVs-1にて−1.1Vから−0.3Vまで電位をスキャンすることによってLSVを実施し、SCEに対して約−0.8Vにてカドミウムストリッピングピークを観察した。このプロトコルの精度を確認するために、標準的な5nM Cd2+の添加量によってCd(NO3)2の“ブラインド”溶液を分析し、[ピーク高さvs.Cd2+濃度]の標準添加量プロットを作成した。
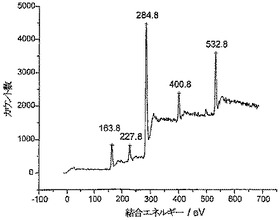
【0047】
図4は、このようにして得られる、Cd2+の量を増大させるためのオーバーレイドLSVボルタンモグラムと標準添加量プロットを示している。Cd2+の濃度は、LSVプロトコルによって20.5nM±0.1nMであると測定された(検出限界(3σ)は0.2nM)。実際のCd2+濃度は20nM±0.1nMであり、LSVプロトコルが、1〜100nMの濃度範囲にわたって微量のCd2+を測定するための正確な方法であることがわかる。
【0048】
PC炭素によってキレート形成されたCd2+の量を測定するために、pH5の酢酸ナトリウム緩衝液中1mMのCd(NO3)2溶液を調製した。この溶液の10μlサンプルを採り、初期のCd2+濃度がLSVプロトコルによって測定できるよう105倍に希釈した。次いで、1mM、2mM、および3mMのCd(NO3)2の10cm3に、それぞれ5mg、10mg、および20mgのPC炭素を加え、10分攪拌した。PC炭素を濾別し、再び濾液の10μlサンプルを採って希釈してから、サンプル中に残留しているCd2+の量を、LSVプロトコルを使用して測定した。PC炭素の各添加量に対して、この手順を3回繰り返した。
【0049】
表1は、量をいろいろ変えたときのPC炭素に対する、キレート化されたCd2+の量を示している。ある時間にわたって実験を繰り返し、10分から12時間まで時間を種々変えて、PC炭素をCd2+と共に攪拌した。PC炭素に対するCd2+の曝露時間を増大させても、キレート化Cd2+の量は増大しなかった。比較のため、ブランクのグラファイト粉末を使用して、同様の実験を行った。ブランクのグラファイト粉末によるCd2+の取り込み量は測定不可であった。表1に記載の結果から、PC炭素が、PC炭素1g当たり1218μモル±200μモルのCd2+をキレート化する、ということを算出することができた。
【0050】
【表1】
【0051】
PC炭素によるCd2+の取り込みは、ポリシステインを他の基質に連結させている従来の研究(「Jurbergs et al,Anal.Chem.,1997,69,1893」;「Malachowski et al,Pure Appl.Chem.,2004,76,777」;「Johnson et al,Anal.Chem.2005,77,30」;および「Howard et al,J.Anal.At.Spectrom.,1999,14,1209」)と比較して、1g当たり最大で100倍大きいことがわかった。特定の理論で拘束されるつもりはないが、これは、グラファイト粉末の表面積が大きいこと、および炭素表面上の多くのエッジ・プレイン様(edge-plane-like)欠陥部位に連結させることができる4-ニトロフェニル基の割合が大きいこと(これにより、他の固体担体に対するよりもはるかに多くのポリシステインをグラファイト粉末に連結させることができる)によるものであると考えられる。さらに、PC炭素をカドミウム(II)溶液に曝露することで、PC炭素によるCd2+イオンの定量的な滴定を迅速に行える。
【0052】
従来の研究により、チオール基との単純なプロトン交換よりむしろ第三立体構造変化(tertiary conformational changes)の結果として、硝酸を使用してポリシステインからCd2+を定量的に回収することができる、ということが実証されている(「Howard et al,J.Anal.At.Spectrom.,1999,14,1209」;および「Miller et al,Anal.Chem.,2001,73,4087」)。カドミウムイオンは、濾過したPC炭素サンプルを1MのHNO3中にて攪拌することによって、PC炭素から回収された。PC炭素の各サンプルを、1.0MのHNO310cm3中にて30分または5時間攪拌した後に、懸濁液を濾過した。濾液の10μlサンプルを取り出し、pH5の緩衝液中に希釈してから、サンプル中に残留しているCd2+の量を、LSVプロトコルを使用して測定した。いずれの場合も、サンプルを30分処理したか、あるいは5時間処理したかに関係なく、キレート化Cd2+の40%±10%が回収された。このことは、Howardらの研究(ポリシステインが、Cd2+に対して弱い結合部位と強い結合部位の両方を示す)と一致している。
【0053】
実施例3:チロシンによるグラファイト粉末とMWCNTの誘導体化
実施例1に記載のジアゾニウム塩化学により、グラファイトとMWCNTに4-ニトロフェニル基を連結した。ニトロ基をSn/HClで還元して、アニリン変性炭素とアニリン変性MWCNTを得た。次いでアニリン基をジアゾ化し、チロシンに連結して、金属をキレート化できる物質を、そしてさらに、アミノ酸またはチオール含有分子をチロシン変性炭素とチロシン変性MWCNTに連結させるための経路を得た。さらに、誘導体化炭素と誘導体化MWCNTの表面上のアニリン部分のアミン基を、金属キレート化/回収に使用すべくチオール基に転化させた。
【0054】
実施例4:L-システインメチルエステルによるガラス質炭素粉末の誘導体化
2gのガラス質炭素球形粉末(GC,直径10〜20μm,タイプI,Alfa Aesar)を10cm3のSOCl2と共に1時間攪拌してから、乾燥CH3Clで洗浄した。これにより、カルボキシル表面基が塩化アシル類縁体に転化された。次いで、この物質と0.5gのL-システイン-メチルエステル塩酸塩(シグマ-アルドリッチ社)とを、10cm3の乾燥CH2Cl2中にて、攪拌しながら、そして0.27cm3のEt3Nを徐々に加えながら反応させた。反応混合物を12時間(一晩)攪拌して、L-システインメチルエステル誘導体化GC球形粉末(“CysMeO-GC”)を得た。このプロセスをスキーム3に示す。
【0055】
【化3】
【0056】
同様の手順にて、ガラス質炭素球形粉末と、グルタチオン(還元形,<99%,アルドリッチ社)およびシステアミン塩酸塩(アクロス・オーガニックス社)とを連結した。
【0057】
実施例5:CysMeO-GC粉末の使用による水からのカドミウムの除去
カドミウムの検出
使用した線形掃引ボルタンメトリー(LSV)ストリッピングプロトコルは、従来の検出プロトコル(Kruusman et al,Electroanalysis,2004,16,399)に基づいた。ホウ素ドープダイヤモンド電極(BDD,直径3mm,ウインザー・サイエンティフィック社)を作用電極として使用し、白金コイルと飽和カロメル電極が、それぞれカウンター電極および参照電極として作用した。電気化学的実験は、pH5.04の0.05M酢酸ナトリウム緩衝液中にて0.1MのKClを支持電解質として加えて、コンピュータ制御のポテンシオスタット(μオートラブ社)を使用して行った。
【0058】
Cd(II)のLSV検出は、下記のパラメーターを使用して行った。酢酸ナトリウム緩衝液10cm3に、試験しようとするサンプルの10μlアリコートを加えた。攪拌しながら、SCEに対して−1.5Vの電位にて60秒間、BDD電極上にカドミウムを堆積させた。電位を、100mVs-1にてSCEに対して−1.1V〜−0.6Vまで掃引し、SCEに対して約−0.780Vにてカドミウムのストリッピングピークが観察された。0.1μMのCd(II)の標準添加量を0.1〜1.0μMの範囲にわたって加え、対応する添加プロットを作成し、これを使用して、最初のサンプル中におけるバックグラウンドCd(II)濃度を算出した。
【0059】
川水からのカドミウムの除去
オックスフォードのチャーウェル川から川水のサンプルを採取した(未処理)。この川水の10cm3サンプルをスパイクして、有毒カドミウム廃棄物の環境的にひどい流出をシミュレートするよう、約1.5mMのカドミウム(II)濃度を生成させた。このコネクションは、ネバ川(ロシアのサンクト・ペテルスブルグを流れており、極めて汚染されていることがよく知られている)における算出された平均Cd(II)濃度である。この“実際の(real)”マトリックスサンプルに10mgのCysMeO-GC粉末を加え、攪拌した。サンプルを濾過し、分析のために10μlアリコートを、上記のLSV Cd(II)ストリッピングプロトコルを使用して5分後に、次いで10分間隔ごとに1時間にわたって取り出した。
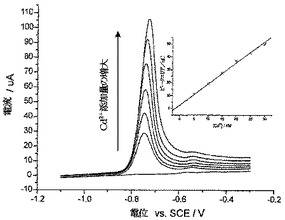
【0060】
図5は、こうして得られたCd(II)濃度プロフィールを示している。10mgのCysMeO-GC粉末によって、サンプルからCd(II)の約87%が除去されたことがわかる。残留Cd(II)の濃度は、サンクト・ペテルスブルグにおける蛇口からの供給上水中のCd(II)の算出飲用水濃度の約半分であった(それでもまだ、WHO、EU、およびEPAのガイドラインより高い)。CysMeO-GC粉末は、環境浄化および/または金属イオン封鎖に使用するための、安価で極めて有効な物質として使用することができる。
【0061】
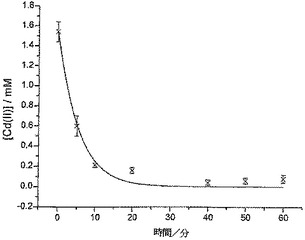
ミネラルウォーターからのカドミウムの除去
50ppb(飲用水に対するEPA勧告最大限度の10倍)のCd(II)濃度が得られるよう、エヴィアン・ミネラルウォーターの10cm3サンプルにCd(II)をスパイクすることによって、飲用上水の汚染をシミュレートした。次いで、この“実際の”マトリックスを10mgのCysMeO-GC粉末と共に攪拌し、前述のように分析した。Cd(II)の除去結果を図6に示す。
【0062】
CysMeO-GC粉末に曝露してから10分以内に、ミネラルウォーター中のCd(II)濃度は、EPA勧告最大限度(5ppb)未満になった。したがってCys-GCは、有毒重金属〔Cd(II)など〕を除去すべく飲用水の濾過に使用するための優れた物質である。
【0063】
実施例6:CysMeO-GC粉末の使用による、水からの銅の除去
使用したスクエアウェーブボルタンメトリー(square wave voltammetry)(SWV)ストリッピングプロトコルは、従来の検出プロトコル(Banks et al,Phys.Chem.Chem.Phys.,2003,5,1652)に基づいた。直径50μmの金ディスク電極(<99.99%,グッドフェロー社)を作用電極として使用し、白金コイルと飽和カロメル電極(SCE,ラジオメーター社)が、それぞれカウンター電極および参照電極として作用した。電気化学的実験は、pH2.00の0.1Mリン酸(H3PO4)緩衝液中にて0.1MのKClを支持電解質として加えて、コンピュータ制御のポテンシオスタット(μオートラブ社)を使用して行った。
【0064】
Cu(II)のSWV検出は、下記のパラメーターを使用して行った:周波数50Hz,ステップ電位2mV,振幅25mV。リン酸緩衝液9.5cm3に、試験しようとするサンプルの0.5cm3アリコートを加えた。攪拌しながら、SCEに対して−1.5Vの電位にて15秒間、作用電極上に銅を堆積させた。電位を、SCEに対して−1.0V〜+0.6Vまで掃引し、SCEに対して約−0.05Vにて銅のストリッピングピークが観察された。1.0μMのCu(II)の標準添加量を1.0〜10.0μMの範囲にわたって加え、対応する添加プロットを作成し、これを使用して、最初のサンプル中におけるバックグラウンドCu(II)濃度を算出した。
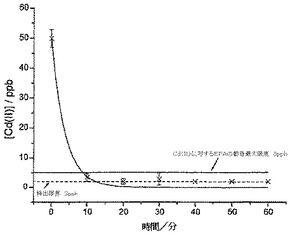
【0065】
川水からの銅の除去
チャーウェル川から川水のサンプル10cm3(未処理)を、上記のSWV銅ストリッピングプロトコルを使用して分析した。約30μMのCu(II)濃度(1.3mgL-1または20.1μMというEPA限度より少し上)を有することが見出され、したがってCu(II)濃度をスパイクせずに使用した。この場合も、サンプルを10mgのCysMeO-GCに曝露し、種々の間隔で1時間にわたって分析して、残留しているCu(II)濃度を測定した。
図7は、サンプルからのCu(II)の除去結果を示している。
【0066】
実施例7:PC炭素粉末およびCysMeO-GC粉末の使用による、水からのヒ素の除去
試剤と化学物質
使用した化学物質は全て分析用グレードであり、受け入れたままの状態にて、さらなる精製を施さずに使用した。これらの化学物質は、(メタ)亜ヒ酸ナトリウム(フルカ社、+99.0%)と硝酸(アルドリッチ社、二重蒸留したPPB/テフロン(登録商標)グレード、ICP-MSによる測定によれば金属不純物はpptレベル)であった。溶液は全て、18.2MΩcm以上の抵抗率を有する脱イオン水(ビベンジウォーターシステムズ社)を使用して調製した。飲用水のサンプルはバングラデシュから入手した。
【0067】
計測
ボルタンメトリー測定は、μオートラブIII(ECO-ケミー社)ポテンシオスタットを使用して行った。測定は全て、3電極セルを使用して行った。作用電極は、金マイクロディスク電極(直径1mm)であり、テフロン(登録商標)ハウジング中に金ワイヤをシールすることによって社内で作製した。カウンター電極は、光沢のある白金ワイヤであり、飽和カロメル電極(ラジオメーター社)が参照電極として使用されている。金電極は、軟らかなラップパッド上にて0.1μmのアルミナスラリーを使用して研磨した。
【0068】
直径3mmのチタン合金マイクロチップ(ジェンコンズ社)を取り付けた、20kHzの周波数で作動する超音波ホーン〔モデルCV26(ソニックス&マテリアルズ社)〕を使用してソノボルタンメトリーによる検討(sonovoltammetric studies)を行った。超音波の強さを熱量測定法により測定し(「Banks et al,Phys.Chem.Chem.Phys.2004,6,3147」;「Magulis et al,Russ.J.Phys.Chem.1969,43,592」;および「Magulis et al,Ultrasonic.Sonochem.,2003,10,343」)、10%にて57Wcm-2であることがわかった。作用電極を超音波ホーンに対して正面向き配列で配置し、超音波ホーンを段付きチップ(stepped tip)のショルダーの向こう側に沈めて、超音波が溶液に確実に効率的に加えられるようにした。ヒ素の検出に対しては、オートラブソフトウェア(三次多項式補正を使用する)を使用してボルタンメトリー曲線をベースライン補正した。
【0069】
PC炭素の使用によるヒ素の除去
ポリシステイン誘導体化炭素粉末を、純水中においてAs(III)と錯形成する能力に関して試験した。堆積プロセス時に超音波によってアシストされる金電極にて、陽極ストリッピングボルタンメトリー(ASV)を使用してAs(III)の濃度を測定した。超音波を強めることで、ASVを金電極にて使用するヒ素検出の感度が大幅に高まった。「Simm et al,Electroanalysis 2005,17,335」に記載の最適化された条件を使用した。各サンプルを錯形成リガンドに曝露する前に対照実験を行って、標準添加量法(standard additions method)によって測定されるAs(III)の濃度が、本方法の検出限度内に入るよう確実に正確になるようにした。
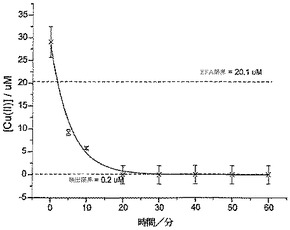
【0070】
(メタ)亜ヒ酸ナトリウムを超純水中に溶解してAs(III)の1.1mM溶液(pH5.4)を調製し、本溶液25mlをフラスコ中に入れて攪拌し、これに10mgのポリシステイン炭素粉末(PC炭素)を加えた。10分、30分、および60分の間隔にて溶液から50μlのサンプルを採取し、次いでこれを0.1Mの硝酸中に希釈し、レベルを追跡して分析を行った。この分析は、金電極をSCEに対して−0.6Vに60秒間保持することによって行い、この時間中、超音波をホーンにて使用して、20mmの距離と5%の振幅をチップした。次いで電位を、堆積電位から1V(SCEに対して)まで100mV/秒のスキャン速度でポジテイブに掃引し、約0.1V(SCEに対して)でのヒ素のストリッピングシグナルを明らかにした。各分析に対し、この初期値を3回測定し、平均値を算出した。それぞれの測定に対して2.4×10-7MのAs(III)を加え、標準添加量法によって存在するAs(III)の最初の濃度を測定するために、これを3回繰り返した。
【0071】
図8は、時間の経過に対するAs(III)濃度の減少を示しており、攪拌60分後にAs(III)の濃度は1.1mMから0.7mMまで減少(36%の減少)し、ポイントを通して一次指数関数的減衰ラインが適合した。本溶液を、さらなる攪拌を行うことなく20日間にわたって静置した後、濃度が0.55mMに減少したことがわかった。
【0072】
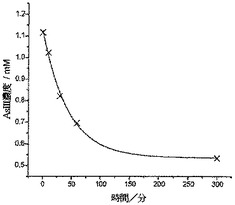
CysMeO-GC粉末の使用によるヒ素の除去
(メタ)亜ヒ酸ナトリウムを超純水中に溶解してAs(III)の0.98mM溶液(pH5.4)を調製し、本溶液25mlをフラスコ中に入れて攪拌し、これに10mgのCys-GC粉末を加えた。10分、20分、および60分の間隔にて溶液から50μlのサンプルを採取し、次いでこれを0.1Mの硝酸中に希釈し、レベルを追跡して分析を行った。
【0073】
図9は、時間の経過に対するAs(III)濃度の減少を示しており、攪拌60分後にAs(III)の濃度は0.98mMから0.7mMまで減少(28.6%の減少)した。本溶液を、さらなる攪拌を行うことなく3日間にわたって静置したが、ヒ素濃度のさらなる減少は見られなかった。
【0074】
次いで、バングラデシュ等の地域からの飲用水中に見られると思われる程度のトレースレベルにて実験を行った(Anawar et al,Environment International 2002,27,597)。サンプルは、50ppbというバングラデシュの限度より4倍大きい200ppb(2.66μM)のAs(III)レベルに調製した。200mgのCysMeO-GC粉末を25mlのサンプル中に混合し、次いでこれを所定時間攪拌してから、システインによるAs(III)の錯形成を停止させるために、濾紙を使用してCysMeO-GC粉末を濾過した。次いでサンプルを0.1Mの硝酸溶液中に1:1に希釈して分析に供した。
【0075】
図10は、わずか10分後にヒ素濃度が200ppbから77ppbに大幅に減少し、そして30分後にレベルが55ppbに減少した、ということを示している。60分での分析結果は、ヒ素の濃度がこのレベルで一定となり(73%の減少)、As(III)が、バングラデシュの飲用安全限度よりやや高い濃度にて存在している、ということを示している。
【0076】
次いで実際のサンプルを使用して、CysMeO-GC粉末が、真正のバングラデシュ井戸水サンプル中のヒ素と錯形成する能力を調べた。サンプルを最初にASV法によって試験して、存在するAs(III)の濃度を測定した。しかしながら、As(III)の濃度が検出可能限界(1×10-8M)未満であることがわかり、したがって実験に使用すべく、水サンプルを120ppbの値にスパイクした。上記実験のように、25mlの水サンプルに200mgのCysMeO-GC粉末を加え、これを所定時間(5分、10分、30分、および45分)攪拌してから濾過して、溶液から粉末を除去した。再び、サンプルを0.1Mの硝酸中に1:1に希釈して分析実験に供した。
【0077】
図11は、一次指数関数的減衰に適合した分析結果を示している。わずか5分の攪拌後に、存在するヒ素の濃度が47%ほど減少し(64ppb)、10分の攪拌後では、濃度がさらに69%ほど減少している(38ppb;すなわち、バングラデシュ飲用安全限度より12ppb少ない)ことがわかる。45分後、濃度が34ppb(すなわち、初期値の28%)で横ばい状態になった。純水ではなく実際のサンプルで分析を行ったので、実験では、バングラデシュの上水中に一般的に見られる多くの微量金属に曝露された(銅、鉛、および水銀など;Anawar et al,Environment International 2002,27,597)。図12は、30分サンプルの分析からのASVプロットを示しており、SCEに対して約0.4Vにて、これら汚染物質のうちの1種による大きなストリッピングウェーブを見ることができる。
【0078】
実施例8:L-システインメチルエステルによる炭素粉末の誘導体化
試剤と装置
ガラス質炭素ミクロ球形粉末(Alfa Aesar,タイプI,直径10〜20μm)と塩化カリウム(Riedel de Haen)を除いて、試剤は全てアルドリッチ社から購入し、市販の最高グレードであり、さらなる精製を行わずに使用した。水溶液は全て、18.2MΩcm以上の抵抗率を有する脱イオン水(ビベンジウォーターシステムズ社)を使用して調製した。pHの測定は、ハンナ・インスツルメントpH213pHメーターを使用して行った。
【0079】
X線光電子スペクトロスコピー(XPS)は、VGクラム4MCDアナライザシステムを使用し、アルミニウムKαバンド(hν=1486.7eV)からのX線照射を使用して行った。XPS実験は全て、90°のテイクオフ角度を有する100eVのアナライザエネルギーを使用して記録した。分析チャンバー中のベース圧を2.0×10-9ミリバール以下に保持した。各炭素粉末サンプルを、両面接着テープを使用してスタブ上に貼り付け、次いでスペクトロメーターの超高真空分析チャンバー中に配置した。X線照射を受けたときに、光電子の放出によって正に帯電するのを防ぐために、スペクトロメーター分析チャンバー内の“フラッドガン”からの電子ビーム(10eV)で、サンプルの表面に衝撃を与えた。フラッドガンがスペクトル線のピーク位置に及ぼす影響を明らかにする上で、記載のピーク位置が、286.6eVというC1s文献値に関して補正されていない点に留意しておかねばならない。得られたスペクトルの分析は、マイクロカル・オリジン6.0を使用して行った。スペクトルピークの帰属は、UKSAFデータベースとNISTデータベースを使用して行った。
【0080】
CysOMe炭素のサンプルに対する燃焼分析は、標準的な方法と装置を使用してC、N、およびSの元素含量(%)を測定することによって行った。
【0081】
炭素粉末へのL-システインメチルエステルの連結
酸素含有表面基(例えば、ヒドロキシル部分やキノニル部分)(グラファイト表面上のエッジ・プレイン欠陥部位をデコレートすることが知られている)を酸化することによって、およびグラファイト粉末を濃硝酸(HNO3)中で18時間攪拌することによって、グラファイト表面にカルボキシル部分を導入した。粉末サンプルから硝酸を除去するために、酸化されたグラファイト粉末を、洗浄液が中性になるまで多量の純水で洗浄した。
【0082】
次いでグラファイト粉末の変性を下記のように達成した。表面カルボキシル基を対応する塩化アシル部分に転化させるために、2gの酸化グラファイト粉末を10cm3の塩化チオニル(SO2Cl2)中にて90分攪拌し、その後、得られた物質を乾燥クロロホルムで洗浄して、未反応の塩化チオニル不純物を除去した。次いで、0.5gのシステインメチルエステル塩酸塩を含有する10cm3の乾燥クロロホルム中に粉末を懸濁させた。この懸濁液に0.27cm3の乾燥トリエチルアミンを滴下し、反応混合物を、不活性アルゴン雰囲気下にて室温で12時間攪拌した。最後に、得られた変性グラファイト粉末(“CysOMe-炭素”)を、多量のクロロホルム、アセトニトリル、アセトン、および純水で洗浄して未反応の化学種を除去した。
【0083】
CysOMe-炭素粉末の特性決定
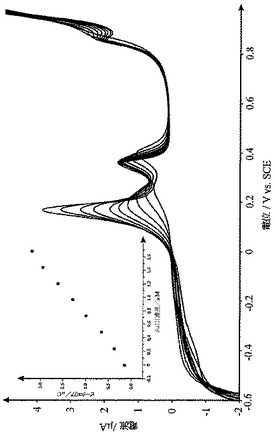
XPSを使用して、どの程度のCysOMeがグラファイの表面に結合しているかを調べた。CysOMe-炭素粉末のサンプルをXPSスペクトロメーター中に据え付け、図13に示すように、スキャンを0eVから1200eVまで行った。UKSAFデータベースとNISTデータベースを使用して、ピークの帰属を行った。
【0084】
各元素の個別のX線断面積によって調整される広域スペクトルにおける各ピークの下の面積から表面の元素組成(%)を算出した。種々の元素に対する関連した原子感度ファクターを考慮にいれると、CysOMeが表面元素の約10%を構成しており、異なったサンプル作製間の変動は±3%である、ということが見出された。この表面被覆率は、燃焼分析を使用して得られる表面被覆率とよく一致している(燃焼分析は、10〜14%のCysOMeの表面被覆率をもたらし、この値は、CysOMe-GC粉末に対する値の約2倍である)。
【0085】
CuII溶液、CdII溶液、またはAsIII溶液に、金属イオンの取り込みが完全となるよう十分な時間にわたって曝露した後のCysOMe-炭素粉末のサンプルに対して、さらにXPS分析を行った(下記のセクションを参照)。図14は、As3sスペクトル、As3p3/2スペクトル、S2sスペクトル、およびS2p3/2スペクトルが観察される区域に対してAsIIIに曝露した後のCysOMe-炭素のXPSスペクトルを示している。AsIII対CysOMeの比(イオウのスペクトル線エリアによって測定)は、相対原子感度ファクターを考慮に入れると約1:1であることがわかった。検討した他の金属に対するXPSの結果は、同様の化学量論関係を示す。
【0086】
実施例9:CysOMe-炭素粉末の使用による、種々の金属イオンの検出と除去
試剤と装置
μAutolabコンピュータ制御ポテンシオスタット(エコケミー社)を使用して電気化学的測定を行った。全体を通して、10cm3の溶液を含んだ3電極セルを使用した。作用電極は、ガラス質炭素電極(GC,直径3mm,BAS)、正方形のホウ素ドープダイヤモンド電極(BDD,3mm×3mm,ウィンザー・サイエンティフィック社)、または金マクロディスク電極(直径1mm,グッドフェロー社)で構成された。光沢のある白金ワイヤ(99.99%,グッドフェロー社)をカウンター電極として作動させ、銀ワイヤの偽電極(99.99%,グッドフェロー社)または飽和カロメル電極(SCE,ラジオメーター社)を参照電極として作動させて3電極集成体を仕上げた。溶液は全て、高純度N2(BOCガス)を使用して20分にわたって脱気し、その後に電気化学的実験を行った。
【0087】
溶液中のAsIII濃度に対する誘導結合プラズマ原子発光分光法(ICPAES)による分析を、パーキンエルマーオプティマ5300DVエミッションICP機器を使用して行った。推奨発光波長は188.979nmであり、最良の検出を得るためにはアキシアル・ビュー(axial view)が推奨される。これは200nmの限界点未満であるので、光学機器をアルゴンの高速流れでパージして、水と空気による光の吸収を最小限に抑えた。
【0088】
5ポイント(0ppb,50ppb,100ppb,150ppb,および200ppb)を使用するAsIIIキャリブレーションは0.9993の相関係数をもたらし、検出限度(ブランクの標準偏差の3倍として定義され、それぞれ3回の繰り返しにて測定された4ブランクチェックからの平均)は9.78ppbすなわち0.0098ppmであることが見出された。パーキンエルマーによる期待値は、この波長に対して1〜10ppbであり、したがって感度は受け入れ可能である。ブランクチェック(blank check)の溶液は、4チェックに対して2.0〜4.5ppbをもたらした。
【0089】
CysOMe-炭素粉末の使用による、CuII除去とCdII除去の熱力学と反応速度論
重金属イオン(CuIIイオン、CdIIイオン、およびAsIIIイオンなど)の除去に対するCysOMe-炭素粉末の有効性を調べた。25mgの変性炭素粉末を、種々の濃度の溶液25cm3中にて種々の時間にわたって攪拌することによって、pH2.0溶液からのCuIIの除去、またはpH5.0溶液からのCdIIの除去に対する濃度-時間プロフィールを作成した。使用した濃度範囲を5μM〜500μMの範囲で変え、このとき正確な濃度(exact solution)は、グラファイト粉末による実験を始める前にLSASV分析を使用して測定した。攪拌時間は、連続状態にて2〜30分であった。
【0090】
CysOMe-炭素とCysOMe-GCとの、CuII取り込み又はCdII取り込みに関する濃度-時間プロフィールの比較から、いずれの場合も変性炭素粉末が、図15に示すように、より多くの量の金属イオンをより速やかな仕方にて除去した、ということがわかる。これは、ガラス質炭素より、CysOMeを含んだグラファイト粉末のほうが、表面積がより大きいことによるものであると考えることができる。
【0091】
ラングミュア等温線モデルとフロイントリッヒ等温線モデルを使用して、実験データを解析した。熱力学的パラメーター、K’とn(CuIIとCdIIの取り込みに関して、CysOMe-炭素とCysOMe-GCの両方に対して得た)を表2に示す。K’とnは、最大吸着容量に関係したフロイントリッヒ定数であり、K’の値が大きくなるほど、そしてnの値が小さくなるほど、被吸着体(adsorbens)に対する吸着剤(adsorbent)の親和性が高くなる。
【0092】
【表2】
【0093】
CysOMe-炭素による金属イオン吸着速度を、検討した各濃度において、対応する濃度-時間プロフィールからの初期金属イオン吸着速度を使用して調べた。比較のため、CysOMe-GCとCysOMe-炭素によるCuIIとCdIIの平均吸着速度定数(kads)を表3に示す。
【0094】
【表3】
【0095】
CysOMe-GC粉末と比較してCysOMe-炭素粉末の吸着速度がより速い(GCミクロ球体の吸着速度の約2倍)のは、グラファイト粒子に対するCysOMeの表面被覆率が増大していることを示している。
【0096】
CysOMe-炭素粉末によるAsIIIイオンの吸着
CysOMe-炭素粉末によるAsIIIイオンの取り込みを下記のように測定した。種々の濃度(10〜150μM)のヒ素を含有する20cm3溶液に40mgの変性炭素粉末を加え、数分〜数時間の範囲にわたって攪拌した。粉末を濾別し、LSASVを使用して溶液を分析して、残留しているAsIIIの濃度を調べた。LSASV法によって分析した一組のサンプルを、次いでICP-AESを使用してAsIII濃度に関して分析した。ICP-AES分析の結果は、LSASVによって得られる結果とよく一致している(5%以内)ことが見出され、このことは、電気分析プロトコルによって正確で信頼できる結果が得られたことを示している。
【0097】
CysMeO-炭素粉末の使用による、微量のAsIIIの除去
初期のAsIII濃度が約70ppbであることがわかっている20cm3溶液中に40mgのCysOMe-炭素粉末を加え、最大30分までの種々の時間にわたって攪拌し、溶液中に残留しているAsIIIの濃度を、前述の微量分析プロトコルを使用してモニターした。
【0098】
図16は、濃度-時間プロフィールの結果を示している。AsIIIの初期濃度は、少量のCysOMe-炭素に曝露してから10分以内にWHO限度(10ppb)未満にまで減少し、20分の曝露後に、本方法の検出限界未満にまで減少した。
【0099】
CysMeO-炭素粉末によるCdII取り込み量の測定
CysOMe-炭素粉末への曝露後にサンプル中に残留しているCdIIの濃度を、pH5.0の酢酸ナトリウム緩衝液中において、Bankら(Talanta 2004,62,279)によって開発されたホウ素ドープダイヤモンジ電極(BDD)にてLSASVプロトコルを使用して測定した。LSASV分析は、下記のパラメーターを使用して行った。BDD電極は、攪拌しながら、SCEに対して−1.5Vの堆積電位に60秒間保持した。次いで、0.1Vs-1のスキャン速度にて、SCEに対して−1.2Vから−0.1Vまで電位を掃引した。カドミウムストリッピングピークは、SCEに対して−0.8Vにて観察された。
【0100】
未知のCdII濃度を有するサンプルを分析する前に、ブランクの酢酸塩緩衝液からなるサンプルに対して標準添加量法を使用して線形範囲を測定した。得られた結果から、LSASV分析プロトコルにより、1〜20μMの線形検出範囲(3σに基づいた検出限界は0.96μM)が得られたことがわかる。必要に応じて、CdIIの濃度が線形範囲内に入るよう、分析の前にサンプルを希釈した。
【0101】
分析しようとするサンプルに標準的な1μM CdII添加量を加え、図17に示すように、標準添加量プロットを作成することによって未知のCdII濃度を決定した。この分析を3回繰り返し、サンプル中に残留しているCdIIの濃度を、3つの結果の平均として算出した。
【0102】
CysMeO-炭素粉末によるCdII取り込み量の測定
サンプル中のCuII濃度を、下記のプロトコルにしたがって、上記の標準添加量法とLSASVプロトコルを使用して測定した。CuIIの分析は、電着プロセス時に塩化銅(I)沈殿物が形成されるのを避けるために(SCE参照電極が使用され、LSASV分析に対して問題がある場合は、沈殿物が形成されることがある)、GC作用電極とAg偽参照電極を使用して0.1MのH3PO4(pH2.0)中にて行った。銅ストリッピングピークは、Agに対して約−0.1Vにおいて観察することができた。1μM CuIIの標準添加量を使用する線形分析濃度範囲は2〜20μMであることがわかった。したがってサンプルはいずれも、この範囲内にはいるよう必要に応じて希釈した。LSASVは、Agに対して−1.5Vの堆積電位、30秒の堆積時間、100mVs-1のスキャン速度、およびAgに対して−1.5Vから+0.8Vまでのスキャン、を使用して行った。
【0103】
CysMeO-炭素粉末によるAsIII取り込み量の測定
LSASVは、参照電極として作用するSCEを含んだ金作用電極(直径1mm)を使用して、0.1M HClの10cm3溶液(pH1.0)中にて行った。LSASV分析は、比較的高い濃度のサンプルに対して、下記のパラメーターを使用して行った:SCEに対して−0.3Vの堆積電位、最初の5秒攪拌して60秒の堆積時間。次いでLSASVボルタンメトリーを、100mVs-1のスキャン速度にて、5mVのステップ電位にて、SCEに対して−0.3Vから+0.4Vまで行った。標準的な2.2μM溶液添加量(5μlの4.4mM標準溶液)を加え、測定した未知サンプル濃度を使用して、標準添加量プロットを作成した。AsIII検出に対する線形範囲は、2〜20μMであることがわかった(3σ値に基づいた検出限界は1.25μM)。濃度がこの範囲内に入るよう、分析の前に、必要に応じて溶液を希釈した。
【0104】
微量分析実験のため、プロトコルを若干変えた。他のパラメーターを上記したものと同一にして、60秒の堆積時間全体にわたって溶液を攪拌した。分析サンプルに加える5μlアリコートが0.22μM標準添加量に相当するよう、標準AsIII溶液を希釈した。得られたボルタンメトリーを図18に示す。線形範囲は0〜2.2μMであることが決定され(検出限界は0.03μM)、したがって分析の前にサンプルを希釈する必要はなかった。
【図面の簡単な説明】
【0105】
【図1a】pH6.8の緩衝液中における4-ニトロフェニル誘導体化炭素(“NP炭素”)のレスポンスを示している連続的なボルタンモグラムである。
【図1b】0.1Mの過塩素酸テトラブチルアンモニウム(TBAP)を担持用電解質として含有するアセトニトリル中における、ブランクのグラファイト粉末とアニリン誘導体化炭素(“AN炭素”)のオーバーレイドのボルタンモグラムである。
【図1c】pH6.8の緩衝液中における4-ニトロ安息香酸誘導体化炭素(“NBAN炭素”)のレスポンスを示している連続的なボルタンモグラムである。
【図2a】AN炭素のX線光電子分光法(XPS)スペクトルのN1s領域を示している。
【図2b】NBAN炭素のXPSスペクトルのN1s領域を示している。
【図3a】ポリ-S-ベンジル-L-システイン誘導体化炭素(“PSBC炭素”)のXPSワイドスペクトルである。
【図3b】ポリ-L-システイン誘導体化炭素(“PC炭素”)のXPSワイドスペクトルである。
【図4】標準的なCd2+添加量の場合の、Cd2+検出に対する線形掃引ストリッピングボルタンモグラムである。差し込み図は、対応する標準添加量プロットを示している。
【図5】10mgのシステインメチルエステル誘導体化ガラス質炭素(“CysMeO-GC”)に曝露した後の、川の水の10cm3サンプル〔当初のCd(II)濃度は約1.5mM〕中に残留しているカドミウムの濃度プロフィールである。
【図6】10mgのCysMeO-GCに曝露した後の、ミネラルウォーターの10cm3サンプル〔当初のCd(II)濃度は50ppb〕中に残留しているカドミウムの濃度プロフィールである。
【図7】10mgのCysMeO-GCに種々の時間にわたって曝露した後の、川の水の10cm3サンプル中に残留している銅の濃度プロフィールである。
【図8】10mgのPC炭素粉末に曝露して、所定時間にわたって攪拌した後に残留しているAs(III)の濃度を示している。この曲線は、一次指数関数的減衰がデータに適合していることを示している。
【図9】10mgのCysMeo-GC粉末に曝露して、所定時間にわたって攪拌した後に残留しているAs(III)の濃度を示している。この曲線は、一次指数関数的減衰がデータに適合していることを示している。
【図10】2000ppbのAs(III)溶液に対し、200mgのCysMeo-GC粉末に曝露して、所定時間にわたって攪拌した後に残留しているAs(III)の濃度を示している。この曲線は、一次指数関数的減衰がデータに適合していることを示している。
【図11】バングラデシュの水サンプルである120ppbのAs(III)溶液に対し、200mgのCysMeo-GC粉末に曝露して、所定時間にわたって攪拌した後に残留しているAs(III)の濃度を示している。この曲線は、一次指数関数的減衰がデータに適合していることを示している。
【図12】200mgのCysMeo-GC球形粉末に曝露して、30分攪拌した後の、120ppbのAs(III)バングラデシュ水サンプルの陽極ストリッピングボルタンモグラムである。線形掃引ボルタンメトリー(LSV)を100mV/sにて行い、2.4×10-7Mの標準添加量を使用した。
【図13】L-システインメチルエステル変性炭素粉末(“CysOMe-炭素”)のXPSスペクトルである。
【図14】AsIIIに曝露した後のCysOMe-炭素粉末の、ベースライン補正したXPSスペクトルである。重要な区域が120〜260eVであることがわかる。点線は、マイクロカル・オリジン・ソトウェア・パッケージ(MicroCal Origin software package)を使用して行われたガウス分布ピークの適合性を示している。
【図15】Cd(NO3)2をpH5.0の酢酸塩緩衝液中に溶解して得られる約55μM溶液からのCdIIの除去に対する、CysOMe-GC粉末吸着剤とCysOMe-炭素粉末吸着剤とを比較しているオーバーレイドの濃度-時間プロフィールである。
【図16】痕跡量のAsIIIのWHO勧告限度未満(10ppb)までの除去に対する濃度-時間プロフィールである。
【図17】標準的なCd2+添加量を1μMずつ増大(0〜20μM)させた場合の、CdII線形掃引陽極ストリッピングボルタンメトリー(LSASV)によるオーバーレイドのボルタンモグラムである。差し込み図は、対応する標準添加量プロットを示している。
【図18】標準的なAsIII添加量を0.22μMずつ増大(0〜2.2μM)させた場合の、オーバーレイドのAsIIILSASVボルタンモグラムである。差し込み図は、対応する標準添加量プロットを示している。
【特許請求の範囲】
【請求項1】
アミノ酸もしくはその誘導体が炭素に結合している誘導体化炭素。
【請求項2】
アミノ酸もしくはその誘導体が前記炭素上のカルボキシル基に結合している、請求項1に記載の誘導体化炭素。
【請求項3】
前記アミノ酸もしくはその誘導体で置換されたフェニルアミン基が前記炭素に結合している、請求項1に記載の誘導体化炭素。
【請求項4】
アミノ酸がイオウ含有アミノ酸である、請求項1〜3のいずれか一項に記載の誘導体化炭素。
【請求項5】
アミノ酸が、システイン、グルタチオン、チロシン、またはこれらの誘導体である、請求項4に記載の誘導体化炭素。
【請求項6】
アミノ酸誘導体がオリゴマーまたはポリマーである、請求項1〜5のいずれか一項に記載の誘導体化炭素。
【請求項7】
アミノ酸誘導体がポリ-S-ベンジル-L-システインである、請求項6に記載の誘導体化炭素。
【請求項8】
炭素が、グラファイト粉末またはガラス質炭素球形粉末である、請求項1〜7のいずれか一項に記載の誘導体化炭素。
【請求項9】
炭素が、ガラス質炭素球形粉末または熱分解グラファイトである、請求項1〜8のいずれか一項に記載の誘導体化炭素。
【請求項10】
炭素がガラス質炭素球形粉末であって、アミノ酸もしくはその誘導体が、システイン、グルタチオン、チロシン、もしくはシステアミンである、請求項9に記載の誘導体化炭素。
【請求項11】
炭素が熱分解グラファイトであって、アミノ酸もしくはその誘導体が、ポリシステインもしくはポリグルタチオンである、請求項9に記載の誘導体化炭素。
【請求項12】
炭素がグラファイト粉末またはガラス質炭素球形粉末であって、アミノ酸が、システインまたはその誘導体である、請求項8または9に記載の誘導体化炭素。
【請求項13】
アミノ酸が、システイン、システインメチルエステル、またはポリ-S-ベンジルL-システインである、請求項12に記載の誘導体化炭素。
【請求項14】
ニトロフェニル誘導体化炭素が得られるような条件下にて、炭素とニトロベンゼンジアゾニウム化合物とを接触させる、誘導体化炭素の製造方法。
【請求項15】
次亜リン酸の存在下にて、炭素とニトロベンゼンジアゾニウム化合物とを接触させる、請求項14に記載の製造方法。
【請求項16】
ニトロフェニル誘導体化炭素を還元して、アニリン誘導体化炭素を形成させることをさらに含む、請求項14または15に記載の製造方法。
【請求項17】
アニリン誘導体化炭素とある化学種とを反応させて、置換されたアニリン誘導体化炭素を得ることをさらに含む、請求項16に記載の製造方法。
【請求項18】
アニリン誘導体化炭素とアミノ酸もしくはその誘導体とを反応させる、請求項17に記載の製造方法。
【請求項19】
アミノ酸もしくはその誘導体および/または炭素が、請求項4〜13のいずれか一項に規定されているとおりである、請求項18に記載の製造方法。
【請求項20】
炭素を、炭素の表面のカルボキシル基を介してアミノ酸もしくはその誘導体に直接結合させる、という誘導体化炭素の製造方法であって、このとき炭素の表面のカルボキシル基をハロゲン化アシル基に転化させること、次いで得られる生成物とアミノ酸もしくはその誘導体とを接触させることを含む、前記誘導体化炭素の製造方法。
【請求項21】
ハロゲン化アシルが塩化アシルである、請求項20に記載の製造方法。
【請求項22】
アミノ酸もしくはその誘導体および/または炭素が、請求項4〜13のいずれか一項に規定されているとおりである、請求項20または21に記載の製造方法。
【請求項23】
請求項1〜13のいずれか一項に記載の誘導体化炭素を含んだ炭素電極。
【請求項24】
請求項23に記載の電極を含んだ電気化学的デバイス。
【請求項25】
請求項1〜13のいずれか一項に記載の誘導体化炭素と液体媒体とを接触させることを含む、液体媒体から金属イオンを除去する方法。
【請求項26】
金属イオンが、Cd(II)イオン、Pb(II)イオン、Zn(II)イオン、Cu(II)イオン、およびAs(III)イオンから選択される、請求項25に記載の方法。
【請求項27】
請求項24に記載の電気化学的デバイスを使用して、液体媒体をボルタンメトリー分析にかけることを含む、液体媒体中における金属イオンの存在を検出する方法。
【請求項28】
液体媒体が水性媒体である、請求項25〜27のいずれか一項に記載の方法。
【請求項1】
アミノ酸もしくはその誘導体が炭素に結合している誘導体化炭素。
【請求項2】
アミノ酸もしくはその誘導体が前記炭素上のカルボキシル基に結合している、請求項1に記載の誘導体化炭素。
【請求項3】
前記アミノ酸もしくはその誘導体で置換されたフェニルアミン基が前記炭素に結合している、請求項1に記載の誘導体化炭素。
【請求項4】
アミノ酸がイオウ含有アミノ酸である、請求項1〜3のいずれか一項に記載の誘導体化炭素。
【請求項5】
アミノ酸が、システイン、グルタチオン、チロシン、またはこれらの誘導体である、請求項4に記載の誘導体化炭素。
【請求項6】
アミノ酸誘導体がオリゴマーまたはポリマーである、請求項1〜5のいずれか一項に記載の誘導体化炭素。
【請求項7】
アミノ酸誘導体がポリ-S-ベンジル-L-システインである、請求項6に記載の誘導体化炭素。
【請求項8】
炭素が、グラファイト粉末またはガラス質炭素球形粉末である、請求項1〜7のいずれか一項に記載の誘導体化炭素。
【請求項9】
炭素が、ガラス質炭素球形粉末または熱分解グラファイトである、請求項1〜8のいずれか一項に記載の誘導体化炭素。
【請求項10】
炭素がガラス質炭素球形粉末であって、アミノ酸もしくはその誘導体が、システイン、グルタチオン、チロシン、もしくはシステアミンである、請求項9に記載の誘導体化炭素。
【請求項11】
炭素が熱分解グラファイトであって、アミノ酸もしくはその誘導体が、ポリシステインもしくはポリグルタチオンである、請求項9に記載の誘導体化炭素。
【請求項12】
炭素がグラファイト粉末またはガラス質炭素球形粉末であって、アミノ酸が、システインまたはその誘導体である、請求項8または9に記載の誘導体化炭素。
【請求項13】
アミノ酸が、システイン、システインメチルエステル、またはポリ-S-ベンジルL-システインである、請求項12に記載の誘導体化炭素。
【請求項14】
ニトロフェニル誘導体化炭素が得られるような条件下にて、炭素とニトロベンゼンジアゾニウム化合物とを接触させる、誘導体化炭素の製造方法。
【請求項15】
次亜リン酸の存在下にて、炭素とニトロベンゼンジアゾニウム化合物とを接触させる、請求項14に記載の製造方法。
【請求項16】
ニトロフェニル誘導体化炭素を還元して、アニリン誘導体化炭素を形成させることをさらに含む、請求項14または15に記載の製造方法。
【請求項17】
アニリン誘導体化炭素とある化学種とを反応させて、置換されたアニリン誘導体化炭素を得ることをさらに含む、請求項16に記載の製造方法。
【請求項18】
アニリン誘導体化炭素とアミノ酸もしくはその誘導体とを反応させる、請求項17に記載の製造方法。
【請求項19】
アミノ酸もしくはその誘導体および/または炭素が、請求項4〜13のいずれか一項に規定されているとおりである、請求項18に記載の製造方法。
【請求項20】
炭素を、炭素の表面のカルボキシル基を介してアミノ酸もしくはその誘導体に直接結合させる、という誘導体化炭素の製造方法であって、このとき炭素の表面のカルボキシル基をハロゲン化アシル基に転化させること、次いで得られる生成物とアミノ酸もしくはその誘導体とを接触させることを含む、前記誘導体化炭素の製造方法。
【請求項21】
ハロゲン化アシルが塩化アシルである、請求項20に記載の製造方法。
【請求項22】
アミノ酸もしくはその誘導体および/または炭素が、請求項4〜13のいずれか一項に規定されているとおりである、請求項20または21に記載の製造方法。
【請求項23】
請求項1〜13のいずれか一項に記載の誘導体化炭素を含んだ炭素電極。
【請求項24】
請求項23に記載の電極を含んだ電気化学的デバイス。
【請求項25】
請求項1〜13のいずれか一項に記載の誘導体化炭素と液体媒体とを接触させることを含む、液体媒体から金属イオンを除去する方法。
【請求項26】
金属イオンが、Cd(II)イオン、Pb(II)イオン、Zn(II)イオン、Cu(II)イオン、およびAs(III)イオンから選択される、請求項25に記載の方法。
【請求項27】
請求項24に記載の電気化学的デバイスを使用して、液体媒体をボルタンメトリー分析にかけることを含む、液体媒体中における金属イオンの存在を検出する方法。
【請求項28】
液体媒体が水性媒体である、請求項25〜27のいずれか一項に記載の方法。
【図1a】

【図1b】
【図1c】
【図2a】
【図2b】
【図3a】
【図3b】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
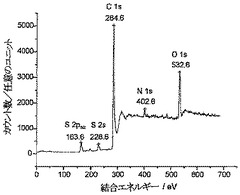
【図14】
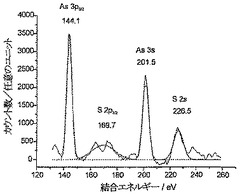
【図15】
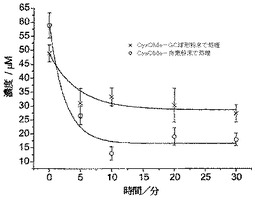
【図16】
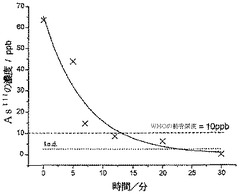
【図17】
【図18】
【図1b】
【図1c】
【図2a】
【図2b】
【図3a】
【図3b】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【公表番号】特表2008−542197(P2008−542197A)
【公表日】平成20年11月27日(2008.11.27)
【国際特許分類】
【出願番号】特願2008−509509(P2008−509509)
【出願日】平成18年5月5日(2006.5.5)
【国際出願番号】PCT/GB2006/001643
【国際公開番号】WO2006/120396
【国際公開日】平成18年11月16日(2006.11.16)
【出願人】(500056231)アイシス・イノベーション・リミテッド (17)
【氏名又は名称原語表記】ISIS INNOVATION LIMITED
【Fターム(参考)】
【公表日】平成20年11月27日(2008.11.27)
【国際特許分類】
【出願日】平成18年5月5日(2006.5.5)
【国際出願番号】PCT/GB2006/001643
【国際公開番号】WO2006/120396
【国際公開日】平成18年11月16日(2006.11.16)
【出願人】(500056231)アイシス・イノベーション・リミテッド (17)
【氏名又は名称原語表記】ISIS INNOVATION LIMITED
【Fターム(参考)】
[ Back to top ]