
調節放出クロピドグレル製剤
クロピドグレル50mg〜150mgに相当するクロピドグレルまたはクロピドグレルの塩を含む1日1回の剤形が開示される。前記剤形は、下記の(a)約3時間以上の平均Tmax、(b)1000ピコグラム/ml以下の平均Cmax、(c)2500ピコグラム/ml/時間超の平均AUC0〜49時間から選択されるクロピドグレルのin vivo血漿プロファイルの少なくとも1つを提供する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、クロピドグレルおよび/またはその薬学的に許容される塩/溶媒和物の医薬組成物に関する。具体的には、本発明は、クロピドグレルおよび/またはその薬学的に許容される塩/溶媒和物の調節放出製剤に関する。本発明は、クロピドグレルおよび/またはその薬学的に許容される塩/溶媒和物の調節放出製剤を調製する方法をさらに提供する。本発明は、クロピドグレル50mg〜150mgに相当するクロピドグレルまたはクロピドグレルの塩を含む1日1回の剤形をさらに提供し、前記剤形は、クロピドグレルについて平均Tmaxが約3時間以上であり、平均Cmaxが1000ピコグラム/ml以下である血漿プロファイルを提供する。クロピドグレルの本発明の製剤は、Sanofi社から市販されているブランド製品であるPlavix(登録商標)クロピドグレル75mg錠と比較して、実質的により低いレベルの不活性クロピドグレルカルボキシ酸代謝物を提供する。
【0002】
クロピドグレルは、メチル(+)−(S)−α−(o−クロロフェニル)−6,7−ジヒドロチエノ(3,2−c)ピリジン−5(4H)−アセテートとして化学的に公知であり、下記の式(I)の構造によって表される。
【0003】
【化1】
【背景技術】
【0004】
クロピドグレルは、血小板凝集阻害剤である。血小板機能を阻害する種々の薬物が、脳卒中または一過性脳虚血発作、心筋梗塞、不安定狭心症、あるいは血管バイパスまたは血管形成術の必要性によって明らかとなる確定した心血管アテローム性動脈硬化症を有する人々における病的事象を減少させることが示されてきた。これは、血小板がこれらの事象の開始および/または進展に関与し、それらを阻害すると事象の割合を減少させることができることを示す。
【0005】
クロピドグレルは、アデノシン二リン酸(ADP)とその血小板受容体の結合、およびそれに続く糖タンパク質GPIIb/IIIa複合体のADP媒介性の活性化を選択的に阻害し、それによって血小板凝集を阻害する。クロピドグレルが生体内変化することは、血小板凝集の阻害を生じさせるために必要である。クロピドグレルはまた、ADP以外のアゴニストによって誘発される血小板凝集も、放出されたADPによる血小板活性化の増幅を遮断することによって阻害する。クロピドグレルは、ホスホジエステラーゼ活性を阻害しない。
【0006】
クロピドグレルは、血小板ADP受容体を不可逆的に修飾することによって作用する。結果的に、クロピドグレルに曝された血小板は、その寿命の残りの期間影響を受ける。
【0007】
血小板凝集の用量依存的阻害を、クロピドグレルの1回の経口投与の2時間後に見ることができる。クロピドグレル75mg/日の反復投与は、第1日目にADP誘導性血小板凝集を阻害し、3日目〜7日目に阻害は定常状態に達する。定常状態では、クロピドグレル75mg/日の用量によって観察される平均阻害レベルは、40%〜60%であった。血小板凝集および出血時間は、治療が中止された後、一般に約5日でベースライン値に徐々に戻る。
【0008】
クロピドグレルは、結晶性重硫酸塩、すなわち重硫酸クロピドグレルの形態で市販されており、ブランド名Plavix(登録商標)で販売されている。
【0009】
血小板凝集阻害剤および抗血栓剤としてのクロピドグレルの治療への応用およびその調製は、米国特許第4,529,596号に開示されている。
【0010】
US6,429,210には、形態IIとして示される硫酸水素クロピドグレルの結晶形態が開示されている。硫酸水素クロピドグレルの調製のためのEP281459に記載されている方法は、結晶形態(形態1)をもたらす。市販品では、結晶性硫酸水素クロピドグレルであるPlavix(登録商標)が、医薬組成物に使用された。
【0011】
US2006/0264636には、クロピドグレルの新規な塩、すなわちクロピドグレルメシラート、クロピドグレルベシラートおよびクロピドグレルトシラート、それらの調製方法、ならびにそれらを含有する医薬組成物が開示されている。
【0012】
US2007/0048370には、クロピドグレル、あるいはその溶媒和物または水和物の製剤が開示されている(ただし、塩はクロピドグレルヨウ化水素酸塩ではなく、錠剤はイオン性および/または塩基性打錠賦形剤、ならびにポリエチレングリコール6000を含有しない)。
【0013】
クロピドグレルは、プロドラッグである。活性代謝物であるチオール誘導体は、クロピドグレルの2−オキソ−クロピドグレルへの酸化、ついで加水分解により形成される。酸化ステップは、主にシトクロムP450アイソエンザイム2B6および3A4、ならびにより少ない程度で1A1、1A2および2C19に制御されている。活性チオール代謝物は、in vitroで単離されているが、速やかに血小板受容体に不可逆的に結合することにより血小板凝集を阻害する。この代謝物は、血漿中では検出されていない。
【0014】
主要循環代謝物の薬物動態は、クロピドグレル50〜150mgの用量範囲において線形性であった(血漿濃度は用量に比例して上昇した)。参照文献:2006年9月15日付け、IPHA Electronic Medicines compendium。
【0015】
血小板は核を欠いており、最も小さい循環ヒト細胞であるが、生理学的および病態生理学的の両方で、血栓症の過程において不可欠で複雑な役割を果たす。血小板の活性化および凝集は、(1)急性冠動脈症候群(ACS)の心筋虚血または梗塞をもたらすアテローム動脈硬化性プラークの自然発生的崩壊、あるいは(2)経皮冠動脈インターベンション(PCI)からもたらされる機械的崩壊後の冠内血栓の増殖において中心的役割を果たす。血小板は最初に、崩壊したプラーク部位においてコラーゲンおよびフォンウィルブランド因子に付着し、最初の血小板の単層をもたらす。活性化の後、血小板はトロンボキサンA2およびアデノシン二リン酸(ADP)などの二次アゴニストを放出し、それが凝固カスケードによって生じたトロンビンと組み合わさり、さらなる血小板の刺激および動員をもたらす。この病態生理学的背景によって、抗血小板療法が、ACSを有する患者、特にPCIを受ける患者の管理の基礎であることは驚くことではない。ADPへの反応について光学的血小板凝集測定法によって測定する場合、研究によってクロピドグレルに反応する用量および時間依存的変動性が示された。Gurbelらによる研究では、選択的冠動脈ステント留置術を受けた96人の患者を、標準的クロピドグレル治療(300mgの初期用量、次いで75mg/日)前、および治療後の複数の時点でモニターした。クロピドグレル耐性(経験的に治療前の値と比較してADP5μmol/Lに反応した凝集の<10%減少と定義付けられる)は、2時間で患者の63%で、24時間で31%に、5日で31%に、30日で15%に見られた。
【0016】
最も高い治療前の値を有する患者は、最初の5日間に亘って最も低い抗血栓性保護を有した。他の報告でMullerらは、24時間でこの治療計画に対してさらなる効果が見られず、クロピドグレル600mgの投与4時間後にADPに対する血小板凝集が<10%の減少である場合を非反応者と定義し、10%〜29%の減少である場合を準反応者と定義した。ADP5μmol/Lに対して、5%が非反応者であり、9%が準反応者であり、ADP20μmol/Lに対して、11%が非反応者であり、26%が準反応者であることをこの研究は見出した。臨床転帰を評価するように設計されていなかったが、Muller研究における興味深い知見は、(試験した105人のうち)2人の患者が亜急性ステント血栓症を起こし、両者ともクロピドグレル非反応の定義に合致したことであった。さらなる報告は狭心症クラスを血小板阻害と関連付け、ADP450mgの投与後により高い狭心症クラスを示す患者は血小板凝集の阻害がより少ないことを見出した。
【0017】
クロピドグレル耐性については、いくつかの機構が可能性を有する。外因性機構には、クロピドグレルの不適切な投与または不十分な投与、およびクロピドグレルとアトロバスタチンとの可能性のある相互作用を含めた薬物・薬物相互作用が挙げられる。クロピドグレル反応とCYP3A4活性(エリスロマイシン呼気検査により測定)とに正相関があり、これは、重要な機構が活性代謝物への可変性変換であり得ることを示唆する。他の可能性がある外因性機構には、プロドラッグの可変性吸収または活性代謝物のクリアランスが含まれるであろう。固有の機構には、P2Y12受容体の可変性、受容体数の増加、ADP放出の増加、または他の血小板活性化経路の上方制御を挙げることができる。参照文献:Circulation 2004、109:3064~3067頁、American Heart Association, Inc.
【0018】
最近、新しい臨床データが明らかになり、Plavixの標準的用量は、患者の約25%に適切な血小板阻害をもたらさないことが示された。Plavix耐性に対する可能性のある説明はいくつかあり、Plavix耐性が実質的現象であるのか、または効果のない投与の結果であるのかはまだ明らかではない。Plavix「耐性」は、より高い初期用量によって実質的に減少できることが、最近の研究によって示された。「Plavix耐性」のこの問題は、心臓病専門医の間でより注目を集めており、2004年のCirculationにおいて公表された研究によって、Plavixからの恩恵が最も少ない患者は[血小板凝集についてのアデノシン二リン酸(ADP)誘導性の百分率の変化によって決定される]、心血管事象の危険がはるかに高いことが示された。参照文献:Circulation.2004、109:3171~3175頁、2004 American Heart Association, Inc.
【0019】
血小板凝集は、血栓形成過程の重要な部分である。アデノシン5−二リン酸、エピネフリンおよび様々な血小板因子が血小板顆粒から押し出され、血小板凝集において主要な役割を果たしているトロンボキサンが形成されることによって、この血小板凝集現象が開始する。血小板動員の血栓形成を妨げることのできる薬剤は、アテローム性動脈硬化症、血栓症および急性冠動脈症候群の治療において重要な役割を果たすことができる。動物から得た多血小板血漿中のin vivoの血小板凝集の測定は、血小板研究において基礎手段である。ADPは、通常の血小板機能のin vitro/ex vivoのスクリーニングのための凝集剤であり、出血性障害の原因を特定するための重要な診断手段に相当する。
【0020】
重硫酸クロピドグレルの投与量は、投与後1〜3時間にクロピドグレルおよびその活性代謝物のピーク濃度をもたらすことが報告されている。これは、投与後直後に高濃度の活性代謝物を、および投与後24時間にはより低い濃度を提供するであろう。さらに、吸収後のクロピドグレルの85%は、血中のエステラーゼによって不活性化され、15%のみが、肝臓での活性代謝物への変換を受ける。要約すれば、患者は、定常状態においてピークレベルで出血の危険に、定常状態においてトラフレベルで乏しい血小板阻害に曝されている。
【0021】
より最近では、様々な研究グループおよび臨床医が、クロピドグレル耐性の明らかになりつつある問題を報告してきた。これは、再発する心血管事象の危険性を増加させ得る。本発明者らは、驚いたことに、クロピドグレルおよびその薬学的に許容される塩の調節放出製剤が、クロピドグレル耐性の問題に対処することができ、したがって反応率を向上させることを見出した。さらに、調節放出製剤はまた、クロピドグレル治療の安全性、利便性および服薬率(compliance)を改善する可能性も提供する。クロピドグレル投与に続く過量は、長引く出血時間、次いで出血性合併症をもたらし得る。出血が観察される場合、適切な治療を考慮すべきである。要するに、クロピドグレルの調節放出製剤は、より良好な抗血小板療法を必要としている数百万の人々の治療を改善するであろう。
【0022】
本発明は、医薬品産業の実現されていない要求に応えるものである。
【0023】
調節放出は、遅延放出、延長放出、パルス放出または持続放出などでよい。放出の調節は、胃出血などの薬物の副作用の最小化のため、または一生続くクロピドグレルの治療が必要な患者におけるクロピドグレル耐性の進行を予防するためなど、たくさんの理由によって所望され得る。
【特許文献1】米国特許第4,529,596号
【特許文献2】US6,429,210
【特許文献3】EP281459
【特許文献4】US2006/0264636
【特許文献5】US2007/0048370
【非特許文献1】2006年9月15日付け
【非特許文献2】Circulation 2004、109:3064~3067頁、American Heart Association, Inc.
【非特許文献3】Circulation.2004、109:3171~3175頁、2004 American Heart Association, Inc.
【発明の開示】
【発明が解決しようとする課題】
【0024】
本発明の重要な目的の1つは、クロピドグレルまたはその薬学的に許容される塩/溶媒和物の調節放出医薬組成物を提供することである。
【0025】
本発明の他の目的は、1000ピコグラム/ml以下のCmaxおよび3時間超のTmaxを有する適切な血漿プロファイルを提供する、クロピドグレル50mg〜150mgに相当するクロピドグレルまたはその薬学的に許容される塩/溶媒和物を使用する調節放出医薬組成物を提供することである。
【0026】
本発明のさらに他の目的は、Sanofi社から市販されているブランド製品であるPlavix(登録商標)クロピドグレル75mg錠と比較して、実質的により低いレベルの不活性クロピドグレルカルボキシ酸代謝物を提供するクロピドグレルの製剤を提供することである。
【0027】
本発明のさらに他の目的は、クロピドグレルまたはその薬学的に許容される塩/溶媒和物の調節放出製剤の製造方法を提供することである。
【課題を解決するための手段】
【0028】
本発明の上記および他の目的は、クロピドグレル50mg〜150mgに相当するクロピドグレルまたはクロピドグレルの塩を含む1日1回の剤形を提供することによって達成され、前記剤形は、
(a)約3時間以上の平均Tmax、
(b)1000ピコグラム/ml以下の平均Cmax、
(c)2500ピコグラム/ml/時間超の平均AUC0〜49時間
から選択されるクロピドグレルのin vivo血漿プロファイルの少なくとも1つを提供する。
【0029】
好ましくは、1日1回の剤形は、クロピドグレル50mg〜150mgに相当するクロピドグレルまたはクロピドグレルの塩を含有し、前記剤形は、1400ng/ml以下の平均Cmaxを有するクロピドグレルカルボキシ酸代謝物のin vivo血漿プロファイルを提供する。
【0030】
好ましくは、剤形は、クロピドグレル75mgに相当するクロピドグレルまたはクロピドグレルの塩を含む。
【0031】
好ましくは、前記平均Cmaxは、約900ピコグラム/ml未満である。
【0032】
好ましくは、前記平均Cmaxは、約900ピコグラム/ml未満である。
【0033】
好ましくは、前記平均Cmaxは、約800ピコグラム/ml未満である。
【0034】
好ましくは、前記平均Cmaxは、1200ng/ml以下、より好ましくは、1100ng/ml以下である。
【発明を実施するための最良の形態】
【0035】
本発明によると、本明細書において下記で使用されるクロピドグレルは、塩酸、臭化水素酸、硫酸、硫酸水素酸、硝酸、およびリン酸などの酸の付加塩などのその塩;または酢酸、プロピオン酸、ヒドロキシ酢酸、乳酸、ピルビン酸、シュウ酸、マロン酸、琥珀酸、マレイン酸、フマル酸、リンゴ酸、酒石酸、クエン酸、メタンスルホン酸、エタンスルホン酸、ベンゼンスルホン酸、p−トルエンスルホン酸、ナプタンスルホン酸(napthane sulfonic)、シクラミン酸、サリチル酸、p−アミノサリチル酸、およびパモン酸から選択される有機酸との塩が含まれることを意味する。さらに、クロピドグレルは、例えば水または有機溶媒との溶媒和物の形態としても使用することができ、その形態を含めることができる。
【0036】
本発明によると、クロピドグレルは、本明細書で使用する場合、結晶性、部分結晶性または実質的に非晶形の形態である場合がある。
【0037】
本発明によると、クロピドグレルは、それを必要としている患者に治療有効量で使用することができる。
【0038】
本発明の第1の態様では、クロピドグレル50mg〜150mgに相当するクロピドグレルまたはクロピドグレルの塩を含む1日1回の剤形が提供され、前記剤形は、(a)約3時間以上の平均Tmax、(b)1000ピコグラム/ml以下の平均Cmax、(c)2500ピコグラム/ml/時間超の平均AUC0〜49時間から選択されるクロピドグレルのin vivo血漿プロファイルの少なくとも1つを提供する。
【0039】
本発明の他の態様では、クロピドグレル50mg〜150mgに相当するクロピドグレルまたはクロピドグレルの塩を含む1日1回の剤形を提供し、前記剤形は、1400ng/ml以下の平均Cmaxを有するクロピドグレルカルボキシ酸代謝物を提供する。
【0040】
本発明の好ましい実施形態では、クロピドグレルの前記平均Cmaxは、約800ピコグラム/ml未満である。
【0041】
本発明者らは、驚いたことに、クロピドグレルの調節放出製剤が、血小板凝集のより安定した阻害を提供できることを見出した。
【0042】
本発明の他の態様では、クロピドグレルおよび少なくとも1種の律速ポリマーを含むクロピドグレルの調節放出製剤を提供する。律速ポリマーは、本質的に親水性または疎水性である。律速材料またはさらなる材料の混合物もまた、本発明の製剤を提供するために使用される。
【0043】
好ましい実施形態では、クロピドグレルの修飾された製剤は、クロピドグレル、1種または複数の親水性ポリマー、任意選択で腸溶性ポリマー、および薬学的に許容される賦形剤を含むマトリックスの形態である。本発明の製剤は、錠剤、カプセル剤、ペレット、顆粒剤、微小錠剤などの経口剤形の形態であることができる。製剤は、錠剤に圧縮されるか、またはカプセル剤に顆粒化および充填されることが好ましい。
【0044】
本発明に使用されるクロピドグレルは、組成物の5〜50重量%の範囲である場合がある。本発明に使用される律速ポリマーは、組成物の10〜90重量%の範囲である場合がある。
【0045】
適切な親水性ポリマーには、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルエチルセルロース、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロースなどのセルロースエーテル;あるいはカルボキシメチルセルロースナトリウム、イナゴマメガム、キサンタンガム、アカシア、トラガカントガム、グアーガム、カラヤガム、アルギン酸塩、ゼラチン、アルブミン、カルボマー、アルギン酸塩、ポリビニルピロリドンまたはその混合物などの他の水溶性または膨潤性ポリマーが挙げられる。これらの親水性ポリマーには、アリルスクロースまたはアリルペンタエリトリトールと架橋したアクリル酸を主成分とするホモポリマー、あるいはアリルペンタエリトリトールと架橋したアクリル酸および長鎖(C10〜C30)アクリル酸アリルを主成分とするコポリマーなどのポリアクリレートポリマーもまた挙げられる。ポリアクリレートポリマーは、単独で使用しても、またはメチルセルロース、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロース、ヒドロキシエチルセルロースなどのセルロースエーテルと混合して使用してもよい。本発明によると、親水性ポリマーは、系の約10重量%〜約70重量%の範囲の量で存在する。
【0046】
好ましい親水性ポリマーは、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロース、メチルセルロースおよびこれらの混合物などのセルロースエーテルからなる群から選択される。
【0047】
本発明の好ましい実施形態では、親水性ポリマーは、2重量%の水溶液が10,000cps超の粘度を有するヒドロキシプロピルメチルセルロースと、2重量%の水溶液が5000cps未満の粘度を有するヒドロキシプロピルセルロースとの混合物である。ヒドロキシプロピルメチルセルロースは、系の総重量に対して約5重量%〜70重量%、好ましくは約20重量%〜45重量%の量で存在し得る。
【0048】
ヒドロキシプロピルセルロースは、系の約5重量%〜50重量%、好ましくは約15重量%〜25重量%の量で存在し得る。
【0049】
本発明に使用することができるヒドロキシプロピルメチルセルロースポリマーの例には、Methocel K4M(登録商標)、Methocel K15M(登録商標)、Methocel K100M(登録商標)、Methocel K100LV(登録商標)などのMethocelが挙げられる。本発明に使用することができるヒドロキシプロピルセルロースポリマーには、例えば、HPC−L、HPC−MなどのHPC、Klucel GF(登録商標)、Klucel JF(登録商標)、Klucel HF(登録商標)、Klucel HX(登録商標)FなどのKlucel(登録商標)が挙げられる。
【0050】
本発明に使用することができる腸溶性ポリマーには、メタクリル酸コポリマーなどのポリアクリレートコポリマー;酢酸フタル酸セルロース、フタル酸ヒドロキシプロピルメチルセルロース、酢酸コハク酸ヒドロキシプロピルメチルセルロースなどのセルロース誘導体;および酢酸フタル酸ポリビニルなどが挙げられる。
【0051】
好ましくは、腸溶性ポリマーは、ポリメタクリレートでよい。ポリメタクリレートは、メタクリル部分を有するモノマーを主成分とした、カチオン性、アニオン性または中性のポリマーあるいはコポリマーである場合がある。このようなポリマーまたはコポリマーの混合物もまた使用することもできる。
【0052】
好ましくは、ポリメタクリレートは、メタクリル酸ジメチルアミノエチル、メタクリル酸およびメタクリル酸エステルの少なくとも1つを主成分とするポリマーまたはコポリマーである。ポリメタクリレートは、メタクリル酸コポリマー、またはメタクリル酸ジメチルアミノエチルおよびメタクリル酸エステルのコポリマーであることが特に好ましい。
【0053】
様々な異なるタイプのポリメタクリレートは、例えば商品名Eudragitで市販されている。本発明による組成物において使用するための好ましい材料は、メタクリル酸コポリマーであるEudargit L(登録商標)、またはEudragit EPOである。特に好ましいものは、Eudragit EPO(登録商標)である。
【0054】
本発明の他の実施形態では、腸溶性ポリマーは、ポリアクリレート腸溶性ポリマーである場合がある。腸溶性ポリマーは、系の約0.5重量%〜30重量%の量で存在し得る。
【0055】
本発明の好ましい実施形態では、クロピドグレルは、親水性ポリマー中に分散しており、薬物の放出は、当業者には公知の方法によって達成される、薬物の拡散、または親水性ポリマーの周囲の媒体への表面浸蝕、または2つの方法の組合せによって主として制御される。
【0056】
本発明の他の態様では、クロピドグレル、疎水性律速ポリマー単独、または疎水性および親水性律速ポリマーの混合物を含む調節放出または延長放出製剤を提供する。疎水性律速ポリマーは、剤形に組み込まれているか、または剤形上にコーティングされ、調節放出製剤を提供する。これに制限はされないが、疎水性律速ポリマーが単独で使用される場合は、剤形に組み込まれ、親水性律速ポリマーと共に使用される場合は、剤形上にコーティングされ、調節放出製剤を提供することが好ましい。
【0057】
最も好ましい疎水性律速ポリマーは、エチルセルロース、ポリメタクリレートおよびこれらの混合物から選択される。最も好ましいポリメタクリレートは、ポリエチルアクリレート、ポリメチルメタクリレート、ポリトリメチルアミノエチルメタクリレートおよびこれらの混合物である。
【0058】
本発明の他の実施形態では、クロピドグレルの調節放出製剤は、薬学的に許容される賦形剤をさらに含む。
【0059】
薬学的に許容される賦形剤には、崩壊剤、希釈剤、安定剤、結合剤、滑沢剤、潤滑剤、着色剤、安定剤、マスキング剤、界面活性剤、可溶化剤、水分捕捉剤、抗酸化剤、緩衝剤、吸着剤、付着剤などが挙げられ、それらのいずれかから選択することができる。
【0060】
崩壊剤は、リン酸カルシウム(一塩基/二塩基/三塩基);カルシウム、ナトリウムなどのカルボキシメチルセルロース塩;セルロース、微結晶性セルロース、キトサン、二酸化シリコーン、クロスカルメロースナトリウム、商品名クロスポビドンとして市販されている、本発明で使用される架橋ポリビニルピロリドン、グアーガム、ヒドロキシプロピルセルロース、ケイ酸アルミウニムマグネシウム、メチルセルロース、微結晶性セルロース、ポビドン、デンプングリコール酸ナトリウム、デンプンまたはその混合物、あるいは当業者に知られている周知の崩壊剤から選択することができる。
【0061】
希釈剤は、炭酸カルシウム、リン酸カルシウム、硫酸カルシウム、セルロース、酢酸セルロース、デキストレート、デキストリン、デキストロース、エチルセルロース、微結晶性セルロース、ポリデキストロース、ポリメタクリレート、スクロース、ラクトース、デンプン、マンニトールまたはその混合物、あるいは当業者に知られている周知の希釈剤から選択することができる。
【0062】
結合剤は、アカシア、アルギン酸、カルボマー、カルボキシメチルセルロースナトリウム、キトサン、デキストレート、デキストリン、デキストロース、エチルセルロース、ゼラチン、グルコース、グアーガム、ヒドロキシエチルセルロース、ヒドロキシエチルメチルセルロース、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ヒプロメロース、ケイ酸マグネシウム、ケイ酸アルミニウム、マルトデキストリン、マルトース、微結晶性セルロース、ポロキサマー、ポリデキストロース、ポリエチレンオキシド、ポビドン、アルギン酸ナトリウム、デンプン、スクロースまたはその混合物、あるいは当業者に知られている周知の結合剤から選択することができる。
【0063】
安定剤は、クエン酸ナトリウム、NaCl、K2HPO4、メグルミン、アスコルビン酸ナトリウム、KCl、亜硫酸ナトリウム、ポロキサマー188/407、ポリエチレングリコール、モノオレイン酸グリセリル、アルギン酸、アルブミン、アルギン酸アンモニウム、アスコルビン酸、パルミチン酸アスコルビル、ベントナイト、ブチルヒドロキシトルエン、アルギン酸カルシウム、カルシウム状態(calcium state)、カルボキシメチルセルロースカルシウム、カルボキシメチルセルロースナトリウム、カラギーナン、イナゴマメ属、コロイド状二酸化ケイ素、シクロデキストリン、ジエタノールアミン、エデト酸塩、パルミトステアリン酸エチレングリコール、モノステアリン酸グリセリン、グアーガム、ケイ酸アルミニウムマグネシウム、レシチン、ヒプロメロース、ヒドロキシプロピルセルロース、ポラクリリンカリウム、ペクチン、ポロキサマー、ポリビニルアルコール、没食子酸プロピル、プロピレングリコール、キシリトール、酢酸亜鉛、ラフィノース、ホウ酸ナトリウム、トレハロース、アルギン酸プロピレングリコール、スルホブチルエーテルβ−シクロデキストリンまたはその混合物、あるいは当業者に知られている周知の安定剤から選択することができる。
【0064】
流動促進剤は、二酸化ケイ素、タルク、ステアリン酸カルシウム、ステアリン酸マグネシウムおよびステアリン酸アルミニウム、またはその混合物からなる群から選択することができる。
【0065】
滑沢剤は、ステアリン酸マグネシウム、ラウリル硫酸マグネシウム、ラウリル硫酸ナトリウム、ステアリルフマル酸ナトリウム、ステアリン酸カルシウム、微結晶性セルロースおよび二酸化シリコーン、硬化綿実油、硬化ヒマシ油、ステアリン酸、ステアリン酸亜鉛またはその混合物から選択することができる。
【0066】
緩衝剤は、アンモニア溶液、炭酸カルシウム、リン酸カルシウム、クエン酸、リン酸ナトリウム、ジエタノールアミン、リンゴ酸、グルタミン酸一ナトリウム、リン酸、クエン酸カリウム、酢酸ナトリウム、重炭酸ナトリウム、ホウ酸ナトリウム、クエン酸ナトリウム、水酸化ナトリウム、乳酸ナトリウム、トリエタノールアミンまたはその混合物、あるいは当業者に知られている周知の緩衝剤から選択することができる。
【0067】
クロピドグレルの調節放出製剤は、湿式造粒法、乾式造粒法、融解造粒法または当業者には公知の方法から選択される任意の方法を用いて調製することができる。
【0068】
本発明の態様の一実施形態では、クロピドグレル単独で、または任意選択で他の賦形剤と共に、ポリマーとブレンドし、錠剤へと圧縮する。
【0069】
あるいはブレンドを押し固めて顆粒を生成し、それをさらに錠剤へと圧縮する。
【0070】
他の実施形態では、ポリマーを適切な溶媒に溶解または分散し、この溶液または分散液を、クロピドグレル単独、または他の賦形剤とブレンドしたクロピドグレルを造粒するために使用する。この実施形態では、これに制限はされないが、クロピドグレルを単独で顆粒化する場合は、スプレーコーティングまたは流動層コーティングが好ましく、一方クロピドグレルを賦形剤とブレンドし顆粒化する場合は、高剪断造粒機を使用した顆粒化が好ましい。
【0071】
さらに他の実施形態では、クロピドグレルのコアは、予め形成したペレット上への薬物の層化によって、またはクロピドグレルを適切な賦形剤とブレンドし、湿式造粒することによって調製する。湿式造粒物を、押し出し、球状化して、クロピドグレルの球状コアを生成する。上記の方法のいずれかによって調製したコアを、次いでエチルセルロースで、あるいはエチルセルロースと、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロース、ヒドロキシエチルセルロース、ポリビニルピロリドン、アルギン酸塩、ガム、カルボマーまたはその混合物からなる群から選択される親水性物質との混合物でコーティングする。
【0072】
本発明による好ましい疎水性ポリマーは、エチルセルロースである。
【0073】
約7cps〜約100cpsの粘度のエチルセルロースが好ましい。これらの中で、約10cps〜約50cpsの粘度のエチルセルロースが最も好ましい。組み込まれるエチルセルロースの量は、選択したグレードによって変化する。約0.5重量パーセント〜約30重量パーセントの量が好ましい。約5重量パーセント〜約15重量パーセントの量が最も好ましい。
【0074】
疎水性ポリマーが親水性律速ポリマーと共に使用される場合、これに制限はされないが、低粘度グレードの親水性物質、例えば3cps〜15cpsのヒドロキシプロピルメチルセルロース、PVPK−12〜PVPK−90のポリビニルピロリドンが好ましい。
【0075】
本発明の他の態様は、クロピドグレルの融点よりも低い融点を有する疎水性律速ポリマーの使用によってクロピドグレルの調節放出製剤を提供する。クロピドグレルおよび低融点疎水性律速ポリマーの融点の差異は、少なくとも30℃であるべきである。この低融点疎水性材料を、流体粘稠性にまで溶解し、クロピドグレルを均一に分散するまでそれに分散させる。次いで融解した固まりを、ふるいにかけ、および/または乾燥して、顆粒を生成し、顆粒をさらに錠剤へと圧縮する。ここで圧縮の前に任意選択で、顆粒を適切な賦形剤とブレンドする。低融点疎水性律速ポリマーは、セチルアルコール、セトステアリルアルコール、白ろう、黄ろう、マイクロクリスタリンワックスおよびこれらの混合物からなる群から選択される。
【0076】
さらに他の実施形態では、クロピドグレルを、ヒプロメロース、カルボポール、ポビドン、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース、エチルセルロースなどの1種または複数のポリマー;ならびに充填剤/希釈剤、結合剤、崩壊剤および滑沢剤などの他の適切な賦形剤とブレンドし、錠剤に圧縮する。あるいは、上記のように調製されたクロピドグレルを有する賦形剤混合物を乾式造粒し、次いで圧縮をしてもよい。
【0077】
本発明の調節放出製剤は、任意選択でコーティングされてもよい。
【0078】
クロピドグレルの調節放出剤形で観察される服薬率の増加によって、これに限定されないが、最近の脳卒中、最近の心筋梗塞、または確定した末梢動脈疾患によって実証されるアテローム性動脈硬化症を有する患者におけるアテローム動脈硬化性事象(心筋梗塞、脳卒中、および血管死)の減少を含めて、クロピドグレルによる治療を必要としている患者の適切な管理を改善することができる。
【0079】
本発明に記載されるクロピドグレルの調節放出製剤およびその方法を、下記に例示する実施例において示す。これらの実施例は、例示としてのみ提供し、したがって本発明の範囲を限定するものと解釈されるべきではない。
【実施例】
【0080】
[実施例1〜4]
【0081】
【表1】
【0082】
実施例1〜4を下記の手順によって調製した。
【0083】
クロピドグレルベシラート、微結晶性セルロース、ヒプロメロース、カルボポールおよびクロスポビドンを、拡散ブレンダー中で混合した。コロイド状二酸化ケイ素および硬化綿実油を、拡散ブレンダー中の上記混合物に加え、潤滑ブレンドを得た。潤滑ブレンドを錠剤に圧縮した。
[実施例5]
【0084】
【表2】
【0085】
実施例5を、下記の手順によって調製した。
【0086】
クロピドグレルベシラート、微結晶性セルロース、ポリエチレンオキシド、カルボポールおよびクロスポビドンを、拡散ブレンダー中で混合した。コロイド状二酸化ケイ素および硬化綿実油を、拡散ブレンダー中の上記混合物に加え、潤滑ブレンドを得た。潤滑ブレンドを、平均重量500mgの錠剤に圧縮した。
[実施例6〜7]
【0087】
【表3】
【0088】
実施例6〜7を、下記の手順によって調製した。
【0089】
クロピドグレルベシラート、微結晶性セルロースおよびポビドンを、拡散ブレンダー中で混合した。コロイド状二酸化ケイ素およびステアリン酸マグネシウムを、拡散ブレンダー中の上記混合物に加え、潤滑ブレンドを得た。潤滑ブレンドを、平均重量450mgの錠剤に圧縮した。
[実施例8]
【0090】
【表4】
【0091】
実施例8を、下記の手順によって調製した。
【0092】
クロピドグレルベシラート、ラクトース、ヒプロメロースおよびカルボポールを、拡散ブレンダー中で混合した。コロイド状二酸化ケイ素、硬化綿実油およびタルクを、拡散ブレンダー中の上記混合物に加え、潤滑ブレンドを得た。潤滑ブレンドを、平均重量290mgの錠剤に圧縮した。
[実施例9]
【0093】
【表5】
【0094】
実施例9を、下記の手順によって調製した。
【0095】
クロピドグレルベシラート、ポビドン90、微結晶性セルロースおよびコロイド状二酸化ケイ素を、#40のふるいにかけた。ステアリン酸亜鉛を#60のふるいにかけた。ふるいにかけたステアリン酸亜鉛を、残りのふるいにかけた賦形剤に加え、ダブルコーンブレンダーで10分間ブレンドした。潤滑ブレンドを、ローラーコンパクターを使用して4〜6kpの硬度で強打した。振動造粒機を使用して#20のふるいにかけ、スラグを完全に強打した。極粒状のポビドン90およびコロイド状二酸化ケイ素を、#30のふるいにかけ、大きさを揃えた顆粒に加え、ブレンドした。ステアリン酸亜鉛を、#60のふるいにかけ、上記混合物とブレンドし、潤滑ブレンドを得た。潤滑ブレンドを、平均重量450mgの錠剤に圧縮した。
[実施例10]
【0096】
【表6】
【0097】
実施例10を、下記の手順によって調製した。
【0098】
クロピドグレルベシラート、微結晶性セルロースおよびポビドンを、拡散ブレンダー中で混合した。コロイド状二酸化ケイ素およびステアリン酸亜鉛を、拡散ブレンダー中の上記混合物に加え、潤滑ブレンドを得た。潤滑ブレンドを、平均重量450mgの錠剤に圧縮した。
[実施例11]
【0099】
【表7】
【0100】
実施例11を、下記の手順によって調製した。
【0101】
重硫酸クロピドグレル、微結晶性セルロースおよびポビドンを、拡散ブレンダー中で混合した。コロイド状二酸化ケイ素およびステアリン酸亜鉛を、拡散ブレンダー中の上記の混合物に加え、潤滑ブレンドを得た。潤滑ブレンドを、平均重量350mgの錠剤に圧縮した。
【0102】
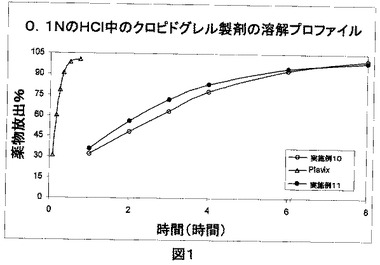
実施例1〜11の錠剤を、0.1NのHCl900ml中で8時間まで50rpmにてUSP Type II装置を使用して溶解プロファイルについて試験した。実施例1〜11により調製した錠剤およびPlavix(登録商標)の溶解プロファイルを、下記の表1および2に表で示す。図1は、0.1NのHCl900ml中で8時間まで50rpmにてUSP Type II装置を使用したPlavix(登録商標)、実施例10および11の比較溶解プロファイルを示す。
【0103】
【表8】
【0104】
【表9】
【0105】
0.1NのHC1中で試験した本発明の組成物の溶解プロファイルは、1時間以内にほぼ完全に薬物を放出するブランド製品であるPlavix(登録商標)と比較して、2〜8時間のより遅く持続する作用を示す。
【0106】
0.1NのHCl900ml中で50rpmにてUSP Type II装置を使用したPlavix(登録商標)、実施例10および11の比較溶解プロファイル(図1):
ex−vivoでのビーグル犬からの多血小板血漿中の抗血小板凝集活性について、実施例4の錠剤を、ブランド製品Plavix(登録商標)重硫酸クロピドグレル75mg錠と比較した。
【0107】
15〜20kgの範囲の体重の生後9〜12カ月の雄イヌで研究を行った。動物を一晩絶食させた。実施例4のクロピドグレル錠剤の単回用量を一群に投与し、他の群にはPlavix(登録商標)を与えた。0時間(薬物投与前)、ならびに薬物投与後0.5時間、2時間、6時間、10時間、24時間、48時間、72時間、96時間、120時間、168時間、192時間および216時間に、動物から採血した。血液採取の216時間後、クロピドグレルの第2の用量を両方の群に投与し、0時間(第1の用量後216時間)、ならびに第2の用量投与後6時間、24時間、72時間、240時間に、動物から採血した。
【0108】
約4mlの血液を、プラスチックシリンジ中に回収し、4%クエン酸三ナトリウム溶液(1:9、クエン酸溶液:血液)を含有するプラスチックチューブに移し、抗凝血剤と穏やかに混合した。血液試料を150gで10分間遠心分離し、多血小板血漿(PRP)を得た。PRPの分離後に、試料を1500gで15分間遠心分離し、乏血小板血漿(PPP)を得た。PRPの血小板数を、Cell−Dyne3700Hematology Analyzerを使用して推定し、ADP誘導性凝集アッセイのためにPPPで3×108/mlに調節した。
【0109】
個々の試料の調節したPRP180μlを、加えたADP試薬(20μM、最終濃度)20μLと共にインキュベートし、光学濃度(OD)を、SPECTRAMAX190マイクロプレートリーダーを使用して10分間30秒毎に連続的に560nmでモニターした。全ての試料を3連で試験した。
【0110】
データ分析:
下記の式を使用して、%凝集を計算した。
%凝集=[{(補正OD(0分)}−{補正OD(10分)}]*100/補正OD(0分)
【0111】
補正OD=PRPの吸光度−PPPの吸光度
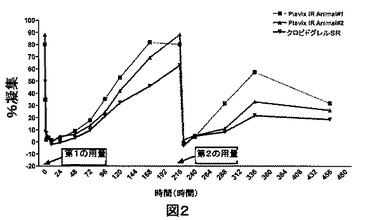
ビーグル犬においてex−vivoで研究したPlavixおよびクロピドグレル75mg錠についての%凝集の比較データを、表3に表で示し、図2に示す。
【0112】
【表10】
【0113】
Plavix、クロピドグレル75mg錠、実施例4の比較血小板凝集作用、ビーグル犬でのEx−vivo研究(図2):
表3および図2に示されるように、クロピドグレルのすべての製剤は、ADP誘導性血小板凝集の有意な阻害をもたらした。
【0114】
Plavix(登録商標)錠剤と比較して、実施例4のクロピドグレル錠剤は、より長い期間持続する作用を示した。Plavix(登録商標)製剤で処理されたイヌは、約8〜9日で作用の完全な逆転を示し、一方実施例4のクロピドグレル錠剤で処理されたイヌは、9日目でも抗血小板凝集作用を示した。第2の用量は血小板凝集のさらなる減少をもたらし、実施例4の錠剤のクロピドグレル錠剤についての作用は、Plavix(登録商標)錠剤よりも良好であった。
【0115】
薬力学的研究を、DiaMed Impact−Rによって抗凝固処理全血における血小板機能を決定することによって、実施例10の通りに調製した錠剤について、Plavix(登録商標)と比較して行った。試験方法は、血小板減少症を含めた原発性止血異常(例えば、フォンウィルブランド病)をスクリーニングし、その治療をモニタリングする血小板機能の研究を目的とする。それは、血小板の機能低下および機能亢進の両方を試験するために使用することができる。さらにそれは、様々な抗血小板薬に対する反応をモニターするための迅速な方法を提供する。この試験を、抗凝固処理全血における血小板機能を決定するために行い、Plavix(登録商標)とクロピドグレルの調節放出製剤(実施例10)とを比較した。
【0116】
血小板凝集アッセイの目的のために、全部で10の血液試料を回収した。静脈血試料4.5m1を、研究期間中−10.00時間、0.00時間(投与前)、ならびに薬物投与後0.50時間、2.00時間、4.00時間、6.00時間、8.00時間、10.00時間、24.00時間および48.00時間に採取する。
【0117】
新しい静脈穿刺によって血液試料を回収した。それが新しい静脈穿刺である場合、新しい静脈穿刺のために血小板凝集因子が存在する可能性があるため、血液2〜3mlを廃棄する。血液試料を、使い捨て注射器を使用して前腕静脈に入れた留置カニューレによって回収した。各サンプリング時点で、血液試料4.5mlを採取し、クエン酸ナトリウム緩衝液を含有する試料回収チューブに移す。
【0118】
クエン酸ナトリウムを含有するチューブ中に試験をする全血試料を回収し、室温(18〜25℃)で保存した。クエン酸塩添加血試料(130μL)をウェルの中央に置き、定義済みパラメーターによって回転処理を施した。ウェルを洗浄し、May−Gruenwald染色液によって染色した。
【0119】
表面被覆率(%)として表す血小板で覆われた総面積率として血小板粘着を評価し、平均サイズ(μm2)として表す凝集物への表面結合の平均サイズとして凝集を評価した。
【0120】
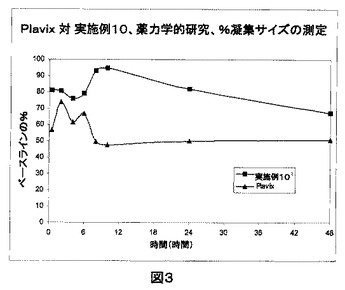
Plavix(登録商標)対クロピドグレル錠剤(実施例10および11)の2種の製剤における血小板の%凝集サイズの比較を、図3および4に示す。
【0121】
Plavix(登録商標)対クロピドグレル錠剤(実施例10)の2種の製剤における血小板の%凝集サイズの比較(図3)。
Plavix(登録商標)と比較して実施例10の製剤においては血小板のさらなる脱凝集があるため、図3に表される通り、抗血小板作用は実施例10においてPlavix(登録商標)よりも良好であることが見出された。
【0122】
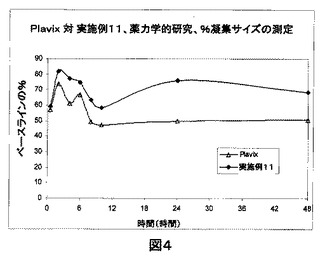
Plavix(登録商標)対クロピドグレル錠剤(実施例11)の2種の製剤における血小板の%凝集サイズの比較(図4)。
Plavix(登録商標)と比較して実施例11の製剤においては血小板のさらなる脱凝集があるため、図4に表される通り、抗血小板作用は実施例11においてPlavix(登録商標)よりも良好であることが見出された。
【0123】
クロピドグレル75mgに相当するクロピドグレルベシラート錠剤(実施例10)、クロピドグレル75mgに相当する重硫酸クロピドグレル錠剤(実施例11)および市販の重硫酸クロピドグレル錠剤であるPlavix(登録商標)を、空腹条件下の健康な大人の男性ヒト対象において非盲検、バランス化、無作為化、3種の処置(three−treatment)、単一期間、単回用量、平行、比較経口薬物動態学的研究を行った。研究は、36対象(各処置について12対象)で行った。
【0124】
研究期間中試料の投与11時間以上前から投与後24時間まで、対象を臨床施設に収容した。対象は、投与前に少なくとも10時間一晩絶食した。投与を除いて、投与前1時間から投与後2時間まで飲料水を禁止した。他の時間は、飲料水は自由に与えられた。各期間において投与の約4時間後、およびそれ以降検査終了まで特定の間隔で食事を提供した。研究期間中全部で23の血液試料が回収された。静脈血試料5.5ml(廃棄したヘパリン添加血液0.5mlを含めて)を、研究期間の間、投与前の時点(投与前)、ならびに薬物投与後0.25時間、0.50時間、0.75時間、1.00時間、1.25時間、1.50時間、1.75時間、2.00時間、2.50時間、3.00時間、4.00時間、5.00時間、6.00時間、7.00時間、8.00時間、9.00時間、10.00時間、12.00時間、16.00時間、24.00時間、36.00時間および48.00時間に採取した。使い捨て注射器を使用して、前腕静脈に入れた留置カニューレから血液試料を回収した。各サンプリング時点で、血液試料(廃棄したヘパリン添加血液0.5mlを含めて)5.5mlを採取し、抗凝血剤としてEDTAを含有する試料回収チューブに移した。遠心分離後、1時間以内に血液試料から血漿を分離し、血漿試料を分析のために取り出すまで−70±5℃で保存した。血漿中のクロピドグレル濃度を、有効なLCMS/MSを使用して定量化した。
【0125】
ピーク血漿濃度(Cmax)、ピーク血漿濃度に到達する時間(Tmax)、最終時点までの血漿濃度−時間曲線下面積(AUC0−t)、無限大まで外挿する血漿濃度−時間曲線下面積(AUC0−∞)、残りの面積率(AUC_%Extrap)、浄化(D)、分布容積(Vd)、血漿排出半減期(t1/2)および排出速度定数(λZ)を、クロピドグレルについて計算した。
【0126】
クロピドグレルおよびクロピドグレルカルボキシ酸代謝物について、Plavix(登録商標)、実施例10および11によって調製したクロピドグレル錠剤で試験した様々な時点の平均血漿濃度を、表4および5に各々表で示す。薬物動態パラメーターの平均値を、表6および7に表で示す。
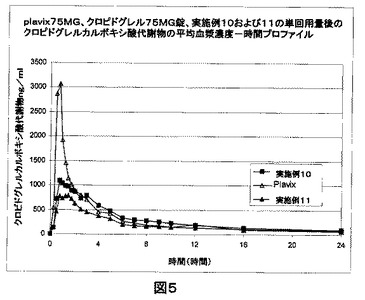
【0127】
図5は、空腹条件下でPlavix(登録商標)、クロピドグレル75mg錠、実施例10および11の単回用量後の、クロピドグレルカルボキシ酸代謝物(ng/mL)の平均血漿濃度−時間プロファイルを示す。
【0128】
【表11】
【0129】
【表12】
【0130】
空腹条件下でPlavix(登録商標)、クロピドグレル75mg錠、実施例10および11の単回用量後の、クロピドグレルカルボキシ酸代謝物(ng/mL)の平均血漿濃度−時間プロファイル(図5)。
【0131】
【表13】
【0132】
【表14】
【0133】
クロピドグレルは、プロドラッグである。活性代謝物であるチオール誘導体は、クロピドグレルの2−オキソ−クロピドグレルへの酸化、ついで加水分解により形成される。主要な循環代謝物は、不活性のカルボキシ酸誘導体である。Plavix(登録商標)における高い血漿レベルによって観察されるように、カルボキシ酸誘導体の急速な形成は、短期間の高濃度のクロピドグレルとの曝露と関連付けられるであろう。本発明のクロピドグレルの修飾された製剤である実施例10の場合、クロピドグレルカルボキシ酸の血漿濃度は、2.5〜8時間の間制御され持続された様式であることを見ることができる。これは活性代謝物の緩慢な形成を示し、それは次にカルボキシ酸誘導体に緩慢に変換される。本発明の製剤は、ブランド製品であるPlavix(登録商標)と比較してカルボキシ酸代謝物について有意に低いCmaxを示すことを観察することができる。本発明の組成物は、Plavix(登録商標)と比較してクロピドグレルについてより高いAUC値を示す。クロピドグレルについてのより高いAUC値およびより大きいTmaxは、不活性カルボキシ酸代謝物についてのCmaxの有意に低い値と共に、より長い期間における薬物のより良好な有効性と関連付けることができる。
【0134】
本発明の特定の好ましい他の実施形態は、本発明を開示する目的のために記載したが、開示された実施形態への修正は、当業者であれば思いつくことができる。したがって、この明細書は、本発明の趣旨および範囲から逸脱しない本発明の全ての実施形態およびその修正を包含することを意図している。
【図面の簡単な説明】
【0135】
【図1】Plavix(登録商標)の溶解プロファイルを示す比較図である。
【図2】Plavix、クロピドグレル75mg錠の血小板凝集作用を示す比較図である。
【図3】Plavix(登録商標)対クロピドグレル錠剤の2種の製剤における血小板の%凝集サイズを示す比較図である。
【図4】Plavix(登録商標)対クロピドグレル錠剤の2種の製剤における血小板の%凝集サイズを示す比較図である。
【図5】Plavix(登録商標)、クロピドグレル75mg錠の単回用量後のクロピドグレルカルボキシ酸代謝物(ng/mL)の平均血漿濃度−時間プロファイルを示す図である。
【技術分野】
【0001】
本発明は、クロピドグレルおよび/またはその薬学的に許容される塩/溶媒和物の医薬組成物に関する。具体的には、本発明は、クロピドグレルおよび/またはその薬学的に許容される塩/溶媒和物の調節放出製剤に関する。本発明は、クロピドグレルおよび/またはその薬学的に許容される塩/溶媒和物の調節放出製剤を調製する方法をさらに提供する。本発明は、クロピドグレル50mg〜150mgに相当するクロピドグレルまたはクロピドグレルの塩を含む1日1回の剤形をさらに提供し、前記剤形は、クロピドグレルについて平均Tmaxが約3時間以上であり、平均Cmaxが1000ピコグラム/ml以下である血漿プロファイルを提供する。クロピドグレルの本発明の製剤は、Sanofi社から市販されているブランド製品であるPlavix(登録商標)クロピドグレル75mg錠と比較して、実質的により低いレベルの不活性クロピドグレルカルボキシ酸代謝物を提供する。
【0002】
クロピドグレルは、メチル(+)−(S)−α−(o−クロロフェニル)−6,7−ジヒドロチエノ(3,2−c)ピリジン−5(4H)−アセテートとして化学的に公知であり、下記の式(I)の構造によって表される。
【0003】
【化1】
【背景技術】
【0004】
クロピドグレルは、血小板凝集阻害剤である。血小板機能を阻害する種々の薬物が、脳卒中または一過性脳虚血発作、心筋梗塞、不安定狭心症、あるいは血管バイパスまたは血管形成術の必要性によって明らかとなる確定した心血管アテローム性動脈硬化症を有する人々における病的事象を減少させることが示されてきた。これは、血小板がこれらの事象の開始および/または進展に関与し、それらを阻害すると事象の割合を減少させることができることを示す。
【0005】
クロピドグレルは、アデノシン二リン酸(ADP)とその血小板受容体の結合、およびそれに続く糖タンパク質GPIIb/IIIa複合体のADP媒介性の活性化を選択的に阻害し、それによって血小板凝集を阻害する。クロピドグレルが生体内変化することは、血小板凝集の阻害を生じさせるために必要である。クロピドグレルはまた、ADP以外のアゴニストによって誘発される血小板凝集も、放出されたADPによる血小板活性化の増幅を遮断することによって阻害する。クロピドグレルは、ホスホジエステラーゼ活性を阻害しない。
【0006】
クロピドグレルは、血小板ADP受容体を不可逆的に修飾することによって作用する。結果的に、クロピドグレルに曝された血小板は、その寿命の残りの期間影響を受ける。
【0007】
血小板凝集の用量依存的阻害を、クロピドグレルの1回の経口投与の2時間後に見ることができる。クロピドグレル75mg/日の反復投与は、第1日目にADP誘導性血小板凝集を阻害し、3日目〜7日目に阻害は定常状態に達する。定常状態では、クロピドグレル75mg/日の用量によって観察される平均阻害レベルは、40%〜60%であった。血小板凝集および出血時間は、治療が中止された後、一般に約5日でベースライン値に徐々に戻る。
【0008】
クロピドグレルは、結晶性重硫酸塩、すなわち重硫酸クロピドグレルの形態で市販されており、ブランド名Plavix(登録商標)で販売されている。
【0009】
血小板凝集阻害剤および抗血栓剤としてのクロピドグレルの治療への応用およびその調製は、米国特許第4,529,596号に開示されている。
【0010】
US6,429,210には、形態IIとして示される硫酸水素クロピドグレルの結晶形態が開示されている。硫酸水素クロピドグレルの調製のためのEP281459に記載されている方法は、結晶形態(形態1)をもたらす。市販品では、結晶性硫酸水素クロピドグレルであるPlavix(登録商標)が、医薬組成物に使用された。
【0011】
US2006/0264636には、クロピドグレルの新規な塩、すなわちクロピドグレルメシラート、クロピドグレルベシラートおよびクロピドグレルトシラート、それらの調製方法、ならびにそれらを含有する医薬組成物が開示されている。
【0012】
US2007/0048370には、クロピドグレル、あるいはその溶媒和物または水和物の製剤が開示されている(ただし、塩はクロピドグレルヨウ化水素酸塩ではなく、錠剤はイオン性および/または塩基性打錠賦形剤、ならびにポリエチレングリコール6000を含有しない)。
【0013】
クロピドグレルは、プロドラッグである。活性代謝物であるチオール誘導体は、クロピドグレルの2−オキソ−クロピドグレルへの酸化、ついで加水分解により形成される。酸化ステップは、主にシトクロムP450アイソエンザイム2B6および3A4、ならびにより少ない程度で1A1、1A2および2C19に制御されている。活性チオール代謝物は、in vitroで単離されているが、速やかに血小板受容体に不可逆的に結合することにより血小板凝集を阻害する。この代謝物は、血漿中では検出されていない。
【0014】
主要循環代謝物の薬物動態は、クロピドグレル50〜150mgの用量範囲において線形性であった(血漿濃度は用量に比例して上昇した)。参照文献:2006年9月15日付け、IPHA Electronic Medicines compendium。
【0015】
血小板は核を欠いており、最も小さい循環ヒト細胞であるが、生理学的および病態生理学的の両方で、血栓症の過程において不可欠で複雑な役割を果たす。血小板の活性化および凝集は、(1)急性冠動脈症候群(ACS)の心筋虚血または梗塞をもたらすアテローム動脈硬化性プラークの自然発生的崩壊、あるいは(2)経皮冠動脈インターベンション(PCI)からもたらされる機械的崩壊後の冠内血栓の増殖において中心的役割を果たす。血小板は最初に、崩壊したプラーク部位においてコラーゲンおよびフォンウィルブランド因子に付着し、最初の血小板の単層をもたらす。活性化の後、血小板はトロンボキサンA2およびアデノシン二リン酸(ADP)などの二次アゴニストを放出し、それが凝固カスケードによって生じたトロンビンと組み合わさり、さらなる血小板の刺激および動員をもたらす。この病態生理学的背景によって、抗血小板療法が、ACSを有する患者、特にPCIを受ける患者の管理の基礎であることは驚くことではない。ADPへの反応について光学的血小板凝集測定法によって測定する場合、研究によってクロピドグレルに反応する用量および時間依存的変動性が示された。Gurbelらによる研究では、選択的冠動脈ステント留置術を受けた96人の患者を、標準的クロピドグレル治療(300mgの初期用量、次いで75mg/日)前、および治療後の複数の時点でモニターした。クロピドグレル耐性(経験的に治療前の値と比較してADP5μmol/Lに反応した凝集の<10%減少と定義付けられる)は、2時間で患者の63%で、24時間で31%に、5日で31%に、30日で15%に見られた。
【0016】
最も高い治療前の値を有する患者は、最初の5日間に亘って最も低い抗血栓性保護を有した。他の報告でMullerらは、24時間でこの治療計画に対してさらなる効果が見られず、クロピドグレル600mgの投与4時間後にADPに対する血小板凝集が<10%の減少である場合を非反応者と定義し、10%〜29%の減少である場合を準反応者と定義した。ADP5μmol/Lに対して、5%が非反応者であり、9%が準反応者であり、ADP20μmol/Lに対して、11%が非反応者であり、26%が準反応者であることをこの研究は見出した。臨床転帰を評価するように設計されていなかったが、Muller研究における興味深い知見は、(試験した105人のうち)2人の患者が亜急性ステント血栓症を起こし、両者ともクロピドグレル非反応の定義に合致したことであった。さらなる報告は狭心症クラスを血小板阻害と関連付け、ADP450mgの投与後により高い狭心症クラスを示す患者は血小板凝集の阻害がより少ないことを見出した。
【0017】
クロピドグレル耐性については、いくつかの機構が可能性を有する。外因性機構には、クロピドグレルの不適切な投与または不十分な投与、およびクロピドグレルとアトロバスタチンとの可能性のある相互作用を含めた薬物・薬物相互作用が挙げられる。クロピドグレル反応とCYP3A4活性(エリスロマイシン呼気検査により測定)とに正相関があり、これは、重要な機構が活性代謝物への可変性変換であり得ることを示唆する。他の可能性がある外因性機構には、プロドラッグの可変性吸収または活性代謝物のクリアランスが含まれるであろう。固有の機構には、P2Y12受容体の可変性、受容体数の増加、ADP放出の増加、または他の血小板活性化経路の上方制御を挙げることができる。参照文献:Circulation 2004、109:3064~3067頁、American Heart Association, Inc.
【0018】
最近、新しい臨床データが明らかになり、Plavixの標準的用量は、患者の約25%に適切な血小板阻害をもたらさないことが示された。Plavix耐性に対する可能性のある説明はいくつかあり、Plavix耐性が実質的現象であるのか、または効果のない投与の結果であるのかはまだ明らかではない。Plavix「耐性」は、より高い初期用量によって実質的に減少できることが、最近の研究によって示された。「Plavix耐性」のこの問題は、心臓病専門医の間でより注目を集めており、2004年のCirculationにおいて公表された研究によって、Plavixからの恩恵が最も少ない患者は[血小板凝集についてのアデノシン二リン酸(ADP)誘導性の百分率の変化によって決定される]、心血管事象の危険がはるかに高いことが示された。参照文献:Circulation.2004、109:3171~3175頁、2004 American Heart Association, Inc.
【0019】
血小板凝集は、血栓形成過程の重要な部分である。アデノシン5−二リン酸、エピネフリンおよび様々な血小板因子が血小板顆粒から押し出され、血小板凝集において主要な役割を果たしているトロンボキサンが形成されることによって、この血小板凝集現象が開始する。血小板動員の血栓形成を妨げることのできる薬剤は、アテローム性動脈硬化症、血栓症および急性冠動脈症候群の治療において重要な役割を果たすことができる。動物から得た多血小板血漿中のin vivoの血小板凝集の測定は、血小板研究において基礎手段である。ADPは、通常の血小板機能のin vitro/ex vivoのスクリーニングのための凝集剤であり、出血性障害の原因を特定するための重要な診断手段に相当する。
【0020】
重硫酸クロピドグレルの投与量は、投与後1〜3時間にクロピドグレルおよびその活性代謝物のピーク濃度をもたらすことが報告されている。これは、投与後直後に高濃度の活性代謝物を、および投与後24時間にはより低い濃度を提供するであろう。さらに、吸収後のクロピドグレルの85%は、血中のエステラーゼによって不活性化され、15%のみが、肝臓での活性代謝物への変換を受ける。要約すれば、患者は、定常状態においてピークレベルで出血の危険に、定常状態においてトラフレベルで乏しい血小板阻害に曝されている。
【0021】
より最近では、様々な研究グループおよび臨床医が、クロピドグレル耐性の明らかになりつつある問題を報告してきた。これは、再発する心血管事象の危険性を増加させ得る。本発明者らは、驚いたことに、クロピドグレルおよびその薬学的に許容される塩の調節放出製剤が、クロピドグレル耐性の問題に対処することができ、したがって反応率を向上させることを見出した。さらに、調節放出製剤はまた、クロピドグレル治療の安全性、利便性および服薬率(compliance)を改善する可能性も提供する。クロピドグレル投与に続く過量は、長引く出血時間、次いで出血性合併症をもたらし得る。出血が観察される場合、適切な治療を考慮すべきである。要するに、クロピドグレルの調節放出製剤は、より良好な抗血小板療法を必要としている数百万の人々の治療を改善するであろう。
【0022】
本発明は、医薬品産業の実現されていない要求に応えるものである。
【0023】
調節放出は、遅延放出、延長放出、パルス放出または持続放出などでよい。放出の調節は、胃出血などの薬物の副作用の最小化のため、または一生続くクロピドグレルの治療が必要な患者におけるクロピドグレル耐性の進行を予防するためなど、たくさんの理由によって所望され得る。
【特許文献1】米国特許第4,529,596号
【特許文献2】US6,429,210
【特許文献3】EP281459
【特許文献4】US2006/0264636
【特許文献5】US2007/0048370
【非特許文献1】2006年9月15日付け
【非特許文献2】Circulation 2004、109:3064~3067頁、American Heart Association, Inc.
【非特許文献3】Circulation.2004、109:3171~3175頁、2004 American Heart Association, Inc.
【発明の開示】
【発明が解決しようとする課題】
【0024】
本発明の重要な目的の1つは、クロピドグレルまたはその薬学的に許容される塩/溶媒和物の調節放出医薬組成物を提供することである。
【0025】
本発明の他の目的は、1000ピコグラム/ml以下のCmaxおよび3時間超のTmaxを有する適切な血漿プロファイルを提供する、クロピドグレル50mg〜150mgに相当するクロピドグレルまたはその薬学的に許容される塩/溶媒和物を使用する調節放出医薬組成物を提供することである。
【0026】
本発明のさらに他の目的は、Sanofi社から市販されているブランド製品であるPlavix(登録商標)クロピドグレル75mg錠と比較して、実質的により低いレベルの不活性クロピドグレルカルボキシ酸代謝物を提供するクロピドグレルの製剤を提供することである。
【0027】
本発明のさらに他の目的は、クロピドグレルまたはその薬学的に許容される塩/溶媒和物の調節放出製剤の製造方法を提供することである。
【課題を解決するための手段】
【0028】
本発明の上記および他の目的は、クロピドグレル50mg〜150mgに相当するクロピドグレルまたはクロピドグレルの塩を含む1日1回の剤形を提供することによって達成され、前記剤形は、
(a)約3時間以上の平均Tmax、
(b)1000ピコグラム/ml以下の平均Cmax、
(c)2500ピコグラム/ml/時間超の平均AUC0〜49時間
から選択されるクロピドグレルのin vivo血漿プロファイルの少なくとも1つを提供する。
【0029】
好ましくは、1日1回の剤形は、クロピドグレル50mg〜150mgに相当するクロピドグレルまたはクロピドグレルの塩を含有し、前記剤形は、1400ng/ml以下の平均Cmaxを有するクロピドグレルカルボキシ酸代謝物のin vivo血漿プロファイルを提供する。
【0030】
好ましくは、剤形は、クロピドグレル75mgに相当するクロピドグレルまたはクロピドグレルの塩を含む。
【0031】
好ましくは、前記平均Cmaxは、約900ピコグラム/ml未満である。
【0032】
好ましくは、前記平均Cmaxは、約900ピコグラム/ml未満である。
【0033】
好ましくは、前記平均Cmaxは、約800ピコグラム/ml未満である。
【0034】
好ましくは、前記平均Cmaxは、1200ng/ml以下、より好ましくは、1100ng/ml以下である。
【発明を実施するための最良の形態】
【0035】
本発明によると、本明細書において下記で使用されるクロピドグレルは、塩酸、臭化水素酸、硫酸、硫酸水素酸、硝酸、およびリン酸などの酸の付加塩などのその塩;または酢酸、プロピオン酸、ヒドロキシ酢酸、乳酸、ピルビン酸、シュウ酸、マロン酸、琥珀酸、マレイン酸、フマル酸、リンゴ酸、酒石酸、クエン酸、メタンスルホン酸、エタンスルホン酸、ベンゼンスルホン酸、p−トルエンスルホン酸、ナプタンスルホン酸(napthane sulfonic)、シクラミン酸、サリチル酸、p−アミノサリチル酸、およびパモン酸から選択される有機酸との塩が含まれることを意味する。さらに、クロピドグレルは、例えば水または有機溶媒との溶媒和物の形態としても使用することができ、その形態を含めることができる。
【0036】
本発明によると、クロピドグレルは、本明細書で使用する場合、結晶性、部分結晶性または実質的に非晶形の形態である場合がある。
【0037】
本発明によると、クロピドグレルは、それを必要としている患者に治療有効量で使用することができる。
【0038】
本発明の第1の態様では、クロピドグレル50mg〜150mgに相当するクロピドグレルまたはクロピドグレルの塩を含む1日1回の剤形が提供され、前記剤形は、(a)約3時間以上の平均Tmax、(b)1000ピコグラム/ml以下の平均Cmax、(c)2500ピコグラム/ml/時間超の平均AUC0〜49時間から選択されるクロピドグレルのin vivo血漿プロファイルの少なくとも1つを提供する。
【0039】
本発明の他の態様では、クロピドグレル50mg〜150mgに相当するクロピドグレルまたはクロピドグレルの塩を含む1日1回の剤形を提供し、前記剤形は、1400ng/ml以下の平均Cmaxを有するクロピドグレルカルボキシ酸代謝物を提供する。
【0040】
本発明の好ましい実施形態では、クロピドグレルの前記平均Cmaxは、約800ピコグラム/ml未満である。
【0041】
本発明者らは、驚いたことに、クロピドグレルの調節放出製剤が、血小板凝集のより安定した阻害を提供できることを見出した。
【0042】
本発明の他の態様では、クロピドグレルおよび少なくとも1種の律速ポリマーを含むクロピドグレルの調節放出製剤を提供する。律速ポリマーは、本質的に親水性または疎水性である。律速材料またはさらなる材料の混合物もまた、本発明の製剤を提供するために使用される。
【0043】
好ましい実施形態では、クロピドグレルの修飾された製剤は、クロピドグレル、1種または複数の親水性ポリマー、任意選択で腸溶性ポリマー、および薬学的に許容される賦形剤を含むマトリックスの形態である。本発明の製剤は、錠剤、カプセル剤、ペレット、顆粒剤、微小錠剤などの経口剤形の形態であることができる。製剤は、錠剤に圧縮されるか、またはカプセル剤に顆粒化および充填されることが好ましい。
【0044】
本発明に使用されるクロピドグレルは、組成物の5〜50重量%の範囲である場合がある。本発明に使用される律速ポリマーは、組成物の10〜90重量%の範囲である場合がある。
【0045】
適切な親水性ポリマーには、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルエチルセルロース、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロースなどのセルロースエーテル;あるいはカルボキシメチルセルロースナトリウム、イナゴマメガム、キサンタンガム、アカシア、トラガカントガム、グアーガム、カラヤガム、アルギン酸塩、ゼラチン、アルブミン、カルボマー、アルギン酸塩、ポリビニルピロリドンまたはその混合物などの他の水溶性または膨潤性ポリマーが挙げられる。これらの親水性ポリマーには、アリルスクロースまたはアリルペンタエリトリトールと架橋したアクリル酸を主成分とするホモポリマー、あるいはアリルペンタエリトリトールと架橋したアクリル酸および長鎖(C10〜C30)アクリル酸アリルを主成分とするコポリマーなどのポリアクリレートポリマーもまた挙げられる。ポリアクリレートポリマーは、単独で使用しても、またはメチルセルロース、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロース、ヒドロキシエチルセルロースなどのセルロースエーテルと混合して使用してもよい。本発明によると、親水性ポリマーは、系の約10重量%〜約70重量%の範囲の量で存在する。
【0046】
好ましい親水性ポリマーは、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロース、メチルセルロースおよびこれらの混合物などのセルロースエーテルからなる群から選択される。
【0047】
本発明の好ましい実施形態では、親水性ポリマーは、2重量%の水溶液が10,000cps超の粘度を有するヒドロキシプロピルメチルセルロースと、2重量%の水溶液が5000cps未満の粘度を有するヒドロキシプロピルセルロースとの混合物である。ヒドロキシプロピルメチルセルロースは、系の総重量に対して約5重量%〜70重量%、好ましくは約20重量%〜45重量%の量で存在し得る。
【0048】
ヒドロキシプロピルセルロースは、系の約5重量%〜50重量%、好ましくは約15重量%〜25重量%の量で存在し得る。
【0049】
本発明に使用することができるヒドロキシプロピルメチルセルロースポリマーの例には、Methocel K4M(登録商標)、Methocel K15M(登録商標)、Methocel K100M(登録商標)、Methocel K100LV(登録商標)などのMethocelが挙げられる。本発明に使用することができるヒドロキシプロピルセルロースポリマーには、例えば、HPC−L、HPC−MなどのHPC、Klucel GF(登録商標)、Klucel JF(登録商標)、Klucel HF(登録商標)、Klucel HX(登録商標)FなどのKlucel(登録商標)が挙げられる。
【0050】
本発明に使用することができる腸溶性ポリマーには、メタクリル酸コポリマーなどのポリアクリレートコポリマー;酢酸フタル酸セルロース、フタル酸ヒドロキシプロピルメチルセルロース、酢酸コハク酸ヒドロキシプロピルメチルセルロースなどのセルロース誘導体;および酢酸フタル酸ポリビニルなどが挙げられる。
【0051】
好ましくは、腸溶性ポリマーは、ポリメタクリレートでよい。ポリメタクリレートは、メタクリル部分を有するモノマーを主成分とした、カチオン性、アニオン性または中性のポリマーあるいはコポリマーである場合がある。このようなポリマーまたはコポリマーの混合物もまた使用することもできる。
【0052】
好ましくは、ポリメタクリレートは、メタクリル酸ジメチルアミノエチル、メタクリル酸およびメタクリル酸エステルの少なくとも1つを主成分とするポリマーまたはコポリマーである。ポリメタクリレートは、メタクリル酸コポリマー、またはメタクリル酸ジメチルアミノエチルおよびメタクリル酸エステルのコポリマーであることが特に好ましい。
【0053】
様々な異なるタイプのポリメタクリレートは、例えば商品名Eudragitで市販されている。本発明による組成物において使用するための好ましい材料は、メタクリル酸コポリマーであるEudargit L(登録商標)、またはEudragit EPOである。特に好ましいものは、Eudragit EPO(登録商標)である。
【0054】
本発明の他の実施形態では、腸溶性ポリマーは、ポリアクリレート腸溶性ポリマーである場合がある。腸溶性ポリマーは、系の約0.5重量%〜30重量%の量で存在し得る。
【0055】
本発明の好ましい実施形態では、クロピドグレルは、親水性ポリマー中に分散しており、薬物の放出は、当業者には公知の方法によって達成される、薬物の拡散、または親水性ポリマーの周囲の媒体への表面浸蝕、または2つの方法の組合せによって主として制御される。
【0056】
本発明の他の態様では、クロピドグレル、疎水性律速ポリマー単独、または疎水性および親水性律速ポリマーの混合物を含む調節放出または延長放出製剤を提供する。疎水性律速ポリマーは、剤形に組み込まれているか、または剤形上にコーティングされ、調節放出製剤を提供する。これに制限はされないが、疎水性律速ポリマーが単独で使用される場合は、剤形に組み込まれ、親水性律速ポリマーと共に使用される場合は、剤形上にコーティングされ、調節放出製剤を提供することが好ましい。
【0057】
最も好ましい疎水性律速ポリマーは、エチルセルロース、ポリメタクリレートおよびこれらの混合物から選択される。最も好ましいポリメタクリレートは、ポリエチルアクリレート、ポリメチルメタクリレート、ポリトリメチルアミノエチルメタクリレートおよびこれらの混合物である。
【0058】
本発明の他の実施形態では、クロピドグレルの調節放出製剤は、薬学的に許容される賦形剤をさらに含む。
【0059】
薬学的に許容される賦形剤には、崩壊剤、希釈剤、安定剤、結合剤、滑沢剤、潤滑剤、着色剤、安定剤、マスキング剤、界面活性剤、可溶化剤、水分捕捉剤、抗酸化剤、緩衝剤、吸着剤、付着剤などが挙げられ、それらのいずれかから選択することができる。
【0060】
崩壊剤は、リン酸カルシウム(一塩基/二塩基/三塩基);カルシウム、ナトリウムなどのカルボキシメチルセルロース塩;セルロース、微結晶性セルロース、キトサン、二酸化シリコーン、クロスカルメロースナトリウム、商品名クロスポビドンとして市販されている、本発明で使用される架橋ポリビニルピロリドン、グアーガム、ヒドロキシプロピルセルロース、ケイ酸アルミウニムマグネシウム、メチルセルロース、微結晶性セルロース、ポビドン、デンプングリコール酸ナトリウム、デンプンまたはその混合物、あるいは当業者に知られている周知の崩壊剤から選択することができる。
【0061】
希釈剤は、炭酸カルシウム、リン酸カルシウム、硫酸カルシウム、セルロース、酢酸セルロース、デキストレート、デキストリン、デキストロース、エチルセルロース、微結晶性セルロース、ポリデキストロース、ポリメタクリレート、スクロース、ラクトース、デンプン、マンニトールまたはその混合物、あるいは当業者に知られている周知の希釈剤から選択することができる。
【0062】
結合剤は、アカシア、アルギン酸、カルボマー、カルボキシメチルセルロースナトリウム、キトサン、デキストレート、デキストリン、デキストロース、エチルセルロース、ゼラチン、グルコース、グアーガム、ヒドロキシエチルセルロース、ヒドロキシエチルメチルセルロース、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ヒプロメロース、ケイ酸マグネシウム、ケイ酸アルミニウム、マルトデキストリン、マルトース、微結晶性セルロース、ポロキサマー、ポリデキストロース、ポリエチレンオキシド、ポビドン、アルギン酸ナトリウム、デンプン、スクロースまたはその混合物、あるいは当業者に知られている周知の結合剤から選択することができる。
【0063】
安定剤は、クエン酸ナトリウム、NaCl、K2HPO4、メグルミン、アスコルビン酸ナトリウム、KCl、亜硫酸ナトリウム、ポロキサマー188/407、ポリエチレングリコール、モノオレイン酸グリセリル、アルギン酸、アルブミン、アルギン酸アンモニウム、アスコルビン酸、パルミチン酸アスコルビル、ベントナイト、ブチルヒドロキシトルエン、アルギン酸カルシウム、カルシウム状態(calcium state)、カルボキシメチルセルロースカルシウム、カルボキシメチルセルロースナトリウム、カラギーナン、イナゴマメ属、コロイド状二酸化ケイ素、シクロデキストリン、ジエタノールアミン、エデト酸塩、パルミトステアリン酸エチレングリコール、モノステアリン酸グリセリン、グアーガム、ケイ酸アルミニウムマグネシウム、レシチン、ヒプロメロース、ヒドロキシプロピルセルロース、ポラクリリンカリウム、ペクチン、ポロキサマー、ポリビニルアルコール、没食子酸プロピル、プロピレングリコール、キシリトール、酢酸亜鉛、ラフィノース、ホウ酸ナトリウム、トレハロース、アルギン酸プロピレングリコール、スルホブチルエーテルβ−シクロデキストリンまたはその混合物、あるいは当業者に知られている周知の安定剤から選択することができる。
【0064】
流動促進剤は、二酸化ケイ素、タルク、ステアリン酸カルシウム、ステアリン酸マグネシウムおよびステアリン酸アルミニウム、またはその混合物からなる群から選択することができる。
【0065】
滑沢剤は、ステアリン酸マグネシウム、ラウリル硫酸マグネシウム、ラウリル硫酸ナトリウム、ステアリルフマル酸ナトリウム、ステアリン酸カルシウム、微結晶性セルロースおよび二酸化シリコーン、硬化綿実油、硬化ヒマシ油、ステアリン酸、ステアリン酸亜鉛またはその混合物から選択することができる。
【0066】
緩衝剤は、アンモニア溶液、炭酸カルシウム、リン酸カルシウム、クエン酸、リン酸ナトリウム、ジエタノールアミン、リンゴ酸、グルタミン酸一ナトリウム、リン酸、クエン酸カリウム、酢酸ナトリウム、重炭酸ナトリウム、ホウ酸ナトリウム、クエン酸ナトリウム、水酸化ナトリウム、乳酸ナトリウム、トリエタノールアミンまたはその混合物、あるいは当業者に知られている周知の緩衝剤から選択することができる。
【0067】
クロピドグレルの調節放出製剤は、湿式造粒法、乾式造粒法、融解造粒法または当業者には公知の方法から選択される任意の方法を用いて調製することができる。
【0068】
本発明の態様の一実施形態では、クロピドグレル単独で、または任意選択で他の賦形剤と共に、ポリマーとブレンドし、錠剤へと圧縮する。
【0069】
あるいはブレンドを押し固めて顆粒を生成し、それをさらに錠剤へと圧縮する。
【0070】
他の実施形態では、ポリマーを適切な溶媒に溶解または分散し、この溶液または分散液を、クロピドグレル単独、または他の賦形剤とブレンドしたクロピドグレルを造粒するために使用する。この実施形態では、これに制限はされないが、クロピドグレルを単独で顆粒化する場合は、スプレーコーティングまたは流動層コーティングが好ましく、一方クロピドグレルを賦形剤とブレンドし顆粒化する場合は、高剪断造粒機を使用した顆粒化が好ましい。
【0071】
さらに他の実施形態では、クロピドグレルのコアは、予め形成したペレット上への薬物の層化によって、またはクロピドグレルを適切な賦形剤とブレンドし、湿式造粒することによって調製する。湿式造粒物を、押し出し、球状化して、クロピドグレルの球状コアを生成する。上記の方法のいずれかによって調製したコアを、次いでエチルセルロースで、あるいはエチルセルロースと、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロース、ヒドロキシエチルセルロース、ポリビニルピロリドン、アルギン酸塩、ガム、カルボマーまたはその混合物からなる群から選択される親水性物質との混合物でコーティングする。
【0072】
本発明による好ましい疎水性ポリマーは、エチルセルロースである。
【0073】
約7cps〜約100cpsの粘度のエチルセルロースが好ましい。これらの中で、約10cps〜約50cpsの粘度のエチルセルロースが最も好ましい。組み込まれるエチルセルロースの量は、選択したグレードによって変化する。約0.5重量パーセント〜約30重量パーセントの量が好ましい。約5重量パーセント〜約15重量パーセントの量が最も好ましい。
【0074】
疎水性ポリマーが親水性律速ポリマーと共に使用される場合、これに制限はされないが、低粘度グレードの親水性物質、例えば3cps〜15cpsのヒドロキシプロピルメチルセルロース、PVPK−12〜PVPK−90のポリビニルピロリドンが好ましい。
【0075】
本発明の他の態様は、クロピドグレルの融点よりも低い融点を有する疎水性律速ポリマーの使用によってクロピドグレルの調節放出製剤を提供する。クロピドグレルおよび低融点疎水性律速ポリマーの融点の差異は、少なくとも30℃であるべきである。この低融点疎水性材料を、流体粘稠性にまで溶解し、クロピドグレルを均一に分散するまでそれに分散させる。次いで融解した固まりを、ふるいにかけ、および/または乾燥して、顆粒を生成し、顆粒をさらに錠剤へと圧縮する。ここで圧縮の前に任意選択で、顆粒を適切な賦形剤とブレンドする。低融点疎水性律速ポリマーは、セチルアルコール、セトステアリルアルコール、白ろう、黄ろう、マイクロクリスタリンワックスおよびこれらの混合物からなる群から選択される。
【0076】
さらに他の実施形態では、クロピドグレルを、ヒプロメロース、カルボポール、ポビドン、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース、エチルセルロースなどの1種または複数のポリマー;ならびに充填剤/希釈剤、結合剤、崩壊剤および滑沢剤などの他の適切な賦形剤とブレンドし、錠剤に圧縮する。あるいは、上記のように調製されたクロピドグレルを有する賦形剤混合物を乾式造粒し、次いで圧縮をしてもよい。
【0077】
本発明の調節放出製剤は、任意選択でコーティングされてもよい。
【0078】
クロピドグレルの調節放出剤形で観察される服薬率の増加によって、これに限定されないが、最近の脳卒中、最近の心筋梗塞、または確定した末梢動脈疾患によって実証されるアテローム性動脈硬化症を有する患者におけるアテローム動脈硬化性事象(心筋梗塞、脳卒中、および血管死)の減少を含めて、クロピドグレルによる治療を必要としている患者の適切な管理を改善することができる。
【0079】
本発明に記載されるクロピドグレルの調節放出製剤およびその方法を、下記に例示する実施例において示す。これらの実施例は、例示としてのみ提供し、したがって本発明の範囲を限定するものと解釈されるべきではない。
【実施例】
【0080】
[実施例1〜4]
【0081】
【表1】
【0082】
実施例1〜4を下記の手順によって調製した。
【0083】
クロピドグレルベシラート、微結晶性セルロース、ヒプロメロース、カルボポールおよびクロスポビドンを、拡散ブレンダー中で混合した。コロイド状二酸化ケイ素および硬化綿実油を、拡散ブレンダー中の上記混合物に加え、潤滑ブレンドを得た。潤滑ブレンドを錠剤に圧縮した。
[実施例5]
【0084】
【表2】
【0085】
実施例5を、下記の手順によって調製した。
【0086】
クロピドグレルベシラート、微結晶性セルロース、ポリエチレンオキシド、カルボポールおよびクロスポビドンを、拡散ブレンダー中で混合した。コロイド状二酸化ケイ素および硬化綿実油を、拡散ブレンダー中の上記混合物に加え、潤滑ブレンドを得た。潤滑ブレンドを、平均重量500mgの錠剤に圧縮した。
[実施例6〜7]
【0087】
【表3】
【0088】
実施例6〜7を、下記の手順によって調製した。
【0089】
クロピドグレルベシラート、微結晶性セルロースおよびポビドンを、拡散ブレンダー中で混合した。コロイド状二酸化ケイ素およびステアリン酸マグネシウムを、拡散ブレンダー中の上記混合物に加え、潤滑ブレンドを得た。潤滑ブレンドを、平均重量450mgの錠剤に圧縮した。
[実施例8]
【0090】
【表4】
【0091】
実施例8を、下記の手順によって調製した。
【0092】
クロピドグレルベシラート、ラクトース、ヒプロメロースおよびカルボポールを、拡散ブレンダー中で混合した。コロイド状二酸化ケイ素、硬化綿実油およびタルクを、拡散ブレンダー中の上記混合物に加え、潤滑ブレンドを得た。潤滑ブレンドを、平均重量290mgの錠剤に圧縮した。
[実施例9]
【0093】
【表5】
【0094】
実施例9を、下記の手順によって調製した。
【0095】
クロピドグレルベシラート、ポビドン90、微結晶性セルロースおよびコロイド状二酸化ケイ素を、#40のふるいにかけた。ステアリン酸亜鉛を#60のふるいにかけた。ふるいにかけたステアリン酸亜鉛を、残りのふるいにかけた賦形剤に加え、ダブルコーンブレンダーで10分間ブレンドした。潤滑ブレンドを、ローラーコンパクターを使用して4〜6kpの硬度で強打した。振動造粒機を使用して#20のふるいにかけ、スラグを完全に強打した。極粒状のポビドン90およびコロイド状二酸化ケイ素を、#30のふるいにかけ、大きさを揃えた顆粒に加え、ブレンドした。ステアリン酸亜鉛を、#60のふるいにかけ、上記混合物とブレンドし、潤滑ブレンドを得た。潤滑ブレンドを、平均重量450mgの錠剤に圧縮した。
[実施例10]
【0096】
【表6】
【0097】
実施例10を、下記の手順によって調製した。
【0098】
クロピドグレルベシラート、微結晶性セルロースおよびポビドンを、拡散ブレンダー中で混合した。コロイド状二酸化ケイ素およびステアリン酸亜鉛を、拡散ブレンダー中の上記混合物に加え、潤滑ブレンドを得た。潤滑ブレンドを、平均重量450mgの錠剤に圧縮した。
[実施例11]
【0099】
【表7】
【0100】
実施例11を、下記の手順によって調製した。
【0101】
重硫酸クロピドグレル、微結晶性セルロースおよびポビドンを、拡散ブレンダー中で混合した。コロイド状二酸化ケイ素およびステアリン酸亜鉛を、拡散ブレンダー中の上記の混合物に加え、潤滑ブレンドを得た。潤滑ブレンドを、平均重量350mgの錠剤に圧縮した。
【0102】
実施例1〜11の錠剤を、0.1NのHCl900ml中で8時間まで50rpmにてUSP Type II装置を使用して溶解プロファイルについて試験した。実施例1〜11により調製した錠剤およびPlavix(登録商標)の溶解プロファイルを、下記の表1および2に表で示す。図1は、0.1NのHCl900ml中で8時間まで50rpmにてUSP Type II装置を使用したPlavix(登録商標)、実施例10および11の比較溶解プロファイルを示す。
【0103】
【表8】
【0104】
【表9】
【0105】
0.1NのHC1中で試験した本発明の組成物の溶解プロファイルは、1時間以内にほぼ完全に薬物を放出するブランド製品であるPlavix(登録商標)と比較して、2〜8時間のより遅く持続する作用を示す。
【0106】
0.1NのHCl900ml中で50rpmにてUSP Type II装置を使用したPlavix(登録商標)、実施例10および11の比較溶解プロファイル(図1):
ex−vivoでのビーグル犬からの多血小板血漿中の抗血小板凝集活性について、実施例4の錠剤を、ブランド製品Plavix(登録商標)重硫酸クロピドグレル75mg錠と比較した。
【0107】
15〜20kgの範囲の体重の生後9〜12カ月の雄イヌで研究を行った。動物を一晩絶食させた。実施例4のクロピドグレル錠剤の単回用量を一群に投与し、他の群にはPlavix(登録商標)を与えた。0時間(薬物投与前)、ならびに薬物投与後0.5時間、2時間、6時間、10時間、24時間、48時間、72時間、96時間、120時間、168時間、192時間および216時間に、動物から採血した。血液採取の216時間後、クロピドグレルの第2の用量を両方の群に投与し、0時間(第1の用量後216時間)、ならびに第2の用量投与後6時間、24時間、72時間、240時間に、動物から採血した。
【0108】
約4mlの血液を、プラスチックシリンジ中に回収し、4%クエン酸三ナトリウム溶液(1:9、クエン酸溶液:血液)を含有するプラスチックチューブに移し、抗凝血剤と穏やかに混合した。血液試料を150gで10分間遠心分離し、多血小板血漿(PRP)を得た。PRPの分離後に、試料を1500gで15分間遠心分離し、乏血小板血漿(PPP)を得た。PRPの血小板数を、Cell−Dyne3700Hematology Analyzerを使用して推定し、ADP誘導性凝集アッセイのためにPPPで3×108/mlに調節した。
【0109】
個々の試料の調節したPRP180μlを、加えたADP試薬(20μM、最終濃度)20μLと共にインキュベートし、光学濃度(OD)を、SPECTRAMAX190マイクロプレートリーダーを使用して10分間30秒毎に連続的に560nmでモニターした。全ての試料を3連で試験した。
【0110】
データ分析:
下記の式を使用して、%凝集を計算した。
%凝集=[{(補正OD(0分)}−{補正OD(10分)}]*100/補正OD(0分)
【0111】
補正OD=PRPの吸光度−PPPの吸光度
ビーグル犬においてex−vivoで研究したPlavixおよびクロピドグレル75mg錠についての%凝集の比較データを、表3に表で示し、図2に示す。
【0112】
【表10】
【0113】
Plavix、クロピドグレル75mg錠、実施例4の比較血小板凝集作用、ビーグル犬でのEx−vivo研究(図2):
表3および図2に示されるように、クロピドグレルのすべての製剤は、ADP誘導性血小板凝集の有意な阻害をもたらした。
【0114】
Plavix(登録商標)錠剤と比較して、実施例4のクロピドグレル錠剤は、より長い期間持続する作用を示した。Plavix(登録商標)製剤で処理されたイヌは、約8〜9日で作用の完全な逆転を示し、一方実施例4のクロピドグレル錠剤で処理されたイヌは、9日目でも抗血小板凝集作用を示した。第2の用量は血小板凝集のさらなる減少をもたらし、実施例4の錠剤のクロピドグレル錠剤についての作用は、Plavix(登録商標)錠剤よりも良好であった。
【0115】
薬力学的研究を、DiaMed Impact−Rによって抗凝固処理全血における血小板機能を決定することによって、実施例10の通りに調製した錠剤について、Plavix(登録商標)と比較して行った。試験方法は、血小板減少症を含めた原発性止血異常(例えば、フォンウィルブランド病)をスクリーニングし、その治療をモニタリングする血小板機能の研究を目的とする。それは、血小板の機能低下および機能亢進の両方を試験するために使用することができる。さらにそれは、様々な抗血小板薬に対する反応をモニターするための迅速な方法を提供する。この試験を、抗凝固処理全血における血小板機能を決定するために行い、Plavix(登録商標)とクロピドグレルの調節放出製剤(実施例10)とを比較した。
【0116】
血小板凝集アッセイの目的のために、全部で10の血液試料を回収した。静脈血試料4.5m1を、研究期間中−10.00時間、0.00時間(投与前)、ならびに薬物投与後0.50時間、2.00時間、4.00時間、6.00時間、8.00時間、10.00時間、24.00時間および48.00時間に採取する。
【0117】
新しい静脈穿刺によって血液試料を回収した。それが新しい静脈穿刺である場合、新しい静脈穿刺のために血小板凝集因子が存在する可能性があるため、血液2〜3mlを廃棄する。血液試料を、使い捨て注射器を使用して前腕静脈に入れた留置カニューレによって回収した。各サンプリング時点で、血液試料4.5mlを採取し、クエン酸ナトリウム緩衝液を含有する試料回収チューブに移す。
【0118】
クエン酸ナトリウムを含有するチューブ中に試験をする全血試料を回収し、室温(18〜25℃)で保存した。クエン酸塩添加血試料(130μL)をウェルの中央に置き、定義済みパラメーターによって回転処理を施した。ウェルを洗浄し、May−Gruenwald染色液によって染色した。
【0119】
表面被覆率(%)として表す血小板で覆われた総面積率として血小板粘着を評価し、平均サイズ(μm2)として表す凝集物への表面結合の平均サイズとして凝集を評価した。
【0120】
Plavix(登録商標)対クロピドグレル錠剤(実施例10および11)の2種の製剤における血小板の%凝集サイズの比較を、図3および4に示す。
【0121】
Plavix(登録商標)対クロピドグレル錠剤(実施例10)の2種の製剤における血小板の%凝集サイズの比較(図3)。
Plavix(登録商標)と比較して実施例10の製剤においては血小板のさらなる脱凝集があるため、図3に表される通り、抗血小板作用は実施例10においてPlavix(登録商標)よりも良好であることが見出された。
【0122】
Plavix(登録商標)対クロピドグレル錠剤(実施例11)の2種の製剤における血小板の%凝集サイズの比較(図4)。
Plavix(登録商標)と比較して実施例11の製剤においては血小板のさらなる脱凝集があるため、図4に表される通り、抗血小板作用は実施例11においてPlavix(登録商標)よりも良好であることが見出された。
【0123】
クロピドグレル75mgに相当するクロピドグレルベシラート錠剤(実施例10)、クロピドグレル75mgに相当する重硫酸クロピドグレル錠剤(実施例11)および市販の重硫酸クロピドグレル錠剤であるPlavix(登録商標)を、空腹条件下の健康な大人の男性ヒト対象において非盲検、バランス化、無作為化、3種の処置(three−treatment)、単一期間、単回用量、平行、比較経口薬物動態学的研究を行った。研究は、36対象(各処置について12対象)で行った。
【0124】
研究期間中試料の投与11時間以上前から投与後24時間まで、対象を臨床施設に収容した。対象は、投与前に少なくとも10時間一晩絶食した。投与を除いて、投与前1時間から投与後2時間まで飲料水を禁止した。他の時間は、飲料水は自由に与えられた。各期間において投与の約4時間後、およびそれ以降検査終了まで特定の間隔で食事を提供した。研究期間中全部で23の血液試料が回収された。静脈血試料5.5ml(廃棄したヘパリン添加血液0.5mlを含めて)を、研究期間の間、投与前の時点(投与前)、ならびに薬物投与後0.25時間、0.50時間、0.75時間、1.00時間、1.25時間、1.50時間、1.75時間、2.00時間、2.50時間、3.00時間、4.00時間、5.00時間、6.00時間、7.00時間、8.00時間、9.00時間、10.00時間、12.00時間、16.00時間、24.00時間、36.00時間および48.00時間に採取した。使い捨て注射器を使用して、前腕静脈に入れた留置カニューレから血液試料を回収した。各サンプリング時点で、血液試料(廃棄したヘパリン添加血液0.5mlを含めて)5.5mlを採取し、抗凝血剤としてEDTAを含有する試料回収チューブに移した。遠心分離後、1時間以内に血液試料から血漿を分離し、血漿試料を分析のために取り出すまで−70±5℃で保存した。血漿中のクロピドグレル濃度を、有効なLCMS/MSを使用して定量化した。
【0125】
ピーク血漿濃度(Cmax)、ピーク血漿濃度に到達する時間(Tmax)、最終時点までの血漿濃度−時間曲線下面積(AUC0−t)、無限大まで外挿する血漿濃度−時間曲線下面積(AUC0−∞)、残りの面積率(AUC_%Extrap)、浄化(D)、分布容積(Vd)、血漿排出半減期(t1/2)および排出速度定数(λZ)を、クロピドグレルについて計算した。
【0126】
クロピドグレルおよびクロピドグレルカルボキシ酸代謝物について、Plavix(登録商標)、実施例10および11によって調製したクロピドグレル錠剤で試験した様々な時点の平均血漿濃度を、表4および5に各々表で示す。薬物動態パラメーターの平均値を、表6および7に表で示す。
【0127】
図5は、空腹条件下でPlavix(登録商標)、クロピドグレル75mg錠、実施例10および11の単回用量後の、クロピドグレルカルボキシ酸代謝物(ng/mL)の平均血漿濃度−時間プロファイルを示す。
【0128】
【表11】
【0129】
【表12】
【0130】
空腹条件下でPlavix(登録商標)、クロピドグレル75mg錠、実施例10および11の単回用量後の、クロピドグレルカルボキシ酸代謝物(ng/mL)の平均血漿濃度−時間プロファイル(図5)。
【0131】
【表13】
【0132】
【表14】
【0133】
クロピドグレルは、プロドラッグである。活性代謝物であるチオール誘導体は、クロピドグレルの2−オキソ−クロピドグレルへの酸化、ついで加水分解により形成される。主要な循環代謝物は、不活性のカルボキシ酸誘導体である。Plavix(登録商標)における高い血漿レベルによって観察されるように、カルボキシ酸誘導体の急速な形成は、短期間の高濃度のクロピドグレルとの曝露と関連付けられるであろう。本発明のクロピドグレルの修飾された製剤である実施例10の場合、クロピドグレルカルボキシ酸の血漿濃度は、2.5〜8時間の間制御され持続された様式であることを見ることができる。これは活性代謝物の緩慢な形成を示し、それは次にカルボキシ酸誘導体に緩慢に変換される。本発明の製剤は、ブランド製品であるPlavix(登録商標)と比較してカルボキシ酸代謝物について有意に低いCmaxを示すことを観察することができる。本発明の組成物は、Plavix(登録商標)と比較してクロピドグレルについてより高いAUC値を示す。クロピドグレルについてのより高いAUC値およびより大きいTmaxは、不活性カルボキシ酸代謝物についてのCmaxの有意に低い値と共に、より長い期間における薬物のより良好な有効性と関連付けることができる。
【0134】
本発明の特定の好ましい他の実施形態は、本発明を開示する目的のために記載したが、開示された実施形態への修正は、当業者であれば思いつくことができる。したがって、この明細書は、本発明の趣旨および範囲から逸脱しない本発明の全ての実施形態およびその修正を包含することを意図している。
【図面の簡単な説明】
【0135】
【図1】Plavix(登録商標)の溶解プロファイルを示す比較図である。
【図2】Plavix、クロピドグレル75mg錠の血小板凝集作用を示す比較図である。
【図3】Plavix(登録商標)対クロピドグレル錠剤の2種の製剤における血小板の%凝集サイズを示す比較図である。
【図4】Plavix(登録商標)対クロピドグレル錠剤の2種の製剤における血小板の%凝集サイズを示す比較図である。
【図5】Plavix(登録商標)、クロピドグレル75mg錠の単回用量後のクロピドグレルカルボキシ酸代謝物(ng/mL)の平均血漿濃度−時間プロファイルを示す図である。
【特許請求の範囲】
【請求項1】
クロピドグレル50mg〜150mgに相当するクロピドグレルまたはクロピドグレルの塩を含む1日1回の剤形であって、
(a)約3時間以上の平均Tmax、
(b)1000ピコグラム/ml以下の平均Cmax、
(c)2500ピコグラム/ml/時間超の平均AUC0〜49時間
から選択されるクロピドグレルのin vivo血漿プロファイルの少なくとも1つを提供する剤形。
【請求項2】
クロピドグレル50mg〜150mgに相当するクロピドグレルまたはクロピドグレルの塩を含む1日1回の剤形であって、1400ng/ml以下の平均Cmaxを有するクロピドグレルカルボキシ酸代謝物のin vivo血漿プロファイルを提供する剤形。
【請求項3】
クロピドグレル75mgに相当するクロピドグレルまたはクロピドグレルの塩を含む、請求項1または2に記載の1日1回の剤形。
【請求項4】
前記平均Cmaxが、約900ピコグラム/ml未満である、請求項1に記載の1日1回の剤形。
【請求項5】
前記平均Cmaxが、約900ピコグラム/ml未満である、請求項1または3に記載の1日1回の剤形。
【請求項6】
前記平均Cmaxが、約800ピコグラム/ml未満である、請求項5に記載の1日1回の剤形。
【請求項7】
前記平均Cmaxが、1200ng/ml以下、好ましくは1100ng/ml以下である、請求項2に記載の1日1回の剤形。
【請求項1】
クロピドグレル50mg〜150mgに相当するクロピドグレルまたはクロピドグレルの塩を含む1日1回の剤形であって、
(a)約3時間以上の平均Tmax、
(b)1000ピコグラム/ml以下の平均Cmax、
(c)2500ピコグラム/ml/時間超の平均AUC0〜49時間
から選択されるクロピドグレルのin vivo血漿プロファイルの少なくとも1つを提供する剤形。
【請求項2】
クロピドグレル50mg〜150mgに相当するクロピドグレルまたはクロピドグレルの塩を含む1日1回の剤形であって、1400ng/ml以下の平均Cmaxを有するクロピドグレルカルボキシ酸代謝物のin vivo血漿プロファイルを提供する剤形。
【請求項3】
クロピドグレル75mgに相当するクロピドグレルまたはクロピドグレルの塩を含む、請求項1または2に記載の1日1回の剤形。
【請求項4】
前記平均Cmaxが、約900ピコグラム/ml未満である、請求項1に記載の1日1回の剤形。
【請求項5】
前記平均Cmaxが、約900ピコグラム/ml未満である、請求項1または3に記載の1日1回の剤形。
【請求項6】
前記平均Cmaxが、約800ピコグラム/ml未満である、請求項5に記載の1日1回の剤形。
【請求項7】
前記平均Cmaxが、1200ng/ml以下、好ましくは1100ng/ml以下である、請求項2に記載の1日1回の剤形。
【図1】

【図2】
【図3】
【図4】
【図5】
【図2】
【図3】
【図4】
【図5】
【公表番号】特表2009−532462(P2009−532462A)
【公表日】平成21年9月10日(2009.9.10)
【国際特許分類】
【出願番号】特願2009−503742(P2009−503742)
【出願日】平成19年4月3日(2007.4.3)
【国際出願番号】PCT/IN2007/000142
【国際公開番号】WO2007/113857
【国際公開日】平成19年10月11日(2007.10.11)
【出願人】(507365927)カディラ・ヘルスケア・リミテッド (26)
【Fターム(参考)】
【公表日】平成21年9月10日(2009.9.10)
【国際特許分類】
【出願日】平成19年4月3日(2007.4.3)
【国際出願番号】PCT/IN2007/000142
【国際公開番号】WO2007/113857
【国際公開日】平成19年10月11日(2007.10.11)
【出願人】(507365927)カディラ・ヘルスケア・リミテッド (26)
【Fターム(参考)】
[ Back to top ]