
貝類由来ADPリボシル化タンパク質、およびその利用
【課題】本発明は、ADPリボシル化活性を有する新規貝類由来タンパク質、および該タンパク質の用途の提供を課題とする。より詳しくは本発明は、ADPリボシル化活性を有する新規貝類由来タンパク質、および該タンパク質を成分とする抗癌剤もしくはアポトーシス誘導剤、並びに、該タンパク質を用いてDNAをADPリボシル化する方法、および、細胞に対してアポトーシス誘導する方法等の提供を課題とする。
【解決手段】貝類からDNAを基質としてADPリボシル化する活性を有する新規タンパク質が同定された。該タンパク質を電気穿孔法により癌細胞株であるHeLa細胞、TMK-1細胞内へ導入することにより、HeLa細胞、TMK-1細胞のアポトーシスが誘導され、細胞死に至ることが判明した。貝類から見出されたADPリボシル化活性を有するタンパク質は、アポトーシス誘導剤もしくは抗癌剤として有用である。
【解決手段】貝類からDNAを基質としてADPリボシル化する活性を有する新規タンパク質が同定された。該タンパク質を電気穿孔法により癌細胞株であるHeLa細胞、TMK-1細胞内へ導入することにより、HeLa細胞、TMK-1細胞のアポトーシスが誘導され、細胞死に至ることが判明した。貝類から見出されたADPリボシル化活性を有するタンパク質は、アポトーシス誘導剤もしくは抗癌剤として有用である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ADPリボシル化活性を有する貝類由来タンパク質、およびその用途に関する。
【背景技術】
【0002】
ADPリボシル化は、β-NADのADP-リボース部分がアクセプター分子に転移される翻訳後修飾であることが知られている。ADP-リボシルトランスフェラーゼは、その反応産物によって分類されている。モノADP-リボシルトランスフェラーゼは、単一のADP-リボースの標的分子への転移を触媒するのに対し、PARPsと呼ばれるポリ(ADP-リボース)ポリメラーゼは、最初にADP-リボースを核タンパク質に転移させた後、ADP-リボース残基の重合化を触媒する(非特許文献1および2参照)。
【0003】
モノADP-リボシルトランスフェラーゼは、細胞外感染症を引き起こす細菌において認められる。コレラ毒素(CT)および百日咳毒素(PT)はそれぞれ、G-タンパク質のGs-およびGi-型αサブユニットを標的とするのに対し、ジフテリア毒素(DT)は、延長因子-2(非特許文献3参照)のジフタミドを改変する。同様に、哺乳類および鳥類においてモノADP-リボシルトランスフェラーゼに関する報告がある。例えば、アルギニン特異的ADP-リボシルトランスフェラーゼは、グリコシルホスファチジルイノシトール(GPI)結合型または可溶性型として存在することが示されており、それらが様々な標的タンパク質を改変することが示唆されている(非特許文献2および4参照)。
【0004】
ピエリシン-1は当初、モンシロチョウの被蛹からの細胞障害性物質として同定された(非特許文献5〜7参照)。ピエリシン-1は、CTおよびPTのようなADP-リボシルトランスフェラーゼとそのN-末端領域において配列類似性を有し、C-末端領域においてリシンスーパーファミリーのレクチンドメインと配列類似性を有する(非特許文献8〜10参照)。他のADP-リボシルトランスフェラーゼとは異なり、ピエリシン-1は、DNAにおけるグアニン残基のN2アミノ基を標的としてN2-(ADP-リボス-1-イル)-2'-デオキシグアノシン(非特許文献11参照)を生成する。C-末端ドメインは、グロボトリアオシルセラミド(Gb3)およびグロボテトラオシルセラミド(Gb4)のようなグリコスフィンゴリピッド受容体に対する結合能を有し、哺乳類細胞表面でのその作用によってピエリシン-1を細胞に組み入れることに関与している(非特許文献9および10参照)。ピエリシン-1は、多様なヒト癌細胞株に対して細胞障害性であり、IC50値は、調べた細胞株13個において0.043〜270 ng/mlの範囲である(非特許文献9、12、13参照)。細胞におけるGb3およびGb4の量はその感度を大きく左右して(非特許文献10参照)、ピエリシン-1によって、Bcl-2およびカスパーゼを含むミトコンドリア経路によって、アポトーシスが誘導される(非特許文献14参照)。その上、CHL細胞におけるHPRT遺伝子の変異は、ピエリシン-1の低用量処置によって誘導することができる(非特許文献15参照)。興味深いことに、ピエリシン-1のmRNAは、後期幼虫において高度に発現されて、そのタンパク質は脂肪体に蓄積され、これは蛹化の際も持続し、このことはピエリシンがモンシロチョウにおける変態の際に機能する可能性があることを示唆している(非特許文献16参照)。同じシロチョウ属に属するオオモンシロチョウ(Pieris brassicae)およびエゾスジグロシロチョウ(P. napi)の蛹は、哺乳類細胞株に対してモンシロチョウと類似の細胞障害活性を含む(非特許文献5参照)。その上、ピエリシン-1と91%同一であるアミノ酸配列を有するピエリシン-2が、オオモンシロチョウから同定されている(非特許文献13参照)。ピエリシン-2はまた、DNAを標的として、ピエリシン-2によって産生されたDNA付加物の構造はピエリシン-1によって産生された付加物と同じである(非特許文献17参照)。
また、以下のリストに記載された文献が報告されている。
【0005】
【特許文献1】特開2001−25390
【非特許文献1】Sugimura, T.著、「Poly(adenosine diphosphate ribose).」、Prog. Nucleic Acid Res. Mol. Biol.、1973年、Vol.13、 p.127-151.
【非特許文献2】Seman, M.外3名著、「Ecto -ADP-ribosyltransferases (ARTs): emerging actors in cell communication and signaling.」、Curr. Med. Chem.、2004年、 Vol.11、p.857-872.
【非特許文献3】Krueger,.K.M. および Barbieri, J.T.著、「The family of bacterial ADP-ribosylating exotoxins.」、Clin. Microbiol. Rev.、1995年、Vol.8、p.347.
【非特許文献4】Okazaki, I.J. および Moss, J.著、「Characterization of glycosylphosphatidylinositiol-anchored, secreted, and intracellular vertebrate mono -ADP-ribosyltransferases.」、Annu. Rev. Nutr.、1999年、Vol.19、p.485-509.
【非特許文献5】Koyama, K.外8名著、「Presence in Pieris rapae of cytotoxic activity against human carcinoma cells.」、Jpn. J. Cancer Res.、1996年、Vol.87、p.1259-1262.
【非特許文献6】Watanabe, M.外4名著、「Purification of pierisin, an inducer of apoptosis in human gastric carcinoma cells, from cabbage butterfly, Pieris rapae.」、Jpn. J. Cancer Res.、1998年、Vol.89、p.556-561.
【非特許文献7】Sugimura, T.著、「Serendipitous discoveries from sudden inspirations and the joy of being a scientist.」、Biochem. Biophys. Res. Commun.、2002年、Vol. 296、p.1037-1038.
【非特許文献8】Watanabe, M.外8名著、「Molecular cloning of an apoptosis-inducing protein, pierisin, from cabbage butterfly: possible involvement of ADP-ribosylation in its activity.」、Proc. Natl. Acad. Sci. USA、1999年、Vol.96、p.10608-10613.
【非特許文献9】Kanazawa, T.外7名著、「Distinct roles for the N- and C-terminal regions in the cytotoxicity of pierisin-1, a putative ADP-ribosylating toxin from cabbage butterfly, against mammalian cells.」、Proc. Natl. Acad. Sci. USA、2001年、Vol.98、p.2226-2231.
【非特許文献10】Matsushima-Hibiya, Y.外8名著、「Identification of glycosphingolipid receptors for pierisin-1, a guanine-specific ADP-ribosylating toxin from the cabbage butterfly.」、J. Biol. Chem.、2003年、Vol.278、p.9972-9978.
【非特許文献11】Takamura-Enya, T.外7名著、「Mono(ADP-ribosyl)ation of 2'-deoxyguanosine residue in DNA by an apoptosis-inducing protein, pierisin-1, from cabbage butterfly.」、Proc. Natl. Acad. Sci. USA、2001年、Vol.98、p.12414-12419.
【非特許文献12】Kono, T.外6名著、「Cytotoxic activity of pierisin, from the cabbage butterfly, Pieris rapae, in various human cancer cell lines.」、Cancer Lett.、1999年、Vol.137、p. 75-81.
【非特許文献13】Matsushima-Hibiya, Y.外6名著、「Purification and cloning of pierisin-2, an apoptosis-inducing protein from the cabbage butterfly, Pieris brassicae.」、Eur. J. Biochem.、2000年、Vol.267、p.5742-5750.
【非特許文献14】Kanazawa, T.外8名著、「Bcl-2 blocks apoptosis caused by pierisin-1, a guanine-specific ADP-ribosylating toxin from the cabbage butterfly.」、Biochem. Biophys. Res. Commun.、2002年、Vol.296、p.20-25.
【非特許文献15】Totsuka, Y.外8名著、「Analysis of HPRT and supF mutations caused by pierisin-1, a guanine specific ADP-ribosylating toxin derived from the cabbage butterfly.」、Chem. Res. Toxicol.、2003年、Vol.16、p.945-952.
【非特許文献16】Watanabe, M.外9名著、「Developmental stage-specific expression and tissue distribution of pierisin-1, a guanine-specific ADP-ribosylating toxin, in Pieris rapae.」、Comp. Biochem. Physiol. A、2004年、Vol.139、p.125-131.
【非特許文献17】Takamura-Enya, T.外4名著、「Mono(ADP-ribosyl)ation of the N2 amino groups of guanine residues in DNA by pierisin-2, from the cabbage butterfly, Pieris brassicae.」、Biochem. Biophys. Res. Commun.、2004年、Vol.323、p.579-582.
【非特許文献18】山崎正利 外2名著、「海洋軟体動物アメフラシ類の抗がん・抗菌蛋白質」、蛋白質核酸酵素、平成13年、Vol. 46、p.382-387.
【発明の開示】
【発明が解決しようとする課題】
【0006】
本発明は、ADPリボシル化活性を有する新規貝類由来タンパク質、および該タンパク質の用途の提供を課題とする。より詳しくは本発明は、ADPリボシル化活性を有する新規貝類由来タンパク質、および該タンパク質を成分とする抗癌剤もしくはアポトーシス誘導剤、並びに、該タンパク質を用いてDNAをADPリボシル化する方法、および、細胞に対してアポトーシス誘導する方法等の提供を課題とする。
【課題を解決するための手段】
【0007】
上記課題を解決すべく本発明者らは鋭意研究を行った。これまでのところ、貝類においてDNAを基質とするADPリボシル化活性を有するタンパク質(ADPリボシル化酵素)は知られていない。本発明者らはチョウセンハマグリ(学名Meretrix lamarckii)、アサリ(学名Ruditapes philippinarum)、およびヤマトシジミ(学名Corbicula japonica)の抽出液に、試験管内で仔牛胸腺DNAと反応させることにより、DNAをADPリボシル化し、DNA付加体 N2-(ADP-ribos-1-yl)-2'-deoxyguanosineを生成させる活性を有するタンパク質を見出すことに成功した。
【0008】
本発明者らは、チョウセンハマグリの抽出液中より分子量20 kDaのタンパク質CARP-1を精製し、精製タンパク質の部分アミノ酸配列を元にcDNAクローニングを行った。クローニングされたcDNAについて解析を行った結果、該cDNAは853塩基対からなり、オープンリーディングフレームは182残基のアミノ酸からなるタンパク質をコードし、分子量は20332と算出された。試験管内で発現させたCARP-1タンパク質には、DNAをADPリボシル化する活性があることが確認された。
【0009】
CARP-1タンパク質のアミノ酸配列に関する相同性解析の結果、ピエリシン-1およびピエリシン-2を含む既知のいかなるタンパク質とも相同性が見出されなかった。しかし、CARP-1タンパク質のアミノ酸配列には、これまでに知られているADPリボシル化酵素の酵素活性に必須なグルタミン酸と予測されるアミノ酸(E128)が含まれることが見出された。そこで、ADPリボシル化酵素に必須と考えられるグルタミン酸に相当するアミノ酸残基(E128)を、同じ酸性アミノ酸であるアスパラギン酸に変異させたところ、活性の95%が喪失することが分かった。
【0010】
さらに本発明者らは、該タンパク質を電気穿孔法により癌細胞株であるHeLa細胞およびTMK-1細胞内へ導入することにより、これらの細胞のアポトーシスが誘導され、細胞死に至ることを見出した。
【0011】
従って、本発明の新規ADPリボシル化タンパク質は、癌細胞に対するアポトーシス誘導剤もしくは抗癌剤としての用途を有することが判明した。
【0012】
上述の如く本発明者らは、DNAをADPリボシル化する活性を有する新規タンパク質を貝類において初めて単離することに成功し、さらに、該タンパク質について上述の新規な用途を初めて見出すことに成功し本発明を完成させた。
【0013】
本発明は、ADPリボシル化活性を有する新規貝類由来タンパク質、および該タンパク質を成分とする抗癌剤もしくはアポトーシス誘導剤、並びに、該タンパク質を用いてDNAをADPリボシル化する方法、および、細胞に対してアポトーシス誘導する方法に関し、より具体的には、
〔1〕 下記(a)〜(d)のいずれかに記載のポリヌクレオチドであって、ADPリボシル化活性を有するポリペプチドをコードするポリヌクレオチド、
(a)配列番号:2に記載のアミノ酸配列からなるポリペプチドをコードするポリヌクレオチド
(b)配列番号:1に記載の塩基配列のコード領域を含むポリヌクレオチド
(c)配列番号:2に記載のアミノ酸配列において、1もしくは複数のアミノ酸が置換、欠失、挿入、および/または付加したアミノ酸配列からなるポリペプチドをコードするポリヌクレオチド
(d)配列番号:1に記載の塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズするポリヌクレオチド
〔2〕 〔1〕に記載のポリヌクレオチドによりコードされるポリペプチド、
〔3〕 〔2〕に記載のポリペプチドもしくはその部分ポリペプチドと、細胞レセプターを認識するタンパク質とが融合した構造のポリペプチド、
〔4〕 〔1〕に記載のポリヌクレオチド、または、〔3〕に記載のポリペプチドをコードするポリヌクレオチドが挿入されたベクター、
〔5〕 〔1〕に記載のポリヌクレオチド、または〔4〕に記載のベクターを保持する宿主細胞、
〔6〕 〔5〕に記載の宿主細胞を培養し、該宿主細胞またはその培養上清から、産生させたポリペプチドを回収する工程を含む、〔2〕または〔3〕に記載のポリペプチドの製造方法、
〔7〕 〔1〕に記載のポリヌクレオチドと特異的にハイブリダイズするポリヌクレオチドであって、少なくとも15ヌクレオチドの鎖長を持つポリヌクレオチド、
〔8〕 〔1〕に記載のポリヌクレオチドまたはその一部に対するアンチセンスポリヌクレオチド、
〔9〕 〔2〕または〔3〕に記載のポリペプチドに結合する抗体、
〔10〕 〔2〕もしくは〔3〕に記載のポリペプチド、または、〔4〕に記載のベクターを有効成分として含む、アポトーシス誘導剤、
〔11〕 〔2〕もしくは〔3〕に記載のポリペプチド、または、〔4〕に記載のベクターを有効成分として含む、抗癌剤、
〔12〕 〔2〕もしくは〔3〕に記載のポリペプチドを有効成分として含む、ADPリボシル化試薬、
〔13〕 CARP-1タンパク質の発現もしくは機能阻害物質を有効成分とする、貝類毒性除去剤、
〔14〕 〔2〕に記載ポリペプチドとDNAとを接触させる工程を含む、DNAをADPリボシル化する方法、
〔15〕 以下の工程(a)または(b)のいずれかの工程を含む、細胞をアポトーシス誘導する方法、
(a)〔2〕に記載のポリペプチド、または〔4〕に記載のベクターを細胞へ導入する工程
(b)〔3〕に記載のポリペプチドと細胞とを接触させる工程
〔16〕 以下の工程(a)〜(c)を含む、抗癌剤のスクリーニング方法、
(a)〔2〕に記載のポリペプチドもしくは該ポリペプチドを発現する細胞と、被検化合物を接触させる工程
(b)前記ポリペプチドの発現量または活性を測定する工程
(c)被検化合物を接触させない場合と比較して、前記発現量または活性を上昇させる化合物を選択する工程
〔17〕 以下の工程(a)〜(c)を含む、貝類毒性除去剤のスクリーニング方法、
(a)〔2〕に記載のポリペプチドもしくは該ポリペプチドを発現する細胞と、被検化合物を接触させる工程
(b)前記ポリペプチドの発現量または活性を測定する工程
(c)被検化合物を接触させない場合と比較して、前記発現量または活性を低下させる化合物を選択する工程
を、提供するものである。
【発明の効果】
【0014】
本発明者らは、多くの国において一般的に消費されている貝類におけるDNA ADP-リボシルトランスフェラーゼの有無を調べ、ハマグリ等の二枚貝がDNA ADP-リボシルトランスフェラーゼ活性を有するタンパク質を含むことを発見した。
【0015】
具体的には、チョウセンハマグリ、アサリ、およびヤマトシジミには、上記の酵素活性を有するタンパク質を含み、モンシロチョウのピエリシン-1および-2の場合と同様に、β-NADおよびDNAからN2-(ADP-リボース-1-イル)-2'-デオキシグアノシンを産生することが証明された。また、CARP-1と呼ばれる20 kDaタンパク質がチョウセンハマグリから精製され、クローニングされたcDNAを用いてインビトロにて翻訳させることにより、CARP-1タンパク質を取得した。該タンパク質は、DNA ADP-リボシルトランスフェラーゼ活性を示した。チョウセンハマグリからの粗抽出物のDNA ADP-リボシルトランスフェラーゼ活性は、6900 PSL/μgであり、この値はそれぞれ、モンシロチョウの幼虫および蛹の値の約1/2および1/20である。CARP-1の推定アミノ酸配列は、ピエリシン-1および-2とわずかな相同性しか示さなかった。
【0016】
NAD結合部位と推定される128位のグルタミン酸についての部位特異的変異誘発実験によって、CARP-1が新規DNA ADP-リボシルトランスフェラーゼであることが証明された。
【0017】
ADP-リボシルトランスフェラーゼの反応メカニズムは、CT、PT、DT、緑膿菌(Pseudomonas aeruginosa)のエンドトキシンA(ETA)、および大腸菌の熱不安定エンテロトキシン(LT)においてほとんどが研究されている(Aktories, K. and Just, I. (2000) Bacterial Protein Toxins. Springer-Verlag, Berlin.;Krueger, K.M. and Barbieri, J.T. (1995) The family of bacterial ADP-ribosylating exotoxins. Clin. Microbiol. Rev., 8, 34-47.)。これらの毒素のX-線結晶学から、それらがその反応中心において類似の構造を共有することが判明した。既知の全てのADP-リボシルトランスフェラーゼにおいて保存されており、活性に必須と考えられるグルタミン酸残基は、NAD-結合部位として作用する。高度に保存されたアルギニン残基およびSer-Thr-Ser/Thrモチーフも同様に、CT、PTおよびLTに存在する。アルギニン残基は、反応ポケットの構造を維持すると考えられており、Ser-Thr-Ser/Thrモチーフはβ鎖-α-ヘリックス構造を構築して、反応腔を維持すると考えられている(Domenighini, M. and Rappuoli, R. (1996) Three conserved consensus sequences identify the NAD-binding site of ADP-ribosylating enzymes, expressed by eukaryotes, bacteria and T-even bacteriophages. Mol. Microbiol., 21, 667-674.;Pallen, M.J., Lam, A.C., Loman, N.J. and McBride, A. (2001) An abundance of bacterial ADP-ribosyltransferases-implications for the origin of exotoxins and their human homologues. Trends Microbiol., 9, 302-307; discussion 308.)。
【0018】
CARP-1と他のADP-リボシルトランスフェラーゼとの間の全体的な配列類似性は非常に低く、その基質の多様性が反映されていることを示唆される。CARP-1においてGlu-128、Arg-12および46〜48位でのSer-Thr-Thrが存在することは、CT、PT、およびLTのようなADP-リボシルトランスフェラーゼの反応腔構造が保存されていることを示唆する。また、CARP-1とピエリシン-1または-2とのアミノ酸相同性が非常に低いということは、CARP-1とピエリシンがいずれも、ある特定のグアニン特異的ADP-リボシルトランスフェラーゼに由来しないことを示唆している。進化的に異なるDNA ADP-リボシルトランスフェラーゼが存在することは、他の種類のDNA ADP-リボシルトランスフェラーゼも同様に種々の生物に存在する可能性があることが、今回の実験によって示唆された。
【0019】
CARP-1の特徴の一つとして、受容体結合ドメインを欠損している点が挙げられる。細菌のADP-リボシル化毒素は、触媒および受容体結合ドメインが同じまたは異なる分子に存在するかどうか、または、結合ドメインが存在するか否かに基づいて三つの群に分類されている。それぞれ、「A/B毒素」、「バイナリ毒素」、および「Aのみの毒素」である。「Aのみの毒素」は、細胞内で作用するために標的細胞における組込みのメカニズムが必要である。ピエリシン-1は典型的なA/B-毒素構造を有する。このタンパク質のC-末端71-kDaドメインは、リシンスーパーファミリーのレクチンドメインと相同性を有する。発現されたN-末端27-kDaドメイン自身は、HeLa細胞とTMK-1細胞に対して細胞障害性を示さないが、N-末端部分が電気穿孔によってHeLa細胞またはTMK-1細胞に転移されると毒性を示すようになる。CARP-1の場合、cDNA配列の解析から、CARP-1タンパク質は、受容体結合ドメインを有さないADP-リボシルトランスフェラーゼであることが示唆された。Sephacryl S-100ゲル濾過によって推定される上記タンパク質の分子量は約28 kDaであり、このタンパク質の天然型には受容体結合サブユニットが存在しないことが示唆された。さらに、チョウセンハマグリからの粗抽出物、および精製されたCARP-1は、電気穿孔法によってHeLa細胞およびTMK-1細胞に導入した場合に細胞障害性を示した。本発明者らはまた、CM52カラムクロマトグラフィーにおける流出分画においてチョウセンハマグリのDNA ADP-リボシルトランスフェラーゼ活性を発見したが、ゲル濾過実験から、この流出分画における活性タンパク質の分子量が精製CARP-1の分子量とほぼ同じであることが示された。これらの結果から、チョウセンハマグリは、哺乳類細胞に入るために必要な受容体結合ドメインを持たないDNA ADP-リボシルトランスフェラーゼを含有することが示唆された。
【0020】
本発明において単離されたcDNAは、ポリアデニル化シグナルおよび伸長部を有する典型的な真核細胞構造を示したことから、単離されたCARP-1遺伝子が海水からの原核微生物のゲノムに存在していた可能性は低い。また、海水中の微生物を含むと予想される鰓、外套膜、および中腸腺から調製した粗抽出物の酵素活性は、軟組織よりはるかに弱く、CARP-1がハマグリ自身から産生されることを示唆している。タンパク質に受容体結合ドメインが存在しないことは、CARP-1が細胞内DNAに作用してハマグリ自身の細胞死を誘導する可能性があることが示唆された。
【0021】
また、癌細胞表面に特異的に提示されているレセプターを認識するタンパク質とのキメラ酵素を作製することにより、癌細胞を選択的に死滅させる貝類由来の新規抗癌剤の提供が可能である。
【発明を実施するための最良の形態】
【0022】
本発明者らによって、ADPリボシル化活性を有する貝類由来のタンパク質、および該タンパク質をコードするポリヌクレオチド(遺伝子)が同定された。
【0023】
本発明のタンパク質は、ADPリボシル化活性を有するタンパク質であって、貝類に由来するタンパク質であれば、その由来する貝の種類は特に制限されず、例えば、チョウセンハマグリ、アサリ、またはヤマトシジミ等の貝類に由来するタンパク質が挙げられる。
【0024】
本発明の上記タンパク質(遺伝子)の好ましい態様としては、例えば、チョウセンハマグリに由来するCARP-1タンパク質(遺伝子)が挙げられる。該CARP-1タンパク質は、DNAを基質とするADPリボシル化活性を有する。
【0025】
上述の貝以外の貝類に由来するADPリボシル化活性を有するタンパク質(例えば、CARP-1タンパク質のホモログ・オルソログ等)もまた本発明のタンパク質に含まれる。
【0026】
本発明において「貝類」とは、特に制限されないが、好ましくは二枚貝である。本発明における貝類として具体的には、チョウセンハマグリ、アサリ、ヤマトシジミ、ウバガイ、ムラサキイガイ、マテガイ、ハマグリ(Meretrix lusoria)、シナハマグリ(Meretrix pethechialis)等を例示することができる。
【0027】
チョウセンハマグリのCARP-1遺伝子のDNA配列を配列番号:1に、該DNAによってコードされるタンパク質のアミノ酸配列を配列番号:2に記載する。
【0028】
ただし、本発明の遺伝子は、必ずしも、配列表に具体的に記載された配列からなるDNAに限定されない。また、本発明のタンパク質は、必ずしも、配列表に具体的に記載されたアミノ酸配列からなるタンパク質に限定されない。
【0029】
上記以外のタンパク質であっても、例えば配列表に記載された配列と高い相同性(通常70%以上、好ましくは80%以上、より好ましくは90%以上、最も好ましくは95%以上)を有し、かつ、本発明のタンパク質が有する機能(例えば、ADPリボシル化活性)を持つタンパク質は、本発明のタンパク質に含まれる。
【0030】
上記タンパク質とは、例えば、配列番号:2に記載のアミノ酸配列において、1以上のアミノ酸が付加、欠失、置換、挿入されたアミノ酸配列からなるタンパク質であって、通常変化するアミノ酸数が30アミノ酸以内、好ましくは10アミノ酸以内、より好ましくは5アミノ酸以内、最も好ましくは3アミノ酸以内である。
【0031】
本発明の遺伝子には、例えば、配列番号:2に記載の塩基配列からなるDNAに対応する他の貝類における内在性の遺伝子(チョウセンハマグリのCARP-1遺伝子のホモログ等)が含まれる。
【0032】
また、配列番号:1に記載の塩基配列からなるDNAに対応する他の貝類の内在性のDNAは、一般的に、配列番号:1に記載のDNAと高い相同性を有する。高い相同性とは、50%以上、好ましくは70%以上、さらに好ましくは80%以上、より好ましくは90%以上(例えば、95%以上、さらには96%、97%、98%または99%以上)の相同性を意味する。この相同性は、mBLASTアルゴリズム(Altschul et al. (1990) Proc. Natl. Acad. Sci. USA 87: 2264-8; Karlin and Altschul (1993) Proc. Natl. Acad. Sci. USA 90: 5873-7)によって決定することができる。また、該DNAは、生体内から単離した場合、配列番号:1に記載のDNAとストリンジェントな条件下でハイブリダイズすると考えられる。ここで「ストリンジェントな条件」としては、例えば「2×SSC、0.1%SDS、50℃」、「2×SSC、0.1%SDS、42℃」、「1×SSC、0.1%SDS、37℃」、よりストリンジェントな条件として「2×SSC、0.1%SDS、65℃」、「0.5×SSC、0.1%SDS、42℃」および「0.2×SSC、0.1%SDS、65℃」の条件を挙げることができる。当業者においては、他の貝類における本発明の遺伝子に相当する内在性の遺伝子を、本発明の遺伝子の塩基配列を基に適宜取得することが可能である。なお、本明細書においては、チョウセンハマグリ以外の貝類におけるCARP-1タンパク質(遺伝子)に相当するタンパク質(遺伝子)、または、該タンパク質と機能的に同等なタンパク質(遺伝子)を、単に「本発明のタンパク質(遺伝子)」と記載する場合がある。
【0033】
本発明のタンパク質は、天然のタンパク質のほか、遺伝子組み換え技術を利用した組換えタンパク質として調製することができる。天然のタンパク質は、例えば本発明のタンパク質が発現していると考えられる貝類由来の細胞(組織)の抽出液に対し、本発明のタンパク質に対する抗体を用いたアフィニティークロマトグラフィーを用いる方法により調製することが可能である。一方、組換えタンパク質は、本発明のタンパク質をコードするDNAで形質転換した細胞を培養することにより調製することが可能である。
【0034】
本発明の好ましい態様としては、下記(a)〜(d)のいずれかに記載のポリヌクレオチドであって、ADPリボシル化活性を有するポリペプチドをコードするポリヌクレオチドを提供する。
(a)配列番号:2に記載のアミノ酸配列からなるポリペプチドをコードするポリヌクレオチド
(b)配列番号:1に記載の塩基配列のコード領域を含むポリヌクレオチド
(c)配列番号:2に記載のアミノ酸配列において、1もしくは複数のアミノ酸が置換、欠失、挿入、および/または付加したアミノ酸配列からなるポリペプチドをコードするポリヌクレオチド
(d)配列番号:1に記載の塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズするポリヌクレオチド
【0035】
また、上記ポリヌクレオチドによってコードされるポリペプチド(タンパク質)もまた、本発明に含まれる。
【0036】
本発明における「ポリヌクレオチド」には、「DNA」および該DNAと相同もしくは相補的な配列からなる「RNA」が含まれる。従って本発明の「ポリヌクレオチド」には、例えば、本発明のタンパク質をコードする遺伝子DNA、該DNAからの転写産物であるRNAが含まれる。また、本発明の「ポリヌクレオチド」には、一本鎖DNAもしくは一本鎖RNA、および二本鎖DNAもしくは二本鎖RNA、並びに、DNAとRNAとのハイブリッド分子が含まれる。
【0037】
本明細書におけるポリペプチドとは、複数のアミノ酸からなる重合体を意味し、そのアミノ酸の長さは特に制限されない。従って、本発明のタンパク質には、所謂「ペプチド」、「オリゴペプチド」、「タンパク質」等も含まれる。本発明のポリペプチドは、天然に存在する状態から修飾されていないもの、および修飾されているものの双方を含む。修飾としては、アセチル化、アシル化、ADP-リボシル化、アミド化、フラビンの共有結合、ヘム部分の共有結合、ヌクレオチドまたはヌクレオチド誘導体の共有結合、脂質または脂質誘導体の共有結合、ホスファチジルイノシトールの共有結合、架橋、環化、ジスルフィド結合の形成、脱メチル化、共有架橋の形成、シスチンの形成、ピログルタメートの形成、ホルミル化、γ-カルボキシル化、グリコシル化、GPIアンカー形成、ヒドロキシル化、ヨウ素化、メチル化、ミリストイル化、酸化、タンパク質分解処理、リン酸化、プレニル化、ラセミ化、セレノイル化、硫酸化、アルギニル化のようなタンパク質へのアミノ酸の転移RNA媒介付加、ユビキチン化等が含まれる。
【0038】
本発明のポリペプチドは、そのアミノ酸配列に従って、一般的な化学合成法により製造することが可能であり、該方法には、通常の液相法および固相法によるペプチド合成法が包含される。かかるペプチド合成法は、より詳しくはアミノ酸配列の情報に基づいて、各アミノ酸を1個ずつ逐次合成させて鎖を延長していくステップワイズエロンゲーション法と、アミノ酸数個からなるフラグメントを予め合成し、次いで各フラグメントをカップリング反応させるフラグメント・コンデンセーション法を包含し、本発明のポリペプチドの合成は、いずれの方法を用いてもよい。
【0039】
このようなペプチド合成法にて用いられる縮合法も、各種方法に従って行うことができる。その具体例としては、例えばアジド法、混合酸無水物法、DCC法、活性エステル法、酸化還元法、DPPA(ジフェニルホスホリルアジド)法、ウッドワード法等を例示できる。
【0040】
これら各種方法に利用できる溶媒もまた、一般的に使用されるものを適宜利用することができる。その例としては、例えばジメチルホルムアミド(DMF)、ジメチルスルホキシド(DMSO)、ヘキサホスホロアミド、ジオキサン、テトラヒドロフラン(THF)、酢酸エチル等及びこれらの混合溶媒等を挙げることができる。なお、上記ペプチド合成反応に際して、反応に関与しないアミノ酸およびペプチドにおけるカルボキシル基は、一般にはエステル化により、例えばメチルエステル、エチルエステル、第三級ブチルエステル等の低級アルキルエステル、例えばベンジルエステル、P−メトキシベンジルエステル、P−ニトロベンジルエステルアラルキルエステル等として保護することができる。また、側鎖に官能基を有するアミノ酸、例えばTyrの水酸基は、アセチル基、ベンジル基、ベンジルオキシカルボニル基、第三級ブチル基等で保護されてもよいが、必ずしもかかる保護は必須ではない。また、例えば、Argのグアニジノ基は、ニトロ基、トシル基、2−メトキシベンゼンスルホニル基、メチシレン−2−スルホニル基、ベンジルオキシカルボニル基、イソボルニルオキシカルボニル基、アダマンチルオキシカルボニル基等の適当な保護基により保護することができる。
【0041】
上記のようにして得ることが可能な本発明のポリペプチドは、通常の方法に従って、例えばイオン交換樹脂、分配クロマトグラフィー、ゲルクロマトグラフィー、アフィニティークロマトグラフィー、高速液体クロマトグラフィー(HPLC)、向流分配法等のペプチド化学の分野で汎用されている方法に従って、適宜、精製を行うことができる。
本発明のポリペプチドは、例えば、配列番号:2に記載のポリペプチド、または配列番号:1に記載のDNA核酸分子を合成し、次いで適当な発現ベクターへ導入した後、宿主細胞内において発現させる遺伝子工学的手法によっても取得することができる。
【0042】
本発明のポリペプチドには、例えば、本発明のタンパク質(例えば、CARP-1タンパク質)と機能的に同等なタンパク質(ポリペプチド)が含まれる。ここで「機能的に同等」とは、対象となるポリペプチドが本発明のタンパク質と同様の(同等の)生物学的あるいは生化学的機能(活性)を有することを指す。このような機能としては、例えば、DNAを基質としたADPリボシル化活性を挙げることができる。
【0043】
あるポリペプチドが本発明のADPリボシル化活性を有するポリペプチドであるか否かは、例えば、後述の実施例に記載された迅速高感度検出法、あるいは、DNA付加体構造解析等によって適宜評価することができる。DNA付加体構造解析は、通常、多量のポリペプチドとヌクレアーゼを要するため、迅速高感度検出法を用いて実施することが好ましいが、いずれの方法によっても好適に実施することができる。
【0044】
一例を示せば、まず、適切な緩衝液及び塩の存在下(例えば50 mM トリス塩酸pH 7.5、1 mMエチレンジアミン四酢酸、50 mM 塩化ナトリウム、1 mM ジチオスレイトール)にて、仔牛胸腺DNA、beta-NAD(微量の32P-NADを含む)と、当該ペプチドを混合し、通常摂氏37度、1時間インキュベートする。インキュベート後、フェノール処理及びクロロホルム抽出によりペプチドを除去し、酢酸アンモニウム塩を用いたアルコール沈澱によって未反応のbeta-NADを除去しつつDNAを回収する(Sambrook, J et al., Molecular Cloning, 2nd ed. 等に記載される常法)。回収したDNAをマイクロコッカルヌクレアーゼおよびホスホジエステラーゼIIを用いて3'-リン酸型デオキシリボヌクレオチドに分解し(非特許文献11)、PEI-セルロース薄層クロマトグラフィーシートを用いて展開する。6 mol/l 酢酸、0.1 mol/l 塩化リチウム、3 mol/l 尿素で展開し、オートラジオグラフィー(X線フィルムよりも富士フィルムのBASシステムの方が、高感度で望ましい)で検出する。ADP-リボシル化デオキシリボヌクレオチド3'-リン酸は他の核酸やNADとは異なり、殆ど展開されずRf値0.05付近に留まる事から検出することができる。
【0045】
なお、本発明において「ADPリボシル化活性」は、ADPリボシルトランスフェラーゼ活性とも表現される。本発明のADPリボシル化活性は、好ましくは、DNAを基質とするものであり、より具体的には、DNAのグアニン残基のN2位のアミノ基をADPリボシル化する活性を指す。
【0046】
本発明のタンパク質を基に改変されたポリペプチド(タンパク質)について、本発明のタンパク質と同等の生物学的あるいは生化学的な機能(活性)を保持しているか否かは、例えば、上述のADPリボシル化活性についての評価方法以外に、例えば、対象となる(被検)ポリペプチドを癌細胞へ導入し、アポトーシスの誘導の有無を観察することによっても評価することが可能である。
【0047】
あるポリペプチドと機能的に同等なポリペプチドを調製するための、当業者によく知られた方法としては、例えばポリペプチド中のアミノ酸配列に変異を導入する方法が挙げられる。具体的には当業者であれば部位特異的変異誘発法(Hashimoto-Gotoh, T. et al. (1995) Gene 152, 271-275、Zoller, MJ, and Smith, M.(1983) Methods Enzymol. 100, 468-500、Kramer, W. et al. (1984) Nucleic Acids Res. 12, 9441-9456、Kramer W, and Fritz HJ(1987) Methods. Enzymol. 154, 350-367、Kunkel,TA(1985) Proc Natl Acad Sci USA. 82, 488-492、Kunkel (1988) Methods Enzymol. 85, 2763-2766)などを用いて、配列番号:2に記載のアミノ酸配列に適宜変異を導入することにより、該ポリペプチドと機能的に同等なポリペプチドを調製することができる。また、ポリペプチド中のアミノ酸の変異は自然に生じることもある。このように、人工的か自然に生じたものかを問わず、本発明者らにより同定されたタンパク質(配列番号:2)のアミノ酸配列において1もしくは複数のアミノ酸配列が変異したアミノ酸配列を有し、該ポリペプチドと機能的に同等なポリペプチドは、本発明のタンパク質(ポリペプチド)に含まれる。
【0048】
上記変異体における、変異するアミノ酸数は、本発明のポリペプチドの有する機能が保持される限り制限はないが、通常15アミノ酸以内であり、好ましくは10アミノ酸以内であり、より好ましくは5アミノ酸以内であり、さらに好ましくは1〜4アミノ酸である。
【0049】
変異するアミノ酸残基としては、アミノ酸側鎖の性質が保存されている別のアミノ酸に変異されることが望ましい。例えばアミノ酸側鎖の性質としては、疎水性アミノ酸(A、I、L、M、F、P、W、Y、V)、親水性アミノ酸(R、D、N、C、E、Q、G、H、K、S、T)、脂肪族側鎖を有するアミノ酸(G、A、V、L、I、P)、水酸基含有側鎖を有するアミノ酸(S、T、Y)、硫黄原子含有側鎖を有するアミノ酸(C、M)、カルボン酸及びアミド含有側鎖を有するアミノ酸(D、N、E、Q)、塩基含有側鎖を有するアミノ離(R、K、H)、芳香族含有側鎖を有するアミノ酸(H、F、Y、W)を挙げることができる(括弧内はいずれもアミノ酸の一文字表記を表す)。
【0050】
あるアミノ酸配列に対する1または複数個のアミノ酸残基の欠失、付加及び/又は他のアミノ酸による置換により修飾されたアミノ酸配列を有するポリペプチドがその生物学的機能(活性)を維持し得ることはすでに知られている(Mark, D. F. et al., Proc. Natl. Acad. Sci. USA (1984) 81, 5662-5666 、Zoller, M. J. & Smith, M. Nucleic Acids Research (1982) 10, 6487-6500 、Wang, A. et al., Science 224, 1431-1433 、Dalbadie-McFarland, G. et al., Proc. Natl. Acad. Sci. USA (1982) 79, 6409-6413)。
【0051】
具体的なアミノ酸配列(例えば、配列番号:2)が開示された場合においては、当業者であれば、これらアミノ酸配列を基に、適宜アミノ酸が改変された配列からなるポリペプチドを作製し、当該ポリペプチドについて、上述の機能を有するか否かを評価し、本発明のタンパク質(ポリペプチド)を適宜選択することが可能である。
【0052】
本発明者らは、CARP-1タンパク質においてADPリボシル化に必須と考えられるグルタミン酸を見出した。該グルタミン酸は、CARP-1タンパク質(配列番号:2)のアミノ酸配列において128位のグルタミン酸に相当する。従って、本発明におけるアミノ酸残基の改変は、このグルタミン酸以外の部位についての改変であることが好ましい。即ち本発明の好ましい態様においては、配列番号:2に記載のアミノ酸配列において、128位のグルタミン酸以外の部位における1もしくは複数のアミノ酸が、置換、欠失、挿入、および/または付加されたアミノ酸配列からなるポリペプチドをコードするポリヌクレオチドである。
【0053】
本発明のCARP-1タンパク質には、コレラ毒素等の立体構造より導かれた保存性の高いアミノ酸が存在する。例えば、該アミノ酸としては、図4に示されるアミノ酸配列において12位のアルギニン(活性に必須の水素結合を提供)が挙げられる。また、47〜49位のセリン-スレオニン-スレオニンからなる配列も活性中心の空間の保持に重要な役割を果たしているものと考えられる。従って、本発明のポリペプチドは、これらのアミノ酸残基を含むことが好ましい。
【0054】
また、本発明において改変されるポリペプチド領域は、必ずしも制限されるものではないが、例えば、上記のアミノ酸以外の領域、即ち、配列番号:2に記載のアミノ酸配列の12位のアルギニン、または、47〜49位のセリン-スレオニン-スレオニン配列以外の領域であることが好ましい。これらの領域の改変であれば、依然としてCARP-1タンパク質の機能が保持されている可能性が高い。
【0055】
本発明のポリペプチドのアミノ酸配列に複数個のアミノ酸残基が付加されたポリペプチドには、これらポリペプチドを含む融合ポリペプチドが含まれる。融合ポリペプチドは、これらポリペプチドと他のペプチド又はポリペプチドとが融合したものである。融合ポリペプチドを作製する方法は、本発明のポリペプチド(例えば、配列番号:2)をコードするDNA(例えば、配列番号:1)と他のペプチド又はポリペプチドをコードするDNAをフレームが一致するように連結してこれを発現ベクターに導入し、宿主で発現させればよく、当業者に公知の手法を用いることができる。本発明のポリペプチドとの融合に付される他のペプチド又はポリペプチドは、特に制限されない。
【0056】
本発明のポリペプチドとの融合に付される他のペプチドとしては、例えば、GST(グルタチオン−S−トランスフェラーゼ)、イムノグロブリン定常領域、β−ガラクトシダーゼ、MBP(マルトース結合タンパク質)等が挙げられる。市販されているこれらペプチドまたはポリペプチドをコードするポリヌクレオチドを本発明のポリペプチドをコードするポリヌクレオチドと融合させ、これにより調製された融合ポリヌクレオチドを発現させることにより、融合ポリペプチドを調製することができる。
【0057】
また、本発明のポリペプチドの好ましい態様としては、本発明のポリペプチドもしくはその部分ポリペプチドと、細胞レセプターを認識するタンパク質とが融合した構造のポリペプチドが挙げられる。
【0058】
例えば、癌細胞表面に特異的に提示されているレセプターを認識するタンパク質と、本発明のポリペプチドとのキメラ酵素は、癌細胞に対して選択的なアポトーシス誘導作用を有することが期待される。
【0059】
上記「レセプターを認識するタンパク質」として具体的には、以下のタンパク質を例示することができるが、これらのタンパク質に限定されない。
(a)ヒト子宮頸癌培養細胞HeLa等に提示されたglobotriaosylceramide (Gb3)、または、globotetraosylceramide (Gb4)を認識するピエリシン-1タンパク質C末端側ドメイン(Kanazawa, T., et. Al., (2001) Proc. Natl. Acad. Sci. USA, 98, 2226-2231; Matsushima-Hibiya, Y., et. Al., (2003) J. Biol. Chem., 278, 9972-9978)
(b)腸管上皮細胞に提示されたganglioside GM1を認識する、コレラ毒素Bサブユニット
【0060】
また、癌細胞表面に特異的に結合する抗体を作製し、該抗体分子の可変領域重鎖および軽鎖が本発明のポリペプチドと融合した構造のポリペプチドを調製することも可能である。融合タンパク質による臨床応用については、公知文献(永田諭志、「蛋白質核酸酵素」、46巻4号、540-546頁、平成13年)を参照することができる。
【0061】
またあるポリペプチドと機能的に同等なポリペプチドを調製する当業者によく知られた他の方法としては、ハイブリダイゼーション技術(Sambrook,J et al., Molecular Cloning 2nd ed., 9.47-9.58, Cold Spring Harbor Lab. press, 1989)を利用する方法が挙げられる。即ち、当業者であれば、本発明のポリペプチドをコードするDNA(例えば、配列番号:1に記載の塩基配列)もしくはその一部をもとに、同種または異種生物由来のDNA試料から、これと相同性の高いDNAを単離して、該DNAから本発明のポリペプチドと機能的に同等なポリペプチドを単離することも通常行いうることである。
【0062】
本発明には、本発明のポリペプチドをコードするDNAとハイブリダイズするDNAによってコードされるポリペプチドであって、本発明のポリペプチドと機能的に同等なポリペプチドが含まれる。このようなポリペプチドとしては、例えば、ハマグリあるいはその他の貝類に内在するADPリボシル化活性を有するタンパク質(例えば、CARP-1のホモログ等)が挙げられる。
【0063】
本発明のポリペプチドと機能的に同等なポリペプチドをコードするDNAを単離するためのハイブリダイゼーションの条件は、当業者であれば適宜選択することができる。ハイブリダイゼーションの条件としては、例えば、低ストリンジェントな条件が挙げられる。低ストリンジェントな条件とは、ハイブリダイゼーション後の洗浄において、例えば42℃、0.1×SSC、0.1%SDSの条件であり、好ましくは50℃、0.1×SSC、0.1%SDSの条件である。より好ましいハイブリダイゼーションの条件としては、高ストリンジェントな条件が挙げられる。高ストリンジェントな条件とは、例えば65℃、5×SSC及び0.1%SDSの条件である。これらの条件において、温度を上げる程に高い相同性を有するDNAが効率的に得られることが期待できる。但し、ハイブリダイゼーションのストリンジェンシーに影響する要素としては温度や塩濃度など複数の要素が考えられ、当業者であればこれら要素を適宜選択することで同様のストリンジェンシーを実現することが可能である。
【0064】
また、ハイブリダイゼーションにかえて、遺伝子増幅技術(PCR)(Current protocols in Molecular Biology edit. Ausubel et al. (1987) Publish. John Wiley&Sons Section 6.1-6.4)を用いて、本発明のポリペプチドをコードするDNA(例えば、配列番号:2または5)の一部を基にプライマーを設計し、本発明者らにより同定されたタンパク質をコードするDNAと相同性の高いDNA断片を単離し、該DNAを基に本発明者らにより同定されたタンパク質と機能的に同等なタンパク質を取得することも可能である。
【0065】
本発明のポリペプチドは「成熟」ポリペプチドの形であっても、融合ポリペプチドのような、より大きいポリペプチドの一部であってもよい。本発明のポリペプチドには、リーダー配列、プロ配列、多重ヒスチジン残基のような精製に役立つ配列、または組換え生産の際の安定性を確保する付加的配列などが含まれていてもよい。
【0066】
これらハイブリダイゼーション技術や遺伝子増幅技術により単離されるDNAによってコードされる、本発明のポリペプチドと機能的に同等なポリペプチドは、通常、本発明のポリペプチド(例えば、配列番号:2)とアミノ酸配列において高い相同性を有する。本発明のポリペプチドには、本発明のタンパク質と機能的に同等であり、かつ該タンパク質のアミノ酸配列と高い相同性を有するポリペプチドも含まれる。高い相同性とは、アミノ酸レベルにおいて、通常、少なくとも50%以上の同一性、好ましくは75%以上の同一性、さらに好ましくは85%以上の同一性、さらに好ましくは95%以上(例えば、96%以上、97%以上、98%以上、99%以上)の同一性を指す。ポリペプチドの相同性を決定するには、文献(Wilbur, W. J. and Lipman, D. J. Proc. Natl. Acad. Sci. USA (1983) 80, 726-730)に記載のアルゴリズムに従えばよい。
【0067】
アミノ酸配列の同一性は、例えば、Karlin and Altschul によるアルゴリズムBLAST (Proc. Natl. Acad. Sci. USA 87:2264-2268, 1990、Proc. Natl. Acad. Sci. USA 90:5873-5877, 1993)によって決定することができる。このアルゴリズムに基づいて、BLASTXと呼ばれるプログラムが開発されている(Altschul et al. J. Mol. Biol.215: 403-410, 1990)。BLASTXによってアミノ酸配列を解析する場合には、パラメーターは例えば、score = 50、wordlength = 3とする。BLASTとGapped BLASTプログラムを用いる場合には、各プログラムのデフォルトパラメーターを用いる。これらの解析方法の具体的な手法は公知である(http://www.ncbi.nlm.nih.gov.)。
【0068】
本発明のポリペプチドは、当業者に公知の方法により、組み換えポリペプチドとして、また天然のポリペプチドとして調製することが可能である。組み換えポリペプチドであれば、例えば、本発明のポリペプチドをコードするDNA(例えば、配列番号:1に記載のDNA)を、適当な発現ベクターに組み込み、これを適当な宿主細胞に導入して得た形質転換体を回収し、抽出物を得た後、イオン交換、逆相、ゲル濾過などのクロマトグラフィー、あるいは本発明のポリペプチドに対する抗体をカラムに固定したアフィニティークロマトグラフィーにかけることにより、または、さらにこれらのカラムを複数組み合わせることにより精製し、調製することが可能である。
【0069】
また、本発明のポリペプチドをグルタチオンS-トランスフェラーゼタンパク質との融合ポリペプチドとして、あるいはヒスチジンを複数付加させた組み換えポリペプチドとして宿主細胞(例えば、動物細胞や微生物細胞等)内で発現させた場合には、発現させた組み換えポリペプチドはグルタチオンカラムあるいはニッケルカラムを用いて精製することができる。融合ポリペプチドの精製後、必要に応じて融合ポリペプチドのうち、目的のポリペプチド以外の領域を、トロンビンまたはファクターXaなどにより切断し、除去することも可能である。
【0070】
天然のタンパク質であれば、当業者に周知の方法、例えば本発明のポリペプチドを発現している組織や細胞の抽出物に対し、本発明のポリペプチドと親和性を有する抗体が結合したアフィニティーカラムを作用させて精製することにより単離することができる。抗体はポリクローナル抗体であっても、モノクローナル抗体であってもよい。
【0071】
また、本発明のポリペプチドは例えば、本発明のポリペプチドを認識する抗体の作製等に利用することが可能である。
【0072】
上述のようにして取得される組換えポリペプチドを用いれば、これに結合する抗体を調製することができる。例えば、ポリクローナル抗体は、精製した本発明のポリペプチドもしくはその一部のペプチドをウサギなどの免疫動物に免疫し、一定期間の後に血液を採取し、血ぺいを除去することにより調製することが可能である。また、モノクローナル抗体は、上記ポリペプチド若しくはペプチドで免疫した動物の抗体産生細胞と骨腫瘍細胞とを融合させ、目的とする抗体を産生する単一クローンの細胞(ハイブリドーマ)を単離し、該細胞から抗体を得ることにより調製することができる。これにより得られた抗体は、本発明のポリペプチドの精製や検出などに利用することが可能である。本発明には、本発明のポリペプチドに結合する抗体が含まれる。これらの抗体を用いることにより、所望の生物もしくは細胞が本発明のポリペプチドを発現するか否かの判別を行うことが可能である。本発明のポリペプチドに結合する抗体もまた本発明に含まれる。
【0073】
本発明のポリヌクレオチドは、本発明のポリペプチドをコードし得るものであればいかなる形態でもよい。即ち、mRNAから合成されたcDNAであるか、ゲノムDNAであるか、化学合成DNAであるかなどを問わない。また、本発明のポリペプチドをコードしうる限り、遺伝暗号の縮重に基づく任意の塩基配列を有するDNAが含まれる。
【0074】
本発明のポリヌクレオチドは、当業者に公知の方法により調製することができる。例えば、本発明のポリペプチドを発現している細胞よりcDNAライブラリーを作製し、本発明のDNA(例えば、配列番号:1に記載の塩基配列)の一部をプローブとしてハイブリダイゼーションを行うことにより調製できる。cDNAライブラリーは、例えば、文献(Sambrook, J. et al., Molecular Cloning、Cold Spring Harbor Laboratory Press (1989))に記載の方法により調製してもよく、あるいは市販のDNAライブラリーを用いてもよい。また、本発明のポリペプチドを発現している細胞よりRNAを調製し、逆転写酵素によりcDNAを合成後、本発明のDNA(例えば、配列番号:1に記載の塩基配列)に基づいてオリゴDNAを合成し、これをプライマーとして用いてPCR反応を行い、本発明のポリペプチドをコードするcDNAを増幅させることにより調製することも可能である。
【0075】
また、得られたcDNAの塩基配列を決定することにより、それがコードする翻訳領域を決定でき、本発明のポリペプチドのアミノ酸配列を得ることができる。また、得られたcDNAをプローブとしてゲノムDNAライブラリーをスクリーニングすることにより、ゲノムDNAを単離することができる。
【0076】
具体的には、次のようにすればよい。まず、本発明のポリペプチドを発現する細胞、組織、器官からmRNAを単離する。mRNAの単離は、公知の方法、例えば、グアニジン超遠心法(Chirgwin, J. M. et al., Biochemistry (1979) 18, 5294-5299)、AGPC法(Chomczynski, P. and Sacchi, N., Anal. Biochem. (1987) 162, 156-159)等により全RNAを調製し、mRNA Purification Kit (Pharmacia) 等を使用して全RNAからmRNAを精製する。また、QuickPrep mRNA Purification Kit (Pharmacia) を用いることによりmRNAを直接調製することもできる。
【0077】
得られたmRNAから逆転写酵素を用いてcDNAを合成する。cDNAの合成は、AMV Reverse Transcriptase First-strand cDNA Synthesis Kit (生化学工業)等を用いて行うこともできる。また、本明細書に記載されたプライマー等を用いて、5'-Ampli FINDER RACE Kit (Clontech製)およびポリメラーゼ連鎖反応 (polymerase chain reaction ; PCR)を用いた5'-RACE法(Frohman, M. A. et al., Proc. Natl. Acad. Sci. U.S.A. (1988) 85, 8998-9002 ; Belyavsky, A. et al., Nucleic Acids Res. (1989) 17, 2919-2932) に従い、cDNAの合成および増幅を行うことができる。得られたPCR産物から目的とするDNA断片を調製し、ベクターDNAと連結する。さらに、これより組換えベクターを作製し、大腸菌等に導入してコロニーを選択して所望の組換えベクターを調製する。目的とするDNAの塩基配列は、公知の方法、例えば、ジデオキシヌクレオチドチェインターミネーション法により確認することができる。
【0078】
また、本発明のDNAの作成においては、発現に使用する宿主のコドン使用頻度を考慮し、より発現効率の高い塩基配列を設計することができる(Grantham, R. et al., Nucelic Acids Research (1981) 9, r43-74)。また、本発明のDNAは、市販のキットや公知の方法によって改変することができる。改変としては、例えば、制限酵素による消化、合成オリゴヌクレオチドや適当なDNAフラグメントの挿入、リンカーの付加、開始コドン(ATG)及び/又は終止コドン(TAA、TGA、又はTAG)の挿入等が挙げられる。
【0079】
本発明のポリヌクレオチドを増幅もしくは検出するためのプライマーおよびプローブもまた、本発明に含まれる。
即ち本発明の好ましい態様としては、以下のポリヌクレオチドを提供する。
(a)本発明のポリヌクレオチドと特異的にハイブリダイズするポリヌクレオチドであって、少なくとも15ヌクレオチドの鎖長を持つポリヌクレオチド
(b)本発明のポリヌクレオチドまたはその一部に対するアンチセンスポリヌクレオチド
【0080】
また本発明は、本発明のポリヌクレオチド、または、本発明のポリペプチドをコードするポリヌクレオチドが挿入されたベクターを提供する。本発明のベクターとしては、組み換えタンパク質の生産に用いるベクターの他、形質転換体作製のために各種細胞内で本発明のポリヌクレオチドを発現させるためのベクターも含まれる。
【0081】
さらに本発明は、本発明の上記ベクターを保持する宿主細胞、および該宿主細胞を利用した本発明のポリペプチドの製造方法を提供する。
【0082】
本発明のベクターとしては、挿入したDNAを安定に保持するものであれば特に制限されず、例えば宿主に大腸菌を用いるのであれば、クローニング用ベクターとしてはpBluescriptベクター(Stratagene社製)などが好ましい。本発明のポリペプチドを生産する目的においてベクターを用いる場合には、特に発現ベクターが有用である。発現ベクターとしては、試験管内、大腸菌内、培養細胞内、生物個体内でポリペプチドを発現するベクターであれば特に制限されないが、例えば、試験管内発現であればpBESTベクター(プロメガ社製)、大腸菌であればpETベクター(Invitrogen社製)、培養細胞であればpME18S-FL3ベクター、生物個体であればpME18Sベクター(Mol Cell Biol. 8:466-472(1988))などが好ましい。ベクターへの本発明のDNAの挿入は、常法により、例えば、制限酵素サイトを用いたリガーゼ反応により行うことができる(Current protocols in Molecular Biology edit. Ausubel et al. (1987) Publish. John Wiley & Sons.Section 11.4-11.11)。
【0083】
本発明のベクターが導入される宿主細胞としては特に制限はなく、目的に応じて種々の宿主細胞が用いられる。ポリペプチドを発現させるための細胞としては、例えば、細菌細胞(例:ストレプトコッカス、スタフィロコッカス、大腸菌、ストレプトミセス、枯草菌)、真菌細胞(例:酵母、アスペルギルス)、昆虫細胞(例:ドロソフィラS2、スポドプテラSF9)、動物細胞(例:CHO、COS、HeLa、C127、3T3、BHK、HEK293、Bowes メラノーマ細胞)および植物細胞を例示することができる。宿主細胞へのベクター導入は、例えば、リン酸カルシウム沈殿法、電気パルス穿孔法(Current protocols in Molecular Biology edit. Ausubel et al. (1987) Publish. John Wiley & Sons.Section 9.1-9.9)、リポフェクタミン法(GIBCO-BRL社製)、マイクロインジェクション法などの公知の方法で行うことが可能である。
【0084】
宿主細胞において発現したポリペプチドを小胞体の内腔に、細胞周辺腔に、または細胞外の環境に分泌させるために、適当な分泌シグナルを目的のポリペプチドに組み込むことができる。これらのシグナルは目的のポリペプチドに対して内因性であっても、異種シグナルであってもよい。
【0085】
本発明のポリペプチドの回収は、本発明のポリペプチドが培地に分泌される場合は、培地を回収する。本発明のポリペプチドが細胞内に産生される場合は、その細胞をまず溶解し、その後にポリペプチドを回収する。
【0086】
組換え細胞培養物から本発明のポリペプチドを回収し精製するには、硫酸アンモニウムまたはエタノール沈殿、酸抽出、アニオンまたはカチオン交換クロマトグラフィー、ホスホセルロースクロマトグラフィー、疎水性相互作用クロマトグラフィー、アフィニティークロマトグラフィー、ヒドロキシルアパタイトクロマトグラフィーおよびレクチンクロマトグラフィーを含めた公知の方法を用いることができる。
【0087】
また本発明は、本発明のポリペプチド、または、本発明のベクターを有効成分として含む、アポトーシス誘導剤を提供する。
【0088】
上記ベクターとしては、通常、本発明のポリペプチドをコードするポリペプチドを担持するプラスミドもしくはウイルスベクターが一般的である。
【0089】
当業者においては、所望のポリヌクレオチドを有するベクターを、一般的な遺伝子工学技術によって、適宜、作製することが可能である。通常、市販の種々のベクターを利用することができる。
【0090】
本発明のベクターは、宿主細胞内において本発明のポリヌクレオチドを保持したり、本発明のポリペプチドを発現させるためにも有用である。
【0091】
本発明におけるポリヌクレオチドは、通常、適当なベクターへ担持(挿入)され、宿主細胞へ導入される。該ベクターとしては、挿入したポリヌクレオチドを安定に保持するものであれば特に制限されず、例えば宿主に大腸菌を用いるのであれば、クローニング用ベクターとしてpBluescriptベクター(Stratagene社製)などが好ましいが、市販の種々のベクターを利用することができる。本発明のポリペプチドを生産する目的としてベクターを用いる場合には、特に発現ベクターが有用である。発現ベクターとしては、試験管内、大腸菌内、培養細胞内、生物個体内でポリペプチドを発現するベクターであれば特に制限されないが、例えば、試験管内発現であればpBESTベクター(プロメガ社製)、大腸菌であればpETベクター(Invitrogen社製)、培養細胞であればpME18S-FL3ベクター、生物個体であればpME18Sベクター(Mol Cell Biol. 8:466-472(1988))などを例示することができる。ベクターへの本発明のポリヌクレオチドの挿入は、常法により、例えば、制限酵素サイトを用いたリガーゼ反応により行うことができる。
【0092】
上記宿主細胞としては特に制限はなく、目的に応じて種々の宿主細胞が用いられる。ポリペプチドを発現させるための細胞としては、例えば、細菌細胞(例:ストレプトコッカス、スタフィロコッカス、大腸菌、ストレプトミセス、枯草菌)、昆虫細胞(例:ドロソフィラS2、スポドプテラSF9)、動物細胞(例:CHO、COS、HeLa、C127、3T3、BHK、HEK293、Bowes メラノーマ細胞)および植物細胞を例示することができる。宿主細胞へのベクター導入は、例えば、リン酸カルシウム沈殿法、電気パルス穿孔法(Current protocols in Molecular Biology edit. Ausubel et al. (1987) Publish. John Wiley & Sons.Section 9.1-9.9)、リポフェクション法、マイクロインジェクション法などの公知の方法で行うことが可能である。
【0093】
宿主細胞において発現したポリペプチドを小胞体の内腔に、細胞周辺腔に、または細胞外の環境に分泌させるために、適当な分泌シグナルを目的のポリペプチドに組み込むことができる。これらのシグナルは目的のポリペプチドに対して内在性であっても、異種シグナルであってもよい。
【0094】
上記製造方法におけるポリペプチドの回収は、本発明のポリペプチドが培地に分泌される場合は、培地を回収する。本発明のポリペプチドが細胞内に産生される場合は、その細胞をまず溶解し、その後にポリペプチドを回収する。
【0095】
組換え細胞培養物から本発明のポリペプチドを回収し精製するには、硫酸アンモニウムまたはエタノール沈殿、酸抽出、アニオンまたはカチオン交換クロマトグラフィー、ホスホセルロースクロマトグラフィー、疎水性相互作用クロマトグラフィー、アフィニティクロマトグラフィー、ヒドロキシルアパタイトクロマトグラフィーおよびレクチンクロマトグラフィーを含めた公知の方法を用いることができる。
【0096】
また、動物の生体内で本発明のポリヌクレオチド(DNA)を発現させる方法としては、本発明のDNAを適当なベクターに組み込み、例えば、レトロウィルス法、リポソーム法、カチオニックリポソーム法、アデノウィルス法などにより生体内に導入する方法などが挙げられる。これにより、癌治療を行うことが可能である。用いられるベクターとしては、例えば、アデノウイルスベクターやレトロウイルスベクターなどが挙げられるが、これらに制限されない。ベクターへの本発明のDNAの挿入などの一般的な遺伝子操作は、常法に従って行うことが可能である(Molecular Cloning ,5.61-5.63)。生体内への投与は、ex vivo法であっても、in vivo法であってもよい。
【0097】
本発明のポリペプチドもしくはベクターを用いて細胞をアポトーシス誘導する方法もまた本発明に含まれる。該方法の好ましい態様においては、以下の工程(a)または(b)を含む方法である。
(a)本発明のポリペプチド、または本発明のベクターを細胞へ導入する工程
(b)本発明のポリペプチドと細胞とを接触させる工程
【0098】
上記工程(b)におけるポリペプチドは、細胞レセプターを認識するタンパク質との融合ポリペプチド(キメラ酵素)であることが好ましい。
【0099】
本発明のアポトーシス誘導剤は、癌細胞に対してアポトーシスを誘導することにより、癌細胞の増殖を抑制(阻止)もしくは癌細胞を死滅させる作用を有する。従って、本発明は、本発明のアポトーシス誘導剤を有効成分とする、抗癌剤(抗腫瘍剤)を提供する。
【0100】
本発明のタンパク質(ポリペプチド)がDNAのグアニン残基をADPリボシル化することにより、癌細胞のアポトーシスあるいは癌細胞における遺伝子の突然変異が誘発され、その結果、癌の発生または増殖を抑制するものと考えられる。
【0101】
本発明の上記アポトーシス誘導剤もしくは抗癌剤が適用可能な細胞の生物類は特に制限されないが、例えば、ヒト、マウス、ラット、イヌ等を挙げることができる。
【0102】
本発明の「抗癌剤」は、「癌細胞増殖抑制剤」、「癌細胞発生抑制剤」、「腫瘍増殖抑制剤」、「癌治療剤」、あるいは「制癌剤」等と表現することも可能である。また、本発明の薬剤は、「医薬品」、「医薬組成物」、「治療用医薬」等と表現することもできる。
【0103】
なお、本発明における「治療」には、癌の発生を予め抑制し得る予防的な効果も含まれる。また、癌細胞(組織)に対して、必ずしも、完全な治療効果を有する場合に限定されず、部分的な効果もしくは改善効果を有する場合であってもよい。
【0104】
また、食用貝の持つ毒性の原因は、本発明のタンパク質(例えば、CARP-1)の有するADPリボシル化活性に起因するものと考えられる。従って、本発明のタンパク質の発現を抑制する物質、または本発明のタンパク質の有するADPリボシル化活性を阻害する物質を貝類に施すことにより、毒性が除去された貝類を取得することが可能である。
【0105】
従って本発明は、本発明のタンパク質の発現もしくは機能阻害物質を有効成分とする、貝類毒性除去剤を提供する。
【0106】
上記発現阻害物質としては、本発明の遺伝子からの転写、または該転写による転写産物からの翻訳を阻害する物質が含まれる。
【0107】
上記発現阻害物質としては例えば、本発明のポリペプチドをコードする遺伝子の発現を抑制するポリヌクレオチドが挙げられる。このようなポリヌクレオチドには、アンチセンスポリヌクレオチド(アンチセンスDNA/RNA;本発明のポリペプチドをコードする遺伝子の転写産物と相補的なアンチセンスRNA、および該RNAをコードするDNA)やリボザイム(本発明のポリペプチドをコードする遺伝子の転写産物を特異的に開裂するリボザイム活性を有するRNAをコードするDNA)、およびRNAi効果を有する核酸(siRNA)が含まれる。
【0108】
アンチセンスポリヌクレオチドが標的遺伝子の発現を抑制する作用としては、以下のような複数の要因が存在する。すなわち、三重鎖形成による転写開始阻害、RNAポリメラーゼによって局部的に開状ループ構造がつくられた部位とのハイブリッド形成による転写抑制、合成の進みつつあるRNAとのハイブリッド形成による転写阻害、イントロンとエキソンとの接合点でのハイブリッド形成によるスプライシング抑制、スプライソソーム形成部位とのハイブリッド形成によるスプライシング抑制、mRNAとのハイブリッド形成による核から細胞質への移行抑制、キャッピング部位やポリ(A)付加部位とのハイブリッド形成によるスプライシング抑制、翻訳開始因子結合部位とのハイブリッド形成による翻訳開始抑制、開始コドン近傍のリボソーム結合部位とのハイブリッド形成による翻訳抑制、mRNAの翻訳領域やポリソーム結合部位とのハイブリッド形成によるペプチド鎖の伸長阻止、および核酸とタンパク質との相互作用部位とのハイブリッド形成による遺伝子発現抑制などである。これらは、転写、スプライシング、または翻訳の過程を阻害して、標的遺伝子の発現を抑制する(平島および井上「新生化学実験講座2 核酸IV 遺伝子の複製と発現」,日本生化学会編,東京化学同人,pp.319-347,1993)。
【0109】
本発明で用いられるアンチセンス核酸(ポリヌクレオチド)は、上記のいずれの作用で標的遺伝子の発現を抑制してもよい。一つの態様としては、遺伝子のmRNAの5'端近傍の非翻訳領域に相補的なアンチセンス配列を設計すれば、遺伝子の翻訳阻害に効果的と考えられる。しかし、コード領域もしくは3'側の非翻訳領域に相補的な配列も使用し得る。このように、遺伝子の翻訳領域だけでなく非翻訳領域の配列のアンチセンス配列を含むポリヌクレオチドも、本発明で利用されるアンチセンスポリヌクレオチドに含まれる。使用されるアンチセンスポリヌクレオチドは、適当なプロモーターの下流に連結され、好ましくは3'側に転写終結シグナルを含む配列が連結される。アンチセンスポリヌクレオチド配列は、標的遺伝子またはその一部と相補的な配列であることが好ましいが、遺伝子の発現を有効に阻害できる限り、完全に相補的でなくてもよい。転写されたRNAは、標的とする遺伝子の転写産物に対して好ましくは90%以上、最も好ましくは95%以上の相補性を有する。アンチセンス配列を用いて、効果的に標的遺伝子の発現を阻害するには、アンチセンスポリヌクレオチドは、アンチセンス効果を引き起こすために、少なくとも15ヌクレオチド以上、好ましくは100ヌクレオチド、さらに好ましくは500ヌクレオチド以上の鎖長を有し、通常、3000ヌクレオチド以内、好ましくは2000ヌクレオチド以内の鎖長を有する。
【0110】
該アンチセンスポリヌクレオチドは、例えば、本発明のポリペプチドをコードするポリヌクレオチド(例えば、配列番号:1)の配列情報を基にホスホロチオネート法(Stein, 1988 Physicochemical properties of phosphorothioate oligodeoxynucleotides. Nucleic Acids Res 16, 3209-21 (1988))などにより調製することが可能である。
【0111】
内在性遺伝子の発現の抑制は、また、リボザイムをコードする核酸(ポリヌクレオチド)を利用して行うことも可能である。リボザイムとは触媒活性を有するRNA分子のことをいう。リボザイムには種々の活性を有するものがあるが、中でもRNAを切断する酵素としてのリボザイムの研究により、RNAの部位特異的な切断を目的とするリボザイムの設計が可能となった。リボザイムには、グループIイントロン型や、RNasePに含まれるM1RNAのように400ヌクレオチド以上の大きさのものもあるが、ハンマーヘッド型やヘアピン型と呼ばれる40ヌクレオチド程度の活性ドメインを有するものもある(小泉誠および大塚栄子, (1990) 蛋白質核酸酵素,35:2191)。
【0112】
例えば、ハンマーヘッド型リボザイムの自己切断ドメインは、G13U14C15のC15の3'側を切断するが、活性にはU14が9位のAと塩基対を形成することが重要とされ、15位の塩基はCの他にAまたはUでも切断されることが示されている(M.Koizumiら,(1988) FEBS Lett.228:225)。リボザイムの基質結合部を標的部位近傍のRNA配列と相補的になるように設計すれば、標的RNA中のUC、UUまたはUAという配列を認識する制限酵素的なRNA切断リボザイムを作出することが可能である(M.Koizumiら,(1988) FEBS Lett. 239:285、小泉誠および大塚栄子,(1990) 蛋白質核酸酵素,35:2191、 M.Koizumiら, (1989) Nucleic Acids Res. 17:7059)。
【0113】
また、ヘアピン型リボザイムも、本発明の目的のために有用である。ヘアピン型リボザイムは、例えばタバコリングスポットウイルスのサテライトRNAのマイナス鎖に見出される(J.M.Buzayan Nature 323:349,1986)。このリボザイムも、標的特異的なRNA切断を起こすように設計できることが示されている(Y.Kikuchi およびN.Sasaki (1992) Nucleic Acids Res. 19:6751、 菊池洋, (1992) 化学と生物 30:112)。
【0114】
また、本発明の上記siRNAの好ましい態様としては、本発明の遺伝子に対してRNAi(RNA interference;RNA干渉)効果を有する二本鎖RNA(siRNA)を挙げることができる。より具体的には、配列番号:1に記載の塩基配列の部分配列に対するセンスRNAおよびアンチセンスRNAからなる二本鎖RNA(siRNA)を挙げることができる。
【0115】
さらに、本発明の上記RNAを発現し得るDNA(ベクター)もまた、本発明の遺伝子の発現を抑制し得る化合物の好ましい態様に含まれる。例えば、本発明の上記二本鎖RNAを発現し得るDNA(ベクター)は、該二本鎖RNAの一方の鎖をコードするDNA、および該二本鎖RNAの他方の鎖をコードするDNAが、それぞれ発現し得るようにプロモーターと連結した構造を有するDNAである。本発明の上記DNAは、当業者においては、一般的な遺伝子工学技術により、適宜作製することができる。より具体的には、本発明のRNAをコードするDNAを公知の種々の発現ベクターへ適宜挿入することによって、本発明の発現ベクターを作製することが可能である。
【0116】
また、本発明のタンパク質のADPリボシル化酵素活性を阻害する活性を有する化合物には、貝類毒性除去剤としての新たな用途があることが、本発明者によって見出された。従って、本発明は、ADPリボシル化酵素活性を阻害する活性を有する化合物を有効成分として含む、貝類毒性除去剤を提供する。
【0117】
ベンズアミド、3-アミノベンズアミド、5-アミノ-2,3-ジヒドロ-1,4-フタルアジンジオンは、DNA ADPリボシル化酵素ピエリシン-1および-2の活性も阻害することが知られている(非特許文献8、13)ことから、これらの化合物は、本発明の貝類毒性除去剤として有用である。
【0118】
また、ガングリオシドG(Q1bα)にはNADの立体構造と類似する部分が有るため百日咳毒素の酵素活性を阻害すると報告されており(横山三紀、実験医学, 14 (2), 409-41 1, 1996)、本発明のタンパク質の機能を阻害するものと考えられる。従って、本発明の好ましい態様としては、ベンズアミド、3-アミノベンズアミド、5-アミノ-2,3-ジヒドロ-1,4-フタルアジンジオン、および、ガングリオシドG(Q1bα)からなる群より選択される少なくとも1以上の化合物を有効成分として含む、貝類毒性除去剤を提供する。
【0119】
また、本発明のポリペプチドは、ADPリボシル化活性を有することから、ADPリボシル化のための研究用試薬としても有用である。従って本発明は、本発明のポリペプチドを有効成分として含むADPリボシル化試薬を提供する。
【0120】
さらに、本発明のポリペプチドもしくは上記ADPリボシル化試薬と、ポリヌクレオチド(DNA)とを接触させる工程を含む、該ポリヌクレオチドをADPリボシル化する方法もまた本発明に含まれる。
【0121】
本発明の方法によってADPリボシル化されたグアニン残基(N2-ADPリボシル化dG付加体)は、この部位でポリメラーゼの伸長反応を停止させる。その結果、該部位において突然変異が起こるものと考えられる。即ち、本発明の方法によってADPリボシル化されたグアニン残基を有するプラスミドを、通常のDNA修復システムを有する細胞へ導入することにより、DNAに対して突然変異を導入させることができる(Shiotani, B., et. Al., (2005) Mutat. Res. 512, 150-155.)。従って、本発明の方法は、DNAに対して突然変異を導入するために利用することも可能である。
【0122】
本発明の薬剤は、生理学的に許容される担体、賦形剤、あるいは希釈剤等と混合し、医薬組成物として経口、あるいは非経口的に投与することができる。経口剤としては、顆粒剤、散剤、錠剤、カプセル剤、溶剤、乳剤、あるいは懸濁剤等の剤型とすることができる。非経口剤としては、注射剤、点滴剤、外用薬剤、あるいは座剤等の剤型を選択することができる。注射剤には、皮下注射剤、筋肉注射剤、あるいは腹腔内注射剤等を示すことができる。外用薬剤には、経鼻投与剤、あるいは軟膏剤等を示すことができる。主成分である本発明の薬剤を含むように、上記の剤型とする製剤技術は公知である。
【0123】
例えば、経口投与用の錠剤は、本発明の薬剤に賦形剤、崩壊剤、結合剤、および滑沢剤等を加えて混合し、圧縮整形することにより製造することができる。賦形剤には、乳糖、デンプン、あるいはマンニトール等が一般に用いられる。崩壊剤としては、炭酸カルシウムやカルボキシメチルセルロースカルシウム等が一般に用いられる。結合剤には、アラビアゴム、カルボキシメチルセルロース、あるいはポリビニルピロリドンが用いられる。滑沢剤としては、タルクやステアリン酸マグネシウム等が公知である。
【0124】
本発明の薬剤を含む錠剤は、マスキングや、腸溶性製剤とするために、公知のコーティングを施すことができる。コーティング剤には、エチルセルロースやポリオキシエチレングリコール等を用いることができる。
【0125】
また注射剤は、主成分である本発明の薬剤を適当な分散剤とともに溶解、分散媒に溶解、あるいは分散させることにより得ることができる。分散媒の選択により、水性溶剤と油性溶剤のいずれの剤型とすることもできる。水性溶剤とするには、蒸留水、生理食塩水、あるいはリンゲル液等を分散媒とする。油性溶剤では、各種植物油やプロピレングリコール等を分散媒に利用する。このとき、必要に応じてパラベン等の保存剤を添加することもできる。また注射剤中には、塩化ナトリウムやブドウ糖等の公知の等張化剤を加えることができる。更に、塩化ベンザルコニウムや塩酸プロカインのような無痛化剤を添加することができる。
【0126】
また、本発明の薬剤を固形、液状、あるいは半固形状の組成物とすることにより外用剤とすることができる。固形、あるいは液状の組成物については、先に述べたものと同様の組成物とすることで外用剤とすることができる。半固形状の組成物は、適当な溶剤に必要に応じて増粘剤を加えて調製することができる。溶剤には、水、エチルアルコール、あるいはポリエチレングリコール等を用いることができる。増粘剤には、一般にベントナイト、ポリビニルアルコール、アクリル酸、メタクリル酸、あるいはポリビニルピロリドン等が用いられる。この組成物には、塩化ベンザルコニウム等の保存剤を加えることができる。また、担体としてカカオ脂のような油性基材、あるいはセルロース誘導体のような水性ゲル基材を組み合わせることにより、座剤とすることもできる。
【0127】
本発明の薬剤を遺伝子治療剤として使用する場合は、本発明の薬剤を注射により直接投与する方法のほか、核酸が組込まれたベクターを投与する方法が挙げられる。上記ベクターとしては、アデノウイルスベクター、アデノ随伴ウイルスベクター、ヘルペスウイルスベクター、ワクシニアウイルスベクター、レトロウイルスベクター、レンチウイルスベクター等が挙げられ、これらのウイルスベクターを用いることにより効率よく投与することができる。
【0128】
また、本発明の薬剤をリポソームなどのリン脂質小胞体に導入し、その小胞体を投与することも可能である。例えば、本発明のポリペプチドもしくはベクターを保持させた小胞体をリポフェクション法により所定の細胞に導入する。そして、得られる細胞を例えば静脈内、動脈内等に全身投与する。癌組織等に局所的に投与することもできる。
【0129】
本発明の薬剤は、安全とされている投与量の範囲内において、ヒトを含む哺乳動物に対して、必要量(有効量)が投与される。本発明の薬剤の投与量は、剤型の種類、投与方法、患者の年齢や体重、患者の症状等を考慮して、最終的には医師または獣医師の判断により適宜決定することができる。
【0130】
また本発明は、本発明のアポトーシス誘導剤もしくは抗癌剤、または該薬剤の候補化合物のスクリーニング方法を提供する。
【0131】
本発明のスクリーニング方法の好ましい態様においては、本発明のタンパク質(例えば、CARP-1タンパク質)の発現量または活性を指標とする方法である。即ち、本発明のタンパク質の発現もしくは活性を上昇させる化合物は、アポトーシス誘導作用を有し、抗癌活性を有することが期待される。
【0132】
本発明の上記方法においては、まず、本発明のタンパク質もしくは本発明のタンパク質を発現する細胞と、被検化合物を接触させる。
【0133】
本方法に用いる「細胞」は、特に制限されないが、好ましくは貝類由来の細胞である。「本発明のタンパク質を発現する細胞」としては、内在性の本発明のタンパク質を発現している細胞、または外来性の本発明の遺伝子が導入され、該遺伝子が発現している細胞を利用することができる。外来性の本発明の遺伝子が発現した細胞は、通常、本発明の遺伝子が挿入された発現ベクターを宿主細胞へ導入することにより作製することができる。該発現ベクターは、一般的な遺伝子工学技術によって作製することができる。
【0134】
本発明のスクリーニング方法に供する被検化合物としては、特に制限はない。例えば、天然化合物、有機化合物、無機化合物、タンパク質、ペプチドなどの単一化合物、並びに、化合物ライブラリー、遺伝子ライブラリーの発現産物、細胞抽出物、細胞培養上清、発酵微生物産生物、海洋生物抽出物、植物抽出物等が挙げられるが、これらに限定されない。
【0135】
また、これらの被検化合物は必要に応じて適宜標識して用いることができる。標識としては、例えば、放射標識、蛍光標識等を挙げることができる。
【0136】
本発明のタンパク質を発現する細胞への被検化合物の「接触」は、通常、本発明のタンパク質を発現する細胞の培養液に被検化合物を添加することによって行うが、この方法に限定されない。被検化合物がタンパク質等の場合には、該タンパク質を発現するDNAベクターを、該細胞へ導入することにより、「接触」を行うことができる。
【0137】
本方法においては、次いで、本発明のタンパク質を発現する細胞における本発明のタンパク質の発現量または活性を測定する。発現量の測定は、当業者が簡便に行い得るものであり、一般的な方法、例えばノーザンブロット法、ウェスタンブロット法等により適宜実施することが可能である。例えば、本発明のタンパク質を発現する細胞に接触させた被検化合物に付した標識を指標にして測定することも可能である。
【0138】
尚、本発明において「発現」とは、タンパク質をコードする遺伝子からの転写(mRNAの生成)、または該遺伝子の転写産物からの翻訳のいずれの場合をも意味する。
【0139】
また、本発明のタンパク質の活性を指標にして上記スクリーニング方法を実施する場合には、本発明のタンパク質の上述の機能(活性)、例えば、本発明のタンパク質の有するADPリボシル化活性を指標とすることができる。本発明のタンパク質の活性の測定は、当業者においては、公知の方法によって適宜実施することができる。
【0140】
本方法においては、次いで、被検化合物を接触させない場合と比較して、本発明のタンパク質の発現量または活性を上昇させる化合物を選択する。
【0141】
上記スクリーニング方法によって取得される化合物は、例えば、アポトーシス誘導剤、または抗癌剤となることが期待される。
【0142】
また、本発明のタンパク質(例えば、CARP-1タンパク質)の発現量または活性を低下させる化合物は、貝類毒性除去作用を有することが期待される。
【0143】
従って本発明は、本発明のタンパク質の発現量または活性を低下させる化合物を選択することを特徴とする、貝類毒性除去剤のスクリーニング方法を提供する。
【0144】
上記スクリーニング方法の好ましい態様としては、以下の工程(a)〜(c)を含む方法である。
(a)本発明のタンパク質もしくは本発明のタンパク質を発現する細胞と、被検化合物を接触させる工程
(b)本発明のタンパク質の発現量または活性を測定する工程
(c)被検化合物を接触させない場合と比較して、前記発現量または活性を低下させる化合物を選択する工程
【実施例】
【0145】
以下本発明を実施例により詳細に説明するが、本発明はこれら実施例により制限されるものではない。なお、本実験に際し、下記の材料・試薬等を用いた。
【0146】
日本の太平洋沿岸から採取された貝類は全て、東京の市場から得て、生きたまま研究室に輸送された。mRNA抽出用途の場合には、輸送されたチョウセンハマグリを滅菌人工海水中で生きたまま保存し、1時間以内に用いた。ホスホジエステラーゼIIおよびミクロコッカスヌクレアーゼは、Worthington(米国ニュージャージー州レイクウッド)から購入し、仔ウシ胸腺DNA、細菌アルカリホスファターゼ、およびβ-NADはSigma(米国ミズーリ州セントルイス)から購入し、β-[アデニレート-32P]NAD(32P-NAD)は、PerkinElmer(米国マサチューセッツ州ボストン)から購入した。ExTaqポリメラーゼは、タカラバイオ(滋賀県大津市)から購入した。KOD-Plusポリメラーゼは東洋紡(大阪市)から購入した。トリプシンおよびオリゴヌクレオチドは、Invitrogen(米国カリフォルニア州カールスバッド)から、CEL 300 PEIポリエチレンイミン-セルロースTLCシートは、Macherey-Nagel(ドイツ、デューレン)から購入した。トリプシン処置ピエリシン-1は、既報(Watanabeら、2004a)に従い調製した。特に明記していない限り、他の材料は全て和光純薬工業(大阪市)から購入した。
【0147】
〔実施例1〕種々の貝類からの抽出物におけるDNA ADP-リボシルトランスフェラーゼのスクリーニング
14種類の貝類の各標本の全軟組織を乳鉢と乳棒によって氷上で、1 mM DTTを含む50 mMトリス-塩酸、pH 7.5の3倍量と共にホモジナイズした後、1分間超音波処理した。このホモジネートを14,000 x gで5分間遠心して、粗抽出物と呼ばれる上清を-80℃で保存した。仔ウシ胸腺DNA 5μg、18.5 kBq 32P-NADおよび10μM β-NADを、反応緩衝液(50 mMトリス-塩酸、pH 7.5、1 mM EDTA、50 mM NaCl、および1 mM DTT)50μlにおいて、当初の軟組織0.03〜1.5 mgに対応する貝類からの粗抽出物1〜50μgタンパク質と共にインキュベートした。プロテナーゼK(和光)1μgを反応物に加えて、37℃でさらに30分間インキュベートした。フェノール抽出、クロロホルム抽出、および酢酸アンモニウム塩によるエタノール沈殿によってDNAを回収した。回収したDNAを、5 mM CaCl2、ミクロコッカスヌクレアーゼ2 U、およびホスホジエステラーゼII 0.02 Uを含む20 mMビストリス-塩酸(pH 6.5)において10μlの容量で処理した。バックグラウンドのノイズを検出するために、DNA混合物3μlをPEI-セルロースTLCシート上に直ちにスポットした。DNA混合物の残りを、37℃で12時間インキュベートすることによってデオキシリボヌクレオチド3'リン酸まで消化して、混合物の3μlをTLCシートにスポットした。6 M酢酸、0.1 M LiClおよび3 M尿素を用いて展開した後、TLCプレートをフジイメージングプレート(富士写真フィルム、東京)に露出して、バイオイメージアナライザ(BAS-2500;富士写真フィルム)によってDNA付加物を検出した。
【0148】
検出された付加物のスポットの放射活性のデータを表1に示し、ADP-リボシル化DNA付加物の形成の典型的な例を図1に示す。
【0149】
【表1】

【0150】
上記表1における相対活性(PSL)は、タンパク質マイクログラム当たりのDNA付加物の比重分析によって計算したものである。PSL(photo stimulated luminescence)はBio Image Analyzer用の放射活性の単位である。ピエリジン-1の相対活性は約10 PSL/pg タンパク質であった。腹足類動物(Gastropods)については短剣印で示す。
【0151】
チョウセンハマグリ(M. lamarckii)、アサリ(R. philippinarum)およびヤマトシジミ(C. japonica)からの粗抽出物をDNAおよび32P-NADと反応させると、付加物のスポットはTLCシート上で明らかに検出され、1790〜6900 PSL/μgタンパク質を示し、スポットのRf値は0.05であり、これはピエリシン-1によって産生された付加物のRf値と同じであった。他のデオキシリボヌクレオチド、ADP-リボースおよびNADのRf値は0.30〜0.95であった。これらの粗抽出物を、DNAおよび32P-NADとの反応の前に、50μgプロナーゼまたは50μgプロテナーゼKによって処置すると、付加物のスポットは検出されず、これらの付加物が酵素反応の結果であったことを示唆している。
【0152】
マテガイ(Solen strictus)、ウバガイ(Spisula sachalinensis)、およびムラサキイガイ(Mytilus galloprovincialis)からの粗抽出物の場合、付加物のスポットも同様に、同じRf値で検出された。これらのPSL値は、189〜450/μgタンパク質であった。エゾバイ(Buccinum middendorffi)、マガキ(Crassostrea gigas)、およびタイラギ(Atrina pinnata)からの粗抽出物の場合、同じRf値でかすかな付加物スポットを生じた。サザエ(Turbo cornutus)、バカガイ(Mactra chinensis、hen clam)およびミルクイ(Tresus keenae)をタンパク質源として用いた場合には、検出可能なシグナルは出現しなかった。
【0153】
〔実施例2〕二枚貝の酵素の存在下でのDNAおよびβ-NADのインキュベーションによって形成された反応産物の分析
実施例1のように、チョウセンハマグリからの抽出物試料は最高の活性を示し、TLCによって検出されたデオキシリボヌクレオチド付加物を生じたことから、以下に示すように生成された付加物を、細菌のアルカリホスファターゼによって脱リン酸化後にHPLCによってさらに分析した。
【0154】
詳しくは、チョウセンハマグリ、アサリおよびヤマトシジミからの粗抽出物に硫酸アンモニウムを60〜90%飽和となるように加えて、遠心した。50 mMトリス-塩酸、pH 7.5に対して透析した後、硫酸アンモニウム沈殿試料10 mgを仔ウシ胸腺DNA 1 mgおよびβ-NAD 20μmolと共に50 mMトリス-塩酸、pH 7.5(アサリに関してはpH 8.5)、1 mM EDTA、および50 mM NaClにおいて全量10 mlにおいて37℃で4時間インキュベートした。DNAを回収して、ミクロコッカスヌクレアーゼ200 U、ホスホジエステラーゼII 2 U、および5 mM CaCl2によって、20 mMビストリス-塩酸(pH 6.5)2 mlにおいて37℃で4時間、デオキシリボヌクレオチド3'リン酸まで消化した。細菌アルカリホスファターゼ(最終濃度、1.5 U/ml)、トリス塩基(40 mM)およびZnCl2(5 mM)を加えて、37℃で4時間インキュベートすることによって、試料をデオキシリボヌクレオシドまでさらに消化した。
【0155】
遠心後、反応混合物の少量に、Shimadzu SPD 10Avp光ダイオードアレイ検出器およびDevelosil RPAQUEOUSカラム(4.6×250 mm;野村化学、愛知県瀬戸市)を備えたShimadzu LC-10Aシステム(島津製作所、京都市)によって、HPLCを40℃で流速1 ml/分で行った。溶媒系は、以下の通りであった:0.25%トリエチルアミン-酢酸(pH 7.0)中で3.5%アセトニトリルを0〜15分、その後0.25%トリエチルアミン-酢酸(pH 7.0)においてアセトニトリル16.8%まで直線勾配で45分。
【0156】
反応混合物の少量を、HP1000 HPLCシステム(Hewlett-Packard、米国カリフォルニア州パロアルト)を備えたMicromass ZQ 2000機器(英国、マンチェスター)エレクトロスプレーイオン化質量分析(ESI-MS)によっても分析した。
【0157】
その結果、図2Aに示すように、dGピークの高さは減少し、そのかわりに二つのピークが保持時間21.5および22.5分で検出され、これは、ピエリシン-1によって形成されたN2-(ADP-リボス-1-イル)-2'-デオキシグアノシンのα体およびβ体の保持時間と一致した。一方、dC、dA、およびTの量は不変のままであった。保持時間21.5および22.5分でのピーク分画のUVスペクトルは、N2-(ADP-リボース-1-イル)-2'-デオキシグアノシンの保持時間とまさに同じであった(図2B)。さらに、LC-ESI-MS分析から、これらのピーク分画における化合物が、m/z 809で分子イオンピークを有すること、デオキシリボース部分の喪失によって生じるm/z693でイオンピークを有すること、およびHPLC溶出剤に由来するトリエチルアミン付加に対応するm/z 910、およびm/z 809で親質量に対応するイオンピークを有することを示した(図6AB)。
【0158】
このように、N2-(ADP-リボース-1-イル)-2'-デオキシグアノシンは、チョウセンハマグリの酵素の存在下で仔ウシ胸腺DNAおよびβ-NADの反応を通して産生されることが証明された。その上、N2-(ADP-リボス-1-イル)-2'-デオキシグアノシンは、アサリおよびヤマトシジミからの抽出物試料によって類似の条件で形成されることが示された。
【0159】
〔実施例3〕チョウセンハマグリからのDNA ADP-リボシルトランスフェラーゼの精製
チョウセンハマグリ(鹿島産)標本5個の、かなり低い酵素活性を有する鰓、外套膜および中腸を除く軟組織(150 g)から調製した粗抽出物を、上述したように硫酸アンモニウム沈殿によって分画した。
【0160】
その結果、DNA付加物形成活性のほとんどは、硫酸アンモニウム60〜90%飽和で得られた沈殿物において検出された。この分画の付加物の形成活性は37℃でpH 7.5〜8.5でほぼ同じレベルで認められた。活性は、4時間まで時間依存的に増加した。
【0161】
次に、活性分画をCM52カラムにおいて精製した。即ち、沈殿物を20 mMリン酸ナトリウム緩衝液、pH 6.0に対して透析して、CM52カルボキシメチル-セルロースカラム(25×150 mm、Whatman、イギリス、ケント)に適用した。カラムを20 mMリン酸ナトリウム緩衝液によって溶出して、保持されたタンパク質を20 mMリン酸ナトリウム緩衝液において0〜400 mM NaClの直線勾配1000 mlによって流速80 ml/時間で溶出した。
【0162】
その結果、開始材料の酵素活性の15%が、20 mMリン酸ナトリウム緩衝液によって溶出し、活性の残りの43%はその後20 mMリン酸ナトリウム緩衝液において0〜400 mM NaClによって回収された。20 mMリン酸ナトリウム緩衝液において0〜400 mM NaClで溶出された酵素活性を含む分画(286〜319 ml)を合わせて、Amicon Ultra-10k(Millipore、ベッドフォード、マサチューセッツ州)によって濃縮して、HiPrep Sephacryl S-100ゲル濾過カラム(16×600 mm、Pharmacia、スウェーデン、ウプサラ)に適用しさらに精製した。タンパク質を、150 mM NaClを含む20 mMリン酸ナトリウム緩衝液によって、流速30 ml/時間で分離して、酵素活性を含む分画(64〜74 ml)を合わせて、Amicon Ultra-10kによって濃縮した後、Mono-S HR 5/5(Pharmacia)を備えたFPLCを流速1 ml/分で行った。溶媒システムは以下の通りであった:0.25 M NaClから1 Mまでの直線勾配を20 mMリン酸ナトリウム緩衝液、pH 6.0において42.5分間。活性分画(12〜13 ml)をプールして、Amicon Ultra-10kによって0.2 mlまで濃縮した。上記の技法は全て4℃未満で行い、精製酵素を-80℃で保存した。
【0163】
DNA ADP-リボシルトランスフェラーゼ活性の同定および精製酵素の分子量の推定のために、既に記述されたとおりにゲル内酵素アッセイを行った(Watanabe, M., Enomoto, S., Takamura-Enya, T., Nakano, T., Koyama, K., Sugimura, T. and Wakabayashi, K. (2004a) Enzymatic properties of pierisin-1 and its N-terminal domain, a guanine-specific ADP-ribosyltransferase from the cabbage butterfly. J. Biochem., 135, 471-477.)。精製タンパク質の電気穿孔は、既に記述されたとおりに行った(Kanazawa, T., Watanabe, M., Matsushima-Hibiya, Y., Kono, T., Tanaka, N., Koyama, K., Sugimura, T. and Wakabayashi, K. (2001) Distinct roles for the N- and C-terminal regions in the cytotoxicity of pierisin-1, a putative ADP-ribosylating toxin from cabbage butterfly, against mammalian cells. Proc. Natl. Acad. Sci. USA, 98, 2226-2231.)。
【0164】
その結果、精製タンパク質のSDS-PAGEおよびゲル内酵素活性アッセイによって、分子量約20 kDaに対応する単一の主なバンドが明らかとなった(図3)。SephacrylおよびMono-Sカラムクロマトグラフィーの段階において、付加物形成活性は、チョウセンハマグリの20 kDaタンパク質を含む分画に限って認められ、他の分画では認められなかった。ハマグリ5個から、精製タンパク質0.48 mgが得られ、付加物形成活性は、抽出物と比較して870倍上昇した。その上、精製酵素によって形成された付加物は、HPLC、UVスペクトル、およびLC-MS分析によってN2-(ADP-リボス-1-イル)-2’-デオキシグアノシンであることが確認された。これらのデータは、精製された酵素がハマグリ抽出物に存在する主要な活性成分であることを示している。
【0165】
ハマグリは一般的にDNA ADP-リボシルトランスフェラーゼを含む可能性が最も高いことから、本発明者らはチョウセンハマグリから精製された本発明の酵素をCARP-1と命名した。
【0166】
なお、以下の表2は、チョウセンハマグリのDNA ADP-リボシルトランスフェラーゼの精製段階を要約したものである。
【0167】
【表2】
【0168】
上記表2において、回収および精製の値は、32P-NADからDNAへ放射活性を組み入れる能力によって決定される酵素活性から算出した。
【0169】
〔実施例4〕ハマグリの酵素をコードするcDNAのPCR増幅、シークエンシング、およびクローニング
精製ハマグリ酵素20μgをトリプシンによって消化して、消化したペプチドを、TSKゲルODS-80Tsカラムを備えた逆相HPLC(東ソー、山口県周南市)によって分離した。単離されたペプチドのアミノ酸配列はProcise 494 HTタンパク質シークエンサー(Perkin-Elmer)によって決定した。総RNAを、Isogen(ニッポン・ジーン、富山県富山市)を用いてチョウセンハマグリの足筋から抽出した。チョウセンハマグリからのmRNAの完全長のcDNAを、GeneRacerキット(Invitrogen)を用いて総RNA調製物(2μg)から合成した。次に、合成されたcDNAに、ハマグリの酵素ペプチドHAAQAFYWLSVK(配列番号:3)およびEIHLAALTDTESSSEGYKENDYDVDT(配列番号:4)に対応する1μMセンスおよびアンチセンス縮重プライマー、またはセンス縮重プライマーおよび1μM 3'アンカープライマー(5'- CGCTACGTAACGGCATGACAGTG -3'/配列番号:5、GeneRacerキット(Invitrogen))を用いて、ExTaq DNAポリメラーゼによってハマグリの酵素特異的配列に関するPCR増幅を行った。5’末端cDNA断片を得るために、各1μMの5’アンカープライマー(5'- GGACACTGACATGGACTGAAGGAGTA -3'/配列番号:6、GeneRacerキット(Invitrogen))およびアンチセンスプライマー(5'- GCTTTTGTTCTCTTTTATCAGTGTATCT -3'/配列番号:7)を用いて、PCRを行った;配列は内部cDNA断片から選択した。得られた5’ cDNA断片にPCR直接シークエンシングを行った。完全長のハマグリ酵素cDNAを、5’アンカープライマーに隣接する配列である5’プライマー(5'- AGCGTTTACTTCCTCTTTCTCTTT -3'/配列番号:8)、およびポリ(A)伸長部に隣接する配列である3'プライマー(5'- GACTATCGTCGTTGTTTATTTTGA -3'/配列番号:9)を用いて、KOD-プラスDNAポリメラーゼによるPCR増幅によって得た。増幅された0.9 kb断片にPCR直接シークエンシングを行って、完全なcDNA配列を決定した。
【0170】
上述のようにチョウセンハマグリの足筋からのRNAおよび消化したペプチドの内部アミノ酸配列を用いて、本発明者らは完全なCARP-1をコードするcDNAを得た。単離されたcDNAのヌクレオチド配列を図4に示す。
【0171】
完全長のcDNAは、5’アンカー配列およびポリアデニル化伸長部を伴う853 bpからなった。最初のATGはヌクレオチド65位で始まり、ヌクレオチド611位のTAG終止コドンで終止した。ポリアデニル化シグナルの可能性がある配列(AATAAA)が、ポリ(A)配列に近いヌクレオチド834〜839位で認められ、真核細胞配列であることを示唆している。ORFによってコードされる推定のタンパク質は、計算分子量20,332を有するアミノ酸182個を含んだ。その上、精製ハマグリADP-リボシルトランスフェラーゼの消化物のペプチド配列9個は、ORFの推定アミノ酸配列の一部と同一であることが判明した(図4)。これらの結果から、単離されたcDNAにおけるORFは、CARP-1遺伝子のコード領域であると結論された。
【0172】
またクローンを構築するために、増幅された0.9 kb PCR断片を鋳型として用いた。PCR増幅CARP-1コード配列をpMAL-p2xプラスミドベクター(New England Biolabs、米国マサチューセッツ州ビバリー)に挿入した。ハマグリの酵素の遺伝子のコード領域を3'-隣接領域の217 bpと共に増幅して、プライマー対5'- CCTAAGCTTATGCCTGCTGGAAAATACCTATAC -3'(配列番号:10)および5'- GGAGAATTCGACTATCGTCGTTGTTTATTTTGA -3'(配列番号:11)を用いて、HindIIIおよびEcoRI制限部位をそれぞれ、5’および3’末端に導入した。ピエリシン-1および-2の場合からCARP-1遺伝子の産物は大腸菌に対して非常に毒性が強いことが予想されたことから、本発明者らは、PCR増幅配列をベクターのtacプロモーターとは反対方向にして、HindIII-EcoRI部位に挿入した。クローン10個をシークエンシングして、いずれのクローンも如何なるヌクレオチド置換も有しなかったことから、これをインビトロでCARP-1発現のために用いた。
【0173】
〔実施例5〕クローニングされたcDNAのインビトロ発現
酵素のcDNAのインビトロ発現は、既報に倣って実施した(Matsushima-Hibiya, Y., Watanabe, M., Kono, T., Kanazawa, T., Koyama, K., Sugimura, T. and Wakabayashi, K. (2000) Purification and cloning of pierisin-2, an apoptosis-inducing protein from the cabbage butterfly, Pieris brassicae. Eur. J. Biochem., 267, 5742-5750.)。簡単に説明すると、コード領域の5'末端にT7プロモーター配列を結合した5'プライマー(5'- TAATACGACTCACTATAGGGAGACCACCATGCCTGCTGGAAAATACCTATACCG -3'/配列番号:12)、および3'プライマー(5'- GACTATCGTCGTTGTTTATTTTGA -3'/配列番号:13)をPCRに用いて、酵素コード配列の上流にT7プロモーター配列を導入した。T7プロモーター配列を有するcDNAの増幅断片は、MEGAscript(Ambion、米国テキサス州オースチン)によって転写され、その後ウサギ網状赤血球溶解物によって翻訳された(Ambion)。
【0174】
インビトロ発現タンパク質の分子量はSDS-PAGEにおいて約20 kDaであり、精製された天然のCARP-1の分子量と同じであった。発現されたタンパク質をDNAおよび32P-NADと反応させた場合、付加物のスポットがTLCシートのRf値0.05において明らかに検出され、これは精製された天然のタンパク質によって産生された付加物のRf値と同じであった。その上、DNA付加物を細菌のアルカリホスファターゼによる脱リン酸化後にHPLCによってさらに分析すると、ほとんどの放射活性は保持時間21.0〜23.0分で回収され、N2-(ADP-リボス-1-イル)-2'-デオキシグアノシン(図7)の保持時間と一致した。このように、発現されたタンパク質は、明らかに、N2-(ADP-リボス-1-イル)-2'-デオキシグアノシンを産生した。この結果は、単離されたcDNAクローンから機能的に活性なタンパク質がインビトロで発現されたことを示している。
【0175】
精製天然タンパク質はHeLa細胞において20μg/mlで如何なる細胞障害作用も示さなかったが、タンパク質3、5および12.5μg/mlを電気穿孔によって導入した場合、細胞増殖速度はそれぞれ、77、58、および26%明らかに減少した。同様に、インビトロ発現CARP-1タンパク質は、10μg/mlでHeLa細胞に対して電気穿孔によって細胞障害性を示した。電気穿孔を行わない場合には細胞障害性を示さなかった。同様の結果はTMK-1細胞を用いても得られている。
【0176】
酵素とDDBJデータベースのタンパク質との全体的な相同性は、BLASTおよびFASTAの検索によって検出されなかったが、GLu-128、Arg-12、および46〜48位のSer-Thr-ThrがCARP-1遺伝子において検出され、これらはADP-リボシルトランスフェラーゼ活性のモチーフであると示唆されている(図5)。
【0177】
〔実施例6〕部位特異的変異誘発
さらに本発明者らは、ADP-リボシル化が、DNA付加物の形成に関与しているか否かを調べるために、以下に示すように127または128位のグルタミン酸残基を、部位特異的変異誘発によってアスパラギン酸(それぞれ、E127D、またはE128D)に置換して、変異したまたは変異していないクローンをインビトロで転写および翻訳した。
【0178】
即ち、所望の位置で変化した配列を含むDNA断片をオーバーラップ伸長PCR技術によって無傷の酵素cDNAサブクローンから増幅した。重なり合った5'および3'断片を得るために、5'断片に関してコード領域の5’末端にT7プロモーター配列を結合した5’プライマー(5'- TAATACGACTCACTATAGGGAGACCACCATGCCTGCTGGAAAATACCTATACCG -3'/配列番号:14)、および3'プライマー(プライマーA、5'- CCTTAACCAAAACGTCTTCATATT -3'/配列番号:15)、ならびに3'断片に関して5'プライマー(プライマーB、5'- AATATGAAGACGTTTTGGTTAAGG -3'/配列番号:16)および3'プライマー(5'- GACTATCGTCGTTGTTTATTTTGA -3'/配列番号:17)を用いて二つの異なるPCR反応を行った。プライマーAおよびBは互いに相補的であり、Glu128Aspに対応するヌクレオチドで変異を含む。Recochip(タカラバイオ)を用いて精製した後、精製5'-および3'-断片を互いに混合して、これを第二ラウンドのPCRの鋳型として用いて完全長の変異DNA断片を得た。このPCR産物をシークエンシングして、予定した変異の導入を確認した、Glu127Aspに対応するヌクレオチドで変異を含むDNA断片を得るために、プライマーAおよびBの配列がそれぞれ、5'- AACCAAAACTTCGTCATATTTTGC -3'(配列番号:18)および5'- GCAAAATATGACGAAGTTTTGGTT -3'(配列番号:19)であったことを除き、上記と同じプロトコールを用いた。得られたDNA断片を、上記のインビトロ発現系の鋳型として用いた。
【0179】
その結果、非変異対照クローンおよび変異クローンからのタンパク質発現効率は、[35S]メチオニンによる翻訳後にSDS-PAGEによって判断すると、ほぼ同じであった。E127D変異体は非変異DNAによる当初の活性の94%を維持したが、E128D変異体は当初の活性の99%を失った事から、Glu-128がDNA ADP-リボシルトランスフェラーゼ活性にとって必須のグルタミン酸であることが示唆された。
【図面の簡単な説明】
【0180】
【図1】図1は、様々な貝類の粗抽出物で形成されたADP-リボシル化DNA付加物の検出について示す写真である。仔ウシ胸腺DNAを粗抽出タンパク質1μg(グループ3-7)の存在下で32P-NADと反応させた。回収した後、ヌクレアーゼ分解の前(左レーン)および後(右レーン)にDNAサンプルをTLCシート上にスポットし、展開し、オートラジオグラフを行った。グループ1:タンパク質無し、グループ2:ピエリシン-1(10 pg:ポジティブコントロール)、グループ3:チョウセンハマグリ、グループ4:アサリ、グループ5:シジミ、グループ6:ムラサキイガイ。矢印はRf値0.05を示す。
【図2A】図2は、β-NADの存在下でチョウセンハマグリ由来の蛋白質精製物およびDNAのインキュベーションによって形成される反応物の分析について示すグラフである。(A)ピエリシン-1とインキュベートしたDNAの加水分解産物のHPLC溶出パターン(上段)、チョウセンハマグリ由来の蛋白質精製物とインキュベートしたDNAの加水分解産物のHPLC溶出パターン(中段)、あるいは蛋白質精製物無しでインキュベートしたDNAの加水分解産物のHPLC溶出パターン(下段)。サンプルはDevelosil RPAQUEOUSカラムに注入し、溶出液は262 nmでのUV吸光度を計測した。矢印は、N2-(ADP-リボス-1-イル)-2'-デオキシグアノシンに相当するピークを指す。
【図2B】図2Bは、図2Aに示す21.5および22.5分の保持時間で、ピーク分画における化合物について、フォトダイオードアレイ検出機を用いたUV吸収スペクトルを示すグラフである。
【図3】図3は、精製したCARP-1のSDS-PAGEの結果を示す写真である。精製したタンパク質1μgを15%ポリアクリルアミドゲルエレクトロポレーションによって分離し、クーマシーブリリアントブルーR-250で染色した(左レーン)。あるいはゲル内ADPリボシルトランスフェラーゼアッセイで評価した(右レーン)。
【図4】図4は、PCRダイレクトスクリーニングによって決定したCARP-1に対するcDNAのヌクレオチド配列および推定アミノ酸配列を示す図である。オープンリーディングフレームは65-613位にある。ポリアデニル化シグナルと予測される配列(834-839位)を、シャープ印によって示す。保存される可能性のあるアミノ酸は太字で示した。
【図5】図5は、ピエリシン-1およびADP-リボシル化トキシンと、CARP-1の相同性領域の推定アミノ酸配列をアライメントした図である。PT-1:PT(GenBank = P04977)、Pierisin-1(Watanabe, M., Kono, T., Matsushima-Hibiya, Y., Kanazawa, T., Nishisaka, N., Kishimoto, T., Koyama, K., Sugimura, T. and Wakabayashi, K. (1999) Molecular cloning of an apoptosis-inducing protein, pierisin, from cabbage butterfly: possible involvement of ADP-ribosylation in its activity. Proc. Natl. Acad. Sci. USA, 96, 10608-10613.)、CT-A:コレラトキシン(GenBank = 1001196A)のAサブユニット。保存されたアルギニン、Ser-Thr-Ser/Thrモチーフおよびグルタミン酸残基を枠で囲んだ。完全に保存されたアミノ酸にはアスタリスク(*)を付けた。
【図6A】図6は、β-NAD存在下において、DNAおよびチョウセンハマグリ由来の酵素のインキュベーションによって形成される反応物のLC-ESI-MS分析の結果を示す図である。(A)HPLCプロフィール(UV)、およびチョウセンハマグリ由来の酵素とインキュベートしたDNAの加水分解産物のイオンクロマトグラム(m/z 809、693および910)を示す。ピエリシン-1とインキュベーションすることによって形成される反応物の解析でも、同様の結果が得られた。
【図6B】図6Bは、図6Aに示される25.5分(上段)および26分(下段)の保持時間で、ピーク分画における化合物のマススペクトラムを示す。
【図7】図7は、32PNADの存在下において、DNAおよびインビトロ翻訳されたCARP-1のインキュベーションによって形成される反応物のHPLC溶出パターンを示す図である。放射活性を有する反応物は、β-NADの存在下においてチョウセンハマグリ由来の酵素とインキュベーションしたDNAと混合した。DNAは加水分解し、Develosil RPAQUEOUSカラムに注入した。溶出液については、262 nmでのUV吸光度を測定した(上段)。1分間隔で回収された各分画の放射活性は、Bio Imaging Analyzer(下段)により測定した。
【技術分野】
【0001】
本発明は、ADPリボシル化活性を有する貝類由来タンパク質、およびその用途に関する。
【背景技術】
【0002】
ADPリボシル化は、β-NADのADP-リボース部分がアクセプター分子に転移される翻訳後修飾であることが知られている。ADP-リボシルトランスフェラーゼは、その反応産物によって分類されている。モノADP-リボシルトランスフェラーゼは、単一のADP-リボースの標的分子への転移を触媒するのに対し、PARPsと呼ばれるポリ(ADP-リボース)ポリメラーゼは、最初にADP-リボースを核タンパク質に転移させた後、ADP-リボース残基の重合化を触媒する(非特許文献1および2参照)。
【0003】
モノADP-リボシルトランスフェラーゼは、細胞外感染症を引き起こす細菌において認められる。コレラ毒素(CT)および百日咳毒素(PT)はそれぞれ、G-タンパク質のGs-およびGi-型αサブユニットを標的とするのに対し、ジフテリア毒素(DT)は、延長因子-2(非特許文献3参照)のジフタミドを改変する。同様に、哺乳類および鳥類においてモノADP-リボシルトランスフェラーゼに関する報告がある。例えば、アルギニン特異的ADP-リボシルトランスフェラーゼは、グリコシルホスファチジルイノシトール(GPI)結合型または可溶性型として存在することが示されており、それらが様々な標的タンパク質を改変することが示唆されている(非特許文献2および4参照)。
【0004】
ピエリシン-1は当初、モンシロチョウの被蛹からの細胞障害性物質として同定された(非特許文献5〜7参照)。ピエリシン-1は、CTおよびPTのようなADP-リボシルトランスフェラーゼとそのN-末端領域において配列類似性を有し、C-末端領域においてリシンスーパーファミリーのレクチンドメインと配列類似性を有する(非特許文献8〜10参照)。他のADP-リボシルトランスフェラーゼとは異なり、ピエリシン-1は、DNAにおけるグアニン残基のN2アミノ基を標的としてN2-(ADP-リボス-1-イル)-2'-デオキシグアノシン(非特許文献11参照)を生成する。C-末端ドメインは、グロボトリアオシルセラミド(Gb3)およびグロボテトラオシルセラミド(Gb4)のようなグリコスフィンゴリピッド受容体に対する結合能を有し、哺乳類細胞表面でのその作用によってピエリシン-1を細胞に組み入れることに関与している(非特許文献9および10参照)。ピエリシン-1は、多様なヒト癌細胞株に対して細胞障害性であり、IC50値は、調べた細胞株13個において0.043〜270 ng/mlの範囲である(非特許文献9、12、13参照)。細胞におけるGb3およびGb4の量はその感度を大きく左右して(非特許文献10参照)、ピエリシン-1によって、Bcl-2およびカスパーゼを含むミトコンドリア経路によって、アポトーシスが誘導される(非特許文献14参照)。その上、CHL細胞におけるHPRT遺伝子の変異は、ピエリシン-1の低用量処置によって誘導することができる(非特許文献15参照)。興味深いことに、ピエリシン-1のmRNAは、後期幼虫において高度に発現されて、そのタンパク質は脂肪体に蓄積され、これは蛹化の際も持続し、このことはピエリシンがモンシロチョウにおける変態の際に機能する可能性があることを示唆している(非特許文献16参照)。同じシロチョウ属に属するオオモンシロチョウ(Pieris brassicae)およびエゾスジグロシロチョウ(P. napi)の蛹は、哺乳類細胞株に対してモンシロチョウと類似の細胞障害活性を含む(非特許文献5参照)。その上、ピエリシン-1と91%同一であるアミノ酸配列を有するピエリシン-2が、オオモンシロチョウから同定されている(非特許文献13参照)。ピエリシン-2はまた、DNAを標的として、ピエリシン-2によって産生されたDNA付加物の構造はピエリシン-1によって産生された付加物と同じである(非特許文献17参照)。
また、以下のリストに記載された文献が報告されている。
【0005】
【特許文献1】特開2001−25390
【非特許文献1】Sugimura, T.著、「Poly(adenosine diphosphate ribose).」、Prog. Nucleic Acid Res. Mol. Biol.、1973年、Vol.13、 p.127-151.
【非特許文献2】Seman, M.外3名著、「Ecto -ADP-ribosyltransferases (ARTs): emerging actors in cell communication and signaling.」、Curr. Med. Chem.、2004年、 Vol.11、p.857-872.
【非特許文献3】Krueger,.K.M. および Barbieri, J.T.著、「The family of bacterial ADP-ribosylating exotoxins.」、Clin. Microbiol. Rev.、1995年、Vol.8、p.347.
【非特許文献4】Okazaki, I.J. および Moss, J.著、「Characterization of glycosylphosphatidylinositiol-anchored, secreted, and intracellular vertebrate mono -ADP-ribosyltransferases.」、Annu. Rev. Nutr.、1999年、Vol.19、p.485-509.
【非特許文献5】Koyama, K.外8名著、「Presence in Pieris rapae of cytotoxic activity against human carcinoma cells.」、Jpn. J. Cancer Res.、1996年、Vol.87、p.1259-1262.
【非特許文献6】Watanabe, M.外4名著、「Purification of pierisin, an inducer of apoptosis in human gastric carcinoma cells, from cabbage butterfly, Pieris rapae.」、Jpn. J. Cancer Res.、1998年、Vol.89、p.556-561.
【非特許文献7】Sugimura, T.著、「Serendipitous discoveries from sudden inspirations and the joy of being a scientist.」、Biochem. Biophys. Res. Commun.、2002年、Vol. 296、p.1037-1038.
【非特許文献8】Watanabe, M.外8名著、「Molecular cloning of an apoptosis-inducing protein, pierisin, from cabbage butterfly: possible involvement of ADP-ribosylation in its activity.」、Proc. Natl. Acad. Sci. USA、1999年、Vol.96、p.10608-10613.
【非特許文献9】Kanazawa, T.外7名著、「Distinct roles for the N- and C-terminal regions in the cytotoxicity of pierisin-1, a putative ADP-ribosylating toxin from cabbage butterfly, against mammalian cells.」、Proc. Natl. Acad. Sci. USA、2001年、Vol.98、p.2226-2231.
【非特許文献10】Matsushima-Hibiya, Y.外8名著、「Identification of glycosphingolipid receptors for pierisin-1, a guanine-specific ADP-ribosylating toxin from the cabbage butterfly.」、J. Biol. Chem.、2003年、Vol.278、p.9972-9978.
【非特許文献11】Takamura-Enya, T.外7名著、「Mono(ADP-ribosyl)ation of 2'-deoxyguanosine residue in DNA by an apoptosis-inducing protein, pierisin-1, from cabbage butterfly.」、Proc. Natl. Acad. Sci. USA、2001年、Vol.98、p.12414-12419.
【非特許文献12】Kono, T.外6名著、「Cytotoxic activity of pierisin, from the cabbage butterfly, Pieris rapae, in various human cancer cell lines.」、Cancer Lett.、1999年、Vol.137、p. 75-81.
【非特許文献13】Matsushima-Hibiya, Y.外6名著、「Purification and cloning of pierisin-2, an apoptosis-inducing protein from the cabbage butterfly, Pieris brassicae.」、Eur. J. Biochem.、2000年、Vol.267、p.5742-5750.
【非特許文献14】Kanazawa, T.外8名著、「Bcl-2 blocks apoptosis caused by pierisin-1, a guanine-specific ADP-ribosylating toxin from the cabbage butterfly.」、Biochem. Biophys. Res. Commun.、2002年、Vol.296、p.20-25.
【非特許文献15】Totsuka, Y.外8名著、「Analysis of HPRT and supF mutations caused by pierisin-1, a guanine specific ADP-ribosylating toxin derived from the cabbage butterfly.」、Chem. Res. Toxicol.、2003年、Vol.16、p.945-952.
【非特許文献16】Watanabe, M.外9名著、「Developmental stage-specific expression and tissue distribution of pierisin-1, a guanine-specific ADP-ribosylating toxin, in Pieris rapae.」、Comp. Biochem. Physiol. A、2004年、Vol.139、p.125-131.
【非特許文献17】Takamura-Enya, T.外4名著、「Mono(ADP-ribosyl)ation of the N2 amino groups of guanine residues in DNA by pierisin-2, from the cabbage butterfly, Pieris brassicae.」、Biochem. Biophys. Res. Commun.、2004年、Vol.323、p.579-582.
【非特許文献18】山崎正利 外2名著、「海洋軟体動物アメフラシ類の抗がん・抗菌蛋白質」、蛋白質核酸酵素、平成13年、Vol. 46、p.382-387.
【発明の開示】
【発明が解決しようとする課題】
【0006】
本発明は、ADPリボシル化活性を有する新規貝類由来タンパク質、および該タンパク質の用途の提供を課題とする。より詳しくは本発明は、ADPリボシル化活性を有する新規貝類由来タンパク質、および該タンパク質を成分とする抗癌剤もしくはアポトーシス誘導剤、並びに、該タンパク質を用いてDNAをADPリボシル化する方法、および、細胞に対してアポトーシス誘導する方法等の提供を課題とする。
【課題を解決するための手段】
【0007】
上記課題を解決すべく本発明者らは鋭意研究を行った。これまでのところ、貝類においてDNAを基質とするADPリボシル化活性を有するタンパク質(ADPリボシル化酵素)は知られていない。本発明者らはチョウセンハマグリ(学名Meretrix lamarckii)、アサリ(学名Ruditapes philippinarum)、およびヤマトシジミ(学名Corbicula japonica)の抽出液に、試験管内で仔牛胸腺DNAと反応させることにより、DNAをADPリボシル化し、DNA付加体 N2-(ADP-ribos-1-yl)-2'-deoxyguanosineを生成させる活性を有するタンパク質を見出すことに成功した。
【0008】
本発明者らは、チョウセンハマグリの抽出液中より分子量20 kDaのタンパク質CARP-1を精製し、精製タンパク質の部分アミノ酸配列を元にcDNAクローニングを行った。クローニングされたcDNAについて解析を行った結果、該cDNAは853塩基対からなり、オープンリーディングフレームは182残基のアミノ酸からなるタンパク質をコードし、分子量は20332と算出された。試験管内で発現させたCARP-1タンパク質には、DNAをADPリボシル化する活性があることが確認された。
【0009】
CARP-1タンパク質のアミノ酸配列に関する相同性解析の結果、ピエリシン-1およびピエリシン-2を含む既知のいかなるタンパク質とも相同性が見出されなかった。しかし、CARP-1タンパク質のアミノ酸配列には、これまでに知られているADPリボシル化酵素の酵素活性に必須なグルタミン酸と予測されるアミノ酸(E128)が含まれることが見出された。そこで、ADPリボシル化酵素に必須と考えられるグルタミン酸に相当するアミノ酸残基(E128)を、同じ酸性アミノ酸であるアスパラギン酸に変異させたところ、活性の95%が喪失することが分かった。
【0010】
さらに本発明者らは、該タンパク質を電気穿孔法により癌細胞株であるHeLa細胞およびTMK-1細胞内へ導入することにより、これらの細胞のアポトーシスが誘導され、細胞死に至ることを見出した。
【0011】
従って、本発明の新規ADPリボシル化タンパク質は、癌細胞に対するアポトーシス誘導剤もしくは抗癌剤としての用途を有することが判明した。
【0012】
上述の如く本発明者らは、DNAをADPリボシル化する活性を有する新規タンパク質を貝類において初めて単離することに成功し、さらに、該タンパク質について上述の新規な用途を初めて見出すことに成功し本発明を完成させた。
【0013】
本発明は、ADPリボシル化活性を有する新規貝類由来タンパク質、および該タンパク質を成分とする抗癌剤もしくはアポトーシス誘導剤、並びに、該タンパク質を用いてDNAをADPリボシル化する方法、および、細胞に対してアポトーシス誘導する方法に関し、より具体的には、
〔1〕 下記(a)〜(d)のいずれかに記載のポリヌクレオチドであって、ADPリボシル化活性を有するポリペプチドをコードするポリヌクレオチド、
(a)配列番号:2に記載のアミノ酸配列からなるポリペプチドをコードするポリヌクレオチド
(b)配列番号:1に記載の塩基配列のコード領域を含むポリヌクレオチド
(c)配列番号:2に記載のアミノ酸配列において、1もしくは複数のアミノ酸が置換、欠失、挿入、および/または付加したアミノ酸配列からなるポリペプチドをコードするポリヌクレオチド
(d)配列番号:1に記載の塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズするポリヌクレオチド
〔2〕 〔1〕に記載のポリヌクレオチドによりコードされるポリペプチド、
〔3〕 〔2〕に記載のポリペプチドもしくはその部分ポリペプチドと、細胞レセプターを認識するタンパク質とが融合した構造のポリペプチド、
〔4〕 〔1〕に記載のポリヌクレオチド、または、〔3〕に記載のポリペプチドをコードするポリヌクレオチドが挿入されたベクター、
〔5〕 〔1〕に記載のポリヌクレオチド、または〔4〕に記載のベクターを保持する宿主細胞、
〔6〕 〔5〕に記載の宿主細胞を培養し、該宿主細胞またはその培養上清から、産生させたポリペプチドを回収する工程を含む、〔2〕または〔3〕に記載のポリペプチドの製造方法、
〔7〕 〔1〕に記載のポリヌクレオチドと特異的にハイブリダイズするポリヌクレオチドであって、少なくとも15ヌクレオチドの鎖長を持つポリヌクレオチド、
〔8〕 〔1〕に記載のポリヌクレオチドまたはその一部に対するアンチセンスポリヌクレオチド、
〔9〕 〔2〕または〔3〕に記載のポリペプチドに結合する抗体、
〔10〕 〔2〕もしくは〔3〕に記載のポリペプチド、または、〔4〕に記載のベクターを有効成分として含む、アポトーシス誘導剤、
〔11〕 〔2〕もしくは〔3〕に記載のポリペプチド、または、〔4〕に記載のベクターを有効成分として含む、抗癌剤、
〔12〕 〔2〕もしくは〔3〕に記載のポリペプチドを有効成分として含む、ADPリボシル化試薬、
〔13〕 CARP-1タンパク質の発現もしくは機能阻害物質を有効成分とする、貝類毒性除去剤、
〔14〕 〔2〕に記載ポリペプチドとDNAとを接触させる工程を含む、DNAをADPリボシル化する方法、
〔15〕 以下の工程(a)または(b)のいずれかの工程を含む、細胞をアポトーシス誘導する方法、
(a)〔2〕に記載のポリペプチド、または〔4〕に記載のベクターを細胞へ導入する工程
(b)〔3〕に記載のポリペプチドと細胞とを接触させる工程
〔16〕 以下の工程(a)〜(c)を含む、抗癌剤のスクリーニング方法、
(a)〔2〕に記載のポリペプチドもしくは該ポリペプチドを発現する細胞と、被検化合物を接触させる工程
(b)前記ポリペプチドの発現量または活性を測定する工程
(c)被検化合物を接触させない場合と比較して、前記発現量または活性を上昇させる化合物を選択する工程
〔17〕 以下の工程(a)〜(c)を含む、貝類毒性除去剤のスクリーニング方法、
(a)〔2〕に記載のポリペプチドもしくは該ポリペプチドを発現する細胞と、被検化合物を接触させる工程
(b)前記ポリペプチドの発現量または活性を測定する工程
(c)被検化合物を接触させない場合と比較して、前記発現量または活性を低下させる化合物を選択する工程
を、提供するものである。
【発明の効果】
【0014】
本発明者らは、多くの国において一般的に消費されている貝類におけるDNA ADP-リボシルトランスフェラーゼの有無を調べ、ハマグリ等の二枚貝がDNA ADP-リボシルトランスフェラーゼ活性を有するタンパク質を含むことを発見した。
【0015】
具体的には、チョウセンハマグリ、アサリ、およびヤマトシジミには、上記の酵素活性を有するタンパク質を含み、モンシロチョウのピエリシン-1および-2の場合と同様に、β-NADおよびDNAからN2-(ADP-リボース-1-イル)-2'-デオキシグアノシンを産生することが証明された。また、CARP-1と呼ばれる20 kDaタンパク質がチョウセンハマグリから精製され、クローニングされたcDNAを用いてインビトロにて翻訳させることにより、CARP-1タンパク質を取得した。該タンパク質は、DNA ADP-リボシルトランスフェラーゼ活性を示した。チョウセンハマグリからの粗抽出物のDNA ADP-リボシルトランスフェラーゼ活性は、6900 PSL/μgであり、この値はそれぞれ、モンシロチョウの幼虫および蛹の値の約1/2および1/20である。CARP-1の推定アミノ酸配列は、ピエリシン-1および-2とわずかな相同性しか示さなかった。
【0016】
NAD結合部位と推定される128位のグルタミン酸についての部位特異的変異誘発実験によって、CARP-1が新規DNA ADP-リボシルトランスフェラーゼであることが証明された。
【0017】
ADP-リボシルトランスフェラーゼの反応メカニズムは、CT、PT、DT、緑膿菌(Pseudomonas aeruginosa)のエンドトキシンA(ETA)、および大腸菌の熱不安定エンテロトキシン(LT)においてほとんどが研究されている(Aktories, K. and Just, I. (2000) Bacterial Protein Toxins. Springer-Verlag, Berlin.;Krueger, K.M. and Barbieri, J.T. (1995) The family of bacterial ADP-ribosylating exotoxins. Clin. Microbiol. Rev., 8, 34-47.)。これらの毒素のX-線結晶学から、それらがその反応中心において類似の構造を共有することが判明した。既知の全てのADP-リボシルトランスフェラーゼにおいて保存されており、活性に必須と考えられるグルタミン酸残基は、NAD-結合部位として作用する。高度に保存されたアルギニン残基およびSer-Thr-Ser/Thrモチーフも同様に、CT、PTおよびLTに存在する。アルギニン残基は、反応ポケットの構造を維持すると考えられており、Ser-Thr-Ser/Thrモチーフはβ鎖-α-ヘリックス構造を構築して、反応腔を維持すると考えられている(Domenighini, M. and Rappuoli, R. (1996) Three conserved consensus sequences identify the NAD-binding site of ADP-ribosylating enzymes, expressed by eukaryotes, bacteria and T-even bacteriophages. Mol. Microbiol., 21, 667-674.;Pallen, M.J., Lam, A.C., Loman, N.J. and McBride, A. (2001) An abundance of bacterial ADP-ribosyltransferases-implications for the origin of exotoxins and their human homologues. Trends Microbiol., 9, 302-307; discussion 308.)。
【0018】
CARP-1と他のADP-リボシルトランスフェラーゼとの間の全体的な配列類似性は非常に低く、その基質の多様性が反映されていることを示唆される。CARP-1においてGlu-128、Arg-12および46〜48位でのSer-Thr-Thrが存在することは、CT、PT、およびLTのようなADP-リボシルトランスフェラーゼの反応腔構造が保存されていることを示唆する。また、CARP-1とピエリシン-1または-2とのアミノ酸相同性が非常に低いということは、CARP-1とピエリシンがいずれも、ある特定のグアニン特異的ADP-リボシルトランスフェラーゼに由来しないことを示唆している。進化的に異なるDNA ADP-リボシルトランスフェラーゼが存在することは、他の種類のDNA ADP-リボシルトランスフェラーゼも同様に種々の生物に存在する可能性があることが、今回の実験によって示唆された。
【0019】
CARP-1の特徴の一つとして、受容体結合ドメインを欠損している点が挙げられる。細菌のADP-リボシル化毒素は、触媒および受容体結合ドメインが同じまたは異なる分子に存在するかどうか、または、結合ドメインが存在するか否かに基づいて三つの群に分類されている。それぞれ、「A/B毒素」、「バイナリ毒素」、および「Aのみの毒素」である。「Aのみの毒素」は、細胞内で作用するために標的細胞における組込みのメカニズムが必要である。ピエリシン-1は典型的なA/B-毒素構造を有する。このタンパク質のC-末端71-kDaドメインは、リシンスーパーファミリーのレクチンドメインと相同性を有する。発現されたN-末端27-kDaドメイン自身は、HeLa細胞とTMK-1細胞に対して細胞障害性を示さないが、N-末端部分が電気穿孔によってHeLa細胞またはTMK-1細胞に転移されると毒性を示すようになる。CARP-1の場合、cDNA配列の解析から、CARP-1タンパク質は、受容体結合ドメインを有さないADP-リボシルトランスフェラーゼであることが示唆された。Sephacryl S-100ゲル濾過によって推定される上記タンパク質の分子量は約28 kDaであり、このタンパク質の天然型には受容体結合サブユニットが存在しないことが示唆された。さらに、チョウセンハマグリからの粗抽出物、および精製されたCARP-1は、電気穿孔法によってHeLa細胞およびTMK-1細胞に導入した場合に細胞障害性を示した。本発明者らはまた、CM52カラムクロマトグラフィーにおける流出分画においてチョウセンハマグリのDNA ADP-リボシルトランスフェラーゼ活性を発見したが、ゲル濾過実験から、この流出分画における活性タンパク質の分子量が精製CARP-1の分子量とほぼ同じであることが示された。これらの結果から、チョウセンハマグリは、哺乳類細胞に入るために必要な受容体結合ドメインを持たないDNA ADP-リボシルトランスフェラーゼを含有することが示唆された。
【0020】
本発明において単離されたcDNAは、ポリアデニル化シグナルおよび伸長部を有する典型的な真核細胞構造を示したことから、単離されたCARP-1遺伝子が海水からの原核微生物のゲノムに存在していた可能性は低い。また、海水中の微生物を含むと予想される鰓、外套膜、および中腸腺から調製した粗抽出物の酵素活性は、軟組織よりはるかに弱く、CARP-1がハマグリ自身から産生されることを示唆している。タンパク質に受容体結合ドメインが存在しないことは、CARP-1が細胞内DNAに作用してハマグリ自身の細胞死を誘導する可能性があることが示唆された。
【0021】
また、癌細胞表面に特異的に提示されているレセプターを認識するタンパク質とのキメラ酵素を作製することにより、癌細胞を選択的に死滅させる貝類由来の新規抗癌剤の提供が可能である。
【発明を実施するための最良の形態】
【0022】
本発明者らによって、ADPリボシル化活性を有する貝類由来のタンパク質、および該タンパク質をコードするポリヌクレオチド(遺伝子)が同定された。
【0023】
本発明のタンパク質は、ADPリボシル化活性を有するタンパク質であって、貝類に由来するタンパク質であれば、その由来する貝の種類は特に制限されず、例えば、チョウセンハマグリ、アサリ、またはヤマトシジミ等の貝類に由来するタンパク質が挙げられる。
【0024】
本発明の上記タンパク質(遺伝子)の好ましい態様としては、例えば、チョウセンハマグリに由来するCARP-1タンパク質(遺伝子)が挙げられる。該CARP-1タンパク質は、DNAを基質とするADPリボシル化活性を有する。
【0025】
上述の貝以外の貝類に由来するADPリボシル化活性を有するタンパク質(例えば、CARP-1タンパク質のホモログ・オルソログ等)もまた本発明のタンパク質に含まれる。
【0026】
本発明において「貝類」とは、特に制限されないが、好ましくは二枚貝である。本発明における貝類として具体的には、チョウセンハマグリ、アサリ、ヤマトシジミ、ウバガイ、ムラサキイガイ、マテガイ、ハマグリ(Meretrix lusoria)、シナハマグリ(Meretrix pethechialis)等を例示することができる。
【0027】
チョウセンハマグリのCARP-1遺伝子のDNA配列を配列番号:1に、該DNAによってコードされるタンパク質のアミノ酸配列を配列番号:2に記載する。
【0028】
ただし、本発明の遺伝子は、必ずしも、配列表に具体的に記載された配列からなるDNAに限定されない。また、本発明のタンパク質は、必ずしも、配列表に具体的に記載されたアミノ酸配列からなるタンパク質に限定されない。
【0029】
上記以外のタンパク質であっても、例えば配列表に記載された配列と高い相同性(通常70%以上、好ましくは80%以上、より好ましくは90%以上、最も好ましくは95%以上)を有し、かつ、本発明のタンパク質が有する機能(例えば、ADPリボシル化活性)を持つタンパク質は、本発明のタンパク質に含まれる。
【0030】
上記タンパク質とは、例えば、配列番号:2に記載のアミノ酸配列において、1以上のアミノ酸が付加、欠失、置換、挿入されたアミノ酸配列からなるタンパク質であって、通常変化するアミノ酸数が30アミノ酸以内、好ましくは10アミノ酸以内、より好ましくは5アミノ酸以内、最も好ましくは3アミノ酸以内である。
【0031】
本発明の遺伝子には、例えば、配列番号:2に記載の塩基配列からなるDNAに対応する他の貝類における内在性の遺伝子(チョウセンハマグリのCARP-1遺伝子のホモログ等)が含まれる。
【0032】
また、配列番号:1に記載の塩基配列からなるDNAに対応する他の貝類の内在性のDNAは、一般的に、配列番号:1に記載のDNAと高い相同性を有する。高い相同性とは、50%以上、好ましくは70%以上、さらに好ましくは80%以上、より好ましくは90%以上(例えば、95%以上、さらには96%、97%、98%または99%以上)の相同性を意味する。この相同性は、mBLASTアルゴリズム(Altschul et al. (1990) Proc. Natl. Acad. Sci. USA 87: 2264-8; Karlin and Altschul (1993) Proc. Natl. Acad. Sci. USA 90: 5873-7)によって決定することができる。また、該DNAは、生体内から単離した場合、配列番号:1に記載のDNAとストリンジェントな条件下でハイブリダイズすると考えられる。ここで「ストリンジェントな条件」としては、例えば「2×SSC、0.1%SDS、50℃」、「2×SSC、0.1%SDS、42℃」、「1×SSC、0.1%SDS、37℃」、よりストリンジェントな条件として「2×SSC、0.1%SDS、65℃」、「0.5×SSC、0.1%SDS、42℃」および「0.2×SSC、0.1%SDS、65℃」の条件を挙げることができる。当業者においては、他の貝類における本発明の遺伝子に相当する内在性の遺伝子を、本発明の遺伝子の塩基配列を基に適宜取得することが可能である。なお、本明細書においては、チョウセンハマグリ以外の貝類におけるCARP-1タンパク質(遺伝子)に相当するタンパク質(遺伝子)、または、該タンパク質と機能的に同等なタンパク質(遺伝子)を、単に「本発明のタンパク質(遺伝子)」と記載する場合がある。
【0033】
本発明のタンパク質は、天然のタンパク質のほか、遺伝子組み換え技術を利用した組換えタンパク質として調製することができる。天然のタンパク質は、例えば本発明のタンパク質が発現していると考えられる貝類由来の細胞(組織)の抽出液に対し、本発明のタンパク質に対する抗体を用いたアフィニティークロマトグラフィーを用いる方法により調製することが可能である。一方、組換えタンパク質は、本発明のタンパク質をコードするDNAで形質転換した細胞を培養することにより調製することが可能である。
【0034】
本発明の好ましい態様としては、下記(a)〜(d)のいずれかに記載のポリヌクレオチドであって、ADPリボシル化活性を有するポリペプチドをコードするポリヌクレオチドを提供する。
(a)配列番号:2に記載のアミノ酸配列からなるポリペプチドをコードするポリヌクレオチド
(b)配列番号:1に記載の塩基配列のコード領域を含むポリヌクレオチド
(c)配列番号:2に記載のアミノ酸配列において、1もしくは複数のアミノ酸が置換、欠失、挿入、および/または付加したアミノ酸配列からなるポリペプチドをコードするポリヌクレオチド
(d)配列番号:1に記載の塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズするポリヌクレオチド
【0035】
また、上記ポリヌクレオチドによってコードされるポリペプチド(タンパク質)もまた、本発明に含まれる。
【0036】
本発明における「ポリヌクレオチド」には、「DNA」および該DNAと相同もしくは相補的な配列からなる「RNA」が含まれる。従って本発明の「ポリヌクレオチド」には、例えば、本発明のタンパク質をコードする遺伝子DNA、該DNAからの転写産物であるRNAが含まれる。また、本発明の「ポリヌクレオチド」には、一本鎖DNAもしくは一本鎖RNA、および二本鎖DNAもしくは二本鎖RNA、並びに、DNAとRNAとのハイブリッド分子が含まれる。
【0037】
本明細書におけるポリペプチドとは、複数のアミノ酸からなる重合体を意味し、そのアミノ酸の長さは特に制限されない。従って、本発明のタンパク質には、所謂「ペプチド」、「オリゴペプチド」、「タンパク質」等も含まれる。本発明のポリペプチドは、天然に存在する状態から修飾されていないもの、および修飾されているものの双方を含む。修飾としては、アセチル化、アシル化、ADP-リボシル化、アミド化、フラビンの共有結合、ヘム部分の共有結合、ヌクレオチドまたはヌクレオチド誘導体の共有結合、脂質または脂質誘導体の共有結合、ホスファチジルイノシトールの共有結合、架橋、環化、ジスルフィド結合の形成、脱メチル化、共有架橋の形成、シスチンの形成、ピログルタメートの形成、ホルミル化、γ-カルボキシル化、グリコシル化、GPIアンカー形成、ヒドロキシル化、ヨウ素化、メチル化、ミリストイル化、酸化、タンパク質分解処理、リン酸化、プレニル化、ラセミ化、セレノイル化、硫酸化、アルギニル化のようなタンパク質へのアミノ酸の転移RNA媒介付加、ユビキチン化等が含まれる。
【0038】
本発明のポリペプチドは、そのアミノ酸配列に従って、一般的な化学合成法により製造することが可能であり、該方法には、通常の液相法および固相法によるペプチド合成法が包含される。かかるペプチド合成法は、より詳しくはアミノ酸配列の情報に基づいて、各アミノ酸を1個ずつ逐次合成させて鎖を延長していくステップワイズエロンゲーション法と、アミノ酸数個からなるフラグメントを予め合成し、次いで各フラグメントをカップリング反応させるフラグメント・コンデンセーション法を包含し、本発明のポリペプチドの合成は、いずれの方法を用いてもよい。
【0039】
このようなペプチド合成法にて用いられる縮合法も、各種方法に従って行うことができる。その具体例としては、例えばアジド法、混合酸無水物法、DCC法、活性エステル法、酸化還元法、DPPA(ジフェニルホスホリルアジド)法、ウッドワード法等を例示できる。
【0040】
これら各種方法に利用できる溶媒もまた、一般的に使用されるものを適宜利用することができる。その例としては、例えばジメチルホルムアミド(DMF)、ジメチルスルホキシド(DMSO)、ヘキサホスホロアミド、ジオキサン、テトラヒドロフラン(THF)、酢酸エチル等及びこれらの混合溶媒等を挙げることができる。なお、上記ペプチド合成反応に際して、反応に関与しないアミノ酸およびペプチドにおけるカルボキシル基は、一般にはエステル化により、例えばメチルエステル、エチルエステル、第三級ブチルエステル等の低級アルキルエステル、例えばベンジルエステル、P−メトキシベンジルエステル、P−ニトロベンジルエステルアラルキルエステル等として保護することができる。また、側鎖に官能基を有するアミノ酸、例えばTyrの水酸基は、アセチル基、ベンジル基、ベンジルオキシカルボニル基、第三級ブチル基等で保護されてもよいが、必ずしもかかる保護は必須ではない。また、例えば、Argのグアニジノ基は、ニトロ基、トシル基、2−メトキシベンゼンスルホニル基、メチシレン−2−スルホニル基、ベンジルオキシカルボニル基、イソボルニルオキシカルボニル基、アダマンチルオキシカルボニル基等の適当な保護基により保護することができる。
【0041】
上記のようにして得ることが可能な本発明のポリペプチドは、通常の方法に従って、例えばイオン交換樹脂、分配クロマトグラフィー、ゲルクロマトグラフィー、アフィニティークロマトグラフィー、高速液体クロマトグラフィー(HPLC)、向流分配法等のペプチド化学の分野で汎用されている方法に従って、適宜、精製を行うことができる。
本発明のポリペプチドは、例えば、配列番号:2に記載のポリペプチド、または配列番号:1に記載のDNA核酸分子を合成し、次いで適当な発現ベクターへ導入した後、宿主細胞内において発現させる遺伝子工学的手法によっても取得することができる。
【0042】
本発明のポリペプチドには、例えば、本発明のタンパク質(例えば、CARP-1タンパク質)と機能的に同等なタンパク質(ポリペプチド)が含まれる。ここで「機能的に同等」とは、対象となるポリペプチドが本発明のタンパク質と同様の(同等の)生物学的あるいは生化学的機能(活性)を有することを指す。このような機能としては、例えば、DNAを基質としたADPリボシル化活性を挙げることができる。
【0043】
あるポリペプチドが本発明のADPリボシル化活性を有するポリペプチドであるか否かは、例えば、後述の実施例に記載された迅速高感度検出法、あるいは、DNA付加体構造解析等によって適宜評価することができる。DNA付加体構造解析は、通常、多量のポリペプチドとヌクレアーゼを要するため、迅速高感度検出法を用いて実施することが好ましいが、いずれの方法によっても好適に実施することができる。
【0044】
一例を示せば、まず、適切な緩衝液及び塩の存在下(例えば50 mM トリス塩酸pH 7.5、1 mMエチレンジアミン四酢酸、50 mM 塩化ナトリウム、1 mM ジチオスレイトール)にて、仔牛胸腺DNA、beta-NAD(微量の32P-NADを含む)と、当該ペプチドを混合し、通常摂氏37度、1時間インキュベートする。インキュベート後、フェノール処理及びクロロホルム抽出によりペプチドを除去し、酢酸アンモニウム塩を用いたアルコール沈澱によって未反応のbeta-NADを除去しつつDNAを回収する(Sambrook, J et al., Molecular Cloning, 2nd ed. 等に記載される常法)。回収したDNAをマイクロコッカルヌクレアーゼおよびホスホジエステラーゼIIを用いて3'-リン酸型デオキシリボヌクレオチドに分解し(非特許文献11)、PEI-セルロース薄層クロマトグラフィーシートを用いて展開する。6 mol/l 酢酸、0.1 mol/l 塩化リチウム、3 mol/l 尿素で展開し、オートラジオグラフィー(X線フィルムよりも富士フィルムのBASシステムの方が、高感度で望ましい)で検出する。ADP-リボシル化デオキシリボヌクレオチド3'-リン酸は他の核酸やNADとは異なり、殆ど展開されずRf値0.05付近に留まる事から検出することができる。
【0045】
なお、本発明において「ADPリボシル化活性」は、ADPリボシルトランスフェラーゼ活性とも表現される。本発明のADPリボシル化活性は、好ましくは、DNAを基質とするものであり、より具体的には、DNAのグアニン残基のN2位のアミノ基をADPリボシル化する活性を指す。
【0046】
本発明のタンパク質を基に改変されたポリペプチド(タンパク質)について、本発明のタンパク質と同等の生物学的あるいは生化学的な機能(活性)を保持しているか否かは、例えば、上述のADPリボシル化活性についての評価方法以外に、例えば、対象となる(被検)ポリペプチドを癌細胞へ導入し、アポトーシスの誘導の有無を観察することによっても評価することが可能である。
【0047】
あるポリペプチドと機能的に同等なポリペプチドを調製するための、当業者によく知られた方法としては、例えばポリペプチド中のアミノ酸配列に変異を導入する方法が挙げられる。具体的には当業者であれば部位特異的変異誘発法(Hashimoto-Gotoh, T. et al. (1995) Gene 152, 271-275、Zoller, MJ, and Smith, M.(1983) Methods Enzymol. 100, 468-500、Kramer, W. et al. (1984) Nucleic Acids Res. 12, 9441-9456、Kramer W, and Fritz HJ(1987) Methods. Enzymol. 154, 350-367、Kunkel,TA(1985) Proc Natl Acad Sci USA. 82, 488-492、Kunkel (1988) Methods Enzymol. 85, 2763-2766)などを用いて、配列番号:2に記載のアミノ酸配列に適宜変異を導入することにより、該ポリペプチドと機能的に同等なポリペプチドを調製することができる。また、ポリペプチド中のアミノ酸の変異は自然に生じることもある。このように、人工的か自然に生じたものかを問わず、本発明者らにより同定されたタンパク質(配列番号:2)のアミノ酸配列において1もしくは複数のアミノ酸配列が変異したアミノ酸配列を有し、該ポリペプチドと機能的に同等なポリペプチドは、本発明のタンパク質(ポリペプチド)に含まれる。
【0048】
上記変異体における、変異するアミノ酸数は、本発明のポリペプチドの有する機能が保持される限り制限はないが、通常15アミノ酸以内であり、好ましくは10アミノ酸以内であり、より好ましくは5アミノ酸以内であり、さらに好ましくは1〜4アミノ酸である。
【0049】
変異するアミノ酸残基としては、アミノ酸側鎖の性質が保存されている別のアミノ酸に変異されることが望ましい。例えばアミノ酸側鎖の性質としては、疎水性アミノ酸(A、I、L、M、F、P、W、Y、V)、親水性アミノ酸(R、D、N、C、E、Q、G、H、K、S、T)、脂肪族側鎖を有するアミノ酸(G、A、V、L、I、P)、水酸基含有側鎖を有するアミノ酸(S、T、Y)、硫黄原子含有側鎖を有するアミノ酸(C、M)、カルボン酸及びアミド含有側鎖を有するアミノ酸(D、N、E、Q)、塩基含有側鎖を有するアミノ離(R、K、H)、芳香族含有側鎖を有するアミノ酸(H、F、Y、W)を挙げることができる(括弧内はいずれもアミノ酸の一文字表記を表す)。
【0050】
あるアミノ酸配列に対する1または複数個のアミノ酸残基の欠失、付加及び/又は他のアミノ酸による置換により修飾されたアミノ酸配列を有するポリペプチドがその生物学的機能(活性)を維持し得ることはすでに知られている(Mark, D. F. et al., Proc. Natl. Acad. Sci. USA (1984) 81, 5662-5666 、Zoller, M. J. & Smith, M. Nucleic Acids Research (1982) 10, 6487-6500 、Wang, A. et al., Science 224, 1431-1433 、Dalbadie-McFarland, G. et al., Proc. Natl. Acad. Sci. USA (1982) 79, 6409-6413)。
【0051】
具体的なアミノ酸配列(例えば、配列番号:2)が開示された場合においては、当業者であれば、これらアミノ酸配列を基に、適宜アミノ酸が改変された配列からなるポリペプチドを作製し、当該ポリペプチドについて、上述の機能を有するか否かを評価し、本発明のタンパク質(ポリペプチド)を適宜選択することが可能である。
【0052】
本発明者らは、CARP-1タンパク質においてADPリボシル化に必須と考えられるグルタミン酸を見出した。該グルタミン酸は、CARP-1タンパク質(配列番号:2)のアミノ酸配列において128位のグルタミン酸に相当する。従って、本発明におけるアミノ酸残基の改変は、このグルタミン酸以外の部位についての改変であることが好ましい。即ち本発明の好ましい態様においては、配列番号:2に記載のアミノ酸配列において、128位のグルタミン酸以外の部位における1もしくは複数のアミノ酸が、置換、欠失、挿入、および/または付加されたアミノ酸配列からなるポリペプチドをコードするポリヌクレオチドである。
【0053】
本発明のCARP-1タンパク質には、コレラ毒素等の立体構造より導かれた保存性の高いアミノ酸が存在する。例えば、該アミノ酸としては、図4に示されるアミノ酸配列において12位のアルギニン(活性に必須の水素結合を提供)が挙げられる。また、47〜49位のセリン-スレオニン-スレオニンからなる配列も活性中心の空間の保持に重要な役割を果たしているものと考えられる。従って、本発明のポリペプチドは、これらのアミノ酸残基を含むことが好ましい。
【0054】
また、本発明において改変されるポリペプチド領域は、必ずしも制限されるものではないが、例えば、上記のアミノ酸以外の領域、即ち、配列番号:2に記載のアミノ酸配列の12位のアルギニン、または、47〜49位のセリン-スレオニン-スレオニン配列以外の領域であることが好ましい。これらの領域の改変であれば、依然としてCARP-1タンパク質の機能が保持されている可能性が高い。
【0055】
本発明のポリペプチドのアミノ酸配列に複数個のアミノ酸残基が付加されたポリペプチドには、これらポリペプチドを含む融合ポリペプチドが含まれる。融合ポリペプチドは、これらポリペプチドと他のペプチド又はポリペプチドとが融合したものである。融合ポリペプチドを作製する方法は、本発明のポリペプチド(例えば、配列番号:2)をコードするDNA(例えば、配列番号:1)と他のペプチド又はポリペプチドをコードするDNAをフレームが一致するように連結してこれを発現ベクターに導入し、宿主で発現させればよく、当業者に公知の手法を用いることができる。本発明のポリペプチドとの融合に付される他のペプチド又はポリペプチドは、特に制限されない。
【0056】
本発明のポリペプチドとの融合に付される他のペプチドとしては、例えば、GST(グルタチオン−S−トランスフェラーゼ)、イムノグロブリン定常領域、β−ガラクトシダーゼ、MBP(マルトース結合タンパク質)等が挙げられる。市販されているこれらペプチドまたはポリペプチドをコードするポリヌクレオチドを本発明のポリペプチドをコードするポリヌクレオチドと融合させ、これにより調製された融合ポリヌクレオチドを発現させることにより、融合ポリペプチドを調製することができる。
【0057】
また、本発明のポリペプチドの好ましい態様としては、本発明のポリペプチドもしくはその部分ポリペプチドと、細胞レセプターを認識するタンパク質とが融合した構造のポリペプチドが挙げられる。
【0058】
例えば、癌細胞表面に特異的に提示されているレセプターを認識するタンパク質と、本発明のポリペプチドとのキメラ酵素は、癌細胞に対して選択的なアポトーシス誘導作用を有することが期待される。
【0059】
上記「レセプターを認識するタンパク質」として具体的には、以下のタンパク質を例示することができるが、これらのタンパク質に限定されない。
(a)ヒト子宮頸癌培養細胞HeLa等に提示されたglobotriaosylceramide (Gb3)、または、globotetraosylceramide (Gb4)を認識するピエリシン-1タンパク質C末端側ドメイン(Kanazawa, T., et. Al., (2001) Proc. Natl. Acad. Sci. USA, 98, 2226-2231; Matsushima-Hibiya, Y., et. Al., (2003) J. Biol. Chem., 278, 9972-9978)
(b)腸管上皮細胞に提示されたganglioside GM1を認識する、コレラ毒素Bサブユニット
【0060】
また、癌細胞表面に特異的に結合する抗体を作製し、該抗体分子の可変領域重鎖および軽鎖が本発明のポリペプチドと融合した構造のポリペプチドを調製することも可能である。融合タンパク質による臨床応用については、公知文献(永田諭志、「蛋白質核酸酵素」、46巻4号、540-546頁、平成13年)を参照することができる。
【0061】
またあるポリペプチドと機能的に同等なポリペプチドを調製する当業者によく知られた他の方法としては、ハイブリダイゼーション技術(Sambrook,J et al., Molecular Cloning 2nd ed., 9.47-9.58, Cold Spring Harbor Lab. press, 1989)を利用する方法が挙げられる。即ち、当業者であれば、本発明のポリペプチドをコードするDNA(例えば、配列番号:1に記載の塩基配列)もしくはその一部をもとに、同種または異種生物由来のDNA試料から、これと相同性の高いDNAを単離して、該DNAから本発明のポリペプチドと機能的に同等なポリペプチドを単離することも通常行いうることである。
【0062】
本発明には、本発明のポリペプチドをコードするDNAとハイブリダイズするDNAによってコードされるポリペプチドであって、本発明のポリペプチドと機能的に同等なポリペプチドが含まれる。このようなポリペプチドとしては、例えば、ハマグリあるいはその他の貝類に内在するADPリボシル化活性を有するタンパク質(例えば、CARP-1のホモログ等)が挙げられる。
【0063】
本発明のポリペプチドと機能的に同等なポリペプチドをコードするDNAを単離するためのハイブリダイゼーションの条件は、当業者であれば適宜選択することができる。ハイブリダイゼーションの条件としては、例えば、低ストリンジェントな条件が挙げられる。低ストリンジェントな条件とは、ハイブリダイゼーション後の洗浄において、例えば42℃、0.1×SSC、0.1%SDSの条件であり、好ましくは50℃、0.1×SSC、0.1%SDSの条件である。より好ましいハイブリダイゼーションの条件としては、高ストリンジェントな条件が挙げられる。高ストリンジェントな条件とは、例えば65℃、5×SSC及び0.1%SDSの条件である。これらの条件において、温度を上げる程に高い相同性を有するDNAが効率的に得られることが期待できる。但し、ハイブリダイゼーションのストリンジェンシーに影響する要素としては温度や塩濃度など複数の要素が考えられ、当業者であればこれら要素を適宜選択することで同様のストリンジェンシーを実現することが可能である。
【0064】
また、ハイブリダイゼーションにかえて、遺伝子増幅技術(PCR)(Current protocols in Molecular Biology edit. Ausubel et al. (1987) Publish. John Wiley&Sons Section 6.1-6.4)を用いて、本発明のポリペプチドをコードするDNA(例えば、配列番号:2または5)の一部を基にプライマーを設計し、本発明者らにより同定されたタンパク質をコードするDNAと相同性の高いDNA断片を単離し、該DNAを基に本発明者らにより同定されたタンパク質と機能的に同等なタンパク質を取得することも可能である。
【0065】
本発明のポリペプチドは「成熟」ポリペプチドの形であっても、融合ポリペプチドのような、より大きいポリペプチドの一部であってもよい。本発明のポリペプチドには、リーダー配列、プロ配列、多重ヒスチジン残基のような精製に役立つ配列、または組換え生産の際の安定性を確保する付加的配列などが含まれていてもよい。
【0066】
これらハイブリダイゼーション技術や遺伝子増幅技術により単離されるDNAによってコードされる、本発明のポリペプチドと機能的に同等なポリペプチドは、通常、本発明のポリペプチド(例えば、配列番号:2)とアミノ酸配列において高い相同性を有する。本発明のポリペプチドには、本発明のタンパク質と機能的に同等であり、かつ該タンパク質のアミノ酸配列と高い相同性を有するポリペプチドも含まれる。高い相同性とは、アミノ酸レベルにおいて、通常、少なくとも50%以上の同一性、好ましくは75%以上の同一性、さらに好ましくは85%以上の同一性、さらに好ましくは95%以上(例えば、96%以上、97%以上、98%以上、99%以上)の同一性を指す。ポリペプチドの相同性を決定するには、文献(Wilbur, W. J. and Lipman, D. J. Proc. Natl. Acad. Sci. USA (1983) 80, 726-730)に記載のアルゴリズムに従えばよい。
【0067】
アミノ酸配列の同一性は、例えば、Karlin and Altschul によるアルゴリズムBLAST (Proc. Natl. Acad. Sci. USA 87:2264-2268, 1990、Proc. Natl. Acad. Sci. USA 90:5873-5877, 1993)によって決定することができる。このアルゴリズムに基づいて、BLASTXと呼ばれるプログラムが開発されている(Altschul et al. J. Mol. Biol.215: 403-410, 1990)。BLASTXによってアミノ酸配列を解析する場合には、パラメーターは例えば、score = 50、wordlength = 3とする。BLASTとGapped BLASTプログラムを用いる場合には、各プログラムのデフォルトパラメーターを用いる。これらの解析方法の具体的な手法は公知である(http://www.ncbi.nlm.nih.gov.)。
【0068】
本発明のポリペプチドは、当業者に公知の方法により、組み換えポリペプチドとして、また天然のポリペプチドとして調製することが可能である。組み換えポリペプチドであれば、例えば、本発明のポリペプチドをコードするDNA(例えば、配列番号:1に記載のDNA)を、適当な発現ベクターに組み込み、これを適当な宿主細胞に導入して得た形質転換体を回収し、抽出物を得た後、イオン交換、逆相、ゲル濾過などのクロマトグラフィー、あるいは本発明のポリペプチドに対する抗体をカラムに固定したアフィニティークロマトグラフィーにかけることにより、または、さらにこれらのカラムを複数組み合わせることにより精製し、調製することが可能である。
【0069】
また、本発明のポリペプチドをグルタチオンS-トランスフェラーゼタンパク質との融合ポリペプチドとして、あるいはヒスチジンを複数付加させた組み換えポリペプチドとして宿主細胞(例えば、動物細胞や微生物細胞等)内で発現させた場合には、発現させた組み換えポリペプチドはグルタチオンカラムあるいはニッケルカラムを用いて精製することができる。融合ポリペプチドの精製後、必要に応じて融合ポリペプチドのうち、目的のポリペプチド以外の領域を、トロンビンまたはファクターXaなどにより切断し、除去することも可能である。
【0070】
天然のタンパク質であれば、当業者に周知の方法、例えば本発明のポリペプチドを発現している組織や細胞の抽出物に対し、本発明のポリペプチドと親和性を有する抗体が結合したアフィニティーカラムを作用させて精製することにより単離することができる。抗体はポリクローナル抗体であっても、モノクローナル抗体であってもよい。
【0071】
また、本発明のポリペプチドは例えば、本発明のポリペプチドを認識する抗体の作製等に利用することが可能である。
【0072】
上述のようにして取得される組換えポリペプチドを用いれば、これに結合する抗体を調製することができる。例えば、ポリクローナル抗体は、精製した本発明のポリペプチドもしくはその一部のペプチドをウサギなどの免疫動物に免疫し、一定期間の後に血液を採取し、血ぺいを除去することにより調製することが可能である。また、モノクローナル抗体は、上記ポリペプチド若しくはペプチドで免疫した動物の抗体産生細胞と骨腫瘍細胞とを融合させ、目的とする抗体を産生する単一クローンの細胞(ハイブリドーマ)を単離し、該細胞から抗体を得ることにより調製することができる。これにより得られた抗体は、本発明のポリペプチドの精製や検出などに利用することが可能である。本発明には、本発明のポリペプチドに結合する抗体が含まれる。これらの抗体を用いることにより、所望の生物もしくは細胞が本発明のポリペプチドを発現するか否かの判別を行うことが可能である。本発明のポリペプチドに結合する抗体もまた本発明に含まれる。
【0073】
本発明のポリヌクレオチドは、本発明のポリペプチドをコードし得るものであればいかなる形態でもよい。即ち、mRNAから合成されたcDNAであるか、ゲノムDNAであるか、化学合成DNAであるかなどを問わない。また、本発明のポリペプチドをコードしうる限り、遺伝暗号の縮重に基づく任意の塩基配列を有するDNAが含まれる。
【0074】
本発明のポリヌクレオチドは、当業者に公知の方法により調製することができる。例えば、本発明のポリペプチドを発現している細胞よりcDNAライブラリーを作製し、本発明のDNA(例えば、配列番号:1に記載の塩基配列)の一部をプローブとしてハイブリダイゼーションを行うことにより調製できる。cDNAライブラリーは、例えば、文献(Sambrook, J. et al., Molecular Cloning、Cold Spring Harbor Laboratory Press (1989))に記載の方法により調製してもよく、あるいは市販のDNAライブラリーを用いてもよい。また、本発明のポリペプチドを発現している細胞よりRNAを調製し、逆転写酵素によりcDNAを合成後、本発明のDNA(例えば、配列番号:1に記載の塩基配列)に基づいてオリゴDNAを合成し、これをプライマーとして用いてPCR反応を行い、本発明のポリペプチドをコードするcDNAを増幅させることにより調製することも可能である。
【0075】
また、得られたcDNAの塩基配列を決定することにより、それがコードする翻訳領域を決定でき、本発明のポリペプチドのアミノ酸配列を得ることができる。また、得られたcDNAをプローブとしてゲノムDNAライブラリーをスクリーニングすることにより、ゲノムDNAを単離することができる。
【0076】
具体的には、次のようにすればよい。まず、本発明のポリペプチドを発現する細胞、組織、器官からmRNAを単離する。mRNAの単離は、公知の方法、例えば、グアニジン超遠心法(Chirgwin, J. M. et al., Biochemistry (1979) 18, 5294-5299)、AGPC法(Chomczynski, P. and Sacchi, N., Anal. Biochem. (1987) 162, 156-159)等により全RNAを調製し、mRNA Purification Kit (Pharmacia) 等を使用して全RNAからmRNAを精製する。また、QuickPrep mRNA Purification Kit (Pharmacia) を用いることによりmRNAを直接調製することもできる。
【0077】
得られたmRNAから逆転写酵素を用いてcDNAを合成する。cDNAの合成は、AMV Reverse Transcriptase First-strand cDNA Synthesis Kit (生化学工業)等を用いて行うこともできる。また、本明細書に記載されたプライマー等を用いて、5'-Ampli FINDER RACE Kit (Clontech製)およびポリメラーゼ連鎖反応 (polymerase chain reaction ; PCR)を用いた5'-RACE法(Frohman, M. A. et al., Proc. Natl. Acad. Sci. U.S.A. (1988) 85, 8998-9002 ; Belyavsky, A. et al., Nucleic Acids Res. (1989) 17, 2919-2932) に従い、cDNAの合成および増幅を行うことができる。得られたPCR産物から目的とするDNA断片を調製し、ベクターDNAと連結する。さらに、これより組換えベクターを作製し、大腸菌等に導入してコロニーを選択して所望の組換えベクターを調製する。目的とするDNAの塩基配列は、公知の方法、例えば、ジデオキシヌクレオチドチェインターミネーション法により確認することができる。
【0078】
また、本発明のDNAの作成においては、発現に使用する宿主のコドン使用頻度を考慮し、より発現効率の高い塩基配列を設計することができる(Grantham, R. et al., Nucelic Acids Research (1981) 9, r43-74)。また、本発明のDNAは、市販のキットや公知の方法によって改変することができる。改変としては、例えば、制限酵素による消化、合成オリゴヌクレオチドや適当なDNAフラグメントの挿入、リンカーの付加、開始コドン(ATG)及び/又は終止コドン(TAA、TGA、又はTAG)の挿入等が挙げられる。
【0079】
本発明のポリヌクレオチドを増幅もしくは検出するためのプライマーおよびプローブもまた、本発明に含まれる。
即ち本発明の好ましい態様としては、以下のポリヌクレオチドを提供する。
(a)本発明のポリヌクレオチドと特異的にハイブリダイズするポリヌクレオチドであって、少なくとも15ヌクレオチドの鎖長を持つポリヌクレオチド
(b)本発明のポリヌクレオチドまたはその一部に対するアンチセンスポリヌクレオチド
【0080】
また本発明は、本発明のポリヌクレオチド、または、本発明のポリペプチドをコードするポリヌクレオチドが挿入されたベクターを提供する。本発明のベクターとしては、組み換えタンパク質の生産に用いるベクターの他、形質転換体作製のために各種細胞内で本発明のポリヌクレオチドを発現させるためのベクターも含まれる。
【0081】
さらに本発明は、本発明の上記ベクターを保持する宿主細胞、および該宿主細胞を利用した本発明のポリペプチドの製造方法を提供する。
【0082】
本発明のベクターとしては、挿入したDNAを安定に保持するものであれば特に制限されず、例えば宿主に大腸菌を用いるのであれば、クローニング用ベクターとしてはpBluescriptベクター(Stratagene社製)などが好ましい。本発明のポリペプチドを生産する目的においてベクターを用いる場合には、特に発現ベクターが有用である。発現ベクターとしては、試験管内、大腸菌内、培養細胞内、生物個体内でポリペプチドを発現するベクターであれば特に制限されないが、例えば、試験管内発現であればpBESTベクター(プロメガ社製)、大腸菌であればpETベクター(Invitrogen社製)、培養細胞であればpME18S-FL3ベクター、生物個体であればpME18Sベクター(Mol Cell Biol. 8:466-472(1988))などが好ましい。ベクターへの本発明のDNAの挿入は、常法により、例えば、制限酵素サイトを用いたリガーゼ反応により行うことができる(Current protocols in Molecular Biology edit. Ausubel et al. (1987) Publish. John Wiley & Sons.Section 11.4-11.11)。
【0083】
本発明のベクターが導入される宿主細胞としては特に制限はなく、目的に応じて種々の宿主細胞が用いられる。ポリペプチドを発現させるための細胞としては、例えば、細菌細胞(例:ストレプトコッカス、スタフィロコッカス、大腸菌、ストレプトミセス、枯草菌)、真菌細胞(例:酵母、アスペルギルス)、昆虫細胞(例:ドロソフィラS2、スポドプテラSF9)、動物細胞(例:CHO、COS、HeLa、C127、3T3、BHK、HEK293、Bowes メラノーマ細胞)および植物細胞を例示することができる。宿主細胞へのベクター導入は、例えば、リン酸カルシウム沈殿法、電気パルス穿孔法(Current protocols in Molecular Biology edit. Ausubel et al. (1987) Publish. John Wiley & Sons.Section 9.1-9.9)、リポフェクタミン法(GIBCO-BRL社製)、マイクロインジェクション法などの公知の方法で行うことが可能である。
【0084】
宿主細胞において発現したポリペプチドを小胞体の内腔に、細胞周辺腔に、または細胞外の環境に分泌させるために、適当な分泌シグナルを目的のポリペプチドに組み込むことができる。これらのシグナルは目的のポリペプチドに対して内因性であっても、異種シグナルであってもよい。
【0085】
本発明のポリペプチドの回収は、本発明のポリペプチドが培地に分泌される場合は、培地を回収する。本発明のポリペプチドが細胞内に産生される場合は、その細胞をまず溶解し、その後にポリペプチドを回収する。
【0086】
組換え細胞培養物から本発明のポリペプチドを回収し精製するには、硫酸アンモニウムまたはエタノール沈殿、酸抽出、アニオンまたはカチオン交換クロマトグラフィー、ホスホセルロースクロマトグラフィー、疎水性相互作用クロマトグラフィー、アフィニティークロマトグラフィー、ヒドロキシルアパタイトクロマトグラフィーおよびレクチンクロマトグラフィーを含めた公知の方法を用いることができる。
【0087】
また本発明は、本発明のポリペプチド、または、本発明のベクターを有効成分として含む、アポトーシス誘導剤を提供する。
【0088】
上記ベクターとしては、通常、本発明のポリペプチドをコードするポリペプチドを担持するプラスミドもしくはウイルスベクターが一般的である。
【0089】
当業者においては、所望のポリヌクレオチドを有するベクターを、一般的な遺伝子工学技術によって、適宜、作製することが可能である。通常、市販の種々のベクターを利用することができる。
【0090】
本発明のベクターは、宿主細胞内において本発明のポリヌクレオチドを保持したり、本発明のポリペプチドを発現させるためにも有用である。
【0091】
本発明におけるポリヌクレオチドは、通常、適当なベクターへ担持(挿入)され、宿主細胞へ導入される。該ベクターとしては、挿入したポリヌクレオチドを安定に保持するものであれば特に制限されず、例えば宿主に大腸菌を用いるのであれば、クローニング用ベクターとしてpBluescriptベクター(Stratagene社製)などが好ましいが、市販の種々のベクターを利用することができる。本発明のポリペプチドを生産する目的としてベクターを用いる場合には、特に発現ベクターが有用である。発現ベクターとしては、試験管内、大腸菌内、培養細胞内、生物個体内でポリペプチドを発現するベクターであれば特に制限されないが、例えば、試験管内発現であればpBESTベクター(プロメガ社製)、大腸菌であればpETベクター(Invitrogen社製)、培養細胞であればpME18S-FL3ベクター、生物個体であればpME18Sベクター(Mol Cell Biol. 8:466-472(1988))などを例示することができる。ベクターへの本発明のポリヌクレオチドの挿入は、常法により、例えば、制限酵素サイトを用いたリガーゼ反応により行うことができる。
【0092】
上記宿主細胞としては特に制限はなく、目的に応じて種々の宿主細胞が用いられる。ポリペプチドを発現させるための細胞としては、例えば、細菌細胞(例:ストレプトコッカス、スタフィロコッカス、大腸菌、ストレプトミセス、枯草菌)、昆虫細胞(例:ドロソフィラS2、スポドプテラSF9)、動物細胞(例:CHO、COS、HeLa、C127、3T3、BHK、HEK293、Bowes メラノーマ細胞)および植物細胞を例示することができる。宿主細胞へのベクター導入は、例えば、リン酸カルシウム沈殿法、電気パルス穿孔法(Current protocols in Molecular Biology edit. Ausubel et al. (1987) Publish. John Wiley & Sons.Section 9.1-9.9)、リポフェクション法、マイクロインジェクション法などの公知の方法で行うことが可能である。
【0093】
宿主細胞において発現したポリペプチドを小胞体の内腔に、細胞周辺腔に、または細胞外の環境に分泌させるために、適当な分泌シグナルを目的のポリペプチドに組み込むことができる。これらのシグナルは目的のポリペプチドに対して内在性であっても、異種シグナルであってもよい。
【0094】
上記製造方法におけるポリペプチドの回収は、本発明のポリペプチドが培地に分泌される場合は、培地を回収する。本発明のポリペプチドが細胞内に産生される場合は、その細胞をまず溶解し、その後にポリペプチドを回収する。
【0095】
組換え細胞培養物から本発明のポリペプチドを回収し精製するには、硫酸アンモニウムまたはエタノール沈殿、酸抽出、アニオンまたはカチオン交換クロマトグラフィー、ホスホセルロースクロマトグラフィー、疎水性相互作用クロマトグラフィー、アフィニティクロマトグラフィー、ヒドロキシルアパタイトクロマトグラフィーおよびレクチンクロマトグラフィーを含めた公知の方法を用いることができる。
【0096】
また、動物の生体内で本発明のポリヌクレオチド(DNA)を発現させる方法としては、本発明のDNAを適当なベクターに組み込み、例えば、レトロウィルス法、リポソーム法、カチオニックリポソーム法、アデノウィルス法などにより生体内に導入する方法などが挙げられる。これにより、癌治療を行うことが可能である。用いられるベクターとしては、例えば、アデノウイルスベクターやレトロウイルスベクターなどが挙げられるが、これらに制限されない。ベクターへの本発明のDNAの挿入などの一般的な遺伝子操作は、常法に従って行うことが可能である(Molecular Cloning ,5.61-5.63)。生体内への投与は、ex vivo法であっても、in vivo法であってもよい。
【0097】
本発明のポリペプチドもしくはベクターを用いて細胞をアポトーシス誘導する方法もまた本発明に含まれる。該方法の好ましい態様においては、以下の工程(a)または(b)を含む方法である。
(a)本発明のポリペプチド、または本発明のベクターを細胞へ導入する工程
(b)本発明のポリペプチドと細胞とを接触させる工程
【0098】
上記工程(b)におけるポリペプチドは、細胞レセプターを認識するタンパク質との融合ポリペプチド(キメラ酵素)であることが好ましい。
【0099】
本発明のアポトーシス誘導剤は、癌細胞に対してアポトーシスを誘導することにより、癌細胞の増殖を抑制(阻止)もしくは癌細胞を死滅させる作用を有する。従って、本発明は、本発明のアポトーシス誘導剤を有効成分とする、抗癌剤(抗腫瘍剤)を提供する。
【0100】
本発明のタンパク質(ポリペプチド)がDNAのグアニン残基をADPリボシル化することにより、癌細胞のアポトーシスあるいは癌細胞における遺伝子の突然変異が誘発され、その結果、癌の発生または増殖を抑制するものと考えられる。
【0101】
本発明の上記アポトーシス誘導剤もしくは抗癌剤が適用可能な細胞の生物類は特に制限されないが、例えば、ヒト、マウス、ラット、イヌ等を挙げることができる。
【0102】
本発明の「抗癌剤」は、「癌細胞増殖抑制剤」、「癌細胞発生抑制剤」、「腫瘍増殖抑制剤」、「癌治療剤」、あるいは「制癌剤」等と表現することも可能である。また、本発明の薬剤は、「医薬品」、「医薬組成物」、「治療用医薬」等と表現することもできる。
【0103】
なお、本発明における「治療」には、癌の発生を予め抑制し得る予防的な効果も含まれる。また、癌細胞(組織)に対して、必ずしも、完全な治療効果を有する場合に限定されず、部分的な効果もしくは改善効果を有する場合であってもよい。
【0104】
また、食用貝の持つ毒性の原因は、本発明のタンパク質(例えば、CARP-1)の有するADPリボシル化活性に起因するものと考えられる。従って、本発明のタンパク質の発現を抑制する物質、または本発明のタンパク質の有するADPリボシル化活性を阻害する物質を貝類に施すことにより、毒性が除去された貝類を取得することが可能である。
【0105】
従って本発明は、本発明のタンパク質の発現もしくは機能阻害物質を有効成分とする、貝類毒性除去剤を提供する。
【0106】
上記発現阻害物質としては、本発明の遺伝子からの転写、または該転写による転写産物からの翻訳を阻害する物質が含まれる。
【0107】
上記発現阻害物質としては例えば、本発明のポリペプチドをコードする遺伝子の発現を抑制するポリヌクレオチドが挙げられる。このようなポリヌクレオチドには、アンチセンスポリヌクレオチド(アンチセンスDNA/RNA;本発明のポリペプチドをコードする遺伝子の転写産物と相補的なアンチセンスRNA、および該RNAをコードするDNA)やリボザイム(本発明のポリペプチドをコードする遺伝子の転写産物を特異的に開裂するリボザイム活性を有するRNAをコードするDNA)、およびRNAi効果を有する核酸(siRNA)が含まれる。
【0108】
アンチセンスポリヌクレオチドが標的遺伝子の発現を抑制する作用としては、以下のような複数の要因が存在する。すなわち、三重鎖形成による転写開始阻害、RNAポリメラーゼによって局部的に開状ループ構造がつくられた部位とのハイブリッド形成による転写抑制、合成の進みつつあるRNAとのハイブリッド形成による転写阻害、イントロンとエキソンとの接合点でのハイブリッド形成によるスプライシング抑制、スプライソソーム形成部位とのハイブリッド形成によるスプライシング抑制、mRNAとのハイブリッド形成による核から細胞質への移行抑制、キャッピング部位やポリ(A)付加部位とのハイブリッド形成によるスプライシング抑制、翻訳開始因子結合部位とのハイブリッド形成による翻訳開始抑制、開始コドン近傍のリボソーム結合部位とのハイブリッド形成による翻訳抑制、mRNAの翻訳領域やポリソーム結合部位とのハイブリッド形成によるペプチド鎖の伸長阻止、および核酸とタンパク質との相互作用部位とのハイブリッド形成による遺伝子発現抑制などである。これらは、転写、スプライシング、または翻訳の過程を阻害して、標的遺伝子の発現を抑制する(平島および井上「新生化学実験講座2 核酸IV 遺伝子の複製と発現」,日本生化学会編,東京化学同人,pp.319-347,1993)。
【0109】
本発明で用いられるアンチセンス核酸(ポリヌクレオチド)は、上記のいずれの作用で標的遺伝子の発現を抑制してもよい。一つの態様としては、遺伝子のmRNAの5'端近傍の非翻訳領域に相補的なアンチセンス配列を設計すれば、遺伝子の翻訳阻害に効果的と考えられる。しかし、コード領域もしくは3'側の非翻訳領域に相補的な配列も使用し得る。このように、遺伝子の翻訳領域だけでなく非翻訳領域の配列のアンチセンス配列を含むポリヌクレオチドも、本発明で利用されるアンチセンスポリヌクレオチドに含まれる。使用されるアンチセンスポリヌクレオチドは、適当なプロモーターの下流に連結され、好ましくは3'側に転写終結シグナルを含む配列が連結される。アンチセンスポリヌクレオチド配列は、標的遺伝子またはその一部と相補的な配列であることが好ましいが、遺伝子の発現を有効に阻害できる限り、完全に相補的でなくてもよい。転写されたRNAは、標的とする遺伝子の転写産物に対して好ましくは90%以上、最も好ましくは95%以上の相補性を有する。アンチセンス配列を用いて、効果的に標的遺伝子の発現を阻害するには、アンチセンスポリヌクレオチドは、アンチセンス効果を引き起こすために、少なくとも15ヌクレオチド以上、好ましくは100ヌクレオチド、さらに好ましくは500ヌクレオチド以上の鎖長を有し、通常、3000ヌクレオチド以内、好ましくは2000ヌクレオチド以内の鎖長を有する。
【0110】
該アンチセンスポリヌクレオチドは、例えば、本発明のポリペプチドをコードするポリヌクレオチド(例えば、配列番号:1)の配列情報を基にホスホロチオネート法(Stein, 1988 Physicochemical properties of phosphorothioate oligodeoxynucleotides. Nucleic Acids Res 16, 3209-21 (1988))などにより調製することが可能である。
【0111】
内在性遺伝子の発現の抑制は、また、リボザイムをコードする核酸(ポリヌクレオチド)を利用して行うことも可能である。リボザイムとは触媒活性を有するRNA分子のことをいう。リボザイムには種々の活性を有するものがあるが、中でもRNAを切断する酵素としてのリボザイムの研究により、RNAの部位特異的な切断を目的とするリボザイムの設計が可能となった。リボザイムには、グループIイントロン型や、RNasePに含まれるM1RNAのように400ヌクレオチド以上の大きさのものもあるが、ハンマーヘッド型やヘアピン型と呼ばれる40ヌクレオチド程度の活性ドメインを有するものもある(小泉誠および大塚栄子, (1990) 蛋白質核酸酵素,35:2191)。
【0112】
例えば、ハンマーヘッド型リボザイムの自己切断ドメインは、G13U14C15のC15の3'側を切断するが、活性にはU14が9位のAと塩基対を形成することが重要とされ、15位の塩基はCの他にAまたはUでも切断されることが示されている(M.Koizumiら,(1988) FEBS Lett.228:225)。リボザイムの基質結合部を標的部位近傍のRNA配列と相補的になるように設計すれば、標的RNA中のUC、UUまたはUAという配列を認識する制限酵素的なRNA切断リボザイムを作出することが可能である(M.Koizumiら,(1988) FEBS Lett. 239:285、小泉誠および大塚栄子,(1990) 蛋白質核酸酵素,35:2191、 M.Koizumiら, (1989) Nucleic Acids Res. 17:7059)。
【0113】
また、ヘアピン型リボザイムも、本発明の目的のために有用である。ヘアピン型リボザイムは、例えばタバコリングスポットウイルスのサテライトRNAのマイナス鎖に見出される(J.M.Buzayan Nature 323:349,1986)。このリボザイムも、標的特異的なRNA切断を起こすように設計できることが示されている(Y.Kikuchi およびN.Sasaki (1992) Nucleic Acids Res. 19:6751、 菊池洋, (1992) 化学と生物 30:112)。
【0114】
また、本発明の上記siRNAの好ましい態様としては、本発明の遺伝子に対してRNAi(RNA interference;RNA干渉)効果を有する二本鎖RNA(siRNA)を挙げることができる。より具体的には、配列番号:1に記載の塩基配列の部分配列に対するセンスRNAおよびアンチセンスRNAからなる二本鎖RNA(siRNA)を挙げることができる。
【0115】
さらに、本発明の上記RNAを発現し得るDNA(ベクター)もまた、本発明の遺伝子の発現を抑制し得る化合物の好ましい態様に含まれる。例えば、本発明の上記二本鎖RNAを発現し得るDNA(ベクター)は、該二本鎖RNAの一方の鎖をコードするDNA、および該二本鎖RNAの他方の鎖をコードするDNAが、それぞれ発現し得るようにプロモーターと連結した構造を有するDNAである。本発明の上記DNAは、当業者においては、一般的な遺伝子工学技術により、適宜作製することができる。より具体的には、本発明のRNAをコードするDNAを公知の種々の発現ベクターへ適宜挿入することによって、本発明の発現ベクターを作製することが可能である。
【0116】
また、本発明のタンパク質のADPリボシル化酵素活性を阻害する活性を有する化合物には、貝類毒性除去剤としての新たな用途があることが、本発明者によって見出された。従って、本発明は、ADPリボシル化酵素活性を阻害する活性を有する化合物を有効成分として含む、貝類毒性除去剤を提供する。
【0117】
ベンズアミド、3-アミノベンズアミド、5-アミノ-2,3-ジヒドロ-1,4-フタルアジンジオンは、DNA ADPリボシル化酵素ピエリシン-1および-2の活性も阻害することが知られている(非特許文献8、13)ことから、これらの化合物は、本発明の貝類毒性除去剤として有用である。
【0118】
また、ガングリオシドG(Q1bα)にはNADの立体構造と類似する部分が有るため百日咳毒素の酵素活性を阻害すると報告されており(横山三紀、実験医学, 14 (2), 409-41 1, 1996)、本発明のタンパク質の機能を阻害するものと考えられる。従って、本発明の好ましい態様としては、ベンズアミド、3-アミノベンズアミド、5-アミノ-2,3-ジヒドロ-1,4-フタルアジンジオン、および、ガングリオシドG(Q1bα)からなる群より選択される少なくとも1以上の化合物を有効成分として含む、貝類毒性除去剤を提供する。
【0119】
また、本発明のポリペプチドは、ADPリボシル化活性を有することから、ADPリボシル化のための研究用試薬としても有用である。従って本発明は、本発明のポリペプチドを有効成分として含むADPリボシル化試薬を提供する。
【0120】
さらに、本発明のポリペプチドもしくは上記ADPリボシル化試薬と、ポリヌクレオチド(DNA)とを接触させる工程を含む、該ポリヌクレオチドをADPリボシル化する方法もまた本発明に含まれる。
【0121】
本発明の方法によってADPリボシル化されたグアニン残基(N2-ADPリボシル化dG付加体)は、この部位でポリメラーゼの伸長反応を停止させる。その結果、該部位において突然変異が起こるものと考えられる。即ち、本発明の方法によってADPリボシル化されたグアニン残基を有するプラスミドを、通常のDNA修復システムを有する細胞へ導入することにより、DNAに対して突然変異を導入させることができる(Shiotani, B., et. Al., (2005) Mutat. Res. 512, 150-155.)。従って、本発明の方法は、DNAに対して突然変異を導入するために利用することも可能である。
【0122】
本発明の薬剤は、生理学的に許容される担体、賦形剤、あるいは希釈剤等と混合し、医薬組成物として経口、あるいは非経口的に投与することができる。経口剤としては、顆粒剤、散剤、錠剤、カプセル剤、溶剤、乳剤、あるいは懸濁剤等の剤型とすることができる。非経口剤としては、注射剤、点滴剤、外用薬剤、あるいは座剤等の剤型を選択することができる。注射剤には、皮下注射剤、筋肉注射剤、あるいは腹腔内注射剤等を示すことができる。外用薬剤には、経鼻投与剤、あるいは軟膏剤等を示すことができる。主成分である本発明の薬剤を含むように、上記の剤型とする製剤技術は公知である。
【0123】
例えば、経口投与用の錠剤は、本発明の薬剤に賦形剤、崩壊剤、結合剤、および滑沢剤等を加えて混合し、圧縮整形することにより製造することができる。賦形剤には、乳糖、デンプン、あるいはマンニトール等が一般に用いられる。崩壊剤としては、炭酸カルシウムやカルボキシメチルセルロースカルシウム等が一般に用いられる。結合剤には、アラビアゴム、カルボキシメチルセルロース、あるいはポリビニルピロリドンが用いられる。滑沢剤としては、タルクやステアリン酸マグネシウム等が公知である。
【0124】
本発明の薬剤を含む錠剤は、マスキングや、腸溶性製剤とするために、公知のコーティングを施すことができる。コーティング剤には、エチルセルロースやポリオキシエチレングリコール等を用いることができる。
【0125】
また注射剤は、主成分である本発明の薬剤を適当な分散剤とともに溶解、分散媒に溶解、あるいは分散させることにより得ることができる。分散媒の選択により、水性溶剤と油性溶剤のいずれの剤型とすることもできる。水性溶剤とするには、蒸留水、生理食塩水、あるいはリンゲル液等を分散媒とする。油性溶剤では、各種植物油やプロピレングリコール等を分散媒に利用する。このとき、必要に応じてパラベン等の保存剤を添加することもできる。また注射剤中には、塩化ナトリウムやブドウ糖等の公知の等張化剤を加えることができる。更に、塩化ベンザルコニウムや塩酸プロカインのような無痛化剤を添加することができる。
【0126】
また、本発明の薬剤を固形、液状、あるいは半固形状の組成物とすることにより外用剤とすることができる。固形、あるいは液状の組成物については、先に述べたものと同様の組成物とすることで外用剤とすることができる。半固形状の組成物は、適当な溶剤に必要に応じて増粘剤を加えて調製することができる。溶剤には、水、エチルアルコール、あるいはポリエチレングリコール等を用いることができる。増粘剤には、一般にベントナイト、ポリビニルアルコール、アクリル酸、メタクリル酸、あるいはポリビニルピロリドン等が用いられる。この組成物には、塩化ベンザルコニウム等の保存剤を加えることができる。また、担体としてカカオ脂のような油性基材、あるいはセルロース誘導体のような水性ゲル基材を組み合わせることにより、座剤とすることもできる。
【0127】
本発明の薬剤を遺伝子治療剤として使用する場合は、本発明の薬剤を注射により直接投与する方法のほか、核酸が組込まれたベクターを投与する方法が挙げられる。上記ベクターとしては、アデノウイルスベクター、アデノ随伴ウイルスベクター、ヘルペスウイルスベクター、ワクシニアウイルスベクター、レトロウイルスベクター、レンチウイルスベクター等が挙げられ、これらのウイルスベクターを用いることにより効率よく投与することができる。
【0128】
また、本発明の薬剤をリポソームなどのリン脂質小胞体に導入し、その小胞体を投与することも可能である。例えば、本発明のポリペプチドもしくはベクターを保持させた小胞体をリポフェクション法により所定の細胞に導入する。そして、得られる細胞を例えば静脈内、動脈内等に全身投与する。癌組織等に局所的に投与することもできる。
【0129】
本発明の薬剤は、安全とされている投与量の範囲内において、ヒトを含む哺乳動物に対して、必要量(有効量)が投与される。本発明の薬剤の投与量は、剤型の種類、投与方法、患者の年齢や体重、患者の症状等を考慮して、最終的には医師または獣医師の判断により適宜決定することができる。
【0130】
また本発明は、本発明のアポトーシス誘導剤もしくは抗癌剤、または該薬剤の候補化合物のスクリーニング方法を提供する。
【0131】
本発明のスクリーニング方法の好ましい態様においては、本発明のタンパク質(例えば、CARP-1タンパク質)の発現量または活性を指標とする方法である。即ち、本発明のタンパク質の発現もしくは活性を上昇させる化合物は、アポトーシス誘導作用を有し、抗癌活性を有することが期待される。
【0132】
本発明の上記方法においては、まず、本発明のタンパク質もしくは本発明のタンパク質を発現する細胞と、被検化合物を接触させる。
【0133】
本方法に用いる「細胞」は、特に制限されないが、好ましくは貝類由来の細胞である。「本発明のタンパク質を発現する細胞」としては、内在性の本発明のタンパク質を発現している細胞、または外来性の本発明の遺伝子が導入され、該遺伝子が発現している細胞を利用することができる。外来性の本発明の遺伝子が発現した細胞は、通常、本発明の遺伝子が挿入された発現ベクターを宿主細胞へ導入することにより作製することができる。該発現ベクターは、一般的な遺伝子工学技術によって作製することができる。
【0134】
本発明のスクリーニング方法に供する被検化合物としては、特に制限はない。例えば、天然化合物、有機化合物、無機化合物、タンパク質、ペプチドなどの単一化合物、並びに、化合物ライブラリー、遺伝子ライブラリーの発現産物、細胞抽出物、細胞培養上清、発酵微生物産生物、海洋生物抽出物、植物抽出物等が挙げられるが、これらに限定されない。
【0135】
また、これらの被検化合物は必要に応じて適宜標識して用いることができる。標識としては、例えば、放射標識、蛍光標識等を挙げることができる。
【0136】
本発明のタンパク質を発現する細胞への被検化合物の「接触」は、通常、本発明のタンパク質を発現する細胞の培養液に被検化合物を添加することによって行うが、この方法に限定されない。被検化合物がタンパク質等の場合には、該タンパク質を発現するDNAベクターを、該細胞へ導入することにより、「接触」を行うことができる。
【0137】
本方法においては、次いで、本発明のタンパク質を発現する細胞における本発明のタンパク質の発現量または活性を測定する。発現量の測定は、当業者が簡便に行い得るものであり、一般的な方法、例えばノーザンブロット法、ウェスタンブロット法等により適宜実施することが可能である。例えば、本発明のタンパク質を発現する細胞に接触させた被検化合物に付した標識を指標にして測定することも可能である。
【0138】
尚、本発明において「発現」とは、タンパク質をコードする遺伝子からの転写(mRNAの生成)、または該遺伝子の転写産物からの翻訳のいずれの場合をも意味する。
【0139】
また、本発明のタンパク質の活性を指標にして上記スクリーニング方法を実施する場合には、本発明のタンパク質の上述の機能(活性)、例えば、本発明のタンパク質の有するADPリボシル化活性を指標とすることができる。本発明のタンパク質の活性の測定は、当業者においては、公知の方法によって適宜実施することができる。
【0140】
本方法においては、次いで、被検化合物を接触させない場合と比較して、本発明のタンパク質の発現量または活性を上昇させる化合物を選択する。
【0141】
上記スクリーニング方法によって取得される化合物は、例えば、アポトーシス誘導剤、または抗癌剤となることが期待される。
【0142】
また、本発明のタンパク質(例えば、CARP-1タンパク質)の発現量または活性を低下させる化合物は、貝類毒性除去作用を有することが期待される。
【0143】
従って本発明は、本発明のタンパク質の発現量または活性を低下させる化合物を選択することを特徴とする、貝類毒性除去剤のスクリーニング方法を提供する。
【0144】
上記スクリーニング方法の好ましい態様としては、以下の工程(a)〜(c)を含む方法である。
(a)本発明のタンパク質もしくは本発明のタンパク質を発現する細胞と、被検化合物を接触させる工程
(b)本発明のタンパク質の発現量または活性を測定する工程
(c)被検化合物を接触させない場合と比較して、前記発現量または活性を低下させる化合物を選択する工程
【実施例】
【0145】
以下本発明を実施例により詳細に説明するが、本発明はこれら実施例により制限されるものではない。なお、本実験に際し、下記の材料・試薬等を用いた。
【0146】
日本の太平洋沿岸から採取された貝類は全て、東京の市場から得て、生きたまま研究室に輸送された。mRNA抽出用途の場合には、輸送されたチョウセンハマグリを滅菌人工海水中で生きたまま保存し、1時間以内に用いた。ホスホジエステラーゼIIおよびミクロコッカスヌクレアーゼは、Worthington(米国ニュージャージー州レイクウッド)から購入し、仔ウシ胸腺DNA、細菌アルカリホスファターゼ、およびβ-NADはSigma(米国ミズーリ州セントルイス)から購入し、β-[アデニレート-32P]NAD(32P-NAD)は、PerkinElmer(米国マサチューセッツ州ボストン)から購入した。ExTaqポリメラーゼは、タカラバイオ(滋賀県大津市)から購入した。KOD-Plusポリメラーゼは東洋紡(大阪市)から購入した。トリプシンおよびオリゴヌクレオチドは、Invitrogen(米国カリフォルニア州カールスバッド)から、CEL 300 PEIポリエチレンイミン-セルロースTLCシートは、Macherey-Nagel(ドイツ、デューレン)から購入した。トリプシン処置ピエリシン-1は、既報(Watanabeら、2004a)に従い調製した。特に明記していない限り、他の材料は全て和光純薬工業(大阪市)から購入した。
【0147】
〔実施例1〕種々の貝類からの抽出物におけるDNA ADP-リボシルトランスフェラーゼのスクリーニング
14種類の貝類の各標本の全軟組織を乳鉢と乳棒によって氷上で、1 mM DTTを含む50 mMトリス-塩酸、pH 7.5の3倍量と共にホモジナイズした後、1分間超音波処理した。このホモジネートを14,000 x gで5分間遠心して、粗抽出物と呼ばれる上清を-80℃で保存した。仔ウシ胸腺DNA 5μg、18.5 kBq 32P-NADおよび10μM β-NADを、反応緩衝液(50 mMトリス-塩酸、pH 7.5、1 mM EDTA、50 mM NaCl、および1 mM DTT)50μlにおいて、当初の軟組織0.03〜1.5 mgに対応する貝類からの粗抽出物1〜50μgタンパク質と共にインキュベートした。プロテナーゼK(和光)1μgを反応物に加えて、37℃でさらに30分間インキュベートした。フェノール抽出、クロロホルム抽出、および酢酸アンモニウム塩によるエタノール沈殿によってDNAを回収した。回収したDNAを、5 mM CaCl2、ミクロコッカスヌクレアーゼ2 U、およびホスホジエステラーゼII 0.02 Uを含む20 mMビストリス-塩酸(pH 6.5)において10μlの容量で処理した。バックグラウンドのノイズを検出するために、DNA混合物3μlをPEI-セルロースTLCシート上に直ちにスポットした。DNA混合物の残りを、37℃で12時間インキュベートすることによってデオキシリボヌクレオチド3'リン酸まで消化して、混合物の3μlをTLCシートにスポットした。6 M酢酸、0.1 M LiClおよび3 M尿素を用いて展開した後、TLCプレートをフジイメージングプレート(富士写真フィルム、東京)に露出して、バイオイメージアナライザ(BAS-2500;富士写真フィルム)によってDNA付加物を検出した。
【0148】
検出された付加物のスポットの放射活性のデータを表1に示し、ADP-リボシル化DNA付加物の形成の典型的な例を図1に示す。
【0149】
【表1】
【0150】
上記表1における相対活性(PSL)は、タンパク質マイクログラム当たりのDNA付加物の比重分析によって計算したものである。PSL(photo stimulated luminescence)はBio Image Analyzer用の放射活性の単位である。ピエリジン-1の相対活性は約10 PSL/pg タンパク質であった。腹足類動物(Gastropods)については短剣印で示す。
【0151】
チョウセンハマグリ(M. lamarckii)、アサリ(R. philippinarum)およびヤマトシジミ(C. japonica)からの粗抽出物をDNAおよび32P-NADと反応させると、付加物のスポットはTLCシート上で明らかに検出され、1790〜6900 PSL/μgタンパク質を示し、スポットのRf値は0.05であり、これはピエリシン-1によって産生された付加物のRf値と同じであった。他のデオキシリボヌクレオチド、ADP-リボースおよびNADのRf値は0.30〜0.95であった。これらの粗抽出物を、DNAおよび32P-NADとの反応の前に、50μgプロナーゼまたは50μgプロテナーゼKによって処置すると、付加物のスポットは検出されず、これらの付加物が酵素反応の結果であったことを示唆している。
【0152】
マテガイ(Solen strictus)、ウバガイ(Spisula sachalinensis)、およびムラサキイガイ(Mytilus galloprovincialis)からの粗抽出物の場合、付加物のスポットも同様に、同じRf値で検出された。これらのPSL値は、189〜450/μgタンパク質であった。エゾバイ(Buccinum middendorffi)、マガキ(Crassostrea gigas)、およびタイラギ(Atrina pinnata)からの粗抽出物の場合、同じRf値でかすかな付加物スポットを生じた。サザエ(Turbo cornutus)、バカガイ(Mactra chinensis、hen clam)およびミルクイ(Tresus keenae)をタンパク質源として用いた場合には、検出可能なシグナルは出現しなかった。
【0153】
〔実施例2〕二枚貝の酵素の存在下でのDNAおよびβ-NADのインキュベーションによって形成された反応産物の分析
実施例1のように、チョウセンハマグリからの抽出物試料は最高の活性を示し、TLCによって検出されたデオキシリボヌクレオチド付加物を生じたことから、以下に示すように生成された付加物を、細菌のアルカリホスファターゼによって脱リン酸化後にHPLCによってさらに分析した。
【0154】
詳しくは、チョウセンハマグリ、アサリおよびヤマトシジミからの粗抽出物に硫酸アンモニウムを60〜90%飽和となるように加えて、遠心した。50 mMトリス-塩酸、pH 7.5に対して透析した後、硫酸アンモニウム沈殿試料10 mgを仔ウシ胸腺DNA 1 mgおよびβ-NAD 20μmolと共に50 mMトリス-塩酸、pH 7.5(アサリに関してはpH 8.5)、1 mM EDTA、および50 mM NaClにおいて全量10 mlにおいて37℃で4時間インキュベートした。DNAを回収して、ミクロコッカスヌクレアーゼ200 U、ホスホジエステラーゼII 2 U、および5 mM CaCl2によって、20 mMビストリス-塩酸(pH 6.5)2 mlにおいて37℃で4時間、デオキシリボヌクレオチド3'リン酸まで消化した。細菌アルカリホスファターゼ(最終濃度、1.5 U/ml)、トリス塩基(40 mM)およびZnCl2(5 mM)を加えて、37℃で4時間インキュベートすることによって、試料をデオキシリボヌクレオシドまでさらに消化した。
【0155】
遠心後、反応混合物の少量に、Shimadzu SPD 10Avp光ダイオードアレイ検出器およびDevelosil RPAQUEOUSカラム(4.6×250 mm;野村化学、愛知県瀬戸市)を備えたShimadzu LC-10Aシステム(島津製作所、京都市)によって、HPLCを40℃で流速1 ml/分で行った。溶媒系は、以下の通りであった:0.25%トリエチルアミン-酢酸(pH 7.0)中で3.5%アセトニトリルを0〜15分、その後0.25%トリエチルアミン-酢酸(pH 7.0)においてアセトニトリル16.8%まで直線勾配で45分。
【0156】
反応混合物の少量を、HP1000 HPLCシステム(Hewlett-Packard、米国カリフォルニア州パロアルト)を備えたMicromass ZQ 2000機器(英国、マンチェスター)エレクトロスプレーイオン化質量分析(ESI-MS)によっても分析した。
【0157】
その結果、図2Aに示すように、dGピークの高さは減少し、そのかわりに二つのピークが保持時間21.5および22.5分で検出され、これは、ピエリシン-1によって形成されたN2-(ADP-リボス-1-イル)-2'-デオキシグアノシンのα体およびβ体の保持時間と一致した。一方、dC、dA、およびTの量は不変のままであった。保持時間21.5および22.5分でのピーク分画のUVスペクトルは、N2-(ADP-リボース-1-イル)-2'-デオキシグアノシンの保持時間とまさに同じであった(図2B)。さらに、LC-ESI-MS分析から、これらのピーク分画における化合物が、m/z 809で分子イオンピークを有すること、デオキシリボース部分の喪失によって生じるm/z693でイオンピークを有すること、およびHPLC溶出剤に由来するトリエチルアミン付加に対応するm/z 910、およびm/z 809で親質量に対応するイオンピークを有することを示した(図6AB)。
【0158】
このように、N2-(ADP-リボース-1-イル)-2'-デオキシグアノシンは、チョウセンハマグリの酵素の存在下で仔ウシ胸腺DNAおよびβ-NADの反応を通して産生されることが証明された。その上、N2-(ADP-リボス-1-イル)-2'-デオキシグアノシンは、アサリおよびヤマトシジミからの抽出物試料によって類似の条件で形成されることが示された。
【0159】
〔実施例3〕チョウセンハマグリからのDNA ADP-リボシルトランスフェラーゼの精製
チョウセンハマグリ(鹿島産)標本5個の、かなり低い酵素活性を有する鰓、外套膜および中腸を除く軟組織(150 g)から調製した粗抽出物を、上述したように硫酸アンモニウム沈殿によって分画した。
【0160】
その結果、DNA付加物形成活性のほとんどは、硫酸アンモニウム60〜90%飽和で得られた沈殿物において検出された。この分画の付加物の形成活性は37℃でpH 7.5〜8.5でほぼ同じレベルで認められた。活性は、4時間まで時間依存的に増加した。
【0161】
次に、活性分画をCM52カラムにおいて精製した。即ち、沈殿物を20 mMリン酸ナトリウム緩衝液、pH 6.0に対して透析して、CM52カルボキシメチル-セルロースカラム(25×150 mm、Whatman、イギリス、ケント)に適用した。カラムを20 mMリン酸ナトリウム緩衝液によって溶出して、保持されたタンパク質を20 mMリン酸ナトリウム緩衝液において0〜400 mM NaClの直線勾配1000 mlによって流速80 ml/時間で溶出した。
【0162】
その結果、開始材料の酵素活性の15%が、20 mMリン酸ナトリウム緩衝液によって溶出し、活性の残りの43%はその後20 mMリン酸ナトリウム緩衝液において0〜400 mM NaClによって回収された。20 mMリン酸ナトリウム緩衝液において0〜400 mM NaClで溶出された酵素活性を含む分画(286〜319 ml)を合わせて、Amicon Ultra-10k(Millipore、ベッドフォード、マサチューセッツ州)によって濃縮して、HiPrep Sephacryl S-100ゲル濾過カラム(16×600 mm、Pharmacia、スウェーデン、ウプサラ)に適用しさらに精製した。タンパク質を、150 mM NaClを含む20 mMリン酸ナトリウム緩衝液によって、流速30 ml/時間で分離して、酵素活性を含む分画(64〜74 ml)を合わせて、Amicon Ultra-10kによって濃縮した後、Mono-S HR 5/5(Pharmacia)を備えたFPLCを流速1 ml/分で行った。溶媒システムは以下の通りであった:0.25 M NaClから1 Mまでの直線勾配を20 mMリン酸ナトリウム緩衝液、pH 6.0において42.5分間。活性分画(12〜13 ml)をプールして、Amicon Ultra-10kによって0.2 mlまで濃縮した。上記の技法は全て4℃未満で行い、精製酵素を-80℃で保存した。
【0163】
DNA ADP-リボシルトランスフェラーゼ活性の同定および精製酵素の分子量の推定のために、既に記述されたとおりにゲル内酵素アッセイを行った(Watanabe, M., Enomoto, S., Takamura-Enya, T., Nakano, T., Koyama, K., Sugimura, T. and Wakabayashi, K. (2004a) Enzymatic properties of pierisin-1 and its N-terminal domain, a guanine-specific ADP-ribosyltransferase from the cabbage butterfly. J. Biochem., 135, 471-477.)。精製タンパク質の電気穿孔は、既に記述されたとおりに行った(Kanazawa, T., Watanabe, M., Matsushima-Hibiya, Y., Kono, T., Tanaka, N., Koyama, K., Sugimura, T. and Wakabayashi, K. (2001) Distinct roles for the N- and C-terminal regions in the cytotoxicity of pierisin-1, a putative ADP-ribosylating toxin from cabbage butterfly, against mammalian cells. Proc. Natl. Acad. Sci. USA, 98, 2226-2231.)。
【0164】
その結果、精製タンパク質のSDS-PAGEおよびゲル内酵素活性アッセイによって、分子量約20 kDaに対応する単一の主なバンドが明らかとなった(図3)。SephacrylおよびMono-Sカラムクロマトグラフィーの段階において、付加物形成活性は、チョウセンハマグリの20 kDaタンパク質を含む分画に限って認められ、他の分画では認められなかった。ハマグリ5個から、精製タンパク質0.48 mgが得られ、付加物形成活性は、抽出物と比較して870倍上昇した。その上、精製酵素によって形成された付加物は、HPLC、UVスペクトル、およびLC-MS分析によってN2-(ADP-リボス-1-イル)-2’-デオキシグアノシンであることが確認された。これらのデータは、精製された酵素がハマグリ抽出物に存在する主要な活性成分であることを示している。
【0165】
ハマグリは一般的にDNA ADP-リボシルトランスフェラーゼを含む可能性が最も高いことから、本発明者らはチョウセンハマグリから精製された本発明の酵素をCARP-1と命名した。
【0166】
なお、以下の表2は、チョウセンハマグリのDNA ADP-リボシルトランスフェラーゼの精製段階を要約したものである。
【0167】
【表2】
【0168】
上記表2において、回収および精製の値は、32P-NADからDNAへ放射活性を組み入れる能力によって決定される酵素活性から算出した。
【0169】
〔実施例4〕ハマグリの酵素をコードするcDNAのPCR増幅、シークエンシング、およびクローニング
精製ハマグリ酵素20μgをトリプシンによって消化して、消化したペプチドを、TSKゲルODS-80Tsカラムを備えた逆相HPLC(東ソー、山口県周南市)によって分離した。単離されたペプチドのアミノ酸配列はProcise 494 HTタンパク質シークエンサー(Perkin-Elmer)によって決定した。総RNAを、Isogen(ニッポン・ジーン、富山県富山市)を用いてチョウセンハマグリの足筋から抽出した。チョウセンハマグリからのmRNAの完全長のcDNAを、GeneRacerキット(Invitrogen)を用いて総RNA調製物(2μg)から合成した。次に、合成されたcDNAに、ハマグリの酵素ペプチドHAAQAFYWLSVK(配列番号:3)およびEIHLAALTDTESSSEGYKENDYDVDT(配列番号:4)に対応する1μMセンスおよびアンチセンス縮重プライマー、またはセンス縮重プライマーおよび1μM 3'アンカープライマー(5'- CGCTACGTAACGGCATGACAGTG -3'/配列番号:5、GeneRacerキット(Invitrogen))を用いて、ExTaq DNAポリメラーゼによってハマグリの酵素特異的配列に関するPCR増幅を行った。5’末端cDNA断片を得るために、各1μMの5’アンカープライマー(5'- GGACACTGACATGGACTGAAGGAGTA -3'/配列番号:6、GeneRacerキット(Invitrogen))およびアンチセンスプライマー(5'- GCTTTTGTTCTCTTTTATCAGTGTATCT -3'/配列番号:7)を用いて、PCRを行った;配列は内部cDNA断片から選択した。得られた5’ cDNA断片にPCR直接シークエンシングを行った。完全長のハマグリ酵素cDNAを、5’アンカープライマーに隣接する配列である5’プライマー(5'- AGCGTTTACTTCCTCTTTCTCTTT -3'/配列番号:8)、およびポリ(A)伸長部に隣接する配列である3'プライマー(5'- GACTATCGTCGTTGTTTATTTTGA -3'/配列番号:9)を用いて、KOD-プラスDNAポリメラーゼによるPCR増幅によって得た。増幅された0.9 kb断片にPCR直接シークエンシングを行って、完全なcDNA配列を決定した。
【0170】
上述のようにチョウセンハマグリの足筋からのRNAおよび消化したペプチドの内部アミノ酸配列を用いて、本発明者らは完全なCARP-1をコードするcDNAを得た。単離されたcDNAのヌクレオチド配列を図4に示す。
【0171】
完全長のcDNAは、5’アンカー配列およびポリアデニル化伸長部を伴う853 bpからなった。最初のATGはヌクレオチド65位で始まり、ヌクレオチド611位のTAG終止コドンで終止した。ポリアデニル化シグナルの可能性がある配列(AATAAA)が、ポリ(A)配列に近いヌクレオチド834〜839位で認められ、真核細胞配列であることを示唆している。ORFによってコードされる推定のタンパク質は、計算分子量20,332を有するアミノ酸182個を含んだ。その上、精製ハマグリADP-リボシルトランスフェラーゼの消化物のペプチド配列9個は、ORFの推定アミノ酸配列の一部と同一であることが判明した(図4)。これらの結果から、単離されたcDNAにおけるORFは、CARP-1遺伝子のコード領域であると結論された。
【0172】
またクローンを構築するために、増幅された0.9 kb PCR断片を鋳型として用いた。PCR増幅CARP-1コード配列をpMAL-p2xプラスミドベクター(New England Biolabs、米国マサチューセッツ州ビバリー)に挿入した。ハマグリの酵素の遺伝子のコード領域を3'-隣接領域の217 bpと共に増幅して、プライマー対5'- CCTAAGCTTATGCCTGCTGGAAAATACCTATAC -3'(配列番号:10)および5'- GGAGAATTCGACTATCGTCGTTGTTTATTTTGA -3'(配列番号:11)を用いて、HindIIIおよびEcoRI制限部位をそれぞれ、5’および3’末端に導入した。ピエリシン-1および-2の場合からCARP-1遺伝子の産物は大腸菌に対して非常に毒性が強いことが予想されたことから、本発明者らは、PCR増幅配列をベクターのtacプロモーターとは反対方向にして、HindIII-EcoRI部位に挿入した。クローン10個をシークエンシングして、いずれのクローンも如何なるヌクレオチド置換も有しなかったことから、これをインビトロでCARP-1発現のために用いた。
【0173】
〔実施例5〕クローニングされたcDNAのインビトロ発現
酵素のcDNAのインビトロ発現は、既報に倣って実施した(Matsushima-Hibiya, Y., Watanabe, M., Kono, T., Kanazawa, T., Koyama, K., Sugimura, T. and Wakabayashi, K. (2000) Purification and cloning of pierisin-2, an apoptosis-inducing protein from the cabbage butterfly, Pieris brassicae. Eur. J. Biochem., 267, 5742-5750.)。簡単に説明すると、コード領域の5'末端にT7プロモーター配列を結合した5'プライマー(5'- TAATACGACTCACTATAGGGAGACCACCATGCCTGCTGGAAAATACCTATACCG -3'/配列番号:12)、および3'プライマー(5'- GACTATCGTCGTTGTTTATTTTGA -3'/配列番号:13)をPCRに用いて、酵素コード配列の上流にT7プロモーター配列を導入した。T7プロモーター配列を有するcDNAの増幅断片は、MEGAscript(Ambion、米国テキサス州オースチン)によって転写され、その後ウサギ網状赤血球溶解物によって翻訳された(Ambion)。
【0174】
インビトロ発現タンパク質の分子量はSDS-PAGEにおいて約20 kDaであり、精製された天然のCARP-1の分子量と同じであった。発現されたタンパク質をDNAおよび32P-NADと反応させた場合、付加物のスポットがTLCシートのRf値0.05において明らかに検出され、これは精製された天然のタンパク質によって産生された付加物のRf値と同じであった。その上、DNA付加物を細菌のアルカリホスファターゼによる脱リン酸化後にHPLCによってさらに分析すると、ほとんどの放射活性は保持時間21.0〜23.0分で回収され、N2-(ADP-リボス-1-イル)-2'-デオキシグアノシン(図7)の保持時間と一致した。このように、発現されたタンパク質は、明らかに、N2-(ADP-リボス-1-イル)-2'-デオキシグアノシンを産生した。この結果は、単離されたcDNAクローンから機能的に活性なタンパク質がインビトロで発現されたことを示している。
【0175】
精製天然タンパク質はHeLa細胞において20μg/mlで如何なる細胞障害作用も示さなかったが、タンパク質3、5および12.5μg/mlを電気穿孔によって導入した場合、細胞増殖速度はそれぞれ、77、58、および26%明らかに減少した。同様に、インビトロ発現CARP-1タンパク質は、10μg/mlでHeLa細胞に対して電気穿孔によって細胞障害性を示した。電気穿孔を行わない場合には細胞障害性を示さなかった。同様の結果はTMK-1細胞を用いても得られている。
【0176】
酵素とDDBJデータベースのタンパク質との全体的な相同性は、BLASTおよびFASTAの検索によって検出されなかったが、GLu-128、Arg-12、および46〜48位のSer-Thr-ThrがCARP-1遺伝子において検出され、これらはADP-リボシルトランスフェラーゼ活性のモチーフであると示唆されている(図5)。
【0177】
〔実施例6〕部位特異的変異誘発
さらに本発明者らは、ADP-リボシル化が、DNA付加物の形成に関与しているか否かを調べるために、以下に示すように127または128位のグルタミン酸残基を、部位特異的変異誘発によってアスパラギン酸(それぞれ、E127D、またはE128D)に置換して、変異したまたは変異していないクローンをインビトロで転写および翻訳した。
【0178】
即ち、所望の位置で変化した配列を含むDNA断片をオーバーラップ伸長PCR技術によって無傷の酵素cDNAサブクローンから増幅した。重なり合った5'および3'断片を得るために、5'断片に関してコード領域の5’末端にT7プロモーター配列を結合した5’プライマー(5'- TAATACGACTCACTATAGGGAGACCACCATGCCTGCTGGAAAATACCTATACCG -3'/配列番号:14)、および3'プライマー(プライマーA、5'- CCTTAACCAAAACGTCTTCATATT -3'/配列番号:15)、ならびに3'断片に関して5'プライマー(プライマーB、5'- AATATGAAGACGTTTTGGTTAAGG -3'/配列番号:16)および3'プライマー(5'- GACTATCGTCGTTGTTTATTTTGA -3'/配列番号:17)を用いて二つの異なるPCR反応を行った。プライマーAおよびBは互いに相補的であり、Glu128Aspに対応するヌクレオチドで変異を含む。Recochip(タカラバイオ)を用いて精製した後、精製5'-および3'-断片を互いに混合して、これを第二ラウンドのPCRの鋳型として用いて完全長の変異DNA断片を得た。このPCR産物をシークエンシングして、予定した変異の導入を確認した、Glu127Aspに対応するヌクレオチドで変異を含むDNA断片を得るために、プライマーAおよびBの配列がそれぞれ、5'- AACCAAAACTTCGTCATATTTTGC -3'(配列番号:18)および5'- GCAAAATATGACGAAGTTTTGGTT -3'(配列番号:19)であったことを除き、上記と同じプロトコールを用いた。得られたDNA断片を、上記のインビトロ発現系の鋳型として用いた。
【0179】
その結果、非変異対照クローンおよび変異クローンからのタンパク質発現効率は、[35S]メチオニンによる翻訳後にSDS-PAGEによって判断すると、ほぼ同じであった。E127D変異体は非変異DNAによる当初の活性の94%を維持したが、E128D変異体は当初の活性の99%を失った事から、Glu-128がDNA ADP-リボシルトランスフェラーゼ活性にとって必須のグルタミン酸であることが示唆された。
【図面の簡単な説明】
【0180】
【図1】図1は、様々な貝類の粗抽出物で形成されたADP-リボシル化DNA付加物の検出について示す写真である。仔ウシ胸腺DNAを粗抽出タンパク質1μg(グループ3-7)の存在下で32P-NADと反応させた。回収した後、ヌクレアーゼ分解の前(左レーン)および後(右レーン)にDNAサンプルをTLCシート上にスポットし、展開し、オートラジオグラフを行った。グループ1:タンパク質無し、グループ2:ピエリシン-1(10 pg:ポジティブコントロール)、グループ3:チョウセンハマグリ、グループ4:アサリ、グループ5:シジミ、グループ6:ムラサキイガイ。矢印はRf値0.05を示す。
【図2A】図2は、β-NADの存在下でチョウセンハマグリ由来の蛋白質精製物およびDNAのインキュベーションによって形成される反応物の分析について示すグラフである。(A)ピエリシン-1とインキュベートしたDNAの加水分解産物のHPLC溶出パターン(上段)、チョウセンハマグリ由来の蛋白質精製物とインキュベートしたDNAの加水分解産物のHPLC溶出パターン(中段)、あるいは蛋白質精製物無しでインキュベートしたDNAの加水分解産物のHPLC溶出パターン(下段)。サンプルはDevelosil RPAQUEOUSカラムに注入し、溶出液は262 nmでのUV吸光度を計測した。矢印は、N2-(ADP-リボス-1-イル)-2'-デオキシグアノシンに相当するピークを指す。
【図2B】図2Bは、図2Aに示す21.5および22.5分の保持時間で、ピーク分画における化合物について、フォトダイオードアレイ検出機を用いたUV吸収スペクトルを示すグラフである。
【図3】図3は、精製したCARP-1のSDS-PAGEの結果を示す写真である。精製したタンパク質1μgを15%ポリアクリルアミドゲルエレクトロポレーションによって分離し、クーマシーブリリアントブルーR-250で染色した(左レーン)。あるいはゲル内ADPリボシルトランスフェラーゼアッセイで評価した(右レーン)。
【図4】図4は、PCRダイレクトスクリーニングによって決定したCARP-1に対するcDNAのヌクレオチド配列および推定アミノ酸配列を示す図である。オープンリーディングフレームは65-613位にある。ポリアデニル化シグナルと予測される配列(834-839位)を、シャープ印によって示す。保存される可能性のあるアミノ酸は太字で示した。
【図5】図5は、ピエリシン-1およびADP-リボシル化トキシンと、CARP-1の相同性領域の推定アミノ酸配列をアライメントした図である。PT-1:PT(GenBank = P04977)、Pierisin-1(Watanabe, M., Kono, T., Matsushima-Hibiya, Y., Kanazawa, T., Nishisaka, N., Kishimoto, T., Koyama, K., Sugimura, T. and Wakabayashi, K. (1999) Molecular cloning of an apoptosis-inducing protein, pierisin, from cabbage butterfly: possible involvement of ADP-ribosylation in its activity. Proc. Natl. Acad. Sci. USA, 96, 10608-10613.)、CT-A:コレラトキシン(GenBank = 1001196A)のAサブユニット。保存されたアルギニン、Ser-Thr-Ser/Thrモチーフおよびグルタミン酸残基を枠で囲んだ。完全に保存されたアミノ酸にはアスタリスク(*)を付けた。
【図6A】図6は、β-NAD存在下において、DNAおよびチョウセンハマグリ由来の酵素のインキュベーションによって形成される反応物のLC-ESI-MS分析の結果を示す図である。(A)HPLCプロフィール(UV)、およびチョウセンハマグリ由来の酵素とインキュベートしたDNAの加水分解産物のイオンクロマトグラム(m/z 809、693および910)を示す。ピエリシン-1とインキュベーションすることによって形成される反応物の解析でも、同様の結果が得られた。
【図6B】図6Bは、図6Aに示される25.5分(上段)および26分(下段)の保持時間で、ピーク分画における化合物のマススペクトラムを示す。
【図7】図7は、32PNADの存在下において、DNAおよびインビトロ翻訳されたCARP-1のインキュベーションによって形成される反応物のHPLC溶出パターンを示す図である。放射活性を有する反応物は、β-NADの存在下においてチョウセンハマグリ由来の酵素とインキュベーションしたDNAと混合した。DNAは加水分解し、Develosil RPAQUEOUSカラムに注入した。溶出液については、262 nmでのUV吸光度を測定した(上段)。1分間隔で回収された各分画の放射活性は、Bio Imaging Analyzer(下段)により測定した。
【特許請求の範囲】
【請求項1】
下記(a)〜(d)のいずれかに記載のポリヌクレオチドであって、ADPリボシル化活性を有するポリペプチドをコードするポリヌクレオチド。
(a)配列番号:2に記載のアミノ酸配列からなるポリペプチドをコードするポリヌクレオチド
(b)配列番号:1に記載の塩基配列のコード領域を含むポリヌクレオチド
(c)配列番号:2に記載のアミノ酸配列において、1もしくは複数のアミノ酸が置換、欠失、挿入、および/または付加したアミノ酸配列からなるポリペプチドをコードするポリヌクレオチド
(d)配列番号:1に記載の塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズするポリヌクレオチド
【請求項2】
請求項1に記載のポリヌクレオチドによりコードされるポリペプチド。
【請求項3】
請求項2に記載のポリペプチドもしくはその部分ポリペプチドと、細胞レセプターを認識するタンパク質とが融合した構造のポリペプチド。
【請求項4】
請求項1に記載のポリヌクレオチド、または、請求項3に記載のポリペプチドをコードするポリヌクレオチドが挿入されたベクター。
【請求項5】
請求項1に記載のポリヌクレオチド、または請求項4に記載のベクターを保持する宿主細胞。
【請求項6】
請求項5に記載の宿主細胞を培養し、該宿主細胞またはその培養上清から、産生させたポリペプチドを回収する工程を含む、請求項2または3に記載のポリペプチドの製造方法。
【請求項7】
請求項1に記載のポリヌクレオチドと特異的にハイブリダイズするポリヌクレオチドであって、少なくとも15ヌクレオチドの鎖長を持つポリヌクレオチド。
【請求項8】
請求項1に記載のポリヌクレオチドまたはその一部に対するアンチセンスポリヌクレオチド。
【請求項9】
請求項2または3に記載のポリペプチドに結合する抗体。
【請求項10】
請求項2もしくは3に記載のポリペプチド、または、請求項4に記載のベクターを有効成分として含む、アポトーシス誘導剤。
【請求項11】
請求項2もしくは3に記載のポリペプチド、または、請求項4に記載のベクターを有効成分として含む、抗癌剤。
【請求項12】
請求項2もしくは3に記載のポリペプチドを有効成分として含む、ADPリボシル化試薬。
【請求項13】
CARP-1タンパク質の発現もしくは機能阻害物質を有効成分とする、貝類毒性除去剤。
【請求項14】
請求項2に記載ポリペプチドとDNAとを接触させる工程を含む、DNAをADPリボシル化する方法。
【請求項15】
以下の工程(a)または(b)のいずれかの工程を含む、細胞をアポトーシス誘導する方法。
(a)請求項2に記載のポリペプチド、または請求項4に記載のベクターを細胞へ導入する工程
(b)請求項3に記載のポリペプチドと細胞とを接触させる工程
【請求項16】
以下の工程(a)〜(c)を含む、抗癌剤のスクリーニング方法。
(a)請求項2に記載のポリペプチドもしくは該ポリペプチドを発現する細胞と、被検化合物を接触させる工程
(b)前記ポリペプチドの発現量または活性を測定する工程
(c)被検化合物を接触させない場合と比較して、前記発現量または活性を上昇させる化合物を選択する工程
【請求項17】
以下の工程(a)〜(c)を含む、貝類毒性除去剤のスクリーニング方法。
(a)請求項2に記載のポリペプチドもしくは該ポリペプチドを発現する細胞と、被検化合物を接触させる工程
(b)前記ポリペプチドの発現量または活性を測定する工程
(c)被検化合物を接触させない場合と比較して、前記発現量または活性を低下させる化合物を選択する工程
【請求項1】
下記(a)〜(d)のいずれかに記載のポリヌクレオチドであって、ADPリボシル化活性を有するポリペプチドをコードするポリヌクレオチド。
(a)配列番号:2に記載のアミノ酸配列からなるポリペプチドをコードするポリヌクレオチド
(b)配列番号:1に記載の塩基配列のコード領域を含むポリヌクレオチド
(c)配列番号:2に記載のアミノ酸配列において、1もしくは複数のアミノ酸が置換、欠失、挿入、および/または付加したアミノ酸配列からなるポリペプチドをコードするポリヌクレオチド
(d)配列番号:1に記載の塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズするポリヌクレオチド
【請求項2】
請求項1に記載のポリヌクレオチドによりコードされるポリペプチド。
【請求項3】
請求項2に記載のポリペプチドもしくはその部分ポリペプチドと、細胞レセプターを認識するタンパク質とが融合した構造のポリペプチド。
【請求項4】
請求項1に記載のポリヌクレオチド、または、請求項3に記載のポリペプチドをコードするポリヌクレオチドが挿入されたベクター。
【請求項5】
請求項1に記載のポリヌクレオチド、または請求項4に記載のベクターを保持する宿主細胞。
【請求項6】
請求項5に記載の宿主細胞を培養し、該宿主細胞またはその培養上清から、産生させたポリペプチドを回収する工程を含む、請求項2または3に記載のポリペプチドの製造方法。
【請求項7】
請求項1に記載のポリヌクレオチドと特異的にハイブリダイズするポリヌクレオチドであって、少なくとも15ヌクレオチドの鎖長を持つポリヌクレオチド。
【請求項8】
請求項1に記載のポリヌクレオチドまたはその一部に対するアンチセンスポリヌクレオチド。
【請求項9】
請求項2または3に記載のポリペプチドに結合する抗体。
【請求項10】
請求項2もしくは3に記載のポリペプチド、または、請求項4に記載のベクターを有効成分として含む、アポトーシス誘導剤。
【請求項11】
請求項2もしくは3に記載のポリペプチド、または、請求項4に記載のベクターを有効成分として含む、抗癌剤。
【請求項12】
請求項2もしくは3に記載のポリペプチドを有効成分として含む、ADPリボシル化試薬。
【請求項13】
CARP-1タンパク質の発現もしくは機能阻害物質を有効成分とする、貝類毒性除去剤。
【請求項14】
請求項2に記載ポリペプチドとDNAとを接触させる工程を含む、DNAをADPリボシル化する方法。
【請求項15】
以下の工程(a)または(b)のいずれかの工程を含む、細胞をアポトーシス誘導する方法。
(a)請求項2に記載のポリペプチド、または請求項4に記載のベクターを細胞へ導入する工程
(b)請求項3に記載のポリペプチドと細胞とを接触させる工程
【請求項16】
以下の工程(a)〜(c)を含む、抗癌剤のスクリーニング方法。
(a)請求項2に記載のポリペプチドもしくは該ポリペプチドを発現する細胞と、被検化合物を接触させる工程
(b)前記ポリペプチドの発現量または活性を測定する工程
(c)被検化合物を接触させない場合と比較して、前記発現量または活性を上昇させる化合物を選択する工程
【請求項17】
以下の工程(a)〜(c)を含む、貝類毒性除去剤のスクリーニング方法。
(a)請求項2に記載のポリペプチドもしくは該ポリペプチドを発現する細胞と、被検化合物を接触させる工程
(b)前記ポリペプチドの発現量または活性を測定する工程
(c)被検化合物を接触させない場合と比較して、前記発現量または活性を低下させる化合物を選択する工程
【図2A】

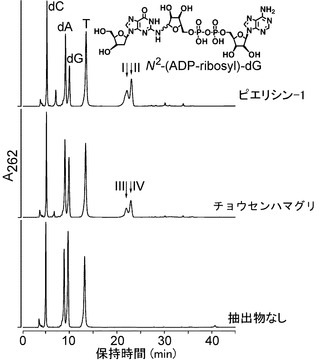
【図2B】
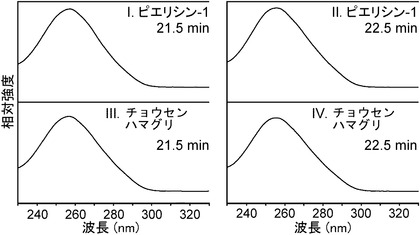
【図4】

【図5】

【図6A】
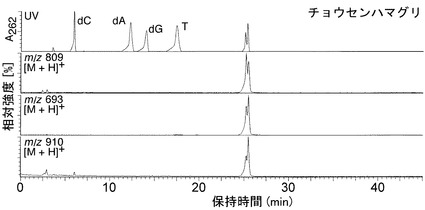
【図6B】
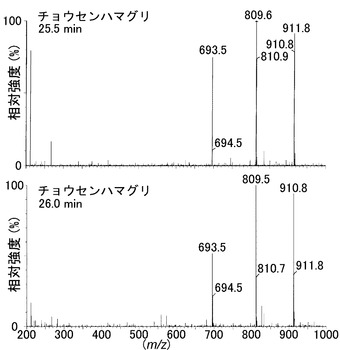
【図7】
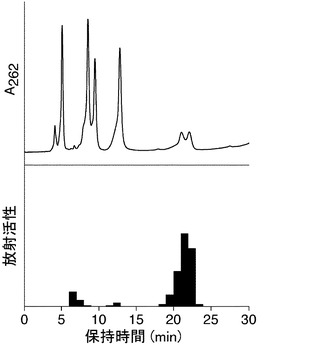
【図1】

【図3】
【図2B】
【図4】
【図5】
【図6A】
【図6B】
【図7】
【図1】
【図3】
【公開番号】特開2008−43(P2008−43A)
【公開日】平成20年1月10日(2008.1.10)
【国際特許分類】
【出願番号】特願2006−171094(P2006−171094)
【出願日】平成18年6月21日(2006.6.21)
【出願人】(803000056)財団法人ヒューマンサイエンス振興財団 (341)
【Fターム(参考)】
【公開日】平成20年1月10日(2008.1.10)
【国際特許分類】
【出願日】平成18年6月21日(2006.6.21)
【出願人】(803000056)財団法人ヒューマンサイエンス振興財団 (341)
【Fターム(参考)】
[ Back to top ]