
質量スペクトル分析を用いた、治療薬投与のための癌患者のセレクション
質量分析データおよび分類アルゴリズムにより、EGFRおよび/またはHER2を標的とした治療薬のような、AktまたはERK/JNK/p38またはPKCでの、またはそれらの上流でのMAPK(マイトジェン活性化プロテインキナーゼ)経路またはPKC(プロテインキナーゼC)経路に関わる受容体のアゴニスト、受容体もしくはタンパク質を標的にする治療薬または治療薬の組み合わせによる治療が固形上皮腫瘍癌患者に有効である可能性が高いかどうかを決定することが可能となる。該方法により、EGFRを標的とした治療薬およびCOX2を標的とした治療薬の組み合わせが該癌患者に有用である可能性が高いかどうか;あるいはNF−κB阻害剤による治療が該癌患者に有用である可能性が高いかどうかを決定することも可能となる。

【発明の詳細な説明】
【技術分野】
【0001】
(関連出願の相互参照)
本願は米国仮出願第61/338,938号(2010年2月24日)の35 U.S.C.119(e)下の利益を主張し、その内容を引用により本明細書中に取り込む。
【0002】
本発明は、あるタイプまたはクラスの薬剤、および/またはそれらの組み合わせが癌患者に有効である可能性が高いかどうかを予測する方法およびシステムに関連する。該方法およびシステムは、患者の血液由来サンプルから得た質量分析データの使用およびその質量分析データに基づき作動する分類子として設定されたコンピューターに関連する。
【背景技術】
【0003】
本発明の出願人であるBiodesix, Incは、上皮成長因子受容体(EGFR)経路を標的とした薬剤の治療が非小細胞性肺癌(NSCLC)患者に有効である可能性が高いかどうかを予測するVeriStratとして知られる検査を開発した。該検査は米国公開番号第7,736,905号に開示され、その内容を引用により本明細書中に取り込む。該検査はまた、Taguchi F. et al.1においても記載され、その内容を引用により本明細書に取り込む。該検査の出願は米国公開番号第7,858,390;7,858,389および7,867,775号においても開示されており、それらの内容を引用により本明細書中に取り込む。
【0004】
簡単に述べると、VeriStrat検査は癌患者の血清および/または血漿サンプルに基づくものである。MALDI-TOF質量分析およびコンピューターで実行されるデータ解析アルゴリズムの組み合わせにより、所定のm/z範囲における8個の積分ピーク強度のセットが訓練コホートのものと比較され、患者サンプルのクラス標識が作製される:VeriStrat「良」、VeriStrat「不良」、またはVeriStrat「未定義」。複数の臨床評価試験において、前処理血清/血漿がVeriStrat「良」であった患者は、上皮成長因子受容体阻害剤で治療された際に、VeriStrat「不良」であった患者と比べて有意に良好なアウトカムを有していた。少数のケース(2%未満)では決定することが出来ず、VeriStrat「未定義」と標識された。VeriStratは本発明の出願人であるBiodesix, Inc.から市販されており、非小細胞性肺癌患者の治療セレクションに用いられる。
【0005】
現代の殆どのバイオマーカーに基づく検査は、腫瘍のタイプおよび組織学、特定の治療介入、ならびに臨床病理学的因子に関して非常に特異的である。例えば、腫瘍組織を用いた遺伝子検査、例えば、EGFRドメインにおける突然変異、KRAS変異の検査、および蛍光In-Situハイブリダイセーション(FISH)を用いた遺伝子コピー数検査は、非常に特殊な適応においてのみ有効である。EGFR変異から腺癌を伴う第一線NSCLC癌におけるゲフィチニブ応答に関する示唆は得られるが、扁平上皮癌ではこのタイプのNSCLCにおけるこれらの変異は非常に稀であるため、同様の有用性を示さない。KRAS変異は結腸直腸癌におけるセツキシマブに対する応答と相関し得るが、これをNSCLCに転用しようとする試みは失敗であった。頭頚部扁平上皮癌(SCCHN)におけるEGFR阻害剤(EGFRI)の有効性に関する既知のマーカーは存在しない。遺伝子検査におけるこれらの制限は、腫瘍新生の複雑なメカニズムの小さなパートにしか過ぎない非常に特殊な変異にそれらが焦点を絞っていることと関係するのかもしれない。また、これらの検査の全ては還元主義的な視点に基づくものであり、即ち、腫瘍生物学を腫瘍細胞のみに還元し、血管支持システムの内皮細胞、細胞外マトリックスおよび免疫系の成分(癌と関連する慢性的な炎症メカニズムに関与する炎症性細胞ならびにケモカインおよびサイトカインなど)からなる腫瘍微小環境と腫瘍細胞との相互作用を無視したものである。
【発明の概要】
【0006】
本明細書中において、腫瘍細胞におけるどの経路がVeriStrat「不良」である上皮腫瘍の独特の性質に関わるかについての我々の理解およびその根拠を提供する。本明細書で提示される理解についての証拠は、いくつかのソース、例えば、臨床的証拠、現象論的証拠、文献的解析、および癌患者の血清サンプルの質量スペクトル分析に基づく分子学的証拠によるものである。本明細書で開示される認識の結果は、以下に記載されるある種の薬剤またはその組み合わせが癌患者に有効である可能性が高いか低いかを予測するための新たな方法(即ち、臨床検査)の形態を取り得る。
【0007】
簡単に述べると、VeriStrat「不良」と識別された患者では、VeriStrat検査はEGFRなどの成長因子および生存因子の受容体の1つまたはそれ以上の下流の経路の活性化状態を測定するが、適当な候補となる経路は、例えば、古典的および非古典的MAPK(マイトジェン活性化プロテインキナーゼ)、AktならびにPKC(プロテインキナーゼC)で制御される反応(図2参照)である。化学療法およびプラセボ対照群のアウトカムにおける多様性により、これらの経路のこれら自身による活性化が予後不良に繋がり得ることが示唆され、NF−κB(活性化B細胞の核内因子κ−軽鎖エンハンサー)(細胞応答を制御し、炎症および免疫応答、ならびに細胞増殖および生存の制御に重要な役割を果たす重要な転写因子である)の関与が指摘される。それが化学療法に対する応答に関与することも知られている。
【0008】
一般的な事柄として、VeriStrat検査は予後不良の群のサブセットを識別し、AktまたはERK/JNK/p38またはPKCでの、またはそれらの上流でのMAPK経路またはPKC経路に関わる受容体のアゴニスト、受容体もしくはタンパク質を標的にする治療薬または治療薬の組み合わせによる治療の固形上皮腫瘍癌患者における異なる利益を予測する。EGFR阻害剤はかかる薬剤の例である。抗EGFR薬が有効である可能性が高いと予測される患者はVeriStrat「良」標識で識別される;逆に抗EGFR薬が有効である可能性が低いと予測される患者はVeriStrat「不良」標識で識別される。用語MAPK(マイトジェン活性化プロテインキナーゼ)は、単一の酵素ではなく少なくとも3個の関連するカスケードの名称として本明細書で用いられる(図2参照)。
【0009】
上記の結果として、VeriStrat「不良」標識と関連する患者では、VeriStrat検査は「不良」の患者を予後不良の癌患者のサブグループとして診断する。実際、VeriStrat「不良」患者はVeriStrat「良」患者とは異なる疾患状態を有すると見做される。
【0010】
さらに、VeriStrat「良」標識を有する癌患者では、MAPK経路が関与する受容体のアゴニスト、受容体またはタンパク質を標的とした薬剤またはその組み合わせによる治療がより有効である可能性がより高く;VeriStrat「不良」標識を有する患者ではかかる治療薬による治療から臨床利益を得る可能性は低い;一方で、VeriStrat「不良」では、これらの受容体、これらの経路の活性化には依存しない下流を阻害する療法または療法の組み合わせが有効である可能性が高い。
【0011】
この認識の臨床への適用は、付属の請求項に反映されるように、いくつかの形態を取り得る。該方法は癌患者の血液由来サンプルの質量分析データを得ること、および分類子として機能するプログラムされたコンピューターを用いたスペクトルの解析に関連する。一形態において、固形上皮腫瘍癌患者を、AktまたはERK/JNK/p38またはPKCでの、またはそれらの上流でのMAPK経路またはPKC(プロテインキナーゼC)経路に関わる受容体のアゴニスト、受容体もしくはタンパク質を標的にする治療薬または治療薬の組み合わせによる治療が有効である可能性が高い患者として、あるいは治療薬または治療薬の組み合わせによる治療が有効である可能性が低い患者として識別する方法であって、a)固形上皮腫瘍癌患者の血液由来サンプルから質量スペクトルを得;b)a)で得た質量スペクトルにおいて1つまたはそれ以上の所定の前処理工程を行い;c)b)の質量スペクトルにおける前処理工程を行った後、1つまたはそれ以上の所定のm/z範囲において該スペクトルにおける選択された特徴の積分強度値を得;d)患者を治療薬または治療薬の組み合わせが有効である可能性が高い、または有効である可能性が低い患者として識別するために、別の固形腫瘍患者の血液由来サンプルから得たクラス標識スペクトルを含む訓練セットを用いた分類アルゴリズムにc)で得た値を用いることを特徴とする方法が開示される。
【0012】
別の一実施態様において、COX阻害剤およびEGFR阻害剤の組み合わせの投与が癌患者に有効である可能性が高いかどうかを予測する方法であって、
a)該癌患者の血液由来サンプルから質量スペクトルを得;
b)工程a)で得られた質量スペクトルにおいて1つまたはそれ以上の所定の前処理工程を行い;
c)工程b)の質量スペクトルにおける前処理工程を行った後、1つまたはそれ以上のm/z範囲において該スペクトルの選択された特徴の積分値を得;
d)該患者をCOX2阻害剤およびEGFR阻害剤の組み合わせの投与による治療が有効である可能性が高いまたは低い患者として識別するために、別の固形上皮腫瘍患者の血液由来サンプルから得たクラス標識スペクトルを含む訓練セットを用いた分類アルゴリズムに工程c)で得た値を用いる
ことを特徴とする方法が開示される。
【図面の簡単な説明】
【0013】
【図1】は患者の血液由来サンプルでVeriStrat検査を行う工程を示すフローチャートである。
【0014】
【図2】はヒトの細胞における選択されたシグナル伝達経路を示すチャートである。
【0015】
【図3】は血清アミロイドA(SAA)アイソフォームの選択された生物活性ならびに癌の進行および治療に対する耐性におけるその役割を表す図である。
【0016】
【図4】はEGFRシグナル伝達経路、それらの相互作用および推定されるSAAによる活性化のポイントを表す図である。
【0017】
【図5】はErbBファミリーの成長因子受容体、例えば、EGFRおよびそれらの阻害剤の説明であり、Yarden Y, Shilo BZ. SnapShot: EGFR signaling pathway. Cell 2007; 131:1018の引用である。
【0018】
【図6】は全ての公表されたVeriStrat分析における治療群によるVeriStrat「良」およびVeriStrat「不良」患者間のハザード比を示すフォレストプロットである。
【0019】
【図7】は異なる化学療法を受けた患者の全生存率(OS)とこれらの患者のVeriStrat標識(「良」および「不良」)のカプラン−マイヤープロットを表したものである。
【0020】
【図8】は異なる濃度のゲフィチニブの存在下におけるゲフィチニブ感受性細胞株HCC4006とゲフィチニブ耐性細胞株A549のVeriStrat「不良」およびVeriStrat「良」血清中での増殖のプロットである。
【0021】
(発明の詳細な説明)
定義
本明細書で用いられるように、単数形「a」、「an」、および「the」は、文脈から特に明確に指定されない限り、複数に関する言及を包含する。
【0022】
本明細書で用いられるように。用語「固形上皮腫瘍」は、例えば、限定される必要はないが、NSCLC、SCCHN、乳癌、腎臓癌、膵臓癌、黒色腫および直腸結腸癌(CRC)である。
【0023】
本明細書で用いられるように、「AktまたはERK/JNK/p38またはPKCでの、またはそれらの上流でのMAPK経路またはPKC経路に関わる受容体のアゴニスト、受容体もしくはタンパク質を標的にする治療薬または治療薬の組み合わせ」は、例えば、限定されないが、erbB受容体受容体ファミリー(例えば、EGFR(HER1)、HER2、HER3、およびHER4)、VEGF受容体(VEGFR2)、肝細胞増殖因子受容体(HGFRまたはMET)、Gタンパク質共役型受容体、インスリン様増殖因子(IGF)受容体、VEGF、成長因子(例えば、TGFαおよびEGF)、およびAktまたはERK/JNK/p38 MAPKまたはPKC経路の、またはそれらの上流の任意の別のタンパク質を標的とした治療薬または薬剤である。さらに、本明細書で用いられるように、用語「AktまたはERK/JNK/p38またはPKCでの、またはそれらの上流でのMAPK経路またはPKC(プロテインキナーゼC)経路に関わる受容体のアゴニスト、受容体もしくはタンパク質を標的にする治療薬または治療薬の組み合わせ」は、既知の治療薬、ならびにこれらのタンパク質を標的とした未だ発見または開示されていない治療薬を包含する。さらに、治療薬の組み合わせは、それらが既に固形上皮腫瘍の治療に用いられているか否かにかかわらず、治療薬の任意の組み合わせを包含する。ある薬剤が特定のタンパク質または経路の阻害剤として同定されたとしても、かかる分類はその作用メカニズムの記載を表すと意図されるものではなく、それはこれらの薬剤の多くの作用メカニズムは完全には理解されていないからであることは記しておくべきであろう。例えば、排他的なリストとして意図されるものではないが、これらの治療薬は以下を含む:
(1)TKI(チロシンキナーゼ阻害剤):小分子チロシンキナーゼ阻害剤(TKI)に分類される多くの薬剤が現在市場に出回っており、I−III相の治験が行われている。TKIは特定の分子の受容体、例えば、上皮増殖因子受容体(EGFR)を標的とすることができ、複数の受容体を標的とすることもできる(「マルチプルキナーゼ阻害剤」と呼ばれる)。これらは、例えば、限定されないが、エルロチニブ、ゲフィチニブ、ソラフェニブ、スニチニブ、パゾパニブ、イマチニブ、ニロチニブ、ラパチニブである。
抗体に基づく阻害剤は、例えば、セツキシマブ(抗EGFR)、パニツムマブ(抗EGFR)、トラスツズマブ(抗−Her2)である。
(2)HGFRまたはMET阻害剤:現在、I−II相の治験中にある長いリストのMETまたはPI3K(METの下流のシグナル伝達酵素である)を阻害する薬剤が存在し、様々な段階において調査されているが、現時点では臨床的には用いられていない。例えば、XL880はMETおよびVEGFR2の強力な阻害剤である。本明細書で用いられるように、用語「MET阻害剤」は、例えば、限定されないが、AMG 208、AMG 102、ARQ 197、AV−299、MetMab、GSK 1363089(XL880)、EMD 1214063、EMD 1204831、MGCD265、クリゾタニブ(PF−02341066)、PF−04217903、MP470である。
(3)COX2阻害剤:本明細書で用いられるように、用語「COX2阻害剤」は、例えば、限定されないが、選択的COX2阻害剤:セレコキシブ、ロフェコキシブ、バルデコキシブ、ルミラコキシブである。
(4)COX1およびCOX2の両方を阻害する別の非ステロイド性抗炎症剤(NSAID)は、イブプロフェン、アスピリン、インドメタシンおよびスリンダクである。かかる薬剤はNF−κB活性化も抑制することが示されている。
(5)別のNF−κB阻害剤:本明細書中で用いられるように、用語「NF−κB阻害剤」は、例えば、限定されないが、三酸化ヒ素(ATO)、サリドマイドおよびそのアナログ、レスベラトロールである。さらに、COX2阻害剤はNF−κB経路にも阻害効果を有する。故に、イブプロフェン、アスピリン、インドメタシンおよびスリンダクといったNSAIDはNF−κBの活性化を抑制することが示されたため、NF−κB阻害剤と見做される。
【0024】
本明細書で用いられるように、用語「VEGF阻害剤」は、例えば、限定されないが、ベバシズマブ、セジラニブ、アキシチニブ、モテサニブ、BIBF 1120、ラムシルマブ、VEGF Trap、リニファニブ(ABT869)、チボザニブ、BMS−690514、XL880、スニチニブ、ソラフェニブ、ブリバニブ、XL−184、パゾパニブである。
【0025】
本明細書で用いられるように、「標的とした治療」は、特定の酵素のモノクローナル抗体または小分子阻害剤といった薬剤または他の物質を受容体といった特定の分子を同定し攻撃するために用いるようなタイプの治療を意味する。これらの例は、EGFR−TKI(エルロチニブ、ゲフィチニブ)、セツキシマブ、ベバシズマブなどである。
【0026】
本明細書で用いられるように、用語「非標的化学療法」または「化学療法」は、DNAに干渉(アルキル化試薬、例えば、シスプラチン、カルボプラチン、オキサリプラチンなど、または代謝拮抗薬、例えば、5−フルオロウラシルまたはペメトレキセド、またはイリノテカンなどのトポイソメラーゼ阻害剤)、または細胞分裂に干渉(ビノレルビン、ドセタキセル、パクリタキセルなど)することにより、急速に分裂する細胞に干渉する治療を意味する。
【0027】
本明細書で用いられるように、「予後」は、治療を受けない場合または標準的な治療を適用された場合の臨床的アウトカムに関連する因子または測定結果を意味する。それは、疾患の自然な経過の測定結果と考えることが出来る。
【0028】
用語「予測の」は、特定の治療方法が有効であるか有効ではないかに関連する因子または測定結果を意味する。予測因子により、予測マーカーの状態に依存して治療方法から異なる利益が得られることが示唆される2。本明細書で用いられるように、用語「疾患状態」は、異なる予後および/または異なる治療応答および/または特定の分子および/または代謝的特徴により特徴付けすることが出来る診断された病状の特定のサブタイプを意味する。
【0029】
ディスカッション
VeriStrat検査は血清サンプルの質量分析データから得た特徴に基づくため、現行の殆どのバイオマーカーを用いた検査とは異なり、癌に関連する一般的な因子を測定することが出来ることを我々は発見した。この事実により、以下に議論されるVeriStrat検査を用いた治療のためのセレクションへの新たな臨床的適用が可能となる。特に、VeriStrat検査により、EGFR阻害のメカニズムにかかわらず、VeriStrat「良」およびVeriStrat「不良」と識別された患者間で同様に生存曲線を区分することが出来る。我々の先の研究において、酵素のATP結合部位をブロックすることにより受容体を阻害する小分子EGFRチロシンキナーゼ阻害剤ゲフィチニブ(Iressa)およびエルロチニブ(Tarceva)1で治療した患者のサンプルセットにVeriStrat検査を行った。NSCLCおよび直腸結腸癌(CRC)の両方において、EGFRを標的とした他の治療薬セツキシマブ(Erbitux)に関してもVeriStrat「良」およびVeriStrat「不良」と識別された患者間で同様の区分が見られた3。セツキシマブはEGF受容体を直接ブロックする抗体である。
【0030】
さらに、VeriStrat検査により、VeriStrat「良」およびVeriStrat「不良」と識別された患者間で、臨床病理学的特徴を超えて同様に区分することが出来る。例えば、VeriStrat検査は、腫瘍が腺癌である患者でも腫瘍が扁平上皮癌である患者でも用いることができる。
【0031】
また、VeriStrat検査は、様々の固形腫瘍において、VeriStrat「良」およびVeriStrat「不良」と識別された患者間の区分を示す。NSCLC、頭頚部扁平細胞癌(SCCHN)、およびCRCにおいてこれが見られた3。
【0032】
さらに、非標的化学療法で治療された患者のVeriStrat検査による生存曲線の区分は、人種、介入治療のタイプ、および腫瘍タイプに依存して異なることが見出された。いくつかの非標的化学療法治療群において区分の証拠があるが、他では区分が存在しない。プラセボ群(即ち、介入治療を受けていない群)においても明確な区分が存在し、VeriStrat検査が予測的な要素を有することが示唆される。
【0033】
図6のフォレストプロットは、現在までに刊行または公表された全てのVeriStrat検査の分析を要約する。分析された各治療群のVeriStrat「良」およびVeriStrat「不良」患者間の全生存率のハザード比(HR)が示される。データは治療タイプに応じて分類されると考えられる。得られたハザード比の範囲によりVeriStratが実際に特定の治療タイプの結果としての良好または不良なアウトカムを示唆し、故に予測能を有することが示される。
【0034】
図6において、治療は、B=ベバシズマブ、C=セツキシマブ、CT=化学療法、E=エルロチニブ、G=ゲフィチニブである。刊行物/公表物は、[1] D. Carbone, 2nd European Lung Cancer Conference, April 2010、 [2] F. Taguchi et al., J Natl Cancer Inst. 2007 Jun6;99(11):838-8461 からアップデートしたBiodesixのファイル, [3] C. Chung et al., Cancer Epidemiol Biomarkers Prev. 2010 Feb;19(2):358-653、 [4] D. Carbone et al., Lung Cancer 2010 Sept; 69(3):337-3404である。
【0035】
非標的化学療法治療群の再分析により、タキセン治療群では明確な区分が見られなかったが、タキセンを含まない化学療法レジメン群ではVeriStrat「良」およびVeriStrat「不良」群間で区分が存在した(図7参照)。
【0036】
検査がこのような広い適用範囲を有することは稀である。
【0037】
まとめると、前記のディスカッションならびに図6および7から以下の結論が導かれる:
1.VeriStrat検査により、EGFR阻害剤(EGFRI)単独療法において、VeriStrat良および不良の患者間で約.45のハザード比における区分が示され、それは、
−EGFRIの作用メカニズム、例えば、小分子TKI(エルロチニブ、ゲフィチニブ)および抗体(受容体)阻害剤に基づくEGFRI(例えば、セツキシマブ)であるかに依存せず、
−組織学的タイプ、例えば、腺癌、および扁平細胞癌であるかに依存せず、
−組織、例えば、NSCLC、SCCHN、およびCRCであるかに依存しない。
2.他の集団的特徴の有意な相関は観察されなかった:
−遺伝子マーカー、例えば、EGFRの変異状態またはKRASの状態とは相関せず、
−性別および人種といった集団的特徴とは相関しない。
3.VeriStratは、治療を受けていないVeriStrat不良およびVeristrat良のサブグループ間の区分で示される強い予測的要素を有する。
−しかしながら、VeriStrat「不良」サブグループにおけるEGFRI単独療法による明確な治療利益は存在しない、即ち、VeriStrat不良サブグループにおけるエルロチニブによる治療はプラセボによる治療と実質的に同効果である一方、VeriStrat「良」サブグループにおいては、EGFRIによる大きな治療利益が存在し、
−組み合わせ療法の効果は特定の薬剤の組み合わせおよび相互作用する経路に対するそれらの効果に依存する。
【0038】
サンプルの質量スペクトルにおける特定のピークがVeriStrat「不良」群においてのみ観察された結果とこれらの事実を全て合わせると、VeriStratは固形上皮腫瘍において臨床的意義(不良アウトカム)を有する新たな疾患状態を定義するという結論が導かれる。観察された現象により、VeriStrat「不良」の腫瘍の分子学的状態に関するいくつかの仮説を立てることができる:EGFRIがこの種の患者に有効ではないため、ならびに、効果がTKIおよび抗体療法と同程度であるため、VeriStrat「不良」患者において、該受容体および該チロシンキナーゼドメインの下流の経路がVeriStrat「良」患者と異なっている、即ちアップレギュレートされている可能性が高い。KRASの変異状態との相関は見られなかったため、侵されている経路はRASの下流であることがさらに結論される。
【0039】
前記の観察、文献的解析および他の一連の根拠に基づき、VeriStrat「不良」腫瘍細胞の異なる特徴に腫瘍細胞のどの経路が関与しているかに関する我々の理解が本明細書で提供される。簡単に述べると、VeriStrat「不良」と識別された患者においてVeriStrat試験はEGFの受容体の下流の1つまたはそれ以上の経路の活性化を評価していることを我々は提案する;候補の可能性がある経路は、古典的および非古典的MAPK、PI3K/AktならびにPKCで制御される経路である(図2の200Aおよび200Bを参照)。化学療法およびプラセボ対照群のアウトカムの多様性により、これらの経路のこれら自身による活性化が予後不良に繋がり得ることが示唆され、NF−κB転写因子(細胞生存の重要な制御因子であり、炎症過程および癌の進行に重要な役割を果たし、化学療法への応答に関与する)の関与が示唆され得る。
【0040】
一般的な事柄として、VeriStrat検査は予後不良の群のサブセットを識別し、AktまたはERK/JNK/p38またはPKCでの、またはそれらの上流でのMAPK経路またはPKC(プロテインキナーゼC)経路に関わる受容体のアゴニスト、受容体もしくはタンパク質を標的にする治療薬または治療薬の組み合わせによる治療から固形上皮腫瘍癌患者が受ける恩恵を予測する。抗EGFR薬が有効である可能性が高いと予測される患者はVeriStrat「良」標識で識別される;逆に抗EGFR薬が有効である可能性が低いと予測される患者はVeriStrat「不良」標識で識別される。VeriStrat「不良」標識を有する患者がMAPK経路を活性化する受容体を標的とした治療薬による治療から臨床利益を受ける可能性は低く;一方で、VeriStrat「不良」患者では、該受容体と独立したこれらの経路の活性化を阻害する療法または療法の組み合わせから臨床利益が得られる可能性が高い。
【0041】
用語MAPK(マイトジェン活性化プロテインキナーゼ)は、単一の酵素ではなく少なくとも3個の関連するカスケードの名称として本明細書で用いられる(図2参照)。
【0042】
前記の主張の結果として、VeriStrat「不良」標識と関連する患者では、VeriStrat検査は「不良」患者を予後不良の癌患者のサブグループとして予測する。
【0043】
認識の結論は、いくつかのタイプの薬剤が癌患者に有効である可能性が高いか低いかを予測するための新しい方法、即ち臨床検査の形態を取り得る。
【0044】
臨床適用の1つにおいて、本発明は、固形上皮腫瘍癌患者を、AktまたはERK/JNK/p38またはPKCでの、またはそれらの上流でのMAPK経路またはPKC経路に関わる受容体のアゴニスト、受容体もしくはタンパク質を標的にする治療薬または治療薬による治療が有効である可能性が高いか、該治療薬または治療薬の組み合わせによる治療が有効である可能性が低いか識別する方法であって、
a)固形上皮腫瘍癌患者の血液由来サンプルから質量スペクトルを得;
b)a)で得た質量スペクトルにおいて1つまたはそれ以上の所定の前処理工程を行い(例えば、バックグランドの除去、ノイズの推定、ノーマライゼーションおよびスペクトルのアラインメント);
c)b)の質量スペクトルにおける前処理工程を行った後、1つまたはそれ以上の所定のm/z範囲(および好ましくは下の表1で説明されるm/zピークに対応する下記のm/z範囲)において該スペクトルにおける選択された特徴の積分強度値を得;
d)患者を治療薬または治療薬の組み合わせが有効である可能性が高い、または有効である可能性が低い患者として識別するために、別の固形腫瘍患者の血液由来サンプルから得たクラス標識スペクトルを含む訓練セットを用いた分類アルゴリズム(例えば、k近傍法)にc)で得た値を用いる
ことを特徴とする方法であると見做すことが出来る。
【0045】
VeriStrat「不良」患者の標的治療に対する耐性の打開の具体例として、COX2阻害剤、例えば、セレコキシブまたはロフェコキシブを治療レジメンとしてEGFRIに追加することにより、EGFRIに対してVeriStrat「不良」標識を有する患者の耐性を打開することが出来る可能性がある。故に、VeriStrat検査はCOX2阻害剤およびEGFRIを含む組み合わせ療法の処方のインジケーターとして用いられ得る。
【0046】
別の一具体例として、VeriStrat「不良」標識はNF−κBの特異的な活性化と関連すると考えられ、故に、該検査はNF−κB阻害剤から最も恩恵を受ける患者を選択し、それ故に不必要な治療およびそれに関連する死亡率を低減させるために用いることができる。
【0047】
別の一具体例として、VeriStrat「不良」標識は特定の非標的化学療法、特にDNA複製および遺伝子発現に干渉する薬剤(シスプラチン、ゲムシタビンまたはペメトレキセド)の治療利益が小さいこと(おそらくは、この過程におけるNF−κB因子の関与による)に関連すると考えられる。
【0048】
VeriStrat「不良」と標識された患者では、(1)受容体と独立してMAPK経路の下流の活性化を阻害する薬剤、例えば、COX2阻害剤など、または(2)炎症性のホスト応答を最小化する薬剤の添加、あるいは、クロストーク経路の活性化を阻害する別の標的薬剤の添加により、標的薬剤に対する耐性を打開することが出来る。
【0049】
VeriStrat検査
いくつかの治療薬または治療薬の組み合わせ、例えば、AktまたはERK/JNK/p38またはPKCでの、またはそれらの上流でのMAPK経路またはPKC経路に関わる受容体のアゴニスト、受容体もしくはタンパク質を標的にする治療薬による治療用にかかる患者を選択するために固形上皮腫瘍癌患者の血液由来サンプルを検査する本開示による方法が、図1のフローチャートに工程100として説明される。
【0050】
工程102において、血清または血漿サンプルを患者から得る。一実施態様において、血清サンプルを3つのアリコートに分け、質量分析および後続の工程104、106(サブ工程108、110および112を含む)、114、116および118を書く折コートについて独立して行う。アリコートの数は、例えば4、5または10アリコートのように異なっていてもよく、各アリコートは後続の工程に用いられる。
【0051】
104において、該サンプル(アリコート)は質量分析で分析される。好ましい質量分析法は、マトリックス支援レーザー脱離イオン化(MALDI)飛行時間型(TOF)質量分析であるが、他の方法でもよい。質量分析により、当該分野で周知のように、多数の質量/電荷(m/z)値における強度値を表すデータ点が得られる。一実施例において、該サンプルを解凍し、4℃、1500rpmで5分間遠心する。さらに、該血清サンプルは1:10、または1:5にMilliQ水で希釈してもよい。希釈サンプルはMALDIプレートのランダムに割り当てられた位置にトリプリケート(即ち、3つの異なるMALDIターゲット上に)スポットしてもよい。0.75μlの希釈した血清をMALDIプレート上にスポットした後、0.75μlの35mg/ml シナピン酸/(50% アセトニトリルおよび0.1%トリフルオロ酢酸(TFA))を加え、5回ピペッティングして混合してもよい。プレートは室温で乾燥することが出来る。本発明の本質に従って血清を調製および加工するために別の技法および方法が用いられ得ることは明らかである。
【0052】
質量スペクトルは、自動または手動でスペクトルを収集するVoyager DE-PROまたはDE-STR MALDI TOF質量分析計を用いてリニアモードの陽イオンで獲得してもよい。各血清サンプルについて525または500個のスペクトルを得るために、75または100個のスペクトルが各MALDIスポットの7または5個の位置から収集される。スペクトルはタンパク質スタンダードの混合物(インスリン(ウシ)、チオレドキシン(E. coli)、およびアポミオグロビン(ウマ))を用いて外部校正される。
【0053】
工程106では、1つまたはそれ以上の所定の前処理工程を工程104で得たスペクトルについて行う。前処理工程106は、工程104で得た質量分析データ上で作動するソフトウェアの指示を用いた一般的な目的のコンピューターで実行される。前処理工程106は、バックグラウンドの除去(工程108)、ノーマライゼーション(工程110)およびアラインメント(工程112)を含む。バックグラウンドの除去工程は、好ましくはバックグラウンドのロバスト、非対称な推定を行うことに関連し、スペクトルからバックグラウンドが差し引かれる。工程108は米国特許番号第7,736,905 B2号および米国公開番号第2005/0267689号(引用により本明細書中に取り込む)に記載されるバックグラウンド除去法を用いる。ノーマライゼーション工程110はバックグラウンドを差し引いたスペクトルのノーマライゼーションに関連する。ノーマライゼーションは部分イオン電流ノーマライゼーションの形態でもよく、あるいは、米国特許番号第7,736,905に記載されるように、全イオン電流ノーマライゼーションでもよい。工程112では、米国特許番号第7,736,905に記載されるように、ノーマライズし、バックグラウンドを差し引いたスペクトルを所定の質量スケール(分類子を用いた訓練セットの調査により得られる)にアラインする。
【0054】
前処理工程106が行われると、工程100は、所定のm/z範囲におけるスペクトルの選択された特徴(ピーク)の値を得る工程114に進む。ピーク検出アルゴリズムのピーク幅設定を用い、ノーマライズおよびバックグラウンド除去を行った強度をこれらのm/z範囲において積分し、この積分値(即ち、特徴の幅の間の曲線下面積)を特徴に割り当てることができる。このm/z範囲においてピークが検出されなかったスペクトルについては、積分範囲は、その時点のm/z位置のピーク幅に対応する幅を有するこの特徴の平均m/z位置周囲の間隔として定義することが出来る。この工程は米国特許番号第7,736,905にさらに詳しく記載される。
【0055】
工程114では、米国特許番号第7,736,905に記載されるように、該スペクトルにおける特徴の積分値は1つまたはそれ以上の以下のm/z範囲から得られる:
5732から5795
5811から5875
6398から6469
11376から11515
11459から11599
11614から11756
11687から11831
11830から11976
12375から12529
23183から23525
23279から23622
65902から67502.
【0056】
好ましい実施態様において、値は表1に示される8個のこれらのm/z範囲から得られ、適宜、12個全ての範囲から得られてもよい。これらのピークの重要性および検出方法は米国特許番号第7,736,905号に記載される。
【0057】
工程116において、工程114で得た値を分類子に供給する(図示された実施態様ではk近傍法(KNN)分類子である)。該分類子は多数の他の患者(NSCLC癌患者でもよく、あるいは、別の固形上皮癌、例えば、HNSCC、乳癌の患者でもよい)からのクラス標識スペクトルの訓練セットを利用する。114の値および訓練セットへのKNN分類アルゴリズムの適用は米国特許番号第7,736,905号で説明される。別の分類子、例えば、確率的KNN分類子または他の分類子が用いられてもよい。
【0058】
工程118において、分類子によりスペクトルに「良」、「不良」または「未定義」が作製される。前述したように、工程104−118は、与えられたサンプルの3個の別々のアリコート(または用いられた任意のアリコート数)について並行して行われる。工程120において、全てのアリコートから同じクラス標識が得られたかどうかのチェックが行われる。もし同じでなかった場合、工程122で示されるように未定義の結果に変更される。全てのアリコートから同じ標識が得られた場合、工程124で示されるようにその標識が記録される。
【0059】
本開示に記載されるように、工程124で報告される新たかつ予想外のクラス標識が開示される。
【0060】
工程106、114、116および118が、典型的には、工程106の前処理工程、工程114でスペクトル値を得ること、工程116のK−NN分類アルゴリズムの適用、および工程118のクラス標識の作製をコードするソフトウェアを用いるプログラムされた一般的なコンピューターで行われることは明らかである。工程116で用いられるクラス標識スペクトルはコンピューターの記憶装置またはコンピューターがアクセス可能な記憶装置に記憶される。
【0061】
我々の先行特許出願公報米国特許番号第7,736,905号に記載されるように、該方法およびプログラムされたコンピューターは臨床検査センターで実行されてもよい。
【0062】
VeriStrat検査の作動メカニズムおよびその実際の結果に関する理解は、このセクションでさらに記載されるいくつかのソースに由来する。
【0063】
タンパク質IDからの直接的証拠
VeriStratは血清または血漿のMALDI−TOF MSのピーク強度を測定する。一実施態様において、VeriStratのシグナチャーは後述の表1に記載される8個の質量分析のピークを含む。分類は、所定のm/z範囲(前述のリストおよび表1を参照)においてサンプルの質量スペクトルを積分することにより強度、即ち特徴値を推定し、観察された8個の特徴値のセットを訓練サンプルのものと7近傍分類アルゴリズムを用いて関連付けすることにより行われる。この方法は非線形結合の特徴値を用いるものであり、一次元スコアの定義を行うことは出来ない。特徴値の線形結合のスコア関数を作製しようとする試みは常に失敗に終わり、常にパフォーマンスの低下を招いてきた。これら8個の特徴の全てまたは殆どが臨床的実用性を得るために有用であると考えられる。
【0064】
用いた特徴値のペプチド含量を決定することによりVeriStrat検査の作動メカニズムを理解することが出来るかもしれないと考えられていた。しかしながら、装置のm/z解像度はそのm/z範囲において1つのタンパク質またはペプチド鎖のみが存在することを保証するに足るほど高いものではないという事実により、これは困難となった。また、8個以上のペプチドが8個のピークシグナチャーを形成すると考えられ、そのいくつかは同じアミノ酸配列が翻訳後修飾を受けたものまたは酸化された形態であり、他のものは依然として未知のペプチドである可能性がある。加えて、特徴値、即ち推定されたピーク強度は、単にその被分析物がサンプル中に多く含まれることに対応するだけではない。これはMALDIのイオン化の複雑さによるものであり、即ち、検出器にヒットするイオン数は被分析物の量およびイオン化確率の両方に相関する。この半定量的な様式のピーク(特徴値)の比較により、タンパク質IDの標準的な方法による比較が困難なままとなる(LC−MS/MS)。
【表1】
【0065】
これらの困難な事柄にもかかわらず、我々は表1のピークの3つが血清アミロイドA(SAA)アイソフォームと関連している強い証拠を得た。我々はプールしたVeriStrat「良」およびVeriStrat「不良」サンプル間でディファレンス(DIGE)分析を行い、m/z11529および11685のピークを十分な配列包括度で分離することに成功し、それらをSAA 19−122およびSAA 20−122と同定した。理論的質量は実測されたm/z値とよく一致した。ゲル上で観測された0.4のPIシフトも理論的予測値とよく一致する。さらに、m/z5843におけるピークは11685のピークが2価に荷電したものと考えられる。これらのピークは、他でも観察されている5(Ducet, et al. Electrophoresis 1996, 17, 866-876、Kiernan et al. FEBS Letters 2003, 537, 166-170)。11445のピークが、親SAAタンパク質のC末端からの切断物の配列に関連するSAAアイソフォームである可能性もある。
【0066】
別のタンパク質またはタンパク質のアイソフォームがVeriStratシグナチャーに存在することは明らかであるが、SAAアイソフォームがVeriStrat検査の作動メカニズムにおいて重要な役割を果たすことも考えられる。以下のセクションでは、SAAがVeriStrat「不良」シグナチャーの少なくとも3つのピークの主要な要素であるという発見に基づくVeriStrat検査の作動メカニズムの推定される理論;SAAといくつかの受容体の相互作用およびこれらの相互作用の生物学的帰結に関する既知の情報、ならびに様々な癌細胞におけるSAAと機能的に結合するこれらの受容体の存在に関する情報が提供される。しかしながら、本発明は必ずしもこの理論に基づく必要はなく、かかる理論は本発明を限定する意図は持たない。
【0067】
癌のバイオマーカーとしてのSAAに関する先行技術文献は、引用文献6−16を参照すること。
【0068】
SAA:生物学的機能および腫瘍発症への関与
機能
SAAファミリーの決定的重要性は、SAAが進化を通して高度に保存された配列である17という事実、およびSAAの発現が感染、外傷または病的過程に応答して劇的に増加することにより示唆される。しかしながら、SAAの正確な生物学的機能は未だ完全には解明されていない。SAAはHDLの成分として脂質の運搬および代謝に関与し、おそらくは疾患の急性期において保護的な役割を果たす18が、慢性的な条件ではSAAは有害因子となる。SAAの発現が高い状態が続くと、関節リウマチなどのいくつかの疾患ではアミロイドAアミロイドーシスが誘発される19。しかしながら、SAAの臨床的に重要な機能の範囲はかなり広く、慢性的な炎症および腫瘍形成への関与も含まれる。後者の2つは密接に関係しており、Vlasova and Moshkovskii20 および Malle et al21のレビューで詳述される。
【0069】
SAAの腫瘍形成への関与は、その多面的な生物学的活性:炎症への関与、例えば、炎症促進性遺伝子の発現の活性化およびサイトカインの制御による慢性過程の補助、細胞外マトリックス分解への関与、抗アポトーシス性の性質、ならびに腫瘍形成に複雑に関与するマイトジェン活性化プロテインキナーゼ(MAPK)を含む特定の経路の活性化に起因する。
【0070】
SAAは細胞外マトリックス(ECM)の接着タンパクとして作用し得ること22、ECMの分解およびリモデリングに重要な役割を果たし、腫瘍新生、転移、および腫瘍浸潤に関与する24,25マトリックスメタロプロテアーゼ(MMP)を誘導すること18,23が示されている。
【0071】
SAAの免疫に関連する機能は、そのサイトカイン様活性により定義される。SAAはIL−8、TNF−αおよびIL−1βの産生を促進し26,27(これはおそらくは、SAA発現のポジティブフィードバックに繋がる)、同様に、細胞性免疫応答に重要な役割を果たすIL−12およびIl−23の産生も促進する28。SAAがPI3Kおよびp38 MAPKを活性化し得ることも示されている。
【0072】
SAAの炎症の制御への関与は、NF−κBおよびMAPK経路の活性化を同時に伴うCOX2発現誘導能と関連する29,30。癌と炎症の重要な相互関係は数多くの研究およびレビューの対象である31−37。近年の大量のデータから、SAAは2つの過程間のメディエーターの1つとして重要な役割を果たすことが示唆されているが、これは、SAAが重要な炎症性および腫瘍形成経路、例えば、古典的および非古典的MAPK経路、転写因子NF−κBのものを活性化することが出来ること、およびおそらくはそれらのクロストークに関わることによる。VeriStratシグナチャーと関連するSAAレベルの上昇は、該経路の活性化を測定に有用なものとして用いることができる。
【0073】
SAAの生物学的活性と関連する受容体および経路
NF−κB転写因子は多くの上皮および血液性の悪性病変で恒常的に活性化していることが知られており、抗アポトーシス性およびアポトーシス性の標的遺伝子、マトリックスメタロプロテアーゼの発現、血管新生および細胞周期を制御することにより41、炎症を伴う癌の進行に重要な役割を果たすと考えられている38,39,40。他方、NF−κBはアポトーシス促進性の遺伝子活性を発揮すること、および腫瘍抑制因子p53と協働してアポトーシスを誘導することも可能である42。実際の作用は刺激、細胞種、および関与するサブユニットに依存する43。Rel/NF−κB因子の抗アポトーシス性およびアポトーシス性作用は必ずしも対立するものではないが、同じ標的遺伝子のアップレギュレーションにより同じ細胞で起こり得る44。NF−κBはおそらくは炎症と癌をリンクする因子の主な1つであるが、これは、IL−6およびTNF−αといった炎症性サイトカイン、ならびにMMPおよびCOX−2を誘導するサイトカインの誘導に関与することによる35,45,46。NF−κBの活性化はEGFにより誘導されることがあり:EGFの刺激はNF−κB活性化を介したデスレセプター誘導性アポトーシスを阻害する。
【0074】
COX−2の過剰な発現は、広範囲の前悪性、悪性および転移性のヒト上皮癌47、例えば、肺癌48で見られる。COX2は、プロスタグランジンE2(PGE2)を介して細胞増殖、血管新生、アポトーシス、および細胞遊走を仲介し、マイトジェン活性化プロテインキナーゼ(MAPK)カスケードの腫瘍原性シグナリングをトランス活性化する49,50。COX2はErkを介してMAPKをトランス活性化する49,92。その関係は相反的なものであり:MAPK経路を介して作用する上皮増殖因子(EGF)はいくつかの上皮細胞においてCOX2活性を劇的に誘導する51。TGFαによるEGFRの活性化はCOX2を刺激し、PGE2放出の増加および有糸分裂誘発の増加を引き起こす52。
【0075】
マイトジェン活性化プロテインキナーゼ(MAPK)カスケードは、活性化した成長因子受容体からの成長刺激シグナルを伝達するため、通常のセルバイオロジーならびに癌の発症に重要な役割を果たす。MAPKシグナル伝達はしばしば、成長因子の1つがそのチロシンキナーゼ型受容体(RTK)である膜受容体に結合することにより開始され、Raf、MEKおよび細胞外シグナル制御キナーゼ(ERK)の作動を引き起こす。近年の研究により、RTKからERKへのシグナリングは単なる直線的なRas依存性経路よりもはるかに複雑なものであることが示され、ETKが仲介するERKシグナリングの強度、期間および細胞局在の決定に重要な役割を果たす様々なシグナリングモジュレーターが同定される必要がある50。SAAは様々な上皮細胞のいくつかの受容体と機能的に結合し、この結合により、上記および特定の治療に対するVeriStrat「不良」患者の耐性に繋がり得るNF−κBおよびMAPKの両方の下流の活性化が起こる(以下においても議論される)。これらの受容体のいくつかに関する概略を以下に示す。
【0076】
FPRL受容体
FPRL受容体は幹細胞53、腸上皮54および肺55を含む様々な細胞で発現される。SAAは古典的なGタンパク質共役型受容体の1つであるFPRL1と相互作用し、細胞機能および上皮増殖および/またはアポトーシスの制御に重要なシグナリングネットワークを誘起する。SAAのFPRL1への結合により、インターロイキンの活性化および誘導が引き起こされる。FPRLの関与によりプロテインキナーゼC(PKC)および転写因子NF−κB経路が活性化され30、これはアポトーシスの阻害および癌の進行を伴う56,57,41。SAAのFPRL1への結合により、好中球およびリウマチ滑膜細胞におけるアポトーシスのレスキューが起こることも示されているが、これは、MAPK、ERK1/2、PI3K/Aktシグナリングのリン酸化、ならびにSTAT3活性化および細胞内Ca2+の放出により仲介され58,59,60、それにより細胞の増殖および生存が促進される。
【0077】
SR−BI受容体
スカベンジャーレセプターB−I(SR−BI)は、選択的なコレステロール取り込みを仲介する高密度リポタンパク質受容体として同定された61。SR−BIはステロイド産生組織および肝臓で最も多く発現されるが、炎症中のマクロファージおよび単球においても発現が上昇する;SB−BIの発現の上昇は、SAAの存在も特徴であるヒトアテローム硬化病変における脂質含有マクロファージにおいても示されている。SAAはSR−BIが仲介する細胞コレステロールのエフラックスを促進する62。
【0078】
Baranova et al.63では、HeLaおよびTHP1(ヒト急性単球性白血病細胞株)細胞におけるSAAのSR−BIへの特異的結合(おそらくは、HDLと結合した状態で)はERK1/2およびp38 MAPKのリン酸化、ならびにIl−8分泌を伴うことが示された。SR−BI受容体の発現は、ヒト肺癌細胞株を含むさまざまな細胞で示されている64。
【0079】
RAGE
終末糖化産物受容体(RAGE)は肺においてのみ検出可能なレベルで常時発現されているが、炎症部位において、主に炎症性および上皮細胞でその発現は急激に上昇する。上皮細胞のRAGEは、膜結合型または可溶型タンパクとして、ストレスにより大幅にアップレギュレートされる。RAGEを介した恒久的なシグナリングにより生存経路が誘導され、アポトーシスおよび(ATP欠乏による)ネクローシスが減弱された。これにより、多くの例において上皮の悪性化を引き起こす背景となる慢性的な炎症が導かれた65。RAGEの発現量の上昇は前立腺癌、直腸癌および胃癌に伴う;一方で、肺癌および食道癌の進行したステージはRAGE発現量の低下を特徴とする66。口腔扁平細胞癌では、RAGEの発現は癌の進行および再発と強く相関し、RAGE陽性患者は著しく短い無病生存期間を示した。数多くの様々なリガンドの内、SAAは終末糖化産物受容体(RAGE)と結合し、ERK1/2およびp38 MAPK経路を介してNF−κBを誘導する(COX経路を誘導することはない)ことが見出された67。
【0080】
TLR
近年の発見により、SAAがToll様レセプター(TLR)であるTLR4およびTLR2の内因性のリガンドとして作用し得ることが示された21。TLR4はいくつかのヒトの癌細胞で発現していることが見出された68,69。肺癌では、TLR4の活性化により免疫抑制性サイトカインTGF−ベータ、血管新生促進性ケモカインIL−8およびVEGFの産生が促進されることが示された。VEGFおよびIL−8分泌促進はp38 MAPK活性化を伴う70。SAAによるTLR4の活性化はp42/44およびp38 MAPKの活性化を必要とした71。
【0081】
TLR2はまた、SAAの機能的受容体であることも示された。TLR2を発現するHeLa細胞はNF−κBの強い活性化を伴ってSAAに応答した;SAAによる刺激は、TLR2−HeLa細胞において、ERK1/2(P−ERK1/2)、p38 MAPK(P−p38)、およびJNK(P−JNK) MAPKのリン酸化の亢進を引き起こし、IκBα(NFκB阻害因子)分解の促進を加速した72。SAAによる特異的な活性化の結果としてのNF−κBの刺激は、マクロファージにおいも示されている73。
【0082】
癌の発症および治療への耐性におけるSAAの予想される相互作用およびその生物学的効果の簡単なスキームを図3に示す。これから見て取れるように、SAAの生物学的機能は、SAAと様々な受容体の相互作用を引き金とする多数の経路のクロストークの視点から見ることができ、それは最終的には少なくとも1つの主要なMAPK経路:ERK、p38およびJNK21,41および/またはNF−κB活性化に収束する。これらの相互作用のいくつかは図4のEGFR伝達経路のスキームにおいて示される。
【0083】
EGFRはいくつかの主要な下流のシグナリング経路、例えば、Ras−Raf−Mek、ならびにホスホイノシチド3−キナーゼ(PI3K)、Akt、およびPKCからなる経路を活性化するチロシンキナーゼ型受容体(TKR)である。次いで、これは、NF−κB転写活性化経路および炎症性経路、例えば、COX2により誘導されるものとの複数のクロストークの関係を介し、増殖、生存、浸潤、転移速度、および腫瘍血管新生に対する効果を有し得る。SAAはこれらの経路を、チロシンキナーゼ型受容体と独立して活性化することができる(幅広の矢印で示す)。
【0084】
EGFRの発現量の上昇および/または恒常的な活性化は数多くの癌、例えば、脳癌、乳癌、腸癌および肺癌に伴う。そのカスケードの成分の変動はその経路の活性化に繋がり、癌の発症および進行に関連すると考えられている。例えば、EGFRキナーゼドメイン(非喫煙者)またはKRAS(喫煙者)における活性化変異は肺癌の早期発症に関連する74,75。Rasタンパクは約25%の腫瘍で恒常的に活性化しており、上流の制御とは独立して分裂促進シグナリングを誘起する76,77。新たに蓄積された大量のデータにより、非直線的なシグナリングおよびトランス活性化が癌の発症および進行に重要な役割を果たすことが示唆される。
【0085】
SAA相互作用と抗癌療法に対する耐性
化学療法、放射線療法および抗炎症療法
前で議論し、図3および4に示されるように、SAAと数多くの受容体との相互作用は抗癌療法に対する耐性と関連する経路の活性化に繋がる。化学療法および放射線療法に対する耐性におけるNF−κBの役割は既に議論される41。NF−κBを阻害すると、アポトーシス応答亢進により、放射線療法78,79、およびデス(death)サイトカイン80に対する感受性が上昇する。同時に、放射線およびいくつかの化学療法剤への曝露はNF−κBの活性化および後続するアポトーシスへの耐性を引き起こす81,79。化学療法(ゲムシタビン)誘発性のNF−κB活性化を阻害することにより、NSCLC細胞株の化学療法誘発性アポトーシスへの感受性が回復することが示されている82,81。他方で、いくつかの場合において、NF−κBは化学療法に対する感受性に関連することが示されており、例えば、NF−κBはパクリタキセル誘発性細胞死に必要であることが示唆されている82。
【0086】
これらの情報を考慮すると、VeriStrat「不良」患者に特徴的な血漿または血清中のSAA濃度の上昇から導かれる1つの結論は、SAAの上昇によりNF−κB転写因子およびMAPK経路の活性化が引き起こされる可能性があるというものである。これは、主に放射線療法に耐性を有する癌と関連する可能性、および患者の化学療法に対する応答に影響する可能性がある。しかしながら、各治療タイプおよび患者のコホートについて個々に評価されるべき数多くの因子が存在する。
【0087】
三酸化ヒ素、クルクミン、サリドマイドといったNF−κB阻害剤は、数多くの臨床治験の対象とされてきた。しかしながら、NF−κB阻害剤は正常な造血前駆細胞の化学療法誘発性アポトーシスも亢進するため、NF−κB阻害剤を化学療法の佐剤として用いることにより骨髄の回復が遅れる可能性があった。NF−κBは自然免疫応答および獲得免疫応答の活性化に重要な役割を有するため、阻害剤の長期間の使用は免疫不全のリスクを伴う可能性が高い41ことは考慮されるべきである。
【0088】
もし、VeriStrat「不良」シグナチャーが実際にNF−κBの特異的な活性化と相関しているならば、このシグナチャーはNF−κB阻害剤が最も有効である患者の選択に用いることができ、不要な治療およびそれに伴う病的状態を減弱できる可能性がある。
【0089】
受容体型チロシンキナーゼを標的とする治療
erbB受容体およびMAPK経路
EGFRおよびHER2は、4つのメンバー(EGFR(HER1)、erbB4(HER4)、erbB3(HER3)、およびerbB2(HER2))からなる上皮増殖因子受容体(EGFR)ファミリーに属する。上皮癌の大半は上皮増殖因子受容体(EGFR)およびHER2受容体の異常な活性化を示すため、これらの受容体の特異的な阻害は標的抗癌療法のストラテジーとなり、多くの研究の対象とされる。
【0090】
リガンドの非存在下において、EGFR受容体はキナーゼ活性が抑制されるような構造で存在する。リガンドの結合により、「二量体化ループ」を露出させるような構造変化が開始され、受容体の二量体化が誘起される。これらの転移は細胞膜を横切って中継され、キナーゼドメインを活性化する。この活性化スキームの多様性はErbBファミリーで見られる。ErbB−3は機能的なキナーゼではないが二量体のパートナーをトランス活性化することができ、他方で、HER2/ErbB−2は活性化構造に「ロックされた」リガンド不在の発癌受容体である。
【0091】
この二量体化によりチロシンキナーゼ機能の活性化が起こり、3つの主要なシグナリング経路を介したシグナル伝達に繋がり、最終的には、アポトーシスの回避、血管新生の持続、抗増殖シグナルへの耐性、増殖シグナルの自給自足、および転移が引き起こされる77,83。
【0092】
該カスケードの構成要素の変化は該経路の活性化を引き起こし、癌の誘発および進行と関連すると考えられており、例えば、EGFRキナーゼドメイン(非喫煙者)またはKRAS(喫煙者)の活性化変異は肺癌の早期発症と関連する74,72。Rasタンパクは約25%の腫瘍で恒常的に活性化しており、上流の制御とは独立した分裂促進シグナリングを誘導する76,77。
【0093】
いくつかのチロシンキナーゼ阻害剤(2つの小分子EGFRチロシンキナーゼ阻害剤−エルロチニブおよびゲフィチニブ、ならびにEGFRおよびHER2のデュアル阻害剤ラパチニブを含む)は、臨床現場で様々な固形腫瘍に現在使用される。ヒト化したモノクローナル抗HER2抗体トラスツズマブ、ならびに2つの抗EGFR抗体−セツキシマブおよびパニツムマブも臨床適用が認可されている。
【0094】
数多くの刊行物でレビューされるチロシンキナーゼ阻害剤(小分子ならびにモノクローナル抗体)に対する自然耐性ならびに獲得耐性は、活性化KRAS変異、met−癌原遺伝子の増幅84、およびT790M変異といった様々な因子に起因する。癌の多様性、および標的薬剤に対して耐性の複数の経路を示す能力により、単剤による治癒的療法の見込みは困難なものとなるが84、これは、多くの理由の中でも取り分け、リガンドとその受容体の相互作用の通常上流にあるものとは独立してシグナリングの活性化が起こる可能性があることによる。複数のチロシンキナーゼの共発現、受容体下流の経路のクロストーク、および伝達カスケードの下流の活性化の重要性を示す証拠が増加している。
【0095】
経路のトランス活性化は、複数の研究において耐性のメカニズムの1つであることが示唆された。例えば、インスリン様成長因子−I受容体(IGF−1R)シグナリングは、ヒト乳癌および前立腺癌細胞株においてゲフィチニブによりブロックされたEGFRを代償できることが示された85。発癌性のPIK3CAまたは別のRTKによる代替の下流のシグナリング、特に、Aktの活性化を介したものは、NSCLCにおけるTKIに対する耐性のメカニズムの1つであると記載される86。
【0096】
Cappuzzoら88は、NSCLC患者のゲフィチニブに対する感受性はAktが活性化された場合に非常に低くなる一方で、EGFRの発現はネガティブであることを観察し、EGFR非依存的活性化がゲフィチニブに対する耐性に繋がり得ることを確認した。
【0097】
我々は、VeriStrat検査により測定されるようなSAAの相互作用がRTK非依存的なMAPKカスケードの活性化を引き起こし、その結果として、TKI耐性となることを提案する。このSAAの活性化のメカニズムは、直接的でも間接的でもよい。SAAの直接的な作用はRAGEまたはTLR2およびTLR4受容体へのその結合により仲介され、古典的MAPK経路の活性化を引き起こし得る(JNKおよびp38の活性化による)。様々な癌細胞および癌細胞と関連する細胞の表面におけるこれらの受容体の存在およびその相互作用は、Malle, et al21でレビューされる。TLR受容体の活性化の結果としてのEGFRの活性化を直接証明する証拠が存在する66。
【0098】
SAAの間接的な作用はFPRL受容体を介して作用し、インターロイキンIl6、およびIl8の放出を引き起こし、次いで、Gタンパク質共役型受容体と反応し、PKCを活性化することで説明することができる。(PKCの活性化により細胞増殖および血管透過性が亢進され、MAPK経路におけるMEKの活性化が引き起こされる86)。加えて、それはVEGFの発現を誘導する。
【0099】
SAAは、肺上皮細胞およびマクロファージにおけるTRL4のリガンドである。所要細胞で発現されるTLRのライゲーションによってもVEGFレベルが上昇することが報告されている70。
【0100】
この情報により、SAAによる主要な3つの全てのMAPK経路の下流の活性化に関与するメカニズムが存在する証拠が提供される。MAPK経路の下流の活性化はRTK非依存的であり、「交差する」チェックポイントから上流の標的阻害に対する耐性を引き起こす可能性がある。
【0101】
前記の観点から、VeriStrat検査を用いる併用療法を含む特定の治療に最も適した患者のセレクションは、薬剤耐性のいくつかのタイプを克服する道具となり得る。
【0102】
併用療法およびVeriStratシグナチャー
TKIおよびCOX2阻害剤
前記で議論したように、SAAはCOX2の発現を誘導することができる。肺癌におけるCOX2の過剰な発現は、Huang et al.87により最初に報告され、約70%の腺癌で観察され88、他の数多くの研究により確認された。
【0103】
数多くの治験によりCOX2およびEGFRシグナリング経路のクロストークが示されている。前記で議論したように、MAPK経路を介して作用する上皮増殖因子(EGF)は、いくつかの上皮細胞においてCOX2活性を著しく誘導する47。TGFαによるEGFRの活性化はCOX2を刺激し、PGE2の放出および有糸分裂誘発の亢進を引き起こす48。他方、COX2の産物であるプロスタグランジンE2(PGE2)は、EGF受容体をトランス活性化することができる45。NSCLCにおいて、PGE2が、EGFR非依存的な様式の細胞内クロストークによりMAPK/Erk経路を活性化することが示され;この効果はGタンパク質共役型受容体およびプロテインキナーゼC(PKC)を通して仲介され、EGFR−TKI耐性に寄与する可能性がある89。
【0104】
他方で、COX2阻害剤はNF−κB経路を阻害することが示されている:セレコキシブはAktおよびIKKの抑制を介してその効果を発揮する。ヒト非小細胞性肺癌において、セレコキシブはNF−κB、ならびにTNF誘導性のJNK、p38 MAPK、およびERKの活性化をIKKおよびAktの抑制を介して抑制し、COX2、ならびに炎症、増殖および腫瘍新生に必要な他の遺伝子の合成のダウンレギュレーションを引き起こす46,90。アスピリンおよびイブプロフェンを含む他のNSAIDは、IKK活性化およびIκBα分解の抑制により作用することが示された。合わせると、これらの考察により標準的な抗癌療法にCOX2を追加することの強力な理論的根拠が提供される。
【0105】
NSCLCにおける抗炎症療法およびチロシンキナーゼ受容体標的療法の併用療法に関する研究およびそれらがEGFR−TKIを克服する可能性は以前の文献でレビューされている90,91。治験の結果はネガティブであった:ゲフィチニブおよびセレコキシブの併用療法における応答率および患者の生存率、ならびにロフェコキシブおよびエルロチニブで治療した患者の病勢コントロール率92,93は単剤療法におけるものと同様であることが分かった。
【0106】
COX阻害剤の添加の効果が、この経路において示唆されるSAAのアップレギュレーション効果により、VeriStrat「不良」患者においてより顕著となる可能性がある。しかしながら、下流のMAPK活性およびNF−κBならびにそれらの相互作用に対するCOX2経路の阻害効果の度合いは不明であるため、該効果の度合いを予測することは困難である。この仮説はさらに研究されるべきである。
【0107】
細胞株に基づく証明(図8)
我々は、VeriStrat「不良」血清が腫瘍細胞に生物学的効果を及ぼし得ること、特に、薬剤感受性細胞株においてそれがゲフィチニブに対する耐性を増強し得ることを示してきた。ゲフィチニブ感受性細胞株HCC4006(エクソン19が欠損したEGFRを有する)および耐性株A549(EGFR野生型)を用いて実験を行った。ステージIIIB/IVであり、Veristrad「良」および「不良」と識別されたNSCLC患者からヒト血清を得た。各分類の血清を合わせてプールを作製し、増殖阻害アッセイに用いた。2種類の組成の培地(RPMI/10%「良」血清またはRPMI/10%「不良」血清)を用いて細胞を播種した(10複製/薬剤濃度;2,000細胞/ウェル)。24時間後、ゲフィチニブを加え、プレートを6日間インキュベートした。増殖阻害の測定にはMTTアッセイを用いた。結果を下記の表2および図8に示す。
【表2】
【0108】
図8は、異なる濃度のゲフィチニブの存在下における、Veristrad「良」およびVeristrad「不良」血清中のゲフィチニブ感受性細胞株HCC4006およびゲフィチニブ耐性細胞株A549の増殖を示すグラフである。図8において、%コントロールは、対応する増殖培地中における所定のゲフィチニブ濃度の吸光度の薬剤非存在下における吸光度の平均値に対する比から算出した。エラーバーは正規化した測定値の標準偏差を表す。
【0109】
VeriStrat「不良」血清中で増殖させた場合、感受性細胞において阻害の相対的な減弱が見られたが、耐性腫瘍細胞では有意な変化は見られなかった。この結果は、VeriStrat「不良」血清が腫瘍細胞に対し直接生物学的効果を有することを示し、VeriStrat「良」血清の効果とは異なるものである。これらの結果は、VeriStratメカニズム、それとホスト−腫瘍相互作用、および患者集団における標的療法の相対的な有効性との関係の我々の仮説を支持するものである。
【0110】
化学療法におけるVeriStrat
図7に示されるように、VeriStrat「不良」シグナチャーはいくつかの非標的療法に対する応答不良に関連するが、他のものには関連しない。VeriStrat分類は、DNA複製またはNF−κBで制御される遺伝子の転写に干渉する化学療法(シスプラチン、ゲムシタビンなど)におけるアウトカムと相関する可能性が高いが、非標的療法におけるVeriStratの有用性の具体的な範囲は実験的に決定する必要がある。
【0111】
かくして、VeriStrat検査の臨床における適用の例は、いくつかの非標的化学療法レジメン(DNAの複製および/またはNF−κB転写因子で制御される遺伝子の活性化と相互作用するものなど)が癌患者に有効である可能性が低いかどうかを予測するための方法であり、サンプル(図1)にVeriStrat検査を行い、もし結果が「不良」のクラス標識であれば該患者には有効である可能性が低いことを特徴とするものになろう。
【0112】
SAAがNF−κB転写因子の活性化の原因であること、ならびに癌の進行および様々な療法への応答におけるNF−κB活性化の役割に関する文献的情報を考慮に入れると、VeriStratシグナチャーは主に放射線療法に耐性を有する癌、ならびに化学療法に対する患者の応答と相関する可能性がある。
【0113】
三酸化ヒ素、クルクミン、サリドマイドといったNF−κB阻害剤は、抗癌剤として臨床治験で評価の対象となっている。しかしながら、それらの有用性は、これらの薬剤に対する応答のバイオマーカーが存在しないこと、ならびにそれらの副作用により、限定されたものとなっている。VeriStratはNF−κBの活性化の亢進のバイオマーカーとして有用であり得、故に、NF−κB阻害剤が最も有効である患者(おそらくは、VeriStrat「不良」)のセレクションに有用となり得る。
【0114】
前記の全てをまとめると、本発明は図1のVeriStrat検査のさらなる使用を包含する。一般的な事柄として、VeriStrat検査は、AktまたはERK/JNK/p38またはPKCでの、またはそれらの上流でのMAPK経路またはPKC(プロテインキナーゼC)経路に関わる受容体のアゴニスト、受容体もしくはタンパク質を標的にする治療薬または治療薬の組み合わせによる治療が患者に有効であるかを予測する。予測の度合いは、特定の薬剤または薬剤の組み合わせに依存するであろう。VeriStrat検査は、下流の制御を標的とする薬剤の効果は予測しない。
【0115】
一実施態様において、本発明は、固形上皮腫瘍癌患者を、AktまたはERK/JNK/p38またはPKCでの、またはそれらの上流でのMAPK経路またはPKC経路に関わる受容体のアゴニスト、受容体もしくはタンパク質を標的にする治療薬または治療薬の組み合わせによる治療が有効である可能性が高い患者として、あるいは治療薬または治療薬の組み合わせによる治療が有効である可能性が低い患者として識別する方法であって:
a)固形上皮腫瘍癌患者の血液由来サンプルから質量スペクトルを得;
b)a)で得た質量スペクトルにおいて1つまたはそれ以上の所定の前処理工程(バックグラウンドの除去、ノーマライゼーションおよびスペクトルのアラインメント)を行い;
c)b)の質量スペクトルにおける前処理工程を行った後、1つまたはそれ以上の所定のm/z範囲(および、好ましくは前記の表1で示されるm/z範囲に対応するm/z範囲)において該スペクトルにおける選択された特徴の積分強度値を得;
d)患者を治療薬または治療薬の組み合わせが有効である可能性が高い、または有効である可能性が低い患者として識別するために、別の固形腫瘍患者の血液由来サンプルから得たクラス標識スペクトルを含む訓練セットを用いた分類アルゴリズム(例えば、k近傍法)にc)で得た値を用いる
ことを特徴とする方法と見做すことができる。
【0116】
特定の例として、EGFR−IにMAPK経路の下流の活性化を遮断する標的薬剤を加えることにより、VeriStrat「不良」シグナチャーを有する患者のEGFR−Iに対する耐性を克服できる可能性がある。
【0117】
別の特定の例として、COX2阻害剤、セレコキシブおよびロフェコキシブをEGFR−Iに治療レジメンとして加えることにより、VeriStrat「不良」シグナチャーを有する患者のEGFR−Iに対する耐性を克服できる可能性がある。VeriStrat検査は、故に、COX2阻害剤およびEGFRIを含む併用療法を処方するためのインジケーターとして用いることができる。特定の実施態様において、COX2阻害剤およびEGFRIの投与が癌患者に有効である可能性が高いかどうかを予測する方法は、a)癌患者の血液由来サンプルから質量スペクトルを得;b)a)で得た質量スペクトルにおいて1つまたはそれ以上の所定の前処理工程(バックグラウンドの除去、ノーマライゼーションおよびスペクトルのアラインメント)を行い;c)b)の質量スペクトルにおける前処理工程を行った後、1つまたはそれ以上の所定のm/z範囲(および、好ましくは前記の表1で示されるm/z範囲に対応するm/z範囲)において該スペクトルにおける選択された特徴の積分強度値を得;d)患者をCOX2阻害剤およびEGFR−Iの投与による治療が有効である可能性が高い、または有効である可能性が低い患者として識別するために、別の固形腫瘍患者の血液由来サンプルから得たクラス標識スペクトルを含む訓練セットを用いた分類アルゴリズム(例えば、k近傍法)にc)で得た値を用いることを特徴とする。特に、クラス標識が「不良」の場合、該患者には有効である可能性が高いことが示唆される。
【0118】
別の特定の例として、VeriStrat「不良」シグナチャーはNF−κBの特異的な活性化と相関すると考えられ、故に、該検査は、NF−κB阻害剤、およびCOX2阻害剤の標準的な化学療法への追加が最も有効である患者の選択に用いることができ、同時に、不必要な治療およびそれに関連する病的状態の軽減にも有用であろう。
【0119】
本開示の方法は、癌患者の血液由来サンプル(またはこれらのサンプルからの質量分析データ)を受け取り、これらの質量分析データを機械で読み取り可能な記憶媒体に蓄え、図1に示される工程および分類工程を機械で行う、例えば、クラス標識(VeriStrat「良」または「不良」)を作製するようプログラムされたコンピューターを用い、故に前記の薬剤または薬剤の組み合わせが有効である可能性が高いとしての識別を予測する臨床試験センターで実行されてもよい。別の一実施態様として、本発明は、COX2阻害剤およびEGFR阻害剤の組み合わせの投与が癌患者に有効である可能性が高いかどうかを識別または予測するよう構成された装置として設定することができる。該装置は、癌患者の血液由来サンプルの質量スペクトルを記憶する記憶装置、コンピューターの記憶媒体またはデータベース、ならびにa)質量スペクトルにおいて1つまたはそれ以上の所定の前処理工程を行い(図1参照);b)1つまたはそれ以上の所定のm/z範囲において該質量スペクトルにおける特徴の積分強度値を得(例えば、表1のピークのリストに含まれる範囲または前記のm/z範囲において);c)患者をCOX2阻害剤およびEGFR阻害剤の組み合わせの投与による治療が有効である可能性が高い、または低い患者として識別するために、別の固形腫瘍患者の血液由来サンプルから得たクラス標識スペクトルを含む訓練セットを用いた分類アルゴリズム(例えば、KNN分類アルゴリズム)にb)で得た値を用いるよう設定されたソフトウェアの指示を実行するプロセッサー(例えば、一般的な目的でプログラムされたコンピューターの一般的なCPU)の組み合わせを含む。
【0120】
別の一例において、本発明は、固形上皮腫瘍癌患者を、AktまたはERK/JNK/p38またはPKCでの、またはそれらの上流でのMAPK(マイトジェン活性化プロテインキナーゼ)経路またはPKC(プロテインキナーゼC)経路に関わる受容体のアゴニスト、受容体もしくはタンパク質を標的にする治療薬または治療薬の組み合わせによる治療が有効である可能性が高い患者として、あるいは治療薬または治療薬の組み合わせによる治療が有効である可能性が低い患者として識別するよう設定された装置として具体化することができる。該装置は、固形上皮腫瘍癌患者の血液由来サンプルの質量スペクトルを記憶する記憶装置、およびa)質量スペクトルにおいて1つまたはそれ以上の所定の前処理工程を行い(図1参照)、b)1つまたはそれ以上の所定のm/z範囲において該質量スペクトルにおける特徴の積分強度値を得(例えば、表1のピークのリストに含まれる範囲または前記のm/z範囲において)、c)患者を治療薬または治療薬の組み合わせが有効である可能性が高い、または低い患者として識別するために、別の固形腫瘍患者の血液由来サンプルから得たクラス標識スペクトルを含む訓練セットを用いた分類アルゴリズムにb)で得た値を用いるよう設定されたソフトウェアの指示を実行するプロセッサーの形態を取る。
【0121】
開示される発明のさらなる例は付属の請求項で説明される。
【0122】
付録
引用文献
1. Taguchi F, Solomon B, Gregorc V, et al. Mass spectrometry to classify non-small-cell lung cancer patients for clinical outcome after treatment with epidermal growth factor receptor tyrosine kinase inhibitors: a multicohort cross-institutional study. J Natl Cancer Inst 2007;99:838-46.
2. Clark GM, Zborowski DM, Culbertson JL, et al. Clinical utility of epidermal growth factor receptor expression for selecting patients with advanced non-small cell lung cancer for treatment with erlotinib. J Thorac Oncol 2006;1:837-46.
3. Chung CH, Seeley EH, Roder H, et al. Detection of tumor epidermal growth factor receptor pathway dependence by serum mass spectrometry in cancer patients. Cancer Epidemiol Biomarkers Prev 2010;19:358-65.
4. Carbone DP, Salmon JS, Billheimer D, et al. VeriStrat((R)) classifier for survival and time to progression in non-small cell lung cancer (NSCLC) patients treated with erlotinib and bevacizumab. Lung Cancer 2009.
5. Kiernan UA, Tubbs KA, Nedelkov D, Niederkofler EE, Nelson RW. Detection of novel truncated forms of human serum amyloid A protein in human plasma. FEBS Lett 2003;537:166-70.
6. Cremona M, Calabro E, Randi G, et al. Elevated levels of the acute-phase serum amyloid are associated with heightened lung cancer risk. Cancer 2010.
7. Benson MD, Eyanson S, Fineberg NS. Serum amyloid A in carcinoma of the lung. Cancer 1986;57:1783-7.
8. Biran H, Friedman N, Neumann L, Pras M, Shainkin-Kestenbaum R. Serum amyloid A (SAA) variations in patients with cancer: correlation with disease activity, stage, primary site, and prognosis. J Clin Pathol 1986;39:794-7.
9. Khan N, Cromer CJ, Campa M, Patz EF, Jr. Clinical utility of serum amyloid A and macrophage migration inhibitory factor as serum biomarkers for the detection of nonsmall cell lung carcinoma. Cancer 2004;101:379-84.
10. Cho WC, Yip TT, Yip C, et al. Identification of serum amyloid a protein as a potentially useful biomarker to monitor relapse of nasopharyngeal cancer by serum proteomic profiling. Clin Cancer Res 2004;10:43-52.
11. Yokoi K, Shih LC, Kobayashi R, et al. Serum amyloid A as a tumor marker in sera of nude mice with orthotopic human pancreatic cancer and in plasma of patients with pancreatic cancer. Int J Oncol 2005;27:1361-9.
12. Gutfeld O, Prus D, Ackerman Z, et al. Expression of serum amyloid A, in normal, dysplastic, and neoplastic human colonic mucosa: implication for a role in colonic tumorigenesis. J Histochem Cytochem 2006;54:63-73.
13. Engwegen JY, Mehra N, Haanen JB, et al. Validation of SELDI-TOF MS serum protein profiles for renal cell carcinoma in new populations. Lab Invest 2007;87:161-72.
14. Dai S, Wang X, Liu L, et al. Discovery and identification of Serum Amyloid A protein elevated in lung cancer serum. Sci China C Life Sci 2007;50:305-11.
15. Liu DH, Wang XM, Zhang LJ, et al. Serum amyloid A protein: a potential biomarker correlated with clinical stage of lung cancer. Biomed Environ Sci 2007;20:33-40.
16. Michaeli A, Finci-Yeheskel Z, Dishon S, Linke RP, Levin M, Urieli-Shoval S. Serum amyloid A enhances plasminogen activation: implication for a role in colon cancer. Biochem Biophys Res Commun 2008;368:368-73.
17. Uhlar CM, Burgess CJ, Sharp PM, Whitehead AS. Evolution of the serum amyloid A (SAA) protein superfamily. Genomics 1994;19:228-35.
18. Uhlar CM, Whitehead AS. Serum amyloid A, the major vertebrate acute-phase reactant. Eur J Biochem 1999;265:501-23.
19. Sipe JD. Amyloidosis. Annu Rev Biochem 1992;61:947-75.
20. Vlasova MA, Moshkovskii SA. Molecular interactions of acute phase serum amyloid A: possible involvement in carcinogenesis. Biochemistry (Mosc) 2006;71:1051-9.
21. Malle E, Sodin-Semrl S, Kovacevic A. Serum amyloid A: an acute-phase protein involved in tumour pathogenesis. Cell Mol Life Sci 2009;66:9-26.
22. Preciado-Patt L, Levartowsky D, Prass M, Hershkoviz R, Lider O, Fridkin M. Inhibition of cell adhesion to glycoproteins of the extracellular matrix by peptides corresponding to serum amyloid A. Toward understanding the physiological role of an enigmatic protein. Eur J Biochem 1994;223:35-42.
23. Migita K, Kawabe Y, Tominaga M, Origuchi T, Aoyagi T, Eguchi K. Serum amyloid A protein induces production of matrix metalloproteinases by human synovial fibroblasts. Lab Invest 1998;78:535-9.
24. Hynes RO. The extracellular matrix: not just pretty fibrils. Science 2009;326:1216-9.
25. Vihinen P, Ala-aho R, Kahari VM. Matrix metalloproteinases as therapeutic targets in cancer. Curr Cancer Drug Targets 2005;5:203-20.
26. Furlaneto CJ, Campa A. A novel function of serum amyloid A: a potent stimulus for the release of tumor necrosis factor-alpha, interleukin-1beta, and interleukin-8 by human blood neutrophil. Biochem Biophys Res Commun 2000;268:405-8.
27. Patel H, Fellowes R, Coade S, Woo P. Human serum amyloid A has cytokine-like properties. Scand J Immunol 1998;48:410-8.
28. He R, Shepard LW, Chen J, Pan ZK, Ye RD. Serum amyloid A is an endogenous ligand that differentially induces IL-12 and IL-23. J Immunol 2006;177:4072-9.
29. Malle E, Bollmann A, Steinmetz A, Gemsa D, Leis HJ, Sattler W. Serum amyloid A (SAA) protein enhances formation of cyclooxygenase metabolites of activated human monocytes. FEBS Lett 1997;419:215-9.
30. Jijon HB, Madsen KL, Walker JW, Allard B, Jobin C. Serum amyloid A activates NF-kappaB and proinflammatory gene expression in human and murine intestinal epithelial cells. Eur J Immunol 2005;35:718-26.
31. Coussens LM, Werb Z. Inflammation and cancer. Nature 2002;420:860-7.
32. Farrow B, Sugiyama Y, Chen A, Uffort E, Nealon W, Mark Evers B. Inflammatory mechanisms contributing to pancreatic cancer development. Ann Surg 2004;239:763-9; discussion 9-71.
33. Ditsworth D, Zong WX. NF-kappaB: key mediator of inflammation-associated cancer. Cancer Biol Ther 2004;3:1214-6.
34. Balkwill F, Coussens LM. Cancer: an inflammatory link. Nature 2004;431:405-6.
35. Lu H, Ouyang W, Huang C. Inflammation, a key event in cancer development. Mol Cancer Res 2006;4:221-33.
36. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature 2008;454:436-44.
37. Lee JM, Yanagawa J, Peebles KA, Sharma S, Mao JT, Dubinett SM. Inflammation in lung carcinogenesis: new targets for lung cancer chemoprevention and treatment. Crit Rev Oncol Hematol 2008;66:208-17.
38. Greten FR, Eckmann L, Greten TF, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004;118:285-96.
39. Pikarsky E, Porat RM, Stein I, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004;431:461-6.
40. Karin M. The IkappaB kinase - a bridge between inflammation and cancer. Cell Res 2008;18:334-42.
41. Lee CH, Jeon YT, Kim SH, Song YS. NF-kappaB as a potential molecular target for cancer therapy. Biofactors 2007;29:19-35.
42. Graham B, Gibson SB. The two faces of NFkappaB in cell survival responses. Cell Cycle 2005;4:1342-5.
43. Kaltschmidt B, Kaltschmidt C, Hofmann TG, Hehner SP, Droge W, Schmitz ML. The pro- or anti-apoptotic function of NF-kappaB is determined by the nature of the apoptotic stimulus. Eur J Biochem 2000;267:3828-35.
44. Bernard D, Monte D, Vandenbunder B, Abbadie C. The c-Rel transcription factor can both induce and inhibit apoptosis in the same cells via the upregulation of MnSOD. Oncogene 2002;21:4392-402.
45. Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol 2002;2:725-34.
46. Shishodia S, Koul D, Aggarwal BB. Cyclooxygenase (COX)-2 inhibitor celecoxib abrogates TNF-induced NF-kappa B activation through inhibition of activation of I kappa B alpha kinase and Akt in human non-small cell lung carcinoma: correlation with suppression of COX-2 synthesis. J Immunol 2004;173:2011-22.
47. Koki AT, Khan NK, Woerner BM, et al. Characterization of cyclooxygenase-2 (COX-2) during tumorigenesis in human epithelial cancers: evidence for potential clinical utility of COX-2 inhibitors in epithelial cancers. Prostaglandins Leukot Essent Fatty Acids 2002;66:13-8.
48. Soslow RA, Dannenberg AJ, Rush D, et al. COX-2 is expressed in human pulmonary, colonic, and mammary tumors. Cancer 2000;89:2637-45.
49. Pai R, Soreghan B, Szabo IL, Pavelka M, Baatar D, Tarnawski AS. Prostaglandin E2 transactivates EGF receptor: a novel mechanism for promoting colon cancer growth and gastrointestinal hypertrophy. Nat Med 2002;8:289-93.
50. McKay MM, Morrison DK. Integrating signals from RTKs to ERK/MAPK. Oncogene 2007;26:3113-21.
51. Richards JA, Petrel TA, Brueggemeier RW. Signaling pathways regulating aromatase and cyclooxygenases in normal and malignant breast cells. J Steroid Biochem Mol Biol 2002;80:203-12.
52. Coffey RJ, Hawkey CJ, Damstrup L, et al. Epidermal growth factor receptor activation induces nuclear targeting of cyclooxygenase-2, basolateral release of prostaglandins, and mitogenesis in polarizing colon cancer cells. Proc Natl Acad Sci U S A 1997;94:657-62.
53. Prossnitz ER, Ye RD. The N-formyl peptide receptor: a model for the study of chemoattractant receptor structure and function. Pharmacol Ther 1997;74:73-102.
54. Babbin BA, Lee WY, Parkos CA, et al. Annexin I regulates SKCO-15 cell invasion by signaling through formyl peptide receptors. J Biol Chem 2006;281:19588-99.
55. Rescher U, Danielczyk A, Markoff A, Gerke V. Functional activation of the formyl peptide receptor by a new endogenous ligand in human lung A549 cells. J Immunol 2002;169:1500-4.
56. Su SB, Gong W, Gao JL, et al. A seven-transmembrane, G protein-coupled receptor, FPRL1, mediates the chemotactic activity of serum amyloid A for human phagocytic cells. J Exp Med 1999;189:395-402.
57. Biswas DK, Martin KJ, McAlister C, et al. Apoptosis caused by chemotherapeutic inhibition of nuclear factor-kappaB activation. Cancer Res 2003;63:290-5.
58. El Kebir D, Jozsef L, Khreiss T, et al. Aspirin-triggered lipoxins override the apoptosis-delaying action of serum amyloid A in human neutrophils: a novel mechanism for resolution of inflammation. J Immunol 2007;179:616-22.
59. Lee HY, Kim MK, Park KS, et al. Serum amyloid A induces contrary immune responses via formyl peptide receptor-like 1 in human monocytes. Mol Pharmacol 2006;70:241-8.
60. Lee MS, Yoo SA, Cho CS, Suh PG, Kim WU, Ryu SH. Serum amyloid A binding to formyl peptide receptor-like 1 induces synovial hyperplasia and angiogenesis. J Immunol 2006;177:5585-94.
61. Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 1996;271:518-20.
62. van der Westhuyzen DR, Cai L, de Beer MC, de Beer FC. Serum amyloid A promotes cholesterol efflux mediated by scavenger receptor B-I. J Biol Chem 2005;280:35890-5.
63. Baranova IN, Vishnyakova TG, Bocharov AV, et al. Serum amyloid A binding to CLA-1 (CD36 and LIMPII analogous-1) mediates serum amyloid A protein-induced activation of ERK1/2 and p38 mitogen-activated protein kinases. J Biol Chem 2005;280:8031-40.
64. Hrzenjak A, Reicher H, Wintersperger A, et al. Inhibition of lung carcinoma cell growth by high density lipoprotein-associated alpha-tocopheryl-succinate. Cell Mol Life Sci 2004;61:1520-31.
65. Sparvero LJ, Asafu-Adjei D, Kang R, et al. RAGE (Receptor for Advanced Glycation Endproducts), RAGE ligands, and their role in cancer and inflammation. J Transl Med 2009;7:17.
66. Franklin WA. RAGE in lung tumors. Am J Respir Crit Care Med 2007;175:106-7.
67. Cai H, Song C, Endoh I, et al. Serum amyloid A induces monocyte tissue factor. J Immunol 2007;178:1852-60.
68. Wang L, Liu Q, Sun Q, Zhang C, Chen T, Cao X. TLR4 signaling in cancer cells promotes chemoattraction of immature dendritic cells via autocrine CCL20. Biochem Biophys Res Commun 2008;366:852-6.
69. Fukata M, Chen A, Vamadevan AS, et al. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology 2007;133:1869-81.
70. He W, Liu Q, Wang L, Chen W, Li N, Cao X. TLR4 signaling promotes immune escape of human lung cancer cells by inducing immunosuppressive cytokines and apoptosis resistance. Mol Immunol 2007;44:2850-9.
71. Sandri S, Rodriguez D, Gomes E, Monteiro HP, Russo M, Campa A. Is serum amyloid A an endogenous TLR4 agonist? J Leukoc Biol 2008;83:1174-80.
72. Cheng N, He R, Tian J, Ye PP, Ye RD. Cutting edge: TLR2 is a functional receptor for acute-phase serum amyloid A. J Immunol 2008;181:22-6.
73. He RL, Zhou J, Hanson CZ, Chen J, Cheng N, Ye RD. Serum amyloid A induces G-CSF expression and neutrophilia via Toll-like receptor 2. Blood 2009;113:429-37.
74. Westra WH. Early glandular neoplasia of the lung. Respir Res 2000;1:163-9.
75. Tang X, Shigematsu H, Bekele BN, et al. EGFR tyrosine kinase domain mutations are detected in histologically normal respiratory epithelium in lung cancer patients. Cancer Res 2005;65:7568-72.
76. Medema RH, Bos JL. The role of p21ras in receptor tyrosine kinase signaling. Crit Rev Oncog 1993;4:615-61.
77. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100:57-70.
78. Shao R, Karunagaran D, Zhou BP, et al. Inhibition of nuclear factor-kappaB activity is involved in E1A-mediated sensitization of radiation-induced apoptosis. J Biol Chem 1997;272:32739-42.
79. Yamagishi N, Miyakoshi J, Takebe H. Enhanced radiosensitivity by inhibition of nuclear factor kappa B activation in human malignant glioma cells. Int J Radiat Biol 1997;72:157-62.
80. Luo JL, Kamata H, Karin M. The anti-death machinery in IKK/NF-kappaB signaling. J Clin Immunol 2005;25:541-50.
81. Brach MA, Hass R, Sherman ML, Gunji H, Weichselbaum R, Kufe D. Ionizing radiation induces expression and binding activity of the nuclear factor kappa B. J Clin Invest 1991;88:691-5.
82. Jones DR, Broad RM, Madrid LV, Baldwin AS, Jr., Mayo MW. Inhibition of NF-kappaB sensitizes non-small cell lung cancer cells to chemotherapy-induced apoptosis. Ann Thorac Surg 2000;70:930-6; discussion 6-7.
83. Gazdar AF. Personalized Medicine and Inhibition of EGFR Signaling in Lung Cancer. N Engl J Med 2009.
84. Lynch TJ, Jr., Blumenschein GR, Jr., Engelman JA, et al. Summary statement novel agents in the treatment of lung cancer: Fifth Cambridge Conference assessing opportunities for combination therapy. J Thorac Oncol 2008;3:S107-12.
85. Jones HE, Goddard L, Gee JM, et al. Insulin-like growth factor-I receptor signalling and acquired resistance to gefitinib (ZD1839; Iressa) in human breast and prostate cancer cells. Endocr Relat Cancer 2004;11:793-814.
86. Engelman JA, Janne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res 2008;14:2895-9.
87. Huang M, Stolina M, Sharma S, et al. Non-small cell lung cancer cyclooxygenase-2-dependent regulation of cytokine balance in lymphocytes and macrophages: up-regulation of interleukin 10 and down-regulation of interleukin 12 production. Cancer Res 1998;58:1208-16.
88. Hida T, Yatabe Y, Achiwa H, et al. Increased expression of cyclooxygenase 2 occurs frequently in human lung cancers, specifically in adenocarcinomas. Cancer Res 1998;58:3761-4.
89. Krysan K, Reckamp KL, Dalwadi H, et al. Prostaglandin E2 activates mitogen-activated protein kinase/Erk pathway signaling and cell proliferation in non-small cell lung cancer cells in an epidermal growth factor receptor-independent manner. Cancer Res 2005;65:6275-81.
90. Krysan K, Reckamp KL, Sharma S, Dubinett SM. The potential and rationale for COX-2 inhibitors in lung cancer. Anticancer Agents Med Chem 2006;6:209-20.
91. Reckamp KL, Gardner BK, Figlin RA, et al. Tumor response to combination celecoxib and erlotinib therapy in non-small cell lung cancer is associated with a low baseline matrix metalloproteinase-9 and a decline in serum-soluble E-cadherin. J Thorac Oncol 2008;3:117-24.
92. Gadgeel SM, Ruckdeschel JC, Heath EI, Heilbrun LK, Venkatramanamoorthy R, Wozniak A. Phase II study of gefitinib, an epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI), and celecoxib, a cyclooxygenase-2 (COX-2) inhibitor, in patients with platinum refractory non-small cell lung cancer (NSCLC). J Thorac Oncol 2007;2:299-305.
93. O’Byrne KJ, Danson S, Dunlop D, et al. Combination therapy with gefitinib and rofecoxib in patients with platinum-pretreated relapsed non small-cell lung cancer. J Clin Oncol 2007;25:3266-73.
【技術分野】
【0001】
(関連出願の相互参照)
本願は米国仮出願第61/338,938号(2010年2月24日)の35 U.S.C.119(e)下の利益を主張し、その内容を引用により本明細書中に取り込む。
【0002】
本発明は、あるタイプまたはクラスの薬剤、および/またはそれらの組み合わせが癌患者に有効である可能性が高いかどうかを予測する方法およびシステムに関連する。該方法およびシステムは、患者の血液由来サンプルから得た質量分析データの使用およびその質量分析データに基づき作動する分類子として設定されたコンピューターに関連する。
【背景技術】
【0003】
本発明の出願人であるBiodesix, Incは、上皮成長因子受容体(EGFR)経路を標的とした薬剤の治療が非小細胞性肺癌(NSCLC)患者に有効である可能性が高いかどうかを予測するVeriStratとして知られる検査を開発した。該検査は米国公開番号第7,736,905号に開示され、その内容を引用により本明細書中に取り込む。該検査はまた、Taguchi F. et al.1においても記載され、その内容を引用により本明細書に取り込む。該検査の出願は米国公開番号第7,858,390;7,858,389および7,867,775号においても開示されており、それらの内容を引用により本明細書中に取り込む。
【0004】
簡単に述べると、VeriStrat検査は癌患者の血清および/または血漿サンプルに基づくものである。MALDI-TOF質量分析およびコンピューターで実行されるデータ解析アルゴリズムの組み合わせにより、所定のm/z範囲における8個の積分ピーク強度のセットが訓練コホートのものと比較され、患者サンプルのクラス標識が作製される:VeriStrat「良」、VeriStrat「不良」、またはVeriStrat「未定義」。複数の臨床評価試験において、前処理血清/血漿がVeriStrat「良」であった患者は、上皮成長因子受容体阻害剤で治療された際に、VeriStrat「不良」であった患者と比べて有意に良好なアウトカムを有していた。少数のケース(2%未満)では決定することが出来ず、VeriStrat「未定義」と標識された。VeriStratは本発明の出願人であるBiodesix, Inc.から市販されており、非小細胞性肺癌患者の治療セレクションに用いられる。
【0005】
現代の殆どのバイオマーカーに基づく検査は、腫瘍のタイプおよび組織学、特定の治療介入、ならびに臨床病理学的因子に関して非常に特異的である。例えば、腫瘍組織を用いた遺伝子検査、例えば、EGFRドメインにおける突然変異、KRAS変異の検査、および蛍光In-Situハイブリダイセーション(FISH)を用いた遺伝子コピー数検査は、非常に特殊な適応においてのみ有効である。EGFR変異から腺癌を伴う第一線NSCLC癌におけるゲフィチニブ応答に関する示唆は得られるが、扁平上皮癌ではこのタイプのNSCLCにおけるこれらの変異は非常に稀であるため、同様の有用性を示さない。KRAS変異は結腸直腸癌におけるセツキシマブに対する応答と相関し得るが、これをNSCLCに転用しようとする試みは失敗であった。頭頚部扁平上皮癌(SCCHN)におけるEGFR阻害剤(EGFRI)の有効性に関する既知のマーカーは存在しない。遺伝子検査におけるこれらの制限は、腫瘍新生の複雑なメカニズムの小さなパートにしか過ぎない非常に特殊な変異にそれらが焦点を絞っていることと関係するのかもしれない。また、これらの検査の全ては還元主義的な視点に基づくものであり、即ち、腫瘍生物学を腫瘍細胞のみに還元し、血管支持システムの内皮細胞、細胞外マトリックスおよび免疫系の成分(癌と関連する慢性的な炎症メカニズムに関与する炎症性細胞ならびにケモカインおよびサイトカインなど)からなる腫瘍微小環境と腫瘍細胞との相互作用を無視したものである。
【発明の概要】
【0006】
本明細書中において、腫瘍細胞におけるどの経路がVeriStrat「不良」である上皮腫瘍の独特の性質に関わるかについての我々の理解およびその根拠を提供する。本明細書で提示される理解についての証拠は、いくつかのソース、例えば、臨床的証拠、現象論的証拠、文献的解析、および癌患者の血清サンプルの質量スペクトル分析に基づく分子学的証拠によるものである。本明細書で開示される認識の結果は、以下に記載されるある種の薬剤またはその組み合わせが癌患者に有効である可能性が高いか低いかを予測するための新たな方法(即ち、臨床検査)の形態を取り得る。
【0007】
簡単に述べると、VeriStrat「不良」と識別された患者では、VeriStrat検査はEGFRなどの成長因子および生存因子の受容体の1つまたはそれ以上の下流の経路の活性化状態を測定するが、適当な候補となる経路は、例えば、古典的および非古典的MAPK(マイトジェン活性化プロテインキナーゼ)、AktならびにPKC(プロテインキナーゼC)で制御される反応(図2参照)である。化学療法およびプラセボ対照群のアウトカムにおける多様性により、これらの経路のこれら自身による活性化が予後不良に繋がり得ることが示唆され、NF−κB(活性化B細胞の核内因子κ−軽鎖エンハンサー)(細胞応答を制御し、炎症および免疫応答、ならびに細胞増殖および生存の制御に重要な役割を果たす重要な転写因子である)の関与が指摘される。それが化学療法に対する応答に関与することも知られている。
【0008】
一般的な事柄として、VeriStrat検査は予後不良の群のサブセットを識別し、AktまたはERK/JNK/p38またはPKCでの、またはそれらの上流でのMAPK経路またはPKC経路に関わる受容体のアゴニスト、受容体もしくはタンパク質を標的にする治療薬または治療薬の組み合わせによる治療の固形上皮腫瘍癌患者における異なる利益を予測する。EGFR阻害剤はかかる薬剤の例である。抗EGFR薬が有効である可能性が高いと予測される患者はVeriStrat「良」標識で識別される;逆に抗EGFR薬が有効である可能性が低いと予測される患者はVeriStrat「不良」標識で識別される。用語MAPK(マイトジェン活性化プロテインキナーゼ)は、単一の酵素ではなく少なくとも3個の関連するカスケードの名称として本明細書で用いられる(図2参照)。
【0009】
上記の結果として、VeriStrat「不良」標識と関連する患者では、VeriStrat検査は「不良」の患者を予後不良の癌患者のサブグループとして診断する。実際、VeriStrat「不良」患者はVeriStrat「良」患者とは異なる疾患状態を有すると見做される。
【0010】
さらに、VeriStrat「良」標識を有する癌患者では、MAPK経路が関与する受容体のアゴニスト、受容体またはタンパク質を標的とした薬剤またはその組み合わせによる治療がより有効である可能性がより高く;VeriStrat「不良」標識を有する患者ではかかる治療薬による治療から臨床利益を得る可能性は低い;一方で、VeriStrat「不良」では、これらの受容体、これらの経路の活性化には依存しない下流を阻害する療法または療法の組み合わせが有効である可能性が高い。
【0011】
この認識の臨床への適用は、付属の請求項に反映されるように、いくつかの形態を取り得る。該方法は癌患者の血液由来サンプルの質量分析データを得ること、および分類子として機能するプログラムされたコンピューターを用いたスペクトルの解析に関連する。一形態において、固形上皮腫瘍癌患者を、AktまたはERK/JNK/p38またはPKCでの、またはそれらの上流でのMAPK経路またはPKC(プロテインキナーゼC)経路に関わる受容体のアゴニスト、受容体もしくはタンパク質を標的にする治療薬または治療薬の組み合わせによる治療が有効である可能性が高い患者として、あるいは治療薬または治療薬の組み合わせによる治療が有効である可能性が低い患者として識別する方法であって、a)固形上皮腫瘍癌患者の血液由来サンプルから質量スペクトルを得;b)a)で得た質量スペクトルにおいて1つまたはそれ以上の所定の前処理工程を行い;c)b)の質量スペクトルにおける前処理工程を行った後、1つまたはそれ以上の所定のm/z範囲において該スペクトルにおける選択された特徴の積分強度値を得;d)患者を治療薬または治療薬の組み合わせが有効である可能性が高い、または有効である可能性が低い患者として識別するために、別の固形腫瘍患者の血液由来サンプルから得たクラス標識スペクトルを含む訓練セットを用いた分類アルゴリズムにc)で得た値を用いることを特徴とする方法が開示される。
【0012】
別の一実施態様において、COX阻害剤およびEGFR阻害剤の組み合わせの投与が癌患者に有効である可能性が高いかどうかを予測する方法であって、
a)該癌患者の血液由来サンプルから質量スペクトルを得;
b)工程a)で得られた質量スペクトルにおいて1つまたはそれ以上の所定の前処理工程を行い;
c)工程b)の質量スペクトルにおける前処理工程を行った後、1つまたはそれ以上のm/z範囲において該スペクトルの選択された特徴の積分値を得;
d)該患者をCOX2阻害剤およびEGFR阻害剤の組み合わせの投与による治療が有効である可能性が高いまたは低い患者として識別するために、別の固形上皮腫瘍患者の血液由来サンプルから得たクラス標識スペクトルを含む訓練セットを用いた分類アルゴリズムに工程c)で得た値を用いる
ことを特徴とする方法が開示される。
【図面の簡単な説明】
【0013】
【図1】は患者の血液由来サンプルでVeriStrat検査を行う工程を示すフローチャートである。
【0014】
【図2】はヒトの細胞における選択されたシグナル伝達経路を示すチャートである。
【0015】
【図3】は血清アミロイドA(SAA)アイソフォームの選択された生物活性ならびに癌の進行および治療に対する耐性におけるその役割を表す図である。
【0016】
【図4】はEGFRシグナル伝達経路、それらの相互作用および推定されるSAAによる活性化のポイントを表す図である。
【0017】
【図5】はErbBファミリーの成長因子受容体、例えば、EGFRおよびそれらの阻害剤の説明であり、Yarden Y, Shilo BZ. SnapShot: EGFR signaling pathway. Cell 2007; 131:1018の引用である。
【0018】
【図6】は全ての公表されたVeriStrat分析における治療群によるVeriStrat「良」およびVeriStrat「不良」患者間のハザード比を示すフォレストプロットである。
【0019】
【図7】は異なる化学療法を受けた患者の全生存率(OS)とこれらの患者のVeriStrat標識(「良」および「不良」)のカプラン−マイヤープロットを表したものである。
【0020】
【図8】は異なる濃度のゲフィチニブの存在下におけるゲフィチニブ感受性細胞株HCC4006とゲフィチニブ耐性細胞株A549のVeriStrat「不良」およびVeriStrat「良」血清中での増殖のプロットである。
【0021】
(発明の詳細な説明)
定義
本明細書で用いられるように、単数形「a」、「an」、および「the」は、文脈から特に明確に指定されない限り、複数に関する言及を包含する。
【0022】
本明細書で用いられるように。用語「固形上皮腫瘍」は、例えば、限定される必要はないが、NSCLC、SCCHN、乳癌、腎臓癌、膵臓癌、黒色腫および直腸結腸癌(CRC)である。
【0023】
本明細書で用いられるように、「AktまたはERK/JNK/p38またはPKCでの、またはそれらの上流でのMAPK経路またはPKC経路に関わる受容体のアゴニスト、受容体もしくはタンパク質を標的にする治療薬または治療薬の組み合わせ」は、例えば、限定されないが、erbB受容体受容体ファミリー(例えば、EGFR(HER1)、HER2、HER3、およびHER4)、VEGF受容体(VEGFR2)、肝細胞増殖因子受容体(HGFRまたはMET)、Gタンパク質共役型受容体、インスリン様増殖因子(IGF)受容体、VEGF、成長因子(例えば、TGFαおよびEGF)、およびAktまたはERK/JNK/p38 MAPKまたはPKC経路の、またはそれらの上流の任意の別のタンパク質を標的とした治療薬または薬剤である。さらに、本明細書で用いられるように、用語「AktまたはERK/JNK/p38またはPKCでの、またはそれらの上流でのMAPK経路またはPKC(プロテインキナーゼC)経路に関わる受容体のアゴニスト、受容体もしくはタンパク質を標的にする治療薬または治療薬の組み合わせ」は、既知の治療薬、ならびにこれらのタンパク質を標的とした未だ発見または開示されていない治療薬を包含する。さらに、治療薬の組み合わせは、それらが既に固形上皮腫瘍の治療に用いられているか否かにかかわらず、治療薬の任意の組み合わせを包含する。ある薬剤が特定のタンパク質または経路の阻害剤として同定されたとしても、かかる分類はその作用メカニズムの記載を表すと意図されるものではなく、それはこれらの薬剤の多くの作用メカニズムは完全には理解されていないからであることは記しておくべきであろう。例えば、排他的なリストとして意図されるものではないが、これらの治療薬は以下を含む:
(1)TKI(チロシンキナーゼ阻害剤):小分子チロシンキナーゼ阻害剤(TKI)に分類される多くの薬剤が現在市場に出回っており、I−III相の治験が行われている。TKIは特定の分子の受容体、例えば、上皮増殖因子受容体(EGFR)を標的とすることができ、複数の受容体を標的とすることもできる(「マルチプルキナーゼ阻害剤」と呼ばれる)。これらは、例えば、限定されないが、エルロチニブ、ゲフィチニブ、ソラフェニブ、スニチニブ、パゾパニブ、イマチニブ、ニロチニブ、ラパチニブである。
抗体に基づく阻害剤は、例えば、セツキシマブ(抗EGFR)、パニツムマブ(抗EGFR)、トラスツズマブ(抗−Her2)である。
(2)HGFRまたはMET阻害剤:現在、I−II相の治験中にある長いリストのMETまたはPI3K(METの下流のシグナル伝達酵素である)を阻害する薬剤が存在し、様々な段階において調査されているが、現時点では臨床的には用いられていない。例えば、XL880はMETおよびVEGFR2の強力な阻害剤である。本明細書で用いられるように、用語「MET阻害剤」は、例えば、限定されないが、AMG 208、AMG 102、ARQ 197、AV−299、MetMab、GSK 1363089(XL880)、EMD 1214063、EMD 1204831、MGCD265、クリゾタニブ(PF−02341066)、PF−04217903、MP470である。
(3)COX2阻害剤:本明細書で用いられるように、用語「COX2阻害剤」は、例えば、限定されないが、選択的COX2阻害剤:セレコキシブ、ロフェコキシブ、バルデコキシブ、ルミラコキシブである。
(4)COX1およびCOX2の両方を阻害する別の非ステロイド性抗炎症剤(NSAID)は、イブプロフェン、アスピリン、インドメタシンおよびスリンダクである。かかる薬剤はNF−κB活性化も抑制することが示されている。
(5)別のNF−κB阻害剤:本明細書中で用いられるように、用語「NF−κB阻害剤」は、例えば、限定されないが、三酸化ヒ素(ATO)、サリドマイドおよびそのアナログ、レスベラトロールである。さらに、COX2阻害剤はNF−κB経路にも阻害効果を有する。故に、イブプロフェン、アスピリン、インドメタシンおよびスリンダクといったNSAIDはNF−κBの活性化を抑制することが示されたため、NF−κB阻害剤と見做される。
【0024】
本明細書で用いられるように、用語「VEGF阻害剤」は、例えば、限定されないが、ベバシズマブ、セジラニブ、アキシチニブ、モテサニブ、BIBF 1120、ラムシルマブ、VEGF Trap、リニファニブ(ABT869)、チボザニブ、BMS−690514、XL880、スニチニブ、ソラフェニブ、ブリバニブ、XL−184、パゾパニブである。
【0025】
本明細書で用いられるように、「標的とした治療」は、特定の酵素のモノクローナル抗体または小分子阻害剤といった薬剤または他の物質を受容体といった特定の分子を同定し攻撃するために用いるようなタイプの治療を意味する。これらの例は、EGFR−TKI(エルロチニブ、ゲフィチニブ)、セツキシマブ、ベバシズマブなどである。
【0026】
本明細書で用いられるように、用語「非標的化学療法」または「化学療法」は、DNAに干渉(アルキル化試薬、例えば、シスプラチン、カルボプラチン、オキサリプラチンなど、または代謝拮抗薬、例えば、5−フルオロウラシルまたはペメトレキセド、またはイリノテカンなどのトポイソメラーゼ阻害剤)、または細胞分裂に干渉(ビノレルビン、ドセタキセル、パクリタキセルなど)することにより、急速に分裂する細胞に干渉する治療を意味する。
【0027】
本明細書で用いられるように、「予後」は、治療を受けない場合または標準的な治療を適用された場合の臨床的アウトカムに関連する因子または測定結果を意味する。それは、疾患の自然な経過の測定結果と考えることが出来る。
【0028】
用語「予測の」は、特定の治療方法が有効であるか有効ではないかに関連する因子または測定結果を意味する。予測因子により、予測マーカーの状態に依存して治療方法から異なる利益が得られることが示唆される2。本明細書で用いられるように、用語「疾患状態」は、異なる予後および/または異なる治療応答および/または特定の分子および/または代謝的特徴により特徴付けすることが出来る診断された病状の特定のサブタイプを意味する。
【0029】
ディスカッション
VeriStrat検査は血清サンプルの質量分析データから得た特徴に基づくため、現行の殆どのバイオマーカーを用いた検査とは異なり、癌に関連する一般的な因子を測定することが出来ることを我々は発見した。この事実により、以下に議論されるVeriStrat検査を用いた治療のためのセレクションへの新たな臨床的適用が可能となる。特に、VeriStrat検査により、EGFR阻害のメカニズムにかかわらず、VeriStrat「良」およびVeriStrat「不良」と識別された患者間で同様に生存曲線を区分することが出来る。我々の先の研究において、酵素のATP結合部位をブロックすることにより受容体を阻害する小分子EGFRチロシンキナーゼ阻害剤ゲフィチニブ(Iressa)およびエルロチニブ(Tarceva)1で治療した患者のサンプルセットにVeriStrat検査を行った。NSCLCおよび直腸結腸癌(CRC)の両方において、EGFRを標的とした他の治療薬セツキシマブ(Erbitux)に関してもVeriStrat「良」およびVeriStrat「不良」と識別された患者間で同様の区分が見られた3。セツキシマブはEGF受容体を直接ブロックする抗体である。
【0030】
さらに、VeriStrat検査により、VeriStrat「良」およびVeriStrat「不良」と識別された患者間で、臨床病理学的特徴を超えて同様に区分することが出来る。例えば、VeriStrat検査は、腫瘍が腺癌である患者でも腫瘍が扁平上皮癌である患者でも用いることができる。
【0031】
また、VeriStrat検査は、様々の固形腫瘍において、VeriStrat「良」およびVeriStrat「不良」と識別された患者間の区分を示す。NSCLC、頭頚部扁平細胞癌(SCCHN)、およびCRCにおいてこれが見られた3。
【0032】
さらに、非標的化学療法で治療された患者のVeriStrat検査による生存曲線の区分は、人種、介入治療のタイプ、および腫瘍タイプに依存して異なることが見出された。いくつかの非標的化学療法治療群において区分の証拠があるが、他では区分が存在しない。プラセボ群(即ち、介入治療を受けていない群)においても明確な区分が存在し、VeriStrat検査が予測的な要素を有することが示唆される。
【0033】
図6のフォレストプロットは、現在までに刊行または公表された全てのVeriStrat検査の分析を要約する。分析された各治療群のVeriStrat「良」およびVeriStrat「不良」患者間の全生存率のハザード比(HR)が示される。データは治療タイプに応じて分類されると考えられる。得られたハザード比の範囲によりVeriStratが実際に特定の治療タイプの結果としての良好または不良なアウトカムを示唆し、故に予測能を有することが示される。
【0034】
図6において、治療は、B=ベバシズマブ、C=セツキシマブ、CT=化学療法、E=エルロチニブ、G=ゲフィチニブである。刊行物/公表物は、[1] D. Carbone, 2nd European Lung Cancer Conference, April 2010、 [2] F. Taguchi et al., J Natl Cancer Inst. 2007 Jun6;99(11):838-8461 からアップデートしたBiodesixのファイル, [3] C. Chung et al., Cancer Epidemiol Biomarkers Prev. 2010 Feb;19(2):358-653、 [4] D. Carbone et al., Lung Cancer 2010 Sept; 69(3):337-3404である。
【0035】
非標的化学療法治療群の再分析により、タキセン治療群では明確な区分が見られなかったが、タキセンを含まない化学療法レジメン群ではVeriStrat「良」およびVeriStrat「不良」群間で区分が存在した(図7参照)。
【0036】
検査がこのような広い適用範囲を有することは稀である。
【0037】
まとめると、前記のディスカッションならびに図6および7から以下の結論が導かれる:
1.VeriStrat検査により、EGFR阻害剤(EGFRI)単独療法において、VeriStrat良および不良の患者間で約.45のハザード比における区分が示され、それは、
−EGFRIの作用メカニズム、例えば、小分子TKI(エルロチニブ、ゲフィチニブ)および抗体(受容体)阻害剤に基づくEGFRI(例えば、セツキシマブ)であるかに依存せず、
−組織学的タイプ、例えば、腺癌、および扁平細胞癌であるかに依存せず、
−組織、例えば、NSCLC、SCCHN、およびCRCであるかに依存しない。
2.他の集団的特徴の有意な相関は観察されなかった:
−遺伝子マーカー、例えば、EGFRの変異状態またはKRASの状態とは相関せず、
−性別および人種といった集団的特徴とは相関しない。
3.VeriStratは、治療を受けていないVeriStrat不良およびVeristrat良のサブグループ間の区分で示される強い予測的要素を有する。
−しかしながら、VeriStrat「不良」サブグループにおけるEGFRI単独療法による明確な治療利益は存在しない、即ち、VeriStrat不良サブグループにおけるエルロチニブによる治療はプラセボによる治療と実質的に同効果である一方、VeriStrat「良」サブグループにおいては、EGFRIによる大きな治療利益が存在し、
−組み合わせ療法の効果は特定の薬剤の組み合わせおよび相互作用する経路に対するそれらの効果に依存する。
【0038】
サンプルの質量スペクトルにおける特定のピークがVeriStrat「不良」群においてのみ観察された結果とこれらの事実を全て合わせると、VeriStratは固形上皮腫瘍において臨床的意義(不良アウトカム)を有する新たな疾患状態を定義するという結論が導かれる。観察された現象により、VeriStrat「不良」の腫瘍の分子学的状態に関するいくつかの仮説を立てることができる:EGFRIがこの種の患者に有効ではないため、ならびに、効果がTKIおよび抗体療法と同程度であるため、VeriStrat「不良」患者において、該受容体および該チロシンキナーゼドメインの下流の経路がVeriStrat「良」患者と異なっている、即ちアップレギュレートされている可能性が高い。KRASの変異状態との相関は見られなかったため、侵されている経路はRASの下流であることがさらに結論される。
【0039】
前記の観察、文献的解析および他の一連の根拠に基づき、VeriStrat「不良」腫瘍細胞の異なる特徴に腫瘍細胞のどの経路が関与しているかに関する我々の理解が本明細書で提供される。簡単に述べると、VeriStrat「不良」と識別された患者においてVeriStrat試験はEGFの受容体の下流の1つまたはそれ以上の経路の活性化を評価していることを我々は提案する;候補の可能性がある経路は、古典的および非古典的MAPK、PI3K/AktならびにPKCで制御される経路である(図2の200Aおよび200Bを参照)。化学療法およびプラセボ対照群のアウトカムの多様性により、これらの経路のこれら自身による活性化が予後不良に繋がり得ることが示唆され、NF−κB転写因子(細胞生存の重要な制御因子であり、炎症過程および癌の進行に重要な役割を果たし、化学療法への応答に関与する)の関与が示唆され得る。
【0040】
一般的な事柄として、VeriStrat検査は予後不良の群のサブセットを識別し、AktまたはERK/JNK/p38またはPKCでの、またはそれらの上流でのMAPK経路またはPKC(プロテインキナーゼC)経路に関わる受容体のアゴニスト、受容体もしくはタンパク質を標的にする治療薬または治療薬の組み合わせによる治療から固形上皮腫瘍癌患者が受ける恩恵を予測する。抗EGFR薬が有効である可能性が高いと予測される患者はVeriStrat「良」標識で識別される;逆に抗EGFR薬が有効である可能性が低いと予測される患者はVeriStrat「不良」標識で識別される。VeriStrat「不良」標識を有する患者がMAPK経路を活性化する受容体を標的とした治療薬による治療から臨床利益を受ける可能性は低く;一方で、VeriStrat「不良」患者では、該受容体と独立したこれらの経路の活性化を阻害する療法または療法の組み合わせから臨床利益が得られる可能性が高い。
【0041】
用語MAPK(マイトジェン活性化プロテインキナーゼ)は、単一の酵素ではなく少なくとも3個の関連するカスケードの名称として本明細書で用いられる(図2参照)。
【0042】
前記の主張の結果として、VeriStrat「不良」標識と関連する患者では、VeriStrat検査は「不良」患者を予後不良の癌患者のサブグループとして予測する。
【0043】
認識の結論は、いくつかのタイプの薬剤が癌患者に有効である可能性が高いか低いかを予測するための新しい方法、即ち臨床検査の形態を取り得る。
【0044】
臨床適用の1つにおいて、本発明は、固形上皮腫瘍癌患者を、AktまたはERK/JNK/p38またはPKCでの、またはそれらの上流でのMAPK経路またはPKC経路に関わる受容体のアゴニスト、受容体もしくはタンパク質を標的にする治療薬または治療薬による治療が有効である可能性が高いか、該治療薬または治療薬の組み合わせによる治療が有効である可能性が低いか識別する方法であって、
a)固形上皮腫瘍癌患者の血液由来サンプルから質量スペクトルを得;
b)a)で得た質量スペクトルにおいて1つまたはそれ以上の所定の前処理工程を行い(例えば、バックグランドの除去、ノイズの推定、ノーマライゼーションおよびスペクトルのアラインメント);
c)b)の質量スペクトルにおける前処理工程を行った後、1つまたはそれ以上の所定のm/z範囲(および好ましくは下の表1で説明されるm/zピークに対応する下記のm/z範囲)において該スペクトルにおける選択された特徴の積分強度値を得;
d)患者を治療薬または治療薬の組み合わせが有効である可能性が高い、または有効である可能性が低い患者として識別するために、別の固形腫瘍患者の血液由来サンプルから得たクラス標識スペクトルを含む訓練セットを用いた分類アルゴリズム(例えば、k近傍法)にc)で得た値を用いる
ことを特徴とする方法であると見做すことが出来る。
【0045】
VeriStrat「不良」患者の標的治療に対する耐性の打開の具体例として、COX2阻害剤、例えば、セレコキシブまたはロフェコキシブを治療レジメンとしてEGFRIに追加することにより、EGFRIに対してVeriStrat「不良」標識を有する患者の耐性を打開することが出来る可能性がある。故に、VeriStrat検査はCOX2阻害剤およびEGFRIを含む組み合わせ療法の処方のインジケーターとして用いられ得る。
【0046】
別の一具体例として、VeriStrat「不良」標識はNF−κBの特異的な活性化と関連すると考えられ、故に、該検査はNF−κB阻害剤から最も恩恵を受ける患者を選択し、それ故に不必要な治療およびそれに関連する死亡率を低減させるために用いることができる。
【0047】
別の一具体例として、VeriStrat「不良」標識は特定の非標的化学療法、特にDNA複製および遺伝子発現に干渉する薬剤(シスプラチン、ゲムシタビンまたはペメトレキセド)の治療利益が小さいこと(おそらくは、この過程におけるNF−κB因子の関与による)に関連すると考えられる。
【0048】
VeriStrat「不良」と標識された患者では、(1)受容体と独立してMAPK経路の下流の活性化を阻害する薬剤、例えば、COX2阻害剤など、または(2)炎症性のホスト応答を最小化する薬剤の添加、あるいは、クロストーク経路の活性化を阻害する別の標的薬剤の添加により、標的薬剤に対する耐性を打開することが出来る。
【0049】
VeriStrat検査
いくつかの治療薬または治療薬の組み合わせ、例えば、AktまたはERK/JNK/p38またはPKCでの、またはそれらの上流でのMAPK経路またはPKC経路に関わる受容体のアゴニスト、受容体もしくはタンパク質を標的にする治療薬による治療用にかかる患者を選択するために固形上皮腫瘍癌患者の血液由来サンプルを検査する本開示による方法が、図1のフローチャートに工程100として説明される。
【0050】
工程102において、血清または血漿サンプルを患者から得る。一実施態様において、血清サンプルを3つのアリコートに分け、質量分析および後続の工程104、106(サブ工程108、110および112を含む)、114、116および118を書く折コートについて独立して行う。アリコートの数は、例えば4、5または10アリコートのように異なっていてもよく、各アリコートは後続の工程に用いられる。
【0051】
104において、該サンプル(アリコート)は質量分析で分析される。好ましい質量分析法は、マトリックス支援レーザー脱離イオン化(MALDI)飛行時間型(TOF)質量分析であるが、他の方法でもよい。質量分析により、当該分野で周知のように、多数の質量/電荷(m/z)値における強度値を表すデータ点が得られる。一実施例において、該サンプルを解凍し、4℃、1500rpmで5分間遠心する。さらに、該血清サンプルは1:10、または1:5にMilliQ水で希釈してもよい。希釈サンプルはMALDIプレートのランダムに割り当てられた位置にトリプリケート(即ち、3つの異なるMALDIターゲット上に)スポットしてもよい。0.75μlの希釈した血清をMALDIプレート上にスポットした後、0.75μlの35mg/ml シナピン酸/(50% アセトニトリルおよび0.1%トリフルオロ酢酸(TFA))を加え、5回ピペッティングして混合してもよい。プレートは室温で乾燥することが出来る。本発明の本質に従って血清を調製および加工するために別の技法および方法が用いられ得ることは明らかである。
【0052】
質量スペクトルは、自動または手動でスペクトルを収集するVoyager DE-PROまたはDE-STR MALDI TOF質量分析計を用いてリニアモードの陽イオンで獲得してもよい。各血清サンプルについて525または500個のスペクトルを得るために、75または100個のスペクトルが各MALDIスポットの7または5個の位置から収集される。スペクトルはタンパク質スタンダードの混合物(インスリン(ウシ)、チオレドキシン(E. coli)、およびアポミオグロビン(ウマ))を用いて外部校正される。
【0053】
工程106では、1つまたはそれ以上の所定の前処理工程を工程104で得たスペクトルについて行う。前処理工程106は、工程104で得た質量分析データ上で作動するソフトウェアの指示を用いた一般的な目的のコンピューターで実行される。前処理工程106は、バックグラウンドの除去(工程108)、ノーマライゼーション(工程110)およびアラインメント(工程112)を含む。バックグラウンドの除去工程は、好ましくはバックグラウンドのロバスト、非対称な推定を行うことに関連し、スペクトルからバックグラウンドが差し引かれる。工程108は米国特許番号第7,736,905 B2号および米国公開番号第2005/0267689号(引用により本明細書中に取り込む)に記載されるバックグラウンド除去法を用いる。ノーマライゼーション工程110はバックグラウンドを差し引いたスペクトルのノーマライゼーションに関連する。ノーマライゼーションは部分イオン電流ノーマライゼーションの形態でもよく、あるいは、米国特許番号第7,736,905に記載されるように、全イオン電流ノーマライゼーションでもよい。工程112では、米国特許番号第7,736,905に記載されるように、ノーマライズし、バックグラウンドを差し引いたスペクトルを所定の質量スケール(分類子を用いた訓練セットの調査により得られる)にアラインする。
【0054】
前処理工程106が行われると、工程100は、所定のm/z範囲におけるスペクトルの選択された特徴(ピーク)の値を得る工程114に進む。ピーク検出アルゴリズムのピーク幅設定を用い、ノーマライズおよびバックグラウンド除去を行った強度をこれらのm/z範囲において積分し、この積分値(即ち、特徴の幅の間の曲線下面積)を特徴に割り当てることができる。このm/z範囲においてピークが検出されなかったスペクトルについては、積分範囲は、その時点のm/z位置のピーク幅に対応する幅を有するこの特徴の平均m/z位置周囲の間隔として定義することが出来る。この工程は米国特許番号第7,736,905にさらに詳しく記載される。
【0055】
工程114では、米国特許番号第7,736,905に記載されるように、該スペクトルにおける特徴の積分値は1つまたはそれ以上の以下のm/z範囲から得られる:
5732から5795
5811から5875
6398から6469
11376から11515
11459から11599
11614から11756
11687から11831
11830から11976
12375から12529
23183から23525
23279から23622
65902から67502.
【0056】
好ましい実施態様において、値は表1に示される8個のこれらのm/z範囲から得られ、適宜、12個全ての範囲から得られてもよい。これらのピークの重要性および検出方法は米国特許番号第7,736,905号に記載される。
【0057】
工程116において、工程114で得た値を分類子に供給する(図示された実施態様ではk近傍法(KNN)分類子である)。該分類子は多数の他の患者(NSCLC癌患者でもよく、あるいは、別の固形上皮癌、例えば、HNSCC、乳癌の患者でもよい)からのクラス標識スペクトルの訓練セットを利用する。114の値および訓練セットへのKNN分類アルゴリズムの適用は米国特許番号第7,736,905号で説明される。別の分類子、例えば、確率的KNN分類子または他の分類子が用いられてもよい。
【0058】
工程118において、分類子によりスペクトルに「良」、「不良」または「未定義」が作製される。前述したように、工程104−118は、与えられたサンプルの3個の別々のアリコート(または用いられた任意のアリコート数)について並行して行われる。工程120において、全てのアリコートから同じクラス標識が得られたかどうかのチェックが行われる。もし同じでなかった場合、工程122で示されるように未定義の結果に変更される。全てのアリコートから同じ標識が得られた場合、工程124で示されるようにその標識が記録される。
【0059】
本開示に記載されるように、工程124で報告される新たかつ予想外のクラス標識が開示される。
【0060】
工程106、114、116および118が、典型的には、工程106の前処理工程、工程114でスペクトル値を得ること、工程116のK−NN分類アルゴリズムの適用、および工程118のクラス標識の作製をコードするソフトウェアを用いるプログラムされた一般的なコンピューターで行われることは明らかである。工程116で用いられるクラス標識スペクトルはコンピューターの記憶装置またはコンピューターがアクセス可能な記憶装置に記憶される。
【0061】
我々の先行特許出願公報米国特許番号第7,736,905号に記載されるように、該方法およびプログラムされたコンピューターは臨床検査センターで実行されてもよい。
【0062】
VeriStrat検査の作動メカニズムおよびその実際の結果に関する理解は、このセクションでさらに記載されるいくつかのソースに由来する。
【0063】
タンパク質IDからの直接的証拠
VeriStratは血清または血漿のMALDI−TOF MSのピーク強度を測定する。一実施態様において、VeriStratのシグナチャーは後述の表1に記載される8個の質量分析のピークを含む。分類は、所定のm/z範囲(前述のリストおよび表1を参照)においてサンプルの質量スペクトルを積分することにより強度、即ち特徴値を推定し、観察された8個の特徴値のセットを訓練サンプルのものと7近傍分類アルゴリズムを用いて関連付けすることにより行われる。この方法は非線形結合の特徴値を用いるものであり、一次元スコアの定義を行うことは出来ない。特徴値の線形結合のスコア関数を作製しようとする試みは常に失敗に終わり、常にパフォーマンスの低下を招いてきた。これら8個の特徴の全てまたは殆どが臨床的実用性を得るために有用であると考えられる。
【0064】
用いた特徴値のペプチド含量を決定することによりVeriStrat検査の作動メカニズムを理解することが出来るかもしれないと考えられていた。しかしながら、装置のm/z解像度はそのm/z範囲において1つのタンパク質またはペプチド鎖のみが存在することを保証するに足るほど高いものではないという事実により、これは困難となった。また、8個以上のペプチドが8個のピークシグナチャーを形成すると考えられ、そのいくつかは同じアミノ酸配列が翻訳後修飾を受けたものまたは酸化された形態であり、他のものは依然として未知のペプチドである可能性がある。加えて、特徴値、即ち推定されたピーク強度は、単にその被分析物がサンプル中に多く含まれることに対応するだけではない。これはMALDIのイオン化の複雑さによるものであり、即ち、検出器にヒットするイオン数は被分析物の量およびイオン化確率の両方に相関する。この半定量的な様式のピーク(特徴値)の比較により、タンパク質IDの標準的な方法による比較が困難なままとなる(LC−MS/MS)。
【表1】
【0065】
これらの困難な事柄にもかかわらず、我々は表1のピークの3つが血清アミロイドA(SAA)アイソフォームと関連している強い証拠を得た。我々はプールしたVeriStrat「良」およびVeriStrat「不良」サンプル間でディファレンス(DIGE)分析を行い、m/z11529および11685のピークを十分な配列包括度で分離することに成功し、それらをSAA 19−122およびSAA 20−122と同定した。理論的質量は実測されたm/z値とよく一致した。ゲル上で観測された0.4のPIシフトも理論的予測値とよく一致する。さらに、m/z5843におけるピークは11685のピークが2価に荷電したものと考えられる。これらのピークは、他でも観察されている5(Ducet, et al. Electrophoresis 1996, 17, 866-876、Kiernan et al. FEBS Letters 2003, 537, 166-170)。11445のピークが、親SAAタンパク質のC末端からの切断物の配列に関連するSAAアイソフォームである可能性もある。
【0066】
別のタンパク質またはタンパク質のアイソフォームがVeriStratシグナチャーに存在することは明らかであるが、SAAアイソフォームがVeriStrat検査の作動メカニズムにおいて重要な役割を果たすことも考えられる。以下のセクションでは、SAAがVeriStrat「不良」シグナチャーの少なくとも3つのピークの主要な要素であるという発見に基づくVeriStrat検査の作動メカニズムの推定される理論;SAAといくつかの受容体の相互作用およびこれらの相互作用の生物学的帰結に関する既知の情報、ならびに様々な癌細胞におけるSAAと機能的に結合するこれらの受容体の存在に関する情報が提供される。しかしながら、本発明は必ずしもこの理論に基づく必要はなく、かかる理論は本発明を限定する意図は持たない。
【0067】
癌のバイオマーカーとしてのSAAに関する先行技術文献は、引用文献6−16を参照すること。
【0068】
SAA:生物学的機能および腫瘍発症への関与
機能
SAAファミリーの決定的重要性は、SAAが進化を通して高度に保存された配列である17という事実、およびSAAの発現が感染、外傷または病的過程に応答して劇的に増加することにより示唆される。しかしながら、SAAの正確な生物学的機能は未だ完全には解明されていない。SAAはHDLの成分として脂質の運搬および代謝に関与し、おそらくは疾患の急性期において保護的な役割を果たす18が、慢性的な条件ではSAAは有害因子となる。SAAの発現が高い状態が続くと、関節リウマチなどのいくつかの疾患ではアミロイドAアミロイドーシスが誘発される19。しかしながら、SAAの臨床的に重要な機能の範囲はかなり広く、慢性的な炎症および腫瘍形成への関与も含まれる。後者の2つは密接に関係しており、Vlasova and Moshkovskii20 および Malle et al21のレビューで詳述される。
【0069】
SAAの腫瘍形成への関与は、その多面的な生物学的活性:炎症への関与、例えば、炎症促進性遺伝子の発現の活性化およびサイトカインの制御による慢性過程の補助、細胞外マトリックス分解への関与、抗アポトーシス性の性質、ならびに腫瘍形成に複雑に関与するマイトジェン活性化プロテインキナーゼ(MAPK)を含む特定の経路の活性化に起因する。
【0070】
SAAは細胞外マトリックス(ECM)の接着タンパクとして作用し得ること22、ECMの分解およびリモデリングに重要な役割を果たし、腫瘍新生、転移、および腫瘍浸潤に関与する24,25マトリックスメタロプロテアーゼ(MMP)を誘導すること18,23が示されている。
【0071】
SAAの免疫に関連する機能は、そのサイトカイン様活性により定義される。SAAはIL−8、TNF−αおよびIL−1βの産生を促進し26,27(これはおそらくは、SAA発現のポジティブフィードバックに繋がる)、同様に、細胞性免疫応答に重要な役割を果たすIL−12およびIl−23の産生も促進する28。SAAがPI3Kおよびp38 MAPKを活性化し得ることも示されている。
【0072】
SAAの炎症の制御への関与は、NF−κBおよびMAPK経路の活性化を同時に伴うCOX2発現誘導能と関連する29,30。癌と炎症の重要な相互関係は数多くの研究およびレビューの対象である31−37。近年の大量のデータから、SAAは2つの過程間のメディエーターの1つとして重要な役割を果たすことが示唆されているが、これは、SAAが重要な炎症性および腫瘍形成経路、例えば、古典的および非古典的MAPK経路、転写因子NF−κBのものを活性化することが出来ること、およびおそらくはそれらのクロストークに関わることによる。VeriStratシグナチャーと関連するSAAレベルの上昇は、該経路の活性化を測定に有用なものとして用いることができる。
【0073】
SAAの生物学的活性と関連する受容体および経路
NF−κB転写因子は多くの上皮および血液性の悪性病変で恒常的に活性化していることが知られており、抗アポトーシス性およびアポトーシス性の標的遺伝子、マトリックスメタロプロテアーゼの発現、血管新生および細胞周期を制御することにより41、炎症を伴う癌の進行に重要な役割を果たすと考えられている38,39,40。他方、NF−κBはアポトーシス促進性の遺伝子活性を発揮すること、および腫瘍抑制因子p53と協働してアポトーシスを誘導することも可能である42。実際の作用は刺激、細胞種、および関与するサブユニットに依存する43。Rel/NF−κB因子の抗アポトーシス性およびアポトーシス性作用は必ずしも対立するものではないが、同じ標的遺伝子のアップレギュレーションにより同じ細胞で起こり得る44。NF−κBはおそらくは炎症と癌をリンクする因子の主な1つであるが、これは、IL−6およびTNF−αといった炎症性サイトカイン、ならびにMMPおよびCOX−2を誘導するサイトカインの誘導に関与することによる35,45,46。NF−κBの活性化はEGFにより誘導されることがあり:EGFの刺激はNF−κB活性化を介したデスレセプター誘導性アポトーシスを阻害する。
【0074】
COX−2の過剰な発現は、広範囲の前悪性、悪性および転移性のヒト上皮癌47、例えば、肺癌48で見られる。COX2は、プロスタグランジンE2(PGE2)を介して細胞増殖、血管新生、アポトーシス、および細胞遊走を仲介し、マイトジェン活性化プロテインキナーゼ(MAPK)カスケードの腫瘍原性シグナリングをトランス活性化する49,50。COX2はErkを介してMAPKをトランス活性化する49,92。その関係は相反的なものであり:MAPK経路を介して作用する上皮増殖因子(EGF)はいくつかの上皮細胞においてCOX2活性を劇的に誘導する51。TGFαによるEGFRの活性化はCOX2を刺激し、PGE2放出の増加および有糸分裂誘発の増加を引き起こす52。
【0075】
マイトジェン活性化プロテインキナーゼ(MAPK)カスケードは、活性化した成長因子受容体からの成長刺激シグナルを伝達するため、通常のセルバイオロジーならびに癌の発症に重要な役割を果たす。MAPKシグナル伝達はしばしば、成長因子の1つがそのチロシンキナーゼ型受容体(RTK)である膜受容体に結合することにより開始され、Raf、MEKおよび細胞外シグナル制御キナーゼ(ERK)の作動を引き起こす。近年の研究により、RTKからERKへのシグナリングは単なる直線的なRas依存性経路よりもはるかに複雑なものであることが示され、ETKが仲介するERKシグナリングの強度、期間および細胞局在の決定に重要な役割を果たす様々なシグナリングモジュレーターが同定される必要がある50。SAAは様々な上皮細胞のいくつかの受容体と機能的に結合し、この結合により、上記および特定の治療に対するVeriStrat「不良」患者の耐性に繋がり得るNF−κBおよびMAPKの両方の下流の活性化が起こる(以下においても議論される)。これらの受容体のいくつかに関する概略を以下に示す。
【0076】
FPRL受容体
FPRL受容体は幹細胞53、腸上皮54および肺55を含む様々な細胞で発現される。SAAは古典的なGタンパク質共役型受容体の1つであるFPRL1と相互作用し、細胞機能および上皮増殖および/またはアポトーシスの制御に重要なシグナリングネットワークを誘起する。SAAのFPRL1への結合により、インターロイキンの活性化および誘導が引き起こされる。FPRLの関与によりプロテインキナーゼC(PKC)および転写因子NF−κB経路が活性化され30、これはアポトーシスの阻害および癌の進行を伴う56,57,41。SAAのFPRL1への結合により、好中球およびリウマチ滑膜細胞におけるアポトーシスのレスキューが起こることも示されているが、これは、MAPK、ERK1/2、PI3K/Aktシグナリングのリン酸化、ならびにSTAT3活性化および細胞内Ca2+の放出により仲介され58,59,60、それにより細胞の増殖および生存が促進される。
【0077】
SR−BI受容体
スカベンジャーレセプターB−I(SR−BI)は、選択的なコレステロール取り込みを仲介する高密度リポタンパク質受容体として同定された61。SR−BIはステロイド産生組織および肝臓で最も多く発現されるが、炎症中のマクロファージおよび単球においても発現が上昇する;SB−BIの発現の上昇は、SAAの存在も特徴であるヒトアテローム硬化病変における脂質含有マクロファージにおいても示されている。SAAはSR−BIが仲介する細胞コレステロールのエフラックスを促進する62。
【0078】
Baranova et al.63では、HeLaおよびTHP1(ヒト急性単球性白血病細胞株)細胞におけるSAAのSR−BIへの特異的結合(おそらくは、HDLと結合した状態で)はERK1/2およびp38 MAPKのリン酸化、ならびにIl−8分泌を伴うことが示された。SR−BI受容体の発現は、ヒト肺癌細胞株を含むさまざまな細胞で示されている64。
【0079】
RAGE
終末糖化産物受容体(RAGE)は肺においてのみ検出可能なレベルで常時発現されているが、炎症部位において、主に炎症性および上皮細胞でその発現は急激に上昇する。上皮細胞のRAGEは、膜結合型または可溶型タンパクとして、ストレスにより大幅にアップレギュレートされる。RAGEを介した恒久的なシグナリングにより生存経路が誘導され、アポトーシスおよび(ATP欠乏による)ネクローシスが減弱された。これにより、多くの例において上皮の悪性化を引き起こす背景となる慢性的な炎症が導かれた65。RAGEの発現量の上昇は前立腺癌、直腸癌および胃癌に伴う;一方で、肺癌および食道癌の進行したステージはRAGE発現量の低下を特徴とする66。口腔扁平細胞癌では、RAGEの発現は癌の進行および再発と強く相関し、RAGE陽性患者は著しく短い無病生存期間を示した。数多くの様々なリガンドの内、SAAは終末糖化産物受容体(RAGE)と結合し、ERK1/2およびp38 MAPK経路を介してNF−κBを誘導する(COX経路を誘導することはない)ことが見出された67。
【0080】
TLR
近年の発見により、SAAがToll様レセプター(TLR)であるTLR4およびTLR2の内因性のリガンドとして作用し得ることが示された21。TLR4はいくつかのヒトの癌細胞で発現していることが見出された68,69。肺癌では、TLR4の活性化により免疫抑制性サイトカインTGF−ベータ、血管新生促進性ケモカインIL−8およびVEGFの産生が促進されることが示された。VEGFおよびIL−8分泌促進はp38 MAPK活性化を伴う70。SAAによるTLR4の活性化はp42/44およびp38 MAPKの活性化を必要とした71。
【0081】
TLR2はまた、SAAの機能的受容体であることも示された。TLR2を発現するHeLa細胞はNF−κBの強い活性化を伴ってSAAに応答した;SAAによる刺激は、TLR2−HeLa細胞において、ERK1/2(P−ERK1/2)、p38 MAPK(P−p38)、およびJNK(P−JNK) MAPKのリン酸化の亢進を引き起こし、IκBα(NFκB阻害因子)分解の促進を加速した72。SAAによる特異的な活性化の結果としてのNF−κBの刺激は、マクロファージにおいも示されている73。
【0082】
癌の発症および治療への耐性におけるSAAの予想される相互作用およびその生物学的効果の簡単なスキームを図3に示す。これから見て取れるように、SAAの生物学的機能は、SAAと様々な受容体の相互作用を引き金とする多数の経路のクロストークの視点から見ることができ、それは最終的には少なくとも1つの主要なMAPK経路:ERK、p38およびJNK21,41および/またはNF−κB活性化に収束する。これらの相互作用のいくつかは図4のEGFR伝達経路のスキームにおいて示される。
【0083】
EGFRはいくつかの主要な下流のシグナリング経路、例えば、Ras−Raf−Mek、ならびにホスホイノシチド3−キナーゼ(PI3K)、Akt、およびPKCからなる経路を活性化するチロシンキナーゼ型受容体(TKR)である。次いで、これは、NF−κB転写活性化経路および炎症性経路、例えば、COX2により誘導されるものとの複数のクロストークの関係を介し、増殖、生存、浸潤、転移速度、および腫瘍血管新生に対する効果を有し得る。SAAはこれらの経路を、チロシンキナーゼ型受容体と独立して活性化することができる(幅広の矢印で示す)。
【0084】
EGFRの発現量の上昇および/または恒常的な活性化は数多くの癌、例えば、脳癌、乳癌、腸癌および肺癌に伴う。そのカスケードの成分の変動はその経路の活性化に繋がり、癌の発症および進行に関連すると考えられている。例えば、EGFRキナーゼドメイン(非喫煙者)またはKRAS(喫煙者)における活性化変異は肺癌の早期発症に関連する74,75。Rasタンパクは約25%の腫瘍で恒常的に活性化しており、上流の制御とは独立して分裂促進シグナリングを誘起する76,77。新たに蓄積された大量のデータにより、非直線的なシグナリングおよびトランス活性化が癌の発症および進行に重要な役割を果たすことが示唆される。
【0085】
SAA相互作用と抗癌療法に対する耐性
化学療法、放射線療法および抗炎症療法
前で議論し、図3および4に示されるように、SAAと数多くの受容体との相互作用は抗癌療法に対する耐性と関連する経路の活性化に繋がる。化学療法および放射線療法に対する耐性におけるNF−κBの役割は既に議論される41。NF−κBを阻害すると、アポトーシス応答亢進により、放射線療法78,79、およびデス(death)サイトカイン80に対する感受性が上昇する。同時に、放射線およびいくつかの化学療法剤への曝露はNF−κBの活性化および後続するアポトーシスへの耐性を引き起こす81,79。化学療法(ゲムシタビン)誘発性のNF−κB活性化を阻害することにより、NSCLC細胞株の化学療法誘発性アポトーシスへの感受性が回復することが示されている82,81。他方で、いくつかの場合において、NF−κBは化学療法に対する感受性に関連することが示されており、例えば、NF−κBはパクリタキセル誘発性細胞死に必要であることが示唆されている82。
【0086】
これらの情報を考慮すると、VeriStrat「不良」患者に特徴的な血漿または血清中のSAA濃度の上昇から導かれる1つの結論は、SAAの上昇によりNF−κB転写因子およびMAPK経路の活性化が引き起こされる可能性があるというものである。これは、主に放射線療法に耐性を有する癌と関連する可能性、および患者の化学療法に対する応答に影響する可能性がある。しかしながら、各治療タイプおよび患者のコホートについて個々に評価されるべき数多くの因子が存在する。
【0087】
三酸化ヒ素、クルクミン、サリドマイドといったNF−κB阻害剤は、数多くの臨床治験の対象とされてきた。しかしながら、NF−κB阻害剤は正常な造血前駆細胞の化学療法誘発性アポトーシスも亢進するため、NF−κB阻害剤を化学療法の佐剤として用いることにより骨髄の回復が遅れる可能性があった。NF−κBは自然免疫応答および獲得免疫応答の活性化に重要な役割を有するため、阻害剤の長期間の使用は免疫不全のリスクを伴う可能性が高い41ことは考慮されるべきである。
【0088】
もし、VeriStrat「不良」シグナチャーが実際にNF−κBの特異的な活性化と相関しているならば、このシグナチャーはNF−κB阻害剤が最も有効である患者の選択に用いることができ、不要な治療およびそれに伴う病的状態を減弱できる可能性がある。
【0089】
受容体型チロシンキナーゼを標的とする治療
erbB受容体およびMAPK経路
EGFRおよびHER2は、4つのメンバー(EGFR(HER1)、erbB4(HER4)、erbB3(HER3)、およびerbB2(HER2))からなる上皮増殖因子受容体(EGFR)ファミリーに属する。上皮癌の大半は上皮増殖因子受容体(EGFR)およびHER2受容体の異常な活性化を示すため、これらの受容体の特異的な阻害は標的抗癌療法のストラテジーとなり、多くの研究の対象とされる。
【0090】
リガンドの非存在下において、EGFR受容体はキナーゼ活性が抑制されるような構造で存在する。リガンドの結合により、「二量体化ループ」を露出させるような構造変化が開始され、受容体の二量体化が誘起される。これらの転移は細胞膜を横切って中継され、キナーゼドメインを活性化する。この活性化スキームの多様性はErbBファミリーで見られる。ErbB−3は機能的なキナーゼではないが二量体のパートナーをトランス活性化することができ、他方で、HER2/ErbB−2は活性化構造に「ロックされた」リガンド不在の発癌受容体である。
【0091】
この二量体化によりチロシンキナーゼ機能の活性化が起こり、3つの主要なシグナリング経路を介したシグナル伝達に繋がり、最終的には、アポトーシスの回避、血管新生の持続、抗増殖シグナルへの耐性、増殖シグナルの自給自足、および転移が引き起こされる77,83。
【0092】
該カスケードの構成要素の変化は該経路の活性化を引き起こし、癌の誘発および進行と関連すると考えられており、例えば、EGFRキナーゼドメイン(非喫煙者)またはKRAS(喫煙者)の活性化変異は肺癌の早期発症と関連する74,72。Rasタンパクは約25%の腫瘍で恒常的に活性化しており、上流の制御とは独立した分裂促進シグナリングを誘導する76,77。
【0093】
いくつかのチロシンキナーゼ阻害剤(2つの小分子EGFRチロシンキナーゼ阻害剤−エルロチニブおよびゲフィチニブ、ならびにEGFRおよびHER2のデュアル阻害剤ラパチニブを含む)は、臨床現場で様々な固形腫瘍に現在使用される。ヒト化したモノクローナル抗HER2抗体トラスツズマブ、ならびに2つの抗EGFR抗体−セツキシマブおよびパニツムマブも臨床適用が認可されている。
【0094】
数多くの刊行物でレビューされるチロシンキナーゼ阻害剤(小分子ならびにモノクローナル抗体)に対する自然耐性ならびに獲得耐性は、活性化KRAS変異、met−癌原遺伝子の増幅84、およびT790M変異といった様々な因子に起因する。癌の多様性、および標的薬剤に対して耐性の複数の経路を示す能力により、単剤による治癒的療法の見込みは困難なものとなるが84、これは、多くの理由の中でも取り分け、リガンドとその受容体の相互作用の通常上流にあるものとは独立してシグナリングの活性化が起こる可能性があることによる。複数のチロシンキナーゼの共発現、受容体下流の経路のクロストーク、および伝達カスケードの下流の活性化の重要性を示す証拠が増加している。
【0095】
経路のトランス活性化は、複数の研究において耐性のメカニズムの1つであることが示唆された。例えば、インスリン様成長因子−I受容体(IGF−1R)シグナリングは、ヒト乳癌および前立腺癌細胞株においてゲフィチニブによりブロックされたEGFRを代償できることが示された85。発癌性のPIK3CAまたは別のRTKによる代替の下流のシグナリング、特に、Aktの活性化を介したものは、NSCLCにおけるTKIに対する耐性のメカニズムの1つであると記載される86。
【0096】
Cappuzzoら88は、NSCLC患者のゲフィチニブに対する感受性はAktが活性化された場合に非常に低くなる一方で、EGFRの発現はネガティブであることを観察し、EGFR非依存的活性化がゲフィチニブに対する耐性に繋がり得ることを確認した。
【0097】
我々は、VeriStrat検査により測定されるようなSAAの相互作用がRTK非依存的なMAPKカスケードの活性化を引き起こし、その結果として、TKI耐性となることを提案する。このSAAの活性化のメカニズムは、直接的でも間接的でもよい。SAAの直接的な作用はRAGEまたはTLR2およびTLR4受容体へのその結合により仲介され、古典的MAPK経路の活性化を引き起こし得る(JNKおよびp38の活性化による)。様々な癌細胞および癌細胞と関連する細胞の表面におけるこれらの受容体の存在およびその相互作用は、Malle, et al21でレビューされる。TLR受容体の活性化の結果としてのEGFRの活性化を直接証明する証拠が存在する66。
【0098】
SAAの間接的な作用はFPRL受容体を介して作用し、インターロイキンIl6、およびIl8の放出を引き起こし、次いで、Gタンパク質共役型受容体と反応し、PKCを活性化することで説明することができる。(PKCの活性化により細胞増殖および血管透過性が亢進され、MAPK経路におけるMEKの活性化が引き起こされる86)。加えて、それはVEGFの発現を誘導する。
【0099】
SAAは、肺上皮細胞およびマクロファージにおけるTRL4のリガンドである。所要細胞で発現されるTLRのライゲーションによってもVEGFレベルが上昇することが報告されている70。
【0100】
この情報により、SAAによる主要な3つの全てのMAPK経路の下流の活性化に関与するメカニズムが存在する証拠が提供される。MAPK経路の下流の活性化はRTK非依存的であり、「交差する」チェックポイントから上流の標的阻害に対する耐性を引き起こす可能性がある。
【0101】
前記の観点から、VeriStrat検査を用いる併用療法を含む特定の治療に最も適した患者のセレクションは、薬剤耐性のいくつかのタイプを克服する道具となり得る。
【0102】
併用療法およびVeriStratシグナチャー
TKIおよびCOX2阻害剤
前記で議論したように、SAAはCOX2の発現を誘導することができる。肺癌におけるCOX2の過剰な発現は、Huang et al.87により最初に報告され、約70%の腺癌で観察され88、他の数多くの研究により確認された。
【0103】
数多くの治験によりCOX2およびEGFRシグナリング経路のクロストークが示されている。前記で議論したように、MAPK経路を介して作用する上皮増殖因子(EGF)は、いくつかの上皮細胞においてCOX2活性を著しく誘導する47。TGFαによるEGFRの活性化はCOX2を刺激し、PGE2の放出および有糸分裂誘発の亢進を引き起こす48。他方、COX2の産物であるプロスタグランジンE2(PGE2)は、EGF受容体をトランス活性化することができる45。NSCLCにおいて、PGE2が、EGFR非依存的な様式の細胞内クロストークによりMAPK/Erk経路を活性化することが示され;この効果はGタンパク質共役型受容体およびプロテインキナーゼC(PKC)を通して仲介され、EGFR−TKI耐性に寄与する可能性がある89。
【0104】
他方で、COX2阻害剤はNF−κB経路を阻害することが示されている:セレコキシブはAktおよびIKKの抑制を介してその効果を発揮する。ヒト非小細胞性肺癌において、セレコキシブはNF−κB、ならびにTNF誘導性のJNK、p38 MAPK、およびERKの活性化をIKKおよびAktの抑制を介して抑制し、COX2、ならびに炎症、増殖および腫瘍新生に必要な他の遺伝子の合成のダウンレギュレーションを引き起こす46,90。アスピリンおよびイブプロフェンを含む他のNSAIDは、IKK活性化およびIκBα分解の抑制により作用することが示された。合わせると、これらの考察により標準的な抗癌療法にCOX2を追加することの強力な理論的根拠が提供される。
【0105】
NSCLCにおける抗炎症療法およびチロシンキナーゼ受容体標的療法の併用療法に関する研究およびそれらがEGFR−TKIを克服する可能性は以前の文献でレビューされている90,91。治験の結果はネガティブであった:ゲフィチニブおよびセレコキシブの併用療法における応答率および患者の生存率、ならびにロフェコキシブおよびエルロチニブで治療した患者の病勢コントロール率92,93は単剤療法におけるものと同様であることが分かった。
【0106】
COX阻害剤の添加の効果が、この経路において示唆されるSAAのアップレギュレーション効果により、VeriStrat「不良」患者においてより顕著となる可能性がある。しかしながら、下流のMAPK活性およびNF−κBならびにそれらの相互作用に対するCOX2経路の阻害効果の度合いは不明であるため、該効果の度合いを予測することは困難である。この仮説はさらに研究されるべきである。
【0107】
細胞株に基づく証明(図8)
我々は、VeriStrat「不良」血清が腫瘍細胞に生物学的効果を及ぼし得ること、特に、薬剤感受性細胞株においてそれがゲフィチニブに対する耐性を増強し得ることを示してきた。ゲフィチニブ感受性細胞株HCC4006(エクソン19が欠損したEGFRを有する)および耐性株A549(EGFR野生型)を用いて実験を行った。ステージIIIB/IVであり、Veristrad「良」および「不良」と識別されたNSCLC患者からヒト血清を得た。各分類の血清を合わせてプールを作製し、増殖阻害アッセイに用いた。2種類の組成の培地(RPMI/10%「良」血清またはRPMI/10%「不良」血清)を用いて細胞を播種した(10複製/薬剤濃度;2,000細胞/ウェル)。24時間後、ゲフィチニブを加え、プレートを6日間インキュベートした。増殖阻害の測定にはMTTアッセイを用いた。結果を下記の表2および図8に示す。
【表2】
【0108】
図8は、異なる濃度のゲフィチニブの存在下における、Veristrad「良」およびVeristrad「不良」血清中のゲフィチニブ感受性細胞株HCC4006およびゲフィチニブ耐性細胞株A549の増殖を示すグラフである。図8において、%コントロールは、対応する増殖培地中における所定のゲフィチニブ濃度の吸光度の薬剤非存在下における吸光度の平均値に対する比から算出した。エラーバーは正規化した測定値の標準偏差を表す。
【0109】
VeriStrat「不良」血清中で増殖させた場合、感受性細胞において阻害の相対的な減弱が見られたが、耐性腫瘍細胞では有意な変化は見られなかった。この結果は、VeriStrat「不良」血清が腫瘍細胞に対し直接生物学的効果を有することを示し、VeriStrat「良」血清の効果とは異なるものである。これらの結果は、VeriStratメカニズム、それとホスト−腫瘍相互作用、および患者集団における標的療法の相対的な有効性との関係の我々の仮説を支持するものである。
【0110】
化学療法におけるVeriStrat
図7に示されるように、VeriStrat「不良」シグナチャーはいくつかの非標的療法に対する応答不良に関連するが、他のものには関連しない。VeriStrat分類は、DNA複製またはNF−κBで制御される遺伝子の転写に干渉する化学療法(シスプラチン、ゲムシタビンなど)におけるアウトカムと相関する可能性が高いが、非標的療法におけるVeriStratの有用性の具体的な範囲は実験的に決定する必要がある。
【0111】
かくして、VeriStrat検査の臨床における適用の例は、いくつかの非標的化学療法レジメン(DNAの複製および/またはNF−κB転写因子で制御される遺伝子の活性化と相互作用するものなど)が癌患者に有効である可能性が低いかどうかを予測するための方法であり、サンプル(図1)にVeriStrat検査を行い、もし結果が「不良」のクラス標識であれば該患者には有効である可能性が低いことを特徴とするものになろう。
【0112】
SAAがNF−κB転写因子の活性化の原因であること、ならびに癌の進行および様々な療法への応答におけるNF−κB活性化の役割に関する文献的情報を考慮に入れると、VeriStratシグナチャーは主に放射線療法に耐性を有する癌、ならびに化学療法に対する患者の応答と相関する可能性がある。
【0113】
三酸化ヒ素、クルクミン、サリドマイドといったNF−κB阻害剤は、抗癌剤として臨床治験で評価の対象となっている。しかしながら、それらの有用性は、これらの薬剤に対する応答のバイオマーカーが存在しないこと、ならびにそれらの副作用により、限定されたものとなっている。VeriStratはNF−κBの活性化の亢進のバイオマーカーとして有用であり得、故に、NF−κB阻害剤が最も有効である患者(おそらくは、VeriStrat「不良」)のセレクションに有用となり得る。
【0114】
前記の全てをまとめると、本発明は図1のVeriStrat検査のさらなる使用を包含する。一般的な事柄として、VeriStrat検査は、AktまたはERK/JNK/p38またはPKCでの、またはそれらの上流でのMAPK経路またはPKC(プロテインキナーゼC)経路に関わる受容体のアゴニスト、受容体もしくはタンパク質を標的にする治療薬または治療薬の組み合わせによる治療が患者に有効であるかを予測する。予測の度合いは、特定の薬剤または薬剤の組み合わせに依存するであろう。VeriStrat検査は、下流の制御を標的とする薬剤の効果は予測しない。
【0115】
一実施態様において、本発明は、固形上皮腫瘍癌患者を、AktまたはERK/JNK/p38またはPKCでの、またはそれらの上流でのMAPK経路またはPKC経路に関わる受容体のアゴニスト、受容体もしくはタンパク質を標的にする治療薬または治療薬の組み合わせによる治療が有効である可能性が高い患者として、あるいは治療薬または治療薬の組み合わせによる治療が有効である可能性が低い患者として識別する方法であって:
a)固形上皮腫瘍癌患者の血液由来サンプルから質量スペクトルを得;
b)a)で得た質量スペクトルにおいて1つまたはそれ以上の所定の前処理工程(バックグラウンドの除去、ノーマライゼーションおよびスペクトルのアラインメント)を行い;
c)b)の質量スペクトルにおける前処理工程を行った後、1つまたはそれ以上の所定のm/z範囲(および、好ましくは前記の表1で示されるm/z範囲に対応するm/z範囲)において該スペクトルにおける選択された特徴の積分強度値を得;
d)患者を治療薬または治療薬の組み合わせが有効である可能性が高い、または有効である可能性が低い患者として識別するために、別の固形腫瘍患者の血液由来サンプルから得たクラス標識スペクトルを含む訓練セットを用いた分類アルゴリズム(例えば、k近傍法)にc)で得た値を用いる
ことを特徴とする方法と見做すことができる。
【0116】
特定の例として、EGFR−IにMAPK経路の下流の活性化を遮断する標的薬剤を加えることにより、VeriStrat「不良」シグナチャーを有する患者のEGFR−Iに対する耐性を克服できる可能性がある。
【0117】
別の特定の例として、COX2阻害剤、セレコキシブおよびロフェコキシブをEGFR−Iに治療レジメンとして加えることにより、VeriStrat「不良」シグナチャーを有する患者のEGFR−Iに対する耐性を克服できる可能性がある。VeriStrat検査は、故に、COX2阻害剤およびEGFRIを含む併用療法を処方するためのインジケーターとして用いることができる。特定の実施態様において、COX2阻害剤およびEGFRIの投与が癌患者に有効である可能性が高いかどうかを予測する方法は、a)癌患者の血液由来サンプルから質量スペクトルを得;b)a)で得た質量スペクトルにおいて1つまたはそれ以上の所定の前処理工程(バックグラウンドの除去、ノーマライゼーションおよびスペクトルのアラインメント)を行い;c)b)の質量スペクトルにおける前処理工程を行った後、1つまたはそれ以上の所定のm/z範囲(および、好ましくは前記の表1で示されるm/z範囲に対応するm/z範囲)において該スペクトルにおける選択された特徴の積分強度値を得;d)患者をCOX2阻害剤およびEGFR−Iの投与による治療が有効である可能性が高い、または有効である可能性が低い患者として識別するために、別の固形腫瘍患者の血液由来サンプルから得たクラス標識スペクトルを含む訓練セットを用いた分類アルゴリズム(例えば、k近傍法)にc)で得た値を用いることを特徴とする。特に、クラス標識が「不良」の場合、該患者には有効である可能性が高いことが示唆される。
【0118】
別の特定の例として、VeriStrat「不良」シグナチャーはNF−κBの特異的な活性化と相関すると考えられ、故に、該検査は、NF−κB阻害剤、およびCOX2阻害剤の標準的な化学療法への追加が最も有効である患者の選択に用いることができ、同時に、不必要な治療およびそれに関連する病的状態の軽減にも有用であろう。
【0119】
本開示の方法は、癌患者の血液由来サンプル(またはこれらのサンプルからの質量分析データ)を受け取り、これらの質量分析データを機械で読み取り可能な記憶媒体に蓄え、図1に示される工程および分類工程を機械で行う、例えば、クラス標識(VeriStrat「良」または「不良」)を作製するようプログラムされたコンピューターを用い、故に前記の薬剤または薬剤の組み合わせが有効である可能性が高いとしての識別を予測する臨床試験センターで実行されてもよい。別の一実施態様として、本発明は、COX2阻害剤およびEGFR阻害剤の組み合わせの投与が癌患者に有効である可能性が高いかどうかを識別または予測するよう構成された装置として設定することができる。該装置は、癌患者の血液由来サンプルの質量スペクトルを記憶する記憶装置、コンピューターの記憶媒体またはデータベース、ならびにa)質量スペクトルにおいて1つまたはそれ以上の所定の前処理工程を行い(図1参照);b)1つまたはそれ以上の所定のm/z範囲において該質量スペクトルにおける特徴の積分強度値を得(例えば、表1のピークのリストに含まれる範囲または前記のm/z範囲において);c)患者をCOX2阻害剤およびEGFR阻害剤の組み合わせの投与による治療が有効である可能性が高い、または低い患者として識別するために、別の固形腫瘍患者の血液由来サンプルから得たクラス標識スペクトルを含む訓練セットを用いた分類アルゴリズム(例えば、KNN分類アルゴリズム)にb)で得た値を用いるよう設定されたソフトウェアの指示を実行するプロセッサー(例えば、一般的な目的でプログラムされたコンピューターの一般的なCPU)の組み合わせを含む。
【0120】
別の一例において、本発明は、固形上皮腫瘍癌患者を、AktまたはERK/JNK/p38またはPKCでの、またはそれらの上流でのMAPK(マイトジェン活性化プロテインキナーゼ)経路またはPKC(プロテインキナーゼC)経路に関わる受容体のアゴニスト、受容体もしくはタンパク質を標的にする治療薬または治療薬の組み合わせによる治療が有効である可能性が高い患者として、あるいは治療薬または治療薬の組み合わせによる治療が有効である可能性が低い患者として識別するよう設定された装置として具体化することができる。該装置は、固形上皮腫瘍癌患者の血液由来サンプルの質量スペクトルを記憶する記憶装置、およびa)質量スペクトルにおいて1つまたはそれ以上の所定の前処理工程を行い(図1参照)、b)1つまたはそれ以上の所定のm/z範囲において該質量スペクトルにおける特徴の積分強度値を得(例えば、表1のピークのリストに含まれる範囲または前記のm/z範囲において)、c)患者を治療薬または治療薬の組み合わせが有効である可能性が高い、または低い患者として識別するために、別の固形腫瘍患者の血液由来サンプルから得たクラス標識スペクトルを含む訓練セットを用いた分類アルゴリズムにb)で得た値を用いるよう設定されたソフトウェアの指示を実行するプロセッサーの形態を取る。
【0121】
開示される発明のさらなる例は付属の請求項で説明される。
【0122】
付録
引用文献
1. Taguchi F, Solomon B, Gregorc V, et al. Mass spectrometry to classify non-small-cell lung cancer patients for clinical outcome after treatment with epidermal growth factor receptor tyrosine kinase inhibitors: a multicohort cross-institutional study. J Natl Cancer Inst 2007;99:838-46.
2. Clark GM, Zborowski DM, Culbertson JL, et al. Clinical utility of epidermal growth factor receptor expression for selecting patients with advanced non-small cell lung cancer for treatment with erlotinib. J Thorac Oncol 2006;1:837-46.
3. Chung CH, Seeley EH, Roder H, et al. Detection of tumor epidermal growth factor receptor pathway dependence by serum mass spectrometry in cancer patients. Cancer Epidemiol Biomarkers Prev 2010;19:358-65.
4. Carbone DP, Salmon JS, Billheimer D, et al. VeriStrat((R)) classifier for survival and time to progression in non-small cell lung cancer (NSCLC) patients treated with erlotinib and bevacizumab. Lung Cancer 2009.
5. Kiernan UA, Tubbs KA, Nedelkov D, Niederkofler EE, Nelson RW. Detection of novel truncated forms of human serum amyloid A protein in human plasma. FEBS Lett 2003;537:166-70.
6. Cremona M, Calabro E, Randi G, et al. Elevated levels of the acute-phase serum amyloid are associated with heightened lung cancer risk. Cancer 2010.
7. Benson MD, Eyanson S, Fineberg NS. Serum amyloid A in carcinoma of the lung. Cancer 1986;57:1783-7.
8. Biran H, Friedman N, Neumann L, Pras M, Shainkin-Kestenbaum R. Serum amyloid A (SAA) variations in patients with cancer: correlation with disease activity, stage, primary site, and prognosis. J Clin Pathol 1986;39:794-7.
9. Khan N, Cromer CJ, Campa M, Patz EF, Jr. Clinical utility of serum amyloid A and macrophage migration inhibitory factor as serum biomarkers for the detection of nonsmall cell lung carcinoma. Cancer 2004;101:379-84.
10. Cho WC, Yip TT, Yip C, et al. Identification of serum amyloid a protein as a potentially useful biomarker to monitor relapse of nasopharyngeal cancer by serum proteomic profiling. Clin Cancer Res 2004;10:43-52.
11. Yokoi K, Shih LC, Kobayashi R, et al. Serum amyloid A as a tumor marker in sera of nude mice with orthotopic human pancreatic cancer and in plasma of patients with pancreatic cancer. Int J Oncol 2005;27:1361-9.
12. Gutfeld O, Prus D, Ackerman Z, et al. Expression of serum amyloid A, in normal, dysplastic, and neoplastic human colonic mucosa: implication for a role in colonic tumorigenesis. J Histochem Cytochem 2006;54:63-73.
13. Engwegen JY, Mehra N, Haanen JB, et al. Validation of SELDI-TOF MS serum protein profiles for renal cell carcinoma in new populations. Lab Invest 2007;87:161-72.
14. Dai S, Wang X, Liu L, et al. Discovery and identification of Serum Amyloid A protein elevated in lung cancer serum. Sci China C Life Sci 2007;50:305-11.
15. Liu DH, Wang XM, Zhang LJ, et al. Serum amyloid A protein: a potential biomarker correlated with clinical stage of lung cancer. Biomed Environ Sci 2007;20:33-40.
16. Michaeli A, Finci-Yeheskel Z, Dishon S, Linke RP, Levin M, Urieli-Shoval S. Serum amyloid A enhances plasminogen activation: implication for a role in colon cancer. Biochem Biophys Res Commun 2008;368:368-73.
17. Uhlar CM, Burgess CJ, Sharp PM, Whitehead AS. Evolution of the serum amyloid A (SAA) protein superfamily. Genomics 1994;19:228-35.
18. Uhlar CM, Whitehead AS. Serum amyloid A, the major vertebrate acute-phase reactant. Eur J Biochem 1999;265:501-23.
19. Sipe JD. Amyloidosis. Annu Rev Biochem 1992;61:947-75.
20. Vlasova MA, Moshkovskii SA. Molecular interactions of acute phase serum amyloid A: possible involvement in carcinogenesis. Biochemistry (Mosc) 2006;71:1051-9.
21. Malle E, Sodin-Semrl S, Kovacevic A. Serum amyloid A: an acute-phase protein involved in tumour pathogenesis. Cell Mol Life Sci 2009;66:9-26.
22. Preciado-Patt L, Levartowsky D, Prass M, Hershkoviz R, Lider O, Fridkin M. Inhibition of cell adhesion to glycoproteins of the extracellular matrix by peptides corresponding to serum amyloid A. Toward understanding the physiological role of an enigmatic protein. Eur J Biochem 1994;223:35-42.
23. Migita K, Kawabe Y, Tominaga M, Origuchi T, Aoyagi T, Eguchi K. Serum amyloid A protein induces production of matrix metalloproteinases by human synovial fibroblasts. Lab Invest 1998;78:535-9.
24. Hynes RO. The extracellular matrix: not just pretty fibrils. Science 2009;326:1216-9.
25. Vihinen P, Ala-aho R, Kahari VM. Matrix metalloproteinases as therapeutic targets in cancer. Curr Cancer Drug Targets 2005;5:203-20.
26. Furlaneto CJ, Campa A. A novel function of serum amyloid A: a potent stimulus for the release of tumor necrosis factor-alpha, interleukin-1beta, and interleukin-8 by human blood neutrophil. Biochem Biophys Res Commun 2000;268:405-8.
27. Patel H, Fellowes R, Coade S, Woo P. Human serum amyloid A has cytokine-like properties. Scand J Immunol 1998;48:410-8.
28. He R, Shepard LW, Chen J, Pan ZK, Ye RD. Serum amyloid A is an endogenous ligand that differentially induces IL-12 and IL-23. J Immunol 2006;177:4072-9.
29. Malle E, Bollmann A, Steinmetz A, Gemsa D, Leis HJ, Sattler W. Serum amyloid A (SAA) protein enhances formation of cyclooxygenase metabolites of activated human monocytes. FEBS Lett 1997;419:215-9.
30. Jijon HB, Madsen KL, Walker JW, Allard B, Jobin C. Serum amyloid A activates NF-kappaB and proinflammatory gene expression in human and murine intestinal epithelial cells. Eur J Immunol 2005;35:718-26.
31. Coussens LM, Werb Z. Inflammation and cancer. Nature 2002;420:860-7.
32. Farrow B, Sugiyama Y, Chen A, Uffort E, Nealon W, Mark Evers B. Inflammatory mechanisms contributing to pancreatic cancer development. Ann Surg 2004;239:763-9; discussion 9-71.
33. Ditsworth D, Zong WX. NF-kappaB: key mediator of inflammation-associated cancer. Cancer Biol Ther 2004;3:1214-6.
34. Balkwill F, Coussens LM. Cancer: an inflammatory link. Nature 2004;431:405-6.
35. Lu H, Ouyang W, Huang C. Inflammation, a key event in cancer development. Mol Cancer Res 2006;4:221-33.
36. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature 2008;454:436-44.
37. Lee JM, Yanagawa J, Peebles KA, Sharma S, Mao JT, Dubinett SM. Inflammation in lung carcinogenesis: new targets for lung cancer chemoprevention and treatment. Crit Rev Oncol Hematol 2008;66:208-17.
38. Greten FR, Eckmann L, Greten TF, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004;118:285-96.
39. Pikarsky E, Porat RM, Stein I, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004;431:461-6.
40. Karin M. The IkappaB kinase - a bridge between inflammation and cancer. Cell Res 2008;18:334-42.
41. Lee CH, Jeon YT, Kim SH, Song YS. NF-kappaB as a potential molecular target for cancer therapy. Biofactors 2007;29:19-35.
42. Graham B, Gibson SB. The two faces of NFkappaB in cell survival responses. Cell Cycle 2005;4:1342-5.
43. Kaltschmidt B, Kaltschmidt C, Hofmann TG, Hehner SP, Droge W, Schmitz ML. The pro- or anti-apoptotic function of NF-kappaB is determined by the nature of the apoptotic stimulus. Eur J Biochem 2000;267:3828-35.
44. Bernard D, Monte D, Vandenbunder B, Abbadie C. The c-Rel transcription factor can both induce and inhibit apoptosis in the same cells via the upregulation of MnSOD. Oncogene 2002;21:4392-402.
45. Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol 2002;2:725-34.
46. Shishodia S, Koul D, Aggarwal BB. Cyclooxygenase (COX)-2 inhibitor celecoxib abrogates TNF-induced NF-kappa B activation through inhibition of activation of I kappa B alpha kinase and Akt in human non-small cell lung carcinoma: correlation with suppression of COX-2 synthesis. J Immunol 2004;173:2011-22.
47. Koki AT, Khan NK, Woerner BM, et al. Characterization of cyclooxygenase-2 (COX-2) during tumorigenesis in human epithelial cancers: evidence for potential clinical utility of COX-2 inhibitors in epithelial cancers. Prostaglandins Leukot Essent Fatty Acids 2002;66:13-8.
48. Soslow RA, Dannenberg AJ, Rush D, et al. COX-2 is expressed in human pulmonary, colonic, and mammary tumors. Cancer 2000;89:2637-45.
49. Pai R, Soreghan B, Szabo IL, Pavelka M, Baatar D, Tarnawski AS. Prostaglandin E2 transactivates EGF receptor: a novel mechanism for promoting colon cancer growth and gastrointestinal hypertrophy. Nat Med 2002;8:289-93.
50. McKay MM, Morrison DK. Integrating signals from RTKs to ERK/MAPK. Oncogene 2007;26:3113-21.
51. Richards JA, Petrel TA, Brueggemeier RW. Signaling pathways regulating aromatase and cyclooxygenases in normal and malignant breast cells. J Steroid Biochem Mol Biol 2002;80:203-12.
52. Coffey RJ, Hawkey CJ, Damstrup L, et al. Epidermal growth factor receptor activation induces nuclear targeting of cyclooxygenase-2, basolateral release of prostaglandins, and mitogenesis in polarizing colon cancer cells. Proc Natl Acad Sci U S A 1997;94:657-62.
53. Prossnitz ER, Ye RD. The N-formyl peptide receptor: a model for the study of chemoattractant receptor structure and function. Pharmacol Ther 1997;74:73-102.
54. Babbin BA, Lee WY, Parkos CA, et al. Annexin I regulates SKCO-15 cell invasion by signaling through formyl peptide receptors. J Biol Chem 2006;281:19588-99.
55. Rescher U, Danielczyk A, Markoff A, Gerke V. Functional activation of the formyl peptide receptor by a new endogenous ligand in human lung A549 cells. J Immunol 2002;169:1500-4.
56. Su SB, Gong W, Gao JL, et al. A seven-transmembrane, G protein-coupled receptor, FPRL1, mediates the chemotactic activity of serum amyloid A for human phagocytic cells. J Exp Med 1999;189:395-402.
57. Biswas DK, Martin KJ, McAlister C, et al. Apoptosis caused by chemotherapeutic inhibition of nuclear factor-kappaB activation. Cancer Res 2003;63:290-5.
58. El Kebir D, Jozsef L, Khreiss T, et al. Aspirin-triggered lipoxins override the apoptosis-delaying action of serum amyloid A in human neutrophils: a novel mechanism for resolution of inflammation. J Immunol 2007;179:616-22.
59. Lee HY, Kim MK, Park KS, et al. Serum amyloid A induces contrary immune responses via formyl peptide receptor-like 1 in human monocytes. Mol Pharmacol 2006;70:241-8.
60. Lee MS, Yoo SA, Cho CS, Suh PG, Kim WU, Ryu SH. Serum amyloid A binding to formyl peptide receptor-like 1 induces synovial hyperplasia and angiogenesis. J Immunol 2006;177:5585-94.
61. Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 1996;271:518-20.
62. van der Westhuyzen DR, Cai L, de Beer MC, de Beer FC. Serum amyloid A promotes cholesterol efflux mediated by scavenger receptor B-I. J Biol Chem 2005;280:35890-5.
63. Baranova IN, Vishnyakova TG, Bocharov AV, et al. Serum amyloid A binding to CLA-1 (CD36 and LIMPII analogous-1) mediates serum amyloid A protein-induced activation of ERK1/2 and p38 mitogen-activated protein kinases. J Biol Chem 2005;280:8031-40.
64. Hrzenjak A, Reicher H, Wintersperger A, et al. Inhibition of lung carcinoma cell growth by high density lipoprotein-associated alpha-tocopheryl-succinate. Cell Mol Life Sci 2004;61:1520-31.
65. Sparvero LJ, Asafu-Adjei D, Kang R, et al. RAGE (Receptor for Advanced Glycation Endproducts), RAGE ligands, and their role in cancer and inflammation. J Transl Med 2009;7:17.
66. Franklin WA. RAGE in lung tumors. Am J Respir Crit Care Med 2007;175:106-7.
67. Cai H, Song C, Endoh I, et al. Serum amyloid A induces monocyte tissue factor. J Immunol 2007;178:1852-60.
68. Wang L, Liu Q, Sun Q, Zhang C, Chen T, Cao X. TLR4 signaling in cancer cells promotes chemoattraction of immature dendritic cells via autocrine CCL20. Biochem Biophys Res Commun 2008;366:852-6.
69. Fukata M, Chen A, Vamadevan AS, et al. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology 2007;133:1869-81.
70. He W, Liu Q, Wang L, Chen W, Li N, Cao X. TLR4 signaling promotes immune escape of human lung cancer cells by inducing immunosuppressive cytokines and apoptosis resistance. Mol Immunol 2007;44:2850-9.
71. Sandri S, Rodriguez D, Gomes E, Monteiro HP, Russo M, Campa A. Is serum amyloid A an endogenous TLR4 agonist? J Leukoc Biol 2008;83:1174-80.
72. Cheng N, He R, Tian J, Ye PP, Ye RD. Cutting edge: TLR2 is a functional receptor for acute-phase serum amyloid A. J Immunol 2008;181:22-6.
73. He RL, Zhou J, Hanson CZ, Chen J, Cheng N, Ye RD. Serum amyloid A induces G-CSF expression and neutrophilia via Toll-like receptor 2. Blood 2009;113:429-37.
74. Westra WH. Early glandular neoplasia of the lung. Respir Res 2000;1:163-9.
75. Tang X, Shigematsu H, Bekele BN, et al. EGFR tyrosine kinase domain mutations are detected in histologically normal respiratory epithelium in lung cancer patients. Cancer Res 2005;65:7568-72.
76. Medema RH, Bos JL. The role of p21ras in receptor tyrosine kinase signaling. Crit Rev Oncog 1993;4:615-61.
77. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100:57-70.
78. Shao R, Karunagaran D, Zhou BP, et al. Inhibition of nuclear factor-kappaB activity is involved in E1A-mediated sensitization of radiation-induced apoptosis. J Biol Chem 1997;272:32739-42.
79. Yamagishi N, Miyakoshi J, Takebe H. Enhanced radiosensitivity by inhibition of nuclear factor kappa B activation in human malignant glioma cells. Int J Radiat Biol 1997;72:157-62.
80. Luo JL, Kamata H, Karin M. The anti-death machinery in IKK/NF-kappaB signaling. J Clin Immunol 2005;25:541-50.
81. Brach MA, Hass R, Sherman ML, Gunji H, Weichselbaum R, Kufe D. Ionizing radiation induces expression and binding activity of the nuclear factor kappa B. J Clin Invest 1991;88:691-5.
82. Jones DR, Broad RM, Madrid LV, Baldwin AS, Jr., Mayo MW. Inhibition of NF-kappaB sensitizes non-small cell lung cancer cells to chemotherapy-induced apoptosis. Ann Thorac Surg 2000;70:930-6; discussion 6-7.
83. Gazdar AF. Personalized Medicine and Inhibition of EGFR Signaling in Lung Cancer. N Engl J Med 2009.
84. Lynch TJ, Jr., Blumenschein GR, Jr., Engelman JA, et al. Summary statement novel agents in the treatment of lung cancer: Fifth Cambridge Conference assessing opportunities for combination therapy. J Thorac Oncol 2008;3:S107-12.
85. Jones HE, Goddard L, Gee JM, et al. Insulin-like growth factor-I receptor signalling and acquired resistance to gefitinib (ZD1839; Iressa) in human breast and prostate cancer cells. Endocr Relat Cancer 2004;11:793-814.
86. Engelman JA, Janne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res 2008;14:2895-9.
87. Huang M, Stolina M, Sharma S, et al. Non-small cell lung cancer cyclooxygenase-2-dependent regulation of cytokine balance in lymphocytes and macrophages: up-regulation of interleukin 10 and down-regulation of interleukin 12 production. Cancer Res 1998;58:1208-16.
88. Hida T, Yatabe Y, Achiwa H, et al. Increased expression of cyclooxygenase 2 occurs frequently in human lung cancers, specifically in adenocarcinomas. Cancer Res 1998;58:3761-4.
89. Krysan K, Reckamp KL, Dalwadi H, et al. Prostaglandin E2 activates mitogen-activated protein kinase/Erk pathway signaling and cell proliferation in non-small cell lung cancer cells in an epidermal growth factor receptor-independent manner. Cancer Res 2005;65:6275-81.
90. Krysan K, Reckamp KL, Sharma S, Dubinett SM. The potential and rationale for COX-2 inhibitors in lung cancer. Anticancer Agents Med Chem 2006;6:209-20.
91. Reckamp KL, Gardner BK, Figlin RA, et al. Tumor response to combination celecoxib and erlotinib therapy in non-small cell lung cancer is associated with a low baseline matrix metalloproteinase-9 and a decline in serum-soluble E-cadherin. J Thorac Oncol 2008;3:117-24.
92. Gadgeel SM, Ruckdeschel JC, Heath EI, Heilbrun LK, Venkatramanamoorthy R, Wozniak A. Phase II study of gefitinib, an epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI), and celecoxib, a cyclooxygenase-2 (COX-2) inhibitor, in patients with platinum refractory non-small cell lung cancer (NSCLC). J Thorac Oncol 2007;2:299-305.
93. O’Byrne KJ, Danson S, Dunlop D, et al. Combination therapy with gefitinib and rofecoxib in patients with platinum-pretreated relapsed non small-cell lung cancer. J Clin Oncol 2007;25:3266-73.
【特許請求の範囲】
【請求項1】
固形上皮腫瘍癌患者を、AktまたはERK/JNK/p38またはPKCでの、またはそれらの上流でのMAPK(マイトジェン活性化プロテインキナーゼ)経路またはPKC(プロテインキナーゼC)経路に関わる受容体のアゴニスト、受容体もしくはタンパク質を標的にする治療薬または治療薬の組み合わせによる治療が有効である可能性が高い患者として、あるいは治療薬または治療薬の組み合わせによる治療が有効である可能性が低い患者として識別する方法であって:
a)固形上皮腫瘍癌患者の血液由来サンプルから質量スペクトルを得;
b)a)で得た質量スペクトルにおいて1つまたはそれ以上の所定の前処理工程を行い;
c)b)の質量スペクトルにおける前処理工程を行った後、1つまたはそれ以上の所定のm/z範囲において該スペクトルにおける選択された特徴の積分強度値を得;
d)患者を治療薬または治療薬の組み合わせが有効である可能性が高い、または有効である可能性が低い患者として識別するために、別の固形腫瘍患者の血液由来サンプルから得たクラス標識スペクトルを含む訓練セットを用いた分類アルゴリズムにc)で得た値を用いる
ことを特徴とする方法。
【請求項2】
治療薬または治療薬の組み合わせがEGFRおよびVEGFを標的としたものである、請求項1の方法。
【請求項3】
治療薬または治療薬の組み合わせがHER2を標的としたものである、請求項1の方法。
【請求項4】
治療薬または治療薬の組合せがトラスツズマブを含む、請求項1の方法。
【請求項5】
治療薬または治療薬の組み合わせがEGFRを標的としたものであり、分類アルゴリズムにより、EGFRを標的とした治療薬との組み合わせにおけるCOX2阻害剤による治療が有効である可能性が高く、EGFRを標的とした治療薬のみによる治療が有効である可能性が低い患者として該患者を識別するクラス標識が得られる、請求項1の方法。
【請求項6】
COX2阻害剤がセレコキシブを含む、請求項5の方法。
【請求項7】
COX2阻害剤がロフェコキシブを含む、請求項5の方法。
【請求項8】
クラス標識により該患者がNF−κB阻害剤による治療が有効である可能性が高い患者としてさらに識別される、請求項1の方法。
【請求項9】
1つまたはそれ以上の所定のm/z範囲が、
5732から5795
5811から5875
6398から6469
11376から11515
11459から11599
11614から11756
11687から11831
11830から11976
12375から12529
23183から23525
23279から23622
65902から67502
を含むm/z範囲の群から選択される、請求項1の方法。
【請求項10】
固形上皮腫瘍癌患者を、AktまたはERK/JNK/p38またはPKCでの、またはそれらの上流でのMAPK(マイトジェン活性化プロテインキナーゼ)経路またはPKC(プロテインキナーゼC)経路に関わる受容体のアゴニスト、受容体もしくはタンパク質を標的にする治療薬または治療薬の組み合わせによる治療が有効である可能性が高い患者として、あるいは治療薬または治療薬の組み合わせによる治療が有効である可能性が低い患者として識別するよう設定された装置であって、
固形上皮腫瘍癌患者の血液由来サンプルの質量スペクトルを記憶する記憶装置、および
a)質量スペクトルにおいて1つまたはそれ以上の所定の前処理工程を行い、b)1つまたはそれ以上の所定のm/z範囲において該質量スペクトルにおける特徴の積分強度値を得、c)患者を治療薬または治療薬の組み合わせが有効である可能性が高い、または低い患者として識別するために、別の固形腫瘍患者の血液由来サンプルから得たクラス標識スペクトルを含む訓練セットを用いた分類アルゴリズムにb)で得た値を用いるよう設定されたソフトウェアの指示を実行するプロセッサーを含む装置。
【請求項11】
治療薬または治療薬の組合せがHER2を標的としたものである、請求項10の装置。
【請求項12】
治療薬または治療薬の組合せがEGFRおよびVEGFを標的としたものである、請求項10の装置。
【請求項13】
治療薬または治療薬の組み合わせがトラスツズマブを含む、請求項10の装置。
【請求項14】
治療薬がEGFRを標的としたものであり、分類アルゴリズムにより、EGFRを標的とした治療薬との組み合わせにおけるCOX2阻害剤による治療が有効である可能性が高く、EGFRを標的とした治療薬のみによる治療が有効である可能性が低い患者として該患者を識別するクラス標識が得られる、請求項10の装置。
【請求項15】
COX2阻害剤がセレコキシブを含む、請求項14の装置。
【請求項16】
COX2阻害剤がロフェコキシブを含む、請求項14の装置。
【請求項17】
NF−κB阻害剤による治療が有効である可能性が高いとして患者がさらに識別される請求項10の装置。
【請求項18】
所定のm/z範囲が、
5732から5795
5811から5875
6398から6469
11376から11515
11459から11599
11614から11756
11687から11831
11830から11976
12375から12529
23183から23525
23279から23622
65902から67502
からなるm/zの群より選択される、請求項10の装置。
【請求項19】
COX阻害剤およびEGFR阻害剤の組み合わせの治療が癌患者に有効である可能性が高いかどうかを予測する方法であって、
a)該癌患者の血液由来サンプルから質量スペクトルを得;
b)工程a)で得られた質量スペクトルにおいて1つまたはそれ以上の所定の前処理工程を行い;
c)工程b)の質量スペクトルにおける前処理工程を行った後、1つまたはそれ以上の所定のm/z範囲において該スペクトルの選択された特徴の積分値を得;
d)該患者をCOX2阻害剤およびEGFR阻害剤の組み合わせの投与による治療が有効である可能性が高いまたは低い患者として識別するために、別の固形上皮腫瘍患者の血液由来サンプルから得たクラス標識スペクトルを含む訓練セットを用いた分類アルゴリズムに工程c)で得た値を用いる
ことを特徴とする方法。
【請求項20】
所定のm/z範囲が、
5732から5795
5811から5875
6398から6469
11376から11515
11459から11599
11614から11756
11687から11831
11830から11976
12375から12529
23183から23525
23279から23622
65902から67502
からなるm/zの群より選択される、請求項19の方法。
【請求項21】
該方法が臨床検査センターで実施されるものである、請求項1の方法。
【請求項22】
該方法が臨床検査センターで実施されるものである、請求項19の方法。
【請求項23】
COX2阻害剤およびEGFR阻害剤の組み合わせの投与が癌患者に有効である可能性が高いかどうかを予測するよう設定された装置であって、
該癌患者の血液由来サンプルの質量スペクトルを記憶する記憶装置、および
a)質量スペクトルにおいて1つまたはそれ以上の所定の前処理工程を行い、b)工程a)における質量スペクトルの前処理工程を行った後、1つまたはそれ以上の所定のm/z範囲において該質量スペクトルにおける選択された特徴の積分強度値を得、c)患者をCOX2阻害剤およびEGFR阻害剤の組み合わせの投与による治療が有効である可能性が高いまたは低い患者として識別するために、別の固形腫瘍患者の血液由来サンプルから得たクラス標識スペクトルを含む訓練セットを用いた分類アルゴリズムにb)で得た値を用いるよう設定されたソフトウェアの指示を実行するプロセッサーを含む装置。
【請求項1】
固形上皮腫瘍癌患者を、AktまたはERK/JNK/p38またはPKCでの、またはそれらの上流でのMAPK(マイトジェン活性化プロテインキナーゼ)経路またはPKC(プロテインキナーゼC)経路に関わる受容体のアゴニスト、受容体もしくはタンパク質を標的にする治療薬または治療薬の組み合わせによる治療が有効である可能性が高い患者として、あるいは治療薬または治療薬の組み合わせによる治療が有効である可能性が低い患者として識別する方法であって:
a)固形上皮腫瘍癌患者の血液由来サンプルから質量スペクトルを得;
b)a)で得た質量スペクトルにおいて1つまたはそれ以上の所定の前処理工程を行い;
c)b)の質量スペクトルにおける前処理工程を行った後、1つまたはそれ以上の所定のm/z範囲において該スペクトルにおける選択された特徴の積分強度値を得;
d)患者を治療薬または治療薬の組み合わせが有効である可能性が高い、または有効である可能性が低い患者として識別するために、別の固形腫瘍患者の血液由来サンプルから得たクラス標識スペクトルを含む訓練セットを用いた分類アルゴリズムにc)で得た値を用いる
ことを特徴とする方法。
【請求項2】
治療薬または治療薬の組み合わせがEGFRおよびVEGFを標的としたものである、請求項1の方法。
【請求項3】
治療薬または治療薬の組み合わせがHER2を標的としたものである、請求項1の方法。
【請求項4】
治療薬または治療薬の組合せがトラスツズマブを含む、請求項1の方法。
【請求項5】
治療薬または治療薬の組み合わせがEGFRを標的としたものであり、分類アルゴリズムにより、EGFRを標的とした治療薬との組み合わせにおけるCOX2阻害剤による治療が有効である可能性が高く、EGFRを標的とした治療薬のみによる治療が有効である可能性が低い患者として該患者を識別するクラス標識が得られる、請求項1の方法。
【請求項6】
COX2阻害剤がセレコキシブを含む、請求項5の方法。
【請求項7】
COX2阻害剤がロフェコキシブを含む、請求項5の方法。
【請求項8】
クラス標識により該患者がNF−κB阻害剤による治療が有効である可能性が高い患者としてさらに識別される、請求項1の方法。
【請求項9】
1つまたはそれ以上の所定のm/z範囲が、
5732から5795
5811から5875
6398から6469
11376から11515
11459から11599
11614から11756
11687から11831
11830から11976
12375から12529
23183から23525
23279から23622
65902から67502
を含むm/z範囲の群から選択される、請求項1の方法。
【請求項10】
固形上皮腫瘍癌患者を、AktまたはERK/JNK/p38またはPKCでの、またはそれらの上流でのMAPK(マイトジェン活性化プロテインキナーゼ)経路またはPKC(プロテインキナーゼC)経路に関わる受容体のアゴニスト、受容体もしくはタンパク質を標的にする治療薬または治療薬の組み合わせによる治療が有効である可能性が高い患者として、あるいは治療薬または治療薬の組み合わせによる治療が有効である可能性が低い患者として識別するよう設定された装置であって、
固形上皮腫瘍癌患者の血液由来サンプルの質量スペクトルを記憶する記憶装置、および
a)質量スペクトルにおいて1つまたはそれ以上の所定の前処理工程を行い、b)1つまたはそれ以上の所定のm/z範囲において該質量スペクトルにおける特徴の積分強度値を得、c)患者を治療薬または治療薬の組み合わせが有効である可能性が高い、または低い患者として識別するために、別の固形腫瘍患者の血液由来サンプルから得たクラス標識スペクトルを含む訓練セットを用いた分類アルゴリズムにb)で得た値を用いるよう設定されたソフトウェアの指示を実行するプロセッサーを含む装置。
【請求項11】
治療薬または治療薬の組合せがHER2を標的としたものである、請求項10の装置。
【請求項12】
治療薬または治療薬の組合せがEGFRおよびVEGFを標的としたものである、請求項10の装置。
【請求項13】
治療薬または治療薬の組み合わせがトラスツズマブを含む、請求項10の装置。
【請求項14】
治療薬がEGFRを標的としたものであり、分類アルゴリズムにより、EGFRを標的とした治療薬との組み合わせにおけるCOX2阻害剤による治療が有効である可能性が高く、EGFRを標的とした治療薬のみによる治療が有効である可能性が低い患者として該患者を識別するクラス標識が得られる、請求項10の装置。
【請求項15】
COX2阻害剤がセレコキシブを含む、請求項14の装置。
【請求項16】
COX2阻害剤がロフェコキシブを含む、請求項14の装置。
【請求項17】
NF−κB阻害剤による治療が有効である可能性が高いとして患者がさらに識別される請求項10の装置。
【請求項18】
所定のm/z範囲が、
5732から5795
5811から5875
6398から6469
11376から11515
11459から11599
11614から11756
11687から11831
11830から11976
12375から12529
23183から23525
23279から23622
65902から67502
からなるm/zの群より選択される、請求項10の装置。
【請求項19】
COX阻害剤およびEGFR阻害剤の組み合わせの治療が癌患者に有効である可能性が高いかどうかを予測する方法であって、
a)該癌患者の血液由来サンプルから質量スペクトルを得;
b)工程a)で得られた質量スペクトルにおいて1つまたはそれ以上の所定の前処理工程を行い;
c)工程b)の質量スペクトルにおける前処理工程を行った後、1つまたはそれ以上の所定のm/z範囲において該スペクトルの選択された特徴の積分値を得;
d)該患者をCOX2阻害剤およびEGFR阻害剤の組み合わせの投与による治療が有効である可能性が高いまたは低い患者として識別するために、別の固形上皮腫瘍患者の血液由来サンプルから得たクラス標識スペクトルを含む訓練セットを用いた分類アルゴリズムに工程c)で得た値を用いる
ことを特徴とする方法。
【請求項20】
所定のm/z範囲が、
5732から5795
5811から5875
6398から6469
11376から11515
11459から11599
11614から11756
11687から11831
11830から11976
12375から12529
23183から23525
23279から23622
65902から67502
からなるm/zの群より選択される、請求項19の方法。
【請求項21】
該方法が臨床検査センターで実施されるものである、請求項1の方法。
【請求項22】
該方法が臨床検査センターで実施されるものである、請求項19の方法。
【請求項23】
COX2阻害剤およびEGFR阻害剤の組み合わせの投与が癌患者に有効である可能性が高いかどうかを予測するよう設定された装置であって、
該癌患者の血液由来サンプルの質量スペクトルを記憶する記憶装置、および
a)質量スペクトルにおいて1つまたはそれ以上の所定の前処理工程を行い、b)工程a)における質量スペクトルの前処理工程を行った後、1つまたはそれ以上の所定のm/z範囲において該質量スペクトルにおける選択された特徴の積分強度値を得、c)患者をCOX2阻害剤およびEGFR阻害剤の組み合わせの投与による治療が有効である可能性が高いまたは低い患者として識別するために、別の固形腫瘍患者の血液由来サンプルから得たクラス標識スペクトルを含む訓練セットを用いた分類アルゴリズムにb)で得た値を用いるよう設定されたソフトウェアの指示を実行するプロセッサーを含む装置。
【図1】

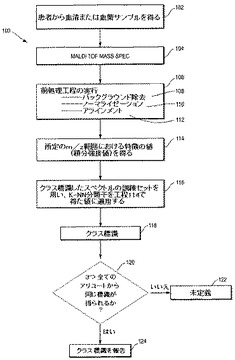
【図2】
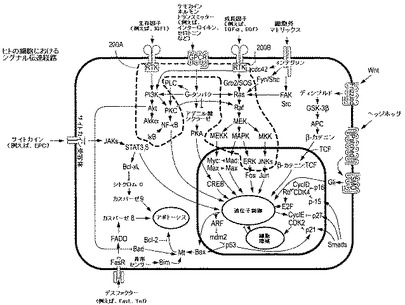
【図3】
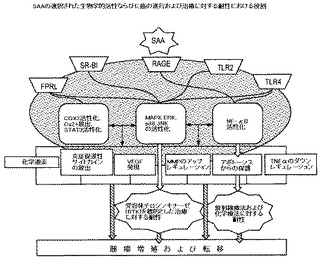
【図4】
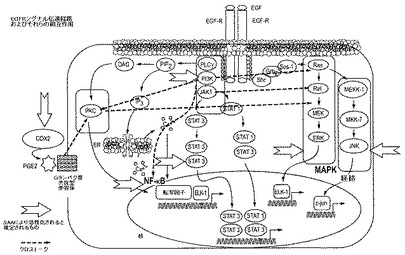
【図5】
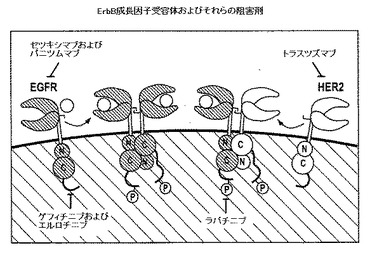
【図6】
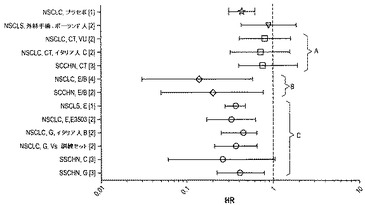
【図7】
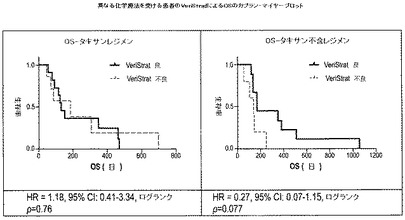
【図8】
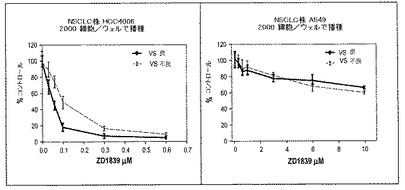
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公表番号】特表2013−520681(P2013−520681A)
【公表日】平成25年6月6日(2013.6.6)
【国際特許分類】
【出願番号】特願2012−555001(P2012−555001)
【出願日】平成23年2月22日(2011.2.22)
【国際出願番号】PCT/US2011/000323
【国際公開番号】WO2011/106084
【国際公開日】平成23年9月1日(2011.9.1)
【出願人】(508293966)バイオデシックス・インコーポレイテッド (3)
【氏名又は名称原語表記】BIODESIX INC
【Fターム(参考)】
【公表日】平成25年6月6日(2013.6.6)
【国際特許分類】
【出願日】平成23年2月22日(2011.2.22)
【国際出願番号】PCT/US2011/000323
【国際公開番号】WO2011/106084
【国際公開日】平成23年9月1日(2011.9.1)
【出願人】(508293966)バイオデシックス・インコーポレイテッド (3)
【氏名又は名称原語表記】BIODESIX INC
【Fターム(参考)】
[ Back to top ]