
質量分析法を用いたペプチドのアミノ酸配列決定方法、該方法に使用されるペプチド誘導体化試薬、及び試薬キット
【課題】本発明の目的は、質量分析法により、簡便且つ効率的にペプチドのアミノ酸配列を完全に決定できる方法、及び当該方法に使用されるペプチド誘導体化試薬及び試薬キットを提供することである。
【解決手段】質量分析法を用いたペプチドのアミノ酸配列を決定する方法において、(1)ペプチドのN末端アミノ基に下記一般式(I)で示す基を結合させることによりペプチドを誘導体化する工程、(2)前記工程(1)で誘導体化されたペプチドに対して、フラグメントイオンの質量スペクトルを取得する工程、及び(3)前記工程(2)で得られた質量スペクトルに基づいて、ペプチドのアミノ酸配列を決定する工程を順次実施する。

【解決手段】質量分析法を用いたペプチドのアミノ酸配列を決定する方法において、(1)ペプチドのN末端アミノ基に下記一般式(I)で示す基を結合させることによりペプチドを誘導体化する工程、(2)前記工程(1)で誘導体化されたペプチドに対して、フラグメントイオンの質量スペクトルを取得する工程、及び(3)前記工程(2)で得られた質量スペクトルに基づいて、ペプチドのアミノ酸配列を決定する工程を順次実施する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、質量分析法により、簡便且つ効率的にペプチドのアミノ酸配列を決定する方法に関する。更に、本発明は、当該方法に使用されるペプチド誘導体化試薬及び試薬キットに関する。
【背景技術】
【0002】
タンパク質及び生理活性ペプチドの機能を解明し、その構造を同定する上で、アミノ酸配列の決定は不可欠である。また、ガン等の疾病のバイオマーカーペプチドの探索においても、ペプチドのアミノ酸配列の決定は不可欠である。従来、ペプチドのアミノ酸配列の決定には、エドマン分解法が一般的に採用されていた。しかしながら、エドマン分解法では、測定感度や効率が低いという問題点に加えて、分析時間やコストの点でも欠点があった。そこで、近年、エドマン分解法に代わるアミノ酸配列の決定法として、質量分析法を用いる方法が注目を浴びている。
【0003】
質量分析法によるアミノ酸配列の決定では、衝突誘起解離等の処理によってペプチドを断片化し、断片化されたペプチドの質量スペクトルを取得し、当該質量スペクトルに基づいてアミノ酸配列が同定される。しかしながら、質量分析法によるアミノ酸配列の決定では、衝突誘起解離によるペプチドの断片化が複雑に、また不完全に起こることが多く、得られる質量スペクトルの解析が不可能又は困難であるという欠点がある。そこで、ペプチドの断片化をより効率的に行う方法として、ペプチドのN末端に特定の化合物を結合させることによってペプチドを誘導化する技術が提案されている(非特許文献1〜3参照)。而るに、従来の方法でペプチドのN末端を誘導化しても、依然としてペプチドの断片化が不十分であり、アミノ酸配列を完全に決定することが困難であり、アミノ酸配列を部分的にしか決定できない。そのため、従来の質量分析法によるアミノ酸配列の決定では、得られた質量スペクトルを、データベースに登録されている公知のペプチドのスペクトルに照合することにより、当該公知のペプチドの配列に該当するか否かを決定するに止まっているのが現状であった。
【0004】
このような従来技術を背景として、質量分析法により、簡便且つ効率的にペプチドのアミノ酸配列を完全に決定する方法の確立が望まれている。
【非特許文献1】Ademczyk, M., et al., Rapid Commun. Mass Spectrom., 13, 1413 (1999)
【非特許文献2】Bao, J., et al., J. Mass Spectrom., 40, 772 (2005)
【非特許文献3】Miyagi, M., et al., Rapid Commun. Mass Spectrom., 12, 603 (1998)
【発明の開示】
【発明が解決しようとする課題】
【0005】
本発明は、上記従来技術の課題を解決することを目的とする。具体的には、本発明は、質量分析法により、簡便且つ効率的にペプチドのアミノ酸配列を完全に決定できる方法、及び当該方法に使用されるペプチド誘導体化試薬及び試薬キットを提供することを目的とする。
【課題を解決するための手段】
【0006】
本発明者らは、上記課題を解決すべく鋭意検討したところ、解析対象となるペプチドのN末端のアミノ基を下記一般式(I)で示す基を結合させることによりペプチドを誘導体化しておくことによって、質量分析法におけるペプチドの断片化をより完全に実施できるので、解析を容易に行える質量スペクトルを取得でき、未知のペプチドのアミノ酸配列を完全に決定可能であることを見出した。本発明は、かかる知見に基づいて更に検討を重ねることによって完成したものである。
【0007】
即ち、本発明は、下記に掲げる態様の発明を提供する。
項1. 質量分析法を用いたペプチドのアミノ酸配列を決定する方法であって、
(1)ペプチドのN末端アミノ基に下記一般式(I)で示す基を結合させることによりペプチドを誘導体化する工程、
【0008】
【化1】
【0009】
[式(I)中、mは0又は1の整数を示し、n1及びn2は、同一又は異なって0〜5の整数を示す。また、式(I)中、Xは下記の基を示す。]
【0010】
【化2】
【0011】
(2)前記工程(1)で誘導体化されたペプチドに対して、フラグメントイオンの質量スペクトルを取得する工程、及び
(3)前記工程(2)で得られた質量スペクトルに基づいて、ペプチドのアミノ酸配列を決定する工程
を含むことを特徴とする、アミノ酸配列の決定方法。
項2. 前記工程(1)が、下記一般式(II)で示す化合物とペプチドを縮合反応させることにより、ペプチドを誘導体化する工程である、項1に記載のアミノ酸配列の決定方法。
【0012】
【化3】
【0013】
[式(II)中、m、n1及びn2は、前記と同じ。]
項3. 前記工程(2)において、エレクトロスプレーイオン化法によりペプチドのイオン化を行う、項1又は2に記載のアミノ酸配列の決定方法。
項4. 前記工程(2)において、衝突誘起解離法によりペプチドの断片化を行う、項1乃至3のいずれかに記載のアミノ酸配列の決定方法。
項5. 質量分析法を用いてペプチドのアミノ酸配列を決定するために使用されるペプチド誘導体化試薬であって、ペプチドのN末端アミノ基に対して下記一般式(I)で示す基を結合させることによりペプチドを誘導体化できる化合物を含有することを特徴とする、ペプチド誘導体化試薬。
【0014】
【化4】
【0015】
[式(I)中、m、n1、n2及びXは前記と同じ。]
項6. 項5に記載のペプチド誘導体化試薬を含有する、質量分析法を用いてペプチドのアミノ酸配列を決定するための試薬キット。
【発明の効果】
【0016】
本発明のアミノ酸配列の決定方法によれば、質量分析法により、ペプチドのアミノ酸配列を解析する上で指標となるbイオン系列及びyイオン系列を詳細に得ることができるので、未知のペプチドであってもアミノ酸配列を完全に決定することが可能である。
【0017】
本発明は、高感度・高効率な未知タンパク質・ペプチドのアミノ酸配列決定法を提供するものであり、アミノ酸配列データベースの乏しいマイナー生物種から単離されることの多い生理活性ペプチドの単離同定研究や、迅速且つ好感度な質量分析計の利点を生かす必要のあるガン等の疾病バイオマーカーペプチドの探索研究等の分野で有用である。
【発明を実施するための最良の形態】
【0018】
1.アミノ酸配列の決定方法
本発明のアミノ酸配列の決定方法において、解析対象となるペプチドは、通常、分子量が2,000以下、好ましくは500〜1000のものが使用される。また、上記分子量よりも大きいペプチド及びタンパク質に対しては、上記分子量になるように断片化した後に、本発明に用いることもできる。ここで、ペプチド及びタンパク質の断片化方法としては、広く知られている手法を採用すればよく、例えば、BrCNを用いた化学的切断方法;トリプシン、キモトリプシン、エンドプロテイナーゼLys-C等のプロテアーゼを用いた酵素切断方法等が例示される。
【0019】
以下、本発明のアミノ酸配列の決定方法は、ペプチドのN末端を誘導体化する工程(以下、工程(1)と表記する)、誘導体化されたペプチドの質量分析を行う工程(以下、工程(2)と表記する)、及び質量スペクトルからアミノ酸配列を決定する工程(以下、工程(3)と表記する)を含むものである。以下、本発明のアミノ酸配列の決定方法について、工程毎に詳述する。
工程(1)
本工程(1)では、ペプチドのN末端アミノ基に下記一般式(I)で示す基を結合させることにより、ペプチドを誘導体化する。
【0020】
【化5】
【0021】
式(I)中、mは0又は1であり、好ましくは0である。また、式(I)中、n1及びn2は、同一又は異なって、0〜5の整数、好ましくは0〜2の整数、更に好ましくは0又は1、特に好ましくは0を示す。式(I)で示す基の中でも、m、n1及びn2が、全て0である場合には、質量分析法におけるペプチドの断片化を一層効率的に行うことができる。
【0022】
式(I)中、Xは、下記の基を示す。
【0023】
【化6】
【0024】
上記Xの基の中でも、基−CO−が好適である。
【0025】
また、一般式(I)で示す基において、上記Aで示される基は、上記Bで示される基に対する相対位置関係で、オルト位、メタ位又はパラ位のいずれに存在していてもよいが、好ましくはパラ位である。
【0026】
上記一般式(I)で示す基において、質量分析法におけるペプチドの断片化をより効果的に行うという観点から、好ましくは下記一般式(I')で示される基、特に好ましくは下記一般式(I'')で示される基が例示される。
【0027】
【化7】
【0028】
【化8】
【0029】
本工程(1)では、ペプチドのN末端アミノ基に対して一般式(I)で示す基を結合させることによりペプチドを誘導体化できる化合物(以下、誘導体化化合物と表記する)を、ペプチドと反応させることにより実施される。本工程(1)で使用される誘導体化化合物としては、一般式(I)で示す基をペプチドのN末端アミノ基に対して結合させ得ることを限度として特に制限されないが、好適な一例として、下記の一般式(II)〜(IV)で示す化合物が挙げられる。当該一般式(II)で示す化合物によれば、当該化合物のカルボキシル基とペプチドのN末端のアミノ基とを通常のペプチド合成で用いられる簡便な縮合反応により、ペプチドのN末端のアミノ基に一般式(I)で示す基を結合させ、誘導体化できるという利点がある。
【0030】
【化9】
【0031】
【化10】
【0032】
【化11】
【0033】
式(II)〜(IV)中、m、n1及びn2は、前記と同様である。なお、一般式(II)〜(IV)で示す化合物は、公知化合物、又は公知化合物から容易に導かれる化合物である。
【0034】
また、上記誘導体化化合物として、一般式(I)で示す基が活性エステル化された化合物、例えば、一般式(I)で示す基にスクシンイミドが結合した化合物、一般式(I)で示す基にパラニトロフェノールが結合した化合物等を使用することもできる。
【0035】
本工程(1)において、ペプチドのN末端アミノ基に対して一般式(I)で示す基を結合させて、誘導体化させる方法については、誘導体化化合物の種類等に応じて適宜設定される。
【0036】
例えば、誘導体化化合物として一般式(II)で示す化合物を使用する場合であれば、次の条件が例示される。ペプチド1モルに対して、誘導体化化合物を通常5〜100モル、好ましくは10モル使用して、溶媒中で縮合反応を行う。溶媒としては、縮合反応を阻害しないものであればよく、例えば、ジメチルホルムアミド(DMF)、ジメチルスルホキシド(DMSO)、アセトニトリル等の非プロトン性極性溶媒及びこれらの混合溶媒等が使用される。これらの溶媒は、水が含まれていてもよい。また、当該反応では、4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride等の脱水縮合剤を用いることが望ましい。当該脱水縮合剤の添加量としては、ペプチド1モルに対して、例えば5〜100モル、好ましくは10モルが例示される。また、ペプチドと誘導体化化合物との反応は、通常10〜40℃、好ましくは25℃で、2〜12時間、好ましくは12時間攪拌を続けることにより行われる。
【0037】
また、例えば、誘導体化化合物として一般式(III)及び(IV)で示す化合物を使用する場合であれば、次の条件が例示される。ペプチド1モルに対して、誘導体化化合物を通常5〜100モル、好ましくは10モル使用して、溶媒中で縮合反応を行う。溶媒としては、例えば、炭酸水素ナトリウム緩衝液(pH 9.5)とジメチルホルムアミド(DMF)、ジメチルスルホキシド(DMSO)、アセトニトリル等の非プロトン性極性溶媒との混合溶媒等が使用される。また、ペプチドと誘導体化化合物との反応は、通常10〜40℃、好ましくは25℃で、2〜12時間、好ましくは12時間攪拌を続けることにより行われる。
【0038】
また、例えば、誘導体化化合物として一般式(I)で示す基が活性エステル化された化合物を使用する場合であれば、次の条件が例示される。ペプチド1モルに対して、一般式(I)で示す基が活性エステル化された化合物を通常5〜100モル、好ましくは10モルを添加して、通常10〜40℃、好ましくは25℃で、2〜12時間、好ましくは12時間攪拌する。当該反応は、水、リン酸ナトリウム緩衝液、アセトニトリル水溶液等の溶媒中で行うことができる。
【0039】
工程(2)
本工程(2)では、前記(1)工程で誘導体化されたペプチドに対して、フラグメントイオンの質量スペクトルを取得する。
【0040】
本工程(2)において、質量スペクトルの取得に使用される質量分析には、例えば、磁場偏向型、四重極型、イオントラップ型、飛行時間型、フーリエ変換イオンサイクロトロン共鳴型、及びこれらのハイブリッド型等のタイプの質量分析計を採用することができる。これらの中でも、イオントラップ−飛行時間ハイブリッド型の質量分析計は、特定の質量のイオン分子を選別した後に断片化し、生成したイオン分子を質量分析する測定を、同じ起源の分子に対して連続して行うことが可能で、且つ各々の測定精度が高いという利点があり、本発明に好適に使用される。
【0041】
本工程(2)の質量スペクトルの取得において、前記(1)工程で誘導体化されたペプチドをイオン化する方法については、例えば、エレクトロスプレーイオン化法(ESI法)、マトリックス支援レーザー脱離イオン化法(MALDI法)、高速原子衝突法(FAB法)等が挙げられる。これらの中でも、ESI法は、溶液状態の試料をそのままイオン化する方法であるが故、一般的なペプチドの分離精製手段である高速液体クロマトグラフィーに直結できるという利点があり、本工程(2)において好ましいイオン化方法である。
【0042】
また、本工程(2)の質量スペクトルの取得において、ペプチドをフラグメント化(断片化)する方法についても、特に制限されないが、例えば、衝突誘起解離法(CID法)、ポストソース分解法(PSD法)、インソース分解法(ISD法)、電子捕獲解離法(ECD法)、電子移動解離法(ETD法)等が挙げられる。これらのフラグメント化方法の内、CID法は最も汎用され、解離効率が高いという利点があり望ましい。
【0043】
本工程(2)において、前記(1)工程で誘導体化されたペプチドに対して、MS分析、又はMSn分析を行うことにより、アミノ酸配列の解析の指標になるbイオン及びyイオンが顕著に増加して検出され、アミノ酸配列を容易に解析できる質量スペクトルが取得される。ここで、MSn分析とはn乗のMS分析を意味し、具体的には、MSn-1分析で得られたイオン分子を断片化した後に、更に特定のイオン分子をプリカーサーとして分析を行うことを意味する。例えばMS2分析はMS/MS分析に相当する。本工程(2)において、MS分析の乗数については、分析対象のペプチドの種類等に応じて適宜設定すればよいが、通常MS2分析〜MS4分析、好ましくはMS2分析〜MS3分析が採用される。
【0044】
工程(3)
本工程(3)では、前記(2)工程で得られた質量スペクトルに基づいて、ペプチドのアミノ酸配列を決定する。
【0045】
前記(2)工程で得られた質量スペクトルには、アミノ酸配列の解析の指標になるbイオン系列(ペプチドのN末端を含むフラグメント)及びyイオン系列(ペプチドのC末端を含むフラグメント)が高精度で検出されている。当該質量スペクトル上で、各々のbイオン及びyイオンを質量順に特定し、その質量差に基づいて該当するアミノ酸及びその配列順序を同定することにより、ペプチドのアミノ酸配列が決定される。
【0046】
2.ペプチド誘導体化試薬、及び試薬キット
更に、本発明は、上記のアミノ酸配列の決定方法を実施するためのペプチド誘導体化試薬を提供する。当該ペプチド誘導体化試薬は、ペプチドのN末端アミノ基に対して一般式(I)で示す基を結合させることによりペプチドを誘導体化できる化合物(誘導体化化合物)を含むことを特徴とするものである。当該誘導体化化合物については、前述する通りである。
【0047】
また、本発明は、上記のアミノ酸配列の決定方法を実施するための試薬キットを提供する。当該試薬キットは、上記ペプチド誘導体化試薬を含むことを特徴とするものである。また、当該試薬キットは、更に、上記アミノ酸配列の決定方法に使用される試薬や上記アミノ酸配列の決定方法のプロトコールを記した手順書が含まれていてもよい。
【実施例】
【0048】
以下、実施例を挙げて本発明を説明するが、本発明はこれらの実施例に限定されるものではない。
1.アミジノ安息香酸の調製
アミジノベンズアミド・塩酸塩(1.5 g)に6N HCl(45 ml)と12N 酢酸(8 ml)を加え、110℃で6時間撹拌した。室温に戻すと白い結晶が析出した。これを濾集し、H2Oで洗浄した後、減圧乾燥させ、白色固体のアミジノ安息香酸・塩酸塩(0.45 g、収率30%)を得た。同定はNMR分析及びLC-MS分析により行った。
1H-NMR(DMSO-d6)δ 7.91 (d, J=8.4, 2H), 8.12 (d, J=8.3, 2H), 9.18 (s, 2H), 9.48 (s, 2H), 13.51 (s, 1H), ESI-MS m/z calcd for [M+H]+ : 165.1; found: 165.1
【0049】
2.アミジノ安息香酸のペプチドへの導入法1
ペプチド1(AAGLQIA:配列番号1)(2 μmol)のDMSO溶液に、アミジノ安息香酸(10 eq)及び脱水縮合剤4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride(DMT-MM、国産化学社製、10 eq)のDMSO溶液を加え、終夜撹拌した。反応終了後、逆相HPLC(溶離液: H2O/アセトニトリル(0.1%TFAを含む)、カラム:Vydac C18, 4.6×250mm)を用いて分取・精製し、アミジノ安息香酸誘導体化ペプチド1を得た。
ESI-MS m/z calcd for [M+H]+: 789.4; found: 789.4.
【0050】
3.アミジノ安息香酸のペプチドへの導入法2
アミジノ安息香酸(2 g)の50% H2O/アセトニトリル溶液に1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride(10eq)とN-hydroxysuccinimide(10 eq)を加え、終夜撹拌した。反応終了後、逆相HPLC(溶離液:H2O/アセトニトリル(0.1%TFAを含む)、カラム:Vydac C18, 4.6×250mm)を用いて分取・精製し、アミジノ安息香酸スクシンイミドエステル体を得た。同定はNMR分析およびLC-MS分析により行った。
1H-NMR(DMSO-d6)δ 2.92 (s, 4H), 8.04 (d, J=8.5, 2H), 8.31 (d, J=8.5, 2H), 9.41 (s, 2H), 9.57 (s, 2H), ESI-MS m/z calcd for [M+H]+ : 262.1; found: 262.1.
次いで、50 mMリン酸ナトリウム緩衝液(pH8.5)に溶解したペプチド1(2 μmol)に、H2Oに溶解したアミジノ安息香酸スクシンイミドエステル体(10 eq)を加え、終夜攪拌した。反応終了後、逆相HPLC(溶離液:H2O/アセトニトリル(0.1%TFAを含む)、カラム:Vydac C18, 4.6×250mm)を用いて分取・精製し、アミジノ安息香酸誘導体化ペプチド1を得た。
ESI-MS m/z calcd for [M+H]+: 789.4; found: 789.4.
【0051】
4.アミジノ安息香酸導入ペプチドのアミノ酸配列解析
上記「2.アミジノ安息香酸のペプチドへの導入法1」で得られたアミジノ安息香酸誘導体化ペプチドを、イオントラップ-飛行時間ハイブリッド型質量分析計(LCMS-IT-TOF、島津製作所社製)を用いたMSn分析に供した。分析はflow-injection法又はnanospray法(溶媒:50% H2O/アセトニトリル、0.1%ギ酸を含む)で行ない、0.5 μg(0.3-0.8 nmol) の試料を質量分析計に導入した。また、比較のためにアミジノ安息香酸によって誘導体化していないペプチドの測定も行なった。
【0052】
5.ペプチド2〜7のアミノ酸配列解析
以下に示すペプチド2〜7についても、上記「2.アミジノ安息香酸のペプチドへの導入法1」と同様の方法によって誘導体化を行い、次いで上記「4.アミジノ安息香酸導入ペプチドのアミノ酸配列解析」と同様の方法でMSn分析を行った。
ペプチド2(DRVYIHPFHL:配列番号2)
ペプチド3(RPPGFSPFR:配列番号3)
ペプチド4(SYANGFSATSASL:配列番号4)
ペプチド5(ATQQTAAYKTLVS:配列番号5)
ペプチド6(TDVNGDGRHAL:配列番号6)
ペプチド7(DYPVDIYYLMD:配列番号7)
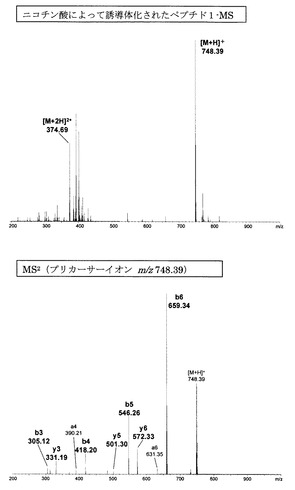
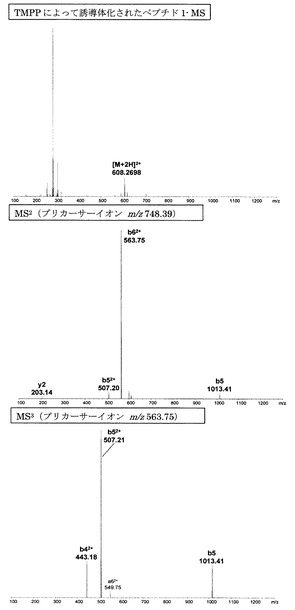
また、従来、ペプチドのフラグメンテーションを改善することが報告されているニコチン酸及びN-tris(2,4,6-trimethoxyphenyl)phosphine-acetic acid(TMPP)を用いてペプチド1の誘導体化を行い、そのMSnスペクトルを比較した。ニコチン酸及びTMPPのペプチドへの導入は文献記載の方法に従って実施した。なお、当該文献は以下の通りである:
Ademczyk, M., et al., Rapid Commun. Mass Spectrom., 13, 1413 (1999)
Cohen, B., et al., Biochemistry, 36, 9035 (1997)
Miyagi, M., et al., Rapid Commun. Mass Spectrom., 12, 603 (1998)
【0053】
6.結果
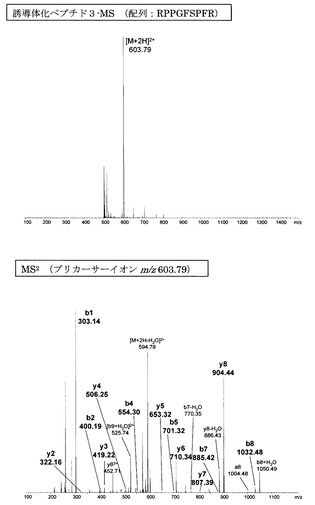
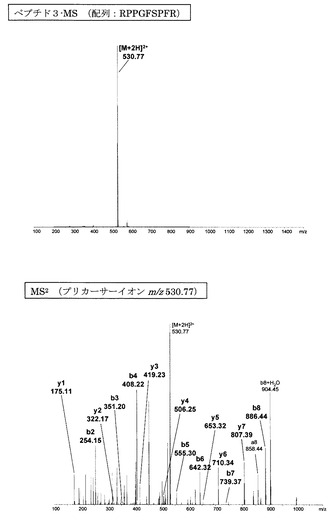
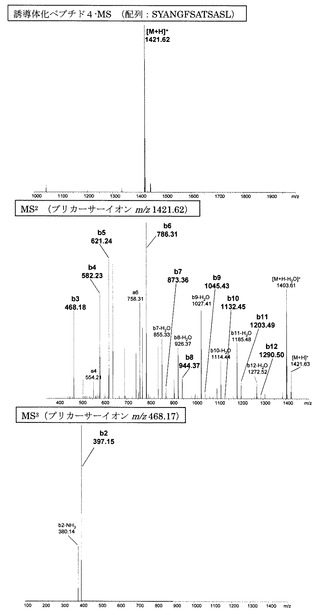
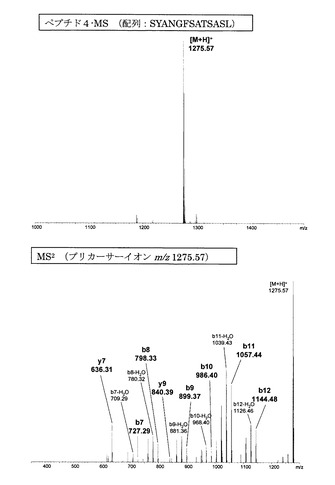
図1〜14にアミジノ安息香酸で誘導体化したペプチド、及び誘導体化していないペプチドのMS、MS2、MS3スペクトルを示す。また、スペクトル中に検出されたb系列およびy系列イオンを、表1に示す。誘導体化していないペプチドにおいては、いずれも配列解析に重要となるb系列およびy系列イオンの連続した生成が少なかったため、アミノ酸配列の決定は困難であった。一方、アミジノ安息香酸によって誘導体化したペプチドにおいては、いずれにおいても、ほぼ全てのb系列イオンと一部のy系列イオンが連続的に検出され、配列の決定が誘導体化前に比べて容易に行えることが明らかとなった。
【0054】
【表1】
【0055】
更に、ニコチン酸及びTMPPによって誘導体化されたペプチドとの比較においても(図15、16及び表2参照)、アミジノ安息香酸による誘導体化の方が連続したb系列イオンの生成が観測されたことから、既知誘導体化試薬と比べてもフラグメント化を促進する効果が高いことが示された。
【0056】
【表2】
【0057】
以上の結果から、アミジノ安息香酸による誘導体化は、質量分析法によるペプチドのアミノ酸配列解析に有用であることが明らかとなった。
【図面の簡単な説明】
【0058】
【図1】アミジノ安息香酸誘導体化ペプチド1のMS分析、MS2分析、及びMS3分析により得られた質量スペクトルを示す図である。
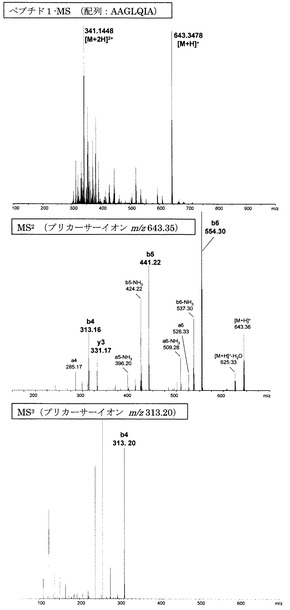
【図2】誘導体化していないペプチド1のMS分析、MS2分析、及びMS3分析により得られた質量スペクトルを示す図である。
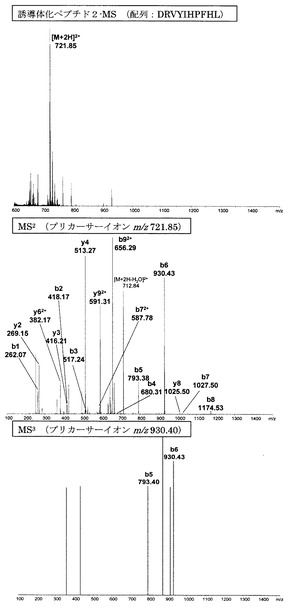
【図3】アミジノ安息香酸誘導体化ペプチド2のMS分析、MS2分析、及びMS3分析により得られた質量スペクトルを示す図である。
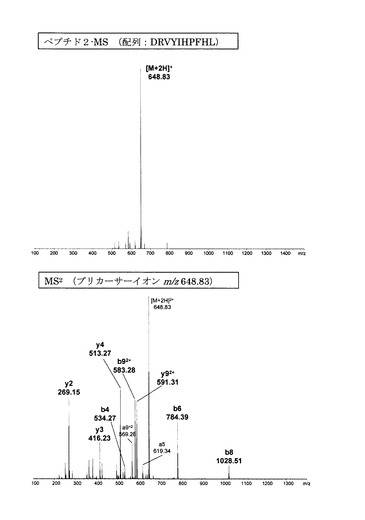
【図4】誘導体化していないペプチド2のMS分析及びMS2分析により得られた質量スペクトルを示す図である。
【図5】アミジノ安息香酸誘導体化ペプチド3のMS分析及びMS2分析により得られた質量スペクトルを示す図である。
【図6】誘導体化していないペプチド3のMS分析及びMS2分析により得られた質量スペクトルを示す図である。
【図7】アミジノ安息香酸誘導体化ペプチド4のMS分析、MS2分析、及びMS3分析により得られた質量スペクトルを示す図である。
【図8】誘導体化していないペプチド4のMS分析及びMS2分析により得られた質量スペクトルを示す図である。
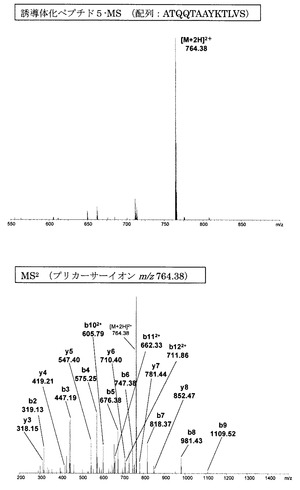
【図9】アミジノ安息香酸誘導体化ペプチド5のMS分析及びMS2分析により得られた質量スペクトルを示す図である。
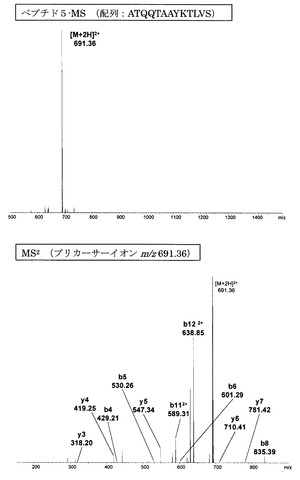
【図10】誘導体化していないペプチド5のMS分析及びMS2分析により得られた質量スペクトルを示す図である。
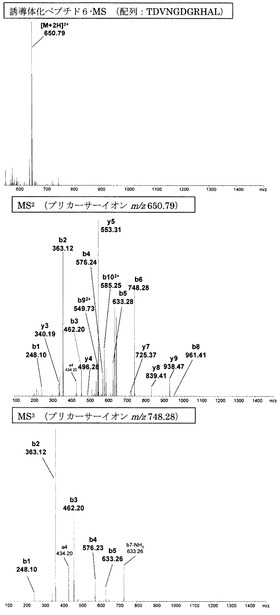
【図11】アミジノ安息香酸誘導体化ペプチド6のMS分析、MS2分析、及びMS3分析により得られた質量スペクトルを示す図である。
【図12】誘導体化していないペプチド6のMS分析及びMS2分析により得られた質量スペクトルを示す図である。
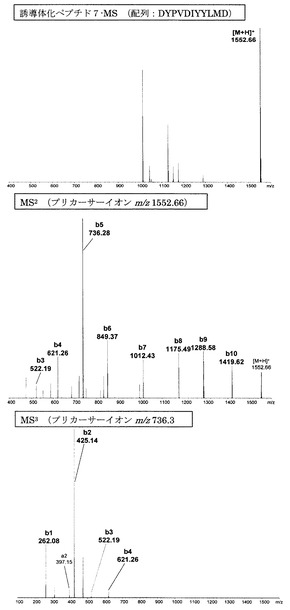
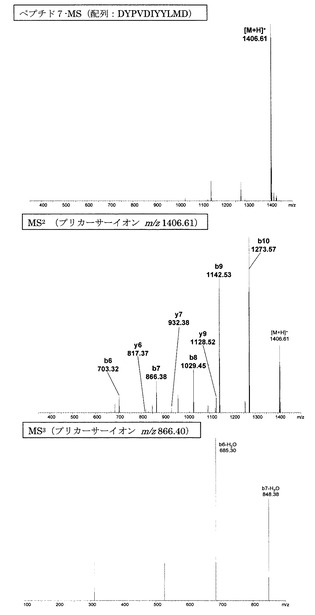
【図13】アミジノ安息香酸誘導体化ペプチド7のMS分析、MS2分析、及びMS3分析により得られた質量スペクトルを示す図である。
【図14】誘導体化していないペプチド7のMS分析、MS2分析、及びMS3分析により得られた質量スペクトルを示す図である。
【図15】ニコチン酸によって誘導体化されたペプチド1のMS分析及びMS2分析により得られた質量スペクトルを示す図である。
【図16】TMPPによって誘導体化されたペプチド1のMS分析、MS2分析、及びMS3分析により得られた質量スペクトルを示す図である。
【技術分野】
【0001】
本発明は、質量分析法により、簡便且つ効率的にペプチドのアミノ酸配列を決定する方法に関する。更に、本発明は、当該方法に使用されるペプチド誘導体化試薬及び試薬キットに関する。
【背景技術】
【0002】
タンパク質及び生理活性ペプチドの機能を解明し、その構造を同定する上で、アミノ酸配列の決定は不可欠である。また、ガン等の疾病のバイオマーカーペプチドの探索においても、ペプチドのアミノ酸配列の決定は不可欠である。従来、ペプチドのアミノ酸配列の決定には、エドマン分解法が一般的に採用されていた。しかしながら、エドマン分解法では、測定感度や効率が低いという問題点に加えて、分析時間やコストの点でも欠点があった。そこで、近年、エドマン分解法に代わるアミノ酸配列の決定法として、質量分析法を用いる方法が注目を浴びている。
【0003】
質量分析法によるアミノ酸配列の決定では、衝突誘起解離等の処理によってペプチドを断片化し、断片化されたペプチドの質量スペクトルを取得し、当該質量スペクトルに基づいてアミノ酸配列が同定される。しかしながら、質量分析法によるアミノ酸配列の決定では、衝突誘起解離によるペプチドの断片化が複雑に、また不完全に起こることが多く、得られる質量スペクトルの解析が不可能又は困難であるという欠点がある。そこで、ペプチドの断片化をより効率的に行う方法として、ペプチドのN末端に特定の化合物を結合させることによってペプチドを誘導化する技術が提案されている(非特許文献1〜3参照)。而るに、従来の方法でペプチドのN末端を誘導化しても、依然としてペプチドの断片化が不十分であり、アミノ酸配列を完全に決定することが困難であり、アミノ酸配列を部分的にしか決定できない。そのため、従来の質量分析法によるアミノ酸配列の決定では、得られた質量スペクトルを、データベースに登録されている公知のペプチドのスペクトルに照合することにより、当該公知のペプチドの配列に該当するか否かを決定するに止まっているのが現状であった。
【0004】
このような従来技術を背景として、質量分析法により、簡便且つ効率的にペプチドのアミノ酸配列を完全に決定する方法の確立が望まれている。
【非特許文献1】Ademczyk, M., et al., Rapid Commun. Mass Spectrom., 13, 1413 (1999)
【非特許文献2】Bao, J., et al., J. Mass Spectrom., 40, 772 (2005)
【非特許文献3】Miyagi, M., et al., Rapid Commun. Mass Spectrom., 12, 603 (1998)
【発明の開示】
【発明が解決しようとする課題】
【0005】
本発明は、上記従来技術の課題を解決することを目的とする。具体的には、本発明は、質量分析法により、簡便且つ効率的にペプチドのアミノ酸配列を完全に決定できる方法、及び当該方法に使用されるペプチド誘導体化試薬及び試薬キットを提供することを目的とする。
【課題を解決するための手段】
【0006】
本発明者らは、上記課題を解決すべく鋭意検討したところ、解析対象となるペプチドのN末端のアミノ基を下記一般式(I)で示す基を結合させることによりペプチドを誘導体化しておくことによって、質量分析法におけるペプチドの断片化をより完全に実施できるので、解析を容易に行える質量スペクトルを取得でき、未知のペプチドのアミノ酸配列を完全に決定可能であることを見出した。本発明は、かかる知見に基づいて更に検討を重ねることによって完成したものである。
【0007】
即ち、本発明は、下記に掲げる態様の発明を提供する。
項1. 質量分析法を用いたペプチドのアミノ酸配列を決定する方法であって、
(1)ペプチドのN末端アミノ基に下記一般式(I)で示す基を結合させることによりペプチドを誘導体化する工程、
【0008】
【化1】
【0009】
[式(I)中、mは0又は1の整数を示し、n1及びn2は、同一又は異なって0〜5の整数を示す。また、式(I)中、Xは下記の基を示す。]
【0010】
【化2】
【0011】
(2)前記工程(1)で誘導体化されたペプチドに対して、フラグメントイオンの質量スペクトルを取得する工程、及び
(3)前記工程(2)で得られた質量スペクトルに基づいて、ペプチドのアミノ酸配列を決定する工程
を含むことを特徴とする、アミノ酸配列の決定方法。
項2. 前記工程(1)が、下記一般式(II)で示す化合物とペプチドを縮合反応させることにより、ペプチドを誘導体化する工程である、項1に記載のアミノ酸配列の決定方法。
【0012】
【化3】
【0013】
[式(II)中、m、n1及びn2は、前記と同じ。]
項3. 前記工程(2)において、エレクトロスプレーイオン化法によりペプチドのイオン化を行う、項1又は2に記載のアミノ酸配列の決定方法。
項4. 前記工程(2)において、衝突誘起解離法によりペプチドの断片化を行う、項1乃至3のいずれかに記載のアミノ酸配列の決定方法。
項5. 質量分析法を用いてペプチドのアミノ酸配列を決定するために使用されるペプチド誘導体化試薬であって、ペプチドのN末端アミノ基に対して下記一般式(I)で示す基を結合させることによりペプチドを誘導体化できる化合物を含有することを特徴とする、ペプチド誘導体化試薬。
【0014】
【化4】
【0015】
[式(I)中、m、n1、n2及びXは前記と同じ。]
項6. 項5に記載のペプチド誘導体化試薬を含有する、質量分析法を用いてペプチドのアミノ酸配列を決定するための試薬キット。
【発明の効果】
【0016】
本発明のアミノ酸配列の決定方法によれば、質量分析法により、ペプチドのアミノ酸配列を解析する上で指標となるbイオン系列及びyイオン系列を詳細に得ることができるので、未知のペプチドであってもアミノ酸配列を完全に決定することが可能である。
【0017】
本発明は、高感度・高効率な未知タンパク質・ペプチドのアミノ酸配列決定法を提供するものであり、アミノ酸配列データベースの乏しいマイナー生物種から単離されることの多い生理活性ペプチドの単離同定研究や、迅速且つ好感度な質量分析計の利点を生かす必要のあるガン等の疾病バイオマーカーペプチドの探索研究等の分野で有用である。
【発明を実施するための最良の形態】
【0018】
1.アミノ酸配列の決定方法
本発明のアミノ酸配列の決定方法において、解析対象となるペプチドは、通常、分子量が2,000以下、好ましくは500〜1000のものが使用される。また、上記分子量よりも大きいペプチド及びタンパク質に対しては、上記分子量になるように断片化した後に、本発明に用いることもできる。ここで、ペプチド及びタンパク質の断片化方法としては、広く知られている手法を採用すればよく、例えば、BrCNを用いた化学的切断方法;トリプシン、キモトリプシン、エンドプロテイナーゼLys-C等のプロテアーゼを用いた酵素切断方法等が例示される。
【0019】
以下、本発明のアミノ酸配列の決定方法は、ペプチドのN末端を誘導体化する工程(以下、工程(1)と表記する)、誘導体化されたペプチドの質量分析を行う工程(以下、工程(2)と表記する)、及び質量スペクトルからアミノ酸配列を決定する工程(以下、工程(3)と表記する)を含むものである。以下、本発明のアミノ酸配列の決定方法について、工程毎に詳述する。
工程(1)
本工程(1)では、ペプチドのN末端アミノ基に下記一般式(I)で示す基を結合させることにより、ペプチドを誘導体化する。
【0020】
【化5】
【0021】
式(I)中、mは0又は1であり、好ましくは0である。また、式(I)中、n1及びn2は、同一又は異なって、0〜5の整数、好ましくは0〜2の整数、更に好ましくは0又は1、特に好ましくは0を示す。式(I)で示す基の中でも、m、n1及びn2が、全て0である場合には、質量分析法におけるペプチドの断片化を一層効率的に行うことができる。
【0022】
式(I)中、Xは、下記の基を示す。
【0023】
【化6】
【0024】
上記Xの基の中でも、基−CO−が好適である。
【0025】
また、一般式(I)で示す基において、上記Aで示される基は、上記Bで示される基に対する相対位置関係で、オルト位、メタ位又はパラ位のいずれに存在していてもよいが、好ましくはパラ位である。
【0026】
上記一般式(I)で示す基において、質量分析法におけるペプチドの断片化をより効果的に行うという観点から、好ましくは下記一般式(I')で示される基、特に好ましくは下記一般式(I'')で示される基が例示される。
【0027】
【化7】
【0028】
【化8】
【0029】
本工程(1)では、ペプチドのN末端アミノ基に対して一般式(I)で示す基を結合させることによりペプチドを誘導体化できる化合物(以下、誘導体化化合物と表記する)を、ペプチドと反応させることにより実施される。本工程(1)で使用される誘導体化化合物としては、一般式(I)で示す基をペプチドのN末端アミノ基に対して結合させ得ることを限度として特に制限されないが、好適な一例として、下記の一般式(II)〜(IV)で示す化合物が挙げられる。当該一般式(II)で示す化合物によれば、当該化合物のカルボキシル基とペプチドのN末端のアミノ基とを通常のペプチド合成で用いられる簡便な縮合反応により、ペプチドのN末端のアミノ基に一般式(I)で示す基を結合させ、誘導体化できるという利点がある。
【0030】
【化9】
【0031】
【化10】
【0032】
【化11】
【0033】
式(II)〜(IV)中、m、n1及びn2は、前記と同様である。なお、一般式(II)〜(IV)で示す化合物は、公知化合物、又は公知化合物から容易に導かれる化合物である。
【0034】
また、上記誘導体化化合物として、一般式(I)で示す基が活性エステル化された化合物、例えば、一般式(I)で示す基にスクシンイミドが結合した化合物、一般式(I)で示す基にパラニトロフェノールが結合した化合物等を使用することもできる。
【0035】
本工程(1)において、ペプチドのN末端アミノ基に対して一般式(I)で示す基を結合させて、誘導体化させる方法については、誘導体化化合物の種類等に応じて適宜設定される。
【0036】
例えば、誘導体化化合物として一般式(II)で示す化合物を使用する場合であれば、次の条件が例示される。ペプチド1モルに対して、誘導体化化合物を通常5〜100モル、好ましくは10モル使用して、溶媒中で縮合反応を行う。溶媒としては、縮合反応を阻害しないものであればよく、例えば、ジメチルホルムアミド(DMF)、ジメチルスルホキシド(DMSO)、アセトニトリル等の非プロトン性極性溶媒及びこれらの混合溶媒等が使用される。これらの溶媒は、水が含まれていてもよい。また、当該反応では、4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride等の脱水縮合剤を用いることが望ましい。当該脱水縮合剤の添加量としては、ペプチド1モルに対して、例えば5〜100モル、好ましくは10モルが例示される。また、ペプチドと誘導体化化合物との反応は、通常10〜40℃、好ましくは25℃で、2〜12時間、好ましくは12時間攪拌を続けることにより行われる。
【0037】
また、例えば、誘導体化化合物として一般式(III)及び(IV)で示す化合物を使用する場合であれば、次の条件が例示される。ペプチド1モルに対して、誘導体化化合物を通常5〜100モル、好ましくは10モル使用して、溶媒中で縮合反応を行う。溶媒としては、例えば、炭酸水素ナトリウム緩衝液(pH 9.5)とジメチルホルムアミド(DMF)、ジメチルスルホキシド(DMSO)、アセトニトリル等の非プロトン性極性溶媒との混合溶媒等が使用される。また、ペプチドと誘導体化化合物との反応は、通常10〜40℃、好ましくは25℃で、2〜12時間、好ましくは12時間攪拌を続けることにより行われる。
【0038】
また、例えば、誘導体化化合物として一般式(I)で示す基が活性エステル化された化合物を使用する場合であれば、次の条件が例示される。ペプチド1モルに対して、一般式(I)で示す基が活性エステル化された化合物を通常5〜100モル、好ましくは10モルを添加して、通常10〜40℃、好ましくは25℃で、2〜12時間、好ましくは12時間攪拌する。当該反応は、水、リン酸ナトリウム緩衝液、アセトニトリル水溶液等の溶媒中で行うことができる。
【0039】
工程(2)
本工程(2)では、前記(1)工程で誘導体化されたペプチドに対して、フラグメントイオンの質量スペクトルを取得する。
【0040】
本工程(2)において、質量スペクトルの取得に使用される質量分析には、例えば、磁場偏向型、四重極型、イオントラップ型、飛行時間型、フーリエ変換イオンサイクロトロン共鳴型、及びこれらのハイブリッド型等のタイプの質量分析計を採用することができる。これらの中でも、イオントラップ−飛行時間ハイブリッド型の質量分析計は、特定の質量のイオン分子を選別した後に断片化し、生成したイオン分子を質量分析する測定を、同じ起源の分子に対して連続して行うことが可能で、且つ各々の測定精度が高いという利点があり、本発明に好適に使用される。
【0041】
本工程(2)の質量スペクトルの取得において、前記(1)工程で誘導体化されたペプチドをイオン化する方法については、例えば、エレクトロスプレーイオン化法(ESI法)、マトリックス支援レーザー脱離イオン化法(MALDI法)、高速原子衝突法(FAB法)等が挙げられる。これらの中でも、ESI法は、溶液状態の試料をそのままイオン化する方法であるが故、一般的なペプチドの分離精製手段である高速液体クロマトグラフィーに直結できるという利点があり、本工程(2)において好ましいイオン化方法である。
【0042】
また、本工程(2)の質量スペクトルの取得において、ペプチドをフラグメント化(断片化)する方法についても、特に制限されないが、例えば、衝突誘起解離法(CID法)、ポストソース分解法(PSD法)、インソース分解法(ISD法)、電子捕獲解離法(ECD法)、電子移動解離法(ETD法)等が挙げられる。これらのフラグメント化方法の内、CID法は最も汎用され、解離効率が高いという利点があり望ましい。
【0043】
本工程(2)において、前記(1)工程で誘導体化されたペプチドに対して、MS分析、又はMSn分析を行うことにより、アミノ酸配列の解析の指標になるbイオン及びyイオンが顕著に増加して検出され、アミノ酸配列を容易に解析できる質量スペクトルが取得される。ここで、MSn分析とはn乗のMS分析を意味し、具体的には、MSn-1分析で得られたイオン分子を断片化した後に、更に特定のイオン分子をプリカーサーとして分析を行うことを意味する。例えばMS2分析はMS/MS分析に相当する。本工程(2)において、MS分析の乗数については、分析対象のペプチドの種類等に応じて適宜設定すればよいが、通常MS2分析〜MS4分析、好ましくはMS2分析〜MS3分析が採用される。
【0044】
工程(3)
本工程(3)では、前記(2)工程で得られた質量スペクトルに基づいて、ペプチドのアミノ酸配列を決定する。
【0045】
前記(2)工程で得られた質量スペクトルには、アミノ酸配列の解析の指標になるbイオン系列(ペプチドのN末端を含むフラグメント)及びyイオン系列(ペプチドのC末端を含むフラグメント)が高精度で検出されている。当該質量スペクトル上で、各々のbイオン及びyイオンを質量順に特定し、その質量差に基づいて該当するアミノ酸及びその配列順序を同定することにより、ペプチドのアミノ酸配列が決定される。
【0046】
2.ペプチド誘導体化試薬、及び試薬キット
更に、本発明は、上記のアミノ酸配列の決定方法を実施するためのペプチド誘導体化試薬を提供する。当該ペプチド誘導体化試薬は、ペプチドのN末端アミノ基に対して一般式(I)で示す基を結合させることによりペプチドを誘導体化できる化合物(誘導体化化合物)を含むことを特徴とするものである。当該誘導体化化合物については、前述する通りである。
【0047】
また、本発明は、上記のアミノ酸配列の決定方法を実施するための試薬キットを提供する。当該試薬キットは、上記ペプチド誘導体化試薬を含むことを特徴とするものである。また、当該試薬キットは、更に、上記アミノ酸配列の決定方法に使用される試薬や上記アミノ酸配列の決定方法のプロトコールを記した手順書が含まれていてもよい。
【実施例】
【0048】
以下、実施例を挙げて本発明を説明するが、本発明はこれらの実施例に限定されるものではない。
1.アミジノ安息香酸の調製
アミジノベンズアミド・塩酸塩(1.5 g)に6N HCl(45 ml)と12N 酢酸(8 ml)を加え、110℃で6時間撹拌した。室温に戻すと白い結晶が析出した。これを濾集し、H2Oで洗浄した後、減圧乾燥させ、白色固体のアミジノ安息香酸・塩酸塩(0.45 g、収率30%)を得た。同定はNMR分析及びLC-MS分析により行った。
1H-NMR(DMSO-d6)δ 7.91 (d, J=8.4, 2H), 8.12 (d, J=8.3, 2H), 9.18 (s, 2H), 9.48 (s, 2H), 13.51 (s, 1H), ESI-MS m/z calcd for [M+H]+ : 165.1; found: 165.1
【0049】
2.アミジノ安息香酸のペプチドへの導入法1
ペプチド1(AAGLQIA:配列番号1)(2 μmol)のDMSO溶液に、アミジノ安息香酸(10 eq)及び脱水縮合剤4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride(DMT-MM、国産化学社製、10 eq)のDMSO溶液を加え、終夜撹拌した。反応終了後、逆相HPLC(溶離液: H2O/アセトニトリル(0.1%TFAを含む)、カラム:Vydac C18, 4.6×250mm)を用いて分取・精製し、アミジノ安息香酸誘導体化ペプチド1を得た。
ESI-MS m/z calcd for [M+H]+: 789.4; found: 789.4.
【0050】
3.アミジノ安息香酸のペプチドへの導入法2
アミジノ安息香酸(2 g)の50% H2O/アセトニトリル溶液に1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride(10eq)とN-hydroxysuccinimide(10 eq)を加え、終夜撹拌した。反応終了後、逆相HPLC(溶離液:H2O/アセトニトリル(0.1%TFAを含む)、カラム:Vydac C18, 4.6×250mm)を用いて分取・精製し、アミジノ安息香酸スクシンイミドエステル体を得た。同定はNMR分析およびLC-MS分析により行った。
1H-NMR(DMSO-d6)δ 2.92 (s, 4H), 8.04 (d, J=8.5, 2H), 8.31 (d, J=8.5, 2H), 9.41 (s, 2H), 9.57 (s, 2H), ESI-MS m/z calcd for [M+H]+ : 262.1; found: 262.1.
次いで、50 mMリン酸ナトリウム緩衝液(pH8.5)に溶解したペプチド1(2 μmol)に、H2Oに溶解したアミジノ安息香酸スクシンイミドエステル体(10 eq)を加え、終夜攪拌した。反応終了後、逆相HPLC(溶離液:H2O/アセトニトリル(0.1%TFAを含む)、カラム:Vydac C18, 4.6×250mm)を用いて分取・精製し、アミジノ安息香酸誘導体化ペプチド1を得た。
ESI-MS m/z calcd for [M+H]+: 789.4; found: 789.4.
【0051】
4.アミジノ安息香酸導入ペプチドのアミノ酸配列解析
上記「2.アミジノ安息香酸のペプチドへの導入法1」で得られたアミジノ安息香酸誘導体化ペプチドを、イオントラップ-飛行時間ハイブリッド型質量分析計(LCMS-IT-TOF、島津製作所社製)を用いたMSn分析に供した。分析はflow-injection法又はnanospray法(溶媒:50% H2O/アセトニトリル、0.1%ギ酸を含む)で行ない、0.5 μg(0.3-0.8 nmol) の試料を質量分析計に導入した。また、比較のためにアミジノ安息香酸によって誘導体化していないペプチドの測定も行なった。
【0052】
5.ペプチド2〜7のアミノ酸配列解析
以下に示すペプチド2〜7についても、上記「2.アミジノ安息香酸のペプチドへの導入法1」と同様の方法によって誘導体化を行い、次いで上記「4.アミジノ安息香酸導入ペプチドのアミノ酸配列解析」と同様の方法でMSn分析を行った。
ペプチド2(DRVYIHPFHL:配列番号2)
ペプチド3(RPPGFSPFR:配列番号3)
ペプチド4(SYANGFSATSASL:配列番号4)
ペプチド5(ATQQTAAYKTLVS:配列番号5)
ペプチド6(TDVNGDGRHAL:配列番号6)
ペプチド7(DYPVDIYYLMD:配列番号7)
また、従来、ペプチドのフラグメンテーションを改善することが報告されているニコチン酸及びN-tris(2,4,6-trimethoxyphenyl)phosphine-acetic acid(TMPP)を用いてペプチド1の誘導体化を行い、そのMSnスペクトルを比較した。ニコチン酸及びTMPPのペプチドへの導入は文献記載の方法に従って実施した。なお、当該文献は以下の通りである:
Ademczyk, M., et al., Rapid Commun. Mass Spectrom., 13, 1413 (1999)
Cohen, B., et al., Biochemistry, 36, 9035 (1997)
Miyagi, M., et al., Rapid Commun. Mass Spectrom., 12, 603 (1998)
【0053】
6.結果
図1〜14にアミジノ安息香酸で誘導体化したペプチド、及び誘導体化していないペプチドのMS、MS2、MS3スペクトルを示す。また、スペクトル中に検出されたb系列およびy系列イオンを、表1に示す。誘導体化していないペプチドにおいては、いずれも配列解析に重要となるb系列およびy系列イオンの連続した生成が少なかったため、アミノ酸配列の決定は困難であった。一方、アミジノ安息香酸によって誘導体化したペプチドにおいては、いずれにおいても、ほぼ全てのb系列イオンと一部のy系列イオンが連続的に検出され、配列の決定が誘導体化前に比べて容易に行えることが明らかとなった。
【0054】
【表1】
【0055】
更に、ニコチン酸及びTMPPによって誘導体化されたペプチドとの比較においても(図15、16及び表2参照)、アミジノ安息香酸による誘導体化の方が連続したb系列イオンの生成が観測されたことから、既知誘導体化試薬と比べてもフラグメント化を促進する効果が高いことが示された。
【0056】
【表2】
【0057】
以上の結果から、アミジノ安息香酸による誘導体化は、質量分析法によるペプチドのアミノ酸配列解析に有用であることが明らかとなった。
【図面の簡単な説明】
【0058】
【図1】アミジノ安息香酸誘導体化ペプチド1のMS分析、MS2分析、及びMS3分析により得られた質量スペクトルを示す図である。
【図2】誘導体化していないペプチド1のMS分析、MS2分析、及びMS3分析により得られた質量スペクトルを示す図である。
【図3】アミジノ安息香酸誘導体化ペプチド2のMS分析、MS2分析、及びMS3分析により得られた質量スペクトルを示す図である。
【図4】誘導体化していないペプチド2のMS分析及びMS2分析により得られた質量スペクトルを示す図である。
【図5】アミジノ安息香酸誘導体化ペプチド3のMS分析及びMS2分析により得られた質量スペクトルを示す図である。
【図6】誘導体化していないペプチド3のMS分析及びMS2分析により得られた質量スペクトルを示す図である。
【図7】アミジノ安息香酸誘導体化ペプチド4のMS分析、MS2分析、及びMS3分析により得られた質量スペクトルを示す図である。
【図8】誘導体化していないペプチド4のMS分析及びMS2分析により得られた質量スペクトルを示す図である。
【図9】アミジノ安息香酸誘導体化ペプチド5のMS分析及びMS2分析により得られた質量スペクトルを示す図である。
【図10】誘導体化していないペプチド5のMS分析及びMS2分析により得られた質量スペクトルを示す図である。
【図11】アミジノ安息香酸誘導体化ペプチド6のMS分析、MS2分析、及びMS3分析により得られた質量スペクトルを示す図である。
【図12】誘導体化していないペプチド6のMS分析及びMS2分析により得られた質量スペクトルを示す図である。
【図13】アミジノ安息香酸誘導体化ペプチド7のMS分析、MS2分析、及びMS3分析により得られた質量スペクトルを示す図である。
【図14】誘導体化していないペプチド7のMS分析、MS2分析、及びMS3分析により得られた質量スペクトルを示す図である。
【図15】ニコチン酸によって誘導体化されたペプチド1のMS分析及びMS2分析により得られた質量スペクトルを示す図である。
【図16】TMPPによって誘導体化されたペプチド1のMS分析、MS2分析、及びMS3分析により得られた質量スペクトルを示す図である。
【特許請求の範囲】
【請求項1】
質量分析法を用いたペプチドのアミノ酸配列を決定する方法であって、
(1)ペプチドのN末端アミノ基に下記一般式(I)で示す基を結合させることによりペプチドを誘導体化する工程、
【化1】
[式(I)中、mは0又は1の整数を示し、n1及びn2は、同一又は異なって0〜5の整数を示す。また、式(I)中、Xは下記の基を示す。]
【化2】
(2)前記工程(1)で誘導体化されたペプチドに対して、フラグメントイオンの質量スペクトルを取得する工程、及び
(3)前記工程(2)で得られた質量スペクトルに基づいて、ペプチドのアミノ酸配列を決定する工程
を含むことを特徴とする、アミノ酸配列の決定方法。
【請求項2】
前記工程(1)が、下記一般式(II)で示す化合物とペプチドを縮合反応させることにより、ペプチドを誘導体化する工程である、請求項1に記載のアミノ酸配列の決定方法。
【化3】
[式(II)中、m、n1及びn2は、前記と同じ。]
【請求項3】
前記工程(2)において、エレクトロスプレーイオン化法によりペプチドのイオン化を行う、請求項1又は2に記載のアミノ酸配列の決定方法。
【請求項4】
前記工程(2)において、衝突誘起解離法によりペプチドの断片化を行う、請求項1乃至3のいずれかに記載のアミノ酸配列の決定方法。
【請求項5】
質量分析法を用いてペプチドのアミノ酸配列を決定するために使用されるペプチド誘導体化試薬であって、ペプチドのN末端アミノ基に対して下記一般式(I)で示す基を結合させることによりペプチドを誘導体化できる化合物を含有することを特徴とする、ペプチド誘導体化試薬。
【化4】
[式(I)中、m、n1、n2及びXは前記と同じ。]
【請求項6】
請求項5に記載のペプチド誘導体化試薬を含有する、質量分析法を用いてペプチドのアミノ酸配列を決定するための試薬キット。
【請求項1】
質量分析法を用いたペプチドのアミノ酸配列を決定する方法であって、
(1)ペプチドのN末端アミノ基に下記一般式(I)で示す基を結合させることによりペプチドを誘導体化する工程、
【化1】
[式(I)中、mは0又は1の整数を示し、n1及びn2は、同一又は異なって0〜5の整数を示す。また、式(I)中、Xは下記の基を示す。]
【化2】
(2)前記工程(1)で誘導体化されたペプチドに対して、フラグメントイオンの質量スペクトルを取得する工程、及び
(3)前記工程(2)で得られた質量スペクトルに基づいて、ペプチドのアミノ酸配列を決定する工程
を含むことを特徴とする、アミノ酸配列の決定方法。
【請求項2】
前記工程(1)が、下記一般式(II)で示す化合物とペプチドを縮合反応させることにより、ペプチドを誘導体化する工程である、請求項1に記載のアミノ酸配列の決定方法。
【化3】
[式(II)中、m、n1及びn2は、前記と同じ。]
【請求項3】
前記工程(2)において、エレクトロスプレーイオン化法によりペプチドのイオン化を行う、請求項1又は2に記載のアミノ酸配列の決定方法。
【請求項4】
前記工程(2)において、衝突誘起解離法によりペプチドの断片化を行う、請求項1乃至3のいずれかに記載のアミノ酸配列の決定方法。
【請求項5】
質量分析法を用いてペプチドのアミノ酸配列を決定するために使用されるペプチド誘導体化試薬であって、ペプチドのN末端アミノ基に対して下記一般式(I)で示す基を結合させることによりペプチドを誘導体化できる化合物を含有することを特徴とする、ペプチド誘導体化試薬。
【化4】
[式(I)中、m、n1、n2及びXは前記と同じ。]
【請求項6】
請求項5に記載のペプチド誘導体化試薬を含有する、質量分析法を用いてペプチドのアミノ酸配列を決定するための試薬キット。
【図1】

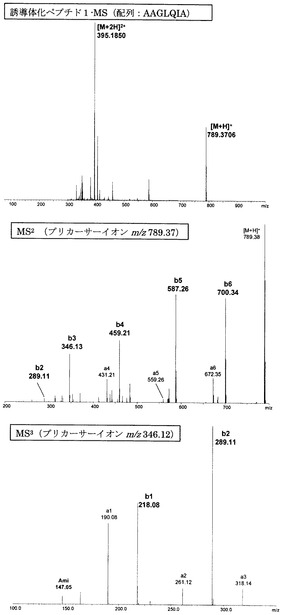
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】

【図13】
【図14】
【図15】
【図16】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【公開番号】特開2008−196965(P2008−196965A)
【公開日】平成20年8月28日(2008.8.28)
【国際特許分類】
【出願番号】特願2007−32209(P2007−32209)
【出願日】平成19年2月13日(2007.2.13)
【出願人】(504132272)国立大学法人京都大学 (1,269)
【Fターム(参考)】
【公開日】平成20年8月28日(2008.8.28)
【国際特許分類】
【出願日】平成19年2月13日(2007.2.13)
【出願人】(504132272)国立大学法人京都大学 (1,269)
【Fターム(参考)】
[ Back to top ]