
超ハイスループットキャプチャーリフトスクリーニング法
本発明は、1つのリガンドに対して選択的アフィニティを有する結合分子を同定しかつ単離する方法に関する。さらに特定すれば、本発明は、パニングなどの他のハイスループット スクリーニング 方法のバイアスおよび制限なしに、数十億個の独立クローンを含有する発現するライブラリーから結合分子を超ハイスループット スクリーニングする方法を提供する。さらに 本発明は、非機能性分子をコードするクローンを本質的に含まない発現ライブラリーを生産する方法を提供する。

【発明の詳細な説明】
【技術分野】
【0001】
1. 発明の分野
本発明は、大きいコンビナトリアルライブラリーから結合タンパク質(例えば、抗体フラグメント)を超ハイスループット選択するためのスクリーニング方法に関する。本明細書に開示されたスクリーニング方法は、初めて、結合タンパク質の非常に大きいライブラリーの迅速なスクリーニング方法を、他のスクリーニング方法(例えば、パニング)がもつバイアスおよび制限なしに提供する。本発明はさらに、非機能性分子をコードするクローンを実質的に含まない分子(例えば、結合分子)の発現ライブラリーを作製する方法に関する。
【背景技術】
【0002】
2. 発明の背景
現在、コンビナトリアル組換えDNA技法を用いて、結合分子について大きい発現ライブラリーを作製することが可能である。とりわけ抗体エンジニアリングの分野ではまさにその通りであり、その場合、組換え抗体ライブラリーは日常的に109個を超えるユニークなクローンを含有する。結合分子の大きいライブラリーが利用可能であるのでほとんどのリガンドに対してほぼ無制限な結合物質の供給源を提供しうる一方、かかる多様なライブラリーを効率的かつ迅速にスクリーニングするスクリーニング方法の開発は遅れている。通常のスクリーニング方法は、FACS分析および96-ウエルプレートを用いるELISAスクリーニングなどの手間と時間のかかる技法に依存してきた。たとえ、自動ロボット機構(ほとんどの研究室の予算をいくらか超えるが)を用いても、これらのスクリーニング方法はこれらの通常の技法を用いた場合、る多様なライブラリーのクローンの小部分のスクリーニングを可能にするだけである。
【0003】
表面ディスプレイライブラリーは、結合分子を提示する生物(例えば、ファージおよび酵母)を固定化リガンド上で連続的な選択(パニング)にかけることにより特定の結合クローンの濃縮を可能にする(総括については、Trends Biotechnol 9: 408-414;Coomberら, 2002, Methods Mol Biol 178: 133-45;Kretzschmarら, 2002, Curr Opin Biotechol 13: 598-602;Fernandez-Gacioら, 2003;Leeら, 2003, Trends Biotechnol 21: 45-52;およびKondoら, 2004, Appl Microbiol Biotechnol 64: 28-40を参照)。パニング技法は非常に多様なライブラリーの迅速なスクリーニングを可能にする一方、ほとんどのパニング技術の繰返し選択サイクルは、一般に、最高の結合アフィニティ(濃縮バイアス)または最高のディスプレイ効率(ディスプレイバイアス)を有する少数の優性クローンが単離されるので、多様性の低下をもたらす。パニング方法の他の欠点は、この方法がしばしばリガンド上の単一優性エピトープだけを認識する結合タンパク質の単離をもたらすことである。さらに、より遅い増殖速度を有するクローンは、パニング中にその結合特性に関係なく集団から速やかに排除される(増殖バイアス)。表面ディスプレイライブラリーの他の制限は、これらが一般に、結合分子と表面を標的化するディスプレイ分子との間の融合の発生に依存することである。かかる融合は、結合特異性の改変されたまたは失った結合分子すら生じうる。さらに、パニングは固定したリガンドを必要とするので、リガンドのコンフォメーションを改変しおよび/または好ましい結合タンパク質認識部位をマスクして、リガンドの不自然な状態またはあまり好ましくない部位と結合する結合分子の単離をもたらしうる。従って、パニング法はしばしば選択バイアスを生じ、無関係なリガンド部位を認識しうる類似の結合特性をもつ少数のクローンだけを単離する一方、多数のそして潜在的にもっと有用なクローンを置き去りにする。
【0004】
表面ディスプレイ・パニング方法のいくつかの制限を克服する2通りのフィルターベースの一般様式のスクリーニング法が開発されていて、一般名で「キャプチャーリフト(capture lift)」と呼ばれている。これらの方法は、結合分子を表面上に提示するのとは逆に、可溶性結合分子を生産することができる発現ライブラリーを利用する。第1のフィルターベースのスクリーニング法は、発現した可溶性結合分子をキャプチャーする所望のリガンドを用いてコーティングしたフィルターを使用する(例えば、Rodenburgら, 1998, Hybridoma 17: 1-8;de Wildtら, 2000, Nat Biotechnol 18: 989-994.;Giovannoniら, 2001, Nucl Acids Res 29: E27を参照)。第2のフィルターベースのスクリーニング法は、結合分子の共通特性(例えば、内因性または遺伝子操作で作られたエピトープタグまたは固有の構造特性または分子の性質)を認識する一般的なキャプチャー分子を用いてコーティングしたフィルター上で、発現した可溶性結合分子をキャプチャーし、その後にキャプチャーされた結合分子を可溶性リガンドを用いて探索する(例えば、Skerraら, 1991, Anal Biochem 196: 151-155;Watkinsら, 1998, Anal Biochem 256: 169-177)。これらの方法は、選択、増殖およびディスプレイバイアスの複雑度を低減しうるが、それでも制限されている。第1の方法は、なお、固定されたリガンドの使用から生じる制限を受け、そして両方とも、比較的手間のかかる方法であって、小さい発現ライブラリー(多様性が106程度のクローン)の徹底したスクリーニングしか行うことはできない(Wuら, 2002, Cancer Immunol Immunother 51: 79-90)。
【0005】
全ての現行ライブラリースクリーニング法がもつ他の制限は、発現ライブラリーの品質にある。発現ライブラリーを作製する現行の方法は様々な増幅およびクローニング技法を利用するので、非機能性分子を発現する人為産物を生産するライブラリークローンを生じる。例えば、PCRおよび関係技法を用いるライブラリーは、これらのDNA複製法の固有の誤差率に応じて非機能性分子を発現する多数のクローンを含有しうる。例えば、PCR法は1塩基対当たり1.6x10-6〜1.1x10-4誤差の誤差率を有する(例えば、Lundberg ら, 1991 Gene 108:1-6.、TindallおよびKunkel, 1988, Biochemistry 27:6008-13を参照)。同様に、分子集団をコードするポリヌクレオチドと表面ディスプレイ用ポリペプチドをコードするポリヌクレオチドとの融合により作製される表面ディスプレイライブラリーでは、多数のクローン(利用する方法に応じて3分の2)が非生産性リーディングフレームにライゲートされたポリヌクレオチドを含有しうる。かかる有害な配列人為産物を含有するクローンは機能性分子を生産し得ない一方、通常、スクリーニングしなければならない生クローン(例えば、ファージまたは細菌)を生産するので、それにより、所望の分子(例えば、結合分子)を同定するためにスクリーニングしなければならない全クローン数は有意に増加する。
【0006】
従って、ファージディスプレイのハイスループット特性を有すると共に、表面ディスプレイ融合タンパク質の使用、リガンドの固定、および複数回のストリンジェントな選択に応じて生じるバイアスから起こる問題を排除するライブラリースクリーニングの方法が真に必要である。かかるスクリーニングの方法は結合分子の表面ディスプレイを避けて、可溶形態の結合分子を発現できるクローンの直接同定を可能にするであろう。理想的には、かかるスクリーニング方法は、さらに非常に大きい発現ライブラリー(109クローン超)の迅速かつ徹底的スクリーニングを可能にするであろう。さらに、非機能性分子(例えば、フレームシフトまたは停止コドン突然変異を有する分子)を生じる有害な配列人為産物を含有するクローンを本質的に含まない分子(例えば、結合分子)の発現ライブラリーを作製する方法は、スクリーニング技法の効率を向上させるであろう。
【0007】
本明細書における参考文献の引用または考察は、これが本発明の先行技術であることを承認すると解釈してはならない。
【発明の開示】
【0008】
3. 発明の概要
本発明は10億個の独立クローンを含有する発現ライブラリーから結合分子を超ハイスループットスクリーニングする方法に関する。上記スクリーニング方法は、1)高密度にまいた発現ライブラリーから結合分子の大集団を発現させるステップ、2)発現した結合分子の集団を固体支持体上に固定するステップ、3)固定した結合分子を少なくとも1つのリガンドと接触させるステップ、4)固定した結合分子と選択的に結合したリガンドを可視化するステップ、および5)少なくとも1つのリガンドを認識しかつ結合する結合分子を発現するクローンを単離するステップを含むかまたは代わりに本質的に上記ステップから成る。このスクリーニング方法は、少なくとも1人当たり1日当たり10億個の結合分子クローンのスクリーニングを可能にしかつ現行スクリーニング技法のいくつかの制限を克服する。さらにこのスクリーニング方法は高価な自動機械の使用を必要としない。
【0009】
一実施形態においては、結合分子は可溶性である。他の実施形態においては、超ハイスループットスクリーニング方法(以後「本発明のスクリーニング方法」または単に「スクリーニング方法」と呼ぶ)を用いて結合分子を発現するライブラリーをスクリーニングする。さらに他の実施形態においては、上記スクリーニング方法を用いてファージ、結合分子を発現する細菌または酵母ライブラリーをスクリーニングする。特定の実施形態においては、上記スクリーニング方法を用いて抗体またはそのフラグメントを発現するライブラリーをスクリーニングする。他の実施形態においては上記スクリーニング方法を用いてレセプター分子を発現するライブラリーをスクリーニングする。さらに他の実施形態においては、上記スクリーニング方法を用いてヌクレオチド-結合分子を発現するライブラリーをスクリーニングする。
【0010】
一実施形態においては、結合分子の発現ライブラリーを高密度でまく。一実施形態においては、結合分子の発現ライブラリーを1mm2当たり10よりも大きい、または100よりも大きい、または1,000よりも大きい、または10,000よりも大きい、または100,000よりも大きい結合分子発現クローンの密度でまく。他の実施形態においては、結合分子発現クローンを1mm2当たり1,000〜15,000クローンの密度でまく。
【0011】
一実施形態においては、結合分子の集団を固体支持体上に固定する。特に、発現した結合分子の集団を、固体支持体上の薬剤との特異的相互作用を介して固体支持体上に選択的に固定する。かかる薬剤としては、限定されるものでないが、化合物、テザー(tether)、リンカー、およびポリペプチド結合ドメインが挙げられる。結合分子の固有の性質が固定を容易にしうることも考えられる。例えば、結合分子の疎水性ドメインはそれらをプラスチック支持体に固定することを可能にしうる。本発明の固体支持体としては、限定されるものでないが、膜、プラスチック、ガラスおよびコーティングしたガラスが挙げられる。
【0012】
一実施形態においては、リガンドは結合分子と選択的に結合できるいずれの分子であってもよく、それらの分子としては、限定されるものでないが、ペプチド、ポリペプチド、核酸、炭水化物、脂質、または有機化合物が挙げられる。他の実施形態においては、リガンドは可溶性である。さらに他の実施形態においては、リガンドは検出ドメインと融合している。検出ドメインは検出シグナルの増幅を可能にしうることを具体的に意図している(下記)。本発明の検出ドメインとしては、限定されるものでないが、チオレドキシン、BSA、ロイシンジッパー、Fcドメインおよびそれらの断片が挙げられる。ある特定の実施形態においては、リガンドが複数の検出ドメインと融合している。さらに、特定の検出ドメインはまた、リガンド-結合分子相互作用の結合活性を増加してスクリーニング方法の結合、特異性および/または感受性の改善をもたらすリガンド二量体(例えば、Fcドメインおよびロイシンジッパードメイン)の形成を可能にしうることも意図する。
【0013】
一実施形態においては、固定した結合分子と選択的に結合したリガンドを検出する。結合したリガンドは直接的なまたは間接的方法により検出しうることを具体的に意図する。リガンドの直接的検出は、多数の技法により実施することができて、それらとしては、限定されるものでないが、リガンドの容易に検出可能な部分(例えば、放射性標識、色素生産検出用酵素)との共有結合性の修飾が挙げられる。リガンドの間接的検出は、当技術分野で周知の方法により実施することができ、それらとしては、限定されるものでないが、リガンドと相互作用することが公知の第2の分子(例えば、抗体)を用いる方法が挙げられる。第2の分子はそれ自体が直接的または間接的方法により検出されてもよい。
【0014】
一実施形態においては、リガンドを認識しかつそれと結合する結合分子クローンを単離する。固体支持体は、リガンドを認識しかつそれと結合する結合分子クローンを含有する結合分子クローンのサブセットを単離するためのテンプレートを提供しうることを具体的に意図する。このより小さい集団を次いで本発明のスクリーニング方法の改変法を用いてスクリーニングすることができる。改変法は、クローンのサブセットをより低い密度で播く、および/または固定するステップを含みうる。クローンのサブセットを、単一クローンが単離されるには十分低いが、サブセット中に存在する各クローンが固体支持体上で少なくとも1回表されるには十分高い密度で播く、および/または固定することを意図する。リガンドを認識しかつそれと結合する結合分子クローンを含有する結合分子クローンのサブセットを、当業者に周知の代わりの方法によりスクリーニングしうることも意図する。代わりの方法としては、限定されるものでないが、ELISAアッセイおよびFACS分析が挙げられる。本発明の方法の1以上の態様は自動化しうることも具体的に意図される。
【0015】
本発明はさらに、非機能性分子を生じる有害な配列人為産物(例えば、フレームシフトまたは停止コドン突然変異を有する分子)を含む分子を発現するクローンを少ししか含有しない発現ライブラリー(例えば、結合分子のライブラリー)を作る方法に関する。上記発現ライブラリーを生産する方法は、1)発現ベクター中に、機能性分子を発現するクローンの選択に有用な少なくとも1つの選択マーカーをコードするポリヌクレオチドとライゲートした分子をコードするポリヌクレオチドを含むクローンのライブラリーを作製するステップ;2)機能性分子を発現するクローンを選択する条件下でステップ(1)において作製したクローンのライブラリーを増やすステップ;ならびに、任意に、3)ステップ(2)の選択されたライブラリーからの機能性分子をコードするポリヌクレオチドを、特定の所望の機能性クローンの同定および/または単離に有用な代わりのベクター中にサブクローニングするステップを含むか、あるいは本質的に上記ステップからなる。
【0016】
本明細書に開示した発現ライブラリーを作る方法は、非機能性分子を生じる有害な配列人為産物を含む分子を発現するクローンを少ししか含有しない発現ライブラリーを作ることを可能にし、それにより、スクリーニングすべき全クローン数を低減することを可能にする。一実施形態においては、上記ライブラリー生産方法を用いて、ファージ、細菌、酵母、植物または哺乳動物系においてスクリーニングするための発現ライブラリーを作製する。
【0017】
一実施形態においては、クローンのライブラリーがコードしかつ発現する分子としては、それから1以上の単一分子の単離が所望される分子の集団が挙げられる。他の実施形態においては、上記発現ライブラリーの生産方法(以後、「本発明のライブラリー生産」または単に「ライブラリーの生産方法」と呼ぶ)を用いて結合分子の集団を発現するライブラリーを作製する。さらに他の実施形態においては、上記ライブラリー生産方法を用いてリガンドの集団を発現するライブラリーを作製する。発現ライブラリーを作製して本明細書に開示したいずれかの結合分子および/またはリガンドの集団を発現しうることが意図される。
【0018】
特定の実施形態においては、上記ライブラリー生産方法を用いて抗体またはそのフラグメントを発現するライブラリーを作製する。他の実施形態においては、上記ライブラリー生産方法を用いてレセプター分子を発現するライブラリーを作製する。さらに他の実施形態においては、上記ライブラリー生産方法を用いてヌクレオチド-結合分子を発現するライブラリーを作製する。
【0019】
非機能性分子を生じる有害な配列人為産物をコードするクローンの排除を容易にするため、分子をコードするポリヌクレオチドを少なくとも1つの選択マーカーをコードするポリヌクレオチドとライゲートする。多数の選択マーカーが当技術分野では周知であり、それには、限定されるものでないが、薬物耐性マーカー(例えば、除草剤および抗生物質耐性遺伝子)、代謝性/栄養要求性マーカー(例えば、必須代謝物の生産に必要な酵素に対する遺伝子)、スクリーニング可能な/精製マーカー(例えば、色素生産検出または精製ドメイン用の酵素をコーディングする遺伝子)が挙げられる。クローンのライブラリーを、有害な配列人為産物を有しない分子を発現させるクローンを選択する条件のもとで増やす。機能性分子をコードするクローンの選択に対して用いられる増殖条件は用いる選択マーカーに応じて変わりうることが理解されるであろう。例えば、薬物耐性遺伝子をコードするポリヌクレオチドを選択に用いるときは、例えば、クローンを適当な薬物の存在のもとで増殖させるであろう。代謝遺伝子をコードするクローンを選択に用いるときは、適当な代謝物の非存在で増やすであろう。
【0020】
本発明のライブラリー生産方法のステップ(1)および(2)で作製しかつ選択した発現ライブラリーを、ライブラリーのスクリーニングに利用しうる代わりのベクター中にサブクローニングしてもよいことが意図される。
【0021】
本発明のライブラリー生産方法を用いて作製した発現ライブラリーを、当技術分野で周知の多数の方法を用いてスクリーニングすることができる。1つの特定の実施形態においては、本発明のスクリーニング方法が利用される。
【0022】
4. 図面の簡単な説明
図面の簡単な説明については下記参照。
【発明を実施するための最良の形態】
【0023】
5. 発明の詳細な説明
本発明は10億個の独立クローンを含有する発現ライブラリーから結合分子を迅速かつ効率的に超ハイスループットスクリーニングする方法を提供する。このスクリーニング方法は、少なくとも1人、1日当たり10億個の結合分子クローンのスクリーニングを可能にする点で有利である。さらに、本発明のスクリーニング方法は、現行ハイスループットスクリーニング技法のいくつかの制限(限定されるものでないが、増殖バイアス、ディスプレイバイアス、濃縮バイアス、ならびに固定したリガンドと結合分子融合体の使用から生じる選択バイアスを含む)を克服する。本発明のスクリーニング技法はさらに、結合分子とそのリガンドの間の特異的相互作用の検出を増強する方法を提供する。さらに、本発明のスクリーニング技法を用いて複数のリガンドに対してスクリーニングすることができる。本発明のスクリーニング方法はそれ故に、非常に多くの結合分子ライブラリーからヒト疾患の診断および治療に用いる特異的結合分子の発見に応用することができる。本発明のスクリーニング方法は、例外的に大きい集団の結合分子のスクリーニングに特に好適であるが、結合分子の大集団と小集団の両方のスクリーニングに利用することができる。本発明のスクリーニング方法の1以上の態様(例えば、検出シグナルの分析、インキュベーションおよび洗浄)は自動化できることも具体的に意図される。
【0024】
上記スクリーニング方法は、1)高密度に播いた発現ライブラリーから結合分子の大集団を発現させるステップ、2)発現した結合分子の集団を固体支持体上に固定するステップ、3)固定した結合分子を少なくとも1つのリガンドと接触させるステップ、4)固定した結合分子と選択的に結合したリガンドを可視化するステップ、および5)少なくとも1つのリガンドを認識しかつ結合する結合分子を発現するクローンを単離するステップを含むか、あるいは本質的に上記ステップから成る。
【0025】
一実施形態においては結合分子は可溶性である。他の実施形態においては、上記スクリーニング方法を用いて、結合分子を発現するライブラリーをスクリーニングする。さらに他の実施形態においては、上記スクリーニング方法を用いて、結合分子を発現するファージ、細菌または酵母ライブラリーをスクリーニングする。本発明のスクリーニング方法を用いてスクリーニングすることができる結合分子のライブラリーとしては、限定されるものでないが、抗体またはそのフラグメントを発現するライブラリー、レセプター分子を発現するライブラリー、ヌクレオチド結合分子を発現するライブラリーおよびランダムペプチドを発現するライブラリーが挙げられる。
【0026】
一実施形態においては、結合分子の発現ライブラリークローンを高密度で播く。特定の一実施形態においては、発現ライブラリークローンを1mm2当たり10よりも大きい、または100よりも大きい、または1,000よりも大きい、または10,000よりも大きい、または100,000よりも大きいクローンの密度でまく。他の実施形態においては、発現ライブラリークローンを1mm2当たり1,000〜15,000クローンの密度でまく。
【0027】
一実施形態においては、発現された可溶性結合分子の集団は固体支持体上に固定されている。発現された可溶性結合分子の集団を、固体支持体(例えば、抗体、化合物)上の薬剤による特定の相互作用を介して、固体支持体上に選択的に固定することを具体的に意図する。結合分子集団の固有の性質が固定化を容易にしうることも意図する。本発明の固体支持体としては、限定されるものでないが、膜、プラスチック、グラスおよびコーティングされたグラスが挙げられる。
【0028】
一実施形態においては、リガンドは結合分子(限定されるものでないが、ペプチド、ポリペプチド、核酸、炭水化物、脂質、または有機化合物を含む)が選択的に結合しうるいずれの分子であってもよい。一実施形態においては、リガンドにはチロシンキナーゼのドメインまたはチロシンキナーゼリガンドが含まれる。意図するチロシンキナーゼおよびチロシンキナーゼリガンドには、限定されるものでないが、レセプターチロシンキナーゼおよび非レセプターチロシンキナーゼが含まれる。他の実施形態においては、リガンドは可溶性である。さらに他の実施形態においては、リガンドは検出ドメインと融合している。検出ドメインは検出シグナル(下記)の増幅を可能にしうることを具体的に意図する。ある特定の実施形態においては、リガンドは複数の検出ドメインと融合している。さらに特定の検出ドメインはまた、リガンド-結合分子相互作用の結合活性を増加してスクリーニング方法の結合、特異性および/または感受性の改善をもたらしうるリガンド-二量体の形成を容易にしうることも意図する。
【0029】
一実施形態においては、固定した結合分子に選択的に結合されたリガンドを検出する。結合されたリガンドを直接的または間接的方法により検出しうることを具体的に意図する。リガンドの直接的検出は多数の技法(限定されるものでないが、放射性標識付きリガンドまたは色素生産検出用酵素による共有結合修飾を含む)により実施することができる。リガンドの間接的検出は、例えば、直接的または間接的方法によりそれ自体検出されるリガンドと相互作用することが公知の抗体を用いて、当技術分野で周知の方法により実施することができる。
【0030】
一実施形態においては、リガンドを認識しかつ結合する結合分子クローンを単離する。固体支持体が、リガンドを認識しかつ結合する結合分子クローンを含有する結合分子クローンのサブセットを単離するためのテンプレートを供給しうることを具体的に意図する。次いでこのより小さい集団を、本発明のスクリーニング方法の改変法を用いてスクリーニングすることができる。上記改変法はクローンのサブセットをより低い密度でまくステップを含んでなる。結合分子クローンのサブセットを、単一クローンが単離されるには十分低いが、サブセット中に存在する各クローンが固体支持体上で少なくとも1回表されるには十分高い密度で播くことを意図する。リガンドを認識し、かつ結合する結合分子を発現するクローンを含有するクローンのサブセットを、当業者に公知の代わりの方法によりスクリーニングしうることも意図する。代わりの方法としては、限定されるものでないが、ELISAアッセイおよびFACS分析が挙げられる。本発明のスクリーニング方法の1以上の態様は自動化できることも具体的に意図する。例えば、非特異的相互作用を排除するために用いるインキュベーションおよび洗浄ステップは、市販の機器を用いて日常的に自動化されているし(例えば、Stovall Washer、カタログ番号WMAA115S、Stovall Life Sciences Inc、Greensboro、NC)、固体支持体上のシグナルの分析も容易に自動化することができる(例えば、Proteome Works Plus Spot Cutterカタログ番号165-7064、Bio-Rad Laboratories Inc.、Hercules、CA)。
【0031】
本発明はまた、非機能性分子を生じる有害な配列人為産物を有する分子をコードするクローンを含まない分子(例えば、結合分子)の発現ライブラリーを生産する方法も提供する。非機能性分子の発現を生じる有害な配列人為産物には、限定されるものでないが、未熟停止コドン、欠失、挿入、フレームシフト突然変異、ノンセンス突然変異およびミスセンス突然変異を含む分子が含まれる。従って、本明細書に使用される用語「非機能性分子」には、限定されるものでないが、有害な配列人為産物(例えば、未熟停止コドン、フレームシフト突然変異、ノンセンス突然変異およびミスセンス突然変異)などを含む分子が挙げられる。非機能性分子を発現するクローンはまた、本明細書では「非機能性クローン」とも呼ばれる。非機能性クローンは一般に生存可能であり、従って、スクリーニング方法により排除しなければならないネガティブクローンを代表する。非機能性クローンの存在は、所望のクローンを同定するためにスクリーニングしなければならないライブラリークローンの全数を非常に増加しうる。本明細書に提供される発現ライブラリーを生産する方法(以後、「本発明のライブラリー生産方法」または単に「ライブラリー生産方法」と呼ぶ)は、本方法がたとえ非機能性分子を発現するクローンをいくらか含むとしても少ししか含まない発現ライブラリーの生産を可能にし、それによりスクリーニングすべきクローンの全数を低減する点で有利である。
【0032】
一実施形態においては、発現ライブラリーを生産する方法は、1)発現ベクター中に、機能性分子を発現するクローンの選択に有用な少なくとも1つの選択マーカーをコードするポリヌクレオチドとライゲートした分子をコードするポリヌクレオチドを含むクローンのライブラリーを作製するステップ;2)機能性分子を発現するクローンを選択する条件下でステップ(1)において作製したクローンのライブラリーを増殖させるステップ;ならびに、任意に、3)ステップ(2)の選択されたライブラリーからの機能性分子をコードするポリヌクレオチドを、特定の所望の機能性クローンの同定および/または単離に有用な代わりのベクター中にサブクローニングするステップを含むかまたは代わりに上記ステップから本質的になる。
【0033】
分子をコードするポリヌクレオチドは個々に少なくとも1つの選択マーカーをコードするポリヌクレオチドとライゲートしていることを意図する。しかし、当業者は、分子をコードする複数のポリヌクレオチドが少なくとも1つの選択マーカーをコードする単一のポリヌクレオチドと直線上にライゲートしていてもよいことを認識するであろう。
【0034】
本発明のライブラリー生産方法を用いて作製されかつ選択された発現ライブラリーは、上記選択されたライブラリーをスクリーニングするために有用な発現ベクター中にあってもよいことを意図する。しかしまた、発現ライブラリーは、上記作製されかつ選択されたライブラリーをスクリーニングするために有用でないが上記ライブラリーを作製しかつ選択するために有用な発現ベクター中に作製されかつ選択されてもよいことを意図する。従って、一実施形態においては、本発明のライブラリー生産方法を用いて作製されかつ選択された発現ライブラリーは、次いで、上記ライブラリーをスクリーニングするために有用な代わりのベクター中にサブクローンされる。
【0035】
従って、ある特定の実施形態においては、ライブラリー生産方法は、1)発現ベクター中に、機能性分子を発現するクローンの選択に有用な少なくとも1つの選択マーカーをコードするポリヌクレオチドとライゲートした分子をコードするポリヌクレオチドを含むクローンのライブラリーを作製するステップ、および2)機能性分子を発現するクローンを選択する条件下でステップ(1)において作製したクローンのライブラリーを増殖させるステップを含んでなる。
【0036】
他の実施形態においては、ライブラリー生産方法は、1)発現ベクター中に、機能性分子を発現するクローンの選択に有用な少なくとも1つの選択マーカーをコードするポリヌクレオチドとライゲートした分子をコードするポリヌクレオチドを含むクローンのライブラリーを作製するステップ;2)機能性分子を発現するクローンを選択する条件下でステップ(1)において作製したクローンのライブラリーを増殖させるステップ;ならびに、3)ステップ(2)の選択されたライブラリーからの機能性分子をコードするポリヌクレオチドを、特定の所望の機能性クローンの同定および/または単離に有用な代わりのベクター中にサブクローニングするステップを含んでなる。
【0037】
発現ライブラリーをスクリーニングするために有用なベクターは当技術分野で周知でありかつ以下に記載され(例えば、表題「発現ライブラリーおよび発現ベクター」の節を参照)、本発明のライブラリー生産方法のステップ(1)および(2)ならびにステップ(3)に有用な具体的なベクターは実施例3〜5に詳述されている(下記)。特定の実施形態においては、作製されかつ選択されたライブラリーからの機能性分子をコードするポリヌクレオチドを、ファージ発現ベクター中にサブクローニングする。
【0038】
本発明のライブラリー生産方法を、種々の系で使用する様々な発現ライブラリーの作製に利用することができる。一実施形態においては、限定されるものでないが、本発明のライブラリー生産方法を用いて、ファージ、細菌、酵母、植物および哺乳動物の系を含む系におけるスクリーニングのための発現ライブラリーを作製する。本発明のライブラリー生産方法を用いて作製した発現ライブラリーは、当業者に周知の多数の方法を用いてスクリーニングすることができる。特定の実施形態においては、本発明のスクリーニング方法が利用される。
【0039】
一実施形態においては、分子をコードするポリヌクレオチドは分子の集団をコードする。他の実施形態においては、ポリヌクレオチドはそれから1以上の単一分子の単離が所望される分子の集団をコードする。例えば、発現ライブラリーを、細胞、組織または生物が発現したメッセンジャーRNAの全集団から作製することができる。あるいは、発現ライブラリーを、例えば、所望のアミノ酸モチーフなどの特定の特徴を有する分子の集団をコードするポリヌクレオチドから作製することができる。分子をコードするポリヌクレオチドの集団を、天然の供給源(例えば、細胞から単離したメッセンジャーRNA)から単離もしくは誘導してもよいし、または新規に作成してもよい(例えば、ランダムポリペプチドをコードするポリヌクレオチド配列)ことを意図する。一実施形態においては、本発明のライブラリー生産方法を用いて、結合分子の集団を発現する発現ライブラリーを作製する。他の実施形態においては、上記ライブラリー生産方法を用いて、リガンドの集団を発現する発現ライブラリーを作製する。さらに他の実施形態においては、上記ライブラリー生産方法を用いて、本明細書に開示したいずれかの結合分子および/またはリガンドの集団を発現する発現ライブラリーを作製する。
【0040】
特定の実施形態においては、上記ライブラリー生産方法を用いて、抗体またはそのフラグメントの集団を発現するライブラリーを作製する。他の実施形態においては上記ライブラリー生産方法を用いて、レセプター分子またはその断片の集団を発現するライブラリーを作製する。さらに他の実施形態においては、上記ライブラリー生産方法を用いて、ヌクレオチド-結合分子またはその断片の集団を発現するライブラリーを作製する。さらに他の特定の実施形態においては、上記ライブラリー生産方法を用いて、ランダムポリペプチドの集団を発現するライブラリーを作製する。
【0041】
非機能性分子をコードするクローンの排除を容易にするため、分子をコードするポリヌクレオチドを少なくとも1つの選択マーカーをコードするポリヌクレオチドとライゲートする。多数の選択マーカーが当技術分野で周知であり、それらとしては、限定されるものでないが、薬物耐性マーカー(例えば、除草剤および抗生物質耐性遺伝子)、代謝性/栄養要求性マーカー(例えば、必須代謝物の生産に必要な酵素に対する遺伝子)、スクリーニング可能な/精製マーカー(例えば、色素生産検出のための酵素または精製ドメインをコードする遺伝子)が挙げられる。クローンのライブラリーを次いで、機能性分子を発現するクローンを選択する条件のもとで増殖させる。当業者は、利用される増殖条件が利用する選択マーカーに応じて変わりうることを理解するであろう。例えば、薬物耐性遺伝子をコードするポリヌクレオチドを選択に用いるときは、例えば、クローンを適当な薬物の存在のもとで増殖しうる。または代謝遺伝子をコードするクローンを選択に用いるときは、適当な代謝物の非存在で増殖しうる。
【0042】
分子をコードするポリヌクレオチドを、固定化および/または検出に有用な少なくとも1つのエピトープタグ配列(本明細書では「マーカー」配列または単に「タグ」配列とも呼ぶ)にライゲートすることを(少なくとも1つの選択マーカーをコードするポリヌクレオチドとライゲートするのに加えて)さらに意図する。多数のマーカーおよび/またはタグ配列、例えば、限定するのではないが、ヘキサ-ヒスチジンペプチド、赤血球凝集素「HA」タグ、および「FLAG」タグが当技術分野では公知である。これらのタグおよびかかるタグの組込みに有用な方法に関するさらなる詳細は、以下の表題「結合分子」および「実施例」の節に記載している。
【0043】
5.1 発現ライブラリーおよび発現ベクター
発現ライブラリーを作製する一般的方法は、当技術分野で周知であり、多数の文献(例えば、Current Protocols in Molecular Biology, F.M. Ausubelら, 編, John Wiley & Sons (Chichester, England, 1998), 第5章および第6章など)から利用しうる。cDNA発現ライブラリーを構築する一般的方法の具体例としては、Chenら, Nature (1995) 377:428-431、Akopianら, Nature (1996) 379:257-262(ポリアデニル化RNAからcDNA発現ライブラリーを作製するための好適な方法を記載する)が挙げられる。ランダムペプチドライブラリーはHrubyら、1997、Curr Opin Chem Biol 1:483-490 に総括され、全ゲノム発現ライブラリーは、例えば、Preussら, 2002, Immunol Rev188: 43-50に記載され、そして核酸および小分子化合物のライブラリーは、Gray, 2001, Curr Opin Neurobiol 11:608-614に総括されている。
【0044】
以下に記載のような分子の集団を発現する発現ライブラリーは、本明細書(実施例3〜5を参照)に開示したような適当な発現ベクター中に構築することができる。特定の実施形態においては、本発明のライブラリー生産方法は、発現ベクターpUCKA中に、少なくともβ-ラクタマーゼ遺伝子をコードするポリヌクレオチドとライゲートした分子をコードするポリヌクレオチドを含有するクローンのライブラリーを作製する第1ステップを含んでなる。従って、本発明は、発現クローンのライブラリーを生産しかつ選択する上で有用な発現ベクターpUCKAを提供する。pUCKAの重要な特徴としては、2つの薬物選択マーカー:該ベクターを含有する細胞を選択/維持するためのカナマイシン耐性遺伝子、および非機能性分子を発現するクローンを選択して除去するためのβ-ラクタマーゼ遺伝子(アンピシリン/カルベニシリン耐性を与える)、複製起点、プロモーター、ならびにシグナル配列、クローニング部位(SfiI)の5'およびクローニング部位の3'、β-ラクタマーゼ遺伝子とフレーム内でライゲートしたFLAGおよびHIS6エピトープタグが挙げられる。本明細書の開示に基づいて、当業者は、pUCKAの変異体および他の代わりの発現ベクターを作製し(例えば、実施例3および5を参照)そして/または本発明のライブラリー生産方法を利用しうることを理解するであろう。他の実施形態においては、本発明は1以上の次の特徴の変化を含むpUCKAの変異体を提供する:エピトープタグ、分子のライブラリーをコードするポリヌクレオチドを挿入するためのクローニング部位、分子のライブラリーをコードするポリヌクレオチドの融合のための選択マーカー、複製起点、シグナル配列、プロモーター、発現ベクターを含む細胞を維持/選択するためのさらなる選択マーカー、細胞においてベクターを発現および/または維持するために必要な他の特殊化した成分の付加。
【0045】
他のベクターは容易に入手しうるし、かつ当業者に周知である。本発明の方法に有用である発現ベクターは、RNA合成を指示するための適当な発現制御配列(プロモーター)を含有しうる。例えば、細菌プロモーターにはlacI、lacZ、T3、T7、gpt、λPR、PLおよびtrpが含まれる。真核生物のプロモーターにはCMV最初期、HSVチミジンキナーゼ、初期および後期SV40、レトロウイルスからのLTR、ならびにマウスメタロチオネイン-Iが含まれる。適当な発現ベクターおよびプロモーターの選択は当業者の技術レベルで十分可能である。発現ベクターはまた、翻訳開始用のリボソーム結合部位および転写ターミネーターも含有しうる。ベクターはまた、発現を増幅するための適当な配列も含みうる。加えて、発現ベクターはさらに上記発現ベクターを含有する形質転換宿主細胞を選択するための表現型形質を与える選択マーカー遺伝子をコードするポリヌクレオチドを含有しうる。選択マーカーのいくつかの例としては、真核細胞培養に対するジヒドロ葉酸レダクターゼまたはネオマイシン耐性、または大腸菌におけるテトラサイクリンまたはアンピシリン耐性が挙げられる。当業者は、ライブラリークローンを含む形質転換宿主細胞を選択および/または維持するために必要な選択マーカーに加えて、機能性クローンを発現するクローンを選択するために第2の選択マーカーが必要でありうることを理解するであろう。
【0046】
利用しうる発現ベクターの代表例としては、限定されるものでないが、ウイルス粒子、バキュロウイルス、ファージ、プラスミド、ファージミド、コスミド、フォスミド、細菌人工染色体、ウイルスDNA(例えば、ワクシニアウイルス、アデノウイルス、ファウルポックスウイルス、仮性狂犬病およびSV40の誘導体)、P1-系人工染色体、酵母プラスミド、酵母人工染色体、および目的の具体的な宿主に特異的な他のベクター(例えば、大腸菌、バシラス菌、アスペルギルス真菌、昆虫、植物、酵母、哺乳動物の細胞、など)が挙げられる。かかるベクターは、染色体性、非染色体性および合成DNA配列を含む。多数の好適なベクター当業者に公知でありかつ市販されている。例示として、次のベクターが提供される:細菌/ファージ:pQEベクター(Qiagen)、pBluescriptプラスミド、pNHベクター、λ DASH(登録商標)IIベクター、pTRG XR、ZAPベクター(Stratagene);ptrc99a、pKK223-3、pDR540、pRIT2T(Pharmacia);真核生物:pXT1、pSG5、pCMV-Script(登録商標)(Stratagene)、pSVK3、pBPV、pMSG、pSVLSV40(Pharmacia)。しかし、いずれの他のプラスミドまたは他のベクターも、それらが宿主中で複製可能でありかつ生存可能である限り使用することができる。一般に、組換え発現ベクターは1以上の宿主にとって適当な複製起点を含みうる。当業者は、現行の開示に基づいて、適当な発現ベクター(例えば、発現を駆動するプロモーターおよび選択のための選択マーカーを含有するもの)を、市販ベクターを改変することにより作製するかまたは本明細書に記載のように、複製、維持、発現、選択および他の所望の形質に必要である適当な成分を組み合わせることにより新規に作製できることを理解するであろう。
【0047】
このように、機能性分子を発現するクローンを選択するために有用な選択マーカーをコードするポリヌクレオチドとライゲートした分子をコードするポリヌクレオチドのライブラリーを、分子を発現するために有用な様々な発現ベクターにおいて作製することができる。
【0048】
5.2 結合分子
本明細書で使用する用語「結合分子」は、リガンドと選択的に結合するのに十分なサイズと複雑度を有する分子を意味することを意図している。かかる分子は一般に高分子であって、限定されるものでないが、それにはポリペプチド、核酸、炭水化物および脂質が含まれる。しかし、具体的には、誘導体、類似体および模倣化合物ならびに小有機化合物も、この用語の定義内に含まれることを意図している。結合分子のサイズは、該分子がリガンドとの選択的結合を示すかまたは示すようにすることができる限り、重要でない。
【0049】
本明細書に使用する用語「選択的な」および「選択性」は、本明細書に用いられるリガンドとの結合分子に言及する場合、欲しないか、または非特異的相互作用から識別することができる相互作用を意味する。識別は、例えば、親和性または結合活性に基づくものであってよく、そして多数の低親和性相互作用または少数の高親和性相互作用から誘導されうる。高親和性相互作用は一般にほぼ10-8 M〜ほぼ10-9 Mより大きいものである。
【0050】
一実施形態においては、結合分子はリガンドとほぼ10-4 Mを超えるアフィニティで結合しうる。他の実施形態においては、リガンドに対してほぼ10-4M、またはほぼ10-5M、またはほぼ10-6M、またはほぼ10-7M、またはほぼ10-8M、またはほぼ10-9M、またはほぼ10-10Mを超えるアフィニティを有しうる。
【0051】
結合分子には、例えば、抗体および他のレセプターまたは免疫系のリガンド結合ポリペプチドが含まれ、限定されるものでないが、T細胞レセプター(TCR)、主要組織適合性複合体(MHC)、CD4レセプター、およびCD8レセプター、その他のCD分子(限定されるものでないが、CD2、CD3、CD19、CD20、CD22を含む)などが含まれうる。具体的に意図される他の結合分子には、限定されるものでないが、細胞表面レセプター、(例えば、インテグリン、成長因子レセプターおよびサイトカインレセプター)、細胞質レセプター(例えば、ステロイドホルモンレセプター)、DNA結合ポリペプチド(例えば、転写因子およびDNA複製因子)が含まれる。結合分子にはまた、上記結合分子の変異体および/またはリガンド結合に恐らく間違いなく寄与しうる上記結合分子の一部分を含有する融合分子も含まれる。さらに、ランダムおよびコンビナトリアルライブラリーから選択された結合分子集団(例えば、ポリペプチド、核酸、アプタマーおよび化合物)も、かかる分子がリガンドと選択的結合活性を示すか示すようにすることができる限り、含まれると意図している。
【0052】
結合分子集団の選択は、所望の結合分子のタイプ、最後の選択される結合分子の必要性および利用意図に依存しうる。1つの手法は、結合分子として機能することが公知であるかまたは結合活性を示すかもしくは示す能力のあることが公知の分子から結合分子集団を作製することである。例えば、抗体および免疫レパートリーの他のレセプターは、本質的に無数の色々な抗原およびリガンドと結合することができる結合分子として機能することがわかっている。それ故に、抗体レパートリーから多様な結合分子の集団を作製すると、本質的に所望のリガンドに対する結合分子の同定を可能にしうる。
【0053】
第2の手法は未知分子の大集団を作製することである。上記集団は配列または構造の十分な多様性を含有し、目的のリガンド組成と結合しうる分子を含有するように作製しなければならない。この手法の利点は、配列、構造または機能について前もっての知識を必要としないことである。その代わり、必要なことの全ては、上記集団がたまたまリガンド複合体と特異的結合相互作用を示す高い確率を有しうるように、十分なサイズと複雑度の集団を作製することである。かかる集団の具体例は、ペプチド、核酸および小分子化合物のランダムライブラリーである。当業者は、どのタイプの手法とどのタイプの結合分子 集団が意図する目的および所望の必要性に応用可能であるかを熟知するかまたは決定することができるであろう。
【0054】
使用する結合分子集団のサイズと多様性は複数の因子により決定されうるのであって、それらの因子には、限定されるものでないが、リガンド集団または組成、所望のアフィニティの範囲、結合分子の複雑度、ならびに所望の結合分子の数とタイプが含まれる。同定すべき結合分子の所望の数が増加すると、結合分子の集団のサイズと多様性も増加する。同様に、結合分子のライブラリー変異体(例えば、抗体またはそのフラグメント)をスクリーニングするとき、集団のサイズは結合分子自体の複雑度とともに増加する。さらに、結合分子の集団のサイズは同様に、リガンドの数または複雑度が増加するとともに増加しうる。
【0055】
小サイズの集団は数百および数千の異なる結合分子からなり、中サイズの集団は数万および数十万からなる一方、大集団は数百万および数十億の異なる結合分子からなるであろう。本発明のスクリーニング方法を用いていずれのサイズの結合分子の集団をスクリーニングすることもできる一方、特徴として、数百万および数十億の異なる結合分子からなる大きいかつ多様な集団、具体的には、ほぼ106、107、108、109、1010、1011またはそれより多い異なる結合分子を含む集団をスクリーニングするのに好適である。同様に、本発明のライブラリー生産方法を用いていずれのサイズの分子の集団のライブラリーを作製することもできるが、数百万および数十億の異なる分子からなる大きいかつ多様な集団、具体的には、ほぼ106、107、108、109、1010、1011またはそれより多い異なる結合分子を含む分子の集団の発現ライブラリーを作製するのに好適である。当業者は、所望の結合分子の数をスクリーニングおよび/または同定するのに十分でありうる結合分子の集団の近似的な多様性を熟知するであろう。
【0056】
結合分子の組換え体ライブラリーが一般に利用されるが、それは結合分子の大きいかつ多様な集団を速やかに作製することができるからである。遺伝子組換えの方法は、集団の固体支持体との選択的固定化のための特徴を固有に含有する天然のレパートリーから、大きい数の結合分子集団を生産することを可能にする。さらに、発現されたポリペプチドまたは核酸の組換えライブラリーは、結合分子集団の固体支持体との選択的固定化を容易にするかまたは直接に機能するように、多数の方法で遺伝子操作することができる。本発明のライブラリー生産方法は結合分子の生産ライブラリーに利用できることを具体的に意図している。
【0057】
結合分子の集団は、その集団が十分に多様であって、その集団が所望のリガンドと選択的に結合する結合分子を少なくとも1つ含有する確率が非常に高い限り、本質的にいずれの供給源からでも生産または誘導することができる。結合分子の集団は、結合分子として機能するかまたは結合活性を示すことがわかっている分子から作製することができ、かかる分子には、限定されるものでないが、抗体、そのフラグメント、免疫系の他のレセプター、レセプター、ヌクレオチド結合タンパク質およびレクチンが含まれる。あるいは、結合分子の集団は未知の分子、例えば、ランダムペプチドライブラリー(Hrubyら, 1997, Curr Opin Chem Biol 1:483-490の総括を参照)、全ゲノム発現ライブラリー(例えば、Preussら, 2002, Immunol Rev188: 43-50)、核酸および小分子化合物(Gray, 2001, Curr Opin Neurobiol 11:608-614の総括を参照)から作製することができる。
【0058】
結合分子集団の選択は所望の結合分子のタイプに依存しうる。例えば、もし高アフィニティ結合分子が所望であれば、抗体結合分子の集団を利用することができる。同様に、類似した異型遺伝子性を示す他の免疫系の分子(例えば、T細胞レセプターおよび主要組織適合性複合体レセプターCD4およびCD8)から結合分子集団を誘導することができる。かかる分子のこれらの正常機能は無限の数の色々な抗原および/またはリガンドと本質的に結合する(Kuby、J.(編), 1997, Immunology, Third Ed., New York, W.H. Freeman & Co.)。それ故に、これらの分子から多様な結合分子の集団を作製すると、いずれかの所望のリガンドに対する結合分子を同定することが可能になりうる。
【0059】
特定の生物学的効果をもつ結合分子を結合分子の集団から同定しうることを具体的に意図する。例えば、結合分子は、標的分子の1以上の生物学的活性を阻害することができるアンタゴニストであってもよい。アンタゴニストは、レセプターのリガンドとの結合(逆もまたしかり)を妨害することにより、かつリガンドにより活性化された受精不能または死滅細胞により、および/またはレセプターもしくはリガンド活性化(例えば、チロシンキナーゼ活性化)または細胞レセプターとのリガンド結合後のシグナル伝達により作用しうる。アンタゴニストは、レセプター-リガンド相互作用を完全にブロックしうるか、またはかかる相互作用を実質的に低下させうる。あるいは、結合分子は、標的分子の1以上の生物学的活性を活性化することができるアゴニストであってもよい。アゴニストは、例えば、標的分子を活性化することにより、および/またはシグナル伝達に介在することにより作用しうる。結合分子の生物学的効果を確認するアッセイは当業者に周知である。
【0060】
公知のまたは固有の結合機能を示す他の結合分子には、本発明のライブラリー生産方法を用いる発現ライブラリーの作製、または本発明のスクリーニング方法における出発集団としての使用に受け入れられるものとして、限定されるものでないが、細胞表面、細胞質および核レセプターを含む様々なレセプターが含まれる。細胞表面レセプターの例には、限定されるものでないが、細胞外マトリックス成分(例えば、インテグリン)、成長因子(例えば、EGFR、FGFR)、ホルモン、インスリンおよびインスリン様タンパク質(IR、IGF-R)、サイトカイン(例えば、IL-4R、IL-13)、レセプターチロシンキナーゼ(例えば、ALK、EphA2、EphA3、EphA4、EphA5、EphA6、EphA7、EphA8、EphB1、EphB2、EphB3、EphB4、EphB5、EphB6)、サイトカイン(例えば、IFNAR)およびケモカイン(例えば、CXC-R、CC-R)に対するレセプターが含まれる。細胞質および核レセプター(Mangelsdorf ら, 1995, Cell 83:835の総括を参照)の例には、限定されるものでないが、ステロイドホルモンレセプター(Kumarら, 1999, Steroids 64:310)、PPARレセプター(Wilsonら, 2000, J Med Chem 43:527)、ビタミンレセプター、および核酸結合タンパク質(de Miguelら, 1998, Curr Opin Chem Biol 2:417-421;および McEwan, I.(編), 2004, Essays in Biochemistry, Volume 40, London, Portland Press Ltd.)が含まれる。
【0061】
結合分子ライブラリーは未知の配列または構造のランダムライブラリーから誘導しうることも具体的に意図される。かかるライブラリーは、当技術分野で公知の標準組換え技法を用いて容易に作製することができる(Leblら、1997、Methods Enzymol 289:336-392 および Shustaら、1999、Curr Opin Biotechnol 10:117-122の総括を参照)。
【0062】
特定の実施形態においては、結合分子の集団はさらに結合分子と融合またはコンジュゲートした異種ポリペプチドを組み込む。異種ポリペプチドには、限定されるものでないが、固定化および/または検出のために有用であるマーカーおよび/またはタグ配列が含まれる。例えば、とりわけ、pQEベクターで提供されるタグ(QIAGEN、Inc.、9259 Eton Avenue、Chatsworth、Calif.、91311)のようなヘキサ-ヒスチジンペプチド(その多くは市販されている)は検出および固定化に有用である(Gentzら、1989、Proc. Natl. Acad. Sci. USA 86:821-824)。検出および固定化の両方に有用な、他のペプチドタグには、限定されるものでないが、インフルエンザ赤血球凝集素タンパク質から誘導されたエピトープに対応する赤血球凝集素「HA」タグ(Wilsonら, 1984, Cell 37:767)が含まれる。ポリペプチド、タンパク質および融合タンパク質は、標準組換えDNA技法によりまたはタンパク質合成技法により、例えば、ペプチド合成機を用いて生産することができる。例えば、ペプチド、ポリペプチド、タンパク質または融合タンパク質をコードする核酸分子は、自動化DNA合成機を含む通常の技法により合成することができる。あるいは、遺伝子断片のPCR増幅は、2つの継続する遺伝子断片間に相補的オーバーハングを生じるアンカープライマーを用い、次いでアニーリングし、そして再増幅してキメラ遺伝子配列を作製することにより行うことができる(例えば、Current Protocols in Molecular Biology, F.M. Ausubelら, 編, John Wiley & Sons (Chichester, England, 1998)およびMolecular Cloning: A Laboratory Manual, 3rd Edition, J. Sambrookら, 編, Cold Spring Harbor Laboratory Press (Cold Spring Harbor, NY, 2001)を参照)。
【0063】
他の実施形態においては、結合分子の集団はさらに、検出および固定化の両方に有用であるビオチンおよび/またはハプテン分子タグを組み込む。結合分子の集団をビオチン化(バイオテクノロジーにおけるビオチンタグの総括は、Diamandisら、1991、Clin Chem 37:625-636を参照)および/またはハプテン化(ハプテンおよび方法の総括は、Molecular Probes: Handbook of Fluorescent Probes and Research Chemicals、R. P. Haugland、第9版、Molecular Probes、(Eugene OR、2004)の第4章を参照)することができる。
【0064】
一実施形態においては、結合分子の集団は可溶性である。いくつかの結合分子は固有で可溶性である一方、他の結合分子は、限定されるものでないが、それらを可溶化するためのさらなる成分(例えば、界面活性剤、カオトロピック剤)の導入を含むさらなる操作を必要とする。可溶性結合分子集団の使用は、固体支持体および/またはリガンドと相互作用することができない不溶性結合分子凝集物の形成を防止することにより、スクリーニングを容易にしうることが具体的に意図される。自然の結合分子を所望する場合、分子を可溶化するために必要な操作はコンフォメーション改変をもたらして自然の状態でリガンドを認識しない結合分子を同定しうるので、結合分子の集団は固有に可溶性であることを期待している。
【0065】
組換え発現ライブラリーから可溶性分子を生産するためには多数の方法がある。方法としては、限定されるものでないが、可溶性タンパク質との融合(下記表題「リガンドと検出」の節を参照)、宿主生物から発現されたポリペプチドの特異的分泌に対するシグナル配列の利用、および細菌溶解を引き起こして細菌により生産されたポリペプチド配列の放出をもたらす溶原性ファージ発現ライブラリーの使用(例えば、Current Protocols in Molecular Biology, F.M. Ausubelら, 編, John Wiley & Sons (Chichester, England, 1998)およびMolecular Cloning: A Laboratory Manual、第3版、J. Sambrookら、編、Cold Spring Harbor Laboratory Press(Cold Spring Harbor、NY、2001)を参照)が挙げられる。当業者は具体的な目的に応用可能なライブラリーのタイプを熟知するかまたは決定することができるであろう。
【0066】
一実施形態においては、本発明のライブラリー生産方法を用いて、可溶性結合分子を発現する組換えファージ、細菌または酵母ライブラリーを作製する。他の実施形態においては、スクリーニング方法を用いて可溶性結合分子を発現する組換えファージ、細菌または酵母ライブラリーをスクリーニングする。ファージ組換えライブラリーの具体例としては、溶原性ファージが細菌により発現された結合ポリペプチドの放出を引き起こすライブラリーおよび結合分子が細胞の溶解なしに周辺質空間中に分泌されるライブラリーが挙げられる。
【0067】
特定の実施形態においては、ライブラリー生産方法を用いて、抗体またはそのフラグメントを発現するライブラリーを作製する。他の実施形態においては、スクリーニング方法を用いて抗体またはそのフラグメントを発現するライブラリーをスクリーニングする。抗体またはそのフラグメントを発現するライブラリーは、本明細書に開示される方法を含む当業者が熟知する様々な方法により作製することができる。例えば、ポリメラーゼ連鎖反応(PCR)を用いて特定の生物の本質的に全ての抗体レパートリーを増幅し、そしてそれを重鎖および軽鎖、その機能性フラグメントの多様な組合せの組換え体集団として、または融合タンパク質として発現させることができる。具体的な方法については、下記実施例1を参照されたい。抗体の機能性フラグメントには、限定されるものでないが、Fab、Fv、scFv、およびCDR領域が含まれる。特に、抗体には、免疫グロブリン分子および免疫グロブリン分子の免疫学的活性フラグメント、すなわち、抗原結合部位を含有する分子が含まれ、これらのフラグメントは、限定されるものでないが、Fc領域またはそのフラグメントを含む他の免疫グロブリンドメインと融合していてもしていなくてもよい。当業者はさらに、限定されるものでないが、scFv-Fc融合、可変領域(例えば、VLおよびVH)-Fc融合およびscFv-scFv-Fc融合を含む他の融合産物を作成できることを理解するであろう。免疫グロブリン分子はいずれのタイプ(例えば、IgG、IgE、IgM、IgD、IgAおよびIgY)、クラス(例えば、IgG1、IgG2、IgG3、IgG4、IgA1およびIgA2)またはサブクラスであってもよい。
【0068】
本発明のライブラリー生産方法を用いて単一特異性、二重特異性またはさらに多重特異性の抗体分子の集団を発現するライブラリーを作製しうることを具体的に意図する。本発明のスクリーニング方法を用いて単一特異性、二重特異性またはさらに多重特異性の抗体分子の集団をスクリーニングすることも具体的に意図する。例えば、PCT公報 WO 93/17715、WO 92/08802、WO 91/00360、およびWO 92/05793;Tuttら、J. Immunol. 147:60-69(1991);米国特許第4,474,893号、第4,714,681号、第4,925,648号、第5,573,920号、および第5,601,819号、;ならびにKostelnyら、J. Immunol. 148:1547-1553(1992)を参照されたい。
【0069】
組換え抗体発現ライブラリーを作製するための具体的な方法およびプロトコルの例は、本明細書(例えば、実施例1、3〜5を参照)ならびに、とりわけ、Huseら, 1989, Science 246: 1275-1281;McCaffertyら, 1990, Nature 348:552-554;Rosokら, 1996, J Biol Chem 271:22611-22618;Bacaら, 1997, J Biol Chem 272:10678-10684;Wuら, 1998, Anal Biochem 256:169-177;Sheetsら, 1998, PNAS USA 95:6157-6162;de Haardら, 1999, J Biol Chem 274:18218-18230;Knappikら, 2000, J Mol Biol 296:57-86;Soderlindら, 2000, Nature Biotechnol 18:852-856;およびAzriel-Rosenfeldら, 2004, J Mol Biol 335:177-192に見出すことができる。
【0070】
また、天然または合成化合物ライブラリーなどの他の化合物の大集団も、それらを固体支持体上に固定することができる限り、本発明のスクリーニング方法を用いてスクリーニングできることも具体的に意図する。かかる他の化合物の大集団を固体支持体上に固定し、次いで1以上の選択のリガンドと選択的に結合する分子を同定するためにスクリーニングすることができる。一実施形態においては、かかる化合物の大集団を選択的に固定することができる。大きい結合分子の集団は、当業者に公知のコンビナトリアル化学の方法により生産した合成化合物であってもよい。
【0071】
5.3 固定化
所望のリガンドに対して選択的結合アフィニティを有する結合ポリペプチドを同定する目的で、スクリーニングすべき結合分子の集団を固定化し、次いで結合分子を所望のリガンドと接触させることが必要である。一実施形態においては、結合分子の集団を固定化する。他の実施形態においては、結合分子の集団を選択的に固定化する。
【0072】
結合分子の固体支持体との固定化に言及するときに本明細書で使用する用語「選択的な」または「選択的に」は、欲しない相互作用から区別することができるある相互作用を意味する。区別は、例えば、アフィニティまたは結合活性に基づくものであってもよく、そして多数の低アフィニティ相互作用または少数の高アフィニティ相互作用から誘導されるものであってもよい。本明細書で使用する用語「選択的固定化」および「選択的に固定化された」は、特異的相互作用(例えば、結合分子上に存在するエピトープに特異的である結合分子の抗体との相互作用など)および結合分子の固有の性質に由来する相互作用(例えば、疎水性ドメインのプラスチック表面との相互作用ならびに架橋剤などの化学部分が介在する相互作用など)の両方の相互作用を包含することを意図する。
【0073】
特定の理論または機構により束縛されるのを欲することなく、結合分子の集団の選択的固定化は、測定される結合相互作用(例えば、結合分子-リガンド相互作用)の感受性を増加するように機能することを意図する。結合分子の集団を固体支持体へ選択的固定化することは、結合分子集団を反応物中に存在しうる無関係なおよび/または汚染性分子から分離する働きをする。従って、結合分子集団の固定化は、結合分子集団を有意に濃縮し、次いで、スクリーニングされる結合分子の集団の一部分でない無関係および/または汚染性分子との非特異的結合相互作用を低下させる。
【0074】
当業者は、結合相互作用を測定することの困難は、結合アッセイの複雑度および結合反応物内の色々な種の数と多様性とともに増加することを理解するであろう。本明細書に開示したスクリーニング方法は、結合分子の集団の選択的固定化が反応物から無関係および/または汚染性分子から除去することにより実質的に欲しないか、および/または無関係な結合相互作用の数を低下させるので、これらの困難を減ずる。従って、本発明のスクリーニング方法は、固体支持体上の結合分子の集団の選択的固定化を介して、検出の感受性と特異性を改善する。
【0075】
結合分子の選択的固定化を利用してアフィニティ技法により分離した集団などの実質的に精製したまたは濃縮した結合分子の集団から、ならびに不均一集団(例えば、細胞抽出物、順化培地)から、結合分子をスクリーニングすることができる。
【0076】
特定の実施形態においては、発現された可溶性結合分子の集団を選択的に固定化する。特に発現された可溶性結合分子の集団を、固体支持体と結合した、および/またはカップリングした作用薬との特異的相互作用を介して固体支持体上に選択的に固定化する。かかる作用薬としては、限定されるものでないが、抗体、ポリペプチド、アプタマー、繋索、リンカー、および結合分子の集団を固体支持体に保持するのに十分な共有結合または非共有結合相互作用を可能にする化学部分が挙げられる。結合分子の固有の性質が選択的固定化を容易にすることも意図する。例えば、結合分子の疎水性ドメインはプラスチック支持体との選択的固定化を可能にする。
【0077】
特定の実施形態においては、結合分子の集団上に存在する1エピトープを認識する1一抗体を固体支持体と結合および/またはカップリングする。例えば、定常ドメインを認識する抗体を用いて抗体結合分子の集団を固定化することができる。あるいは、特定のエピトープタグ(例えば、HA、FLAG、Myc、6xHisエピトープタグ、上記)を認識する抗体を用いて、上記エピトープタグを含むように遺伝子操作で作られた結合分子の集団を固定化することができる。同様に、ビオチンまたはアビジンを用いて他の結合対(上記)のパートナーを含有するように遺伝子操作で作られた結合分子の集団を固定化することができる。例えば、ビオチンを固体支持体とカップリング/結合させ、結合分子をアビジンを含有するように遺伝子操作で作ることができる。あるいは、アビジンを固体支持体とカップリング/結合させる一方、結合分子の集団をビオチンを用いて標識することができる。
【0078】
他の実施形態においては、結合分子の集団上に存在するエピトープおよび/またはドメインを認識するアプタマーを固体支持体と結合および/またはカップリングさせることができる。具体的な標的に特異的なアプタマーを選択する方法は当技術分野では周知である。(Jayasena, S.D., 1999, Clin. Chem. 45:1628-1650のアプタマー技法の総括を参照).
固体支持体を使用することにより、高濃度の結合分子の固定化が可能になる。固定された高濃度の結合分子は、非常に大きい数の分子の迅速なスクリーニングを可能にし、かつさらに、リガンドとの低いアフィニティ相互作用を検出する能力を増強して低濃度のリガンドを検出する働きをする。
【0079】
本質的にいずれの固体支持体も本発明のスクリーニング方法における使用に受け入れられる。当業者は、具体的な必要性に適合しかつ結合分子の集団の全て、または統計上代表的な数を固定化する能力を有するために必要な支持体を熟知するであろう。固体支持体は、固定化結合分子のより高い密度の達成を可能にする多孔材料にすることができる。さらに固体支持体を、スクリーニング方法に必要とされる操作(例えば、洗浄、インキュベーション、可視化方法)に適合しうる一方、結合分子集団を保持する能力を維持する特徴をもつものを選ぶことができる。かかる操作は、無結合のリガンド集団の除去および非特異的相互作用物を除去するための洗浄ならびにリガンド-結合分子相互作用の可視化に重要でありうる。上記操作が容易であることは、多数の操作を必要とすることが多い超ハイスループットスクリーニングを実施するときに有利である。本発明の固体支持体には、限定されるものでないが、ニトロセルロース、ナイロン、二フッ化ポリビニリデン、プラスチック、ガラス、ポリアクリルアミドおよびアガロースなどの膜が含まれる。固体支持体は、結合分子の集団(例えば、ビーズ)の固定化を支持する限り、本質的にいずれのサイズまたは形状で作ることもできる。
【0080】
本発明のスクリーニング方法で使用される固体支持体は改変しうることを具体的に意図する。例えば、非常に様々な官能基を固体支持体の表面と結合および/またはカップリングさせて、結合分子の集団の固定化を容易にするかまたは他のスクリーニング方法の態様(例えば、検出、洗浄)を増強することができる。固体支持体と結合させることができる官能基には、限定されるものでないが、化学部分(例えば、架橋剤)、アプタマー(例えば、RNA、DNA)、ポリペプチド(例えば、抗体、ストレプトアビジン)および他の生体分子(例えば、ビオチン、脂質)が含まれる。官能基は、限定されるものでないが、共有結合性、非共有結合性、加水分解性、光分解性、光活性化性、可逆的および非可逆的を含むいくつかの相互作用により結合分子の集団の固定化に介在することができる。
【0081】
非常に大きな結合分子の集団の迅速なスクリーニングを可能にするために、本発明は、固体支持体1mm2当たり少なくとも3,800の独立クローンのスクリーニングを可能にする高密度で播く方法を利用する。従来の方法では、典型的には、固体支持体1mm2当たりたかだかほぼ1〜6の独立クローンのスクリーニングが可能である。例えば、抗体分子の多様なライブラリーは容易に109の独立クローンを超えうる。通常の方法を用いると、このサイズのライブラリーは、全集団をスクリーニングするために、例えば、少なくとも20,000のフィルター(直径83mmで)を必要としうる。本発明のスクリーニング方法を用いると、全ライブラリーをわずか33のフィルターを用いてスクリーニングすることができる。従って、本発明は従来の方法より少なくとも600倍の改善を提供する。
【0082】
一実施形態においては、発現ライブラリークローンを高密度でまく。特定の実施形態においては、発現ライブラリークローンを1mm2当たり、ほぼ10を超える、またはほぼ100を超える、またはほぼ1,000を超える、またはほぼ2,000を超える、またはほぼ3,000を超える、またはほぼ4,000を超える、またはほぼ5,000を超える、またはほぼ6,000を超える、またはほぼ7,000を超える、またはほぼ8,000を超える、またはほぼ9,000を超える、またはほぼ10,000を超える、またはほぼ25,000を超える、またはほぼ50,000を超える、またはほぼ75,000を超える、またはほぼ100,000クローンを超える密度でまく。他の実施形態においては、発現ライブラリークローンを1mm2当たり、10を超える、または100を超える、または1,000を超える、または2,000を超える、または3,000を超える、または4,000を超える、または5,000を超える、または6,000を超える、または7,000を超える、または8,000を超える、または9,000を超える、または10,000を超える、または25,000を超える、または50,000を超える、または75,000を超える、または100,000クローンを超える密度でまく。さらに他の実施形態においては、発現ライブラリークローンを1mm2当たり、ほぼ1,000〜ほぼ10,000クローンの密度でまく。なおさらに他の実施形態においては、発現ライブラリークローンを1mm2当たり、1,000〜10,000クローンの密度でまく。上記クローンは可溶性結合分子の集団を発現する個々の細胞(例えば、酵母または細菌)であってもよいしまたは可溶性結合分子の集団をコードするファージに感染させた細菌細胞であってもよい。具体的な実施形態においては、上記クローンは可溶性結合分子の集団をコードする溶菌性ファージに感染させた細胞である。可溶性結合分子を細胞から分泌させてもよいしまたは溶解後の細胞から放出させてもよいことを具体的に意図する。溶解は、限定されるものでないが、化学的方法(例えば、アルカリ溶解)および 生物学的 方法(例えば、溶菌性ファージによる感染)を含む、当業者に公知のいくつかの方法により促進することができる。可溶性結合分子のクローンは、いずれかの供給源(例
えば、ランダムペプチドライブラリーおよびコンビナトリアル化学ライブラリー)から誘導されかつ上記の密度で固体支持体に固定化された個々の分子のプールでありうることも意図する。
【0083】
5.4 リガンドおよび検出
本明細書で使用する用語「リガンド」には、リガンドに対して結合アフィニティを有する結合分子により認識されうるいずれかの分子が含まれる。リガンドはタンパク質、DNA、脂質、炭水化物または小分子であってもよい。一実施形態においては、可溶性リガンドは、限定されるものでないが、ペプチド、ポリペプチド、核酸、炭水化物、脂質、または有機化合物を含む、結合分子と選択的に結合することができるいずれの分子であってもよい。当業者は、先に結合分子として考察した分子はリガンドであってもよいことを理解するであろう。例えば、細胞表面レセプター(例えば、インテグリン、成長因子レセプターまたはサイトカインレセプター)をリガンドとして用いて、結合分子の集団(例えば、ランダムペプチド、抗体またはコンビナトリアル化学ライブラリー)をスクリーニングし、これらのレセプターに対するアゴニストまたはアンタゴニストとして利用することができる結合分子を同定することができる。
【0084】
一実施形態においては、リガンドは少なくとも1つの細胞表面タンパク質(例えば、グリコシル化表面タンパク質)由来のドメインまたはペプチド、癌関連タンパク質、サイトカイン、ケモカイン、ペプチドホルモン、神経伝達物質、細胞表面レセプター(例えば、細胞表面レセプターキナーゼ、7回膜貫通レセプター、ウイルスレセプターおよびコ-レセプター)、細胞外マトリックス結合タンパク質、細胞-結合タンパク質、病原体の抗原(例えば、細菌抗原、マラリア抗原など)を含む。他の実施形態においては、リガンドは、チロシンキナーゼまたはチロシンキナーゼリガンド由来の少なくとも1つのドメインまたはペプチドを含む。意図するチロシンキナーゼおよびチロシンキナーゼリガンドには、限定されるものでないが、レセプターチロシンキナーゼ(例えば、EGFR/上皮性成長因子、Eph/Ephrin、FGF/繊維芽細胞成長因子、FN/フィブロネクチンインスリン、IGF/インスリン様成長因子、NGF/神経成長因子、PDGF/血小板由来の成長因子、およびTie/アンギオポエチンレセプターファミリー)および非レセプターチロシンキナーゼ(例えば、Src、Tec、JAK、Fes、Abl、FAK、Csk、およびSykファミリー)が含まれる。
【0085】
一実施形態においては、本発明のスクリーニング方法を用いて単一リガンドに対して選択的アフィニティを示す結合分子をスクリーニングする。さらに、本発明のスクリーニング方法を用いて複数のリガンドと結合する結合分子の集団をスクリーニングしうることを具体的に意図する。
【0086】
リガンドおよび/またはリガンド集団は、結合分子の必要性および意図する用途ならびにリガンドまたはリガンド組成物の特徴に応じて選択しうる。例えば、本発明のスクリーニング方法を用いて、単一リガンドに対してまたは全細胞もしくは組織と同じように複雑なリガンド集団ならびにほんの僅かな種の単一リガンド集団に対して選択的アフィニティを示す結合分子を同定することができる。
【0087】
リガンドまたはリガンド集団は実質的に精製されていてもまたは様々な量の他の無関係な種を含有してもよい。例えば、単一リガンドは、よく特徴づけられて高度に精製された分子(例えば、組換えタンパク質)であってもよい一方、分子の集団はいくつかの数の供給源(例えば、1以上のリガンドの部分的にまたは実質的に精製された調製物、または細胞ライセートまたはホモジネートの粗調製物を含む)由来であってもよい。さらに本発明はまた、単一リガンド、またはリガンドの集団(ここで、リガンドは細胞ライセート中のポリペプチドまたは他の高分子である)に対して、選択的なアフィニティを有する結合分子を同定するスクリーニング方法も提供する。生化学的によく特徴づけられていないリガンド(例えば、細胞ライセート)を本発明の方法で使用しうることを具体的に意図する。さらに、本発明のスクリーニング方法を用いて、ある集団の1つまたは少数のメンバーに対して選択性のある結合分子を同定することができる。例えば、もしあるリガンドの集団のいずれかのメンバーに対して選択性のある結合分子を生産するのが所望であれば、それぞれの個々のメンバーを単一集団中に組み合わせて、本発明のスクリーニング方法を用いて同時にスクリーニングすることができる。リガンドの単一集団は、限定されるものでないが、単一細胞内にいくつかの異なる分子を一緒に発現させることによりいくつかの異なるリガンド調製物を一緒にプールすることを含むいくつかの方法により作製することができる。
【0088】
スクリーニング方法に用いるリガンド集団は、色々なサイズの実質的に精製した分子または粗細胞調製物またはその他の複合体組成物から成ってもよい。一般に、単一リガンドまたは2つの異なるリガンド種の単純な集団を使用する。しかし、3、4、5、6、7、8、9、10以上の異なるリガンドから構成された単純なリガンド集団を使用してもよい。本発明のスクリーニング方法を、細胞内の十〜数百の異なるリガンド種を含有する中度のリガンド集団、ならびにほぼ数万の異なるリガンド種、例えば、細胞内の多数の異なる分子を含有する複雑なリガンド集団に対して使用しうることも具体的に意図する。集団サイズとタイプの選択は、結合分子の必要性および意図する用途に依存しうる。当業者は、具体的な必要性にとって好適である集団のサイズとタイプを熟知するであろう。全ての場合に、リガンドおよび/またはリガンド集団は、実質的に精製されてもよいしまたは様々な量の他の無関係な種を含有してもよい。
【0089】
一実施形態においては、リガンドおよび/またはリガンド集団は可溶性である。いくつかのリガンドは固有に可溶性である一方、他のリガンドはさらなる操作を必要としうるのであって、それらの操作としては、限定されるものでないが、それらを可溶化するためのさらなる成分(例えば、界面活性剤、カオトロピック剤)の導入が挙げられる。可溶性リガンドの使用は、結合分子と相互作用することができない不溶性リガンド凝集物の生成を防止することにより、固定化された結合分子の集団の効率的なスクリーニングを容易にし得ることを具体的に意図する。その自然の状態でリガンドと相互作用する結合分子を所望する場合、リガンドを可溶化するために必要な操作はコンフォメーション改変をもたらしてリガンドの自然の状態を認識しない結合分子を同定しうるので、目的のリガンドは固有に可溶性であることを期待している。
【0090】
本発明は、少なくとも1つのリガンドに対して選択的なアフィニティを示す少なくとも1つの結合分子を同定するために、結合分子の集団の超ハイスループットスクリーニングを行うスクリーニング方法を提供する。結合分子は、結合分子の集団を少なくとも1つのリガンドと接触させることにより同定する。次いでリガンド自体を適当な検出方法により検出する。
【0091】
一実施形態においては、固定された結合分子と選択的に結合したリガンドを検出する。結合したリガンドを、例えば、発光、放射性同位体、発色の検出に関わりうる直接的または間接的方法、またはリガンドの検出を可能にするいずれかの方法により検出しうることを具体的に意図する。リガンドの直接検出は、限定されるものでないが、リガンドの容易に検出可能な部分による共有結合性修飾、例えば、ヨウ素化などの放射性同位体を用いる化学的修飾を含む当業者に馴染みのある多数の技法により実施することができる。直接的方法はまた、適当な検出分子のリガンドとの融合にも関わり、例えば、リガンドをルシフェラーゼと融合して発光により検出してもよいし、または酵素(例えば、lacZ、西洋わさびペルオキシダーゼ、アルカリホスファターゼおよび下記)と融合して適当な比色測定により検出してもよい。
【0092】
リガンドの間接的な検出は、限定されるものでないが、リガンドと相互作用することが公知の第2の分子を用いることを含む当業者に周知の方法により実施することができる。第2の分子はそれ自体が直接的または間接的方法で検出される。例えば、リガンドをビオチン化して、適当に標識したアビジン分子を用いて検出する。ハプテン分子を類似の方法(上記)に利用することができる。さらに、リガンドに特異的な抗体をリガンドに特異的な第1抗体と相互作用しうる第2抗体を用いて検出してもよく、この場合、再び直接的検出について先に記載した検出方法を用いる。
【0093】
直接的および間接的検出は両方とも、リガンドまたは間接的検出に用いる第2分子を検出可能な基質とカップリングすることにより実施することができる。検出可能な基質の例としては、様々な酵素、補欠分子族、蛍光物質、発光物質、生物発光物質、放射性物質、ポジトロン放出金属、および非放射性常磁性金属イオンが挙げられる。例えば、当技術分野で公知の技法を用いて、検出可能な物質をリガンドを認識する抗体(またはそのフラグメント)と直接、また中間物質(例えば、当技術分野で公知のリンカー)を介して間接にカップリングまたはコンジュゲートすることができる。例えば、本発明による診断に用いる抗体とコンジュゲートできる金属イオンに対する米国特許第号4,741,900を参照されたい。好適な酵素の例としては、西洋わさびペルオキシダーゼ、アルカリホスファターゼ、β-ガラクトシダーゼ、またはアセチルコリンエステラーゼ;好適な補欠分子族複合体の例としてはストレプトアビジン/ビオチンおよびアビジン/ビオチン;好適な蛍光材料の例としてはウンベリフェロン、フルオレセイン、フルオレセインイソチオシアナート、ローダミン、ジクロロトリアジニルアミンフルオレセイン、ダンシルクロリドまたはフィコエリトリン;発光物質の例としてはルミノール;生物発光物質の例としてはルシフェラーゼ、ルシフェリン、およびエクロリン;そして好適な放射性材料の例としては125I、131I、111Inまたは99Tcが挙げられる。
【0094】
他の実施形態においては、リガンドはさらに検出ドメインを含む。検出ドメインはリガンドの検出を容易にするいずれの分子であってもよい。一実施形態においては、検出ドメインは検出シグナルの増幅を可能にして検出の感受性をより高くしうる。例えば、検出ドメインは、リガンドと相互作用して次いでそれ自体が検出されて検出シグナルの増幅をもたらす第2分子(例えば、ビオチン分子または抗体)の数を拡大する働きをなしうる。
【0095】
特定の実施形態においては、ポリペプチドリガンドをその検出方法が存在する1以上のドメインを含むように遺伝子組換えによって作る。例えば、ベクターとしては、限定されるものでないが、lac Z-融合タンパク質が生産されるように、リガンドコード配列が個々にlac Zコード領域のフレーム内でベクターとライゲートされている大腸菌発現ベクターpUR278(Rutherら、1983、EMBO 12:1791)、pINベクター(InouyeおよびInouye、1985、Nucleic Acids Res. 13:3101-3109; Van Heeke & Schuster、1989、J. Biol. Chem. 24:5503-5509)などが挙げられる。lac Zタンパク質は、比色検出方法を用いて直接検出してもまたはlac Zを特異的に認識する抗体を用いて間接に検出してもよい。さらにlac Zに特異的な抗体を、検出シグナルの増幅をもたらすリガンドを認識する抗体と組み合わせてもよい。
【0096】
不溶性ポリペプチドリガンドの場合、pGEXベクターを用いてリガンドポリペプチドをグルタチオン5-トランスフェラーゼ(GST)との融合タンパク質として発現させてもよい。一般に、リガンドだけでは可溶性でなくとも、リガンド-GST融合タンパク質は可溶性でありうる。さらにリガンド-GST融合タンパク質は、溶解した細胞からマトリックスグルタチオンアガロースビーズへの吸着と結合およびその後の遊離グルタチオンの存在のもとでの溶出により容易に精製することができる。GSTドメインも検出ドメインとして機能する。リガンド-GST、リガンド-lacZおよび他の類似のリガンド-検出ドメイン融合物の作製は、大きい容易に検出される分子を加えずに検出するのが困難である小ペプチドリガンドにとって特に有利である。
【0097】
他の実施形態においては、リガンドはさらに、リガンドの多量体形成を可能にする検出ドメインを含む。例えばリガンドを、二量体化を促進しうる抗体Fcドメインと融合することができる。ポリペプチドを抗体の定常領域と融合またはコンジュゲートする方法は当技術分野で公知である。例えば、米国特許第5,336,603号、第5,622,929号、第5,359,046号、第5,349,053号、第5,447,851号、第5,723,125号、第5,783,18号、1第5,908,626号、第5,844,095号、,および第5,112,946号;EP 307,434; EP 367,166;EP 394,827;国際特許出願公開WO 91/06570、WO 96/04388、WO 96/22024、WO 97/34631、およびWO 99/04813;Ashkenaziら, 1991, Proc. Natl. Acad. Sci. USA 88: 10535-10539;Trauneckerら, 1988, Nature, 331:84-86;Zhengら, 1995, J. Immunol. 154:5590-5600;およびVilら, 1992, Proc. Natl. Acad. Sci. USA 89:11337- 11341を参照されたい。あるいは、リガンドをロイシンジッパードメインと融合してもよい。可溶性オリゴマータンパク質を生産するための好適なロイシンジッパードメインの例は、PCT出願WO94/10308に記載され、そして肺界面活性タンパク質D(SPD)から誘導されたロイシンジッパーはHoppeら(FEBS Letters 344:191, 1994)に記載されている。それと融合した異種タンパク質の安定な三量体化を可能にする修飾されたロイシンジッパーの使用は、Fanslowら(Semin. Immunol. 6:267-278, 1994)に記載されている。ロイシンジッパーペプチドと融合した可溶性ポリペプチドを含む組換え融合タンパク質を好適な宿主細胞に発現させ、そして生成する可溶性オリゴマーを培養上清から回収する。ある特定のロイシンジッパー部分が優先的に三量体を形成する。一例は肺界面活性剤タンパク質D(SPD)から誘導されるロイシンジッパーであって、Hoppeら(FEBS Letters 344:191, 1994)および米国特許第号5,716,805に記載されている。
【0098】
一実施形態においては、リガンドを認識して結合する結合分子を発現するクローンを単離する。固体支持体は、リガンドを認識して結合する結合分子を発現するクローンを含有するクローンのサブセットを単離するためのテンプレートを提供しうることを具体的に意図する。このより小さい集団は、次いで本発明のスクリーニング方法の改変法を用いてスクリーニングすることができる。上記改変はクローンのサブセットをより低い密度でまくステップを含んでなる。特定の実施形態においては、クローンのサブセットを、単一クローンが単離されるには十分低いがサブセット中に存在する各クローンが固体支持体上で少なくとも1回、表されるには十分高い密度でまく。リガンドを認識して結合する結合分子を発現するクローンを含有するクローンのサブセットを当業者に公知の代わりの方法によりスクリーニングしうることも意図する。代わりの方法としては、限定されるものでないが、ELISAアッセイおよびFACS分析が挙げられる。
【0099】
5.5 選択マーカーと選択
本発明のライブラリー生産方法は、選択マーカーをコードするポリヌクレオチドとライゲートした分子をコードするポリヌクレオチドを含むクローンのライブラリーを作製する第1ステップを含んでなる。選択マーカーは一般に以下に記載されたものでありそしてある特定の具体例を実施例3に詳述している(上記)。選択マーカーをコードするポリヌクレオチドは、ライブラリークローンを含む形質転換した宿主細胞の選択および/または維持のために必要なポリヌクレオチドと同じであってもよいし、または第2の選択マーカーをコードする第2のポリヌクレオチドを利用してもよいことを具体的に意図する。
【0100】
選択マーカーは、容易に選択し、および/またはスクリーニングすることができる所望の形質をコードする多様な遺伝子のグループを包含する。選択マーカーは3つの一般的カテゴリーに分類することができる;1)特定の薬物に対する耐性の形質を与える薬物耐性マーカー遺伝子。例えば、β-ラクタマーゼ遺伝子はアンピシリンおよびカルベニシリンに対する耐性を与え、ネオマイシンホスホトランスフェラーゼII型(NPT II)はネオマイシン/カナマイシン/G418に対する耐性を与え(Colberre-Garapinら, 1981, J. Mol. Biol. 150:1)、クロラムフェニコールアセチルトランスフェラーゼはクロラムフェニコールに対する耐性を与え(Herrera-Estrellaら, 1983, Nature 303, 209-213)、ジヒドロ葉酸レダクターゼ(dhfr)はメトトレキセートに対する耐性を与え(Wiglerら, 1980, Natl. Acad. Sci. USA 77:3567;O’Hareら, 1981, Proc. Natl. Acad. Sci. USA 78:1527)、gptはミコフェノール酸に対する耐性を与え(Mulligan & Berg, 1981, Proc. Natl. Acad. Sci. USA 78:2072)、そしてハイグロマイシンホスホトランスフェラーゼ(hygro)はハイグロマイシンに対する耐性を与える(Santerreら, 1984, Gene 30:147)。2)形質転換された細胞が必須代謝物(通常、アミノ酸)を合成できるようにする代謝または栄養要求マーカー遺伝子(細胞は上記代謝物を生産できないが、増殖のために必要であり、上記代謝物の非存在のもとで細胞が増殖することを可能にする遺伝子)。例えば、HIS3、LEU2、TRP1およびURA3は、それぞれ、ヒスチジン、ロイシン、トリプトファンおよびウラシルを欠く培地においてある特定の栄養要求酵母菌株が増殖する能力を与える。3)タンパク質またはタンパク質ドメインをコードするスクリーニング可能なまたは精製マーカー遺伝子であって、様々な研究室の試験または精製技法を介して同定することができるかまたはタンパク質ドメインを発現するクローンの精製/同定を容易にする遺伝子。例えば、X-galを用いて検出されるβ-ガラクトシダーゼ(beta-gal)(Helmerら、1984、Bio/Technology 2、520-527)、炭化水素化合物を用いて検出されるルシフェラーゼ(lux)(Konczら、1987、PNAS 84、131-135)、またはカルモジュリンアフィニティ精製を用いて精製されるカルモジュリン結合ドメインなどのタンパク質ドメイン。
【0101】
当業者は、機能性分子を発現しないクローン(例えば、未熟停止コドンまたはフレームシフト突然変異を有する分子)は恐らく融合タンパク質を作製しないかまたは非機能性融合タンパク質を作製しうることを理解しうる。かかる非機能性分子を発現するクローンは、機能性融合タンパク質を発現しないクローンが排除される選択条件下でクローンのライブラリーを増殖することにより選択することができる。従って、本発明のライブラリー生産方法はさらに、機能性分子を発現するクローンを選択する条件下でクローンのライブラリーを増殖する第2のステップを含んでなる。
【0102】
当業者には、本発明のライブラリー生産方法に使用する選択マーカーおよび選択条件は明らかであろう。選択マーカーおよび選択条件は、作製するライブラリーの選択ならびに利用する発現ベクターおよび宿主細胞に応じて選ばれるであろう。
【実施例】
【0103】
6. 実施例
以下、本発明は、次の実施例を参考にして説明される。これらの実施例は、説明するためだけに与えられたものであって、本発明は、これらの実施例に限定されると決して推量してはならないが、むしろ、本明細書に記載の開示の結果として明らかになるいずれかおよび全ての変化を包含すると推量されるべきである。
【0104】
6.1 実施例1
ヒトscFvライブラリーの超ハイスループットスクリーニング
ほぼ2x109メンバーを含有する大きいファージscFv発現ライブラリーをヒトリンパ節および脾臓組織(図2を参照)から作製した。全ライブラリーをビオチン化EphA4-Fc融合タンパク質を用いてスクリーニングした(図1、3および4を参照)。3回のスクリーニングの後、24クローンのパネルを単離すると、そのうちの20クローンはELISAアッセイにおいて強い結合を示した(図4参照)。
【0105】
6.1.1 材料および方法
ナイーヴなヒトscFvライブラリーの生産: ヒトscFvライブラリーの作製は本質的に、Gaoら, 1999, J. Am. Chem. Soc. 121:6517-6518;およびMaoら, 1999, Proc Natl Acad Sci USA 96:6953-8に記載の通りであるが、次の改変を加えた:固定化ドメインとして作用しうるエピトープタグ(図2Bを参照)を補ったクローニング部位を遺伝子操作でM13中に作製し、そして発現ベクターとして用いた。さらにここで用いたscFvはその自然型(すなわち、M13コートタンパク質と融合されていない)である。概要を説明すると、リンパ節および脾臓組織から得たヒトmRNAを逆転写し、そしてVHおよびVL配列をPCRにより増幅した。オーバーラップPCRを利用してVHおよびVL配列を接合し、scFv遺伝子をコードするポリヌクレオチドを生成させた。次いでscFv遺伝子をM13ファージ発現ベクター中にライゲートし、そして大腸菌中に形質転換した。模式図は図2Aを参照されたい。ライブラリーの複雑度はほぼ2x109である。
【0106】
リガンドのビオチン標識化: 市販のEphA4-Fc(R&D System、Cat# 369-EA-200)をPBS(pH7.2)に対して透析した。透析後のEphA4-Fcの最終濃度は0.83mg/mlであった。1mlのEphA4-Fcをビオチン化に用いた。使用直前に、Sulfo-NHS-LC-ビオチン(PIERCE、Cat#: 21335)の10mM溶液を水で調製した。16.6μlの10mMビオチン試薬溶液を1mlの透析したEphA4-Fcに加え(ビオチンのモル過剰倍率は20である)、そして反応混合物を氷上で2時間インキュベートした。未反応ビオチンは、PBS(pH7.2)に対する透析により、ビオチン化EphA4-Fcから除去した。
【0107】
超ハイスループットスクリーニング:第1日 午後に、M9プレートからのTG1の単一コロニーを用いて2XYTに接種した。培養物を30℃で一晩インキュベートした。
【0108】
第2日 TG1を接種する:TG1-Blueの一晩培養物を2XYT中に1:100で希釈し、37℃にて250rpmでほぼ1.5〜2時間インキュベートした(OD600nm=0.5〜0.8)。
【0109】
TG1をファージに感染させる:ファージをPBS中に、キャプチャーリフトアッセイ用にほぼ1010pfu/ml、そしてファージ滴定用にほぼ105pfu/mlの濃度で希釈した。上層寒天溶液(SB+0.7%寒天)を12mlチューブ中にアリコート(6ml/チューブ)をとり、そして50℃水浴中で使用まで保持した。次の混合物を調製した:
a. ステップ1からのTG1:800μl、
b. IPTG(1M):6μl、および
c. 希釈したファージ溶液:6μl。
【0110】
400μlの上記混合物を6mlの上層寒天に加え、混合し、次いで静かに100mmプレーンLB寒天プレートの上層に注ぎ、そして室温にて10分間静置した。次いでまいたプレートを37℃にてほぼ5〜6時間インキュベートした。
【0111】
フィルターを調製する: プレートのインキュベーションの間、83mm NC膜の片側を鉛筆で標識をつけた。抗Flag抗体(M2Ab、4.9mg/ml、Sigmaより入手)をPBS(pH7.2)を用いて1:500に希釈し、そして100mmペトリ皿中に置いた。標識した膜を、標識した側を上にして抗体溶液の表面上に置いて、3時間、室温にて遅い速度のプラットフォーム振とう機上でインキュベートした。次いでフィルターを裏返し、さらに30分間インキュベートした。抗体溶液を除去し、そしてフィルターをPBS中の4%脱脂乳(Bio-Radから入手)を用いて2時間室温にてブロックした。次いでフィルターを3回PBSを用いてすすぎ洗い、化学フード内で乾燥した。
【0112】
細菌/ファージローン(lawn)上でのフィルターのインキュベーション: 細菌/ファージプレートをインキュベーターから取出した。単離したプラークが滴定プレート(ここでは108希釈プレート)上に見られた。乾燥したフィルターを標識した側を上にして、細菌/ファージローンの表面上に静かに敷き、フィルター位置を、フィルターの孔に針を突っ込んでマークした。次いでプレートを室温(25℃)にて一晩(湿容器内で)インキュベートした。
【0113】
第3日 フィルターを注意深くプレートから剥がし、PBSにより2回、次いでTPBS(PBS+0.1% Tween20)により6回(標識した側を上にして3回および標識した側を下にしてさらに3回)、真空洗浄器を用いて洗浄した。次いで膜を、スーパーブロッキングバッファー(PIERCE)中の1%BSAに1:1000で希釈したEphA4-FC-ビオチンを用いて、標識した側を下にしてインキュベートした(4ml/フィルター)。EphA4-FC-ビオチンのストック濃度は0.8mg/mlであった。次いで膜を3時間、室温にてロッキングプラットフォーム上で振とうしながらインキュベートした。次いで膜を6回(標識した側を上にして3回および標識した側を下にしてさらに3回)、TPBSにより真空洗浄器を用いて洗浄した。次いで洗浄したフィルターをスーパーブロッキングバッファー(PIERCE、10ml/フィルター)中の1%BSAに1:4000で希釈したアビジン-AP溶液を用いて、振とうしながら、1時間インキュベートした。この最後のインキュベーションの後に、フィルターを次いで上記のように洗浄し、3〜5回、PBSによりすすぎ洗いした。洗浄バッファーを取り除いて、NBT/BCIT溶液(PIERCE、5ml/フィルター)をフィルターを含有するプレートに加えた。発色させ(3〜5分)、発色したフィルターを乾燥し、プレートを整列し、そして第2回スクリーニングに対するポジティブプラークを拾った。
【0114】
抗体スクリーニング/Elisa(2.5mlスケール):第1日- 午後に、M9プレートからのTG1の単一コロニーを用いて2XYTに接種した。培養物を30℃で一晩インキュベートした。
【0115】
第2日 TG1を接種する:TG1-Blueの一晩培養物を2XYT中に1:100で希釈し、37℃にて250rpmでほぼ1.5〜2時間インキュベートし(OD600nm=0.5〜0.8)、次いでチューブ(2.5ml/チューブ)にアリコートをとった。
【0116】
高力価ファージ調製: よく単離されたプラークを拾い、これを用いて上記からの2.5ml培養物に接種した。チューブを一晩、37℃にて、250-300rpmで振とうしながらインキュベートした。
【0117】
コート抗原: 抗原(ここでは、EphA4-His融合タンパク質およびネガティブ対照としてSynagisを用いた)をコーティングバッファー(炭酸塩バッファー、pH9.6)でほぼ5-10μg/mlに希釈し、マイクロタイタープレートに25μl/ウエルを加え、そして4℃にて一晩インキュベートした。
【0118】
第3日 ELISA-ブロッキング: 過剰のコーティング溶液を除去し、ウエルをPBS(ブロッキングバッファー)中の4%脱脂乳を用いて50μl/ウエルで1時間、37℃にてブロックした。一晩インキュベーションした細胞培養物を3500rpm x 20分の遠心分離により採集した。過剰のブロッキング溶液を除去し、25μl/ウエルの一晩培養物上清を加えた。37℃にて1時間、インキュベートした。プレートをプレート洗浄器を用いて洗浄し、そして25μl/ウエルの抗Flag M2抗体(ブロッキングバッファー中に1:1000希釈)を各ウエルに加えた。次いでプレートを30分間37℃にてインキュベートした。プレートを再びプレート洗浄器を用いて洗浄しそして25μl/ウエルのヤギ抗-マウス(IgG-H+L)-HRPコンジュゲート(ブロッキングバッファーに1:1000希釈)を各ウエルに加えた。プレートを30分間、37℃にてインキュベートした。プレートをプレート洗浄器を用いて洗浄し、次いでピペット(ピペットのアップアンドダウンを20回)により1回、および蒸留水によりさらに10回洗浄した。発色: 25〜35μl/ウエルのTMB基質を各ウエルに加え、ほぼ5分間発色させた。反応を、等容積の0.18M H2SO4を加えることにより停止しそして吸収を450nmにて読み取った。
【0119】
抗体精製(800mlスケール):第1日 午後に、M9プレートからのTG1の単一コロニーを用いて2XYTに接種した。培養物を30℃で一晩インキュベートした。
【0120】
第2日 2mlの一晩細胞培養物を200mlの2XYT培地中に希釈し、そして振とう機内で37℃にて250rpmでOD600nm〜0.8まで増殖した。250μlの一晩培養の高力価ファージ(典型的にはほぼ1012pfu/ml)を用いて200ml細胞に15分間、室温にて感染させた。600mlの2XYT培地を200mlの感染した細胞に加えた。感染した細胞を37℃にて1時間、次いで30℃にて 一晩増殖した。
【0121】
第3日 細胞を、8000rpm(Beckman JA-10回転子)で20分間、遠心分離によって取り出し、そして上清を0.45μmフィルターを通過させることにより濾過した。濾過した上清を、PBSで前洗浄した抗FLAG M2アガロースアフィニティカラムに供給した。次いでカラムを少なくとも10カラム体積のPBSを用いて洗浄した。次いで抗体(scFv)をカラムから溶出バッファー(100mMグリシン、pHを3に調節)を用いて溶出した。1M Tris-HCl(pH 8.0)を溶出した抗体に加えて中和した。中和した抗体を次いでPBSに対して透析してほぼ1mlに濃縮した。
【0122】
6.1.2 考察
キャプチャーリフトスクリーニング方法は通常、ハイスループットスクリーニング方法に固有のいくつかのバイアスを避けるために使用される。しかし、大きい多様なライブラリーに対しては価値が限られているので、その使用は小ライブラリー集団のスクリーニングに限られている。キャプチャーリフトスクリーニングは、高いアフィニティをもつ抗体クローンの選択に対して、ヒト化抗体の選択に対して、そして、さらに最近には、小ライブラリー(多様性ほぼ105)からの新規抗体分子の発見に対して首尾よく使用されている。本明細書において、本発明者らは初めて、非常に大きい集団からの稀なクローンの同定を可能にする非常に大きい結合分子ライブラリーの迅速かつ効率的なスクリーニングを可能にする著しく改善されたキャプチャーリフト手法を記載する。
【0123】
ほぼ2x109メンバーを含有する大きいファージscFv発現ライブラリーを、ヒトリンパ節および脾臓組織から作製した。そのライブラリーをほぼ3800クローン/mm2の超高密度でまいた。図3は、第1回(3A)、第2回(3B)、ならびに第3回および最終回(3C)スクリーニングからのポジティブクローンを含有する代表的フィルターを示す。次いでそのライブラリーを、ビオチン化EphA4-Fc融合タンパク質を用いてスクリーニングした。EphA4-Fc融合体の使用は、より大きい数のビオチン分子が各リガンドと結合することを可能にし、アビジン-APによる検出時にシグナルの有意な増幅をもたらす。化学発光に基づく検出方法(例えば、Salernoら, 2003, J Chromatogr B Analyt Technol Biomed Life Sci 793: 75;Mattsonら, 1996, Anal Biochem 240: 306;Kricka, 1991, Clin Chem 37: 1472を参照)の使用は、検出用2次分子と融合したリガンドの使用の有無に関わらず検出シグナルの有意な増幅を可能にしうるし、かつさらに多数のクローン数を各プレート上にまくことを可能にするであろう。
【0124】
提示した手法を用いると、少なくとも3000万クローンを単一の82mmニトロセルロース膜上でスクリーニングすることができる。これは、従来記載されているキャプチャーリフト方法よりフィルター1枚当たり300倍多いクローンである。伝統的なキャプチャーリフト方法は、このサイズをすっかりスクリーニングするために1ライブラリーに対して20,000〜40,000プレート(100mm)を必要としよう。本明細書に記載の超高密度プレーティングおよびスクリーニング方法を用いると、全ライブラリーをスクリーニングするために、わずかほぼ67プレートしか必要としない。
【0125】
6.2 実施例2
超ハイスループットスクリーニングによる抗イディオタイプscFvの単離
実施例1で作製したライブラリーを、MEDI-AAA、抗インターフェロンα抗体の抗原結合ドメインを特異的に認識する抗イディオタイプ抗体を同定しかつ単離するためにも利用した。該ライブラリーをビオチン化MEDI-AAA(Fab)2フラグメント(図5)を用いてスクリーニングした。2回のスクリーニングの後に4クローンを単離し、その1つはELISAアッセイでMEDI-AAA抗体と強い結合を示す一方、いくつかの無関係な抗体と結合しなかった(図6参照)。
【0126】
6.2.1 材料と方法
MEDI-AAA(Fab)2の調製: MEDI-AAA(Fab)2を、固定化したペプシン試薬を用いて調製した(Pierce cat. 20341)。製造業者の指示に従って、500μgのMEDI-AAA抗体を消化した。消化した抗体を、遠心分離によりペプシン樹脂から分離し、そして溶出剤をタンパク質Aカラム上に流して抗体Fc断片を除去した。精製した(Fab)2をPierce濃縮溶液を用いて、製造業者の推奨に従って濃縮し、その後、1 X PBS中にpH7.2で4℃にて一晩透析した。最後のタンパク質を10%Bis-Trisタンパク質ゲル(図5)上でMOPSバッファー(Invitrogen cat. NP0001)を用いて分析した。
【0127】
MEDI-AAA(Fab)2ビオチン化: 200μgのMEDI-AAA(Fab)2を、NHS-LC-ビオチン試薬を用いて製造業者の取扱説明書(Pierce cat.21338)に従いビオチン化した。20ビオチン/(Fab)2分子の比を用いて、発色過程の高い感度を確実にした。組み込まれなかったビオチンはNAP5 脱塩カラム(Pierce cat. 17-0853-01)を用いて除去した。最後の標識した(Fab)2を、上記のように、ならびに無処置IgGおよび無標識(Fab)2とともに変性および非変性条件下で分析した(図5)。
【0128】
ファージ培養物: 最小培地M9(Teknova cat. M2100)プレート上で増殖したTG1細菌の単一コロニーを用いて1mlの2XYT(Teknova cat. Y0167)に接種し、30℃にて一晩インキュベートした。一晩TG1培養物を用いて2XYT中の培養を0.1 OD600 37℃、250 rpmで開始して0.5〜0.8 OD600の中対数(mid log)に到達するまで続けた。上層寒天(スーパーブロス、0.7%寒天)をマイクロ波で融解しそして15mlファルコンチューブ中にアリコート(6ml/チューブ)をとり、チューブを50℃水浴で必要なだけインキュベートした。キャプチャーリフトファージ発現ライブラリー(2.08x109pfu/μl)をそれぞれ、1:40、1:400および1:4x106に希釈して、ほぼ1x107pfu/μl、1x106pfu/μlおよび1x102pfu/μlを得た。TG-1細菌を感染させるために、1μlの1x107pfu/μlおよび1μlの1x106pfu/μl希釈したファージを、800μlの中対数(mid log)TG-1とともに6μlの1M IPTGを含有する5つのエッペンドルフチューブに加えた。TG1の1つのチューブだけは1x106pfu/μl希釈(滴定プレート)で感染させ、そしてこのTG1の残りのチューブはネガティブ対照として用いた。感染させたTG-1培養物を上層寒天のチューブと組み合わせて、予熱したLBプレート(100mm)上に注いだ。上層寒天を10分間室温にて固化させた後に、プレートを37℃にて5〜6時間インキュベートした。
【0129】
フィルター調製: 10枚のニトロセルロース膜(Protron Bioscience cat. 10401116、82 mm)の片側を、鉛筆を用いて抗原および膜数を記入して標識した。抗フラグM2抗体(sigma cat.)を、100mlの1X PBS pH7.2中に1:500で希釈し、この溶液の10mlを10cmペトリ皿のそれぞれに加えた。ニトロセルロース膜を、標識した側を上にして抗フラグM2抗体(Sigma Cat. F3165)中で3時間、低速度振とう機上で室温にてインキュベートした。最初のインキュベーション後に、ニトロセルロース膜を逆転してさらに30分間インキュベートした。膜を簡単にすすぎ洗いし、10 mlの1X PBS pH 7.2中の4%脱脂乳(Bio-Rad cat. 170-6404)を用いて2時間ブロックした。乳溶液を取出した後に、膜を3回、1X PBS pH 7.2中で洗浄し、そしてフード内で乾燥させた。
【0130】
キャプチャーリフト選択: 滴定プレート(1x102希釈)上にプラークが現れた後に、プレートをインキュベーターから取り出した。ニトロセルロース膜を、scFvをキャプチャーする1x107および1x106プレートの上層寒天の表面上に注意深く重ねた。21ゲージ針を用いて12時の位置に1つ、4時の位置に2つそして8時の位置に3つの孔を開けて、プレート上のフィルターの方向と位置をマークした。次いでプレートを25℃にて一晩インキュベートした。1:4x106ライブラリー希釈プレートを用いて実ライブラリー力価をチェックした。サイドアームの付いたKonteの4Lフラスコとフィルターホルダー(Fisher cat. K953840-4090)を用いてフィルターを洗った。最初にフィルターを寒天表面から注意深く取り出し、そして両側の膜を3回PBST(1X PSB、0.1% Tween 20)を用いて、洗浄ボトルからのTPBSを供給する一方、ホルダーを真空に接続することによりすすぎ洗った。ハイブリダイゼーション溶液を作るために、0.1μg/mlのビオチン化ニュートロステンシン(neutrostensin)、4μgのビオチン化MEDI-AAA(Fab)2および100μgのGEA44抗体を、スーパーブロック溶液(Piercecat. 37515)中の1%BSAに加えた。それぞれの膜を、10cmペトリ皿に入った4mlのハイブリダイゼーション溶液中で静かに攪拌しながら3時間室温にてインキュベートした。フィルターホルダーと真空を用いて、膜の両側を3回、PBSTにより洗浄した。フィルターを、10mlのブロッキングバッファー中の1:4000アビジン-AP(Pierce Cat. 31002)を用いて、1時間室温にて静かに振とうしながらインキュベートした。フィルターを5mlのNBT/BCIT(Piercecat.34042)溶液により発色する前に、フィルターを3回両側をPBSTにより洗浄した。褐色のポジティブドットが現れた後に、フィルターを水を用いてすすぎ洗いして反応を停止した。フィルターをフード内で乾燥しそして透明シート上で光コピーをとった。位置と方向をマークするフィルターに作った孔を用いて、プラークプレートを透明シート上に置き、寒天の針孔を透明シート上の針穴と整列させた。ポジティブスポット上のプラークを大
きいオリフィスマイクロピペット(VWR、Cat#:53503-614)のチップを用いて拾い、そして溶出バッファー(10mM Tris、100mM Nacl、pH 7.4)へ移した。溶出ファージ(ほぼ2〜5 x 102pfu/μl)で出発し、1:10希釈物(ほぼ2〜5 x 104pfu/μl)を用いて、先に記載の第2回のキャプチャーリフト用のプラークプレートを調製した。拾ったポジティブプラークのそれぞれに対して1枚のフィルターを使用した。
【0131】
ファージ生産: 第2回のキャプチャーリフトから溶出したポジティブプラークを1:200(1〜3 x 102pfu/μl)に希釈し、先の通り、10cmペトリ皿上にまいた。個々のプラークを250μlの溶出バッファーが入った96ウエルプレート中に拾った。プラークを寒天から溶出させた後に、200ulの溶出したファージを用いて、深いウエルプレート中の500μlの中対数(mid-log)TGIに接種した。ファージ生産のために、培養物を一晩、30℃にてインキュベートした。
【0132】
ELISAスクリーニング: ELISAマイクロタイタープレートを一晩、4℃にて50μlのMEDI-AAA(l5μg/ml)および対照抗体を用いてコーティングした。対照抗体には、市販ポリクローナルヒトIgG(Jackson Immunoresearch Lab、Cat. 009-000-003)、Synagis(登録商標)およびMEDI-AAAと高度の相同性を共有する軽鎖フレームワークを有する(フレームワーク残基81のうち77が同一である)無関係な抗体が含まれた。プレートをELISAブロッキング溶液(1 X PBST中に2%乳)を用いて1時間、室温にてブロックした。また48μlの一晩培養ファージ上清も、12μlの5XELISAブロッキング溶液を用いて1時間室温にてブロックした。インキュベーションの後、プレートを5回、1 X PBSTにより、Elx405プレート洗浄器を用いて洗浄し、そして50μlの前ブロックしたファージ上清を各ウエルに加えた。室温での1時間インキュベーションの後、プレートを再び、前記の通り洗浄し、そしてブロッキング溶液中の1:2000抗フラグM2(Sigma)抗体希釈液50μlを各ウエルに加えた。次いでプレートを室温にて1時間インキュベートし、そして5回、Elx405プレート洗浄器を用いて洗浄した。ELISAサンドイッチを完了させるために、ブロッキング溶液中の1:4000ヤギ抗-マウスHRP抗体(Pierce cat. 31164)50μlを各ウエルに加え、室温にて1時間インキュベートした。プレートを10回、PBSTにより、Elx405プレート洗浄器を用いて洗浄し、そして50μlのTMB基質(KPL cat. 52-00-03)を加えた。5〜10分間のインキュベーションの後、50μlの0.2M H2SO4を加えることにより反応を停止した。プレートを450nmにて読み取り、そして各抗体に対するシグナルをプロットした(図6AおよびB)。
【0133】
6.2.2 結果
実施例1で作製したファージscFv発現ライブラリーを、超ハイスループット(ほぼ126-1267クローン/mm2の密度でまいた)によりスクリーニングしかつビオチン化MEDI-AAA(Fab)2フラグメントを用いてスクリーニングして(図5参照)、抗イディオタイプscFvクローンを同定した。2回のスクリーニングの後、4クローンを単離し(データは示してない)、それらの1つはELISAアッセイでMEDI-AAA抗インターフェロンα抗体と強い結合を示す一方、BSAまたはいくつかの無関係の抗体と結合しなかった(図6参照)。特に抗イディオタイプscFvはヒトポリクローナル抗体調製物(図6B)または高度に関係した軽鎖を有する無関係なヒト抗体(対照)(図6AおよびB)と有意な結合を示さなかった。超ハイスループットスクリーニング方法の使用は、抗イディオタイプ抗体の迅速な同定を可能にした。ほぼ5.5x107のクローンを丁度10枚のプレート上で超ハイスループットスクリーニング方法を用いて速やかにスクリーニングしたが、伝統的方法は同じ数のクローンをスクリーニングするには少なくとも550枚のプレートを必要としたであろう。
【0134】
6.3 実施例3
発現可能な抗体ライブラリー構築と非機能性クローンの排除
プラスミドpUCKAを作製して、β-ラクタマーゼ遺伝子(アンピシリン/カルベニシリン耐性を与える)をコードするポリヌクレオチドとライゲートした3'-FLAGおよびHIS6エピトープタグの両方によるscFvのライブラリーのクローニングを容易にした。停止コドンを伴ってまたは伴わないでクローニングしたいくつかのscFvは、停止コドンを欠くクローンだけがカルベニシリン耐性であることを実証した。全ライブラリーをpUCKAベクター中にクローニングし、そして選択前の非機能性クローンの数はほぼ25%であることがわかった。非機能性タンパク質をコードするクローンを除去する選択の後に構築したファージライブラリーは、5x108を超える複雑度を有することがわかった。
【0135】
発現ベクターpUCKAの構築: ベクターpUCKAをpUC19から誘導した。プライマー対KanaFor/KanaRev、pUCFor/EcoRIRev、およびpUCRev/EcoRIForを利用し、pET-27b(Novagen)およびpUC19をそれぞれテンプレートとして、カナマイシン遺伝子およびpUC19主鎖を増幅した。3つのPCR断片をゲル精製しかつプライマーEcoRIFor/EcoRIRevを用いるオーバーラップPCRにより組み立てた。PCR産物をEcoR Iにより消化し、自己ライゲートし、そしてカナマイシンの選択のもとでXL1-Blue中に形質転換して、ベクターpUCK(示してない)を作製し、そのβ-ラクタマーゼ遺伝子をカナマイシンにより置き換えた。ポリリンカー(リボソーム結合部位、p3リーダー配列、Flag-タグ、およびHis-6-タグを含む)を、ファージ発現ベクターpMD102からプライマーHindIIIFor/EBNSRevを用いて増幅し、そしてHindIII/EcoRI部位中にクローニングしてベクターpUCK-1を作った。開始コドン、ATGのないβ-ラクタマーゼ遺伝子を、pUC19からプライマーAmpFor/AmpRevを用いて増幅し、そしてpUCK-1のSpeI/EcoRI部位中にクローニングしてベクターpUCKAを作製した。プライマー配列については表1を参照されたい。
【表1】
【0136】
クローンscFv F9、LX2、EA20、およびEA44: F9、LX2、EA20、およびEA44と名付けられたいくつかのscFvをコードし、そのC末端に開始コドンをもつかまたはもたないポリヌクレオチドを、pETHis(T7プロモーターの制御下のscFv発現ベクター)から切除し、pUCKAのSfi I部位中にクローニングし、XL1-Blue中に形質転換し、そしてLB-寒天/30μg/mlカナマイシンを含有するカナマイシンプレート上で拡大した。そのコロニーを拾い、カルベニシリンを伴うまたは伴わないLB/2XYT培地中に100μg/mlで接種した。scFv遺伝子の末端に停止コドンのないクローンだけがカルベニシリンの選択のもとで生存することができる(データは示してない)。このデータに基づくと、このベクターは、抗体ライブラリーから所望でない停止コドンまたはフレームワークを伴う抗体遺伝子を除去するように装備することができる。停止コドンをもたないscFvを発現するこれらのクローンの増殖速度をまた、色々なカルベニシリンの濃度で評価した(表2)。
【0137】
表2: 停止コドンのないscFv分子を発現するクローンの増殖速度
【表2】
【0138】
scFv発現ライブラリーの構築: 2x109メンバーをもつ本来のファージ発現ベクター(上記実施例1を参照)中のscFv遺伝子ライブラリーを、Sfi Iを用いて消化し、アガロース上でゲル精製し、そして、同じ制限酵素を用いて切断しておいたpUCKAベクター中にライゲートした。ライゲートした生成物を大腸菌XL1-Blue(テトラサイクリン耐性菌株)コンピテント細胞中にエレクトロポレートしてほぼ5×109の多様性を有する独立した形質転換体を得た。エレクトロポレーションの後、細胞を50枚のディッシュ(150mm×10mm;Nunc)中のLB寒天(2%グルコース、50μg/mlカナマイシン、および20μg/mlテトラサイクリンを含有する)上にまいて、一晩、30℃にてインキュベートした。クローンをプレートから300mlの2XYT培地中に掻き出した。掻き出した細菌を100μg/mlカルベニシリン、50μg/mlカナマイシン、および2%グルコースを含有する1リットルの2XYT培地に接種した。細胞を37℃にて4〜6時間、振とうしながら増殖してフレームシフトまたは停止コドンをもつライブラリー中の抗体遺伝子を除去し、発現可能な抗体ライブラリーを構築した。次いでグリセロールを残りの細菌に加えて最終濃度10%にして-80℃にて貯蔵した。
【0139】
系効率の試験: 一晩、30℃にて増殖した後、形質転換したライブラリーのLB-寒天ディッシュから192クローンを拾い、そして100μg/mlカルベニシリン、50μg/mlカナマイシン、および2%グルコースを含有する100μl2XYT培地の入った96プレート2枚中に接種した。37℃にて8時間、振とうしながら増殖した後、各ウエルから5μlを、各ウエル中に0.5ml 2XYTを含有する4つの深いウエルプレート(4つのうち、2つはカルベニシリン100μg/mlを含み、2つは含まない)中に移した。プレートを37℃にて一晩増殖し、そして直接観察により増殖(ポジティブ/ネガティブ)のスコアを付けた。全てのクローンはカルベニシリンを含まないと非常によく増殖することができる。しかし、100μg/mlカルベニシリンの選択のもとでは、75%(192クローンのうち144)しか生存できない。
【0140】
発現可能なファージ抗体発現ライブラリーの構築: 100μg/mlカルベニシリンの選択のもとで生存した細菌ライブラリーから、Maxiプラスミド単離キット(Qiagen)を用いて、プラスミドを抽出した。発現可能なscFvライブラリー遺伝子をSfiIを用いて消化し、ゲル精製し、そして同じくSfiIにより切断したファージ発現ベクターpMD102中にライゲートした。ライゲートした混合物、scFvライブラリーをXL1-Blueコンピテント細胞中に15個のキュベット(Bio-Rad)を用いてエレクトロポレートし、2XYT上層寒天と混合し、40枚のLB寒天ディッシュ(150mm x 10mm;Nunc)上にまいて、30℃にて一晩インキュベートした。このライブラリーの多様性は、プラーク数としてカウントして5x108を超えた。ファージライブラリーを、上層寒天から200mlのファージ溶出バッファー(10mM Tris-HCl、150mM NaCl、pH7.5)を用いて溶出した。PEGおよびNaClを加えて、それぞれ4%および3%の最終濃度として、ファージを沈降させた。ファージを、8%のグリセロールを含有するPBSバッファー中に再懸濁し、アリコートをとり、そして-80℃にて貯蔵した。
【0141】
6.3.1 結果
選択マーカーをコードするポリヌクレオチドとライゲートした分子をコードするポリヌクレオチドのライブラリーの作製を容易にするために、標準の分子クローニング技法を用いてプラスミドpUCKAを作製した。図7はプラスミドマップであり、図8はライブラリークローニング部位周りのヌクレオチド配列の詳細である。pUCKAの重要な特徴としては、2つの薬物選択マーカー(該ベクターを含有する細胞を選択/維持するためのカナマイシン耐性遺伝子および非機能性分子を発現するクローンを除去する選択のためのアンピシリン/カルベニシリン耐性を与えるβ-ラクタマーゼ遺伝子)、複製起点、クローニング部位(SfiI)の5'およびクローニング部位の3'のプロモーターおよびシグナル配列、ならびにβ-ラクタマーゼ遺伝子のフレーム内にライゲートされたFLAGおよびHIS6エピトープタグが挙げられる。β-ラクタマーゼ遺伝子は開始コドンを欠き、従って機能性融合タンパク質をコードする転写物だけがβ-ラクタマーゼを発現する細胞を生じうることに注意されたい。従って、正しいリーディングフレーム内にないかまたは停止コドンを生じる突然変異を含有するかまたはフレームシフト突然変異を含有するSfi I部位中にクローニングされたポリヌクレオチドは、機能性融合タンパク質(すなわち、機能性β-ラクタマーゼを発現する)をコードする転写物を作製しないしかつアンピシリン/カルベニシリンに対する耐性がないであろう。
【0142】
ベクターを試験するために、いくつかのscFvをSfi I部位中に(適当なリーディングフレーム内に)、停止コドンとともにまたはなしでクローニングした。停止コドンなしでscFvをコードするクローンだけが機能性融合タンパク質をコードする転写物を作製するはずである。実際、停止コドンを欠くscFvをコードするクローンだけがカルベニシリンの存在のもとで増殖することができた。停止コドンを欠くscFvをコードするいくつかのクローンの増殖速度を試験した(表2参照)。低濃度のカルベニシリン(50〜100μg/ml)においては、クローン間の差は少ししかなかったが、高濃度(200〜400μg/ml)においては、恐らくこれらのクローンの発現レベルが低いことを反映して、いくつかのクローンは増殖が遅かった。さらに高濃度のカルベニシリンにおいては、どのscFvクローンも増殖しなかった。これらのデータは、低発現クローン選択条件の損失を避けるために、最小濃度のネガティブ選択剤、薬物を用いることが必要であるか、または、もし栄養要求性選択マーカーを利用するのであれば、選択の時間を増加することが必要であることを示した。
【0143】
非機能性分子を発現する(すなわち、pUCKA中にクローニングしたときに機能性融合タンパク質をコードする転写物を作製しないであろう)クローンの存在量を決定するために、全ライブラリーをpUCKAベクター中にクローニングして大腸菌中に形質転換し、そしてカナマイシン中で増殖して形質転換細胞を選択した。192の個々のコロニーを単離し、そして100μg/mlのカルベニシリンとともにまたは無しで増殖させた。カルベニシリンの存在のもとで、192のうち144だけが増殖し、選択前の非機能性クローンの数はほぼ25%であることを示した。従って、pUCKAなどのベクター中のライブラリーを作製して機能性分子をコードするクローンだけを選択することにより、非機能性クローンの有意な減少を達成することができる。
【0144】
pUCKA中にクローニングしたライブラリーを次いでカルベニシリンの存在のもとで増殖して非機能性クローンを排除した。選択後に、scFv遺伝子を含有するSfi I断片をプラスミドpMD102中にサブクローニングすることにより、ファージライブラリーを構築した(図9)。得られるファージライブラリーは5x108より高い複雑度を有することがわかった。
【0145】
6.4 実施例4
ナイーブなヒトscFv発現ライブラリーの直接にpUCKA中への構築
pUCKAにおけるナイーブなヒトscFv発現ライブラリーの生産: 選択のためのヒトscFvライブラリーの作製は、scFvを直接pUCKA中にクローニングすることを除いて、本質的に実施例1に記載の通り実施することができる。概要を説明すると、リンパ節および脾臓組織から得たヒトmRNAを逆転写し、そしてVHおよびVL配列をPCRにより増幅した。オーバーラップPCRを次に利用してVHとVL配列を接合し、scFv遺伝子を生成させた。次いでscFv遺伝子をpUCKA発現ベクター中にSfi I制限酵素切断部位においてライゲートし、そして大腸菌中に形質転換した。形質転換した大腸菌を、全クローンの増殖を許容する条件のもとで(例えば、カナマイシンを含有するがアンピシリン/カルベニシリンを含有しない培地で)増殖するか、またはβ-ラクタマーゼ遺伝子とライゲートした機能性scFvをコードするクローンだけを増殖する選択条件のもとで(例えば、カナマイシンおよびアンピシリン/カルベニシリンの両方を含有するまたはただアンピシリン/カルベニシリンだけを含有する培地で)増殖することができる。得られる形質転換大腸菌細胞を許容条件のもとで増殖させる場合、得られる培養物の一部分または全ては従って、選択条件のもとで増殖することができる。いくつかの具体的な抗生物質濃度は実施例1および3に記載されている。得られる培養物はスクリーニングしてもよいしまたは(例えば、凍結グリセロールストックのように)後日の使用のために貯蔵してもよい。
【0146】
スクリーニングのためにはscFvフラグメントを、アンピシリン/カルベニシリン中で増殖しうるクローンの選択(すなわち、β-ラクタマーゼ遺伝子と融合した機能性scFvをコードするクローンの選択)後に、ファージ発現またはファージディスプレイに好適な代わりのベクター中にサブクローニングすることが望ましい。上記ハイスループットスクリーニング方法の利用を容易にするためには、scFvフラグメントをpMD102などのファージ発現に好適なベクター中にサブクローニングする。
【0147】
6.5 実施例5
さらなる発現ベクター
発現ベクター、さらなる発現ベクターの構築: さらなる発現ベクターを、pUCKAの構築に用いたのと類似の方法で、pUC19または他のベクターから誘導することができる。あるいは、さらなる発現ベクターを、新規に、所望の特徴をコードするポリヌクレオチドのライゲーションにより作製することができる。さらなる発現ベクターを生産するために有用なクローニング方法は当技術分野で公知である。例えば、Current Protocols in Molecular Biology, F.M. Ausubelら, 編, John Wiley & Sons(Chichester, England, 1998)およびMolecular Cloning: A Laboratory Manual、第3版, J. Sambrookら, 編, Cold Spring Harbor Laboratory Press(Cold Spring Harbor, NY, 2001)を参照されたい。
【0148】
作製されるさらなるベクターはpUCKAと異なる1以上の特徴を有することができ、それらの特徴としては、限定されるものでないが、エピトープタグ、分子のライブラリーをコードするポリヌクレオチドを挿入するクローニング部位、分子のライブラリーをコードするポリヌクレオチドを融合するための選択マーカー、複製起点、シグナル配列、プロモーター、発現ベクターを含む細胞を維持/選択するためのさらなる選択マーカー、細胞においてベクターを発現および/または維持するために必要なその他の特殊な成分が挙げられる。
【0149】
上記発明は、明確性と理解を目的として若干詳細にわたって記載したが、本開示を読むことから、本発明の真の範囲から逸脱することなく形態および詳細の様々な変化をなしうることが当業者に明らかであろう。例えば、上記の全ての技法と装置は様々な組合せで用いることができる。本出願に引用した全ての刊行物、特許、特許出願、またはその他の文書は参照によりその全文が全ての目的に対して、あたかもそれぞれの個々の刊行物、特許、特許出願、またはその他の文書が個々に参照により全ての目的に対して組み入れられると示されたのと同程度に、組み入れられる。さらに、米国予備特許出願第60/623,240号(2004年11月1日出願)は参照によりその全文が全ての目的のために組み入れられる
【図面の簡単な説明】
【0150】
【図1】使用するキャプチャーリフトの全体スキームの概観図である。キャプチャー試薬をコーティングしかつブロックしたフィルターを、細菌ローン(lawn)中のファージ発現scFvライブラリーを含有するプレート上に置いた。そのフィルターを持ち上げて(またはリフトして(lift))、ビオチン標識した抗原、アビジン酵素コンジュゲート、および検出用現像薬を逐次用いてインキュベートする。実験の詳細は実施例1を参照されたい。
【図2A】ヒトscFvライブラリーを作製する全体スキームの概観図である。可変重鎖(VH)および軽鎖(VL)を逆転写し、増幅し、次いでオーバーラップPCRにより接合する。得られるscFv遺伝子を次いでファージ発現ベクター(クローニング詳細は図2Bを参照)中にライゲートし、そしてスクリーニングのために大腸菌中に形質転換する(A)。実験の詳細は実施例1を参照されたい。。
【図2B】ヒトscFvライブラリーを作製する全体スキームの概観図である。可変重鎖(VH)および軽鎖(VL)を逆転写し、増幅し、次いでオーバーラップPCRにより接合する。得られるscFv遺伝子を次いでファージ発現ベクター(クローニング詳細は図2Bを参照)中にライゲートし、そしてスクリーニングのために大腸菌中に形質転換する。実験の詳細は実施例1を参照されたい。M13ファージ発現ベクターのサブクローン領域(配列番号:1-2)を用いて、scFvライブラリーを作製しかつ大腸菌において発現させる(B)。
【図3A】第1回のスクリーニングからのフィルター上の複数のポジティブクローンシグナルの写真であり、ほぼ3820クローン/mm2(ほぼ3x107pfu/フィルター)を含有する。フィルターの一部分を拡大レンズを介して写真撮影し、ポジティブクローンの詳細を拡大した(矢印で示した)(A)。第2回のスクリーニングからのフィルター上のポジティブクローンの写真、ほぼ105pfu/フィルターを含有する(B)。第3回のスクリーニングからのフィルター上のポジティブクローンの写真(C)。
【図3B】第1回のスクリーニングからのフィルター上の複数のポジティブクローンシグナルの写真であり、ほぼ3820クローン/mm2(ほぼ3x107pfu/フィルター)を含有する。第2回のスクリーニングからのフィルター上のポジティブクローンの写真、ほぼ105pfu/フィルターを含有する(B)。
【図3C】第1回のスクリーニングからのフィルター上の複数のポジティブクローンシグナルの写真であり、ほぼ3820クローン/mm2(ほぼ3x107pfu/フィルター)を含有する。第3回のスクリーニングからのフィルター上のポジティブクローンの写真(C)。
【図4】超ハイスループットスクリーニングにより単一ライブラリーから単離した24個の独立クローンのELISA分析である。24クローンのうち、EphA4-His抗原と特異的に結合した22クローンを単離してスクリーニングに使用した。
【図5】抗イディオタイプscFvクローン単離のために調製したビオチン化MEDI-AAA(Fab)2のクーマシーブルー染色である。調製したビオチン化MEDI-AAA(Fab)2(レーンAおよびD)、無標識MEDI-AAA(Fab)2(レーンBおよびE)、およびMEDI-AAA IgG(レーンCおよびG)を、PAGEにより未変性(レーンA、BおよびC)または変性(レーンD、E、FおよびG)条件のもとで分離して泳動させた。レーンGはSeeBlue2マーカーである。
【図6A】抗イディオタイプscFvクローンはMEDI-AAA IgG CDRに対して特異的である。抗イディオタイプscFvを発現するクローンの相対的結合活性を、ELISAにより全長MEDI-AAA IgG、密接に関係する軽鎖フレームワークと同一のFc領域を有する無関係なIgG、およびBSAに対して試験した(複数の実験において)(パネルA)。抗イディオタイプscFvをさらに、市販ポリクローナル抗体調製物および似ていないフレームワークを有する他の無関係なIgG(Syn)に対して試験した(パネルB)。抗イディオタイプscFvクローンはMEDI-AAA IgGと強固な結合を示す一方、無関係なIgGまたはBSAとは少ししか結合しなかった。
【図6B】抗イディオタイプscFvクローンはMEDI-AAA IgG CDRに対して特異的である。抗イディオタイプscFvを発現するクローンの相対的結合活性を、ELISAにより全長MEDI-AAA IgG、密接に関係する軽鎖フレームワークと同一のFc領域を有する無関係なIgG、およびBSAに対して試験した(複数の実験において)(パネルA)。抗イディオタイプscFvをさらに、市販ポリクローナル抗体調製物および似ていないフレームワークを有する他の無関係なIgG(Syn)に対して試験した(パネルB)。抗イディオタイプscFvクローンはMEDI-AAA IgGと強固な結合を示す一方、無関係なIgGまたはBSAとは少ししか結合しなかった。
【図7】pUCKAのプラスミドマップである。いずれかのポリヌクレオチドを、3'-FLAGおよびHis6エピトープタグとともに、アンピシリンr(β-ラクタマーゼ遺伝子)選択マーカーとの融合タンパク質としてクローニングすることができる。また、プラスミド上にカナマイシンr選択マーカーも存在する。停止コドンまたはフレームシフト突然変異を含有するコード領域はβ-ラクタマーゼをコードするタンパク質を生産しないので、得られるクローンはアンピシリン耐性ではないであろう。VHおよびVLコード領域をpUCKAベクター中にクローニングして、scFvを3'-FLAGおよびHis6エピトープタグとともにアンピシリンr選択マーカーとの融合タンパク質として生産させる配置を示した。
【図8】pUCKAのクローニング領域の詳細である。アンピシリン選択に用いた発現プラスミドの3'-クローニング領域のヌクレオチド配列(配列番号:3-4)を示す。FLAGおよびHis6エピトープタグコード領域およびアミノ酸配列も示した(それぞれ赤色と青色の矢印)。アンピシリン耐性に関わるβ-ラクタマーゼ遺伝子のシグナル配列も示した。
【図9】pMD102のプラスミドマップである。選択したライブラリーからのVHおよびVLコード領域を、3'-FLAGおよびHis6エピトープタグのフレーム内でpMD102中にクローニングすることができる(なお、pMD102は実施例1および2に記載したハイスループットキャプチャーリフト方法によるスクリーニングのためのscFvファージ発現に必要な全遺伝子を含有する)。
【技術分野】
【0001】
1. 発明の分野
本発明は、大きいコンビナトリアルライブラリーから結合タンパク質(例えば、抗体フラグメント)を超ハイスループット選択するためのスクリーニング方法に関する。本明細書に開示されたスクリーニング方法は、初めて、結合タンパク質の非常に大きいライブラリーの迅速なスクリーニング方法を、他のスクリーニング方法(例えば、パニング)がもつバイアスおよび制限なしに提供する。本発明はさらに、非機能性分子をコードするクローンを実質的に含まない分子(例えば、結合分子)の発現ライブラリーを作製する方法に関する。
【背景技術】
【0002】
2. 発明の背景
現在、コンビナトリアル組換えDNA技法を用いて、結合分子について大きい発現ライブラリーを作製することが可能である。とりわけ抗体エンジニアリングの分野ではまさにその通りであり、その場合、組換え抗体ライブラリーは日常的に109個を超えるユニークなクローンを含有する。結合分子の大きいライブラリーが利用可能であるのでほとんどのリガンドに対してほぼ無制限な結合物質の供給源を提供しうる一方、かかる多様なライブラリーを効率的かつ迅速にスクリーニングするスクリーニング方法の開発は遅れている。通常のスクリーニング方法は、FACS分析および96-ウエルプレートを用いるELISAスクリーニングなどの手間と時間のかかる技法に依存してきた。たとえ、自動ロボット機構(ほとんどの研究室の予算をいくらか超えるが)を用いても、これらのスクリーニング方法はこれらの通常の技法を用いた場合、る多様なライブラリーのクローンの小部分のスクリーニングを可能にするだけである。
【0003】
表面ディスプレイライブラリーは、結合分子を提示する生物(例えば、ファージおよび酵母)を固定化リガンド上で連続的な選択(パニング)にかけることにより特定の結合クローンの濃縮を可能にする(総括については、Trends Biotechnol 9: 408-414;Coomberら, 2002, Methods Mol Biol 178: 133-45;Kretzschmarら, 2002, Curr Opin Biotechol 13: 598-602;Fernandez-Gacioら, 2003;Leeら, 2003, Trends Biotechnol 21: 45-52;およびKondoら, 2004, Appl Microbiol Biotechnol 64: 28-40を参照)。パニング技法は非常に多様なライブラリーの迅速なスクリーニングを可能にする一方、ほとんどのパニング技術の繰返し選択サイクルは、一般に、最高の結合アフィニティ(濃縮バイアス)または最高のディスプレイ効率(ディスプレイバイアス)を有する少数の優性クローンが単離されるので、多様性の低下をもたらす。パニング方法の他の欠点は、この方法がしばしばリガンド上の単一優性エピトープだけを認識する結合タンパク質の単離をもたらすことである。さらに、より遅い増殖速度を有するクローンは、パニング中にその結合特性に関係なく集団から速やかに排除される(増殖バイアス)。表面ディスプレイライブラリーの他の制限は、これらが一般に、結合分子と表面を標的化するディスプレイ分子との間の融合の発生に依存することである。かかる融合は、結合特異性の改変されたまたは失った結合分子すら生じうる。さらに、パニングは固定したリガンドを必要とするので、リガンドのコンフォメーションを改変しおよび/または好ましい結合タンパク質認識部位をマスクして、リガンドの不自然な状態またはあまり好ましくない部位と結合する結合分子の単離をもたらしうる。従って、パニング法はしばしば選択バイアスを生じ、無関係なリガンド部位を認識しうる類似の結合特性をもつ少数のクローンだけを単離する一方、多数のそして潜在的にもっと有用なクローンを置き去りにする。
【0004】
表面ディスプレイ・パニング方法のいくつかの制限を克服する2通りのフィルターベースの一般様式のスクリーニング法が開発されていて、一般名で「キャプチャーリフト(capture lift)」と呼ばれている。これらの方法は、結合分子を表面上に提示するのとは逆に、可溶性結合分子を生産することができる発現ライブラリーを利用する。第1のフィルターベースのスクリーニング法は、発現した可溶性結合分子をキャプチャーする所望のリガンドを用いてコーティングしたフィルターを使用する(例えば、Rodenburgら, 1998, Hybridoma 17: 1-8;de Wildtら, 2000, Nat Biotechnol 18: 989-994.;Giovannoniら, 2001, Nucl Acids Res 29: E27を参照)。第2のフィルターベースのスクリーニング法は、結合分子の共通特性(例えば、内因性または遺伝子操作で作られたエピトープタグまたは固有の構造特性または分子の性質)を認識する一般的なキャプチャー分子を用いてコーティングしたフィルター上で、発現した可溶性結合分子をキャプチャーし、その後にキャプチャーされた結合分子を可溶性リガンドを用いて探索する(例えば、Skerraら, 1991, Anal Biochem 196: 151-155;Watkinsら, 1998, Anal Biochem 256: 169-177)。これらの方法は、選択、増殖およびディスプレイバイアスの複雑度を低減しうるが、それでも制限されている。第1の方法は、なお、固定されたリガンドの使用から生じる制限を受け、そして両方とも、比較的手間のかかる方法であって、小さい発現ライブラリー(多様性が106程度のクローン)の徹底したスクリーニングしか行うことはできない(Wuら, 2002, Cancer Immunol Immunother 51: 79-90)。
【0005】
全ての現行ライブラリースクリーニング法がもつ他の制限は、発現ライブラリーの品質にある。発現ライブラリーを作製する現行の方法は様々な増幅およびクローニング技法を利用するので、非機能性分子を発現する人為産物を生産するライブラリークローンを生じる。例えば、PCRおよび関係技法を用いるライブラリーは、これらのDNA複製法の固有の誤差率に応じて非機能性分子を発現する多数のクローンを含有しうる。例えば、PCR法は1塩基対当たり1.6x10-6〜1.1x10-4誤差の誤差率を有する(例えば、Lundberg ら, 1991 Gene 108:1-6.、TindallおよびKunkel, 1988, Biochemistry 27:6008-13を参照)。同様に、分子集団をコードするポリヌクレオチドと表面ディスプレイ用ポリペプチドをコードするポリヌクレオチドとの融合により作製される表面ディスプレイライブラリーでは、多数のクローン(利用する方法に応じて3分の2)が非生産性リーディングフレームにライゲートされたポリヌクレオチドを含有しうる。かかる有害な配列人為産物を含有するクローンは機能性分子を生産し得ない一方、通常、スクリーニングしなければならない生クローン(例えば、ファージまたは細菌)を生産するので、それにより、所望の分子(例えば、結合分子)を同定するためにスクリーニングしなければならない全クローン数は有意に増加する。
【0006】
従って、ファージディスプレイのハイスループット特性を有すると共に、表面ディスプレイ融合タンパク質の使用、リガンドの固定、および複数回のストリンジェントな選択に応じて生じるバイアスから起こる問題を排除するライブラリースクリーニングの方法が真に必要である。かかるスクリーニングの方法は結合分子の表面ディスプレイを避けて、可溶形態の結合分子を発現できるクローンの直接同定を可能にするであろう。理想的には、かかるスクリーニング方法は、さらに非常に大きい発現ライブラリー(109クローン超)の迅速かつ徹底的スクリーニングを可能にするであろう。さらに、非機能性分子(例えば、フレームシフトまたは停止コドン突然変異を有する分子)を生じる有害な配列人為産物を含有するクローンを本質的に含まない分子(例えば、結合分子)の発現ライブラリーを作製する方法は、スクリーニング技法の効率を向上させるであろう。
【0007】
本明細書における参考文献の引用または考察は、これが本発明の先行技術であることを承認すると解釈してはならない。
【発明の開示】
【0008】
3. 発明の概要
本発明は10億個の独立クローンを含有する発現ライブラリーから結合分子を超ハイスループットスクリーニングする方法に関する。上記スクリーニング方法は、1)高密度にまいた発現ライブラリーから結合分子の大集団を発現させるステップ、2)発現した結合分子の集団を固体支持体上に固定するステップ、3)固定した結合分子を少なくとも1つのリガンドと接触させるステップ、4)固定した結合分子と選択的に結合したリガンドを可視化するステップ、および5)少なくとも1つのリガンドを認識しかつ結合する結合分子を発現するクローンを単離するステップを含むかまたは代わりに本質的に上記ステップから成る。このスクリーニング方法は、少なくとも1人当たり1日当たり10億個の結合分子クローンのスクリーニングを可能にしかつ現行スクリーニング技法のいくつかの制限を克服する。さらにこのスクリーニング方法は高価な自動機械の使用を必要としない。
【0009】
一実施形態においては、結合分子は可溶性である。他の実施形態においては、超ハイスループットスクリーニング方法(以後「本発明のスクリーニング方法」または単に「スクリーニング方法」と呼ぶ)を用いて結合分子を発現するライブラリーをスクリーニングする。さらに他の実施形態においては、上記スクリーニング方法を用いてファージ、結合分子を発現する細菌または酵母ライブラリーをスクリーニングする。特定の実施形態においては、上記スクリーニング方法を用いて抗体またはそのフラグメントを発現するライブラリーをスクリーニングする。他の実施形態においては上記スクリーニング方法を用いてレセプター分子を発現するライブラリーをスクリーニングする。さらに他の実施形態においては、上記スクリーニング方法を用いてヌクレオチド-結合分子を発現するライブラリーをスクリーニングする。
【0010】
一実施形態においては、結合分子の発現ライブラリーを高密度でまく。一実施形態においては、結合分子の発現ライブラリーを1mm2当たり10よりも大きい、または100よりも大きい、または1,000よりも大きい、または10,000よりも大きい、または100,000よりも大きい結合分子発現クローンの密度でまく。他の実施形態においては、結合分子発現クローンを1mm2当たり1,000〜15,000クローンの密度でまく。
【0011】
一実施形態においては、結合分子の集団を固体支持体上に固定する。特に、発現した結合分子の集団を、固体支持体上の薬剤との特異的相互作用を介して固体支持体上に選択的に固定する。かかる薬剤としては、限定されるものでないが、化合物、テザー(tether)、リンカー、およびポリペプチド結合ドメインが挙げられる。結合分子の固有の性質が固定を容易にしうることも考えられる。例えば、結合分子の疎水性ドメインはそれらをプラスチック支持体に固定することを可能にしうる。本発明の固体支持体としては、限定されるものでないが、膜、プラスチック、ガラスおよびコーティングしたガラスが挙げられる。
【0012】
一実施形態においては、リガンドは結合分子と選択的に結合できるいずれの分子であってもよく、それらの分子としては、限定されるものでないが、ペプチド、ポリペプチド、核酸、炭水化物、脂質、または有機化合物が挙げられる。他の実施形態においては、リガンドは可溶性である。さらに他の実施形態においては、リガンドは検出ドメインと融合している。検出ドメインは検出シグナルの増幅を可能にしうることを具体的に意図している(下記)。本発明の検出ドメインとしては、限定されるものでないが、チオレドキシン、BSA、ロイシンジッパー、Fcドメインおよびそれらの断片が挙げられる。ある特定の実施形態においては、リガンドが複数の検出ドメインと融合している。さらに、特定の検出ドメインはまた、リガンド-結合分子相互作用の結合活性を増加してスクリーニング方法の結合、特異性および/または感受性の改善をもたらすリガンド二量体(例えば、Fcドメインおよびロイシンジッパードメイン)の形成を可能にしうることも意図する。
【0013】
一実施形態においては、固定した結合分子と選択的に結合したリガンドを検出する。結合したリガンドは直接的なまたは間接的方法により検出しうることを具体的に意図する。リガンドの直接的検出は、多数の技法により実施することができて、それらとしては、限定されるものでないが、リガンドの容易に検出可能な部分(例えば、放射性標識、色素生産検出用酵素)との共有結合性の修飾が挙げられる。リガンドの間接的検出は、当技術分野で周知の方法により実施することができ、それらとしては、限定されるものでないが、リガンドと相互作用することが公知の第2の分子(例えば、抗体)を用いる方法が挙げられる。第2の分子はそれ自体が直接的または間接的方法により検出されてもよい。
【0014】
一実施形態においては、リガンドを認識しかつそれと結合する結合分子クローンを単離する。固体支持体は、リガンドを認識しかつそれと結合する結合分子クローンを含有する結合分子クローンのサブセットを単離するためのテンプレートを提供しうることを具体的に意図する。このより小さい集団を次いで本発明のスクリーニング方法の改変法を用いてスクリーニングすることができる。改変法は、クローンのサブセットをより低い密度で播く、および/または固定するステップを含みうる。クローンのサブセットを、単一クローンが単離されるには十分低いが、サブセット中に存在する各クローンが固体支持体上で少なくとも1回表されるには十分高い密度で播く、および/または固定することを意図する。リガンドを認識しかつそれと結合する結合分子クローンを含有する結合分子クローンのサブセットを、当業者に周知の代わりの方法によりスクリーニングしうることも意図する。代わりの方法としては、限定されるものでないが、ELISAアッセイおよびFACS分析が挙げられる。本発明の方法の1以上の態様は自動化しうることも具体的に意図される。
【0015】
本発明はさらに、非機能性分子を生じる有害な配列人為産物(例えば、フレームシフトまたは停止コドン突然変異を有する分子)を含む分子を発現するクローンを少ししか含有しない発現ライブラリー(例えば、結合分子のライブラリー)を作る方法に関する。上記発現ライブラリーを生産する方法は、1)発現ベクター中に、機能性分子を発現するクローンの選択に有用な少なくとも1つの選択マーカーをコードするポリヌクレオチドとライゲートした分子をコードするポリヌクレオチドを含むクローンのライブラリーを作製するステップ;2)機能性分子を発現するクローンを選択する条件下でステップ(1)において作製したクローンのライブラリーを増やすステップ;ならびに、任意に、3)ステップ(2)の選択されたライブラリーからの機能性分子をコードするポリヌクレオチドを、特定の所望の機能性クローンの同定および/または単離に有用な代わりのベクター中にサブクローニングするステップを含むか、あるいは本質的に上記ステップからなる。
【0016】
本明細書に開示した発現ライブラリーを作る方法は、非機能性分子を生じる有害な配列人為産物を含む分子を発現するクローンを少ししか含有しない発現ライブラリーを作ることを可能にし、それにより、スクリーニングすべき全クローン数を低減することを可能にする。一実施形態においては、上記ライブラリー生産方法を用いて、ファージ、細菌、酵母、植物または哺乳動物系においてスクリーニングするための発現ライブラリーを作製する。
【0017】
一実施形態においては、クローンのライブラリーがコードしかつ発現する分子としては、それから1以上の単一分子の単離が所望される分子の集団が挙げられる。他の実施形態においては、上記発現ライブラリーの生産方法(以後、「本発明のライブラリー生産」または単に「ライブラリーの生産方法」と呼ぶ)を用いて結合分子の集団を発現するライブラリーを作製する。さらに他の実施形態においては、上記ライブラリー生産方法を用いてリガンドの集団を発現するライブラリーを作製する。発現ライブラリーを作製して本明細書に開示したいずれかの結合分子および/またはリガンドの集団を発現しうることが意図される。
【0018】
特定の実施形態においては、上記ライブラリー生産方法を用いて抗体またはそのフラグメントを発現するライブラリーを作製する。他の実施形態においては、上記ライブラリー生産方法を用いてレセプター分子を発現するライブラリーを作製する。さらに他の実施形態においては、上記ライブラリー生産方法を用いてヌクレオチド-結合分子を発現するライブラリーを作製する。
【0019】
非機能性分子を生じる有害な配列人為産物をコードするクローンの排除を容易にするため、分子をコードするポリヌクレオチドを少なくとも1つの選択マーカーをコードするポリヌクレオチドとライゲートする。多数の選択マーカーが当技術分野では周知であり、それには、限定されるものでないが、薬物耐性マーカー(例えば、除草剤および抗生物質耐性遺伝子)、代謝性/栄養要求性マーカー(例えば、必須代謝物の生産に必要な酵素に対する遺伝子)、スクリーニング可能な/精製マーカー(例えば、色素生産検出または精製ドメイン用の酵素をコーディングする遺伝子)が挙げられる。クローンのライブラリーを、有害な配列人為産物を有しない分子を発現させるクローンを選択する条件のもとで増やす。機能性分子をコードするクローンの選択に対して用いられる増殖条件は用いる選択マーカーに応じて変わりうることが理解されるであろう。例えば、薬物耐性遺伝子をコードするポリヌクレオチドを選択に用いるときは、例えば、クローンを適当な薬物の存在のもとで増殖させるであろう。代謝遺伝子をコードするクローンを選択に用いるときは、適当な代謝物の非存在で増やすであろう。
【0020】
本発明のライブラリー生産方法のステップ(1)および(2)で作製しかつ選択した発現ライブラリーを、ライブラリーのスクリーニングに利用しうる代わりのベクター中にサブクローニングしてもよいことが意図される。
【0021】
本発明のライブラリー生産方法を用いて作製した発現ライブラリーを、当技術分野で周知の多数の方法を用いてスクリーニングすることができる。1つの特定の実施形態においては、本発明のスクリーニング方法が利用される。
【0022】
4. 図面の簡単な説明
図面の簡単な説明については下記参照。
【発明を実施するための最良の形態】
【0023】
5. 発明の詳細な説明
本発明は10億個の独立クローンを含有する発現ライブラリーから結合分子を迅速かつ効率的に超ハイスループットスクリーニングする方法を提供する。このスクリーニング方法は、少なくとも1人、1日当たり10億個の結合分子クローンのスクリーニングを可能にする点で有利である。さらに、本発明のスクリーニング方法は、現行ハイスループットスクリーニング技法のいくつかの制限(限定されるものでないが、増殖バイアス、ディスプレイバイアス、濃縮バイアス、ならびに固定したリガンドと結合分子融合体の使用から生じる選択バイアスを含む)を克服する。本発明のスクリーニング技法はさらに、結合分子とそのリガンドの間の特異的相互作用の検出を増強する方法を提供する。さらに、本発明のスクリーニング技法を用いて複数のリガンドに対してスクリーニングすることができる。本発明のスクリーニング方法はそれ故に、非常に多くの結合分子ライブラリーからヒト疾患の診断および治療に用いる特異的結合分子の発見に応用することができる。本発明のスクリーニング方法は、例外的に大きい集団の結合分子のスクリーニングに特に好適であるが、結合分子の大集団と小集団の両方のスクリーニングに利用することができる。本発明のスクリーニング方法の1以上の態様(例えば、検出シグナルの分析、インキュベーションおよび洗浄)は自動化できることも具体的に意図される。
【0024】
上記スクリーニング方法は、1)高密度に播いた発現ライブラリーから結合分子の大集団を発現させるステップ、2)発現した結合分子の集団を固体支持体上に固定するステップ、3)固定した結合分子を少なくとも1つのリガンドと接触させるステップ、4)固定した結合分子と選択的に結合したリガンドを可視化するステップ、および5)少なくとも1つのリガンドを認識しかつ結合する結合分子を発現するクローンを単離するステップを含むか、あるいは本質的に上記ステップから成る。
【0025】
一実施形態においては結合分子は可溶性である。他の実施形態においては、上記スクリーニング方法を用いて、結合分子を発現するライブラリーをスクリーニングする。さらに他の実施形態においては、上記スクリーニング方法を用いて、結合分子を発現するファージ、細菌または酵母ライブラリーをスクリーニングする。本発明のスクリーニング方法を用いてスクリーニングすることができる結合分子のライブラリーとしては、限定されるものでないが、抗体またはそのフラグメントを発現するライブラリー、レセプター分子を発現するライブラリー、ヌクレオチド結合分子を発現するライブラリーおよびランダムペプチドを発現するライブラリーが挙げられる。
【0026】
一実施形態においては、結合分子の発現ライブラリークローンを高密度で播く。特定の一実施形態においては、発現ライブラリークローンを1mm2当たり10よりも大きい、または100よりも大きい、または1,000よりも大きい、または10,000よりも大きい、または100,000よりも大きいクローンの密度でまく。他の実施形態においては、発現ライブラリークローンを1mm2当たり1,000〜15,000クローンの密度でまく。
【0027】
一実施形態においては、発現された可溶性結合分子の集団は固体支持体上に固定されている。発現された可溶性結合分子の集団を、固体支持体(例えば、抗体、化合物)上の薬剤による特定の相互作用を介して、固体支持体上に選択的に固定することを具体的に意図する。結合分子集団の固有の性質が固定化を容易にしうることも意図する。本発明の固体支持体としては、限定されるものでないが、膜、プラスチック、グラスおよびコーティングされたグラスが挙げられる。
【0028】
一実施形態においては、リガンドは結合分子(限定されるものでないが、ペプチド、ポリペプチド、核酸、炭水化物、脂質、または有機化合物を含む)が選択的に結合しうるいずれの分子であってもよい。一実施形態においては、リガンドにはチロシンキナーゼのドメインまたはチロシンキナーゼリガンドが含まれる。意図するチロシンキナーゼおよびチロシンキナーゼリガンドには、限定されるものでないが、レセプターチロシンキナーゼおよび非レセプターチロシンキナーゼが含まれる。他の実施形態においては、リガンドは可溶性である。さらに他の実施形態においては、リガンドは検出ドメインと融合している。検出ドメインは検出シグナル(下記)の増幅を可能にしうることを具体的に意図する。ある特定の実施形態においては、リガンドは複数の検出ドメインと融合している。さらに特定の検出ドメインはまた、リガンド-結合分子相互作用の結合活性を増加してスクリーニング方法の結合、特異性および/または感受性の改善をもたらしうるリガンド-二量体の形成を容易にしうることも意図する。
【0029】
一実施形態においては、固定した結合分子に選択的に結合されたリガンドを検出する。結合されたリガンドを直接的または間接的方法により検出しうることを具体的に意図する。リガンドの直接的検出は多数の技法(限定されるものでないが、放射性標識付きリガンドまたは色素生産検出用酵素による共有結合修飾を含む)により実施することができる。リガンドの間接的検出は、例えば、直接的または間接的方法によりそれ自体検出されるリガンドと相互作用することが公知の抗体を用いて、当技術分野で周知の方法により実施することができる。
【0030】
一実施形態においては、リガンドを認識しかつ結合する結合分子クローンを単離する。固体支持体が、リガンドを認識しかつ結合する結合分子クローンを含有する結合分子クローンのサブセットを単離するためのテンプレートを供給しうることを具体的に意図する。次いでこのより小さい集団を、本発明のスクリーニング方法の改変法を用いてスクリーニングすることができる。上記改変法はクローンのサブセットをより低い密度でまくステップを含んでなる。結合分子クローンのサブセットを、単一クローンが単離されるには十分低いが、サブセット中に存在する各クローンが固体支持体上で少なくとも1回表されるには十分高い密度で播くことを意図する。リガンドを認識し、かつ結合する結合分子を発現するクローンを含有するクローンのサブセットを、当業者に公知の代わりの方法によりスクリーニングしうることも意図する。代わりの方法としては、限定されるものでないが、ELISAアッセイおよびFACS分析が挙げられる。本発明のスクリーニング方法の1以上の態様は自動化できることも具体的に意図する。例えば、非特異的相互作用を排除するために用いるインキュベーションおよび洗浄ステップは、市販の機器を用いて日常的に自動化されているし(例えば、Stovall Washer、カタログ番号WMAA115S、Stovall Life Sciences Inc、Greensboro、NC)、固体支持体上のシグナルの分析も容易に自動化することができる(例えば、Proteome Works Plus Spot Cutterカタログ番号165-7064、Bio-Rad Laboratories Inc.、Hercules、CA)。
【0031】
本発明はまた、非機能性分子を生じる有害な配列人為産物を有する分子をコードするクローンを含まない分子(例えば、結合分子)の発現ライブラリーを生産する方法も提供する。非機能性分子の発現を生じる有害な配列人為産物には、限定されるものでないが、未熟停止コドン、欠失、挿入、フレームシフト突然変異、ノンセンス突然変異およびミスセンス突然変異を含む分子が含まれる。従って、本明細書に使用される用語「非機能性分子」には、限定されるものでないが、有害な配列人為産物(例えば、未熟停止コドン、フレームシフト突然変異、ノンセンス突然変異およびミスセンス突然変異)などを含む分子が挙げられる。非機能性分子を発現するクローンはまた、本明細書では「非機能性クローン」とも呼ばれる。非機能性クローンは一般に生存可能であり、従って、スクリーニング方法により排除しなければならないネガティブクローンを代表する。非機能性クローンの存在は、所望のクローンを同定するためにスクリーニングしなければならないライブラリークローンの全数を非常に増加しうる。本明細書に提供される発現ライブラリーを生産する方法(以後、「本発明のライブラリー生産方法」または単に「ライブラリー生産方法」と呼ぶ)は、本方法がたとえ非機能性分子を発現するクローンをいくらか含むとしても少ししか含まない発現ライブラリーの生産を可能にし、それによりスクリーニングすべきクローンの全数を低減する点で有利である。
【0032】
一実施形態においては、発現ライブラリーを生産する方法は、1)発現ベクター中に、機能性分子を発現するクローンの選択に有用な少なくとも1つの選択マーカーをコードするポリヌクレオチドとライゲートした分子をコードするポリヌクレオチドを含むクローンのライブラリーを作製するステップ;2)機能性分子を発現するクローンを選択する条件下でステップ(1)において作製したクローンのライブラリーを増殖させるステップ;ならびに、任意に、3)ステップ(2)の選択されたライブラリーからの機能性分子をコードするポリヌクレオチドを、特定の所望の機能性クローンの同定および/または単離に有用な代わりのベクター中にサブクローニングするステップを含むかまたは代わりに上記ステップから本質的になる。
【0033】
分子をコードするポリヌクレオチドは個々に少なくとも1つの選択マーカーをコードするポリヌクレオチドとライゲートしていることを意図する。しかし、当業者は、分子をコードする複数のポリヌクレオチドが少なくとも1つの選択マーカーをコードする単一のポリヌクレオチドと直線上にライゲートしていてもよいことを認識するであろう。
【0034】
本発明のライブラリー生産方法を用いて作製されかつ選択された発現ライブラリーは、上記選択されたライブラリーをスクリーニングするために有用な発現ベクター中にあってもよいことを意図する。しかしまた、発現ライブラリーは、上記作製されかつ選択されたライブラリーをスクリーニングするために有用でないが上記ライブラリーを作製しかつ選択するために有用な発現ベクター中に作製されかつ選択されてもよいことを意図する。従って、一実施形態においては、本発明のライブラリー生産方法を用いて作製されかつ選択された発現ライブラリーは、次いで、上記ライブラリーをスクリーニングするために有用な代わりのベクター中にサブクローンされる。
【0035】
従って、ある特定の実施形態においては、ライブラリー生産方法は、1)発現ベクター中に、機能性分子を発現するクローンの選択に有用な少なくとも1つの選択マーカーをコードするポリヌクレオチドとライゲートした分子をコードするポリヌクレオチドを含むクローンのライブラリーを作製するステップ、および2)機能性分子を発現するクローンを選択する条件下でステップ(1)において作製したクローンのライブラリーを増殖させるステップを含んでなる。
【0036】
他の実施形態においては、ライブラリー生産方法は、1)発現ベクター中に、機能性分子を発現するクローンの選択に有用な少なくとも1つの選択マーカーをコードするポリヌクレオチドとライゲートした分子をコードするポリヌクレオチドを含むクローンのライブラリーを作製するステップ;2)機能性分子を発現するクローンを選択する条件下でステップ(1)において作製したクローンのライブラリーを増殖させるステップ;ならびに、3)ステップ(2)の選択されたライブラリーからの機能性分子をコードするポリヌクレオチドを、特定の所望の機能性クローンの同定および/または単離に有用な代わりのベクター中にサブクローニングするステップを含んでなる。
【0037】
発現ライブラリーをスクリーニングするために有用なベクターは当技術分野で周知でありかつ以下に記載され(例えば、表題「発現ライブラリーおよび発現ベクター」の節を参照)、本発明のライブラリー生産方法のステップ(1)および(2)ならびにステップ(3)に有用な具体的なベクターは実施例3〜5に詳述されている(下記)。特定の実施形態においては、作製されかつ選択されたライブラリーからの機能性分子をコードするポリヌクレオチドを、ファージ発現ベクター中にサブクローニングする。
【0038】
本発明のライブラリー生産方法を、種々の系で使用する様々な発現ライブラリーの作製に利用することができる。一実施形態においては、限定されるものでないが、本発明のライブラリー生産方法を用いて、ファージ、細菌、酵母、植物および哺乳動物の系を含む系におけるスクリーニングのための発現ライブラリーを作製する。本発明のライブラリー生産方法を用いて作製した発現ライブラリーは、当業者に周知の多数の方法を用いてスクリーニングすることができる。特定の実施形態においては、本発明のスクリーニング方法が利用される。
【0039】
一実施形態においては、分子をコードするポリヌクレオチドは分子の集団をコードする。他の実施形態においては、ポリヌクレオチドはそれから1以上の単一分子の単離が所望される分子の集団をコードする。例えば、発現ライブラリーを、細胞、組織または生物が発現したメッセンジャーRNAの全集団から作製することができる。あるいは、発現ライブラリーを、例えば、所望のアミノ酸モチーフなどの特定の特徴を有する分子の集団をコードするポリヌクレオチドから作製することができる。分子をコードするポリヌクレオチドの集団を、天然の供給源(例えば、細胞から単離したメッセンジャーRNA)から単離もしくは誘導してもよいし、または新規に作成してもよい(例えば、ランダムポリペプチドをコードするポリヌクレオチド配列)ことを意図する。一実施形態においては、本発明のライブラリー生産方法を用いて、結合分子の集団を発現する発現ライブラリーを作製する。他の実施形態においては、上記ライブラリー生産方法を用いて、リガンドの集団を発現する発現ライブラリーを作製する。さらに他の実施形態においては、上記ライブラリー生産方法を用いて、本明細書に開示したいずれかの結合分子および/またはリガンドの集団を発現する発現ライブラリーを作製する。
【0040】
特定の実施形態においては、上記ライブラリー生産方法を用いて、抗体またはそのフラグメントの集団を発現するライブラリーを作製する。他の実施形態においては上記ライブラリー生産方法を用いて、レセプター分子またはその断片の集団を発現するライブラリーを作製する。さらに他の実施形態においては、上記ライブラリー生産方法を用いて、ヌクレオチド-結合分子またはその断片の集団を発現するライブラリーを作製する。さらに他の特定の実施形態においては、上記ライブラリー生産方法を用いて、ランダムポリペプチドの集団を発現するライブラリーを作製する。
【0041】
非機能性分子をコードするクローンの排除を容易にするため、分子をコードするポリヌクレオチドを少なくとも1つの選択マーカーをコードするポリヌクレオチドとライゲートする。多数の選択マーカーが当技術分野で周知であり、それらとしては、限定されるものでないが、薬物耐性マーカー(例えば、除草剤および抗生物質耐性遺伝子)、代謝性/栄養要求性マーカー(例えば、必須代謝物の生産に必要な酵素に対する遺伝子)、スクリーニング可能な/精製マーカー(例えば、色素生産検出のための酵素または精製ドメインをコードする遺伝子)が挙げられる。クローンのライブラリーを次いで、機能性分子を発現するクローンを選択する条件のもとで増殖させる。当業者は、利用される増殖条件が利用する選択マーカーに応じて変わりうることを理解するであろう。例えば、薬物耐性遺伝子をコードするポリヌクレオチドを選択に用いるときは、例えば、クローンを適当な薬物の存在のもとで増殖しうる。または代謝遺伝子をコードするクローンを選択に用いるときは、適当な代謝物の非存在で増殖しうる。
【0042】
分子をコードするポリヌクレオチドを、固定化および/または検出に有用な少なくとも1つのエピトープタグ配列(本明細書では「マーカー」配列または単に「タグ」配列とも呼ぶ)にライゲートすることを(少なくとも1つの選択マーカーをコードするポリヌクレオチドとライゲートするのに加えて)さらに意図する。多数のマーカーおよび/またはタグ配列、例えば、限定するのではないが、ヘキサ-ヒスチジンペプチド、赤血球凝集素「HA」タグ、および「FLAG」タグが当技術分野では公知である。これらのタグおよびかかるタグの組込みに有用な方法に関するさらなる詳細は、以下の表題「結合分子」および「実施例」の節に記載している。
【0043】
5.1 発現ライブラリーおよび発現ベクター
発現ライブラリーを作製する一般的方法は、当技術分野で周知であり、多数の文献(例えば、Current Protocols in Molecular Biology, F.M. Ausubelら, 編, John Wiley & Sons (Chichester, England, 1998), 第5章および第6章など)から利用しうる。cDNA発現ライブラリーを構築する一般的方法の具体例としては、Chenら, Nature (1995) 377:428-431、Akopianら, Nature (1996) 379:257-262(ポリアデニル化RNAからcDNA発現ライブラリーを作製するための好適な方法を記載する)が挙げられる。ランダムペプチドライブラリーはHrubyら、1997、Curr Opin Chem Biol 1:483-490 に総括され、全ゲノム発現ライブラリーは、例えば、Preussら, 2002, Immunol Rev188: 43-50に記載され、そして核酸および小分子化合物のライブラリーは、Gray, 2001, Curr Opin Neurobiol 11:608-614に総括されている。
【0044】
以下に記載のような分子の集団を発現する発現ライブラリーは、本明細書(実施例3〜5を参照)に開示したような適当な発現ベクター中に構築することができる。特定の実施形態においては、本発明のライブラリー生産方法は、発現ベクターpUCKA中に、少なくともβ-ラクタマーゼ遺伝子をコードするポリヌクレオチドとライゲートした分子をコードするポリヌクレオチドを含有するクローンのライブラリーを作製する第1ステップを含んでなる。従って、本発明は、発現クローンのライブラリーを生産しかつ選択する上で有用な発現ベクターpUCKAを提供する。pUCKAの重要な特徴としては、2つの薬物選択マーカー:該ベクターを含有する細胞を選択/維持するためのカナマイシン耐性遺伝子、および非機能性分子を発現するクローンを選択して除去するためのβ-ラクタマーゼ遺伝子(アンピシリン/カルベニシリン耐性を与える)、複製起点、プロモーター、ならびにシグナル配列、クローニング部位(SfiI)の5'およびクローニング部位の3'、β-ラクタマーゼ遺伝子とフレーム内でライゲートしたFLAGおよびHIS6エピトープタグが挙げられる。本明細書の開示に基づいて、当業者は、pUCKAの変異体および他の代わりの発現ベクターを作製し(例えば、実施例3および5を参照)そして/または本発明のライブラリー生産方法を利用しうることを理解するであろう。他の実施形態においては、本発明は1以上の次の特徴の変化を含むpUCKAの変異体を提供する:エピトープタグ、分子のライブラリーをコードするポリヌクレオチドを挿入するためのクローニング部位、分子のライブラリーをコードするポリヌクレオチドの融合のための選択マーカー、複製起点、シグナル配列、プロモーター、発現ベクターを含む細胞を維持/選択するためのさらなる選択マーカー、細胞においてベクターを発現および/または維持するために必要な他の特殊化した成分の付加。
【0045】
他のベクターは容易に入手しうるし、かつ当業者に周知である。本発明の方法に有用である発現ベクターは、RNA合成を指示するための適当な発現制御配列(プロモーター)を含有しうる。例えば、細菌プロモーターにはlacI、lacZ、T3、T7、gpt、λPR、PLおよびtrpが含まれる。真核生物のプロモーターにはCMV最初期、HSVチミジンキナーゼ、初期および後期SV40、レトロウイルスからのLTR、ならびにマウスメタロチオネイン-Iが含まれる。適当な発現ベクターおよびプロモーターの選択は当業者の技術レベルで十分可能である。発現ベクターはまた、翻訳開始用のリボソーム結合部位および転写ターミネーターも含有しうる。ベクターはまた、発現を増幅するための適当な配列も含みうる。加えて、発現ベクターはさらに上記発現ベクターを含有する形質転換宿主細胞を選択するための表現型形質を与える選択マーカー遺伝子をコードするポリヌクレオチドを含有しうる。選択マーカーのいくつかの例としては、真核細胞培養に対するジヒドロ葉酸レダクターゼまたはネオマイシン耐性、または大腸菌におけるテトラサイクリンまたはアンピシリン耐性が挙げられる。当業者は、ライブラリークローンを含む形質転換宿主細胞を選択および/または維持するために必要な選択マーカーに加えて、機能性クローンを発現するクローンを選択するために第2の選択マーカーが必要でありうることを理解するであろう。
【0046】
利用しうる発現ベクターの代表例としては、限定されるものでないが、ウイルス粒子、バキュロウイルス、ファージ、プラスミド、ファージミド、コスミド、フォスミド、細菌人工染色体、ウイルスDNA(例えば、ワクシニアウイルス、アデノウイルス、ファウルポックスウイルス、仮性狂犬病およびSV40の誘導体)、P1-系人工染色体、酵母プラスミド、酵母人工染色体、および目的の具体的な宿主に特異的な他のベクター(例えば、大腸菌、バシラス菌、アスペルギルス真菌、昆虫、植物、酵母、哺乳動物の細胞、など)が挙げられる。かかるベクターは、染色体性、非染色体性および合成DNA配列を含む。多数の好適なベクター当業者に公知でありかつ市販されている。例示として、次のベクターが提供される:細菌/ファージ:pQEベクター(Qiagen)、pBluescriptプラスミド、pNHベクター、λ DASH(登録商標)IIベクター、pTRG XR、ZAPベクター(Stratagene);ptrc99a、pKK223-3、pDR540、pRIT2T(Pharmacia);真核生物:pXT1、pSG5、pCMV-Script(登録商標)(Stratagene)、pSVK3、pBPV、pMSG、pSVLSV40(Pharmacia)。しかし、いずれの他のプラスミドまたは他のベクターも、それらが宿主中で複製可能でありかつ生存可能である限り使用することができる。一般に、組換え発現ベクターは1以上の宿主にとって適当な複製起点を含みうる。当業者は、現行の開示に基づいて、適当な発現ベクター(例えば、発現を駆動するプロモーターおよび選択のための選択マーカーを含有するもの)を、市販ベクターを改変することにより作製するかまたは本明細書に記載のように、複製、維持、発現、選択および他の所望の形質に必要である適当な成分を組み合わせることにより新規に作製できることを理解するであろう。
【0047】
このように、機能性分子を発現するクローンを選択するために有用な選択マーカーをコードするポリヌクレオチドとライゲートした分子をコードするポリヌクレオチドのライブラリーを、分子を発現するために有用な様々な発現ベクターにおいて作製することができる。
【0048】
5.2 結合分子
本明細書で使用する用語「結合分子」は、リガンドと選択的に結合するのに十分なサイズと複雑度を有する分子を意味することを意図している。かかる分子は一般に高分子であって、限定されるものでないが、それにはポリペプチド、核酸、炭水化物および脂質が含まれる。しかし、具体的には、誘導体、類似体および模倣化合物ならびに小有機化合物も、この用語の定義内に含まれることを意図している。結合分子のサイズは、該分子がリガンドとの選択的結合を示すかまたは示すようにすることができる限り、重要でない。
【0049】
本明細書に使用する用語「選択的な」および「選択性」は、本明細書に用いられるリガンドとの結合分子に言及する場合、欲しないか、または非特異的相互作用から識別することができる相互作用を意味する。識別は、例えば、親和性または結合活性に基づくものであってよく、そして多数の低親和性相互作用または少数の高親和性相互作用から誘導されうる。高親和性相互作用は一般にほぼ10-8 M〜ほぼ10-9 Mより大きいものである。
【0050】
一実施形態においては、結合分子はリガンドとほぼ10-4 Mを超えるアフィニティで結合しうる。他の実施形態においては、リガンドに対してほぼ10-4M、またはほぼ10-5M、またはほぼ10-6M、またはほぼ10-7M、またはほぼ10-8M、またはほぼ10-9M、またはほぼ10-10Mを超えるアフィニティを有しうる。
【0051】
結合分子には、例えば、抗体および他のレセプターまたは免疫系のリガンド結合ポリペプチドが含まれ、限定されるものでないが、T細胞レセプター(TCR)、主要組織適合性複合体(MHC)、CD4レセプター、およびCD8レセプター、その他のCD分子(限定されるものでないが、CD2、CD3、CD19、CD20、CD22を含む)などが含まれうる。具体的に意図される他の結合分子には、限定されるものでないが、細胞表面レセプター、(例えば、インテグリン、成長因子レセプターおよびサイトカインレセプター)、細胞質レセプター(例えば、ステロイドホルモンレセプター)、DNA結合ポリペプチド(例えば、転写因子およびDNA複製因子)が含まれる。結合分子にはまた、上記結合分子の変異体および/またはリガンド結合に恐らく間違いなく寄与しうる上記結合分子の一部分を含有する融合分子も含まれる。さらに、ランダムおよびコンビナトリアルライブラリーから選択された結合分子集団(例えば、ポリペプチド、核酸、アプタマーおよび化合物)も、かかる分子がリガンドと選択的結合活性を示すか示すようにすることができる限り、含まれると意図している。
【0052】
結合分子集団の選択は、所望の結合分子のタイプ、最後の選択される結合分子の必要性および利用意図に依存しうる。1つの手法は、結合分子として機能することが公知であるかまたは結合活性を示すかもしくは示す能力のあることが公知の分子から結合分子集団を作製することである。例えば、抗体および免疫レパートリーの他のレセプターは、本質的に無数の色々な抗原およびリガンドと結合することができる結合分子として機能することがわかっている。それ故に、抗体レパートリーから多様な結合分子の集団を作製すると、本質的に所望のリガンドに対する結合分子の同定を可能にしうる。
【0053】
第2の手法は未知分子の大集団を作製することである。上記集団は配列または構造の十分な多様性を含有し、目的のリガンド組成と結合しうる分子を含有するように作製しなければならない。この手法の利点は、配列、構造または機能について前もっての知識を必要としないことである。その代わり、必要なことの全ては、上記集団がたまたまリガンド複合体と特異的結合相互作用を示す高い確率を有しうるように、十分なサイズと複雑度の集団を作製することである。かかる集団の具体例は、ペプチド、核酸および小分子化合物のランダムライブラリーである。当業者は、どのタイプの手法とどのタイプの結合分子 集団が意図する目的および所望の必要性に応用可能であるかを熟知するかまたは決定することができるであろう。
【0054】
使用する結合分子集団のサイズと多様性は複数の因子により決定されうるのであって、それらの因子には、限定されるものでないが、リガンド集団または組成、所望のアフィニティの範囲、結合分子の複雑度、ならびに所望の結合分子の数とタイプが含まれる。同定すべき結合分子の所望の数が増加すると、結合分子の集団のサイズと多様性も増加する。同様に、結合分子のライブラリー変異体(例えば、抗体またはそのフラグメント)をスクリーニングするとき、集団のサイズは結合分子自体の複雑度とともに増加する。さらに、結合分子の集団のサイズは同様に、リガンドの数または複雑度が増加するとともに増加しうる。
【0055】
小サイズの集団は数百および数千の異なる結合分子からなり、中サイズの集団は数万および数十万からなる一方、大集団は数百万および数十億の異なる結合分子からなるであろう。本発明のスクリーニング方法を用いていずれのサイズの結合分子の集団をスクリーニングすることもできる一方、特徴として、数百万および数十億の異なる結合分子からなる大きいかつ多様な集団、具体的には、ほぼ106、107、108、109、1010、1011またはそれより多い異なる結合分子を含む集団をスクリーニングするのに好適である。同様に、本発明のライブラリー生産方法を用いていずれのサイズの分子の集団のライブラリーを作製することもできるが、数百万および数十億の異なる分子からなる大きいかつ多様な集団、具体的には、ほぼ106、107、108、109、1010、1011またはそれより多い異なる結合分子を含む分子の集団の発現ライブラリーを作製するのに好適である。当業者は、所望の結合分子の数をスクリーニングおよび/または同定するのに十分でありうる結合分子の集団の近似的な多様性を熟知するであろう。
【0056】
結合分子の組換え体ライブラリーが一般に利用されるが、それは結合分子の大きいかつ多様な集団を速やかに作製することができるからである。遺伝子組換えの方法は、集団の固体支持体との選択的固定化のための特徴を固有に含有する天然のレパートリーから、大きい数の結合分子集団を生産することを可能にする。さらに、発現されたポリペプチドまたは核酸の組換えライブラリーは、結合分子集団の固体支持体との選択的固定化を容易にするかまたは直接に機能するように、多数の方法で遺伝子操作することができる。本発明のライブラリー生産方法は結合分子の生産ライブラリーに利用できることを具体的に意図している。
【0057】
結合分子の集団は、その集団が十分に多様であって、その集団が所望のリガンドと選択的に結合する結合分子を少なくとも1つ含有する確率が非常に高い限り、本質的にいずれの供給源からでも生産または誘導することができる。結合分子の集団は、結合分子として機能するかまたは結合活性を示すことがわかっている分子から作製することができ、かかる分子には、限定されるものでないが、抗体、そのフラグメント、免疫系の他のレセプター、レセプター、ヌクレオチド結合タンパク質およびレクチンが含まれる。あるいは、結合分子の集団は未知の分子、例えば、ランダムペプチドライブラリー(Hrubyら, 1997, Curr Opin Chem Biol 1:483-490の総括を参照)、全ゲノム発現ライブラリー(例えば、Preussら, 2002, Immunol Rev188: 43-50)、核酸および小分子化合物(Gray, 2001, Curr Opin Neurobiol 11:608-614の総括を参照)から作製することができる。
【0058】
結合分子集団の選択は所望の結合分子のタイプに依存しうる。例えば、もし高アフィニティ結合分子が所望であれば、抗体結合分子の集団を利用することができる。同様に、類似した異型遺伝子性を示す他の免疫系の分子(例えば、T細胞レセプターおよび主要組織適合性複合体レセプターCD4およびCD8)から結合分子集団を誘導することができる。かかる分子のこれらの正常機能は無限の数の色々な抗原および/またはリガンドと本質的に結合する(Kuby、J.(編), 1997, Immunology, Third Ed., New York, W.H. Freeman & Co.)。それ故に、これらの分子から多様な結合分子の集団を作製すると、いずれかの所望のリガンドに対する結合分子を同定することが可能になりうる。
【0059】
特定の生物学的効果をもつ結合分子を結合分子の集団から同定しうることを具体的に意図する。例えば、結合分子は、標的分子の1以上の生物学的活性を阻害することができるアンタゴニストであってもよい。アンタゴニストは、レセプターのリガンドとの結合(逆もまたしかり)を妨害することにより、かつリガンドにより活性化された受精不能または死滅細胞により、および/またはレセプターもしくはリガンド活性化(例えば、チロシンキナーゼ活性化)または細胞レセプターとのリガンド結合後のシグナル伝達により作用しうる。アンタゴニストは、レセプター-リガンド相互作用を完全にブロックしうるか、またはかかる相互作用を実質的に低下させうる。あるいは、結合分子は、標的分子の1以上の生物学的活性を活性化することができるアゴニストであってもよい。アゴニストは、例えば、標的分子を活性化することにより、および/またはシグナル伝達に介在することにより作用しうる。結合分子の生物学的効果を確認するアッセイは当業者に周知である。
【0060】
公知のまたは固有の結合機能を示す他の結合分子には、本発明のライブラリー生産方法を用いる発現ライブラリーの作製、または本発明のスクリーニング方法における出発集団としての使用に受け入れられるものとして、限定されるものでないが、細胞表面、細胞質および核レセプターを含む様々なレセプターが含まれる。細胞表面レセプターの例には、限定されるものでないが、細胞外マトリックス成分(例えば、インテグリン)、成長因子(例えば、EGFR、FGFR)、ホルモン、インスリンおよびインスリン様タンパク質(IR、IGF-R)、サイトカイン(例えば、IL-4R、IL-13)、レセプターチロシンキナーゼ(例えば、ALK、EphA2、EphA3、EphA4、EphA5、EphA6、EphA7、EphA8、EphB1、EphB2、EphB3、EphB4、EphB5、EphB6)、サイトカイン(例えば、IFNAR)およびケモカイン(例えば、CXC-R、CC-R)に対するレセプターが含まれる。細胞質および核レセプター(Mangelsdorf ら, 1995, Cell 83:835の総括を参照)の例には、限定されるものでないが、ステロイドホルモンレセプター(Kumarら, 1999, Steroids 64:310)、PPARレセプター(Wilsonら, 2000, J Med Chem 43:527)、ビタミンレセプター、および核酸結合タンパク質(de Miguelら, 1998, Curr Opin Chem Biol 2:417-421;および McEwan, I.(編), 2004, Essays in Biochemistry, Volume 40, London, Portland Press Ltd.)が含まれる。
【0061】
結合分子ライブラリーは未知の配列または構造のランダムライブラリーから誘導しうることも具体的に意図される。かかるライブラリーは、当技術分野で公知の標準組換え技法を用いて容易に作製することができる(Leblら、1997、Methods Enzymol 289:336-392 および Shustaら、1999、Curr Opin Biotechnol 10:117-122の総括を参照)。
【0062】
特定の実施形態においては、結合分子の集団はさらに結合分子と融合またはコンジュゲートした異種ポリペプチドを組み込む。異種ポリペプチドには、限定されるものでないが、固定化および/または検出のために有用であるマーカーおよび/またはタグ配列が含まれる。例えば、とりわけ、pQEベクターで提供されるタグ(QIAGEN、Inc.、9259 Eton Avenue、Chatsworth、Calif.、91311)のようなヘキサ-ヒスチジンペプチド(その多くは市販されている)は検出および固定化に有用である(Gentzら、1989、Proc. Natl. Acad. Sci. USA 86:821-824)。検出および固定化の両方に有用な、他のペプチドタグには、限定されるものでないが、インフルエンザ赤血球凝集素タンパク質から誘導されたエピトープに対応する赤血球凝集素「HA」タグ(Wilsonら, 1984, Cell 37:767)が含まれる。ポリペプチド、タンパク質および融合タンパク質は、標準組換えDNA技法によりまたはタンパク質合成技法により、例えば、ペプチド合成機を用いて生産することができる。例えば、ペプチド、ポリペプチド、タンパク質または融合タンパク質をコードする核酸分子は、自動化DNA合成機を含む通常の技法により合成することができる。あるいは、遺伝子断片のPCR増幅は、2つの継続する遺伝子断片間に相補的オーバーハングを生じるアンカープライマーを用い、次いでアニーリングし、そして再増幅してキメラ遺伝子配列を作製することにより行うことができる(例えば、Current Protocols in Molecular Biology, F.M. Ausubelら, 編, John Wiley & Sons (Chichester, England, 1998)およびMolecular Cloning: A Laboratory Manual, 3rd Edition, J. Sambrookら, 編, Cold Spring Harbor Laboratory Press (Cold Spring Harbor, NY, 2001)を参照)。
【0063】
他の実施形態においては、結合分子の集団はさらに、検出および固定化の両方に有用であるビオチンおよび/またはハプテン分子タグを組み込む。結合分子の集団をビオチン化(バイオテクノロジーにおけるビオチンタグの総括は、Diamandisら、1991、Clin Chem 37:625-636を参照)および/またはハプテン化(ハプテンおよび方法の総括は、Molecular Probes: Handbook of Fluorescent Probes and Research Chemicals、R. P. Haugland、第9版、Molecular Probes、(Eugene OR、2004)の第4章を参照)することができる。
【0064】
一実施形態においては、結合分子の集団は可溶性である。いくつかの結合分子は固有で可溶性である一方、他の結合分子は、限定されるものでないが、それらを可溶化するためのさらなる成分(例えば、界面活性剤、カオトロピック剤)の導入を含むさらなる操作を必要とする。可溶性結合分子集団の使用は、固体支持体および/またはリガンドと相互作用することができない不溶性結合分子凝集物の形成を防止することにより、スクリーニングを容易にしうることが具体的に意図される。自然の結合分子を所望する場合、分子を可溶化するために必要な操作はコンフォメーション改変をもたらして自然の状態でリガンドを認識しない結合分子を同定しうるので、結合分子の集団は固有に可溶性であることを期待している。
【0065】
組換え発現ライブラリーから可溶性分子を生産するためには多数の方法がある。方法としては、限定されるものでないが、可溶性タンパク質との融合(下記表題「リガンドと検出」の節を参照)、宿主生物から発現されたポリペプチドの特異的分泌に対するシグナル配列の利用、および細菌溶解を引き起こして細菌により生産されたポリペプチド配列の放出をもたらす溶原性ファージ発現ライブラリーの使用(例えば、Current Protocols in Molecular Biology, F.M. Ausubelら, 編, John Wiley & Sons (Chichester, England, 1998)およびMolecular Cloning: A Laboratory Manual、第3版、J. Sambrookら、編、Cold Spring Harbor Laboratory Press(Cold Spring Harbor、NY、2001)を参照)が挙げられる。当業者は具体的な目的に応用可能なライブラリーのタイプを熟知するかまたは決定することができるであろう。
【0066】
一実施形態においては、本発明のライブラリー生産方法を用いて、可溶性結合分子を発現する組換えファージ、細菌または酵母ライブラリーを作製する。他の実施形態においては、スクリーニング方法を用いて可溶性結合分子を発現する組換えファージ、細菌または酵母ライブラリーをスクリーニングする。ファージ組換えライブラリーの具体例としては、溶原性ファージが細菌により発現された結合ポリペプチドの放出を引き起こすライブラリーおよび結合分子が細胞の溶解なしに周辺質空間中に分泌されるライブラリーが挙げられる。
【0067】
特定の実施形態においては、ライブラリー生産方法を用いて、抗体またはそのフラグメントを発現するライブラリーを作製する。他の実施形態においては、スクリーニング方法を用いて抗体またはそのフラグメントを発現するライブラリーをスクリーニングする。抗体またはそのフラグメントを発現するライブラリーは、本明細書に開示される方法を含む当業者が熟知する様々な方法により作製することができる。例えば、ポリメラーゼ連鎖反応(PCR)を用いて特定の生物の本質的に全ての抗体レパートリーを増幅し、そしてそれを重鎖および軽鎖、その機能性フラグメントの多様な組合せの組換え体集団として、または融合タンパク質として発現させることができる。具体的な方法については、下記実施例1を参照されたい。抗体の機能性フラグメントには、限定されるものでないが、Fab、Fv、scFv、およびCDR領域が含まれる。特に、抗体には、免疫グロブリン分子および免疫グロブリン分子の免疫学的活性フラグメント、すなわち、抗原結合部位を含有する分子が含まれ、これらのフラグメントは、限定されるものでないが、Fc領域またはそのフラグメントを含む他の免疫グロブリンドメインと融合していてもしていなくてもよい。当業者はさらに、限定されるものでないが、scFv-Fc融合、可変領域(例えば、VLおよびVH)-Fc融合およびscFv-scFv-Fc融合を含む他の融合産物を作成できることを理解するであろう。免疫グロブリン分子はいずれのタイプ(例えば、IgG、IgE、IgM、IgD、IgAおよびIgY)、クラス(例えば、IgG1、IgG2、IgG3、IgG4、IgA1およびIgA2)またはサブクラスであってもよい。
【0068】
本発明のライブラリー生産方法を用いて単一特異性、二重特異性またはさらに多重特異性の抗体分子の集団を発現するライブラリーを作製しうることを具体的に意図する。本発明のスクリーニング方法を用いて単一特異性、二重特異性またはさらに多重特異性の抗体分子の集団をスクリーニングすることも具体的に意図する。例えば、PCT公報 WO 93/17715、WO 92/08802、WO 91/00360、およびWO 92/05793;Tuttら、J. Immunol. 147:60-69(1991);米国特許第4,474,893号、第4,714,681号、第4,925,648号、第5,573,920号、および第5,601,819号、;ならびにKostelnyら、J. Immunol. 148:1547-1553(1992)を参照されたい。
【0069】
組換え抗体発現ライブラリーを作製するための具体的な方法およびプロトコルの例は、本明細書(例えば、実施例1、3〜5を参照)ならびに、とりわけ、Huseら, 1989, Science 246: 1275-1281;McCaffertyら, 1990, Nature 348:552-554;Rosokら, 1996, J Biol Chem 271:22611-22618;Bacaら, 1997, J Biol Chem 272:10678-10684;Wuら, 1998, Anal Biochem 256:169-177;Sheetsら, 1998, PNAS USA 95:6157-6162;de Haardら, 1999, J Biol Chem 274:18218-18230;Knappikら, 2000, J Mol Biol 296:57-86;Soderlindら, 2000, Nature Biotechnol 18:852-856;およびAzriel-Rosenfeldら, 2004, J Mol Biol 335:177-192に見出すことができる。
【0070】
また、天然または合成化合物ライブラリーなどの他の化合物の大集団も、それらを固体支持体上に固定することができる限り、本発明のスクリーニング方法を用いてスクリーニングできることも具体的に意図する。かかる他の化合物の大集団を固体支持体上に固定し、次いで1以上の選択のリガンドと選択的に結合する分子を同定するためにスクリーニングすることができる。一実施形態においては、かかる化合物の大集団を選択的に固定することができる。大きい結合分子の集団は、当業者に公知のコンビナトリアル化学の方法により生産した合成化合物であってもよい。
【0071】
5.3 固定化
所望のリガンドに対して選択的結合アフィニティを有する結合ポリペプチドを同定する目的で、スクリーニングすべき結合分子の集団を固定化し、次いで結合分子を所望のリガンドと接触させることが必要である。一実施形態においては、結合分子の集団を固定化する。他の実施形態においては、結合分子の集団を選択的に固定化する。
【0072】
結合分子の固体支持体との固定化に言及するときに本明細書で使用する用語「選択的な」または「選択的に」は、欲しない相互作用から区別することができるある相互作用を意味する。区別は、例えば、アフィニティまたは結合活性に基づくものであってもよく、そして多数の低アフィニティ相互作用または少数の高アフィニティ相互作用から誘導されるものであってもよい。本明細書で使用する用語「選択的固定化」および「選択的に固定化された」は、特異的相互作用(例えば、結合分子上に存在するエピトープに特異的である結合分子の抗体との相互作用など)および結合分子の固有の性質に由来する相互作用(例えば、疎水性ドメインのプラスチック表面との相互作用ならびに架橋剤などの化学部分が介在する相互作用など)の両方の相互作用を包含することを意図する。
【0073】
特定の理論または機構により束縛されるのを欲することなく、結合分子の集団の選択的固定化は、測定される結合相互作用(例えば、結合分子-リガンド相互作用)の感受性を増加するように機能することを意図する。結合分子の集団を固体支持体へ選択的固定化することは、結合分子集団を反応物中に存在しうる無関係なおよび/または汚染性分子から分離する働きをする。従って、結合分子集団の固定化は、結合分子集団を有意に濃縮し、次いで、スクリーニングされる結合分子の集団の一部分でない無関係および/または汚染性分子との非特異的結合相互作用を低下させる。
【0074】
当業者は、結合相互作用を測定することの困難は、結合アッセイの複雑度および結合反応物内の色々な種の数と多様性とともに増加することを理解するであろう。本明細書に開示したスクリーニング方法は、結合分子の集団の選択的固定化が反応物から無関係および/または汚染性分子から除去することにより実質的に欲しないか、および/または無関係な結合相互作用の数を低下させるので、これらの困難を減ずる。従って、本発明のスクリーニング方法は、固体支持体上の結合分子の集団の選択的固定化を介して、検出の感受性と特異性を改善する。
【0075】
結合分子の選択的固定化を利用してアフィニティ技法により分離した集団などの実質的に精製したまたは濃縮した結合分子の集団から、ならびに不均一集団(例えば、細胞抽出物、順化培地)から、結合分子をスクリーニングすることができる。
【0076】
特定の実施形態においては、発現された可溶性結合分子の集団を選択的に固定化する。特に発現された可溶性結合分子の集団を、固体支持体と結合した、および/またはカップリングした作用薬との特異的相互作用を介して固体支持体上に選択的に固定化する。かかる作用薬としては、限定されるものでないが、抗体、ポリペプチド、アプタマー、繋索、リンカー、および結合分子の集団を固体支持体に保持するのに十分な共有結合または非共有結合相互作用を可能にする化学部分が挙げられる。結合分子の固有の性質が選択的固定化を容易にすることも意図する。例えば、結合分子の疎水性ドメインはプラスチック支持体との選択的固定化を可能にする。
【0077】
特定の実施形態においては、結合分子の集団上に存在する1エピトープを認識する1一抗体を固体支持体と結合および/またはカップリングする。例えば、定常ドメインを認識する抗体を用いて抗体結合分子の集団を固定化することができる。あるいは、特定のエピトープタグ(例えば、HA、FLAG、Myc、6xHisエピトープタグ、上記)を認識する抗体を用いて、上記エピトープタグを含むように遺伝子操作で作られた結合分子の集団を固定化することができる。同様に、ビオチンまたはアビジンを用いて他の結合対(上記)のパートナーを含有するように遺伝子操作で作られた結合分子の集団を固定化することができる。例えば、ビオチンを固体支持体とカップリング/結合させ、結合分子をアビジンを含有するように遺伝子操作で作ることができる。あるいは、アビジンを固体支持体とカップリング/結合させる一方、結合分子の集団をビオチンを用いて標識することができる。
【0078】
他の実施形態においては、結合分子の集団上に存在するエピトープおよび/またはドメインを認識するアプタマーを固体支持体と結合および/またはカップリングさせることができる。具体的な標的に特異的なアプタマーを選択する方法は当技術分野では周知である。(Jayasena, S.D., 1999, Clin. Chem. 45:1628-1650のアプタマー技法の総括を参照).
固体支持体を使用することにより、高濃度の結合分子の固定化が可能になる。固定された高濃度の結合分子は、非常に大きい数の分子の迅速なスクリーニングを可能にし、かつさらに、リガンドとの低いアフィニティ相互作用を検出する能力を増強して低濃度のリガンドを検出する働きをする。
【0079】
本質的にいずれの固体支持体も本発明のスクリーニング方法における使用に受け入れられる。当業者は、具体的な必要性に適合しかつ結合分子の集団の全て、または統計上代表的な数を固定化する能力を有するために必要な支持体を熟知するであろう。固体支持体は、固定化結合分子のより高い密度の達成を可能にする多孔材料にすることができる。さらに固体支持体を、スクリーニング方法に必要とされる操作(例えば、洗浄、インキュベーション、可視化方法)に適合しうる一方、結合分子集団を保持する能力を維持する特徴をもつものを選ぶことができる。かかる操作は、無結合のリガンド集団の除去および非特異的相互作用物を除去するための洗浄ならびにリガンド-結合分子相互作用の可視化に重要でありうる。上記操作が容易であることは、多数の操作を必要とすることが多い超ハイスループットスクリーニングを実施するときに有利である。本発明の固体支持体には、限定されるものでないが、ニトロセルロース、ナイロン、二フッ化ポリビニリデン、プラスチック、ガラス、ポリアクリルアミドおよびアガロースなどの膜が含まれる。固体支持体は、結合分子の集団(例えば、ビーズ)の固定化を支持する限り、本質的にいずれのサイズまたは形状で作ることもできる。
【0080】
本発明のスクリーニング方法で使用される固体支持体は改変しうることを具体的に意図する。例えば、非常に様々な官能基を固体支持体の表面と結合および/またはカップリングさせて、結合分子の集団の固定化を容易にするかまたは他のスクリーニング方法の態様(例えば、検出、洗浄)を増強することができる。固体支持体と結合させることができる官能基には、限定されるものでないが、化学部分(例えば、架橋剤)、アプタマー(例えば、RNA、DNA)、ポリペプチド(例えば、抗体、ストレプトアビジン)および他の生体分子(例えば、ビオチン、脂質)が含まれる。官能基は、限定されるものでないが、共有結合性、非共有結合性、加水分解性、光分解性、光活性化性、可逆的および非可逆的を含むいくつかの相互作用により結合分子の集団の固定化に介在することができる。
【0081】
非常に大きな結合分子の集団の迅速なスクリーニングを可能にするために、本発明は、固体支持体1mm2当たり少なくとも3,800の独立クローンのスクリーニングを可能にする高密度で播く方法を利用する。従来の方法では、典型的には、固体支持体1mm2当たりたかだかほぼ1〜6の独立クローンのスクリーニングが可能である。例えば、抗体分子の多様なライブラリーは容易に109の独立クローンを超えうる。通常の方法を用いると、このサイズのライブラリーは、全集団をスクリーニングするために、例えば、少なくとも20,000のフィルター(直径83mmで)を必要としうる。本発明のスクリーニング方法を用いると、全ライブラリーをわずか33のフィルターを用いてスクリーニングすることができる。従って、本発明は従来の方法より少なくとも600倍の改善を提供する。
【0082】
一実施形態においては、発現ライブラリークローンを高密度でまく。特定の実施形態においては、発現ライブラリークローンを1mm2当たり、ほぼ10を超える、またはほぼ100を超える、またはほぼ1,000を超える、またはほぼ2,000を超える、またはほぼ3,000を超える、またはほぼ4,000を超える、またはほぼ5,000を超える、またはほぼ6,000を超える、またはほぼ7,000を超える、またはほぼ8,000を超える、またはほぼ9,000を超える、またはほぼ10,000を超える、またはほぼ25,000を超える、またはほぼ50,000を超える、またはほぼ75,000を超える、またはほぼ100,000クローンを超える密度でまく。他の実施形態においては、発現ライブラリークローンを1mm2当たり、10を超える、または100を超える、または1,000を超える、または2,000を超える、または3,000を超える、または4,000を超える、または5,000を超える、または6,000を超える、または7,000を超える、または8,000を超える、または9,000を超える、または10,000を超える、または25,000を超える、または50,000を超える、または75,000を超える、または100,000クローンを超える密度でまく。さらに他の実施形態においては、発現ライブラリークローンを1mm2当たり、ほぼ1,000〜ほぼ10,000クローンの密度でまく。なおさらに他の実施形態においては、発現ライブラリークローンを1mm2当たり、1,000〜10,000クローンの密度でまく。上記クローンは可溶性結合分子の集団を発現する個々の細胞(例えば、酵母または細菌)であってもよいしまたは可溶性結合分子の集団をコードするファージに感染させた細菌細胞であってもよい。具体的な実施形態においては、上記クローンは可溶性結合分子の集団をコードする溶菌性ファージに感染させた細胞である。可溶性結合分子を細胞から分泌させてもよいしまたは溶解後の細胞から放出させてもよいことを具体的に意図する。溶解は、限定されるものでないが、化学的方法(例えば、アルカリ溶解)および 生物学的 方法(例えば、溶菌性ファージによる感染)を含む、当業者に公知のいくつかの方法により促進することができる。可溶性結合分子のクローンは、いずれかの供給源(例
えば、ランダムペプチドライブラリーおよびコンビナトリアル化学ライブラリー)から誘導されかつ上記の密度で固体支持体に固定化された個々の分子のプールでありうることも意図する。
【0083】
5.4 リガンドおよび検出
本明細書で使用する用語「リガンド」には、リガンドに対して結合アフィニティを有する結合分子により認識されうるいずれかの分子が含まれる。リガンドはタンパク質、DNA、脂質、炭水化物または小分子であってもよい。一実施形態においては、可溶性リガンドは、限定されるものでないが、ペプチド、ポリペプチド、核酸、炭水化物、脂質、または有機化合物を含む、結合分子と選択的に結合することができるいずれの分子であってもよい。当業者は、先に結合分子として考察した分子はリガンドであってもよいことを理解するであろう。例えば、細胞表面レセプター(例えば、インテグリン、成長因子レセプターまたはサイトカインレセプター)をリガンドとして用いて、結合分子の集団(例えば、ランダムペプチド、抗体またはコンビナトリアル化学ライブラリー)をスクリーニングし、これらのレセプターに対するアゴニストまたはアンタゴニストとして利用することができる結合分子を同定することができる。
【0084】
一実施形態においては、リガンドは少なくとも1つの細胞表面タンパク質(例えば、グリコシル化表面タンパク質)由来のドメインまたはペプチド、癌関連タンパク質、サイトカイン、ケモカイン、ペプチドホルモン、神経伝達物質、細胞表面レセプター(例えば、細胞表面レセプターキナーゼ、7回膜貫通レセプター、ウイルスレセプターおよびコ-レセプター)、細胞外マトリックス結合タンパク質、細胞-結合タンパク質、病原体の抗原(例えば、細菌抗原、マラリア抗原など)を含む。他の実施形態においては、リガンドは、チロシンキナーゼまたはチロシンキナーゼリガンド由来の少なくとも1つのドメインまたはペプチドを含む。意図するチロシンキナーゼおよびチロシンキナーゼリガンドには、限定されるものでないが、レセプターチロシンキナーゼ(例えば、EGFR/上皮性成長因子、Eph/Ephrin、FGF/繊維芽細胞成長因子、FN/フィブロネクチンインスリン、IGF/インスリン様成長因子、NGF/神経成長因子、PDGF/血小板由来の成長因子、およびTie/アンギオポエチンレセプターファミリー)および非レセプターチロシンキナーゼ(例えば、Src、Tec、JAK、Fes、Abl、FAK、Csk、およびSykファミリー)が含まれる。
【0085】
一実施形態においては、本発明のスクリーニング方法を用いて単一リガンドに対して選択的アフィニティを示す結合分子をスクリーニングする。さらに、本発明のスクリーニング方法を用いて複数のリガンドと結合する結合分子の集団をスクリーニングしうることを具体的に意図する。
【0086】
リガンドおよび/またはリガンド集団は、結合分子の必要性および意図する用途ならびにリガンドまたはリガンド組成物の特徴に応じて選択しうる。例えば、本発明のスクリーニング方法を用いて、単一リガンドに対してまたは全細胞もしくは組織と同じように複雑なリガンド集団ならびにほんの僅かな種の単一リガンド集団に対して選択的アフィニティを示す結合分子を同定することができる。
【0087】
リガンドまたはリガンド集団は実質的に精製されていてもまたは様々な量の他の無関係な種を含有してもよい。例えば、単一リガンドは、よく特徴づけられて高度に精製された分子(例えば、組換えタンパク質)であってもよい一方、分子の集団はいくつかの数の供給源(例えば、1以上のリガンドの部分的にまたは実質的に精製された調製物、または細胞ライセートまたはホモジネートの粗調製物を含む)由来であってもよい。さらに本発明はまた、単一リガンド、またはリガンドの集団(ここで、リガンドは細胞ライセート中のポリペプチドまたは他の高分子である)に対して、選択的なアフィニティを有する結合分子を同定するスクリーニング方法も提供する。生化学的によく特徴づけられていないリガンド(例えば、細胞ライセート)を本発明の方法で使用しうることを具体的に意図する。さらに、本発明のスクリーニング方法を用いて、ある集団の1つまたは少数のメンバーに対して選択性のある結合分子を同定することができる。例えば、もしあるリガンドの集団のいずれかのメンバーに対して選択性のある結合分子を生産するのが所望であれば、それぞれの個々のメンバーを単一集団中に組み合わせて、本発明のスクリーニング方法を用いて同時にスクリーニングすることができる。リガンドの単一集団は、限定されるものでないが、単一細胞内にいくつかの異なる分子を一緒に発現させることによりいくつかの異なるリガンド調製物を一緒にプールすることを含むいくつかの方法により作製することができる。
【0088】
スクリーニング方法に用いるリガンド集団は、色々なサイズの実質的に精製した分子または粗細胞調製物またはその他の複合体組成物から成ってもよい。一般に、単一リガンドまたは2つの異なるリガンド種の単純な集団を使用する。しかし、3、4、5、6、7、8、9、10以上の異なるリガンドから構成された単純なリガンド集団を使用してもよい。本発明のスクリーニング方法を、細胞内の十〜数百の異なるリガンド種を含有する中度のリガンド集団、ならびにほぼ数万の異なるリガンド種、例えば、細胞内の多数の異なる分子を含有する複雑なリガンド集団に対して使用しうることも具体的に意図する。集団サイズとタイプの選択は、結合分子の必要性および意図する用途に依存しうる。当業者は、具体的な必要性にとって好適である集団のサイズとタイプを熟知するであろう。全ての場合に、リガンドおよび/またはリガンド集団は、実質的に精製されてもよいしまたは様々な量の他の無関係な種を含有してもよい。
【0089】
一実施形態においては、リガンドおよび/またはリガンド集団は可溶性である。いくつかのリガンドは固有に可溶性である一方、他のリガンドはさらなる操作を必要としうるのであって、それらの操作としては、限定されるものでないが、それらを可溶化するためのさらなる成分(例えば、界面活性剤、カオトロピック剤)の導入が挙げられる。可溶性リガンドの使用は、結合分子と相互作用することができない不溶性リガンド凝集物の生成を防止することにより、固定化された結合分子の集団の効率的なスクリーニングを容易にし得ることを具体的に意図する。その自然の状態でリガンドと相互作用する結合分子を所望する場合、リガンドを可溶化するために必要な操作はコンフォメーション改変をもたらしてリガンドの自然の状態を認識しない結合分子を同定しうるので、目的のリガンドは固有に可溶性であることを期待している。
【0090】
本発明は、少なくとも1つのリガンドに対して選択的なアフィニティを示す少なくとも1つの結合分子を同定するために、結合分子の集団の超ハイスループットスクリーニングを行うスクリーニング方法を提供する。結合分子は、結合分子の集団を少なくとも1つのリガンドと接触させることにより同定する。次いでリガンド自体を適当な検出方法により検出する。
【0091】
一実施形態においては、固定された結合分子と選択的に結合したリガンドを検出する。結合したリガンドを、例えば、発光、放射性同位体、発色の検出に関わりうる直接的または間接的方法、またはリガンドの検出を可能にするいずれかの方法により検出しうることを具体的に意図する。リガンドの直接検出は、限定されるものでないが、リガンドの容易に検出可能な部分による共有結合性修飾、例えば、ヨウ素化などの放射性同位体を用いる化学的修飾を含む当業者に馴染みのある多数の技法により実施することができる。直接的方法はまた、適当な検出分子のリガンドとの融合にも関わり、例えば、リガンドをルシフェラーゼと融合して発光により検出してもよいし、または酵素(例えば、lacZ、西洋わさびペルオキシダーゼ、アルカリホスファターゼおよび下記)と融合して適当な比色測定により検出してもよい。
【0092】
リガンドの間接的な検出は、限定されるものでないが、リガンドと相互作用することが公知の第2の分子を用いることを含む当業者に周知の方法により実施することができる。第2の分子はそれ自体が直接的または間接的方法で検出される。例えば、リガンドをビオチン化して、適当に標識したアビジン分子を用いて検出する。ハプテン分子を類似の方法(上記)に利用することができる。さらに、リガンドに特異的な抗体をリガンドに特異的な第1抗体と相互作用しうる第2抗体を用いて検出してもよく、この場合、再び直接的検出について先に記載した検出方法を用いる。
【0093】
直接的および間接的検出は両方とも、リガンドまたは間接的検出に用いる第2分子を検出可能な基質とカップリングすることにより実施することができる。検出可能な基質の例としては、様々な酵素、補欠分子族、蛍光物質、発光物質、生物発光物質、放射性物質、ポジトロン放出金属、および非放射性常磁性金属イオンが挙げられる。例えば、当技術分野で公知の技法を用いて、検出可能な物質をリガンドを認識する抗体(またはそのフラグメント)と直接、また中間物質(例えば、当技術分野で公知のリンカー)を介して間接にカップリングまたはコンジュゲートすることができる。例えば、本発明による診断に用いる抗体とコンジュゲートできる金属イオンに対する米国特許第号4,741,900を参照されたい。好適な酵素の例としては、西洋わさびペルオキシダーゼ、アルカリホスファターゼ、β-ガラクトシダーゼ、またはアセチルコリンエステラーゼ;好適な補欠分子族複合体の例としてはストレプトアビジン/ビオチンおよびアビジン/ビオチン;好適な蛍光材料の例としてはウンベリフェロン、フルオレセイン、フルオレセインイソチオシアナート、ローダミン、ジクロロトリアジニルアミンフルオレセイン、ダンシルクロリドまたはフィコエリトリン;発光物質の例としてはルミノール;生物発光物質の例としてはルシフェラーゼ、ルシフェリン、およびエクロリン;そして好適な放射性材料の例としては125I、131I、111Inまたは99Tcが挙げられる。
【0094】
他の実施形態においては、リガンドはさらに検出ドメインを含む。検出ドメインはリガンドの検出を容易にするいずれの分子であってもよい。一実施形態においては、検出ドメインは検出シグナルの増幅を可能にして検出の感受性をより高くしうる。例えば、検出ドメインは、リガンドと相互作用して次いでそれ自体が検出されて検出シグナルの増幅をもたらす第2分子(例えば、ビオチン分子または抗体)の数を拡大する働きをなしうる。
【0095】
特定の実施形態においては、ポリペプチドリガンドをその検出方法が存在する1以上のドメインを含むように遺伝子組換えによって作る。例えば、ベクターとしては、限定されるものでないが、lac Z-融合タンパク質が生産されるように、リガンドコード配列が個々にlac Zコード領域のフレーム内でベクターとライゲートされている大腸菌発現ベクターpUR278(Rutherら、1983、EMBO 12:1791)、pINベクター(InouyeおよびInouye、1985、Nucleic Acids Res. 13:3101-3109; Van Heeke & Schuster、1989、J. Biol. Chem. 24:5503-5509)などが挙げられる。lac Zタンパク質は、比色検出方法を用いて直接検出してもまたはlac Zを特異的に認識する抗体を用いて間接に検出してもよい。さらにlac Zに特異的な抗体を、検出シグナルの増幅をもたらすリガンドを認識する抗体と組み合わせてもよい。
【0096】
不溶性ポリペプチドリガンドの場合、pGEXベクターを用いてリガンドポリペプチドをグルタチオン5-トランスフェラーゼ(GST)との融合タンパク質として発現させてもよい。一般に、リガンドだけでは可溶性でなくとも、リガンド-GST融合タンパク質は可溶性でありうる。さらにリガンド-GST融合タンパク質は、溶解した細胞からマトリックスグルタチオンアガロースビーズへの吸着と結合およびその後の遊離グルタチオンの存在のもとでの溶出により容易に精製することができる。GSTドメインも検出ドメインとして機能する。リガンド-GST、リガンド-lacZおよび他の類似のリガンド-検出ドメイン融合物の作製は、大きい容易に検出される分子を加えずに検出するのが困難である小ペプチドリガンドにとって特に有利である。
【0097】
他の実施形態においては、リガンドはさらに、リガンドの多量体形成を可能にする検出ドメインを含む。例えばリガンドを、二量体化を促進しうる抗体Fcドメインと融合することができる。ポリペプチドを抗体の定常領域と融合またはコンジュゲートする方法は当技術分野で公知である。例えば、米国特許第5,336,603号、第5,622,929号、第5,359,046号、第5,349,053号、第5,447,851号、第5,723,125号、第5,783,18号、1第5,908,626号、第5,844,095号、,および第5,112,946号;EP 307,434; EP 367,166;EP 394,827;国際特許出願公開WO 91/06570、WO 96/04388、WO 96/22024、WO 97/34631、およびWO 99/04813;Ashkenaziら, 1991, Proc. Natl. Acad. Sci. USA 88: 10535-10539;Trauneckerら, 1988, Nature, 331:84-86;Zhengら, 1995, J. Immunol. 154:5590-5600;およびVilら, 1992, Proc. Natl. Acad. Sci. USA 89:11337- 11341を参照されたい。あるいは、リガンドをロイシンジッパードメインと融合してもよい。可溶性オリゴマータンパク質を生産するための好適なロイシンジッパードメインの例は、PCT出願WO94/10308に記載され、そして肺界面活性タンパク質D(SPD)から誘導されたロイシンジッパーはHoppeら(FEBS Letters 344:191, 1994)に記載されている。それと融合した異種タンパク質の安定な三量体化を可能にする修飾されたロイシンジッパーの使用は、Fanslowら(Semin. Immunol. 6:267-278, 1994)に記載されている。ロイシンジッパーペプチドと融合した可溶性ポリペプチドを含む組換え融合タンパク質を好適な宿主細胞に発現させ、そして生成する可溶性オリゴマーを培養上清から回収する。ある特定のロイシンジッパー部分が優先的に三量体を形成する。一例は肺界面活性剤タンパク質D(SPD)から誘導されるロイシンジッパーであって、Hoppeら(FEBS Letters 344:191, 1994)および米国特許第号5,716,805に記載されている。
【0098】
一実施形態においては、リガンドを認識して結合する結合分子を発現するクローンを単離する。固体支持体は、リガンドを認識して結合する結合分子を発現するクローンを含有するクローンのサブセットを単離するためのテンプレートを提供しうることを具体的に意図する。このより小さい集団は、次いで本発明のスクリーニング方法の改変法を用いてスクリーニングすることができる。上記改変はクローンのサブセットをより低い密度でまくステップを含んでなる。特定の実施形態においては、クローンのサブセットを、単一クローンが単離されるには十分低いがサブセット中に存在する各クローンが固体支持体上で少なくとも1回、表されるには十分高い密度でまく。リガンドを認識して結合する結合分子を発現するクローンを含有するクローンのサブセットを当業者に公知の代わりの方法によりスクリーニングしうることも意図する。代わりの方法としては、限定されるものでないが、ELISAアッセイおよびFACS分析が挙げられる。
【0099】
5.5 選択マーカーと選択
本発明のライブラリー生産方法は、選択マーカーをコードするポリヌクレオチドとライゲートした分子をコードするポリヌクレオチドを含むクローンのライブラリーを作製する第1ステップを含んでなる。選択マーカーは一般に以下に記載されたものでありそしてある特定の具体例を実施例3に詳述している(上記)。選択マーカーをコードするポリヌクレオチドは、ライブラリークローンを含む形質転換した宿主細胞の選択および/または維持のために必要なポリヌクレオチドと同じであってもよいし、または第2の選択マーカーをコードする第2のポリヌクレオチドを利用してもよいことを具体的に意図する。
【0100】
選択マーカーは、容易に選択し、および/またはスクリーニングすることができる所望の形質をコードする多様な遺伝子のグループを包含する。選択マーカーは3つの一般的カテゴリーに分類することができる;1)特定の薬物に対する耐性の形質を与える薬物耐性マーカー遺伝子。例えば、β-ラクタマーゼ遺伝子はアンピシリンおよびカルベニシリンに対する耐性を与え、ネオマイシンホスホトランスフェラーゼII型(NPT II)はネオマイシン/カナマイシン/G418に対する耐性を与え(Colberre-Garapinら, 1981, J. Mol. Biol. 150:1)、クロラムフェニコールアセチルトランスフェラーゼはクロラムフェニコールに対する耐性を与え(Herrera-Estrellaら, 1983, Nature 303, 209-213)、ジヒドロ葉酸レダクターゼ(dhfr)はメトトレキセートに対する耐性を与え(Wiglerら, 1980, Natl. Acad. Sci. USA 77:3567;O’Hareら, 1981, Proc. Natl. Acad. Sci. USA 78:1527)、gptはミコフェノール酸に対する耐性を与え(Mulligan & Berg, 1981, Proc. Natl. Acad. Sci. USA 78:2072)、そしてハイグロマイシンホスホトランスフェラーゼ(hygro)はハイグロマイシンに対する耐性を与える(Santerreら, 1984, Gene 30:147)。2)形質転換された細胞が必須代謝物(通常、アミノ酸)を合成できるようにする代謝または栄養要求マーカー遺伝子(細胞は上記代謝物を生産できないが、増殖のために必要であり、上記代謝物の非存在のもとで細胞が増殖することを可能にする遺伝子)。例えば、HIS3、LEU2、TRP1およびURA3は、それぞれ、ヒスチジン、ロイシン、トリプトファンおよびウラシルを欠く培地においてある特定の栄養要求酵母菌株が増殖する能力を与える。3)タンパク質またはタンパク質ドメインをコードするスクリーニング可能なまたは精製マーカー遺伝子であって、様々な研究室の試験または精製技法を介して同定することができるかまたはタンパク質ドメインを発現するクローンの精製/同定を容易にする遺伝子。例えば、X-galを用いて検出されるβ-ガラクトシダーゼ(beta-gal)(Helmerら、1984、Bio/Technology 2、520-527)、炭化水素化合物を用いて検出されるルシフェラーゼ(lux)(Konczら、1987、PNAS 84、131-135)、またはカルモジュリンアフィニティ精製を用いて精製されるカルモジュリン結合ドメインなどのタンパク質ドメイン。
【0101】
当業者は、機能性分子を発現しないクローン(例えば、未熟停止コドンまたはフレームシフト突然変異を有する分子)は恐らく融合タンパク質を作製しないかまたは非機能性融合タンパク質を作製しうることを理解しうる。かかる非機能性分子を発現するクローンは、機能性融合タンパク質を発現しないクローンが排除される選択条件下でクローンのライブラリーを増殖することにより選択することができる。従って、本発明のライブラリー生産方法はさらに、機能性分子を発現するクローンを選択する条件下でクローンのライブラリーを増殖する第2のステップを含んでなる。
【0102】
当業者には、本発明のライブラリー生産方法に使用する選択マーカーおよび選択条件は明らかであろう。選択マーカーおよび選択条件は、作製するライブラリーの選択ならびに利用する発現ベクターおよび宿主細胞に応じて選ばれるであろう。
【実施例】
【0103】
6. 実施例
以下、本発明は、次の実施例を参考にして説明される。これらの実施例は、説明するためだけに与えられたものであって、本発明は、これらの実施例に限定されると決して推量してはならないが、むしろ、本明細書に記載の開示の結果として明らかになるいずれかおよび全ての変化を包含すると推量されるべきである。
【0104】
6.1 実施例1
ヒトscFvライブラリーの超ハイスループットスクリーニング
ほぼ2x109メンバーを含有する大きいファージscFv発現ライブラリーをヒトリンパ節および脾臓組織(図2を参照)から作製した。全ライブラリーをビオチン化EphA4-Fc融合タンパク質を用いてスクリーニングした(図1、3および4を参照)。3回のスクリーニングの後、24クローンのパネルを単離すると、そのうちの20クローンはELISAアッセイにおいて強い結合を示した(図4参照)。
【0105】
6.1.1 材料および方法
ナイーヴなヒトscFvライブラリーの生産: ヒトscFvライブラリーの作製は本質的に、Gaoら, 1999, J. Am. Chem. Soc. 121:6517-6518;およびMaoら, 1999, Proc Natl Acad Sci USA 96:6953-8に記載の通りであるが、次の改変を加えた:固定化ドメインとして作用しうるエピトープタグ(図2Bを参照)を補ったクローニング部位を遺伝子操作でM13中に作製し、そして発現ベクターとして用いた。さらにここで用いたscFvはその自然型(すなわち、M13コートタンパク質と融合されていない)である。概要を説明すると、リンパ節および脾臓組織から得たヒトmRNAを逆転写し、そしてVHおよびVL配列をPCRにより増幅した。オーバーラップPCRを利用してVHおよびVL配列を接合し、scFv遺伝子をコードするポリヌクレオチドを生成させた。次いでscFv遺伝子をM13ファージ発現ベクター中にライゲートし、そして大腸菌中に形質転換した。模式図は図2Aを参照されたい。ライブラリーの複雑度はほぼ2x109である。
【0106】
リガンドのビオチン標識化: 市販のEphA4-Fc(R&D System、Cat# 369-EA-200)をPBS(pH7.2)に対して透析した。透析後のEphA4-Fcの最終濃度は0.83mg/mlであった。1mlのEphA4-Fcをビオチン化に用いた。使用直前に、Sulfo-NHS-LC-ビオチン(PIERCE、Cat#: 21335)の10mM溶液を水で調製した。16.6μlの10mMビオチン試薬溶液を1mlの透析したEphA4-Fcに加え(ビオチンのモル過剰倍率は20である)、そして反応混合物を氷上で2時間インキュベートした。未反応ビオチンは、PBS(pH7.2)に対する透析により、ビオチン化EphA4-Fcから除去した。
【0107】
超ハイスループットスクリーニング:第1日 午後に、M9プレートからのTG1の単一コロニーを用いて2XYTに接種した。培養物を30℃で一晩インキュベートした。
【0108】
第2日 TG1を接種する:TG1-Blueの一晩培養物を2XYT中に1:100で希釈し、37℃にて250rpmでほぼ1.5〜2時間インキュベートした(OD600nm=0.5〜0.8)。
【0109】
TG1をファージに感染させる:ファージをPBS中に、キャプチャーリフトアッセイ用にほぼ1010pfu/ml、そしてファージ滴定用にほぼ105pfu/mlの濃度で希釈した。上層寒天溶液(SB+0.7%寒天)を12mlチューブ中にアリコート(6ml/チューブ)をとり、そして50℃水浴中で使用まで保持した。次の混合物を調製した:
a. ステップ1からのTG1:800μl、
b. IPTG(1M):6μl、および
c. 希釈したファージ溶液:6μl。
【0110】
400μlの上記混合物を6mlの上層寒天に加え、混合し、次いで静かに100mmプレーンLB寒天プレートの上層に注ぎ、そして室温にて10分間静置した。次いでまいたプレートを37℃にてほぼ5〜6時間インキュベートした。
【0111】
フィルターを調製する: プレートのインキュベーションの間、83mm NC膜の片側を鉛筆で標識をつけた。抗Flag抗体(M2Ab、4.9mg/ml、Sigmaより入手)をPBS(pH7.2)を用いて1:500に希釈し、そして100mmペトリ皿中に置いた。標識した膜を、標識した側を上にして抗体溶液の表面上に置いて、3時間、室温にて遅い速度のプラットフォーム振とう機上でインキュベートした。次いでフィルターを裏返し、さらに30分間インキュベートした。抗体溶液を除去し、そしてフィルターをPBS中の4%脱脂乳(Bio-Radから入手)を用いて2時間室温にてブロックした。次いでフィルターを3回PBSを用いてすすぎ洗い、化学フード内で乾燥した。
【0112】
細菌/ファージローン(lawn)上でのフィルターのインキュベーション: 細菌/ファージプレートをインキュベーターから取出した。単離したプラークが滴定プレート(ここでは108希釈プレート)上に見られた。乾燥したフィルターを標識した側を上にして、細菌/ファージローンの表面上に静かに敷き、フィルター位置を、フィルターの孔に針を突っ込んでマークした。次いでプレートを室温(25℃)にて一晩(湿容器内で)インキュベートした。
【0113】
第3日 フィルターを注意深くプレートから剥がし、PBSにより2回、次いでTPBS(PBS+0.1% Tween20)により6回(標識した側を上にして3回および標識した側を下にしてさらに3回)、真空洗浄器を用いて洗浄した。次いで膜を、スーパーブロッキングバッファー(PIERCE)中の1%BSAに1:1000で希釈したEphA4-FC-ビオチンを用いて、標識した側を下にしてインキュベートした(4ml/フィルター)。EphA4-FC-ビオチンのストック濃度は0.8mg/mlであった。次いで膜を3時間、室温にてロッキングプラットフォーム上で振とうしながらインキュベートした。次いで膜を6回(標識した側を上にして3回および標識した側を下にしてさらに3回)、TPBSにより真空洗浄器を用いて洗浄した。次いで洗浄したフィルターをスーパーブロッキングバッファー(PIERCE、10ml/フィルター)中の1%BSAに1:4000で希釈したアビジン-AP溶液を用いて、振とうしながら、1時間インキュベートした。この最後のインキュベーションの後に、フィルターを次いで上記のように洗浄し、3〜5回、PBSによりすすぎ洗いした。洗浄バッファーを取り除いて、NBT/BCIT溶液(PIERCE、5ml/フィルター)をフィルターを含有するプレートに加えた。発色させ(3〜5分)、発色したフィルターを乾燥し、プレートを整列し、そして第2回スクリーニングに対するポジティブプラークを拾った。
【0114】
抗体スクリーニング/Elisa(2.5mlスケール):第1日- 午後に、M9プレートからのTG1の単一コロニーを用いて2XYTに接種した。培養物を30℃で一晩インキュベートした。
【0115】
第2日 TG1を接種する:TG1-Blueの一晩培養物を2XYT中に1:100で希釈し、37℃にて250rpmでほぼ1.5〜2時間インキュベートし(OD600nm=0.5〜0.8)、次いでチューブ(2.5ml/チューブ)にアリコートをとった。
【0116】
高力価ファージ調製: よく単離されたプラークを拾い、これを用いて上記からの2.5ml培養物に接種した。チューブを一晩、37℃にて、250-300rpmで振とうしながらインキュベートした。
【0117】
コート抗原: 抗原(ここでは、EphA4-His融合タンパク質およびネガティブ対照としてSynagisを用いた)をコーティングバッファー(炭酸塩バッファー、pH9.6)でほぼ5-10μg/mlに希釈し、マイクロタイタープレートに25μl/ウエルを加え、そして4℃にて一晩インキュベートした。
【0118】
第3日 ELISA-ブロッキング: 過剰のコーティング溶液を除去し、ウエルをPBS(ブロッキングバッファー)中の4%脱脂乳を用いて50μl/ウエルで1時間、37℃にてブロックした。一晩インキュベーションした細胞培養物を3500rpm x 20分の遠心分離により採集した。過剰のブロッキング溶液を除去し、25μl/ウエルの一晩培養物上清を加えた。37℃にて1時間、インキュベートした。プレートをプレート洗浄器を用いて洗浄し、そして25μl/ウエルの抗Flag M2抗体(ブロッキングバッファー中に1:1000希釈)を各ウエルに加えた。次いでプレートを30分間37℃にてインキュベートした。プレートを再びプレート洗浄器を用いて洗浄しそして25μl/ウエルのヤギ抗-マウス(IgG-H+L)-HRPコンジュゲート(ブロッキングバッファーに1:1000希釈)を各ウエルに加えた。プレートを30分間、37℃にてインキュベートした。プレートをプレート洗浄器を用いて洗浄し、次いでピペット(ピペットのアップアンドダウンを20回)により1回、および蒸留水によりさらに10回洗浄した。発色: 25〜35μl/ウエルのTMB基質を各ウエルに加え、ほぼ5分間発色させた。反応を、等容積の0.18M H2SO4を加えることにより停止しそして吸収を450nmにて読み取った。
【0119】
抗体精製(800mlスケール):第1日 午後に、M9プレートからのTG1の単一コロニーを用いて2XYTに接種した。培養物を30℃で一晩インキュベートした。
【0120】
第2日 2mlの一晩細胞培養物を200mlの2XYT培地中に希釈し、そして振とう機内で37℃にて250rpmでOD600nm〜0.8まで増殖した。250μlの一晩培養の高力価ファージ(典型的にはほぼ1012pfu/ml)を用いて200ml細胞に15分間、室温にて感染させた。600mlの2XYT培地を200mlの感染した細胞に加えた。感染した細胞を37℃にて1時間、次いで30℃にて 一晩増殖した。
【0121】
第3日 細胞を、8000rpm(Beckman JA-10回転子)で20分間、遠心分離によって取り出し、そして上清を0.45μmフィルターを通過させることにより濾過した。濾過した上清を、PBSで前洗浄した抗FLAG M2アガロースアフィニティカラムに供給した。次いでカラムを少なくとも10カラム体積のPBSを用いて洗浄した。次いで抗体(scFv)をカラムから溶出バッファー(100mMグリシン、pHを3に調節)を用いて溶出した。1M Tris-HCl(pH 8.0)を溶出した抗体に加えて中和した。中和した抗体を次いでPBSに対して透析してほぼ1mlに濃縮した。
【0122】
6.1.2 考察
キャプチャーリフトスクリーニング方法は通常、ハイスループットスクリーニング方法に固有のいくつかのバイアスを避けるために使用される。しかし、大きい多様なライブラリーに対しては価値が限られているので、その使用は小ライブラリー集団のスクリーニングに限られている。キャプチャーリフトスクリーニングは、高いアフィニティをもつ抗体クローンの選択に対して、ヒト化抗体の選択に対して、そして、さらに最近には、小ライブラリー(多様性ほぼ105)からの新規抗体分子の発見に対して首尾よく使用されている。本明細書において、本発明者らは初めて、非常に大きい集団からの稀なクローンの同定を可能にする非常に大きい結合分子ライブラリーの迅速かつ効率的なスクリーニングを可能にする著しく改善されたキャプチャーリフト手法を記載する。
【0123】
ほぼ2x109メンバーを含有する大きいファージscFv発現ライブラリーを、ヒトリンパ節および脾臓組織から作製した。そのライブラリーをほぼ3800クローン/mm2の超高密度でまいた。図3は、第1回(3A)、第2回(3B)、ならびに第3回および最終回(3C)スクリーニングからのポジティブクローンを含有する代表的フィルターを示す。次いでそのライブラリーを、ビオチン化EphA4-Fc融合タンパク質を用いてスクリーニングした。EphA4-Fc融合体の使用は、より大きい数のビオチン分子が各リガンドと結合することを可能にし、アビジン-APによる検出時にシグナルの有意な増幅をもたらす。化学発光に基づく検出方法(例えば、Salernoら, 2003, J Chromatogr B Analyt Technol Biomed Life Sci 793: 75;Mattsonら, 1996, Anal Biochem 240: 306;Kricka, 1991, Clin Chem 37: 1472を参照)の使用は、検出用2次分子と融合したリガンドの使用の有無に関わらず検出シグナルの有意な増幅を可能にしうるし、かつさらに多数のクローン数を各プレート上にまくことを可能にするであろう。
【0124】
提示した手法を用いると、少なくとも3000万クローンを単一の82mmニトロセルロース膜上でスクリーニングすることができる。これは、従来記載されているキャプチャーリフト方法よりフィルター1枚当たり300倍多いクローンである。伝統的なキャプチャーリフト方法は、このサイズをすっかりスクリーニングするために1ライブラリーに対して20,000〜40,000プレート(100mm)を必要としよう。本明細書に記載の超高密度プレーティングおよびスクリーニング方法を用いると、全ライブラリーをスクリーニングするために、わずかほぼ67プレートしか必要としない。
【0125】
6.2 実施例2
超ハイスループットスクリーニングによる抗イディオタイプscFvの単離
実施例1で作製したライブラリーを、MEDI-AAA、抗インターフェロンα抗体の抗原結合ドメインを特異的に認識する抗イディオタイプ抗体を同定しかつ単離するためにも利用した。該ライブラリーをビオチン化MEDI-AAA(Fab)2フラグメント(図5)を用いてスクリーニングした。2回のスクリーニングの後に4クローンを単離し、その1つはELISAアッセイでMEDI-AAA抗体と強い結合を示す一方、いくつかの無関係な抗体と結合しなかった(図6参照)。
【0126】
6.2.1 材料と方法
MEDI-AAA(Fab)2の調製: MEDI-AAA(Fab)2を、固定化したペプシン試薬を用いて調製した(Pierce cat. 20341)。製造業者の指示に従って、500μgのMEDI-AAA抗体を消化した。消化した抗体を、遠心分離によりペプシン樹脂から分離し、そして溶出剤をタンパク質Aカラム上に流して抗体Fc断片を除去した。精製した(Fab)2をPierce濃縮溶液を用いて、製造業者の推奨に従って濃縮し、その後、1 X PBS中にpH7.2で4℃にて一晩透析した。最後のタンパク質を10%Bis-Trisタンパク質ゲル(図5)上でMOPSバッファー(Invitrogen cat. NP0001)を用いて分析した。
【0127】
MEDI-AAA(Fab)2ビオチン化: 200μgのMEDI-AAA(Fab)2を、NHS-LC-ビオチン試薬を用いて製造業者の取扱説明書(Pierce cat.21338)に従いビオチン化した。20ビオチン/(Fab)2分子の比を用いて、発色過程の高い感度を確実にした。組み込まれなかったビオチンはNAP5 脱塩カラム(Pierce cat. 17-0853-01)を用いて除去した。最後の標識した(Fab)2を、上記のように、ならびに無処置IgGおよび無標識(Fab)2とともに変性および非変性条件下で分析した(図5)。
【0128】
ファージ培養物: 最小培地M9(Teknova cat. M2100)プレート上で増殖したTG1細菌の単一コロニーを用いて1mlの2XYT(Teknova cat. Y0167)に接種し、30℃にて一晩インキュベートした。一晩TG1培養物を用いて2XYT中の培養を0.1 OD600 37℃、250 rpmで開始して0.5〜0.8 OD600の中対数(mid log)に到達するまで続けた。上層寒天(スーパーブロス、0.7%寒天)をマイクロ波で融解しそして15mlファルコンチューブ中にアリコート(6ml/チューブ)をとり、チューブを50℃水浴で必要なだけインキュベートした。キャプチャーリフトファージ発現ライブラリー(2.08x109pfu/μl)をそれぞれ、1:40、1:400および1:4x106に希釈して、ほぼ1x107pfu/μl、1x106pfu/μlおよび1x102pfu/μlを得た。TG-1細菌を感染させるために、1μlの1x107pfu/μlおよび1μlの1x106pfu/μl希釈したファージを、800μlの中対数(mid log)TG-1とともに6μlの1M IPTGを含有する5つのエッペンドルフチューブに加えた。TG1の1つのチューブだけは1x106pfu/μl希釈(滴定プレート)で感染させ、そしてこのTG1の残りのチューブはネガティブ対照として用いた。感染させたTG-1培養物を上層寒天のチューブと組み合わせて、予熱したLBプレート(100mm)上に注いだ。上層寒天を10分間室温にて固化させた後に、プレートを37℃にて5〜6時間インキュベートした。
【0129】
フィルター調製: 10枚のニトロセルロース膜(Protron Bioscience cat. 10401116、82 mm)の片側を、鉛筆を用いて抗原および膜数を記入して標識した。抗フラグM2抗体(sigma cat.)を、100mlの1X PBS pH7.2中に1:500で希釈し、この溶液の10mlを10cmペトリ皿のそれぞれに加えた。ニトロセルロース膜を、標識した側を上にして抗フラグM2抗体(Sigma Cat. F3165)中で3時間、低速度振とう機上で室温にてインキュベートした。最初のインキュベーション後に、ニトロセルロース膜を逆転してさらに30分間インキュベートした。膜を簡単にすすぎ洗いし、10 mlの1X PBS pH 7.2中の4%脱脂乳(Bio-Rad cat. 170-6404)を用いて2時間ブロックした。乳溶液を取出した後に、膜を3回、1X PBS pH 7.2中で洗浄し、そしてフード内で乾燥させた。
【0130】
キャプチャーリフト選択: 滴定プレート(1x102希釈)上にプラークが現れた後に、プレートをインキュベーターから取り出した。ニトロセルロース膜を、scFvをキャプチャーする1x107および1x106プレートの上層寒天の表面上に注意深く重ねた。21ゲージ針を用いて12時の位置に1つ、4時の位置に2つそして8時の位置に3つの孔を開けて、プレート上のフィルターの方向と位置をマークした。次いでプレートを25℃にて一晩インキュベートした。1:4x106ライブラリー希釈プレートを用いて実ライブラリー力価をチェックした。サイドアームの付いたKonteの4Lフラスコとフィルターホルダー(Fisher cat. K953840-4090)を用いてフィルターを洗った。最初にフィルターを寒天表面から注意深く取り出し、そして両側の膜を3回PBST(1X PSB、0.1% Tween 20)を用いて、洗浄ボトルからのTPBSを供給する一方、ホルダーを真空に接続することによりすすぎ洗った。ハイブリダイゼーション溶液を作るために、0.1μg/mlのビオチン化ニュートロステンシン(neutrostensin)、4μgのビオチン化MEDI-AAA(Fab)2および100μgのGEA44抗体を、スーパーブロック溶液(Piercecat. 37515)中の1%BSAに加えた。それぞれの膜を、10cmペトリ皿に入った4mlのハイブリダイゼーション溶液中で静かに攪拌しながら3時間室温にてインキュベートした。フィルターホルダーと真空を用いて、膜の両側を3回、PBSTにより洗浄した。フィルターを、10mlのブロッキングバッファー中の1:4000アビジン-AP(Pierce Cat. 31002)を用いて、1時間室温にて静かに振とうしながらインキュベートした。フィルターを5mlのNBT/BCIT(Piercecat.34042)溶液により発色する前に、フィルターを3回両側をPBSTにより洗浄した。褐色のポジティブドットが現れた後に、フィルターを水を用いてすすぎ洗いして反応を停止した。フィルターをフード内で乾燥しそして透明シート上で光コピーをとった。位置と方向をマークするフィルターに作った孔を用いて、プラークプレートを透明シート上に置き、寒天の針孔を透明シート上の針穴と整列させた。ポジティブスポット上のプラークを大
きいオリフィスマイクロピペット(VWR、Cat#:53503-614)のチップを用いて拾い、そして溶出バッファー(10mM Tris、100mM Nacl、pH 7.4)へ移した。溶出ファージ(ほぼ2〜5 x 102pfu/μl)で出発し、1:10希釈物(ほぼ2〜5 x 104pfu/μl)を用いて、先に記載の第2回のキャプチャーリフト用のプラークプレートを調製した。拾ったポジティブプラークのそれぞれに対して1枚のフィルターを使用した。
【0131】
ファージ生産: 第2回のキャプチャーリフトから溶出したポジティブプラークを1:200(1〜3 x 102pfu/μl)に希釈し、先の通り、10cmペトリ皿上にまいた。個々のプラークを250μlの溶出バッファーが入った96ウエルプレート中に拾った。プラークを寒天から溶出させた後に、200ulの溶出したファージを用いて、深いウエルプレート中の500μlの中対数(mid-log)TGIに接種した。ファージ生産のために、培養物を一晩、30℃にてインキュベートした。
【0132】
ELISAスクリーニング: ELISAマイクロタイタープレートを一晩、4℃にて50μlのMEDI-AAA(l5μg/ml)および対照抗体を用いてコーティングした。対照抗体には、市販ポリクローナルヒトIgG(Jackson Immunoresearch Lab、Cat. 009-000-003)、Synagis(登録商標)およびMEDI-AAAと高度の相同性を共有する軽鎖フレームワークを有する(フレームワーク残基81のうち77が同一である)無関係な抗体が含まれた。プレートをELISAブロッキング溶液(1 X PBST中に2%乳)を用いて1時間、室温にてブロックした。また48μlの一晩培養ファージ上清も、12μlの5XELISAブロッキング溶液を用いて1時間室温にてブロックした。インキュベーションの後、プレートを5回、1 X PBSTにより、Elx405プレート洗浄器を用いて洗浄し、そして50μlの前ブロックしたファージ上清を各ウエルに加えた。室温での1時間インキュベーションの後、プレートを再び、前記の通り洗浄し、そしてブロッキング溶液中の1:2000抗フラグM2(Sigma)抗体希釈液50μlを各ウエルに加えた。次いでプレートを室温にて1時間インキュベートし、そして5回、Elx405プレート洗浄器を用いて洗浄した。ELISAサンドイッチを完了させるために、ブロッキング溶液中の1:4000ヤギ抗-マウスHRP抗体(Pierce cat. 31164)50μlを各ウエルに加え、室温にて1時間インキュベートした。プレートを10回、PBSTにより、Elx405プレート洗浄器を用いて洗浄し、そして50μlのTMB基質(KPL cat. 52-00-03)を加えた。5〜10分間のインキュベーションの後、50μlの0.2M H2SO4を加えることにより反応を停止した。プレートを450nmにて読み取り、そして各抗体に対するシグナルをプロットした(図6AおよびB)。
【0133】
6.2.2 結果
実施例1で作製したファージscFv発現ライブラリーを、超ハイスループット(ほぼ126-1267クローン/mm2の密度でまいた)によりスクリーニングしかつビオチン化MEDI-AAA(Fab)2フラグメントを用いてスクリーニングして(図5参照)、抗イディオタイプscFvクローンを同定した。2回のスクリーニングの後、4クローンを単離し(データは示してない)、それらの1つはELISAアッセイでMEDI-AAA抗インターフェロンα抗体と強い結合を示す一方、BSAまたはいくつかの無関係の抗体と結合しなかった(図6参照)。特に抗イディオタイプscFvはヒトポリクローナル抗体調製物(図6B)または高度に関係した軽鎖を有する無関係なヒト抗体(対照)(図6AおよびB)と有意な結合を示さなかった。超ハイスループットスクリーニング方法の使用は、抗イディオタイプ抗体の迅速な同定を可能にした。ほぼ5.5x107のクローンを丁度10枚のプレート上で超ハイスループットスクリーニング方法を用いて速やかにスクリーニングしたが、伝統的方法は同じ数のクローンをスクリーニングするには少なくとも550枚のプレートを必要としたであろう。
【0134】
6.3 実施例3
発現可能な抗体ライブラリー構築と非機能性クローンの排除
プラスミドpUCKAを作製して、β-ラクタマーゼ遺伝子(アンピシリン/カルベニシリン耐性を与える)をコードするポリヌクレオチドとライゲートした3'-FLAGおよびHIS6エピトープタグの両方によるscFvのライブラリーのクローニングを容易にした。停止コドンを伴ってまたは伴わないでクローニングしたいくつかのscFvは、停止コドンを欠くクローンだけがカルベニシリン耐性であることを実証した。全ライブラリーをpUCKAベクター中にクローニングし、そして選択前の非機能性クローンの数はほぼ25%であることがわかった。非機能性タンパク質をコードするクローンを除去する選択の後に構築したファージライブラリーは、5x108を超える複雑度を有することがわかった。
【0135】
発現ベクターpUCKAの構築: ベクターpUCKAをpUC19から誘導した。プライマー対KanaFor/KanaRev、pUCFor/EcoRIRev、およびpUCRev/EcoRIForを利用し、pET-27b(Novagen)およびpUC19をそれぞれテンプレートとして、カナマイシン遺伝子およびpUC19主鎖を増幅した。3つのPCR断片をゲル精製しかつプライマーEcoRIFor/EcoRIRevを用いるオーバーラップPCRにより組み立てた。PCR産物をEcoR Iにより消化し、自己ライゲートし、そしてカナマイシンの選択のもとでXL1-Blue中に形質転換して、ベクターpUCK(示してない)を作製し、そのβ-ラクタマーゼ遺伝子をカナマイシンにより置き換えた。ポリリンカー(リボソーム結合部位、p3リーダー配列、Flag-タグ、およびHis-6-タグを含む)を、ファージ発現ベクターpMD102からプライマーHindIIIFor/EBNSRevを用いて増幅し、そしてHindIII/EcoRI部位中にクローニングしてベクターpUCK-1を作った。開始コドン、ATGのないβ-ラクタマーゼ遺伝子を、pUC19からプライマーAmpFor/AmpRevを用いて増幅し、そしてpUCK-1のSpeI/EcoRI部位中にクローニングしてベクターpUCKAを作製した。プライマー配列については表1を参照されたい。
【表1】
【0136】
クローンscFv F9、LX2、EA20、およびEA44: F9、LX2、EA20、およびEA44と名付けられたいくつかのscFvをコードし、そのC末端に開始コドンをもつかまたはもたないポリヌクレオチドを、pETHis(T7プロモーターの制御下のscFv発現ベクター)から切除し、pUCKAのSfi I部位中にクローニングし、XL1-Blue中に形質転換し、そしてLB-寒天/30μg/mlカナマイシンを含有するカナマイシンプレート上で拡大した。そのコロニーを拾い、カルベニシリンを伴うまたは伴わないLB/2XYT培地中に100μg/mlで接種した。scFv遺伝子の末端に停止コドンのないクローンだけがカルベニシリンの選択のもとで生存することができる(データは示してない)。このデータに基づくと、このベクターは、抗体ライブラリーから所望でない停止コドンまたはフレームワークを伴う抗体遺伝子を除去するように装備することができる。停止コドンをもたないscFvを発現するこれらのクローンの増殖速度をまた、色々なカルベニシリンの濃度で評価した(表2)。
【0137】
表2: 停止コドンのないscFv分子を発現するクローンの増殖速度
【表2】
【0138】
scFv発現ライブラリーの構築: 2x109メンバーをもつ本来のファージ発現ベクター(上記実施例1を参照)中のscFv遺伝子ライブラリーを、Sfi Iを用いて消化し、アガロース上でゲル精製し、そして、同じ制限酵素を用いて切断しておいたpUCKAベクター中にライゲートした。ライゲートした生成物を大腸菌XL1-Blue(テトラサイクリン耐性菌株)コンピテント細胞中にエレクトロポレートしてほぼ5×109の多様性を有する独立した形質転換体を得た。エレクトロポレーションの後、細胞を50枚のディッシュ(150mm×10mm;Nunc)中のLB寒天(2%グルコース、50μg/mlカナマイシン、および20μg/mlテトラサイクリンを含有する)上にまいて、一晩、30℃にてインキュベートした。クローンをプレートから300mlの2XYT培地中に掻き出した。掻き出した細菌を100μg/mlカルベニシリン、50μg/mlカナマイシン、および2%グルコースを含有する1リットルの2XYT培地に接種した。細胞を37℃にて4〜6時間、振とうしながら増殖してフレームシフトまたは停止コドンをもつライブラリー中の抗体遺伝子を除去し、発現可能な抗体ライブラリーを構築した。次いでグリセロールを残りの細菌に加えて最終濃度10%にして-80℃にて貯蔵した。
【0139】
系効率の試験: 一晩、30℃にて増殖した後、形質転換したライブラリーのLB-寒天ディッシュから192クローンを拾い、そして100μg/mlカルベニシリン、50μg/mlカナマイシン、および2%グルコースを含有する100μl2XYT培地の入った96プレート2枚中に接種した。37℃にて8時間、振とうしながら増殖した後、各ウエルから5μlを、各ウエル中に0.5ml 2XYTを含有する4つの深いウエルプレート(4つのうち、2つはカルベニシリン100μg/mlを含み、2つは含まない)中に移した。プレートを37℃にて一晩増殖し、そして直接観察により増殖(ポジティブ/ネガティブ)のスコアを付けた。全てのクローンはカルベニシリンを含まないと非常によく増殖することができる。しかし、100μg/mlカルベニシリンの選択のもとでは、75%(192クローンのうち144)しか生存できない。
【0140】
発現可能なファージ抗体発現ライブラリーの構築: 100μg/mlカルベニシリンの選択のもとで生存した細菌ライブラリーから、Maxiプラスミド単離キット(Qiagen)を用いて、プラスミドを抽出した。発現可能なscFvライブラリー遺伝子をSfiIを用いて消化し、ゲル精製し、そして同じくSfiIにより切断したファージ発現ベクターpMD102中にライゲートした。ライゲートした混合物、scFvライブラリーをXL1-Blueコンピテント細胞中に15個のキュベット(Bio-Rad)を用いてエレクトロポレートし、2XYT上層寒天と混合し、40枚のLB寒天ディッシュ(150mm x 10mm;Nunc)上にまいて、30℃にて一晩インキュベートした。このライブラリーの多様性は、プラーク数としてカウントして5x108を超えた。ファージライブラリーを、上層寒天から200mlのファージ溶出バッファー(10mM Tris-HCl、150mM NaCl、pH7.5)を用いて溶出した。PEGおよびNaClを加えて、それぞれ4%および3%の最終濃度として、ファージを沈降させた。ファージを、8%のグリセロールを含有するPBSバッファー中に再懸濁し、アリコートをとり、そして-80℃にて貯蔵した。
【0141】
6.3.1 結果
選択マーカーをコードするポリヌクレオチドとライゲートした分子をコードするポリヌクレオチドのライブラリーの作製を容易にするために、標準の分子クローニング技法を用いてプラスミドpUCKAを作製した。図7はプラスミドマップであり、図8はライブラリークローニング部位周りのヌクレオチド配列の詳細である。pUCKAの重要な特徴としては、2つの薬物選択マーカー(該ベクターを含有する細胞を選択/維持するためのカナマイシン耐性遺伝子および非機能性分子を発現するクローンを除去する選択のためのアンピシリン/カルベニシリン耐性を与えるβ-ラクタマーゼ遺伝子)、複製起点、クローニング部位(SfiI)の5'およびクローニング部位の3'のプロモーターおよびシグナル配列、ならびにβ-ラクタマーゼ遺伝子のフレーム内にライゲートされたFLAGおよびHIS6エピトープタグが挙げられる。β-ラクタマーゼ遺伝子は開始コドンを欠き、従って機能性融合タンパク質をコードする転写物だけがβ-ラクタマーゼを発現する細胞を生じうることに注意されたい。従って、正しいリーディングフレーム内にないかまたは停止コドンを生じる突然変異を含有するかまたはフレームシフト突然変異を含有するSfi I部位中にクローニングされたポリヌクレオチドは、機能性融合タンパク質(すなわち、機能性β-ラクタマーゼを発現する)をコードする転写物を作製しないしかつアンピシリン/カルベニシリンに対する耐性がないであろう。
【0142】
ベクターを試験するために、いくつかのscFvをSfi I部位中に(適当なリーディングフレーム内に)、停止コドンとともにまたはなしでクローニングした。停止コドンなしでscFvをコードするクローンだけが機能性融合タンパク質をコードする転写物を作製するはずである。実際、停止コドンを欠くscFvをコードするクローンだけがカルベニシリンの存在のもとで増殖することができた。停止コドンを欠くscFvをコードするいくつかのクローンの増殖速度を試験した(表2参照)。低濃度のカルベニシリン(50〜100μg/ml)においては、クローン間の差は少ししかなかったが、高濃度(200〜400μg/ml)においては、恐らくこれらのクローンの発現レベルが低いことを反映して、いくつかのクローンは増殖が遅かった。さらに高濃度のカルベニシリンにおいては、どのscFvクローンも増殖しなかった。これらのデータは、低発現クローン選択条件の損失を避けるために、最小濃度のネガティブ選択剤、薬物を用いることが必要であるか、または、もし栄養要求性選択マーカーを利用するのであれば、選択の時間を増加することが必要であることを示した。
【0143】
非機能性分子を発現する(すなわち、pUCKA中にクローニングしたときに機能性融合タンパク質をコードする転写物を作製しないであろう)クローンの存在量を決定するために、全ライブラリーをpUCKAベクター中にクローニングして大腸菌中に形質転換し、そしてカナマイシン中で増殖して形質転換細胞を選択した。192の個々のコロニーを単離し、そして100μg/mlのカルベニシリンとともにまたは無しで増殖させた。カルベニシリンの存在のもとで、192のうち144だけが増殖し、選択前の非機能性クローンの数はほぼ25%であることを示した。従って、pUCKAなどのベクター中のライブラリーを作製して機能性分子をコードするクローンだけを選択することにより、非機能性クローンの有意な減少を達成することができる。
【0144】
pUCKA中にクローニングしたライブラリーを次いでカルベニシリンの存在のもとで増殖して非機能性クローンを排除した。選択後に、scFv遺伝子を含有するSfi I断片をプラスミドpMD102中にサブクローニングすることにより、ファージライブラリーを構築した(図9)。得られるファージライブラリーは5x108より高い複雑度を有することがわかった。
【0145】
6.4 実施例4
ナイーブなヒトscFv発現ライブラリーの直接にpUCKA中への構築
pUCKAにおけるナイーブなヒトscFv発現ライブラリーの生産: 選択のためのヒトscFvライブラリーの作製は、scFvを直接pUCKA中にクローニングすることを除いて、本質的に実施例1に記載の通り実施することができる。概要を説明すると、リンパ節および脾臓組織から得たヒトmRNAを逆転写し、そしてVHおよびVL配列をPCRにより増幅した。オーバーラップPCRを次に利用してVHとVL配列を接合し、scFv遺伝子を生成させた。次いでscFv遺伝子をpUCKA発現ベクター中にSfi I制限酵素切断部位においてライゲートし、そして大腸菌中に形質転換した。形質転換した大腸菌を、全クローンの増殖を許容する条件のもとで(例えば、カナマイシンを含有するがアンピシリン/カルベニシリンを含有しない培地で)増殖するか、またはβ-ラクタマーゼ遺伝子とライゲートした機能性scFvをコードするクローンだけを増殖する選択条件のもとで(例えば、カナマイシンおよびアンピシリン/カルベニシリンの両方を含有するまたはただアンピシリン/カルベニシリンだけを含有する培地で)増殖することができる。得られる形質転換大腸菌細胞を許容条件のもとで増殖させる場合、得られる培養物の一部分または全ては従って、選択条件のもとで増殖することができる。いくつかの具体的な抗生物質濃度は実施例1および3に記載されている。得られる培養物はスクリーニングしてもよいしまたは(例えば、凍結グリセロールストックのように)後日の使用のために貯蔵してもよい。
【0146】
スクリーニングのためにはscFvフラグメントを、アンピシリン/カルベニシリン中で増殖しうるクローンの選択(すなわち、β-ラクタマーゼ遺伝子と融合した機能性scFvをコードするクローンの選択)後に、ファージ発現またはファージディスプレイに好適な代わりのベクター中にサブクローニングすることが望ましい。上記ハイスループットスクリーニング方法の利用を容易にするためには、scFvフラグメントをpMD102などのファージ発現に好適なベクター中にサブクローニングする。
【0147】
6.5 実施例5
さらなる発現ベクター
発現ベクター、さらなる発現ベクターの構築: さらなる発現ベクターを、pUCKAの構築に用いたのと類似の方法で、pUC19または他のベクターから誘導することができる。あるいは、さらなる発現ベクターを、新規に、所望の特徴をコードするポリヌクレオチドのライゲーションにより作製することができる。さらなる発現ベクターを生産するために有用なクローニング方法は当技術分野で公知である。例えば、Current Protocols in Molecular Biology, F.M. Ausubelら, 編, John Wiley & Sons(Chichester, England, 1998)およびMolecular Cloning: A Laboratory Manual、第3版, J. Sambrookら, 編, Cold Spring Harbor Laboratory Press(Cold Spring Harbor, NY, 2001)を参照されたい。
【0148】
作製されるさらなるベクターはpUCKAと異なる1以上の特徴を有することができ、それらの特徴としては、限定されるものでないが、エピトープタグ、分子のライブラリーをコードするポリヌクレオチドを挿入するクローニング部位、分子のライブラリーをコードするポリヌクレオチドを融合するための選択マーカー、複製起点、シグナル配列、プロモーター、発現ベクターを含む細胞を維持/選択するためのさらなる選択マーカー、細胞においてベクターを発現および/または維持するために必要なその他の特殊な成分が挙げられる。
【0149】
上記発明は、明確性と理解を目的として若干詳細にわたって記載したが、本開示を読むことから、本発明の真の範囲から逸脱することなく形態および詳細の様々な変化をなしうることが当業者に明らかであろう。例えば、上記の全ての技法と装置は様々な組合せで用いることができる。本出願に引用した全ての刊行物、特許、特許出願、またはその他の文書は参照によりその全文が全ての目的に対して、あたかもそれぞれの個々の刊行物、特許、特許出願、またはその他の文書が個々に参照により全ての目的に対して組み入れられると示されたのと同程度に、組み入れられる。さらに、米国予備特許出願第60/623,240号(2004年11月1日出願)は参照によりその全文が全ての目的のために組み入れられる
【図面の簡単な説明】
【0150】
【図1】使用するキャプチャーリフトの全体スキームの概観図である。キャプチャー試薬をコーティングしかつブロックしたフィルターを、細菌ローン(lawn)中のファージ発現scFvライブラリーを含有するプレート上に置いた。そのフィルターを持ち上げて(またはリフトして(lift))、ビオチン標識した抗原、アビジン酵素コンジュゲート、および検出用現像薬を逐次用いてインキュベートする。実験の詳細は実施例1を参照されたい。
【図2A】ヒトscFvライブラリーを作製する全体スキームの概観図である。可変重鎖(VH)および軽鎖(VL)を逆転写し、増幅し、次いでオーバーラップPCRにより接合する。得られるscFv遺伝子を次いでファージ発現ベクター(クローニング詳細は図2Bを参照)中にライゲートし、そしてスクリーニングのために大腸菌中に形質転換する(A)。実験の詳細は実施例1を参照されたい。。
【図2B】ヒトscFvライブラリーを作製する全体スキームの概観図である。可変重鎖(VH)および軽鎖(VL)を逆転写し、増幅し、次いでオーバーラップPCRにより接合する。得られるscFv遺伝子を次いでファージ発現ベクター(クローニング詳細は図2Bを参照)中にライゲートし、そしてスクリーニングのために大腸菌中に形質転換する。実験の詳細は実施例1を参照されたい。M13ファージ発現ベクターのサブクローン領域(配列番号:1-2)を用いて、scFvライブラリーを作製しかつ大腸菌において発現させる(B)。
【図3A】第1回のスクリーニングからのフィルター上の複数のポジティブクローンシグナルの写真であり、ほぼ3820クローン/mm2(ほぼ3x107pfu/フィルター)を含有する。フィルターの一部分を拡大レンズを介して写真撮影し、ポジティブクローンの詳細を拡大した(矢印で示した)(A)。第2回のスクリーニングからのフィルター上のポジティブクローンの写真、ほぼ105pfu/フィルターを含有する(B)。第3回のスクリーニングからのフィルター上のポジティブクローンの写真(C)。
【図3B】第1回のスクリーニングからのフィルター上の複数のポジティブクローンシグナルの写真であり、ほぼ3820クローン/mm2(ほぼ3x107pfu/フィルター)を含有する。第2回のスクリーニングからのフィルター上のポジティブクローンの写真、ほぼ105pfu/フィルターを含有する(B)。
【図3C】第1回のスクリーニングからのフィルター上の複数のポジティブクローンシグナルの写真であり、ほぼ3820クローン/mm2(ほぼ3x107pfu/フィルター)を含有する。第3回のスクリーニングからのフィルター上のポジティブクローンの写真(C)。
【図4】超ハイスループットスクリーニングにより単一ライブラリーから単離した24個の独立クローンのELISA分析である。24クローンのうち、EphA4-His抗原と特異的に結合した22クローンを単離してスクリーニングに使用した。
【図5】抗イディオタイプscFvクローン単離のために調製したビオチン化MEDI-AAA(Fab)2のクーマシーブルー染色である。調製したビオチン化MEDI-AAA(Fab)2(レーンAおよびD)、無標識MEDI-AAA(Fab)2(レーンBおよびE)、およびMEDI-AAA IgG(レーンCおよびG)を、PAGEにより未変性(レーンA、BおよびC)または変性(レーンD、E、FおよびG)条件のもとで分離して泳動させた。レーンGはSeeBlue2マーカーである。
【図6A】抗イディオタイプscFvクローンはMEDI-AAA IgG CDRに対して特異的である。抗イディオタイプscFvを発現するクローンの相対的結合活性を、ELISAにより全長MEDI-AAA IgG、密接に関係する軽鎖フレームワークと同一のFc領域を有する無関係なIgG、およびBSAに対して試験した(複数の実験において)(パネルA)。抗イディオタイプscFvをさらに、市販ポリクローナル抗体調製物および似ていないフレームワークを有する他の無関係なIgG(Syn)に対して試験した(パネルB)。抗イディオタイプscFvクローンはMEDI-AAA IgGと強固な結合を示す一方、無関係なIgGまたはBSAとは少ししか結合しなかった。
【図6B】抗イディオタイプscFvクローンはMEDI-AAA IgG CDRに対して特異的である。抗イディオタイプscFvを発現するクローンの相対的結合活性を、ELISAにより全長MEDI-AAA IgG、密接に関係する軽鎖フレームワークと同一のFc領域を有する無関係なIgG、およびBSAに対して試験した(複数の実験において)(パネルA)。抗イディオタイプscFvをさらに、市販ポリクローナル抗体調製物および似ていないフレームワークを有する他の無関係なIgG(Syn)に対して試験した(パネルB)。抗イディオタイプscFvクローンはMEDI-AAA IgGと強固な結合を示す一方、無関係なIgGまたはBSAとは少ししか結合しなかった。
【図7】pUCKAのプラスミドマップである。いずれかのポリヌクレオチドを、3'-FLAGおよびHis6エピトープタグとともに、アンピシリンr(β-ラクタマーゼ遺伝子)選択マーカーとの融合タンパク質としてクローニングすることができる。また、プラスミド上にカナマイシンr選択マーカーも存在する。停止コドンまたはフレームシフト突然変異を含有するコード領域はβ-ラクタマーゼをコードするタンパク質を生産しないので、得られるクローンはアンピシリン耐性ではないであろう。VHおよびVLコード領域をpUCKAベクター中にクローニングして、scFvを3'-FLAGおよびHis6エピトープタグとともにアンピシリンr選択マーカーとの融合タンパク質として生産させる配置を示した。
【図8】pUCKAのクローニング領域の詳細である。アンピシリン選択に用いた発現プラスミドの3'-クローニング領域のヌクレオチド配列(配列番号:3-4)を示す。FLAGおよびHis6エピトープタグコード領域およびアミノ酸配列も示した(それぞれ赤色と青色の矢印)。アンピシリン耐性に関わるβ-ラクタマーゼ遺伝子のシグナル配列も示した。
【図9】pMD102のプラスミドマップである。選択したライブラリーからのVHおよびVLコード領域を、3'-FLAGおよびHis6エピトープタグのフレーム内でpMD102中にクローニングすることができる(なお、pMD102は実施例1および2に記載したハイスループットキャプチャーリフト方法によるスクリーニングのためのscFvファージ発現に必要な全遺伝子を含有する)。
【特許請求の範囲】
【請求項1】
結合分子のライブラリーについて超ハイスループットスクリーニングを行い、1つのリガンドに対して選択的アフィニティを有する結合分子を同定する方法であって、
a)上記結合分子のライブラリーを10〜15,000クローン/mm2の密度でまくステップ;
b)上記結合分子の集団を固体支持体に固定化するステップ;
c)上記固定化されたライブラリーを少なくとも1つのリガンドと接触させるステップ;および
d)上記リガンドの少なくとも1つと選択的に結合する少なくとも1つの結合分子を同定するステップ
を含んでなる上記方法。
【請求項2】
上記結合分子が可溶性である、請求項1に記載の方法。
【請求項3】
上記結合分子が選択的に固定化される、請求項1に記載の方法。
【請求項4】
上記結合分子のライブラリーが発現ライブラリーにおいて生産される、請求項1に記載の方法。
【請求項5】
上記発現ライブラリーが抗体ライブラリーである、請求項4に記載の方法。
【請求項6】
上記抗体ライブラリーが固定化ドメインと融合した抗体を発現する、請求項5に記載の方法。
【請求項7】
上記リガンドが可溶性である、請求項1に記載の方法。
【請求項8】
上記リガンドがポリペプチドである、請求項7に記載の方法。
【請求項9】
上記ポリペプチドが検出ドメインと融合されている、請求項8に記載の方法。
【請求項10】
結合分子のライブラリーを1000〜5,000クローン/mm2の密度でまく、請求項1に記載の方法。
【請求項11】
1以上の結合分子の少なくとも1つのリガンドとの相互作用の検出を増強する方法であって、
a)上記結合分子を固定化するステップ;
b)上記固定化された結合分子を少なくとも1つのリガンドと接触させるステップ;および
c)少なくとも1つのリガンドの上記固定された結合分子との相互作用を検出するステップ
を含んでなる上記方法。
【請求項12】
上記結合分子が可溶性である、請求項11に記載の方法。
【請求項13】
上記リガンドが検出ドメインと融合されている、請求項13に記載の方法。
【請求項14】
a) 発現ベクターにおいて、機能性分子を発現するクローンの選択に有用な選択マーカーをコードするポリヌクレオチドとライゲートした、分子をコードするポリヌクレオチドを含むクローンのライブラリーを作製するステップ、および
b) a)で作製した上記クローンのライブラリーを、機能性分子を発現するクローンを選択する条件のもとで増殖するステップ
を含んでなる、発現ライブラリーを作製する方法。
【請求項15】
ステップb)の上記選択したライブラリーからの機能性分子をコードするポリヌクレオチドを、特定の所望の機能性クローンの同定および/または単離に有用な代わりのベクター中にサブクローニングするステップをさらに含んでなる、請求項14に記載の方法。
【請求項16】
上記発現ベクターが大腸菌発現ベクターである、請求項14に記載の方法。
【請求項17】
上記大腸菌発現ベクターがpUCKAである、請求項16に記載の方法。
【請求項18】
上記クローンのライブラリーが、結合分子の集団をコードするポリヌクレオチドを含む、請求項14に記載の方法。
【請求項19】
上記結合分子の集団が抗体である、請求項18に記載の方法。
【請求項20】
上記代わりのベクターがファージ発現ベクターである、請求項15に記載の方法。
【請求項1】
結合分子のライブラリーについて超ハイスループットスクリーニングを行い、1つのリガンドに対して選択的アフィニティを有する結合分子を同定する方法であって、
a)上記結合分子のライブラリーを10〜15,000クローン/mm2の密度でまくステップ;
b)上記結合分子の集団を固体支持体に固定化するステップ;
c)上記固定化されたライブラリーを少なくとも1つのリガンドと接触させるステップ;および
d)上記リガンドの少なくとも1つと選択的に結合する少なくとも1つの結合分子を同定するステップ
を含んでなる上記方法。
【請求項2】
上記結合分子が可溶性である、請求項1に記載の方法。
【請求項3】
上記結合分子が選択的に固定化される、請求項1に記載の方法。
【請求項4】
上記結合分子のライブラリーが発現ライブラリーにおいて生産される、請求項1に記載の方法。
【請求項5】
上記発現ライブラリーが抗体ライブラリーである、請求項4に記載の方法。
【請求項6】
上記抗体ライブラリーが固定化ドメインと融合した抗体を発現する、請求項5に記載の方法。
【請求項7】
上記リガンドが可溶性である、請求項1に記載の方法。
【請求項8】
上記リガンドがポリペプチドである、請求項7に記載の方法。
【請求項9】
上記ポリペプチドが検出ドメインと融合されている、請求項8に記載の方法。
【請求項10】
結合分子のライブラリーを1000〜5,000クローン/mm2の密度でまく、請求項1に記載の方法。
【請求項11】
1以上の結合分子の少なくとも1つのリガンドとの相互作用の検出を増強する方法であって、
a)上記結合分子を固定化するステップ;
b)上記固定化された結合分子を少なくとも1つのリガンドと接触させるステップ;および
c)少なくとも1つのリガンドの上記固定された結合分子との相互作用を検出するステップ
を含んでなる上記方法。
【請求項12】
上記結合分子が可溶性である、請求項11に記載の方法。
【請求項13】
上記リガンドが検出ドメインと融合されている、請求項13に記載の方法。
【請求項14】
a) 発現ベクターにおいて、機能性分子を発現するクローンの選択に有用な選択マーカーをコードするポリヌクレオチドとライゲートした、分子をコードするポリヌクレオチドを含むクローンのライブラリーを作製するステップ、および
b) a)で作製した上記クローンのライブラリーを、機能性分子を発現するクローンを選択する条件のもとで増殖するステップ
を含んでなる、発現ライブラリーを作製する方法。
【請求項15】
ステップb)の上記選択したライブラリーからの機能性分子をコードするポリヌクレオチドを、特定の所望の機能性クローンの同定および/または単離に有用な代わりのベクター中にサブクローニングするステップをさらに含んでなる、請求項14に記載の方法。
【請求項16】
上記発現ベクターが大腸菌発現ベクターである、請求項14に記載の方法。
【請求項17】
上記大腸菌発現ベクターがpUCKAである、請求項16に記載の方法。
【請求項18】
上記クローンのライブラリーが、結合分子の集団をコードするポリヌクレオチドを含む、請求項14に記載の方法。
【請求項19】
上記結合分子の集団が抗体である、請求項18に記載の方法。
【請求項20】
上記代わりのベクターがファージ発現ベクターである、請求項15に記載の方法。
【図1】

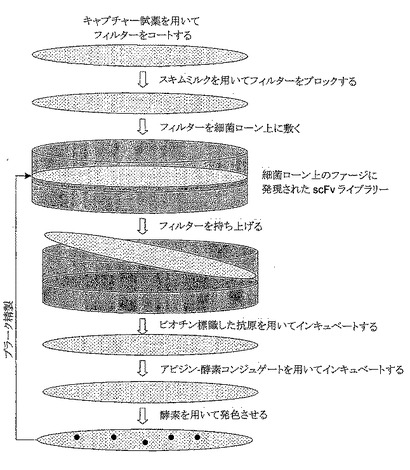
【図2A】
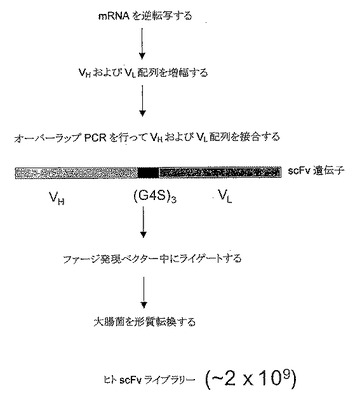
【図2B】

【図3A】

【図3B】

【図3C】

【図4】

【図5】
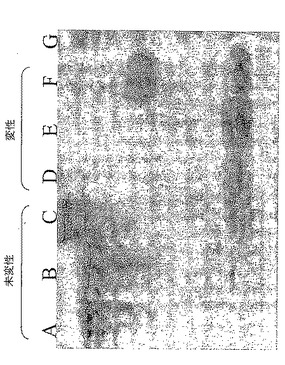
【図6A】

【図6B】

【図7】
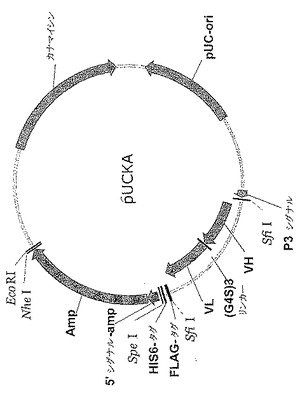
【図8】
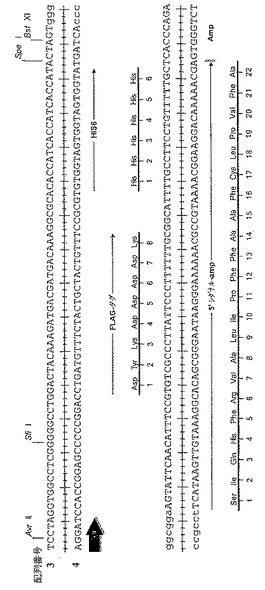
【図9】
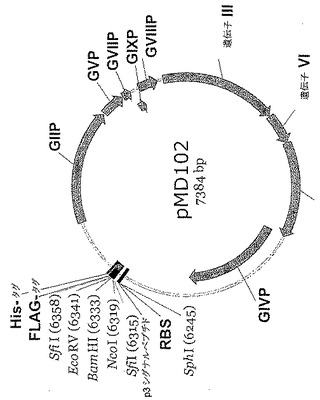
【図2A】
【図2B】
【図3A】
【図3B】
【図3C】
【図4】
【図5】
【図6A】
【図6B】
【図7】
【図8】
【図9】
【公表番号】特表2008−519265(P2008−519265A)
【公表日】平成20年6月5日(2008.6.5)
【国際特許分類】
【出願番号】特願2007−539292(P2007−539292)
【出願日】平成17年10月31日(2005.10.31)
【国際出願番号】PCT/US2005/039436
【国際公開番号】WO2006/050346
【国際公開日】平成18年5月11日(2006.5.11)
【出願人】(504333972)メディミューン,インコーポレーテッド (108)
【Fターム(参考)】
【公表日】平成20年6月5日(2008.6.5)
【国際特許分類】
【出願日】平成17年10月31日(2005.10.31)
【国際出願番号】PCT/US2005/039436
【国際公開番号】WO2006/050346
【国際公開日】平成18年5月11日(2006.5.11)
【出願人】(504333972)メディミューン,インコーポレーテッド (108)
【Fターム(参考)】
[ Back to top ]