
超変異性生物の作製法
【課題】細胞を超変異性にするための方法、および遺伝学的、並びに表現型的に改変された細胞系を提供する。
【解決手段】ヒトミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子の、超変異性の細胞及び生物を作製するための使用。前記これらの遺伝子を細胞及びトランスジェニック動物へ導入する、新規かつ有用な特性を有する新たな細胞系及び動物変種が、自然な変異の速度に頼るよりも効率的に調製されうる前記方法。増強された変異の速度をさらに強化するための、変異原の使用。
【解決手段】ヒトミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子の、超変異性の細胞及び生物を作製するための使用。前記これらの遺伝子を細胞及びトランスジェニック動物へ導入する、新規かつ有用な特性を有する新たな細胞系及び動物変種が、自然な変異の速度に頼るよりも効率的に調製されうる前記方法。増強された変異の速度をさらに強化するための、変異原の使用。
【発明の詳細な説明】
【技術分野】
【0001】
本出願は、2000年5月17日出願の60/204,769に基づく優先権を主張するものであり、その開示は本明細書に明示的に組み込まれる。
【0002】
発明の技術分野
本発明は、ミスマッチ修復遺伝子の領域に関する。特に、本発明は、変異誘発の分野に関する。
【背景技術】
【0003】
発明の背景
過去4年間に、リンチ(Lynch)症候群IIとしても既知の遺伝性非ポリポーシス性大腸癌症候群(Hereditary Nonpolyposis Colorectal Cancer Syndrome/HNPCC)の遺伝学的原因が、その疾患に罹患した血族の大部分について確認された(13)。HNPCCの分子的基礎は、欠陥ミスマッチ修復(MMR)に起因する遺伝学的不安定性を含む。mutSホモログGTBP、hMSH2、及びhMSH3、並びにmutLホモログhMLH1、hPMS1、及びhPMS2を含む、MMR過程に参与していると考えられるタンパク質をコードする多くの遺伝子が、齧歯類及びヒトにおいて同定されている(2,7,11,17,20,21,22,24)。これらの遺伝子のうちの4つ(hMSH2、hMLH1、hPMS1、及びhPMS2)の生殖系列変異が、HNPCC家系において同定された(2,11,13,17,24)。MMR欠損により生じる変異誘発欠陥(mutator defect)は、任意のDNA配列に影響を与えうるが、MMR異常に対する特に高い感受性を有するのはミクロサテライト配列である(14)。従って、ミクロサテライトの不安定性は、欠陥MMRの有用な指標である。ミクロサテライトの不安定性は、HNPCC患者に生じる事実上全ての腫瘍において発生するのに加え、別個の分子的特性及び表現型的特性を有する散発性腫瘍の極一部にも見出される(27)。
【0004】
HNPCCは、常染色体優性遺伝するため、罹患家族メンバーの正常細胞は、(罹患している親から遺伝した)関連MMR遺伝子の変異対立遺伝子1個と、(罹患していない親から遺伝した)野生型対立遺伝子1個とを含有している。しかしながら、腫瘍発生の初期の段階において、野生型対立遺伝子が体細胞性変異によって不活化されて、機能性MMR遺伝子を有しない細胞が残され、MMR活性の著しい欠陥がもたらされる。腫瘍細胞における欠陥MMRを作製するためには、生殖系列変異に加え体細胞性変異が必要とされるため、他の遺伝性癌を惹起する腫瘍抑制遺伝子の二対立遺伝子不活化と類似した、このメカニズムは、一般的に、2ヒットを含むものと呼ばれる(11,13,25)。この2ヒットメカニズムと一致して、HNPCC患者の非新生物細胞は、一般に、野生型対立遺伝子の存在のため、ほぼ正常なレベルのMMR活性を保持している。
【0005】
欠陥MMRを有する広範囲の生物が、ゲノム全体に拡がった遺伝子変異を有することが見出されている。全ての場合に、これらの生物は、特定のMMR遺伝子の両コピーに生殖系列変異を有している。最近、ニコライデス(Nicolaides)らの研究により、MMR遺伝子のドミナントネガティブ対立遺伝子を導入することにより、高等生物由来の細胞においてMMRの減少が達成されうることが示された。これらのデータは、そのような手法の使用によって、新たな出力形質を生成させるため遺伝学的に改変された生物が作製されうることを示唆している。当分野においては、遺伝学的な多様性を作製するためのさらなる方法が必要とされている。
【発明の概要】
【0006】
発明の概要
本発明の目的は、細胞を超変異性にするための方法を提供することである。
【0007】
本発明の別の目的は、遺伝学的に改変された細胞系を提供することである。
【0008】
本発明の別の目的は、表現型的に改変された細胞系を提供することである。
【0009】
本発明のさらに別の目的は、細胞において遺伝学的超変異の速度を増強するための方法を提供することである。
【0010】
本発明のさらなる目的は、細胞において関心対象の(1個以上の)遺伝子を変異させる方法を提供することである。
【0011】
本発明のさらなる目的は、遺伝学的に改変された細菌プリンホスホリラーゼのための材料の組成物を、特許請求の範囲に記載することである。
【0012】
本発明のさらなる目的は、真核細胞のミスマッチ修復欠損をモニタリングするための診断道具としての、遺伝学的に改変された細菌プリンホスホリラーゼのための材料の組成物を、特許請求の範囲に記載することである。
【0013】
本発明のさらなる目的は、真核細胞における改変されたミスマッチ修復を測定するための、ポリモノヌクレオチド鎖(tract)を組み込むことにより遺伝学的に改変された遺伝子を作製するための材料の組成物を、特許請求の範囲に記載することである。
【0014】
本発明のさらに別の目的は、新たな表現型を有する細胞を作出する方法を提供することである。
【0015】
本発明のさらに別の目的は、新たな表現型及び安定なゲノムを有する細胞を作出する方法を提供することである。
【0016】
本発明のさらに別の目的は、細胞又は生物のゲノムの遺伝学的安定性を制御する方法を提供することである。
【0017】
本発明のさらなる目的は、ドミナントネガティブミスマッチ修復遺伝子変異体を含有する誘導可能ベクターを使用して超変異性細胞系を作製することである。
【0018】
本発明のさらなる目的は、誘導された遺伝子発現条件下で、ドミナントネガティブミスマッチ修復遺伝子変異体を有する誘導可能ベクターを含有している超変異性細胞系をスクリーニングすることである。
【0019】
本発明のさらなる目的は、誘導された遺伝子発現条件下で、ドミナントネガティブミスマッチ修復遺伝子変異体を有する誘導可能ベクターを含有している超変異性細胞系を、改変された遺伝子構造及び/又は新たな表現型に関してスクリーニングすることである。
【0020】
本発明のさらなる目的は、ゲノムの安定性を回復させるため、構造的に改変された標的遺伝子及び/又は新たな表現型を含有している細胞におけるドミナントネガティブMMR遺伝子の発現をオフ状態にすることである。
【0021】
本発明のさらなる目的は、化学変異原又は電離放射線の存在下で、誘導条件下で、ドミナントネガティブミスマッチ修復遺伝子変異体を含む誘導可能ベクターを含有している超変異性細胞系を、構造的に改変された標的遺伝子及び/又は新たな表現型に関してスクリーニングすることである。次いで、改変された遺伝子構造及び/又は新たな表現型を含有している細胞を誘導分子から離し、遺伝学的安定性を回復させる。
【0022】
本発明のこれら及びその他の目的は、下記の態様のうちの一つ以上によって提供される。本発明の一つの態様において、超変異性細胞を作成するための方法が提供される。ミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子をコードするポリヌクレオチドが細胞へ導入される。細胞は、遺伝子の導入の結果として超変異性となる。
【0023】
本発明の別の態様において、単離された超変異性細胞が提供されるであろう。細胞は、ミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子を含む。細胞をDNAアルキル化剤に曝す。細胞は増強された超変異速度を示す。
【0024】
本発明の別の態様において、関心対象の遺伝子に変異を導入するための方法が提供される。ミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子をコードするポリヌクレオチドが、細胞へ導入される。細胞は、遺伝子の導入の結果として超変異性となる。細胞は、関心対象の遺伝子をさらに含む。細胞を増殖させる。関心対象の遺伝子が変異を保持しているか否かを決定するため、細胞が試験される。
【0025】
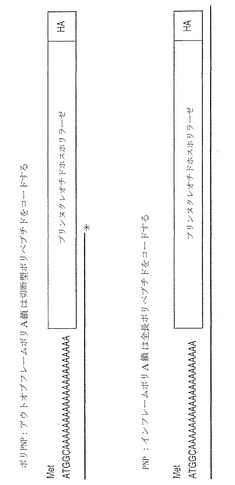
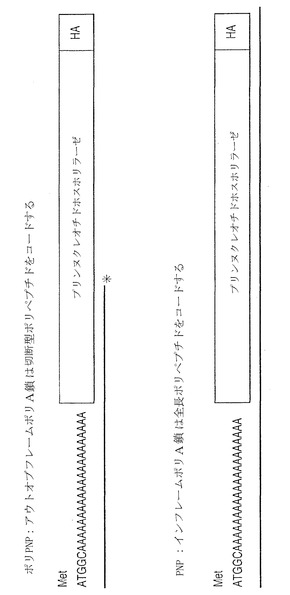
本発明の別の態様において、真核細胞のミスマッチ修復活性を測定するため、ポリモノヌクレオチド鎖を遺伝子に挿入するための方法が提供される。ポリモノヌクレオチド鎖が、遺伝子又はcDNAのコーディング領域にアウトオブフレーム(out-of-frame)で挿入される。その遺伝子が、細胞へ導入される。ポリモノヌクレオチド鎖が、ミスマッチ修復欠損により改変される。インフレーム(in-frame)の改変された遺伝子が作製される。
【0026】
本発明の別の態様において、細胞の新たな表現型を生成させるための方法が提供される。ミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子をコードするポリヌクレオチドが、細胞へ導入される。細胞は、遺伝子の導入の結果として超変異性となる。細胞を増殖させる。新たな表現型の発現に関して細胞が試験される。本発明の別の態様は、改変された標的遺伝子及び/又は新たな表現型のための、化学変異原又は電離放射線の存在下での、誘導条件下での、ドミナントネガティブミスマッチ修復遺伝子変異体からなる誘導可能ベクターを含有している細胞の使用である。次いで、改変された遺伝子構造及び/又は新たな表現型を含有している細胞を誘導分子から離し、遺伝学的安定性を回復させる。ここで、細胞は、組換え製造及び/又は遺伝子発見(これらに限定はされない)のような商業的な特性のために使用される。
【0027】
本発明の別の態様は、改変された標的遺伝子及び/又は新たな表現型のための、化学変異原又は電離放射線の存在下での、誘導条件下での、関心対象の遺伝子を含有しているMMR欠陥細胞の使用である。次いで、改変された遺伝子構造及び/又は新たな表現型を含有している細胞は、野生型MMR補足遺伝子で安定的に形質導入し、遺伝学的安定性を回復させる。ここで、細胞は、組換え製造又は遺伝子発見のために使用される。
【0028】
本発明の別の態様において、欠陥ミスマッチ修復遺伝子を有する細胞において、遺伝学的安定性を回復させるための方法が提供される。ミスマッチ修復過程の活性を回復させ、そのゲノムは安定となる。
【0029】
本発明の別の態様において、欠陥ミスマッチ修復活性及び新たに選択された表現型を有する細胞において、遺伝学的安定性を回復させるための方法が提供される。MMR欠損は、ゲノム変異を介した内因性MMR遺伝子の不活化により、又はドミナントネガティブMMR遺伝子対立遺伝子を生成させる真核発現ベクターの導入により、起こりうる。内因性MMR遺伝子の欠陥により内因性MMR遺伝子を欠いている細胞の場合には、細胞が、新たな表現型、又は改変された遺伝子、RNA、もしくはポリペプチドに関して選択される。細胞は、宿主細胞のゲノム欠陥を補足する正常な機能性MMR遺伝子の導入によって、遺伝学的に安定なものとなる。この補足群は、ミスマッチ修復欠損に参与していることが知られている任意の遺伝子の使用を含みうる。ドミナントネガティブミスマッチ修復遺伝子の発現が、DNA超変異性を誘導するために使用される場合には、ドミナントネガティブMMR遺伝子発現は、誘導分子の除去により、又は当業者によって使用されている標準的な遺伝子ノックアウト技術を使用して、ドミナントネガティブ遺伝子対立遺伝子の発現をノックアウトすることにより抑制されるであろう(Waldman,T.ら、Cancer Res 55:5187-5190,1995)。いずれの場合にも、細胞の遺伝学的安定性が回復し、新たな表現型は安定なものとなる。
【0030】
本発明のこれら及びその他の態様は、生物、細胞、及び動物において増強された変異性を作製しうる方法を当分野に提供し、潜在的に有用なゲノム改変を保持している安定な生物、細胞、及び動物を提供する。
【0031】
ドミナントネガティブMMR遺伝子対立遺伝子の使用は、制御された様式で、宿主生物のゲノム全体に全体的な変異を作製する上で重要である。ドミナントネガティブ対立遺伝子の使用は、広範囲の宿主(細菌、酵母、哺乳動物、植物)において全体的な変異誘発を誘導しうることが以前に証明されているが、ゲノムワイドな変異を作製するため、ドミナントネガティブMMR遺伝子変異体をオン状態にした後、新たな生物学的出力形質(例えば、化学変異原に対する耐性)を選択し、遺伝学的安定性を回復させるためドミナントネガティブMMR遺伝子対立遺伝子をオフ状態にするための、誘導可能ベクターの使用は、本発明の新たな局面である。ここで、この方法は、新たな表現型に関与している遺伝子の遺伝学的変異に関してスクリーニングされうる、遺伝学的に多様な原核細胞、真核細胞、及び哺乳動物細胞の作製に適している。さらに、本出願は、誘導可能発現因子の調節下にあるドミナントネガティブMMR遺伝子対立遺伝子の、MMR正常細胞への導入の使用を開示する。次いで、安定的、又は一過性的形質導入された細胞が、ドミナントネガティブMMR遺伝子の発現をもたらす誘導分子へと曝される。ドミナントネガティブ産物の発現は、内因性MMR機構を妨害し、それにより遺伝学的に多様な亜系をもたらす遺伝学的不安定性を引き起こす。次いで、これらの細胞は、特別な選択アッセイ下に置かれ、新たな表現型及び/又は改変された遺伝子構造に関してスクリーニングされる。改変された標的遺伝子及び/又は新たな表現型を含有している亜系の樹立の後、次いで、誘導分子の除去により、細胞が遺伝学的に安定なものとなり、ここで、改変された遺伝子を含有している、かつ/又は新たな表現型を示す安定な細胞系が生成する。この細胞系は、遺伝子発見、薬物標的発見、組換え遺伝子変異誘発、及び/又は組換えタンパク質産生のために使用されうる。
【0032】
MMR欠損生物が、ゲノムワイドな又は遺伝子座特異的な変異誘発の増強されたレベルをもたらすアルキル化剤又は電離放射線のようなDNA損傷剤に対する比較的高い耐性を有することはよく確立されている。本明細書において、本発明者らは、誘導可能発現因子の調節下にあるドミナントネガティブMMRを発現している細胞系の、増強されたゲノムワイドな変異誘発をもたらしうるDNA損傷剤への曝露の使用を開示する。次いで、細胞系は、標的遺伝子内の変異に関してスクリーニングされるか、又は新規表現型に関してスクリーニングされる。次いで、遺伝学的安定性を回復させるため、ドミナントネガティブMMR遺伝子対立遺伝子を「オフ状態」にするため、改変された遺伝子又は表現型を有する亜系は、誘導剤から除去される。この細胞系は、遺伝子発見、薬物標的発見、組換え遺伝子変異誘発、及び/又は組換えタンパク質産生のために使用されうる。
【0033】
最後に、天然にMMRの欠陥を有する哺乳動物細胞系の使用が、関心対象の遺伝子を含有しているプラスミドを導入するために使用されうる。遺伝子を導入し、一過的又は安定的に発現させる。ここで、細胞を増殖させ、構造及び/又は機能の改変を有するものを同定するため、導入された遺伝子の構造及び/又は機能をスクリーニングする。変異速度を増強するため、宿主におけるゲノムワイドな変異の速度を増強するための、アルキル化化学変異原又は電離放射線(これらに限定はされない)のようなDNA損傷剤へとさらに細胞を曝すこともできる。関心対象の遺伝子に変異を含有している(1個以上の)細胞が作製された後、細胞は内因性MMR欠陥を補足する遺伝子で安定的に形質導入される。ここで、細胞系は遺伝学的に安定なものとなり、細胞系は、遺伝子発見、組換え遺伝子変異誘発、及び/又は組換えタンパク質産生のための改変された遺伝子産物の作製に適したものとなる。
【図面の簡単な説明】
【0034】
【図1】PMS2発現ベクター(図1A)及びpCARレポーター(図1B)の図。
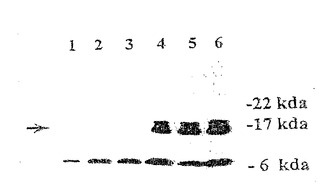
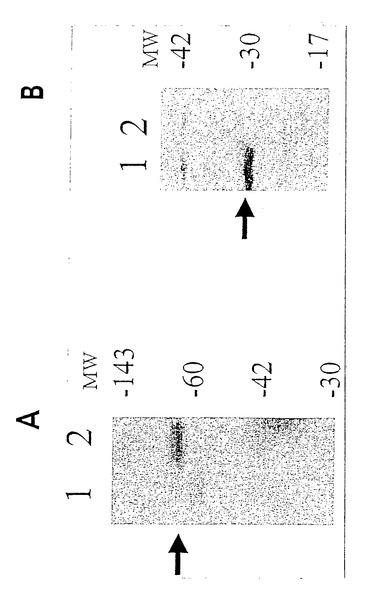
【図2A】薬物選択の17日後の、pCARレポーター及びPMS2発現ベクターで同時トランスフェクトされたSH細胞。(図2A)トランスフェクトされていないSH細胞(レーン1)、又はPMS2NOT(レーン2)もしくはPMS2WT(レーン3)でトランスフェクトされたSH細胞に由来する溶解物のウェスタンブロット。矢印は、hPMS2と予想される110kDタンパク質を示す。A及びBは、いずれも、hPMS2のN末端に対して作製された抗体を用いて探索された。Aにおける上方のポリペプチド及びBにおける下方のポリペプチドは、交差反応性ハムスタータンパク質を表す。
【図2B】薬物選択の17日後の、pCARレポーター及びPMS2発現ベクターで同時トランスフェクトされたSH細胞。(図2B)トランスフェクトされていないSH細胞(レーン1)、又はPMS2NOT(レーン2)もしくはPMS2134(レーン3)でトランスフェクトされたSH細胞に由来する溶解物のウェスタンブロット。矢印は、hPMS-134と予想される14kDタンパク質を示す。A及びBは、いずれも、hPMS2のN末端に対して作製された抗体を用いて探索された。Aにおける上方のポリペプチド及びBにおける下方のポリペプチドは、交差反応性ハムスタータンパク質を表す。
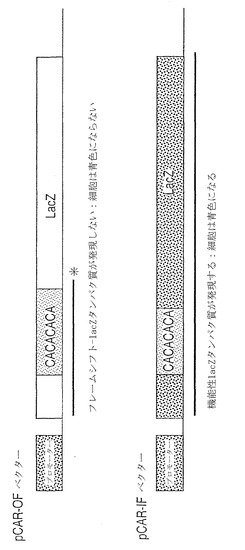
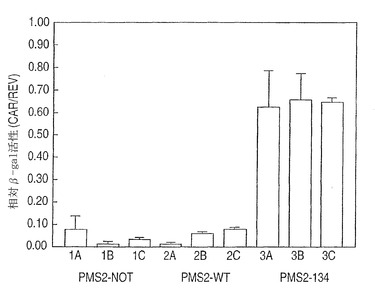
【図2C】薬物選択の17日後の、pCARレポーター及びPMS2発現ベクターで同時トランスフェクトされたSH細胞。(図2C)PMS2NOT(レーン1)、PMS2WT(レーン2)、又はPMS2134(レーン3)とレポータープラスミドとで共トランスフェクトされたSH細胞に由来する溶解物におけるβ-ガラクトシダーゼ活性。相対β-ガラクトシダーゼ活性とは、pCARIFでトランスフェクトされた細胞における活性と比較された、pCAROFでトランスフェクトされた細胞におけるβ-ガラクトシダーゼ活性の比として定義される。この規準化により、トランスフェクション効率がコントロールされ、様々なPMS2エフェクター遺伝子を発現している細胞におけるβ-ガラクトシダーゼ活性がコントロールされた。
【図3】PMS2WT発現ベクター(図3A)又はPMS2134発現ベクター(図3B)で安定的に形質導入され、次いでpCAROFで再トランスフェクトされたSH細胞のプールされたクローンのインサイチューβ-ガラクトシダーゼ活性。薬物選択の17日後に、コロニーをプールし、培養し、β-ガラクトシダーゼ活性に関して染色した。pCAROFに由来するβ-ガラクトシダーゼを発現しているPMS2134形質導入SH細胞のプールされた培養物が、図3Bに可視である。図示された各視野は、3連の実験において見出されたものの代表である。
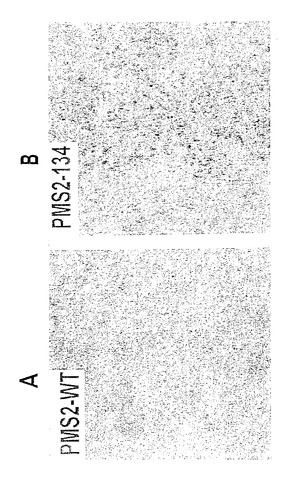
【図4】ドミナントネガティブミスマッチ修復遺伝子対立遺伝子を含有している誘導可能哺乳動物発現ベクターの作製。C末端にV5エピトープを含む、又は含まないPMS134 cDNAを、エクジソンステロイドにより制御されたpIND哺乳動物発現ベクターへとクローニングした。PMS134 cDNAは、熱ショック最小プロモーターに対しセンス方向でベクターの唯一のBamHI部位へクローニングされた。得られたベクターは、それぞれ、pINDPMS134V5又はpINDPMS134と呼ばれる。pINDベクターは、選択可能マーカーとしてネオマイシン耐性遺伝子を含有している。
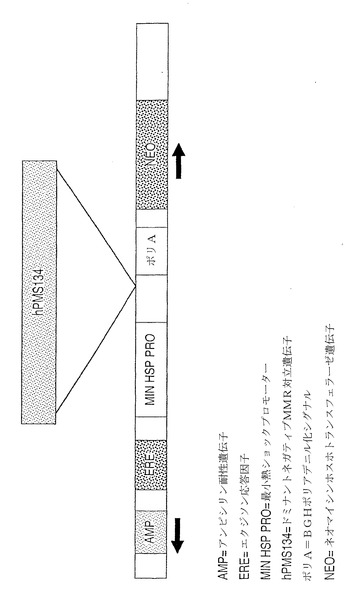
【図5】PMS134の誘導による改変された遺伝子配列の作製。オープンリーディングフレームを崩壊させ、フレームシフトを生じさせるポリCAリピートをβ-gal遺伝子のN末端に含むβ-ガラクトシダーゼレポータープラスミドを含有しているpCAR-OFプラスミドで、空のpINDベクター又はpINDPMS134を含有している細胞をトランスフェクトした。プラスミドは、安定な系を選択するためのハイグロマイシン耐性遺伝子も含有している。G418/ハイグロマイシン耐性であった細胞を、1μMエクジソンの存在下又は非存在下で10日間増幅・増殖させた。14日後、機能性β-galを産生した増殖巣に関して、細胞をインサイチューで染色した。示されているように、ステロイド誘導剤の存在下で増殖させた細胞においては相当数(1視野当たり25個)のβ-gal陽性増殖巣が観察されたが、誘導剤分子の非存在下で増殖させた培養物においてはほとんど観察されなかった。
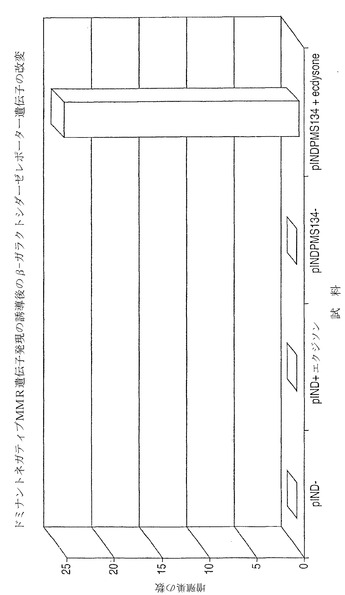
【図6】選択後の遺伝学的に安定な細胞の再樹立。化学誘導剤の除去(及びその後のドミナントネガティブMMR対立遺伝子の抑止)の後、クローンが遺伝学的に安定であるか否かを決定するため、pINDPMS134/pCAR-OFクローンを単離し、機能性β-gal活性に関して試験した。β-gal発現を有するクローンを、クローンを含むウェルが1ディッシュ当たり45個となるよう限界希釈して96穴プレートに播いた。クローンを、再度、エクジソンの存在下又は非存在下で14代(1代/日)にわたり増殖させ、β-gal活性に関してインサイチューで染色した。示されているように、ステロイド誘導剤の非存在下で増殖させた細胞においては相当数のβ-gal陽性クローンが観察されたが(45ウェル中42ウェルがβ-gal陽性であった)、定常的な誘導剤曝露下ではより多数のクローンがβ-gal活性を失った(45ウェル中18ウェルがβ-gal陽性であった)。これらのデータは、MMRの遮断によって、インビボで遺伝学的に改変されたクローンにおいて、遺伝学的安定性を回復させることが可能であることを例証している。
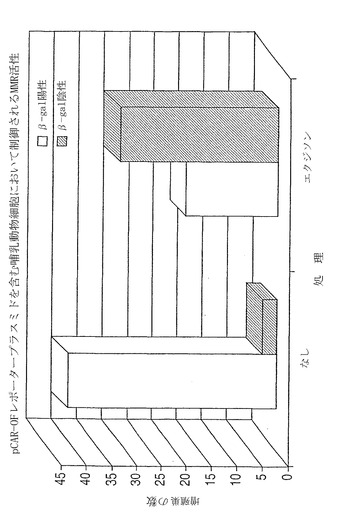
【図7】N末端に挿入されたアウトオブフレームのポリA鎖を有する遺伝学的に改変されたプリンホスホリラーゼ(PNP)遺伝子(ポリPNPと呼ばれる)の図。この遺伝子は、非機能性PNP遺伝子をコードする。ポリA鎖が、欠陥MMRによってランダムに改変された場合、鎖はシフトし、機能性PNP遺伝子の生成を可能にする。PNPは非毒性プロドラッグ9-(β-D-2-デオキシエリスロペントフラノシル)-6-メチル-プリン(MPDと呼ばれる)を毒性6-メチルプリンアナログ(MPと呼ばれる)へと変換しうる。構築物は、ウェスタンブロット分析のため、C末端にヘマグルチニン(HA)タグを有している。
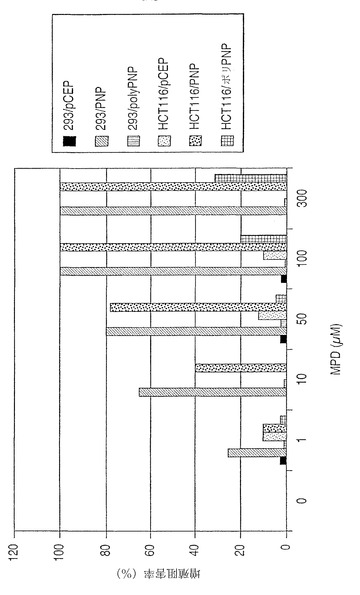
【図8A】MPDへ曝された場合、又は曝されない場合の、ポリPNPを発現しているMMR欠陥細胞又はMMR正常細胞の毒性アッセイ。(図8A)グラフは、MMR正常細胞と対照的に、ポリPNPを発現しているMMR欠陥細胞においては、MPDプロドラッグの存在下で細胞が死滅することを示している。
【図8B】MPDへ曝された場合、又は曝されない場合の、ポリPNPを発現しているMMR欠陥細胞又はMMR正常細胞の毒性アッセイ。(図8B)MMR正常細胞と対照的な、MMR欠陥細胞におけるポリPNP-HAタグ化ポリペプチドの産生を示すウェスタンブロット。
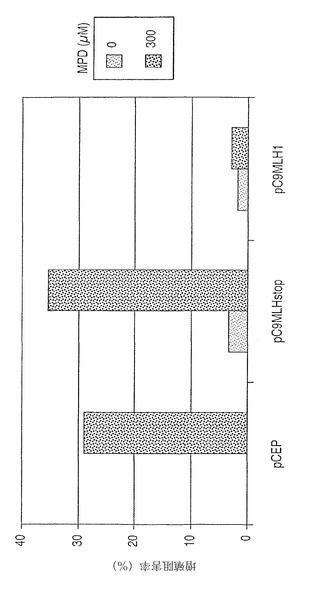
【図9】MPDへ曝された場合、又は曝されない場合の、ポリPNPを発現しているMMR欠陥細胞又はMMR正常細胞の毒性アッセイ法。グラフは、MMR欠陥HCT116細胞(MLH1が遺伝学的に欠損している)において、切断型(非機能性)MLH1 cDNA(pC9MLHstop)でトランスフェクトされたHCT116細胞において見られるような活性型へとポリPNP遺伝子が変換されないという事実により示されるように、機能性MLH1遺伝子の導入によって、細胞の遺伝学的安定性が回復することを示している。これらのデータは、MMR欠損が機能性MMR遺伝子(HCT116/pC9MLH1)によって補足され、従って、改変された遺伝子又は遺伝子座のゲノム完全性が維持されうることを例証している。
【図10】MLH1発現構築物及びポリPNP遺伝子でトランスフェクトされた細胞のウェスタンブロット。(図10A)MLHstop発現ベクター(レーン1)又はMLH1ベクター(レーン2)でトランスフェクトされた細胞に由来する細胞溶解物を溶解させ、HCT116細胞におけるMLH1タンパク質発現に関して探索した。図10Aに示されるように、MLH1全長発現構築物でトランスフェクトされた細胞は、予想分子量のポリペプチド(矢印)を産生した。(図10B)MLHstop発現ベクター(レーン1)又はMLH1ベクター(レーン2)とポリPNP遺伝子とでトランスフェクトされたHCT116細胞に由来する細胞溶解物を溶解させ、PNPタンパク質のC末端のHAタグを検出することができる抗HAモノクローナル抗体を使用してポリPNPに関して探索した。図10Bに示されるように、細胞のゲノム安定性を回復させ、従ってポリPNP遺伝子のゲノム構造を維持した、機能性MLH1 cDNAでトランスフェクトされた細胞とは対照的に、MLHstop発現構築物でトランスフェクトされた細胞は、予想分子量のポリペプチド(矢印)を産生した。
【発明を実施するための形態】
【0035】
[発明の詳細な説明]
本発明者らは、ミスマッチ修復を欠損している細胞を利用することにより、超変異性の細胞を開発し、改変された遺伝子、RNA、ポリペプチドおよび細胞、または新しい表現型を有する生物体全体を創造するための方法を発見した。そのような遺伝子のドミナントネガティブ対立遺伝子は、細胞またはトランスジェニック動物へ導入される場合、DNA修復の効率性が低下することにより自然突然変異の割合が増加し、それにより、細胞または動物を超変異性にする。超変異性細胞または動物は、その後、遺伝子に新しい突然変異を発生させて、宿主細胞または生物体の新しい出力形質を生み出すために利用されうる。本発明者らは、DNAに損傷を引き起こす化学薬品の使用により、ドミナントネガティブミスマッチ修復遺伝子の対立遺伝子を発現している細胞において、超変異性の割合を増幅させうることを示す。本発明者らはまた、改変された遺伝子の選択およびMMR(ミスマッチ修復)を回復することによる宿主細胞または生物体の遺伝子的安定性の回復が、改変された遺伝子、RNA、またはポリペプチドからなる安定的生物学的生産物へと導くことができることも示す。
【0036】
細菌から哺乳動物細胞の範囲に及ぶ細胞におけるタンパク質複合体は、ミスマッチ修復、またはミスマッチプルーフリーディングと呼ばれる過程を実行する。ミスマッチ修復遺伝子とは、そのようなミスマッチ修復複合体のタンパク質の一つをコードする遺伝子である。何か特定の作用機構の理論に結びつける必要はないが、ミスマッチ修復複合体はヌクレオチド塩基の非相補的対合から生じるDNAヘリックスの歪みを検出するものと考えられている。新しい方のDNA鎖における非相補的塩基が切除され、その切除された塩基が古い方のDNA鎖に相補的である適切な塩基に置換される。このようにして、細胞は、DNA複製における誤りの結果として生じる多くの突然変異を排除する。
【0037】
ドミナントネガティブ対立遺伝子は、同じ細胞に野生型対立遺伝子が存在している場合でさえも、ミスマッチ修復の欠損した表現型を引き起こす。ミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子の例には、コドン134において切断突然変異を起こすヒト遺伝子hPMS2-134がある。その突然変異は、この遺伝子の産生物を134番目のアミノ酸の位置で異常に終結させ、その結果として、N末端の133個のアミノ酸を含む短縮されたポリペプチドを生じる。そのような突然変異は、DNA複製後細胞に蓄積される突然変異の割合の増加を引き起こす。ミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子の発現は、野生型対立遺伝子の存在下でさえも、ミスマッチ修復活性の障害を生じる。そのような効果を生じる任意の対立遺伝子を本発明において使用することができる。
【0038】
ミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子を、ヒト、動物、酵母、細菌、または他の生物体の細胞から得ることができる。欠損したミスマッチ修復活性について細胞をスクリーニングすることにより、そのような対立遺伝子を同定することができる。癌を有する動物またはヒト由来の細胞を、欠損したミスマッチ修復についてスクリーニングすることができる。大腸癌患者由来の細胞は特に有用でありうる。ミスマッチ修復タンパク質をコードする任意の細胞由来のゲノムDNA、cDNA、またはmRNAを野生型配列からの変異として分析することができる。ミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子をまた、例えば、hPMS2-134対立遺伝子または他のミスマッチ修復遺伝子の変異体を生成することにより、人工的に作成することができる。部位特異的突然変異誘発の様々な技術を使用することができる。超変異性細胞または動物を発生させることにおける使用について、自然的であろうと人工的であろうと、そのような対立遺伝子の適合性は、それがドミナントネガティブ対立遺伝子であるかどうかを決定するために、1つまたは複数の野生型対立遺伝子の存在下においてその対立遺伝子により引き起こされるミスマッチ修復活性を試験することにより、評価することができる。
【0039】
ミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子を導入された細胞、生物体、または動物は、超変異性になるものと思われる。これは、そのような細胞または動物の自然突然変異の割合がそのような対立遺伝子を含有しない細胞または動物と比較して上昇していることを意味する。自然突然変異の割合の上昇程度は、正常の細胞または動物の自然突然変異の割合の少なくとも2倍、5倍、10倍、20倍、50倍、100倍、200倍、500倍、または1000倍となりうる。
【0040】
本発明の1つの局面に従って、ミスマッチ修復タンパク質のドミナントネガティブ型をコードするポリヌクレオチドを任意の真核細胞またはトランスジェニック動物へ導入する。遺伝子は、ミスマッチ修復複合体、例えば、PMS2、PMS1、MLH1、GTBP、MSH3またはMSH2の部分であるタンパク質をコードする任意のドミナントネガティブ対立遺伝子であってもよい。ドミナントネガティブ対立遺伝子は、天然に存在しているか、または実験室で作成されうる。ポリヌクレオチドはゲノムDNA、cDNA、RNA、または化学的合成ポリヌクレオチドの形であってもよい。ポリヌクレオチドは、常時活性型プロモーターセグメント(限定されるものではないが、CMV、SV40、EF-1□またはLTR配列のような)を含む発現ベクターへ、またはドミナントネガティブミスマッチ修復遺伝子の発現を制御することができる、テトラサイクリン、またはエクジソン/グルココルチコイド誘導性ベクターのような誘導性プロモーター配列へクローニングすることができる。ポリヌクレオチドをトランスフェクションにより細胞へ導入することができる。
【0041】
トランスフェクションとは、ポリヌクレオチドを細胞へ導入する任意の過程である。トランスフェクションの過程を、生きている動物に、例えば、遺伝子治療用のベクターを使って行うことができる、またはインビトロで、例えば、培養における1つまたは複数の単離された細胞の懸濁液を使って行うことができる。細胞は、例えば、ヒトまたは他の霊長類、哺乳動物または他の脊椎動物、無脊椎動物、および原生動物または酵母のような単細胞型生物体から単離された細胞を含む、真核細胞の任意の型であってもよい。
【0042】
一般的に、トランスフェクションは、細胞の懸濁液、または単細胞を使って実行されるものと思われるが、処理された細胞または組織の画分が、トランスフェクトされた細胞を増殖させてかつ使用できるように、そのポリヌクレオチドを組み入れるのに十分である限りは、他の方法もまた適用されうる。そのポリヌクレオチドのタンパク質産物は、細胞において一過性にまたは安定的に発現されてもよい。トランスフェクションについての技術はよく知られている。ポリヌクレオチドを導入するために利用可能な技術は、限定されるものではないが、エレクトロポレーション、形質導入、細胞融合、塩化カルシウムの使用、および対象の細胞との融合のための脂質とともにポリヌクレオチドをパッキングする方法を含む。いったん、細胞をミスマッチ修復遺伝子をトランスフェクトしたならば、細胞を培養において増殖かつ複製することができる。トランスフェクションが安定的な場合には、多くの細胞産生について遺伝子が一貫したレベルで発現されるような、その後に細胞系が生じる。
【0043】
単離された細胞は、ヒトおよび動物の組織から、酵素、例えば、コラゲナーゼまたはトリプシンなどでその組織を前処理するまたはしないのいずれかで、個々の細胞を機械的に分離して取り出し、かつそれらを適当な細胞培養培地へ移行することにより得られる細胞である。そのような単離された細胞は、典型的には、他の細胞型の非存在下で培養される。ミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子の導入について選択される細胞は、初代細胞培養もしくは不死化した細胞系の形態における真核細胞由来であってもよいし、または単細胞型生物体の懸濁液由来であってもよい。
【0044】
ミスマッチ修復タンパク質のドミナントネガティブ型をコードするポリヌクレオチドは、トランスジェニック動物を作成することにより動物のゲノムへ導入されうる。動物は、トランスジェニック動物を作成するために適当な技術が利用可能である任意の種であってもよい。例として、トランスジェニック動物は、例えばウシ、ブタ、ヒツジ、ヤギ、ウマなどの家畜から;例えば、ミルクに組換えタンパク質を発現するウシ、ブタ、またはヤギのような組み換えタンパク質の生産に使用される動物から;または、例えば、マウス、ラット、ハムスター、モルモット、ウサギなどの研究または生産物試験用の実験動物から調製されうる。
【0045】
当技術分野において公知のトランスジェニック動物を作成するための任意の方法を使用することができる。トランスジェニック動物を作成する一つの過程に従って、ポリヌクレオチドは動物の受精卵へ注入され、その注入された卵は、偽妊娠のメスへ置かれる。卵は、ポリヌクレオチドが組み入れられかつ発現している成熟動物へと発育する。受精卵は、卵および偽妊娠のメスと同じ種の供与体動物の精子からインビトロで作成され、偽妊娠のメスは、受精卵を受けて妊娠するようにホルモン処理によって調製される。トランスジェニック動物を作成する代替方法は、ポリヌクレオチドを注入またはトランスフェクションにより胚性細胞へ導入することおよびその胚性細胞を発育胚へ再導入することを含む。この方法では、しかしながら、ポリヌクレオチドが生殖系列細胞へ組み入れられない場合には、その遺伝子は子孫へ伝えられないと思われる。それゆえ、この方法により作成されたトランスジェニック動物は、その遺伝子が動物の生殖細胞へ組み入れられているかどうかを決定するために評価しなければならない。いったん、トランスジェニック動物が作成されると、それらは生殖年齢まで成長することができ、そして交尾して出産し、トランスジェニック動物の群体が維持されうる。
【0046】
いったん、トランスフェクションされた細胞系またはトランスジェニック動物の群体が作成されれば、それを1つまたは複数の対象の遺伝子において新しい突然変異を発生させるために使用することができる。対象の遺伝子は、細胞系もしくはトランスジェニック動物に本来所有されている、または細胞系もしくはトランスジェニック動物へ導入されている任意の遺伝子でありうる。突然変異を導入するためにそのような細胞または動物を使用することの利点は、その細胞または動物が商業的に有益な生物学的生産物を生産しうる遺伝的改変の広いスペクトルをもちうることである。超変異性動物をその後、育て、新しい所望の出力形質(ミルク生産、ペスト抵抗性などのような)について選択することができる。いったん、新しい形質が同定されれば、ドミナントネガティブ対立遺伝子は、当業者に使用される技術によりその対立遺伝子を直接的にノックアウトすること、または所望の形質および安定的ゲノムを有する子孫を選択するためにそのドミナントネガティブ対立遺伝子を欠損している仲間へ交配することにより除去されうる。もう一つの代替法は、CRE-LOX発現系を使用することであり、それにより、いったん新しい出力形質が確立されれば、ドミナントネガティブ対立遺伝子が動物のゲノムからスプライスされる。
【0047】
本発明のもう一つの局面は、宿主のゲノムにおいて突然変異の割合の増幅を引き起こすためのMMRを欠損している細胞(変異された内因性MMR遺伝子による、またはドミナントネガティブMMR遺伝子の導入を通して)および化学的変異誘発物質の使用である。MMR活性の欠損は、DNA損傷試薬の毒性効果に対して、より抵抗性の細胞を作成することで知られていた。本発明は、正常性(proficient)MMR細胞を作成すること;ドミナントネガティブMMR遺伝子の対立遺伝子の発現およびその後DNA変異誘発物質の使用でゲノムの超変異性を増幅することによるミスマッチ修復欠損の利用について開示する。本出願はまた、新しい遺伝的構造および/または新しい表現型を有する細胞を発生させるゲノムの変化の割合を増加させるために本来MMRを欠損している細胞を用いることおよび化学的変異誘発物質の曝露の有用性についても開示する。化学的変異誘発物質は、化学的性質、例えば、アルキル化剤、架橋結合剤などにより分類できる。他のものは本明細書に列挙されていないのだが、以下の化学的変異誘発物質は、本発明に従って有用である。N-エチル-N-ニトロソ尿素(ENU)、N-メチル-N-ニトロソ尿素(MNU)、プロカルバジン塩酸塩、クロラムブシル、シクロホスファミド、メチルメタンスルホン酸(MMS)、エチルメタンスルホン酸(EMS)、硫酸ジエチル、アクリルアミドモノマー、トリエチレンメラミン(TEM)、メルファラン、ナイトロジェンマスタード、ビンクリスチン、ジメチルニトロソアミン、N-メチル-N'-ニトロ-ニトロソグアニジン(MNNG)、7,12-ジメチルベンズアントラセン(dimethilbenz(a)anthracene、DMBA)、酸化エチレン、ヘキサメチルホスホルアミド、ブスルファン。本発明の好ましい局面において、100個の遺伝子ごとに1つの突然変異;1突然変異/1,000遺伝子の範囲の突然変異の割合を与える、突然変異誘発技術が利用される。そのような組み合わせ(MMR欠損および化学的変異誘発物質)の使用により、各特定の試薬によって優先的に誘発されるゲノムの変化(限定されるものではないが、遺伝子のコード領域、遺伝子のイントロンの領域、または5'もしくは3'の基部および/もしくは末端領域の関連内のDNAセグメントの伸展または欠失、点突然変異、改変反復配列のような)の広いアレイでの発生を可能にするものと思われる。
【0048】
突然変異は、例えば、ゲノムDNA、cDNA、メッセンジャーRNA、または対象の遺伝子に関連するアミノ酸の配列を調べることによって、細胞または動物の遺伝子型における変化について分析することにより、検出されうる。突然変異はまた、遺伝子の表現型をスクリーニングすることにより検出されうる。改変された表現型は、突然変異遺伝子によりコードされるタンパク質の電気泳動易動度、分光学的性質、または他の物理学的もしくは構造的特性における変化を同定することにより検出されうる。タンパク質の改変された機能について、インサイチューにおいて、単離された形において、またはモデル系においてスクリーニングすることもできる。限定されるものではないが、タンパク質分泌、薬品耐性、病原体耐性などのような、対象の遺伝子の機能に関連する細胞または動物の任意の性質の変化についてスクリーニングすることができる。
【0049】
本出願のもう一つの発明は、ドミナントネガティブの発現および正常に機能するMMR遺伝子の発現を制御する誘導性ベクターの使用である。本出願は、いったん、新しい出力形質、改変された遺伝子、RNAまたはポリペプチドを示す宿主細胞または生物体が、化学的変異誘発物質の組み合わせの有りまたは無しで、形質選択を通して発生したならば、この細胞または生物体の遺伝的安定なバージョンを確立するために、DNA安定性を回復する、そのようなストラテジーを使用することの有用性について開示する。ドミナントネガティブMMR遺伝子の対立遺伝子を異所性に発現した結果としてのMMR欠損細胞の場合において、MMR活性は、細胞培養または生物体の環境から誘導物質分子を除去することにより、減少または完全に無くなる。さらに、ドミナントネガティブMMR遺伝子の発現は、生殖細胞または体細胞におけるDNAノックアウト技術分野での当業者に標準的な方法を使って、MMR遺伝子の対立遺伝子をノックアウトすることにより、抑制させることができる(Waldman, T.ら、Cancer Res. 55:5187-5190, 1995)。
【0050】
さらにもう一つの発明は、HCT116、DLD-1などのようなMMR欠損細胞系において、それらによる細胞が化学的変異誘発物質で処理され、病原体耐性、薬品耐性などのような新しい出力形質について選択されており、MMR活性を回復することの利用について開示する。細胞は、その後、内因性MMR欠損を補足し、かつ新しい出力形質、改変された遺伝子配列、改変されたRNA発現および/または改変されたタンパク質発現を示す細胞または生物体のDNA安定性を回復する野生型MMR遺伝子のコピーでトランスフェクトされる。
【0051】
上記の開示は、一般的に本発明を説明している。より完全な理解は、以下の明細な実施例を参照することにより得ることができ、実施例は、例示の目的としてのみ本明細書に提供され、本発明の範囲を限定するものではない。
【実施例】
【0052】
実施例1:ミスマッチ修復正常性細胞において超変異性を引き起こすドミナントネガティブミスマッチ修復タンパク質の使用
著明なMMRの欠陥は、2人のHNPCC患者の正常細胞で見出された。この欠陥がインビボで操作的であることは、そのような患者の非腫瘍性細胞においてマイクロサテライトの不安定性の広範な存在により証明された。2人の患者の1人は、hPMS2遺伝子のコドン134での生殖系列切断突然変異(hPMS2134突然変異)を有し、一方、もう一人の患者はhMLH1遺伝子内の小さな生殖系列欠失を有していた(26)。これらのデータは、このように、HNPCC患者の生化学的および生物学的特徴を説明するものとして一般的に信じられている2つ対象のモデルに矛盾した。これらの患者の正常細胞におけるこのMMR欠損についての根拠は不明瞭であり、いくつかの可能性のある解釈が提示された。例えば、関連しているMMR遺伝子の第二の対立遺伝子が未発見の機構を通してこれらの患者の生殖系列において不活性化されているという可能性、または、MMR過程に関与する他の遺伝子の未知の突然変異が既知の生殖系列の突然変異と協力して存在しているという可能性があった。MMR欠損が正常な増殖および発生と矛盾しないことは、マウスでのノックアウト実験から明らかであり、これらの可能性(1、3、6)を支持している。または、変異誘発遺伝子がドミナントネガティブ効果を発揮し、その結果として、対応するMMR遺伝子およびMMR過程に関与するすべての他の遺伝子の野生型対立遺伝子の存在下でさえもMMR欠損を生じるという可能性があった。これらの可能性の間を区別するために、本発明者らは、MMR正常性細胞系においてhPMS2134突然変異によりコードされる切断型ポリペプチドを発現し、かつ細胞のMMR活性に対するその影響を分析した。その結果より、この突然変異体が真にドミナントネガティブの効果を発揮することができ、その結果としてMMR欠損の生化学的および遺伝学的顕在化を生じたことが示された。
【0053】
MMR正常性シリアンハムスターTKts13細胞系(以後、SH細胞と呼ぶ)に、様々なhPMS2発現プラスミドと、MMR活性を評価するためのレポーター構築物とを同時トランスフェクトした。hPMS2発現プラスミドは、正常なhPMS2遺伝子産物または上記の患者において同定された切断型hPMS2遺伝子を含んでいた(それぞれ、PMS2WTおよびPMS2134、図1A)。hPMS2配列を欠いている「空の」ベクター(PMS2NOT、図1A)が追加の対照として果たした。レポーター構築物pCAROF(out of frame)は、ハイグロマイシン耐性遺伝子と、コード領域の5'末端に29個の塩基対のアウトオブフレームポリCA鎖を含むβ-ガラクトシダーゼ遺伝子とを含んでいた。レポーター構築物pCARIF(in frame)は、ポリCA鎖が27個の塩基対である点を除いて一致しており、それゆえ、β-ガラクトシダーゼのリーディングフレームを妨害しなかった(図1B)。pCAROFレポーターは、トランスフェクションに引き続いてフレーム回復突然変異(すなわち、挿入または欠失)が起こらないかぎりは、β-ガラクトシダーゼ活性を生じないであろう。
【0054】
異なるトランスフェクションスキームはSH細胞に対するPMS2134突然変異の効果を評価するために使用された。第一のスキームにおいて、発現ベクターと、レポーターとを共に同時トランスフェクションした。17日間のハイグロマイシンでの選択の結果として100クローン以上含有するプールおよび個々のクローンが生成され、ウェスタンブロット法およびβ-ガラクトシダーゼアッセイ法のために収集された。PMS2WTおよびPMS2134で形質導入されたSH細胞は、ウェスタンブロット上に抗PMS2抗体で評価されたことだが、予想されたサイズのポリペプチドを合成した(図2Aおよび図2B)。予想されたように、事実上、β-ガラクトシダーゼ活性は、pCAROFレポーターと、PMS2NOTとをトランスフェクトしたSH細胞において、観察されなかった(図2C)。しかしながら、PMS2134をトランスフェクトしたSH細胞は、PMS2WTをトランスフェクトした細胞以上にβ-ガラクトシダーゼ活性を有意に発現した(図2C)。これらの結果より、PMS2134構築物によりコードされる切断型ポリペプチドが内因性MMR機構を撹乱させ、その結果として、リーディングフレームを回復する欠失または挿入を生じたことが示唆された。レポーター構築物の多コピーが本発明者らの条件下で形質導入されたため、これらの推定される欠失または挿入の正確な性質を評価することができず、かつ野生型β-ガラクトシダーゼ配列は予想された突然変異体を遙かに越える量があり、直接的シーケンシングによるそれらの証明は除外された。
【0055】
第二のスキームにおいて、SH細胞を各PMS2発現ベクターと、ハイグロマイシン耐性プラスミドpLHL4とを同時トランスフェクトした。100個以上のクローンを含むハイグロマイシン耐性培養物はプールされかつ増量された。これらの培養物は、その後、pCARIFまたはpCAROFレポーターと、ジェネテシン選択が可能な別々のプラスミドとを同時トランスフェクトした。2週間後、ハイグロマイシンおよびジェネテシンの両方に耐性である100個以上のコロニーをそれぞれ含む、プールされた細胞をβ-ガラクトシダーゼ活性を評価するためにXgalで染色した。図3に示されるように、PMS2134をトランスフェクトした培養物(パネルB)は多くの青色の細胞を含んでいたが、一方、PMS2WTをトランスフェクトした培養物(パネルA)においては、事実上、どの細胞も青色ではなかった。各場合において、トランスフェクション効率は、pCARIFを用いる平行トランスフェクションにより調整され、その平行トランスフェクションはまた、様々なPMS2効果遺伝子を発現する細胞のβ-ガラクトシダーゼ活性についての対照としての役割を果たし、すべての場合において類似したβ-ガラクトシダーゼ発現レベルであるという結果であった(示されず)。同様の実験プロトコールをMMR正常性ヒト胚性腎臓細胞系293に適用した場合、PMS2WTトランスフェクションと比較してPMS2134トランスフェクション後のβ-ガラクトシダーゼ活性において増加もまた観察された。これらの細胞は、pCAROFと、様々なPMS2効果プラスミドとを同時トランスフェクトし、ハイグロマイシンで17日間選択した。17日目に、コロニーをβ-ガラクトシダーゼ活性を評価するためにXgalで染色し、β-ガラクトシダーゼ発現細胞について評点をつけた。表1に示されるように、PMS2134ポリペプチドを発現するそれらの細胞のみが検出可能なβ-ガラクトシダーゼ活性を発現した。これらのデータは、げっ歯類系およびヒト系の両方において、類似したhPMS2134タンパク質のドミナントネガティブ効果を証明し、かつこれらの研究においてげっ歯類系の有用性を確証している。
【0056】
(表1) pCAROFレポーター構築物と、PMS2効果プラスミドとをトランスフェクトした293クローンのβ-ガラクトシダーゼ発現。293細胞は、pCAROFβ-ガラクトシダーゼレポータープラスミドと、PMS2NOT、PMS2WT、またはPMS2134効果プラスミドとを同時トランスフェクトした。トランスフェクトされた細胞は、ハイグロマイシンで17日間選択し、β-ガラクトシダーゼ活性のためにXgalで染色した(青色の細胞)。下記の結果は、3連の実験の平均値 +/ 標準偏差を示す。

【0057】
プラスミド
完全長野生型hPMS2 cDNAは、記載されているように、ヒトHeLa cDNAライブラリーから得られた(18)。アミノ酸134位に終止コドンを含むhPMS2 cDNAは、突然変異が発見された患者からRTPCR法により得られた(9)。cDNA断片は、SV40ポリアデニル化シグナルが続いているSV40プロモーターを含むpSG5ベクターへBamHI部位にクローニングされた(8)。図1に記載されているpCARレポーターベクターは、参照文献21および25に記載されているように、構築された。
【0058】
細胞系およびトランスフェクション
シリアンハムスター線維芽細胞Tkts13およびヒトHEK293細胞は、ATCCから得られ、記載されているように培養された(15)。hPMS2を発現する安定的にトランスフェクションされた細胞系は、PMS2発現ベクターおよび3:1(pCAR:pLHL4)の割合でハイグロマイシン耐性遺伝子をコードするpLHL4プラスミドの同時トランスフェクションにより作製され、かつハイグロマイシンで選択された。pCARレポーターを含む安定的にトランスフェクトされた細胞系は、pCARベクターをネオマイシン耐性プラスミドをコードするpNTKプラスミドまたはpLHL4のいずれかとともに同時トランスフェクションにより作製された。すべてのトランスフェクションは、以前に記載されているように、リン酸カルシウムを用いて行われた(15)。
【0059】
β-ガラクトシダーゼアッセイ法
pCARでのトランスフェクションに続いて17日間目に、β-ガラクトシダーゼアッセイ法を45 mM 2-メルカプトエタノール、1 mM MgCl2、0.1 M NaPO4および0.6 mg/ml クロロフェノールレッドβ-D-ガラクトピラノシド(CPRG、Boehringer Mannheim)中の20 gのタンパク質を用いて行った。反応は、1時間インキュベートされ、0.5 M Na2CO3の添加により停止させ、かつ576 nmでの分光測光法により分析された(16)。インサイチューβ-ガラクトシダーゼ染色のために、細胞をPBS中の1 % グルタルアルデヒドで固定化し、0.15 M NaCl、1 mM MgCl2、3.3 mM K4Fe(CN)6、0.2 % Xgal中で2時間、37℃でインキュベートした。
【0060】
ウェスタンブロット法
PMS2についてのウェスタンブロットは、実施例5に記載されているように、ヒト完全長ポリペプチドのコドン1〜20に対して産生したポリクローナルな抗ヒトPMS2を用いて行われた。
【0061】
実施例2:ドミナントネガティブミスマッチ修復遺伝子の対立遺伝子はMMR活性に欠陥を引き起こす
PMS2WTおよびPMS2134をトランスフェクトされた細胞の間でのβ-ガラクトシダーゼ活性における違いについてもっとも可能性が高い解釈は、PMS2134タンパク質がMMR活性を妨害し、その結果として、pCAROFレポーター内の高頻度の突然変異およびORFの再確立を起こすということである。MMRは変化するという仮説を直接的に試験するために、本発明者らは、実施例1に記載されているように、PMS2-WT、PMS2-134またはPMS2-NOTの空のベクターを含む細胞からの個々のクローンでのMMRについて生化学的アッセイ法を使用した。核抽出物は、クローンから調製され、以前に記載されている条件下で、/CA\挿入欠失またはG/Tミスマッチのいずれかを含むヘテロ二重鎖基質とともにインキュベートされた。/CA\およびG/Tのヘテロ二重鎖は、それぞれ、3'方向および5'方向からの修復を試験するために使用された。これらのアッセイにおいて、PMS2-134発現クローンとその他のクローンとの間には劇的な違いがあった(表2A)。すべてのクローンは基質を3'方向から修復したが(/CA\ヘテロ二重鎖)、PMS2134ポリペプチドを発現する細胞は、ほとんど5'修復活性をもたなかった。PMS2134トランスフェクションから生じるプールされたクローンについて、またはヘテロ二重鎖が24塩基対ループを含む場合、ミスマッチ修復における同様の方向上の欠陥は明らかであり、それらの例は表2Bに示されている。3'/CA\PMS2-WT修復アッセイにおいて、MMR活性に少量の減少が観察されたが、おそらく、PMS2タンパク質の過剰発現による生化学的アッセイ法における干渉の結果であろう。インサイチューβ-ガラクトシダーゼアッセイ法において、PMS2-WTにより有意な活性を生じなかったが(図3;表1)、インビボの条件を反映している可能性がより高い結果といえる。
【0062】
(表2) SHクローン(A)またはプールされた培養物(B)由来の核抽出物のミスマッチ修復活性。抽出物を24 fmolのヘテロ二重鎖でのMMR活性について試験した。*これらのデータは、5つ以上の独立した実験から導かれた類似の結果を表している。
【0063】
ミスマッチ修復についての生化学的アッセイ法
核抽出物におけるMMR活性は、記載されているように、24 fmolの基質を用いて行われた(12、25)。相補性アッセイ法は、最終KCl濃度を100 mMへ調整しながら、〜100 ngの精製されたMutLαまたはMutSα成分を100 μgの核抽出物へ添加することにより行われた(4、10、30)。これらの実験に用いられる基質は、ミスマッチまで5'側181ヌクレオチドまたは3'側125ヌクレオチドの鎖切断を含む。値は少なくとも二連で行われた実験を代表している。
【0064】
実施例3:遺伝子座における突然変異を増幅するためのMMR欠損細胞および化学的変異誘発物質の使用
遺伝的突然変異の割合を増幅しかつ改変された遺伝子、RNA発現またはポリペプチドを有する細胞を産生するために、MMR欠損および化学的変異誘発物質の使用は、そのような多様性を生じるための強力な道具である。MMR欠損細胞を使用することの利点は、細胞をそのような化合物の毒性効果に対してなお、より抵抗性にさせているこの活性が減少することにより、宿主生物体または細胞の遺伝子および表現型の変化における増加が見込まれることである。以下の実験は、本発明の有用性を証明するために行われる。限定されるものではないが、HCT116、DLD-1などのようなMMRについて遺伝的に欠損している細胞、またはドミナントネガティブ対立遺伝子の異所性発現によりMMR欠損にさせられている実施例1および実施例2に記載されているもののような細胞は、本発明の下で網羅される。簡単に言えば、MMR正常性および欠損性細胞を増殖培地において、化学的変異誘発物質の範囲濃度(1 nm〜1 mM)で、1時間〜24時間、37℃、5% CO2でインキュベートする。インキュベーションが完了した後、化学的変異誘発物質を培地から洗浄し、以前に記載され(Walker, VEら、Mutant Res. 17:371-388, 1999)かつ当業者に知られているように、細胞をヒポキサンチン、アミノプテリン、およびチミンの存在下で増殖させてHPRT突然変異細胞について評点をつける。細胞を10 cmの組織培養皿に1 X 105細胞/mlで播き、増殖培地において14日間、37℃、5 % CO2で増殖させる。14日後、HAT耐性コロニー数は、顕微鏡下においてカウントすることにより測定された。典型的実験により、化学的に処理されたMMR欠損細胞において、対照細胞よりも、有意により多くの数のHAT耐性コロニーが形成されることが明らかにされ、宿主細胞/生物体の内因性遺伝子内に突然変異を増加させる能力を証明するものと思われる。MMR欠損細胞、化学的または電離放射線への曝露を加えた使用はまた、トランスフェクションおよび一過的または安定的細胞系の選択を経て導入された標的遺伝子内にインビボで遺伝的突然変異を増幅させるために用いることができる。形質導入された標的遺伝子内に遺伝的突然変異を増幅させるためのMMR欠損に加えて化学的変異誘発物質の能力を証明するために、本発明者らは、上記の細胞の使用により、pCAROFベクター(実施例1参照)をHCT116細胞へトランスフェクションした。細胞をハイグロマイシン耐性によりpCAR-OF陽性クローンについて選択した。ハイグロマイシン耐性細胞を集密まで増殖させ、100,000個の細胞を10 μMのエチル-メタン-スルホン酸(EMS)アルキル化化合物に8時間曝し、増殖培地へ戻した。その後、細胞を一晩増殖させ、それから、3連の10 cmのディッシュに、1,000細胞/プレートの密度で播いた。細胞を10日間増殖させ、実施例1に記載されている方法を用いて、β-ガラクトシダーゼ活性について評点をつけた。その結果より、その化合物の非存在下で増殖した細胞において、β-ガラクトシダーゼ陽性増殖巣数は92+/-10 /ディッシュであることが示された。対照的に、EMSに曝した細胞は、β-ガラクトシダーゼ陽性細胞数において、有意な増加を生じた(205+/-18)。これらのデータは、インビボで標的遺伝子に遺伝的突然変異を発生させるためのMMR欠損細胞に化学的変異誘発物質を加えた有用性を証明している。この方法は、商業的目的として標的遺伝子に遺伝的多様性を生じるために有用である。
【0065】
実施例4:誘導性ベクターによるドミナントネガティブMMR遺伝子の対立遺伝子を発現するミスマッチ修復欠損細胞の回復されたDNA安定性
ドミナントネガティブMMR遺伝子の対立遺伝子の異所性発現を用いてDNA超変異性を誘発する能力は、遺伝的な多様性サブタイプを有する真核細胞を産生することへの多くの重要な商業的適用がある。以下の実験は、実施例1および実施例2に記載されているようなMMR欠損細胞のゲノムにおける遺伝子的変化をそのMMR正常性を回復することにより永久的に刷り込みする能力を証明している。まず、PMS2-134ドミナントネガティブ対立遺伝子をpcDNA4/TO/PMS134Sと呼ばれる真核の誘導性ベクター系pcDNA4/TO(テトラサイクリン誘導性ベクター)(Invitrogen)、pIND/PMS134Sと呼ばれるpIND/V5-Hisグルココルチコイド誘導性ベクター(Invitrogen)へクローニングした。下記のように、Tk-ts13またはHEK293細胞を各ベクターと、pCAR-OF(選択マーカーとしてハイグロマイシン耐性遺伝子を含む)とを同時トランスフェクトした。空のベクターは、各組み合わせについての対照として使用された。トランスフェクションされた細胞は、ゼオシン/ハイグロマイシン(Z/H)耐性細胞またはネオマイシン/ハイグロマイシン(N/H)耐性細胞について10日間〜14日間選択された。クローンは選び取られ、個々のクローンまたはプールとして増量された。細胞は増量され、誘導化学物質(pcDNA4/TO/PMS134S用に1 μg/mlのテトラサイクリンおよびpIND/PMS134S用に1 μMのエクジソン)の含有または非含有の増殖培地(10 %ウシ胎児血清を添加したDMEM)で、6ウェルの組織培養プレートに1 X 105細胞/mlで播かれた。細胞培養物は収集し、37℃、5 % CO2での培養の24時間後、タンパク質発現を誘導されたPMS2-134についてウェスタン分析により分析した(実施例5に記載されているように)。pIND/PMS134S細胞の抽出物のウェスタン分析により、エクジソンの存在下で増殖された場合、〜17 kdのタンパク質の生成が明らかになったが、一方、エクジソン非含有で増殖されたそれらは検出可能なレベルをもたなかった。誘導性PMS2-134発現を有するクローンは増量され、エクジソンまたはテトラサイクリンの存在下で24時間増殖された。誘導物質の非存在下でウェスタンブロットによりPMS2-134発現の損失の動力学を同定するために、細胞は72時間収集された。これらのデータより、72時間後タンパク質は検出不可能なレベルであることが証明された。
【0066】
誘導性ベクター系を用いて遺伝子的不安定性を誘発する能力を証明するために、pIND/PMS134SまたはpIND/V5-Hisを含む細胞をエクジソンの含有または非含有で14日間増殖させ、記載されているように(MCB論文)、インサイチューでβ-ガラクトシダーゼ活性を測定するために染色した。図5に示されるように、観察可能な青色増殖巣をもたなかった空のベクター対照とは対照的に、エクジソンの存在下で増殖されたpIND/PMS134S細胞において、有意の数の小細胞増殖巣が陽性の青色に着色した(倒立顕微鏡の評価下での観察では25個の細胞/視野)。対照的に、誘発物質の非存在下で増殖されたpIND/PMS134S細胞もpIND/V5-His細胞のどちらも陽性に着色しなかった。これらのデータは、遺伝子においてゲノムの不安定性および遺伝的多様性を発生させるために誘導性ベクターの制御下でドミナントネガティブMMR遺伝子を用いて、改変された生化学的機能および/または新しい表現型を生じる能力を証明している。
【0067】
これらの細胞において、PMS2-134発現を抑圧することでMMR正常性を回復することができることを証明するために、以下の実験を行った。細胞をZ/HまたはN/Hを加えた誘導性培地中で14日間維持した。各クローンまたはプールのサブセットを24ウェルのファルコンのディッシュへ、5X104細胞/mlで播いた。細胞を37℃、5 % CO2で一晩増殖させた。次の日、細胞を以前に記載されているように、β-ガラクトシダーゼ発現についてインビボで染色した(Nicolaidesら、Mol. Cell Biol. 18:1635-1641, 1998)。青色に変化する細胞は、MMR機構においてPMS2-134のドミナントネガティブの効果による内因性MMR活性における減少のために、そのようになった。これらの細胞は、誘導物質分子の存在下または非存在下において、96ウェルのプレートに限界希釈によりサブクローニングされた。誘導物質剤が除去されているクローンにおいて復帰細胞(青色ではない細胞)の数がより少ないことが見出された(クローンが誘導物質での常時曝露下であるプレート(45ウェルのうち18ウェル)とは対照的に、45ウェルのうち42ウェル)場合、PMS2-134発現クローンにおいて回復された遺伝子的安定性が証明された。これらのデータより、ゲノムの安定性および遺伝的進化を制御する能力が、制御されたMMR遺伝子発現を用いて証明される。
【0068】
上記の誘導性MMR遺伝子ストラテジーと組み合わせての実施例3に記載されている化学的変異誘発物質の使用はまた、新しい表現型を有する遺伝的多様性宿主生物体を発生させるための、および/または改変された遺伝子発現の安定的生産のための方法としての適用において仕込まれる。この効果を証明するために、誘導性ドミナントネガティブ発現を含む細胞を誘導物質分子に曝し、かつ引き続いて化学的変異誘発物質または電離放射線に曝す。その後、細胞を誘導物質分子の存在下で増量し、配列分析または生化学的活性により決定されるような新しい表現型および/または改変された遺伝子構造を有する細胞について培養物を選択する。改変された遺伝子または表現型を有する細胞をその後、誘導物質分子からはずし、遺伝子的安定性および表現型を回復させる。
【0069】
トランスフェクション
pIND/PMS134SまたはpcDNA/TO/PMS134S誘導性ベクターを有するエクジソン受容体含有安定的HEK293細胞の産生。HEK293-エクジソンR細胞をリポフェクタミン2000(Gibco/BRL)を用いて、pINDもしくはpcDNA/TOの空のベクターまたはpIND/PMS134SもしくはpcDNA/TO/PMS134Sベクターでトランスフェクションした。細胞は、選択的マーカー耐性について選択され、クローンおよびプールは増量された。その後、安定的系を1 μMのエクジソンに48時間曝し、抽出物を単離し、下記のPMS134ポリペプチドのN末端に特異的な抗血清を用いて、誘導性PMS134発現を有するクローンを同定するためにウェスタンブロット法により分析した。
【0070】
プラスミド
PMS2-134は、pSG5PMS134(実施例1に記載)ベクター由来のBamHI断片として、以下の誘導性発現ベクターへクローニングされた。テトラサイクリン誘導性ベクター(pcDNA4/TO/PMS134S)は、EM-7プロモーターおよびSV40ポリAの配列の制御下のゼオシン(zeocin)選択的マーカーを含む。プラスミドの構造はエンドヌクレアーゼ制限分析およびシーケンシングにより確認された。グルココルチコイド誘導性ベクター(pIND/PMS134S)は、SV40初期プロモーターおよびポリAの配列の制御下のネオマイシン選択的マーカーを含む。ベクターの模式図は図4に示されている。
【0071】
トランスフェクション
誘導性発現ベクターは、単独、または実施例1に記載されているようなpCAR-OFベクターとの組み合わせのどちらかで上記の方法に従い、Tk-ts13細胞およびHEK293細胞へ同時トランスフェクションされた。記載されているように(参照文献15、Grassoら、J. Biol. Chem. 273:24016-24024, 1998)、細胞をゼオシン/ハイグロマイシン(pcDNA4/TO/PMS134S)またはネオマイシン/ハイグロマイシン(pIND/PMS134S)耐性クローンについて選択された。耐性クローンを選び取る、および/またはプールする、かつタンパク質分析のために増量させた。
【0072】
実施例5:MMR遺伝子の補足遺伝子を発現することによるMMRの回復および遺伝子的安定性の回復および固定されたゲノム構造の確立
限定されるものではないが、HCT116、DLD-1、およびHEC-1-A細胞系(参照文献12、25およびKondoら、J. Biochem. 125:818-825, 1999)のような内因性遺伝子の欠陥による欠損MMR活性を有する細胞の使用が、遺伝子の遺伝的構造を改変して、新規な抗菌剤、生理活性成長因子またはホルモン、改変された抗体構造などのような商業的に実行可能な様々な分子を生産することに役に立つことができる。そのような細胞の有用性および価値は、いったん改変された遺伝子構造が生成されれば、この遺伝子変化の完全性は、機能的補足MMR遺伝子の導入により細胞を遺伝子的に安定させることにより細胞のゲノムに保存されうることである。この例は、遺伝子的に改変された細菌のプリンヌクレオチドホスホリラーゼ(PNP)遺伝子で、その遺伝子のN末端にアウトオブフレームのポリA鎖が挿入されている(ポリPNPと呼ばれる)遺伝子の導入により、MMR欠損細胞において遺伝子的に改変することができ、また、補足MMR遺伝子の直接的発現によりMMR欠損細胞をMMR正常性にする場合、遺伝子的安定性となりうる。ポリPNP遺伝子は非機能的PNP遺伝子としてコードしている。ポリA鎖が欠損MMRのための遺伝子的改変によりランダムに変化する場合、その鎖はランダムに変化して、機能的PNP遺伝子およびポリペプチドの産生が可能になる。PNPは、無毒性の9-(β-D-2-デオキシ-エリスロ-ペント-フラノシル)-6-メチル-プリンのプロドラッグ(MPDと呼ばれる)基質を毒性の6-メチルプリン類似体(MPと呼ばれる)に変換する(Sorscher, EJら、Gene Therapy 1:233-238, 1994)。ポリPNP遺伝子は、抗HA抗体を用いてのウェスタンブロット分析によるコードポリペプチドの検出を容易にするために、C末端に血球凝集素エピトープタグを含むように操作された。ポリPNP遺伝子は、選択のためのハイグロマイシン耐性(Hyg)遺伝子を有するpCEP4発現ベクターへクローニングされた。この遺伝子を示す模式図は図7に示されている。PNPと呼ばれる相同遺伝子群はまた、インフレームポリA鎖がPNP活性についての陽性対照としてその遺伝子のN末端へクローニングされているように作成された。簡単には、MMR欠損HCT116細胞系およびMMR正常性HEK293細胞系をポリPNP、PNP発現ベクター、または空のpCEP4ベクターでトランスフェクションした。その後、細胞はHyg耐性について選択され、クローンは単離された。増量された細胞を漸増的量のMPD(0 μM、1 μM、10 μM、50 μM、100 μM、300 μM)の存在下で10日間増殖させた。処理期間後、細胞を血球計算器およびトリパンブルー排除によりカウントした。図8Aに示されているように、HCT116/ポリPNP細胞を100 μMまたは300 μMのMPD、それぞれの存在下において培養した場合、細胞数に20 %および30 %の減少が観察された。対照的に、MMR正常性HEK293/ポリPNP細胞については、使用されるMPDの最高濃度(300 μM)においてさえ、細胞増殖に減少は観察されなかった。両方の細胞系について、細胞が50 μM〜300 μMのMPDの存在下において増殖する場合、PNPの発現は、その結果として100 %の増殖抑制を生じ、両方の細胞系への変換されたMPの毒性効果を証明した。ウェスタンブロットにより、HAエピトープを含むポリペプチドが実際に、HCT116/ポリPNP細胞において生成されていることが確認され、このように、ポリPNP遺伝子構造が変化して機能的かつ完全長のPNP酵素を産生したことが証明された(図8B)。
【0073】
遺伝子的安定性の回復および改変された遺伝子座の引き続く刷り込みは、実行可能な生物学的生産物を生産するための本出願の重要な発明であり、それらにより、所望の改変された出力形質を有する改変された生体分子、細胞または生物体全体を長期間の使用のために遺伝子的に安定に作成される。化学的変異誘発物質に曝されたまたは曝されていない、かつ所望の遺伝子的変化について選択された、安定的MMR欠損細胞系を産生するために、突然変異内因性MMR遺伝子座の代わりになることができる補足MMR遺伝子の導入が本出願において開示されている。これは、ヒトMutL相同体MLH1(12、24、25)について遺伝子的に欠損しているHCT116細胞を用いる例により証明される。この例において、哺乳動物発現ベクターで、機能的MLH1ポリペプチドについてコードする(pC9MLH1)または未熟な停止コドンを有するMLH1 cDNAについてコードする発現ベクター(pC9MLHstop)が使用される。これらの発現ベクターはこのベクターを含む細胞の選択を可能にするネオマイシン(neo)耐性遺伝子を含む。MMR活性を補足する能力をそうではないMMR欠損細胞において証明するため、および遺伝子座の改変された構造を永久的に刷り込みするために、ポリPNPおよびpC9MLH構築物をHCT116細胞へ同時トランスフェクションした。細胞を10日間、neoおよびHygにおいて選択し、耐性クローンを単離しかつ増量させた。その後、細胞をMPDの存在下で培養し、10日後増殖についてカウントした。図9において証明されているように、MLH1野生型cDNAをトランスフェクトした細胞は、MLHstopをトランスフェクトした細胞とは対照的に、ウェスタン法による測定では、MLH1を発現していた。さらに、細胞を300 μMのMPDの存在下で増殖した場合、MLH1を発現しているそれらの細胞は、培地のみで増殖した細胞と比較して、全細胞増殖において2 %の減少を示し、一方、MLHstopまたは空のベクターおよびポリPNPをトランスフェクトした細胞は、培地のみで増殖した細胞と比較して、細胞増殖において35 %の低下があった。これらのデータは、異所的に発現されている野生型MMR遺伝子またはcDNAでMMR欠陥を補足することにより、MMR欠損細胞系のゲノム安定性を確立し、かつ生物学的活性または不活性のPNPのような、新しい出力形質および/または改変されたゲノムまたはポリペプチド構造を生産するために、選択された長期間安定的系を確立することができることを証明している。
【0074】
プラスミド
完全長野生型hMLH1 cDNAは、記載されているように、ヒトHela cDNAライブラリーから得られた(18)。終止コドンを含むMLH1 cDNAは、突然変異が発見された患者からRTPCRにより得られた(24)。cDNA断片は、SV40ポリアデニル化シグナル(8)およびネオマイシン耐性についてコードする遺伝子が続くCMVプロモーターを含む、pCEP9ベクター(Invitrogen)のXhoI部位へクローニングされた。pC9MLH1ベクターは完全長機能MLH1タンパク質を産生するが、一方pC9MLH1stopは非機能性の切断型MLH1ポリペプチドを産生する。ポリPNPおよびPNPのベクターは図4に記載されている。ポリPNPは、結果として切断型ポリペプチドを生じる、細菌のPNP遺伝子のコドン2の後に挿入される21塩基のアウトオブフレームポリA鎖を含む(Sorscher, EJら、Gene Therapy 1:233-238, 1994)。PNPは、結果として完全長の機能的に活性なPNPタンパク質を生じる、細菌のPNP遺伝子のコドン2の後に挿入される20塩基のインフレームポリA鎖を含む。ポリPNPおよびPNP遺伝子の両方とも、終止コドンが続くC末端にインフレームで融合された血球凝集素(HA)エピトープを有する。ポリPNPおよびPNP遺伝子は、センスプライマー:
を用いてポリメラーゼ連鎖反応により構築され、ポリA鎖は下線を施されているが、PNP用のプライマーはポリA鎖において1つ少ないAを含む。両方の構築物についてのアンチセンスプライマーは、
である。DH5α細菌のDNAを増幅のための鋳型として使用した。改変されたPNP遺伝子は、以前に記載されているように、緩衝液中、95℃を30秒間、54℃を1分間、72℃を1分間の25サイクルで増幅して産生した(19)。増幅されたゲノムの挿入部はTテイル化ベクター(TAクローニング、Invitrogen)へクローニングされ、組換えクローンは正しいヌクレオチド配列を有するベクターを同定するためにシーケンシングされた。その後、PNP断片はTAクローニングベクターポリリンカーからの部位を用いてpCEP4ベクター(Invitrogen)のKpnI-XhoI部位へサブクローニングされた。組換えPNP発現ベクターは、内部プライマー配列を用いる配列の確実性を保証するためにシーケンシングされた。
【0075】
細胞系およびトランスフェクション
ヒトHCT116およびHEK293細胞はATCCから得られ、ベンダーにより提案されているように、10 %ウシ胎児血清を添加したRPMI中で培養された。細胞は、メーカーのプロトコール(Gibco/BRL)に従いリポソームを用いてPNPおよび/またはMLH1発現ベクターでトランスフェクションされた。トランスフェクション、続いてハイグロマイシンでの選択により、空のベクター、PNPまたはポリPNPを発現する安定的にトランスフェクションされた細胞系が産生された。補足的実験として、HCT116細胞をPNP/MLH1、PNP/MLH1stop、ポリPNP/MLH1またはポリPNP/MLH1stopで、各プラスミドの5 μgを用いて1:1の割合で、トランスフェクションし、ハイグロマイシンおよびネオマイシン耐性について選択した。10日後、薬剤耐性コロニーが観察され、分析のために採取した。
【0076】
MPD殺害性アッセイ法
MPD殺害性アッセイ法のために、細胞を2X104 細胞/mlで播き、かつ1 mlの等分量を24ウェルコースター組織培養皿に播いた。殺害性アッセイ法のために、細胞を0 μM、1 μM、10 μM、50 μM、100 μM、および300 μMのMPDに3連で播いた。細胞をトリプシン処理して10日間増殖させ、トリパンブルー排除を用いて血球計算器でカウントした。データは、各研究について平均値+/-SDとして示されている。
【0077】
ウェスタンブロット
カウント後、各0 μM MPDで処理された細胞から同じ細胞数を試料緩衝液(60 mM トリス、pH 6.8、2 % SDS、10 % グリセロール、0.1 M 2-メルカプトエタノール、0.001 % ブロモフェノールブルー)に直接溶解し、5分間煮沸した。タンパク質可溶化液を18 % トリス-グリシンゲル(Novex)上での電気泳動により分離した。ゲルを48 mM トリス、40 mM グリシン、0.0375 % SDS、20 % メタノールにおいてイモビロン-P(Immobilon-P)(Millipore)上へ電気的ブロットし、0.05 % Tween20および5 % 練乳を加えたトリス緩衝生理食塩水において、室温で1時間ブロックした。フィルターをヒトMLH1または血球凝集素(HA)(Boehringer Manheim)に対して産生されたモノクローナル抗体(αMLH14)および西洋わさびペルオキシダーゼ結合型ウサギ抗-マウス第二次抗体で探索し、検出のために化学ルミネッセンス(Pierce)を使用した。マウスIgGは、第一次抗体の非特異的抗体との相互作用について評価し、かつ使用された抗血清が期待されたタンパク質を検出していることを保証するために、すべての実験について対照として使用された。
【0078】
参照文献
【技術分野】
【0001】
本出願は、2000年5月17日出願の60/204,769に基づく優先権を主張するものであり、その開示は本明細書に明示的に組み込まれる。
【0002】
発明の技術分野
本発明は、ミスマッチ修復遺伝子の領域に関する。特に、本発明は、変異誘発の分野に関する。
【背景技術】
【0003】
発明の背景
過去4年間に、リンチ(Lynch)症候群IIとしても既知の遺伝性非ポリポーシス性大腸癌症候群(Hereditary Nonpolyposis Colorectal Cancer Syndrome/HNPCC)の遺伝学的原因が、その疾患に罹患した血族の大部分について確認された(13)。HNPCCの分子的基礎は、欠陥ミスマッチ修復(MMR)に起因する遺伝学的不安定性を含む。mutSホモログGTBP、hMSH2、及びhMSH3、並びにmutLホモログhMLH1、hPMS1、及びhPMS2を含む、MMR過程に参与していると考えられるタンパク質をコードする多くの遺伝子が、齧歯類及びヒトにおいて同定されている(2,7,11,17,20,21,22,24)。これらの遺伝子のうちの4つ(hMSH2、hMLH1、hPMS1、及びhPMS2)の生殖系列変異が、HNPCC家系において同定された(2,11,13,17,24)。MMR欠損により生じる変異誘発欠陥(mutator defect)は、任意のDNA配列に影響を与えうるが、MMR異常に対する特に高い感受性を有するのはミクロサテライト配列である(14)。従って、ミクロサテライトの不安定性は、欠陥MMRの有用な指標である。ミクロサテライトの不安定性は、HNPCC患者に生じる事実上全ての腫瘍において発生するのに加え、別個の分子的特性及び表現型的特性を有する散発性腫瘍の極一部にも見出される(27)。
【0004】
HNPCCは、常染色体優性遺伝するため、罹患家族メンバーの正常細胞は、(罹患している親から遺伝した)関連MMR遺伝子の変異対立遺伝子1個と、(罹患していない親から遺伝した)野生型対立遺伝子1個とを含有している。しかしながら、腫瘍発生の初期の段階において、野生型対立遺伝子が体細胞性変異によって不活化されて、機能性MMR遺伝子を有しない細胞が残され、MMR活性の著しい欠陥がもたらされる。腫瘍細胞における欠陥MMRを作製するためには、生殖系列変異に加え体細胞性変異が必要とされるため、他の遺伝性癌を惹起する腫瘍抑制遺伝子の二対立遺伝子不活化と類似した、このメカニズムは、一般的に、2ヒットを含むものと呼ばれる(11,13,25)。この2ヒットメカニズムと一致して、HNPCC患者の非新生物細胞は、一般に、野生型対立遺伝子の存在のため、ほぼ正常なレベルのMMR活性を保持している。
【0005】
欠陥MMRを有する広範囲の生物が、ゲノム全体に拡がった遺伝子変異を有することが見出されている。全ての場合に、これらの生物は、特定のMMR遺伝子の両コピーに生殖系列変異を有している。最近、ニコライデス(Nicolaides)らの研究により、MMR遺伝子のドミナントネガティブ対立遺伝子を導入することにより、高等生物由来の細胞においてMMRの減少が達成されうることが示された。これらのデータは、そのような手法の使用によって、新たな出力形質を生成させるため遺伝学的に改変された生物が作製されうることを示唆している。当分野においては、遺伝学的な多様性を作製するためのさらなる方法が必要とされている。
【発明の概要】
【0006】
発明の概要
本発明の目的は、細胞を超変異性にするための方法を提供することである。
【0007】
本発明の別の目的は、遺伝学的に改変された細胞系を提供することである。
【0008】
本発明の別の目的は、表現型的に改変された細胞系を提供することである。
【0009】
本発明のさらに別の目的は、細胞において遺伝学的超変異の速度を増強するための方法を提供することである。
【0010】
本発明のさらなる目的は、細胞において関心対象の(1個以上の)遺伝子を変異させる方法を提供することである。
【0011】
本発明のさらなる目的は、遺伝学的に改変された細菌プリンホスホリラーゼのための材料の組成物を、特許請求の範囲に記載することである。
【0012】
本発明のさらなる目的は、真核細胞のミスマッチ修復欠損をモニタリングするための診断道具としての、遺伝学的に改変された細菌プリンホスホリラーゼのための材料の組成物を、特許請求の範囲に記載することである。
【0013】
本発明のさらなる目的は、真核細胞における改変されたミスマッチ修復を測定するための、ポリモノヌクレオチド鎖(tract)を組み込むことにより遺伝学的に改変された遺伝子を作製するための材料の組成物を、特許請求の範囲に記載することである。
【0014】
本発明のさらに別の目的は、新たな表現型を有する細胞を作出する方法を提供することである。
【0015】
本発明のさらに別の目的は、新たな表現型及び安定なゲノムを有する細胞を作出する方法を提供することである。
【0016】
本発明のさらに別の目的は、細胞又は生物のゲノムの遺伝学的安定性を制御する方法を提供することである。
【0017】
本発明のさらなる目的は、ドミナントネガティブミスマッチ修復遺伝子変異体を含有する誘導可能ベクターを使用して超変異性細胞系を作製することである。
【0018】
本発明のさらなる目的は、誘導された遺伝子発現条件下で、ドミナントネガティブミスマッチ修復遺伝子変異体を有する誘導可能ベクターを含有している超変異性細胞系をスクリーニングすることである。
【0019】
本発明のさらなる目的は、誘導された遺伝子発現条件下で、ドミナントネガティブミスマッチ修復遺伝子変異体を有する誘導可能ベクターを含有している超変異性細胞系を、改変された遺伝子構造及び/又は新たな表現型に関してスクリーニングすることである。
【0020】
本発明のさらなる目的は、ゲノムの安定性を回復させるため、構造的に改変された標的遺伝子及び/又は新たな表現型を含有している細胞におけるドミナントネガティブMMR遺伝子の発現をオフ状態にすることである。
【0021】
本発明のさらなる目的は、化学変異原又は電離放射線の存在下で、誘導条件下で、ドミナントネガティブミスマッチ修復遺伝子変異体を含む誘導可能ベクターを含有している超変異性細胞系を、構造的に改変された標的遺伝子及び/又は新たな表現型に関してスクリーニングすることである。次いで、改変された遺伝子構造及び/又は新たな表現型を含有している細胞を誘導分子から離し、遺伝学的安定性を回復させる。
【0022】
本発明のこれら及びその他の目的は、下記の態様のうちの一つ以上によって提供される。本発明の一つの態様において、超変異性細胞を作成するための方法が提供される。ミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子をコードするポリヌクレオチドが細胞へ導入される。細胞は、遺伝子の導入の結果として超変異性となる。
【0023】
本発明の別の態様において、単離された超変異性細胞が提供されるであろう。細胞は、ミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子を含む。細胞をDNAアルキル化剤に曝す。細胞は増強された超変異速度を示す。
【0024】
本発明の別の態様において、関心対象の遺伝子に変異を導入するための方法が提供される。ミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子をコードするポリヌクレオチドが、細胞へ導入される。細胞は、遺伝子の導入の結果として超変異性となる。細胞は、関心対象の遺伝子をさらに含む。細胞を増殖させる。関心対象の遺伝子が変異を保持しているか否かを決定するため、細胞が試験される。
【0025】
本発明の別の態様において、真核細胞のミスマッチ修復活性を測定するため、ポリモノヌクレオチド鎖を遺伝子に挿入するための方法が提供される。ポリモノヌクレオチド鎖が、遺伝子又はcDNAのコーディング領域にアウトオブフレーム(out-of-frame)で挿入される。その遺伝子が、細胞へ導入される。ポリモノヌクレオチド鎖が、ミスマッチ修復欠損により改変される。インフレーム(in-frame)の改変された遺伝子が作製される。
【0026】
本発明の別の態様において、細胞の新たな表現型を生成させるための方法が提供される。ミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子をコードするポリヌクレオチドが、細胞へ導入される。細胞は、遺伝子の導入の結果として超変異性となる。細胞を増殖させる。新たな表現型の発現に関して細胞が試験される。本発明の別の態様は、改変された標的遺伝子及び/又は新たな表現型のための、化学変異原又は電離放射線の存在下での、誘導条件下での、ドミナントネガティブミスマッチ修復遺伝子変異体からなる誘導可能ベクターを含有している細胞の使用である。次いで、改変された遺伝子構造及び/又は新たな表現型を含有している細胞を誘導分子から離し、遺伝学的安定性を回復させる。ここで、細胞は、組換え製造及び/又は遺伝子発見(これらに限定はされない)のような商業的な特性のために使用される。
【0027】
本発明の別の態様は、改変された標的遺伝子及び/又は新たな表現型のための、化学変異原又は電離放射線の存在下での、誘導条件下での、関心対象の遺伝子を含有しているMMR欠陥細胞の使用である。次いで、改変された遺伝子構造及び/又は新たな表現型を含有している細胞は、野生型MMR補足遺伝子で安定的に形質導入し、遺伝学的安定性を回復させる。ここで、細胞は、組換え製造又は遺伝子発見のために使用される。
【0028】
本発明の別の態様において、欠陥ミスマッチ修復遺伝子を有する細胞において、遺伝学的安定性を回復させるための方法が提供される。ミスマッチ修復過程の活性を回復させ、そのゲノムは安定となる。
【0029】
本発明の別の態様において、欠陥ミスマッチ修復活性及び新たに選択された表現型を有する細胞において、遺伝学的安定性を回復させるための方法が提供される。MMR欠損は、ゲノム変異を介した内因性MMR遺伝子の不活化により、又はドミナントネガティブMMR遺伝子対立遺伝子を生成させる真核発現ベクターの導入により、起こりうる。内因性MMR遺伝子の欠陥により内因性MMR遺伝子を欠いている細胞の場合には、細胞が、新たな表現型、又は改変された遺伝子、RNA、もしくはポリペプチドに関して選択される。細胞は、宿主細胞のゲノム欠陥を補足する正常な機能性MMR遺伝子の導入によって、遺伝学的に安定なものとなる。この補足群は、ミスマッチ修復欠損に参与していることが知られている任意の遺伝子の使用を含みうる。ドミナントネガティブミスマッチ修復遺伝子の発現が、DNA超変異性を誘導するために使用される場合には、ドミナントネガティブMMR遺伝子発現は、誘導分子の除去により、又は当業者によって使用されている標準的な遺伝子ノックアウト技術を使用して、ドミナントネガティブ遺伝子対立遺伝子の発現をノックアウトすることにより抑制されるであろう(Waldman,T.ら、Cancer Res 55:5187-5190,1995)。いずれの場合にも、細胞の遺伝学的安定性が回復し、新たな表現型は安定なものとなる。
【0030】
本発明のこれら及びその他の態様は、生物、細胞、及び動物において増強された変異性を作製しうる方法を当分野に提供し、潜在的に有用なゲノム改変を保持している安定な生物、細胞、及び動物を提供する。
【0031】
ドミナントネガティブMMR遺伝子対立遺伝子の使用は、制御された様式で、宿主生物のゲノム全体に全体的な変異を作製する上で重要である。ドミナントネガティブ対立遺伝子の使用は、広範囲の宿主(細菌、酵母、哺乳動物、植物)において全体的な変異誘発を誘導しうることが以前に証明されているが、ゲノムワイドな変異を作製するため、ドミナントネガティブMMR遺伝子変異体をオン状態にした後、新たな生物学的出力形質(例えば、化学変異原に対する耐性)を選択し、遺伝学的安定性を回復させるためドミナントネガティブMMR遺伝子対立遺伝子をオフ状態にするための、誘導可能ベクターの使用は、本発明の新たな局面である。ここで、この方法は、新たな表現型に関与している遺伝子の遺伝学的変異に関してスクリーニングされうる、遺伝学的に多様な原核細胞、真核細胞、及び哺乳動物細胞の作製に適している。さらに、本出願は、誘導可能発現因子の調節下にあるドミナントネガティブMMR遺伝子対立遺伝子の、MMR正常細胞への導入の使用を開示する。次いで、安定的、又は一過性的形質導入された細胞が、ドミナントネガティブMMR遺伝子の発現をもたらす誘導分子へと曝される。ドミナントネガティブ産物の発現は、内因性MMR機構を妨害し、それにより遺伝学的に多様な亜系をもたらす遺伝学的不安定性を引き起こす。次いで、これらの細胞は、特別な選択アッセイ下に置かれ、新たな表現型及び/又は改変された遺伝子構造に関してスクリーニングされる。改変された標的遺伝子及び/又は新たな表現型を含有している亜系の樹立の後、次いで、誘導分子の除去により、細胞が遺伝学的に安定なものとなり、ここで、改変された遺伝子を含有している、かつ/又は新たな表現型を示す安定な細胞系が生成する。この細胞系は、遺伝子発見、薬物標的発見、組換え遺伝子変異誘発、及び/又は組換えタンパク質産生のために使用されうる。
【0032】
MMR欠損生物が、ゲノムワイドな又は遺伝子座特異的な変異誘発の増強されたレベルをもたらすアルキル化剤又は電離放射線のようなDNA損傷剤に対する比較的高い耐性を有することはよく確立されている。本明細書において、本発明者らは、誘導可能発現因子の調節下にあるドミナントネガティブMMRを発現している細胞系の、増強されたゲノムワイドな変異誘発をもたらしうるDNA損傷剤への曝露の使用を開示する。次いで、細胞系は、標的遺伝子内の変異に関してスクリーニングされるか、又は新規表現型に関してスクリーニングされる。次いで、遺伝学的安定性を回復させるため、ドミナントネガティブMMR遺伝子対立遺伝子を「オフ状態」にするため、改変された遺伝子又は表現型を有する亜系は、誘導剤から除去される。この細胞系は、遺伝子発見、薬物標的発見、組換え遺伝子変異誘発、及び/又は組換えタンパク質産生のために使用されうる。
【0033】
最後に、天然にMMRの欠陥を有する哺乳動物細胞系の使用が、関心対象の遺伝子を含有しているプラスミドを導入するために使用されうる。遺伝子を導入し、一過的又は安定的に発現させる。ここで、細胞を増殖させ、構造及び/又は機能の改変を有するものを同定するため、導入された遺伝子の構造及び/又は機能をスクリーニングする。変異速度を増強するため、宿主におけるゲノムワイドな変異の速度を増強するための、アルキル化化学変異原又は電離放射線(これらに限定はされない)のようなDNA損傷剤へとさらに細胞を曝すこともできる。関心対象の遺伝子に変異を含有している(1個以上の)細胞が作製された後、細胞は内因性MMR欠陥を補足する遺伝子で安定的に形質導入される。ここで、細胞系は遺伝学的に安定なものとなり、細胞系は、遺伝子発見、組換え遺伝子変異誘発、及び/又は組換えタンパク質産生のための改変された遺伝子産物の作製に適したものとなる。
【図面の簡単な説明】
【0034】
【図1】PMS2発現ベクター(図1A)及びpCARレポーター(図1B)の図。
【図2A】薬物選択の17日後の、pCARレポーター及びPMS2発現ベクターで同時トランスフェクトされたSH細胞。(図2A)トランスフェクトされていないSH細胞(レーン1)、又はPMS2NOT(レーン2)もしくはPMS2WT(レーン3)でトランスフェクトされたSH細胞に由来する溶解物のウェスタンブロット。矢印は、hPMS2と予想される110kDタンパク質を示す。A及びBは、いずれも、hPMS2のN末端に対して作製された抗体を用いて探索された。Aにおける上方のポリペプチド及びBにおける下方のポリペプチドは、交差反応性ハムスタータンパク質を表す。
【図2B】薬物選択の17日後の、pCARレポーター及びPMS2発現ベクターで同時トランスフェクトされたSH細胞。(図2B)トランスフェクトされていないSH細胞(レーン1)、又はPMS2NOT(レーン2)もしくはPMS2134(レーン3)でトランスフェクトされたSH細胞に由来する溶解物のウェスタンブロット。矢印は、hPMS-134と予想される14kDタンパク質を示す。A及びBは、いずれも、hPMS2のN末端に対して作製された抗体を用いて探索された。Aにおける上方のポリペプチド及びBにおける下方のポリペプチドは、交差反応性ハムスタータンパク質を表す。
【図2C】薬物選択の17日後の、pCARレポーター及びPMS2発現ベクターで同時トランスフェクトされたSH細胞。(図2C)PMS2NOT(レーン1)、PMS2WT(レーン2)、又はPMS2134(レーン3)とレポータープラスミドとで共トランスフェクトされたSH細胞に由来する溶解物におけるβ-ガラクトシダーゼ活性。相対β-ガラクトシダーゼ活性とは、pCARIFでトランスフェクトされた細胞における活性と比較された、pCAROFでトランスフェクトされた細胞におけるβ-ガラクトシダーゼ活性の比として定義される。この規準化により、トランスフェクション効率がコントロールされ、様々なPMS2エフェクター遺伝子を発現している細胞におけるβ-ガラクトシダーゼ活性がコントロールされた。
【図3】PMS2WT発現ベクター(図3A)又はPMS2134発現ベクター(図3B)で安定的に形質導入され、次いでpCAROFで再トランスフェクトされたSH細胞のプールされたクローンのインサイチューβ-ガラクトシダーゼ活性。薬物選択の17日後に、コロニーをプールし、培養し、β-ガラクトシダーゼ活性に関して染色した。pCAROFに由来するβ-ガラクトシダーゼを発現しているPMS2134形質導入SH細胞のプールされた培養物が、図3Bに可視である。図示された各視野は、3連の実験において見出されたものの代表である。
【図4】ドミナントネガティブミスマッチ修復遺伝子対立遺伝子を含有している誘導可能哺乳動物発現ベクターの作製。C末端にV5エピトープを含む、又は含まないPMS134 cDNAを、エクジソンステロイドにより制御されたpIND哺乳動物発現ベクターへとクローニングした。PMS134 cDNAは、熱ショック最小プロモーターに対しセンス方向でベクターの唯一のBamHI部位へクローニングされた。得られたベクターは、それぞれ、pINDPMS134V5又はpINDPMS134と呼ばれる。pINDベクターは、選択可能マーカーとしてネオマイシン耐性遺伝子を含有している。
【図5】PMS134の誘導による改変された遺伝子配列の作製。オープンリーディングフレームを崩壊させ、フレームシフトを生じさせるポリCAリピートをβ-gal遺伝子のN末端に含むβ-ガラクトシダーゼレポータープラスミドを含有しているpCAR-OFプラスミドで、空のpINDベクター又はpINDPMS134を含有している細胞をトランスフェクトした。プラスミドは、安定な系を選択するためのハイグロマイシン耐性遺伝子も含有している。G418/ハイグロマイシン耐性であった細胞を、1μMエクジソンの存在下又は非存在下で10日間増幅・増殖させた。14日後、機能性β-galを産生した増殖巣に関して、細胞をインサイチューで染色した。示されているように、ステロイド誘導剤の存在下で増殖させた細胞においては相当数(1視野当たり25個)のβ-gal陽性増殖巣が観察されたが、誘導剤分子の非存在下で増殖させた培養物においてはほとんど観察されなかった。
【図6】選択後の遺伝学的に安定な細胞の再樹立。化学誘導剤の除去(及びその後のドミナントネガティブMMR対立遺伝子の抑止)の後、クローンが遺伝学的に安定であるか否かを決定するため、pINDPMS134/pCAR-OFクローンを単離し、機能性β-gal活性に関して試験した。β-gal発現を有するクローンを、クローンを含むウェルが1ディッシュ当たり45個となるよう限界希釈して96穴プレートに播いた。クローンを、再度、エクジソンの存在下又は非存在下で14代(1代/日)にわたり増殖させ、β-gal活性に関してインサイチューで染色した。示されているように、ステロイド誘導剤の非存在下で増殖させた細胞においては相当数のβ-gal陽性クローンが観察されたが(45ウェル中42ウェルがβ-gal陽性であった)、定常的な誘導剤曝露下ではより多数のクローンがβ-gal活性を失った(45ウェル中18ウェルがβ-gal陽性であった)。これらのデータは、MMRの遮断によって、インビボで遺伝学的に改変されたクローンにおいて、遺伝学的安定性を回復させることが可能であることを例証している。
【図7】N末端に挿入されたアウトオブフレームのポリA鎖を有する遺伝学的に改変されたプリンホスホリラーゼ(PNP)遺伝子(ポリPNPと呼ばれる)の図。この遺伝子は、非機能性PNP遺伝子をコードする。ポリA鎖が、欠陥MMRによってランダムに改変された場合、鎖はシフトし、機能性PNP遺伝子の生成を可能にする。PNPは非毒性プロドラッグ9-(β-D-2-デオキシエリスロペントフラノシル)-6-メチル-プリン(MPDと呼ばれる)を毒性6-メチルプリンアナログ(MPと呼ばれる)へと変換しうる。構築物は、ウェスタンブロット分析のため、C末端にヘマグルチニン(HA)タグを有している。
【図8A】MPDへ曝された場合、又は曝されない場合の、ポリPNPを発現しているMMR欠陥細胞又はMMR正常細胞の毒性アッセイ。(図8A)グラフは、MMR正常細胞と対照的に、ポリPNPを発現しているMMR欠陥細胞においては、MPDプロドラッグの存在下で細胞が死滅することを示している。
【図8B】MPDへ曝された場合、又は曝されない場合の、ポリPNPを発現しているMMR欠陥細胞又はMMR正常細胞の毒性アッセイ。(図8B)MMR正常細胞と対照的な、MMR欠陥細胞におけるポリPNP-HAタグ化ポリペプチドの産生を示すウェスタンブロット。
【図9】MPDへ曝された場合、又は曝されない場合の、ポリPNPを発現しているMMR欠陥細胞又はMMR正常細胞の毒性アッセイ法。グラフは、MMR欠陥HCT116細胞(MLH1が遺伝学的に欠損している)において、切断型(非機能性)MLH1 cDNA(pC9MLHstop)でトランスフェクトされたHCT116細胞において見られるような活性型へとポリPNP遺伝子が変換されないという事実により示されるように、機能性MLH1遺伝子の導入によって、細胞の遺伝学的安定性が回復することを示している。これらのデータは、MMR欠損が機能性MMR遺伝子(HCT116/pC9MLH1)によって補足され、従って、改変された遺伝子又は遺伝子座のゲノム完全性が維持されうることを例証している。
【図10】MLH1発現構築物及びポリPNP遺伝子でトランスフェクトされた細胞のウェスタンブロット。(図10A)MLHstop発現ベクター(レーン1)又はMLH1ベクター(レーン2)でトランスフェクトされた細胞に由来する細胞溶解物を溶解させ、HCT116細胞におけるMLH1タンパク質発現に関して探索した。図10Aに示されるように、MLH1全長発現構築物でトランスフェクトされた細胞は、予想分子量のポリペプチド(矢印)を産生した。(図10B)MLHstop発現ベクター(レーン1)又はMLH1ベクター(レーン2)とポリPNP遺伝子とでトランスフェクトされたHCT116細胞に由来する細胞溶解物を溶解させ、PNPタンパク質のC末端のHAタグを検出することができる抗HAモノクローナル抗体を使用してポリPNPに関して探索した。図10Bに示されるように、細胞のゲノム安定性を回復させ、従ってポリPNP遺伝子のゲノム構造を維持した、機能性MLH1 cDNAでトランスフェクトされた細胞とは対照的に、MLHstop発現構築物でトランスフェクトされた細胞は、予想分子量のポリペプチド(矢印)を産生した。
【発明を実施するための形態】
【0035】
[発明の詳細な説明]
本発明者らは、ミスマッチ修復を欠損している細胞を利用することにより、超変異性の細胞を開発し、改変された遺伝子、RNA、ポリペプチドおよび細胞、または新しい表現型を有する生物体全体を創造するための方法を発見した。そのような遺伝子のドミナントネガティブ対立遺伝子は、細胞またはトランスジェニック動物へ導入される場合、DNA修復の効率性が低下することにより自然突然変異の割合が増加し、それにより、細胞または動物を超変異性にする。超変異性細胞または動物は、その後、遺伝子に新しい突然変異を発生させて、宿主細胞または生物体の新しい出力形質を生み出すために利用されうる。本発明者らは、DNAに損傷を引き起こす化学薬品の使用により、ドミナントネガティブミスマッチ修復遺伝子の対立遺伝子を発現している細胞において、超変異性の割合を増幅させうることを示す。本発明者らはまた、改変された遺伝子の選択およびMMR(ミスマッチ修復)を回復することによる宿主細胞または生物体の遺伝子的安定性の回復が、改変された遺伝子、RNA、またはポリペプチドからなる安定的生物学的生産物へと導くことができることも示す。
【0036】
細菌から哺乳動物細胞の範囲に及ぶ細胞におけるタンパク質複合体は、ミスマッチ修復、またはミスマッチプルーフリーディングと呼ばれる過程を実行する。ミスマッチ修復遺伝子とは、そのようなミスマッチ修復複合体のタンパク質の一つをコードする遺伝子である。何か特定の作用機構の理論に結びつける必要はないが、ミスマッチ修復複合体はヌクレオチド塩基の非相補的対合から生じるDNAヘリックスの歪みを検出するものと考えられている。新しい方のDNA鎖における非相補的塩基が切除され、その切除された塩基が古い方のDNA鎖に相補的である適切な塩基に置換される。このようにして、細胞は、DNA複製における誤りの結果として生じる多くの突然変異を排除する。
【0037】
ドミナントネガティブ対立遺伝子は、同じ細胞に野生型対立遺伝子が存在している場合でさえも、ミスマッチ修復の欠損した表現型を引き起こす。ミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子の例には、コドン134において切断突然変異を起こすヒト遺伝子hPMS2-134がある。その突然変異は、この遺伝子の産生物を134番目のアミノ酸の位置で異常に終結させ、その結果として、N末端の133個のアミノ酸を含む短縮されたポリペプチドを生じる。そのような突然変異は、DNA複製後細胞に蓄積される突然変異の割合の増加を引き起こす。ミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子の発現は、野生型対立遺伝子の存在下でさえも、ミスマッチ修復活性の障害を生じる。そのような効果を生じる任意の対立遺伝子を本発明において使用することができる。
【0038】
ミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子を、ヒト、動物、酵母、細菌、または他の生物体の細胞から得ることができる。欠損したミスマッチ修復活性について細胞をスクリーニングすることにより、そのような対立遺伝子を同定することができる。癌を有する動物またはヒト由来の細胞を、欠損したミスマッチ修復についてスクリーニングすることができる。大腸癌患者由来の細胞は特に有用でありうる。ミスマッチ修復タンパク質をコードする任意の細胞由来のゲノムDNA、cDNA、またはmRNAを野生型配列からの変異として分析することができる。ミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子をまた、例えば、hPMS2-134対立遺伝子または他のミスマッチ修復遺伝子の変異体を生成することにより、人工的に作成することができる。部位特異的突然変異誘発の様々な技術を使用することができる。超変異性細胞または動物を発生させることにおける使用について、自然的であろうと人工的であろうと、そのような対立遺伝子の適合性は、それがドミナントネガティブ対立遺伝子であるかどうかを決定するために、1つまたは複数の野生型対立遺伝子の存在下においてその対立遺伝子により引き起こされるミスマッチ修復活性を試験することにより、評価することができる。
【0039】
ミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子を導入された細胞、生物体、または動物は、超変異性になるものと思われる。これは、そのような細胞または動物の自然突然変異の割合がそのような対立遺伝子を含有しない細胞または動物と比較して上昇していることを意味する。自然突然変異の割合の上昇程度は、正常の細胞または動物の自然突然変異の割合の少なくとも2倍、5倍、10倍、20倍、50倍、100倍、200倍、500倍、または1000倍となりうる。
【0040】
本発明の1つの局面に従って、ミスマッチ修復タンパク質のドミナントネガティブ型をコードするポリヌクレオチドを任意の真核細胞またはトランスジェニック動物へ導入する。遺伝子は、ミスマッチ修復複合体、例えば、PMS2、PMS1、MLH1、GTBP、MSH3またはMSH2の部分であるタンパク質をコードする任意のドミナントネガティブ対立遺伝子であってもよい。ドミナントネガティブ対立遺伝子は、天然に存在しているか、または実験室で作成されうる。ポリヌクレオチドはゲノムDNA、cDNA、RNA、または化学的合成ポリヌクレオチドの形であってもよい。ポリヌクレオチドは、常時活性型プロモーターセグメント(限定されるものではないが、CMV、SV40、EF-1□またはLTR配列のような)を含む発現ベクターへ、またはドミナントネガティブミスマッチ修復遺伝子の発現を制御することができる、テトラサイクリン、またはエクジソン/グルココルチコイド誘導性ベクターのような誘導性プロモーター配列へクローニングすることができる。ポリヌクレオチドをトランスフェクションにより細胞へ導入することができる。
【0041】
トランスフェクションとは、ポリヌクレオチドを細胞へ導入する任意の過程である。トランスフェクションの過程を、生きている動物に、例えば、遺伝子治療用のベクターを使って行うことができる、またはインビトロで、例えば、培養における1つまたは複数の単離された細胞の懸濁液を使って行うことができる。細胞は、例えば、ヒトまたは他の霊長類、哺乳動物または他の脊椎動物、無脊椎動物、および原生動物または酵母のような単細胞型生物体から単離された細胞を含む、真核細胞の任意の型であってもよい。
【0042】
一般的に、トランスフェクションは、細胞の懸濁液、または単細胞を使って実行されるものと思われるが、処理された細胞または組織の画分が、トランスフェクトされた細胞を増殖させてかつ使用できるように、そのポリヌクレオチドを組み入れるのに十分である限りは、他の方法もまた適用されうる。そのポリヌクレオチドのタンパク質産物は、細胞において一過性にまたは安定的に発現されてもよい。トランスフェクションについての技術はよく知られている。ポリヌクレオチドを導入するために利用可能な技術は、限定されるものではないが、エレクトロポレーション、形質導入、細胞融合、塩化カルシウムの使用、および対象の細胞との融合のための脂質とともにポリヌクレオチドをパッキングする方法を含む。いったん、細胞をミスマッチ修復遺伝子をトランスフェクトしたならば、細胞を培養において増殖かつ複製することができる。トランスフェクションが安定的な場合には、多くの細胞産生について遺伝子が一貫したレベルで発現されるような、その後に細胞系が生じる。
【0043】
単離された細胞は、ヒトおよび動物の組織から、酵素、例えば、コラゲナーゼまたはトリプシンなどでその組織を前処理するまたはしないのいずれかで、個々の細胞を機械的に分離して取り出し、かつそれらを適当な細胞培養培地へ移行することにより得られる細胞である。そのような単離された細胞は、典型的には、他の細胞型の非存在下で培養される。ミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子の導入について選択される細胞は、初代細胞培養もしくは不死化した細胞系の形態における真核細胞由来であってもよいし、または単細胞型生物体の懸濁液由来であってもよい。
【0044】
ミスマッチ修復タンパク質のドミナントネガティブ型をコードするポリヌクレオチドは、トランスジェニック動物を作成することにより動物のゲノムへ導入されうる。動物は、トランスジェニック動物を作成するために適当な技術が利用可能である任意の種であってもよい。例として、トランスジェニック動物は、例えばウシ、ブタ、ヒツジ、ヤギ、ウマなどの家畜から;例えば、ミルクに組換えタンパク質を発現するウシ、ブタ、またはヤギのような組み換えタンパク質の生産に使用される動物から;または、例えば、マウス、ラット、ハムスター、モルモット、ウサギなどの研究または生産物試験用の実験動物から調製されうる。
【0045】
当技術分野において公知のトランスジェニック動物を作成するための任意の方法を使用することができる。トランスジェニック動物を作成する一つの過程に従って、ポリヌクレオチドは動物の受精卵へ注入され、その注入された卵は、偽妊娠のメスへ置かれる。卵は、ポリヌクレオチドが組み入れられかつ発現している成熟動物へと発育する。受精卵は、卵および偽妊娠のメスと同じ種の供与体動物の精子からインビトロで作成され、偽妊娠のメスは、受精卵を受けて妊娠するようにホルモン処理によって調製される。トランスジェニック動物を作成する代替方法は、ポリヌクレオチドを注入またはトランスフェクションにより胚性細胞へ導入することおよびその胚性細胞を発育胚へ再導入することを含む。この方法では、しかしながら、ポリヌクレオチドが生殖系列細胞へ組み入れられない場合には、その遺伝子は子孫へ伝えられないと思われる。それゆえ、この方法により作成されたトランスジェニック動物は、その遺伝子が動物の生殖細胞へ組み入れられているかどうかを決定するために評価しなければならない。いったん、トランスジェニック動物が作成されると、それらは生殖年齢まで成長することができ、そして交尾して出産し、トランスジェニック動物の群体が維持されうる。
【0046】
いったん、トランスフェクションされた細胞系またはトランスジェニック動物の群体が作成されれば、それを1つまたは複数の対象の遺伝子において新しい突然変異を発生させるために使用することができる。対象の遺伝子は、細胞系もしくはトランスジェニック動物に本来所有されている、または細胞系もしくはトランスジェニック動物へ導入されている任意の遺伝子でありうる。突然変異を導入するためにそのような細胞または動物を使用することの利点は、その細胞または動物が商業的に有益な生物学的生産物を生産しうる遺伝的改変の広いスペクトルをもちうることである。超変異性動物をその後、育て、新しい所望の出力形質(ミルク生産、ペスト抵抗性などのような)について選択することができる。いったん、新しい形質が同定されれば、ドミナントネガティブ対立遺伝子は、当業者に使用される技術によりその対立遺伝子を直接的にノックアウトすること、または所望の形質および安定的ゲノムを有する子孫を選択するためにそのドミナントネガティブ対立遺伝子を欠損している仲間へ交配することにより除去されうる。もう一つの代替法は、CRE-LOX発現系を使用することであり、それにより、いったん新しい出力形質が確立されれば、ドミナントネガティブ対立遺伝子が動物のゲノムからスプライスされる。
【0047】
本発明のもう一つの局面は、宿主のゲノムにおいて突然変異の割合の増幅を引き起こすためのMMRを欠損している細胞(変異された内因性MMR遺伝子による、またはドミナントネガティブMMR遺伝子の導入を通して)および化学的変異誘発物質の使用である。MMR活性の欠損は、DNA損傷試薬の毒性効果に対して、より抵抗性の細胞を作成することで知られていた。本発明は、正常性(proficient)MMR細胞を作成すること;ドミナントネガティブMMR遺伝子の対立遺伝子の発現およびその後DNA変異誘発物質の使用でゲノムの超変異性を増幅することによるミスマッチ修復欠損の利用について開示する。本出願はまた、新しい遺伝的構造および/または新しい表現型を有する細胞を発生させるゲノムの変化の割合を増加させるために本来MMRを欠損している細胞を用いることおよび化学的変異誘発物質の曝露の有用性についても開示する。化学的変異誘発物質は、化学的性質、例えば、アルキル化剤、架橋結合剤などにより分類できる。他のものは本明細書に列挙されていないのだが、以下の化学的変異誘発物質は、本発明に従って有用である。N-エチル-N-ニトロソ尿素(ENU)、N-メチル-N-ニトロソ尿素(MNU)、プロカルバジン塩酸塩、クロラムブシル、シクロホスファミド、メチルメタンスルホン酸(MMS)、エチルメタンスルホン酸(EMS)、硫酸ジエチル、アクリルアミドモノマー、トリエチレンメラミン(TEM)、メルファラン、ナイトロジェンマスタード、ビンクリスチン、ジメチルニトロソアミン、N-メチル-N'-ニトロ-ニトロソグアニジン(MNNG)、7,12-ジメチルベンズアントラセン(dimethilbenz(a)anthracene、DMBA)、酸化エチレン、ヘキサメチルホスホルアミド、ブスルファン。本発明の好ましい局面において、100個の遺伝子ごとに1つの突然変異;1突然変異/1,000遺伝子の範囲の突然変異の割合を与える、突然変異誘発技術が利用される。そのような組み合わせ(MMR欠損および化学的変異誘発物質)の使用により、各特定の試薬によって優先的に誘発されるゲノムの変化(限定されるものではないが、遺伝子のコード領域、遺伝子のイントロンの領域、または5'もしくは3'の基部および/もしくは末端領域の関連内のDNAセグメントの伸展または欠失、点突然変異、改変反復配列のような)の広いアレイでの発生を可能にするものと思われる。
【0048】
突然変異は、例えば、ゲノムDNA、cDNA、メッセンジャーRNA、または対象の遺伝子に関連するアミノ酸の配列を調べることによって、細胞または動物の遺伝子型における変化について分析することにより、検出されうる。突然変異はまた、遺伝子の表現型をスクリーニングすることにより検出されうる。改変された表現型は、突然変異遺伝子によりコードされるタンパク質の電気泳動易動度、分光学的性質、または他の物理学的もしくは構造的特性における変化を同定することにより検出されうる。タンパク質の改変された機能について、インサイチューにおいて、単離された形において、またはモデル系においてスクリーニングすることもできる。限定されるものではないが、タンパク質分泌、薬品耐性、病原体耐性などのような、対象の遺伝子の機能に関連する細胞または動物の任意の性質の変化についてスクリーニングすることができる。
【0049】
本出願のもう一つの発明は、ドミナントネガティブの発現および正常に機能するMMR遺伝子の発現を制御する誘導性ベクターの使用である。本出願は、いったん、新しい出力形質、改変された遺伝子、RNAまたはポリペプチドを示す宿主細胞または生物体が、化学的変異誘発物質の組み合わせの有りまたは無しで、形質選択を通して発生したならば、この細胞または生物体の遺伝的安定なバージョンを確立するために、DNA安定性を回復する、そのようなストラテジーを使用することの有用性について開示する。ドミナントネガティブMMR遺伝子の対立遺伝子を異所性に発現した結果としてのMMR欠損細胞の場合において、MMR活性は、細胞培養または生物体の環境から誘導物質分子を除去することにより、減少または完全に無くなる。さらに、ドミナントネガティブMMR遺伝子の発現は、生殖細胞または体細胞におけるDNAノックアウト技術分野での当業者に標準的な方法を使って、MMR遺伝子の対立遺伝子をノックアウトすることにより、抑制させることができる(Waldman, T.ら、Cancer Res. 55:5187-5190, 1995)。
【0050】
さらにもう一つの発明は、HCT116、DLD-1などのようなMMR欠損細胞系において、それらによる細胞が化学的変異誘発物質で処理され、病原体耐性、薬品耐性などのような新しい出力形質について選択されており、MMR活性を回復することの利用について開示する。細胞は、その後、内因性MMR欠損を補足し、かつ新しい出力形質、改変された遺伝子配列、改変されたRNA発現および/または改変されたタンパク質発現を示す細胞または生物体のDNA安定性を回復する野生型MMR遺伝子のコピーでトランスフェクトされる。
【0051】
上記の開示は、一般的に本発明を説明している。より完全な理解は、以下の明細な実施例を参照することにより得ることができ、実施例は、例示の目的としてのみ本明細書に提供され、本発明の範囲を限定するものではない。
【実施例】
【0052】
実施例1:ミスマッチ修復正常性細胞において超変異性を引き起こすドミナントネガティブミスマッチ修復タンパク質の使用
著明なMMRの欠陥は、2人のHNPCC患者の正常細胞で見出された。この欠陥がインビボで操作的であることは、そのような患者の非腫瘍性細胞においてマイクロサテライトの不安定性の広範な存在により証明された。2人の患者の1人は、hPMS2遺伝子のコドン134での生殖系列切断突然変異(hPMS2134突然変異)を有し、一方、もう一人の患者はhMLH1遺伝子内の小さな生殖系列欠失を有していた(26)。これらのデータは、このように、HNPCC患者の生化学的および生物学的特徴を説明するものとして一般的に信じられている2つ対象のモデルに矛盾した。これらの患者の正常細胞におけるこのMMR欠損についての根拠は不明瞭であり、いくつかの可能性のある解釈が提示された。例えば、関連しているMMR遺伝子の第二の対立遺伝子が未発見の機構を通してこれらの患者の生殖系列において不活性化されているという可能性、または、MMR過程に関与する他の遺伝子の未知の突然変異が既知の生殖系列の突然変異と協力して存在しているという可能性があった。MMR欠損が正常な増殖および発生と矛盾しないことは、マウスでのノックアウト実験から明らかであり、これらの可能性(1、3、6)を支持している。または、変異誘発遺伝子がドミナントネガティブ効果を発揮し、その結果として、対応するMMR遺伝子およびMMR過程に関与するすべての他の遺伝子の野生型対立遺伝子の存在下でさえもMMR欠損を生じるという可能性があった。これらの可能性の間を区別するために、本発明者らは、MMR正常性細胞系においてhPMS2134突然変異によりコードされる切断型ポリペプチドを発現し、かつ細胞のMMR活性に対するその影響を分析した。その結果より、この突然変異体が真にドミナントネガティブの効果を発揮することができ、その結果としてMMR欠損の生化学的および遺伝学的顕在化を生じたことが示された。
【0053】
MMR正常性シリアンハムスターTKts13細胞系(以後、SH細胞と呼ぶ)に、様々なhPMS2発現プラスミドと、MMR活性を評価するためのレポーター構築物とを同時トランスフェクトした。hPMS2発現プラスミドは、正常なhPMS2遺伝子産物または上記の患者において同定された切断型hPMS2遺伝子を含んでいた(それぞれ、PMS2WTおよびPMS2134、図1A)。hPMS2配列を欠いている「空の」ベクター(PMS2NOT、図1A)が追加の対照として果たした。レポーター構築物pCAROF(out of frame)は、ハイグロマイシン耐性遺伝子と、コード領域の5'末端に29個の塩基対のアウトオブフレームポリCA鎖を含むβ-ガラクトシダーゼ遺伝子とを含んでいた。レポーター構築物pCARIF(in frame)は、ポリCA鎖が27個の塩基対である点を除いて一致しており、それゆえ、β-ガラクトシダーゼのリーディングフレームを妨害しなかった(図1B)。pCAROFレポーターは、トランスフェクションに引き続いてフレーム回復突然変異(すなわち、挿入または欠失)が起こらないかぎりは、β-ガラクトシダーゼ活性を生じないであろう。
【0054】
異なるトランスフェクションスキームはSH細胞に対するPMS2134突然変異の効果を評価するために使用された。第一のスキームにおいて、発現ベクターと、レポーターとを共に同時トランスフェクションした。17日間のハイグロマイシンでの選択の結果として100クローン以上含有するプールおよび個々のクローンが生成され、ウェスタンブロット法およびβ-ガラクトシダーゼアッセイ法のために収集された。PMS2WTおよびPMS2134で形質導入されたSH細胞は、ウェスタンブロット上に抗PMS2抗体で評価されたことだが、予想されたサイズのポリペプチドを合成した(図2Aおよび図2B)。予想されたように、事実上、β-ガラクトシダーゼ活性は、pCAROFレポーターと、PMS2NOTとをトランスフェクトしたSH細胞において、観察されなかった(図2C)。しかしながら、PMS2134をトランスフェクトしたSH細胞は、PMS2WTをトランスフェクトした細胞以上にβ-ガラクトシダーゼ活性を有意に発現した(図2C)。これらの結果より、PMS2134構築物によりコードされる切断型ポリペプチドが内因性MMR機構を撹乱させ、その結果として、リーディングフレームを回復する欠失または挿入を生じたことが示唆された。レポーター構築物の多コピーが本発明者らの条件下で形質導入されたため、これらの推定される欠失または挿入の正確な性質を評価することができず、かつ野生型β-ガラクトシダーゼ配列は予想された突然変異体を遙かに越える量があり、直接的シーケンシングによるそれらの証明は除外された。
【0055】
第二のスキームにおいて、SH細胞を各PMS2発現ベクターと、ハイグロマイシン耐性プラスミドpLHL4とを同時トランスフェクトした。100個以上のクローンを含むハイグロマイシン耐性培養物はプールされかつ増量された。これらの培養物は、その後、pCARIFまたはpCAROFレポーターと、ジェネテシン選択が可能な別々のプラスミドとを同時トランスフェクトした。2週間後、ハイグロマイシンおよびジェネテシンの両方に耐性である100個以上のコロニーをそれぞれ含む、プールされた細胞をβ-ガラクトシダーゼ活性を評価するためにXgalで染色した。図3に示されるように、PMS2134をトランスフェクトした培養物(パネルB)は多くの青色の細胞を含んでいたが、一方、PMS2WTをトランスフェクトした培養物(パネルA)においては、事実上、どの細胞も青色ではなかった。各場合において、トランスフェクション効率は、pCARIFを用いる平行トランスフェクションにより調整され、その平行トランスフェクションはまた、様々なPMS2効果遺伝子を発現する細胞のβ-ガラクトシダーゼ活性についての対照としての役割を果たし、すべての場合において類似したβ-ガラクトシダーゼ発現レベルであるという結果であった(示されず)。同様の実験プロトコールをMMR正常性ヒト胚性腎臓細胞系293に適用した場合、PMS2WTトランスフェクションと比較してPMS2134トランスフェクション後のβ-ガラクトシダーゼ活性において増加もまた観察された。これらの細胞は、pCAROFと、様々なPMS2効果プラスミドとを同時トランスフェクトし、ハイグロマイシンで17日間選択した。17日目に、コロニーをβ-ガラクトシダーゼ活性を評価するためにXgalで染色し、β-ガラクトシダーゼ発現細胞について評点をつけた。表1に示されるように、PMS2134ポリペプチドを発現するそれらの細胞のみが検出可能なβ-ガラクトシダーゼ活性を発現した。これらのデータは、げっ歯類系およびヒト系の両方において、類似したhPMS2134タンパク質のドミナントネガティブ効果を証明し、かつこれらの研究においてげっ歯類系の有用性を確証している。
【0056】
(表1) pCAROFレポーター構築物と、PMS2効果プラスミドとをトランスフェクトした293クローンのβ-ガラクトシダーゼ発現。293細胞は、pCAROFβ-ガラクトシダーゼレポータープラスミドと、PMS2NOT、PMS2WT、またはPMS2134効果プラスミドとを同時トランスフェクトした。トランスフェクトされた細胞は、ハイグロマイシンで17日間選択し、β-ガラクトシダーゼ活性のためにXgalで染色した(青色の細胞)。下記の結果は、3連の実験の平均値 +/ 標準偏差を示す。
【0057】
プラスミド
完全長野生型hPMS2 cDNAは、記載されているように、ヒトHeLa cDNAライブラリーから得られた(18)。アミノ酸134位に終止コドンを含むhPMS2 cDNAは、突然変異が発見された患者からRTPCR法により得られた(9)。cDNA断片は、SV40ポリアデニル化シグナルが続いているSV40プロモーターを含むpSG5ベクターへBamHI部位にクローニングされた(8)。図1に記載されているpCARレポーターベクターは、参照文献21および25に記載されているように、構築された。
【0058】
細胞系およびトランスフェクション
シリアンハムスター線維芽細胞Tkts13およびヒトHEK293細胞は、ATCCから得られ、記載されているように培養された(15)。hPMS2を発現する安定的にトランスフェクションされた細胞系は、PMS2発現ベクターおよび3:1(pCAR:pLHL4)の割合でハイグロマイシン耐性遺伝子をコードするpLHL4プラスミドの同時トランスフェクションにより作製され、かつハイグロマイシンで選択された。pCARレポーターを含む安定的にトランスフェクトされた細胞系は、pCARベクターをネオマイシン耐性プラスミドをコードするpNTKプラスミドまたはpLHL4のいずれかとともに同時トランスフェクションにより作製された。すべてのトランスフェクションは、以前に記載されているように、リン酸カルシウムを用いて行われた(15)。
【0059】
β-ガラクトシダーゼアッセイ法
pCARでのトランスフェクションに続いて17日間目に、β-ガラクトシダーゼアッセイ法を45 mM 2-メルカプトエタノール、1 mM MgCl2、0.1 M NaPO4および0.6 mg/ml クロロフェノールレッドβ-D-ガラクトピラノシド(CPRG、Boehringer Mannheim)中の20 gのタンパク質を用いて行った。反応は、1時間インキュベートされ、0.5 M Na2CO3の添加により停止させ、かつ576 nmでの分光測光法により分析された(16)。インサイチューβ-ガラクトシダーゼ染色のために、細胞をPBS中の1 % グルタルアルデヒドで固定化し、0.15 M NaCl、1 mM MgCl2、3.3 mM K4Fe(CN)6、0.2 % Xgal中で2時間、37℃でインキュベートした。
【0060】
ウェスタンブロット法
PMS2についてのウェスタンブロットは、実施例5に記載されているように、ヒト完全長ポリペプチドのコドン1〜20に対して産生したポリクローナルな抗ヒトPMS2を用いて行われた。
【0061】
実施例2:ドミナントネガティブミスマッチ修復遺伝子の対立遺伝子はMMR活性に欠陥を引き起こす
PMS2WTおよびPMS2134をトランスフェクトされた細胞の間でのβ-ガラクトシダーゼ活性における違いについてもっとも可能性が高い解釈は、PMS2134タンパク質がMMR活性を妨害し、その結果として、pCAROFレポーター内の高頻度の突然変異およびORFの再確立を起こすということである。MMRは変化するという仮説を直接的に試験するために、本発明者らは、実施例1に記載されているように、PMS2-WT、PMS2-134またはPMS2-NOTの空のベクターを含む細胞からの個々のクローンでのMMRについて生化学的アッセイ法を使用した。核抽出物は、クローンから調製され、以前に記載されている条件下で、/CA\挿入欠失またはG/Tミスマッチのいずれかを含むヘテロ二重鎖基質とともにインキュベートされた。/CA\およびG/Tのヘテロ二重鎖は、それぞれ、3'方向および5'方向からの修復を試験するために使用された。これらのアッセイにおいて、PMS2-134発現クローンとその他のクローンとの間には劇的な違いがあった(表2A)。すべてのクローンは基質を3'方向から修復したが(/CA\ヘテロ二重鎖)、PMS2134ポリペプチドを発現する細胞は、ほとんど5'修復活性をもたなかった。PMS2134トランスフェクションから生じるプールされたクローンについて、またはヘテロ二重鎖が24塩基対ループを含む場合、ミスマッチ修復における同様の方向上の欠陥は明らかであり、それらの例は表2Bに示されている。3'/CA\PMS2-WT修復アッセイにおいて、MMR活性に少量の減少が観察されたが、おそらく、PMS2タンパク質の過剰発現による生化学的アッセイ法における干渉の結果であろう。インサイチューβ-ガラクトシダーゼアッセイ法において、PMS2-WTにより有意な活性を生じなかったが(図3;表1)、インビボの条件を反映している可能性がより高い結果といえる。
【0062】
(表2) SHクローン(A)またはプールされた培養物(B)由来の核抽出物のミスマッチ修復活性。抽出物を24 fmolのヘテロ二重鎖でのMMR活性について試験した。*これらのデータは、5つ以上の独立した実験から導かれた類似の結果を表している。
【0063】
ミスマッチ修復についての生化学的アッセイ法
核抽出物におけるMMR活性は、記載されているように、24 fmolの基質を用いて行われた(12、25)。相補性アッセイ法は、最終KCl濃度を100 mMへ調整しながら、〜100 ngの精製されたMutLαまたはMutSα成分を100 μgの核抽出物へ添加することにより行われた(4、10、30)。これらの実験に用いられる基質は、ミスマッチまで5'側181ヌクレオチドまたは3'側125ヌクレオチドの鎖切断を含む。値は少なくとも二連で行われた実験を代表している。
【0064】
実施例3:遺伝子座における突然変異を増幅するためのMMR欠損細胞および化学的変異誘発物質の使用
遺伝的突然変異の割合を増幅しかつ改変された遺伝子、RNA発現またはポリペプチドを有する細胞を産生するために、MMR欠損および化学的変異誘発物質の使用は、そのような多様性を生じるための強力な道具である。MMR欠損細胞を使用することの利点は、細胞をそのような化合物の毒性効果に対してなお、より抵抗性にさせているこの活性が減少することにより、宿主生物体または細胞の遺伝子および表現型の変化における増加が見込まれることである。以下の実験は、本発明の有用性を証明するために行われる。限定されるものではないが、HCT116、DLD-1などのようなMMRについて遺伝的に欠損している細胞、またはドミナントネガティブ対立遺伝子の異所性発現によりMMR欠損にさせられている実施例1および実施例2に記載されているもののような細胞は、本発明の下で網羅される。簡単に言えば、MMR正常性および欠損性細胞を増殖培地において、化学的変異誘発物質の範囲濃度(1 nm〜1 mM)で、1時間〜24時間、37℃、5% CO2でインキュベートする。インキュベーションが完了した後、化学的変異誘発物質を培地から洗浄し、以前に記載され(Walker, VEら、Mutant Res. 17:371-388, 1999)かつ当業者に知られているように、細胞をヒポキサンチン、アミノプテリン、およびチミンの存在下で増殖させてHPRT突然変異細胞について評点をつける。細胞を10 cmの組織培養皿に1 X 105細胞/mlで播き、増殖培地において14日間、37℃、5 % CO2で増殖させる。14日後、HAT耐性コロニー数は、顕微鏡下においてカウントすることにより測定された。典型的実験により、化学的に処理されたMMR欠損細胞において、対照細胞よりも、有意により多くの数のHAT耐性コロニーが形成されることが明らかにされ、宿主細胞/生物体の内因性遺伝子内に突然変異を増加させる能力を証明するものと思われる。MMR欠損細胞、化学的または電離放射線への曝露を加えた使用はまた、トランスフェクションおよび一過的または安定的細胞系の選択を経て導入された標的遺伝子内にインビボで遺伝的突然変異を増幅させるために用いることができる。形質導入された標的遺伝子内に遺伝的突然変異を増幅させるためのMMR欠損に加えて化学的変異誘発物質の能力を証明するために、本発明者らは、上記の細胞の使用により、pCAROFベクター(実施例1参照)をHCT116細胞へトランスフェクションした。細胞をハイグロマイシン耐性によりpCAR-OF陽性クローンについて選択した。ハイグロマイシン耐性細胞を集密まで増殖させ、100,000個の細胞を10 μMのエチル-メタン-スルホン酸(EMS)アルキル化化合物に8時間曝し、増殖培地へ戻した。その後、細胞を一晩増殖させ、それから、3連の10 cmのディッシュに、1,000細胞/プレートの密度で播いた。細胞を10日間増殖させ、実施例1に記載されている方法を用いて、β-ガラクトシダーゼ活性について評点をつけた。その結果より、その化合物の非存在下で増殖した細胞において、β-ガラクトシダーゼ陽性増殖巣数は92+/-10 /ディッシュであることが示された。対照的に、EMSに曝した細胞は、β-ガラクトシダーゼ陽性細胞数において、有意な増加を生じた(205+/-18)。これらのデータは、インビボで標的遺伝子に遺伝的突然変異を発生させるためのMMR欠損細胞に化学的変異誘発物質を加えた有用性を証明している。この方法は、商業的目的として標的遺伝子に遺伝的多様性を生じるために有用である。
【0065】
実施例4:誘導性ベクターによるドミナントネガティブMMR遺伝子の対立遺伝子を発現するミスマッチ修復欠損細胞の回復されたDNA安定性
ドミナントネガティブMMR遺伝子の対立遺伝子の異所性発現を用いてDNA超変異性を誘発する能力は、遺伝的な多様性サブタイプを有する真核細胞を産生することへの多くの重要な商業的適用がある。以下の実験は、実施例1および実施例2に記載されているようなMMR欠損細胞のゲノムにおける遺伝子的変化をそのMMR正常性を回復することにより永久的に刷り込みする能力を証明している。まず、PMS2-134ドミナントネガティブ対立遺伝子をpcDNA4/TO/PMS134Sと呼ばれる真核の誘導性ベクター系pcDNA4/TO(テトラサイクリン誘導性ベクター)(Invitrogen)、pIND/PMS134Sと呼ばれるpIND/V5-Hisグルココルチコイド誘導性ベクター(Invitrogen)へクローニングした。下記のように、Tk-ts13またはHEK293細胞を各ベクターと、pCAR-OF(選択マーカーとしてハイグロマイシン耐性遺伝子を含む)とを同時トランスフェクトした。空のベクターは、各組み合わせについての対照として使用された。トランスフェクションされた細胞は、ゼオシン/ハイグロマイシン(Z/H)耐性細胞またはネオマイシン/ハイグロマイシン(N/H)耐性細胞について10日間〜14日間選択された。クローンは選び取られ、個々のクローンまたはプールとして増量された。細胞は増量され、誘導化学物質(pcDNA4/TO/PMS134S用に1 μg/mlのテトラサイクリンおよびpIND/PMS134S用に1 μMのエクジソン)の含有または非含有の増殖培地(10 %ウシ胎児血清を添加したDMEM)で、6ウェルの組織培養プレートに1 X 105細胞/mlで播かれた。細胞培養物は収集し、37℃、5 % CO2での培養の24時間後、タンパク質発現を誘導されたPMS2-134についてウェスタン分析により分析した(実施例5に記載されているように)。pIND/PMS134S細胞の抽出物のウェスタン分析により、エクジソンの存在下で増殖された場合、〜17 kdのタンパク質の生成が明らかになったが、一方、エクジソン非含有で増殖されたそれらは検出可能なレベルをもたなかった。誘導性PMS2-134発現を有するクローンは増量され、エクジソンまたはテトラサイクリンの存在下で24時間増殖された。誘導物質の非存在下でウェスタンブロットによりPMS2-134発現の損失の動力学を同定するために、細胞は72時間収集された。これらのデータより、72時間後タンパク質は検出不可能なレベルであることが証明された。
【0066】
誘導性ベクター系を用いて遺伝子的不安定性を誘発する能力を証明するために、pIND/PMS134SまたはpIND/V5-Hisを含む細胞をエクジソンの含有または非含有で14日間増殖させ、記載されているように(MCB論文)、インサイチューでβ-ガラクトシダーゼ活性を測定するために染色した。図5に示されるように、観察可能な青色増殖巣をもたなかった空のベクター対照とは対照的に、エクジソンの存在下で増殖されたpIND/PMS134S細胞において、有意の数の小細胞増殖巣が陽性の青色に着色した(倒立顕微鏡の評価下での観察では25個の細胞/視野)。対照的に、誘発物質の非存在下で増殖されたpIND/PMS134S細胞もpIND/V5-His細胞のどちらも陽性に着色しなかった。これらのデータは、遺伝子においてゲノムの不安定性および遺伝的多様性を発生させるために誘導性ベクターの制御下でドミナントネガティブMMR遺伝子を用いて、改変された生化学的機能および/または新しい表現型を生じる能力を証明している。
【0067】
これらの細胞において、PMS2-134発現を抑圧することでMMR正常性を回復することができることを証明するために、以下の実験を行った。細胞をZ/HまたはN/Hを加えた誘導性培地中で14日間維持した。各クローンまたはプールのサブセットを24ウェルのファルコンのディッシュへ、5X104細胞/mlで播いた。細胞を37℃、5 % CO2で一晩増殖させた。次の日、細胞を以前に記載されているように、β-ガラクトシダーゼ発現についてインビボで染色した(Nicolaidesら、Mol. Cell Biol. 18:1635-1641, 1998)。青色に変化する細胞は、MMR機構においてPMS2-134のドミナントネガティブの効果による内因性MMR活性における減少のために、そのようになった。これらの細胞は、誘導物質分子の存在下または非存在下において、96ウェルのプレートに限界希釈によりサブクローニングされた。誘導物質剤が除去されているクローンにおいて復帰細胞(青色ではない細胞)の数がより少ないことが見出された(クローンが誘導物質での常時曝露下であるプレート(45ウェルのうち18ウェル)とは対照的に、45ウェルのうち42ウェル)場合、PMS2-134発現クローンにおいて回復された遺伝子的安定性が証明された。これらのデータより、ゲノムの安定性および遺伝的進化を制御する能力が、制御されたMMR遺伝子発現を用いて証明される。
【0068】
上記の誘導性MMR遺伝子ストラテジーと組み合わせての実施例3に記載されている化学的変異誘発物質の使用はまた、新しい表現型を有する遺伝的多様性宿主生物体を発生させるための、および/または改変された遺伝子発現の安定的生産のための方法としての適用において仕込まれる。この効果を証明するために、誘導性ドミナントネガティブ発現を含む細胞を誘導物質分子に曝し、かつ引き続いて化学的変異誘発物質または電離放射線に曝す。その後、細胞を誘導物質分子の存在下で増量し、配列分析または生化学的活性により決定されるような新しい表現型および/または改変された遺伝子構造を有する細胞について培養物を選択する。改変された遺伝子または表現型を有する細胞をその後、誘導物質分子からはずし、遺伝子的安定性および表現型を回復させる。
【0069】
トランスフェクション
pIND/PMS134SまたはpcDNA/TO/PMS134S誘導性ベクターを有するエクジソン受容体含有安定的HEK293細胞の産生。HEK293-エクジソンR細胞をリポフェクタミン2000(Gibco/BRL)を用いて、pINDもしくはpcDNA/TOの空のベクターまたはpIND/PMS134SもしくはpcDNA/TO/PMS134Sベクターでトランスフェクションした。細胞は、選択的マーカー耐性について選択され、クローンおよびプールは増量された。その後、安定的系を1 μMのエクジソンに48時間曝し、抽出物を単離し、下記のPMS134ポリペプチドのN末端に特異的な抗血清を用いて、誘導性PMS134発現を有するクローンを同定するためにウェスタンブロット法により分析した。
【0070】
プラスミド
PMS2-134は、pSG5PMS134(実施例1に記載)ベクター由来のBamHI断片として、以下の誘導性発現ベクターへクローニングされた。テトラサイクリン誘導性ベクター(pcDNA4/TO/PMS134S)は、EM-7プロモーターおよびSV40ポリAの配列の制御下のゼオシン(zeocin)選択的マーカーを含む。プラスミドの構造はエンドヌクレアーゼ制限分析およびシーケンシングにより確認された。グルココルチコイド誘導性ベクター(pIND/PMS134S)は、SV40初期プロモーターおよびポリAの配列の制御下のネオマイシン選択的マーカーを含む。ベクターの模式図は図4に示されている。
【0071】
トランスフェクション
誘導性発現ベクターは、単独、または実施例1に記載されているようなpCAR-OFベクターとの組み合わせのどちらかで上記の方法に従い、Tk-ts13細胞およびHEK293細胞へ同時トランスフェクションされた。記載されているように(参照文献15、Grassoら、J. Biol. Chem. 273:24016-24024, 1998)、細胞をゼオシン/ハイグロマイシン(pcDNA4/TO/PMS134S)またはネオマイシン/ハイグロマイシン(pIND/PMS134S)耐性クローンについて選択された。耐性クローンを選び取る、および/またはプールする、かつタンパク質分析のために増量させた。
【0072】
実施例5:MMR遺伝子の補足遺伝子を発現することによるMMRの回復および遺伝子的安定性の回復および固定されたゲノム構造の確立
限定されるものではないが、HCT116、DLD-1、およびHEC-1-A細胞系(参照文献12、25およびKondoら、J. Biochem. 125:818-825, 1999)のような内因性遺伝子の欠陥による欠損MMR活性を有する細胞の使用が、遺伝子の遺伝的構造を改変して、新規な抗菌剤、生理活性成長因子またはホルモン、改変された抗体構造などのような商業的に実行可能な様々な分子を生産することに役に立つことができる。そのような細胞の有用性および価値は、いったん改変された遺伝子構造が生成されれば、この遺伝子変化の完全性は、機能的補足MMR遺伝子の導入により細胞を遺伝子的に安定させることにより細胞のゲノムに保存されうることである。この例は、遺伝子的に改変された細菌のプリンヌクレオチドホスホリラーゼ(PNP)遺伝子で、その遺伝子のN末端にアウトオブフレームのポリA鎖が挿入されている(ポリPNPと呼ばれる)遺伝子の導入により、MMR欠損細胞において遺伝子的に改変することができ、また、補足MMR遺伝子の直接的発現によりMMR欠損細胞をMMR正常性にする場合、遺伝子的安定性となりうる。ポリPNP遺伝子は非機能的PNP遺伝子としてコードしている。ポリA鎖が欠損MMRのための遺伝子的改変によりランダムに変化する場合、その鎖はランダムに変化して、機能的PNP遺伝子およびポリペプチドの産生が可能になる。PNPは、無毒性の9-(β-D-2-デオキシ-エリスロ-ペント-フラノシル)-6-メチル-プリンのプロドラッグ(MPDと呼ばれる)基質を毒性の6-メチルプリン類似体(MPと呼ばれる)に変換する(Sorscher, EJら、Gene Therapy 1:233-238, 1994)。ポリPNP遺伝子は、抗HA抗体を用いてのウェスタンブロット分析によるコードポリペプチドの検出を容易にするために、C末端に血球凝集素エピトープタグを含むように操作された。ポリPNP遺伝子は、選択のためのハイグロマイシン耐性(Hyg)遺伝子を有するpCEP4発現ベクターへクローニングされた。この遺伝子を示す模式図は図7に示されている。PNPと呼ばれる相同遺伝子群はまた、インフレームポリA鎖がPNP活性についての陽性対照としてその遺伝子のN末端へクローニングされているように作成された。簡単には、MMR欠損HCT116細胞系およびMMR正常性HEK293細胞系をポリPNP、PNP発現ベクター、または空のpCEP4ベクターでトランスフェクションした。その後、細胞はHyg耐性について選択され、クローンは単離された。増量された細胞を漸増的量のMPD(0 μM、1 μM、10 μM、50 μM、100 μM、300 μM)の存在下で10日間増殖させた。処理期間後、細胞を血球計算器およびトリパンブルー排除によりカウントした。図8Aに示されているように、HCT116/ポリPNP細胞を100 μMまたは300 μMのMPD、それぞれの存在下において培養した場合、細胞数に20 %および30 %の減少が観察された。対照的に、MMR正常性HEK293/ポリPNP細胞については、使用されるMPDの最高濃度(300 μM)においてさえ、細胞増殖に減少は観察されなかった。両方の細胞系について、細胞が50 μM〜300 μMのMPDの存在下において増殖する場合、PNPの発現は、その結果として100 %の増殖抑制を生じ、両方の細胞系への変換されたMPの毒性効果を証明した。ウェスタンブロットにより、HAエピトープを含むポリペプチドが実際に、HCT116/ポリPNP細胞において生成されていることが確認され、このように、ポリPNP遺伝子構造が変化して機能的かつ完全長のPNP酵素を産生したことが証明された(図8B)。
【0073】
遺伝子的安定性の回復および改変された遺伝子座の引き続く刷り込みは、実行可能な生物学的生産物を生産するための本出願の重要な発明であり、それらにより、所望の改変された出力形質を有する改変された生体分子、細胞または生物体全体を長期間の使用のために遺伝子的に安定に作成される。化学的変異誘発物質に曝されたまたは曝されていない、かつ所望の遺伝子的変化について選択された、安定的MMR欠損細胞系を産生するために、突然変異内因性MMR遺伝子座の代わりになることができる補足MMR遺伝子の導入が本出願において開示されている。これは、ヒトMutL相同体MLH1(12、24、25)について遺伝子的に欠損しているHCT116細胞を用いる例により証明される。この例において、哺乳動物発現ベクターで、機能的MLH1ポリペプチドについてコードする(pC9MLH1)または未熟な停止コドンを有するMLH1 cDNAについてコードする発現ベクター(pC9MLHstop)が使用される。これらの発現ベクターはこのベクターを含む細胞の選択を可能にするネオマイシン(neo)耐性遺伝子を含む。MMR活性を補足する能力をそうではないMMR欠損細胞において証明するため、および遺伝子座の改変された構造を永久的に刷り込みするために、ポリPNPおよびpC9MLH構築物をHCT116細胞へ同時トランスフェクションした。細胞を10日間、neoおよびHygにおいて選択し、耐性クローンを単離しかつ増量させた。その後、細胞をMPDの存在下で培養し、10日後増殖についてカウントした。図9において証明されているように、MLH1野生型cDNAをトランスフェクトした細胞は、MLHstopをトランスフェクトした細胞とは対照的に、ウェスタン法による測定では、MLH1を発現していた。さらに、細胞を300 μMのMPDの存在下で増殖した場合、MLH1を発現しているそれらの細胞は、培地のみで増殖した細胞と比較して、全細胞増殖において2 %の減少を示し、一方、MLHstopまたは空のベクターおよびポリPNPをトランスフェクトした細胞は、培地のみで増殖した細胞と比較して、細胞増殖において35 %の低下があった。これらのデータは、異所的に発現されている野生型MMR遺伝子またはcDNAでMMR欠陥を補足することにより、MMR欠損細胞系のゲノム安定性を確立し、かつ生物学的活性または不活性のPNPのような、新しい出力形質および/または改変されたゲノムまたはポリペプチド構造を生産するために、選択された長期間安定的系を確立することができることを証明している。
【0074】
プラスミド
完全長野生型hMLH1 cDNAは、記載されているように、ヒトHela cDNAライブラリーから得られた(18)。終止コドンを含むMLH1 cDNAは、突然変異が発見された患者からRTPCRにより得られた(24)。cDNA断片は、SV40ポリアデニル化シグナル(8)およびネオマイシン耐性についてコードする遺伝子が続くCMVプロモーターを含む、pCEP9ベクター(Invitrogen)のXhoI部位へクローニングされた。pC9MLH1ベクターは完全長機能MLH1タンパク質を産生するが、一方pC9MLH1stopは非機能性の切断型MLH1ポリペプチドを産生する。ポリPNPおよびPNPのベクターは図4に記載されている。ポリPNPは、結果として切断型ポリペプチドを生じる、細菌のPNP遺伝子のコドン2の後に挿入される21塩基のアウトオブフレームポリA鎖を含む(Sorscher, EJら、Gene Therapy 1:233-238, 1994)。PNPは、結果として完全長の機能的に活性なPNPタンパク質を生じる、細菌のPNP遺伝子のコドン2の後に挿入される20塩基のインフレームポリA鎖を含む。ポリPNPおよびPNP遺伝子の両方とも、終止コドンが続くC末端にインフレームで融合された血球凝集素(HA)エピトープを有する。ポリPNPおよびPNP遺伝子は、センスプライマー:
を用いてポリメラーゼ連鎖反応により構築され、ポリA鎖は下線を施されているが、PNP用のプライマーはポリA鎖において1つ少ないAを含む。両方の構築物についてのアンチセンスプライマーは、
である。DH5α細菌のDNAを増幅のための鋳型として使用した。改変されたPNP遺伝子は、以前に記載されているように、緩衝液中、95℃を30秒間、54℃を1分間、72℃を1分間の25サイクルで増幅して産生した(19)。増幅されたゲノムの挿入部はTテイル化ベクター(TAクローニング、Invitrogen)へクローニングされ、組換えクローンは正しいヌクレオチド配列を有するベクターを同定するためにシーケンシングされた。その後、PNP断片はTAクローニングベクターポリリンカーからの部位を用いてpCEP4ベクター(Invitrogen)のKpnI-XhoI部位へサブクローニングされた。組換えPNP発現ベクターは、内部プライマー配列を用いる配列の確実性を保証するためにシーケンシングされた。
【0075】
細胞系およびトランスフェクション
ヒトHCT116およびHEK293細胞はATCCから得られ、ベンダーにより提案されているように、10 %ウシ胎児血清を添加したRPMI中で培養された。細胞は、メーカーのプロトコール(Gibco/BRL)に従いリポソームを用いてPNPおよび/またはMLH1発現ベクターでトランスフェクションされた。トランスフェクション、続いてハイグロマイシンでの選択により、空のベクター、PNPまたはポリPNPを発現する安定的にトランスフェクションされた細胞系が産生された。補足的実験として、HCT116細胞をPNP/MLH1、PNP/MLH1stop、ポリPNP/MLH1またはポリPNP/MLH1stopで、各プラスミドの5 μgを用いて1:1の割合で、トランスフェクションし、ハイグロマイシンおよびネオマイシン耐性について選択した。10日後、薬剤耐性コロニーが観察され、分析のために採取した。
【0076】
MPD殺害性アッセイ法
MPD殺害性アッセイ法のために、細胞を2X104 細胞/mlで播き、かつ1 mlの等分量を24ウェルコースター組織培養皿に播いた。殺害性アッセイ法のために、細胞を0 μM、1 μM、10 μM、50 μM、100 μM、および300 μMのMPDに3連で播いた。細胞をトリプシン処理して10日間増殖させ、トリパンブルー排除を用いて血球計算器でカウントした。データは、各研究について平均値+/-SDとして示されている。
【0077】
ウェスタンブロット
カウント後、各0 μM MPDで処理された細胞から同じ細胞数を試料緩衝液(60 mM トリス、pH 6.8、2 % SDS、10 % グリセロール、0.1 M 2-メルカプトエタノール、0.001 % ブロモフェノールブルー)に直接溶解し、5分間煮沸した。タンパク質可溶化液を18 % トリス-グリシンゲル(Novex)上での電気泳動により分離した。ゲルを48 mM トリス、40 mM グリシン、0.0375 % SDS、20 % メタノールにおいてイモビロン-P(Immobilon-P)(Millipore)上へ電気的ブロットし、0.05 % Tween20および5 % 練乳を加えたトリス緩衝生理食塩水において、室温で1時間ブロックした。フィルターをヒトMLH1または血球凝集素(HA)(Boehringer Manheim)に対して産生されたモノクローナル抗体(αMLH14)および西洋わさびペルオキシダーゼ結合型ウサギ抗-マウス第二次抗体で探索し、検出のために化学ルミネッセンス(Pierce)を使用した。マウスIgGは、第一次抗体の非特異的抗体との相互作用について評価し、かつ使用された抗血清が期待されたタンパク質を検出していることを保証するために、すべての実験について対照として使用された。
【0078】
参照文献
【特許請求の範囲】
【請求項1】
関心対象の遺伝子において変異を作製するための方法であって、
関心対象の遺伝子と、誘導可能転写制御因子の調節下にあるミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子とを含む超変異性哺乳動物細胞を増殖させる工程;
関心対象の遺伝子が変異を保持しているか否かを決定するため、細胞を試験する工程;及び
ドミナントネガティブ対立遺伝子の発現を減少させることにより、ミスマッチ修復活性を細胞に回復させる工程を含む、方法。
【請求項2】
試験工程が、関心対象の遺伝子のヌクレオチド配列を分析することを含む、請求項1記載の方法。
【請求項3】
試験工程が、関心対象の遺伝子から転写されたmRNAを分析することを含む、請求項1記載の方法。
【請求項4】
試験工程が、関心対象の遺伝子によりコードされたタンパク質を分析することを含む、請求項1記載の方法。
【請求項5】
試験工程が、細胞の表現型を分析することを含む、請求項1記載の方法。
【請求項6】
哺乳動物細胞が、細胞が超変異性となるよう、ミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子を含むポリヌクレオチドを哺乳動物細胞へ導入する過程により作成される、請求項1記載の方法。
【請求項7】
リーティングフレームシフトを引き起こすポリモノヌクレオチド鎖(tract)が介在しているレポーター遺伝子が、哺乳動物細胞へ導入され、超変異性をモニタリングさせる、請求項6記載の方法。
【請求項8】
哺乳動物において変異を作製するための方法であって、
誘導可能転写制御因子の調節下にあるミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子を含む哺乳動物1個体以上を誘導条件下で成長させる工程;
成長工程中に獲得された新たな形質を有する哺乳動物1個体以上を選択する工程;
哺乳動物を非誘導条件に供することにより、遺伝子の安定性を哺乳動物に回復させる工程を含む、方法。
【請求項9】
新たな形質が、ヌクレオチド配列を分析することにより同定される、請求項8記載の方法。
【請求項10】
新たな形質が、mRNAを分析することにより同定される、請求項8記載の方法。
【請求項11】
新たな形質が、タンパク質を分析することにより同定される、請求項8記載の方法。
【請求項12】
新たな形質が、表現型を分析することにより同定される、請求項8記載の方法。
【請求項13】
請求項8記載の方法により作成されたトランスジェニック哺乳動物。
【請求項14】
ミスマッチ修復遺伝子がPMS2である、請求項13記載のトランスジェニック哺乳動物。
【請求項15】
ミスマッチ修復遺伝子がヒトPMS2である、請求項13記載のトランスジェニック哺乳動物。
【請求項16】
対立遺伝子が切断変異を含む、請求項13記載のトランスジェニック哺乳動物。
【請求項17】
対立遺伝子がコドン134に切断変異を含む、請求項15記載のトランスジェニック哺乳動物。
【請求項18】
切断変異が野生型PMS2のヌクレオチド424のチミジンである、請求項17記載のトランスジェニック哺乳動物。
【請求項19】
関心対象の遺伝子において変異を作製するための方法であって、
(a)関心対象の遺伝子と、(b)誘導可能転写制御因子の調節下にあるミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子とを含む哺乳動物細胞を誘導条件下で増殖させる工程;
細胞を変異原と接触させる工程;
改変された遺伝子、改変されたRNA、改変されたポリペプチド、又は改変された表 現型的形質を含む細胞1個以上を選択する工程を含む、方法。
【請求項20】
非誘導条件下で培養することにより、選択された1個以上の細胞におけるドミナントネガティブ対立遺伝子の発現を減少させる工程をさらに含む、請求項19記載の方法。
【請求項21】
ドミナントネガティブ対立遺伝子の発現を、ドミナントネガティブ対立遺伝子の部位特異的変異誘発により減少させる、請求項19記載の方法。
【請求項22】
関心対象の遺伝子において変異を作製するための方法であって、
(a)関心対象の遺伝子と、(b)ミスマッチ修復遺伝子の遺伝学的欠陥とを含む細胞を、変異原で処理する工程;
改変された遺伝子、RNA、ポリペプチド、又は表現型的形質を含む細胞1個以上を選択する工程を含む、方法。
【請求項23】
ミスマッチ修復遺伝子の遺伝学的欠陥が、PMS2に存在する、請求項22記載の方法。
【請求項24】
ミスマッチ修復遺伝子の遺伝学的欠陥が、PMS1に存在する、請求項22記載の方法。
【請求項25】
ミスマッチ修復遺伝子の遺伝学的欠陥が、MLH1に存在する、請求項22記載の方法。
【請求項26】
ミスマッチ修復遺伝子の遺伝学的欠陥が、MSH2に存在する、請求項22記載の方法。
【請求項27】
ミスマッチ修復遺伝子の遺伝学的欠陥が、GTBP/MSH6に存在する、請求項22記載の方法。
【請求項28】
ミスマッチ修復遺伝子の遺伝学的欠陥が、MSH3に存在する、請求項22記載の方法。
【請求項29】
遺伝学的欠陥がドミナントネガティブ変異である、請求項22記載の方法。
【請求項30】
遺伝学的欠陥がドミナントネガティブ変異である、請求項23記載の方法。
【請求項31】
遺伝学的安定性が回復するよう、補足ミスマッチ修復遺伝子を1個以上の選択された細胞に導入する工程をさらに含む、請求項22記載の方法。
【請求項32】
補足ミスマッチ修復遺伝子が、1個以上の選択された細胞において構成性の活性を有する、請求項31記載の方法。
【請求項33】
補足ミスマッチ修復遺伝子が誘導可能に制御される、請求項31記載の方法。
【請求項34】
補足ミスマッチ修復遺伝子がPMS2である、請求項31記載の方法。
【請求項35】
補足ミスマッチ修復遺伝子がPMS1である、請求項31記載の方法。
【請求項36】
補足ミスマッチ修復遺伝子がMLH1である、請求項31記載の方法。
【請求項37】
補足ミスマッチ修復遺伝子がMSH2である、請求項31記載の方法。
【請求項38】
補足ミスマッチ修復遺伝子がGTBP/MSH6である、請求項31記載の方法。
【請求項39】
補足ミスマッチ修復遺伝子がMSH3である、請求項31記載の方法。
【請求項40】
補足ミスマッチ修復遺伝子が、ミスマッチ修復正常細胞との細胞−細胞融合により、1個以上の選択された細胞に導入される、請求項31記載の方法。
【請求項41】
細胞のミスマッチ修復活性を測定するための方法であって、
ポリモノヌクレオチド鎖の下流の遺伝子のリーティングフレームを崩壊させるポリモノヌクレオチド鎖をコーディング領域に含んでいる遺伝子の細胞における機能(遺伝子の機能は細胞の減少したミスマッチ修復活性と相関している)をアッセイする工程を含む、方法。
【請求項42】
遺伝子のリーティングフレームを崩壊させないポリモノヌクレオチド鎖を遺伝子内に有する細胞が、対照として使用される、請求項41記載の方法。
【請求項43】
細胞がミスマッチ修復の欠陥を有する、請求項41記載の方法。
【請求項44】
ミスマッチ修復正常性(proficient)細胞が対照として使用される、請求項43記載の方法。
【請求項45】
誘導可能転写制御因子の調節下にあるミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子を含む哺乳動物。
【請求項46】
ミスマッチ修復遺伝子がPMS2である、請求項45記載の哺乳動物。
【請求項47】
ミスマッチ修復遺伝子がヒトPMS2である、請求項45記載の哺乳動物。
【請求項48】
対立遺伝子が切断変異を含む、請求項45記載の哺乳動物。
【請求項49】
対立遺伝子がコドン134に切断変異を含む、請求項48記載の哺乳動物。
【請求項50】
切断変異が野生型PMS2のヌクレオチド424のチミジンである、請求項49記載の哺乳動物。
【請求項1】
関心対象の遺伝子において変異を作製するための方法であって、
関心対象の遺伝子と、誘導可能転写制御因子の調節下にあるミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子とを含む超変異性哺乳動物細胞を増殖させる工程;
関心対象の遺伝子が変異を保持しているか否かを決定するため、細胞を試験する工程;及び
ドミナントネガティブ対立遺伝子の発現を減少させることにより、ミスマッチ修復活性を細胞に回復させる工程を含む、方法。
【請求項2】
試験工程が、関心対象の遺伝子のヌクレオチド配列を分析することを含む、請求項1記載の方法。
【請求項3】
試験工程が、関心対象の遺伝子から転写されたmRNAを分析することを含む、請求項1記載の方法。
【請求項4】
試験工程が、関心対象の遺伝子によりコードされたタンパク質を分析することを含む、請求項1記載の方法。
【請求項5】
試験工程が、細胞の表現型を分析することを含む、請求項1記載の方法。
【請求項6】
哺乳動物細胞が、細胞が超変異性となるよう、ミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子を含むポリヌクレオチドを哺乳動物細胞へ導入する過程により作成される、請求項1記載の方法。
【請求項7】
リーティングフレームシフトを引き起こすポリモノヌクレオチド鎖(tract)が介在しているレポーター遺伝子が、哺乳動物細胞へ導入され、超変異性をモニタリングさせる、請求項6記載の方法。
【請求項8】
哺乳動物において変異を作製するための方法であって、
誘導可能転写制御因子の調節下にあるミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子を含む哺乳動物1個体以上を誘導条件下で成長させる工程;
成長工程中に獲得された新たな形質を有する哺乳動物1個体以上を選択する工程;
哺乳動物を非誘導条件に供することにより、遺伝子の安定性を哺乳動物に回復させる工程を含む、方法。
【請求項9】
新たな形質が、ヌクレオチド配列を分析することにより同定される、請求項8記載の方法。
【請求項10】
新たな形質が、mRNAを分析することにより同定される、請求項8記載の方法。
【請求項11】
新たな形質が、タンパク質を分析することにより同定される、請求項8記載の方法。
【請求項12】
新たな形質が、表現型を分析することにより同定される、請求項8記載の方法。
【請求項13】
請求項8記載の方法により作成されたトランスジェニック哺乳動物。
【請求項14】
ミスマッチ修復遺伝子がPMS2である、請求項13記載のトランスジェニック哺乳動物。
【請求項15】
ミスマッチ修復遺伝子がヒトPMS2である、請求項13記載のトランスジェニック哺乳動物。
【請求項16】
対立遺伝子が切断変異を含む、請求項13記載のトランスジェニック哺乳動物。
【請求項17】
対立遺伝子がコドン134に切断変異を含む、請求項15記載のトランスジェニック哺乳動物。
【請求項18】
切断変異が野生型PMS2のヌクレオチド424のチミジンである、請求項17記載のトランスジェニック哺乳動物。
【請求項19】
関心対象の遺伝子において変異を作製するための方法であって、
(a)関心対象の遺伝子と、(b)誘導可能転写制御因子の調節下にあるミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子とを含む哺乳動物細胞を誘導条件下で増殖させる工程;
細胞を変異原と接触させる工程;
改変された遺伝子、改変されたRNA、改変されたポリペプチド、又は改変された表 現型的形質を含む細胞1個以上を選択する工程を含む、方法。
【請求項20】
非誘導条件下で培養することにより、選択された1個以上の細胞におけるドミナントネガティブ対立遺伝子の発現を減少させる工程をさらに含む、請求項19記載の方法。
【請求項21】
ドミナントネガティブ対立遺伝子の発現を、ドミナントネガティブ対立遺伝子の部位特異的変異誘発により減少させる、請求項19記載の方法。
【請求項22】
関心対象の遺伝子において変異を作製するための方法であって、
(a)関心対象の遺伝子と、(b)ミスマッチ修復遺伝子の遺伝学的欠陥とを含む細胞を、変異原で処理する工程;
改変された遺伝子、RNA、ポリペプチド、又は表現型的形質を含む細胞1個以上を選択する工程を含む、方法。
【請求項23】
ミスマッチ修復遺伝子の遺伝学的欠陥が、PMS2に存在する、請求項22記載の方法。
【請求項24】
ミスマッチ修復遺伝子の遺伝学的欠陥が、PMS1に存在する、請求項22記載の方法。
【請求項25】
ミスマッチ修復遺伝子の遺伝学的欠陥が、MLH1に存在する、請求項22記載の方法。
【請求項26】
ミスマッチ修復遺伝子の遺伝学的欠陥が、MSH2に存在する、請求項22記載の方法。
【請求項27】
ミスマッチ修復遺伝子の遺伝学的欠陥が、GTBP/MSH6に存在する、請求項22記載の方法。
【請求項28】
ミスマッチ修復遺伝子の遺伝学的欠陥が、MSH3に存在する、請求項22記載の方法。
【請求項29】
遺伝学的欠陥がドミナントネガティブ変異である、請求項22記載の方法。
【請求項30】
遺伝学的欠陥がドミナントネガティブ変異である、請求項23記載の方法。
【請求項31】
遺伝学的安定性が回復するよう、補足ミスマッチ修復遺伝子を1個以上の選択された細胞に導入する工程をさらに含む、請求項22記載の方法。
【請求項32】
補足ミスマッチ修復遺伝子が、1個以上の選択された細胞において構成性の活性を有する、請求項31記載の方法。
【請求項33】
補足ミスマッチ修復遺伝子が誘導可能に制御される、請求項31記載の方法。
【請求項34】
補足ミスマッチ修復遺伝子がPMS2である、請求項31記載の方法。
【請求項35】
補足ミスマッチ修復遺伝子がPMS1である、請求項31記載の方法。
【請求項36】
補足ミスマッチ修復遺伝子がMLH1である、請求項31記載の方法。
【請求項37】
補足ミスマッチ修復遺伝子がMSH2である、請求項31記載の方法。
【請求項38】
補足ミスマッチ修復遺伝子がGTBP/MSH6である、請求項31記載の方法。
【請求項39】
補足ミスマッチ修復遺伝子がMSH3である、請求項31記載の方法。
【請求項40】
補足ミスマッチ修復遺伝子が、ミスマッチ修復正常細胞との細胞−細胞融合により、1個以上の選択された細胞に導入される、請求項31記載の方法。
【請求項41】
細胞のミスマッチ修復活性を測定するための方法であって、
ポリモノヌクレオチド鎖の下流の遺伝子のリーティングフレームを崩壊させるポリモノヌクレオチド鎖をコーディング領域に含んでいる遺伝子の細胞における機能(遺伝子の機能は細胞の減少したミスマッチ修復活性と相関している)をアッセイする工程を含む、方法。
【請求項42】
遺伝子のリーティングフレームを崩壊させないポリモノヌクレオチド鎖を遺伝子内に有する細胞が、対照として使用される、請求項41記載の方法。
【請求項43】
細胞がミスマッチ修復の欠陥を有する、請求項41記載の方法。
【請求項44】
ミスマッチ修復正常性(proficient)細胞が対照として使用される、請求項43記載の方法。
【請求項45】
誘導可能転写制御因子の調節下にあるミスマッチ修復遺伝子のドミナントネガティブ対立遺伝子を含む哺乳動物。
【請求項46】
ミスマッチ修復遺伝子がPMS2である、請求項45記載の哺乳動物。
【請求項47】
ミスマッチ修復遺伝子がヒトPMS2である、請求項45記載の哺乳動物。
【請求項48】
対立遺伝子が切断変異を含む、請求項45記載の哺乳動物。
【請求項49】
対立遺伝子がコドン134に切断変異を含む、請求項48記載の哺乳動物。
【請求項50】
切断変異が野生型PMS2のヌクレオチド424のチミジンである、請求項49記載の哺乳動物。
【図1】

【図2A】

【図2B】
【図2C】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8A】
【図8B】

【図9】
【図10】
【図2A】
【図2B】
【図2C】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8A】
【図8B】
【図9】
【図10】
【公開番号】特開2013−31445(P2013−31445A)
【公開日】平成25年2月14日(2013.2.14)
【国際特許分類】
【出願番号】特願2012−193802(P2012−193802)
【出願日】平成24年9月4日(2012.9.4)
【分割の表示】特願2001−584574(P2001−584574)の分割
【原出願日】平成13年5月14日(2001.5.14)
【出願人】(500238848)ザ ジョンズ ホプキンス ユニバーシティ (1)
【出願人】(503092205)モルフォテック・インコーポレーテッド (1)
【出願人】(304029697)
【出願人】(304029701)
【出願人】(304029723)
【出願人】(304029734)
【出願人】(304029745)
【Fターム(参考)】
【公開日】平成25年2月14日(2013.2.14)
【国際特許分類】
【出願日】平成24年9月4日(2012.9.4)
【分割の表示】特願2001−584574(P2001−584574)の分割
【原出願日】平成13年5月14日(2001.5.14)
【出願人】(500238848)ザ ジョンズ ホプキンス ユニバーシティ (1)
【出願人】(503092205)モルフォテック・インコーポレーテッド (1)
【出願人】(304029697)
【出願人】(304029701)
【出願人】(304029723)
【出願人】(304029734)
【出願人】(304029745)
【Fターム(参考)】
[ Back to top ]