
軟骨疾患修復治療用組成物
【課題】再生能力が極めて低く、一度損傷するとその修復は殆ど不可能に近いと考えられている軟骨の疾患の修復治療を可能にする薬物療法および/または医薬品を提供する。
【解決手段】ホスホジエステラーゼ(PDE)4阻害作用を有するPDE4阻害剤(例えば下式の化合物)を有効成分とする軟骨疾患修復治療用組成物。

【解決手段】ホスホジエステラーゼ(PDE)4阻害作用を有するPDE4阻害剤(例えば下式の化合物)を有効成分とする軟骨疾患修復治療用組成物。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、軟骨疾患修復治療用組成物、さらに詳しくは、ホスホジエステラーゼ(PDE)4阻害作用を有する化合物を有効成分とする、変形性関節症、軟骨形成異常症、変形性椎間板症、半月板損傷等の軟骨疾患を修復治療するための医薬組成物に関する。
【背景技術】
【0002】
軟骨は、骨と共に骨格を形成したり、内臓を保護する役目を持つ、弾性力に富む組織であり、軟骨組織は軟骨細胞とそれを取り囲む軟骨基質とからなる。
軟骨は間葉由来の軟骨芽細胞が分裂増殖しながら周囲に基質を産生したものである。一方、軟骨基質は無定形基質と繊維成分とからなり、それらの組成比率の違いにより、(1)硝子軟骨(関節軟骨、肋軟骨、甲状軟骨等)、(2)線維軟骨(椎間円板、恥骨結合)、(3)弾性軟骨(咽頭蓋軟骨、外耳道軟骨、耳介軟骨等)の3種に分類される[医学大辞典(18版;南山堂発行)第1542頁]。
【0003】
軟骨基質の主成分は、プロテオグリカンおよびコラーゲン(II型、IX型等)であり、プロテオグリカンは軟骨組織特有の膨潤性に関与し、コラーゲン線維は軟骨の張力および剪断力に対する剛性に関与することが知られている。
軟骨基質中のプロテオグリカンでは、コンドロイチン硫酸、ケラタン硫酸等のグルコサミノグリカンが分子量約22万のコア蛋白に結合して巨大分子を形成したものとなっており、グルコサミノグリカンが多くの水分子を水和していることが、軟骨の膨潤性に寄与していると考えられている[ザ・ボーン(The Bone)第4巻第8頁(1994年)]。
【0004】
このうち、関節軟骨は骨組織との移行部に石灰化層を有し、成長が完了した後には、血液の直接的な供給を殆ど受けることなく、軟骨細胞の栄養は関節液から供給される。また、関節軟骨は細胞分化度の高い硝子軟骨から形成されており、再生能力の極度に低いデリケートな組織である。
この関節軟骨の表面を粘稠性の高い滑液が覆い、ヒアルロン酸・蛋白複合体を主役とした潤滑液による潤滑機構が働き、滑らかな関節運動が維持されているが、関節軟骨には所謂耐用年数が存在すると考えられており、老化に伴う関節変化は必発する避けがたい生理的現象である。
【0005】
軟骨の障害に起因する疾患としては、例えば、変形性関節症をはじめとして、軟骨形成異常症、変形性椎間板症、半月板損傷等が知られている。
このうち、変形性関節症は、関節を構成する組織、主に、関節軟骨の退行性変化を基に、骨・軟骨の増殖性変化を来し、最終的には関節の形態が著明に変化するに至る疾患であるが、人口の高齢化に伴って増加しており、中でも膝関節症は病態の進行と共に、立位保持、歩行動作が障害されて日常動作能力(Ability of Daily Life; ADL)の著しい低下を引き起こし、寝たきり状態の原因となることもある。
【0006】
変形性関節症の治療は大きく、保存的治療と手術療法に分けられており、保存的治療として、(1)非ステロイド性消炎鎮痛剤の投与、(2)温熱療法、(3)体重のコントロール、(4)装具療法、(5)ステロイド性消炎鎮痛剤の関節内注入、(6)ヒアルロン酸製剤の関節内注入等が行われている。また、保存療法が奏効しない症例や進行例、末期例に対しては、手術療法として、(1)関節鏡視下洗浄、(2)脛骨高位骨切り術、(3)人工関節置換等が行われている[老化と疾患第10巻、第2号、61〜69頁(1997年)&第6号、第66〜77頁(1997年)]。
一方、PDE4阻害作用を有する化合物は種々知られており、PDE4の作用を阻害することにより、炎症性メディエイターの放出を阻害できる[ジャーナル・オブ・モレキュラー・アンド・セルラー・カーディオロジー(J. Mol. Cell. Cardiol.)12(Suppl. II), S61 (1989年)]。
【0007】
また、PDE4阻害作用を有する化合物が、免疫刺激剤に応答して単核食細胞により放出されるサイトカインであるTNF-αの産生を抑制し、TNF−αによって引き起こされる各種炎症性疾患の治療に有用であることが記載されている〔特表2000−503678号、特表2000−502724号、特表2000−510105号、特表2000−514804号、特表2000−502350号、特表2000−501741号〕。しかし、PDE4阻害剤が軟骨疾患の修復治療に有効であることは全く知られていない。
【発明の概要】
【発明が解決しようとする課題】
【0008】
上述したように、軟骨は再生能力が極めて低く、一度損傷するとその修復は殆ど不可能に近いと考えられており、従来の薬物療法はその進行を抑制する保存的治療に過ぎなかった。従って、その軟骨疾患の修復治療を可能にする薬物療法および/または医薬品の開発が永年求められている。
【課題を解決するための手段】
【0009】
本発明者らは、軟骨細胞においてPDE4が産生されていることを見出し、PDE4阻害作用を有する化合物が、軟骨疾患に対して活性を示すことを知り、研究を重ねたところ、該PDE4阻害作用を有する化合物が軟骨疾患の修復治療に有用であることを見出し、本発明を完成した。
すなわち、本発明は、PDE4阻害剤を有効成分とする軟骨疾患修復治療用組成物を提供するものである。本発明は、特に、軟骨疾患部位に局所的に適用するのに適した製剤、特にマイクロスフェア形態の、軟骨疾患修復治療用組成物を提供するものである。
【0010】
本発明の軟骨疾患修復治療用組成物は、軟骨、とりわけ、再生能力が極度に低い関節軟骨に対し、軟骨基質蛋白の遺伝子発現を増大させることにより、優れた基質産生促進作用を有し、軟骨の修復により、軟骨の疾患を治療することができる。
ここに、軟骨疾患の修復治療とは、軟骨疾患の悪化を抑制するのみならず、疾患、傷害等により、変形・磨耗した軟骨を元の形に修復する治療を意味するものである。
【0011】
本発明の医薬組成物は、PDE4阻害剤を有効成分とし、これに通常の医薬用賦形剤または希釈剤を配合して調製される。好ましい医薬組成物は、PDE4阻害剤と生体内適合性かつ生体内分解性ポリマーとを含有する徐放性の局所投与用の組成物である。該局所投与性組成物は、デポ剤(depot formulation)の形態とするのが好ましく、さらに、マイクロスフェア形態とすることが好ましく、そのマイクロスフェアは注射剤の形態とすることもできる。
【0012】
本発明の医薬組成物において、活性成分として用いられるPDE4阻害剤としては、PDE4阻害活性を有する化合物がすべて含まれ、例えば、特開平5−229987号、特開平9−59255号、特開平10−226685号、欧州公開No.158380、国際公開No.94/25437、米国特許No.5223504、国際公開No.95/4045、欧州公開No.497564、欧州公開No.569414、欧州公開No.623607、欧州公開No.163965、米国特許No.5605914、国際公開No.95/35282、国際公開No.96/215、米国特許No.5804588、米国特許No.5552438、国際公開No.93/9118、国際公開No.96/31485、欧州公開No.459505、国際公開No.97/22585、欧州公開No.738715、国際公開No.91/16314、国際公開No.96/218、国際公開No.97/18208、欧州公開No.158380、国際公開No.99/50270、欧州公開No.260817、国際公開No.98/11113、国際公開No.94/22852、欧州公開No.432856、米国特許No.4193926、国際公開No.98/13348、国際公開No.96/6843、特表2000−503678号(=国際公開No.98/14432)、特表2000−502724号(=国際公開No.98/9961)、特表2000−510105号(=国際公開No.97/40032)、特表2000−514804号(=国際公開No.98/2440)、特表2000−502350号(=国際公開No.97/23457)、特表2000−501741号(=国際公開No.97/2585)等に記載の化合物が挙げられる。
【0013】
本発明の軟骨疾患修復治療用組成物に適したPDE4阻害剤は、トレンズ・イン・ファーマコロジカル・サイエンシーズ(Trends in Pharmacological Sciences)11巻150〜155頁の記載に従って分類したPDE1〜5のうち、PDE4に対して他のPDE(PDE1〜3および5)に対するよりもより強い阻害作用を有する選択的PDE4阻害剤が好ましく、PDE4に対する阻害作用が他のPDEに対するよりも10倍以上強いものが好ましい。より好ましいものは、他のPDEに対するよりも50倍以上、さらに好ましくは100倍以上の阻害作用を有するものである。
好ましいPDE4阻害剤は、アドバンシーズ・イン・サイクリック・ヌクレオチド・リサーチ(Advances in Cyclic Nucleotide Research)10巻、69〜92頁[1979年レイベン・プレス(Raven Press)発行]記載の方法に準じて測定したPDE4阻害活性のIC50が0.1〜1000nM、好ましくは0.1〜100nMの化合物である。より好ましくは、IC50は100nM未満である。
【0014】
選択的PDE4阻害剤の具体例としては、下記構造で示される化合物番号(1)〜(57)の化合物またはその薬理的に許容し得る塩が挙げられる。
【化1】
【化2】
【化3】
【化4】
【化5】
【化6】
【化7】
【化8】
【化9】
【化10】
【化11】
【化12】
【化13】
【化14】
【化15】
【化16】
【化17】
【化18】
【化19】
【化20】
【化21】
【化22】
【化23】
【化24】
【化25】
【化26】
【化27】
【化28】
【化29】
【0015】
PDE4阻害活性を有する化合物は、化学構造上から、次の(A)〜(D)に分けることが出来、それらのうちから適宜選択されるが、本発明におけるPDE4阻害剤としては、(A)および(B)の化合物が好ましく、(A)の化合物がとりわけ好ましい。
(A) ナフタレン骨格またはこれに類似する部分構造を有する化合物[例えば、化合物番号(1)、(2)、(38)、(47)、(52)〜(57)]、
(B) 3−シクロペンチルオキシ−4−メトキシフェニル構造またはこれに類似する部分構造を有する化合物[例えば、化合物番号(6)、(9)、(11)、(12)、(14)、(17)、(19)、(20)、(21)、(24)、(25)、(26)、(27)、(33)、(34)、(35)、(39)、(40)、(44)、(49)、(50)、(51)]、
(C) キサンチン骨格またはこれに類似する部分構造を有する化合物[例えば、化合物番号(5)、(7)、(28)、(29)、(30)、(31)、(32)、(36)、(37)、(41)、(43)、(46)]、および
(D) 上記(A)〜(C)以外の構造を有する化合物[例えば、化合物番号(3)、(4)、(8)、(10)、(13)、(15)、(16)、(18)、(22)、(23)、(42)、(45)、(48)]
【0016】
上記(A)の化合物としては、例えば、下記一般式(I)〜(III)で示される化合物およびそれらの薬理的に許容し得る塩を挙げることができる。
【化30】
(式中、R1およびR2は同一または異なって、水素原子、水酸基、シクロ低級アルキルオキシ基、または置換基を有していてもよい低級アルコキシ基を表すか、或いは、互いに末端で結合して低級アルキレンジオキシ基を形成し、R3は置換基を有していてもよい含窒素複素6員環式基、−OR4およびOR5は同一または異なって、保護されていてもよい水酸基を表す)(特開平5−229987号)
【0017】
【化31】
(式中、R1'およびR2'は同一または異なって水素原子または保護されていてもよい水酸基を表し、R3'およびR4'のいずれか一方が、保護されていてもよい水酸基置換メチル基、他方が水素原子、低級アルキル基または保護されていてもよい水酸基置換メチル基であり、R5'およびR6'は同一または異なって、水素原子、置換されていてもよい低級アルキル基、置換されていてもよいフェニル基または保護されていてもよいアミノ基を表すか、あるいは互いに末端で結合して隣接する窒素原子とともに置換されていてもよい複素環式基を形成している)(特開平9−59255号)
【0018】
【化32】
(式中、Aは式:
【化33】
から選ばれるいずれか1つの基[但し、R1”およびR2”は同一または異なって、水素原子または保護されていてもよいヒドロキシ基、R31は保護されていてもよいヒドロキシメチル基、R32は水素原子、低級アルキル基または保護されていてもよいヒドロキシメチル基、R33は置換基を有していてもよい低級アルキル基、R41は保護されていてもよいヒドロキシメチル基、R42は保護されていてもよいヒドロキシメチル基、点線は二重結合の存在または非存在を表す]を表し、R5”およびR6”は同一または異なって、水素原子または保護されていてもよいアミノ基を表すか、あるいは互いに末端で結合して隣接する窒素原子と共に置換されていてもよい複素環式基を形成している。)(特開平10−226685号)
【0019】
本発明の軟骨疾患修復治療用組成物の有効成分であるPDE4阻害剤としては、(A)の化合物のうち、ナフタレン骨格またはイソキノリン骨格を有する化合物またはその薬理的に許容し得る塩がとりわけ好ましく、化合物番号(1)および(2)の化合物またはその薬理的に許容し得る塩がとりわけ好ましい。
PDE4阻害剤が全身的に作用した場合には、投与量によっては嘔吐や胃酸分泌を引き起こすこともあるため[セルラー・シグナリング(Cellular Signaling)9(3−4)227〜236頁(1997年)]、本発明の軟骨疾患修復治療用組成物は疾患部位の近傍(とりわけ、関節軟骨の近傍)に、局所投与され、全身的な薬物の血中濃度を上昇させず、軟骨疾患部位での薬物濃度を維持するようにするのが望ましい。また、このような目的を達成するためには、徐放性とするのが望ましく、徐放性とすることにより、投与回数を少なくし、患者負担の軽減を図ることもできる。
【0020】
本発明の組成物の好ましい形態としては、局所投与で薬物を徐々に放出するデポ剤(例えばペレット製剤、ゲル製剤、マトリックス製剤、マイクロスフェア製剤、生体内適合性かつ生体内分解性ポリマーの水溶液に薬物を添加して徐放化した製剤、投与時には溶液であるが、生体内に投与されることによってゲルを形成するように設計された製剤、一般に整形外科の領域での使用が報告されている種々基剤に封入した製剤等)が挙げられる。
ペレット剤としては、例えば、末端カルボキシル基がアルコールによりエステル化された乳酸−グリコール酸共重合体微粒子と薬物とを圧縮成型して得られる長期徐放性製剤(特開2001−187749号)等を挙げることができる。
【0021】
ゲル製剤としては、例えば、ジャーナル・オブ・コントロールド・リリース(Journal of Controlled Release) 59 (1999) 77-86記載の、ポリエチレングリコールを化学的に結合させたヒアルロン酸と薬物とをリン酸緩衝液に溶解させたゲル製剤等を挙げることができる。
マトリックス製剤としては、例えば、コラーゲンの粒状物質中または繊維膜中に薬物を含浸した製剤、コラーゲンの粒状物質中または繊維膜の調製中に薬物を添加して含有させた製剤(特開平10−182499号、特開平6−305983号)等を挙げることが出来る。
【0022】
生体内適合性かつ生体内分解性ポリマーの水溶液に薬物を添加して徐放化した製剤としては、例えば、ヒアルロン酸ナトリウムの水溶液に薬物を添加して徐放化した製剤等が考えられる。
投与時には溶液であるが、生体内に投与されることによってゲルを形成するように設計された製剤としては、例えば、ジャーナル・オブ・コントロールド・リリース(Journal of Controlled Release)33 (1995) 237-243 記載の、乳酸−グリコール酸共重合体と薬物とをN−メチル−2−ピロリドンに溶解させた製剤、同 27 (1993) 139-147 記載の、乳酸−グリコール酸共重合体とポリエチレングリコールとのブロック共重合体等の低温では溶液状態で存在するが、体温ではゲルとなる高分子と薬物を含有する製剤を挙げることができる。
【0023】
一般に整形外科の領域で報告されている種々の基剤に封入した製剤としては、例えば、基剤(例えば、水難溶性の生体内適合性かつ生体内分解性ポリマー、ポリメチルメタクリレート、ヒドロキシアパタイト、トリカルシウムホスフェート等)と薬物とを混合して製造される製剤[バイオマテリアルズ(Biomaterials)21巻、2405−2412頁(2000年)、インターナショナル・ジャーナル・オブ・ファーマシューティクス(International Journal of Pharmaceutics)206巻1−12頁(2000年)]を挙げることができる。
軟骨疾患修復治療に要する期間における投与回数を少なくする上からは、局所投与により、有効量のPDE4阻害剤を徐々に障害を有する軟骨の近傍(とりわけ、関節軟骨の近傍)に放出するものが好ましい。
【0024】
デポ剤の内、注射剤の形で局所に投与し易いマイクロスフェアの場合、注射針を通過する粒径であるのが好ましく、粒子径が0.01〜150μm、とりわけ0.1〜100μmの範囲であるものが疾患部位への刺激を抑制できる点では好ましい。
本発明のPDE4阻害剤を有効成分とする軟骨疾患修復治療用組成物は、障害を有する軟骨の近傍(とりわけ、関節軟骨の近傍)に、局所的に投与することを考えれば、投与量を小さくするのが好ましく、組成物(例えばマイクロスフェア製剤)中のPDE4阻害剤の量は0.0001〜80%重量%とするのが好ましく、さらに好ましくは0.001〜50%重量、さらに好ましくは0.01〜50%重量である。有効成分としてのPDE4の投与量は、用いるPDE4阻害剤の種類、対象の体重、年齢、症状、適用部位等に応じて医師により適宜決定されるが、局所適用の場合、通常、疾患部位あたり1ng〜1gの範囲である。
【0025】
本発明の軟骨疾患修復治療用組成物は、PDE4阻害剤と薬理的に許容される賦形剤または希釈剤とから、常法に従って調製されるが、好ましい組成物は、PDE4阻害剤と生体内適合性かつ生体内分解性ポリマーとを配合して調製される。
このうち、水難溶性の生体内適合性かつ生体内分解性ポリマーは、1gを溶解するのに、水(25℃)が1000ml以上必要となるような生体内適合性かつ生体内分解性ポリマーであり、具体的には、ヒドロキシ脂肪酸ポリエステルおよびその誘導体(例えば、ポリ乳酸、ポリグリコール酸、ポリクエン酸、ポリリンゴ酸、ポリ−β−ヒドロキシ酪酸、ε−カプロラクトン開環重合体、乳酸−グリコール酸共重合体、2−ヒドロキシ酪酸−グリコール酸共重合体、ポリ乳酸とポリエチレングリコールとのブロック共重合体、ポリグリコール酸とポリエチレングリコールとのブロック共重合体、乳酸−グリコール酸共重合体とポリエチレングリコールとのブロック共重合体など)、α−シアノアクリル酸アルキルエステルのポリマー(例えば、ポリブチル−2−シアノアクリレートなど)、ポリアルキレンオキサレート(例えば、ポリトリメチレンオキサレート、ポリテトラメチレンオキサレートなど)、ポリオルソエステル、ポリカーボネート(例えば、ポリエチレンカーボネート、ポリエチレンプロピレンカーボネートなど)、ポリオルソカーボネート、ポリアミノ酸(例えば、ポリ−γ−L−アラニン、ポリ−γ−ベンジル−L−グルタミン酸、ポリ−γ−メチル−L−グルタミン酸など)、ヒアルロン酸エステルなどが挙げられ、これらの1種または2種以上を用いることができる。他の使用可能な生体内適合性かつ生体内分解性ポリマーとしては、ヒアルロン酸ナトリウム、コンドロイチン硫酸、コラーゲン、ゼラチン、フィブリン等が挙げられる。
【0026】
水難溶性の生体内適合性かつ生体内分解性ポリマーのうち、特に好ましいものはヒドロキシ脂肪酸のポリエステルである。それらは平均分子量が2000〜約800000の範囲内、より好ましくは2000〜約200000の範囲内のものが特に好適であり、平均分子量が5000〜50000の範囲のものが最も好適である。
また、上記ヒドロキシ脂肪酸のポリエステルのうち、更に好ましいのは、ポリ乳酸、乳酸−グリコール酸共重合体、2−ヒドロキシ酪酸−グリコール酸共重合体である。乳酸−グリコール酸共重合体における乳酸/グリコール酸のモル比は、好ましくは90/10〜30/70、より好ましくは80/20〜40/60であり、2−ヒドロキシ酪酸−グリコール酸共重合体における2−ヒドロキシ酪酸/グリコール酸のモル比は、好ましくは90/10〜30/70、より好ましくは80/20〜40/60である。
【0027】
前記PDE4阻害剤をデポ剤とするには、その形態に応じて、適宜製剤化することができ、必要に応じて、製剤化に先立ち、予めPDE4阻害剤を微粒子化してもよい。
PDE4阻害剤を微粒子化するには、慣用の微粒子製法を適宜使用することができ、ジェットミル粉砕、ハンマーミル粉砕、回転ボールミル粉砕、振動ボールミル粉砕、ビーズミル粉砕、シェーカーミル粉砕、ロッドミル粉砕、チューブミル粉砕等により、物理的に粉砕する粉砕法や、薬物を一旦溶媒に溶解後、pH調整、温度変化、溶媒組成の変更等を行って、晶析させ、遠心分離あるいは濾過等の方法で回収するいわゆる晶析法を採用することができる。
【0028】
本発明の医薬組成物の上記各種製剤を製造するには、PDE4阻害剤に合わせて、既知の製法を適宜適用することができる。
例えば、マイクロスフェア製剤を製造するには次の方法が用いられる。また、PDE4阻害剤が塩を形成しているために、マイクロスフェアへの取り込み率が悪い場合には、酸または塩基を用いて遊離の形に変換した後、マイクロスフェア化してもよい。
【0029】
(1)水中乾燥法:
沸点が水より低く水と非混和性である有機溶媒に水難溶性の生体内適合性かつ生体内分解性ポリマーを溶解させた溶液(水難溶性ポリマー溶液)に薬物を含有させ、かくして得られる有機相を水相中に分散してO/W型エマルションを調製後に、該有機溶媒を除去する方法であり、例えば、特公昭56−19324号、特開昭63−91325号、特開平8−151321号、ジャインらの文献(Kajeev Jainら、"Controlled Drug Delivery by Biodegradable Poly(Ester) Devices: Different Preparative Approaches", Drug Development and Industrial Pharmacy、24(8)巻、703−727頁、1998年)、特開昭60−100516号、特開昭62−201816号、特開平9−221417号、および特開平6−211648号に記載の方法と同様にして行われる。
【0030】
(2)相分離法:
水難溶性の生体内適合性かつ生体内分解性ポリマーの有機溶媒溶液に、薬物もしくは薬物水溶液を溶解・分散させ、これに硬化剤を撹拌下徐々に加え、析出固化させる方法であり、例えば特開昭60−67417号、米国特許第5503851号、米国特許第5000886号、Eur. J. Pharm. Biopharm. 42(1)巻、16〜24頁(1996年)および前記ジャインらの文献に記載の方法と同様にして行われる。
【0031】
(3)噴霧乾燥法:
水難溶性の生体内適合性かつ生体内分解性ポリマーの有機溶媒溶液に、薬物を溶解・分散させるか、または薬物水溶液を分散させ、これをスプレーノズルでスプレードライヤー(噴霧乾燥器)の乾燥室内へ噴霧し、きわめて短時間に噴霧液滴内の有機溶媒を揮発させる方法であり、例えば、特開平1−155942号、特開平5−194200号、特開平5−70363号、特開平8−151321号、特開平9−221417号、米国特許第5922253号、「Spray Drying Handbook」(John Wiley & Sons, New York 1984)、ディージィの文献[Partick B. Deasy、「Microcapsulation and Related Drug Processes」(Marcel Dekker, Inc.、New York 1984)]、および前記ジャインらの文献などに記載の方法と同様にして行われる。
【0032】
(4)溶媒拡散法:
薬物および水難溶解性の生体内適合性かつ生体内分解性ポリマーを溶解させた水混和性有機溶媒の溶液を、保護コロイド剤の水溶液に添加し、攪拌乳化して微粒子を得る方法であり、例えば、特開平5−58882号、特開平9−110678号、インターナショナル・ジャーナル・オブ・ファーマシューティクス(International Journal of Pharmaceutics)187巻、143〜152(1999)に記載の方法と同様にして行われる。
上記「水中乾燥法」では、有機相の形態によって調製法が異なるが、いずれも常法にしたがって行うことができる。該有機相の形態としては以下のものが含まれる。
【0033】
(a)水難溶性の生体内適合性かつ生体内分解性ポリマー溶液に、薬物が直接溶解もしくは分散されている有機相。これを水相中に分散するとO/W型エマルションとなる(特公昭56−19324号、特開昭63−91325号、特開平6−32732号、特開平8−151321号、特開平6−32732号、前記ジャインらの文献など)。
(b)水難溶性の生体内適合性かつ生体内分解性ポリマー溶液に、薬物水溶液が分散されているW/O型エマルションからなる有機相。そのW/O型エマルションを水相中に分散すると、(W/O)/W型エマルションとなる(特開昭60−100516号、特開昭62−201816号、特開平9−221417号、前記ジャインらの文献など)。
【0034】
(c)2種以上の水難溶性の生体内適合性かつ生体内分解性ポリマーを用い、一方のポリマー溶液中に分散されている他方のポリマー溶液中に、薬物が溶解もしくは分散しているO/O型エマルションからなる有機相。そのO/O型エマルションを水相中に分散すると(O/O)/W型エマルションとなる(特開平6−211648号)。
上記いずれの形態の有機相においても常法により、例えば、断続振盪法、プロペラ型またはタービン型撹拌機を用いる混和法、コロイドミル法、ホモジナイザーを用いる方法、超音波照射法などによって、エマルションを形成させる。
【0035】
これらの方法において用いられる有機溶媒としては、ハロゲン化炭化水素系溶媒(塩化メチレン、クロロホルム、四塩化炭素、クロロエタン、ジクロロエタン、トリクロロエタンなど)、脂肪酸エステル系溶媒(酢酸エチル、酢酸ブチルなど)、芳香族炭化水素系溶媒(ベンゼン)、脂肪族炭化水素(n−ヘキサン、n−ペンタン、シクロヘキサンなど)、ケトン系溶媒(メチルエチルケトンなど)、エーテル系溶媒(ジエチルエーテル、ジイソプロピルエーテル、メチルイソブチルエーテルなど)が挙げられる。
上記のエマルションを形成させる際、エマルションを安定化するために乳化剤、例えばアニオン性界面活性剤(オレイン酸ナトリウム、ステアリン酸ナトリウム、ラウリル硫酸ナトリウム等)、非イオン性界面活性剤(ポリオキシエチレンソルビタン脂肪酸エステル[Tween80、Tween60(日光ケミカルズ製)等]、ポリエチレンヒマシ油誘導体[HCO−60、HCO−50(日光ケミカルズ製)]、ポリビニルピロリドン、ポリビニルアルコール、カルボキシメチルセルロース、メチルセルロース、レシチン、ゼラチン等)を水相に添加してもよい。
【0036】
また、PDE4阻害剤に加えて、他の成分を配合する場合は、上記O/W型エマルションを形成する際に、有機溶媒相にそれらを添加するのが好ましい。なお、薬効成分含量の高いマイクロスフェア製剤を得るためには、有機相の調製に際して薬効成分濃度を高くしておく必要があり、この場合、薬効成分の水相への流出を防ぐために、水相中に浸透圧調節剤を添加することもできる(日本特許第2608245号を参照)。
上記のようにして得られるO/W型エマルションを水中乾燥法に付すことにより、エマルションに含まれる有機溶媒を除去してマイクロスフェアを製造する。
【0037】
有機溶媒を除去するには、そのエマルション系を加温したり、減圧下に置くか、または気体を吹きつける方法などの慣用の方法が採用され、たとえば、開放系で溶媒を留去する方法(特公昭56−19324号、特開昭63−91325号、特開平8−151321号、特開平6−211648号)、閉鎖系で溶媒留去する方法(特開平9−221418号)などが採用され得る。また、多量の外水相を用いて溶媒を抽出・除去する方法(日本特許第2582186号)によっても行うことができる。
また次の方法も、PDE4阻害剤の種類に応じて、適宜適用することができる。
【0038】
薬物、生体内分解性ポリマーおよび水と混和する前記ポリマーの良溶媒(溶媒A:アセトン、テトラヒドロフラン等)を含む溶液を、溶媒Aと混和する前記ポリマーの貧溶媒(溶媒B:水、エタノール等)および溶媒Aと混和しない前記ポリマーの貧溶媒(溶媒C:グリセリン等)を含む均一混合液中に添加して乳化することにより、ポリマー溶液が分散相、均一混合液が連続相を形成するエマルションを調製し、分散相から溶媒Aを除去する方法(国際公開No.01/80835 )。
水より沸点の低い有機溶媒(塩化メチレン、酢酸エチル等)および水難溶性ポリマーを含む有機相が水相に乳化したエマルションから水中乾燥法でマイクロスフェアを製造するにあたり、(1)気体分離膜(浸透気化膜、多孔性膜等)を備えた装置を用い、(2)水中乾燥に付されるエマルションを気体分離膜の一方の側に供給し、(3)気体分離膜の他方の側へエマルションに含まれる有機溶媒を留去する方法(国際公開No.01/83594)。
【0039】
さらに、マイクロスフェアを水相中で有機溶媒の沸点以上に加温し(特開2000−239152号)、または高融点添加物でマイクロスフェアを覆った後、加温乾燥する(特開平9−221417号)ことにより、マイクロスフェアに残存する有機溶媒を除去することもできる。
このようにして得られるマイクロスフェアは遠心分離、濾過或いは篩などで回収し、その表面に付着した水相添加物等を洗浄除去し、所望により、マイクロスフェア同士の凝集を防止するために糖あるいは糖アルコール、無機塩等、好ましくは、ラクトース、マンニトール、ソルビトールなどの凝集防止剤を添加した後、凍結乾燥に付す。この際、所望の粒子径のマイクロスフェアを得るために、篩にかけることが好ましく、特にマイクロスフェア製剤を注射剤として用いる場合に、通針性を向上させるために、例えば150μmまたはそれ以下の径で篩過を行うのが好ましい。
【0040】
「相分離法」によってマイクロスフェアを製造するためには、前記水中乾燥法で用いたものと同様の有機溶媒の他、アセトン、アセトニトリル、テトラヒドロフラン、ジオキサンなどの両親媒性溶媒を使用することもできる。これらの有機溶媒を用いた水難溶性ポリマーの有機溶媒溶液にPDE4阻害剤および所望により他の成分、またはそれらの水溶液を加えて溶解または分散させ、有機相を形成させる。この有機相を上記有機溶媒と混和しない溶媒(分散媒)、たとえばシリコンオイル類、流動パラフィン、ゴマ油、大豆油、コーン油、綿実油、ココナッツ油、アマニ油などに撹拌下徐々に添加してO/O型エマルションを形成させる。所望により分散媒には界面活性剤を添加してもよい。このエマルションを冷却して水難溶性ポリマーを固化させる、もしくは加熱して有機相中の溶媒を蒸発させることで水難溶性ポリマーを固化させる。または攪拌下このエマルションに硬化剤、たとえばヘキサン、シクロヘキサン、メチルエチルケトン、オクタメチルシクロテトラシロキサン等を徐々に添加する、もしくは硬化剤にエマルションを徐々に添加することによって、水難溶性ポリマーを析出させることでマイクロスフェアを形成させる。
【0041】
このようにして形成されたマイクロスフェアは遠心分離、濾過あるいは篩などで回収し、その表面に付着した溶媒や添加剤をヘキサンや精製水などで洗浄除去し、所望により通風乾燥、減圧乾燥、凍結乾燥などに付す。あるいは前記水中乾燥法の場合と同様に、凝集防止剤などを添加した後、凍結乾燥に付す。
なお、相分離法における内部有機相の形態としては以下のものが含まれる。
(a)水難溶性の生体内適合性かつ生体内分解性ポリマー溶液に、薬物が直接溶解もしくは分散されている有機相。
(b)水難溶性の生体内適合性かつ生体内分解性ポリマー溶液に、薬物水溶液が分散されているW/O型エマルションからなる有機相。
(c)2種以上の水難溶性の生体内適合性かつ生体内分解性ポリマーを用い、一方のポリマー溶液中に分散されている他方のポリマー溶液中に、薬物が溶解もしくは分散もしくは薬物溶液が分散しているO/O型エマルションからなる有機相。
【0042】
また「噴霧乾燥法」によってマイクロスフェアを製造するには、前記相分離法で用いたものと同様の有機溶媒を用い、これに水難溶性の生体内適合性かつ生体内分解性ポリマーを溶かし、その有機溶媒溶液に、PDE4阻害剤および所望により他の成分を溶解、分散させるか、またはそれら薬物の水溶液を分散させ、その分散液をスプレーノズルを通して噴霧乾燥器内に噴霧して有機溶媒を揮発させてマイクロスフェアを形成させる。
用いられる噴霧乾燥器は市販の製品、例えばパルビス・ミニ・スプレイ(Pulvis Mini Spray)GS31(ヤマト製)、ミニスプレードライヤー(柴田科学製)などがいずれも使用され得る。
【0043】
このようにして得られるマイクロスフェアは水中乾燥法で得られるものと同様に後処理に付されて所望のマイクロスフェア製剤が得られる。
「溶媒拡散法」において、水混和性有機溶媒としてはアセトン、メタノール、エタノール、これらの混合溶媒が挙げることができ、必要に応じて薬物を溶解し得る揮発性溶媒(塩化メチレン、クロロホルム)を加えてもよい。保護コロイド剤としてはポリビニルアルコールを挙げることが出来る。
【0044】
本発明のPDE4阻害剤を有効成分とする軟骨疾患修復治療用組成物をマイクロスフェアの形で疾患部位またはその近傍に投与する場合には、局所的に投与するのが好ましく、局所、とりわけ関節内への注射または埋め込みが好ましい。
マイクロスフェアを注射剤とするには、分散剤を含有する水溶液に本発明で得られるマイクロスフェアを、0.0001〜1000mg/ml、好ましくは0.0005〜800mg/ml、さらに好ましくは0.001〜500mg/mlの濃度となるように分散して調製することができる。
【0045】
用いられる分散剤としては、ポリオキシエチレンソルビタン脂肪酸エステル(Tween80、Tween60(日光ケミカルズ製)など)、ポリエチレンヒマシ油(HCO−60、HCO−50(日光ケミカルズ製)など)等の非イオン界面活性剤、カルボキシメチルセルロースナトリウム等のセルロース系分散剤、アルギン酸ナトリウム、デキストラン、ヒアルロン酸ナトリウムが挙げられる。これらの分散剤は、マイクロスフェアの分散性を向上させ、薬効成分の溶出を安定化させる働きがあり、通常、0.01〜2重量%、好ましくは0.05〜1重量%で添加される。
上記注射剤には、適宜、保存剤(メチルパラベン、プロピルパラベン、ベンジルアルコール、クロロブタノール、ソルビン酸、ホウ酸、アミノ酸、ポリエチレングリコール類など)、等張化剤(塩化ナトリウム、グリセリン、ソルビトール、ブドウ糖、マンニトールなど)、pH調節剤(水酸化ナトリウム、水酸化カリウム、塩酸、リン酸、クエン酸、シュウ酸、炭酸、酢酸、アルギニン、リジンなど)、緩衝剤(リン酸水素ナトリウム、リン酸水素カリウムなど)が配合される。
【0046】
注射剤には、更に、必要に応じて、ステロイド系消炎鎮痛剤、非ステロイド系消炎鎮痛剤を溶解・分散してもよい。ステロイド系消炎鎮痛剤としては、例えば、デキサメタゾン、トリアムシノロン、トリアムシノロンアセトニド、ハロプレドン、パラメタゾン、ヒドロコルチゾン、プレドニゾロン、メチルプレドニゾロン、ベタメタゾン等をあげることができる。また、非ステロイド系消炎鎮痛剤としては、イブプロフェン、ケトプロフェン、インドメタシン、ナプロキセン、ピロキシカム等を挙げることができる。
また、PDE4阻害剤を含有するマイクロスフェアの注射剤は、上記懸濁液のほか、用時調製用に、凝集防止剤およびマイクロスフェアを含有する固形製剤と分散剤および注射用蒸留水を組み合わせた注射剤キットとすることもできる。
【0047】
キットに使用する固形製剤は、凝集防止剤を含む水溶液にマイクロスフェアを懸濁後、凍結乾燥、減圧乾燥、噴霧乾燥等することにより、調製することができ、とりわけ、凍結乾燥で調製するのが好ましい。
固形製剤の製造に際しては、注射用蒸留水への再分散性を向上させるために、凝集防止剤(マンニトール、ソルビトール、ラクトース、ブドウ糖、キシリトール、マルトース、ガラクトース、シュクロース等)を含む水溶液に分散剤を添加することもでき、分散性のよい固形剤とすることができる。必要に応じて、分散剤と共に、ステロイド系消炎鎮痛剤、非ステロイド系消炎鎮痛剤を組合わせた注射剤キットとすることもできる。
【0048】
本発明のPDE4阻害剤を有効成分とする軟骨疾患修復治療用組成物は、各種温血哺乳動物、例えば、ヒト、家畜動物(ウマ、ウシ、ヒツジ、ブタ)、愛玩動物(イヌ、ネコ)等に用いることができ、軟骨疾患修復治療用組成物は、これらの動物における変形性関節症、軟骨形成異常症、変形性椎間板症、半月板損傷等の各種軟骨疾患の修復治療に使用することができ、とりわけ、変形性関節症の修復治療に好適に使用することができる。
【図面の簡単な説明】
【0049】
【図1】ウサギ関節軟骨細胞抽出液をMonoQセファロースカラムクロマトグラフィーで分離した各画分におけるcAMP加水分解活性を示すグラフである(化合物番号(44)存在下:○;化合物番号(44)非存在下:●)。
【図2】化合物番号(1)含有マイクロスフェアまたは非含有マイクロスフェアの存在下での、老齢ウサギ関節軟骨の修復を観察した顕微鏡写真の模写図である。
【図3】ヒト関節軟骨細胞抽出液をMonoQセファロースカラムクロマトグラフィーで分離した各画分におけるcAMPまたはcGMP加水分解活性を示すグラフである。
【図4】図3においてcAMPに対し強い加水分解活性を示した画分(フラクション番号28〜30 に対するPDE4阻害剤の阻害活性(IC50)を示すグラフである。
【図5】実施例1、実施例2および実施例3のマイクロスフェアのインビトロでの薬物溶出特性を示すグラフである。
【図6】化合物番号(1)をラットに静脈内投与した場合の、血漿中濃度の経時変化を示すグラフである。データは平均値±標準偏差(例数:3)で示した。
【図7】実施例1−(5)、実施例2−(2)および実施例3−(2)で得られたマイクロスフェア分散液をラットに皮下注射した場合の血漿中濃度の経時変化を示すグラフである。データは平均値±標準偏差(例数:5)で示した。
【図8】実施例2−(2)で得られたマイクロスフェア分散液をラットに皮下注射した場合の製剤中に残存している化合物番号(1)の経時変化を示すグラフである。データは平均値±標準偏差(例数:5)で示した。
【図9】実施例6−(5)および実施例7−(2)で得られたマイクロスフェア分散液をラットに皮下注射した場合の製剤中に残存している化合物番号(2)の経時変化を示すグラフである。データは平均値±標準偏差(例数:4)で示した。
【発明を実施するための形態】
【実施例】
【0050】
つぎに、実験例、実施例および試験例を挙げて本発明をさらに具体的に説明する。なお、以下の実施例等において用いる化合物番号は前記化学構造式で示した好ましいPDE4阻害剤の具体例の化合物番号を意味する。
実験例1(関節軟骨細胞の基質産生増加)
(試験化合物)
化合物番号(1)(10−5Mまたは10−4M);
化合物番号(2)(10−6Mまたは10−5M);
化合物番号(9)(10−6Mまたは10−5M);
化合物番号(11)(10−6M);
化合物番号(21)(10−6Mまたは10−5M);
化合物番号(27)(10−6Mまたは10−5M);
化合物番号(44)(10−5Mまたは10−4M);
【0051】
(関節軟骨細胞の分離・維持)
NZW系ウサギ(北山ラベス;雄性;4週齡)4羽をエーテル麻酔後、放血致死させ、無菌的に大腿骨側の膝関節を得た。これをグルコース0.2%含有リン酸緩衝液(pH7.2)中に保存し、膝関節部の表層のみをメスで削り取り、グルコース0.2%含有リン酸緩衝液(pH7.2)を入れた50mlチューブに集めた。集められた関節表層をシャーレ上でカミソリを用いてできるだけ細かく切り刻み、10xトリプシン−エチレンジアミンテトラ酢酸(EDTA)・4Na(GIBCO製;カタログ番号15400−054)を含むグルコース0.2%含有リン酸緩衝液(50ml、トリプシン100mg含有、EDTA・4Na 40mg含有;pH7.2)中、37℃で15分間振盪した。振盪後、遠心分離(1,400rpm)により、沈殿物を分取後、グルコース0.2%含有リン酸緩衝液40mlで2回洗浄した。洗浄後の沈殿物に細胞分散用コラゲナーゼ(和光純薬製;カタログ番号034−10533)60mgを溶解した無血清α最小必須培地(MEM:minimum essential medium)(GIBCO製;カタログ番号12571−063)40mlを添加し、これを滅菌済スターラー・バー入りの100mlビーカーに移した。スターラー・バーを回転させながら、37℃のCO2インキュベーターで約1時間コラゲナーゼ処理した。コラゲナーゼ処理物から40μMセルストレーナー(FALCON製;カタログ番号2340)により軟骨片を除去し、除去後の処理物に10%ウシ胎仔血清(FCS:fetal calf serum)含有αMEM培地10mlを添加し、遠心分離(1,400rpm)した。沈殿物を10%FCS−αMEM培地で2回洗浄し、同培地で適当な容量に懸濁し、この懸濁液を1ウェル当たり細胞数2万個となるようにコラーゲンタイプII(和光純薬製;カタログ番号033−13901)コートプレート(48ウェル)に播種した。翌日10%FCS−αMEM培地で培地交換した。
【0052】
(基質産生増加)
上記操作で培地交換後、細胞がコンフルエントになった段階で、検体添加群には試験化合物含有培地(ビヒクルとしてジメチルスルホキシドを0.1%含有)で培地交換した。試験化合物を添加する際に用いる培地には、0.2mMアスコルビン酸含有10%FCS−αMEM培地を用いた。最初の検体含有培地添加日をday1とし、day3に再度同培地で培地交換し、day5まで培養を継続した。一方、検体非添加群には、試験化合物を含有しないこと(ビヒクルのみ含有)以外は全く同一の培地を同時期に添加し、同様に培養を行った。培養終了後、培養物から上清を除去し、パラホルムアルデヒドを4%の濃度に溶解した中性緩衝液0.25mlを添加して2時間細胞を固定した。リン酸緩衝液(pH7.2)1mlで3回洗浄後、軟骨基質(プロテオグリカン)を選択的に着色するアルシアンブルー8GX(シグマ製;カタログ番号A3157)を0.1%濃度に溶解した0.1M塩酸で4時間染色した。染色後、リン酸緩衝液(pH7.2)1mlで3回洗浄し、6M塩酸グアニジン水溶液0.25mlで、軟骨基質を染色したアルシアンブルーを溶解し、その一部を用いて620nmでの吸光度を測定した。この吸光度から染色に用いられたアルシアンブルー量を算出し、この量から基質(プロテオグリカン)の量を推定した。その結果を表1に示す。
【表1】
【0053】
上記表1に示すとおり、PDE4阻害活性を有する試験化合物(1)、(2)、(9)、(11)、(21)、(27)および(44)のすべてに基質産生増加作用が認められた。
実験例2(関節軟骨中に発現するcAMP分解性PDEの分画)
NZW系ウサギ(北山ラベス;雄性;4週齢)4羽をエーテル麻酔後、放血致死させ、無菌的に大腿骨側の膝関節を得た。これをグルコース0.2%含有リン酸緩衝液(pH7.2)中に保存し、膝関節部の表層のみをメスで削り取り、グルコース0.2%含有リン酸緩衝液(pH7.2)を入れた50mlチューブに集めた。集められた関節表層をシャーレ上でカミソリを用いてできるだけ細かく切り刻み、氷冷したリン酸塩緩衝液で洗浄した後、氷冷ホモジナイズ用緩衝液(20mM Tris−HCl、pH7.4、2mM酢酸マグネシウム、0.3mM塩化カルシウム、1mMジチオスレイトール、40μMロイペプチン、1.3mMベンズアミジン、0.2mMフェニルメチルスルホニルフルオリド、1mMアジ化ナトリウム)中でホモジナイザー(Polytoron, Kinematica製)を用いて破砕した。得られたホモジネートを遠心(100000×g、60分間)し、上清を分取した。
【0054】
溶出緩衝液(20mM Tris−HCl、pH7.4、1mM塩化カルシウム、1mMジチオトレイトール、2μMロイペプチン、5mMベンズアミジン)で平衡化したMonoQセファロース高速カラム(Amersham Pharmacia Biotech社製)に、前記で得た上清を供した。カラムを溶出緩衝液20mlで洗浄した後、塩化ナトリウム勾配で蛋白を溶出し、氷冷下1mlずつ分画した。各画分について、cAMPを基質とする加水分解活性(PDE活性)を測定した。
PDE活性の測定は、ラジオラベル核酸法により行った。すなわち、1μMの非標識cAMPおよび22nMの[3H]−cAMP(Amersham Pharmacia Biotech社製)を含むアッセイ用緩衝液(50mM Tris−HCl、pH8.0、5mM塩化マグネシウム、4mM 2−メルカプトエタノール、0.33mg/mlウシ血清アルブミン(脂肪酸不含、シグマ社製)、1mMエチレングリコールビス(β−アミノエチルエーテル)−N,N,N',N'−4酢酸)500μl中に10〜30μlの溶出画分を添加して反応を開始した。薬物非投与群は薬物を添加せず、薬物投与群には1×10−5Mになるように化合物番号(44)を添加した。37℃で30分間保温して反応を行った後、1.5分間煮沸して反応を停止させ、さらに1mg/mlのヘビ毒(Crtalus atrox snake venom)100μlを添加して37℃で30分間保温した。ついで、500μlのメタノールを添加し、反応液をDowexカラム(1x8−400)で処理した後、各溶出液に液体シンチレーションカクテルを加え、ラジオ活性を測定した。その結果を図1に示す。
【0055】
図1に示されるとおり、cAMPに対する強い加水分解活性の4つのピークが認められた。これらの4つのピークは、(1)cAMPを選択的に加水分解すること、(2)そのcAMP加水分解活性がcGMPによる影響を受けないこと、(3)選択的PDE4阻害薬である化合物番号(44)により強く阻害されること、という性質をみたすことから、PDE4に由来するピークであると考えられた。PDE4にはPDE4A、PDE4B、PDE4C、PDE4Dの4つのサブタイプが報告されており(サルドウら、セルラー・シグナリング[Cellular Signaling]、10巻、427〜440頁、1998年)、4つのピークはこれらのサブタイプあるいはそれらから由来するスプラシンング変異体であると考えられた。
【0056】
実験例3(軟骨基質タンパク遺伝子の発現)
最終濃度1×10−4および1×10−5Mの化合物番号(1)の存在下で実験例1と同じ方法によって4日間培養したウサギ膝関節軟骨細胞より、ISOGEN(ニッポンジーン社製)によって抽出した全RNA15μgを4.5μlの滅菌水に溶解した。この溶液と2μlの5×MOPS緩衝液、3.5μlのホルムアルデヒド、10μlのホルムアミドとを混合し、90℃、15分間熱変性した後、ホルムアルデヒド存在下で1%アガロースゲル上で電気泳動した。電気泳動終了後、RNAをナイロン膜(アマシャムファルマシア社製)へキャピラリー法にて一晩トランスファーを行った。UV架橋によりRNAをナイロン膜に固定した後、50mlのハイブリダイゼーション溶液(6×SSC、5×Denhardt's溶液、(50%ホルムアミドはなし)、0.5%SDS、100μg/ml熱変性サケ精子DNA)中にて60℃、2時間プレハイブリダイゼーションを行った。
【0057】
次いでマウスII型コラーゲン遺伝子、ヒトアグリカン(プロテオグリカンを構成する代表的な蛋白)遺伝子のDNAプローブを各々、α[32P]dCTPを用い、ランダムプライムラベリングキットVer.2(アマシャムファルマシア社製)によって放射性標識した。各プローブ1×108dpmを各々、5mlのハイブリダイゼーション溶液と共にプレハイブリダイゼーション処理したナイロン膜に加え、密封した後、60℃で一晩ハイブリダイゼーションを行った。ナイロン膜の洗浄は0.2×SSC、0.2%硫酸ドデシルナトリウムを含む溶液中で、60℃、40分間の洗浄操作を3回繰り返し行った。ナイロン膜オートラジオグラフィーに供し、X線フィルムをLAS−1000(富士写真フィルム社製)によって撮影した。各RNAの相対量をImage Gause(富士写真フィルム社製)によって測定した後、28SRNA(内因性RNA:内部標準)によって補正し、非投与群の遺伝子発現の量を100(%)として、試験化合物投与群の遺伝子発現の量を算出した。その結果を表2に示す。
【表2】
【0058】
上記表2に示すとおり、PDE4阻害薬である試験化合物(1)の添加群においては、II型コラーゲン、アグリカン遺伝子の発現量は濃度依存的に増大していたことから、PDE4阻害薬は関節軟骨細胞に作用し、関節軟骨基質の主要な成分であるII型コラーゲン、アグリカンの遺伝子発現を増大させることにより、軟骨基質の産生を促進することがわかった。
【0059】
実験例4(老齢ウサギ膝関節軟骨基質増加)
(予備飼育)
老齢のJW系ウサギ(北山ラベス;雄性;37週齢)を室温(23±2℃)、湿度(50±20%)で飼育した。市販の餌(オリエンタルバイオ製;CE−2)を自由摂取させた。
(関節軟骨基質増加)
上記ウサギをネンブタール(ダイナボット社製;50mg/kg/ml)を耳静脈に注射し、麻酔した。左膝関節部の毛を剃り、70%水性エタノールで消毒後、検体投与群には、関節内に、実施例2−(3)で製造した検体含有マイクロスフェア分散液250μl(薬物量:2.5mg)を18ゲージの注射針(テルモ製)を用いて注入した。一方、検体非投与群には、関節内に、対照例1−(2)で製造した検体非含有マイクロスフェア分散液250μlを注入した。投与から14日後に、ウサギをネンブタール麻酔下で放血致死させ、膝関節を摘出し、ホルムアルデヒドを10%の濃度に溶解した中性緩衝液で固定後、0.5M EDTA−4Na水溶液による脱灰処理を施し、切片を作成した。切片に軟骨基質(プロテオグリカン)を選択的に着色するアルシアンブルー8GX(シグマ製;A3157)を0.10%濃度に溶解した0.1M塩酸で染色し、検体投与群と検体非投与群の間の染色性を顕微鏡にて比較した。その結果、アルシアンブルーで染色される基質(プロテオグリカン)の層の厚さが検体投与群では検体非投与群の層の厚さの3倍以上となっていた。
【0060】
実験例5(老齢ウサギ膝関節軟骨修復)
(予備飼育)
老齢のJW系ウサギ(北山ラベス;雄性;37週齢)を、ウサギ専用ケージ(C type:W370×D520×H330)中、室温(23±2℃)、湿度(50±20%)で飼育した。市販の餌(オリエンタルバイオ製;CE−2)を自由摂取させた。
(関節軟骨修復)
上記ウサギをネンブタール(アボット社製;50mg/kg/ml)を耳静脈に注射し、麻酔した。左膝関節部の毛を剃り、70%水性エタノールで消毒後、左膝正中靭帯を切開し、大腿骨頭および半月板を露出させ、周囲の血液は滅菌綿で止血した。大腿骨骨頭中間部の窪み(非荷重部)に直径2mm、深さ3mmの穴をドリル(東洋アソシエイツ製;ミスターマイスター)を用いてあけた。滅菌生理食塩水で穿孔中に生じた骨片などを洗浄除去後、関節包、正中靭帯を絹糸で縫合し、滅菌綿で止血、消毒した。9日後、検体投与群には、関節内に、実施例2−(3)で製造した検体含有マイクロスフェア分散液250μl(薬物量:2.5mg)を18ゲージの注射針(テルモ製)を用いて注入した。一方、検体非投与群には、関節内に、対照例1−(2)で製造した検体非含有マイクロスフェア分散液250μlを注入した。投与から14日後に、ウサギをネンブタール麻酔下で放血致死させ、膝関節を摘出し、ホルムアルデヒドを10%の濃度に溶解した中性緩衝液で固定後、0.5M EDTA−4Na水溶液による脱灰処理を施し、切片を作成した。孔の修復度を顕微鏡で観察した。その結果を図2に示す。図2に示されるとおり、検体投与群は検体非投与群に比べ明らかに孔の修復が進んでいることが確認された。
【0061】
実験例6(パパイン誘発変形性膝関節症に対する修復治癒効果)
(予備飼育)
日本白色種ウサギ(北山ラベス;雄性;13週齡)を室温(23±2℃)、湿度(50±20%)で8日間飼育した。飼育期間中、市販の餌(RC4 オリエンタル酵母)を1日約140gの割合で摂取させた。
(修復治癒効果)
上記ウサギにネンブタール(アボット:lot 791102)を耳静脈から注射し、麻酔した。ウサギの両膝部分の毛をかり、70%エタノールで消毒した。両膝に0.8%パパイン(Merck EC3.4.22.2 lot 587644 019)生理食塩水溶液0.5mlを5日間隔で2回注射した。二回目の注射から1週間後、検体投与群(左膝;1群4〜6匹)には、実施例2−(3)で製造したマイクロスフェア分散液(化合物番号(1)の化合物を0.2mgまたは2mg含有)を関節内に注射した。一方、検体非投与群(右膝;1群4〜6匹)には対照例1−(2)で製造した薬物非含有マイクロスフェア分散液を検体投与群の分散液と同量関節内に注射した。また、アルツ投与群(左膝;1群2匹)には、1%ヒアルロン酸ナトリウム水溶液(科研製薬製アルツ)0.3mlを関節内に注射した。一方、アルツ非投与群(右膝;1群2匹)には、0.3mlの生理食塩水を関節内に注射した。アルツ投与群およびアルツ非投与群には、同様の関節内注射を1週間毎に、計4回ずつ行った。最終の注射から4週間後、ウサギをエーテル麻酔下で放血により安楽死させ、膝を摘出し、ホルムアルデヒドを10%の濃度に溶解した中性緩衝液にて固定した。その結果、パパイン処置によって、関節軟骨表面の不整化、ヘマトキシリン・エオジン染色性の低下、軟骨基質の繊維化、関節軟骨細胞の消失などを特徴とする関節軟骨の変性が見られた。アルツ処理では、ヘマトキシリン・エオジン染色で若干の染色性低下の抑制はみられたものの、大幅な回復効果は見られなかった。一方、化合物番号(1)含有マイクロスフェア投与では、上記病理症状が著しく改善した。なお、検体非投与群およびアルツ非投与群のいずれにおいても回復効果は全く認められなかった。
【0062】
実験例7(ヒト関節軟骨中に発現するcAMP分解性PDEの分画)
ヒト変形性関節症患者の手術の際に摘出した膝関節軟骨から軟骨部分をメスで削り取り、氷冷したリン酸緩衝液で洗浄した後、−80℃で保存した。軟骨を−80℃で粉砕後、氷冷ホモジナイド緩衝液[20mM Tris-HCl、pH8.0、1mM エチレングリコールビス(β−アミノエチルエーテル)―N,N,N',N’−テトラ酢酸、1mM ジチオスレイトール、10μg/ml ロイペプチン、5mM ベンズアミジン、0.2mM フェニルメチルスルホニルフルオリド、1mM アジ化ナトリウム、5mM メルカプトエタノール]中で、ホモジナイザー(Kinematica製、Polytoron)を用いて粉砕した。得られたホモジネートを遠心分離(100000×g;30分)して、上清を分取した。
溶出緩衝液[20mM Tris-HCl、pH8.0、1mM エチレングリコールビス(β−アミノエチルエーテル)―N,N,N',N’−テトラ酢酸、1mM ジチオスレイトール、2μg/ml ロイペプチン、5mM ベンズアミジン]で平衡化したMonoQセファロース高速カラム(Amasham Pharmacia Biotech社製)に、上記上清を導通した。カラムを溶出緩衝液20mlで洗浄後、塩化ナトリウム水溶液(濃度勾配:0〜1000mM、70ml)でタンパクを溶出し、氷冷下で1mlずつ分画した。各分画について、cAMP及びcGMPを基質とする加水分解活性(PDE活性)を測定した。
【0063】
PDE活性の測定は、ラジオラベル核酸法により行った。すなわち、1μMの非標識cAMPおよび22nMの[3H]−cAMP(Amasham Pharmacia Biotech製)を含むアッセイ用緩衝液(50mM Tris−HCl、pH8.0、5mM塩化マグネシウム、4mM 2−メルカプトエタノール)500μl中に10〜50μlの溶出画分を添加して反応を開始した。
37℃で30分間保温した後、1.5分間煮沸して反応を停止させ、更に、1mg/mlのヘビ毒(Crtalus atrox snake venom)100μlを添加して、37℃で30分間保温した。ついで、500μlのメタノールを添加し、反応液をDowexカラム(1×8−400)で処理した後、溶液に液体シンチレーションカクテルを加え、ラジオ活性を測定した。結果を図3に示す。
【0064】
図3に示されるように、ヒト変形性関節症患者の軟骨の処理液には、cGMPに対する加水分解活性を有する画分は含まれていないが、cAMPに対する強い加水分解活性を有する画分が含まれていることが判明した。
cAMPに対する強い加水分解活性を有する画分(フラクション番号:28〜30に対するPDE4阻害剤の阻害活性を、前述のラジオラベル核酸法で測定した。試験化合物は、化合物番号(1)、化合物番号(2)、化合物番号(11)、化合物番号(44)、化合物番号(27)である。
実験の結果、該画分の加水分解活性は化合物番号(1)および(2)、特に化合物番号(2)により強く阻害を受けることが確認された。また、Journal of Medicinal Chemistry vol. 42, 1088-1099 (1999)記載の方法で測定した、この画分に対する化合物番号(1)および(2)のIC50は同文献に記載されたPDE4阻害作用のIC50と一致した。結果を図4に示す。
【0065】
実験例8(ヒト関節軟骨細胞における細胞内cAMP上昇)
(関節軟骨細胞の分離)
ヒト関節軟骨(変形性股関節症患者の関節軟骨)をリン酸緩衝液(pH7.2)中に浸し、関節部の表層のみをメスで削り取り、同緩衝液を入れた50mlチューブに集めた。集められた関節表層をシャーレ上でカミソリを用いてできるだけ細かく切り刻み、遠心管に移した。
【0066】
ヒアルロニダーゼ(シグマ社製;カタログ番号H−3506)を1mg/mlの濃度で含むリン酸緩衝液(pH7.2)を遠心管に入れ、37℃で15分間振盪した。遠心分離(2000rpm、5分間)により沈殿物を分取後、トリプシンを0.25%の濃度で含むHank’s balanced salt solution(GIBCO社製;カタログ番号15050−065)に入れ、37℃で30分間振盪した。
遠心分離(2000rpm、5分間)により沈殿物を分取後、沈殿物に細胞分散用コラーゲナーゼ(和光純薬製;カタログ番号034−10533)を0.25%の濃度で、ウシ胎仔血清(GIBCO社製;fetal calf serum;カタログ番号10099−141)を10%の濃度で含有するα最小必須培地(GIBCO社製;カタログ番号12571−063)を添加し、37℃で1晩振盪した。
【0067】
40μmセルストレーナー(FALCON社製;カタログ番号2340)により軟骨片を除去し、除去後のコラーゲナーゼ処理物に、ウシ胎仔血清を10%の濃度で含有するα最小必須培地を添加して遠心分離(1400rpm、10分間)した。
ウシ胎仔血清を10%の濃度で含有するα最小必須培地で沈殿物を3回洗浄後、同培地で適当な容量に懸濁し、この懸濁液を1ウエル当たり細胞数50000個となるようにプレート(48ウエル)に播種した。翌日、ウシ胎仔血清を10%の濃度で含有するα最小必須培地で培地交換した。α最小必須培地は、抗生物質(100U/mlペニシリンG、100μg/ml硫酸ストレプトマイシン)、抗真菌剤(0.25μg/mlアンフォテリシンB)(GIBCO社製;カタログ番号15240−062)を添加したものを用いた。
【0068】
(細胞内cAMP濃度上昇)
培地の交換後、細胞がコンフルエントになった段階で、検体添加群では、PGE2(シグマ社製、カタログ番号P−0409)を1μMの濃度で、検体化合物を10−6M又は10−5M濃度で添加した、ウシ胎仔血清を10%の濃度で含有するα最小必須培地(ジメチルスルホキシド0.1%を含有)で培地交換した。一方、検体非添加群では、PGE2(シグマ社製、カタログ番号P−0409)を1μMの濃度で添加した、ウシ胎仔血清を10%の濃度で含有するα最小必須培地(ジメチルスルホキシド0.1%を含有)で培地交換した。
30分間培養後、培地を捨て、リン酸緩衝液(pH7.2)で洗浄し、50%エタノール水溶液で30分間処理後、エタノールを回収し、エタノール抽出物を蒸発乾固した。これをcAMP EIAシステム(Amasham Pharmacia Biotech社製;カタログ番号RPN225)に添付されたアッセイ用緩衝液に溶解し、同システムでcAMP濃度を測定した。その結果を表3に示す。
【表3】
【0069】
実験例9(ウサギ関節軟骨細胞のIL−1存在下での基質産生)
NZW系ウサギ(北山ラベス;雄性;4週齢)4羽をエーテル麻酔後、放血致死させ、無菌的に大腿骨側の膝関節を得た。これから、グルコースを0.2%含有するリン酸緩衝液(pH7.2)中で、膝関節部の表層のみをメスで削り取り、グルコースを0.2%含有するリン酸緩衝液(pH7.2)を入れたチューブに集めた。集められた関節表層をシャーレ上でカミソリを用いてできるだけ細かく切り刻み、10xトリプシン−エチレンジアミンテトラ酢酸4ナトリウム(EDTA・4Na)(GIBCO社製;カタログ番号15400−054)を添加した、グルコースを0.2%の濃度で含有するリン酸緩衝液(トリプシン100mg、EDTA・4Na40mg含有;pH7.2)50mlに加えて、37℃で15分間振盪した。
振盪後、遠心分離(1400rpm)して沈殿物を分取し、これをグルコースを0.2%含有するリン酸緩衝液(pH7.2)40mlで2回洗浄した。
【0070】
洗浄後の沈殿物に細胞分散用コラーゲナーゼ(和光純薬製;カタログ番号034−10533)60mgを溶解した、抗生物質(200U/mlペニシリンG、200μg/ml硫酸ストレプトマイシン)(GIBCO社製;カタログ番号15140−122)を含有するα最小必須培地(GIBCO社製;カタログ番号12571−063)40mlを添加し、これを滅菌済スターラー・バー入りの100mlビーカーに移した。
スターラー・バーを回転させながら、37℃のCO2インキュベーターで約30分間培養後、デオキシリボヌクレアーゼI(宝酒造製;カタログ番号2210A)を70U/mlとなるように添加し、同条件で、更に30分間培養した。
培養物の上清を別容器に移し取り、残った軟骨片を新たに調製した、コラーゲナーゼ60mgを含み、デオキシリボヌクレアーゼIを70U/mlの濃度で含有するα最小必須培地で、約30分間培養した。
【0071】
先に移し取った培養物上清及び最終培養物から40μmセルストレーナー(Falcon製;カタログ番号2340)で軟骨片を除去し、除去後の培養物に、ウシ胎仔血清(GIBCO社製;fetal calf serum;カタログ番号10099−141)を10%の濃度で含み、抗生物質(200U/mlペニシリンG、200μg/ml硫酸ストレプトマイシン)(GIBCO社製;カタログ番号15140−122)を含有するα最小必須培地(GIBCO社製;カタログ番号12571−063)10mlを添加し、遠心分離(1400rpm,10分間)を行った。
沈殿物を同様のウシ胎仔血清、抗生物質含有α最小必須培地で2回洗浄し、同培地で適当な容量に懸濁し、この懸濁液を1ウエル当たり細胞数20000個となるようにプレート(48ウエル)に播種した。翌日、同培地で培地交換を行った。
【0072】
(基質産生増加)
上記培地交換後、細胞がコンフルエントになった段階で、検体投与群には、1ng/mlのリコンビナントヒトIL−1β(PEPRO TECH製;カタログ番号200−01B)と検体化合物を含有した培地(ジメチルスルホキシドを0.1%の濃度で含有)で培地交換した。この培地にはアスコルビン酸を0.2mMの濃度で含む、前記ウシ胎仔血清、抗生物質含有α最小必須培地を用いた。一方、検体非投与群は、検体化合物を含有しない以外は検体投与群と同様の培地で培地交換した。
最初のIL−1β含有培地添加日をday1とし、day3まで培養した。
【0073】
培養後、培養物から上清を除去し、10%中性緩衝ホルマリン液(和光純薬製;カタログ番号060−01667)0.25mlを添加して30分間細胞を固定した。
固定した細胞をリン酸緩衝液(pH7.2)1mlで3回洗浄後、基質(プロテオグリカン)を選択的に染色するアルシアンブルー8GX(シグマ社製;カタログ番号A3157)を0.1%の濃度で含む0.1M塩酸で4時間染色した。
染色後、リン酸緩衝液(pH7.2)1mlで3回洗浄し、6M塩酸グアニジン水溶液0.25mlで、軟骨基質を染色したアルシアンブルーを溶解した。その一部を用いて、620nmでの吸光度を測定し、この吸光度から染色に用いられたアルシアンブルー量を算出した。このアルシアンブルー量から基質(プロテオグリカン)量を推定した。検体を添加しない場合のプロテオグリカン産生量を100%として、相対比率を示した。結果を表4に示す。
【表4】
【0074】
表4に示すとおり、IL−1の存在下、PDE4阻害作用を有する試験化合物は基質産生を促進した。IL−1は、変形性関節症の滑液や軟骨細胞に発現していること、および軟骨基質であるプロテオグリカン等の基質分解酵素であるMMP(matrix metalloproteinase)の産生や合成を誘導することなどから、軟骨基質分解に重要な役割を果たしていると考えられている(The Journal of Pharmacology and Experimental Therapeutics, vol 277, p1672-1675 (1996);Journal of Biochemistry, vol 123, p431-439 (1998); Arthritis & Rheumatism, vol 44, p585-594 (2001)。従って、上記の結果は本発明組成物の有効成分であるPDE4阻害剤が、IL−1が関与する軟骨基質分解を阻害しうることを示唆している。
【0075】
実験例10(内側半月板部分切除/両側側副靭帯切除により誘発される変形性膝関節症に対する修復治療効果)
(予備飼育)
日本白色ウサギ(雄性;10週齢;1群7匹)を、室温(23±2℃)、湿度(55±15%)で16日間飼育した。飼育期間中、市販の餌(オリエンタルバイオ製;LRC4)を自由摂取させた。
【0076】
(変形性膝関節症治癒)
エーテル麻酔下で、上記ウサギ右膝関節を切開し、内側半月板の1/2部分を両鋭小直剪刀を用いて切除した。同時に両側側副靭帯を切断した。術後筋肉と表皮を縫合し、消毒を行った。施術後2週間後に、エーテル麻酔下、検体投与群(1群:7匹)には、それぞれ,化合物番号(2)1μgを含む実施例7−(1)で製造したマイクロスフェアを関節腔内に投与した。検体非投与群(1群:7匹)には、対照例2で製造した薬物非含有マイクロスフェアを同量投与した。術後6週間後に、もう一度、上記薬物含有及び非含有マイクロスフェアを投与した。施術後、10週間後にエーテル麻酔下で開腹、放血により安楽死させ脛骨近位端を摘出した。摘出した脛骨近位端に墨汁を塗布して10%中性緩衝ホルマリン溶液に浸漬固定した。
【0077】
(実験結果)
ホルマリンで固定した脛骨近位端の余分な墨汁を抜き取り、真上方向から実体顕微鏡(オリンパス社製、モデルSZX12-3111)を用いて脛骨表面の画像を画像解析装置(IMAGING RESEARCH 社製 MCIDイメージングアナライザー)に取込んだ。同解析装置を用いて、内側の墨汁が残存している部分の面積(変性部面積)を測定した。同時に、内側全体の面積を測定し、変性部面積の内側全体の面積に対する割合(変性部面積率)を百分率で算出した。
結果を表5に示す。
【表5】
【0078】
実施例1
(1)化合物番号(1)0.1gおよび乳酸−グリコール酸共重合体(乳酸/グリコール酸=50/50;平均分子量20000;PLGA5020:和光純薬製)1.9gに塩化メチレン4.0gを添加し、30分間振盪溶解することにより油相(O)を形成した。
(2)油相を0.5%ポリビニルアルコール(ポバールPVA−220C:クラレ製)水溶液8mlに加え、ホモジナイザー(Polytoron:Kinematica製)を用いて25℃で5分間乳化することにより、水相に油相が分散した乳化液(O/W)を製造した。
(3)この乳化液を精製水1000mlに添加し、スリーワンモーター(新東科学製)を用いて400rpmで攪拌しながら、25℃で3時間液中乾燥することにより塩化メチレンを除去した。
(4)生成するマイクロスフェア懸濁液から、目開き150μmのフィルターを通して凝集物を除去し、目開き20μmのフィルターで吸引濾過することにより水相を除去した。得られたマイクロスフェアに少量の精製水を添加し、凍結乾燥することにより、マイクロスフェア1.6gを得た。
【0079】
得られたマイクロスフェア10mgをアセトニトリル3mlに溶解し、0.5M塩化ナトリウム水溶液7mlを加えてミキサー(Touch mixer MT-51:Yamato製)にて攪拌後、2000rpmで5分遠心分離し、上清を分取した。上清の一部をFL−HPLC(カラム;Hypersil 5-ODS 直径4mm、長さ300mm:ジーエルサイエンス製、励起波長:315nm、蛍光波長:465nm)にかけ、別途作成した薬物溶液の検量線からこれに含まれる薬物量を測定し、これと上清量とからマイクロスフェア中の薬物含有量を算出した。その結果、薬物の含有量は4.21%であった。
また、得られたマイクロスフェアをポリオキシエチレンソルビタン脂肪酸エステル(Tween80:日光ケミカルズ製)の希薄溶液に適量分散させ、粒度分布測定装置SALD−1100(島津製)にて粒度分布を測定し、平均粒子径を算出した。その結果、平均粒子径は57μmであった。
【0080】
(5)上記(4)で得られたマイクロスフェアを、カルボキシメチルセルロースナトリウム(ニチリン化学工業製)を0.5%、ポリオキシエチレンソルビタン脂肪酸エステル(Tween80:日光ケミカルズ製)を0.1%含む生理食塩水(分散媒)に2.5mg/mlの薬物割合となるように添加し、ミキサー(Touch mixer MT-51:Yamato製)にて、十分攪拌することにより、マイクロスフェア分散液を調製した。
【0081】
実施例2
(1)乳酸−グリコール酸共重合体(乳酸/グリコール酸=50/50;平均分子量20000;PLGA5020:和光純薬)0.57gと乳酸重合体(平均分子量20000;PLA0020:和光純薬製)1.33gを混合して用いること以外は、実施例1−(1)〜(4)と同様にして、マイクロスフェア1.6gを得た。
実施例1−(4)と同様にしてマイクロスフェア中の薬物含有量および平均粒子径を測定した。その結果、薬物含有量は3.70%、平均粒子径は47.7μmであった。
(2)上記(1)で得られたマイクロスフェアを実施例1−(5)と同様に処理してマイクロスフェア分散液(薬物割合:2.5mg/ml)を調製した。
(3)上記(1)で得られたマイクロスフェアを実施例1−(5)と同様にしてマイクロスフェア分散液(薬物割合:10.0mg/ml)を調製した。
【0082】
実施例3
(1)乳酸重合体(平均分子量20000;PLA0020:和光純薬製))を用いること以外は、実施例1−(1)〜(4)と同様にして、マイクロスフェア1.5gを得た。
実施例1−(4)と同様にしてマイクロスフェア中の薬物含有量および平均粒子径を測定した。その結果、薬物含有量は3.73%、平均粒子径は52.2μmであった。
(2)上記(1)で得られたマイクロスフェアを実施例1−(5)と同様に処理してマイクロスフェア分散液(薬物割合:2.5mg/ml)を調製した。
【0083】
実施例4
(1)化合物番号(1)0.2gおよび乳酸重合体(平均分子量20000;PLA0020:和光純薬製)0.3gに塩化メチレン1.0gを添加し、ミキサー(Touch mixer MT-51:Yamato製)にて十分攪拌、溶解することにより、油相(O)を形成した。
(2)油相を0.25%メチルセルロース(メトローズ:信越化学社製)水溶液4mlに加え、ホモジナイザー(Polytoron:Kinematica製)を用いて25℃で5分間乳化することにより、水相に油相が分散した乳化液(O/W)を製造した。
(3)この乳化液を精製水400mlに添加し、スリーワンモーター(新東科学製)を用いて400rpmで攪拌しながら、25℃で3時間液中乾燥することにより塩化メチレンを除去した。
(4)生成するマイクロスフェア懸濁液から、目開き150μmのフィルターを通して凝集物を除去し、目開き20μmのフィルターで吸引濾過することにより水相を除去した。得られたマイクロスフェアに少量の精製水を添加して凍結乾燥することでマイクロスフェアを得た。実施例1−(4)と同様にしてマイクロスフェア中の薬物含有量および平均粒子径を測定した。その結果、薬物含有量は39.6%、平均粒子径は33.4μmであった。
【0084】
実施例5
(1)化合物番号(1)0.05gおよび乳酸−グリコール酸共重合体(乳酸/グリコール酸=50/50;平均分子量20000;R202H:ベーリンガーインゲルハイム社製)0.45gに塩化メチレン1.0gを添加し、ミキサー(Touch mixer MT-51:Yamato製)にて十分攪拌、溶解することにより、油相(O)を形成した。
(2)油相を0.5%ポリビニルアルコール(ゴセノールEG−40:日本合成化学工業製)水溶液40mlに加え、ホモジナイザー(Polytoron:Kinematica製)を用いて25℃で4分間乳化することにより、水相に油相が分散した乳化液(O/W)を製造した。
(3)この乳化液を予め400mlの精製水を入れた円筒状密閉容器(内径110mm;内容積1000ml)に注ぎ、スリーワンモーター(BL-600;HEIDON社製)に装着した直径50mmの4枚攪拌羽根(プロペラR型;HEIDON社製)により25℃、400rpmで攪拌すると同時に、容器内に挿入した円筒型シリコーンゴム製中空糸膜モジュール(永柳工業株式会社製)を用い、中空糸の内側に窒素ガスを通気して容器内から塩化メチレンを除去した。この際の窒素ガス通気速度は2L/分とした。この操作を1時間行った。
【0085】
なお、円筒型シリコーンゴム製中空膜糸モジュールとしては、次の仕様を有するNAGASEP M60-1800円筒型を使用した。
円筒の直径 :100mm
円筒の長さ :120mm×120mm
中空糸膜の膜厚 :60μm
中空糸膜の内径 :200μm
中空糸膜の外径 :320μm
中空糸の本数 :1800
中空糸膜の有効膜面積:0.15m2
【0086】
(4)生成するマイクロスフェア懸濁液から、目開き150μmのフィルターを通して凝集物を除去し、目開き20μmのフィルターで吸引濾過することにより水相を除去した。得られたマイクロスフェアに少量の精製水を添加して凍結乾燥することでマイクロスフェアを0.26g得た。実施例1−(4)と同様にしてマイクロスフェア中の薬物含有量および平均粒子径を測定した。その結果、薬物含有量は3.07%、平均粒子径は71.7μmであった。
【0087】
実施例6
(1)化合物番号(2)0.05gおよび乳酸−グリコール酸共重合体(乳酸/グリコール酸=50/50;平均分子量20000;RG502H:ベーリンガーインゲルハイム社製)0.45gに塩化メチレン2.5gを添加し、ミキサー(Touch mixer MT-51:Yamato製)にて十分攪拌、溶解することにより、油相(O)を形成した。
(2)油相を0.5%ポリビニルアルコール(ポバールPVA−220C:クラレ製)水溶液3mlに加え、ホモジナイザー(Polytoron:Kinematica製)を用いて22℃で5分間乳化することにより、水相に油相が分散した乳化液(O/W)を製造した。
(3)上記(1)および(2)の操作を5回繰り返し、得られた乳化液(5回分)を併せた上で、精製水1000mlに添加し、スリーワンモーター(新東科学製)を用いて400rpmで攪拌しながら、25℃で1時間30分、次いで40℃で1時間、25℃で30分間液中乾燥することにより塩化メチレンを除去した。
(4)生成するマイクロスフェア懸濁液から、目開き150μmのフィルターを通して凝集物を除去し、目開き20μmのフィルターで吸引濾過することにより水相を除去した。得られたマイクロスフェアに少量の精製水を添加して凍結乾燥することでマイクロスフェア2.3gを採取した。
【0088】
得られたマイクロスフェア10mgをアセトニトリル3mlに溶解し、0.5M塩化ナトリウム水溶液6mlを加えてミキサー(Touch mixer MT-51:Yamato製)にて攪拌後、2000rpmで5分遠心分離し、上清を分取した。上清の一部をUV−HPLC(カラム;Hypersil 5-ODS 直径4mm、長さ300mm:ジーエルサイエンス製、測定波長:240nm)にかけ、別途作成した薬物溶液の検量線からこれに含まれる薬物量を測定し、これと上清量とからマイクロスフェア中の薬物含有量を算出した。また、実施例1−(4)と同様にして平均粒子径を測定した。その結果、薬物含有量は9.9%、平均粒子径は26.4μmであった。
(5)上記(4)で得られたマイクロスフェアを実施例1−(5)と同様に処理してマイクロスフェア分散液(薬物割合:0.1mg/ml)を調製した。
【0089】
実施例7
(1)乳酸−グリコール酸共重合体(乳酸/グリコール酸=75/25;平均分子量20000;PLGA7520:和光純薬製)を用いること、ならびに塩化メチレンの添加量を2.0gとする以外は、実施例6−(1)〜(4)と同様にして、マイクロスフェア2.2gを得た。
実施例6−(4)と同様にしてマイクロスフェア中の薬物含有量および平均粒子径を測定した。その結果、薬物含有量は10.1%、平均粒子径は27.0μmであった。
(2)上記(1)で得られたマイクロスフェアを実施例6−(5)と同様に処理してマイクロスフェア分散液(薬物割合:0.1mg/ml)を調製した。
【0090】
対照例1(実施例2の対照)
(1) 乳酸−グリコール酸共重合体(乳酸/グリコール酸=50/50;平均分子量20000;PLGA5020:和光純薬)0.6gと乳酸重合体(平均分子量20000)1.4gに塩化メチレン4.0gを添加し、30分間振盪溶解することにより油相(O)を形成する。以下、実施例1−(1)〜(4)と同様にして、薬物を含まないマイクロスフェア1.7gを得た。
(2)上記(1)で得られたマイクロスフェアを実施例1−(5)と同様にして、マイクロスフェア分散液(分散液中のマイクロスフェア分散濃度は実施例2−(3)と同一)を調製した。
対照例2(実施例7の対照)
乳酸−グリコール酸共重合体(乳酸/グリコール酸=75/25;平均分子量20000;PLGA7520:和光純薬製)0.45gに塩化メチレン2.0gを添加し、ミキサー(Touch mixer MT-51:Yamato製)にて十分攪拌、溶解することにより、油相を形成した。以下、実施例6−(2)〜(4)と同様にして、薬物を含まないマイクロスフェア2.2gを得た。
【0091】
試験例1
試験管にマイクロスフェア10mgを入れ、0.05% Tween80含有リン酸緩衝液(pH7.4)の10mlを添加し、37℃の空気恒温庫中の回転培養機にて25rpmで攪拌した。攪拌開始時から一定時間後に、溶出液を遠心分離(2000rpm、5分)し、上清から9mlをサンプリングして、FL−HPLC(カラム;Hypersil 5-ODS 直径4mm、長さ300mm:ジーエルサイエンス製、励起波長:315nm、蛍光波長:465nm)にかけ、別途作成した薬物溶液の検量線からこれに含まれる薬物量を測定し、これとサンプリングの量とから薬物溶出量を算出した。
また、サンプリング後の試験管に、pH7.4リン酸緩衝液9mlを添加し、同様の条件で攪拌、サンプリングして薬物溶出量を算出する操作を経時的に繰り返した。
【0092】
最終のサンプリング後、試験管から残りの溶出液を除去し、残存するマイクロスフェア中に含まれる薬物量を実施例1−(4)の方法で測定した。
この操作を実施例1、実施例2ならびに実施例3で得られたマイクロスフェアについて行った。その結果を図5に示す。
なお、溶出した薬物量とマイクロスフェアに残存した薬物量の合計を100%として、溶出率を算出した。
【0093】
試験例2
SD系雄性ラット(7週令、1群3匹、日本SLC)を12時間照明、室温:23±2℃、自由摂水・摂餌の条件下で1週間の馴化期間後、ポリエチレングリコール400(和光純薬社製)を10%含んだ生理食塩水に溶解した化合物番号(1)(1mg/ml)を1匹当たり0.5ml(総薬物投与量:0.5mg/ラット)の割合で大腿静脈から急速投与した。
薬物投与後、経時的にエーテル麻酔下で頚静脈よりヘパリンを添加した注射筒で血液を採取し遠心分離により血漿を得た。血漿0.1mlに内部標準溶液と1Mのリン酸水素2カリウム0.2mlを添加し、クロロホルム7.0mlを添加後、10分間振とう、5分間遠心分離して5mlの有機相を分取した。採取した有機相を窒素気流下、40℃で蒸発乾固し、移動相の0.5mlで再溶解後にFL−HPLC(カラム;Hypersil 5-ODS 直径4mm、長さ300mm:ジーエルサイエンス製、励起波長:315nm、蛍光波長:465nm)により血漿中濃度を測定した。その結果を図6に示す。
【0094】
試験例3
SD系雄性ラット(7週令、1群5匹、日本SLC)を12時間照明、室温:23±2℃、自由摂水・摂餌の条件下で1週間の馴化期間後、実施例1−(5)、実施例2−(2)、実施例3−(2)で得られたマイクロスフェア分散液を1匹当たり2ml(総薬物投与量:5mg/ラット)の割合で背部皮下に投与した。薬物投与後、経時的にエーテル麻酔下で頚静脈よりヘパリンを添加した注射筒で血液を採取し遠心分離により血漿を得た。血漿中の化合物濃度は試験例2と同様の方法で測定した。PDE4阻害剤をマイクロスフェア化することにより、PDE4阻害剤を1/10量しか含まない生理食塩水溶液を静脈注射した場合(試験例2)と比べても、PDE4阻害剤の最高血漿中濃度を1/25の1〜1/100に抑制することが出来た。その結果を図7に示す。
【0095】
試験例4
SD系雄性ラット(7週令、1群5匹、日本SLC)を12時間照明、室温:23±2℃、自由摂水・摂餌の条件下で1週間の馴化期間後、実施例2−(2)で得られたマイクロスフェア分散液を1匹当たり2ml(総薬物投与量:5mg/ラット)の割合で背部皮下に投与した。
薬物投与3、7、10、14、21、35日後、投与部位よりマイクロスフェアを回収した。回収したマイクロスフェアに内部標準物質を含有したアセトニトリル5mlを添加して、ホモジナイザー(Polytoron:Kinematica製)で溶解した。3000rpm、5分間遠心分離後の上清3mlを採取し、0.5M塩化ナトリウム水溶液7mlを加えてミキサー(Touch mixer MT-51:Yamato製)にて攪拌後、2000rpmで5分遠心分離し、上清を分取した。上清の一部をKCプレップオムニ13(片山化学社製)で濾過してFL−HPLC(カラム;Hypersil 5-ODS 直径4mm、長さ300mm:ジーエルサイエンス製、励起波長:315nm、蛍光波長:465nm)にかけ、別途作成した薬物溶液の検量線からこれに含まれる薬物量を測定し、これと上清量とからマイクロスフェア中の薬物残存量を算出した。その結果を図8に示す。
【0096】
試験例5
SD系雄性ラット(7週令、日本SLC)を12時間照明、室温:23±2℃、自由摂水・摂餌の条件下で1週間の馴化期間後、実施例6−(5)ならびに7−(2)で得られた化合物番号(2)含有マイクロスフェア分散液を1匹当たり1ml(総薬物投与量:0.1mg/ラット)の割合で背部皮下に投与した。
経時的に投与部位よりマイクロスフェアを回収し、アセトニトリル10mlを添加してホモジナイザー(Polytoron:Kinematica製)で溶解した。3000rpm、5分間遠心分離後の上清3mlを採取し、0.5M塩化ナトリウム水溶液6mlを加えてミキサー(Touch mixer MT-51:Yamato製)にて攪拌後、2000rpmで5分遠心分離し、上清を分取した。上清の一部をKCプレップオムニ13(片山化学社製)で濾過してUV−HPLC(カラム;Hypersil 5-ODS 直径4mm、長さ300mm:ジーエルサイエンス製、測定波長:240nm)にかけ、別途作成した薬物溶液の検量線からこれに含まれる薬物量を測定し、これと上清量とからマイクロスフェア中の薬物残存量を算出した。その結果を図9に示す。
【産業上の利用可能性】
【0097】
従来、軟骨疾患については薬物療法では保存的治療しか行えないと考えられていたにも拘わらず、本発明の軟骨疾患修復治療用組成物は、PDE4阻害剤を有効成分とし、特に、軟骨疾患部位に局所投与することにより、PDE4阻害剤の全身作用による副作用を伴うことなく、軟骨を修復することができ、軟骨疾患、特に変形性関節症に修復治療効果を発揮する。また、PDE4阻害剤と生体内適合性かつ生体内分解性ポリマーを含有する組成物をデポ剤の形とし、特にマイクロスフェア形態で注射剤として軟骨疾患部位に局所的に注射することにより持続的に効果を発揮することができ、さらに優れた効果が得られる。
【技術分野】
【0001】
本発明は、軟骨疾患修復治療用組成物、さらに詳しくは、ホスホジエステラーゼ(PDE)4阻害作用を有する化合物を有効成分とする、変形性関節症、軟骨形成異常症、変形性椎間板症、半月板損傷等の軟骨疾患を修復治療するための医薬組成物に関する。
【背景技術】
【0002】
軟骨は、骨と共に骨格を形成したり、内臓を保護する役目を持つ、弾性力に富む組織であり、軟骨組織は軟骨細胞とそれを取り囲む軟骨基質とからなる。
軟骨は間葉由来の軟骨芽細胞が分裂増殖しながら周囲に基質を産生したものである。一方、軟骨基質は無定形基質と繊維成分とからなり、それらの組成比率の違いにより、(1)硝子軟骨(関節軟骨、肋軟骨、甲状軟骨等)、(2)線維軟骨(椎間円板、恥骨結合)、(3)弾性軟骨(咽頭蓋軟骨、外耳道軟骨、耳介軟骨等)の3種に分類される[医学大辞典(18版;南山堂発行)第1542頁]。
【0003】
軟骨基質の主成分は、プロテオグリカンおよびコラーゲン(II型、IX型等)であり、プロテオグリカンは軟骨組織特有の膨潤性に関与し、コラーゲン線維は軟骨の張力および剪断力に対する剛性に関与することが知られている。
軟骨基質中のプロテオグリカンでは、コンドロイチン硫酸、ケラタン硫酸等のグルコサミノグリカンが分子量約22万のコア蛋白に結合して巨大分子を形成したものとなっており、グルコサミノグリカンが多くの水分子を水和していることが、軟骨の膨潤性に寄与していると考えられている[ザ・ボーン(The Bone)第4巻第8頁(1994年)]。
【0004】
このうち、関節軟骨は骨組織との移行部に石灰化層を有し、成長が完了した後には、血液の直接的な供給を殆ど受けることなく、軟骨細胞の栄養は関節液から供給される。また、関節軟骨は細胞分化度の高い硝子軟骨から形成されており、再生能力の極度に低いデリケートな組織である。
この関節軟骨の表面を粘稠性の高い滑液が覆い、ヒアルロン酸・蛋白複合体を主役とした潤滑液による潤滑機構が働き、滑らかな関節運動が維持されているが、関節軟骨には所謂耐用年数が存在すると考えられており、老化に伴う関節変化は必発する避けがたい生理的現象である。
【0005】
軟骨の障害に起因する疾患としては、例えば、変形性関節症をはじめとして、軟骨形成異常症、変形性椎間板症、半月板損傷等が知られている。
このうち、変形性関節症は、関節を構成する組織、主に、関節軟骨の退行性変化を基に、骨・軟骨の増殖性変化を来し、最終的には関節の形態が著明に変化するに至る疾患であるが、人口の高齢化に伴って増加しており、中でも膝関節症は病態の進行と共に、立位保持、歩行動作が障害されて日常動作能力(Ability of Daily Life; ADL)の著しい低下を引き起こし、寝たきり状態の原因となることもある。
【0006】
変形性関節症の治療は大きく、保存的治療と手術療法に分けられており、保存的治療として、(1)非ステロイド性消炎鎮痛剤の投与、(2)温熱療法、(3)体重のコントロール、(4)装具療法、(5)ステロイド性消炎鎮痛剤の関節内注入、(6)ヒアルロン酸製剤の関節内注入等が行われている。また、保存療法が奏効しない症例や進行例、末期例に対しては、手術療法として、(1)関節鏡視下洗浄、(2)脛骨高位骨切り術、(3)人工関節置換等が行われている[老化と疾患第10巻、第2号、61〜69頁(1997年)&第6号、第66〜77頁(1997年)]。
一方、PDE4阻害作用を有する化合物は種々知られており、PDE4の作用を阻害することにより、炎症性メディエイターの放出を阻害できる[ジャーナル・オブ・モレキュラー・アンド・セルラー・カーディオロジー(J. Mol. Cell. Cardiol.)12(Suppl. II), S61 (1989年)]。
【0007】
また、PDE4阻害作用を有する化合物が、免疫刺激剤に応答して単核食細胞により放出されるサイトカインであるTNF-αの産生を抑制し、TNF−αによって引き起こされる各種炎症性疾患の治療に有用であることが記載されている〔特表2000−503678号、特表2000−502724号、特表2000−510105号、特表2000−514804号、特表2000−502350号、特表2000−501741号〕。しかし、PDE4阻害剤が軟骨疾患の修復治療に有効であることは全く知られていない。
【発明の概要】
【発明が解決しようとする課題】
【0008】
上述したように、軟骨は再生能力が極めて低く、一度損傷するとその修復は殆ど不可能に近いと考えられており、従来の薬物療法はその進行を抑制する保存的治療に過ぎなかった。従って、その軟骨疾患の修復治療を可能にする薬物療法および/または医薬品の開発が永年求められている。
【課題を解決するための手段】
【0009】
本発明者らは、軟骨細胞においてPDE4が産生されていることを見出し、PDE4阻害作用を有する化合物が、軟骨疾患に対して活性を示すことを知り、研究を重ねたところ、該PDE4阻害作用を有する化合物が軟骨疾患の修復治療に有用であることを見出し、本発明を完成した。
すなわち、本発明は、PDE4阻害剤を有効成分とする軟骨疾患修復治療用組成物を提供するものである。本発明は、特に、軟骨疾患部位に局所的に適用するのに適した製剤、特にマイクロスフェア形態の、軟骨疾患修復治療用組成物を提供するものである。
【0010】
本発明の軟骨疾患修復治療用組成物は、軟骨、とりわけ、再生能力が極度に低い関節軟骨に対し、軟骨基質蛋白の遺伝子発現を増大させることにより、優れた基質産生促進作用を有し、軟骨の修復により、軟骨の疾患を治療することができる。
ここに、軟骨疾患の修復治療とは、軟骨疾患の悪化を抑制するのみならず、疾患、傷害等により、変形・磨耗した軟骨を元の形に修復する治療を意味するものである。
【0011】
本発明の医薬組成物は、PDE4阻害剤を有効成分とし、これに通常の医薬用賦形剤または希釈剤を配合して調製される。好ましい医薬組成物は、PDE4阻害剤と生体内適合性かつ生体内分解性ポリマーとを含有する徐放性の局所投与用の組成物である。該局所投与性組成物は、デポ剤(depot formulation)の形態とするのが好ましく、さらに、マイクロスフェア形態とすることが好ましく、そのマイクロスフェアは注射剤の形態とすることもできる。
【0012】
本発明の医薬組成物において、活性成分として用いられるPDE4阻害剤としては、PDE4阻害活性を有する化合物がすべて含まれ、例えば、特開平5−229987号、特開平9−59255号、特開平10−226685号、欧州公開No.158380、国際公開No.94/25437、米国特許No.5223504、国際公開No.95/4045、欧州公開No.497564、欧州公開No.569414、欧州公開No.623607、欧州公開No.163965、米国特許No.5605914、国際公開No.95/35282、国際公開No.96/215、米国特許No.5804588、米国特許No.5552438、国際公開No.93/9118、国際公開No.96/31485、欧州公開No.459505、国際公開No.97/22585、欧州公開No.738715、国際公開No.91/16314、国際公開No.96/218、国際公開No.97/18208、欧州公開No.158380、国際公開No.99/50270、欧州公開No.260817、国際公開No.98/11113、国際公開No.94/22852、欧州公開No.432856、米国特許No.4193926、国際公開No.98/13348、国際公開No.96/6843、特表2000−503678号(=国際公開No.98/14432)、特表2000−502724号(=国際公開No.98/9961)、特表2000−510105号(=国際公開No.97/40032)、特表2000−514804号(=国際公開No.98/2440)、特表2000−502350号(=国際公開No.97/23457)、特表2000−501741号(=国際公開No.97/2585)等に記載の化合物が挙げられる。
【0013】
本発明の軟骨疾患修復治療用組成物に適したPDE4阻害剤は、トレンズ・イン・ファーマコロジカル・サイエンシーズ(Trends in Pharmacological Sciences)11巻150〜155頁の記載に従って分類したPDE1〜5のうち、PDE4に対して他のPDE(PDE1〜3および5)に対するよりもより強い阻害作用を有する選択的PDE4阻害剤が好ましく、PDE4に対する阻害作用が他のPDEに対するよりも10倍以上強いものが好ましい。より好ましいものは、他のPDEに対するよりも50倍以上、さらに好ましくは100倍以上の阻害作用を有するものである。
好ましいPDE4阻害剤は、アドバンシーズ・イン・サイクリック・ヌクレオチド・リサーチ(Advances in Cyclic Nucleotide Research)10巻、69〜92頁[1979年レイベン・プレス(Raven Press)発行]記載の方法に準じて測定したPDE4阻害活性のIC50が0.1〜1000nM、好ましくは0.1〜100nMの化合物である。より好ましくは、IC50は100nM未満である。
【0014】
選択的PDE4阻害剤の具体例としては、下記構造で示される化合物番号(1)〜(57)の化合物またはその薬理的に許容し得る塩が挙げられる。
【化1】
【化2】
【化3】
【化4】
【化5】
【化6】
【化7】
【化8】
【化9】
【化10】
【化11】
【化12】
【化13】
【化14】
【化15】
【化16】
【化17】
【化18】
【化19】
【化20】
【化21】
【化22】
【化23】
【化24】
【化25】
【化26】
【化27】
【化28】
【化29】
【0015】
PDE4阻害活性を有する化合物は、化学構造上から、次の(A)〜(D)に分けることが出来、それらのうちから適宜選択されるが、本発明におけるPDE4阻害剤としては、(A)および(B)の化合物が好ましく、(A)の化合物がとりわけ好ましい。
(A) ナフタレン骨格またはこれに類似する部分構造を有する化合物[例えば、化合物番号(1)、(2)、(38)、(47)、(52)〜(57)]、
(B) 3−シクロペンチルオキシ−4−メトキシフェニル構造またはこれに類似する部分構造を有する化合物[例えば、化合物番号(6)、(9)、(11)、(12)、(14)、(17)、(19)、(20)、(21)、(24)、(25)、(26)、(27)、(33)、(34)、(35)、(39)、(40)、(44)、(49)、(50)、(51)]、
(C) キサンチン骨格またはこれに類似する部分構造を有する化合物[例えば、化合物番号(5)、(7)、(28)、(29)、(30)、(31)、(32)、(36)、(37)、(41)、(43)、(46)]、および
(D) 上記(A)〜(C)以外の構造を有する化合物[例えば、化合物番号(3)、(4)、(8)、(10)、(13)、(15)、(16)、(18)、(22)、(23)、(42)、(45)、(48)]
【0016】
上記(A)の化合物としては、例えば、下記一般式(I)〜(III)で示される化合物およびそれらの薬理的に許容し得る塩を挙げることができる。
【化30】
(式中、R1およびR2は同一または異なって、水素原子、水酸基、シクロ低級アルキルオキシ基、または置換基を有していてもよい低級アルコキシ基を表すか、或いは、互いに末端で結合して低級アルキレンジオキシ基を形成し、R3は置換基を有していてもよい含窒素複素6員環式基、−OR4およびOR5は同一または異なって、保護されていてもよい水酸基を表す)(特開平5−229987号)
【0017】
【化31】
(式中、R1'およびR2'は同一または異なって水素原子または保護されていてもよい水酸基を表し、R3'およびR4'のいずれか一方が、保護されていてもよい水酸基置換メチル基、他方が水素原子、低級アルキル基または保護されていてもよい水酸基置換メチル基であり、R5'およびR6'は同一または異なって、水素原子、置換されていてもよい低級アルキル基、置換されていてもよいフェニル基または保護されていてもよいアミノ基を表すか、あるいは互いに末端で結合して隣接する窒素原子とともに置換されていてもよい複素環式基を形成している)(特開平9−59255号)
【0018】
【化32】
(式中、Aは式:
【化33】
から選ばれるいずれか1つの基[但し、R1”およびR2”は同一または異なって、水素原子または保護されていてもよいヒドロキシ基、R31は保護されていてもよいヒドロキシメチル基、R32は水素原子、低級アルキル基または保護されていてもよいヒドロキシメチル基、R33は置換基を有していてもよい低級アルキル基、R41は保護されていてもよいヒドロキシメチル基、R42は保護されていてもよいヒドロキシメチル基、点線は二重結合の存在または非存在を表す]を表し、R5”およびR6”は同一または異なって、水素原子または保護されていてもよいアミノ基を表すか、あるいは互いに末端で結合して隣接する窒素原子と共に置換されていてもよい複素環式基を形成している。)(特開平10−226685号)
【0019】
本発明の軟骨疾患修復治療用組成物の有効成分であるPDE4阻害剤としては、(A)の化合物のうち、ナフタレン骨格またはイソキノリン骨格を有する化合物またはその薬理的に許容し得る塩がとりわけ好ましく、化合物番号(1)および(2)の化合物またはその薬理的に許容し得る塩がとりわけ好ましい。
PDE4阻害剤が全身的に作用した場合には、投与量によっては嘔吐や胃酸分泌を引き起こすこともあるため[セルラー・シグナリング(Cellular Signaling)9(3−4)227〜236頁(1997年)]、本発明の軟骨疾患修復治療用組成物は疾患部位の近傍(とりわけ、関節軟骨の近傍)に、局所投与され、全身的な薬物の血中濃度を上昇させず、軟骨疾患部位での薬物濃度を維持するようにするのが望ましい。また、このような目的を達成するためには、徐放性とするのが望ましく、徐放性とすることにより、投与回数を少なくし、患者負担の軽減を図ることもできる。
【0020】
本発明の組成物の好ましい形態としては、局所投与で薬物を徐々に放出するデポ剤(例えばペレット製剤、ゲル製剤、マトリックス製剤、マイクロスフェア製剤、生体内適合性かつ生体内分解性ポリマーの水溶液に薬物を添加して徐放化した製剤、投与時には溶液であるが、生体内に投与されることによってゲルを形成するように設計された製剤、一般に整形外科の領域での使用が報告されている種々基剤に封入した製剤等)が挙げられる。
ペレット剤としては、例えば、末端カルボキシル基がアルコールによりエステル化された乳酸−グリコール酸共重合体微粒子と薬物とを圧縮成型して得られる長期徐放性製剤(特開2001−187749号)等を挙げることができる。
【0021】
ゲル製剤としては、例えば、ジャーナル・オブ・コントロールド・リリース(Journal of Controlled Release) 59 (1999) 77-86記載の、ポリエチレングリコールを化学的に結合させたヒアルロン酸と薬物とをリン酸緩衝液に溶解させたゲル製剤等を挙げることができる。
マトリックス製剤としては、例えば、コラーゲンの粒状物質中または繊維膜中に薬物を含浸した製剤、コラーゲンの粒状物質中または繊維膜の調製中に薬物を添加して含有させた製剤(特開平10−182499号、特開平6−305983号)等を挙げることが出来る。
【0022】
生体内適合性かつ生体内分解性ポリマーの水溶液に薬物を添加して徐放化した製剤としては、例えば、ヒアルロン酸ナトリウムの水溶液に薬物を添加して徐放化した製剤等が考えられる。
投与時には溶液であるが、生体内に投与されることによってゲルを形成するように設計された製剤としては、例えば、ジャーナル・オブ・コントロールド・リリース(Journal of Controlled Release)33 (1995) 237-243 記載の、乳酸−グリコール酸共重合体と薬物とをN−メチル−2−ピロリドンに溶解させた製剤、同 27 (1993) 139-147 記載の、乳酸−グリコール酸共重合体とポリエチレングリコールとのブロック共重合体等の低温では溶液状態で存在するが、体温ではゲルとなる高分子と薬物を含有する製剤を挙げることができる。
【0023】
一般に整形外科の領域で報告されている種々の基剤に封入した製剤としては、例えば、基剤(例えば、水難溶性の生体内適合性かつ生体内分解性ポリマー、ポリメチルメタクリレート、ヒドロキシアパタイト、トリカルシウムホスフェート等)と薬物とを混合して製造される製剤[バイオマテリアルズ(Biomaterials)21巻、2405−2412頁(2000年)、インターナショナル・ジャーナル・オブ・ファーマシューティクス(International Journal of Pharmaceutics)206巻1−12頁(2000年)]を挙げることができる。
軟骨疾患修復治療に要する期間における投与回数を少なくする上からは、局所投与により、有効量のPDE4阻害剤を徐々に障害を有する軟骨の近傍(とりわけ、関節軟骨の近傍)に放出するものが好ましい。
【0024】
デポ剤の内、注射剤の形で局所に投与し易いマイクロスフェアの場合、注射針を通過する粒径であるのが好ましく、粒子径が0.01〜150μm、とりわけ0.1〜100μmの範囲であるものが疾患部位への刺激を抑制できる点では好ましい。
本発明のPDE4阻害剤を有効成分とする軟骨疾患修復治療用組成物は、障害を有する軟骨の近傍(とりわけ、関節軟骨の近傍)に、局所的に投与することを考えれば、投与量を小さくするのが好ましく、組成物(例えばマイクロスフェア製剤)中のPDE4阻害剤の量は0.0001〜80%重量%とするのが好ましく、さらに好ましくは0.001〜50%重量、さらに好ましくは0.01〜50%重量である。有効成分としてのPDE4の投与量は、用いるPDE4阻害剤の種類、対象の体重、年齢、症状、適用部位等に応じて医師により適宜決定されるが、局所適用の場合、通常、疾患部位あたり1ng〜1gの範囲である。
【0025】
本発明の軟骨疾患修復治療用組成物は、PDE4阻害剤と薬理的に許容される賦形剤または希釈剤とから、常法に従って調製されるが、好ましい組成物は、PDE4阻害剤と生体内適合性かつ生体内分解性ポリマーとを配合して調製される。
このうち、水難溶性の生体内適合性かつ生体内分解性ポリマーは、1gを溶解するのに、水(25℃)が1000ml以上必要となるような生体内適合性かつ生体内分解性ポリマーであり、具体的には、ヒドロキシ脂肪酸ポリエステルおよびその誘導体(例えば、ポリ乳酸、ポリグリコール酸、ポリクエン酸、ポリリンゴ酸、ポリ−β−ヒドロキシ酪酸、ε−カプロラクトン開環重合体、乳酸−グリコール酸共重合体、2−ヒドロキシ酪酸−グリコール酸共重合体、ポリ乳酸とポリエチレングリコールとのブロック共重合体、ポリグリコール酸とポリエチレングリコールとのブロック共重合体、乳酸−グリコール酸共重合体とポリエチレングリコールとのブロック共重合体など)、α−シアノアクリル酸アルキルエステルのポリマー(例えば、ポリブチル−2−シアノアクリレートなど)、ポリアルキレンオキサレート(例えば、ポリトリメチレンオキサレート、ポリテトラメチレンオキサレートなど)、ポリオルソエステル、ポリカーボネート(例えば、ポリエチレンカーボネート、ポリエチレンプロピレンカーボネートなど)、ポリオルソカーボネート、ポリアミノ酸(例えば、ポリ−γ−L−アラニン、ポリ−γ−ベンジル−L−グルタミン酸、ポリ−γ−メチル−L−グルタミン酸など)、ヒアルロン酸エステルなどが挙げられ、これらの1種または2種以上を用いることができる。他の使用可能な生体内適合性かつ生体内分解性ポリマーとしては、ヒアルロン酸ナトリウム、コンドロイチン硫酸、コラーゲン、ゼラチン、フィブリン等が挙げられる。
【0026】
水難溶性の生体内適合性かつ生体内分解性ポリマーのうち、特に好ましいものはヒドロキシ脂肪酸のポリエステルである。それらは平均分子量が2000〜約800000の範囲内、より好ましくは2000〜約200000の範囲内のものが特に好適であり、平均分子量が5000〜50000の範囲のものが最も好適である。
また、上記ヒドロキシ脂肪酸のポリエステルのうち、更に好ましいのは、ポリ乳酸、乳酸−グリコール酸共重合体、2−ヒドロキシ酪酸−グリコール酸共重合体である。乳酸−グリコール酸共重合体における乳酸/グリコール酸のモル比は、好ましくは90/10〜30/70、より好ましくは80/20〜40/60であり、2−ヒドロキシ酪酸−グリコール酸共重合体における2−ヒドロキシ酪酸/グリコール酸のモル比は、好ましくは90/10〜30/70、より好ましくは80/20〜40/60である。
【0027】
前記PDE4阻害剤をデポ剤とするには、その形態に応じて、適宜製剤化することができ、必要に応じて、製剤化に先立ち、予めPDE4阻害剤を微粒子化してもよい。
PDE4阻害剤を微粒子化するには、慣用の微粒子製法を適宜使用することができ、ジェットミル粉砕、ハンマーミル粉砕、回転ボールミル粉砕、振動ボールミル粉砕、ビーズミル粉砕、シェーカーミル粉砕、ロッドミル粉砕、チューブミル粉砕等により、物理的に粉砕する粉砕法や、薬物を一旦溶媒に溶解後、pH調整、温度変化、溶媒組成の変更等を行って、晶析させ、遠心分離あるいは濾過等の方法で回収するいわゆる晶析法を採用することができる。
【0028】
本発明の医薬組成物の上記各種製剤を製造するには、PDE4阻害剤に合わせて、既知の製法を適宜適用することができる。
例えば、マイクロスフェア製剤を製造するには次の方法が用いられる。また、PDE4阻害剤が塩を形成しているために、マイクロスフェアへの取り込み率が悪い場合には、酸または塩基を用いて遊離の形に変換した後、マイクロスフェア化してもよい。
【0029】
(1)水中乾燥法:
沸点が水より低く水と非混和性である有機溶媒に水難溶性の生体内適合性かつ生体内分解性ポリマーを溶解させた溶液(水難溶性ポリマー溶液)に薬物を含有させ、かくして得られる有機相を水相中に分散してO/W型エマルションを調製後に、該有機溶媒を除去する方法であり、例えば、特公昭56−19324号、特開昭63−91325号、特開平8−151321号、ジャインらの文献(Kajeev Jainら、"Controlled Drug Delivery by Biodegradable Poly(Ester) Devices: Different Preparative Approaches", Drug Development and Industrial Pharmacy、24(8)巻、703−727頁、1998年)、特開昭60−100516号、特開昭62−201816号、特開平9−221417号、および特開平6−211648号に記載の方法と同様にして行われる。
【0030】
(2)相分離法:
水難溶性の生体内適合性かつ生体内分解性ポリマーの有機溶媒溶液に、薬物もしくは薬物水溶液を溶解・分散させ、これに硬化剤を撹拌下徐々に加え、析出固化させる方法であり、例えば特開昭60−67417号、米国特許第5503851号、米国特許第5000886号、Eur. J. Pharm. Biopharm. 42(1)巻、16〜24頁(1996年)および前記ジャインらの文献に記載の方法と同様にして行われる。
【0031】
(3)噴霧乾燥法:
水難溶性の生体内適合性かつ生体内分解性ポリマーの有機溶媒溶液に、薬物を溶解・分散させるか、または薬物水溶液を分散させ、これをスプレーノズルでスプレードライヤー(噴霧乾燥器)の乾燥室内へ噴霧し、きわめて短時間に噴霧液滴内の有機溶媒を揮発させる方法であり、例えば、特開平1−155942号、特開平5−194200号、特開平5−70363号、特開平8−151321号、特開平9−221417号、米国特許第5922253号、「Spray Drying Handbook」(John Wiley & Sons, New York 1984)、ディージィの文献[Partick B. Deasy、「Microcapsulation and Related Drug Processes」(Marcel Dekker, Inc.、New York 1984)]、および前記ジャインらの文献などに記載の方法と同様にして行われる。
【0032】
(4)溶媒拡散法:
薬物および水難溶解性の生体内適合性かつ生体内分解性ポリマーを溶解させた水混和性有機溶媒の溶液を、保護コロイド剤の水溶液に添加し、攪拌乳化して微粒子を得る方法であり、例えば、特開平5−58882号、特開平9−110678号、インターナショナル・ジャーナル・オブ・ファーマシューティクス(International Journal of Pharmaceutics)187巻、143〜152(1999)に記載の方法と同様にして行われる。
上記「水中乾燥法」では、有機相の形態によって調製法が異なるが、いずれも常法にしたがって行うことができる。該有機相の形態としては以下のものが含まれる。
【0033】
(a)水難溶性の生体内適合性かつ生体内分解性ポリマー溶液に、薬物が直接溶解もしくは分散されている有機相。これを水相中に分散するとO/W型エマルションとなる(特公昭56−19324号、特開昭63−91325号、特開平6−32732号、特開平8−151321号、特開平6−32732号、前記ジャインらの文献など)。
(b)水難溶性の生体内適合性かつ生体内分解性ポリマー溶液に、薬物水溶液が分散されているW/O型エマルションからなる有機相。そのW/O型エマルションを水相中に分散すると、(W/O)/W型エマルションとなる(特開昭60−100516号、特開昭62−201816号、特開平9−221417号、前記ジャインらの文献など)。
【0034】
(c)2種以上の水難溶性の生体内適合性かつ生体内分解性ポリマーを用い、一方のポリマー溶液中に分散されている他方のポリマー溶液中に、薬物が溶解もしくは分散しているO/O型エマルションからなる有機相。そのO/O型エマルションを水相中に分散すると(O/O)/W型エマルションとなる(特開平6−211648号)。
上記いずれの形態の有機相においても常法により、例えば、断続振盪法、プロペラ型またはタービン型撹拌機を用いる混和法、コロイドミル法、ホモジナイザーを用いる方法、超音波照射法などによって、エマルションを形成させる。
【0035】
これらの方法において用いられる有機溶媒としては、ハロゲン化炭化水素系溶媒(塩化メチレン、クロロホルム、四塩化炭素、クロロエタン、ジクロロエタン、トリクロロエタンなど)、脂肪酸エステル系溶媒(酢酸エチル、酢酸ブチルなど)、芳香族炭化水素系溶媒(ベンゼン)、脂肪族炭化水素(n−ヘキサン、n−ペンタン、シクロヘキサンなど)、ケトン系溶媒(メチルエチルケトンなど)、エーテル系溶媒(ジエチルエーテル、ジイソプロピルエーテル、メチルイソブチルエーテルなど)が挙げられる。
上記のエマルションを形成させる際、エマルションを安定化するために乳化剤、例えばアニオン性界面活性剤(オレイン酸ナトリウム、ステアリン酸ナトリウム、ラウリル硫酸ナトリウム等)、非イオン性界面活性剤(ポリオキシエチレンソルビタン脂肪酸エステル[Tween80、Tween60(日光ケミカルズ製)等]、ポリエチレンヒマシ油誘導体[HCO−60、HCO−50(日光ケミカルズ製)]、ポリビニルピロリドン、ポリビニルアルコール、カルボキシメチルセルロース、メチルセルロース、レシチン、ゼラチン等)を水相に添加してもよい。
【0036】
また、PDE4阻害剤に加えて、他の成分を配合する場合は、上記O/W型エマルションを形成する際に、有機溶媒相にそれらを添加するのが好ましい。なお、薬効成分含量の高いマイクロスフェア製剤を得るためには、有機相の調製に際して薬効成分濃度を高くしておく必要があり、この場合、薬効成分の水相への流出を防ぐために、水相中に浸透圧調節剤を添加することもできる(日本特許第2608245号を参照)。
上記のようにして得られるO/W型エマルションを水中乾燥法に付すことにより、エマルションに含まれる有機溶媒を除去してマイクロスフェアを製造する。
【0037】
有機溶媒を除去するには、そのエマルション系を加温したり、減圧下に置くか、または気体を吹きつける方法などの慣用の方法が採用され、たとえば、開放系で溶媒を留去する方法(特公昭56−19324号、特開昭63−91325号、特開平8−151321号、特開平6−211648号)、閉鎖系で溶媒留去する方法(特開平9−221418号)などが採用され得る。また、多量の外水相を用いて溶媒を抽出・除去する方法(日本特許第2582186号)によっても行うことができる。
また次の方法も、PDE4阻害剤の種類に応じて、適宜適用することができる。
【0038】
薬物、生体内分解性ポリマーおよび水と混和する前記ポリマーの良溶媒(溶媒A:アセトン、テトラヒドロフラン等)を含む溶液を、溶媒Aと混和する前記ポリマーの貧溶媒(溶媒B:水、エタノール等)および溶媒Aと混和しない前記ポリマーの貧溶媒(溶媒C:グリセリン等)を含む均一混合液中に添加して乳化することにより、ポリマー溶液が分散相、均一混合液が連続相を形成するエマルションを調製し、分散相から溶媒Aを除去する方法(国際公開No.01/80835 )。
水より沸点の低い有機溶媒(塩化メチレン、酢酸エチル等)および水難溶性ポリマーを含む有機相が水相に乳化したエマルションから水中乾燥法でマイクロスフェアを製造するにあたり、(1)気体分離膜(浸透気化膜、多孔性膜等)を備えた装置を用い、(2)水中乾燥に付されるエマルションを気体分離膜の一方の側に供給し、(3)気体分離膜の他方の側へエマルションに含まれる有機溶媒を留去する方法(国際公開No.01/83594)。
【0039】
さらに、マイクロスフェアを水相中で有機溶媒の沸点以上に加温し(特開2000−239152号)、または高融点添加物でマイクロスフェアを覆った後、加温乾燥する(特開平9−221417号)ことにより、マイクロスフェアに残存する有機溶媒を除去することもできる。
このようにして得られるマイクロスフェアは遠心分離、濾過或いは篩などで回収し、その表面に付着した水相添加物等を洗浄除去し、所望により、マイクロスフェア同士の凝集を防止するために糖あるいは糖アルコール、無機塩等、好ましくは、ラクトース、マンニトール、ソルビトールなどの凝集防止剤を添加した後、凍結乾燥に付す。この際、所望の粒子径のマイクロスフェアを得るために、篩にかけることが好ましく、特にマイクロスフェア製剤を注射剤として用いる場合に、通針性を向上させるために、例えば150μmまたはそれ以下の径で篩過を行うのが好ましい。
【0040】
「相分離法」によってマイクロスフェアを製造するためには、前記水中乾燥法で用いたものと同様の有機溶媒の他、アセトン、アセトニトリル、テトラヒドロフラン、ジオキサンなどの両親媒性溶媒を使用することもできる。これらの有機溶媒を用いた水難溶性ポリマーの有機溶媒溶液にPDE4阻害剤および所望により他の成分、またはそれらの水溶液を加えて溶解または分散させ、有機相を形成させる。この有機相を上記有機溶媒と混和しない溶媒(分散媒)、たとえばシリコンオイル類、流動パラフィン、ゴマ油、大豆油、コーン油、綿実油、ココナッツ油、アマニ油などに撹拌下徐々に添加してO/O型エマルションを形成させる。所望により分散媒には界面活性剤を添加してもよい。このエマルションを冷却して水難溶性ポリマーを固化させる、もしくは加熱して有機相中の溶媒を蒸発させることで水難溶性ポリマーを固化させる。または攪拌下このエマルションに硬化剤、たとえばヘキサン、シクロヘキサン、メチルエチルケトン、オクタメチルシクロテトラシロキサン等を徐々に添加する、もしくは硬化剤にエマルションを徐々に添加することによって、水難溶性ポリマーを析出させることでマイクロスフェアを形成させる。
【0041】
このようにして形成されたマイクロスフェアは遠心分離、濾過あるいは篩などで回収し、その表面に付着した溶媒や添加剤をヘキサンや精製水などで洗浄除去し、所望により通風乾燥、減圧乾燥、凍結乾燥などに付す。あるいは前記水中乾燥法の場合と同様に、凝集防止剤などを添加した後、凍結乾燥に付す。
なお、相分離法における内部有機相の形態としては以下のものが含まれる。
(a)水難溶性の生体内適合性かつ生体内分解性ポリマー溶液に、薬物が直接溶解もしくは分散されている有機相。
(b)水難溶性の生体内適合性かつ生体内分解性ポリマー溶液に、薬物水溶液が分散されているW/O型エマルションからなる有機相。
(c)2種以上の水難溶性の生体内適合性かつ生体内分解性ポリマーを用い、一方のポリマー溶液中に分散されている他方のポリマー溶液中に、薬物が溶解もしくは分散もしくは薬物溶液が分散しているO/O型エマルションからなる有機相。
【0042】
また「噴霧乾燥法」によってマイクロスフェアを製造するには、前記相分離法で用いたものと同様の有機溶媒を用い、これに水難溶性の生体内適合性かつ生体内分解性ポリマーを溶かし、その有機溶媒溶液に、PDE4阻害剤および所望により他の成分を溶解、分散させるか、またはそれら薬物の水溶液を分散させ、その分散液をスプレーノズルを通して噴霧乾燥器内に噴霧して有機溶媒を揮発させてマイクロスフェアを形成させる。
用いられる噴霧乾燥器は市販の製品、例えばパルビス・ミニ・スプレイ(Pulvis Mini Spray)GS31(ヤマト製)、ミニスプレードライヤー(柴田科学製)などがいずれも使用され得る。
【0043】
このようにして得られるマイクロスフェアは水中乾燥法で得られるものと同様に後処理に付されて所望のマイクロスフェア製剤が得られる。
「溶媒拡散法」において、水混和性有機溶媒としてはアセトン、メタノール、エタノール、これらの混合溶媒が挙げることができ、必要に応じて薬物を溶解し得る揮発性溶媒(塩化メチレン、クロロホルム)を加えてもよい。保護コロイド剤としてはポリビニルアルコールを挙げることが出来る。
【0044】
本発明のPDE4阻害剤を有効成分とする軟骨疾患修復治療用組成物をマイクロスフェアの形で疾患部位またはその近傍に投与する場合には、局所的に投与するのが好ましく、局所、とりわけ関節内への注射または埋め込みが好ましい。
マイクロスフェアを注射剤とするには、分散剤を含有する水溶液に本発明で得られるマイクロスフェアを、0.0001〜1000mg/ml、好ましくは0.0005〜800mg/ml、さらに好ましくは0.001〜500mg/mlの濃度となるように分散して調製することができる。
【0045】
用いられる分散剤としては、ポリオキシエチレンソルビタン脂肪酸エステル(Tween80、Tween60(日光ケミカルズ製)など)、ポリエチレンヒマシ油(HCO−60、HCO−50(日光ケミカルズ製)など)等の非イオン界面活性剤、カルボキシメチルセルロースナトリウム等のセルロース系分散剤、アルギン酸ナトリウム、デキストラン、ヒアルロン酸ナトリウムが挙げられる。これらの分散剤は、マイクロスフェアの分散性を向上させ、薬効成分の溶出を安定化させる働きがあり、通常、0.01〜2重量%、好ましくは0.05〜1重量%で添加される。
上記注射剤には、適宜、保存剤(メチルパラベン、プロピルパラベン、ベンジルアルコール、クロロブタノール、ソルビン酸、ホウ酸、アミノ酸、ポリエチレングリコール類など)、等張化剤(塩化ナトリウム、グリセリン、ソルビトール、ブドウ糖、マンニトールなど)、pH調節剤(水酸化ナトリウム、水酸化カリウム、塩酸、リン酸、クエン酸、シュウ酸、炭酸、酢酸、アルギニン、リジンなど)、緩衝剤(リン酸水素ナトリウム、リン酸水素カリウムなど)が配合される。
【0046】
注射剤には、更に、必要に応じて、ステロイド系消炎鎮痛剤、非ステロイド系消炎鎮痛剤を溶解・分散してもよい。ステロイド系消炎鎮痛剤としては、例えば、デキサメタゾン、トリアムシノロン、トリアムシノロンアセトニド、ハロプレドン、パラメタゾン、ヒドロコルチゾン、プレドニゾロン、メチルプレドニゾロン、ベタメタゾン等をあげることができる。また、非ステロイド系消炎鎮痛剤としては、イブプロフェン、ケトプロフェン、インドメタシン、ナプロキセン、ピロキシカム等を挙げることができる。
また、PDE4阻害剤を含有するマイクロスフェアの注射剤は、上記懸濁液のほか、用時調製用に、凝集防止剤およびマイクロスフェアを含有する固形製剤と分散剤および注射用蒸留水を組み合わせた注射剤キットとすることもできる。
【0047】
キットに使用する固形製剤は、凝集防止剤を含む水溶液にマイクロスフェアを懸濁後、凍結乾燥、減圧乾燥、噴霧乾燥等することにより、調製することができ、とりわけ、凍結乾燥で調製するのが好ましい。
固形製剤の製造に際しては、注射用蒸留水への再分散性を向上させるために、凝集防止剤(マンニトール、ソルビトール、ラクトース、ブドウ糖、キシリトール、マルトース、ガラクトース、シュクロース等)を含む水溶液に分散剤を添加することもでき、分散性のよい固形剤とすることができる。必要に応じて、分散剤と共に、ステロイド系消炎鎮痛剤、非ステロイド系消炎鎮痛剤を組合わせた注射剤キットとすることもできる。
【0048】
本発明のPDE4阻害剤を有効成分とする軟骨疾患修復治療用組成物は、各種温血哺乳動物、例えば、ヒト、家畜動物(ウマ、ウシ、ヒツジ、ブタ)、愛玩動物(イヌ、ネコ)等に用いることができ、軟骨疾患修復治療用組成物は、これらの動物における変形性関節症、軟骨形成異常症、変形性椎間板症、半月板損傷等の各種軟骨疾患の修復治療に使用することができ、とりわけ、変形性関節症の修復治療に好適に使用することができる。
【図面の簡単な説明】
【0049】
【図1】ウサギ関節軟骨細胞抽出液をMonoQセファロースカラムクロマトグラフィーで分離した各画分におけるcAMP加水分解活性を示すグラフである(化合物番号(44)存在下:○;化合物番号(44)非存在下:●)。
【図2】化合物番号(1)含有マイクロスフェアまたは非含有マイクロスフェアの存在下での、老齢ウサギ関節軟骨の修復を観察した顕微鏡写真の模写図である。
【図3】ヒト関節軟骨細胞抽出液をMonoQセファロースカラムクロマトグラフィーで分離した各画分におけるcAMPまたはcGMP加水分解活性を示すグラフである。
【図4】図3においてcAMPに対し強い加水分解活性を示した画分(フラクション番号28〜30 に対するPDE4阻害剤の阻害活性(IC50)を示すグラフである。
【図5】実施例1、実施例2および実施例3のマイクロスフェアのインビトロでの薬物溶出特性を示すグラフである。
【図6】化合物番号(1)をラットに静脈内投与した場合の、血漿中濃度の経時変化を示すグラフである。データは平均値±標準偏差(例数:3)で示した。
【図7】実施例1−(5)、実施例2−(2)および実施例3−(2)で得られたマイクロスフェア分散液をラットに皮下注射した場合の血漿中濃度の経時変化を示すグラフである。データは平均値±標準偏差(例数:5)で示した。
【図8】実施例2−(2)で得られたマイクロスフェア分散液をラットに皮下注射した場合の製剤中に残存している化合物番号(1)の経時変化を示すグラフである。データは平均値±標準偏差(例数:5)で示した。
【図9】実施例6−(5)および実施例7−(2)で得られたマイクロスフェア分散液をラットに皮下注射した場合の製剤中に残存している化合物番号(2)の経時変化を示すグラフである。データは平均値±標準偏差(例数:4)で示した。
【発明を実施するための形態】
【実施例】
【0050】
つぎに、実験例、実施例および試験例を挙げて本発明をさらに具体的に説明する。なお、以下の実施例等において用いる化合物番号は前記化学構造式で示した好ましいPDE4阻害剤の具体例の化合物番号を意味する。
実験例1(関節軟骨細胞の基質産生増加)
(試験化合物)
化合物番号(1)(10−5Mまたは10−4M);
化合物番号(2)(10−6Mまたは10−5M);
化合物番号(9)(10−6Mまたは10−5M);
化合物番号(11)(10−6M);
化合物番号(21)(10−6Mまたは10−5M);
化合物番号(27)(10−6Mまたは10−5M);
化合物番号(44)(10−5Mまたは10−4M);
【0051】
(関節軟骨細胞の分離・維持)
NZW系ウサギ(北山ラベス;雄性;4週齡)4羽をエーテル麻酔後、放血致死させ、無菌的に大腿骨側の膝関節を得た。これをグルコース0.2%含有リン酸緩衝液(pH7.2)中に保存し、膝関節部の表層のみをメスで削り取り、グルコース0.2%含有リン酸緩衝液(pH7.2)を入れた50mlチューブに集めた。集められた関節表層をシャーレ上でカミソリを用いてできるだけ細かく切り刻み、10xトリプシン−エチレンジアミンテトラ酢酸(EDTA)・4Na(GIBCO製;カタログ番号15400−054)を含むグルコース0.2%含有リン酸緩衝液(50ml、トリプシン100mg含有、EDTA・4Na 40mg含有;pH7.2)中、37℃で15分間振盪した。振盪後、遠心分離(1,400rpm)により、沈殿物を分取後、グルコース0.2%含有リン酸緩衝液40mlで2回洗浄した。洗浄後の沈殿物に細胞分散用コラゲナーゼ(和光純薬製;カタログ番号034−10533)60mgを溶解した無血清α最小必須培地(MEM:minimum essential medium)(GIBCO製;カタログ番号12571−063)40mlを添加し、これを滅菌済スターラー・バー入りの100mlビーカーに移した。スターラー・バーを回転させながら、37℃のCO2インキュベーターで約1時間コラゲナーゼ処理した。コラゲナーゼ処理物から40μMセルストレーナー(FALCON製;カタログ番号2340)により軟骨片を除去し、除去後の処理物に10%ウシ胎仔血清(FCS:fetal calf serum)含有αMEM培地10mlを添加し、遠心分離(1,400rpm)した。沈殿物を10%FCS−αMEM培地で2回洗浄し、同培地で適当な容量に懸濁し、この懸濁液を1ウェル当たり細胞数2万個となるようにコラーゲンタイプII(和光純薬製;カタログ番号033−13901)コートプレート(48ウェル)に播種した。翌日10%FCS−αMEM培地で培地交換した。
【0052】
(基質産生増加)
上記操作で培地交換後、細胞がコンフルエントになった段階で、検体添加群には試験化合物含有培地(ビヒクルとしてジメチルスルホキシドを0.1%含有)で培地交換した。試験化合物を添加する際に用いる培地には、0.2mMアスコルビン酸含有10%FCS−αMEM培地を用いた。最初の検体含有培地添加日をday1とし、day3に再度同培地で培地交換し、day5まで培養を継続した。一方、検体非添加群には、試験化合物を含有しないこと(ビヒクルのみ含有)以外は全く同一の培地を同時期に添加し、同様に培養を行った。培養終了後、培養物から上清を除去し、パラホルムアルデヒドを4%の濃度に溶解した中性緩衝液0.25mlを添加して2時間細胞を固定した。リン酸緩衝液(pH7.2)1mlで3回洗浄後、軟骨基質(プロテオグリカン)を選択的に着色するアルシアンブルー8GX(シグマ製;カタログ番号A3157)を0.1%濃度に溶解した0.1M塩酸で4時間染色した。染色後、リン酸緩衝液(pH7.2)1mlで3回洗浄し、6M塩酸グアニジン水溶液0.25mlで、軟骨基質を染色したアルシアンブルーを溶解し、その一部を用いて620nmでの吸光度を測定した。この吸光度から染色に用いられたアルシアンブルー量を算出し、この量から基質(プロテオグリカン)の量を推定した。その結果を表1に示す。
【表1】
【0053】
上記表1に示すとおり、PDE4阻害活性を有する試験化合物(1)、(2)、(9)、(11)、(21)、(27)および(44)のすべてに基質産生増加作用が認められた。
実験例2(関節軟骨中に発現するcAMP分解性PDEの分画)
NZW系ウサギ(北山ラベス;雄性;4週齢)4羽をエーテル麻酔後、放血致死させ、無菌的に大腿骨側の膝関節を得た。これをグルコース0.2%含有リン酸緩衝液(pH7.2)中に保存し、膝関節部の表層のみをメスで削り取り、グルコース0.2%含有リン酸緩衝液(pH7.2)を入れた50mlチューブに集めた。集められた関節表層をシャーレ上でカミソリを用いてできるだけ細かく切り刻み、氷冷したリン酸塩緩衝液で洗浄した後、氷冷ホモジナイズ用緩衝液(20mM Tris−HCl、pH7.4、2mM酢酸マグネシウム、0.3mM塩化カルシウム、1mMジチオスレイトール、40μMロイペプチン、1.3mMベンズアミジン、0.2mMフェニルメチルスルホニルフルオリド、1mMアジ化ナトリウム)中でホモジナイザー(Polytoron, Kinematica製)を用いて破砕した。得られたホモジネートを遠心(100000×g、60分間)し、上清を分取した。
【0054】
溶出緩衝液(20mM Tris−HCl、pH7.4、1mM塩化カルシウム、1mMジチオトレイトール、2μMロイペプチン、5mMベンズアミジン)で平衡化したMonoQセファロース高速カラム(Amersham Pharmacia Biotech社製)に、前記で得た上清を供した。カラムを溶出緩衝液20mlで洗浄した後、塩化ナトリウム勾配で蛋白を溶出し、氷冷下1mlずつ分画した。各画分について、cAMPを基質とする加水分解活性(PDE活性)を測定した。
PDE活性の測定は、ラジオラベル核酸法により行った。すなわち、1μMの非標識cAMPおよび22nMの[3H]−cAMP(Amersham Pharmacia Biotech社製)を含むアッセイ用緩衝液(50mM Tris−HCl、pH8.0、5mM塩化マグネシウム、4mM 2−メルカプトエタノール、0.33mg/mlウシ血清アルブミン(脂肪酸不含、シグマ社製)、1mMエチレングリコールビス(β−アミノエチルエーテル)−N,N,N',N'−4酢酸)500μl中に10〜30μlの溶出画分を添加して反応を開始した。薬物非投与群は薬物を添加せず、薬物投与群には1×10−5Mになるように化合物番号(44)を添加した。37℃で30分間保温して反応を行った後、1.5分間煮沸して反応を停止させ、さらに1mg/mlのヘビ毒(Crtalus atrox snake venom)100μlを添加して37℃で30分間保温した。ついで、500μlのメタノールを添加し、反応液をDowexカラム(1x8−400)で処理した後、各溶出液に液体シンチレーションカクテルを加え、ラジオ活性を測定した。その結果を図1に示す。
【0055】
図1に示されるとおり、cAMPに対する強い加水分解活性の4つのピークが認められた。これらの4つのピークは、(1)cAMPを選択的に加水分解すること、(2)そのcAMP加水分解活性がcGMPによる影響を受けないこと、(3)選択的PDE4阻害薬である化合物番号(44)により強く阻害されること、という性質をみたすことから、PDE4に由来するピークであると考えられた。PDE4にはPDE4A、PDE4B、PDE4C、PDE4Dの4つのサブタイプが報告されており(サルドウら、セルラー・シグナリング[Cellular Signaling]、10巻、427〜440頁、1998年)、4つのピークはこれらのサブタイプあるいはそれらから由来するスプラシンング変異体であると考えられた。
【0056】
実験例3(軟骨基質タンパク遺伝子の発現)
最終濃度1×10−4および1×10−5Mの化合物番号(1)の存在下で実験例1と同じ方法によって4日間培養したウサギ膝関節軟骨細胞より、ISOGEN(ニッポンジーン社製)によって抽出した全RNA15μgを4.5μlの滅菌水に溶解した。この溶液と2μlの5×MOPS緩衝液、3.5μlのホルムアルデヒド、10μlのホルムアミドとを混合し、90℃、15分間熱変性した後、ホルムアルデヒド存在下で1%アガロースゲル上で電気泳動した。電気泳動終了後、RNAをナイロン膜(アマシャムファルマシア社製)へキャピラリー法にて一晩トランスファーを行った。UV架橋によりRNAをナイロン膜に固定した後、50mlのハイブリダイゼーション溶液(6×SSC、5×Denhardt's溶液、(50%ホルムアミドはなし)、0.5%SDS、100μg/ml熱変性サケ精子DNA)中にて60℃、2時間プレハイブリダイゼーションを行った。
【0057】
次いでマウスII型コラーゲン遺伝子、ヒトアグリカン(プロテオグリカンを構成する代表的な蛋白)遺伝子のDNAプローブを各々、α[32P]dCTPを用い、ランダムプライムラベリングキットVer.2(アマシャムファルマシア社製)によって放射性標識した。各プローブ1×108dpmを各々、5mlのハイブリダイゼーション溶液と共にプレハイブリダイゼーション処理したナイロン膜に加え、密封した後、60℃で一晩ハイブリダイゼーションを行った。ナイロン膜の洗浄は0.2×SSC、0.2%硫酸ドデシルナトリウムを含む溶液中で、60℃、40分間の洗浄操作を3回繰り返し行った。ナイロン膜オートラジオグラフィーに供し、X線フィルムをLAS−1000(富士写真フィルム社製)によって撮影した。各RNAの相対量をImage Gause(富士写真フィルム社製)によって測定した後、28SRNA(内因性RNA:内部標準)によって補正し、非投与群の遺伝子発現の量を100(%)として、試験化合物投与群の遺伝子発現の量を算出した。その結果を表2に示す。
【表2】
【0058】
上記表2に示すとおり、PDE4阻害薬である試験化合物(1)の添加群においては、II型コラーゲン、アグリカン遺伝子の発現量は濃度依存的に増大していたことから、PDE4阻害薬は関節軟骨細胞に作用し、関節軟骨基質の主要な成分であるII型コラーゲン、アグリカンの遺伝子発現を増大させることにより、軟骨基質の産生を促進することがわかった。
【0059】
実験例4(老齢ウサギ膝関節軟骨基質増加)
(予備飼育)
老齢のJW系ウサギ(北山ラベス;雄性;37週齢)を室温(23±2℃)、湿度(50±20%)で飼育した。市販の餌(オリエンタルバイオ製;CE−2)を自由摂取させた。
(関節軟骨基質増加)
上記ウサギをネンブタール(ダイナボット社製;50mg/kg/ml)を耳静脈に注射し、麻酔した。左膝関節部の毛を剃り、70%水性エタノールで消毒後、検体投与群には、関節内に、実施例2−(3)で製造した検体含有マイクロスフェア分散液250μl(薬物量:2.5mg)を18ゲージの注射針(テルモ製)を用いて注入した。一方、検体非投与群には、関節内に、対照例1−(2)で製造した検体非含有マイクロスフェア分散液250μlを注入した。投与から14日後に、ウサギをネンブタール麻酔下で放血致死させ、膝関節を摘出し、ホルムアルデヒドを10%の濃度に溶解した中性緩衝液で固定後、0.5M EDTA−4Na水溶液による脱灰処理を施し、切片を作成した。切片に軟骨基質(プロテオグリカン)を選択的に着色するアルシアンブルー8GX(シグマ製;A3157)を0.10%濃度に溶解した0.1M塩酸で染色し、検体投与群と検体非投与群の間の染色性を顕微鏡にて比較した。その結果、アルシアンブルーで染色される基質(プロテオグリカン)の層の厚さが検体投与群では検体非投与群の層の厚さの3倍以上となっていた。
【0060】
実験例5(老齢ウサギ膝関節軟骨修復)
(予備飼育)
老齢のJW系ウサギ(北山ラベス;雄性;37週齢)を、ウサギ専用ケージ(C type:W370×D520×H330)中、室温(23±2℃)、湿度(50±20%)で飼育した。市販の餌(オリエンタルバイオ製;CE−2)を自由摂取させた。
(関節軟骨修復)
上記ウサギをネンブタール(アボット社製;50mg/kg/ml)を耳静脈に注射し、麻酔した。左膝関節部の毛を剃り、70%水性エタノールで消毒後、左膝正中靭帯を切開し、大腿骨頭および半月板を露出させ、周囲の血液は滅菌綿で止血した。大腿骨骨頭中間部の窪み(非荷重部)に直径2mm、深さ3mmの穴をドリル(東洋アソシエイツ製;ミスターマイスター)を用いてあけた。滅菌生理食塩水で穿孔中に生じた骨片などを洗浄除去後、関節包、正中靭帯を絹糸で縫合し、滅菌綿で止血、消毒した。9日後、検体投与群には、関節内に、実施例2−(3)で製造した検体含有マイクロスフェア分散液250μl(薬物量:2.5mg)を18ゲージの注射針(テルモ製)を用いて注入した。一方、検体非投与群には、関節内に、対照例1−(2)で製造した検体非含有マイクロスフェア分散液250μlを注入した。投与から14日後に、ウサギをネンブタール麻酔下で放血致死させ、膝関節を摘出し、ホルムアルデヒドを10%の濃度に溶解した中性緩衝液で固定後、0.5M EDTA−4Na水溶液による脱灰処理を施し、切片を作成した。孔の修復度を顕微鏡で観察した。その結果を図2に示す。図2に示されるとおり、検体投与群は検体非投与群に比べ明らかに孔の修復が進んでいることが確認された。
【0061】
実験例6(パパイン誘発変形性膝関節症に対する修復治癒効果)
(予備飼育)
日本白色種ウサギ(北山ラベス;雄性;13週齡)を室温(23±2℃)、湿度(50±20%)で8日間飼育した。飼育期間中、市販の餌(RC4 オリエンタル酵母)を1日約140gの割合で摂取させた。
(修復治癒効果)
上記ウサギにネンブタール(アボット:lot 791102)を耳静脈から注射し、麻酔した。ウサギの両膝部分の毛をかり、70%エタノールで消毒した。両膝に0.8%パパイン(Merck EC3.4.22.2 lot 587644 019)生理食塩水溶液0.5mlを5日間隔で2回注射した。二回目の注射から1週間後、検体投与群(左膝;1群4〜6匹)には、実施例2−(3)で製造したマイクロスフェア分散液(化合物番号(1)の化合物を0.2mgまたは2mg含有)を関節内に注射した。一方、検体非投与群(右膝;1群4〜6匹)には対照例1−(2)で製造した薬物非含有マイクロスフェア分散液を検体投与群の分散液と同量関節内に注射した。また、アルツ投与群(左膝;1群2匹)には、1%ヒアルロン酸ナトリウム水溶液(科研製薬製アルツ)0.3mlを関節内に注射した。一方、アルツ非投与群(右膝;1群2匹)には、0.3mlの生理食塩水を関節内に注射した。アルツ投与群およびアルツ非投与群には、同様の関節内注射を1週間毎に、計4回ずつ行った。最終の注射から4週間後、ウサギをエーテル麻酔下で放血により安楽死させ、膝を摘出し、ホルムアルデヒドを10%の濃度に溶解した中性緩衝液にて固定した。その結果、パパイン処置によって、関節軟骨表面の不整化、ヘマトキシリン・エオジン染色性の低下、軟骨基質の繊維化、関節軟骨細胞の消失などを特徴とする関節軟骨の変性が見られた。アルツ処理では、ヘマトキシリン・エオジン染色で若干の染色性低下の抑制はみられたものの、大幅な回復効果は見られなかった。一方、化合物番号(1)含有マイクロスフェア投与では、上記病理症状が著しく改善した。なお、検体非投与群およびアルツ非投与群のいずれにおいても回復効果は全く認められなかった。
【0062】
実験例7(ヒト関節軟骨中に発現するcAMP分解性PDEの分画)
ヒト変形性関節症患者の手術の際に摘出した膝関節軟骨から軟骨部分をメスで削り取り、氷冷したリン酸緩衝液で洗浄した後、−80℃で保存した。軟骨を−80℃で粉砕後、氷冷ホモジナイド緩衝液[20mM Tris-HCl、pH8.0、1mM エチレングリコールビス(β−アミノエチルエーテル)―N,N,N',N’−テトラ酢酸、1mM ジチオスレイトール、10μg/ml ロイペプチン、5mM ベンズアミジン、0.2mM フェニルメチルスルホニルフルオリド、1mM アジ化ナトリウム、5mM メルカプトエタノール]中で、ホモジナイザー(Kinematica製、Polytoron)を用いて粉砕した。得られたホモジネートを遠心分離(100000×g;30分)して、上清を分取した。
溶出緩衝液[20mM Tris-HCl、pH8.0、1mM エチレングリコールビス(β−アミノエチルエーテル)―N,N,N',N’−テトラ酢酸、1mM ジチオスレイトール、2μg/ml ロイペプチン、5mM ベンズアミジン]で平衡化したMonoQセファロース高速カラム(Amasham Pharmacia Biotech社製)に、上記上清を導通した。カラムを溶出緩衝液20mlで洗浄後、塩化ナトリウム水溶液(濃度勾配:0〜1000mM、70ml)でタンパクを溶出し、氷冷下で1mlずつ分画した。各分画について、cAMP及びcGMPを基質とする加水分解活性(PDE活性)を測定した。
【0063】
PDE活性の測定は、ラジオラベル核酸法により行った。すなわち、1μMの非標識cAMPおよび22nMの[3H]−cAMP(Amasham Pharmacia Biotech製)を含むアッセイ用緩衝液(50mM Tris−HCl、pH8.0、5mM塩化マグネシウム、4mM 2−メルカプトエタノール)500μl中に10〜50μlの溶出画分を添加して反応を開始した。
37℃で30分間保温した後、1.5分間煮沸して反応を停止させ、更に、1mg/mlのヘビ毒(Crtalus atrox snake venom)100μlを添加して、37℃で30分間保温した。ついで、500μlのメタノールを添加し、反応液をDowexカラム(1×8−400)で処理した後、溶液に液体シンチレーションカクテルを加え、ラジオ活性を測定した。結果を図3に示す。
【0064】
図3に示されるように、ヒト変形性関節症患者の軟骨の処理液には、cGMPに対する加水分解活性を有する画分は含まれていないが、cAMPに対する強い加水分解活性を有する画分が含まれていることが判明した。
cAMPに対する強い加水分解活性を有する画分(フラクション番号:28〜30に対するPDE4阻害剤の阻害活性を、前述のラジオラベル核酸法で測定した。試験化合物は、化合物番号(1)、化合物番号(2)、化合物番号(11)、化合物番号(44)、化合物番号(27)である。
実験の結果、該画分の加水分解活性は化合物番号(1)および(2)、特に化合物番号(2)により強く阻害を受けることが確認された。また、Journal of Medicinal Chemistry vol. 42, 1088-1099 (1999)記載の方法で測定した、この画分に対する化合物番号(1)および(2)のIC50は同文献に記載されたPDE4阻害作用のIC50と一致した。結果を図4に示す。
【0065】
実験例8(ヒト関節軟骨細胞における細胞内cAMP上昇)
(関節軟骨細胞の分離)
ヒト関節軟骨(変形性股関節症患者の関節軟骨)をリン酸緩衝液(pH7.2)中に浸し、関節部の表層のみをメスで削り取り、同緩衝液を入れた50mlチューブに集めた。集められた関節表層をシャーレ上でカミソリを用いてできるだけ細かく切り刻み、遠心管に移した。
【0066】
ヒアルロニダーゼ(シグマ社製;カタログ番号H−3506)を1mg/mlの濃度で含むリン酸緩衝液(pH7.2)を遠心管に入れ、37℃で15分間振盪した。遠心分離(2000rpm、5分間)により沈殿物を分取後、トリプシンを0.25%の濃度で含むHank’s balanced salt solution(GIBCO社製;カタログ番号15050−065)に入れ、37℃で30分間振盪した。
遠心分離(2000rpm、5分間)により沈殿物を分取後、沈殿物に細胞分散用コラーゲナーゼ(和光純薬製;カタログ番号034−10533)を0.25%の濃度で、ウシ胎仔血清(GIBCO社製;fetal calf serum;カタログ番号10099−141)を10%の濃度で含有するα最小必須培地(GIBCO社製;カタログ番号12571−063)を添加し、37℃で1晩振盪した。
【0067】
40μmセルストレーナー(FALCON社製;カタログ番号2340)により軟骨片を除去し、除去後のコラーゲナーゼ処理物に、ウシ胎仔血清を10%の濃度で含有するα最小必須培地を添加して遠心分離(1400rpm、10分間)した。
ウシ胎仔血清を10%の濃度で含有するα最小必須培地で沈殿物を3回洗浄後、同培地で適当な容量に懸濁し、この懸濁液を1ウエル当たり細胞数50000個となるようにプレート(48ウエル)に播種した。翌日、ウシ胎仔血清を10%の濃度で含有するα最小必須培地で培地交換した。α最小必須培地は、抗生物質(100U/mlペニシリンG、100μg/ml硫酸ストレプトマイシン)、抗真菌剤(0.25μg/mlアンフォテリシンB)(GIBCO社製;カタログ番号15240−062)を添加したものを用いた。
【0068】
(細胞内cAMP濃度上昇)
培地の交換後、細胞がコンフルエントになった段階で、検体添加群では、PGE2(シグマ社製、カタログ番号P−0409)を1μMの濃度で、検体化合物を10−6M又は10−5M濃度で添加した、ウシ胎仔血清を10%の濃度で含有するα最小必須培地(ジメチルスルホキシド0.1%を含有)で培地交換した。一方、検体非添加群では、PGE2(シグマ社製、カタログ番号P−0409)を1μMの濃度で添加した、ウシ胎仔血清を10%の濃度で含有するα最小必須培地(ジメチルスルホキシド0.1%を含有)で培地交換した。
30分間培養後、培地を捨て、リン酸緩衝液(pH7.2)で洗浄し、50%エタノール水溶液で30分間処理後、エタノールを回収し、エタノール抽出物を蒸発乾固した。これをcAMP EIAシステム(Amasham Pharmacia Biotech社製;カタログ番号RPN225)に添付されたアッセイ用緩衝液に溶解し、同システムでcAMP濃度を測定した。その結果を表3に示す。
【表3】
【0069】
実験例9(ウサギ関節軟骨細胞のIL−1存在下での基質産生)
NZW系ウサギ(北山ラベス;雄性;4週齢)4羽をエーテル麻酔後、放血致死させ、無菌的に大腿骨側の膝関節を得た。これから、グルコースを0.2%含有するリン酸緩衝液(pH7.2)中で、膝関節部の表層のみをメスで削り取り、グルコースを0.2%含有するリン酸緩衝液(pH7.2)を入れたチューブに集めた。集められた関節表層をシャーレ上でカミソリを用いてできるだけ細かく切り刻み、10xトリプシン−エチレンジアミンテトラ酢酸4ナトリウム(EDTA・4Na)(GIBCO社製;カタログ番号15400−054)を添加した、グルコースを0.2%の濃度で含有するリン酸緩衝液(トリプシン100mg、EDTA・4Na40mg含有;pH7.2)50mlに加えて、37℃で15分間振盪した。
振盪後、遠心分離(1400rpm)して沈殿物を分取し、これをグルコースを0.2%含有するリン酸緩衝液(pH7.2)40mlで2回洗浄した。
【0070】
洗浄後の沈殿物に細胞分散用コラーゲナーゼ(和光純薬製;カタログ番号034−10533)60mgを溶解した、抗生物質(200U/mlペニシリンG、200μg/ml硫酸ストレプトマイシン)(GIBCO社製;カタログ番号15140−122)を含有するα最小必須培地(GIBCO社製;カタログ番号12571−063)40mlを添加し、これを滅菌済スターラー・バー入りの100mlビーカーに移した。
スターラー・バーを回転させながら、37℃のCO2インキュベーターで約30分間培養後、デオキシリボヌクレアーゼI(宝酒造製;カタログ番号2210A)を70U/mlとなるように添加し、同条件で、更に30分間培養した。
培養物の上清を別容器に移し取り、残った軟骨片を新たに調製した、コラーゲナーゼ60mgを含み、デオキシリボヌクレアーゼIを70U/mlの濃度で含有するα最小必須培地で、約30分間培養した。
【0071】
先に移し取った培養物上清及び最終培養物から40μmセルストレーナー(Falcon製;カタログ番号2340)で軟骨片を除去し、除去後の培養物に、ウシ胎仔血清(GIBCO社製;fetal calf serum;カタログ番号10099−141)を10%の濃度で含み、抗生物質(200U/mlペニシリンG、200μg/ml硫酸ストレプトマイシン)(GIBCO社製;カタログ番号15140−122)を含有するα最小必須培地(GIBCO社製;カタログ番号12571−063)10mlを添加し、遠心分離(1400rpm,10分間)を行った。
沈殿物を同様のウシ胎仔血清、抗生物質含有α最小必須培地で2回洗浄し、同培地で適当な容量に懸濁し、この懸濁液を1ウエル当たり細胞数20000個となるようにプレート(48ウエル)に播種した。翌日、同培地で培地交換を行った。
【0072】
(基質産生増加)
上記培地交換後、細胞がコンフルエントになった段階で、検体投与群には、1ng/mlのリコンビナントヒトIL−1β(PEPRO TECH製;カタログ番号200−01B)と検体化合物を含有した培地(ジメチルスルホキシドを0.1%の濃度で含有)で培地交換した。この培地にはアスコルビン酸を0.2mMの濃度で含む、前記ウシ胎仔血清、抗生物質含有α最小必須培地を用いた。一方、検体非投与群は、検体化合物を含有しない以外は検体投与群と同様の培地で培地交換した。
最初のIL−1β含有培地添加日をday1とし、day3まで培養した。
【0073】
培養後、培養物から上清を除去し、10%中性緩衝ホルマリン液(和光純薬製;カタログ番号060−01667)0.25mlを添加して30分間細胞を固定した。
固定した細胞をリン酸緩衝液(pH7.2)1mlで3回洗浄後、基質(プロテオグリカン)を選択的に染色するアルシアンブルー8GX(シグマ社製;カタログ番号A3157)を0.1%の濃度で含む0.1M塩酸で4時間染色した。
染色後、リン酸緩衝液(pH7.2)1mlで3回洗浄し、6M塩酸グアニジン水溶液0.25mlで、軟骨基質を染色したアルシアンブルーを溶解した。その一部を用いて、620nmでの吸光度を測定し、この吸光度から染色に用いられたアルシアンブルー量を算出した。このアルシアンブルー量から基質(プロテオグリカン)量を推定した。検体を添加しない場合のプロテオグリカン産生量を100%として、相対比率を示した。結果を表4に示す。
【表4】
【0074】
表4に示すとおり、IL−1の存在下、PDE4阻害作用を有する試験化合物は基質産生を促進した。IL−1は、変形性関節症の滑液や軟骨細胞に発現していること、および軟骨基質であるプロテオグリカン等の基質分解酵素であるMMP(matrix metalloproteinase)の産生や合成を誘導することなどから、軟骨基質分解に重要な役割を果たしていると考えられている(The Journal of Pharmacology and Experimental Therapeutics, vol 277, p1672-1675 (1996);Journal of Biochemistry, vol 123, p431-439 (1998); Arthritis & Rheumatism, vol 44, p585-594 (2001)。従って、上記の結果は本発明組成物の有効成分であるPDE4阻害剤が、IL−1が関与する軟骨基質分解を阻害しうることを示唆している。
【0075】
実験例10(内側半月板部分切除/両側側副靭帯切除により誘発される変形性膝関節症に対する修復治療効果)
(予備飼育)
日本白色ウサギ(雄性;10週齢;1群7匹)を、室温(23±2℃)、湿度(55±15%)で16日間飼育した。飼育期間中、市販の餌(オリエンタルバイオ製;LRC4)を自由摂取させた。
【0076】
(変形性膝関節症治癒)
エーテル麻酔下で、上記ウサギ右膝関節を切開し、内側半月板の1/2部分を両鋭小直剪刀を用いて切除した。同時に両側側副靭帯を切断した。術後筋肉と表皮を縫合し、消毒を行った。施術後2週間後に、エーテル麻酔下、検体投与群(1群:7匹)には、それぞれ,化合物番号(2)1μgを含む実施例7−(1)で製造したマイクロスフェアを関節腔内に投与した。検体非投与群(1群:7匹)には、対照例2で製造した薬物非含有マイクロスフェアを同量投与した。術後6週間後に、もう一度、上記薬物含有及び非含有マイクロスフェアを投与した。施術後、10週間後にエーテル麻酔下で開腹、放血により安楽死させ脛骨近位端を摘出した。摘出した脛骨近位端に墨汁を塗布して10%中性緩衝ホルマリン溶液に浸漬固定した。
【0077】
(実験結果)
ホルマリンで固定した脛骨近位端の余分な墨汁を抜き取り、真上方向から実体顕微鏡(オリンパス社製、モデルSZX12-3111)を用いて脛骨表面の画像を画像解析装置(IMAGING RESEARCH 社製 MCIDイメージングアナライザー)に取込んだ。同解析装置を用いて、内側の墨汁が残存している部分の面積(変性部面積)を測定した。同時に、内側全体の面積を測定し、変性部面積の内側全体の面積に対する割合(変性部面積率)を百分率で算出した。
結果を表5に示す。
【表5】
【0078】
実施例1
(1)化合物番号(1)0.1gおよび乳酸−グリコール酸共重合体(乳酸/グリコール酸=50/50;平均分子量20000;PLGA5020:和光純薬製)1.9gに塩化メチレン4.0gを添加し、30分間振盪溶解することにより油相(O)を形成した。
(2)油相を0.5%ポリビニルアルコール(ポバールPVA−220C:クラレ製)水溶液8mlに加え、ホモジナイザー(Polytoron:Kinematica製)を用いて25℃で5分間乳化することにより、水相に油相が分散した乳化液(O/W)を製造した。
(3)この乳化液を精製水1000mlに添加し、スリーワンモーター(新東科学製)を用いて400rpmで攪拌しながら、25℃で3時間液中乾燥することにより塩化メチレンを除去した。
(4)生成するマイクロスフェア懸濁液から、目開き150μmのフィルターを通して凝集物を除去し、目開き20μmのフィルターで吸引濾過することにより水相を除去した。得られたマイクロスフェアに少量の精製水を添加し、凍結乾燥することにより、マイクロスフェア1.6gを得た。
【0079】
得られたマイクロスフェア10mgをアセトニトリル3mlに溶解し、0.5M塩化ナトリウム水溶液7mlを加えてミキサー(Touch mixer MT-51:Yamato製)にて攪拌後、2000rpmで5分遠心分離し、上清を分取した。上清の一部をFL−HPLC(カラム;Hypersil 5-ODS 直径4mm、長さ300mm:ジーエルサイエンス製、励起波長:315nm、蛍光波長:465nm)にかけ、別途作成した薬物溶液の検量線からこれに含まれる薬物量を測定し、これと上清量とからマイクロスフェア中の薬物含有量を算出した。その結果、薬物の含有量は4.21%であった。
また、得られたマイクロスフェアをポリオキシエチレンソルビタン脂肪酸エステル(Tween80:日光ケミカルズ製)の希薄溶液に適量分散させ、粒度分布測定装置SALD−1100(島津製)にて粒度分布を測定し、平均粒子径を算出した。その結果、平均粒子径は57μmであった。
【0080】
(5)上記(4)で得られたマイクロスフェアを、カルボキシメチルセルロースナトリウム(ニチリン化学工業製)を0.5%、ポリオキシエチレンソルビタン脂肪酸エステル(Tween80:日光ケミカルズ製)を0.1%含む生理食塩水(分散媒)に2.5mg/mlの薬物割合となるように添加し、ミキサー(Touch mixer MT-51:Yamato製)にて、十分攪拌することにより、マイクロスフェア分散液を調製した。
【0081】
実施例2
(1)乳酸−グリコール酸共重合体(乳酸/グリコール酸=50/50;平均分子量20000;PLGA5020:和光純薬)0.57gと乳酸重合体(平均分子量20000;PLA0020:和光純薬製)1.33gを混合して用いること以外は、実施例1−(1)〜(4)と同様にして、マイクロスフェア1.6gを得た。
実施例1−(4)と同様にしてマイクロスフェア中の薬物含有量および平均粒子径を測定した。その結果、薬物含有量は3.70%、平均粒子径は47.7μmであった。
(2)上記(1)で得られたマイクロスフェアを実施例1−(5)と同様に処理してマイクロスフェア分散液(薬物割合:2.5mg/ml)を調製した。
(3)上記(1)で得られたマイクロスフェアを実施例1−(5)と同様にしてマイクロスフェア分散液(薬物割合:10.0mg/ml)を調製した。
【0082】
実施例3
(1)乳酸重合体(平均分子量20000;PLA0020:和光純薬製))を用いること以外は、実施例1−(1)〜(4)と同様にして、マイクロスフェア1.5gを得た。
実施例1−(4)と同様にしてマイクロスフェア中の薬物含有量および平均粒子径を測定した。その結果、薬物含有量は3.73%、平均粒子径は52.2μmであった。
(2)上記(1)で得られたマイクロスフェアを実施例1−(5)と同様に処理してマイクロスフェア分散液(薬物割合:2.5mg/ml)を調製した。
【0083】
実施例4
(1)化合物番号(1)0.2gおよび乳酸重合体(平均分子量20000;PLA0020:和光純薬製)0.3gに塩化メチレン1.0gを添加し、ミキサー(Touch mixer MT-51:Yamato製)にて十分攪拌、溶解することにより、油相(O)を形成した。
(2)油相を0.25%メチルセルロース(メトローズ:信越化学社製)水溶液4mlに加え、ホモジナイザー(Polytoron:Kinematica製)を用いて25℃で5分間乳化することにより、水相に油相が分散した乳化液(O/W)を製造した。
(3)この乳化液を精製水400mlに添加し、スリーワンモーター(新東科学製)を用いて400rpmで攪拌しながら、25℃で3時間液中乾燥することにより塩化メチレンを除去した。
(4)生成するマイクロスフェア懸濁液から、目開き150μmのフィルターを通して凝集物を除去し、目開き20μmのフィルターで吸引濾過することにより水相を除去した。得られたマイクロスフェアに少量の精製水を添加して凍結乾燥することでマイクロスフェアを得た。実施例1−(4)と同様にしてマイクロスフェア中の薬物含有量および平均粒子径を測定した。その結果、薬物含有量は39.6%、平均粒子径は33.4μmであった。
【0084】
実施例5
(1)化合物番号(1)0.05gおよび乳酸−グリコール酸共重合体(乳酸/グリコール酸=50/50;平均分子量20000;R202H:ベーリンガーインゲルハイム社製)0.45gに塩化メチレン1.0gを添加し、ミキサー(Touch mixer MT-51:Yamato製)にて十分攪拌、溶解することにより、油相(O)を形成した。
(2)油相を0.5%ポリビニルアルコール(ゴセノールEG−40:日本合成化学工業製)水溶液40mlに加え、ホモジナイザー(Polytoron:Kinematica製)を用いて25℃で4分間乳化することにより、水相に油相が分散した乳化液(O/W)を製造した。
(3)この乳化液を予め400mlの精製水を入れた円筒状密閉容器(内径110mm;内容積1000ml)に注ぎ、スリーワンモーター(BL-600;HEIDON社製)に装着した直径50mmの4枚攪拌羽根(プロペラR型;HEIDON社製)により25℃、400rpmで攪拌すると同時に、容器内に挿入した円筒型シリコーンゴム製中空糸膜モジュール(永柳工業株式会社製)を用い、中空糸の内側に窒素ガスを通気して容器内から塩化メチレンを除去した。この際の窒素ガス通気速度は2L/分とした。この操作を1時間行った。
【0085】
なお、円筒型シリコーンゴム製中空膜糸モジュールとしては、次の仕様を有するNAGASEP M60-1800円筒型を使用した。
円筒の直径 :100mm
円筒の長さ :120mm×120mm
中空糸膜の膜厚 :60μm
中空糸膜の内径 :200μm
中空糸膜の外径 :320μm
中空糸の本数 :1800
中空糸膜の有効膜面積:0.15m2
【0086】
(4)生成するマイクロスフェア懸濁液から、目開き150μmのフィルターを通して凝集物を除去し、目開き20μmのフィルターで吸引濾過することにより水相を除去した。得られたマイクロスフェアに少量の精製水を添加して凍結乾燥することでマイクロスフェアを0.26g得た。実施例1−(4)と同様にしてマイクロスフェア中の薬物含有量および平均粒子径を測定した。その結果、薬物含有量は3.07%、平均粒子径は71.7μmであった。
【0087】
実施例6
(1)化合物番号(2)0.05gおよび乳酸−グリコール酸共重合体(乳酸/グリコール酸=50/50;平均分子量20000;RG502H:ベーリンガーインゲルハイム社製)0.45gに塩化メチレン2.5gを添加し、ミキサー(Touch mixer MT-51:Yamato製)にて十分攪拌、溶解することにより、油相(O)を形成した。
(2)油相を0.5%ポリビニルアルコール(ポバールPVA−220C:クラレ製)水溶液3mlに加え、ホモジナイザー(Polytoron:Kinematica製)を用いて22℃で5分間乳化することにより、水相に油相が分散した乳化液(O/W)を製造した。
(3)上記(1)および(2)の操作を5回繰り返し、得られた乳化液(5回分)を併せた上で、精製水1000mlに添加し、スリーワンモーター(新東科学製)を用いて400rpmで攪拌しながら、25℃で1時間30分、次いで40℃で1時間、25℃で30分間液中乾燥することにより塩化メチレンを除去した。
(4)生成するマイクロスフェア懸濁液から、目開き150μmのフィルターを通して凝集物を除去し、目開き20μmのフィルターで吸引濾過することにより水相を除去した。得られたマイクロスフェアに少量の精製水を添加して凍結乾燥することでマイクロスフェア2.3gを採取した。
【0088】
得られたマイクロスフェア10mgをアセトニトリル3mlに溶解し、0.5M塩化ナトリウム水溶液6mlを加えてミキサー(Touch mixer MT-51:Yamato製)にて攪拌後、2000rpmで5分遠心分離し、上清を分取した。上清の一部をUV−HPLC(カラム;Hypersil 5-ODS 直径4mm、長さ300mm:ジーエルサイエンス製、測定波長:240nm)にかけ、別途作成した薬物溶液の検量線からこれに含まれる薬物量を測定し、これと上清量とからマイクロスフェア中の薬物含有量を算出した。また、実施例1−(4)と同様にして平均粒子径を測定した。その結果、薬物含有量は9.9%、平均粒子径は26.4μmであった。
(5)上記(4)で得られたマイクロスフェアを実施例1−(5)と同様に処理してマイクロスフェア分散液(薬物割合:0.1mg/ml)を調製した。
【0089】
実施例7
(1)乳酸−グリコール酸共重合体(乳酸/グリコール酸=75/25;平均分子量20000;PLGA7520:和光純薬製)を用いること、ならびに塩化メチレンの添加量を2.0gとする以外は、実施例6−(1)〜(4)と同様にして、マイクロスフェア2.2gを得た。
実施例6−(4)と同様にしてマイクロスフェア中の薬物含有量および平均粒子径を測定した。その結果、薬物含有量は10.1%、平均粒子径は27.0μmであった。
(2)上記(1)で得られたマイクロスフェアを実施例6−(5)と同様に処理してマイクロスフェア分散液(薬物割合:0.1mg/ml)を調製した。
【0090】
対照例1(実施例2の対照)
(1) 乳酸−グリコール酸共重合体(乳酸/グリコール酸=50/50;平均分子量20000;PLGA5020:和光純薬)0.6gと乳酸重合体(平均分子量20000)1.4gに塩化メチレン4.0gを添加し、30分間振盪溶解することにより油相(O)を形成する。以下、実施例1−(1)〜(4)と同様にして、薬物を含まないマイクロスフェア1.7gを得た。
(2)上記(1)で得られたマイクロスフェアを実施例1−(5)と同様にして、マイクロスフェア分散液(分散液中のマイクロスフェア分散濃度は実施例2−(3)と同一)を調製した。
対照例2(実施例7の対照)
乳酸−グリコール酸共重合体(乳酸/グリコール酸=75/25;平均分子量20000;PLGA7520:和光純薬製)0.45gに塩化メチレン2.0gを添加し、ミキサー(Touch mixer MT-51:Yamato製)にて十分攪拌、溶解することにより、油相を形成した。以下、実施例6−(2)〜(4)と同様にして、薬物を含まないマイクロスフェア2.2gを得た。
【0091】
試験例1
試験管にマイクロスフェア10mgを入れ、0.05% Tween80含有リン酸緩衝液(pH7.4)の10mlを添加し、37℃の空気恒温庫中の回転培養機にて25rpmで攪拌した。攪拌開始時から一定時間後に、溶出液を遠心分離(2000rpm、5分)し、上清から9mlをサンプリングして、FL−HPLC(カラム;Hypersil 5-ODS 直径4mm、長さ300mm:ジーエルサイエンス製、励起波長:315nm、蛍光波長:465nm)にかけ、別途作成した薬物溶液の検量線からこれに含まれる薬物量を測定し、これとサンプリングの量とから薬物溶出量を算出した。
また、サンプリング後の試験管に、pH7.4リン酸緩衝液9mlを添加し、同様の条件で攪拌、サンプリングして薬物溶出量を算出する操作を経時的に繰り返した。
【0092】
最終のサンプリング後、試験管から残りの溶出液を除去し、残存するマイクロスフェア中に含まれる薬物量を実施例1−(4)の方法で測定した。
この操作を実施例1、実施例2ならびに実施例3で得られたマイクロスフェアについて行った。その結果を図5に示す。
なお、溶出した薬物量とマイクロスフェアに残存した薬物量の合計を100%として、溶出率を算出した。
【0093】
試験例2
SD系雄性ラット(7週令、1群3匹、日本SLC)を12時間照明、室温:23±2℃、自由摂水・摂餌の条件下で1週間の馴化期間後、ポリエチレングリコール400(和光純薬社製)を10%含んだ生理食塩水に溶解した化合物番号(1)(1mg/ml)を1匹当たり0.5ml(総薬物投与量:0.5mg/ラット)の割合で大腿静脈から急速投与した。
薬物投与後、経時的にエーテル麻酔下で頚静脈よりヘパリンを添加した注射筒で血液を採取し遠心分離により血漿を得た。血漿0.1mlに内部標準溶液と1Mのリン酸水素2カリウム0.2mlを添加し、クロロホルム7.0mlを添加後、10分間振とう、5分間遠心分離して5mlの有機相を分取した。採取した有機相を窒素気流下、40℃で蒸発乾固し、移動相の0.5mlで再溶解後にFL−HPLC(カラム;Hypersil 5-ODS 直径4mm、長さ300mm:ジーエルサイエンス製、励起波長:315nm、蛍光波長:465nm)により血漿中濃度を測定した。その結果を図6に示す。
【0094】
試験例3
SD系雄性ラット(7週令、1群5匹、日本SLC)を12時間照明、室温:23±2℃、自由摂水・摂餌の条件下で1週間の馴化期間後、実施例1−(5)、実施例2−(2)、実施例3−(2)で得られたマイクロスフェア分散液を1匹当たり2ml(総薬物投与量:5mg/ラット)の割合で背部皮下に投与した。薬物投与後、経時的にエーテル麻酔下で頚静脈よりヘパリンを添加した注射筒で血液を採取し遠心分離により血漿を得た。血漿中の化合物濃度は試験例2と同様の方法で測定した。PDE4阻害剤をマイクロスフェア化することにより、PDE4阻害剤を1/10量しか含まない生理食塩水溶液を静脈注射した場合(試験例2)と比べても、PDE4阻害剤の最高血漿中濃度を1/25の1〜1/100に抑制することが出来た。その結果を図7に示す。
【0095】
試験例4
SD系雄性ラット(7週令、1群5匹、日本SLC)を12時間照明、室温:23±2℃、自由摂水・摂餌の条件下で1週間の馴化期間後、実施例2−(2)で得られたマイクロスフェア分散液を1匹当たり2ml(総薬物投与量:5mg/ラット)の割合で背部皮下に投与した。
薬物投与3、7、10、14、21、35日後、投与部位よりマイクロスフェアを回収した。回収したマイクロスフェアに内部標準物質を含有したアセトニトリル5mlを添加して、ホモジナイザー(Polytoron:Kinematica製)で溶解した。3000rpm、5分間遠心分離後の上清3mlを採取し、0.5M塩化ナトリウム水溶液7mlを加えてミキサー(Touch mixer MT-51:Yamato製)にて攪拌後、2000rpmで5分遠心分離し、上清を分取した。上清の一部をKCプレップオムニ13(片山化学社製)で濾過してFL−HPLC(カラム;Hypersil 5-ODS 直径4mm、長さ300mm:ジーエルサイエンス製、励起波長:315nm、蛍光波長:465nm)にかけ、別途作成した薬物溶液の検量線からこれに含まれる薬物量を測定し、これと上清量とからマイクロスフェア中の薬物残存量を算出した。その結果を図8に示す。
【0096】
試験例5
SD系雄性ラット(7週令、日本SLC)を12時間照明、室温:23±2℃、自由摂水・摂餌の条件下で1週間の馴化期間後、実施例6−(5)ならびに7−(2)で得られた化合物番号(2)含有マイクロスフェア分散液を1匹当たり1ml(総薬物投与量:0.1mg/ラット)の割合で背部皮下に投与した。
経時的に投与部位よりマイクロスフェアを回収し、アセトニトリル10mlを添加してホモジナイザー(Polytoron:Kinematica製)で溶解した。3000rpm、5分間遠心分離後の上清3mlを採取し、0.5M塩化ナトリウム水溶液6mlを加えてミキサー(Touch mixer MT-51:Yamato製)にて攪拌後、2000rpmで5分遠心分離し、上清を分取した。上清の一部をKCプレップオムニ13(片山化学社製)で濾過してUV−HPLC(カラム;Hypersil 5-ODS 直径4mm、長さ300mm:ジーエルサイエンス製、測定波長:240nm)にかけ、別途作成した薬物溶液の検量線からこれに含まれる薬物量を測定し、これと上清量とからマイクロスフェア中の薬物残存量を算出した。その結果を図9に示す。
【産業上の利用可能性】
【0097】
従来、軟骨疾患については薬物療法では保存的治療しか行えないと考えられていたにも拘わらず、本発明の軟骨疾患修復治療用組成物は、PDE4阻害剤を有効成分とし、特に、軟骨疾患部位に局所投与することにより、PDE4阻害剤の全身作用による副作用を伴うことなく、軟骨を修復することができ、軟骨疾患、特に変形性関節症に修復治療効果を発揮する。また、PDE4阻害剤と生体内適合性かつ生体内分解性ポリマーを含有する組成物をデポ剤の形とし、特にマイクロスフェア形態で注射剤として軟骨疾患部位に局所的に注射することにより持続的に効果を発揮することができ、さらに優れた効果が得られる。
【特許請求の範囲】
【請求項1】
下記化合物番号(1)、(2)、(9)、(11)、(21)、(27)、(44)、(52)、(53)、(56)または(57)またはその薬理的に許容しうる塩から選ばれる1種または2種以上のPDE4阻害剤を有効成分とする軟骨疾患修復治療用組成物。
【化1】
【化2】
【化3】
【化4】
【化5】
【化6】
【化7】
【化8】
【化9】
【化10】
【請求項2】
PDE4阻害剤が、下記化合物番号(1)、(2)、(52)、(53)、(56)または(57)またはその薬理的に許容しうる塩から選ばれる1種または2種以上である請求項1に記載の組成物。
【化11】
【化12】
【化13】
【化14】
【化15】
【請求項3】
PDE4阻害剤が、下記化合物番号(1)または(2)から選ばれる化合物またはその薬理的に許容しうる塩である請求項1記載の組成物。
【化16】
【請求項4】
PDE4阻害剤が、下記化合物番号(1)またはその薬理的に許容しうる塩である請求項2記載の組成物。
【化17】
【請求項5】
PDE4阻害剤が、下記化合物番号(2)またはその薬理的に許容しうる塩である請求項2記載の組成物。
【化18】
【請求項6】
局所投与用である請求項1〜5のいずれか1項記載の組成物。
【請求項1】
下記化合物番号(1)、(2)、(9)、(11)、(21)、(27)、(44)、(52)、(53)、(56)または(57)またはその薬理的に許容しうる塩から選ばれる1種または2種以上のPDE4阻害剤を有効成分とする軟骨疾患修復治療用組成物。
【化1】
【化2】
【化3】
【化4】
【化5】
【化6】
【化7】
【化8】
【化9】
【化10】
【請求項2】
PDE4阻害剤が、下記化合物番号(1)、(2)、(52)、(53)、(56)または(57)またはその薬理的に許容しうる塩から選ばれる1種または2種以上である請求項1に記載の組成物。
【化11】
【化12】
【化13】
【化14】
【化15】
【請求項3】
PDE4阻害剤が、下記化合物番号(1)または(2)から選ばれる化合物またはその薬理的に許容しうる塩である請求項1記載の組成物。
【化16】
【請求項4】
PDE4阻害剤が、下記化合物番号(1)またはその薬理的に許容しうる塩である請求項2記載の組成物。
【化17】
【請求項5】
PDE4阻害剤が、下記化合物番号(2)またはその薬理的に許容しうる塩である請求項2記載の組成物。
【化18】
【請求項6】
局所投与用である請求項1〜5のいずれか1項記載の組成物。
【図1】

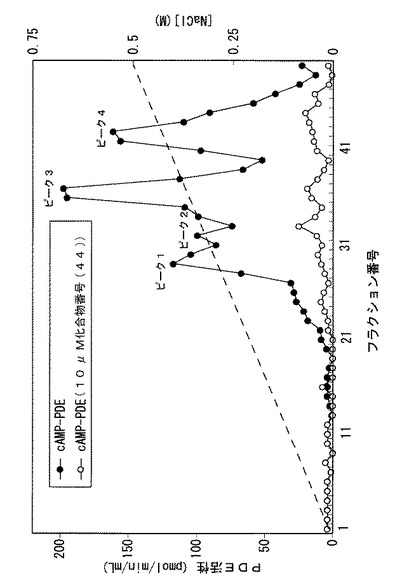
【図2】

【図3】
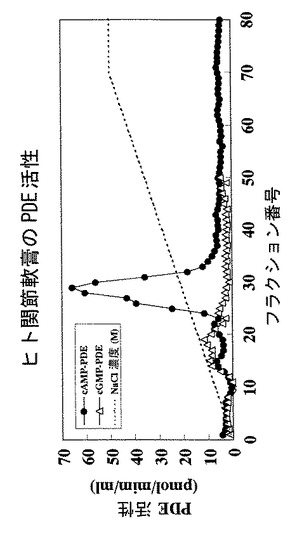
【図4】
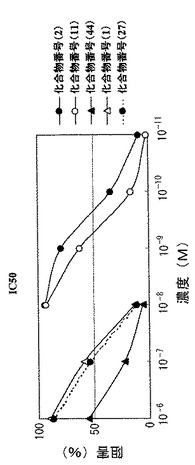
【図5】
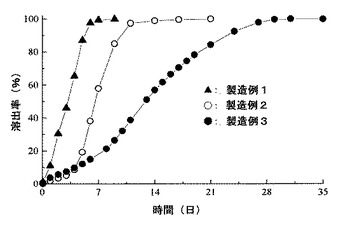
【図6】
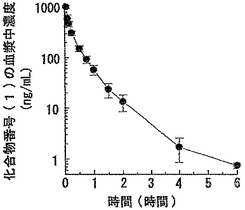
【図7】
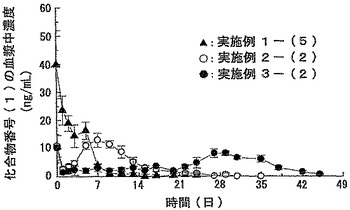
【図8】
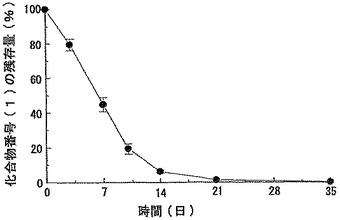
【図9】
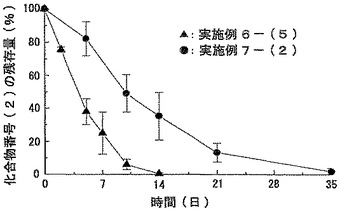
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公開番号】特開2010−155850(P2010−155850A)
【公開日】平成22年7月15日(2010.7.15)
【国際特許分類】
【出願番号】特願2010−32748(P2010−32748)
【出願日】平成22年2月17日(2010.2.17)
【分割の表示】特願2002−591036(P2002−591036)の分割
【原出願日】平成14年5月22日(2002.5.22)
【出願人】(000002956)田辺三菱製薬株式会社 (225)
【Fターム(参考)】
【公開日】平成22年7月15日(2010.7.15)
【国際特許分類】
【出願日】平成22年2月17日(2010.2.17)
【分割の表示】特願2002−591036(P2002−591036)の分割
【原出願日】平成14年5月22日(2002.5.22)
【出願人】(000002956)田辺三菱製薬株式会社 (225)
【Fターム(参考)】
[ Back to top ]