
転移性骨癌の治療のための受容体アンタゴニスト
【課題】転移性骨癌に対する新しい治療法が、早急に必要とされる。
【解決手段】本発明は、IGF-IRアンタゴニスト及び/又はPDGFRαアンタゴニストを投与することにより、骨癌、特に転移性骨癌を治療する方法を提供する。本発明は、ヒトPDGFRαに結合し、受容体の活性化を中和する抗体も提供する。本発明は、PDGFRαの活性化を中和する方法、及び抗体単独又は他の薬剤と併用して使用することにより、腫瘍疾患を患う哺乳動物を治療する方法を、さらに提供する。
【解決手段】本発明は、IGF-IRアンタゴニスト及び/又はPDGFRαアンタゴニストを投与することにより、骨癌、特に転移性骨癌を治療する方法を提供する。本発明は、ヒトPDGFRαに結合し、受容体の活性化を中和する抗体も提供する。本発明は、PDGFRαの活性化を中和する方法、及び抗体単独又は他の薬剤と併用して使用することにより、腫瘍疾患を患う哺乳動物を治療する方法を、さらに提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本出願は、米国仮出願第60/691,920号の利益を享受するものである。
【0002】
本発明は、IGF-IRアンタゴニスト及び/又はPDGFRαアンタゴニストを投与することにより、骨癌、特に転移性骨癌を治療する方法を提供する。本発明は、ヒトPDGFRαに結合し、受容体の活性化を中和する抗体も提供する。本発明は、PDGFRαの活性化を中和する方法、及び抗体を単独又は他の薬剤と組み合わせて使用することにより、腫瘍性疾患を患う哺乳動物を治療する方法を、さらに提供する。
【背景技術】
【0003】
前立腺癌は、男性では最も一般的な癌で、米国では毎年約220,000人が罹患し、29,000人が死亡する。前立腺癌と診断された男性の大半は、転移性疾患を有する。さらに、転移は、手術や放射線療法による治療をよそに、多くの他の前立腺癌患者において、最終的には進行する。骨は、前立腺癌転移が最も多い部位であり、乳癌及び肺癌が頻繁に転移する部位でもある。ほとんどの前立腺癌転移はアンドロゲン依存性であるので、外科又は内科的去勢で迅速な効果を認めるが、ほぼ全ての患者において、最終的には腫瘍がアンドロゲン非依存性となるので、重大な罹患率及び死亡率を導く。いったん骨転移が生じると、現在利用可能な療法では限られた効果しか有さない。転移性前立腺癌に関して報告されているもののうち、最も有効な認可された治療法(ドセタキセルの投与)は、生存期間の中央値を約3ヶ月延長する(Petrylak et al., 2004, N. Engl. J. Med. 351:1513; Tannock et al., 2004, N. Engl. J. Med. 351:1502)。従って、転移性骨癌に対する新しい治療法が、早急に必要とされる。
【0004】
インスリン様成長因子受容体(IGF-IR)は、正常胎児並びに出生後の成長及び発育にとって不可欠な、広範に分布する膜貫通型チロシンキナーゼ受容体である。IGF-IRは、ほとんどの細胞型の細胞表面に存在し、成長因子IGF-I及びIGF-II(以降は、まとめてIGFと称する)のシグナル伝達分子として機能する。IGF-IRは、細胞増殖、細胞分化、細胞サイズの変化を促進し、またアポトーシスから細胞を防御することができる。また、細胞の形質転換に準必須であるとも考えられてきた(Adams et al., Cell. Mol. Life Sci. 57:1050-93 (2000); Baserga, Oncogene 19:5574-81 (2000)でレビューされる)。IGF-IRの高レベルの発現が、前立腺癌の骨転移巣から得た組織サンプルで報告されている。骨は、体内で最も多くのIGF貯蔵を含む。
【0005】
IGF-IRは、ジスルフィド結合により共有結合した2つのα鎖及び2つのβ鎖を含む予め形成されたヘテロ四量体である。受容体サブユニットは、180 kdの単一のポリペプチド鎖の一部として合成された後、タンパク分解酵素によるプロセシングによってα(130kd)及びβ(95kd)サブユニットになる。α鎖全体は細胞外に存在し、リガンド結合するための部位を含んでいる。β鎖は膜貫通ドメイン、チロシンキナーゼドメイン、及び細胞分化及び形質転換に不可欠なC末端の伸長を備えるが、マイトジェンのシグナル伝達及びアポトーシスからの防御には必要である。
【0006】
IGF-IRは、インスリン受容体(IR)と、特にβ鎖配列において(70%の相同性)、非常に類似している。この相同性のおかげで、これらの受容体が、1つのIRダイマー及び1つのIGF-IRダイマーを含んでなるハイブリッドを形成できることが最近の研究で実証された(Pandini et al., Clin. Canc. Res. 5:1935-19 (1999))。ハイブリッド形成は、正常細胞及び形質転換した細胞の両方で生じ、ハイブリッドの組成は、細胞中の2つのホモダイマー受容体(IR及びIGF-IR)の濃度によって決まる。ハイブリッド受容体はIR及びIGF-IRの対からなるが、該ハイブリッドはIGF-IRに対する場合と同程度の親和性でIGFに選択的に結合し、インスリンとは弱くしか結合しない(Siddle and Soos, The IGF System. Humana Press, pp. 199-225. 1999)。従って、これらのハイブリッドは、正常細胞及び形質転換した細胞の両方において、IGFと結合してシグナルを伝達することができる。
【0007】
第2のIGF受容体であるIGF-IIR又はマンノース-6-ホスフェート(M6P)受容体も、IGF-IIのリガンドと高い親和性で結合するが、チロシンキナーゼ活性を欠く(Oates et al., Breast Cancer Res. Treat. 47:269-81 (1998))。従って、IGF-IIの分解が生じるので、IGF-IIのくぼみ(sink)は、このリガンドの成長促進効果を抑制すると考えられる。腫瘍細胞中のIGF-IIRが失われると、IGF-IIとIGF-IRとの結合に対する拮抗効果が解除されることにより、成長能力が増強され得る(Byrd et al., J. Biol. Chem. 274:24408-16 (1999))。
【0008】
血小板由来成長因子受容体α及びβ(PDGFRα及びPDGFRβ)は、III型の受容体チロシンキナーゼである。PDGFRαは発育に関して必須であり、成長期において重要な機能を果たす。例えば、ヌル突然変異させたホモ接合マウスは、胚形成期に死亡する。発生の後期段階において、PDGFRαは多くの間充織構造中で発現し、隣接する上皮細胞は血小板由来成長因子(PDGF)を産生する。正常前立腺又は過形成前立腺から得た組織サンプルは、検査ではPDGFRαに対し陰性であるが、適合被験体から得た原発性前立腺腫瘍及び骨本体は、PDGFRαを発現する。さらに、様々な転移性部位から得た前立腺細胞株のうち、骨転移由来のPC3細胞では、PDGFRαが観察されるが、リンパ節(LNCaP)及び脳(DU-145)転移巣から得た細胞株では、PDGFRαは観察されない。
【0009】
成長因子のうち、血小板由来成長因子ファミリーは、ジスルフィド結合した5種類の異なるダイマー、PDGF-AA、-BB、-AB、-CC、及び-DDからなり、これらはPDGFRα及びPDGFRβを介して作用する。これらの成長因子は、2つの受容体タンパク質と同時に結合し、受容体の二量体化、自己リン酸化、及び細胞内シグナル伝達を誘導するジスルフィド結合したポリペプチド鎖からなるダイマー分子である。PDGFRα及びPDGFRβは構造的に類似しており、ホモダイマーだけでなく、ヘテロダイマーも形成できる。PDGFRβは、高親和性ではPDGF-A鎖に結合しないので、PDGF-AAはαα受容体ダイマーのみを活性化するのに対し、PDGF-AB及びPDGF-CCはαα及びαβ受容体ヘテロダイマーを活性化する。
【先行技術文献】
【非特許文献】
【0010】
【非特許文献1】Petrylak et al., 2004, N. Engl. J. Med. 351:1513
【非特許文献2】Tannock et al., 2004, N. Engl. J. Med. 351:1502
【非特許文献3】Adams et al., Cell. Mol. Life Sci. 57:1050-93 (2000)
【非特許文献4】Baserga, Oncogene 19:5574-81 (2000)
【非特許文献5】Pandini et al., Clin. Canc. Res. 5:1935-19 (1999)
【非特許文献6】Siddle 及び Soos, The IGF System. Humana Press, pp. 199-225. 1999
【非特許文献7】Oates et al., Breast Cancer Res. Treat. 47:269-81 (1998)
【非特許文献8】Byrd et al., J. Biol. Chem. 274:24408-16 (1999)
【発明の概要】
【0011】
本発明は、原発性及び転移性骨腫瘍の治療に関連し、該骨腫瘍には前立腺、乳房又は肺から発生し、インスリン様成長因子-I受容体(IGF-IR)及び/又はα血小板由来成長因子受容体(PDGFRα)を発現する腫瘍が含まれる。
【0012】
治療対象となる腫瘍は、ホルモン/アンドロゲン依存性又はホルモン/アンドロゲン非依存性であり、例えば、前立腺、乳房、又は肺から発生したものである。
【0013】
本発明は、骨腫瘍を患う被験体を治療する方法、及び骨腫瘍の増大を阻害する方法を提供する。本発明の方法は、有効量のIGF-IRアンタゴニスト又は有効量のPDGFRαアンタゴニストを投与することを含む。受容体アンタゴニストには、抗体及び抗体フラグメントだけでなく、細胞内小分子阻害剤が含まれる。
【0014】
本発明は、それらの標的受容体と結合して、リガンド結合を阻害する抗IGF-IR又は抗PDGFRα抗体を提供する。本発明は、IGF-IR又はPDGFRαの活性化を中和する抗体及び他のアンタゴニストも提供する。さらに、ある抗体は、例えば内在化及び/又は分解によって、それらの標的受容体のダウンレギュレーションを促進する。従って、抗体及び小分子アンタゴニストは、下流のシグナル伝達分子、例えばAkt、p42/p44、及びMAPKの活性化を阻害するように機能する。
本発明の方法は、IGF-IR又はPDGFRαアンタゴニストを、単独で、互いに組み合わせて、又は他の癌療法、例えば化学療法剤及び放射線照射と組み合わせて、使用することを含む。
【0015】
本発明は、PDGFRαに結合する抗体及び抗体フラグメントだけでなく、該抗体産生のためのヌクレオチド及び宿主細胞も提供する。本発明の抗体は、リガンド結合を遮断し、受容体の活性化を中和する。本発明は、抗体の単独使用、他の受容体アンタゴニスト若しくは抗腫瘍剤と組み合わせた使用、又は腫瘍性疾患を治療するためのコンジュゲートとしての使用も提供する。抗PDGFRα抗体は、例えば、卵巣癌、乳癌、肺癌、肝細胞癌、胃腸間質腫瘍、メラノーマ、腎細胞癌、前立腺癌、及び柔組織肉腫を治療するために使用される。
【図面の簡単な説明】
【0016】
【図1】図1は、腫瘍が150〜200 mm3に達した際に開始された治療期間中における、去勢したSCIDマウス中のLuCaP35V皮下移植片腫瘍の増大を示す。パネルA:未治療の対照;パネルB:被験動物をドセタキセル(10 mg/kg又は20 mg/kgのいずれか)単独又は抗IGF-IR抗体(40 mg/kg IMC-A12)との併用で4週間治療した;パネルC:皮下LuCaP35V移植片腫瘍を有する未治療及び治療したSCIDマウスにおける血清PSAレベル。治療したマウスは、ドセタキセル(20 mg/kg)単独又はドセタキセル(10 mg/kg又は20 mg/kgのいずれか)と抗IGF-IR抗体(40 mg/kg IMC-A12)の併用治療を受けた。腫瘍が150〜200 mm3に達した際に治療を開始し、4週間後に治療を終了した。
【図2】図2は、ドセタキセル(10 mg/kg)単独(パネルA)又は抗IGF-IR抗体(40 mg/kg IMC-A12) (パネルB)との併用で治療したLuCaP35V移植片腫瘍の単一細胞の懸濁液を示す。R1で標識した領域は、断片化したDNAを有するアポトーシス細胞に相当する(FITC標識が増加している)。
【図3】図3は、ドセタキセル(10 mg/kg又は20 mg/kg)単独及び抗IGF-IR抗体(40 mg/kg IMC-A12)との併用による治療終了後の、腫瘍移植変片におけるDNA合成(BrDu取り込み)を示す。
【図4】図4は、ドセタキセル及びAl2並びにドセタキセル単独による治療に反応した前立腺腫瘍細胞における、前立腺腫瘍の悪性度(TUBB)、抗アンドロゲン療法(BIRC5)に対する耐性、及びアポトーシス誘導(IGFBP3)と関係する遺伝子の示差発現を示す。
【図5】図5は、治療終了後のA12血清レベルを示す。
【図6】図6は、ドセタキセル(10 mg/kg又は20 mg/kgのいずれか)単独又は抗IGF-IR抗体(40 mg/kg IMC-A12)との併用により継続して治療した、疾患を有さない被験動物の体重(総合的な細胞毒性評価)を示す。
【図7】図7は、LuCaP23.1細胞を移植したSCIDマウス中で移植片が産生するPSAに対する、抗IGF-IR抗体(IMC-A12)の治療効果を示す。
【図8】図8は、LuCaP23.1細胞を移植したSCIDマウスの一連のX線写真を示す。A12マウスには、40 mg/ml IMC-A12(i.p.)を6週間にわたり週3回投与した。X線写真は屠殺時に撮影した。
【図9】図9は、LuCaP23.1ヒト前立腺細胞の頚骨内移植片を有するSCIDマウスのPSAレベル(a)及び代表的なX線写真(b)を示す。
【図10】図10は、前立腺癌細胞中のAkt活性に対するヒト骨髄穿刺液の効果を示す。細胞溶解物をSDS-PAGEに供した。ウェスタンブロット解析のために、ホスホAkt(Ser-473;cell signaling Technology)、PDGFRα(R&D Systems)及びアクチン(Sigma)を標的とする抗体を用いて、膜にブロットした。一次抗体の結合を、HRP-コンジュゲートプロテインA又はプロテインG(Sigma)を用いて検出した。
【図11】図11は、PC3-ML細胞におけるAktリン酸化の誘導及び阻害を示す。パネルAは、30 ng/ml PDFG-BBにさらした細胞中でのAktリン酸化のAG-1296用量依存的阻害を示す。パネルBは骨穿刺液のAktリン酸化及び20μMのAG-1296による阻害を示す。パネルCは、Aktリン酸化の誘導に関して、100 pg/ml PDGF-AA及び100 pg/ml PDGF-BBの併用の効力と比べた場合の、骨髄穿刺液の効力を示す。パネルDは、骨髄穿刺液誘導性のAkt-リン酸化、AG-1296による骨髄誘導性のAkt-リン酸化の阻害、及びPDFG-AA+PDFG-BBにより誘導されるAkt-リン酸化の強度を比較する。
【図12】図12は、PDGFRαアンタゴニストによるPC3-ML細胞中のAktリン酸化の阻害を示す。パネルAは、30 ng/mlのPDGF-BBにより誘導されたAktリン酸化に対する、モノクローナル抗体IMC-3G3の用量依存的効果を示す。パネルB及びCは、骨髄が誘導するAktリン酸化に対するIMC-3G3及びAG1296の効果の比較を提供する。パネルDは、Aktリン酸化の阻害が、IMC-3G3プレインキュベーション時間に依存的であることを示す。
【図13】図13は、PDGFRαに対する抗体の結合を示す。A:固定したPDGFRαの細胞外ドメインに対する抗PDGFRα抗体の直接結合。B:固定したPDGFRαと[125I]PDGF-AAとの結合の阻害。
【図14】図14は、PDGFRα及び下流エフェクター分子のリン酸化の特異的阻害を示す。
【図15】図15は、PAE Rα細胞におけるPDGF-AAで刺激した[3H]チミジン取り込みのmAbによる阻害を示す。
【図16】図16は、SKLMS-1(A)及びU118(B)細胞における、PDGF-AAが誘導した下流エフェクター分子の活性化の阻害を示す。
【図17】図17は、U118細胞(A)及びSKLMS-1細胞(B)における、mAbによるPDGF-AA刺激性[3H]チミジン取り込みの阻害を示す。SKLMS-1(C)及びU118(D)細胞について、PDGF-BB刺激性[3H]チミジン取り込みの阻害も示す。
【図18】図18は、ヌードマウス中で定着したU118(神経膠芽腫;パネルA)及びSKLMS-1(平滑筋肉腫;パネルB)腫瘍移植片の治療での用量依存的効果を示す。
【図19】図19は、非特異的ヒトIgGによる処置と比較した場合の、抗PDGFRα抗体で処置したU118腫瘍におけるin vivoのPDGFRαリン酸化の減少を示す。
【図20】図20は、ハイブリドーマ由来のヒトVH及びVκ可変領域遺伝子のクローニング並びに完全ヒト重鎖(IgG1)及び軽鎖タンパク質の発現に使用されるGS発現ベクターを示す。実施例で説明するとおり、2つのベクターを組換えて、該組合わせたベクターをNSO細胞にトランスフェクトした。
【発明を実施するための形態】
【0017】
本発明は、インスリン様成長因子-I受容体(IGF-IR)に結合する抗体又は抗体フラグメントによる骨腫瘍の治療に関する。IGF-Iのエンドクリン発現は、主に成長ホルモンにより制御され、肝臓で産生されるが、他の種類の組織もIGF-1を発現することができ、該組織には成長因子の大きな貯蔵を含む骨が含まれる。腫瘍細胞型により、IGF-Iはエンドクリン、パラクリン、及び/又はオートクリン制御に関与する(Yu, H. and Rohan, J., J. Natl. Cancer Inst. 92:1472-89 (2000))。
【0018】
IGF-IRに結合する抗体が、IGF-IRを発現する骨腫瘍の治療法において有用であることが見出された。該抗体は、単独で、又は他の癌療法剤、特に化学療法剤と組み合わせて使用できる。抗IGF-IR療法は、単独で又は1以上の抗腫瘍剤(例えば、化学療法又は放射線療法等)と組み合わせて、顕著な治療的効果を有する。腫瘍増大の抑制は、アポトーシスの増加を伴うことが多いので、全ての治療を中止した後も持続する。しかしながら、化学療法単独で処置した動物では、腫瘍が再増殖し始める。
【0019】
PDGFRαは、骨腫瘍の増大において重要な役割を果たすことも見出された。例えば、PDGFRαを発現する特定の腫瘍細胞株は、骨に選択的に転移する。かかる細胞株は、骨髄中に存在する可溶性因子に反応して、PDGFRαの活性化及び下流シグナル伝達分子のリン酸化を増加させる。骨髄によるPDGFRαの活性化は、PDGFRαアンタゴニストにより、低下又は完全に阻害される。また、PDGFRα及び他の受容体チロシンキナーゼ系を介したシグナル伝達によって通常活性化される下流シグナル伝達分子のリン酸化は、非常に減少する。PI3K/Akt生存経路は、直接的にPDGFRαを活性化するリガンドによるだけでなく、受容体のトランス活性化を生じさせる骨髄中に存在する因子によっても、PDGFRαシグナル伝達により活性化されることをあるデータは示唆する。
【0020】
本発明の治療対象となる原発性骨腫瘍には、骨肉腫、軟骨肉腫、線維肉腫、及び血管肉腫が含まれるが、これに限定されない。特に、悪性二次性(転移性)腫瘍が、原発性骨腫瘍よりもはるかに多い。本発明の治療対象となる転移性骨腫瘍は、多様な原発巣(source)から発生し、そのうち最も多いのは前立腺、乳房、又は肺である。転移性骨癌の原発巣は通常、患者の既往歴から明らかとなる。腫瘍は、骨芽球性又は骨溶解性であり得る。腫瘍は、発生時にIGF-IR刺激に依存的であるか、または、IGF-IR依存性に転化する場合がある。例えば、初期の段階ではホルモン/アンドロゲン依存性であってもアンドロゲン又はホルモン産生を抑制する物理療法又は化学療法によって制御可能である前立腺癌又は前立腺癌の転移は、IGF-IRを介した刺激への感受性増大によって、ホルモン/アンドロゲン非依存性になる場合がある。そのため、本発明はさらに、ホルモン/アンドロゲン非依存性腫瘍の治療の提供に加えて、アンドロゲン又はホルモン産生の抑制にはよらずに、例えば、抗腫瘍剤と共にIGF-IR抗体を同時投与することによって、ホルモン/アンドロゲン依存性骨腫瘍を治療するのにも有用である。かかる腫瘍には、IGFが豊富な骨環境においてIFG-IRを介して刺激される転移性骨腫瘍が含まれる。該腫瘍は、ホルモン刺激に対し感受性があるが、IGF環境が関与しなくても増殖できる程に感受性はない。よって、かかる腫瘍にとって、ホルモン除去は必須でないかもしれない。
【0021】
PDGF依存性の骨腫瘍だけでなく、「骨髄」依存性の腫瘍も、本発明により治療できる。骨髄依存性の腫瘍は、骨髄中に存在する可溶性因子に反応して、PDGFRαの活性化を提示する。例えば、本明細書で例示するとおり、ヒト転移性PDGFRαを発現する癌細胞株は、骨髄穿刺液への曝露により、PDGFRα活性化及びAkt+のリン酸化を受ける。抗PDGFRα抗体及び小分子PDGFRαアンタゴニストは、該細胞株において、PDGFRα活性化及びAkt+リン酸化をそれぞれ阻害する。PDGFRαを活性化する可溶性骨髄因子には、PDGF-AA及び-BBが含まれるが、これに限定されない。
【0022】
かかる骨髄依存性は、PDGFRαを介したシグナル伝達に関係するが、PDGFRαリガンドとPDGFRαとの結合だけに関与しているわけではない。例えば、本明細書で説明するとおり、所定のリガンド(PDGF-AA又は-BB)によるPDGFRαの活性化は、骨髄穿刺液による活性化よりも弱いことに留意されたい。さらに、骨髄穿刺液の存在下では、インキュベーション時間の増加により、Akt+のリン酸化が減少するのが観察される。纏めると、これらの結果は、PDGFの結合に反応することに加えて、他の骨髄成分に対し感受性のある他のシグナル伝達因子(例えば、他の受容体チロシンキナーゼ)によって、PDGFRαがトランス活性化(リン酸化)され得ることを示唆する。いずれにせよ、骨での転移性増殖に適した細胞株(すなわち、骨に選択的に転移する細胞株)では、骨髄依存性PDGFRαの活性化が観察され、該活性化は、PDGFRαアンタゴニストにより阻害される。さらに、PDGFRαアンタゴニストによる治療は、PI3K/Akt抗アポトーシス経路及びマイトジェン活性化プロテインキナーゼ(MAPK)の骨髄誘導性刺激を阻害する。
【0023】
PDGFRαアンタゴニストにより治療される骨腫瘍は、前立腺癌細胞の転移として発生するか、上記のとおりホルモン/アンドロゲン依存性であるか、又はホルモン/アンドロゲン非依存性に転化したものである。かかる腫瘍は、前立腺癌以外の転移としても発生し得る。当業者であれば、公知の従来の試験法を用いて、かかる状態及び障害を容易に診断できるだろう。
【0024】
治療とは、動物における疾患の任意の治療を意味し、以下:(1)哺乳動物において、疾患が発症するのを予防すること、すなわち疾患が誘発されてもよいが、疾患の症状は経験も提示もしないこと、例えば、臨床症状の出現の防止;(2)疾患を抑制すること、例えば、疾患進行の停止;又は(3)疾患を軽減すること、例えば、疾患症状の消失が含まれる。腫瘍増殖の阻害には、増殖を減速又は停止させることだけでなく、腫瘍を消退させることが含まれる。疾患治療における有効量とは、必要とする哺乳動物に投与した際に、上記に定義したとおり、その疾患治療に十分に有効な量を意味する。本発明のIGF-IRアンタゴニスト及びPDGFRαアンタゴニストは、単独で、互いに組み合わせて、又は1以上の抗腫瘍剤、例えば化学療法剤又は放射線照射などと組み合わせて、投与できる。
【0025】
本発明の実施形態では、治療対象となる腫瘍におけるIGF-IR及び/又はPDGFRαの発現レベルを測定することが望ましい。そのような場合、腫瘍の生検検体は、当技術分野で周知の方法により、収集し及び分析することができる。本発明の別の実施形態では、該当する受容体が、特定の種類の腫瘍において共通して発現若しくは活性化されるか、又は疾患が進行した際に非特異的に発現若しくは活性化されることに基づいて、IGF-IRアンタゴニスト又はPDGFRαアンタゴニストが投与される。
【0026】
IGF-IRアンタゴニストは、細胞外アンタゴニスト又は細胞内アンタゴニストであり、2以上のアンタゴニストを適用することもできる。細胞外アンタゴニストには、IGF-IRに結合するタンパク質若しくは他の生体分子又は1以上のそのリガンド(例えば、IGF-I及びIGF-IIはIGF-IRの天然のリガンドである)が含まれるが、これに限定されない。本発明の実施形態では、細胞外アンタゴニストは、IGF-IRがそのリガンドと結合するのを阻害する。1実施形態では、アンタゴニストは、例えばIMC-A12のような抗IGF-IR抗体である。別の実施形態では、アンタゴニストは、IGF-IRのフラグメントに結合する可溶性リガンドである。細胞内IGF-IRアンタゴニストは生体分子でもよいが、通常は小さな分子である。例としては、チロシンキナーゼ阻害剤AGl024(Calbiochem)、インスリン様成長因子-I受容体キナーゼ阻害剤NVP-AEW541 (Novartis)、及びインスリン様成長因子-I/インスリン受容体阻害剤BMS-554417 (Bristol Myers Squibb)が含まれるが、これに限定されない。本発明での使用に有用な小分子はIGF-IRの阻害剤であるが、IGF-IRに完全に特異的である必要はないことを理解されたい。
【0027】
本発明で使用される抗IGF-IR抗体は、次の特性のうち1以上を示す:
1)抗体はIGF-IRの外部ドメインに結合し、IGF-I又はIGF-IIがIGF-IRに結合するのを阻害する。阻害は、例えば、精製された受容体又は膜結合受容体を用いて、直接的な結合アッセイにより測定できる。この実施形態では、本発明の抗体又はそのフラグメントは、好ましくは、IGF-IRの天然のリガンド(IGF-I及びIGF-II)と少なくとも同じ強さで、IGF-IRに結合する。
【0028】
2)抗体は、IGF-IRを中和する。IGF-IRの外部の細胞外ドメインに対するリガンド(例えば、IGF-I又はIGF-II)の結合は、βサブユニット並びにMAPK、Akt、及びIRS-Iを含む下流のシグナル伝達分子の自己リン酸化を刺激する。
【0029】
IGF-IRの中和には、通常シグナル伝達に関係する当該活性のうち1以上を阻害、減少、不活化及び/又は遮断することが含まれる。中和は、例えば、組織、培養細胞、又は精製した細胞成分を用いて、in vivo、ex vivo、又はin vitroで測定できる。中和には、IGF-IR/IRヘテロダイマーだけでなく、IGF-IRホモダイマーの阻害も含まれる。従って、IGF-IRの中和は様々な効果を有しており、該効果には、成長(増殖及び分化)、血管新生(血管の補強、浸潤、及び転移)、並びに細胞の移動性及び転移(細胞接着及び浸潤)の阻害、減少、不活化及び/又は遮断が含まれる。
【0030】
IGF-IRの中和の1つの指標は、受容体のチロシンキナーゼ活性の阻害である。チロシンキナーゼ阻害は、周知の方法を用いて;例えば、組換えキナーゼ受容体の自己リン酸化レベル及び/又は天然若しくは合成の基質のリン酸化を測定することにより、測定できる。従って、リン酸化アッセイは、本発明に関して中和抗体を決定するのに有用である。リン酸化は、例えばELISAアッセイ又はウェスタンブロットにおいて、ホスホチロシンに特異的な抗体を用いて検出できる。チロシンキナーゼ活性についての幾つかのアッセイが、Panek et al., J. Pharmacol. Exp. Them. 283: 1433-44 (1997)及びBatley et al., Life Sd. 62:143-50 (1998)で報告されている。本発明の抗体は、リガンドと反応する細胞において、IGF-IRのチロシンリン酸化を、少なくとも約75%、好ましくは少なくとも約85%、及びより好ましくは少なくとも約90%減少させる。
【0031】
IGF-IR中和の別の指標は、IGF-IR下流の基質のリン酸化の阻害である。よって、MAPK、Akt、又はIRS-Iのリン酸化レベルが測定される。リン酸化の減少は、少なくとも約40%であり、少なくとも約60%、又は少なくとも約80%であり得る。
【0032】
さらに、測定されるタンパク質がIGF-IRチロシンキナーゼ活性によって制御される場合には、タンパク質発現の検出方法を利用して、IGF-IR中和を測定できる。これらの方法には、タンパク質発現の検出のための免疫組織化学的検査(IHC)、遺伝子増幅の検出のための蛍光in situハイブリダーゼーション(FISH)、競合的放射性リガンド結合アッセイ、固相マトリックスブロット法、例えば、ノーザン及びサザンブロット、逆転写酵素ポリメラーゼ連鎖反応(RT-PCR)及びELISAが含まれる。例えば、Grandis et al., Cancer, 78:1284-92 (1996); Shimizu et al., Japan J. Cancer Res., 85:567-71 (1994); Sauter et al., Am. J. Path., 148:1047-53 (1996); Collins, GHa 15:289-96 (1995); Radinsky et al., Clin. Cancer Res. 1:19-31 (1995); Petrides et al., Cancer Res. 50:3934-39 (1990); Hoffmann et al., AntiCancer Res. 17:4419-26 (1997); Wikstrand et al., Cancer Res. 55:3140-48 (1995)を参照されたい。
【0033】
Ex vivoアッセイを利用して、IGF-IR中和を測定することもできる。例えば、受容体チロシンキナーゼ阻害は、阻害剤の存在下及び不在下で、受容体リガンドにより刺激された細胞株を使用するマイトジェンアッセイによって、観察できる。MCF7乳癌株(American Type Culture Collection (ATCC), Rockville, MD)は、IGF-IRを発現するような細胞株であり、IGF-I又はIGF-IIにより刺激される。別の方法は、IGF-IRを発現する腫瘍細胞又はIGF-IRを発現するように形質導入された細胞の成長の阻害について、試験することも含む。腫瘍モデル、例えば、ヒト腫瘍細胞を注入されたマウスを用いて、阻害を観察することもできる。
【0034】
本発明の抗体は、IGF-IR中和のいかなる特定機構によっても制限されない。本発明の抗IGF-IR抗体は、IGF-IR細胞表面受容体に外側から結合でき、リガンド(例えば、IGF-I又はIGF-II)の結合及び受容体と結合したチロシンキナーゼにより介在される後続のシグナル伝達を遮断でき、さらにIGF-IR及びシグナル伝達カスケード下流の他のタンパク質のリン酸化を防止できる。
【0035】
3)抗体は、IGF-IRを下方調節する。細胞の表面上に存在するIGF-IRの量は、受容体タンパク質の産生、内在化、及び分解によって決まる。細胞の表面上に存在するIGF-IRの量は、受容体又は該受容体に結合した分子の内在化を検出することにより、間接的に測定できる。例えば、受容体の内在化は、IGF-IRを発現する細胞を、標識した抗体に接触させることにより測定できる。その後、膜に結合した抗体を剥離し、回収して、数える。内在化した抗体は、細胞を溶解させ、溶解物中の標識を検出することにより、測定できる。
【0036】
別の方法は、抗IGF-IR抗体又は他の物質による治療後に、細胞上に存在する受容体の量を、例えばIGF-IRの表面発現について染色した細胞の蛍光活性化細胞選別分析法により、直接的に測定することである。染色した細胞を37℃でインキュベートし、蛍光強度をじっくりと測定する。対照では、染色した集団の一部を4℃(受容体内在化が停止する条件)で、インキュベートし得る。
【0037】
細胞表面IGF-IRは、IGF-IRに特異的で、かつ、試験される抗体の結合を遮断しない又は試験される抗体の結合と競合しない様々な抗体を用いて、検出及び測定できる(Burtrum, et al. Cancer Res. 63:8912-21 (2003))。本発明の抗体によるIGF-IRを発現する細胞の処置は、細胞表面IGF-IRの減少をもたらす。好ましい実施形態では、本発明の抗体による治療に反応して、少なくとも約70%、より好ましくは少なくとも約80%、さらにより好ましくは少なくとも約90%が減少する。顕著な減少が、わずか4時間で観察できる。
【0038】
下方調節の別の指標は、細胞中に存在する受容体タンパク質総数の減少であり、これは内部受容体の分解を反映する。よって、本発明の抗体による細胞(特に、癌細胞)の処置は、細胞性IGF-IR総数の減少をもたらす。好ましい実施形態では、この減少は、少なくとも約70%、より好ましくは少なくとも約80%、さらにより好ましくは少なくとも約90%である。
【0039】
ヒト被験体の治療では、本発明の抗体は、好ましくはヒト抗体である。他には、抗体は、非ヒトの霊長類若しくは他の哺乳動物に由来し、又はヒト化抗体若しくはキメラ抗体である。本発明の実施形態では、抗IGF-IR抗体は、配列番号35、配列番号37、配列番号39、配列番号45、配列番号47、及び配列番号49(それぞれCDR1H、CDR2H、CDR3H、CDR1L、CDR2L、CDR3L)からなる群から選択される1、2、3、4、5、及び/又は6の相補性決定領域(CDR)を含む。別の実施形態では、抗IGF-IR抗体は、配列番号35、配列番号37、配列番号39、配列番号55、配列番号57、及び配列番号59(それぞれCDR1H、CDR2H、CDR3H、CDR1L、CDR2L、CDR3L)からなる群から選択される1、2、3、4、5、及び/又は6の相補性決定領域(CDR)を含む。好ましくは、本発明の抗体(又はそのフラグメント)は、配列番号35、配列番号37及び配列番号39で表される重鎖CDRを有する。他にまた好ましくは、本発明の抗体(そのフラグメントを含む)は、配列番号45、配列番号47及び配列番号49又は配列番号55、配列番号57及び配列番号59で表される軽鎖CDRを有する。かかる抗IGF-IR抗体の1つは、配列番号41で示される重鎖可変ドメイン及び配列番号51で示される軽鎖可変ドメインを有するヒトIgG1抗体IMC-A12 (WO2005016970)である。別の好ましいヒト抗体は、IMC-A12と同一の重鎖可変ドメイン及び配列番号61で示される軽鎖可変ドメインを有するIMC-2F8 (WO2005016970)である。有用な抗体には、IGF-IRへの結合についてMC-A12又はIMC-2F8と競合する抗IGF-IR抗体だけでなく、他のエピトープに結合する抗体(すなわち、他のエピトープに結合し、前述の特性、例えばリガンドの遮断、受容体の内在化等を提示するが、IMC-A12又はIMC-2F8とは競合しない抗体)が、さらに含まれる。
【0040】
本発明では、PDGFRαアンタゴニストを治療のために使用することもできる。PDGFRαアンタゴニストは、細胞外アンタゴニスト又は細胞内アンタゴニストであり、及び2以上のアンタゴニストを適用してもよい。細胞外アンタゴニストには、PDGFRαと結合するタンパク質若しくは他の生体分子又は1以上のそのリガンド(例えば、PDGF-AA、-AB、-BB、-CC)が含まれるが、これに限定されない。本発明の実施形態では、PDGFRαにそのリガンドが結合するのを細胞外アンタゴニストが阻害する。1実施形態では、アンタゴニストは例えば、IMC-3G3のような抗PDGFRα抗体である。別の実施形態では、該結合するタンパク質は、PDGFRαの可溶性リガンドに結合するフラグメントである。細胞内IGF-IRアンタゴニストは生体分子でもよいが、通常は小さな分子である。1実施形態では、細胞内PDGFRαアンタゴニストはAG1296である。AG1296(Calbiochem)は、PDGFα、PDGFβs、及びc-KITの阻害剤であり、Flt3とも反応する。PDGFRを標的とする他の小分子には、STI-571(イマチニブメシレート、Gleevec(登録商標), Novartis)及びSUl1248(スニチニブマーレート、SUTENT(登録商標)、Pfizer)が含まれる。
【0041】
本発明の実施形態では、抗PDGFRα抗体は、配列番号2、配列番号4、配列番号6、配列番号10、配列番号12、及び配列番号14(それぞれCDRlH、CDR2H、CDR3H、CDRlL、CDR2L、CDR3L)からなる群から選択される1、2、3、4、5、及び/又は6の相補性決定領域(CDR)を含む。好ましくは、本発明の抗体(又はそのフラグメント)は、配列番号2、配列番号4及び配列番号6で表されるCDRを有する。他にまた好ましくは、本発明の抗体又はそのフラグメントは、配列番号10、配列番号12及び配列番号14で表されるCDRを有する。CDRのアミノ酸配列は、下記表1に記載のとおりである。
【表1】

【0042】
別の実施形態では、抗PDGFRα抗体又はそのフラグメントは、配列番号8で表されるヒト重鎖可変領域及び/又は配列番号16で表されるヒト軽鎖可変領域を有する。IMC-3G3は、そのような抗体であり、本発明で説明される。
【0043】
好ましくは、本発明の抗体又はそのフラグメントは、PDGFRαを中和する。PDGFRαの細胞外ドメインと、リガンド(例えばPDGF-AA、PDGF-AB、PDGF-BB又はPDGF-CC)との結合は、受容体の二量体化、自己リン酸化、受容体内部の細胞質チロシンキナーゼドメインの活性化、並びにDNA合成(遺伝子の活性化)の制御及び細胞周期の進行又は細胞分裂周期の制御と関係する複数のシグナル伝達経路やトランス活性化経路の開始を刺激する。抗PDGFRα抗体は、典型的には、リガンド結合及び/又は受容体の二量体化を遮断し、自己リン酸化、チロシンキナーゼ活性の活性化及びシグナル伝達のうち1以上を阻害する。本発明の抗PDGFRα抗体は、PDGFRαの細胞外リガンド結合領域に特異的であり得、リガンドがPDGFRαと結合するのを阻害できる。好ましくは、かかる抗PDGFRα抗体又はそのフラグメントは、PDGFRαの天然のリガンドと少なくとも同じ強さで、PDGFRαに結合する。代わりに又はさらに、抗体は、受容体ダイマーの境界面を別途形成し得る受容体モノマーの領域に特異的であり得る。かかる抗体は、受容体モノマーへのリガンドの結合が遮断されても遮断されなくとも、二量体形成を遮断する。
【0044】
抗IGF-IR抗体について上記したとおり、受容体の中和は、様々なin vivo、in vitro、及びex vivo方法によって測定できる。本発明の1実施形態では、抗PDGFRα抗体は、PDGFRαのリン酸化を少なくとも約75%減少させる。他の実施形態では、リン酸化を少なくとも約85%又は少なくとも約90%減少させる。本発明の実施形態では、PDGFRαシグナル伝達の阻害の結果、リン酸化又は下流のシグナル伝達経路因子(Akt、p42/p44等)が、少なくとも約40%、少なくとも約60%、又は少なくとも約80%減少する。受容体の中和は、所定のリガンド(例えば、PDGF-AA、-AB、-BB、-CC)、かかるリガンドの混合物又はPDGFを含有する骨髄穿刺液といった調製物だけでなく、他の成長促進因子を用いても、測定できる。
【0045】
PDGFRαの中和は、通常シグナル伝達に関係する当該活性のうち1以上の阻害、減少、不活化及び/又は遮断を含む。従って、PDGFRαの中和は、成長(増殖及び分化)、血管新生(血管の補強、浸潤、及び転移)、並びに細胞の移動性及び転移(細胞接着及び浸潤)の阻害、減少、不活化及び/又は遮断を含む、様々な効果を有する。
【0046】
上記のとおり、ex vivoアッセイを利用して、PDGFRαの中和を測定することができる。例えば、PDGF-AAにより刺激されたヒトSKLMS-1平滑筋肉腫細胞(American Type Culture Collection (ATCC), Rockville, MD; ATCC HTB-88TM)又はU118神経膠芽腫細胞(ATCC HTB-15TM)を使用して、PDGFRα阻害をアッセイできる。成長の阻害は、PDGFRαを発現するヒト腫瘍細胞を注入したSCIDマウスを用いて、確認することができる。
【0047】
本発明は、PDGFRα中和のいかなる特定機構によっても制限されない。本発明の抗PDGFRα抗体は、PDGFRα細胞表面受容体に外側から結合でき、リガンド(例えば、PDGF-AA、PDGF-AB、PDGF-BB、PDGF-CC)の結合を阻止でき、PDGFRαのリン酸化を阻害でき、受容体と結合したチロシンキナーゼによって介在されるシグナル伝達を阻害でき、並びにシグナル伝達下流の因子の活性を調節することができる。受容体-抗体複合体は、中和及び分解されることによって、細胞表面受容体のダウンレギュレーションをもたらすこともできる。マトリックスメタロプロテイナーゼは、腫瘍細胞の浸潤及び転移において機能を果たすが、該プロテインは、本発明の抗体によってもダウンレギュレートされる。そのうえ、本発明の抗体は、成長因子の産生及び血管新生の阻害を示せる。
【0048】
上記のとおり、本発明のPDGFRαアンタゴニストは、転移性骨腫瘍も含めた骨腫瘍を治療するのに有用である。PDGFRαを発現し、かつ、本発明で治療できる他の腫瘍の種類には、卵巣癌、乳癌、肺癌、肝細胞癌、胃腸間質腫瘍、メラノーマ、腎細胞癌、前立腺癌、及び柔組織肉腫が含まれるが、これに限定されない。柔組織肉腫は、脂肪、筋肉、神経、腱、並びに血管及びリンパ管といった組織で発生する。典型的には、腫瘍細胞はPDGFRαを発現する。PDGFRαの発現は、例えば組織化学的解析又はRNA解析により測定できる。例えば、放射線標識したIMC-3G3と、U118細胞及びSKLMS-1腫瘍細胞との結合のスキャッチャード解析によれば、該細胞上のPDGFRα分子の数がそれぞれ約500及び2500個であることがわかる。
【0049】
PDGFRαアンタゴニストは、腫瘍細胞自体の上で発現したPDGFRαによるシグナル伝達を阻害することによって、又は周辺のストローマ細胞上でPDGFRαが発現するのを阻害して、腫瘍細胞から発現したPDGFによってパラクリン刺激を受けるのを避けることにより、機能する。従って、IMC-3G3及び他のPDGFRαアンタゴニストといった抗体は、PDGFRαのオートクリン及び/又はパラクリン刺激を特徴とする腫瘍を治療するのに、有用である。
【0050】
本発明の抗体フラグメントは、全抗体を切断することにより、又は該フラグメントをコードするDNAを発現させることにより作製できる。抗体のフラグメントは、Lamoyi et al., J. Immunol. methods, 56: 235-243 (1983)及びParham, J. Immunol. 131 : 2895-2902 (1983)により記載された方法によって、調製できる。かかるフラグメントは、Fabフラグメント又はF(ab')2フラグメントの片方又は両方を含み得る。かかるフラグメントには、一本鎖フラグメント可変領域抗体、すなわちscFv、dibodies、又は他の抗体フラグメントも含まれる。かかる機能性等価物の作製方法は、PCT出願WO 93/21319、欧州特許出願EP 239400;PCT出願WO 89/09622;欧州特許出願EP 338745;及び欧州特許出願EP 332424に開示されている。
【0051】
ベクターの形質転換及び本発明の抗体の発現に好ましい宿主細胞は、哺乳動物の細胞、例えばCOS-7細胞、チャイニーズハムスター卵巣(CHO)細胞、及びリンパ系起源の細胞株、例えば、リンパ腫細胞、骨髄腫(例えば、NSO)細胞、又はハイブリドーマ細胞である。他の真核性宿主、例えば酵母を代わりに利用することができる。
【0052】
酵母中で遺伝子構築物を発現させたい場合に、酵母中での使用に好適な選択遺伝子は、酵母プラスミドYRp7中に存在するtrp1遺伝子である。Stinchcomb et al. Nature, 282: 39 (1979); Kingsman et al., Gene, 7: 141 (1979)。trp1遺伝子は、トリプトファン中で成長する能力を欠く酵母の変異株、例えば、ATCC番号44076又はPEP4-1の選択マーカーとなる(Jones, Genetics, 85: 12 (1977))。従って、酵母宿主細胞ゲノム中のtrp1欠陥の存在によって、トリプトファン不在下での成長による形質転換を検出するのに有効な環境がもたらされる。同様に、Leu2-欠損酵母株(ATCC 20,622又は38,626)は、Leu遺伝子を含有する公知のプラスミドによって、補完される。
【0053】
形質転換した宿主細胞は、同化可能な炭素源(糖質、例えばグルコース又はラクトース)、窒素源(アミノ酸、ペプチド、タンパク質又はそれらの分解産物、例えばペプトン、アンモニウム塩など)、及び無機塩源(ナトリウム、カリウム、マグネシウム、及びカルシウムの硫黄塩、リン酸塩及び/又は炭酸塩)を含有する液状培地中で、当技術分野で公知の方法により培養される。該培地には、例えば微量元素(例えば、鉄、亜鉛、マンガンなど)のような成長促進物質がさらに含まれる。
【0054】
本発明の高親和性抗PDGFRα抗体及び抗IGF-IR抗体は、ヒト重鎖及び軽鎖可変領域遺伝子から構築されたファージディスプレイライブラリーから単離できる。例えば、本発明の可変ドメインは、再構成された可変領域遺伝子を含む末梢血リンパ球から入手することができる。他には、可変ドメイン部分、例えばCDR及びFW領域は、様々な起源から得ることができ、組換えることができる。さらに、可変ドメインの一部(例えば、FW領域)は合成コンセンサス配列であってもよい。
【0055】
本発明の抗体及び抗体フラグメントは、例えば、天然の抗体、又はFab若しくはscFvファージディスプレイライブラリーから入手できる。VH及びVLドメインを含む抗体からシングルドメイン抗体を作製して、結合、発現又は可溶性を増強するためには、CDRの外側で特定のアミノ酸を置換することが望ましいことが知られている。例えば、他には、VH-VL界面に埋め込まれたアミノ酸残基を改変することが望ましい。
【0056】
さらに、本発明の抗体及び抗体フラグメントは、ヒト免疫グロブリンγ重鎖及びκ軽鎖を産生するトランスジェニックマウス(例えば、San Jose, Califから入手したKMマウス)を用いて、標準的なハイブリドーマ技術(Harlow & Lane, ed., 抗体: A Laboratory Manual, Cold Spring Harbor, 211-213 (1998);引用により本明細書に取り込まれる)により入手できる。好ましい実施形態では、ヒト抗体を産生するゲノムの本質的部分を、マウスのゲノムに挿入し、内因性マウス抗体を産生できなくさせる。かかるマウスは、PDGFRαを皮下投与し(s.c.)、(通常は完全フロインドアジュバント中で)必要に応じてブーストして、免疫できる。免疫方法は、当技術分野では周知である。
【0057】
本発明のIGF-IR結合性抗体を同定するために使用されるタンパク質は、好ましくはIGF-IRであり、より好ましくはIGF-IRの細胞外ドメインである。本発明のPDGFRα結合性抗体を同定するために使用されるタンパク質は、好ましくはPDGFRαであり、より好ましくはPDGFRαの細胞外ドメインである。かかる細胞外ドメインは、遊離型であるか、他の分子と結合している。
【0058】
本発明は、前記の抗体又はそのフラグメントをコードする単離されたポリヌクレオチドも提供する。IMC-A12抗IGF-IR抗体の詳細は、WO2005016970に開示されている。表2は、IMC-3G3の核酸配列を示す。
【表2】
【0059】
ヒト抗体をコードするDNAは、対応するヒト抗体領域から実質的又は排他的に誘導された、CDR以外のヒト定常領域及び可変領域をコードするDNAとヒト由来のCDRをコードするDNA(重鎖可変ドメインCDRを表す配列番号1、3、及び5、並びに軽鎖可変ドメインCDRを表す配列番号9、11、及び13)を組換えることにより、調製できる。
【0060】
抗体のフラグメントをコードするDNAの好適な供給源には、ハイブリドーマ及び脾臓細胞といった任意の細胞であって、完全長の抗体を発現するものが含まれる。フラグメントは、そのもの自体を抗体等価物として使用でき、又は上記のとおり等価物に組換えて使用できる。このセクションに記載のDNA欠失及び組換えは、公知の方法、例えば抗体等価物に関して上記に挙げた文献に記載の方法、並びに/又は他の標準的DNA組換え技術、例えば下記の方法により実施できる。DNAの別の供給源は、当技術分野で公知のファージディスプレイライブラリーから産生される一本鎖抗体である。
【0061】
さらに、本発明は、発現配列、プロモーター及びエンハンサー配列と機能し得るように連結された前記ポリヌクレオチド配列を含む発現ベクターを提供する。細菌のような原核生物系及び真核生物系(酵母及び哺乳動物細胞培養系を含むがこれに限定されない)中で抗体ポリペプチドの効率的な合成に使用される多様なベクターが、開発されている。本発明のベクターは、染色体DNA、非染色体DNA、及び合成DNA配列のセグメントを含み得る。
【0062】
任意の好適な発現ベクターを利用できる。例えば、原核生物のクローニングベクターには、E. coli、例えばcolEl、pCRl、pBR322、pMB9、pUC、pKSM及びRP4由来のプラスミドが含まれる。原核生物ベクターには、ファージDNA、例えばMl3及び他のフィラメント状一本鎖DNAファージの誘導体も含まれる。酵母中で使用するのに有用なベクターは、例えば2μプラスミドである。哺乳動物細胞中での発現に好適なベクターには、SV40、アデノウイルス、レトロウイルス由来のDNA配列の周知の誘導体、哺乳動物の機能性ベクターの組み合わせにより作製したシャトルベクター、例えば上記のもの、並びに機能性プラスミド及びファージDNAが含まれる。
【0063】
さらなる真核生物の発現ベクターが、当技術分野では知られている(例えば、PJ. Southern and P. Berg, J. Mol. Appl. Genet., 1, 327-341 (1982); Subramani et al., Mol. Cell. Biol., 1: 854-864 (1981); Kaufmann and Sharp, 「Amplification And expression of Sequences Cotransfected with a Modular Dihydrofolate Reductase Complementary DNA Gene」, J. Mol. Biol. 159, 601-621 (1982); Kaufmann and Sharp, Mol. Cell. Biol. 159, 601-664 (1982); Scahill et al., 「Expression And Characterization Of The Product Of A human Immune Interferon DNA Gene In Chinese Hamster Ovary Cells」, Proc. Nat'l Acad. Sci. USA 80, 4654-4659 (1983); Urlaub and Chasin, Proc. Nat'l Acad. Sci. USA 77, 4216-4220, (1980))。
【0064】
本発明で有用な発現ベクターには、発現されるDNA配列又はフラグメントに機能し得るように連結された少なくとも1つの発現制御配列が含まれる。制御配列をベクターに挿入することにより、クローニングしたDNA配列の発現を制御及び調節することができる。有用な発現制御配列は、例えばlac系、trp系、tac系、trc系、λファージの主要なオペレーター及びプロモーター領域、fdコートタンパク質の制御領域、酵母の解糖性プロモーター、例えば3-ホスホグリセリン酸キナーゼのプロモーター、酵母の酸性ホスファターゼのプロモーター、例えばPho5、酵母α-接合因子のプロモーター、並びにポリオーマウイルス、アデノウイルス、レトロウイルス、及びシミアンウイルス由来のプロモーター、例えば初期及び後期プロモーター又はSV40、並びに原核生物又は真核生物細胞及びそれらのウイルス又はそれらの組み合わせの遺伝子の発現を制御することが知られているその他の配列である。
【0065】
本発明は、前記の発現ベクターを含む組換え宿主細胞も提供する。本発明の抗体は、ハイブリドーマ以外の細胞株中でも発現できる。本発明のポリペプチドをコードする配列を含む核酸を、好適な哺乳動物の宿主細胞の形質転換のために使用できる。
【0066】
特定の所望の細胞株は、所望のタンパク質の高レベル発現、構成的発現、及び最小レベルの発現に基づいて、宿主タンパク質から選別される。発現のために宿主として利用できる哺乳動物の細胞株が当技術分野では周知であり、多くの不死化細胞株、例えば、NSO細胞、チャイニーズハムスター卵巣(CHO)細胞、ベビーハムスター腎(BHK)細胞及び多くの他の細胞が含まれるが、これに限定されない。さらなる好適な真核生物細胞には、酵母及び他の真菌が含まれる。有用な原核生物の宿主には、例えば、E. coli SG-936、E. coli HB 101、E. coli W3110、E. coli X1776、E. coli X2282、E. coli DHI、及び E. coli MRC1のような大腸菌(E.coli)、シュードモナス(Pseudomonas)、枯草菌(Bacillus subtilis)のようなバチルス(Bacillus)、及びストレプトミセス(Streptomyces)が含まれる。
【0067】
これらの本発明の組換え宿主細胞を使用することにより、抗体又はそのフラグメントを発現させることができる条件下で該細胞を培養し、宿主細胞又は宿主細胞周辺の培地から抗体又はフラグメントを精製することにより、抗体又はそのフラグメントを産生させることができる。組換え宿主細胞中での分泌のための発現した抗体又はフラグメントの指向化は、シグナル又は分泌性リーダーペプチドをコードする配列(Shokri et al., Appl Microbiol Biotechnol. 60(6):654- 64 (2003), Nielsen et al., Prot. Eng. 10:1-6 (1997)及びvon Heinje et al., Nucl. Acids Res. 14:4683-4690 (1986)を参照されたい)を、所望の抗体をコードする遺伝子の5'末端に挿入することにより、促進される。これらの分泌性リーダーペプチド因子は、原核生物又は真核生物のいずれかの配列から誘導できる。よって好適には、分泌性リーダーペプチドを使用して、アミノ酸をポリペプチドのN末端に結合することにより、宿主細胞サイトゾルから生じたポリペプチドの移動及び培地への分泌を指向させる。
【0068】
この発明の抗体は、さらなるアミノ酸残基と融合し得る。かかるアミノ酸残基は、おそらくは単離を促進するペプチドタグである。特定の臓器又は組織への帰巣(homing)のために、他のアミノ酸残基を補完することもできる。
【0069】
別の実施形態では、トランスジェニック動物中で抗体をコードする核酸を発現させることにより、本発明の抗体を作製する。その場合、該抗体を発現させて、回収することができる。例えば、回収及び単離を促進する組織特異的な方法で、抗体を発現させることができる。そのような実施形態の1つでは、本発明の抗体は、授乳期間中に分泌用の乳腺中で発現する。トランスジェニック動物には、マウス、ヤギ及びウサギが含まれるが、これに限定されない。
【0070】
本発明で使用できる抗体には、完全免疫グロブリン、免疫グロブリンの抗原結合性フラグメントだけでなく、免疫グロブリンの抗原結合性ドメインを含む抗原結合性タンパク質が含まれる。免疫グロブリンの抗原結合性フラグメントには、例えば、Fab、Fab'、及びF(ab')2が含まれる。他の抗体の形態は、結合特異性を維持するが、例えば、二特異性、多価性(3以上の結合部位)、コンパクトなサイズ(例えば、結合ドメインのみ)を望ましくは含むその他の特性を有するように、改良されている。
【0071】
一本鎖抗体は、その抗体が由来する全抗体の定常ドメインの一部又は全てを欠く。従って、一本鎖抗体は、全抗体の使用に付随する問題のいくつかを解決できる。例えば、一本鎖抗体は、重鎖定常領域及び他の生体分子間の特定の望ましからざる相互作用が存在しない場合が多い。さらに、一本鎖抗体は全抗体よりもかなり小さく、全抗体よりも優れた透過性を有する場合があり、より効率的に標的抗原結合部位に局在化し、結合することができる。そのうえ、一本鎖抗体はサイズが比較的小さいが故に、レシピエントにおける望ましからざる免疫応答が、全抗体よりも誘発されにくい。
【0072】
多重一本鎖抗体は、それぞれの一本鎖が、第1のペプチドリンカーにより共有結合された1つのVH及び1つのVLドメインを有しており、該抗体は少なくとも1以上のペプチドリンカーにより共有結合されることによって、多価一本鎖抗体を形成することができ、それにより単特異性又は複数特異性となり得る。多価一本鎖抗体のそれぞれの鎖は、可変軽鎖フラグメント及び可変重鎖フラグメントを含んでおり、少なくとも1つの他の鎖とペプチドリンカーにより連結されている。ペプチドリンカーは、少なくとも15アミノ酸残基からなる。アミノ酸残基の最大数は、約100である。
【0073】
2つの一本鎖抗体を結合させて、二価性ダイマーとしても知られるジアボディ(diabody)を形成させることができる。ジアボディは、2つの鎖及び2つの結合部位を有し、単特異性又は二特異性であり得る。ジアボディのそれぞれの鎖は、VLドメインに連結されたVHドメインを含む。ドメインは、同一鎖上にあるドメイン間のペア形成を阻害するのに十分に短いリンカーによって、連結される。これにより、異なる鎖上の相補性ドメイン間のペア形成が促進され、2つの抗原結合部位が再生成される。
【0074】
3つの一本鎖抗体を結合させて、三価性トリマーとしても知られるトリアボディ(triabody)を形成することができる。トリアボディはVL又はVHドメインのアミノ末端を、VL又はVHドメインのカルボキシ末端に直接、すなわちいかなるリンカー配列も使用せずに、融合させて構築する。トリアボディは、環状の、頭-尾様式で配置されたポリペプチドを具備した3つのFv頭部を有する。トリアボディがとり得るコンホメーションは、互いに120度の角度で平面に配置された3つの結合部位を有する平面的なものである。トリアボディは、単特異性、二特異性又は三特異性である。
【0075】
従って、本発明の抗体及びそのフラグメントには、天然の抗体、二価性フラグメント、例えば(Fab')2、一価性フラグメント、例えばFab、一本鎖抗体、一本鎖Fv(scFv)、シングルドメイン抗体、多価性一本鎖抗体、ジアボディ、トリアボディ、及び抗原に特異的に結合するようなものが含まれるが、これに限定されない。
【0076】
抗IGF-IR抗体及び抗PDGFRα抗体又は抗体フラグメントは、IGF-IR(WO2005016970)又はPDGFRαを有する細胞に結合することによって内在化され得、抗癌剤に化学的に又は生合成的に連結させることができる。かかる抗体に連結される抗癌剤には、抗体が結合した腫瘍又は抗体が結合した細胞周辺にある腫瘍を、破壊又は損傷させる任意の薬剤が含まれる。例えば、抗癌剤は、化学療法剤又はラジオアイソトープのような毒性剤である。好適な化学療法剤は、当業者には公知であり、アントラサイクリン系(例えば、ダウノマイシン及びドキソルビシン)、メトトレキサート、ビンデシン、ネオカルチノスタチン、シスプラチン、クロラムブシル、サイトシンアラビノサイド、5-フルオロウリジン、メルファラン、リシン及びカリケアマイシンが含まれる。化学療法剤は、従来技術を用いて抗体に結合される(例えば、Hermentin and Seiler, Behring Inst. Mitt. 82:197- 215(1988)を参照されたい)。
【0077】
抗癌剤として使用するのに好適なラジオアイソトープも、当業者には公知である。例えば、131I又は211Atが使用される。これらのアイソトープは、従来技術を用いて、抗体に結合される(例えば、Pedley et al., Br. J. Cancer 68, 69- 73(1993)を参照されたい)。
【0078】
他には、抗体と結合される抗癌剤は、プロドラッグを活性化する酵素である。この場合、標的部位に到達するまでは不活性型を維持するプロドラッグが投与され、該標的部位でその細胞毒性型に変換される。実際には、抗体-酵素コンジュゲートを患者に投与して、治療対象となる組織の領域に局在化させることができる。従って、プロドラッグを患者に投与することにより、治療される組織領域で細胞毒性薬剤に変換させる。
【0079】
他の抗癌剤には、サイトカイン、例えばインターロイキン-2(IL-2)、インターロイキン-4(IL-4)又は腫瘍壊死因子α(TNF-α)が含まれる。該抗体は、サイトカインを腫瘍に指向させることにより、サイトカインは他の組織に影響を与えることなく腫瘍の破壊又は損傷を媒介することができる。サイトカインは、従来のDNA組換え技術を用いることにより、DNAレベルで、抗体に結合させることができる。
【0080】
本発明のある実施形態では、1以上の抗腫瘍剤と組み合わせて抗IGF-IR又は抗PDGFRα抗体を投与する。併用療法の例については、例えば米国特許第6,217,866号(Schlessinger et al.) (抗腫瘍剤と併用する抗-EGFR抗体);WO 99/60023 (Waksal et al.) (放射線療法と併用する抗-EGFR抗体)を参照されたい。任意の好適な抗腫瘍剤、例えば、化学療法剤、放射線療法又はそれらの組み合わせを使用することができる。抗腫瘍剤は、アルキル化剤又は抗代謝剤であり得る。アルキル化剤の例には、シスプラチン、シクロホスファミド、メルファラン、及びダカルバジンが含まれるが、これに限定されない。抗代謝剤の例には、ドキソルビシン、ダウノルビシン、及びパクリタキセル、ゲムシタビンが含まれるが、これに限定されない。
【0081】
有用な抗腫瘍剤には、有糸分裂阻害剤、例えばタキサン類、ドセタキセル及びパクリタキセルも含まれる。トポイソメラーゼ阻害剤は、本発明の抗体と組み合わせて使用できる別のクラスの抗腫瘍剤である。これらには、トポイソメラーゼI又はトポイソメラーゼIIの阻害剤も含まれる。トポイソメラーゼI阻害剤には、イリノテカン(CPT-11)、アミノカンプトテシン、カンプトテシン、DX-8951f、トポテカンが含まれる。トポイソメラーゼII阻害剤には、エトポシド(VP-16)及びテニポシド(VM-26)が含まれる。他の物質については、トポイソメラーゼ阻害活性及び抗腫瘍剤としての有効性について、現在評価中である。好ましい実施形態では、トポイソメラーゼ阻害剤はイリノテカン(CPT-11)である。
【0082】
本発明の特別の実施形態では、ドセタキセルと組み合わせて抗IGF-IR抗体を投与する。本発明の別の実施形態では、ドキソルビシンと組み合わせて抗PDGFRα抗体を投与する。
【0083】
抗腫瘍剤が放射線照射である場合、放射線源は、治療対象となる患者の外部(外部照射療法-EBRT)又は内部(近接照射療法-BT)にある。投与される抗腫瘍剤の用量は、数多くの因子によって決まり、該因子には例えば、薬剤の種類、治療される腫瘍の種類及び重症度並びに薬剤の投与経路が含まれる。しかしながら、本発明は、いかなる特定の用量によっても制限されないことを強調したい。
【0084】
抗体(抗IGF-IR又は抗PDGFRα)療法及び抗体+抗腫瘍剤療法は、(例えば、乳癌のための)補助ホルモン療法又は(例えば、前立腺癌のための)アンドロゲン抑制療法を受ける患者でも使用できる。
【0085】
本発明の抗IGF-IRアンタゴニスト及び抗PDGFRαアンタゴニストは、腫瘍増殖又は血管新生に関与する他の受容体を中和する受容体アンタゴニストと同時に、又は共に投与できる。例えば、本発明の実施形態では、抗IGF-IR抗体及び抗PDGFRα抗体を同時投与する。1実施形態において、標的腫瘍細胞がIGF-IR及びPDGFRαの両方を発現する場合には、それぞれの受容体を介するシグナル伝達によって、共通するシグナル伝達因子が活性化される。1つの受容体の阻害は、共通する下流因子の活性化を通常は低下させるが、両方の受容体を阻害すれば、活性化をさらに低下させるであろう。別の実施形態では、腫瘍又は周辺組織中の特定の細胞は相当量の受容体を発現し、他の細胞は相当量の第2の受容体を発現する。アンタゴニストの同時投与は、腫瘍細胞の増殖及び周辺細胞のパラクリン刺激を減少させる。
【0086】
同時投与の代わりに、二特異性(二重特異性)抗体を投与することもできる。様々な所望の特徴を取り入れるように設計された多様な二特異性抗体が存在する。例えば、二特異性ジアボディは、最も小さいサイズを有する。4つの抗原結合部位(それぞれの結合特異性につき2つ)を備えた二特異性抗体は、対応する天然の抗体と同程度の結合活性を有する。ある二特異性抗体は、Fc領域を取り込むことによって、天然の抗体のエフェクター機能(例えば、補体依存性細胞毒性(CDC)及び抗体依存性細胞毒性(ADCC))を維持する。WO 01/90192はIgG-様の四価抗体を記載しており、WO 2006/020258は2つのジアボディを包含し、エフェクター機能を維持する四価抗体を記載している。
【0087】
別の実施形態では、抗IGF-IR抗体又は抗PDGFRα抗体又はその他のアンタゴニストを、上皮成長因子受容体(例えば、EGFR、Her2/erbB2、erbB3、erbB4)に特異的に結合する受容体アンタゴニストと組み合わせて使用する。特に好ましいのは、EGFRの細胞外ドメインに結合し、そのリガンドのうち1以上の結合を遮断し、さらに/又はリガンドにより誘発されるEGFRの活性化を中和する抗原結合性タンパク質である。EGFRアンタゴニストには、EGFRのリガンドに結合し、EGFRとそのリガンドとの結合を阻害する抗体も含まれる。EGFRのリガンドには、例えば、EGF、TGF-α、アンフィレグリン、ヘパリン結合性EGF(HB-EGF)及びベタセルリンが含まれる。EGF及びTGF-αは、EGFR介在性刺激をもたらす主な内因性リガンドであると考えられているが、TGF-αは、血管新生を促進する能力のほうが高いことが証明されている。EGFRアンタゴニストには、他のEGFR受容体サブユニット(すなわち、EGFRホモダイマー)とのEGFRの二量体化、又は他の成長因子受容体(例えば、HER2)とのヘテロ二量体化を阻害する物質も含まれる。EGFRアンタゴニストには、生体分子及び小分子、例えばEGFRの細胞質ドメインに直接作用し、EGFR介在性シグナル伝達を阻害する合成キナーゼ阻害剤がさらに含まれる。Erbitux(登録商標)(セツキシマブ)は、EGFRに結合しリガンド結合を遮断するEGFRアンタゴニストの一例である。小分子EGFRアンタゴニストの一例はIRESSATM(ZD1939)であり、これはEGFRを阻害するためにATP擬似体(mimetic)として機能するキノザリン(quinozaline)誘導体である。米国特許第5,616,582号(Zeneca Limited);WO 96/33980 (Zeneca Limited)の4ページを参照されたい。また、Rowinsky et al, ASCOの第37回年次会議で発表された抄録5, San Francisco, CA, 12-15 May 2001; Anido et al, ASCOの第37回年次会議で発表された抄録1712, San Francisco, CA, 12-15 May 2001も参照されたい。小分子EGFRアンタゴニストの別の例は、Tarceva(登録商標)(OSI-774)であり、これは4-(置換フェニルアミノ)キノザリン誘導体[6,7-ビス(2-メトキシ-エトキシ)-キナゾリン-4-イル]-(3-エチニル-フェニル)アミン塩酸塩]EGFR阻害剤である。WO 96/30347(Pfizer Inc.)の例えば2ページ12行目〜4ページ34行目及び、19ページ14〜17行目を参照されたい。また、Moyer et al., Cancer Res., 57: 4838-48 (1997); Pollack et al., J. Pharmacol, 291: 739-48 (1999)も参照されたい。Tarceva(登録商標)は、EGFRのリン酸化及びその下流のPI3/Akt及びMAP(マイトジェン活性プロテイン)キナーゼシグナル伝達経路を阻害することによって機能し、p27を仲介する細胞周期の停止をもたらすことができる。Hidalgo et al, ASCOの第37回年次会議で発表された抄録281, San Francisco, CA, 12-15 May 2001を参照されたい。
【0088】
他の小分子もEGFRを阻害することが報告されており、その多くはEGFRのチロシンキナーゼドメインに特異的であると考えられている。かかる小分子EGFRアンタゴニストのいくつかの例は、WO 91/116051、WO 96/30347、WO 96/33980、WO 97/27199 (Zeneca Limited)、WO 97/30034 (Zeneca Limited)、WO 97/42187 (Zeneca Limited)、WO 97/49688 (Pfizer Inc.)、WO 98/33798 (Warner Lambert Company)、WO 00/18761 (American Cyanamid Company)、及びWO 00/31048 (Warner Lambert Company)に記載されている。特異的な小分子EGFRアンタゴニストの例にはCl-1033(Pfizer)が含まれ、これはキノザリン(N-[4-(3-クロロ-4-フルオロ-フェニルアミノ)-7-(3-モルホリン4-イル-プロポキシ)-キナゾリン-6-イル]-アクリルアミド)である、チロシンキナーゼ、特にEGFRの阻害剤であり、WO 00/31048の8ページ22〜26行目に記載されている。また、PKIl66 (Novartis)はEGFRのピロロピリミジン阻害剤であって、WO 97/27199の10〜12ページに記載されており;GW2016 (GlaxoSmithKline)はEGFR及びHER2の阻害剤であり;EKB569 (Wyeth)はEGFR及びHER2を過剰発現する腫瘍細胞の増殖をin vitro及びin vivoで阻害することが報告されており;AG-1478 (Tryphostin)は、EGFR及びerbB-2の両方からのシグナル伝達を阻害するキナゾリン小分子であり;AG-1478 (Sugen)はプロテインキナーゼCK2も阻害する2つの基質の阻害剤であり;PD 153035 (Parke-Davis)は、EGFRキナーゼ活性及び腫瘍増殖を阻害することが報告されており、培養下の細胞中でアポトーシスを誘導し、細胞障害性化学療法剤の細胞毒性を増強し;SPM-924 (Schwarz Pharma)は前立腺癌の治療を目的としたチロシンキナーゼ阻害剤であり;CP-546,989 (OSI Pharmaceuticals)は、固形癌の治療において血管新生の阻害剤であることが報告されており;ADL-681は癌の治療を目的とするEGFRキナーゼ阻害剤であり;PD 158780は、A4431移植片の腫瘍増殖速度を抑制することがマウスで報告されているピリドピリミジンであり;CP-358,774は、HN5異種移植片中で自己リン酸化を阻害することがマウスで報告されているキンゾリン(quinzoline)であり;ZD1839は外陰癌、NSCLC、前立腺癌、前立腺癌、卵巣癌、及び大腸癌を含むマウス移植片モデルで、抗腫瘍活性を有することが報告されているキンゾリンであり;CGP 59326Aは、EGFR陽性移植片の成長を阻害することがマウスで報告されているピロロピリミジンであり;PD 165557 (Pfizer);CGP54211及びCGP53353 (Novartis)はジアニルナフタルイミドである。天然由来のEGFRチロシンキナーゼ阻害剤には、ゲニステイン、ハービマイシンA、クエルセチン、及びエルブスタチンが含まれる。
【0089】
EGFRを阻害することが報告されている故に本発明の範囲にはいる更なる小分子には、三環式化合物、例えば米国特許第5,679,683号に記載の化合物;キナゾリン誘導体、例えば米国特許第5,616,582号に記載の化合物;及びインドール化合物、例えば米国特許第5,196,446号に記載の化合物がある。
【0090】
IGF-IR又はPDGFRαと共に標的化され得る別の受容体は、血管内皮増殖因子受容体(VEGFR)である。本発明の実施形態では、VEGFRアンタゴニストと組み合わせて抗IGF-IR抗体又は抗PDGFRα抗体を使用する。1実施形態では、VEGFR-1/FIt-1受容体に特異的に結合するアンタゴニストを使用する。別の実施形態では、VEGFRアンタゴニストはVEGFR-2/KDR受容体に特異的に結合する。特に好ましいのは、VEGFR-1又はVEGFR-2の細胞外ドメインに結合し、それらのリガンド(VEGFR-2はVEGFによって最も強力に刺激され;VEGFR-1はPlGFによって最も強力に刺激されるが、VEGFによっても刺激される)による結合を遮断し、及び/又はリガンドによって誘発される活性化を中和する抗原結合性タンパク質である。例えば、IMC-1121はVEGFR-2に結合し、中和するヒト抗体である(WO 03/075840; Zhu)。別の例は、可溶性の細胞表面で発現されるVEGFR-1に結合するMAb6.12である。ScFv 6.12は、マウスモノクローナル抗体MAb6.12のVL及びVHドメインを含む。MAb 6.12を産生するハイブリドーマ細胞株は、特許手続上の微生物の寄託の国際的承認に関するブダペスト条約(ブダペスト条約)の規制下で、ATCC番号PTA-3344として寄託されている。別の実施形態では、VEGFRアンタゴニストは、VEGFRリガンドに結合し、リガンドによるVEGFRの活性化を遮断する。例えば、Avastin(登録商標)(ベバシツマブ)は、VEGFと結合する抗体である。
【0091】
腫瘍形成に関与する成長因子受容体の他の例は、神経成長因子(NGFR)及び線維芽細胞成長因子(FGFR)である。
【0092】
更なる他の実施形態では、抗IGF-IR及び抗PDGFRα抗体は、例えば、サイトカイン(例えば、IL-10及びIL-13)のような好適なアジュバント、又はこれらに限定されないが、ケモカイン、腫瘍関連抗原、及びペプチドのようなその他の免疫賦活剤のうち1以上と組み合わせて、投与できる。例えば、Larrivee et al., 前掲を参照されたい。しかしながら、抗IGF-IR又は抗PDGFRα抗体の投与だけでも、十分に、治療上有効に腫瘍の増殖を防止、阻害、又は減少し得ることを理解されたい。
【0093】
併用療法では、抗IGF-IR又は抗PDGFRα抗体を、別の薬剤による治療の開始前、治療中、又は治療後に投与するだけでなく、それらを任意で組み合わせて、すなわち、抗腫瘍剤療法の開始前及び治療中、開始前及び治療後、治療中及び治療後、又は開始前、治療中、及び治療後に投与できる。例えば、抗体は、放射線療法を開始する前1〜30日間、好ましくは3〜20日間、より好ましくは5〜12日間投与できる。本発明の好ましい実施形態では、抗体療法と同時に、又はより好ましくは抗体療法後に化学療法剤を投与される。
【0094】
本発明では、任意の好適な方法又は経路を使用して、本発明の抗体を投与することができ、場合によっては、抗腫瘍剤及び/又はその他の受容体のアンタゴニストを同時投与することができる。本発明で利用される抗腫瘍剤レジメンには、患者の腫瘍症状の治療にとって最適と考えられる任意のレジメンが含まれる。多様な悪性腫瘍が、特異的な抗腫瘍抗体及び特異的な抗腫瘍剤の使用を必要としており、該使用は患者ごとに決定されるだろう。投与経路には、例えば、経口投与、静脈内投与、腹腔内投与、皮下投与、又は筋肉内投与が含まれる。投与されるアンタゴニストの用量は、例えば、アンタゴニストの種類、治療される腫瘍の種類及び重症度、並びにアンタゴニストの投与経路を含む様々な因子によって決定される。しかしながら、本発明は、いかなる特定の投与方法又は投与経路によっても限定されないことを強調したい。
【0095】
当業者であれば、治療の用量及び頻度は、それぞれの患者の認容性、並びに使用される遮断剤又は阻害剤の薬力学的特性及び薬物動態学的特性によって、決まることがわかるであろう。理想的には、使用薬剤の飽和した薬物動態をもたらすことが望ましい。抗IGF-IR及び抗PDGFRα抗体の初回投与量はいずれも、例えば、約10〜約1000 mg/m2であり、好ましくは約200〜約400 mg/m2である。この後、いくつかの1日量又は週用量、例えば約200〜約400 mg/m2で、薬剤をさらに投与する。副作用について患者をモニターし、かかる副作用が重篤である場合には、治療を中止する。
【0096】
当業者は、有効用量を決定するために、治療の進捗をモニターする方法も知っている。前立腺癌からの骨転移では、かかる方法の1つはPSAレベルをモニターすることである。骨転移をモニターするその他の方法には、骨スキャン及びMRIが含まれる。
【0097】
癌治療誘発性の骨喪失(CTIBL)のリスクがあるか、又はCTIBLが問題となる患者(例えば、乳癌に対する補助ホルモン療法を受ける患者又は前立腺癌に対するアンドロゲン抑制両方を受ける患者)では、前述した任意の治療を、CTIBLの予防用薬剤、例えばビスホスホネートの投与で補完してもよい。ビスホスホネートには、例えばクロドロネート、リセドロネート及びゾレドロン酸が含まれる。
【0098】
本出願全体を通して、様々な文献、参照テキスト、教科書、技術マニュアル、特許、及び特許出願が引用されている。これらの文献、特許、特許出願及びその他の文献の教示及び開示は、引用によりその全体が本出願に包含され、本発明に付随する技術態様をより十分に説明する。
【0099】
本明細書に開示の発明の原理範囲内における変更が、当業者によりなされることを理解および予測される。そして、それらの変更は、本発明の範囲内に包含されることが意図される。
【0100】
以下の実施例により本発明をさらに説明されるが、いかなる方法によっても、本発明の範囲は限定されるべきでない。従来方法、例えばベクター及びプラススミドの構築並びに抗体及び抗体フラグメントの発現で利用される方法の詳細な説明は、数多くの文献から得ることができ、該文献にはSambrook, J et al., (1989) Molecular Cloning: A Laboratory Manual, 2nd ed., Cold Spring Harbor Laboratory Press; Coligan, J. et al. (1994) Current Protocols in Immunology, Wiley & Sons, Incorporated; Enna, SJ. et al. (1991) Current Protocols in Pharmacology, Wiley & Sons, Bonifacino, J.S. et al. (1999) Current Protocols in Cell Biology, Wiley & Sonsが含まれる。本明細書に記載の全ての文献は、その全体が本明細書に組込まれる。
【実施例】
【0101】
実施例1
IMC-A12及びドセタキセルの腫瘍増殖に対する効果
アンドロゲン非依存性(AI)LuCaP35Vの腫瘍小片(20〜30 mm3)を、32匹の6週令の去勢したSCIDマウスに、前述のとおり皮下に(s.c.)それぞれ移植した。移植した腫瘍の体積が150〜200 mm3に達するまで観察し、被験動物を治療試験のために、4つのグループに無作為割付けした。グループ1の被験動物には、用量20 mg/kgのドセタキセルによる治療を行った。グループ2の被験動物には、用量10 mg/kgのドセタキセルによる治療を行った。グループ3の被験動物には、10 mg/kgのドセタキセル及び40 mg/kgのA12の併用療法を行った。グループ4の被験動物には、20 mg/kgのドセタキセル及び40 mg/kgのA12の併用療法を行った。すべての治療は、腹腔内(ip)投与した。ドセタキセルは、週に1回投与した。Al2は週に3回投与した。すべての被験動物を4週間治療し、安楽死させる前にさらに4週間モニターした。1週間に2回腫瘍を測定して、式:体積 = L X W2/2を用いて腫瘍体積を算出した。我々ワシントン大学のIACUCによって承認された動物プロトコールに従って、腫瘍体積が1000 mm3に達するよりも前に、又は被験動物の体重減少が最初の体重の20%を超える前に、被験動物を安楽死させた。被験動物の体重は、週に2回測定した。血液サンプルを、眼窩洞から毎週採取した。血清を分離し、IMxトータルPSAアッセイ(Abott Laboratories, Abott Park, IL)を用いて、PSAレベルを測定した。in vivoでの腫瘍細胞増殖速度の評価のために被験動物を安楽死させる1時間前に、BrdUを腫瘍に注射した。
【0102】
安楽死させた後に、腫瘍を回収し二つに分けた。腫瘍の一部は10%中性緩衝ホルマリン(NFB)中で固定し、パラフィン包埋した。免疫組織化学(IHC)染色のために、5ミクロンの切片を作製した。腫瘍の残りの部分は、ミンチして70μmのナイロンシーブで濾過することにより、機械的に単一の細胞に分離させた。
【0103】
図1に示すとおり、LuCaP 35V移植片は、治療をしない場合には、362.0±72.0 mm3/週の平均成長速度で、マウスで急激に成長した。未治療群の被験動物は全て、試験群における治療開始後3週間以内に、腫瘍体積が1000 mm3を超えたので屠殺する必要があった。40μg/kgのA12単独で被験動物を治療した場合には、腫瘍成長速度は、治療期間中は192.7±35.6 mm3/週まで低下した。ドセタキセルを用量10 mg/kgで被験動物に投与した場合には、LuCaP 35V腫瘍の成長速度は、平均で29.6 ±6.1 mm3/週まで低下した。Al2治療と組み合わせてドセタキセルを与えた場合には、LuCaP 35V腫瘍の成長速度は平均7.9 ±1.0 mm3 /週までさらに低下した(図1b)。A12を併用したドセタキセルの阻害効果は、治療終了後4週間以上持続した。より高い用量のドセタキセル(20mg/kg)を被験動物に投与した場合には、A12治療の併用の有無に関わらず、腫瘍体積は4週間の治療期間中は増加しなかった;一方で、腫瘍体積が減少する傾向を観察した。しかしながら、A12を併用してドセタキセルで治療した被験動物群では、治療終了後4週間は、腫瘍体積の減少が持続した。対照的に、ドセタキセル単独で治療した被験動物群では、腫瘍体積が平均速度27.0±16.1 mm3/週で増加した。これらの結果は、A12を併用した所与の用量のドセタキセルが、治療中又は治療追跡後に、腫瘍増殖に対するドセタキセルの阻害効果を増強できることを示している。
【0104】
PSAは、前立腺腫瘍の増殖を評価するための使用される一般的な臨床パラメーターである。血清PSAレベルを、治療中及び治療後に被験動物で測定した。図1cに示すとおり、A12及びドセタキセル、又は20 mg/kgドセタキセル単独で治療した被験動物では、4週間の治療期間は、PSAの血清レベルの顕著な変化は見られなかった。このことは、腫瘍増殖の抑制と一貫性がある。治療終了後、ドセタキセル単独で治療した被験動物では、血清PSAレベルの増加が示された。一方、A12を併用してドセタキセルで治療した被験動物では、血清PSAレベルは一定であるか又はむしろ減少した。これらのデータは、ドセタキセル及びA12で治療した被験動物では、治療後に腫瘍増殖の阻害が持続したことと一致する。
【0105】
抗IGF-IR抗体と組み合わせたドセタキセルによるアポトーシスの誘導
治療終了時において、ドセタキセル及びA12の併用治療の細胞周期及び細胞生存に対するin vivo効果を、Apop-Direct kit (BD BioScience)を用いて、末端デオキシヌクレオチジルトランスフェラーゼ介在切断末端標識(TUNEL)アッセイ及びプロピジウム(PI)染色によって、既報のとおり測定した。簡潔にいうと、単一細胞の懸濁液から得た1×106細胞を10%中性緩衝ホルマリン(NBF)にいれた後、70%エタノールアルコールに−2O℃で30分間いれて固定した。数回の洗浄後に、細胞を0.1%Triton X-100で溶解し、FITCとコンジュゲートしたdUTP及び末端デオキシヌクレオチジルトランスフェラーゼ酵素(TdT)と共に37℃で1時間インキュベートした後、PI/RNase緩衝液(100μg/mlのPI、50μg/ml RNase)と共に室温で60分間インキュベートした。BD FACscanを用いて、サンプルをフローサイトメトリーで解析した。データは、CellQuestPROソフトウェアで解析した。
【0106】
治療終了後4週間、A12を併用してドセタキセルで治療した被験動物から得た腫瘍のかなりの割合(10 mg/kgのドセタキセルで治療したグループでは66.7%、20mg/kgのドセタキセルで治療したグループでは77.8%)で、使用したドセタキセルの用量と関係なく、アポトーシスを検出した(図2b及び表1)。これらの腫瘍におけるアポトーシス事象は、平均で15.0±4.3%の割合で発生した。ドセタキセル単独で治療した被験動物では、腫瘍においてアポトーシスを検出しなかった。代わりに、腫瘍の大部分(10 mg/kgのドセタキセルで治療したグループでは88%、20 mg/kgのドセタキセルで治療したグループでは100%)で、正常の細胞周期が進行した(図2a及び表3)。
【表3】
【0107】
様々な治療終了後に、腫瘍細胞の増殖能力をさらに評価するために、パラフィン切片を抗BrDu抗体で染色した。腫瘍サンプルを10%NBF中で固定し、パラフィン包埋し、及びスライド上に5μmで切片化した。脱パラフィン化及び再水和の後に、95℃で2×5分間で0.01 Mクエン酸(pH 6.0)を用いて抗原を回収した。スライドを30分間冷却した後、PBSで逐次すすいだ。メタノール中で15分間、0.3%H2O2でインキュベートすることにより、内因性ペルオキシダーゼ活性をクエンチングした。0.05%Tween 20(PBST)を含むPBS中で、1.5%正常ヤギ血清で1時間ブロックした後、スライドをマウス抗BrdU抗体(1μg/ml)で1時間インキュベートした。その後、ビオチン化したヤギ抗マウスIgGと連続して30分間インキュベートし、ペルオキシダーゼ標識したアビジン(Santa Cruz Biotechnology)と30分間インキュベートし、ジアミノベンジジン(DAB)/過酸化水素クロモーゲン基質(Vector Laboratories, Burlingame, CA)と5〜10分間インキュベートした。すべてのインキュベーションステップは室温で行った。スライドはヘマトキシリン(Sigma)を用いて対比染色し、パーマウント(Fisher Scientific, Fair Lawn, New Jersey)を用いてマウントした。陰性対照として、抗BrdU一次抗体の代わりにマウスIgG(Vector Laboratories)を使用した。Zeiss顕微鏡下でスライドを観察し、デジタル画像を入手した。数多くのBrdUで標識した核小体及び総核小体は、各切片につき10個の無作為視野から集めた。増殖指標は、BrdU陽性の核小体数を、総核小体数で割ることにより算出した。スライドにつき、10区域をカウントした。ヘマトキシリン及びエオシン(Richard Allen, Kalamazoo, MI)を用いることにより、H&E染色を行った。
【0108】
ドセタキセル及びA12で治療した被験動物では、BrDu取り込みが、同一用量のドセタキセル単独で治療したものよりも顕しく減少した(図3)。BrDu取り込みに関するこれらのデータは、細胞周期及びアポトーシスに関する上記所見とも一致しており、Al2がドセタキセルの細胞毒性を顕しく増強したことを示唆する。
【0109】
抗IGF-IR抗体と組み合わせたドセタキセルvs.ドセタキセル単独で治療した腫瘍における遺伝子発現の示差的制御
Al2によるドセタキセルの効果の著しい増強でとり得るメカニズムを調べるために、免疫組織化学分析及びフローサイトメトリー分析により、採取した腫瘍の全てにおいて、IGF-IR発現を調べた。全ての治療群で、又は対照群(データは示してない)と比較して、表面IGF-IR発現の差はなかった。20 mg/kgのドセタキセル及びA12を併用して20 mg/kgのドセタキセルを投与した患者から得た腫瘍において、cDNAマイクロアレイ分析を用いて、治療後の遺伝子発現を調べた。SAM解析では、ドセタキセル及びA12の併用療法を受けた腫瘍において、ドセタキセル単独療法を受けた腫瘍と比較して、示差的に発現したものとして49個の遺伝子を同定した。この際、発現レベルの差は2倍を超えており、過誤発見率(FDR)は10%未満であった(データを示していない)。13個の遺伝子が、アポトーシス又は細胞周期の制御に関与している可能性があることを同定した(表4)。13個の遺伝子は全て、2つの治療間で少なくとも2倍異なっており、FDRは0.02%未満であった。ドセタキセル単独で治療した腫瘍と比べ、ドセタキセル及びA12で治療した腫瘍では、9個の遺伝子がダウンレギュレートされており、4個の遺伝子がアップレギュレートされていた。
【表4】
【0110】
選択した遺伝子について、リアルタイムRT-PCRで結果を確認した。標的cDNAの基準となるPCR断片を精製した。リアルタイムRT-PCRのために、10 ng/μl〜103 pg/μlの一連の標準希釈液を使用して、標準曲線を作成した。プールした腫瘍の各グループから得た1μgの総RNAを、Superscript第一鎖合成システム(Invitrogen)による第一鎖cDNA合成のために使用した。1μlのcDNA第一鎖、特異的なプライマーセット、及びLightcycler FastStart DNA Master Plus SYBR Greenを含有する反応混合物20μl中で、製造業者の指示書(Roche, Nutley, NJ)に従いRoche Lightcyclerを用いて、リアルタイムRT-PCRを行った。Lightcycler software v3.5を用いて、RT-PCR産物の融解曲線分析を行った。アンプリコンサイズは、アガロースゲル電気泳動で確認した。それぞれのサンプルは、2回ずつアッセイした。結果を図4に示す。
【0111】
ダウンレギュレートされた遺伝子のうち、TUBBはドセタキセル耐性をもたらすことがわかっており(Tanaka et al., 2004, Int. J. Cancer 111, 617-26)、BIRC5 (survivin)の発現増加は、強力な前立腺癌及び抗アンドロゲン療法に対する耐性と関係することがわかっている(de Angelis et al., 2004, Int. J. Oncol. 24, 1279-88; Zhang et al., 2005, Oncogene 24, 2474-82)。そのうえ、TUBBはIGF-IR介在型形質転換に関与するIGF-IR制御遺伝子である(Loughran et al., 2005, Oncogene 24, 6185-93)。4つのアップレギュレートされた遺伝子のうち、IGFBP3はIGF-リガンドのシグナル伝達を阻害するだけでなく、リガンド依存的な様式で前立腺腫瘍細胞におけるアポトーシスを誘導することがわかっている(Grimberg et al., 2000, J. Cell. Physiol. 183, 1-9)。
【0112】
治療後のA12の血清レベル
A12を併用してドセタキセルを投与した被験動物において、A12の血清レベルを測定した。血清A12レベルは、治療終了後2週では100倍減少し、治療終了後4週では、非常に低いレベルで検出した(図5)。
【0113】
総合的な細胞毒性
ドセタキセル及びIMC-A12の同時投与による細胞毒性を調べた。A12は、マウスIGF-IRと95%超の交差反応性を有するが、薬剤の併用又はドセタキセル単独で治療した被験動物では、対照群の腫瘍を有する被験動物と比べて、異常な日常活動や挙動の変化は全く観察しなかった。細胞周期及びアポトーシスアッセイの両方では、どの治療群でも腎細胞に対する有意な影響を認めなかった(データは示していない)。体重変化の有意な変化も、治療群間で認めなかった(図6)。
【0114】
骨転移に対する抗IGF-IR抗体療法
骨中の前立腺癌細胞の転移増殖に対する抗IGF-IR抗体による治療の効果を、SCIDマウスの頸骨に直接注射した前立腺癌細胞を用いて評価した。この方法によって、循環系からの化学走化性依存性浸潤には頼らずに、転移性腫瘍を直接定着させた。多様な腫瘍株を、骨転移を定着させるために利用することができる。これらには、PC-3、LuCaP35、骨融解性病変を産生するLnCaP細胞、及び造骨性病変を産生するLuCaP 23.1が含まれる。
【0115】
LuCaP23.1細胞は、IGF-IRを発現する細胞であるが、骨環境下では80%以下の取り込み率を有しており、造骨性反応をもたらす。予備実験において、LuCaP23.1サンプルでは、腫瘍における骨体積対組織体積(%BV/TV)が、対照群の頸骨の場合と比べ有意に増加した(対照群の254〜503%, p=0.024)。頸骨におけるLuCaP23.1腫瘍は全て、正常サンプルでは存在しなかった新しい骨梁及び正常の骨髄と置き換えられた多数の腫瘍病巣を提示した。いくつかの被験体では、腫瘍及び骨成長がもとの骨の外側に伸長した。LuCaP23.1サンプルの%BV/TVの増加は、去勢術後にも観察した;腫瘍化した頸骨の%BV/TVは、腫瘍化していない頸骨の212〜354%であった(p=0.024)。LuCaP23.1の内部頸骨の異種移植片で観察された結果は、腫瘍細胞により新規の骨形成が促進されることを示す。さらに、腫瘍は、骨芽球性骨転移のヒトサンプルと多くの類似点を示し、該類似点には多数の腫瘍病巣及び食塩骨基質量の増加が含まれる。
【0116】
IMC-Al2による治療の効果を評価するために、LuCaP23.1異種移植片腫瘍をSCIDマウスに移植し、血清PSAレベルを隔週で測定して腫瘍増殖を評価した。頸骨腫瘍細胞の移植2週間前に、全ての被験動物を去勢させた。試験マウスへのIMC-A12の投与は、血清PSAレベルが5〜10 ng/mlに達した(腫瘍が定着したことを示す)時に、開始した。40 mg/kgのIMC-A12を、6週間かけて週に3回、腹腔内注射(i.p.)した。
【0117】
腫瘍化した頸骨及び腫瘍のない対側の頸骨の骨塩濃度(BMD)を、二重X線吸収分析法(PIXImus Lunar densitometer)で測定した。該方法は、腫瘍細胞注射部位の2.5 mm×2.5 mmのエリアで行うか、又は対側頸骨のうち移植部位に対応する部位で行った。隔週の病変の評価は、血清PSAの測定によって行った。去勢後の血清PSAレベルに基づいて対照群で骨病変の再発を確認した(LuCaP35は>60 ng/ml、LuCaP23.1は>500 ng/ml)時、X線撮影で骨病変が出現した時、又は被験動物が易感染性となった時に、全ての被験動物を屠殺した。被験動物を屠殺する1時間前に、腫瘍細胞の増殖をモニターするために、BrdUを注射した。屠殺する前にX線撮影(FaxitronX-ray MX-20)を行い、屠殺時に両方の頸骨のBMDを測定した。
【表5】
【0118】
IMC-Al2で治療したマウスでは、血清PSAレベルが有意に低かった(図7)。また、骨芽球性転移腫瘍の増殖に関係するBMDの増加も有意に減少した(表5)。腫瘍化していない下肢のBMD測定により、IMC-A12治療では骨密度を喪失(骨粗しょう症)させないことがわかった。IMC-Al2で治療したマウス及び治療していないマウスのX線撮影画像は、治療したマウスでは、腫瘍増悪を有意に減少又は予防したことを示す(図8)。
【0119】
骨転移に対する抗IGF-IR抗体及びドセタキセルの併用
頸骨腫瘍注射の2週間前に、SCIDマウスを去勢させた。LuCaP23.1前立腺癌細胞をマウスの頸骨に直接注射することにより骨転移を発生させ、造骨性病変に成長させた。異種移植片はIGF-IRを発現する。腫瘍増殖を評価するために、隔週で血清PSAレベルを測定する。血清PSAレベルが5〜10 ng/mlに達し、腫瘍の定着が示された時に、被験動物を4つのグループに無作為割付けした。
【0120】
2つのグループでは、40 mg/kgのIMC-A12を6週間にわたり、週に3回腹腔内注射した。そして、1つのグループにはIMC-Al2+ドセタキセル20 mg/kg(i.p.)を6週間にわたり週に1回投与し、第2のグループには、IMC-A12+ドセタキセル10 mg(i.p.)を6週間にわたり週に3回投与した。対照群には、IMC-A12なしで10又は20 mgのドセタキセル(i.p.)を投与した。
【0121】
被験動物は、PSAを毎週測定してモニターした。治療終了後は、ドセタキセル単独グループの腫瘍が腫瘍の再増大を示すまで、毎週PSA測定をして被験動物のモニターを続けた。ドセタキセル単独グループにおいて、(未治療の被験動物よりも遅い速度ではあるが)PSA値が上昇した時にも、IMC-A12+ドセタキセルで治療したマウスにおけるPSAレベルは安定状態にあり、いくつかの被験動物では低下し始める。治療終了から6週間後でさえ、PSAレベルの低下を継続して観察した。
【0122】
上記のとおり、移植時及び屠殺時にBMD測定を行い、屠殺する直前にX線撮影を行う。IMC-A12+ドセタキセルで治療したグループでは、少量のBMDの増加を示すか、又はBMDが全く増加せず、X線撮影画像では骨芽細胞活性の兆候がわずかであるか、又は全くないことを示す。
【0123】
骨転移に対する抗IGF-IR抗体及びドセタキセルの併用
LuCaP23.1ヒト前立腺腫瘍小片(20〜30 mm3)を機械的に消化した。2〜5×105個のLuCaP23.1生細胞を、6〜8週令のSCIDマウスの頸骨に注射した。3つのグループに無作為割付した21匹のマウスを、この試験では使用した。腫瘍注射後に、血清PSAを毎週モニターした。血清PSAレベルが5〜10 ng/mlに達し、腫瘍増殖が示された時に治療を開始した。グループ1には、対照溶媒である生理食塩水緩衝液を投与した。グループ2には、20 mg/kgのドセタキセルを4週間かけて週に1回投与(i.p.)した。グループ3には、4週間にわたり、20 mg/kgのドセタキセルを週に1回及び40mg/kgのAl2を週3回投与(i.p.)した。治療反応が、骨芽球性であるか又は骨融解性であるか決定するために、全ての治療終了時に、被験動物のDexaスキャン及びX線撮影によってBMDを測定した。
【0124】
ドセタキセル単独又はA12を併用したドセタキセルは、LuCaP23.1腫瘍増殖を有意に阻害した。この結果は、血清PSAレベルの抑制に反映されており(図9a)、2つの治療間で有意な差は認めなかった。しかしながら、ドセタキセル単独で治療した被験動物では、治療終了後に血清PSAが上昇し始め、腫瘍の再増殖を示した;一方、併用療法を受けた被験動物では、血清PSAレベルの持続した抑制を観察し、治療後に長時間の腫瘍静止が示された。血清PSAレベルは、骨密度(BMD)及びX線撮影でみた腫瘍化した骨のサイズと相関することが示された(図9b)。対照群、ドセタキセル20、及びA12を併用したドセタキセル20で治療した被験動物における5週後の平均骨密度は、それぞれ0.112±0.01、0.09±0.02、及び0.05±0.009(平均±SEM)であった。治療群では、明らかに骨密度が減少傾向にあった。
【0125】
実施例2
骨髄穿刺液により誘導されるAktリン酸化
Cambrex (PoieticsTM Donor Program)より、健常男性ドナー(18〜45歳)から入手した骨髄サンプルの供給を受けた。サンプルを1,500 rpmで遠心分離することにより、可溶性相と細胞性相を分離した。連続して0.8μm及び0.22μmのフィルターを用いて、上澄液を濾過した。50μlの骨髄穿刺液を1 mlの溶媒を用いて(最終希釈1:20)、細胞に投与した。
【0126】
血清の存在下で行った実験では、細胞を、10%FBS及び50μg/mlのゲンタマイシンを補填したDMEM中で、骨髄に曝露する前に24時間インキュベートした。血清の不在下(飢餓細胞)での実験では、細胞をPBSで2回洗浄し、成長培地を無血清DMEMで置き換え、細胞を、骨髄調製物に曝露する前に4時間インキュベートした。使用時に、PDGF受容体の特異的阻害剤であるAG-1296(Rice et al., 1999, Amer. J. Path. 155, 213- 21)を添加して、骨髄穿刺液にさらす前に30分間培養した。IMC-3G3抗体を、下記の治療期間の前に投与した。
【0127】
Aktによる骨髄活性化を、PDGFRαを発現するPC3-ML細胞中で検出したが、受容体を欠くDU-145細胞では検出しなかった。1つの実験では、Akt活性化に対する血清成分の影響を最小限とするために、細胞を、無血清培地中で4時間、前もってインキュベートした。骨髄抽出物の添加により、PC3-ML細胞では強力なAktリン酸化が生じたが、DU-145細胞ではAktリン酸化が生じなかった(図10A)。反応の有意性を評価するために、血清を用いて第2の実験を行った。PC3-ML細胞中では、骨髄穿刺液によるAktリン酸化の強力な刺激を、血清の存在下でも観察した(図10B)。DU-145細胞では、ほんの小さな反応しか現れなかった。
【0128】
PDGFRα介在性Aktリン酸化
骨芽細胞及び破骨細胞は、PDGF-AA及びPDGF-BBの両方を分泌するが、骨髄の可溶性環境では、これらの成長因子を与えると考えられている。骨髄抽出物に対するPC3-ML細胞の反応性が、PDGFRαを介したシグナル伝達と関係するか否かを決定するために、20μMのAG-1296の不在下又は存在下で、PC3-ML細胞を骨髄穿刺液にさらした。この濃度のAG-1296は、PDGF-BB誘導性のAkt活性化を完全に阻害した(図11A)。AG-1296は、骨髄穿刺液誘導性のAkt活性化を、40%を超えて阻害した(図11B及びD)。このことは、骨髄誘導性Akt活性化の大半においてPDGFRαシグナル伝達が役割と果たすことを示している。
【0129】
PDGFRαシグナル伝達に対するPDGF-AA及び-BBの直接的な関与についても、骨髄穿刺液のその他の成分と比較して評価した。3名の異なるドナーから入手した骨髄穿刺液中のPDGF-AA及び-BBの濃度を測定したところ、400 pg/ml〜2 ng/mlであった。骨髄穿刺液の20倍希釈液を得て、20〜100 pg/ml濃度のPDGF-AA及び-BBに、実際に試験細胞をさらした。PC3-ML細胞を、100 pg/mlのPDGF-AA及び-BBでそれぞれ適切に処理した。Aktリン酸化は、骨髄穿刺液で得たものの10%未満であった(図3C及びD)。従って、PDGFRαシグナル伝達によるAkt経路の活性化は、PDGF-AA及び-BB以外のPDGFRαリガンド並びに/又はリガンドの直接結合によるPDGFRαの活性化以外の機構と、関係することが明らかである。
【0130】
抗PDGFRα抗体によるAktリン酸化の阻害
中和抗体IMC-3G3は、ヒトPDGFRαに特異的であるが、PC3-ML細胞のAktリン酸化を阻害するその能力についても試験した。30分間のプレインキュベーション及び濃度20μg/mlのIMC-3G3で、30 ng/mlのPDGF-BBによる刺激効果を中和した(図12A)。抗体による処理も、骨髄誘導性のAktリン酸化を約40%阻害した(図12B及びC)。Aktリン酸化に対するIMC-3G3の阻害効果は、プレインキュベーション時間に依存しており、30分間のインキュベーション(12B及びC)よりも、120分間のインキュベーションのほうが、著しく有効であることも観察した(図12D)。1つの考えられる説明は、IMC-3G3がPDGFRαの内在化を誘導しており、その阻害効果は、リガンド結合の遮断だけでなく、細胞膜からの受容体の除去にも関係しているというものである。
【0131】
実施例3
ヒト抗PDGFRα抗体の単離
ヒト抗PDGFRαモノクローナル抗体を、標準的ハイブリドーマ技術(Harlow & Lane, ed., Antibody: A Laboratory Manual, Cold Spring Harbor, 211-213 (1998);これらは、引用により本明細書に包含される)により、ヒトγ重鎖及びκ軽鎖免疫グロブリンを発現するトランスジェニックマウス(Medarex Inc., Sunnyvale, CA)を用いて作製した。ヒトPDGFRα細胞外ドメイン(ECD)は、R&D Systems (Minneapolis, MN)から購入した。PDGFRαを安定して発現する3×107個のブタ大動脈内皮細胞(PAE Rα)を皮下投与(s.c.)して、KMマウスを免疫した。4週間後、マウスを、完全フロインドアジュバント中50μgのPDGFRαECD(s.c.)でブーストし、更に3×107のPAE Rα細胞(i.p.)を加えた。3週間毎にさらに2回、不完全フロインドアジュバント中25μgのPDGFRαECDで、マウスをブーストした。
【0132】
高い血清結合力価及び遮断力価を有するマウス由来の脾臓細胞を単離し、骨髄腫細胞と融合させた。遮断活性を提示するハイブリドーマ培養物をサブクローニングし、これらのハイブリドーマ由来の抗体を、プロテインGクロマトグラフィーで精製した。
【0133】
PDGFRαとの結合について、直接結合アッセイでIgGを評価した。PBSに溶解したPDGFRαECDを、96-ウェルプレート上に固定した(100 ng/ウェル)。その後、プレートをPBST(PBS+0.05%Tween 20)で洗浄し、PBSM(PBS中3%ミルク;200μL/ウェル)で、25℃で2時間ブロックした。PBSM中で希釈したIgGを、固定したPDGFRαECDと25℃で1時間インキュベートした後、プレートをPBSTで洗浄した。PBSM中1:5,000に希釈した第2抗体(ヤギF(ab')2 抗ヒトIgG-セイヨウワサビペルオキシダーゼコンジュゲート;BioSource International, Camarillo, CA)を25℃で1時間加えた。PBSTでプレートを洗浄した後、TMBペルオキシダーゼ基質(KPL, Gaithersburg, MD)を加え、100μLの1 mol/L H2SO4で反応を停止させた。マイクロプレートリーダー(Molecular Devices, Sunnyvale, CA)を用いて、A450 nmでプレートを読み取った。
【0134】
固相PDGF遮断アッセイ(Duan et al., 1991, J. Biol. Chem. 266:413-8を参照されたい:該文献は、引用により本明細書に包含される)を利用して、PDGFの遮断を評価した。PDGFRαECDをPBS中で希釈し、これで96-ウェルマイクロタイタープレート(放射線照射したタンパク質結合性ポリスチレンのImmulon 2HB 1×12 Removawell平坦ストリップ; Dynex Technologies, Chantilly, VA)上を被覆した。それぞれのウェルは、総体積100μL中60 ngのPDGFRαで、25℃で3時間被覆した。その後、プレートを2回洗浄し、25 mmol/L HEPES (pH 7.45)、0.5%ゼラチン、100 mmol/L NaCl、及び0.1%Tween 20を用いて、4℃で一晩遮断した。その後、プレートを25℃で20分間温め、結合緩衝液(25 mmol/L HEPES (pH 7.45)、0.3%ゼラチン、100 mmol/L NaCl、0.01% Tween 20)で1回洗浄した。50力価のIgGをそれぞれのウェルに加え、25℃で30分間インキュベートした。ヨウ化PDGFを結合緩衝液で希釈し、それぞれのウェルに加えた(50μLの1 nmol/L溶液)プレートを25℃で2時間インキュベートした後、結合緩衝液で5回洗浄した。それぞれのウェルはガンマカウンタでカウントした。細胞ベースの遮断アッセイを、Heldin et al., 1988, EMBO J. 7, 1387-93に記載のとおり行った。
【0135】
PDGFRαとの抗体結合反応速度を、BIAcore 3000装置(BIAcore, Inc., Piscataway, NJ)を用いて測定した。PDGFRαECDをセンサーチップ上に固定し、様々な濃度で抗体を注入した。それぞれの濃度でセンサーグラムを得て、BIA Evaluation 2.0 programを用いて評価を行い、速度定数を決定した。親和性定数Kdは速度定数の比Koff/Konから算出した。
【0136】
図13は、ELISAでの、固定したPDGFRαECDに対するヒトモノクローナル抗体IMC-3G3の用量依存的結合を示す。PDGFRαECDに対する最大結合の50%に必要な抗体濃度は、0.06 nmol/Lであった(表6)。BIAcore装置上で表面プラズモン共鳴により測定したところ、ED50は抗体におけるKdと一致した(表6)。モノクローナル抗体は、[125I]PDGF-BBが固定した受容体と結合するのを、IC50 0.43 nmol/Lで遮断した。PDGFRα上でのPDGF-AAとPDGF-BBの結合部位は、構造上同一の場所ではない。この結果は、3G3のエピトープが、両方の成長因子の結合部位と空間的に重複していることを示唆している。
【表6】
【0137】
受容体のリン酸化及び下流エフェクター分子の活性化の阻害
IMC-3G3によるPDGF誘導性細胞内シグナル伝達に対する効果を、PAE Rα細胞を用いて調べた。細胞を6-ウェルファルコン組織培養プレート(250,000細胞/ウェル)に播種し、一晩成長させた。その後ウェルをすすぎ、無血清培地中でインキュベートした。細胞を静止させるために一晩インキュベートした後、該細胞を抗体で、37℃で30分間処理した。その後、PDGF-AA又はPDGF-BBを加え、37℃でさらに10分間インキュベートした。そして、細胞を剥離し、200μLの溶解緩衝液(50 mmol/L Tris-HCl (pH 8.0)、1%Triton X-100、150 mmol/L NaCl、1 mmol/L EDTA、0.1%SDS、1 mmol/L o-バナジウム酸ナトリウム及びプロテアーゼ阻害剤(Complete Mini、Roche、Mannheim、Germany))中で溶解した。細胞溶解物を、増強された化学ルミネセンス試薬及びHyperfilm(Amersham Biosciences)を用いて、SDS-PAGE及びウェスタンブロット法により解析した。
【0138】
リガンド誘導性の受容体チロシンリン酸化を阻害する能力について、抗体を試験した。PDGF-AA及びPDGF-BBは、PDGFRαチロシンリン酸化を、それぞれ1 nmol/Lの濃度及び3 nmol/Lの濃度で約5倍増加させる。高濃度(10 nmol/L)のリガンドは、おそらくはリガンド誘導性の分解によって、リン酸化した受容体を減少させる。該抗体は、PDGF-BB誘導性の受容体を阻害して、バックグラウンドに近づけた(図14Aの一番上の列)。受容体のリン酸化を誘導するPDGF-AAを用いた場合にも、同様のデータが得た。
【0139】
PDGFは、有糸分裂シグナルを伝達させて、下流エフェクタータンパク質を介して受容体を発現する細胞上で抗アポトーシス作用を発揮する。そのため、MAPK p44/p42及びAkt(それぞれ、細胞成長及び抗アポトーシス経路に関与する)の活性化を阻害するその能力について、モノクローナル抗体を調べた。抗PDGFRα抗体は、PDGF-BB (図2A)及びPDGF-AA(図示してない)に応答したMAPK及びAktの両方のリン酸化を阻害した。PDGFRαのリン酸化の阻害は用量依存的であり、0.25 nmol/Lで50%を阻害した(図14B)。
【0140】
抗有糸分裂活性
抗PDGFRαモノクローナル抗体を、PDGFR-AAによって誘導されるPAE Rα細胞の有糸分裂を遮断するその能力について、試験した。細胞を96-ウェル組織培養プレート(1×104細胞/ウェル)に播種し、100μL培地/ウェル中で一晩生育した。その後、ウェルを無血清培養液ですすぎ、それぞれのウェルに75μL無血清培地を加え、細胞を一晩、血清欠乏状態とした。IgGを加え(25μL/ウェル)、プレートを30分間、37℃でインキュベートした。その後、PDGF-AA又はPDGF-BB (25μL/ウェル)を加え、プレートを18〜20時間、37℃でインキュベートした。それぞれのウェルに0.25μCi[3H]チミジン(25μL/ウェル)を加えた後、プレートをさらに4時間インキュベートした。抗体、PDGF、及び[3H]チミジンは、全て無血清培地中で希釈した。その後、1%ウシ血清アルブミンを加えたPBSで細胞を洗浄し、トリプシン(100μL/ウェル)による処理で剥離した。フィルター上に細胞を集め、MACH III細胞回収機(Tomtec, Inc., Hamden, CT)を用いて、再蒸留水でスライスを洗浄した。濾過処理した後、DNAに取り込まれた放射能をシンチレーションカウンタ(Wallac Microbeta, model 1450)で計測した。
【0141】
IMC-3G3を血清欠乏状態のPAE Rα細胞に加えた場合、PDGF-AA誘導性のチミジン取り込みは、EC50 8.3 nmol/Lで特異的に阻害された(図15)。該抗体は、3 nmol/L PDGF-BBによって誘導性されるPAE Rα細胞の有糸分裂も、EC50 1.25 nmol/Lで阻害した(データは示していない)。
【0142】
PDGFRαを発現するヒト腫瘍細胞株の増殖阻害
PDGFRαを発現するヒト腫瘍細胞株を試験して、悪性増殖に対するヒト抗PDGFRα抗体の効果をin vitro及びin vivo系で測定した。フローサイトメトリーで測定した際にPDGFRαを発現するような2つの腫瘍細胞株は、SKLMS-1(平滑筋肉腫)及びU118(神経膠芽腫)である。これらの細胞株も、有糸分裂アッセイにおいてリガンドと反応し、マウスにおいて腫瘍を形成する。SKLMS-1は、パラクリン刺激だけでなく、オートクリン刺激も行う可能性がある。SKLMS-1は、培養して生育した際に、定量的サンドウィッチ酵素イムノアッセイ法(R&D Systems)により、PDGF-AAタンパク質を発現することがわかった。
【0143】
図16Aからわかるように、IMC-3G3は、SKLMS-1細胞のPDGF-AA刺激に応答したAkt及びMAPKの両方のリン酸化を阻害した。Aktリン酸化の阻害は100%であり、MAPKの阻害は約80%であった。該抗体は、U118細胞におけるリン酸化の有効な阻害剤でもある(図16B)。リガンド誘導性の腫瘍細胞の有糸分裂も遮断した。抗PDGFRα抗体を血清欠乏状態のU118細胞に加えた場合、PDGF-AA誘導性のチミジン取り込みは、EC50 3.4 nmol/Lで特異的に阻害された(図17A)。該抗体は、SKLMS-1細胞のPDGF-AA誘導性の有糸分裂応答をEC50 5 nmol/L(図17B)で阻害するだけでなく、PDGF-BB刺激性有糸分裂応答も阻害する(図17C)。U118細胞では、PDGF-BB刺激性有糸分裂応答のわずかな部分的阻害(66 nmol/Lで40%;図17D)を観察した。これは、それらの細胞において、PDGFRα及びPDGFRβの両方が発現しているためである(データは示していない)。
【0144】
腫瘍異種移植片の成長阻害
無胸腺ヌードマウス中、神経膠芽腫(U118)及び平滑筋肉腫(SKLMS-1)皮下(s.c.)異種移植片モデルにおいて、IMC-3G3をin vivoで試験した。皮下の腫瘍異種移植片は、Matrigel (Collaborative Research Biochemicals, Bedford, MA)中で混合した10×106 SKLMS-1又はU118細胞を、雌の無胸腺ヌードマウス(Crl:NU/NU-nuBR, Charles River Laboratories, Wilmington, MA)に注射することにより、定着させた。腫瘍は、平均腫瘍体積(π/6×最長径×垂直方向の高さ2)が約400 mm3になるまで成長させた。マウスを5つのグループに無作為割付けし(n = 12)、試験期間中は週に2回i.p.注射して処置した。グループ1のマウスは、対照溶媒(0.9%NaCl;洗浄用のUSP, B/Braun)で処置した。グループ2〜4のマウスは、6、20、及び60 mg/kgの当該抗PDGFRα抗体で処置した。グループ5のマウスは、60 mg/kgのヒトIgG(Sigma)で処置した。6、20、又は60 mg/kgの抗PDGFRα抗体又はヒトIgGで処置したグループでは、初回用量をそれぞれ21.4、71.4、及び 214 mg/kgで与えた。初回用量は、週2回の投与レジュメンを用いて、第1の用量(消失半減期7日間)から定常状態の血清濃度を達成するように計算した。週に2回腫瘍体積を評価し、治療群における腫瘍増殖を、繰り返しANOVAを測定して比較した。
【0145】
図18Aに示すとおり、生理食塩水で処置したマウス(P = 0.74)と比べ、ヒトIgGは神経膠芽腫の成長に対し何も効果を及ぼさなかった。一方、抗PDGFRα抗体は、用量6 mg/kg(P = 0.06)、20 mg/kg(P = 0.03)、及び60 mg/kg(P =0.0004)で、腫瘍増殖を有意に阻害した。U118試験の最後において、%T/C [(試験終了時に3G3で処置したグループの平均腫瘍体積/治療開始時の平均腫瘍体積)/(試験終了時に未処理のグループの平均腫瘍体積/試験開始時の平均腫瘍体積)×100]値は、用量6、20、及び60 mg/kgの3G3で処置したグループでは、それぞれ67%、63%、及び35%であった。さらに、6、20、及び60 mg/kgの処置グループにおいて、4/12、5/11、10/12匹の被験動物で腫瘍の消退を観察した。一方、いずれの対照群でも、腫瘍の消退は認めなかった。
【0146】
図18Bは、6 mg/kg(P = 0.02)、20 mg/kg(P = 0.003)、及び60 mg/kg(P < 0.0001)の処置により、平滑筋肉腫の成長も有意に阻害されたことを示す。最終的な%T/C体積の値は、6、20、及び60 mg/kgの処置群において、それぞれ66%、57%、及び31%であり、腫瘍消退は認めなかった。
【0147】
試験終了時における異種移植片の組織学的検査は、対照療法を受けた被験動物から得た腫瘍と比べ、処置した被験動物から得た腫瘍では明らかなに差を示した。切除した腫瘍を、QDL固定液中で、4℃で24時間固定した。パラフィン包埋した後、4μmに切片化し、ホルマリン固定した切片をMayer's H&E(Richard Allen, Kalamazoo, MI)で染色した。
【0148】
最も高い用量(60 mg/kg)で処置したU118グループでは、生存している腫瘍細胞は非常にわずかしか認めなかった。また、生理食塩水の対照群と比べ、実質的により多くの細胞低密度領域が存在した(図18C)。25日目の処置したSKLMS-1異種移植片では、生理食塩水の対照群と比べ、生存している腫瘍細胞量及び細胞密度の減少も認めた(図18D)。
【0149】
神経膠芽腫株のPDGFRα介在性刺激のin vitro阻害
U118腫瘍における受容体ホスホチロシンのレベルを、抗PDGFRα抗体又はヒトIgGで処置してから1週間後に評価した。定着したU118腫瘍(500 mm3)を有するマウスを、初回用量214 mg/kgで処置し、72時間後に維持用量60 mg/kgの抗体で処置した。第1の抗体注射から1週間(168時間)後に(平均して、腫瘍消退が観察される前の時点に;図18Aを参照されたい)、マウスから腫瘍を採取し、リン酸化アッセイ溶解緩衝液中でホモジナイズした(上記参照)。溶解物を14,000 rpmで2回遠心分離し、回収した上澄液のタンパク質濃度を測定した(Bio-Radタンパク質アッセイ, Bio-Rad, Hercules, CA)。それぞれのサンプルからの溶解物(4 mg)を、抗PDGFRα抗体を用いて免疫沈降させた。その後、免疫沈降させたヒトPDGFRαを、抗PDGFR又は抗ホスホチロシン抗体のいずれかでイムノブロットした。図19は、これらの腫瘍において、抗PDGFRα抗体の投与が、ヒトIgG対照と比べPDGFRαホスホチロシンのレベルを減少させることを示す。
【0150】
細胞株作製技術
はじめに、ヒト抗PDGFRα抗体の重鎖及び軽鎖可変ドメインをコードする遺伝子をクローニングし、配列決定した。一連のプライマーは、MEDAREX由来のハイブリドーマ中で、ヒト免疫グロブリン可変領域配列の5'及び3'フランキング配列にアニーリングするMEDAREXから得た。重鎖可変領域は、プライマー対AB88(フォワード)及びAB90(リバース)(表7)を用いて増幅した。軽鎖産物は、フォワードプライマーAB182及びリバースプライマーAB16(表7)を含むプライマー対を用いて増幅した。これらの0.4kb反応産物をベクターZeroBlunt (Invitrogen)にクローニングして、AB88-1(VH)及びAB182-3(Vκ)を作製し、挿入物をユニバーサルT7及びM13Rプライマーを用いて配列決定した。
【表7】
【0151】
完全IgG1抗体を発現するプラスミドベクターを作製するために、クローニングした可変領域をPCR増幅し、定常領域遺伝子を含有する発現ベクターに2段階でライゲートした。最初のPCR重鎖増幅では、プライマーIPHF5(フォワード)及びIPHR5(リバース)の鋳型として、25 ngのプラスミドAB88-1を利用した。第2のPCR重鎖増幅では、鋳型として5μlの一次反応物並びにプライマーOPSIF及びIPHR5を利用した。効果的な免疫グロブリンのプロセシング及び分泌のために、2つのフォワードプライマーを併用して、19アミノ酸のマウス重鎖遺伝子シグナル配列(MGWSCIILFLVATATGVHS;配列番号24)をコードする免疫グロブリン遺伝子の5’末端に57塩基対を付加する。さらに、フォワードプライマーOPSIFは、哺乳動物細胞中でのこれらの遺伝子の効率的な翻訳開始のためにコンセンサス「Kozak」配列(J MoI. Biol. 196:947)を付加し、増幅産物のクローニングのために5'HindIII制限エンドヌクレアーゼ部位を、好適な発現ベクターに付加する。重鎖リバースプライマーは、定常領域ベクターにクローニングするためのフレーム内NheI部位を包含する。
【0152】
PCRは、Expand PCRキット(Boehringer Mannheim Inc.)を製造業者の説明書に従い用いて、Expand緩衝液システム#3を、以下のサイクリング条件で50μlの反応物中で利用し、2段階行った。
【0153】
1サイクル 94℃ 2分
5サイクル 94℃ 20秒
48℃ 60秒
68℃ 2分
20サイクル 94℃ 20秒
65℃ 60秒
68℃ 2分
1サイクル 68℃ 5分
【0154】
2ラウンドのPCR後に、PCR産物をアガロースゲル電気泳動した後に精製し、HindIII-Nhel消化断片としてベクターpDFcにクローニングした(図8)。該ベクターには、ヒトγ1定常領域が含まれる。
【0155】
最初のPCR軽鎖増幅では、鋳型として25 ngのpAB182-3プラスミド、プライマーIPLF4(フォワード)及びIPLR2(リバース)を利用した。第2のPCR軽鎖増幅では、鋳型として5μlの一次反応物並びにプライマーOPSIF及びIPLR2を利用した。重鎖では、2つのフォワードプライマーが分泌シグナル配列をもたらす。軽鎖リバースプライマーは、κ定常領域ベクターpLckにクローニングするために、フレーム内BsiWI部位を含む(図8)。上記の重鎖について、PCR反応を行った。2ラウンドのPCR後、アガロースゲル電気泳動した後にPCR産物を精製し、pLckにクローニングした。該ベクターには、ヒトκ軽鎖定常領域が含まれる。
【表8】
【0156】
安定したトランスフェクションを目的とした単一のプラスミドベクターを作製するために、CMVプロモーター、重鎖コード領域、及びポリA要素を含む重鎖発現カセットを、NotI-SalI断片としての軽鎖ベクターにクローニングした(図20)。
【0157】
その後、この構築物を利用して、骨髄腫細胞株NSO細胞中で、安定した産生株を作製した。BioRad Gene PulserIIを用いて、エレクトロポーレーションを行って、発現プラスミドをNSO細胞にトランスフェクトさせた。トランスフェクション前に、プラスミドDNAをPvulで線状化し、エタノール沈降させ、濃度0.4 mg/ml(lOOμlのdH2O中40μg)で懸濁した。最終体積800μl中で、250ボルトの単一パルス400μFdにより、40μgのDNAを細胞にエレクトロポーレートした。エレクトロポーレートした細胞を、50μlのアリコートで、10%透析ウシ胎児血清(dFCS) (Hyclone, Lot#: AHA7675)及び2 mMグルタミン(Invitrogen/Life Technologies)を含有するDMEM培地(JRH Biosciences Inc.)中に、5,000〜10,000細胞/ウェル濃度で約18個の96-ウェルプレートのウェルに分散させた。グルタミン合成酵素(GS)陽性トランスフェクション体の選択は、10%dFCSを含有し1×GS補填剤(JRH Biosciences Inc.)を補填した無グルタミンDMEMを添加してから2時間後に開始した。コロニーの成長及び拡大を可能とするために、細胞を37℃、5%CO2雰囲気下で2〜4時間後インキュベートした。300個を超えるコロニーを、抗ヒトFc(γ)ELISA (A450 nmでのセイヨウワサビペルオキシダーゼ検出)を用いて、スクリーニングした。3〜5日間かけて培養し、抗体発現コロニー(58%)を拡大させて、再現性について再び試験した。細胞を無血清培地に適用するために、それぞれの継代培養で等しい体積の無血清GS-OS培養液を添加して、陽性細胞株を拡大させた。3日-サブコンフルエントな24ウェル培養物中で25μg/ml以上を産生する強陽性物を、さらなる分析のために拡張して、無血清培地に完全に適合させた。
【0158】
本明細書に開示した発明の原理の範囲内での変更が、当業者によりなされ得ることが理解され、また期待される。また、そういった変更は、本発明の範囲内に包含することが意図される。
【技術分野】
【0001】
本出願は、米国仮出願第60/691,920号の利益を享受するものである。
【0002】
本発明は、IGF-IRアンタゴニスト及び/又はPDGFRαアンタゴニストを投与することにより、骨癌、特に転移性骨癌を治療する方法を提供する。本発明は、ヒトPDGFRαに結合し、受容体の活性化を中和する抗体も提供する。本発明は、PDGFRαの活性化を中和する方法、及び抗体を単独又は他の薬剤と組み合わせて使用することにより、腫瘍性疾患を患う哺乳動物を治療する方法を、さらに提供する。
【背景技術】
【0003】
前立腺癌は、男性では最も一般的な癌で、米国では毎年約220,000人が罹患し、29,000人が死亡する。前立腺癌と診断された男性の大半は、転移性疾患を有する。さらに、転移は、手術や放射線療法による治療をよそに、多くの他の前立腺癌患者において、最終的には進行する。骨は、前立腺癌転移が最も多い部位であり、乳癌及び肺癌が頻繁に転移する部位でもある。ほとんどの前立腺癌転移はアンドロゲン依存性であるので、外科又は内科的去勢で迅速な効果を認めるが、ほぼ全ての患者において、最終的には腫瘍がアンドロゲン非依存性となるので、重大な罹患率及び死亡率を導く。いったん骨転移が生じると、現在利用可能な療法では限られた効果しか有さない。転移性前立腺癌に関して報告されているもののうち、最も有効な認可された治療法(ドセタキセルの投与)は、生存期間の中央値を約3ヶ月延長する(Petrylak et al., 2004, N. Engl. J. Med. 351:1513; Tannock et al., 2004, N. Engl. J. Med. 351:1502)。従って、転移性骨癌に対する新しい治療法が、早急に必要とされる。
【0004】
インスリン様成長因子受容体(IGF-IR)は、正常胎児並びに出生後の成長及び発育にとって不可欠な、広範に分布する膜貫通型チロシンキナーゼ受容体である。IGF-IRは、ほとんどの細胞型の細胞表面に存在し、成長因子IGF-I及びIGF-II(以降は、まとめてIGFと称する)のシグナル伝達分子として機能する。IGF-IRは、細胞増殖、細胞分化、細胞サイズの変化を促進し、またアポトーシスから細胞を防御することができる。また、細胞の形質転換に準必須であるとも考えられてきた(Adams et al., Cell. Mol. Life Sci. 57:1050-93 (2000); Baserga, Oncogene 19:5574-81 (2000)でレビューされる)。IGF-IRの高レベルの発現が、前立腺癌の骨転移巣から得た組織サンプルで報告されている。骨は、体内で最も多くのIGF貯蔵を含む。
【0005】
IGF-IRは、ジスルフィド結合により共有結合した2つのα鎖及び2つのβ鎖を含む予め形成されたヘテロ四量体である。受容体サブユニットは、180 kdの単一のポリペプチド鎖の一部として合成された後、タンパク分解酵素によるプロセシングによってα(130kd)及びβ(95kd)サブユニットになる。α鎖全体は細胞外に存在し、リガンド結合するための部位を含んでいる。β鎖は膜貫通ドメイン、チロシンキナーゼドメイン、及び細胞分化及び形質転換に不可欠なC末端の伸長を備えるが、マイトジェンのシグナル伝達及びアポトーシスからの防御には必要である。
【0006】
IGF-IRは、インスリン受容体(IR)と、特にβ鎖配列において(70%の相同性)、非常に類似している。この相同性のおかげで、これらの受容体が、1つのIRダイマー及び1つのIGF-IRダイマーを含んでなるハイブリッドを形成できることが最近の研究で実証された(Pandini et al., Clin. Canc. Res. 5:1935-19 (1999))。ハイブリッド形成は、正常細胞及び形質転換した細胞の両方で生じ、ハイブリッドの組成は、細胞中の2つのホモダイマー受容体(IR及びIGF-IR)の濃度によって決まる。ハイブリッド受容体はIR及びIGF-IRの対からなるが、該ハイブリッドはIGF-IRに対する場合と同程度の親和性でIGFに選択的に結合し、インスリンとは弱くしか結合しない(Siddle and Soos, The IGF System. Humana Press, pp. 199-225. 1999)。従って、これらのハイブリッドは、正常細胞及び形質転換した細胞の両方において、IGFと結合してシグナルを伝達することができる。
【0007】
第2のIGF受容体であるIGF-IIR又はマンノース-6-ホスフェート(M6P)受容体も、IGF-IIのリガンドと高い親和性で結合するが、チロシンキナーゼ活性を欠く(Oates et al., Breast Cancer Res. Treat. 47:269-81 (1998))。従って、IGF-IIの分解が生じるので、IGF-IIのくぼみ(sink)は、このリガンドの成長促進効果を抑制すると考えられる。腫瘍細胞中のIGF-IIRが失われると、IGF-IIとIGF-IRとの結合に対する拮抗効果が解除されることにより、成長能力が増強され得る(Byrd et al., J. Biol. Chem. 274:24408-16 (1999))。
【0008】
血小板由来成長因子受容体α及びβ(PDGFRα及びPDGFRβ)は、III型の受容体チロシンキナーゼである。PDGFRαは発育に関して必須であり、成長期において重要な機能を果たす。例えば、ヌル突然変異させたホモ接合マウスは、胚形成期に死亡する。発生の後期段階において、PDGFRαは多くの間充織構造中で発現し、隣接する上皮細胞は血小板由来成長因子(PDGF)を産生する。正常前立腺又は過形成前立腺から得た組織サンプルは、検査ではPDGFRαに対し陰性であるが、適合被験体から得た原発性前立腺腫瘍及び骨本体は、PDGFRαを発現する。さらに、様々な転移性部位から得た前立腺細胞株のうち、骨転移由来のPC3細胞では、PDGFRαが観察されるが、リンパ節(LNCaP)及び脳(DU-145)転移巣から得た細胞株では、PDGFRαは観察されない。
【0009】
成長因子のうち、血小板由来成長因子ファミリーは、ジスルフィド結合した5種類の異なるダイマー、PDGF-AA、-BB、-AB、-CC、及び-DDからなり、これらはPDGFRα及びPDGFRβを介して作用する。これらの成長因子は、2つの受容体タンパク質と同時に結合し、受容体の二量体化、自己リン酸化、及び細胞内シグナル伝達を誘導するジスルフィド結合したポリペプチド鎖からなるダイマー分子である。PDGFRα及びPDGFRβは構造的に類似しており、ホモダイマーだけでなく、ヘテロダイマーも形成できる。PDGFRβは、高親和性ではPDGF-A鎖に結合しないので、PDGF-AAはαα受容体ダイマーのみを活性化するのに対し、PDGF-AB及びPDGF-CCはαα及びαβ受容体ヘテロダイマーを活性化する。
【先行技術文献】
【非特許文献】
【0010】
【非特許文献1】Petrylak et al., 2004, N. Engl. J. Med. 351:1513
【非特許文献2】Tannock et al., 2004, N. Engl. J. Med. 351:1502
【非特許文献3】Adams et al., Cell. Mol. Life Sci. 57:1050-93 (2000)
【非特許文献4】Baserga, Oncogene 19:5574-81 (2000)
【非特許文献5】Pandini et al., Clin. Canc. Res. 5:1935-19 (1999)
【非特許文献6】Siddle 及び Soos, The IGF System. Humana Press, pp. 199-225. 1999
【非特許文献7】Oates et al., Breast Cancer Res. Treat. 47:269-81 (1998)
【非特許文献8】Byrd et al., J. Biol. Chem. 274:24408-16 (1999)
【発明の概要】
【0011】
本発明は、原発性及び転移性骨腫瘍の治療に関連し、該骨腫瘍には前立腺、乳房又は肺から発生し、インスリン様成長因子-I受容体(IGF-IR)及び/又はα血小板由来成長因子受容体(PDGFRα)を発現する腫瘍が含まれる。
【0012】
治療対象となる腫瘍は、ホルモン/アンドロゲン依存性又はホルモン/アンドロゲン非依存性であり、例えば、前立腺、乳房、又は肺から発生したものである。
【0013】
本発明は、骨腫瘍を患う被験体を治療する方法、及び骨腫瘍の増大を阻害する方法を提供する。本発明の方法は、有効量のIGF-IRアンタゴニスト又は有効量のPDGFRαアンタゴニストを投与することを含む。受容体アンタゴニストには、抗体及び抗体フラグメントだけでなく、細胞内小分子阻害剤が含まれる。
【0014】
本発明は、それらの標的受容体と結合して、リガンド結合を阻害する抗IGF-IR又は抗PDGFRα抗体を提供する。本発明は、IGF-IR又はPDGFRαの活性化を中和する抗体及び他のアンタゴニストも提供する。さらに、ある抗体は、例えば内在化及び/又は分解によって、それらの標的受容体のダウンレギュレーションを促進する。従って、抗体及び小分子アンタゴニストは、下流のシグナル伝達分子、例えばAkt、p42/p44、及びMAPKの活性化を阻害するように機能する。
本発明の方法は、IGF-IR又はPDGFRαアンタゴニストを、単独で、互いに組み合わせて、又は他の癌療法、例えば化学療法剤及び放射線照射と組み合わせて、使用することを含む。
【0015】
本発明は、PDGFRαに結合する抗体及び抗体フラグメントだけでなく、該抗体産生のためのヌクレオチド及び宿主細胞も提供する。本発明の抗体は、リガンド結合を遮断し、受容体の活性化を中和する。本発明は、抗体の単独使用、他の受容体アンタゴニスト若しくは抗腫瘍剤と組み合わせた使用、又は腫瘍性疾患を治療するためのコンジュゲートとしての使用も提供する。抗PDGFRα抗体は、例えば、卵巣癌、乳癌、肺癌、肝細胞癌、胃腸間質腫瘍、メラノーマ、腎細胞癌、前立腺癌、及び柔組織肉腫を治療するために使用される。
【図面の簡単な説明】
【0016】
【図1】図1は、腫瘍が150〜200 mm3に達した際に開始された治療期間中における、去勢したSCIDマウス中のLuCaP35V皮下移植片腫瘍の増大を示す。パネルA:未治療の対照;パネルB:被験動物をドセタキセル(10 mg/kg又は20 mg/kgのいずれか)単独又は抗IGF-IR抗体(40 mg/kg IMC-A12)との併用で4週間治療した;パネルC:皮下LuCaP35V移植片腫瘍を有する未治療及び治療したSCIDマウスにおける血清PSAレベル。治療したマウスは、ドセタキセル(20 mg/kg)単独又はドセタキセル(10 mg/kg又は20 mg/kgのいずれか)と抗IGF-IR抗体(40 mg/kg IMC-A12)の併用治療を受けた。腫瘍が150〜200 mm3に達した際に治療を開始し、4週間後に治療を終了した。
【図2】図2は、ドセタキセル(10 mg/kg)単独(パネルA)又は抗IGF-IR抗体(40 mg/kg IMC-A12) (パネルB)との併用で治療したLuCaP35V移植片腫瘍の単一細胞の懸濁液を示す。R1で標識した領域は、断片化したDNAを有するアポトーシス細胞に相当する(FITC標識が増加している)。
【図3】図3は、ドセタキセル(10 mg/kg又は20 mg/kg)単独及び抗IGF-IR抗体(40 mg/kg IMC-A12)との併用による治療終了後の、腫瘍移植変片におけるDNA合成(BrDu取り込み)を示す。
【図4】図4は、ドセタキセル及びAl2並びにドセタキセル単独による治療に反応した前立腺腫瘍細胞における、前立腺腫瘍の悪性度(TUBB)、抗アンドロゲン療法(BIRC5)に対する耐性、及びアポトーシス誘導(IGFBP3)と関係する遺伝子の示差発現を示す。
【図5】図5は、治療終了後のA12血清レベルを示す。
【図6】図6は、ドセタキセル(10 mg/kg又は20 mg/kgのいずれか)単独又は抗IGF-IR抗体(40 mg/kg IMC-A12)との併用により継続して治療した、疾患を有さない被験動物の体重(総合的な細胞毒性評価)を示す。
【図7】図7は、LuCaP23.1細胞を移植したSCIDマウス中で移植片が産生するPSAに対する、抗IGF-IR抗体(IMC-A12)の治療効果を示す。
【図8】図8は、LuCaP23.1細胞を移植したSCIDマウスの一連のX線写真を示す。A12マウスには、40 mg/ml IMC-A12(i.p.)を6週間にわたり週3回投与した。X線写真は屠殺時に撮影した。
【図9】図9は、LuCaP23.1ヒト前立腺細胞の頚骨内移植片を有するSCIDマウスのPSAレベル(a)及び代表的なX線写真(b)を示す。
【図10】図10は、前立腺癌細胞中のAkt活性に対するヒト骨髄穿刺液の効果を示す。細胞溶解物をSDS-PAGEに供した。ウェスタンブロット解析のために、ホスホAkt(Ser-473;cell signaling Technology)、PDGFRα(R&D Systems)及びアクチン(Sigma)を標的とする抗体を用いて、膜にブロットした。一次抗体の結合を、HRP-コンジュゲートプロテインA又はプロテインG(Sigma)を用いて検出した。
【図11】図11は、PC3-ML細胞におけるAktリン酸化の誘導及び阻害を示す。パネルAは、30 ng/ml PDFG-BBにさらした細胞中でのAktリン酸化のAG-1296用量依存的阻害を示す。パネルBは骨穿刺液のAktリン酸化及び20μMのAG-1296による阻害を示す。パネルCは、Aktリン酸化の誘導に関して、100 pg/ml PDGF-AA及び100 pg/ml PDGF-BBの併用の効力と比べた場合の、骨髄穿刺液の効力を示す。パネルDは、骨髄穿刺液誘導性のAkt-リン酸化、AG-1296による骨髄誘導性のAkt-リン酸化の阻害、及びPDFG-AA+PDFG-BBにより誘導されるAkt-リン酸化の強度を比較する。
【図12】図12は、PDGFRαアンタゴニストによるPC3-ML細胞中のAktリン酸化の阻害を示す。パネルAは、30 ng/mlのPDGF-BBにより誘導されたAktリン酸化に対する、モノクローナル抗体IMC-3G3の用量依存的効果を示す。パネルB及びCは、骨髄が誘導するAktリン酸化に対するIMC-3G3及びAG1296の効果の比較を提供する。パネルDは、Aktリン酸化の阻害が、IMC-3G3プレインキュベーション時間に依存的であることを示す。
【図13】図13は、PDGFRαに対する抗体の結合を示す。A:固定したPDGFRαの細胞外ドメインに対する抗PDGFRα抗体の直接結合。B:固定したPDGFRαと[125I]PDGF-AAとの結合の阻害。
【図14】図14は、PDGFRα及び下流エフェクター分子のリン酸化の特異的阻害を示す。
【図15】図15は、PAE Rα細胞におけるPDGF-AAで刺激した[3H]チミジン取り込みのmAbによる阻害を示す。
【図16】図16は、SKLMS-1(A)及びU118(B)細胞における、PDGF-AAが誘導した下流エフェクター分子の活性化の阻害を示す。
【図17】図17は、U118細胞(A)及びSKLMS-1細胞(B)における、mAbによるPDGF-AA刺激性[3H]チミジン取り込みの阻害を示す。SKLMS-1(C)及びU118(D)細胞について、PDGF-BB刺激性[3H]チミジン取り込みの阻害も示す。
【図18】図18は、ヌードマウス中で定着したU118(神経膠芽腫;パネルA)及びSKLMS-1(平滑筋肉腫;パネルB)腫瘍移植片の治療での用量依存的効果を示す。
【図19】図19は、非特異的ヒトIgGによる処置と比較した場合の、抗PDGFRα抗体で処置したU118腫瘍におけるin vivoのPDGFRαリン酸化の減少を示す。
【図20】図20は、ハイブリドーマ由来のヒトVH及びVκ可変領域遺伝子のクローニング並びに完全ヒト重鎖(IgG1)及び軽鎖タンパク質の発現に使用されるGS発現ベクターを示す。実施例で説明するとおり、2つのベクターを組換えて、該組合わせたベクターをNSO細胞にトランスフェクトした。
【発明を実施するための形態】
【0017】
本発明は、インスリン様成長因子-I受容体(IGF-IR)に結合する抗体又は抗体フラグメントによる骨腫瘍の治療に関する。IGF-Iのエンドクリン発現は、主に成長ホルモンにより制御され、肝臓で産生されるが、他の種類の組織もIGF-1を発現することができ、該組織には成長因子の大きな貯蔵を含む骨が含まれる。腫瘍細胞型により、IGF-Iはエンドクリン、パラクリン、及び/又はオートクリン制御に関与する(Yu, H. and Rohan, J., J. Natl. Cancer Inst. 92:1472-89 (2000))。
【0018】
IGF-IRに結合する抗体が、IGF-IRを発現する骨腫瘍の治療法において有用であることが見出された。該抗体は、単独で、又は他の癌療法剤、特に化学療法剤と組み合わせて使用できる。抗IGF-IR療法は、単独で又は1以上の抗腫瘍剤(例えば、化学療法又は放射線療法等)と組み合わせて、顕著な治療的効果を有する。腫瘍増大の抑制は、アポトーシスの増加を伴うことが多いので、全ての治療を中止した後も持続する。しかしながら、化学療法単独で処置した動物では、腫瘍が再増殖し始める。
【0019】
PDGFRαは、骨腫瘍の増大において重要な役割を果たすことも見出された。例えば、PDGFRαを発現する特定の腫瘍細胞株は、骨に選択的に転移する。かかる細胞株は、骨髄中に存在する可溶性因子に反応して、PDGFRαの活性化及び下流シグナル伝達分子のリン酸化を増加させる。骨髄によるPDGFRαの活性化は、PDGFRαアンタゴニストにより、低下又は完全に阻害される。また、PDGFRα及び他の受容体チロシンキナーゼ系を介したシグナル伝達によって通常活性化される下流シグナル伝達分子のリン酸化は、非常に減少する。PI3K/Akt生存経路は、直接的にPDGFRαを活性化するリガンドによるだけでなく、受容体のトランス活性化を生じさせる骨髄中に存在する因子によっても、PDGFRαシグナル伝達により活性化されることをあるデータは示唆する。
【0020】
本発明の治療対象となる原発性骨腫瘍には、骨肉腫、軟骨肉腫、線維肉腫、及び血管肉腫が含まれるが、これに限定されない。特に、悪性二次性(転移性)腫瘍が、原発性骨腫瘍よりもはるかに多い。本発明の治療対象となる転移性骨腫瘍は、多様な原発巣(source)から発生し、そのうち最も多いのは前立腺、乳房、又は肺である。転移性骨癌の原発巣は通常、患者の既往歴から明らかとなる。腫瘍は、骨芽球性又は骨溶解性であり得る。腫瘍は、発生時にIGF-IR刺激に依存的であるか、または、IGF-IR依存性に転化する場合がある。例えば、初期の段階ではホルモン/アンドロゲン依存性であってもアンドロゲン又はホルモン産生を抑制する物理療法又は化学療法によって制御可能である前立腺癌又は前立腺癌の転移は、IGF-IRを介した刺激への感受性増大によって、ホルモン/アンドロゲン非依存性になる場合がある。そのため、本発明はさらに、ホルモン/アンドロゲン非依存性腫瘍の治療の提供に加えて、アンドロゲン又はホルモン産生の抑制にはよらずに、例えば、抗腫瘍剤と共にIGF-IR抗体を同時投与することによって、ホルモン/アンドロゲン依存性骨腫瘍を治療するのにも有用である。かかる腫瘍には、IGFが豊富な骨環境においてIFG-IRを介して刺激される転移性骨腫瘍が含まれる。該腫瘍は、ホルモン刺激に対し感受性があるが、IGF環境が関与しなくても増殖できる程に感受性はない。よって、かかる腫瘍にとって、ホルモン除去は必須でないかもしれない。
【0021】
PDGF依存性の骨腫瘍だけでなく、「骨髄」依存性の腫瘍も、本発明により治療できる。骨髄依存性の腫瘍は、骨髄中に存在する可溶性因子に反応して、PDGFRαの活性化を提示する。例えば、本明細書で例示するとおり、ヒト転移性PDGFRαを発現する癌細胞株は、骨髄穿刺液への曝露により、PDGFRα活性化及びAkt+のリン酸化を受ける。抗PDGFRα抗体及び小分子PDGFRαアンタゴニストは、該細胞株において、PDGFRα活性化及びAkt+リン酸化をそれぞれ阻害する。PDGFRαを活性化する可溶性骨髄因子には、PDGF-AA及び-BBが含まれるが、これに限定されない。
【0022】
かかる骨髄依存性は、PDGFRαを介したシグナル伝達に関係するが、PDGFRαリガンドとPDGFRαとの結合だけに関与しているわけではない。例えば、本明細書で説明するとおり、所定のリガンド(PDGF-AA又は-BB)によるPDGFRαの活性化は、骨髄穿刺液による活性化よりも弱いことに留意されたい。さらに、骨髄穿刺液の存在下では、インキュベーション時間の増加により、Akt+のリン酸化が減少するのが観察される。纏めると、これらの結果は、PDGFの結合に反応することに加えて、他の骨髄成分に対し感受性のある他のシグナル伝達因子(例えば、他の受容体チロシンキナーゼ)によって、PDGFRαがトランス活性化(リン酸化)され得ることを示唆する。いずれにせよ、骨での転移性増殖に適した細胞株(すなわち、骨に選択的に転移する細胞株)では、骨髄依存性PDGFRαの活性化が観察され、該活性化は、PDGFRαアンタゴニストにより阻害される。さらに、PDGFRαアンタゴニストによる治療は、PI3K/Akt抗アポトーシス経路及びマイトジェン活性化プロテインキナーゼ(MAPK)の骨髄誘導性刺激を阻害する。
【0023】
PDGFRαアンタゴニストにより治療される骨腫瘍は、前立腺癌細胞の転移として発生するか、上記のとおりホルモン/アンドロゲン依存性であるか、又はホルモン/アンドロゲン非依存性に転化したものである。かかる腫瘍は、前立腺癌以外の転移としても発生し得る。当業者であれば、公知の従来の試験法を用いて、かかる状態及び障害を容易に診断できるだろう。
【0024】
治療とは、動物における疾患の任意の治療を意味し、以下:(1)哺乳動物において、疾患が発症するのを予防すること、すなわち疾患が誘発されてもよいが、疾患の症状は経験も提示もしないこと、例えば、臨床症状の出現の防止;(2)疾患を抑制すること、例えば、疾患進行の停止;又は(3)疾患を軽減すること、例えば、疾患症状の消失が含まれる。腫瘍増殖の阻害には、増殖を減速又は停止させることだけでなく、腫瘍を消退させることが含まれる。疾患治療における有効量とは、必要とする哺乳動物に投与した際に、上記に定義したとおり、その疾患治療に十分に有効な量を意味する。本発明のIGF-IRアンタゴニスト及びPDGFRαアンタゴニストは、単独で、互いに組み合わせて、又は1以上の抗腫瘍剤、例えば化学療法剤又は放射線照射などと組み合わせて、投与できる。
【0025】
本発明の実施形態では、治療対象となる腫瘍におけるIGF-IR及び/又はPDGFRαの発現レベルを測定することが望ましい。そのような場合、腫瘍の生検検体は、当技術分野で周知の方法により、収集し及び分析することができる。本発明の別の実施形態では、該当する受容体が、特定の種類の腫瘍において共通して発現若しくは活性化されるか、又は疾患が進行した際に非特異的に発現若しくは活性化されることに基づいて、IGF-IRアンタゴニスト又はPDGFRαアンタゴニストが投与される。
【0026】
IGF-IRアンタゴニストは、細胞外アンタゴニスト又は細胞内アンタゴニストであり、2以上のアンタゴニストを適用することもできる。細胞外アンタゴニストには、IGF-IRに結合するタンパク質若しくは他の生体分子又は1以上のそのリガンド(例えば、IGF-I及びIGF-IIはIGF-IRの天然のリガンドである)が含まれるが、これに限定されない。本発明の実施形態では、細胞外アンタゴニストは、IGF-IRがそのリガンドと結合するのを阻害する。1実施形態では、アンタゴニストは、例えばIMC-A12のような抗IGF-IR抗体である。別の実施形態では、アンタゴニストは、IGF-IRのフラグメントに結合する可溶性リガンドである。細胞内IGF-IRアンタゴニストは生体分子でもよいが、通常は小さな分子である。例としては、チロシンキナーゼ阻害剤AGl024(Calbiochem)、インスリン様成長因子-I受容体キナーゼ阻害剤NVP-AEW541 (Novartis)、及びインスリン様成長因子-I/インスリン受容体阻害剤BMS-554417 (Bristol Myers Squibb)が含まれるが、これに限定されない。本発明での使用に有用な小分子はIGF-IRの阻害剤であるが、IGF-IRに完全に特異的である必要はないことを理解されたい。
【0027】
本発明で使用される抗IGF-IR抗体は、次の特性のうち1以上を示す:
1)抗体はIGF-IRの外部ドメインに結合し、IGF-I又はIGF-IIがIGF-IRに結合するのを阻害する。阻害は、例えば、精製された受容体又は膜結合受容体を用いて、直接的な結合アッセイにより測定できる。この実施形態では、本発明の抗体又はそのフラグメントは、好ましくは、IGF-IRの天然のリガンド(IGF-I及びIGF-II)と少なくとも同じ強さで、IGF-IRに結合する。
【0028】
2)抗体は、IGF-IRを中和する。IGF-IRの外部の細胞外ドメインに対するリガンド(例えば、IGF-I又はIGF-II)の結合は、βサブユニット並びにMAPK、Akt、及びIRS-Iを含む下流のシグナル伝達分子の自己リン酸化を刺激する。
【0029】
IGF-IRの中和には、通常シグナル伝達に関係する当該活性のうち1以上を阻害、減少、不活化及び/又は遮断することが含まれる。中和は、例えば、組織、培養細胞、又は精製した細胞成分を用いて、in vivo、ex vivo、又はin vitroで測定できる。中和には、IGF-IR/IRヘテロダイマーだけでなく、IGF-IRホモダイマーの阻害も含まれる。従って、IGF-IRの中和は様々な効果を有しており、該効果には、成長(増殖及び分化)、血管新生(血管の補強、浸潤、及び転移)、並びに細胞の移動性及び転移(細胞接着及び浸潤)の阻害、減少、不活化及び/又は遮断が含まれる。
【0030】
IGF-IRの中和の1つの指標は、受容体のチロシンキナーゼ活性の阻害である。チロシンキナーゼ阻害は、周知の方法を用いて;例えば、組換えキナーゼ受容体の自己リン酸化レベル及び/又は天然若しくは合成の基質のリン酸化を測定することにより、測定できる。従って、リン酸化アッセイは、本発明に関して中和抗体を決定するのに有用である。リン酸化は、例えばELISAアッセイ又はウェスタンブロットにおいて、ホスホチロシンに特異的な抗体を用いて検出できる。チロシンキナーゼ活性についての幾つかのアッセイが、Panek et al., J. Pharmacol. Exp. Them. 283: 1433-44 (1997)及びBatley et al., Life Sd. 62:143-50 (1998)で報告されている。本発明の抗体は、リガンドと反応する細胞において、IGF-IRのチロシンリン酸化を、少なくとも約75%、好ましくは少なくとも約85%、及びより好ましくは少なくとも約90%減少させる。
【0031】
IGF-IR中和の別の指標は、IGF-IR下流の基質のリン酸化の阻害である。よって、MAPK、Akt、又はIRS-Iのリン酸化レベルが測定される。リン酸化の減少は、少なくとも約40%であり、少なくとも約60%、又は少なくとも約80%であり得る。
【0032】
さらに、測定されるタンパク質がIGF-IRチロシンキナーゼ活性によって制御される場合には、タンパク質発現の検出方法を利用して、IGF-IR中和を測定できる。これらの方法には、タンパク質発現の検出のための免疫組織化学的検査(IHC)、遺伝子増幅の検出のための蛍光in situハイブリダーゼーション(FISH)、競合的放射性リガンド結合アッセイ、固相マトリックスブロット法、例えば、ノーザン及びサザンブロット、逆転写酵素ポリメラーゼ連鎖反応(RT-PCR)及びELISAが含まれる。例えば、Grandis et al., Cancer, 78:1284-92 (1996); Shimizu et al., Japan J. Cancer Res., 85:567-71 (1994); Sauter et al., Am. J. Path., 148:1047-53 (1996); Collins, GHa 15:289-96 (1995); Radinsky et al., Clin. Cancer Res. 1:19-31 (1995); Petrides et al., Cancer Res. 50:3934-39 (1990); Hoffmann et al., AntiCancer Res. 17:4419-26 (1997); Wikstrand et al., Cancer Res. 55:3140-48 (1995)を参照されたい。
【0033】
Ex vivoアッセイを利用して、IGF-IR中和を測定することもできる。例えば、受容体チロシンキナーゼ阻害は、阻害剤の存在下及び不在下で、受容体リガンドにより刺激された細胞株を使用するマイトジェンアッセイによって、観察できる。MCF7乳癌株(American Type Culture Collection (ATCC), Rockville, MD)は、IGF-IRを発現するような細胞株であり、IGF-I又はIGF-IIにより刺激される。別の方法は、IGF-IRを発現する腫瘍細胞又はIGF-IRを発現するように形質導入された細胞の成長の阻害について、試験することも含む。腫瘍モデル、例えば、ヒト腫瘍細胞を注入されたマウスを用いて、阻害を観察することもできる。
【0034】
本発明の抗体は、IGF-IR中和のいかなる特定機構によっても制限されない。本発明の抗IGF-IR抗体は、IGF-IR細胞表面受容体に外側から結合でき、リガンド(例えば、IGF-I又はIGF-II)の結合及び受容体と結合したチロシンキナーゼにより介在される後続のシグナル伝達を遮断でき、さらにIGF-IR及びシグナル伝達カスケード下流の他のタンパク質のリン酸化を防止できる。
【0035】
3)抗体は、IGF-IRを下方調節する。細胞の表面上に存在するIGF-IRの量は、受容体タンパク質の産生、内在化、及び分解によって決まる。細胞の表面上に存在するIGF-IRの量は、受容体又は該受容体に結合した分子の内在化を検出することにより、間接的に測定できる。例えば、受容体の内在化は、IGF-IRを発現する細胞を、標識した抗体に接触させることにより測定できる。その後、膜に結合した抗体を剥離し、回収して、数える。内在化した抗体は、細胞を溶解させ、溶解物中の標識を検出することにより、測定できる。
【0036】
別の方法は、抗IGF-IR抗体又は他の物質による治療後に、細胞上に存在する受容体の量を、例えばIGF-IRの表面発現について染色した細胞の蛍光活性化細胞選別分析法により、直接的に測定することである。染色した細胞を37℃でインキュベートし、蛍光強度をじっくりと測定する。対照では、染色した集団の一部を4℃(受容体内在化が停止する条件)で、インキュベートし得る。
【0037】
細胞表面IGF-IRは、IGF-IRに特異的で、かつ、試験される抗体の結合を遮断しない又は試験される抗体の結合と競合しない様々な抗体を用いて、検出及び測定できる(Burtrum, et al. Cancer Res. 63:8912-21 (2003))。本発明の抗体によるIGF-IRを発現する細胞の処置は、細胞表面IGF-IRの減少をもたらす。好ましい実施形態では、本発明の抗体による治療に反応して、少なくとも約70%、より好ましくは少なくとも約80%、さらにより好ましくは少なくとも約90%が減少する。顕著な減少が、わずか4時間で観察できる。
【0038】
下方調節の別の指標は、細胞中に存在する受容体タンパク質総数の減少であり、これは内部受容体の分解を反映する。よって、本発明の抗体による細胞(特に、癌細胞)の処置は、細胞性IGF-IR総数の減少をもたらす。好ましい実施形態では、この減少は、少なくとも約70%、より好ましくは少なくとも約80%、さらにより好ましくは少なくとも約90%である。
【0039】
ヒト被験体の治療では、本発明の抗体は、好ましくはヒト抗体である。他には、抗体は、非ヒトの霊長類若しくは他の哺乳動物に由来し、又はヒト化抗体若しくはキメラ抗体である。本発明の実施形態では、抗IGF-IR抗体は、配列番号35、配列番号37、配列番号39、配列番号45、配列番号47、及び配列番号49(それぞれCDR1H、CDR2H、CDR3H、CDR1L、CDR2L、CDR3L)からなる群から選択される1、2、3、4、5、及び/又は6の相補性決定領域(CDR)を含む。別の実施形態では、抗IGF-IR抗体は、配列番号35、配列番号37、配列番号39、配列番号55、配列番号57、及び配列番号59(それぞれCDR1H、CDR2H、CDR3H、CDR1L、CDR2L、CDR3L)からなる群から選択される1、2、3、4、5、及び/又は6の相補性決定領域(CDR)を含む。好ましくは、本発明の抗体(又はそのフラグメント)は、配列番号35、配列番号37及び配列番号39で表される重鎖CDRを有する。他にまた好ましくは、本発明の抗体(そのフラグメントを含む)は、配列番号45、配列番号47及び配列番号49又は配列番号55、配列番号57及び配列番号59で表される軽鎖CDRを有する。かかる抗IGF-IR抗体の1つは、配列番号41で示される重鎖可変ドメイン及び配列番号51で示される軽鎖可変ドメインを有するヒトIgG1抗体IMC-A12 (WO2005016970)である。別の好ましいヒト抗体は、IMC-A12と同一の重鎖可変ドメイン及び配列番号61で示される軽鎖可変ドメインを有するIMC-2F8 (WO2005016970)である。有用な抗体には、IGF-IRへの結合についてMC-A12又はIMC-2F8と競合する抗IGF-IR抗体だけでなく、他のエピトープに結合する抗体(すなわち、他のエピトープに結合し、前述の特性、例えばリガンドの遮断、受容体の内在化等を提示するが、IMC-A12又はIMC-2F8とは競合しない抗体)が、さらに含まれる。
【0040】
本発明では、PDGFRαアンタゴニストを治療のために使用することもできる。PDGFRαアンタゴニストは、細胞外アンタゴニスト又は細胞内アンタゴニストであり、及び2以上のアンタゴニストを適用してもよい。細胞外アンタゴニストには、PDGFRαと結合するタンパク質若しくは他の生体分子又は1以上のそのリガンド(例えば、PDGF-AA、-AB、-BB、-CC)が含まれるが、これに限定されない。本発明の実施形態では、PDGFRαにそのリガンドが結合するのを細胞外アンタゴニストが阻害する。1実施形態では、アンタゴニストは例えば、IMC-3G3のような抗PDGFRα抗体である。別の実施形態では、該結合するタンパク質は、PDGFRαの可溶性リガンドに結合するフラグメントである。細胞内IGF-IRアンタゴニストは生体分子でもよいが、通常は小さな分子である。1実施形態では、細胞内PDGFRαアンタゴニストはAG1296である。AG1296(Calbiochem)は、PDGFα、PDGFβs、及びc-KITの阻害剤であり、Flt3とも反応する。PDGFRを標的とする他の小分子には、STI-571(イマチニブメシレート、Gleevec(登録商標), Novartis)及びSUl1248(スニチニブマーレート、SUTENT(登録商標)、Pfizer)が含まれる。
【0041】
本発明の実施形態では、抗PDGFRα抗体は、配列番号2、配列番号4、配列番号6、配列番号10、配列番号12、及び配列番号14(それぞれCDRlH、CDR2H、CDR3H、CDRlL、CDR2L、CDR3L)からなる群から選択される1、2、3、4、5、及び/又は6の相補性決定領域(CDR)を含む。好ましくは、本発明の抗体(又はそのフラグメント)は、配列番号2、配列番号4及び配列番号6で表されるCDRを有する。他にまた好ましくは、本発明の抗体又はそのフラグメントは、配列番号10、配列番号12及び配列番号14で表されるCDRを有する。CDRのアミノ酸配列は、下記表1に記載のとおりである。
【表1】
【0042】
別の実施形態では、抗PDGFRα抗体又はそのフラグメントは、配列番号8で表されるヒト重鎖可変領域及び/又は配列番号16で表されるヒト軽鎖可変領域を有する。IMC-3G3は、そのような抗体であり、本発明で説明される。
【0043】
好ましくは、本発明の抗体又はそのフラグメントは、PDGFRαを中和する。PDGFRαの細胞外ドメインと、リガンド(例えばPDGF-AA、PDGF-AB、PDGF-BB又はPDGF-CC)との結合は、受容体の二量体化、自己リン酸化、受容体内部の細胞質チロシンキナーゼドメインの活性化、並びにDNA合成(遺伝子の活性化)の制御及び細胞周期の進行又は細胞分裂周期の制御と関係する複数のシグナル伝達経路やトランス活性化経路の開始を刺激する。抗PDGFRα抗体は、典型的には、リガンド結合及び/又は受容体の二量体化を遮断し、自己リン酸化、チロシンキナーゼ活性の活性化及びシグナル伝達のうち1以上を阻害する。本発明の抗PDGFRα抗体は、PDGFRαの細胞外リガンド結合領域に特異的であり得、リガンドがPDGFRαと結合するのを阻害できる。好ましくは、かかる抗PDGFRα抗体又はそのフラグメントは、PDGFRαの天然のリガンドと少なくとも同じ強さで、PDGFRαに結合する。代わりに又はさらに、抗体は、受容体ダイマーの境界面を別途形成し得る受容体モノマーの領域に特異的であり得る。かかる抗体は、受容体モノマーへのリガンドの結合が遮断されても遮断されなくとも、二量体形成を遮断する。
【0044】
抗IGF-IR抗体について上記したとおり、受容体の中和は、様々なin vivo、in vitro、及びex vivo方法によって測定できる。本発明の1実施形態では、抗PDGFRα抗体は、PDGFRαのリン酸化を少なくとも約75%減少させる。他の実施形態では、リン酸化を少なくとも約85%又は少なくとも約90%減少させる。本発明の実施形態では、PDGFRαシグナル伝達の阻害の結果、リン酸化又は下流のシグナル伝達経路因子(Akt、p42/p44等)が、少なくとも約40%、少なくとも約60%、又は少なくとも約80%減少する。受容体の中和は、所定のリガンド(例えば、PDGF-AA、-AB、-BB、-CC)、かかるリガンドの混合物又はPDGFを含有する骨髄穿刺液といった調製物だけでなく、他の成長促進因子を用いても、測定できる。
【0045】
PDGFRαの中和は、通常シグナル伝達に関係する当該活性のうち1以上の阻害、減少、不活化及び/又は遮断を含む。従って、PDGFRαの中和は、成長(増殖及び分化)、血管新生(血管の補強、浸潤、及び転移)、並びに細胞の移動性及び転移(細胞接着及び浸潤)の阻害、減少、不活化及び/又は遮断を含む、様々な効果を有する。
【0046】
上記のとおり、ex vivoアッセイを利用して、PDGFRαの中和を測定することができる。例えば、PDGF-AAにより刺激されたヒトSKLMS-1平滑筋肉腫細胞(American Type Culture Collection (ATCC), Rockville, MD; ATCC HTB-88TM)又はU118神経膠芽腫細胞(ATCC HTB-15TM)を使用して、PDGFRα阻害をアッセイできる。成長の阻害は、PDGFRαを発現するヒト腫瘍細胞を注入したSCIDマウスを用いて、確認することができる。
【0047】
本発明は、PDGFRα中和のいかなる特定機構によっても制限されない。本発明の抗PDGFRα抗体は、PDGFRα細胞表面受容体に外側から結合でき、リガンド(例えば、PDGF-AA、PDGF-AB、PDGF-BB、PDGF-CC)の結合を阻止でき、PDGFRαのリン酸化を阻害でき、受容体と結合したチロシンキナーゼによって介在されるシグナル伝達を阻害でき、並びにシグナル伝達下流の因子の活性を調節することができる。受容体-抗体複合体は、中和及び分解されることによって、細胞表面受容体のダウンレギュレーションをもたらすこともできる。マトリックスメタロプロテイナーゼは、腫瘍細胞の浸潤及び転移において機能を果たすが、該プロテインは、本発明の抗体によってもダウンレギュレートされる。そのうえ、本発明の抗体は、成長因子の産生及び血管新生の阻害を示せる。
【0048】
上記のとおり、本発明のPDGFRαアンタゴニストは、転移性骨腫瘍も含めた骨腫瘍を治療するのに有用である。PDGFRαを発現し、かつ、本発明で治療できる他の腫瘍の種類には、卵巣癌、乳癌、肺癌、肝細胞癌、胃腸間質腫瘍、メラノーマ、腎細胞癌、前立腺癌、及び柔組織肉腫が含まれるが、これに限定されない。柔組織肉腫は、脂肪、筋肉、神経、腱、並びに血管及びリンパ管といった組織で発生する。典型的には、腫瘍細胞はPDGFRαを発現する。PDGFRαの発現は、例えば組織化学的解析又はRNA解析により測定できる。例えば、放射線標識したIMC-3G3と、U118細胞及びSKLMS-1腫瘍細胞との結合のスキャッチャード解析によれば、該細胞上のPDGFRα分子の数がそれぞれ約500及び2500個であることがわかる。
【0049】
PDGFRαアンタゴニストは、腫瘍細胞自体の上で発現したPDGFRαによるシグナル伝達を阻害することによって、又は周辺のストローマ細胞上でPDGFRαが発現するのを阻害して、腫瘍細胞から発現したPDGFによってパラクリン刺激を受けるのを避けることにより、機能する。従って、IMC-3G3及び他のPDGFRαアンタゴニストといった抗体は、PDGFRαのオートクリン及び/又はパラクリン刺激を特徴とする腫瘍を治療するのに、有用である。
【0050】
本発明の抗体フラグメントは、全抗体を切断することにより、又は該フラグメントをコードするDNAを発現させることにより作製できる。抗体のフラグメントは、Lamoyi et al., J. Immunol. methods, 56: 235-243 (1983)及びParham, J. Immunol. 131 : 2895-2902 (1983)により記載された方法によって、調製できる。かかるフラグメントは、Fabフラグメント又はF(ab')2フラグメントの片方又は両方を含み得る。かかるフラグメントには、一本鎖フラグメント可変領域抗体、すなわちscFv、dibodies、又は他の抗体フラグメントも含まれる。かかる機能性等価物の作製方法は、PCT出願WO 93/21319、欧州特許出願EP 239400;PCT出願WO 89/09622;欧州特許出願EP 338745;及び欧州特許出願EP 332424に開示されている。
【0051】
ベクターの形質転換及び本発明の抗体の発現に好ましい宿主細胞は、哺乳動物の細胞、例えばCOS-7細胞、チャイニーズハムスター卵巣(CHO)細胞、及びリンパ系起源の細胞株、例えば、リンパ腫細胞、骨髄腫(例えば、NSO)細胞、又はハイブリドーマ細胞である。他の真核性宿主、例えば酵母を代わりに利用することができる。
【0052】
酵母中で遺伝子構築物を発現させたい場合に、酵母中での使用に好適な選択遺伝子は、酵母プラスミドYRp7中に存在するtrp1遺伝子である。Stinchcomb et al. Nature, 282: 39 (1979); Kingsman et al., Gene, 7: 141 (1979)。trp1遺伝子は、トリプトファン中で成長する能力を欠く酵母の変異株、例えば、ATCC番号44076又はPEP4-1の選択マーカーとなる(Jones, Genetics, 85: 12 (1977))。従って、酵母宿主細胞ゲノム中のtrp1欠陥の存在によって、トリプトファン不在下での成長による形質転換を検出するのに有効な環境がもたらされる。同様に、Leu2-欠損酵母株(ATCC 20,622又は38,626)は、Leu遺伝子を含有する公知のプラスミドによって、補完される。
【0053】
形質転換した宿主細胞は、同化可能な炭素源(糖質、例えばグルコース又はラクトース)、窒素源(アミノ酸、ペプチド、タンパク質又はそれらの分解産物、例えばペプトン、アンモニウム塩など)、及び無機塩源(ナトリウム、カリウム、マグネシウム、及びカルシウムの硫黄塩、リン酸塩及び/又は炭酸塩)を含有する液状培地中で、当技術分野で公知の方法により培養される。該培地には、例えば微量元素(例えば、鉄、亜鉛、マンガンなど)のような成長促進物質がさらに含まれる。
【0054】
本発明の高親和性抗PDGFRα抗体及び抗IGF-IR抗体は、ヒト重鎖及び軽鎖可変領域遺伝子から構築されたファージディスプレイライブラリーから単離できる。例えば、本発明の可変ドメインは、再構成された可変領域遺伝子を含む末梢血リンパ球から入手することができる。他には、可変ドメイン部分、例えばCDR及びFW領域は、様々な起源から得ることができ、組換えることができる。さらに、可変ドメインの一部(例えば、FW領域)は合成コンセンサス配列であってもよい。
【0055】
本発明の抗体及び抗体フラグメントは、例えば、天然の抗体、又はFab若しくはscFvファージディスプレイライブラリーから入手できる。VH及びVLドメインを含む抗体からシングルドメイン抗体を作製して、結合、発現又は可溶性を増強するためには、CDRの外側で特定のアミノ酸を置換することが望ましいことが知られている。例えば、他には、VH-VL界面に埋め込まれたアミノ酸残基を改変することが望ましい。
【0056】
さらに、本発明の抗体及び抗体フラグメントは、ヒト免疫グロブリンγ重鎖及びκ軽鎖を産生するトランスジェニックマウス(例えば、San Jose, Califから入手したKMマウス)を用いて、標準的なハイブリドーマ技術(Harlow & Lane, ed., 抗体: A Laboratory Manual, Cold Spring Harbor, 211-213 (1998);引用により本明細書に取り込まれる)により入手できる。好ましい実施形態では、ヒト抗体を産生するゲノムの本質的部分を、マウスのゲノムに挿入し、内因性マウス抗体を産生できなくさせる。かかるマウスは、PDGFRαを皮下投与し(s.c.)、(通常は完全フロインドアジュバント中で)必要に応じてブーストして、免疫できる。免疫方法は、当技術分野では周知である。
【0057】
本発明のIGF-IR結合性抗体を同定するために使用されるタンパク質は、好ましくはIGF-IRであり、より好ましくはIGF-IRの細胞外ドメインである。本発明のPDGFRα結合性抗体を同定するために使用されるタンパク質は、好ましくはPDGFRαであり、より好ましくはPDGFRαの細胞外ドメインである。かかる細胞外ドメインは、遊離型であるか、他の分子と結合している。
【0058】
本発明は、前記の抗体又はそのフラグメントをコードする単離されたポリヌクレオチドも提供する。IMC-A12抗IGF-IR抗体の詳細は、WO2005016970に開示されている。表2は、IMC-3G3の核酸配列を示す。
【表2】
【0059】
ヒト抗体をコードするDNAは、対応するヒト抗体領域から実質的又は排他的に誘導された、CDR以外のヒト定常領域及び可変領域をコードするDNAとヒト由来のCDRをコードするDNA(重鎖可変ドメインCDRを表す配列番号1、3、及び5、並びに軽鎖可変ドメインCDRを表す配列番号9、11、及び13)を組換えることにより、調製できる。
【0060】
抗体のフラグメントをコードするDNAの好適な供給源には、ハイブリドーマ及び脾臓細胞といった任意の細胞であって、完全長の抗体を発現するものが含まれる。フラグメントは、そのもの自体を抗体等価物として使用でき、又は上記のとおり等価物に組換えて使用できる。このセクションに記載のDNA欠失及び組換えは、公知の方法、例えば抗体等価物に関して上記に挙げた文献に記載の方法、並びに/又は他の標準的DNA組換え技術、例えば下記の方法により実施できる。DNAの別の供給源は、当技術分野で公知のファージディスプレイライブラリーから産生される一本鎖抗体である。
【0061】
さらに、本発明は、発現配列、プロモーター及びエンハンサー配列と機能し得るように連結された前記ポリヌクレオチド配列を含む発現ベクターを提供する。細菌のような原核生物系及び真核生物系(酵母及び哺乳動物細胞培養系を含むがこれに限定されない)中で抗体ポリペプチドの効率的な合成に使用される多様なベクターが、開発されている。本発明のベクターは、染色体DNA、非染色体DNA、及び合成DNA配列のセグメントを含み得る。
【0062】
任意の好適な発現ベクターを利用できる。例えば、原核生物のクローニングベクターには、E. coli、例えばcolEl、pCRl、pBR322、pMB9、pUC、pKSM及びRP4由来のプラスミドが含まれる。原核生物ベクターには、ファージDNA、例えばMl3及び他のフィラメント状一本鎖DNAファージの誘導体も含まれる。酵母中で使用するのに有用なベクターは、例えば2μプラスミドである。哺乳動物細胞中での発現に好適なベクターには、SV40、アデノウイルス、レトロウイルス由来のDNA配列の周知の誘導体、哺乳動物の機能性ベクターの組み合わせにより作製したシャトルベクター、例えば上記のもの、並びに機能性プラスミド及びファージDNAが含まれる。
【0063】
さらなる真核生物の発現ベクターが、当技術分野では知られている(例えば、PJ. Southern and P. Berg, J. Mol. Appl. Genet., 1, 327-341 (1982); Subramani et al., Mol. Cell. Biol., 1: 854-864 (1981); Kaufmann and Sharp, 「Amplification And expression of Sequences Cotransfected with a Modular Dihydrofolate Reductase Complementary DNA Gene」, J. Mol. Biol. 159, 601-621 (1982); Kaufmann and Sharp, Mol. Cell. Biol. 159, 601-664 (1982); Scahill et al., 「Expression And Characterization Of The Product Of A human Immune Interferon DNA Gene In Chinese Hamster Ovary Cells」, Proc. Nat'l Acad. Sci. USA 80, 4654-4659 (1983); Urlaub and Chasin, Proc. Nat'l Acad. Sci. USA 77, 4216-4220, (1980))。
【0064】
本発明で有用な発現ベクターには、発現されるDNA配列又はフラグメントに機能し得るように連結された少なくとも1つの発現制御配列が含まれる。制御配列をベクターに挿入することにより、クローニングしたDNA配列の発現を制御及び調節することができる。有用な発現制御配列は、例えばlac系、trp系、tac系、trc系、λファージの主要なオペレーター及びプロモーター領域、fdコートタンパク質の制御領域、酵母の解糖性プロモーター、例えば3-ホスホグリセリン酸キナーゼのプロモーター、酵母の酸性ホスファターゼのプロモーター、例えばPho5、酵母α-接合因子のプロモーター、並びにポリオーマウイルス、アデノウイルス、レトロウイルス、及びシミアンウイルス由来のプロモーター、例えば初期及び後期プロモーター又はSV40、並びに原核生物又は真核生物細胞及びそれらのウイルス又はそれらの組み合わせの遺伝子の発現を制御することが知られているその他の配列である。
【0065】
本発明は、前記の発現ベクターを含む組換え宿主細胞も提供する。本発明の抗体は、ハイブリドーマ以外の細胞株中でも発現できる。本発明のポリペプチドをコードする配列を含む核酸を、好適な哺乳動物の宿主細胞の形質転換のために使用できる。
【0066】
特定の所望の細胞株は、所望のタンパク質の高レベル発現、構成的発現、及び最小レベルの発現に基づいて、宿主タンパク質から選別される。発現のために宿主として利用できる哺乳動物の細胞株が当技術分野では周知であり、多くの不死化細胞株、例えば、NSO細胞、チャイニーズハムスター卵巣(CHO)細胞、ベビーハムスター腎(BHK)細胞及び多くの他の細胞が含まれるが、これに限定されない。さらなる好適な真核生物細胞には、酵母及び他の真菌が含まれる。有用な原核生物の宿主には、例えば、E. coli SG-936、E. coli HB 101、E. coli W3110、E. coli X1776、E. coli X2282、E. coli DHI、及び E. coli MRC1のような大腸菌(E.coli)、シュードモナス(Pseudomonas)、枯草菌(Bacillus subtilis)のようなバチルス(Bacillus)、及びストレプトミセス(Streptomyces)が含まれる。
【0067】
これらの本発明の組換え宿主細胞を使用することにより、抗体又はそのフラグメントを発現させることができる条件下で該細胞を培養し、宿主細胞又は宿主細胞周辺の培地から抗体又はフラグメントを精製することにより、抗体又はそのフラグメントを産生させることができる。組換え宿主細胞中での分泌のための発現した抗体又はフラグメントの指向化は、シグナル又は分泌性リーダーペプチドをコードする配列(Shokri et al., Appl Microbiol Biotechnol. 60(6):654- 64 (2003), Nielsen et al., Prot. Eng. 10:1-6 (1997)及びvon Heinje et al., Nucl. Acids Res. 14:4683-4690 (1986)を参照されたい)を、所望の抗体をコードする遺伝子の5'末端に挿入することにより、促進される。これらの分泌性リーダーペプチド因子は、原核生物又は真核生物のいずれかの配列から誘導できる。よって好適には、分泌性リーダーペプチドを使用して、アミノ酸をポリペプチドのN末端に結合することにより、宿主細胞サイトゾルから生じたポリペプチドの移動及び培地への分泌を指向させる。
【0068】
この発明の抗体は、さらなるアミノ酸残基と融合し得る。かかるアミノ酸残基は、おそらくは単離を促進するペプチドタグである。特定の臓器又は組織への帰巣(homing)のために、他のアミノ酸残基を補完することもできる。
【0069】
別の実施形態では、トランスジェニック動物中で抗体をコードする核酸を発現させることにより、本発明の抗体を作製する。その場合、該抗体を発現させて、回収することができる。例えば、回収及び単離を促進する組織特異的な方法で、抗体を発現させることができる。そのような実施形態の1つでは、本発明の抗体は、授乳期間中に分泌用の乳腺中で発現する。トランスジェニック動物には、マウス、ヤギ及びウサギが含まれるが、これに限定されない。
【0070】
本発明で使用できる抗体には、完全免疫グロブリン、免疫グロブリンの抗原結合性フラグメントだけでなく、免疫グロブリンの抗原結合性ドメインを含む抗原結合性タンパク質が含まれる。免疫グロブリンの抗原結合性フラグメントには、例えば、Fab、Fab'、及びF(ab')2が含まれる。他の抗体の形態は、結合特異性を維持するが、例えば、二特異性、多価性(3以上の結合部位)、コンパクトなサイズ(例えば、結合ドメインのみ)を望ましくは含むその他の特性を有するように、改良されている。
【0071】
一本鎖抗体は、その抗体が由来する全抗体の定常ドメインの一部又は全てを欠く。従って、一本鎖抗体は、全抗体の使用に付随する問題のいくつかを解決できる。例えば、一本鎖抗体は、重鎖定常領域及び他の生体分子間の特定の望ましからざる相互作用が存在しない場合が多い。さらに、一本鎖抗体は全抗体よりもかなり小さく、全抗体よりも優れた透過性を有する場合があり、より効率的に標的抗原結合部位に局在化し、結合することができる。そのうえ、一本鎖抗体はサイズが比較的小さいが故に、レシピエントにおける望ましからざる免疫応答が、全抗体よりも誘発されにくい。
【0072】
多重一本鎖抗体は、それぞれの一本鎖が、第1のペプチドリンカーにより共有結合された1つのVH及び1つのVLドメインを有しており、該抗体は少なくとも1以上のペプチドリンカーにより共有結合されることによって、多価一本鎖抗体を形成することができ、それにより単特異性又は複数特異性となり得る。多価一本鎖抗体のそれぞれの鎖は、可変軽鎖フラグメント及び可変重鎖フラグメントを含んでおり、少なくとも1つの他の鎖とペプチドリンカーにより連結されている。ペプチドリンカーは、少なくとも15アミノ酸残基からなる。アミノ酸残基の最大数は、約100である。
【0073】
2つの一本鎖抗体を結合させて、二価性ダイマーとしても知られるジアボディ(diabody)を形成させることができる。ジアボディは、2つの鎖及び2つの結合部位を有し、単特異性又は二特異性であり得る。ジアボディのそれぞれの鎖は、VLドメインに連結されたVHドメインを含む。ドメインは、同一鎖上にあるドメイン間のペア形成を阻害するのに十分に短いリンカーによって、連結される。これにより、異なる鎖上の相補性ドメイン間のペア形成が促進され、2つの抗原結合部位が再生成される。
【0074】
3つの一本鎖抗体を結合させて、三価性トリマーとしても知られるトリアボディ(triabody)を形成することができる。トリアボディはVL又はVHドメインのアミノ末端を、VL又はVHドメインのカルボキシ末端に直接、すなわちいかなるリンカー配列も使用せずに、融合させて構築する。トリアボディは、環状の、頭-尾様式で配置されたポリペプチドを具備した3つのFv頭部を有する。トリアボディがとり得るコンホメーションは、互いに120度の角度で平面に配置された3つの結合部位を有する平面的なものである。トリアボディは、単特異性、二特異性又は三特異性である。
【0075】
従って、本発明の抗体及びそのフラグメントには、天然の抗体、二価性フラグメント、例えば(Fab')2、一価性フラグメント、例えばFab、一本鎖抗体、一本鎖Fv(scFv)、シングルドメイン抗体、多価性一本鎖抗体、ジアボディ、トリアボディ、及び抗原に特異的に結合するようなものが含まれるが、これに限定されない。
【0076】
抗IGF-IR抗体及び抗PDGFRα抗体又は抗体フラグメントは、IGF-IR(WO2005016970)又はPDGFRαを有する細胞に結合することによって内在化され得、抗癌剤に化学的に又は生合成的に連結させることができる。かかる抗体に連結される抗癌剤には、抗体が結合した腫瘍又は抗体が結合した細胞周辺にある腫瘍を、破壊又は損傷させる任意の薬剤が含まれる。例えば、抗癌剤は、化学療法剤又はラジオアイソトープのような毒性剤である。好適な化学療法剤は、当業者には公知であり、アントラサイクリン系(例えば、ダウノマイシン及びドキソルビシン)、メトトレキサート、ビンデシン、ネオカルチノスタチン、シスプラチン、クロラムブシル、サイトシンアラビノサイド、5-フルオロウリジン、メルファラン、リシン及びカリケアマイシンが含まれる。化学療法剤は、従来技術を用いて抗体に結合される(例えば、Hermentin and Seiler, Behring Inst. Mitt. 82:197- 215(1988)を参照されたい)。
【0077】
抗癌剤として使用するのに好適なラジオアイソトープも、当業者には公知である。例えば、131I又は211Atが使用される。これらのアイソトープは、従来技術を用いて、抗体に結合される(例えば、Pedley et al., Br. J. Cancer 68, 69- 73(1993)を参照されたい)。
【0078】
他には、抗体と結合される抗癌剤は、プロドラッグを活性化する酵素である。この場合、標的部位に到達するまでは不活性型を維持するプロドラッグが投与され、該標的部位でその細胞毒性型に変換される。実際には、抗体-酵素コンジュゲートを患者に投与して、治療対象となる組織の領域に局在化させることができる。従って、プロドラッグを患者に投与することにより、治療される組織領域で細胞毒性薬剤に変換させる。
【0079】
他の抗癌剤には、サイトカイン、例えばインターロイキン-2(IL-2)、インターロイキン-4(IL-4)又は腫瘍壊死因子α(TNF-α)が含まれる。該抗体は、サイトカインを腫瘍に指向させることにより、サイトカインは他の組織に影響を与えることなく腫瘍の破壊又は損傷を媒介することができる。サイトカインは、従来のDNA組換え技術を用いることにより、DNAレベルで、抗体に結合させることができる。
【0080】
本発明のある実施形態では、1以上の抗腫瘍剤と組み合わせて抗IGF-IR又は抗PDGFRα抗体を投与する。併用療法の例については、例えば米国特許第6,217,866号(Schlessinger et al.) (抗腫瘍剤と併用する抗-EGFR抗体);WO 99/60023 (Waksal et al.) (放射線療法と併用する抗-EGFR抗体)を参照されたい。任意の好適な抗腫瘍剤、例えば、化学療法剤、放射線療法又はそれらの組み合わせを使用することができる。抗腫瘍剤は、アルキル化剤又は抗代謝剤であり得る。アルキル化剤の例には、シスプラチン、シクロホスファミド、メルファラン、及びダカルバジンが含まれるが、これに限定されない。抗代謝剤の例には、ドキソルビシン、ダウノルビシン、及びパクリタキセル、ゲムシタビンが含まれるが、これに限定されない。
【0081】
有用な抗腫瘍剤には、有糸分裂阻害剤、例えばタキサン類、ドセタキセル及びパクリタキセルも含まれる。トポイソメラーゼ阻害剤は、本発明の抗体と組み合わせて使用できる別のクラスの抗腫瘍剤である。これらには、トポイソメラーゼI又はトポイソメラーゼIIの阻害剤も含まれる。トポイソメラーゼI阻害剤には、イリノテカン(CPT-11)、アミノカンプトテシン、カンプトテシン、DX-8951f、トポテカンが含まれる。トポイソメラーゼII阻害剤には、エトポシド(VP-16)及びテニポシド(VM-26)が含まれる。他の物質については、トポイソメラーゼ阻害活性及び抗腫瘍剤としての有効性について、現在評価中である。好ましい実施形態では、トポイソメラーゼ阻害剤はイリノテカン(CPT-11)である。
【0082】
本発明の特別の実施形態では、ドセタキセルと組み合わせて抗IGF-IR抗体を投与する。本発明の別の実施形態では、ドキソルビシンと組み合わせて抗PDGFRα抗体を投与する。
【0083】
抗腫瘍剤が放射線照射である場合、放射線源は、治療対象となる患者の外部(外部照射療法-EBRT)又は内部(近接照射療法-BT)にある。投与される抗腫瘍剤の用量は、数多くの因子によって決まり、該因子には例えば、薬剤の種類、治療される腫瘍の種類及び重症度並びに薬剤の投与経路が含まれる。しかしながら、本発明は、いかなる特定の用量によっても制限されないことを強調したい。
【0084】
抗体(抗IGF-IR又は抗PDGFRα)療法及び抗体+抗腫瘍剤療法は、(例えば、乳癌のための)補助ホルモン療法又は(例えば、前立腺癌のための)アンドロゲン抑制療法を受ける患者でも使用できる。
【0085】
本発明の抗IGF-IRアンタゴニスト及び抗PDGFRαアンタゴニストは、腫瘍増殖又は血管新生に関与する他の受容体を中和する受容体アンタゴニストと同時に、又は共に投与できる。例えば、本発明の実施形態では、抗IGF-IR抗体及び抗PDGFRα抗体を同時投与する。1実施形態において、標的腫瘍細胞がIGF-IR及びPDGFRαの両方を発現する場合には、それぞれの受容体を介するシグナル伝達によって、共通するシグナル伝達因子が活性化される。1つの受容体の阻害は、共通する下流因子の活性化を通常は低下させるが、両方の受容体を阻害すれば、活性化をさらに低下させるであろう。別の実施形態では、腫瘍又は周辺組織中の特定の細胞は相当量の受容体を発現し、他の細胞は相当量の第2の受容体を発現する。アンタゴニストの同時投与は、腫瘍細胞の増殖及び周辺細胞のパラクリン刺激を減少させる。
【0086】
同時投与の代わりに、二特異性(二重特異性)抗体を投与することもできる。様々な所望の特徴を取り入れるように設計された多様な二特異性抗体が存在する。例えば、二特異性ジアボディは、最も小さいサイズを有する。4つの抗原結合部位(それぞれの結合特異性につき2つ)を備えた二特異性抗体は、対応する天然の抗体と同程度の結合活性を有する。ある二特異性抗体は、Fc領域を取り込むことによって、天然の抗体のエフェクター機能(例えば、補体依存性細胞毒性(CDC)及び抗体依存性細胞毒性(ADCC))を維持する。WO 01/90192はIgG-様の四価抗体を記載しており、WO 2006/020258は2つのジアボディを包含し、エフェクター機能を維持する四価抗体を記載している。
【0087】
別の実施形態では、抗IGF-IR抗体又は抗PDGFRα抗体又はその他のアンタゴニストを、上皮成長因子受容体(例えば、EGFR、Her2/erbB2、erbB3、erbB4)に特異的に結合する受容体アンタゴニストと組み合わせて使用する。特に好ましいのは、EGFRの細胞外ドメインに結合し、そのリガンドのうち1以上の結合を遮断し、さらに/又はリガンドにより誘発されるEGFRの活性化を中和する抗原結合性タンパク質である。EGFRアンタゴニストには、EGFRのリガンドに結合し、EGFRとそのリガンドとの結合を阻害する抗体も含まれる。EGFRのリガンドには、例えば、EGF、TGF-α、アンフィレグリン、ヘパリン結合性EGF(HB-EGF)及びベタセルリンが含まれる。EGF及びTGF-αは、EGFR介在性刺激をもたらす主な内因性リガンドであると考えられているが、TGF-αは、血管新生を促進する能力のほうが高いことが証明されている。EGFRアンタゴニストには、他のEGFR受容体サブユニット(すなわち、EGFRホモダイマー)とのEGFRの二量体化、又は他の成長因子受容体(例えば、HER2)とのヘテロ二量体化を阻害する物質も含まれる。EGFRアンタゴニストには、生体分子及び小分子、例えばEGFRの細胞質ドメインに直接作用し、EGFR介在性シグナル伝達を阻害する合成キナーゼ阻害剤がさらに含まれる。Erbitux(登録商標)(セツキシマブ)は、EGFRに結合しリガンド結合を遮断するEGFRアンタゴニストの一例である。小分子EGFRアンタゴニストの一例はIRESSATM(ZD1939)であり、これはEGFRを阻害するためにATP擬似体(mimetic)として機能するキノザリン(quinozaline)誘導体である。米国特許第5,616,582号(Zeneca Limited);WO 96/33980 (Zeneca Limited)の4ページを参照されたい。また、Rowinsky et al, ASCOの第37回年次会議で発表された抄録5, San Francisco, CA, 12-15 May 2001; Anido et al, ASCOの第37回年次会議で発表された抄録1712, San Francisco, CA, 12-15 May 2001も参照されたい。小分子EGFRアンタゴニストの別の例は、Tarceva(登録商標)(OSI-774)であり、これは4-(置換フェニルアミノ)キノザリン誘導体[6,7-ビス(2-メトキシ-エトキシ)-キナゾリン-4-イル]-(3-エチニル-フェニル)アミン塩酸塩]EGFR阻害剤である。WO 96/30347(Pfizer Inc.)の例えば2ページ12行目〜4ページ34行目及び、19ページ14〜17行目を参照されたい。また、Moyer et al., Cancer Res., 57: 4838-48 (1997); Pollack et al., J. Pharmacol, 291: 739-48 (1999)も参照されたい。Tarceva(登録商標)は、EGFRのリン酸化及びその下流のPI3/Akt及びMAP(マイトジェン活性プロテイン)キナーゼシグナル伝達経路を阻害することによって機能し、p27を仲介する細胞周期の停止をもたらすことができる。Hidalgo et al, ASCOの第37回年次会議で発表された抄録281, San Francisco, CA, 12-15 May 2001を参照されたい。
【0088】
他の小分子もEGFRを阻害することが報告されており、その多くはEGFRのチロシンキナーゼドメインに特異的であると考えられている。かかる小分子EGFRアンタゴニストのいくつかの例は、WO 91/116051、WO 96/30347、WO 96/33980、WO 97/27199 (Zeneca Limited)、WO 97/30034 (Zeneca Limited)、WO 97/42187 (Zeneca Limited)、WO 97/49688 (Pfizer Inc.)、WO 98/33798 (Warner Lambert Company)、WO 00/18761 (American Cyanamid Company)、及びWO 00/31048 (Warner Lambert Company)に記載されている。特異的な小分子EGFRアンタゴニストの例にはCl-1033(Pfizer)が含まれ、これはキノザリン(N-[4-(3-クロロ-4-フルオロ-フェニルアミノ)-7-(3-モルホリン4-イル-プロポキシ)-キナゾリン-6-イル]-アクリルアミド)である、チロシンキナーゼ、特にEGFRの阻害剤であり、WO 00/31048の8ページ22〜26行目に記載されている。また、PKIl66 (Novartis)はEGFRのピロロピリミジン阻害剤であって、WO 97/27199の10〜12ページに記載されており;GW2016 (GlaxoSmithKline)はEGFR及びHER2の阻害剤であり;EKB569 (Wyeth)はEGFR及びHER2を過剰発現する腫瘍細胞の増殖をin vitro及びin vivoで阻害することが報告されており;AG-1478 (Tryphostin)は、EGFR及びerbB-2の両方からのシグナル伝達を阻害するキナゾリン小分子であり;AG-1478 (Sugen)はプロテインキナーゼCK2も阻害する2つの基質の阻害剤であり;PD 153035 (Parke-Davis)は、EGFRキナーゼ活性及び腫瘍増殖を阻害することが報告されており、培養下の細胞中でアポトーシスを誘導し、細胞障害性化学療法剤の細胞毒性を増強し;SPM-924 (Schwarz Pharma)は前立腺癌の治療を目的としたチロシンキナーゼ阻害剤であり;CP-546,989 (OSI Pharmaceuticals)は、固形癌の治療において血管新生の阻害剤であることが報告されており;ADL-681は癌の治療を目的とするEGFRキナーゼ阻害剤であり;PD 158780は、A4431移植片の腫瘍増殖速度を抑制することがマウスで報告されているピリドピリミジンであり;CP-358,774は、HN5異種移植片中で自己リン酸化を阻害することがマウスで報告されているキンゾリン(quinzoline)であり;ZD1839は外陰癌、NSCLC、前立腺癌、前立腺癌、卵巣癌、及び大腸癌を含むマウス移植片モデルで、抗腫瘍活性を有することが報告されているキンゾリンであり;CGP 59326Aは、EGFR陽性移植片の成長を阻害することがマウスで報告されているピロロピリミジンであり;PD 165557 (Pfizer);CGP54211及びCGP53353 (Novartis)はジアニルナフタルイミドである。天然由来のEGFRチロシンキナーゼ阻害剤には、ゲニステイン、ハービマイシンA、クエルセチン、及びエルブスタチンが含まれる。
【0089】
EGFRを阻害することが報告されている故に本発明の範囲にはいる更なる小分子には、三環式化合物、例えば米国特許第5,679,683号に記載の化合物;キナゾリン誘導体、例えば米国特許第5,616,582号に記載の化合物;及びインドール化合物、例えば米国特許第5,196,446号に記載の化合物がある。
【0090】
IGF-IR又はPDGFRαと共に標的化され得る別の受容体は、血管内皮増殖因子受容体(VEGFR)である。本発明の実施形態では、VEGFRアンタゴニストと組み合わせて抗IGF-IR抗体又は抗PDGFRα抗体を使用する。1実施形態では、VEGFR-1/FIt-1受容体に特異的に結合するアンタゴニストを使用する。別の実施形態では、VEGFRアンタゴニストはVEGFR-2/KDR受容体に特異的に結合する。特に好ましいのは、VEGFR-1又はVEGFR-2の細胞外ドメインに結合し、それらのリガンド(VEGFR-2はVEGFによって最も強力に刺激され;VEGFR-1はPlGFによって最も強力に刺激されるが、VEGFによっても刺激される)による結合を遮断し、及び/又はリガンドによって誘発される活性化を中和する抗原結合性タンパク質である。例えば、IMC-1121はVEGFR-2に結合し、中和するヒト抗体である(WO 03/075840; Zhu)。別の例は、可溶性の細胞表面で発現されるVEGFR-1に結合するMAb6.12である。ScFv 6.12は、マウスモノクローナル抗体MAb6.12のVL及びVHドメインを含む。MAb 6.12を産生するハイブリドーマ細胞株は、特許手続上の微生物の寄託の国際的承認に関するブダペスト条約(ブダペスト条約)の規制下で、ATCC番号PTA-3344として寄託されている。別の実施形態では、VEGFRアンタゴニストは、VEGFRリガンドに結合し、リガンドによるVEGFRの活性化を遮断する。例えば、Avastin(登録商標)(ベバシツマブ)は、VEGFと結合する抗体である。
【0091】
腫瘍形成に関与する成長因子受容体の他の例は、神経成長因子(NGFR)及び線維芽細胞成長因子(FGFR)である。
【0092】
更なる他の実施形態では、抗IGF-IR及び抗PDGFRα抗体は、例えば、サイトカイン(例えば、IL-10及びIL-13)のような好適なアジュバント、又はこれらに限定されないが、ケモカイン、腫瘍関連抗原、及びペプチドのようなその他の免疫賦活剤のうち1以上と組み合わせて、投与できる。例えば、Larrivee et al., 前掲を参照されたい。しかしながら、抗IGF-IR又は抗PDGFRα抗体の投与だけでも、十分に、治療上有効に腫瘍の増殖を防止、阻害、又は減少し得ることを理解されたい。
【0093】
併用療法では、抗IGF-IR又は抗PDGFRα抗体を、別の薬剤による治療の開始前、治療中、又は治療後に投与するだけでなく、それらを任意で組み合わせて、すなわち、抗腫瘍剤療法の開始前及び治療中、開始前及び治療後、治療中及び治療後、又は開始前、治療中、及び治療後に投与できる。例えば、抗体は、放射線療法を開始する前1〜30日間、好ましくは3〜20日間、より好ましくは5〜12日間投与できる。本発明の好ましい実施形態では、抗体療法と同時に、又はより好ましくは抗体療法後に化学療法剤を投与される。
【0094】
本発明では、任意の好適な方法又は経路を使用して、本発明の抗体を投与することができ、場合によっては、抗腫瘍剤及び/又はその他の受容体のアンタゴニストを同時投与することができる。本発明で利用される抗腫瘍剤レジメンには、患者の腫瘍症状の治療にとって最適と考えられる任意のレジメンが含まれる。多様な悪性腫瘍が、特異的な抗腫瘍抗体及び特異的な抗腫瘍剤の使用を必要としており、該使用は患者ごとに決定されるだろう。投与経路には、例えば、経口投与、静脈内投与、腹腔内投与、皮下投与、又は筋肉内投与が含まれる。投与されるアンタゴニストの用量は、例えば、アンタゴニストの種類、治療される腫瘍の種類及び重症度、並びにアンタゴニストの投与経路を含む様々な因子によって決定される。しかしながら、本発明は、いかなる特定の投与方法又は投与経路によっても限定されないことを強調したい。
【0095】
当業者であれば、治療の用量及び頻度は、それぞれの患者の認容性、並びに使用される遮断剤又は阻害剤の薬力学的特性及び薬物動態学的特性によって、決まることがわかるであろう。理想的には、使用薬剤の飽和した薬物動態をもたらすことが望ましい。抗IGF-IR及び抗PDGFRα抗体の初回投与量はいずれも、例えば、約10〜約1000 mg/m2であり、好ましくは約200〜約400 mg/m2である。この後、いくつかの1日量又は週用量、例えば約200〜約400 mg/m2で、薬剤をさらに投与する。副作用について患者をモニターし、かかる副作用が重篤である場合には、治療を中止する。
【0096】
当業者は、有効用量を決定するために、治療の進捗をモニターする方法も知っている。前立腺癌からの骨転移では、かかる方法の1つはPSAレベルをモニターすることである。骨転移をモニターするその他の方法には、骨スキャン及びMRIが含まれる。
【0097】
癌治療誘発性の骨喪失(CTIBL)のリスクがあるか、又はCTIBLが問題となる患者(例えば、乳癌に対する補助ホルモン療法を受ける患者又は前立腺癌に対するアンドロゲン抑制両方を受ける患者)では、前述した任意の治療を、CTIBLの予防用薬剤、例えばビスホスホネートの投与で補完してもよい。ビスホスホネートには、例えばクロドロネート、リセドロネート及びゾレドロン酸が含まれる。
【0098】
本出願全体を通して、様々な文献、参照テキスト、教科書、技術マニュアル、特許、及び特許出願が引用されている。これらの文献、特許、特許出願及びその他の文献の教示及び開示は、引用によりその全体が本出願に包含され、本発明に付随する技術態様をより十分に説明する。
【0099】
本明細書に開示の発明の原理範囲内における変更が、当業者によりなされることを理解および予測される。そして、それらの変更は、本発明の範囲内に包含されることが意図される。
【0100】
以下の実施例により本発明をさらに説明されるが、いかなる方法によっても、本発明の範囲は限定されるべきでない。従来方法、例えばベクター及びプラススミドの構築並びに抗体及び抗体フラグメントの発現で利用される方法の詳細な説明は、数多くの文献から得ることができ、該文献にはSambrook, J et al., (1989) Molecular Cloning: A Laboratory Manual, 2nd ed., Cold Spring Harbor Laboratory Press; Coligan, J. et al. (1994) Current Protocols in Immunology, Wiley & Sons, Incorporated; Enna, SJ. et al. (1991) Current Protocols in Pharmacology, Wiley & Sons, Bonifacino, J.S. et al. (1999) Current Protocols in Cell Biology, Wiley & Sonsが含まれる。本明細書に記載の全ての文献は、その全体が本明細書に組込まれる。
【実施例】
【0101】
実施例1
IMC-A12及びドセタキセルの腫瘍増殖に対する効果
アンドロゲン非依存性(AI)LuCaP35Vの腫瘍小片(20〜30 mm3)を、32匹の6週令の去勢したSCIDマウスに、前述のとおり皮下に(s.c.)それぞれ移植した。移植した腫瘍の体積が150〜200 mm3に達するまで観察し、被験動物を治療試験のために、4つのグループに無作為割付けした。グループ1の被験動物には、用量20 mg/kgのドセタキセルによる治療を行った。グループ2の被験動物には、用量10 mg/kgのドセタキセルによる治療を行った。グループ3の被験動物には、10 mg/kgのドセタキセル及び40 mg/kgのA12の併用療法を行った。グループ4の被験動物には、20 mg/kgのドセタキセル及び40 mg/kgのA12の併用療法を行った。すべての治療は、腹腔内(ip)投与した。ドセタキセルは、週に1回投与した。Al2は週に3回投与した。すべての被験動物を4週間治療し、安楽死させる前にさらに4週間モニターした。1週間に2回腫瘍を測定して、式:体積 = L X W2/2を用いて腫瘍体積を算出した。我々ワシントン大学のIACUCによって承認された動物プロトコールに従って、腫瘍体積が1000 mm3に達するよりも前に、又は被験動物の体重減少が最初の体重の20%を超える前に、被験動物を安楽死させた。被験動物の体重は、週に2回測定した。血液サンプルを、眼窩洞から毎週採取した。血清を分離し、IMxトータルPSAアッセイ(Abott Laboratories, Abott Park, IL)を用いて、PSAレベルを測定した。in vivoでの腫瘍細胞増殖速度の評価のために被験動物を安楽死させる1時間前に、BrdUを腫瘍に注射した。
【0102】
安楽死させた後に、腫瘍を回収し二つに分けた。腫瘍の一部は10%中性緩衝ホルマリン(NFB)中で固定し、パラフィン包埋した。免疫組織化学(IHC)染色のために、5ミクロンの切片を作製した。腫瘍の残りの部分は、ミンチして70μmのナイロンシーブで濾過することにより、機械的に単一の細胞に分離させた。
【0103】
図1に示すとおり、LuCaP 35V移植片は、治療をしない場合には、362.0±72.0 mm3/週の平均成長速度で、マウスで急激に成長した。未治療群の被験動物は全て、試験群における治療開始後3週間以内に、腫瘍体積が1000 mm3を超えたので屠殺する必要があった。40μg/kgのA12単独で被験動物を治療した場合には、腫瘍成長速度は、治療期間中は192.7±35.6 mm3/週まで低下した。ドセタキセルを用量10 mg/kgで被験動物に投与した場合には、LuCaP 35V腫瘍の成長速度は、平均で29.6 ±6.1 mm3/週まで低下した。Al2治療と組み合わせてドセタキセルを与えた場合には、LuCaP 35V腫瘍の成長速度は平均7.9 ±1.0 mm3 /週までさらに低下した(図1b)。A12を併用したドセタキセルの阻害効果は、治療終了後4週間以上持続した。より高い用量のドセタキセル(20mg/kg)を被験動物に投与した場合には、A12治療の併用の有無に関わらず、腫瘍体積は4週間の治療期間中は増加しなかった;一方で、腫瘍体積が減少する傾向を観察した。しかしながら、A12を併用してドセタキセルで治療した被験動物群では、治療終了後4週間は、腫瘍体積の減少が持続した。対照的に、ドセタキセル単独で治療した被験動物群では、腫瘍体積が平均速度27.0±16.1 mm3/週で増加した。これらの結果は、A12を併用した所与の用量のドセタキセルが、治療中又は治療追跡後に、腫瘍増殖に対するドセタキセルの阻害効果を増強できることを示している。
【0104】
PSAは、前立腺腫瘍の増殖を評価するための使用される一般的な臨床パラメーターである。血清PSAレベルを、治療中及び治療後に被験動物で測定した。図1cに示すとおり、A12及びドセタキセル、又は20 mg/kgドセタキセル単独で治療した被験動物では、4週間の治療期間は、PSAの血清レベルの顕著な変化は見られなかった。このことは、腫瘍増殖の抑制と一貫性がある。治療終了後、ドセタキセル単独で治療した被験動物では、血清PSAレベルの増加が示された。一方、A12を併用してドセタキセルで治療した被験動物では、血清PSAレベルは一定であるか又はむしろ減少した。これらのデータは、ドセタキセル及びA12で治療した被験動物では、治療後に腫瘍増殖の阻害が持続したことと一致する。
【0105】
抗IGF-IR抗体と組み合わせたドセタキセルによるアポトーシスの誘導
治療終了時において、ドセタキセル及びA12の併用治療の細胞周期及び細胞生存に対するin vivo効果を、Apop-Direct kit (BD BioScience)を用いて、末端デオキシヌクレオチジルトランスフェラーゼ介在切断末端標識(TUNEL)アッセイ及びプロピジウム(PI)染色によって、既報のとおり測定した。簡潔にいうと、単一細胞の懸濁液から得た1×106細胞を10%中性緩衝ホルマリン(NBF)にいれた後、70%エタノールアルコールに−2O℃で30分間いれて固定した。数回の洗浄後に、細胞を0.1%Triton X-100で溶解し、FITCとコンジュゲートしたdUTP及び末端デオキシヌクレオチジルトランスフェラーゼ酵素(TdT)と共に37℃で1時間インキュベートした後、PI/RNase緩衝液(100μg/mlのPI、50μg/ml RNase)と共に室温で60分間インキュベートした。BD FACscanを用いて、サンプルをフローサイトメトリーで解析した。データは、CellQuestPROソフトウェアで解析した。
【0106】
治療終了後4週間、A12を併用してドセタキセルで治療した被験動物から得た腫瘍のかなりの割合(10 mg/kgのドセタキセルで治療したグループでは66.7%、20mg/kgのドセタキセルで治療したグループでは77.8%)で、使用したドセタキセルの用量と関係なく、アポトーシスを検出した(図2b及び表1)。これらの腫瘍におけるアポトーシス事象は、平均で15.0±4.3%の割合で発生した。ドセタキセル単独で治療した被験動物では、腫瘍においてアポトーシスを検出しなかった。代わりに、腫瘍の大部分(10 mg/kgのドセタキセルで治療したグループでは88%、20 mg/kgのドセタキセルで治療したグループでは100%)で、正常の細胞周期が進行した(図2a及び表3)。
【表3】
【0107】
様々な治療終了後に、腫瘍細胞の増殖能力をさらに評価するために、パラフィン切片を抗BrDu抗体で染色した。腫瘍サンプルを10%NBF中で固定し、パラフィン包埋し、及びスライド上に5μmで切片化した。脱パラフィン化及び再水和の後に、95℃で2×5分間で0.01 Mクエン酸(pH 6.0)を用いて抗原を回収した。スライドを30分間冷却した後、PBSで逐次すすいだ。メタノール中で15分間、0.3%H2O2でインキュベートすることにより、内因性ペルオキシダーゼ活性をクエンチングした。0.05%Tween 20(PBST)を含むPBS中で、1.5%正常ヤギ血清で1時間ブロックした後、スライドをマウス抗BrdU抗体(1μg/ml)で1時間インキュベートした。その後、ビオチン化したヤギ抗マウスIgGと連続して30分間インキュベートし、ペルオキシダーゼ標識したアビジン(Santa Cruz Biotechnology)と30分間インキュベートし、ジアミノベンジジン(DAB)/過酸化水素クロモーゲン基質(Vector Laboratories, Burlingame, CA)と5〜10分間インキュベートした。すべてのインキュベーションステップは室温で行った。スライドはヘマトキシリン(Sigma)を用いて対比染色し、パーマウント(Fisher Scientific, Fair Lawn, New Jersey)を用いてマウントした。陰性対照として、抗BrdU一次抗体の代わりにマウスIgG(Vector Laboratories)を使用した。Zeiss顕微鏡下でスライドを観察し、デジタル画像を入手した。数多くのBrdUで標識した核小体及び総核小体は、各切片につき10個の無作為視野から集めた。増殖指標は、BrdU陽性の核小体数を、総核小体数で割ることにより算出した。スライドにつき、10区域をカウントした。ヘマトキシリン及びエオシン(Richard Allen, Kalamazoo, MI)を用いることにより、H&E染色を行った。
【0108】
ドセタキセル及びA12で治療した被験動物では、BrDu取り込みが、同一用量のドセタキセル単独で治療したものよりも顕しく減少した(図3)。BrDu取り込みに関するこれらのデータは、細胞周期及びアポトーシスに関する上記所見とも一致しており、Al2がドセタキセルの細胞毒性を顕しく増強したことを示唆する。
【0109】
抗IGF-IR抗体と組み合わせたドセタキセルvs.ドセタキセル単独で治療した腫瘍における遺伝子発現の示差的制御
Al2によるドセタキセルの効果の著しい増強でとり得るメカニズムを調べるために、免疫組織化学分析及びフローサイトメトリー分析により、採取した腫瘍の全てにおいて、IGF-IR発現を調べた。全ての治療群で、又は対照群(データは示してない)と比較して、表面IGF-IR発現の差はなかった。20 mg/kgのドセタキセル及びA12を併用して20 mg/kgのドセタキセルを投与した患者から得た腫瘍において、cDNAマイクロアレイ分析を用いて、治療後の遺伝子発現を調べた。SAM解析では、ドセタキセル及びA12の併用療法を受けた腫瘍において、ドセタキセル単独療法を受けた腫瘍と比較して、示差的に発現したものとして49個の遺伝子を同定した。この際、発現レベルの差は2倍を超えており、過誤発見率(FDR)は10%未満であった(データを示していない)。13個の遺伝子が、アポトーシス又は細胞周期の制御に関与している可能性があることを同定した(表4)。13個の遺伝子は全て、2つの治療間で少なくとも2倍異なっており、FDRは0.02%未満であった。ドセタキセル単独で治療した腫瘍と比べ、ドセタキセル及びA12で治療した腫瘍では、9個の遺伝子がダウンレギュレートされており、4個の遺伝子がアップレギュレートされていた。
【表4】
【0110】
選択した遺伝子について、リアルタイムRT-PCRで結果を確認した。標的cDNAの基準となるPCR断片を精製した。リアルタイムRT-PCRのために、10 ng/μl〜103 pg/μlの一連の標準希釈液を使用して、標準曲線を作成した。プールした腫瘍の各グループから得た1μgの総RNAを、Superscript第一鎖合成システム(Invitrogen)による第一鎖cDNA合成のために使用した。1μlのcDNA第一鎖、特異的なプライマーセット、及びLightcycler FastStart DNA Master Plus SYBR Greenを含有する反応混合物20μl中で、製造業者の指示書(Roche, Nutley, NJ)に従いRoche Lightcyclerを用いて、リアルタイムRT-PCRを行った。Lightcycler software v3.5を用いて、RT-PCR産物の融解曲線分析を行った。アンプリコンサイズは、アガロースゲル電気泳動で確認した。それぞれのサンプルは、2回ずつアッセイした。結果を図4に示す。
【0111】
ダウンレギュレートされた遺伝子のうち、TUBBはドセタキセル耐性をもたらすことがわかっており(Tanaka et al., 2004, Int. J. Cancer 111, 617-26)、BIRC5 (survivin)の発現増加は、強力な前立腺癌及び抗アンドロゲン療法に対する耐性と関係することがわかっている(de Angelis et al., 2004, Int. J. Oncol. 24, 1279-88; Zhang et al., 2005, Oncogene 24, 2474-82)。そのうえ、TUBBはIGF-IR介在型形質転換に関与するIGF-IR制御遺伝子である(Loughran et al., 2005, Oncogene 24, 6185-93)。4つのアップレギュレートされた遺伝子のうち、IGFBP3はIGF-リガンドのシグナル伝達を阻害するだけでなく、リガンド依存的な様式で前立腺腫瘍細胞におけるアポトーシスを誘導することがわかっている(Grimberg et al., 2000, J. Cell. Physiol. 183, 1-9)。
【0112】
治療後のA12の血清レベル
A12を併用してドセタキセルを投与した被験動物において、A12の血清レベルを測定した。血清A12レベルは、治療終了後2週では100倍減少し、治療終了後4週では、非常に低いレベルで検出した(図5)。
【0113】
総合的な細胞毒性
ドセタキセル及びIMC-A12の同時投与による細胞毒性を調べた。A12は、マウスIGF-IRと95%超の交差反応性を有するが、薬剤の併用又はドセタキセル単独で治療した被験動物では、対照群の腫瘍を有する被験動物と比べて、異常な日常活動や挙動の変化は全く観察しなかった。細胞周期及びアポトーシスアッセイの両方では、どの治療群でも腎細胞に対する有意な影響を認めなかった(データは示していない)。体重変化の有意な変化も、治療群間で認めなかった(図6)。
【0114】
骨転移に対する抗IGF-IR抗体療法
骨中の前立腺癌細胞の転移増殖に対する抗IGF-IR抗体による治療の効果を、SCIDマウスの頸骨に直接注射した前立腺癌細胞を用いて評価した。この方法によって、循環系からの化学走化性依存性浸潤には頼らずに、転移性腫瘍を直接定着させた。多様な腫瘍株を、骨転移を定着させるために利用することができる。これらには、PC-3、LuCaP35、骨融解性病変を産生するLnCaP細胞、及び造骨性病変を産生するLuCaP 23.1が含まれる。
【0115】
LuCaP23.1細胞は、IGF-IRを発現する細胞であるが、骨環境下では80%以下の取り込み率を有しており、造骨性反応をもたらす。予備実験において、LuCaP23.1サンプルでは、腫瘍における骨体積対組織体積(%BV/TV)が、対照群の頸骨の場合と比べ有意に増加した(対照群の254〜503%, p=0.024)。頸骨におけるLuCaP23.1腫瘍は全て、正常サンプルでは存在しなかった新しい骨梁及び正常の骨髄と置き換えられた多数の腫瘍病巣を提示した。いくつかの被験体では、腫瘍及び骨成長がもとの骨の外側に伸長した。LuCaP23.1サンプルの%BV/TVの増加は、去勢術後にも観察した;腫瘍化した頸骨の%BV/TVは、腫瘍化していない頸骨の212〜354%であった(p=0.024)。LuCaP23.1の内部頸骨の異種移植片で観察された結果は、腫瘍細胞により新規の骨形成が促進されることを示す。さらに、腫瘍は、骨芽球性骨転移のヒトサンプルと多くの類似点を示し、該類似点には多数の腫瘍病巣及び食塩骨基質量の増加が含まれる。
【0116】
IMC-Al2による治療の効果を評価するために、LuCaP23.1異種移植片腫瘍をSCIDマウスに移植し、血清PSAレベルを隔週で測定して腫瘍増殖を評価した。頸骨腫瘍細胞の移植2週間前に、全ての被験動物を去勢させた。試験マウスへのIMC-A12の投与は、血清PSAレベルが5〜10 ng/mlに達した(腫瘍が定着したことを示す)時に、開始した。40 mg/kgのIMC-A12を、6週間かけて週に3回、腹腔内注射(i.p.)した。
【0117】
腫瘍化した頸骨及び腫瘍のない対側の頸骨の骨塩濃度(BMD)を、二重X線吸収分析法(PIXImus Lunar densitometer)で測定した。該方法は、腫瘍細胞注射部位の2.5 mm×2.5 mmのエリアで行うか、又は対側頸骨のうち移植部位に対応する部位で行った。隔週の病変の評価は、血清PSAの測定によって行った。去勢後の血清PSAレベルに基づいて対照群で骨病変の再発を確認した(LuCaP35は>60 ng/ml、LuCaP23.1は>500 ng/ml)時、X線撮影で骨病変が出現した時、又は被験動物が易感染性となった時に、全ての被験動物を屠殺した。被験動物を屠殺する1時間前に、腫瘍細胞の増殖をモニターするために、BrdUを注射した。屠殺する前にX線撮影(FaxitronX-ray MX-20)を行い、屠殺時に両方の頸骨のBMDを測定した。
【表5】
【0118】
IMC-Al2で治療したマウスでは、血清PSAレベルが有意に低かった(図7)。また、骨芽球性転移腫瘍の増殖に関係するBMDの増加も有意に減少した(表5)。腫瘍化していない下肢のBMD測定により、IMC-A12治療では骨密度を喪失(骨粗しょう症)させないことがわかった。IMC-Al2で治療したマウス及び治療していないマウスのX線撮影画像は、治療したマウスでは、腫瘍増悪を有意に減少又は予防したことを示す(図8)。
【0119】
骨転移に対する抗IGF-IR抗体及びドセタキセルの併用
頸骨腫瘍注射の2週間前に、SCIDマウスを去勢させた。LuCaP23.1前立腺癌細胞をマウスの頸骨に直接注射することにより骨転移を発生させ、造骨性病変に成長させた。異種移植片はIGF-IRを発現する。腫瘍増殖を評価するために、隔週で血清PSAレベルを測定する。血清PSAレベルが5〜10 ng/mlに達し、腫瘍の定着が示された時に、被験動物を4つのグループに無作為割付けした。
【0120】
2つのグループでは、40 mg/kgのIMC-A12を6週間にわたり、週に3回腹腔内注射した。そして、1つのグループにはIMC-Al2+ドセタキセル20 mg/kg(i.p.)を6週間にわたり週に1回投与し、第2のグループには、IMC-A12+ドセタキセル10 mg(i.p.)を6週間にわたり週に3回投与した。対照群には、IMC-A12なしで10又は20 mgのドセタキセル(i.p.)を投与した。
【0121】
被験動物は、PSAを毎週測定してモニターした。治療終了後は、ドセタキセル単独グループの腫瘍が腫瘍の再増大を示すまで、毎週PSA測定をして被験動物のモニターを続けた。ドセタキセル単独グループにおいて、(未治療の被験動物よりも遅い速度ではあるが)PSA値が上昇した時にも、IMC-A12+ドセタキセルで治療したマウスにおけるPSAレベルは安定状態にあり、いくつかの被験動物では低下し始める。治療終了から6週間後でさえ、PSAレベルの低下を継続して観察した。
【0122】
上記のとおり、移植時及び屠殺時にBMD測定を行い、屠殺する直前にX線撮影を行う。IMC-A12+ドセタキセルで治療したグループでは、少量のBMDの増加を示すか、又はBMDが全く増加せず、X線撮影画像では骨芽細胞活性の兆候がわずかであるか、又は全くないことを示す。
【0123】
骨転移に対する抗IGF-IR抗体及びドセタキセルの併用
LuCaP23.1ヒト前立腺腫瘍小片(20〜30 mm3)を機械的に消化した。2〜5×105個のLuCaP23.1生細胞を、6〜8週令のSCIDマウスの頸骨に注射した。3つのグループに無作為割付した21匹のマウスを、この試験では使用した。腫瘍注射後に、血清PSAを毎週モニターした。血清PSAレベルが5〜10 ng/mlに達し、腫瘍増殖が示された時に治療を開始した。グループ1には、対照溶媒である生理食塩水緩衝液を投与した。グループ2には、20 mg/kgのドセタキセルを4週間かけて週に1回投与(i.p.)した。グループ3には、4週間にわたり、20 mg/kgのドセタキセルを週に1回及び40mg/kgのAl2を週3回投与(i.p.)した。治療反応が、骨芽球性であるか又は骨融解性であるか決定するために、全ての治療終了時に、被験動物のDexaスキャン及びX線撮影によってBMDを測定した。
【0124】
ドセタキセル単独又はA12を併用したドセタキセルは、LuCaP23.1腫瘍増殖を有意に阻害した。この結果は、血清PSAレベルの抑制に反映されており(図9a)、2つの治療間で有意な差は認めなかった。しかしながら、ドセタキセル単独で治療した被験動物では、治療終了後に血清PSAが上昇し始め、腫瘍の再増殖を示した;一方、併用療法を受けた被験動物では、血清PSAレベルの持続した抑制を観察し、治療後に長時間の腫瘍静止が示された。血清PSAレベルは、骨密度(BMD)及びX線撮影でみた腫瘍化した骨のサイズと相関することが示された(図9b)。対照群、ドセタキセル20、及びA12を併用したドセタキセル20で治療した被験動物における5週後の平均骨密度は、それぞれ0.112±0.01、0.09±0.02、及び0.05±0.009(平均±SEM)であった。治療群では、明らかに骨密度が減少傾向にあった。
【0125】
実施例2
骨髄穿刺液により誘導されるAktリン酸化
Cambrex (PoieticsTM Donor Program)より、健常男性ドナー(18〜45歳)から入手した骨髄サンプルの供給を受けた。サンプルを1,500 rpmで遠心分離することにより、可溶性相と細胞性相を分離した。連続して0.8μm及び0.22μmのフィルターを用いて、上澄液を濾過した。50μlの骨髄穿刺液を1 mlの溶媒を用いて(最終希釈1:20)、細胞に投与した。
【0126】
血清の存在下で行った実験では、細胞を、10%FBS及び50μg/mlのゲンタマイシンを補填したDMEM中で、骨髄に曝露する前に24時間インキュベートした。血清の不在下(飢餓細胞)での実験では、細胞をPBSで2回洗浄し、成長培地を無血清DMEMで置き換え、細胞を、骨髄調製物に曝露する前に4時間インキュベートした。使用時に、PDGF受容体の特異的阻害剤であるAG-1296(Rice et al., 1999, Amer. J. Path. 155, 213- 21)を添加して、骨髄穿刺液にさらす前に30分間培養した。IMC-3G3抗体を、下記の治療期間の前に投与した。
【0127】
Aktによる骨髄活性化を、PDGFRαを発現するPC3-ML細胞中で検出したが、受容体を欠くDU-145細胞では検出しなかった。1つの実験では、Akt活性化に対する血清成分の影響を最小限とするために、細胞を、無血清培地中で4時間、前もってインキュベートした。骨髄抽出物の添加により、PC3-ML細胞では強力なAktリン酸化が生じたが、DU-145細胞ではAktリン酸化が生じなかった(図10A)。反応の有意性を評価するために、血清を用いて第2の実験を行った。PC3-ML細胞中では、骨髄穿刺液によるAktリン酸化の強力な刺激を、血清の存在下でも観察した(図10B)。DU-145細胞では、ほんの小さな反応しか現れなかった。
【0128】
PDGFRα介在性Aktリン酸化
骨芽細胞及び破骨細胞は、PDGF-AA及びPDGF-BBの両方を分泌するが、骨髄の可溶性環境では、これらの成長因子を与えると考えられている。骨髄抽出物に対するPC3-ML細胞の反応性が、PDGFRαを介したシグナル伝達と関係するか否かを決定するために、20μMのAG-1296の不在下又は存在下で、PC3-ML細胞を骨髄穿刺液にさらした。この濃度のAG-1296は、PDGF-BB誘導性のAkt活性化を完全に阻害した(図11A)。AG-1296は、骨髄穿刺液誘導性のAkt活性化を、40%を超えて阻害した(図11B及びD)。このことは、骨髄誘導性Akt活性化の大半においてPDGFRαシグナル伝達が役割と果たすことを示している。
【0129】
PDGFRαシグナル伝達に対するPDGF-AA及び-BBの直接的な関与についても、骨髄穿刺液のその他の成分と比較して評価した。3名の異なるドナーから入手した骨髄穿刺液中のPDGF-AA及び-BBの濃度を測定したところ、400 pg/ml〜2 ng/mlであった。骨髄穿刺液の20倍希釈液を得て、20〜100 pg/ml濃度のPDGF-AA及び-BBに、実際に試験細胞をさらした。PC3-ML細胞を、100 pg/mlのPDGF-AA及び-BBでそれぞれ適切に処理した。Aktリン酸化は、骨髄穿刺液で得たものの10%未満であった(図3C及びD)。従って、PDGFRαシグナル伝達によるAkt経路の活性化は、PDGF-AA及び-BB以外のPDGFRαリガンド並びに/又はリガンドの直接結合によるPDGFRαの活性化以外の機構と、関係することが明らかである。
【0130】
抗PDGFRα抗体によるAktリン酸化の阻害
中和抗体IMC-3G3は、ヒトPDGFRαに特異的であるが、PC3-ML細胞のAktリン酸化を阻害するその能力についても試験した。30分間のプレインキュベーション及び濃度20μg/mlのIMC-3G3で、30 ng/mlのPDGF-BBによる刺激効果を中和した(図12A)。抗体による処理も、骨髄誘導性のAktリン酸化を約40%阻害した(図12B及びC)。Aktリン酸化に対するIMC-3G3の阻害効果は、プレインキュベーション時間に依存しており、30分間のインキュベーション(12B及びC)よりも、120分間のインキュベーションのほうが、著しく有効であることも観察した(図12D)。1つの考えられる説明は、IMC-3G3がPDGFRαの内在化を誘導しており、その阻害効果は、リガンド結合の遮断だけでなく、細胞膜からの受容体の除去にも関係しているというものである。
【0131】
実施例3
ヒト抗PDGFRα抗体の単離
ヒト抗PDGFRαモノクローナル抗体を、標準的ハイブリドーマ技術(Harlow & Lane, ed., Antibody: A Laboratory Manual, Cold Spring Harbor, 211-213 (1998);これらは、引用により本明細書に包含される)により、ヒトγ重鎖及びκ軽鎖免疫グロブリンを発現するトランスジェニックマウス(Medarex Inc., Sunnyvale, CA)を用いて作製した。ヒトPDGFRα細胞外ドメイン(ECD)は、R&D Systems (Minneapolis, MN)から購入した。PDGFRαを安定して発現する3×107個のブタ大動脈内皮細胞(PAE Rα)を皮下投与(s.c.)して、KMマウスを免疫した。4週間後、マウスを、完全フロインドアジュバント中50μgのPDGFRαECD(s.c.)でブーストし、更に3×107のPAE Rα細胞(i.p.)を加えた。3週間毎にさらに2回、不完全フロインドアジュバント中25μgのPDGFRαECDで、マウスをブーストした。
【0132】
高い血清結合力価及び遮断力価を有するマウス由来の脾臓細胞を単離し、骨髄腫細胞と融合させた。遮断活性を提示するハイブリドーマ培養物をサブクローニングし、これらのハイブリドーマ由来の抗体を、プロテインGクロマトグラフィーで精製した。
【0133】
PDGFRαとの結合について、直接結合アッセイでIgGを評価した。PBSに溶解したPDGFRαECDを、96-ウェルプレート上に固定した(100 ng/ウェル)。その後、プレートをPBST(PBS+0.05%Tween 20)で洗浄し、PBSM(PBS中3%ミルク;200μL/ウェル)で、25℃で2時間ブロックした。PBSM中で希釈したIgGを、固定したPDGFRαECDと25℃で1時間インキュベートした後、プレートをPBSTで洗浄した。PBSM中1:5,000に希釈した第2抗体(ヤギF(ab')2 抗ヒトIgG-セイヨウワサビペルオキシダーゼコンジュゲート;BioSource International, Camarillo, CA)を25℃で1時間加えた。PBSTでプレートを洗浄した後、TMBペルオキシダーゼ基質(KPL, Gaithersburg, MD)を加え、100μLの1 mol/L H2SO4で反応を停止させた。マイクロプレートリーダー(Molecular Devices, Sunnyvale, CA)を用いて、A450 nmでプレートを読み取った。
【0134】
固相PDGF遮断アッセイ(Duan et al., 1991, J. Biol. Chem. 266:413-8を参照されたい:該文献は、引用により本明細書に包含される)を利用して、PDGFの遮断を評価した。PDGFRαECDをPBS中で希釈し、これで96-ウェルマイクロタイタープレート(放射線照射したタンパク質結合性ポリスチレンのImmulon 2HB 1×12 Removawell平坦ストリップ; Dynex Technologies, Chantilly, VA)上を被覆した。それぞれのウェルは、総体積100μL中60 ngのPDGFRαで、25℃で3時間被覆した。その後、プレートを2回洗浄し、25 mmol/L HEPES (pH 7.45)、0.5%ゼラチン、100 mmol/L NaCl、及び0.1%Tween 20を用いて、4℃で一晩遮断した。その後、プレートを25℃で20分間温め、結合緩衝液(25 mmol/L HEPES (pH 7.45)、0.3%ゼラチン、100 mmol/L NaCl、0.01% Tween 20)で1回洗浄した。50力価のIgGをそれぞれのウェルに加え、25℃で30分間インキュベートした。ヨウ化PDGFを結合緩衝液で希釈し、それぞれのウェルに加えた(50μLの1 nmol/L溶液)プレートを25℃で2時間インキュベートした後、結合緩衝液で5回洗浄した。それぞれのウェルはガンマカウンタでカウントした。細胞ベースの遮断アッセイを、Heldin et al., 1988, EMBO J. 7, 1387-93に記載のとおり行った。
【0135】
PDGFRαとの抗体結合反応速度を、BIAcore 3000装置(BIAcore, Inc., Piscataway, NJ)を用いて測定した。PDGFRαECDをセンサーチップ上に固定し、様々な濃度で抗体を注入した。それぞれの濃度でセンサーグラムを得て、BIA Evaluation 2.0 programを用いて評価を行い、速度定数を決定した。親和性定数Kdは速度定数の比Koff/Konから算出した。
【0136】
図13は、ELISAでの、固定したPDGFRαECDに対するヒトモノクローナル抗体IMC-3G3の用量依存的結合を示す。PDGFRαECDに対する最大結合の50%に必要な抗体濃度は、0.06 nmol/Lであった(表6)。BIAcore装置上で表面プラズモン共鳴により測定したところ、ED50は抗体におけるKdと一致した(表6)。モノクローナル抗体は、[125I]PDGF-BBが固定した受容体と結合するのを、IC50 0.43 nmol/Lで遮断した。PDGFRα上でのPDGF-AAとPDGF-BBの結合部位は、構造上同一の場所ではない。この結果は、3G3のエピトープが、両方の成長因子の結合部位と空間的に重複していることを示唆している。
【表6】
【0137】
受容体のリン酸化及び下流エフェクター分子の活性化の阻害
IMC-3G3によるPDGF誘導性細胞内シグナル伝達に対する効果を、PAE Rα細胞を用いて調べた。細胞を6-ウェルファルコン組織培養プレート(250,000細胞/ウェル)に播種し、一晩成長させた。その後ウェルをすすぎ、無血清培地中でインキュベートした。細胞を静止させるために一晩インキュベートした後、該細胞を抗体で、37℃で30分間処理した。その後、PDGF-AA又はPDGF-BBを加え、37℃でさらに10分間インキュベートした。そして、細胞を剥離し、200μLの溶解緩衝液(50 mmol/L Tris-HCl (pH 8.0)、1%Triton X-100、150 mmol/L NaCl、1 mmol/L EDTA、0.1%SDS、1 mmol/L o-バナジウム酸ナトリウム及びプロテアーゼ阻害剤(Complete Mini、Roche、Mannheim、Germany))中で溶解した。細胞溶解物を、増強された化学ルミネセンス試薬及びHyperfilm(Amersham Biosciences)を用いて、SDS-PAGE及びウェスタンブロット法により解析した。
【0138】
リガンド誘導性の受容体チロシンリン酸化を阻害する能力について、抗体を試験した。PDGF-AA及びPDGF-BBは、PDGFRαチロシンリン酸化を、それぞれ1 nmol/Lの濃度及び3 nmol/Lの濃度で約5倍増加させる。高濃度(10 nmol/L)のリガンドは、おそらくはリガンド誘導性の分解によって、リン酸化した受容体を減少させる。該抗体は、PDGF-BB誘導性の受容体を阻害して、バックグラウンドに近づけた(図14Aの一番上の列)。受容体のリン酸化を誘導するPDGF-AAを用いた場合にも、同様のデータが得た。
【0139】
PDGFは、有糸分裂シグナルを伝達させて、下流エフェクタータンパク質を介して受容体を発現する細胞上で抗アポトーシス作用を発揮する。そのため、MAPK p44/p42及びAkt(それぞれ、細胞成長及び抗アポトーシス経路に関与する)の活性化を阻害するその能力について、モノクローナル抗体を調べた。抗PDGFRα抗体は、PDGF-BB (図2A)及びPDGF-AA(図示してない)に応答したMAPK及びAktの両方のリン酸化を阻害した。PDGFRαのリン酸化の阻害は用量依存的であり、0.25 nmol/Lで50%を阻害した(図14B)。
【0140】
抗有糸分裂活性
抗PDGFRαモノクローナル抗体を、PDGFR-AAによって誘導されるPAE Rα細胞の有糸分裂を遮断するその能力について、試験した。細胞を96-ウェル組織培養プレート(1×104細胞/ウェル)に播種し、100μL培地/ウェル中で一晩生育した。その後、ウェルを無血清培養液ですすぎ、それぞれのウェルに75μL無血清培地を加え、細胞を一晩、血清欠乏状態とした。IgGを加え(25μL/ウェル)、プレートを30分間、37℃でインキュベートした。その後、PDGF-AA又はPDGF-BB (25μL/ウェル)を加え、プレートを18〜20時間、37℃でインキュベートした。それぞれのウェルに0.25μCi[3H]チミジン(25μL/ウェル)を加えた後、プレートをさらに4時間インキュベートした。抗体、PDGF、及び[3H]チミジンは、全て無血清培地中で希釈した。その後、1%ウシ血清アルブミンを加えたPBSで細胞を洗浄し、トリプシン(100μL/ウェル)による処理で剥離した。フィルター上に細胞を集め、MACH III細胞回収機(Tomtec, Inc., Hamden, CT)を用いて、再蒸留水でスライスを洗浄した。濾過処理した後、DNAに取り込まれた放射能をシンチレーションカウンタ(Wallac Microbeta, model 1450)で計測した。
【0141】
IMC-3G3を血清欠乏状態のPAE Rα細胞に加えた場合、PDGF-AA誘導性のチミジン取り込みは、EC50 8.3 nmol/Lで特異的に阻害された(図15)。該抗体は、3 nmol/L PDGF-BBによって誘導性されるPAE Rα細胞の有糸分裂も、EC50 1.25 nmol/Lで阻害した(データは示していない)。
【0142】
PDGFRαを発現するヒト腫瘍細胞株の増殖阻害
PDGFRαを発現するヒト腫瘍細胞株を試験して、悪性増殖に対するヒト抗PDGFRα抗体の効果をin vitro及びin vivo系で測定した。フローサイトメトリーで測定した際にPDGFRαを発現するような2つの腫瘍細胞株は、SKLMS-1(平滑筋肉腫)及びU118(神経膠芽腫)である。これらの細胞株も、有糸分裂アッセイにおいてリガンドと反応し、マウスにおいて腫瘍を形成する。SKLMS-1は、パラクリン刺激だけでなく、オートクリン刺激も行う可能性がある。SKLMS-1は、培養して生育した際に、定量的サンドウィッチ酵素イムノアッセイ法(R&D Systems)により、PDGF-AAタンパク質を発現することがわかった。
【0143】
図16Aからわかるように、IMC-3G3は、SKLMS-1細胞のPDGF-AA刺激に応答したAkt及びMAPKの両方のリン酸化を阻害した。Aktリン酸化の阻害は100%であり、MAPKの阻害は約80%であった。該抗体は、U118細胞におけるリン酸化の有効な阻害剤でもある(図16B)。リガンド誘導性の腫瘍細胞の有糸分裂も遮断した。抗PDGFRα抗体を血清欠乏状態のU118細胞に加えた場合、PDGF-AA誘導性のチミジン取り込みは、EC50 3.4 nmol/Lで特異的に阻害された(図17A)。該抗体は、SKLMS-1細胞のPDGF-AA誘導性の有糸分裂応答をEC50 5 nmol/L(図17B)で阻害するだけでなく、PDGF-BB刺激性有糸分裂応答も阻害する(図17C)。U118細胞では、PDGF-BB刺激性有糸分裂応答のわずかな部分的阻害(66 nmol/Lで40%;図17D)を観察した。これは、それらの細胞において、PDGFRα及びPDGFRβの両方が発現しているためである(データは示していない)。
【0144】
腫瘍異種移植片の成長阻害
無胸腺ヌードマウス中、神経膠芽腫(U118)及び平滑筋肉腫(SKLMS-1)皮下(s.c.)異種移植片モデルにおいて、IMC-3G3をin vivoで試験した。皮下の腫瘍異種移植片は、Matrigel (Collaborative Research Biochemicals, Bedford, MA)中で混合した10×106 SKLMS-1又はU118細胞を、雌の無胸腺ヌードマウス(Crl:NU/NU-nuBR, Charles River Laboratories, Wilmington, MA)に注射することにより、定着させた。腫瘍は、平均腫瘍体積(π/6×最長径×垂直方向の高さ2)が約400 mm3になるまで成長させた。マウスを5つのグループに無作為割付けし(n = 12)、試験期間中は週に2回i.p.注射して処置した。グループ1のマウスは、対照溶媒(0.9%NaCl;洗浄用のUSP, B/Braun)で処置した。グループ2〜4のマウスは、6、20、及び60 mg/kgの当該抗PDGFRα抗体で処置した。グループ5のマウスは、60 mg/kgのヒトIgG(Sigma)で処置した。6、20、又は60 mg/kgの抗PDGFRα抗体又はヒトIgGで処置したグループでは、初回用量をそれぞれ21.4、71.4、及び 214 mg/kgで与えた。初回用量は、週2回の投与レジュメンを用いて、第1の用量(消失半減期7日間)から定常状態の血清濃度を達成するように計算した。週に2回腫瘍体積を評価し、治療群における腫瘍増殖を、繰り返しANOVAを測定して比較した。
【0145】
図18Aに示すとおり、生理食塩水で処置したマウス(P = 0.74)と比べ、ヒトIgGは神経膠芽腫の成長に対し何も効果を及ぼさなかった。一方、抗PDGFRα抗体は、用量6 mg/kg(P = 0.06)、20 mg/kg(P = 0.03)、及び60 mg/kg(P =0.0004)で、腫瘍増殖を有意に阻害した。U118試験の最後において、%T/C [(試験終了時に3G3で処置したグループの平均腫瘍体積/治療開始時の平均腫瘍体積)/(試験終了時に未処理のグループの平均腫瘍体積/試験開始時の平均腫瘍体積)×100]値は、用量6、20、及び60 mg/kgの3G3で処置したグループでは、それぞれ67%、63%、及び35%であった。さらに、6、20、及び60 mg/kgの処置グループにおいて、4/12、5/11、10/12匹の被験動物で腫瘍の消退を観察した。一方、いずれの対照群でも、腫瘍の消退は認めなかった。
【0146】
図18Bは、6 mg/kg(P = 0.02)、20 mg/kg(P = 0.003)、及び60 mg/kg(P < 0.0001)の処置により、平滑筋肉腫の成長も有意に阻害されたことを示す。最終的な%T/C体積の値は、6、20、及び60 mg/kgの処置群において、それぞれ66%、57%、及び31%であり、腫瘍消退は認めなかった。
【0147】
試験終了時における異種移植片の組織学的検査は、対照療法を受けた被験動物から得た腫瘍と比べ、処置した被験動物から得た腫瘍では明らかなに差を示した。切除した腫瘍を、QDL固定液中で、4℃で24時間固定した。パラフィン包埋した後、4μmに切片化し、ホルマリン固定した切片をMayer's H&E(Richard Allen, Kalamazoo, MI)で染色した。
【0148】
最も高い用量(60 mg/kg)で処置したU118グループでは、生存している腫瘍細胞は非常にわずかしか認めなかった。また、生理食塩水の対照群と比べ、実質的により多くの細胞低密度領域が存在した(図18C)。25日目の処置したSKLMS-1異種移植片では、生理食塩水の対照群と比べ、生存している腫瘍細胞量及び細胞密度の減少も認めた(図18D)。
【0149】
神経膠芽腫株のPDGFRα介在性刺激のin vitro阻害
U118腫瘍における受容体ホスホチロシンのレベルを、抗PDGFRα抗体又はヒトIgGで処置してから1週間後に評価した。定着したU118腫瘍(500 mm3)を有するマウスを、初回用量214 mg/kgで処置し、72時間後に維持用量60 mg/kgの抗体で処置した。第1の抗体注射から1週間(168時間)後に(平均して、腫瘍消退が観察される前の時点に;図18Aを参照されたい)、マウスから腫瘍を採取し、リン酸化アッセイ溶解緩衝液中でホモジナイズした(上記参照)。溶解物を14,000 rpmで2回遠心分離し、回収した上澄液のタンパク質濃度を測定した(Bio-Radタンパク質アッセイ, Bio-Rad, Hercules, CA)。それぞれのサンプルからの溶解物(4 mg)を、抗PDGFRα抗体を用いて免疫沈降させた。その後、免疫沈降させたヒトPDGFRαを、抗PDGFR又は抗ホスホチロシン抗体のいずれかでイムノブロットした。図19は、これらの腫瘍において、抗PDGFRα抗体の投与が、ヒトIgG対照と比べPDGFRαホスホチロシンのレベルを減少させることを示す。
【0150】
細胞株作製技術
はじめに、ヒト抗PDGFRα抗体の重鎖及び軽鎖可変ドメインをコードする遺伝子をクローニングし、配列決定した。一連のプライマーは、MEDAREX由来のハイブリドーマ中で、ヒト免疫グロブリン可変領域配列の5'及び3'フランキング配列にアニーリングするMEDAREXから得た。重鎖可変領域は、プライマー対AB88(フォワード)及びAB90(リバース)(表7)を用いて増幅した。軽鎖産物は、フォワードプライマーAB182及びリバースプライマーAB16(表7)を含むプライマー対を用いて増幅した。これらの0.4kb反応産物をベクターZeroBlunt (Invitrogen)にクローニングして、AB88-1(VH)及びAB182-3(Vκ)を作製し、挿入物をユニバーサルT7及びM13Rプライマーを用いて配列決定した。
【表7】
【0151】
完全IgG1抗体を発現するプラスミドベクターを作製するために、クローニングした可変領域をPCR増幅し、定常領域遺伝子を含有する発現ベクターに2段階でライゲートした。最初のPCR重鎖増幅では、プライマーIPHF5(フォワード)及びIPHR5(リバース)の鋳型として、25 ngのプラスミドAB88-1を利用した。第2のPCR重鎖増幅では、鋳型として5μlの一次反応物並びにプライマーOPSIF及びIPHR5を利用した。効果的な免疫グロブリンのプロセシング及び分泌のために、2つのフォワードプライマーを併用して、19アミノ酸のマウス重鎖遺伝子シグナル配列(MGWSCIILFLVATATGVHS;配列番号24)をコードする免疫グロブリン遺伝子の5’末端に57塩基対を付加する。さらに、フォワードプライマーOPSIFは、哺乳動物細胞中でのこれらの遺伝子の効率的な翻訳開始のためにコンセンサス「Kozak」配列(J MoI. Biol. 196:947)を付加し、増幅産物のクローニングのために5'HindIII制限エンドヌクレアーゼ部位を、好適な発現ベクターに付加する。重鎖リバースプライマーは、定常領域ベクターにクローニングするためのフレーム内NheI部位を包含する。
【0152】
PCRは、Expand PCRキット(Boehringer Mannheim Inc.)を製造業者の説明書に従い用いて、Expand緩衝液システム#3を、以下のサイクリング条件で50μlの反応物中で利用し、2段階行った。
【0153】
1サイクル 94℃ 2分
5サイクル 94℃ 20秒
48℃ 60秒
68℃ 2分
20サイクル 94℃ 20秒
65℃ 60秒
68℃ 2分
1サイクル 68℃ 5分
【0154】
2ラウンドのPCR後に、PCR産物をアガロースゲル電気泳動した後に精製し、HindIII-Nhel消化断片としてベクターpDFcにクローニングした(図8)。該ベクターには、ヒトγ1定常領域が含まれる。
【0155】
最初のPCR軽鎖増幅では、鋳型として25 ngのpAB182-3プラスミド、プライマーIPLF4(フォワード)及びIPLR2(リバース)を利用した。第2のPCR軽鎖増幅では、鋳型として5μlの一次反応物並びにプライマーOPSIF及びIPLR2を利用した。重鎖では、2つのフォワードプライマーが分泌シグナル配列をもたらす。軽鎖リバースプライマーは、κ定常領域ベクターpLckにクローニングするために、フレーム内BsiWI部位を含む(図8)。上記の重鎖について、PCR反応を行った。2ラウンドのPCR後、アガロースゲル電気泳動した後にPCR産物を精製し、pLckにクローニングした。該ベクターには、ヒトκ軽鎖定常領域が含まれる。
【表8】
【0156】
安定したトランスフェクションを目的とした単一のプラスミドベクターを作製するために、CMVプロモーター、重鎖コード領域、及びポリA要素を含む重鎖発現カセットを、NotI-SalI断片としての軽鎖ベクターにクローニングした(図20)。
【0157】
その後、この構築物を利用して、骨髄腫細胞株NSO細胞中で、安定した産生株を作製した。BioRad Gene PulserIIを用いて、エレクトロポーレーションを行って、発現プラスミドをNSO細胞にトランスフェクトさせた。トランスフェクション前に、プラスミドDNAをPvulで線状化し、エタノール沈降させ、濃度0.4 mg/ml(lOOμlのdH2O中40μg)で懸濁した。最終体積800μl中で、250ボルトの単一パルス400μFdにより、40μgのDNAを細胞にエレクトロポーレートした。エレクトロポーレートした細胞を、50μlのアリコートで、10%透析ウシ胎児血清(dFCS) (Hyclone, Lot#: AHA7675)及び2 mMグルタミン(Invitrogen/Life Technologies)を含有するDMEM培地(JRH Biosciences Inc.)中に、5,000〜10,000細胞/ウェル濃度で約18個の96-ウェルプレートのウェルに分散させた。グルタミン合成酵素(GS)陽性トランスフェクション体の選択は、10%dFCSを含有し1×GS補填剤(JRH Biosciences Inc.)を補填した無グルタミンDMEMを添加してから2時間後に開始した。コロニーの成長及び拡大を可能とするために、細胞を37℃、5%CO2雰囲気下で2〜4時間後インキュベートした。300個を超えるコロニーを、抗ヒトFc(γ)ELISA (A450 nmでのセイヨウワサビペルオキシダーゼ検出)を用いて、スクリーニングした。3〜5日間かけて培養し、抗体発現コロニー(58%)を拡大させて、再現性について再び試験した。細胞を無血清培地に適用するために、それぞれの継代培養で等しい体積の無血清GS-OS培養液を添加して、陽性細胞株を拡大させた。3日-サブコンフルエントな24ウェル培養物中で25μg/ml以上を産生する強陽性物を、さらなる分析のために拡張して、無血清培地に完全に適合させた。
【0158】
本明細書に開示した発明の原理の範囲内での変更が、当業者によりなされ得ることが理解され、また期待される。また、そういった変更は、本発明の範囲内に包含することが意図される。
【特許請求の範囲】
【請求項1】
配列番号2で表されるCDRH1;配列番号4で表されるCDRH2;配列番号6で表されるCDRH3;配列番号10で表されるCDRL1;配列番号12で表されるCDRL2;及び配列番号14で表されるCDRL3からなる群から選択される1以上の相補性決定領域を含み、PDGFRαに特異的な、単離されたヒト抗体又は抗体フラグメント。
【請求項2】
配列番号2で表されるCDRH1;配列番号4で表されるCDRH2;及び配列番号6で表されるCDRH3を含む、請求項1に記載の抗体又は抗体フラグメント。
【請求項3】
配列番号8を含む、請求項1に記載の抗体又は抗体フラグメント。
【請求項4】
配列番号10で表されるCDRL1;配列番号12で表されるCDRL2;及び配列番号14で表されるCDRL3を含む、請求項1に記載の抗体又は抗体フラグメント。
【請求項5】
配列番号16を含む、請求項1に記載の抗体又は抗体フラグメント。
【請求項6】
配列番号2で表されるCDRH1;配列番号4で表されるCDRH2;配列番号6で表されるCDRH3;配列番号10で表されるCDRL1;配列番号12で表されるCDRL2;及び配列番号14で表されるCDRL3を含む、請求項1に記載の抗体又は抗体フラグメント。
【請求項7】
配列番号8及び配列番号16を含む、請求項1に記載の抗体又は抗体フラグメント。
【請求項8】
PDGFRαと選択的に結合する、請求項1〜7のいずれか1項に記載の抗体又は抗体フラグメント。
【請求項9】
PDGFRαが、PDGFRαのリガンドに結合するのを阻害する、請求項1〜7のいずれか1項に記載の抗体又は抗体フラグメント。
【請求項10】
PDGFRαを中和する、請求項1〜7のいずれか1項に記載の抗体又は抗体フラグメント。
【請求項11】
一本鎖抗体、Fab、一本鎖Fv、ジアボディ(diabody)、及びトリアボディ(triabody)からなる群から選択される、請求項1〜7のいずれか1項に記載の抗体又は抗体フラグメント。
【請求項12】
請求項1〜11のいずれか1項に記載の抗体又は抗体フラグメントのコンジュゲート。
【請求項13】
抗腫瘍剤、標的分子又はレポーター分子を含む、請求項12に記載のコンジュゲート。
【請求項14】
抗体又は抗体フラグメントをコードし、配列番号1で表されるCDRH1;配列番号3で表されるCDRH2;配列番号5で表されるCDRH3;配列番号9で表されるCDRL1;配列番号11で表されるCDRL2;及び配列番号13で表されるCDRL3からなる群から選択される1以上のヌクレオチド配列を含む、単離されたポリヌクレオチド。
【請求項15】
配列番号7を含む、請求項14に記載の単離されたポリヌクレオチド。
【請求項16】
配列番号15を含む、請求項14に記載の単離されたポリヌクレオチド。
【請求項17】
請求項14〜16のいずれか1項に記載のポリヌクレオチドを含む発現ベクター。
【請求項18】
請求項17に記載の発現ベクターを含む、組換え宿主細胞。
【請求項19】
配列番号8を含むポリペプチド及び配列番号16を含むポリペプチドを産生する、請求項18に記載の組換え宿主細胞。
【請求項20】
配列番号8及び配列番号16を含むポリペプチドを産生する、請求項18に記載の組換え宿主細胞。
【請求項21】
哺乳動物においてPDGFRαの活性化を中和する方法であって、有効量の請求項1〜11のいずれか1項に記載の抗体を投与することを含む、前記方法。
【請求項22】
哺乳動物において腫瘍の増殖を阻害する方法であって、治療上有効量の請求項1〜11のいずれか1項に記載の抗体を投与することを含む、前記方法。
【請求項23】
腫瘍がPDGFRαを発現する、請求項22に記載の方法。
【請求項24】
腫瘍がPDGFRαを過剰発現する、請求項22に記載の方法。
【請求項25】
腫瘍が原発性腫瘍である、請求項22に記載の方法。
【請求項26】
腫瘍が転移性腫瘍である、請求項22に記載の方法。
【請求項27】
腫瘍が治療抵抗性の腫瘍である、請求項22に記載の方法。
【請求項28】
腫瘍が血管形成腫瘍である、請求項22に記載の方法。
【請求項29】
腫瘍が、卵巣癌、乳癌、肺癌、肝細胞癌、胃腸間質腫瘍、メラノーマ、腎細胞癌、前立腺癌、及び柔組織肉腫からなる群から選択される、請求項22に記載の方法。
【請求項30】
抗体又は抗体フラグメントが、抗腫瘍剤と組み合わせて投与される、請求項22〜29のいずれか1項に記載の方法。
【請求項31】
抗腫瘍剤が化学療法剤である、請求項30に記載の方法。
【請求項32】
抗腫瘍剤がドキソルビシンである、請求項30に記載の方法。
【請求項33】
抗腫瘍剤が放射線療法である、請求項30に記載の方法。
【請求項34】
抗体又は抗体フラグメントが、第2のPDGFRαアンタゴニストと共に投与される、請求項22〜33のいずれか1項に記載の方法。
【請求項35】
PDGFRαアンタゴニストが、細胞内PDGFRαアンタゴニストである、請求項34に記載の方法。
【請求項36】
治療上有効量の上皮成長因子受容体(EGFR)アンタゴニストを投与することをさらに含む、請求項22〜35のいずれか1項に記載の方法。
【請求項37】
治療上有効量のインスリン様成長因子受容体(IGF-IR)アンタゴニストを投与することをさらに含む、請求項22〜36のいずれか1項に記載の方法。
【請求項1】
配列番号2で表されるCDRH1;配列番号4で表されるCDRH2;配列番号6で表されるCDRH3;配列番号10で表されるCDRL1;配列番号12で表されるCDRL2;及び配列番号14で表されるCDRL3からなる群から選択される1以上の相補性決定領域を含み、PDGFRαに特異的な、単離されたヒト抗体又は抗体フラグメント。
【請求項2】
配列番号2で表されるCDRH1;配列番号4で表されるCDRH2;及び配列番号6で表されるCDRH3を含む、請求項1に記載の抗体又は抗体フラグメント。
【請求項3】
配列番号8を含む、請求項1に記載の抗体又は抗体フラグメント。
【請求項4】
配列番号10で表されるCDRL1;配列番号12で表されるCDRL2;及び配列番号14で表されるCDRL3を含む、請求項1に記載の抗体又は抗体フラグメント。
【請求項5】
配列番号16を含む、請求項1に記載の抗体又は抗体フラグメント。
【請求項6】
配列番号2で表されるCDRH1;配列番号4で表されるCDRH2;配列番号6で表されるCDRH3;配列番号10で表されるCDRL1;配列番号12で表されるCDRL2;及び配列番号14で表されるCDRL3を含む、請求項1に記載の抗体又は抗体フラグメント。
【請求項7】
配列番号8及び配列番号16を含む、請求項1に記載の抗体又は抗体フラグメント。
【請求項8】
PDGFRαと選択的に結合する、請求項1〜7のいずれか1項に記載の抗体又は抗体フラグメント。
【請求項9】
PDGFRαが、PDGFRαのリガンドに結合するのを阻害する、請求項1〜7のいずれか1項に記載の抗体又は抗体フラグメント。
【請求項10】
PDGFRαを中和する、請求項1〜7のいずれか1項に記載の抗体又は抗体フラグメント。
【請求項11】
一本鎖抗体、Fab、一本鎖Fv、ジアボディ(diabody)、及びトリアボディ(triabody)からなる群から選択される、請求項1〜7のいずれか1項に記載の抗体又は抗体フラグメント。
【請求項12】
請求項1〜11のいずれか1項に記載の抗体又は抗体フラグメントのコンジュゲート。
【請求項13】
抗腫瘍剤、標的分子又はレポーター分子を含む、請求項12に記載のコンジュゲート。
【請求項14】
抗体又は抗体フラグメントをコードし、配列番号1で表されるCDRH1;配列番号3で表されるCDRH2;配列番号5で表されるCDRH3;配列番号9で表されるCDRL1;配列番号11で表されるCDRL2;及び配列番号13で表されるCDRL3からなる群から選択される1以上のヌクレオチド配列を含む、単離されたポリヌクレオチド。
【請求項15】
配列番号7を含む、請求項14に記載の単離されたポリヌクレオチド。
【請求項16】
配列番号15を含む、請求項14に記載の単離されたポリヌクレオチド。
【請求項17】
請求項14〜16のいずれか1項に記載のポリヌクレオチドを含む発現ベクター。
【請求項18】
請求項17に記載の発現ベクターを含む、組換え宿主細胞。
【請求項19】
配列番号8を含むポリペプチド及び配列番号16を含むポリペプチドを産生する、請求項18に記載の組換え宿主細胞。
【請求項20】
配列番号8及び配列番号16を含むポリペプチドを産生する、請求項18に記載の組換え宿主細胞。
【請求項21】
哺乳動物においてPDGFRαの活性化を中和する方法であって、有効量の請求項1〜11のいずれか1項に記載の抗体を投与することを含む、前記方法。
【請求項22】
哺乳動物において腫瘍の増殖を阻害する方法であって、治療上有効量の請求項1〜11のいずれか1項に記載の抗体を投与することを含む、前記方法。
【請求項23】
腫瘍がPDGFRαを発現する、請求項22に記載の方法。
【請求項24】
腫瘍がPDGFRαを過剰発現する、請求項22に記載の方法。
【請求項25】
腫瘍が原発性腫瘍である、請求項22に記載の方法。
【請求項26】
腫瘍が転移性腫瘍である、請求項22に記載の方法。
【請求項27】
腫瘍が治療抵抗性の腫瘍である、請求項22に記載の方法。
【請求項28】
腫瘍が血管形成腫瘍である、請求項22に記載の方法。
【請求項29】
腫瘍が、卵巣癌、乳癌、肺癌、肝細胞癌、胃腸間質腫瘍、メラノーマ、腎細胞癌、前立腺癌、及び柔組織肉腫からなる群から選択される、請求項22に記載の方法。
【請求項30】
抗体又は抗体フラグメントが、抗腫瘍剤と組み合わせて投与される、請求項22〜29のいずれか1項に記載の方法。
【請求項31】
抗腫瘍剤が化学療法剤である、請求項30に記載の方法。
【請求項32】
抗腫瘍剤がドキソルビシンである、請求項30に記載の方法。
【請求項33】
抗腫瘍剤が放射線療法である、請求項30に記載の方法。
【請求項34】
抗体又は抗体フラグメントが、第2のPDGFRαアンタゴニストと共に投与される、請求項22〜33のいずれか1項に記載の方法。
【請求項35】
PDGFRαアンタゴニストが、細胞内PDGFRαアンタゴニストである、請求項34に記載の方法。
【請求項36】
治療上有効量の上皮成長因子受容体(EGFR)アンタゴニストを投与することをさらに含む、請求項22〜35のいずれか1項に記載の方法。
【請求項37】
治療上有効量のインスリン様成長因子受容体(IGF-IR)アンタゴニストを投与することをさらに含む、請求項22〜36のいずれか1項に記載の方法。
【図1】

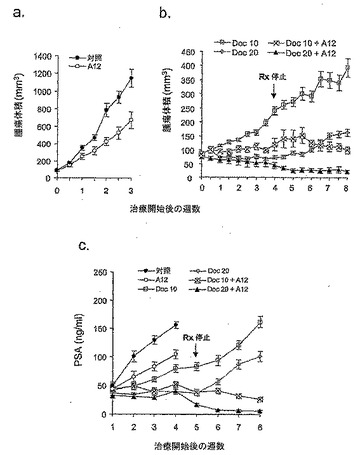
【図2】

【図3】
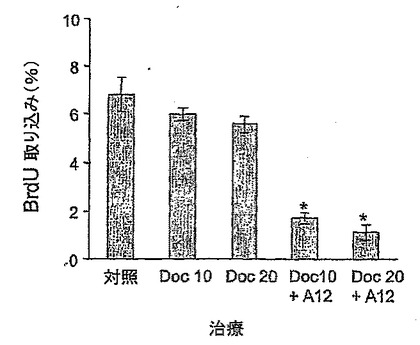
【図4】

【図5】
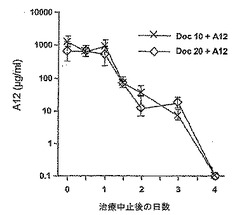
【図6】
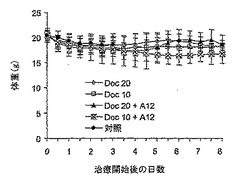
【図7】

【図8】
【図9】
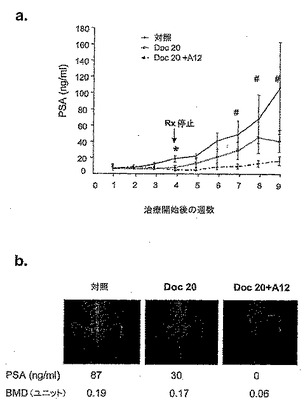
【図10】
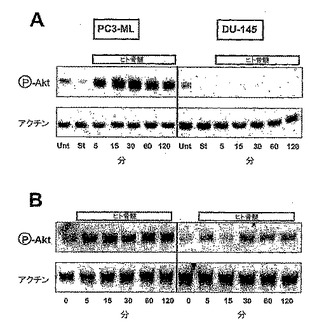
【図11】
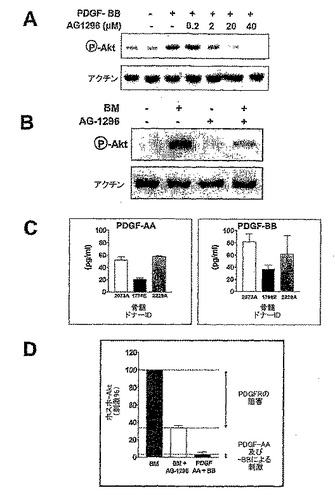
【図12】
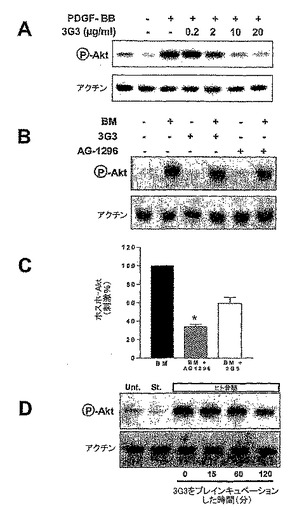
【図13】
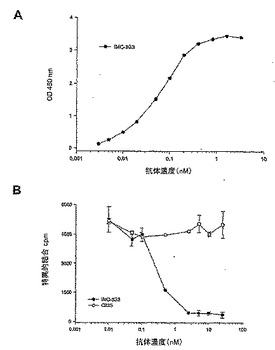
【図14】
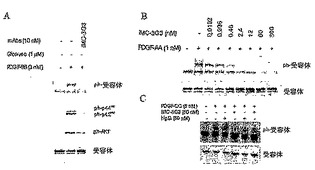
【図15】

【図16】
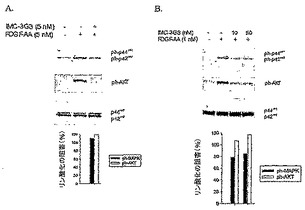
【図17】
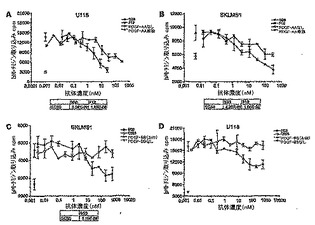
【図18】
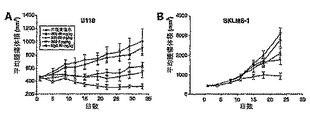
【図19】
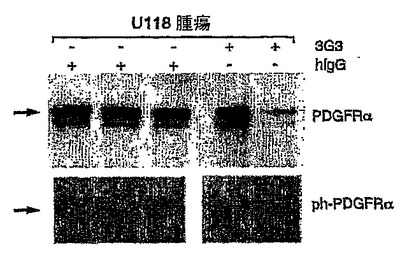
【図20】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【公開番号】特開2012−179060(P2012−179060A)
【公開日】平成24年9月20日(2012.9.20)
【国際特許分類】
【出願番号】特願2012−134040(P2012−134040)
【出願日】平成24年6月13日(2012.6.13)
【分割の表示】特願2009−33517(P2009−33517)の分割
【原出願日】平成18年6月19日(2006.6.19)
【出願人】(508188662)イムクローン・リミテッド・ライアビリティ・カンパニー (23)
【氏名又は名称原語表記】Imclone LLC
【Fターム(参考)】
【公開日】平成24年9月20日(2012.9.20)
【国際特許分類】
【出願日】平成24年6月13日(2012.6.13)
【分割の表示】特願2009−33517(P2009−33517)の分割
【原出願日】平成18年6月19日(2006.6.19)
【出願人】(508188662)イムクローン・リミテッド・ライアビリティ・カンパニー (23)
【氏名又は名称原語表記】Imclone LLC
【Fターム(参考)】
[ Back to top ]