
送達剤のマイクロ粒子またはナノ粒子を含む医薬製剤
【課題】本発明は、送達剤および/または活性剤を含む、マイクロ粒子および/またはナノ粒子に関する。本発明はまた、活性剤および送達剤の固体の放出制御剤形を含む、医薬製剤および固体剤形に関する。
【解決手段】(a)インスリンの治療有効量;および(b)送達剤、を含む固体剤形であって、胃で実質的に崩壊または溶解しないが、小腸では崩壊または溶解する固体剤形。
【解決手段】(a)インスリンの治療有効量;および(b)送達剤、を含む固体剤形であって、胃で実質的に崩壊または溶解しないが、小腸では崩壊または溶解する固体剤形。
【発明の詳細な説明】
【技術分野】
【0001】
本出願は、2004年9月23日に出願した米国仮出願第60/612810号および2004年8月13日に出願した米国仮出願第60/601258号の優先権を主張するものである。両米国仮出願は、参照により本明細書に援用する。
【背景技術】
【0002】
本発明は、医薬製剤および同医薬製剤を調製するための方法に関する。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】米国特許公開番号2005/0143424
【特許文献2】米国仮出願第60/569476号
【特許文献3】国際公開番号WO03/057650
【特許文献4】米国特許第4421685号
【特許文献5】米国特許第5474978号
【特許文献6】米国特許第5534488号
【特許文献7】米国特許第5650386号
【特許文献8】米国特許第5866536号
【特許文献9】国際公開番号WO94/23767
【特許文献10】国際公開番号WO95/11690
【特許文献11】国際公開番号WO95/28920
【特許文献12】国際公開番号WO95/28838
【特許文献13】国際公開番号WO96/10396
【特許文献14】国際公開番号WO96/09813
【特許文献15】国際公開番号WO96/12473
【特許文献16】国際公開番号WO96/12475
【特許文献17】国際公開番号WO96/30036
【特許文献18】国際公開番号WO96/33699
【特許文献19】国際公開番号WO97/131938
【特許文献20】国際公開番号WO97/36480
【特許文献21】国際公開番号WO98/21951
【特許文献22】国際公開番号WO98/25589
【特許文献23】国際公開番号WO98/34632
【特許文献24】国際公開番号WO98/49135
【特許文献25】国際公開番号WO99/16427
【特許文献26】国際公開番号WO00/06534
【特許文献27】国際公開番号WO00/07979
【特許文献28】国際公開番号WO00/40203
【特許文献29】国際公開番号WO00/46182
【特許文献30】国際公開番号WO00/47188
【特許文献31】国際公開番号WO00/48589
【特許文献32】国際公開番号WO00/50386
【特許文献33】国際公開番号WO00/59863
【特許文献34】国際公開番号WO00/59480
【特許文献35】国際公開番号WO01/32130
【特許文献36】国際公開番号WO01/32596
【特許文献37】国際公開番号WO01/34114
【特許文献38】国際公開番号WO01/44199
【特許文献39】国際公開番号WO01/51454
【特許文献40】国際公開番号WO01/70219
【特許文献41】国際公開番号WO01/92206
【特許文献42】国際公開番号WO02/02509
【特許文献43】国際公開番号WO02/15959
【特許文献44】国際公開番号WO02/16309
【特許文献45】国際公開番号WO02/20466
【特許文献46】国際公開番号WO02/19969
【特許文献47】国際公開番号WO02/070438
【特許文献48】国際公開番号WO03/026582
【特許文献49】国際公開番号WO02/100338
【特許文献50】国際公開番号WO03/045306
【特許文献51】国際公開番号WO03/26582
【特許文献52】国際公開番号WO03/057170
【特許文献53】米国特許第5851579号
【特許文献54】米国特許第6458383号
【非特許文献】
【0004】
【非特許文献1】Physicians' Desk Reference(58th Ed., 2004, Medical Economics Company, Inc.、モントベール、ニュージャージー州)
【発明の開示】
【発明が解決しようとする課題】
【0005】
インスリンなどの薬物のための、改良された経口送達システムは、絶えず必要とされている。
【課題を解決するための手段】
【0006】
本発明は、送達剤化合物単独または送達剤化合物と活性剤との組合せを含む、経口投与用のマイクロ粒子および/またはナノ粒子に関する。これらの粒子を含む製剤(ならびに送達剤化合物および活性剤のみを含む粒子のための製剤)は、送達剤化合物と活性剤との単純な混合物を、粉剤、錠剤またはカプセル剤として経口投与するのに比べて、変化の少ない、活性剤の極めて優れたバイオアベイラビリティーを提供する。特定のいずれの理論によっても制限されることなく、少なくともいくつかの実施形態において考えられていることによると、この改良は、(1)胃から、幽門(通常、1000〜2000μmの直径を有する)を経て、粒子の溶解および送達剤を介した薬物の吸収が最もよく起こると考えられている小腸へと移行することが可能になるような小サイズのマイクロ粒子またはナノ粒子によるもの、ならびに(2)送達剤化合物が吸収部位において活性剤と共に存在することを確実にする、粒子中での送達剤化合物と活性剤との間の親密な接触によるものであり得る。従来の錠剤またはカプセル剤が幽門を通って小腸へと入っていくためには最初に溶解して十分に小さい粒子になる必要があるのとは異なり、マイクロ粒子およびナノ粒子は自由に幽門を通って小腸へと入っていくので、錠剤の崩壊に起因する変動、および胃運動によって変わる胃通過が最小限に抑えられる。
【0007】
一実施形態によると、送達剤化合物および活性剤を含む粒子は、約900または1000μm未満の中央粒径(メジアンサイズ)を有する。例えば、中央粒径は、約45〜約850μm、約45〜約150μm、約150〜約250μm、約250〜約425μm、約425〜約850μm、約100〜約1000nmまたは約500〜約1000nmの範囲であることができる。別の実施形態によると、粒子は、約1μm未満の中央粒径を有する。いくつかの実施形態では、粒子は、約1ナノメートルと小さく、約999マイクロメートルと大きい可能性がある。例えば、粒子は、約999マイクロメートル未満、約1ナノメートル〜約999マイクロメートル、約1〜約999マイクロメートル、約1〜約999ナノメートル、約45〜約850マイクロメートル、約45〜約150マイクロメートル、約150〜約250マイクロメートル、約250〜約425マイクロメートル、約425〜約850マイクロメートル、約100〜約1000ナノメートルまたは約500〜約1000ナノメートルの中央粒径を有することができる。
【0008】
別の実施形態は、送達剤化合物および活性剤を含む医薬製剤であり、送達剤化合物は、粒子の形である。粒子は、約999マイクロメートル未満、約1ナノメートル〜約999マイクロメートル、約1〜約999ナノメートルまたは約7〜約16マイクロメートルの中央粒径(メジアンサイズ)を有することができる。任意選択により、活性剤は粒子の形であってもよい。例えば、活性剤粒子の中央粒径は、約999マイクロメートル未満、約1ナノメートル〜約999マイクロメートル、約1〜約999マイクロメートルまたは約1〜約999ナノメートルであることができる。一実施形態によると、送達剤化合物粒子および活性剤粒子はどちらも、約1〜約999マイクロメートルの中央粒径を有する。別の実施形態によると、送達剤化合物粒子および活性剤粒子はどちらも、約1〜約999ナノメートルの中央粒径を有する。
【0009】
さらに別の実施形態は、送達剤および活性剤を含む医薬製剤であり、活性剤は、約999マイクロメートル未満の中央粒径(メジアンサイズ)を有する粒子の形である。一実施形態によると、活性剤粒子の中央粒径は、約1ナノメートル〜約999マイクロメートル、約1〜約999マイクロメートルまたは約1〜約999ナノメートルである。
【0010】
粒子は、微細顆粒またはマイクロビーズ(例えば、丸/ボール形状かつ約0.2mm〜約2.0mmの直径を有するビーズ)の形であることができる。マイクロビーズは、圧縮によって形成され得る。一実施形態では、医薬製剤は、送達剤化合物を含むマイクロビーズを含有し、送達剤化合物は、インスリンまたはヘパリンなどの活性剤で被覆されている。マイクロビーズは、約0.2mm〜約2.0mmの範囲にある直径を有することができる。
【0011】
粒子は、セルロース誘導体(例えば、CMCナトリウム(デラウェア州ウィルミントンのAqualonから入手可能))またはポリアクリル酸(例えば、オハイオ州クリーブランドのB.F.Goodrichから入手可能なCarbopol(登録商標))などの粘膜付着剤を含有してもよい。粘膜付着剤は、(1)粘膜(胃腸管を含む)への付着を促進することによって、送達剤-活性剤の粘膜への接触を延長させること、(2)活性剤(例えば、インスリンの場合)を安定化させ、保護すること、および(3)生体膜(粘膜を含む)の浸透性を増加させることによって、送達を改良し、活性剤のバイオアベイラビリティーを向上させること、が可能である。
【0012】
インスリンを送達剤化合物と同時に、胃で分解しないが腸では分解する固体の経口剤形で経口投与することは、インスリンの極めて優れたバイオアベイラビリティーをもたらすということもまた発見されている。インスリンまたは異なる活性剤を含むそのような固体の経口剤形は、胃で分解する形態および送達剤化合物を含まない形態に比べて、極めて優れたバイオアベイラビリティーを提供する。特定のいずれの理論によっても制限されることなく、この改良は、インスリンおよび他の活性剤の、胃液中に存在する酵素または酸による分解に対する感受性によるものと考えられている。固体の経口剤形は胃で分解しないので、インスリンおよび他の活性剤は、腸に届くまで分解から保護される。
【0013】
本発明の別の実施形態は、治療有効量の活性剤および送達剤を含む医薬製剤(固体の経口剤形など)であり、医薬製剤は、経口投与の場合、約250秒〜約650秒の崩壊時間を有する。別の実施形態では、経口投与の場合、崩壊時間は約350秒〜約550秒である。さらに別の実施形態では、経口投与の場合、崩壊時間は約60秒を超える。さらに別の実施形態では、経口投与の場合、崩壊時間は約400秒を超える。崩壊時間は、米国薬局方<701>に記載されている方法を用いて、水中で37±2℃にて測定することができる。崩壊時間は、約1秒〜約24時間以上もの範囲に及ぶ可能性があり、これは多くの要因に依存する。かかる要因には、特定の活性剤(1種または複数)、送達剤化合物(1種または複数)、および医薬製剤中に含まれる賦形剤が含まれるが、これらに限定されない。
【0014】
別の実施形態は、治療有効量の活性剤および送達剤を含む医薬製剤(固体の経口剤形など)であり、この固体の経口剤形は、胃で実質的に崩壊または溶解しないが、腸で実質的には崩壊または溶解する。好ましい実施形態では、活性剤はインスリンである。別の好ましい実施形態では、活性剤はインスリン誘導体である。
【0015】
別の実施形態では、該医薬製剤は、胃での崩壊を遅延させるために腸溶性コーティングで被覆されている固体の経口剤形である。腸溶性コーティングには、フタル酸ヒドロキシプロピルメチルセルロース、酢酸コハク酸ヒドロキシプロピルメチルセルロース、フタル酸酢酸ポリビニル、酢酸トリメリット酸セルロース、酢酸フタル酸セルロース、ポリ(メタクリル酸-アクリル酸エチル)、およびポリ(メタクリル酸-メタクリル酸メチル)が含まれるが、これらに限定されない。
【0016】
さらに別の実施形態では、本医薬製剤は、崩壊よりもむしろ剤形の表面から侵食されるように製剤し得る。
【0017】
本医薬製剤は、医薬製剤中の活性剤が酵素によって分解されるのを抑制するために、酵素阻害剤を含むことができる。
【0018】
一実施形態では、送達剤は、以下の構造を有する化合物またはこの塩である:
【化1】

【0019】
上記式中、
Arは、フェニルまたはナフチルであり;
Arは、1個もしくは複数の-OH、ハロゲン、C1〜C4アルキル、C1〜C4アルケニル、C1〜C4アルコキシまたはC1〜C4ハロアルコキシで任意選択で置換されていてもよく;
R1は、C3〜C20アルキル、C4〜C20アルケニル、フェニル、ナフチル、(C1〜C10アルキル)フェニル、(C1〜C10アルケニル)フェニル、(C1〜C10アルキル)ナフチル、(C1〜C10アルケニル)ナフチル、フェニル(C1〜C10アルキル)、フェニル(C1〜C10アルケニル)、ナフチル(C1〜C10アルキル)、またはナフチル(C1〜C10アルケニル)であり;
R1は、C1〜C4アルキル、C2〜C4アルケニル、C1〜C4アルコキシ、C1〜C4ハロアルコキシ、-OH、-SH、-CO2R8、またはこれらのいずれかの組合せで、任意選択により置換されていてもよく;
R2は、水素、C1〜C4アルキル、またはC2〜C4アルケニルであり;
R1は、酸素、窒素、硫黄、またはこれらのいずれかの組合せで任意選択により中断されていてもよい。
式A中の用語「2-OH-Ar」は、2位にヒドロキシル基を有するフェニル基またはナフチル基を指す。
【0020】
一実施形態によると、上記化合物は、酸性基に対してアルファ位にあるアミノ基で置換されていない。
【0021】
好ましくは、Arはハロゲンで置換されている。
【0022】
好ましくは、R2は水素である。
【0023】
好ましくは、R1は非置換である。
【0024】
好ましくは、R1は中断されていない。
【0025】
好ましくは、R1はC1〜10、C3〜9、C3〜7、C3、C7またはC9アルキルである。一実施形態によると、R1は分岐していない。
【0026】
好ましい送達剤化合物には、N-(8-[2-ヒドロキシベンゾイル]アミノ)カプリル酸(SNACの遊離酸)、N-(10-[2-ヒドロキシベンゾイル]アミノ)デカン酸(SNADの遊離酸)、4-[(2-ヒドロキシ-4-クロロベンゾイル)-アミノ]ブタン酸(4-CNABの遊離酸)、およびこれらの塩、ならびにこれらの溶媒和物および水和物が含まれるが、これらに限定されない。この塩は、例えば、モノナトリウム塩(すなわちSNAC、SNADもしくは4-CNAB)またはジナトリウム塩などのナトリウム塩であってよい。
【0027】
別の実施形態では、送達剤は、以下の構造を有する化合物またはこの塩である:
【化2】
【0028】
上記式中、
R1、R2、R3およびR4は独立に、H、-OH、ハロゲン、C1〜C4アルキル、C2〜C4アルケニル、C1〜C4アルコキシ、-C(O)R8、-NO2、-NR9R10、または-N+R9R10R11(R12)-であり;
R5は、H、-OH、-NO2、ハロゲン、-CF3、-NR14R15、-N+R14R15R16(R13)-、アミド、C1〜C12アルコキシ、C1〜C12アルキル、C2〜C12アルケニル、カルバミン酸エステル、炭酸エステル、尿素、または-C(O)R18であり;
R5は、ハロゲン、-OH、-SH、または-COOHで任意選択で置換されていてもよく;
R5は、O、N、Sまたは-C(O)-で任意選択により中断されていてもよく;
R6は、C1〜C12アルキレン、C2〜C12アルケニレンまたはアリーレンであり;
R6は、C1〜C4アルキル、C2〜C4アルケニル、C1〜C4アルコキシ、-OH、-SH、ハロゲン、-NH2または-CO2R8で任意選択により置換されていてもよく;
R6は、OまたはNで任意選択により妨害されていてもよく;
R7は、結合またはアリーレンであり;
R7は、-OH、ハロゲン、-C(O)CH3、-NR10R11、または-N+R10R11R12(R13)-で任意選択により置換されていてもよく;
R8は、H、C1〜C4アルキル、C2〜C4アルケニル、または-NH2であり;
R9、R10、R11およびR12は独立に、HまたはC1〜C10アルキルであり;
R13は、ハライド、ヒドロキシド、サルフェート、テトラフルオロボレートまたはホスフェートであり;
R14、R15およびR16は独立に、H、C1〜C10アルキル、-COOHで置換されているC1〜C10アルキル、C2〜C12アルケニル、-COOHまたは-C(O)R17で置換されているC2〜C12アルケニルであり;
R17は、-OH、C1〜C10アルキル、またはC2〜C12アルケニルであり;かつ
R18は、H、C1〜C6アルキル、-OH、-NR14R15、N+R14R15R16(R13)-である。
【0029】
さらに別の実施形態では、送達剤は、以下の構造を有する化合物またはこの塩である:
【化3】
【0030】
上記式中、
R1、R2、R3、R4およびR5は独立に、H、-CN、-OH、-OCH3、またはハロゲンであり、少なくともR1、R2、R3、R4およびR5のうちの1つは-CNであり;
R6は、C1〜C12の直鎖または分枝の、アルキレン、アルケニレン、アリーレン、アルキル(アリーレン)またはアリール(アルキレン)である。
【0031】
さらに別の実施形態では、送達剤は、以下の構造を有する化合物またはこの塩である:
【化4】
【0032】
上記式中、
Xは各々、水素、ハロゲン、ヒドロキシル、またはC1〜C3アルコキシであり、
Rは、置換もしくは非置換のC1〜C3アルキレン、または置換もしくは非置換のC2〜C3アルケニレンであり、
nは、1から4までの整数である。
【0033】
さらに別の実施形態では、送達剤は、以下の構造を有する化合物またはこの塩である:
【化5】
【0034】
上記式中、
Xは、ハロゲンであり、Rは、置換もしくは非置換のC1〜C3アルキレン、または置換もしくは非置換のC2〜C3アルケニレンである。
【0035】
好ましい送達剤化合物には、N-(8-[2-ヒドロキシベンゾイル]-アミノ)カプリル酸、N-(10-[2-ヒドロキシベンゾイル]-アミノ)デカン酸、8-(2-ヒドロキシ-4-メトキシベンゾイルアミノ)オクタン酸、8-(2-ヒドロキシ-5-クロロベンゾイルアミノ)-オクタン酸、4-[(2-ヒドロキシ-4-クロロベンゾイル)アミノ]ブタン酸、および医薬として許容できるこれらの塩が含まれるが、これらに限定されない。本発明の医薬製剤は、上記送達剤化合物のいずれか、または他の送達剤化合物のいずれかを、単独で、または1種もしくは複数の追加の送達剤化合物と組み合わせて含むことができる。
【0036】
好適な活性剤には、タンパク質、ポリペプチド、ペプチド、ホルモン、多糖類およびこれらの合成由来、天然由来または組換え由来物:すなわち、成長ホルモン;成長ホルモン放出ホルモン;成長ホルモン放出因子、インターフェロン;インターロイキン-1;インターロイキン-2;亜鉛、ナトリウム、カルシウムおよびアンモニウムを含む対イオンを任意選択で有していてもよいインスリン;インスリン様成長因子;ヘパリン;カルシトニン;エリスロポエチン;心房性ナトリウム利尿因子;抗原;モノクローナル抗体;ソマトスタチン;プロテアーゼ阻害剤;副腎皮質刺激ホルモン、ゴナドトロピン放出ホルモン;オキシトシン;黄体形成ホルモン放出ホルモン;卵胞刺激ホルモン;グルコセレブロシダーゼ;トロンボポエチン;フィルグラスチム;プロスタグランジン;サイクロスポリン;バソプレシン;クロモリンナトリウム;バンコマイシン;デスフェリオキサミン;ビスホスホネート;副甲状腺ホルモン;抗片頭痛剤;グルカゴン様ペプチド1(GLP-1);抗菌薬;ビタミン;およびこれらの化合物の、類似体、フラグメント、模倣体もしくはポリエチレングリコール(PEG)修飾誘導体;またはこれらの任意の組合せが含まれるが、これらに限定されない。好ましい活性剤には、インスリンおよびヘパリン(未分画ヘパリンおよび低分子量ヘパリンを含むが、これらに限定されない)が含まれるが、これらに限定されない。
【0037】
本発明の一実施形態では、活性剤はインスリンである。本発明の含インスリン医薬製剤はまた、第2の血糖降下薬、腎ブドウ糖再吸収阻害剤、または上述の任意の組合せ(例えば、参照により本明細書に援用する米国特許公開番号2005/0143424に記載されているものなど)を含むことができる。好適な第2の血糖降下薬には、インスリン分泌促進薬、インスリン抵抗性改善薬、インスリン模倣体、α-グルコシダーゼ阻害剤、糖形成抑制剤および上述のいずれかの任意の組合せが含まれるが、これらに限定されない。一実施形態によると、固体剤形には、スルホニル尿素、メグリチニド類似体、ビグアニド(好ましくはメトホルミン)、または前記いずれかの任意の組合せが含まれる。好ましい実施形態によると、固体剤形はメトホルミンを含む。
【0038】
また、本発明のマイクロ粒子またはナノ粒子を含み、および/または上記の崩壊時間を有する、固体単位剤形などの医薬製剤も提供される。単位剤形は、錠剤、カプセル剤、粉剤またはサシェの形態であることが可能である。単位剤形は、1種もしくは複数の、腸溶性コーティング、崩壊剤、スーパー崩壊剤(デンプングリコール酸ナトリウムまたはクロスカルメロースナトリウムなど)および超粒子スーパー崩壊剤を、単独でまたは組み合わせて有することができる。
【0039】
一実施形態では、固体の経口単位剤形は、速崩壊性錠剤である。別の実施形態では、固体単位剤形は、放出制御または遅延放出を有する。
【0040】
一実施形態によると、本発明は、上記粒子および崩壊剤を含む錠剤を提供する。一実施形態では、崩壊剤は、デンプングリコール酸ナトリウム(英国サウスハンバーサイド州のAzebe UK Ltd.から入手可能なPrimojel(登録商標))、クロスカルメロースナトリウム(英国サウスハンバーサイド州のAzebe UK Ltd.から入手可能なPrimellose(登録商標))または超粒子スーパー崩壊剤などのスーパー崩壊剤である。
【0041】
別の実施形態は、治療有効量のインスリンおよび送達剤化合物を含む固体剤形であり、固体剤形は、経口投与した場合、少なくとも60秒の崩壊時間を有する。固体剤形は、腸溶性コーティングを有するか、または表面侵食製剤であってよい。固体剤形は、1種または複数の酵素阻害剤をさらに含むことができる。
【0042】
さらに別の実施形態は、治療有効量のインスリンおよび送達剤化合物を含む固体剤形であり、固体剤形は、胃で実質的に崩壊または溶解しないが、小腸では崩壊または溶解する。固体剤形は、腸溶性コーティングを有するか、または表面侵食製剤であってよい。固体剤形は、1種または複数の酵素阻害剤をさらに含むことができる。
【0043】
別の実施形態は、活性剤を、動物、特にその活性剤を必要としている動物に投与するための方法であり、本発明のマイクロ粒子またはナノ粒子および/または上記の崩壊時間を有するもの(すなわち、放出制御もしくは持続放出を有するもの)を含む医薬製剤を投与することによる方法である。経口投与が好ましい投与経路である。
【0044】
さらに別の実施形態は、本発明のマイクロ粒子またはナノ粒子を含む固体単位剤形、および/または上記の崩壊時間を有する(すなわち、放出制御もしくは持続放出を有する)固体単位剤形を含有する本発明の医薬製剤を投与することによって、動物における、疾患を治療する方法または所望の生理学的効果を得るための方法である。さらに別の実施形態は、本発明の医薬製剤を経口投与することによって、経口による活性剤のバイオアベイラビリティーを向上させる方法である。
【0045】
さらに別の実施形態は、哺乳動物に、本発明の含インスリン医薬製剤、例えば本発明のマイクロ粒子またはナノ粒子を含むものおよび/または上記の崩壊時間を有するものの治療有効量を投与することによって、哺乳動物(例えばヒト、特にこれを必要としているヒト)における、糖尿病を治療する方法および/またはインスリンの連用に伴う全身性高インスリン血症の発生率を減少させる方法である。一実施形態では、送達剤化合物は、4-CNABの遊離酸または医薬として許容できるその塩である。本医薬製剤は、連用を基礎として投与され得る。
【0046】
さらに別の実施形態は、哺乳動物に、本発明の含インスリン医薬製剤、例えば本発明のマイクロ粒子またはナノ粒子を含む医薬製剤および/または上記の崩壊時間を有する医薬製剤などの治療有効量を投与することによって、哺乳動物(例えばヒト、特にこれを必要としているヒト)における、耐糖能の低下、初期の糖尿病もしくは後期の糖尿病を治療する方法、またはグルコース恒常性を得る方法である。一実施形態では、送達剤化合物は、4-CNABの遊離酸または医薬として許容できるその塩である。本医薬製剤は、連用を基礎として投与され得る。
【0047】
さらに別の実施形態は、本明細書に記載されている、含インスリン医薬製剤の治療有効量を、ヒト糖尿病患者に連用を基礎として経口投与することによって、ヒト糖尿病患者を治療する方法である。
【0048】
さらに別の実施形態は、送達剤化合物および活性剤の溶液を、例えば固体が形成されるまで乾燥させることによって、ならびに任意選択で粒子を単離することによって、本発明のマイクロ粒子およびナノ粒子を調製する方法である。好ましくは、混合物は均質である(例えば、送達剤化合物および活性剤は、混合物中で均一に分散されている)。この方法は、送達剤化合物、活性剤および溶媒の混合物の同時乾燥を含む。好適な溶媒には、ヒドロキシル系溶媒、水、およびこれらの混合物が含まれるが、これらに限定されない。一実施形態によると、混合物は、約10〜約40℃で(例えば室温で)乾燥される。好ましくは、乾燥は制御された温度で行う。一実施形態によると、乾燥は不活性ガス(好ましくは窒素ガス)上で行う。乾燥した材料は、所望の粒径を得るために、任意選択で粉砕および/または篩にかけることも可能である。この方法により、送達剤化合物および活性剤の均質な混合物を含む粒子を得ることができる。
【0049】
本発明のマイクロ粒子およびナノ粒子を調製する別の方法は、送達剤化合物、活性剤および溶媒の混合物を凍結乾燥することによるものである。好適な溶媒には、ヒドロキシル系溶媒、水、およびこれらの混合物が含まれるが、これらに限定されない。
【0050】
本発明のマイクロ粒子およびナノ粒子を調製するさらに別の方法は、(1)送達剤化合物および活性剤を超臨界流体中に溶解すること、ならびに(2) 系の圧力を低下させて、送達剤化合物および活性剤を極めて微細な粒子として析出させることによるものである。この析出は、超臨界溶液が急速膨張した結果である。
【0051】
以下の実施形態は、「固体の医薬組成物の実施形態」として本明細書でまとめて参照される。
【0052】
本発明の上記特徴および付随する他の多くの利点は、以下の詳細な説明を、添付図面と併せて参照することによって、よりよく理解されるようになるであろう。
【図面の簡単な説明】
【0053】
【図1】胃および空腸への直接投与について描いた概略図である。
【図2】共処理したマイクロ粒子を胃および空腸に直接投与した後のインスリン濃度(±SEM)を経時的に示すグラフである。
【図3】共処理したマイクロ粒子を胃および空腸に直接投与した後の血糖値の変化(±SEM)を経時的に示すグラフである。
【図4】以下の3種の異なる剤形から強制経口投与した後の血糖の変化(±SEM)を経時的に示すグラフである:1)インスリンおよび担体を圧縮することによって製造した錠剤、2)共処理したインスリンおよび担体のマイクロ粒子を含むカプセル剤、ならびに3)インスリンおよび担体の単純混合物を含むカプセル剤。
【図5】以下の3種の異なる剤形から強制経口投与した後のインスリン濃度(±SEM)を経時的に示すグラフである:1)インスリンおよび担体を圧縮することによって製造した錠剤、2)共処理したインスリンおよび担体のマイクロ粒子を含むカプセル剤、ならびに3)インスリンおよび担体の単純混合物を含むカプセル剤。
【図6】共処理したインスリンおよび担体のマイクロ粒子を含むカプセル剤を強制経口投与した後のインスリン濃度(±SEM)を経時的に示すグラフである。ラット10匹中2匹が極めて高いインスリン吸収を示した。これら2匹の高い応答例を含めた平均値(N=10)および含めなかった平均値(N=8)についてグラフに描く。
【図7】共処理したインスリンおよび担体のマイクロ粒子を含むカプセル剤を強制経口投与した後の血糖の変化(±SEM)を経時的に示すグラフである。ラット10匹中2匹が極めて高いインスリン吸収を示した。これら2匹の高い応答例を含めた平均値(N=10)および含めなかった平均値(N=8)についてグラフに描く。
【図8】共処理したインスリンおよび担体を胃および空腸にin situ投与して得られた推定絶対的バイオアベイラビリティー(±SEM)を示すグラフである。以下の2種の組成物を評価した:1)インスリン(0.25mg/kg)+送達剤(37.5mg/kg)、および2)インスリン(0.5mg/kg)+送達剤(75mg/kg)。
【図9】1)皮下投与、2)胃への直接投与、3)空腸への直接投与、4)インスリンおよび担体を圧縮することによって製造した錠剤、5)共処理したインスリンおよび担体のマイクロ粒子を含むカプセル剤(2匹の高い応答例を含めたものおよび含めなかったもの)、ならびに6)インスリンおよび担体の単純混合物を含むカプセル剤、から得られたインスリン濃度の推定絶対的バイオアベイラビリティー(±SEM)を示すグラフである。
【図10】1)胃への直接投与、2)空腸への直接投与、3)インスリンおよび担体を圧縮することによって製造した錠剤、4)共処理したインスリンおよび担体のマイクロ粒子を含むカプセル剤(2匹の高い応答例を含めたものおよび含めなかったもの)、ならびに5)インスリンおよび担体の単純混合物を含むカプセル剤、から得られた、門脈におけるインスリン濃度の推定バイオアベイラビリティー(±SEM)を示すグラフである。
【図11】1)胃への直接投与、2)空腸への直接投与、3)インスリンおよび担体を圧縮することによって製造した錠剤、4)共処理したインスリンおよび担体のマイクロ粒子を含むカプセル剤(2匹の高い応答例を含めたものおよび含めなかったもの)、ならびに5)インスリンおよび担体の単純混合物を含むカプセル剤から得られた、皮下投与と比較したインスリンの推定バイオアベイラビリティー(±SEM)を示すグラフである。
【図12】1)胃への直接投与、2)空腸への直接投与、3)インスリンおよび担体を圧縮することによって製造した錠剤、4)共処理したインスリンおよび担体のマイクロ粒子を含むカプセル剤(2匹の高い応答例を含めたものおよび含めなかったもの)、ならびに5)インスリンおよび担体の単純混合物を含むカプセル剤、から得られた、門脈におけるインスリンの、皮下投与と比較した推定バイオアベイラビリティー(±SEM)を示すグラフである。
【図13】共処理したインスリンおよび担体のマイクロ粒子を含むカプセル剤を強制経口投与した後の個別のインスリン濃度を経時的に示すグラフである。ラット14およびラット17が極めて高いインスリン吸収を示した。これら2匹の高い応答例を含めた平均値(N=10)および含めなかった平均値(N=8)をグラフに描く。
【図14】共処理したインスリンおよび担体のマイクロ粒子を含むカプセル剤を強制経口投与した後の個別の血糖の変化を経時的に示すグラフである。ラット14およびラット17が極めて高いインスリン吸収を示した。これら2匹の高い応答例を含めた平均値(N=10)および含めなかった平均値(N=8)をグラフに描く。
【図15】共処理したインスリンおよび担体のマイクロ粒子を含むカプセル剤を強制経口投与した後の個別のインスリン濃度を経時的に示すグラフである。ラット14およびラット17を除外した。N=8から得られた平均値をグラフに描く。
【図16】共処理したインスリンおよび担体のマイクロ粒子を含むカプセル剤を強制経口投与した後の個別の血糖の変化を経時的に示すグラフである。ラット14およびラット17を除外した。N=8から得られた平均値についてグラフに描く。
【図17】インスリンおよび送達剤を含む、実施例の製剤1〜6を摂取させたアカゲザルにおける血清ブドウ糖濃度の変化を経時的に描くグラフである。これらの製剤は異なる崩壊時間を有する。
【図18】インスリンおよび送達剤を含む、実施例の製剤1〜6を摂取させたアカゲザルにおける血清インスリン濃度の変化を経時的に描くグラフである。これらの製剤は異なる崩壊時間を有する。
【図19】実施例10に記載したSNAD/ヘパリン製剤を投与した後の、サルにおける抗因子Xa活性(U/ml)対時間を示すグラフである。
【発明を実施するための最良の形態】
【0054】
[定義]
本明細書に記載されている「粒子」、「マイクロビーズ」、および「顆粒」は、任意の形状であってよく、送達剤化合物および/または活性剤に加えて、1種または複数の成分を含んでいてもよい。所定の任意の粒子、マイクロビーズ、または顆粒の具体的な成分は、用いる方法によって決まることもあり、1バッチ分由来の、個々の粒子、マイクロビーズまたは顆粒において、必ずしも同一であるわけではない。
【0055】
例えば、活性剤の粒子、マイクロビーズ、または顆粒を、送達剤化合物の粒子、マイクロビーズ、または顆粒と別々に調製する場合は、活性剤の粒子、マイクロビーズ、または顆粒は、一般に送達剤化合物を含まず、送達剤化合物の粒子、マイクロビーズ、または顆粒は、一般に活性剤を含まない。ただし、各粒子、マイクロビーズ、または顆粒は、本明細書に開示されているように他の成分を含んでもよい。
【0056】
他の実施形態では、粒子、マイクロビーズ、または顆粒は、溶液、懸濁液、または混合物から形成されることができ、この溶液、懸濁液、または混合物は、限定するものではないが、液体または乾燥形態であって、少なくとも活性剤および送達剤化合物を含む。したがって、例えば所定の任意の粒子、マイクロビーズ、または顆粒は、活性剤および送達剤化合物の両方を含み、1種または複数の他の成分をさらに含んでもよい。
【0057】
用語「直径」および「中央粒径(メジアン粒径)」は、一般に、粒子、マイクロビーズ、および顆粒の寸法を指すために使用される。「中央粒径(メジアン粒径)」または「直径」は、実施例8、9、10について、以下の通り測定した。
機器: Mastersizer 2000 (EQ202、モデルMS2K、製造番号34315-67)
製造会社: MALVERN instruments、英国
ソフトウェア: Mastersizer 2000
付属品: Scirocco 2000 (A) (モデルADA2000、製造番号34270/73)
分散: 乾式分散
解析モデル: 汎用
粒子RI: 1.520
曇り度: 1〜6%
基準: サンプル分散ユニットのためのMalvern品質監査基準
【0058】
Malvern Mastersizer 2000では、レーザー回折およびモデルフィッティングによって粒径が測定される。任意の2相系(例えば、粉末、懸濁液、または乳液)に良好に分散させたサンプルを、サンプル中に存在する粒径に適した長さのレンズで集光させたHe-Neレーザーの通り道に導入する。レーザーの通り道にある粒子の散乱パターンが、多数配置された検出器で測定される。各検出器によって、特定の範囲の角度からのデータが測定される。
【0059】
Malvern装置では、測定される粒子が完全な球形であると仮定されている。非球形粒子については、結果として得られる粒径分布は、他の原理に基づく方法で得られる粒径分布とは異なる可能性がある。コンピュータによる測定は、これに伴って顕微鏡による調査を行い、調査している粒子の種類を決定しなければならないことがよくある。不均整に成形された粒子については、Mastersizer 2000から得られた粒径データは、測定された粒子と同体積の仮想球体の直径として解釈される。(注:d(0.1)は、サンプルの10%がこのサイズ未満の粒径であり、d(0.5)は、サンプルの50%がこのサイズ未満の粒径であり、d(0.9)は、サンプルの90%がこのサイズ未満の粒径である。)
【0060】
一般に、この装置は例えば1個の粒子がレーザーを通り過ぎる最中に、1つの寸法を測定する;すなわち、この装置は粒子を貫通する直線の長さを測定する。不均整な粒子については、この装置を用いると、レーザーに対する粒子の配向により、個々の粒子の最長寸法、最短寸法または他の寸法のいずれかを1回しか測定しないことになるため、結果にばらつきが生じる。しかし、多数の粒子が採寸され、平均直径または中央粒径が算出される。したがって、「粒径」または「直径」の数字は、粒子の平均の「粒径」または「直径」の推定値である。別の方法として、「直径」または「粒径」は、実施例1に記載の篩法によって測定される。「直径」は、ある実施形態では、例えば粒子は端が丸くなっているか、概ね球状を有する可能性があるが、球状または丸い寸法を必ずしも意味するわけではないものと解釈されるべきである。
【0061】
本発明は、「粒径」または「直径」という狭い範囲内に入る粒子、マイクロビーズまたは顆粒に限定されないということも理解されるべきである。したがって、例えば、いくつかの実施形態では、少なくとも使用される成分および方法に応じて、例えば、同バッチにおいて、ナノメートルスケールとマイクロメートルスケールの両方の範囲内に入る多少の粒子を含む可能性がある。実際の個々の粒子の「粒径」または「直径」は、比較的狭い範囲内、または比較的広い範囲内に入る可能性がある。
【0062】
本明細書および添付の特許請求の範囲で使用する場合、単数形(英文では、「a」、「an」および「the」)は、文脈が他に特に明確に指示しない限り、複数の指示対象を含む。したがって、例えば、「粒子」という言及は1種または複数の粒子を含み、「活性剤」は1種または複数のかかる活性剤を含み、「送達剤」は1種または複数の送達剤を含む。
【0063】
用語「約」は一般に、所定の値または範囲の10%以内、好ましくは5%以内、およびより好ましくは1%以内を意味する。
【0064】
本明細書で使用する用語「水和物」には、(i)分子の形で結合した水を含む物質、および(ii)1個もしくは複数の結晶水分子を含む結晶質、または自由水を含む結晶性物質が含まれるが、これらに限定されない。
【0065】
本明細書で使用する用語「溶媒和物」には、溶媒の分子またはイオンと、送達剤化合物もしくはこれらの塩、もしくはこれらの溶媒和物、もしくはこれらの水和物の分子またはイオンとの分子錯体またはイオン錯体が含まれるが、これらに限定されない。
【0066】
用語「送達剤」は、本明細書に開示されている任意の送達剤化合物を指す。
【0067】
用語「SNAC」は、N-(8-[2-ヒドロキシベンゾイル]-アミノ)カプリル酸の一ナトリウム塩を指し、他に特に指示がない限り、2004年5月6日に出願した米国仮出願第60/569476号(参照により本明細書に援用する)に記載されている一ナトリウム塩の種々の多形体を含む。
【0068】
用語「SNAD」は、他に特に指示がない限り、N-(10-[2-ヒドロキシベンゾイル]-アミノ)デカン酸の一ナトリウム塩を指す。用語「SNADのジナトリウム塩」は、N-(10-[2-ヒドロキシベンゾイル]-アミノ)デカン酸の二ナトリウム塩を指す。
【0069】
用語「5-CNAC」は、他に特に指示がない限り、N-(8-[2-ヒドロキシ-5-クロロベンゾイル]-アミノ)オクタン酸の一ナトリウム塩を指す。
【0070】
用語「4-CNAB」は、N-4-[(2-ヒドロキシ-4-クロロベンゾイル)アミノ]ブタン酸の一ナトリウム塩を指し、他に特に指示がない限り、国際公開番号WO03/057650(参照により本明細書に援用する)に記載されている、これらの無水物、一水和物およびイソプロパノール溶媒和物ならびに一ナトリウム塩の種々の多形体を含む。
【0071】
「活性剤の有効量」とは、ある期間にわたって活性剤が投与される生体の状態を治療または予防するのに有効である活性剤の量であり、例えば、所望の投与間隔の間に治療効果を与える活性剤の量である。
【0072】
用語「インスリン」は、インスリンの全形態を指し、それぞれ参照によって全体を本明細書に援用する、米国特許第4421685号、第5474978号および第5534488号に記載されているものなどの、天然由来のインスリンおよびインスリンの合成形態を含むが、これらに限定されない。
【0073】
用語「インスリン誘導体」は、インスリン作用を有するインスリン由来のタンパク質およびペプチドを指し、例えば、リスプロ、B10Asp、およびHOE-901を含む。
【0074】
「送達剤の有効量」とは、任意の投与経路(例えば本出願で議論されている、経口(例えば、胃腸管の生体膜を越えて)、経鼻、経肺、経皮、経頬、経膣、および/または経眼を含むが、これらに限定されない経路)によって、所望量の活性剤の吸収を可能および/または促進する送達剤の量である。
【0075】
本明細書で使用する用語「アルキル」および「アルケニル」には、直鎖および分枝の、アルキルおよびアルケニル置換基がそれぞれ含まれる。
【0076】
「医薬として許容できる」という言葉は、哺乳動物に投与する場合に生理的に耐容できる、添加剤または組成物を指す。
【0077】
「実質的に崩壊する」という言葉は、錠剤の全体積の約75%〜約95%が、粉々に壊れ、この構成成分(例えば、不溶性の被覆粒子、不溶性の崩壊剤など)に分解し、小さい凝集体を除けば、錠剤はもはや原型をとどめていないということを意味する。
【0078】
「表面侵食製剤」は、崩壊はしないが、その代わりに侵食される製剤を指す。例えば、製剤は、所定の期間にわたって表面から溶解し、錠剤は一般に、原型を保ったままこの全体的な形を維持する。表面侵食製剤では、所定の期間にわたって活性剤の持続放出が可能となる。
【0079】
用語「微粉化する」および「微粉化された」は一般に、直径/サイズが、マイクロ粒子および/またはナノ粒子の一般的な範囲内であるように加工処理すること、または加工処理されている粒子を指す。
【0080】
用語「マイクロ粒子」は一般に、約1〜約999マイクロメートル(ミクロン、μm)の範囲にある直径を有する粒子を含む。
【0081】
用語「ナノ粒子」は一般に、約1〜約999ナノメートル(nm)の範囲にある直径を有する粒子を含む。
【0082】
用語「インスリン誘導体」は、インスリン作用を有する、インスリン由来のタンパク質およびペプチドを含み、例えば、リスプロ、B10Asp、およびHOE-901を含む。
【0083】
「インスリン分泌促進薬」は、主に膵臓β-細胞に作用して血液中へのインスリン分泌を促進することで血糖降下作用を示し、例えば、スルホニル尿素(例えば、トルブタミド、クロルプロパミド、グリベンクラミド(グリブリド)、グリピジド、グリメペリド、およびグリクラジド);ならびにメグリチニド類似体(例えばレパグリニド、ナテグリニド、メグリチニド、およびミチグリニド(KAD-1229))が含まれる。他のインスリン分泌促進薬は、例えば、K+-ATPチャネル阻害剤(例えば、BTS-67-582)、グルカゴン様ペプチド-1受容体アゴニスト(例えば、グルカゴン様ペプチド-1、エキセンディン-4、およびNN-2211)ならびにグルカゴン様ペプチド-1の作用を高める効果を有するジペプチジルペプチダーゼ-IV阻害剤である。一実施形態によると、インスリン分泌促進薬は、スルホニル尿素またはメグリチニド類似体である。
【0084】
用語「インスリン抵抗性改善薬」には、標的組織中でインスリンの作用を高めることで、血糖降下作用を示す薬剤が含まれ、例えば、ペルオキシソーム増殖剤応答性受容体(PPAR)-γアゴニスト(例として、ピオグリタゾン、ロシグリタゾン、およびシグリタゾンなどのチアゾリジンをベースとした化合物;またはGI-262570、JTT-501、YM-440、NN-622、およびKRP-297などの非チアゾリジンをベースとした化合物)、PPAR-γアンタゴニストならびにプロテインチロシンホスファターゼ阻害剤が含まれる。インスリン抵抗性改善薬には、例えば、インスリン抵抗性を改善する作用を有する医薬品が含まれ、例えば、ビグアニド(例として、メトホルミン、フェンホルミン、およびブホルミン、好ましくはメトホルミン)、PPAR-αアゴニスト(シンフィブラート、クロフィブラート、ベザフィブラート、およびクリノフィブラートなどのフィブラート系化合物ならびに非フィブラート系化合物)、抗肥満薬(例として、シブトラミンなどの5-ヒドロキシトリプタミン(5-HT)再取り込み阻害剤、オルリスタットなどのリパーゼ阻害剤、およびAJ-9677などのアドレナリンβ-受容体アゴニスト)が挙げられる。好ましいインスリン抵抗性改善薬には、メトホルミンなどのビグアニドが含まれるが、これらに限定されない。
【0085】
用語「インスリン模倣体」は、生理学的なインスリン作用、すなわち、多かれ少なかれインスリンとは独立してブドウ糖の細胞への取り込みを促進する作用を通じて血糖降下作用を発現する、インスリン誘導体以外の薬剤を指し、例えば、インスリン受容体活性化剤(例としてCLX-0901およびL-783281)ならびにバナジウムが含まれる。
【0086】
用語「α-グルコシダーゼ阻害剤」は、ブドウ糖の体内への吸収を抑制することを通じて、主に、腸管においてα-グルコシダーゼを阻害することによって、血糖降下作用を発現する薬剤を指し、例えば、アカルボース、ボグリボース、およびミグリトールが含まれる。
【0087】
用語「糖形成抑制剤」は、主に糖形成を阻害することを通じて、血糖降下作用を発現する薬剤を指し、例えば、グルカゴン分泌抑制剤(例として、M&B-39890Aおよびオクトレオチド)、脂肪酸分解阻害剤(例として、ニコチン酸誘導体およびカルニチンパルミトイルトランスフェラーゼ-1阻害剤)、ならびにグルコース-6-ホスファターゼ阻害剤が含まれる。
【0088】
用語「腎ブドウ糖再吸収阻害剤」は、尿細管においてブドウ糖再吸収を阻害する薬剤を指す。腎ブドウ糖再吸収阻害剤の主要な作用は、標的組織細胞への取り込み促進、腸管からの吸収抑制、または組織中の合成抑制による血糖降下作用を伴わない。好適な腎ブドウ糖再吸収阻害剤には、参照により本明細書に援用する米国特許公開番号2005/0143424に記載されているものが含まれるが、これらに限定されない。
【0089】
[送達剤化合物]
送達剤化合物は、参照により本明細書に援用する、米国特許第5650386号および第5866536号ならびに国際公開番号WO94/23767、WO95/11690、WO95/28920、WO95/28838、WO96/10396、WO96/09813、WO96/12473、WO96/12475、WO96/30036、WO96/33699、WO97/31938、WO97/36480、WO98/21951、WO98/25589、WO98/34632、WO98/49135、WO99/16427、WO00/06534、WO00/07979、WO00/40203、WO00/46182、WO00/47188、WO00/48589、WO00/50386、WO00/59863、WO00/59480、WO01/32130、WO01/32596、WO01/34114、WO01/44199、WO01/51454、WO01/70219、WO01/92206、WO02/02509、WO02/15959、WO02/16309、WO02/20466、WO02/19969、WO02/070438、WO03/026582、WO02/100338、WO03/045306、WO03/26582、およびWO03/057170に記載されているもののいずれかであってよい。
【0090】
送達剤化合物の非限定例には、N-(8-[2-ヒドロキシベンゾイル]アミノ)カプリル酸、N-(10-[2-ヒドロキシベンゾイル]アミノ)デカン酸、8-(2-ヒドロキシ-4-メトキシベンゾイルアミノ)オクタン酸、8-(2-ヒドロキシ-5-クロロベンゾイル-アミノ)オクタン酸、4-[(2-ヒドロキシ-4-クロロベンゾイル)アミノ]ブタン酸、およびこれらの塩が含まれる。好ましい塩には、一ナトリウム塩(モノナトリウム塩)および二ナトリウム塩(ジナトリウム塩)が含まれるが、これらに限定されない。
【0091】
一実施形態によると、送達剤化合物は、N-(8-[2-ヒドロキシベンゾイル]アミノ)カプリル酸または医薬として許容できるその塩である。
【0092】
別の実施形態によると、送達剤化合物は、N-(10-[2-ヒドロキシベンゾイル]アミノ)デカン酸またはこ医薬として許容できるその塩である。
【0093】
別の実施形態によると、送達剤化合物は、4-[(2-ヒドロキシ-4-クロロベンゾイル)アミノ]ブタン酸または医薬として許容できるその塩である。
【0094】
別の実施形態によると、送達剤化合物は、8-(2-ヒドロキシ-5-クロロベンゾイルアミノ)オクタン酸または医薬として許容できるその塩である。
【0095】
送達剤化合物は、カルボン酸またはそのナトリウム塩などの医薬として許容できる塩、ならびにこれらの水和物および溶媒和物の形であることが可能である。塩は、1価または多価の塩であってよく、例えばモノナトリウム塩およびジナトリウム塩(例えば、8-(2-ヒドロキシ-5-クロロベンゾイルアミノ)-オクタン酸のジナトリウム塩、N-(8-[2-ヒドロキシベンゾイル]アミノ)カプリル酸のジナトリウム塩、N-(10-[2-ヒドロキシベンゾイル]アミノ)デカン酸のジナトリウム塩)などが挙げられる。例として、参照により本明細書に援用する国際公開番号WO00/59863を参照されたい。送達剤化合物は、例えば、担体の溶解プロフィールの変更に及ぼす影響に応じて選択された異なる対イオンを含むことができる。
【0096】
送達剤化合物は、当技術分野において公知の方法、例えば、上記公開(例として、国際公開番号WO98/34632、WO00/07979、WO01/44199、WO01/32596、WO02/02509、WO02/20466およびWO03/045306)で議論されている方法で調製することができる。SNAC、SNAD、4-CNABならびにこれらの遊離酸およびその他の塩は、当技術分野において公知の方法、例えば、それぞれ参照により本明細書に援用する、米国特許第5650386号および第5866536号ならびに国際公開番号WO02/02509に記載されている方法で調製することができる。
【0097】
本発明の送達剤化合物の塩は、当技術分野において公知の方法で調製することができる。例えば、ナトリウム塩は、送達剤化合物をエタノールに溶解させ、水酸化ナトリウム水溶液を加えることによって調製することができる。
【0098】
送達剤化合物は、再結晶によって、または単独のもしくは縦一列に連結した、1つもしくは複数の固体クロマトグラフ担体上での分画によって精製することができる。好適な再結晶溶媒系には、アセトニトリル、メタノール、およびテトラヒドロフランが含まれるが、これらに限定されない。分画は、アルミナなどの適切なクロマトグラフ担体上で、移動相としてメタノール/n-プロパノール混合物を使用して;逆相クロマトグラフィーで、移動相としてトリフルオロ酢酸/アセトニトリル混合物を使用して;およびイオン交換クロマトグラフィーで、移動相として水または適切な緩衝液を使用して、行うことができる。陰イオン交換クロマトグラフィーを行う場合は、0〜500mMの塩化ナトリウム勾配を使用することが好ましい。
【0099】
送達剤は、-NHC(O)NH-、-C(O)NH-、-NHC(O)、-OOC-、-COO-、-NHC(O)O-、-OC(O)NH-、-CH2NH-NHCH2-、-CH2NHC(O)O-、-OC(O)NHCH2-、-CH2NHCOCH2O-、-OCH2C(O)NHCH2-、-NHC(O)CH2O-、-OCH2C(O)NH-、-NH-、-O-、および炭素-炭素結合からなる群から選択される結合基によって、送達剤に結合されたポリマーを含んでよい。ただし、ポリマーの送達剤はポリペプチドでもポリアミノ酸でもないことを条件とする。ポリマーは、哺乳動物に使用して安全な、交互コポリマー、ブロックコポリマー、およびランダムコポリマーを含む任意のポリマーであることができるが、これらに限定されない。好ましいポリマーには、ポリエチレン;ポリアクリレート;ポリメタクリレート;ポリ(オキシエチレン);ポリ(プロピレン);ポリプロピレングリコール;ポリエチレングリコール(PEG);およびこれらの誘導体、およびこれらの組合せを含むが、これらに限定されない。ポリマーの分子量は、通常約100〜約200,000ダルトンの範囲である。ポリマーの分子量は、好ましくは約200〜約10,000ダルトンの範囲である。一実施形態では、ポリマーの分子量は、約200〜約600ダルトンの範囲であり、より好ましくは約300〜約550ダルトンの範囲である。
【0100】
[活性剤]
本発明における使用に適した活性剤には、生物学的活性剤および化学的活性剤が含まれ、これには殺虫剤、薬物および治療薬が含まれるが、これらに限定されない。好適な活性剤には、胃腸管において、酸加水分解、酵素などによって効果が減少するもの、無効になるもの、または破壊されるものが含まれる。同様に、好適な活性剤としては、経口投与した場合に、サイズ、構造または電荷などの生理化学的特性が、吸収を禁止または妨害する高分子薬剤が含まれる。
【0101】
例えば、本発明における使用に適した生物学的または化学的活性剤には、以下のものが含まれるが、これらに限定されない。すなわち、タンパク質;ポリペプチド;ペプチド;ホルモン;多糖類、および特にムコ多糖類の混合物;炭水化物;脂質;極性が小さい有機分子(すなわち、500ダルトン以下の分子量を有する極性有機分子);その他の有機化合物;ならびに特に、それ自身では胃腸粘膜を通過しない(もしくは投与量の一部だけが通過する)化合物、および/または胃腸管において、酸および酵素によって化学開裂され易い化合物;あるいはこれらの任意の組合せが挙げられる。
【0102】
さらなる例としては、これらの合成由来、天然由来または組換え由来を含む以下のものが含まれるが、これらに限定されない:すなわち、ヒト成長ホルモン(hGH)、組換えヒト成長ホルモン(rhGH)、ウシ成長ホルモン、およびブタ成長ホルモンを含む、成長ホルモン;成長ホルモン放出ホルモン;成長ホルモン放出因子、α(例えば、インターフェロンアルファコン-1(カリフォルニア州ブリスベーンのInterMune,Inc.からInfergen(登録商標)として入手可能))、β、およびγを含むインターフェロン;インターロイキン-1;インターロイキン-2;亜鉛、ナトリウム、カルシウム、およびアンモニウムを含む対イオンを任意選択で有していてもよい、ブタ、ウシ、ヒト、およびヒト組換え型を含むインスリン;IGF-1を含むインスリン様成長因子;未分画ヘパリン、ヘパリン類似物質、デルマタン、コンドロイチン、低分子量ヘパリン、極低分子量ヘパリン、超低分子量ヘパリンを含むヘパリン;サケ、ウナギ、ブタ、およびヒトを含むカルシトニン;エリスロポエチン;心房性ナトリウム利尿因子;抗原;モノクローナル抗体;ソマトスタチン;プロテアーゼ阻害剤;副腎皮質刺激ホルモン、ゴナドトロピン放出ホルモン;オキシトシン;黄体形成ホルモン放出ホルモン;卵胞刺激ホルモン;グルコセレブロシダーゼ;トロンボポエチン;フィルグラスチム;プロスタグランジン;サイクロスポリン;バソプレシン;クロモリンナトリウム(クロモグリク酸ナトリウムもしくはクロモグリク酸二ナトリウム);バンコマイシン;デスフェリオキサミン(DFO);アレンドロネート、チルドロネート、エチドロネート、クロドロネート、パミドロネート、オルパドロネート、およびインカドロネートなどのビスホスホネート;そのフラグメントを含めた副甲状腺ホルモン(PTH);BIBN-4096BSおよび他のカルシトニン遺伝子関連タンパク質アンタゴニストなどの抗片頭痛剤;グルカゴン様ペプチド1(GLP-1);抗生物質、抗細菌薬、および抗真菌薬を含めた抗菌薬;ビタミン;これらの化合物の類似体、フラグメント、模倣体、もしくはポリエチレングリコール(PEG)修飾誘導体;またはこれらの任意の組合せ、が挙げられる。抗生物質の非限定例としては、グラム陽性作用を有する、殺菌性の、リポペプチド系、および環状ペプチド系の抗生物質、例えばダプトマイシン、ならびにこれらの類似体が含まれる。
【0103】
一実施形態によると、活性剤はインスリンである。
【0104】
別の実施形態によると、活性剤は、未分画ヘパリンまたは低分子量ヘパリンなどのヘパリンである。
【0105】
本発明の医薬組成物または単位剤形において使用する活性剤の量は、標的とする適応症を治療するために有効な量である。しかし、この量は、組成物を単位剤形で使用する場合の量に比べて少ない可能性がある。この理由として、単位剤形は複数種の送達剤/活性剤を含むことがあり、そのような組成物は分割された有効量を含むことがあるためである。したがって、総有効量は、合計で活性剤の有効量を含む累積単位で投与される可能性がある。その上、活性剤の有効量は、動物の年齢および体重、動物の健康状態ならびにその他の因子を含む多くの因子によって変化するものであることを当業者は認識するであろう。
【0106】
使用すべき活性剤の総量は、当業者に公知の方法で決定することができる。しかし、本発明の組成物は、上記送達剤なしで活性剤を含む組成物に比べて、より効果的に活性剤を送達することができ、従来の単位剤形または送達システムにおいて使用される活性剤量よりも、より低い量の活性剤を被験者に投与することができ、しかも、同一の血中濃度および/または治療効果を得ることが可能である。
【0107】
一実施形態によると、インスリンは、(活性剤の重量に基づいて)1日に被験者の体重1kg当たり約0.025〜約1.0mg(mg/kg/day)、約0.06〜約0.25mg/kg/day、または約0.09〜約0.19mg/kg/dayの用量で投与される。所望の用量を、単回投与または分割投与のどちらかとして投与することができる。
【0108】
一般に、活性剤の送達を促進する送達剤の有効量は、活性剤と共に投与される。一実施形態によると、送達剤量と活性剤量は、モルベースで、約100:1〜約1:1、約80:1〜約2:1、または約20:1〜約10:1の範囲である。送達剤対活性剤のモルベースの範囲は、送達剤と活性剤との特定の組合せに関しては、100:1を超える可能性がある。また、送達剤対活性剤の範囲は、送達剤と活性剤との特定の組合せの場合には、約1:1以下、例えば0.1:1以下である可能性がある。
【0109】
単位剤形はまた、賦形剤、崩壊剤、潤滑剤、可塑剤、着色剤、着香剤、矯味剤、糖類、甘味剤、および塩類のいずれか1つまたはいずれかの組合せを含むこともできる。
【0110】
本発明の組成物は、生物学的または化学的活性剤を、任意の動物に投与するのに有用である。動物には、ニワトリなどの鳥類、昆虫、魚類、爬虫類、哺乳動物(齧歯動物、水生哺乳動物、イヌおよびネコなどの家庭動物、ヒツジ、ブタ、ウシ、およびウマなどの家畜、ならびに好ましくはヒト、を含むが、これらに限定されない)が含まれるが、これらに限定されない。
【0111】
本発明の別の実施形態は、本発明の粒子を投与することによって、動物において、下記の表1に掲載したものなどの疾患を治療もしくは予防するための方法、または所望の生理学的効果を得るための方法である。好ましくは、所望の疾患の治療もしくは予防のための、または所望の生理学的効果を得るための粒子の有効量を投与する。活性剤に対する具体的な適応症は、参照により本明細書に援用するPhysicians' Desk Reference (58th Ed., 2004, Medical Economics Company, Inc.、モントベール、ニュージャージー州)に見い出すことができる。下記の表における活性剤には、これらの類似体、フラグメント、模倣体、およびポリエチレングリコール修飾誘導体が含まれる。
【0112】
【表1】
【0113】
【表2】
【0114】
【表3】
【0115】
【表4】
【0116】
【表5】
【0117】
【表6】
【0118】
[放出制御製剤もしくは持続放出製剤]
本発明の固体剤形は、胃での崩壊を抑制または遅延するように製剤し得る。本発明における使用に適した放出制御製剤は、例えば、腸溶性コーティングを含むか、または表面から侵食されるように製剤し得る。
【0119】
一実施形態によると、固体の経口剤形は、治療有効量の活性剤および送達剤を含み、固体の経口剤形は、経口投与した場合に、約250秒〜約650秒の崩壊時間を有する。別の実施形態では、経口投与した場合に、崩壊時間は約350秒〜約550秒である。一実施形態では、経口投与した場合に、崩壊時間は約60秒を超える。別の実施形態では、経口投与した場合に、崩壊時間は約400秒を超える。崩壊時間は、米国薬局方<701>に記載されている方法を用いて、水中で37±2℃にて測定することができる。
【0120】
本発明の固体剤形は、腸溶性コーティングで被覆してもよい。腸溶性コーティングは、固体剤形における薬物組成物または複数薬物組成物の放出を遅延させるための主要な制御としての役割を果たすことができる。腸溶性コーティングは、胃で原型を保ったままとどまり、固体剤形の状態で、胃への放出を抑制または遅延する。活性剤の放出は、固体剤形が腸に届くまで遅延される。ひとたび腸に入るとすぐに、高pHによって活性剤が放出される。腸溶性コーティングには、フタル酸ヒドロキシプロピルメチルセルロース、酢酸コハク酸ヒドロキシプロピルメチルセルロース、フタル酸酢酸ポリビニル、酢酸トリメリット酸セルロース、酢酸フタル酸セルロース、ポリ(メタクリル酸-アクリル酸エチル)、およびポリ(メタクリル酸-メタクリル酸メチル)が含まれるが、これらに限定されない。本発明に従って使用することができる他の腸溶性コーティングは、参照により本明細書に援用する米国特許第5851579号に記載されている。
【0121】
本発明の一実施形態では、腸溶性コーティングは、全錠剤または他の剤形に適用される。一実施形態では、腸溶性コーティングは、上記のマイクロ粒子および/またはナノ粒子を含む系などの多粒子系に適用される。
【0122】
本発明の固体剤形は、錠剤(もしくは他の剤形)の表面から、または多粒子系(例えば、上記のマイクロ粒子を含む系)の表面から侵食されるように製剤し得る。これらの表面侵食製剤は、崩壊というよりもむしろ表面からゆっくりと溶解する。表面侵食の速度を制御することによって、固体剤形の活性剤および薬物組成物の放出を遅延させることができる。表面侵食製剤は、固体の経口剤形が腸に届くまで、活性剤または薬物組成物の実質的な放出が起こらないように製剤し得る。
【0123】
[酵素阻害剤]
本発明の固体剤形(本発明のマイクロ粒子またはナノ粒子を含み、かつ/または上記の崩壊時間を有する)は、酵素阻害剤を含んでもよい。固体単位剤形に組み込まれる酵素阻害剤は、酵素による分解を受け易い可能性のあるインスリンまたは他の活性剤の崩壊を抑制することができる。酵素阻害剤は、参照により本明細書に援用する米国特許第6458383号に記載されている。
【0124】
一般に、阻害剤は以下の種類に分類することができる:すなわち、P-アミノベンズアミジン、FK-448、メシル酸カモスタット、およびグリココール酸ナトリウムを含めた、アミノ酸をベースとしない阻害剤;アミノボロン酸誘導体およびn-アセチルシステインを含めた、アミノ酸および修飾アミノ酸;バシトラシン、ホスフィン酸ジペプチド誘導体、ペプスタチン、アンチパイン、ロイペプチン、キモスタチン、エラスタチン、ベスタチン、ホスホラミンドン、ピューロマイシン、サイトカラシンポテトカルボキシペプチダーゼ阻害剤、およびアマスタチンを含めた、ペプチドおよび修飾ペプチド;アプロチニン(ウシ膵臓トリプシンインヒビター)、Bowman-Birkインヒビターおよび大豆トリプシンインヒビター、トリ卵白トリプシンインヒビター、トリオボインヒビター、およびヒト膵臓トリプシンインヒビターを含めた、ポリペプチドプロテアーゼ阻害剤;EDTA、EGTA、1,10-フェナントロリン、およびヒドロキシキノリンを含めた、錯化剤;ならびにポリアクリレート誘導体、キトサン、セルロース誘導体、キトサン-EDTA、キトサン-EDTA-アンチパイン、ポリアクリル酸-バシトラシン、カルボキシメチルセルロース-ペプスタチン、ポリアクリル酸-Bowman-Birkインヒビターを含めた、粘膜付着性ポリマーおよびポリマー-阻害剤結合体、である。
【0125】
酵素阻害剤の選択および濃度は、毒性、プロテアーゼの特異性および阻害剤の有効性に基づくものであり、当業者には明らかであろう。
【0126】
理論によって制限されることを所望することなく、阻害剤は、単独でまたは組み合わせて機能することができると考えられる:阻害剤は、例えば、酵素の基質結合部位で結合し、これによって基質への接近を阻止することによる競合阻害剤(この機序によって作用すると考えられる阻害剤の例は、アンチパイン、エラスタチン、およびBowman Birkインヒビターである。);基質に加えて酵素部位で同時に結合することができ、これらの結合部位が同一ではない非競合阻害剤;および/または、酵素構造の外部へ必須金属イオンが奪われることによって引き起こされる酵素活性の損失にもとづく錯化剤、が挙げられる。
【0127】
(実施例)
以下の実施例は、制限することなく本発明を例示するものである。他に特に指示がない限り、割合はすべて重量で示す。
【0128】
(実施例1)
1. 試験品
a. 部位特異的in situ実験および強制経口投与実験に使用する、共処理したインスリン/送達剤マイクロ粒子
組換えヒト亜鉛インスリン(50mg)および4-CNABナトリウム(7.5g)を脱イオン水50mlに溶かした。この澄明溶液を窒素気流で、室温にて24時間乾燥させた。得られた共処理ケーキを粉砕して微粒子にし、次いで40/60メッシュスクリーンを通して篩にかけ、特定の粒径範囲のマイクロ粒子を得た。本試験において使用したマイクロ粒子の粒径は、250〜420μmの範囲であった。これらのマイクロ粒子には、0.55重量%のインスリン、9.5重量%の水、および89.5重量%の送達剤が含まれていた。合計約90%(w/w)のインスリンを、この工程から回収した。
【0129】
粒子は、異なるサイズの目開き(850μm、425μm、250μm、150μm、45μm)を有する篩を通過させることによって測定した。この方法を用いて、中央粒径は、約45〜約850μm、約45〜約150μm、約150〜約250μm、約250〜約425μm、または約425〜約850μmの範囲であると決定することができる。
【0130】
マイクロ粒子中のインスリン含量を、逆相HPLC(Phenomenex column:Luna 3u C18(2) 100Å、150×4.6mm、3マイクロ;移動相:A、水中0.1%TFA;B、アセトニトリル中0.1%TFA;検出器:UV280nm)を用いて測定した。粒子の含水量を、737KF電量計を用いて測定した。
【0131】
b. マイクロ粒子を充填した強制経口投与用カプセル剤
ゼラチンカプセル剤(サイズ#9)をラットの試験に使用した。ラットの平均体重350mgに基づいて、マイクロ粒子の必要量を決定し、ゼラチンカプセル内に手で充填した。充填した各カプセル剤には、約16mg(インスリン0.0875mgに相当)のマイクロ粒子が入っていた。
【0132】
c. 強制経口投与実験用インスリン/送達剤の小型錠剤
インスリンと送達剤とを1:150(w/w)の比率でよく混合した。これは、0.67%(w/w)のインスリンに相当した。ラットの平均体重350mgに基づいて、インスリン0.175mgと送達剤26.26mgとを含む混合粉末の総量26.43mgを、Carverプレス中、1000psiの圧力下で直接圧縮して錠剤にした。円柱形の小型錠剤は、直径2mmおよび高さ6mmであった。
【0133】
d. インスリン/送達剤の物理学的混合物を充填した強制経口投与実験用カプセル剤
インスリンと送達剤とを1:150(w/w)の比率でよく混合した。ラットの平均体重350mgに基づいて、インスリンと送達剤との混合物の量を決定し、ゼラチンカプセル(サイズ#9)内に手で充填した。各カプセル剤には、26.43mgの混合物(インスリン0.175mgに相当)が入っていた。
【0134】
2. In Situ実験用の直接投与手法
直接投与手法についての概略図を図1に示す。清潔な白衣、マスク、安全保護眼鏡、手袋、および手術帽を使用して、汚染されていない環境で手術を行った。Sprague Dawleyラットに導入濃度として5%イソフルランを用いて麻酔状態を誘発させ、試験が完了するまで純酸素中2%イソフルランで麻酔を維持した。
【0135】
a. 胃への直接投与
血液採取用に右頚静脈にカテーテルを挿入した後、食道および気管上の皮膚を切開し、顎二腹筋の吻側腹部(保護用筋束)を確認し、食道へ向かって接近するように切開した。食道を一部切開し、6〜9cmある食道の切片に対して12cmのPE204管を挿入した。投与製剤を、この管を通じて導入し、先端の尖っていない鋼線を使用してマイクロ粒子を押し込んだ。投与後、胃から何も漏出しないようにするために、食道を3-0絹縫合糸で縛った。
【0136】
b. 空腸への直接投与
血液採取用に右頚静脈にカテーテルを挿入した後、白線を胸骨に向かって切開することによって腹腔を開き、次いで、剣状軟骨を露出した。空腸に最も近い部分を最初に確認した。近接した空腸の血管部分を一部挟み、投与管を遠位端に向かって導入した。投与後、投与管を除去し、2cmのPE206管を押し込んで、挟み傷が2cm管の両端の中央に位置するように設置した。縫合糸を管に巻きつけて両端で空腸と共に結び、少量のvetbond(登録商標)組織接着剤(ミネソタ州、セントポールの3Mから入手可能)で傷を閉じた。
【0137】
3. 強制経口投与手法
Sprague Dawleyラット(体重は約350グラムであった)において、強制経口投与によって試験を行った。小型錠剤またはカプセル剤を、套管針を有する改変された強制給餌用のチューブを使用して、ラットに経口投与した。ラットを約24時間絶食させ、ケタミン(44mg/kg)およびソラジン(1.5mg/kg)を筋肉内投与することによって麻酔した。所定の時間間隔で、血液サンプルを尾動脈から採取し、ブドウ糖およびインスリンのバイオアッセイ用の血漿または血清として、適切に調製した。この動物を実験終了時に屠殺し、局所毒性の何らかの徴候について、ラット胃腸粘膜を観察した。
【0138】
4. バイオアッセイ手法
ラットにおけるインスリンの血清濃度を、インスリンELISAテストキット(DSL Inc.)を使用して測定した。定量限界(LOQ)は、12.5μU/mLに規定されており、本アッセイの較正された線形範囲は最大250μU/mLである。グルコメーターを用いて、血糖値の変化を測定した。
【0139】
5. 結果
a. 部位特異的実験(In Situ)の結果
共処理したマイクロ粒子を胃および空腸に直接投与した後のインスリン濃度および血糖値の変化をそれぞれ図2および3に示す。個々のデータの一覧を表2〜5に示す。
【0140】
空腸への投与から得られたインスリン濃度は、各製剤からの最初のサンプリング点で最大値に達した(tmax=15分)。対応するブドウ糖のtminは、その後約30分で現れた。
【0141】
【表7】
【0142】
【表8】
【0143】
【表9】
【0144】
【表10】
【0145】
b. 錠剤およびカプセル剤を使用した強制経口投与実験からの結果
3種の被験製剤から得られたブドウ糖およびインスリンのデータをそれぞれ図4および5に示す。個々のデータの一覧を表6〜7に示す。胃および空腸への直接投与試験から得られた結果を、比較のために入れている。インスリンおよび送達剤の単純混合物についてのブドウ糖およびインスリンの個々のデータを表8に示す。
【0146】
共処理したインスリンおよび担体のマイクロ粒子を含むカプセル剤を投与したラット10匹の群において、平均最小ブドウ糖低下は、30分で基準線から70%であった。ラット1匹が15〜30分で死亡したが、低血糖症によるものと思われる。ラット6匹を30分でデキストロースを用いて救済した。さらにラット1匹を60分で救済し、30分で救済した6匹中2匹は、60分後死亡した。実験後のラットの剖検から、強制経口投与手法による胃腸刺激または胃腸損傷の徴候は見られなかった。同量のインスリンおよび担体を含む錠剤から得られた平均最小ブドウ糖低下は50%であった。
【0147】
対応するインスリン濃度を図5に示す。インスリン濃度は、カプセル中に共処理したマイクロ粒子を含むものが最も高く、後に錠剤および単純混合のカプセル剤が続く。
【0148】
共処理したマイクロ粒子を含むカプセル剤を使用した強制経口投与試験において、ラット2匹(10匹中)は高いインスリン吸収を示すことがわかった。保存サンプルを再分析したところ、上述の表6(3)に示すように、インスリン濃度はオリジナルサンプルから得たものとほぼ同一であった。2匹の高い応答例を含めたインスリン濃度および含めなかったインスリン濃度をそれぞれ図6および7に示す。N=10およびN=8から得られた、個々のおよび平均の、インスリンおよびブドウ糖プロフィールを図13〜16に示す。
【0149】
【表11】
【0150】
【表12】
【0151】
【表13】
【0152】
(実施例2)
1. 静脈、門脈および皮下実験の概要
a. 実験
齧歯動物において、静脈内投与、門脈内投与、および皮下投与を行い、絶対的バイオアベイラビリティー、インスリンの門脈内吸収、および皮下投与に対する相対的バイオアベイラビリティーを推定した。データを表9〜11にまとめる。静脈内投与から得られた平均インスリンAUC0→∞/投与量は、0.0093 min.kg/mlであった。絶対的バイオアベイラビリティーの推定値において、この値は一定であると仮定した。
【0153】
【表14】
【0154】
【表15】
【0155】
【表16】
【0156】
【表17】
【0157】
b. 結果
門脈に対する全身のインスリン比率は、約0.62(表10のデータから算出した)であることがわかった。したがって、門脈におけるバイオアベイラビリティーは、絶対的バイオアベイラビリティーを0.62で除することによって算出することができる。門脈のバイオアベイラビリティーから、経口送達による薬物吸収の推定値が得られる。皮下投与から得られた平均インスリンAUC0→t/投与量は、0.00516min.kg/mlであった。この値を使用して、皮下に対するバイオアベイラビリティーを推定する。静脈内のデータを除外して、t=0から最終サンプリング時点までの全AUC(すなわちAUC0→t)を算出した。
【0158】
ラットモデルにおいて、門脈内投与から得られたこれらの結果から、インスリンの最大絶対的バイオアベイラビリティーは、経口送達から、またはインスリンが100%胃腸(GI)吸収され門脈へと移行する他のいずれの手段によっても、約60%であると示唆される。次に、皮下(SC)から得られた絶対的バイオアベイラビリティーは、約56%である。
【0159】
バイオアベイラビリティーの推定値(絶対的バイオアベイラビリティー、門脈のバイオアベイラビリティー、皮下に対する相対的バイオアベイラビリティー、および皮下に対する門脈の相対的バイオアベイラビリティー)を図8〜12および表12にまとめる。胃および空腸へのin situ投与から得られた推定絶対的バイオアベイラビリティーを図8に示す。共処理したインスリン(0.5mg/kg)および送達剤(75mg/kg)を含むマイクロ粒子から得られたバイオアベイラビリティー値は、胃内投与の場合には5%、空腸内投与の場合には18%であった。
【0160】
インスリン0.5mg/kgおよび送達剤75mg/kgを含む製剤を使用して、ラットに強制経口投与した錠剤およびカプセル製剤から得られた推定絶対的バイオアベイラビリティーを図9に示す。バイオアベイラビリティー値は、インスリンおよび担体の単純混合物を含む錠剤で投与した場合には6%、カプセル剤で投与した場合には1.6%であった。
【0161】
【表18】
【0162】
【表19】
【0163】
【表20】
【0164】
【表21】
【0165】
(実施例3)
[模擬胃液中でのインスリンおよび4-CNABの安定性]
模擬胃液(SGF)中でのインスリンの安定性を、4-CNABの有無で評価した。インスリン(1 mg/ml)を含み、4-CNABモノナトリウム(1 mg/ml)を含む溶液と含まない溶液とを調製した。
【0166】
胃の酵素であるペプシンを含むSGFと含まないSGFとを調製した。SGF pH1.2を、米国薬局方NF26ガイドラインにより調製した。塩化ナトリウム2gおよびペプシン3.2gを計量して適切な容器に加え、脱イオン水を加えて体積を1リットルにした。必要であれば、濃HClまたは濃NaOHを加えることにより、pHを1.2に調整した。ペプシンを除外した第二のSGF溶液も調製した。
【0167】
SGFの50mlサンプル4つ(ペプシン有り2つおよびペプシン無し2つ)を、37℃に設定した循環水浴に連結されているジャケット付き容器中に置いた。この溶液を、磁気攪拌子を用いて10分間攪拌して溶液を37℃にし、熱平衡に達するようにした。50mgの4-CNABを、ペプシンを含むサンプル1つおよびペプシンを含まないサンプル1つに加え、これらの溶液を2〜3分間攪拌して4-CNABを溶かした。50mgのインスリンをこれらの各サンプルに加えた。インスリンの溶解後、溶液のサンプルを所定の時間間隔で採取し、濾過して直ちに、インスリンおよび4-CNABの含量についてHPLCで分析した。インスリンがすべて溶解した後に抜き取った最初のサンプルを、時間ゼロ(0)で採取したものとした。結果を表13に示す。
【0168】
【表22】
【0169】
本明細書で使用する用語「理論上の%」は、サンプルを採取した時点での、抜き取った溶液の濃度(mg/mL)を、実験のための測定成分の理論上の濃度(mg/mL)と比較したパーセントを意味する。HPLC分析の標準偏差は±5%であった。これらの結果は以下のことを示している。すなわち、インスリンは、ペプシンを含むSGF中で不安定である。これは、最初のサンプリング点でインスリンがわずか3.0%しか残存していなかった(97%のインスリンが分解した)ためである。一方、ペプシンを含まないSGF中では、インスリンは、少なくとも2時間まで安定である。
【0170】
(実施例4)
[模擬腸液中でのインスリンの安定性]
模擬腸液(SIF)中でのインスリンの安定性を、4-CNABの存在下および不存在下で評価した。
【0171】
膵臓の酵素を含むSIFと含まないSIFとを調製した。SIF pH7.5を、米国薬局方NF26ガイドラインにより調製した。SIFの調製は、リン酸二水素カリウム6.8gおよびパンクレアチン10gを適切な容器に加え、脱イオン水を加えて体積を1リットルにすることによって行った。必要であれば、0.2N水酸化ナトリウムを加えることにより、pHを7.5に調整した。膵臓の酵素であるパンクレアチンを除いた第二のSIF溶液も調製した。
【0172】
SIFの50mlサンプル4つ(パンクレアチン有り2つおよびパンクレアチン無し2つ)を、37℃に設定した循環水浴に連結されているジャケット付き容器中に置いた。この溶液を、磁気攪拌子を用いて10分間攪拌して溶液を37℃にし、熱平衡に達するようにした。50mgの4-CNABを、パンクレアチンを含むサンプル1つおよびパンクレアチンを含まないサンプル1つに加え、これらの溶液を2〜3分間攪拌して4-CNABを溶かした。50mgのインスリンをこれらの各サンプルに加えた。インスリンの溶解後、溶液のサンプルを所定の時間間隔で採取し、直ちに、インスリンおよび4-CNABの含量についてHPLCで分析した。結果を表14に示す。
【0173】
【表23】
【0174】
これらの結果は、インスリンが、パンクレアチンを含まないSIF中では安定であり、酵素が存在すると分解するということを示している。インスリンは、酵素を含むSGF中および含まないSGF中に比べて、酵素を含むSIF中および含まないSIF中で、より安定である。最初のサンプリング時点(0分)で、酵素を含むSGF中にはインスリンが3.0%しか残存していなかったが、SIF中にはインスリンが58.9%および66.9%残存していた。
【0175】
(実施例5)
[製剤がインスリンの吸収および作用に及ぼす影響]
表15に示す6種のインスリン含有製剤を以下のように調製した。
【0176】
【表24】
【0177】
Polyplasdone XLは、デラウェア州ウィルミントンのInternational Specialty Productsから入手可能である;Emcocel HD90、Prosolv HD90、Emcompress、およびAnhydrous Emcompressは、ニューヨーク州パターソンのJRS Pharmaから入手可能である。
【0178】
これらの製剤をアカゲザルに、100mg/kgの4-CNABおよび13U/kgインスリンを含む投与量で摂取させた。各群4匹(雄2匹および雌2匹)のアカゲザルを、少なくとも投与約12時間前から投与4時間後まで絶食させた。水は、投与約1時間前から投与2時間後まで与えず、その後は自由に摂取させた。投与後は水5mlで流し込んだ。投与15分前、ならびに投与5、10、15、20、30、45分後および1、1.5、2、3、4時間後に、静脈穿刺で血液サンプル(各約2mlずつ)を採取した。各血液サンプルを2部に分けた。1部は、室温で凝固させ、2〜8℃で10分間、3000rpmで遠心分離した。得られた血清を2等分し、発送まで-70℃で保存した。一方のサンプルは、ELISAによるインスリン分析用に、ドライアイスを入れてEmisphereに発送し、もう一方は血清中ブドウ糖分析用にCROによって保管された。血液のもう1部は、氷水浴上で30分までの間静置し、2〜8℃で10分間、3000rpmで遠心分離した。得られた血漿を、HPLCによる4-CNABの含量分析用に、ドライアイスを入れてEmisphereに発送した。各製剤をアカゲザル4匹に投与した。ただし、製剤1は、アカゲザル8匹に投与した。血液サンプルを上記のように所定の間隔で採取し、インスリンおよびブドウ糖濃度について分析した。結果を表16ならびに図17および18に示す。
【0179】
【表25】
【0180】
崩壊時間を、米国薬局方<701>に記載されている方法を用いて、水中で37±2℃にて測定した。水を入れた複数の管を、37±2℃に維持した水浴中に浸した組み立て式バスケットラックに置いた。組み立て式バスケットラックは一定周期で管を昇降する。錠剤を管に置き、完全に崩壊したかどうかを決定するために定期的に調べた。各錠剤を異なる6本の管で試験した。1つまたは2つの錠剤がなかなか崩壊しない場合には、追加の錠剤でこの手法を繰り返した。インスリンの平均最大濃度(Cmax)を、上記のように測定したインスリンの血清濃度に基づいて、各群について測定した。実験中、これらの霊長類における血糖値が非常に低くなる場合(<1mmol/L)には、血糖を安全値まで上昇させるために、これらにデキストロースを投与する。各群についての平均Cmaxおよび救済したアカゲザルの数を表17に示す。
【0181】
【表26】
【0182】
(実施例6)
[腸溶性錠剤の調製]
カプセル剤の製造は、150単位インスリン、200mg 4-CNAB、0.4%w/wポビドン、〜29.1%w/w Emcompress、1%w/w SLS、および1%w/wステアリン酸マグネシウムを含む製剤300mgを、サイズ2の白色不透明カプセルに封入することによって行った。これらのカプセルをまず、Opadry clearを含むサブコートで被覆して5%増量し、次いで腸溶性コートで被覆して20%増量し、カプセルにおける全増量分を25%にした。
【0183】
錠剤については、上記の製剤300mgを押圧することによって錠剤を製造した。10%増量腸溶性コートを適用した。サブコートおよび腸溶性コートの処方を下表18に示す。
【0184】
【表27】
【0185】
Opadry(登録商標) Clearは、ペンシルベニア州ウエストポイントのColorconから入手可能である。
【0186】
Milli Q Waterは高度に精製された水であり、マサチューセッツ州ビラリカのMilliporeから入手可能である。
【0187】
Eudragit L30D55は、ニュージャージー州パーシッパニーのDegussa AGから入手可能である。
【0188】
腸溶性コートの有効性を確認するために、被覆したカプセル剤および錠剤を、0.1N HCl中に2時間、またはpH6.8リン酸緩衝液中に1時間置いた。被覆したカプセル剤および錠剤は、0.1N HClに溶けなかったが、pH6.8リン酸緩衝液には溶けた。
【0189】
(実施例7)
[ヘパリンで被覆したSNACマイクロビーズ]
5gのSNACおよび0.5gのステアリン酸マグネシウムを混合した。混合粉末0.02gをダイに送り込んだ。SNACおよびステアリン酸マグネシウムの小ビーズを1200 PSI barの圧力で製造した。このビーズは、約0.2mm〜約2.0mmのサイズの丸/ボール形状を有していた。次いで、SNACビーズを液体形態のヘパリン2.5gで回転方式によって被覆し、真空オーブンで40℃にて10時間乾燥させた。
【0190】
(実施例8)
[ヘパリン含有微粉化SNAD]
SNADを、35メッシュTyler標準篩を通して選別した。SNADの粉砕は、250mLステンレス製粉砕ジャーが装備されたGlen Mills, Model S100遠心ボールミル(ニュージャージー州クリフトン)を使用して行い、かつ直径30mm(440c)ステンレス製ボールを使用した。検討した加工処理条件は、(1)使用するボールの数、(2)粉砕持続時間、(3)粉砕速度、および(4)粉砕ジャーへの全投入量であった。Scirocco 2000乾燥用付属品が装備されたMalvern Mastersizer 2000を、粒径測定に使用した。2θ範囲が5〜40°2θで走査する、Kratos XRD 6000(バージョン4.1)X線粉末回折装置を、結晶化度の変化をモニターするために使用した。発散スリットは1°、散乱スリットは1°および受光スリットは0.3mmであった。Brinkmann737KF電量計を含水量の測定に使用し、さらにQuantachrome Nova 3000 Series Surface Area Analyzerを比表面積の測定に使用した。
【0191】
これらの結果から、事前選別したSNADの粒径分布はd(0.1)=1.6μm、d(0.5)=10.5μm、およびd(0.9)=314.9μmであることが示された。1〜5個の範囲にある種々の数のボールを使用して得られたデータから、投入するボールの最適数は2個であることが示された。2個のボールを使用することにより、d(0.1)=1.1μm、d(0.5)=12.0μm、およびd(0.9)=154.3μmの粒径が得られた。
【0192】
ボールの数および投入量を固定して、粉砕時間の効果を評価したところ、120分という粉砕時間が最適であり、d(0.1)=2.0μm、d(0.5)=15.4、およびd(0.9)=62.9μmの粒径分布が得られることが示された。
【0193】
粉砕速度100、300、および500rpmについて評価したところ、最適粉砕は300rpmで得られることが示された。この速度によって、粒径分布は、未粉砕のSNADのd(0.9)=314.9μmと比較して、d(0.9)=62.9μmが得られた。
【0194】
250mL粉砕ジャーに対して、投入量37mLの場合に、75および112mLに比べて、より良い粉砕を実現した。X線粉末回折分析から、SNADに関しては、粉砕によって結晶化度の変化が起こらないことが示された。カールフィッシャー含水量測定により、含水量の著しい変化は全くないことが示された。
【0195】
次いで、上記SNADをヘパリンと混合した。
【0196】
(実施例9)
[微粉化ヘパリン含有微粉化SNAC]
SNACおよびヘパリンを、実施例8に記載の手法で、2個のボールを用いて200rpmで120分間、別々に微粉化し、次いで一緒に混合した。微粉化SNACは、7.574μmのd(0.5)を有していた。下表19に示す製剤を含むSNAC/ヘパリンカプセル剤の調製は、これらの製剤をハードゼラチンカプセルに手で詰め込むことによって行った。
【0197】
【表28】
【0198】
ヘパリン、SNAC、およびラウリル硫酸ナトリウムを混合した。これとは別に、PEG300、モノカプリル酸プロピレングリコール、および水(製剤Bについて)を混合した。液体のPEG300/モノカプリル酸プロピレングリコール混合物の50%を乳鉢に移した。ヘパリン、SNAC、およびラウリル硫酸ナトリウムを混合した粉末を少しずつ加え、液体と共に乳鉢および乳棒で粉砕した。次いで、得られた混合物でカプセルを充填した。
【0199】
(実施例10)
[微粉化SNAC/ヘパリン]
ヘパリン(118.5mg/投与1回分(22,500rpm))およびSNAC(125mg/投与1回分)を乾式混合し、35メッシュスクリーンを通して選別し、ボールミルで約4分間粉砕した。混合物をカプセル(Capsugel Size 1カプセル(サウスカロライナ州グリーンウッド))に詰め込んだ。
【0200】
これらのカプセル剤をアカゲザル(サル1匹につき2カプセル)に以下の手法で投与した。体重3.5〜5.0kgのアカゲザルを、実験前に一晩絶食させ、投与約2時間後に飼料を元に戻した。水は、投与に使用した量を除いて、投与30分前から投与30分後まで与えなかった。ピルガンを使用して口の奥に各剤形を供給した。剤形放出後、逆浸透水5mlを口腔内に投与し、嚥下し易くした。供給後、カプセルが嚥下されたことを保証するために口腔を検査した。血液サンプルからの抗因子Xaを6時間にわたって測定した。
【0201】
結果を図19に示す。
【0202】
(実施例11)
[微粉化SNAC/ヘパリン]
下表20に示す微粉化SNAC/ヘパリンを含むカプセル剤を以下のように調製した。
【0203】
ヘパリンおよびSNACの溶液を以下のように調製した。ヘパリンおよびSNACの必要量を量り分け、予め水酸化ナトリウムを用いてpHを約8に調整しておいた水を加えた。得られた溶液のpHは、約7.3〜7.5の範囲であった。水酸化ナトリウムを用いて溶液のpHを約8のpHに調整した。次いで、この溶液をRotoVap装置内で真空下、50℃にて乾燥させた。蒸発は、以下に概略を述べるプログラムを使用して行った。
1. 760から200torrへの真空の迅速な減少
2. 2分かけて200から100torrへの真空圧の減少
3. 2分かけて100から50torrへの真空圧の減少
4. 4分かけて50から25torrへの真空圧の減少
5. 4分かけて25から15torrへの真空圧の減少
6. 2分かけて15から10torrへの真空圧の減少
7. 30分かけて10±2torrおよび70rpmでの蒸発
8. 手動で50rpmに切り替え、4時間蒸発を継続
【0204】
サンプルを一晩真空乾燥させた。次いで、得られた粉末を微粉化し、カプセルに充填して所望する用量とした。
【0205】
【表29】
【0206】
本発明は、本明細書に記載されている特定の実施形態によって範囲を限定されない。実際に、本明細書に記載されているものに加えて、本発明の種々の変更例は、上述の説明および添付の図から、当業者には明らかになるであろう。かかる変更例は、添付の特許請求の範囲内に含まれることが意図される。
【0207】
特許、特許出願、公開、製品に関する説明およびプロトコルが本出願を通じて引用されているが、これらの開示はすべての目的のために参照により本明細書に全体を援用する。
【技術分野】
【0001】
本出願は、2004年9月23日に出願した米国仮出願第60/612810号および2004年8月13日に出願した米国仮出願第60/601258号の優先権を主張するものである。両米国仮出願は、参照により本明細書に援用する。
【背景技術】
【0002】
本発明は、医薬製剤および同医薬製剤を調製するための方法に関する。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】米国特許公開番号2005/0143424
【特許文献2】米国仮出願第60/569476号
【特許文献3】国際公開番号WO03/057650
【特許文献4】米国特許第4421685号
【特許文献5】米国特許第5474978号
【特許文献6】米国特許第5534488号
【特許文献7】米国特許第5650386号
【特許文献8】米国特許第5866536号
【特許文献9】国際公開番号WO94/23767
【特許文献10】国際公開番号WO95/11690
【特許文献11】国際公開番号WO95/28920
【特許文献12】国際公開番号WO95/28838
【特許文献13】国際公開番号WO96/10396
【特許文献14】国際公開番号WO96/09813
【特許文献15】国際公開番号WO96/12473
【特許文献16】国際公開番号WO96/12475
【特許文献17】国際公開番号WO96/30036
【特許文献18】国際公開番号WO96/33699
【特許文献19】国際公開番号WO97/131938
【特許文献20】国際公開番号WO97/36480
【特許文献21】国際公開番号WO98/21951
【特許文献22】国際公開番号WO98/25589
【特許文献23】国際公開番号WO98/34632
【特許文献24】国際公開番号WO98/49135
【特許文献25】国際公開番号WO99/16427
【特許文献26】国際公開番号WO00/06534
【特許文献27】国際公開番号WO00/07979
【特許文献28】国際公開番号WO00/40203
【特許文献29】国際公開番号WO00/46182
【特許文献30】国際公開番号WO00/47188
【特許文献31】国際公開番号WO00/48589
【特許文献32】国際公開番号WO00/50386
【特許文献33】国際公開番号WO00/59863
【特許文献34】国際公開番号WO00/59480
【特許文献35】国際公開番号WO01/32130
【特許文献36】国際公開番号WO01/32596
【特許文献37】国際公開番号WO01/34114
【特許文献38】国際公開番号WO01/44199
【特許文献39】国際公開番号WO01/51454
【特許文献40】国際公開番号WO01/70219
【特許文献41】国際公開番号WO01/92206
【特許文献42】国際公開番号WO02/02509
【特許文献43】国際公開番号WO02/15959
【特許文献44】国際公開番号WO02/16309
【特許文献45】国際公開番号WO02/20466
【特許文献46】国際公開番号WO02/19969
【特許文献47】国際公開番号WO02/070438
【特許文献48】国際公開番号WO03/026582
【特許文献49】国際公開番号WO02/100338
【特許文献50】国際公開番号WO03/045306
【特許文献51】国際公開番号WO03/26582
【特許文献52】国際公開番号WO03/057170
【特許文献53】米国特許第5851579号
【特許文献54】米国特許第6458383号
【非特許文献】
【0004】
【非特許文献1】Physicians' Desk Reference(58th Ed., 2004, Medical Economics Company, Inc.、モントベール、ニュージャージー州)
【発明の開示】
【発明が解決しようとする課題】
【0005】
インスリンなどの薬物のための、改良された経口送達システムは、絶えず必要とされている。
【課題を解決するための手段】
【0006】
本発明は、送達剤化合物単独または送達剤化合物と活性剤との組合せを含む、経口投与用のマイクロ粒子および/またはナノ粒子に関する。これらの粒子を含む製剤(ならびに送達剤化合物および活性剤のみを含む粒子のための製剤)は、送達剤化合物と活性剤との単純な混合物を、粉剤、錠剤またはカプセル剤として経口投与するのに比べて、変化の少ない、活性剤の極めて優れたバイオアベイラビリティーを提供する。特定のいずれの理論によっても制限されることなく、少なくともいくつかの実施形態において考えられていることによると、この改良は、(1)胃から、幽門(通常、1000〜2000μmの直径を有する)を経て、粒子の溶解および送達剤を介した薬物の吸収が最もよく起こると考えられている小腸へと移行することが可能になるような小サイズのマイクロ粒子またはナノ粒子によるもの、ならびに(2)送達剤化合物が吸収部位において活性剤と共に存在することを確実にする、粒子中での送達剤化合物と活性剤との間の親密な接触によるものであり得る。従来の錠剤またはカプセル剤が幽門を通って小腸へと入っていくためには最初に溶解して十分に小さい粒子になる必要があるのとは異なり、マイクロ粒子およびナノ粒子は自由に幽門を通って小腸へと入っていくので、錠剤の崩壊に起因する変動、および胃運動によって変わる胃通過が最小限に抑えられる。
【0007】
一実施形態によると、送達剤化合物および活性剤を含む粒子は、約900または1000μm未満の中央粒径(メジアンサイズ)を有する。例えば、中央粒径は、約45〜約850μm、約45〜約150μm、約150〜約250μm、約250〜約425μm、約425〜約850μm、約100〜約1000nmまたは約500〜約1000nmの範囲であることができる。別の実施形態によると、粒子は、約1μm未満の中央粒径を有する。いくつかの実施形態では、粒子は、約1ナノメートルと小さく、約999マイクロメートルと大きい可能性がある。例えば、粒子は、約999マイクロメートル未満、約1ナノメートル〜約999マイクロメートル、約1〜約999マイクロメートル、約1〜約999ナノメートル、約45〜約850マイクロメートル、約45〜約150マイクロメートル、約150〜約250マイクロメートル、約250〜約425マイクロメートル、約425〜約850マイクロメートル、約100〜約1000ナノメートルまたは約500〜約1000ナノメートルの中央粒径を有することができる。
【0008】
別の実施形態は、送達剤化合物および活性剤を含む医薬製剤であり、送達剤化合物は、粒子の形である。粒子は、約999マイクロメートル未満、約1ナノメートル〜約999マイクロメートル、約1〜約999ナノメートルまたは約7〜約16マイクロメートルの中央粒径(メジアンサイズ)を有することができる。任意選択により、活性剤は粒子の形であってもよい。例えば、活性剤粒子の中央粒径は、約999マイクロメートル未満、約1ナノメートル〜約999マイクロメートル、約1〜約999マイクロメートルまたは約1〜約999ナノメートルであることができる。一実施形態によると、送達剤化合物粒子および活性剤粒子はどちらも、約1〜約999マイクロメートルの中央粒径を有する。別の実施形態によると、送達剤化合物粒子および活性剤粒子はどちらも、約1〜約999ナノメートルの中央粒径を有する。
【0009】
さらに別の実施形態は、送達剤および活性剤を含む医薬製剤であり、活性剤は、約999マイクロメートル未満の中央粒径(メジアンサイズ)を有する粒子の形である。一実施形態によると、活性剤粒子の中央粒径は、約1ナノメートル〜約999マイクロメートル、約1〜約999マイクロメートルまたは約1〜約999ナノメートルである。
【0010】
粒子は、微細顆粒またはマイクロビーズ(例えば、丸/ボール形状かつ約0.2mm〜約2.0mmの直径を有するビーズ)の形であることができる。マイクロビーズは、圧縮によって形成され得る。一実施形態では、医薬製剤は、送達剤化合物を含むマイクロビーズを含有し、送達剤化合物は、インスリンまたはヘパリンなどの活性剤で被覆されている。マイクロビーズは、約0.2mm〜約2.0mmの範囲にある直径を有することができる。
【0011】
粒子は、セルロース誘導体(例えば、CMCナトリウム(デラウェア州ウィルミントンのAqualonから入手可能))またはポリアクリル酸(例えば、オハイオ州クリーブランドのB.F.Goodrichから入手可能なCarbopol(登録商標))などの粘膜付着剤を含有してもよい。粘膜付着剤は、(1)粘膜(胃腸管を含む)への付着を促進することによって、送達剤-活性剤の粘膜への接触を延長させること、(2)活性剤(例えば、インスリンの場合)を安定化させ、保護すること、および(3)生体膜(粘膜を含む)の浸透性を増加させることによって、送達を改良し、活性剤のバイオアベイラビリティーを向上させること、が可能である。
【0012】
インスリンを送達剤化合物と同時に、胃で分解しないが腸では分解する固体の経口剤形で経口投与することは、インスリンの極めて優れたバイオアベイラビリティーをもたらすということもまた発見されている。インスリンまたは異なる活性剤を含むそのような固体の経口剤形は、胃で分解する形態および送達剤化合物を含まない形態に比べて、極めて優れたバイオアベイラビリティーを提供する。特定のいずれの理論によっても制限されることなく、この改良は、インスリンおよび他の活性剤の、胃液中に存在する酵素または酸による分解に対する感受性によるものと考えられている。固体の経口剤形は胃で分解しないので、インスリンおよび他の活性剤は、腸に届くまで分解から保護される。
【0013】
本発明の別の実施形態は、治療有効量の活性剤および送達剤を含む医薬製剤(固体の経口剤形など)であり、医薬製剤は、経口投与の場合、約250秒〜約650秒の崩壊時間を有する。別の実施形態では、経口投与の場合、崩壊時間は約350秒〜約550秒である。さらに別の実施形態では、経口投与の場合、崩壊時間は約60秒を超える。さらに別の実施形態では、経口投与の場合、崩壊時間は約400秒を超える。崩壊時間は、米国薬局方<701>に記載されている方法を用いて、水中で37±2℃にて測定することができる。崩壊時間は、約1秒〜約24時間以上もの範囲に及ぶ可能性があり、これは多くの要因に依存する。かかる要因には、特定の活性剤(1種または複数)、送達剤化合物(1種または複数)、および医薬製剤中に含まれる賦形剤が含まれるが、これらに限定されない。
【0014】
別の実施形態は、治療有効量の活性剤および送達剤を含む医薬製剤(固体の経口剤形など)であり、この固体の経口剤形は、胃で実質的に崩壊または溶解しないが、腸で実質的には崩壊または溶解する。好ましい実施形態では、活性剤はインスリンである。別の好ましい実施形態では、活性剤はインスリン誘導体である。
【0015】
別の実施形態では、該医薬製剤は、胃での崩壊を遅延させるために腸溶性コーティングで被覆されている固体の経口剤形である。腸溶性コーティングには、フタル酸ヒドロキシプロピルメチルセルロース、酢酸コハク酸ヒドロキシプロピルメチルセルロース、フタル酸酢酸ポリビニル、酢酸トリメリット酸セルロース、酢酸フタル酸セルロース、ポリ(メタクリル酸-アクリル酸エチル)、およびポリ(メタクリル酸-メタクリル酸メチル)が含まれるが、これらに限定されない。
【0016】
さらに別の実施形態では、本医薬製剤は、崩壊よりもむしろ剤形の表面から侵食されるように製剤し得る。
【0017】
本医薬製剤は、医薬製剤中の活性剤が酵素によって分解されるのを抑制するために、酵素阻害剤を含むことができる。
【0018】
一実施形態では、送達剤は、以下の構造を有する化合物またはこの塩である:
【化1】
【0019】
上記式中、
Arは、フェニルまたはナフチルであり;
Arは、1個もしくは複数の-OH、ハロゲン、C1〜C4アルキル、C1〜C4アルケニル、C1〜C4アルコキシまたはC1〜C4ハロアルコキシで任意選択で置換されていてもよく;
R1は、C3〜C20アルキル、C4〜C20アルケニル、フェニル、ナフチル、(C1〜C10アルキル)フェニル、(C1〜C10アルケニル)フェニル、(C1〜C10アルキル)ナフチル、(C1〜C10アルケニル)ナフチル、フェニル(C1〜C10アルキル)、フェニル(C1〜C10アルケニル)、ナフチル(C1〜C10アルキル)、またはナフチル(C1〜C10アルケニル)であり;
R1は、C1〜C4アルキル、C2〜C4アルケニル、C1〜C4アルコキシ、C1〜C4ハロアルコキシ、-OH、-SH、-CO2R8、またはこれらのいずれかの組合せで、任意選択により置換されていてもよく;
R2は、水素、C1〜C4アルキル、またはC2〜C4アルケニルであり;
R1は、酸素、窒素、硫黄、またはこれらのいずれかの組合せで任意選択により中断されていてもよい。
式A中の用語「2-OH-Ar」は、2位にヒドロキシル基を有するフェニル基またはナフチル基を指す。
【0020】
一実施形態によると、上記化合物は、酸性基に対してアルファ位にあるアミノ基で置換されていない。
【0021】
好ましくは、Arはハロゲンで置換されている。
【0022】
好ましくは、R2は水素である。
【0023】
好ましくは、R1は非置換である。
【0024】
好ましくは、R1は中断されていない。
【0025】
好ましくは、R1はC1〜10、C3〜9、C3〜7、C3、C7またはC9アルキルである。一実施形態によると、R1は分岐していない。
【0026】
好ましい送達剤化合物には、N-(8-[2-ヒドロキシベンゾイル]アミノ)カプリル酸(SNACの遊離酸)、N-(10-[2-ヒドロキシベンゾイル]アミノ)デカン酸(SNADの遊離酸)、4-[(2-ヒドロキシ-4-クロロベンゾイル)-アミノ]ブタン酸(4-CNABの遊離酸)、およびこれらの塩、ならびにこれらの溶媒和物および水和物が含まれるが、これらに限定されない。この塩は、例えば、モノナトリウム塩(すなわちSNAC、SNADもしくは4-CNAB)またはジナトリウム塩などのナトリウム塩であってよい。
【0027】
別の実施形態では、送達剤は、以下の構造を有する化合物またはこの塩である:
【化2】
【0028】
上記式中、
R1、R2、R3およびR4は独立に、H、-OH、ハロゲン、C1〜C4アルキル、C2〜C4アルケニル、C1〜C4アルコキシ、-C(O)R8、-NO2、-NR9R10、または-N+R9R10R11(R12)-であり;
R5は、H、-OH、-NO2、ハロゲン、-CF3、-NR14R15、-N+R14R15R16(R13)-、アミド、C1〜C12アルコキシ、C1〜C12アルキル、C2〜C12アルケニル、カルバミン酸エステル、炭酸エステル、尿素、または-C(O)R18であり;
R5は、ハロゲン、-OH、-SH、または-COOHで任意選択で置換されていてもよく;
R5は、O、N、Sまたは-C(O)-で任意選択により中断されていてもよく;
R6は、C1〜C12アルキレン、C2〜C12アルケニレンまたはアリーレンであり;
R6は、C1〜C4アルキル、C2〜C4アルケニル、C1〜C4アルコキシ、-OH、-SH、ハロゲン、-NH2または-CO2R8で任意選択により置換されていてもよく;
R6は、OまたはNで任意選択により妨害されていてもよく;
R7は、結合またはアリーレンであり;
R7は、-OH、ハロゲン、-C(O)CH3、-NR10R11、または-N+R10R11R12(R13)-で任意選択により置換されていてもよく;
R8は、H、C1〜C4アルキル、C2〜C4アルケニル、または-NH2であり;
R9、R10、R11およびR12は独立に、HまたはC1〜C10アルキルであり;
R13は、ハライド、ヒドロキシド、サルフェート、テトラフルオロボレートまたはホスフェートであり;
R14、R15およびR16は独立に、H、C1〜C10アルキル、-COOHで置換されているC1〜C10アルキル、C2〜C12アルケニル、-COOHまたは-C(O)R17で置換されているC2〜C12アルケニルであり;
R17は、-OH、C1〜C10アルキル、またはC2〜C12アルケニルであり;かつ
R18は、H、C1〜C6アルキル、-OH、-NR14R15、N+R14R15R16(R13)-である。
【0029】
さらに別の実施形態では、送達剤は、以下の構造を有する化合物またはこの塩である:
【化3】
【0030】
上記式中、
R1、R2、R3、R4およびR5は独立に、H、-CN、-OH、-OCH3、またはハロゲンであり、少なくともR1、R2、R3、R4およびR5のうちの1つは-CNであり;
R6は、C1〜C12の直鎖または分枝の、アルキレン、アルケニレン、アリーレン、アルキル(アリーレン)またはアリール(アルキレン)である。
【0031】
さらに別の実施形態では、送達剤は、以下の構造を有する化合物またはこの塩である:
【化4】
【0032】
上記式中、
Xは各々、水素、ハロゲン、ヒドロキシル、またはC1〜C3アルコキシであり、
Rは、置換もしくは非置換のC1〜C3アルキレン、または置換もしくは非置換のC2〜C3アルケニレンであり、
nは、1から4までの整数である。
【0033】
さらに別の実施形態では、送達剤は、以下の構造を有する化合物またはこの塩である:
【化5】
【0034】
上記式中、
Xは、ハロゲンであり、Rは、置換もしくは非置換のC1〜C3アルキレン、または置換もしくは非置換のC2〜C3アルケニレンである。
【0035】
好ましい送達剤化合物には、N-(8-[2-ヒドロキシベンゾイル]-アミノ)カプリル酸、N-(10-[2-ヒドロキシベンゾイル]-アミノ)デカン酸、8-(2-ヒドロキシ-4-メトキシベンゾイルアミノ)オクタン酸、8-(2-ヒドロキシ-5-クロロベンゾイルアミノ)-オクタン酸、4-[(2-ヒドロキシ-4-クロロベンゾイル)アミノ]ブタン酸、および医薬として許容できるこれらの塩が含まれるが、これらに限定されない。本発明の医薬製剤は、上記送達剤化合物のいずれか、または他の送達剤化合物のいずれかを、単独で、または1種もしくは複数の追加の送達剤化合物と組み合わせて含むことができる。
【0036】
好適な活性剤には、タンパク質、ポリペプチド、ペプチド、ホルモン、多糖類およびこれらの合成由来、天然由来または組換え由来物:すなわち、成長ホルモン;成長ホルモン放出ホルモン;成長ホルモン放出因子、インターフェロン;インターロイキン-1;インターロイキン-2;亜鉛、ナトリウム、カルシウムおよびアンモニウムを含む対イオンを任意選択で有していてもよいインスリン;インスリン様成長因子;ヘパリン;カルシトニン;エリスロポエチン;心房性ナトリウム利尿因子;抗原;モノクローナル抗体;ソマトスタチン;プロテアーゼ阻害剤;副腎皮質刺激ホルモン、ゴナドトロピン放出ホルモン;オキシトシン;黄体形成ホルモン放出ホルモン;卵胞刺激ホルモン;グルコセレブロシダーゼ;トロンボポエチン;フィルグラスチム;プロスタグランジン;サイクロスポリン;バソプレシン;クロモリンナトリウム;バンコマイシン;デスフェリオキサミン;ビスホスホネート;副甲状腺ホルモン;抗片頭痛剤;グルカゴン様ペプチド1(GLP-1);抗菌薬;ビタミン;およびこれらの化合物の、類似体、フラグメント、模倣体もしくはポリエチレングリコール(PEG)修飾誘導体;またはこれらの任意の組合せが含まれるが、これらに限定されない。好ましい活性剤には、インスリンおよびヘパリン(未分画ヘパリンおよび低分子量ヘパリンを含むが、これらに限定されない)が含まれるが、これらに限定されない。
【0037】
本発明の一実施形態では、活性剤はインスリンである。本発明の含インスリン医薬製剤はまた、第2の血糖降下薬、腎ブドウ糖再吸収阻害剤、または上述の任意の組合せ(例えば、参照により本明細書に援用する米国特許公開番号2005/0143424に記載されているものなど)を含むことができる。好適な第2の血糖降下薬には、インスリン分泌促進薬、インスリン抵抗性改善薬、インスリン模倣体、α-グルコシダーゼ阻害剤、糖形成抑制剤および上述のいずれかの任意の組合せが含まれるが、これらに限定されない。一実施形態によると、固体剤形には、スルホニル尿素、メグリチニド類似体、ビグアニド(好ましくはメトホルミン)、または前記いずれかの任意の組合せが含まれる。好ましい実施形態によると、固体剤形はメトホルミンを含む。
【0038】
また、本発明のマイクロ粒子またはナノ粒子を含み、および/または上記の崩壊時間を有する、固体単位剤形などの医薬製剤も提供される。単位剤形は、錠剤、カプセル剤、粉剤またはサシェの形態であることが可能である。単位剤形は、1種もしくは複数の、腸溶性コーティング、崩壊剤、スーパー崩壊剤(デンプングリコール酸ナトリウムまたはクロスカルメロースナトリウムなど)および超粒子スーパー崩壊剤を、単独でまたは組み合わせて有することができる。
【0039】
一実施形態では、固体の経口単位剤形は、速崩壊性錠剤である。別の実施形態では、固体単位剤形は、放出制御または遅延放出を有する。
【0040】
一実施形態によると、本発明は、上記粒子および崩壊剤を含む錠剤を提供する。一実施形態では、崩壊剤は、デンプングリコール酸ナトリウム(英国サウスハンバーサイド州のAzebe UK Ltd.から入手可能なPrimojel(登録商標))、クロスカルメロースナトリウム(英国サウスハンバーサイド州のAzebe UK Ltd.から入手可能なPrimellose(登録商標))または超粒子スーパー崩壊剤などのスーパー崩壊剤である。
【0041】
別の実施形態は、治療有効量のインスリンおよび送達剤化合物を含む固体剤形であり、固体剤形は、経口投与した場合、少なくとも60秒の崩壊時間を有する。固体剤形は、腸溶性コーティングを有するか、または表面侵食製剤であってよい。固体剤形は、1種または複数の酵素阻害剤をさらに含むことができる。
【0042】
さらに別の実施形態は、治療有効量のインスリンおよび送達剤化合物を含む固体剤形であり、固体剤形は、胃で実質的に崩壊または溶解しないが、小腸では崩壊または溶解する。固体剤形は、腸溶性コーティングを有するか、または表面侵食製剤であってよい。固体剤形は、1種または複数の酵素阻害剤をさらに含むことができる。
【0043】
別の実施形態は、活性剤を、動物、特にその活性剤を必要としている動物に投与するための方法であり、本発明のマイクロ粒子またはナノ粒子および/または上記の崩壊時間を有するもの(すなわち、放出制御もしくは持続放出を有するもの)を含む医薬製剤を投与することによる方法である。経口投与が好ましい投与経路である。
【0044】
さらに別の実施形態は、本発明のマイクロ粒子またはナノ粒子を含む固体単位剤形、および/または上記の崩壊時間を有する(すなわち、放出制御もしくは持続放出を有する)固体単位剤形を含有する本発明の医薬製剤を投与することによって、動物における、疾患を治療する方法または所望の生理学的効果を得るための方法である。さらに別の実施形態は、本発明の医薬製剤を経口投与することによって、経口による活性剤のバイオアベイラビリティーを向上させる方法である。
【0045】
さらに別の実施形態は、哺乳動物に、本発明の含インスリン医薬製剤、例えば本発明のマイクロ粒子またはナノ粒子を含むものおよび/または上記の崩壊時間を有するものの治療有効量を投与することによって、哺乳動物(例えばヒト、特にこれを必要としているヒト)における、糖尿病を治療する方法および/またはインスリンの連用に伴う全身性高インスリン血症の発生率を減少させる方法である。一実施形態では、送達剤化合物は、4-CNABの遊離酸または医薬として許容できるその塩である。本医薬製剤は、連用を基礎として投与され得る。
【0046】
さらに別の実施形態は、哺乳動物に、本発明の含インスリン医薬製剤、例えば本発明のマイクロ粒子またはナノ粒子を含む医薬製剤および/または上記の崩壊時間を有する医薬製剤などの治療有効量を投与することによって、哺乳動物(例えばヒト、特にこれを必要としているヒト)における、耐糖能の低下、初期の糖尿病もしくは後期の糖尿病を治療する方法、またはグルコース恒常性を得る方法である。一実施形態では、送達剤化合物は、4-CNABの遊離酸または医薬として許容できるその塩である。本医薬製剤は、連用を基礎として投与され得る。
【0047】
さらに別の実施形態は、本明細書に記載されている、含インスリン医薬製剤の治療有効量を、ヒト糖尿病患者に連用を基礎として経口投与することによって、ヒト糖尿病患者を治療する方法である。
【0048】
さらに別の実施形態は、送達剤化合物および活性剤の溶液を、例えば固体が形成されるまで乾燥させることによって、ならびに任意選択で粒子を単離することによって、本発明のマイクロ粒子およびナノ粒子を調製する方法である。好ましくは、混合物は均質である(例えば、送達剤化合物および活性剤は、混合物中で均一に分散されている)。この方法は、送達剤化合物、活性剤および溶媒の混合物の同時乾燥を含む。好適な溶媒には、ヒドロキシル系溶媒、水、およびこれらの混合物が含まれるが、これらに限定されない。一実施形態によると、混合物は、約10〜約40℃で(例えば室温で)乾燥される。好ましくは、乾燥は制御された温度で行う。一実施形態によると、乾燥は不活性ガス(好ましくは窒素ガス)上で行う。乾燥した材料は、所望の粒径を得るために、任意選択で粉砕および/または篩にかけることも可能である。この方法により、送達剤化合物および活性剤の均質な混合物を含む粒子を得ることができる。
【0049】
本発明のマイクロ粒子およびナノ粒子を調製する別の方法は、送達剤化合物、活性剤および溶媒の混合物を凍結乾燥することによるものである。好適な溶媒には、ヒドロキシル系溶媒、水、およびこれらの混合物が含まれるが、これらに限定されない。
【0050】
本発明のマイクロ粒子およびナノ粒子を調製するさらに別の方法は、(1)送達剤化合物および活性剤を超臨界流体中に溶解すること、ならびに(2) 系の圧力を低下させて、送達剤化合物および活性剤を極めて微細な粒子として析出させることによるものである。この析出は、超臨界溶液が急速膨張した結果である。
【0051】
以下の実施形態は、「固体の医薬組成物の実施形態」として本明細書でまとめて参照される。
【0052】
本発明の上記特徴および付随する他の多くの利点は、以下の詳細な説明を、添付図面と併せて参照することによって、よりよく理解されるようになるであろう。
【図面の簡単な説明】
【0053】
【図1】胃および空腸への直接投与について描いた概略図である。
【図2】共処理したマイクロ粒子を胃および空腸に直接投与した後のインスリン濃度(±SEM)を経時的に示すグラフである。
【図3】共処理したマイクロ粒子を胃および空腸に直接投与した後の血糖値の変化(±SEM)を経時的に示すグラフである。
【図4】以下の3種の異なる剤形から強制経口投与した後の血糖の変化(±SEM)を経時的に示すグラフである:1)インスリンおよび担体を圧縮することによって製造した錠剤、2)共処理したインスリンおよび担体のマイクロ粒子を含むカプセル剤、ならびに3)インスリンおよび担体の単純混合物を含むカプセル剤。
【図5】以下の3種の異なる剤形から強制経口投与した後のインスリン濃度(±SEM)を経時的に示すグラフである:1)インスリンおよび担体を圧縮することによって製造した錠剤、2)共処理したインスリンおよび担体のマイクロ粒子を含むカプセル剤、ならびに3)インスリンおよび担体の単純混合物を含むカプセル剤。
【図6】共処理したインスリンおよび担体のマイクロ粒子を含むカプセル剤を強制経口投与した後のインスリン濃度(±SEM)を経時的に示すグラフである。ラット10匹中2匹が極めて高いインスリン吸収を示した。これら2匹の高い応答例を含めた平均値(N=10)および含めなかった平均値(N=8)についてグラフに描く。
【図7】共処理したインスリンおよび担体のマイクロ粒子を含むカプセル剤を強制経口投与した後の血糖の変化(±SEM)を経時的に示すグラフである。ラット10匹中2匹が極めて高いインスリン吸収を示した。これら2匹の高い応答例を含めた平均値(N=10)および含めなかった平均値(N=8)についてグラフに描く。
【図8】共処理したインスリンおよび担体を胃および空腸にin situ投与して得られた推定絶対的バイオアベイラビリティー(±SEM)を示すグラフである。以下の2種の組成物を評価した:1)インスリン(0.25mg/kg)+送達剤(37.5mg/kg)、および2)インスリン(0.5mg/kg)+送達剤(75mg/kg)。
【図9】1)皮下投与、2)胃への直接投与、3)空腸への直接投与、4)インスリンおよび担体を圧縮することによって製造した錠剤、5)共処理したインスリンおよび担体のマイクロ粒子を含むカプセル剤(2匹の高い応答例を含めたものおよび含めなかったもの)、ならびに6)インスリンおよび担体の単純混合物を含むカプセル剤、から得られたインスリン濃度の推定絶対的バイオアベイラビリティー(±SEM)を示すグラフである。
【図10】1)胃への直接投与、2)空腸への直接投与、3)インスリンおよび担体を圧縮することによって製造した錠剤、4)共処理したインスリンおよび担体のマイクロ粒子を含むカプセル剤(2匹の高い応答例を含めたものおよび含めなかったもの)、ならびに5)インスリンおよび担体の単純混合物を含むカプセル剤、から得られた、門脈におけるインスリン濃度の推定バイオアベイラビリティー(±SEM)を示すグラフである。
【図11】1)胃への直接投与、2)空腸への直接投与、3)インスリンおよび担体を圧縮することによって製造した錠剤、4)共処理したインスリンおよび担体のマイクロ粒子を含むカプセル剤(2匹の高い応答例を含めたものおよび含めなかったもの)、ならびに5)インスリンおよび担体の単純混合物を含むカプセル剤から得られた、皮下投与と比較したインスリンの推定バイオアベイラビリティー(±SEM)を示すグラフである。
【図12】1)胃への直接投与、2)空腸への直接投与、3)インスリンおよび担体を圧縮することによって製造した錠剤、4)共処理したインスリンおよび担体のマイクロ粒子を含むカプセル剤(2匹の高い応答例を含めたものおよび含めなかったもの)、ならびに5)インスリンおよび担体の単純混合物を含むカプセル剤、から得られた、門脈におけるインスリンの、皮下投与と比較した推定バイオアベイラビリティー(±SEM)を示すグラフである。
【図13】共処理したインスリンおよび担体のマイクロ粒子を含むカプセル剤を強制経口投与した後の個別のインスリン濃度を経時的に示すグラフである。ラット14およびラット17が極めて高いインスリン吸収を示した。これら2匹の高い応答例を含めた平均値(N=10)および含めなかった平均値(N=8)をグラフに描く。
【図14】共処理したインスリンおよび担体のマイクロ粒子を含むカプセル剤を強制経口投与した後の個別の血糖の変化を経時的に示すグラフである。ラット14およびラット17が極めて高いインスリン吸収を示した。これら2匹の高い応答例を含めた平均値(N=10)および含めなかった平均値(N=8)をグラフに描く。
【図15】共処理したインスリンおよび担体のマイクロ粒子を含むカプセル剤を強制経口投与した後の個別のインスリン濃度を経時的に示すグラフである。ラット14およびラット17を除外した。N=8から得られた平均値をグラフに描く。
【図16】共処理したインスリンおよび担体のマイクロ粒子を含むカプセル剤を強制経口投与した後の個別の血糖の変化を経時的に示すグラフである。ラット14およびラット17を除外した。N=8から得られた平均値についてグラフに描く。
【図17】インスリンおよび送達剤を含む、実施例の製剤1〜6を摂取させたアカゲザルにおける血清ブドウ糖濃度の変化を経時的に描くグラフである。これらの製剤は異なる崩壊時間を有する。
【図18】インスリンおよび送達剤を含む、実施例の製剤1〜6を摂取させたアカゲザルにおける血清インスリン濃度の変化を経時的に描くグラフである。これらの製剤は異なる崩壊時間を有する。
【図19】実施例10に記載したSNAD/ヘパリン製剤を投与した後の、サルにおける抗因子Xa活性(U/ml)対時間を示すグラフである。
【発明を実施するための最良の形態】
【0054】
[定義]
本明細書に記載されている「粒子」、「マイクロビーズ」、および「顆粒」は、任意の形状であってよく、送達剤化合物および/または活性剤に加えて、1種または複数の成分を含んでいてもよい。所定の任意の粒子、マイクロビーズ、または顆粒の具体的な成分は、用いる方法によって決まることもあり、1バッチ分由来の、個々の粒子、マイクロビーズまたは顆粒において、必ずしも同一であるわけではない。
【0055】
例えば、活性剤の粒子、マイクロビーズ、または顆粒を、送達剤化合物の粒子、マイクロビーズ、または顆粒と別々に調製する場合は、活性剤の粒子、マイクロビーズ、または顆粒は、一般に送達剤化合物を含まず、送達剤化合物の粒子、マイクロビーズ、または顆粒は、一般に活性剤を含まない。ただし、各粒子、マイクロビーズ、または顆粒は、本明細書に開示されているように他の成分を含んでもよい。
【0056】
他の実施形態では、粒子、マイクロビーズ、または顆粒は、溶液、懸濁液、または混合物から形成されることができ、この溶液、懸濁液、または混合物は、限定するものではないが、液体または乾燥形態であって、少なくとも活性剤および送達剤化合物を含む。したがって、例えば所定の任意の粒子、マイクロビーズ、または顆粒は、活性剤および送達剤化合物の両方を含み、1種または複数の他の成分をさらに含んでもよい。
【0057】
用語「直径」および「中央粒径(メジアン粒径)」は、一般に、粒子、マイクロビーズ、および顆粒の寸法を指すために使用される。「中央粒径(メジアン粒径)」または「直径」は、実施例8、9、10について、以下の通り測定した。
機器: Mastersizer 2000 (EQ202、モデルMS2K、製造番号34315-67)
製造会社: MALVERN instruments、英国
ソフトウェア: Mastersizer 2000
付属品: Scirocco 2000 (A) (モデルADA2000、製造番号34270/73)
分散: 乾式分散
解析モデル: 汎用
粒子RI: 1.520
曇り度: 1〜6%
基準: サンプル分散ユニットのためのMalvern品質監査基準
【0058】
Malvern Mastersizer 2000では、レーザー回折およびモデルフィッティングによって粒径が測定される。任意の2相系(例えば、粉末、懸濁液、または乳液)に良好に分散させたサンプルを、サンプル中に存在する粒径に適した長さのレンズで集光させたHe-Neレーザーの通り道に導入する。レーザーの通り道にある粒子の散乱パターンが、多数配置された検出器で測定される。各検出器によって、特定の範囲の角度からのデータが測定される。
【0059】
Malvern装置では、測定される粒子が完全な球形であると仮定されている。非球形粒子については、結果として得られる粒径分布は、他の原理に基づく方法で得られる粒径分布とは異なる可能性がある。コンピュータによる測定は、これに伴って顕微鏡による調査を行い、調査している粒子の種類を決定しなければならないことがよくある。不均整に成形された粒子については、Mastersizer 2000から得られた粒径データは、測定された粒子と同体積の仮想球体の直径として解釈される。(注:d(0.1)は、サンプルの10%がこのサイズ未満の粒径であり、d(0.5)は、サンプルの50%がこのサイズ未満の粒径であり、d(0.9)は、サンプルの90%がこのサイズ未満の粒径である。)
【0060】
一般に、この装置は例えば1個の粒子がレーザーを通り過ぎる最中に、1つの寸法を測定する;すなわち、この装置は粒子を貫通する直線の長さを測定する。不均整な粒子については、この装置を用いると、レーザーに対する粒子の配向により、個々の粒子の最長寸法、最短寸法または他の寸法のいずれかを1回しか測定しないことになるため、結果にばらつきが生じる。しかし、多数の粒子が採寸され、平均直径または中央粒径が算出される。したがって、「粒径」または「直径」の数字は、粒子の平均の「粒径」または「直径」の推定値である。別の方法として、「直径」または「粒径」は、実施例1に記載の篩法によって測定される。「直径」は、ある実施形態では、例えば粒子は端が丸くなっているか、概ね球状を有する可能性があるが、球状または丸い寸法を必ずしも意味するわけではないものと解釈されるべきである。
【0061】
本発明は、「粒径」または「直径」という狭い範囲内に入る粒子、マイクロビーズまたは顆粒に限定されないということも理解されるべきである。したがって、例えば、いくつかの実施形態では、少なくとも使用される成分および方法に応じて、例えば、同バッチにおいて、ナノメートルスケールとマイクロメートルスケールの両方の範囲内に入る多少の粒子を含む可能性がある。実際の個々の粒子の「粒径」または「直径」は、比較的狭い範囲内、または比較的広い範囲内に入る可能性がある。
【0062】
本明細書および添付の特許請求の範囲で使用する場合、単数形(英文では、「a」、「an」および「the」)は、文脈が他に特に明確に指示しない限り、複数の指示対象を含む。したがって、例えば、「粒子」という言及は1種または複数の粒子を含み、「活性剤」は1種または複数のかかる活性剤を含み、「送達剤」は1種または複数の送達剤を含む。
【0063】
用語「約」は一般に、所定の値または範囲の10%以内、好ましくは5%以内、およびより好ましくは1%以内を意味する。
【0064】
本明細書で使用する用語「水和物」には、(i)分子の形で結合した水を含む物質、および(ii)1個もしくは複数の結晶水分子を含む結晶質、または自由水を含む結晶性物質が含まれるが、これらに限定されない。
【0065】
本明細書で使用する用語「溶媒和物」には、溶媒の分子またはイオンと、送達剤化合物もしくはこれらの塩、もしくはこれらの溶媒和物、もしくはこれらの水和物の分子またはイオンとの分子錯体またはイオン錯体が含まれるが、これらに限定されない。
【0066】
用語「送達剤」は、本明細書に開示されている任意の送達剤化合物を指す。
【0067】
用語「SNAC」は、N-(8-[2-ヒドロキシベンゾイル]-アミノ)カプリル酸の一ナトリウム塩を指し、他に特に指示がない限り、2004年5月6日に出願した米国仮出願第60/569476号(参照により本明細書に援用する)に記載されている一ナトリウム塩の種々の多形体を含む。
【0068】
用語「SNAD」は、他に特に指示がない限り、N-(10-[2-ヒドロキシベンゾイル]-アミノ)デカン酸の一ナトリウム塩を指す。用語「SNADのジナトリウム塩」は、N-(10-[2-ヒドロキシベンゾイル]-アミノ)デカン酸の二ナトリウム塩を指す。
【0069】
用語「5-CNAC」は、他に特に指示がない限り、N-(8-[2-ヒドロキシ-5-クロロベンゾイル]-アミノ)オクタン酸の一ナトリウム塩を指す。
【0070】
用語「4-CNAB」は、N-4-[(2-ヒドロキシ-4-クロロベンゾイル)アミノ]ブタン酸の一ナトリウム塩を指し、他に特に指示がない限り、国際公開番号WO03/057650(参照により本明細書に援用する)に記載されている、これらの無水物、一水和物およびイソプロパノール溶媒和物ならびに一ナトリウム塩の種々の多形体を含む。
【0071】
「活性剤の有効量」とは、ある期間にわたって活性剤が投与される生体の状態を治療または予防するのに有効である活性剤の量であり、例えば、所望の投与間隔の間に治療効果を与える活性剤の量である。
【0072】
用語「インスリン」は、インスリンの全形態を指し、それぞれ参照によって全体を本明細書に援用する、米国特許第4421685号、第5474978号および第5534488号に記載されているものなどの、天然由来のインスリンおよびインスリンの合成形態を含むが、これらに限定されない。
【0073】
用語「インスリン誘導体」は、インスリン作用を有するインスリン由来のタンパク質およびペプチドを指し、例えば、リスプロ、B10Asp、およびHOE-901を含む。
【0074】
「送達剤の有効量」とは、任意の投与経路(例えば本出願で議論されている、経口(例えば、胃腸管の生体膜を越えて)、経鼻、経肺、経皮、経頬、経膣、および/または経眼を含むが、これらに限定されない経路)によって、所望量の活性剤の吸収を可能および/または促進する送達剤の量である。
【0075】
本明細書で使用する用語「アルキル」および「アルケニル」には、直鎖および分枝の、アルキルおよびアルケニル置換基がそれぞれ含まれる。
【0076】
「医薬として許容できる」という言葉は、哺乳動物に投与する場合に生理的に耐容できる、添加剤または組成物を指す。
【0077】
「実質的に崩壊する」という言葉は、錠剤の全体積の約75%〜約95%が、粉々に壊れ、この構成成分(例えば、不溶性の被覆粒子、不溶性の崩壊剤など)に分解し、小さい凝集体を除けば、錠剤はもはや原型をとどめていないということを意味する。
【0078】
「表面侵食製剤」は、崩壊はしないが、その代わりに侵食される製剤を指す。例えば、製剤は、所定の期間にわたって表面から溶解し、錠剤は一般に、原型を保ったままこの全体的な形を維持する。表面侵食製剤では、所定の期間にわたって活性剤の持続放出が可能となる。
【0079】
用語「微粉化する」および「微粉化された」は一般に、直径/サイズが、マイクロ粒子および/またはナノ粒子の一般的な範囲内であるように加工処理すること、または加工処理されている粒子を指す。
【0080】
用語「マイクロ粒子」は一般に、約1〜約999マイクロメートル(ミクロン、μm)の範囲にある直径を有する粒子を含む。
【0081】
用語「ナノ粒子」は一般に、約1〜約999ナノメートル(nm)の範囲にある直径を有する粒子を含む。
【0082】
用語「インスリン誘導体」は、インスリン作用を有する、インスリン由来のタンパク質およびペプチドを含み、例えば、リスプロ、B10Asp、およびHOE-901を含む。
【0083】
「インスリン分泌促進薬」は、主に膵臓β-細胞に作用して血液中へのインスリン分泌を促進することで血糖降下作用を示し、例えば、スルホニル尿素(例えば、トルブタミド、クロルプロパミド、グリベンクラミド(グリブリド)、グリピジド、グリメペリド、およびグリクラジド);ならびにメグリチニド類似体(例えばレパグリニド、ナテグリニド、メグリチニド、およびミチグリニド(KAD-1229))が含まれる。他のインスリン分泌促進薬は、例えば、K+-ATPチャネル阻害剤(例えば、BTS-67-582)、グルカゴン様ペプチド-1受容体アゴニスト(例えば、グルカゴン様ペプチド-1、エキセンディン-4、およびNN-2211)ならびにグルカゴン様ペプチド-1の作用を高める効果を有するジペプチジルペプチダーゼ-IV阻害剤である。一実施形態によると、インスリン分泌促進薬は、スルホニル尿素またはメグリチニド類似体である。
【0084】
用語「インスリン抵抗性改善薬」には、標的組織中でインスリンの作用を高めることで、血糖降下作用を示す薬剤が含まれ、例えば、ペルオキシソーム増殖剤応答性受容体(PPAR)-γアゴニスト(例として、ピオグリタゾン、ロシグリタゾン、およびシグリタゾンなどのチアゾリジンをベースとした化合物;またはGI-262570、JTT-501、YM-440、NN-622、およびKRP-297などの非チアゾリジンをベースとした化合物)、PPAR-γアンタゴニストならびにプロテインチロシンホスファターゼ阻害剤が含まれる。インスリン抵抗性改善薬には、例えば、インスリン抵抗性を改善する作用を有する医薬品が含まれ、例えば、ビグアニド(例として、メトホルミン、フェンホルミン、およびブホルミン、好ましくはメトホルミン)、PPAR-αアゴニスト(シンフィブラート、クロフィブラート、ベザフィブラート、およびクリノフィブラートなどのフィブラート系化合物ならびに非フィブラート系化合物)、抗肥満薬(例として、シブトラミンなどの5-ヒドロキシトリプタミン(5-HT)再取り込み阻害剤、オルリスタットなどのリパーゼ阻害剤、およびAJ-9677などのアドレナリンβ-受容体アゴニスト)が挙げられる。好ましいインスリン抵抗性改善薬には、メトホルミンなどのビグアニドが含まれるが、これらに限定されない。
【0085】
用語「インスリン模倣体」は、生理学的なインスリン作用、すなわち、多かれ少なかれインスリンとは独立してブドウ糖の細胞への取り込みを促進する作用を通じて血糖降下作用を発現する、インスリン誘導体以外の薬剤を指し、例えば、インスリン受容体活性化剤(例としてCLX-0901およびL-783281)ならびにバナジウムが含まれる。
【0086】
用語「α-グルコシダーゼ阻害剤」は、ブドウ糖の体内への吸収を抑制することを通じて、主に、腸管においてα-グルコシダーゼを阻害することによって、血糖降下作用を発現する薬剤を指し、例えば、アカルボース、ボグリボース、およびミグリトールが含まれる。
【0087】
用語「糖形成抑制剤」は、主に糖形成を阻害することを通じて、血糖降下作用を発現する薬剤を指し、例えば、グルカゴン分泌抑制剤(例として、M&B-39890Aおよびオクトレオチド)、脂肪酸分解阻害剤(例として、ニコチン酸誘導体およびカルニチンパルミトイルトランスフェラーゼ-1阻害剤)、ならびにグルコース-6-ホスファターゼ阻害剤が含まれる。
【0088】
用語「腎ブドウ糖再吸収阻害剤」は、尿細管においてブドウ糖再吸収を阻害する薬剤を指す。腎ブドウ糖再吸収阻害剤の主要な作用は、標的組織細胞への取り込み促進、腸管からの吸収抑制、または組織中の合成抑制による血糖降下作用を伴わない。好適な腎ブドウ糖再吸収阻害剤には、参照により本明細書に援用する米国特許公開番号2005/0143424に記載されているものが含まれるが、これらに限定されない。
【0089】
[送達剤化合物]
送達剤化合物は、参照により本明細書に援用する、米国特許第5650386号および第5866536号ならびに国際公開番号WO94/23767、WO95/11690、WO95/28920、WO95/28838、WO96/10396、WO96/09813、WO96/12473、WO96/12475、WO96/30036、WO96/33699、WO97/31938、WO97/36480、WO98/21951、WO98/25589、WO98/34632、WO98/49135、WO99/16427、WO00/06534、WO00/07979、WO00/40203、WO00/46182、WO00/47188、WO00/48589、WO00/50386、WO00/59863、WO00/59480、WO01/32130、WO01/32596、WO01/34114、WO01/44199、WO01/51454、WO01/70219、WO01/92206、WO02/02509、WO02/15959、WO02/16309、WO02/20466、WO02/19969、WO02/070438、WO03/026582、WO02/100338、WO03/045306、WO03/26582、およびWO03/057170に記載されているもののいずれかであってよい。
【0090】
送達剤化合物の非限定例には、N-(8-[2-ヒドロキシベンゾイル]アミノ)カプリル酸、N-(10-[2-ヒドロキシベンゾイル]アミノ)デカン酸、8-(2-ヒドロキシ-4-メトキシベンゾイルアミノ)オクタン酸、8-(2-ヒドロキシ-5-クロロベンゾイル-アミノ)オクタン酸、4-[(2-ヒドロキシ-4-クロロベンゾイル)アミノ]ブタン酸、およびこれらの塩が含まれる。好ましい塩には、一ナトリウム塩(モノナトリウム塩)および二ナトリウム塩(ジナトリウム塩)が含まれるが、これらに限定されない。
【0091】
一実施形態によると、送達剤化合物は、N-(8-[2-ヒドロキシベンゾイル]アミノ)カプリル酸または医薬として許容できるその塩である。
【0092】
別の実施形態によると、送達剤化合物は、N-(10-[2-ヒドロキシベンゾイル]アミノ)デカン酸またはこ医薬として許容できるその塩である。
【0093】
別の実施形態によると、送達剤化合物は、4-[(2-ヒドロキシ-4-クロロベンゾイル)アミノ]ブタン酸または医薬として許容できるその塩である。
【0094】
別の実施形態によると、送達剤化合物は、8-(2-ヒドロキシ-5-クロロベンゾイルアミノ)オクタン酸または医薬として許容できるその塩である。
【0095】
送達剤化合物は、カルボン酸またはそのナトリウム塩などの医薬として許容できる塩、ならびにこれらの水和物および溶媒和物の形であることが可能である。塩は、1価または多価の塩であってよく、例えばモノナトリウム塩およびジナトリウム塩(例えば、8-(2-ヒドロキシ-5-クロロベンゾイルアミノ)-オクタン酸のジナトリウム塩、N-(8-[2-ヒドロキシベンゾイル]アミノ)カプリル酸のジナトリウム塩、N-(10-[2-ヒドロキシベンゾイル]アミノ)デカン酸のジナトリウム塩)などが挙げられる。例として、参照により本明細書に援用する国際公開番号WO00/59863を参照されたい。送達剤化合物は、例えば、担体の溶解プロフィールの変更に及ぼす影響に応じて選択された異なる対イオンを含むことができる。
【0096】
送達剤化合物は、当技術分野において公知の方法、例えば、上記公開(例として、国際公開番号WO98/34632、WO00/07979、WO01/44199、WO01/32596、WO02/02509、WO02/20466およびWO03/045306)で議論されている方法で調製することができる。SNAC、SNAD、4-CNABならびにこれらの遊離酸およびその他の塩は、当技術分野において公知の方法、例えば、それぞれ参照により本明細書に援用する、米国特許第5650386号および第5866536号ならびに国際公開番号WO02/02509に記載されている方法で調製することができる。
【0097】
本発明の送達剤化合物の塩は、当技術分野において公知の方法で調製することができる。例えば、ナトリウム塩は、送達剤化合物をエタノールに溶解させ、水酸化ナトリウム水溶液を加えることによって調製することができる。
【0098】
送達剤化合物は、再結晶によって、または単独のもしくは縦一列に連結した、1つもしくは複数の固体クロマトグラフ担体上での分画によって精製することができる。好適な再結晶溶媒系には、アセトニトリル、メタノール、およびテトラヒドロフランが含まれるが、これらに限定されない。分画は、アルミナなどの適切なクロマトグラフ担体上で、移動相としてメタノール/n-プロパノール混合物を使用して;逆相クロマトグラフィーで、移動相としてトリフルオロ酢酸/アセトニトリル混合物を使用して;およびイオン交換クロマトグラフィーで、移動相として水または適切な緩衝液を使用して、行うことができる。陰イオン交換クロマトグラフィーを行う場合は、0〜500mMの塩化ナトリウム勾配を使用することが好ましい。
【0099】
送達剤は、-NHC(O)NH-、-C(O)NH-、-NHC(O)、-OOC-、-COO-、-NHC(O)O-、-OC(O)NH-、-CH2NH-NHCH2-、-CH2NHC(O)O-、-OC(O)NHCH2-、-CH2NHCOCH2O-、-OCH2C(O)NHCH2-、-NHC(O)CH2O-、-OCH2C(O)NH-、-NH-、-O-、および炭素-炭素結合からなる群から選択される結合基によって、送達剤に結合されたポリマーを含んでよい。ただし、ポリマーの送達剤はポリペプチドでもポリアミノ酸でもないことを条件とする。ポリマーは、哺乳動物に使用して安全な、交互コポリマー、ブロックコポリマー、およびランダムコポリマーを含む任意のポリマーであることができるが、これらに限定されない。好ましいポリマーには、ポリエチレン;ポリアクリレート;ポリメタクリレート;ポリ(オキシエチレン);ポリ(プロピレン);ポリプロピレングリコール;ポリエチレングリコール(PEG);およびこれらの誘導体、およびこれらの組合せを含むが、これらに限定されない。ポリマーの分子量は、通常約100〜約200,000ダルトンの範囲である。ポリマーの分子量は、好ましくは約200〜約10,000ダルトンの範囲である。一実施形態では、ポリマーの分子量は、約200〜約600ダルトンの範囲であり、より好ましくは約300〜約550ダルトンの範囲である。
【0100】
[活性剤]
本発明における使用に適した活性剤には、生物学的活性剤および化学的活性剤が含まれ、これには殺虫剤、薬物および治療薬が含まれるが、これらに限定されない。好適な活性剤には、胃腸管において、酸加水分解、酵素などによって効果が減少するもの、無効になるもの、または破壊されるものが含まれる。同様に、好適な活性剤としては、経口投与した場合に、サイズ、構造または電荷などの生理化学的特性が、吸収を禁止または妨害する高分子薬剤が含まれる。
【0101】
例えば、本発明における使用に適した生物学的または化学的活性剤には、以下のものが含まれるが、これらに限定されない。すなわち、タンパク質;ポリペプチド;ペプチド;ホルモン;多糖類、および特にムコ多糖類の混合物;炭水化物;脂質;極性が小さい有機分子(すなわち、500ダルトン以下の分子量を有する極性有機分子);その他の有機化合物;ならびに特に、それ自身では胃腸粘膜を通過しない(もしくは投与量の一部だけが通過する)化合物、および/または胃腸管において、酸および酵素によって化学開裂され易い化合物;あるいはこれらの任意の組合せが挙げられる。
【0102】
さらなる例としては、これらの合成由来、天然由来または組換え由来を含む以下のものが含まれるが、これらに限定されない:すなわち、ヒト成長ホルモン(hGH)、組換えヒト成長ホルモン(rhGH)、ウシ成長ホルモン、およびブタ成長ホルモンを含む、成長ホルモン;成長ホルモン放出ホルモン;成長ホルモン放出因子、α(例えば、インターフェロンアルファコン-1(カリフォルニア州ブリスベーンのInterMune,Inc.からInfergen(登録商標)として入手可能))、β、およびγを含むインターフェロン;インターロイキン-1;インターロイキン-2;亜鉛、ナトリウム、カルシウム、およびアンモニウムを含む対イオンを任意選択で有していてもよい、ブタ、ウシ、ヒト、およびヒト組換え型を含むインスリン;IGF-1を含むインスリン様成長因子;未分画ヘパリン、ヘパリン類似物質、デルマタン、コンドロイチン、低分子量ヘパリン、極低分子量ヘパリン、超低分子量ヘパリンを含むヘパリン;サケ、ウナギ、ブタ、およびヒトを含むカルシトニン;エリスロポエチン;心房性ナトリウム利尿因子;抗原;モノクローナル抗体;ソマトスタチン;プロテアーゼ阻害剤;副腎皮質刺激ホルモン、ゴナドトロピン放出ホルモン;オキシトシン;黄体形成ホルモン放出ホルモン;卵胞刺激ホルモン;グルコセレブロシダーゼ;トロンボポエチン;フィルグラスチム;プロスタグランジン;サイクロスポリン;バソプレシン;クロモリンナトリウム(クロモグリク酸ナトリウムもしくはクロモグリク酸二ナトリウム);バンコマイシン;デスフェリオキサミン(DFO);アレンドロネート、チルドロネート、エチドロネート、クロドロネート、パミドロネート、オルパドロネート、およびインカドロネートなどのビスホスホネート;そのフラグメントを含めた副甲状腺ホルモン(PTH);BIBN-4096BSおよび他のカルシトニン遺伝子関連タンパク質アンタゴニストなどの抗片頭痛剤;グルカゴン様ペプチド1(GLP-1);抗生物質、抗細菌薬、および抗真菌薬を含めた抗菌薬;ビタミン;これらの化合物の類似体、フラグメント、模倣体、もしくはポリエチレングリコール(PEG)修飾誘導体;またはこれらの任意の組合せ、が挙げられる。抗生物質の非限定例としては、グラム陽性作用を有する、殺菌性の、リポペプチド系、および環状ペプチド系の抗生物質、例えばダプトマイシン、ならびにこれらの類似体が含まれる。
【0103】
一実施形態によると、活性剤はインスリンである。
【0104】
別の実施形態によると、活性剤は、未分画ヘパリンまたは低分子量ヘパリンなどのヘパリンである。
【0105】
本発明の医薬組成物または単位剤形において使用する活性剤の量は、標的とする適応症を治療するために有効な量である。しかし、この量は、組成物を単位剤形で使用する場合の量に比べて少ない可能性がある。この理由として、単位剤形は複数種の送達剤/活性剤を含むことがあり、そのような組成物は分割された有効量を含むことがあるためである。したがって、総有効量は、合計で活性剤の有効量を含む累積単位で投与される可能性がある。その上、活性剤の有効量は、動物の年齢および体重、動物の健康状態ならびにその他の因子を含む多くの因子によって変化するものであることを当業者は認識するであろう。
【0106】
使用すべき活性剤の総量は、当業者に公知の方法で決定することができる。しかし、本発明の組成物は、上記送達剤なしで活性剤を含む組成物に比べて、より効果的に活性剤を送達することができ、従来の単位剤形または送達システムにおいて使用される活性剤量よりも、より低い量の活性剤を被験者に投与することができ、しかも、同一の血中濃度および/または治療効果を得ることが可能である。
【0107】
一実施形態によると、インスリンは、(活性剤の重量に基づいて)1日に被験者の体重1kg当たり約0.025〜約1.0mg(mg/kg/day)、約0.06〜約0.25mg/kg/day、または約0.09〜約0.19mg/kg/dayの用量で投与される。所望の用量を、単回投与または分割投与のどちらかとして投与することができる。
【0108】
一般に、活性剤の送達を促進する送達剤の有効量は、活性剤と共に投与される。一実施形態によると、送達剤量と活性剤量は、モルベースで、約100:1〜約1:1、約80:1〜約2:1、または約20:1〜約10:1の範囲である。送達剤対活性剤のモルベースの範囲は、送達剤と活性剤との特定の組合せに関しては、100:1を超える可能性がある。また、送達剤対活性剤の範囲は、送達剤と活性剤との特定の組合せの場合には、約1:1以下、例えば0.1:1以下である可能性がある。
【0109】
単位剤形はまた、賦形剤、崩壊剤、潤滑剤、可塑剤、着色剤、着香剤、矯味剤、糖類、甘味剤、および塩類のいずれか1つまたはいずれかの組合せを含むこともできる。
【0110】
本発明の組成物は、生物学的または化学的活性剤を、任意の動物に投与するのに有用である。動物には、ニワトリなどの鳥類、昆虫、魚類、爬虫類、哺乳動物(齧歯動物、水生哺乳動物、イヌおよびネコなどの家庭動物、ヒツジ、ブタ、ウシ、およびウマなどの家畜、ならびに好ましくはヒト、を含むが、これらに限定されない)が含まれるが、これらに限定されない。
【0111】
本発明の別の実施形態は、本発明の粒子を投与することによって、動物において、下記の表1に掲載したものなどの疾患を治療もしくは予防するための方法、または所望の生理学的効果を得るための方法である。好ましくは、所望の疾患の治療もしくは予防のための、または所望の生理学的効果を得るための粒子の有効量を投与する。活性剤に対する具体的な適応症は、参照により本明細書に援用するPhysicians' Desk Reference (58th Ed., 2004, Medical Economics Company, Inc.、モントベール、ニュージャージー州)に見い出すことができる。下記の表における活性剤には、これらの類似体、フラグメント、模倣体、およびポリエチレングリコール修飾誘導体が含まれる。
【0112】
【表1】
【0113】
【表2】
【0114】
【表3】
【0115】
【表4】
【0116】
【表5】
【0117】
【表6】
【0118】
[放出制御製剤もしくは持続放出製剤]
本発明の固体剤形は、胃での崩壊を抑制または遅延するように製剤し得る。本発明における使用に適した放出制御製剤は、例えば、腸溶性コーティングを含むか、または表面から侵食されるように製剤し得る。
【0119】
一実施形態によると、固体の経口剤形は、治療有効量の活性剤および送達剤を含み、固体の経口剤形は、経口投与した場合に、約250秒〜約650秒の崩壊時間を有する。別の実施形態では、経口投与した場合に、崩壊時間は約350秒〜約550秒である。一実施形態では、経口投与した場合に、崩壊時間は約60秒を超える。別の実施形態では、経口投与した場合に、崩壊時間は約400秒を超える。崩壊時間は、米国薬局方<701>に記載されている方法を用いて、水中で37±2℃にて測定することができる。
【0120】
本発明の固体剤形は、腸溶性コーティングで被覆してもよい。腸溶性コーティングは、固体剤形における薬物組成物または複数薬物組成物の放出を遅延させるための主要な制御としての役割を果たすことができる。腸溶性コーティングは、胃で原型を保ったままとどまり、固体剤形の状態で、胃への放出を抑制または遅延する。活性剤の放出は、固体剤形が腸に届くまで遅延される。ひとたび腸に入るとすぐに、高pHによって活性剤が放出される。腸溶性コーティングには、フタル酸ヒドロキシプロピルメチルセルロース、酢酸コハク酸ヒドロキシプロピルメチルセルロース、フタル酸酢酸ポリビニル、酢酸トリメリット酸セルロース、酢酸フタル酸セルロース、ポリ(メタクリル酸-アクリル酸エチル)、およびポリ(メタクリル酸-メタクリル酸メチル)が含まれるが、これらに限定されない。本発明に従って使用することができる他の腸溶性コーティングは、参照により本明細書に援用する米国特許第5851579号に記載されている。
【0121】
本発明の一実施形態では、腸溶性コーティングは、全錠剤または他の剤形に適用される。一実施形態では、腸溶性コーティングは、上記のマイクロ粒子および/またはナノ粒子を含む系などの多粒子系に適用される。
【0122】
本発明の固体剤形は、錠剤(もしくは他の剤形)の表面から、または多粒子系(例えば、上記のマイクロ粒子を含む系)の表面から侵食されるように製剤し得る。これらの表面侵食製剤は、崩壊というよりもむしろ表面からゆっくりと溶解する。表面侵食の速度を制御することによって、固体剤形の活性剤および薬物組成物の放出を遅延させることができる。表面侵食製剤は、固体の経口剤形が腸に届くまで、活性剤または薬物組成物の実質的な放出が起こらないように製剤し得る。
【0123】
[酵素阻害剤]
本発明の固体剤形(本発明のマイクロ粒子またはナノ粒子を含み、かつ/または上記の崩壊時間を有する)は、酵素阻害剤を含んでもよい。固体単位剤形に組み込まれる酵素阻害剤は、酵素による分解を受け易い可能性のあるインスリンまたは他の活性剤の崩壊を抑制することができる。酵素阻害剤は、参照により本明細書に援用する米国特許第6458383号に記載されている。
【0124】
一般に、阻害剤は以下の種類に分類することができる:すなわち、P-アミノベンズアミジン、FK-448、メシル酸カモスタット、およびグリココール酸ナトリウムを含めた、アミノ酸をベースとしない阻害剤;アミノボロン酸誘導体およびn-アセチルシステインを含めた、アミノ酸および修飾アミノ酸;バシトラシン、ホスフィン酸ジペプチド誘導体、ペプスタチン、アンチパイン、ロイペプチン、キモスタチン、エラスタチン、ベスタチン、ホスホラミンドン、ピューロマイシン、サイトカラシンポテトカルボキシペプチダーゼ阻害剤、およびアマスタチンを含めた、ペプチドおよび修飾ペプチド;アプロチニン(ウシ膵臓トリプシンインヒビター)、Bowman-Birkインヒビターおよび大豆トリプシンインヒビター、トリ卵白トリプシンインヒビター、トリオボインヒビター、およびヒト膵臓トリプシンインヒビターを含めた、ポリペプチドプロテアーゼ阻害剤;EDTA、EGTA、1,10-フェナントロリン、およびヒドロキシキノリンを含めた、錯化剤;ならびにポリアクリレート誘導体、キトサン、セルロース誘導体、キトサン-EDTA、キトサン-EDTA-アンチパイン、ポリアクリル酸-バシトラシン、カルボキシメチルセルロース-ペプスタチン、ポリアクリル酸-Bowman-Birkインヒビターを含めた、粘膜付着性ポリマーおよびポリマー-阻害剤結合体、である。
【0125】
酵素阻害剤の選択および濃度は、毒性、プロテアーゼの特異性および阻害剤の有効性に基づくものであり、当業者には明らかであろう。
【0126】
理論によって制限されることを所望することなく、阻害剤は、単独でまたは組み合わせて機能することができると考えられる:阻害剤は、例えば、酵素の基質結合部位で結合し、これによって基質への接近を阻止することによる競合阻害剤(この機序によって作用すると考えられる阻害剤の例は、アンチパイン、エラスタチン、およびBowman Birkインヒビターである。);基質に加えて酵素部位で同時に結合することができ、これらの結合部位が同一ではない非競合阻害剤;および/または、酵素構造の外部へ必須金属イオンが奪われることによって引き起こされる酵素活性の損失にもとづく錯化剤、が挙げられる。
【0127】
(実施例)
以下の実施例は、制限することなく本発明を例示するものである。他に特に指示がない限り、割合はすべて重量で示す。
【0128】
(実施例1)
1. 試験品
a. 部位特異的in situ実験および強制経口投与実験に使用する、共処理したインスリン/送達剤マイクロ粒子
組換えヒト亜鉛インスリン(50mg)および4-CNABナトリウム(7.5g)を脱イオン水50mlに溶かした。この澄明溶液を窒素気流で、室温にて24時間乾燥させた。得られた共処理ケーキを粉砕して微粒子にし、次いで40/60メッシュスクリーンを通して篩にかけ、特定の粒径範囲のマイクロ粒子を得た。本試験において使用したマイクロ粒子の粒径は、250〜420μmの範囲であった。これらのマイクロ粒子には、0.55重量%のインスリン、9.5重量%の水、および89.5重量%の送達剤が含まれていた。合計約90%(w/w)のインスリンを、この工程から回収した。
【0129】
粒子は、異なるサイズの目開き(850μm、425μm、250μm、150μm、45μm)を有する篩を通過させることによって測定した。この方法を用いて、中央粒径は、約45〜約850μm、約45〜約150μm、約150〜約250μm、約250〜約425μm、または約425〜約850μmの範囲であると決定することができる。
【0130】
マイクロ粒子中のインスリン含量を、逆相HPLC(Phenomenex column:Luna 3u C18(2) 100Å、150×4.6mm、3マイクロ;移動相:A、水中0.1%TFA;B、アセトニトリル中0.1%TFA;検出器:UV280nm)を用いて測定した。粒子の含水量を、737KF電量計を用いて測定した。
【0131】
b. マイクロ粒子を充填した強制経口投与用カプセル剤
ゼラチンカプセル剤(サイズ#9)をラットの試験に使用した。ラットの平均体重350mgに基づいて、マイクロ粒子の必要量を決定し、ゼラチンカプセル内に手で充填した。充填した各カプセル剤には、約16mg(インスリン0.0875mgに相当)のマイクロ粒子が入っていた。
【0132】
c. 強制経口投与実験用インスリン/送達剤の小型錠剤
インスリンと送達剤とを1:150(w/w)の比率でよく混合した。これは、0.67%(w/w)のインスリンに相当した。ラットの平均体重350mgに基づいて、インスリン0.175mgと送達剤26.26mgとを含む混合粉末の総量26.43mgを、Carverプレス中、1000psiの圧力下で直接圧縮して錠剤にした。円柱形の小型錠剤は、直径2mmおよび高さ6mmであった。
【0133】
d. インスリン/送達剤の物理学的混合物を充填した強制経口投与実験用カプセル剤
インスリンと送達剤とを1:150(w/w)の比率でよく混合した。ラットの平均体重350mgに基づいて、インスリンと送達剤との混合物の量を決定し、ゼラチンカプセル(サイズ#9)内に手で充填した。各カプセル剤には、26.43mgの混合物(インスリン0.175mgに相当)が入っていた。
【0134】
2. In Situ実験用の直接投与手法
直接投与手法についての概略図を図1に示す。清潔な白衣、マスク、安全保護眼鏡、手袋、および手術帽を使用して、汚染されていない環境で手術を行った。Sprague Dawleyラットに導入濃度として5%イソフルランを用いて麻酔状態を誘発させ、試験が完了するまで純酸素中2%イソフルランで麻酔を維持した。
【0135】
a. 胃への直接投与
血液採取用に右頚静脈にカテーテルを挿入した後、食道および気管上の皮膚を切開し、顎二腹筋の吻側腹部(保護用筋束)を確認し、食道へ向かって接近するように切開した。食道を一部切開し、6〜9cmある食道の切片に対して12cmのPE204管を挿入した。投与製剤を、この管を通じて導入し、先端の尖っていない鋼線を使用してマイクロ粒子を押し込んだ。投与後、胃から何も漏出しないようにするために、食道を3-0絹縫合糸で縛った。
【0136】
b. 空腸への直接投与
血液採取用に右頚静脈にカテーテルを挿入した後、白線を胸骨に向かって切開することによって腹腔を開き、次いで、剣状軟骨を露出した。空腸に最も近い部分を最初に確認した。近接した空腸の血管部分を一部挟み、投与管を遠位端に向かって導入した。投与後、投与管を除去し、2cmのPE206管を押し込んで、挟み傷が2cm管の両端の中央に位置するように設置した。縫合糸を管に巻きつけて両端で空腸と共に結び、少量のvetbond(登録商標)組織接着剤(ミネソタ州、セントポールの3Mから入手可能)で傷を閉じた。
【0137】
3. 強制経口投与手法
Sprague Dawleyラット(体重は約350グラムであった)において、強制経口投与によって試験を行った。小型錠剤またはカプセル剤を、套管針を有する改変された強制給餌用のチューブを使用して、ラットに経口投与した。ラットを約24時間絶食させ、ケタミン(44mg/kg)およびソラジン(1.5mg/kg)を筋肉内投与することによって麻酔した。所定の時間間隔で、血液サンプルを尾動脈から採取し、ブドウ糖およびインスリンのバイオアッセイ用の血漿または血清として、適切に調製した。この動物を実験終了時に屠殺し、局所毒性の何らかの徴候について、ラット胃腸粘膜を観察した。
【0138】
4. バイオアッセイ手法
ラットにおけるインスリンの血清濃度を、インスリンELISAテストキット(DSL Inc.)を使用して測定した。定量限界(LOQ)は、12.5μU/mLに規定されており、本アッセイの較正された線形範囲は最大250μU/mLである。グルコメーターを用いて、血糖値の変化を測定した。
【0139】
5. 結果
a. 部位特異的実験(In Situ)の結果
共処理したマイクロ粒子を胃および空腸に直接投与した後のインスリン濃度および血糖値の変化をそれぞれ図2および3に示す。個々のデータの一覧を表2〜5に示す。
【0140】
空腸への投与から得られたインスリン濃度は、各製剤からの最初のサンプリング点で最大値に達した(tmax=15分)。対応するブドウ糖のtminは、その後約30分で現れた。
【0141】
【表7】
【0142】
【表8】
【0143】
【表9】
【0144】
【表10】
【0145】
b. 錠剤およびカプセル剤を使用した強制経口投与実験からの結果
3種の被験製剤から得られたブドウ糖およびインスリンのデータをそれぞれ図4および5に示す。個々のデータの一覧を表6〜7に示す。胃および空腸への直接投与試験から得られた結果を、比較のために入れている。インスリンおよび送達剤の単純混合物についてのブドウ糖およびインスリンの個々のデータを表8に示す。
【0146】
共処理したインスリンおよび担体のマイクロ粒子を含むカプセル剤を投与したラット10匹の群において、平均最小ブドウ糖低下は、30分で基準線から70%であった。ラット1匹が15〜30分で死亡したが、低血糖症によるものと思われる。ラット6匹を30分でデキストロースを用いて救済した。さらにラット1匹を60分で救済し、30分で救済した6匹中2匹は、60分後死亡した。実験後のラットの剖検から、強制経口投与手法による胃腸刺激または胃腸損傷の徴候は見られなかった。同量のインスリンおよび担体を含む錠剤から得られた平均最小ブドウ糖低下は50%であった。
【0147】
対応するインスリン濃度を図5に示す。インスリン濃度は、カプセル中に共処理したマイクロ粒子を含むものが最も高く、後に錠剤および単純混合のカプセル剤が続く。
【0148】
共処理したマイクロ粒子を含むカプセル剤を使用した強制経口投与試験において、ラット2匹(10匹中)は高いインスリン吸収を示すことがわかった。保存サンプルを再分析したところ、上述の表6(3)に示すように、インスリン濃度はオリジナルサンプルから得たものとほぼ同一であった。2匹の高い応答例を含めたインスリン濃度および含めなかったインスリン濃度をそれぞれ図6および7に示す。N=10およびN=8から得られた、個々のおよび平均の、インスリンおよびブドウ糖プロフィールを図13〜16に示す。
【0149】
【表11】
【0150】
【表12】
【0151】
【表13】
【0152】
(実施例2)
1. 静脈、門脈および皮下実験の概要
a. 実験
齧歯動物において、静脈内投与、門脈内投与、および皮下投与を行い、絶対的バイオアベイラビリティー、インスリンの門脈内吸収、および皮下投与に対する相対的バイオアベイラビリティーを推定した。データを表9〜11にまとめる。静脈内投与から得られた平均インスリンAUC0→∞/投与量は、0.0093 min.kg/mlであった。絶対的バイオアベイラビリティーの推定値において、この値は一定であると仮定した。
【0153】
【表14】
【0154】
【表15】
【0155】
【表16】
【0156】
【表17】
【0157】
b. 結果
門脈に対する全身のインスリン比率は、約0.62(表10のデータから算出した)であることがわかった。したがって、門脈におけるバイオアベイラビリティーは、絶対的バイオアベイラビリティーを0.62で除することによって算出することができる。門脈のバイオアベイラビリティーから、経口送達による薬物吸収の推定値が得られる。皮下投与から得られた平均インスリンAUC0→t/投与量は、0.00516min.kg/mlであった。この値を使用して、皮下に対するバイオアベイラビリティーを推定する。静脈内のデータを除外して、t=0から最終サンプリング時点までの全AUC(すなわちAUC0→t)を算出した。
【0158】
ラットモデルにおいて、門脈内投与から得られたこれらの結果から、インスリンの最大絶対的バイオアベイラビリティーは、経口送達から、またはインスリンが100%胃腸(GI)吸収され門脈へと移行する他のいずれの手段によっても、約60%であると示唆される。次に、皮下(SC)から得られた絶対的バイオアベイラビリティーは、約56%である。
【0159】
バイオアベイラビリティーの推定値(絶対的バイオアベイラビリティー、門脈のバイオアベイラビリティー、皮下に対する相対的バイオアベイラビリティー、および皮下に対する門脈の相対的バイオアベイラビリティー)を図8〜12および表12にまとめる。胃および空腸へのin situ投与から得られた推定絶対的バイオアベイラビリティーを図8に示す。共処理したインスリン(0.5mg/kg)および送達剤(75mg/kg)を含むマイクロ粒子から得られたバイオアベイラビリティー値は、胃内投与の場合には5%、空腸内投与の場合には18%であった。
【0160】
インスリン0.5mg/kgおよび送達剤75mg/kgを含む製剤を使用して、ラットに強制経口投与した錠剤およびカプセル製剤から得られた推定絶対的バイオアベイラビリティーを図9に示す。バイオアベイラビリティー値は、インスリンおよび担体の単純混合物を含む錠剤で投与した場合には6%、カプセル剤で投与した場合には1.6%であった。
【0161】
【表18】
【0162】
【表19】
【0163】
【表20】
【0164】
【表21】
【0165】
(実施例3)
[模擬胃液中でのインスリンおよび4-CNABの安定性]
模擬胃液(SGF)中でのインスリンの安定性を、4-CNABの有無で評価した。インスリン(1 mg/ml)を含み、4-CNABモノナトリウム(1 mg/ml)を含む溶液と含まない溶液とを調製した。
【0166】
胃の酵素であるペプシンを含むSGFと含まないSGFとを調製した。SGF pH1.2を、米国薬局方NF26ガイドラインにより調製した。塩化ナトリウム2gおよびペプシン3.2gを計量して適切な容器に加え、脱イオン水を加えて体積を1リットルにした。必要であれば、濃HClまたは濃NaOHを加えることにより、pHを1.2に調整した。ペプシンを除外した第二のSGF溶液も調製した。
【0167】
SGFの50mlサンプル4つ(ペプシン有り2つおよびペプシン無し2つ)を、37℃に設定した循環水浴に連結されているジャケット付き容器中に置いた。この溶液を、磁気攪拌子を用いて10分間攪拌して溶液を37℃にし、熱平衡に達するようにした。50mgの4-CNABを、ペプシンを含むサンプル1つおよびペプシンを含まないサンプル1つに加え、これらの溶液を2〜3分間攪拌して4-CNABを溶かした。50mgのインスリンをこれらの各サンプルに加えた。インスリンの溶解後、溶液のサンプルを所定の時間間隔で採取し、濾過して直ちに、インスリンおよび4-CNABの含量についてHPLCで分析した。インスリンがすべて溶解した後に抜き取った最初のサンプルを、時間ゼロ(0)で採取したものとした。結果を表13に示す。
【0168】
【表22】
【0169】
本明細書で使用する用語「理論上の%」は、サンプルを採取した時点での、抜き取った溶液の濃度(mg/mL)を、実験のための測定成分の理論上の濃度(mg/mL)と比較したパーセントを意味する。HPLC分析の標準偏差は±5%であった。これらの結果は以下のことを示している。すなわち、インスリンは、ペプシンを含むSGF中で不安定である。これは、最初のサンプリング点でインスリンがわずか3.0%しか残存していなかった(97%のインスリンが分解した)ためである。一方、ペプシンを含まないSGF中では、インスリンは、少なくとも2時間まで安定である。
【0170】
(実施例4)
[模擬腸液中でのインスリンの安定性]
模擬腸液(SIF)中でのインスリンの安定性を、4-CNABの存在下および不存在下で評価した。
【0171】
膵臓の酵素を含むSIFと含まないSIFとを調製した。SIF pH7.5を、米国薬局方NF26ガイドラインにより調製した。SIFの調製は、リン酸二水素カリウム6.8gおよびパンクレアチン10gを適切な容器に加え、脱イオン水を加えて体積を1リットルにすることによって行った。必要であれば、0.2N水酸化ナトリウムを加えることにより、pHを7.5に調整した。膵臓の酵素であるパンクレアチンを除いた第二のSIF溶液も調製した。
【0172】
SIFの50mlサンプル4つ(パンクレアチン有り2つおよびパンクレアチン無し2つ)を、37℃に設定した循環水浴に連結されているジャケット付き容器中に置いた。この溶液を、磁気攪拌子を用いて10分間攪拌して溶液を37℃にし、熱平衡に達するようにした。50mgの4-CNABを、パンクレアチンを含むサンプル1つおよびパンクレアチンを含まないサンプル1つに加え、これらの溶液を2〜3分間攪拌して4-CNABを溶かした。50mgのインスリンをこれらの各サンプルに加えた。インスリンの溶解後、溶液のサンプルを所定の時間間隔で採取し、直ちに、インスリンおよび4-CNABの含量についてHPLCで分析した。結果を表14に示す。
【0173】
【表23】
【0174】
これらの結果は、インスリンが、パンクレアチンを含まないSIF中では安定であり、酵素が存在すると分解するということを示している。インスリンは、酵素を含むSGF中および含まないSGF中に比べて、酵素を含むSIF中および含まないSIF中で、より安定である。最初のサンプリング時点(0分)で、酵素を含むSGF中にはインスリンが3.0%しか残存していなかったが、SIF中にはインスリンが58.9%および66.9%残存していた。
【0175】
(実施例5)
[製剤がインスリンの吸収および作用に及ぼす影響]
表15に示す6種のインスリン含有製剤を以下のように調製した。
【0176】
【表24】
【0177】
Polyplasdone XLは、デラウェア州ウィルミントンのInternational Specialty Productsから入手可能である;Emcocel HD90、Prosolv HD90、Emcompress、およびAnhydrous Emcompressは、ニューヨーク州パターソンのJRS Pharmaから入手可能である。
【0178】
これらの製剤をアカゲザルに、100mg/kgの4-CNABおよび13U/kgインスリンを含む投与量で摂取させた。各群4匹(雄2匹および雌2匹)のアカゲザルを、少なくとも投与約12時間前から投与4時間後まで絶食させた。水は、投与約1時間前から投与2時間後まで与えず、その後は自由に摂取させた。投与後は水5mlで流し込んだ。投与15分前、ならびに投与5、10、15、20、30、45分後および1、1.5、2、3、4時間後に、静脈穿刺で血液サンプル(各約2mlずつ)を採取した。各血液サンプルを2部に分けた。1部は、室温で凝固させ、2〜8℃で10分間、3000rpmで遠心分離した。得られた血清を2等分し、発送まで-70℃で保存した。一方のサンプルは、ELISAによるインスリン分析用に、ドライアイスを入れてEmisphereに発送し、もう一方は血清中ブドウ糖分析用にCROによって保管された。血液のもう1部は、氷水浴上で30分までの間静置し、2〜8℃で10分間、3000rpmで遠心分離した。得られた血漿を、HPLCによる4-CNABの含量分析用に、ドライアイスを入れてEmisphereに発送した。各製剤をアカゲザル4匹に投与した。ただし、製剤1は、アカゲザル8匹に投与した。血液サンプルを上記のように所定の間隔で採取し、インスリンおよびブドウ糖濃度について分析した。結果を表16ならびに図17および18に示す。
【0179】
【表25】
【0180】
崩壊時間を、米国薬局方<701>に記載されている方法を用いて、水中で37±2℃にて測定した。水を入れた複数の管を、37±2℃に維持した水浴中に浸した組み立て式バスケットラックに置いた。組み立て式バスケットラックは一定周期で管を昇降する。錠剤を管に置き、完全に崩壊したかどうかを決定するために定期的に調べた。各錠剤を異なる6本の管で試験した。1つまたは2つの錠剤がなかなか崩壊しない場合には、追加の錠剤でこの手法を繰り返した。インスリンの平均最大濃度(Cmax)を、上記のように測定したインスリンの血清濃度に基づいて、各群について測定した。実験中、これらの霊長類における血糖値が非常に低くなる場合(<1mmol/L)には、血糖を安全値まで上昇させるために、これらにデキストロースを投与する。各群についての平均Cmaxおよび救済したアカゲザルの数を表17に示す。
【0181】
【表26】
【0182】
(実施例6)
[腸溶性錠剤の調製]
カプセル剤の製造は、150単位インスリン、200mg 4-CNAB、0.4%w/wポビドン、〜29.1%w/w Emcompress、1%w/w SLS、および1%w/wステアリン酸マグネシウムを含む製剤300mgを、サイズ2の白色不透明カプセルに封入することによって行った。これらのカプセルをまず、Opadry clearを含むサブコートで被覆して5%増量し、次いで腸溶性コートで被覆して20%増量し、カプセルにおける全増量分を25%にした。
【0183】
錠剤については、上記の製剤300mgを押圧することによって錠剤を製造した。10%増量腸溶性コートを適用した。サブコートおよび腸溶性コートの処方を下表18に示す。
【0184】
【表27】
【0185】
Opadry(登録商標) Clearは、ペンシルベニア州ウエストポイントのColorconから入手可能である。
【0186】
Milli Q Waterは高度に精製された水であり、マサチューセッツ州ビラリカのMilliporeから入手可能である。
【0187】
Eudragit L30D55は、ニュージャージー州パーシッパニーのDegussa AGから入手可能である。
【0188】
腸溶性コートの有効性を確認するために、被覆したカプセル剤および錠剤を、0.1N HCl中に2時間、またはpH6.8リン酸緩衝液中に1時間置いた。被覆したカプセル剤および錠剤は、0.1N HClに溶けなかったが、pH6.8リン酸緩衝液には溶けた。
【0189】
(実施例7)
[ヘパリンで被覆したSNACマイクロビーズ]
5gのSNACおよび0.5gのステアリン酸マグネシウムを混合した。混合粉末0.02gをダイに送り込んだ。SNACおよびステアリン酸マグネシウムの小ビーズを1200 PSI barの圧力で製造した。このビーズは、約0.2mm〜約2.0mmのサイズの丸/ボール形状を有していた。次いで、SNACビーズを液体形態のヘパリン2.5gで回転方式によって被覆し、真空オーブンで40℃にて10時間乾燥させた。
【0190】
(実施例8)
[ヘパリン含有微粉化SNAD]
SNADを、35メッシュTyler標準篩を通して選別した。SNADの粉砕は、250mLステンレス製粉砕ジャーが装備されたGlen Mills, Model S100遠心ボールミル(ニュージャージー州クリフトン)を使用して行い、かつ直径30mm(440c)ステンレス製ボールを使用した。検討した加工処理条件は、(1)使用するボールの数、(2)粉砕持続時間、(3)粉砕速度、および(4)粉砕ジャーへの全投入量であった。Scirocco 2000乾燥用付属品が装備されたMalvern Mastersizer 2000を、粒径測定に使用した。2θ範囲が5〜40°2θで走査する、Kratos XRD 6000(バージョン4.1)X線粉末回折装置を、結晶化度の変化をモニターするために使用した。発散スリットは1°、散乱スリットは1°および受光スリットは0.3mmであった。Brinkmann737KF電量計を含水量の測定に使用し、さらにQuantachrome Nova 3000 Series Surface Area Analyzerを比表面積の測定に使用した。
【0191】
これらの結果から、事前選別したSNADの粒径分布はd(0.1)=1.6μm、d(0.5)=10.5μm、およびd(0.9)=314.9μmであることが示された。1〜5個の範囲にある種々の数のボールを使用して得られたデータから、投入するボールの最適数は2個であることが示された。2個のボールを使用することにより、d(0.1)=1.1μm、d(0.5)=12.0μm、およびd(0.9)=154.3μmの粒径が得られた。
【0192】
ボールの数および投入量を固定して、粉砕時間の効果を評価したところ、120分という粉砕時間が最適であり、d(0.1)=2.0μm、d(0.5)=15.4、およびd(0.9)=62.9μmの粒径分布が得られることが示された。
【0193】
粉砕速度100、300、および500rpmについて評価したところ、最適粉砕は300rpmで得られることが示された。この速度によって、粒径分布は、未粉砕のSNADのd(0.9)=314.9μmと比較して、d(0.9)=62.9μmが得られた。
【0194】
250mL粉砕ジャーに対して、投入量37mLの場合に、75および112mLに比べて、より良い粉砕を実現した。X線粉末回折分析から、SNADに関しては、粉砕によって結晶化度の変化が起こらないことが示された。カールフィッシャー含水量測定により、含水量の著しい変化は全くないことが示された。
【0195】
次いで、上記SNADをヘパリンと混合した。
【0196】
(実施例9)
[微粉化ヘパリン含有微粉化SNAC]
SNACおよびヘパリンを、実施例8に記載の手法で、2個のボールを用いて200rpmで120分間、別々に微粉化し、次いで一緒に混合した。微粉化SNACは、7.574μmのd(0.5)を有していた。下表19に示す製剤を含むSNAC/ヘパリンカプセル剤の調製は、これらの製剤をハードゼラチンカプセルに手で詰め込むことによって行った。
【0197】
【表28】
【0198】
ヘパリン、SNAC、およびラウリル硫酸ナトリウムを混合した。これとは別に、PEG300、モノカプリル酸プロピレングリコール、および水(製剤Bについて)を混合した。液体のPEG300/モノカプリル酸プロピレングリコール混合物の50%を乳鉢に移した。ヘパリン、SNAC、およびラウリル硫酸ナトリウムを混合した粉末を少しずつ加え、液体と共に乳鉢および乳棒で粉砕した。次いで、得られた混合物でカプセルを充填した。
【0199】
(実施例10)
[微粉化SNAC/ヘパリン]
ヘパリン(118.5mg/投与1回分(22,500rpm))およびSNAC(125mg/投与1回分)を乾式混合し、35メッシュスクリーンを通して選別し、ボールミルで約4分間粉砕した。混合物をカプセル(Capsugel Size 1カプセル(サウスカロライナ州グリーンウッド))に詰め込んだ。
【0200】
これらのカプセル剤をアカゲザル(サル1匹につき2カプセル)に以下の手法で投与した。体重3.5〜5.0kgのアカゲザルを、実験前に一晩絶食させ、投与約2時間後に飼料を元に戻した。水は、投与に使用した量を除いて、投与30分前から投与30分後まで与えなかった。ピルガンを使用して口の奥に各剤形を供給した。剤形放出後、逆浸透水5mlを口腔内に投与し、嚥下し易くした。供給後、カプセルが嚥下されたことを保証するために口腔を検査した。血液サンプルからの抗因子Xaを6時間にわたって測定した。
【0201】
結果を図19に示す。
【0202】
(実施例11)
[微粉化SNAC/ヘパリン]
下表20に示す微粉化SNAC/ヘパリンを含むカプセル剤を以下のように調製した。
【0203】
ヘパリンおよびSNACの溶液を以下のように調製した。ヘパリンおよびSNACの必要量を量り分け、予め水酸化ナトリウムを用いてpHを約8に調整しておいた水を加えた。得られた溶液のpHは、約7.3〜7.5の範囲であった。水酸化ナトリウムを用いて溶液のpHを約8のpHに調整した。次いで、この溶液をRotoVap装置内で真空下、50℃にて乾燥させた。蒸発は、以下に概略を述べるプログラムを使用して行った。
1. 760から200torrへの真空の迅速な減少
2. 2分かけて200から100torrへの真空圧の減少
3. 2分かけて100から50torrへの真空圧の減少
4. 4分かけて50から25torrへの真空圧の減少
5. 4分かけて25から15torrへの真空圧の減少
6. 2分かけて15から10torrへの真空圧の減少
7. 30分かけて10±2torrおよび70rpmでの蒸発
8. 手動で50rpmに切り替え、4時間蒸発を継続
【0204】
サンプルを一晩真空乾燥させた。次いで、得られた粉末を微粉化し、カプセルに充填して所望する用量とした。
【0205】
【表29】
【0206】
本発明は、本明細書に記載されている特定の実施形態によって範囲を限定されない。実際に、本明細書に記載されているものに加えて、本発明の種々の変更例は、上述の説明および添付の図から、当業者には明らかになるであろう。かかる変更例は、添付の特許請求の範囲内に含まれることが意図される。
【0207】
特許、特許出願、公開、製品に関する説明およびプロトコルが本出願を通じて引用されているが、これらの開示はすべての目的のために参照により本明細書に全体を援用する。
【特許請求の範囲】
【請求項1】
(a)インスリンの治療有効量;および
(b)送達剤
を含む固体剤形であって、経口投与した場合に、少なくとも60秒の崩壊時間を有する固体剤形。
【請求項2】
腸溶性コーティングをさらに含む、請求項1に記載の固体剤形。
【請求項3】
表面侵食性製剤である、請求項1に記載の固体剤形。
【請求項4】
酵素阻害剤をさらに含む、請求項1から3のいずれか一項に記載の固体剤形。
【請求項5】
999マイクロメートル未満の中央粒径を有する粒子を含む、請求項1から4のいずれか一項に記載の固体剤形。
【請求項6】
請求項1から5のいずれか一項に記載の固体剤形の治療有効量を投与することを含む、ヒトにおける、耐糖能の低下、初期の糖尿病、もしくは後期の糖尿病を治療する方法、またはグルコース恒常性を得る方法。
【請求項7】
前記医薬製剤を、連用を基礎として投与することを含む、請求項6に記載の方法。
【請求項8】
請求項1から5のいずれか一項に記載の固体剤形の治療有効量を、ヒト糖尿病患者に連用を基礎として経口投与することを含む、前記ヒト糖尿病患者を治療する方法。
【請求項9】
請求項1から5のいずれか一項に記載の固体剤形の治療有効量を、患者に連用を基礎として経口投与することを含む、インスリンを必要としている患者における、インスリンの連用に伴う全身性高インスリン血症の発生率を減少させる方法、および糖尿病を治療する方法。
【請求項1】
(a)インスリンの治療有効量;および
(b)送達剤
を含む固体剤形であって、経口投与した場合に、少なくとも60秒の崩壊時間を有する固体剤形。
【請求項2】
腸溶性コーティングをさらに含む、請求項1に記載の固体剤形。
【請求項3】
表面侵食性製剤である、請求項1に記載の固体剤形。
【請求項4】
酵素阻害剤をさらに含む、請求項1から3のいずれか一項に記載の固体剤形。
【請求項5】
999マイクロメートル未満の中央粒径を有する粒子を含む、請求項1から4のいずれか一項に記載の固体剤形。
【請求項6】
請求項1から5のいずれか一項に記載の固体剤形の治療有効量を投与することを含む、ヒトにおける、耐糖能の低下、初期の糖尿病、もしくは後期の糖尿病を治療する方法、またはグルコース恒常性を得る方法。
【請求項7】
前記医薬製剤を、連用を基礎として投与することを含む、請求項6に記載の方法。
【請求項8】
請求項1から5のいずれか一項に記載の固体剤形の治療有効量を、ヒト糖尿病患者に連用を基礎として経口投与することを含む、前記ヒト糖尿病患者を治療する方法。
【請求項9】
請求項1から5のいずれか一項に記載の固体剤形の治療有効量を、患者に連用を基礎として経口投与することを含む、インスリンを必要としている患者における、インスリンの連用に伴う全身性高インスリン血症の発生率を減少させる方法、および糖尿病を治療する方法。
【図1】

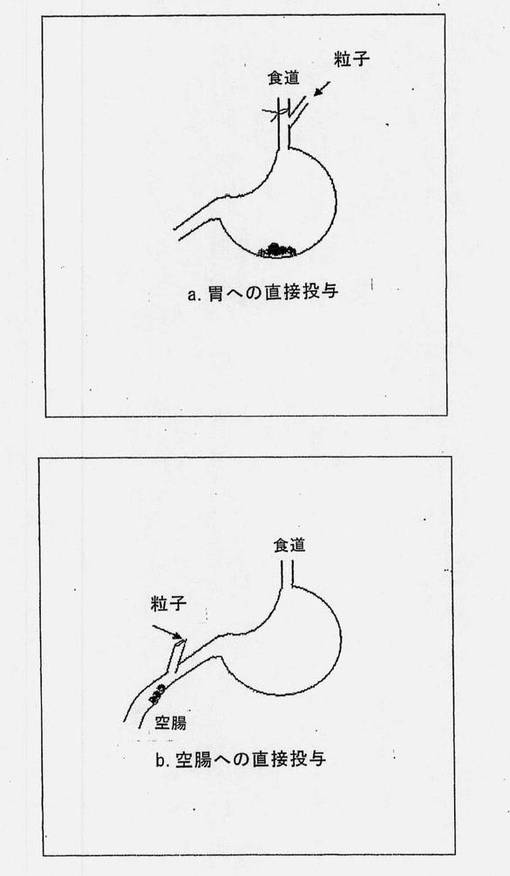
【図2】
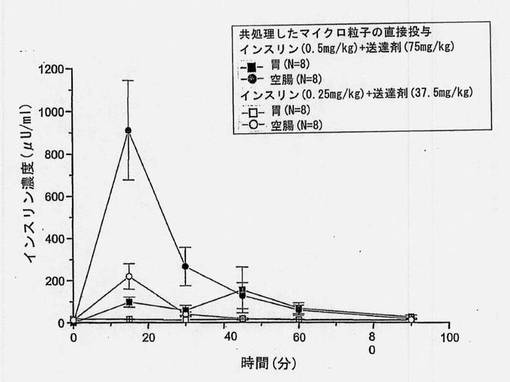
【図3】
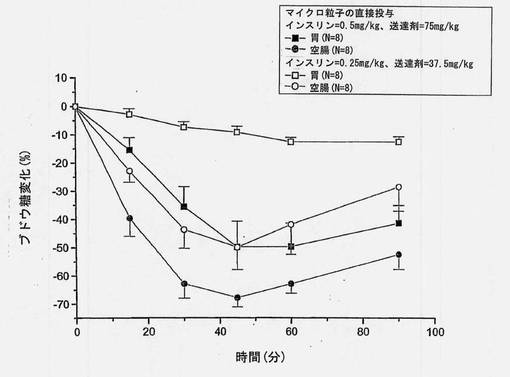
【図4】
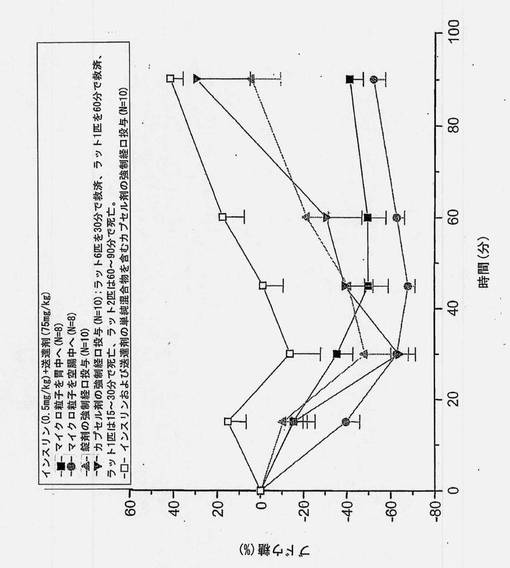
【図5】
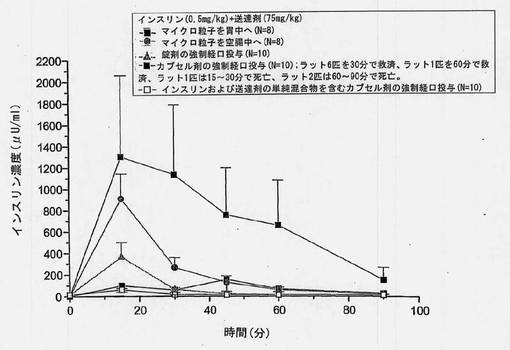
【図6】
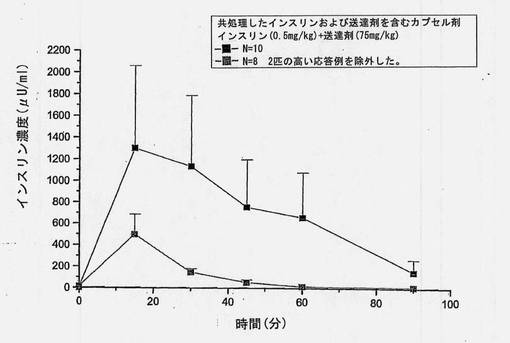
【図7】
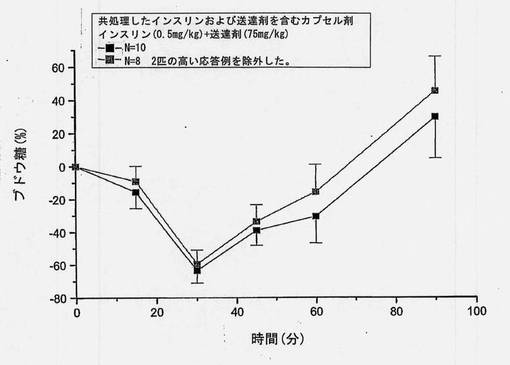
【図8】
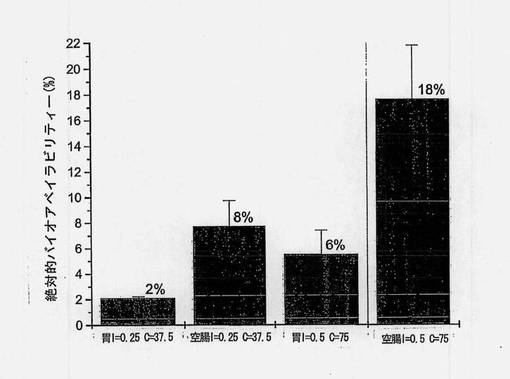
【図9】
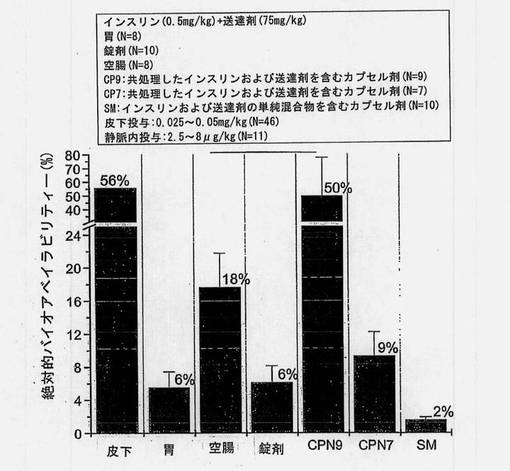
【図10】
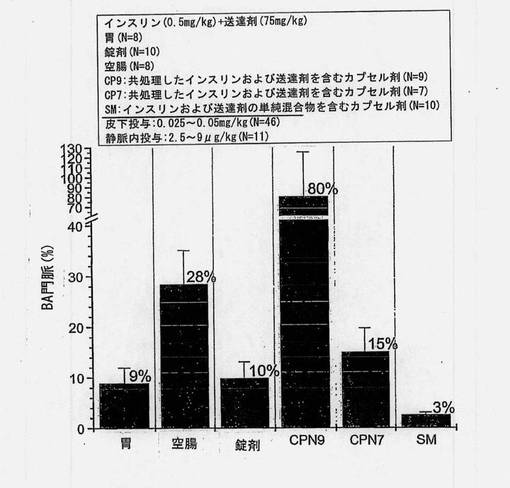
【図11】
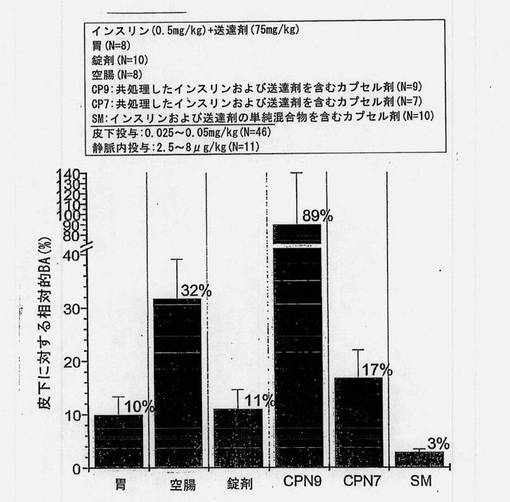
【図12】
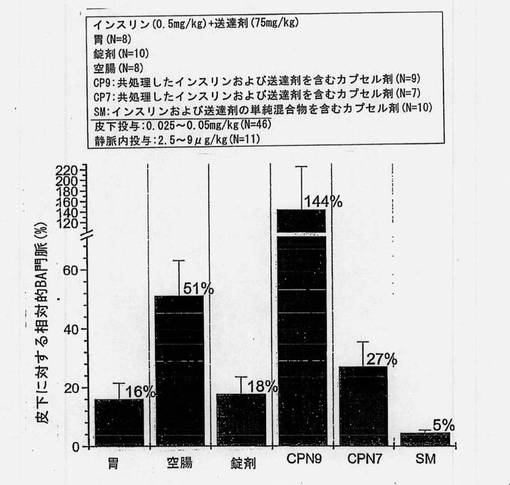
【図13】
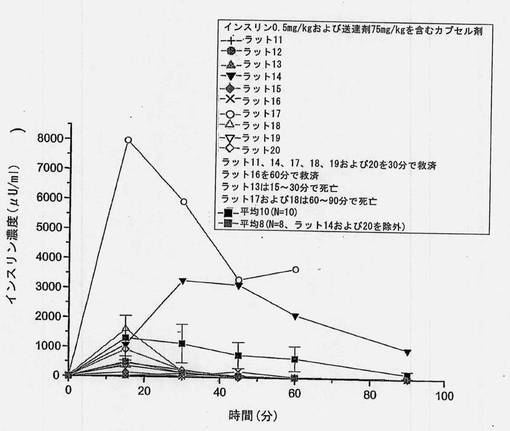
【図14】
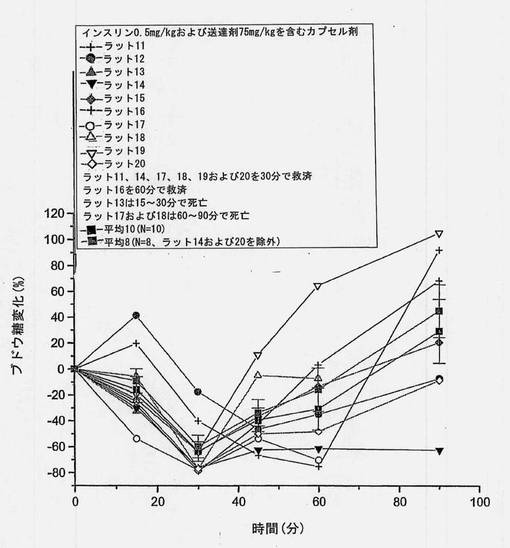
【図15】
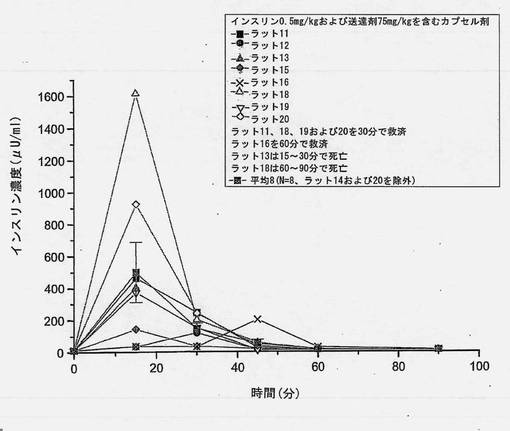
【図16】
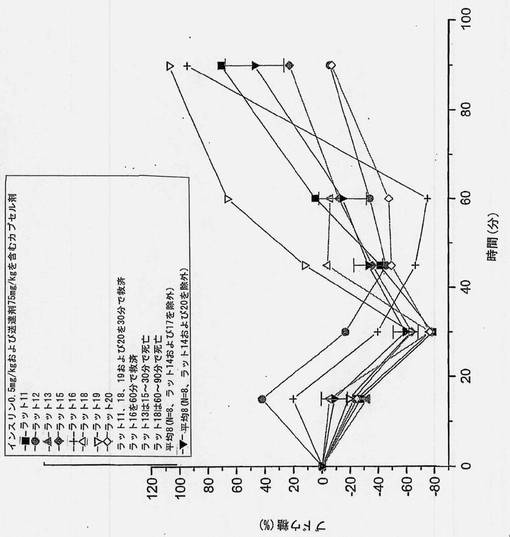
【図17】
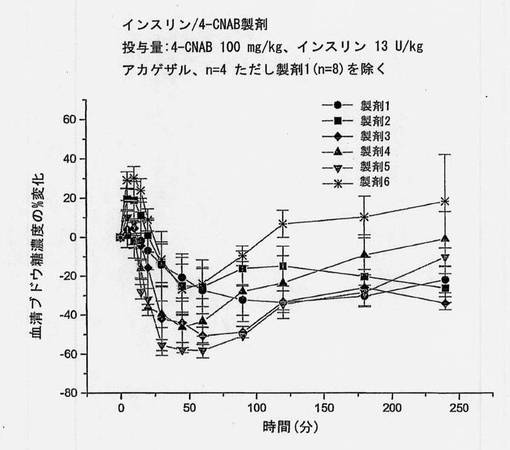
【図18】
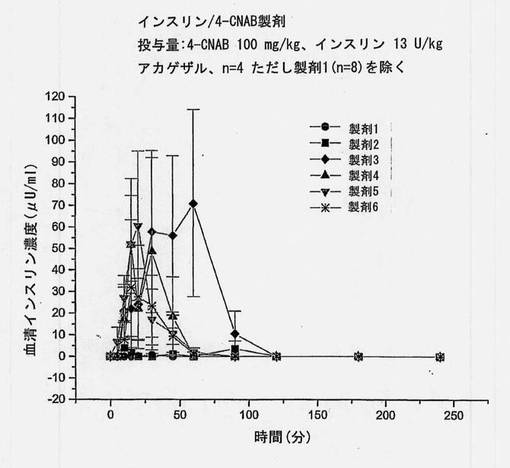
【図19】
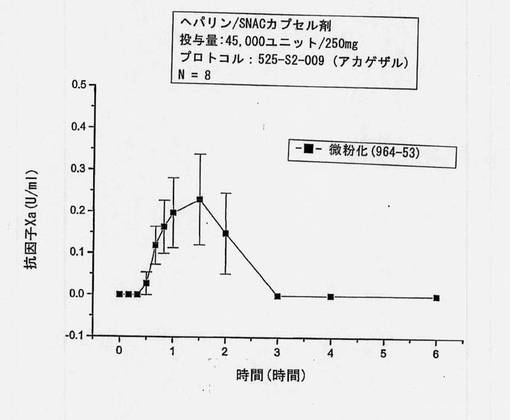
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【公開番号】特開2012−121923(P2012−121923A)
【公開日】平成24年6月28日(2012.6.28)
【国際特許分類】
【出願番号】特願2012−55453(P2012−55453)
【出願日】平成24年3月13日(2012.3.13)
【分割の表示】特願2007−525876(P2007−525876)の分割
【原出願日】平成17年8月15日(2005.8.15)
【出願人】(500139958)エミスフェアー・テクノロジーズ・インク (28)
【Fターム(参考)】
【公開日】平成24年6月28日(2012.6.28)
【国際特許分類】
【出願日】平成24年3月13日(2012.3.13)
【分割の表示】特願2007−525876(P2007−525876)の分割
【原出願日】平成17年8月15日(2005.8.15)
【出願人】(500139958)エミスフェアー・テクノロジーズ・インク (28)
【Fターム(参考)】
[ Back to top ]