
逆相カラムクロマトグラフィーを使用したヘパリンまたは低分子量ヘパリンを構成する特定グループの測定方法
本発明は、第四アンモニウム塩を動的にコートした固定相を使用する高速液体クロマトグラフィーによってヘパリン、低分子量ヘパリン、超低分子量ヘパリン及びオリゴ糖を分析する方法を提供する。本発明の方法はサンプルを前処理なしに分析するため、または、部分解重合もしくは完全解重合させ場合によっては還元させたサンプルを分析するために使用し得る。糖の特異的検出が可能である。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ヘパリン、低分子量ヘパリン(LMWH)または超低分子量ヘパリン(ULMWH)を構成するオリゴ糖の直接分析方法を提供する。
【背景技術】
【0002】
へパリンは、天然ソースから抽出されたグリコサミノグリカンファミリーの生物活性物質であり、有効な抗凝固性及び抗血栓性を有している。これらは術後静脈血栓の治療に特に有用である。
【0003】
ヘパリンソースからLMWHを製造するために、長いヘパリン多糖鎖を低分子量の短い鎖に分割する。これは化学的または酵素的な解重合によって行われる。例えば、エノキサパリンの場合、解重合プロセスは2つの競合的化学反応、即ち、β除去とベンジルエステルの加水分解とを含む。その結果として、平均分子量約4500DaのLMWH多糖鎖が得られる。LMWHは非分割ヘパリンと同様に、いくつかの多糖鎖に沿って分布した特定の5糖配列でATIIIに結合することによって凝固を阻害する。
【0004】
LMWHの認可製品の製造業者は各々で異なる解重合プロセスを使用している。2つの製造業者が同じプロセスを使用することがなければ、このようなプロセスの違いから、異なる化学構造を持ち、従って異なる薬理学的活性及び異なる臨床使用承認表示をもつLMWHが製造される。
【0005】
従って、LMWHはそれらの製造に使用された解重合プロセスによって構造的に識別される(R.J.Linhardtら,Seminars in Thombosis and Hemostatis 1999;25(3 supp.):5−16)。その結果としてLMWHはヘパリンよりも不均質である。異なるプロセスの各々が特有の極めて複雑な構造的修飾を多糖鎖に生じさせる。これらの修飾は、鎖の長さ、鎖の配列、及び、構造フィンガープリントの違いを含む。その結果として、異なるLMWHの各々は、異なる薬理学的プロフィル及び異なる承認臨床表示を有している。
【0006】
米国でLovenoxRTM及び他のいくつかの国々でClexaneRTMという商品名で販売されているエノキサパリンナトリウムを純ヘパリンから製造するプロセスでは、水相アルカリ性解重合プロセスによってオリゴ糖鎖の還元性末端のグルコサミンに部分的ではあるが特徴的な変換が生じる。
【0007】
この変換の第一段階は、グルコサミンとマンノサミンとの間の可逆的エピマー化から成る(T.Toidaら,J.Carbohydrate Chemistry,15(3),351−360(1996))。第二段階は、グルコサミンの6−O−脱硫酸であり、これによって “1,6−無水”と呼ばれる誘導体が形成される(国際特許出願WO01/29055)。
【0008】
【化4】
【0009】
6−O−硫酸化された末端グルコサミンを有しているオリゴ糖鎖の場合にこの種の誘導体が得られる。
【0010】
1,6−無水結合で修飾された末端をもつオリゴ糖鎖のパーセンテージはLovenoxRTM(エノキサパリンナトリウム)のオリゴ糖混合物の構造的特徴である。現在知られている範囲では、LovenoxRTM(エノキサパリンナトリウム)の成分の15%−25%が鎖の還元性末端に1,6−無水構造を有している。
【0011】
最近では、例えば公開された米国特許出願No.2002−0055621A1に記載されているように、強塩基の存在下の解重合を使用するヘパリンフラグメントの新しい製造方法で約1500−約3000ドルトンの範囲の重量平均分子量をもつ超低分子量ヘパリン(ULMWH)が得られるようになっている。該特許の記載内容は参照によって本発明に含まれるものとする。
【0012】
ヘパリン、低分子量ヘパリン、超低分子量ヘパリンが元のままの(intact)分子であるかまたは酵素消化物であるかを分析することは、混合物が多分散性であり、また、分子の官能化度が高くまた変わり易いため、難しい作業である。この問題に取組むためにクロマトグラフィー法及び電気泳動法が開発された。毛管電気泳動(CE)はヘパリンオリゴ糖を分離するために使用される(M.Guerriniら,Glycobiology 12(2002)713−719;R.J.Linhardt.ら,BioMethods(Basel)9(1997)183−197)。しかしながら、CEの選択性は液体クロマトグラフィーに比べてかなり低い。更に、(UVによる)CE検出ではしばしば、不十分な感度が原因で問題が生じる。さらに、この分析方法のために開発された分離を分取技術に適応させることができないこともCEの欠点のひとつである。
【0013】
いくつかの場合には、クロマトグラフィー分離を伴うことなくMALDI−TOFマススペクトロメトリー分析を行うことができ、これを予備的なヘパリンオリゴ糖分析方法及び配列決定ツールとして使用してもよい(H.Sakaguchiら,J.Biochem.129(2001)107−18;A.J.Rhombergら,Proc.Nalt.Acad.Sci.USA 95(1998)4176−4181;L.Sturialeら,Semin.Thromb.Hemost.27(2001)465−472)。しかしながらMALDI−TOFマススペクトロメトリー分析は複合混合物に対する分解能がない。また、費用も高い。
【0014】
これらの理由から、硫酸化オリゴ糖の分離には液体クロマトグラフィーが好ましい。塩化ナトリウムを基剤とする移動相による強アニオン交換(SAX)はヘパリン由来のオリゴ糖を分解するクロマトグラフィー方法の1つである。更に、HPLCまたは低圧クロマトグラフィーによるゲル浸透クロマトグラフィーは、オリゴ糖のサイズ分離及び脱塩に使用できる強力なツールである。これらの方法では移動相に塩化ナトリウムを使用することが2つの重大な欠点を生む。即ち、NaClを含む移動相は塩の腐蝕作用を克服するように特殊処理されたクロマトグラフを必要とする。また、215nmよりも低い波長に強力なUV吸光度を有している。このため、ある種のオリゴ糖に対してはこの検出方法の使用が制限される。
【0015】
このような理由で、SAXクロマトグラフィーの固定相の開発が行き詰っていた。SAXクロマトグラフィーの使用はしばしば5−10未満の硫酸基を含有するオリゴ糖の分離に限定されており、高度に硫酸化した生成物の場合には固定相の開孔への物質移動が少ない結果として重大なピーク拡大が頻繁に生じる。C18またはC8結合シリカ上のイオン対クロマトグラフィーをテトラブチルアンモニウム含有移動相で代替する方法(N.K.Karamanosら,J.Chromatogr.765(1997)169−179;C.F.Moffatら,Eur.J.Biochem.,197(1991 449−459)または等価の方法も、アセトニトリル溶出勾配の終点で観察される選択性の低下を理由として低硫酸化種に限定されている。イオン対クロマトグラフィーの注目を呼ぶ開発が最近Linhardtらによって報告された(J.Chromatogr.,1014(2003)215−223)。この方法はマススペクトロスコピーと容易に組合せることができるであろうが、その使用はやはり高硫酸化オリゴ糖に対する選択性によって限定される。セチルトリメチルアンモニウム(CTA)のようなより親油性のアンモニウム塩とのイオン対合は難しい。その理由は、このような塩は、硫酸化オリゴ糖と共に著しく非極性のイオン対を形成し、これらの多くが水性溶媒に不溶性であるからである。
【0016】
CTAのようなイオン対化剤を予め吸着させた固定相を使用する無機アニオンの分離はItoらによって一度ならず行われた(J.Chromatogr.,549(1991)265−272;Anal.Chem.,63(1991)273−276.;J.Chromatogr.,598(1992) 237−241)。
【発明の開示】
【発明が解決しようとする課題】
【0017】
従って、本発明の目的は、20個までの硫酸基基をもち十二糖という大きさの高電荷のオリゴ糖を分離及び分析できる新規な配列決定方法を提供することである。
【0018】
本発明の別の目的は、ヘパリン、低分子量ヘパリン、超低分子量ヘパリン及びオリゴ糖から選択されたサンプルのアッセイ方法を提供することである。本発明の方法では、第四アンモニウム塩をコートした逆相カラムを使用したオリゴ糖の複合混合物のクロマトグラフィー分離及び分析によってサンプルを分析する。これに関連した本発明の特徴によれば、逆相カラムがC8またはC18カラムである。
【課題を解決するための手段】
【0019】
1つの実施態様では、本発明の方法が、C8またはC18逆相カラムに第四アンモニウム塩溶液を動的にコートすることによって調製した固定相を使用する分析用高速液体クロマトグラフィーを使用する。(逆相がC8またはC18に限定されないこと、他の逆相もこの目的に適合し得ることは理解されよう。)
【0020】
本発明の1つの実施態様では、固定相がセチルトリメチルアンモニウム塩またはセチルピリジニウム塩で動的にコートされたC8またはC18逆相カラムである。逆相カラムは例えばHypersil BDSでよい。
【0021】
本発明において、“ヘパリン”という用語は、粗ヘパリン、高品質化ヘパリン、精製ヘパリンを制限なく含む、ヘパリン生成物以外のすべての形態のヘパリンを意味する。本発明において、“ヘパリン生成物”という用語はLMWH及びULMWHを意味する。
【0022】
本発明において、“低分子量ヘパリン”または“LMWH”は、約3,000ドルトン超で約10,000ドルトン未満の重量平均分子量を有している、ヘパリンから得られた多糖混合物を意味する。
【0023】
本発明において、“超低分子量ヘパリン”または“ULMWH” は、約1,500ドルトンから約3,000ドルトンまでの重量平均分子量を有している、ヘパリンから得られた多糖混合物を意味する。
【0024】
本文中で使用したCTA−SAXクロマトグラフィーは、第四アンモニウム塩で動的にコートされ約pH2−約pH12の範囲で一定の正味正電荷を維持している逆相シリカカラムで行うアニオン交換クロマトグラフィーを意味する。
【0025】
本発明の1つの実施態様では、逆相カラムがオクチルまたはオクタデシルシリカカラムであろう。
【0026】
本発明の1つの実施態様では、第四アンモニウム塩がセチルトリメチルアンモニウムまたはセチルピリジニウムであろう。
【0027】
本発明の1つの実施態様では、CTA−SAXクロマトグラフィーが、セチルトリメチルアンモニウムまたはセチルピリジニウムでコートされたオクチルまたはオクタデシル結合シリカを含む固体支持体を使用する。この実施態様では、慣用のオクチルまたはオクタデシル結合シリカがセチルトリメチルアンモニウム(CTA)またはセチルピリジニウム塩溶液によって“動的にコート”されており、それらのアニオン交換容量は、吸着されたCTAまたはセチルピリジニウムの量によって慎重にコントロールされる。
【0028】
1つの実施態様では、クロマトグラフィーの固定相がセチルトリメチルアンモニウム塩でコートされたHypersil BDS C8またはC18逆相カラムである。
【0029】
本発明の分析方法は以下の段階を含む:C8またはC18逆相カラムに第四アンモニウム塩溶液を動的にコートすることによって調製した固定相を使用する高速液体クロマトグラフィー(例えば、CTA−SAXクロマトグラフィー)によってサンプルを直接分析し、二糖から十二糖までの全てのオリゴ糖を検出する。この場合、オリゴ糖配列は生成物中に存在するままで観察される。これはオリゴ糖を直接検査できる分析である。
【0030】
ゲル浸透クロマトグラフィー(分析すべきサンプルのGPC)による分析とC8またはC18逆相カラムに第四アンモニウム塩溶液を動的にコートすることによって調製した固定相を使用する高速液体クロマトグラフィー(例えばCTA−SAXクロマトグラフィー)とを組合せた分析も、二糖から十二糖までのオリゴ糖を分析するために使用し得る。この二次元分離方法では、分解能の向上、及び、サンプル中のオリゴ糖の直接検査が可能である。さらに、二次元分析はオリゴ糖ピークがオーバーラップする可能性を最小にする。これもまた被験サンプル中に存在するままのオリゴ糖を直接観察できる方法である。
【0031】
C8またはC18逆相カラムに第四アンモニウム塩溶液を動的にコートすることによって調製した固定相を使用する高速液体クロマトグラフィー(例えば、CTA−SAXクロマトグラフィー)の前に、アッセイ対象サンプルを解重合してもよい。本発明のこの実施態様では、ヘパリナーゼの作用によってサンプルを部分的にまたは全体的に解重合し、次いで得られた解重合物を適宜還元させてもよい。次に、例えばCTA−SAXクロマトグラフィーまたはゲル浸透クロマトグラフィーとCTA−SAXクロマトグラフィーとの併用によって解重合物を分析する。
【0032】
1つの具体的な実施態様では、解重合物をNaBH4で処理することによって還元し、次いでCTA−SAXクロマトグラフィーまたはゲル浸透クロマトグラフィーとCTA−SAXクロマトグラフィーとの併用によって分析する。
【0033】
1つの実施態様で、本発明の方法は、ヘパリン、低分子量ヘパリン(LMWH)、超低分子量ヘパリン(ULMWH)及びオリゴ糖を直接分析するために使用でき、C8またはC18逆相カラムの第四アンモニウム塩コーティングを使用してオリゴ糖の複合混合物を分離及び分析し二糖から十二糖までのすべてのオリゴ糖を検出し得る。
【0034】
1つの関連実施態様では、分析をCTA−SAXクロマトグラフィーによって行う。
【0035】
別の実施態様では、ヘパリン、低分子量ヘパリン、超低分子量ヘパリン及びオリゴ糖の分析方法が2つの段階、即ち、(a)ゲル浸透クロマトグラフィーによる第一分離段階、及び、(b)C8またはC18逆相カラムの第四アンモニウム塩コーティングを使用して二糖から十二糖までの画分の各々を分離し、二糖から十二糖までのすべてのオリゴ糖を検出する第二分離段階、を含む。
【0036】
関連実施態様では、上記分析の第二分離段階をCTA−SAXクロマトグラフィーによって行う。
【0037】
本発明の別の実施態様では、ヘパリン、低分子量ヘパリン、超低分子量ヘパリン及びオリゴ糖の分析方法が、例えばヘパリナーゼの作用によってサンプルを部分的または全体的に解重合し、次いで解重合物を適宜還元させる段階を含む。次に、オリゴ糖の複合混合物を分離及び分析するためにC8またはC18逆相カラムの第四アンモニウム塩コーティングを使用して直接分析を行って二糖から十二糖までのすべてのオリゴ糖を検出する。
【0038】
関連実施態様では、上記分析をCTA−SAXクロマトグラフィーによって行う。
【0039】
LMWH、ULMWH及びオリゴ糖の製造に使用する解重合プロセスの選択性を決定するために本発明方法を使用してもよい。十二糖までのオリゴ糖を直接分析することができれば、特定の解重合プロセスによって生成するオリゴ糖配列を決定できる。先に指摘したように、2つ以上の製造業者が同じプロセスを使用しているのでなければ、LMWHの製造業者の各々は異なる解重合プロセスを使用しており、その結果として、異なる化学構造のLMWHが生成し、これらはそれぞれに異なる薬理学的活性及び臨床使用承認表示を有することになる。本発明方法はここに、LMWHまたはULMWHの各々の極めて複雑な特有の構造プロフィルを同定し特性決定するための方法を提供する。
【0040】
本発明の方法はまた、1,6−無水結合(即ち、“1,6−無水基”)または製造プロセスによって生じた他の何らかの特定の構造的修飾によって修飾された末端をもつオリゴ糖鎖の存在を検出及び/または定量するために使用し得る。
【0041】
本発明の1つの実施態様では、アッセイ対象サンプル中の “1,6−無水基”の存在を十二糖までのCTA−SAXクロマトグラフィーによって直接的に(即ち、酵素的または化学的前処理なしで)分析及び/または定量し得る。
【0042】
本発明の別の実施態様では、ゲル浸透クロマトグラフィーを使用してサンプルを分画化し、次いで二糖から十二糖までの各画分のCTA−SAXクロマトグラフィーによってサンプル中の“1,6−無水基”を直接的に分析及び/または定量し得る。本発明の方法によれば、1,6−無水基を含むオリゴ糖を個別に同定できまたサンプル中のこの部分(二糖から十二糖)におけるそれらの分布も同定できる。
【0043】
本発明の別の実施態様では、ヘパリナーゼの混合物で完全に解重合したサンプル中の“1,6−無水基”を分析する。
【0044】
関連実施態様では、ヘパリナーゼ混合物がヘパリナーゼ1(EC 4.2.2.7)、ヘパリナーゼ2(ヘパリンリアーゼII)及びヘパリナーゼ3(EC 4.2.2.8)を含み、各ヘパリナーゼが例えば0.5単位/mlで存在している。(これらの酵素はGrampian Enzymesグループによって上市されている。)
【0045】
本発明の方法は、“1,6−無水”誘導体を含有していない低分子量ヘパリンからLovenoxRTM(エノキサパリンナトリウム)を識別し得る。逆の言い方をすれば、本発明の方法によって、低分子量ヘパリンがLovenoxRTM(エノキサパリンナトリウム)の物理化学的特性をもたないことを確認することが可能である。
【0046】
さらに、本発明方法は、β−除去を含む化学的または酵素的解重合によるLovenoxRTM(エノキサパリンナトリウム)のようなLMWH及びULMWHの製造プロセスを標準化して均一なバッチを得るためにサンプルをインプロセスでコントロールする工業的方法に応用できる。
【0047】
1つの実施態様では、LovenoxRTM(エノキサパリンナトリウム)を特性決定するために本発明の方法を使用し得る。この方法は以下の段階を含む:
LovenoxRTM(エノキサパリンナトリウム)のサンプルをヘパリナーゼ1(EC 4.2.2.7)、ヘパリナーゼ2(ヘパリンリアーゼII)及びヘパリナーゼ3(EC 4.2.2.8)の混合物で解重合させる;
場合によっては、解重合したサンプルを例えば酢酸ナトリウム中のNaBH4溶液で処理することによって還元させる(この段階は、1,6−無水形態でない還元性末端を特異的に還元させることが可能である(国際特許出願WO01/72762に記載の生成物));
次いで、解重合し場合によっては還元したサンプル中の種々のオリゴ糖をHPLC(高速液体クロマトグラフィー)によって分離する。
【0048】
本発明の1つの実施態様では、HPLCがCTA−SAXクロマトグラフィー(上記に定義)でよい。
【0049】
別の実施態様では本発明は、例えば本文中に記載のようなヘパリン、低分子量ヘパリン、超低分子量ヘパリン及びオリゴ糖の分析方法に使用するための、C8またはC18逆相カラムに第四アンモニウム塩を動的にコートする処理を含むHPLC/固定相の調製方法を提供する。特定実施態様では、第四アンモニウム塩がセチルトリメチルアンモニウム硫酸水素塩であり、動的コーティングがH2O−CH3OH溶液中、約45℃のカラム温度で行われる。カラムに吸着されるCTAの量は、カラム通過溶液が含有しているメタノールのパーセンテージに正比例し、従って調節できる。
【0050】
本発明に使用されるCTA−SAXクロマトグラフィーでは、C8またはC18逆相カラムが例えばHypersil BDSカラムまたはHypersil β basic(Thermo Finnigan)でよい。分析作業のためには、C8またはC18逆相カラムが3μmの粒度、約15cmのカラム長さ及び約1mm−約4.6mmの範囲のカラム直径を有していればよい。分取目的には、C8またはC18逆相カラムが、もっと大きい直径、例えば50mm、及び、もっと大きい粒度、例えば8μmを有していればよい。
【0051】
当業者は、カラムに吸着させるCTAの量、粒度、カラム直径及びカラム長さを変更することによって、20個までの硫酸基基をもつ十二糖までの大きさの高電荷オリゴ糖を分離できる。更に、最近のオクタデシル結合シリカの開発は3μm粒子の高効率使用を可能にした。
【0052】
本発明の方法では、硫酸化オリゴ糖の保持時間がカラムに吸着されたセチルトリメチルアンモニウム塩の量に従属して直接的に調整され得る。20個以下の硫酸基を含有しているオリゴ糖は、慣用のSAX分析よりも優れた分解能で分離され得る。カラムコーティングの安定性が数百回の注入を可能にする。
【0053】
本発明はまた、リアーゼによるヘパリン、LMWHまたはULMWHの部分解重合に基づく簡単なオリゴ糖の配列決定方法を提供する。更に、本発明のCTA−SAX法は上述のような分取規模の処理に直接的に移行できる。メタノールまたはアセトニトリルのような有機溶媒を単独でまたは水と混合してカラムに通すとカラムからCTAを脱着させることが可能である。しかしながら水性移動相を使用する限り、CTAはカラムに吸着された状態に維持され、カラムは少なくとも数ヶ月間の使用が可能である。この期間中、特に使用期間の初期には保持の僅かな減少が観察されるかもしれないが、カラム保持は比較的安定に維持される(K.Itoら,J.Chromatogr.,549(1991 265−272A)。
【0054】
本発明のCTA−SAXカラムの追加の特性は、カラムがオリゴ糖の還元性末端のα及びβアノマーを分離する能力を有することである。2つのアノマーのモル比は還元性末端の硫酸化パターンに依存する。還元性末端がN−硫酸化グルコサミンの場合には、アノマーα/アノマーβの比は約95/5であるが、N−アセチル化グルコサミンの場合にはこの比が60/40から40/60の範囲である。同様に、グルコサミンの6位及び3位の硫酸化がアノマーの特徴的な溶出パターンを与える。マンノサミンで終結するオリゴ糖で極めて特異的なアノマーパターンが観察される(例えば、ΔIs−lsidepi)。前述したように、この特殊な構造はLovenoxRTM(エノキサパリンナトリウム)の特徴であり、化学プロセス中にアルカリ性媒体中で還元性末端がエピマー化することによって生じる。
【0055】
使用する装置は溶出勾配を形成できかつUVデテクタを備えているいかなるクロマトグラフでもよい。本発明の1つの実施態様では、UVデテクタは諸成分のUVスペクトルを発生させ、異なる2つの波長の吸光度の差から生じる複合シグナルを記録し得るダイオードアレイである。このようなダイオードアレイデテクタによれば、アセチル化オリゴ糖の特異的検出が容易である。関連実施態様では、約200nm−約400nmのUV範囲に透明なHPLC移動相を使用する。本発明のこの実施態様に使用できる移動相の非限定例は、過塩素酸ナトリウムまたはメタンスルホン酸ナトリウムを基剤とする移動相である。1つの実施態様では、移動相がメタンスルホン酸アンモニウムの水溶液である。
【0056】
従って、アニオン交換クロマトグラフィーによる分離を使用する上記に定義のような分析方法も本発明の目的の1つである。この方法では、約200nM−約400nmのUV領域に透明な移動相が使用される。
【0057】
いくつかの実施態様では、強アニオンクロマトグラフィー分離が約2.0−約7.0のpHで行われる。もっと高いpH条件も使用できる。関連実施態様では、約2.5のpHが使用されるであろう。別の実施態様では、発色性イオンがメタンスルホン酸アンモニウムであり、メタンスルホン酸をアンモニア水溶液でpH2.5に中和することによって調製し得る。
【0058】
代表的なクロマトグラフィー分離条件を以下に与える:
溶媒A:メタンスルホン酸の添加によってpH3に調整したMilli−Q品質の水
溶媒B:pH2.5の2Mのメタンスルホン酸アンモニウム
直線溶出勾配:T=0分:%B=1;T=74分:%B=100
流速:0.22ml/分
カラム温度40℃。
【0059】
本発明の別の目的は、ヘパリン、低分子量ヘパリン(LMWH)または超低分子量ヘパリンに見出される特異的基の検出方法である。
【0060】
1つの実施態様では、この方法が、UVによるヘパリンまたはLMWH基の検出特異性を強化する。非アセチル化多糖はいずれも所与のpHでかなり類似のUVスペクトルを有しているので、非アセチル化糖の吸光度が相殺されるように選択した2つの波長の吸光度の差をシグナルとして取出すことによってアセチル化糖を選択的に検出することが可能である。
【0061】
実施例に後述するように、202nm及び230nmを検出波長及び基準波長として選択し、202−230nmのシグナルを記録するとよい。使用できる波長の選択が移動相のpHに依存することは当業者には理解されよう(上記条件が最適条件になるように数nmの調整が必要になることもあり得る)。
【0062】
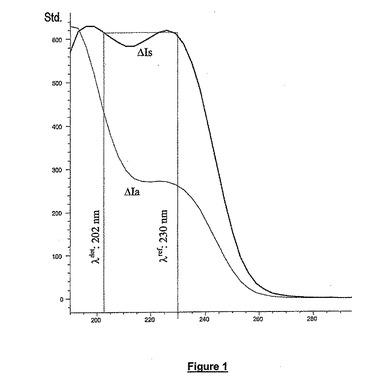
2つ以上の波長の吸光度を同時に測定できるいかなるUVデテクタも使用し得る。本発明の1つの実施態様では、Agilent Technologies社のDAD1100デテクタを使用する。この実施態様では、一方で234nm及び他方で202−230nmで二重検出を行う。アセチル化オリゴ糖の選択的検出の原理を図1に示す。図1では硫酸化二糖ΔIsのUVスペクトルをアセチル化二糖ΔIaのUVスペクトルに比較している。
【0063】
従って、アセチル化糖を選択的に検出することが可能な検出方法を含む分析方法も本発明の目的の1つである。
【0064】
いくつかの実施態様では、分析方法がCTA−SAXクロマトグラフィーによる分離を使用しており、非アセチル化糖の吸光度が相殺されるように選択した2つの波長の吸光度の差を測定することによってアセチル化糖を選択的に検出する。更に本発明の方法では以下に示す4つの1,6−無水残基を定量できるが、このような定量のためには、クロマトグラフィーシステムが混合物の他のすべての成分についても十分な選択性を有することが要求されていた。
【0065】
ピーク分割の原因は、1つはアノマー形態にあり、もっと些少な別の原因はオリゴ糖鎖の末端位置に存在するときのΔIIs、ΔIIIs及びΔIsに部分的に存在するグルコサミン−マンノサミン間の可逆的エピマー化にある。しかしながら、CTA−SAX法はクロマトグラムの全部のオリゴ糖を十分に定量できる分解能を有している。
【0066】
酵素的解重合及び還元性末端の還元の後のLovenoxRTM(エノキサパリンナトリウム)の1,6−無水誘導体は4つの本質的な形態で存在する:
【0067】
【化5】
【0068】
末端二糖ユニットに1,6−無水末端を含有しており該末端二糖のウロン酸に2−O−硫酸基を有していないすべてのオリゴ糖及び多糖は、ヘパリナーゼによって完全に解重合され、二糖1及び2の形態になっている。他方、末端の糖がウロン酸に2−O−硫酸基を含有しており、マンノサミン形態であるとき、1,6−無水誘導体は四糖の形態である(ヘパリナーゼ抵抗性の形態)。
【0069】
混合物中に三糖1(下記参照)も存在し得る。これは以下の構造に導く別の分解プロセスから誘導される(LovenoxRTM(エノキサパリンナトリウム)の化学的解重合中に観察されるピーリング現象)。
【0070】
【化6】
【0071】
混合物の他の成分はLovenoxRTM(エノキサパリンナトリウム)だけの特徴ではない。勿論、ヘパリン鎖の8つの基本的二糖が存在する。これらの8つの基本的二糖は、Sigma社及びその他の会社によって上市されている。
【0072】
混合物中の他の二糖、即ち、二糖ΔIISgal及びΔIVSgalが本発明の方法によって同定された。これらは、−ldoA(2S)−GlcNS(6S)−及び−ldoA(2S)−GlcNS−のアルカリ性2−O−脱硫酸化を起原として有しており、2つのガラクツロン酸の形成に導く。これらはヘパリンの初期構造中に通常は存在していない(U.M.Desaiら,Arch.Biochem.Biophys.,306(2)461−468(1993)。
【0073】
【化7】
【0074】
3−O−硫酸化グルコサミンを含有しているオリゴ糖はヘパリナーゼによる開裂に抵抗性であり、四糖の形態に維持される。
【0075】
たいていの低分子量ヘパリンの場合、ヘパリンがブタの粘膜から抽出され、主な四糖は以下に示すものである。これらの四糖は酵素的解重合に抵抗性であり、抗トロンビンIIIに親和性をもつ配列を含む。これらの四糖は以下の記号で示される:ΔIIa−IIsglu及びΔIIa−IVsglu(S.Yamadaら,J.Biol.Chem.;270(7),4780−4787(1993)。
【0076】
【化8】
【0077】
ヘパリナーゼで開裂された混合物の最終成分はグリコセリン末端ΔGlcA−Gal−Gal−Xyl−Serである(K.Sugaharaら,J.Biol.Chem.;270(39),22914−22923(1995);K.Sugahara,;J.Biol.Chem.;267(3),1528−1533(1992))。
【0078】
【化9】
【0079】
本発明の1つの実施態様によれば、分析したオリゴ糖混合物の二次元プロフィルを得るためにGPCカラムをCTA−SAXカラムに直結させる。関連実施態様では、ヘパリナーゼによるサンプルの部分解重合によって得られたヘパリンオリゴ糖をサイズ及び硫酸化パターン及び消化物のCTA−SAX分析に従って配列決定する方法が、CTA−SAXカラムに直結された分析用GPCカラムを使用して行われ、分析したオリゴ糖混合物の二次元プロフィルが得られる。
【0080】
ヘパリン、LovenoxRTM(エノキサパリンナトリウム)を非限定例とする低分子量ヘパリン、超低分子量ヘパリン及びオリゴ糖(十二糖以下)を分析する本発明の方法は、LovenoxRTM/ClexaneRTMを認定薬物として引用した出願に準じて規制当局の承認を求めるいかなるLMWHにも勿論適用できる。
【0081】
(実施例)
以下の実施例は本発明の種々の特徴を示す。しかしながら本発明は以下に例示の実施例に限定されない。
【0082】
【表1】
【0083】
材料:メタンスルホン酸(99.5%)はAldrichから購入した。Flavobacterium heparinumに由来の酵素リアーゼ(ヘパリナーゼI(EC 4.2.2.7)、ヘパリナーゼII(EC番号なし)ヘパリナーゼIII(EC 4.2.2.8))はすべてGrampian Enzymes(Aberdeen)から入手した。セチルトリメチルアンモニウム硫酸水素塩はMerck(Darmstadt)から購入した。その他の試薬及び化学薬品はいずれも入手できる最高品質のものであった。水はMillipore精製システムで精製した。
【実施例1】
【0084】
酵素消化
以下の一般方法を実施例3−5に適宜使用した。
【0085】
ヘパリンまたはオリゴ糖(0.1mg)の完全消化は、2mMのCa(OAc)2と2mg/mlのBSAとを含有している全量30μlの100mMの酢酸ナトリウムバッファ,pH 7.0中、10mlUのヘパリナーゼI、10mlUのヘパリナーゼII及び10mlUのヘパリナーゼIIIの混合物によって室温で48時間行う。ヘパリナーゼIによるヘパリンまたはオリゴ糖(0.1mg)の部分消化は、200mMのNaClと2mg/mlのBSAとを含有している全量1−30μlの5mMのNaH2PO4,pH7.0中、0.1−10mlUのヘパリナーゼIによって、17℃で1時間から10日間行う。
【実施例2】
【0086】
CTA−SAXクロマトグラフィー
以下の一般方法を実施例3−5に適宜使用した。
【0087】
装置:使用したクロマトグラフィーシステムは、UVダイオードアレイデテクタを備えた慣用のAgilent 1100システムである。
【0088】
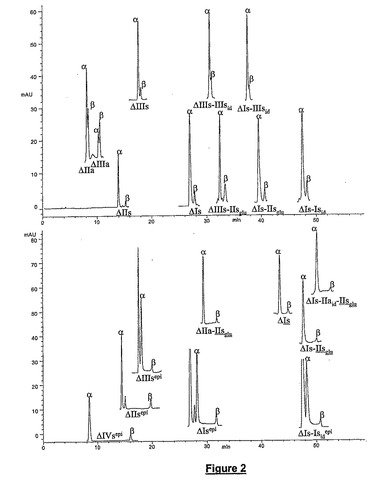
カラムの動的コーテイング:使用した手順は、Ito and Sunhara(Anal.Chem.,63(1991)273−276)に記載された手順と同様である。Hypersil BDS(Thermo Finnigan製)を充填したC8またはC18逆相カラムを使用する(150x2.1mm内径、粒度3μm)。カラムをH2O−CH3OH(68−32)v/v溶液中の1mMのセチルトリメチルアンモニウム硫酸水素塩と共に45℃のカラム温度で240分間平衡させる。カラムに吸着されたCTAの量は、カラム通過溶液が含有していたメタノールのパーセンテージに正比例するので調節できる。
【0089】
クロマトグラフィー条件:使用カラムを既述のようにCTAで動的コーティングする。発色性イオンは、メタンスルホン酸をアンモニア水溶液でpH2.5に中和することによって調製したメタンスルホン酸アンモニウムである。CTA−SAXの溶媒Bは、2Mのメタンスルホン酸アンモニウム,pH2.5である。溶媒Aはメタンスルホン酸の添加によってpH3に調整したMilli−Q品質水である。1%Bから出発して74分で100%Bになる直線勾配を流速0.22ml.分−1で使用する。毎回の処理後に、18分間のリコンディショニング段階を使用する。カラム温度は40℃である。
【0090】
これらの条件を使用すると、オリゴ糖の濃度、サンプルのイオン強度、及び、ピーク拡大を生じないイオンの溶出能に依存して、約1μl−約1mlの範囲の量を注入できる。例えば、分析用GPCから得られた画分を使用する場合(100mMの酢酸アンモニウム)500μ1までの量を注入できる。
【0091】
従って、サンプルが濃NaCl溶液(ほぼ2N)に含有されているときは、注入前に水で4倍または5倍に希釈する必要がある。いずれにしても、水で4倍に希釈した400μlのNaCl溶液を注入するほうが、100μlの初期NaCl溶液を注入するよりも良好な分離結果が得られる。
【0092】
メタンスルホネートがUV透明性なので、ダイオードデテクタアレイを使用して官能基を選択的に検出できる。化学的または酵素的なβ−除去によって得られた不飽和オリゴ糖は特徴的なUVスペクトルを有している。オリゴ糖の構造を記号化するために使用した略号は上記に示した。UVスペクトルは主として、移動相のpH及びグルコサミンのN−アセチル基の存在に依存する。図2に示すように、pH2.5の移動相では、202nm(検出波長)の吸光度から247nm(基準波長)の吸光度を減算することによって、非N−アセチル化オリゴ糖の寄与分をUVスペクトルから除去することが可能である(Huber,Diode array detection in HPLC,Chromatogr.Sci.Ser.,62(1993)363−392)。得られたシグナルにはN−アセチル化オリゴ糖だけが検出されている。ヘパリン性オリゴ糖の場合、UVスペクトルが移動相のpHに依存することは理解されよう。その結果として、pHが2.5でなく6.0である場合には、基準波長が247nmでなく230nmであろう。すべてのクロマトグラムに2つの検出シグナルが与えられる。232nmのUVシグナルはすべての不飽和オリゴ糖を検出し、202−247nmの吸光度の差に対応するシグナルはN−アセチル化オリゴ糖を特異的に検出する。同様に、溶出勾配に起因する基線量のドリフトはブランク処理によって消去される。
【0093】
CTA−SAX法は、粒度5μmのHypersil BDS C18を充填した250x21mmカラムを使用する準分取規模で使用し得る。カラムのCTAコーティングは分析用カラムの場合と同様にカラム温度を45℃に調整して行う。移動相は濃度0−2.5NのNaClの水溶液である。通常は希塩酸を加えてpHを2.5に調整する。分離は約10−約20ml.分−1の範囲の流速を使用して室温で行う。234nmのUV検出を使用する。集めた画分を酢酸で中和し、後述するようにGPCカラムで脱塩する。
【0094】
分析用GPC:分析用ゲル浸透は、1つのガードTSK Super SWプレカラム(35x4.6mm)(TOSOH BIOSEP)の付いた2つのTSK Super SW2000(300x4.6mm)で行う。不飽和オリゴ糖は232nmのUV吸光度の測定によって検出される。移動相は0.1Mの酢酸アンモニウムであり、流速は0.15ml.分−1である。注入量は5μlである。GPCをCTA−SAXクロマトグラフイーに結合させる場合には、サンプル濃度は次段階の検出が可能な十分に高い濃度でなければならない。解重合したサンプルを分析するときは、GPC注入溶液中のサンプル濃度は、サンプル毎に異なるであろうが、約25−約100mg/mlの範囲であればよい。二糖、四糖、六糖、八糖、十糖に対応するピークを集める。250μlまでの各画分をCTA−SAXカラムに直接的に再注入する。
【0095】
脱塩手順:水を溶出剤として使用し、SephadexRTM G−10 Superfine(Pharmacia)を充填したカラムに、脱塩すべきオリゴ糖を通す。カラムサイズは脱塩すべきサンプルの量に応じて選択し得る。この目的には、100x7cm、100x5cm、40x2.6cm、20x1.6cmのカラムセットが好都合である。導電率及び232nmの吸光度を測定することによって二重検出を行う。
【0096】
配列決定手順:150x3mmのCTA−SAXカラムで分離し20x1.6cmのSephadexRTM G−10カラムで脱塩した精製オリゴ糖を集める。あるいは、精製オリゴ糖を水で5倍に希釈し、希NH3を加えてpH7.0に中和する。次に、ヘパリナーゼまたは2−スルファターゼを使用する部分的分画化によって配列決定する(R.Ramanら,J.Biol.Chem.278(2003)12167−12174;G.Venkataramanら,Science 286(1999)537−542;S.Ernstら,Critical Review in Biochemistry and Molecular Biology,30(1995)387−444)。
いくつかの分析では、オリゴ糖鎖の還元性末端に位置する二糖をマークするために100mMの酢酸ナトリウム中のNaBH4によってオリゴ糖を予備還元する。150x2.1mmのCTA−SAXカラムでHPLC注入によって分画化をモニターする。
【0097】
NMRスペクトル:700μlの99%D20にサンプルを溶解した。標準パルス列を使用し、室温、400.13MHzでオリゴ糖の一次元(1D)及び二次元(2D)NMRスペクトル(COSY,TOCSY,ROESY及びHSQC)をBruker DRX400スペクトル計に記録した。2D等核Hartmann−Hahn(TOCSY)実験をスピンロック時間100msで記録した。2D ROESYスペクトルは混合時間400msで得られた。化学シフトは外部標準TSPD4を基準としたppmで与える。プロトン化学シフトはTOCSY実験から測定した。プロトン結合定数は、スペクトル分解能が十分でプロトンシグナルがオーバーラップしないときは1Dスペクトルで直接測定できた。炭素化学シフトはHSQCスペクトルから得られた。構造決定は1H及び13C NMRスペクトロスコピ―をTOCSY,ROESY及びHSQC 1H/13C 2Dスペクトルと共に使用して行った。
【実施例3】
【0098】
ブタ腸ヘパリンの完全解重合
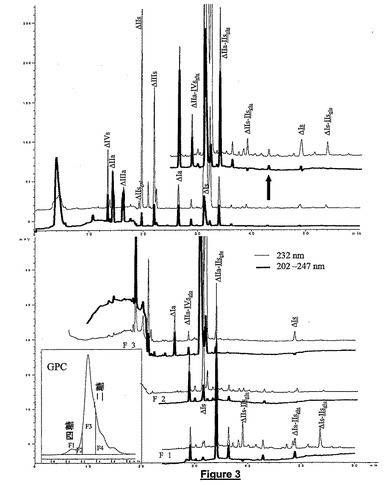
図3は上記に記載のヘパリナーゼ混合物によって完全に解重合したブタ腸ヘパリンのCTA−SAXクロマトグラムを示す。また、GPC及びCTA−SAXカラムの組合せによる本発明の二重検出の利点を示す。ΔIVa以外のすべての基本的二糖が分解された。ΔIVaは硫酸官能基が存在しないので保持されなかった。アセチル化二糖ΔIIa、ΔIIIa及びΔIaは双方のUVシグナルに検出されたが、N−硫酸化二糖は第一のシグナル(232nmのUV吸光度)だけに検出された。本発明方法の3−O硫酸化オリゴ糖の検出能力は、これらのオリゴ糖がATIII結合配列の一部であるという理由から注目に値する。
【0099】
異なる5つの硫酸化度(401<Mw<867)を含む二糖画分の不均一性が原因で、完全に解重合したサンプル中に存在するオリゴ糖すべてを解明することはできなかった。四糖については、重量平均分子量範囲が1066−1330である。例えば、主要な2つの四糖(S.Yamadaら,J.Biol.Chem,270(1995)8696−8705)ΔIIa−IIsglu及びΔIIa−IVsglu(Mw1066及び1168)は高度に硫酸化した二糖に極めて近い分子量を有している。従って、GPC画分F1−F3だけをCTA−FAXカラムに注入した。低硫酸化二糖画分F4はあまり重要でなく、注入されたGPC移動相の酢酸アンモニウムはそれらの溶出範囲に対応してCTA−SAXクロマトグラムの初端を乱した。
【0100】
分離に引き続いて、ヘパリン消化物中の新しい四硫酸化二糖を同定した。分子量767ドルトンがLC/MSによって確認され(結果公表せず)、これはΔIs構造に一致する。この二糖は最近、3−OST−5Hの発現によってヘパリン中でin vitro形成された(Mochizukiら,J.Biol.Chem.,278(2003)26780−26787)。この種の3−O硫酸化二糖は、ウシ腸ヘパリンに由来の完全解重合ヘパリン中に多量に存在する。しかしながら、ウシ腸ヘパリンのサンプル中では本発明の二重UV検出方法で2つの同様の二糖ΔIIs及びΔIIIsが検出され、また、3−O硫酸化四糖ΔIIs−IIsglu、ΔIs−IIsglu及びΔIa−IIsgluも検出できる。不飽和酸に存在する2−O硫酸官能基は、最大吸収波長をシフトさせ、それらの認識を可能にする。極めて過剰な量のヘパリナーゼ混合物でヘパリナーゼ抵抗性の四糖を処理すると、二糖生成物の部分的解重合及びキャラクタリゼーションが可能になった。例えばこれらの条件下でΔIa−IIsgluは部分的にΔIa及びΔIIsに変換された。
【実施例4】
【0101】
ヘパリナーゼ1によるブタ腸ヘパリンの解重合
ヘパリナーゼ1によるブタ腸ヘパリンの解重合を試験し、得られたオリゴ糖の多くを同定した。この混合物はヘパリナーゼ1による開裂が連続的に進行するので極めて分解性(evolutive)であることが証明された。一般には二糖、四糖及び六糖の混合物が反応終了後に回収されるが、その理由は主として、アセチル化部位−Glc(NS,6−OHまたは6S)↓IdoA(2−OHまたは2S)−GlcNAc(6−OHまたは6S)↓GlcA−がヘパリナーゼ1に抵抗性なので六糖が生成し、−GlcNS(6−OHまたは6S)↓GlcA−の抵抗性が四糖を生成するからである。
【0102】
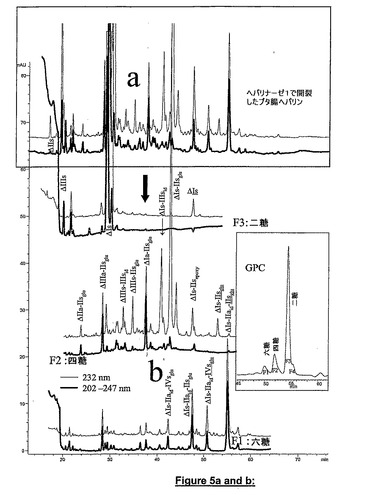
図5aは、ヘパリナーゼ1によって消化されたブタ腸ヘパリンの完全クロマトグラムをその二糖、四糖及び六糖画分に関連するクロマトグラム(図5b)と共に示す。四糖ΔIs−Isidが二糖ΔIsに完全変換したことは、酵素反応がほぼ完全であったことを示す。図示の分離は、CTA−SAXクロマトグラフイーの単独使用では得られなかった。その理由は、ある種の低硫酸化六糖は同じ時間範囲でいくつかの硫酸化四糖として溶出したからである。二糖画分のクロマトグラムは、ΔIs、ΔIIs、ΔIIIs及び3−O硫酸化ΔIsを含んでいた。四糖画分は、ΔIs−IIIsid、ΔIs−IIsid、ΔIIIs−IIIsid及びΔIIIs−IIsgluを含んでおり、これは多くの論文で報告された単離構造に符合した。例えば、H.Mochizukiら,J.Biol.Chem.278(2003)26780−26787参照。
【0103】
本発明では、単離したヘパリナーゼ1抵抗性四糖にヘパリナーゼ2を作用させ開裂後に二糖構成単位の混合物とすることによって構造認識を行った。ΔIs−IIsglu及びΔIIIs−IIsgluの場合、UV吸収の最大とアノマー溶出パターンとを考察して可能なΔIIs−Isid及びΔIIs−IIIsid配列を除去した。IIsのイズロニック立体配置(即ち、GlcA−GlcNS)がヘパリン中にほとんど存在しないので、Lindahlの法則(J.Biol.Chem.265(1990)7292−7300)を応用して、第二の可能性ΔIs−IIsid及びΔIIIs−IIsidを除去した。最も難しいケースはΔIs−IIIsidとΔIIIs−Isidとの識別であった。この識別は、予測された四糖ΔIs−IIIsidをNaBH4で予め還元することによって行った。極めて過剰な量のヘパリナーゼ1、2及び3の混合物を十分に脱塩したサンプルに作用させると、還元四糖が開裂され、ΔIsが還元ΔIIIs(アノマーの不在によって容易に同定される)二糖と共に生じた。完全に解重合したサンプル中と同様にΔIs−IIsgluも存在していた。この混合物中には新しい構造(ΔIs−IIsepoxy)も同定された。
【0104】
【化10】
【0105】
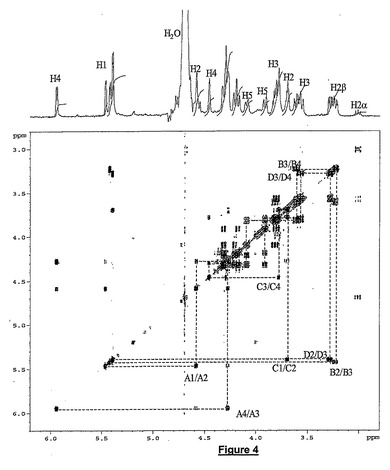
1H及び13CのNMRスペクトロスコピーをCOSY,TOCSY,ROESY及びHSQCの1H/13C 2Dスペクトルと共に使用して構造を決定した。1H及び13Cの全アサインメント(ΔIs−IIsepoxyのプロトン及び炭素の化学シフト)を以下に報告する:
【0106】
【表2】
【0107】
1Dプロトンスペクトルのシグナル(図4)とTOCSYスペクトルの分析とを統合すると、化合物が四糖の特性を有していることが示される。3.01ppmで観察された弱いシグナルは、還元性末端に他のアルファアノマー残基(15%)が存在することを示す。4,5−不飽和ウロン酸のアサインメントを開始するために5.95ppmで観察されたエチレン系プロトンH4を使用する。4つの残基に対応する4つのアノマープロトンがTOCSYスペクトルで明白に同定される。プロトン間結合定数は、公表されたデータと完全に一致する(Abdel−Malikら,Carbohydr.Res.,159(1987)11−23)。β−(1−>4)−O−グルコシド結合の証拠は、ROESYスペクトルで残基間の相関関係から観察される。プロトン及び炭素化学シフトは、観察された2Dスペクトル相関関係と共にΔIs−IIsepoxy適合性である。
【0108】
この四糖ΔIs−IIsepoxyはΔIs−(Isid)nの2−O硫酸化によって生成されるが(R.N.Rejら,Carbohydr.Res.,200(1990)437−447;M.Jasejaら,Can.J.Chem. 67(1989)1449−1456;S.Pianiら,J.Carbohydr.Chem.12(1993)507−21;U.R.Desaiら,Carbohydr.Res.,241(1993)249−259)、アルカリ性条件下(1NのNaOH)、60℃で30分間処理したウシ肺ヘパリンから単離された。ヘパリナーゼ1による解重合は、NMRによるこの成分の単離及びキャラクタリゼーションを可能にした。この中間体は比較的不安定であり、さらに加水分解が進んでL−イズロン酸またはL−ガラクツロン酸部分に分解される(M.Jasejaら,Can.J.Chem.,67(1989)1449−1456;U.R.Desaiら,Carbohydr.Res.,241(1993)249−259)。
【0109】
N−アセチル化四糖ΔIa−IIsglu、ΔIIIa−IIsglu及びΔIIa−IIsgluも同定した。これらは解重合の最終時期にN−アセチル化六糖が式:
【0110】
【化11】
に従って開裂する結果として生成する。
【0111】
イズロン酸の2−O硫酸化は、部位−Glc(NS,6S)↓IdoA(2S)−GlcNAc(6−OHまたは6S)の開裂を促進した。ΔIIa−IIsgluの存在は、反応:
【0112】
【化12】
が可能であるが不完全であることを示した。六糖画分中のΔIs−Iaid−IIsglu及びΔIs−IIIaid−IIsgluの非存在は、それらが完全に開裂されたことを示した。他方、ΔIs−IIaid−IIsgluは六糖画分の主要成分としてまだ存在していた。六糖画分のその他の成分、ΔIs−IIaid−IIsglu、ΔIs−IIaid−IVsglu及びΔIs−IIaid−IVsgluは、我々の配列決定法を使用して先ず同定し、次いで分取クロマトグラフィーによって単離し、それらの一致をNMRスペクトロスコピーによって確認した。3−O硫酸化六糖は、式:
【0113】
【化13】
で示されるような反応を使用するヘパリナーゼ2の作用によって同定された。この分画化スキームを考察すると、ΔIIa−IIsglu−Isid構造の存在の可能性はUVスペクトル及びアノマーパターンの双方を検討することによって排除された。他の2つの六糖、ΔIs−IIaid−IIsglu及びΔIs−IIaid−IVsgluは、ヘパリナーゼ1、ヘパリナーゼ3+ヘパリナーゼ1、次いで3つのヘパリナーゼの混合物の作用を使用する同様の方法で同定した。
【0114】
既述のように、ヘパリナーゼ1の作用はΔIIa−IIsglu及びΔIIa−IVsgluに導く。
【0115】
【化14】
【0116】
ΔIs−IIaidから二糖構成単位への変換がヘパリナーゼ3の作用に起因することに注目されたい。同じ酵素処理後に、ΔIs−IIIaid−IIsgluはΔIs−IIIaid+ΔIIsに分画化されただけであった。この場合、ΔIs↓IIIaid−IIsgluと対照的にΔIs↓IIaidをヘパリナーゼ1で開裂することはできない。ΔIs−IIaidまたはΔIs−IIIaidのような、還元性末端にアセチル化グルコサミンを有している多くのオリゴ糖は、CTA−SAXクロマトグラフィーでΔIIaまたはΔIIIaと同様の特異的アノマーパターンを有している。所望の場合には、これが還元性末端にN−アセチル化部分をもつオリゴ糖のもう1つの同定方法である。結論として、ヘパリナーゼ1によるアセチル化六糖から四糖への変換を導くために、開裂性の低い順に以下の順序が確定した:ΔIs−IIaid−IIsglu ≒ ΔIs−IIaid−IVsglu < ΔIs−IIaid−IIsglu ≒ ΔIs−IIaid−IVsglu < ΔIIIs−IIIaid−IIsglu ≒ ΔIIIs−IIIaid−IVsglu < ΔIs−IIIaid−IIsglu ≒ Δls−IIIaid−IVsglu > ΔIs−Iaid−IIsglu。
【0117】
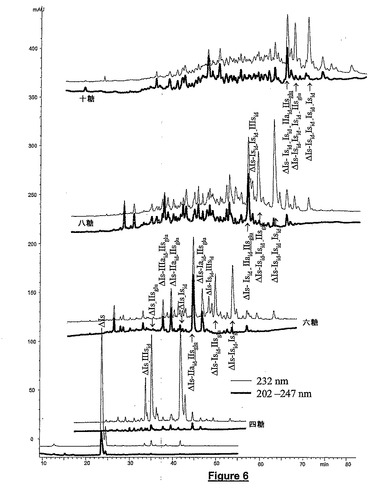
図6はヘパリナーゼ1で部分解重合させることによってブタ腸ヘパリンから得られた画分のCTA−SAXクロマトグラムを示す。この図は、高度に硫酸化したオリゴ糖についてもCTA−SAXの高い分解能が得られることを示す。図3及び図4の四糖及び六糖画分の内容の比較によって、ヘパリナーゼ1によるヘパリン開裂の進行の影響を示す。
【実施例5】
【0118】
配列決定の例
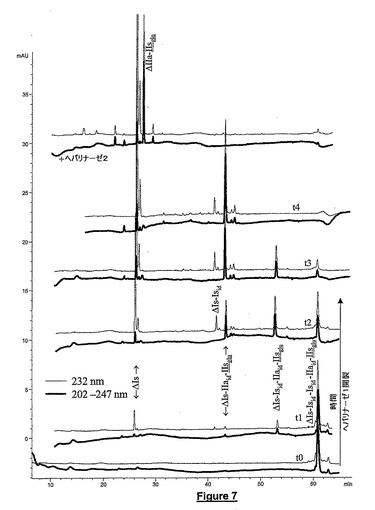
ヘパリナーゼによる管理解重合に基づくオリゴ糖の簡単な配列決定方法が開発された。選択的UV検出に組合せたCTA−SAXの分解能は分画化スキームの同定及び出発材料構造の推定を可能にする。ヘパリナーゼのようなリアーゼによる消化物の分析に基づく方法の弱点は、開裂中の不飽和結合の生成後にウロン酸が消滅するので(β−除去)ウロン酸の立体配置に関する構造が比較的不確実なことである(Y.Kariyaら,J.Biochem.123(1998)240−246)。ヘパリン二糖の立体配置及び結合に関する法則を考察するならばこの不確実性が実際にはかなり小さい。必要な場合には酸の立体配置をNMR法によって確認した。我々の配列決定方法は、ヘパリンオリゴ糖構造に対する我々の知識を向上させた。この方法は、基本的四糖の同定から開始し、次いで六糖及びそれ以上の多糖の同定を漸進的に継続する。図7は、十糖ΔIs−Isid−Isid−IIaid−IIsgluの配列決定例を示す。ヘパリナーゼ1を使用したとき、2つの部位:ΔIs↓Isid↓Isid−IIaid−IIsgluが開裂された。4つのフラグメントΔIs−Isid−IIaid−IIsglu、ΔIs−IIaid−IIsglu、ΔIs−Isid及びΔlsの混合物を観察した。反応がさらに進行するのに伴って、ΔIs−Isid−IIaid−IIsgluがΔIs−IIaid−IIsglu及びΔIsに開裂された。アセチル化画分は二重波長UV検出を使用して容易に検出された。ヘパリナーゼ2を使用すると、3つの部位、ΔIs↓Isid↓Isid↓IIaid−IIsgluが開裂された。これらの部位が開裂されたとき、残りのフラグメントはΔlla−IIsglu及びΔIsであった。同定されたフラグメントの構造を考察すると、配列決定された十糖の可能な唯一の構造は、ΔIs−Isid−Isid−IIaid−IIsgluである。ΔIs−Isid−IIaid−IIsglu−Isidの配列決定は可能でない。その理由はこれらがヘパリナーゼ1開裂によって ΔIs−Isidのフラグメントを与えることができないからである。別の可能配列ΔIs−IIaid−IIsglu−Isid−Isidは、その八糖フラグメントIs−Isid−IIaid−IIsgluの認識及び出発十糖のアノマーパターンの双方によって除去された。
【0119】
本発明による定量方法では、混合物中に含有されていたすべての不飽和オリゴ糖が5500mol−1.l.cm−1に等しい同じモル吸光度を有しているという広く受け入れられている仮説を利用する。従って、出発低分子量ヘパリン中の解重合混合物の全ての構成成分の重量パーセンテージを決定することが可能である。
【図面の簡単な説明】
【0120】
【図1】硫酸化二糖ΔIsのUVスペクトルをアセチル化二糖ΔIaのUVスペクトルに比較したアセチル化オリゴ糖の選択的検出を示す。x−軸は時間を分で示し、y−軸はUVデテクタからのミリアンペアシグナルを表す。
【図2】CTA−SAXカラムにおけるオリゴ糖アノマーの分離を示す。x−軸は時間を分で示し、y−軸はUVデテクタからのミリアンペアシグナルを表す。
【図3】ブタ腸ヘパリンの完全解重合によって得られた混合物中のオリゴ糖を分離するためのGPC/CTA−SAX結合の使用を示す。x−軸は時間を分で示し、y−軸はUVデテクタからのミリアンペアシグナルを表す。
【図4】D20−400MHz中のΔIs−IIsepoxyのProtonスペクトル及びCOSYスペクトルを示す。主なクロスピークを、糖単位に対応する文字及びプロトン数zで標識する。
【図5】5a及び5bは、ブタ腸ヘパリンをヘパリナーゼ1で完全解重合して得られた混合物中のオリゴ糖を分離するためのGPC/CTA−SAX結合の使用を示す。x−軸は時間を分で示し、y−軸はUVデテクタからのミリアンペアシグナルを表す。
【図6】ブタ腸ヘパリンをヘパリナーゼ1で部分解重合して得られた混合物中のオリゴ糖を分離するためのGPC/CTA−SAX結合の使用を示す。x−軸は時間を分で示し、y−軸はUVデテクタからのミリアンペアシグナルを表す。
【図7】ΔIs−Isid−Isid−IIaid−IIsgluの配列決定を示す。x−軸は時間を分で示し、y−軸はUVデテクタからのミリアンペアシグナルを表す。
【技術分野】
【0001】
本発明は、ヘパリン、低分子量ヘパリン(LMWH)または超低分子量ヘパリン(ULMWH)を構成するオリゴ糖の直接分析方法を提供する。
【背景技術】
【0002】
へパリンは、天然ソースから抽出されたグリコサミノグリカンファミリーの生物活性物質であり、有効な抗凝固性及び抗血栓性を有している。これらは術後静脈血栓の治療に特に有用である。
【0003】
ヘパリンソースからLMWHを製造するために、長いヘパリン多糖鎖を低分子量の短い鎖に分割する。これは化学的または酵素的な解重合によって行われる。例えば、エノキサパリンの場合、解重合プロセスは2つの競合的化学反応、即ち、β除去とベンジルエステルの加水分解とを含む。その結果として、平均分子量約4500DaのLMWH多糖鎖が得られる。LMWHは非分割ヘパリンと同様に、いくつかの多糖鎖に沿って分布した特定の5糖配列でATIIIに結合することによって凝固を阻害する。
【0004】
LMWHの認可製品の製造業者は各々で異なる解重合プロセスを使用している。2つの製造業者が同じプロセスを使用することがなければ、このようなプロセスの違いから、異なる化学構造を持ち、従って異なる薬理学的活性及び異なる臨床使用承認表示をもつLMWHが製造される。
【0005】
従って、LMWHはそれらの製造に使用された解重合プロセスによって構造的に識別される(R.J.Linhardtら,Seminars in Thombosis and Hemostatis 1999;25(3 supp.):5−16)。その結果としてLMWHはヘパリンよりも不均質である。異なるプロセスの各々が特有の極めて複雑な構造的修飾を多糖鎖に生じさせる。これらの修飾は、鎖の長さ、鎖の配列、及び、構造フィンガープリントの違いを含む。その結果として、異なるLMWHの各々は、異なる薬理学的プロフィル及び異なる承認臨床表示を有している。
【0006】
米国でLovenoxRTM及び他のいくつかの国々でClexaneRTMという商品名で販売されているエノキサパリンナトリウムを純ヘパリンから製造するプロセスでは、水相アルカリ性解重合プロセスによってオリゴ糖鎖の還元性末端のグルコサミンに部分的ではあるが特徴的な変換が生じる。
【0007】
この変換の第一段階は、グルコサミンとマンノサミンとの間の可逆的エピマー化から成る(T.Toidaら,J.Carbohydrate Chemistry,15(3),351−360(1996))。第二段階は、グルコサミンの6−O−脱硫酸であり、これによって “1,6−無水”と呼ばれる誘導体が形成される(国際特許出願WO01/29055)。
【0008】
【化4】
【0009】
6−O−硫酸化された末端グルコサミンを有しているオリゴ糖鎖の場合にこの種の誘導体が得られる。
【0010】
1,6−無水結合で修飾された末端をもつオリゴ糖鎖のパーセンテージはLovenoxRTM(エノキサパリンナトリウム)のオリゴ糖混合物の構造的特徴である。現在知られている範囲では、LovenoxRTM(エノキサパリンナトリウム)の成分の15%−25%が鎖の還元性末端に1,6−無水構造を有している。
【0011】
最近では、例えば公開された米国特許出願No.2002−0055621A1に記載されているように、強塩基の存在下の解重合を使用するヘパリンフラグメントの新しい製造方法で約1500−約3000ドルトンの範囲の重量平均分子量をもつ超低分子量ヘパリン(ULMWH)が得られるようになっている。該特許の記載内容は参照によって本発明に含まれるものとする。
【0012】
ヘパリン、低分子量ヘパリン、超低分子量ヘパリンが元のままの(intact)分子であるかまたは酵素消化物であるかを分析することは、混合物が多分散性であり、また、分子の官能化度が高くまた変わり易いため、難しい作業である。この問題に取組むためにクロマトグラフィー法及び電気泳動法が開発された。毛管電気泳動(CE)はヘパリンオリゴ糖を分離するために使用される(M.Guerriniら,Glycobiology 12(2002)713−719;R.J.Linhardt.ら,BioMethods(Basel)9(1997)183−197)。しかしながら、CEの選択性は液体クロマトグラフィーに比べてかなり低い。更に、(UVによる)CE検出ではしばしば、不十分な感度が原因で問題が生じる。さらに、この分析方法のために開発された分離を分取技術に適応させることができないこともCEの欠点のひとつである。
【0013】
いくつかの場合には、クロマトグラフィー分離を伴うことなくMALDI−TOFマススペクトロメトリー分析を行うことができ、これを予備的なヘパリンオリゴ糖分析方法及び配列決定ツールとして使用してもよい(H.Sakaguchiら,J.Biochem.129(2001)107−18;A.J.Rhombergら,Proc.Nalt.Acad.Sci.USA 95(1998)4176−4181;L.Sturialeら,Semin.Thromb.Hemost.27(2001)465−472)。しかしながらMALDI−TOFマススペクトロメトリー分析は複合混合物に対する分解能がない。また、費用も高い。
【0014】
これらの理由から、硫酸化オリゴ糖の分離には液体クロマトグラフィーが好ましい。塩化ナトリウムを基剤とする移動相による強アニオン交換(SAX)はヘパリン由来のオリゴ糖を分解するクロマトグラフィー方法の1つである。更に、HPLCまたは低圧クロマトグラフィーによるゲル浸透クロマトグラフィーは、オリゴ糖のサイズ分離及び脱塩に使用できる強力なツールである。これらの方法では移動相に塩化ナトリウムを使用することが2つの重大な欠点を生む。即ち、NaClを含む移動相は塩の腐蝕作用を克服するように特殊処理されたクロマトグラフを必要とする。また、215nmよりも低い波長に強力なUV吸光度を有している。このため、ある種のオリゴ糖に対してはこの検出方法の使用が制限される。
【0015】
このような理由で、SAXクロマトグラフィーの固定相の開発が行き詰っていた。SAXクロマトグラフィーの使用はしばしば5−10未満の硫酸基を含有するオリゴ糖の分離に限定されており、高度に硫酸化した生成物の場合には固定相の開孔への物質移動が少ない結果として重大なピーク拡大が頻繁に生じる。C18またはC8結合シリカ上のイオン対クロマトグラフィーをテトラブチルアンモニウム含有移動相で代替する方法(N.K.Karamanosら,J.Chromatogr.765(1997)169−179;C.F.Moffatら,Eur.J.Biochem.,197(1991 449−459)または等価の方法も、アセトニトリル溶出勾配の終点で観察される選択性の低下を理由として低硫酸化種に限定されている。イオン対クロマトグラフィーの注目を呼ぶ開発が最近Linhardtらによって報告された(J.Chromatogr.,1014(2003)215−223)。この方法はマススペクトロスコピーと容易に組合せることができるであろうが、その使用はやはり高硫酸化オリゴ糖に対する選択性によって限定される。セチルトリメチルアンモニウム(CTA)のようなより親油性のアンモニウム塩とのイオン対合は難しい。その理由は、このような塩は、硫酸化オリゴ糖と共に著しく非極性のイオン対を形成し、これらの多くが水性溶媒に不溶性であるからである。
【0016】
CTAのようなイオン対化剤を予め吸着させた固定相を使用する無機アニオンの分離はItoらによって一度ならず行われた(J.Chromatogr.,549(1991)265−272;Anal.Chem.,63(1991)273−276.;J.Chromatogr.,598(1992) 237−241)。
【発明の開示】
【発明が解決しようとする課題】
【0017】
従って、本発明の目的は、20個までの硫酸基基をもち十二糖という大きさの高電荷のオリゴ糖を分離及び分析できる新規な配列決定方法を提供することである。
【0018】
本発明の別の目的は、ヘパリン、低分子量ヘパリン、超低分子量ヘパリン及びオリゴ糖から選択されたサンプルのアッセイ方法を提供することである。本発明の方法では、第四アンモニウム塩をコートした逆相カラムを使用したオリゴ糖の複合混合物のクロマトグラフィー分離及び分析によってサンプルを分析する。これに関連した本発明の特徴によれば、逆相カラムがC8またはC18カラムである。
【課題を解決するための手段】
【0019】
1つの実施態様では、本発明の方法が、C8またはC18逆相カラムに第四アンモニウム塩溶液を動的にコートすることによって調製した固定相を使用する分析用高速液体クロマトグラフィーを使用する。(逆相がC8またはC18に限定されないこと、他の逆相もこの目的に適合し得ることは理解されよう。)
【0020】
本発明の1つの実施態様では、固定相がセチルトリメチルアンモニウム塩またはセチルピリジニウム塩で動的にコートされたC8またはC18逆相カラムである。逆相カラムは例えばHypersil BDSでよい。
【0021】
本発明において、“ヘパリン”という用語は、粗ヘパリン、高品質化ヘパリン、精製ヘパリンを制限なく含む、ヘパリン生成物以外のすべての形態のヘパリンを意味する。本発明において、“ヘパリン生成物”という用語はLMWH及びULMWHを意味する。
【0022】
本発明において、“低分子量ヘパリン”または“LMWH”は、約3,000ドルトン超で約10,000ドルトン未満の重量平均分子量を有している、ヘパリンから得られた多糖混合物を意味する。
【0023】
本発明において、“超低分子量ヘパリン”または“ULMWH” は、約1,500ドルトンから約3,000ドルトンまでの重量平均分子量を有している、ヘパリンから得られた多糖混合物を意味する。
【0024】
本文中で使用したCTA−SAXクロマトグラフィーは、第四アンモニウム塩で動的にコートされ約pH2−約pH12の範囲で一定の正味正電荷を維持している逆相シリカカラムで行うアニオン交換クロマトグラフィーを意味する。
【0025】
本発明の1つの実施態様では、逆相カラムがオクチルまたはオクタデシルシリカカラムであろう。
【0026】
本発明の1つの実施態様では、第四アンモニウム塩がセチルトリメチルアンモニウムまたはセチルピリジニウムであろう。
【0027】
本発明の1つの実施態様では、CTA−SAXクロマトグラフィーが、セチルトリメチルアンモニウムまたはセチルピリジニウムでコートされたオクチルまたはオクタデシル結合シリカを含む固体支持体を使用する。この実施態様では、慣用のオクチルまたはオクタデシル結合シリカがセチルトリメチルアンモニウム(CTA)またはセチルピリジニウム塩溶液によって“動的にコート”されており、それらのアニオン交換容量は、吸着されたCTAまたはセチルピリジニウムの量によって慎重にコントロールされる。
【0028】
1つの実施態様では、クロマトグラフィーの固定相がセチルトリメチルアンモニウム塩でコートされたHypersil BDS C8またはC18逆相カラムである。
【0029】
本発明の分析方法は以下の段階を含む:C8またはC18逆相カラムに第四アンモニウム塩溶液を動的にコートすることによって調製した固定相を使用する高速液体クロマトグラフィー(例えば、CTA−SAXクロマトグラフィー)によってサンプルを直接分析し、二糖から十二糖までの全てのオリゴ糖を検出する。この場合、オリゴ糖配列は生成物中に存在するままで観察される。これはオリゴ糖を直接検査できる分析である。
【0030】
ゲル浸透クロマトグラフィー(分析すべきサンプルのGPC)による分析とC8またはC18逆相カラムに第四アンモニウム塩溶液を動的にコートすることによって調製した固定相を使用する高速液体クロマトグラフィー(例えばCTA−SAXクロマトグラフィー)とを組合せた分析も、二糖から十二糖までのオリゴ糖を分析するために使用し得る。この二次元分離方法では、分解能の向上、及び、サンプル中のオリゴ糖の直接検査が可能である。さらに、二次元分析はオリゴ糖ピークがオーバーラップする可能性を最小にする。これもまた被験サンプル中に存在するままのオリゴ糖を直接観察できる方法である。
【0031】
C8またはC18逆相カラムに第四アンモニウム塩溶液を動的にコートすることによって調製した固定相を使用する高速液体クロマトグラフィー(例えば、CTA−SAXクロマトグラフィー)の前に、アッセイ対象サンプルを解重合してもよい。本発明のこの実施態様では、ヘパリナーゼの作用によってサンプルを部分的にまたは全体的に解重合し、次いで得られた解重合物を適宜還元させてもよい。次に、例えばCTA−SAXクロマトグラフィーまたはゲル浸透クロマトグラフィーとCTA−SAXクロマトグラフィーとの併用によって解重合物を分析する。
【0032】
1つの具体的な実施態様では、解重合物をNaBH4で処理することによって還元し、次いでCTA−SAXクロマトグラフィーまたはゲル浸透クロマトグラフィーとCTA−SAXクロマトグラフィーとの併用によって分析する。
【0033】
1つの実施態様で、本発明の方法は、ヘパリン、低分子量ヘパリン(LMWH)、超低分子量ヘパリン(ULMWH)及びオリゴ糖を直接分析するために使用でき、C8またはC18逆相カラムの第四アンモニウム塩コーティングを使用してオリゴ糖の複合混合物を分離及び分析し二糖から十二糖までのすべてのオリゴ糖を検出し得る。
【0034】
1つの関連実施態様では、分析をCTA−SAXクロマトグラフィーによって行う。
【0035】
別の実施態様では、ヘパリン、低分子量ヘパリン、超低分子量ヘパリン及びオリゴ糖の分析方法が2つの段階、即ち、(a)ゲル浸透クロマトグラフィーによる第一分離段階、及び、(b)C8またはC18逆相カラムの第四アンモニウム塩コーティングを使用して二糖から十二糖までの画分の各々を分離し、二糖から十二糖までのすべてのオリゴ糖を検出する第二分離段階、を含む。
【0036】
関連実施態様では、上記分析の第二分離段階をCTA−SAXクロマトグラフィーによって行う。
【0037】
本発明の別の実施態様では、ヘパリン、低分子量ヘパリン、超低分子量ヘパリン及びオリゴ糖の分析方法が、例えばヘパリナーゼの作用によってサンプルを部分的または全体的に解重合し、次いで解重合物を適宜還元させる段階を含む。次に、オリゴ糖の複合混合物を分離及び分析するためにC8またはC18逆相カラムの第四アンモニウム塩コーティングを使用して直接分析を行って二糖から十二糖までのすべてのオリゴ糖を検出する。
【0038】
関連実施態様では、上記分析をCTA−SAXクロマトグラフィーによって行う。
【0039】
LMWH、ULMWH及びオリゴ糖の製造に使用する解重合プロセスの選択性を決定するために本発明方法を使用してもよい。十二糖までのオリゴ糖を直接分析することができれば、特定の解重合プロセスによって生成するオリゴ糖配列を決定できる。先に指摘したように、2つ以上の製造業者が同じプロセスを使用しているのでなければ、LMWHの製造業者の各々は異なる解重合プロセスを使用しており、その結果として、異なる化学構造のLMWHが生成し、これらはそれぞれに異なる薬理学的活性及び臨床使用承認表示を有することになる。本発明方法はここに、LMWHまたはULMWHの各々の極めて複雑な特有の構造プロフィルを同定し特性決定するための方法を提供する。
【0040】
本発明の方法はまた、1,6−無水結合(即ち、“1,6−無水基”)または製造プロセスによって生じた他の何らかの特定の構造的修飾によって修飾された末端をもつオリゴ糖鎖の存在を検出及び/または定量するために使用し得る。
【0041】
本発明の1つの実施態様では、アッセイ対象サンプル中の “1,6−無水基”の存在を十二糖までのCTA−SAXクロマトグラフィーによって直接的に(即ち、酵素的または化学的前処理なしで)分析及び/または定量し得る。
【0042】
本発明の別の実施態様では、ゲル浸透クロマトグラフィーを使用してサンプルを分画化し、次いで二糖から十二糖までの各画分のCTA−SAXクロマトグラフィーによってサンプル中の“1,6−無水基”を直接的に分析及び/または定量し得る。本発明の方法によれば、1,6−無水基を含むオリゴ糖を個別に同定できまたサンプル中のこの部分(二糖から十二糖)におけるそれらの分布も同定できる。
【0043】
本発明の別の実施態様では、ヘパリナーゼの混合物で完全に解重合したサンプル中の“1,6−無水基”を分析する。
【0044】
関連実施態様では、ヘパリナーゼ混合物がヘパリナーゼ1(EC 4.2.2.7)、ヘパリナーゼ2(ヘパリンリアーゼII)及びヘパリナーゼ3(EC 4.2.2.8)を含み、各ヘパリナーゼが例えば0.5単位/mlで存在している。(これらの酵素はGrampian Enzymesグループによって上市されている。)
【0045】
本発明の方法は、“1,6−無水”誘導体を含有していない低分子量ヘパリンからLovenoxRTM(エノキサパリンナトリウム)を識別し得る。逆の言い方をすれば、本発明の方法によって、低分子量ヘパリンがLovenoxRTM(エノキサパリンナトリウム)の物理化学的特性をもたないことを確認することが可能である。
【0046】
さらに、本発明方法は、β−除去を含む化学的または酵素的解重合によるLovenoxRTM(エノキサパリンナトリウム)のようなLMWH及びULMWHの製造プロセスを標準化して均一なバッチを得るためにサンプルをインプロセスでコントロールする工業的方法に応用できる。
【0047】
1つの実施態様では、LovenoxRTM(エノキサパリンナトリウム)を特性決定するために本発明の方法を使用し得る。この方法は以下の段階を含む:
LovenoxRTM(エノキサパリンナトリウム)のサンプルをヘパリナーゼ1(EC 4.2.2.7)、ヘパリナーゼ2(ヘパリンリアーゼII)及びヘパリナーゼ3(EC 4.2.2.8)の混合物で解重合させる;
場合によっては、解重合したサンプルを例えば酢酸ナトリウム中のNaBH4溶液で処理することによって還元させる(この段階は、1,6−無水形態でない還元性末端を特異的に還元させることが可能である(国際特許出願WO01/72762に記載の生成物));
次いで、解重合し場合によっては還元したサンプル中の種々のオリゴ糖をHPLC(高速液体クロマトグラフィー)によって分離する。
【0048】
本発明の1つの実施態様では、HPLCがCTA−SAXクロマトグラフィー(上記に定義)でよい。
【0049】
別の実施態様では本発明は、例えば本文中に記載のようなヘパリン、低分子量ヘパリン、超低分子量ヘパリン及びオリゴ糖の分析方法に使用するための、C8またはC18逆相カラムに第四アンモニウム塩を動的にコートする処理を含むHPLC/固定相の調製方法を提供する。特定実施態様では、第四アンモニウム塩がセチルトリメチルアンモニウム硫酸水素塩であり、動的コーティングがH2O−CH3OH溶液中、約45℃のカラム温度で行われる。カラムに吸着されるCTAの量は、カラム通過溶液が含有しているメタノールのパーセンテージに正比例し、従って調節できる。
【0050】
本発明に使用されるCTA−SAXクロマトグラフィーでは、C8またはC18逆相カラムが例えばHypersil BDSカラムまたはHypersil β basic(Thermo Finnigan)でよい。分析作業のためには、C8またはC18逆相カラムが3μmの粒度、約15cmのカラム長さ及び約1mm−約4.6mmの範囲のカラム直径を有していればよい。分取目的には、C8またはC18逆相カラムが、もっと大きい直径、例えば50mm、及び、もっと大きい粒度、例えば8μmを有していればよい。
【0051】
当業者は、カラムに吸着させるCTAの量、粒度、カラム直径及びカラム長さを変更することによって、20個までの硫酸基基をもつ十二糖までの大きさの高電荷オリゴ糖を分離できる。更に、最近のオクタデシル結合シリカの開発は3μm粒子の高効率使用を可能にした。
【0052】
本発明の方法では、硫酸化オリゴ糖の保持時間がカラムに吸着されたセチルトリメチルアンモニウム塩の量に従属して直接的に調整され得る。20個以下の硫酸基を含有しているオリゴ糖は、慣用のSAX分析よりも優れた分解能で分離され得る。カラムコーティングの安定性が数百回の注入を可能にする。
【0053】
本発明はまた、リアーゼによるヘパリン、LMWHまたはULMWHの部分解重合に基づく簡単なオリゴ糖の配列決定方法を提供する。更に、本発明のCTA−SAX法は上述のような分取規模の処理に直接的に移行できる。メタノールまたはアセトニトリルのような有機溶媒を単独でまたは水と混合してカラムに通すとカラムからCTAを脱着させることが可能である。しかしながら水性移動相を使用する限り、CTAはカラムに吸着された状態に維持され、カラムは少なくとも数ヶ月間の使用が可能である。この期間中、特に使用期間の初期には保持の僅かな減少が観察されるかもしれないが、カラム保持は比較的安定に維持される(K.Itoら,J.Chromatogr.,549(1991 265−272A)。
【0054】
本発明のCTA−SAXカラムの追加の特性は、カラムがオリゴ糖の還元性末端のα及びβアノマーを分離する能力を有することである。2つのアノマーのモル比は還元性末端の硫酸化パターンに依存する。還元性末端がN−硫酸化グルコサミンの場合には、アノマーα/アノマーβの比は約95/5であるが、N−アセチル化グルコサミンの場合にはこの比が60/40から40/60の範囲である。同様に、グルコサミンの6位及び3位の硫酸化がアノマーの特徴的な溶出パターンを与える。マンノサミンで終結するオリゴ糖で極めて特異的なアノマーパターンが観察される(例えば、ΔIs−lsidepi)。前述したように、この特殊な構造はLovenoxRTM(エノキサパリンナトリウム)の特徴であり、化学プロセス中にアルカリ性媒体中で還元性末端がエピマー化することによって生じる。
【0055】
使用する装置は溶出勾配を形成できかつUVデテクタを備えているいかなるクロマトグラフでもよい。本発明の1つの実施態様では、UVデテクタは諸成分のUVスペクトルを発生させ、異なる2つの波長の吸光度の差から生じる複合シグナルを記録し得るダイオードアレイである。このようなダイオードアレイデテクタによれば、アセチル化オリゴ糖の特異的検出が容易である。関連実施態様では、約200nm−約400nmのUV範囲に透明なHPLC移動相を使用する。本発明のこの実施態様に使用できる移動相の非限定例は、過塩素酸ナトリウムまたはメタンスルホン酸ナトリウムを基剤とする移動相である。1つの実施態様では、移動相がメタンスルホン酸アンモニウムの水溶液である。
【0056】
従って、アニオン交換クロマトグラフィーによる分離を使用する上記に定義のような分析方法も本発明の目的の1つである。この方法では、約200nM−約400nmのUV領域に透明な移動相が使用される。
【0057】
いくつかの実施態様では、強アニオンクロマトグラフィー分離が約2.0−約7.0のpHで行われる。もっと高いpH条件も使用できる。関連実施態様では、約2.5のpHが使用されるであろう。別の実施態様では、発色性イオンがメタンスルホン酸アンモニウムであり、メタンスルホン酸をアンモニア水溶液でpH2.5に中和することによって調製し得る。
【0058】
代表的なクロマトグラフィー分離条件を以下に与える:
溶媒A:メタンスルホン酸の添加によってpH3に調整したMilli−Q品質の水
溶媒B:pH2.5の2Mのメタンスルホン酸アンモニウム
直線溶出勾配:T=0分:%B=1;T=74分:%B=100
流速:0.22ml/分
カラム温度40℃。
【0059】
本発明の別の目的は、ヘパリン、低分子量ヘパリン(LMWH)または超低分子量ヘパリンに見出される特異的基の検出方法である。
【0060】
1つの実施態様では、この方法が、UVによるヘパリンまたはLMWH基の検出特異性を強化する。非アセチル化多糖はいずれも所与のpHでかなり類似のUVスペクトルを有しているので、非アセチル化糖の吸光度が相殺されるように選択した2つの波長の吸光度の差をシグナルとして取出すことによってアセチル化糖を選択的に検出することが可能である。
【0061】
実施例に後述するように、202nm及び230nmを検出波長及び基準波長として選択し、202−230nmのシグナルを記録するとよい。使用できる波長の選択が移動相のpHに依存することは当業者には理解されよう(上記条件が最適条件になるように数nmの調整が必要になることもあり得る)。
【0062】
2つ以上の波長の吸光度を同時に測定できるいかなるUVデテクタも使用し得る。本発明の1つの実施態様では、Agilent Technologies社のDAD1100デテクタを使用する。この実施態様では、一方で234nm及び他方で202−230nmで二重検出を行う。アセチル化オリゴ糖の選択的検出の原理を図1に示す。図1では硫酸化二糖ΔIsのUVスペクトルをアセチル化二糖ΔIaのUVスペクトルに比較している。
【0063】
従って、アセチル化糖を選択的に検出することが可能な検出方法を含む分析方法も本発明の目的の1つである。
【0064】
いくつかの実施態様では、分析方法がCTA−SAXクロマトグラフィーによる分離を使用しており、非アセチル化糖の吸光度が相殺されるように選択した2つの波長の吸光度の差を測定することによってアセチル化糖を選択的に検出する。更に本発明の方法では以下に示す4つの1,6−無水残基を定量できるが、このような定量のためには、クロマトグラフィーシステムが混合物の他のすべての成分についても十分な選択性を有することが要求されていた。
【0065】
ピーク分割の原因は、1つはアノマー形態にあり、もっと些少な別の原因はオリゴ糖鎖の末端位置に存在するときのΔIIs、ΔIIIs及びΔIsに部分的に存在するグルコサミン−マンノサミン間の可逆的エピマー化にある。しかしながら、CTA−SAX法はクロマトグラムの全部のオリゴ糖を十分に定量できる分解能を有している。
【0066】
酵素的解重合及び還元性末端の還元の後のLovenoxRTM(エノキサパリンナトリウム)の1,6−無水誘導体は4つの本質的な形態で存在する:
【0067】
【化5】
【0068】
末端二糖ユニットに1,6−無水末端を含有しており該末端二糖のウロン酸に2−O−硫酸基を有していないすべてのオリゴ糖及び多糖は、ヘパリナーゼによって完全に解重合され、二糖1及び2の形態になっている。他方、末端の糖がウロン酸に2−O−硫酸基を含有しており、マンノサミン形態であるとき、1,6−無水誘導体は四糖の形態である(ヘパリナーゼ抵抗性の形態)。
【0069】
混合物中に三糖1(下記参照)も存在し得る。これは以下の構造に導く別の分解プロセスから誘導される(LovenoxRTM(エノキサパリンナトリウム)の化学的解重合中に観察されるピーリング現象)。
【0070】
【化6】
【0071】
混合物の他の成分はLovenoxRTM(エノキサパリンナトリウム)だけの特徴ではない。勿論、ヘパリン鎖の8つの基本的二糖が存在する。これらの8つの基本的二糖は、Sigma社及びその他の会社によって上市されている。
【0072】
混合物中の他の二糖、即ち、二糖ΔIISgal及びΔIVSgalが本発明の方法によって同定された。これらは、−ldoA(2S)−GlcNS(6S)−及び−ldoA(2S)−GlcNS−のアルカリ性2−O−脱硫酸化を起原として有しており、2つのガラクツロン酸の形成に導く。これらはヘパリンの初期構造中に通常は存在していない(U.M.Desaiら,Arch.Biochem.Biophys.,306(2)461−468(1993)。
【0073】
【化7】
【0074】
3−O−硫酸化グルコサミンを含有しているオリゴ糖はヘパリナーゼによる開裂に抵抗性であり、四糖の形態に維持される。
【0075】
たいていの低分子量ヘパリンの場合、ヘパリンがブタの粘膜から抽出され、主な四糖は以下に示すものである。これらの四糖は酵素的解重合に抵抗性であり、抗トロンビンIIIに親和性をもつ配列を含む。これらの四糖は以下の記号で示される:ΔIIa−IIsglu及びΔIIa−IVsglu(S.Yamadaら,J.Biol.Chem.;270(7),4780−4787(1993)。
【0076】
【化8】
【0077】
ヘパリナーゼで開裂された混合物の最終成分はグリコセリン末端ΔGlcA−Gal−Gal−Xyl−Serである(K.Sugaharaら,J.Biol.Chem.;270(39),22914−22923(1995);K.Sugahara,;J.Biol.Chem.;267(3),1528−1533(1992))。
【0078】
【化9】
【0079】
本発明の1つの実施態様によれば、分析したオリゴ糖混合物の二次元プロフィルを得るためにGPCカラムをCTA−SAXカラムに直結させる。関連実施態様では、ヘパリナーゼによるサンプルの部分解重合によって得られたヘパリンオリゴ糖をサイズ及び硫酸化パターン及び消化物のCTA−SAX分析に従って配列決定する方法が、CTA−SAXカラムに直結された分析用GPCカラムを使用して行われ、分析したオリゴ糖混合物の二次元プロフィルが得られる。
【0080】
ヘパリン、LovenoxRTM(エノキサパリンナトリウム)を非限定例とする低分子量ヘパリン、超低分子量ヘパリン及びオリゴ糖(十二糖以下)を分析する本発明の方法は、LovenoxRTM/ClexaneRTMを認定薬物として引用した出願に準じて規制当局の承認を求めるいかなるLMWHにも勿論適用できる。
【0081】
(実施例)
以下の実施例は本発明の種々の特徴を示す。しかしながら本発明は以下に例示の実施例に限定されない。
【0082】
【表1】
【0083】
材料:メタンスルホン酸(99.5%)はAldrichから購入した。Flavobacterium heparinumに由来の酵素リアーゼ(ヘパリナーゼI(EC 4.2.2.7)、ヘパリナーゼII(EC番号なし)ヘパリナーゼIII(EC 4.2.2.8))はすべてGrampian Enzymes(Aberdeen)から入手した。セチルトリメチルアンモニウム硫酸水素塩はMerck(Darmstadt)から購入した。その他の試薬及び化学薬品はいずれも入手できる最高品質のものであった。水はMillipore精製システムで精製した。
【実施例1】
【0084】
酵素消化
以下の一般方法を実施例3−5に適宜使用した。
【0085】
ヘパリンまたはオリゴ糖(0.1mg)の完全消化は、2mMのCa(OAc)2と2mg/mlのBSAとを含有している全量30μlの100mMの酢酸ナトリウムバッファ,pH 7.0中、10mlUのヘパリナーゼI、10mlUのヘパリナーゼII及び10mlUのヘパリナーゼIIIの混合物によって室温で48時間行う。ヘパリナーゼIによるヘパリンまたはオリゴ糖(0.1mg)の部分消化は、200mMのNaClと2mg/mlのBSAとを含有している全量1−30μlの5mMのNaH2PO4,pH7.0中、0.1−10mlUのヘパリナーゼIによって、17℃で1時間から10日間行う。
【実施例2】
【0086】
CTA−SAXクロマトグラフィー
以下の一般方法を実施例3−5に適宜使用した。
【0087】
装置:使用したクロマトグラフィーシステムは、UVダイオードアレイデテクタを備えた慣用のAgilent 1100システムである。
【0088】
カラムの動的コーテイング:使用した手順は、Ito and Sunhara(Anal.Chem.,63(1991)273−276)に記載された手順と同様である。Hypersil BDS(Thermo Finnigan製)を充填したC8またはC18逆相カラムを使用する(150x2.1mm内径、粒度3μm)。カラムをH2O−CH3OH(68−32)v/v溶液中の1mMのセチルトリメチルアンモニウム硫酸水素塩と共に45℃のカラム温度で240分間平衡させる。カラムに吸着されたCTAの量は、カラム通過溶液が含有していたメタノールのパーセンテージに正比例するので調節できる。
【0089】
クロマトグラフィー条件:使用カラムを既述のようにCTAで動的コーティングする。発色性イオンは、メタンスルホン酸をアンモニア水溶液でpH2.5に中和することによって調製したメタンスルホン酸アンモニウムである。CTA−SAXの溶媒Bは、2Mのメタンスルホン酸アンモニウム,pH2.5である。溶媒Aはメタンスルホン酸の添加によってpH3に調整したMilli−Q品質水である。1%Bから出発して74分で100%Bになる直線勾配を流速0.22ml.分−1で使用する。毎回の処理後に、18分間のリコンディショニング段階を使用する。カラム温度は40℃である。
【0090】
これらの条件を使用すると、オリゴ糖の濃度、サンプルのイオン強度、及び、ピーク拡大を生じないイオンの溶出能に依存して、約1μl−約1mlの範囲の量を注入できる。例えば、分析用GPCから得られた画分を使用する場合(100mMの酢酸アンモニウム)500μ1までの量を注入できる。
【0091】
従って、サンプルが濃NaCl溶液(ほぼ2N)に含有されているときは、注入前に水で4倍または5倍に希釈する必要がある。いずれにしても、水で4倍に希釈した400μlのNaCl溶液を注入するほうが、100μlの初期NaCl溶液を注入するよりも良好な分離結果が得られる。
【0092】
メタンスルホネートがUV透明性なので、ダイオードデテクタアレイを使用して官能基を選択的に検出できる。化学的または酵素的なβ−除去によって得られた不飽和オリゴ糖は特徴的なUVスペクトルを有している。オリゴ糖の構造を記号化するために使用した略号は上記に示した。UVスペクトルは主として、移動相のpH及びグルコサミンのN−アセチル基の存在に依存する。図2に示すように、pH2.5の移動相では、202nm(検出波長)の吸光度から247nm(基準波長)の吸光度を減算することによって、非N−アセチル化オリゴ糖の寄与分をUVスペクトルから除去することが可能である(Huber,Diode array detection in HPLC,Chromatogr.Sci.Ser.,62(1993)363−392)。得られたシグナルにはN−アセチル化オリゴ糖だけが検出されている。ヘパリン性オリゴ糖の場合、UVスペクトルが移動相のpHに依存することは理解されよう。その結果として、pHが2.5でなく6.0である場合には、基準波長が247nmでなく230nmであろう。すべてのクロマトグラムに2つの検出シグナルが与えられる。232nmのUVシグナルはすべての不飽和オリゴ糖を検出し、202−247nmの吸光度の差に対応するシグナルはN−アセチル化オリゴ糖を特異的に検出する。同様に、溶出勾配に起因する基線量のドリフトはブランク処理によって消去される。
【0093】
CTA−SAX法は、粒度5μmのHypersil BDS C18を充填した250x21mmカラムを使用する準分取規模で使用し得る。カラムのCTAコーティングは分析用カラムの場合と同様にカラム温度を45℃に調整して行う。移動相は濃度0−2.5NのNaClの水溶液である。通常は希塩酸を加えてpHを2.5に調整する。分離は約10−約20ml.分−1の範囲の流速を使用して室温で行う。234nmのUV検出を使用する。集めた画分を酢酸で中和し、後述するようにGPCカラムで脱塩する。
【0094】
分析用GPC:分析用ゲル浸透は、1つのガードTSK Super SWプレカラム(35x4.6mm)(TOSOH BIOSEP)の付いた2つのTSK Super SW2000(300x4.6mm)で行う。不飽和オリゴ糖は232nmのUV吸光度の測定によって検出される。移動相は0.1Mの酢酸アンモニウムであり、流速は0.15ml.分−1である。注入量は5μlである。GPCをCTA−SAXクロマトグラフイーに結合させる場合には、サンプル濃度は次段階の検出が可能な十分に高い濃度でなければならない。解重合したサンプルを分析するときは、GPC注入溶液中のサンプル濃度は、サンプル毎に異なるであろうが、約25−約100mg/mlの範囲であればよい。二糖、四糖、六糖、八糖、十糖に対応するピークを集める。250μlまでの各画分をCTA−SAXカラムに直接的に再注入する。
【0095】
脱塩手順:水を溶出剤として使用し、SephadexRTM G−10 Superfine(Pharmacia)を充填したカラムに、脱塩すべきオリゴ糖を通す。カラムサイズは脱塩すべきサンプルの量に応じて選択し得る。この目的には、100x7cm、100x5cm、40x2.6cm、20x1.6cmのカラムセットが好都合である。導電率及び232nmの吸光度を測定することによって二重検出を行う。
【0096】
配列決定手順:150x3mmのCTA−SAXカラムで分離し20x1.6cmのSephadexRTM G−10カラムで脱塩した精製オリゴ糖を集める。あるいは、精製オリゴ糖を水で5倍に希釈し、希NH3を加えてpH7.0に中和する。次に、ヘパリナーゼまたは2−スルファターゼを使用する部分的分画化によって配列決定する(R.Ramanら,J.Biol.Chem.278(2003)12167−12174;G.Venkataramanら,Science 286(1999)537−542;S.Ernstら,Critical Review in Biochemistry and Molecular Biology,30(1995)387−444)。
いくつかの分析では、オリゴ糖鎖の還元性末端に位置する二糖をマークするために100mMの酢酸ナトリウム中のNaBH4によってオリゴ糖を予備還元する。150x2.1mmのCTA−SAXカラムでHPLC注入によって分画化をモニターする。
【0097】
NMRスペクトル:700μlの99%D20にサンプルを溶解した。標準パルス列を使用し、室温、400.13MHzでオリゴ糖の一次元(1D)及び二次元(2D)NMRスペクトル(COSY,TOCSY,ROESY及びHSQC)をBruker DRX400スペクトル計に記録した。2D等核Hartmann−Hahn(TOCSY)実験をスピンロック時間100msで記録した。2D ROESYスペクトルは混合時間400msで得られた。化学シフトは外部標準TSPD4を基準としたppmで与える。プロトン化学シフトはTOCSY実験から測定した。プロトン結合定数は、スペクトル分解能が十分でプロトンシグナルがオーバーラップしないときは1Dスペクトルで直接測定できた。炭素化学シフトはHSQCスペクトルから得られた。構造決定は1H及び13C NMRスペクトロスコピ―をTOCSY,ROESY及びHSQC 1H/13C 2Dスペクトルと共に使用して行った。
【実施例3】
【0098】
ブタ腸ヘパリンの完全解重合
図3は上記に記載のヘパリナーゼ混合物によって完全に解重合したブタ腸ヘパリンのCTA−SAXクロマトグラムを示す。また、GPC及びCTA−SAXカラムの組合せによる本発明の二重検出の利点を示す。ΔIVa以外のすべての基本的二糖が分解された。ΔIVaは硫酸官能基が存在しないので保持されなかった。アセチル化二糖ΔIIa、ΔIIIa及びΔIaは双方のUVシグナルに検出されたが、N−硫酸化二糖は第一のシグナル(232nmのUV吸光度)だけに検出された。本発明方法の3−O硫酸化オリゴ糖の検出能力は、これらのオリゴ糖がATIII結合配列の一部であるという理由から注目に値する。
【0099】
異なる5つの硫酸化度(401<Mw<867)を含む二糖画分の不均一性が原因で、完全に解重合したサンプル中に存在するオリゴ糖すべてを解明することはできなかった。四糖については、重量平均分子量範囲が1066−1330である。例えば、主要な2つの四糖(S.Yamadaら,J.Biol.Chem,270(1995)8696−8705)ΔIIa−IIsglu及びΔIIa−IVsglu(Mw1066及び1168)は高度に硫酸化した二糖に極めて近い分子量を有している。従って、GPC画分F1−F3だけをCTA−FAXカラムに注入した。低硫酸化二糖画分F4はあまり重要でなく、注入されたGPC移動相の酢酸アンモニウムはそれらの溶出範囲に対応してCTA−SAXクロマトグラムの初端を乱した。
【0100】
分離に引き続いて、ヘパリン消化物中の新しい四硫酸化二糖を同定した。分子量767ドルトンがLC/MSによって確認され(結果公表せず)、これはΔIs構造に一致する。この二糖は最近、3−OST−5Hの発現によってヘパリン中でin vitro形成された(Mochizukiら,J.Biol.Chem.,278(2003)26780−26787)。この種の3−O硫酸化二糖は、ウシ腸ヘパリンに由来の完全解重合ヘパリン中に多量に存在する。しかしながら、ウシ腸ヘパリンのサンプル中では本発明の二重UV検出方法で2つの同様の二糖ΔIIs及びΔIIIsが検出され、また、3−O硫酸化四糖ΔIIs−IIsglu、ΔIs−IIsglu及びΔIa−IIsgluも検出できる。不飽和酸に存在する2−O硫酸官能基は、最大吸収波長をシフトさせ、それらの認識を可能にする。極めて過剰な量のヘパリナーゼ混合物でヘパリナーゼ抵抗性の四糖を処理すると、二糖生成物の部分的解重合及びキャラクタリゼーションが可能になった。例えばこれらの条件下でΔIa−IIsgluは部分的にΔIa及びΔIIsに変換された。
【実施例4】
【0101】
ヘパリナーゼ1によるブタ腸ヘパリンの解重合
ヘパリナーゼ1によるブタ腸ヘパリンの解重合を試験し、得られたオリゴ糖の多くを同定した。この混合物はヘパリナーゼ1による開裂が連続的に進行するので極めて分解性(evolutive)であることが証明された。一般には二糖、四糖及び六糖の混合物が反応終了後に回収されるが、その理由は主として、アセチル化部位−Glc(NS,6−OHまたは6S)↓IdoA(2−OHまたは2S)−GlcNAc(6−OHまたは6S)↓GlcA−がヘパリナーゼ1に抵抗性なので六糖が生成し、−GlcNS(6−OHまたは6S)↓GlcA−の抵抗性が四糖を生成するからである。
【0102】
図5aは、ヘパリナーゼ1によって消化されたブタ腸ヘパリンの完全クロマトグラムをその二糖、四糖及び六糖画分に関連するクロマトグラム(図5b)と共に示す。四糖ΔIs−Isidが二糖ΔIsに完全変換したことは、酵素反応がほぼ完全であったことを示す。図示の分離は、CTA−SAXクロマトグラフイーの単独使用では得られなかった。その理由は、ある種の低硫酸化六糖は同じ時間範囲でいくつかの硫酸化四糖として溶出したからである。二糖画分のクロマトグラムは、ΔIs、ΔIIs、ΔIIIs及び3−O硫酸化ΔIsを含んでいた。四糖画分は、ΔIs−IIIsid、ΔIs−IIsid、ΔIIIs−IIIsid及びΔIIIs−IIsgluを含んでおり、これは多くの論文で報告された単離構造に符合した。例えば、H.Mochizukiら,J.Biol.Chem.278(2003)26780−26787参照。
【0103】
本発明では、単離したヘパリナーゼ1抵抗性四糖にヘパリナーゼ2を作用させ開裂後に二糖構成単位の混合物とすることによって構造認識を行った。ΔIs−IIsglu及びΔIIIs−IIsgluの場合、UV吸収の最大とアノマー溶出パターンとを考察して可能なΔIIs−Isid及びΔIIs−IIIsid配列を除去した。IIsのイズロニック立体配置(即ち、GlcA−GlcNS)がヘパリン中にほとんど存在しないので、Lindahlの法則(J.Biol.Chem.265(1990)7292−7300)を応用して、第二の可能性ΔIs−IIsid及びΔIIIs−IIsidを除去した。最も難しいケースはΔIs−IIIsidとΔIIIs−Isidとの識別であった。この識別は、予測された四糖ΔIs−IIIsidをNaBH4で予め還元することによって行った。極めて過剰な量のヘパリナーゼ1、2及び3の混合物を十分に脱塩したサンプルに作用させると、還元四糖が開裂され、ΔIsが還元ΔIIIs(アノマーの不在によって容易に同定される)二糖と共に生じた。完全に解重合したサンプル中と同様にΔIs−IIsgluも存在していた。この混合物中には新しい構造(ΔIs−IIsepoxy)も同定された。
【0104】
【化10】
【0105】
1H及び13CのNMRスペクトロスコピーをCOSY,TOCSY,ROESY及びHSQCの1H/13C 2Dスペクトルと共に使用して構造を決定した。1H及び13Cの全アサインメント(ΔIs−IIsepoxyのプロトン及び炭素の化学シフト)を以下に報告する:
【0106】
【表2】
【0107】
1Dプロトンスペクトルのシグナル(図4)とTOCSYスペクトルの分析とを統合すると、化合物が四糖の特性を有していることが示される。3.01ppmで観察された弱いシグナルは、還元性末端に他のアルファアノマー残基(15%)が存在することを示す。4,5−不飽和ウロン酸のアサインメントを開始するために5.95ppmで観察されたエチレン系プロトンH4を使用する。4つの残基に対応する4つのアノマープロトンがTOCSYスペクトルで明白に同定される。プロトン間結合定数は、公表されたデータと完全に一致する(Abdel−Malikら,Carbohydr.Res.,159(1987)11−23)。β−(1−>4)−O−グルコシド結合の証拠は、ROESYスペクトルで残基間の相関関係から観察される。プロトン及び炭素化学シフトは、観察された2Dスペクトル相関関係と共にΔIs−IIsepoxy適合性である。
【0108】
この四糖ΔIs−IIsepoxyはΔIs−(Isid)nの2−O硫酸化によって生成されるが(R.N.Rejら,Carbohydr.Res.,200(1990)437−447;M.Jasejaら,Can.J.Chem. 67(1989)1449−1456;S.Pianiら,J.Carbohydr.Chem.12(1993)507−21;U.R.Desaiら,Carbohydr.Res.,241(1993)249−259)、アルカリ性条件下(1NのNaOH)、60℃で30分間処理したウシ肺ヘパリンから単離された。ヘパリナーゼ1による解重合は、NMRによるこの成分の単離及びキャラクタリゼーションを可能にした。この中間体は比較的不安定であり、さらに加水分解が進んでL−イズロン酸またはL−ガラクツロン酸部分に分解される(M.Jasejaら,Can.J.Chem.,67(1989)1449−1456;U.R.Desaiら,Carbohydr.Res.,241(1993)249−259)。
【0109】
N−アセチル化四糖ΔIa−IIsglu、ΔIIIa−IIsglu及びΔIIa−IIsgluも同定した。これらは解重合の最終時期にN−アセチル化六糖が式:
【0110】
【化11】
に従って開裂する結果として生成する。
【0111】
イズロン酸の2−O硫酸化は、部位−Glc(NS,6S)↓IdoA(2S)−GlcNAc(6−OHまたは6S)の開裂を促進した。ΔIIa−IIsgluの存在は、反応:
【0112】
【化12】
が可能であるが不完全であることを示した。六糖画分中のΔIs−Iaid−IIsglu及びΔIs−IIIaid−IIsgluの非存在は、それらが完全に開裂されたことを示した。他方、ΔIs−IIaid−IIsgluは六糖画分の主要成分としてまだ存在していた。六糖画分のその他の成分、ΔIs−IIaid−IIsglu、ΔIs−IIaid−IVsglu及びΔIs−IIaid−IVsgluは、我々の配列決定法を使用して先ず同定し、次いで分取クロマトグラフィーによって単離し、それらの一致をNMRスペクトロスコピーによって確認した。3−O硫酸化六糖は、式:
【0113】
【化13】
で示されるような反応を使用するヘパリナーゼ2の作用によって同定された。この分画化スキームを考察すると、ΔIIa−IIsglu−Isid構造の存在の可能性はUVスペクトル及びアノマーパターンの双方を検討することによって排除された。他の2つの六糖、ΔIs−IIaid−IIsglu及びΔIs−IIaid−IVsgluは、ヘパリナーゼ1、ヘパリナーゼ3+ヘパリナーゼ1、次いで3つのヘパリナーゼの混合物の作用を使用する同様の方法で同定した。
【0114】
既述のように、ヘパリナーゼ1の作用はΔIIa−IIsglu及びΔIIa−IVsgluに導く。
【0115】
【化14】
【0116】
ΔIs−IIaidから二糖構成単位への変換がヘパリナーゼ3の作用に起因することに注目されたい。同じ酵素処理後に、ΔIs−IIIaid−IIsgluはΔIs−IIIaid+ΔIIsに分画化されただけであった。この場合、ΔIs↓IIIaid−IIsgluと対照的にΔIs↓IIaidをヘパリナーゼ1で開裂することはできない。ΔIs−IIaidまたはΔIs−IIIaidのような、還元性末端にアセチル化グルコサミンを有している多くのオリゴ糖は、CTA−SAXクロマトグラフィーでΔIIaまたはΔIIIaと同様の特異的アノマーパターンを有している。所望の場合には、これが還元性末端にN−アセチル化部分をもつオリゴ糖のもう1つの同定方法である。結論として、ヘパリナーゼ1によるアセチル化六糖から四糖への変換を導くために、開裂性の低い順に以下の順序が確定した:ΔIs−IIaid−IIsglu ≒ ΔIs−IIaid−IVsglu < ΔIs−IIaid−IIsglu ≒ ΔIs−IIaid−IVsglu < ΔIIIs−IIIaid−IIsglu ≒ ΔIIIs−IIIaid−IVsglu < ΔIs−IIIaid−IIsglu ≒ Δls−IIIaid−IVsglu > ΔIs−Iaid−IIsglu。
【0117】
図6はヘパリナーゼ1で部分解重合させることによってブタ腸ヘパリンから得られた画分のCTA−SAXクロマトグラムを示す。この図は、高度に硫酸化したオリゴ糖についてもCTA−SAXの高い分解能が得られることを示す。図3及び図4の四糖及び六糖画分の内容の比較によって、ヘパリナーゼ1によるヘパリン開裂の進行の影響を示す。
【実施例5】
【0118】
配列決定の例
ヘパリナーゼによる管理解重合に基づくオリゴ糖の簡単な配列決定方法が開発された。選択的UV検出に組合せたCTA−SAXの分解能は分画化スキームの同定及び出発材料構造の推定を可能にする。ヘパリナーゼのようなリアーゼによる消化物の分析に基づく方法の弱点は、開裂中の不飽和結合の生成後にウロン酸が消滅するので(β−除去)ウロン酸の立体配置に関する構造が比較的不確実なことである(Y.Kariyaら,J.Biochem.123(1998)240−246)。ヘパリン二糖の立体配置及び結合に関する法則を考察するならばこの不確実性が実際にはかなり小さい。必要な場合には酸の立体配置をNMR法によって確認した。我々の配列決定方法は、ヘパリンオリゴ糖構造に対する我々の知識を向上させた。この方法は、基本的四糖の同定から開始し、次いで六糖及びそれ以上の多糖の同定を漸進的に継続する。図7は、十糖ΔIs−Isid−Isid−IIaid−IIsgluの配列決定例を示す。ヘパリナーゼ1を使用したとき、2つの部位:ΔIs↓Isid↓Isid−IIaid−IIsgluが開裂された。4つのフラグメントΔIs−Isid−IIaid−IIsglu、ΔIs−IIaid−IIsglu、ΔIs−Isid及びΔlsの混合物を観察した。反応がさらに進行するのに伴って、ΔIs−Isid−IIaid−IIsgluがΔIs−IIaid−IIsglu及びΔIsに開裂された。アセチル化画分は二重波長UV検出を使用して容易に検出された。ヘパリナーゼ2を使用すると、3つの部位、ΔIs↓Isid↓Isid↓IIaid−IIsgluが開裂された。これらの部位が開裂されたとき、残りのフラグメントはΔlla−IIsglu及びΔIsであった。同定されたフラグメントの構造を考察すると、配列決定された十糖の可能な唯一の構造は、ΔIs−Isid−Isid−IIaid−IIsgluである。ΔIs−Isid−IIaid−IIsglu−Isidの配列決定は可能でない。その理由はこれらがヘパリナーゼ1開裂によって ΔIs−Isidのフラグメントを与えることができないからである。別の可能配列ΔIs−IIaid−IIsglu−Isid−Isidは、その八糖フラグメントIs−Isid−IIaid−IIsgluの認識及び出発十糖のアノマーパターンの双方によって除去された。
【0119】
本発明による定量方法では、混合物中に含有されていたすべての不飽和オリゴ糖が5500mol−1.l.cm−1に等しい同じモル吸光度を有しているという広く受け入れられている仮説を利用する。従って、出発低分子量ヘパリン中の解重合混合物の全ての構成成分の重量パーセンテージを決定することが可能である。
【図面の簡単な説明】
【0120】
【図1】硫酸化二糖ΔIsのUVスペクトルをアセチル化二糖ΔIaのUVスペクトルに比較したアセチル化オリゴ糖の選択的検出を示す。x−軸は時間を分で示し、y−軸はUVデテクタからのミリアンペアシグナルを表す。
【図2】CTA−SAXカラムにおけるオリゴ糖アノマーの分離を示す。x−軸は時間を分で示し、y−軸はUVデテクタからのミリアンペアシグナルを表す。
【図3】ブタ腸ヘパリンの完全解重合によって得られた混合物中のオリゴ糖を分離するためのGPC/CTA−SAX結合の使用を示す。x−軸は時間を分で示し、y−軸はUVデテクタからのミリアンペアシグナルを表す。
【図4】D20−400MHz中のΔIs−IIsepoxyのProtonスペクトル及びCOSYスペクトルを示す。主なクロスピークを、糖単位に対応する文字及びプロトン数zで標識する。
【図5】5a及び5bは、ブタ腸ヘパリンをヘパリナーゼ1で完全解重合して得られた混合物中のオリゴ糖を分離するためのGPC/CTA−SAX結合の使用を示す。x−軸は時間を分で示し、y−軸はUVデテクタからのミリアンペアシグナルを表す。
【図6】ブタ腸ヘパリンをヘパリナーゼ1で部分解重合して得られた混合物中のオリゴ糖を分離するためのGPC/CTA−SAX結合の使用を示す。x−軸は時間を分で示し、y−軸はUVデテクタからのミリアンペアシグナルを表す。
【図7】ΔIs−Isid−Isid−IIaid−IIsgluの配列決定を示す。x−軸は時間を分で示し、y−軸はUVデテクタからのミリアンペアシグナルを表す。
【特許請求の範囲】
【請求項1】
オリゴ糖の複合混合物のクロマトグラフィー分離及び分析のため、サンプルが第四アンモニウム塩をコートした逆相カラムを使用して分析される、ヘパリン、低分子量ヘパリン、超低分子量ヘパリン及びオリゴ糖から選択されたサンプルのアッセイ方法。
【請求項2】
逆相カラムがC8またはC18カラムである請求項1に記載の方法。
【請求項3】
二糖から十二糖までのすべてのオリゴ糖が検出される請求項1に記載の方法。
【請求項4】
第四アンモニウム塩をコートしたC8またはC18逆相カラムを使用してサンプルが前処理なしに分析される請求項2に記載の方法。
【請求項5】
第四アンモニウム塩をコートしたC8またはC18逆相カラムがセチルトリメチルアンモニウム−強アニオン交換(CTA−SAX)カラムである請求項2に記載の方法。
【請求項6】
第四アンモニウム塩をコートしたC8またはC18逆相カラムがCTA−SAXカラムである請求項2に記載の方法。
【請求項7】
第四アンモニウム塩をコートしたC8またはC18逆相カラムを使用するクロマトグラフィー分離に先立ってゲル浸透クロマトグラフィー(GPC)によってサンプルが分画化される請求項2に記載の方法。
【請求項8】
第四アンモニウム塩をコートしたC8またはC18逆相カラムがCTA−SAXカラムである請求項7に記載の方法。
【請求項9】
第四アンモニウム塩をコートしたC8またはC18逆相カラムを使用するクロマトグラフィー分離に先立ってサンプルが部分的にまたは全体的に解重合される請求項2に記載の方法。
【請求項10】
サンプルを解重合した後、第四アンモニウム塩をコートしたC8またはC18逆相カラムを使用してクロマトグラフィー分離する前に、該サンプルが還元される請求項2に記載の方法。
【請求項11】
第四アンモニウム塩をコートしたC8またはC18逆相カラムがCTA−SAXカラムである請求項9に記載の方法。
【請求項12】
第四アンモニウム塩をコートしたC8またはC18逆相カラムがCTA−SAXカラムである請求項10に記載の方法。
【請求項13】
(a)酵素的解重合によってサンプルを解重合させる段階、
(b)解重合したサンプルを還元させる段階、及び
(c)段階(a)及び/または段階(b)のサンプルをCTA−SAXクロマトグラフィーによってアッセイする段階、
を含むヘパリン、低分子量ヘパリン、超低分子量ヘパリン及びオリゴ糖から選択されたサンプルのアッセイ方法。
【請求項14】
少なくとも1つのヘパリナーゼを使用してサンプルが酵素的に解重合される請求項13に記載の方法。
【請求項15】
少なくとも1つのヘパリナーゼがヘパリナーゼ1(EC 4.2.2.7)、ヘパリナーゼ2(ヘパリンリアーゼII)及びヘパリナーゼ3(EC 4.2.2.8)から選択される請求項14に記載の方法。
【請求項16】
ヘパリナーゼ1(EC 4.2.2.7)、ヘパリナーゼ2(ヘパリンリアーゼII)及びヘパリナーゼ3(EC 4.2.2.8)を含むヘパリナーゼ混合物を使用してサンプルが酵素的に解重合される請求項14に記載の方法。
【請求項17】
NaBH4またはホウ水素化アニオンのアルカリ金属塩によってサンプルが還元される請求項10に記載の方法。
【請求項18】
低分子量ヘパリンがエノキサパリンナトリウムである請求項1に記載の方法。
【請求項19】
サンプルがエノキサパリンナトリウムであり、1,6−無水形態でない前記エノキサパリンナトリウムの還元性末端が還元されるように前記サンプルが還元される請求項10に記載の方法。
【請求項20】
クロマトグラフィー分離が約200nmから約400nmの範囲の波長を有するUV光に対し透明な移動相を使用する請求項1に記載の方法。
【請求項21】
クロマトグラフィー分離がメタンスルホネート塩を含む移動相を使用する請求項1に記載の方法。
【請求項22】
クロマトグラフィー分離が、メタンスルホン酸アンモニウムまたはメタンスルホン酸ナトリウムを含む移動相を使用する請求項1に記載の方法。
【請求項23】
第四アンモニウム塩をコートしたC8またはC18逆相カラムにおけるクロマトグラフィーが約2.0から約7.0のpHで行われる請求項2に記載の方法。
【請求項24】
第四アンモニウム塩をコートしたC8またはC18逆相カラムにおけるクロマトグラフィーが約2.5から約3.0のpHで行われる請求項2に記載の方法。
【請求項25】
第四アンモニウム塩をコートしたC8またはC18逆相カラムにおけるクロマトグラフィーが、メタンスルホン酸の添加によってpH約3及び/または2Mのメタンスルホン酸アンモニウムの添加によってpH約2.5に調整された水を含む移動相で行われる請求項12に記載の方法。
【請求項26】
1,6−無水結合によって修飾された末端をもつオリゴ糖鎖の存在を検出する段階をさらに含む請求項1に記載の方法。
【請求項27】
アセチル化糖を検出する段階をさらに含む請求項1に記載の方法。
【請求項28】
アセチル化糖及び非アセチル化糖の双方に吸収される波長で測定した吸光度をアセチル化糖に吸収されるが非アセチル化糖には吸収されない波長で測定した吸光度から減算することによってアセチル化糖が検出される請求項27に記載の方法。
【請求項29】
アセチル化糖が選択的に検出される請求項28に記載の方法。
【請求項30】
検出されたアセチル化糖が、ΔIVa、ΔIIa、ΔIIIa、ΔIa、ΔIIa−IVsglu及びΔIIa−IIsgluから選択された少なくとも1つのアセチル化オリゴ糖である請求項28に記載の方法。
【請求項31】
低分子量ヘパリンが、LovenoxRTM/ClexaneRTM(エノキサパリンナトリウムインジェクション)を認定薬物として引用した出願に準じて規制当局による承認を求めるLMWHのいずれかである請求項1に記載の方法。
【請求項32】
カラムがオリゴ糖の定量用の第四アンモニウム塩をコートしたC8またはC18の逆相樹脂を含んでいる、オリゴ糖の複合混合物を分離するためのクロマトグラフィーカラムの使用。
【請求項33】
C8またはC18の逆相樹脂がHypersil BDSである請求項32に記載の使用。
【請求項34】
第四アンモニウム塩がセチルトリメチルアンモニウム硫酸水素塩である請求項32に記載の使用。
【請求項35】
前記分析が、以下の4つの糖:
【化1】
のいずれかから選択された少なくとも1つの糖の検出に導く請求項1に記載の方法。
【請求項36】
前記分析が、以下の糖:
【化2】
の検出に導く請求項1に記載の方法。
【請求項37】
前記分析が、以下の糖:
【化3】
のいずれかから選択された少なくとも1つの糖の検出に導く請求項1に記載の方法。
【請求項1】
オリゴ糖の複合混合物のクロマトグラフィー分離及び分析のため、サンプルが第四アンモニウム塩をコートした逆相カラムを使用して分析される、ヘパリン、低分子量ヘパリン、超低分子量ヘパリン及びオリゴ糖から選択されたサンプルのアッセイ方法。
【請求項2】
逆相カラムがC8またはC18カラムである請求項1に記載の方法。
【請求項3】
二糖から十二糖までのすべてのオリゴ糖が検出される請求項1に記載の方法。
【請求項4】
第四アンモニウム塩をコートしたC8またはC18逆相カラムを使用してサンプルが前処理なしに分析される請求項2に記載の方法。
【請求項5】
第四アンモニウム塩をコートしたC8またはC18逆相カラムがセチルトリメチルアンモニウム−強アニオン交換(CTA−SAX)カラムである請求項2に記載の方法。
【請求項6】
第四アンモニウム塩をコートしたC8またはC18逆相カラムがCTA−SAXカラムである請求項2に記載の方法。
【請求項7】
第四アンモニウム塩をコートしたC8またはC18逆相カラムを使用するクロマトグラフィー分離に先立ってゲル浸透クロマトグラフィー(GPC)によってサンプルが分画化される請求項2に記載の方法。
【請求項8】
第四アンモニウム塩をコートしたC8またはC18逆相カラムがCTA−SAXカラムである請求項7に記載の方法。
【請求項9】
第四アンモニウム塩をコートしたC8またはC18逆相カラムを使用するクロマトグラフィー分離に先立ってサンプルが部分的にまたは全体的に解重合される請求項2に記載の方法。
【請求項10】
サンプルを解重合した後、第四アンモニウム塩をコートしたC8またはC18逆相カラムを使用してクロマトグラフィー分離する前に、該サンプルが還元される請求項2に記載の方法。
【請求項11】
第四アンモニウム塩をコートしたC8またはC18逆相カラムがCTA−SAXカラムである請求項9に記載の方法。
【請求項12】
第四アンモニウム塩をコートしたC8またはC18逆相カラムがCTA−SAXカラムである請求項10に記載の方法。
【請求項13】
(a)酵素的解重合によってサンプルを解重合させる段階、
(b)解重合したサンプルを還元させる段階、及び
(c)段階(a)及び/または段階(b)のサンプルをCTA−SAXクロマトグラフィーによってアッセイする段階、
を含むヘパリン、低分子量ヘパリン、超低分子量ヘパリン及びオリゴ糖から選択されたサンプルのアッセイ方法。
【請求項14】
少なくとも1つのヘパリナーゼを使用してサンプルが酵素的に解重合される請求項13に記載の方法。
【請求項15】
少なくとも1つのヘパリナーゼがヘパリナーゼ1(EC 4.2.2.7)、ヘパリナーゼ2(ヘパリンリアーゼII)及びヘパリナーゼ3(EC 4.2.2.8)から選択される請求項14に記載の方法。
【請求項16】
ヘパリナーゼ1(EC 4.2.2.7)、ヘパリナーゼ2(ヘパリンリアーゼII)及びヘパリナーゼ3(EC 4.2.2.8)を含むヘパリナーゼ混合物を使用してサンプルが酵素的に解重合される請求項14に記載の方法。
【請求項17】
NaBH4またはホウ水素化アニオンのアルカリ金属塩によってサンプルが還元される請求項10に記載の方法。
【請求項18】
低分子量ヘパリンがエノキサパリンナトリウムである請求項1に記載の方法。
【請求項19】
サンプルがエノキサパリンナトリウムであり、1,6−無水形態でない前記エノキサパリンナトリウムの還元性末端が還元されるように前記サンプルが還元される請求項10に記載の方法。
【請求項20】
クロマトグラフィー分離が約200nmから約400nmの範囲の波長を有するUV光に対し透明な移動相を使用する請求項1に記載の方法。
【請求項21】
クロマトグラフィー分離がメタンスルホネート塩を含む移動相を使用する請求項1に記載の方法。
【請求項22】
クロマトグラフィー分離が、メタンスルホン酸アンモニウムまたはメタンスルホン酸ナトリウムを含む移動相を使用する請求項1に記載の方法。
【請求項23】
第四アンモニウム塩をコートしたC8またはC18逆相カラムにおけるクロマトグラフィーが約2.0から約7.0のpHで行われる請求項2に記載の方法。
【請求項24】
第四アンモニウム塩をコートしたC8またはC18逆相カラムにおけるクロマトグラフィーが約2.5から約3.0のpHで行われる請求項2に記載の方法。
【請求項25】
第四アンモニウム塩をコートしたC8またはC18逆相カラムにおけるクロマトグラフィーが、メタンスルホン酸の添加によってpH約3及び/または2Mのメタンスルホン酸アンモニウムの添加によってpH約2.5に調整された水を含む移動相で行われる請求項12に記載の方法。
【請求項26】
1,6−無水結合によって修飾された末端をもつオリゴ糖鎖の存在を検出する段階をさらに含む請求項1に記載の方法。
【請求項27】
アセチル化糖を検出する段階をさらに含む請求項1に記載の方法。
【請求項28】
アセチル化糖及び非アセチル化糖の双方に吸収される波長で測定した吸光度をアセチル化糖に吸収されるが非アセチル化糖には吸収されない波長で測定した吸光度から減算することによってアセチル化糖が検出される請求項27に記載の方法。
【請求項29】
アセチル化糖が選択的に検出される請求項28に記載の方法。
【請求項30】
検出されたアセチル化糖が、ΔIVa、ΔIIa、ΔIIIa、ΔIa、ΔIIa−IVsglu及びΔIIa−IIsgluから選択された少なくとも1つのアセチル化オリゴ糖である請求項28に記載の方法。
【請求項31】
低分子量ヘパリンが、LovenoxRTM/ClexaneRTM(エノキサパリンナトリウムインジェクション)を認定薬物として引用した出願に準じて規制当局による承認を求めるLMWHのいずれかである請求項1に記載の方法。
【請求項32】
カラムがオリゴ糖の定量用の第四アンモニウム塩をコートしたC8またはC18の逆相樹脂を含んでいる、オリゴ糖の複合混合物を分離するためのクロマトグラフィーカラムの使用。
【請求項33】
C8またはC18の逆相樹脂がHypersil BDSである請求項32に記載の使用。
【請求項34】
第四アンモニウム塩がセチルトリメチルアンモニウム硫酸水素塩である請求項32に記載の使用。
【請求項35】
前記分析が、以下の4つの糖:
【化1】
のいずれかから選択された少なくとも1つの糖の検出に導く請求項1に記載の方法。
【請求項36】
前記分析が、以下の糖:
【化2】
の検出に導く請求項1に記載の方法。
【請求項37】
前記分析が、以下の糖:
【化3】
のいずれかから選択された少なくとも1つの糖の検出に導く請求項1に記載の方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公表番号】特表2007−523352(P2007−523352A)
【公表日】平成19年8月16日(2007.8.16)
【国際特許分類】
【出願番号】特願2007−500189(P2007−500189)
【出願日】平成17年2月18日(2005.2.18)
【国際出願番号】PCT/EP2005/002575
【国際公開番号】WO2005/080438
【国際公開日】平成17年9月1日(2005.9.1)
【出願人】(500152119)アバンテイス・フアルマ・エス・アー (65)
【Fターム(参考)】
【公表日】平成19年8月16日(2007.8.16)
【国際特許分類】
【出願日】平成17年2月18日(2005.2.18)
【国際出願番号】PCT/EP2005/002575
【国際公開番号】WO2005/080438
【国際公開日】平成17年9月1日(2005.9.1)
【出願人】(500152119)アバンテイス・フアルマ・エス・アー (65)
【Fターム(参考)】
[ Back to top ]