
連続発酵によるタンパク質の製造方法
【課題】簡便な操作方法で、長時間にわたり安定して高生産性を維持する連続発酵法によるタンパク質の製造方法の提供。
【解決手段】連続発酵法において、発酵培養液の分離膜に高い透過性と高い細胞阻止率を持ち閉塞しにくい、ポリフッ化ビニリデン系樹脂製等の多孔性膜を用い、低い膜間差圧で濾過処理することにより、微生物が高濃度まで増殖することを可能にし、タンパク質の生産量が向上する連続発酵による、イヌインターフェロンγ等のタンパク質の製造方法。
【解決手段】連続発酵法において、発酵培養液の分離膜に高い透過性と高い細胞阻止率を持ち閉塞しにくい、ポリフッ化ビニリデン系樹脂製等の多孔性膜を用い、低い膜間差圧で濾過処理することにより、微生物が高濃度まで増殖することを可能にし、タンパク質の生産量が向上する連続発酵による、イヌインターフェロンγ等のタンパク質の製造方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、タンパク質を生産する能力を有する微生物の発酵培養液を、分離膜として目詰まりが生じにくい多孔性膜を通して濾過し、未濾過液を発酵培養液に保持または還流し、その微生物の培地を前記の発酵培養液に追加し、かつ微生物菌体内からタンパク質を回収する連続発酵によるタンパク質の製造方法に関するものである。
【背景技術】
【0002】
微生物の培養を伴う物質生産方法である発酵法は、大きく(1)回分発酵法(Batch発酵法)および流加発酵法(Fed−Batch発酵法)と、(2)連続発酵法に分類することができる。
【0003】
上記(1)の回分発酵法および流加発酵法は、設備的には簡素であり、短時間で培養が終了し、雑菌汚染による被害が少ないという利点がある。しかしながら、時間経過とともに発酵培養液中の栄養素が少なくなり、浸透圧あるいは生産物阻害等の影響により菌体増殖および生産性が低下してくる。そのため、長時間にわたり安定して菌体の増殖および高生産性を維持するのが困難である。
【0004】
一方、上記(2)の連続発酵法は、発酵反応槽内で目的物質が高濃度に蓄積されることを回避することによって、長時間にわたって高生産性を維持できるという特徴がある。しかしながら、発酵培養液へ培地の連続的な供給を行うと共に微生物を含んだ発酵培養液を抜き出すために、発酵培養液中の微生物が希釈されることから、生産効率の向上は限定されたものとなる。
【0005】
次に、タンパク質とその製造方法に関する技術背景について説明する。工業的に生産されているタンパク質として、酵素を挙げることができる。多くの工業的に重要な酵素、例えば、デンプン分解酵素およびタンパク分解酵素は、バチルス属の微生物、例えば、B.サチリス(B. subtilis) 、B.アミロリクエファシエンス(B.amyloliquefaciens)、B.リヘニホルミス(B. licheniformis)、B.ステアロセルモフィルス(B. stearothermophilus) 、およびB.コアギュランス(B.coagulans) から生産される。また、大量生産の例としては、酵素ニトリルヒドラターゼを生産し、それからアミド化合物が製造されている例がある(特許文献参照1参照。)。アルデヒドの測定用酵素およびホルムアルデヒドなどの有害な環境汚染アルデヒドの分解、除去用酵素として有用であるアルデヒドオキシダーゼの生産も研究されている(特許文献参照2参照。)。コレステロール低下作用、抗血液凝固作用および学習機能向上作用など多彩な生理作用が報告されているドコサヘキサエン酸を生産する酵素の研究も行われている(特許文献参照3参照。)。
【0006】
これらのタンパク質は、元来、所望のタンパク質の生産能力のある微生物、あるいは所望のタンパク質をコードする遺伝子を導入した組換え微生物を培養する発酵法により生産されている。その発酵法としては、回分式発酵法が用いられている。具体的に、効率的な生産性が必要であるα―アミラーゼの生産(非特許文献1参照。)、β−アミラーゼの生産(非特許文献2、3参照。)、ジヒドロ葉酸還元酵素の生産(非特許文献4参照。)、およびインターフェロンおよびインシュリンの生産(非特許文献5参照。)は、いずれも回分式発酵法が用いられている。しかしながら、回分式発酵法では生産効率の向上は困難である。
【0007】
このように、タンパク質の生産において、効率的なタンパク質の製造を目的とした生産性能の向上に向けた様々な検討が行われているが、更なるタンパク質生産技術革新が望まれていた。
【特許文献1】特開2007−061035号公報
【特許文献2】特開2001−299351号公報
【特許文献3】特開2001−169780号公報
【非特許文献1】Palva, Gene (1982)19:81〜87;シノミヤ他
【非特許文献2】Agric. Biol.Chem.(1981)45;1733〜1735;Gray及びChang
【非特許文献3】J. Bacteril(1981)145:422〜428;Williams他
【非特許文献4】Gene(1981)16:199〜206
【非特許文献5】Palva, Gene (1983)22:229〜235
【発明の開示】
【発明が解決しようとする課題】
【0008】
本発明の目的は、簡便な操作方法で、長時間にわたり安定して高生産性を維持する連続発酵法によるタンパク質の製造方法を提供すること、すなわち、連続発酵法において、微生物の発酵培養液を分離膜で濾過し、未濾過液を発酵培養液に保持または還流し、発酵培養液中の微生物濃度を向上させ、かつ、高く維持させることにより高い物質生産性を維持する連続発酵によるタンパク質の製造方法を提供することにある。
【課題を解決するための手段】
【0009】
本発明者らは、鋭意研究の結果、微生物の分離膜内への侵入が少なく、微生物を膜間差圧が低い条件で発酵培養液を濾過した場合に、膜の目詰まりが著しく抑制されることを見出し、分離膜を用いた連続発酵を行うことにより、課題であった微生物が高濃度まで増殖することを可能にし、タンパク質の生産量が飛躍的に向上することを発見し、本発明を完成した。
【0010】
本発明のタンパク質の製造方法は、タンパク質の生産能力を有する微生物の発酵培養液を分離膜で濾過し、未濾過液を前記の発酵培養液に保持または還流し、その微生物の培地を前記の発酵培養液に追加し、かつ、微生物菌体内からタンパク質を回収する連続発酵によるタンパク質の製造方法であって、前記の分離膜として平均細孔径が0.01μm以上1μm未満の細孔を有する多孔性膜を用い、その膜間差圧を0.1から20kPaの範囲にして濾過処理することを特徴とする連続発酵によるタンパク質の製造方法である。
【0011】
本発明のタンパク質の製造方法の好ましい態様によれば、前記の多孔性膜の純水透過係数は2×10−9m3/m2/s/pa以上6×10−7m3/m2/s/pa以下である。
【0012】
本発明のタンパク質の製造方法の好ましい態様によれば、前記の多孔性膜の平均細孔径は0.01μm以上0.2μm未満であり、かつ、該平均細孔径の標準偏差は0.1μm以下である。
【0013】
本発明のタンパク質の製造方法の好ましい態様によれば、前記の多孔性膜の膜表面粗さは0.1μm以下である。
【0014】
本発明のタンパク質の製造方法の好ましい態様によれば、前記の多孔性膜は多孔性樹脂層を含む多孔性膜である。
【0015】
本発明のタンパク質の製造方法の好ましい態様によれば、前記の多孔性膜の素材がポリフッ化ビニリデン系樹脂である。
【0016】
本発明のタンパク質の製造方法の好ましい態様によれば、前記の微生物の発酵原料は糖類を含むものである。
【0017】
本発明のタンパク質の製造方法の好ましい態様によれば、前記のタンパク質をコードする遺伝子はリン酸プロモーターの下流に連結され、かつ培地に含まれる無機リン酸濃度は1mM以下である。
【0018】
本発明のタンパク質の製造方法の好ましい態様によれば、前記のタンパク質はイヌインターフェロンγである。
【0019】
本発明のタンパク質の製造方法の好ましい態様によれば、前記の微生物は大腸菌である。
【発明の効果】
【0020】
本発明によれば、簡便な操作条件で、目詰まりが生じにくい多孔性膜を使用することにより、微生物の増殖を抑制することなく、発酵培養液中の微生物濃度を向上させ、かつ、高く維持させることにより高い物質生産性を維持するタンパク質の製造が可能になる。
【発明を実施するための最良の形態】
【0021】
本発明は、タンパク質の生産能力を有する微生物の発酵培養液を分離膜で濾過し、未濾過液を前記の発酵培養液に保持または還流し、その微生物の培地を前記の発酵培養液に追加し、かつ、微生物菌体内からタンパク質を回収する連続発酵によるタンパク質の製造方法であって、前記の分離膜として平均細孔径が0.01μm以上1μm未満の細孔を有する多孔性膜を用い、その膜間差圧を0.1から20kPaの範囲にして濾過処理することを特徴とする連続発酵によるタンパク質の製造方法である。
【0022】
本発明で用いられる多孔性膜は、発酵に使用する微生物による目詰まりが起こりにくく、かつ濾過性能が長期間安定に継続するものであることが望ましい。そのため、本発明で使用される多孔性膜は、平均細孔径が、0.01μm以上1μm未満であることが重要である。後述するように、多孔性膜としては、多孔質基材の少なくとも1表面に多孔質樹脂層を有する構造のものが好ましく用いられる。
【0023】
多孔質樹脂層が多孔性膜の両面に存在する場合、少なくとも一方の多孔質樹脂層が、上記の平均細孔径の条件を満たしていればよい。平均細孔径がこの範囲内にあると、菌体や汚泥などがリークすることのない高い排除率と、高い透水性を両立させることができ、さらに目詰まりしにくく、透水性を長時間保持することが、より高い精度と再現性を持って実施することができる。平均細孔径が、この範囲内にあれば、大腸菌や酵母などを用いた場合、目詰まりが少なく、また、細胞の濾液への漏れもなく安定に連続発酵が実施可能である。また、大腸菌や酵母より小さな細菌類を用いた場合は、0.4μm以下の平均細孔径であればよりよく、0.2μm以下の平均細孔径であればなお好適に実施可能である。平均細孔径は、小さすぎると透水量が低下することがあるので、通常は0.02μm以上であることが好ましく、より好ましくは0.04μm以上である。
【0024】
ここで、平均細孔径は、倍率10,000倍の走査型電子顕微鏡観察における、9.2μm×10.4μmの範囲内で観察できる細孔すべての直径を測定し、平均することにより求めることができる。細孔径の標準偏差σは、0.1μm以下であることが好ましい。細孔径の標準偏差σは、上述の9.2μm×10.4μmの範囲内で観察できる細孔数をNとして、測定した各々の直径をXk、細孔直径の平均をX(ave)とした以下式(1)より算出した。
【0025】
【数1】

【0026】
本発明で用いられる多孔性膜において、発酵培養液の透過性が重要点の一つであり、透過性の指標として、使用前の多孔性膜の純水透過係数を用いることができる。多孔性膜の純水透過係数が、逆浸透膜による25℃の温度の精製水を用い、ヘッド高さ1mで透水量を測定し算出したとき、2×10−9m3/m2/s/pa以上であることが好ましく、2×10−9以上6×10−7m3/m2/s/pa以下であれば、実用的に十分な透過水量が得られる。
【0027】
本発明で用いられる多孔性膜の表面粗さは、分離膜の目詰まりに影響を与える因子であり、好ましくは、その表面粗さが0.1μm以下のときに分離膜の剥離係数や膜抵抗を低下させることができ、より低い膜間差圧で連続培養が実施可能である。また、表面粗さが低いことで、微生物の濾過において、膜表面で発生する剪断力を低下させることが期待でき、微生物の破壊が抑制され、多孔性膜の目詰まりも抑制されることにより、長期間安定な濾過が可能になると考えられる。
【0028】
ここで、表面粗さは、下記の原子間力顕微鏡装置(AFM)を使用して、下記の条件で測定することができる。
・装置 原子間力顕微鏡装置(Digital Instruments(株)製Nanoscope IIIa)
・条件 探針 SiNカンチレバー(Digital Instruments(株)製)
走査モード コンタクトモード(気中測定)
水中タッピングモード(水中測定)
走査範囲 10μm、25μm 四方(気中測定)
5μm、10μm 四方(水中測定)
走査解像度 512×512
・試料調製 測定に際し膜サンプルは常温でエタノールに15分浸漬後RO水中に24時間浸漬し洗浄した後風乾し用いた。RO水とは、ろ過膜の一種である逆浸透膜(RO膜)を用いてろ過し、イオンや塩類などの不純物を排除した水を指す。RO膜の孔の大きさは、概ね2nm以下である。
【0029】
膜の表面粗さdroughは、AFMにより各ポイントのZ軸方向の高さから下記の(数2)から算出した。
【0030】
【数2】
【0031】
本発明における多孔性膜は、被処理水の水質や用途に応じた分離性能と透水性能を有するものであり、阻止性能、透水性能や耐汚れ性という分離性能の点からは、多孔質樹脂層を含む多孔性膜であることが好ましい。すなわち、好ましい多孔性膜は、多孔質基材の表面に、分離機能層として作用とする多孔質樹脂層を有している。多孔質基材は、多孔質樹脂層を支持して分離膜に強度を与える。
【0032】
本発明で用いられる多孔性膜が、多孔質基材の表面に多孔質樹脂層を有している場合、多孔質基材に多孔質樹脂層が浸透していても、多孔質基材に多孔質樹脂層が浸透していなくてもどちらでも良く、用途に応じて選択される。
【0033】
多孔質基材の材質としては、有機材料および無機材料等で特に限定されないが、有機繊維が望ましい。好ましい多孔質基材は、セルロース繊維、セルローストリアセテート繊維、ポリエステル繊維、ポリプロピレン繊維およびポリエチレン繊維などの有機繊維を用いてなる織布や不織布であり、なかでも、密度の制御が比較的容易であり、製造も容易で安価な不織布が好ましく用いられる。多孔質基材の平均厚みは、好ましくは50μm以上3000μm以下である。
【0034】
多孔質樹脂層は、上述したように分離機能層として作用するもので、有機高分子膜を好適に使用することができる。有機高分子膜は、基本的に有機ポリマー材料から構成される分離膜のことであり、例えば、有機繊維の不織布やマクロポア構造多孔質有機基材と当該多孔質有機基材の孔径より小さな孔径を有する多孔質樹脂層が複合化された構造を持つ場合が多い。
【0035】
ここで、多孔質樹脂層の材質としては、ポリエチレン系樹脂、ポリプロピレン系樹脂、ポリ塩化ビニル系樹脂、ポリフッ化ビニリデン系樹脂、ポリスルホン系樹脂、ポリエーテルスルホン系樹脂、ポリアクリロニトリル系樹脂、セルロース系樹脂およびセルローストリアセテート系樹脂などが挙げられ、これらの樹脂を主成分とする樹脂の混合物であってもよい。なかでも、溶液による製膜が容易で、物理的耐久性や耐薬品性にも優れているポリ塩化ビニル系樹脂、ポリフッ化ビニリデン系樹脂、ポリスルホン系樹脂、ポリエーテルスルホン系樹脂およびポリアクリロニトリル系樹脂が好ましく用いられる。特に、ポリフッ化ビニリデン系樹脂またはそれを主成分とするものが最も好ましく用いられる。
【0036】
ここで、ポリフッ化ビニリデン系樹脂としては、フッ化ビニリデンの単独重合体が好ましく用いられるが、フッ化ビニリデンの単独重合体の他、フッ化ビニリデンと共重合可能なビニル系単量体との共重合体も好ましく用いられる。このようなビニル系単量体としては、テトラフルオロエチレン、ヘキサフルオロプロピレンおよび三塩化フッ化エチレンなどが例示される。
【0037】
本発明で用いられる多孔性膜は、平膜であっても中空糸膜であっても良い。
【0038】
多孔性膜が平膜の場合、その厚みは用途に応じて選択されるが、好ましくは20μm以上5000μm以下の範囲であり、より好ましくは50μm以上2000μm以下の範囲で選択される。上述のように、多孔性膜は、多孔質基材と多孔質樹脂層とから形成されていても良い。多孔質基材の厚みは、好ましくは50μm以上3000μm以下の範囲で選択される。
【0039】
多孔性膜が中空糸膜の場合、内径は好ましくは200μm以上5000μm以下の範囲で選択され、膜厚は好ましくは20μm以上2000μm以下の範囲で選択される。また、有機繊維または無機繊維を筒状にした織物や編み物を含んでいても良い。
【0040】
まず、多孔性膜のうち、平膜の作成法の概要について説明する。
【0041】
多孔質基材の表面に、樹脂と溶媒とを含む原液の被膜を形成すると共に、その原液を多孔質基材に含浸させる。その後、被膜を有する多孔質基材の被膜側表面のみを、非溶媒を含む凝固浴と接触させて樹脂を凝固させると共に、多孔質基材の表面に多孔質樹脂層を形成する。
【0042】
原液は、樹脂を溶媒に溶解させて調整する。原液の温度は、製膜性の観点から、通常、5〜120℃の範囲内で選定することが好ましい。溶媒は、樹脂を溶解するものであり、樹脂に作用してそれらが多孔質樹脂層を形成するのを促すものである。溶媒としては、N−メチルピロリジノン(NMP)、N,N−ジメチルアセトアミド(DMAc)、N,N−ジメチルホルムアミド(DMF)、ジメチルスルホキシド(DMSO)、N− メチル− 2 − ピロリドン、メチルエチルケトン、テトラヒドロフラン、テトラメチル尿素、リン酸トリメチル、シクロヘキサノン、イソホロン、γ− ブチロラクトン、メチルイソアミルケトン、フタル酸ジメチル、プロピレングリコールメチルエーテール、プロピレンカーボネート、ジアセトンアルコール、グリセロールトリアセテート、アセトンおよびメチルエチルケトンなどを用いることができる。なかでも、樹脂の溶解性の高いN−メチルピロリジノン(NMP)、N,N−ジメチルアセトアミド(DMAc)、N,N−ジメチルホルムアミド(DMF)およびジメチルスルホキシド(DMSO)が好ましく用いられる。これらの溶媒は、単独で用いても良いし2種類以上を混合して用いても良い。原液は、先述の樹脂を好ましくは5重量%以上60重量%以下の濃度で、上述の溶媒に溶解させることにより調製することができる。
【0043】
また、例えば、ポリエチレングリコール、ポリビニルアルコール、ポリビニルピロリドンおよびグリセリンなどの溶媒以外の成分を溶媒に添加しても良い。溶媒に非溶媒を添加することもできる。非溶媒は、樹脂を溶解しない液体である。非溶媒は、樹脂の凝固の速度を制御して細孔の大きさを制御するように作用する。非溶媒としては、水や、メタノールおよびエタノールなどのアルコール類を用いることができる。なかでも、非溶媒として、価格の点から水やメタノールが好ましく用いられる。溶媒以外の成分および非溶媒は、混合物であってもよい。
【0044】
原液には、開孔剤を添加することもできる。開孔剤は、凝固浴に浸漬された際に抽出されて、樹脂層を多孔質にする作用を持つものである。開孔剤を添加することにより、平均細孔径の大きさを制御することができる。開孔剤は、凝固浴への溶解性の高いものであることが好ましい。開孔剤としては、例えば、塩化カルシウムや炭酸カルシウムなどの無機塩を用いることができる。また、開孔剤として、ポリエチレングリコールやポリプロピレングリコールなどのポリオキシアルキレン類や、ポリビニルアルコール、ポリビニルブチラールおよびポリアクリル酸などの水溶性高分子化合物や、グリセリンを用いることができる。
【0045】
次に、多孔性膜のうち、中空糸膜の作成法の概要について説明する。
【0046】
中空糸膜は、樹脂と溶媒からなる原液を二重管式口金の外側の管から吐出すると共に、中空部形成用流体を二重管式口金の内側の管から吐出して、冷却浴中で冷却固化して作製することができる。
【0047】
原液は、上述の平膜の作成法で述べた樹脂を好ましくは20重量%以上60重量%以下の濃度で、上述の平膜の生成法で述べた溶媒に溶解させることにより調整することができる。また、中空部形成用流体には、通常気体もしくは液体を用いることができる。また、得られた中空糸膜の外表面に、新たな多孔性樹脂層をコーティング(積層)することもできる。積層は中空糸膜の性質、例えば、親水性・疎水性あるいは細孔径等を所望の性質に変化させるために行うことができる。積層される新たな多孔性樹脂層は、樹脂を溶媒に溶解させた原液を、非溶媒を含む凝固浴と接触させて樹脂を凝固させることによって作製することができる。その樹脂の材質は、例えば、上述有機高分子膜の材質と同様のものが好ましく用いられる。また、積層方法としては、原液に中空糸膜を浸漬してもよいし、中空糸膜の表面に原液を塗布してもよく、積層後、付着した原液の一部を掻き取ったり、エアナイフを用いて吹き飛ばしすることにより積層量を調整することもできる。
【0048】
本発明で用いられる多孔性膜は、支持体と組み合わせることによって分離膜エレメントとして使用することができる。支持体として支持板を用い、その支持板の少なくとも片面に本発明で用いられる多孔性膜を配した多孔性膜を有する分離膜エレメントは、本発明で用いられる膜エレメントの好適な形態の一つである。この形態では、膜面積を大きくすることが困難なので、透水量を大きくするために、支持板の両面に多孔性膜を配することも好ましい態様である。
【0049】
本発明において、微生物を濾過処理する際の膜間差圧は、微生物および培地成分が容易に目詰まりしない条件であればよいが、膜間差圧を0.1kPa以上20kPa以下の範囲として濾過処理することが重要である。また、使用前の純水透過係数が、逆浸透膜による25℃の温度の精製水を用い、ヘッド高さ1mで透水量を測定し算出したとき、2×10−9m3/m2/s/pa以上の範囲であることが好ましく、さらに2×10−9m3/m2/s/pa以上6×10−7m3/m2/s/pa以下の範囲にあることが好ましい。濾過の駆動力としては、培養液と多孔性膜処理水の液位差(水頭差)を利用したサイホンにより多孔性膜に膜間差圧を発生させることが可能であり、また、濾過の駆動力として多孔性膜処理水側に吸引ポンプを設置してもよいし、多孔性膜の発酵培養液側に加圧ポンプを設置することも可能である。膜間差圧は、発酵培養液と多孔性膜処理水の液位差を変化させることで制御することができる、またポンプを使用する場合には吸引圧力により制御することができ、更に発酵培養液側の圧力を導入する気体または液体の圧力によって制御することができる。これら圧力制御を行う場合には、発酵培養液側の圧力と多孔性膜処理水側の圧力差をもって膜間差圧とし、膜間差圧の制御に用いることができる。
【0050】
本発明で使用される発酵原料としては、発酵培養する微生物の生育を促し、目的とする発酵生産物であるタンパク質を良好に生産させ得るものであれば良い。発酵原料としては、例えば、炭素源、窒素源、無機塩類、および必要に応じてアミノ酸、およびビタミンなどの有機微量栄養素を適宜含有する液体培地等が好ましく用いられる。
【0051】
上記の炭素源としては、グルコース、シュークロース、フラクトース、ガラクトースやラクトース等の糖類、これら糖類を含有する澱粉糖化液、甘藷糖蜜、甜菜糖蜜、ハイテストモラセス、更には酢酸等の有機酸、エタノールなどのアルコール類およびリセリンなども使用される。ここで糖分とは、多価アルコールの最初の酸化生成物であり、アルデヒド基またはケトン基をひとつ持ち、アルデヒド基を持つ糖をアルドース、ケトン基を持つ糖をケトースと分類される炭水化物のことを指す。
【0052】
また上記の窒素源としては、アンモニアガス、アンモニア水、アンモニウム塩類、尿素、硝酸塩類、その他補助的に使用される有機窒素源、例えば油粕類、大豆加水分解液、カゼイン分解物、その他のアミノ酸、ビタミン類、コーンスティープリカー、酵母または酵母エキス、肉エキス、ペプトン等のペプチド類および各種発酵菌体およびその加水分解物などが使用される。
【0053】
また上記の無機塩類としては、リン酸塩、マグネシウム塩、カルシウム塩、鉄塩およびマンガン塩等が使用される。
【0054】
また、アミノ基とカルボキシル基の両方の官能基を持つ、また生体の蛋白質の構成ユニットとなる有機化合物であるアミノ酸としては、中性アミノ酸 、酸性アミノ酸および塩基性アミノ酸が使用される。中性アミノ酸には、グリシン、アラニン、分枝アミノ酸(バリン、ロイシン、イソロイシン)、ヒドロキシアミノ酸(セリン、スレオニン)、含硫アミノ酸 (システイン、メチオニン)、酸アミドアミノ酸(アスパラギン、グルタミン)などの脂肪族アミノ酸 、プロリンなどのイミノ酸 、フェニルアラニン、チロシンおよびトリプトファンなどの芳香族アミノ酸が挙げられる。また、酸性アミノ酸としては、アスパラギン酸、グルタミン酸がある。塩基性アミノ酸には、リシン、アルギニンおよびヒスチジンが使用される。
【0055】
本発明で使用される微生物が生育のために特定の栄養素を必要とする場合には、その栄養物を標品もしくはそれを含有する天然物として添加することができる。また、消泡剤を必要に応じて使用することができる。
【0056】
本発明において、培養液とは、発酵原料に微生物が増殖した結果得られる液のことを言う。追加する発酵原料の組成は、目的とするタンパク質の生産性が高くなるように、培養開始時の発酵原料組成から適宜変更しても良い。
【0057】
本発明で使用されるタンパク質の生産能力のある微生物は、製造するタンパク質をコードする遺伝子を元来有する、あるいは導入されている微生物のことである。本発明で好適に用いることができる微生物としては、例えば、発酵工業においてよく使用されるパン酵母などの酵母、カビなどの真核微生物、大腸菌、枯草菌およびコリネ型細菌などの原核微生物などを挙げることができる。使用される微生物は、自然環境から単離されたものでもよく、また、突然変異や遺伝子組換えによって一部性質が改変されたものであってもよい。本発明においては、特に大腸菌が好ましく用いられる。
【0058】
本発明のタンパク質の製造法で製造されるタンパク質をコードする遺伝子は、タンパク質を発現可能なプロモーターの支配下に連結して用いることが好ましい。プロモーターの種類としては、構成的プロモーターと誘導的プロモーターがあり、本発明ではどちらも使用できる。なかでも、発現能力の高いプロモーターを用いることが望ましく、構成的なプロモーターとしては、Bacillus sp. SK−1株由来のD−アミノ酸アミノトランスフェラーゼのオープンリーディングフレーム(ORF)の上流に局在するプロモーター、カリフラワーモザイクウイルス(CaMV)35S RNA遺伝子のプロモーター、アクチン遺伝子のプロモーター、枯草菌由来のルビスコプロモーターおよびADH1プロモーターなどが挙げられる。
【0059】
また、誘導的プロモーターとしては、T7プロモーター、Tacプロモーター、PLプロモーター、PRプロモーター、PSEプロモーター、トリプトファンプロモーターおよびリン酸プロモーターを例示することができる。また、1つのプロモーターだけではなく、2個または3個のプロモーターを連結したダブル、トリプルプロモーターとしても使用できるし、種の違うプロモーターを連結させたハイブリッドプロモーターとしても使用することができる。
【0060】
ここで、Bacillus sp. SK−1株由来のD−アミノ酸アミノトランスフェラーゼのオープンリーディングフレーム(ORF)の上流に局在するプロモーター、カリフラワーモザイクウイルス(CaMV)35S RNA遺伝子のプロモーター、アクチン遺伝子のプロモーター、枯草菌由来のルビスコプロモーター、およびADH1プロモーターは、構成的に発現が誘導されるプロモーターである。また、T7プロモーター、Tacプロモーター、lacプロモーター、PLプロモーター、PRプロモーターおよびPSEプロモーターは、イソプロピルチオ−β−D−ガラクトシド(IPTG)で発現が誘導されるプロモーターである。トリプトファンプロモーターは、培養液中のトリプトファン濃度が低下することで発現が誘導されるプロモーターであり、またインドール酢酸(IAA)を添加しても発現が誘導される。リン酸プロモーターは、発酵培養液中の無機リン酸の濃度が低下することにより発現が誘導されるプロモーターである。
【0061】
本発明のタンパク質の製造法は、タンパク質の生産能力を有する微生物の発酵培養液を分離膜で濾過し、未濾過液を前記の発酵培養液に保持または還流し、その微生物の培地を前記の発酵培養液に追加し、かつ、微生物菌体内からタンパク質を回収する。本発明では、発酵培養液に追加する培地はタンパク質の生産に適した組成に変更することが可能であることから、高生産性を維持したタンパク質の製造が実現する。すなわち、本発明のタンパク質の製造法は、タンパク質を発現せしめるプロモーターとして、前述Bacillus sp. SK−1株由来のD−アミノ酸アミノトランスフェラーゼのオープンリーディングフレーム(ORF)の上流に局在する構成的なプロモーター、カリフラワーモザイクウイルス(CaMV)35S RNA遺伝子のプロモーター、アクチン遺伝子のプロモーター、枯草菌由来のルビスコプロモーター、およびADH1プロモーターのように構成的に発現可能なプロモーターを用いることができる。また、前述トリプトファンプロモーターやリン酸プロモーターのような、培地中あるいは培養液中の化学品濃度に依存して発現が制御可能なプロモーターを用いることもできる。
【0062】
本発明のタンパク質の製造法では、構成的なプロモーターを用いた場合、微生物の培地は特に制限されることはなく、微生物が生育することにより、発酵培養液中の微生物が高濃度まで生育されることから、生産性の高いタンパク質の製造を行うことができる。T7プロモーター、Tacプロモーター、PLプロモーター、PRプロモーター、PSEプロモーターおよびlacプロモーターの場合、微生物の培地にIPTGを含むことにより、発酵培養液中にIPTGを添加させることができることから、生産性の高いタンパク質の製造を行うことができる。
【0063】
トリプトファンプロモーターの場合、微生物の培地に含まれるトリプトファン濃度を低くすることにより、発酵培養液中のトリプトファン濃度を低下させることができることから生産性の高いタンパク質の製造を行うことができるし、発酵培養液中にIAAを添加することで生産性の高いタンパク質の製造を行うことができる。リン酸プロモーター場合、微生物の培地に含まれる無機リン酸濃度を低くすることにより、発酵培養液中のリン酸濃度を低下させることができることから、生産性の高いタンパク質の製造を行うことができる。
【0064】
発明のタンパク質の製造方法において、前記のリン酸プロモーターを好ましく用いることができる。ここで、リン酸プロモーターに関して説明する。リン酸結合タンパク質(PhoS)遺伝子の上流域をコードするDNAは、得られたゲノムDNAを元に、リン酸結合タンパク遺伝子の上流域の5’および3’末端配列を付加したプライマーを設計してPCRを行うことによって得ることができる。ゲノムDNAは、例えば、次のようにして製造することができる。第一に、大腸菌K−12株由来、中でもKLF48/KL159株からゲノムDNAを抽出する。上記大腸菌よりゲノムDNAを抽出する方法としては、アルカリ法による抽出(ヌクレイック アシッド リサーチ 第7巻、第6号、1513−1523)や、非フェノール性試薬とタンパク質凝集剤を使用し、相配分に基づいて比重の違いにより中間に凝集層を形成させる凝集分配法(新生化学実験講座2 核酸1 分離精製:13−51(1991))などの中から適当な方法を選んで行うことができる。
【0065】
リン酸結合タンパク質は、リン酸の取り込みに関係しており、リン酸が少なくなれば、それを取り込もうとしてリン酸結合タンパク質が合成され、培地中のリン酸に結合して細胞質内に送り込む機能を有している。これらのタンパク質の転写制御は、これらのタンパク質をコードしている遺伝子のさらに上流域に存在し、pho boxと呼ばれているコンセンサス領域ctgtcataaatctgtcacを含むプロモーターである。この領域は、リン酸結合タンパク質をコードしている遺伝子の上流であり約200−1000bpのDNA配列である。このようにして得られたリン酸プロモーターの下流に、所望のタンパク質をコードする遺伝子を連結した遺伝子発現カセットを導入した微生物を用いることができる。
【0066】
微生物の発酵培養液は、リン酸プロモーターが活性化する濃度まで低下させることが好ましい。リン酸プロモーターは、無機リン酸濃度が1mM未満でタンパク質が発現する。そこで、初めの発酵培養液の無機リン酸濃度は、1mM以上であればよい。初めの発酵培養液の無機リン酸濃度は1mM未満でも良いが、菌体の生育が遅くなる傾向を示す。また、無機リン酸濃度が高濃度であった場合、希釈に時間がかかり、効率的にタンパク質を生産できない可能性がある。好ましくは、2〜5mMの濃度で添加された無機リン酸を含む培養液を使用することが好ましい。また、追加する培地は、無機リン酸を1mM以下の濃度で含んだ培地を利用することがよい。効率的に発酵培養液のリン酸が希釈されるので、好ましくは0〜0.5mMの無機リン酸を含んだ培地を利用することが好ましい。
【0067】
タンパク質を生産する能力を有する微生物の培養は、通常、pH3〜9、温度15〜70℃の範囲で行われることが多い。発酵培養液のpHは、無機あるいは有機の酸、アルカリ性物質、さらには尿素、炭酸カルシウム、アンモニアガスなどによって上記範囲内のあらかじめ定められた値に調節することができる。
【0068】
微生物の培養において、酸素の供給速度を上げる必要があれば、空気に酸素を加えて酸素濃度を例えば21%以上に保つ、発酵培養液を加圧する、攪拌速度を上げる、あるいは通気量を上げるなどの手段を用いることができる。逆に、酸素の供給速度を下げる必要があれば、炭酸ガス、窒素およびアルゴンなど酸素を含まないガスを空気に混合して供給することも可能である。
【0069】
本発明の培養初期にBatch培養またはFed−Batch培養を行って微生物濃度を高くした後に連続発酵(引き抜き)を開始しても良いし、高濃度の菌体をシードし、培養開始とともに連続発酵を行っても良い。適当な時期から培地の供給および培養物の引き抜きを行うことが可能である。培地供給と培養物の引き抜きの開始時期は必ずしも同じである必要はない。また、培地の供給と培養物の引き抜きは連続的であってもよいし、間欠的であってもよい。培地には上記に示したような菌体増殖に必要な栄養素を添加し、菌体増殖が連続的に行われるようにすればよい。
【0070】
発酵培養液中の微生物の濃度は、効率よい生産性を得る上で、発酵培養液の環境が微生物の増殖にとって不適切となって死滅する比率が高くならない範囲で、高い状態で維持することが好ましい。また、連続発酵装置の運転上の不都合や生産効率の低下を招かなければ、微生物の濃度の上限値は特に限定されない。
【0071】
タンパク質生産能力のあるフレッシュな菌体を増殖させつつ行う連続発酵操作は、培養管理上、通常は単一の発酵反応槽で行うことが好ましい。しかしながら、菌体を増殖しつつ生産物を生成する連続発酵法であれば、発酵反応槽の数は問わない。発酵反応槽の容量が小さい等の理由から、複数の発酵反応槽を用いることもあり得る。この場合、複数の発酵反応槽を配管で並列または直列に接続して連続発酵を行っても高生産性は得られる。
【0072】
本発明においては、微生物を発酵反応槽に維持したままで、発酵反応槽からの発酵培養液の連続的かつ効率的な抜き出しが可能となることから、微生物を連続的に培養し、十分な増殖を確保した後に培地組成を変更し、目的とするタンパク質を効率よく製造することも可能である。
【0073】
本発明のタンパク質の製造法で好適に製造されるタンパク質としては、産業的価値から考えてヒトや哺乳類が産生するタンパク質、例えば、成長ホルモン、サイトカイン、増殖因子および細胞骨格タンパク質などのタンパク質が挙げられる。また、微生物、植物または昆虫などが産生する酵素や種々タンパク質、ペプチドも本発明で好適に製造されるタンパク質に含まれる。また、これらタンパク質のアミノ酸配列を改変した変異タンパク質も本発明で製造されるタンパク質に含まれる。更に、2種類以上のタンパク質、あるいはペプチドを連結したキメラタンパク質およびキメラペプチドも本発明のタンパク質の製造法で製造可能である。
【0074】
本発明においての、タンパク質の回収について概要を説明する。
【0075】
まず、タンパク質を菌体から回収するとは、発酵培養液を抜き出し、そこに含まれる菌体を回収して後述した方法によりタンパク質を抽出・精製することである。
【0076】
発酵培養液を抜き出すのは、菌体が発酵培養液中に含まれていればどのタイミングで抜き出しても良い。例えば、植菌後すぐの発酵培養液でも構わない。しかしながら、効率よい生産性を得る上で、発酵培養液の環境が微生物の増殖にとって不適切となって死滅する比率が高くならない範囲で、高い状態で維持しているときに回収する好ましい。その時期に発酵培養液を抜き出せば、効率的に高生産でタンパク質が得られる。また、適宜発酵培養液を抜きだして、タンパク質が高発現あるいは高活性な時期に回収するのも良い。
【0077】
本発明においては、培養された微生物菌体内からタンパク質を回収する。具体的には、発酵反応槽から発酵培養液を抜き出し、培養された微生物菌体内からタンパク質を回収する。
【0078】
タンパク質を回収する方法としては、例えば、ホモジナイザー、超音波、ガラスビーズ、ブレンダーおよびブレスなど機械的な方法で菌体を破砕し精製する方法を採用することができる。精製は、カラムクロマトグラフィー、液体クロマトグラフィーおよびイオン交換クロマトグラフィーなどにより精製が可能である。
【0079】
また、タンパク質を回収する方法としては、界面活性剤や変性剤などの化学的な方法を用いることも可能である。この場合、タンパク質が変性されることがあるので、可溶化後、透析や希釈により界面活性剤や変性剤を除去した後精製し、タンパク質を得ることができる。
【0080】
本発明の連続発酵によるタンパク質の製造方法で用いられる連続発酵装置1つの例は、主に、微生物を保持しタンパク質を製造するための発酵反応槽、および微生物と培地を濾過分離するための多孔性膜を含む分離膜エレメントから構成される。分離膜エレメントは、発酵反応槽の内部または外部のいずれに設置されてもよい。
【0081】
本発明の連続発酵によるタンパク質の製造方法は、分離膜に平均細孔径が0.01μm以上1μm未満の多孔性膜を使用し、濾過圧力である膜間差圧を0.1から20kPaの範囲にして濾過処理することを特徴としている。そのため、特別に発酵反応槽内を加圧状態に保つ必要がないことから、濾過分離装置と発酵反応槽間で発酵培養液を循環させる動力手段が不要となり、分離膜エレメントを発酵反応槽内部に設置して発酵装置をコンパクト化することも可能である。
【0082】
本発明の連続発酵によるタンパク質の製造方法で用いられる連続発酵装置のうち、分離膜エレメントが発酵反応槽の内部に設置された代表的な一例を図1の概要図に示す。図1は、本発明の連続発酵によるタンパク質の製造方法で用いられる連続発酵装置の一つの実施の形態を示す概要側面図である。
【0083】
図1において、連続発酵装置は、内部に分離膜エレメント2を備えた発酵反応槽1と水頭差制御装置3で基本的に構成されている。発酵反応槽1内の分離膜エレメント2には、多孔性膜が組み込まれている。この多孔性膜としては、例えば、国際公開第2002/064240号パンフレットに開示されている膜および膜エレメントを使用することができる。分離膜エレメントに関しては、追って詳述する。
【0084】
次に、図1の連続発酵装置による連続発酵の形態について説明する。図1において、培地供給ポンプ7によって培地を発酵反応槽1に連続的もしくは断続的に投入する。培地は発酵反応槽1に投入前に、必要に応じて加熱殺菌、加熱滅菌あるいはフィルターを用いた滅菌を行うことができる。培養時には、必要に応じて攪拌機5で発酵反応槽1内の発酵培養液を攪拌し、また必要に応じて気体供給装置4によって必要とする気体を供給し、また必要に応じてpHセンサ・制御装置9、およびpH調整溶液供給ポンプ8によって発酵培養液のpHを調整し、また必要に応じて温度調節器10によって発酵培養液の温度を調節することにより生産性の高いタンパク質生産を行うことができる。ここでは、計装・制御装置による発酵培養液の物理化学的条件の調節に、pHおよび温度を例示したが、必要に応じて溶存酸素やORPの制御、更にはオンラインケミカルセンサーなどの分析装置により培養液中の様々な物質の濃度を測定し、それを指標とした物理化学的条件の制御を行うことができる。また、培地の連続的もしくは断続的投入の形態に関しては、例えば、上記計装装置による培養液の物理化学的環境の測定値を指標として、培地投入量および速度を適宜調節することができる。
【0085】
発酵培養液は、発酵反応槽1内に設置された分離膜エレメント2によって発酵培養液と濾液に濾過・分離される。発酵培養液には、菌体が含まれる。また、分離膜によって濾過・分離された濾液には菌体は含まれない。分離膜エレメントにより、装置系内に菌体を維持させつつ、菌体増殖時に発生する酢酸等の老廃物を濾液として装置系内から出すことができる。
【0086】
また、濾過・分離された微生物は装置系内にとどまることにより装置系内の微生物濃度を高く維持することができ、生産性の高いタンパク質生産を可能としている。ここで、分離膜エレメント2による濾過・分離は、発酵反応槽1の水面との水頭差圧によって行い、特別な動力は必要ない。また、必要に応じてレベルセンサ6および水頭差圧制御装置3によって、分離膜エレメント2の濾過・分離速度および発酵反応槽内の発酵培養液量を適当に調節することができる。上記の分離膜エレメント2による濾過・分離には水頭差圧によって行うことを例示したが、必要に応じてポンプ等による吸引濾過あるいは装置系内を加圧することにより濾過・分離することもできる。
【0087】
タンパク質を菌体から回収する時期は、基本的にはいつでもかまわない。効率よい生産性を得る上で、発酵培養液の環境が微生物の増殖にとって不適切となって死滅する比率が高くならない範囲で、高い状態で維持しているときに回収することが好ましい。また、適宜発酵培養液を抜きだして、タンパク質が高発現あるいは高活性な時期に回収することが好ましい。
【0088】
タンパク質の回収には、発酵反応槽から発酵培養液を引き抜き、上述した手段により、発酵培養液中に含まれる微生物菌体内からタンパク質を回収することができる。
【0089】
次に、本発明の連続発酵によるタンパク質の製造方法で用いられる連続発酵装置のうち、分離膜エレメントが、発酵反応槽の外部に設置された代表的な一例を図2の概要図に示す。図2は、本発明で用いられる他の連続発酵装置の一つの実施の形態を説明するための概要側面図である。
【0090】
図2において、連続発酵装置は、発酵反応槽1と、分離膜エレメント2を内部に備えた膜分離槽12と、水頭差制御装置3とで基本的に構成されている。膜分離槽12中の分離膜エレメント2には、多孔性膜が組み込まれている。この多孔性膜としては、例えば、国際公開第2002/064240号パンフレットに開示されている分離膜および分離膜エレメントを使用することが好適である。また、膜分離槽12は、発酵培養液循環ポンプ11を介して発酵反応槽1に接続されている。
【0091】
図2において、培地供給ポンプ7によって培地を発酵反応槽1に投入し、必要に応じて、攪拌機5で発酵反応槽1内の発酵培養液を攪拌し、また必要に応じて、気体供給装置4によって必要とする気体を供給し、また必要に応じて、pHセンサ・制御装置9およびよびpH調整溶液供給ポンプ8によって発酵培養液のpHを調整し、また必要に応じて、温度調節器10によって発酵培養液の温度を調節することにより、生産性の高い発酵生産を行うことができる。さらに、装置内の発酵培養液は、発酵培養液循環ポンプ11によって発酵反応槽1と膜分離槽12の間を循環する。発酵培養液は、分離膜エレメント2によって微生物を含む発酵培養液と微生物を含まない濾液に濾過・分離され、濾液は装置系内から取り除かれる。
【0092】
また、濾過・分離された微生物は、装置系内にとどまることで装置系内の微生物濃度を高く維持することができ、生産性の高い発酵生産を可能としている。ここで、分離膜エレメント2による濾過・分離は、膜分離槽12の水面との水頭差圧によって行い、特別な動力を必要としない。また、必要に応じて、レベルセンサ6および水頭差圧制御装置3によって、分離膜エレメント2の濾過・分離速度および装置系内の発酵培養液量を適当に調節することができる。また必要に応じて、気体供給装置4によって必要とする気体を膜分離槽12内に供給することができる。上記のように、分離膜エレメント2による濾過・分離には水頭差圧によって行うことを例示したが、必要に応じて、ポンプ等による吸引濾過あるいは装置系内を加圧することにより濾過・分離することもできる。
【0093】
タンパク質の回収には、適宜発酵培養液を抜きだして、タンパク質が高発現あるいは高活性な時期に回収する。タンパク質が高発現あるいは高活性である発酵培養液を抜き出し、菌体のみ回収する。菌体回収には、例えば、遠心分離機を用いて、遠心操作にて菌体を回収することが可能である。次に、回収した菌体を破砕する。破砕の方法は、例えば、ホモジナイザー、超音波など機械的な方法を用いて行うことができる。破砕した菌体を、例えばリン酸緩衝液などの適当な緩衝液に懸濁し、カラムクロマトグラフィー、液体クロマトグラフィーおよびイオン交換クロマトグラフィーなどを用いて、目的のタンパク質を精製・回収することができる。
【0094】
次に、本発明で用いられる分離膜エレメントについて説明する。
【0095】
本発明で用いられる分離膜エレメントの好適な形態の例である国際公開第2002/064240号パンフレットに開示されている分離膜および分離膜エレメントを、以下、図を用いてその概略を説明する。図3は、本発明で用いられる分離膜エレメントの一つの実施の形態を説明するための概要斜視図である。
【0096】
分離膜エレメントは、図3に示すように、剛性有する支持板13の両面に、流路材14と前記の分離膜15(多孔性膜)とをこの順序で配し構成されている。支持板13は、両面に凹部16を有している。分離膜15は発酵培養液を濾過する。流路材14は、分離膜15で濾過された濾過液を効率よく支持板13に流すためのものである。支持板13に流れた濾過液は、支持板13の凹部16を通って外部に取り出される。外部へは、集水パイプ17を介して連続発酵装置外部に取り出される。
【0097】
図4に示す分離膜エレメントについて説明する。図4は、本発明で用いられる別の分離膜エレメントを例示説明するための概略斜視図である。分離膜エレメントは、図4に示すように、中空糸膜(多孔性膜)で構成された分離膜束18と上部樹脂封止層19と下部樹脂封止層20によって主に構成される。分離膜束18は、上部樹脂封止層19および下部樹脂封止層20よって束状に接着・固定化されている。下部樹脂封止層20による接着・固定化は、分離膜束18の中空糸膜(多孔性膜)の中空部を封止しており、発酵培養液の漏出を防ぐ構造になっている。一方、上部樹脂封止層19は、分離膜束18の中空糸膜(多孔性膜)の内孔を封止しておらず、集水パイプ22に透過水が流れる構造となっている。この分離膜エレメントは、支持フレーム21を介して連続発酵装置内に設置することが可能である。分離膜束18によって濾過された濾過液は、中空糸膜の中空部を通り、集水パイプ22を介して連続発酵装置外部に取り出される。濾過液を取り出すための動力として、水頭差圧、ポンプ、液体や気体等による吸引濾過、あるいは装置系内を加圧するなどの方法を用いることができる。
【0098】
本発明の連続発酵によるタンパク質の製造方法で用いられる連続発酵装置の分離膜エレメントを構成する部材は、高圧蒸気滅菌操作に耐性の部材であることが好ましい。連続発酵装置内が滅菌可能であれば、連続発酵時に好ましくない微生物による汚染の危険を回避することができ、より安定した連続発酵が可能となる。分離膜エレメントを構成する部材は、高圧蒸気滅菌操作の条件である、121℃で15分間に耐性であることが好ましい。
【0099】
分離膜エレメント部材には、例えば、ステンレスやアルミニウムなどの金属、ポリアミド系樹脂、フッ素系樹脂、ポリカーボネート系樹脂、ポリアセタール系樹脂、ポリブチレンテレフタレート系樹脂、PVDF、変性ポリフェニレンエーテル系樹脂およびポリサルホン系樹脂等の樹脂を好ましく選定することができる。
【0100】
本発明の連続発酵によるタンパク質の製造方法で用いられる連続発酵装置では、分離膜エレメントは、図1のように発酵反応槽内に設置しても良いし、図2のように発酵反応槽外に設置しても良い。分離膜エレメントを発酵反応槽外に設置する場合には、別途、膜分離槽を設けてその内部に分離膜エレメントを設置することができ、発酵反応槽と膜分離槽の間を培養液を循環させながら、分離膜エレメントにより培養液を連続的に濾過することができる。
【0101】
本発明の連続発酵によるタンパク質の製造方法で用いられる連続発酵装置では、膜分離槽は、高圧蒸気滅菌可能であることが望ましい。膜分離槽が高圧蒸気滅菌可能であると、雑菌による汚染回避が容易である。
【0102】
本発明の連続発酵によるタンパク質の製造方法に従って、連続発酵を行った場合、従来のバッチ培養と比較して、高い菌体濃度が得られ、極めて効率のよいタンパク質生産が可能となる。ここで、連続培養における生産速度は、次の(式3)で計算される。
・発酵生産速度(g/L/hr)=抜き取り液中の生産物濃度(g/L)×発酵培養液抜き取り速度(L/hr)÷装置の運転液量(L)・・・(式3)
また、バッチ培養での発酵生産速度は、原料炭素源をすべて消費した時点の生産物量(g)を、炭素源の消費に要した時間(h)とその時点の培養液量(L)で除して求められる。
【実施例】
【0103】
以下、本発明をさらに詳細に説明するために、タンパク質としてイヌインターフェロンγ、タンパク質の生産能力のある微生物として大腸菌を選定し、図1および図2の概要図に示す連続発酵装置を用いたタンパク質の製造を行った。本発明は、これら実施例の記載に限定されるものではない。ここで、大腸菌としては、大腸菌MC1061株(ATCC社製)を用いた。大腸菌MC1061株に、イヌインターフェロンγをコードする遺伝子を導入することにより、組換えイヌインターフェロンγ生産能力をもつ大腸菌株を造成し実施した。具体的には、イヌインターフェロンγをコードする遺伝子を大腸菌K−12株由来のリン酸結合タンパク質遺伝子の下流に連結したプラスミドを導入することにより、組換えイヌインターフェロンγ生産能力を持つ形質転換体大腸菌株を造成して使用した。
【0104】
(参考例1)リン酸結合タンパク遺伝子(phoS)の上流域遺伝子を含むベクターの構築
(1)大腸菌K−12株のゲノムDNAの調製
K−12株であるKLF/KL159株(Yale University E.coli. GeneticResourceCenter CGSC Strain#4302)をT培地(バクトトリプトン10g/L、NaCl 5g/L)で終日培養後、遠心操作により沈殿とし、核酸抽出剤「Sepagene」(三光純薬社製)を用いてゲノムDNAを回収した。すなわち、トリス緩衝液で構成される試薬1を沈殿に添加し、撹拌後室温で10分間静置した。次に、チオシアン酸グアニジンで構成される試薬2を添加してゆるやかに混和した後、吸着剤とクロロホルムで構成される試薬3を添加し激しく混和した。12000rpm、15分間遠心操作後、上清を回収し酢酸緩衝液で構成される試薬4を加え、さらにイソプロパノール加え転倒混和した。12000rpm,15分遠心後、上清を除去し沈殿物を滅菌蒸留水に溶解した。
【0105】
(2)リン酸結合タンパク質phoS上流配列遺伝子の取得
大腸菌K12株ゲノム配列情報(MEDLINE)を元に、配列番号1と配列番号2の2種類のプライマーをDNAシンセサイザーにて合成した。上記(1)で得られたゲノムDNAを0.5mLのミクロ遠心チューブに2μL添加し、各プライマーを20pmol,20mMトリス塩酸緩衝液(pH8.0)、1.5mM MgCl2,25mM KCL,100μg/mLゼラチン、50μM各dNTP、4単位 LA−TaqDNAポリメラーゼとなるように各試薬を加え、全量100μLとした。DNAの変性条件を94℃、1分、プライマーのアニーリング条件を55℃、1分、プライマーの伸長条件を72℃、1分の各条件でPerkin−Elmer Cetus社のDNAサーマルサイクラーを用い、35サイクル反応させた。これを1%アガロースゲルにて電気泳動し、約300bpのDNA断片を常法に従って調製した。さらに、配列番号3と配列番号4の2種類のプライマーをDNAシンセサイザーにて合成した。上記アガロースゲルより調製したDNA断片を0.5mLのミクロ遠心チューブに2μL添加し、各プライマーを20pmol,20mMトリス塩酸緩衝液(pH8.0)、1.5mM MgCl2,25mM KCL,100μg/mLゼラチン、50μM各dNTP、4単位 LA−TaqDNAポリメラーゼとなるように各試薬を加え、全量100μLとした。DNAの変性条件を94℃、1分、プライマーのアニーリング条件を55℃、1分、プライマーの伸長条件を72℃、1分の各条件でPerkin−Elmer Cetus社のDNAサーマルサイクラーを用い、35サイクル反応させた。これを1%アガロースゲルにて電気泳動し、約300bpのDNA断片を常法に従って調製した。
【0106】
このようにして得られたDNA断片を、Invitrogen社のT−Vectorに宝酒造(株)のDNA Ligation Kit Ver.1を用いて連結した。これを用いて常法に従い大腸菌を形質転換し、得られた形質転換体からプラスミドDNAを常法により調製した。次に、このプラスミドにPCR断片が挿入されていることを前述と同じ条件のPCRによって確認後、蛍光DNAシーケンサーを用い、その添付プロトコールに従ってパーキンエルマー社のダイターミネーターサイクルシーケンシングキットを用いて得られたDNA断片が大腸菌K12株由来の遺伝子をコードする配列であることを確認した(配列番号5)。
【0107】
(3)リン酸結合タンパク質遺伝子(phoS)上流配列およびSD配列を付加したベクターの構築
上記(2)で調製したリン酸結合タンパク質phoSの上流配列遺伝子を含むベクターを用いて、リン酸結合タンパク質phoSの上流配列遺伝子の3’末にSD配列を付加した。すなわち、センス側のプライマーである配列番号3とアンチセンス側のプライマーすなわちSD配列を有するプライマー配列番号6を作製した。次に、配列番号3と配列番号6のプライマーの組み合わせを用いたPCRを実施した。すなわち、上記ベクターDNAを0.5mLのミクロ遠心チューブに2μL添加し、各プライマーを20pmol,20mMトリス塩酸緩衝液(pH8.0)、1.5mM MgCl2,25mM KCL,100μg/mLゼラチン、50μM各dNTP、4単位 LA−TaqDNAポリメラーゼとなるように各試薬を加え、全量100μLとした。DNAの変性条件を94℃、1分、プライマーのアニーリング条件を55℃、1分、プライマーの伸長条件を72℃、1分の各条件でPerkin−Elmer Cetus社のDNAサーマルサイクラーを用い、35サイクル反応させた。これを1%アガロースゲルにて電気泳動し、約300bpのDNA断片を常法に従って調製した。
【0108】
このDNA断片をInvitrogen社のT−Vectorに宝酒造(株)のDNA Ligation Kit Ver.1を用いて連結した。これを用いて常法に従い大腸菌を形質転換し、得られた形質転換体よりプラスミドDNAを常法により調製した。次に、このプラスミドにPCR断片が挿入されていることを前述と同じ条件のPCRによって確認後、蛍光DNAシーケンサーを用い、その添付プロトコールに従ってパーキンエルマー社のダイターミネーターサイクルシーケンシングキットを用いて得られたDNA断片が大腸菌K12株由来の遺伝子をコードする配列であることを確認した。(配列番号7)
上記(2)で調製したリン酸結合タンパク質phoSの上流配列遺伝子を含むベクターおよびタカラバイオ社のpBR322をそれぞれEcoRIによる制限酵素処理を行い、それぞれをアガロースゲル電気泳動を行い、常法に従いに遺伝子を切り出した。pBR322については、Alkaline Phospatase Shrimp(日本ロシュ社製)による脱リン酸化処理後、リン酸結合タンパク質phoSの上流配列遺伝子と宝酒造(株)のDNA Ligation Kit Ver.1を用いて連結した。これを用いて常法に従い大腸菌を形質転換し、得られた形質転換体よりプラスミドDNAを常法により調製した。次に、このプラスミドにPCR断片が挿入されていることを前述と同じ条件のPCRによって確認後、蛍光DNAシーケンサー(パーキンエルマー社製DNAシーケンサー373S)を用い、その添付プロトコールに従って、パーキンエルマー社のダイターミネーターサイクルシーケンシングキットを用いて、得られたDNA断片がリン酸結合タンパク質phoSの上流配列遺伝子および新たに付加したSD配列をコードする配列であることを確認した。ベクターの名称について、配列番号7をpPho2とする。
【0109】
(参考例2)イヌIFN-γ遺伝子の取得
(1)イヌcDNAの調製
イヌ末梢血よりイヌリンパ球を分離し、フィトヘムアグルチニン(PHA)を50ug/mLの終濃度で48時間刺激した。その後、“ISOGEN”(登録商標)(ニッポンジーン社製)を用いて総RNAを調製した。得られたRNAを1mM EDTAを含む10mM トリス塩酸緩衝液(pH7.5)(以下TEと略する)に溶解し、70℃の温度で5分間処理した後、1M LiClを含むTEで平衡化したオリゴdTセルロースカラムにRNA溶液をアプライし、同緩衝液にて洗浄した。さらに、0.3M LiClを含むTEで洗浄後、0.01%SDSを含む1mM EDTA(pH7.0)で吸着したポリ(A)RNAを溶出した。このようしにて得られたポリ(A)RNAを用いて一本鎖cDNAを合成した。すなわち、滅菌した0.5mLのミクロ遠心チューブに5μgのポリ(A)RNAと0.5μgのオリゴdTプライマー(12−18mer)を入れ、ジエチルピロカルボネート処理滅菌水を加えて12μLにし、70℃の温度で10分間インキュベートした後、氷中に1分間つけた。これに200mM MgCl2を2μL、10mM dNTPを1μLおよび0.1M DTTを2μLそれぞれ加え、42℃で5分間インキュベートした後、200ユニットのGibcoBRL社製Super Script II RTを1μL加え、42℃の温度でさらに50分間インキュベートしてcDNA合成反応を行った。さらに、70℃の温度で15分間インキュベートして反応を停止し、氷上に5分間おいた。この反応液に1μLのE.coli RNaseH(2units/mL)を加え、37℃の温度で20分間インキュベートした。
【0110】
(2)イヌIFN−γ遺伝子の取得
イヌIFN−γのN末端およびC末端の塩基配列をもとに、配列番号8と配列番号9の2種類のプライマーをDNAシンセサイザーにて合成した。上記(1)のイヌリンパ球から得られたcDNAを0.5mLのミクロ遠心チューブに2μL添加し、各プライマーを20pmol,20mMトリス塩酸緩衝液(pH8.0)、1.5mM MgCl2,25mM KCL,100μg/mLゼラチン、50μM各dNTP、4単位 TaqDNAポリメラーゼとなるように各試薬を加え、全量100μLとした。DNAの変性条件を94℃、1分、プライマーのアニーリング条件を55℃、2分、プライマーの伸長条件を72℃、3分の各条件でPerkin−Elmer Cetus社のDNAサーマルサイクラーを用い、35サイクル反応させた。これを1%アガロースゲルにて電気泳動し、約500bpのDNA断片を常法に従って調製した。
【0111】
このようにして得られたDNA断片を、Invitrogen社のT−Vectorに宝酒造(株)のDNA Ligation Kit Ver.1を用いて連結した。これを用いて常法に従い大腸菌を形質転換し、得られた形質転換体からプラスミドDNAを常法により調製した。次に、このプラスミドにPCR断片が挿入されていることを前述と同じ条件のPCRによって確認後、蛍光DNAシーケンサーを用い、その添付プロトコールに従ってパーキンエルマー社のダイターミネーターサイクルシーケンシングキットを用いて、得られたDNA断片がイヌIFN−γをコードする配列であることを確認した。
【0112】
(3)イヌIFN−γ変異体遺伝子の取得
イヌIFN−γ変異体遺伝子は、イヌIFN−γタンパク質のC末端が16残基欠損させたものであり、イヌIFN−γ遺伝子のN末端及びC末端の塩基配列を基に、配列番号10と配列番号11の2種類のプライマーをDNAシンセサイザーにて合成した。上記(1)のイヌリンパ球から得られたcDNAを鋳型として、上記(2)と同様にして約500bpのDNA断片を得、T−Vectorに挿入し、イヌIFN−γをコードする配列であることを確認した(配列番号12)。
【0113】
(参考例3)リン酸結合タンパク遺伝子(phoS)の上流域遺伝子とイヌIFN−γ遺伝子を含むベクターの構築
参考例1および参考例2で調製したpPho2ベクターおよびイヌIFN−γ変異体遺伝子をそれぞれNdeIおよびXhoIによる制限酵素処理を行い、それぞれをアガロースゲル電気泳動を行い、常法に従いに遺伝子を切り出した。その遺伝子を宝酒造(株)のDNA Ligation Kit Ver.1を用いて連結した。これを用いて常法に従い大腸菌JM109を形質転換し、得られた形質転換体よりプラスミドDNAを常法により調製した。さらに次に、このプラスミドDNAを用いて常法に従い大腸菌MC1061(ATCC社製)に形質転換した。
【0114】
(参考例4)多孔性膜の作製(その1)
樹脂としてポリフッ化ビニリデン(PVDF)樹脂を、また溶媒としてN,N−ジメチルアセトアミド(DMAc)をそれぞれ用い、これらを90℃の温度下に十分に攪拌し、次の組成を有する原液を得た。
[原液]
・PVDF:13.0重量%
・DMAc:87.0重量%
次に、上記の原液を25℃の温度に冷却した後、あらかじめガラス板上に貼り付けて置いた、密度が0.48g/cm3で、厚みが220μmのポリエステル繊維製不織布(多孔質基材)に塗布し、直ちに次の組成を有する25℃の温度の凝固浴中に5分間浸漬して、多孔質基材に多孔質樹脂層が形成された多孔性膜を得た。
[凝固浴]
・水 :30.0重量%
・DMAc:70.0重量%
この多孔性膜をガラス板から剥がした後、80℃の温度の熱水に3回浸漬してDMAcを洗い出し、分離膜を得た。多孔質樹脂層表面の9.2μm×10.4μmの範囲内を、倍率10,000倍で走査型電子顕微鏡観察を行ったところ、観察できる細孔すべての直径の平均は0.1μmであった。次に、上記の分離膜について純水透水透過係数を評価したところ、50×10-9m3/m2/s/Paであった。純水透水量の測定は、逆浸透膜による25℃の温度の精製水を用い、ヘッド高さ1mで行った。また、平均細孔径の標準偏差は0.035μmで、膜表面粗さは0.06μmであった。
【0115】
(参考例5)多孔性膜の作製(その2)
重量平均分子量41.7万のフッ化ビニリデンホモポリマーとγ-ブチロラクトンとを、それぞれ38重量%と62重量%の割合で170℃の温度で溶解し原液を作製した。この原液をγ-ブチロラクトンを中空部形成液体として随拌させながら口金から吐出し、温度20℃のγ-ブチロラクトン80重量%水溶液からなる冷却浴中で固化して中空糸膜を作製した。
【0116】
次いで、重量平均分子量28.4万のフッ化ビニリデンホモポリマーを14重量%、セルロースアセテートプロピオネート(イーストマンケミカル社、CAP482−0.5)を1重量%、N-メチル-2-ピロリドンを77重量%、ポリオキシエチレンヤシ油脂肪酸ソルビタン(三洋化成株式会社製、商品名“イオネットT−20C”(登録商標))を5重量%、および水を3重量%の割合で95℃の温度で混合溶解して原液を調整した。この原液を、上記で得られた中空糸膜の表面に均一に塗布し、すぐに水浴中で凝固させた本発明で用いる中空糸膜(多孔性膜)を製作した。得られた中空糸膜(分離膜)の被処理水側表面の平均細孔径は、0.05μmであった。次に、上記の分離膜である中空糸膜について純水透水量を評価したところ、5.5×10-9m3/m2・s・Paであった。透水量の測定は、逆浸透膜による25℃の温度の精製水を用い、ヘッド高さ1mで行った。また、平均細孔径の標準偏差 は0.006μmであった。
【0117】
(比較例1)
微生物を用いた培養形態として最も典型的なバッチ培養を1.5L容のジャーファーメンターを用いて行い、そのタンパク質生産性を評価した。培地は、121℃の温度で20分間高圧蒸気滅菌して用いた。この比較例1では、微生物として大腸菌MC1061−イヌインターフェロンγ株を用い、生産物である組換えイヌインターフェロンγの評価はウエスタンブロッティング法を用いて評価し、グルコース濃度の測定には“グルコーステストワコーC”(登録商標)(和光純薬社製)、リン酸濃度測定には“ホスファC−テストワコー”(登録商標)(和光純薬社製)を用いた。比較例1の運転条件は、下記のとおりである。
・発酵反応槽容量(初発培地量):1.0(L)
・温度調整:37(℃)
・発酵反応槽通気量:1.5(L/min)空気
・発酵反応槽攪拌速度:800(rpm)
・pH調整:2N H2SO4および14N アンモニア水によりpH7.0に調整。
【0118】
まず、大腸菌MC1061−イヌインターフェロンγ株を、試験管で5mlの表1で示した組成の培地にテトラサイクリン(10μg/ml)を添加した培地で一晩振とう培養した(前々培養)。得られた培養液を、新鮮なテトラサイクリン(10μg/ml)を添加した初発培地50mlに植菌し、500ml容坂口フラスコで24時間、37℃の温度で、振幅30cmで、180rpmの条件下で培養を行った(前培養)。前培養液を、ジャーファーメンターの1.0Lの初発培地に植菌した。初発培地を用い、バッチ培養を行った。培養の結果、菌体濃度OD600=7で増殖が止まり、後述する実施例1の結果と比較して1/4以下であった。また、適宜サンプリングしておいた培養液の各フラクションの菌体濃度OD600=10で統一し、その菌体液をサンプルとし、コニカイムノステインHRP(コニカ社製)を用い、添付のプロトコールに従ってイヌインターフェロンγの検出を行った。その結果は、モレキュラーイメージャー(BioRad社製)を用いてシグナル強度を測定した。また、非形質転換大腸菌をネガティブコントロールのサンプルとして使用し、インタードック(東レ株式会社製:イヌインターフェロンγ)をポジティブコントロールのサンプルとして使用した。その結果、イヌインターフェロンγの発現が確認されなかった。
【0119】
(実施例1)タンパク質の製造(その1)
図1の連続発酵装置を稼働させることにより、連続発酵生産系が得られるかどうかを調べるため、表1に示す組成培地と表2に示す組成の追加培地を用い、この装置の連続発酵試験を行った。培地は、121℃の温度で20分間高圧蒸気滅菌し、追加培地は、121℃の温度で60分間高圧蒸気滅菌して用いた。分離膜エレメント部材には、ステンレスおよびポリサルホン樹脂の成形品を用いた。分離膜には、参考例4で作製したポリフッ化ビニリデン(PVDF)を主成分とする多孔性膜を用いた。この実施例1における運転条件は、特に断らない限り下記のとおりである。
・発酵反応槽容量:1.5(L)
・使用分離膜:ポリフッ化ビニリデン濾過膜
・分離膜エレメント有効濾過面積:120平方cm
・温度調整:37(℃)
・発酵反応槽通気量:1.5(L/min)空気
・発酵反応槽攪拌速度:800(rpm)
・pH調整:2N H2SO4および14N アンモニア水によりpH7.0に調整。
・滅菌:分離膜エレメントを含む発酵反応槽、および培地は121℃、20minのオートクレーブにより高圧蒸気滅菌。また、追加培地は、121℃、60minのオートクレーブにより高圧蒸気滅菌。
・膜透過水量制御:発酵反応槽水頭差により流量を制御(水頭差は2m以内で制御した)。
【0120】
微生物として、大腸菌MC1061−イヌインターフェロンγ株を用い、表1に示す組成の培地を用い、追加培地として表2に示す組成の培地を用いた。また、グルコース濃度の測定には“グルコーステストワコーC”(登録商標)(和光純薬社製)、リン酸濃度測定には“ホスファC−テストワコー”(登録商標)(和光純薬社製)を用いた。
【0121】
【表1】
【0122】
【表2】
【0123】
まず、大腸菌MC1061−イヌインターフェロンγ株を、試験管で5mlの表1で示した組成の培地にテトラサイクリン(10μg/ml)を添加した培地で一晩振とう培養した(前々々培養)。得られた培養液を、新鮮なテトラサイクリン(10μg/ml)を添加した表1で示した組成の培地50mlに植菌し、500ml容坂口フラスコで24時間、37℃の温度で、振幅30cmで、180rpmの条件下で培養を行った(前々培養)。
前々培養液を、図1に示した連続発酵装置に1.5Lの表1で示した組成の培地に植菌し、発酵反応槽1を付属の攪拌機5によって800rpmで攪拌し、発酵反応槽1の通気量の調整、温度調整およびpH調整を行い培養を行った。前培養完了後直ちに、表2で示した組成の追加培地の連続供給を行い、連続発酵装置の発酵培養液量を1.5Lとなるように膜透過水量の制御を行いながら連続培養し、リン酸の濃度を低下させていき、連続発酵によるタンパク質の製造を行った。連続発酵試験を行うときの膜透過水量の制御は、水頭差制御装置3により、発酵反応槽水頭を最大2m以内、すなわち膜間差圧が20kPa以内となるように適宜水頭差を変化させることにより行った。適宜、膜透過濾液中のリン酸濃度および残存グルコース濃度を測定した。培養の結果、菌体濃度OD600=30以上まで増殖し、高濃度培養が可能になった。
【0124】
また、適宜発酵培養液をサンプリングしておき、リン酸濃度がどの程度低下した場合にタンパク質が発現しているかウエスタンブロッティング法で確認した。方法としては、各フラクションの菌体濃度OD600=10で統一し、その菌体液をサンプルとし、コニカイムノステインHRP(コニカ社製)を用い、添付のプロトコールに従ってイヌインターフェロンγの検出を行った。すなわち、SDS−PAGE電気泳動し、ブロッティングしたメンブレンをブロッキング溶液(5%スキムミルク・0.1%Tween20・PBS)中で4℃終夜ブロッキングした。メンブレンをTPBS(0.1%Tween20・PBS)で2回洗浄し、TPBSで1000倍希釈した抗イヌインターフェロンγ抗体で室温1時間処理した。メンブレンをTPBSで2回洗浄し、更にTPBSで5分間3回洗浄した。その後TPBSで10000倍希釈した後、HRPラベル抗ラビットIgG抗体(コスモバイオ社製)で室温1時間処理した。メンブレンをTPBSで2回洗浄し、更にTPBSで5分間3回洗浄した後、コニカイムノステインHRP(コニカ社製)の検出試薬(溶液A+溶液B+溶液C+溶液D)を加え、発色させた。その結果は、モレキュラーイメージャー(BioRad社製)を用いてシグナル強度を測定した。また、非形質転換大腸菌をネガティブコントロールのサンプルとして使用し、インタードック(東レ株式会社製:イヌインターフェロンγ)をポジティブコントロールのサンプルとして使用した。
その結果、リン酸濃度1mM以下で、イヌインターフェロンγが大量に発現していることが確認できた。また、上記に示す比較例1と比較して5〜10倍に産生していることを確認した。図1の連続発酵装置を用いることにより、菌体増殖は保ちながら、培地中の成分の濃度を低下させることにより連続発酵によるタンパク質の製造が可能であることを確認することができた。
【0125】
(実施例2)タンパク質の製造(その2)
図2の連続発酵装置を稼働させることにより、連続発酵生産系が得られるかどうかを調べるため、表1に示す組成培地と表2に示す組成の追加培地を用い、この装置の連続発酵試験を行った。培地は、121℃の温度で20分間高圧蒸気滅菌し、追加培地は、121℃の温度で60分間高圧蒸気滅菌して用いた。分離膜エレメント部材には、ステンレスおよびポリサルホン樹脂の成形品を用いた。分離膜には、参考例4で作製したポリフッ化ビニリデン(PVDF)を主成分とする多孔性膜を用いた。この実施例2における運転条件は、特に断らない限り下記のとおりである。
・発酵反応槽容量:1.5(L)
・膜分離槽容量:0.5(L)
・使用分離膜:ポリフッ化ビニリデン濾過膜
・膜分離エレメント有効濾過面積:120平方cm
・温度調整:37(℃)
・発酵反応槽通気量:1.5(L/min)空気
・発酵反応槽攪拌速度:800(rpm)
・膜分離槽通気量:0.3(L/min)
・pH調整:2N H2SO4および14N アンモニア水によりpH7.0に調整
・培養液循環装置による循環液量:0.1(L/min)
・膜透過水量制御:発酵反応槽水頭差により流量を制御(水頭差は、2m以内で制御した)。
【0126】
微生物として大腸菌MC1061-イヌインターフェロンγ株を用い、表1に示す組成の培地を用い、追加培地として表2に示す組成の培地を用いた。また、グルコース濃度の測定には“グルコーステストワコーC”(登録商標)(和光純薬社製)、リン酸濃度測定には“ホスファC−テストワコー”(登録商標)(和光純薬社製)を用いた。
【0127】
まず、大腸菌MC1061−イヌインターフェロンγ株を、試験管で5mlの表1で示した組成の培地にテトラサイクリン(10μg/ml)を添加した培地で一晩振とう培養した(前々々培養)。得られた培養液を、新鮮なテトラサイクリン(10μg/ml)を添加した表1で示した組成の培地50mlに植菌し、500ml容坂口フラスコで24時間、37℃の温度で、振幅30cmで、180rpmの条件下で培養を行った(前々培養)。
前々培養液を、図2に示した連続発酵装置に2Lの表1で示した組成の培地に植菌し、発酵反応槽1を付属の攪拌機5によって800rpmで攪拌し、発酵反応槽1の通気量の調整、温度調整およびpH調整を行い培養を行った。前培養完了後直ちに、表2で示した組成の追加培地の連続供給を行い、連続発酵装置の発酵培養液量を2Lとなるように膜透過水量の制御を行いながら連続培養し、リン酸の濃度を低下させていき、連続発酵によるタンパク質の製造を行った。連続発酵試験を行うときの膜透過水量の制御は、水頭差制御装置3により、発酵反応槽水頭を最大2m以内、すなわち膜間差圧が20kPa以内となるように適宜水頭差を変化させることで行った。適宜、膜透過濾液中のリン酸濃度および残存グルコース濃度を測定した。培養の結果、菌体濃度OD600=30以上まで増殖し、高濃度培養が可能になった。
【0128】
また、適宜発酵培養液をサンプリングしておき、リン酸濃度がどの程度低下した場合にタンパク質が発現しているかウエスタンブロッティング法で確認した。方法としては、各フラクションの菌体濃度OD600=10で統一し、その菌体液をサンプルとし、コニカイムノステインHRP(コニカ社製)を用い、添付のプロトコールに従ってイヌインターフェロンγの検出を行った。すなわち、SDS−PAGE電気泳動し、ブロッティングしたメンブレンをブロッキング溶液(5%スキムミルク・0.1%Tween20・PBS)中で4℃終夜ブロッキングした。メンブレンをTPBS(0.1%Tween20・PBS)で2回洗浄し、TPBSで1000倍希釈した抗イヌインターフェロンγ抗体で室温1時間処理した。メンブレンをTPBSで2回洗浄し、更にTPBSで5分間3回洗浄した。その後TPBSで10000倍希釈した後、HRPラベル抗ラビットIgG抗体(コスモバイオ社製)で室温1時間処理した。メンブレンをTPBSで2回洗浄し、更にTPBSで5分間3回洗浄した後、コニカイムノステインHRP(コニカ社製)の検出試薬(溶液A+溶液B+溶液C+溶液D)を加え、発色させた。その結果は、モレキュラーイメージャー(BioRad社製)を用いてシグナル強度を測定した。また、非形質転換大腸菌をネガティブコントロールのサンプルとして使用し、インタードック(東レ株式会社製:イヌインターフェロンγ)をポジティブコントロールのサンプルとして使用した。
その結果、リン酸濃度1mM以下でイヌインターフェロンγが大量に発現していることが確認できた。また、上記に示す比較例1と比較して5〜10倍に産生していることを確認した。図2に示した連続発酵装置を用いることにより、菌体増殖は保ちながら、培地中の成分の濃度を低下させることで連続発酵によるタンパク質の製造が可能であることを確認することができた。
【0129】
(実施例3)タンパク質の製造(その3)
図1の連続発酵装置を稼働させることにより、連続発酵生産系が得られるかどうかを調べるため、表1に示す組成培地と表2に示す組成の追加培地を用い、この装置の連続発酵試験を行った。培地は、121℃の温度で20分間高圧蒸気滅菌し、追加培地は、121℃の温度で60分間高圧蒸気滅菌して用いた。分離膜エレメント部材には、ステンレスおよびポリサルホン樹脂の成形品を用いた。分離膜には、参考例5で作製したポリフッ化ビニリデン(PVDF)を主成分とする多孔性膜を用いた。この実施例3における運転条件は、特に断らない限り下記のとおりである。
・発酵反応槽容量:1.5(L)
・使用分離膜:ポリフッ化ビニリデン濾過膜
・分離膜エレメント有効濾過面積:120平方cm
・温度調整:37(℃)
・発酵反応槽通気量:1.5(L/min)空気
・発酵反応槽攪拌速度:800(rpm)
・pH調整:2N H2SO4および14N アンモニア水によりpH7.0に調整
・滅菌:分離膜エレメントを含む発酵反応槽、および培地は121℃、20minのオートクレーブにより高圧蒸気滅菌。また、追加培地は121℃、60minのオートクレーブにより高圧蒸気滅菌。
・膜透過水量制御:発酵反応槽水頭差により流量を制御(水頭差は2m以内で制御した)。
【0130】
微生物として大腸菌MC1061−イヌインターフェロンγ株を用い、表1に示す組成の培地を用い、追加培地として表2に示す組成の培地を用いた。また、グルコース濃度の測定には“グルコーステストワコーC”(登録商標)(和光純薬社製)、リン酸濃度測定には“ホスファC−テストワコー”(登録商標)(和光純薬社製)を用いた。
【0131】
まず、大腸菌MC1061−イヌインターフェロンγ株を、試験管で5mlの表1で示した組成の培地にテトラサイクリン(10μg/ml)を添加した培地で一晩振とう培養した(前々々培養)。得られた培養液を、新鮮なテトラサイクリン(10μg/ml)を添加した表1で示した組成の培地50mlに植菌し、500ml容坂口フラスコで24時間、37℃の温度で、振幅30cmで、180rpmの条件下で培養を行った(前々培養)。
前々培養液を、図1に示した連続発酵装置に1.5Lの表1で示した組成の培地に植菌し、発酵反応槽1を付属の攪拌機5によって800rpmで攪拌し、発酵反応槽1の通気量の調整、温度調整およびpH調整を行い培養を行った。前培養完了後直ちに、表2で示した組成の追加培地の連続供給を行い、連続発酵装置の発酵培養液量を1.5Lとなるように膜透過水量の制御を行いながら連続培養し、リン酸の濃度を低下させていき、連続発酵によるタンパク質の製造を行った。連続発酵試験を行うときの膜透過水量の制御は、水頭差制御装置3により、発酵反応槽水頭を最大2m以内、すなわち膜間差圧が20kPa以内となるように適宜水頭差を変化させることで行った。適宜、膜透過濾液中のリン酸濃度および残存グルコース濃度を測定した。培養の結果、菌体濃度OD600=30以上まで増殖し、高濃度培養が可能になった。
【0132】
また、適宜発酵培養液をサンプリングしておき、リン酸濃度がどの程度低下した場合にタンパク質が発現しているかウエスタンブロッティング法で確認した。方法としては、各フラクションの菌体濃度OD600=10で統一し、その菌体液をサンプルとし、コニカイムノステインHRP(コニカ社製)を用い、添付のプロトコールに従ってイヌインターフェロンγの検出を行った。すなわち、SDS−PAGE電気泳動し、ブロッティングしたメンブレンをブロッキング溶液(5%スキムミルク・0.1%Tween20・PBS)中で4℃終夜ブロッキングした。メンブレンをTPBS(0.1%Tween20・PBS)で2回洗浄し、TPBSで1000倍希釈した抗イヌインターフェロンγ抗体で室温1時間処理した。メンブレンをTPBSで2回洗浄し、更にTPBSで5分間3回洗浄した。その後TPBSで10000倍希釈した後、HRPラベル抗ラビットIgG抗体(コスモバイオ社製)で室温1時間処理した。メンブレンをTPBSで2回洗浄し、更にTPBSで5分間3回洗浄した後、コニカイムノステインHRP(コニカ社製)の検出試薬(溶液A+溶液B+溶液C+溶液D)を加え、発色させた。その結果は、モレキュラーイメージャー(BioRad社製)を用いてシグナル強度を測定した。また、非形質転換大腸菌をネガティブコントロールのサンプルとして使用し、インタードック(東レ株式会社製:イヌインターフェロンγ)をポジティブコントロールのサンプルとして使用した。
その結果、リン酸濃度1mM以下でイヌインターフェロンγが大量に発現していることが確認できた。また、上記した比較例1と比較して5〜10倍に産生していることを確認した。図1に示した連続発酵装置を用いることにより、菌体増殖は保ちながら、培地中の成分の濃度を低下させることで連続発酵によるタンパク質の製造が可能であることを確認することができた。
【0133】
(実施例4)タンパク質の製造(その4)
図2の連続発酵装置を稼働させることにより、連続発酵生産系が得られるかどうかを調べるため、表1に示す組成培地と表2に示す組成の追加培地を用い、この装置の連続発酵試験を行った。培地は、121℃の温度で20分間高圧蒸気滅菌し、追加培地は、121℃の温度で60分間高圧蒸気滅菌して用いた。分離膜エレメント部材には、ステンレスおよびポリサルホン樹脂の成形品を用いた。分離膜には、参考例5で作製したポリフッ化ビニリデン(PVDF)を主成分とする多孔性膜を用いた。この実施例4における運転条件は、特に断らない限り下記のとおりである。
・発酵反応槽容量:1.5(L)
・膜分離槽容量:0.5(L)
・使用分離膜:ポリフッ化ビニリデン濾過膜
・膜分離エレメント有効濾過面積:120平方cm
・温度調整:37(℃)
・発酵反応槽通気量:1.5(L/min)空気
・発酵反応槽攪拌速度:800(rpm)
・膜分離槽通気量:0.3(L/min)
・pH調整:2N H2SO4および14N アンモニア水によりpH7.0に調整
・培養液循環装置による循環液量:0.1(L/min)
・膜透過水量制御:発酵反応槽水頭差により流量を制御(水頭差は、2m以内で制御した)。
【0134】
微生物として大腸菌MC1061−イヌインターフェロンγ株を用い、表1に示す組成の培地を用い、追加培地として表2に示す組成の培地を用いた。また、グルコース濃度の測定には“グルコーステストワコーC”(登録商標)(和光純薬社製)、リン酸濃度測定には“ホスファC−テストワコー”(登録商標)(和光純薬社製)を用いた。
【0135】
まず、大腸菌MC1061-イヌインターフェロンγ株を、試験管で5mlの表1で示した組成の培地にテトラサイクリン(10μg/ml)を添加した培地で一晩振とう培養した(前々々培養)。得られた培養液を、新鮮なテトラサイクリン(10μg/ml)を添加した表1で示した組成の培地50mlに植菌し、500ml容坂口フラスコで24時間、37℃の温度で、振幅30cmで、180rpmの条件下で培養を行った(前々培養)。
前々培養液を、図2に示した連続発酵装置に2Lの表1で示した組成の培地に植菌し、発酵反応槽1を付属の攪拌機5によって800rpmで攪拌し、発酵反応槽1の通気量の調整、温度調整およびpH調整を行い培養を行った。前培養完了後直ちに、表2で示した組成の追加培地の連続供給を行い、連続発酵装置の発酵培養液量を2Lとなるように膜透過水量の制御を行いながら連続培養し、リン酸の濃度を低下させていき、連続発酵によるタンパク質の製造を行った。連続発酵試験を行うときの膜透過水量の制御は、水頭差制御装置3により、発酵反応槽水頭を最大2m以内、すなわち膜間差圧が20kPa以内となるように適宜水頭差を変化させることで行った。適宜、膜透過濾液中のリン酸濃度および残存グルコース濃度を測定した。培養の結果、菌体濃度OD600=30以上まで増殖し、高濃度培養が可能になった。
【0136】
また、適宜培養液をサンプリングしておき、リン酸濃度がどの程度低下したらタンパク質が発現しているかウエスタンブロッティング法で確認した。方法としては、各フラクションの菌体濃度OD600=10で統一し、その菌体液をサンプルとし、コニカイムノステインHRP(コニカ社製)を用い、添付のプロトコールに従ってイヌインターフェロンγの検出を行った。すなわち、SDS−PAGE電気泳動し、ブロッティングしたメンブレンをブロッキング溶液(5%スキムミルク・0.1%Tween20・PBS)中で4℃終夜ブロッキングした。メンブレンをTPBS(0.1%Tween20・PBS)で2回洗浄し、TPBSで1000倍希釈した抗イヌインターフェロンγ抗体で室温1時間処理した。メンブレンをTPBSで2回洗浄し、更にTPBSで5分間3回洗浄した。その後TPBSで10000倍希釈した後、HRPラベル抗ラビットIgG抗体(コスモバイオ社製)で室温1時間処理した。メンブレンをTPBSで2回洗浄し、更にTPBSで5分間3回洗浄した後、コニカイムノステインHRP(コニカ社製)の検出試薬(溶液A+溶液B+溶液C+溶液D)を加え、発色させた。その結果は、モレキュラーイメージャー(BioRad社製)を用いてシグナル強度を測定した。また、非形質転換大腸菌をネガティブコントロールのサンプルとして使用し、インタードック(東レ株式会社製:イヌインターフェロンγ)をポジティブコントロールのサンプルとして使用した。
【0137】
その結果、リン酸濃度1mM以下でイヌインターフェロンγが大量に発現していることが確認できた。また、上記に示す比較例1と比較して5〜10倍に産生していることを確認した。本連続発酵装置を用いることにより、菌体増殖は保ちながら、培地中の成分の濃度を低下させることで連続発酵によるタンパク質の製造が可能であることを確認することができた。また、比較例1および実施例1および実施例2の菌体量および発現タンパク質の結果を合わせて表3に、また、比較例1および実施例3および実施例4の菌体量および発現タンパク質の結果を合わせて表4に示した。
【0138】
【表3】
【0139】
【表4】
【0140】
これらの比較結果からわかるように、連続発酵装置を用いることにより、タンパク質の生産が大幅に向上することを明らかにすることができた。本発明によって開示された多孔性膜を組み込んだ連続発酵装置を用い、膜間差圧を制御することにより、発酵培養液を多孔性膜によって濾液と未濾過液に分離し、培地成分の濃度を低下させタンパク質を発現させるとともに、未濾過液を発酵培養液に戻す連続発酵方法を可能とし、微生物量を高く維持しながら、連続発酵によるタンパク質の製造が可能であることが明らかとなった。
【図面の簡単な説明】
【0141】
【図1】図1は、本発明で用いられる膜分離型連続発酵装置の一つの実施の形態を説明するための概要側面図である。
【図2】図2は、本発明で用いられる他の膜分離型連続発酵装置の一つの実施の形態を説明するための概要側面図である。
【図3】図3は、本発明で用いられる分離膜エレメントの一つの実施の形態を説明するための概要斜視図である。
【図4】図4は、本発明で用いられる他の分離膜エレメントの例を説明するための断面説明図である。
【符号の説明】
【0142】
1 発酵反応槽
2 分離膜エレメント
3 水頭差制御装置
4 気体供給装置
5 攪拌機
6 レベルセンサ
7 培地供給ポンプ
8 pH調整溶液供給ポンプ
9 pHセンサ・制御装置
10 温度調節器
11 発酵培養液循環ポンプ
12 膜分離槽
13 支持板
14 流路材
15 分離膜
16 凹部
17 集水パイプ
18 分離膜束
19 上部樹脂封止層
20 下部樹脂封止層
21 支持フレーム
22 集水パイプ
【技術分野】
【0001】
本発明は、タンパク質を生産する能力を有する微生物の発酵培養液を、分離膜として目詰まりが生じにくい多孔性膜を通して濾過し、未濾過液を発酵培養液に保持または還流し、その微生物の培地を前記の発酵培養液に追加し、かつ微生物菌体内からタンパク質を回収する連続発酵によるタンパク質の製造方法に関するものである。
【背景技術】
【0002】
微生物の培養を伴う物質生産方法である発酵法は、大きく(1)回分発酵法(Batch発酵法)および流加発酵法(Fed−Batch発酵法)と、(2)連続発酵法に分類することができる。
【0003】
上記(1)の回分発酵法および流加発酵法は、設備的には簡素であり、短時間で培養が終了し、雑菌汚染による被害が少ないという利点がある。しかしながら、時間経過とともに発酵培養液中の栄養素が少なくなり、浸透圧あるいは生産物阻害等の影響により菌体増殖および生産性が低下してくる。そのため、長時間にわたり安定して菌体の増殖および高生産性を維持するのが困難である。
【0004】
一方、上記(2)の連続発酵法は、発酵反応槽内で目的物質が高濃度に蓄積されることを回避することによって、長時間にわたって高生産性を維持できるという特徴がある。しかしながら、発酵培養液へ培地の連続的な供給を行うと共に微生物を含んだ発酵培養液を抜き出すために、発酵培養液中の微生物が希釈されることから、生産効率の向上は限定されたものとなる。
【0005】
次に、タンパク質とその製造方法に関する技術背景について説明する。工業的に生産されているタンパク質として、酵素を挙げることができる。多くの工業的に重要な酵素、例えば、デンプン分解酵素およびタンパク分解酵素は、バチルス属の微生物、例えば、B.サチリス(B. subtilis) 、B.アミロリクエファシエンス(B.amyloliquefaciens)、B.リヘニホルミス(B. licheniformis)、B.ステアロセルモフィルス(B. stearothermophilus) 、およびB.コアギュランス(B.coagulans) から生産される。また、大量生産の例としては、酵素ニトリルヒドラターゼを生産し、それからアミド化合物が製造されている例がある(特許文献参照1参照。)。アルデヒドの測定用酵素およびホルムアルデヒドなどの有害な環境汚染アルデヒドの分解、除去用酵素として有用であるアルデヒドオキシダーゼの生産も研究されている(特許文献参照2参照。)。コレステロール低下作用、抗血液凝固作用および学習機能向上作用など多彩な生理作用が報告されているドコサヘキサエン酸を生産する酵素の研究も行われている(特許文献参照3参照。)。
【0006】
これらのタンパク質は、元来、所望のタンパク質の生産能力のある微生物、あるいは所望のタンパク質をコードする遺伝子を導入した組換え微生物を培養する発酵法により生産されている。その発酵法としては、回分式発酵法が用いられている。具体的に、効率的な生産性が必要であるα―アミラーゼの生産(非特許文献1参照。)、β−アミラーゼの生産(非特許文献2、3参照。)、ジヒドロ葉酸還元酵素の生産(非特許文献4参照。)、およびインターフェロンおよびインシュリンの生産(非特許文献5参照。)は、いずれも回分式発酵法が用いられている。しかしながら、回分式発酵法では生産効率の向上は困難である。
【0007】
このように、タンパク質の生産において、効率的なタンパク質の製造を目的とした生産性能の向上に向けた様々な検討が行われているが、更なるタンパク質生産技術革新が望まれていた。
【特許文献1】特開2007−061035号公報
【特許文献2】特開2001−299351号公報
【特許文献3】特開2001−169780号公報
【非特許文献1】Palva, Gene (1982)19:81〜87;シノミヤ他
【非特許文献2】Agric. Biol.Chem.(1981)45;1733〜1735;Gray及びChang
【非特許文献3】J. Bacteril(1981)145:422〜428;Williams他
【非特許文献4】Gene(1981)16:199〜206
【非特許文献5】Palva, Gene (1983)22:229〜235
【発明の開示】
【発明が解決しようとする課題】
【0008】
本発明の目的は、簡便な操作方法で、長時間にわたり安定して高生産性を維持する連続発酵法によるタンパク質の製造方法を提供すること、すなわち、連続発酵法において、微生物の発酵培養液を分離膜で濾過し、未濾過液を発酵培養液に保持または還流し、発酵培養液中の微生物濃度を向上させ、かつ、高く維持させることにより高い物質生産性を維持する連続発酵によるタンパク質の製造方法を提供することにある。
【課題を解決するための手段】
【0009】
本発明者らは、鋭意研究の結果、微生物の分離膜内への侵入が少なく、微生物を膜間差圧が低い条件で発酵培養液を濾過した場合に、膜の目詰まりが著しく抑制されることを見出し、分離膜を用いた連続発酵を行うことにより、課題であった微生物が高濃度まで増殖することを可能にし、タンパク質の生産量が飛躍的に向上することを発見し、本発明を完成した。
【0010】
本発明のタンパク質の製造方法は、タンパク質の生産能力を有する微生物の発酵培養液を分離膜で濾過し、未濾過液を前記の発酵培養液に保持または還流し、その微生物の培地を前記の発酵培養液に追加し、かつ、微生物菌体内からタンパク質を回収する連続発酵によるタンパク質の製造方法であって、前記の分離膜として平均細孔径が0.01μm以上1μm未満の細孔を有する多孔性膜を用い、その膜間差圧を0.1から20kPaの範囲にして濾過処理することを特徴とする連続発酵によるタンパク質の製造方法である。
【0011】
本発明のタンパク質の製造方法の好ましい態様によれば、前記の多孔性膜の純水透過係数は2×10−9m3/m2/s/pa以上6×10−7m3/m2/s/pa以下である。
【0012】
本発明のタンパク質の製造方法の好ましい態様によれば、前記の多孔性膜の平均細孔径は0.01μm以上0.2μm未満であり、かつ、該平均細孔径の標準偏差は0.1μm以下である。
【0013】
本発明のタンパク質の製造方法の好ましい態様によれば、前記の多孔性膜の膜表面粗さは0.1μm以下である。
【0014】
本発明のタンパク質の製造方法の好ましい態様によれば、前記の多孔性膜は多孔性樹脂層を含む多孔性膜である。
【0015】
本発明のタンパク質の製造方法の好ましい態様によれば、前記の多孔性膜の素材がポリフッ化ビニリデン系樹脂である。
【0016】
本発明のタンパク質の製造方法の好ましい態様によれば、前記の微生物の発酵原料は糖類を含むものである。
【0017】
本発明のタンパク質の製造方法の好ましい態様によれば、前記のタンパク質をコードする遺伝子はリン酸プロモーターの下流に連結され、かつ培地に含まれる無機リン酸濃度は1mM以下である。
【0018】
本発明のタンパク質の製造方法の好ましい態様によれば、前記のタンパク質はイヌインターフェロンγである。
【0019】
本発明のタンパク質の製造方法の好ましい態様によれば、前記の微生物は大腸菌である。
【発明の効果】
【0020】
本発明によれば、簡便な操作条件で、目詰まりが生じにくい多孔性膜を使用することにより、微生物の増殖を抑制することなく、発酵培養液中の微生物濃度を向上させ、かつ、高く維持させることにより高い物質生産性を維持するタンパク質の製造が可能になる。
【発明を実施するための最良の形態】
【0021】
本発明は、タンパク質の生産能力を有する微生物の発酵培養液を分離膜で濾過し、未濾過液を前記の発酵培養液に保持または還流し、その微生物の培地を前記の発酵培養液に追加し、かつ、微生物菌体内からタンパク質を回収する連続発酵によるタンパク質の製造方法であって、前記の分離膜として平均細孔径が0.01μm以上1μm未満の細孔を有する多孔性膜を用い、その膜間差圧を0.1から20kPaの範囲にして濾過処理することを特徴とする連続発酵によるタンパク質の製造方法である。
【0022】
本発明で用いられる多孔性膜は、発酵に使用する微生物による目詰まりが起こりにくく、かつ濾過性能が長期間安定に継続するものであることが望ましい。そのため、本発明で使用される多孔性膜は、平均細孔径が、0.01μm以上1μm未満であることが重要である。後述するように、多孔性膜としては、多孔質基材の少なくとも1表面に多孔質樹脂層を有する構造のものが好ましく用いられる。
【0023】
多孔質樹脂層が多孔性膜の両面に存在する場合、少なくとも一方の多孔質樹脂層が、上記の平均細孔径の条件を満たしていればよい。平均細孔径がこの範囲内にあると、菌体や汚泥などがリークすることのない高い排除率と、高い透水性を両立させることができ、さらに目詰まりしにくく、透水性を長時間保持することが、より高い精度と再現性を持って実施することができる。平均細孔径が、この範囲内にあれば、大腸菌や酵母などを用いた場合、目詰まりが少なく、また、細胞の濾液への漏れもなく安定に連続発酵が実施可能である。また、大腸菌や酵母より小さな細菌類を用いた場合は、0.4μm以下の平均細孔径であればよりよく、0.2μm以下の平均細孔径であればなお好適に実施可能である。平均細孔径は、小さすぎると透水量が低下することがあるので、通常は0.02μm以上であることが好ましく、より好ましくは0.04μm以上である。
【0024】
ここで、平均細孔径は、倍率10,000倍の走査型電子顕微鏡観察における、9.2μm×10.4μmの範囲内で観察できる細孔すべての直径を測定し、平均することにより求めることができる。細孔径の標準偏差σは、0.1μm以下であることが好ましい。細孔径の標準偏差σは、上述の9.2μm×10.4μmの範囲内で観察できる細孔数をNとして、測定した各々の直径をXk、細孔直径の平均をX(ave)とした以下式(1)より算出した。
【0025】
【数1】
【0026】
本発明で用いられる多孔性膜において、発酵培養液の透過性が重要点の一つであり、透過性の指標として、使用前の多孔性膜の純水透過係数を用いることができる。多孔性膜の純水透過係数が、逆浸透膜による25℃の温度の精製水を用い、ヘッド高さ1mで透水量を測定し算出したとき、2×10−9m3/m2/s/pa以上であることが好ましく、2×10−9以上6×10−7m3/m2/s/pa以下であれば、実用的に十分な透過水量が得られる。
【0027】
本発明で用いられる多孔性膜の表面粗さは、分離膜の目詰まりに影響を与える因子であり、好ましくは、その表面粗さが0.1μm以下のときに分離膜の剥離係数や膜抵抗を低下させることができ、より低い膜間差圧で連続培養が実施可能である。また、表面粗さが低いことで、微生物の濾過において、膜表面で発生する剪断力を低下させることが期待でき、微生物の破壊が抑制され、多孔性膜の目詰まりも抑制されることにより、長期間安定な濾過が可能になると考えられる。
【0028】
ここで、表面粗さは、下記の原子間力顕微鏡装置(AFM)を使用して、下記の条件で測定することができる。
・装置 原子間力顕微鏡装置(Digital Instruments(株)製Nanoscope IIIa)
・条件 探針 SiNカンチレバー(Digital Instruments(株)製)
走査モード コンタクトモード(気中測定)
水中タッピングモード(水中測定)
走査範囲 10μm、25μm 四方(気中測定)
5μm、10μm 四方(水中測定)
走査解像度 512×512
・試料調製 測定に際し膜サンプルは常温でエタノールに15分浸漬後RO水中に24時間浸漬し洗浄した後風乾し用いた。RO水とは、ろ過膜の一種である逆浸透膜(RO膜)を用いてろ過し、イオンや塩類などの不純物を排除した水を指す。RO膜の孔の大きさは、概ね2nm以下である。
【0029】
膜の表面粗さdroughは、AFMにより各ポイントのZ軸方向の高さから下記の(数2)から算出した。
【0030】
【数2】
【0031】
本発明における多孔性膜は、被処理水の水質や用途に応じた分離性能と透水性能を有するものであり、阻止性能、透水性能や耐汚れ性という分離性能の点からは、多孔質樹脂層を含む多孔性膜であることが好ましい。すなわち、好ましい多孔性膜は、多孔質基材の表面に、分離機能層として作用とする多孔質樹脂層を有している。多孔質基材は、多孔質樹脂層を支持して分離膜に強度を与える。
【0032】
本発明で用いられる多孔性膜が、多孔質基材の表面に多孔質樹脂層を有している場合、多孔質基材に多孔質樹脂層が浸透していても、多孔質基材に多孔質樹脂層が浸透していなくてもどちらでも良く、用途に応じて選択される。
【0033】
多孔質基材の材質としては、有機材料および無機材料等で特に限定されないが、有機繊維が望ましい。好ましい多孔質基材は、セルロース繊維、セルローストリアセテート繊維、ポリエステル繊維、ポリプロピレン繊維およびポリエチレン繊維などの有機繊維を用いてなる織布や不織布であり、なかでも、密度の制御が比較的容易であり、製造も容易で安価な不織布が好ましく用いられる。多孔質基材の平均厚みは、好ましくは50μm以上3000μm以下である。
【0034】
多孔質樹脂層は、上述したように分離機能層として作用するもので、有機高分子膜を好適に使用することができる。有機高分子膜は、基本的に有機ポリマー材料から構成される分離膜のことであり、例えば、有機繊維の不織布やマクロポア構造多孔質有機基材と当該多孔質有機基材の孔径より小さな孔径を有する多孔質樹脂層が複合化された構造を持つ場合が多い。
【0035】
ここで、多孔質樹脂層の材質としては、ポリエチレン系樹脂、ポリプロピレン系樹脂、ポリ塩化ビニル系樹脂、ポリフッ化ビニリデン系樹脂、ポリスルホン系樹脂、ポリエーテルスルホン系樹脂、ポリアクリロニトリル系樹脂、セルロース系樹脂およびセルローストリアセテート系樹脂などが挙げられ、これらの樹脂を主成分とする樹脂の混合物であってもよい。なかでも、溶液による製膜が容易で、物理的耐久性や耐薬品性にも優れているポリ塩化ビニル系樹脂、ポリフッ化ビニリデン系樹脂、ポリスルホン系樹脂、ポリエーテルスルホン系樹脂およびポリアクリロニトリル系樹脂が好ましく用いられる。特に、ポリフッ化ビニリデン系樹脂またはそれを主成分とするものが最も好ましく用いられる。
【0036】
ここで、ポリフッ化ビニリデン系樹脂としては、フッ化ビニリデンの単独重合体が好ましく用いられるが、フッ化ビニリデンの単独重合体の他、フッ化ビニリデンと共重合可能なビニル系単量体との共重合体も好ましく用いられる。このようなビニル系単量体としては、テトラフルオロエチレン、ヘキサフルオロプロピレンおよび三塩化フッ化エチレンなどが例示される。
【0037】
本発明で用いられる多孔性膜は、平膜であっても中空糸膜であっても良い。
【0038】
多孔性膜が平膜の場合、その厚みは用途に応じて選択されるが、好ましくは20μm以上5000μm以下の範囲であり、より好ましくは50μm以上2000μm以下の範囲で選択される。上述のように、多孔性膜は、多孔質基材と多孔質樹脂層とから形成されていても良い。多孔質基材の厚みは、好ましくは50μm以上3000μm以下の範囲で選択される。
【0039】
多孔性膜が中空糸膜の場合、内径は好ましくは200μm以上5000μm以下の範囲で選択され、膜厚は好ましくは20μm以上2000μm以下の範囲で選択される。また、有機繊維または無機繊維を筒状にした織物や編み物を含んでいても良い。
【0040】
まず、多孔性膜のうち、平膜の作成法の概要について説明する。
【0041】
多孔質基材の表面に、樹脂と溶媒とを含む原液の被膜を形成すると共に、その原液を多孔質基材に含浸させる。その後、被膜を有する多孔質基材の被膜側表面のみを、非溶媒を含む凝固浴と接触させて樹脂を凝固させると共に、多孔質基材の表面に多孔質樹脂層を形成する。
【0042】
原液は、樹脂を溶媒に溶解させて調整する。原液の温度は、製膜性の観点から、通常、5〜120℃の範囲内で選定することが好ましい。溶媒は、樹脂を溶解するものであり、樹脂に作用してそれらが多孔質樹脂層を形成するのを促すものである。溶媒としては、N−メチルピロリジノン(NMP)、N,N−ジメチルアセトアミド(DMAc)、N,N−ジメチルホルムアミド(DMF)、ジメチルスルホキシド(DMSO)、N− メチル− 2 − ピロリドン、メチルエチルケトン、テトラヒドロフラン、テトラメチル尿素、リン酸トリメチル、シクロヘキサノン、イソホロン、γ− ブチロラクトン、メチルイソアミルケトン、フタル酸ジメチル、プロピレングリコールメチルエーテール、プロピレンカーボネート、ジアセトンアルコール、グリセロールトリアセテート、アセトンおよびメチルエチルケトンなどを用いることができる。なかでも、樹脂の溶解性の高いN−メチルピロリジノン(NMP)、N,N−ジメチルアセトアミド(DMAc)、N,N−ジメチルホルムアミド(DMF)およびジメチルスルホキシド(DMSO)が好ましく用いられる。これらの溶媒は、単独で用いても良いし2種類以上を混合して用いても良い。原液は、先述の樹脂を好ましくは5重量%以上60重量%以下の濃度で、上述の溶媒に溶解させることにより調製することができる。
【0043】
また、例えば、ポリエチレングリコール、ポリビニルアルコール、ポリビニルピロリドンおよびグリセリンなどの溶媒以外の成分を溶媒に添加しても良い。溶媒に非溶媒を添加することもできる。非溶媒は、樹脂を溶解しない液体である。非溶媒は、樹脂の凝固の速度を制御して細孔の大きさを制御するように作用する。非溶媒としては、水や、メタノールおよびエタノールなどのアルコール類を用いることができる。なかでも、非溶媒として、価格の点から水やメタノールが好ましく用いられる。溶媒以外の成分および非溶媒は、混合物であってもよい。
【0044】
原液には、開孔剤を添加することもできる。開孔剤は、凝固浴に浸漬された際に抽出されて、樹脂層を多孔質にする作用を持つものである。開孔剤を添加することにより、平均細孔径の大きさを制御することができる。開孔剤は、凝固浴への溶解性の高いものであることが好ましい。開孔剤としては、例えば、塩化カルシウムや炭酸カルシウムなどの無機塩を用いることができる。また、開孔剤として、ポリエチレングリコールやポリプロピレングリコールなどのポリオキシアルキレン類や、ポリビニルアルコール、ポリビニルブチラールおよびポリアクリル酸などの水溶性高分子化合物や、グリセリンを用いることができる。
【0045】
次に、多孔性膜のうち、中空糸膜の作成法の概要について説明する。
【0046】
中空糸膜は、樹脂と溶媒からなる原液を二重管式口金の外側の管から吐出すると共に、中空部形成用流体を二重管式口金の内側の管から吐出して、冷却浴中で冷却固化して作製することができる。
【0047】
原液は、上述の平膜の作成法で述べた樹脂を好ましくは20重量%以上60重量%以下の濃度で、上述の平膜の生成法で述べた溶媒に溶解させることにより調整することができる。また、中空部形成用流体には、通常気体もしくは液体を用いることができる。また、得られた中空糸膜の外表面に、新たな多孔性樹脂層をコーティング(積層)することもできる。積層は中空糸膜の性質、例えば、親水性・疎水性あるいは細孔径等を所望の性質に変化させるために行うことができる。積層される新たな多孔性樹脂層は、樹脂を溶媒に溶解させた原液を、非溶媒を含む凝固浴と接触させて樹脂を凝固させることによって作製することができる。その樹脂の材質は、例えば、上述有機高分子膜の材質と同様のものが好ましく用いられる。また、積層方法としては、原液に中空糸膜を浸漬してもよいし、中空糸膜の表面に原液を塗布してもよく、積層後、付着した原液の一部を掻き取ったり、エアナイフを用いて吹き飛ばしすることにより積層量を調整することもできる。
【0048】
本発明で用いられる多孔性膜は、支持体と組み合わせることによって分離膜エレメントとして使用することができる。支持体として支持板を用い、その支持板の少なくとも片面に本発明で用いられる多孔性膜を配した多孔性膜を有する分離膜エレメントは、本発明で用いられる膜エレメントの好適な形態の一つである。この形態では、膜面積を大きくすることが困難なので、透水量を大きくするために、支持板の両面に多孔性膜を配することも好ましい態様である。
【0049】
本発明において、微生物を濾過処理する際の膜間差圧は、微生物および培地成分が容易に目詰まりしない条件であればよいが、膜間差圧を0.1kPa以上20kPa以下の範囲として濾過処理することが重要である。また、使用前の純水透過係数が、逆浸透膜による25℃の温度の精製水を用い、ヘッド高さ1mで透水量を測定し算出したとき、2×10−9m3/m2/s/pa以上の範囲であることが好ましく、さらに2×10−9m3/m2/s/pa以上6×10−7m3/m2/s/pa以下の範囲にあることが好ましい。濾過の駆動力としては、培養液と多孔性膜処理水の液位差(水頭差)を利用したサイホンにより多孔性膜に膜間差圧を発生させることが可能であり、また、濾過の駆動力として多孔性膜処理水側に吸引ポンプを設置してもよいし、多孔性膜の発酵培養液側に加圧ポンプを設置することも可能である。膜間差圧は、発酵培養液と多孔性膜処理水の液位差を変化させることで制御することができる、またポンプを使用する場合には吸引圧力により制御することができ、更に発酵培養液側の圧力を導入する気体または液体の圧力によって制御することができる。これら圧力制御を行う場合には、発酵培養液側の圧力と多孔性膜処理水側の圧力差をもって膜間差圧とし、膜間差圧の制御に用いることができる。
【0050】
本発明で使用される発酵原料としては、発酵培養する微生物の生育を促し、目的とする発酵生産物であるタンパク質を良好に生産させ得るものであれば良い。発酵原料としては、例えば、炭素源、窒素源、無機塩類、および必要に応じてアミノ酸、およびビタミンなどの有機微量栄養素を適宜含有する液体培地等が好ましく用いられる。
【0051】
上記の炭素源としては、グルコース、シュークロース、フラクトース、ガラクトースやラクトース等の糖類、これら糖類を含有する澱粉糖化液、甘藷糖蜜、甜菜糖蜜、ハイテストモラセス、更には酢酸等の有機酸、エタノールなどのアルコール類およびリセリンなども使用される。ここで糖分とは、多価アルコールの最初の酸化生成物であり、アルデヒド基またはケトン基をひとつ持ち、アルデヒド基を持つ糖をアルドース、ケトン基を持つ糖をケトースと分類される炭水化物のことを指す。
【0052】
また上記の窒素源としては、アンモニアガス、アンモニア水、アンモニウム塩類、尿素、硝酸塩類、その他補助的に使用される有機窒素源、例えば油粕類、大豆加水分解液、カゼイン分解物、その他のアミノ酸、ビタミン類、コーンスティープリカー、酵母または酵母エキス、肉エキス、ペプトン等のペプチド類および各種発酵菌体およびその加水分解物などが使用される。
【0053】
また上記の無機塩類としては、リン酸塩、マグネシウム塩、カルシウム塩、鉄塩およびマンガン塩等が使用される。
【0054】
また、アミノ基とカルボキシル基の両方の官能基を持つ、また生体の蛋白質の構成ユニットとなる有機化合物であるアミノ酸としては、中性アミノ酸 、酸性アミノ酸および塩基性アミノ酸が使用される。中性アミノ酸には、グリシン、アラニン、分枝アミノ酸(バリン、ロイシン、イソロイシン)、ヒドロキシアミノ酸(セリン、スレオニン)、含硫アミノ酸 (システイン、メチオニン)、酸アミドアミノ酸(アスパラギン、グルタミン)などの脂肪族アミノ酸 、プロリンなどのイミノ酸 、フェニルアラニン、チロシンおよびトリプトファンなどの芳香族アミノ酸が挙げられる。また、酸性アミノ酸としては、アスパラギン酸、グルタミン酸がある。塩基性アミノ酸には、リシン、アルギニンおよびヒスチジンが使用される。
【0055】
本発明で使用される微生物が生育のために特定の栄養素を必要とする場合には、その栄養物を標品もしくはそれを含有する天然物として添加することができる。また、消泡剤を必要に応じて使用することができる。
【0056】
本発明において、培養液とは、発酵原料に微生物が増殖した結果得られる液のことを言う。追加する発酵原料の組成は、目的とするタンパク質の生産性が高くなるように、培養開始時の発酵原料組成から適宜変更しても良い。
【0057】
本発明で使用されるタンパク質の生産能力のある微生物は、製造するタンパク質をコードする遺伝子を元来有する、あるいは導入されている微生物のことである。本発明で好適に用いることができる微生物としては、例えば、発酵工業においてよく使用されるパン酵母などの酵母、カビなどの真核微生物、大腸菌、枯草菌およびコリネ型細菌などの原核微生物などを挙げることができる。使用される微生物は、自然環境から単離されたものでもよく、また、突然変異や遺伝子組換えによって一部性質が改変されたものであってもよい。本発明においては、特に大腸菌が好ましく用いられる。
【0058】
本発明のタンパク質の製造法で製造されるタンパク質をコードする遺伝子は、タンパク質を発現可能なプロモーターの支配下に連結して用いることが好ましい。プロモーターの種類としては、構成的プロモーターと誘導的プロモーターがあり、本発明ではどちらも使用できる。なかでも、発現能力の高いプロモーターを用いることが望ましく、構成的なプロモーターとしては、Bacillus sp. SK−1株由来のD−アミノ酸アミノトランスフェラーゼのオープンリーディングフレーム(ORF)の上流に局在するプロモーター、カリフラワーモザイクウイルス(CaMV)35S RNA遺伝子のプロモーター、アクチン遺伝子のプロモーター、枯草菌由来のルビスコプロモーターおよびADH1プロモーターなどが挙げられる。
【0059】
また、誘導的プロモーターとしては、T7プロモーター、Tacプロモーター、PLプロモーター、PRプロモーター、PSEプロモーター、トリプトファンプロモーターおよびリン酸プロモーターを例示することができる。また、1つのプロモーターだけではなく、2個または3個のプロモーターを連結したダブル、トリプルプロモーターとしても使用できるし、種の違うプロモーターを連結させたハイブリッドプロモーターとしても使用することができる。
【0060】
ここで、Bacillus sp. SK−1株由来のD−アミノ酸アミノトランスフェラーゼのオープンリーディングフレーム(ORF)の上流に局在するプロモーター、カリフラワーモザイクウイルス(CaMV)35S RNA遺伝子のプロモーター、アクチン遺伝子のプロモーター、枯草菌由来のルビスコプロモーター、およびADH1プロモーターは、構成的に発現が誘導されるプロモーターである。また、T7プロモーター、Tacプロモーター、lacプロモーター、PLプロモーター、PRプロモーターおよびPSEプロモーターは、イソプロピルチオ−β−D−ガラクトシド(IPTG)で発現が誘導されるプロモーターである。トリプトファンプロモーターは、培養液中のトリプトファン濃度が低下することで発現が誘導されるプロモーターであり、またインドール酢酸(IAA)を添加しても発現が誘導される。リン酸プロモーターは、発酵培養液中の無機リン酸の濃度が低下することにより発現が誘導されるプロモーターである。
【0061】
本発明のタンパク質の製造法は、タンパク質の生産能力を有する微生物の発酵培養液を分離膜で濾過し、未濾過液を前記の発酵培養液に保持または還流し、その微生物の培地を前記の発酵培養液に追加し、かつ、微生物菌体内からタンパク質を回収する。本発明では、発酵培養液に追加する培地はタンパク質の生産に適した組成に変更することが可能であることから、高生産性を維持したタンパク質の製造が実現する。すなわち、本発明のタンパク質の製造法は、タンパク質を発現せしめるプロモーターとして、前述Bacillus sp. SK−1株由来のD−アミノ酸アミノトランスフェラーゼのオープンリーディングフレーム(ORF)の上流に局在する構成的なプロモーター、カリフラワーモザイクウイルス(CaMV)35S RNA遺伝子のプロモーター、アクチン遺伝子のプロモーター、枯草菌由来のルビスコプロモーター、およびADH1プロモーターのように構成的に発現可能なプロモーターを用いることができる。また、前述トリプトファンプロモーターやリン酸プロモーターのような、培地中あるいは培養液中の化学品濃度に依存して発現が制御可能なプロモーターを用いることもできる。
【0062】
本発明のタンパク質の製造法では、構成的なプロモーターを用いた場合、微生物の培地は特に制限されることはなく、微生物が生育することにより、発酵培養液中の微生物が高濃度まで生育されることから、生産性の高いタンパク質の製造を行うことができる。T7プロモーター、Tacプロモーター、PLプロモーター、PRプロモーター、PSEプロモーターおよびlacプロモーターの場合、微生物の培地にIPTGを含むことにより、発酵培養液中にIPTGを添加させることができることから、生産性の高いタンパク質の製造を行うことができる。
【0063】
トリプトファンプロモーターの場合、微生物の培地に含まれるトリプトファン濃度を低くすることにより、発酵培養液中のトリプトファン濃度を低下させることができることから生産性の高いタンパク質の製造を行うことができるし、発酵培養液中にIAAを添加することで生産性の高いタンパク質の製造を行うことができる。リン酸プロモーター場合、微生物の培地に含まれる無機リン酸濃度を低くすることにより、発酵培養液中のリン酸濃度を低下させることができることから、生産性の高いタンパク質の製造を行うことができる。
【0064】
発明のタンパク質の製造方法において、前記のリン酸プロモーターを好ましく用いることができる。ここで、リン酸プロモーターに関して説明する。リン酸結合タンパク質(PhoS)遺伝子の上流域をコードするDNAは、得られたゲノムDNAを元に、リン酸結合タンパク遺伝子の上流域の5’および3’末端配列を付加したプライマーを設計してPCRを行うことによって得ることができる。ゲノムDNAは、例えば、次のようにして製造することができる。第一に、大腸菌K−12株由来、中でもKLF48/KL159株からゲノムDNAを抽出する。上記大腸菌よりゲノムDNAを抽出する方法としては、アルカリ法による抽出(ヌクレイック アシッド リサーチ 第7巻、第6号、1513−1523)や、非フェノール性試薬とタンパク質凝集剤を使用し、相配分に基づいて比重の違いにより中間に凝集層を形成させる凝集分配法(新生化学実験講座2 核酸1 分離精製:13−51(1991))などの中から適当な方法を選んで行うことができる。
【0065】
リン酸結合タンパク質は、リン酸の取り込みに関係しており、リン酸が少なくなれば、それを取り込もうとしてリン酸結合タンパク質が合成され、培地中のリン酸に結合して細胞質内に送り込む機能を有している。これらのタンパク質の転写制御は、これらのタンパク質をコードしている遺伝子のさらに上流域に存在し、pho boxと呼ばれているコンセンサス領域ctgtcataaatctgtcacを含むプロモーターである。この領域は、リン酸結合タンパク質をコードしている遺伝子の上流であり約200−1000bpのDNA配列である。このようにして得られたリン酸プロモーターの下流に、所望のタンパク質をコードする遺伝子を連結した遺伝子発現カセットを導入した微生物を用いることができる。
【0066】
微生物の発酵培養液は、リン酸プロモーターが活性化する濃度まで低下させることが好ましい。リン酸プロモーターは、無機リン酸濃度が1mM未満でタンパク質が発現する。そこで、初めの発酵培養液の無機リン酸濃度は、1mM以上であればよい。初めの発酵培養液の無機リン酸濃度は1mM未満でも良いが、菌体の生育が遅くなる傾向を示す。また、無機リン酸濃度が高濃度であった場合、希釈に時間がかかり、効率的にタンパク質を生産できない可能性がある。好ましくは、2〜5mMの濃度で添加された無機リン酸を含む培養液を使用することが好ましい。また、追加する培地は、無機リン酸を1mM以下の濃度で含んだ培地を利用することがよい。効率的に発酵培養液のリン酸が希釈されるので、好ましくは0〜0.5mMの無機リン酸を含んだ培地を利用することが好ましい。
【0067】
タンパク質を生産する能力を有する微生物の培養は、通常、pH3〜9、温度15〜70℃の範囲で行われることが多い。発酵培養液のpHは、無機あるいは有機の酸、アルカリ性物質、さらには尿素、炭酸カルシウム、アンモニアガスなどによって上記範囲内のあらかじめ定められた値に調節することができる。
【0068】
微生物の培養において、酸素の供給速度を上げる必要があれば、空気に酸素を加えて酸素濃度を例えば21%以上に保つ、発酵培養液を加圧する、攪拌速度を上げる、あるいは通気量を上げるなどの手段を用いることができる。逆に、酸素の供給速度を下げる必要があれば、炭酸ガス、窒素およびアルゴンなど酸素を含まないガスを空気に混合して供給することも可能である。
【0069】
本発明の培養初期にBatch培養またはFed−Batch培養を行って微生物濃度を高くした後に連続発酵(引き抜き)を開始しても良いし、高濃度の菌体をシードし、培養開始とともに連続発酵を行っても良い。適当な時期から培地の供給および培養物の引き抜きを行うことが可能である。培地供給と培養物の引き抜きの開始時期は必ずしも同じである必要はない。また、培地の供給と培養物の引き抜きは連続的であってもよいし、間欠的であってもよい。培地には上記に示したような菌体増殖に必要な栄養素を添加し、菌体増殖が連続的に行われるようにすればよい。
【0070】
発酵培養液中の微生物の濃度は、効率よい生産性を得る上で、発酵培養液の環境が微生物の増殖にとって不適切となって死滅する比率が高くならない範囲で、高い状態で維持することが好ましい。また、連続発酵装置の運転上の不都合や生産効率の低下を招かなければ、微生物の濃度の上限値は特に限定されない。
【0071】
タンパク質生産能力のあるフレッシュな菌体を増殖させつつ行う連続発酵操作は、培養管理上、通常は単一の発酵反応槽で行うことが好ましい。しかしながら、菌体を増殖しつつ生産物を生成する連続発酵法であれば、発酵反応槽の数は問わない。発酵反応槽の容量が小さい等の理由から、複数の発酵反応槽を用いることもあり得る。この場合、複数の発酵反応槽を配管で並列または直列に接続して連続発酵を行っても高生産性は得られる。
【0072】
本発明においては、微生物を発酵反応槽に維持したままで、発酵反応槽からの発酵培養液の連続的かつ効率的な抜き出しが可能となることから、微生物を連続的に培養し、十分な増殖を確保した後に培地組成を変更し、目的とするタンパク質を効率よく製造することも可能である。
【0073】
本発明のタンパク質の製造法で好適に製造されるタンパク質としては、産業的価値から考えてヒトや哺乳類が産生するタンパク質、例えば、成長ホルモン、サイトカイン、増殖因子および細胞骨格タンパク質などのタンパク質が挙げられる。また、微生物、植物または昆虫などが産生する酵素や種々タンパク質、ペプチドも本発明で好適に製造されるタンパク質に含まれる。また、これらタンパク質のアミノ酸配列を改変した変異タンパク質も本発明で製造されるタンパク質に含まれる。更に、2種類以上のタンパク質、あるいはペプチドを連結したキメラタンパク質およびキメラペプチドも本発明のタンパク質の製造法で製造可能である。
【0074】
本発明においての、タンパク質の回収について概要を説明する。
【0075】
まず、タンパク質を菌体から回収するとは、発酵培養液を抜き出し、そこに含まれる菌体を回収して後述した方法によりタンパク質を抽出・精製することである。
【0076】
発酵培養液を抜き出すのは、菌体が発酵培養液中に含まれていればどのタイミングで抜き出しても良い。例えば、植菌後すぐの発酵培養液でも構わない。しかしながら、効率よい生産性を得る上で、発酵培養液の環境が微生物の増殖にとって不適切となって死滅する比率が高くならない範囲で、高い状態で維持しているときに回収する好ましい。その時期に発酵培養液を抜き出せば、効率的に高生産でタンパク質が得られる。また、適宜発酵培養液を抜きだして、タンパク質が高発現あるいは高活性な時期に回収するのも良い。
【0077】
本発明においては、培養された微生物菌体内からタンパク質を回収する。具体的には、発酵反応槽から発酵培養液を抜き出し、培養された微生物菌体内からタンパク質を回収する。
【0078】
タンパク質を回収する方法としては、例えば、ホモジナイザー、超音波、ガラスビーズ、ブレンダーおよびブレスなど機械的な方法で菌体を破砕し精製する方法を採用することができる。精製は、カラムクロマトグラフィー、液体クロマトグラフィーおよびイオン交換クロマトグラフィーなどにより精製が可能である。
【0079】
また、タンパク質を回収する方法としては、界面活性剤や変性剤などの化学的な方法を用いることも可能である。この場合、タンパク質が変性されることがあるので、可溶化後、透析や希釈により界面活性剤や変性剤を除去した後精製し、タンパク質を得ることができる。
【0080】
本発明の連続発酵によるタンパク質の製造方法で用いられる連続発酵装置1つの例は、主に、微生物を保持しタンパク質を製造するための発酵反応槽、および微生物と培地を濾過分離するための多孔性膜を含む分離膜エレメントから構成される。分離膜エレメントは、発酵反応槽の内部または外部のいずれに設置されてもよい。
【0081】
本発明の連続発酵によるタンパク質の製造方法は、分離膜に平均細孔径が0.01μm以上1μm未満の多孔性膜を使用し、濾過圧力である膜間差圧を0.1から20kPaの範囲にして濾過処理することを特徴としている。そのため、特別に発酵反応槽内を加圧状態に保つ必要がないことから、濾過分離装置と発酵反応槽間で発酵培養液を循環させる動力手段が不要となり、分離膜エレメントを発酵反応槽内部に設置して発酵装置をコンパクト化することも可能である。
【0082】
本発明の連続発酵によるタンパク質の製造方法で用いられる連続発酵装置のうち、分離膜エレメントが発酵反応槽の内部に設置された代表的な一例を図1の概要図に示す。図1は、本発明の連続発酵によるタンパク質の製造方法で用いられる連続発酵装置の一つの実施の形態を示す概要側面図である。
【0083】
図1において、連続発酵装置は、内部に分離膜エレメント2を備えた発酵反応槽1と水頭差制御装置3で基本的に構成されている。発酵反応槽1内の分離膜エレメント2には、多孔性膜が組み込まれている。この多孔性膜としては、例えば、国際公開第2002/064240号パンフレットに開示されている膜および膜エレメントを使用することができる。分離膜エレメントに関しては、追って詳述する。
【0084】
次に、図1の連続発酵装置による連続発酵の形態について説明する。図1において、培地供給ポンプ7によって培地を発酵反応槽1に連続的もしくは断続的に投入する。培地は発酵反応槽1に投入前に、必要に応じて加熱殺菌、加熱滅菌あるいはフィルターを用いた滅菌を行うことができる。培養時には、必要に応じて攪拌機5で発酵反応槽1内の発酵培養液を攪拌し、また必要に応じて気体供給装置4によって必要とする気体を供給し、また必要に応じてpHセンサ・制御装置9、およびpH調整溶液供給ポンプ8によって発酵培養液のpHを調整し、また必要に応じて温度調節器10によって発酵培養液の温度を調節することにより生産性の高いタンパク質生産を行うことができる。ここでは、計装・制御装置による発酵培養液の物理化学的条件の調節に、pHおよび温度を例示したが、必要に応じて溶存酸素やORPの制御、更にはオンラインケミカルセンサーなどの分析装置により培養液中の様々な物質の濃度を測定し、それを指標とした物理化学的条件の制御を行うことができる。また、培地の連続的もしくは断続的投入の形態に関しては、例えば、上記計装装置による培養液の物理化学的環境の測定値を指標として、培地投入量および速度を適宜調節することができる。
【0085】
発酵培養液は、発酵反応槽1内に設置された分離膜エレメント2によって発酵培養液と濾液に濾過・分離される。発酵培養液には、菌体が含まれる。また、分離膜によって濾過・分離された濾液には菌体は含まれない。分離膜エレメントにより、装置系内に菌体を維持させつつ、菌体増殖時に発生する酢酸等の老廃物を濾液として装置系内から出すことができる。
【0086】
また、濾過・分離された微生物は装置系内にとどまることにより装置系内の微生物濃度を高く維持することができ、生産性の高いタンパク質生産を可能としている。ここで、分離膜エレメント2による濾過・分離は、発酵反応槽1の水面との水頭差圧によって行い、特別な動力は必要ない。また、必要に応じてレベルセンサ6および水頭差圧制御装置3によって、分離膜エレメント2の濾過・分離速度および発酵反応槽内の発酵培養液量を適当に調節することができる。上記の分離膜エレメント2による濾過・分離には水頭差圧によって行うことを例示したが、必要に応じてポンプ等による吸引濾過あるいは装置系内を加圧することにより濾過・分離することもできる。
【0087】
タンパク質を菌体から回収する時期は、基本的にはいつでもかまわない。効率よい生産性を得る上で、発酵培養液の環境が微生物の増殖にとって不適切となって死滅する比率が高くならない範囲で、高い状態で維持しているときに回収することが好ましい。また、適宜発酵培養液を抜きだして、タンパク質が高発現あるいは高活性な時期に回収することが好ましい。
【0088】
タンパク質の回収には、発酵反応槽から発酵培養液を引き抜き、上述した手段により、発酵培養液中に含まれる微生物菌体内からタンパク質を回収することができる。
【0089】
次に、本発明の連続発酵によるタンパク質の製造方法で用いられる連続発酵装置のうち、分離膜エレメントが、発酵反応槽の外部に設置された代表的な一例を図2の概要図に示す。図2は、本発明で用いられる他の連続発酵装置の一つの実施の形態を説明するための概要側面図である。
【0090】
図2において、連続発酵装置は、発酵反応槽1と、分離膜エレメント2を内部に備えた膜分離槽12と、水頭差制御装置3とで基本的に構成されている。膜分離槽12中の分離膜エレメント2には、多孔性膜が組み込まれている。この多孔性膜としては、例えば、国際公開第2002/064240号パンフレットに開示されている分離膜および分離膜エレメントを使用することが好適である。また、膜分離槽12は、発酵培養液循環ポンプ11を介して発酵反応槽1に接続されている。
【0091】
図2において、培地供給ポンプ7によって培地を発酵反応槽1に投入し、必要に応じて、攪拌機5で発酵反応槽1内の発酵培養液を攪拌し、また必要に応じて、気体供給装置4によって必要とする気体を供給し、また必要に応じて、pHセンサ・制御装置9およびよびpH調整溶液供給ポンプ8によって発酵培養液のpHを調整し、また必要に応じて、温度調節器10によって発酵培養液の温度を調節することにより、生産性の高い発酵生産を行うことができる。さらに、装置内の発酵培養液は、発酵培養液循環ポンプ11によって発酵反応槽1と膜分離槽12の間を循環する。発酵培養液は、分離膜エレメント2によって微生物を含む発酵培養液と微生物を含まない濾液に濾過・分離され、濾液は装置系内から取り除かれる。
【0092】
また、濾過・分離された微生物は、装置系内にとどまることで装置系内の微生物濃度を高く維持することができ、生産性の高い発酵生産を可能としている。ここで、分離膜エレメント2による濾過・分離は、膜分離槽12の水面との水頭差圧によって行い、特別な動力を必要としない。また、必要に応じて、レベルセンサ6および水頭差圧制御装置3によって、分離膜エレメント2の濾過・分離速度および装置系内の発酵培養液量を適当に調節することができる。また必要に応じて、気体供給装置4によって必要とする気体を膜分離槽12内に供給することができる。上記のように、分離膜エレメント2による濾過・分離には水頭差圧によって行うことを例示したが、必要に応じて、ポンプ等による吸引濾過あるいは装置系内を加圧することにより濾過・分離することもできる。
【0093】
タンパク質の回収には、適宜発酵培養液を抜きだして、タンパク質が高発現あるいは高活性な時期に回収する。タンパク質が高発現あるいは高活性である発酵培養液を抜き出し、菌体のみ回収する。菌体回収には、例えば、遠心分離機を用いて、遠心操作にて菌体を回収することが可能である。次に、回収した菌体を破砕する。破砕の方法は、例えば、ホモジナイザー、超音波など機械的な方法を用いて行うことができる。破砕した菌体を、例えばリン酸緩衝液などの適当な緩衝液に懸濁し、カラムクロマトグラフィー、液体クロマトグラフィーおよびイオン交換クロマトグラフィーなどを用いて、目的のタンパク質を精製・回収することができる。
【0094】
次に、本発明で用いられる分離膜エレメントについて説明する。
【0095】
本発明で用いられる分離膜エレメントの好適な形態の例である国際公開第2002/064240号パンフレットに開示されている分離膜および分離膜エレメントを、以下、図を用いてその概略を説明する。図3は、本発明で用いられる分離膜エレメントの一つの実施の形態を説明するための概要斜視図である。
【0096】
分離膜エレメントは、図3に示すように、剛性有する支持板13の両面に、流路材14と前記の分離膜15(多孔性膜)とをこの順序で配し構成されている。支持板13は、両面に凹部16を有している。分離膜15は発酵培養液を濾過する。流路材14は、分離膜15で濾過された濾過液を効率よく支持板13に流すためのものである。支持板13に流れた濾過液は、支持板13の凹部16を通って外部に取り出される。外部へは、集水パイプ17を介して連続発酵装置外部に取り出される。
【0097】
図4に示す分離膜エレメントについて説明する。図4は、本発明で用いられる別の分離膜エレメントを例示説明するための概略斜視図である。分離膜エレメントは、図4に示すように、中空糸膜(多孔性膜)で構成された分離膜束18と上部樹脂封止層19と下部樹脂封止層20によって主に構成される。分離膜束18は、上部樹脂封止層19および下部樹脂封止層20よって束状に接着・固定化されている。下部樹脂封止層20による接着・固定化は、分離膜束18の中空糸膜(多孔性膜)の中空部を封止しており、発酵培養液の漏出を防ぐ構造になっている。一方、上部樹脂封止層19は、分離膜束18の中空糸膜(多孔性膜)の内孔を封止しておらず、集水パイプ22に透過水が流れる構造となっている。この分離膜エレメントは、支持フレーム21を介して連続発酵装置内に設置することが可能である。分離膜束18によって濾過された濾過液は、中空糸膜の中空部を通り、集水パイプ22を介して連続発酵装置外部に取り出される。濾過液を取り出すための動力として、水頭差圧、ポンプ、液体や気体等による吸引濾過、あるいは装置系内を加圧するなどの方法を用いることができる。
【0098】
本発明の連続発酵によるタンパク質の製造方法で用いられる連続発酵装置の分離膜エレメントを構成する部材は、高圧蒸気滅菌操作に耐性の部材であることが好ましい。連続発酵装置内が滅菌可能であれば、連続発酵時に好ましくない微生物による汚染の危険を回避することができ、より安定した連続発酵が可能となる。分離膜エレメントを構成する部材は、高圧蒸気滅菌操作の条件である、121℃で15分間に耐性であることが好ましい。
【0099】
分離膜エレメント部材には、例えば、ステンレスやアルミニウムなどの金属、ポリアミド系樹脂、フッ素系樹脂、ポリカーボネート系樹脂、ポリアセタール系樹脂、ポリブチレンテレフタレート系樹脂、PVDF、変性ポリフェニレンエーテル系樹脂およびポリサルホン系樹脂等の樹脂を好ましく選定することができる。
【0100】
本発明の連続発酵によるタンパク質の製造方法で用いられる連続発酵装置では、分離膜エレメントは、図1のように発酵反応槽内に設置しても良いし、図2のように発酵反応槽外に設置しても良い。分離膜エレメントを発酵反応槽外に設置する場合には、別途、膜分離槽を設けてその内部に分離膜エレメントを設置することができ、発酵反応槽と膜分離槽の間を培養液を循環させながら、分離膜エレメントにより培養液を連続的に濾過することができる。
【0101】
本発明の連続発酵によるタンパク質の製造方法で用いられる連続発酵装置では、膜分離槽は、高圧蒸気滅菌可能であることが望ましい。膜分離槽が高圧蒸気滅菌可能であると、雑菌による汚染回避が容易である。
【0102】
本発明の連続発酵によるタンパク質の製造方法に従って、連続発酵を行った場合、従来のバッチ培養と比較して、高い菌体濃度が得られ、極めて効率のよいタンパク質生産が可能となる。ここで、連続培養における生産速度は、次の(式3)で計算される。
・発酵生産速度(g/L/hr)=抜き取り液中の生産物濃度(g/L)×発酵培養液抜き取り速度(L/hr)÷装置の運転液量(L)・・・(式3)
また、バッチ培養での発酵生産速度は、原料炭素源をすべて消費した時点の生産物量(g)を、炭素源の消費に要した時間(h)とその時点の培養液量(L)で除して求められる。
【実施例】
【0103】
以下、本発明をさらに詳細に説明するために、タンパク質としてイヌインターフェロンγ、タンパク質の生産能力のある微生物として大腸菌を選定し、図1および図2の概要図に示す連続発酵装置を用いたタンパク質の製造を行った。本発明は、これら実施例の記載に限定されるものではない。ここで、大腸菌としては、大腸菌MC1061株(ATCC社製)を用いた。大腸菌MC1061株に、イヌインターフェロンγをコードする遺伝子を導入することにより、組換えイヌインターフェロンγ生産能力をもつ大腸菌株を造成し実施した。具体的には、イヌインターフェロンγをコードする遺伝子を大腸菌K−12株由来のリン酸結合タンパク質遺伝子の下流に連結したプラスミドを導入することにより、組換えイヌインターフェロンγ生産能力を持つ形質転換体大腸菌株を造成して使用した。
【0104】
(参考例1)リン酸結合タンパク遺伝子(phoS)の上流域遺伝子を含むベクターの構築
(1)大腸菌K−12株のゲノムDNAの調製
K−12株であるKLF/KL159株(Yale University E.coli. GeneticResourceCenter CGSC Strain#4302)をT培地(バクトトリプトン10g/L、NaCl 5g/L)で終日培養後、遠心操作により沈殿とし、核酸抽出剤「Sepagene」(三光純薬社製)を用いてゲノムDNAを回収した。すなわち、トリス緩衝液で構成される試薬1を沈殿に添加し、撹拌後室温で10分間静置した。次に、チオシアン酸グアニジンで構成される試薬2を添加してゆるやかに混和した後、吸着剤とクロロホルムで構成される試薬3を添加し激しく混和した。12000rpm、15分間遠心操作後、上清を回収し酢酸緩衝液で構成される試薬4を加え、さらにイソプロパノール加え転倒混和した。12000rpm,15分遠心後、上清を除去し沈殿物を滅菌蒸留水に溶解した。
【0105】
(2)リン酸結合タンパク質phoS上流配列遺伝子の取得
大腸菌K12株ゲノム配列情報(MEDLINE)を元に、配列番号1と配列番号2の2種類のプライマーをDNAシンセサイザーにて合成した。上記(1)で得られたゲノムDNAを0.5mLのミクロ遠心チューブに2μL添加し、各プライマーを20pmol,20mMトリス塩酸緩衝液(pH8.0)、1.5mM MgCl2,25mM KCL,100μg/mLゼラチン、50μM各dNTP、4単位 LA−TaqDNAポリメラーゼとなるように各試薬を加え、全量100μLとした。DNAの変性条件を94℃、1分、プライマーのアニーリング条件を55℃、1分、プライマーの伸長条件を72℃、1分の各条件でPerkin−Elmer Cetus社のDNAサーマルサイクラーを用い、35サイクル反応させた。これを1%アガロースゲルにて電気泳動し、約300bpのDNA断片を常法に従って調製した。さらに、配列番号3と配列番号4の2種類のプライマーをDNAシンセサイザーにて合成した。上記アガロースゲルより調製したDNA断片を0.5mLのミクロ遠心チューブに2μL添加し、各プライマーを20pmol,20mMトリス塩酸緩衝液(pH8.0)、1.5mM MgCl2,25mM KCL,100μg/mLゼラチン、50μM各dNTP、4単位 LA−TaqDNAポリメラーゼとなるように各試薬を加え、全量100μLとした。DNAの変性条件を94℃、1分、プライマーのアニーリング条件を55℃、1分、プライマーの伸長条件を72℃、1分の各条件でPerkin−Elmer Cetus社のDNAサーマルサイクラーを用い、35サイクル反応させた。これを1%アガロースゲルにて電気泳動し、約300bpのDNA断片を常法に従って調製した。
【0106】
このようにして得られたDNA断片を、Invitrogen社のT−Vectorに宝酒造(株)のDNA Ligation Kit Ver.1を用いて連結した。これを用いて常法に従い大腸菌を形質転換し、得られた形質転換体からプラスミドDNAを常法により調製した。次に、このプラスミドにPCR断片が挿入されていることを前述と同じ条件のPCRによって確認後、蛍光DNAシーケンサーを用い、その添付プロトコールに従ってパーキンエルマー社のダイターミネーターサイクルシーケンシングキットを用いて得られたDNA断片が大腸菌K12株由来の遺伝子をコードする配列であることを確認した(配列番号5)。
【0107】
(3)リン酸結合タンパク質遺伝子(phoS)上流配列およびSD配列を付加したベクターの構築
上記(2)で調製したリン酸結合タンパク質phoSの上流配列遺伝子を含むベクターを用いて、リン酸結合タンパク質phoSの上流配列遺伝子の3’末にSD配列を付加した。すなわち、センス側のプライマーである配列番号3とアンチセンス側のプライマーすなわちSD配列を有するプライマー配列番号6を作製した。次に、配列番号3と配列番号6のプライマーの組み合わせを用いたPCRを実施した。すなわち、上記ベクターDNAを0.5mLのミクロ遠心チューブに2μL添加し、各プライマーを20pmol,20mMトリス塩酸緩衝液(pH8.0)、1.5mM MgCl2,25mM KCL,100μg/mLゼラチン、50μM各dNTP、4単位 LA−TaqDNAポリメラーゼとなるように各試薬を加え、全量100μLとした。DNAの変性条件を94℃、1分、プライマーのアニーリング条件を55℃、1分、プライマーの伸長条件を72℃、1分の各条件でPerkin−Elmer Cetus社のDNAサーマルサイクラーを用い、35サイクル反応させた。これを1%アガロースゲルにて電気泳動し、約300bpのDNA断片を常法に従って調製した。
【0108】
このDNA断片をInvitrogen社のT−Vectorに宝酒造(株)のDNA Ligation Kit Ver.1を用いて連結した。これを用いて常法に従い大腸菌を形質転換し、得られた形質転換体よりプラスミドDNAを常法により調製した。次に、このプラスミドにPCR断片が挿入されていることを前述と同じ条件のPCRによって確認後、蛍光DNAシーケンサーを用い、その添付プロトコールに従ってパーキンエルマー社のダイターミネーターサイクルシーケンシングキットを用いて得られたDNA断片が大腸菌K12株由来の遺伝子をコードする配列であることを確認した。(配列番号7)
上記(2)で調製したリン酸結合タンパク質phoSの上流配列遺伝子を含むベクターおよびタカラバイオ社のpBR322をそれぞれEcoRIによる制限酵素処理を行い、それぞれをアガロースゲル電気泳動を行い、常法に従いに遺伝子を切り出した。pBR322については、Alkaline Phospatase Shrimp(日本ロシュ社製)による脱リン酸化処理後、リン酸結合タンパク質phoSの上流配列遺伝子と宝酒造(株)のDNA Ligation Kit Ver.1を用いて連結した。これを用いて常法に従い大腸菌を形質転換し、得られた形質転換体よりプラスミドDNAを常法により調製した。次に、このプラスミドにPCR断片が挿入されていることを前述と同じ条件のPCRによって確認後、蛍光DNAシーケンサー(パーキンエルマー社製DNAシーケンサー373S)を用い、その添付プロトコールに従って、パーキンエルマー社のダイターミネーターサイクルシーケンシングキットを用いて、得られたDNA断片がリン酸結合タンパク質phoSの上流配列遺伝子および新たに付加したSD配列をコードする配列であることを確認した。ベクターの名称について、配列番号7をpPho2とする。
【0109】
(参考例2)イヌIFN-γ遺伝子の取得
(1)イヌcDNAの調製
イヌ末梢血よりイヌリンパ球を分離し、フィトヘムアグルチニン(PHA)を50ug/mLの終濃度で48時間刺激した。その後、“ISOGEN”(登録商標)(ニッポンジーン社製)を用いて総RNAを調製した。得られたRNAを1mM EDTAを含む10mM トリス塩酸緩衝液(pH7.5)(以下TEと略する)に溶解し、70℃の温度で5分間処理した後、1M LiClを含むTEで平衡化したオリゴdTセルロースカラムにRNA溶液をアプライし、同緩衝液にて洗浄した。さらに、0.3M LiClを含むTEで洗浄後、0.01%SDSを含む1mM EDTA(pH7.0)で吸着したポリ(A)RNAを溶出した。このようしにて得られたポリ(A)RNAを用いて一本鎖cDNAを合成した。すなわち、滅菌した0.5mLのミクロ遠心チューブに5μgのポリ(A)RNAと0.5μgのオリゴdTプライマー(12−18mer)を入れ、ジエチルピロカルボネート処理滅菌水を加えて12μLにし、70℃の温度で10分間インキュベートした後、氷中に1分間つけた。これに200mM MgCl2を2μL、10mM dNTPを1μLおよび0.1M DTTを2μLそれぞれ加え、42℃で5分間インキュベートした後、200ユニットのGibcoBRL社製Super Script II RTを1μL加え、42℃の温度でさらに50分間インキュベートしてcDNA合成反応を行った。さらに、70℃の温度で15分間インキュベートして反応を停止し、氷上に5分間おいた。この反応液に1μLのE.coli RNaseH(2units/mL)を加え、37℃の温度で20分間インキュベートした。
【0110】
(2)イヌIFN−γ遺伝子の取得
イヌIFN−γのN末端およびC末端の塩基配列をもとに、配列番号8と配列番号9の2種類のプライマーをDNAシンセサイザーにて合成した。上記(1)のイヌリンパ球から得られたcDNAを0.5mLのミクロ遠心チューブに2μL添加し、各プライマーを20pmol,20mMトリス塩酸緩衝液(pH8.0)、1.5mM MgCl2,25mM KCL,100μg/mLゼラチン、50μM各dNTP、4単位 TaqDNAポリメラーゼとなるように各試薬を加え、全量100μLとした。DNAの変性条件を94℃、1分、プライマーのアニーリング条件を55℃、2分、プライマーの伸長条件を72℃、3分の各条件でPerkin−Elmer Cetus社のDNAサーマルサイクラーを用い、35サイクル反応させた。これを1%アガロースゲルにて電気泳動し、約500bpのDNA断片を常法に従って調製した。
【0111】
このようにして得られたDNA断片を、Invitrogen社のT−Vectorに宝酒造(株)のDNA Ligation Kit Ver.1を用いて連結した。これを用いて常法に従い大腸菌を形質転換し、得られた形質転換体からプラスミドDNAを常法により調製した。次に、このプラスミドにPCR断片が挿入されていることを前述と同じ条件のPCRによって確認後、蛍光DNAシーケンサーを用い、その添付プロトコールに従ってパーキンエルマー社のダイターミネーターサイクルシーケンシングキットを用いて、得られたDNA断片がイヌIFN−γをコードする配列であることを確認した。
【0112】
(3)イヌIFN−γ変異体遺伝子の取得
イヌIFN−γ変異体遺伝子は、イヌIFN−γタンパク質のC末端が16残基欠損させたものであり、イヌIFN−γ遺伝子のN末端及びC末端の塩基配列を基に、配列番号10と配列番号11の2種類のプライマーをDNAシンセサイザーにて合成した。上記(1)のイヌリンパ球から得られたcDNAを鋳型として、上記(2)と同様にして約500bpのDNA断片を得、T−Vectorに挿入し、イヌIFN−γをコードする配列であることを確認した(配列番号12)。
【0113】
(参考例3)リン酸結合タンパク遺伝子(phoS)の上流域遺伝子とイヌIFN−γ遺伝子を含むベクターの構築
参考例1および参考例2で調製したpPho2ベクターおよびイヌIFN−γ変異体遺伝子をそれぞれNdeIおよびXhoIによる制限酵素処理を行い、それぞれをアガロースゲル電気泳動を行い、常法に従いに遺伝子を切り出した。その遺伝子を宝酒造(株)のDNA Ligation Kit Ver.1を用いて連結した。これを用いて常法に従い大腸菌JM109を形質転換し、得られた形質転換体よりプラスミドDNAを常法により調製した。さらに次に、このプラスミドDNAを用いて常法に従い大腸菌MC1061(ATCC社製)に形質転換した。
【0114】
(参考例4)多孔性膜の作製(その1)
樹脂としてポリフッ化ビニリデン(PVDF)樹脂を、また溶媒としてN,N−ジメチルアセトアミド(DMAc)をそれぞれ用い、これらを90℃の温度下に十分に攪拌し、次の組成を有する原液を得た。
[原液]
・PVDF:13.0重量%
・DMAc:87.0重量%
次に、上記の原液を25℃の温度に冷却した後、あらかじめガラス板上に貼り付けて置いた、密度が0.48g/cm3で、厚みが220μmのポリエステル繊維製不織布(多孔質基材)に塗布し、直ちに次の組成を有する25℃の温度の凝固浴中に5分間浸漬して、多孔質基材に多孔質樹脂層が形成された多孔性膜を得た。
[凝固浴]
・水 :30.0重量%
・DMAc:70.0重量%
この多孔性膜をガラス板から剥がした後、80℃の温度の熱水に3回浸漬してDMAcを洗い出し、分離膜を得た。多孔質樹脂層表面の9.2μm×10.4μmの範囲内を、倍率10,000倍で走査型電子顕微鏡観察を行ったところ、観察できる細孔すべての直径の平均は0.1μmであった。次に、上記の分離膜について純水透水透過係数を評価したところ、50×10-9m3/m2/s/Paであった。純水透水量の測定は、逆浸透膜による25℃の温度の精製水を用い、ヘッド高さ1mで行った。また、平均細孔径の標準偏差は0.035μmで、膜表面粗さは0.06μmであった。
【0115】
(参考例5)多孔性膜の作製(その2)
重量平均分子量41.7万のフッ化ビニリデンホモポリマーとγ-ブチロラクトンとを、それぞれ38重量%と62重量%の割合で170℃の温度で溶解し原液を作製した。この原液をγ-ブチロラクトンを中空部形成液体として随拌させながら口金から吐出し、温度20℃のγ-ブチロラクトン80重量%水溶液からなる冷却浴中で固化して中空糸膜を作製した。
【0116】
次いで、重量平均分子量28.4万のフッ化ビニリデンホモポリマーを14重量%、セルロースアセテートプロピオネート(イーストマンケミカル社、CAP482−0.5)を1重量%、N-メチル-2-ピロリドンを77重量%、ポリオキシエチレンヤシ油脂肪酸ソルビタン(三洋化成株式会社製、商品名“イオネットT−20C”(登録商標))を5重量%、および水を3重量%の割合で95℃の温度で混合溶解して原液を調整した。この原液を、上記で得られた中空糸膜の表面に均一に塗布し、すぐに水浴中で凝固させた本発明で用いる中空糸膜(多孔性膜)を製作した。得られた中空糸膜(分離膜)の被処理水側表面の平均細孔径は、0.05μmであった。次に、上記の分離膜である中空糸膜について純水透水量を評価したところ、5.5×10-9m3/m2・s・Paであった。透水量の測定は、逆浸透膜による25℃の温度の精製水を用い、ヘッド高さ1mで行った。また、平均細孔径の標準偏差 は0.006μmであった。
【0117】
(比較例1)
微生物を用いた培養形態として最も典型的なバッチ培養を1.5L容のジャーファーメンターを用いて行い、そのタンパク質生産性を評価した。培地は、121℃の温度で20分間高圧蒸気滅菌して用いた。この比較例1では、微生物として大腸菌MC1061−イヌインターフェロンγ株を用い、生産物である組換えイヌインターフェロンγの評価はウエスタンブロッティング法を用いて評価し、グルコース濃度の測定には“グルコーステストワコーC”(登録商標)(和光純薬社製)、リン酸濃度測定には“ホスファC−テストワコー”(登録商標)(和光純薬社製)を用いた。比較例1の運転条件は、下記のとおりである。
・発酵反応槽容量(初発培地量):1.0(L)
・温度調整:37(℃)
・発酵反応槽通気量:1.5(L/min)空気
・発酵反応槽攪拌速度:800(rpm)
・pH調整:2N H2SO4および14N アンモニア水によりpH7.0に調整。
【0118】
まず、大腸菌MC1061−イヌインターフェロンγ株を、試験管で5mlの表1で示した組成の培地にテトラサイクリン(10μg/ml)を添加した培地で一晩振とう培養した(前々培養)。得られた培養液を、新鮮なテトラサイクリン(10μg/ml)を添加した初発培地50mlに植菌し、500ml容坂口フラスコで24時間、37℃の温度で、振幅30cmで、180rpmの条件下で培養を行った(前培養)。前培養液を、ジャーファーメンターの1.0Lの初発培地に植菌した。初発培地を用い、バッチ培養を行った。培養の結果、菌体濃度OD600=7で増殖が止まり、後述する実施例1の結果と比較して1/4以下であった。また、適宜サンプリングしておいた培養液の各フラクションの菌体濃度OD600=10で統一し、その菌体液をサンプルとし、コニカイムノステインHRP(コニカ社製)を用い、添付のプロトコールに従ってイヌインターフェロンγの検出を行った。その結果は、モレキュラーイメージャー(BioRad社製)を用いてシグナル強度を測定した。また、非形質転換大腸菌をネガティブコントロールのサンプルとして使用し、インタードック(東レ株式会社製:イヌインターフェロンγ)をポジティブコントロールのサンプルとして使用した。その結果、イヌインターフェロンγの発現が確認されなかった。
【0119】
(実施例1)タンパク質の製造(その1)
図1の連続発酵装置を稼働させることにより、連続発酵生産系が得られるかどうかを調べるため、表1に示す組成培地と表2に示す組成の追加培地を用い、この装置の連続発酵試験を行った。培地は、121℃の温度で20分間高圧蒸気滅菌し、追加培地は、121℃の温度で60分間高圧蒸気滅菌して用いた。分離膜エレメント部材には、ステンレスおよびポリサルホン樹脂の成形品を用いた。分離膜には、参考例4で作製したポリフッ化ビニリデン(PVDF)を主成分とする多孔性膜を用いた。この実施例1における運転条件は、特に断らない限り下記のとおりである。
・発酵反応槽容量:1.5(L)
・使用分離膜:ポリフッ化ビニリデン濾過膜
・分離膜エレメント有効濾過面積:120平方cm
・温度調整:37(℃)
・発酵反応槽通気量:1.5(L/min)空気
・発酵反応槽攪拌速度:800(rpm)
・pH調整:2N H2SO4および14N アンモニア水によりpH7.0に調整。
・滅菌:分離膜エレメントを含む発酵反応槽、および培地は121℃、20minのオートクレーブにより高圧蒸気滅菌。また、追加培地は、121℃、60minのオートクレーブにより高圧蒸気滅菌。
・膜透過水量制御:発酵反応槽水頭差により流量を制御(水頭差は2m以内で制御した)。
【0120】
微生物として、大腸菌MC1061−イヌインターフェロンγ株を用い、表1に示す組成の培地を用い、追加培地として表2に示す組成の培地を用いた。また、グルコース濃度の測定には“グルコーステストワコーC”(登録商標)(和光純薬社製)、リン酸濃度測定には“ホスファC−テストワコー”(登録商標)(和光純薬社製)を用いた。
【0121】
【表1】
【0122】
【表2】
【0123】
まず、大腸菌MC1061−イヌインターフェロンγ株を、試験管で5mlの表1で示した組成の培地にテトラサイクリン(10μg/ml)を添加した培地で一晩振とう培養した(前々々培養)。得られた培養液を、新鮮なテトラサイクリン(10μg/ml)を添加した表1で示した組成の培地50mlに植菌し、500ml容坂口フラスコで24時間、37℃の温度で、振幅30cmで、180rpmの条件下で培養を行った(前々培養)。
前々培養液を、図1に示した連続発酵装置に1.5Lの表1で示した組成の培地に植菌し、発酵反応槽1を付属の攪拌機5によって800rpmで攪拌し、発酵反応槽1の通気量の調整、温度調整およびpH調整を行い培養を行った。前培養完了後直ちに、表2で示した組成の追加培地の連続供給を行い、連続発酵装置の発酵培養液量を1.5Lとなるように膜透過水量の制御を行いながら連続培養し、リン酸の濃度を低下させていき、連続発酵によるタンパク質の製造を行った。連続発酵試験を行うときの膜透過水量の制御は、水頭差制御装置3により、発酵反応槽水頭を最大2m以内、すなわち膜間差圧が20kPa以内となるように適宜水頭差を変化させることにより行った。適宜、膜透過濾液中のリン酸濃度および残存グルコース濃度を測定した。培養の結果、菌体濃度OD600=30以上まで増殖し、高濃度培養が可能になった。
【0124】
また、適宜発酵培養液をサンプリングしておき、リン酸濃度がどの程度低下した場合にタンパク質が発現しているかウエスタンブロッティング法で確認した。方法としては、各フラクションの菌体濃度OD600=10で統一し、その菌体液をサンプルとし、コニカイムノステインHRP(コニカ社製)を用い、添付のプロトコールに従ってイヌインターフェロンγの検出を行った。すなわち、SDS−PAGE電気泳動し、ブロッティングしたメンブレンをブロッキング溶液(5%スキムミルク・0.1%Tween20・PBS)中で4℃終夜ブロッキングした。メンブレンをTPBS(0.1%Tween20・PBS)で2回洗浄し、TPBSで1000倍希釈した抗イヌインターフェロンγ抗体で室温1時間処理した。メンブレンをTPBSで2回洗浄し、更にTPBSで5分間3回洗浄した。その後TPBSで10000倍希釈した後、HRPラベル抗ラビットIgG抗体(コスモバイオ社製)で室温1時間処理した。メンブレンをTPBSで2回洗浄し、更にTPBSで5分間3回洗浄した後、コニカイムノステインHRP(コニカ社製)の検出試薬(溶液A+溶液B+溶液C+溶液D)を加え、発色させた。その結果は、モレキュラーイメージャー(BioRad社製)を用いてシグナル強度を測定した。また、非形質転換大腸菌をネガティブコントロールのサンプルとして使用し、インタードック(東レ株式会社製:イヌインターフェロンγ)をポジティブコントロールのサンプルとして使用した。
その結果、リン酸濃度1mM以下で、イヌインターフェロンγが大量に発現していることが確認できた。また、上記に示す比較例1と比較して5〜10倍に産生していることを確認した。図1の連続発酵装置を用いることにより、菌体増殖は保ちながら、培地中の成分の濃度を低下させることにより連続発酵によるタンパク質の製造が可能であることを確認することができた。
【0125】
(実施例2)タンパク質の製造(その2)
図2の連続発酵装置を稼働させることにより、連続発酵生産系が得られるかどうかを調べるため、表1に示す組成培地と表2に示す組成の追加培地を用い、この装置の連続発酵試験を行った。培地は、121℃の温度で20分間高圧蒸気滅菌し、追加培地は、121℃の温度で60分間高圧蒸気滅菌して用いた。分離膜エレメント部材には、ステンレスおよびポリサルホン樹脂の成形品を用いた。分離膜には、参考例4で作製したポリフッ化ビニリデン(PVDF)を主成分とする多孔性膜を用いた。この実施例2における運転条件は、特に断らない限り下記のとおりである。
・発酵反応槽容量:1.5(L)
・膜分離槽容量:0.5(L)
・使用分離膜:ポリフッ化ビニリデン濾過膜
・膜分離エレメント有効濾過面積:120平方cm
・温度調整:37(℃)
・発酵反応槽通気量:1.5(L/min)空気
・発酵反応槽攪拌速度:800(rpm)
・膜分離槽通気量:0.3(L/min)
・pH調整:2N H2SO4および14N アンモニア水によりpH7.0に調整
・培養液循環装置による循環液量:0.1(L/min)
・膜透過水量制御:発酵反応槽水頭差により流量を制御(水頭差は、2m以内で制御した)。
【0126】
微生物として大腸菌MC1061-イヌインターフェロンγ株を用い、表1に示す組成の培地を用い、追加培地として表2に示す組成の培地を用いた。また、グルコース濃度の測定には“グルコーステストワコーC”(登録商標)(和光純薬社製)、リン酸濃度測定には“ホスファC−テストワコー”(登録商標)(和光純薬社製)を用いた。
【0127】
まず、大腸菌MC1061−イヌインターフェロンγ株を、試験管で5mlの表1で示した組成の培地にテトラサイクリン(10μg/ml)を添加した培地で一晩振とう培養した(前々々培養)。得られた培養液を、新鮮なテトラサイクリン(10μg/ml)を添加した表1で示した組成の培地50mlに植菌し、500ml容坂口フラスコで24時間、37℃の温度で、振幅30cmで、180rpmの条件下で培養を行った(前々培養)。
前々培養液を、図2に示した連続発酵装置に2Lの表1で示した組成の培地に植菌し、発酵反応槽1を付属の攪拌機5によって800rpmで攪拌し、発酵反応槽1の通気量の調整、温度調整およびpH調整を行い培養を行った。前培養完了後直ちに、表2で示した組成の追加培地の連続供給を行い、連続発酵装置の発酵培養液量を2Lとなるように膜透過水量の制御を行いながら連続培養し、リン酸の濃度を低下させていき、連続発酵によるタンパク質の製造を行った。連続発酵試験を行うときの膜透過水量の制御は、水頭差制御装置3により、発酵反応槽水頭を最大2m以内、すなわち膜間差圧が20kPa以内となるように適宜水頭差を変化させることで行った。適宜、膜透過濾液中のリン酸濃度および残存グルコース濃度を測定した。培養の結果、菌体濃度OD600=30以上まで増殖し、高濃度培養が可能になった。
【0128】
また、適宜発酵培養液をサンプリングしておき、リン酸濃度がどの程度低下した場合にタンパク質が発現しているかウエスタンブロッティング法で確認した。方法としては、各フラクションの菌体濃度OD600=10で統一し、その菌体液をサンプルとし、コニカイムノステインHRP(コニカ社製)を用い、添付のプロトコールに従ってイヌインターフェロンγの検出を行った。すなわち、SDS−PAGE電気泳動し、ブロッティングしたメンブレンをブロッキング溶液(5%スキムミルク・0.1%Tween20・PBS)中で4℃終夜ブロッキングした。メンブレンをTPBS(0.1%Tween20・PBS)で2回洗浄し、TPBSで1000倍希釈した抗イヌインターフェロンγ抗体で室温1時間処理した。メンブレンをTPBSで2回洗浄し、更にTPBSで5分間3回洗浄した。その後TPBSで10000倍希釈した後、HRPラベル抗ラビットIgG抗体(コスモバイオ社製)で室温1時間処理した。メンブレンをTPBSで2回洗浄し、更にTPBSで5分間3回洗浄した後、コニカイムノステインHRP(コニカ社製)の検出試薬(溶液A+溶液B+溶液C+溶液D)を加え、発色させた。その結果は、モレキュラーイメージャー(BioRad社製)を用いてシグナル強度を測定した。また、非形質転換大腸菌をネガティブコントロールのサンプルとして使用し、インタードック(東レ株式会社製:イヌインターフェロンγ)をポジティブコントロールのサンプルとして使用した。
その結果、リン酸濃度1mM以下でイヌインターフェロンγが大量に発現していることが確認できた。また、上記に示す比較例1と比較して5〜10倍に産生していることを確認した。図2に示した連続発酵装置を用いることにより、菌体増殖は保ちながら、培地中の成分の濃度を低下させることで連続発酵によるタンパク質の製造が可能であることを確認することができた。
【0129】
(実施例3)タンパク質の製造(その3)
図1の連続発酵装置を稼働させることにより、連続発酵生産系が得られるかどうかを調べるため、表1に示す組成培地と表2に示す組成の追加培地を用い、この装置の連続発酵試験を行った。培地は、121℃の温度で20分間高圧蒸気滅菌し、追加培地は、121℃の温度で60分間高圧蒸気滅菌して用いた。分離膜エレメント部材には、ステンレスおよびポリサルホン樹脂の成形品を用いた。分離膜には、参考例5で作製したポリフッ化ビニリデン(PVDF)を主成分とする多孔性膜を用いた。この実施例3における運転条件は、特に断らない限り下記のとおりである。
・発酵反応槽容量:1.5(L)
・使用分離膜:ポリフッ化ビニリデン濾過膜
・分離膜エレメント有効濾過面積:120平方cm
・温度調整:37(℃)
・発酵反応槽通気量:1.5(L/min)空気
・発酵反応槽攪拌速度:800(rpm)
・pH調整:2N H2SO4および14N アンモニア水によりpH7.0に調整
・滅菌:分離膜エレメントを含む発酵反応槽、および培地は121℃、20minのオートクレーブにより高圧蒸気滅菌。また、追加培地は121℃、60minのオートクレーブにより高圧蒸気滅菌。
・膜透過水量制御:発酵反応槽水頭差により流量を制御(水頭差は2m以内で制御した)。
【0130】
微生物として大腸菌MC1061−イヌインターフェロンγ株を用い、表1に示す組成の培地を用い、追加培地として表2に示す組成の培地を用いた。また、グルコース濃度の測定には“グルコーステストワコーC”(登録商標)(和光純薬社製)、リン酸濃度測定には“ホスファC−テストワコー”(登録商標)(和光純薬社製)を用いた。
【0131】
まず、大腸菌MC1061−イヌインターフェロンγ株を、試験管で5mlの表1で示した組成の培地にテトラサイクリン(10μg/ml)を添加した培地で一晩振とう培養した(前々々培養)。得られた培養液を、新鮮なテトラサイクリン(10μg/ml)を添加した表1で示した組成の培地50mlに植菌し、500ml容坂口フラスコで24時間、37℃の温度で、振幅30cmで、180rpmの条件下で培養を行った(前々培養)。
前々培養液を、図1に示した連続発酵装置に1.5Lの表1で示した組成の培地に植菌し、発酵反応槽1を付属の攪拌機5によって800rpmで攪拌し、発酵反応槽1の通気量の調整、温度調整およびpH調整を行い培養を行った。前培養完了後直ちに、表2で示した組成の追加培地の連続供給を行い、連続発酵装置の発酵培養液量を1.5Lとなるように膜透過水量の制御を行いながら連続培養し、リン酸の濃度を低下させていき、連続発酵によるタンパク質の製造を行った。連続発酵試験を行うときの膜透過水量の制御は、水頭差制御装置3により、発酵反応槽水頭を最大2m以内、すなわち膜間差圧が20kPa以内となるように適宜水頭差を変化させることで行った。適宜、膜透過濾液中のリン酸濃度および残存グルコース濃度を測定した。培養の結果、菌体濃度OD600=30以上まで増殖し、高濃度培養が可能になった。
【0132】
また、適宜発酵培養液をサンプリングしておき、リン酸濃度がどの程度低下した場合にタンパク質が発現しているかウエスタンブロッティング法で確認した。方法としては、各フラクションの菌体濃度OD600=10で統一し、その菌体液をサンプルとし、コニカイムノステインHRP(コニカ社製)を用い、添付のプロトコールに従ってイヌインターフェロンγの検出を行った。すなわち、SDS−PAGE電気泳動し、ブロッティングしたメンブレンをブロッキング溶液(5%スキムミルク・0.1%Tween20・PBS)中で4℃終夜ブロッキングした。メンブレンをTPBS(0.1%Tween20・PBS)で2回洗浄し、TPBSで1000倍希釈した抗イヌインターフェロンγ抗体で室温1時間処理した。メンブレンをTPBSで2回洗浄し、更にTPBSで5分間3回洗浄した。その後TPBSで10000倍希釈した後、HRPラベル抗ラビットIgG抗体(コスモバイオ社製)で室温1時間処理した。メンブレンをTPBSで2回洗浄し、更にTPBSで5分間3回洗浄した後、コニカイムノステインHRP(コニカ社製)の検出試薬(溶液A+溶液B+溶液C+溶液D)を加え、発色させた。その結果は、モレキュラーイメージャー(BioRad社製)を用いてシグナル強度を測定した。また、非形質転換大腸菌をネガティブコントロールのサンプルとして使用し、インタードック(東レ株式会社製:イヌインターフェロンγ)をポジティブコントロールのサンプルとして使用した。
その結果、リン酸濃度1mM以下でイヌインターフェロンγが大量に発現していることが確認できた。また、上記した比較例1と比較して5〜10倍に産生していることを確認した。図1に示した連続発酵装置を用いることにより、菌体増殖は保ちながら、培地中の成分の濃度を低下させることで連続発酵によるタンパク質の製造が可能であることを確認することができた。
【0133】
(実施例4)タンパク質の製造(その4)
図2の連続発酵装置を稼働させることにより、連続発酵生産系が得られるかどうかを調べるため、表1に示す組成培地と表2に示す組成の追加培地を用い、この装置の連続発酵試験を行った。培地は、121℃の温度で20分間高圧蒸気滅菌し、追加培地は、121℃の温度で60分間高圧蒸気滅菌して用いた。分離膜エレメント部材には、ステンレスおよびポリサルホン樹脂の成形品を用いた。分離膜には、参考例5で作製したポリフッ化ビニリデン(PVDF)を主成分とする多孔性膜を用いた。この実施例4における運転条件は、特に断らない限り下記のとおりである。
・発酵反応槽容量:1.5(L)
・膜分離槽容量:0.5(L)
・使用分離膜:ポリフッ化ビニリデン濾過膜
・膜分離エレメント有効濾過面積:120平方cm
・温度調整:37(℃)
・発酵反応槽通気量:1.5(L/min)空気
・発酵反応槽攪拌速度:800(rpm)
・膜分離槽通気量:0.3(L/min)
・pH調整:2N H2SO4および14N アンモニア水によりpH7.0に調整
・培養液循環装置による循環液量:0.1(L/min)
・膜透過水量制御:発酵反応槽水頭差により流量を制御(水頭差は、2m以内で制御した)。
【0134】
微生物として大腸菌MC1061−イヌインターフェロンγ株を用い、表1に示す組成の培地を用い、追加培地として表2に示す組成の培地を用いた。また、グルコース濃度の測定には“グルコーステストワコーC”(登録商標)(和光純薬社製)、リン酸濃度測定には“ホスファC−テストワコー”(登録商標)(和光純薬社製)を用いた。
【0135】
まず、大腸菌MC1061-イヌインターフェロンγ株を、試験管で5mlの表1で示した組成の培地にテトラサイクリン(10μg/ml)を添加した培地で一晩振とう培養した(前々々培養)。得られた培養液を、新鮮なテトラサイクリン(10μg/ml)を添加した表1で示した組成の培地50mlに植菌し、500ml容坂口フラスコで24時間、37℃の温度で、振幅30cmで、180rpmの条件下で培養を行った(前々培養)。
前々培養液を、図2に示した連続発酵装置に2Lの表1で示した組成の培地に植菌し、発酵反応槽1を付属の攪拌機5によって800rpmで攪拌し、発酵反応槽1の通気量の調整、温度調整およびpH調整を行い培養を行った。前培養完了後直ちに、表2で示した組成の追加培地の連続供給を行い、連続発酵装置の発酵培養液量を2Lとなるように膜透過水量の制御を行いながら連続培養し、リン酸の濃度を低下させていき、連続発酵によるタンパク質の製造を行った。連続発酵試験を行うときの膜透過水量の制御は、水頭差制御装置3により、発酵反応槽水頭を最大2m以内、すなわち膜間差圧が20kPa以内となるように適宜水頭差を変化させることで行った。適宜、膜透過濾液中のリン酸濃度および残存グルコース濃度を測定した。培養の結果、菌体濃度OD600=30以上まで増殖し、高濃度培養が可能になった。
【0136】
また、適宜培養液をサンプリングしておき、リン酸濃度がどの程度低下したらタンパク質が発現しているかウエスタンブロッティング法で確認した。方法としては、各フラクションの菌体濃度OD600=10で統一し、その菌体液をサンプルとし、コニカイムノステインHRP(コニカ社製)を用い、添付のプロトコールに従ってイヌインターフェロンγの検出を行った。すなわち、SDS−PAGE電気泳動し、ブロッティングしたメンブレンをブロッキング溶液(5%スキムミルク・0.1%Tween20・PBS)中で4℃終夜ブロッキングした。メンブレンをTPBS(0.1%Tween20・PBS)で2回洗浄し、TPBSで1000倍希釈した抗イヌインターフェロンγ抗体で室温1時間処理した。メンブレンをTPBSで2回洗浄し、更にTPBSで5分間3回洗浄した。その後TPBSで10000倍希釈した後、HRPラベル抗ラビットIgG抗体(コスモバイオ社製)で室温1時間処理した。メンブレンをTPBSで2回洗浄し、更にTPBSで5分間3回洗浄した後、コニカイムノステインHRP(コニカ社製)の検出試薬(溶液A+溶液B+溶液C+溶液D)を加え、発色させた。その結果は、モレキュラーイメージャー(BioRad社製)を用いてシグナル強度を測定した。また、非形質転換大腸菌をネガティブコントロールのサンプルとして使用し、インタードック(東レ株式会社製:イヌインターフェロンγ)をポジティブコントロールのサンプルとして使用した。
【0137】
その結果、リン酸濃度1mM以下でイヌインターフェロンγが大量に発現していることが確認できた。また、上記に示す比較例1と比較して5〜10倍に産生していることを確認した。本連続発酵装置を用いることにより、菌体増殖は保ちながら、培地中の成分の濃度を低下させることで連続発酵によるタンパク質の製造が可能であることを確認することができた。また、比較例1および実施例1および実施例2の菌体量および発現タンパク質の結果を合わせて表3に、また、比較例1および実施例3および実施例4の菌体量および発現タンパク質の結果を合わせて表4に示した。
【0138】
【表3】
【0139】
【表4】
【0140】
これらの比較結果からわかるように、連続発酵装置を用いることにより、タンパク質の生産が大幅に向上することを明らかにすることができた。本発明によって開示された多孔性膜を組み込んだ連続発酵装置を用い、膜間差圧を制御することにより、発酵培養液を多孔性膜によって濾液と未濾過液に分離し、培地成分の濃度を低下させタンパク質を発現させるとともに、未濾過液を発酵培養液に戻す連続発酵方法を可能とし、微生物量を高く維持しながら、連続発酵によるタンパク質の製造が可能であることが明らかとなった。
【図面の簡単な説明】
【0141】
【図1】図1は、本発明で用いられる膜分離型連続発酵装置の一つの実施の形態を説明するための概要側面図である。
【図2】図2は、本発明で用いられる他の膜分離型連続発酵装置の一つの実施の形態を説明するための概要側面図である。
【図3】図3は、本発明で用いられる分離膜エレメントの一つの実施の形態を説明するための概要斜視図である。
【図4】図4は、本発明で用いられる他の分離膜エレメントの例を説明するための断面説明図である。
【符号の説明】
【0142】
1 発酵反応槽
2 分離膜エレメント
3 水頭差制御装置
4 気体供給装置
5 攪拌機
6 レベルセンサ
7 培地供給ポンプ
8 pH調整溶液供給ポンプ
9 pHセンサ・制御装置
10 温度調節器
11 発酵培養液循環ポンプ
12 膜分離槽
13 支持板
14 流路材
15 分離膜
16 凹部
17 集水パイプ
18 分離膜束
19 上部樹脂封止層
20 下部樹脂封止層
21 支持フレーム
22 集水パイプ
【特許請求の範囲】
【請求項1】
タンパク質の生産能力を有する微生物の発酵培養液を分離膜で濾過し、未濾過液を前記の発酵培養液に保持または還流し、その微生物の培地を前記の発酵培養液に追加し、かつ、微生物菌体内からタンパク質を回収する連続発酵によるタンパク質の製造方法であって、前記の分離膜として平均細孔径が0.01μm以上1μm未満の細孔を有する多孔性膜を用い、その膜間差圧を0.1から20kPaの範囲にして濾過処理することを特徴とする連続発酵によるタンパク質の製造方法。
【請求項2】
多孔性膜の純水透過係数が、2×10−9m3/m2/s/pa以上6×10−7m3/m2/s/pa以下である請求項1記載の連続発酵によるタンパク質の製造方法。
【請求項3】
多孔性膜の平均細孔径が0.01μm以上0.2μm未満であり、かつ、該平均細孔径の標準偏差が0.1μm以下である請求項1または2記載の連続発酵によるタンパク質の製造方法。
【請求項4】
多孔性膜の膜表面粗さが0.1μm以下である請求項1から3のいずれかに記載の連続発酵によるタンパク質の製造方法。
【請求項5】
多孔性膜が多孔性樹脂層を含む多孔性膜である請求項1から4のいずれかに記載の連続発酵によるタンパク質の製造方法。
【請求項6】
多孔性膜の素材がポリフッ化ビニリデン系樹脂である請求項1から5のいずれかに記載の連続発酵によるタンパク質の製造方法。
【請求項7】
微生物の発酵原料が糖類を含む請求項1から6のいずれかに記載の連続発酵によるタンパク質の製造方法。
【請求項8】
タンパク質をコードする遺伝子がリン酸プロモーターの下流に連結され、かつ培地に含まれる無機リン酸濃度が1mM以下である請求項1から7のいずれかに記載の連続発酵によるタンパク質の製造方法。
【請求項9】
タンパク質がイヌインターフェロンγである請求項1から8のいずれかに記載の連続発酵によるタンパク質の製造方法。
【請求項10】
微生物が大腸菌である請求項1から9のいずれかに記載の連続発酵によるタンパク質の製造方法。
【請求項1】
タンパク質の生産能力を有する微生物の発酵培養液を分離膜で濾過し、未濾過液を前記の発酵培養液に保持または還流し、その微生物の培地を前記の発酵培養液に追加し、かつ、微生物菌体内からタンパク質を回収する連続発酵によるタンパク質の製造方法であって、前記の分離膜として平均細孔径が0.01μm以上1μm未満の細孔を有する多孔性膜を用い、その膜間差圧を0.1から20kPaの範囲にして濾過処理することを特徴とする連続発酵によるタンパク質の製造方法。
【請求項2】
多孔性膜の純水透過係数が、2×10−9m3/m2/s/pa以上6×10−7m3/m2/s/pa以下である請求項1記載の連続発酵によるタンパク質の製造方法。
【請求項3】
多孔性膜の平均細孔径が0.01μm以上0.2μm未満であり、かつ、該平均細孔径の標準偏差が0.1μm以下である請求項1または2記載の連続発酵によるタンパク質の製造方法。
【請求項4】
多孔性膜の膜表面粗さが0.1μm以下である請求項1から3のいずれかに記載の連続発酵によるタンパク質の製造方法。
【請求項5】
多孔性膜が多孔性樹脂層を含む多孔性膜である請求項1から4のいずれかに記載の連続発酵によるタンパク質の製造方法。
【請求項6】
多孔性膜の素材がポリフッ化ビニリデン系樹脂である請求項1から5のいずれかに記載の連続発酵によるタンパク質の製造方法。
【請求項7】
微生物の発酵原料が糖類を含む請求項1から6のいずれかに記載の連続発酵によるタンパク質の製造方法。
【請求項8】
タンパク質をコードする遺伝子がリン酸プロモーターの下流に連結され、かつ培地に含まれる無機リン酸濃度が1mM以下である請求項1から7のいずれかに記載の連続発酵によるタンパク質の製造方法。
【請求項9】
タンパク質がイヌインターフェロンγである請求項1から8のいずれかに記載の連続発酵によるタンパク質の製造方法。
【請求項10】
微生物が大腸菌である請求項1から9のいずれかに記載の連続発酵によるタンパク質の製造方法。
【図1】

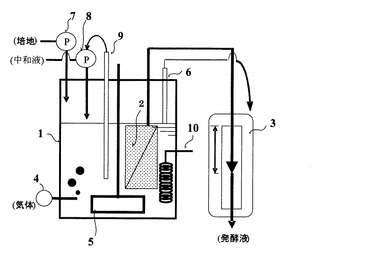
【図2】
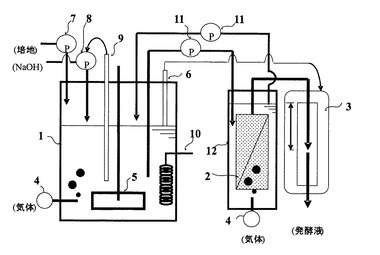
【図3】
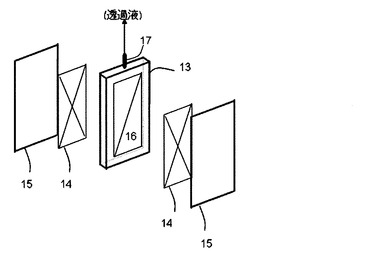
【図4】
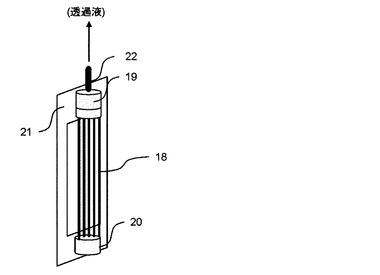
【図2】
【図3】
【図4】
【公開番号】特開2009−39073(P2009−39073A)
【公開日】平成21年2月26日(2009.2.26)
【国際特許分類】
【出願番号】特願2007−209935(P2007−209935)
【出願日】平成19年8月10日(2007.8.10)
【出願人】(000003159)東レ株式会社 (7,677)
【Fターム(参考)】
【公開日】平成21年2月26日(2009.2.26)
【国際特許分類】
【出願日】平成19年8月10日(2007.8.10)
【出願人】(000003159)東レ株式会社 (7,677)
【Fターム(参考)】
[ Back to top ]