
運動障害治療剤
【課題】 少なくとも1種のアデノシンA2A受容体アンタゴニストを投与することを特徴とする運動障害を患っている患者を治療する薬剤を提供すること。
【解決手段】 パーキンソン病の治療においてL-ドーパ療法を受けている患者におけるL-ドーパの副作用を軽減する方法、L-ドーパ治療を1種以上のアデノシンA2A受容体アンタゴニストの有効量と組み合わせることにより、臨床上有効な用量より少ないL-ドーパを用いて、パーキンソン病患者を治療するための方法および組成物、少なくとも1種のアデノシンA2A受容体アンタゴニスト、L-ドーパおよびドーパミンアゴニストならびに/またはCOMT阻害剤および/もしくはMAO阻害剤を同時に投与することによるパーキンソン病の有効な治療方法等を提供する。
【解決手段】 パーキンソン病の治療においてL-ドーパ療法を受けている患者におけるL-ドーパの副作用を軽減する方法、L-ドーパ治療を1種以上のアデノシンA2A受容体アンタゴニストの有効量と組み合わせることにより、臨床上有効な用量より少ないL-ドーパを用いて、パーキンソン病患者を治療するための方法および組成物、少なくとも1種のアデノシンA2A受容体アンタゴニスト、L-ドーパおよびドーパミンアゴニストならびに/またはCOMT阻害剤および/もしくはMAO阻害剤を同時に投与することによるパーキンソン病の有効な治療方法等を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、少なくとも1種のアデノシンA2A受容体アンタゴニストを投与することを特徴とする運動障害を患っている患者を治療する方法に関する。
【背景技術】
【0002】
運動障害とは、運動の不足もしくは欠如(例えば、パーキンソン病等)または過剰運動(例えば、ジストニア(dystonia)、ジスキネジア(dyskinesia)、振戦(tremor)、舞踏病(chorea)、バリズム(ballism)、アカシジア(akathisia)、アテトーシス(athetosis)、運動緩慢(bradykinesia)、すくみ(freezing)、硬直(rigidity)、姿勢不安定(postural instability)、ミオクローヌス(myoclonus)およびチック(tics)もしくはトゥレット症候群(Tourette syndrome)等)のいずれかを特徴とする神経学上の状態である。ワッツ(Watts)およびウィリアム(William)編(1997)ならびにシャルマン(Shulman)およびウェイナー(Weiner)(1997)参照。
【0003】
パーキンソン病および運動合併症
パーキンソン病(振戦麻痺)は、振戦ならびに歩行、運動および協調の困難を特徴とする脳の疾患である。この疾病は筋運動を支配する脳の一部の損傷と関連している。
パーキンソン病は最初、1817年に英国でジェームス・パーキンソン(James Parkinson)によって記載された。この疾病はおよそ1000人のうち2人に影響を及ぼし、ほとんどの場合50歳以降に発症する。症状はまず、平均して、約60歳で表れ、パーキンソン病の症状の重篤度は時間とともに悪化する傾向がある。男性、女性双方に影響を及ぼし、高齢者の最も多く見られる神経疾患の1つである。「パーキンソン症候群(parkinsonism)」という用語は、パーキンソン病において見られる運動の変化の種類の組み合わせを含むいずれかの状態を指す。パーキンソン症候群は遺伝性であることもあるし、その他の疾患によってまたは外部要因によって引き起こされることもある(続発性パーキンソン症候群)。
【0004】
米国では、約100万人がパーキンソン病を患っていると考えられており、毎年、約50,000件の新しい症例が報告されている。通常、症状は高齢期に表れるので、これらの数字は、今後数十年にわたり、集団の平均年齢が上がるにつれ高くなると予測される。この疾患は70代および80代の人々の間で最もよく見られ、女性よりも男性でわずかにより多く見られるようである。
【0005】
黒質緻密部および腹側被蓋野のドーパミン作動性ニューロンは、それぞれ、運動の調節および認知において重大な役割を果たしている。数通りの証拠から、黒質中のドーパミン作動性細胞(すなわち、ドーパミン産生細胞)の変性がパーキンソン病の症状をもたらすことが示唆されている。黒質のその領域に集中しているドーパミン作動性細胞は、体内で最も速く加齢する細胞である。ドーパミン作動性細胞が崩壊すると、運動に対する制御が損なわれ、パーキンソン病が発症する。
【0006】
通常、パーキンソン病の最初の症状は、特に身体を静止しているときの肢の振戦(震えまたは振動)である。振戦は半身で始まることが多く、片側の手において頻繁に起きる。その他のよく起こる症状としては、例えばゆっくりとした動き(運動緩慢)、運動不能(アキネジア)、体肢の硬直、引きずり歩行、前かがみの姿勢等のその他の運動障害が挙げられる。パーキンソン病患者は表情が乏しくなり、静かな声で話すことが多い。この疾病は、鬱病、不安、人格変化、認知障害、痴呆、睡眠障害、言語障害または性的不全といった二次的な症状を引き起こすことがある。パーキンソン病の治療法は知られていない。治療はそれらの症状の制御を目的としている。薬物療法では主として神経伝達物質間の不均衡を制御することによってそれらの症状を制御する。ほとんどのパーキンソン病の初期患者は、ドーパミン補充療法による対症療法に対してよく応答するが、疾病が進行するにつれ能力障害が増加する。
【0007】
用いる薬剤、用量および投与間隔は、症例に応じて変わる。症状変化に合わせて、用いる薬剤の組み合わせを調節する必要があり得る。多くの薬剤は重大な副作用を引き起こすことがあるので、医療提供者によるモニタリングとフォローアップが重要である。
現在利用できるパーキンソン病用薬剤は、一般に、数年間は十分な対症制御をもたらすものの、多くの患者が、運動変動およびジスキネジアを発症し、これが臨床効果を鈍らせる。ラスコール(Rascol)ら(2000)およびパーキンソン研究グループ(2000)。これが起こると、ドーパミン作動性療法を増やすことはジスキネジアを悪化させる恐れがあり、ドーパミン作動性療法を減らすことは運動機能を悪化させ、オフ時間を増加させる恐れがある。この問題を踏まえて、非ドーパミン作動性神経伝達物質系の治療的処置の可能性が注目されることとなった。
【0008】
ほとんどのパーキンソン病の症状は、ドーパミンの不足から生じ、ほとんどの抗パーキンソン薬はドーパミンを元の状態に戻すかドーパミンの作用を模倣するものである。しかし、これらの薬剤はドーパミンを恒久的に元に戻すものではなく、ドーパミンの作用を正確に模倣するものでもない。黒質にドーパミン細胞がないことがパーキンソン病の主な特徴ではあるが、非ドーパミン神経細胞も喪失している。さらに、ドーパミン応答細胞は黒質だけでなく他の脳領域にも存在する。したがって、パーキンソン病において有効な薬剤は、これらの細胞を刺激することにより、例えば悪心、幻覚、錯乱等の副作用を引き起こし得る。
【0009】
L-ドーパは1967年に報告され、以来最も有効な抗パーキンソン薬となっている。L-ドーパの有効性が最も高い症状としては、運動緩慢、硬直、静止時振戦、歩行困難および小書症が挙げられる。L-ドーパの有効性があまり望めない症状としては、姿勢不安定、動作時振戦および嚥下困難が挙げられる。L-ドーパは痴呆を悪化させる可能性がある。L-ドーパは、パーキンソン病において、強くかつ急速な治療上の効果をもたらすが、最終的には、例えばウェアリング・オフ現象、オン・オフ変動、ジスキネジア等の運動合併症をはじめとするドーパミンによって引き起こされる重篤で好ましくない反応が現れる。マースデン(Marsden)ら(1982)。運動合併症は、通常、一度発症すると、L-ドーパまたは他のドーパミン作動性薬剤による処置では制御不能である。
【0010】
パーキンソン病の初期にはL-ドーパを1日3回服用する。脳内でのピーク濃度は投与後1〜2時間で生じる。薬剤の半減期は短い(0.5〜1時間)が、脳内に残存しているドーパミン細胞がドーパミンを貯蔵し、数時間の間その活性を維持するには十分である。パーキンソン病が進行すると、より多くのドーパミン細胞が死滅し、残存する細胞ではその効果を維持するのに十分なドーパミンを貯蔵できなくなり、各投与量での作用持続時間が減少し、患者に対してより高用量なまたはより頻繁な投与が必要となる。2〜5年後には、50〜75%もの患者で、L-ドーパに対する反応、つまりオン/オフ期間に変動が起こる。変動に伴い、患者はジスキネジアを発症する。通常、ジスキネジアはL-ドーパのピーク作用時に生じるが、薬剤の効果が切れるときまたはストレスが多いときにもまた生じる場合がある。変動およびジスキネジアは患者の生活に深刻な影響を及ぼし得る。L-ドーパが連続投与されれば(静脈内ポンプによって)、オン/オフ作用はなくなり、ジスキネジアも減少する。しかし、L-ドーパを静脈内投与することはできない。
【0011】
L-ドーパを単独で服用すると、その一部がドーパ−デカルボキシラーゼによって脳外でドーパミンに変換される。このように生じたドーパミンは脳に入ることができず、例えば悪心、嘔吐、食欲の喪失等の副作用を引き起こす。従って、L-ドーパはカルビドパまたはベンセラジド(benserazide)と組み合わせることが多い。カルビドパは脳外のドーパ−デカルボキシラーゼをブロックすることにより、悪心、嘔吐および食欲の喪失を引き起こすことなく、より多くのL-ドーパが脳に入ることができるようにする。アタメット(Atamet)またはシネメット(Sinemet)はカルビドパとL-ドーパの両方を含有する錠剤である。カルビドパとの組み合わせでは、L-ドーパの半減期は1.2〜2.3時間である。
【0012】
その発見から30年、L-ドーパは依然としてパーキンソン病の最良の治療である。この疾病の初期段階では、患者は通常L-ドーパに対する良好な反応を享受するが、疾病が進行すると、L-ドーパはあまり有用でなくなる傾向がある。これはL-ドーパの効力の喪失によるものではなく、例えばエンド・オブ・ドーズ(end-of-dose)での悪化または「ウェアリング・オフ(wearing-off)」、「オン/オフ変動」、ジスキネジア等の運動応答における逆変動のような運動合併症の発症によるものである。オン/オフ変動とは、薬剤治療における効果(「オン」状態、患者にパーキンソン病の症状が比較的ない期間)が、突然、容認できないほどに失われ、パーキンソン状態(「オフ」状態)を発現することである。ウェアリング・オフ現象はL-ドーパが有効である期間の減少であり、「オフ」状態が徐々に再発することを特徴とし、「オン」状態が短くなる。ジスキネジアは、舞踏病(多動性の、目的のないダンスのような動き)とジストニア(持続性の、異常な筋収縮)に大別することができる。1974年に、ディボアザン(Duvoisin)が最初にこれらの異常な不随意運動に着目し、パーキンソン病の患者の半数以上が治療の6ヶ月以内にジスキネジアを発症するということを見出した。治療期間が長くなるにつれ、ジスキネジアの頻度および重症度も増加する。神経保護に有用であると思われる薬剤のパーキンソン病における効果の可能性について影響を与えた研究−DATATOP試験−では、平均20.5ヶ月間L-ドーパの治療を受けた患者の20〜30%でL-ドーパ誘発性ジスキネジアが観察された。最終的に、L-ドーパ治療を受けた患者のほとんどがジスキネジアを経験し、患者の最大80%で、治療の5年以内にジスキネジアを発症した。パーキンソン研究グループ(1966)およびラスコール(Rascol)ら(2000)。治療に関連したジスキネジアは、単にL-ドーパのみの問題ではなく、ドーパミン受容体アゴニストも同様にジスキネジアを誘発し得る。このように、共通の用語「L-ドーパ誘発性ジスキネジア」は、一般用語でドーパミン治療に関連したジスキネジアを記載するために用いられることもある。ほとんどのジスキネジアは、レボドパまたは他のドーパミン受容体アゴニストが、被殻中の過敏性ドーパミン受容体に対して十分である一定の脳内濃度であるときに生じる(ピーク・ドーズ・ジスキネジア(peak-dose-dyskinesia))。しかし、ジスキネジアはまた、ドーパミン濃度が低い際(オフ・ジストニア)、またはドーパミン濃度が増減する状態(二相性ジスキネジア)でも生じる。また、例えばミオクローヌス、アカシジア等の他の運動障害もL-ドーパ誘発性ジスキネジアの範疇の構成要素であり得る。
【0013】
パーキンソン病におけるL-ドーパによる運動合併症の生物学的機序は、未だ少しも解明されていない。その生物学的機序は、進行性の疾病であることおよび黒質のニューロンが持続的に喪失されることだけではなく、ドーパミン受容体の感受性およびタンパク質のそれらの下流発現の変化、ならびにその一連の発現がL-ドーパまたはドーパミンアゴニストの用量および投与方法と少なくともある程度関係している遺伝子に関与している可能性が示唆されている。例えばグルタメート作動性神経伝達、GABA作動性神経伝達、オピオイドペプチド作動性伝達等の非ドーパミン系における変化もまた、パーキンソン病におけるL-ドーパによる運動合併症の根底にあるニューロンのメカニズムに関与している可能性がある。ベザート(Bezard)ら(2001)。特に、ドーパミン作動性薬剤の短い血漿半減期とその結果としての短い作用期間およびドーパミン作動性薬剤によるドーパミン受容体の拍動性刺激が、運動変動およびピーク・ドーズ・ジスキネジアと関連していると思われる。これらの事象はすべてが相まって、大脳基底核と皮質の間に信号を伝える発火パターンの変化をもたらす。
【0014】
当初、変動のある患者にL-ドーパの補助的療法として導入されたドーパミンアゴニストは、今では、初期の患者における単独療法としてますます提唱されるようになっている。しかし、通常、ドーパミンアゴニストの抗パーキンソン病作用はL-ドーパのものよりも少なく、2〜4年後にはその効果が減弱する。より強力な治療が必要な場合には、低用量のL-ドーパをアゴニストに「追加する」ことができる。別の戦略として、最初からアゴニストを低用量のL-ドーパと組み合わせることもある。両戦略ともL-ドーパと同程度に有効であり、運動変動およびジスキネジアの危険性を大きく減少させるという効果を有するとされている。しかし、これらの主張は少数の試験的研究に基づいたものであり、そのすべてには方法論的な欠点がある。
【0015】
さらに、ドーパミン受容体アゴニストもまたジスキネジアを誘発し得る。ドーパミンアゴニストもまた、先にL-ドーパに曝されていたパーキンソン病の動物でジスキネジアを誘発する。神経精神医学的副作用、特に、幻覚および精神病により、ドーパミンアゴニストの使用が制限されることも多い。ドーパミンアゴニストの補助的使用によってもたらされる有力な利点があるにもかかわらず、このようにL-ドーパによる運動合併症を制御するのは極めて困難であるか、不可能でさえあり得る。オラノー(Olanow)、ワッツ(Watts)およびコラー(Koller)編(2001)参照。最後に、ドーパミンアゴニストは、進行したパーキンソン病および重度の運動変動およびジスキネジアの患者においてL-ドーパの代わりとして単独療法で用いられることがある。
【0016】
より最近では、例えばトルカポン(tolcapone)、エンタカポン(entacapone)等のカテコールアミン−O−メチルトランスフェラーゼ(COMT)阻害剤が、L-ドーパの補助的療法として提唱されている。これらの化合物は、Cmaxを大きく高めることなく、L-ドーパの血漿半減期を延長する。したがって、これらはウェアリング・オフの期間を減少させるが、ピーク・ドーズ・ジスキネジアをはじめとするピーク・ドーズの副作用の強度を高める傾向がある。トルカポンは少数の患者では重篤な肝臓毒性を誘発するようである。
【0017】
例えばトリヘキシフェニジル(アーテン(artane))、ビペリジン(コゲンチン(cogentin))等の抗コリン作用薬は、脳においてアセチルコリンの作用をブロックする。この結果、例えば流延、振戦等の症状が軽度〜中程度改善され得る。65歳を超える患者では、抗コリン作用薬を用いて治療した場合、例えば口渇、かすみ目、便秘、錯乱、幻覚等の副作用が起こりやすい。
【0018】
ジストニア
ジストニアという用語は、持続的に異常な姿勢をもたらす持続性の筋収縮を特徴とする運動障害を指す。この定義に基づいて、いくつかのジストニア症候群があり、これらは、全身性(すべての身体部分に影響を及ぼす)、髄節性(隣接する身体部分に影響を及ぼす)または局所性(単一の身体部分に限定された)のようなその臨床上の特徴にしたがって細分化することができる。局所性ジストニアとしては、痙攣性斜頚(spasmodic torticollis)、眼瞼痙攣(blepharospasm)、半側顔面痙攣(hemifacial spasm)、開口ジストニア(oromandibular dystonia)、痙攣性発声障害(spasmodic dysphonia)およびジストニア性書痙(dystonic writer's cramp)が挙げられる。
【0019】
さまざまな程度のジストニアがある。比較的正常なライフスタイルを維持できる人もあれば、持続的に障害があり、フルタイムの介護を必要とすることが多い人もいる。
症状は局所的であることもあれば、身体のある領域、例えば、首または腕または脚に限定されていることもある。様々な種類の局所性ジストニアがある。眼瞼痙攣は瞼の運動を制御する筋肉の不随意収縮を特徴とする。症状は、断続的なもの、無痛なもの、休みなく続くまでに増えた瞬き、有痛なもの、機能上の盲目をもたらす閉眼まで様々であり得る。痙攣性斜頚としても知られる頸部ジストニア(CD)の患者では、頭頸部の筋痙攣は有痛であり、首をねじれさせることもある。これらの有痛の痙攣は断続的であることもあるし、休みなく続くこともある。開口ジストニアおよび舌ジストニアは、口の開閉を引き起こす顔下部の強力な収縮を特徴とする。咀嚼や異常な舌の動きも起こり得る。喉頭ジストニアとしても知られる痙攣性発声障害(SD)では、発声器(喉頭)の筋が影響を受ける。SDは声帯を開けることか閉じることのいずれかの困難を特徴とし、これによって声が緊張した、しわがれた、のどの詰まったようなまたはささやくような音質のいずれかになる。体肢ジストニアでは、上肢、手、下肢または足の1以上の筋の不随意収縮がある。これらの種類の局所性ジストニアとしては、書痙および他の職業性ジストニアが挙げられる。
【0020】
分節性であるか、または例えば頭と首、上肢と躯幹等の身体の2つの隣接する領域に関与する症状を有する患者もいる。他の患者では、症状が多巣性であったり、例えば両腕、上肢と下肢等の互いに隣接していない身体の2つの領域に現れたりすることもある。全身性ジストニアでは、症状は上肢または下肢において始まり、進行してより広範なものとなる。最終的に躯幹および身体の残りの部分にまで及ぶ。
【0021】
原発性または特発性ジストニアのほとんどの症例は、遺伝性であり、遺伝子の欠陥の結果として起こると考えられている。これらの患者では、ジストニアは単一の症状として起こり、潜在的疾患を伴わない。例えば、早発性原発性ジストニアのほとんどの場合が、DYT-1遺伝子における突然変異によるものである。この疾病遺伝子の結果として起こる早発性ジストニアは、最も多く見られかつ重症型の遺伝性ジストニアである。他の遺伝的原因が原発性ジストニアを引き起こすことは稀である。
【0022】
ジストニアが関与する疾病としては、遺伝性痙性対麻痺(hereditary spastic paraplegia (HSP))、つまり両下肢の進行性の脱力およびこわばりを特徴とする脊髄の遺伝性変性疾患の一群;ハンチントン病(Huntington's disease (HD))、つまり情緒的、行動的および精神医学的異常ならびに運動異常の発症を特徴とする遺伝性進行性神経変性疾患;多系統萎縮症(multiple system atrophy (MSA))、つまり運動、血圧およびその他の身体機能に影響を及ぼす症状の組み合わせを特徴とする神経変性疾患;病的ミオクローヌス(pathologic myoclonus);進行性核上性麻痺(progressive supranuclear palsy);レストレスレッグズ症候群(restless legs syndrome);レット症候群(Rett syndrome);痙縮(spasticity);シデナム舞踏病(Sydenham's chorea);トゥレット症候群(Tourette syndrome)およびウィルソン病(Wilson disease)が挙げられる。
【0023】
ジストニアは、例えばウィルソン病、多発性硬化症、脳卒中等の別の潜在的疾病過程;例えば自動車事故での損傷または誕生時の酸欠等の脳の外傷;または薬物療法の副作用として起こることもある。この種のジストニアは続発性または症候性ジストニアと呼ばれる。成人で、最もよく見られる続発性ジストニアの種類は、遅発性ジストニア(tardive dystonia)であり、これは特定の神経遮断薬または抗精神病薬(精神疾患の治療に用いられる)の使用の結果として起こる。これらの薬剤としては、ハロペリドール(ハルドール(Haldol)(登録商標))またはクロルプロマジン(トラジン(Thorazine)(登録商標))が挙げられる。中枢神経のドーパミン受容体をブロックする他の薬剤もまた遅発性ジストニアを引き起こし得る。ほとんどの患者で、薬剤に曝露され続けた後、症状はいずれそのうち生じる。表1にジストニアを引き起こし得る薬剤のリストを示す。
【0024】
【表1】

【0025】
ジストニアを治療するためには、いくつかの選択肢を利用できる。薬剤は単独で用いてもよいし、組み合わせて用いてもよい。さらに、その他の治療形態と組み合わせてもよい。現在使用されている薬剤としては、ボツリヌス毒素(BTX)、ベンゾジアゼピン、バクロフェン、抗コリン作用薬およびドーパミン遮断薬/ドーパミン枯渇剤が挙げられる。外科的処置もまた有用であり、これには視床手術、淡蒼球手術、深脳刺激、心筋切除術(筋切開)、ラミセクトミー(ramisectomy)、神経根切断術および末梢神経除去が含まれる。
【0026】
遅発性ジスキネジアおよびその他の錐体外路症候群
神経系の錐体外路系は、大脳基底核を中心としており、錐体路を通って、一般に視床への入力により、運動制御に影響を及ぼす。錐体外路系が障害を受けると、運動制御が影響を受け、患者は錐体外路症候群にさいなまれる。これらは、振戦、舞踏病、アテトーシスおよびジストニアを含む神経学的な影響の組み合わせである。これは、神経遮断薬の一般的な副作用である。これらの反応を引き起こすことが知られているその他の薬剤としては、ハロペリドール、モリンドン、ペルフェナジンおよびアミノトリプチリン、ロキサピン、ピモジドおよびまれにではあるがベンゾジアゼピンが挙げられる。
【0027】
遅発性ジスキネジアは、不随意神経運動障害である。発症の型式に応じて、鑑別診断には、シデナム舞踏病(Sydenham's chorea)、ハンチントン舞踏病、先天性捻転ジストニア(congenital torsion dystonia)、病的興奮、および統合失調症の常同的行為もしくは癖が含まれ得る。米国神経精神薬理学会(American College of Neuropsychopharmacology)−FDAタスクフォース(1973)。遅発性ジスキネジアは、特定の精神状態または胃腸の状態を治療するために処方された神経遮断薬の使用の結果として引き起こされる。これらの薬剤の長期服用によって、線条体に生化学的異常が生じることがある。遅発性ジストニアは、遅発性ジスキネジアのより重度の形であると考えられている。
【0028】
その他の密接に関連する処置不能の神経疾患は、現在、遅発性ジスキネジアの変種として認識されている。遅発性アカシジアは、精神的緊張および不安という精神的な苦痛ならびに体を動かすことへの強迫的な衝動を伴う。極端な場合、患者は内面的な苦痛を受けて、もはや静かに座ったままでいることができない。遅発性ジストニアは、顔、首および肩の頻繁な筋痙攣を伴い、あまりにも頻繁であるため、外観を損ない、身体に障害を引き起こし、苦痛を伴うことがある。
【0029】
遅発性ジスキネジアの治療は、満足するまでに至っていない。抗精神病薬の廃止がしばしば提唱されるが(バルデサリニ(Baldessarini)(1990))、より重度な形の運動障害に至ってしまうことが多くある。種々の医薬が試みられ、いくつかのものでは成功が報告されている。この分野における初期の研究者たちは、ドーパミンレベルを枯渇させることが知られている化合物であるレセルピン(セルパシル(Serpasil)(登録商標))に注目した。レセルピンとα−メチルドーパ(アルドメット(Aldomet)(登録商標))は、長期にわたる遅発性ジスキネジアの治療において、両化合物とも総合的症状の軽減においてプラセボよりも統計的に有効であることを示した。ファン(Huang)ら(1981)。しかしながら、別の研究では、例えばα−メチルドーパ等のカテコールアミン合成ブロッカーが遅発性ジスキネジアに対して有効性を示さなかったことが示された。チロシンヒドロキシラーゼ、ドーパミンおよびノルエピネフィリンの合成の律速段階を阻害する試験段階の薬剤であるAMPTはジスキネジアを一部軽減した。
【0030】
これまで、遅発性ジスキネジアは、神経遮断薬の用量を増加することによって治療されることが多かった。これによってまず、遅発性ジスキネジアの病態生理が治療されるが、さらなる除神経およびその後の過敏症によって病因を悪化させ得る。そのため、最初は動きが減少するか、または見られなくなるが、その後に再発する。外観を損なうような遅発性ジスキネジアの患者が神経遮断薬による治療の代替治療を必要とするような特定の状況では、非定型神経遮断薬、クロザピンの使用が有用であり得る。
【0031】
リチウムは、CNSに対するその他の作用も有するばかりでなくモノアミンのシナプス前放出を妨げる。2つの研究で、リチウムによる遅発性ジスキネジアの軽度の改善が報告されているが、その2つ以外では、改善も悪化も報告されていない。テッパー(Tepper)およびハス(Haas)(1979)。
経口用ピモジドは、運動度の改善をもたらした。クラベリア(Claveria)ら(1975)。部分セロトニン受容体アゴニストであるブスピロン(ブスパー(BuSpar)(登録商標))もまたこの状態の治療に有用であり得る。モス(Moss)ら(1993)。ラットでは、ブスピロンが、長期にわたる神経遮断薬の投与によって誘発されたDA受容体弱感受性を逆転させるが、この作用はD2受容体で部分アゴニスト作用によりヒトでも起こり得るものである。報告では、遅発性ジスキネジアをレセルピン、テトラベナジン、メトクロプラミド、三環系抗鬱薬、ベンズトロピン、フェニトインおよびアンフェタミンと関連づけている。
【0032】
神経遮断薬以外で、ジスキネジアを定期的に引き起こす薬剤はL-ドーパおよび他のドーパミン作動性薬剤であり、パーキンソン病患者はパーキンソン病用のこれらの薬剤を服用している。実際には、L-ドーパは、神経遮断薬誘発性の遅発性ジスキネジアを改善することができる。
遅発性ジスキネジアには一般に認められた治療がない。ケーシー(Casey)(1999)。差障りのある抗精神病薬の投与を中止するか、または患者への投与を非定型抗精神病薬(リスペリドンはおそらく除かれる)へと切り替えることで運動障害が緩和されることがある。遅発性ジスキネジアの治療は、近年、再検討されてきた。イーガン(Egan)ら(1997)。ほとんどの薬剤治療の戦略は、ドーパミン活性を低下させることまたはCNSコリン作
動性効果を高めることに向けられている。遅発性ジスキネジアの病因が長期のドーパミン作動性受容体部位の遮断に関連している場合、および病態生理が除神経による過敏症に関連している場合には、理論的にはこの筋道を遮断する薬剤が潜在的に有益である可能性がある。
【0033】
神経遮断薬誘発性の遅発性ジスキネジアの治療には、数多くの薬物が試されてきた。患者の症例数、研究設計、および使用される薬剤の用量には差があるため、個々の薬剤の結果は相反している。バルデサリニ(Baldessarini)およびタルシー(Tarsy)(1978);ならびにクローアンズ(Klawans)ら(1980)。
例えばレセルピンおよびテトラベナジン等のアミン枯渇剤は、ドーパミン、ノルエピネフリンおよびセロトニンのシナプス前ニューロン蓄積媒介物への再取り込みをブロックし、それによって、脳におけるこれらの物質を枯渇させることによって作用する。これらの薬剤を用いた研究では、遅発性ジスキネジアの改善が示されたが、副作用によってその使用が制限されており、それらの研究は短期間の場合についてのものである。神経遮断薬を用いて報告されたように、短期間の鎮静が起こり得る。
【0034】
いくつかのコリン作動性アゴニストが遅発性ジスキネジアの患者に投与されてきた。経口的に生物学的利用可能なアセチルコリンの前駆体である塩化コリンおよびホスファチジルコリン(レシチン)は、短期間の研究において有用であると報告されている。デアノール アセトアミノベンゾエートは、当初、遅発性ジスキネジアの治療に有効であると報告されたが、他の研究では、これらの知見は確認されなかった。ゲレンバーグ(Gelenberg)ら(1990)。
【0035】
中枢GABAの機構を強化すると考えられている薬物で遅発性ジスキネジアを治療する試みがいくつかなされてきた。サカー(Thaker)ら(1990)。10人の遅発性ジスキネジア患者に関する6ヶ月を超える研究では、ベンズトロピン2mg IVにより7患者でジスキネジア的な動きが増え、残る3患者ではそれが減少した。ムーア(Moore)およびバウアーズ(Bowers)(1980)。予備的報告では、30〜60mg/日の用量のβ−アドレナリン遮断薬であるプロプラノロール(インデラル(登録商標))での治療により、1〜10日以内に4患者で遅発性ジスキネジアの著しい軽減がもたらされた。ウィルバー(Wilbur)およびクリック(Kulik)(1980)。
【0036】
いくつかの研究では、遅発性ジスキネジアのビタミンEでの治療の有効性を調べている。アドラー(Adler)ら(1999);レーア(Lohr)およびカリギウリ(Caligiuri)(1996);レーア(Lohr)ら(1988);エルカシェフ(Elkashef)ら(1990);シュリキ(Shriqui)ら(1992);イーガン(Egan)ら(1992);アドラー(Adler)ら(1993a);アドラー(Adler)ら(1993b);ゴールドバーグ(Goldberg)(1996);マクレジー(McCreadie)ら(1994);ダビリ(Dabiri)ら(1993);ビショット(Bischot)ら(1993);アフタル(Akhtar)ら(1993);およびダビリ(Dabiri)ら(1994)。
【0037】
これまで、遅発性ジスキネジアは、大部分の患者において永続的なものまたは回復不可能であるものと考えられていた。しかしながら、必ずしもそうであるとは限らない。より早期に遅発性ジスキネジアが診断されて神経遮断薬の投与が中止されるほど、疾患の回復の予後が良くなる。若年成人では、遅発性ジスキネジアが早期に投薬を中止した後、数週間内に消失する。ウルブランド(Uhrbrand)およびファルバイ(Faurbye)(1960);イトウ(Itoh)ら(1981);ドリーセンス(Driesens)(1988);およびガルドス(Gardos)ら(1994)。
【0038】
表2は、遅発性ジスキネジアの治療に用いられてきた種々の薬剤をまとめたものである。
【0039】
【表2】
【0040】
神経遮断薬の錐体外路系への作用によって引き起こされるその他の運動症候群としては、薬剤誘発性パーキンソン症候群、アカシジア、ジストニア、注視発症(oculogyric crisis)および後弓反張(opisthotonus)が挙げられる。アカシジアは、運動的に落ち着きのないことを特徴とする状態であり、その状態は不安を感じる程度から、静かに横たわることまたは座ることができない、または眠ることができない程度までさまざまである。考えられる原因としては、例えばフェノチアジン等の神経遮断薬による中毒反応が挙げられる。注視発症は、発作性の眼の不随意性上方偏視である。眼瞼は縮まっている場合が多い。発作は数分間〜数時間続く。フェノチアジン、ハロペリドールおよびメトクロプラミドに敏感な患者に起こる可能性がある。後弓反張は、頭、首および脊椎が後方に反り返る形の痙攣である。
【0041】
アデノシンA2A受容体
アデノシンは、4種の主要な受容体サブタイプ、A1、A2A、A2B、A3(これらはその一次配列によって特性決定されている)を介して作用することがわかっている。フレドホルム(Fredholm)ら(1994)。アデノシンA2受容体はさらに、A2A(高親和性)とA2B(低親和性)のサブタイプに分けられる。ダリー(Daly)ら(1983);およびバーンズ(Burns)ら(1986)。A1、A2BおよびA3受容体が脳内に広範囲にわたって分布しているのに対し、A2A受容体は大脳基底核、とりわけ、尾状-被殻(caudate-putamen)(線条体)、側坐核および淡蒼球、ならびに嗅結節に高度に局在している。ジャーヴィス(Jarvis)ら(1989);およびシッフマン(Schiffmann)(1991b)。大脳基底核は、終脳に局在し、いくつかの相互接続されている核:線条体、淡蒼球外節(GPe)、淡蒼球内節(GPi)、黒質緻密部(SNc)、黒質網様部(SNr)および視床下核(STN)からなる。大脳基底核は、運動行動を起こすための運動感覚(sensorimotor)、連合および辺縁系情報の統合に関与する皮質下回路(subcortical circuits)の重要な要素である。大脳基底核の主要構成要素は線条体であり、ここではGABA作動性の中型の有棘ニューロンが唯一の投射ニューロンであり、これは線条体ニューロン群の90%以上に相当する。
【0042】
中型の有棘ニューロンは、皮質および視床から膨大なグルタメート作動性入力を受け、それらのGABA作動性出力を大脳基底核の主要な出力核、つまりGPiおよびSNrに、「間接経路」で線条体−淡蒼球系の中型の有棘ニューロン(striatopallidal medium spiny neurons)を介して、および「直接経路」で線条体−黒質系の中型の有棘ニューロン(striatonigral medium spiny neurons)を介して投射する。アレキサンダー(Alexander)ら(1990);ゲーフェン(Gerfen)(1992);およびグライベル(Graybiel)(1990)。中型の有棘ニューロンは、また線条体内GABA作動性、コリン作動性および黒質線条体ドーパミン作動性の調節入力を受ける。線条体黒質系の直接経路のニューロンは、GABAだけでなくサブスタンスP/ダイノルフィンも含有しており、線条体からGPi/SNrへと直接投射する。これらのニューロンは、GPi/SNrニューロンに対して直接的な阻害作用を与える。線条体淡蒼球系の間接経路の線条体ニューロンは、GABAだけでなくエンケファリンも含有しており、GPeおよびSTNでのシナプス結合を介して線条体とGPi/SNrとをつなぐ。これらのニューロンでは、アデノシンA2A受容体が、ほぼ例外なく、間接経路の線条体と淡蒼球にある線条体−淡蒼球系の中型の有棘ニューロン[シッフマン(Schiffmann)ら(1991a)]および線条体にあるアセチルコリンを含有する大型の無棘介在ニューロン[ディクソン(Dixon)ら(1996)]に局在し、GABA、アセチルコリンおよびグルタメートの神経伝達を調節することがわかってきた。クロカワ(Kurokawa)ら(1996);モリ(Mori)ら(1996);シンドウ(Shindou)ら(2001);オチ(Ochi)ら(2000);リチャードソン(Richardson)ら(1997);およびカセ(Kase)(2001)。
【0043】
神経科学の近年の進歩は、アデノシンA2A受容体に選択的な薬剤の開発とともに、アデノシンおよびアデノシンA2A受容体についての認識の拡大を促した。アデノシンA2A受容体アンタゴニストが数種類のパーキンソン病の動物モデル(例えば、MPTP処置したサル)の運動機能障害を改善するが、またドーパミン作動性薬剤とは異なるアデノシンA2A受容体アンタゴニストの特徴も示すことを、行動研究は示している。リチャードソン(Richardson)ら(1997);カセ(Kase)ら(2000);およびカセ(Kase)(2001)。
【0044】
選択的アデノシンA2A受容体アンタゴニストであるKW-6002の抗パーキンソン病作用は、MPTP処置したマーモセットおよびカニクイザルを用いて研究されてきた。カンダ(Kanda)ら(1998a);グロンジン(Grondin)ら(1999);およびカンダ(Kanda)ら(2000)(非特許文献1〜3参照)。MPTP処置マーモセットでは、KW-6002の経口投与により、用量依存的に自発運動の増加が誘発され、最大11時間まで持続した。カンダ(Kanda)ら(1998a)。自発運動は正常動物で認められるレベルまで増加したが、L-ドーパでは運動亢進が誘発された。さらに、L-ドーパを前投与したMPTP処置マーモセットでは、21日間のKW-6002を用いた処置により、ジスキネジアがほとんどまたは全く誘発されなかったが、同じ条件下での、L-ドーパを用いた処置では、顕著なジスキネジアが誘発された。ジスキネジアを発症するように処置されたMPTP処置マーモセットに、KW-6002(20mg/kg)を閾値のL-ドーパとともに1日1回、5日間投与した場合、ジスキネジアを増加させることなく抗パーキンソン病活性が増強された。カンダ(Kanda)ら(2000)。KW-6002は、またさらにキナピロール(quinpirole)、ドーパミンD2受容体アゴニストの抗パーキンソン病作用を増強したが、SKF80723、ドーパミンD1受容体アゴニストの作用は増強しなかった。総合すれば、これらの研究結果は、アデノシンA2Aアンタゴニストが、パーキンソン病の初期の患者に単独療法として抗パーキンソン病効果をもたらす可能性があること、およびL-ドーパ治療を受けた運動合併症患者では、ジスキネジアを増加させることなく抗パーキンソン病作用を改善させる可能性があることを示唆している。
【0045】
アデノシンA2Aアンタゴニストが抗パーキンソン病作用を発揮するメカニズムは依然として十分には解明されていないが、現在、次のメカニズムが提唱されている。
霊長類のパーキンソン病またはMPTP処置のいずれにおいても、黒質−線条体ドーパミン作動性経路が破壊された後の最も関連のある変化は、線条体淡蒼球系の経路の活動亢進である。このような活動亢進の原因は、直接的な線条体黒質系の経路および間接的な線条体淡蒼球系の経路間の不均衡にあり、これがパーキンソン病状態を引き起こす。デロング(DeLong)(1990);およびオベソ(Obeso)ら(2000)。アデノシンA2A受容体は、線条体黒質の中型の有棘ニューロンではなく、中型の有棘ニューロンのサブポピュレーション(subpopulation)である線条体淡蒼球系の中型の有棘ニューロンで特に発現されているということが知られている。
【0046】
線条体淡蒼球系の中型の有棘のGABA作動性投射ニューロンは、アデノシンA2A受容体媒介性調節の主要な標的ニューロンの1つであるとわかっている。カセ(Kase)(2001)。そのため、線条体では、アデノシンA2A受容体が線条体内のGABA作動性のフィードバック/フィードフォワード(feedback/feedforward)阻害のネットワークを通じて投射ニューロンの興奮性を制御し[モリ(Mori)ら(1996)]、淡蒼球(GPe)では、アデノシンA2A受容体の活性化によって神経末端からのGABA放出が促進され、視床下核(STN)へと投射するGPe投射ニューロンの興奮性を抑制している可能性がある[シンドウ(Shindou)ら(2001)]。アデノシンA2A受容体アンタゴニストは、線条体淡蒼球系のこの二重調節メカニズムを選択的にブロックし、線条体淡蒼球系の中型の有棘のニューロンにおける過度の活性化の抑制をもたらす。このことによって、線条体淡蒼球系/線条体黒質系ニューロンの不均衡が正常な状態にシフトし、その結果、パーキンソン病状態における運動機能の回復がもたらされる可能性がある。オチ(Ochi)ら(2000);カセ(Kase)(2001)、アオヤマ(Aoyama)ら(2002)。
【0047】
アデノシンA2A受容体を媒介した作用メカニズムは、線条体淡蒼球系の中型の有棘ニューロンにあるアデノシンA2A受容体とともに局在しているドーパミンD2受容体とは関係なく機能する可能性がある(アオヤマ(Aoyama)ら(2000))。ゲーフェン(Gerfen)ら(1990)。D2受容体ノックアウトマウス(D2R-/-)は、パーキンソン病と類似した運動表現型を示し、線条体の中型の有棘ニューロンで発現される神経ペプチド遺伝子のレベルが著しく変化した。D2R-/-マウスおよび野生型マウス間では、アデノシンA2A受容体mRNAの分布および発現レベルならびに受容体の結合特性に差は見られず、これは、D2受容体がないということがアデノシンA2A受容体特性に影響を与えないことを示唆している。KW-6002によるアデノシンA2A受容体の遮断によって、自発運動および運動の協調性が回復し、線条体エンケファリンの発現レベルが正常なマウスのレベルまで低下した。アオヤマ(Aoyama)ら(2000)。これらの結果は、アデノシンA2A受容体およびD2受容体が、大脳基底核での神経および運動機能の制御において、相反する活性ではあるが独立した活性を有していることを示唆している。ドーパミン作動性の系とは無関係なアデノシンA2A受容体の独立した機能が、アデノシンA2A受容体およびD2受容体ノックアウトマウスを用いた研究によって確認された。チェン(Chen)ら(2001b)。
【0048】
パーキンソン病のL-ドーパによる運動合併症におけるアデノシンA2A受容体の生理学的機能および病態生理学的機能は、全く分かっていない。L-ドーパ誘発性ジスキネジアの神経メカニズムは、一般に直接経路ではなくむしろ間接経路と関係していると考えられている。クロスマン(Crossman)(1990)。L-ドーパ誘発性ジスキネジアは、STNまたはGPiにおける活性がGPeからの過度の阻害の結果として与えられる一定のレベルを下回る場合に起こる。オベソ(Obeso)ら(1997)。主として、直接経路の異常が、L-ドーパ誘発性ジスキネジアの発症に大きく寄与する可能性があるというもう1つの仮説が提唱されている。
【0049】
アデノシンA2A受容体アンタゴニストの神経保護作用が、ラットおよびマウスならびにアデノシンA2A受容体ノックアウトマウスにおける神経毒(MPTPまたは6−ヒドロキシドーパミン)誘発性のドーパミン作動性神経変性により示されている。イケダ(Ikeda)ら(2002);およびチェン(Chen)ら(2001a)。今日まで、ドーパミン作動性ニューロンを死に至らしめる基本的な発病のメカニズムの抑止に成功した治療はなかった。
【0050】
それゆえ、アデノシンA2A受容体の遮断効果を有する非ドーパミン作動性薬剤療法が、パーキンソン病を治療するための手段として提供される。さらに、代表的なドーパミン作動薬の副作用、つまり運動合併症を増加させる危険もしくは発症させる危険がほとんどまたは全くない、抗パーキンソン病作用を提供するアデノシンA2A受容体アンタゴニストが望ましい。
【0051】
いくつかのキサンチン化合物は、アデノシンA2A受容体アンタゴニスト作用、抗パーキンソン病作用、抗鬱作用、神経変性に対する阻害活性等を示すことが知られている(特許文献1〜4等参照)。
【特許文献1】米国特許第5,484,920号明細書
【特許文献2】米国特許第5,587,378号明細書
【特許文献3】米国特許第5,543,415号明細書
【特許文献4】欧州特許公開第1016407号明細書
【非特許文献1】「アナルズ・オブ・ニューロロジー(Ann. Neurol. )」、第43巻、507-513、1998年
【非特許文献2】「エクスペリメンタル・ニューロロジー( Exp. Neurol. )」、第162巻、321-327、2000年
【非特許文献3】「ニューロロジー(Neurology)」、第52巻、1673-1677、1999年
【発明の開示】
【課題を解決するための手段】
【0052】
本発明は、パーキンソン病患者に1種以上のアデノシンA2A受容体アンタゴニストを投与することまたは併用投与することを特徴とするL-ドーパ療法の副作用を軽減または抑制する方法を提供する。このような治療は、例えばL-ドーパまたは他のドーパミン作動性薬剤で誘発される運動合併症を患っている患者を治療して、オフ時間を減少させるおよび/またはジスキネジアを改善するのに有効であり得る。
【0053】
本発明は、さらにL-ドーパを減量して治療するための方法および組成物を提供する。この方法は、臨床上有効な量より少ないL-ドーパと、該L-ドーパの効果を発揮させるために有効な量の1種以上のアデノシンA2A受容体アンタゴニストとの組み合わせを、それらを必要とする患者に投与することを特徴とする。
本発明は、さらに少なくとも1種のアデノシンA2A受容体アンタゴニストの有効量を、COMT阻害剤およびに/またはDAおよび/もしくはMAO阻害剤と組み合わせて投与することを特徴とするパーキンソン病および/またはL-ドーパによる運動合併症を治療する方法を提供する。
【0054】
本発明は、また、患者が追加のL-ドーパ療法を必要とすることを遅らせるか、または完全になくさせるように、L-ドーパまたはドーパミン作動性薬剤の前投与または後投与なしに、アデノシンA2A受容体アンタゴニストまたはアデノシンA2A受容体アンタゴニストとドーパミンアゴニストとの組み合わせのいずれかを、それらを必要とする患者に投与し、L-ドーパによる運動合併症の発症を遅らせるかまたはその進行を妨げることを特徴とするパーキンソン病治療の有効時間を延長する方法を提供する。
【0055】
本発明は、また、少なくとも1種のアデノシンA2A受容体アンタゴニストの有効量を、それを必要とする患者に投与することを特徴とする運動障害を治療する方法も包含する。このような治療は、例えば振戦、運動緩徐、歩行、ジストニア、ジスキネジア、遅発性ジスキネジアまたはその他の錐体外路症候群を治療するのに有効であるか、または運動障害を引き起こす薬剤の作用を防ぐかもしくは減少させるのに有効であり得る。
発明の詳細
本発明は、次の(1)〜(50)に関する。
【0056】
(1)パーキンソン病患者に少なくとも1種のアデノシンA2A受容体アンタゴニストの有効量を投与することを特徴とするL-ドーパおよび/またはドーパミンアゴニスト療法の副作用を軽減または抑制する方法。
(2)患者がL-ドーパまたは他のドーパミン作動性薬剤で誘発される運動合併症を患っている上記(1)に記載の方法。
【0057】
(3)運動変動のオフ時間を減少させる上記(2)に記載の方法。
(4)運動合併症におけるジスキネジアが改善される上記(2)に記載の方法。
(5)アデノシンA2A受容体アンタゴニストがキサンチン誘導体またはその薬理学上許容される塩である上記(1)に記載の方法。
(6)アデノシンA2A受容体アンタゴニストが式(I):
【0058】
【化65】
【0059】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【0060】
【化66】
【0061】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【0062】
【化67】
【0063】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
で表される上記(1)に記載の方法。
(7) アデノシンA2A受容体アンタゴニストが式(I-A):
【0064】
【化68】
【0065】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【0066】
【化69】
【0067】
(式中、R7、R8およびR9のうち少なくとも1つは低級アルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R10は水素原子または低級アルキルを表す)または
【0068】
【化70】
【0069】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
で表される上記(1)に記載の方法。
(8) アデノシンA2A受容体アンタゴニストが式(I-B):
【0070】
【化71】
【0071】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【0072】
【化72】
【0073】
(式中、R6およびmは前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
で表される上記(1)に記載の方法。
(9) アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである上記(1)に記載の方法。
【0074】
(10) 臨床上有効な量より少ないL-ドーパと、該L-ドーパの効果を発揮させるために有効な量の1種以上のアデノシンA2A受容体アンタゴニストとの組み合わせを、それらを必要とする患者に投与することを特徴とするL-ドーパを減量して治療する(L-DOPA sparing treatment)方法。
(11) アデノシンA2A受容体アンタゴニストがキサンチン誘導体またはその薬理学上許容される塩である上記(10)に記載の方法。
【0075】
(12) アデノシンA2A受容体アンタゴニストが式(I):
【0076】
【化73】
【0077】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【0078】
【化74】
【0079】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【0080】
【化75】
【0081】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
で表される上記(10)に記載の方法。
(13) アデノシンA2A受容体アンタゴニストが式(I-A):
【0082】
【化76】
【0083】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【0084】
【化77】
【0085】
(式中、R7、R8およびR9のうち少なくとも1つは低級アルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R10は水素原子または低級アルキルを表す)または
【0086】
【化78】
【0087】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
で表される上記(10)に記載の方法。
(14) アデノシンA2A受容体アンタゴニストが式(I-B):
【0088】
【化79】
【0089】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【0090】
【化80】
【0091】
(式中、R6およびmは前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
で表される上記(10)に記載の方法。
(15) アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである上記(10)に記載の方法。
【0092】
(16) 臨床上有効な量より少ないL-ドーパと、該L-ドーパの効果を発揮させるために有効な量の1種以上のアデノシンA2A受容体アンタゴニストとを含有するL-ドーパを減量した治療(L-DOPA sparing treatment)のための組成物。
(17) アデノシンA2A受容体アンタゴニストがキサンチン誘導体またはその薬理学上許容される塩である上記(16)に記載の組成物。
【0093】
(18) アデノシンA2A受容体アンタゴニストが式(I):
【0094】
【化81】
【0095】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【0096】
【化82】
【0097】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【0098】
【化83】
【0099】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
で表される上記(16)に記載の組成物。
(19) アデノシンA2A受容体アンタゴニストが式(I-A):
【0100】
【化84】
【0101】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【0102】
【化85】
【0103】
(式中、R7、R8およびR9のうち少なくとも1つは低級アルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R10は水素原子または低級アルキルを表す)または
【0104】
【化86】
【0105】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
で表される上記(16)に記載の組成物。
(20) アデノシンA2A受容体アンタゴニストが式(I-B):
【0106】
【化87】
【0107】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【0108】
【化88】
【0109】
(式中、R6およびmは前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
で表される上記(16)に記載の組成物。
(21) アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである上記(16)に記載の組成物。
【0110】
(22) 少なくとも1種のアデノシンA2A受容体アンタゴニストの有効量を、COMT阻害剤ならびに/またはDAおよび/もしくはMAO阻害剤と組み合わせて、それらを必要とする患者に投与すること特徴とするパーキンソン病および/またはL-ドーパによる運動合併症を治療する方法。
(23) アデノシンA2A受容体アンタゴニストがキサンチン誘導体またはその薬理学上許容される塩である上記(22)に記載の方法。
【0111】
(24) アデノシンA2A受容体アンタゴニストが式(I):
【0112】
【化89】
【0113】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【0114】
【化90】
【0115】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【0116】
【化91】
【0117】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
で表される上記(22)に記載の方法。
(25) アデノシンA2A受容体アンタゴニストが式(I-A):
【0118】
【化92】
【0119】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【0120】
【化93】
【0121】
(式中、R7、R8およびR9のうち少なくとも1つは低級アルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R10は水素原子または低級アルキルを表す)または
【0122】
【化94】
【0123】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
で表される上記(22)に記載の方法。
(26) アデノシンA2A受容体アンタゴニストが式(I-B):
【0124】
【化95】
【0125】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【0126】
【化96】
【0127】
(式中、R6およびmは前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
で表される上記(22)に記載の方法。
(27) アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである上記(22)に記載の方法。
【0128】
(28) 少なくとも1種のアデノシンA2A受容体アンタゴニストの有効量と、COMT阻害剤ならびに/またはDAおよび/もしくはMAO阻害剤とを含有するパーキンソン病の治療のための組成物。
(29) アデノシンA2A受容体アンタゴニストがキサンチン誘導体またはその薬理学上許容される塩である上記(28)に記載の組成物。
【0129】
(30) アデノシンA2A受容体アンタゴニストが式(I):
【0130】
【化97】
【0131】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【0132】
【化98】
【0133】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【0134】
【化99】
【0135】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
で表される上記(28)に記載の組成物。
(31) アデノシンA2A受容体アンタゴニストが式(I-A):
【0136】
【化100】
【0137】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【0138】
【化101】
【0139】
(式中、R7、R8およびR9のうち少なくとも1つは低級アルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R10は水素原子または低級アルキルを表す)または
【0140】
【化102】
【0141】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
で表される上記(28)に記載の組成物。
(32) アデノシンA2A受容体アンタゴニストが式(I-B):
【0142】
【化103】
【0143】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【0144】
【化104】
【0145】
(式中、R6およびmは前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
で表される上記(28)に記載の組成物。
(33) アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである上記(28)に記載の組成物。
【0146】
(34) 患者が追加のL-ドーパ療法を必要とすることを遅らせるか、またはなくさせるために有効な量の、アデノシンA2A受容体アンタゴニストまたはアデノシンA2A受容体アンタゴニストとドーパミンアゴニストとの組み合わせのいずれかを、それらを必要とする患者に投与することを特徴とするパーキンソン病治療の有効時間を延長する方法。
(35) 運動合併症の発症を遅らせる上記(34)に記載の方法。
【0147】
(36) 患者がL-ドーパまたはドーパミン作動性薬剤の前投与を受けていない上記(34)に記載の方法。
(37) 患者がL-ドーパまたはドーパミン作動性薬剤の後投与を受けない上記(34)に記載の方法。
(38) アデノシンA2A受容体アンタゴニストがキサンチン誘導体またはその薬理学上許容される塩である上記(34)に記載の方法。
【0148】
(39) アデノシンA2A受容体アンタゴニストが式(I):
【0149】
【化105】
【0150】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【0151】
【化106】
【0152】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【0153】
【化107】
【0154】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
で表される上記(34)に記載の方法。
(40) アデノシンA2A受容体アンタゴニストが式(I-A):
【0155】
【化108】
【0156】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【0157】
【化109】
【0158】
(式中、R7、R8およびR9のうち少なくとも1つは低級アルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R10は水素原子または低級アルキルを表す)または
【0159】
【化110】
【0160】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
で表される上記(34)に記載の方法。
(41) アデノシンA2A受容体アンタゴニストが式(I-B):
【0161】
【化111】
【0162】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【0163】
【化112】
【0164】
(式中、R6およびmは前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
で表される上記(34)に記載の方法。
(42) アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである上記(34)に記載の方法。
【0165】
(43) 少なくとも1種のアデノシンA2A受容体アンタゴニストの有効量を、それを必要とする患者に投与することを特徴とする運動障害を治療する方法。
(44) 患者が振戦、運動緩慢、歩行、ジストニア、ジスキネジア、遅発性ジスキネジアまたはその他の錐体外路症候群を患っている上記(43)に記載の方法。
(45) アデノシンA2A受容体アンタゴニストが、運動障害を引き起こす薬剤の作用を減少させる上記(43)に記載の方法。
【0166】
(46) アデノシンA2A受容体アンタゴニストがキサンチン誘導体またはその薬理学上許容される塩である上記(43)に記載の方法。
(47) アデノシンA2A受容体アンタゴニストが式(I):
【0167】
【化113】
【0168】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【0169】
【化114】
【0170】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【0171】
【化115】
【0172】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
で表される上記(43)に記載の方法。
(48) アデノシンA2A受容体アンタゴニストが式(I-A):
【0173】
【化116】
【0174】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【0175】
【化117】
【0176】
(式中、R7、R8およびR9のうち少なくとも1つは低級アルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R5は水素原子または低級アルキルを表す)または
【0177】
【化118】
【0178】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
で表される上記(43)に記載の方法。
(49) アデノシンA2A受容体アンタゴニストが式(I-B):
【0179】
【化119】
【0180】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【0181】
【化120】
【0182】
(式中、R6およびmは前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
で表される上記(43)に記載の方法。
(50) アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである上記(43)に記載の方法。
【0183】
さらに、本発明は、次の(51)〜(60)に関する。
(51) アデノシンA2A受容体アンタゴニストを含有するL-ドーパおよび/またはドーパミンアゴニスト療法の副作用を軽減または抑制する薬剤。
(52) L-ドーパおよび/またはドーパミンアゴニスト療法の副作用を軽減または抑制する薬剤の製造のためのアデノシンA2A受容体アンタゴニストの使用。
【0184】
(53) 臨床上有効な量より少ないL-ドーパと、該L-ドーパの効果を発揮させるために有効な量の1種以上のアデノシンA2A受容体アンタゴニストとを含有するL-ドーパを減量して治療するための薬剤。L-ドーパおよびアデノシンA2A受容体アンタゴニストは、単一の投与形態で存在してもよいし、または別々の投与形態で存在してもよい。
(54) L-ドーパを減量して治療するための薬剤の製造のための、臨床上有効な量より少ないL-ドーパと、該L-ドーパの効果を発揮させるために有効な量の1種以上のアデノシンA2A受容体アンタゴニストとの組み合わせの使用。
【0185】
(55) アデノシンA2A受容体アンタゴニストならびにCOMT阻害剤および/またはDAおよび/またはMAO阻害剤を含有するパーキンソン病および/またはL-ドーパによる運動合併症を治療する薬剤。アデノシンA2A受容体アンタゴニストならびにCOMT阻害剤および/またはDAおよび/またはMAO阻害剤は、単一の投与形態で存在してもよいし、または別々の投与形態で存在してもよい。
【0186】
(56) パーキンソン病および/またはL-ドーパによる運動合併症を治療する薬剤の製造のためのアデノシンA2A受容体アンタゴニストならびにCOMT阻害剤および/またはDAおよび/またはMAO阻害剤の使用。
(57) アデノシンA2A受容体アンタゴニストまたはアデノシンA2A受容体アンタゴニストとドーパミンアゴニストとの組み合わせのいずれかを含有する患者が追加のL-ドーパ療法を必要とすることを遅らせるか、またはなくさせることにより、パーキンソン病治療の有効時間を延長する薬剤。アデノシンA2A受容体アンタゴニストとドーパミンアゴニストとの組み合わせを用いる場合、アデノシンA2A受容体アンタゴニストおよびドーパミンアゴニストは、単一の投与形態で存在してもよいし、または別々の投与形態で存在してもよい。
【0187】
(58) 患者が追加のL-ドーパ療法を必要とすることを遅らせるか、またはなくさせることにより、パーキンソン病治療の有効時間を延長する薬剤の製造のためのアデノシンA2A受容体アンタゴニストまたはアデノシンA2A受容体アンタゴニストとドーパミンアゴニストとの組み合わせのいずれかの使用。
(59) アデノシンA2A受容体アンタゴニストを含有する運動障害を治療する薬剤。
【0188】
(60) 運動障害を治療する薬剤の製造のためのアデノシンA2A受容体アンタゴニストの使用。
本発明は、1種以上のアデノシンA2A受容体アンタゴニストを投与することを特徴とする運動障害を患っている患者を治療する方法を対象とする。「アデノシンA2A受容体アンタゴニスト」とは、例えばアデノシンA2A受容体と結合すること、アデノシンの受容体との結合を妨害することまたは阻止することによって、少なくとも1種のアデノシンで媒介される生理活性を阻害、抑制または消滅させる化合物を意味する。
【0189】
本発明では、アデノシンA2A受容体が、例えば間接経路または大脳基底核出力核活性を制御する際に機能することから、アデノシンA2A受容体アンタゴニストを運動障害の治療に用いることができると考えている。アデノシンA2A受容体はまた運動行動または運動機能障害の制御にも関係していると考えられる。
アデノシンA2A受容体アンタゴニストは、いくつかの様式で機能する。このアンタゴニストは、アデノシンがアデノシンA2A受容体と結合することを実質的に妨害するか、ブロックするかまたは防ぐために十分な親和性および特異性により、アデノシンと結合するかまたはアデノシンを隔離することができ、それによって、例えば間接経路の線条体GABA作動性出力の調節、例えばSNr等の大脳基底核での出力核の活性等の1種以上のアデノシンA2A受容体で媒介される生理機能を阻害、抑制または消滅させ、それによって、大脳基底核
において運動行動を制御する。本発明では、アデノシンA2A受容体アンタゴニストの抗パーキンソン病活性がこの活性に起因すると考えている。本発明では、さらにパーキンソン病患者におけるL-ドーパおよび/またはドーパミンアゴニスト療法の副作用を軽減または抑制するアデノシンA2A受容体アンタゴニストの能力がこの活性に起因すると考えている。本発明では、さらにL-ドーパおよび/またはドーパミンアゴニストに誘発される運動合併症の発症におけるアデノシン受容体アンタゴニストの関与がこの活性に起因すると考えている。また、アデノシンA2A受容体アンタゴニストは、例えば6-OHDA(6−ヒドロキシドーパミン)、1−メチル−4−フェニル−1,2,3,6−テトラヒドロピリジン(MPTP)等のドーパミン作動性神経毒によって誘発されるニューロン変性カスケードおよびグリア細胞を介したドーパミン作動性神経毒の産生を阻害し得る。従って、アデノシンA2A受容体アンタゴニストは、神経毒のドーパミン作動性神経変性作用を阻害する方法およびグリオーシス(gliosis)を阻害する方法を提供する。アデノシンA2A受容体アンタゴニストのこれらの特徴が、パーキンソン病の進行を阻止する。このように、アデノシンA2A受容体アンタゴニストを使用することによって、患者が追加のL-ドーパ療法を必要とするのを遅らせるかまたは完全になくさせるような療法を提供するか、またはL-ドーパによる運動合併症の発症を遅らせるかもしくはその進行を妨げる療法を提供する。
【0190】
このように、本発明のアデノシンA2A受容体アンタゴニストは、1種以上のアデノシンA2A受容体アンタゴニストの有効量を投与することによって、パーキンソン病患者および運動障害を患っている他の患者を治療する方法を対象とする。また、本発明のアデノシンA2A受容体アンタゴニストは、パーキンソン病の治療におけるL-ドーパによる運動合併症をはじめとするL-ドーパ療法の副作用を軽減または抑制する方法においても有用である。さらに、アデノシンA2A受容体アンタゴニストを用いたパーキンソン病の治療では、L-ドーパを用いた治療の必要性を回避することができ、例えば悪心、活動亢進、例えばウェアリング・オフ、オン・オフ変動等の運動変動、ジスキネジア等の副作用なしにまたは軽減しながら、パーキンソン病を有効に治療するために必要なL-ドーパの量を減らすことができる。さらに、本発明は、患者がL-ドーパ療法を必要とすることを遅らせるかまたは完全になくさせて、L-ドーパによる運動合併症の発症を遅らせるかまたはその進行を妨げるように、アデノシンA2A受容体アンタゴニストを投与することによりパーキンソン病患者を治療する方法を提供する。さらに、本発明は、その他の運動障害を患っている患者における振戦、運動緩徐、歩行、ジストニアならびに遅発性ジスキネジアおよびその他の錐体外路症候群を治療する方法を提供する。
【0191】
本発明に用いるアデノシンA2A受容体アンタゴニストは、アデノシンA2A受容体アンタゴニスト活性を有している限り、限定されない。その例としては、US 5,484,920、US 5,703,085、WO 92/06976、WO 94/01114、US 5,565,460、WO 98/42711、WO 00/17201、WO 99/43678、WO 01/92264、WO 99/35147、WO 00/13682、WO 00/13681、WO 00/69464、WO 01/40230、WO 01/02409、WO 01/02400、EP 1054012、WO 01/62233、WO 01/17999、WO 01/80893、WO 02/14282、WO 01/97786等に開示された化合物が挙げられる。より具体的には、例えば、
(1)次式(I)で表される化合物
【0192】
【化121】
【0193】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【0194】
【化122】
【0195】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【0196】
【化123】
【0197】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
(2)次式(I-A)で表される化合物
【0198】
【化124】
【0199】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【0200】
【化125】
【0201】
(式中、R7、R8およびR9のうち少なくとも1つはアルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R10は水素原子または低級アルキルを表す)または
【0202】
【化126】
【0203】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
および
(3)次式(I-B)で表される化合物
【0204】
【化127】
【0205】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【0206】
【化128】
【0207】
(式中、R6およびmはそれぞれ前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
ならびにそれらの薬理学上許容される塩が挙げられる。
式(I)、式(I-A)および式(I-B)の基の定義において、低級アルキルおよび低級アルコキシの低級アルキル部分は、1〜6個の炭素原子を有する直鎖状または分枝状のアルキル基、例えばメチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec-ブチル、tert-ブチル、ペンチル、ネオペンチル、ヘキシ等を意味する。低級アルケニルは、2〜6個の炭素原子を有する直鎖状または分枝状アルケニル基、例えばビニル、アリル、メタクリル、クロチル、3−ブテニル、2−ペンテニル、4−ペンテニル、2−ヘキセニル、5−ヘキセニル等を意味する。低級アルキニルは、2〜6個の炭素原子を有する直鎖状または分枝状アルキニル基、例えばエチニル、プロパルギル、2−ブチニル、3−ブチニル、2−ペンチニル、4−ペンチニル、2−ヘキシニル、5−ヘキシニル、4−メチル−2−ペンチニル等を意味する。アリールは、フェニルまたはナフチルを意味する。シクロアルキルは、3〜8個の炭素原子を有するシクロアルキル基、例えばシクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル等を意味する。複素環基としては、フリル、チエニル、ピロリル、ピラニル、チオピラニル、ピリジル、チアゾリル、イミダゾリル、ピリミジル、トリアジニル、インドリル、キノリル、プリニル、ベンゾチアゾリル等が例示される。ハロゲンとしては、フッ素、塩素、臭素およびヨウ素が包含される。
【0208】
置換アリール、置換複素環基および置換ナフチルは、それぞれ1〜4個の独立して選択された置換基を有する。置換基としては、低級アルキル、ヒドロキシ、置換もしくは非置換の低級アルコキシ、ハロゲン、ニトロ、アミノ、低級アルキルアミノ、ジ(低級アルキル)アミノ、トリフルオロメチル、トリフルオロメトキシ、ベンジルオキシ、フェニル、フェノキシ等が例示される。低級アルキルならびに低級アルコキシ、低級アルキルアミノおよびジ(低級アルキル)アミノの低級アルキル部分は、前記低級アルキルと同義である。ハロゲンは、前記ハロゲンと同義である。置換低級アルコキシにおける置換基としては、ヒドロキシ、低級アルコキシ、ハロゲン、アミノ、アジド、カルボキシ、低級アルコキシカルボニル等が例示される。低級アルコキシおよび低級アルコキシカルボニルの低級アルキル部分は、前記低級アルキルと同義であり、ハロゲンは、前記ハロゲンと同義である。
【0209】
上述した化合物(I)、化合物(I-A)および化合物(I-B)の薬理学上許容される塩としては、薬理学上許容される酸付加塩、金属塩、アンモニウム塩、有機アミン付加塩、アミノ酸付加塩等が例示される。
薬理学上許容される酸付加塩としては、例えば塩酸塩、硫酸塩、リン酸塩等の無機酸付加塩、例えば酢酸塩、マレイン酸塩、フマル酸塩、酒石酸塩、クエン酸塩等の有機酸付加塩等が例示される。薬理学上許容される金属塩としては、例えばナトリウム塩、カリウム塩等のアルカリ金属塩、例えばマグネシウム塩、カルシウム塩等のアルカリ土類金属塩、アルミニウム塩、亜鉛塩等が例示される。薬理学上許容されるアンモニウム塩としては、アンモニウム、テトラメチルアンモニウム等が例示される。薬理学上許容される有機アミン付加塩としては、モルホリン付加塩、ピペリジン付加塩等が例示される。薬理学上許容されるアミノ酸付加塩としては、リジン付加塩、グリシン付加塩、フェニルアラニン付加塩等が例示される。
【0210】
式(I)、式(I-A)および式(I-B)で表される化合物は、米国特許第5,543,415号;同5,587,378号;および同5,484,920号に記載されており、それらに記載の方法に従って合成される。
本発明の方法に有用な好ましくアデノシンA2A受容体アンタゴニストは、(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチン(次式(II))を含有する。
【0211】
【化129】
【0212】
また本発明では、式IIはKW-6002と称される。
「L-ドーパによる副作用を軽減または抑制する」とは、本発明によれば、本発明の化合物が患者の覚醒時間(awake time)中の「オフ」状態の量を減少させることを意味すると解される。オフ状態とは、本発明によれば、パーキンソン病用薬剤の服用による治療効果が薄れ、その結果、患者が、例えばパーキンソン病統一スケール(UPDRS)ならびにヘーン(Hoehn)およびヤール(Yahr)(HY)尺度等によって分類されるようなパーキンソン病の症状を起こす期間を意味すると解される。
【0213】
本発明は、また、患者の覚醒時間中の「オン」状態の割合を増やすことによってL-ドーパによる副作用を軽減することを対象とする。オン状態とは、パーキンソン病用薬剤の服用後、UPDRSおよびHY尺度によって分類されるパーキンソン病の症状が、患者に比較的みられない期間を意味する。本発明の方法によって治療可能な患者としては、パーキンソン・ジスキネジア・スケール(PDS)によって判定される運動合併症を伴うかまたは伴わない、初期段階、中間段階および進行した段階のパーキンソン病の患者が挙げられる。ジスキネジアは、UPDRS、改良ゲッツジスキネジア統一スケール(modified Goetz Dyskinesia Rating Scale)(MGDRS)および/または異常不随意運動評価尺度(AIMS)によって個別に判定することができる。本発明に係る治療は、進行したパーキンソン病を患っている患者に特に有効である。
【0214】
本発明によれば、本発明のアデノシンA2A受容体アンタゴニストは、L-ドーパまたはドーパミンアゴニストと併用投与することができる、すなわち、実質的に同時に投与することができる。また、アデノシンA2A受容体アンタゴニストは、患者がある用量のL-ドーパまたはドーパミンアゴニストを服用する前後のいずれかに、単独投与できることも想定される。L-ドーパの必要性の実質的な減少および/またはL-ドーパ療法の一般的な副作用の減少または抑制は、とりわけ、運動変動およびジスキネジアの症状においてKW-6002を投
与した場合に見られる。このように、本発明は、運動変動、ジスキネジア、悪心、およびL-ドーパ療法のその他の一般的な副作用を引き起こすL-ドーパとともにKW-6002を投与することによって、ヒトのパーキンソン病を治療する改良方法を提供する。
【0215】
さらに本発明は、L-ドーパの前投与または後投与なしに、アデノシンA2A受容体アンタゴニストまたはアデノシンA2A受容体アンタゴニストとドーパミンアゴニストとの組み合わせのいずれかを投与すること特徴とするパーキンソン病治療の有効時間を延長する方法を提供する。L-ドーパの必要性はなくなるか、または少なくとも実質的に減少すると同時に、付随するL-ドーパ療法の副作用が回避される。アデノシンA2A受容体アンタゴニストとドーパミンアゴニストとの「組み合わせ」は、同時にまたは少なくとも生理活性を重複させるように患者に与えられる。本発明のアデノシンA2A受容体アンタゴニストは、L-ドーパによる運動合併症の発症を妨げ、またドーパミン作動性神経変性も予防することから、アデノシンA2A受容体アンタゴニストは、単独でまたはドーパミンアゴニストとともに投与されると、L-ドーパによる運動合併症の発症を遅らせるかまたはその進行を妨げることができる。
【0216】
本発明によれば、アデノシンA2A受容体アンタゴニストは、単独でまたは例えばブロモクリプチン、カベルゴリン、プラミペキソール、ロピネロール(ropinerole)、ペルゴリド等のドーパミンアゴニストとともに投与することができ、それによってL-ドーパの必要性が現れるのを回避するか、または少なくともL-ドーパの必要性が現れるまでの時間が延長される。
【0217】
本発明はさらに、パーキンソン病患者をL-ドーパを減量して治療する方法を提供する。すなわち、臨床上有効な量より少ないL-ドーパを用いる治療であり、臨床上有効な量より少ないL-ドーパの効果が維持される。この方法は、臨床上有効な量より少ないL-ドーパと、アデノシンA2A受容体アンタゴニストの有効量とを用いて、患者を治療することを特徴とする。臨床上有効な量より少ないL-ドーパとは、個々の患者の治療に有効ではないL-ドーパの量を意味する。一般に、L-ドーパは、1日当たり100mg〜1gの用量を分割(通常、250mgを1日4回)して投与する。耐え難い副作用、通常は運動障害を発症するまで、1日当たり100〜750mgで、3〜7日の間隔をおいて用量を徐々に増やす。カルビドパと併用投与する場合には、L-ドーパの有効量は減る。個々の患者に向けたL-ドーパの臨床上有効な量より少ない量を決定することおよびアデノシンA2A受容体アンタゴニストの存在下でのその量を適宜調整することは、十分に当業者の技術の範囲内のものである。
【0218】
臨床上有効な量より少ないL-ドーパと、必要に応じてアデノシンA2A受容体アンタゴニストと、必要に応じてドーパミンアンタゴニストとを含有する組成物は、当技術分野でよく知られた方法および本明細書に記載した方法によって製造される。カルビドパおよびその他の有効成分の追加量もまた、当業者であれば決定できる。
本発明は、さらに、少なくとも1種のアデノシンA2A受容体アンタゴニストと少なくとも1種のCOMT阻害剤またはMAO-B阻害剤とを用いてパーキンソン病を治療する方法を提供する。該組成物は、当技術分野でよく知られた任意の方法により同時にまたは連続的に投与することができる。このような組成物の製造方法および投与方法は、当技術分野ではよく知られている。適当なCOMT阻害剤およびMAO阻害剤については、本明細書に記載されており、当技術分野では周知である。これらとしては、限定されるものではないが、エンタカポンおよびトルカポン、ならびにデプレニルが挙げられる。以下で示すように、アデノシンA2A受容体アンタゴニストとCOMTまたはMAO-B阻害剤との併用療法では、副作用は増加しない。
【0219】
「治療の有効時間を延長する」とは、患者がL-ドーパ療法を必要とするのを遅らせるかまたは完全になくさせるように、主観的に、またはUPDRS、AIMS、PDS、HYおよび/またはMGDRSに従って客観的に、患者のパーキンソン病の症状および運動合併症を軽減または抑制することを意味する。
本発明は、また、それを必要とする患者に、少なくとも1種のアデノシンA2A受容体アンタゴニストの有効量を投与することを特徴とする運動障害を治療する方法を包含する。このような治療は、振戦、運動緩徐、歩行、ジストニア、または遅発性ジスキネジアもしくはその他の錐体外路症候群を治療するのに有効であるか、または運動障害を引き起こす薬剤の作用を防ぐかもしくは減少させるのに有効であり得る。このような薬剤としては、当技術分野では公知であり、限定されるものではないが、表1に記載するものが挙げられる。
【0220】
「運動障害を治療する」とは、限定されるものではないが、振戦、ジストニア、ジスキネジア、痙縮といった症状を止めるかまたは弱めることを意味する。症状の変化は、限定されるものではないが、UPDRS、AIMS、PDS、HYおよび/またはMGDRSといった当技術分野では公知のいずれかの方法によって判定することができる。
「治療」または「治療する」という用語は、運動機能障害を改善するか、あるいは疾病または疾患の発症を妨げるかもしくは遅らせるか、その進行を遅らせるかまたはその症状を改善するためのアデノシン活性の有効な阻害、抑制または停止を指す。
【0221】
このように、本発明は、本発明のアデノシンA2A受容体アンタゴニストを使用することによって、アデノシンとアデノシンA2A受容体との相互作用または結合を妨害するか、ブロックするかまたは防ぐ方法を提供する。
本発明の投与するための医薬組成物は、少なくとも1種のアデノシンA2A受容体アンタゴニストを、薬理学上許容される担体との任意の組み合わせを特徴とする。これらの組成物は、それらの意図する目的を達成するいずれの方法によって投与してもよい。本発明の組成物の投与に関する量および計画は、パーキンソン病患者の治療の分野に従事する当業者ならば、容易に決めることができる。
【0222】
本明細書に記載した組成物は、限定されるものではないが、経口;経鼻;経肺;例えば皮下、静脈内、筋肉内、腹腔内等の非経口;十二指腸内;経皮的;または口腔内をはじめ、適していればいずれの方法でも投与することができる。
投与量は有効量であり、患者の年齢、健康状態および体重、これまでに治療または並行して行っている治療がある場合には、その種類、治療の頻度および望む効果の性質に応じて変わる。適当な用量を決定する場合には、通常、いくつかの因子を配慮する。これらの因子としては、患者の年齢、性別および体重、治療状態、状態の重篤度および投与されている薬剤の形態が挙げられる。
【0223】
「有効な量」とは、有益なまたは所望の臨床結果を達成するのに十分な量である。有効な量は、1以上の用量で投与してもよい。治療の観点から言えば、有効な量は、疾病または疾患の進行を軽減する、改善する、安定させる、回復させるもしくは遅らせる、または疾病または疾患の病理学的影響を減少させるのに十分な量である。有効な量は、一般に医師によって個別に判断され、当業者の技量の範囲内である。
【0224】
本発明の組成物は、また、薬理学上活性な化合物に加えて、活性化合物の薬理学上許容される製剤への加工を容易にする賦形剤を含む適当な薬理学上許容される担体を含有することができる。好ましくは、製剤、特に経口投与が可能で、好ましい投与形態に用いることができる製剤、例えば錠剤、トローチ剤およびカプセル剤、また例えば坐剤等の直腸投与が可能な製剤等、ならびに注射剤による投与に適当な溶液は、約0.1〜99%、好ましくは約20〜85%の活性化合物を賦形剤とともに含有する。薬理学上許容される液体組成物は、例えば、本明細書で実施された化合物を、水、生理食塩水、デキストロース、グリセロール、エタノール等の液体賦形剤に溶解または分散させることによって調製することができる。組成物は、また、その他の薬剤(medicinal agents)、医薬品(pharmaceutical agents)、担体および例えば湿潤剤または乳化剤、pH緩衝剤等の補助剤を含有することができる。
【0225】
本発明の医薬組成物は、組成物の形態に適当な様式で投与する。一般的な経路としては、皮下、筋肉内、腹腔内、皮内、経口、経鼻および経肺(すなわち、エアロゾールによる)が挙げられる。ヒトに用いるための本発明の医薬組成物は、通常、経口的に投与される。
経口、経鼻または局所投与用の医薬組成物は、錠剤、カプセル剤、散剤、液剤および懸濁剤といった固体、半固体または液体の形態で提供することができる。注射剤用の組成物は、液状溶液もしくは懸濁液として、エマルションとして、または注射剤とするために液体へ溶解または懸濁するのに適当な固体形態として提供することができる。気道を経由する投与には、適当なエアロゾール化装置を用いる場合には、固体、粉末または液体のエアロゾールを提供する組成物が好ましい。必ずしも必要ではないが、医薬組成物は、正確な量を投与するのに適当な単位投与形態で提供されることが好ましい。また、本発明は徐放性または持続性形態も開示するものであり、これによって、比較的一貫したレベルの活性化合物が長期間にわたって提供される。
【0226】
アデノシンA2A受容体アンタゴニストは、体重1キログラム当たり約0.001〜約20.0mgの量で投与できることが好ましい。投与量が、体重1キログラム当たり約0.01〜約10mgの範囲であることがより好ましい。本発明のアデノシンA2A受容体アンタゴニスト組成物は、最終的には血中から除かれるため、投与に関しては、該組成物が示され、また好ましい。
アデノシンA2A受容体アンタゴニストは、投与製剤に応じて、かつ治療上有効であるような量で投与することができる。全身投与量は、患者の年齢、体重および状態ならびに投与経路に応じて変わる。
【0227】
本発明の方法に有用な医薬製剤は、公知の方法で製造される。医薬組成物の調製は、一般的に認められた医薬製剤の調製方法に従って実施される。例えば、レミングトンズ・ファーマシューティカル・サイエンシス(Remington’s Pharmaceutical Sciences)第18版(1990)、マーチン(Martin)編、マック・パブリッシング・コーポレーション(Mack Publishing Co.)、ペンシルバニア(PA)参照。意図する用途および投与様式に応じて、医薬組成物の調製において有効成分をさらに加工することが望ましいこともある。適当な加工法としては、滅菌、適当な非毒性および非干渉成分との混合、投与単位への分割ならびに送達容器への封入が挙げられる。
【0228】
経口用医薬製剤は、活性化合物を固形賦形剤と混合し、得られた混合物を場合によって粉砕し、所望または必要に応じて、適当な助剤を添加した後、顆粒混合物を加工することにより、錠剤を得ることができる。
適当な賦形剤としては、サッカライドなどの充填剤に限定されるものではないが、例えばラクトースまたはスクロース、マンニトールまたはソルビトール;セルロース誘導体;亜鉛化合物;リン酸三カルシウムまたはリン酸三カルシウムまたはリン酸水素カルシウム等のリン酸水素カルシウム等のリン酸カルシウム;またはリン酸水素カルシウム等のリン酸三カルシウム、またはリン酸水素カルシウム等のリン酸カルシウム等の充填剤、ならびに例えばトウモロコシデンプン、コムギデンプン、コメデンプン、ジャガイモデンプンを用いたデンプンペースト;ゼラチン;トラガカントガム;および/またはポリビニルピロリドン等の結合剤が挙げられる。
【0229】
助剤としては、流動調節剤およびシリカ、タルク、ステアリン酸またはそれらの塩、および/またはポリエチレングリコール等の滑沢剤が挙げられる。錠剤、カプレット剤またはカプセル剤の核部は、所望により、胃液に耐えるような適当なコーティングが施される。この目的には、濃縮糖類溶液を用いることができ、場合によって、これはアラビアゴム、タルク、ポリビニルピロリドン、ポリエチレングリコールおよび/または二酸化チタン、ラッカー溶液および適当な有機溶媒もしくは混合溶媒を含有することができる。胃液に耐えるコーティング、すなわち腸溶性コーティングを施すためには、アセチルセルロースフタレートまたはヒドロキシプロピルメチルセルロースフタレート等の適当なセルロース調製物の溶液が用いられる。色素または顔料を、例えば識別のためにまたは活性化合物用量の組み合わせを明示するために、錠剤またはコーティングに添加することができる。
【0230】
経口的に用いることができるその他の医薬製剤としては、ゼラチン製の押し込み型カプセル剤、ならびにゼラチンとグリセロールまたはソルビトール等の可塑剤とでできた密閉型軟カプセル剤が挙げられる。押し込み型カプセル剤は、活性化合物を顆粒の形態で含むことができ、この顆粒はラクトース等の充填剤、デンプン等の結合剤、および/またはタルクまたはステアリン酸マグネシウム等の滑沢剤、および場合によって安定化剤と混合することができる。軟カプセル剤では、活性化合物を脂肪油または流動パラフィン等の適当な液体に溶解するかまたは懸濁することが好ましい。さらに、安定化剤を添加してもよい。
【0231】
本発明のアデノシンA2A受容体アンタゴニストは、また生物分解性の徐放性担体と混合
する場合にはインプラント剤の形態で投与することもできる。また、有効成分の持続放出を目的として、有効成分を経皮パッチとして製剤することもできる。インプラント剤およびパッチ剤の製造方法は、当技術分野では周知である。レミングトンズ・ファーマシューティカル・サイエンス(Remington’s Pharmaceutical Sciences) 第18版(1990);およびキドニュウズ(Kydonieus)編(1992)トゥリータス・オン・コントロールド・ドラッグ・デリバリー(Treatise on controlled drug delivery)、マーセル・デッカー(Marcel Dekker)、ニューヨーク(NY)。
【0232】
本発明を以下の実施例によってさらに例示するが、本発明は実施例によって何ら限定されない。本明細書に記載した全ての参照文献は、参照により本明細書に組み入れられる。
【実施例1】
【0233】
アデノシンA2A受容体アンタゴニストであるKW-6002の、L-ドーパ関連の運動合併症によって悪化したパーキンソン病の治療としての安全性および有効性を、12週間の多施設試験研究(multicenter, exploratory study)にて検討した。運動合併症を伴うPD被験者を、無作為かつ盲目的に、3つのパラレル治療群(parallel treatment arms)のうちの1つに振り分けた:プラセボ(n=29);KW-6002 最大20mg/日(n=26);KW-6002 最大40mg/日(n=28)。2つの主要な有効性指標を設定した:1) 8時間の来院中に治験担当医師によって判定される「オフ」時間の変化、および2) 被験者の自宅での運動日誌によって判定される「オフ」時間の変化。
【0234】
83人の登録被験者のうち65人が試験を完了した;中止脱落率は、治療群間において同様であった。KW-6002治療は、患者の覚醒時間中の「オフ」状態で過ごす割合の減少において、プラセボ治療よりも有意に効果的であった。自宅での日誌により評価した場合には、覚醒時間中のオフ状態で過ごす割合が、プラセボ群では2.2%増加したのと比較して、KW-6002に振り分けられた被験者では7.1%減少した(p=0.008)。プラセボ群よりもKW-6002群におけるオフ時間の減少が1.7時間多かった(p=0.004)。治験担当医師による8時間のオン/オフ判定の結果では、統計学的な有意性に達した(p=0.054)。KW-6002で治療された患者が過ごした「オフ」状態の時間は、プラセボ群の患者のものよりも0.51時間少なかった(p=0.061)。
【0235】
試験では、またプラセボ群と比較して、ベースラインから12週までKW-6002で治療した患者における早朝ジストニアの減少が示された。
方法
これは、運動合併症を伴うL-ドーパで治療されたPD患者における補助療法としてのKW-6002の安全性および有効性についての、12週間の、二重盲検の、プラセボ対照の、無作為の、パラレル群の多施設試験研究である。適格患者は、英国パーキンソン病協会(UnitedKingdom PD Society(UKPDS))脳バンク診断基準(ダニエル(Daniel)ら(1993))を満たし、少なくとも1年間、L-ドーパ/カルビドパを服用しており、1日当たり少なくとも4回、L-ドーパ/カルビドパを服用しており、かつエンド・オブ・ドーズ(end-of-dose)でのウェアリング・オフ(wearing-off)をはじめとする運動合併症を起こしている患者とした。
【0236】
インフォームド・コンセントを行った後、被験者は4〜8週間のスクリーニングを受けた。訪問の4週前には、投薬の用量用法を一定にした。この訪問時に、被験者は自宅での日誌の全ての記入に関する指導を受けた。
ベースライン時に、被験者は、8時間の院内(in-office)評価を受けた。この評価に先立ち、被験者はPD用薬を差し控え、午前零時から絶食をした。最初の評価の後、その日初回のPD用薬を投与し、その後の用量は、被験者の通常の投与間隔で投与した。評価は特別指導を受けた、有害事象(adverse events)および臨床検査の結果を知らされていない評価者(blinded rater)によって行われた。被験者には、無作為化を適格なものとするために、8時間の評価中、PD用薬の投与後に少なくとも90分のオフ時間を示すことが求められた。
【0237】
スクリーニングおよびベースラインの評価を無事終了した被験者を、KW-6002の2つの投与用量用法群または対応するプラセボのうちの1群に、1:1:1の比で無作為に振り分けた。KW-6002群に無作為に振り分けられた患者は、1週〜4週の期間に5mg/日、5週〜8週の期間に10mg/日および9週〜12週の期間に20mg/日(5/10/20群)、または1週〜4週の期間に10mg/日、5週〜8週の期間に20mg/日および5週〜9週の期間に40mg/日(10/20/40群)のいずれかで投与を受けた(図1)。試験薬は、毎日、被験者の通常の朝食とともに、1回量として服用した。
【0238】
その後の評価は、2、4、6、8、10および12週目に行った。被験者は、各訪問の前の週中に自宅で3種類の日誌の全ての記入を行った。各訪問時に、有害事象を評価した。8時間の院内評価は、4、8および12週目に行った。臨床血液検査およびECGは、ベースライン時、4週、8週および12週目に行った。
試験遂行中、治験担当医師は、L-ドーパ関連の有害事象を改善するよう、L-ドーパの1日の全投与量を減らすことができた。L-ドーパ投与の間隔を変更することは、認められなかった。
結果
83人の被験者を無作為に振り分けた。
【0239】
試験群間では、人口統計的およびベースライン特性の顕著な差は見られなかった。
3治療群全ての被験者は、それらの試験薬をピル数に基づく99%を服薬した。試験中、いずれの治療群においてもまたは上記2つのKW-6002群を併せた群とプラセボ群とを比較しても、L-ドーパの平均1日投与量に有意な変化はなかった。
自宅での日誌で評価した場合、上記2つのKW-6002群に無作為に振り分けられた被験者は、プラセボ群に無作為に振り分けられた被験者と比較して、オフ時間における有意な減少があった(図1)。覚醒時間中のオフ状態で過ごす割合が、プラセボ群では2.2%増加したのと比較して、KW-6002に振り分けられた被験者では7.1%減少した(p=0.008)。上記2つのKW-6002投与群はいずれも、プラセボ群と比較して、オフ時間の割合において有意な減少を示した。同様に、各KW-6002群と同様に上記2つのKW-6002群を併せた群では、オフ時間の合計時間において、有意な減少を示した。オフ時間において、プラセボ群での0.5時間増加したのと比較して、KW-6002に振り分けられた被験者では1.2時間減少した(p=0.004)(図1)。
【0240】
上記2つのKW-6002群を併せた群では、プラセボ群と比較して、オフ時間におけるより大きな減少傾向があることが、8時間の院内評価での医師によるオフ時間の判定で確認された。オフ時間が、プラセボ群では3.3%減少したのと比較して、KW-6002に振り分けられた被験者では10.0%の減少を示した(p=0.05)。同様に、オフ時間が、プラセボ群では0.3時間の減少したのと比較して、KW-6002に振り分けられた被験者では0.8時間の減少を示した(p=0.06)。高用量KW-6002群(10/20/40群)でのオフ時間の減少は有意なものであった(P=0.02)。
【0241】
KW-6002で治療した患者の早朝ジストニアは、プラセボ群と比較して、ベースラインから12週まで減少した。
全般的な有害事象プロフィールでは、KW-6002で治療した被験者とプラセボで治療した被験者とで差がなかった。重篤な有害事象の全般的な発生は、試験群全体にわたり同様に分布した。総中止脱落数および有害事象による中止脱落数は、KW-6002群とプラセボ群間で同様であった。KW-6002群とプラセボ群間では、最大血圧または最小血圧、心拍数、呼吸数、体重、ECG、および尿検査もしくは血液化学検査の平均値における顕著な変化または差は認められず、臨床参照範囲内であった。
【0242】
本試験では、ドーパミンアゴニスト(例えばプラミペキソール、ペルゴリド、ロピニロール、ブロモクリプチン)、COMT阻害剤(例えばエンタカポン、トルカポン)およびMAO阻害剤であるセレギリンとの種々の医薬の併用下で、KW-6002はオフ時間を有意に減少させ、かつ安全性および優れた忍容性を示した。
この試験での結果に基づき、アデノシンA2A受容体アンタゴニストであるKW-6002は、L-ドーパによる運動合併症を伴うパーキンソン病患者のオフ時間を安全にかつ有効に減少させることができる。
【0243】
本試験はまた、アデノシンA2A受容体アンタゴニストであるKW-6002が、L-ドーパとドーパミンアゴニストおよび/またはCOMT阻害剤および/またはMAO阻害剤との医薬の併用により治療したパーキンソン病患者におけるオフ時間を有意に減少させたことを示している。
本試験はまた、KW-6002がパーキンソン病患者の早朝ジストニアを減少させることも示している。
【0244】
前述の発明を、明確にし、理解するために、例示および実施例によって細部にわたって記載してきたが、当業者には、特定の変更および改変を行うことができることは明らかであろう。よって、説明および実施例は、本発明の範囲を限定するものではない。
【実施例2】
【0245】
この二重盲検プラセボ比較試験への参加には、中程度〜重症のパーキンソン病患者16人が同意した。全ての患者を、KW-6002または対応するプラセボカプセル剤のいずれかに無作為に振り分けた。この試験は、6週間の増量の投与用法用量(40および80mg/日)で実施した。パーキンソン症候群をUPDRS パートIII 運動能力検査により評価した。第二のブラインドされた評価者(blinded rater)によるその後のオフライン評定のため、全ての評価をビデオテープに録画した。
【0246】
KW-6002単独または各患者の至適L-ドーパ用量の静脈内持続注入(a steady-state intravenous infusion)との組み合わせでは、パーキンソン病の重篤度には全く影響がなかった。注入した閾値のL-ドーパでは、KW-6002は、抗パーキンソン病作用を38%増強した(p < 0.05)。医学上有意な薬物毒性は認められなかった。
閾値のL-ドーパと組み合わせたKW-6002は、至適L-ドーパ用量単独の場合と同程度、運動能力の(UPDRS III 運動能力検査尺度を用いて評価した)項目を改善させた。
【0247】
このように、本発明は、L-ドーパによる治療を1種以上のアデノシンA2A受容体アンタゴニストの有効量と組み合わせることによって、臨床上有効な量より少ないL-ドーパ(すなわち、L-ドーパ減量効果)でパーキンソン病患者を治療するための方法および組成物を提供する。
本試験では、安静時およびイスからの立ち上がった時の振戦の平均評価点が、ベースラインおよびプラセボ群に関して、4週および6週で実質的に改善されることが示された。KW-6002で治療した患者では、ベースラインおよびプラセボ治療群に対し、歩行および身体運動緩徐の平均評価点が6週でかなりの改善が認められた。このことは、KW-6002がまた、パーキンソン病患者およびその他の運動疾患を有する患者の両方における振戦および歩行の治療に有効であることを意味する。
【0248】
このように、本発明は、振戦、運動緩徐、歩行および寡動に関連する運動障害を有効治療する方法を提供する。
実施例1および2の試験結果から、アデノシンA2A受容体のメカニズムが、パーキンソン病および運動合併症の症状の発現においてある役割を果たしていること、およびこの受容体を選択的にブロックすることが可能な薬剤が、L-ドーパ治療を受けたこの疾患を伴う患者に治療効果を与えることが確認される。
【0249】
すなわち、本発明は、それを必要とする患者に、1種以上のアデノシンA2A受容体アンタゴニストの有効量を投与することにより運動障害を治療する方法、ならびにパーキンソン病の治療においてL-ドーパ療法を受けている患者のL-ドーパの副作用を減少させるか抑制する方法を提供する。
【実施例3】
【0250】
6−ヒドロキシドーパミン障害処置ラットおよび6−ヒドロキシドーパミン障害処置後にL-ドーパによる長期処置を施したラットにおいて、大脳基底核の出力核、黒質緻密部(SNr)のGABAおよびグルタメート濃度を測定する。アデノシンA2A受容体選択的アンタゴニストの、SNrのGABAおよびグルタメートレベルおよびジスキネジアに及ぼす影響を試験した。
方法:
6−ヒドロキシドーパミン(8μg)をラットの左内側前脳束に注入した。障害処置の1週間後、アポモルヒネを注入(0.1mg/kg 皮下)によるラットの逆側回転(contralateralturning)について試験した。強い逆側回転を示す動物のみを、その後の試験に用いた。アポモルヒネ試験の3日後、L-ドーパを20mg/kgの用量で、1日2回、1〜3週間経口投与した。
【0251】
L-ドーパ誘発性ジスキネジアを認定するため、ラットを個別に観察し、歩行運動、軸性、四肢および口舌のAIMをはじめとする異常不随意時間(abnormal involuntary moments)(AIM)の重症度尺度を記録し、AIMが現れている時間/モニタリング期間の比率によって4つのAIMサブタイプそれぞれに0〜4の評点を付けた。L-ドーパによる長期治療中、AIMの重症度尺度の記録を行った。さらに、微量透析試験中、四肢および軸性のAIMのそれぞれの振幅による尺度を記録した。四肢または軸性のAIMの振幅評点(それぞれ0〜4までの範囲)を、動物の手足/四肢の転位および遠位筋と近位筋群との目に見える関与の両方の大きさ、または動物の首および胴体のその身体の縦軸からの側方変位(または捻転)それぞれに基づき評価した。
SNrにおけるGABAおよびグルタメートを、6−ヒドロキシドーパミン障害処置後とL-ドーパによる反復処置が終了してから4日後、in vivo微量透析技術によって測定した。ラットを各試験チャンバーに入れ、SNrに挿入した微量透析プローブを、自由に動かすことが可能な(持続的な回転行動も可能)液体旋回装置(TCS2-23,Eicom)に取り付けた。プローブを改変リンゲル溶液(1.2mmol/L CaCl2、2.7mmol/L KCl、148mmol/L NaClおよび0.85mmol/L MgCl2;pH7、人工脳脊髄液)とともに、マイクロインジェクションポンプ(CMA/100,Carnegie Medicin AB)を介して2μL/分の速度で連続的に潅流した。放出の基礎レベルを3〜4時間安定化させた後、2時間の潅流中に分画収集装置(CMA/140,Carnegie Medicin)を用いて4サンプル(各60μL)を収集した。各サンプルにあたり60μLの潅水(30分間のもの)をサンプリングチューブ(自動サンプリングインジェクター 231XL,Eicom用のサンプル容器)に2x30μLに分割し、各サンプル中のGABAおよびグルタメートの濃度を測定した。サンプルは直ちにアッセイするかまたは急速冷凍(-80℃)し、保存した後、アッセイした。GABAおよびグルタメートは、オルトフタルジアルデヒド試薬によるアミノ酸のプレカラム誘導体化を行った後、蛍光検出を用いた逆相高速液体クロマトグラフィーにより解析した。LindrothおよびMopper(1979)。
結果:
KW-6002(1mg/kg 経口)は、6−ヒドロキシドーパミン障害処置ラットのSNrにおいて、GABAおよびグルタメートレベルの、著しい、持続的な増加を引き起こした(図2、3)。L-ドーパはまた、6−ヒドロキシドーパミン障害処置ラットによる黒質GABAおよびグルタメートレベルの増加を誘導した(図4、5)。
【0252】
1週間、毎日、L-ドーパによる反復処置をした場合のAIMは、個々のラットによってなお様々であり、最大重症度等級を短時間維持した。2〜3週間のL-ドーパによる長期処置で、動物は安定したAIMを示し、L-ドーパ投与後10分間〜3時間、平均最大AIM評点(9)を維持した。
表3に示すように、黒質グルタメートの基礎濃度は、L-ドーパによる長期処置の2週間の間、一定レベルを維持し、3週間で飛躍的に増加した。一方、黒質GABAレベルは期間を通じて変化がなかった。表3は、6−ヒドロキシドーパミン障害処置後にL-ドーパによる長期処置を施したラットにおける黒質GABAおよびグルタメートの基礎レベルを示している。
【0253】
【表3】
【0254】
L-ドーパは、著しいAIM(四肢および軸性AIMの振幅評点合計)を誘導した。一方、KW-6002は、長期処置したラットにおいてほとんどまたは全くAIMを誘導しなかった(図6)。
L-ドーパは、黒質GABAレベルに影響を及ぼすことなく、グルタメートレベルを高めた。一方、KW-6002は、黒質GABAおよびグルタメートレベルに対して、全くまたはほとんど影響を及ぼさなかった(図7、8)。
【0255】
L-ドーパ誘発性AIMの振幅増大は、L-ドーパ誘発性黒質グルタメートレベルの高まりとともに経時変化した(図6および8)。
【実施例4】
【0256】
MPTPの反復注入によってパーキンソン病にしたL-ドーパまたはドーパミン作動性薬剤を受けたことが全くないMPTPサルにおける、L-ドーパ単独またはKW-6002またはプラセボと
の組み合わせでの長期治療の効果の比較。
動物:3〜5kgの間の重量の8匹の雌の薬剤未投与のカニクイザルを用いた。これらを、明らかなパーキンソン症候群(我々の尺度の6以上に当たる身体障害の評点で扱われる運動不能、前かがみの姿勢および振戦)の発症までMPTP(毎日0.5mg)を皮下注入することによってパーキンソン病にした。必要な累積用量は変動した:3.5〜23.5mg。
【0257】
著しく運動不能であることから早期に処置する必要のあった動物を除き、動物を1ヶ月以上回復させた。少なくとも、1日1回、これらに評点を付けた。身体障害評点は、その期間を通じて安定した状態を保っていた。
処置:全ての動物を、1日1回、L-ドーパ/ベンセラジド 100/25mg(総用量)で処置した。特別なカプセル剤ハンドラ(capsule handler)を用いて、薬剤を経口投与した。KW-6002群の動物はまた、この化合物(90mg/kg)も経口経路によって服用した。動物を毎日そのケージ内でワンウェイスクリーンを通して観察し(月曜日から金曜日まで)、重大な事象(異常行動−ジスキネジア)にはビデオ録画を行った。その薬剤の効能が及ぶ前およびその間、それらに身体障害尺度、実際にはジスキネジア評価尺度で評点を付けた。L-ドーパによる処置を1ヶ月間続けた。
結果
L-ドーパに対する抗パーキンソン病効果は、4週間の間でのパーキンソン病評点の改善という点から言えば、安定しており、L-ドーパ単独群と併用(L-ドーパ+KW-6002)治療群とでは同等であった(図9)。
【0258】
自発運動数は、併用治療群において、より高レベルへと増加し、そのレベルは4週間にわたり維持された(図10)。
ジスキネジアは、併用治療群よりもL-ドーパ群で急速に増え、より高いレベルに達した。このように、ジスキネジアの発症は、KW-6002の存在下で遅らされた。ジスキネジアが現れた後でさえ(3週および4週)、KW-6002治療群では、L-ドーパ単独群よりもジスキネジアの発症は少なかった(図11)。
【0259】
1ヶ月の処置期間の終了時に、全ての薬剤を中止した。その翌日、KW-6002群の動物に、標準用量のL-ドーパ/ベンセラジド(100/25mg)の経口投与を試みた。ジスキネジアをすでに示している3動物では、その組み合わせに対して同様の応答があった。
つまり、これまでに薬剤未投与であるパーキンソン病のサルの1ヶ月間の治療において、KW-6002をL-ドーパへ加えることによって、ジスキネジアの発症が遅らされ、ジスキネジアの発症が少なくなり、それと同時に、より強い自発運動応答およびパーキンソン病評点においてL-ドーパ単独群と同等の改善がもたらされた。
【実施例5】
【0260】
ジスキネジアを示すよう予めL-ドーパを前投与したMPTP処置コモンマーモセットにおいて、L-ドーパ誘発性ジスキネジアに及ぼすKW-6002の影響を調べた。
方法:MPTP(Sigma-Aldrich,St.Louis,MO,USA)を生理食塩水に溶かし、毎日、2.0mg/kgの用量で、5日間皮下投与した。次いで、MPTP 2mg/kgをさらに約3週間投与した。MPTP曝露の8週間後、動物は、基本的な自発運動の著しい減少、連係動作の遅延および減少、身体部位の異常な状態、およびチェック動作(checking movement)および目の瞬きの減少などの慢性パーキンソン病の症状を示した。慢性パーキンソン病の十分な症状を示す動物を、本試験に選抜した。
【0261】
次いで、L-ドーパ(10mg/kg 経口)とともにベンセラジド(2.5mg/kg 経口)を、MPTP処置マーモセットに1日2回、28日間投与し、ジスキネジアを誘導した。動物のジスキネジアに表4に記載の評価尺度を用いて評点を付けた。各L-ドーパ投与によって、高いジスキネジア評点である最大値8を示した動物を本研究に用いた。MPTP処置マーモセットにて、L-ドーパ(10mg/kg 経口、ベンセラジド 2.5mg/kg 経口とともに)によって誘導されたジスキネジアに評点を付けた。評点をL-ドーパ前値として算出した。その翌日、投与媒体(Vehicle)対照値を得るために、動物にVehicleを投与した。1日後、それらにL-ドーパ(2.5mg/kg、経口)を投与し、L-ドーパ対照値を得た。次いで、KW-6002の、L-ドーパ誘発性ジスキネジアに及ぼす影響を観察した。L-ドーパ(2.5mg/kg、経口)と組み合わせたKW-6002(10mg/kg 経口)の投与をその翌日(1日目)に開始し、1日1回、21日間繰り返し、その後、1週間ウォッシュアウト(washout)期間をおいた。動物におけるジスキネジアを、評価尺度に従って、1日、3日、5日、7日、14日、21日および28日目に評価した。さらに、35日目にL-ドーパ(10mg/kg 経口)をマーモセットに投与して、L-ドーパ後値を得た。
【0262】
表4は、四肢ジストニア、舞踏病および舞踏病アテトーゼジスキネジアならびに常同症の存在を数値で示した結果を示している。例えば口腔顔面運動、ミオクローヌスおよび複雑な常同行為(例えば入念なチェック、強迫的グルーミング)等の異常な動作は、ジスキネジア評価から除外される。
【0263】
【表4】
【0264】
ジスキネジアについての備考
ジストニア(腕、脚および胴体):異常な持続的な姿勢(例 脚の挙上)。
常同的なリーチング(stereotypic reaching)(腕)
アテトーシス(腕および脚):くねくねと捻る動作。
舞踏病(腕および脚):四肢の異常な急速な(ダンス様の)動作。
アカシジア:運動性不穏状態。
ジスキネジア評点は、ジスキネジアの重症度に応じて高くなり、最高評点は4点である。
【0265】
結果:結果を図12に示した。L-ドーパ(2.5mg/kg)の経口投与により、ジスキネジアを示すよう予めL-ドーパを前投与したMPTP処置コモンマーモセットに軽度ジスキネジアが誘導された。L-ドーパ(2.5mg/kg 経口)による誘発性ジスキネジアは、L-ドーパ単独の対照と比較して、21日間、KW-6002(10mg/kg 経口)では変化がなかったか、または減少する傾向があった。21日目に、KW-6002は、2.5mg/kgのL-ドーパ単独と比較して、L-ドーパ誘発性ジスキネジアの有意な減少を示した。L-ドーパ誘発性ジスキネジアの、KW-6002によってもたらされる有意な減少は、KW-6002およびL-ドーパの21日間の反復投与後の、KW-6002(10mg/kg)のL-ドーパ(2.5mg/kg)をともなう1週間の急性投与によって認められた。
【0266】
つまり、これらの試験の結果は、KW-6002がL-ドーパ誘発性ジスキネジアを抑制することを示している。
調製例1:錠剤
次の組成の錠剤を従来の方法で調製する。
KW-6002(40g)を286.8gのラクトースおよび60gのジャガイモデンプンと混合し、続いて、120gの10%ヒドロキシプロピルセルロース水溶液の添加を行う。得られた混合物を混練し、造粒した後、従来の方法により乾燥させる。この顆粒を精製し、錠剤を作製するのに用いる顆粒を得る。顆粒を1.2gのステアリン酸マグネシウムと混合した後、混合物を、8mm径の乳棒を備えた錠剤製造機(RT-15型,菊水)を用いて、それぞれが20mgの有効成分を含む錠剤に成形する。
【0267】
この処方を表5に示す。
表5
化合物(I) 20 mg
ラクトース 143.4mg
ジャガイモデンプン 30 mg
ヒドロキシプロピルセルロース 6 mg
ステアリン酸マグネシウム 0.6mg
200 mg
調製例2:カプセル剤
次の組成のカプセル剤を従来の方法で調製する。
【0268】
KW-6002(200g)を995gのアビセル(Avicel)および5gのステアリン酸マグネシウムと混合する。混合物を、カプセル充填機(LZ-64型,Zanashi)を用いて、それぞれが120mgの容量を有する硬カプセルNo.4の中に入れ、それぞれの有効成分を含むカプセル剤を得る。
この処方を表6に示す。
【0269】
表6
化合物(I) 20 mg
アビセル 99.5mg
ステアリン酸マグネシウム 0.5mg
120 mg
調製例3:注射剤
次の組成の注射剤を従来の方法で調製する。
【0270】
KW-6002(1g)を100gの精製大豆油に溶かし、続いて、12gの精製卵黄レシチンおよび25gの注射用グリセリンの添加を行う。得られた混合物を注射用蒸留水で最大1,000mlにし、十分に混合し、従来の方法により乳化させる。得られた分散液を0.2μmの使い捨て薄膜フィルター を用いて、無菌濾過に付した後、2ml分量で、ガラスバイアル中に無菌状態で入れ、各バイアルが2mgの有効成分を含む注射剤を得る。
【0271】
この処方を表7に示す。
表7
化合物(I) 2 mg
精製大豆油 200 mg
精製卵黄レシチン 24 mg
注射用グリセリン 50 mg
注射用蒸留水 1.72ml
2.00ml
前述の発明を、明確にし、理解するために、例示および実施例によって細部にわたって記載してきたが、当業者には、特定の変更および改変を行うことができることは明らかであろう。よって、説明および実施例は、本発明の範囲を限定するものではない。
参照文献一覧
US Patent No. 5,484,920
US Patent No. 5,543,415
US Patent No. 5,587,378
Adler et al. (1993a) “Vitamin E treatment of tardive dyskinesia” Am. J. Psych. 150:1405-1407
Adler et al. (1993b) “Vitamin E in tardive dyskinesia: time course of effect after placebo substitution” Psychopharmacol. Bull. 29:371-374
Adler et al. (1999) “Vitamin E treatment for tardive dyskinesia” Arch. Gen. Psych. 56:836-841
Akhtar et al. (1993) “Vitamin E in the treatment of tardive dyskinesia” J. Neural. Transm. Gen. Sect. 92:197-201
Alexander et al. (1990) “Functional architecture of basal ganglia circuits: neuronal substrates of processing” Trends Neurosci. 13:266-271
American College of Neuropsychopharmacology-FDA Task Force (1973) “Neurologic syndromes associated with neuroleptic drug use” N. Engl. J. Med. 289:20-23
Aoyama et al. (2000) “Rescue of locomotor impairment in dopamine D2receptor-deficient mice by an adenosine A2Areceptor antagonist” J. Neurosci. 20:5848-5852
Aoyama et al. (2002) “Distribution of adenosine A2Areceptor antagonist KW-6002 and its effect on gene expression in the rat brain” Brain Res. 953:119-25
Arvidsson et al. (1997) J. Comp. Neurol. 378:454-467
Augood et al. (1994) “adenosine A2Areceptor mRNA is expressed by enkephalin cells but not by somatostatin cells in rat striatum: co-expression study” Mol. Brain Res. 22:204-210
Baik et al. (1995) “Parkinsonian-like locomotor impairment in mice lacking dopamine D2 receptors” Nature 377:424-428
Baldessarini and Tarsy (1978) “Tardive dyskinesia” In: Psychopharmacology: a generation of progress. Lipton et al. eds. NY Raven Press pp. 995-1004
Baldessarini (1990) “Drugs and the treatment of psychiatric disorders” Goodman and Gilman's The Pharmacological Basis of Therapeutics, Gilman et al. (eds). New York, Pergamon Press, 8th Ed pp. 383-435
Bartholini (1983) “GABA system, GABA receptor agonist and dyskinesia” In: Modern problems in pharmacopsychiatry. Ban et al. eds. New York: Karger 21:143-154
Bezard et al. (2001) “Pathophysiology of levodopa-induced dyskinesia: Potential for new therapies” Nature Neurosci. Rev. 577-588
Bischot et al. (1993) “Vitamin E in extrapyramidal disorders” J. Postgrad. Med. 39:124-126
Blurn and Korczyn (1983) “Peptide neurotransmitters and their implications for the treatment of tardive dyskinesia” In: Modern problems in pharmacopsychiatry. Ban et al. eds. New York: Karger 21:187-95
Brown et al. (1991) “Striatal A2receptor regulates apomorphine-induced turning in rats with unilateral dopamine denervation” Psychopharmacol. 103:78-82
Burns et al. (1986) “Characterization of the A2adenosine receptor labeled by [3H]NECA in rat striatal membranes” Mol. Pharmacol. 29:331-346
Casey (1999) “Tardive dyskinesia and atypical antipsychotic drugs” Schizophrenia Res. 35:561-566
Chen et al. (2001a) J. Neurosci. 21:RC143(1-6)
Chen et al. (2001b) “The role of the D2 dopamine receptor (D2R) in A2A adenosine receptor (A2AR)-mediated behavioral and cellular responses as revealed by A2A and D2receptor knockout mice” Proc. Natl. Acad. Sci. USA 98:1970-1975
Chergui et al. (2000) “Functional GluR6 kinase receptors in the striatum: indirect down-regulation of synaptic transmission” J. Neurosci. 20:2175-2182
Claveria et al. (1975) “Tardive dyskinesia treated with pimozide” J. Neurol. Sci.; 24:393-401
Cochiolo et al. (2000) J. Neuroscience Res.59:126-135
Crossman (1990) “A hypothesis on the pathophysiological mechanisms that underlie levodopa or dopamine-agonist-induced dyskinesia in Parkinson’s disease: implications for future strategies in treatment” Mov. Disord. 5:100-108
Dabiri et al. (1993) “Effectiveness of vitamin E for treatment of long-term tardive dyskinesia” Am. J. Psych. 150:1405-1407
Dabiri et al. (1994) “Effectiveness of vitamin E for treatment of long-term tardive dyskinesia” Am. J. Psych. 151:925-926
Daly et al. (1983) “Subclasses of adenosine receptors in the central nervous system: interaction with caffeine and related methylxanthines” Cell. Mol. Neurobiol. 3:69-80
Daniel et al. (1993) “Parkinson's disease society brain bank, London: overview and research” J. Neural Transm. 39(suppl):165-172
DeLong (1990) “Primate model of movement disorders of basal ganglia origin” Trends Neurosci. 13:281-285
Dixon et al. (1996) “Tissue distribution of adenosine A2A receptor mRNAs in therat” Bri. J. Pharmacol. 118:1461-1468
Driesens (1988) “Neuroleptic medication facilitates the natural occurrence of tardive dyskinesia. A critical review” Acta-Psychiatr.-Belg. 88:195-205
Duvoisin (1974) “Variations in the ‘on-off’ phenomenon” Neurology 24:431-441Egan et al. (1992) “Treatment of tardive dyskinesia with vitamin E” Am. J. Psych. 149:773-777
Egan et al. (1997) “Treatment of tardive dyskinesia” Schiz. Bull. 23:583-609
Elkashef et al. (1990) “Vitamin E in the treatment of tardive dyskinesia” Am. J. Psych. 147:505-506
Fahn et al. (1987) “Members of the UPDRS development committee. Unified Parkinson's disease rating scale” in: Fahn et al. eds., Recent developments in Parkinson's disease, Florham Park, NJ Macmillan Health Care Information 2:153-163 and 293-304
Ferre et al. (1991) “Stimulation of adenosine A2receptors induces catalepsy” Neurosci Lett. 57:1062-1067
Ferre et al. (1996) “Dopamine D1receptor-mediated facilitation of GABAergic neurotransmission in the rat strioentopeduncular pathways and its modulation by adenosine A1receptor-mediated mechanisms” Eur. J. Neurosci. 8:1545-1553
Fink et al. (1992) “Molecular cloning of the rat A2Aadenosine receptor: selective co-expression with D2 dopamine receptors in rat striatum” Mol. Brain Res. 14:186-195
Florio et al. (1999) J. Pharmacol. Exp. Ther.290:817-824
Fredholm et al. (1994) “Action of caffeine in the brain with special reference to dependence” Pharmacol. Rev. 46:143-156
Gardos et al. (1994) “Ten-Year outcome of tardive dyskinesia” Am. J. Psych. 151:836-841
Gelenberg et al. (1990) “A crossover study of lecithin treatment of tardive dyskinesia” J. Clin. Psych. 51:149-153
Gerfen et al. (1990) “D1 and D2dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons” Science 250:1429-1432
Gerfen (1992) “The neostriatal mosaic: multiple levels of compartmental organization in the basal ganglia” Ann. Rev. Neurosci. 15:285-320
Goetz et al. (1994) “Utility of an objective dyskinesia rating scale for Parkinson's disease: inter- and intra-rater reliability assessment” Movement Dis. 9:390-394
Goldberg (1996) “The Use of Vitamin E to Treat People with Tardive Dyskinesia” J. Clin. Psych. 57:167-173
Graybiel (1990) “Neurotransmitters and neuromodulators in the basal ganglia” Trends Neurosci. 13:244-254
Grondin et al. (1999) “Antiparkinsonian effect of a new selective adenosine A2A receptor antagonist in MPTP-treated monkeys” Neurology 52:1673-1677
Groves (1983) “A theory of the functional organization of the neostriatum and the neostriatal control of voluntary movement” Brain Res. Rev. 5:109-132
Hoehn et al. (1967) “Parkinsonism: onset, progression and mortality” Neurol. 17:427-442
Hauser et al. (2000) “A home diary to assess functional status in Parkinson's disease patients with motor fluctuations and dyskinesia” Clin. Neuropharmacol. 23:75-81
Huang et al. (1981) “Reserpine and α-methyldopa in the treatment of tardive dyskinesia” Psychopharmacology 73:359-362
Hubble et al. (2002a) “Kyowa 6002-US-001 Study. Group. A novel adenosine antagonist (KW-6002) as a treatment for advanced Parkinson's disease (PD) with motor complications” Neurol. 54(suppl 6):S21.001
Hubble et al. (2002b) “A novel adenosine antagonist (KW-6002) as a treatment for advanced Parkinson's disease with motor complications” Neurol. 58(suppl 3):A162
Huntinger et al. (2001) “Ultrastructural localization of adenosine A2Areceptors suggests multiple cellular sites for modulation of GABAergic neurons in rat striatum” J. Comparative Neurol. 431:331-356
Ikeda et al. (2002) “Neuroprotection by adenosine A2Areceptor blockade in experimental models of Parkinson's disease” J. Neurochem. 80:262-270
Isaacson et al. (1995) “Counting quanta: direct measurements of transmitter release at a central synapse” Neuron 15:875-884
Itoh et al. (1981) “Drug-induced tardive dyskinesia” In: Current developments in psychopharmacology. Essman, Valzelli, eds. Jamaica, NY: Spectrum 93-126
Jaeger et al. (1994) “Surround inhibition among projection neurons is weak or nonexistent in the rat neostriatum” J. Neurophysiol. 72:2555-2558
Jarvis et al. (1989) “Direct autographic localization of adenosine A2receptors in rat brain using the A2selective agonist [3H]CGS21680” Eur. J. Pharmacol. 168:243-246
Kanda et al. (1994) “KF 17837: a novel selective adenosine A2Areceptor antagonist with anticataleptic activity” Eur. J. Pharmacol. 52:48-54
Kanda et al. (1998a) “Adenosine A2Aantagonist: a novel antiparkinsonian agent that does not provoke dyskinesia in parkinsonian monkeys” Ann. Neurol. 43:507-513
Kanda et al. (1998b) NeuroReport 9:2857-2860
Kanda et al. (2000) “Combined use of the adenosine A2A antagonist KW-6002 with L-DOPA or with selective D1or D2 dopamine agonists increases antiparkinsonian activity but not dyskinesia in MPTP-treated monkeys” Exp. Neurol. 162:321-327
Karp et al. (1981) “Metoclopramide treatment of tardive dyskinesia” JAMA 246:1934-1935
Kase et al. (2000) “Adenosine receptors and Parkinson's disease” Academic Press, San Diego
Kase (2001) “New aspects of physiological and pathophysiological functions of adenosine A2A receptor in basal ganglia” Biosci. Biotechnol. Biochem. 65:1447-1457
Kawaguchi (1989) “Intracellular recording of identified neostriatal patch and matrix spiny cells in a slice preparation preserving cortical inputs” J. Neurophysiol. 62:1052-1068
Kawaguchi et al. (1990) “Projection subtypes of rat neostriatal matrix cells revealed by intracellular injection of biocytin” J. Neurosci. 10:3421-3438
Kawaguchi et al. (1995) “Striatal interneurons: chemical, physiological and morphological characterization” Trends Neurosci. 18:527-535
Kawaguchi (1997) “Neostriatal cell subtypes and their functional roles” Neurosci. Res. 27:1-8
Kirk et al. (1994) “Adenosine A2Areceptor-mediated modulation of striatal [3H]GABA and [3H]acetylcholine release” J. Neurochem. 62:960-966
Kita et al. (1983) “Pallidal inputs to subthalamus: intracellular analysis” Brain Res. 264:255-265
Kita (1993) “GABAergic circuits of the striatum” in: Arbuthnott et al. eds., Progress in Brain Research, Elsevier Science Publishers B.V. 99:51-72
Kita (1994) “Parvalbumin-immunopositive neurons in rat globus pallidus: a light and electron microscope study” Brain Res. 657:31-41
Kita (1996) “Glutamatergic and GABAergic postsynaptic responses of striatal spiny neurons to intrastriatal and cortical stimulation recorded in slice preparations” Neurosci. 70:925-940
Klawans et al. (1980) “Tardive dyskinesia: Review and update” Am. J. Psych. 137:900-908
Koga et al. (2000) “Adenosine A2Areceptor antagonists KF17837 and KW-6002 potentiate rotation induced by dopaminergic drugs in hemi-parkinsonian rats” Eur. J. Pharmacol. 408:249-255
Koos (1999) “Inhibitory control of neurostriatal projection neurons by GABAergic interneurons” Nature Neurosci. 2:467-472
Kurokawa et al. (1994) “Inhibition by KF17837 of adenosine A2Areceptor-mediated modulation of striatal GABA and Ach release” BR. J. Pharmacol. 113:43-48
Kurokawa et al. (1996) J. Neurochem. 66:1882-1888
Kuwana et al. (1999) Adv. Neurol. 80:121-123
Lindroth and Mopper (1979) “High performance liquid chromatography determination of subpicomole amounts of amino acids by precolumn fluorescence derivatization
with o-phthaldialdehyde” Analyt. Chem. 51:1667-1674
Lohr and Caligiuri (1996) “A double-blind placebo-controlled study of vitamin E treatment of tardive dyskinesia” J. Clin. Psych. 57:167-73
Lohr et al. (1988) “Vitamin E in the treatment of tardive dyskinesia: the possible involvement of free radical mechanisms” Schiz. Bull. 14:291-296
Loopuijt et al. (1985) “Organization of the striatum: collateralization of its efferent axons” Brain Res. 348:86-99
Marsden et al. (1982) “Fluctuations in disability in Parkinson’s disease: clinical aspects” In: Mardsen, CD, Fahn S., eds. Movement disorders. New York: Butterworth Scientific pp. 96-122
McCreadie et al. (1994) “The Nithsdale Schizophrenia Surveys. XIV: Plasma lipid peroxide and serum vitamin E levels in patients with and without tardive dyskinesia, and in normal subjects” Am. J. Psych. 151:925-926
Moore and Bowers (1980) “Identification of a subgroup of tardive dyskinesia patients by pharmacologic probes” Am. J. Psych. 137:1202-1205
Mori et al. (1996) “The role of adenosine A2Areceptors in regulating GABAergic synaptic transmission in striatal medium spiny neurons” J. Neurosci. 16:605-611Mori et al. (2002) “Adenosine A2Areceptor-mediated dual modulation of GABAergic synaptic transmission in the striatopallidal system and its implication in action mechanism of A2Aantagonists for a novel therapy in Parkinson's disease” Neurol. (in press)
Moss et al. (1993) “Buspirone in the treatment of tardive dyskinesia” J. Clin. Psychopharmacol. 13:204-209
Nakanishi et al. (1987) “Electrical membrane properties of rat subthalamic neurons in an in vivo slice preparation” Brain Res. 437:35-44
Nambu et al. (1997) “Morphology of globus pallidus neurons: its correlation with electrophysiology in guinea pig brain slices” J. Comp. Neurol. 377:85-94
Nonaka et al. (1994) “(E)-8-(3,4,5-trimethoxystyryl)-1,3-dipropyl-7-methyl-xanthine, a potent and selective adenosine A2A receptor antagonist” Eur. J. Pharmacol. 267:335-341
Nonaka et al. (2002) “Biochemical characterization of a novel adenosine A2Areceptor antagonist, KW-6002” Seikaguku 74:937 3P663 (Abstract)
Obeso et al. (1997) “Basal ganglia physiology - A critical review” Advances in Neurology (Obeso et al. eds) 74:3-7 Lippincott Raven Publishers, Philadelphia
Obeso et al. (2000) “Pathophysiology of the basal ganglia in Parkinson's disease” Trends Neurosci. 23(Suppl):S8-S19
Ochi et al. (2000) “Systemic adenosine A2Areceptor antagonist decreases GABA release from rat globus pallidus increased by nigrostriatal lesions: A microdialysis study” Neuroscience 100:53-62
Oertel et al. (1984) “Immunocytochemical studies of GABAergic neurons in rat basal ganglia and their relations to other neuronal systems” Neurosci. Lett. 47:233-238
Olanow, Watts and Koller eds.(2001) An Algorithm (Decision Tree) for the Management of Parkinson’s Disease (2001): Treatment Guidelines, Neurology 56, Suppl. 5.
Parkinson Study Group (1996) “Impact of deprenyl and tocopherol treatment on Parkinson’s disease in DATATOP patients requiring levodopa” Ann. Neurol. 39:37-45
Parkinson Study Group (2000) “Pramipexole vs. levodopa as initial treatment for Parkinson's disease: a randomized controlled trial” JAMA 284:1931-1938
Paskevich et al. (1991) “Morphological assessment of neuronal aggregates in the striatum of the rat” J. Comp. Neurol. 305:361-369
Przedborski et al. (1995) Neuroscience 67:631-647
Rascol et al. (2000) “A five-year study of the incidence of dyskinesia in patients with early Parkinson's disease who were treated with ropinirole or levodopa” N. Eng. J. Med. 342:1484-1491
Richardson et al. (1997) “Adenosine A2Areceptor antagonists as new agents for the treatment of Parkinson's disease” Trends Pharmacol. Sci. 18:338-344
Rosin et al. (1998) “Immunohistochemical localization of adenosine A2Areceptors in the rat central nervous system” J. Comp. Neurol. 401:163-186
Schiffmann et al. (1990) Brain Res.519:333-337
Schiffmann et al. (1991a) “Striatal restricted A2receptor (RDC8) is expressed by enkephalin but not by substance P neurons: an in situ hybridization histochemistry study” J. Neurochem. 57:1062-1067
Schiffmann et al. (1991b) “Distribution of adenosine A2 receptor mRNA in the human brain” Neurosci. Lett. 130:177-181
Sherzai et al. (2002a) “Adenosine A2Aantagonist treatment of Parkinson's disease” Neurol. 54(Suppl 6):P06.104
Sherzai et al. (2002b) “Adenosine A2Aantagonist treatment of Parkinson's disease” Neurol. 58(suppl 3):A467
Shimada et al. (1992) “(E)-1,3-dialkyl-7-methyl-8-(3,4,5-trimethoxystyryl)xanthines: potent and selective adenosine A2 antagonists” J. Med. Chem. 35:2342-2345Shimada et al. (1997) “Adenosine A2Aantagonists with potent anti-cataleptic activity” Bio. Med. Chem. Lett. 7:2349-2352
Shindou et al. (2001) “Adenosine A2Areceptor enhances GABAA-mediated IPSCs in the rat globus pallidus” J. Physiol. 532:423-434
Shindou et al. (2002) “Presynaptic adenosine A2Areceptor enhances GABAergic synaptic transmission via cAMP dependent mechanism in the rat globus pallidus” Br.
J. Pharmacol. 136:296-302
Shiozaki et al. (1999) “Action of adenosine A2Areceptor antagonist KW-6002 on drug-induced catalepsy and hypokinesia caused by reserpine or MPTP” Psychopharmacology 147:90-95
Shriqui et al. (1992) “Vitamin E in the treatment of tardive dyskinesia: a double-blind placebo-controlled study” Am. J. Psych. 149:391-143
Shulman and Weiner (1997) Multiple system atrophy. In: Watts and Koller eds. Movement Disorders: Neurological Principles and Practice. New York, NY: McGraw-Hill pp. 297-306
Smith et al. (1987) “Distribution of GABA-immunoreactive neurons in the basal ganglia of the squirrel monkey” J. Comp. Neurol. 259:50-64
Stern et al. (1998) “Membrane potential synchrony of simultaneously recorded striatal spiny neurons in vivo” Nature 394:475-478
Tepper and Haas (1979) “Prevalence of tardive dyskinesia” J. Clin. Psych. 40:508-516
Thaker et al. (1990) “Clonazepam treatment of tardive dyskinesia: a practical GABAmimetic strategy” Am. J. Psych. 147:445-451
Uhrbrand and Faurbye (1960) “Reversible and irreversible dyskinesia after treatment with perphenazine, chlorpromazine, reserpine, ECT therapy” Psychopharmacologia 1:408-418
Watts and William eds. (1997) Movement Disorders: Neurologic Principles and Practice. New York: McGraw-Hill
Wichmann et al. (1993) “Pathophysiology of parkinsonian motor abnormalities” In: Narabayashi et al. eds. Advance in Neurology, Raven Press Ltd., 60:53-61
Wilbur and Kulik (1980) “Propranolol for tardive dyskinesia and EPS from neuroleptics: Possible involvement of β-adrenergic mechanisms” Prog. Neuro-Psychopharmacol. 4:627-632
【図面の簡単な説明】
【0272】
【図1】図1は、プラセボ群およびKW-6002群を併せた群の、自宅での日誌で記録したオフ時間の変化を示すグラフである。KW-6002を用いて治療を受けた被験者では、12週目に、オフの時間が有意に大幅に減少した(*p=0.004)。
【図2】図2は、6−ヒドロキシドーパミン障害処置ラットにおける、黒質GABAのレベルへのKW-6002の影響を示すグラフである。GABAのレベルは、化合物投与前の初期値からの変化率として表されている。KW-6002は、1mg/kg経口で黒質GABAのレベルを有意に高めた。
【図3】図3は、6−ヒドロキシドーパミン障害処置ラットにおける、グルタメートのレベルへのKW-6002の影響を示すグラフである。グルタメートのレベルは、化合物投与前の初期値からの変化率として表されている。KW-6002は、1mg/kg経口でグルタメートのレベルを有意に高めた。
【図4】図4は、6−ヒドロキシドーパミン障害処置ラットにおける、黒質GABAのレベルへのL-ドーパの影響を示すグラフである。L-ドーパは、黒質GABAをKW-6002によるそれらと同様のレベルまでの有意に上昇させた。
【図5】図5は、6−ヒドロキシドーパミン障害処置ラットにおける、グルタメートのレベルへのL-ドーパの影響を示すグラフである。L-ドーパは、グルタメートをKW-6002によるそれらと同様のレベルまでの有意に上昇させた。
【図6】図6は、長期間L-ドーパ処置した6−ヒドロキシドーパミン障害処置ラットにおける、異常不随意運動(AIMs)の総評点へのKW-6002およびL-ドーパの影響の経時変化を示すグラフである。L-ドーパは、著しくAIMsを誘発したが、KW-6002は、AIMsをほとんどまたは全く誘発しなかった。
【図7】図7は、長期間L-ドーパ処置した6−ヒドロキシドーパミン障害処置ラットにおける、黒質GABAのレベルへのKW-6002およびL-ドーパの影響の経時変化を示すグラフである。L-ドーパは、黒質GABAのレベルに影響を及ぼさなかった。KW-6002は、黒質GABAのレベルに、全くまたはほとんど影響を及ぼさなかった。
【図8】図8は、長期間L-ドーパ処置した6−ヒドロキシドーパミン障害処置ラットにおける、グルタメートのレベルへのKW-6002およびL-ドーパの影響の経時変化を示すグラフである。L-ドーパは、黒質GABAのレベルに影響を及ぼすことなく、グルタメートのレベルを高めた。KW-6002は、黒質GABAおよびグルタメートのレベルに、全くまたはほとんど影響を及ぼさなかった。
【図9】図9は、カニクイザルにおけるL-ドーパ単独処置(L-ドーパ/ベンセラジド;100/25mg(総用量)、1日1回)およびL-ドーパとKW-6002の処置(90mg/kg、1日1回)の間の、L-ドーパに対する抗パーキンソン病作用へのKW-6002の影響を示すグラフである。L-ドーパに対する抗パーキンソン病作用は、4週間にわたるパーキンソン病評点の改善という点から言えば、安定しており、2群間では同等であった。
【図10】図10は、カニクイザルにおけるL-ドーパ単独処置(L-ドーパ/ベンセラジド;100/25mg(総用量)、1日1回)およびL-ドーパとKW-6002の処置(90mg/kg、1日1回)の間の、L-ドーパに対する自発運動応答へのKW-6002の影響を示すグラフである。自発運動数は、併用処置群でより高いレベルに増加し、そのレベルは4週間にわたり維持された。
【図11】図11は、カニクイザルにおけるL-ドーパ単独処置(L-ドーパ/ベンセラジド;100/25mg(総用量)、1日1回)およびL-ドーパとKW-6002の処置(90mg/kg、1日1回)の間の、L-ドーパに対するジスキネジア応答へのKW-6002の影響を示すグラフである。ジスキネジアは、併用処置群よりもL-ドーパ群で急速に増加し、より高いレベルに達した。ジスキネジアの発症は、KW-6002の存在下では遅延された。
【図12】図12は、L-ドーパ誘発性ジスキネジアへのKW-6002の影響を示すグラフである。ジスキネジアを発症するようにL-ドーパを前投与した一般的なMPTP処置マーモセットにおいて、L-ドーパ(2.5mg/kg p.o.でベンセラジド0.625mg/kg p.o.とともに)を毎日、21日間投与して、ジスキネジアを誘発させ、同時にKW-6002を投与した。動物には、予めL-ドーパを10mg/kg p.o.でベンセラジド2.5mg/kg p.o.とともに、1日2回、28日間与えた(L-ドーパ)。併用処置では、生じた不随意運動量は増えず、それどころか2.5mg/kgのL-ドーパ単独投与と比べて、21日目に有意に減少した。 KW-6002は、21日間の長期処置により、L-ドーパ誘発性ジスキネジアを有意に減少させた。
【技術分野】
【0001】
本発明は、少なくとも1種のアデノシンA2A受容体アンタゴニストを投与することを特徴とする運動障害を患っている患者を治療する方法に関する。
【背景技術】
【0002】
運動障害とは、運動の不足もしくは欠如(例えば、パーキンソン病等)または過剰運動(例えば、ジストニア(dystonia)、ジスキネジア(dyskinesia)、振戦(tremor)、舞踏病(chorea)、バリズム(ballism)、アカシジア(akathisia)、アテトーシス(athetosis)、運動緩慢(bradykinesia)、すくみ(freezing)、硬直(rigidity)、姿勢不安定(postural instability)、ミオクローヌス(myoclonus)およびチック(tics)もしくはトゥレット症候群(Tourette syndrome)等)のいずれかを特徴とする神経学上の状態である。ワッツ(Watts)およびウィリアム(William)編(1997)ならびにシャルマン(Shulman)およびウェイナー(Weiner)(1997)参照。
【0003】
パーキンソン病および運動合併症
パーキンソン病(振戦麻痺)は、振戦ならびに歩行、運動および協調の困難を特徴とする脳の疾患である。この疾病は筋運動を支配する脳の一部の損傷と関連している。
パーキンソン病は最初、1817年に英国でジェームス・パーキンソン(James Parkinson)によって記載された。この疾病はおよそ1000人のうち2人に影響を及ぼし、ほとんどの場合50歳以降に発症する。症状はまず、平均して、約60歳で表れ、パーキンソン病の症状の重篤度は時間とともに悪化する傾向がある。男性、女性双方に影響を及ぼし、高齢者の最も多く見られる神経疾患の1つである。「パーキンソン症候群(parkinsonism)」という用語は、パーキンソン病において見られる運動の変化の種類の組み合わせを含むいずれかの状態を指す。パーキンソン症候群は遺伝性であることもあるし、その他の疾患によってまたは外部要因によって引き起こされることもある(続発性パーキンソン症候群)。
【0004】
米国では、約100万人がパーキンソン病を患っていると考えられており、毎年、約50,000件の新しい症例が報告されている。通常、症状は高齢期に表れるので、これらの数字は、今後数十年にわたり、集団の平均年齢が上がるにつれ高くなると予測される。この疾患は70代および80代の人々の間で最もよく見られ、女性よりも男性でわずかにより多く見られるようである。
【0005】
黒質緻密部および腹側被蓋野のドーパミン作動性ニューロンは、それぞれ、運動の調節および認知において重大な役割を果たしている。数通りの証拠から、黒質中のドーパミン作動性細胞(すなわち、ドーパミン産生細胞)の変性がパーキンソン病の症状をもたらすことが示唆されている。黒質のその領域に集中しているドーパミン作動性細胞は、体内で最も速く加齢する細胞である。ドーパミン作動性細胞が崩壊すると、運動に対する制御が損なわれ、パーキンソン病が発症する。
【0006】
通常、パーキンソン病の最初の症状は、特に身体を静止しているときの肢の振戦(震えまたは振動)である。振戦は半身で始まることが多く、片側の手において頻繁に起きる。その他のよく起こる症状としては、例えばゆっくりとした動き(運動緩慢)、運動不能(アキネジア)、体肢の硬直、引きずり歩行、前かがみの姿勢等のその他の運動障害が挙げられる。パーキンソン病患者は表情が乏しくなり、静かな声で話すことが多い。この疾病は、鬱病、不安、人格変化、認知障害、痴呆、睡眠障害、言語障害または性的不全といった二次的な症状を引き起こすことがある。パーキンソン病の治療法は知られていない。治療はそれらの症状の制御を目的としている。薬物療法では主として神経伝達物質間の不均衡を制御することによってそれらの症状を制御する。ほとんどのパーキンソン病の初期患者は、ドーパミン補充療法による対症療法に対してよく応答するが、疾病が進行するにつれ能力障害が増加する。
【0007】
用いる薬剤、用量および投与間隔は、症例に応じて変わる。症状変化に合わせて、用いる薬剤の組み合わせを調節する必要があり得る。多くの薬剤は重大な副作用を引き起こすことがあるので、医療提供者によるモニタリングとフォローアップが重要である。
現在利用できるパーキンソン病用薬剤は、一般に、数年間は十分な対症制御をもたらすものの、多くの患者が、運動変動およびジスキネジアを発症し、これが臨床効果を鈍らせる。ラスコール(Rascol)ら(2000)およびパーキンソン研究グループ(2000)。これが起こると、ドーパミン作動性療法を増やすことはジスキネジアを悪化させる恐れがあり、ドーパミン作動性療法を減らすことは運動機能を悪化させ、オフ時間を増加させる恐れがある。この問題を踏まえて、非ドーパミン作動性神経伝達物質系の治療的処置の可能性が注目されることとなった。
【0008】
ほとんどのパーキンソン病の症状は、ドーパミンの不足から生じ、ほとんどの抗パーキンソン薬はドーパミンを元の状態に戻すかドーパミンの作用を模倣するものである。しかし、これらの薬剤はドーパミンを恒久的に元に戻すものではなく、ドーパミンの作用を正確に模倣するものでもない。黒質にドーパミン細胞がないことがパーキンソン病の主な特徴ではあるが、非ドーパミン神経細胞も喪失している。さらに、ドーパミン応答細胞は黒質だけでなく他の脳領域にも存在する。したがって、パーキンソン病において有効な薬剤は、これらの細胞を刺激することにより、例えば悪心、幻覚、錯乱等の副作用を引き起こし得る。
【0009】
L-ドーパは1967年に報告され、以来最も有効な抗パーキンソン薬となっている。L-ドーパの有効性が最も高い症状としては、運動緩慢、硬直、静止時振戦、歩行困難および小書症が挙げられる。L-ドーパの有効性があまり望めない症状としては、姿勢不安定、動作時振戦および嚥下困難が挙げられる。L-ドーパは痴呆を悪化させる可能性がある。L-ドーパは、パーキンソン病において、強くかつ急速な治療上の効果をもたらすが、最終的には、例えばウェアリング・オフ現象、オン・オフ変動、ジスキネジア等の運動合併症をはじめとするドーパミンによって引き起こされる重篤で好ましくない反応が現れる。マースデン(Marsden)ら(1982)。運動合併症は、通常、一度発症すると、L-ドーパまたは他のドーパミン作動性薬剤による処置では制御不能である。
【0010】
パーキンソン病の初期にはL-ドーパを1日3回服用する。脳内でのピーク濃度は投与後1〜2時間で生じる。薬剤の半減期は短い(0.5〜1時間)が、脳内に残存しているドーパミン細胞がドーパミンを貯蔵し、数時間の間その活性を維持するには十分である。パーキンソン病が進行すると、より多くのドーパミン細胞が死滅し、残存する細胞ではその効果を維持するのに十分なドーパミンを貯蔵できなくなり、各投与量での作用持続時間が減少し、患者に対してより高用量なまたはより頻繁な投与が必要となる。2〜5年後には、50〜75%もの患者で、L-ドーパに対する反応、つまりオン/オフ期間に変動が起こる。変動に伴い、患者はジスキネジアを発症する。通常、ジスキネジアはL-ドーパのピーク作用時に生じるが、薬剤の効果が切れるときまたはストレスが多いときにもまた生じる場合がある。変動およびジスキネジアは患者の生活に深刻な影響を及ぼし得る。L-ドーパが連続投与されれば(静脈内ポンプによって)、オン/オフ作用はなくなり、ジスキネジアも減少する。しかし、L-ドーパを静脈内投与することはできない。
【0011】
L-ドーパを単独で服用すると、その一部がドーパ−デカルボキシラーゼによって脳外でドーパミンに変換される。このように生じたドーパミンは脳に入ることができず、例えば悪心、嘔吐、食欲の喪失等の副作用を引き起こす。従って、L-ドーパはカルビドパまたはベンセラジド(benserazide)と組み合わせることが多い。カルビドパは脳外のドーパ−デカルボキシラーゼをブロックすることにより、悪心、嘔吐および食欲の喪失を引き起こすことなく、より多くのL-ドーパが脳に入ることができるようにする。アタメット(Atamet)またはシネメット(Sinemet)はカルビドパとL-ドーパの両方を含有する錠剤である。カルビドパとの組み合わせでは、L-ドーパの半減期は1.2〜2.3時間である。
【0012】
その発見から30年、L-ドーパは依然としてパーキンソン病の最良の治療である。この疾病の初期段階では、患者は通常L-ドーパに対する良好な反応を享受するが、疾病が進行すると、L-ドーパはあまり有用でなくなる傾向がある。これはL-ドーパの効力の喪失によるものではなく、例えばエンド・オブ・ドーズ(end-of-dose)での悪化または「ウェアリング・オフ(wearing-off)」、「オン/オフ変動」、ジスキネジア等の運動応答における逆変動のような運動合併症の発症によるものである。オン/オフ変動とは、薬剤治療における効果(「オン」状態、患者にパーキンソン病の症状が比較的ない期間)が、突然、容認できないほどに失われ、パーキンソン状態(「オフ」状態)を発現することである。ウェアリング・オフ現象はL-ドーパが有効である期間の減少であり、「オフ」状態が徐々に再発することを特徴とし、「オン」状態が短くなる。ジスキネジアは、舞踏病(多動性の、目的のないダンスのような動き)とジストニア(持続性の、異常な筋収縮)に大別することができる。1974年に、ディボアザン(Duvoisin)が最初にこれらの異常な不随意運動に着目し、パーキンソン病の患者の半数以上が治療の6ヶ月以内にジスキネジアを発症するということを見出した。治療期間が長くなるにつれ、ジスキネジアの頻度および重症度も増加する。神経保護に有用であると思われる薬剤のパーキンソン病における効果の可能性について影響を与えた研究−DATATOP試験−では、平均20.5ヶ月間L-ドーパの治療を受けた患者の20〜30%でL-ドーパ誘発性ジスキネジアが観察された。最終的に、L-ドーパ治療を受けた患者のほとんどがジスキネジアを経験し、患者の最大80%で、治療の5年以内にジスキネジアを発症した。パーキンソン研究グループ(1966)およびラスコール(Rascol)ら(2000)。治療に関連したジスキネジアは、単にL-ドーパのみの問題ではなく、ドーパミン受容体アゴニストも同様にジスキネジアを誘発し得る。このように、共通の用語「L-ドーパ誘発性ジスキネジア」は、一般用語でドーパミン治療に関連したジスキネジアを記載するために用いられることもある。ほとんどのジスキネジアは、レボドパまたは他のドーパミン受容体アゴニストが、被殻中の過敏性ドーパミン受容体に対して十分である一定の脳内濃度であるときに生じる(ピーク・ドーズ・ジスキネジア(peak-dose-dyskinesia))。しかし、ジスキネジアはまた、ドーパミン濃度が低い際(オフ・ジストニア)、またはドーパミン濃度が増減する状態(二相性ジスキネジア)でも生じる。また、例えばミオクローヌス、アカシジア等の他の運動障害もL-ドーパ誘発性ジスキネジアの範疇の構成要素であり得る。
【0013】
パーキンソン病におけるL-ドーパによる運動合併症の生物学的機序は、未だ少しも解明されていない。その生物学的機序は、進行性の疾病であることおよび黒質のニューロンが持続的に喪失されることだけではなく、ドーパミン受容体の感受性およびタンパク質のそれらの下流発現の変化、ならびにその一連の発現がL-ドーパまたはドーパミンアゴニストの用量および投与方法と少なくともある程度関係している遺伝子に関与している可能性が示唆されている。例えばグルタメート作動性神経伝達、GABA作動性神経伝達、オピオイドペプチド作動性伝達等の非ドーパミン系における変化もまた、パーキンソン病におけるL-ドーパによる運動合併症の根底にあるニューロンのメカニズムに関与している可能性がある。ベザート(Bezard)ら(2001)。特に、ドーパミン作動性薬剤の短い血漿半減期とその結果としての短い作用期間およびドーパミン作動性薬剤によるドーパミン受容体の拍動性刺激が、運動変動およびピーク・ドーズ・ジスキネジアと関連していると思われる。これらの事象はすべてが相まって、大脳基底核と皮質の間に信号を伝える発火パターンの変化をもたらす。
【0014】
当初、変動のある患者にL-ドーパの補助的療法として導入されたドーパミンアゴニストは、今では、初期の患者における単独療法としてますます提唱されるようになっている。しかし、通常、ドーパミンアゴニストの抗パーキンソン病作用はL-ドーパのものよりも少なく、2〜4年後にはその効果が減弱する。より強力な治療が必要な場合には、低用量のL-ドーパをアゴニストに「追加する」ことができる。別の戦略として、最初からアゴニストを低用量のL-ドーパと組み合わせることもある。両戦略ともL-ドーパと同程度に有効であり、運動変動およびジスキネジアの危険性を大きく減少させるという効果を有するとされている。しかし、これらの主張は少数の試験的研究に基づいたものであり、そのすべてには方法論的な欠点がある。
【0015】
さらに、ドーパミン受容体アゴニストもまたジスキネジアを誘発し得る。ドーパミンアゴニストもまた、先にL-ドーパに曝されていたパーキンソン病の動物でジスキネジアを誘発する。神経精神医学的副作用、特に、幻覚および精神病により、ドーパミンアゴニストの使用が制限されることも多い。ドーパミンアゴニストの補助的使用によってもたらされる有力な利点があるにもかかわらず、このようにL-ドーパによる運動合併症を制御するのは極めて困難であるか、不可能でさえあり得る。オラノー(Olanow)、ワッツ(Watts)およびコラー(Koller)編(2001)参照。最後に、ドーパミンアゴニストは、進行したパーキンソン病および重度の運動変動およびジスキネジアの患者においてL-ドーパの代わりとして単独療法で用いられることがある。
【0016】
より最近では、例えばトルカポン(tolcapone)、エンタカポン(entacapone)等のカテコールアミン−O−メチルトランスフェラーゼ(COMT)阻害剤が、L-ドーパの補助的療法として提唱されている。これらの化合物は、Cmaxを大きく高めることなく、L-ドーパの血漿半減期を延長する。したがって、これらはウェアリング・オフの期間を減少させるが、ピーク・ドーズ・ジスキネジアをはじめとするピーク・ドーズの副作用の強度を高める傾向がある。トルカポンは少数の患者では重篤な肝臓毒性を誘発するようである。
【0017】
例えばトリヘキシフェニジル(アーテン(artane))、ビペリジン(コゲンチン(cogentin))等の抗コリン作用薬は、脳においてアセチルコリンの作用をブロックする。この結果、例えば流延、振戦等の症状が軽度〜中程度改善され得る。65歳を超える患者では、抗コリン作用薬を用いて治療した場合、例えば口渇、かすみ目、便秘、錯乱、幻覚等の副作用が起こりやすい。
【0018】
ジストニア
ジストニアという用語は、持続的に異常な姿勢をもたらす持続性の筋収縮を特徴とする運動障害を指す。この定義に基づいて、いくつかのジストニア症候群があり、これらは、全身性(すべての身体部分に影響を及ぼす)、髄節性(隣接する身体部分に影響を及ぼす)または局所性(単一の身体部分に限定された)のようなその臨床上の特徴にしたがって細分化することができる。局所性ジストニアとしては、痙攣性斜頚(spasmodic torticollis)、眼瞼痙攣(blepharospasm)、半側顔面痙攣(hemifacial spasm)、開口ジストニア(oromandibular dystonia)、痙攣性発声障害(spasmodic dysphonia)およびジストニア性書痙(dystonic writer's cramp)が挙げられる。
【0019】
さまざまな程度のジストニアがある。比較的正常なライフスタイルを維持できる人もあれば、持続的に障害があり、フルタイムの介護を必要とすることが多い人もいる。
症状は局所的であることもあれば、身体のある領域、例えば、首または腕または脚に限定されていることもある。様々な種類の局所性ジストニアがある。眼瞼痙攣は瞼の運動を制御する筋肉の不随意収縮を特徴とする。症状は、断続的なもの、無痛なもの、休みなく続くまでに増えた瞬き、有痛なもの、機能上の盲目をもたらす閉眼まで様々であり得る。痙攣性斜頚としても知られる頸部ジストニア(CD)の患者では、頭頸部の筋痙攣は有痛であり、首をねじれさせることもある。これらの有痛の痙攣は断続的であることもあるし、休みなく続くこともある。開口ジストニアおよび舌ジストニアは、口の開閉を引き起こす顔下部の強力な収縮を特徴とする。咀嚼や異常な舌の動きも起こり得る。喉頭ジストニアとしても知られる痙攣性発声障害(SD)では、発声器(喉頭)の筋が影響を受ける。SDは声帯を開けることか閉じることのいずれかの困難を特徴とし、これによって声が緊張した、しわがれた、のどの詰まったようなまたはささやくような音質のいずれかになる。体肢ジストニアでは、上肢、手、下肢または足の1以上の筋の不随意収縮がある。これらの種類の局所性ジストニアとしては、書痙および他の職業性ジストニアが挙げられる。
【0020】
分節性であるか、または例えば頭と首、上肢と躯幹等の身体の2つの隣接する領域に関与する症状を有する患者もいる。他の患者では、症状が多巣性であったり、例えば両腕、上肢と下肢等の互いに隣接していない身体の2つの領域に現れたりすることもある。全身性ジストニアでは、症状は上肢または下肢において始まり、進行してより広範なものとなる。最終的に躯幹および身体の残りの部分にまで及ぶ。
【0021】
原発性または特発性ジストニアのほとんどの症例は、遺伝性であり、遺伝子の欠陥の結果として起こると考えられている。これらの患者では、ジストニアは単一の症状として起こり、潜在的疾患を伴わない。例えば、早発性原発性ジストニアのほとんどの場合が、DYT-1遺伝子における突然変異によるものである。この疾病遺伝子の結果として起こる早発性ジストニアは、最も多く見られかつ重症型の遺伝性ジストニアである。他の遺伝的原因が原発性ジストニアを引き起こすことは稀である。
【0022】
ジストニアが関与する疾病としては、遺伝性痙性対麻痺(hereditary spastic paraplegia (HSP))、つまり両下肢の進行性の脱力およびこわばりを特徴とする脊髄の遺伝性変性疾患の一群;ハンチントン病(Huntington's disease (HD))、つまり情緒的、行動的および精神医学的異常ならびに運動異常の発症を特徴とする遺伝性進行性神経変性疾患;多系統萎縮症(multiple system atrophy (MSA))、つまり運動、血圧およびその他の身体機能に影響を及ぼす症状の組み合わせを特徴とする神経変性疾患;病的ミオクローヌス(pathologic myoclonus);進行性核上性麻痺(progressive supranuclear palsy);レストレスレッグズ症候群(restless legs syndrome);レット症候群(Rett syndrome);痙縮(spasticity);シデナム舞踏病(Sydenham's chorea);トゥレット症候群(Tourette syndrome)およびウィルソン病(Wilson disease)が挙げられる。
【0023】
ジストニアは、例えばウィルソン病、多発性硬化症、脳卒中等の別の潜在的疾病過程;例えば自動車事故での損傷または誕生時の酸欠等の脳の外傷;または薬物療法の副作用として起こることもある。この種のジストニアは続発性または症候性ジストニアと呼ばれる。成人で、最もよく見られる続発性ジストニアの種類は、遅発性ジストニア(tardive dystonia)であり、これは特定の神経遮断薬または抗精神病薬(精神疾患の治療に用いられる)の使用の結果として起こる。これらの薬剤としては、ハロペリドール(ハルドール(Haldol)(登録商標))またはクロルプロマジン(トラジン(Thorazine)(登録商標))が挙げられる。中枢神経のドーパミン受容体をブロックする他の薬剤もまた遅発性ジストニアを引き起こし得る。ほとんどの患者で、薬剤に曝露され続けた後、症状はいずれそのうち生じる。表1にジストニアを引き起こし得る薬剤のリストを示す。
【0024】
【表1】
【0025】
ジストニアを治療するためには、いくつかの選択肢を利用できる。薬剤は単独で用いてもよいし、組み合わせて用いてもよい。さらに、その他の治療形態と組み合わせてもよい。現在使用されている薬剤としては、ボツリヌス毒素(BTX)、ベンゾジアゼピン、バクロフェン、抗コリン作用薬およびドーパミン遮断薬/ドーパミン枯渇剤が挙げられる。外科的処置もまた有用であり、これには視床手術、淡蒼球手術、深脳刺激、心筋切除術(筋切開)、ラミセクトミー(ramisectomy)、神経根切断術および末梢神経除去が含まれる。
【0026】
遅発性ジスキネジアおよびその他の錐体外路症候群
神経系の錐体外路系は、大脳基底核を中心としており、錐体路を通って、一般に視床への入力により、運動制御に影響を及ぼす。錐体外路系が障害を受けると、運動制御が影響を受け、患者は錐体外路症候群にさいなまれる。これらは、振戦、舞踏病、アテトーシスおよびジストニアを含む神経学的な影響の組み合わせである。これは、神経遮断薬の一般的な副作用である。これらの反応を引き起こすことが知られているその他の薬剤としては、ハロペリドール、モリンドン、ペルフェナジンおよびアミノトリプチリン、ロキサピン、ピモジドおよびまれにではあるがベンゾジアゼピンが挙げられる。
【0027】
遅発性ジスキネジアは、不随意神経運動障害である。発症の型式に応じて、鑑別診断には、シデナム舞踏病(Sydenham's chorea)、ハンチントン舞踏病、先天性捻転ジストニア(congenital torsion dystonia)、病的興奮、および統合失調症の常同的行為もしくは癖が含まれ得る。米国神経精神薬理学会(American College of Neuropsychopharmacology)−FDAタスクフォース(1973)。遅発性ジスキネジアは、特定の精神状態または胃腸の状態を治療するために処方された神経遮断薬の使用の結果として引き起こされる。これらの薬剤の長期服用によって、線条体に生化学的異常が生じることがある。遅発性ジストニアは、遅発性ジスキネジアのより重度の形であると考えられている。
【0028】
その他の密接に関連する処置不能の神経疾患は、現在、遅発性ジスキネジアの変種として認識されている。遅発性アカシジアは、精神的緊張および不安という精神的な苦痛ならびに体を動かすことへの強迫的な衝動を伴う。極端な場合、患者は内面的な苦痛を受けて、もはや静かに座ったままでいることができない。遅発性ジストニアは、顔、首および肩の頻繁な筋痙攣を伴い、あまりにも頻繁であるため、外観を損ない、身体に障害を引き起こし、苦痛を伴うことがある。
【0029】
遅発性ジスキネジアの治療は、満足するまでに至っていない。抗精神病薬の廃止がしばしば提唱されるが(バルデサリニ(Baldessarini)(1990))、より重度な形の運動障害に至ってしまうことが多くある。種々の医薬が試みられ、いくつかのものでは成功が報告されている。この分野における初期の研究者たちは、ドーパミンレベルを枯渇させることが知られている化合物であるレセルピン(セルパシル(Serpasil)(登録商標))に注目した。レセルピンとα−メチルドーパ(アルドメット(Aldomet)(登録商標))は、長期にわたる遅発性ジスキネジアの治療において、両化合物とも総合的症状の軽減においてプラセボよりも統計的に有効であることを示した。ファン(Huang)ら(1981)。しかしながら、別の研究では、例えばα−メチルドーパ等のカテコールアミン合成ブロッカーが遅発性ジスキネジアに対して有効性を示さなかったことが示された。チロシンヒドロキシラーゼ、ドーパミンおよびノルエピネフィリンの合成の律速段階を阻害する試験段階の薬剤であるAMPTはジスキネジアを一部軽減した。
【0030】
これまで、遅発性ジスキネジアは、神経遮断薬の用量を増加することによって治療されることが多かった。これによってまず、遅発性ジスキネジアの病態生理が治療されるが、さらなる除神経およびその後の過敏症によって病因を悪化させ得る。そのため、最初は動きが減少するか、または見られなくなるが、その後に再発する。外観を損なうような遅発性ジスキネジアの患者が神経遮断薬による治療の代替治療を必要とするような特定の状況では、非定型神経遮断薬、クロザピンの使用が有用であり得る。
【0031】
リチウムは、CNSに対するその他の作用も有するばかりでなくモノアミンのシナプス前放出を妨げる。2つの研究で、リチウムによる遅発性ジスキネジアの軽度の改善が報告されているが、その2つ以外では、改善も悪化も報告されていない。テッパー(Tepper)およびハス(Haas)(1979)。
経口用ピモジドは、運動度の改善をもたらした。クラベリア(Claveria)ら(1975)。部分セロトニン受容体アゴニストであるブスピロン(ブスパー(BuSpar)(登録商標))もまたこの状態の治療に有用であり得る。モス(Moss)ら(1993)。ラットでは、ブスピロンが、長期にわたる神経遮断薬の投与によって誘発されたDA受容体弱感受性を逆転させるが、この作用はD2受容体で部分アゴニスト作用によりヒトでも起こり得るものである。報告では、遅発性ジスキネジアをレセルピン、テトラベナジン、メトクロプラミド、三環系抗鬱薬、ベンズトロピン、フェニトインおよびアンフェタミンと関連づけている。
【0032】
神経遮断薬以外で、ジスキネジアを定期的に引き起こす薬剤はL-ドーパおよび他のドーパミン作動性薬剤であり、パーキンソン病患者はパーキンソン病用のこれらの薬剤を服用している。実際には、L-ドーパは、神経遮断薬誘発性の遅発性ジスキネジアを改善することができる。
遅発性ジスキネジアには一般に認められた治療がない。ケーシー(Casey)(1999)。差障りのある抗精神病薬の投与を中止するか、または患者への投与を非定型抗精神病薬(リスペリドンはおそらく除かれる)へと切り替えることで運動障害が緩和されることがある。遅発性ジスキネジアの治療は、近年、再検討されてきた。イーガン(Egan)ら(1997)。ほとんどの薬剤治療の戦略は、ドーパミン活性を低下させることまたはCNSコリン作
動性効果を高めることに向けられている。遅発性ジスキネジアの病因が長期のドーパミン作動性受容体部位の遮断に関連している場合、および病態生理が除神経による過敏症に関連している場合には、理論的にはこの筋道を遮断する薬剤が潜在的に有益である可能性がある。
【0033】
神経遮断薬誘発性の遅発性ジスキネジアの治療には、数多くの薬物が試されてきた。患者の症例数、研究設計、および使用される薬剤の用量には差があるため、個々の薬剤の結果は相反している。バルデサリニ(Baldessarini)およびタルシー(Tarsy)(1978);ならびにクローアンズ(Klawans)ら(1980)。
例えばレセルピンおよびテトラベナジン等のアミン枯渇剤は、ドーパミン、ノルエピネフリンおよびセロトニンのシナプス前ニューロン蓄積媒介物への再取り込みをブロックし、それによって、脳におけるこれらの物質を枯渇させることによって作用する。これらの薬剤を用いた研究では、遅発性ジスキネジアの改善が示されたが、副作用によってその使用が制限されており、それらの研究は短期間の場合についてのものである。神経遮断薬を用いて報告されたように、短期間の鎮静が起こり得る。
【0034】
いくつかのコリン作動性アゴニストが遅発性ジスキネジアの患者に投与されてきた。経口的に生物学的利用可能なアセチルコリンの前駆体である塩化コリンおよびホスファチジルコリン(レシチン)は、短期間の研究において有用であると報告されている。デアノール アセトアミノベンゾエートは、当初、遅発性ジスキネジアの治療に有効であると報告されたが、他の研究では、これらの知見は確認されなかった。ゲレンバーグ(Gelenberg)ら(1990)。
【0035】
中枢GABAの機構を強化すると考えられている薬物で遅発性ジスキネジアを治療する試みがいくつかなされてきた。サカー(Thaker)ら(1990)。10人の遅発性ジスキネジア患者に関する6ヶ月を超える研究では、ベンズトロピン2mg IVにより7患者でジスキネジア的な動きが増え、残る3患者ではそれが減少した。ムーア(Moore)およびバウアーズ(Bowers)(1980)。予備的報告では、30〜60mg/日の用量のβ−アドレナリン遮断薬であるプロプラノロール(インデラル(登録商標))での治療により、1〜10日以内に4患者で遅発性ジスキネジアの著しい軽減がもたらされた。ウィルバー(Wilbur)およびクリック(Kulik)(1980)。
【0036】
いくつかの研究では、遅発性ジスキネジアのビタミンEでの治療の有効性を調べている。アドラー(Adler)ら(1999);レーア(Lohr)およびカリギウリ(Caligiuri)(1996);レーア(Lohr)ら(1988);エルカシェフ(Elkashef)ら(1990);シュリキ(Shriqui)ら(1992);イーガン(Egan)ら(1992);アドラー(Adler)ら(1993a);アドラー(Adler)ら(1993b);ゴールドバーグ(Goldberg)(1996);マクレジー(McCreadie)ら(1994);ダビリ(Dabiri)ら(1993);ビショット(Bischot)ら(1993);アフタル(Akhtar)ら(1993);およびダビリ(Dabiri)ら(1994)。
【0037】
これまで、遅発性ジスキネジアは、大部分の患者において永続的なものまたは回復不可能であるものと考えられていた。しかしながら、必ずしもそうであるとは限らない。より早期に遅発性ジスキネジアが診断されて神経遮断薬の投与が中止されるほど、疾患の回復の予後が良くなる。若年成人では、遅発性ジスキネジアが早期に投薬を中止した後、数週間内に消失する。ウルブランド(Uhrbrand)およびファルバイ(Faurbye)(1960);イトウ(Itoh)ら(1981);ドリーセンス(Driesens)(1988);およびガルドス(Gardos)ら(1994)。
【0038】
表2は、遅発性ジスキネジアの治療に用いられてきた種々の薬剤をまとめたものである。
【0039】
【表2】
【0040】
神経遮断薬の錐体外路系への作用によって引き起こされるその他の運動症候群としては、薬剤誘発性パーキンソン症候群、アカシジア、ジストニア、注視発症(oculogyric crisis)および後弓反張(opisthotonus)が挙げられる。アカシジアは、運動的に落ち着きのないことを特徴とする状態であり、その状態は不安を感じる程度から、静かに横たわることまたは座ることができない、または眠ることができない程度までさまざまである。考えられる原因としては、例えばフェノチアジン等の神経遮断薬による中毒反応が挙げられる。注視発症は、発作性の眼の不随意性上方偏視である。眼瞼は縮まっている場合が多い。発作は数分間〜数時間続く。フェノチアジン、ハロペリドールおよびメトクロプラミドに敏感な患者に起こる可能性がある。後弓反張は、頭、首および脊椎が後方に反り返る形の痙攣である。
【0041】
アデノシンA2A受容体
アデノシンは、4種の主要な受容体サブタイプ、A1、A2A、A2B、A3(これらはその一次配列によって特性決定されている)を介して作用することがわかっている。フレドホルム(Fredholm)ら(1994)。アデノシンA2受容体はさらに、A2A(高親和性)とA2B(低親和性)のサブタイプに分けられる。ダリー(Daly)ら(1983);およびバーンズ(Burns)ら(1986)。A1、A2BおよびA3受容体が脳内に広範囲にわたって分布しているのに対し、A2A受容体は大脳基底核、とりわけ、尾状-被殻(caudate-putamen)(線条体)、側坐核および淡蒼球、ならびに嗅結節に高度に局在している。ジャーヴィス(Jarvis)ら(1989);およびシッフマン(Schiffmann)(1991b)。大脳基底核は、終脳に局在し、いくつかの相互接続されている核:線条体、淡蒼球外節(GPe)、淡蒼球内節(GPi)、黒質緻密部(SNc)、黒質網様部(SNr)および視床下核(STN)からなる。大脳基底核は、運動行動を起こすための運動感覚(sensorimotor)、連合および辺縁系情報の統合に関与する皮質下回路(subcortical circuits)の重要な要素である。大脳基底核の主要構成要素は線条体であり、ここではGABA作動性の中型の有棘ニューロンが唯一の投射ニューロンであり、これは線条体ニューロン群の90%以上に相当する。
【0042】
中型の有棘ニューロンは、皮質および視床から膨大なグルタメート作動性入力を受け、それらのGABA作動性出力を大脳基底核の主要な出力核、つまりGPiおよびSNrに、「間接経路」で線条体−淡蒼球系の中型の有棘ニューロン(striatopallidal medium spiny neurons)を介して、および「直接経路」で線条体−黒質系の中型の有棘ニューロン(striatonigral medium spiny neurons)を介して投射する。アレキサンダー(Alexander)ら(1990);ゲーフェン(Gerfen)(1992);およびグライベル(Graybiel)(1990)。中型の有棘ニューロンは、また線条体内GABA作動性、コリン作動性および黒質線条体ドーパミン作動性の調節入力を受ける。線条体黒質系の直接経路のニューロンは、GABAだけでなくサブスタンスP/ダイノルフィンも含有しており、線条体からGPi/SNrへと直接投射する。これらのニューロンは、GPi/SNrニューロンに対して直接的な阻害作用を与える。線条体淡蒼球系の間接経路の線条体ニューロンは、GABAだけでなくエンケファリンも含有しており、GPeおよびSTNでのシナプス結合を介して線条体とGPi/SNrとをつなぐ。これらのニューロンでは、アデノシンA2A受容体が、ほぼ例外なく、間接経路の線条体と淡蒼球にある線条体−淡蒼球系の中型の有棘ニューロン[シッフマン(Schiffmann)ら(1991a)]および線条体にあるアセチルコリンを含有する大型の無棘介在ニューロン[ディクソン(Dixon)ら(1996)]に局在し、GABA、アセチルコリンおよびグルタメートの神経伝達を調節することがわかってきた。クロカワ(Kurokawa)ら(1996);モリ(Mori)ら(1996);シンドウ(Shindou)ら(2001);オチ(Ochi)ら(2000);リチャードソン(Richardson)ら(1997);およびカセ(Kase)(2001)。
【0043】
神経科学の近年の進歩は、アデノシンA2A受容体に選択的な薬剤の開発とともに、アデノシンおよびアデノシンA2A受容体についての認識の拡大を促した。アデノシンA2A受容体アンタゴニストが数種類のパーキンソン病の動物モデル(例えば、MPTP処置したサル)の運動機能障害を改善するが、またドーパミン作動性薬剤とは異なるアデノシンA2A受容体アンタゴニストの特徴も示すことを、行動研究は示している。リチャードソン(Richardson)ら(1997);カセ(Kase)ら(2000);およびカセ(Kase)(2001)。
【0044】
選択的アデノシンA2A受容体アンタゴニストであるKW-6002の抗パーキンソン病作用は、MPTP処置したマーモセットおよびカニクイザルを用いて研究されてきた。カンダ(Kanda)ら(1998a);グロンジン(Grondin)ら(1999);およびカンダ(Kanda)ら(2000)(非特許文献1〜3参照)。MPTP処置マーモセットでは、KW-6002の経口投与により、用量依存的に自発運動の増加が誘発され、最大11時間まで持続した。カンダ(Kanda)ら(1998a)。自発運動は正常動物で認められるレベルまで増加したが、L-ドーパでは運動亢進が誘発された。さらに、L-ドーパを前投与したMPTP処置マーモセットでは、21日間のKW-6002を用いた処置により、ジスキネジアがほとんどまたは全く誘発されなかったが、同じ条件下での、L-ドーパを用いた処置では、顕著なジスキネジアが誘発された。ジスキネジアを発症するように処置されたMPTP処置マーモセットに、KW-6002(20mg/kg)を閾値のL-ドーパとともに1日1回、5日間投与した場合、ジスキネジアを増加させることなく抗パーキンソン病活性が増強された。カンダ(Kanda)ら(2000)。KW-6002は、またさらにキナピロール(quinpirole)、ドーパミンD2受容体アゴニストの抗パーキンソン病作用を増強したが、SKF80723、ドーパミンD1受容体アゴニストの作用は増強しなかった。総合すれば、これらの研究結果は、アデノシンA2Aアンタゴニストが、パーキンソン病の初期の患者に単独療法として抗パーキンソン病効果をもたらす可能性があること、およびL-ドーパ治療を受けた運動合併症患者では、ジスキネジアを増加させることなく抗パーキンソン病作用を改善させる可能性があることを示唆している。
【0045】
アデノシンA2Aアンタゴニストが抗パーキンソン病作用を発揮するメカニズムは依然として十分には解明されていないが、現在、次のメカニズムが提唱されている。
霊長類のパーキンソン病またはMPTP処置のいずれにおいても、黒質−線条体ドーパミン作動性経路が破壊された後の最も関連のある変化は、線条体淡蒼球系の経路の活動亢進である。このような活動亢進の原因は、直接的な線条体黒質系の経路および間接的な線条体淡蒼球系の経路間の不均衡にあり、これがパーキンソン病状態を引き起こす。デロング(DeLong)(1990);およびオベソ(Obeso)ら(2000)。アデノシンA2A受容体は、線条体黒質の中型の有棘ニューロンではなく、中型の有棘ニューロンのサブポピュレーション(subpopulation)である線条体淡蒼球系の中型の有棘ニューロンで特に発現されているということが知られている。
【0046】
線条体淡蒼球系の中型の有棘のGABA作動性投射ニューロンは、アデノシンA2A受容体媒介性調節の主要な標的ニューロンの1つであるとわかっている。カセ(Kase)(2001)。そのため、線条体では、アデノシンA2A受容体が線条体内のGABA作動性のフィードバック/フィードフォワード(feedback/feedforward)阻害のネットワークを通じて投射ニューロンの興奮性を制御し[モリ(Mori)ら(1996)]、淡蒼球(GPe)では、アデノシンA2A受容体の活性化によって神経末端からのGABA放出が促進され、視床下核(STN)へと投射するGPe投射ニューロンの興奮性を抑制している可能性がある[シンドウ(Shindou)ら(2001)]。アデノシンA2A受容体アンタゴニストは、線条体淡蒼球系のこの二重調節メカニズムを選択的にブロックし、線条体淡蒼球系の中型の有棘のニューロンにおける過度の活性化の抑制をもたらす。このことによって、線条体淡蒼球系/線条体黒質系ニューロンの不均衡が正常な状態にシフトし、その結果、パーキンソン病状態における運動機能の回復がもたらされる可能性がある。オチ(Ochi)ら(2000);カセ(Kase)(2001)、アオヤマ(Aoyama)ら(2002)。
【0047】
アデノシンA2A受容体を媒介した作用メカニズムは、線条体淡蒼球系の中型の有棘ニューロンにあるアデノシンA2A受容体とともに局在しているドーパミンD2受容体とは関係なく機能する可能性がある(アオヤマ(Aoyama)ら(2000))。ゲーフェン(Gerfen)ら(1990)。D2受容体ノックアウトマウス(D2R-/-)は、パーキンソン病と類似した運動表現型を示し、線条体の中型の有棘ニューロンで発現される神経ペプチド遺伝子のレベルが著しく変化した。D2R-/-マウスおよび野生型マウス間では、アデノシンA2A受容体mRNAの分布および発現レベルならびに受容体の結合特性に差は見られず、これは、D2受容体がないということがアデノシンA2A受容体特性に影響を与えないことを示唆している。KW-6002によるアデノシンA2A受容体の遮断によって、自発運動および運動の協調性が回復し、線条体エンケファリンの発現レベルが正常なマウスのレベルまで低下した。アオヤマ(Aoyama)ら(2000)。これらの結果は、アデノシンA2A受容体およびD2受容体が、大脳基底核での神経および運動機能の制御において、相反する活性ではあるが独立した活性を有していることを示唆している。ドーパミン作動性の系とは無関係なアデノシンA2A受容体の独立した機能が、アデノシンA2A受容体およびD2受容体ノックアウトマウスを用いた研究によって確認された。チェン(Chen)ら(2001b)。
【0048】
パーキンソン病のL-ドーパによる運動合併症におけるアデノシンA2A受容体の生理学的機能および病態生理学的機能は、全く分かっていない。L-ドーパ誘発性ジスキネジアの神経メカニズムは、一般に直接経路ではなくむしろ間接経路と関係していると考えられている。クロスマン(Crossman)(1990)。L-ドーパ誘発性ジスキネジアは、STNまたはGPiにおける活性がGPeからの過度の阻害の結果として与えられる一定のレベルを下回る場合に起こる。オベソ(Obeso)ら(1997)。主として、直接経路の異常が、L-ドーパ誘発性ジスキネジアの発症に大きく寄与する可能性があるというもう1つの仮説が提唱されている。
【0049】
アデノシンA2A受容体アンタゴニストの神経保護作用が、ラットおよびマウスならびにアデノシンA2A受容体ノックアウトマウスにおける神経毒(MPTPまたは6−ヒドロキシドーパミン)誘発性のドーパミン作動性神経変性により示されている。イケダ(Ikeda)ら(2002);およびチェン(Chen)ら(2001a)。今日まで、ドーパミン作動性ニューロンを死に至らしめる基本的な発病のメカニズムの抑止に成功した治療はなかった。
【0050】
それゆえ、アデノシンA2A受容体の遮断効果を有する非ドーパミン作動性薬剤療法が、パーキンソン病を治療するための手段として提供される。さらに、代表的なドーパミン作動薬の副作用、つまり運動合併症を増加させる危険もしくは発症させる危険がほとんどまたは全くない、抗パーキンソン病作用を提供するアデノシンA2A受容体アンタゴニストが望ましい。
【0051】
いくつかのキサンチン化合物は、アデノシンA2A受容体アンタゴニスト作用、抗パーキンソン病作用、抗鬱作用、神経変性に対する阻害活性等を示すことが知られている(特許文献1〜4等参照)。
【特許文献1】米国特許第5,484,920号明細書
【特許文献2】米国特許第5,587,378号明細書
【特許文献3】米国特許第5,543,415号明細書
【特許文献4】欧州特許公開第1016407号明細書
【非特許文献1】「アナルズ・オブ・ニューロロジー(Ann. Neurol. )」、第43巻、507-513、1998年
【非特許文献2】「エクスペリメンタル・ニューロロジー( Exp. Neurol. )」、第162巻、321-327、2000年
【非特許文献3】「ニューロロジー(Neurology)」、第52巻、1673-1677、1999年
【発明の開示】
【課題を解決するための手段】
【0052】
本発明は、パーキンソン病患者に1種以上のアデノシンA2A受容体アンタゴニストを投与することまたは併用投与することを特徴とするL-ドーパ療法の副作用を軽減または抑制する方法を提供する。このような治療は、例えばL-ドーパまたは他のドーパミン作動性薬剤で誘発される運動合併症を患っている患者を治療して、オフ時間を減少させるおよび/またはジスキネジアを改善するのに有効であり得る。
【0053】
本発明は、さらにL-ドーパを減量して治療するための方法および組成物を提供する。この方法は、臨床上有効な量より少ないL-ドーパと、該L-ドーパの効果を発揮させるために有効な量の1種以上のアデノシンA2A受容体アンタゴニストとの組み合わせを、それらを必要とする患者に投与することを特徴とする。
本発明は、さらに少なくとも1種のアデノシンA2A受容体アンタゴニストの有効量を、COMT阻害剤およびに/またはDAおよび/もしくはMAO阻害剤と組み合わせて投与することを特徴とするパーキンソン病および/またはL-ドーパによる運動合併症を治療する方法を提供する。
【0054】
本発明は、また、患者が追加のL-ドーパ療法を必要とすることを遅らせるか、または完全になくさせるように、L-ドーパまたはドーパミン作動性薬剤の前投与または後投与なしに、アデノシンA2A受容体アンタゴニストまたはアデノシンA2A受容体アンタゴニストとドーパミンアゴニストとの組み合わせのいずれかを、それらを必要とする患者に投与し、L-ドーパによる運動合併症の発症を遅らせるかまたはその進行を妨げることを特徴とするパーキンソン病治療の有効時間を延長する方法を提供する。
【0055】
本発明は、また、少なくとも1種のアデノシンA2A受容体アンタゴニストの有効量を、それを必要とする患者に投与することを特徴とする運動障害を治療する方法も包含する。このような治療は、例えば振戦、運動緩徐、歩行、ジストニア、ジスキネジア、遅発性ジスキネジアまたはその他の錐体外路症候群を治療するのに有効であるか、または運動障害を引き起こす薬剤の作用を防ぐかもしくは減少させるのに有効であり得る。
発明の詳細
本発明は、次の(1)〜(50)に関する。
【0056】
(1)パーキンソン病患者に少なくとも1種のアデノシンA2A受容体アンタゴニストの有効量を投与することを特徴とするL-ドーパおよび/またはドーパミンアゴニスト療法の副作用を軽減または抑制する方法。
(2)患者がL-ドーパまたは他のドーパミン作動性薬剤で誘発される運動合併症を患っている上記(1)に記載の方法。
【0057】
(3)運動変動のオフ時間を減少させる上記(2)に記載の方法。
(4)運動合併症におけるジスキネジアが改善される上記(2)に記載の方法。
(5)アデノシンA2A受容体アンタゴニストがキサンチン誘導体またはその薬理学上許容される塩である上記(1)に記載の方法。
(6)アデノシンA2A受容体アンタゴニストが式(I):
【0058】
【化65】
【0059】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【0060】
【化66】
【0061】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【0062】
【化67】
【0063】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
で表される上記(1)に記載の方法。
(7) アデノシンA2A受容体アンタゴニストが式(I-A):
【0064】
【化68】
【0065】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【0066】
【化69】
【0067】
(式中、R7、R8およびR9のうち少なくとも1つは低級アルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R10は水素原子または低級アルキルを表す)または
【0068】
【化70】
【0069】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
で表される上記(1)に記載の方法。
(8) アデノシンA2A受容体アンタゴニストが式(I-B):
【0070】
【化71】
【0071】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【0072】
【化72】
【0073】
(式中、R6およびmは前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
で表される上記(1)に記載の方法。
(9) アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである上記(1)に記載の方法。
【0074】
(10) 臨床上有効な量より少ないL-ドーパと、該L-ドーパの効果を発揮させるために有効な量の1種以上のアデノシンA2A受容体アンタゴニストとの組み合わせを、それらを必要とする患者に投与することを特徴とするL-ドーパを減量して治療する(L-DOPA sparing treatment)方法。
(11) アデノシンA2A受容体アンタゴニストがキサンチン誘導体またはその薬理学上許容される塩である上記(10)に記載の方法。
【0075】
(12) アデノシンA2A受容体アンタゴニストが式(I):
【0076】
【化73】
【0077】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【0078】
【化74】
【0079】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【0080】
【化75】
【0081】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
で表される上記(10)に記載の方法。
(13) アデノシンA2A受容体アンタゴニストが式(I-A):
【0082】
【化76】
【0083】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【0084】
【化77】
【0085】
(式中、R7、R8およびR9のうち少なくとも1つは低級アルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R10は水素原子または低級アルキルを表す)または
【0086】
【化78】
【0087】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
で表される上記(10)に記載の方法。
(14) アデノシンA2A受容体アンタゴニストが式(I-B):
【0088】
【化79】
【0089】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【0090】
【化80】
【0091】
(式中、R6およびmは前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
で表される上記(10)に記載の方法。
(15) アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである上記(10)に記載の方法。
【0092】
(16) 臨床上有効な量より少ないL-ドーパと、該L-ドーパの効果を発揮させるために有効な量の1種以上のアデノシンA2A受容体アンタゴニストとを含有するL-ドーパを減量した治療(L-DOPA sparing treatment)のための組成物。
(17) アデノシンA2A受容体アンタゴニストがキサンチン誘導体またはその薬理学上許容される塩である上記(16)に記載の組成物。
【0093】
(18) アデノシンA2A受容体アンタゴニストが式(I):
【0094】
【化81】
【0095】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【0096】
【化82】
【0097】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【0098】
【化83】
【0099】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
で表される上記(16)に記載の組成物。
(19) アデノシンA2A受容体アンタゴニストが式(I-A):
【0100】
【化84】
【0101】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【0102】
【化85】
【0103】
(式中、R7、R8およびR9のうち少なくとも1つは低級アルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R10は水素原子または低級アルキルを表す)または
【0104】
【化86】
【0105】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
で表される上記(16)に記載の組成物。
(20) アデノシンA2A受容体アンタゴニストが式(I-B):
【0106】
【化87】
【0107】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【0108】
【化88】
【0109】
(式中、R6およびmは前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
で表される上記(16)に記載の組成物。
(21) アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである上記(16)に記載の組成物。
【0110】
(22) 少なくとも1種のアデノシンA2A受容体アンタゴニストの有効量を、COMT阻害剤ならびに/またはDAおよび/もしくはMAO阻害剤と組み合わせて、それらを必要とする患者に投与すること特徴とするパーキンソン病および/またはL-ドーパによる運動合併症を治療する方法。
(23) アデノシンA2A受容体アンタゴニストがキサンチン誘導体またはその薬理学上許容される塩である上記(22)に記載の方法。
【0111】
(24) アデノシンA2A受容体アンタゴニストが式(I):
【0112】
【化89】
【0113】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【0114】
【化90】
【0115】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【0116】
【化91】
【0117】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
で表される上記(22)に記載の方法。
(25) アデノシンA2A受容体アンタゴニストが式(I-A):
【0118】
【化92】
【0119】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【0120】
【化93】
【0121】
(式中、R7、R8およびR9のうち少なくとも1つは低級アルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R10は水素原子または低級アルキルを表す)または
【0122】
【化94】
【0123】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
で表される上記(22)に記載の方法。
(26) アデノシンA2A受容体アンタゴニストが式(I-B):
【0124】
【化95】
【0125】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【0126】
【化96】
【0127】
(式中、R6およびmは前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
で表される上記(22)に記載の方法。
(27) アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである上記(22)に記載の方法。
【0128】
(28) 少なくとも1種のアデノシンA2A受容体アンタゴニストの有効量と、COMT阻害剤ならびに/またはDAおよび/もしくはMAO阻害剤とを含有するパーキンソン病の治療のための組成物。
(29) アデノシンA2A受容体アンタゴニストがキサンチン誘導体またはその薬理学上許容される塩である上記(28)に記載の組成物。
【0129】
(30) アデノシンA2A受容体アンタゴニストが式(I):
【0130】
【化97】
【0131】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【0132】
【化98】
【0133】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【0134】
【化99】
【0135】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
で表される上記(28)に記載の組成物。
(31) アデノシンA2A受容体アンタゴニストが式(I-A):
【0136】
【化100】
【0137】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【0138】
【化101】
【0139】
(式中、R7、R8およびR9のうち少なくとも1つは低級アルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R10は水素原子または低級アルキルを表す)または
【0140】
【化102】
【0141】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
で表される上記(28)に記載の組成物。
(32) アデノシンA2A受容体アンタゴニストが式(I-B):
【0142】
【化103】
【0143】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【0144】
【化104】
【0145】
(式中、R6およびmは前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
で表される上記(28)に記載の組成物。
(33) アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである上記(28)に記載の組成物。
【0146】
(34) 患者が追加のL-ドーパ療法を必要とすることを遅らせるか、またはなくさせるために有効な量の、アデノシンA2A受容体アンタゴニストまたはアデノシンA2A受容体アンタゴニストとドーパミンアゴニストとの組み合わせのいずれかを、それらを必要とする患者に投与することを特徴とするパーキンソン病治療の有効時間を延長する方法。
(35) 運動合併症の発症を遅らせる上記(34)に記載の方法。
【0147】
(36) 患者がL-ドーパまたはドーパミン作動性薬剤の前投与を受けていない上記(34)に記載の方法。
(37) 患者がL-ドーパまたはドーパミン作動性薬剤の後投与を受けない上記(34)に記載の方法。
(38) アデノシンA2A受容体アンタゴニストがキサンチン誘導体またはその薬理学上許容される塩である上記(34)に記載の方法。
【0148】
(39) アデノシンA2A受容体アンタゴニストが式(I):
【0149】
【化105】
【0150】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【0151】
【化106】
【0152】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【0153】
【化107】
【0154】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
で表される上記(34)に記載の方法。
(40) アデノシンA2A受容体アンタゴニストが式(I-A):
【0155】
【化108】
【0156】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【0157】
【化109】
【0158】
(式中、R7、R8およびR9のうち少なくとも1つは低級アルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R10は水素原子または低級アルキルを表す)または
【0159】
【化110】
【0160】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
で表される上記(34)に記載の方法。
(41) アデノシンA2A受容体アンタゴニストが式(I-B):
【0161】
【化111】
【0162】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【0163】
【化112】
【0164】
(式中、R6およびmは前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
で表される上記(34)に記載の方法。
(42) アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである上記(34)に記載の方法。
【0165】
(43) 少なくとも1種のアデノシンA2A受容体アンタゴニストの有効量を、それを必要とする患者に投与することを特徴とする運動障害を治療する方法。
(44) 患者が振戦、運動緩慢、歩行、ジストニア、ジスキネジア、遅発性ジスキネジアまたはその他の錐体外路症候群を患っている上記(43)に記載の方法。
(45) アデノシンA2A受容体アンタゴニストが、運動障害を引き起こす薬剤の作用を減少させる上記(43)に記載の方法。
【0166】
(46) アデノシンA2A受容体アンタゴニストがキサンチン誘導体またはその薬理学上許容される塩である上記(43)に記載の方法。
(47) アデノシンA2A受容体アンタゴニストが式(I):
【0167】
【化113】
【0168】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【0169】
【化114】
【0170】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【0171】
【化115】
【0172】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
で表される上記(43)に記載の方法。
(48) アデノシンA2A受容体アンタゴニストが式(I-A):
【0173】
【化116】
【0174】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【0175】
【化117】
【0176】
(式中、R7、R8およびR9のうち少なくとも1つは低級アルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R5は水素原子または低級アルキルを表す)または
【0177】
【化118】
【0178】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
で表される上記(43)に記載の方法。
(49) アデノシンA2A受容体アンタゴニストが式(I-B):
【0179】
【化119】
【0180】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【0181】
【化120】
【0182】
(式中、R6およびmは前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
で表される上記(43)に記載の方法。
(50) アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである上記(43)に記載の方法。
【0183】
さらに、本発明は、次の(51)〜(60)に関する。
(51) アデノシンA2A受容体アンタゴニストを含有するL-ドーパおよび/またはドーパミンアゴニスト療法の副作用を軽減または抑制する薬剤。
(52) L-ドーパおよび/またはドーパミンアゴニスト療法の副作用を軽減または抑制する薬剤の製造のためのアデノシンA2A受容体アンタゴニストの使用。
【0184】
(53) 臨床上有効な量より少ないL-ドーパと、該L-ドーパの効果を発揮させるために有効な量の1種以上のアデノシンA2A受容体アンタゴニストとを含有するL-ドーパを減量して治療するための薬剤。L-ドーパおよびアデノシンA2A受容体アンタゴニストは、単一の投与形態で存在してもよいし、または別々の投与形態で存在してもよい。
(54) L-ドーパを減量して治療するための薬剤の製造のための、臨床上有効な量より少ないL-ドーパと、該L-ドーパの効果を発揮させるために有効な量の1種以上のアデノシンA2A受容体アンタゴニストとの組み合わせの使用。
【0185】
(55) アデノシンA2A受容体アンタゴニストならびにCOMT阻害剤および/またはDAおよび/またはMAO阻害剤を含有するパーキンソン病および/またはL-ドーパによる運動合併症を治療する薬剤。アデノシンA2A受容体アンタゴニストならびにCOMT阻害剤および/またはDAおよび/またはMAO阻害剤は、単一の投与形態で存在してもよいし、または別々の投与形態で存在してもよい。
【0186】
(56) パーキンソン病および/またはL-ドーパによる運動合併症を治療する薬剤の製造のためのアデノシンA2A受容体アンタゴニストならびにCOMT阻害剤および/またはDAおよび/またはMAO阻害剤の使用。
(57) アデノシンA2A受容体アンタゴニストまたはアデノシンA2A受容体アンタゴニストとドーパミンアゴニストとの組み合わせのいずれかを含有する患者が追加のL-ドーパ療法を必要とすることを遅らせるか、またはなくさせることにより、パーキンソン病治療の有効時間を延長する薬剤。アデノシンA2A受容体アンタゴニストとドーパミンアゴニストとの組み合わせを用いる場合、アデノシンA2A受容体アンタゴニストおよびドーパミンアゴニストは、単一の投与形態で存在してもよいし、または別々の投与形態で存在してもよい。
【0187】
(58) 患者が追加のL-ドーパ療法を必要とすることを遅らせるか、またはなくさせることにより、パーキンソン病治療の有効時間を延長する薬剤の製造のためのアデノシンA2A受容体アンタゴニストまたはアデノシンA2A受容体アンタゴニストとドーパミンアゴニストとの組み合わせのいずれかの使用。
(59) アデノシンA2A受容体アンタゴニストを含有する運動障害を治療する薬剤。
【0188】
(60) 運動障害を治療する薬剤の製造のためのアデノシンA2A受容体アンタゴニストの使用。
本発明は、1種以上のアデノシンA2A受容体アンタゴニストを投与することを特徴とする運動障害を患っている患者を治療する方法を対象とする。「アデノシンA2A受容体アンタゴニスト」とは、例えばアデノシンA2A受容体と結合すること、アデノシンの受容体との結合を妨害することまたは阻止することによって、少なくとも1種のアデノシンで媒介される生理活性を阻害、抑制または消滅させる化合物を意味する。
【0189】
本発明では、アデノシンA2A受容体が、例えば間接経路または大脳基底核出力核活性を制御する際に機能することから、アデノシンA2A受容体アンタゴニストを運動障害の治療に用いることができると考えている。アデノシンA2A受容体はまた運動行動または運動機能障害の制御にも関係していると考えられる。
アデノシンA2A受容体アンタゴニストは、いくつかの様式で機能する。このアンタゴニストは、アデノシンがアデノシンA2A受容体と結合することを実質的に妨害するか、ブロックするかまたは防ぐために十分な親和性および特異性により、アデノシンと結合するかまたはアデノシンを隔離することができ、それによって、例えば間接経路の線条体GABA作動性出力の調節、例えばSNr等の大脳基底核での出力核の活性等の1種以上のアデノシンA2A受容体で媒介される生理機能を阻害、抑制または消滅させ、それによって、大脳基底核
において運動行動を制御する。本発明では、アデノシンA2A受容体アンタゴニストの抗パーキンソン病活性がこの活性に起因すると考えている。本発明では、さらにパーキンソン病患者におけるL-ドーパおよび/またはドーパミンアゴニスト療法の副作用を軽減または抑制するアデノシンA2A受容体アンタゴニストの能力がこの活性に起因すると考えている。本発明では、さらにL-ドーパおよび/またはドーパミンアゴニストに誘発される運動合併症の発症におけるアデノシン受容体アンタゴニストの関与がこの活性に起因すると考えている。また、アデノシンA2A受容体アンタゴニストは、例えば6-OHDA(6−ヒドロキシドーパミン)、1−メチル−4−フェニル−1,2,3,6−テトラヒドロピリジン(MPTP)等のドーパミン作動性神経毒によって誘発されるニューロン変性カスケードおよびグリア細胞を介したドーパミン作動性神経毒の産生を阻害し得る。従って、アデノシンA2A受容体アンタゴニストは、神経毒のドーパミン作動性神経変性作用を阻害する方法およびグリオーシス(gliosis)を阻害する方法を提供する。アデノシンA2A受容体アンタゴニストのこれらの特徴が、パーキンソン病の進行を阻止する。このように、アデノシンA2A受容体アンタゴニストを使用することによって、患者が追加のL-ドーパ療法を必要とするのを遅らせるかまたは完全になくさせるような療法を提供するか、またはL-ドーパによる運動合併症の発症を遅らせるかもしくはその進行を妨げる療法を提供する。
【0190】
このように、本発明のアデノシンA2A受容体アンタゴニストは、1種以上のアデノシンA2A受容体アンタゴニストの有効量を投与することによって、パーキンソン病患者および運動障害を患っている他の患者を治療する方法を対象とする。また、本発明のアデノシンA2A受容体アンタゴニストは、パーキンソン病の治療におけるL-ドーパによる運動合併症をはじめとするL-ドーパ療法の副作用を軽減または抑制する方法においても有用である。さらに、アデノシンA2A受容体アンタゴニストを用いたパーキンソン病の治療では、L-ドーパを用いた治療の必要性を回避することができ、例えば悪心、活動亢進、例えばウェアリング・オフ、オン・オフ変動等の運動変動、ジスキネジア等の副作用なしにまたは軽減しながら、パーキンソン病を有効に治療するために必要なL-ドーパの量を減らすことができる。さらに、本発明は、患者がL-ドーパ療法を必要とすることを遅らせるかまたは完全になくさせて、L-ドーパによる運動合併症の発症を遅らせるかまたはその進行を妨げるように、アデノシンA2A受容体アンタゴニストを投与することによりパーキンソン病患者を治療する方法を提供する。さらに、本発明は、その他の運動障害を患っている患者における振戦、運動緩徐、歩行、ジストニアならびに遅発性ジスキネジアおよびその他の錐体外路症候群を治療する方法を提供する。
【0191】
本発明に用いるアデノシンA2A受容体アンタゴニストは、アデノシンA2A受容体アンタゴニスト活性を有している限り、限定されない。その例としては、US 5,484,920、US 5,703,085、WO 92/06976、WO 94/01114、US 5,565,460、WO 98/42711、WO 00/17201、WO 99/43678、WO 01/92264、WO 99/35147、WO 00/13682、WO 00/13681、WO 00/69464、WO 01/40230、WO 01/02409、WO 01/02400、EP 1054012、WO 01/62233、WO 01/17999、WO 01/80893、WO 02/14282、WO 01/97786等に開示された化合物が挙げられる。より具体的には、例えば、
(1)次式(I)で表される化合物
【0192】
【化121】
【0193】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【0194】
【化122】
【0195】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【0196】
【化123】
【0197】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
(2)次式(I-A)で表される化合物
【0198】
【化124】
【0199】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【0200】
【化125】
【0201】
(式中、R7、R8およびR9のうち少なくとも1つはアルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R10は水素原子または低級アルキルを表す)または
【0202】
【化126】
【0203】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
および
(3)次式(I-B)で表される化合物
【0204】
【化127】
【0205】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【0206】
【化128】
【0207】
(式中、R6およびmはそれぞれ前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
ならびにそれらの薬理学上許容される塩が挙げられる。
式(I)、式(I-A)および式(I-B)の基の定義において、低級アルキルおよび低級アルコキシの低級アルキル部分は、1〜6個の炭素原子を有する直鎖状または分枝状のアルキル基、例えばメチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec-ブチル、tert-ブチル、ペンチル、ネオペンチル、ヘキシ等を意味する。低級アルケニルは、2〜6個の炭素原子を有する直鎖状または分枝状アルケニル基、例えばビニル、アリル、メタクリル、クロチル、3−ブテニル、2−ペンテニル、4−ペンテニル、2−ヘキセニル、5−ヘキセニル等を意味する。低級アルキニルは、2〜6個の炭素原子を有する直鎖状または分枝状アルキニル基、例えばエチニル、プロパルギル、2−ブチニル、3−ブチニル、2−ペンチニル、4−ペンチニル、2−ヘキシニル、5−ヘキシニル、4−メチル−2−ペンチニル等を意味する。アリールは、フェニルまたはナフチルを意味する。シクロアルキルは、3〜8個の炭素原子を有するシクロアルキル基、例えばシクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル等を意味する。複素環基としては、フリル、チエニル、ピロリル、ピラニル、チオピラニル、ピリジル、チアゾリル、イミダゾリル、ピリミジル、トリアジニル、インドリル、キノリル、プリニル、ベンゾチアゾリル等が例示される。ハロゲンとしては、フッ素、塩素、臭素およびヨウ素が包含される。
【0208】
置換アリール、置換複素環基および置換ナフチルは、それぞれ1〜4個の独立して選択された置換基を有する。置換基としては、低級アルキル、ヒドロキシ、置換もしくは非置換の低級アルコキシ、ハロゲン、ニトロ、アミノ、低級アルキルアミノ、ジ(低級アルキル)アミノ、トリフルオロメチル、トリフルオロメトキシ、ベンジルオキシ、フェニル、フェノキシ等が例示される。低級アルキルならびに低級アルコキシ、低級アルキルアミノおよびジ(低級アルキル)アミノの低級アルキル部分は、前記低級アルキルと同義である。ハロゲンは、前記ハロゲンと同義である。置換低級アルコキシにおける置換基としては、ヒドロキシ、低級アルコキシ、ハロゲン、アミノ、アジド、カルボキシ、低級アルコキシカルボニル等が例示される。低級アルコキシおよび低級アルコキシカルボニルの低級アルキル部分は、前記低級アルキルと同義であり、ハロゲンは、前記ハロゲンと同義である。
【0209】
上述した化合物(I)、化合物(I-A)および化合物(I-B)の薬理学上許容される塩としては、薬理学上許容される酸付加塩、金属塩、アンモニウム塩、有機アミン付加塩、アミノ酸付加塩等が例示される。
薬理学上許容される酸付加塩としては、例えば塩酸塩、硫酸塩、リン酸塩等の無機酸付加塩、例えば酢酸塩、マレイン酸塩、フマル酸塩、酒石酸塩、クエン酸塩等の有機酸付加塩等が例示される。薬理学上許容される金属塩としては、例えばナトリウム塩、カリウム塩等のアルカリ金属塩、例えばマグネシウム塩、カルシウム塩等のアルカリ土類金属塩、アルミニウム塩、亜鉛塩等が例示される。薬理学上許容されるアンモニウム塩としては、アンモニウム、テトラメチルアンモニウム等が例示される。薬理学上許容される有機アミン付加塩としては、モルホリン付加塩、ピペリジン付加塩等が例示される。薬理学上許容されるアミノ酸付加塩としては、リジン付加塩、グリシン付加塩、フェニルアラニン付加塩等が例示される。
【0210】
式(I)、式(I-A)および式(I-B)で表される化合物は、米国特許第5,543,415号;同5,587,378号;および同5,484,920号に記載されており、それらに記載の方法に従って合成される。
本発明の方法に有用な好ましくアデノシンA2A受容体アンタゴニストは、(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチン(次式(II))を含有する。
【0211】
【化129】
【0212】
また本発明では、式IIはKW-6002と称される。
「L-ドーパによる副作用を軽減または抑制する」とは、本発明によれば、本発明の化合物が患者の覚醒時間(awake time)中の「オフ」状態の量を減少させることを意味すると解される。オフ状態とは、本発明によれば、パーキンソン病用薬剤の服用による治療効果が薄れ、その結果、患者が、例えばパーキンソン病統一スケール(UPDRS)ならびにヘーン(Hoehn)およびヤール(Yahr)(HY)尺度等によって分類されるようなパーキンソン病の症状を起こす期間を意味すると解される。
【0213】
本発明は、また、患者の覚醒時間中の「オン」状態の割合を増やすことによってL-ドーパによる副作用を軽減することを対象とする。オン状態とは、パーキンソン病用薬剤の服用後、UPDRSおよびHY尺度によって分類されるパーキンソン病の症状が、患者に比較的みられない期間を意味する。本発明の方法によって治療可能な患者としては、パーキンソン・ジスキネジア・スケール(PDS)によって判定される運動合併症を伴うかまたは伴わない、初期段階、中間段階および進行した段階のパーキンソン病の患者が挙げられる。ジスキネジアは、UPDRS、改良ゲッツジスキネジア統一スケール(modified Goetz Dyskinesia Rating Scale)(MGDRS)および/または異常不随意運動評価尺度(AIMS)によって個別に判定することができる。本発明に係る治療は、進行したパーキンソン病を患っている患者に特に有効である。
【0214】
本発明によれば、本発明のアデノシンA2A受容体アンタゴニストは、L-ドーパまたはドーパミンアゴニストと併用投与することができる、すなわち、実質的に同時に投与することができる。また、アデノシンA2A受容体アンタゴニストは、患者がある用量のL-ドーパまたはドーパミンアゴニストを服用する前後のいずれかに、単独投与できることも想定される。L-ドーパの必要性の実質的な減少および/またはL-ドーパ療法の一般的な副作用の減少または抑制は、とりわけ、運動変動およびジスキネジアの症状においてKW-6002を投
与した場合に見られる。このように、本発明は、運動変動、ジスキネジア、悪心、およびL-ドーパ療法のその他の一般的な副作用を引き起こすL-ドーパとともにKW-6002を投与することによって、ヒトのパーキンソン病を治療する改良方法を提供する。
【0215】
さらに本発明は、L-ドーパの前投与または後投与なしに、アデノシンA2A受容体アンタゴニストまたはアデノシンA2A受容体アンタゴニストとドーパミンアゴニストとの組み合わせのいずれかを投与すること特徴とするパーキンソン病治療の有効時間を延長する方法を提供する。L-ドーパの必要性はなくなるか、または少なくとも実質的に減少すると同時に、付随するL-ドーパ療法の副作用が回避される。アデノシンA2A受容体アンタゴニストとドーパミンアゴニストとの「組み合わせ」は、同時にまたは少なくとも生理活性を重複させるように患者に与えられる。本発明のアデノシンA2A受容体アンタゴニストは、L-ドーパによる運動合併症の発症を妨げ、またドーパミン作動性神経変性も予防することから、アデノシンA2A受容体アンタゴニストは、単独でまたはドーパミンアゴニストとともに投与されると、L-ドーパによる運動合併症の発症を遅らせるかまたはその進行を妨げることができる。
【0216】
本発明によれば、アデノシンA2A受容体アンタゴニストは、単独でまたは例えばブロモクリプチン、カベルゴリン、プラミペキソール、ロピネロール(ropinerole)、ペルゴリド等のドーパミンアゴニストとともに投与することができ、それによってL-ドーパの必要性が現れるのを回避するか、または少なくともL-ドーパの必要性が現れるまでの時間が延長される。
【0217】
本発明はさらに、パーキンソン病患者をL-ドーパを減量して治療する方法を提供する。すなわち、臨床上有効な量より少ないL-ドーパを用いる治療であり、臨床上有効な量より少ないL-ドーパの効果が維持される。この方法は、臨床上有効な量より少ないL-ドーパと、アデノシンA2A受容体アンタゴニストの有効量とを用いて、患者を治療することを特徴とする。臨床上有効な量より少ないL-ドーパとは、個々の患者の治療に有効ではないL-ドーパの量を意味する。一般に、L-ドーパは、1日当たり100mg〜1gの用量を分割(通常、250mgを1日4回)して投与する。耐え難い副作用、通常は運動障害を発症するまで、1日当たり100〜750mgで、3〜7日の間隔をおいて用量を徐々に増やす。カルビドパと併用投与する場合には、L-ドーパの有効量は減る。個々の患者に向けたL-ドーパの臨床上有効な量より少ない量を決定することおよびアデノシンA2A受容体アンタゴニストの存在下でのその量を適宜調整することは、十分に当業者の技術の範囲内のものである。
【0218】
臨床上有効な量より少ないL-ドーパと、必要に応じてアデノシンA2A受容体アンタゴニストと、必要に応じてドーパミンアンタゴニストとを含有する組成物は、当技術分野でよく知られた方法および本明細書に記載した方法によって製造される。カルビドパおよびその他の有効成分の追加量もまた、当業者であれば決定できる。
本発明は、さらに、少なくとも1種のアデノシンA2A受容体アンタゴニストと少なくとも1種のCOMT阻害剤またはMAO-B阻害剤とを用いてパーキンソン病を治療する方法を提供する。該組成物は、当技術分野でよく知られた任意の方法により同時にまたは連続的に投与することができる。このような組成物の製造方法および投与方法は、当技術分野ではよく知られている。適当なCOMT阻害剤およびMAO阻害剤については、本明細書に記載されており、当技術分野では周知である。これらとしては、限定されるものではないが、エンタカポンおよびトルカポン、ならびにデプレニルが挙げられる。以下で示すように、アデノシンA2A受容体アンタゴニストとCOMTまたはMAO-B阻害剤との併用療法では、副作用は増加しない。
【0219】
「治療の有効時間を延長する」とは、患者がL-ドーパ療法を必要とするのを遅らせるかまたは完全になくさせるように、主観的に、またはUPDRS、AIMS、PDS、HYおよび/またはMGDRSに従って客観的に、患者のパーキンソン病の症状および運動合併症を軽減または抑制することを意味する。
本発明は、また、それを必要とする患者に、少なくとも1種のアデノシンA2A受容体アンタゴニストの有効量を投与することを特徴とする運動障害を治療する方法を包含する。このような治療は、振戦、運動緩徐、歩行、ジストニア、または遅発性ジスキネジアもしくはその他の錐体外路症候群を治療するのに有効であるか、または運動障害を引き起こす薬剤の作用を防ぐかもしくは減少させるのに有効であり得る。このような薬剤としては、当技術分野では公知であり、限定されるものではないが、表1に記載するものが挙げられる。
【0220】
「運動障害を治療する」とは、限定されるものではないが、振戦、ジストニア、ジスキネジア、痙縮といった症状を止めるかまたは弱めることを意味する。症状の変化は、限定されるものではないが、UPDRS、AIMS、PDS、HYおよび/またはMGDRSといった当技術分野では公知のいずれかの方法によって判定することができる。
「治療」または「治療する」という用語は、運動機能障害を改善するか、あるいは疾病または疾患の発症を妨げるかもしくは遅らせるか、その進行を遅らせるかまたはその症状を改善するためのアデノシン活性の有効な阻害、抑制または停止を指す。
【0221】
このように、本発明は、本発明のアデノシンA2A受容体アンタゴニストを使用することによって、アデノシンとアデノシンA2A受容体との相互作用または結合を妨害するか、ブロックするかまたは防ぐ方法を提供する。
本発明の投与するための医薬組成物は、少なくとも1種のアデノシンA2A受容体アンタゴニストを、薬理学上許容される担体との任意の組み合わせを特徴とする。これらの組成物は、それらの意図する目的を達成するいずれの方法によって投与してもよい。本発明の組成物の投与に関する量および計画は、パーキンソン病患者の治療の分野に従事する当業者ならば、容易に決めることができる。
【0222】
本明細書に記載した組成物は、限定されるものではないが、経口;経鼻;経肺;例えば皮下、静脈内、筋肉内、腹腔内等の非経口;十二指腸内;経皮的;または口腔内をはじめ、適していればいずれの方法でも投与することができる。
投与量は有効量であり、患者の年齢、健康状態および体重、これまでに治療または並行して行っている治療がある場合には、その種類、治療の頻度および望む効果の性質に応じて変わる。適当な用量を決定する場合には、通常、いくつかの因子を配慮する。これらの因子としては、患者の年齢、性別および体重、治療状態、状態の重篤度および投与されている薬剤の形態が挙げられる。
【0223】
「有効な量」とは、有益なまたは所望の臨床結果を達成するのに十分な量である。有効な量は、1以上の用量で投与してもよい。治療の観点から言えば、有効な量は、疾病または疾患の進行を軽減する、改善する、安定させる、回復させるもしくは遅らせる、または疾病または疾患の病理学的影響を減少させるのに十分な量である。有効な量は、一般に医師によって個別に判断され、当業者の技量の範囲内である。
【0224】
本発明の組成物は、また、薬理学上活性な化合物に加えて、活性化合物の薬理学上許容される製剤への加工を容易にする賦形剤を含む適当な薬理学上許容される担体を含有することができる。好ましくは、製剤、特に経口投与が可能で、好ましい投与形態に用いることができる製剤、例えば錠剤、トローチ剤およびカプセル剤、また例えば坐剤等の直腸投与が可能な製剤等、ならびに注射剤による投与に適当な溶液は、約0.1〜99%、好ましくは約20〜85%の活性化合物を賦形剤とともに含有する。薬理学上許容される液体組成物は、例えば、本明細書で実施された化合物を、水、生理食塩水、デキストロース、グリセロール、エタノール等の液体賦形剤に溶解または分散させることによって調製することができる。組成物は、また、その他の薬剤(medicinal agents)、医薬品(pharmaceutical agents)、担体および例えば湿潤剤または乳化剤、pH緩衝剤等の補助剤を含有することができる。
【0225】
本発明の医薬組成物は、組成物の形態に適当な様式で投与する。一般的な経路としては、皮下、筋肉内、腹腔内、皮内、経口、経鼻および経肺(すなわち、エアロゾールによる)が挙げられる。ヒトに用いるための本発明の医薬組成物は、通常、経口的に投与される。
経口、経鼻または局所投与用の医薬組成物は、錠剤、カプセル剤、散剤、液剤および懸濁剤といった固体、半固体または液体の形態で提供することができる。注射剤用の組成物は、液状溶液もしくは懸濁液として、エマルションとして、または注射剤とするために液体へ溶解または懸濁するのに適当な固体形態として提供することができる。気道を経由する投与には、適当なエアロゾール化装置を用いる場合には、固体、粉末または液体のエアロゾールを提供する組成物が好ましい。必ずしも必要ではないが、医薬組成物は、正確な量を投与するのに適当な単位投与形態で提供されることが好ましい。また、本発明は徐放性または持続性形態も開示するものであり、これによって、比較的一貫したレベルの活性化合物が長期間にわたって提供される。
【0226】
アデノシンA2A受容体アンタゴニストは、体重1キログラム当たり約0.001〜約20.0mgの量で投与できることが好ましい。投与量が、体重1キログラム当たり約0.01〜約10mgの範囲であることがより好ましい。本発明のアデノシンA2A受容体アンタゴニスト組成物は、最終的には血中から除かれるため、投与に関しては、該組成物が示され、また好ましい。
アデノシンA2A受容体アンタゴニストは、投与製剤に応じて、かつ治療上有効であるような量で投与することができる。全身投与量は、患者の年齢、体重および状態ならびに投与経路に応じて変わる。
【0227】
本発明の方法に有用な医薬製剤は、公知の方法で製造される。医薬組成物の調製は、一般的に認められた医薬製剤の調製方法に従って実施される。例えば、レミングトンズ・ファーマシューティカル・サイエンシス(Remington’s Pharmaceutical Sciences)第18版(1990)、マーチン(Martin)編、マック・パブリッシング・コーポレーション(Mack Publishing Co.)、ペンシルバニア(PA)参照。意図する用途および投与様式に応じて、医薬組成物の調製において有効成分をさらに加工することが望ましいこともある。適当な加工法としては、滅菌、適当な非毒性および非干渉成分との混合、投与単位への分割ならびに送達容器への封入が挙げられる。
【0228】
経口用医薬製剤は、活性化合物を固形賦形剤と混合し、得られた混合物を場合によって粉砕し、所望または必要に応じて、適当な助剤を添加した後、顆粒混合物を加工することにより、錠剤を得ることができる。
適当な賦形剤としては、サッカライドなどの充填剤に限定されるものではないが、例えばラクトースまたはスクロース、マンニトールまたはソルビトール;セルロース誘導体;亜鉛化合物;リン酸三カルシウムまたはリン酸三カルシウムまたはリン酸水素カルシウム等のリン酸水素カルシウム等のリン酸カルシウム;またはリン酸水素カルシウム等のリン酸三カルシウム、またはリン酸水素カルシウム等のリン酸カルシウム等の充填剤、ならびに例えばトウモロコシデンプン、コムギデンプン、コメデンプン、ジャガイモデンプンを用いたデンプンペースト;ゼラチン;トラガカントガム;および/またはポリビニルピロリドン等の結合剤が挙げられる。
【0229】
助剤としては、流動調節剤およびシリカ、タルク、ステアリン酸またはそれらの塩、および/またはポリエチレングリコール等の滑沢剤が挙げられる。錠剤、カプレット剤またはカプセル剤の核部は、所望により、胃液に耐えるような適当なコーティングが施される。この目的には、濃縮糖類溶液を用いることができ、場合によって、これはアラビアゴム、タルク、ポリビニルピロリドン、ポリエチレングリコールおよび/または二酸化チタン、ラッカー溶液および適当な有機溶媒もしくは混合溶媒を含有することができる。胃液に耐えるコーティング、すなわち腸溶性コーティングを施すためには、アセチルセルロースフタレートまたはヒドロキシプロピルメチルセルロースフタレート等の適当なセルロース調製物の溶液が用いられる。色素または顔料を、例えば識別のためにまたは活性化合物用量の組み合わせを明示するために、錠剤またはコーティングに添加することができる。
【0230】
経口的に用いることができるその他の医薬製剤としては、ゼラチン製の押し込み型カプセル剤、ならびにゼラチンとグリセロールまたはソルビトール等の可塑剤とでできた密閉型軟カプセル剤が挙げられる。押し込み型カプセル剤は、活性化合物を顆粒の形態で含むことができ、この顆粒はラクトース等の充填剤、デンプン等の結合剤、および/またはタルクまたはステアリン酸マグネシウム等の滑沢剤、および場合によって安定化剤と混合することができる。軟カプセル剤では、活性化合物を脂肪油または流動パラフィン等の適当な液体に溶解するかまたは懸濁することが好ましい。さらに、安定化剤を添加してもよい。
【0231】
本発明のアデノシンA2A受容体アンタゴニストは、また生物分解性の徐放性担体と混合
する場合にはインプラント剤の形態で投与することもできる。また、有効成分の持続放出を目的として、有効成分を経皮パッチとして製剤することもできる。インプラント剤およびパッチ剤の製造方法は、当技術分野では周知である。レミングトンズ・ファーマシューティカル・サイエンス(Remington’s Pharmaceutical Sciences) 第18版(1990);およびキドニュウズ(Kydonieus)編(1992)トゥリータス・オン・コントロールド・ドラッグ・デリバリー(Treatise on controlled drug delivery)、マーセル・デッカー(Marcel Dekker)、ニューヨーク(NY)。
【0232】
本発明を以下の実施例によってさらに例示するが、本発明は実施例によって何ら限定されない。本明細書に記載した全ての参照文献は、参照により本明細書に組み入れられる。
【実施例1】
【0233】
アデノシンA2A受容体アンタゴニストであるKW-6002の、L-ドーパ関連の運動合併症によって悪化したパーキンソン病の治療としての安全性および有効性を、12週間の多施設試験研究(multicenter, exploratory study)にて検討した。運動合併症を伴うPD被験者を、無作為かつ盲目的に、3つのパラレル治療群(parallel treatment arms)のうちの1つに振り分けた:プラセボ(n=29);KW-6002 最大20mg/日(n=26);KW-6002 最大40mg/日(n=28)。2つの主要な有効性指標を設定した:1) 8時間の来院中に治験担当医師によって判定される「オフ」時間の変化、および2) 被験者の自宅での運動日誌によって判定される「オフ」時間の変化。
【0234】
83人の登録被験者のうち65人が試験を完了した;中止脱落率は、治療群間において同様であった。KW-6002治療は、患者の覚醒時間中の「オフ」状態で過ごす割合の減少において、プラセボ治療よりも有意に効果的であった。自宅での日誌により評価した場合には、覚醒時間中のオフ状態で過ごす割合が、プラセボ群では2.2%増加したのと比較して、KW-6002に振り分けられた被験者では7.1%減少した(p=0.008)。プラセボ群よりもKW-6002群におけるオフ時間の減少が1.7時間多かった(p=0.004)。治験担当医師による8時間のオン/オフ判定の結果では、統計学的な有意性に達した(p=0.054)。KW-6002で治療された患者が過ごした「オフ」状態の時間は、プラセボ群の患者のものよりも0.51時間少なかった(p=0.061)。
【0235】
試験では、またプラセボ群と比較して、ベースラインから12週までKW-6002で治療した患者における早朝ジストニアの減少が示された。
方法
これは、運動合併症を伴うL-ドーパで治療されたPD患者における補助療法としてのKW-6002の安全性および有効性についての、12週間の、二重盲検の、プラセボ対照の、無作為の、パラレル群の多施設試験研究である。適格患者は、英国パーキンソン病協会(UnitedKingdom PD Society(UKPDS))脳バンク診断基準(ダニエル(Daniel)ら(1993))を満たし、少なくとも1年間、L-ドーパ/カルビドパを服用しており、1日当たり少なくとも4回、L-ドーパ/カルビドパを服用しており、かつエンド・オブ・ドーズ(end-of-dose)でのウェアリング・オフ(wearing-off)をはじめとする運動合併症を起こしている患者とした。
【0236】
インフォームド・コンセントを行った後、被験者は4〜8週間のスクリーニングを受けた。訪問の4週前には、投薬の用量用法を一定にした。この訪問時に、被験者は自宅での日誌の全ての記入に関する指導を受けた。
ベースライン時に、被験者は、8時間の院内(in-office)評価を受けた。この評価に先立ち、被験者はPD用薬を差し控え、午前零時から絶食をした。最初の評価の後、その日初回のPD用薬を投与し、その後の用量は、被験者の通常の投与間隔で投与した。評価は特別指導を受けた、有害事象(adverse events)および臨床検査の結果を知らされていない評価者(blinded rater)によって行われた。被験者には、無作為化を適格なものとするために、8時間の評価中、PD用薬の投与後に少なくとも90分のオフ時間を示すことが求められた。
【0237】
スクリーニングおよびベースラインの評価を無事終了した被験者を、KW-6002の2つの投与用量用法群または対応するプラセボのうちの1群に、1:1:1の比で無作為に振り分けた。KW-6002群に無作為に振り分けられた患者は、1週〜4週の期間に5mg/日、5週〜8週の期間に10mg/日および9週〜12週の期間に20mg/日(5/10/20群)、または1週〜4週の期間に10mg/日、5週〜8週の期間に20mg/日および5週〜9週の期間に40mg/日(10/20/40群)のいずれかで投与を受けた(図1)。試験薬は、毎日、被験者の通常の朝食とともに、1回量として服用した。
【0238】
その後の評価は、2、4、6、8、10および12週目に行った。被験者は、各訪問の前の週中に自宅で3種類の日誌の全ての記入を行った。各訪問時に、有害事象を評価した。8時間の院内評価は、4、8および12週目に行った。臨床血液検査およびECGは、ベースライン時、4週、8週および12週目に行った。
試験遂行中、治験担当医師は、L-ドーパ関連の有害事象を改善するよう、L-ドーパの1日の全投与量を減らすことができた。L-ドーパ投与の間隔を変更することは、認められなかった。
結果
83人の被験者を無作為に振り分けた。
【0239】
試験群間では、人口統計的およびベースライン特性の顕著な差は見られなかった。
3治療群全ての被験者は、それらの試験薬をピル数に基づく99%を服薬した。試験中、いずれの治療群においてもまたは上記2つのKW-6002群を併せた群とプラセボ群とを比較しても、L-ドーパの平均1日投与量に有意な変化はなかった。
自宅での日誌で評価した場合、上記2つのKW-6002群に無作為に振り分けられた被験者は、プラセボ群に無作為に振り分けられた被験者と比較して、オフ時間における有意な減少があった(図1)。覚醒時間中のオフ状態で過ごす割合が、プラセボ群では2.2%増加したのと比較して、KW-6002に振り分けられた被験者では7.1%減少した(p=0.008)。上記2つのKW-6002投与群はいずれも、プラセボ群と比較して、オフ時間の割合において有意な減少を示した。同様に、各KW-6002群と同様に上記2つのKW-6002群を併せた群では、オフ時間の合計時間において、有意な減少を示した。オフ時間において、プラセボ群での0.5時間増加したのと比較して、KW-6002に振り分けられた被験者では1.2時間減少した(p=0.004)(図1)。
【0240】
上記2つのKW-6002群を併せた群では、プラセボ群と比較して、オフ時間におけるより大きな減少傾向があることが、8時間の院内評価での医師によるオフ時間の判定で確認された。オフ時間が、プラセボ群では3.3%減少したのと比較して、KW-6002に振り分けられた被験者では10.0%の減少を示した(p=0.05)。同様に、オフ時間が、プラセボ群では0.3時間の減少したのと比較して、KW-6002に振り分けられた被験者では0.8時間の減少を示した(p=0.06)。高用量KW-6002群(10/20/40群)でのオフ時間の減少は有意なものであった(P=0.02)。
【0241】
KW-6002で治療した患者の早朝ジストニアは、プラセボ群と比較して、ベースラインから12週まで減少した。
全般的な有害事象プロフィールでは、KW-6002で治療した被験者とプラセボで治療した被験者とで差がなかった。重篤な有害事象の全般的な発生は、試験群全体にわたり同様に分布した。総中止脱落数および有害事象による中止脱落数は、KW-6002群とプラセボ群間で同様であった。KW-6002群とプラセボ群間では、最大血圧または最小血圧、心拍数、呼吸数、体重、ECG、および尿検査もしくは血液化学検査の平均値における顕著な変化または差は認められず、臨床参照範囲内であった。
【0242】
本試験では、ドーパミンアゴニスト(例えばプラミペキソール、ペルゴリド、ロピニロール、ブロモクリプチン)、COMT阻害剤(例えばエンタカポン、トルカポン)およびMAO阻害剤であるセレギリンとの種々の医薬の併用下で、KW-6002はオフ時間を有意に減少させ、かつ安全性および優れた忍容性を示した。
この試験での結果に基づき、アデノシンA2A受容体アンタゴニストであるKW-6002は、L-ドーパによる運動合併症を伴うパーキンソン病患者のオフ時間を安全にかつ有効に減少させることができる。
【0243】
本試験はまた、アデノシンA2A受容体アンタゴニストであるKW-6002が、L-ドーパとドーパミンアゴニストおよび/またはCOMT阻害剤および/またはMAO阻害剤との医薬の併用により治療したパーキンソン病患者におけるオフ時間を有意に減少させたことを示している。
本試験はまた、KW-6002がパーキンソン病患者の早朝ジストニアを減少させることも示している。
【0244】
前述の発明を、明確にし、理解するために、例示および実施例によって細部にわたって記載してきたが、当業者には、特定の変更および改変を行うことができることは明らかであろう。よって、説明および実施例は、本発明の範囲を限定するものではない。
【実施例2】
【0245】
この二重盲検プラセボ比較試験への参加には、中程度〜重症のパーキンソン病患者16人が同意した。全ての患者を、KW-6002または対応するプラセボカプセル剤のいずれかに無作為に振り分けた。この試験は、6週間の増量の投与用法用量(40および80mg/日)で実施した。パーキンソン症候群をUPDRS パートIII 運動能力検査により評価した。第二のブラインドされた評価者(blinded rater)によるその後のオフライン評定のため、全ての評価をビデオテープに録画した。
【0246】
KW-6002単独または各患者の至適L-ドーパ用量の静脈内持続注入(a steady-state intravenous infusion)との組み合わせでは、パーキンソン病の重篤度には全く影響がなかった。注入した閾値のL-ドーパでは、KW-6002は、抗パーキンソン病作用を38%増強した(p < 0.05)。医学上有意な薬物毒性は認められなかった。
閾値のL-ドーパと組み合わせたKW-6002は、至適L-ドーパ用量単独の場合と同程度、運動能力の(UPDRS III 運動能力検査尺度を用いて評価した)項目を改善させた。
【0247】
このように、本発明は、L-ドーパによる治療を1種以上のアデノシンA2A受容体アンタゴニストの有効量と組み合わせることによって、臨床上有効な量より少ないL-ドーパ(すなわち、L-ドーパ減量効果)でパーキンソン病患者を治療するための方法および組成物を提供する。
本試験では、安静時およびイスからの立ち上がった時の振戦の平均評価点が、ベースラインおよびプラセボ群に関して、4週および6週で実質的に改善されることが示された。KW-6002で治療した患者では、ベースラインおよびプラセボ治療群に対し、歩行および身体運動緩徐の平均評価点が6週でかなりの改善が認められた。このことは、KW-6002がまた、パーキンソン病患者およびその他の運動疾患を有する患者の両方における振戦および歩行の治療に有効であることを意味する。
【0248】
このように、本発明は、振戦、運動緩徐、歩行および寡動に関連する運動障害を有効治療する方法を提供する。
実施例1および2の試験結果から、アデノシンA2A受容体のメカニズムが、パーキンソン病および運動合併症の症状の発現においてある役割を果たしていること、およびこの受容体を選択的にブロックすることが可能な薬剤が、L-ドーパ治療を受けたこの疾患を伴う患者に治療効果を与えることが確認される。
【0249】
すなわち、本発明は、それを必要とする患者に、1種以上のアデノシンA2A受容体アンタゴニストの有効量を投与することにより運動障害を治療する方法、ならびにパーキンソン病の治療においてL-ドーパ療法を受けている患者のL-ドーパの副作用を減少させるか抑制する方法を提供する。
【実施例3】
【0250】
6−ヒドロキシドーパミン障害処置ラットおよび6−ヒドロキシドーパミン障害処置後にL-ドーパによる長期処置を施したラットにおいて、大脳基底核の出力核、黒質緻密部(SNr)のGABAおよびグルタメート濃度を測定する。アデノシンA2A受容体選択的アンタゴニストの、SNrのGABAおよびグルタメートレベルおよびジスキネジアに及ぼす影響を試験した。
方法:
6−ヒドロキシドーパミン(8μg)をラットの左内側前脳束に注入した。障害処置の1週間後、アポモルヒネを注入(0.1mg/kg 皮下)によるラットの逆側回転(contralateralturning)について試験した。強い逆側回転を示す動物のみを、その後の試験に用いた。アポモルヒネ試験の3日後、L-ドーパを20mg/kgの用量で、1日2回、1〜3週間経口投与した。
【0251】
L-ドーパ誘発性ジスキネジアを認定するため、ラットを個別に観察し、歩行運動、軸性、四肢および口舌のAIMをはじめとする異常不随意時間(abnormal involuntary moments)(AIM)の重症度尺度を記録し、AIMが現れている時間/モニタリング期間の比率によって4つのAIMサブタイプそれぞれに0〜4の評点を付けた。L-ドーパによる長期治療中、AIMの重症度尺度の記録を行った。さらに、微量透析試験中、四肢および軸性のAIMのそれぞれの振幅による尺度を記録した。四肢または軸性のAIMの振幅評点(それぞれ0〜4までの範囲)を、動物の手足/四肢の転位および遠位筋と近位筋群との目に見える関与の両方の大きさ、または動物の首および胴体のその身体の縦軸からの側方変位(または捻転)それぞれに基づき評価した。
SNrにおけるGABAおよびグルタメートを、6−ヒドロキシドーパミン障害処置後とL-ドーパによる反復処置が終了してから4日後、in vivo微量透析技術によって測定した。ラットを各試験チャンバーに入れ、SNrに挿入した微量透析プローブを、自由に動かすことが可能な(持続的な回転行動も可能)液体旋回装置(TCS2-23,Eicom)に取り付けた。プローブを改変リンゲル溶液(1.2mmol/L CaCl2、2.7mmol/L KCl、148mmol/L NaClおよび0.85mmol/L MgCl2;pH7、人工脳脊髄液)とともに、マイクロインジェクションポンプ(CMA/100,Carnegie Medicin AB)を介して2μL/分の速度で連続的に潅流した。放出の基礎レベルを3〜4時間安定化させた後、2時間の潅流中に分画収集装置(CMA/140,Carnegie Medicin)を用いて4サンプル(各60μL)を収集した。各サンプルにあたり60μLの潅水(30分間のもの)をサンプリングチューブ(自動サンプリングインジェクター 231XL,Eicom用のサンプル容器)に2x30μLに分割し、各サンプル中のGABAおよびグルタメートの濃度を測定した。サンプルは直ちにアッセイするかまたは急速冷凍(-80℃)し、保存した後、アッセイした。GABAおよびグルタメートは、オルトフタルジアルデヒド試薬によるアミノ酸のプレカラム誘導体化を行った後、蛍光検出を用いた逆相高速液体クロマトグラフィーにより解析した。LindrothおよびMopper(1979)。
結果:
KW-6002(1mg/kg 経口)は、6−ヒドロキシドーパミン障害処置ラットのSNrにおいて、GABAおよびグルタメートレベルの、著しい、持続的な増加を引き起こした(図2、3)。L-ドーパはまた、6−ヒドロキシドーパミン障害処置ラットによる黒質GABAおよびグルタメートレベルの増加を誘導した(図4、5)。
【0252】
1週間、毎日、L-ドーパによる反復処置をした場合のAIMは、個々のラットによってなお様々であり、最大重症度等級を短時間維持した。2〜3週間のL-ドーパによる長期処置で、動物は安定したAIMを示し、L-ドーパ投与後10分間〜3時間、平均最大AIM評点(9)を維持した。
表3に示すように、黒質グルタメートの基礎濃度は、L-ドーパによる長期処置の2週間の間、一定レベルを維持し、3週間で飛躍的に増加した。一方、黒質GABAレベルは期間を通じて変化がなかった。表3は、6−ヒドロキシドーパミン障害処置後にL-ドーパによる長期処置を施したラットにおける黒質GABAおよびグルタメートの基礎レベルを示している。
【0253】
【表3】
【0254】
L-ドーパは、著しいAIM(四肢および軸性AIMの振幅評点合計)を誘導した。一方、KW-6002は、長期処置したラットにおいてほとんどまたは全くAIMを誘導しなかった(図6)。
L-ドーパは、黒質GABAレベルに影響を及ぼすことなく、グルタメートレベルを高めた。一方、KW-6002は、黒質GABAおよびグルタメートレベルに対して、全くまたはほとんど影響を及ぼさなかった(図7、8)。
【0255】
L-ドーパ誘発性AIMの振幅増大は、L-ドーパ誘発性黒質グルタメートレベルの高まりとともに経時変化した(図6および8)。
【実施例4】
【0256】
MPTPの反復注入によってパーキンソン病にしたL-ドーパまたはドーパミン作動性薬剤を受けたことが全くないMPTPサルにおける、L-ドーパ単独またはKW-6002またはプラセボと
の組み合わせでの長期治療の効果の比較。
動物:3〜5kgの間の重量の8匹の雌の薬剤未投与のカニクイザルを用いた。これらを、明らかなパーキンソン症候群(我々の尺度の6以上に当たる身体障害の評点で扱われる運動不能、前かがみの姿勢および振戦)の発症までMPTP(毎日0.5mg)を皮下注入することによってパーキンソン病にした。必要な累積用量は変動した:3.5〜23.5mg。
【0257】
著しく運動不能であることから早期に処置する必要のあった動物を除き、動物を1ヶ月以上回復させた。少なくとも、1日1回、これらに評点を付けた。身体障害評点は、その期間を通じて安定した状態を保っていた。
処置:全ての動物を、1日1回、L-ドーパ/ベンセラジド 100/25mg(総用量)で処置した。特別なカプセル剤ハンドラ(capsule handler)を用いて、薬剤を経口投与した。KW-6002群の動物はまた、この化合物(90mg/kg)も経口経路によって服用した。動物を毎日そのケージ内でワンウェイスクリーンを通して観察し(月曜日から金曜日まで)、重大な事象(異常行動−ジスキネジア)にはビデオ録画を行った。その薬剤の効能が及ぶ前およびその間、それらに身体障害尺度、実際にはジスキネジア評価尺度で評点を付けた。L-ドーパによる処置を1ヶ月間続けた。
結果
L-ドーパに対する抗パーキンソン病効果は、4週間の間でのパーキンソン病評点の改善という点から言えば、安定しており、L-ドーパ単独群と併用(L-ドーパ+KW-6002)治療群とでは同等であった(図9)。
【0258】
自発運動数は、併用治療群において、より高レベルへと増加し、そのレベルは4週間にわたり維持された(図10)。
ジスキネジアは、併用治療群よりもL-ドーパ群で急速に増え、より高いレベルに達した。このように、ジスキネジアの発症は、KW-6002の存在下で遅らされた。ジスキネジアが現れた後でさえ(3週および4週)、KW-6002治療群では、L-ドーパ単独群よりもジスキネジアの発症は少なかった(図11)。
【0259】
1ヶ月の処置期間の終了時に、全ての薬剤を中止した。その翌日、KW-6002群の動物に、標準用量のL-ドーパ/ベンセラジド(100/25mg)の経口投与を試みた。ジスキネジアをすでに示している3動物では、その組み合わせに対して同様の応答があった。
つまり、これまでに薬剤未投与であるパーキンソン病のサルの1ヶ月間の治療において、KW-6002をL-ドーパへ加えることによって、ジスキネジアの発症が遅らされ、ジスキネジアの発症が少なくなり、それと同時に、より強い自発運動応答およびパーキンソン病評点においてL-ドーパ単独群と同等の改善がもたらされた。
【実施例5】
【0260】
ジスキネジアを示すよう予めL-ドーパを前投与したMPTP処置コモンマーモセットにおいて、L-ドーパ誘発性ジスキネジアに及ぼすKW-6002の影響を調べた。
方法:MPTP(Sigma-Aldrich,St.Louis,MO,USA)を生理食塩水に溶かし、毎日、2.0mg/kgの用量で、5日間皮下投与した。次いで、MPTP 2mg/kgをさらに約3週間投与した。MPTP曝露の8週間後、動物は、基本的な自発運動の著しい減少、連係動作の遅延および減少、身体部位の異常な状態、およびチェック動作(checking movement)および目の瞬きの減少などの慢性パーキンソン病の症状を示した。慢性パーキンソン病の十分な症状を示す動物を、本試験に選抜した。
【0261】
次いで、L-ドーパ(10mg/kg 経口)とともにベンセラジド(2.5mg/kg 経口)を、MPTP処置マーモセットに1日2回、28日間投与し、ジスキネジアを誘導した。動物のジスキネジアに表4に記載の評価尺度を用いて評点を付けた。各L-ドーパ投与によって、高いジスキネジア評点である最大値8を示した動物を本研究に用いた。MPTP処置マーモセットにて、L-ドーパ(10mg/kg 経口、ベンセラジド 2.5mg/kg 経口とともに)によって誘導されたジスキネジアに評点を付けた。評点をL-ドーパ前値として算出した。その翌日、投与媒体(Vehicle)対照値を得るために、動物にVehicleを投与した。1日後、それらにL-ドーパ(2.5mg/kg、経口)を投与し、L-ドーパ対照値を得た。次いで、KW-6002の、L-ドーパ誘発性ジスキネジアに及ぼす影響を観察した。L-ドーパ(2.5mg/kg、経口)と組み合わせたKW-6002(10mg/kg 経口)の投与をその翌日(1日目)に開始し、1日1回、21日間繰り返し、その後、1週間ウォッシュアウト(washout)期間をおいた。動物におけるジスキネジアを、評価尺度に従って、1日、3日、5日、7日、14日、21日および28日目に評価した。さらに、35日目にL-ドーパ(10mg/kg 経口)をマーモセットに投与して、L-ドーパ後値を得た。
【0262】
表4は、四肢ジストニア、舞踏病および舞踏病アテトーゼジスキネジアならびに常同症の存在を数値で示した結果を示している。例えば口腔顔面運動、ミオクローヌスおよび複雑な常同行為(例えば入念なチェック、強迫的グルーミング)等の異常な動作は、ジスキネジア評価から除外される。
【0263】
【表4】
【0264】
ジスキネジアについての備考
ジストニア(腕、脚および胴体):異常な持続的な姿勢(例 脚の挙上)。
常同的なリーチング(stereotypic reaching)(腕)
アテトーシス(腕および脚):くねくねと捻る動作。
舞踏病(腕および脚):四肢の異常な急速な(ダンス様の)動作。
アカシジア:運動性不穏状態。
ジスキネジア評点は、ジスキネジアの重症度に応じて高くなり、最高評点は4点である。
【0265】
結果:結果を図12に示した。L-ドーパ(2.5mg/kg)の経口投与により、ジスキネジアを示すよう予めL-ドーパを前投与したMPTP処置コモンマーモセットに軽度ジスキネジアが誘導された。L-ドーパ(2.5mg/kg 経口)による誘発性ジスキネジアは、L-ドーパ単独の対照と比較して、21日間、KW-6002(10mg/kg 経口)では変化がなかったか、または減少する傾向があった。21日目に、KW-6002は、2.5mg/kgのL-ドーパ単独と比較して、L-ドーパ誘発性ジスキネジアの有意な減少を示した。L-ドーパ誘発性ジスキネジアの、KW-6002によってもたらされる有意な減少は、KW-6002およびL-ドーパの21日間の反復投与後の、KW-6002(10mg/kg)のL-ドーパ(2.5mg/kg)をともなう1週間の急性投与によって認められた。
【0266】
つまり、これらの試験の結果は、KW-6002がL-ドーパ誘発性ジスキネジアを抑制することを示している。
調製例1:錠剤
次の組成の錠剤を従来の方法で調製する。
KW-6002(40g)を286.8gのラクトースおよび60gのジャガイモデンプンと混合し、続いて、120gの10%ヒドロキシプロピルセルロース水溶液の添加を行う。得られた混合物を混練し、造粒した後、従来の方法により乾燥させる。この顆粒を精製し、錠剤を作製するのに用いる顆粒を得る。顆粒を1.2gのステアリン酸マグネシウムと混合した後、混合物を、8mm径の乳棒を備えた錠剤製造機(RT-15型,菊水)を用いて、それぞれが20mgの有効成分を含む錠剤に成形する。
【0267】
この処方を表5に示す。
表5
化合物(I) 20 mg
ラクトース 143.4mg
ジャガイモデンプン 30 mg
ヒドロキシプロピルセルロース 6 mg
ステアリン酸マグネシウム 0.6mg
200 mg
調製例2:カプセル剤
次の組成のカプセル剤を従来の方法で調製する。
【0268】
KW-6002(200g)を995gのアビセル(Avicel)および5gのステアリン酸マグネシウムと混合する。混合物を、カプセル充填機(LZ-64型,Zanashi)を用いて、それぞれが120mgの容量を有する硬カプセルNo.4の中に入れ、それぞれの有効成分を含むカプセル剤を得る。
この処方を表6に示す。
【0269】
表6
化合物(I) 20 mg
アビセル 99.5mg
ステアリン酸マグネシウム 0.5mg
120 mg
調製例3:注射剤
次の組成の注射剤を従来の方法で調製する。
【0270】
KW-6002(1g)を100gの精製大豆油に溶かし、続いて、12gの精製卵黄レシチンおよび25gの注射用グリセリンの添加を行う。得られた混合物を注射用蒸留水で最大1,000mlにし、十分に混合し、従来の方法により乳化させる。得られた分散液を0.2μmの使い捨て薄膜フィルター を用いて、無菌濾過に付した後、2ml分量で、ガラスバイアル中に無菌状態で入れ、各バイアルが2mgの有効成分を含む注射剤を得る。
【0271】
この処方を表7に示す。
表7
化合物(I) 2 mg
精製大豆油 200 mg
精製卵黄レシチン 24 mg
注射用グリセリン 50 mg
注射用蒸留水 1.72ml
2.00ml
前述の発明を、明確にし、理解するために、例示および実施例によって細部にわたって記載してきたが、当業者には、特定の変更および改変を行うことができることは明らかであろう。よって、説明および実施例は、本発明の範囲を限定するものではない。
参照文献一覧
US Patent No. 5,484,920
US Patent No. 5,543,415
US Patent No. 5,587,378
Adler et al. (1993a) “Vitamin E treatment of tardive dyskinesia” Am. J. Psych. 150:1405-1407
Adler et al. (1993b) “Vitamin E in tardive dyskinesia: time course of effect after placebo substitution” Psychopharmacol. Bull. 29:371-374
Adler et al. (1999) “Vitamin E treatment for tardive dyskinesia” Arch. Gen. Psych. 56:836-841
Akhtar et al. (1993) “Vitamin E in the treatment of tardive dyskinesia” J. Neural. Transm. Gen. Sect. 92:197-201
Alexander et al. (1990) “Functional architecture of basal ganglia circuits: neuronal substrates of processing” Trends Neurosci. 13:266-271
American College of Neuropsychopharmacology-FDA Task Force (1973) “Neurologic syndromes associated with neuroleptic drug use” N. Engl. J. Med. 289:20-23
Aoyama et al. (2000) “Rescue of locomotor impairment in dopamine D2receptor-deficient mice by an adenosine A2Areceptor antagonist” J. Neurosci. 20:5848-5852
Aoyama et al. (2002) “Distribution of adenosine A2Areceptor antagonist KW-6002 and its effect on gene expression in the rat brain” Brain Res. 953:119-25
Arvidsson et al. (1997) J. Comp. Neurol. 378:454-467
Augood et al. (1994) “adenosine A2Areceptor mRNA is expressed by enkephalin cells but not by somatostatin cells in rat striatum: co-expression study” Mol. Brain Res. 22:204-210
Baik et al. (1995) “Parkinsonian-like locomotor impairment in mice lacking dopamine D2 receptors” Nature 377:424-428
Baldessarini and Tarsy (1978) “Tardive dyskinesia” In: Psychopharmacology: a generation of progress. Lipton et al. eds. NY Raven Press pp. 995-1004
Baldessarini (1990) “Drugs and the treatment of psychiatric disorders” Goodman and Gilman's The Pharmacological Basis of Therapeutics, Gilman et al. (eds). New York, Pergamon Press, 8th Ed pp. 383-435
Bartholini (1983) “GABA system, GABA receptor agonist and dyskinesia” In: Modern problems in pharmacopsychiatry. Ban et al. eds. New York: Karger 21:143-154
Bezard et al. (2001) “Pathophysiology of levodopa-induced dyskinesia: Potential for new therapies” Nature Neurosci. Rev. 577-588
Bischot et al. (1993) “Vitamin E in extrapyramidal disorders” J. Postgrad. Med. 39:124-126
Blurn and Korczyn (1983) “Peptide neurotransmitters and their implications for the treatment of tardive dyskinesia” In: Modern problems in pharmacopsychiatry. Ban et al. eds. New York: Karger 21:187-95
Brown et al. (1991) “Striatal A2receptor regulates apomorphine-induced turning in rats with unilateral dopamine denervation” Psychopharmacol. 103:78-82
Burns et al. (1986) “Characterization of the A2adenosine receptor labeled by [3H]NECA in rat striatal membranes” Mol. Pharmacol. 29:331-346
Casey (1999) “Tardive dyskinesia and atypical antipsychotic drugs” Schizophrenia Res. 35:561-566
Chen et al. (2001a) J. Neurosci. 21:RC143(1-6)
Chen et al. (2001b) “The role of the D2 dopamine receptor (D2R) in A2A adenosine receptor (A2AR)-mediated behavioral and cellular responses as revealed by A2A and D2receptor knockout mice” Proc. Natl. Acad. Sci. USA 98:1970-1975
Chergui et al. (2000) “Functional GluR6 kinase receptors in the striatum: indirect down-regulation of synaptic transmission” J. Neurosci. 20:2175-2182
Claveria et al. (1975) “Tardive dyskinesia treated with pimozide” J. Neurol. Sci.; 24:393-401
Cochiolo et al. (2000) J. Neuroscience Res.59:126-135
Crossman (1990) “A hypothesis on the pathophysiological mechanisms that underlie levodopa or dopamine-agonist-induced dyskinesia in Parkinson’s disease: implications for future strategies in treatment” Mov. Disord. 5:100-108
Dabiri et al. (1993) “Effectiveness of vitamin E for treatment of long-term tardive dyskinesia” Am. J. Psych. 150:1405-1407
Dabiri et al. (1994) “Effectiveness of vitamin E for treatment of long-term tardive dyskinesia” Am. J. Psych. 151:925-926
Daly et al. (1983) “Subclasses of adenosine receptors in the central nervous system: interaction with caffeine and related methylxanthines” Cell. Mol. Neurobiol. 3:69-80
Daniel et al. (1993) “Parkinson's disease society brain bank, London: overview and research” J. Neural Transm. 39(suppl):165-172
DeLong (1990) “Primate model of movement disorders of basal ganglia origin” Trends Neurosci. 13:281-285
Dixon et al. (1996) “Tissue distribution of adenosine A2A receptor mRNAs in therat” Bri. J. Pharmacol. 118:1461-1468
Driesens (1988) “Neuroleptic medication facilitates the natural occurrence of tardive dyskinesia. A critical review” Acta-Psychiatr.-Belg. 88:195-205
Duvoisin (1974) “Variations in the ‘on-off’ phenomenon” Neurology 24:431-441Egan et al. (1992) “Treatment of tardive dyskinesia with vitamin E” Am. J. Psych. 149:773-777
Egan et al. (1997) “Treatment of tardive dyskinesia” Schiz. Bull. 23:583-609
Elkashef et al. (1990) “Vitamin E in the treatment of tardive dyskinesia” Am. J. Psych. 147:505-506
Fahn et al. (1987) “Members of the UPDRS development committee. Unified Parkinson's disease rating scale” in: Fahn et al. eds., Recent developments in Parkinson's disease, Florham Park, NJ Macmillan Health Care Information 2:153-163 and 293-304
Ferre et al. (1991) “Stimulation of adenosine A2receptors induces catalepsy” Neurosci Lett. 57:1062-1067
Ferre et al. (1996) “Dopamine D1receptor-mediated facilitation of GABAergic neurotransmission in the rat strioentopeduncular pathways and its modulation by adenosine A1receptor-mediated mechanisms” Eur. J. Neurosci. 8:1545-1553
Fink et al. (1992) “Molecular cloning of the rat A2Aadenosine receptor: selective co-expression with D2 dopamine receptors in rat striatum” Mol. Brain Res. 14:186-195
Florio et al. (1999) J. Pharmacol. Exp. Ther.290:817-824
Fredholm et al. (1994) “Action of caffeine in the brain with special reference to dependence” Pharmacol. Rev. 46:143-156
Gardos et al. (1994) “Ten-Year outcome of tardive dyskinesia” Am. J. Psych. 151:836-841
Gelenberg et al. (1990) “A crossover study of lecithin treatment of tardive dyskinesia” J. Clin. Psych. 51:149-153
Gerfen et al. (1990) “D1 and D2dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons” Science 250:1429-1432
Gerfen (1992) “The neostriatal mosaic: multiple levels of compartmental organization in the basal ganglia” Ann. Rev. Neurosci. 15:285-320
Goetz et al. (1994) “Utility of an objective dyskinesia rating scale for Parkinson's disease: inter- and intra-rater reliability assessment” Movement Dis. 9:390-394
Goldberg (1996) “The Use of Vitamin E to Treat People with Tardive Dyskinesia” J. Clin. Psych. 57:167-173
Graybiel (1990) “Neurotransmitters and neuromodulators in the basal ganglia” Trends Neurosci. 13:244-254
Grondin et al. (1999) “Antiparkinsonian effect of a new selective adenosine A2A receptor antagonist in MPTP-treated monkeys” Neurology 52:1673-1677
Groves (1983) “A theory of the functional organization of the neostriatum and the neostriatal control of voluntary movement” Brain Res. Rev. 5:109-132
Hoehn et al. (1967) “Parkinsonism: onset, progression and mortality” Neurol. 17:427-442
Hauser et al. (2000) “A home diary to assess functional status in Parkinson's disease patients with motor fluctuations and dyskinesia” Clin. Neuropharmacol. 23:75-81
Huang et al. (1981) “Reserpine and α-methyldopa in the treatment of tardive dyskinesia” Psychopharmacology 73:359-362
Hubble et al. (2002a) “Kyowa 6002-US-001 Study. Group. A novel adenosine antagonist (KW-6002) as a treatment for advanced Parkinson's disease (PD) with motor complications” Neurol. 54(suppl 6):S21.001
Hubble et al. (2002b) “A novel adenosine antagonist (KW-6002) as a treatment for advanced Parkinson's disease with motor complications” Neurol. 58(suppl 3):A162
Huntinger et al. (2001) “Ultrastructural localization of adenosine A2Areceptors suggests multiple cellular sites for modulation of GABAergic neurons in rat striatum” J. Comparative Neurol. 431:331-356
Ikeda et al. (2002) “Neuroprotection by adenosine A2Areceptor blockade in experimental models of Parkinson's disease” J. Neurochem. 80:262-270
Isaacson et al. (1995) “Counting quanta: direct measurements of transmitter release at a central synapse” Neuron 15:875-884
Itoh et al. (1981) “Drug-induced tardive dyskinesia” In: Current developments in psychopharmacology. Essman, Valzelli, eds. Jamaica, NY: Spectrum 93-126
Jaeger et al. (1994) “Surround inhibition among projection neurons is weak or nonexistent in the rat neostriatum” J. Neurophysiol. 72:2555-2558
Jarvis et al. (1989) “Direct autographic localization of adenosine A2receptors in rat brain using the A2selective agonist [3H]CGS21680” Eur. J. Pharmacol. 168:243-246
Kanda et al. (1994) “KF 17837: a novel selective adenosine A2Areceptor antagonist with anticataleptic activity” Eur. J. Pharmacol. 52:48-54
Kanda et al. (1998a) “Adenosine A2Aantagonist: a novel antiparkinsonian agent that does not provoke dyskinesia in parkinsonian monkeys” Ann. Neurol. 43:507-513
Kanda et al. (1998b) NeuroReport 9:2857-2860
Kanda et al. (2000) “Combined use of the adenosine A2A antagonist KW-6002 with L-DOPA or with selective D1or D2 dopamine agonists increases antiparkinsonian activity but not dyskinesia in MPTP-treated monkeys” Exp. Neurol. 162:321-327
Karp et al. (1981) “Metoclopramide treatment of tardive dyskinesia” JAMA 246:1934-1935
Kase et al. (2000) “Adenosine receptors and Parkinson's disease” Academic Press, San Diego
Kase (2001) “New aspects of physiological and pathophysiological functions of adenosine A2A receptor in basal ganglia” Biosci. Biotechnol. Biochem. 65:1447-1457
Kawaguchi (1989) “Intracellular recording of identified neostriatal patch and matrix spiny cells in a slice preparation preserving cortical inputs” J. Neurophysiol. 62:1052-1068
Kawaguchi et al. (1990) “Projection subtypes of rat neostriatal matrix cells revealed by intracellular injection of biocytin” J. Neurosci. 10:3421-3438
Kawaguchi et al. (1995) “Striatal interneurons: chemical, physiological and morphological characterization” Trends Neurosci. 18:527-535
Kawaguchi (1997) “Neostriatal cell subtypes and their functional roles” Neurosci. Res. 27:1-8
Kirk et al. (1994) “Adenosine A2Areceptor-mediated modulation of striatal [3H]GABA and [3H]acetylcholine release” J. Neurochem. 62:960-966
Kita et al. (1983) “Pallidal inputs to subthalamus: intracellular analysis” Brain Res. 264:255-265
Kita (1993) “GABAergic circuits of the striatum” in: Arbuthnott et al. eds., Progress in Brain Research, Elsevier Science Publishers B.V. 99:51-72
Kita (1994) “Parvalbumin-immunopositive neurons in rat globus pallidus: a light and electron microscope study” Brain Res. 657:31-41
Kita (1996) “Glutamatergic and GABAergic postsynaptic responses of striatal spiny neurons to intrastriatal and cortical stimulation recorded in slice preparations” Neurosci. 70:925-940
Klawans et al. (1980) “Tardive dyskinesia: Review and update” Am. J. Psych. 137:900-908
Koga et al. (2000) “Adenosine A2Areceptor antagonists KF17837 and KW-6002 potentiate rotation induced by dopaminergic drugs in hemi-parkinsonian rats” Eur. J. Pharmacol. 408:249-255
Koos (1999) “Inhibitory control of neurostriatal projection neurons by GABAergic interneurons” Nature Neurosci. 2:467-472
Kurokawa et al. (1994) “Inhibition by KF17837 of adenosine A2Areceptor-mediated modulation of striatal GABA and Ach release” BR. J. Pharmacol. 113:43-48
Kurokawa et al. (1996) J. Neurochem. 66:1882-1888
Kuwana et al. (1999) Adv. Neurol. 80:121-123
Lindroth and Mopper (1979) “High performance liquid chromatography determination of subpicomole amounts of amino acids by precolumn fluorescence derivatization
with o-phthaldialdehyde” Analyt. Chem. 51:1667-1674
Lohr and Caligiuri (1996) “A double-blind placebo-controlled study of vitamin E treatment of tardive dyskinesia” J. Clin. Psych. 57:167-73
Lohr et al. (1988) “Vitamin E in the treatment of tardive dyskinesia: the possible involvement of free radical mechanisms” Schiz. Bull. 14:291-296
Loopuijt et al. (1985) “Organization of the striatum: collateralization of its efferent axons” Brain Res. 348:86-99
Marsden et al. (1982) “Fluctuations in disability in Parkinson’s disease: clinical aspects” In: Mardsen, CD, Fahn S., eds. Movement disorders. New York: Butterworth Scientific pp. 96-122
McCreadie et al. (1994) “The Nithsdale Schizophrenia Surveys. XIV: Plasma lipid peroxide and serum vitamin E levels in patients with and without tardive dyskinesia, and in normal subjects” Am. J. Psych. 151:925-926
Moore and Bowers (1980) “Identification of a subgroup of tardive dyskinesia patients by pharmacologic probes” Am. J. Psych. 137:1202-1205
Mori et al. (1996) “The role of adenosine A2Areceptors in regulating GABAergic synaptic transmission in striatal medium spiny neurons” J. Neurosci. 16:605-611Mori et al. (2002) “Adenosine A2Areceptor-mediated dual modulation of GABAergic synaptic transmission in the striatopallidal system and its implication in action mechanism of A2Aantagonists for a novel therapy in Parkinson's disease” Neurol. (in press)
Moss et al. (1993) “Buspirone in the treatment of tardive dyskinesia” J. Clin. Psychopharmacol. 13:204-209
Nakanishi et al. (1987) “Electrical membrane properties of rat subthalamic neurons in an in vivo slice preparation” Brain Res. 437:35-44
Nambu et al. (1997) “Morphology of globus pallidus neurons: its correlation with electrophysiology in guinea pig brain slices” J. Comp. Neurol. 377:85-94
Nonaka et al. (1994) “(E)-8-(3,4,5-trimethoxystyryl)-1,3-dipropyl-7-methyl-xanthine, a potent and selective adenosine A2A receptor antagonist” Eur. J. Pharmacol. 267:335-341
Nonaka et al. (2002) “Biochemical characterization of a novel adenosine A2Areceptor antagonist, KW-6002” Seikaguku 74:937 3P663 (Abstract)
Obeso et al. (1997) “Basal ganglia physiology - A critical review” Advances in Neurology (Obeso et al. eds) 74:3-7 Lippincott Raven Publishers, Philadelphia
Obeso et al. (2000) “Pathophysiology of the basal ganglia in Parkinson's disease” Trends Neurosci. 23(Suppl):S8-S19
Ochi et al. (2000) “Systemic adenosine A2Areceptor antagonist decreases GABA release from rat globus pallidus increased by nigrostriatal lesions: A microdialysis study” Neuroscience 100:53-62
Oertel et al. (1984) “Immunocytochemical studies of GABAergic neurons in rat basal ganglia and their relations to other neuronal systems” Neurosci. Lett. 47:233-238
Olanow, Watts and Koller eds.(2001) An Algorithm (Decision Tree) for the Management of Parkinson’s Disease (2001): Treatment Guidelines, Neurology 56, Suppl. 5.
Parkinson Study Group (1996) “Impact of deprenyl and tocopherol treatment on Parkinson’s disease in DATATOP patients requiring levodopa” Ann. Neurol. 39:37-45
Parkinson Study Group (2000) “Pramipexole vs. levodopa as initial treatment for Parkinson's disease: a randomized controlled trial” JAMA 284:1931-1938
Paskevich et al. (1991) “Morphological assessment of neuronal aggregates in the striatum of the rat” J. Comp. Neurol. 305:361-369
Przedborski et al. (1995) Neuroscience 67:631-647
Rascol et al. (2000) “A five-year study of the incidence of dyskinesia in patients with early Parkinson's disease who were treated with ropinirole or levodopa” N. Eng. J. Med. 342:1484-1491
Richardson et al. (1997) “Adenosine A2Areceptor antagonists as new agents for the treatment of Parkinson's disease” Trends Pharmacol. Sci. 18:338-344
Rosin et al. (1998) “Immunohistochemical localization of adenosine A2Areceptors in the rat central nervous system” J. Comp. Neurol. 401:163-186
Schiffmann et al. (1990) Brain Res.519:333-337
Schiffmann et al. (1991a) “Striatal restricted A2receptor (RDC8) is expressed by enkephalin but not by substance P neurons: an in situ hybridization histochemistry study” J. Neurochem. 57:1062-1067
Schiffmann et al. (1991b) “Distribution of adenosine A2 receptor mRNA in the human brain” Neurosci. Lett. 130:177-181
Sherzai et al. (2002a) “Adenosine A2Aantagonist treatment of Parkinson's disease” Neurol. 54(Suppl 6):P06.104
Sherzai et al. (2002b) “Adenosine A2Aantagonist treatment of Parkinson's disease” Neurol. 58(suppl 3):A467
Shimada et al. (1992) “(E)-1,3-dialkyl-7-methyl-8-(3,4,5-trimethoxystyryl)xanthines: potent and selective adenosine A2 antagonists” J. Med. Chem. 35:2342-2345Shimada et al. (1997) “Adenosine A2Aantagonists with potent anti-cataleptic activity” Bio. Med. Chem. Lett. 7:2349-2352
Shindou et al. (2001) “Adenosine A2Areceptor enhances GABAA-mediated IPSCs in the rat globus pallidus” J. Physiol. 532:423-434
Shindou et al. (2002) “Presynaptic adenosine A2Areceptor enhances GABAergic synaptic transmission via cAMP dependent mechanism in the rat globus pallidus” Br.
J. Pharmacol. 136:296-302
Shiozaki et al. (1999) “Action of adenosine A2Areceptor antagonist KW-6002 on drug-induced catalepsy and hypokinesia caused by reserpine or MPTP” Psychopharmacology 147:90-95
Shriqui et al. (1992) “Vitamin E in the treatment of tardive dyskinesia: a double-blind placebo-controlled study” Am. J. Psych. 149:391-143
Shulman and Weiner (1997) Multiple system atrophy. In: Watts and Koller eds. Movement Disorders: Neurological Principles and Practice. New York, NY: McGraw-Hill pp. 297-306
Smith et al. (1987) “Distribution of GABA-immunoreactive neurons in the basal ganglia of the squirrel monkey” J. Comp. Neurol. 259:50-64
Stern et al. (1998) “Membrane potential synchrony of simultaneously recorded striatal spiny neurons in vivo” Nature 394:475-478
Tepper and Haas (1979) “Prevalence of tardive dyskinesia” J. Clin. Psych. 40:508-516
Thaker et al. (1990) “Clonazepam treatment of tardive dyskinesia: a practical GABAmimetic strategy” Am. J. Psych. 147:445-451
Uhrbrand and Faurbye (1960) “Reversible and irreversible dyskinesia after treatment with perphenazine, chlorpromazine, reserpine, ECT therapy” Psychopharmacologia 1:408-418
Watts and William eds. (1997) Movement Disorders: Neurologic Principles and Practice. New York: McGraw-Hill
Wichmann et al. (1993) “Pathophysiology of parkinsonian motor abnormalities” In: Narabayashi et al. eds. Advance in Neurology, Raven Press Ltd., 60:53-61
Wilbur and Kulik (1980) “Propranolol for tardive dyskinesia and EPS from neuroleptics: Possible involvement of β-adrenergic mechanisms” Prog. Neuro-Psychopharmacol. 4:627-632
【図面の簡単な説明】
【0272】
【図1】図1は、プラセボ群およびKW-6002群を併せた群の、自宅での日誌で記録したオフ時間の変化を示すグラフである。KW-6002を用いて治療を受けた被験者では、12週目に、オフの時間が有意に大幅に減少した(*p=0.004)。
【図2】図2は、6−ヒドロキシドーパミン障害処置ラットにおける、黒質GABAのレベルへのKW-6002の影響を示すグラフである。GABAのレベルは、化合物投与前の初期値からの変化率として表されている。KW-6002は、1mg/kg経口で黒質GABAのレベルを有意に高めた。
【図3】図3は、6−ヒドロキシドーパミン障害処置ラットにおける、グルタメートのレベルへのKW-6002の影響を示すグラフである。グルタメートのレベルは、化合物投与前の初期値からの変化率として表されている。KW-6002は、1mg/kg経口でグルタメートのレベルを有意に高めた。
【図4】図4は、6−ヒドロキシドーパミン障害処置ラットにおける、黒質GABAのレベルへのL-ドーパの影響を示すグラフである。L-ドーパは、黒質GABAをKW-6002によるそれらと同様のレベルまでの有意に上昇させた。
【図5】図5は、6−ヒドロキシドーパミン障害処置ラットにおける、グルタメートのレベルへのL-ドーパの影響を示すグラフである。L-ドーパは、グルタメートをKW-6002によるそれらと同様のレベルまでの有意に上昇させた。
【図6】図6は、長期間L-ドーパ処置した6−ヒドロキシドーパミン障害処置ラットにおける、異常不随意運動(AIMs)の総評点へのKW-6002およびL-ドーパの影響の経時変化を示すグラフである。L-ドーパは、著しくAIMsを誘発したが、KW-6002は、AIMsをほとんどまたは全く誘発しなかった。
【図7】図7は、長期間L-ドーパ処置した6−ヒドロキシドーパミン障害処置ラットにおける、黒質GABAのレベルへのKW-6002およびL-ドーパの影響の経時変化を示すグラフである。L-ドーパは、黒質GABAのレベルに影響を及ぼさなかった。KW-6002は、黒質GABAのレベルに、全くまたはほとんど影響を及ぼさなかった。
【図8】図8は、長期間L-ドーパ処置した6−ヒドロキシドーパミン障害処置ラットにおける、グルタメートのレベルへのKW-6002およびL-ドーパの影響の経時変化を示すグラフである。L-ドーパは、黒質GABAのレベルに影響を及ぼすことなく、グルタメートのレベルを高めた。KW-6002は、黒質GABAおよびグルタメートのレベルに、全くまたはほとんど影響を及ぼさなかった。
【図9】図9は、カニクイザルにおけるL-ドーパ単独処置(L-ドーパ/ベンセラジド;100/25mg(総用量)、1日1回)およびL-ドーパとKW-6002の処置(90mg/kg、1日1回)の間の、L-ドーパに対する抗パーキンソン病作用へのKW-6002の影響を示すグラフである。L-ドーパに対する抗パーキンソン病作用は、4週間にわたるパーキンソン病評点の改善という点から言えば、安定しており、2群間では同等であった。
【図10】図10は、カニクイザルにおけるL-ドーパ単独処置(L-ドーパ/ベンセラジド;100/25mg(総用量)、1日1回)およびL-ドーパとKW-6002の処置(90mg/kg、1日1回)の間の、L-ドーパに対する自発運動応答へのKW-6002の影響を示すグラフである。自発運動数は、併用処置群でより高いレベルに増加し、そのレベルは4週間にわたり維持された。
【図11】図11は、カニクイザルにおけるL-ドーパ単独処置(L-ドーパ/ベンセラジド;100/25mg(総用量)、1日1回)およびL-ドーパとKW-6002の処置(90mg/kg、1日1回)の間の、L-ドーパに対するジスキネジア応答へのKW-6002の影響を示すグラフである。ジスキネジアは、併用処置群よりもL-ドーパ群で急速に増加し、より高いレベルに達した。ジスキネジアの発症は、KW-6002の存在下では遅延された。
【図12】図12は、L-ドーパ誘発性ジスキネジアへのKW-6002の影響を示すグラフである。ジスキネジアを発症するようにL-ドーパを前投与した一般的なMPTP処置マーモセットにおいて、L-ドーパ(2.5mg/kg p.o.でベンセラジド0.625mg/kg p.o.とともに)を毎日、21日間投与して、ジスキネジアを誘発させ、同時にKW-6002を投与した。動物には、予めL-ドーパを10mg/kg p.o.でベンセラジド2.5mg/kg p.o.とともに、1日2回、28日間与えた(L-ドーパ)。併用処置では、生じた不随意運動量は増えず、それどころか2.5mg/kgのL-ドーパ単独投与と比べて、21日目に有意に減少した。 KW-6002は、21日間の長期処置により、L-ドーパ誘発性ジスキネジアを有意に減少させた。
【特許請求の範囲】
【請求項1】
少なくとも1種のアデノシンA2A受容体アンタゴニストを含有するL-ドーパおよび/またはドーパミンアゴニスト療法の副作用を減少させる、あるいは軽減または抑制する薬剤。
【請求項2】
副作用がL-ドーパまたは他のドーパミン作動性薬剤で誘発される運動合併症である請求項1に記載の薬剤。
【請求項3】
運動変動のオフ時間を減少させる請求項2に記載の薬剤。
【請求項4】
運動合併症がジスキネジアである請求項2に記載の薬剤。
【請求項5】
アデノシンA2A受容体アンタゴニストがキサンチン誘導体またはその薬理学上許容される塩である請求項1〜4のいずれかに記載の薬剤。
【請求項6】
アデノシンA2A受容体アンタゴニストが式(I):
【化1】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【化2】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【化3】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
で表される請求項1〜4のいずれかに記載の薬剤。
【請求項7】
アデノシンA2A受容体アンタゴニストが式(I-A):
【化4】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【化5】
(式中、R7、R8およびR9のうち少なくとも1つは低級アルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R10は水素原子または低級アルキルを表す)または
【化6】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
で表される請求項1〜4のいずれかに記載の薬剤。
【請求項8】
アデノシンA2A受容体アンタゴニストが式(I-B):
【化7】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【化8】
(式中、R6およびmは前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
で表される請求項1〜4のいずれかに記載の薬剤。
【請求項9】
アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである請求項1、2または4に記載の薬剤。
【請求項10】
臨床上有効な量より少ないL-ドーパと、該L-ドーパの効果を発揮させるために有効な量の1種以上のアデノシンA2A受容体アンタゴニストを含有するL-ドーパを減量して治療する(L-DOPA sparing treatment)ための薬剤。
【請求項11】
アデノシンA2A受容体アンタゴニストがキサンチン誘導体またはその薬理学上許容される塩である請求項10に記載の薬剤。
【請求項12】
アデノシンA2A受容体アンタゴニストが式(I):
【化9】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【化10】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【化11】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
で表される請求項10に記載の薬剤。
【請求項13】
アデノシンA2A受容体アンタゴニストが式(I-A):
【化12】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【化13】
(式中、R7、R8およびR9のうち少なくとも1つは低級アルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R10は水素原子または低級アルキルを表す)または
【化14】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
で表される請求項10に記載の薬剤。
【請求項14】
アデノシンA2A受容体アンタゴニストが式(I-B):
【化15】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【化16】
(式中、R6およびmは前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
で表される請求項10に記載の薬剤。
【請求項15】
アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである請求項10に記載の薬剤。
【請求項16】
臨床上有効な量より少ないL-ドーパと、該L-ドーパの効果を発揮させるために有効な量の1種以上のアデノシンA2A受容体アンタゴニストとを含有するL-ドーパを減量した治療(L-DOPA sparing treatment)のための組成物。
【請求項17】
アデノシンA2A受容体アンタゴニストがキサンチン誘導体またはその薬理学上許容される塩である請求項16に記載の組成物。
【請求項18】
アデノシンA2A受容体アンタゴニストが式(I):
【化17】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【化18】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【化19】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
で表される請求項16に記載の組成物。
【請求項19】
アデノシンA2A受容体アンタゴニストが式(I-A):
【化20】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【化21】
(式中、R7、R8およびR9のうち少なくとも1つは低級アルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R10は水素原子または低級アルキルを表す)または
【化22】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
で表される請求項16に記載の組成物。
【請求項20】
アデノシンA2A受容体アンタゴニストが式(I-B):
【化23】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【化24】
(式中、R6およびmは前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
で表される請求項16に記載の組成物。
【請求項21】
アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである請求項16に記載の組成物。
【請求項22】
少なくとも1種のアデノシンA2A受容体アンタゴニストと、COMT阻害剤ならびに/またはDAおよび/もしくはMAO阻害剤とを含有するパーキンソン病および/またはL-ドーパによる運動合併症を治療するための薬剤。
【請求項23】
アデノシンA2A受容体アンタゴニストがキサンチン誘導体またはその薬理学上許容される塩である請求項22に記載の薬剤。
【請求項24】
アデノシンA2A受容体アンタゴニストが式(I):
【化25】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【化26】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【化27】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
で表される請求項22に記載の薬剤。
【請求項25】
アデノシンA2A受容体アンタゴニストが式(I-A):
【化28】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【化29】
(式中、R7、R8およびR9のうち少なくとも1つは低級アルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R10は水素原子または低級アルキルを表す)または
【化30】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
で表される請求項22に記載の薬剤。
【請求項26】
アデノシンA2A受容体アンタゴニストが式(I-B):
【化31】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【化32】
(式中、R6およびmは前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
で表される請求項22に記載の薬剤。
【請求項27】
アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである請求項22に記載の薬剤。
【請求項28】
少なくとも1種のアデノシンA2A受容体アンタゴニストの有効量と、COMT阻害剤ならびに/またはDAおよび/もしくはMAO阻害剤とを含有するパーキンソン病の治療のための組成物。
【請求項29】
アデノシンA2A受容体アンタゴニストがキサンチン誘導体またはその薬理学上許容される塩である請求項28に記載の組成物。
【請求項30】
アデノシンA2A受容体アンタゴニストが式(I):
【化33】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【化34】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【化35】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
で表される請求項28に記載の組成物。
【請求項31】
アデノシンA2A受容体アンタゴニストが式(I-A):
【化36】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【化37】
(式中、R7、R8およびR9のうち少なくとも1つは低級アルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R10は水素原子または低級アルキルを表す)または
【化38】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
で表される請求項28に記載の組成物。
【請求項32】
アデノシンA2A受容体アンタゴニストが式(I-B):
【化39】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【化40】
(式中、R6およびmは前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
で表される請求項28に記載の組成物。
【請求項33】
アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである請求項28に記載の組成物。
【請求項34】
アデノシンA2A受容体アンタゴニストまたはアデノシンA2A受容体アンタゴニストとドーパミンアゴニストとの組み合わせのいずれかを含有する、患者が追加のL-ドーパ療法を必要とすることを遅らせるか、またはなくさせることにより、パーキンソン病治療の有効時間を延長する薬剤。
【請求項35】
運動合併症の発症を遅らせる請求項34に記載の薬剤。
【請求項36】
患者がL-ドーパまたはドーパミン作動性薬剤の前投与を受けていない場合に有効な請求項34に記載の薬剤。
【請求項37】
患者がL-ドーパまたはドーパミン作動性薬剤の後投与を受けない場合に有効な請求項34に記載の薬剤。
【請求項38】
アデノシンA2A受容体アンタゴニストがキサンチン誘導体またはその薬理学上許容される塩である請求項34〜37のいずれかに記載の薬剤。
【請求項39】
アデノシンA2A受容体アンタゴニストが式(I):
【化41】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【化42】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【化43】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
で表される請求項34〜37のいずれかに記載の薬剤。
【請求項40】
アデノシンA2A受容体アンタゴニストが式(I-A):
【化44】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【化45】
(式中、R7、R8およびR9のうち少なくとも1つは低級アルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R10は水素原子または低級アルキルを表す)または
【化46】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
で表される請求項34〜37のいずれかに記載の薬剤。
【請求項41】
アデノシンA2A受容体アンタゴニストが式(I-B):
【化47】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【化48】
(式中、R6およびmは前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
で表される請求項34〜37のいずれかに記載の薬剤。
【請求項42】
アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである請求項34〜37のいずれかに記載の薬剤。
【請求項43】
少なくとも1種のアデノシンA2A受容体アンタゴニストの有効量を含有する運動障害を治療する薬剤。
【請求項44】
運動障害が振戦、運動緩慢、歩行障害、ジストニア、ジスキネジア、遅発性ジスキネジアまたはその他の錐体外路症候群である請求項43に記載の薬剤。
【請求項45】
アデノシンA2A受容体アンタゴニストが、運動障害を引き起こす薬剤の作用を減少させる請求項43または44に記載の方法。
【請求項46】
アデノシンA2A受容体アンタゴニストがキサンチン誘導体またはその薬理学上許容される塩である請求項43または44に記載の方法。
【請求項47】
アデノシンA2A受容体アンタゴニストが式(I):
【化49】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【化50】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【化51】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
で表される請求項43または44に記載の方法。
【請求項48】
アデノシンA2A受容体アンタゴニストが式(I-A):
【化52】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【化53】
(式中、R7、R8およびR9のうち少なくとも1つは低級アルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R10は水素原子または低級アルキルを表す)または
【化54】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
で表される請求項43または44に記載の薬剤。
【請求項49】
アデノシンA2A受容体アンタゴニストが式(I-B):
【化55】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【化56】
(式中、R6およびmは前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
で表される請求項43または44に記載の薬剤。
【請求項50】
アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである請求項43または44に記載の薬剤。
【請求項51】
少なくとも1種のアデノシンA2A受容体アンタゴニストの有効量を含有する過剰運動を治療する薬剤。
【請求項52】
少なくとも1種のアデノシンA2A受容体アンタゴニストの有効量を含有する薬剤誘発性の過剰運動を治療する薬剤。
【請求項53】
アデノシンA2A受容体アンタゴニストがキサンチン誘導体またはその薬理学上許容される塩である請求項51または52に記載の薬剤。
【請求項54】
アデノシンA2A受容体アンタゴニストが式(I):
【化57】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【化58】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【化59】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
で表される請求項51または52に記載の薬剤。
【請求項55】
アデノシンA2A受容体アンタゴニストが式(I-A):
【化60】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【化61】
(式中、R7、R8およびR9のうち少なくとも1つは低級アルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R10は水素原子または低級アルキルを表す)または
【化62】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
で表される請求項51または52に記載の薬剤。
【請求項56】
アデノシンA2A受容体アンタゴニストが式(I-B):
【化63】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【化64】
(式中、R6およびmは前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
で表される請求項51または52に記載の薬剤。
【請求項57】
アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである請求項51または52に記載の薬剤。
【請求項58】
アデノシンA2A受容体アンタゴニストおよびL−ドーパを含有するパーキンソン病の治療剤。
【請求項59】
L−ドーパと同時にまたは時間を置いて別々に投与するためのアデノシンA2A受容体アンタゴニストを含有するパーキンソン病の治療剤。
【請求項60】
アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである請求項58または59に記載の治療剤。
【請求項1】
少なくとも1種のアデノシンA2A受容体アンタゴニストを含有するL-ドーパおよび/またはドーパミンアゴニスト療法の副作用を減少させる、あるいは軽減または抑制する薬剤。
【請求項2】
副作用がL-ドーパまたは他のドーパミン作動性薬剤で誘発される運動合併症である請求項1に記載の薬剤。
【請求項3】
運動変動のオフ時間を減少させる請求項2に記載の薬剤。
【請求項4】
運動合併症がジスキネジアである請求項2に記載の薬剤。
【請求項5】
アデノシンA2A受容体アンタゴニストがキサンチン誘導体またはその薬理学上許容される塩である請求項1〜4のいずれかに記載の薬剤。
【請求項6】
アデノシンA2A受容体アンタゴニストが式(I):
【化1】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【化2】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【化3】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
で表される請求項1〜4のいずれかに記載の薬剤。
【請求項7】
アデノシンA2A受容体アンタゴニストが式(I-A):
【化4】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【化5】
(式中、R7、R8およびR9のうち少なくとも1つは低級アルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R10は水素原子または低級アルキルを表す)または
【化6】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
で表される請求項1〜4のいずれかに記載の薬剤。
【請求項8】
アデノシンA2A受容体アンタゴニストが式(I-B):
【化7】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【化8】
(式中、R6およびmは前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
で表される請求項1〜4のいずれかに記載の薬剤。
【請求項9】
アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである請求項1、2または4に記載の薬剤。
【請求項10】
臨床上有効な量より少ないL-ドーパと、該L-ドーパの効果を発揮させるために有効な量の1種以上のアデノシンA2A受容体アンタゴニストを含有するL-ドーパを減量して治療する(L-DOPA sparing treatment)ための薬剤。
【請求項11】
アデノシンA2A受容体アンタゴニストがキサンチン誘導体またはその薬理学上許容される塩である請求項10に記載の薬剤。
【請求項12】
アデノシンA2A受容体アンタゴニストが式(I):
【化9】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【化10】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【化11】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
で表される請求項10に記載の薬剤。
【請求項13】
アデノシンA2A受容体アンタゴニストが式(I-A):
【化12】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【化13】
(式中、R7、R8およびR9のうち少なくとも1つは低級アルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R10は水素原子または低級アルキルを表す)または
【化14】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
で表される請求項10に記載の薬剤。
【請求項14】
アデノシンA2A受容体アンタゴニストが式(I-B):
【化15】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【化16】
(式中、R6およびmは前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
で表される請求項10に記載の薬剤。
【請求項15】
アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである請求項10に記載の薬剤。
【請求項16】
臨床上有効な量より少ないL-ドーパと、該L-ドーパの効果を発揮させるために有効な量の1種以上のアデノシンA2A受容体アンタゴニストとを含有するL-ドーパを減量した治療(L-DOPA sparing treatment)のための組成物。
【請求項17】
アデノシンA2A受容体アンタゴニストがキサンチン誘導体またはその薬理学上許容される塩である請求項16に記載の組成物。
【請求項18】
アデノシンA2A受容体アンタゴニストが式(I):
【化17】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【化18】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【化19】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
で表される請求項16に記載の組成物。
【請求項19】
アデノシンA2A受容体アンタゴニストが式(I-A):
【化20】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【化21】
(式中、R7、R8およびR9のうち少なくとも1つは低級アルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R10は水素原子または低級アルキルを表す)または
【化22】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
で表される請求項16に記載の組成物。
【請求項20】
アデノシンA2A受容体アンタゴニストが式(I-B):
【化23】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【化24】
(式中、R6およびmは前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
で表される請求項16に記載の組成物。
【請求項21】
アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである請求項16に記載の組成物。
【請求項22】
少なくとも1種のアデノシンA2A受容体アンタゴニストと、COMT阻害剤ならびに/またはDAおよび/もしくはMAO阻害剤とを含有するパーキンソン病および/またはL-ドーパによる運動合併症を治療するための薬剤。
【請求項23】
アデノシンA2A受容体アンタゴニストがキサンチン誘導体またはその薬理学上許容される塩である請求項22に記載の薬剤。
【請求項24】
アデノシンA2A受容体アンタゴニストが式(I):
【化25】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【化26】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【化27】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
で表される請求項22に記載の薬剤。
【請求項25】
アデノシンA2A受容体アンタゴニストが式(I-A):
【化28】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【化29】
(式中、R7、R8およびR9のうち少なくとも1つは低級アルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R10は水素原子または低級アルキルを表す)または
【化30】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
で表される請求項22に記載の薬剤。
【請求項26】
アデノシンA2A受容体アンタゴニストが式(I-B):
【化31】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【化32】
(式中、R6およびmは前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
で表される請求項22に記載の薬剤。
【請求項27】
アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである請求項22に記載の薬剤。
【請求項28】
少なくとも1種のアデノシンA2A受容体アンタゴニストの有効量と、COMT阻害剤ならびに/またはDAおよび/もしくはMAO阻害剤とを含有するパーキンソン病の治療のための組成物。
【請求項29】
アデノシンA2A受容体アンタゴニストがキサンチン誘導体またはその薬理学上許容される塩である請求項28に記載の組成物。
【請求項30】
アデノシンA2A受容体アンタゴニストが式(I):
【化33】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【化34】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【化35】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
で表される請求項28に記載の組成物。
【請求項31】
アデノシンA2A受容体アンタゴニストが式(I-A):
【化36】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【化37】
(式中、R7、R8およびR9のうち少なくとも1つは低級アルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R10は水素原子または低級アルキルを表す)または
【化38】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
で表される請求項28に記載の組成物。
【請求項32】
アデノシンA2A受容体アンタゴニストが式(I-B):
【化39】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【化40】
(式中、R6およびmは前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
で表される請求項28に記載の組成物。
【請求項33】
アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである請求項28に記載の組成物。
【請求項34】
アデノシンA2A受容体アンタゴニストまたはアデノシンA2A受容体アンタゴニストとドーパミンアゴニストとの組み合わせのいずれかを含有する、患者が追加のL-ドーパ療法を必要とすることを遅らせるか、またはなくさせることにより、パーキンソン病治療の有効時間を延長する薬剤。
【請求項35】
運動合併症の発症を遅らせる請求項34に記載の薬剤。
【請求項36】
患者がL-ドーパまたはドーパミン作動性薬剤の前投与を受けていない場合に有効な請求項34に記載の薬剤。
【請求項37】
患者がL-ドーパまたはドーパミン作動性薬剤の後投与を受けない場合に有効な請求項34に記載の薬剤。
【請求項38】
アデノシンA2A受容体アンタゴニストがキサンチン誘導体またはその薬理学上許容される塩である請求項34〜37のいずれかに記載の薬剤。
【請求項39】
アデノシンA2A受容体アンタゴニストが式(I):
【化41】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【化42】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【化43】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
で表される請求項34〜37のいずれかに記載の薬剤。
【請求項40】
アデノシンA2A受容体アンタゴニストが式(I-A):
【化44】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【化45】
(式中、R7、R8およびR9のうち少なくとも1つは低級アルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R10は水素原子または低級アルキルを表す)または
【化46】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
で表される請求項34〜37のいずれかに記載の薬剤。
【請求項41】
アデノシンA2A受容体アンタゴニストが式(I-B):
【化47】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【化48】
(式中、R6およびmは前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
で表される請求項34〜37のいずれかに記載の薬剤。
【請求項42】
アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである請求項34〜37のいずれかに記載の薬剤。
【請求項43】
少なくとも1種のアデノシンA2A受容体アンタゴニストの有効量を含有する運動障害を治療する薬剤。
【請求項44】
運動障害が振戦、運動緩慢、歩行障害、ジストニア、ジスキネジア、遅発性ジスキネジアまたはその他の錐体外路症候群である請求項43に記載の薬剤。
【請求項45】
アデノシンA2A受容体アンタゴニストが、運動障害を引き起こす薬剤の作用を減少させる請求項43または44に記載の方法。
【請求項46】
アデノシンA2A受容体アンタゴニストがキサンチン誘導体またはその薬理学上許容される塩である請求項43または44に記載の方法。
【請求項47】
アデノシンA2A受容体アンタゴニストが式(I):
【化49】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【化50】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【化51】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
で表される請求項43または44に記載の方法。
【請求項48】
アデノシンA2A受容体アンタゴニストが式(I-A):
【化52】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【化53】
(式中、R7、R8およびR9のうち少なくとも1つは低級アルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R10は水素原子または低級アルキルを表す)または
【化54】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
で表される請求項43または44に記載の薬剤。
【請求項49】
アデノシンA2A受容体アンタゴニストが式(I-B):
【化55】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【化56】
(式中、R6およびmは前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
で表される請求項43または44に記載の薬剤。
【請求項50】
アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである請求項43または44に記載の薬剤。
【請求項51】
少なくとも1種のアデノシンA2A受容体アンタゴニストの有効量を含有する過剰運動を治療する薬剤。
【請求項52】
少なくとも1種のアデノシンA2A受容体アンタゴニストの有効量を含有する薬剤誘発性の過剰運動を治療する薬剤。
【請求項53】
アデノシンA2A受容体アンタゴニストがキサンチン誘導体またはその薬理学上許容される塩である請求項51または52に記載の薬剤。
【請求項54】
アデノシンA2A受容体アンタゴニストが式(I):
【化57】
<式中、R1、R2およびR3は独立して水素原子、低級アルキル、低級アルケニルまたは低級アルキニルを表し、R4はシクロアルキル、−(CH2)n−R5(式中、R5は置換もしくは非置換のアリールまたは置換もしくは非置換の複素環基を表し、nは0〜4の整数である)または
【化58】
[式中、Y1およびY2は独立して水素原子、ハロゲンまたは低級アルキルを表し、Zは置換もしくは非置換のアリールまたは
【化59】
(式中、R6は水素原子、ヒドロキシ、低級アルキル、低級アルコキシ、ハロゲン、ニトロまたはアミノを表し、mは1〜3の整数を表す)を表す]を表し、X1およびX2は独立してOまたはSを表す>
で表される請求項51または52に記載の薬剤。
【請求項55】
アデノシンA2A受容体アンタゴニストが式(I-A):
【化60】
[式中、R1aおよびR2aは独立してメチルまたはエチルを表し、R3aは水素原子または低級アルキルを表し、Zaは
【化61】
(式中、R7、R8およびR9のうち少なくとも1つは低級アルキルまたは低級アルコキシを表し、その他のものは水素原子を表し、R10は水素原子または低級アルキルを表す)または
【化62】
(式中、R6およびmはそれぞれ前記と同義である)を表す]
で表される請求項51または52に記載の薬剤。
【請求項56】
アデノシンA2A受容体アンタゴニストが式(I-B):
【化63】
[式中、R1bおよびR2bは独立して水素原子、プロピル、ブチル、低級アルケニルまたは低級アルキニルを表し、R3bは水素原子または低級アルキルを表し、Zbは置換もしくは非置換のナフチルまたは
【化64】
(式中、R6およびmは前記と同義である)を表し、Y1およびY2はそれぞれ前記と同義である]
で表される請求項51または52に記載の薬剤。
【請求項57】
アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである請求項51または52に記載の薬剤。
【請求項58】
アデノシンA2A受容体アンタゴニストおよびL−ドーパを含有するパーキンソン病の治療剤。
【請求項59】
L−ドーパと同時にまたは時間を置いて別々に投与するためのアデノシンA2A受容体アンタゴニストを含有するパーキンソン病の治療剤。
【請求項60】
アデノシンA2A受容体アンタゴニストが(E)−8−(3,4−ジメトキシスチリル)−1,3−ジエチル−7−メチルキサンチンである請求項58または59に記載の治療剤。
【図1】

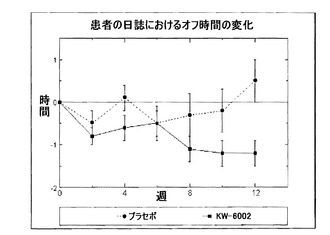
【図2】
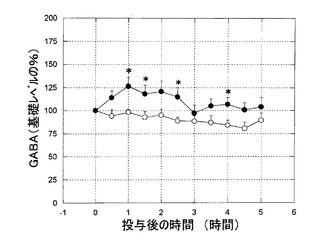
【図3】
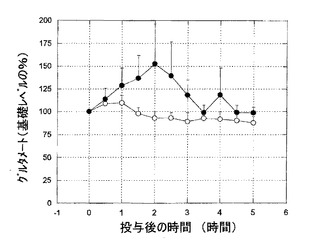
【図4】
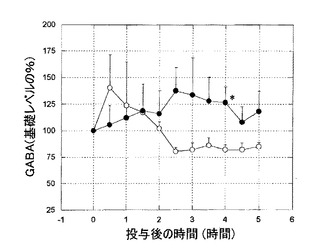
【図5】
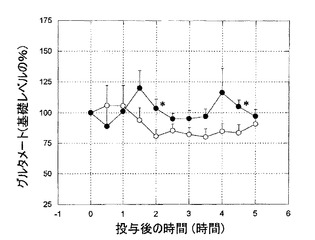
【図6】
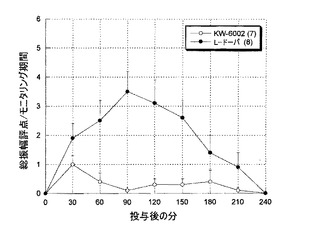
【図7】
【図8】
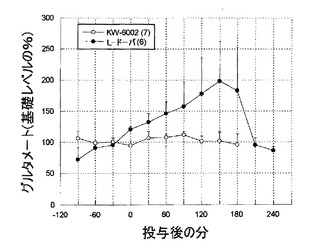
【図9】
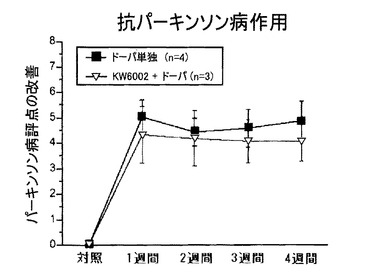
【図10】
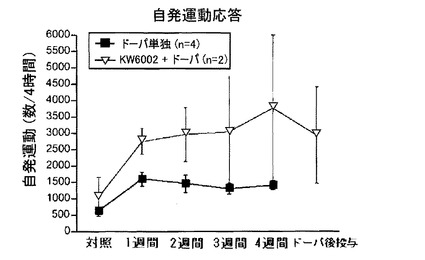
【図11】
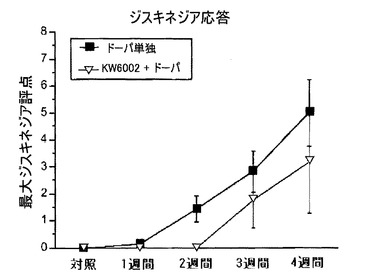
【図12】
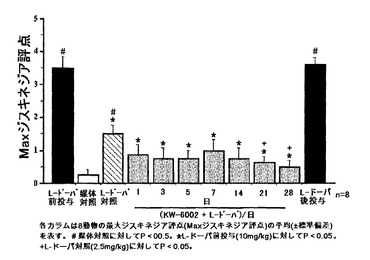
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【公開番号】特開2009−67807(P2009−67807A)
【公開日】平成21年4月2日(2009.4.2)
【国際特許分類】
【出願番号】特願2008−282859(P2008−282859)
【出願日】平成20年11月4日(2008.11.4)
【分割の表示】特願2003−563566(P2003−563566)の分割
【原出願日】平成15年1月28日(2003.1.28)
【出願人】(000001029)協和発酵キリン株式会社 (276)
【Fターム(参考)】
【公開日】平成21年4月2日(2009.4.2)
【国際特許分類】
【出願日】平成20年11月4日(2008.11.4)
【分割の表示】特願2003−563566(P2003−563566)の分割
【原出願日】平成15年1月28日(2003.1.28)
【出願人】(000001029)協和発酵キリン株式会社 (276)
【Fターム(参考)】
[ Back to top ]