
選択された生物のための人工プロモーターライブラリー及び該ライブラリー由来のプロモーター
【課題】新規な人工プロモーターライブラリーの提供。
【解決手段】選択された生物又は生物の群のための人工のプロモーターライブラリーは、そのセンス鎖が前記生物又は生物の群からの有効なプロモーターの少なくとも2の共通配列又はその各々の少なくとも半分を含む一部分、及び少くとも7のヌクレオチドがヌクレオベースA,T,C及びGの中から無作偽に選択された不定の長さの周囲の又は介在するヌクレオチド配列(スペーサー)を含む二本鎖 DNAフラグメントの混合物として作製される。
【解決手段】選択された生物又は生物の群のための人工のプロモーターライブラリーは、そのセンス鎖が前記生物又は生物の群からの有効なプロモーターの少なくとも2の共通配列又はその各々の少なくとも半分を含む一部分、及び少くとも7のヌクレオチドがヌクレオベースA,T,C及びGの中から無作偽に選択された不定の長さの周囲の又は介在するヌクレオチド配列(スペーサー)を含む二本鎖 DNAフラグメントの混合物として作製される。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、人工プロモーターライブラリー及び所定の生物又は生物の群のための人工プロモーターライブラリーを構築する方法に関する。本発明は、これらのライブラリー由来の個々の新規のプロモーターにも関する。更に、本発明は、生物のための人工プロモーターライブラリーからのプロモーターの使用により所定の生物中の遺伝子の発現を最適化する方法に関する。原則として、本発明による人工プロモーターライブラリーは、いずれかの生きている生物において用いるために作製することができるが、現在、それらは、微生物の遺伝子発現を調節するために最も価値あるものである。本発明において、用語“微生物”とは、細菌のような原核生物、並びにイーストのような他の真核微生物、他の真菌及び高等生物の細胞系を含むよう広く解釈されるであろう。
【背景技術】
【0002】
生きている生物の代謝工学技術は、ここ10年超の間、遺伝子工学が実行可能になってきたという事実にかかわらず、工業上の適用に関してはまだ初期である。かなりの程度、これは、株の能力を改良することに関する多くの試みが失敗に終わった結果によるものである。代謝量を増加させる試みの否定的な結果については2つの理由がある:
【0003】
1つは、遺伝子工学者が、細胞代謝の制御及び調節の繊細さを見のがす傾向があることである。速度制限があると予想される酵素の発現は、例えばその遺伝子を高複製数プラスミドにおくことにより、10〜100 倍、増加する。又は、経路における分枝の流れは遺伝子を削除することにより排除される。極めて多くの場合、これは、例えば、細胞代謝の他の部分(例えば、生物の成長に本質的である過程)に本質的である代謝産物を低下させることにより、代謝に二次的効果を与えるであろうし、その最終的な結果は、要求される産物に関するその細胞の全体の能力を減少させる結果となり得る。その代わりとして、正常な発現レベルに関する関連遺伝子の発現を調節すること、並びに例えば、その流れの量を最大又は最小にするレベルとして、最適な発現レベルを決定することが必要である。
【0004】
否定的な結果についての第2の理由は、それ自体の速度制限コンセプトにあり:代謝調節理論(Kacser及びBurns, 1973)及び経路における個々のステップによる調節の実験による測定(Schaffら、1989; Jeasenら、1993) は、特定の流れに関する速度制限であると予想される反応ステップがその流れにわたって全く調節していないかほとんど調節していないことが判明したことを示した。かわりに、細胞代謝の調節及び制御は、経路中のいくつかの酵素に分配されることが判明し、より高い流量を得るためにいくつかの酵素の発現を増強することが必要であり得る。
【0005】
代謝調節理論によれば、経路における全ての酵素により発揮される全体の流量制御は、常に1に合計されるはずである。それゆえ、1つの酵素濃度が最適化された後、流量制御は、他の酵素に移り、そして次に更に流量を増加させるために更なる回数の酵素最適化を行うことが有用であり得る。
要約すると、流量最適化は、1)何部もの過剰発現でなく酵素濃度の細かい調節及びしばしば2)速度制限ステップを見つけ出すのでなく経路中のいくつかの酵素のレベルの最適化を必要とする。
【0006】
現在、1000倍超、遺伝子発現を増加させ、及び/又は(例えば温度誘導システムを用いて)発酵過程の間の特定の時点において遺伝子発現に向けることに利用できる多くのシステムがある。発酵槽において定常状態の遺伝子発現、即ち正常発現レベルの 150%ないし70%に向かわせることに関して、より困難性が増加する。原則として、関心の遺伝子の前に lac型プロモーターを用い、そして次に、lacシステムの所定量のインデューサー、例えばIPTG(イソプロピル−β−D−チオガラクトシド)を加え、又は正確な温度で温度感受性システムを用いることができる。これらの可能性は、しばしば、大規模な工業的適用について実用的でない。かわりの方法は、正確に正しい強さを有するプロモーターを用いることである。しかしながら、このようなプロモーターはあまり利用できず、そして更に、第1の場所において遺伝子の発現を最適にするために所定範囲のプロモーター活性を必要とする。
【0007】
過去20年間、微生物の共通の配列を規定し、最適化するために多くの研究がなされてきた。多くの原核生物において、転写のための開始部位に対しておおよその位置−10及び−35において2つ程度の保存された DNA配列、各々TATAAT及びTTGACAが、それら2つの間の約17塩基対と共にしばしば見出されている。この分野における定説は、これらの要素を含むことにより、生じたプロモーターが強力になる傾向があるであろうことである。実際、比較的まれな出来事であるプロモーター上昇変異は、通常、上述の共通配列により適合するが、下降変異は、共通配列にあまり適合せず、これらの間のより最適でない経路となる。更に、ランダム DNA配列を2つの共通配列の1つの場所にクローン化する場合、生じたプロモーターの強さは、通常、共通配列に対する相同性の程度と相関する。
【0008】
原則として、プロモーターの強さの調節は、共通配列中の塩基対変化により、又はこれらの間のスペーサーの長さの変化により達成することができよう。しかしながら、プロモーターの長さのこのような変化の影響は大きくなる傾向があるであろうし(本発明の実施例1を参照のこと)、それゆえ共通配列の塩基対変化による強さの調節の小さなステップを行うことは容易でない。
【0009】
2つの共通配列を分離するスペーサーの長さはプロモーターの強さのために重要な役割を果たすことが知られているが、その共通配列間のスペーサーの配列は、通常、プロモーターの強さについてほとんど重要でないと考えられており、そして変異誘発によるスペーサー内の更なる共通配列を同定するための試みは、実際成功していない。これまで、共通配列を維持し、かつスペーサーの長さを比較的一定にしながら、スペーサーを無作偽化する試みを行った者はいない。
【0010】
共通配列のうちの少なくとも1つを無作偽化する実験を含む、微生物の共通配列を規定し、最適化するための多くの実験が行われている。これらの実験のいくつかにおいて、共通配列の周囲のヌクレオチドの一部も、17bpと異なるスペーサーの長さのプロモーターの形成を許容するため、及び/又は共通配列の周囲の可能性ある新規の共通モチーフを見い出すために、無作偽化された。これは、有効なプロモーターを形成するであろう機会が極めて小さく、そして共通配列に対する相同性が十分に高くて弱いプロモーターを生ずるまれな場合を見出すために、選択を行わなければならない。
【発明の開示】
【0011】
発明の目的
我々が目指すプロモーターライブラリーは、特定の種の工学技術のための関心の活性になり得る全体の範囲のプロモーター活性、好ましくは検出できる最も弱いプロモーターから可能な最も強力なプロモーターをカバーするはずである。更に、我々は、小さなステップにおいてこの広範な活性範囲をカバーすること、即ち、上述のような流量最適化の目的に適したものであるために、ステップ当り50〜100 %だけ活性を増加させることを目指す。
【0012】
本発明において、我々は、共通配列の間のスペーサーの配列が以前に認められているよりもかなり重要であることを示す。スペーサー配列は、1)スペーサー配列の主要な部分が種々であると同時にランダムであり、そして2)同時に、共通配列の少くとも半分が一定に維持されている場合、プロモーターの強さに強い影響力を有し得る。我々は、これら2つの条件が満たされるなら、我々の方法は、極めて強力なプロモーターを含む、広範囲の活性をカバーするプロモーターを作り出すのに用いることができる。プロモーター活性の範囲は代謝の工学技術の目的のために極めて適したプロモーターを作る活性変化の小さなステップにおいてカバーされる。更に、我々は、その方法が、広範囲の生物のため、かつ異常に高い頻度においてプロモーターを作り出すのに用いることができる。
【0013】
発明の概要
本発明は、所定の生物又は生物の群のための人工的なプロモーターライブラリーであって、それらのセンス鎖が前記生物もしくは生物の群からの有効なプロモーターの少なくとも2の共通配列を含む二本鎖 DNAフラグメントの混合物、又は各々の少なくとも半分を含むその一部と、少なくとも7のヌクレオチドがヌクレオベースA,T,C及びGから無作偽に選択された種々の長さの周囲の又は介在するヌクレオチド配列(スペーサー)と、を含む人工プロモーターライブラリーを供する。但し、以前から周知であるプロモーター配列及び天然のソースから単離されたプロモーター配列は含まれない。
【0014】
プロモーターの長さの最も広範囲のバリエーションは、ヌクレオベースA,T,C及びGの中から、スペーサー配列内の少なくとも10、好ましくは少なくとも12、そしてより好ましくは少なくとも14のヌクレオチドがランダムに選択される時に得られる。
二本鎖 DNAフラグメントのセンス鎖は、ライブラリーのプロモーターに特定の調節特性を与える調節 DNA配列も含み得る。このような特定の調節特性は、好ましくは、増殖条件の変化、例えばpH、浸透度、温度又は増殖相の変化による活性化である。
【0015】
クローニング目的のために、二本鎖 DNAフラグメントは、通常、それらの端の一方又は両方に加えられた制限エンドヌクレアーゼのための1又は複数の認識部位を含む配列;制限エンドヌクレアーゼのための多重の認識部位(多重クローニング部位MCS)を特定する最も便利な配列を有する。
選択される生物又は生物のグループは、原核生物から及び真核生物から、特に、原核及び真核微生物、例えばイースト、他の真菌及び高等生物の細胞系から選択することができる。
【0016】
例えば乳製品工業における、原核生物の興味あるグループは、ラクトコッカス(Lactococcus )、ストレプトコッカス(Streptococcus )、エンテロコッカス(Enterococcus)、ラクトバチルス(Lactobacillus )及びロイコノストック(Leuconostoc )属の乳酸バクテリア、特にラクトコッカス・ラクチス(Lactococcus lactis)及びストレプトコッカス・サーモフィルス(Streptococcus thermophilus)の株からなる。他の興味ある原核生物は、エスケリキア(Escherichia )、バチルス(Bacillus)及びシュードモナス(Pseudomonas )属、特に大腸菌、バチルス・サブチリス(Bacillus subtilis )及びシュードモナス・プチダ(Pseudomonas putida)種に属するバクテリアである。
【0017】
原核生物のための人工プロモーターライブラリーにおいて、前記共通配列は、例えば、−10シグナル(−12〜−7):TATAAT及び各々少なくとも3の保存されたヌクレオチドを含む−10シグナル又はその一部の上流の少なくとも1のアクティベータータンパク質結合部位を含み得る。
最も多くの場合、原核生物のための人工プロモーターライブラリー内に保持された共通配列は、−35シグナル(−35〜−30):TTGACA及び−10シグナル(−12〜−7):TATAAT又は各々の少なくとも3の保存されたヌクレオチドを含む両方の一部を含むであろう。
【0018】
より有効なプロモーターは、通常、前記共通配列が−35シグナルの4〜6の保存されたヌクレオチド及び−10シグナルの4〜6の保存されたヌクレオチド、好ましくは各々5又は6のヌクレオチド、より好ましくは各々全部で6のヌクレオチドを含む場合に得られる。最も有効なプロモーターは、介在する保存されたモチーフ、例えば保存されたモチーフ−44〜−41:AGTT、−40〜−36:TATTC 、−15〜−14:TG、及び+1〜+8:GTACTGTTから選択されたモチーフを更に含む場合に得られる。
【0019】
このようなプロモーターにおいて、−35シグナルと−10シグナルとの間のスペーサーの長さは14〜23bp、好ましくは16〜18bp、より好ましくは17bpである。これは、そのシグナル内のヌクレオチドのうちのいくつかが変異された場合でさえも、そのヘキサマーシグナル間のスペーサーの長さを意味することが理解されるはずである。
真核生物において、前記共通配列は、TATAボックス及び少なくとも1の上流活性化配列(UAS) を含むべきである。
【0020】
興味ある真核微生物は、通常のパン酵母である。イースト種サッカロマイセス・セレビシアエ(Saccharomyces cerevisiae)である。サッカロマイセスに用いられるプロモーターにおいて、共通配列は、サッカロマイセス・セレビシアエにおいて機能する転写開始シグナル(T1ボックス)を更に含む。
【0021】
サッカロマイセス・セレビシアエのための本発明による人工プロモーターライブラリーの特定の実施形態において、前記共通配列は、TATAボックス:TATAAA、 UASGCN4p :TGACTCA 、及びアルギニンレプレッサーのための結合部位argRとしても機能するT1ボックス:CTCTTAAGTGCAAGTGACTGCGA を含む。
【0022】
上述の人工プロモーターライブラリーの個々のプロモーターも本発明に含まれる。以下の実施例に従って作製された特定のプロモーターは、以下の配列番号:5〜58に記載されるものである。本発明は、本発明の人工プロモーターライブラリーにより規定されるプロモーター由来の人工プロモーターを更に含む。
【0023】
本発明は、所定の生物又は生物のグループのための人工プロモーターライブラリーを作製する方法であって、前記生物又は生物の群から有効なプロモーターの少なくとも2の共通配列を選択し;該共通配列又はその各々の少なくとも半分を含む一部、並びに少なくとも7のヌクレオチドがヌクレオベースA,T,C及びGの中から無作偽に選択された不定の長さの周囲の又は介在するヌクレオチド配列(スペーサー)を含む一本鎖 DNA配列の混合物を合成し;そして該一本鎖 DNA配列を第2鎖の合成により二本鎖 DNAフラグメントに変換することを含む方法も供する。
【0024】
上述の通り、最も広範囲のバリエーションは、スペーサー配列内の少なくとも10、好ましくは少なくとも12、より好ましくは少なくとも14のヌクレオチドがヌクレオベースA,T,C及びGの中から無作偽に選択される場合に得られる。
調節可能な人工プロモーターライブラリーを得るために、合成される一本鎖 DNA配列は、特定の調節特性をライブラリーのプロモーターに与える調節 DNA配列を含み得る。このような特定の調節特性は、好ましくは、増殖条件の変化、例えばpH、浸透性、温度又は増殖相の変化による活性化である。
【0025】
また、クローニングのために適した人工プロモーターライブラリーを得るために、制限エンドヌクレアーゼのための1又は複数の認識部位を特定する配列を、合成において一本鎖 DNA配列の一端又は両端に加えてもよく、又はこのような制限部位を含むリンカーを二本鎖 DNAフラグメントの一端又は両端に連結してもよい。最も便利なのは、このような配列が、制限エンドヌクレアーゼのための多重認識部位(多重クローニング部位MCS)を特定することである。
【0026】
本発明による方法により人工プロモーターライブラリーを調製することができる選択される生物、及び特定の生物のプロモーターライブラリーのために選択される種々の縮重した配列は、人工プロモーターライブラリー自体について上述されるのと同じである。
【0027】
上述の人工プロモーターライブラリーの可能な使用に関して、本発明は、微生物中の遺伝子の発現を最適化する方法であって、a)請求項1〜26のいずれか一に記載の人工プロモーターライブラリー又は請求項30〜55のいずれかに記載の方法により作製された人工プロモーターライブラリーから、活性変化の比較的小さなステップにおけるプロモーター活性の範囲を包含する一セットのプロモーターを選択し、
b)該一セットのプロモーターを、前記遺伝子を各々のクローン内で該セットからの少なくとも1のプロモーターの制御下におくように、前記生物内にクローニングし、
c)該選択されたクローンを増殖させ、そして産物形成の最適な流れを示すものを見い出すためにそれらをスクリーニングすることを含む方法を更に供する。
【0028】
この方法は、好ましくは、原核及び真核微生物、例えばバクテリア、イースト、他の真菌及び哺乳動物細胞系からなる群から選択される生物と共に用いられる。
【0029】
図面の簡単な記載
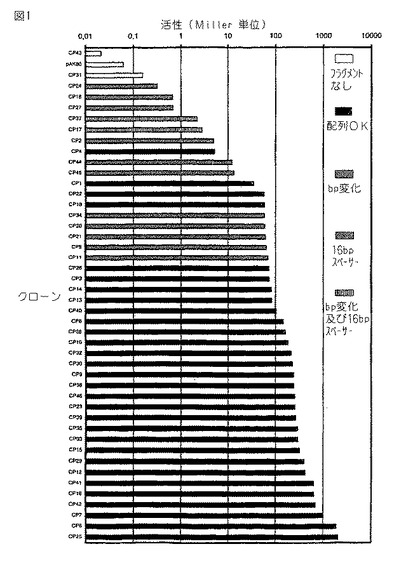
図1。実施例1からのL.ラクチス(L. lactis )のための人工プロモーターのライブラリー。プロモーター活性(Miller単位)を、2μg/mlエリトロマイシンを補給したGM17培地中で増殖した株MG1363におけるプロモータークローニングベクター pAK80上での異なる合成プロモータークローンから転写されたb−ガラクトシダーゼをコードするリポーター遺伝子(lacLM )の発現からアッセイした。データ点のパターンは、−35もしくは−10共通配列のいずれかにおいて又はこれら2つの共通配列の間のスペーサーの長さにおいてプロモータークローンが異なることを示す。
【0030】
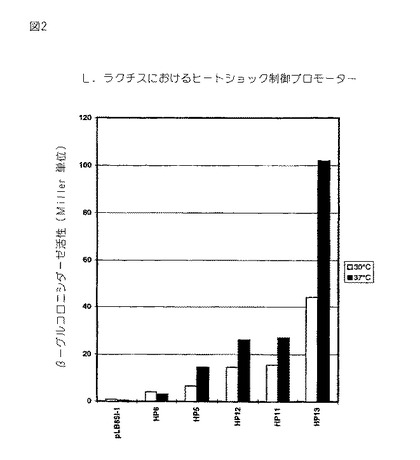
図2。実施例2からのL.ラクチスのための人工のヒートショック調節プロモーターのライブラリー。プロモーター活性(Miller単位)を、株 LB436内の染色体上の、異なる合成プロモータークローンから転写されたβ−グルクロニダーゼをコードするリポーター遺伝子(gusA)の発現からアッセイした。その細胞は、2μg/mlエリトロマイシンを補給したGM17培地中で、2つの異なる温度30及び37℃で増殖させた。β−グルクロニダーゼについてのアッセイを、X-glucを基質として用いたことを除いてβ−ガラクトシダーゼアッセイ(実施例1を参照のこと)と平行して行った。
【0031】
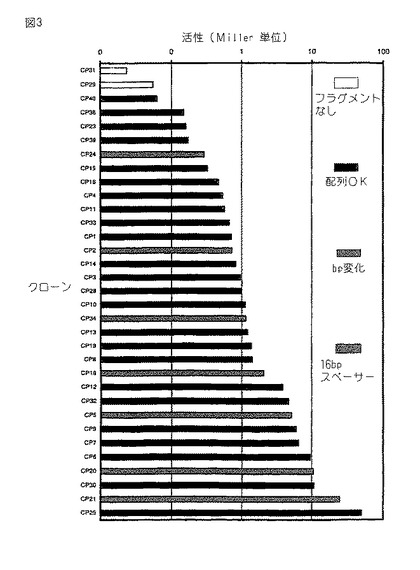
図3。大腸菌内の活性についてアッセイした実施例1からの人工プロモーターのライブラリー。プロモーター活性(Miller単位)を、 200μg/mlエリトロマイシンを補給したLB培地中で増殖させた株BOE270中での、プロモータークローニングベクター pAK80上で、異なる合成プロモータークローンから転写されたβ−ガラクトシダーゼをコードするリポーター遺伝子(lacLM )の発現からアッセイした。データ点のパターンは、−35もしくは−10共通配列のいずれかにおいて、又はこれら2つの共通配列の間のスペーサーの長さにおいてプロモータークローンが異なることを示す。
【0032】
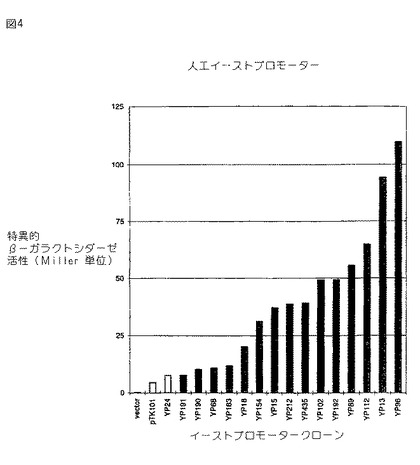
図4。実施例7からのS.セレビシアエ(S. cerevisiae )のための人工プロモーターのライブラリー。プロモーター活性(Miller単位)を、2%グルコースを補給したSD最小培地中で増殖させたS.セレビシアエ株SG24(URA3-52) 中のプロモータークローニングベクターpYLZ-2上で、異なる合成プロモータークローンから転写されたβ−ガラクトシダーゼをコードするリポーター遺伝子(lacZ)の発現からアッセイした。YP24及び YP435は、 UASGCN4p 結合部位内に、各々1bp欠失及び点変異を有する。pTK101は、 UAS配列が削除され、TATAボックスが存在するプロモーターを含む。
【0033】
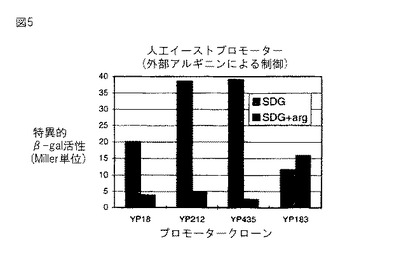
図5。実施例7からの外来アルギニンによる人工イーストプロモーターの制御。プロモーター活性(Miller単位)を、1g/lアルギニンあり(SDG) 又はなし(SDG+arg)で2%グルコースを補給したSD最小培地中で増殖させたS.セレビシアエ株SG24(URA3-52) におけるプロモータークローニングベクター上で、異なる合成プロモータークローンから転写されたβ−ガラクトシダーゼをコードするリポーター遺伝子(lacZ)の発現からアッセイした。 YP183は、argRレプレッサーのための結合部位内に13bpの欠失を有する。
【0034】
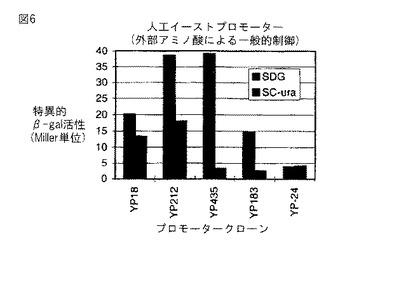
図6。実施例7からの外来アミノ酸による人工イーストプロモーターの制御。プロモーター活性(Miller単位)を、2%グルコースを補給したSD最小培地(SDG) 又はウラシルを含まない(アミノ酸を含む)複合培地(SC-ura)内で増殖させたS.セレビシアエ株SG24(URA3-52) におけるプロモータークローニングベクターpYLZ-2上で、異なる合成プロモータークローンから転写されたβ−ガラクトシダーゼをコードするリポーター遺伝子(lacZ)の発現からアッセイした。YR24は UASGCN4p 結合部位内に1bp欠失を有する。
【発明を実施するための最良の形態】
【0035】
我々の方法において、縮重したオリゴヌクレオチドは、プロモーターライブラリーが作製されるべき生物又は生物の群のために合成される。オリゴヌクレオチドの配列は、特定の生物内でプロモーター機能を有効にする特徴に基づき、文献から利用できる知識を組み合わせることにより記述される。オリゴヌクレオチド内に固定されるのに必要とされる情報の量は、異なる生物の間で幾分か種々である。例えば大腸菌において、−35及び−10共通配列内のより少ない完全性の適合により、及び17bpからずれたこれらの配列(各々TTGACA及びTATAAT)の間の間隔により形成することができるが、L.ラクチス内の強力なプロモーターの要求はより厳格であるようである。
【0036】
第2に、一本鎖オリゴヌクレオチドは、二本鎖 DNAフラグメントに変換され、プロモータープロビングベクターにクローニングされる。そのプロモーター含有クローンは、例えばインジケータープレート上の種々の程度の色のコロニーを供する能力により同定される。これは、原則として、我々に極めて強力なプロモーターのみを供するが、我々は、共通配列の間のスペーサー配列をランダムに変化させることにより、その生じたプロモーターの強度が調節されることを発見した。実際、この方法を用いて、我々は、活性変化の小さなステップにおいて、関心のものになるような全体の範囲のプロモーターに及ぶプロモーターライブラリーを得た。
【0037】
次に、遺伝子発現の最適化は次のように達成されよう:1)プロモーターライブラリーから、例えば野生型プロモーターの強さの25%、50%、 100%、 200%及び 400%を有するプロモーターを選択する。2)次に、これらのプロモーターを、関心の遺伝子の上流の野生型プロモーターの位置にクローン化する。3)次にこれら5つの構成物各々で得られた最適化されるべき変数(例えば経路を通る流量)の大きさを測定し、そして最適な構成物が生産生物として直接用いられる。更に発現を微妙に調節すること又はプロモーター活性の範囲を拡げることが必要であり得る。上述の誘導システムに優るこのシステムの直接の利点は、最適なプロモーター活性を決定した後、株は、原則として、発酵過程に直ちに用いることができることである。
【0038】
1つの好ましい実施形態において、本発明のランダムスペーサー法は、グラム陽性バクテリア、ラクトコッカス・ラクチス(Lactococcus lactis)のための一連の構成的プロモーターを作るのに用いられる。他の好ましい実施形態において、我々は、本発明のランダムスペーサー法により作られたプロモーターがグラム陰性バクテリア、大腸菌及びシュードモナス(Pseudomonas )の少くとも2種において及びグラム陽性バクテリアバチルス・サブチルス(Bacillus subtilis)において機能的であることを示す。我々は、個々のプロモーターの強度が用いる生物に依存すること、即ち特定の生物において特定のプロモーターが強力であり、他の生物においてそれは弱いが、テストした全ての生物において、プロモーターは活性変化の小さなステップにおいて広範囲の活性を包含することを示す。
【0039】
しばしば、例えば発現される遺伝子産物が細胞の増殖を阻害するので、遺伝子発現を特定の程度まで、及び発酵の特定の段階で、活性化することが要求される。プロモーターがpH、温度又は増殖相の変化により活性化されるであろうように、特定の調節メカニズムで異なる強度のプロモーターを得るために上述の技術を組み合わせることが有用である。
【0040】
これにより、他の好ましい実施形態において、本発明のランダムスペーサー法は、一連の特異的に調節されるプロモーターを作り出すのに用いられる。実施例2に詳述するように、先のアプローチは、グラム陽性バクテリア、ラクトコッカス・ラクチスのためのヒートショック調節プロモーターのライブラリーを作り出すために特定の調節 DNAと組み合わせて用いられる。
【0041】
原核生物に加えて、真核微生物(イースト及び他の真菌並びに哺乳動物細胞系)は所定範囲の有機化合物及び種々のタンパク質の生産のための重要な微生物である。それゆえ、更にこれらの生物において遺伝子発現を調節するために上述のアプローチを開発することに関心がある。これにより、実施例7に示すような更に他の好ましい実施形態において、本発明のランダムスペーサー法は、パン酵母、サッカロマイセス・セレビシアエ(Saccharomyces cerevisiae)のための一連のプロモーターを作り出すために用いられる。ここで、それらプロモーターは、 GCN4p及びARGR調節を備える。
【0042】
真核細胞における転写開始の調節は、原核生物と比べて幾分かより複雑である。転写開始部位は、通常、共通配列TATAAA又はその一部を含むが、原核生物のものと似ていないいわゆるTATAボックスの後にあり、そのTATAボックスから転写開始部位への距離はほとんど規定されていない。サッカロマイセス・セレビシアエにおいて、この距離は、典型的には、40〜120 ヌクレオチドである(Oliver及びWarmington, 1989) 。多くの原核生物プロモーター内で見出されるいわゆる−35共通ヘキサマーはサッカロマイセス・セレビシアエ内に存在しない。
【0043】
かわりに、いわゆる上流活性化配列(UAS) が転写開始部位の上流に見出される。これら UASは、後に転写開始のアクティベーターとして機能し得る特定の DNA結合タンパク質により認識される。例えば、アミノ酸生合成に関連する遺伝子の上流に見い出される UAS配列 UASGCN4p は、これらの遺伝子の転写を活性化するGCN4タンパク質のための結合部位を特定する DNA配列からなる(Hinnebusch, 1992)。プロモーター要素間の距離がプロモーターの強度のために重大であるようである(実施例1を参照のこと)原核生物と対照的に、真核生物プロモーター内のTATAボックスと UAS配列との間の距離は極めて可変性であり、約1000bpまでであり得る。特定の遺伝子は UASの1超の複製さえ含むが、活性化のためには1つで十分であるようである。
【0044】
イーストから知られている UAS配列の1つは、GCN4タンパク質のための結合部位である。GCN4タンパク質のための結合部位を含むプロモーターは、増殖培地中のアミノ酸供給の状態により調節されるはずであり;アミノ酸の欠如下において、GCN4タンパク質が形成され、特定の UAS配列に結合してアミノ酸の生合成に関連するプロモーターにおいて転写を刺激する。GCN4タンパク質結合部位(UASGCN4p ) のための共通配列は、短い逆反復 TGACTCAである。
【0045】
GCN4タンパク質により活性化されるサッカロマイセス・セレビシアエにおけるプロモーターはARG8プロモーターである。このプロモーターにおいて、唯一の UASGCN4p 配列の複製があり、そしてそれはTATAボックスから59bpに位置する(我々はこの距離をスペーサー1と呼ぶ)。転写開始はTATAボックスの下流約40〜60ヌクレオチドでおこる。ARG8プロモーターは、アルギニンレプレッサー、argRのための結合部位として機能する DNA配列も含み(Crabeelら、1995) 、それは外来アルギニンによりプロモーターを4倍抑制する。
【0046】
これにより、この場合、プロモーターは 136bp内に位置し、そしてそれは、先の例に概説されるように本発明のランダムスペーサー法によりプロモーターライブラリーを開発するために、そのシステムを魅力あるものにする2つの調節特性を含む。しかしながら、その方法はこのモデルシステムに限られず;原則として、約1000ヌクレオチドより小さいスペーサーにより分離されたTATAボックス、 UAS配列、レプレッサー結合部位等のいずれかの組合せがこの方法のための出発点として適するはずである。
【実施例】
【0047】
実施例1. L.ラクチスプロモーターライブラリーのための縮重したオリゴヌクレオチドのデザイン
文献によれば(de Vos及びSimons, 1994の報告を参照のこと)、L.ラクチスにおける強力なプロモーターは共通して次のヌクレオチド配列(数字は番号+1で与えられる転写開始部位に対する位置を示す):−12〜−7:TATAAT;−15〜−14:TG;−35〜−30:TTGACAを有する傾向がある。−10と−35との間のスペーシングは17ヌクレオチドであるようである。しかしながら、L.ラクチスについて出版されているプロモーター配列の親密な比較は、上述の1つを除くいくつかの位置において、ヌクレオチドが多少よく保存されていることを示す。
【0048】
これらの位置のいくつかは:−1:A;−3:A又はT(=W);−6:A;−13:A又はG(=R);−40〜−36:TATTC である。更に、 Nilsson及びJohansen (1994, BBA)は、L.ラクチスからの比較的強力なプロモーター(転移 RNA及びリボソーム RNAオペロンのためのプロモーター)の間でよく保存されているようである2つのモチーフ:+1〜+8:GTACTGTT、及び−44〜−41:AGTTを指摘した。これらのモチーフは、そのプロモーターから強力かつ増殖比依存の発現を与え得る。
【0049】
これらの付加的なモチーフが含まれる場合、L.ラクチスにおける有効なプロモーターのための以下の53ヌクレオベースの縮重した配列に達する。これら53ヌクレオベースのうち、34塩基が保存され、2つが半保存性であり(R及びW)、そして17が4つのヌクレオチドの間で極めてランダムに変化している。
【0050】
【化1】

【0051】
更に、この縮重した配列は、制限エンドヌクレアーゼのための多重認識部位(多重クローニング部位MCS)を特定する配列に隣接し、合成されるべき実際のオリゴヌクレオチド混合物は、配列番号:1
【0052】
【化2】
に示される縮重した配列を有する。
【0053】
本明細書によるオリゴヌクレオチドの混合物は、 Hobolth DNA合成により合成した。
このオリゴヌクレオチド混合物は、最初は一本鎖であり、クローニングの目的のために二本鎖 DNAフラグメントに変換されなければならない。これは、プロモーターオリゴヌクレオチドの3′末端に相補的な配列を有する10bpオリゴヌクレオチドを試験管内で合成することにより行った。次にこのオリゴヌクレオチドをdATP, dCTP, dGTP及びdTTPの存在下での DNAポリメラーゼIのクレノウフラグメントによる第2鎖合成のためのプライマーとして用いた。この方法において第2 DNA鎖は正確に第1 DNAに相補的になった。
【0054】
その結果物は3′及び5′端の両方に制限エンドヌクレアーゼのための多重認識部位を含む 100bp二本鎖 DNAフラグメントの混合物である。次にこれらの DNAフラグメントを、ベクター DNAフラグメントpAK80 (Israelsenら、1995)の端に適合する適切な“粘着”末端を作るために制限エンドヌクレアーゼで切断した。 pAK80は、大腸菌及びL.ラクチスの両方において繁殖のための複製源を有することを意味するシャトルベクターである。この方法において、クローニングステップは、便利には、大腸菌内で行うことができ、次にL.ラクチス内で生理的実験を行うことができる。更に、 pAK80は、多重クローニング部位の下流にプロモーターのないβ−ガラクトシダーゼリポーター遺伝子シテム(lacLM )を有する。これにより、プロモーターが多重クローニング部位内に挿入されなければ、 pAK80は lacLM遺伝子を発現しない。
【0055】
二本鎖 DNAフラグメントの混合物をクローニングベクター pAK80内にクローニングするために2つのクローニングストラテジーを用いた。
1)混合物をBamHI及び PstIで消化し、ベクター pAK80を PstI及び BglII(BglIIはBamHIに適合する)で消化した。
【0056】
2)混合物を SspI及びHincIIで消化し、ベクター pAK80を SmaIで消化した(全部の3つの酵素は平滑端 DNAフラグメントを作り出す)。
両方の場合、ベクター DNAを、クローニングベクターの再連結を防ぐために子ウシインテスチンホスファターゼ(CIP) で更に処理した。次に、そのフラグメント混合物及びベクター DNAを、T4 DNAリガーゼ及び標準連結条件を用いて16℃で一晩、連結した。
【0057】
連結混合物を、エリトロマイシン耐性についての選択と共に、大腸菌 K-12 Δlac に形質転換した。大腸菌K-12株の細胞 MT102を、Ca2+イオンでの標準的処理(Maniatisら、1982)を用いてコンピテントにした。次に連結混合物を、標準的な形質転換手順(Maniatisら、1982)を用いてこれらの細胞に形質転換し、そして生じたクローンを、X-gal(5−ブロモ−4−クロロ−3−インドリル−β−D−ガラクトシド)を含むプレート上で青いコロニーを作るであろうβ−ガラクトシダーゼ活性についてスクリーニングした。その形質転換混合物を、 200μg/mlエリトロマイシン、1%グリセロール、及び 100μg/ml X-galを含むLBプレート上にプレートした。 150のエリトロマイシン耐性形質転換体を得た。全て最初は白かったが、長期のインキュベーション(4℃で2週間)の後、これらのコロニーのうち46は明るい青色になった。これにより、ストラテジー1)を用いて、我々は17の青色コロニー(CP30〜CP46)を見出し、そしてストラテジー2)を用いて、我々は29の青色コロニー(CP1〜CP29)を見出した。
【0058】
これらのコロニー(CP1〜CP46)の各々からプラスミド DNAを単離し、制限酵素マッピングにより分析した。ほぼ全てのプラスミド(CP31及びCP43を除く)が、このベクター上の、さもなければプロモーターのない lacLM遺伝子の転写を指図するであろう方向で、 pAK80の MCS内に挿入されたプロモーターフラグメントを含んでいた。
【0059】
次に、これら46のプラスミド DNA調製物を、エリトロマイシン耐性についての選択と共に、L.ラクチス亜種ラクチスMG1363に形質転換した。L.ラクチス亜種ラクチス株の細胞MG1363(Gasson, 1983)を、Holo及びNes(1989) により記載されるように、2%グリシンを含む SGM17培地中で一晩、その細胞を増殖させることによりコンピテントにした。次に、上述の46のクローンの各々からのプラスミド DNAを、エレクトロポレーション法(Holo及びNes, 1989)を用いてこれらの細胞内に形質転換した。再生後、細胞を2μg/mlエリトロマイシンを含むSRプレート上にプレートした。次の X-galプレート上での青色についてのスクリーニングは、46のクローンの間でβ−ガラクトシダーゼ活性の大きな差を示し;いくつかのクローンは24時間のインキュベーション後に暗い青色のコロニーになり、他のものは1週間超のインキュベーション後に明るい青色のコロニーになった。
【0060】
次に、MG1363における46のコロニーの液体培養物のβ−ガラクトシダーゼ活性を、Miller (1972) により記載されるように決定し、 Israelsenら (1995) により改良した。各々 pAK80の46のプラスミド誘導体のうち1つを有する株MG1363の培養物を、30℃で一晩、1%グルコースを補給したM17培地中で増殖させた。次にこれらの培養物25〜100 μlを、最も弱いプロモータークローンの場合に2mlの培養物を(遠心による20倍の濃縮の後に)用いたことを除いて、後のβ−ガラクトシダーゼアッセイに用いた。これらの結果を図1に示す。明らかに、クローン化したプロモーターフラグメントの有効性において極めて大きな差があると共に、これらのクローンは、 0.3ユニットのβ−ガラクトシダーゼ活性からこの生物について知られているおそらく最も強力なプロモーターである2000ユニットまでの範囲のプロモーター活性をカバーする。
【0061】
更に、広い範囲が活性の小さな変化によりカバーされ、それゆえこれらのプロモーターフラグメントにより、我々は、L.ラクチスにおいて遺伝子の広範囲の発現を得ることができるばかりでなく、流量最適化の目的のために小さなステップにおいてL.ラクチスにおいて遺伝子の発現を調節することもできる。
上述の46のクローンの DNA配列決定は、挿入されたプロモーターフラグメントのほとんどが、オリゴヌクレオチドデザインについてもとから特定された DNA配列を有していたが(上述を参照のこと)、残りのフラグメントの配列はその配列から少しずれていた。
【0062】
β−ガラクトシダーゼアッセイにおいて低い活性を与えた(70ユニット又はそれ未満のβ−ガラクトシダーゼアッセイ)プロモーターフラグメントのほとんどは、共通配列のうちの1つ又は共通配列の間の16bpスペーサー内の誤りを有した。この結果は、定説に従う、即ち、共通配列の変化は所定のプロモーターの活性に大きな影響を与え、活性変化の小さなステップにおいて所定範囲の活性をカバーするプロモーターライブラリーを作り出すためにより繊細なアプローチが必要とされるという事実を強調する。明らかに、我々がスペーサーの配列をランダムに変えるかわりに、共通配列内の変化及び/又はスペーサーの長さの変化を許容するであろうなら、かなり弱いプロモータークローンのみが生じ、そして生じたライブラリーは代謝工学に適さないであろう。
【0063】
一般に、高い活性(70ユニット超)を与えるクローンはオリゴヌクレオチドにより特定して同じ配列を有した。全体として、完全な共通配列及び17塩基対スペーサー長を有するクローンの活性は、5ユニット(CP4)から2000ユニット(CP25)の活性の範囲である。それゆえ、これらの結果は、共通配列を一定に維持しながらスペーサーをランダムにすることにより、プロモーター活性の少くとも 400倍の変化が観察されたことを示す。
【0064】
通常、代謝工学目的のために、比較的強力なプロモーターが要求される。しかしながら、むしろ弱いプロモーターが必要とされる場合もあり得る。先のオリゴヌクレオチド混合物の合成の間におこった比較的少い誤りはプロモーターフラグメント中に存在することを意図せず;我々のデータは、共通配列中の低い割合の誤りを有するオリゴヌクレオチドの混合物を計画的に作り出すのに役立ち得ることを示唆する。
先の種々の酵素反応に用いられる酵素は、 Pharmacia及びBoehringerから得て、それらにより推奨されるように用いた。
【0065】
実施例2. 温度制御されるL.ラクチスプロモーターのライブラリーのための縮重したオリゴヌクレオチドのデザイン
この例は、L.ラクチスのための温度制御されたプロモーターライブラリーの開発を示す。L.ラクチスのヒートショック応答に関連することが示されている8塩基対逆反復を含む調節因子を、−35配列の数塩基対上流に挿入する。これらの調節因子の最小の範囲は、9(又はそれ未満の)塩基対により分離された9bp(又はそれより長い)逆反復(IR)を含む27塩基対:
【0066】
【化3】
【0067】
であるようである。それゆえ、異なる強度の構成的プロモーターを得るためのアプローチを、この逆反復と組み合わせ、これにより培養培地の温度を変化させることにより数倍に誘導することができる種々の基底活性の一連のプロモーターを得ることが可能であるはずである。
【0068】
それゆえ、先の一連の構成的プロモーターからその配列のコア部分(−35〜+6)を含むオリゴヌクレオチドをデザインした(実施例1及び配列番号:1を参照のこと)。−35ヘキサマーの上流の配列を、上述の逆反復配列で置換し、そして位置+6の下流の配列も、実施例1と比べて2つの保存された領域を除去するように改変した(増殖率制御に関連しているが、L.ラクチスのための強力なプロモーターを作り出すことに関して重要でないことが判明している+1〜+8:GTACTGTT及び−44〜−41:AGTT、実施例1を参照のこと)。
【0069】
逆反復内の2つの逆 DNA配列間のスペーサー(sp.1)の配列を、これが生じたプロモーターの温度制御にいずれの効果を与えるかを見るため、例えばそれが温度を変えることにより何倍誘導され得たかを見るために、ランダムに変化させた。逆反復及び−35ヘキサマーの間のスペーシング(sp.2)の重要性は知られていないが、原則として、この管理はプロモーターのヒートショック応答に寄与し又はそれを調節することができる。しかしながら、パラメータの数を制限するために、我々は、天然の形状(L.ラクチスからのdnaJプロモーター由来のもの; van Asseldonkら、1993):5倍のTからなる短いスペーサー配列を含むように選択した。
【0070】
これらの配列を組み合わせた時、以下の、L.ラクチスの温度制御プロモーターのための73bp共通配列に達する。これら73bpのうち、45が保存されており、2つは半保存性であり(R及びW)そして26は4つのヌクレオベースの間でランダムに変化している。
【0071】
【化4】
【0072】
更に、この縮重した配列は、制限エンドヌクレアーゼのための多重認識部位(多重クローニング部位MCS)を特定する配列に隣接させ、そして合成された実際のオリゴヌクレオチド混合物は、以下の配列番号:2:
【0073】
【化5】
に示される縮重した配列を有する。
【0074】
このオリゴヌクレオチド混合物を二本鎖 DNAフラグメント(DSDF)に変換し、プロモータークローニングベクターpLB85iにクローニングした。pLB85iは大腸菌における複製の起源であるがL.ラクチスにおいてそうではない複製の起源及び両生物のための選択マーカーを有する。L.ラクチスのための複製の起源のかわりに、それは、 int遺伝子産物がトランスにおいて供されるなら、L.ラクチス染色体へのプラスミドの挿入を推し進めることができるattP配列を含む。それゆえ、この例は、ランダムスペーサー法により作られたプロモーターを用いる染色体にコードされた遺伝子の調節も示す。
【0075】
pLB51は、プロモータークローニングベクターでもあり、プロモーターのないgusA遺伝子の上流の多重クローニング部位を含む。gusAは、その基質が X-galでなくX-glucであることを除いて他の例に用いたlacZ及び lacLMスクリーニングシステムに似ている。それを、ヒートショック制御プロモーターが分析されると思われるこの特定の適用のためのリポーター遺伝子として選択した。これは、gusA遺伝子産物が実施例1に用いた lacLM遺伝子に関連する問題となり得る熱不安定性でないからである。
【0076】
次に、DSDF混合物を XbaI及び AseIで消化し、ベクターpLB85iを XbaI及び NdeI(NdeIは AseIと適合する)で消化しそして更にアルカリホスファターゼで処理してベクターから5のリン酸基を除去した。DFDS混合物及びベクターの連結の後、その連結混合物を、アンピシリン耐性についての選択と共にgusA陰性大腸菌株である株KW1に形質転換した。これは、そのコロニーの80%が異なる青色コロニー強度を有する約 100のコロニーを生じた。このことは、これらのコロニーが予想されるヒートショックプロモーターフラグメントを有していることを示す。更なる分析のために20の青色コロニーをとった。
【0077】
これらのコロニーからのプラスミド調製物の制限分析は、それらがおおよそ正しい大きさの挿入物を有していることを示した。次にこれらのプラスミド調製物のうちの6つを LB436を形質転換するのに用いた(L.ラクチス染色体上のattB部位へのプラスミドの組込みのための必要な int遺伝子を供する第2のプラスミド pLB81を含むL.ラクチスMG1363の誘導体)。それら形質転換体の各々から、その構成物の、L.ラクチス染色体上のattB結合部位への予想される組込みを有するコロニーを単離した。それら構成物の組込みは、attB領域内の1つのプライマー及びpLB85i領域内の1つのプライマーを用いて、標準的なコロニー PCRにより確認した。
【0078】
次にそれらコロニーを、各々30℃及び37℃においてエリトロマイシン及び 100μg/ml X-gluc を供したGM17培地上で青色コロニーを形成する能力についてテストし、そしてそれらコロニーは、2つの温度において色の強度の明らかな差を示した。このことは、プロモーター活性が温度制御下であることを示す。次に、ヒートショックプロモーターからのgusA発現を、5つの選択されたヒートショック制御プロモーターについて、2つの温度において液体培養物中で測定した(図2を参照のこと)。
【0079】
それらコロニーは異なる活性を有し、30℃において広範囲のプロモーター活性をカバーし、そして5つのクローンのうち4つ(HP6を除く全て)は37℃においてより高いプロモーター活性を供した。このことは、プロモーターが、実際に、温度制御されたことを示す。興味あることに、プロモーターは、ほぼ同じ倍率だけ、即ち30℃から37℃の温度シフトにより 1.7〜2.3 倍だけ制御された。このことは、逆反復中のスペーサー(スペーサー1)がこれらの人工プロモーターの誘導の倍率を決定することに関して小さな重要性を有することを示す。
【0080】
我々は、蓄積したgusA活性を調べた。データは、プロモーターが温度制御されることを明らかに示すが、遺伝子発現を分析する当業者は、このことが、例えば温度のような外部パラメータの特定の変化によりもたらされるプロモーター活性の変化を観察するのをより困難にすることを認めるであろう。更に、ヒートショック制御プロモーターの活性は、図2に示すように、定常状態レベルを比べた時に見られるより温度のシフト後直ちに一時的に10倍高くなることを示す。プロモーター活性の変化をより注意深く観察するために、温度の乱れの前及びその後種々の時間においてタンパク質又はmRNA合成の比率を調べるべきである。それゆえ我々は、30℃から37℃への温度の変化の後、種々の時間において5つのクローンから RNAを単離し、標準的なノーザンブロッティング(Maniatisら、1983)によりgusA mRNA を視覚化し、何倍の個々のプロモータークローンが温度変化により誘導されたかを分析した。
【0081】
実施例3.
グラム陽性バクテリアバチルス・サブチリスは、所定範囲の異種タンパク質の生産のための工業用バイオリアクターとして広範囲に用いられる。それゆえ、本発明のランダムスペーサー法がこの生物のためのプロモーターライブラリーを作るのにも用いることができるか否かをテストすることに関心がある。バチルス・サブチリスのための共通配列は、大腸菌及びL.ラクチスのための共通配列に極めて類似しており、それゆえ、我々は、そのアプローチがバチルス・サブチリスにあてはまるか否かを、いくつかのCPプロモーターをこのバチルス・サブチリスのためのプロモータークローニングベクターにサブクローニングすることによりテストし、次に1)CPプロモーターがバチルス・サブチリス内で活性であるか否か及び2)更に、この生物内で、共通配列間のスペーサーがプロモーターの強さのために重要な役割を果たすか否かを問うことができよう。
【0082】
我々は、バチルス・サブチリス染色体上の amy座へのlacZへのプロモーター融合物の組込みのためにデザインしたプロモータークローニングベクターpDG268を用いることを選んだ。そのベクターは、アンピシリン及びネオマイシン耐性を与え、それは最初のクローニング目的のために大腸菌内で複製できるがバチルス・サブチリス内ではできないであろう。そのベクターの直鎖形態をバチルス・サブチリスに形質転換した時、それはバチルス・サブチリス染色体上の amy座に挿入されるであろう。それゆえ、この例は、染色体によりコードされた遺伝子の調節のためのランダムスペーサー法を通して作られたプロモーターの使用の例としても機能する。
【0083】
CPプロモーターは、実際は、バチルス・サブチリス内でも活性であり、そして更に、そのプロモーターの個々の強度は互いに極めて異なった。プロモーターライブラリーがバチルス・サブチリス内の広範囲のプロモーター活性もカバーしたという事実は、本ランダムスペーサー法がこの生物にもあてはまることを示す。
【0084】
実施例4.
実施例1のCPプロモーターを、L.ラクチスにおける使用のためにデザインしたが、大腸菌プロモーターの共通配列は、実施例1に供されるオリゴヌクレオチドの配列内に含まれる。更に、実施例1においてCPプロモーターライブラリーを作るために用いたベクターは、L.ラクチス及び大腸菌のためのシャトルベクターであり、これは、我々がグラム陰性バクテリア宿主大腸菌内のCPプロモーターの活性を分析するのを許容する。図3は、大腸菌内のCPプロモーターの33の活性を示す。明らかに、個々のCPプロモーターの活性も極めて異なり、そして同時に、プロモーターは広範囲の活性をカバーする。興味あることに、大腸菌及びL.ラクチスにおける個々のプロモーターの強さの間の相関はあまり強くなく;L.ラクチス内で強力なプロモーターであることが見出されたいくつかのプロモーターは大腸菌内で弱くその逆も成り立つことが見出された。
【0085】
プロモーターの活性は、β−ガラクトシダーゼ活性に関して、L.ラクチスにおいて見出された活性より一般にかなり低かった。これはおそらく、プロモータークローニングベクター pAK80がグラム陽性バクテリアL.ラクチスに用いるためにデザインされ、最適化され、それゆえ大腸菌内で翻訳効率が低くなり得るためである。それゆえ、我々は、CPプロモーターのうちの3つ(CP20, CP22及びCP25)を、β−ガラクトシダーゼをコードするプロモーターのないlacZ遺伝子の上流における大腸菌プロモーターをクローニングするためにデザインしたプロモータークローニングベクターpCB267にサブクローニングした。
【0086】
このプロモーターシステムにおいて、CPプロモーターは極めて有効なプロモーターであることが判明したが、それら3つのプロモーターの間の強さの相対的な差は保存された。これにより、CP25プロモーターは、大腸菌において用いるための周知の最も強力なプロモーターの中の1つであると考えられるハイブリッドプロモーターptacより 2.5倍高い活性を供した。それゆえ、これらのデータは、我々のアプローチの強さを更に証明する:本発明のランダムスペーサー法により得られた比較的少数のプロモータークローンを分析することにより、我々は、大腸菌及びL.ラクチスの両方において最も強力なプロモーターのいくつかにうまく到達した。
【0087】
実施例5.
グラム陰性種に属するバクテリア、シュードモナスは、それらの適用において、例えば汚染された土壌中の化学老廃物のバイオデグラデーションにおける適用のため、次第に重要になってきている。しかしながら、ここでも、遺伝子工学は適切なプロモーター及び発現システムの欠如により妨げられている。シュードモナスプロモーターに関する文献は、シュードモナスのための共通配列は大腸菌、L.ラクチス及びバチルス・サブチリスのものほど十分には規定されていないことを示す。
【0088】
これにより、−35領域においては、しばしば保存されたTTGR(R=A又はG)が見出されるが、−35共通配列の残りは異なるプロモーター間で種々である。−10共通配列はおそらくTATRATである。TTGRモチーフと−10配列との間のスペーシングは18〜19bpであり、それは大腸菌においてしばしば見出される16〜17bpスペーサーの等価物である。この生物中の生長プロモーターのための共通配列は大腸菌及びL.ラクチスのための共通配列に極めて近い。
【0089】
それゆえ我々は、プロモーターのないβ−ガラクトシダーゼ遺伝子を含むクローニングベクターにプロモーターをクローニングすることにより、シュードモナス・プチダにおいて、実施例1からの所定範囲のCPプロモーターをテストした。更に、CPプロモーターの活性は、広範囲のプロモーター活性にわたって強さにおいて異なった。これらの結果は、この種においても、本発明のランダムスペーサー法は、相対的に強力なプロモーター及び活性変化の小さなステップにおいてカバーされる広範囲のプロモーター活性の両方を作るのに用いることができるであろうことを示す。
【0090】
実施例6.
我々は、実施例5に記載される共通配列に基づくオリゴヌクレオチドをデザインし、実施例1に記載されるように多重クローニング部位を組み込んだ。次のオリゴヌクレオチドを合成した。
MCS-(N)8-TTGR-N19-TATRAT-(N)8-MCS
【0091】
そのオリゴヌクレオチドを、そのオリゴヌクレオチドの3′末端に相補的なプライマーを用いて二本鎖 DNAに変換し、そしてプロモータープローブベクター上のプロモーターのないβ−ガラクトシダーゼ遺伝子の上流にクローニングした。その連結混合物を、そのプラスミドが有する抗体物質耐性のための選択と共に、 X-galを含むプレート上でシュードモナス・プチダに直接形質転換した。これは、種々の青色強度の約 100のクローンを生じた。次に、30のクローンを上述のようにβ−ガラクトシダーゼ活性について分析した。これらの結果は、この種においても、本発明のランダムスペーサー法は、相対的に強力なプロモーター及び活性変化の小さなステップにおいてカバーされる広範囲のプロモーターの両方を作るのに用いることができるであろうことを示す。
【0092】
実施例7. サッカロマイセス・セレビシアエ(Saccharomyces cerevisiae)プロモーターライブラリーのための縮重したオリゴヌクレオチドのデザイン
5′端からはじめて:EcoRI制限部位(後のクローニングストラテジーに用いるためのもの、以下を参照のこと)、共通な UASGCN4p 、59bpスペーサー(スペーサー1)、共通なTATAAA配列(TATAボックス)、38bpスペーサー(スペーサー2)、23bpレプレッサー結合配列(転写が開始される領域でもある)、T1ボックスとARG8遺伝子の最初のコドンとの間のスペーサー配列(このスペーサーはARG8遺伝子の最初のコドン、 ATGも含むARG8遺伝子からのネイティブ配列である)、並びに最後にBamHI部位を含む 199bpオリゴヌクレオチドをデザインした。これは、β−ガラクトシダーゼリポーター遺伝子との枠内融合物を供するようにデザインされる(以下を参照のこと)。
【0093】
スペーサー1及びスペーサー2の配列はランダムに変化させ、そして合成された実際のオリゴヌクレオチド混合物は配列番号:3に示される以下の縮重した配列:
【0094】
【化6】
を有した。
【0095】
このオリゴヌクレオチド混合物を、実施例1に記載される 199bp縮重オリゴヌクレオチドの3′端の最後の23bpに相補的なオリゴヌクレオチドを用いて、二本鎖 DNAフラグメント(DSDF)に変換した。次に、それを次の通り、2つのプロモータークローニングベクター、pYLZ-2及びpYLZ-6(Hermannら、1992) のいずれかにクローニングした:DSDF混合物をEcoRI及びBamHIで二重消化し、そしてDSDFをベクター DNAに連結した。その連結混合物を、アンピシリン耐性についての選択と共に、実施例1に記載されるように、大腸菌内に形質転換した。プラスミド DNAを 500の個々のクローンから単離し、制限酵素EcoRI及びBamHIでの消化により予想されるプロモーターフラグメントの存在についてスクリーニングした。
【0096】
17のクローンが約 200bpのEcoRI−BamHI挿入物を有することが見い出された。これら17のクローンからのプラスミド DNAを、それらプラスミドが有するURA3マーカーについての選択と共にS.セレビシアエに形質転換し、そしてβ−ガラクトシダーゼの生産についてアッセイした。図4は、それら17のクローンについてのβ−ガラクトシダーゼの生じた活性を示す。全てのプロモーターが、プロモーターフラグメントの挿入のないクローニングベクター自体より高い活性を有する(pYLZ-2)。しかしながら、より重要なのは、この場合においても、それらクローンは活性変化の少いステップにおいて所定範囲のプロモーター活性をカバーすることである。
【0097】
配列分析は、上述の17のクローンがスペーサー1とスペーサー2との間に完全なTATAボックス(TATAAA)を有したが、それら17のクローンのうちの2つ、YP24及び YP435の各々は UASGCN4p 内に欠失を有することを示した。しかしながら、 YP435の活性は39ユニットであり、それは YP212で得た値に近い。これらのデータは、スペーサーのランダム配列のプロモーターの強さへの影響が UASGCN4p 結合部位の状態の影響より強いことを示唆する。
【0098】
上述の通り、人工イーストプロモーターは、2つの異なる制御特性で構築された。1つは、それらプロモーターは増殖培地中でアルギニンの存在により制御されることである。人工イーストプロモーターがアルギニンによっても制御されるか否かをテストするため、我々は、アルギニンあり及びなし(SD+2%グルコース;SD+2%グルコース+1g/lアルギニン)で、最小培地中でいくつかのクローンを増殖させた。図5は、これらの実験の結果を示す。クローンYP18, YP212, YP435は、実際に、アルギニンの存在により、各々5,8,15倍制御された。 YP183はアルギニンにより制御されず、そして配列分析は、このプロモータークローンがアルギニンレプレッサー結合部位内に13bp欠失を有することを確認した。
【0099】
我々は、それらプロモーターが増殖培地中の外部アミノ酸により制御されるか否かも、最小培地(SD+2%グルコース)及び豊富な培地(SD+2%グルコース−URA)中で、イーストプロモータークローンのいくつかのプロモーター活性を分析することによりテストした。図6は、これらの実験の結果を示す。実際、クローンYP18, YP212, YP435、及び YP183中に存在するプロモーターは、アミノ酸の存在により、2〜10倍に制御された。YP24は、このクローン上の UASGCN4p 部位内におきた誤りにより制御されなかった(上述の記載を参照のこと)。
アミノ酸及び特定のアルギニン制御に基づく結果は、本発明のランダムスペーサー法は、広範囲のプロモーター活性をカバーし、そして外部シグナルにより制御することができるプロモーターを作るのに用いることができることを証明する。本発明の制御態様は、アミノ酸及びアルギニン制御により例示されるが、これらに限られない。
【0100】
引用文献
Crabeel, M., de Rijcke, M., Seneca, S., Heimberg, H., Pfeiffer, I., and Matisova, A., 1995. Further definition of the sequence and position requirements of the arginine control element that mediates repression and induction by arginine in Saccharomyces cerevisiae. Yeast 11: 1367-1380
de Vos, W.M., and Simons, G., 1994. Gene cloning and expression systems in lactococci, p.52-105. In M.J.Gasson and W.M. de Vos (eds.), Genetics and biotechnology of lactic acid bacteria. Blackie Academic & Professional, Glasgow, United Kingdom.
【0101】
Gasson, M.J. 1983. Plasmid complements of Streptococcus lactis NCDO 712 and other lactic streptococci after protoplast-induced curing. J.Bacteriol., 154, 1-9.Guarents, L., 1983, Yeast promoters and lacZ fusions designed to study expression of cloned genes in yeast. Methods Enzymol. 101, 181-191.
Hermann, H., Hacker, U., Bandlow, W., and Magdolen, V., 1992. pYLZ vectors: Saccharomyces cerevisiae/Escherichia coli shuttle plasmids to analyze yeast promoters. Gene 119: 137-41.
【0102】
Hinnebusch, A.G., 1992. General and Pathway-specific Regulatory Mechanisms Controlling the Synthesis of Amino Acid Biosynthetic Enzymes in Saccharomyces cerevisiae, p.319-414. In E.W.Jones, J.R.Pringle and J.R.Broach (eds.), The Molecular and Cellular Biology of the Yeast Saccharomyces: Gene Expression, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
Holo, H., and Nes, I.F., 1989. High frequency transformation, by electroporation, of Lactococcus lactis subsp. cremoris grown with glycine in osmotically stabilized media. Appl.Environ.Microbiol., 55, 3119-3123.
【0103】
Israelsen, H. 1995. Cloning and partial characterization of regulated promoters from Lactococcus lactis Tn917-lacZ integrants with the new promoter probe vector, pAK80. Appl.Environ.Microbiol., 61, 2540-2547.
Jensen, P.R., Westerhoff, H.V., and Michelsen, O., 1993. Excess capacity of H + -ATPase and inverse respiratory control in Escherichia coli. EMBO J., 12, 1277-1282. Kacser, H. and Burns, J.A. 1973. The control of flux. Symp.Soc.Exp.Biol., 27: 65-104.
【0104】
Maniatis, T., Fritsch, E.F., and Sambrook, J., 1982. Molecular cloning. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
Miller, J.H., 1972. Experiments in molecular genetics, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
Nilsson, D., and Johansen, E., 1994. A conserved sequence in tRNA and rRNA promoters of Lactococcus lactis. Biochim.Biophys.Acta, 1219: 141-144.
【0105】
Oliver, S.G., and Warmington, J.R., 1989. Transcription. In The yeasts, volume 3, Rose and Harrison (Eds.). Academic Press, London, 117-160.
Schaaff, I., Heinisch, J., Zimmermann, F.K., 1989. Overproduction of glycolytic enzymes in yeast. Yeast, 5: 285-290.
van Asseldonk, M., Simons, A., Visser, H., de Vos, W.M., and Simons, G., 1993. Cloning, nucleotide sequence, and regulatory analysis of the Lactococcus lactis dnaJ gene. J.Bacteriol. 175, 1637-1644.
【図面の簡単な説明】
【0106】
【図1】図1は、実施例1からのL.ラクチス(L. lactis )のための人工プロモーターのライブラリーを示す。
【図2】図2は、実施例2からのL.ラクチスのための人工のヒートショック調節プロモーターのライブラリーを示す。
【図3】図3は、大腸菌内の活性についてアッセイした実施例1からの人工プロモーターのライブラリーを示す。
【0107】
【図4】図4は、実施例7からのS.セレビシアエ(S. cerevisiae )のための人工プロモーターのライブラリーを示す。
【図5】図5は、実施例7からの外来アルギニンによる人工イーストプロモーターの制御を示す。
【図6】図6は、実施例7からの外来アミノ酸による人工イーストプロモーターの制御を示す。
【技術分野】
【0001】
本発明は、人工プロモーターライブラリー及び所定の生物又は生物の群のための人工プロモーターライブラリーを構築する方法に関する。本発明は、これらのライブラリー由来の個々の新規のプロモーターにも関する。更に、本発明は、生物のための人工プロモーターライブラリーからのプロモーターの使用により所定の生物中の遺伝子の発現を最適化する方法に関する。原則として、本発明による人工プロモーターライブラリーは、いずれかの生きている生物において用いるために作製することができるが、現在、それらは、微生物の遺伝子発現を調節するために最も価値あるものである。本発明において、用語“微生物”とは、細菌のような原核生物、並びにイーストのような他の真核微生物、他の真菌及び高等生物の細胞系を含むよう広く解釈されるであろう。
【背景技術】
【0002】
生きている生物の代謝工学技術は、ここ10年超の間、遺伝子工学が実行可能になってきたという事実にかかわらず、工業上の適用に関してはまだ初期である。かなりの程度、これは、株の能力を改良することに関する多くの試みが失敗に終わった結果によるものである。代謝量を増加させる試みの否定的な結果については2つの理由がある:
【0003】
1つは、遺伝子工学者が、細胞代謝の制御及び調節の繊細さを見のがす傾向があることである。速度制限があると予想される酵素の発現は、例えばその遺伝子を高複製数プラスミドにおくことにより、10〜100 倍、増加する。又は、経路における分枝の流れは遺伝子を削除することにより排除される。極めて多くの場合、これは、例えば、細胞代謝の他の部分(例えば、生物の成長に本質的である過程)に本質的である代謝産物を低下させることにより、代謝に二次的効果を与えるであろうし、その最終的な結果は、要求される産物に関するその細胞の全体の能力を減少させる結果となり得る。その代わりとして、正常な発現レベルに関する関連遺伝子の発現を調節すること、並びに例えば、その流れの量を最大又は最小にするレベルとして、最適な発現レベルを決定することが必要である。
【0004】
否定的な結果についての第2の理由は、それ自体の速度制限コンセプトにあり:代謝調節理論(Kacser及びBurns, 1973)及び経路における個々のステップによる調節の実験による測定(Schaffら、1989; Jeasenら、1993) は、特定の流れに関する速度制限であると予想される反応ステップがその流れにわたって全く調節していないかほとんど調節していないことが判明したことを示した。かわりに、細胞代謝の調節及び制御は、経路中のいくつかの酵素に分配されることが判明し、より高い流量を得るためにいくつかの酵素の発現を増強することが必要であり得る。
【0005】
代謝調節理論によれば、経路における全ての酵素により発揮される全体の流量制御は、常に1に合計されるはずである。それゆえ、1つの酵素濃度が最適化された後、流量制御は、他の酵素に移り、そして次に更に流量を増加させるために更なる回数の酵素最適化を行うことが有用であり得る。
要約すると、流量最適化は、1)何部もの過剰発現でなく酵素濃度の細かい調節及びしばしば2)速度制限ステップを見つけ出すのでなく経路中のいくつかの酵素のレベルの最適化を必要とする。
【0006】
現在、1000倍超、遺伝子発現を増加させ、及び/又は(例えば温度誘導システムを用いて)発酵過程の間の特定の時点において遺伝子発現に向けることに利用できる多くのシステムがある。発酵槽において定常状態の遺伝子発現、即ち正常発現レベルの 150%ないし70%に向かわせることに関して、より困難性が増加する。原則として、関心の遺伝子の前に lac型プロモーターを用い、そして次に、lacシステムの所定量のインデューサー、例えばIPTG(イソプロピル−β−D−チオガラクトシド)を加え、又は正確な温度で温度感受性システムを用いることができる。これらの可能性は、しばしば、大規模な工業的適用について実用的でない。かわりの方法は、正確に正しい強さを有するプロモーターを用いることである。しかしながら、このようなプロモーターはあまり利用できず、そして更に、第1の場所において遺伝子の発現を最適にするために所定範囲のプロモーター活性を必要とする。
【0007】
過去20年間、微生物の共通の配列を規定し、最適化するために多くの研究がなされてきた。多くの原核生物において、転写のための開始部位に対しておおよその位置−10及び−35において2つ程度の保存された DNA配列、各々TATAAT及びTTGACAが、それら2つの間の約17塩基対と共にしばしば見出されている。この分野における定説は、これらの要素を含むことにより、生じたプロモーターが強力になる傾向があるであろうことである。実際、比較的まれな出来事であるプロモーター上昇変異は、通常、上述の共通配列により適合するが、下降変異は、共通配列にあまり適合せず、これらの間のより最適でない経路となる。更に、ランダム DNA配列を2つの共通配列の1つの場所にクローン化する場合、生じたプロモーターの強さは、通常、共通配列に対する相同性の程度と相関する。
【0008】
原則として、プロモーターの強さの調節は、共通配列中の塩基対変化により、又はこれらの間のスペーサーの長さの変化により達成することができよう。しかしながら、プロモーターの長さのこのような変化の影響は大きくなる傾向があるであろうし(本発明の実施例1を参照のこと)、それゆえ共通配列の塩基対変化による強さの調節の小さなステップを行うことは容易でない。
【0009】
2つの共通配列を分離するスペーサーの長さはプロモーターの強さのために重要な役割を果たすことが知られているが、その共通配列間のスペーサーの配列は、通常、プロモーターの強さについてほとんど重要でないと考えられており、そして変異誘発によるスペーサー内の更なる共通配列を同定するための試みは、実際成功していない。これまで、共通配列を維持し、かつスペーサーの長さを比較的一定にしながら、スペーサーを無作偽化する試みを行った者はいない。
【0010】
共通配列のうちの少なくとも1つを無作偽化する実験を含む、微生物の共通配列を規定し、最適化するための多くの実験が行われている。これらの実験のいくつかにおいて、共通配列の周囲のヌクレオチドの一部も、17bpと異なるスペーサーの長さのプロモーターの形成を許容するため、及び/又は共通配列の周囲の可能性ある新規の共通モチーフを見い出すために、無作偽化された。これは、有効なプロモーターを形成するであろう機会が極めて小さく、そして共通配列に対する相同性が十分に高くて弱いプロモーターを生ずるまれな場合を見出すために、選択を行わなければならない。
【発明の開示】
【0011】
発明の目的
我々が目指すプロモーターライブラリーは、特定の種の工学技術のための関心の活性になり得る全体の範囲のプロモーター活性、好ましくは検出できる最も弱いプロモーターから可能な最も強力なプロモーターをカバーするはずである。更に、我々は、小さなステップにおいてこの広範な活性範囲をカバーすること、即ち、上述のような流量最適化の目的に適したものであるために、ステップ当り50〜100 %だけ活性を増加させることを目指す。
【0012】
本発明において、我々は、共通配列の間のスペーサーの配列が以前に認められているよりもかなり重要であることを示す。スペーサー配列は、1)スペーサー配列の主要な部分が種々であると同時にランダムであり、そして2)同時に、共通配列の少くとも半分が一定に維持されている場合、プロモーターの強さに強い影響力を有し得る。我々は、これら2つの条件が満たされるなら、我々の方法は、極めて強力なプロモーターを含む、広範囲の活性をカバーするプロモーターを作り出すのに用いることができる。プロモーター活性の範囲は代謝の工学技術の目的のために極めて適したプロモーターを作る活性変化の小さなステップにおいてカバーされる。更に、我々は、その方法が、広範囲の生物のため、かつ異常に高い頻度においてプロモーターを作り出すのに用いることができる。
【0013】
発明の概要
本発明は、所定の生物又は生物の群のための人工的なプロモーターライブラリーであって、それらのセンス鎖が前記生物もしくは生物の群からの有効なプロモーターの少なくとも2の共通配列を含む二本鎖 DNAフラグメントの混合物、又は各々の少なくとも半分を含むその一部と、少なくとも7のヌクレオチドがヌクレオベースA,T,C及びGから無作偽に選択された種々の長さの周囲の又は介在するヌクレオチド配列(スペーサー)と、を含む人工プロモーターライブラリーを供する。但し、以前から周知であるプロモーター配列及び天然のソースから単離されたプロモーター配列は含まれない。
【0014】
プロモーターの長さの最も広範囲のバリエーションは、ヌクレオベースA,T,C及びGの中から、スペーサー配列内の少なくとも10、好ましくは少なくとも12、そしてより好ましくは少なくとも14のヌクレオチドがランダムに選択される時に得られる。
二本鎖 DNAフラグメントのセンス鎖は、ライブラリーのプロモーターに特定の調節特性を与える調節 DNA配列も含み得る。このような特定の調節特性は、好ましくは、増殖条件の変化、例えばpH、浸透度、温度又は増殖相の変化による活性化である。
【0015】
クローニング目的のために、二本鎖 DNAフラグメントは、通常、それらの端の一方又は両方に加えられた制限エンドヌクレアーゼのための1又は複数の認識部位を含む配列;制限エンドヌクレアーゼのための多重の認識部位(多重クローニング部位MCS)を特定する最も便利な配列を有する。
選択される生物又は生物のグループは、原核生物から及び真核生物から、特に、原核及び真核微生物、例えばイースト、他の真菌及び高等生物の細胞系から選択することができる。
【0016】
例えば乳製品工業における、原核生物の興味あるグループは、ラクトコッカス(Lactococcus )、ストレプトコッカス(Streptococcus )、エンテロコッカス(Enterococcus)、ラクトバチルス(Lactobacillus )及びロイコノストック(Leuconostoc )属の乳酸バクテリア、特にラクトコッカス・ラクチス(Lactococcus lactis)及びストレプトコッカス・サーモフィルス(Streptococcus thermophilus)の株からなる。他の興味ある原核生物は、エスケリキア(Escherichia )、バチルス(Bacillus)及びシュードモナス(Pseudomonas )属、特に大腸菌、バチルス・サブチリス(Bacillus subtilis )及びシュードモナス・プチダ(Pseudomonas putida)種に属するバクテリアである。
【0017】
原核生物のための人工プロモーターライブラリーにおいて、前記共通配列は、例えば、−10シグナル(−12〜−7):TATAAT及び各々少なくとも3の保存されたヌクレオチドを含む−10シグナル又はその一部の上流の少なくとも1のアクティベータータンパク質結合部位を含み得る。
最も多くの場合、原核生物のための人工プロモーターライブラリー内に保持された共通配列は、−35シグナル(−35〜−30):TTGACA及び−10シグナル(−12〜−7):TATAAT又は各々の少なくとも3の保存されたヌクレオチドを含む両方の一部を含むであろう。
【0018】
より有効なプロモーターは、通常、前記共通配列が−35シグナルの4〜6の保存されたヌクレオチド及び−10シグナルの4〜6の保存されたヌクレオチド、好ましくは各々5又は6のヌクレオチド、より好ましくは各々全部で6のヌクレオチドを含む場合に得られる。最も有効なプロモーターは、介在する保存されたモチーフ、例えば保存されたモチーフ−44〜−41:AGTT、−40〜−36:TATTC 、−15〜−14:TG、及び+1〜+8:GTACTGTTから選択されたモチーフを更に含む場合に得られる。
【0019】
このようなプロモーターにおいて、−35シグナルと−10シグナルとの間のスペーサーの長さは14〜23bp、好ましくは16〜18bp、より好ましくは17bpである。これは、そのシグナル内のヌクレオチドのうちのいくつかが変異された場合でさえも、そのヘキサマーシグナル間のスペーサーの長さを意味することが理解されるはずである。
真核生物において、前記共通配列は、TATAボックス及び少なくとも1の上流活性化配列(UAS) を含むべきである。
【0020】
興味ある真核微生物は、通常のパン酵母である。イースト種サッカロマイセス・セレビシアエ(Saccharomyces cerevisiae)である。サッカロマイセスに用いられるプロモーターにおいて、共通配列は、サッカロマイセス・セレビシアエにおいて機能する転写開始シグナル(T1ボックス)を更に含む。
【0021】
サッカロマイセス・セレビシアエのための本発明による人工プロモーターライブラリーの特定の実施形態において、前記共通配列は、TATAボックス:TATAAA、 UASGCN4p :TGACTCA 、及びアルギニンレプレッサーのための結合部位argRとしても機能するT1ボックス:CTCTTAAGTGCAAGTGACTGCGA を含む。
【0022】
上述の人工プロモーターライブラリーの個々のプロモーターも本発明に含まれる。以下の実施例に従って作製された特定のプロモーターは、以下の配列番号:5〜58に記載されるものである。本発明は、本発明の人工プロモーターライブラリーにより規定されるプロモーター由来の人工プロモーターを更に含む。
【0023】
本発明は、所定の生物又は生物のグループのための人工プロモーターライブラリーを作製する方法であって、前記生物又は生物の群から有効なプロモーターの少なくとも2の共通配列を選択し;該共通配列又はその各々の少なくとも半分を含む一部、並びに少なくとも7のヌクレオチドがヌクレオベースA,T,C及びGの中から無作偽に選択された不定の長さの周囲の又は介在するヌクレオチド配列(スペーサー)を含む一本鎖 DNA配列の混合物を合成し;そして該一本鎖 DNA配列を第2鎖の合成により二本鎖 DNAフラグメントに変換することを含む方法も供する。
【0024】
上述の通り、最も広範囲のバリエーションは、スペーサー配列内の少なくとも10、好ましくは少なくとも12、より好ましくは少なくとも14のヌクレオチドがヌクレオベースA,T,C及びGの中から無作偽に選択される場合に得られる。
調節可能な人工プロモーターライブラリーを得るために、合成される一本鎖 DNA配列は、特定の調節特性をライブラリーのプロモーターに与える調節 DNA配列を含み得る。このような特定の調節特性は、好ましくは、増殖条件の変化、例えばpH、浸透性、温度又は増殖相の変化による活性化である。
【0025】
また、クローニングのために適した人工プロモーターライブラリーを得るために、制限エンドヌクレアーゼのための1又は複数の認識部位を特定する配列を、合成において一本鎖 DNA配列の一端又は両端に加えてもよく、又はこのような制限部位を含むリンカーを二本鎖 DNAフラグメントの一端又は両端に連結してもよい。最も便利なのは、このような配列が、制限エンドヌクレアーゼのための多重認識部位(多重クローニング部位MCS)を特定することである。
【0026】
本発明による方法により人工プロモーターライブラリーを調製することができる選択される生物、及び特定の生物のプロモーターライブラリーのために選択される種々の縮重した配列は、人工プロモーターライブラリー自体について上述されるのと同じである。
【0027】
上述の人工プロモーターライブラリーの可能な使用に関して、本発明は、微生物中の遺伝子の発現を最適化する方法であって、a)請求項1〜26のいずれか一に記載の人工プロモーターライブラリー又は請求項30〜55のいずれかに記載の方法により作製された人工プロモーターライブラリーから、活性変化の比較的小さなステップにおけるプロモーター活性の範囲を包含する一セットのプロモーターを選択し、
b)該一セットのプロモーターを、前記遺伝子を各々のクローン内で該セットからの少なくとも1のプロモーターの制御下におくように、前記生物内にクローニングし、
c)該選択されたクローンを増殖させ、そして産物形成の最適な流れを示すものを見い出すためにそれらをスクリーニングすることを含む方法を更に供する。
【0028】
この方法は、好ましくは、原核及び真核微生物、例えばバクテリア、イースト、他の真菌及び哺乳動物細胞系からなる群から選択される生物と共に用いられる。
【0029】
図面の簡単な記載
図1。実施例1からのL.ラクチス(L. lactis )のための人工プロモーターのライブラリー。プロモーター活性(Miller単位)を、2μg/mlエリトロマイシンを補給したGM17培地中で増殖した株MG1363におけるプロモータークローニングベクター pAK80上での異なる合成プロモータークローンから転写されたb−ガラクトシダーゼをコードするリポーター遺伝子(lacLM )の発現からアッセイした。データ点のパターンは、−35もしくは−10共通配列のいずれかにおいて又はこれら2つの共通配列の間のスペーサーの長さにおいてプロモータークローンが異なることを示す。
【0030】
図2。実施例2からのL.ラクチスのための人工のヒートショック調節プロモーターのライブラリー。プロモーター活性(Miller単位)を、株 LB436内の染色体上の、異なる合成プロモータークローンから転写されたβ−グルクロニダーゼをコードするリポーター遺伝子(gusA)の発現からアッセイした。その細胞は、2μg/mlエリトロマイシンを補給したGM17培地中で、2つの異なる温度30及び37℃で増殖させた。β−グルクロニダーゼについてのアッセイを、X-glucを基質として用いたことを除いてβ−ガラクトシダーゼアッセイ(実施例1を参照のこと)と平行して行った。
【0031】
図3。大腸菌内の活性についてアッセイした実施例1からの人工プロモーターのライブラリー。プロモーター活性(Miller単位)を、 200μg/mlエリトロマイシンを補給したLB培地中で増殖させた株BOE270中での、プロモータークローニングベクター pAK80上で、異なる合成プロモータークローンから転写されたβ−ガラクトシダーゼをコードするリポーター遺伝子(lacLM )の発現からアッセイした。データ点のパターンは、−35もしくは−10共通配列のいずれかにおいて、又はこれら2つの共通配列の間のスペーサーの長さにおいてプロモータークローンが異なることを示す。
【0032】
図4。実施例7からのS.セレビシアエ(S. cerevisiae )のための人工プロモーターのライブラリー。プロモーター活性(Miller単位)を、2%グルコースを補給したSD最小培地中で増殖させたS.セレビシアエ株SG24(URA3-52) 中のプロモータークローニングベクターpYLZ-2上で、異なる合成プロモータークローンから転写されたβ−ガラクトシダーゼをコードするリポーター遺伝子(lacZ)の発現からアッセイした。YP24及び YP435は、 UASGCN4p 結合部位内に、各々1bp欠失及び点変異を有する。pTK101は、 UAS配列が削除され、TATAボックスが存在するプロモーターを含む。
【0033】
図5。実施例7からの外来アルギニンによる人工イーストプロモーターの制御。プロモーター活性(Miller単位)を、1g/lアルギニンあり(SDG) 又はなし(SDG+arg)で2%グルコースを補給したSD最小培地中で増殖させたS.セレビシアエ株SG24(URA3-52) におけるプロモータークローニングベクター上で、異なる合成プロモータークローンから転写されたβ−ガラクトシダーゼをコードするリポーター遺伝子(lacZ)の発現からアッセイした。 YP183は、argRレプレッサーのための結合部位内に13bpの欠失を有する。
【0034】
図6。実施例7からの外来アミノ酸による人工イーストプロモーターの制御。プロモーター活性(Miller単位)を、2%グルコースを補給したSD最小培地(SDG) 又はウラシルを含まない(アミノ酸を含む)複合培地(SC-ura)内で増殖させたS.セレビシアエ株SG24(URA3-52) におけるプロモータークローニングベクターpYLZ-2上で、異なる合成プロモータークローンから転写されたβ−ガラクトシダーゼをコードするリポーター遺伝子(lacZ)の発現からアッセイした。YR24は UASGCN4p 結合部位内に1bp欠失を有する。
【発明を実施するための最良の形態】
【0035】
我々の方法において、縮重したオリゴヌクレオチドは、プロモーターライブラリーが作製されるべき生物又は生物の群のために合成される。オリゴヌクレオチドの配列は、特定の生物内でプロモーター機能を有効にする特徴に基づき、文献から利用できる知識を組み合わせることにより記述される。オリゴヌクレオチド内に固定されるのに必要とされる情報の量は、異なる生物の間で幾分か種々である。例えば大腸菌において、−35及び−10共通配列内のより少ない完全性の適合により、及び17bpからずれたこれらの配列(各々TTGACA及びTATAAT)の間の間隔により形成することができるが、L.ラクチス内の強力なプロモーターの要求はより厳格であるようである。
【0036】
第2に、一本鎖オリゴヌクレオチドは、二本鎖 DNAフラグメントに変換され、プロモータープロビングベクターにクローニングされる。そのプロモーター含有クローンは、例えばインジケータープレート上の種々の程度の色のコロニーを供する能力により同定される。これは、原則として、我々に極めて強力なプロモーターのみを供するが、我々は、共通配列の間のスペーサー配列をランダムに変化させることにより、その生じたプロモーターの強度が調節されることを発見した。実際、この方法を用いて、我々は、活性変化の小さなステップにおいて、関心のものになるような全体の範囲のプロモーターに及ぶプロモーターライブラリーを得た。
【0037】
次に、遺伝子発現の最適化は次のように達成されよう:1)プロモーターライブラリーから、例えば野生型プロモーターの強さの25%、50%、 100%、 200%及び 400%を有するプロモーターを選択する。2)次に、これらのプロモーターを、関心の遺伝子の上流の野生型プロモーターの位置にクローン化する。3)次にこれら5つの構成物各々で得られた最適化されるべき変数(例えば経路を通る流量)の大きさを測定し、そして最適な構成物が生産生物として直接用いられる。更に発現を微妙に調節すること又はプロモーター活性の範囲を拡げることが必要であり得る。上述の誘導システムに優るこのシステムの直接の利点は、最適なプロモーター活性を決定した後、株は、原則として、発酵過程に直ちに用いることができることである。
【0038】
1つの好ましい実施形態において、本発明のランダムスペーサー法は、グラム陽性バクテリア、ラクトコッカス・ラクチス(Lactococcus lactis)のための一連の構成的プロモーターを作るのに用いられる。他の好ましい実施形態において、我々は、本発明のランダムスペーサー法により作られたプロモーターがグラム陰性バクテリア、大腸菌及びシュードモナス(Pseudomonas )の少くとも2種において及びグラム陽性バクテリアバチルス・サブチルス(Bacillus subtilis)において機能的であることを示す。我々は、個々のプロモーターの強度が用いる生物に依存すること、即ち特定の生物において特定のプロモーターが強力であり、他の生物においてそれは弱いが、テストした全ての生物において、プロモーターは活性変化の小さなステップにおいて広範囲の活性を包含することを示す。
【0039】
しばしば、例えば発現される遺伝子産物が細胞の増殖を阻害するので、遺伝子発現を特定の程度まで、及び発酵の特定の段階で、活性化することが要求される。プロモーターがpH、温度又は増殖相の変化により活性化されるであろうように、特定の調節メカニズムで異なる強度のプロモーターを得るために上述の技術を組み合わせることが有用である。
【0040】
これにより、他の好ましい実施形態において、本発明のランダムスペーサー法は、一連の特異的に調節されるプロモーターを作り出すのに用いられる。実施例2に詳述するように、先のアプローチは、グラム陽性バクテリア、ラクトコッカス・ラクチスのためのヒートショック調節プロモーターのライブラリーを作り出すために特定の調節 DNAと組み合わせて用いられる。
【0041】
原核生物に加えて、真核微生物(イースト及び他の真菌並びに哺乳動物細胞系)は所定範囲の有機化合物及び種々のタンパク質の生産のための重要な微生物である。それゆえ、更にこれらの生物において遺伝子発現を調節するために上述のアプローチを開発することに関心がある。これにより、実施例7に示すような更に他の好ましい実施形態において、本発明のランダムスペーサー法は、パン酵母、サッカロマイセス・セレビシアエ(Saccharomyces cerevisiae)のための一連のプロモーターを作り出すために用いられる。ここで、それらプロモーターは、 GCN4p及びARGR調節を備える。
【0042】
真核細胞における転写開始の調節は、原核生物と比べて幾分かより複雑である。転写開始部位は、通常、共通配列TATAAA又はその一部を含むが、原核生物のものと似ていないいわゆるTATAボックスの後にあり、そのTATAボックスから転写開始部位への距離はほとんど規定されていない。サッカロマイセス・セレビシアエにおいて、この距離は、典型的には、40〜120 ヌクレオチドである(Oliver及びWarmington, 1989) 。多くの原核生物プロモーター内で見出されるいわゆる−35共通ヘキサマーはサッカロマイセス・セレビシアエ内に存在しない。
【0043】
かわりに、いわゆる上流活性化配列(UAS) が転写開始部位の上流に見出される。これら UASは、後に転写開始のアクティベーターとして機能し得る特定の DNA結合タンパク質により認識される。例えば、アミノ酸生合成に関連する遺伝子の上流に見い出される UAS配列 UASGCN4p は、これらの遺伝子の転写を活性化するGCN4タンパク質のための結合部位を特定する DNA配列からなる(Hinnebusch, 1992)。プロモーター要素間の距離がプロモーターの強度のために重大であるようである(実施例1を参照のこと)原核生物と対照的に、真核生物プロモーター内のTATAボックスと UAS配列との間の距離は極めて可変性であり、約1000bpまでであり得る。特定の遺伝子は UASの1超の複製さえ含むが、活性化のためには1つで十分であるようである。
【0044】
イーストから知られている UAS配列の1つは、GCN4タンパク質のための結合部位である。GCN4タンパク質のための結合部位を含むプロモーターは、増殖培地中のアミノ酸供給の状態により調節されるはずであり;アミノ酸の欠如下において、GCN4タンパク質が形成され、特定の UAS配列に結合してアミノ酸の生合成に関連するプロモーターにおいて転写を刺激する。GCN4タンパク質結合部位(UASGCN4p ) のための共通配列は、短い逆反復 TGACTCAである。
【0045】
GCN4タンパク質により活性化されるサッカロマイセス・セレビシアエにおけるプロモーターはARG8プロモーターである。このプロモーターにおいて、唯一の UASGCN4p 配列の複製があり、そしてそれはTATAボックスから59bpに位置する(我々はこの距離をスペーサー1と呼ぶ)。転写開始はTATAボックスの下流約40〜60ヌクレオチドでおこる。ARG8プロモーターは、アルギニンレプレッサー、argRのための結合部位として機能する DNA配列も含み(Crabeelら、1995) 、それは外来アルギニンによりプロモーターを4倍抑制する。
【0046】
これにより、この場合、プロモーターは 136bp内に位置し、そしてそれは、先の例に概説されるように本発明のランダムスペーサー法によりプロモーターライブラリーを開発するために、そのシステムを魅力あるものにする2つの調節特性を含む。しかしながら、その方法はこのモデルシステムに限られず;原則として、約1000ヌクレオチドより小さいスペーサーにより分離されたTATAボックス、 UAS配列、レプレッサー結合部位等のいずれかの組合せがこの方法のための出発点として適するはずである。
【実施例】
【0047】
実施例1. L.ラクチスプロモーターライブラリーのための縮重したオリゴヌクレオチドのデザイン
文献によれば(de Vos及びSimons, 1994の報告を参照のこと)、L.ラクチスにおける強力なプロモーターは共通して次のヌクレオチド配列(数字は番号+1で与えられる転写開始部位に対する位置を示す):−12〜−7:TATAAT;−15〜−14:TG;−35〜−30:TTGACAを有する傾向がある。−10と−35との間のスペーシングは17ヌクレオチドであるようである。しかしながら、L.ラクチスについて出版されているプロモーター配列の親密な比較は、上述の1つを除くいくつかの位置において、ヌクレオチドが多少よく保存されていることを示す。
【0048】
これらの位置のいくつかは:−1:A;−3:A又はT(=W);−6:A;−13:A又はG(=R);−40〜−36:TATTC である。更に、 Nilsson及びJohansen (1994, BBA)は、L.ラクチスからの比較的強力なプロモーター(転移 RNA及びリボソーム RNAオペロンのためのプロモーター)の間でよく保存されているようである2つのモチーフ:+1〜+8:GTACTGTT、及び−44〜−41:AGTTを指摘した。これらのモチーフは、そのプロモーターから強力かつ増殖比依存の発現を与え得る。
【0049】
これらの付加的なモチーフが含まれる場合、L.ラクチスにおける有効なプロモーターのための以下の53ヌクレオベースの縮重した配列に達する。これら53ヌクレオベースのうち、34塩基が保存され、2つが半保存性であり(R及びW)、そして17が4つのヌクレオチドの間で極めてランダムに変化している。
【0050】
【化1】
【0051】
更に、この縮重した配列は、制限エンドヌクレアーゼのための多重認識部位(多重クローニング部位MCS)を特定する配列に隣接し、合成されるべき実際のオリゴヌクレオチド混合物は、配列番号:1
【0052】
【化2】
に示される縮重した配列を有する。
【0053】
本明細書によるオリゴヌクレオチドの混合物は、 Hobolth DNA合成により合成した。
このオリゴヌクレオチド混合物は、最初は一本鎖であり、クローニングの目的のために二本鎖 DNAフラグメントに変換されなければならない。これは、プロモーターオリゴヌクレオチドの3′末端に相補的な配列を有する10bpオリゴヌクレオチドを試験管内で合成することにより行った。次にこのオリゴヌクレオチドをdATP, dCTP, dGTP及びdTTPの存在下での DNAポリメラーゼIのクレノウフラグメントによる第2鎖合成のためのプライマーとして用いた。この方法において第2 DNA鎖は正確に第1 DNAに相補的になった。
【0054】
その結果物は3′及び5′端の両方に制限エンドヌクレアーゼのための多重認識部位を含む 100bp二本鎖 DNAフラグメントの混合物である。次にこれらの DNAフラグメントを、ベクター DNAフラグメントpAK80 (Israelsenら、1995)の端に適合する適切な“粘着”末端を作るために制限エンドヌクレアーゼで切断した。 pAK80は、大腸菌及びL.ラクチスの両方において繁殖のための複製源を有することを意味するシャトルベクターである。この方法において、クローニングステップは、便利には、大腸菌内で行うことができ、次にL.ラクチス内で生理的実験を行うことができる。更に、 pAK80は、多重クローニング部位の下流にプロモーターのないβ−ガラクトシダーゼリポーター遺伝子シテム(lacLM )を有する。これにより、プロモーターが多重クローニング部位内に挿入されなければ、 pAK80は lacLM遺伝子を発現しない。
【0055】
二本鎖 DNAフラグメントの混合物をクローニングベクター pAK80内にクローニングするために2つのクローニングストラテジーを用いた。
1)混合物をBamHI及び PstIで消化し、ベクター pAK80を PstI及び BglII(BglIIはBamHIに適合する)で消化した。
【0056】
2)混合物を SspI及びHincIIで消化し、ベクター pAK80を SmaIで消化した(全部の3つの酵素は平滑端 DNAフラグメントを作り出す)。
両方の場合、ベクター DNAを、クローニングベクターの再連結を防ぐために子ウシインテスチンホスファターゼ(CIP) で更に処理した。次に、そのフラグメント混合物及びベクター DNAを、T4 DNAリガーゼ及び標準連結条件を用いて16℃で一晩、連結した。
【0057】
連結混合物を、エリトロマイシン耐性についての選択と共に、大腸菌 K-12 Δlac に形質転換した。大腸菌K-12株の細胞 MT102を、Ca2+イオンでの標準的処理(Maniatisら、1982)を用いてコンピテントにした。次に連結混合物を、標準的な形質転換手順(Maniatisら、1982)を用いてこれらの細胞に形質転換し、そして生じたクローンを、X-gal(5−ブロモ−4−クロロ−3−インドリル−β−D−ガラクトシド)を含むプレート上で青いコロニーを作るであろうβ−ガラクトシダーゼ活性についてスクリーニングした。その形質転換混合物を、 200μg/mlエリトロマイシン、1%グリセロール、及び 100μg/ml X-galを含むLBプレート上にプレートした。 150のエリトロマイシン耐性形質転換体を得た。全て最初は白かったが、長期のインキュベーション(4℃で2週間)の後、これらのコロニーのうち46は明るい青色になった。これにより、ストラテジー1)を用いて、我々は17の青色コロニー(CP30〜CP46)を見出し、そしてストラテジー2)を用いて、我々は29の青色コロニー(CP1〜CP29)を見出した。
【0058】
これらのコロニー(CP1〜CP46)の各々からプラスミド DNAを単離し、制限酵素マッピングにより分析した。ほぼ全てのプラスミド(CP31及びCP43を除く)が、このベクター上の、さもなければプロモーターのない lacLM遺伝子の転写を指図するであろう方向で、 pAK80の MCS内に挿入されたプロモーターフラグメントを含んでいた。
【0059】
次に、これら46のプラスミド DNA調製物を、エリトロマイシン耐性についての選択と共に、L.ラクチス亜種ラクチスMG1363に形質転換した。L.ラクチス亜種ラクチス株の細胞MG1363(Gasson, 1983)を、Holo及びNes(1989) により記載されるように、2%グリシンを含む SGM17培地中で一晩、その細胞を増殖させることによりコンピテントにした。次に、上述の46のクローンの各々からのプラスミド DNAを、エレクトロポレーション法(Holo及びNes, 1989)を用いてこれらの細胞内に形質転換した。再生後、細胞を2μg/mlエリトロマイシンを含むSRプレート上にプレートした。次の X-galプレート上での青色についてのスクリーニングは、46のクローンの間でβ−ガラクトシダーゼ活性の大きな差を示し;いくつかのクローンは24時間のインキュベーション後に暗い青色のコロニーになり、他のものは1週間超のインキュベーション後に明るい青色のコロニーになった。
【0060】
次に、MG1363における46のコロニーの液体培養物のβ−ガラクトシダーゼ活性を、Miller (1972) により記載されるように決定し、 Israelsenら (1995) により改良した。各々 pAK80の46のプラスミド誘導体のうち1つを有する株MG1363の培養物を、30℃で一晩、1%グルコースを補給したM17培地中で増殖させた。次にこれらの培養物25〜100 μlを、最も弱いプロモータークローンの場合に2mlの培養物を(遠心による20倍の濃縮の後に)用いたことを除いて、後のβ−ガラクトシダーゼアッセイに用いた。これらの結果を図1に示す。明らかに、クローン化したプロモーターフラグメントの有効性において極めて大きな差があると共に、これらのクローンは、 0.3ユニットのβ−ガラクトシダーゼ活性からこの生物について知られているおそらく最も強力なプロモーターである2000ユニットまでの範囲のプロモーター活性をカバーする。
【0061】
更に、広い範囲が活性の小さな変化によりカバーされ、それゆえこれらのプロモーターフラグメントにより、我々は、L.ラクチスにおいて遺伝子の広範囲の発現を得ることができるばかりでなく、流量最適化の目的のために小さなステップにおいてL.ラクチスにおいて遺伝子の発現を調節することもできる。
上述の46のクローンの DNA配列決定は、挿入されたプロモーターフラグメントのほとんどが、オリゴヌクレオチドデザインについてもとから特定された DNA配列を有していたが(上述を参照のこと)、残りのフラグメントの配列はその配列から少しずれていた。
【0062】
β−ガラクトシダーゼアッセイにおいて低い活性を与えた(70ユニット又はそれ未満のβ−ガラクトシダーゼアッセイ)プロモーターフラグメントのほとんどは、共通配列のうちの1つ又は共通配列の間の16bpスペーサー内の誤りを有した。この結果は、定説に従う、即ち、共通配列の変化は所定のプロモーターの活性に大きな影響を与え、活性変化の小さなステップにおいて所定範囲の活性をカバーするプロモーターライブラリーを作り出すためにより繊細なアプローチが必要とされるという事実を強調する。明らかに、我々がスペーサーの配列をランダムに変えるかわりに、共通配列内の変化及び/又はスペーサーの長さの変化を許容するであろうなら、かなり弱いプロモータークローンのみが生じ、そして生じたライブラリーは代謝工学に適さないであろう。
【0063】
一般に、高い活性(70ユニット超)を与えるクローンはオリゴヌクレオチドにより特定して同じ配列を有した。全体として、完全な共通配列及び17塩基対スペーサー長を有するクローンの活性は、5ユニット(CP4)から2000ユニット(CP25)の活性の範囲である。それゆえ、これらの結果は、共通配列を一定に維持しながらスペーサーをランダムにすることにより、プロモーター活性の少くとも 400倍の変化が観察されたことを示す。
【0064】
通常、代謝工学目的のために、比較的強力なプロモーターが要求される。しかしながら、むしろ弱いプロモーターが必要とされる場合もあり得る。先のオリゴヌクレオチド混合物の合成の間におこった比較的少い誤りはプロモーターフラグメント中に存在することを意図せず;我々のデータは、共通配列中の低い割合の誤りを有するオリゴヌクレオチドの混合物を計画的に作り出すのに役立ち得ることを示唆する。
先の種々の酵素反応に用いられる酵素は、 Pharmacia及びBoehringerから得て、それらにより推奨されるように用いた。
【0065】
実施例2. 温度制御されるL.ラクチスプロモーターのライブラリーのための縮重したオリゴヌクレオチドのデザイン
この例は、L.ラクチスのための温度制御されたプロモーターライブラリーの開発を示す。L.ラクチスのヒートショック応答に関連することが示されている8塩基対逆反復を含む調節因子を、−35配列の数塩基対上流に挿入する。これらの調節因子の最小の範囲は、9(又はそれ未満の)塩基対により分離された9bp(又はそれより長い)逆反復(IR)を含む27塩基対:
【0066】
【化3】
【0067】
であるようである。それゆえ、異なる強度の構成的プロモーターを得るためのアプローチを、この逆反復と組み合わせ、これにより培養培地の温度を変化させることにより数倍に誘導することができる種々の基底活性の一連のプロモーターを得ることが可能であるはずである。
【0068】
それゆえ、先の一連の構成的プロモーターからその配列のコア部分(−35〜+6)を含むオリゴヌクレオチドをデザインした(実施例1及び配列番号:1を参照のこと)。−35ヘキサマーの上流の配列を、上述の逆反復配列で置換し、そして位置+6の下流の配列も、実施例1と比べて2つの保存された領域を除去するように改変した(増殖率制御に関連しているが、L.ラクチスのための強力なプロモーターを作り出すことに関して重要でないことが判明している+1〜+8:GTACTGTT及び−44〜−41:AGTT、実施例1を参照のこと)。
【0069】
逆反復内の2つの逆 DNA配列間のスペーサー(sp.1)の配列を、これが生じたプロモーターの温度制御にいずれの効果を与えるかを見るため、例えばそれが温度を変えることにより何倍誘導され得たかを見るために、ランダムに変化させた。逆反復及び−35ヘキサマーの間のスペーシング(sp.2)の重要性は知られていないが、原則として、この管理はプロモーターのヒートショック応答に寄与し又はそれを調節することができる。しかしながら、パラメータの数を制限するために、我々は、天然の形状(L.ラクチスからのdnaJプロモーター由来のもの; van Asseldonkら、1993):5倍のTからなる短いスペーサー配列を含むように選択した。
【0070】
これらの配列を組み合わせた時、以下の、L.ラクチスの温度制御プロモーターのための73bp共通配列に達する。これら73bpのうち、45が保存されており、2つは半保存性であり(R及びW)そして26は4つのヌクレオベースの間でランダムに変化している。
【0071】
【化4】
【0072】
更に、この縮重した配列は、制限エンドヌクレアーゼのための多重認識部位(多重クローニング部位MCS)を特定する配列に隣接させ、そして合成された実際のオリゴヌクレオチド混合物は、以下の配列番号:2:
【0073】
【化5】
に示される縮重した配列を有する。
【0074】
このオリゴヌクレオチド混合物を二本鎖 DNAフラグメント(DSDF)に変換し、プロモータークローニングベクターpLB85iにクローニングした。pLB85iは大腸菌における複製の起源であるがL.ラクチスにおいてそうではない複製の起源及び両生物のための選択マーカーを有する。L.ラクチスのための複製の起源のかわりに、それは、 int遺伝子産物がトランスにおいて供されるなら、L.ラクチス染色体へのプラスミドの挿入を推し進めることができるattP配列を含む。それゆえ、この例は、ランダムスペーサー法により作られたプロモーターを用いる染色体にコードされた遺伝子の調節も示す。
【0075】
pLB51は、プロモータークローニングベクターでもあり、プロモーターのないgusA遺伝子の上流の多重クローニング部位を含む。gusAは、その基質が X-galでなくX-glucであることを除いて他の例に用いたlacZ及び lacLMスクリーニングシステムに似ている。それを、ヒートショック制御プロモーターが分析されると思われるこの特定の適用のためのリポーター遺伝子として選択した。これは、gusA遺伝子産物が実施例1に用いた lacLM遺伝子に関連する問題となり得る熱不安定性でないからである。
【0076】
次に、DSDF混合物を XbaI及び AseIで消化し、ベクターpLB85iを XbaI及び NdeI(NdeIは AseIと適合する)で消化しそして更にアルカリホスファターゼで処理してベクターから5のリン酸基を除去した。DFDS混合物及びベクターの連結の後、その連結混合物を、アンピシリン耐性についての選択と共にgusA陰性大腸菌株である株KW1に形質転換した。これは、そのコロニーの80%が異なる青色コロニー強度を有する約 100のコロニーを生じた。このことは、これらのコロニーが予想されるヒートショックプロモーターフラグメントを有していることを示す。更なる分析のために20の青色コロニーをとった。
【0077】
これらのコロニーからのプラスミド調製物の制限分析は、それらがおおよそ正しい大きさの挿入物を有していることを示した。次にこれらのプラスミド調製物のうちの6つを LB436を形質転換するのに用いた(L.ラクチス染色体上のattB部位へのプラスミドの組込みのための必要な int遺伝子を供する第2のプラスミド pLB81を含むL.ラクチスMG1363の誘導体)。それら形質転換体の各々から、その構成物の、L.ラクチス染色体上のattB結合部位への予想される組込みを有するコロニーを単離した。それら構成物の組込みは、attB領域内の1つのプライマー及びpLB85i領域内の1つのプライマーを用いて、標準的なコロニー PCRにより確認した。
【0078】
次にそれらコロニーを、各々30℃及び37℃においてエリトロマイシン及び 100μg/ml X-gluc を供したGM17培地上で青色コロニーを形成する能力についてテストし、そしてそれらコロニーは、2つの温度において色の強度の明らかな差を示した。このことは、プロモーター活性が温度制御下であることを示す。次に、ヒートショックプロモーターからのgusA発現を、5つの選択されたヒートショック制御プロモーターについて、2つの温度において液体培養物中で測定した(図2を参照のこと)。
【0079】
それらコロニーは異なる活性を有し、30℃において広範囲のプロモーター活性をカバーし、そして5つのクローンのうち4つ(HP6を除く全て)は37℃においてより高いプロモーター活性を供した。このことは、プロモーターが、実際に、温度制御されたことを示す。興味あることに、プロモーターは、ほぼ同じ倍率だけ、即ち30℃から37℃の温度シフトにより 1.7〜2.3 倍だけ制御された。このことは、逆反復中のスペーサー(スペーサー1)がこれらの人工プロモーターの誘導の倍率を決定することに関して小さな重要性を有することを示す。
【0080】
我々は、蓄積したgusA活性を調べた。データは、プロモーターが温度制御されることを明らかに示すが、遺伝子発現を分析する当業者は、このことが、例えば温度のような外部パラメータの特定の変化によりもたらされるプロモーター活性の変化を観察するのをより困難にすることを認めるであろう。更に、ヒートショック制御プロモーターの活性は、図2に示すように、定常状態レベルを比べた時に見られるより温度のシフト後直ちに一時的に10倍高くなることを示す。プロモーター活性の変化をより注意深く観察するために、温度の乱れの前及びその後種々の時間においてタンパク質又はmRNA合成の比率を調べるべきである。それゆえ我々は、30℃から37℃への温度の変化の後、種々の時間において5つのクローンから RNAを単離し、標準的なノーザンブロッティング(Maniatisら、1983)によりgusA mRNA を視覚化し、何倍の個々のプロモータークローンが温度変化により誘導されたかを分析した。
【0081】
実施例3.
グラム陽性バクテリアバチルス・サブチリスは、所定範囲の異種タンパク質の生産のための工業用バイオリアクターとして広範囲に用いられる。それゆえ、本発明のランダムスペーサー法がこの生物のためのプロモーターライブラリーを作るのにも用いることができるか否かをテストすることに関心がある。バチルス・サブチリスのための共通配列は、大腸菌及びL.ラクチスのための共通配列に極めて類似しており、それゆえ、我々は、そのアプローチがバチルス・サブチリスにあてはまるか否かを、いくつかのCPプロモーターをこのバチルス・サブチリスのためのプロモータークローニングベクターにサブクローニングすることによりテストし、次に1)CPプロモーターがバチルス・サブチリス内で活性であるか否か及び2)更に、この生物内で、共通配列間のスペーサーがプロモーターの強さのために重要な役割を果たすか否かを問うことができよう。
【0082】
我々は、バチルス・サブチリス染色体上の amy座へのlacZへのプロモーター融合物の組込みのためにデザインしたプロモータークローニングベクターpDG268を用いることを選んだ。そのベクターは、アンピシリン及びネオマイシン耐性を与え、それは最初のクローニング目的のために大腸菌内で複製できるがバチルス・サブチリス内ではできないであろう。そのベクターの直鎖形態をバチルス・サブチリスに形質転換した時、それはバチルス・サブチリス染色体上の amy座に挿入されるであろう。それゆえ、この例は、染色体によりコードされた遺伝子の調節のためのランダムスペーサー法を通して作られたプロモーターの使用の例としても機能する。
【0083】
CPプロモーターは、実際は、バチルス・サブチリス内でも活性であり、そして更に、そのプロモーターの個々の強度は互いに極めて異なった。プロモーターライブラリーがバチルス・サブチリス内の広範囲のプロモーター活性もカバーしたという事実は、本ランダムスペーサー法がこの生物にもあてはまることを示す。
【0084】
実施例4.
実施例1のCPプロモーターを、L.ラクチスにおける使用のためにデザインしたが、大腸菌プロモーターの共通配列は、実施例1に供されるオリゴヌクレオチドの配列内に含まれる。更に、実施例1においてCPプロモーターライブラリーを作るために用いたベクターは、L.ラクチス及び大腸菌のためのシャトルベクターであり、これは、我々がグラム陰性バクテリア宿主大腸菌内のCPプロモーターの活性を分析するのを許容する。図3は、大腸菌内のCPプロモーターの33の活性を示す。明らかに、個々のCPプロモーターの活性も極めて異なり、そして同時に、プロモーターは広範囲の活性をカバーする。興味あることに、大腸菌及びL.ラクチスにおける個々のプロモーターの強さの間の相関はあまり強くなく;L.ラクチス内で強力なプロモーターであることが見出されたいくつかのプロモーターは大腸菌内で弱くその逆も成り立つことが見出された。
【0085】
プロモーターの活性は、β−ガラクトシダーゼ活性に関して、L.ラクチスにおいて見出された活性より一般にかなり低かった。これはおそらく、プロモータークローニングベクター pAK80がグラム陽性バクテリアL.ラクチスに用いるためにデザインされ、最適化され、それゆえ大腸菌内で翻訳効率が低くなり得るためである。それゆえ、我々は、CPプロモーターのうちの3つ(CP20, CP22及びCP25)を、β−ガラクトシダーゼをコードするプロモーターのないlacZ遺伝子の上流における大腸菌プロモーターをクローニングするためにデザインしたプロモータークローニングベクターpCB267にサブクローニングした。
【0086】
このプロモーターシステムにおいて、CPプロモーターは極めて有効なプロモーターであることが判明したが、それら3つのプロモーターの間の強さの相対的な差は保存された。これにより、CP25プロモーターは、大腸菌において用いるための周知の最も強力なプロモーターの中の1つであると考えられるハイブリッドプロモーターptacより 2.5倍高い活性を供した。それゆえ、これらのデータは、我々のアプローチの強さを更に証明する:本発明のランダムスペーサー法により得られた比較的少数のプロモータークローンを分析することにより、我々は、大腸菌及びL.ラクチスの両方において最も強力なプロモーターのいくつかにうまく到達した。
【0087】
実施例5.
グラム陰性種に属するバクテリア、シュードモナスは、それらの適用において、例えば汚染された土壌中の化学老廃物のバイオデグラデーションにおける適用のため、次第に重要になってきている。しかしながら、ここでも、遺伝子工学は適切なプロモーター及び発現システムの欠如により妨げられている。シュードモナスプロモーターに関する文献は、シュードモナスのための共通配列は大腸菌、L.ラクチス及びバチルス・サブチリスのものほど十分には規定されていないことを示す。
【0088】
これにより、−35領域においては、しばしば保存されたTTGR(R=A又はG)が見出されるが、−35共通配列の残りは異なるプロモーター間で種々である。−10共通配列はおそらくTATRATである。TTGRモチーフと−10配列との間のスペーシングは18〜19bpであり、それは大腸菌においてしばしば見出される16〜17bpスペーサーの等価物である。この生物中の生長プロモーターのための共通配列は大腸菌及びL.ラクチスのための共通配列に極めて近い。
【0089】
それゆえ我々は、プロモーターのないβ−ガラクトシダーゼ遺伝子を含むクローニングベクターにプロモーターをクローニングすることにより、シュードモナス・プチダにおいて、実施例1からの所定範囲のCPプロモーターをテストした。更に、CPプロモーターの活性は、広範囲のプロモーター活性にわたって強さにおいて異なった。これらの結果は、この種においても、本発明のランダムスペーサー法は、相対的に強力なプロモーター及び活性変化の小さなステップにおいてカバーされる広範囲のプロモーター活性の両方を作るのに用いることができるであろうことを示す。
【0090】
実施例6.
我々は、実施例5に記載される共通配列に基づくオリゴヌクレオチドをデザインし、実施例1に記載されるように多重クローニング部位を組み込んだ。次のオリゴヌクレオチドを合成した。
MCS-(N)8-TTGR-N19-TATRAT-(N)8-MCS
【0091】
そのオリゴヌクレオチドを、そのオリゴヌクレオチドの3′末端に相補的なプライマーを用いて二本鎖 DNAに変換し、そしてプロモータープローブベクター上のプロモーターのないβ−ガラクトシダーゼ遺伝子の上流にクローニングした。その連結混合物を、そのプラスミドが有する抗体物質耐性のための選択と共に、 X-galを含むプレート上でシュードモナス・プチダに直接形質転換した。これは、種々の青色強度の約 100のクローンを生じた。次に、30のクローンを上述のようにβ−ガラクトシダーゼ活性について分析した。これらの結果は、この種においても、本発明のランダムスペーサー法は、相対的に強力なプロモーター及び活性変化の小さなステップにおいてカバーされる広範囲のプロモーターの両方を作るのに用いることができるであろうことを示す。
【0092】
実施例7. サッカロマイセス・セレビシアエ(Saccharomyces cerevisiae)プロモーターライブラリーのための縮重したオリゴヌクレオチドのデザイン
5′端からはじめて:EcoRI制限部位(後のクローニングストラテジーに用いるためのもの、以下を参照のこと)、共通な UASGCN4p 、59bpスペーサー(スペーサー1)、共通なTATAAA配列(TATAボックス)、38bpスペーサー(スペーサー2)、23bpレプレッサー結合配列(転写が開始される領域でもある)、T1ボックスとARG8遺伝子の最初のコドンとの間のスペーサー配列(このスペーサーはARG8遺伝子の最初のコドン、 ATGも含むARG8遺伝子からのネイティブ配列である)、並びに最後にBamHI部位を含む 199bpオリゴヌクレオチドをデザインした。これは、β−ガラクトシダーゼリポーター遺伝子との枠内融合物を供するようにデザインされる(以下を参照のこと)。
【0093】
スペーサー1及びスペーサー2の配列はランダムに変化させ、そして合成された実際のオリゴヌクレオチド混合物は配列番号:3に示される以下の縮重した配列:
【0094】
【化6】
を有した。
【0095】
このオリゴヌクレオチド混合物を、実施例1に記載される 199bp縮重オリゴヌクレオチドの3′端の最後の23bpに相補的なオリゴヌクレオチドを用いて、二本鎖 DNAフラグメント(DSDF)に変換した。次に、それを次の通り、2つのプロモータークローニングベクター、pYLZ-2及びpYLZ-6(Hermannら、1992) のいずれかにクローニングした:DSDF混合物をEcoRI及びBamHIで二重消化し、そしてDSDFをベクター DNAに連結した。その連結混合物を、アンピシリン耐性についての選択と共に、実施例1に記載されるように、大腸菌内に形質転換した。プラスミド DNAを 500の個々のクローンから単離し、制限酵素EcoRI及びBamHIでの消化により予想されるプロモーターフラグメントの存在についてスクリーニングした。
【0096】
17のクローンが約 200bpのEcoRI−BamHI挿入物を有することが見い出された。これら17のクローンからのプラスミド DNAを、それらプラスミドが有するURA3マーカーについての選択と共にS.セレビシアエに形質転換し、そしてβ−ガラクトシダーゼの生産についてアッセイした。図4は、それら17のクローンについてのβ−ガラクトシダーゼの生じた活性を示す。全てのプロモーターが、プロモーターフラグメントの挿入のないクローニングベクター自体より高い活性を有する(pYLZ-2)。しかしながら、より重要なのは、この場合においても、それらクローンは活性変化の少いステップにおいて所定範囲のプロモーター活性をカバーすることである。
【0097】
配列分析は、上述の17のクローンがスペーサー1とスペーサー2との間に完全なTATAボックス(TATAAA)を有したが、それら17のクローンのうちの2つ、YP24及び YP435の各々は UASGCN4p 内に欠失を有することを示した。しかしながら、 YP435の活性は39ユニットであり、それは YP212で得た値に近い。これらのデータは、スペーサーのランダム配列のプロモーターの強さへの影響が UASGCN4p 結合部位の状態の影響より強いことを示唆する。
【0098】
上述の通り、人工イーストプロモーターは、2つの異なる制御特性で構築された。1つは、それらプロモーターは増殖培地中でアルギニンの存在により制御されることである。人工イーストプロモーターがアルギニンによっても制御されるか否かをテストするため、我々は、アルギニンあり及びなし(SD+2%グルコース;SD+2%グルコース+1g/lアルギニン)で、最小培地中でいくつかのクローンを増殖させた。図5は、これらの実験の結果を示す。クローンYP18, YP212, YP435は、実際に、アルギニンの存在により、各々5,8,15倍制御された。 YP183はアルギニンにより制御されず、そして配列分析は、このプロモータークローンがアルギニンレプレッサー結合部位内に13bp欠失を有することを確認した。
【0099】
我々は、それらプロモーターが増殖培地中の外部アミノ酸により制御されるか否かも、最小培地(SD+2%グルコース)及び豊富な培地(SD+2%グルコース−URA)中で、イーストプロモータークローンのいくつかのプロモーター活性を分析することによりテストした。図6は、これらの実験の結果を示す。実際、クローンYP18, YP212, YP435、及び YP183中に存在するプロモーターは、アミノ酸の存在により、2〜10倍に制御された。YP24は、このクローン上の UASGCN4p 部位内におきた誤りにより制御されなかった(上述の記載を参照のこと)。
アミノ酸及び特定のアルギニン制御に基づく結果は、本発明のランダムスペーサー法は、広範囲のプロモーター活性をカバーし、そして外部シグナルにより制御することができるプロモーターを作るのに用いることができることを証明する。本発明の制御態様は、アミノ酸及びアルギニン制御により例示されるが、これらに限られない。
【0100】
引用文献
Crabeel, M., de Rijcke, M., Seneca, S., Heimberg, H., Pfeiffer, I., and Matisova, A., 1995. Further definition of the sequence and position requirements of the arginine control element that mediates repression and induction by arginine in Saccharomyces cerevisiae. Yeast 11: 1367-1380
de Vos, W.M., and Simons, G., 1994. Gene cloning and expression systems in lactococci, p.52-105. In M.J.Gasson and W.M. de Vos (eds.), Genetics and biotechnology of lactic acid bacteria. Blackie Academic & Professional, Glasgow, United Kingdom.
【0101】
Gasson, M.J. 1983. Plasmid complements of Streptococcus lactis NCDO 712 and other lactic streptococci after protoplast-induced curing. J.Bacteriol., 154, 1-9.Guarents, L., 1983, Yeast promoters and lacZ fusions designed to study expression of cloned genes in yeast. Methods Enzymol. 101, 181-191.
Hermann, H., Hacker, U., Bandlow, W., and Magdolen, V., 1992. pYLZ vectors: Saccharomyces cerevisiae/Escherichia coli shuttle plasmids to analyze yeast promoters. Gene 119: 137-41.
【0102】
Hinnebusch, A.G., 1992. General and Pathway-specific Regulatory Mechanisms Controlling the Synthesis of Amino Acid Biosynthetic Enzymes in Saccharomyces cerevisiae, p.319-414. In E.W.Jones, J.R.Pringle and J.R.Broach (eds.), The Molecular and Cellular Biology of the Yeast Saccharomyces: Gene Expression, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
Holo, H., and Nes, I.F., 1989. High frequency transformation, by electroporation, of Lactococcus lactis subsp. cremoris grown with glycine in osmotically stabilized media. Appl.Environ.Microbiol., 55, 3119-3123.
【0103】
Israelsen, H. 1995. Cloning and partial characterization of regulated promoters from Lactococcus lactis Tn917-lacZ integrants with the new promoter probe vector, pAK80. Appl.Environ.Microbiol., 61, 2540-2547.
Jensen, P.R., Westerhoff, H.V., and Michelsen, O., 1993. Excess capacity of H + -ATPase and inverse respiratory control in Escherichia coli. EMBO J., 12, 1277-1282. Kacser, H. and Burns, J.A. 1973. The control of flux. Symp.Soc.Exp.Biol., 27: 65-104.
【0104】
Maniatis, T., Fritsch, E.F., and Sambrook, J., 1982. Molecular cloning. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
Miller, J.H., 1972. Experiments in molecular genetics, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
Nilsson, D., and Johansen, E., 1994. A conserved sequence in tRNA and rRNA promoters of Lactococcus lactis. Biochim.Biophys.Acta, 1219: 141-144.
【0105】
Oliver, S.G., and Warmington, J.R., 1989. Transcription. In The yeasts, volume 3, Rose and Harrison (Eds.). Academic Press, London, 117-160.
Schaaff, I., Heinisch, J., Zimmermann, F.K., 1989. Overproduction of glycolytic enzymes in yeast. Yeast, 5: 285-290.
van Asseldonk, M., Simons, A., Visser, H., de Vos, W.M., and Simons, G., 1993. Cloning, nucleotide sequence, and regulatory analysis of the Lactococcus lactis dnaJ gene. J.Bacteriol. 175, 1637-1644.
【図面の簡単な説明】
【0106】
【図1】図1は、実施例1からのL.ラクチス(L. lactis )のための人工プロモーターのライブラリーを示す。
【図2】図2は、実施例2からのL.ラクチスのための人工のヒートショック調節プロモーターのライブラリーを示す。
【図3】図3は、大腸菌内の活性についてアッセイした実施例1からの人工プロモーターのライブラリーを示す。
【0107】
【図4】図4は、実施例7からのS.セレビシアエ(S. cerevisiae )のための人工プロモーターのライブラリーを示す。
【図5】図5は、実施例7からの外来アルギニンによる人工イーストプロモーターの制御を示す。
【図6】図6は、実施例7からの外来アミノ酸による人工イーストプロモーターの制御を示す。
【特許請求の範囲】
【請求項1】
選択された生物又は生物の群のための人工のプロモーターライブラリーであって、二本鎖 DNAフラグメントのセンス鎖が、前記生物もしくは生物の群からの有効なプロモーターの少くとも2の共通配列又は該共通配列各々の少くとも半分を含む一部分と、ヌクレオベースA,T,C及びGの中から無作偽に少くとも7のヌクレオチドが選択された不定の長さの周囲の又は介在するヌクレオチド配列(スペーサー)と、を含む二本鎖 DNAフラグメントの混合物を含み、但し、以前から周知のプロモーター配列及び天然のソースから単離されたプロモーター配列を含まない、人工のプロモーターライブラリー。
【請求項2】
前記スペーサー配列中の少くとも10、好ましくは少くとも12、より好ましくは少くとも14のヌクレオチドがヌクレオベースA,T,C及びGの中から無作偽に選択されることを特徴とする請求項1に記載の人工のプロモーターライブラリー。
【請求項3】
前記二本鎖 DNAフラグメントのセンス鎖が、前記ライブラリーのプロモーターに特定の調節特性を与える調節 DNA配列を含むことを特徴とする請求項1又は2に記載の人工のプロモーターライブラリー。
【請求項4】
前記特定の調節特性が、増殖条件の変化による活性化であることを特徴とする請求項3に記載の人工のプロモーターライブラリー。
【請求項5】
前記二本鎖 DNAフラグメントが、両端の一方又は両方に加えられた制限エンドヌクレアーゼのための1又は複数の認識部位を含む配列を有する請求項1〜4のいずれか一に記載の人工のプロモーターライブラリー。
【請求項6】
前記二本鎖 DNAフラグメントが、両端の一方又は両方に加えられた制限エンドヌクレアーゼのための多重認識部位を特定する配列を有することを特徴とする請求項5に記載の人工のプロモーターライブラリー。
【請求項7】
前記選択された生物又は生物の群が、原核生物からなる群から選択されることを特徴とする請求項1〜6のいずれか一に記載の人工のプロモーターライブラリー。
【請求項8】
前記原核微生物が、ラクトコッカス(Lactococcus )、ストレプトコッカス(Streptococcus )、エンテロコッカス(Enterococcus)、ラクトバチルス(Lactobacillus )、ロイコノストック(Leuconostoc )、エスケリキア(Escherichia )、バチルス(Bacillus)及びシュードモナス(Pseudomonas )属に属するバクテリアからなる群から選択されることを特徴とする請求項7に記載の人工のプロモーターライブラリー。
【請求項9】
前記原核微生物が、ラクトコッカス・ラクチス(Lactococcus lactis)、ストレプトコッカス・サーモフィルス(Streptococcus thermophilus)、エスケリキア・コリ(Escherichia coli)、バチルス・サブチリス(Bacillus subtilis )及びシュードモナス・プチダ(Pseudomonas putida)からなる群から選択されることを特徴とする請求項8に記載の人工のプロモーターライブラリー。
【請求項10】
前記共通配列が、−10シグナル(−12〜−7):TATAAT及び該−10シグナルの上流の少くとも1のアクティベータータンパク質結合部位又はその各々の少くとも3の保存されたヌクレオチドを含む一部分を含むことを特徴とする請求項7〜9のいずれか一に記載の人工のプロモーターライブラリー。
【請求項11】
前記共通配列が、−35シグナル(−35〜−30):TTGACA及び−10シグナル(−12〜−7):TATAAT又はその各々の少くとも3の保存されたヌクレオチドを含む両方の一部分を含むことを特徴とする請求項7〜9のいずれか一に記載の人工のプロモーターライブラリー。
【請求項12】
前記共通配列が、−35シグナルの4〜6の保存されたヌクレオチド及び−10シグナルの4〜6の保存されたヌクレオチド、好ましくは各々5又は6、より好ましくは各々全部で6のヌクレオチドを含むことを特徴とする請求項11に記載の人工のプロモーターライブラリー。
【請求項13】
前記共通配列が、介在する保存されたモチーフを更に含むことを特徴とする請求項10〜12のいずれか一に記載の人工のプロモーターライブラリー。
【請求項14】
前記介在する保存されたモチーフが、保存されたモチーフ−44〜−41:AGTT、−40〜−36:TATTC 、−15〜−14:TG、及び+1〜+8:GTACTGTTからなる群から選択されることを特徴とする請求項13に記載の人工のプロモーターライブラリー。
【請求項15】
前記−35シグナルと−10シグナルとの間のスペーサーの長さが14〜23bp、好ましくは16〜18bp、より好ましくは17bpであることを特徴とする請求項11〜14のいずれか一に記載の人工のプロモーターライブラリー。
【請求項16】
前記二本鎖 DNAフラグメントのセンス鎖が、前記共通配列及びスペーサー長の少数の変異を伴う配列番号:1に記載の縮重した配列を有することを特徴とする請求項11〜15のいずれか一に記載の人工のプロモーターライブラリー。
【請求項17】
上流温度調節 DNA配列を含む前記二本鎖 DNAフラグメントのセンス鎖が、前記共通配列及びスペーサー長の少数の変異を伴う配列番号:2に示される縮重した配列を有することを特徴とする請求項11〜15のいずれか一に記載の人工のプロモーターライブラリー。
【請求項18】
前記選択された生物又は生物の群が、真核生物からなる群から選択されることを特徴とする請求項1〜6のいずれか一に記載の人工のプロモーターライブラリー。
【請求項19】
前記真核生物又は生物の群が、真核微生物からなる群から選択されることを特徴とする請求項18に記載の人工のプロモーターライブラリー。
【請求項20】
前記真核微生物又は微生物の群が、イースト、他の真菌及び哺乳動物細胞系からなる群から選択されることを特徴とする請求項19に記載の人工のプロモーターライブラリー。
【請求項21】
前記真核微生物が、種サッカロマイセス・セレビシアエのイーストであることを特徴とする請求項20に記載の人工のプロモーターライブラリー。
【請求項22】
前記共通配列が、TATAボックス及び少くとも1の上流活性化配列(UAS) を含むことを特徴とする請求項18〜21のいずれか一に記載の人工のプロモーターライブラリー。
【請求項23】
前記共通配列が、サッカロマイセス・セレビシアエ内で機能する転写開始シグナル(TIボックス)を更に含むことを特徴とする請求項22に記載の人工のプロモーターライブラリー。
【請求項24】
前記共通配列が、アルギニンレセプターのための結合部位argRとしても機能する、TATAボックス:TATAAA、 UASGCN4p :TGACTCA 、及びTIボックス:CTCTTAAGTGCAAGTGACTGCGA を含むことを特徴とする請求項23に記載の人工のプロモーターライブラリー。
【請求項25】
TATAボックスと第1の UASとの間及び存在するなら該第1の UASと次の UASとの間のスペーサーの長さが、1000bpまで、好ましくは10〜500bp 、より好ましくは15〜150bp であることを特徴とする請求項22〜24のいずれか一に記載の人工のプロモーターライブラリー。
【請求項26】
前記二本鎖 DNAフラグメントのセンス鎖が、前記共通配列及びスペーサー長の少数の変異を伴い配列番号:3に示される縮重した配列を有することを特徴とする請求項22〜25のいずれか一に記載の人工のプロモーターライブラリー。
【請求項27】
請求項1〜26のいずれか一に記載の人工のプロモーターから選択された人工のプロモーター。
【請求項28】
配列番号:5〜58からなる群から選択された DNA配列を含む請求項27に記載のプロモーター。
【請求項29】
請求項27又は28に記載の人工のプロモーター由来の人工のプロモーター。
【請求項30】
選択された生物又は生物の群のための人工のプロモーターライブラリーを作製する方法であって、該生物又は生物の群からの有効なプロモーターの少くとも2の共通配列を選択し;該共通配列又はその各々の少くとも半分を含む一部分と、少くとも7のヌクレオチドがヌクレオベースA,T,C及びGの中から無作偽に選択された不定の長さの周囲の又は介在するヌクレオチド配列(スペーサー)と、を含む一本鎖 DNA配列の混合物を合成し;そして該一本鎖 DNA配列を、第2鎖合成により二本鎖 DNAフラグメントに変換することを含む方法。
【請求項31】
前記スペーサー配列中の少くとも10、好ましくは少くとも12、より好ましくは少くとも14のヌクレオチドがヌクレオベースA,T,C及びGの中から無作偽に選択されることを特徴とする請求項30に記載の方法。
【請求項32】
前記一本鎖 DNA配列が、前記ライブラリーのプロモーターに特定の調節特性を与える調節 DNA配列を含むことを特徴とする請求項30又は31に記載の方法。
【請求項33】
前記特定の調節特性が、増殖条件の変化による活性化であることを特徴とする請求項32に記載の方法。
【請求項34】
制限エンドヌクレアーゼのための1又は複数の認識部位を特定する配列が前記合成において前記一本鎖 DNA配列の両端の一方もしくは両方に加えられ、又は前記制限部位を含むリンカーが前記二本鎖 DNAフラグメントの両端の一方もしくは両方に連結されることを特徴とする請求項30〜34のいずれか一に記載の方法。
【請求項35】
制限エンドヌクレアーゼのための複数の認識部位を特定する配列が前記合成において前記一本鎖 DNA配列の両端の一方もしくは両方に加えられ、又は前記多重クローニング部位(MCS) を含むリンカーが前記二本鎖 DNAフラグメントの両端の一方もしくは両方に連結されることを特徴とする請求項34に記載の方法。
【請求項36】
前記選択された生物又は生物の群が、原核生物からなる群から選択されることを特徴とする請求項30〜35のいずれか一に記載の方法。
【請求項37】
前記原核微生物が、ラクトコッカス(Lactococcus )、エスケリキア(Escherichia )、バチルス(Bacillus)及びシュードモナス(Pseudomonas )属に属するバクテリアからなる群から選択されることを特徴とする請求項36に記載の方法。
【請求項38】
前記原核微生物が、ラクトコッカス・ラクチス(Lactococcus lactis)、エスケリキア・コリ(Escherichia coli)、バチルス・サブチリス(Bacillus subtilis )及びシュードモナス・プチダ(Pseudomonas putida)からなる群から選択されることを特徴とする請求項37に記載の方法。
【請求項39】
前記共通配列が、−10シグナル(−12〜−7):TATAAT及び該−10シグナルの上流の少くとも1のアクティベータータンパク質結合部位又はその各々の少くとも3の保存されたヌクレオチドを含む一部分を含むことを特徴とする請求項36〜38のいずれか一に記載の方法。
【請求項40】
前記共通配列が、−35シグナル(−35〜−30):TTGACA及び−10シグナル(−12〜−7):TATAAT又はその各々の少くとも3の保存されたヌクレオチドを含む両方の一部分を含むことを特徴とする請求項36〜38のいずれか一に記載の方法。
【請求項41】
前記共通配列が、−35シグナルの4〜6の保存されたヌクレオチド及び−10シグナルの4〜6の保存されたヌクレオチド、好ましくは各々5又は6、より好ましくは各々全部で6のヌクレオチドを含むことを特徴とする請求項40に記載の方法。
【請求項42】
前記共通配列が、介在する保存されたモチーフを更に含むことを特徴とする請求項39〜41のいずれか一に記載の方法。
【請求項43】
前記介在する保存されたモチーフが、保存されたモチーフ−44〜−41:AGTT、−40〜−36:TATTC 、−15〜−14:TG、及び+1〜+8:GTACTGTTからなる群から選択されることを特徴とする請求項42に記載の方法。
【請求項44】
前記−35シグナルと−10シグナルとの間のスペーサーの長さが14〜23bp、好ましくは16〜18bp、より好ましくは17bpであることを特徴とする請求項40〜43のいずれか一に記載の方法。
【請求項45】
前記一本鎖 DNA配列の混合物が、前記共通配列及びスペーサー長の少数の変異を伴う配列番号:1に記載の縮重した配列を有することを特徴とする請求項40〜44のいずれか一に記載の方法。
【請求項46】
上流温度調節 DNA配列を含む前記一本鎖 DNA配列の混合物が、前記共通配列及びスペーサー長の少数の変異を伴う配列番号:2に示される縮重した配列を有することを特徴とする請求項40〜42のいずれか一に記載の方法。
【請求項47】
前記選択された生物又は生物の群が、真核生物からなる群から選択されることを特徴とする請求項30〜35のいずれか一に記載の方法。
【請求項48】
前記真核生物又は生物の群が、真核微生物からなる群から選択されることを特徴とする請求項47に記載の方法。
【請求項49】
前記真核微生物又は微生物の群が、イースト、他の真菌及び哺乳動物細胞系からなる群から選択されることを特徴とする請求項48に記載の方法。
【請求項50】
前記真核微生物が、種サッカロマイセス・セレビシアエのイーストであることを特徴とする請求項49に記載の方法。
【請求項51】
前記共通配列が、TATAボックス及び少くとも1の上流活性化配列(UAS) を含むことを特徴とする請求項47〜50のいずれか一に記載の方法。
【請求項52】
前記共通配列が、サッカロマイセス・セレビシアエ内で機能する転写開始シグナル(TIボックス)を更に含むことを特徴とする請求項51に記載の方法。
【請求項53】
前記共通配列が、アルギニンレセプターのための結合部位argRとしても機能する、TATAボックス:TATAAA、 UASGCN4p :TGACTCA 、及びTIボックス:CTCTTAAGTGCAAGTGACTGCGA を含むことを特徴とする請求項52に記載の方法。
【請求項54】
TATAボックスと第1の UASとの間及び存在するなら該第1の UASと次の UASとの間のスペーサーの長さが、1000bpまで、好ましくは10〜500bp 、より好ましくは15〜150bp であることを特徴とする請求項51〜53のいずれか一に記載の方法。
【請求項55】
前記二本鎖 DNAフラグメントのセンス鎖が、前記共通配列及びスペーサー長の少数の変異を伴い配列番号:3に示される縮重した配列を有することを特徴とする請求項51〜54のいずれか一に記載の方法。
【請求項56】
微生物内の遺伝子の発現を最適化する方法であって、
a)請求項1〜26のいずれか一に記載の人工のプロモーターライブラリー又は請求項30〜55のいずれか一に記載の方法により作製された人工のプロモーターライブラリーから、活性変化の比較的少ないステップにおいて所定範囲のプロモーター活性をカバーする一セットのプロモーターを選択し;
b)該プロモーターのセットを、前記生物内に、各々のクローン内で前記遺伝子を前記セットからの少なくとも1のプロモーターの制御下におくようにクローニングし、
c)前記選択されたクローンを増殖させ、そしてそれを、産物形成の最適な流れを示すものを見出すためにスクリーニングすること
を含む方法。
【請求項57】
前記選択された生物が、原核及び真核微生物からなる群から選択されることを特徴とする請求項56に記載の方法。
【請求項1】
選択された生物又は生物の群のための人工のプロモーターライブラリーであって、二本鎖 DNAフラグメントのセンス鎖が、前記生物もしくは生物の群からの有効なプロモーターの少くとも2の共通配列又は該共通配列各々の少くとも半分を含む一部分と、ヌクレオベースA,T,C及びGの中から無作偽に少くとも7のヌクレオチドが選択された不定の長さの周囲の又は介在するヌクレオチド配列(スペーサー)と、を含む二本鎖 DNAフラグメントの混合物を含み、但し、以前から周知のプロモーター配列及び天然のソースから単離されたプロモーター配列を含まない、人工のプロモーターライブラリー。
【請求項2】
前記スペーサー配列中の少くとも10、好ましくは少くとも12、より好ましくは少くとも14のヌクレオチドがヌクレオベースA,T,C及びGの中から無作偽に選択されることを特徴とする請求項1に記載の人工のプロモーターライブラリー。
【請求項3】
前記二本鎖 DNAフラグメントのセンス鎖が、前記ライブラリーのプロモーターに特定の調節特性を与える調節 DNA配列を含むことを特徴とする請求項1又は2に記載の人工のプロモーターライブラリー。
【請求項4】
前記特定の調節特性が、増殖条件の変化による活性化であることを特徴とする請求項3に記載の人工のプロモーターライブラリー。
【請求項5】
前記二本鎖 DNAフラグメントが、両端の一方又は両方に加えられた制限エンドヌクレアーゼのための1又は複数の認識部位を含む配列を有する請求項1〜4のいずれか一に記載の人工のプロモーターライブラリー。
【請求項6】
前記二本鎖 DNAフラグメントが、両端の一方又は両方に加えられた制限エンドヌクレアーゼのための多重認識部位を特定する配列を有することを特徴とする請求項5に記載の人工のプロモーターライブラリー。
【請求項7】
前記選択された生物又は生物の群が、原核生物からなる群から選択されることを特徴とする請求項1〜6のいずれか一に記載の人工のプロモーターライブラリー。
【請求項8】
前記原核微生物が、ラクトコッカス(Lactococcus )、ストレプトコッカス(Streptococcus )、エンテロコッカス(Enterococcus)、ラクトバチルス(Lactobacillus )、ロイコノストック(Leuconostoc )、エスケリキア(Escherichia )、バチルス(Bacillus)及びシュードモナス(Pseudomonas )属に属するバクテリアからなる群から選択されることを特徴とする請求項7に記載の人工のプロモーターライブラリー。
【請求項9】
前記原核微生物が、ラクトコッカス・ラクチス(Lactococcus lactis)、ストレプトコッカス・サーモフィルス(Streptococcus thermophilus)、エスケリキア・コリ(Escherichia coli)、バチルス・サブチリス(Bacillus subtilis )及びシュードモナス・プチダ(Pseudomonas putida)からなる群から選択されることを特徴とする請求項8に記載の人工のプロモーターライブラリー。
【請求項10】
前記共通配列が、−10シグナル(−12〜−7):TATAAT及び該−10シグナルの上流の少くとも1のアクティベータータンパク質結合部位又はその各々の少くとも3の保存されたヌクレオチドを含む一部分を含むことを特徴とする請求項7〜9のいずれか一に記載の人工のプロモーターライブラリー。
【請求項11】
前記共通配列が、−35シグナル(−35〜−30):TTGACA及び−10シグナル(−12〜−7):TATAAT又はその各々の少くとも3の保存されたヌクレオチドを含む両方の一部分を含むことを特徴とする請求項7〜9のいずれか一に記載の人工のプロモーターライブラリー。
【請求項12】
前記共通配列が、−35シグナルの4〜6の保存されたヌクレオチド及び−10シグナルの4〜6の保存されたヌクレオチド、好ましくは各々5又は6、より好ましくは各々全部で6のヌクレオチドを含むことを特徴とする請求項11に記載の人工のプロモーターライブラリー。
【請求項13】
前記共通配列が、介在する保存されたモチーフを更に含むことを特徴とする請求項10〜12のいずれか一に記載の人工のプロモーターライブラリー。
【請求項14】
前記介在する保存されたモチーフが、保存されたモチーフ−44〜−41:AGTT、−40〜−36:TATTC 、−15〜−14:TG、及び+1〜+8:GTACTGTTからなる群から選択されることを特徴とする請求項13に記載の人工のプロモーターライブラリー。
【請求項15】
前記−35シグナルと−10シグナルとの間のスペーサーの長さが14〜23bp、好ましくは16〜18bp、より好ましくは17bpであることを特徴とする請求項11〜14のいずれか一に記載の人工のプロモーターライブラリー。
【請求項16】
前記二本鎖 DNAフラグメントのセンス鎖が、前記共通配列及びスペーサー長の少数の変異を伴う配列番号:1に記載の縮重した配列を有することを特徴とする請求項11〜15のいずれか一に記載の人工のプロモーターライブラリー。
【請求項17】
上流温度調節 DNA配列を含む前記二本鎖 DNAフラグメントのセンス鎖が、前記共通配列及びスペーサー長の少数の変異を伴う配列番号:2に示される縮重した配列を有することを特徴とする請求項11〜15のいずれか一に記載の人工のプロモーターライブラリー。
【請求項18】
前記選択された生物又は生物の群が、真核生物からなる群から選択されることを特徴とする請求項1〜6のいずれか一に記載の人工のプロモーターライブラリー。
【請求項19】
前記真核生物又は生物の群が、真核微生物からなる群から選択されることを特徴とする請求項18に記載の人工のプロモーターライブラリー。
【請求項20】
前記真核微生物又は微生物の群が、イースト、他の真菌及び哺乳動物細胞系からなる群から選択されることを特徴とする請求項19に記載の人工のプロモーターライブラリー。
【請求項21】
前記真核微生物が、種サッカロマイセス・セレビシアエのイーストであることを特徴とする請求項20に記載の人工のプロモーターライブラリー。
【請求項22】
前記共通配列が、TATAボックス及び少くとも1の上流活性化配列(UAS) を含むことを特徴とする請求項18〜21のいずれか一に記載の人工のプロモーターライブラリー。
【請求項23】
前記共通配列が、サッカロマイセス・セレビシアエ内で機能する転写開始シグナル(TIボックス)を更に含むことを特徴とする請求項22に記載の人工のプロモーターライブラリー。
【請求項24】
前記共通配列が、アルギニンレセプターのための結合部位argRとしても機能する、TATAボックス:TATAAA、 UASGCN4p :TGACTCA 、及びTIボックス:CTCTTAAGTGCAAGTGACTGCGA を含むことを特徴とする請求項23に記載の人工のプロモーターライブラリー。
【請求項25】
TATAボックスと第1の UASとの間及び存在するなら該第1の UASと次の UASとの間のスペーサーの長さが、1000bpまで、好ましくは10〜500bp 、より好ましくは15〜150bp であることを特徴とする請求項22〜24のいずれか一に記載の人工のプロモーターライブラリー。
【請求項26】
前記二本鎖 DNAフラグメントのセンス鎖が、前記共通配列及びスペーサー長の少数の変異を伴い配列番号:3に示される縮重した配列を有することを特徴とする請求項22〜25のいずれか一に記載の人工のプロモーターライブラリー。
【請求項27】
請求項1〜26のいずれか一に記載の人工のプロモーターから選択された人工のプロモーター。
【請求項28】
配列番号:5〜58からなる群から選択された DNA配列を含む請求項27に記載のプロモーター。
【請求項29】
請求項27又は28に記載の人工のプロモーター由来の人工のプロモーター。
【請求項30】
選択された生物又は生物の群のための人工のプロモーターライブラリーを作製する方法であって、該生物又は生物の群からの有効なプロモーターの少くとも2の共通配列を選択し;該共通配列又はその各々の少くとも半分を含む一部分と、少くとも7のヌクレオチドがヌクレオベースA,T,C及びGの中から無作偽に選択された不定の長さの周囲の又は介在するヌクレオチド配列(スペーサー)と、を含む一本鎖 DNA配列の混合物を合成し;そして該一本鎖 DNA配列を、第2鎖合成により二本鎖 DNAフラグメントに変換することを含む方法。
【請求項31】
前記スペーサー配列中の少くとも10、好ましくは少くとも12、より好ましくは少くとも14のヌクレオチドがヌクレオベースA,T,C及びGの中から無作偽に選択されることを特徴とする請求項30に記載の方法。
【請求項32】
前記一本鎖 DNA配列が、前記ライブラリーのプロモーターに特定の調節特性を与える調節 DNA配列を含むことを特徴とする請求項30又は31に記載の方法。
【請求項33】
前記特定の調節特性が、増殖条件の変化による活性化であることを特徴とする請求項32に記載の方法。
【請求項34】
制限エンドヌクレアーゼのための1又は複数の認識部位を特定する配列が前記合成において前記一本鎖 DNA配列の両端の一方もしくは両方に加えられ、又は前記制限部位を含むリンカーが前記二本鎖 DNAフラグメントの両端の一方もしくは両方に連結されることを特徴とする請求項30〜34のいずれか一に記載の方法。
【請求項35】
制限エンドヌクレアーゼのための複数の認識部位を特定する配列が前記合成において前記一本鎖 DNA配列の両端の一方もしくは両方に加えられ、又は前記多重クローニング部位(MCS) を含むリンカーが前記二本鎖 DNAフラグメントの両端の一方もしくは両方に連結されることを特徴とする請求項34に記載の方法。
【請求項36】
前記選択された生物又は生物の群が、原核生物からなる群から選択されることを特徴とする請求項30〜35のいずれか一に記載の方法。
【請求項37】
前記原核微生物が、ラクトコッカス(Lactococcus )、エスケリキア(Escherichia )、バチルス(Bacillus)及びシュードモナス(Pseudomonas )属に属するバクテリアからなる群から選択されることを特徴とする請求項36に記載の方法。
【請求項38】
前記原核微生物が、ラクトコッカス・ラクチス(Lactococcus lactis)、エスケリキア・コリ(Escherichia coli)、バチルス・サブチリス(Bacillus subtilis )及びシュードモナス・プチダ(Pseudomonas putida)からなる群から選択されることを特徴とする請求項37に記載の方法。
【請求項39】
前記共通配列が、−10シグナル(−12〜−7):TATAAT及び該−10シグナルの上流の少くとも1のアクティベータータンパク質結合部位又はその各々の少くとも3の保存されたヌクレオチドを含む一部分を含むことを特徴とする請求項36〜38のいずれか一に記載の方法。
【請求項40】
前記共通配列が、−35シグナル(−35〜−30):TTGACA及び−10シグナル(−12〜−7):TATAAT又はその各々の少くとも3の保存されたヌクレオチドを含む両方の一部分を含むことを特徴とする請求項36〜38のいずれか一に記載の方法。
【請求項41】
前記共通配列が、−35シグナルの4〜6の保存されたヌクレオチド及び−10シグナルの4〜6の保存されたヌクレオチド、好ましくは各々5又は6、より好ましくは各々全部で6のヌクレオチドを含むことを特徴とする請求項40に記載の方法。
【請求項42】
前記共通配列が、介在する保存されたモチーフを更に含むことを特徴とする請求項39〜41のいずれか一に記載の方法。
【請求項43】
前記介在する保存されたモチーフが、保存されたモチーフ−44〜−41:AGTT、−40〜−36:TATTC 、−15〜−14:TG、及び+1〜+8:GTACTGTTからなる群から選択されることを特徴とする請求項42に記載の方法。
【請求項44】
前記−35シグナルと−10シグナルとの間のスペーサーの長さが14〜23bp、好ましくは16〜18bp、より好ましくは17bpであることを特徴とする請求項40〜43のいずれか一に記載の方法。
【請求項45】
前記一本鎖 DNA配列の混合物が、前記共通配列及びスペーサー長の少数の変異を伴う配列番号:1に記載の縮重した配列を有することを特徴とする請求項40〜44のいずれか一に記載の方法。
【請求項46】
上流温度調節 DNA配列を含む前記一本鎖 DNA配列の混合物が、前記共通配列及びスペーサー長の少数の変異を伴う配列番号:2に示される縮重した配列を有することを特徴とする請求項40〜42のいずれか一に記載の方法。
【請求項47】
前記選択された生物又は生物の群が、真核生物からなる群から選択されることを特徴とする請求項30〜35のいずれか一に記載の方法。
【請求項48】
前記真核生物又は生物の群が、真核微生物からなる群から選択されることを特徴とする請求項47に記載の方法。
【請求項49】
前記真核微生物又は微生物の群が、イースト、他の真菌及び哺乳動物細胞系からなる群から選択されることを特徴とする請求項48に記載の方法。
【請求項50】
前記真核微生物が、種サッカロマイセス・セレビシアエのイーストであることを特徴とする請求項49に記載の方法。
【請求項51】
前記共通配列が、TATAボックス及び少くとも1の上流活性化配列(UAS) を含むことを特徴とする請求項47〜50のいずれか一に記載の方法。
【請求項52】
前記共通配列が、サッカロマイセス・セレビシアエ内で機能する転写開始シグナル(TIボックス)を更に含むことを特徴とする請求項51に記載の方法。
【請求項53】
前記共通配列が、アルギニンレセプターのための結合部位argRとしても機能する、TATAボックス:TATAAA、 UASGCN4p :TGACTCA 、及びTIボックス:CTCTTAAGTGCAAGTGACTGCGA を含むことを特徴とする請求項52に記載の方法。
【請求項54】
TATAボックスと第1の UASとの間及び存在するなら該第1の UASと次の UASとの間のスペーサーの長さが、1000bpまで、好ましくは10〜500bp 、より好ましくは15〜150bp であることを特徴とする請求項51〜53のいずれか一に記載の方法。
【請求項55】
前記二本鎖 DNAフラグメントのセンス鎖が、前記共通配列及びスペーサー長の少数の変異を伴い配列番号:3に示される縮重した配列を有することを特徴とする請求項51〜54のいずれか一に記載の方法。
【請求項56】
微生物内の遺伝子の発現を最適化する方法であって、
a)請求項1〜26のいずれか一に記載の人工のプロモーターライブラリー又は請求項30〜55のいずれか一に記載の方法により作製された人工のプロモーターライブラリーから、活性変化の比較的少ないステップにおいて所定範囲のプロモーター活性をカバーする一セットのプロモーターを選択し;
b)該プロモーターのセットを、前記生物内に、各々のクローン内で前記遺伝子を前記セットからの少なくとも1のプロモーターの制御下におくようにクローニングし、
c)前記選択されたクローンを増殖させ、そしてそれを、産物形成の最適な流れを示すものを見出すためにスクリーニングすること
を含む方法。
【請求項57】
前記選択された生物が、原核及び真核微生物からなる群から選択されることを特徴とする請求項56に記載の方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2009−22293(P2009−22293A)
【公開日】平成21年2月5日(2009.2.5)
【国際特許分類】
【外国語出願】
【出願番号】特願2008−232486(P2008−232486)
【出願日】平成20年9月10日(2008.9.10)
【分割の表示】特願平10−510287の分割
【原出願日】平成9年8月25日(1997.8.25)
【出願人】(508242931)
【Fターム(参考)】
【公開日】平成21年2月5日(2009.2.5)
【国際特許分類】
【出願番号】特願2008−232486(P2008−232486)
【出願日】平成20年9月10日(2008.9.10)
【分割の表示】特願平10−510287の分割
【原出願日】平成9年8月25日(1997.8.25)
【出願人】(508242931)
【Fターム(参考)】
[ Back to top ]