
選択マーカー遺伝子
本発明は、本明細書においてDSM-2と呼ばれる新規の遺伝子に関する。この遺伝子は、ストレプトマイセス・セリカラーA3において同定された。DSM-2タンパク質は、PATおよびBARと遠縁である。本発明はまた、DSM-2タンパク質をコードする植物に最適化された遺伝子を提供し、DSM-2は、除草剤グルホシネートおよびビアロホスに対する耐性を植物および植物細胞に賦与するためのトランスジェニック形質として使用され得る。本発明の遺伝子の1つの好ましい使用は、選択マーカーとしての使用である。細菌系における選択マーカーとしてのこの遺伝子の使用は、植物の形質転換の効率を上昇させることができる。唯一の選択マーカーとしてのDSM-2の使用により、クローニング中のさらなる(アンピシリン耐性などの)医療用抗生物質マーカーが不要となる。本発明によれば、様々なその他の使用も可能である。
【発明の詳細な説明】
【背景技術】
【0001】
発明の背景
選択マーカーとは、同定され追跡され得る検出可能な遺伝形質またはDNAのセグメントである。マーカー遺伝子は、典型的には、標的遺伝子とも呼ばれるもう一つの遺伝子のためのフラッグとして役立つ。マーカー遺伝子は、典型的には、標的細胞を形質転換するために使用される標的遺伝子と共に使用される。標的遺伝子を遺伝的に受容する標的細胞は、マーカー遺伝子も発現する細胞を選択することにより同定され得る。2つの遺伝子(マーカー遺伝子および標的遺伝子)が遺伝連鎖し、通常一緒に遺伝されるよう、マーカー遺伝子は標的遺伝子に十分に近く存在する。選択マーカーの現在の標準物は、ホスフィノトリシンアセチルトランスフェラーゼと呼ばれる酵素をコードする「pat」遺伝子である。
【0002】
グルタミンシンセターゼ(「GS」)は、多くの植物において、植物細胞の発達および生命にとって必須の酵素である。GSはグルタミン酸をグルタミンに変換する。GSはアンモニア同化および窒素代謝にも関与している。GSは、大部分の植物において、硝酸塩還元によって放出されるアンモニアの解毒のための経路に関与している。従って、GSの強力な阻害剤は、植物細胞にとって極めて毒性である。植物の内部での除草剤の分解または修飾は、抵抗性をもたらし得る。
【0003】
特定のクラスの除草剤が、植物におけるGSの阻害による毒性効果に基づき開発されている。ビアラホスおよびホスフィノトリシンは、2つのそのようなGS作用の阻害剤であり、優れた除草特性を保有している。これらの2つの除草剤は非選択的であり;土壌に存在する様々な植物種すべての生長を阻害し、従って、それらの完全な駆除を引き起こす。
【0004】
ビアラホスも、広いスペクトルを有する除草剤である。ビアラホスは、ホスフィノトリシン(PPTまたはPTC;2-アミノ-4-メチルホスフィノ酪酸)、L-グルタミン酸のアナログ、および2個のL-アラニン残基から構成されている。従って、PPTとビアラホスとの間の構造的な違いは、PPTの場合には2個のアラニンアミノ酸が存在しない点にある。細胞内ペプチダーゼによるビアラホスのL-アラニン残基の除去によって、PPTが放出される。PPTはGSの強力な阻害剤である。PPTによる植物におけるGSの阻害は、アンモニアの迅速な蓄積および植物細胞の死を引き起こす。
【0005】
ビアラホスは、最初、抗生物質特性を有することが開示され、そのため、殺虫薬または殺真菌薬として使用され得た。米国特許第3,832,394号(特許文献1)は、ストレプトマイセス・ハイグロスコピカス(Streptomyces hygroscopicus)の培養、およびその培養培地からのビアラホスの回収に関する。しかしながら、ストレプトマイセス・ビリドクロモジェネス(viridochromogenes)のようなその他の株も、この化合物を産生する。ホサラシン(phosalacin)のような、PPT部分を含有しているその他のトリペプチド抗生物質も、自然界に存在することが公知である。PPTは、化学合成によっても入手され、市販されている。
【0006】
ビアラホスを産生するストレプトマイセス・ハイグロスコピカスおよびストレプトマイセス・ビリドクロモジェネスは、ホスフィノトリシンアセチルトランスフェラーゼ活性を有する酵素によってPPT毒性から保護される。Plant Physiol, April 2001, Vol. 125, pp. 1585-1590(「Expression of bar in the Plastid Genome Confers Herbicide Resistance」, Lutz et al.)(非特許文献1)。これらの抗生物質を産生するストレプトマイセス種は、もしこれらの抗生物質に対する自己防御機序を有していなければ、それ自体破壊されるであろう。この自己防御機序は、いくつかの事例において、抗生物質効果を阻害することができる酵素を含むことが見出されている。
【0007】
ホスフィノトリシンアセチルトランスフェラーゼは、bar遺伝子(ビアラホス抵抗性;Thompson et al., 1987)またはpat遺伝子(ホスフィノトリシンアセチルトランスフェラーゼ;Strauch et al., 1988)のいずれかによってコードされ、PPTの遊離アミノ基のアセチル化によってPPTを解毒する。これらの2つの遺伝子によってコードされた酵素は、機能的に同一であり、アミノ酸レベルで85%の同一性を示す(Wohlleben et al., 1988; Wehrmann et al., 1996)。核遺伝子からのキメラbarまたはpat遺伝子の細胞質における発現により、PPT抵抗性作物が入手されている。タバコ(ニコチアナ・タバカム品種プティ・ハバナ(Nicotiana tabacum cv Petit Havana))、ジャガイモ、セイヨウアブラナ(Brassica napus)、ブラッシカ・オレラセア(Brassica oleracea)(De Block et al., 1987; De Block et al., 1989)、トウモロコシ(Spencer et al., 1990)、およびイネ(Cao etal., 1992)においては、PPT抵抗性についての直接選択によって、除草剤抵抗性の系が獲得されている。
【0008】
遺伝子(bar)が、ストレプトマイセス・セリカラー(coelicolor)A3においてhrdDシグマ因子遺伝子に隣接して同定された。予測されたbar産物は、ビアラホス産生種ストレプトマイセス・ビリドクロモジェネスおよびストレプトマイセス・ハイグロスコピカスのpat遺伝子およびbar遺伝子のものとそれぞれ32.2%および30.4%の同一性を示した。S.セリカラーbar遺伝子は、高コピー数ベクターによりS.セリカラーにおいてクローニングされた場合、ビアラホスに対する抵抗性を付与した。Bedford et al., Gene, 1991 Jul 31; 104(1):39-45, 「Characterization of a gene conferring bialaphos resistance in Streptomyces coelicolor A3(2)」(非特許文献2)。その他の微生物におけるこの遺伝子の異種発現、またはこの遺伝子の植物への形質転換は、これまでに報告されていない。
【0009】
除草剤抵抗性形質の使用は、pat遺伝子がストレプトマイセス・ビリドクロモジェネスから単離されるDE 3642 829 A(特許文献2)ならびに米国特許第5,879,903号(特許文献3)(ならびに第5,637,489号(特許文献4);第5,276,268号(特許文献5);および第5,273,894号(特許文献6))において言及されている。WO 87/05629(特許文献7)ならびに米国特許第5,648,477号(特許文献8)(ならびに第5,646,024号(特許文献9)および第5,561,236号(特許文献10))は、(PPTのような)グルタミンシンセターゼ阻害剤から植物細胞および植物体を保護するためのS.ハイグロスコピカス由来のbar遺伝子の使用、ならびに植物体における除草剤抵抗性の発達に言及している。除草剤BASTA(Hoechstホスフィノトリシン)またはハービエース(Herbiace)(Meiji Seikaビアラホス)に対する抵抗性をコードする遺伝子が、アグロバクテリウム感染によって、タバコ(ニコチアナ・タバカム品種プティ・ハバン(Havan)SR1)、ジャガイモ(ソラナム・ツベロサム品種ベノリマ(Solanum tuberosum cv Benolima))、およびトマト(リコパーシカム・エスカレンタム(Lycopersicum esculentum))植物へ導入され、除草剤抵抗性を付与した。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】米国特許第3,832,394号
【特許文献2】DE 3642 829 A
【特許文献3】米国特許第5,879,903号
【特許文献4】米国特許第5,637,489号
【特許文献5】米国特許第5,276,268号
【特許文献6】米国特許第5,273,894号
【特許文献7】WO 87/05629
【特許文献8】米国特許第5,648,477号
【特許文献9】米国特許第5,646,024号
【特許文献10】米国特許第5,561,236号
【非特許文献】
【0011】
【非特許文献1】Plant Physiol, April 2001, Vol. 125, pp. 1585-1590(「Expression of bar in the Plastid Genome Confers Herbicide Resistance」, Lutz et al.)
【非特許文献2】Bedford et al., Gene, 1991 Jul 31; 104(1):39-45,「Characterization of a gene conferring bialaphos resistance in Streptomyces coelicolor A3(2)」
【発明の概要】
【0012】
本発明は、本明細書においてDSM-2と呼ばれる新規の遺伝子に関する。この遺伝子はストレプトマイセス・セリカラー(A3)において同定された。DSM-2タンパク質は、PATおよびBARの遠縁である。本発明はまた、DSM-2タンパク質をコードする、植物に最適化された遺伝子を提供する。DSM-2は、微生物、植物細胞、および植物体において、除草抗菌分子、グルホシネート、ホスフィノトリシン、および/またはビアラホスに対する耐性を付与するためのトランスジェニック形質として使用され得る。
【0013】
アラビドプシス(Arabidopsis)への形質転換は、グルホシネートの高い割合の回収を可能にする。導入された後、DSM-2遺伝子は、有意な選択および完全な植物体の抵抗性を提供する能力を有する。
【0014】
本発明の遺伝子には、単にバイオテクノロジープロジェクトのための選択マーカーとして、高い本質的な価値がある。いくつかの好ましい態様において、本発明の遺伝子は、培養物中の形質転換に成功した細胞を選択するため、ならびに温室および圃場において完全な植物体を選択するためのマーカーとして使用され得る。この遺伝子および類似ホモログは、patおよび/またはbarの代わりに使用され得る。
【0015】
細菌形質転換系における選択マーカーとしてのこの遺伝子の使用は、植物のすべての形質転換の効率を増加させることができる。唯一の選択マーカーとしてのDSM-2の使用は、クローニング中のさらなる(アンピシリン抵抗性のような)医療用抗生物質マーカーの必要性を排除する。
【0016】
本発明によれば、様々なその他の使用も可能である。DSM-2は、除草剤グルホシネートおよびビアラホスに対する耐性を植物に付与するトランスジェニック形質として有用であり得る。
【0017】
この遺伝子は、修飾されたアグロバクテリウム株と合わせて、新規の植物形質転換系のための基礎としても使用され得る。タンパク質作製および非医療用抗生物質抵抗性マーカー遺伝子の組み入れのために使用されるシュードモナス・フルオレセンス(Pseudomonas fluorescens)の新規の株またはその他の微生物株も、本発明により作製され得る。医療用抗生物質抵抗性要素からのフラグメント精製が排除されることによる、クローニングおよび形質転換の過程および効率の改善も、有益であるかもしれない。
【0018】
除草剤耐性作物(HTC)形質に加えて、本発明の遺伝子によってトランスジェニック作物において除草剤耐性が作出される、除草剤を使用して雑草を制御する方法も、本発明の範囲内である。本発明のHTC形質の組み合わせも、他のHTC形質(グリホセート耐性および2,4-D耐性を含むが、これらに限定はされない)と組み合わせられた場合、特に、(グリホセートのような)除草剤に対する新たに獲得された抵抗性または本質的な耐性を有する種を制御するために、有益である。さらに、(世界的にますます普及してきている)グリホセート耐性作物を他のグリホセート耐性作物と輪作する場合、グリホセート抵抗性の自生植物の制御が困難である場合がある。従って、作物へ重ね合わせられた(stacked)、または個々に形質転換されたこれらのトランスジェニック形質の使用は、他のHTC自生植物の制御のための手段を提供することができる。
【図面の簡単な説明】
【0019】
【図1】DSM2によって媒介されるN-アセチル化によるグルホシネートの不活性化を示す図である。
【発明を実施するための形態】
【0020】
発明の詳細な説明
本発明は、本明細書においてDSM-2と呼ばれる新規の遺伝子に関する。この遺伝子はストレプトマイセス・セリカラー(A3)において同定された。DSM-2タンパク質は、PATおよびBARの遠縁である(それぞれ30%および28%のアミノ酸同一性)。本発明はまた、例えば、ヘミコット・バイアス(hemicot bias)を有する、DSM-2タンパク質をコードする、植物に最適化された遺伝子を提供する。DSM-2は、植物体および植物細胞において、除草剤グルホシネート、ホスフィノトリシン、およびビアラホスに対する耐性を付与するためのトランスジェニック形質として使用され得る。
【0021】
本発明の遺伝子には、単にバイオテクノロジープロジェクトのための選択マーカーとして、高い本質的な価値がある。しかしながら、本発明によれば、様々なその他の使用が可能である。
【0022】
DSM-2は、除草剤グルホシネート、ホスフィノトリシン、およびビアラホスに対する耐性を植物に付与するトランスジェニック形質として有用であり得る。
【0023】
いくつかの好ましい態様において、(除草剤耐性作物HTCとは別に、またはそれに加えて)培養物、温室、および圃場における形質転換に成功した細胞ならびに完全な植物体を選択するためのマーカーとしてである。この遺伝子および類似ホモログは、patおよび/またはbarの代わりに使用され得る。本明細書において例示される1つの態様は、NT-1タバコ細胞における選択マーカーとしてのDSM-2の使用である。本発明の遺伝子は、トウモロコシおよびイネのようなその他の植物系においても選択マーカーとして使用され得る。
【0024】
細菌系における選択マーカーとしてのこの遺伝子の使用は、すべての植物形質転換の効率を増加させることができる。唯一の選択マーカーとしてのDSM-2の使用は、クローニング中のさらなる(アンピシリン抵抗性のような)医療用抗生物質マーカーの必要性を排除する。
【0025】
実験は、大腸菌(Escherichia coli)細胞系BL21-Star(DE3)(Invitrogenカタログ#C6010-03)が、100μg/mlという濃度のグルホシネートアンモニウム(Basta)を含有している最少培地で阻害されることを証明した。BL21 Star細胞系におけるDSM-2の発現は、400μg/mlのグルホシネートを含有している最少培地で抵抗性を補完した。これらの実験は、DSM-2の発現が、グルホシネートによって阻害される細菌において、クローニングの適用のための非医療用抗生物質選択マーカーとして使用され得ることを示している。
【0026】
さらなる実験は、植物プロモーター、シロイヌナズナ(Arabidopsis thaliana)PolyUbiquitin 10(At Ubi10)およびウイルスプロモーター、キャッサバ葉脈モザイクウイルス(CsVMV)が、大腸菌株BL21-Star(DE3)において機能性であることを証明した。両プロモーターは、200μg/mlのグルホシネートを含有している最少培地に対する抵抗性を提供するために充分なDSM-2タンパク質を発現した。これらの植物プロモーターは、大腸菌において非医療用抗生物質選択マーカーとしてDSM-2の発現を駆動するために使用され得る。細菌および植物の両方において単一の植物プロモーターが機能性であれば、各々の種のための別々の選択マーカーの必要性が排除される。
【0027】
この遺伝子は、修飾されたアグロバクテリウム株と合わせて、新規の植物形質転換系のための基礎としても使用され得る。非医療用抗生物質抵抗性マーカー遺伝子を使用したタンパク質作製のためのシュードモナス・フルオレセンスの新規の株またはその他の微生物株も、本発明により作製され得る。医療用抗生物質抵抗性要素からのフラグメント精製が排除されることによる、クローニングおよび形質転換の過程および効率の改善も、有益であり得る。
【0028】
HTC形質に加えて、本発明の遺伝子によって除草剤耐性がトランスジェニック作物において作出される、除草剤を使用して雑草を制御する方法も、本発明の範囲内である。本発明のHTC形質の組み合わせも、他のHTC形質(グリホセート耐性および2,4-D耐性を含むが、これらに限定はされない)と組み合わせられた場合、特に、(グリホセートのような)除草剤に対する新たに獲得された抵抗性または本質的な耐性を有する種の制御のため、有益である。さらに、(世界的にますます普及してきている)グリホセート耐性作物を他のグリホセート耐性作物と輪作する場合、グリホセート抵抗性自生植物の制御が困難である場合がある。従って、作物へ重ね合わせられた、または個々に形質転換されたトランスジェニック形質の使用は、他のHTC自生植物の制御のための手段を提供することができる。
【0029】
本発明のタンパク質(および供給源の単離物)
本発明は機能的タンパク質を提供する。「機能的活性」(または「活性な」)とは、本明細書では、本発明による使用のためのタンパク質/酵素が、除草剤を分解するか、またはその活性を減少する能力を有する(単独でまたは他のタンパク質と組み合わせて)ことを意味する。本発明のタンパク質を産生する植物は、該植物が除草剤で処理される場合に、タンパク質発現のレベルが、該植物を、除草剤に対して完全もしくは部分的に抵抗性に、または耐性にするための十分であるような、タンパク質の「有効量」を好ましく産生する(他に特定されない限り典型的な割合において;典型的な適用割合は、例えば、周知のHerbicide Handbook(Weed Science Society of America, Eighth Edition, 2002)において見出され得る)。除草剤は、通常の圃場使用割合および濃度において、正常に標的植物を殺傷する割合で適用され得る(本発明のために、レベルおよび/または濃度は、以前に使用されたものよりも任意により高くあり得る)。好ましくは、本発明の植物細胞および植物は、除草剤処理によって引き起こされる生育阻害または損傷に対して保護される。本発明の形質転換植物および植物細胞は、好ましくは、本明細書で考察されるように、除草剤に対して抵抗性または耐性にされ、このことは、形質転換植物および植物細胞が、本明細書で考察されるように、有効量の1種または複数の除草剤の存在下で生育することができることを意味する。本発明の好ましいタンパク質は、1種または複数のアリールオキシアルカノエート化合物を代謝するための触媒活性を有する。「抵抗性」という用語を容易に論じることはできないし、「耐性である」という動詞または「耐性の」という形容詞を容易に使用することはできない。産業界は、除草剤抵抗性作物(HRC)対除草剤耐性作物(HTC)の議論に、膨大な時間を費やしてきた。HTCは産業界において好まれる用語である。しかし、米国雑草科学会(Weed Science Society of America)による公式の抵抗性の定義は、「野生型に対して通常は致死的である除草剤の用量への曝露後、植物の、遺伝性の、生存および再生する能力である。植物においては、抵抗性は、天然に存在するか、または組織培養もしくは変異誘発によって産生される変種の遺伝子操作または遺伝子選択のような技術によって誘導される可能性がある」というものである。本明細書で使用される場合、他に示されない限り、除草剤「抵抗性」は、本開示の出願時における現行版のThe Herbicide Handbookによって示唆されるように、遺伝性であり、かつ所定の植物のための除草剤による、典型的な有効な除草性処理の存在下で、植物が生長および再生することを可能にする。当業者によって認識されるように、たとえ除草剤曝露によるある程度の植物損傷が明白であっても、植物は、なお「抵抗性である」と見なされる可能性がある。本明細書で使用される場合、「耐性」という用語は、「抵抗性」という用語よりも広く、本明細書で定義されるような「抵抗性」を含み、かつ同じ除草剤用量において同じ遺伝子型の野生型植物を典型的には生じる、除草剤によって誘導された種々の程度の損傷に対して特定の植物が耐える能力の改善を含む。
【0030】
植物または細菌の系への機能的活性の移入は、そのベクターが存在する宿主に対して適切であるタンパク質発現ベクターに組み込まれた、本発明のタンパク質についてのアミノ酸配列をコードする核酸配列を含むことができる。機能的活性を有するタンパク質をコードする核酸配列を得るための1つの方法は、本明細書で考察されるように、タンパク質のアミノ酸配列から推定される情報を使用して、関心対象のタンパク質を産生する細菌種からのネイティブな遺伝物質を単離することである。このネイティブ配列は、例えば、以下でより詳細に考察されるように、植物での発現のために最適化することができる。最適化されたポリヌクレオチドはまた、タンパク質配列に基づいて設計することもできる。
【0031】
これらのタンパク質のクラスおよびこれらをコードするポリヌクレオチドを特徴付けするための1つの方法は、一連の特定化された条件下で、例示されたヌクレオチド配列(その相補体および/またはいずれかの鎖に由来するプローブ)とハイブリダイズするその能力によってポリヌクレオチドを定義することによるか、および/または例示された配列に由来するプライマーを使用するPCRによって増幅されるそれらの能力による。
【0032】
本発明による使用のためのタンパク質を入手するための多数の方法が存在する。例えば、本明細書に開示されるタンパク質に対する抗体が、タンパク質の混合物から他のタンパク質を同定および単離するために使用され得る。詳細には、抗体は、他の関連するタンパク質を比較した場合に、最も保存されているか、または最も区別できるタンパク質の一部分に対して惹起されてもよい。次いで、これらの抗体は、免疫沈殿、酵素結合免疫吸着アッセイ法(ELISA)、またはイムノブロッティングによって、特徴的な活性を有する等価なタンパク質を特異的に同定するために使用することができる。本明細書に開示されるタンパク質、または等価なタンパク質、またはこれらのタンパク質のフラグメントに対する抗体が、標準的な手順を使用して容易に調製され得る。このような抗体は本発明の1つの局面である。本発明の抗体は、モノクローナル抗体およびポリクローナル抗体を含み、好ましくは、例示または示唆されたタンパク質に応答して産生される。
【0033】
本開示の恩典により、本発明のタンパク質および遺伝子は、例えば、組換え細菌および/または野生型細菌のような多様な微生物を含む多様な起源から入手され得る。
【0034】
細菌からの単離物の変異体は、当技術分野において周知である手法によって作製することができる。例えば、胞子非形成変異体は、単離物のエチルメタンスルホネート(EMS)変異誘発を通して入手することができる。これらの変異体は、当技術分野において周知の手法によって、紫外光およびニトロソグアニジンを使用して作製することができる。
【0035】
本明細書で言及または示唆される対象の単離物のいずれか「からの」またはそれ「から入手可能である」タンパク質は、そのタンパク質(または同様のタンパク質)が、別の細菌株または植物などの単離物またはある他の供給源から入手可能であることを意味する。「から入手可能である」はまた、この暗示的意味を有し、例えば、植物での発現のために改変されている、所定の型の細菌から入手可能であるタンパク質を含む。当業者は、細菌の遺伝子およびタンパク質の開示が与えられることで、植物がそのタンパク質を産生するために操作され得ることを容易に認識する。抗体調製物、核酸プローブ(例えば、DNA、RNA、またはPNA)などは、本明細書に開示されるポリヌクレオチドおよび/またはアミノ酸配列を使用して調製され得、かつ他の(天然の)供給源から他の関連する遺伝子をスクリーニングおよび回収するために使用され得る。
【0036】
標準的な分子生物学技術は、本明細書に記載されるタンパク質および遺伝子をクローニングおよび配列決定するために使用されてもよい。さらなる情報は、参照により本明細書に組み入れられる、Sambrook et al., 1989において見出され得る。
【0037】
ポリヌクレオチドおよびプローブ
本発明はさらに、本発明による使用のためのタンパク質をコードする核酸配列を提供する。本発明はさらに、所望の除草剤活性を有するタンパク質をコードする遺伝子を同定および特徴付けする方法を提供する。1つの態様において、本発明は、ハイブリダイゼーションプローブおよび/またはPCR技術のためのプライマーとして有用である特有のヌクレオチド配列を提供する。これらのプライマーは、関心対象の特定の遺伝子の同定、特徴付け、および/または単離において有用であり得る特徴的な遺伝子フラグメントを産生する。本発明のヌクレオチド配列は、以前に記載されているタンパク質から区別できるタンパク質をコードする。
【0038】
本発明のポリヌクレオチドは、所望の宿主細胞中でタンパク質またはペプチドをコードするために完全な「遺伝子」を形成するために使用することができる。例えば、当業者が容易に認識するように、対象のポリヌクレオチドは、当技術分野において容易に公知であるように、関心対象の宿主中で、プロモーターの制御下に適切に配置され得る。遺伝子発現および一過性/組織特異的発現のレベルは、本発明の有用性に非常に影響を与えることができる。一般的に、分解可能な遺伝子のタンパク質発現のレベルがより大きいと、基質(この場合は標的除草剤)の完全な分解がより多くなる。プロモーターは、高い発現が植物の健康に結果的に負の影響を有さない限り、高いレベルで標的遺伝子を発現することが所望される。典型的には、すべての生育段階において植物の完全な保護のためにすべての組織中で構成的に発現されるDSM-2遺伝子を有することが望まれる。しかし、代替として、植物的において発現される抵抗性遺伝子を使用してもよい。これは、雑草制御のための作物における標的除草剤の使用を可能にし、開花段階の間の適用によって標的作物の有性生殖を引き続き制御する。
【0039】
当業者に公知であるように、DNAは、典型的には、二本鎖型で存在する。この配列において、一方の鎖は他方の鎖に対して相補的であり、逆もまた同様である。(例えば)DNAは植物中で複製されるので、DNAのさらなる相補鎖が産生される。「コード鎖」は、アンチセンス鎖と結合する鎖をいうために当技術分野において頻繁に使用される。mRNAは、DNAの「アンチセンス」鎖から転写される。「センス」鎖または「コード」鎖は、オープンリーディングフレーム(ORF)として読み取られることができ、関心対象のタンパク質またはペプチドを形成する一連のコドン(コドンは、特定のアミノ酸を特定するための3残基単位として読まれ得る3つのヌクレオチドである)を有する。インビボでタンパク質を産生するために、DNAの鎖は、典型的には、タンパク質のための鋳型として使用されるmRNAの相補鎖に転写される。従って、本発明は、相補鎖を含む、添付の配列表および/または等価物に示される例示されるポリヌクレオチドの使用を含む。例示されたDNA分子に対して機能的に等価であるRNAおよびPNA(ペプチド核酸)は本発明に含まれる。
【0040】
本発明の1つの態様において、細菌からの単離物は、微生物の高い増殖を生じる条件下で培養され得る。一本鎖ゲノム核酸を提供するために微生物を処理した後、DNAは、本発明のプライマーと接触され得、PCR増幅に供され得る。関心対象の遺伝子の特徴的なフラグメントは、この手法によって増幅され、それにより、関心対象の遺伝子の存在を同定する。
【0041】
本発明のさらなる局面は、本明細書に開示される方法およびヌクレオチド配列を使用して同定された遺伝子および単離物を含む。このように同定された遺伝子は、本発明の除草剤抵抗性タンパク質をコードすることができる。
【0042】
本発明によるタンパク質および遺伝子は、例えば、オリゴヌクレオチドプローブを使用することによって同定および入手することができる。これらのプローブは、適切な標識によって検出可能であり得るか、または国際出願番号WO 93/16094において記載されているように、固有に蛍光性に作製されてもよい、検出可能なヌクレオチド配列である。これらのプローブ(および本発明のポリヌクレオチド)は、DNA、RNA、またはPNAであってもよい。アデニン(A)、シトシン(C)、グアニン(G)、チミン(T)、およびウラシル(U;RNA分子用)に加えて、本発明の合成プローブ(およびポリヌクレオチド)はまた、イノシン(4種すべての塩基と対合することが可能な中性塩基;時折、合成プローブ中で4種すべての塩基の混合物の代わりに使用される)および/または合成(非天然)塩基を有することができる。従って、合成の、縮重オリゴヌクレオチドが本明細書で言及される場合、「N」または「n」が総称的に使用され、「N」または「n」は、G、A、T、C、またはイノシンであり得る。曖昧なコードは、本明細書で使用されるように、本願の出願の時点の標準IUPAC命名法の慣例に従っている(例えば、RはAまたはGを意味し、YはCまたはTを意味する、など)。
【0043】
当技術分野において周知であるように、プローブ分子が核酸試料にハイブリダイズする場合には、プローブおよび試料が実質的な相同性/類似性/同一性を有することが合理的に推測され得る。好ましくは、ポリヌクレオチドのハイブリダイゼーションは、例えば、Keller, G. H., M. M. Manak(1987)DNA Probes, Stockton Press, New York, NY, pp. 169-170によって記載されるような、当該分野において周知の技術によって、ポリヌクレオチドのハイブリダイゼーションが最初に行われ、続いて、低ストリンジェンシー、中程度のストリンジェンシー、または高ストリンジェンシーの条件下での洗浄が行われる。例えば、そこに言及されているように、低ストリンジェンシー条件は、2×SSC(標準生理食塩水クエン酸)/0.1% SDS(ドデシル硫酸ナトリウム)を用いる、室温にて15分間の最初の洗浄によって達成され得る。2回の洗浄が典型的には実行される。次いで、より高いストリンジェンシーが、塩濃度を低下させることによって、および/または温度を上昇させることによって達成され得る。例えば、上記に記載される洗浄は、各々、0.1×SSC/0.1% SDSを用いる室温における15分間の2回の洗浄が続き得、これには、各々、0.1×SSC/0.1% SDSを用いる55℃における30分間の洗浄が続く。これらの温度は、本明細書に示され、かつ当業者に公知であるような他のハイブリダイゼーションおよび洗浄のプロトコールとともに使用することができる(例えば、SSPEは、SSCの代わりに塩として使用することができる)。2×SSC/0.1% SDSは、445mlの水に、50mlの20×SSCおよび5mlの10% SDSを加えることによって調製することができる。20×SSCは、NaCl(175.3g/0.150M)、クエン酸ナトリウム(88.2g/0.015M)、および水を合わせ、10N NaOHでpHを7.0に調整し、次いで、体積を1リットルに調整することによって調製することができる。10% SDSは、オートクレーブした50mlの水中に10gのSDSを溶解すること、次いで100mlに希釈することによって調製することができる。
【0044】
プローブの検出は、ハイブリダイゼーションが維持されたか否かを公知の様式で判定するための手段を提供する。このようなプローブ分析は、本明細書の遺伝子を同定するための迅速な方法を提供する。本発明によるプローブとして使用されるヌクレオチドセグメントは、DNAシンセサイザーおよび標準的な手法を使用して合成することができる。これらのヌクレオチド配列はまた、本発明の遺伝子を増幅するためのPCRプライマーとして使用することができる。
【0045】
分子のハイブリダイゼーション特性は、本発明のポリヌクレオチドを規定するために使用することができる。従って、本発明は、本明細書で例示されるポリヌクレオチドとハイブリダイズするポリヌクレオチド(および/またはそれらの相補体、好ましくはそれらの完全な相補体)を含む。すなわち、遺伝子(およびそれがコードするタンパク質)を規定するための1つの方法は、例えば、公知のまたは特に例示される遺伝子と(本明細書に具体的に開示されるいずれかの条件下で)ハイブリダイズするその能力による。
【0046】
本明細書で使用されるように、ハイブリダイゼーションのための「ストリンジェントな」条件とは、本出願者によって使用される条件と同じかまたはほぼ同じである、ハイブリダイゼーションの特異性の程度を達成する条件をいう。具体的には、32P-標識された遺伝子特異的プローブとの、サザンブロット上に固定化されたDNAのハイブリダイゼーションは、標準的な方法によって実行することができる(例えば、Maniatis et al. 1982を参照されたい)。一般的に、ハイブリダイゼーションおよび引き続く洗浄は、標的配列の検出を可能にする条件下で実行することができる。二本鎖DNA遺伝子プローブについては、ハイブリダイゼーションは、6×SSPE、5×Denhardt's溶液、0.1% SDS、0.1mg/ml変性DNA中で、DNAハイブリッドの融解温度(Tm)よりも20〜25℃下で、一晩実行することができる。融解温度は以下の数式によって記載される(Beltz et al. 1983)。
Tm = 81.5 C + 16.6 Log [Na+] + 0.41(%G+C)- 0.61(ホルムアルデヒド%)- 600/塩基対中の二重鎖の長さ。
【0047】
洗浄は、典型的には、以下のように実行することができる。
(1)1×SSPE、0.1% SDS中での室温で15分間、2回(低ストリンジェンシー洗浄)。
(2)0.2×SSPE、0.1% SDS中でのTm-20℃で15分間、1回(中程度ストリンジェンシー洗浄)。
【0048】
オリゴヌクレオチドプローブについては、ハイブリダイゼーションは、6×SSPE、5×Denhardt's溶液、0.1% SDS、0.1mg/ml変性DNA中で、ハイブリッドの融解温度(Tm)よりも10〜20℃下で、一晩実行することができる。融解温度は以下の数式によって決定することができる。
Tm(℃)= 2(T/A塩基対の数)+ 4(G/C塩基対の数)(Suggs et al., 1981)。
【0049】
洗浄は、典型的には、以下のように実行することができる。
(1)1×SSPE、0.1% SDS中での室温で15分間、2回(低ストリンジェンシー洗浄)。
(2)1×SSPE、0.1% SDS中でのハイブリダイゼーション温度で15分間、1回(中程度ストリンジェンシー洗浄)。
【0050】
一般的に、塩および/または温度は、ストリンジェンシーを変化させるために変更することができる。>70程度の塩基長の標識したDNAフラグメントを用いる場合、以下の条件が使用され得る。
低:1または2×SPPE、室温
低:1または2×SSPE、42℃
中程度:0.2×または1×SSPE、65℃
高:0.1×SSPE、65℃。
【0051】
二重鎖の形成および安定性は、ハイブリッドの2本の鎖の間の実質的な相補性に依存し、上記に記述されるように、特定の程度のミスマッチが許容され得る。それゆえに、本発明のプローブ配列は、記載された配列の変異(単一と複数の両方)、欠失、挿入、およびこれらの組み合わせを含み、ここで、該変異、挿入、および欠失は、関心対象の標的ポリヌクレオチドとの安定なハイブリッドの形成を許容する。変異、挿入、および欠失は、多くの方法で所定のポリヌクレオチド配列中で産生され得るが、これらの方法は当業者に公知である。他の方法は将来公知になる可能性がある。
【0052】
PCR技術
ポリメラーゼ連鎖反応(PCR)は、反復性の、酵素的な、プライマーを用いる、核酸配列の合成である。この手法は周知であり、当業者に一般的に公知である(Mullis, 米国特許第4,683,195号、同第4,683,202号、および同第4,800,159号; Saiki et al., 1985を参照されたい)。PCRは、標的配列の反対の鎖にハイブリダイズする2つのオリゴヌクレオチドプライマーによって隣接される関心対象のDNAフラグメントの酵素的増幅に基づく。これらのプライマーは、好ましくは、3'末端が互いに対して向けられて配置される。鋳型の熱変性、それらの相補的配列へのプライマーのアニーリング、およびDNAポリメラーゼを用いてのアニールされたプライマーの伸長は、PCRプライマーの5'末端によって規定されるセグメントの増幅を生じる。各プライマーの伸長産物は、他のプライマーのための鋳型として働くことができ、従って、各サイクルは、以前のサイクルで産生されたDNAフラグメントの量を本質的に倍加させる。これにより、特定の標的フラグメントの指数関数的な蓄積が、数時間以内に数百万倍にまでになる。耐熱性細菌サーマス アクアティカス(Thermus aquaticus)から単離されたTaqポリメラーゼなどの耐熱性DNAポリメラーゼを使用することによって、この増幅プロセスは、完全に自動化することができる。使用することができる他の酵素は、当業者に公知である。
【0053】
例示されるDNA配列、またはそのセグメントは、PCR増幅のためのプライマーとして使用することができる。PCR増幅を実行する際に、特定の程度のミスマッチが、プライマーと鋳型の間に許容され得る。それゆえに、例示されたプライマーの変異、欠失、および挿入(とりわけ、5'末端へのヌクレオチドの付加)は、本発明の範囲内にある。変異、欠失、および挿入は、当業者に公知である方法によって、所定のプライマー中で作製することができる。
【0054】
遺伝子およびタンパク質の改変
対象の遺伝子およびタンパク質は、キメラタンパク質または融合タンパク質を産生するために、他の遺伝子およびタンパク質に融合され得る。本発明に従って有用な遺伝子およびタンパク質は、特に例示された全長配列のみならず、これらの配列、これらの配列の変種、変異体、キメラ、ならびに融合物の一部分、セグメント、および/またはフラグメント(隣接するフラグメント、ならびに全長分子と比較して内部および/または末端の欠失を含む)を含む。本発明のタンパク質は、これらが所望の機能的活性を保持する限りは、置換されたアミノ酸を有することができる。「変種」遺伝子は、例示されたタンパク質に対して等価であるかまたは類似している活性を有する、同じタンパク質または等価なタンパク質をコードするヌクレオチド配列を有する。「変種タンパク質」および「等価なタンパク質」という用語は、標的基質に対して同じかまたは本質的に同じ生物学的/機能的活性、および例示されたタンパク質と等価な配列を有するタンパク質をいう。本明細書で使用されるように、「等価な」配列との言及は、有意な程度にまで活性を改善し、または活性に有害な影響を与えない、アミノ酸の置換、欠失、付加、または挿入を有する配列をいう。活性を保持しているフラグメントもまた、この定義の中に含まれる。例示されたタンパク質の対応するフラグメントと同じかまたは同様の機能または活性を保持するフラグメントおよび他の等価物は、本発明の範囲内にある。アミノ酸の置換または付加などの変化は、例えば、(タンパク質の機能的活性を具体的に/実質的に減少することなく)タンパク質のプロテアーゼ安定性を増加すること(もしくは減少すること)、または制限部位の付加などの種々の目的のために作製することができる。遺伝子のバリエーションは、例えば、点変異を作製するための標準的な技術を使用して容易に構築されうる。
【0055】
加えて、例えば、米国特許第5,605,793号は、ランダムなまたは的を絞ったフラグメント化後にDNA再アセンブリーを使用することによって、さらなる分子の多様性を生成するための方法を記載している。これは、「遺伝子シャッフリング」と呼ぶことができ、これは、典型的には、複数の異なるDNA分子のフラグメントを混合する工程、続いて、再生の反復ラウンドを含む。これは、開始遺伝子によってコードされるタンパク質の活性を改善することができる。この結果は、改善された活性、変化した基質特異性、増加した酵素安定性、変化した立体特異性、または他の特徴を有するキメラタンパク質である。
【0056】
「シャッフリング」は、関心対象タンパク質の原子3D(三次元)配位および結晶構造を入手および精査後に、設計および標的化され得る。従って、「集中シャッフリング」は、表面に露出しているセグメントなどの、改変のために理想的であるタンパク質の特定のセグメントに向けることができ、好ましくは、タンパク質フォールディングおよび必須の3D構造の完全性と関連する内部セグメントではない。
【0057】
変種遺伝子は、変種タンパク質を産生するために使用することができる。組換え宿主は、変種タンパク質を産生するために使用することができる。「遺伝子シャッフリング」技術および他の技術を使用して、本発明に例示される任意の配列の特定の残基(アミノ酸またはヌクレオチド)を有する特定のセグメントを含む、等価な遺伝子およびタンパク質が構築され得る。このような技術は、例示されたかまたは示唆された配列(またはその相補体(完全な相補体))のいずれかにおける(同じサイズの)セグメントに対応する、例えば、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99、100、101、102、103、104、105、106、107、108、109、110、111、112、113、114、115、116、117、118、119、120、121、122、123、124、125、126、127、128、129、130、131、132、133、134、135、136、137、138、139、140、141、142、143、144、145、146、147、148、149、150、151、152、153、154、155、156、157、158、159、160、161、162、163、164、165、166、167、168、169、および170個の連続するアミノ酸残基を有する、等価な/機能的に活性なタンパク質を得るために調整することができる。特に関心対象の領域について、そのようなセグメントをコードするポリヌクレオチドも、本発明に含まれ、特に保存された領域のため、プローブおよび/またはプライマーとしても使用され得る。
【0058】
全長遺伝子のフラグメントは、標準的な手法に従って、市販のエキソヌクレアーゼまたはエンドヌクレアーゼを使用して作製することができる。例えば、Bal31などの酵素または部位特異的変異誘発が、これらの遺伝子の末端からヌクレオチドを体系的に切断するために使用することができる。また、活性フラグメントをコードする遺伝子は、種々の制限酵素を使用して入手されてもよい。これらの活性フラグメントを直接的に入手するためにプロテアーゼを使用してもよい。
【0059】
タンパク質が短縮され、なお機能的活性を保持し得ることは、本明細書に開示されるように、本発明の範囲内にある。「短縮型タンパク質」は、残りの短縮されたタンパク質が、切断後に所望の活性を保持し、かつその活性を示しながら、タンパク質の一部分が切断可能であることを意味する。切断は、種々のプロテアーゼによって達成することができる。さらに、効率的に切断されたタンパク質は、分子生物学的技術を使用して産生することができ、ここで、該タンパク質をコードするDNA塩基は、制限エンドヌクレアーゼまたは当業者に使用可能である他の技術のいずれかを通して除去される。短縮化後、該タンパク質は、大腸菌、バキュロウイルス、植物に基づくウイルス系、酵母などのような異種系において発現することができ、次いで、活性を測定するために本明細書に開示されるように昆虫アッセイ法に配置することができる。短縮型タンパク質は、これらが完全な全長配列よりも短いものを有しながら、機能的活性を保持するように首尾よく産生され得ることは、当技術分野において周知である。例えば、B.t.タンパク質は、短縮(コアタンパク質)型で使用することができる(例えば、Hofte et al.(1989)、およびAdang et al.(1985)を参照されたい)。本明細書で使用されるように、「タンパク質」という用語は、機能的に活性な短縮型を含むことができる。
【0060】
ある場合において、とりわけ、植物における発現のために、短縮型タンパク質を発現する短縮型遺伝子を使用することが有利であり得る。好ましい短縮型遺伝子は、典型的には、全長タンパク質の40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、または99%をコードする。
【0061】
本明細書の特定のタンパク質は、具体的に本明細書に例示されてきた。これらのタンパク質は本発明のタンパク質の単なる例であるので、本発明が、例示されたタンパク質の同じかまたは同様の活性を有する変種または等価なタンパク質(およびその等価物をコードするヌクレオチド配列)を含むことは容易に明らかとなるはずである。等価なタンパク質は、例示されるタンパク質とアミノ酸類似性(および/または相同性)を有する。アミノ酸同一性は、典型的には、少なくとも60%、好ましくは少なくとも75%、より好ましくは少なくとも80%、さらにより好ましくは少なくとも90%であり、および少なくとも95%であり得る。好ましい本発明のタンパク質はまた、より特定には同一性および/または類似性の範囲によって定義することができる。例えば、同一性および/または類似性は、本明細書で例示または示唆される配列と比較して、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、または99%であり得る。上記に列挙した任意の数字が、上限および下限を定義するために使用され得る。
【0062】
他に特定されない限り、本明細書で使用されるように、2つの核酸の配列同一性パーセントおよび/または配列類似性パーセントは、Karlin and Altschul 1993において改変された、Karlin and Altschul, 1990のアルゴリズムを使用して決定される。このようなアルゴリズムは、Altschul et al., 1990のNBLASTプログラムおよびXBLASTプログラムに組み込まれている。BLASTヌクレオチド検索は、NBLASTプログラム、スコア=100、ワード長=12を用いて実行される。ギャップ付きBLASTは、Altschul et al., 1997において記載されるように使用され得る。BLASTプログラムおよびギャップ付きBLASTプログラムを使用する場合、それぞれのプログラム(NBLASTおよびXBLAST)のデフォルトパラメーターが使用される。NCBI/NIHウェブサイトを参照されたい。比較目的のためにギャップ付きアラインメントを得るために、Vector NTI Suite 8(InforMax, Inc., North Bethesda, MD, U.S.A.)のAlignX機能が、デフォルトパラメーターを用いて使用された。これらは、15のギャップオープニングペナルティー、6.66のギャップ伸長ペナルティー、および8のギャップ分離ペナルティーであった。
【0063】
タンパク質の種々の特性および三次元的な特徴もまた、タンパク質の活性/機能性に有害な影響を与えることなく、変化させることができる。保存性アミノ酸置換は、分子の活性および/または三次元配置に有害な影響を与えないように、許容/作製することができる。アミノ酸は、以下のクラスに位置付けることができる:非極性、非荷電極性、塩基性、および酸性。1つのクラスのアミノ酸が同じ型の別のアミノ酸で置換される保存性置換は、その置換が化合物の生物学的活性に有害でない限り、本発明の範囲内にある。表2は、各クラスに属するアミノ酸の例の列挙を提供する。
【0064】
(表2)

【0065】
ある例において、非保存性置換もまた、作製することができる。しかし、好ましい置換は、タンパク質の機能的/生物学的活性を有意に損なわない。
【0066】
本明細書で使用されるように、「単離された」ポリヌクレオチドおよび/または「精製された」タンパク質との言及は、それらが天然に見出されるのではない状態である場合のこれらの分子をいう。従って、「単離された」および/または「精製された」との言及は、本明細書に記載されるように、「人の手」の関与を意味する。例えば、発現のために植物に入れた本発明における細菌「遺伝子」は、「単離されたポリヌクレオチド」である。同様に、細菌タンパク質に由来し、かつ植物によって産生されるタンパク質は、「単離されたタンパク質」である。
【0067】
遺伝コードの縮重/冗長性のために、種々の異なるDNA配列が、本明細書に開示されるアミノ酸配列をコードし得る。同じ、または本質的に同じタンパク質をコードする代替的なDNA配列を作製することは、十分に当業者の技量の範囲内にある。これらの変種DNA配列は本発明の範囲内にある。これはまた、「植物における発現のための配列の最適化」と題された以下の節においてより詳細に考察される。
【0068】
植物における発現のための配列の最適化
植物での異種遺伝子の高発現を得るために、遺伝子が植物細胞(の細胞質)においてより効率的に発現されるように、その遺伝子を再操作することが一般的に好ましい。トウモロコシは、このような植物の1つであり、ここでは、該植物において遺伝子の発現レベルを増加させるために、形質転換の前に異種遺伝子を再設計することが好ましい場合がある。それゆえに、細菌タンパク質をコードする遺伝子の設計における追加の工程は、双子葉植物種または単子葉植物種に関わらず、標的植物配列とより密接に整列されるコドンの偏りを使用する、最適な発現のための異種遺伝子の再操作である。配列はまた、本明細書の別の箇所で考察される、任意のより特定な型の植物における発現のために最適化することができる。
【0069】
トランスジェニック宿主
本発明のタンパク質をコードする遺伝子は、広範な種々の微生物または植物の宿主に導入することができる。本発明は、トランスジェニック植物細胞およびトランスジェニック植物を含む。好ましい植物(および植物細胞)は、トウモロコシ、アラビドプシス、タバコ、ダイズ、ワタ、アブラナ、イネ、コムギ、芝草、および牧草などである。他の型のトランスジェニック植物、例えば、果物、野菜、観賞用植物、および木もまた、本発明に従って作製することができる。より一般的には、双子葉植物および/または単子葉植物は、本発明の種々の局面において使用することができる。
【0070】
従って、本発明は、単子葉植物および双子葉植物を含む維管束植物および非維管束植物、針葉樹、蘚苔類、藻類、真菌、ならびに細菌と共に使用するために適応し得る。動物細胞および動物細胞培養物も可能である。
【0071】
好ましい態様において、遺伝子の発現は、直接的または間接的に、関心対象のタンパク質の細胞内産生(および維持)を生じる。植物は、この様式で、除草剤抵抗性にされ得る。このような宿主は、トランスジェニック、組換え、形質転換された、および/またはトランスフェクトされた、宿主および/または細胞と呼ばれ得る。本発明のある局面において(例えば、関心対象の遺伝子をクローニングおよび調製する場合)、微生物(好ましくは、細菌)細胞が、本開示の恩典を伴って、標準的な技術に従って、産生および使用され得る。
【0072】
本発明のポリヌクレオチドでトランスフェクトされた植物細胞は、再生して完全な植物体となることができる。本発明は、組織細胞培養、液体培養、およびプレート培養を含む、細胞培養を含む。本発明の植物を生成するために産生および/または使用される種子もまた、本発明の範囲内に含まれる。他の植物組織および部分もまた、本発明に含まれる。本発明は、同様に、本発明のポリヌクレオチドを含む植物または細胞を産生する方法を含む。このような植物を産生する1つの好ましい方法は、本発明の種子を植えることによる。
【0073】
トランスジェニック宿主を形成するための遺伝子の挿入
本発明の種子の1つの局面は、本発明のタンパク質を発現する本発明のポリヌクレオチドを用いる、植物、植物種子、および他の宿主細胞の形質転換/トランスフェクションである。この様式で形質転換された植物は、異なる作用の様式を有する種々の除草剤に対して抵抗性にされ得る。
【0074】
広範な種々の方法が、遺伝子の安定な維持および発現を可能にする条件下で、標的宿主に、所望のタンパク質をコードする遺伝子を導入するために使用可能である。これらの方法は当業者に周知であり、例えば、米国特許第5,135,867号において記載されている。
【0075】
DSM-2ポリヌクレオチドを含むベクターは本発明の範囲に含まれる。例えば、大腸菌における複製系および形質転換細胞の選択を可能にするマーカーを含む多数のクローニングベクターが、高等植物への外来性遺伝子の挿入の調製のために使用可能である。これらのベクターには、例えば、pBR322、pUCシリーズ、M13mpシリーズ、pACYC184などが含まれる。従って、タンパク質をコードする配列は、適切な制限部位でベクターに挿入することができる。得られるプラスミドは、大腸菌への形質転換のために使用される。大腸菌細胞は、適切な栄養培地中で培養され、次いで収集および溶解される。プラスミドは、ゲノムDNAからの精製によって回収される。配列分析、制限分析、電気泳動、および他の生化学的-分子生物学的方法が、分析の方法として一般的に実行される。各操作の後、使用されるDNA配列は、制限消化し、かつ次のDNA配列に連結することができる。各プラスミド配列は、同じまたは他のプラスミドにクローニングすることができる。植物に所望の遺伝子を挿入する方法に依存して、他のDNA配列が必要である可能性がある。例えば、TiまたはRiプラスミドが植物細胞の形質転換のために使用される場合には、TiまたはRiプラスミドT-DNAの少なくともライトボーダー、しかし往々にして、ライトボーダーおよびレフトボーダーが、挿入される遺伝子の隣接領域として連結されなければならない。植物細胞の形質転換のためのT-DNAの使用は、EP 120 516; Hoekema(1985); Fraley et al.(1986);およびAn et al.(1985)において徹底的に調査および記載されてきた。
【0076】
多数の技術が、植物宿主細胞にDNAを挿入するために使用可能である。これらの技術には、形質転換因子としてのアグロバクテリウム・ツメファシエンス(Agrobacterium tumefaciens)またはアグロバクテリウム・リゾゲネス(Agrobacterium rhizogenes)使用するT-DNAを用いる形質転換、融合、注入、微粒子銃(微粒子射撃)、シリコンカーバイトウィスカー、エアロゾルビーム、PEG、またはエレクトロポレーション、ならびに他の可能な方法が含まれる。アグロバクテリアが形質転換のために使用される場合、挿入されるDNAは、特別なプラスミド、すなわち、中間体ベクターまたはバイナリーベクターのいずれかにクローニングされなければならない。この中間体ベクターは、T-DNA中の配列に対して相同である配列による相同組換えによって、TiまたはRiプラスミドに組み込まれ得る。TiまたはRiプラスミドはまた、T-DNAの移入のために必要であるvir領域を含む。中間体ベクターは、アグロバクテリア中でそれ自体複製することはできない。中間体ベクターは、ヘルパープラスミド(接合)によって、アグロバクテリウム・ツメファシエンスに移入され得る。バイナリーベクターは、大腸菌とアグロバクテリアの両方の中でそれ自体複製することができる。これらは、選択マーカー遺伝子、ならびに右側および左側の、T-DNAボーダー領域によってフレーム形成される、リンカーまたはポリリンカーを含む。これらは、アグロバクテリアに直接的に形質転換され得る(Holsters, 1978)。宿主細胞として使用されるアグロバクテリウムは、vir領域を有するプラスミドを含む。このvir領域は、植物細胞へのT-DNAの移入のために必要である。さらなるT-DNAが含まれてもよい。このように形質転換された細菌は、植物細胞の形質転換のために使用される。植物移植片は、植物細胞へのDNAの移入のために、アグロバクテリウム・ツメファシエンスまたはアグロバクテリウム・リゾゲネスとともに有利に培養することができる。次いで、選択のために抗生物質または殺生物剤を含んでもよい適切な培地中で、感染された植物材料(例えば、葉の小片、茎のセグメント、根であるが、しかしプロトプラストまたは懸濁培養細胞もまた)から完全な植物体が再生され得る。次いで、このように得られた植物は、挿入されたDNAの存在について試験することができる。特別な要求は、注入およびエレクトロポレーションの場合にはなされない。通常のプラスミド、例えば、pUC誘導体などを使用することが可能である。
【0077】
形質転換細胞は、通常の様式で植物の内部で成長する。これらは、胚細胞を形成し、子孫の植物に形質転換形質を伝達することができる。このような植物は、通常の様式で生育させることができ、同じ形質転換された遺伝性因子または他の遺伝性因子を有する植物と交雑させることができる。得られる雑種個体は、対応する表現型特性を有する。
【0078】
本発明のある好ましい態様において、細菌タンパク質をコードする遺伝子は、植物ゲノムに挿入された転写単位から発現される。好ましくは、該転写単位は、植物ゲノムへの安定な組み込みが可能である組換えベクターであり、タンパク質をコードするmRNAを発現する形質転換された植物系統の選択を可能にする。
【0079】
挿入されたDNAが一旦ゲノムに組み込まれたら、それはそこで比較的安定である(かつそこから再び出ることはない)。これは、殺生物剤または抗生物質、とりわけ、カナマイシン、G418、ブレオマイシン、ハイグロマイシン、またはクロラムフェニコールなどに対する抵抗性を、形質転換植物細胞に付与する選択マーカーを通常含む。植物選択マーカーはまた、典型的には、グルホシネート、(PAT)、グリホセート(EPSPS)、イマゼチアピル(AHAS)などのような種々の除草剤に対する抵抗性を提供することができる。個々に使用されるマーカーは、従って、挿入されたDNAを含まない細胞以外の形質転換細胞の選択を可能にするはずである。関心対象の遺伝子は、植物細胞において、構成的または誘導性のいずれかのプロモーターによって好ましく発現される。一旦発現されると、mRNAはタンパク質に翻訳され、それによって、関心対象のアミノ酸がタンパク質に組み込まれる。植物細胞において発現されるタンパク質をコードする遺伝子は、構成的プロモーター、組織特異的プロモーター、または誘導性プロモーターの制御下にあり得る。
【0080】
植物細胞に外来性の組換えベクターを導入するため、および導入された遺伝子を安定に維持および発現する植物を入手するためのいくつかの技術が存在している。このような技術には、微粒子上にコートされた遺伝物質の細胞への直接的な導入が含まれる(Cornellに対する米国特許第4,945,050号、およびDowElanco、現在はDow AgroSciences, LLCに対する米国特許第5,141,131号)。加えて、植物は、アグロバクテリウム技術(以下を参照されたい:University of Toledoに対する米国特許第5,177,010;Texas A&Mに対する米国特許第5,104,310号;欧州特許出願第0131624B1号;Schilperootに対する欧州特許出願第120516号、同第159418Bl号および同第176,112号;Schilperootに対する米国特許第5,149,645、同第5,469,976号、同第5,464,763号および同第4,940,838号および同第4,693,976号;すべてMax Planckに対する欧州特許出願第116718号、同第290799号、同第320500号;日本たばこ産業(Japan Tobacco)に対する欧州特許出願第604662号および同第627752号、ならびに米国特許第5,591,616号;Ciba Geigy、現在Syngentaに対する欧州特許出願第0267159号および同第0292435号、ならびに米国特許第5,231,019号;両方ともCalgeneに対する米国特許第5,463,174号および同第4,762,785号;ならびに両方ともAgracetusに対する米国特許第5,004,863号および同第5,159,135号)を使用して形質転換されてもよい。他の形質転換技術には、ウィスカー技術が含まれる。両方ともZeneca、現在Syngentaに対する米国特許第5,302,523号および同第5,464,765号を参照されたい。他の直接DNA輸送形質転換技術には、エアロゾルビーム技術が含まれる。米国特許第6,809,232を参照されたい。エレクトロポレーション技術もまた、植物を形質転換するために使用されてきた。Boyce Thompson Instituteに対するWO 87/06614;両方ともDekalbに対する米国特許第5,472,869号および同第5,384,253号;ならびに両方ともPlant Genetic Systemsに対するWO 92/09696およびWO93/21335を参照されたい。さらに、ウイルスベクターもまた、関心対象のタンパク質を発現するトランスジェニック植物を産生するために使用することができる。例えば、単子葉植物は、Mycogen Plant ScienceおよびCiba-Geigy(現在はSyngenta)に対する米国特許第5,569,597号ならびに両方ともBiosource、現在Large Scale Biologyに対する米国特許第5,589,367号および同第5,316,931号に記載されている方法を使用して、ウイルスベクターで形質転換することができる。
【0081】
以前に言及したように、DNA構築物が植物宿主に導入される様式は、本発明にとって決定的ではない。効率的な形質転換を提供する任意の方法が使用されてもよい。例えば、植物細胞形質転換のための種々の方法が本明細書に記載され、これには、TiまたはRi-プラスミド、およびアグロバクテリウム媒介形質転換を実行するための同様のものの使用が含まれる。多くの場合において、T-DNAボーダー、より特定にはライトボーダーによって、一方または両方の側で境界を接する、形質転換のために使用される構築物を有することが望ましい。これは、構築物が、形質転換のための様式としてアグロバクテリウム・ツメファシエンスまたはアグロバクテリウム・リゾゲネスを使用する場合に特に有用であるが、T-DNAボーダーは、形質転換の他の様式を用いる使用を見出し得る。アグロバクテリウムが植物細胞形質転換のために使用される場合、宿主中に存在するT-DNAまたはTiもしくはRiプラスミドとの相同組換えのために宿主に導入され得るベクターが使用され得る。ベクターの導入は、エレクトロポレーション、トリペアレンタルメイティング、および当業者に公知であるグラム陰性細菌を形質転換するための他の技術を介して実行されてもよい。アグロバクテリウム宿主へのベクター形質転換の様式は、本発明にとって決定的ではない。組換えのためのT-DNAを含むTiまたはRiプラスミドは、こぶ形成を引き起こすことが可能であるか、または可能でなくてもよく、vir遺伝子が該宿主中に存在している限り、本発明には決定的ではない。
【0082】
アグロバクテリウムが形質転換のために使用されるいくつかの場合において、T-DNAボーダー中にある発現構築物は、Ditta et al.(1980)およびEPO 0 120 515に記載されるようなpRK2またはその誘導体などの広い範囲のベクターに挿入される。形質転換されたアグロバクテリウムおよび形質転換された植物細胞の選択を可能にする、本明細書に記載されるような1種または複数のマーカーが、発現構築物およびT-DNAに含まれる。使用される特定のマーカーは、本発明にとって本質的ではなく、好ましいマーカーは、使用される宿主および構築物に依存する。
【0083】
アグロバクテリウムを使用する植物細胞の形質転換のために、移植片は、その形質転換を可能にするために十分な時間の間、形質転換されたアグロバクテリウムと合わされ、かつインキュベートされてもよい。形質転換後、アグロバクテリアは適切な抗生物質を用いる選択によって殺傷され、植物細胞は適切な選択培地を用いて培養される。一旦、カルスが形成されると、シュート形成は、植物組織培養および植物再生の分野で周知である方法に従って、適切な植物ホルモンを使用することによって促進され得る。しかし、カルスの中間段階が常に必要であるわけではない。シュート形成後、該植物細胞は、シュート形成を促進する培地に移すことができ、それによって、植物再生を完了する。次いで、植物は生育して種子になり、該種子は、将来の世代を樹立するために使用され得る。形質転換技術に関わらず、細菌タンパク質をコードする遺伝子は、ベクター中に植物プロモーター調節エレメント、ならびにNosなどのような3'-非翻訳転写終結領域を含めることによって、植物細胞において該遺伝子を発現するように適合された遺伝子移入ベクターに好ましく組み込まれる。
【0084】
植物を形質転換するための多数の技術に加えて、外来性遺伝子を接触される組織の型は、同様に変化してもよい。このような組織には、胚形成組織、I、II、およびIII型カルス組織、胚軸、成長点、根組織、師部における発現のための組織などが含まれるがこれらに限定されない。ほぼすべての植物組織が、本明細書に記載される適切な技術を使用して、脱分化の間に形質転換されてもよい。
【0085】
選択マーカーに加えて、レポーター遺伝子を加えることが望ましくあり得る。いくつかの例において、レポーター遺伝子は、選択マーカーとともに、または選択マーカーなしで使用されてもよい。レポーター遺伝子は、典型的には、レシピエントの生物または組織には存在せず、かつ典型的には、ある表現型の変化または酵素的特性を生じるタンパク質をコードする遺伝子である。このような遺伝子の例は、Weising et al., 1988において提供される。好ましいレポーター遺伝子には、大腸菌のuidA遺伝子座のβ-グルクロニダーゼ(GUS)、大腸菌のTn9からのクロラムフェニコールアセチルトランスフェラーゼ遺伝子、生物発光クラゲ、エクオレア・ビクトリア(Aequorea victoria)からの緑色蛍光タンパク質、およびホタル、フォチヌス・ピラリス(Photinus pyralis)からのルシフェラーゼ遺伝子が含まれる。次いで、レポーター遺伝子発現を検出するためのアッセイ法は、該遺伝子がレシピエント細胞に導入された後の適切な時点で実行され得る。好ましいこのようなアッセイ法は、形質転換細胞を同定するために、Jefferson et al.,(1987)によって記載されるような、大腸菌のuidA遺伝子座のβ-グルクロニダーゼ(GUS)をコードする遺伝子の使用を必要とする。
【0086】
植物プロモーター調節エレメントに加えて、種々の供給源からのプロモーター調節エレメントが、外来性遺伝子を発現するために、植物細胞において効率的に使用され得る。例えば、細菌起源のプロモーター調節エレメント、例えば、オクトピンシンターゼプロモーター、ノパリンシンターゼプロモーター、マンノピンシンターゼプロモーター;ウイルス起源のプロモーター、例えば、カリフラワーモザイクウイルス(35Sおよび19S)、35T(これは、再操作された35Sプロモーターである、米国特許第6,166,302号、とりわけ実施例7Eを参照されたい)などが使用されてもよい。植物プロモーター調節エレメントには、リブロース-1,6-ビスリン酸(RUBP)カルボキシラーゼスモールサブユニット(ssu)、β-コングリシニンプロモーター、β-ファセオニリンプロモーター、ADHプロモーター、熱ショックプロモーター、および組織特異的プロモーターが含まれるがこれらに限定されない。他のエレメント、例えば、マトリックス結合領域、足場付着領域、イントロン、エンハンサー、ポリアデニル化配列などが存在してもよく、従って、転写効率またはDNA組み込みを改善してもよい。このようなエレメントは、DNA機能のために必要であるかもしれないし、必要でないかもしれないが、これらは、転写、mRNA安定性などに影響を与えることによって、より良好な発現またはDNAの機能を提供することができる。このようなエレメントは、植物での形質転換されたDNAの最適な性能を得るために、必要に応じてDNAに含まれてもよい。典型的なエレメントには、Adh-イントロン1、Adh-イントロン6、アルファルファモザイクウイルスコートタンパク質リーダー配列、オスモチンUTR配列、トウモロコシストリークウイルスコートタンパク質リーダー配列、ならびに当業者に使用可能であるその他が含まれるがこれらに限定されない。構成的プロモーター調節エレメントもまた使用され、すべての細胞型およびすべての時点での連続的な遺伝子発現を方向付けることができる(例えば、アクチン、ユビキチン、CaMV 35Sなど)。組織特異的プロモーター調節エレメントは、特定の組織または組織型、例えば、葉または種子における遺伝子発現の原因であり(例えば、ゼイン、オレオシン、ナピン、ACP、グロブリンなど)、これらもまた使用されてもよい。
【0087】
プロモーター調節エレメントはまた、植物の発生の特定の段階の間に活性であり(または不活性であり)、かつ植物の組織および器官において活性であってもよい。このような例には、花粉特異的、胚特異的、トウモロコシ毛特異的、ワタ繊維特異的、根特異的、種子胚乳特異的、または栄養相特異的なプロモーターエレメントなどが含まれるがこれらに限定されない。特定の状況下で、特定のシグナルに応答する遺伝子、例えば:物理的刺激(熱ショック遺伝子)、光(RUBPカルボキシラーゼ)、ホルモン(Em)、代謝物、化学物質(テトラサイクリン応答性)、およびストレスに対して応答する遺伝子の発現の原因である誘導性プロモーター調節エレメントを使用することが所望であってもよい。植物において機能する他の所望の転写および翻訳エレメントが使用されてもよい。多数の植物特異的遺伝子移入ベクターが当技術分野で公知である。
【0088】
植物RNAウイルスを用いる系もまた、細菌タンパク質を発現するために使用され得る。そうすることにおいて、タンパク質を発現する遺伝子は、関心対象の宿主植物に感染する適切な植物ウイルスのコートプロモーター領域に挿入され得る。次いで、このタンパク質が発現され得、従って、除草剤による損傷からの植物の保護を提供する。植物RNAウイルスを用いる系は、Mycogen Plant Sciences, Inc. に対する米国特許第5,500,360号ならびにBiosource、現在はLarge Scale Biologyに対する米国特許第5,316,931号および同第5,589,367号に記載されている。
【0089】
選択剤
グルホシネートおよびビアラホスに加え、本発明に従って使用され得る選択剤には、本発明のDSM-2遺伝子によって媒介されるアセチルトランスフェラーゼ機序によって不活性化され得るすべての合成および天然のアナログが含まれる。例えば、図1を参照されたい。
【0090】
具体的に示されない限り、または暗示されない限り、1つの(「a」「an」)およびその(「the」)という用語は、本明細書で使用される場合、「少なくとも1つの」を意味する。
【0091】
本明細書で言及されるか、または引用されるすべての特許、特許出願、特許仮出願、および刊行物は、それらが本明細書の明示的な教示と矛盾しない範囲で、それらの全体が参照により本明細書に組み入れられる。
【0092】
以下は、本発明を実施するための手法を例証する実施例である。これらは、限定として解釈されるべきではない。他に注記されない限り、すべてのパーセンテージは重量によっており、すべての溶媒混合物の割合は体積によっている。
【0093】
実施例1-植物においてグルホシネートに対する抵抗性を付与する遺伝子を同定するための方法
植物または細胞培養物において除草剤分解活性を有する遺伝子を同定するための方法として、NCBI(National Center for Biotechnology Information)などの現在の公的なデータベースを調査することが可能である。このプロセスを開始するために、所望の特性(すなわち、ホスフィノトリシンアセチルトランスフェラーゼ)を有するタンパク質をコードする、すでに同定された機能的遺伝子配列を持っていることが必要である。次いで、このタンパク質配列は、使用可能である寄託されたNCBIタンパク質配列に対して比較するために、BLAST(基礎局所的アラインメント検索ツール(Basic Local Alignment Search Tool))(Altschul et al., 1997)アルゴリズムのためのインプットとして使用する。デフォルト設定を使用すると、この検索は、様々なレベルで100個までの相同タンパク質配列を回答する。これらは、アミノ酸レベルが、高い同一性(85〜98%)から非常に低い同一性(23〜32%)までの範囲である。伝統的には、高い相同性を有する配列のみが、インプット配列と同様の特性を保持していることが予測される。この場合、≦50%相同性を有する配列のみが選択される。本明細書において例示されるように、わずか30%のアミノ酸保存性(ストレプトマイセス・ハイグロスコピカス由来のpatと比較して)を有するホモログのクローニングおよび組換えによる発現は、形質転換していない植物細胞培養物から形質転換した植物細胞培養物を選択するために使用できる。
【0094】
DSM-2を、patに対して30%のみ、barに対し28%のみのアミノ酸同一性を有するホモログとしてNCBIデータベース(the ncbi.nlm.nih.gov ウェブサイト;アクセッション番号AAA26705)から同定した。同一性パーセントは、データベース中に寄託されたヌクレオチド配列を最初にタンパク質に翻訳すること、次いで複数配列のアラインメントを実行するためのVectorNTIソフトウェアパッケージ中のClustalWを使用することによって決定した。
【0095】
実施例2-植物および細菌における発現のための配列の最適化
2.1-バックグラウンド
植物における異種遺伝子のより高いレベルの発現を得るために、これらが植物細胞でより効率的に発現されるように、タンパク質をコードする遺伝子を再操作することが好ましい場合がある。トウモロコシは、このような植物の1つであり、該植物でのその発現レベルおよびコード化タンパク質のレベルを増加させるために、形質転換の前に、異種のタンパク質コード領域を再設計することが好ましい場合がある。それゆえに、細菌タンパク質をコードする遺伝子の設計におけるさらなる工程は、最適な発現のために異種遺伝子の再操作である。例えば、Kawabe et al.(2003), 「Patterns of Codon Usage Bias in Three Dicot and Four Monocot Plant Species」, Genes Genet. Syst., pp.343-352;およびIkemura et al.(1993), 「Plant Molecular Biology Labfax」, Croy, ed., Bios Scientific Publishers Ltd., p. 3748)、ならびにその中に引用されたすべての関連のある参照を参照されたい。
【0096】
トウモロコシにおける発現のための細菌タンパク質の再操作の1つの理由は、例えば、ネイティブ遺伝子の最適でないG+C含量に起因する。例えば、多くのネイティブな細菌遺伝子の非常に低いG+C含量(および高いA+T含量に向かう結果としての傾斜)は、きわめてA+Tリッチであることが知られている植物遺伝子制御配列を模倣または複製する配列の生成をもたらす。植物に導入される遺伝子のDNA中のあるA+Tリッチ配列(例えば、遺伝子プロモーター中に通常見出されるTATAボックス領域)の存在は、遺伝子の異常な転写を生じる可能性がある。他方、転写されたmRNAに存在する他の調節配列(例えば、ポリアデニル化シグナル配列(AAUAAA)またはプレmRNAスプライシングに関与する核内低分子RNAに相補的である配列)の存在は、RNAの不安定化をもたらす可能性がある。したがって、より好ましくは植物に最適化された遺伝子と呼ばれる、トウモロコシにおける発現のために細菌タンパク質をコードする遺伝子の設計における1つの目的は、より高いG+C含量を有するDNA配列、および好ましくは、代謝酵素をコードするトウモロコシ遺伝子の配列に近いものを生成することである。細菌タンパク質をコードする植物に最適化された遺伝子の設計の別の目的は、配列の改変が翻訳を妨害しないDNA配列を生成することである。
【0097】
表3は、どのように高G+C含量がトウモロコシにおいて存在するかを例証する。表3におけるデータより、遺伝子のコード領域は、GenBank(リリース71)から抽出し、かつ塩基組成は、MacVector(商標)プログラムを使用して計算した(Accelerys, San Diego, California)。イントロン配列は計算において無視した。
【0098】
(表3)トウモロコシ遺伝子のタンパク質コード領域のG+C含量の集計
a 遺伝子の数を()内に示す。
b 標準偏差を()内に示す。
c 合わせた群の平均は平均の計算において無視した。
【0099】
遺伝コードの冗長性/縮重によって与えられる柔軟性に起因して(すなわち、あるアミノ酸は1つより多くのコドンによって特定される)、異なる生物または生物のクラスにおけるゲノムの進化は、重複するコドンの示差的な使用を生じてきた。この「コドンの偏り」は、タンパク質コード領域の平均塩基組成を反映している。例えば、比較的低いG+C含量を有する生物は、冗長性コドンの第3の位置においてAまたはTを有するコドンを使用するのに対して、より高いG+C含量を有するものは第3の位置においてGまたはCを有するコドンを使用する。mRNA中の「マイナーな」コドンの存在は、とりわけ、そのマイナーなコドンに対応する荷電したtRNAの相対的な豊富さが低い場合に、そのmRNAの絶対的な翻訳速度を減少する可能性があると考えられている。このことの拡張は、個々のマイナーなコドンによる翻訳速度の減少が、複数のマイナーなコドンについて少なくとも相加的であることである。それゆえに、マイナーなコドンの高い相対含量を有するmRNAは、対応して低い翻訳速度を有する。この速度は、引き続く低レベルのコードされるタンパク質によって反映される。
【0100】
トウモロコシ(トウモロコシ、または他の植物、例えば、ワタ、ダイズ、コムギ、アブラナ属/アブラナ、イネ;またはより一般的には油料穀物、一般に、単子葉植物、双子葉植物、および半子葉(hemicot)植物)での発現のために細菌タンパク質をコードする遺伝子を操作する際に、植物のコドンの偏りが決定された。トウモロコシについてのこのコドンの偏りは、そのタンパク質をコードするために植物が使用する、統計学的なコドンの分布であり、好ましいコドン使用頻度は表4に示す。偏りを決定した後に、関心対象の遺伝子におけるコドンの頻度パーセントが決定される。植物によって好まれる第1のコドンが、ならびに複数の選択が存在する場合には好ましいコドンの第2、第3、および第4の選択が、決定されるべきである。次いで、細菌タンパク質のアミノ酸配列をコードする新規なDNA配列が設計され得るが、この新規な配列は、タンパク質のアミノ酸配列中の各位置におけるアミノ酸を特定するために、ネイティブなDNA配列(タンパク質をコードしている)とは、植物の(第1に好ましい、第2に好ましい、第3に好ましい、または第4に好ましい)コドンの置換によって異なっている。次いで、この新規な配列は、改変によって作製されたかもしれない制限酵素部位について分析する。同定された部位は、コドンを、第1の、第2の、第3の、または第4の選択の好ましいコドンで置き換えることによってさらに改変される。関心対象の遺伝子の転写または翻訳に影響を与え得る配列中の他の部位は、エキソン:イントロン連結(5'または3')、ポリA付加シグナル、またはRNAポリメラーゼ終結シグナルである。配列はさらに、TAまたはGCダブレットの頻度を減少するように分析および改変される。ダブレットに加えて、同じである約4個より多くの残基を有するGまたはC配列ブロックは、配列の転写に影響を与え得る。それゆえに、これらのブロックもまた、第1または第2の選択などのコドンを、次に好ましい選択のコドンで置き換えることによって改変される。
【0101】
(表4)トウモロコシにおいて発現されるタンパク質についての好ましいアミノ酸コドン
【0102】
細菌タンパク質をコードする植物に最適化された遺伝子は、約63%の第1の選択のコドン、約22%〜約37%の間の第2の選択のコドン、および約15%〜約0%の間の第3または第4の選択のコドンを含み、全体のパーセンテージが100%であることが好ましい。最も好ましい植物に最適化された遺伝子は、約63%の第1の選択のコドン、少なくとも約22%の第2の選択のコドン、約7.5%の第3の選択のコドン、および約7.5%の第4の選択のコドンを含み、全体のパーセンテージは100%である。上記の方法は、遺伝子が植物で最適に発現されるように、特定の植物に対して外来性である遺伝子を改変することを当業者に可能にする。この方法はさらに、PCT出願WO 97/13402において例証されている。
【0103】
従って、細菌タンパク質をコードする植物に最適化された遺伝子を設計するために、DNA配列は、特定の植物のために遺伝子配列から確立されたコドンの偏りの表から確立された冗長な遺伝コードを使用して、該タンパク質のアミノ酸配列をコードするように設計される。得られるDNA配列は、より高い程度のコドンの多様性、望ましい塩基組成を有し、戦略的に配置された制限酵素認識部位を含むことができ、遺伝子の転写、または生成物mRNAの翻訳を妨害するかもしれない配列を欠いている。従って、本発明のタンパク質/遺伝子に対して機能的に等価である合成遺伝子は、植物を含む宿主を形質転換するために使用することができる。合成遺伝子の産生に関するさらなる手引きは、例えば、米国特許第5,380,831号において見出すことができる。
【0104】
2.2-DSM-2植物再構築分析
ネイティブDSM-2コード領域の513塩基対(bp)のDNA配列(SEQ ID NO:1)の広範な分析は、最適な植物発現に対して有害であると考えられているいくつかの配列モチーフの存在、ならびに最適でないコドン組成を明らかにした。SEQ ID NO:1によってコードされるタンパク質はSEQ ID NO:2として提示される。単子葉植物ならびに双子葉植物における組換えタンパク質の産生を改善するために、SEQ ID NO:2として開示されるネイティブ配列と同一である1つのタンパク質(SEQ ID NO:4)をコードする「植物に最適化された」DNA配列DSM-2 v2(SEQ ID NO:3)を開発した。対照的に、ネイティブのコード領域のDNA配列および植物に最適化されたコード領域(v1)のDNA配列は78.3%だけ同一である。表5は、ネイティブ配列(カラムAおよびD)および植物に最適化された配列(カラムBおよびE)のコドン組成の違いを示し、ならびに理論的な植物に最適化された配列(カラムCおよびF)に対する比較を可能にする。
【0105】
(表5)ネイティブDSM-2、植物に最適化されたバージョン(v2)、および理論的な植物最適化バージョンのコード領域のコドン組成の比較
【0106】
表5の検討から、ネイティブのコード領域および植物に最適化されたコード領域は、同一のタンパク質をコードする一方で、実質的に互いに異なることが明白である。植物に最適化されたバージョン(v1)は、DSM-2タンパク質をコードする理論的な植物に最適化されたコード領域のコドン組成を密接に模倣する。
【0107】
実施例3-形質転換ベクターのクローニング
3.1-DSM-2(v2)を含有しているバイナリープラスミドの構築
DSM-2(v2)コドン最適化遺伝子をコードする配列(DASPICO45)を、制限酵素BbsI(New England Biolabs, Inc., Beverly MA, cat #R0539s)およびSacI(New England Biolabs, Inc., cat #R0156s)で切断した。得られたフラグメントを、対応する制限部位NcoI(New England Biolabs, cat #R0193s)およびSacIでpDAB773にライゲーションした。陽性コロニーを制限酵素消化を介して同定した。得られたクローンは、Rb7 MAR v3 // At Ubi10プロモーターv2 // 関心対象の遺伝子 // Atu Orf 1 3'UTR v3を含有していた。関心対象の遺伝子としてDSM-2(v2)を含有していたプラスミドを、pDAB3774と名付けた。
【0108】
Rb7 MAR v3 // AtUbi10プロモーターv2 // 関心対象の遺伝子 // Atu Orf 1 3'UTR v3カセットを、AgeI(New England Biolabs, Inc., cat #R0552s)制限フラグメントとしてバイナリーベクターpDAB3736へクローニングした。バイナリープラスミドの左側および右側の境界部の間にこのカセットをクローニングした。陽性コロニーを制限酵素消化および配列決定反応を介して同定した。Rb7 MAR v3 // AtUbi10プロモーターv2 // DSM-2 v2 // Atu Orf 1 3'UTR v3を含有している構築物を、pDAB3778と名付けた。
【0109】
Rb7 MAR v3 // AtUbi10プロモーターv2 // PATv3 // Atu Orf 1 3'UTR v3カセットを含有している対照構築物を、pDAB3736からGateWay attRデスティネーションカセットを除去することにより完成させた。pDAB3736を、PacI(New England Biolabs, Inc., cat #R0547s)制限酵素により消化した。PacIは、pDAB3736においてGateWay attRデスティネーションカセットに隣接している。PacIで消化されたプラスミドを、自己ライゲーションし、大腸菌Top10細胞(Invitrogen, Carlsbad CA, cat# C4040-10)に形質転換した。陽性コロニーを、制限酵素消化および配列決定反応を介して同定した。得られた構築物を、pDAB3779と名付けた。
【0110】
3.2-付加的な形質転換構築物のクローニング
適切な植物種への形質転換のために作出されたすべてのその他の構築物が、本明細書中に既に記載されたような類似した手法、およびその他の標準的な分子クローニング法(Maniatis et al., 1982)を使用して構築された。表6は、適切なプロモーターおよび定義された特色、ならびに形質転換された作物と共に、使用されたすべての形質転換構築物を列挙する。
【0111】
(表6)様々な植物種の形質転換において使用された構築物
*A=アラビドプシス
T=タバコ
R=イネ
Cn=トウモロコシ
CsVMV=キャッサバ葉脈モザイクウイルスプロモーター
AtUbi10=シロイヌナズナユビキチン10プロモーター
RB7 Mar v2=ニコチアナ・タバカム・マトリックス関連領域(MAR)
NtOsm=ニコチアナ・タバカム・オスモチン5'および3'非翻訳領域
ZmUbi1=ジー・メイズ(Zea mays)ユビキチン1プロモーター
【0112】
実施例4-原核生物および真核生物のプロモーターを使用したDSM-2(v2)による感受性大腸菌(BL-21)の補完
4.1-DSM-2(v2)を含有している大腸菌発現プラスミドの構築
DSM-2(v2)コドン最適化配列を、制限酵素BbsIおよびSacIで消化した。得られたフラグメントを、NcoIおよびSacIの対応する制限部位でpDAB779へクローニングした。pDAB779はpET28a(+)発現ベクターである(Novagen, Madison WI, cat# 69864-3)。DSM-2(v2)遺伝子コーディング配列を含有している陽性コロニーを、制限酵素消化を介して同定した。DSM-2 // pET28a(+)構築物をpDAB4412と名付けた。
【0113】
発現プラスミドpET(空ベクター対照)、およびpDAB4412を、標準的な方法を使用して、大腸菌T7発現株BL21-Star(DE3)(Invitrogen, Carlsbad CA, cat# C6010-03)に形質転換した。50μg/ml抗生物質および75μM IPTG(イソプロピル-α-D-チオガラクトピラノシド)を含有している250mLのLB培地への10〜200個の新しく形質転換されたコロニーにより、発現培養物を開始させた。培養物を180〜200rpmで24時間28℃で増殖させた。4℃で10分間、3,400×gで、250mlのNalgeneボトルにおける遠心分離によって細胞を収集した。ペレットを4〜4.5mLのバターフィールドリン酸(Butterfield's Phosphate)溶液(Hardy Diagnostics, Santa Maria, CA; 0.3mMリン酸カリウムpH7.2)に懸濁させた。懸濁した細胞を、直径0.1mmのガラスビーズ(Biospec, Bartlesville, OK, catalog number 1107901)1mLを含む50mLポリプロピレンスクリューキャップ遠心管に移した。細胞-ガラスビーズ混合物を氷上で冷却し、次いで、およそ20の出力でBranson Sonifier 250(Danbury CT)を用いて、2mmプローブを使用して、2回の45秒の突発波(突発波間には完全に冷却)を用いた超音波処理によって細胞を溶解させた。溶解物を2mLエッペンドルフチューブに移し、16,000×gで5分間、遠心分離した。上清を収集し、タンパク質濃度を測定した。Bio-Rad Protein Dye Assay ReagentをH2Oで1:5希釈し、1mLを、1:10希釈の各試料10μL、ならびに5、10、15、20、および25μg/mLの濃度のウシ血清アルブミン(BSA)に添加した。Shimadzu UV 160U分光光度計(Kyoto, JP)で595nmの波長で光学濃度を測定する分光光度計で試料を読み取った。各試料に含有されていたタンパク質の量を、BSA標準曲線に対して計算し、リン酸緩衝液で3〜6mg/mLの間に調整した。発現されたタンパク質を可視化するため、溶解物をSDSタンパク質ゲル上で泳動した。
【0114】
4.2-BASTAに対する感受性についての一般的なクローニング株の評価
大腸菌およびアグロバクテリウム・ツメファシエンスの選択された細胞系を、漸増的に増加する濃度のグルホシネート(BASTA)を含有している最少培地上に播種した。細胞系;BL21-Star(DE3)、Top10、DH5α、アグロバクテリウム・ツメファシエンスC58、およびアグロバクテリウム・ツメファシエンスLBA4404sを、まず、複合培地上で増殖させた。大腸菌株はLBで増殖させ、アグロバクテリウム・ツメファシエンス株はYEPで増殖させた。5マイクロリットルの細菌培養物を、様々な濃度のグルホシネートを含有している最少培地プレートへ均一に播種し、分散させた。濃度は、0μg/ml、250μg/ml、500μg/ml、1000μg/ml、2000μg/ml、および4000μg/mlのBASTAからなっていた。さらに、対照として細菌株を複合培地のプレートへ播種した。アグロバクテリウム・ツメファシエンス株はYEP寒天プレート上に播種し、大腸菌株はLB寒天プレート上に播種した。大腸菌株を含有しているプレートは、37℃で24時間インキュベートした。アグロバクテリウム・ツメファシエンス株を含有しているプレートは、25℃で48時間インキュベートした。割り当てられたインキュベーション時間の後、プレートを細菌増殖について観察した。表7は、様々な株の、グルホシネートを含有している最少培地で増殖する能力を例示している。1つの株BL21-Star(DE3)細胞系のみが、グルホシネートによって実質的に阻害された。
【0115】
(表7)最少培地上で増殖させた様々な細菌株のグルホシネートに対する応答
大腸菌は37℃で24時間増殖させた。アグロバクテリウムは25℃で48時間増殖させた。+++ = 激しい菌叢増殖 // ++ = 軽度の菌叢増殖 // + = 斑状の菌叢増殖 // -- = 増殖なし
【0116】
4.3-大腸菌BL21-Star(DE3)の細胞増殖を補完するためのDSM-2(v2)の組み換え発現
PAT(v3)を含有しているpET28a(+)発現プラスミドを、陽性対照として構築した。PAT(v3)を、NcoI-SacIフラグメントとして、pDAB779の対応する制限部位へクローニングした。PAT(v3)遺伝子フラグメントを含有している陽性クローンについて、制限酵素消化を介して確証した。この構築物をpDAB4434と名付けた。
【0117】
プラスミドpDAB4434、pDAB4412、および空pETベクター(対照)を、大腸菌BL21-Star(DE3)細菌細胞に形質転換した。発現培養物を、50μg/mlの抗生物質および75μM IPTG(イソプロピル-α-D-チオガラクトピラノシド)を含有しているLB培地250mLへの10〜200個の新しく形質転換されたコロニーにより開始させた。培養物を180〜200rpmで28℃で24時間増殖させた。5マイクロリットルの培養物を、複合培地対照および漸増的に増加する濃度のグルホシネートおよび20μM IPTGを含有している最少培地へ播種した。培養物を、プレート上に均一に分散させ、28℃で24時間インキュベートした。割り当てられたインキュベーション時間の後、プレートを細菌増殖について観察した。これらの結果は表8に例示される。
【0118】
(表8)
大腸菌は20uM IPTGを含む培地上で28℃で24時間増殖させた。-- = 増殖なし // +++ = 明瞭なコロニー増殖
【0119】
4.4-大腸菌BL21-Star(DE3)細胞におけるDSM-2の組み換え発現を駆動するための植物プロモーターの使用
ウイルスプロモーターCsVMVまたは植物プロモーターAtUbi10のいずれかの下でDSM-2(v2)遺伝子およびPAT遺伝子のコーディング配列が発現されたプラスミド構築物を、グルホシネートを含有している最少培地で補完について分析した。プラスミド3778(Rb7 MARv3 // AtUbi10プロモーター // DSM-2(v2)// Atu Orf 1 3'UTR)、3779(Rb7 MARv3 // AtUbi10プロモーター // PAT // Atu Orf 1 3'UTR)、3264(CsVMVプロモーター // DSM-2 // Atu Orf24 3'UTR)、3037(CsVMVプロモーター // PAT // Atu Orf 25/26 3'UTR)、および770(CsVMVプロモーター // GUS v3 // Atu Orf 24 3'UTRを含有している対照プラスミド)を、大腸菌BL21-Star(DE3)に形質転換し、複合培地中で増殖させた。5マイクロリットルの培養物を、増加する濃度のグルホシネートを含有している最少培地に播種し、37℃で48時間インキュベートした。結果は表9に例示される。これらのデータは、植物およびウイルスのプロモーターが、細菌細胞内で中レベルの機能性を有しており、選択マーカーの発現を駆動するために使用され得ることを示している。
【0120】
(表9)
大腸菌は37℃で48時間増殖させた。++++ = 激しい菌叢増殖;+++ = 菌叢増殖;++ = 多数の明瞭なコロニー;+ = まばらなコロニー増殖
【0121】
実施例5-生化学的特徴決定のためのDSM-2の精製およびウエスタン分析のための抗体作製
5.1-組み換え発現
プラスミドpDAB4412中のコドン最適化DSM-2(v2)遺伝子を保持している(Invitrogen, Carlsbad, CAから購入された)大腸菌BL-21(DE3)Star細胞を、種の調製のため、37℃で一晩、50μg/mlカナマイシンが補足された3mlのLB培地に播種するために使用した。およそ2mlの種培養物を、2.8Lの整流装置付き三角フラスコの中のカナマイシン(50μg/ml)を含有している1Lの新鮮なLBに移した。培養物を、0.8〜1.0に近いOD600を入手するためにおよそ6時間、250RPMでシェーカー(New Brunswick Scientific, Model Innova 44)上で37℃でインキュベートした。イソプロピルβ-D-1-チオガラクトピラノシド(IPTG)を培養物中に最終75μMで添加し、一晩の誘導のため18℃でインキュベートし続けた。4℃で15分間8,000RPMでの遠心分離によって細胞を採集し、細胞ペーストを-80℃で保存するか、または精製のために直ちに処理した。
【0122】
1Lの培養物から湿重量でおよそ5gの大腸菌細胞を解凍し、20mMトリス-HCl、pH 8.0および0.3mlのProtease Inhibitor Cocktail(Sigma, cat# P8465)を含有している300mlの抽出緩衝液に再懸濁させ、超音波処理によって15分間氷上で破砕した。溶解物を20分間24,000RPMで4℃で遠心分離し、上清を0.8μmおよび0.45μmの膜でろ過した。すべてのその後のタンパク質分離は、Pharmacia AKTA Explorer 100を使用して実施し、4℃で操作した。ろ液を、20mMトリス-HCl、pH 8.0緩衝液で平衡化されたQXL Sepharose Fast Flowカラム(Pharmacia HiPrep 16/10、20mlベッドサイズ)に10ml/分で適用した。溶出液のOD280が基線に戻るまで、この緩衝液でカラムを洗浄し、5mlの画分を収集しながら、5ml/分の流速で0.5Lの0〜0.4M NaCl直線勾配によりタンパク質を溶出させた。グルホシネート変換活性にも対応する、見かけの20kDaのバンド(予測されたDSM-2分子量は19.3kDaである)を含むSDS-PAGEによって決定されるような、DSM-2を含有している画分をプールした。試料を4倍容量の5mM DTT、0.5%トリトンX-100、5%グリセロールを含有している20mMトリス-HCl、pH 7.5緩衝液により希釈し、4ml/分でMono Qカラム(Pharmacia 10/100 GL、8mlベッドサイズ)に再適用した。同緩衝液中の0.1〜0.3M NaCl勾配により、タンパク質を溶出させた。DSM-2を含有している主要ピークをプールし、固形硫酸アンモニウムを最終1.0Mで添加し、20mMトリス-HCl、pH 7.5中の1.0M硫酸アンモニウムで平衡化されたPhenyl Fast Flowカラム(Pharmacia HiTrap、5mlベッドサイズ)に適用した。溶出液のOD280が基線に戻るまで、このカラムを4ml/分で平衡緩衝液により洗浄し、次いで、20mMトリス-HCl、pH 7.5中の1.0M〜0 硫酸アンモニウムの直線勾配により、50分(3ml/分)以内に、タンパク質を溶出させ、3mlの画分を収集した。75mS/cmで溶出したDSM-2を含有している主要ピーク画分をプールし、MWCO 10kDa膜遠心濾過装置(Millipore)を使用して、およそ3mg/mlにまで濃縮した。次いで、試料を、1ml/分の流速で、PBS緩衝液により、Superdex 75ゲル濾過カラム(Pharmacia XK 16/60、110mlベッドサイズ)に適用した。純粋なDSM-2を含有しているピーク画分をプールし、-80℃で保存した。標準としてウシ血清アルブミンを使用して、ブラッドフォード(Bradford)アッセイまたは全アミノ酸分析によってタンパク質濃度を決定した。精製されたDSM-2の活性を、ホスフィノトリシンアセチルトランスフェラーゼ(PAT)アッセイのための標準的な手法(Wehrmann et al. 1996)に基づき測定した。
【0123】
5.2-抗体作製
DSM-2に対するウサギポリクローナル抗体を、Invitrogen Antibody Services(South San Francisco, CA, Cat# M0300)によって提供されるRabbit Polyclonal Antibody-Standard Protocolsを使用して作製した。大腸菌において発現され精製されたDSM-2(前記セクションを参照されたい)を、免疫原として供給した。簡単に説明すると、2羽のニュージーランドウサギに、0.25mgのキーホールリンペットヘモシアニンおよび不完全フロイントアジュバント(IFA)により乳化された1mgのDSM-2タンパク質を皮下(SQ)注射した。ウサギを2週間休息させ、3週間の休息期間を空けて、IFAで乳化された0.5mgのDSM-2タンパク質により3回、SQ追加刺激した。最終追加刺激の2週間後、各ウサギから血清を収集し、力価について直接ELISAで試験した(示されないデータ)。特異抗体のより良好な力価を与えた2番のウサギで、付加的な2回の追加刺激および終末採血(Invitrogen Cat#M0311およびM0313)を実施した。
【0124】
5.3-ウエスタンブロッティング分析
およそ100mgのカルス組織を、3個のステンレス鋼BBビーズを含有している2mLの微量遠心管に入れた。250μLの抽出緩衝液(0.1%トリトンX-100、10mM DTT、および1mL当たり5μLのプロテアーゼ阻害剤カクテルを含有しているリン酸緩衝生理食塩水)を添加し、チューブをGeno/Grinder(Model 2000-115, Certiprep, Metuchen, NJ)に固定し、500rpmの1×という設定で6分間振とうした。チューブを10,000×gで10分間遠心分離し、可溶性タンパク質を含有している上清を、別々のチューブにピペットで移し、氷中に保存した。ペレットを上記のようにして抽出し(2回目)、上清を以前の画分と共にプールし、分析した。
【0125】
植物試料から抽出されたタンパク質をレムリ(Laemmli)緩衝液中で変性させ、95℃で10分間インキュベートした。変性タンパク質を、製造業者のプロトコルに従って、Novex 8〜16%Tris-Glycineプレキャストゲル(Invitrogen Cat#EC60452BOX)で分離した後、標準的なプロトコルを使用してニトロセルロース膜に転写した。
【0126】
すべてのウエスタンブロッティングインキュベーション工程を室温で1時間実施した。ブロットを、まず、4%乳を含有しているPBS(PBSM)でブロッキングし、次いで、PBSMで5000倍希釈されたDSM-2特異的ウサギポリクローナル抗体(前記パラグラフを参照されたい)中でインキュベートした。0.05%トゥイーン-20を含有しているPBS(PBST)で5分間3回洗浄した後、ヤギ抗ウサギ抗体/西洋ワサビペルオキシダーゼコンジュゲートをブロット上でインキュベートした。検出されたタンパク質を、化学発光基質(Pierce Biotechnology, Rockford, IL Cat# 32106)およびX線フィルム露光を使用して可視化した。
【0127】
実施例6-アラビドプシスへの形質転換および選択
6.1-アラビドプシス・サリアナ生育条件
野生型アラビドプシス種子を、0.1%アガロース(Sigma Chemical Co., St. Louis, MO)溶液中に懸濁した。懸濁した種子を、4℃で2日間保存し、休止要件を完了し、かつ同調的な種子の発芽を確実にした(層形成)。
【0128】
Sunshine Mix LP5(Sun Gro Horticulture, Bellevue, WA)を、微細なバーミキュライトで覆い、濡れるまで下側からHoagland溶液を与えた。層形成された種子を播種し、湿気ドーム(KORD Products, Bramalea, Ontario, Canada)で7日間覆った。
【0129】
種子を発芽させ、植物を、Conviron(モデルCMP4030およびCMP3244、Controlled Environments Limited, Winnipeg, Manitoba, Canada)中で、定常的な温度(22℃)および湿度(40〜50%)下で、長日条件(16時間光/8時間暗)下で、120〜150μmol/m2秒の光強度で生育させた。植物を、初期にはHoagland溶液で水をやり、次には脱イオン水で水をやり、土壌の湿り気を保つが濡れないように維持した。
【0130】
6.2-アグロバクテリウム形質転換
画線したDH5αコロニーを含み、エリスロマイシン(Sigma Chemical Co., St. Louis, MO)(200mg/L)またはスペクチノマイシン(100mg/L)を含むLB+寒天を使用して、4mlミニプレップ培養(液体LB+エリスロマイシン)に接種するためにコロニーを提供した。この培養物を、定常的に攪拌しながら、37℃で一晩インキュベートした。製造業者の指示に従って実施するQiagen(Valencia, CA)Spin Mini Prepsを使用して、プラスミドDNAを精製した。
【0131】
電気的に形質転換受容性であるアグロバクテリウム・ツメファシエンス(株Z707、EHA101、およびLBA4404)細胞を、Weigel and Glazebrook(2002)のプロトコールを使用して調製した。形質転換受容性アグロバクテリウム細胞を、Weigel and Glazebrook(2002)から適合したエレクトロポレーション法を使用して形質転換した。50μlの形質転換受容性アグロ細胞を氷上で融解し、10〜25ngの所望のプラスミドを細胞に加えた。DNAおよび細胞混合物を、あらかじめ冷やしたエレクトロポレーションキュベット(2mm)に加えた。Eppendorf Electroporator 2510を、以下の条件:電圧: 2.4kV、パルス長: 5m秒を用いて形質転換のために使用した。
【0132】
エレクトロポレーションの後、1mlのYEPブロス(1リットルあたり: 10g酵母抽出物、10gバクト-ペプトン、5g NaCl)をキュベットに加え、細胞-YEP懸濁物を15ml培養チューブに移した。細胞を、定常的に攪拌しながら、ウォーターバス中で28℃にて4時間インキュベートした。インキュベーション後、培養物を、エリスロマイシン(200mg/L)またはスペクチノマイシン(100mg/L)およびストレプトマイシン(Sigma Chemical Co., St. Louis, MO)(250mg/mL)を有するYEP+寒天上に蒔いた。プレートを、2〜4日間、28℃にてインキュベートした。
【0133】
コロニーを選択し、エリスロマイシン(200mg/L)またはスペクチノマイシン(100mg/L)およびストレプトマイシン(250mg/mL)を有する新鮮なYEP+寒天上に画線し、28℃で1〜3日間インキュベートした。コロニーをPCR分析のために選択して、ベクター特異的プライマーを使用することにより、遺伝子インサートの存在を確認した。以下を例外として、製造業者の指示に従って実施するQiagen Spin Mini Prepsを使用して、選択されたアグロバクテリウムコロニーからプラスミドDNAを精製した。DNA精製のために、15mlの一晩ミニプレップ培養物(液体YEP+エリスロマイシン(200mg/L)またはスペクチノマイシン(100mg/L)およびストレプトマイシン(250mg/mL))の4mlアリコートを使用した。Qiagen Spin Mini Prep DNAを使用することの代替として、10μlの水中に懸濁した、形質転換したアグロバクテリウム細胞を、100℃で5分間溶解した。アグロバクテリウム形質転換において使用したバイナリーベクターからのプラスミドDNAを、対照として含めた。PCR反応を、製造業者の指示に従って、0.5×濃度で、Takara Mirus Bio Inc.(Madison, Wisconsin)からのTaq DNAポリメラーゼを使用して完了した。PCR反応を、以下の条件:1)94℃3分間、2)94℃45秒間、3)55℃30秒間、4)72℃1分間、29サイクル、次に72℃で10分間の1サイクルでプログラムされたMJ Research Peltier Thermal Cycler中で実行した。反応物を、サイクリング後に4℃で維持した。増幅を、1%アガロースゲル電気泳動によって分析し、臭化エチジウム染色によって可視化した。そのPCR産物がプラスミド対照と同一であったコロニーを選択した。
【0134】
6.3-アラビドプシス形質転換
アラビドプシスを、フローラルディップ法を使用して形質転換した。選択したコロニーを、エリスロマイシン(200mg/L)またはスペクチノマイシン(100mg/L)およびストレプトマイシン(250mg/mL)を含むYEPブロスの1つまたは複数の15〜30mlのプレ培養物に接種するために使用した。培養物を、220rpmで定常的に攪拌しながら、28℃で一晩インキュベートした。各プレ培養物を、エリスロマイシン(200mg/L)またはスペクチノマイシン(100mg/L)およびストレプトマイシン(250mg/mL)を含むYEPブロスの2つの500ml培養に接種するために使用し、培養物を、定常的に攪拌しながら、28℃で一晩インキュベートした。次いで、細胞を、約8700×gで10分間、室温でペレット化し、得られる上清を廃棄した。この細胞ペレットを、1/2×MurashigeおよびSkoog塩/GamborgのB5ビタミン、10%(w/v)スクロース、0.044μM ベンジルアミノプリン(DMSO中の1mg/ml保存液の10μl/リットル)および300μl/リットルSilwet L-77を含む500mlの浸透培地に穏やかに再懸濁した。約1ヶ月齢の植物を15秒間、培地に浸し、最新の花序を確実に沈める。次いで、植物を、それらの側面を下にして横たえ、24時間覆い(透明または不透明)、次いで水で洗浄し、かつ直立状態で配置した。植物を22℃で、16時間明期/8時間暗期の光周期で生育させた。浸漬の約4週間後、種子を収集した。
【0135】
6.4-形質転換植物の選択
新しく収集したT1種子[DSM-2(v2)遺伝子]を、室温で7日間乾燥させた。T1種子を、26.5×51cm発芽トレイ(T.O. Plastics Inc., Clearwater, MN)中に播種し、各々は、40mlの0.1%アガロース溶液に事前に懸濁された、層形成されたT1種子(約10,000個の種子)の200mgアリコートを受容し、4℃で2日間保存し、休止要件を完了し、かつ同調的な種子の発芽を確実にした。
【0136】
Sunshine Mix LP5(Sun Gro Horticulture Inc., Bellevue, WA)を、微細なバーミキュライトで覆い、濡れるまで下側からHoagland溶液を与え、次いで、重力で水抜きした。各40mlのアリコートの層形成された種子を、ピペットを用いてバーミキュライト上に均一に播種し、湿気ドーム(KORD Products, Bramalea, Ontario, Canada)で4〜5日間覆った。2,4-D出芽後噴霧を使用する初期の形質転換体選択(同時形質転換AAD-12遺伝子の選択;USSN 60/731,044を参照)の1日前にドームを取り外した。
【0137】
播種後(DAP)7日、および11DPAで再度、T1植物(それぞれ子葉および2〜4-lf段階)に、2,4-D除草剤(456g ae/L 2,4-D Amine 4、Dow AgroSciences LLC, Indianapolis, IN)の0.016%溶液を、10ml/トレイの噴霧量で(703L/ha)、適用あたり50 g ae/ha 2,4-D DMAの有効割合を送達するために、DeVilbiss圧縮空気噴霧チップを使用して噴霧した。生き残り(活動的に生育している植物)を、最後の噴霧の4〜7日後に同定し、鉢植え用培地(Metro Mix 360)で調製した3インチポットに個別に移植した。移植した植物を、以前と同様に、湿気ドームで3〜4日覆い、22℃グロースチャンバー中に配置した、または直接温室に移動した。次に、ドームを取り外し、DSM-2(v2)がグルホサミン除草剤抵抗性を提供する能力について試験する少なくとも1日前に、植物を温室(22±5℃、50±30% RH、14時間明期:10時間暗期、最低で500μE/m2s1の天然+追加の光)で栽培した。
【0138】
次いで、T1植物を、様々な割合のグルホサミンにランダムに割り当てた。アラビドプシスについては、140g ai/ha グルホサミンが、意味のあるレベルの抵抗性を有する植物から、感受性の植物を区別するための有効用量である。上昇した割合は、抵抗性の相対レベルを決定するためにも適用した(280、560、または1120g ai/ha)。表10は、アリールオキシアルカノエート除草剤抵抗性遺伝子(AAD-12 v1)に及ぶ比較を示す;USSN 60/731,044を参照。
【0139】
すべてのグルホシネート除草剤適用が、187L/haの噴霧量でトラック噴霧器によって適用された。市販のLiberty(商標)剤(200g ai/L、Bayer Crop Science, Research Triangle Park, NC)。グルホシネートに対する耐性を示したT1植物に、T2世代においてさらにアクセスした。
【0140】
6.5-形質転換植物の選択の結果
最初のアラビドプシス形質転換は、DSM-2(v2)(植物に最適化された遺伝子)を使用して行った。T1形質転換体を、まず、2,4-D DMA選択スキームを使用して、非形質転換種子のバックグラウンドから選択した。100,000個を超えるT1種子をスクリーニングし、260個の2,4-D抵抗性植物(AAD-12遺伝子)を同定した。これは、AAD-12+2,4-Dが選択のために使用される場合の構築物の選択頻度の正常範囲よりわずかに高い0.26%という形質転換/選択頻度に等しかった。上記で選択されたT1植物を、続いて個々のポットに移植し、様々な割合の市販のグルホシネート除草剤を噴霧した。表10は、アラビドプシスT1形質転換体にグルホシネート抵抗性を付与するためのDSM-2(v2)遺伝子および対照遺伝子の応答を比較している。応答は、視覚的損傷% 2WATによって提示される。データは、ほとんどもしくは全く損傷なし(<20%)、中程度の損傷(20〜40%)、または重度の損傷(>40%)を示す個体のヒストグラムとして提示された。各T1は独立した形質転換事象であるので、所定の割合における個々のT1応答の有意な変動が予想される。算術平均および標準偏差が各処理について提示される。非形質転換野生型アラビドプシスは、グルホシネート感受性対照として働いた。DSM-2(v2)遺伝子は、個々のT1アラビドプシス植物に除草剤抵抗性を付与した。所定の処理において、植物応答のレベルは大きく変動した。これは、各植物が独立した形質転換事象を表すという事実に起因し得る。重要な注目点として、140g ai/haグルホシネートより上で、影響を受けなかった個体が存在する一方で、重度に影響を受けたものもあった。割合による集団全体の損傷平均が、DSM-2(v2)で形質転換された植物と、野生型対照またはAAD-12+PAT形質転換対照との間の有意な違いを単に実証するために、表10に提示される。多くのDSM-2(v2)個体は、ほとんどまたは全く損傷なしに、1,120g ai/haグルホシネートに耐えて生存した。
【0141】
(表10)野生型植物およびT1 AAD-12+PAT植物と比較された、発芽後適用された一連のグルホシネート割合に対するT1 DSM-2 v2(植物最適化)形質転換T1アラビドプシスの応答
【0142】
6.6-選択マーカーとしてのDSM-2(v2)
選択剤としてグルホシネートを使用して、選択マーカーとしてDSM-2(v2)を使用する能力を、上記のように形質転換されたアラビドプシスを用いて分析した。DSM-2(v2)を含有しているおよそ100個のT1世代アラビドプシス種子(100〜150個の種子)または2mgのPATを含有しているホモ接合性T5植物を、およそ10,000個の野生型(感受性)種子に加えた(spiked)。植物の各トレイは、以下の処理時点:7 DAPおよび11 DAPで、2回の280g ai/haグルホシネートの適用を受けた。処理は、以前に記載されたようなDeVilbiss噴霧チップを用いて適用された。各々から、別に2mgのT1世代アラビドプシス種子を播き、比較カウントとして噴霧しなかった。17 DAPに、植物を抵抗性または感受性として同定した。処理されたものと未処理のものとの間のカウントは類似していることが表11に示された。これらの結果は、DSM-2(v2)が、集団のための代替的な選択マーカーとして有効に使用可能であることを示す。
【0143】
(表11)播かれた種子の量および280g ai/haグルホシネート処理後に生存していた植物の数
【0144】
6.7-分子分析
6.7.1-組織の収集、DNAの単離および定量Xμl
新鮮な組織をチューブに配置し、4℃で2日間凍結乾燥する。組織を完全に乾燥させた後、タングステンビーズ(Heavy Shot)をチューブ中に配置し、試料を、1分間、Kelcoビーズミルを使用する乾式粉砕に供する。次いで、標準的なDNeasy DNA単離手法に従う(Qiagen, DNeasy 69109)。次いで、抽出されたDNAのアリコートをPico Green(Molecular Probes P7589)で染色し、濃度をng/μlで得るために既知の標準を用いてフルオロメーター(波長485/530-BioTek)で読み取る。
【0145】
6.7.2-インベーダーアッセイ分析Xμl
DNA試料を0.7ng/μlに希釈し、次いで95℃で10分間、サーモサイクラー中のインキュベーションによって変性させる。次いで、インベーダーアッセイ反応混合物を、Third Wave Technologiesにより発表された96穴フォーマット手順に従うことにより調製する。調製された反応混合物7.5μlを96穴プレートの各ウェルに分配し、続いて7.5μlの対照および0.7ng/μlの希釈された変性未知試料を分配する。各ウェルに15μlのミネラルオイル(Sigma)を重層する。次いで、プレートを、63℃で1時間インキュベートし、フルオロメーター(Biotek)上で読み取る。標的プローブについてのバックグラウンドに対するシグナルの%(FAM色素波長560/620)を、内部対照プローブについてのバックグラウンドに対するシグナルの%(RED色素波長485/530)で除する計算は、比率を計算する。その比率は、事象の接合状態を決定するために使用される。
【0146】
6.8-遺伝性
多様なT1事象を、T2種子を産生させるために自家受粉した。これらの種子を、100個のランダムなT2同胞にグルホシネート(200g ai/ha)を適用することによって後代検定した。個々の各T2植物を、3インチ四方のポットに移植した後、噴霧適用した(187L/ha適用割合のトラック噴霧器)。T1ファミリー(T2植物)の63%が、χ二乗検定によって決定されるように(P>0.05)、メンデル遺伝に伴う優性遺伝性単一遺伝子座についての予測された3抵抗性:1感受性モデルに分離した。
【0147】
単一遺伝子座として分離した系の各々からランダムに選択された16の植物体に対して接合状態についてのインベーダー法を実施した。インベーダー法によりホモ接合性と判定されたT2個体から種子を収集した(T3種子)。インベーダー法によりホモ接合性と判定された4つのT2ファミリーの各々からの25のT3同胞を、以前に記載したように後代検定した。ホモ接合性(非分離集団)であると予測されたT2ファミリーのすべてが、非分離性であった。これらのデータは、DSM-2(v2)が安定的に組み込まれ、メンデル様式で少なくとも3世代まで遺伝することを示す。
【0148】
実施例7-除草剤を使用したトウモロコシのウィスカー媒介形質転換
7.1-DSM-2(v2)のクローニング
DSM-2(v2)遺伝子を、Bbs1/Sac1フラグメントとしてDASPICO45ベクターから切り出した。これを、ZmUbi1単子葉植物プロモーターを含有している同様に切断されたpDAB3812ベクターに定方向でライゲーションした。2つのフラグメントを、T4 DNAリガーゼを使用して一緒にライゲーションし、DH5α細胞に形質転換した。QiagenのQIA Spinミニプレップキットを使用して、得られたコロニーに対してミニプレップを実施し、方向について確認するためこれらのコロニーを消化した。ZmUbi1/DSM-2(v2)/ZmPer5を含有している最終構築物をpDAB3250と名付けた。PAT遺伝子を含有している同一の対照ベクターを上記のようにして構築した。この構築物をpDAB3251と名付けた。
【0149】
7.2-カルス/懸濁の開始
カルス培養の開始のために未成熟胚を得るために、温室で生長したHi-II親AおよびBの間のF1交雑を実施した(Armstrong et al. 1991)。胚が1.0〜1.2mmのサイズになったら(受粉後約9〜10日)、穂を収集し、表面を、Liqui-Nox(商標)で磨いて洗浄することによって表面殺菌し、70%エタノールに2〜3分間浸漬し、次いで20%の市販の漂白剤(0.1%次亜塩素酸ナトリウム)に30分間浸漬した。
【0150】
穂を、滅菌した蒸留水ですすぎ、未成熟接合体胚を無菌的に切除し、15Ag10培地(N6培地(Chu et al., 1975)、1.0mg/L 2,4-D、20 g/L スクロース、100mg/L カゼイン加水分解物(酵素消化)、25mM L-プロリン、10 mg/L AgN03、2.5 g/L Gelrite, pH 5.8)上で、胚盤を培地から離れるように向けて2〜3週間培養した。適切な形態(Welter et al., 1995)を示す組織を、約6週間の間、週に2回の間隔で新鮮な15Ag10培地に選択的に移し、次いで、約2ヶ月間の間、週に2回の間隔で4つの培地(N6培地、1.0mg/L 2,4-D、20g/L スクロース、100mg/L カゼイン加水分解物(酵素消化)、6 mM L-プロリン、2.5 g/L Gelrite、 pH 5.8)に移した。
【0151】
胚形成懸濁培養を開始するために、単一の胚から生じたカルス組織の約3mlの充填した細胞量(PCV)を、約30mlのH9CP+液体培地(MS基礎塩混合物(Murashige and Skoog, 1962)、10倍少ないニコチン酸および5倍多いチアミン-HCl、2.0mg/L 2,4-D、2.0mg/L α-ナフタレン酢酸(NAA)、30g/Lスクロース、200mg/L カゼイン加水分解物(酸消化)、100mg/L ミオ-イノシトール、6mM L-プロリン、5% v/v ココナッツ水(継代培養直前に加えた)、pH 6.0を含む改変MSビタミン)に加えた。懸濁培養を、暗条件下で、125ml Erlenmeyerフラスコ中で、125rpmおよび28℃に設定した温度制御シェーカー中で維持した。細胞系統は、典型的には、開始後2ヶ月から3ヶ月以内に樹立されるようになる。樹立の間、ワイドボアピペットを使用して3ml PCVおよび7mlの馴化培地を20mlの新鮮なH9CP+液体培地に加えることによって、懸濁物を3.5日毎に継代培養した。一旦、組織が成長で倍加を開始したら、懸濁物をスケールアップし、500mlフラスコに維持し、それによって、12ml PCVの細胞および28ml 馴化培地を80ml H9CP+培地に移した。一旦懸濁物が完全に樹立されると、将来の使用のために凍結保存した。
【0152】
7.3-凍結保存および懸濁物の融解
継代培養の2日後、4ml PCVの懸濁細胞および4mlの馴化培地を8mlの凍結防止剤(ココナッツ水を含まないH9CP+培地に、1Mグリセロール、1M DMSO、2Mスクロースを溶解、フィルター滅菌する)に加え、125mlフラスコ中、125rpmで、4℃、1時間振盪させた。1時間後、4.5mlを冷やした5.0mlのCorning凍結バイアルに加えた。一旦充填したら、個々のバイアルを、制御速度フリーザー中にて4℃で15分間保持し、次いで、-40℃の最終温度に達するまで、-0.5℃/分の速度で凍結させた。最終温度に到達した後、バイアルを、液体窒素蒸気で満たしたCryoplus 4貯蔵ユニット(Forma Scientific)の内側のラック中の箱に移した。
【0153】
融解するために、バイアルを、貯蔵ユニットから取り出し、密閉したドライアイス容器の中に配置し、次いで、40〜45℃に保持したウォーターバスに、「沸騰」がおさまるまで入れた。融解した場合、内容物を、ふたがある100×25mmペトリ皿中の重ねた滅菌70 mm Whatman濾紙(No. 4)約8枚に注いだ。液体を数分間濾紙に吸収させ、次いで、細胞を含む上端の濾紙をGN6培地(N6培地、2.0 mg/L 2,4-D、30g/Lスクロース、2.5 g/L Gelrite、pH 5.8)の上に1週間移した。1週間後、見込みのある形態を有する組織のみを、濾紙から直接的に新鮮なGN6培地に移した。この組織を、1〜3グラムが、125ml Erlenmeyerフラスコ中の約30mL H9CP+培地への懸濁の開始のために使用可能になるまで、7〜14日毎に継代培養した。以前に記載されたようにその時点で継代培養が行われた、全体で12ml PCVが得られる時点まで、3mlのPCVを新鮮なH9CP+培地に3.5日毎に継代培養した。
【0154】
形質転換の約24時間前に、12ml PCVの以前に凍結した胚形成トウモロコシ懸濁細胞プラス28mlの馴化培地を、500 mlのErlenmeyerフラスコ中の80mlのGN6液体培地(Gelriteを欠くGN6培地)に継代培養し、125rpm、28℃のシェーカー上に配置した。全体で36ml PCVが3つのフラスコに分布するように、同じ細胞系統を使用して、これを2回反復した。24時間後、GN6液体培地を取り出し、細胞に原形質分離を起こさせるために、フラスコあたり72ml GN6 S/M振盪培地(N6培地、2.0 mg/L 2,4-D、30g/L スクロース、45.5g/L ソルビトール、45.5 g/L マンニトール、100mg/L ミオ-イノシトール、pH 6.0)で置き換えた。フラスコを、シェーカー上に30〜35分間配置し、この時間の間、適切な量のGN6 S/M液体培地を、約405mgのあらかじめオートクレーブしたシリコンカーバイドウィスカー(Advanced Composite Materials, Inc.)に加えることによって、シリコンカーバイドウィスカーの50mg/ml懸濁液を調製した。
【0155】
GN6 S/M中でのインキュベーション後、各フラスコの内容物を250ml遠心分子ボトルにプールした、一旦すべての細胞が底にたまると、約14mlのGN6 S/Mを除いて取り出され、後の使用のために滅菌1Lフラスコに収集された。あらかじめ濡らしたウィスカーの懸濁物を、最大速度で60秒間ボルテックスにより激しく撹拌し、8.1mlをボトルに加え、最終段階として170μg DNAをそこに加えた。ボトルを、市販のペイントミキサー、改良Red Devil 5400に直に配置し、10秒間攪拌した。攪拌後、浸透圧を減少するために、細胞のカクテル、培地、ウィスカー、およびDNAを、125mlの新鮮なGN6液体培地とともに、1Lフラスコの内容物に加えた。細胞を、シェーカー上で2時間回収し、その後、室内の真空ラインに接続したガラス細胞コレクターユニットを使用して、Whatman #4濾紙(5.5cm)上で濾過した。
【0156】
約6mlの分散した懸濁液を、真空にするに従って、フィルターにピペットにより移した。フィルターは、GN6培地の60×20mmプレート上に配置した。プレートは、個々のプレートの蒸発を最小化するために単一のプラスチックの層(<2ミル厚)でゆるくシールした暗箱中で、28℃で1週間培養した。
【0157】
1週間後、20枚の濾紙をGN6培地(1ハービエース)の60×20mmプレートに移し、20枚の濾紙をGN6培地(2ハービエース)の60×20mmプレートに移し、20枚の濾紙をGN6培地(4ハービエース)の60×20mmプレートに移した(N6培地、2.0mg/L 2,4-D、30g/Lスクロース、100mg/L myo-イノシトール、1、2、または4mg/Lビアラホス(ハービエース由来)、および2.5g/L Gelrite、pH 5.8)。プレートを箱の中に配置し、さらに1週間培養した。
【0158】
さらに1週間後、すべての濾紙を再び同濃度のGN6+ハービエース培地(1H、2H、および/または4H)に移した。プレートを箱の中に配置し、さらに1週間培養した。
【0159】
形質転換の3週間後、プレート上の細胞の1/2を、ハービエースのビアラホス1、2、または4mg/Lを含む3.0mlの溶解したGN6アガロース培地(121℃で10分間だけオートクレーブされた、N6培地、2.0mg/L 2,4-D、30g/Lスクロース、100mg/L myo-イノシトール、7g/L Sea Plaqueアガロース、pH 5.8)にこすり取ることによって、組織を包埋した。この組織を破壊し、3mLのアガロースおよび組織を、細胞が最初に培養された濃度に応じて、GN6(1H、2H、および/または4H)の100×15mmプレートの表面に均一に注いだ。これを、各プレート上の残りの1/2の細胞を用いて反復した。包埋後は、プレートをNescofilm(登録商標)またはParafilm M(登録商標)で個別にシールし、次いで、暗い箱の中で28℃で約4週間培養した。
【0160】
7.4-植物体再生のためのプロトコール
推定形質転換単離物は、典型的には、形質転換の5〜8週間後に最初に可視となった。可能性のある単離物を包埋されたプレートから除去し、60×20mmプレート内の同濃度の新鮮な選択培地に移した。およそ2週間後に持続的な生長が明白であれば、その事象は抵抗性であると見なされる。次いで、抵抗性事象のサブセットを、分子分析にかけた。
【0161】
再生を、(MS塩およびビタミン、30.0g/L スクロース、5mg/L BAP、0.25mg/L 2,4-D、1mg/L ビアラホス、2.5g/L Gelrite;pH 5.7)を含む、サイトカイニンベースの誘導培地、28(1H)にカルス細胞を移すことによって開始する。細胞を、微小光(13μEm-2s-1)中で1週間増殖させ、次いで、より明るい光(40μEm-2s-1)でもう一週間増殖させ、その後、植物増殖調節因子を欠いている以外は28(1H)と同一である再生培地36(1H)に移す。小さな(3〜5cm)植物体を取り出し、選択なしのSHGA培地(SchenkおよびHildebrandtの基礎塩およびビタミン、1972;1g/L myo-イノシトール、10g/L スクロース、2.0g/L Gelrite,pH 5.8)を含む150×25mm培養チューブに配置する。一旦植物体が十分な根およびシュート系を発達したら、これらは温室の中の土に移植する。
【0162】
7.5-分子分析:トウモロコシ材料および方法
7.5.1-組織の収集、DNAの単離および定量
新鮮な組織をチューブに配置し、4℃で2日間凍結乾燥する。組織を完全に乾燥させた後、タングステンビーズ(Valenite)をチューブ中に配置し、試料を、1分間、Kelcoビーズミルを使用する乾式粉砕に供する。次いで、標準的なDNeasy DNA単離手法に従う(Qiagen, DNeasy 69109)。次いで、抽出したDNAのアリコートをPico Green(Molecular Probes P7589)で染色し、濃度をng/μlで得るために既知の標準を用いてフルオロメーター(BioTek)で読み取る。
【0163】
7.5.2-PATインベーダーアッセイ分析
DNA試料を20ng/μlに希釈し、次いで95℃で10分間、サーモサイクラー中のインキュベーションによって変性させる。次いで、Signal Probeミックスを、提供されるオリゴミックスおよびMgCl2を使用して調製する(Third Wave Technologies)。7.5μlのアリコートをインベーダーアッセイプレートの各ウェルに配置し、続いて対照、標準、および20ng/μlに希釈した未知の試料の7.5μlのアリコートを配置する。各ウェルに15μlのミネラルオイル(Sigma)を重層する。次いで、プレートを、63℃で1時間インキュベートし、フルオロメーター(Biotek)上で読み取る。バックグラウンド内部対照プローブよりも上のシグナル%で除算した、標的プローブについてのバックグラウンドよりも上のシグナル%の計算は、比率を計算する。サザンブロット分析を用いて開発されかつ確証された既知のコピー標準の比率を使用して、未知の事象の見積もったコピーを同定する。
【0164】
7.5.3-PATのポリメラーゼ連鎖反応
全体で100ngの全DNAを鋳型として使用する。20mMの各プライマーを、Takara Ex Taq PCR Polymeraseキット(Mirus TAKRR001A)とともに使用する。PAT PTUのためのプライマーは、
である。試料を、94℃3分間、ならびに94℃30秒間、62℃30秒間、および72℃3分15秒間の35サイクル、続いて72℃10分間に供することによって、9700 Geneampサーモサイクラー(Applied Biosystems)においてPCR反応を実行する。コード領域PCR PATのためのプライマーは、
である。試料を、94℃3分間、ならびに94℃30秒間、65℃30秒間、および72℃1分45秒間の35サイクル、続いて72℃10分間に供することによって、9700 Geneampサーモサイクラー(Applied Biosystems)において、PCR反応を実行する。DSM-2のためのコード領域PCRのためのプライマーは、
である。試料を、94℃3分間、ならびに94℃30秒間、65℃30秒間、および72℃45秒間の35サイクル、続いて72℃10分間に供することによって、9700 Geneampサーモサイクラー(Applied Biosystems)において、PCR反応を実行する。PCR産物を、EtBrで染色される1%アガロースゲル上での電気泳動によって分析する。
【0165】
7.5.4-サザンブロット分析
サザンブロット分析を、Qiagen DNeasyキットから得られた全DNAを用いて実施する。全体で5μgの全ゲノムDNAを、組み込みデータを得るため、NcoIおよびSwaIによる消化に一晩供する。制限酵素SspIによる5μgの消化を、PTUデータを得るために使用した。SspI消化データを分析した後、制限酵素MfeIの方がよい酵素の選択であると考えられたため、MfeIを残りの試料の全てを消化するために使用した。一晩消化の後、約100ngのアリコートを1%ゲル上で泳動し、完全な消化を確実にする。この保証後、試料を、大きな0.85%アガロースゲル上で、40ボルトで一晩泳動させる。次いで、このゲルを、0.2M NaOH、0.6M NaCl中で30分間変性させる。次いで、このゲルを、pH 7.5の0.5M Tris HCl、1.5M NaCl中で30分間中和させる。次いで、20×SSCを含むゲル装置を、ゲルからナイロンメンブレン(Millipore INYC00010)への重力による転写を得るために一晩設定する。一晩の転写後、次いで、このメンブレンを、120,000マイクロジュールで、クロスリンカー(Stratagene UV stratalinker 1800)によりUV光に供する。次いで、このメンブレンを、0.1% SDS、0.1 SSC中で45分間洗浄する。45分間の洗浄後、このメンブレンを80℃で3時間ベーキングし、次いでハイブリダイゼーションまで4℃に保存する。ハイブリダイゼーション鋳型フラグメントは、プラスミドDNAを使用するコード領域PCRを使用して調製する。産物を1%アガロースゲル上で泳動し、切除し、および次いで、Qiagen(28706)ゲル抽出手法を使用してゲル抽出する。次いで、このメンブレンを、Perfect Hyb緩衝液(Sigma H7033)中で1時間、60℃段階のプレハイブリダイゼーションに供する。Prime it RmT dCTP-labeling reaction(Stratagene 300392)手法を使用して、p32に基づくプローブを開発する(Perkin Elmer)。このプローブを、Probe Quant. G50 カラム(Amersham 27-5335-01)を使用して精製する。ハイブリダイゼーション緩衝液1 mlあたり200万カウントCPMを使用して、サザンブロットを一晩ハイブリダイズさせる。一晩のハイブリダイゼーションの後、次いで、このブロットを、0.1% SDS、0.1 SSC中で65℃における2回の20分間の洗浄に供する。次いで、このブロットを、-80℃でインキュベートしてフィルムに一晩露光させる。
【0166】
7.6-結果
以前に記載された2つの構築物(PATおよびDSM-2)の各々を用いて、トウモロコシのウィスカー媒介形質転換を3回実施した。それらの集合的な実験から、230個の単離物が回収された。1および2mg/Lビアラホス(ハービエース由来)を含有している培地では、PATとDSM-2との間の事象回収は極めて類似していたが、4mg/Lビアラホスを含有している培地における事象回収は、DSM-2よりPATの方が高かった。
【0167】
(表Ex.7.6−1)
【0168】
1または2ハービエースで選択されたDSM-2事象のうちの48を、サザンブロットを介した、コピー数分析および完全な植物転写単位(PTU)の存在にかけた。48の事象は、すべて、少なくとも1コピーのDSM-2遺伝子を含有していた。より少数コピーの事象(3以下)からの結果のサブセットを、以下に提示する。
【0169】
(表Ex.7.6−2)
【0170】
1、2、または4ハービエースで選択されたPAT事象のうちの48を、それぞれインベーダーアッセイおよびPCRを介した、コピー数推定および完全な植物転写単位(PTU)の存在にかけた。48の事象は、すべて、少なくとも1コピーのPAT遺伝子を含有していた。より少数コピーの事象(3以下)からの結果のサブセットを、以下に提示する。
【0171】
(表Ex.7.6−3)
【0172】
およそ6〜10個のT0植物体が、15個のDSM-2含有事象の各々から再生し、7〜8個の植物体が、Libertyに対する耐性を査定するために以前にリストされた、7個のPAT含有事象の各々から再生した。
【0173】
31個の異なる事象からのカルス組織試料を、非形質転換カルス(陰性対照)と共に、ウエスタンブロッティング実験を用いて分析した。陰性対照以外のすべての試料について、20kDaというマーカーに対する相対分子量を有する1本のバンドが観察された。それは、19.7kDaというタンパク質の予測されたサイズに一致した。さらに、バンドは、標準、すなわち大腸菌から精製された精製DSM-2タンパク質とも同一のサイズを有する。ゲル上の0.7μg/mLの標準に5(+++++)という任意スコアを与え、異なる事象からのバンドを、相対的に等級付け、以下の表にリストした。
【0174】
(表Ex.7.6−4)
【0175】
7.6.1-T0トウモロコシにおける葉塗布直接比較
T0 DSM-2(v2)植物に、グルホシネート除草剤を滴下にて塗布した。15個のT0事象の各々からの4個の同胞が試験され、個々の各植物体の4枚の葉が、およそV8期でグルホシネートの滴下を受容した。滴下処理を各割合についてランダム化し、個々の葉における処理位置の変動を可能にした。トウモロコシの場合、0.25%v/vグルホシネートが、有意のレベルの抵抗性を有する植物体から感受性植物体を区別するための最小有効用量である。より高い割合も、抵抗性の相対レベルを決定するために適用した(0.5%、1.0%、および2.0%v/v)。グルホシネート処理は、直径およそ2.5cmの処理区域に、先端に綿の付いたアプリケータを使用して適用された。
【0176】
表11は、トウモロコシT0形質転換体にグルホシネート抵抗性を付与するためのDSM-2(v2)遺伝子および対照遺伝子の応答を比較している。応答は、視覚的損傷% 2WATによって提示される。データは、ほとんどもしくは全く損傷なし(<20%)、中程度の損傷(20〜40%)、または重度の損傷(>40%)を示す個体のヒストグラムとして提示される。各T0は独立した形質転換事象であるので、所定の割合における個々のT0応答の有意な変動が予想される。算術平均および標準偏差が各処理について提示される。非形質転換野生型トウモロコシは、グルホシネート感受性対照として働いた。DSM-2(v2)遺伝子は、個々のT0トウモロコシ植物に除草剤抵抗性を付与した。所定の処理において、植物応答のレベルは大きく変動した。これは、各植物が独立した形質転換事象を表すという事実に起因し得る。重要な注目点として、最大2%v/vグルホシネートにおいて、DSM-2(v2)は、PAT形質転換植物より全体的に優れている。割合による集団全体の損傷平均が、単に、DSM-2(v2)で形質転換された植物と、野生型対照またはPAT形質転換対照との間の有意な違いを実証するために、表11に提示される。
【0177】
(表11)野生型植物およびT0 PAT形質転換植物と比較された、発芽後の植物体に葉塗布として適用された一連のグルホシネート割合に対するT0 DSM-2(v2)(植物最適化)形質転換トウモロコシの応答
【0178】
7.6.2-T2トウモロコシにおける高いグルホシネート耐性の立証
T1 DSM-2(v2)×5XH751の交雑からの種子を、Metro Mix培地を含有している4インチのポットに植え、2葉期に、ヌルを除去するために、560g ai/haグルホシネートで187L/haに設定されたトラック噴霧器で噴霧した。7DATで、ヌルを除去し、以下の割合で上記のようなトラック噴霧器で抵抗性植物に噴霧した:0、560、1120、2240、および4480g ai/haグルホシネート。3 DATおよび14 DATで、植物を等級付け、5XH751×Hi II対照植物体と比較した。下記表Ex.7.6.2-1は、20%未満の損傷を有する最大2240g ai/haグルホシネートを提供した個々のDSM-2(v2)植物体が存在することを示す。DSM-2(v2)は、PAT形質転換対照においても4480g ai/haグルホシネートに対する類似した耐性を提供した。
【0179】
(表Ex.7.6.2−1)発芽後(14DAT)に適用された一連のグルホシネート割合に対するT2トウモロコシの応答
【0180】
7.6.3-トウモロコシにおけるDSM-2(v2)の遺伝性
T1 DSM-2(v2)×5XH751の交雑からの種子を、Metro Mix培地を含有している3インチのポットに植え、2葉期に、0、280、560、1120、2240、および4480g ai/haグルホシネートで187L/haに設定されたトラック噴霧器で噴霧した。3 DATおよび14 DATで、植物を等級付け、5XH751×Hi II対照植物と比較した。以前のように、0〜100%の全体的な視覚的損傷により植物を等級付けた。各集団の分離を決定するためには、1120 g ai/ha以上の割合を選んだ。耐性植物および感受性植物を計数したところ、T1ファミリーのすべてが、χ二乗検定によって決定されるように、単一遺伝子座、優性メンデル形質(1R:1S)として分離することが決定された。生存している植物体を、T2世代を作製するために自家受粉した。DSM-2(v2)は、商業的なハイブリッドと相反交雑された場合、複数の種において強固なグルホシネート抵抗性遺伝子として遺伝性である。
【0181】
5つのDSM-2(v2)T2ファミリーにおいて後代検定も実施した。種子を、上記のような3インチのポットに植えた。3葉期に、すべての植物に、以前に記載されたようなトラック噴霧器で560g ai/haグルホシネートを噴霧した。7 DATの後、耐性植物体および感受性植物体を計数した。試験された5つの系のうちの4つが、χ二乗検定によって決定されるように、単一遺伝子座、優性メンデル形質(3R:1S)として分離した。
【0182】
7.6.4-除草剤スペクトルを増加させるためのDSM-2(v2)の重ね合わせ
T1植物のBE1146RRとの交雑を行った。DSM-2(v2)形質転換植物は、従来法により、付加的な関心対象の形質を含有している他のトウモロコシ系と交配され得る。グリホセート耐性形質CP4を含有している近交系(BE1146RR)を、DSM-2(v2)を含有しているT1植物と交雑した。通常であれば致死的である除草剤割合に等しいか、またはそれを超える割合で、逐次的に、または容器での混合物により、グルホシネートおよびグリホセートを適用することにより、両除草剤に対する耐性形質の効力について、後続世代の植物を試験することができる(例えば、280、560、1120g ae/ha、またはそれ以上の両除草剤を植物に噴霧することができる)。これは、除草剤抵抗性管理のため、組み合わせ適用または逐次適用において両除草剤を使用する能力を同定するであろう。
【0183】
実施例8-抗体を介した形質転換植物からのタンパク質検出
8.1-ポリクローナル抗体作製
精製されたDSM-2(前記セクションを参照されたい)を、5ミリグラム、ウサギポリクローナル抗体作製のために、Invitrogen Custom Antibody Services(South San Francisco, CA)に送達した。ウサギは、12週間で4回の注射を受容し、各注射は、1mLの不完全フロイントアジュバントに懸濁した精製されたタンパク質0.5mgを含有していた。血清を、特異性および親和性を確認するため、直接ELISAおよびウエスタンブロッティング実験の両方において試験した。
【0184】
8.2-カルス組織からのDSM-2の抽出
1穴パンチャーを使用した4枚のトウモロコシ(Hi-II)葉ディスクを、2個のステンレス鋼ビーズ(4.5mm;Daisy Co,, Cat# 145462-000)、および500μLの植物抽出緩衝液(0.1%トリトンX-100および1mL当たり5μLのプロテアーゼ阻害剤カクテル(Sigma Cat # P9599)を含有しているPBS)を含有している微量遠心管に入れた。チューブを、Geno/Grinder(Model 2000-115, Certiprep, Metuchen, NJ)に固定し、500rpmの1×の設定で6分間振とうした。チューブを5000×gで10分間遠心分離し、可溶性タンパク質を含有している上清を、DSM-2の存在を検出するために、ウエスタンブロッティング実験において分析した。
【0185】
8.3-ウエスタンブロッティング分析
葉抽出物に様々な濃度の精製されたDSM-2を加え、95℃で10分間レムリ試料緩衝液と共にインキュベートした後、8〜16%Tris-Glycineプレキャストゲルにおける電気泳動分離を行った。次いで、タンパク質を、標準的なプロトコルを使用して、ニトロセルロース膜上に電気的に転写した。4%スキムミルクを含むPBSでブロッキングした後、DSM-2タンパク質を、抗DSM-2抗血清と、それに続くヤギ抗ウサギ/HRPコンジュゲートによって検出した。検出されたタンパク質を、化学発光基質ECL Western Analysis Reagent(Amersham Cat.# RPN 21058)によって可視化した。
【0186】
8.4-結果
大腸菌細胞において発現され、精製されたタンパク質を使用して、DSM-2に対するポリクローナル抗体を作成した。4回の注射の後、抗血清は、直接ELISAで観察されるように、抗DSM-2抗体の高い力価を有していた。100,000倍希釈でも、血清は、バックグラウンドより6倍大きいシグナルを提供した。
【0187】
(野生型トウモロコシ(Hi-II)葉基質におけるDSM-2の)ウエスタンブロッティング分析において、血清は、DSM-2(v2)遺伝子に基づき予測された分子量と比較可能である、およそ22kDaの主要なバンドを検出することができた。試験された最低濃度5ng/mLで抽出物を入れた場合でも、同一のバンドを検出することができた。高いDSM-2濃度ではマイナーなバンドも観察されたが、これらは、より低い濃度では観察されなかったことから、標的タンパク質の凝集物であると考えられる。予測された分子量と比較可能な分子量を有する単一の主要なバンドが観察された。(このゲル上の泳動したレーンは、分子量マーカー、ならびにそれぞれ濃度0.005、0.05、0.5、および5μg/mLでDSM-2タンパク質を含有している葉抽出物であった)。さらに、バックグラウンドシグナルがほとんど観察されなかったように、ポリクローナル抗体は、トウモロコシ葉タンパク質と交差反応しなかった。
【0188】
形質転換タバコカルス組織からのDSM-2の発現を、ウエスタンブロッティング分析を使用して決定した。標準物(およそ22kDa)として比較サイズを有する検出されたバンドが、異なる事象で観察され、このことは、DSM-2がトランスジェニックタバコ組織において発現されたことを示している。
【0189】
実施例9-タバコ(細胞培養物)形質転換
形質転換の4日前に、7日毎に継代培養されていた1週齢のNT-1タバコ懸濁物を、40mlのNT-1 B培地に2mlのNT-1培養物または1mlの濃縮細胞を添加することにより、新鮮な培地へと継代培養した。継代培養された懸濁物を、125rpmのシェーカー上で25±1℃で暗所で維持した。
【0190】
(表Ex.9−1)NT-1 B培地組成
【0191】
関心対象のバイナリーベクターを保持しているアグロバクテリウム・ツメファシエンス[LBA4404株]の50%グリセロールストックを、50〜100mg/Lスペクチノマイシンを含有している30mlのYEP液体(10g/L酵母抽出物、10g/Lペプトン、5g/L NaCl、0〜10g/Lスクロース)に20、100、または500μlを添加することにより、液体の一晩の培養を開始させるために使用した。OD600が1.5±0.2になるまで、細菌培養物を150〜250rpmでインキュベーターシェーカーで28℃で暗所でインキュベートされた。これはおよそ18〜20時間を要した。
【0192】
試験された各ベクターについて、20〜70mLの4日齢の懸濁細胞を、無菌の容器に移し、そこに、適切なODの500〜1750μlのアグロバクテリウム懸濁物を添加した。均一の混合物が達成されることを保証するため、10mlの広い孔径のピペットで5回上下に細胞をピペッティングした。次いで、均一の懸濁物を、リピートピペッターの25mlのバレルに引き上げ、1ウェル当たり250μlの懸濁物を、懸濁物が使い尽くされるまで、24穴プレートに分配し続けた。ウェルプレートをパラフィルムで覆い、3日間の共培養のため振とうすることなく25±1℃で暗所で培養した。
【0193】
共培養後、すべての過剰の液体を、1mLピペットチップで個々のウェルから除去し、残存している細胞を、1mlのNTC液(オートクレーブ処理後に添加された500mg/Lカルベニシリンを含有しているNT-1 B培地)に再懸濁させた。個々のウェルの内容物を、使い捨てのトランスファーピペットを使用して、100×25mmの選択プレートの表面全体に分散させた。選択培地は、オートクレーブ処理後に添加された7.5〜15mg/Lのビアラホスまたは技術的等級のグルホシネートアンモニウムが補足された8g/l TC寒天により凝固したNTC培地からなっていた。すべての選択プレートを、覆うことなく、28℃で暗所で維持した。
【0194】
推定形質転換体が、殺傷された非形質転換細胞のバックグラウンド上にカルスの小さなクラスタとして出現した。形質転換のおよそ2〜6週間後に、カルスを単離した。各カルス単離物を、同一の選択培地を含有している60×20mmプレートに移し、およそ2週間増殖させた後、分析に供した。
【0195】
9.1-結果
DSM-2(v2)をPATと比較する並行実験を完了した。研究において、PAT選択プレートの100%が、10mg/Lビアラホス培地上で少なくとも1つのPCR陽性単離物を産生し、一方、DSM-2(v2)選択プレートの79%が、少なくとも1つのPCR陽性単離物を産生した。すべての事象が、コード領域PCRを介して、DSM-2(v2)遺伝子またはPAT遺伝子の存在について分析され、陽性であることが見出された。
【0196】
(表Ex.9−2)
【0197】
DSM-2(v2)で選択された事象の小さなサブセットにおいてウエスタンブロットを実施したところ、以下のデータで分かるように、3つの陽性事象が同定された。第2の実験において、DSM-2(V2)で処理されたタバコ細胞を、7.5、10、12.5、または15mg/Lビアラホスまたは技術的等級のグルホシネートアンモニウムで選択した。コード領域PCRによる立証後の形質転換頻度(少なくとも1つのカルスを産生する選択プレートの%)を、以下の表にリストする。
【0198】
(表Ex.9−3)
【0199】
実施例10-その他の作物のアグロバクテリウム形質転換
本開示を考慮すれば、当技術分野において公知の技術を使用して、本発明に従い、付加的な作物を形質転換することができる。ライムギのアグロバクテリウム媒介形質転換については、例えば、Popelka and Altpeter(2003)を参照されたい。ダイズのアグロバクテリウム媒介形質転換については、例えば、Hinchee et al.,1988を参照されたい。ソルガムのアグロバクテリウム媒介形質転換については、例えば、Zhao et al., 2000を参照されたい。オオムギのアグロバクテリウム媒介形質転換については、例えば、Tingay et al., 1997を参照されたい。コムギのアグロバクテリウム媒介形質転換については、例えば、Cheng et al., 1997を参照されたい。イネのアグロバクテリウム媒介形質転換については、例えば、Hiei et al., 1997を参照されたい。
【0200】
これらおよびその他の植物のラテン名は以下に与えられる。例えば、トウモロコシ(イネ科ジー・メイズ(Zea mays))、コムギ(イネ科トリチカム(Triticum)種)、イネ(イネ科オリザ(Oryza)種およびマコモ(Zizania)種)、オオムギ(イネ科ホルデウム(Hordeum)種)、ワタ(アブロマ(Abroma)双子葉類アブロマ・オーガスタ(augusta)およびアオイ科ゴシピウム(Gossypium)種)、ダイズ(マメ科グリシン・マックス(Glycine max))、テンサイ(アカザ科ベータ・ブルガリス・アルティッシマ(Beta vulgaris altissima))、サトウキビ(アレンガ・ピナータ(Arenga pinnata))、トマト(ナス科リコパーシコン・エスカレンタム(Lycopersicon esculentum)およびその他の種、フィザリス・イコスカルパ(Physalis ixocarpa)、ソラナム・インカナム(Solanum incanum)およびその他の種、ならびにサイフォマンドラ・ベタセア(Cyphomandra betacea))、ジャガイモ、サツマイモ、ライムギ(イネ科セカレ(Secale)種)、コショウ(ナス科カプシカム・アンナム(Capsicum annuum)、シメンセ(sinense)、およびフルテセンス(frutescens))、レタス(キク科ラクチュカ・サティバ(Lactuca sativa)、ペレニス(perennis)、およびパルチェラ(pulchella))、キャベツ、セロリ(セリ科アピウム・グラベオレンス(Apium graveolens))、ナス(ナス科ソラヌム・メロンジナ(Solanum melongena))、ソルガム(すべてのソルガム(Sorghum)種)、アルファルファ(マメ科メジカーゴ・サティバム(Medicago sativum))、ニンジン(セリ科ダウカス・キャロータ・サティバ(Daucus carota sativa))、マメ(マメ科ファセロラス(Phaseolus)種およびその他の属)、オートムギ(アベナ・サティバ(Avena Sativa)およびストリゴーサ(Strigosa))、エンドウ(マメ科ピサム(Pisum)、ビグナ(Vigna)、およびテトラゴノロバス(Tetragonolobus)種)、ヒマワリ(キク科ヘリアンサス・アニュス(Helianthus annuus))、カボチャ(双子葉類キュカービタ(Cucurbita)種)、キュウリ(双子葉類ジェネラ(genera))、タバコ(ナス科ニコチアナ(Nicotiana)種)、アラビドプシス(アブラナ科シロイヌナズナ)、芝草(ロリウム(Lolium)、アグロスチス(Agrostis)、およびその他の科)、ならびにクローバー(マメ科)を含む(がこれらに限定はされない)これらおよびその他の植物にDSM-2(v1)を形質転換するために、これらおよびその他の(非アグロバクテリウム)形質転換技術が使用され得ることは明白であるはずである。例えば、DSM-2(v2)遺伝子を有するそのような植物は、本発明に含まれる。DSM-2(v2)を用いる形質転換の結果としてグルホシネートまたはビアラホスに対する抵抗性が付与された植物における植生制御は、グルホシネートを選択的に適用することにより改良され得る。
【0201】
DSM-2(v2)は、多くの落葉材木および常緑材木の育成系において季節に合わせて使用するための、DSM-2によって不活化され得る除草剤(例えば、グルホシネート、ビアラホス、および/またはホスフィノトリシン)の適用可能性を増加させる可能性を有する。グルホシネートまたはビアラホスに対して抵抗性の材木種は、損傷の懸念のないこれらの除草剤の過剰使用の柔軟性を増加させるであろう。これらの種には、ハンノキ、トネリコ、アスペン、ブナ、カバノキ、サクランボ、ユーカリ、ヒッコリー、カエデ、オーク、マツ、およびポプラが含まれるであろうが、これらに限定はされない。装飾用の種における選択的制御のためのグルホシネートまたはビアラホスに対する抵抗性の使用も、本発明の範囲内である。例には、バラ、ニシキギ、ペチュニア、ベゴニア、およびマリーゴールドが含まれ得るが、これらに限定はされない。
【0202】
実施例11-任意の作物におけるグリホセート耐性形質と重ね合わされたDSM-2(V2)
北米において栽培されるワタ、アブラナ、およびダイズのエーカー(acre)の大部分がグリホセート耐性形質を含み、GTトウモロコシの採用は上昇している。さらなるGT作物(例えば、例えば、コムギ、イネ、テンサイ、芝草など)が開発中であるが、現在まで商業的に発売されていない。多くの他のグリホセート抵抗性種が、開発段階のために実験中である(例えば、アルファルファ、サトウキビ、ヒマワリ、ビート、エンドウマメ、ニンジン、キュウリ、レタス、タマネギ、イチゴ、トマト、およびタバコ;ポプラおよびスイートガムのような林業種;ならびにマリゴールド、ペチュニア、およびベゴニアなどの園芸種;World Wide Web上のisb.vt.edu/cfdocs/fieldtestsl.cfm , 2005)。GTCは、この系によって提供される制御された雑草の完全な広がりおよび便利さおよびコスト効果のための価値のある手段である。しかし、現在標準であるベース処理としてのグリホセートの有用性は、グリホセート抵抗性雑草を選択することである。さらに、グリホセートが固有に効力が弱い雑草は、グリホセートのみの化学物質プログラムが実施されている場合には、圃場で優勢な種に転じている。従来的な育種を通して、または新規な形質転換事象として一緒にのいずれかで、GT形質とDSM-2(v2)を重ね合わせることによって、雑草制御の効力、柔軟性、ならびに雑草の変化および除草剤抵抗性の発生を管理する能力が改善できる。雑草制御オプションの改善のためのいくつかのシナリオが想定でき、ここで、DSM-2(v2)およびGT形質は、任意の単子葉植物種または双子葉植物種において重ね合わせられる。
a)グリホセートは、大部分のイネ科雑草種および広葉雑草種の制御のために、標準的な発芽後適用割合(420〜2160g ae/ha、好ましくは560〜840g ae/ha)で適用することができる。コニザ カンデンシス(Conyza canadensis)のような広葉雑草またはグリホセートを用いる制御に対して固有に困難である雑草(例えば、コメリナ(Commelina)種、イポメア(Ipomoea)種)の制御のために、280-2240g ae/ha(好ましくは350〜1700g ae/ha)グルホシネートが、効果的な制御を提供するために、連続して、容器で混合して、またはグリホセートと前もって混合して適用することができる。
b)現在、GTCにおいて適用されるグリホセートの割合は、一般的に、適用のタイミングあたり、560〜2240 g ae/haの範囲である。グリホセートは、広葉雑草種よりも、イネ科植物雑草に対してはるかにより効力がある。DSM-2(v2)+GTの重ね合わせた形質は、イネ科植物に有効な割合のグリホセート(105〜840g ae/ha、より好ましくは210〜420g ae/ha)を可能にする。次いで、グルホシネート(280〜2240g ae/ha、より好ましくは350〜1700g ae/ha)を、必要な広葉雑草の制御を提供するために、連続して、容器で混合して、またはイネ科雑草に有効な割合のグリホセートと前もって混合して適用することができる。
【0203】
当業者は、その他の除草剤(例えば、ビアラホス)が、DSM-2(v2)を用いた植物の形質転換によって可能になることを認識するであろう。CPR(Crop Protection Reference)書に編集された除草剤のラベルもしくは類似の編集、オンラインで編集されたラベル(例えば、cdms.net/manuf/manuf.asp)、またはAgriliance(2003)からのCrop Protection Guideのような商業的もしくは学術的な作物保護指針により、具体的な割合は決定され得る。DSM-2(v2)によってHTCにおける使用を可能にされた各代替除草剤は、単独で使用されたとしても、容器で混合されたとしても、または連続的に使用されたとしても、本発明の範囲内にあると見なされる。
【0204】
実施例12-任意の作物においてAHAS形質と重ね合わされたDSM-2(v2)
イミダゾリノン除草剤耐性(AHASなど)は、現在、トウモロコシ、イネ、およびコムギを含むがこれらに限定はされない、北米において栽培されている多数の作物に存在している。さらなるイミダゾリノン耐性作物(例えば、ワタおよびテンサイ)が開発中であるが、現在のところ市販されてはいない。多くのイミダゾリノン除草剤(例えば、イマザモクス、イマゼタピル、イマザキン、およびイマザピク)は、現在、様々な従来的な作物において選択的に使用されている。イマゼタピル、イマザモクス、および非選択的なイマザピル(imazapyr)の使用は、AHASなどのようなイミダゾリノン耐性形質を通して可能にされてきた。現在のところ、市販のイミダゾリノン耐性HTCは、非トランスジェニックであるという利点を有する。この化学クラスは、有意な土壌残留活性を有し、従って、グリホセートまたはグルホシネートに基づく系とは異なり、適用のタイミングを超えて拡張される雑草制御を提供することも可能である。しかし、イミダゾリノン除草剤によって制御される雑草のスペクトルは、グリホセートほど広くない(Agriliance, 2003)。さらに、イミダゾリノン除草剤は、多くの雑草が抵抗性を発達させてきた作用の様式(アセトラクテートシンターゼ、ALSの阻害)を有する(Heap, 2004)。従来的な交配を通して、または新規の形質転換事象として一緒に、イミダゾリノン耐性形質とDSM-2(v2)を重ね合わせることによって、雑草制御の効率、柔軟性、ならびに雑草のシフトおよび除草剤抵抗性の発達を管理する能力が改善され得る。DSM-2(v2)およびイミダゾリノンに対する耐性形質が、単子葉植物種または双子葉植物種において重ね合わせられる、雑草制御オプションの改善のためのいくつかのシナリオが想定できる。
a)イマゼタピルは、多くのイネ科雑草種および広葉雑草種の制御のために、標準的な発芽後適用割合(35〜280g ae/ha、好ましくは70〜140g ae/ha)で適用することができる。
i)アマランサス ルディス(Amaranthus rudis)、アンブロシア トリフィダ(Ambrosia trifida)、チェノポジウム アルブム(Chenopodium album)のようなALS阻害剤抵抗性広葉雑草(とりわけ、Heap, 2004)は、280〜2240g ae/ha、より好ましくは350〜1700g ae/haグルホシネートを容器で混合することによって制御できる。
ii)イポモエア(Ipomoea)種のようなイミダゾリノン除草剤に対して固有により耐性である広葉雑草種は、280〜2240g ae/ha、より好ましくは350〜1700g ae/ha グルホシネートを容器で混合することによってもまた制御できる。
【0205】
雑草制御の分野の当業者は、単独で、または1種もしくは複数の組み合わせでの、種々の市販のイミダゾリノン除草剤、およびグルホシネートベースの除草剤のいずれかの使用が、従来的な育種または遺伝子操作のいずれかによる、DSM-2(v2)形質転換および任意のイミダゾリノン耐性との重ね合わせによって可能であることを認識している。これらの化学物質の典型的な他の除草剤の特定の割合は、CPR(Crop Protection Reference)書籍もしくは同様の編集物、オンラインで編集された表示(例えば、cdms.net/manuf/manuf.asp)、または任意の商業的もしくは学術的な作物保護の手引書、例えば、Crop Protection Guide from Agriliance(2003)において編集されている、除草剤表示によって決定することができる。単独で、容器での混合で、または連続して使用されるかに関わらず、DSM-2(v2)によるHTCにおける使用のために可能にされる各々の代替的な除草剤は、本発明の範囲内にあると見なされる。
【0206】
実施例13-イネにおけるDSM-2(V2)
13.1-培地の説明
使用する培地を1M KOHでpH 5.8に調整し、2.5g/1 Phytagel(Sigma)で固化した。胚形成カルスを、40ml半固形培地を含む100×20mm ペトリ皿中で培養した。細胞懸濁液を、35ml液体培地を含む125mlコニカルフラスコ中で維持し、125rpmで回転させた。胚形成培養の誘導および維持は、暗所にて25〜26℃で行った(Zhang et al. 1996)。
【0207】
胚形成カルスの誘導および維持は以前に記載されたようにNB基礎培地上で行ったが(Li et al. 1993)、500mg/lグルタミンを含むように適合させた。懸濁培養を開始し、マルトースの代わりに30g/lを含めたSZ液体培地(Zhang et al. 1998)中で維持した。浸透培地(NBO)は、各0.256Mのマンニトールおよびソルビトールの添加を伴うNB培地からなった。除草剤耐性カルスを、8mg/lビアラホスを補充したNB培地で、3週間ごとに継代培養しながら9週間選択した。
【0208】
13.2-組織培養物の発展
オリザ サティバ(Oryza sativa)L.ジャポニカ 栽培種タイペイ(L. japonica cv. Taipei)309の成熟乾燥種子を、Zhang et al. 1996において記載されるように殺菌した。胚形成組織を、暗所でNB培地上で殺菌成熟イネ種子を培養することによって誘導した。約1mm直径の一次カルスを胚盤から取り出し、SZ液体培地中で細胞懸濁を開始するために使用した。次いで、懸濁物を、Zhang 1995に記載されるように維持した。懸濁物に由来する胚発生組織を、以前の継代培養の3〜5日後に液体培養から取り出し、NBO浸透培地に移し、ペトリ皿中で約2.5cmの横方向の円を形成して、ボンバードメントの前4時間培養した。ボンバードメントの16〜20時間後、組織を、NBO培地からNBH8除草剤選択培地に移し、ボンバードメント表面が上側を向くことを確実にし、かつ暗所で3時間インキュベートした。新たに形成されたカルスを、3週間に2回、新鮮なNBH8培地へ継代培養した。
【0209】
13.3-微粒子銃ボンバードメント
すべてのボンバードメントは、Biolistic PDS-1000/He(商標)システム(Bio-Rad, Laboratories, Inc.)を用いて実施した。3mgの1.0ミクロン直径の金粒子を1回、100%エタノールで、2回滅菌蒸留水で洗浄し、およびシリコン処理したEppendorfチューブ中の50μl水中に懸濁した。5μgプラスミドDNA、20μlスペルミジン(0.1M)および50μl 塩化カルシウム(2.5M)を金懸濁物に加えた。この混合物を室温で10分間インキュベートし、10000rpmで10秒間ペレット化し、60μl冷100%エタノール中に再懸濁し、かつ8〜9μlを各マイクロキャリアに分配した。組織試料を、Zhang et al.(1996)によって記載されるように、1100psiおよび27inのHg真空でボンバードメントした。
【0210】
実施例14-任意の作物においてAAD-1(v3)と重ね合わされたDSM-2(v2)
グルホシネートは、グリホセートと同様に、比較的非選択的なスペクトルの広いイネ科および広葉樹に対する除草剤である。グルホシネートの作用の様式は、グリホセートと異なる。それは、より即効性であり、除草剤適用後24〜48時間で、処理された葉の脱水および「焼け(burning)」をもたらす。これは、迅速な雑草制御の出現のため有利である。しかしながら、これは、標的植物の分裂組織領域へのグルホシネートの転移も制限し、それが、多くの種における2つの化合物の相対的な雑草制御性能の査定によって証明されるような、より不十分な雑草制御をもたらす(Agriliance, 2003)。
【0211】
従来的な交配を通して、または新規の形質転換事象として一緒に、AAD-1(v3)(USSN 11/587,893;WO 2005/107437を参照されたい)をグルホシネート耐性形質と重ね合わせることにより、雑草制御の効率、柔軟性、ならびに雑草のシフト(shift)および除草剤抵抗性の発達を管理する能力が改善され得る。前記実施例において言及されたように、AAD-1(v3)を用いて作物を形質転換することによって、単子葉作物においてAOPP除草剤を選択的に適用することが可能であり、単子葉作物は、フェノキシオーキシン安全性のより高い限界を有するため、フェノキシオーキシンを双子葉作物に選択的に適用することができる。AAD-1(v3)およびグルホシネートに対する耐性形質が、単子葉作物種または双子葉作物種において重ね合わせられる、雑草制御オプションの改善のためのいくつかのシナリオが想定できる。
a)グルホシネートは、多くのイネ科雑草種および広葉雑草種の制御のために、標準的な発芽後適用割合(200〜1700g ae/ha、好ましくは350〜500g ae/ha)で適用され得る。現在のところ、グルホシネート抵抗性雑草は確認されていないが、グルホシネートは、本質的により耐性の雑草をグリホセートより数多く有する。
i)本質的に耐性のイネ科雑草種(例えば、エキノクロア(Echinochloa)種またはソルガム種)は、10〜200g ae/ha(好ましくは20〜100g ae/ha)のキザロホップを容器で混合することによって制御され得る。
ii)本質に耐性の広葉雑草種(例えば、シルシウム・アーベンシス(Cirsium arvensis)およびアポシナム・カンナビナム(Apocynum cannabinum))は、これらのより制御が困難な多年生種の効率的な制御のため、そして一年生広葉雑草種の制御の強固さを改善するため、280〜2240g ae/ha、より好ましくは560〜2240g ae/haの2,4-Dを容器で混合することによって制御され得る。
b)例えば、グルホシネート(200〜500g ae/ha)+ 2,4-D(280〜1120g ae/ha)+ キザロホップ(10〜100g ae/ha)の三元組み合わせは、より強固な、重複する雑草制御スペクトルを提供することができる。さらに、重複するスペクトルは、除草剤抵抗性雑草の管理および遅延のための付加的なメカニズムを提供する。
【0212】
実施例15-任意の作物においてAAD-12(v2)と重ね合わされたDSM-2(v2)
グルホシネートは、グリホセートと同様に、比較的非選択的なスペクトルの広いイネ科および広葉樹に対する除草剤である。グルホシネートの作用の様式は、グリホセートと異なる。それは、より即効性であり、除草剤適用後24〜48時間で、処理された葉の脱水および「焼け」をもたらす。これは、迅速な雑草制御の出現のため有利である。しかしながら、これは、標的植物の分裂組織領域へのグルホシネートの転移も制限し、それが、多くの種における2つの化合物の相対的な雑草制御性能の査定によって証明されるような、より不十分な雑草制御をもたらす(Agriliance, 2005)。
【0213】
従来的な交配を通して、または新規の形質転換事象として一緒に、AAD-12(v1)をグルホシネート耐性形質と重ね合わせることにより、雑草制御の効率、柔軟性、ならびに雑草のシフトおよび除草剤抵抗性の発達を管理する能力が改善され得る。AAD-12(v1)およびグルホシネートに対する耐性形質が単子葉植物種または双子葉植物種において重ね合わせられる、雑草制御オプションの改善のためのいくつかのシナリオが想定できる。
a)グルホシネートは、多くのイネ科雑草種および広葉雑草種の制御のために、標準的な発芽後適用割合(200〜1700g ae/ha、好ましくは350〜500g ae/ha)で適用され得る。現在のところ、グルホシネート抵抗性雑草は確認されていないが、グルホシネートは本質的により耐性の雑草をグリホセートより数多く有する。
i)本質的に耐性の広葉雑草種(例えば、シルシウム・アーベンシス、アポシナム・カンナビナム、およびコニザ・カンデンシス(Conyza candensis))は、これらのより制御が困難な多年生種の効率的な制御のため、そして一年生広葉雑草種の制御の強固さを改善するため、280〜2240g ae/ha、より好ましくは560〜2240g ae/haの2,4-Dを容器で混合することによって制御され得る。トリクロピルおよびフルロキシピルが、雑草制御計画において考慮することが許容される成分であろう。トリクロピルについては、適用割合は、典型的には70〜1120g ae/ha、より典型的には140〜420g ae/haであろう。フルロキシピルについては、適用割合は、典型的には35〜560g ae/ha、より典型的には70〜280g ae/haの範囲であろう。
b)例えば、グルホシネート(200〜500g ae/ha)+/- 2,4-D(280〜1120g ae/ha)+/- トリクロピルまたはフルロキシピル(上記の割合)の多重組み合わせは、より強固な、重複する雑草制御スペクトルを提供することができる。さらに、重複するスペクトルは、除草剤抵抗性雑草の管理および遅延のための付加的なメカニズムを提供する。
【0214】
実施例16-任意の作物において昆虫抵抗性(IR)または他の提供形質と重ね合わされたDMS-2(v2)
トランスジェニック形質によって供給される作物における昆虫抵抗性は、北米において、または世界中でのトウモロコシおよびワタの生産において普及している(prevelant)。IRおよびHTの合わせた形質を有する市販の製品は、複数の種子企業によって開発されている。これらには、Bt IR形質(例えば、lifesci.sussex.ac.ukウェブサイト, 2006に列挙されるBt毒素)および上述のHTC形質のいずれかまたはそのすべてが含まれる。したがって、この提供物がもたらす価値は、単一の提供物中の遺伝的手段を通して、複数の厄介な問題を制御する能力を含む。雑草制御と昆虫制御とが互いと独立して達成される場合、この提供物の便利さは限定される。単独の、または1つもしくは複数のさらなるHTC形質と重ね合わされたDSM-2(v2)は、従来的な育種を通して、または新たな形質転換事象として合同してのいずれかで、1つもしくは複数のインプット形質(例えば、昆虫抵抗性、真菌抵抗性、またはストレス耐性など)(isb.vt.edu/cfdocs/fieldtestsl. cfm, 2005)と重ね合わせることができる。利点には、DSM-2(v2)および関連する除草剤耐性によって提供される雑草制御の改善に加えて、害虫および/または他の農学的なストレスを管理する能力とともに、上記の実施例11〜15に記載される簡便さおよび柔軟性が含まれる。従って、本発明は、任意の数の農学的な問題を柔軟かつ費用効率よく制御する能力を有する、作物品質の改善の完全な農学的パッケージを提供するために使用することができる。
【0215】
IRおよびHTの合わせた形質は、大部分の農学的および園芸用/観賞用作物および林業における用途を有する。DSM-2(v2)およびその釣り合った除草剤耐性ならびに任意の数のBtまたは非Bt IR遺伝子によって付与される昆虫抵抗性は、実施例10に列挙されたがこれらに限定されない作物種に適用できる。雑草制御の分野の当業者は、対応するHT形質またはIR形質との重ね合わせが、例えば、従来的な育種または遺伝子操作によって達成できることを認識している。
【0216】
実施例17-さらなる遺伝子の組み合わせ
本発明は、より広く、より強固な雑草制御および除草剤抵抗性管理オプションと適合性の除草剤耐性植物を提供するため、グリホセート、ALS(イミダゾリノン、スルホニル尿素)、アリールオキシアルカノエート、HPPD、PPO、およびグルホシネートに対する抵抗性遺伝子を含むがこれらに限定はされない、一つまたは複数の他の除草剤抵抗性遺伝子と共に「重ね合わせられた」本発明の一つまたは複数の酵素を産生する植物も含む。本発明は、さらに、本明細書において例示された遺伝子およびタンパク質のホモログを利用する方法および組成物を含む。
【0217】
いくつかの態様において、本発明は、ビアラホス、ホスフィノトリシン、またはグルホシネートと、一つまたは複数の市販されている除草剤(例えば、グリホセート、グルホシネート、パラコート、ALS阻害剤(例えば、スルホニル尿素、イミダゾリノン、トリアゾロピリミジンスルホンアニリド等)、HPPD阻害剤(例えば、メソトリオン、イソキサフルトル等)、2,4-D、フルロキシピル、トリコプリル、ジカンバ、ブロモキシニル、アリールオキシフェノキシプロピオネート他)とに耐性の単子葉植物および双子葉植物を提供する。そのような除草剤耐性を担う核酸配列を含むベクターも開示され、雑草制御および雑草集団シフトの防止のため、そのような耐性植物および除草剤の組み合わせを使用する方法も開示される。本発明は、除草剤の新規の組み合わせを新たな方式で使用することを可能にする。さらに、本発明は、グリホセートのような一つまたは複数の除草剤に抵抗性の雑草の系統の発達を防止し、それらの系統を制御する新規の方法を提供する。本発明は、(グルホシネートのような)除草剤に対して通常であれば感受性であろう植物のための種子を植え付ける直前の、植え付けられる区域への植え付け前適用を含む、除草剤および作物の新規の組み合わせの新規の使用を可能にする。
【0218】
本発明のDSM-2遺伝子は、当技術分野において周知であるグルホシネート耐性のための付加的な機序のため、一つまたは複数のpat/bar遺伝子と重ね合わせられてもよい。
【0219】
HPPD(ヒドロキシルフェニルピルビン酸ジオキシゲナーゼ)をコードする遺伝子の植物における使用については、例えば、米国特許第6,268,549号および第7,297,541号を参照されたい。そのような「重ね合わせられた」植物は、グルホシネート抵抗性のための他の遺伝子と組み合わせられ得、そのような重ね合わせられた植物(および様々なその他の植物および本発明の重ね合わせられた植物)は、グリホセート抵抗性の発達を防止するために使用され得る。
【0220】
グリホセートN-アセチルトランスフェラーゼ(GAT)活性を有する酵素をコードする遺伝子も、本発明のDSM-2遺伝子と共に使用され得る(重ね合わせられ得る)。例えば、Castle et al.(2004), 「Discovery of Directed Evolution of a Glyphosate Tolerance Gene」, Science Vol. 34, pp. 1151-1154;およびWO 2002/36782を参照されたい。
【0221】
本発明のDSM-2遺伝子は、それぞれWO 2005/107437、WO 2007/053482、およびUSSN 60/928,303のAAD-1遺伝子およびAAD-12遺伝子およびAAD-13遺伝子と重ね合わせられ得、それらに開示されるように、いくつかの好ましい態様において、グリホセート抵抗性に対抗するために使用され得る。
【0222】
配列の簡単な説明
SEQ ID NO:1は、ネイティブDSM-2の配列である。
SEQ ID NO:2は、ネイティブタンパク質の配列である。
SEQ ID NO:3は、ヘミコット(Hemicot)DSM-2(v2)の配列である。
SEQ ID NO:4は、再構築されたタンパク質の配列である。
SEQ ID NO:5は、Pat PTUプライマー(MAS 123)である。
SEQ ID NO:6は、Pat PTUプライマー(Per 5-4)である。
SEQ ID NO:7は、Patコード領域プライマーである。
SEQ ID NO:8は、Patコード領域プライマーである。
SEQ ID NO:9は、DSM-2(v2)コード領域プライマーである。
SEQ ID NO:10は、DSM-2(v2)コード領域プライマーである。
【背景技術】
【0001】
発明の背景
選択マーカーとは、同定され追跡され得る検出可能な遺伝形質またはDNAのセグメントである。マーカー遺伝子は、典型的には、標的遺伝子とも呼ばれるもう一つの遺伝子のためのフラッグとして役立つ。マーカー遺伝子は、典型的には、標的細胞を形質転換するために使用される標的遺伝子と共に使用される。標的遺伝子を遺伝的に受容する標的細胞は、マーカー遺伝子も発現する細胞を選択することにより同定され得る。2つの遺伝子(マーカー遺伝子および標的遺伝子)が遺伝連鎖し、通常一緒に遺伝されるよう、マーカー遺伝子は標的遺伝子に十分に近く存在する。選択マーカーの現在の標準物は、ホスフィノトリシンアセチルトランスフェラーゼと呼ばれる酵素をコードする「pat」遺伝子である。
【0002】
グルタミンシンセターゼ(「GS」)は、多くの植物において、植物細胞の発達および生命にとって必須の酵素である。GSはグルタミン酸をグルタミンに変換する。GSはアンモニア同化および窒素代謝にも関与している。GSは、大部分の植物において、硝酸塩還元によって放出されるアンモニアの解毒のための経路に関与している。従って、GSの強力な阻害剤は、植物細胞にとって極めて毒性である。植物の内部での除草剤の分解または修飾は、抵抗性をもたらし得る。
【0003】
特定のクラスの除草剤が、植物におけるGSの阻害による毒性効果に基づき開発されている。ビアラホスおよびホスフィノトリシンは、2つのそのようなGS作用の阻害剤であり、優れた除草特性を保有している。これらの2つの除草剤は非選択的であり;土壌に存在する様々な植物種すべての生長を阻害し、従って、それらの完全な駆除を引き起こす。
【0004】
ビアラホスも、広いスペクトルを有する除草剤である。ビアラホスは、ホスフィノトリシン(PPTまたはPTC;2-アミノ-4-メチルホスフィノ酪酸)、L-グルタミン酸のアナログ、および2個のL-アラニン残基から構成されている。従って、PPTとビアラホスとの間の構造的な違いは、PPTの場合には2個のアラニンアミノ酸が存在しない点にある。細胞内ペプチダーゼによるビアラホスのL-アラニン残基の除去によって、PPTが放出される。PPTはGSの強力な阻害剤である。PPTによる植物におけるGSの阻害は、アンモニアの迅速な蓄積および植物細胞の死を引き起こす。
【0005】
ビアラホスは、最初、抗生物質特性を有することが開示され、そのため、殺虫薬または殺真菌薬として使用され得た。米国特許第3,832,394号(特許文献1)は、ストレプトマイセス・ハイグロスコピカス(Streptomyces hygroscopicus)の培養、およびその培養培地からのビアラホスの回収に関する。しかしながら、ストレプトマイセス・ビリドクロモジェネス(viridochromogenes)のようなその他の株も、この化合物を産生する。ホサラシン(phosalacin)のような、PPT部分を含有しているその他のトリペプチド抗生物質も、自然界に存在することが公知である。PPTは、化学合成によっても入手され、市販されている。
【0006】
ビアラホスを産生するストレプトマイセス・ハイグロスコピカスおよびストレプトマイセス・ビリドクロモジェネスは、ホスフィノトリシンアセチルトランスフェラーゼ活性を有する酵素によってPPT毒性から保護される。Plant Physiol, April 2001, Vol. 125, pp. 1585-1590(「Expression of bar in the Plastid Genome Confers Herbicide Resistance」, Lutz et al.)(非特許文献1)。これらの抗生物質を産生するストレプトマイセス種は、もしこれらの抗生物質に対する自己防御機序を有していなければ、それ自体破壊されるであろう。この自己防御機序は、いくつかの事例において、抗生物質効果を阻害することができる酵素を含むことが見出されている。
【0007】
ホスフィノトリシンアセチルトランスフェラーゼは、bar遺伝子(ビアラホス抵抗性;Thompson et al., 1987)またはpat遺伝子(ホスフィノトリシンアセチルトランスフェラーゼ;Strauch et al., 1988)のいずれかによってコードされ、PPTの遊離アミノ基のアセチル化によってPPTを解毒する。これらの2つの遺伝子によってコードされた酵素は、機能的に同一であり、アミノ酸レベルで85%の同一性を示す(Wohlleben et al., 1988; Wehrmann et al., 1996)。核遺伝子からのキメラbarまたはpat遺伝子の細胞質における発現により、PPT抵抗性作物が入手されている。タバコ(ニコチアナ・タバカム品種プティ・ハバナ(Nicotiana tabacum cv Petit Havana))、ジャガイモ、セイヨウアブラナ(Brassica napus)、ブラッシカ・オレラセア(Brassica oleracea)(De Block et al., 1987; De Block et al., 1989)、トウモロコシ(Spencer et al., 1990)、およびイネ(Cao etal., 1992)においては、PPT抵抗性についての直接選択によって、除草剤抵抗性の系が獲得されている。
【0008】
遺伝子(bar)が、ストレプトマイセス・セリカラー(coelicolor)A3においてhrdDシグマ因子遺伝子に隣接して同定された。予測されたbar産物は、ビアラホス産生種ストレプトマイセス・ビリドクロモジェネスおよびストレプトマイセス・ハイグロスコピカスのpat遺伝子およびbar遺伝子のものとそれぞれ32.2%および30.4%の同一性を示した。S.セリカラーbar遺伝子は、高コピー数ベクターによりS.セリカラーにおいてクローニングされた場合、ビアラホスに対する抵抗性を付与した。Bedford et al., Gene, 1991 Jul 31; 104(1):39-45, 「Characterization of a gene conferring bialaphos resistance in Streptomyces coelicolor A3(2)」(非特許文献2)。その他の微生物におけるこの遺伝子の異種発現、またはこの遺伝子の植物への形質転換は、これまでに報告されていない。
【0009】
除草剤抵抗性形質の使用は、pat遺伝子がストレプトマイセス・ビリドクロモジェネスから単離されるDE 3642 829 A(特許文献2)ならびに米国特許第5,879,903号(特許文献3)(ならびに第5,637,489号(特許文献4);第5,276,268号(特許文献5);および第5,273,894号(特許文献6))において言及されている。WO 87/05629(特許文献7)ならびに米国特許第5,648,477号(特許文献8)(ならびに第5,646,024号(特許文献9)および第5,561,236号(特許文献10))は、(PPTのような)グルタミンシンセターゼ阻害剤から植物細胞および植物体を保護するためのS.ハイグロスコピカス由来のbar遺伝子の使用、ならびに植物体における除草剤抵抗性の発達に言及している。除草剤BASTA(Hoechstホスフィノトリシン)またはハービエース(Herbiace)(Meiji Seikaビアラホス)に対する抵抗性をコードする遺伝子が、アグロバクテリウム感染によって、タバコ(ニコチアナ・タバカム品種プティ・ハバン(Havan)SR1)、ジャガイモ(ソラナム・ツベロサム品種ベノリマ(Solanum tuberosum cv Benolima))、およびトマト(リコパーシカム・エスカレンタム(Lycopersicum esculentum))植物へ導入され、除草剤抵抗性を付与した。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】米国特許第3,832,394号
【特許文献2】DE 3642 829 A
【特許文献3】米国特許第5,879,903号
【特許文献4】米国特許第5,637,489号
【特許文献5】米国特許第5,276,268号
【特許文献6】米国特許第5,273,894号
【特許文献7】WO 87/05629
【特許文献8】米国特許第5,648,477号
【特許文献9】米国特許第5,646,024号
【特許文献10】米国特許第5,561,236号
【非特許文献】
【0011】
【非特許文献1】Plant Physiol, April 2001, Vol. 125, pp. 1585-1590(「Expression of bar in the Plastid Genome Confers Herbicide Resistance」, Lutz et al.)
【非特許文献2】Bedford et al., Gene, 1991 Jul 31; 104(1):39-45,「Characterization of a gene conferring bialaphos resistance in Streptomyces coelicolor A3(2)」
【発明の概要】
【0012】
本発明は、本明細書においてDSM-2と呼ばれる新規の遺伝子に関する。この遺伝子はストレプトマイセス・セリカラー(A3)において同定された。DSM-2タンパク質は、PATおよびBARの遠縁である。本発明はまた、DSM-2タンパク質をコードする、植物に最適化された遺伝子を提供する。DSM-2は、微生物、植物細胞、および植物体において、除草抗菌分子、グルホシネート、ホスフィノトリシン、および/またはビアラホスに対する耐性を付与するためのトランスジェニック形質として使用され得る。
【0013】
アラビドプシス(Arabidopsis)への形質転換は、グルホシネートの高い割合の回収を可能にする。導入された後、DSM-2遺伝子は、有意な選択および完全な植物体の抵抗性を提供する能力を有する。
【0014】
本発明の遺伝子には、単にバイオテクノロジープロジェクトのための選択マーカーとして、高い本質的な価値がある。いくつかの好ましい態様において、本発明の遺伝子は、培養物中の形質転換に成功した細胞を選択するため、ならびに温室および圃場において完全な植物体を選択するためのマーカーとして使用され得る。この遺伝子および類似ホモログは、patおよび/またはbarの代わりに使用され得る。
【0015】
細菌形質転換系における選択マーカーとしてのこの遺伝子の使用は、植物のすべての形質転換の効率を増加させることができる。唯一の選択マーカーとしてのDSM-2の使用は、クローニング中のさらなる(アンピシリン抵抗性のような)医療用抗生物質マーカーの必要性を排除する。
【0016】
本発明によれば、様々なその他の使用も可能である。DSM-2は、除草剤グルホシネートおよびビアラホスに対する耐性を植物に付与するトランスジェニック形質として有用であり得る。
【0017】
この遺伝子は、修飾されたアグロバクテリウム株と合わせて、新規の植物形質転換系のための基礎としても使用され得る。タンパク質作製および非医療用抗生物質抵抗性マーカー遺伝子の組み入れのために使用されるシュードモナス・フルオレセンス(Pseudomonas fluorescens)の新規の株またはその他の微生物株も、本発明により作製され得る。医療用抗生物質抵抗性要素からのフラグメント精製が排除されることによる、クローニングおよび形質転換の過程および効率の改善も、有益であるかもしれない。
【0018】
除草剤耐性作物(HTC)形質に加えて、本発明の遺伝子によってトランスジェニック作物において除草剤耐性が作出される、除草剤を使用して雑草を制御する方法も、本発明の範囲内である。本発明のHTC形質の組み合わせも、他のHTC形質(グリホセート耐性および2,4-D耐性を含むが、これらに限定はされない)と組み合わせられた場合、特に、(グリホセートのような)除草剤に対する新たに獲得された抵抗性または本質的な耐性を有する種を制御するために、有益である。さらに、(世界的にますます普及してきている)グリホセート耐性作物を他のグリホセート耐性作物と輪作する場合、グリホセート抵抗性の自生植物の制御が困難である場合がある。従って、作物へ重ね合わせられた(stacked)、または個々に形質転換されたこれらのトランスジェニック形質の使用は、他のHTC自生植物の制御のための手段を提供することができる。
【図面の簡単な説明】
【0019】
【図1】DSM2によって媒介されるN-アセチル化によるグルホシネートの不活性化を示す図である。
【発明を実施するための形態】
【0020】
発明の詳細な説明
本発明は、本明細書においてDSM-2と呼ばれる新規の遺伝子に関する。この遺伝子はストレプトマイセス・セリカラー(A3)において同定された。DSM-2タンパク質は、PATおよびBARの遠縁である(それぞれ30%および28%のアミノ酸同一性)。本発明はまた、例えば、ヘミコット・バイアス(hemicot bias)を有する、DSM-2タンパク質をコードする、植物に最適化された遺伝子を提供する。DSM-2は、植物体および植物細胞において、除草剤グルホシネート、ホスフィノトリシン、およびビアラホスに対する耐性を付与するためのトランスジェニック形質として使用され得る。
【0021】
本発明の遺伝子には、単にバイオテクノロジープロジェクトのための選択マーカーとして、高い本質的な価値がある。しかしながら、本発明によれば、様々なその他の使用が可能である。
【0022】
DSM-2は、除草剤グルホシネート、ホスフィノトリシン、およびビアラホスに対する耐性を植物に付与するトランスジェニック形質として有用であり得る。
【0023】
いくつかの好ましい態様において、(除草剤耐性作物HTCとは別に、またはそれに加えて)培養物、温室、および圃場における形質転換に成功した細胞ならびに完全な植物体を選択するためのマーカーとしてである。この遺伝子および類似ホモログは、patおよび/またはbarの代わりに使用され得る。本明細書において例示される1つの態様は、NT-1タバコ細胞における選択マーカーとしてのDSM-2の使用である。本発明の遺伝子は、トウモロコシおよびイネのようなその他の植物系においても選択マーカーとして使用され得る。
【0024】
細菌系における選択マーカーとしてのこの遺伝子の使用は、すべての植物形質転換の効率を増加させることができる。唯一の選択マーカーとしてのDSM-2の使用は、クローニング中のさらなる(アンピシリン抵抗性のような)医療用抗生物質マーカーの必要性を排除する。
【0025】
実験は、大腸菌(Escherichia coli)細胞系BL21-Star(DE3)(Invitrogenカタログ#C6010-03)が、100μg/mlという濃度のグルホシネートアンモニウム(Basta)を含有している最少培地で阻害されることを証明した。BL21 Star細胞系におけるDSM-2の発現は、400μg/mlのグルホシネートを含有している最少培地で抵抗性を補完した。これらの実験は、DSM-2の発現が、グルホシネートによって阻害される細菌において、クローニングの適用のための非医療用抗生物質選択マーカーとして使用され得ることを示している。
【0026】
さらなる実験は、植物プロモーター、シロイヌナズナ(Arabidopsis thaliana)PolyUbiquitin 10(At Ubi10)およびウイルスプロモーター、キャッサバ葉脈モザイクウイルス(CsVMV)が、大腸菌株BL21-Star(DE3)において機能性であることを証明した。両プロモーターは、200μg/mlのグルホシネートを含有している最少培地に対する抵抗性を提供するために充分なDSM-2タンパク質を発現した。これらの植物プロモーターは、大腸菌において非医療用抗生物質選択マーカーとしてDSM-2の発現を駆動するために使用され得る。細菌および植物の両方において単一の植物プロモーターが機能性であれば、各々の種のための別々の選択マーカーの必要性が排除される。
【0027】
この遺伝子は、修飾されたアグロバクテリウム株と合わせて、新規の植物形質転換系のための基礎としても使用され得る。非医療用抗生物質抵抗性マーカー遺伝子を使用したタンパク質作製のためのシュードモナス・フルオレセンスの新規の株またはその他の微生物株も、本発明により作製され得る。医療用抗生物質抵抗性要素からのフラグメント精製が排除されることによる、クローニングおよび形質転換の過程および効率の改善も、有益であり得る。
【0028】
HTC形質に加えて、本発明の遺伝子によって除草剤耐性がトランスジェニック作物において作出される、除草剤を使用して雑草を制御する方法も、本発明の範囲内である。本発明のHTC形質の組み合わせも、他のHTC形質(グリホセート耐性および2,4-D耐性を含むが、これらに限定はされない)と組み合わせられた場合、特に、(グリホセートのような)除草剤に対する新たに獲得された抵抗性または本質的な耐性を有する種の制御のため、有益である。さらに、(世界的にますます普及してきている)グリホセート耐性作物を他のグリホセート耐性作物と輪作する場合、グリホセート抵抗性自生植物の制御が困難である場合がある。従って、作物へ重ね合わせられた、または個々に形質転換されたトランスジェニック形質の使用は、他のHTC自生植物の制御のための手段を提供することができる。
【0029】
本発明のタンパク質(および供給源の単離物)
本発明は機能的タンパク質を提供する。「機能的活性」(または「活性な」)とは、本明細書では、本発明による使用のためのタンパク質/酵素が、除草剤を分解するか、またはその活性を減少する能力を有する(単独でまたは他のタンパク質と組み合わせて)ことを意味する。本発明のタンパク質を産生する植物は、該植物が除草剤で処理される場合に、タンパク質発現のレベルが、該植物を、除草剤に対して完全もしくは部分的に抵抗性に、または耐性にするための十分であるような、タンパク質の「有効量」を好ましく産生する(他に特定されない限り典型的な割合において;典型的な適用割合は、例えば、周知のHerbicide Handbook(Weed Science Society of America, Eighth Edition, 2002)において見出され得る)。除草剤は、通常の圃場使用割合および濃度において、正常に標的植物を殺傷する割合で適用され得る(本発明のために、レベルおよび/または濃度は、以前に使用されたものよりも任意により高くあり得る)。好ましくは、本発明の植物細胞および植物は、除草剤処理によって引き起こされる生育阻害または損傷に対して保護される。本発明の形質転換植物および植物細胞は、好ましくは、本明細書で考察されるように、除草剤に対して抵抗性または耐性にされ、このことは、形質転換植物および植物細胞が、本明細書で考察されるように、有効量の1種または複数の除草剤の存在下で生育することができることを意味する。本発明の好ましいタンパク質は、1種または複数のアリールオキシアルカノエート化合物を代謝するための触媒活性を有する。「抵抗性」という用語を容易に論じることはできないし、「耐性である」という動詞または「耐性の」という形容詞を容易に使用することはできない。産業界は、除草剤抵抗性作物(HRC)対除草剤耐性作物(HTC)の議論に、膨大な時間を費やしてきた。HTCは産業界において好まれる用語である。しかし、米国雑草科学会(Weed Science Society of America)による公式の抵抗性の定義は、「野生型に対して通常は致死的である除草剤の用量への曝露後、植物の、遺伝性の、生存および再生する能力である。植物においては、抵抗性は、天然に存在するか、または組織培養もしくは変異誘発によって産生される変種の遺伝子操作または遺伝子選択のような技術によって誘導される可能性がある」というものである。本明細書で使用される場合、他に示されない限り、除草剤「抵抗性」は、本開示の出願時における現行版のThe Herbicide Handbookによって示唆されるように、遺伝性であり、かつ所定の植物のための除草剤による、典型的な有効な除草性処理の存在下で、植物が生長および再生することを可能にする。当業者によって認識されるように、たとえ除草剤曝露によるある程度の植物損傷が明白であっても、植物は、なお「抵抗性である」と見なされる可能性がある。本明細書で使用される場合、「耐性」という用語は、「抵抗性」という用語よりも広く、本明細書で定義されるような「抵抗性」を含み、かつ同じ除草剤用量において同じ遺伝子型の野生型植物を典型的には生じる、除草剤によって誘導された種々の程度の損傷に対して特定の植物が耐える能力の改善を含む。
【0030】
植物または細菌の系への機能的活性の移入は、そのベクターが存在する宿主に対して適切であるタンパク質発現ベクターに組み込まれた、本発明のタンパク質についてのアミノ酸配列をコードする核酸配列を含むことができる。機能的活性を有するタンパク質をコードする核酸配列を得るための1つの方法は、本明細書で考察されるように、タンパク質のアミノ酸配列から推定される情報を使用して、関心対象のタンパク質を産生する細菌種からのネイティブな遺伝物質を単離することである。このネイティブ配列は、例えば、以下でより詳細に考察されるように、植物での発現のために最適化することができる。最適化されたポリヌクレオチドはまた、タンパク質配列に基づいて設計することもできる。
【0031】
これらのタンパク質のクラスおよびこれらをコードするポリヌクレオチドを特徴付けするための1つの方法は、一連の特定化された条件下で、例示されたヌクレオチド配列(その相補体および/またはいずれかの鎖に由来するプローブ)とハイブリダイズするその能力によってポリヌクレオチドを定義することによるか、および/または例示された配列に由来するプライマーを使用するPCRによって増幅されるそれらの能力による。
【0032】
本発明による使用のためのタンパク質を入手するための多数の方法が存在する。例えば、本明細書に開示されるタンパク質に対する抗体が、タンパク質の混合物から他のタンパク質を同定および単離するために使用され得る。詳細には、抗体は、他の関連するタンパク質を比較した場合に、最も保存されているか、または最も区別できるタンパク質の一部分に対して惹起されてもよい。次いで、これらの抗体は、免疫沈殿、酵素結合免疫吸着アッセイ法(ELISA)、またはイムノブロッティングによって、特徴的な活性を有する等価なタンパク質を特異的に同定するために使用することができる。本明細書に開示されるタンパク質、または等価なタンパク質、またはこれらのタンパク質のフラグメントに対する抗体が、標準的な手順を使用して容易に調製され得る。このような抗体は本発明の1つの局面である。本発明の抗体は、モノクローナル抗体およびポリクローナル抗体を含み、好ましくは、例示または示唆されたタンパク質に応答して産生される。
【0033】
本開示の恩典により、本発明のタンパク質および遺伝子は、例えば、組換え細菌および/または野生型細菌のような多様な微生物を含む多様な起源から入手され得る。
【0034】
細菌からの単離物の変異体は、当技術分野において周知である手法によって作製することができる。例えば、胞子非形成変異体は、単離物のエチルメタンスルホネート(EMS)変異誘発を通して入手することができる。これらの変異体は、当技術分野において周知の手法によって、紫外光およびニトロソグアニジンを使用して作製することができる。
【0035】
本明細書で言及または示唆される対象の単離物のいずれか「からの」またはそれ「から入手可能である」タンパク質は、そのタンパク質(または同様のタンパク質)が、別の細菌株または植物などの単離物またはある他の供給源から入手可能であることを意味する。「から入手可能である」はまた、この暗示的意味を有し、例えば、植物での発現のために改変されている、所定の型の細菌から入手可能であるタンパク質を含む。当業者は、細菌の遺伝子およびタンパク質の開示が与えられることで、植物がそのタンパク質を産生するために操作され得ることを容易に認識する。抗体調製物、核酸プローブ(例えば、DNA、RNA、またはPNA)などは、本明細書に開示されるポリヌクレオチドおよび/またはアミノ酸配列を使用して調製され得、かつ他の(天然の)供給源から他の関連する遺伝子をスクリーニングおよび回収するために使用され得る。
【0036】
標準的な分子生物学技術は、本明細書に記載されるタンパク質および遺伝子をクローニングおよび配列決定するために使用されてもよい。さらなる情報は、参照により本明細書に組み入れられる、Sambrook et al., 1989において見出され得る。
【0037】
ポリヌクレオチドおよびプローブ
本発明はさらに、本発明による使用のためのタンパク質をコードする核酸配列を提供する。本発明はさらに、所望の除草剤活性を有するタンパク質をコードする遺伝子を同定および特徴付けする方法を提供する。1つの態様において、本発明は、ハイブリダイゼーションプローブおよび/またはPCR技術のためのプライマーとして有用である特有のヌクレオチド配列を提供する。これらのプライマーは、関心対象の特定の遺伝子の同定、特徴付け、および/または単離において有用であり得る特徴的な遺伝子フラグメントを産生する。本発明のヌクレオチド配列は、以前に記載されているタンパク質から区別できるタンパク質をコードする。
【0038】
本発明のポリヌクレオチドは、所望の宿主細胞中でタンパク質またはペプチドをコードするために完全な「遺伝子」を形成するために使用することができる。例えば、当業者が容易に認識するように、対象のポリヌクレオチドは、当技術分野において容易に公知であるように、関心対象の宿主中で、プロモーターの制御下に適切に配置され得る。遺伝子発現および一過性/組織特異的発現のレベルは、本発明の有用性に非常に影響を与えることができる。一般的に、分解可能な遺伝子のタンパク質発現のレベルがより大きいと、基質(この場合は標的除草剤)の完全な分解がより多くなる。プロモーターは、高い発現が植物の健康に結果的に負の影響を有さない限り、高いレベルで標的遺伝子を発現することが所望される。典型的には、すべての生育段階において植物の完全な保護のためにすべての組織中で構成的に発現されるDSM-2遺伝子を有することが望まれる。しかし、代替として、植物的において発現される抵抗性遺伝子を使用してもよい。これは、雑草制御のための作物における標的除草剤の使用を可能にし、開花段階の間の適用によって標的作物の有性生殖を引き続き制御する。
【0039】
当業者に公知であるように、DNAは、典型的には、二本鎖型で存在する。この配列において、一方の鎖は他方の鎖に対して相補的であり、逆もまた同様である。(例えば)DNAは植物中で複製されるので、DNAのさらなる相補鎖が産生される。「コード鎖」は、アンチセンス鎖と結合する鎖をいうために当技術分野において頻繁に使用される。mRNAは、DNAの「アンチセンス」鎖から転写される。「センス」鎖または「コード」鎖は、オープンリーディングフレーム(ORF)として読み取られることができ、関心対象のタンパク質またはペプチドを形成する一連のコドン(コドンは、特定のアミノ酸を特定するための3残基単位として読まれ得る3つのヌクレオチドである)を有する。インビボでタンパク質を産生するために、DNAの鎖は、典型的には、タンパク質のための鋳型として使用されるmRNAの相補鎖に転写される。従って、本発明は、相補鎖を含む、添付の配列表および/または等価物に示される例示されるポリヌクレオチドの使用を含む。例示されたDNA分子に対して機能的に等価であるRNAおよびPNA(ペプチド核酸)は本発明に含まれる。
【0040】
本発明の1つの態様において、細菌からの単離物は、微生物の高い増殖を生じる条件下で培養され得る。一本鎖ゲノム核酸を提供するために微生物を処理した後、DNAは、本発明のプライマーと接触され得、PCR増幅に供され得る。関心対象の遺伝子の特徴的なフラグメントは、この手法によって増幅され、それにより、関心対象の遺伝子の存在を同定する。
【0041】
本発明のさらなる局面は、本明細書に開示される方法およびヌクレオチド配列を使用して同定された遺伝子および単離物を含む。このように同定された遺伝子は、本発明の除草剤抵抗性タンパク質をコードすることができる。
【0042】
本発明によるタンパク質および遺伝子は、例えば、オリゴヌクレオチドプローブを使用することによって同定および入手することができる。これらのプローブは、適切な標識によって検出可能であり得るか、または国際出願番号WO 93/16094において記載されているように、固有に蛍光性に作製されてもよい、検出可能なヌクレオチド配列である。これらのプローブ(および本発明のポリヌクレオチド)は、DNA、RNA、またはPNAであってもよい。アデニン(A)、シトシン(C)、グアニン(G)、チミン(T)、およびウラシル(U;RNA分子用)に加えて、本発明の合成プローブ(およびポリヌクレオチド)はまた、イノシン(4種すべての塩基と対合することが可能な中性塩基;時折、合成プローブ中で4種すべての塩基の混合物の代わりに使用される)および/または合成(非天然)塩基を有することができる。従って、合成の、縮重オリゴヌクレオチドが本明細書で言及される場合、「N」または「n」が総称的に使用され、「N」または「n」は、G、A、T、C、またはイノシンであり得る。曖昧なコードは、本明細書で使用されるように、本願の出願の時点の標準IUPAC命名法の慣例に従っている(例えば、RはAまたはGを意味し、YはCまたはTを意味する、など)。
【0043】
当技術分野において周知であるように、プローブ分子が核酸試料にハイブリダイズする場合には、プローブおよび試料が実質的な相同性/類似性/同一性を有することが合理的に推測され得る。好ましくは、ポリヌクレオチドのハイブリダイゼーションは、例えば、Keller, G. H., M. M. Manak(1987)DNA Probes, Stockton Press, New York, NY, pp. 169-170によって記載されるような、当該分野において周知の技術によって、ポリヌクレオチドのハイブリダイゼーションが最初に行われ、続いて、低ストリンジェンシー、中程度のストリンジェンシー、または高ストリンジェンシーの条件下での洗浄が行われる。例えば、そこに言及されているように、低ストリンジェンシー条件は、2×SSC(標準生理食塩水クエン酸)/0.1% SDS(ドデシル硫酸ナトリウム)を用いる、室温にて15分間の最初の洗浄によって達成され得る。2回の洗浄が典型的には実行される。次いで、より高いストリンジェンシーが、塩濃度を低下させることによって、および/または温度を上昇させることによって達成され得る。例えば、上記に記載される洗浄は、各々、0.1×SSC/0.1% SDSを用いる室温における15分間の2回の洗浄が続き得、これには、各々、0.1×SSC/0.1% SDSを用いる55℃における30分間の洗浄が続く。これらの温度は、本明細書に示され、かつ当業者に公知であるような他のハイブリダイゼーションおよび洗浄のプロトコールとともに使用することができる(例えば、SSPEは、SSCの代わりに塩として使用することができる)。2×SSC/0.1% SDSは、445mlの水に、50mlの20×SSCおよび5mlの10% SDSを加えることによって調製することができる。20×SSCは、NaCl(175.3g/0.150M)、クエン酸ナトリウム(88.2g/0.015M)、および水を合わせ、10N NaOHでpHを7.0に調整し、次いで、体積を1リットルに調整することによって調製することができる。10% SDSは、オートクレーブした50mlの水中に10gのSDSを溶解すること、次いで100mlに希釈することによって調製することができる。
【0044】
プローブの検出は、ハイブリダイゼーションが維持されたか否かを公知の様式で判定するための手段を提供する。このようなプローブ分析は、本明細書の遺伝子を同定するための迅速な方法を提供する。本発明によるプローブとして使用されるヌクレオチドセグメントは、DNAシンセサイザーおよび標準的な手法を使用して合成することができる。これらのヌクレオチド配列はまた、本発明の遺伝子を増幅するためのPCRプライマーとして使用することができる。
【0045】
分子のハイブリダイゼーション特性は、本発明のポリヌクレオチドを規定するために使用することができる。従って、本発明は、本明細書で例示されるポリヌクレオチドとハイブリダイズするポリヌクレオチド(および/またはそれらの相補体、好ましくはそれらの完全な相補体)を含む。すなわち、遺伝子(およびそれがコードするタンパク質)を規定するための1つの方法は、例えば、公知のまたは特に例示される遺伝子と(本明細書に具体的に開示されるいずれかの条件下で)ハイブリダイズするその能力による。
【0046】
本明細書で使用されるように、ハイブリダイゼーションのための「ストリンジェントな」条件とは、本出願者によって使用される条件と同じかまたはほぼ同じである、ハイブリダイゼーションの特異性の程度を達成する条件をいう。具体的には、32P-標識された遺伝子特異的プローブとの、サザンブロット上に固定化されたDNAのハイブリダイゼーションは、標準的な方法によって実行することができる(例えば、Maniatis et al. 1982を参照されたい)。一般的に、ハイブリダイゼーションおよび引き続く洗浄は、標的配列の検出を可能にする条件下で実行することができる。二本鎖DNA遺伝子プローブについては、ハイブリダイゼーションは、6×SSPE、5×Denhardt's溶液、0.1% SDS、0.1mg/ml変性DNA中で、DNAハイブリッドの融解温度(Tm)よりも20〜25℃下で、一晩実行することができる。融解温度は以下の数式によって記載される(Beltz et al. 1983)。
Tm = 81.5 C + 16.6 Log [Na+] + 0.41(%G+C)- 0.61(ホルムアルデヒド%)- 600/塩基対中の二重鎖の長さ。
【0047】
洗浄は、典型的には、以下のように実行することができる。
(1)1×SSPE、0.1% SDS中での室温で15分間、2回(低ストリンジェンシー洗浄)。
(2)0.2×SSPE、0.1% SDS中でのTm-20℃で15分間、1回(中程度ストリンジェンシー洗浄)。
【0048】
オリゴヌクレオチドプローブについては、ハイブリダイゼーションは、6×SSPE、5×Denhardt's溶液、0.1% SDS、0.1mg/ml変性DNA中で、ハイブリッドの融解温度(Tm)よりも10〜20℃下で、一晩実行することができる。融解温度は以下の数式によって決定することができる。
Tm(℃)= 2(T/A塩基対の数)+ 4(G/C塩基対の数)(Suggs et al., 1981)。
【0049】
洗浄は、典型的には、以下のように実行することができる。
(1)1×SSPE、0.1% SDS中での室温で15分間、2回(低ストリンジェンシー洗浄)。
(2)1×SSPE、0.1% SDS中でのハイブリダイゼーション温度で15分間、1回(中程度ストリンジェンシー洗浄)。
【0050】
一般的に、塩および/または温度は、ストリンジェンシーを変化させるために変更することができる。>70程度の塩基長の標識したDNAフラグメントを用いる場合、以下の条件が使用され得る。
低:1または2×SPPE、室温
低:1または2×SSPE、42℃
中程度:0.2×または1×SSPE、65℃
高:0.1×SSPE、65℃。
【0051】
二重鎖の形成および安定性は、ハイブリッドの2本の鎖の間の実質的な相補性に依存し、上記に記述されるように、特定の程度のミスマッチが許容され得る。それゆえに、本発明のプローブ配列は、記載された配列の変異(単一と複数の両方)、欠失、挿入、およびこれらの組み合わせを含み、ここで、該変異、挿入、および欠失は、関心対象の標的ポリヌクレオチドとの安定なハイブリッドの形成を許容する。変異、挿入、および欠失は、多くの方法で所定のポリヌクレオチド配列中で産生され得るが、これらの方法は当業者に公知である。他の方法は将来公知になる可能性がある。
【0052】
PCR技術
ポリメラーゼ連鎖反応(PCR)は、反復性の、酵素的な、プライマーを用いる、核酸配列の合成である。この手法は周知であり、当業者に一般的に公知である(Mullis, 米国特許第4,683,195号、同第4,683,202号、および同第4,800,159号; Saiki et al., 1985を参照されたい)。PCRは、標的配列の反対の鎖にハイブリダイズする2つのオリゴヌクレオチドプライマーによって隣接される関心対象のDNAフラグメントの酵素的増幅に基づく。これらのプライマーは、好ましくは、3'末端が互いに対して向けられて配置される。鋳型の熱変性、それらの相補的配列へのプライマーのアニーリング、およびDNAポリメラーゼを用いてのアニールされたプライマーの伸長は、PCRプライマーの5'末端によって規定されるセグメントの増幅を生じる。各プライマーの伸長産物は、他のプライマーのための鋳型として働くことができ、従って、各サイクルは、以前のサイクルで産生されたDNAフラグメントの量を本質的に倍加させる。これにより、特定の標的フラグメントの指数関数的な蓄積が、数時間以内に数百万倍にまでになる。耐熱性細菌サーマス アクアティカス(Thermus aquaticus)から単離されたTaqポリメラーゼなどの耐熱性DNAポリメラーゼを使用することによって、この増幅プロセスは、完全に自動化することができる。使用することができる他の酵素は、当業者に公知である。
【0053】
例示されるDNA配列、またはそのセグメントは、PCR増幅のためのプライマーとして使用することができる。PCR増幅を実行する際に、特定の程度のミスマッチが、プライマーと鋳型の間に許容され得る。それゆえに、例示されたプライマーの変異、欠失、および挿入(とりわけ、5'末端へのヌクレオチドの付加)は、本発明の範囲内にある。変異、欠失、および挿入は、当業者に公知である方法によって、所定のプライマー中で作製することができる。
【0054】
遺伝子およびタンパク質の改変
対象の遺伝子およびタンパク質は、キメラタンパク質または融合タンパク質を産生するために、他の遺伝子およびタンパク質に融合され得る。本発明に従って有用な遺伝子およびタンパク質は、特に例示された全長配列のみならず、これらの配列、これらの配列の変種、変異体、キメラ、ならびに融合物の一部分、セグメント、および/またはフラグメント(隣接するフラグメント、ならびに全長分子と比較して内部および/または末端の欠失を含む)を含む。本発明のタンパク質は、これらが所望の機能的活性を保持する限りは、置換されたアミノ酸を有することができる。「変種」遺伝子は、例示されたタンパク質に対して等価であるかまたは類似している活性を有する、同じタンパク質または等価なタンパク質をコードするヌクレオチド配列を有する。「変種タンパク質」および「等価なタンパク質」という用語は、標的基質に対して同じかまたは本質的に同じ生物学的/機能的活性、および例示されたタンパク質と等価な配列を有するタンパク質をいう。本明細書で使用されるように、「等価な」配列との言及は、有意な程度にまで活性を改善し、または活性に有害な影響を与えない、アミノ酸の置換、欠失、付加、または挿入を有する配列をいう。活性を保持しているフラグメントもまた、この定義の中に含まれる。例示されたタンパク質の対応するフラグメントと同じかまたは同様の機能または活性を保持するフラグメントおよび他の等価物は、本発明の範囲内にある。アミノ酸の置換または付加などの変化は、例えば、(タンパク質の機能的活性を具体的に/実質的に減少することなく)タンパク質のプロテアーゼ安定性を増加すること(もしくは減少すること)、または制限部位の付加などの種々の目的のために作製することができる。遺伝子のバリエーションは、例えば、点変異を作製するための標準的な技術を使用して容易に構築されうる。
【0055】
加えて、例えば、米国特許第5,605,793号は、ランダムなまたは的を絞ったフラグメント化後にDNA再アセンブリーを使用することによって、さらなる分子の多様性を生成するための方法を記載している。これは、「遺伝子シャッフリング」と呼ぶことができ、これは、典型的には、複数の異なるDNA分子のフラグメントを混合する工程、続いて、再生の反復ラウンドを含む。これは、開始遺伝子によってコードされるタンパク質の活性を改善することができる。この結果は、改善された活性、変化した基質特異性、増加した酵素安定性、変化した立体特異性、または他の特徴を有するキメラタンパク質である。
【0056】
「シャッフリング」は、関心対象タンパク質の原子3D(三次元)配位および結晶構造を入手および精査後に、設計および標的化され得る。従って、「集中シャッフリング」は、表面に露出しているセグメントなどの、改変のために理想的であるタンパク質の特定のセグメントに向けることができ、好ましくは、タンパク質フォールディングおよび必須の3D構造の完全性と関連する内部セグメントではない。
【0057】
変種遺伝子は、変種タンパク質を産生するために使用することができる。組換え宿主は、変種タンパク質を産生するために使用することができる。「遺伝子シャッフリング」技術および他の技術を使用して、本発明に例示される任意の配列の特定の残基(アミノ酸またはヌクレオチド)を有する特定のセグメントを含む、等価な遺伝子およびタンパク質が構築され得る。このような技術は、例示されたかまたは示唆された配列(またはその相補体(完全な相補体))のいずれかにおける(同じサイズの)セグメントに対応する、例えば、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99、100、101、102、103、104、105、106、107、108、109、110、111、112、113、114、115、116、117、118、119、120、121、122、123、124、125、126、127、128、129、130、131、132、133、134、135、136、137、138、139、140、141、142、143、144、145、146、147、148、149、150、151、152、153、154、155、156、157、158、159、160、161、162、163、164、165、166、167、168、169、および170個の連続するアミノ酸残基を有する、等価な/機能的に活性なタンパク質を得るために調整することができる。特に関心対象の領域について、そのようなセグメントをコードするポリヌクレオチドも、本発明に含まれ、特に保存された領域のため、プローブおよび/またはプライマーとしても使用され得る。
【0058】
全長遺伝子のフラグメントは、標準的な手法に従って、市販のエキソヌクレアーゼまたはエンドヌクレアーゼを使用して作製することができる。例えば、Bal31などの酵素または部位特異的変異誘発が、これらの遺伝子の末端からヌクレオチドを体系的に切断するために使用することができる。また、活性フラグメントをコードする遺伝子は、種々の制限酵素を使用して入手されてもよい。これらの活性フラグメントを直接的に入手するためにプロテアーゼを使用してもよい。
【0059】
タンパク質が短縮され、なお機能的活性を保持し得ることは、本明細書に開示されるように、本発明の範囲内にある。「短縮型タンパク質」は、残りの短縮されたタンパク質が、切断後に所望の活性を保持し、かつその活性を示しながら、タンパク質の一部分が切断可能であることを意味する。切断は、種々のプロテアーゼによって達成することができる。さらに、効率的に切断されたタンパク質は、分子生物学的技術を使用して産生することができ、ここで、該タンパク質をコードするDNA塩基は、制限エンドヌクレアーゼまたは当業者に使用可能である他の技術のいずれかを通して除去される。短縮化後、該タンパク質は、大腸菌、バキュロウイルス、植物に基づくウイルス系、酵母などのような異種系において発現することができ、次いで、活性を測定するために本明細書に開示されるように昆虫アッセイ法に配置することができる。短縮型タンパク質は、これらが完全な全長配列よりも短いものを有しながら、機能的活性を保持するように首尾よく産生され得ることは、当技術分野において周知である。例えば、B.t.タンパク質は、短縮(コアタンパク質)型で使用することができる(例えば、Hofte et al.(1989)、およびAdang et al.(1985)を参照されたい)。本明細書で使用されるように、「タンパク質」という用語は、機能的に活性な短縮型を含むことができる。
【0060】
ある場合において、とりわけ、植物における発現のために、短縮型タンパク質を発現する短縮型遺伝子を使用することが有利であり得る。好ましい短縮型遺伝子は、典型的には、全長タンパク質の40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、または99%をコードする。
【0061】
本明細書の特定のタンパク質は、具体的に本明細書に例示されてきた。これらのタンパク質は本発明のタンパク質の単なる例であるので、本発明が、例示されたタンパク質の同じかまたは同様の活性を有する変種または等価なタンパク質(およびその等価物をコードするヌクレオチド配列)を含むことは容易に明らかとなるはずである。等価なタンパク質は、例示されるタンパク質とアミノ酸類似性(および/または相同性)を有する。アミノ酸同一性は、典型的には、少なくとも60%、好ましくは少なくとも75%、より好ましくは少なくとも80%、さらにより好ましくは少なくとも90%であり、および少なくとも95%であり得る。好ましい本発明のタンパク質はまた、より特定には同一性および/または類似性の範囲によって定義することができる。例えば、同一性および/または類似性は、本明細書で例示または示唆される配列と比較して、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、または99%であり得る。上記に列挙した任意の数字が、上限および下限を定義するために使用され得る。
【0062】
他に特定されない限り、本明細書で使用されるように、2つの核酸の配列同一性パーセントおよび/または配列類似性パーセントは、Karlin and Altschul 1993において改変された、Karlin and Altschul, 1990のアルゴリズムを使用して決定される。このようなアルゴリズムは、Altschul et al., 1990のNBLASTプログラムおよびXBLASTプログラムに組み込まれている。BLASTヌクレオチド検索は、NBLASTプログラム、スコア=100、ワード長=12を用いて実行される。ギャップ付きBLASTは、Altschul et al., 1997において記載されるように使用され得る。BLASTプログラムおよびギャップ付きBLASTプログラムを使用する場合、それぞれのプログラム(NBLASTおよびXBLAST)のデフォルトパラメーターが使用される。NCBI/NIHウェブサイトを参照されたい。比較目的のためにギャップ付きアラインメントを得るために、Vector NTI Suite 8(InforMax, Inc., North Bethesda, MD, U.S.A.)のAlignX機能が、デフォルトパラメーターを用いて使用された。これらは、15のギャップオープニングペナルティー、6.66のギャップ伸長ペナルティー、および8のギャップ分離ペナルティーであった。
【0063】
タンパク質の種々の特性および三次元的な特徴もまた、タンパク質の活性/機能性に有害な影響を与えることなく、変化させることができる。保存性アミノ酸置換は、分子の活性および/または三次元配置に有害な影響を与えないように、許容/作製することができる。アミノ酸は、以下のクラスに位置付けることができる:非極性、非荷電極性、塩基性、および酸性。1つのクラスのアミノ酸が同じ型の別のアミノ酸で置換される保存性置換は、その置換が化合物の生物学的活性に有害でない限り、本発明の範囲内にある。表2は、各クラスに属するアミノ酸の例の列挙を提供する。
【0064】
(表2)
【0065】
ある例において、非保存性置換もまた、作製することができる。しかし、好ましい置換は、タンパク質の機能的/生物学的活性を有意に損なわない。
【0066】
本明細書で使用されるように、「単離された」ポリヌクレオチドおよび/または「精製された」タンパク質との言及は、それらが天然に見出されるのではない状態である場合のこれらの分子をいう。従って、「単離された」および/または「精製された」との言及は、本明細書に記載されるように、「人の手」の関与を意味する。例えば、発現のために植物に入れた本発明における細菌「遺伝子」は、「単離されたポリヌクレオチド」である。同様に、細菌タンパク質に由来し、かつ植物によって産生されるタンパク質は、「単離されたタンパク質」である。
【0067】
遺伝コードの縮重/冗長性のために、種々の異なるDNA配列が、本明細書に開示されるアミノ酸配列をコードし得る。同じ、または本質的に同じタンパク質をコードする代替的なDNA配列を作製することは、十分に当業者の技量の範囲内にある。これらの変種DNA配列は本発明の範囲内にある。これはまた、「植物における発現のための配列の最適化」と題された以下の節においてより詳細に考察される。
【0068】
植物における発現のための配列の最適化
植物での異種遺伝子の高発現を得るために、遺伝子が植物細胞(の細胞質)においてより効率的に発現されるように、その遺伝子を再操作することが一般的に好ましい。トウモロコシは、このような植物の1つであり、ここでは、該植物において遺伝子の発現レベルを増加させるために、形質転換の前に異種遺伝子を再設計することが好ましい場合がある。それゆえに、細菌タンパク質をコードする遺伝子の設計における追加の工程は、双子葉植物種または単子葉植物種に関わらず、標的植物配列とより密接に整列されるコドンの偏りを使用する、最適な発現のための異種遺伝子の再操作である。配列はまた、本明細書の別の箇所で考察される、任意のより特定な型の植物における発現のために最適化することができる。
【0069】
トランスジェニック宿主
本発明のタンパク質をコードする遺伝子は、広範な種々の微生物または植物の宿主に導入することができる。本発明は、トランスジェニック植物細胞およびトランスジェニック植物を含む。好ましい植物(および植物細胞)は、トウモロコシ、アラビドプシス、タバコ、ダイズ、ワタ、アブラナ、イネ、コムギ、芝草、および牧草などである。他の型のトランスジェニック植物、例えば、果物、野菜、観賞用植物、および木もまた、本発明に従って作製することができる。より一般的には、双子葉植物および/または単子葉植物は、本発明の種々の局面において使用することができる。
【0070】
従って、本発明は、単子葉植物および双子葉植物を含む維管束植物および非維管束植物、針葉樹、蘚苔類、藻類、真菌、ならびに細菌と共に使用するために適応し得る。動物細胞および動物細胞培養物も可能である。
【0071】
好ましい態様において、遺伝子の発現は、直接的または間接的に、関心対象のタンパク質の細胞内産生(および維持)を生じる。植物は、この様式で、除草剤抵抗性にされ得る。このような宿主は、トランスジェニック、組換え、形質転換された、および/またはトランスフェクトされた、宿主および/または細胞と呼ばれ得る。本発明のある局面において(例えば、関心対象の遺伝子をクローニングおよび調製する場合)、微生物(好ましくは、細菌)細胞が、本開示の恩典を伴って、標準的な技術に従って、産生および使用され得る。
【0072】
本発明のポリヌクレオチドでトランスフェクトされた植物細胞は、再生して完全な植物体となることができる。本発明は、組織細胞培養、液体培養、およびプレート培養を含む、細胞培養を含む。本発明の植物を生成するために産生および/または使用される種子もまた、本発明の範囲内に含まれる。他の植物組織および部分もまた、本発明に含まれる。本発明は、同様に、本発明のポリヌクレオチドを含む植物または細胞を産生する方法を含む。このような植物を産生する1つの好ましい方法は、本発明の種子を植えることによる。
【0073】
トランスジェニック宿主を形成するための遺伝子の挿入
本発明の種子の1つの局面は、本発明のタンパク質を発現する本発明のポリヌクレオチドを用いる、植物、植物種子、および他の宿主細胞の形質転換/トランスフェクションである。この様式で形質転換された植物は、異なる作用の様式を有する種々の除草剤に対して抵抗性にされ得る。
【0074】
広範な種々の方法が、遺伝子の安定な維持および発現を可能にする条件下で、標的宿主に、所望のタンパク質をコードする遺伝子を導入するために使用可能である。これらの方法は当業者に周知であり、例えば、米国特許第5,135,867号において記載されている。
【0075】
DSM-2ポリヌクレオチドを含むベクターは本発明の範囲に含まれる。例えば、大腸菌における複製系および形質転換細胞の選択を可能にするマーカーを含む多数のクローニングベクターが、高等植物への外来性遺伝子の挿入の調製のために使用可能である。これらのベクターには、例えば、pBR322、pUCシリーズ、M13mpシリーズ、pACYC184などが含まれる。従って、タンパク質をコードする配列は、適切な制限部位でベクターに挿入することができる。得られるプラスミドは、大腸菌への形質転換のために使用される。大腸菌細胞は、適切な栄養培地中で培養され、次いで収集および溶解される。プラスミドは、ゲノムDNAからの精製によって回収される。配列分析、制限分析、電気泳動、および他の生化学的-分子生物学的方法が、分析の方法として一般的に実行される。各操作の後、使用されるDNA配列は、制限消化し、かつ次のDNA配列に連結することができる。各プラスミド配列は、同じまたは他のプラスミドにクローニングすることができる。植物に所望の遺伝子を挿入する方法に依存して、他のDNA配列が必要である可能性がある。例えば、TiまたはRiプラスミドが植物細胞の形質転換のために使用される場合には、TiまたはRiプラスミドT-DNAの少なくともライトボーダー、しかし往々にして、ライトボーダーおよびレフトボーダーが、挿入される遺伝子の隣接領域として連結されなければならない。植物細胞の形質転換のためのT-DNAの使用は、EP 120 516; Hoekema(1985); Fraley et al.(1986);およびAn et al.(1985)において徹底的に調査および記載されてきた。
【0076】
多数の技術が、植物宿主細胞にDNAを挿入するために使用可能である。これらの技術には、形質転換因子としてのアグロバクテリウム・ツメファシエンス(Agrobacterium tumefaciens)またはアグロバクテリウム・リゾゲネス(Agrobacterium rhizogenes)使用するT-DNAを用いる形質転換、融合、注入、微粒子銃(微粒子射撃)、シリコンカーバイトウィスカー、エアロゾルビーム、PEG、またはエレクトロポレーション、ならびに他の可能な方法が含まれる。アグロバクテリアが形質転換のために使用される場合、挿入されるDNAは、特別なプラスミド、すなわち、中間体ベクターまたはバイナリーベクターのいずれかにクローニングされなければならない。この中間体ベクターは、T-DNA中の配列に対して相同である配列による相同組換えによって、TiまたはRiプラスミドに組み込まれ得る。TiまたはRiプラスミドはまた、T-DNAの移入のために必要であるvir領域を含む。中間体ベクターは、アグロバクテリア中でそれ自体複製することはできない。中間体ベクターは、ヘルパープラスミド(接合)によって、アグロバクテリウム・ツメファシエンスに移入され得る。バイナリーベクターは、大腸菌とアグロバクテリアの両方の中でそれ自体複製することができる。これらは、選択マーカー遺伝子、ならびに右側および左側の、T-DNAボーダー領域によってフレーム形成される、リンカーまたはポリリンカーを含む。これらは、アグロバクテリアに直接的に形質転換され得る(Holsters, 1978)。宿主細胞として使用されるアグロバクテリウムは、vir領域を有するプラスミドを含む。このvir領域は、植物細胞へのT-DNAの移入のために必要である。さらなるT-DNAが含まれてもよい。このように形質転換された細菌は、植物細胞の形質転換のために使用される。植物移植片は、植物細胞へのDNAの移入のために、アグロバクテリウム・ツメファシエンスまたはアグロバクテリウム・リゾゲネスとともに有利に培養することができる。次いで、選択のために抗生物質または殺生物剤を含んでもよい適切な培地中で、感染された植物材料(例えば、葉の小片、茎のセグメント、根であるが、しかしプロトプラストまたは懸濁培養細胞もまた)から完全な植物体が再生され得る。次いで、このように得られた植物は、挿入されたDNAの存在について試験することができる。特別な要求は、注入およびエレクトロポレーションの場合にはなされない。通常のプラスミド、例えば、pUC誘導体などを使用することが可能である。
【0077】
形質転換細胞は、通常の様式で植物の内部で成長する。これらは、胚細胞を形成し、子孫の植物に形質転換形質を伝達することができる。このような植物は、通常の様式で生育させることができ、同じ形質転換された遺伝性因子または他の遺伝性因子を有する植物と交雑させることができる。得られる雑種個体は、対応する表現型特性を有する。
【0078】
本発明のある好ましい態様において、細菌タンパク質をコードする遺伝子は、植物ゲノムに挿入された転写単位から発現される。好ましくは、該転写単位は、植物ゲノムへの安定な組み込みが可能である組換えベクターであり、タンパク質をコードするmRNAを発現する形質転換された植物系統の選択を可能にする。
【0079】
挿入されたDNAが一旦ゲノムに組み込まれたら、それはそこで比較的安定である(かつそこから再び出ることはない)。これは、殺生物剤または抗生物質、とりわけ、カナマイシン、G418、ブレオマイシン、ハイグロマイシン、またはクロラムフェニコールなどに対する抵抗性を、形質転換植物細胞に付与する選択マーカーを通常含む。植物選択マーカーはまた、典型的には、グルホシネート、(PAT)、グリホセート(EPSPS)、イマゼチアピル(AHAS)などのような種々の除草剤に対する抵抗性を提供することができる。個々に使用されるマーカーは、従って、挿入されたDNAを含まない細胞以外の形質転換細胞の選択を可能にするはずである。関心対象の遺伝子は、植物細胞において、構成的または誘導性のいずれかのプロモーターによって好ましく発現される。一旦発現されると、mRNAはタンパク質に翻訳され、それによって、関心対象のアミノ酸がタンパク質に組み込まれる。植物細胞において発現されるタンパク質をコードする遺伝子は、構成的プロモーター、組織特異的プロモーター、または誘導性プロモーターの制御下にあり得る。
【0080】
植物細胞に外来性の組換えベクターを導入するため、および導入された遺伝子を安定に維持および発現する植物を入手するためのいくつかの技術が存在している。このような技術には、微粒子上にコートされた遺伝物質の細胞への直接的な導入が含まれる(Cornellに対する米国特許第4,945,050号、およびDowElanco、現在はDow AgroSciences, LLCに対する米国特許第5,141,131号)。加えて、植物は、アグロバクテリウム技術(以下を参照されたい:University of Toledoに対する米国特許第5,177,010;Texas A&Mに対する米国特許第5,104,310号;欧州特許出願第0131624B1号;Schilperootに対する欧州特許出願第120516号、同第159418Bl号および同第176,112号;Schilperootに対する米国特許第5,149,645、同第5,469,976号、同第5,464,763号および同第4,940,838号および同第4,693,976号;すべてMax Planckに対する欧州特許出願第116718号、同第290799号、同第320500号;日本たばこ産業(Japan Tobacco)に対する欧州特許出願第604662号および同第627752号、ならびに米国特許第5,591,616号;Ciba Geigy、現在Syngentaに対する欧州特許出願第0267159号および同第0292435号、ならびに米国特許第5,231,019号;両方ともCalgeneに対する米国特許第5,463,174号および同第4,762,785号;ならびに両方ともAgracetusに対する米国特許第5,004,863号および同第5,159,135号)を使用して形質転換されてもよい。他の形質転換技術には、ウィスカー技術が含まれる。両方ともZeneca、現在Syngentaに対する米国特許第5,302,523号および同第5,464,765号を参照されたい。他の直接DNA輸送形質転換技術には、エアロゾルビーム技術が含まれる。米国特許第6,809,232を参照されたい。エレクトロポレーション技術もまた、植物を形質転換するために使用されてきた。Boyce Thompson Instituteに対するWO 87/06614;両方ともDekalbに対する米国特許第5,472,869号および同第5,384,253号;ならびに両方ともPlant Genetic Systemsに対するWO 92/09696およびWO93/21335を参照されたい。さらに、ウイルスベクターもまた、関心対象のタンパク質を発現するトランスジェニック植物を産生するために使用することができる。例えば、単子葉植物は、Mycogen Plant ScienceおよびCiba-Geigy(現在はSyngenta)に対する米国特許第5,569,597号ならびに両方ともBiosource、現在Large Scale Biologyに対する米国特許第5,589,367号および同第5,316,931号に記載されている方法を使用して、ウイルスベクターで形質転換することができる。
【0081】
以前に言及したように、DNA構築物が植物宿主に導入される様式は、本発明にとって決定的ではない。効率的な形質転換を提供する任意の方法が使用されてもよい。例えば、植物細胞形質転換のための種々の方法が本明細書に記載され、これには、TiまたはRi-プラスミド、およびアグロバクテリウム媒介形質転換を実行するための同様のものの使用が含まれる。多くの場合において、T-DNAボーダー、より特定にはライトボーダーによって、一方または両方の側で境界を接する、形質転換のために使用される構築物を有することが望ましい。これは、構築物が、形質転換のための様式としてアグロバクテリウム・ツメファシエンスまたはアグロバクテリウム・リゾゲネスを使用する場合に特に有用であるが、T-DNAボーダーは、形質転換の他の様式を用いる使用を見出し得る。アグロバクテリウムが植物細胞形質転換のために使用される場合、宿主中に存在するT-DNAまたはTiもしくはRiプラスミドとの相同組換えのために宿主に導入され得るベクターが使用され得る。ベクターの導入は、エレクトロポレーション、トリペアレンタルメイティング、および当業者に公知であるグラム陰性細菌を形質転換するための他の技術を介して実行されてもよい。アグロバクテリウム宿主へのベクター形質転換の様式は、本発明にとって決定的ではない。組換えのためのT-DNAを含むTiまたはRiプラスミドは、こぶ形成を引き起こすことが可能であるか、または可能でなくてもよく、vir遺伝子が該宿主中に存在している限り、本発明には決定的ではない。
【0082】
アグロバクテリウムが形質転換のために使用されるいくつかの場合において、T-DNAボーダー中にある発現構築物は、Ditta et al.(1980)およびEPO 0 120 515に記載されるようなpRK2またはその誘導体などの広い範囲のベクターに挿入される。形質転換されたアグロバクテリウムおよび形質転換された植物細胞の選択を可能にする、本明細書に記載されるような1種または複数のマーカーが、発現構築物およびT-DNAに含まれる。使用される特定のマーカーは、本発明にとって本質的ではなく、好ましいマーカーは、使用される宿主および構築物に依存する。
【0083】
アグロバクテリウムを使用する植物細胞の形質転換のために、移植片は、その形質転換を可能にするために十分な時間の間、形質転換されたアグロバクテリウムと合わされ、かつインキュベートされてもよい。形質転換後、アグロバクテリアは適切な抗生物質を用いる選択によって殺傷され、植物細胞は適切な選択培地を用いて培養される。一旦、カルスが形成されると、シュート形成は、植物組織培養および植物再生の分野で周知である方法に従って、適切な植物ホルモンを使用することによって促進され得る。しかし、カルスの中間段階が常に必要であるわけではない。シュート形成後、該植物細胞は、シュート形成を促進する培地に移すことができ、それによって、植物再生を完了する。次いで、植物は生育して種子になり、該種子は、将来の世代を樹立するために使用され得る。形質転換技術に関わらず、細菌タンパク質をコードする遺伝子は、ベクター中に植物プロモーター調節エレメント、ならびにNosなどのような3'-非翻訳転写終結領域を含めることによって、植物細胞において該遺伝子を発現するように適合された遺伝子移入ベクターに好ましく組み込まれる。
【0084】
植物を形質転換するための多数の技術に加えて、外来性遺伝子を接触される組織の型は、同様に変化してもよい。このような組織には、胚形成組織、I、II、およびIII型カルス組織、胚軸、成長点、根組織、師部における発現のための組織などが含まれるがこれらに限定されない。ほぼすべての植物組織が、本明細書に記載される適切な技術を使用して、脱分化の間に形質転換されてもよい。
【0085】
選択マーカーに加えて、レポーター遺伝子を加えることが望ましくあり得る。いくつかの例において、レポーター遺伝子は、選択マーカーとともに、または選択マーカーなしで使用されてもよい。レポーター遺伝子は、典型的には、レシピエントの生物または組織には存在せず、かつ典型的には、ある表現型の変化または酵素的特性を生じるタンパク質をコードする遺伝子である。このような遺伝子の例は、Weising et al., 1988において提供される。好ましいレポーター遺伝子には、大腸菌のuidA遺伝子座のβ-グルクロニダーゼ(GUS)、大腸菌のTn9からのクロラムフェニコールアセチルトランスフェラーゼ遺伝子、生物発光クラゲ、エクオレア・ビクトリア(Aequorea victoria)からの緑色蛍光タンパク質、およびホタル、フォチヌス・ピラリス(Photinus pyralis)からのルシフェラーゼ遺伝子が含まれる。次いで、レポーター遺伝子発現を検出するためのアッセイ法は、該遺伝子がレシピエント細胞に導入された後の適切な時点で実行され得る。好ましいこのようなアッセイ法は、形質転換細胞を同定するために、Jefferson et al.,(1987)によって記載されるような、大腸菌のuidA遺伝子座のβ-グルクロニダーゼ(GUS)をコードする遺伝子の使用を必要とする。
【0086】
植物プロモーター調節エレメントに加えて、種々の供給源からのプロモーター調節エレメントが、外来性遺伝子を発現するために、植物細胞において効率的に使用され得る。例えば、細菌起源のプロモーター調節エレメント、例えば、オクトピンシンターゼプロモーター、ノパリンシンターゼプロモーター、マンノピンシンターゼプロモーター;ウイルス起源のプロモーター、例えば、カリフラワーモザイクウイルス(35Sおよび19S)、35T(これは、再操作された35Sプロモーターである、米国特許第6,166,302号、とりわけ実施例7Eを参照されたい)などが使用されてもよい。植物プロモーター調節エレメントには、リブロース-1,6-ビスリン酸(RUBP)カルボキシラーゼスモールサブユニット(ssu)、β-コングリシニンプロモーター、β-ファセオニリンプロモーター、ADHプロモーター、熱ショックプロモーター、および組織特異的プロモーターが含まれるがこれらに限定されない。他のエレメント、例えば、マトリックス結合領域、足場付着領域、イントロン、エンハンサー、ポリアデニル化配列などが存在してもよく、従って、転写効率またはDNA組み込みを改善してもよい。このようなエレメントは、DNA機能のために必要であるかもしれないし、必要でないかもしれないが、これらは、転写、mRNA安定性などに影響を与えることによって、より良好な発現またはDNAの機能を提供することができる。このようなエレメントは、植物での形質転換されたDNAの最適な性能を得るために、必要に応じてDNAに含まれてもよい。典型的なエレメントには、Adh-イントロン1、Adh-イントロン6、アルファルファモザイクウイルスコートタンパク質リーダー配列、オスモチンUTR配列、トウモロコシストリークウイルスコートタンパク質リーダー配列、ならびに当業者に使用可能であるその他が含まれるがこれらに限定されない。構成的プロモーター調節エレメントもまた使用され、すべての細胞型およびすべての時点での連続的な遺伝子発現を方向付けることができる(例えば、アクチン、ユビキチン、CaMV 35Sなど)。組織特異的プロモーター調節エレメントは、特定の組織または組織型、例えば、葉または種子における遺伝子発現の原因であり(例えば、ゼイン、オレオシン、ナピン、ACP、グロブリンなど)、これらもまた使用されてもよい。
【0087】
プロモーター調節エレメントはまた、植物の発生の特定の段階の間に活性であり(または不活性であり)、かつ植物の組織および器官において活性であってもよい。このような例には、花粉特異的、胚特異的、トウモロコシ毛特異的、ワタ繊維特異的、根特異的、種子胚乳特異的、または栄養相特異的なプロモーターエレメントなどが含まれるがこれらに限定されない。特定の状況下で、特定のシグナルに応答する遺伝子、例えば:物理的刺激(熱ショック遺伝子)、光(RUBPカルボキシラーゼ)、ホルモン(Em)、代謝物、化学物質(テトラサイクリン応答性)、およびストレスに対して応答する遺伝子の発現の原因である誘導性プロモーター調節エレメントを使用することが所望であってもよい。植物において機能する他の所望の転写および翻訳エレメントが使用されてもよい。多数の植物特異的遺伝子移入ベクターが当技術分野で公知である。
【0088】
植物RNAウイルスを用いる系もまた、細菌タンパク質を発現するために使用され得る。そうすることにおいて、タンパク質を発現する遺伝子は、関心対象の宿主植物に感染する適切な植物ウイルスのコートプロモーター領域に挿入され得る。次いで、このタンパク質が発現され得、従って、除草剤による損傷からの植物の保護を提供する。植物RNAウイルスを用いる系は、Mycogen Plant Sciences, Inc. に対する米国特許第5,500,360号ならびにBiosource、現在はLarge Scale Biologyに対する米国特許第5,316,931号および同第5,589,367号に記載されている。
【0089】
選択剤
グルホシネートおよびビアラホスに加え、本発明に従って使用され得る選択剤には、本発明のDSM-2遺伝子によって媒介されるアセチルトランスフェラーゼ機序によって不活性化され得るすべての合成および天然のアナログが含まれる。例えば、図1を参照されたい。
【0090】
具体的に示されない限り、または暗示されない限り、1つの(「a」「an」)およびその(「the」)という用語は、本明細書で使用される場合、「少なくとも1つの」を意味する。
【0091】
本明細書で言及されるか、または引用されるすべての特許、特許出願、特許仮出願、および刊行物は、それらが本明細書の明示的な教示と矛盾しない範囲で、それらの全体が参照により本明細書に組み入れられる。
【0092】
以下は、本発明を実施するための手法を例証する実施例である。これらは、限定として解釈されるべきではない。他に注記されない限り、すべてのパーセンテージは重量によっており、すべての溶媒混合物の割合は体積によっている。
【0093】
実施例1-植物においてグルホシネートに対する抵抗性を付与する遺伝子を同定するための方法
植物または細胞培養物において除草剤分解活性を有する遺伝子を同定するための方法として、NCBI(National Center for Biotechnology Information)などの現在の公的なデータベースを調査することが可能である。このプロセスを開始するために、所望の特性(すなわち、ホスフィノトリシンアセチルトランスフェラーゼ)を有するタンパク質をコードする、すでに同定された機能的遺伝子配列を持っていることが必要である。次いで、このタンパク質配列は、使用可能である寄託されたNCBIタンパク質配列に対して比較するために、BLAST(基礎局所的アラインメント検索ツール(Basic Local Alignment Search Tool))(Altschul et al., 1997)アルゴリズムのためのインプットとして使用する。デフォルト設定を使用すると、この検索は、様々なレベルで100個までの相同タンパク質配列を回答する。これらは、アミノ酸レベルが、高い同一性(85〜98%)から非常に低い同一性(23〜32%)までの範囲である。伝統的には、高い相同性を有する配列のみが、インプット配列と同様の特性を保持していることが予測される。この場合、≦50%相同性を有する配列のみが選択される。本明細書において例示されるように、わずか30%のアミノ酸保存性(ストレプトマイセス・ハイグロスコピカス由来のpatと比較して)を有するホモログのクローニングおよび組換えによる発現は、形質転換していない植物細胞培養物から形質転換した植物細胞培養物を選択するために使用できる。
【0094】
DSM-2を、patに対して30%のみ、barに対し28%のみのアミノ酸同一性を有するホモログとしてNCBIデータベース(the ncbi.nlm.nih.gov ウェブサイト;アクセッション番号AAA26705)から同定した。同一性パーセントは、データベース中に寄託されたヌクレオチド配列を最初にタンパク質に翻訳すること、次いで複数配列のアラインメントを実行するためのVectorNTIソフトウェアパッケージ中のClustalWを使用することによって決定した。
【0095】
実施例2-植物および細菌における発現のための配列の最適化
2.1-バックグラウンド
植物における異種遺伝子のより高いレベルの発現を得るために、これらが植物細胞でより効率的に発現されるように、タンパク質をコードする遺伝子を再操作することが好ましい場合がある。トウモロコシは、このような植物の1つであり、該植物でのその発現レベルおよびコード化タンパク質のレベルを増加させるために、形質転換の前に、異種のタンパク質コード領域を再設計することが好ましい場合がある。それゆえに、細菌タンパク質をコードする遺伝子の設計におけるさらなる工程は、最適な発現のために異種遺伝子の再操作である。例えば、Kawabe et al.(2003), 「Patterns of Codon Usage Bias in Three Dicot and Four Monocot Plant Species」, Genes Genet. Syst., pp.343-352;およびIkemura et al.(1993), 「Plant Molecular Biology Labfax」, Croy, ed., Bios Scientific Publishers Ltd., p. 3748)、ならびにその中に引用されたすべての関連のある参照を参照されたい。
【0096】
トウモロコシにおける発現のための細菌タンパク質の再操作の1つの理由は、例えば、ネイティブ遺伝子の最適でないG+C含量に起因する。例えば、多くのネイティブな細菌遺伝子の非常に低いG+C含量(および高いA+T含量に向かう結果としての傾斜)は、きわめてA+Tリッチであることが知られている植物遺伝子制御配列を模倣または複製する配列の生成をもたらす。植物に導入される遺伝子のDNA中のあるA+Tリッチ配列(例えば、遺伝子プロモーター中に通常見出されるTATAボックス領域)の存在は、遺伝子の異常な転写を生じる可能性がある。他方、転写されたmRNAに存在する他の調節配列(例えば、ポリアデニル化シグナル配列(AAUAAA)またはプレmRNAスプライシングに関与する核内低分子RNAに相補的である配列)の存在は、RNAの不安定化をもたらす可能性がある。したがって、より好ましくは植物に最適化された遺伝子と呼ばれる、トウモロコシにおける発現のために細菌タンパク質をコードする遺伝子の設計における1つの目的は、より高いG+C含量を有するDNA配列、および好ましくは、代謝酵素をコードするトウモロコシ遺伝子の配列に近いものを生成することである。細菌タンパク質をコードする植物に最適化された遺伝子の設計の別の目的は、配列の改変が翻訳を妨害しないDNA配列を生成することである。
【0097】
表3は、どのように高G+C含量がトウモロコシにおいて存在するかを例証する。表3におけるデータより、遺伝子のコード領域は、GenBank(リリース71)から抽出し、かつ塩基組成は、MacVector(商標)プログラムを使用して計算した(Accelerys, San Diego, California)。イントロン配列は計算において無視した。
【0098】
(表3)トウモロコシ遺伝子のタンパク質コード領域のG+C含量の集計
a 遺伝子の数を()内に示す。
b 標準偏差を()内に示す。
c 合わせた群の平均は平均の計算において無視した。
【0099】
遺伝コードの冗長性/縮重によって与えられる柔軟性に起因して(すなわち、あるアミノ酸は1つより多くのコドンによって特定される)、異なる生物または生物のクラスにおけるゲノムの進化は、重複するコドンの示差的な使用を生じてきた。この「コドンの偏り」は、タンパク質コード領域の平均塩基組成を反映している。例えば、比較的低いG+C含量を有する生物は、冗長性コドンの第3の位置においてAまたはTを有するコドンを使用するのに対して、より高いG+C含量を有するものは第3の位置においてGまたはCを有するコドンを使用する。mRNA中の「マイナーな」コドンの存在は、とりわけ、そのマイナーなコドンに対応する荷電したtRNAの相対的な豊富さが低い場合に、そのmRNAの絶対的な翻訳速度を減少する可能性があると考えられている。このことの拡張は、個々のマイナーなコドンによる翻訳速度の減少が、複数のマイナーなコドンについて少なくとも相加的であることである。それゆえに、マイナーなコドンの高い相対含量を有するmRNAは、対応して低い翻訳速度を有する。この速度は、引き続く低レベルのコードされるタンパク質によって反映される。
【0100】
トウモロコシ(トウモロコシ、または他の植物、例えば、ワタ、ダイズ、コムギ、アブラナ属/アブラナ、イネ;またはより一般的には油料穀物、一般に、単子葉植物、双子葉植物、および半子葉(hemicot)植物)での発現のために細菌タンパク質をコードする遺伝子を操作する際に、植物のコドンの偏りが決定された。トウモロコシについてのこのコドンの偏りは、そのタンパク質をコードするために植物が使用する、統計学的なコドンの分布であり、好ましいコドン使用頻度は表4に示す。偏りを決定した後に、関心対象の遺伝子におけるコドンの頻度パーセントが決定される。植物によって好まれる第1のコドンが、ならびに複数の選択が存在する場合には好ましいコドンの第2、第3、および第4の選択が、決定されるべきである。次いで、細菌タンパク質のアミノ酸配列をコードする新規なDNA配列が設計され得るが、この新規な配列は、タンパク質のアミノ酸配列中の各位置におけるアミノ酸を特定するために、ネイティブなDNA配列(タンパク質をコードしている)とは、植物の(第1に好ましい、第2に好ましい、第3に好ましい、または第4に好ましい)コドンの置換によって異なっている。次いで、この新規な配列は、改変によって作製されたかもしれない制限酵素部位について分析する。同定された部位は、コドンを、第1の、第2の、第3の、または第4の選択の好ましいコドンで置き換えることによってさらに改変される。関心対象の遺伝子の転写または翻訳に影響を与え得る配列中の他の部位は、エキソン:イントロン連結(5'または3')、ポリA付加シグナル、またはRNAポリメラーゼ終結シグナルである。配列はさらに、TAまたはGCダブレットの頻度を減少するように分析および改変される。ダブレットに加えて、同じである約4個より多くの残基を有するGまたはC配列ブロックは、配列の転写に影響を与え得る。それゆえに、これらのブロックもまた、第1または第2の選択などのコドンを、次に好ましい選択のコドンで置き換えることによって改変される。
【0101】
(表4)トウモロコシにおいて発現されるタンパク質についての好ましいアミノ酸コドン
【0102】
細菌タンパク質をコードする植物に最適化された遺伝子は、約63%の第1の選択のコドン、約22%〜約37%の間の第2の選択のコドン、および約15%〜約0%の間の第3または第4の選択のコドンを含み、全体のパーセンテージが100%であることが好ましい。最も好ましい植物に最適化された遺伝子は、約63%の第1の選択のコドン、少なくとも約22%の第2の選択のコドン、約7.5%の第3の選択のコドン、および約7.5%の第4の選択のコドンを含み、全体のパーセンテージは100%である。上記の方法は、遺伝子が植物で最適に発現されるように、特定の植物に対して外来性である遺伝子を改変することを当業者に可能にする。この方法はさらに、PCT出願WO 97/13402において例証されている。
【0103】
従って、細菌タンパク質をコードする植物に最適化された遺伝子を設計するために、DNA配列は、特定の植物のために遺伝子配列から確立されたコドンの偏りの表から確立された冗長な遺伝コードを使用して、該タンパク質のアミノ酸配列をコードするように設計される。得られるDNA配列は、より高い程度のコドンの多様性、望ましい塩基組成を有し、戦略的に配置された制限酵素認識部位を含むことができ、遺伝子の転写、または生成物mRNAの翻訳を妨害するかもしれない配列を欠いている。従って、本発明のタンパク質/遺伝子に対して機能的に等価である合成遺伝子は、植物を含む宿主を形質転換するために使用することができる。合成遺伝子の産生に関するさらなる手引きは、例えば、米国特許第5,380,831号において見出すことができる。
【0104】
2.2-DSM-2植物再構築分析
ネイティブDSM-2コード領域の513塩基対(bp)のDNA配列(SEQ ID NO:1)の広範な分析は、最適な植物発現に対して有害であると考えられているいくつかの配列モチーフの存在、ならびに最適でないコドン組成を明らかにした。SEQ ID NO:1によってコードされるタンパク質はSEQ ID NO:2として提示される。単子葉植物ならびに双子葉植物における組換えタンパク質の産生を改善するために、SEQ ID NO:2として開示されるネイティブ配列と同一である1つのタンパク質(SEQ ID NO:4)をコードする「植物に最適化された」DNA配列DSM-2 v2(SEQ ID NO:3)を開発した。対照的に、ネイティブのコード領域のDNA配列および植物に最適化されたコード領域(v1)のDNA配列は78.3%だけ同一である。表5は、ネイティブ配列(カラムAおよびD)および植物に最適化された配列(カラムBおよびE)のコドン組成の違いを示し、ならびに理論的な植物に最適化された配列(カラムCおよびF)に対する比較を可能にする。
【0105】
(表5)ネイティブDSM-2、植物に最適化されたバージョン(v2)、および理論的な植物最適化バージョンのコード領域のコドン組成の比較
【0106】
表5の検討から、ネイティブのコード領域および植物に最適化されたコード領域は、同一のタンパク質をコードする一方で、実質的に互いに異なることが明白である。植物に最適化されたバージョン(v1)は、DSM-2タンパク質をコードする理論的な植物に最適化されたコード領域のコドン組成を密接に模倣する。
【0107】
実施例3-形質転換ベクターのクローニング
3.1-DSM-2(v2)を含有しているバイナリープラスミドの構築
DSM-2(v2)コドン最適化遺伝子をコードする配列(DASPICO45)を、制限酵素BbsI(New England Biolabs, Inc., Beverly MA, cat #R0539s)およびSacI(New England Biolabs, Inc., cat #R0156s)で切断した。得られたフラグメントを、対応する制限部位NcoI(New England Biolabs, cat #R0193s)およびSacIでpDAB773にライゲーションした。陽性コロニーを制限酵素消化を介して同定した。得られたクローンは、Rb7 MAR v3 // At Ubi10プロモーターv2 // 関心対象の遺伝子 // Atu Orf 1 3'UTR v3を含有していた。関心対象の遺伝子としてDSM-2(v2)を含有していたプラスミドを、pDAB3774と名付けた。
【0108】
Rb7 MAR v3 // AtUbi10プロモーターv2 // 関心対象の遺伝子 // Atu Orf 1 3'UTR v3カセットを、AgeI(New England Biolabs, Inc., cat #R0552s)制限フラグメントとしてバイナリーベクターpDAB3736へクローニングした。バイナリープラスミドの左側および右側の境界部の間にこのカセットをクローニングした。陽性コロニーを制限酵素消化および配列決定反応を介して同定した。Rb7 MAR v3 // AtUbi10プロモーターv2 // DSM-2 v2 // Atu Orf 1 3'UTR v3を含有している構築物を、pDAB3778と名付けた。
【0109】
Rb7 MAR v3 // AtUbi10プロモーターv2 // PATv3 // Atu Orf 1 3'UTR v3カセットを含有している対照構築物を、pDAB3736からGateWay attRデスティネーションカセットを除去することにより完成させた。pDAB3736を、PacI(New England Biolabs, Inc., cat #R0547s)制限酵素により消化した。PacIは、pDAB3736においてGateWay attRデスティネーションカセットに隣接している。PacIで消化されたプラスミドを、自己ライゲーションし、大腸菌Top10細胞(Invitrogen, Carlsbad CA, cat# C4040-10)に形質転換した。陽性コロニーを、制限酵素消化および配列決定反応を介して同定した。得られた構築物を、pDAB3779と名付けた。
【0110】
3.2-付加的な形質転換構築物のクローニング
適切な植物種への形質転換のために作出されたすべてのその他の構築物が、本明細書中に既に記載されたような類似した手法、およびその他の標準的な分子クローニング法(Maniatis et al., 1982)を使用して構築された。表6は、適切なプロモーターおよび定義された特色、ならびに形質転換された作物と共に、使用されたすべての形質転換構築物を列挙する。
【0111】
(表6)様々な植物種の形質転換において使用された構築物
*A=アラビドプシス
T=タバコ
R=イネ
Cn=トウモロコシ
CsVMV=キャッサバ葉脈モザイクウイルスプロモーター
AtUbi10=シロイヌナズナユビキチン10プロモーター
RB7 Mar v2=ニコチアナ・タバカム・マトリックス関連領域(MAR)
NtOsm=ニコチアナ・タバカム・オスモチン5'および3'非翻訳領域
ZmUbi1=ジー・メイズ(Zea mays)ユビキチン1プロモーター
【0112】
実施例4-原核生物および真核生物のプロモーターを使用したDSM-2(v2)による感受性大腸菌(BL-21)の補完
4.1-DSM-2(v2)を含有している大腸菌発現プラスミドの構築
DSM-2(v2)コドン最適化配列を、制限酵素BbsIおよびSacIで消化した。得られたフラグメントを、NcoIおよびSacIの対応する制限部位でpDAB779へクローニングした。pDAB779はpET28a(+)発現ベクターである(Novagen, Madison WI, cat# 69864-3)。DSM-2(v2)遺伝子コーディング配列を含有している陽性コロニーを、制限酵素消化を介して同定した。DSM-2 // pET28a(+)構築物をpDAB4412と名付けた。
【0113】
発現プラスミドpET(空ベクター対照)、およびpDAB4412を、標準的な方法を使用して、大腸菌T7発現株BL21-Star(DE3)(Invitrogen, Carlsbad CA, cat# C6010-03)に形質転換した。50μg/ml抗生物質および75μM IPTG(イソプロピル-α-D-チオガラクトピラノシド)を含有している250mLのLB培地への10〜200個の新しく形質転換されたコロニーにより、発現培養物を開始させた。培養物を180〜200rpmで24時間28℃で増殖させた。4℃で10分間、3,400×gで、250mlのNalgeneボトルにおける遠心分離によって細胞を収集した。ペレットを4〜4.5mLのバターフィールドリン酸(Butterfield's Phosphate)溶液(Hardy Diagnostics, Santa Maria, CA; 0.3mMリン酸カリウムpH7.2)に懸濁させた。懸濁した細胞を、直径0.1mmのガラスビーズ(Biospec, Bartlesville, OK, catalog number 1107901)1mLを含む50mLポリプロピレンスクリューキャップ遠心管に移した。細胞-ガラスビーズ混合物を氷上で冷却し、次いで、およそ20の出力でBranson Sonifier 250(Danbury CT)を用いて、2mmプローブを使用して、2回の45秒の突発波(突発波間には完全に冷却)を用いた超音波処理によって細胞を溶解させた。溶解物を2mLエッペンドルフチューブに移し、16,000×gで5分間、遠心分離した。上清を収集し、タンパク質濃度を測定した。Bio-Rad Protein Dye Assay ReagentをH2Oで1:5希釈し、1mLを、1:10希釈の各試料10μL、ならびに5、10、15、20、および25μg/mLの濃度のウシ血清アルブミン(BSA)に添加した。Shimadzu UV 160U分光光度計(Kyoto, JP)で595nmの波長で光学濃度を測定する分光光度計で試料を読み取った。各試料に含有されていたタンパク質の量を、BSA標準曲線に対して計算し、リン酸緩衝液で3〜6mg/mLの間に調整した。発現されたタンパク質を可視化するため、溶解物をSDSタンパク質ゲル上で泳動した。
【0114】
4.2-BASTAに対する感受性についての一般的なクローニング株の評価
大腸菌およびアグロバクテリウム・ツメファシエンスの選択された細胞系を、漸増的に増加する濃度のグルホシネート(BASTA)を含有している最少培地上に播種した。細胞系;BL21-Star(DE3)、Top10、DH5α、アグロバクテリウム・ツメファシエンスC58、およびアグロバクテリウム・ツメファシエンスLBA4404sを、まず、複合培地上で増殖させた。大腸菌株はLBで増殖させ、アグロバクテリウム・ツメファシエンス株はYEPで増殖させた。5マイクロリットルの細菌培養物を、様々な濃度のグルホシネートを含有している最少培地プレートへ均一に播種し、分散させた。濃度は、0μg/ml、250μg/ml、500μg/ml、1000μg/ml、2000μg/ml、および4000μg/mlのBASTAからなっていた。さらに、対照として細菌株を複合培地のプレートへ播種した。アグロバクテリウム・ツメファシエンス株はYEP寒天プレート上に播種し、大腸菌株はLB寒天プレート上に播種した。大腸菌株を含有しているプレートは、37℃で24時間インキュベートした。アグロバクテリウム・ツメファシエンス株を含有しているプレートは、25℃で48時間インキュベートした。割り当てられたインキュベーション時間の後、プレートを細菌増殖について観察した。表7は、様々な株の、グルホシネートを含有している最少培地で増殖する能力を例示している。1つの株BL21-Star(DE3)細胞系のみが、グルホシネートによって実質的に阻害された。
【0115】
(表7)最少培地上で増殖させた様々な細菌株のグルホシネートに対する応答
大腸菌は37℃で24時間増殖させた。アグロバクテリウムは25℃で48時間増殖させた。+++ = 激しい菌叢増殖 // ++ = 軽度の菌叢増殖 // + = 斑状の菌叢増殖 // -- = 増殖なし
【0116】
4.3-大腸菌BL21-Star(DE3)の細胞増殖を補完するためのDSM-2(v2)の組み換え発現
PAT(v3)を含有しているpET28a(+)発現プラスミドを、陽性対照として構築した。PAT(v3)を、NcoI-SacIフラグメントとして、pDAB779の対応する制限部位へクローニングした。PAT(v3)遺伝子フラグメントを含有している陽性クローンについて、制限酵素消化を介して確証した。この構築物をpDAB4434と名付けた。
【0117】
プラスミドpDAB4434、pDAB4412、および空pETベクター(対照)を、大腸菌BL21-Star(DE3)細菌細胞に形質転換した。発現培養物を、50μg/mlの抗生物質および75μM IPTG(イソプロピル-α-D-チオガラクトピラノシド)を含有しているLB培地250mLへの10〜200個の新しく形質転換されたコロニーにより開始させた。培養物を180〜200rpmで28℃で24時間増殖させた。5マイクロリットルの培養物を、複合培地対照および漸増的に増加する濃度のグルホシネートおよび20μM IPTGを含有している最少培地へ播種した。培養物を、プレート上に均一に分散させ、28℃で24時間インキュベートした。割り当てられたインキュベーション時間の後、プレートを細菌増殖について観察した。これらの結果は表8に例示される。
【0118】
(表8)
大腸菌は20uM IPTGを含む培地上で28℃で24時間増殖させた。-- = 増殖なし // +++ = 明瞭なコロニー増殖
【0119】
4.4-大腸菌BL21-Star(DE3)細胞におけるDSM-2の組み換え発現を駆動するための植物プロモーターの使用
ウイルスプロモーターCsVMVまたは植物プロモーターAtUbi10のいずれかの下でDSM-2(v2)遺伝子およびPAT遺伝子のコーディング配列が発現されたプラスミド構築物を、グルホシネートを含有している最少培地で補完について分析した。プラスミド3778(Rb7 MARv3 // AtUbi10プロモーター // DSM-2(v2)// Atu Orf 1 3'UTR)、3779(Rb7 MARv3 // AtUbi10プロモーター // PAT // Atu Orf 1 3'UTR)、3264(CsVMVプロモーター // DSM-2 // Atu Orf24 3'UTR)、3037(CsVMVプロモーター // PAT // Atu Orf 25/26 3'UTR)、および770(CsVMVプロモーター // GUS v3 // Atu Orf 24 3'UTRを含有している対照プラスミド)を、大腸菌BL21-Star(DE3)に形質転換し、複合培地中で増殖させた。5マイクロリットルの培養物を、増加する濃度のグルホシネートを含有している最少培地に播種し、37℃で48時間インキュベートした。結果は表9に例示される。これらのデータは、植物およびウイルスのプロモーターが、細菌細胞内で中レベルの機能性を有しており、選択マーカーの発現を駆動するために使用され得ることを示している。
【0120】
(表9)
大腸菌は37℃で48時間増殖させた。++++ = 激しい菌叢増殖;+++ = 菌叢増殖;++ = 多数の明瞭なコロニー;+ = まばらなコロニー増殖
【0121】
実施例5-生化学的特徴決定のためのDSM-2の精製およびウエスタン分析のための抗体作製
5.1-組み換え発現
プラスミドpDAB4412中のコドン最適化DSM-2(v2)遺伝子を保持している(Invitrogen, Carlsbad, CAから購入された)大腸菌BL-21(DE3)Star細胞を、種の調製のため、37℃で一晩、50μg/mlカナマイシンが補足された3mlのLB培地に播種するために使用した。およそ2mlの種培養物を、2.8Lの整流装置付き三角フラスコの中のカナマイシン(50μg/ml)を含有している1Lの新鮮なLBに移した。培養物を、0.8〜1.0に近いOD600を入手するためにおよそ6時間、250RPMでシェーカー(New Brunswick Scientific, Model Innova 44)上で37℃でインキュベートした。イソプロピルβ-D-1-チオガラクトピラノシド(IPTG)を培養物中に最終75μMで添加し、一晩の誘導のため18℃でインキュベートし続けた。4℃で15分間8,000RPMでの遠心分離によって細胞を採集し、細胞ペーストを-80℃で保存するか、または精製のために直ちに処理した。
【0122】
1Lの培養物から湿重量でおよそ5gの大腸菌細胞を解凍し、20mMトリス-HCl、pH 8.0および0.3mlのProtease Inhibitor Cocktail(Sigma, cat# P8465)を含有している300mlの抽出緩衝液に再懸濁させ、超音波処理によって15分間氷上で破砕した。溶解物を20分間24,000RPMで4℃で遠心分離し、上清を0.8μmおよび0.45μmの膜でろ過した。すべてのその後のタンパク質分離は、Pharmacia AKTA Explorer 100を使用して実施し、4℃で操作した。ろ液を、20mMトリス-HCl、pH 8.0緩衝液で平衡化されたQXL Sepharose Fast Flowカラム(Pharmacia HiPrep 16/10、20mlベッドサイズ)に10ml/分で適用した。溶出液のOD280が基線に戻るまで、この緩衝液でカラムを洗浄し、5mlの画分を収集しながら、5ml/分の流速で0.5Lの0〜0.4M NaCl直線勾配によりタンパク質を溶出させた。グルホシネート変換活性にも対応する、見かけの20kDaのバンド(予測されたDSM-2分子量は19.3kDaである)を含むSDS-PAGEによって決定されるような、DSM-2を含有している画分をプールした。試料を4倍容量の5mM DTT、0.5%トリトンX-100、5%グリセロールを含有している20mMトリス-HCl、pH 7.5緩衝液により希釈し、4ml/分でMono Qカラム(Pharmacia 10/100 GL、8mlベッドサイズ)に再適用した。同緩衝液中の0.1〜0.3M NaCl勾配により、タンパク質を溶出させた。DSM-2を含有している主要ピークをプールし、固形硫酸アンモニウムを最終1.0Mで添加し、20mMトリス-HCl、pH 7.5中の1.0M硫酸アンモニウムで平衡化されたPhenyl Fast Flowカラム(Pharmacia HiTrap、5mlベッドサイズ)に適用した。溶出液のOD280が基線に戻るまで、このカラムを4ml/分で平衡緩衝液により洗浄し、次いで、20mMトリス-HCl、pH 7.5中の1.0M〜0 硫酸アンモニウムの直線勾配により、50分(3ml/分)以内に、タンパク質を溶出させ、3mlの画分を収集した。75mS/cmで溶出したDSM-2を含有している主要ピーク画分をプールし、MWCO 10kDa膜遠心濾過装置(Millipore)を使用して、およそ3mg/mlにまで濃縮した。次いで、試料を、1ml/分の流速で、PBS緩衝液により、Superdex 75ゲル濾過カラム(Pharmacia XK 16/60、110mlベッドサイズ)に適用した。純粋なDSM-2を含有しているピーク画分をプールし、-80℃で保存した。標準としてウシ血清アルブミンを使用して、ブラッドフォード(Bradford)アッセイまたは全アミノ酸分析によってタンパク質濃度を決定した。精製されたDSM-2の活性を、ホスフィノトリシンアセチルトランスフェラーゼ(PAT)アッセイのための標準的な手法(Wehrmann et al. 1996)に基づき測定した。
【0123】
5.2-抗体作製
DSM-2に対するウサギポリクローナル抗体を、Invitrogen Antibody Services(South San Francisco, CA, Cat# M0300)によって提供されるRabbit Polyclonal Antibody-Standard Protocolsを使用して作製した。大腸菌において発現され精製されたDSM-2(前記セクションを参照されたい)を、免疫原として供給した。簡単に説明すると、2羽のニュージーランドウサギに、0.25mgのキーホールリンペットヘモシアニンおよび不完全フロイントアジュバント(IFA)により乳化された1mgのDSM-2タンパク質を皮下(SQ)注射した。ウサギを2週間休息させ、3週間の休息期間を空けて、IFAで乳化された0.5mgのDSM-2タンパク質により3回、SQ追加刺激した。最終追加刺激の2週間後、各ウサギから血清を収集し、力価について直接ELISAで試験した(示されないデータ)。特異抗体のより良好な力価を与えた2番のウサギで、付加的な2回の追加刺激および終末採血(Invitrogen Cat#M0311およびM0313)を実施した。
【0124】
5.3-ウエスタンブロッティング分析
およそ100mgのカルス組織を、3個のステンレス鋼BBビーズを含有している2mLの微量遠心管に入れた。250μLの抽出緩衝液(0.1%トリトンX-100、10mM DTT、および1mL当たり5μLのプロテアーゼ阻害剤カクテルを含有しているリン酸緩衝生理食塩水)を添加し、チューブをGeno/Grinder(Model 2000-115, Certiprep, Metuchen, NJ)に固定し、500rpmの1×という設定で6分間振とうした。チューブを10,000×gで10分間遠心分離し、可溶性タンパク質を含有している上清を、別々のチューブにピペットで移し、氷中に保存した。ペレットを上記のようにして抽出し(2回目)、上清を以前の画分と共にプールし、分析した。
【0125】
植物試料から抽出されたタンパク質をレムリ(Laemmli)緩衝液中で変性させ、95℃で10分間インキュベートした。変性タンパク質を、製造業者のプロトコルに従って、Novex 8〜16%Tris-Glycineプレキャストゲル(Invitrogen Cat#EC60452BOX)で分離した後、標準的なプロトコルを使用してニトロセルロース膜に転写した。
【0126】
すべてのウエスタンブロッティングインキュベーション工程を室温で1時間実施した。ブロットを、まず、4%乳を含有しているPBS(PBSM)でブロッキングし、次いで、PBSMで5000倍希釈されたDSM-2特異的ウサギポリクローナル抗体(前記パラグラフを参照されたい)中でインキュベートした。0.05%トゥイーン-20を含有しているPBS(PBST)で5分間3回洗浄した後、ヤギ抗ウサギ抗体/西洋ワサビペルオキシダーゼコンジュゲートをブロット上でインキュベートした。検出されたタンパク質を、化学発光基質(Pierce Biotechnology, Rockford, IL Cat# 32106)およびX線フィルム露光を使用して可視化した。
【0127】
実施例6-アラビドプシスへの形質転換および選択
6.1-アラビドプシス・サリアナ生育条件
野生型アラビドプシス種子を、0.1%アガロース(Sigma Chemical Co., St. Louis, MO)溶液中に懸濁した。懸濁した種子を、4℃で2日間保存し、休止要件を完了し、かつ同調的な種子の発芽を確実にした(層形成)。
【0128】
Sunshine Mix LP5(Sun Gro Horticulture, Bellevue, WA)を、微細なバーミキュライトで覆い、濡れるまで下側からHoagland溶液を与えた。層形成された種子を播種し、湿気ドーム(KORD Products, Bramalea, Ontario, Canada)で7日間覆った。
【0129】
種子を発芽させ、植物を、Conviron(モデルCMP4030およびCMP3244、Controlled Environments Limited, Winnipeg, Manitoba, Canada)中で、定常的な温度(22℃)および湿度(40〜50%)下で、長日条件(16時間光/8時間暗)下で、120〜150μmol/m2秒の光強度で生育させた。植物を、初期にはHoagland溶液で水をやり、次には脱イオン水で水をやり、土壌の湿り気を保つが濡れないように維持した。
【0130】
6.2-アグロバクテリウム形質転換
画線したDH5αコロニーを含み、エリスロマイシン(Sigma Chemical Co., St. Louis, MO)(200mg/L)またはスペクチノマイシン(100mg/L)を含むLB+寒天を使用して、4mlミニプレップ培養(液体LB+エリスロマイシン)に接種するためにコロニーを提供した。この培養物を、定常的に攪拌しながら、37℃で一晩インキュベートした。製造業者の指示に従って実施するQiagen(Valencia, CA)Spin Mini Prepsを使用して、プラスミドDNAを精製した。
【0131】
電気的に形質転換受容性であるアグロバクテリウム・ツメファシエンス(株Z707、EHA101、およびLBA4404)細胞を、Weigel and Glazebrook(2002)のプロトコールを使用して調製した。形質転換受容性アグロバクテリウム細胞を、Weigel and Glazebrook(2002)から適合したエレクトロポレーション法を使用して形質転換した。50μlの形質転換受容性アグロ細胞を氷上で融解し、10〜25ngの所望のプラスミドを細胞に加えた。DNAおよび細胞混合物を、あらかじめ冷やしたエレクトロポレーションキュベット(2mm)に加えた。Eppendorf Electroporator 2510を、以下の条件:電圧: 2.4kV、パルス長: 5m秒を用いて形質転換のために使用した。
【0132】
エレクトロポレーションの後、1mlのYEPブロス(1リットルあたり: 10g酵母抽出物、10gバクト-ペプトン、5g NaCl)をキュベットに加え、細胞-YEP懸濁物を15ml培養チューブに移した。細胞を、定常的に攪拌しながら、ウォーターバス中で28℃にて4時間インキュベートした。インキュベーション後、培養物を、エリスロマイシン(200mg/L)またはスペクチノマイシン(100mg/L)およびストレプトマイシン(Sigma Chemical Co., St. Louis, MO)(250mg/mL)を有するYEP+寒天上に蒔いた。プレートを、2〜4日間、28℃にてインキュベートした。
【0133】
コロニーを選択し、エリスロマイシン(200mg/L)またはスペクチノマイシン(100mg/L)およびストレプトマイシン(250mg/mL)を有する新鮮なYEP+寒天上に画線し、28℃で1〜3日間インキュベートした。コロニーをPCR分析のために選択して、ベクター特異的プライマーを使用することにより、遺伝子インサートの存在を確認した。以下を例外として、製造業者の指示に従って実施するQiagen Spin Mini Prepsを使用して、選択されたアグロバクテリウムコロニーからプラスミドDNAを精製した。DNA精製のために、15mlの一晩ミニプレップ培養物(液体YEP+エリスロマイシン(200mg/L)またはスペクチノマイシン(100mg/L)およびストレプトマイシン(250mg/mL))の4mlアリコートを使用した。Qiagen Spin Mini Prep DNAを使用することの代替として、10μlの水中に懸濁した、形質転換したアグロバクテリウム細胞を、100℃で5分間溶解した。アグロバクテリウム形質転換において使用したバイナリーベクターからのプラスミドDNAを、対照として含めた。PCR反応を、製造業者の指示に従って、0.5×濃度で、Takara Mirus Bio Inc.(Madison, Wisconsin)からのTaq DNAポリメラーゼを使用して完了した。PCR反応を、以下の条件:1)94℃3分間、2)94℃45秒間、3)55℃30秒間、4)72℃1分間、29サイクル、次に72℃で10分間の1サイクルでプログラムされたMJ Research Peltier Thermal Cycler中で実行した。反応物を、サイクリング後に4℃で維持した。増幅を、1%アガロースゲル電気泳動によって分析し、臭化エチジウム染色によって可視化した。そのPCR産物がプラスミド対照と同一であったコロニーを選択した。
【0134】
6.3-アラビドプシス形質転換
アラビドプシスを、フローラルディップ法を使用して形質転換した。選択したコロニーを、エリスロマイシン(200mg/L)またはスペクチノマイシン(100mg/L)およびストレプトマイシン(250mg/mL)を含むYEPブロスの1つまたは複数の15〜30mlのプレ培養物に接種するために使用した。培養物を、220rpmで定常的に攪拌しながら、28℃で一晩インキュベートした。各プレ培養物を、エリスロマイシン(200mg/L)またはスペクチノマイシン(100mg/L)およびストレプトマイシン(250mg/mL)を含むYEPブロスの2つの500ml培養に接種するために使用し、培養物を、定常的に攪拌しながら、28℃で一晩インキュベートした。次いで、細胞を、約8700×gで10分間、室温でペレット化し、得られる上清を廃棄した。この細胞ペレットを、1/2×MurashigeおよびSkoog塩/GamborgのB5ビタミン、10%(w/v)スクロース、0.044μM ベンジルアミノプリン(DMSO中の1mg/ml保存液の10μl/リットル)および300μl/リットルSilwet L-77を含む500mlの浸透培地に穏やかに再懸濁した。約1ヶ月齢の植物を15秒間、培地に浸し、最新の花序を確実に沈める。次いで、植物を、それらの側面を下にして横たえ、24時間覆い(透明または不透明)、次いで水で洗浄し、かつ直立状態で配置した。植物を22℃で、16時間明期/8時間暗期の光周期で生育させた。浸漬の約4週間後、種子を収集した。
【0135】
6.4-形質転換植物の選択
新しく収集したT1種子[DSM-2(v2)遺伝子]を、室温で7日間乾燥させた。T1種子を、26.5×51cm発芽トレイ(T.O. Plastics Inc., Clearwater, MN)中に播種し、各々は、40mlの0.1%アガロース溶液に事前に懸濁された、層形成されたT1種子(約10,000個の種子)の200mgアリコートを受容し、4℃で2日間保存し、休止要件を完了し、かつ同調的な種子の発芽を確実にした。
【0136】
Sunshine Mix LP5(Sun Gro Horticulture Inc., Bellevue, WA)を、微細なバーミキュライトで覆い、濡れるまで下側からHoagland溶液を与え、次いで、重力で水抜きした。各40mlのアリコートの層形成された種子を、ピペットを用いてバーミキュライト上に均一に播種し、湿気ドーム(KORD Products, Bramalea, Ontario, Canada)で4〜5日間覆った。2,4-D出芽後噴霧を使用する初期の形質転換体選択(同時形質転換AAD-12遺伝子の選択;USSN 60/731,044を参照)の1日前にドームを取り外した。
【0137】
播種後(DAP)7日、および11DPAで再度、T1植物(それぞれ子葉および2〜4-lf段階)に、2,4-D除草剤(456g ae/L 2,4-D Amine 4、Dow AgroSciences LLC, Indianapolis, IN)の0.016%溶液を、10ml/トレイの噴霧量で(703L/ha)、適用あたり50 g ae/ha 2,4-D DMAの有効割合を送達するために、DeVilbiss圧縮空気噴霧チップを使用して噴霧した。生き残り(活動的に生育している植物)を、最後の噴霧の4〜7日後に同定し、鉢植え用培地(Metro Mix 360)で調製した3インチポットに個別に移植した。移植した植物を、以前と同様に、湿気ドームで3〜4日覆い、22℃グロースチャンバー中に配置した、または直接温室に移動した。次に、ドームを取り外し、DSM-2(v2)がグルホサミン除草剤抵抗性を提供する能力について試験する少なくとも1日前に、植物を温室(22±5℃、50±30% RH、14時間明期:10時間暗期、最低で500μE/m2s1の天然+追加の光)で栽培した。
【0138】
次いで、T1植物を、様々な割合のグルホサミンにランダムに割り当てた。アラビドプシスについては、140g ai/ha グルホサミンが、意味のあるレベルの抵抗性を有する植物から、感受性の植物を区別するための有効用量である。上昇した割合は、抵抗性の相対レベルを決定するためにも適用した(280、560、または1120g ai/ha)。表10は、アリールオキシアルカノエート除草剤抵抗性遺伝子(AAD-12 v1)に及ぶ比較を示す;USSN 60/731,044を参照。
【0139】
すべてのグルホシネート除草剤適用が、187L/haの噴霧量でトラック噴霧器によって適用された。市販のLiberty(商標)剤(200g ai/L、Bayer Crop Science, Research Triangle Park, NC)。グルホシネートに対する耐性を示したT1植物に、T2世代においてさらにアクセスした。
【0140】
6.5-形質転換植物の選択の結果
最初のアラビドプシス形質転換は、DSM-2(v2)(植物に最適化された遺伝子)を使用して行った。T1形質転換体を、まず、2,4-D DMA選択スキームを使用して、非形質転換種子のバックグラウンドから選択した。100,000個を超えるT1種子をスクリーニングし、260個の2,4-D抵抗性植物(AAD-12遺伝子)を同定した。これは、AAD-12+2,4-Dが選択のために使用される場合の構築物の選択頻度の正常範囲よりわずかに高い0.26%という形質転換/選択頻度に等しかった。上記で選択されたT1植物を、続いて個々のポットに移植し、様々な割合の市販のグルホシネート除草剤を噴霧した。表10は、アラビドプシスT1形質転換体にグルホシネート抵抗性を付与するためのDSM-2(v2)遺伝子および対照遺伝子の応答を比較している。応答は、視覚的損傷% 2WATによって提示される。データは、ほとんどもしくは全く損傷なし(<20%)、中程度の損傷(20〜40%)、または重度の損傷(>40%)を示す個体のヒストグラムとして提示された。各T1は独立した形質転換事象であるので、所定の割合における個々のT1応答の有意な変動が予想される。算術平均および標準偏差が各処理について提示される。非形質転換野生型アラビドプシスは、グルホシネート感受性対照として働いた。DSM-2(v2)遺伝子は、個々のT1アラビドプシス植物に除草剤抵抗性を付与した。所定の処理において、植物応答のレベルは大きく変動した。これは、各植物が独立した形質転換事象を表すという事実に起因し得る。重要な注目点として、140g ai/haグルホシネートより上で、影響を受けなかった個体が存在する一方で、重度に影響を受けたものもあった。割合による集団全体の損傷平均が、DSM-2(v2)で形質転換された植物と、野生型対照またはAAD-12+PAT形質転換対照との間の有意な違いを単に実証するために、表10に提示される。多くのDSM-2(v2)個体は、ほとんどまたは全く損傷なしに、1,120g ai/haグルホシネートに耐えて生存した。
【0141】
(表10)野生型植物およびT1 AAD-12+PAT植物と比較された、発芽後適用された一連のグルホシネート割合に対するT1 DSM-2 v2(植物最適化)形質転換T1アラビドプシスの応答
【0142】
6.6-選択マーカーとしてのDSM-2(v2)
選択剤としてグルホシネートを使用して、選択マーカーとしてDSM-2(v2)を使用する能力を、上記のように形質転換されたアラビドプシスを用いて分析した。DSM-2(v2)を含有しているおよそ100個のT1世代アラビドプシス種子(100〜150個の種子)または2mgのPATを含有しているホモ接合性T5植物を、およそ10,000個の野生型(感受性)種子に加えた(spiked)。植物の各トレイは、以下の処理時点:7 DAPおよび11 DAPで、2回の280g ai/haグルホシネートの適用を受けた。処理は、以前に記載されたようなDeVilbiss噴霧チップを用いて適用された。各々から、別に2mgのT1世代アラビドプシス種子を播き、比較カウントとして噴霧しなかった。17 DAPに、植物を抵抗性または感受性として同定した。処理されたものと未処理のものとの間のカウントは類似していることが表11に示された。これらの結果は、DSM-2(v2)が、集団のための代替的な選択マーカーとして有効に使用可能であることを示す。
【0143】
(表11)播かれた種子の量および280g ai/haグルホシネート処理後に生存していた植物の数
【0144】
6.7-分子分析
6.7.1-組織の収集、DNAの単離および定量Xμl
新鮮な組織をチューブに配置し、4℃で2日間凍結乾燥する。組織を完全に乾燥させた後、タングステンビーズ(Heavy Shot)をチューブ中に配置し、試料を、1分間、Kelcoビーズミルを使用する乾式粉砕に供する。次いで、標準的なDNeasy DNA単離手法に従う(Qiagen, DNeasy 69109)。次いで、抽出されたDNAのアリコートをPico Green(Molecular Probes P7589)で染色し、濃度をng/μlで得るために既知の標準を用いてフルオロメーター(波長485/530-BioTek)で読み取る。
【0145】
6.7.2-インベーダーアッセイ分析Xμl
DNA試料を0.7ng/μlに希釈し、次いで95℃で10分間、サーモサイクラー中のインキュベーションによって変性させる。次いで、インベーダーアッセイ反応混合物を、Third Wave Technologiesにより発表された96穴フォーマット手順に従うことにより調製する。調製された反応混合物7.5μlを96穴プレートの各ウェルに分配し、続いて7.5μlの対照および0.7ng/μlの希釈された変性未知試料を分配する。各ウェルに15μlのミネラルオイル(Sigma)を重層する。次いで、プレートを、63℃で1時間インキュベートし、フルオロメーター(Biotek)上で読み取る。標的プローブについてのバックグラウンドに対するシグナルの%(FAM色素波長560/620)を、内部対照プローブについてのバックグラウンドに対するシグナルの%(RED色素波長485/530)で除する計算は、比率を計算する。その比率は、事象の接合状態を決定するために使用される。
【0146】
6.8-遺伝性
多様なT1事象を、T2種子を産生させるために自家受粉した。これらの種子を、100個のランダムなT2同胞にグルホシネート(200g ai/ha)を適用することによって後代検定した。個々の各T2植物を、3インチ四方のポットに移植した後、噴霧適用した(187L/ha適用割合のトラック噴霧器)。T1ファミリー(T2植物)の63%が、χ二乗検定によって決定されるように(P>0.05)、メンデル遺伝に伴う優性遺伝性単一遺伝子座についての予測された3抵抗性:1感受性モデルに分離した。
【0147】
単一遺伝子座として分離した系の各々からランダムに選択された16の植物体に対して接合状態についてのインベーダー法を実施した。インベーダー法によりホモ接合性と判定されたT2個体から種子を収集した(T3種子)。インベーダー法によりホモ接合性と判定された4つのT2ファミリーの各々からの25のT3同胞を、以前に記載したように後代検定した。ホモ接合性(非分離集団)であると予測されたT2ファミリーのすべてが、非分離性であった。これらのデータは、DSM-2(v2)が安定的に組み込まれ、メンデル様式で少なくとも3世代まで遺伝することを示す。
【0148】
実施例7-除草剤を使用したトウモロコシのウィスカー媒介形質転換
7.1-DSM-2(v2)のクローニング
DSM-2(v2)遺伝子を、Bbs1/Sac1フラグメントとしてDASPICO45ベクターから切り出した。これを、ZmUbi1単子葉植物プロモーターを含有している同様に切断されたpDAB3812ベクターに定方向でライゲーションした。2つのフラグメントを、T4 DNAリガーゼを使用して一緒にライゲーションし、DH5α細胞に形質転換した。QiagenのQIA Spinミニプレップキットを使用して、得られたコロニーに対してミニプレップを実施し、方向について確認するためこれらのコロニーを消化した。ZmUbi1/DSM-2(v2)/ZmPer5を含有している最終構築物をpDAB3250と名付けた。PAT遺伝子を含有している同一の対照ベクターを上記のようにして構築した。この構築物をpDAB3251と名付けた。
【0149】
7.2-カルス/懸濁の開始
カルス培養の開始のために未成熟胚を得るために、温室で生長したHi-II親AおよびBの間のF1交雑を実施した(Armstrong et al. 1991)。胚が1.0〜1.2mmのサイズになったら(受粉後約9〜10日)、穂を収集し、表面を、Liqui-Nox(商標)で磨いて洗浄することによって表面殺菌し、70%エタノールに2〜3分間浸漬し、次いで20%の市販の漂白剤(0.1%次亜塩素酸ナトリウム)に30分間浸漬した。
【0150】
穂を、滅菌した蒸留水ですすぎ、未成熟接合体胚を無菌的に切除し、15Ag10培地(N6培地(Chu et al., 1975)、1.0mg/L 2,4-D、20 g/L スクロース、100mg/L カゼイン加水分解物(酵素消化)、25mM L-プロリン、10 mg/L AgN03、2.5 g/L Gelrite, pH 5.8)上で、胚盤を培地から離れるように向けて2〜3週間培養した。適切な形態(Welter et al., 1995)を示す組織を、約6週間の間、週に2回の間隔で新鮮な15Ag10培地に選択的に移し、次いで、約2ヶ月間の間、週に2回の間隔で4つの培地(N6培地、1.0mg/L 2,4-D、20g/L スクロース、100mg/L カゼイン加水分解物(酵素消化)、6 mM L-プロリン、2.5 g/L Gelrite、 pH 5.8)に移した。
【0151】
胚形成懸濁培養を開始するために、単一の胚から生じたカルス組織の約3mlの充填した細胞量(PCV)を、約30mlのH9CP+液体培地(MS基礎塩混合物(Murashige and Skoog, 1962)、10倍少ないニコチン酸および5倍多いチアミン-HCl、2.0mg/L 2,4-D、2.0mg/L α-ナフタレン酢酸(NAA)、30g/Lスクロース、200mg/L カゼイン加水分解物(酸消化)、100mg/L ミオ-イノシトール、6mM L-プロリン、5% v/v ココナッツ水(継代培養直前に加えた)、pH 6.0を含む改変MSビタミン)に加えた。懸濁培養を、暗条件下で、125ml Erlenmeyerフラスコ中で、125rpmおよび28℃に設定した温度制御シェーカー中で維持した。細胞系統は、典型的には、開始後2ヶ月から3ヶ月以内に樹立されるようになる。樹立の間、ワイドボアピペットを使用して3ml PCVおよび7mlの馴化培地を20mlの新鮮なH9CP+液体培地に加えることによって、懸濁物を3.5日毎に継代培養した。一旦、組織が成長で倍加を開始したら、懸濁物をスケールアップし、500mlフラスコに維持し、それによって、12ml PCVの細胞および28ml 馴化培地を80ml H9CP+培地に移した。一旦懸濁物が完全に樹立されると、将来の使用のために凍結保存した。
【0152】
7.3-凍結保存および懸濁物の融解
継代培養の2日後、4ml PCVの懸濁細胞および4mlの馴化培地を8mlの凍結防止剤(ココナッツ水を含まないH9CP+培地に、1Mグリセロール、1M DMSO、2Mスクロースを溶解、フィルター滅菌する)に加え、125mlフラスコ中、125rpmで、4℃、1時間振盪させた。1時間後、4.5mlを冷やした5.0mlのCorning凍結バイアルに加えた。一旦充填したら、個々のバイアルを、制御速度フリーザー中にて4℃で15分間保持し、次いで、-40℃の最終温度に達するまで、-0.5℃/分の速度で凍結させた。最終温度に到達した後、バイアルを、液体窒素蒸気で満たしたCryoplus 4貯蔵ユニット(Forma Scientific)の内側のラック中の箱に移した。
【0153】
融解するために、バイアルを、貯蔵ユニットから取り出し、密閉したドライアイス容器の中に配置し、次いで、40〜45℃に保持したウォーターバスに、「沸騰」がおさまるまで入れた。融解した場合、内容物を、ふたがある100×25mmペトリ皿中の重ねた滅菌70 mm Whatman濾紙(No. 4)約8枚に注いだ。液体を数分間濾紙に吸収させ、次いで、細胞を含む上端の濾紙をGN6培地(N6培地、2.0 mg/L 2,4-D、30g/Lスクロース、2.5 g/L Gelrite、pH 5.8)の上に1週間移した。1週間後、見込みのある形態を有する組織のみを、濾紙から直接的に新鮮なGN6培地に移した。この組織を、1〜3グラムが、125ml Erlenmeyerフラスコ中の約30mL H9CP+培地への懸濁の開始のために使用可能になるまで、7〜14日毎に継代培養した。以前に記載されたようにその時点で継代培養が行われた、全体で12ml PCVが得られる時点まで、3mlのPCVを新鮮なH9CP+培地に3.5日毎に継代培養した。
【0154】
形質転換の約24時間前に、12ml PCVの以前に凍結した胚形成トウモロコシ懸濁細胞プラス28mlの馴化培地を、500 mlのErlenmeyerフラスコ中の80mlのGN6液体培地(Gelriteを欠くGN6培地)に継代培養し、125rpm、28℃のシェーカー上に配置した。全体で36ml PCVが3つのフラスコに分布するように、同じ細胞系統を使用して、これを2回反復した。24時間後、GN6液体培地を取り出し、細胞に原形質分離を起こさせるために、フラスコあたり72ml GN6 S/M振盪培地(N6培地、2.0 mg/L 2,4-D、30g/L スクロース、45.5g/L ソルビトール、45.5 g/L マンニトール、100mg/L ミオ-イノシトール、pH 6.0)で置き換えた。フラスコを、シェーカー上に30〜35分間配置し、この時間の間、適切な量のGN6 S/M液体培地を、約405mgのあらかじめオートクレーブしたシリコンカーバイドウィスカー(Advanced Composite Materials, Inc.)に加えることによって、シリコンカーバイドウィスカーの50mg/ml懸濁液を調製した。
【0155】
GN6 S/M中でのインキュベーション後、各フラスコの内容物を250ml遠心分子ボトルにプールした、一旦すべての細胞が底にたまると、約14mlのGN6 S/Mを除いて取り出され、後の使用のために滅菌1Lフラスコに収集された。あらかじめ濡らしたウィスカーの懸濁物を、最大速度で60秒間ボルテックスにより激しく撹拌し、8.1mlをボトルに加え、最終段階として170μg DNAをそこに加えた。ボトルを、市販のペイントミキサー、改良Red Devil 5400に直に配置し、10秒間攪拌した。攪拌後、浸透圧を減少するために、細胞のカクテル、培地、ウィスカー、およびDNAを、125mlの新鮮なGN6液体培地とともに、1Lフラスコの内容物に加えた。細胞を、シェーカー上で2時間回収し、その後、室内の真空ラインに接続したガラス細胞コレクターユニットを使用して、Whatman #4濾紙(5.5cm)上で濾過した。
【0156】
約6mlの分散した懸濁液を、真空にするに従って、フィルターにピペットにより移した。フィルターは、GN6培地の60×20mmプレート上に配置した。プレートは、個々のプレートの蒸発を最小化するために単一のプラスチックの層(<2ミル厚)でゆるくシールした暗箱中で、28℃で1週間培養した。
【0157】
1週間後、20枚の濾紙をGN6培地(1ハービエース)の60×20mmプレートに移し、20枚の濾紙をGN6培地(2ハービエース)の60×20mmプレートに移し、20枚の濾紙をGN6培地(4ハービエース)の60×20mmプレートに移した(N6培地、2.0mg/L 2,4-D、30g/Lスクロース、100mg/L myo-イノシトール、1、2、または4mg/Lビアラホス(ハービエース由来)、および2.5g/L Gelrite、pH 5.8)。プレートを箱の中に配置し、さらに1週間培養した。
【0158】
さらに1週間後、すべての濾紙を再び同濃度のGN6+ハービエース培地(1H、2H、および/または4H)に移した。プレートを箱の中に配置し、さらに1週間培養した。
【0159】
形質転換の3週間後、プレート上の細胞の1/2を、ハービエースのビアラホス1、2、または4mg/Lを含む3.0mlの溶解したGN6アガロース培地(121℃で10分間だけオートクレーブされた、N6培地、2.0mg/L 2,4-D、30g/Lスクロース、100mg/L myo-イノシトール、7g/L Sea Plaqueアガロース、pH 5.8)にこすり取ることによって、組織を包埋した。この組織を破壊し、3mLのアガロースおよび組織を、細胞が最初に培養された濃度に応じて、GN6(1H、2H、および/または4H)の100×15mmプレートの表面に均一に注いだ。これを、各プレート上の残りの1/2の細胞を用いて反復した。包埋後は、プレートをNescofilm(登録商標)またはParafilm M(登録商標)で個別にシールし、次いで、暗い箱の中で28℃で約4週間培養した。
【0160】
7.4-植物体再生のためのプロトコール
推定形質転換単離物は、典型的には、形質転換の5〜8週間後に最初に可視となった。可能性のある単離物を包埋されたプレートから除去し、60×20mmプレート内の同濃度の新鮮な選択培地に移した。およそ2週間後に持続的な生長が明白であれば、その事象は抵抗性であると見なされる。次いで、抵抗性事象のサブセットを、分子分析にかけた。
【0161】
再生を、(MS塩およびビタミン、30.0g/L スクロース、5mg/L BAP、0.25mg/L 2,4-D、1mg/L ビアラホス、2.5g/L Gelrite;pH 5.7)を含む、サイトカイニンベースの誘導培地、28(1H)にカルス細胞を移すことによって開始する。細胞を、微小光(13μEm-2s-1)中で1週間増殖させ、次いで、より明るい光(40μEm-2s-1)でもう一週間増殖させ、その後、植物増殖調節因子を欠いている以外は28(1H)と同一である再生培地36(1H)に移す。小さな(3〜5cm)植物体を取り出し、選択なしのSHGA培地(SchenkおよびHildebrandtの基礎塩およびビタミン、1972;1g/L myo-イノシトール、10g/L スクロース、2.0g/L Gelrite,pH 5.8)を含む150×25mm培養チューブに配置する。一旦植物体が十分な根およびシュート系を発達したら、これらは温室の中の土に移植する。
【0162】
7.5-分子分析:トウモロコシ材料および方法
7.5.1-組織の収集、DNAの単離および定量
新鮮な組織をチューブに配置し、4℃で2日間凍結乾燥する。組織を完全に乾燥させた後、タングステンビーズ(Valenite)をチューブ中に配置し、試料を、1分間、Kelcoビーズミルを使用する乾式粉砕に供する。次いで、標準的なDNeasy DNA単離手法に従う(Qiagen, DNeasy 69109)。次いで、抽出したDNAのアリコートをPico Green(Molecular Probes P7589)で染色し、濃度をng/μlで得るために既知の標準を用いてフルオロメーター(BioTek)で読み取る。
【0163】
7.5.2-PATインベーダーアッセイ分析
DNA試料を20ng/μlに希釈し、次いで95℃で10分間、サーモサイクラー中のインキュベーションによって変性させる。次いで、Signal Probeミックスを、提供されるオリゴミックスおよびMgCl2を使用して調製する(Third Wave Technologies)。7.5μlのアリコートをインベーダーアッセイプレートの各ウェルに配置し、続いて対照、標準、および20ng/μlに希釈した未知の試料の7.5μlのアリコートを配置する。各ウェルに15μlのミネラルオイル(Sigma)を重層する。次いで、プレートを、63℃で1時間インキュベートし、フルオロメーター(Biotek)上で読み取る。バックグラウンド内部対照プローブよりも上のシグナル%で除算した、標的プローブについてのバックグラウンドよりも上のシグナル%の計算は、比率を計算する。サザンブロット分析を用いて開発されかつ確証された既知のコピー標準の比率を使用して、未知の事象の見積もったコピーを同定する。
【0164】
7.5.3-PATのポリメラーゼ連鎖反応
全体で100ngの全DNAを鋳型として使用する。20mMの各プライマーを、Takara Ex Taq PCR Polymeraseキット(Mirus TAKRR001A)とともに使用する。PAT PTUのためのプライマーは、
である。試料を、94℃3分間、ならびに94℃30秒間、62℃30秒間、および72℃3分15秒間の35サイクル、続いて72℃10分間に供することによって、9700 Geneampサーモサイクラー(Applied Biosystems)においてPCR反応を実行する。コード領域PCR PATのためのプライマーは、
である。試料を、94℃3分間、ならびに94℃30秒間、65℃30秒間、および72℃1分45秒間の35サイクル、続いて72℃10分間に供することによって、9700 Geneampサーモサイクラー(Applied Biosystems)において、PCR反応を実行する。DSM-2のためのコード領域PCRのためのプライマーは、
である。試料を、94℃3分間、ならびに94℃30秒間、65℃30秒間、および72℃45秒間の35サイクル、続いて72℃10分間に供することによって、9700 Geneampサーモサイクラー(Applied Biosystems)において、PCR反応を実行する。PCR産物を、EtBrで染色される1%アガロースゲル上での電気泳動によって分析する。
【0165】
7.5.4-サザンブロット分析
サザンブロット分析を、Qiagen DNeasyキットから得られた全DNAを用いて実施する。全体で5μgの全ゲノムDNAを、組み込みデータを得るため、NcoIおよびSwaIによる消化に一晩供する。制限酵素SspIによる5μgの消化を、PTUデータを得るために使用した。SspI消化データを分析した後、制限酵素MfeIの方がよい酵素の選択であると考えられたため、MfeIを残りの試料の全てを消化するために使用した。一晩消化の後、約100ngのアリコートを1%ゲル上で泳動し、完全な消化を確実にする。この保証後、試料を、大きな0.85%アガロースゲル上で、40ボルトで一晩泳動させる。次いで、このゲルを、0.2M NaOH、0.6M NaCl中で30分間変性させる。次いで、このゲルを、pH 7.5の0.5M Tris HCl、1.5M NaCl中で30分間中和させる。次いで、20×SSCを含むゲル装置を、ゲルからナイロンメンブレン(Millipore INYC00010)への重力による転写を得るために一晩設定する。一晩の転写後、次いで、このメンブレンを、120,000マイクロジュールで、クロスリンカー(Stratagene UV stratalinker 1800)によりUV光に供する。次いで、このメンブレンを、0.1% SDS、0.1 SSC中で45分間洗浄する。45分間の洗浄後、このメンブレンを80℃で3時間ベーキングし、次いでハイブリダイゼーションまで4℃に保存する。ハイブリダイゼーション鋳型フラグメントは、プラスミドDNAを使用するコード領域PCRを使用して調製する。産物を1%アガロースゲル上で泳動し、切除し、および次いで、Qiagen(28706)ゲル抽出手法を使用してゲル抽出する。次いで、このメンブレンを、Perfect Hyb緩衝液(Sigma H7033)中で1時間、60℃段階のプレハイブリダイゼーションに供する。Prime it RmT dCTP-labeling reaction(Stratagene 300392)手法を使用して、p32に基づくプローブを開発する(Perkin Elmer)。このプローブを、Probe Quant. G50 カラム(Amersham 27-5335-01)を使用して精製する。ハイブリダイゼーション緩衝液1 mlあたり200万カウントCPMを使用して、サザンブロットを一晩ハイブリダイズさせる。一晩のハイブリダイゼーションの後、次いで、このブロットを、0.1% SDS、0.1 SSC中で65℃における2回の20分間の洗浄に供する。次いで、このブロットを、-80℃でインキュベートしてフィルムに一晩露光させる。
【0166】
7.6-結果
以前に記載された2つの構築物(PATおよびDSM-2)の各々を用いて、トウモロコシのウィスカー媒介形質転換を3回実施した。それらの集合的な実験から、230個の単離物が回収された。1および2mg/Lビアラホス(ハービエース由来)を含有している培地では、PATとDSM-2との間の事象回収は極めて類似していたが、4mg/Lビアラホスを含有している培地における事象回収は、DSM-2よりPATの方が高かった。
【0167】
(表Ex.7.6−1)
【0168】
1または2ハービエースで選択されたDSM-2事象のうちの48を、サザンブロットを介した、コピー数分析および完全な植物転写単位(PTU)の存在にかけた。48の事象は、すべて、少なくとも1コピーのDSM-2遺伝子を含有していた。より少数コピーの事象(3以下)からの結果のサブセットを、以下に提示する。
【0169】
(表Ex.7.6−2)
【0170】
1、2、または4ハービエースで選択されたPAT事象のうちの48を、それぞれインベーダーアッセイおよびPCRを介した、コピー数推定および完全な植物転写単位(PTU)の存在にかけた。48の事象は、すべて、少なくとも1コピーのPAT遺伝子を含有していた。より少数コピーの事象(3以下)からの結果のサブセットを、以下に提示する。
【0171】
(表Ex.7.6−3)
【0172】
およそ6〜10個のT0植物体が、15個のDSM-2含有事象の各々から再生し、7〜8個の植物体が、Libertyに対する耐性を査定するために以前にリストされた、7個のPAT含有事象の各々から再生した。
【0173】
31個の異なる事象からのカルス組織試料を、非形質転換カルス(陰性対照)と共に、ウエスタンブロッティング実験を用いて分析した。陰性対照以外のすべての試料について、20kDaというマーカーに対する相対分子量を有する1本のバンドが観察された。それは、19.7kDaというタンパク質の予測されたサイズに一致した。さらに、バンドは、標準、すなわち大腸菌から精製された精製DSM-2タンパク質とも同一のサイズを有する。ゲル上の0.7μg/mLの標準に5(+++++)という任意スコアを与え、異なる事象からのバンドを、相対的に等級付け、以下の表にリストした。
【0174】
(表Ex.7.6−4)
【0175】
7.6.1-T0トウモロコシにおける葉塗布直接比較
T0 DSM-2(v2)植物に、グルホシネート除草剤を滴下にて塗布した。15個のT0事象の各々からの4個の同胞が試験され、個々の各植物体の4枚の葉が、およそV8期でグルホシネートの滴下を受容した。滴下処理を各割合についてランダム化し、個々の葉における処理位置の変動を可能にした。トウモロコシの場合、0.25%v/vグルホシネートが、有意のレベルの抵抗性を有する植物体から感受性植物体を区別するための最小有効用量である。より高い割合も、抵抗性の相対レベルを決定するために適用した(0.5%、1.0%、および2.0%v/v)。グルホシネート処理は、直径およそ2.5cmの処理区域に、先端に綿の付いたアプリケータを使用して適用された。
【0176】
表11は、トウモロコシT0形質転換体にグルホシネート抵抗性を付与するためのDSM-2(v2)遺伝子および対照遺伝子の応答を比較している。応答は、視覚的損傷% 2WATによって提示される。データは、ほとんどもしくは全く損傷なし(<20%)、中程度の損傷(20〜40%)、または重度の損傷(>40%)を示す個体のヒストグラムとして提示される。各T0は独立した形質転換事象であるので、所定の割合における個々のT0応答の有意な変動が予想される。算術平均および標準偏差が各処理について提示される。非形質転換野生型トウモロコシは、グルホシネート感受性対照として働いた。DSM-2(v2)遺伝子は、個々のT0トウモロコシ植物に除草剤抵抗性を付与した。所定の処理において、植物応答のレベルは大きく変動した。これは、各植物が独立した形質転換事象を表すという事実に起因し得る。重要な注目点として、最大2%v/vグルホシネートにおいて、DSM-2(v2)は、PAT形質転換植物より全体的に優れている。割合による集団全体の損傷平均が、単に、DSM-2(v2)で形質転換された植物と、野生型対照またはPAT形質転換対照との間の有意な違いを実証するために、表11に提示される。
【0177】
(表11)野生型植物およびT0 PAT形質転換植物と比較された、発芽後の植物体に葉塗布として適用された一連のグルホシネート割合に対するT0 DSM-2(v2)(植物最適化)形質転換トウモロコシの応答
【0178】
7.6.2-T2トウモロコシにおける高いグルホシネート耐性の立証
T1 DSM-2(v2)×5XH751の交雑からの種子を、Metro Mix培地を含有している4インチのポットに植え、2葉期に、ヌルを除去するために、560g ai/haグルホシネートで187L/haに設定されたトラック噴霧器で噴霧した。7DATで、ヌルを除去し、以下の割合で上記のようなトラック噴霧器で抵抗性植物に噴霧した:0、560、1120、2240、および4480g ai/haグルホシネート。3 DATおよび14 DATで、植物を等級付け、5XH751×Hi II対照植物体と比較した。下記表Ex.7.6.2-1は、20%未満の損傷を有する最大2240g ai/haグルホシネートを提供した個々のDSM-2(v2)植物体が存在することを示す。DSM-2(v2)は、PAT形質転換対照においても4480g ai/haグルホシネートに対する類似した耐性を提供した。
【0179】
(表Ex.7.6.2−1)発芽後(14DAT)に適用された一連のグルホシネート割合に対するT2トウモロコシの応答
【0180】
7.6.3-トウモロコシにおけるDSM-2(v2)の遺伝性
T1 DSM-2(v2)×5XH751の交雑からの種子を、Metro Mix培地を含有している3インチのポットに植え、2葉期に、0、280、560、1120、2240、および4480g ai/haグルホシネートで187L/haに設定されたトラック噴霧器で噴霧した。3 DATおよび14 DATで、植物を等級付け、5XH751×Hi II対照植物と比較した。以前のように、0〜100%の全体的な視覚的損傷により植物を等級付けた。各集団の分離を決定するためには、1120 g ai/ha以上の割合を選んだ。耐性植物および感受性植物を計数したところ、T1ファミリーのすべてが、χ二乗検定によって決定されるように、単一遺伝子座、優性メンデル形質(1R:1S)として分離することが決定された。生存している植物体を、T2世代を作製するために自家受粉した。DSM-2(v2)は、商業的なハイブリッドと相反交雑された場合、複数の種において強固なグルホシネート抵抗性遺伝子として遺伝性である。
【0181】
5つのDSM-2(v2)T2ファミリーにおいて後代検定も実施した。種子を、上記のような3インチのポットに植えた。3葉期に、すべての植物に、以前に記載されたようなトラック噴霧器で560g ai/haグルホシネートを噴霧した。7 DATの後、耐性植物体および感受性植物体を計数した。試験された5つの系のうちの4つが、χ二乗検定によって決定されるように、単一遺伝子座、優性メンデル形質(3R:1S)として分離した。
【0182】
7.6.4-除草剤スペクトルを増加させるためのDSM-2(v2)の重ね合わせ
T1植物のBE1146RRとの交雑を行った。DSM-2(v2)形質転換植物は、従来法により、付加的な関心対象の形質を含有している他のトウモロコシ系と交配され得る。グリホセート耐性形質CP4を含有している近交系(BE1146RR)を、DSM-2(v2)を含有しているT1植物と交雑した。通常であれば致死的である除草剤割合に等しいか、またはそれを超える割合で、逐次的に、または容器での混合物により、グルホシネートおよびグリホセートを適用することにより、両除草剤に対する耐性形質の効力について、後続世代の植物を試験することができる(例えば、280、560、1120g ae/ha、またはそれ以上の両除草剤を植物に噴霧することができる)。これは、除草剤抵抗性管理のため、組み合わせ適用または逐次適用において両除草剤を使用する能力を同定するであろう。
【0183】
実施例8-抗体を介した形質転換植物からのタンパク質検出
8.1-ポリクローナル抗体作製
精製されたDSM-2(前記セクションを参照されたい)を、5ミリグラム、ウサギポリクローナル抗体作製のために、Invitrogen Custom Antibody Services(South San Francisco, CA)に送達した。ウサギは、12週間で4回の注射を受容し、各注射は、1mLの不完全フロイントアジュバントに懸濁した精製されたタンパク質0.5mgを含有していた。血清を、特異性および親和性を確認するため、直接ELISAおよびウエスタンブロッティング実験の両方において試験した。
【0184】
8.2-カルス組織からのDSM-2の抽出
1穴パンチャーを使用した4枚のトウモロコシ(Hi-II)葉ディスクを、2個のステンレス鋼ビーズ(4.5mm;Daisy Co,, Cat# 145462-000)、および500μLの植物抽出緩衝液(0.1%トリトンX-100および1mL当たり5μLのプロテアーゼ阻害剤カクテル(Sigma Cat # P9599)を含有しているPBS)を含有している微量遠心管に入れた。チューブを、Geno/Grinder(Model 2000-115, Certiprep, Metuchen, NJ)に固定し、500rpmの1×の設定で6分間振とうした。チューブを5000×gで10分間遠心分離し、可溶性タンパク質を含有している上清を、DSM-2の存在を検出するために、ウエスタンブロッティング実験において分析した。
【0185】
8.3-ウエスタンブロッティング分析
葉抽出物に様々な濃度の精製されたDSM-2を加え、95℃で10分間レムリ試料緩衝液と共にインキュベートした後、8〜16%Tris-Glycineプレキャストゲルにおける電気泳動分離を行った。次いで、タンパク質を、標準的なプロトコルを使用して、ニトロセルロース膜上に電気的に転写した。4%スキムミルクを含むPBSでブロッキングした後、DSM-2タンパク質を、抗DSM-2抗血清と、それに続くヤギ抗ウサギ/HRPコンジュゲートによって検出した。検出されたタンパク質を、化学発光基質ECL Western Analysis Reagent(Amersham Cat.# RPN 21058)によって可視化した。
【0186】
8.4-結果
大腸菌細胞において発現され、精製されたタンパク質を使用して、DSM-2に対するポリクローナル抗体を作成した。4回の注射の後、抗血清は、直接ELISAで観察されるように、抗DSM-2抗体の高い力価を有していた。100,000倍希釈でも、血清は、バックグラウンドより6倍大きいシグナルを提供した。
【0187】
(野生型トウモロコシ(Hi-II)葉基質におけるDSM-2の)ウエスタンブロッティング分析において、血清は、DSM-2(v2)遺伝子に基づき予測された分子量と比較可能である、およそ22kDaの主要なバンドを検出することができた。試験された最低濃度5ng/mLで抽出物を入れた場合でも、同一のバンドを検出することができた。高いDSM-2濃度ではマイナーなバンドも観察されたが、これらは、より低い濃度では観察されなかったことから、標的タンパク質の凝集物であると考えられる。予測された分子量と比較可能な分子量を有する単一の主要なバンドが観察された。(このゲル上の泳動したレーンは、分子量マーカー、ならびにそれぞれ濃度0.005、0.05、0.5、および5μg/mLでDSM-2タンパク質を含有している葉抽出物であった)。さらに、バックグラウンドシグナルがほとんど観察されなかったように、ポリクローナル抗体は、トウモロコシ葉タンパク質と交差反応しなかった。
【0188】
形質転換タバコカルス組織からのDSM-2の発現を、ウエスタンブロッティング分析を使用して決定した。標準物(およそ22kDa)として比較サイズを有する検出されたバンドが、異なる事象で観察され、このことは、DSM-2がトランスジェニックタバコ組織において発現されたことを示している。
【0189】
実施例9-タバコ(細胞培養物)形質転換
形質転換の4日前に、7日毎に継代培養されていた1週齢のNT-1タバコ懸濁物を、40mlのNT-1 B培地に2mlのNT-1培養物または1mlの濃縮細胞を添加することにより、新鮮な培地へと継代培養した。継代培養された懸濁物を、125rpmのシェーカー上で25±1℃で暗所で維持した。
【0190】
(表Ex.9−1)NT-1 B培地組成
【0191】
関心対象のバイナリーベクターを保持しているアグロバクテリウム・ツメファシエンス[LBA4404株]の50%グリセロールストックを、50〜100mg/Lスペクチノマイシンを含有している30mlのYEP液体(10g/L酵母抽出物、10g/Lペプトン、5g/L NaCl、0〜10g/Lスクロース)に20、100、または500μlを添加することにより、液体の一晩の培養を開始させるために使用した。OD600が1.5±0.2になるまで、細菌培養物を150〜250rpmでインキュベーターシェーカーで28℃で暗所でインキュベートされた。これはおよそ18〜20時間を要した。
【0192】
試験された各ベクターについて、20〜70mLの4日齢の懸濁細胞を、無菌の容器に移し、そこに、適切なODの500〜1750μlのアグロバクテリウム懸濁物を添加した。均一の混合物が達成されることを保証するため、10mlの広い孔径のピペットで5回上下に細胞をピペッティングした。次いで、均一の懸濁物を、リピートピペッターの25mlのバレルに引き上げ、1ウェル当たり250μlの懸濁物を、懸濁物が使い尽くされるまで、24穴プレートに分配し続けた。ウェルプレートをパラフィルムで覆い、3日間の共培養のため振とうすることなく25±1℃で暗所で培養した。
【0193】
共培養後、すべての過剰の液体を、1mLピペットチップで個々のウェルから除去し、残存している細胞を、1mlのNTC液(オートクレーブ処理後に添加された500mg/Lカルベニシリンを含有しているNT-1 B培地)に再懸濁させた。個々のウェルの内容物を、使い捨てのトランスファーピペットを使用して、100×25mmの選択プレートの表面全体に分散させた。選択培地は、オートクレーブ処理後に添加された7.5〜15mg/Lのビアラホスまたは技術的等級のグルホシネートアンモニウムが補足された8g/l TC寒天により凝固したNTC培地からなっていた。すべての選択プレートを、覆うことなく、28℃で暗所で維持した。
【0194】
推定形質転換体が、殺傷された非形質転換細胞のバックグラウンド上にカルスの小さなクラスタとして出現した。形質転換のおよそ2〜6週間後に、カルスを単離した。各カルス単離物を、同一の選択培地を含有している60×20mmプレートに移し、およそ2週間増殖させた後、分析に供した。
【0195】
9.1-結果
DSM-2(v2)をPATと比較する並行実験を完了した。研究において、PAT選択プレートの100%が、10mg/Lビアラホス培地上で少なくとも1つのPCR陽性単離物を産生し、一方、DSM-2(v2)選択プレートの79%が、少なくとも1つのPCR陽性単離物を産生した。すべての事象が、コード領域PCRを介して、DSM-2(v2)遺伝子またはPAT遺伝子の存在について分析され、陽性であることが見出された。
【0196】
(表Ex.9−2)
【0197】
DSM-2(v2)で選択された事象の小さなサブセットにおいてウエスタンブロットを実施したところ、以下のデータで分かるように、3つの陽性事象が同定された。第2の実験において、DSM-2(V2)で処理されたタバコ細胞を、7.5、10、12.5、または15mg/Lビアラホスまたは技術的等級のグルホシネートアンモニウムで選択した。コード領域PCRによる立証後の形質転換頻度(少なくとも1つのカルスを産生する選択プレートの%)を、以下の表にリストする。
【0198】
(表Ex.9−3)
【0199】
実施例10-その他の作物のアグロバクテリウム形質転換
本開示を考慮すれば、当技術分野において公知の技術を使用して、本発明に従い、付加的な作物を形質転換することができる。ライムギのアグロバクテリウム媒介形質転換については、例えば、Popelka and Altpeter(2003)を参照されたい。ダイズのアグロバクテリウム媒介形質転換については、例えば、Hinchee et al.,1988を参照されたい。ソルガムのアグロバクテリウム媒介形質転換については、例えば、Zhao et al., 2000を参照されたい。オオムギのアグロバクテリウム媒介形質転換については、例えば、Tingay et al., 1997を参照されたい。コムギのアグロバクテリウム媒介形質転換については、例えば、Cheng et al., 1997を参照されたい。イネのアグロバクテリウム媒介形質転換については、例えば、Hiei et al., 1997を参照されたい。
【0200】
これらおよびその他の植物のラテン名は以下に与えられる。例えば、トウモロコシ(イネ科ジー・メイズ(Zea mays))、コムギ(イネ科トリチカム(Triticum)種)、イネ(イネ科オリザ(Oryza)種およびマコモ(Zizania)種)、オオムギ(イネ科ホルデウム(Hordeum)種)、ワタ(アブロマ(Abroma)双子葉類アブロマ・オーガスタ(augusta)およびアオイ科ゴシピウム(Gossypium)種)、ダイズ(マメ科グリシン・マックス(Glycine max))、テンサイ(アカザ科ベータ・ブルガリス・アルティッシマ(Beta vulgaris altissima))、サトウキビ(アレンガ・ピナータ(Arenga pinnata))、トマト(ナス科リコパーシコン・エスカレンタム(Lycopersicon esculentum)およびその他の種、フィザリス・イコスカルパ(Physalis ixocarpa)、ソラナム・インカナム(Solanum incanum)およびその他の種、ならびにサイフォマンドラ・ベタセア(Cyphomandra betacea))、ジャガイモ、サツマイモ、ライムギ(イネ科セカレ(Secale)種)、コショウ(ナス科カプシカム・アンナム(Capsicum annuum)、シメンセ(sinense)、およびフルテセンス(frutescens))、レタス(キク科ラクチュカ・サティバ(Lactuca sativa)、ペレニス(perennis)、およびパルチェラ(pulchella))、キャベツ、セロリ(セリ科アピウム・グラベオレンス(Apium graveolens))、ナス(ナス科ソラヌム・メロンジナ(Solanum melongena))、ソルガム(すべてのソルガム(Sorghum)種)、アルファルファ(マメ科メジカーゴ・サティバム(Medicago sativum))、ニンジン(セリ科ダウカス・キャロータ・サティバ(Daucus carota sativa))、マメ(マメ科ファセロラス(Phaseolus)種およびその他の属)、オートムギ(アベナ・サティバ(Avena Sativa)およびストリゴーサ(Strigosa))、エンドウ(マメ科ピサム(Pisum)、ビグナ(Vigna)、およびテトラゴノロバス(Tetragonolobus)種)、ヒマワリ(キク科ヘリアンサス・アニュス(Helianthus annuus))、カボチャ(双子葉類キュカービタ(Cucurbita)種)、キュウリ(双子葉類ジェネラ(genera))、タバコ(ナス科ニコチアナ(Nicotiana)種)、アラビドプシス(アブラナ科シロイヌナズナ)、芝草(ロリウム(Lolium)、アグロスチス(Agrostis)、およびその他の科)、ならびにクローバー(マメ科)を含む(がこれらに限定はされない)これらおよびその他の植物にDSM-2(v1)を形質転換するために、これらおよびその他の(非アグロバクテリウム)形質転換技術が使用され得ることは明白であるはずである。例えば、DSM-2(v2)遺伝子を有するそのような植物は、本発明に含まれる。DSM-2(v2)を用いる形質転換の結果としてグルホシネートまたはビアラホスに対する抵抗性が付与された植物における植生制御は、グルホシネートを選択的に適用することにより改良され得る。
【0201】
DSM-2(v2)は、多くの落葉材木および常緑材木の育成系において季節に合わせて使用するための、DSM-2によって不活化され得る除草剤(例えば、グルホシネート、ビアラホス、および/またはホスフィノトリシン)の適用可能性を増加させる可能性を有する。グルホシネートまたはビアラホスに対して抵抗性の材木種は、損傷の懸念のないこれらの除草剤の過剰使用の柔軟性を増加させるであろう。これらの種には、ハンノキ、トネリコ、アスペン、ブナ、カバノキ、サクランボ、ユーカリ、ヒッコリー、カエデ、オーク、マツ、およびポプラが含まれるであろうが、これらに限定はされない。装飾用の種における選択的制御のためのグルホシネートまたはビアラホスに対する抵抗性の使用も、本発明の範囲内である。例には、バラ、ニシキギ、ペチュニア、ベゴニア、およびマリーゴールドが含まれ得るが、これらに限定はされない。
【0202】
実施例11-任意の作物におけるグリホセート耐性形質と重ね合わされたDSM-2(V2)
北米において栽培されるワタ、アブラナ、およびダイズのエーカー(acre)の大部分がグリホセート耐性形質を含み、GTトウモロコシの採用は上昇している。さらなるGT作物(例えば、例えば、コムギ、イネ、テンサイ、芝草など)が開発中であるが、現在まで商業的に発売されていない。多くの他のグリホセート抵抗性種が、開発段階のために実験中である(例えば、アルファルファ、サトウキビ、ヒマワリ、ビート、エンドウマメ、ニンジン、キュウリ、レタス、タマネギ、イチゴ、トマト、およびタバコ;ポプラおよびスイートガムのような林業種;ならびにマリゴールド、ペチュニア、およびベゴニアなどの園芸種;World Wide Web上のisb.vt.edu/cfdocs/fieldtestsl.cfm , 2005)。GTCは、この系によって提供される制御された雑草の完全な広がりおよび便利さおよびコスト効果のための価値のある手段である。しかし、現在標準であるベース処理としてのグリホセートの有用性は、グリホセート抵抗性雑草を選択することである。さらに、グリホセートが固有に効力が弱い雑草は、グリホセートのみの化学物質プログラムが実施されている場合には、圃場で優勢な種に転じている。従来的な育種を通して、または新規な形質転換事象として一緒にのいずれかで、GT形質とDSM-2(v2)を重ね合わせることによって、雑草制御の効力、柔軟性、ならびに雑草の変化および除草剤抵抗性の発生を管理する能力が改善できる。雑草制御オプションの改善のためのいくつかのシナリオが想定でき、ここで、DSM-2(v2)およびGT形質は、任意の単子葉植物種または双子葉植物種において重ね合わせられる。
a)グリホセートは、大部分のイネ科雑草種および広葉雑草種の制御のために、標準的な発芽後適用割合(420〜2160g ae/ha、好ましくは560〜840g ae/ha)で適用することができる。コニザ カンデンシス(Conyza canadensis)のような広葉雑草またはグリホセートを用いる制御に対して固有に困難である雑草(例えば、コメリナ(Commelina)種、イポメア(Ipomoea)種)の制御のために、280-2240g ae/ha(好ましくは350〜1700g ae/ha)グルホシネートが、効果的な制御を提供するために、連続して、容器で混合して、またはグリホセートと前もって混合して適用することができる。
b)現在、GTCにおいて適用されるグリホセートの割合は、一般的に、適用のタイミングあたり、560〜2240 g ae/haの範囲である。グリホセートは、広葉雑草種よりも、イネ科植物雑草に対してはるかにより効力がある。DSM-2(v2)+GTの重ね合わせた形質は、イネ科植物に有効な割合のグリホセート(105〜840g ae/ha、より好ましくは210〜420g ae/ha)を可能にする。次いで、グルホシネート(280〜2240g ae/ha、より好ましくは350〜1700g ae/ha)を、必要な広葉雑草の制御を提供するために、連続して、容器で混合して、またはイネ科雑草に有効な割合のグリホセートと前もって混合して適用することができる。
【0203】
当業者は、その他の除草剤(例えば、ビアラホス)が、DSM-2(v2)を用いた植物の形質転換によって可能になることを認識するであろう。CPR(Crop Protection Reference)書に編集された除草剤のラベルもしくは類似の編集、オンラインで編集されたラベル(例えば、cdms.net/manuf/manuf.asp)、またはAgriliance(2003)からのCrop Protection Guideのような商業的もしくは学術的な作物保護指針により、具体的な割合は決定され得る。DSM-2(v2)によってHTCにおける使用を可能にされた各代替除草剤は、単独で使用されたとしても、容器で混合されたとしても、または連続的に使用されたとしても、本発明の範囲内にあると見なされる。
【0204】
実施例12-任意の作物においてAHAS形質と重ね合わされたDSM-2(v2)
イミダゾリノン除草剤耐性(AHASなど)は、現在、トウモロコシ、イネ、およびコムギを含むがこれらに限定はされない、北米において栽培されている多数の作物に存在している。さらなるイミダゾリノン耐性作物(例えば、ワタおよびテンサイ)が開発中であるが、現在のところ市販されてはいない。多くのイミダゾリノン除草剤(例えば、イマザモクス、イマゼタピル、イマザキン、およびイマザピク)は、現在、様々な従来的な作物において選択的に使用されている。イマゼタピル、イマザモクス、および非選択的なイマザピル(imazapyr)の使用は、AHASなどのようなイミダゾリノン耐性形質を通して可能にされてきた。現在のところ、市販のイミダゾリノン耐性HTCは、非トランスジェニックであるという利点を有する。この化学クラスは、有意な土壌残留活性を有し、従って、グリホセートまたはグルホシネートに基づく系とは異なり、適用のタイミングを超えて拡張される雑草制御を提供することも可能である。しかし、イミダゾリノン除草剤によって制御される雑草のスペクトルは、グリホセートほど広くない(Agriliance, 2003)。さらに、イミダゾリノン除草剤は、多くの雑草が抵抗性を発達させてきた作用の様式(アセトラクテートシンターゼ、ALSの阻害)を有する(Heap, 2004)。従来的な交配を通して、または新規の形質転換事象として一緒に、イミダゾリノン耐性形質とDSM-2(v2)を重ね合わせることによって、雑草制御の効率、柔軟性、ならびに雑草のシフトおよび除草剤抵抗性の発達を管理する能力が改善され得る。DSM-2(v2)およびイミダゾリノンに対する耐性形質が、単子葉植物種または双子葉植物種において重ね合わせられる、雑草制御オプションの改善のためのいくつかのシナリオが想定できる。
a)イマゼタピルは、多くのイネ科雑草種および広葉雑草種の制御のために、標準的な発芽後適用割合(35〜280g ae/ha、好ましくは70〜140g ae/ha)で適用することができる。
i)アマランサス ルディス(Amaranthus rudis)、アンブロシア トリフィダ(Ambrosia trifida)、チェノポジウム アルブム(Chenopodium album)のようなALS阻害剤抵抗性広葉雑草(とりわけ、Heap, 2004)は、280〜2240g ae/ha、より好ましくは350〜1700g ae/haグルホシネートを容器で混合することによって制御できる。
ii)イポモエア(Ipomoea)種のようなイミダゾリノン除草剤に対して固有により耐性である広葉雑草種は、280〜2240g ae/ha、より好ましくは350〜1700g ae/ha グルホシネートを容器で混合することによってもまた制御できる。
【0205】
雑草制御の分野の当業者は、単独で、または1種もしくは複数の組み合わせでの、種々の市販のイミダゾリノン除草剤、およびグルホシネートベースの除草剤のいずれかの使用が、従来的な育種または遺伝子操作のいずれかによる、DSM-2(v2)形質転換および任意のイミダゾリノン耐性との重ね合わせによって可能であることを認識している。これらの化学物質の典型的な他の除草剤の特定の割合は、CPR(Crop Protection Reference)書籍もしくは同様の編集物、オンラインで編集された表示(例えば、cdms.net/manuf/manuf.asp)、または任意の商業的もしくは学術的な作物保護の手引書、例えば、Crop Protection Guide from Agriliance(2003)において編集されている、除草剤表示によって決定することができる。単独で、容器での混合で、または連続して使用されるかに関わらず、DSM-2(v2)によるHTCにおける使用のために可能にされる各々の代替的な除草剤は、本発明の範囲内にあると見なされる。
【0206】
実施例13-イネにおけるDSM-2(V2)
13.1-培地の説明
使用する培地を1M KOHでpH 5.8に調整し、2.5g/1 Phytagel(Sigma)で固化した。胚形成カルスを、40ml半固形培地を含む100×20mm ペトリ皿中で培養した。細胞懸濁液を、35ml液体培地を含む125mlコニカルフラスコ中で維持し、125rpmで回転させた。胚形成培養の誘導および維持は、暗所にて25〜26℃で行った(Zhang et al. 1996)。
【0207】
胚形成カルスの誘導および維持は以前に記載されたようにNB基礎培地上で行ったが(Li et al. 1993)、500mg/lグルタミンを含むように適合させた。懸濁培養を開始し、マルトースの代わりに30g/lを含めたSZ液体培地(Zhang et al. 1998)中で維持した。浸透培地(NBO)は、各0.256Mのマンニトールおよびソルビトールの添加を伴うNB培地からなった。除草剤耐性カルスを、8mg/lビアラホスを補充したNB培地で、3週間ごとに継代培養しながら9週間選択した。
【0208】
13.2-組織培養物の発展
オリザ サティバ(Oryza sativa)L.ジャポニカ 栽培種タイペイ(L. japonica cv. Taipei)309の成熟乾燥種子を、Zhang et al. 1996において記載されるように殺菌した。胚形成組織を、暗所でNB培地上で殺菌成熟イネ種子を培養することによって誘導した。約1mm直径の一次カルスを胚盤から取り出し、SZ液体培地中で細胞懸濁を開始するために使用した。次いで、懸濁物を、Zhang 1995に記載されるように維持した。懸濁物に由来する胚発生組織を、以前の継代培養の3〜5日後に液体培養から取り出し、NBO浸透培地に移し、ペトリ皿中で約2.5cmの横方向の円を形成して、ボンバードメントの前4時間培養した。ボンバードメントの16〜20時間後、組織を、NBO培地からNBH8除草剤選択培地に移し、ボンバードメント表面が上側を向くことを確実にし、かつ暗所で3時間インキュベートした。新たに形成されたカルスを、3週間に2回、新鮮なNBH8培地へ継代培養した。
【0209】
13.3-微粒子銃ボンバードメント
すべてのボンバードメントは、Biolistic PDS-1000/He(商標)システム(Bio-Rad, Laboratories, Inc.)を用いて実施した。3mgの1.0ミクロン直径の金粒子を1回、100%エタノールで、2回滅菌蒸留水で洗浄し、およびシリコン処理したEppendorfチューブ中の50μl水中に懸濁した。5μgプラスミドDNA、20μlスペルミジン(0.1M)および50μl 塩化カルシウム(2.5M)を金懸濁物に加えた。この混合物を室温で10分間インキュベートし、10000rpmで10秒間ペレット化し、60μl冷100%エタノール中に再懸濁し、かつ8〜9μlを各マイクロキャリアに分配した。組織試料を、Zhang et al.(1996)によって記載されるように、1100psiおよび27inのHg真空でボンバードメントした。
【0210】
実施例14-任意の作物においてAAD-1(v3)と重ね合わされたDSM-2(v2)
グルホシネートは、グリホセートと同様に、比較的非選択的なスペクトルの広いイネ科および広葉樹に対する除草剤である。グルホシネートの作用の様式は、グリホセートと異なる。それは、より即効性であり、除草剤適用後24〜48時間で、処理された葉の脱水および「焼け(burning)」をもたらす。これは、迅速な雑草制御の出現のため有利である。しかしながら、これは、標的植物の分裂組織領域へのグルホシネートの転移も制限し、それが、多くの種における2つの化合物の相対的な雑草制御性能の査定によって証明されるような、より不十分な雑草制御をもたらす(Agriliance, 2003)。
【0211】
従来的な交配を通して、または新規の形質転換事象として一緒に、AAD-1(v3)(USSN 11/587,893;WO 2005/107437を参照されたい)をグルホシネート耐性形質と重ね合わせることにより、雑草制御の効率、柔軟性、ならびに雑草のシフト(shift)および除草剤抵抗性の発達を管理する能力が改善され得る。前記実施例において言及されたように、AAD-1(v3)を用いて作物を形質転換することによって、単子葉作物においてAOPP除草剤を選択的に適用することが可能であり、単子葉作物は、フェノキシオーキシン安全性のより高い限界を有するため、フェノキシオーキシンを双子葉作物に選択的に適用することができる。AAD-1(v3)およびグルホシネートに対する耐性形質が、単子葉作物種または双子葉作物種において重ね合わせられる、雑草制御オプションの改善のためのいくつかのシナリオが想定できる。
a)グルホシネートは、多くのイネ科雑草種および広葉雑草種の制御のために、標準的な発芽後適用割合(200〜1700g ae/ha、好ましくは350〜500g ae/ha)で適用され得る。現在のところ、グルホシネート抵抗性雑草は確認されていないが、グルホシネートは、本質的により耐性の雑草をグリホセートより数多く有する。
i)本質的に耐性のイネ科雑草種(例えば、エキノクロア(Echinochloa)種またはソルガム種)は、10〜200g ae/ha(好ましくは20〜100g ae/ha)のキザロホップを容器で混合することによって制御され得る。
ii)本質に耐性の広葉雑草種(例えば、シルシウム・アーベンシス(Cirsium arvensis)およびアポシナム・カンナビナム(Apocynum cannabinum))は、これらのより制御が困難な多年生種の効率的な制御のため、そして一年生広葉雑草種の制御の強固さを改善するため、280〜2240g ae/ha、より好ましくは560〜2240g ae/haの2,4-Dを容器で混合することによって制御され得る。
b)例えば、グルホシネート(200〜500g ae/ha)+ 2,4-D(280〜1120g ae/ha)+ キザロホップ(10〜100g ae/ha)の三元組み合わせは、より強固な、重複する雑草制御スペクトルを提供することができる。さらに、重複するスペクトルは、除草剤抵抗性雑草の管理および遅延のための付加的なメカニズムを提供する。
【0212】
実施例15-任意の作物においてAAD-12(v2)と重ね合わされたDSM-2(v2)
グルホシネートは、グリホセートと同様に、比較的非選択的なスペクトルの広いイネ科および広葉樹に対する除草剤である。グルホシネートの作用の様式は、グリホセートと異なる。それは、より即効性であり、除草剤適用後24〜48時間で、処理された葉の脱水および「焼け」をもたらす。これは、迅速な雑草制御の出現のため有利である。しかしながら、これは、標的植物の分裂組織領域へのグルホシネートの転移も制限し、それが、多くの種における2つの化合物の相対的な雑草制御性能の査定によって証明されるような、より不十分な雑草制御をもたらす(Agriliance, 2005)。
【0213】
従来的な交配を通して、または新規の形質転換事象として一緒に、AAD-12(v1)をグルホシネート耐性形質と重ね合わせることにより、雑草制御の効率、柔軟性、ならびに雑草のシフトおよび除草剤抵抗性の発達を管理する能力が改善され得る。AAD-12(v1)およびグルホシネートに対する耐性形質が単子葉植物種または双子葉植物種において重ね合わせられる、雑草制御オプションの改善のためのいくつかのシナリオが想定できる。
a)グルホシネートは、多くのイネ科雑草種および広葉雑草種の制御のために、標準的な発芽後適用割合(200〜1700g ae/ha、好ましくは350〜500g ae/ha)で適用され得る。現在のところ、グルホシネート抵抗性雑草は確認されていないが、グルホシネートは本質的により耐性の雑草をグリホセートより数多く有する。
i)本質的に耐性の広葉雑草種(例えば、シルシウム・アーベンシス、アポシナム・カンナビナム、およびコニザ・カンデンシス(Conyza candensis))は、これらのより制御が困難な多年生種の効率的な制御のため、そして一年生広葉雑草種の制御の強固さを改善するため、280〜2240g ae/ha、より好ましくは560〜2240g ae/haの2,4-Dを容器で混合することによって制御され得る。トリクロピルおよびフルロキシピルが、雑草制御計画において考慮することが許容される成分であろう。トリクロピルについては、適用割合は、典型的には70〜1120g ae/ha、より典型的には140〜420g ae/haであろう。フルロキシピルについては、適用割合は、典型的には35〜560g ae/ha、より典型的には70〜280g ae/haの範囲であろう。
b)例えば、グルホシネート(200〜500g ae/ha)+/- 2,4-D(280〜1120g ae/ha)+/- トリクロピルまたはフルロキシピル(上記の割合)の多重組み合わせは、より強固な、重複する雑草制御スペクトルを提供することができる。さらに、重複するスペクトルは、除草剤抵抗性雑草の管理および遅延のための付加的なメカニズムを提供する。
【0214】
実施例16-任意の作物において昆虫抵抗性(IR)または他の提供形質と重ね合わされたDMS-2(v2)
トランスジェニック形質によって供給される作物における昆虫抵抗性は、北米において、または世界中でのトウモロコシおよびワタの生産において普及している(prevelant)。IRおよびHTの合わせた形質を有する市販の製品は、複数の種子企業によって開発されている。これらには、Bt IR形質(例えば、lifesci.sussex.ac.ukウェブサイト, 2006に列挙されるBt毒素)および上述のHTC形質のいずれかまたはそのすべてが含まれる。したがって、この提供物がもたらす価値は、単一の提供物中の遺伝的手段を通して、複数の厄介な問題を制御する能力を含む。雑草制御と昆虫制御とが互いと独立して達成される場合、この提供物の便利さは限定される。単独の、または1つもしくは複数のさらなるHTC形質と重ね合わされたDSM-2(v2)は、従来的な育種を通して、または新たな形質転換事象として合同してのいずれかで、1つもしくは複数のインプット形質(例えば、昆虫抵抗性、真菌抵抗性、またはストレス耐性など)(isb.vt.edu/cfdocs/fieldtestsl. cfm, 2005)と重ね合わせることができる。利点には、DSM-2(v2)および関連する除草剤耐性によって提供される雑草制御の改善に加えて、害虫および/または他の農学的なストレスを管理する能力とともに、上記の実施例11〜15に記載される簡便さおよび柔軟性が含まれる。従って、本発明は、任意の数の農学的な問題を柔軟かつ費用効率よく制御する能力を有する、作物品質の改善の完全な農学的パッケージを提供するために使用することができる。
【0215】
IRおよびHTの合わせた形質は、大部分の農学的および園芸用/観賞用作物および林業における用途を有する。DSM-2(v2)およびその釣り合った除草剤耐性ならびに任意の数のBtまたは非Bt IR遺伝子によって付与される昆虫抵抗性は、実施例10に列挙されたがこれらに限定されない作物種に適用できる。雑草制御の分野の当業者は、対応するHT形質またはIR形質との重ね合わせが、例えば、従来的な育種または遺伝子操作によって達成できることを認識している。
【0216】
実施例17-さらなる遺伝子の組み合わせ
本発明は、より広く、より強固な雑草制御および除草剤抵抗性管理オプションと適合性の除草剤耐性植物を提供するため、グリホセート、ALS(イミダゾリノン、スルホニル尿素)、アリールオキシアルカノエート、HPPD、PPO、およびグルホシネートに対する抵抗性遺伝子を含むがこれらに限定はされない、一つまたは複数の他の除草剤抵抗性遺伝子と共に「重ね合わせられた」本発明の一つまたは複数の酵素を産生する植物も含む。本発明は、さらに、本明細書において例示された遺伝子およびタンパク質のホモログを利用する方法および組成物を含む。
【0217】
いくつかの態様において、本発明は、ビアラホス、ホスフィノトリシン、またはグルホシネートと、一つまたは複数の市販されている除草剤(例えば、グリホセート、グルホシネート、パラコート、ALS阻害剤(例えば、スルホニル尿素、イミダゾリノン、トリアゾロピリミジンスルホンアニリド等)、HPPD阻害剤(例えば、メソトリオン、イソキサフルトル等)、2,4-D、フルロキシピル、トリコプリル、ジカンバ、ブロモキシニル、アリールオキシフェノキシプロピオネート他)とに耐性の単子葉植物および双子葉植物を提供する。そのような除草剤耐性を担う核酸配列を含むベクターも開示され、雑草制御および雑草集団シフトの防止のため、そのような耐性植物および除草剤の組み合わせを使用する方法も開示される。本発明は、除草剤の新規の組み合わせを新たな方式で使用することを可能にする。さらに、本発明は、グリホセートのような一つまたは複数の除草剤に抵抗性の雑草の系統の発達を防止し、それらの系統を制御する新規の方法を提供する。本発明は、(グルホシネートのような)除草剤に対して通常であれば感受性であろう植物のための種子を植え付ける直前の、植え付けられる区域への植え付け前適用を含む、除草剤および作物の新規の組み合わせの新規の使用を可能にする。
【0218】
本発明のDSM-2遺伝子は、当技術分野において周知であるグルホシネート耐性のための付加的な機序のため、一つまたは複数のpat/bar遺伝子と重ね合わせられてもよい。
【0219】
HPPD(ヒドロキシルフェニルピルビン酸ジオキシゲナーゼ)をコードする遺伝子の植物における使用については、例えば、米国特許第6,268,549号および第7,297,541号を参照されたい。そのような「重ね合わせられた」植物は、グルホシネート抵抗性のための他の遺伝子と組み合わせられ得、そのような重ね合わせられた植物(および様々なその他の植物および本発明の重ね合わせられた植物)は、グリホセート抵抗性の発達を防止するために使用され得る。
【0220】
グリホセートN-アセチルトランスフェラーゼ(GAT)活性を有する酵素をコードする遺伝子も、本発明のDSM-2遺伝子と共に使用され得る(重ね合わせられ得る)。例えば、Castle et al.(2004), 「Discovery of Directed Evolution of a Glyphosate Tolerance Gene」, Science Vol. 34, pp. 1151-1154;およびWO 2002/36782を参照されたい。
【0221】
本発明のDSM-2遺伝子は、それぞれWO 2005/107437、WO 2007/053482、およびUSSN 60/928,303のAAD-1遺伝子およびAAD-12遺伝子およびAAD-13遺伝子と重ね合わせられ得、それらに開示されるように、いくつかの好ましい態様において、グリホセート抵抗性に対抗するために使用され得る。
【0222】
配列の簡単な説明
SEQ ID NO:1は、ネイティブDSM-2の配列である。
SEQ ID NO:2は、ネイティブタンパク質の配列である。
SEQ ID NO:3は、ヘミコット(Hemicot)DSM-2(v2)の配列である。
SEQ ID NO:4は、再構築されたタンパク質の配列である。
SEQ ID NO:5は、Pat PTUプライマー(MAS 123)である。
SEQ ID NO:6は、Pat PTUプライマー(Per 5-4)である。
SEQ ID NO:7は、Patコード領域プライマーである。
SEQ ID NO:8は、Patコード領域プライマーである。
SEQ ID NO:9は、DSM-2(v2)コード領域プライマーである。
SEQ ID NO:10は、DSM-2(v2)コード領域プライマーである。
【特許請求の範囲】
【請求項1】
ホスフィノトリシンアセチルトランスフェラーゼ活性を有するタンパク質をコードするポリヌクレオチドを含むトランスジェニック植物細胞であって、該ポリヌクレオチドが、65℃で6×SSCという条件の下で、SEQ ID NO:2をコードする核酸プローブの完全な相補体とハイブリダイズする、トランスジェニック植物細胞。
【請求項2】
プローブがSEQ ID NO:1およびSEQ ID NO:3からなる群より選択される配列を含む、請求項1記載の細胞。
【請求項3】
タンパク質がSEQ ID NO:2のアミノ酸配列と少なくとも95%の配列同一性を有する、請求項1記載の細胞。
【請求項4】
植物細胞において機能的なプロモーターおよび該プロモーターに機能的に連結されたポリヌクレオチドを含むベクターであって、該ポリヌクレオチドが、ホスフィノトリシンアセチルトランスフェラーゼ活性を有するタンパク質をコードし、かつ該ポリヌクレオチドが、65℃で6×SSCという条件の下で、SEQ ID NO:2のアミノ酸配列をコードする核酸プローブの完全な相補体とハイブリダイズする、ベクター。
【請求項5】
複数の植物細胞に請求項4記載のベクターを提供する工程、および該ベクターを含まない細胞を殺傷しながらまたはそれらの増殖を阻害しながら、前記ポリヌクレオチドを発現する細胞が増殖することを可能にする除草剤の濃度において該複数の細胞を増殖させる工程を含む、請求項1記載の植物細胞を選択する方法であって、該除草剤がホスフィノトリシンを含む、方法。
【請求項6】
除草剤がビアロホスおよびグルホシネートからなる群より選択される、請求項5記載の方法。
【請求項7】
形質転換植物細胞を同定しかつ再生させる工程をさらに含む、請求項5記載の方法。
【請求項8】
複数の請求項1記載の細胞を含む植物。
【請求項9】
請求項8記載の植物の種子。
【請求項10】
バチルス・チューリンゲンシス(Bacillus thuringiensis)、フォトラブダス(Photorhabdus)、およびゼノラブダス(Xenorhabdus)からなる群より選択される生物に由来する昆虫抵抗性遺伝子をさらに含む、請求項1記載の植物細胞。
【請求項11】
第2の除草剤抵抗性遺伝子をさらに含む、請求項1記載の植物細胞。
【請求項12】
前記タンパク質を産生する、請求項8記載の植物。
【請求項13】
タンパク質がSEQ ID NO:2を含む、請求項12記載の植物。
【請求項14】
請求項4のベクターを用いる形質転換に複数の植物細胞を供する工程、培地中で該細胞を培養する工程、該細胞をホスフィノトリシンに曝す工程、および該細胞がホスフィノトリシンに対して抵抗性であるか否かを判定する工程を含む、植物細胞における抵抗性マーカーとしてホスフィノトリシン抵抗性遺伝子を使用する方法。
【請求項15】
請求項4記載のベクターを用いて植物細胞を形質転換する工程、および形質転換植物細胞を繁殖物を産生する植物体へと再生させる工程を含む、ホスフィノトリシン抵抗性の植物細胞、植物体、およびそれらの繁殖物を作成する方法。
【請求項16】
植物のゲノムに請求項4記載のベクターを組み入れる工程、およびホスフィノトリシン抵抗性について選択する工程を含む、ホスフィノトリシン抵抗性植物の作製の方法。
【請求項17】
(a)出発植物細胞のポリメラーゼによって認識されるプロモーター、および
(b)グルタミンシンセターゼ阻害剤を産生するストレプトマイセス・セリカラー(Streptomyces coelicolor)A3微生物に由来するDNAフラグメントを含むコード領域
を含む組換えDNAを出発植物細胞の核ゲノムに組み入れる工程を含む、ホスフィノトリシンまたはホスフィノトリシン部分を有する化合物を含むグルタミンシンセターゼ阻害剤の除草活性に対して耐性である植物細胞を作製する方法であって、該DNAフラグメントが、該グルタミンシンセターゼ阻害剤に対するアセチルトランスフェラーゼ活性を有するタンパク質をコードする、方法。
【請求項18】
ホスフィノトリシンまたはホスフィノトリシン部分を有する化合物を含むグルタミンシンセターゼ阻害剤の除草活性に対して耐性である植物を作製する方法であって、
(a)請求項1記載の植物細胞を作製する工程、および
(b)該細胞から、核ゲノムに前記ポリヌクレオチドを含む植物体を再生させる工程
を含む、方法。
【請求項19】
雑草を駆除することにより、圃場における栽培植物の群を保護する方法であって、該植物が、その細胞のゲノムに請求項4記載のベクターが組み入れられており、該雑草が、活性成分としてグルタミンシンセターゼ阻害剤を含む除草剤の適用によって駆除される、方法。
【請求項20】
(i)
(a)該出発植物細胞のポリメラーゼによって認識されるプロモーター、および
(b)ホスフィノトリシンまたはホスフィノトリシン部分を有する化合物を含むグルタミンシンセターゼ阻害剤を産生するストレプトマイセス・セリカラーA3微生物に由来するDNAフラグメントを含むコード領域
を含む外来DNAを用いて、植物細胞培養物中の出発植物細胞を形質転換する工程;ならびに
(ii)非形質転換植物細胞を殺傷するために十分な濃度のグルタミンシンセターゼ阻害剤を植物細胞培養物に適用することにより形質転換植物細胞を選択する工程
を含む、核ゲノムに組み入れられた外来DNAを有する形質転換植物細胞の純粋培養物を作製する方法。
【請求項21】
変異タンパク質を作製するためにSEQ ID NO:2を含むタンパク質を修飾する工程、およびホスフィノトリシンアセチルトランスフェラーゼ活性について該変異タンパク質を分析する工程を含む、ホスフィノトリシンアセチルトランスフェラーゼ活性を有する合成タンパク質を作製する方法。
【請求項22】
複数の修飾ポリヌクレオチドを作出するためにSEQ ID NO:1およびSEQ ID NO:3からなる群より選択されるポリヌクレオチドを修飾する工程、ならびに該修飾ポリヌクレオチドを遺伝子シャッフリング手法において使用する工程を含む、請求項21記載の方法。
【請求項23】
SEQ ID NO:1およびSEQ ID NO:3からなる群より選択されるポリヌクレオチドをエラープローンPCRにより修飾する工程を含む、請求項21記載の方法。
【請求項24】
前記アミノ酸配列のうちの少なくとも一つをコードする少なくとも一つのポリヌクレオチドを修飾する工程を含む、請求項21記載の方法。
【請求項25】
請求項4記載のベクターを含む、ストレプトマイセス以外の細菌細胞。
【請求項26】
(a)複数の植物細胞に請求項4記載のベクターを提供する工程;および
(b)該ベクターを欠く細胞を殺傷しながらまたはそれらの増殖を阻害しながら該ポリヌクレオチドを発現する細胞が増殖することを可能にする除草剤の濃度において該複数の細胞を増殖させる工程
を含む、請求項4記載のベクターを含む植物細胞を選択する方法であって、
該除草剤がホスフィノトリシンを含む、方法。
【請求項27】
発現カセットが低コピー数バイナリーベクターの中に存在する、請求項26記載の方法。
【請求項28】
除草剤がビアロホスおよびグルホシネートからなる群より選択される、請求項26記載の方法。
【請求項29】
細菌発現系、植物細胞培養物発現系、藻類発現系、動物細胞発現系、ウイルス発現系、真菌発現系、および酵母発現系からなる群より選択される発現系において使用される、請求項26記載の方法。
【請求項30】
発現系がシュードモナス・フルオレセンス(Pseudomonas fluorescens)発現系である、請求項29記載の方法。
【請求項31】
AAD-1およびAAD-12からなる群より選択される除草剤抵抗性遺伝子をさらに含む、請求項1記載のトランスジェニック植物細胞。
【請求項32】
圃場にグリホセートを適用する工程をさらに含む方法であって、植物がグリホセート抵抗性遺伝子をさらに含む、請求項19記載の方法。
【請求項33】
圃場にイミダゾリノン除草剤を適用する工程をさらに含む方法であって、植物がイミダゾリノン抵抗性遺伝子をさらに含む、請求項19記載の方法。
【請求項34】
圃場に2,4-Dを適用する工程をさらに含む方法であって、植物がAAD-1およびAAD-12からなる群より選択される除草剤抵抗性遺伝子をさらに含む、請求項19記載の方法。
【請求項35】
圃場にアリールオキシフェノキシプロピオネート除草剤を適用する工程をさらに含む方法であって、植物がAAD-1除草剤抵抗性遺伝子をさらに含む、請求項19記載の方法。
【請求項36】
圃場にピリジルオキシアセテート除草剤を適用する工程をさらに含む方法であって、植物がAAD-12除草剤抵抗性遺伝子をさらに含む、請求項19記載の方法。
【請求項37】
DSM-2遺伝子を含有している個々の植物または作物植物の群を、該遺伝子を含有していない同一種の植物の集団から選択または区別する方法。
【請求項38】
植物の集合にグルホシネートまたはビアロホスを適用する工程を含む、請求項37記載の方法。
【請求項39】
温室、グロースチャンバー、または圃場において実施される、請求項37記載の方法。
【請求項40】
少なくとも1種類の請求項12記載のトランスジェニック植物の種子を圃場に植える工程を含み、
該圃場の少なくとも一部に、グルホシネート、ホスフィノトリシン、およびビアロホスからなる群より選択される第1の除草剤を適用する工程;ならびに
該圃場の少なくとも一部に少なくとも1種類の他の除草剤を適用する工程
をさらに含む、
圃場において少なくとも1種類の雑草を制御する方法であって、
該植物が、
グルタミンシンセターゼ阻害除草剤に対する抵抗性を付与する酵素をコードする異種ポリヌクレオチド、および
少なくとも1種類の他の除草剤に対する抵抗性を付与する酵素をコードする第2の異種ポリヌクレオチド
を含む、方法。
【請求項41】
除草剤が連続的にまたは同時に適用される、請求項40記載の方法。
【請求項42】
第1の除草剤がグルタミンシンセターゼ阻害除草剤である、請求項40記載の方法。
【請求項43】
少なくとも1種類の他の除草剤が、2,4-D、アセトクロル、アシフルオルフェン、アロキシジム、アミドスルフロン、アミノピラリド、アトラジン、ベフルブタミド、ビスピリバック、ブタフェナシル、カフェンストロール、カルフェントラゾン、クロリムロン、クロロトルロン、シニドン-エチル、クレトジム、クロジナホップ、クロマゾン、クロプロキシジム(cloproxydim)、クロピラリド、クロランスラム、シアナジン、シクロスルファムロン、シクロキシジム、シハロホップ、ダイムロン、ジカンバ、ジクロホップ、ジクロルプロップ、ジクロスラム、ジフルフェニカン、ジメテナミド、ジクワット、ジチオピル、ジウロン、エタルフルラリン、フェノキサプロップ、フラザスルフロン、フロラスラム、フルアジホップ、フルカルバゾン、フルフェナセット、フルフェニカン、フルフェンピル(flufenpyr)、フルメツラム、フルミクロラック、フルミオキサジン、フルロキシピル、フルチアセト、ホメサフェン、ホラムスルフロン、グルホシネート、グリホセート、ハロサフェン(halosafen)、ハロスルフロン、ハロキシホップ、イマザメタベンズ、イマザモクス、イマザピク、イマザピル、イマザキン、イマゼタピル、イマゾスルフロン、ヨードスルフロン、イオキシニル、イソキサベン、イソキサフルトール、ラクトフェン、リヌロン、MCPA、メコプロップ、メフェナセット、メフルイジド、メソスルフロン、メソトリオン、メタミホップ、メタザクロル、メトスラム、メトリブジン、MSMA、ナプロパミド、ニコスルフロン、ノルフルラゾン、オリザリン、オキサジアゾン、オキシフルオルフェン、パラコート、ペブラート、ペンジメタリン、ペノクススラム、ピクロラム、ピコリナフェン(picolinafen)、ピノキサデン(pinoxaden)、プリミスルフロン、プロホキシジム、プロパニル、ピラフルフェン、ピラゾスルフロン、ピリベンゾキシム、ピリミノバック、ピリチオバック、ピロキサスルホン、ピロクススラム(pyroxsulam)、キンクロラック、キンメラック、キザロホップ、リムスルフロン、サフルフェナシル、セトキシジム、シマジン、スルコトリオン、スルフェントラゾン、スルホメツロン、テフリルトリオン(tefuryltrione)、テムボトリオン(tembotrione)、テプラロキシジム、テルバシル、チアゾピル、チジアズロン、チエンカルバゾン(thiencarbazone)、チフェンスルフロン、チオベンカルブ、トプラメゾン、トラルコキシジム、トリアスルフロン、トリベヌロン、トリクロピル、トリフロキシスルフロン、トリフルラリン、トリフルスルフロン、およびトリトスルフロン(tritosulfuron)からなる群より選択される、請求項40記載の方法。
【請求項44】
植物から試料を収集する工程、および前記ポリヌクレオチドの存在について該試料を分析する工程を含む、植物が請求項8記載のポリヌクレオチドを含むか否かを検出する方法。
【請求項45】
前記ポリヌクレオチドによってコードされたタンパク質の存在について試料を分析する工程を含む、請求項44記載の方法。
【請求項46】
前記ポリヌクレオチドの存在について検出するためにPCRプライマーまたはプローブを使用する工程を含む、請求項44記載の方法。
【請求項47】
前記タンパク質の存在について検出するために抗体を使用する工程を含む、請求項44記載の方法。
【請求項48】
バチルス・チューリンゲンシス、フォトラブダス、およびゼノラブダスからなる群より選択される生物に由来する昆虫抵抗性遺伝子をさらに含む、請求項8記載の植物。
【請求項49】
真菌抵抗性、ストレス耐性、収量の増加、油の性質の改善、繊維品質の改善、ウイルス抵抗性、熟成の遅延、低温耐性、および塩耐性からなる群より選択される農業形質のための遺伝子をさらに含む、請求項8記載の植物。
【請求項50】
少なくとも1種類の請求項8記載の植物を圃場において生長させる工程、および該圃場の少なくとも一部にグルタミンシンセターゼ阻害除草剤を適用する工程を含む、圃場において少なくとも1種類の雑草を制御する方法。
【請求項51】
除草剤グルホシネート、ビアロホス、およびホスフィノトリシンをさらに含む、請求項50記載の方法。
【請求項52】
植物が、グリホセート、グルホシネート、2,4-D、キザロホップ、イマゼタピル、クロルスルフロン、ジカンバ、メソトリオン、イソキサフルトール、およびブタフェナシルからなる群より選択される除草剤に対して抵抗性である、請求項50記載の方法。
【請求項53】
植物が単子葉植物である、請求項24記載の方法。
【請求項54】
単子葉植物が、トウモロコシ、イネ、コムギ、オオムギ、ライムギ、サトウキビ、暖地型および寒地型の芝草、オートムギ、ソルガム、および牧草からなる群より選択される、請求項53記載の方法。
【請求項55】
植物が双子葉植物である、請求項24記載の方法。
【請求項56】
温室、グロースチャンバー、または圃場において実施される方法であって、DSM-2遺伝子が選択マーカーとして機能する、請求項37記載の方法。
【請求項57】
ポリヌクレオチドが、植物における発現を増加させるためのコドン使用頻度を有する、請求項12記載の植物。
【請求項58】
ホスフィノトリシンアセチルトランスフェラーゼ活性を有するタンパク質をコードするポリヌクレオチドを含むトランスジェニックダイズ植物であって、該ポリヌクレオチドが、65℃で6×SSCという条件の下で、SEQ ID NO:2をコードする核酸プローブの完全な相補体とハイブリダイズする、トランスジェニックダイズ植物。
【請求項59】
ホスフィノトリシンアセチルトランスフェラーゼ活性を有するタンパク質をコードするポリヌクレオチドを含むトランスジェニックトウモロコシ植物であって、該ポリヌクレオチドが、65℃で6×SSCという条件の下で、SEQ ID NO:2をコードする核酸プローブの完全な相補体とハイブリダイズする、トランスジェニックトウモロコシ植物。
【請求項60】
請求項58または59記載のトランスジェニック植物を作製するために、植物における発現を増加させるためのコドン使用頻度を有するポリヌクレオチドを使用する方法。
【請求項61】
作物植物にポリヌクレオチドを組み込む工程を含む、植物に除草剤耐性を伝達するためにタンパク質を使用する方法であって、該ポリヌクレオチドが該タンパク質をコードし、かつ該タンパク質がホスフィノトリシンアセチルトランスフェラーゼ活性を有し、該ポリヌクレオチドが、65℃で6×SSCという条件の下で、SEQ ID NO:2をコードする核酸プローブの完全な相補体とハイブリダイズし、かつ該ポリヌクレオチドの発現および該タンパク質の産生が該植物をホスフィノトリシン除草剤耐性にする、方法。
【請求項1】
ホスフィノトリシンアセチルトランスフェラーゼ活性を有するタンパク質をコードするポリヌクレオチドを含むトランスジェニック植物細胞であって、該ポリヌクレオチドが、65℃で6×SSCという条件の下で、SEQ ID NO:2をコードする核酸プローブの完全な相補体とハイブリダイズする、トランスジェニック植物細胞。
【請求項2】
プローブがSEQ ID NO:1およびSEQ ID NO:3からなる群より選択される配列を含む、請求項1記載の細胞。
【請求項3】
タンパク質がSEQ ID NO:2のアミノ酸配列と少なくとも95%の配列同一性を有する、請求項1記載の細胞。
【請求項4】
植物細胞において機能的なプロモーターおよび該プロモーターに機能的に連結されたポリヌクレオチドを含むベクターであって、該ポリヌクレオチドが、ホスフィノトリシンアセチルトランスフェラーゼ活性を有するタンパク質をコードし、かつ該ポリヌクレオチドが、65℃で6×SSCという条件の下で、SEQ ID NO:2のアミノ酸配列をコードする核酸プローブの完全な相補体とハイブリダイズする、ベクター。
【請求項5】
複数の植物細胞に請求項4記載のベクターを提供する工程、および該ベクターを含まない細胞を殺傷しながらまたはそれらの増殖を阻害しながら、前記ポリヌクレオチドを発現する細胞が増殖することを可能にする除草剤の濃度において該複数の細胞を増殖させる工程を含む、請求項1記載の植物細胞を選択する方法であって、該除草剤がホスフィノトリシンを含む、方法。
【請求項6】
除草剤がビアロホスおよびグルホシネートからなる群より選択される、請求項5記載の方法。
【請求項7】
形質転換植物細胞を同定しかつ再生させる工程をさらに含む、請求項5記載の方法。
【請求項8】
複数の請求項1記載の細胞を含む植物。
【請求項9】
請求項8記載の植物の種子。
【請求項10】
バチルス・チューリンゲンシス(Bacillus thuringiensis)、フォトラブダス(Photorhabdus)、およびゼノラブダス(Xenorhabdus)からなる群より選択される生物に由来する昆虫抵抗性遺伝子をさらに含む、請求項1記載の植物細胞。
【請求項11】
第2の除草剤抵抗性遺伝子をさらに含む、請求項1記載の植物細胞。
【請求項12】
前記タンパク質を産生する、請求項8記載の植物。
【請求項13】
タンパク質がSEQ ID NO:2を含む、請求項12記載の植物。
【請求項14】
請求項4のベクターを用いる形質転換に複数の植物細胞を供する工程、培地中で該細胞を培養する工程、該細胞をホスフィノトリシンに曝す工程、および該細胞がホスフィノトリシンに対して抵抗性であるか否かを判定する工程を含む、植物細胞における抵抗性マーカーとしてホスフィノトリシン抵抗性遺伝子を使用する方法。
【請求項15】
請求項4記載のベクターを用いて植物細胞を形質転換する工程、および形質転換植物細胞を繁殖物を産生する植物体へと再生させる工程を含む、ホスフィノトリシン抵抗性の植物細胞、植物体、およびそれらの繁殖物を作成する方法。
【請求項16】
植物のゲノムに請求項4記載のベクターを組み入れる工程、およびホスフィノトリシン抵抗性について選択する工程を含む、ホスフィノトリシン抵抗性植物の作製の方法。
【請求項17】
(a)出発植物細胞のポリメラーゼによって認識されるプロモーター、および
(b)グルタミンシンセターゼ阻害剤を産生するストレプトマイセス・セリカラー(Streptomyces coelicolor)A3微生物に由来するDNAフラグメントを含むコード領域
を含む組換えDNAを出発植物細胞の核ゲノムに組み入れる工程を含む、ホスフィノトリシンまたはホスフィノトリシン部分を有する化合物を含むグルタミンシンセターゼ阻害剤の除草活性に対して耐性である植物細胞を作製する方法であって、該DNAフラグメントが、該グルタミンシンセターゼ阻害剤に対するアセチルトランスフェラーゼ活性を有するタンパク質をコードする、方法。
【請求項18】
ホスフィノトリシンまたはホスフィノトリシン部分を有する化合物を含むグルタミンシンセターゼ阻害剤の除草活性に対して耐性である植物を作製する方法であって、
(a)請求項1記載の植物細胞を作製する工程、および
(b)該細胞から、核ゲノムに前記ポリヌクレオチドを含む植物体を再生させる工程
を含む、方法。
【請求項19】
雑草を駆除することにより、圃場における栽培植物の群を保護する方法であって、該植物が、その細胞のゲノムに請求項4記載のベクターが組み入れられており、該雑草が、活性成分としてグルタミンシンセターゼ阻害剤を含む除草剤の適用によって駆除される、方法。
【請求項20】
(i)
(a)該出発植物細胞のポリメラーゼによって認識されるプロモーター、および
(b)ホスフィノトリシンまたはホスフィノトリシン部分を有する化合物を含むグルタミンシンセターゼ阻害剤を産生するストレプトマイセス・セリカラーA3微生物に由来するDNAフラグメントを含むコード領域
を含む外来DNAを用いて、植物細胞培養物中の出発植物細胞を形質転換する工程;ならびに
(ii)非形質転換植物細胞を殺傷するために十分な濃度のグルタミンシンセターゼ阻害剤を植物細胞培養物に適用することにより形質転換植物細胞を選択する工程
を含む、核ゲノムに組み入れられた外来DNAを有する形質転換植物細胞の純粋培養物を作製する方法。
【請求項21】
変異タンパク質を作製するためにSEQ ID NO:2を含むタンパク質を修飾する工程、およびホスフィノトリシンアセチルトランスフェラーゼ活性について該変異タンパク質を分析する工程を含む、ホスフィノトリシンアセチルトランスフェラーゼ活性を有する合成タンパク質を作製する方法。
【請求項22】
複数の修飾ポリヌクレオチドを作出するためにSEQ ID NO:1およびSEQ ID NO:3からなる群より選択されるポリヌクレオチドを修飾する工程、ならびに該修飾ポリヌクレオチドを遺伝子シャッフリング手法において使用する工程を含む、請求項21記載の方法。
【請求項23】
SEQ ID NO:1およびSEQ ID NO:3からなる群より選択されるポリヌクレオチドをエラープローンPCRにより修飾する工程を含む、請求項21記載の方法。
【請求項24】
前記アミノ酸配列のうちの少なくとも一つをコードする少なくとも一つのポリヌクレオチドを修飾する工程を含む、請求項21記載の方法。
【請求項25】
請求項4記載のベクターを含む、ストレプトマイセス以外の細菌細胞。
【請求項26】
(a)複数の植物細胞に請求項4記載のベクターを提供する工程;および
(b)該ベクターを欠く細胞を殺傷しながらまたはそれらの増殖を阻害しながら該ポリヌクレオチドを発現する細胞が増殖することを可能にする除草剤の濃度において該複数の細胞を増殖させる工程
を含む、請求項4記載のベクターを含む植物細胞を選択する方法であって、
該除草剤がホスフィノトリシンを含む、方法。
【請求項27】
発現カセットが低コピー数バイナリーベクターの中に存在する、請求項26記載の方法。
【請求項28】
除草剤がビアロホスおよびグルホシネートからなる群より選択される、請求項26記載の方法。
【請求項29】
細菌発現系、植物細胞培養物発現系、藻類発現系、動物細胞発現系、ウイルス発現系、真菌発現系、および酵母発現系からなる群より選択される発現系において使用される、請求項26記載の方法。
【請求項30】
発現系がシュードモナス・フルオレセンス(Pseudomonas fluorescens)発現系である、請求項29記載の方法。
【請求項31】
AAD-1およびAAD-12からなる群より選択される除草剤抵抗性遺伝子をさらに含む、請求項1記載のトランスジェニック植物細胞。
【請求項32】
圃場にグリホセートを適用する工程をさらに含む方法であって、植物がグリホセート抵抗性遺伝子をさらに含む、請求項19記載の方法。
【請求項33】
圃場にイミダゾリノン除草剤を適用する工程をさらに含む方法であって、植物がイミダゾリノン抵抗性遺伝子をさらに含む、請求項19記載の方法。
【請求項34】
圃場に2,4-Dを適用する工程をさらに含む方法であって、植物がAAD-1およびAAD-12からなる群より選択される除草剤抵抗性遺伝子をさらに含む、請求項19記載の方法。
【請求項35】
圃場にアリールオキシフェノキシプロピオネート除草剤を適用する工程をさらに含む方法であって、植物がAAD-1除草剤抵抗性遺伝子をさらに含む、請求項19記載の方法。
【請求項36】
圃場にピリジルオキシアセテート除草剤を適用する工程をさらに含む方法であって、植物がAAD-12除草剤抵抗性遺伝子をさらに含む、請求項19記載の方法。
【請求項37】
DSM-2遺伝子を含有している個々の植物または作物植物の群を、該遺伝子を含有していない同一種の植物の集団から選択または区別する方法。
【請求項38】
植物の集合にグルホシネートまたはビアロホスを適用する工程を含む、請求項37記載の方法。
【請求項39】
温室、グロースチャンバー、または圃場において実施される、請求項37記載の方法。
【請求項40】
少なくとも1種類の請求項12記載のトランスジェニック植物の種子を圃場に植える工程を含み、
該圃場の少なくとも一部に、グルホシネート、ホスフィノトリシン、およびビアロホスからなる群より選択される第1の除草剤を適用する工程;ならびに
該圃場の少なくとも一部に少なくとも1種類の他の除草剤を適用する工程
をさらに含む、
圃場において少なくとも1種類の雑草を制御する方法であって、
該植物が、
グルタミンシンセターゼ阻害除草剤に対する抵抗性を付与する酵素をコードする異種ポリヌクレオチド、および
少なくとも1種類の他の除草剤に対する抵抗性を付与する酵素をコードする第2の異種ポリヌクレオチド
を含む、方法。
【請求項41】
除草剤が連続的にまたは同時に適用される、請求項40記載の方法。
【請求項42】
第1の除草剤がグルタミンシンセターゼ阻害除草剤である、請求項40記載の方法。
【請求項43】
少なくとも1種類の他の除草剤が、2,4-D、アセトクロル、アシフルオルフェン、アロキシジム、アミドスルフロン、アミノピラリド、アトラジン、ベフルブタミド、ビスピリバック、ブタフェナシル、カフェンストロール、カルフェントラゾン、クロリムロン、クロロトルロン、シニドン-エチル、クレトジム、クロジナホップ、クロマゾン、クロプロキシジム(cloproxydim)、クロピラリド、クロランスラム、シアナジン、シクロスルファムロン、シクロキシジム、シハロホップ、ダイムロン、ジカンバ、ジクロホップ、ジクロルプロップ、ジクロスラム、ジフルフェニカン、ジメテナミド、ジクワット、ジチオピル、ジウロン、エタルフルラリン、フェノキサプロップ、フラザスルフロン、フロラスラム、フルアジホップ、フルカルバゾン、フルフェナセット、フルフェニカン、フルフェンピル(flufenpyr)、フルメツラム、フルミクロラック、フルミオキサジン、フルロキシピル、フルチアセト、ホメサフェン、ホラムスルフロン、グルホシネート、グリホセート、ハロサフェン(halosafen)、ハロスルフロン、ハロキシホップ、イマザメタベンズ、イマザモクス、イマザピク、イマザピル、イマザキン、イマゼタピル、イマゾスルフロン、ヨードスルフロン、イオキシニル、イソキサベン、イソキサフルトール、ラクトフェン、リヌロン、MCPA、メコプロップ、メフェナセット、メフルイジド、メソスルフロン、メソトリオン、メタミホップ、メタザクロル、メトスラム、メトリブジン、MSMA、ナプロパミド、ニコスルフロン、ノルフルラゾン、オリザリン、オキサジアゾン、オキシフルオルフェン、パラコート、ペブラート、ペンジメタリン、ペノクススラム、ピクロラム、ピコリナフェン(picolinafen)、ピノキサデン(pinoxaden)、プリミスルフロン、プロホキシジム、プロパニル、ピラフルフェン、ピラゾスルフロン、ピリベンゾキシム、ピリミノバック、ピリチオバック、ピロキサスルホン、ピロクススラム(pyroxsulam)、キンクロラック、キンメラック、キザロホップ、リムスルフロン、サフルフェナシル、セトキシジム、シマジン、スルコトリオン、スルフェントラゾン、スルホメツロン、テフリルトリオン(tefuryltrione)、テムボトリオン(tembotrione)、テプラロキシジム、テルバシル、チアゾピル、チジアズロン、チエンカルバゾン(thiencarbazone)、チフェンスルフロン、チオベンカルブ、トプラメゾン、トラルコキシジム、トリアスルフロン、トリベヌロン、トリクロピル、トリフロキシスルフロン、トリフルラリン、トリフルスルフロン、およびトリトスルフロン(tritosulfuron)からなる群より選択される、請求項40記載の方法。
【請求項44】
植物から試料を収集する工程、および前記ポリヌクレオチドの存在について該試料を分析する工程を含む、植物が請求項8記載のポリヌクレオチドを含むか否かを検出する方法。
【請求項45】
前記ポリヌクレオチドによってコードされたタンパク質の存在について試料を分析する工程を含む、請求項44記載の方法。
【請求項46】
前記ポリヌクレオチドの存在について検出するためにPCRプライマーまたはプローブを使用する工程を含む、請求項44記載の方法。
【請求項47】
前記タンパク質の存在について検出するために抗体を使用する工程を含む、請求項44記載の方法。
【請求項48】
バチルス・チューリンゲンシス、フォトラブダス、およびゼノラブダスからなる群より選択される生物に由来する昆虫抵抗性遺伝子をさらに含む、請求項8記載の植物。
【請求項49】
真菌抵抗性、ストレス耐性、収量の増加、油の性質の改善、繊維品質の改善、ウイルス抵抗性、熟成の遅延、低温耐性、および塩耐性からなる群より選択される農業形質のための遺伝子をさらに含む、請求項8記載の植物。
【請求項50】
少なくとも1種類の請求項8記載の植物を圃場において生長させる工程、および該圃場の少なくとも一部にグルタミンシンセターゼ阻害除草剤を適用する工程を含む、圃場において少なくとも1種類の雑草を制御する方法。
【請求項51】
除草剤グルホシネート、ビアロホス、およびホスフィノトリシンをさらに含む、請求項50記載の方法。
【請求項52】
植物が、グリホセート、グルホシネート、2,4-D、キザロホップ、イマゼタピル、クロルスルフロン、ジカンバ、メソトリオン、イソキサフルトール、およびブタフェナシルからなる群より選択される除草剤に対して抵抗性である、請求項50記載の方法。
【請求項53】
植物が単子葉植物である、請求項24記載の方法。
【請求項54】
単子葉植物が、トウモロコシ、イネ、コムギ、オオムギ、ライムギ、サトウキビ、暖地型および寒地型の芝草、オートムギ、ソルガム、および牧草からなる群より選択される、請求項53記載の方法。
【請求項55】
植物が双子葉植物である、請求項24記載の方法。
【請求項56】
温室、グロースチャンバー、または圃場において実施される方法であって、DSM-2遺伝子が選択マーカーとして機能する、請求項37記載の方法。
【請求項57】
ポリヌクレオチドが、植物における発現を増加させるためのコドン使用頻度を有する、請求項12記載の植物。
【請求項58】
ホスフィノトリシンアセチルトランスフェラーゼ活性を有するタンパク質をコードするポリヌクレオチドを含むトランスジェニックダイズ植物であって、該ポリヌクレオチドが、65℃で6×SSCという条件の下で、SEQ ID NO:2をコードする核酸プローブの完全な相補体とハイブリダイズする、トランスジェニックダイズ植物。
【請求項59】
ホスフィノトリシンアセチルトランスフェラーゼ活性を有するタンパク質をコードするポリヌクレオチドを含むトランスジェニックトウモロコシ植物であって、該ポリヌクレオチドが、65℃で6×SSCという条件の下で、SEQ ID NO:2をコードする核酸プローブの完全な相補体とハイブリダイズする、トランスジェニックトウモロコシ植物。
【請求項60】
請求項58または59記載のトランスジェニック植物を作製するために、植物における発現を増加させるためのコドン使用頻度を有するポリヌクレオチドを使用する方法。
【請求項61】
作物植物にポリヌクレオチドを組み込む工程を含む、植物に除草剤耐性を伝達するためにタンパク質を使用する方法であって、該ポリヌクレオチドが該タンパク質をコードし、かつ該タンパク質がホスフィノトリシンアセチルトランスフェラーゼ活性を有し、該ポリヌクレオチドが、65℃で6×SSCという条件の下で、SEQ ID NO:2をコードする核酸プローブの完全な相補体とハイブリダイズし、かつ該ポリヌクレオチドの発現および該タンパク質の産生が該植物をホスフィノトリシン除草剤耐性にする、方法。
【図1】

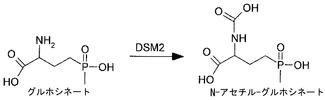
【公表番号】特表2010−512153(P2010−512153A)
【公表日】平成22年4月22日(2010.4.22)
【国際特許分類】
【出願番号】特願2009−540502(P2009−540502)
【出願日】平成19年12月7日(2007.12.7)
【国際出願番号】PCT/US2007/086813
【国際公開番号】WO2008/070845
【国際公開日】平成20年6月12日(2008.6.12)
【出願人】(505412443)ダウ アグロサイエンス リミテッド ライアビリティー カンパニー (10)
【Fターム(参考)】
【公表日】平成22年4月22日(2010.4.22)
【国際特許分類】
【出願日】平成19年12月7日(2007.12.7)
【国際出願番号】PCT/US2007/086813
【国際公開番号】WO2008/070845
【国際公開日】平成20年6月12日(2008.6.12)
【出願人】(505412443)ダウ アグロサイエンス リミテッド ライアビリティー カンパニー (10)
【Fターム(参考)】
[ Back to top ]