
選択的な制限断片増幅:一般的なDNAフィンガプリント法
【課題】本発明は、所定の特定の制限エンドヌクレアーゼに対する制限部位を複数含むと共に、その核酸の少なくとも一部分は未知である出発DNAの少なくとも一部分を制御増幅する方法に係わる。本発明方法は、ヒト、動物または植物のDNAフィンガープリント形成、制限断片長さ多形性の同定に適用される。本発明を適用するためのキットも提供される。
【解決手段】本発明は、生物のDNAを少なくとも1種の制限酵素で切断した後に得られる制限断片をPCR法により増幅させる新規な方法を提供する。この新規なPCR法の使用において、使用するオリゴヌクレオチドは公知のDNA配列には指向せずに制限断片の末端を識別するように設計される。
【解決手段】本発明は、生物のDNAを少なくとも1種の制限酵素で切断した後に得られる制限断片をPCR法により増幅させる新規な方法を提供する。この新規なPCR法の使用において、使用するオリゴヌクレオチドは公知のDNA配列には指向せずに制限断片の末端を識別するように設計される。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、限定はしないが植物および動物の育種、品種もしくは栽培変種の同定、診断医薬、動物および植物における病気の診断、ヒトにおける遺伝病の同定、家族関係の分析、法医学的分析および微生物分類を包含する種々異なる多くの分野におけるDNAフィンガプリント法およびDNAマーカーの使用に関する。
【0002】
より詳細には本発明は、DNAフィンガプリント法(DNA fingerprinting)および微生物から高等な植物、動物およびヒトに至る範囲のゲノムにおける特定DNAマーカーの検出方法に関する。さらに本発明は、種々異なる応用分野にて本発明の方法に使用される合成DNA分子およびそれに基づく生産物に関する。
【背景技術】
【0003】
1.DNAフィンガプリント法
DNAフィンガプリントまたはDNA分類、並びに他の遺伝子型分類や特性化やDNA同定の方法は個人、変種もしくは種族または品種の遺伝子構成もしくはゲノムにおける類似性または1つ以上の区別的な特徴を特性化することを意味する。一般的規則は遺伝子関係が近縁であるほど同一性が大となり或いはゲノムの類似性が一層充分となり、その結果ゲノムにおける区別的な特徴がより稀となる。これらの類似した或いは区別的な特徴は、DNAを制限エンドヌクレアーゼで切断した後に生物のDNAを分析して示すことができる。制限エンドヌクレアーゼは、短いヌクレオチド配列(一般に長さ4〜8塩基)を認識すると共に2個のDNA鎖を切断して別個の長さを有するDNA断片を生成させる酵素である。制限エンドヌクレアーゼは、その高度の配列特異性により、DNA分子を極めて特異的に切断する。その結果、再現しうるDNA断片群が生成される。DNA断片は、その長さに応じ多孔質マトリックスもしくはゲルで分画して典型的なバンドパターンを生じ、生物の遺伝子構成におけるDNAフィンガプリントを構成することができる。
【0004】
2.DNA多形性
極めて近縁の品種、変種もしくは種族のフィンガプリントを比較すると、DNAフィンガプリントは同一であるか或いは極めて類似する。同一のDNAフィンガプリントにも拘らず相違点が観察される場合、これら相違点はDNA多形性と呼ばれる。これらは、フィンガプリントに出現する新たなDNA断片である。DNAはその位置にて多形性であると言われ、この新規なDNA断片はDNAマーカーとして使用することができる。制限酵素切断により得られるDNAフィンガプリントで検出されるDNA多形性は、制限エンドヌクレアーゼ標的部位を破壊する突然変異、新規な標的部位を形成する突然変異、挿入、欠失または2つの制限部位の間の逆位のようなDNA配列における変化から生じうる。
【0005】
この種のDNA多形性は一般にRFLP、すなわち制限断片長さ多形性と呼ばれる。この種の突然変異的変化は、メンデルの法則で遺伝する場合、真正な遺伝子マーカーとして挙動する。その結果、DNA多形性を他の遺伝子マーカーと全く同様に血統分析、形質の遺伝に関する遺伝子研究、個体の同定などに遺伝子マーカーとして使用することができる。
【0006】
3.DNAフィンガプリント技術
ウィルスを除く殆ど全ての生存する生物につき、これら生物の全ゲノムDNAの制限消化物は、個々のバンドを評価することができないほど多くのバンドをもたらす。したがって、全てのDNAフィンガプリント法は、小割合のDNA断片のみを可視化させてDNAフィンガプリントを構成する単純なバンドパターンを得ると言う原理に基づいている。
【0007】
最も広く使用されている方法は、生物のDNAを制限エンドヌクレアーゼで消化し、制限断片をゲル電気泳動により分画し、分画されたDNA断片を膜上に移動させると共に結合させ、次いでこの膜を特定DNA断片(「プローブ」)とハイブリダイズさせることを含む。DNA断片は二本鎖DNA分子を形成すると共に、膜上のDNA断片は相補的ヌクレオチド配列を有する。プローブを可視化可能なマーカーで標識すれば、プローブが結合したDNA断片を可視化することができる。この手法は一般に「サザンハイブリダイゼーション」と呼ばれる。近縁のゲノムDNA分子においてプローブが結合した対応の制限断片の寸法に差が観察されれば、これらの差はDNA多形性、より詳細には制限断片長さ多形性と呼ばれる。制限断片長さの差は、DNAプローブにより認識される遺伝子座の種々異なる対立型に対応する。DNAフィンガプリント法のためのサザンハイブリダイゼーションも広く使用されているが、この方法は面倒であって時間がかかる。
【0008】
さらに、この方法は解像能が低く、せいぜい単一の反応における単一の遺伝子座もしくは2〜3の遺伝子座を評価するにしか使用することができない。
【0009】
4.ポリメラーゼ連鎖反応
ポリメラーゼ連鎖反応(PCR)技術は、インビトロにて特定DNA断片を合成するための方法である。この方法は、DNA分子上の独特の配列に結合する特定のオリゴヌクレオチドおよび熱安定性のDNAポリメラーゼを使用することに基づく。オリゴヌクレオチドは、DNAの対向鎖にアニールしてそれぞれ新たなDNA鎖の合成を指令するようDNA合成反応でプライマとして作用するように設計される。したがって、1ラウンドの合成においてはプライマー間でDNA分子の完全コピーが作成され、プライマー間のDNAが複製される。各ラウンドのDNA合成の結果、二倍量のDNAが生じて2個のプライマー間で構成されるDNAの増幅をもたらす。その結果、PCR技術は少量の「基質DNA」を用いて正確なDNAセグメントを合成することを可能にする。
【発明の開示】
【発明が解決しようとする課題】
【0010】
本発明は、生物のDNAを少なくとも1種の制限酵素で切断した後に得られる制限断片をPCR法により増幅させる新規な方法を提供する。この新規なPCR法の使用において、使用するオリゴヌクレオチドは公知のDNA配列には指向せずに制限断片の末端を識別するように設計される。このためには、オリゴヌクレオチドリンカー(もしくはアダプタ)を制限断片の末端に付加させることにより該末端を改変する必要がある。この理由は、制限酵素の末端が一般に極く僅かのヌクレオチド(すなわち2〜8個のヌクレオチド)しか持たず、PCR増幅用のプライマーを設計するために使用するには短か過ぎるからである。
【0011】
本発明は、ゲノムDNA分子を制限エンドヌクレアーゼで消化して得られたDNA断片の複雑な混合物から1個もしくはそれ以上の制限断片を増幅させるためのポリメラーゼ連鎖反応技術(PCR)の新規な使用に基づく。本発明の1つの特定の利点は、制限断片の5’および3’末端におけるヌクレオチド配列が決定されない状況においてDNA制限断片の増幅を可能にすることである。このような場合、増幅すべき制限断片の各鎖にハイブリダイズする通常の配列特異的プライマーは規定することができず、したがって増幅目的に当業界で知られた方法を使用することができない。
【0012】
本発明の方法はたとえば2種の異なる方法で使用することができ、2種の異なる使用をもたらす:
(1)PCR技術により増幅すべき1個もしくはそれ以上の制限断片のサブセットをランダムに選択することによるゲノムのDNAフィンガプリント法。さらに本発明は、上記方法に用いるための合成オリゴヌクレオチドをも包含し、さらにこれら方法は法医学的型分類、微生物同定、変種同定、血統分析、および遺伝子形質に関連したDNAマーカーのスクリーニング等に使用とすることができる。
(2)多形性でありうる1種もしくはそれ以上の予備選択されたDNA断片をPCR増幅により同定するための方法。さらに本発明は、前記方法に使用するための特定の合成オリゴヌクレオチドをも包含し、さらにこれら方法はヒトの遺伝病のスクリーニング、植物および動物の育種における耕種学的形質の遺伝の監視、および病気における感染物質の検出等に使用することができる。
【課題を解決するための手段】
【0013】
1.定義
以下の説明および実施例においては多くの用語を使用する。これら用語に与えるべき範囲を含め本明細書および請求の範囲を明瞭かつ一貫して理解するため、次の定義を設ける。
−制限エンドヌクレアーゼ:制限エンドヌクレアーゼもしくは制限酵素は二本鎖DNA分子における特定塩基配列(標的部位)を認識する酵素であって、各標的部位にてDNA分子の両ストランドを切断する。
−制限断片:制限エンドヌクレアーゼによる切断により産生されるDNA分子を制限断片と称する。任意所定のゲノムは特定の制限エンドヌクレアーゼにより個々の制限断片群に消化される。制限エンドヌクレアーゼ切断によって生ずるDNA断片を分離して、ゲル電気泳動により検出する。
−制限断片長さ多形性(RFLP):たとえば2種の近縁生物のゲノムDNAは、多くの部位でそのヌクレオチド配列組成の相違点を示す。これらの相違点が制限エンドヌクレアーゼに関する標的部位に存在すれば、改変された標的部位はその箇所で切断されないだろう。同様に、ヌクレオチド配列変化は他の生物には存在しない新たな標的部位を導入し、その箇所で制限酵素によるDNAの切断を引き起こす。或いは、1種の生物で生ずる、制限エンドヌクレアーゼ用の2つの標的部位の間でのヌクレオチドの挿入もしくは欠失は、これら標的部位の間の間隔を改変する。このために、2種の生物DNAの消化により異なる長さを有する制限断片が生成する。2種の生物のDNAの消化によって生ずる制限断片の長さに多形性が生ずる。
【0014】
−ゲル電気泳動:制限断片を検出するため、寸法を基礎とした二本鎖DNA分子を分画する分析法が必要とされる。この種の分画を達成する最も一般的に用いられる技術は電気泳動である。この種のゲル内でDNA断片が移動する速度はその寸法に依存する。したがって移動距離は、断片長が増大するにつれて減少する。ゲル電気泳動により分画されたDNA断片は、パターンに含まれる断片の個数が少なければ染色法によって直接可視化することができる。
【0015】
−合成オリゴヌクレオチド:好ましくは約10〜約50塩基を有する化学的に合成しうる一本鎖DNA分子を合成オリゴヌクレオチドと称する。一般に、これら合成DNA分子は独特のヌクレオチド配列を有するよう設計されるが、関連配列を有すると共にヌクレオチド配列内の特定位置に異なるヌクレオチド組成を有する分子ファミリーを合成することもできる。合成オリゴヌクレオチドと言う用語は、独特なヌクレオチド配列を有するDNA分子を意味するよう用いられる。混合合成オリゴヌクレオチドと言う用語は、関連する合成オリゴヌクレオチドのファミリーを意味すべく用いられる。
【0016】
−連結:2個の二本鎖DNA分子を互いに共有結合させる酵素リガーゼによって触媒される酵素反応を連結(ligation)と称する。一般に両DNA鎖は互いに共有結合するが、一方の末端の化学的もしくは酵素的改変により2個のストランドの一方の連結を防止することもできる。その場合、2個のDNA鎖の一方にのみ共有結合が生ずる。
【0017】
−アダプター:たとえば長さ10〜30塩基対のような限られた個数の塩基対を有する、制限断片の末端に連結しうるよう設計される短い二本鎖DNA分子をアダプターと称する。これらアダプターは、部分的に互いに相補的であるヌクレオチド配列を持った2個の合成オリゴヌクレオチドで構成される。2個の合成オリゴヌクレオチドを混合すれば、適当な条件下で溶液中で二本鎖構造が形成されるだろう。アダプター分子の一方の末端は制限断片の末端に連結しうるよう設計され、他方の末端は連結しえないよう設計される。
【0018】
−ポリメラーゼ連鎖反応(PCR):インビトロにて基質DNAからDNA断片を合成する酵素反応をPCRと称する。この反応は、数百〜数千塩基対という短い間隔で分離されるDNA分子におけるヌクレオチド配列に相補的な2個の合成オリゴヌクレオチドの使用、および熱安定性DNAポリメラーゼの使用を含む。連鎖反応は、たとえば10〜30サイクルのシリーズで構成される。各サイクルでは、先ず最初に基質DNAを高温度で変性させる。冷却した後、大過剰で存在する合成オリゴヌクレオチドは、溶液中の基質DNA分子と、相補的ヌクレオチド配列を有する基質DNA分子上の特定部位で二本鎖構造を形成する。次いでオリゴヌクレオチド−基質DNA複合体はDNAポリメラーゼにより触媒されるDNA合成反応の開始部位として作用し、その結果、基質DNA鎖に相補的な新たなDNA鎖が合成される。
【0019】
−DNA増幅:DNA増幅と言う用語は、ポリメラーゼ連鎖反応(PCR)を用いるインビトロでの二本鎖DNA分子の合成を意味すべく使用される。PCR反応の生成物を増幅DNA断片と称する。
【0020】
−プライマー:一般に、プライマーと言う用語はDNAの合成を開始させうるDNA鎖を意味する。DNAポリメラーゼはプライマーなしには新たにDNAを合成することができない。これらは存在するDNA鎖を、組立てるべきヌクレオチドの順序を指令する鋳型として相補鎖が使用される反応において伸長させうるだけである。PCR反応に使用される合成オリゴヌクレオチド分子をプライマーと呼ぶ。
【0021】
−サザンハイブリダイゼーション:サザンブロッティングとも称するサザンハイブリダイゼーションの目的は、アガロースゲル電気泳動により分画されたDNAを、分画過程から生じたDNA断片の相対位置を保持させながら、たとえばナイロン膜もしくはニトロセルロース紙のような支持体上に物理的に移動させることにある。アガロースゲルから支持体への移動を達成すべく用いられる方法は、DNAをゲルから支持体中へ毛細管作用により移動させることである。
【0022】
−核酸ハイブリダイゼーション:核酸ハイブリダイゼーションは、たとえばナイロン膜もしくはニトロセルロース紙のような支持体上で一本鎖DNAのハイブリダイゼーションにより関連DNA配列を検出するために用いられる。相補的塩基配列を有する核酸分子は、適切な条件下で溶液中にて混合すれば二本鎖構造を再形成する。二本鎖構造は、たとえ一方が支持体に固定化されたとしても2個の相補的一本鎖核酸の間で形成される。サザンハイブリダイゼーション法においては後者が生ずる。
【0023】
−ハイブリダイゼーションプローブ:サザンハイブリダイゼーション法で特定のDNA配列を検出するには、標識されたDNA分子またはハイブリダイゼーションプローブをたとえばナイロン膜もしくはニトロセルロースフィルターのような支持体に結合した分画DNAに対し反応させる。標識されたDNAプローブに相補的なDNA配列を有するフィルター上の領域は、再アニーリング反応の結果としてそれ自身も標識される。次いでこの種の標識を示すフィルターの領域を、用いる標識の種類に応じて検出することができる。ハイブリダイゼーションプローブは一般に、トウモロコシ(maize)ゲノムからの特定DNA配列の分子クローニングによって生成される。
【0024】
[発明の実施の形態]
2.好適実施態様の説明
本発明はより詳細には、ポリメラーゼ連鎖反応(PCR)を長さ多形性(length polymorphisms)を含む制限断片多形性(RFP)の検出に用いうる方法および手段に関する。本発明はRFPを検出する方法、本発明の方法に使用するための合成オリゴヌクレオチド、RFPを検出するための手段を含むキット、並びに植物および動物の育種、遺伝病の診断、生物の同定、および法医学的型分類などへの本発明の方法および手法の使用を包含する。
【0025】
詳細には本発明は、個々のゲノム制限断片または、任意の生物、微生物、植物、動物もしくはヒトからの個々に1個もしくはそれ以上の特定の形質に遺伝子的に関連し或いは総合的に生物、変種もしくは個体を同定すべく使用しうるゲノムのフィンガプリントを与える1群のゲノム制限断片を同定するための手段を提供する。
【0026】
制限断片の産生および同定のための本発明による一般的方法は制限エンドヌクレアーゼの使用、制限断片に対する合成オリゴヌクレオチドの結合、および制限断片のPCR増幅(図1)を含む。制限エンドヌクレアーゼは特定部位、すなわち標的部位でゲノムDNA分子を切断することにより制限断片を生成させる。
【0027】
制限断片における末端のヌクレオチド配列が既知であってもなくても制限断片のPCR増幅は、本発明によれば、先ず最初に合成オリゴヌクレオチド(アダプター)を制限断片の末端に結合させてPCR増幅で使用されるプライマーのアンカー塩基として作用する2個の共通標識を各制限断片に付与することにより達成することができる。
【0028】
典型的には、制限酵素は両ストランドの末端ヌクレオチドが塩基対形成するフラッシュ末端(flush ends)、または2本のストランドの一方が突出して短い一本鎖伸長を与えるスタッガー末端(staggered ends)をもたらす(図2)。フラッシュ末端を有する制限断片の場合は、アダプターを1フラッシュ末端と共に使用する。スタッガー末端を有する制限断片の場合は、制限断片の一本鎖伸長に相補的である一本鎖伸長を持ったアダプターを使用する。その結果、それぞれの種類の制限断片につき特定アダプターを使用し、これらアタプターは一方の末端においてのみ相違してアダプターを制限断片に連結させうるようにする。典型的には、用いるアダプターは互いに部分的に相補的な2種の合成オリゴヌクレオチドで構成され、これらオリゴヌクレオチドは一般に長さ約10〜30ヌクレオチド、好ましくは長さ12〜22ヌクレオチドであると共に、溶液中で互いに混合すれば二本鎖構造を形成する。酵素リガーゼを用いて各アダプターを制限断片の混合物に連結させる。制限断片に対し大モル過剰のアダプターを用いれば、全制限断片が両末端でアダプターを保持して終端するよう確保される。この方法により作成される制限断片を標識制限断片と称し、この方法をさらに制限断片標識化と称する。
【0029】
これらアダプターはかくして、後続のPCR増幅反応に用いる上記特徴を備えたプライマーの鋳型として使用することができる。本発明の好適具体例において、制限断片はその両末端に同じアタプターを有し、単一のPCRプライマーを用いて図3に示すように制限断片を増幅することができる。この種の場合、全制限断片が同様に標識されるので、標識制限断片の混合物のPCR増幅は同時様式(synchronous fashion)で全制限断片を増幅することが明らかである。2種の異なる制限酵素を用いてDNAを切断する他の具体例においては、2種の異なるアダプターを制限断片の各末端に連結させる。この場合、2種の異なるPCRプライマーを用いてこの種の制限断片を増幅することができる。2種の制限酵素を用いる他の好適具体例においては、一方の酵素末端に対するアダプターをビオチニル化する。これは制限断片の複雑な混合物からこの制限酵素用に少なくとも1端部を有する制限断片を選択することを可能にし、その際ビオチニル化された分子を分離する常法を用いる。この工程は制限断片の出発混合物の複雑性を減少させると共にPCR増幅に先行する濃縮工程を構成し、或る場合にはこれによってバックグランドを減少させる。数種の異なる断片の同時的増幅はしばしば多重PCRと呼ばれる。多重制限断片増幅の原理については図4に示す。
【0030】
さらに本発明は制御された増幅が可能となるようPCR増幅反応を指令するための特異的に設計されたプライマーおよび特定の方法に基づくものであって、本発明の特定具体例によれば小サブセットの標識制限断片のみが増幅されるようにする。
【0031】
一般にゲノムDNA、特に動物、植物もしくはヒトのゲノムDNAの制限エンドヌクレアーゼ消化物は極めて多数の制限断片をもたらす。制限断片の個数はゲノムの寸法、並びにゲノムにおける制限エンドヌクレアーゼ標的部位の存在頻度に依存し、この存在頻度は次いで主として標的部位におけるヌクレオチドの数により決定される。一般的に使用される制限エンドヌクレアーゼの標的部位におけるヌクレオチドの数は4〜8個の範囲である。生物のゲノム寸法は、微生物の場合における数百万塩基対から動物および植物における数千万塩基対の範囲で広範囲に変化することができる。したがって、制限酵素によりゲノムDNA分子を切断した後に得られる制限断片の個数は数百〜数百万の範囲で変化することができる。一般に制限断片の個数は、ゲル電気泳動により分画されたゲノムDNA消化物において個々の制限断片を同定しえないほど大である。この種の消化物は一般にバンドの汚れをもたらす。
【0032】
したがって標識制限断片のPCR増幅は、全断片が同時的にPCR反応で同時増幅するため、バンドの汚れをもたらす。大寸法のゲノムDNAに対し適用しうる本発明の好適具体例においては、増幅すべき制限断片の個数を制限する一般的原理を用いた。これは、比較的少数の標識制限断片のみがPCR増幅反応に際し増幅されるようサブセットの標識制限断片を予備選択することにより行われる。
【0033】
本発明のこの具体例で規定される選択的原理は、図5に示すように、PCR増幅用のプライマーとして使用されるオリゴヌクレオチドの設計である。
【0034】
標識制限断片は次の一般的構造を有する:逆DNA配列(一定配列)により両側にフランキングする可変DNA配列(標識前の制限断片に対応)。逆DNA配列(一定DNA配列)は、制限エンドヌクレアーゼの標的配列の部分と制限断片の両末端に結合したアダプターの配列の部分で構成される。一定DNA配列の間に含まれる制限断片の可変配列は一般に未知であり、したがってランダム配列組成を有する。その結果、一定DNA配列にフランキングするヌクレオチド配列は制限断片の大混合物中で全体的にランダムである。
【0035】
したがって本発明は一定ヌクレオチド配列部分を含む特定PCRプライマーをも提供し、さらに得られる制限断片の制限サブセットの増幅に依存する本発明の実施態様においては可変配列部分をも含む。一定配列部分においてヌクレオチド配列は、プライマーが制限断片の末端における一方のDNA鎖の一定DNA配列と完全に塩基対形成するよう設計される。可変配列部分は、選択した1〜10塩基の範囲のランダムに選択したヌクレオチド配列で構成される。
【0036】
「可変配列(variable sequence)」と言う用語は一層正確には、サブセットの制限断片を増幅する目的で一定に留まる配列を形成する選択ヌクレオチドよりなる配列を意味する。本発明の特定具体例においては、選択塩基よりなる数種の配列を用いて数種の別個のプライマーを規定することができる。この種の場合、プライマーは同じ一定配列と、形成されるプライマー間で異なる選択塩基により構成される可変配列とを有することができる。
【0037】
PCR工程で増幅される標識制限断片の予備選択を指令するのは、プライマーの3’末端へのこれら可変(選択)配列の付加である。PCR反応を適する条件下で行う場合、プライマはこれら標識制限断片に対しDNA合成のみを開始させ、ここで可変DNA配列は図5に示したように標識制限断片の鋳型ストランドと完全に塩基対を形成することができる。
【0038】
選択は、プライマーの可変配列部分に存在するヌクレオチドの数により決定される。プライマーの選択性は可変(選択)配列部分におけるヌクレオチド数に伴って増加する。さらに選択性塩基と言う用語は可変配列部分におけるヌクレオチドを意味し、これら塩基の選択がプライマーを選択性にすることを示す。使用したプライマーの選択性塩基が断片の末端にて両相補的配列を認識する場合、標識制限断片のみが増幅されることを理解されねばならない。プライマーが1末端にのみ適合(match)してアニーリングする場合、増幅は対数的でなく直線的となり、生成物は未検出のままとなろう。
【0039】
予め異なる個数の選択性塩基を有する可変配列で得られる選択性の程度を推定することができ、一般式42n[ここでnは選択性塩基の個数に等しい]を用いる。1個の選択性塩基を用いて16個の標識断片のうち1個が増幅され、2個の選択性塩基を用いて256個のうち1個が増幅され、3個の選択性塩基を用いて4,096個のうち1個が増幅され、さらに4個の選択性塩基を用いて65,536個のうち1個が増幅され、以下同様である。本発明の1好適具体例は、用いる制限酵素により生成される断片の個数とは無関係に、ランダムサブセットの標識制限断片を任意のゲノムDNA消化物から選択的に増幅することを可能にする。
【0040】
好適具体例において、選択性ヌクレオチドの個数は、増幅される制限断片の個数を5〜200個に制限するよう選択される。この個数は断片の個数を42nで割算して計算しうるが、正確な予測は可能でない。何故なら、必ずしも全ての制限断片を等しい効率で増幅しえないからである。したがって、実際には理論的に予想されるよりも少ない断片が増幅される。さらに、2種(もしくはそれ以上)のプライマーの混合物を用いうることも指摘すべきである。これは各プライマーにより認識される断片の増幅を可能にし、さらに2個のプライマーにより認識される断片の増幅を可能にする。最後に、プライマーの選択性ヌクレオチドと相補的鋳型との間の塩基対形式に基づく選択は、PCR反応におけるアニーリング工程において選択される温度により強く影響を受けることも指摘すべきであり、この温度がプライマー/鋳型複合体の融解温度よりも低く或いはそれに近接すれば、プライマーは不完全な組合わせで鋳型配列にアニーリングして、複合体に誤った組合わせ(mismatch)を生ぜしめる。これは、予想よりも多い断片の増幅をもたらして一層多い変動結果を生ぜしめるため回避されるべきである。
【0041】
本発明により得られるPCR生成物は、寸法によりDNA分子を分離した後にDNA分子を適する試薬で染色する標準的な分画法を用いて同定することができる。或いは、PCR増幅に用いるプライマーを適する放射性標識もしくは蛍光性発色団で標識して、サイズ分画の後に反応生成物を同定することもできる。本発明の好適具体例において、PCR生成物は限定はしないがたとえばアガロース、ポリアクリルアミドまたは混合アガロース/ポリアクリルアミドのような標準的ゲルマトリックスを用いるゲル電気泳動により分画される。本発明により得られるPCR生成物は、増幅制限断片(ARF)と言う用語でも示される。
【0042】
本発明の手段および方法を用いて、任意の複雑なゲノムの制限消化物から1群のARFを生成させることができる。本発明は、得られる制限断片の個数をARFを分離すべく使用するゲル分画システムの解像力に応じて調節することができる。1特定具体例において、選択的プライマーは5〜10個のARFを生成するよう設計され、次いでこれらをアガロースゲル電気泳動により分離する。他の特定具体例は、20〜50個のARFを生成するよう設計された選択性プライマーの使用を含み、次いでこれらARFを限定はしないがたとえばポリアクリルアミドゲルもしくは混合アクリルアミド−アガロースゲルのような高分解能のゲル電気泳動システムによって分離する。
【0043】
1好適具体例において、制限酵素は20〜1000塩基対の寸法範囲にて制限断片をもたらすよう選択される。何故なら、PCR増幅に関して一般的に知られるように、この断片寸法範囲が最も効果的に増幅されるからである。各種の標準ゲルマトリックスでずっと多くの断片を分画しうるが、DNA配列決定に関して現在使用されているような変性ポリアクリルアミドゲル系での分画により最も良好な結果が得られる。
【0044】
本発明によれば、PCR増幅反応にてそれぞれ異なる選択性プライマーを用い種々異なる群のARFが得られる。分離後に同定されるARFのパターンは、ゲノムDNAの独特な完全に再現性のあるフィンガプリントを構成する。この種のフィンガプリントは、限定はしないがたとえば法医学的分類、生物の診断的同定、並びに品種、種族、変種もしくは個体の確認のような数種の用途を有する。同定のレベルは、特定群の種々異なる構成員により示される類似性の程度(変動性の程度)により決定される。変動性もしくは類似性は、関連ゲノムにおけるヌクレオチド組成の変動程度によって決定される。本発明の基礎となる原理は、各増幅制限断片にて図9に示すように所定間隔で互いに分離された2種のヌクレオチド配列を検出する点にある。2種のヌクレオチド配列のそれぞれは2つの部分で構成される:すなわち(a)制限エンドヌクレアーゼのための標的部位、および(b)選択性プライマーに含まれる標的部位に隣接したヌクレオチド配列。関連する生物、品種、変種、種族もしくは個体において、これら配列要素およびその相対的間隔はより大きい或いはより小さい程度で保持される。したがってフィンガプリントは、ゲノム間の配列関係の程度を決定するための基礎を構成する。他方、ARFパターンにおける差を用いて、各ゲノムを互いに区別することもできる。ゲノムの他のフィンガプリント法に優る本発明の特定の利点は、この方法で得られる高い分解能(resolution)である。数十もしくは数百のARFを同時に比較することができる。
【0045】
本発明の他の特定応用は、制限断片多形性(RFP)のスクリーニングおよび同定を含む。ゲノムDNAのヌクレオチド組成の変化は、しばしば制限断片の多形性をもたらす。挿入もしくは欠失はこれらを有する制限断片の寸法に影響を及ぼし(図10)、ヌクレオチド変化は制限エンドヌクレアーゼ標的部位の除去または新たな制限エンドヌクレアーゼ標的部位の形成(図11)をもたらしうる。この種の変化を確認するため最も一般的に使用される技術、はクローン化されたDNAプローブを用いるサザンブロッティング実験であり、すなわち一般に制限断片長さ多形性(RFLP)検出と呼ばれる技術である。この技術は、種々異なるゲノムにおける関連RFLPのためのサザンブロッティング実験におけるランダムにクローン化されたDNA断片の徹底的なスクリーニングを含む。本発明の方法によれば、RFPを種々異なるゲノムから得られたARFを比較して直接に同定することができる。原理的に、本発明の方法はRFPを検出するための感度がより大である。何故なら、制限エンドヌクレアーゼの標的部位における差が検出されるだけでなく、隣接ヌクレオチド配列における差が選択性PCRプライマーに含まれるからである。その結果、本発明の方法はRFLPを検出するためのずっと優秀な方法を構成する。
【0046】
RFLPは法医学的型分類、ヒトにおける遺伝病の監視、並びに植物および動物の育種における耕種学的形質の遺伝の監視を含む数種の用途に現在使用されている。基礎となる原理は、特定の遺伝子特性に密接に関連した或る種のDNA多形性を用いて特定の遺伝子特性の存在もしくは不存在を監視しうる点にある。
【0047】
本発明の方法によれば、ARFパターンの分析を用いて特定の遺伝子特性に対する多形性ARFの遺伝的関係を規定することができる。さらに、この種の多形性ARFは増幅断片長さ多形性(AFLP)と呼ばれ、これらをクローン化DNAプローブを用いてサザンブロッティング実験で検出されるRFLP型DNA多形性から区別することができる。
【0048】
本発明の1特定用途は、特定遺伝子特性に関連したAFLPの検出を含む。この用途は、特定遺伝子特性における差を示す近縁個体のゲノムDNAの制限消化物における異なる選択性プライマーで得られたARFパターンの分析、並びに1種もしくはそれ以上のAFLPの遺伝と特定遺伝子特性により示される表現型との間の相関関係を見出しうる分析技術の使用を包含する。
【0049】
第2の好適な本発明の実施態様は、1種もしくはそれ以上の特定制限断片を同定するための本発明方法の使用を含む。1つの特定制限断片は、先ず最初に制限断片の各末端における最初の8〜12塩基のヌクレオチド配列を決定することにより標識制限断片の複雑な混合物から増幅させることができる。これら配列に基づき2種のプライマーを設計することができ、それぞれ5〜10個の選択性ヌクレオチドは制限断片の相補鎖の制限部位にフランキングする配列に相補的な配列を示す。各群のプライマーを用いて、PCR増幅の後に単一の増幅断片を得ることができる。この方法に用いる制限断片はクローン化制限断片または増幅制限断片のいずれかとすることができる。必ずしも多くの制限断片は極めて効率的に増幅しえないので、多形性DNAマーカーを同定するための本発明の好適方法は先ず最初にランダムに選択された断片群を増幅させ、次いでPCR増幅の後に強いバンドをもたらすAFLPを同定する。これらAFLPは、制限断片特異性プライマーを開発するべく配列決定して特性化することができる。典型的には、AFLPは対応のDNAバンドをゲルから切除し、次いで両末端におけるヌクレオチド配列を決定して制限エンドヌクレアーゼ標的部位に隣接した最初の5〜10ヌクレオチドの配列を確認することにより分離される。これらヌクレオチド配列が知られた後、ゲノムDNA消化物から単一の制限断片のみを増幅する制限断片特異性プライマーを設計することができる。本発明によるこの特定具体例において、2種の異なる選択性プライマーのうち1組を用いて特定制限断片を検出することができる。1組の2種の選択性プライマーのそれぞれにおいて、選択塩基は制限エンドヌクレアーゼ標的部位に隣接したヌクレオチド配列に相補的となるよう選択され、これを図8に示す。各プライマーに含ませる選択塩基の個数は、制限エンドヌクレアーゼ断片混合物の複雑性に依存する。
【0050】
PCR技術は過去数年間にわたり著しく発展し、ヒトの健康管理における最も広く用いられる診断法の1つに急速になりつつある。その用途は特に感染症の検出および遺伝病の検出を含む。各診断試験は2種の特定合成オリゴヌクレオチドの使用に基づくものであり、これらオリゴヌクレオチドをPCR反応におけるプライマーとして用いることにより特定長さの1種もしくはそれ以上のDNA断片を得る。病気検出において試験は1試料当り1個程度に少ないDNA分子の存在を検出して、特徴的DNA断片を得る。遺伝病の場合には、生成物を正常の対立遺伝子と病気の対立遺伝子を区別しうるようプライマーを設計する。区別は、プライマーに対し相補的なゲノム内のDNAセグメントの配列差に依存するか或いは2種のプライマー間における間隔差に依存する。
【0051】
プライマーは極めて高程度の特異性を示すので種々異なる病気を同時に監視することができ、しばしば多重PCRと呼ばれる方法である。しかしながら多重PCR法は、一般に僅か数種(5〜8)の異なる形質しか同時に監視しえない点で限界を有する。この限界の科学的根拠は、PCR増幅の最適条件(アニーリング温度、Mg+濃度、プライマー濃度など)が用いるプライマー対に応じて相当に変化する点である。多重PCRにおいては、全プライマー対が検出可能な生成物を生成する妥協条件を確立せねばならない。さらに、この現象に加えて、種々異なる断片の増幅効率に強力な差が生じる現象がある。その結果、しばしば或る種のプライマー対の生成物が多重PCR反応では検出しえないという問題に遭遇する。
【0052】
本発明の方法は本質的に多重PCRのこれら限界を解消する。何故なら、本発明に用いる全プライマーはそのヌクレオチド配列の実質的部分を共通して有するからである。さらに、AFLPを選択することにより、等しい効率で増幅されるDNAマーカーが選択される。したがって、異なる選択性プライマのための最適なPCR増幅条件は、一般的に使用される配列特異性プライマーで観察されるよりもずっと低い変動を示す。本質的に、合成オリゴヌクレオチドにおける塩基(これら塩基は複雑なゲノムにおける所定寸法の単一DNA断片を検出する所要の特異性を得るのに必要である)の個数(これは上記のように計算する)と、効率的なPCR増幅にとって最適であるオリゴヌクレオチドの長さおよび組成との間の理想的妥協点が存在する。したがって、本発明の方法は多重PCRのかなり優秀な方法を提供する。
【0053】
本発明は、任意のゲノムからDNAマーカーを分離すると共にこの種のDNAマーカーをあらゆる可能なDNAフィンガプリント法の用途に使用するための一般的方法を提供する。
【0054】
以下、限定はしないが実施例および図面により本発明をさらに説明する。
【実施例】
【0055】
実施例1:PSTIを使用するトマトDNAの選択的制限フラグメント増幅
A)DNAの単離及び修飾
全トマトDNA(Lycopersicon esculentum c.v.Moneymaker)をBernatzski及びTanksley(Theor.Appl.Genet.72,314−321)に記載の如く幼葉から単離した。典型的な収量は、新鮮な葉物質1グラム当たりDNA 50−100μgであった。DNAをPstI(Pharmacia)で制限し、二重鎖(ds)PstIアダプターを以下に記載の方法に従って制限フラグメントに連結した。これらのアダプターは以下の構造:
5- CTCGTAGACTGCGTACATGCA -3
3- CATCTGACGCATGT -5
を有していた。これらのアダプター内の3’TGCA−の突出端をPstIにより作った付着端にアニールする。5’C−残基がAに置き換わっているので、PstI認識配列CTGCAGは、このアダプターの連結により修復されない。連結反応は、得られる末端が殆どDNAフラグメント−アダプター分子であるような方法で設計されていた。これは、
1.アダプター−アダプターの連結を排除する非ホスホリル化アダプターを使用する、
2.同時に連結及び制限反応を実施する
ことにより実施した。後者の方法により、任意のフラグメント−フラグメントの連結産物が制限でき、これによりこれらの産物を殆ど完全に除去できた。PstI認識配列はこれらの産物中で修復されないので、アダプター−フラグメント連結産物は制限酵素により制限され得ない。アダプター連結に使用する反応条件は以下の通りであった。
【0056】
2μg トマトDNA
0.2μg アダプター
20単位 PstI
1単位 T4 DNA−リガーゼ
10mM Tris.HAc pH 7.5,10mM MgAc,50mM KAc,
2mM ジチオトレイトール,0.5mM ATP
連結反応を反応容積20μl中37℃で3時間実施した。アダプター連結後、非連結アダプターを選択的に沈殿させることにより除去した。この目的のため、反応混合物を100μlに増やし、NH4Acを終濃度2.5Mで添加した。−20℃のエタノール(100μl)を添加し、混合物を室温で5分間インキュベートした。4℃に冷却したエッペンドルフ遠心分離機中、14000rpmで10分間遠心分離してDNAを集めた。DNAペレットを室温で70%エタノール0.5mlで1回洗浄し、T0.1E(10mM Tris.HCl pH 8.0,0.1mM EDTA)40μlに溶解させた。DNAを−20℃で貯蔵した。本明細書中に記載する選択的沈殿法により、反応混合物から非連結アダプターを効率良く除去したが、小さなDNAフラグメント(<200bp)も失ってしまった。
【0057】
B)増幅方法
上記方法で製造したDNAをPstIフラグメントを増幅するための鋳型として使用した。PCRの反応混合物は、
1ng 鋳型DNA
150ng プライマー
1単位 Taq DNAポリメラーゼ(Perkin Elmer)
200μM 全4dNTP
10mM Tris.HCl pH 8.5,1.5mM MgCl2,50mM KCl
H2O (全容量が50μlになるまで)
を含んでいた。増幅反応中の蒸発を防ぐために、反応混合物を軽質鉱油(light mineral oil)20μlで被覆した。以下のサイクルプロフィール:94℃で1分、60℃で1分、60℃から72℃に温度を1℃/5秒の速度で上昇及び72℃で2.5分を使用してPerkin Elmer DNA Thermal Cycler上でPCRを実施した。全部で33サイクル実施した。反応後、クロロホルム20μl及び充填染料10μl、この場合染料Orange G(Merck)0.1% w/vと一緒に50%蔗糖を添加した。次いでこれを反応混合物と十分に混合し、充填染料を補った反応混合物から有機相(鉱油及びクロロホルム)を分離するために軽く遠心分離した。この反応混合物20μlを1.0%アガロースゲル上で分析した。
【0058】
C)選択性の高いプライマーを使用するトマトDNAの増幅
PstIで制限し、次いでPstIアダプターをつけたトマトDNAを上記条件を使用して増幅した。以下の配列:
1. 5-CTCGTAGACTGCGTACA-3
2. 5-GACTGCGTACAtgcagA-3
3. 5-GACTGCGTACAtgcagAC-3
4. 5-GACTGCGTACAtgcagACC-3
をもつ4つのプライマーを選択した。プライマー1はDNAを修飾するのに使用するアダプターのトップストランドの一部なので、全PstIフラグメントを増幅しなければならない。プライマー2はアダプター配列の一部、PstI認識配列(小文字)及び1個の選択的ヌクレオチド(大文字)を含むので、理論的には全PstIフラグメントの約1/16部分を増幅しなければならない。プライマー3及び4はプライマー2に似ているが、選択的ヌクレオチドをそれぞれ2つ及び3つ含んでいるので、PstIフラグメントの約1/256及び1/4096を増幅すると予想される。反応混合物の一部を1.0%アガロースゲル上で分析し、結果を図11に示す。この図面のレーン1及び6はDNAマーカーを含んでおり、そのサイズを左側に示す。レーン2、3、4及び5は各々プライマー1、2、3及び4で得られたPCRを含んでいる。この結果は、3つの選択的ヌクレオチドを使用したプライマーの場合にのみ、はっきりしたバンドパターンの増幅フラグメント数が得られることを示している。他の3つのプライマーはアガロースゲル上に溶解できなかったバンドパターンを与えたが、これは多くのPCR産物が生成したためである。これらの多くのPCR産物内でも、常に幾つかのフラグメントが目立っており、他のPCR産物の不鮮明なバックグラウンド上でもバンドとして検出される。恐らくこれらの強い産物は、トマト遺伝子上に高コピー数で存在するか、または他の産物よりもずっと効率的に増幅するのだろう。トマト遺伝子DNAのPstIフラグメントの総数(20,000〜100,000)により、3つの選択的ヌクレオチドをもつプライマーが使用されてアガロースゲル上で鮮明なバンドパターンを作ったに違いないと予想された。
【0059】
D)サザンブロット上の増幅フラグメントの分析
増幅フラグメントが同一サイズの真正制限フラグメントに対応することを立証するために、これらの増幅フラグメントをサザンブロット上で試験した。この目的のために、プライマー4で得られた4つの個々のフラグメントをアガロースゲルから切り出した。これらのゲル切片からDNAをガラスビーズ(Gene Clean,製造業者Bio 101)に吸着させることにより精製し、精製DNAの一部を再増幅して4つのDNAフラグメントを各々約1μg得た。続いて再増幅反応を1.0%分取アガロースゲル上で電気泳動させ、所望のDNAフラグメントを精製した。各フラグメント200ngを、製造業者(Boehringer Mannheim)により推奨された方法に従ってランダムヘキサマー標識キットを使用して(α−32P)dATPで標識した。全トマトDNAをPstIで制限し、1.0%アガロースゲル上で電気泳動にかけた。制限DNAを約3μg含む4つのはっきりと分離したレーンを使用した。次に、アガロースゲルを、製造業者(New England Nuclear)により示された遺伝子スクリーン+ハイブリダイゼーション膜にブロットした。ブロット後、ゲルをPstIで制限したトマトDNAを1レーン含む4つの切片に切断した。これらの4つの切片をKlein−Lankhorstら(Theor.Apll.Genet.81,661−667)に記載の方法に従って4つのDNAプローブの一つに各々ハイブリダイズさせた。ハイブリダイズしたブロットをKodak XAR5フィルムを使用してオートラジオグラフに40時間かけた。得られた結果は、4つのDNAプローブにより識別された全てのゲノムDNAフラグメントはこれらのプローブと同一の長さを持つことを示した。これは、プローブとして使用した増幅フラグメントはブロット上で検出されたフラグメント由来であることを示していた。
【0060】
E)単一制限フラグメントの選択的増幅
プライマーの3セットをトマト遺伝子DNA由来の3つの対応するランダムPstI−フラグメント用にデザインしたが、その中でもPstI−認識配列に隣接する配列が公知であった。5つの選択的ヌクレオチドを持つプライマーのセットを以下に示すように製造した。
【0061】
プライマーセット1:
配列1:
5-ctgcagCAGTACCAGC----CCGGCACCTGctgcag-3
5-TGCGTAACATtgcagCAGTA-3 3-TGGACgacgtACATGCGT-5
プライマー1.1 プライマー1.2
【0062】
プライマーセット2:
配列2:
5-ctgcagCCGAATCTCT-----AGTGAGTTAGctgcag-3
5-TGCGTACAtgcagCCGAA-3 3-CAATCgacgtACATGCGT-5
プライマー2.1 プライマー2.2
【0063】
プライマーセット3:
配列1:
5-ctgcagAATACCAAGA-----GCAAGCACAGctgcag-3
5-TGCGTACAtgcagTTATG-3 3-GTGTCgacgtACATGCGT-5
プライマー3.1 プライマー3.2
【0064】
トマトDNAをPstIで消化し、アダプターを上記の如く制限フラグメントの末端に連結した。このDNAを上記セクションの条件のひとつを使用して、プライマーセット1または2または3を使用するPCRの鋳型として使用した。各PCRの反応産物を1.0%アガロースゲル上で分析した。このゲルは図12に示されている。図12は13個のレーンを示しているが、レーン1、2、12及び13はDNAマーカーである。これらのマーカーのkbの大きさをゲルの両側に示す。レーン3、6及び9は、ベクターフラグメント、pUC18(Yanisch−Perronら,Gene 33,103−119)及び挿入PstIフラグメントを産生する、PstIで制限した3つのPstIフラグメントを各々含むプラスミドDNAを示す。レーン4及び5は、各々全ゲノムDNA1ng及び対応するプラスミドDNA5fgのプライマーセット1での増幅を示す。レーン7及び8は、全ゲノムDNA及びプラスミドDNAのプライマーセット2での増幅を示し、レーン10及び11は、プライマーセット3での増幅を示す。これらの結果は、5つの選択的ヌクレオチドをもつプライマーでの選択的制限フラグメント増幅方法を使用すると少なくとも20,000フラグメントの混合物から単一のPstIフラグメントを増幅し得ることを示している。
【0065】
F)SRFAを使用するDNA多型現象の識別
先のセクションにおいて、選択的制限フラグメント増幅法を使用すれば、配列情報が入手可能な場合、ランダムまたは特定のフラグメントのいずれかにおいて制限フラグメントを増幅し得ることが明示された。従って、同一種の2つの個体間で制限部位多型現象に関して調査可能である。非常に近縁であるが系の一方にはネコブセンチュウ耐性遺伝子、Miが存在するという点が異なる2つのトマト系に関してこのことを以下に記載する。このMi−遺伝子はLycopersicon peruvianum、食用トマトL.esculentumの遠縁種に由来する。これを、交雑、次いでL.esculentum親に12回戻し交雑し、さらにMi−遺伝子の存在に関して子孫を選択することによりL.esculentum系に導入した。従って、2つのトマト系は、その遺伝子物質の小さな部分だけ、即ちMi−遺伝子及び周囲領域が異なる。古典的遺伝学方法を使用して、Mi−領域がこの系のゲノムの<1%を構築すると計算した。
【0066】
DNAを2種類のトマト系(系83M−71392,Mi−感受性及び系83M−71398,Mi−耐性;De Ruiter Seeds,Bleiswijk,オランダ)から単離し、続いてPstIで制限し、上記の如くアダプターをつけた。選択的ヌクレオチドの伸長が異なるプライマーを使用して増幅反応を多数回実施した。3つの選択的ヌクレオチドを使用し、単一プライマーの他に異なる2つのプライマーの組み合わせを使用した。反応物を混合ポリアクリルアミド/アガロースゲル上で分析した。その際、アクリルアミド対ビスアクリルアミド20:1の割合で2.5%ポリアクリルアミド及び1.0%アガロースを使用した。1.5mmのスペーサーを使用してProtean IIゲルユニット(Biorad)上にゲルを流した。全部で16の異なるプライマーを使用すると、単一プライマーに関しては16の反応となり、2つのプライマーの考え得る全部の組み合わせに関しては120の反応となる。これらの組み合わせの6つを使用するゲルの典型例を図13に示す。このゲルのレーン1及び14はDNAマーカーを含んでおり、そのサイズをゲルの右側にkbで示す。レーン2及び3、4及び5、6及び7などは、2つのトマト系のプライマー対または特異的プライマーでの増幅を含んでいる。制限部位多型現象に関するスクリーニングから多くのフラグメントが産生したが、そのうちの3つは非常に顕著であり、これらは図13のレーン9、11及び12に示されている(小さな丸で示されている)。同一プライマーが両方の反応に存在するので(違いはレーン11の第2プライマーの存在である)、レーン9及び11の多型現象のバンドは同一であると予測した。レーン11及び12の2つの多型現象フラグメントをゲルから切り出し、ゲル切片を18ゲージ針に押し通して壊し、DNAを100mM Tris.HCl pH 8.0,10mM EDTAの200μl中で拡散させることにより溶離してゲル切片から溶出させた。上記の如くこれらのフラグメントの再増幅用に2μlを使用した。T4 DNAポリメラーゼを使用して各フラグメント200ngをブラント端とし、続いてSmaIで制限したプラスミドベクターpUC18(Yanisch−Perronら,Gene 33,103−109)100ngに連結した。連結混合物をE.coliに形質転換し、各フラグメントに関して1つの組換えE.coliクローンを配列分析用に選択した。これらの取り扱い操作は全て、Sambrook、Fritsch及びManiatis:Molecular Cloning,A Laboratory Manual(Cold Spring Harbor Laboratory Press,New York)に記載の標準法を使用して実施した。
【0067】
6つの選択的ヌクレオチドを持つプライマー2セットを、上記の如く2つのフラグメントの配列をベースとして合成した。本発明者は、これらのプライマーセットを使用して各フラグメントを特異的に増幅し得た。トマト系からフラグメント(トマト系由来)だけを増幅した。しかしながら、これらのプライマーセットは、この多型現象を発見するのに使用した3つの選択的ヌクレオチドを持つプライマーに関して当初知見されたのと同一の多型現象を示した。
【0068】
実施例2:2つの制限酵素を使用するトマトDNAの選択的制限フラグメント増幅
実施例1では、選択的制限フラグメント増幅(SRDA)の原理は、トマトDNA及び制限酵素PstIを用いて例証されている。本実施例では、2つの制限酵素、PstI及びMseIを使用してSRFAを説明する。
【0069】
DNAの単離及び修飾
全トマトDNAを実施例1に記載の如く、幼葉から単離した。所謂、同一遺伝子系の2対、GemR及びGemS、各々GCR26及びGCR151[これらの系は、以下の文献に記載されている:Denby及びWilliams,(1962),Can.J.Plant Sci.42,681−685,Smith及びRitchie,(1983),Plant Mol.Biol.Rep.1,41−45]をDNA源として使用した。同一遺伝子の各対の2つの個体は遺伝子的に非常に似ているが、真菌病原体Verticillium albo−atratumに対する耐性を与える特徴の存在が異なる。
【0070】
DNAの修飾の第1段階は、2つの酵素PstI及びMseIでDNAを制限することを含んでいた。DNAの制限及び続くDNAフラグメントへのアダプターの連結を、RL−緩衝液(制限−連結緩衝液)と名付けられ、かつ10mM Tris.HAc/10mM MgAc/50mM KAc/5mM DTT, pH 7.5を含む同一緩衝液中で実施した。
【0071】
PstI及びMseIによるDNAの制限
2.5μg DNA
12.5単位 PstI(Pharmacia,10単位/μl)
12.5単位 MseI(N.E.Biolabs,4単位/μl)
5μl 10×RL−緩衝液
50μlになるまでの量のH2O
インキュベーションを37℃で1時間実施した。
【0072】
DNAの修飾の次段階は、DNAフラグメントの末端へのアダプター分子の連結である。まず、好適な二重鎖アダプター分子を製造しなければならなかった。
【0073】
アダプターの製造
MseIアダプター: 5-GACGATGAGTCCTGAG-3
3-TACTCAGGACTCAT-5
【0074】
このアダプター50pMole/μlの溶液を製造するために、
16マー、5-GACGATGAGTCCTGAG-3 8μg(1430 pMoles)を全容量H2O 28.6μl中の14マー、5-TACTCAGGACTCAT-3 7μg(1430 pMoles)と混合した。
PstIアダプター:5-ビオ-CTCGTAGACTGCGTACATGCA-3
3-CATCTGACGCATGT-5
【0075】
このアダプター5pMoles/μlの溶液を製造するために、ビオチニル化21マー、5-ビオ-CTCGTAGACTGCGTACATGCA-3 5.25μg(715 pMoles)を全容量H2O 143μl中の14マー、5-TGTACGCAGTCTAC-3 3.5μg(715 pMoles)と混合した。
【0076】
アダプター分子の連結
制限DNAに、以下のもの:
1μl PstI ビオ−アダプター(=5 pMol)
1μl MseI アダプター(=50 pMol)
1.2μl 10mM ATP
1μl 10×RL−緩衝液
1単位 T4 DNAリガーゼ(Pharmacia,5単位/μl)
10μlになるまでの量のH2O
を含む混合物10μlを添加した。
【0077】
得られた反応混合物60μlを37℃で3時間インキュベートした。
【0078】
制限部位が連結後に修復されないようにアダプターをデザインした。制限酵素が連結反応時にまだ活性であるから、フラグメントコンカテマーが制限され、故に、この方法ではフラグメント−フラグメント連結を避けられた。アダプターはホスホリル化されていなかったので、アダプター−アダプター連結はできなかった(実施例1参照)。
【0079】
ビオチニル化DNAフラグメントの選択
2つの制限酵素を使用するSRFA用の鋳型−DNAの製造は、通常、単一酵素を用いるSRFAを使用する際には使用しない追加の段階を含んでいた。この段階において、ビオチニル化アダプターに連結するDNAフラグメントを他のすべてのフラグメントから分離した。
【0080】
ビオチニル化フラグメントをこの段階で非−ビオチニル化フラグメント(MseI−MseI−フラグメント)から常磁性ストレプトアビジンビーズ(Dynal)に結合させることにより分離した。10μlビーズを100μl STEX(100mM NaCl/10mM Tris.HCl/1mM EDTA/0.1% Triton X−100 pH 8.0)中で洗浄し、140μl STEX中に再懸濁させた。次いでビーズを連結混合物に添加すると、終容量200μlとなった。これを緩やかに撹拌しながら室温で30分間インキュベートすると、ビオチニル化DNAフラグメントが正しくビーズに結合できた。マグネットをビーズを含むチューブを近づけることによってこのビーズを集めた。こうすると、上清を他のチューブにピペットで移す際にビーズが移されない。ビーズを1度洗浄し、続いて新しいチューブに移した。次いでビーズを200μl STEXで3回洗浄した。最終的にビーズを200μl T01.E(10mM Tris/0.1mM EDTA,pH 8.0)中に再懸濁させ、新しいチューブに移した。DNAを4℃で保持した。
【0081】
制限酵素で制限し、アダプターをつけ、常磁性ストレプトアビジンビーズにつけ、上記の如く製造したMseI−MseIフラグメントから精製したDNAを、以下の段階で鋳型DNAと呼称する。
【0082】
PstI−MseIフラグメントの増幅
上記の如く製造した鋳型DNAは、記載のトマト系からの全PstI−MseIフラグメント、さらに全く内部MseI−フラグメントを含まない少量のPstI−PstI−フラグメントを含まなければならない。この実験において、これらの多くのPstI−MseIフラグメントが、実施例1に記載の如く本質的に増幅により可視化された。この実施例に記載の方法により得られたフラグメントの種類は実施例1に記載のフラグメントよりもずっと小さかったので、増幅産生物のゲル分析は変性アクリルアミドゲル(Maxam及びGilvert,Proc.Natl.Acad.Sci.U.S.A.74,560−564)上で実施した。さらにこの種のゲルはレーン1つあたり100個以上のバンドに分離できた。これは、実施例1に記載のアガロースゲルの約10倍以上であった。フラグメントは、(γ−32P)ATP及びポリヌクレオチドキナーゼで5’末端でPCRプライマーの一つを標識することにより可視化できた。
【0083】
PCRプライマーの標識
標識用に選択したプライマーは、MseIプライマー1と名付けられた19マー、5−GATGAGTCCTGAGTAAgaa−3であり、選択的ヌクレオチドは小文字で表されている。標識は、以下の方法により実施した。
【0084】
3.0μl 18マー(50ng/μl=150ngの溶液から)
5.0μl (γ−32P)ATP(10μCi/μl=50μCiの溶液から)
3.0μl 250mM Tris.HCl/100mM MgCl2/50mM DTT,pH 7.5
0.5μl T4−キナーゼ(Pharmacia 10単位/μl)
18.5μl H2O
【0085】
これにより総容量は30μlとなり、これを37℃で30分間インキュベートした。各PCRに関しては、この5’標識プライマー1μlを添加した。
【0086】
全部で28回PCRを実施し、4つの鋳型DNAを各々7つのプライマーの組み合わせで増幅した。各プライマーの組み合わせは同一MseIプライマー(上記のMseIプライマー1)を含んでいたが、PstIプライマーの選択に応じて変動した。全部で7つのプライマーを選択した(MseIプライマーに関しては、選択的ヌクレオチドは小文字で表されている)。
【0087】
PstIプライマー1:5-GACTGCGTACATGCAGga-3
PstIプライマー2:5-GACTGCGTACATGCAGgt-3
PstIプライマー3:5-GACTGCGTACATGCAGgg-3
PstIプライマー4:5-GACTGCGTACATGCAGag-3
PstIプライマー5:5-GACTGCGTACATGCAGat-3
PstIプライマー6:5-GACTGCGTACATGCAGct-3
PstIプライマー7:5-GACTGCGTACATGCAGta-3
全PSRプライマーは、濃度50ng/μlでH2Oに溶解した。
【0088】
増幅反応
PCR混合物は、以下のものを含んでいた。
2.0μl 鋳型DNA
1.0μl 5’標識MseIプライマー(5ng)
0.5μl 未標識MseIプライマー(25ng)
0.6μl PstIプライマー(30ng)
2.0μl 100mM Tris.HCl/15mM MgCl2/500mM KCl,pH 8.5
0.8μl 5mM dNTP
0.1μl Taq ポリメラーゼ(Cetus Perkin Elmer,5単位/μl)13.0μl H2O
全ての反応成分を添加し、十分に混合し、PCRの重要成分、通常酵素を最後に添加した。続いて反応をできるだけ迅速に開始した。
【0089】
増幅をPerkin Elmer 9600 thermal cycler上で実施した。サイクルプロフィールは以下の通りであった。
1サイクル:変性: 94℃で30秒
アニーリング: 65℃で30秒
伸長: 72℃で60秒
11サイクル:変性: 94℃で30秒
アニーリング温度を各サイクルごとに0.7℃ずつ下げて行く。64.3℃,63.6℃,62.9℃,62.2℃,61.5℃,60.8℃,60.1℃,59.4℃,58.7℃,58.0℃,57.3℃.各温度で30秒ずつインキュベートする。
【0090】
伸長: 72℃で60秒
23サイクル:変性: 94℃で30秒
アニーリング: 56℃で30秒
伸長: 72℃で60秒
【0091】
増幅フラグメントのゲル分析
反応産物を4.5%変性ポリアクリルアミドゲル上で実施した。50×38cmゲルを使用し、これらのゲルを製造するためにゲルカセットをBioradから入手した。4.5% w/v アクリルアミド/0.225% w/v ビスアクリルアミド/7.5M 尿素/50mM Tris/50mM ホウ酸/1mM EDTA,pH 8.3を含むゲル溶液100mlを使用した。ゲルにキャストする直前に、100mlゲル溶液を500μl 10% 過硫酸アンモニウム及び100μl TEMEDと混合した。Tris/ホウ酸/EDTA−緩衝液を電気泳動緩衝液として使用した。これらは100mM Tris/100mM ホウ酸/2mM EDTA,pH 8.3を含んでいた。反応混合物を98% ホルムアミド/10mM EDTA/0.01% w/vブロモフェノールブルー/0.01% w/vキシレンシアノールの等量(20μl)と混合した。得られた混合物を95℃で3分間加熱し、次いですぐに氷冷した。各サンプル2μlをゲル上に載せた。電気泳動時、ゲルに一定の電圧110ワットをかけると、一定の発熱(thermal development)があった。これらの条件下のゲルの磁場強度は、40〜50ボルト/cmに相当した。
【0092】
SRFA反応の結果を図14に示す。レーンを1〜28まで番号付け、各々、7つのプライマー組み合わせの1つを持つ4種類のトマト系を含んでいる。ゲル上のトマト系の順番は、1.GCR26、2.GCR151、3.GemR、4.GemSであった。レーン1〜4はMseIプライマー1及びPstIプライマー1で増幅したこれらのDNAを含み、レーン5〜8はMseIプライマー1及びPstIプライマー2で増幅したこれらのDNAを含み、レーンを9〜12はMseIプライマー1及びPstIプライマー3で増幅したこれらのDNAを含み、レーン13〜16はMseIプライマー1及びPstIプライマー4で増幅したこれらのDNAを含み、レーン17〜20はMseIプライマー1及びPstIプライマー5で増幅したこれらのDNAを含み、レーン21〜24はMseIプライマー1及びPstIプライマー6で増幅したこれらのDNAを含み、並びにレーン25〜28はMseIプライマー1及びPstIプライマー7で増幅したこれらのDNAを含む。ゲルはサイズマーカーを含まないが、DNAフラグメントを図の下部に±200ヌクレオチド、上部に±500ヌクレオチドに対応して可視化させた。
【0093】
実施例3
2種の制限酵素を用いた種々のLACTUCA種のDNAの選択的制限フラグメント増幅
実施例2ではトマトDNAにおいて、2種の制限酵素を使用する選択的制限フラグメント(SRFA)増幅の原理を例証した。この実施例においては、同じ2種の制限酵素PstI及びMseIを使用して種々のLactuca種のDNAについても同様の結果が得られることを示す。
【0094】
DNAの単離及び修飾
種々のLactuca種の幼葉材料を使用し、実施例1に記載のごとくDNAを単離した。下記に示すように、かかる植物には市販用レタス品種(L.sativa)と、2種の野生Lactuca種L.saligna及びL.virosaとが含まれていた。植物は下記のごとき恣意的番号で示す。
1.L.saligna,nr.21,植物1
2.L.saligna,nr.21,植物2
3.L.saligna,nr.22,植物1
4.L.saligna,nr.22,植物2
5.L.virosa, nr.01,植物1
6.L.virosa, nr.01,植物2
7.L.virosa, nr.02
8.L.virosa, nr.03,植物1
9.L.virosa, nr.03,植物2
10.L.sativa,市販用バターヘッド(butterhead)品種
【0095】
分析した遺伝物質は、4種の植物で2つの異なる個体を含み、6種の植物タイプを示した。
【0096】
LactucaDNAを修飾してSRFAの鋳型を生成するのは、実施例2に記載の方法と同様に実施した。
【0097】
PstI−MseIフラグメントの増幅
上述のごとく調製したDNAをSRFA反応の鋳型として使用した。1つのMseIプライマーと2つの異なるPstIプライマーとを用い、2種のプライマーの組合せを使用した。かかるプライマーは下記の通りである(選択的ヌクレオチドは小文字で示した):
MseIプライマー :5-GATGAGTCCTGAGTAAaca-3
PstIプライマー1:5-GACTGCGTACATGCAGaa-3
PstIプライマー2:5-GACTGCGTACATGCAGca-3
【0098】
上記プライマーを使用したPstI−MseIフラグメントの増幅は、実施例2に記載したのと全く同様に実施し、実施例2に記載のごとく変性ポリアクリルアミドゲル上で、生成されたフラグメントを可視化した。得られたバンドパターンを図15に示す。レーン1〜10は、PstIプライマー1と組合わせたMseIプライマーを用いて増幅したDNA 1〜10を示し、レーン11〜20は、PstIプライマー2と組合わせたMseIプライマーを用いて増幅したDNA 1〜10を示す。ヌクレオチド内のサイズマーカー(この図では見えない)はゲルの右側に示されている。バンドパターンの差異は、種々の植物の類縁関係の差異を反映している。
【0099】
実施例4:種々の制限酵素の組合せによるトウモロコシ同系交配系の選択的制限フラグメント増幅
実施例2及び3ではそれぞれトマトDNA及びレタス(Lactuca種)DNAを使用し、2種の制限酵素を使用する選択的制限フラグメント(SRFA)増幅の原理を例証した。この実施例においては、トウモロコシ(Zeaトウモロコシ)系においても同様の結果が得られることを示す。更に、種々の制限酵素の組合せを使用し、この場合にはトウモロコシ系のDNAフィンガープリントが得られることも示す。
【0100】
DNAの単離及び増幅
番号1及び2を付した2種のトウモロコシ同系交配系を使用した。かかるトウモロコシ系源は、本発明者らの経験によれば選択した任意の系がSRFAを使用して優れたDNAフィンガープリントを与えるので、重要でない。Saghai−Mahoof et al.,(1984),Proc.Natl.Acad.Sci.U.S.A.81,8014−8018に記載のごとく、幼葉材料からかかる系のDNAを単離した。以下の制限酵素の組合せ(EKs)を使用して鋳型DNAを作製した:PstI/TaqI、EcoRI/TaqI、AseI/TaqI、Sse8387−I/TaqI。全ての酵素はPharmaciaから購入したが、但しAseIはNew England Biolabsから購入し、Sse8387IはAmershamから購入した。鋳型DNAは実質的に実施例2及び3に記載のごとく調製したが、但し下記の点が異なる。
【0101】
まずTaqIと一緒に65℃で1時間インキュベートし、次いで第2の酵素、即ちPstI、AseI、EcoRIまたはSse8387Iと一緒に37℃で更に1時間インキュベートすることにより、DNAの制限を実施した。アダプターの連結は、下記のアダプターを使用して実施例2に記載のごとく実施した:
TaqIアダプター:5-GACGATGAGTCCTGAC-3
3-TACTCAGGACTGGC-5
PatI及びSse8387Iアダプター:
5-bio-CTCGTAGACTGCGTACATGCA-3
3-CATCTGACGCATGT-5
AseIアダプター:5-bio-CTCGTAGACTGCGTACC-3
3-CTGACGCATGGAT-5
EcoRIアダプター:5-bio-CTCGTAGACTGCGTACC-3
3-CTGACGCATGGTTAA-5
【0102】
制限フラグメントの増幅
実施例2に記載のごとく制限フラグメントを増幅した。増幅産物を標識するために選択したプライマーは、(小文字で示した)3つの選択的ヌクレオチドを有する下記のTaqIプライマーであった:
TaqIプライマー 1.5-TGAGTCCTGACCGAacc-3
(5’標識) 2.5-TGAGTCCTGACCGAaca-3
3.5-TGAGTCCTGACCGAcaa-3
4.5-TGAGTCCTGACCGAcac-3
【0103】
上記4つのプライマーを、4種類全ての酵素の組合せを用いた増幅産物の検出に使用した。各酵素の組合せにおいて、他方の酵素に対して4つのプライマーを選択し、各酵素に対して全部で16種の組合せを与えた。これらのプライマーを下記に示す(選択的ヌクレオチドは小文字で示す)。EcoRI及びAseIに対しては3つの選択的ヌクレオチドを含むプライマーを選択し、PstIに対しては2つの選択ヌクレオチドを含むプライマーを選択し、SseIに対しては単一の選択ヌクレオチドを含むプライマーを選択した。トウモロコシゲノムDNAを切断する頻度の少ない酵素に対しては、選択的ヌクレオチドのより少ない伸長部を含むプライマーを選択した。
EcoRIプライマー:1.5-CTGCGTTACCAATTCcaa-3
2.5-CTGCGTTACCAATTCaca-3
3.5-CTGCGTTACCAATTCaac-3
4.5-CTGCGTTACCAATTCcag-3
AseIプライマー: 1.5-GACTGCGTACCTAATaac-3
2.5-GACTGCGTACCTAATaag-3
3.5-GACTGCGTACCTAATacc-3
4.5-GACTGCGTACCTAATgaa-3
PstIプライマー: 1.5-GACTGCGTACATGCAGac-3
2.5-GACTGCGTACATGCAGaa-3
3.5-GACTGCGTACATGCAGca-3
4.5-GACTGCGTACATGCAGcc-3
Sse8387Iプライマー:1.5-GACTGCGTACATGCAGGa-3
2.5-GACTGCGTACATGCAGGg-3
3.5-GACTGCGTACATGCAGGc-3
4.5-GACTGCGTACATGCAGGt-3
【0104】
実施例2に記載の方法に従って全部で128種のPCRを実施した(2DNA×4酵素の組合せ×16プライマーの組合せ)。これらのPCR反応産物を実施例2に記載のごとき(48レーン/ゲルを含む)3種のゲルにおいて分析した。全てのプライマーの組合せで、1レーン当たり50〜100バンドのDNAフィンガープリントが得られたが、但しSseI/TaqIの組合せにおいては1レーン当たり10〜15のバンドしか得られなかった。1つのゲルの例を図16に示す。この図は、PstI/TaqI及びEcoRI/TaqIの酵素の組合せを用いて得られたDNAフィンガープリントの分析におけるゲルの一部を示している。レーン1〜8はそれぞれTaqIプライマー3とPstIプライマー1、2、3及び4とを用いたSRFAにより得られた2種のトウモロコシDNAのDNAフィンガープリントを示しており、レーン9〜16はそれぞれTaqIプライマー4とPstIプライマー1、2、3及び4とを用いたSRFAにより得られた2種のトウモロコシDNAのDNAフィンガープリントを示しており、レーン17はPstIを用いて制限したサイズマーカーλDNAを示しており〔ヌクレオチド内のフラグメントの幾つかのサイズは右側に示してある〕、レーン18〜25はそれぞれTaqIプライマー1とEcoRIプライマー1、2、3及び4とを用いたSRFAにより得られた2種のトウモロコシDNAのDNAフィンガープリントを示している。
【0105】
実施例5:細菌DNAの選択的制限フラグメント
実施例2、3及び4においてはそれぞれトマト、レタス(Lactuca種)及びトウモロコシのDNAにおいて、2種の制限酵素を使用する選択的制限フラグメント(SRFA)増幅の原理を例証した。この実施例においては、この方法を使用して細菌DNAを特性分析し得ることを示す。細菌における本発明方法の有用性を示すため、ベルギーのGentにあるthe Laboratory of Microbiologyから多数のXanthomonas campestris株を入手した。
【0106】
DNAの単離及び増幅
全てのDNAは、種々の資源、ほとんどは感染植物から単離したXanthomonas campestris株から調製した。番号1〜26を付したこれらの株を下記に列挙する。これらの株はベルギーのGhentにあるthe Laboratory of Microbiologyから入手し得る。
【0107】
DNA 亜種 pathovar 単離体
1. albilineans 494
2. fragariae 708
3. oryzae oryzae 5047
4. oryzae populi 5743
5. maltophilia 958
6. campestris campestris 568
7. campestris alfalfae 497
8. campestris coracanae 686
9. campestris citri 8655
10. campestris citri 9658
11. campestris citri 9181
12. campestris citri 8657
13. campestris citri 8654
14. campestris citri 8650
15. campestris citri 682
16. campestris citri 681
17. campestris citri 9325
18. campestris citri 9321
19. campestris citri 9176
20. campestris citri 9671
21. campestris citri 9665
22. campestris citri 9182
23. campestris citri 568
24. campestris citri 9167
25. campestris citri 9175
26. campestris citri 9160
【0108】
上記細菌株のDNAをMarmur(J.Mol.Biol.3,208〜218)によって記載されているように単離した。実質的に実施例4に記載のごとくDNAを制限したが、但し制限酵素としてTaqI及びApaIを選択した。アダプターの連結は、下記のアダプターを使用して実施例4に記載のごとく実施した:
TaqIアダプター:5-GACGATGAGTCCTGAC-3
3-TACTCAGGACTGGC-5
ApaIアダプター:5-bio-TCGTAGACTGCGTACAGGCC-3
3-CATCTGACGCATGT-5
【0109】
制限フラグメントの増幅
実施例2に記載のごとく制限フラグメントを増幅した。SRFAに選択したプライマーは、TaqIプライマー 5-CGATGAGTCCTGACCGAg-3(小文字で示した1つの選択的ヌクレオチドを有する)と、ApaIプライマー 5-GACTGCGTACAGGCCCg-3(小文字で示した1つの選択的ヌクレオチドを有する)であった。実施例2に記載のごとく増幅フラグメントを検出するために、ApaIプライマーの5’末端を標識した。
【0110】
上述のプライマーを使用して26種のDNAの各々を増幅した。増幅条件は実施例2に記載の通りであったが、但し、実施例2、3及び4の植物DNAと比較してDNAの複雑性がより低いため、PCRの最後の9サイクルは省略した。
【0111】
この実施例に記載の細菌DNAを用いて得られたDNAフィンガープリントを図17に示す。レーン1〜26は細菌DNA1〜26を示す。ヌクレオチド内のマーカーDNAのサイズ(ゲル上には見えない)はゲルの右側に示されている。この図は、細菌株の類縁関係がバンドパターンの類似性に反映されることを明らかに示している。
【0112】
実施例6
2種の制限酵素を用いた種々の動物のDNAの選択的制限フラグメント増幅
先の実施例では種々の資源の植物DNAにおいて、選択的制限フラグメント増幅(SRFA)を例証した。ここでは、種々の家畜から得られるランダムなDNA試料を使用し、本発明方法の有効性を説明する。試験した動物種は:Gallus domesticus(ニワトリ);Susscrofa domestica L.(ブタ);Bos taurus(ウシ);Equus caballus(ウマ)であった。使用した制限酵素はSse8387I及びMseIであった。
【0113】
DNAの単離及び修飾
Maniatisら(1989)によって記載された方法に従い、血液試料からDNAを単離した。DNA試料1〜3(ニワトリ)、4〜7(ブタ)、8〜11(ウシ)及び12〜15(ウマ)を制限酵素Sse8387I及びMseIによって消化した。実施例2に記載のごとく、DNAフラグメントをアダプターに連結した。制限酵素Sse8387I及びPstIは互換性のある3’突出部を生成するので、本発明者らは実施例2に記載のごときPstI及びMseIアダプターを使用し得た。
【0114】
制限フラグメントの増幅
上記のごとく番号を与え、実施例2に記載のごとく調製した鋳型DNAはSRFA反応における鋳型として作用した。使用したプライマーの組合せは、1つのMseIプライマーと異なるSseIプライマーからなった:
MseIプライマー: 5-GATGAGTCCTGAGTAAtac-3
Sse8387Iプライマー1:5-GACTGCGTACATGCAGGaa-3
Sse8387Iプライマー2:5-GACTGCGTACATGCAGGag-3
【0115】
上述のプライマー対を使用したSse8387I−MseIフラグメントの増幅は、実施例2に記載の方法を使用して実施した。反応生成物を、実施例2に記載の変性ポリアクリルアミドゲルにおいて試験した。上記試料のフィンガープリントを示すオートラジオグラフを図18に示す。レーン1〜15は、Sse8387Iプライマー1と組合わせたMseIプライマーを用いて増幅したDNA1〜15のフィンガープリントを示し、レーン16〜30は、Sse8387Iプライマー2と組合わせたMseIプライマーを用いて得られた同様のパターンを示している。同一種の動物間のフィンガープリントの差異は動物集団内の不均一性(heterogeneity)を反映しているが、総合的なパターンは特定の種に特徴的である。
【0116】
特定の実施態様において、本発明は、所定の特定の(特異的)制限エンドヌクレアーゼに対する制限部位を複数含むと共に、その核酸配列の少なくとも一部分が未知である出発DNAの少なくとも一部分を制御増幅する方法であって、
(a)前記出発DNAを前記特定の制限エンドヌクレアーゼを用いて消化し、それを、それぞれ5’末端及び3’末端を含む対応する一連の制限フラグメントに断片化し、
(b)後述する5’及び3’アダプターがまだ独立した形態になっていないならば、前記特定のエンドヌクレアーゼに対するただ1つの部位をそのヌクレオチド配列内に含む所定の二重鎖オリゴヌクレオチドリンカーを、前記特定のエンドヌクレアーゼを用いて消化し、それぞれかかる5’及び3’アダプターに切断し、
(c)出発DNAから得られた制限フラグメントを、それらの5’及び3’末端で、それぞれ前記3’及び5’アダプターに連結し、それによって、タグをそれぞれ5’及び3’末端に有する、出発DNAのタグ付き制限フラグメント〔そのタグのヌクレオチド配列は、前記特定の制限部位内に含まれるヌクレオチドを含む3’及び5’アダプター配列を含有する〕を生成し、
(d)プライマーに適した鋳型を与えるのに適当であれば、先の連結に先立って前記5’及び3’アダプターが、所定の一定配列のオリゴヌクレオチドセグメントをそれぞれ5’及び3’末端に付加することにより伸長されていない場合、同じ目的で適当であれば、前記タグ付き制限フラグメントの対応末端を前記オリゴヌクレオチドを用いて伸長させ、それによって、前記一定配列によって両末端が伸長されたタグ付き制限フラグメントを得、
(e)前記タグ付きまたは適当であれば伸長された制限フラグメントをハイブリダイジング条件下で2種のオリゴヌクレオチドプライマーと接触させ、
(f)前記プライマーが、前記タグ付きまたは適当であれば伸長された制限フラグメントの5’及び3’末端の鎖〔それ自体は前記プライマーの鋳型として作用する鎖と相補的である〕の末端部分と同じヌクレオチド配列を有する配列を含む場合、前記プライマーはそれぞれ、鋳型鎖の前記所定の特定の制限エンドヌクレアーゼに対する部位の形成に関与するものと相補的なヌクレオチドを含んでおり、
(g)前記プライマーにハイブリダイズした伸長制限フラグメントを、要求されるヌクレオチド及びポリメラーゼの存在下にPCRまたは同様の方法により増幅し、ハイブリダイズしたプライマーを、該プライマーが最初にハイブリダイズした出発DNAの制限フラグメントに沿ってそれらの全長にわたって伸長させ、
(h)前記最後に述べた制限フラグメントを同定または回収する
ことからなる。
【0117】
本発明方法の特定の実施態様においては、少なくとも1つの前記プライマーの求められる伸長方向で最後のヌクレオチドが、前記特定のエンドヌクレアーゼに対する制限部位内に含まれる最後のヌクレオチドに対応しており、該方法は、増幅された出発DNAの制限フラグメントを同定または回収することを含む。
【0118】
本発明の別の特定の実施態様においては、少なくとも1つの前記プライマーは、増幅ステップの間、対応制限フラグメント内で、前記特定のエンドヌクレアーゼに対する制限部位内に含まれる最後のヌクレオチドを超えて、それ自身の伸長の方向に伸びる所定数(1つまたは幾つか)のヌクレオチドからなる選択された配列を含む。
【0119】
上述の方法の特定の実施態様においては、二重鎖DNAリンカーは、全てが相互に異なる特定のエンドヌクレアーゼに対する部位を幾つか含んでおり、該方法は、同じ出発DNAにおいて、かかる制限エンドヌクレアーゼの1種と、もう1種の特定のエンドヌクレアーゼとを使用し、他の特定のエンドヌクレアーゼについては、上記説明において定義したようにヌクレオチド配列が選択されたプライマーを使用し、上述のステップを繰り返すことからなる。
【0120】
上述の方法または本発明のオリゴヌクレオチドは、同じ生体種由来の所定のDNA、例えば細菌、植物、もしくはヒトを含む動物のゲノムDNAまたはそのフラグメントにおいて、それらのなかでのまたは対応する所定の標準DNAに対しての多型現象の同定に適している。これは、調査対象DNAに該方法を実施する、即ち増幅または伸長反応し得る条件下に該オリゴヌクレオチドと接触させ、前記各DNA及び必要によっては前記標準DNAから出発して得られた制限パターンを比較し、DNA多型現象の存在及び適当であればその位置を、種々のDNAの制限フラグメントのサイズ間に認められ差異と相関させることからなる。
【0121】
本発明は更に、その種々のフラグメントが、未切断出発DNAの初期消化物〔これは、同じ所定の特定のエンドヌクレアーゼを用いて当該DNAから生成される〕に対応する配列を有するフラグメント化DNAであって、前記フラグメントの全てが5’及び3’末端にそれぞれ、所定アダプター〔つまり、前記特定のエンドヌクレアーゼに対するただ1つの制限部位を当初含んでいた同じ出発DNAリンカーの切断部分に対応し、更に必要によっては所定の一定配列を用いて伸長された、所定の3’及び5’アダプター〕によってタグが付けられていることを特徴とするフラグメント化DNAに係わる。フラグメント化DNAは、フラグメントを電場の作用下に最初に移動させた適当な支持体、例えばゲル支持体上の移動バンドパターンの形態であり得る。
【0122】
フラグメント化DNAは更に、5’末端から出発して下記の組成を特徴とするオリゴヌクレオチドを含む末端部分を含み得る:
(i)アダプターとして使用される所定のDNA配列と相補的な、10塩基以上且つ30塩基以下のヌクレオチド配列(一定配列)、この直ぐ後に、
(ii)ヌクレオチド配列またはその一部が(ii)に含まれない限り、ステップ(a)で使用された特定の制限エンドヌクレアーゼのターゲット部位と相補的なヌクレオチド配列、この直ぐ後に、
(iii)選択された1以上且つ10未満、例えば長さ1〜5のヌクレオチド配列。
【0123】
本発明は、少なくとも1つの特定の制限エンドヌクレアーゼによって所定のDNAをフラグメントに断片化し、かかるフラグメントを分析するためのキットであって、次のものを含む。
【0124】
−特定の制限エンドヌクレアーゼ;
−前記特定のエンドヌクレアーゼに対するただ1つの部位をそのヌクレオチド配列内に含み、それによって、それぞれ対応する5’及び3’アダプターに切断される二重鎖DNAオリゴヌクレオチドリンカーであって、あとでこのキットのPCRプライマー用鋳型を与え得る5’及び3’部分を与えるに十分なサイズを有する二重鎖DNAオリゴヌクレオチドリンカー;
−一方では、あとで該プライマーの鋳型として作用する鎖と相補的な5’及び3’アダプターの鎖と同じ配列をそれぞれ有するPCRプライマーであって、更に、鋳型鎖内の前記所定の特定の制限部位に対する部位の形成に関与するものと相補的なヌクレオチドを含むプライマー;
−前記リンカーを前記特定の制限エンドヌクレアーゼによって消化してそれぞれ前記5’及び3’アダプターを作製する前に、前記5’アダプターの5’末端もしくは前記3’アダプターの3’末端または両方を伸長するために、或いは、前記5’及び3’アダプターを出発DNAのフラグメントの末端に連結してその後得られたタグ付きフラグメントの伸長のため、前記プライマーを用いたハイブリダイゼーションに十分な長さの部位を生成するための所定(一定)配列のオリゴヌクレオチドセグメント(適当な場合);
−前記特定のエンドヌクレアーゼを用いて消化することにより得られる、断片化試験を実施する所定のDNAに対応するフラグメント化標準DNA(必要による)。
【0125】
上記キットの特定の実施態様は、前記5’及び3’アダプターまたはタグ付きDNAフラグメントの5’及び3’末端の伸長のための前記オリゴヌクレオチドセグメントが同一のヌクレオチド配列を有するものである。
【0126】
別の実施態様においては、該キットのリンカーは、全てが相互に異なる特定のエンドヌクレアーゼに対する固有の部位を幾つか含んでおり、該キットは更に、前記種々の特定のエンドヌクレアーゼを用いて前記リンカーを切断することによりそれぞれ形成される3’及び5アダプターの各々に対応するプライマーを含んでいる。これらのプライマーは、前記リンカーにおいて、前記特定のエンドヌクレアーゼの各々により切断することにより生成される3’及び5’アダプターに関して、請求項8に記載のようなものである。
【0127】
特定の実施態様においては、キットは対応する特定の制限エンドヌクレアーゼに関して上述のごときフラグメント化標準DNAを含み得、各フラグメント化標準DNAは、所定の特定の制限酵素の各々に関係している。
【図面の簡単な説明】
【0128】
【図1】ゲノムDNA分子を制限酵素で切断して得られた標識制限断片の一般的なPCR増幅法の概略図である。
【図2】制限断片の異なる末端、すなわちフラッシュ末端およびスタッガー末端に対するアダプターの結合を示す。
【図3】標識制限断片のPCR増幅を示し、ボックス領域は制限断片に結合するアダプターとPCR増幅に用いるプライマーとを示し、矢印はDNA合成の方向を示す。
【図4】標識制限断片のPCR増幅に関する概略図を示す。
【図5】標識制限断片のPCR増幅に用いる選択的プライマーの一般的設計を示し、矢印は制限断片の各末端における逆反復配列を示し、プライマーの選択性についてはそれぞれ選択的塩基配列と制限断片鋳型DNAの配列との間に完全な整合および全体的な不整合が存在する2つの例で示す。
【図6】アダプター配列に隣接してトリヌクレオチド配列を有する鋳型DNA分子を選択するPCRプライマーを用いた選択的PCR増幅の原理を示す。
【図7】標識制限断片の選択的PCR増幅を示す。
【図8】それぞれ6個の選択的塩基からなる2個のPCRプライマーの組合せ物を用いた断片特異的増幅の原理を示し、各プライマーは制限断片の異なる鎖中に二本鎖構造を形成し、これによってDNA合成を開始させうるプライマー/鋳型複合体を形成する(矢印で示す)。
【図9】選択的制限断片増幅の方法により認識される一般的な配列要素を示し、図はさらに、認識される2つのヌクレオチド配列およびこれら2つの配列を分離する間隔を含む。
【図10】増幅された断片長さ多形性の確認方法で検出されるヌクレオチド配列変化の種類を示す。
【図11】選択性を増大するプライマーを用いるPstIで制限したトマトDNAの増幅結果の分析を含む1.0%アガロースゲルを示す。
【図12】図12は、断片特異的プライマーを用いるトマトDNAの3種の異なるPstI断片に関する特異的増幅結果の分析を含む1.0%アガロースゲルを示す。
【図13】2種のトマト品種の選択的制限断片増幅により得られるDNAフィンガプリントを有する2.5%ポリアクリルアミド/1%アガロースゲルを示す。
【図14】酵素組合せPstI/MseIと共にSRFAを用いる4種のトマト品種のDNAフィンガプリントを有する4.5%変性ポリアクリルアミドゲル部分を示す。
【図15】酵素組合せPstI/MseIと共にSRFAを用いる10種のLactuca品種のDNAフィンガプリントを有する4.5%変性ポリアクリルアミドゲル部分を示す。
【図16】酵素組合せPstI/TaqIおよびEcoRI/TaqIと共にSRFAを用いる2種のコーン品種のDNAフィンガプリントを有する4.5%変性ポリアクリルアミドゲル部分を示す。
【図17】酵素組合せApaI/TaqIと共にSRFAを用いる26種のキサントモナス・キャンペストリス(Xanthomonas campestris)株のDNAフィンガプリントを有する4.5%変性ポリアクリルアミドゲル部分を示す。
【図18】酵素組合せSseI/MseIと共にSRFAを用いる4種の家畜動物、すなわちニワトリ、ブタ、ウシおよびウマの種々異なる個体のDNAフィンガプリントを有する4.5%変性ポリアクリルアミドゲル部分を示す。
【技術分野】
【0001】
本発明は、限定はしないが植物および動物の育種、品種もしくは栽培変種の同定、診断医薬、動物および植物における病気の診断、ヒトにおける遺伝病の同定、家族関係の分析、法医学的分析および微生物分類を包含する種々異なる多くの分野におけるDNAフィンガプリント法およびDNAマーカーの使用に関する。
【0002】
より詳細には本発明は、DNAフィンガプリント法(DNA fingerprinting)および微生物から高等な植物、動物およびヒトに至る範囲のゲノムにおける特定DNAマーカーの検出方法に関する。さらに本発明は、種々異なる応用分野にて本発明の方法に使用される合成DNA分子およびそれに基づく生産物に関する。
【背景技術】
【0003】
1.DNAフィンガプリント法
DNAフィンガプリントまたはDNA分類、並びに他の遺伝子型分類や特性化やDNA同定の方法は個人、変種もしくは種族または品種の遺伝子構成もしくはゲノムにおける類似性または1つ以上の区別的な特徴を特性化することを意味する。一般的規則は遺伝子関係が近縁であるほど同一性が大となり或いはゲノムの類似性が一層充分となり、その結果ゲノムにおける区別的な特徴がより稀となる。これらの類似した或いは区別的な特徴は、DNAを制限エンドヌクレアーゼで切断した後に生物のDNAを分析して示すことができる。制限エンドヌクレアーゼは、短いヌクレオチド配列(一般に長さ4〜8塩基)を認識すると共に2個のDNA鎖を切断して別個の長さを有するDNA断片を生成させる酵素である。制限エンドヌクレアーゼは、その高度の配列特異性により、DNA分子を極めて特異的に切断する。その結果、再現しうるDNA断片群が生成される。DNA断片は、その長さに応じ多孔質マトリックスもしくはゲルで分画して典型的なバンドパターンを生じ、生物の遺伝子構成におけるDNAフィンガプリントを構成することができる。
【0004】
2.DNA多形性
極めて近縁の品種、変種もしくは種族のフィンガプリントを比較すると、DNAフィンガプリントは同一であるか或いは極めて類似する。同一のDNAフィンガプリントにも拘らず相違点が観察される場合、これら相違点はDNA多形性と呼ばれる。これらは、フィンガプリントに出現する新たなDNA断片である。DNAはその位置にて多形性であると言われ、この新規なDNA断片はDNAマーカーとして使用することができる。制限酵素切断により得られるDNAフィンガプリントで検出されるDNA多形性は、制限エンドヌクレアーゼ標的部位を破壊する突然変異、新規な標的部位を形成する突然変異、挿入、欠失または2つの制限部位の間の逆位のようなDNA配列における変化から生じうる。
【0005】
この種のDNA多形性は一般にRFLP、すなわち制限断片長さ多形性と呼ばれる。この種の突然変異的変化は、メンデルの法則で遺伝する場合、真正な遺伝子マーカーとして挙動する。その結果、DNA多形性を他の遺伝子マーカーと全く同様に血統分析、形質の遺伝に関する遺伝子研究、個体の同定などに遺伝子マーカーとして使用することができる。
【0006】
3.DNAフィンガプリント技術
ウィルスを除く殆ど全ての生存する生物につき、これら生物の全ゲノムDNAの制限消化物は、個々のバンドを評価することができないほど多くのバンドをもたらす。したがって、全てのDNAフィンガプリント法は、小割合のDNA断片のみを可視化させてDNAフィンガプリントを構成する単純なバンドパターンを得ると言う原理に基づいている。
【0007】
最も広く使用されている方法は、生物のDNAを制限エンドヌクレアーゼで消化し、制限断片をゲル電気泳動により分画し、分画されたDNA断片を膜上に移動させると共に結合させ、次いでこの膜を特定DNA断片(「プローブ」)とハイブリダイズさせることを含む。DNA断片は二本鎖DNA分子を形成すると共に、膜上のDNA断片は相補的ヌクレオチド配列を有する。プローブを可視化可能なマーカーで標識すれば、プローブが結合したDNA断片を可視化することができる。この手法は一般に「サザンハイブリダイゼーション」と呼ばれる。近縁のゲノムDNA分子においてプローブが結合した対応の制限断片の寸法に差が観察されれば、これらの差はDNA多形性、より詳細には制限断片長さ多形性と呼ばれる。制限断片長さの差は、DNAプローブにより認識される遺伝子座の種々異なる対立型に対応する。DNAフィンガプリント法のためのサザンハイブリダイゼーションも広く使用されているが、この方法は面倒であって時間がかかる。
【0008】
さらに、この方法は解像能が低く、せいぜい単一の反応における単一の遺伝子座もしくは2〜3の遺伝子座を評価するにしか使用することができない。
【0009】
4.ポリメラーゼ連鎖反応
ポリメラーゼ連鎖反応(PCR)技術は、インビトロにて特定DNA断片を合成するための方法である。この方法は、DNA分子上の独特の配列に結合する特定のオリゴヌクレオチドおよび熱安定性のDNAポリメラーゼを使用することに基づく。オリゴヌクレオチドは、DNAの対向鎖にアニールしてそれぞれ新たなDNA鎖の合成を指令するようDNA合成反応でプライマとして作用するように設計される。したがって、1ラウンドの合成においてはプライマー間でDNA分子の完全コピーが作成され、プライマー間のDNAが複製される。各ラウンドのDNA合成の結果、二倍量のDNAが生じて2個のプライマー間で構成されるDNAの増幅をもたらす。その結果、PCR技術は少量の「基質DNA」を用いて正確なDNAセグメントを合成することを可能にする。
【発明の開示】
【発明が解決しようとする課題】
【0010】
本発明は、生物のDNAを少なくとも1種の制限酵素で切断した後に得られる制限断片をPCR法により増幅させる新規な方法を提供する。この新規なPCR法の使用において、使用するオリゴヌクレオチドは公知のDNA配列には指向せずに制限断片の末端を識別するように設計される。このためには、オリゴヌクレオチドリンカー(もしくはアダプタ)を制限断片の末端に付加させることにより該末端を改変する必要がある。この理由は、制限酵素の末端が一般に極く僅かのヌクレオチド(すなわち2〜8個のヌクレオチド)しか持たず、PCR増幅用のプライマーを設計するために使用するには短か過ぎるからである。
【0011】
本発明は、ゲノムDNA分子を制限エンドヌクレアーゼで消化して得られたDNA断片の複雑な混合物から1個もしくはそれ以上の制限断片を増幅させるためのポリメラーゼ連鎖反応技術(PCR)の新規な使用に基づく。本発明の1つの特定の利点は、制限断片の5’および3’末端におけるヌクレオチド配列が決定されない状況においてDNA制限断片の増幅を可能にすることである。このような場合、増幅すべき制限断片の各鎖にハイブリダイズする通常の配列特異的プライマーは規定することができず、したがって増幅目的に当業界で知られた方法を使用することができない。
【0012】
本発明の方法はたとえば2種の異なる方法で使用することができ、2種の異なる使用をもたらす:
(1)PCR技術により増幅すべき1個もしくはそれ以上の制限断片のサブセットをランダムに選択することによるゲノムのDNAフィンガプリント法。さらに本発明は、上記方法に用いるための合成オリゴヌクレオチドをも包含し、さらにこれら方法は法医学的型分類、微生物同定、変種同定、血統分析、および遺伝子形質に関連したDNAマーカーのスクリーニング等に使用とすることができる。
(2)多形性でありうる1種もしくはそれ以上の予備選択されたDNA断片をPCR増幅により同定するための方法。さらに本発明は、前記方法に使用するための特定の合成オリゴヌクレオチドをも包含し、さらにこれら方法はヒトの遺伝病のスクリーニング、植物および動物の育種における耕種学的形質の遺伝の監視、および病気における感染物質の検出等に使用することができる。
【課題を解決するための手段】
【0013】
1.定義
以下の説明および実施例においては多くの用語を使用する。これら用語に与えるべき範囲を含め本明細書および請求の範囲を明瞭かつ一貫して理解するため、次の定義を設ける。
−制限エンドヌクレアーゼ:制限エンドヌクレアーゼもしくは制限酵素は二本鎖DNA分子における特定塩基配列(標的部位)を認識する酵素であって、各標的部位にてDNA分子の両ストランドを切断する。
−制限断片:制限エンドヌクレアーゼによる切断により産生されるDNA分子を制限断片と称する。任意所定のゲノムは特定の制限エンドヌクレアーゼにより個々の制限断片群に消化される。制限エンドヌクレアーゼ切断によって生ずるDNA断片を分離して、ゲル電気泳動により検出する。
−制限断片長さ多形性(RFLP):たとえば2種の近縁生物のゲノムDNAは、多くの部位でそのヌクレオチド配列組成の相違点を示す。これらの相違点が制限エンドヌクレアーゼに関する標的部位に存在すれば、改変された標的部位はその箇所で切断されないだろう。同様に、ヌクレオチド配列変化は他の生物には存在しない新たな標的部位を導入し、その箇所で制限酵素によるDNAの切断を引き起こす。或いは、1種の生物で生ずる、制限エンドヌクレアーゼ用の2つの標的部位の間でのヌクレオチドの挿入もしくは欠失は、これら標的部位の間の間隔を改変する。このために、2種の生物DNAの消化により異なる長さを有する制限断片が生成する。2種の生物のDNAの消化によって生ずる制限断片の長さに多形性が生ずる。
【0014】
−ゲル電気泳動:制限断片を検出するため、寸法を基礎とした二本鎖DNA分子を分画する分析法が必要とされる。この種の分画を達成する最も一般的に用いられる技術は電気泳動である。この種のゲル内でDNA断片が移動する速度はその寸法に依存する。したがって移動距離は、断片長が増大するにつれて減少する。ゲル電気泳動により分画されたDNA断片は、パターンに含まれる断片の個数が少なければ染色法によって直接可視化することができる。
【0015】
−合成オリゴヌクレオチド:好ましくは約10〜約50塩基を有する化学的に合成しうる一本鎖DNA分子を合成オリゴヌクレオチドと称する。一般に、これら合成DNA分子は独特のヌクレオチド配列を有するよう設計されるが、関連配列を有すると共にヌクレオチド配列内の特定位置に異なるヌクレオチド組成を有する分子ファミリーを合成することもできる。合成オリゴヌクレオチドと言う用語は、独特なヌクレオチド配列を有するDNA分子を意味するよう用いられる。混合合成オリゴヌクレオチドと言う用語は、関連する合成オリゴヌクレオチドのファミリーを意味すべく用いられる。
【0016】
−連結:2個の二本鎖DNA分子を互いに共有結合させる酵素リガーゼによって触媒される酵素反応を連結(ligation)と称する。一般に両DNA鎖は互いに共有結合するが、一方の末端の化学的もしくは酵素的改変により2個のストランドの一方の連結を防止することもできる。その場合、2個のDNA鎖の一方にのみ共有結合が生ずる。
【0017】
−アダプター:たとえば長さ10〜30塩基対のような限られた個数の塩基対を有する、制限断片の末端に連結しうるよう設計される短い二本鎖DNA分子をアダプターと称する。これらアダプターは、部分的に互いに相補的であるヌクレオチド配列を持った2個の合成オリゴヌクレオチドで構成される。2個の合成オリゴヌクレオチドを混合すれば、適当な条件下で溶液中で二本鎖構造が形成されるだろう。アダプター分子の一方の末端は制限断片の末端に連結しうるよう設計され、他方の末端は連結しえないよう設計される。
【0018】
−ポリメラーゼ連鎖反応(PCR):インビトロにて基質DNAからDNA断片を合成する酵素反応をPCRと称する。この反応は、数百〜数千塩基対という短い間隔で分離されるDNA分子におけるヌクレオチド配列に相補的な2個の合成オリゴヌクレオチドの使用、および熱安定性DNAポリメラーゼの使用を含む。連鎖反応は、たとえば10〜30サイクルのシリーズで構成される。各サイクルでは、先ず最初に基質DNAを高温度で変性させる。冷却した後、大過剰で存在する合成オリゴヌクレオチドは、溶液中の基質DNA分子と、相補的ヌクレオチド配列を有する基質DNA分子上の特定部位で二本鎖構造を形成する。次いでオリゴヌクレオチド−基質DNA複合体はDNAポリメラーゼにより触媒されるDNA合成反応の開始部位として作用し、その結果、基質DNA鎖に相補的な新たなDNA鎖が合成される。
【0019】
−DNA増幅:DNA増幅と言う用語は、ポリメラーゼ連鎖反応(PCR)を用いるインビトロでの二本鎖DNA分子の合成を意味すべく使用される。PCR反応の生成物を増幅DNA断片と称する。
【0020】
−プライマー:一般に、プライマーと言う用語はDNAの合成を開始させうるDNA鎖を意味する。DNAポリメラーゼはプライマーなしには新たにDNAを合成することができない。これらは存在するDNA鎖を、組立てるべきヌクレオチドの順序を指令する鋳型として相補鎖が使用される反応において伸長させうるだけである。PCR反応に使用される合成オリゴヌクレオチド分子をプライマーと呼ぶ。
【0021】
−サザンハイブリダイゼーション:サザンブロッティングとも称するサザンハイブリダイゼーションの目的は、アガロースゲル電気泳動により分画されたDNAを、分画過程から生じたDNA断片の相対位置を保持させながら、たとえばナイロン膜もしくはニトロセルロース紙のような支持体上に物理的に移動させることにある。アガロースゲルから支持体への移動を達成すべく用いられる方法は、DNAをゲルから支持体中へ毛細管作用により移動させることである。
【0022】
−核酸ハイブリダイゼーション:核酸ハイブリダイゼーションは、たとえばナイロン膜もしくはニトロセルロース紙のような支持体上で一本鎖DNAのハイブリダイゼーションにより関連DNA配列を検出するために用いられる。相補的塩基配列を有する核酸分子は、適切な条件下で溶液中にて混合すれば二本鎖構造を再形成する。二本鎖構造は、たとえ一方が支持体に固定化されたとしても2個の相補的一本鎖核酸の間で形成される。サザンハイブリダイゼーション法においては後者が生ずる。
【0023】
−ハイブリダイゼーションプローブ:サザンハイブリダイゼーション法で特定のDNA配列を検出するには、標識されたDNA分子またはハイブリダイゼーションプローブをたとえばナイロン膜もしくはニトロセルロースフィルターのような支持体に結合した分画DNAに対し反応させる。標識されたDNAプローブに相補的なDNA配列を有するフィルター上の領域は、再アニーリング反応の結果としてそれ自身も標識される。次いでこの種の標識を示すフィルターの領域を、用いる標識の種類に応じて検出することができる。ハイブリダイゼーションプローブは一般に、トウモロコシ(maize)ゲノムからの特定DNA配列の分子クローニングによって生成される。
【0024】
[発明の実施の形態]
2.好適実施態様の説明
本発明はより詳細には、ポリメラーゼ連鎖反応(PCR)を長さ多形性(length polymorphisms)を含む制限断片多形性(RFP)の検出に用いうる方法および手段に関する。本発明はRFPを検出する方法、本発明の方法に使用するための合成オリゴヌクレオチド、RFPを検出するための手段を含むキット、並びに植物および動物の育種、遺伝病の診断、生物の同定、および法医学的型分類などへの本発明の方法および手法の使用を包含する。
【0025】
詳細には本発明は、個々のゲノム制限断片または、任意の生物、微生物、植物、動物もしくはヒトからの個々に1個もしくはそれ以上の特定の形質に遺伝子的に関連し或いは総合的に生物、変種もしくは個体を同定すべく使用しうるゲノムのフィンガプリントを与える1群のゲノム制限断片を同定するための手段を提供する。
【0026】
制限断片の産生および同定のための本発明による一般的方法は制限エンドヌクレアーゼの使用、制限断片に対する合成オリゴヌクレオチドの結合、および制限断片のPCR増幅(図1)を含む。制限エンドヌクレアーゼは特定部位、すなわち標的部位でゲノムDNA分子を切断することにより制限断片を生成させる。
【0027】
制限断片における末端のヌクレオチド配列が既知であってもなくても制限断片のPCR増幅は、本発明によれば、先ず最初に合成オリゴヌクレオチド(アダプター)を制限断片の末端に結合させてPCR増幅で使用されるプライマーのアンカー塩基として作用する2個の共通標識を各制限断片に付与することにより達成することができる。
【0028】
典型的には、制限酵素は両ストランドの末端ヌクレオチドが塩基対形成するフラッシュ末端(flush ends)、または2本のストランドの一方が突出して短い一本鎖伸長を与えるスタッガー末端(staggered ends)をもたらす(図2)。フラッシュ末端を有する制限断片の場合は、アダプターを1フラッシュ末端と共に使用する。スタッガー末端を有する制限断片の場合は、制限断片の一本鎖伸長に相補的である一本鎖伸長を持ったアダプターを使用する。その結果、それぞれの種類の制限断片につき特定アダプターを使用し、これらアタプターは一方の末端においてのみ相違してアダプターを制限断片に連結させうるようにする。典型的には、用いるアダプターは互いに部分的に相補的な2種の合成オリゴヌクレオチドで構成され、これらオリゴヌクレオチドは一般に長さ約10〜30ヌクレオチド、好ましくは長さ12〜22ヌクレオチドであると共に、溶液中で互いに混合すれば二本鎖構造を形成する。酵素リガーゼを用いて各アダプターを制限断片の混合物に連結させる。制限断片に対し大モル過剰のアダプターを用いれば、全制限断片が両末端でアダプターを保持して終端するよう確保される。この方法により作成される制限断片を標識制限断片と称し、この方法をさらに制限断片標識化と称する。
【0029】
これらアダプターはかくして、後続のPCR増幅反応に用いる上記特徴を備えたプライマーの鋳型として使用することができる。本発明の好適具体例において、制限断片はその両末端に同じアタプターを有し、単一のPCRプライマーを用いて図3に示すように制限断片を増幅することができる。この種の場合、全制限断片が同様に標識されるので、標識制限断片の混合物のPCR増幅は同時様式(synchronous fashion)で全制限断片を増幅することが明らかである。2種の異なる制限酵素を用いてDNAを切断する他の具体例においては、2種の異なるアダプターを制限断片の各末端に連結させる。この場合、2種の異なるPCRプライマーを用いてこの種の制限断片を増幅することができる。2種の制限酵素を用いる他の好適具体例においては、一方の酵素末端に対するアダプターをビオチニル化する。これは制限断片の複雑な混合物からこの制限酵素用に少なくとも1端部を有する制限断片を選択することを可能にし、その際ビオチニル化された分子を分離する常法を用いる。この工程は制限断片の出発混合物の複雑性を減少させると共にPCR増幅に先行する濃縮工程を構成し、或る場合にはこれによってバックグランドを減少させる。数種の異なる断片の同時的増幅はしばしば多重PCRと呼ばれる。多重制限断片増幅の原理については図4に示す。
【0030】
さらに本発明は制御された増幅が可能となるようPCR増幅反応を指令するための特異的に設計されたプライマーおよび特定の方法に基づくものであって、本発明の特定具体例によれば小サブセットの標識制限断片のみが増幅されるようにする。
【0031】
一般にゲノムDNA、特に動物、植物もしくはヒトのゲノムDNAの制限エンドヌクレアーゼ消化物は極めて多数の制限断片をもたらす。制限断片の個数はゲノムの寸法、並びにゲノムにおける制限エンドヌクレアーゼ標的部位の存在頻度に依存し、この存在頻度は次いで主として標的部位におけるヌクレオチドの数により決定される。一般的に使用される制限エンドヌクレアーゼの標的部位におけるヌクレオチドの数は4〜8個の範囲である。生物のゲノム寸法は、微生物の場合における数百万塩基対から動物および植物における数千万塩基対の範囲で広範囲に変化することができる。したがって、制限酵素によりゲノムDNA分子を切断した後に得られる制限断片の個数は数百〜数百万の範囲で変化することができる。一般に制限断片の個数は、ゲル電気泳動により分画されたゲノムDNA消化物において個々の制限断片を同定しえないほど大である。この種の消化物は一般にバンドの汚れをもたらす。
【0032】
したがって標識制限断片のPCR増幅は、全断片が同時的にPCR反応で同時増幅するため、バンドの汚れをもたらす。大寸法のゲノムDNAに対し適用しうる本発明の好適具体例においては、増幅すべき制限断片の個数を制限する一般的原理を用いた。これは、比較的少数の標識制限断片のみがPCR増幅反応に際し増幅されるようサブセットの標識制限断片を予備選択することにより行われる。
【0033】
本発明のこの具体例で規定される選択的原理は、図5に示すように、PCR増幅用のプライマーとして使用されるオリゴヌクレオチドの設計である。
【0034】
標識制限断片は次の一般的構造を有する:逆DNA配列(一定配列)により両側にフランキングする可変DNA配列(標識前の制限断片に対応)。逆DNA配列(一定DNA配列)は、制限エンドヌクレアーゼの標的配列の部分と制限断片の両末端に結合したアダプターの配列の部分で構成される。一定DNA配列の間に含まれる制限断片の可変配列は一般に未知であり、したがってランダム配列組成を有する。その結果、一定DNA配列にフランキングするヌクレオチド配列は制限断片の大混合物中で全体的にランダムである。
【0035】
したがって本発明は一定ヌクレオチド配列部分を含む特定PCRプライマーをも提供し、さらに得られる制限断片の制限サブセットの増幅に依存する本発明の実施態様においては可変配列部分をも含む。一定配列部分においてヌクレオチド配列は、プライマーが制限断片の末端における一方のDNA鎖の一定DNA配列と完全に塩基対形成するよう設計される。可変配列部分は、選択した1〜10塩基の範囲のランダムに選択したヌクレオチド配列で構成される。
【0036】
「可変配列(variable sequence)」と言う用語は一層正確には、サブセットの制限断片を増幅する目的で一定に留まる配列を形成する選択ヌクレオチドよりなる配列を意味する。本発明の特定具体例においては、選択塩基よりなる数種の配列を用いて数種の別個のプライマーを規定することができる。この種の場合、プライマーは同じ一定配列と、形成されるプライマー間で異なる選択塩基により構成される可変配列とを有することができる。
【0037】
PCR工程で増幅される標識制限断片の予備選択を指令するのは、プライマーの3’末端へのこれら可変(選択)配列の付加である。PCR反応を適する条件下で行う場合、プライマはこれら標識制限断片に対しDNA合成のみを開始させ、ここで可変DNA配列は図5に示したように標識制限断片の鋳型ストランドと完全に塩基対を形成することができる。
【0038】
選択は、プライマーの可変配列部分に存在するヌクレオチドの数により決定される。プライマーの選択性は可変(選択)配列部分におけるヌクレオチド数に伴って増加する。さらに選択性塩基と言う用語は可変配列部分におけるヌクレオチドを意味し、これら塩基の選択がプライマーを選択性にすることを示す。使用したプライマーの選択性塩基が断片の末端にて両相補的配列を認識する場合、標識制限断片のみが増幅されることを理解されねばならない。プライマーが1末端にのみ適合(match)してアニーリングする場合、増幅は対数的でなく直線的となり、生成物は未検出のままとなろう。
【0039】
予め異なる個数の選択性塩基を有する可変配列で得られる選択性の程度を推定することができ、一般式42n[ここでnは選択性塩基の個数に等しい]を用いる。1個の選択性塩基を用いて16個の標識断片のうち1個が増幅され、2個の選択性塩基を用いて256個のうち1個が増幅され、3個の選択性塩基を用いて4,096個のうち1個が増幅され、さらに4個の選択性塩基を用いて65,536個のうち1個が増幅され、以下同様である。本発明の1好適具体例は、用いる制限酵素により生成される断片の個数とは無関係に、ランダムサブセットの標識制限断片を任意のゲノムDNA消化物から選択的に増幅することを可能にする。
【0040】
好適具体例において、選択性ヌクレオチドの個数は、増幅される制限断片の個数を5〜200個に制限するよう選択される。この個数は断片の個数を42nで割算して計算しうるが、正確な予測は可能でない。何故なら、必ずしも全ての制限断片を等しい効率で増幅しえないからである。したがって、実際には理論的に予想されるよりも少ない断片が増幅される。さらに、2種(もしくはそれ以上)のプライマーの混合物を用いうることも指摘すべきである。これは各プライマーにより認識される断片の増幅を可能にし、さらに2個のプライマーにより認識される断片の増幅を可能にする。最後に、プライマーの選択性ヌクレオチドと相補的鋳型との間の塩基対形式に基づく選択は、PCR反応におけるアニーリング工程において選択される温度により強く影響を受けることも指摘すべきであり、この温度がプライマー/鋳型複合体の融解温度よりも低く或いはそれに近接すれば、プライマーは不完全な組合わせで鋳型配列にアニーリングして、複合体に誤った組合わせ(mismatch)を生ぜしめる。これは、予想よりも多い断片の増幅をもたらして一層多い変動結果を生ぜしめるため回避されるべきである。
【0041】
本発明により得られるPCR生成物は、寸法によりDNA分子を分離した後にDNA分子を適する試薬で染色する標準的な分画法を用いて同定することができる。或いは、PCR増幅に用いるプライマーを適する放射性標識もしくは蛍光性発色団で標識して、サイズ分画の後に反応生成物を同定することもできる。本発明の好適具体例において、PCR生成物は限定はしないがたとえばアガロース、ポリアクリルアミドまたは混合アガロース/ポリアクリルアミドのような標準的ゲルマトリックスを用いるゲル電気泳動により分画される。本発明により得られるPCR生成物は、増幅制限断片(ARF)と言う用語でも示される。
【0042】
本発明の手段および方法を用いて、任意の複雑なゲノムの制限消化物から1群のARFを生成させることができる。本発明は、得られる制限断片の個数をARFを分離すべく使用するゲル分画システムの解像力に応じて調節することができる。1特定具体例において、選択的プライマーは5〜10個のARFを生成するよう設計され、次いでこれらをアガロースゲル電気泳動により分離する。他の特定具体例は、20〜50個のARFを生成するよう設計された選択性プライマーの使用を含み、次いでこれらARFを限定はしないがたとえばポリアクリルアミドゲルもしくは混合アクリルアミド−アガロースゲルのような高分解能のゲル電気泳動システムによって分離する。
【0043】
1好適具体例において、制限酵素は20〜1000塩基対の寸法範囲にて制限断片をもたらすよう選択される。何故なら、PCR増幅に関して一般的に知られるように、この断片寸法範囲が最も効果的に増幅されるからである。各種の標準ゲルマトリックスでずっと多くの断片を分画しうるが、DNA配列決定に関して現在使用されているような変性ポリアクリルアミドゲル系での分画により最も良好な結果が得られる。
【0044】
本発明によれば、PCR増幅反応にてそれぞれ異なる選択性プライマーを用い種々異なる群のARFが得られる。分離後に同定されるARFのパターンは、ゲノムDNAの独特な完全に再現性のあるフィンガプリントを構成する。この種のフィンガプリントは、限定はしないがたとえば法医学的分類、生物の診断的同定、並びに品種、種族、変種もしくは個体の確認のような数種の用途を有する。同定のレベルは、特定群の種々異なる構成員により示される類似性の程度(変動性の程度)により決定される。変動性もしくは類似性は、関連ゲノムにおけるヌクレオチド組成の変動程度によって決定される。本発明の基礎となる原理は、各増幅制限断片にて図9に示すように所定間隔で互いに分離された2種のヌクレオチド配列を検出する点にある。2種のヌクレオチド配列のそれぞれは2つの部分で構成される:すなわち(a)制限エンドヌクレアーゼのための標的部位、および(b)選択性プライマーに含まれる標的部位に隣接したヌクレオチド配列。関連する生物、品種、変種、種族もしくは個体において、これら配列要素およびその相対的間隔はより大きい或いはより小さい程度で保持される。したがってフィンガプリントは、ゲノム間の配列関係の程度を決定するための基礎を構成する。他方、ARFパターンにおける差を用いて、各ゲノムを互いに区別することもできる。ゲノムの他のフィンガプリント法に優る本発明の特定の利点は、この方法で得られる高い分解能(resolution)である。数十もしくは数百のARFを同時に比較することができる。
【0045】
本発明の他の特定応用は、制限断片多形性(RFP)のスクリーニングおよび同定を含む。ゲノムDNAのヌクレオチド組成の変化は、しばしば制限断片の多形性をもたらす。挿入もしくは欠失はこれらを有する制限断片の寸法に影響を及ぼし(図10)、ヌクレオチド変化は制限エンドヌクレアーゼ標的部位の除去または新たな制限エンドヌクレアーゼ標的部位の形成(図11)をもたらしうる。この種の変化を確認するため最も一般的に使用される技術、はクローン化されたDNAプローブを用いるサザンブロッティング実験であり、すなわち一般に制限断片長さ多形性(RFLP)検出と呼ばれる技術である。この技術は、種々異なるゲノムにおける関連RFLPのためのサザンブロッティング実験におけるランダムにクローン化されたDNA断片の徹底的なスクリーニングを含む。本発明の方法によれば、RFPを種々異なるゲノムから得られたARFを比較して直接に同定することができる。原理的に、本発明の方法はRFPを検出するための感度がより大である。何故なら、制限エンドヌクレアーゼの標的部位における差が検出されるだけでなく、隣接ヌクレオチド配列における差が選択性PCRプライマーに含まれるからである。その結果、本発明の方法はRFLPを検出するためのずっと優秀な方法を構成する。
【0046】
RFLPは法医学的型分類、ヒトにおける遺伝病の監視、並びに植物および動物の育種における耕種学的形質の遺伝の監視を含む数種の用途に現在使用されている。基礎となる原理は、特定の遺伝子特性に密接に関連した或る種のDNA多形性を用いて特定の遺伝子特性の存在もしくは不存在を監視しうる点にある。
【0047】
本発明の方法によれば、ARFパターンの分析を用いて特定の遺伝子特性に対する多形性ARFの遺伝的関係を規定することができる。さらに、この種の多形性ARFは増幅断片長さ多形性(AFLP)と呼ばれ、これらをクローン化DNAプローブを用いてサザンブロッティング実験で検出されるRFLP型DNA多形性から区別することができる。
【0048】
本発明の1特定用途は、特定遺伝子特性に関連したAFLPの検出を含む。この用途は、特定遺伝子特性における差を示す近縁個体のゲノムDNAの制限消化物における異なる選択性プライマーで得られたARFパターンの分析、並びに1種もしくはそれ以上のAFLPの遺伝と特定遺伝子特性により示される表現型との間の相関関係を見出しうる分析技術の使用を包含する。
【0049】
第2の好適な本発明の実施態様は、1種もしくはそれ以上の特定制限断片を同定するための本発明方法の使用を含む。1つの特定制限断片は、先ず最初に制限断片の各末端における最初の8〜12塩基のヌクレオチド配列を決定することにより標識制限断片の複雑な混合物から増幅させることができる。これら配列に基づき2種のプライマーを設計することができ、それぞれ5〜10個の選択性ヌクレオチドは制限断片の相補鎖の制限部位にフランキングする配列に相補的な配列を示す。各群のプライマーを用いて、PCR増幅の後に単一の増幅断片を得ることができる。この方法に用いる制限断片はクローン化制限断片または増幅制限断片のいずれかとすることができる。必ずしも多くの制限断片は極めて効率的に増幅しえないので、多形性DNAマーカーを同定するための本発明の好適方法は先ず最初にランダムに選択された断片群を増幅させ、次いでPCR増幅の後に強いバンドをもたらすAFLPを同定する。これらAFLPは、制限断片特異性プライマーを開発するべく配列決定して特性化することができる。典型的には、AFLPは対応のDNAバンドをゲルから切除し、次いで両末端におけるヌクレオチド配列を決定して制限エンドヌクレアーゼ標的部位に隣接した最初の5〜10ヌクレオチドの配列を確認することにより分離される。これらヌクレオチド配列が知られた後、ゲノムDNA消化物から単一の制限断片のみを増幅する制限断片特異性プライマーを設計することができる。本発明によるこの特定具体例において、2種の異なる選択性プライマーのうち1組を用いて特定制限断片を検出することができる。1組の2種の選択性プライマーのそれぞれにおいて、選択塩基は制限エンドヌクレアーゼ標的部位に隣接したヌクレオチド配列に相補的となるよう選択され、これを図8に示す。各プライマーに含ませる選択塩基の個数は、制限エンドヌクレアーゼ断片混合物の複雑性に依存する。
【0050】
PCR技術は過去数年間にわたり著しく発展し、ヒトの健康管理における最も広く用いられる診断法の1つに急速になりつつある。その用途は特に感染症の検出および遺伝病の検出を含む。各診断試験は2種の特定合成オリゴヌクレオチドの使用に基づくものであり、これらオリゴヌクレオチドをPCR反応におけるプライマーとして用いることにより特定長さの1種もしくはそれ以上のDNA断片を得る。病気検出において試験は1試料当り1個程度に少ないDNA分子の存在を検出して、特徴的DNA断片を得る。遺伝病の場合には、生成物を正常の対立遺伝子と病気の対立遺伝子を区別しうるようプライマーを設計する。区別は、プライマーに対し相補的なゲノム内のDNAセグメントの配列差に依存するか或いは2種のプライマー間における間隔差に依存する。
【0051】
プライマーは極めて高程度の特異性を示すので種々異なる病気を同時に監視することができ、しばしば多重PCRと呼ばれる方法である。しかしながら多重PCR法は、一般に僅か数種(5〜8)の異なる形質しか同時に監視しえない点で限界を有する。この限界の科学的根拠は、PCR増幅の最適条件(アニーリング温度、Mg+濃度、プライマー濃度など)が用いるプライマー対に応じて相当に変化する点である。多重PCRにおいては、全プライマー対が検出可能な生成物を生成する妥協条件を確立せねばならない。さらに、この現象に加えて、種々異なる断片の増幅効率に強力な差が生じる現象がある。その結果、しばしば或る種のプライマー対の生成物が多重PCR反応では検出しえないという問題に遭遇する。
【0052】
本発明の方法は本質的に多重PCRのこれら限界を解消する。何故なら、本発明に用いる全プライマーはそのヌクレオチド配列の実質的部分を共通して有するからである。さらに、AFLPを選択することにより、等しい効率で増幅されるDNAマーカーが選択される。したがって、異なる選択性プライマのための最適なPCR増幅条件は、一般的に使用される配列特異性プライマーで観察されるよりもずっと低い変動を示す。本質的に、合成オリゴヌクレオチドにおける塩基(これら塩基は複雑なゲノムにおける所定寸法の単一DNA断片を検出する所要の特異性を得るのに必要である)の個数(これは上記のように計算する)と、効率的なPCR増幅にとって最適であるオリゴヌクレオチドの長さおよび組成との間の理想的妥協点が存在する。したがって、本発明の方法は多重PCRのかなり優秀な方法を提供する。
【0053】
本発明は、任意のゲノムからDNAマーカーを分離すると共にこの種のDNAマーカーをあらゆる可能なDNAフィンガプリント法の用途に使用するための一般的方法を提供する。
【0054】
以下、限定はしないが実施例および図面により本発明をさらに説明する。
【実施例】
【0055】
実施例1:PSTIを使用するトマトDNAの選択的制限フラグメント増幅
A)DNAの単離及び修飾
全トマトDNA(Lycopersicon esculentum c.v.Moneymaker)をBernatzski及びTanksley(Theor.Appl.Genet.72,314−321)に記載の如く幼葉から単離した。典型的な収量は、新鮮な葉物質1グラム当たりDNA 50−100μgであった。DNAをPstI(Pharmacia)で制限し、二重鎖(ds)PstIアダプターを以下に記載の方法に従って制限フラグメントに連結した。これらのアダプターは以下の構造:
5- CTCGTAGACTGCGTACATGCA -3
3- CATCTGACGCATGT -5
を有していた。これらのアダプター内の3’TGCA−の突出端をPstIにより作った付着端にアニールする。5’C−残基がAに置き換わっているので、PstI認識配列CTGCAGは、このアダプターの連結により修復されない。連結反応は、得られる末端が殆どDNAフラグメント−アダプター分子であるような方法で設計されていた。これは、
1.アダプター−アダプターの連結を排除する非ホスホリル化アダプターを使用する、
2.同時に連結及び制限反応を実施する
ことにより実施した。後者の方法により、任意のフラグメント−フラグメントの連結産物が制限でき、これによりこれらの産物を殆ど完全に除去できた。PstI認識配列はこれらの産物中で修復されないので、アダプター−フラグメント連結産物は制限酵素により制限され得ない。アダプター連結に使用する反応条件は以下の通りであった。
【0056】
2μg トマトDNA
0.2μg アダプター
20単位 PstI
1単位 T4 DNA−リガーゼ
10mM Tris.HAc pH 7.5,10mM MgAc,50mM KAc,
2mM ジチオトレイトール,0.5mM ATP
連結反応を反応容積20μl中37℃で3時間実施した。アダプター連結後、非連結アダプターを選択的に沈殿させることにより除去した。この目的のため、反応混合物を100μlに増やし、NH4Acを終濃度2.5Mで添加した。−20℃のエタノール(100μl)を添加し、混合物を室温で5分間インキュベートした。4℃に冷却したエッペンドルフ遠心分離機中、14000rpmで10分間遠心分離してDNAを集めた。DNAペレットを室温で70%エタノール0.5mlで1回洗浄し、T0.1E(10mM Tris.HCl pH 8.0,0.1mM EDTA)40μlに溶解させた。DNAを−20℃で貯蔵した。本明細書中に記載する選択的沈殿法により、反応混合物から非連結アダプターを効率良く除去したが、小さなDNAフラグメント(<200bp)も失ってしまった。
【0057】
B)増幅方法
上記方法で製造したDNAをPstIフラグメントを増幅するための鋳型として使用した。PCRの反応混合物は、
1ng 鋳型DNA
150ng プライマー
1単位 Taq DNAポリメラーゼ(Perkin Elmer)
200μM 全4dNTP
10mM Tris.HCl pH 8.5,1.5mM MgCl2,50mM KCl
H2O (全容量が50μlになるまで)
を含んでいた。増幅反応中の蒸発を防ぐために、反応混合物を軽質鉱油(light mineral oil)20μlで被覆した。以下のサイクルプロフィール:94℃で1分、60℃で1分、60℃から72℃に温度を1℃/5秒の速度で上昇及び72℃で2.5分を使用してPerkin Elmer DNA Thermal Cycler上でPCRを実施した。全部で33サイクル実施した。反応後、クロロホルム20μl及び充填染料10μl、この場合染料Orange G(Merck)0.1% w/vと一緒に50%蔗糖を添加した。次いでこれを反応混合物と十分に混合し、充填染料を補った反応混合物から有機相(鉱油及びクロロホルム)を分離するために軽く遠心分離した。この反応混合物20μlを1.0%アガロースゲル上で分析した。
【0058】
C)選択性の高いプライマーを使用するトマトDNAの増幅
PstIで制限し、次いでPstIアダプターをつけたトマトDNAを上記条件を使用して増幅した。以下の配列:
1. 5-CTCGTAGACTGCGTACA-3
2. 5-GACTGCGTACAtgcagA-3
3. 5-GACTGCGTACAtgcagAC-3
4. 5-GACTGCGTACAtgcagACC-3
をもつ4つのプライマーを選択した。プライマー1はDNAを修飾するのに使用するアダプターのトップストランドの一部なので、全PstIフラグメントを増幅しなければならない。プライマー2はアダプター配列の一部、PstI認識配列(小文字)及び1個の選択的ヌクレオチド(大文字)を含むので、理論的には全PstIフラグメントの約1/16部分を増幅しなければならない。プライマー3及び4はプライマー2に似ているが、選択的ヌクレオチドをそれぞれ2つ及び3つ含んでいるので、PstIフラグメントの約1/256及び1/4096を増幅すると予想される。反応混合物の一部を1.0%アガロースゲル上で分析し、結果を図11に示す。この図面のレーン1及び6はDNAマーカーを含んでおり、そのサイズを左側に示す。レーン2、3、4及び5は各々プライマー1、2、3及び4で得られたPCRを含んでいる。この結果は、3つの選択的ヌクレオチドを使用したプライマーの場合にのみ、はっきりしたバンドパターンの増幅フラグメント数が得られることを示している。他の3つのプライマーはアガロースゲル上に溶解できなかったバンドパターンを与えたが、これは多くのPCR産物が生成したためである。これらの多くのPCR産物内でも、常に幾つかのフラグメントが目立っており、他のPCR産物の不鮮明なバックグラウンド上でもバンドとして検出される。恐らくこれらの強い産物は、トマト遺伝子上に高コピー数で存在するか、または他の産物よりもずっと効率的に増幅するのだろう。トマト遺伝子DNAのPstIフラグメントの総数(20,000〜100,000)により、3つの選択的ヌクレオチドをもつプライマーが使用されてアガロースゲル上で鮮明なバンドパターンを作ったに違いないと予想された。
【0059】
D)サザンブロット上の増幅フラグメントの分析
増幅フラグメントが同一サイズの真正制限フラグメントに対応することを立証するために、これらの増幅フラグメントをサザンブロット上で試験した。この目的のために、プライマー4で得られた4つの個々のフラグメントをアガロースゲルから切り出した。これらのゲル切片からDNAをガラスビーズ(Gene Clean,製造業者Bio 101)に吸着させることにより精製し、精製DNAの一部を再増幅して4つのDNAフラグメントを各々約1μg得た。続いて再増幅反応を1.0%分取アガロースゲル上で電気泳動させ、所望のDNAフラグメントを精製した。各フラグメント200ngを、製造業者(Boehringer Mannheim)により推奨された方法に従ってランダムヘキサマー標識キットを使用して(α−32P)dATPで標識した。全トマトDNAをPstIで制限し、1.0%アガロースゲル上で電気泳動にかけた。制限DNAを約3μg含む4つのはっきりと分離したレーンを使用した。次に、アガロースゲルを、製造業者(New England Nuclear)により示された遺伝子スクリーン+ハイブリダイゼーション膜にブロットした。ブロット後、ゲルをPstIで制限したトマトDNAを1レーン含む4つの切片に切断した。これらの4つの切片をKlein−Lankhorstら(Theor.Apll.Genet.81,661−667)に記載の方法に従って4つのDNAプローブの一つに各々ハイブリダイズさせた。ハイブリダイズしたブロットをKodak XAR5フィルムを使用してオートラジオグラフに40時間かけた。得られた結果は、4つのDNAプローブにより識別された全てのゲノムDNAフラグメントはこれらのプローブと同一の長さを持つことを示した。これは、プローブとして使用した増幅フラグメントはブロット上で検出されたフラグメント由来であることを示していた。
【0060】
E)単一制限フラグメントの選択的増幅
プライマーの3セットをトマト遺伝子DNA由来の3つの対応するランダムPstI−フラグメント用にデザインしたが、その中でもPstI−認識配列に隣接する配列が公知であった。5つの選択的ヌクレオチドを持つプライマーのセットを以下に示すように製造した。
【0061】
プライマーセット1:
配列1:
5-ctgcagCAGTACCAGC----CCGGCACCTGctgcag-3
5-TGCGTAACATtgcagCAGTA-3 3-TGGACgacgtACATGCGT-5
プライマー1.1 プライマー1.2
【0062】
プライマーセット2:
配列2:
5-ctgcagCCGAATCTCT-----AGTGAGTTAGctgcag-3
5-TGCGTACAtgcagCCGAA-3 3-CAATCgacgtACATGCGT-5
プライマー2.1 プライマー2.2
【0063】
プライマーセット3:
配列1:
5-ctgcagAATACCAAGA-----GCAAGCACAGctgcag-3
5-TGCGTACAtgcagTTATG-3 3-GTGTCgacgtACATGCGT-5
プライマー3.1 プライマー3.2
【0064】
トマトDNAをPstIで消化し、アダプターを上記の如く制限フラグメントの末端に連結した。このDNAを上記セクションの条件のひとつを使用して、プライマーセット1または2または3を使用するPCRの鋳型として使用した。各PCRの反応産物を1.0%アガロースゲル上で分析した。このゲルは図12に示されている。図12は13個のレーンを示しているが、レーン1、2、12及び13はDNAマーカーである。これらのマーカーのkbの大きさをゲルの両側に示す。レーン3、6及び9は、ベクターフラグメント、pUC18(Yanisch−Perronら,Gene 33,103−119)及び挿入PstIフラグメントを産生する、PstIで制限した3つのPstIフラグメントを各々含むプラスミドDNAを示す。レーン4及び5は、各々全ゲノムDNA1ng及び対応するプラスミドDNA5fgのプライマーセット1での増幅を示す。レーン7及び8は、全ゲノムDNA及びプラスミドDNAのプライマーセット2での増幅を示し、レーン10及び11は、プライマーセット3での増幅を示す。これらの結果は、5つの選択的ヌクレオチドをもつプライマーでの選択的制限フラグメント増幅方法を使用すると少なくとも20,000フラグメントの混合物から単一のPstIフラグメントを増幅し得ることを示している。
【0065】
F)SRFAを使用するDNA多型現象の識別
先のセクションにおいて、選択的制限フラグメント増幅法を使用すれば、配列情報が入手可能な場合、ランダムまたは特定のフラグメントのいずれかにおいて制限フラグメントを増幅し得ることが明示された。従って、同一種の2つの個体間で制限部位多型現象に関して調査可能である。非常に近縁であるが系の一方にはネコブセンチュウ耐性遺伝子、Miが存在するという点が異なる2つのトマト系に関してこのことを以下に記載する。このMi−遺伝子はLycopersicon peruvianum、食用トマトL.esculentumの遠縁種に由来する。これを、交雑、次いでL.esculentum親に12回戻し交雑し、さらにMi−遺伝子の存在に関して子孫を選択することによりL.esculentum系に導入した。従って、2つのトマト系は、その遺伝子物質の小さな部分だけ、即ちMi−遺伝子及び周囲領域が異なる。古典的遺伝学方法を使用して、Mi−領域がこの系のゲノムの<1%を構築すると計算した。
【0066】
DNAを2種類のトマト系(系83M−71392,Mi−感受性及び系83M−71398,Mi−耐性;De Ruiter Seeds,Bleiswijk,オランダ)から単離し、続いてPstIで制限し、上記の如くアダプターをつけた。選択的ヌクレオチドの伸長が異なるプライマーを使用して増幅反応を多数回実施した。3つの選択的ヌクレオチドを使用し、単一プライマーの他に異なる2つのプライマーの組み合わせを使用した。反応物を混合ポリアクリルアミド/アガロースゲル上で分析した。その際、アクリルアミド対ビスアクリルアミド20:1の割合で2.5%ポリアクリルアミド及び1.0%アガロースを使用した。1.5mmのスペーサーを使用してProtean IIゲルユニット(Biorad)上にゲルを流した。全部で16の異なるプライマーを使用すると、単一プライマーに関しては16の反応となり、2つのプライマーの考え得る全部の組み合わせに関しては120の反応となる。これらの組み合わせの6つを使用するゲルの典型例を図13に示す。このゲルのレーン1及び14はDNAマーカーを含んでおり、そのサイズをゲルの右側にkbで示す。レーン2及び3、4及び5、6及び7などは、2つのトマト系のプライマー対または特異的プライマーでの増幅を含んでいる。制限部位多型現象に関するスクリーニングから多くのフラグメントが産生したが、そのうちの3つは非常に顕著であり、これらは図13のレーン9、11及び12に示されている(小さな丸で示されている)。同一プライマーが両方の反応に存在するので(違いはレーン11の第2プライマーの存在である)、レーン9及び11の多型現象のバンドは同一であると予測した。レーン11及び12の2つの多型現象フラグメントをゲルから切り出し、ゲル切片を18ゲージ針に押し通して壊し、DNAを100mM Tris.HCl pH 8.0,10mM EDTAの200μl中で拡散させることにより溶離してゲル切片から溶出させた。上記の如くこれらのフラグメントの再増幅用に2μlを使用した。T4 DNAポリメラーゼを使用して各フラグメント200ngをブラント端とし、続いてSmaIで制限したプラスミドベクターpUC18(Yanisch−Perronら,Gene 33,103−109)100ngに連結した。連結混合物をE.coliに形質転換し、各フラグメントに関して1つの組換えE.coliクローンを配列分析用に選択した。これらの取り扱い操作は全て、Sambrook、Fritsch及びManiatis:Molecular Cloning,A Laboratory Manual(Cold Spring Harbor Laboratory Press,New York)に記載の標準法を使用して実施した。
【0067】
6つの選択的ヌクレオチドを持つプライマー2セットを、上記の如く2つのフラグメントの配列をベースとして合成した。本発明者は、これらのプライマーセットを使用して各フラグメントを特異的に増幅し得た。トマト系からフラグメント(トマト系由来)だけを増幅した。しかしながら、これらのプライマーセットは、この多型現象を発見するのに使用した3つの選択的ヌクレオチドを持つプライマーに関して当初知見されたのと同一の多型現象を示した。
【0068】
実施例2:2つの制限酵素を使用するトマトDNAの選択的制限フラグメント増幅
実施例1では、選択的制限フラグメント増幅(SRDA)の原理は、トマトDNA及び制限酵素PstIを用いて例証されている。本実施例では、2つの制限酵素、PstI及びMseIを使用してSRFAを説明する。
【0069】
DNAの単離及び修飾
全トマトDNAを実施例1に記載の如く、幼葉から単離した。所謂、同一遺伝子系の2対、GemR及びGemS、各々GCR26及びGCR151[これらの系は、以下の文献に記載されている:Denby及びWilliams,(1962),Can.J.Plant Sci.42,681−685,Smith及びRitchie,(1983),Plant Mol.Biol.Rep.1,41−45]をDNA源として使用した。同一遺伝子の各対の2つの個体は遺伝子的に非常に似ているが、真菌病原体Verticillium albo−atratumに対する耐性を与える特徴の存在が異なる。
【0070】
DNAの修飾の第1段階は、2つの酵素PstI及びMseIでDNAを制限することを含んでいた。DNAの制限及び続くDNAフラグメントへのアダプターの連結を、RL−緩衝液(制限−連結緩衝液)と名付けられ、かつ10mM Tris.HAc/10mM MgAc/50mM KAc/5mM DTT, pH 7.5を含む同一緩衝液中で実施した。
【0071】
PstI及びMseIによるDNAの制限
2.5μg DNA
12.5単位 PstI(Pharmacia,10単位/μl)
12.5単位 MseI(N.E.Biolabs,4単位/μl)
5μl 10×RL−緩衝液
50μlになるまでの量のH2O
インキュベーションを37℃で1時間実施した。
【0072】
DNAの修飾の次段階は、DNAフラグメントの末端へのアダプター分子の連結である。まず、好適な二重鎖アダプター分子を製造しなければならなかった。
【0073】
アダプターの製造
MseIアダプター: 5-GACGATGAGTCCTGAG-3
3-TACTCAGGACTCAT-5
【0074】
このアダプター50pMole/μlの溶液を製造するために、
16マー、5-GACGATGAGTCCTGAG-3 8μg(1430 pMoles)を全容量H2O 28.6μl中の14マー、5-TACTCAGGACTCAT-3 7μg(1430 pMoles)と混合した。
PstIアダプター:5-ビオ-CTCGTAGACTGCGTACATGCA-3
3-CATCTGACGCATGT-5
【0075】
このアダプター5pMoles/μlの溶液を製造するために、ビオチニル化21マー、5-ビオ-CTCGTAGACTGCGTACATGCA-3 5.25μg(715 pMoles)を全容量H2O 143μl中の14マー、5-TGTACGCAGTCTAC-3 3.5μg(715 pMoles)と混合した。
【0076】
アダプター分子の連結
制限DNAに、以下のもの:
1μl PstI ビオ−アダプター(=5 pMol)
1μl MseI アダプター(=50 pMol)
1.2μl 10mM ATP
1μl 10×RL−緩衝液
1単位 T4 DNAリガーゼ(Pharmacia,5単位/μl)
10μlになるまでの量のH2O
を含む混合物10μlを添加した。
【0077】
得られた反応混合物60μlを37℃で3時間インキュベートした。
【0078】
制限部位が連結後に修復されないようにアダプターをデザインした。制限酵素が連結反応時にまだ活性であるから、フラグメントコンカテマーが制限され、故に、この方法ではフラグメント−フラグメント連結を避けられた。アダプターはホスホリル化されていなかったので、アダプター−アダプター連結はできなかった(実施例1参照)。
【0079】
ビオチニル化DNAフラグメントの選択
2つの制限酵素を使用するSRFA用の鋳型−DNAの製造は、通常、単一酵素を用いるSRFAを使用する際には使用しない追加の段階を含んでいた。この段階において、ビオチニル化アダプターに連結するDNAフラグメントを他のすべてのフラグメントから分離した。
【0080】
ビオチニル化フラグメントをこの段階で非−ビオチニル化フラグメント(MseI−MseI−フラグメント)から常磁性ストレプトアビジンビーズ(Dynal)に結合させることにより分離した。10μlビーズを100μl STEX(100mM NaCl/10mM Tris.HCl/1mM EDTA/0.1% Triton X−100 pH 8.0)中で洗浄し、140μl STEX中に再懸濁させた。次いでビーズを連結混合物に添加すると、終容量200μlとなった。これを緩やかに撹拌しながら室温で30分間インキュベートすると、ビオチニル化DNAフラグメントが正しくビーズに結合できた。マグネットをビーズを含むチューブを近づけることによってこのビーズを集めた。こうすると、上清を他のチューブにピペットで移す際にビーズが移されない。ビーズを1度洗浄し、続いて新しいチューブに移した。次いでビーズを200μl STEXで3回洗浄した。最終的にビーズを200μl T01.E(10mM Tris/0.1mM EDTA,pH 8.0)中に再懸濁させ、新しいチューブに移した。DNAを4℃で保持した。
【0081】
制限酵素で制限し、アダプターをつけ、常磁性ストレプトアビジンビーズにつけ、上記の如く製造したMseI−MseIフラグメントから精製したDNAを、以下の段階で鋳型DNAと呼称する。
【0082】
PstI−MseIフラグメントの増幅
上記の如く製造した鋳型DNAは、記載のトマト系からの全PstI−MseIフラグメント、さらに全く内部MseI−フラグメントを含まない少量のPstI−PstI−フラグメントを含まなければならない。この実験において、これらの多くのPstI−MseIフラグメントが、実施例1に記載の如く本質的に増幅により可視化された。この実施例に記載の方法により得られたフラグメントの種類は実施例1に記載のフラグメントよりもずっと小さかったので、増幅産生物のゲル分析は変性アクリルアミドゲル(Maxam及びGilvert,Proc.Natl.Acad.Sci.U.S.A.74,560−564)上で実施した。さらにこの種のゲルはレーン1つあたり100個以上のバンドに分離できた。これは、実施例1に記載のアガロースゲルの約10倍以上であった。フラグメントは、(γ−32P)ATP及びポリヌクレオチドキナーゼで5’末端でPCRプライマーの一つを標識することにより可視化できた。
【0083】
PCRプライマーの標識
標識用に選択したプライマーは、MseIプライマー1と名付けられた19マー、5−GATGAGTCCTGAGTAAgaa−3であり、選択的ヌクレオチドは小文字で表されている。標識は、以下の方法により実施した。
【0084】
3.0μl 18マー(50ng/μl=150ngの溶液から)
5.0μl (γ−32P)ATP(10μCi/μl=50μCiの溶液から)
3.0μl 250mM Tris.HCl/100mM MgCl2/50mM DTT,pH 7.5
0.5μl T4−キナーゼ(Pharmacia 10単位/μl)
18.5μl H2O
【0085】
これにより総容量は30μlとなり、これを37℃で30分間インキュベートした。各PCRに関しては、この5’標識プライマー1μlを添加した。
【0086】
全部で28回PCRを実施し、4つの鋳型DNAを各々7つのプライマーの組み合わせで増幅した。各プライマーの組み合わせは同一MseIプライマー(上記のMseIプライマー1)を含んでいたが、PstIプライマーの選択に応じて変動した。全部で7つのプライマーを選択した(MseIプライマーに関しては、選択的ヌクレオチドは小文字で表されている)。
【0087】
PstIプライマー1:5-GACTGCGTACATGCAGga-3
PstIプライマー2:5-GACTGCGTACATGCAGgt-3
PstIプライマー3:5-GACTGCGTACATGCAGgg-3
PstIプライマー4:5-GACTGCGTACATGCAGag-3
PstIプライマー5:5-GACTGCGTACATGCAGat-3
PstIプライマー6:5-GACTGCGTACATGCAGct-3
PstIプライマー7:5-GACTGCGTACATGCAGta-3
全PSRプライマーは、濃度50ng/μlでH2Oに溶解した。
【0088】
増幅反応
PCR混合物は、以下のものを含んでいた。
2.0μl 鋳型DNA
1.0μl 5’標識MseIプライマー(5ng)
0.5μl 未標識MseIプライマー(25ng)
0.6μl PstIプライマー(30ng)
2.0μl 100mM Tris.HCl/15mM MgCl2/500mM KCl,pH 8.5
0.8μl 5mM dNTP
0.1μl Taq ポリメラーゼ(Cetus Perkin Elmer,5単位/μl)13.0μl H2O
全ての反応成分を添加し、十分に混合し、PCRの重要成分、通常酵素を最後に添加した。続いて反応をできるだけ迅速に開始した。
【0089】
増幅をPerkin Elmer 9600 thermal cycler上で実施した。サイクルプロフィールは以下の通りであった。
1サイクル:変性: 94℃で30秒
アニーリング: 65℃で30秒
伸長: 72℃で60秒
11サイクル:変性: 94℃で30秒
アニーリング温度を各サイクルごとに0.7℃ずつ下げて行く。64.3℃,63.6℃,62.9℃,62.2℃,61.5℃,60.8℃,60.1℃,59.4℃,58.7℃,58.0℃,57.3℃.各温度で30秒ずつインキュベートする。
【0090】
伸長: 72℃で60秒
23サイクル:変性: 94℃で30秒
アニーリング: 56℃で30秒
伸長: 72℃で60秒
【0091】
増幅フラグメントのゲル分析
反応産物を4.5%変性ポリアクリルアミドゲル上で実施した。50×38cmゲルを使用し、これらのゲルを製造するためにゲルカセットをBioradから入手した。4.5% w/v アクリルアミド/0.225% w/v ビスアクリルアミド/7.5M 尿素/50mM Tris/50mM ホウ酸/1mM EDTA,pH 8.3を含むゲル溶液100mlを使用した。ゲルにキャストする直前に、100mlゲル溶液を500μl 10% 過硫酸アンモニウム及び100μl TEMEDと混合した。Tris/ホウ酸/EDTA−緩衝液を電気泳動緩衝液として使用した。これらは100mM Tris/100mM ホウ酸/2mM EDTA,pH 8.3を含んでいた。反応混合物を98% ホルムアミド/10mM EDTA/0.01% w/vブロモフェノールブルー/0.01% w/vキシレンシアノールの等量(20μl)と混合した。得られた混合物を95℃で3分間加熱し、次いですぐに氷冷した。各サンプル2μlをゲル上に載せた。電気泳動時、ゲルに一定の電圧110ワットをかけると、一定の発熱(thermal development)があった。これらの条件下のゲルの磁場強度は、40〜50ボルト/cmに相当した。
【0092】
SRFA反応の結果を図14に示す。レーンを1〜28まで番号付け、各々、7つのプライマー組み合わせの1つを持つ4種類のトマト系を含んでいる。ゲル上のトマト系の順番は、1.GCR26、2.GCR151、3.GemR、4.GemSであった。レーン1〜4はMseIプライマー1及びPstIプライマー1で増幅したこれらのDNAを含み、レーン5〜8はMseIプライマー1及びPstIプライマー2で増幅したこれらのDNAを含み、レーンを9〜12はMseIプライマー1及びPstIプライマー3で増幅したこれらのDNAを含み、レーン13〜16はMseIプライマー1及びPstIプライマー4で増幅したこれらのDNAを含み、レーン17〜20はMseIプライマー1及びPstIプライマー5で増幅したこれらのDNAを含み、レーン21〜24はMseIプライマー1及びPstIプライマー6で増幅したこれらのDNAを含み、並びにレーン25〜28はMseIプライマー1及びPstIプライマー7で増幅したこれらのDNAを含む。ゲルはサイズマーカーを含まないが、DNAフラグメントを図の下部に±200ヌクレオチド、上部に±500ヌクレオチドに対応して可視化させた。
【0093】
実施例3
2種の制限酵素を用いた種々のLACTUCA種のDNAの選択的制限フラグメント増幅
実施例2ではトマトDNAにおいて、2種の制限酵素を使用する選択的制限フラグメント(SRFA)増幅の原理を例証した。この実施例においては、同じ2種の制限酵素PstI及びMseIを使用して種々のLactuca種のDNAについても同様の結果が得られることを示す。
【0094】
DNAの単離及び修飾
種々のLactuca種の幼葉材料を使用し、実施例1に記載のごとくDNAを単離した。下記に示すように、かかる植物には市販用レタス品種(L.sativa)と、2種の野生Lactuca種L.saligna及びL.virosaとが含まれていた。植物は下記のごとき恣意的番号で示す。
1.L.saligna,nr.21,植物1
2.L.saligna,nr.21,植物2
3.L.saligna,nr.22,植物1
4.L.saligna,nr.22,植物2
5.L.virosa, nr.01,植物1
6.L.virosa, nr.01,植物2
7.L.virosa, nr.02
8.L.virosa, nr.03,植物1
9.L.virosa, nr.03,植物2
10.L.sativa,市販用バターヘッド(butterhead)品種
【0095】
分析した遺伝物質は、4種の植物で2つの異なる個体を含み、6種の植物タイプを示した。
【0096】
LactucaDNAを修飾してSRFAの鋳型を生成するのは、実施例2に記載の方法と同様に実施した。
【0097】
PstI−MseIフラグメントの増幅
上述のごとく調製したDNAをSRFA反応の鋳型として使用した。1つのMseIプライマーと2つの異なるPstIプライマーとを用い、2種のプライマーの組合せを使用した。かかるプライマーは下記の通りである(選択的ヌクレオチドは小文字で示した):
MseIプライマー :5-GATGAGTCCTGAGTAAaca-3
PstIプライマー1:5-GACTGCGTACATGCAGaa-3
PstIプライマー2:5-GACTGCGTACATGCAGca-3
【0098】
上記プライマーを使用したPstI−MseIフラグメントの増幅は、実施例2に記載したのと全く同様に実施し、実施例2に記載のごとく変性ポリアクリルアミドゲル上で、生成されたフラグメントを可視化した。得られたバンドパターンを図15に示す。レーン1〜10は、PstIプライマー1と組合わせたMseIプライマーを用いて増幅したDNA 1〜10を示し、レーン11〜20は、PstIプライマー2と組合わせたMseIプライマーを用いて増幅したDNA 1〜10を示す。ヌクレオチド内のサイズマーカー(この図では見えない)はゲルの右側に示されている。バンドパターンの差異は、種々の植物の類縁関係の差異を反映している。
【0099】
実施例4:種々の制限酵素の組合せによるトウモロコシ同系交配系の選択的制限フラグメント増幅
実施例2及び3ではそれぞれトマトDNA及びレタス(Lactuca種)DNAを使用し、2種の制限酵素を使用する選択的制限フラグメント(SRFA)増幅の原理を例証した。この実施例においては、トウモロコシ(Zeaトウモロコシ)系においても同様の結果が得られることを示す。更に、種々の制限酵素の組合せを使用し、この場合にはトウモロコシ系のDNAフィンガープリントが得られることも示す。
【0100】
DNAの単離及び増幅
番号1及び2を付した2種のトウモロコシ同系交配系を使用した。かかるトウモロコシ系源は、本発明者らの経験によれば選択した任意の系がSRFAを使用して優れたDNAフィンガープリントを与えるので、重要でない。Saghai−Mahoof et al.,(1984),Proc.Natl.Acad.Sci.U.S.A.81,8014−8018に記載のごとく、幼葉材料からかかる系のDNAを単離した。以下の制限酵素の組合せ(EKs)を使用して鋳型DNAを作製した:PstI/TaqI、EcoRI/TaqI、AseI/TaqI、Sse8387−I/TaqI。全ての酵素はPharmaciaから購入したが、但しAseIはNew England Biolabsから購入し、Sse8387IはAmershamから購入した。鋳型DNAは実質的に実施例2及び3に記載のごとく調製したが、但し下記の点が異なる。
【0101】
まずTaqIと一緒に65℃で1時間インキュベートし、次いで第2の酵素、即ちPstI、AseI、EcoRIまたはSse8387Iと一緒に37℃で更に1時間インキュベートすることにより、DNAの制限を実施した。アダプターの連結は、下記のアダプターを使用して実施例2に記載のごとく実施した:
TaqIアダプター:5-GACGATGAGTCCTGAC-3
3-TACTCAGGACTGGC-5
PatI及びSse8387Iアダプター:
5-bio-CTCGTAGACTGCGTACATGCA-3
3-CATCTGACGCATGT-5
AseIアダプター:5-bio-CTCGTAGACTGCGTACC-3
3-CTGACGCATGGAT-5
EcoRIアダプター:5-bio-CTCGTAGACTGCGTACC-3
3-CTGACGCATGGTTAA-5
【0102】
制限フラグメントの増幅
実施例2に記載のごとく制限フラグメントを増幅した。増幅産物を標識するために選択したプライマーは、(小文字で示した)3つの選択的ヌクレオチドを有する下記のTaqIプライマーであった:
TaqIプライマー 1.5-TGAGTCCTGACCGAacc-3
(5’標識) 2.5-TGAGTCCTGACCGAaca-3
3.5-TGAGTCCTGACCGAcaa-3
4.5-TGAGTCCTGACCGAcac-3
【0103】
上記4つのプライマーを、4種類全ての酵素の組合せを用いた増幅産物の検出に使用した。各酵素の組合せにおいて、他方の酵素に対して4つのプライマーを選択し、各酵素に対して全部で16種の組合せを与えた。これらのプライマーを下記に示す(選択的ヌクレオチドは小文字で示す)。EcoRI及びAseIに対しては3つの選択的ヌクレオチドを含むプライマーを選択し、PstIに対しては2つの選択ヌクレオチドを含むプライマーを選択し、SseIに対しては単一の選択ヌクレオチドを含むプライマーを選択した。トウモロコシゲノムDNAを切断する頻度の少ない酵素に対しては、選択的ヌクレオチドのより少ない伸長部を含むプライマーを選択した。
EcoRIプライマー:1.5-CTGCGTTACCAATTCcaa-3
2.5-CTGCGTTACCAATTCaca-3
3.5-CTGCGTTACCAATTCaac-3
4.5-CTGCGTTACCAATTCcag-3
AseIプライマー: 1.5-GACTGCGTACCTAATaac-3
2.5-GACTGCGTACCTAATaag-3
3.5-GACTGCGTACCTAATacc-3
4.5-GACTGCGTACCTAATgaa-3
PstIプライマー: 1.5-GACTGCGTACATGCAGac-3
2.5-GACTGCGTACATGCAGaa-3
3.5-GACTGCGTACATGCAGca-3
4.5-GACTGCGTACATGCAGcc-3
Sse8387Iプライマー:1.5-GACTGCGTACATGCAGGa-3
2.5-GACTGCGTACATGCAGGg-3
3.5-GACTGCGTACATGCAGGc-3
4.5-GACTGCGTACATGCAGGt-3
【0104】
実施例2に記載の方法に従って全部で128種のPCRを実施した(2DNA×4酵素の組合せ×16プライマーの組合せ)。これらのPCR反応産物を実施例2に記載のごとき(48レーン/ゲルを含む)3種のゲルにおいて分析した。全てのプライマーの組合せで、1レーン当たり50〜100バンドのDNAフィンガープリントが得られたが、但しSseI/TaqIの組合せにおいては1レーン当たり10〜15のバンドしか得られなかった。1つのゲルの例を図16に示す。この図は、PstI/TaqI及びEcoRI/TaqIの酵素の組合せを用いて得られたDNAフィンガープリントの分析におけるゲルの一部を示している。レーン1〜8はそれぞれTaqIプライマー3とPstIプライマー1、2、3及び4とを用いたSRFAにより得られた2種のトウモロコシDNAのDNAフィンガープリントを示しており、レーン9〜16はそれぞれTaqIプライマー4とPstIプライマー1、2、3及び4とを用いたSRFAにより得られた2種のトウモロコシDNAのDNAフィンガープリントを示しており、レーン17はPstIを用いて制限したサイズマーカーλDNAを示しており〔ヌクレオチド内のフラグメントの幾つかのサイズは右側に示してある〕、レーン18〜25はそれぞれTaqIプライマー1とEcoRIプライマー1、2、3及び4とを用いたSRFAにより得られた2種のトウモロコシDNAのDNAフィンガープリントを示している。
【0105】
実施例5:細菌DNAの選択的制限フラグメント
実施例2、3及び4においてはそれぞれトマト、レタス(Lactuca種)及びトウモロコシのDNAにおいて、2種の制限酵素を使用する選択的制限フラグメント(SRFA)増幅の原理を例証した。この実施例においては、この方法を使用して細菌DNAを特性分析し得ることを示す。細菌における本発明方法の有用性を示すため、ベルギーのGentにあるthe Laboratory of Microbiologyから多数のXanthomonas campestris株を入手した。
【0106】
DNAの単離及び増幅
全てのDNAは、種々の資源、ほとんどは感染植物から単離したXanthomonas campestris株から調製した。番号1〜26を付したこれらの株を下記に列挙する。これらの株はベルギーのGhentにあるthe Laboratory of Microbiologyから入手し得る。
【0107】
DNA 亜種 pathovar 単離体
1. albilineans 494
2. fragariae 708
3. oryzae oryzae 5047
4. oryzae populi 5743
5. maltophilia 958
6. campestris campestris 568
7. campestris alfalfae 497
8. campestris coracanae 686
9. campestris citri 8655
10. campestris citri 9658
11. campestris citri 9181
12. campestris citri 8657
13. campestris citri 8654
14. campestris citri 8650
15. campestris citri 682
16. campestris citri 681
17. campestris citri 9325
18. campestris citri 9321
19. campestris citri 9176
20. campestris citri 9671
21. campestris citri 9665
22. campestris citri 9182
23. campestris citri 568
24. campestris citri 9167
25. campestris citri 9175
26. campestris citri 9160
【0108】
上記細菌株のDNAをMarmur(J.Mol.Biol.3,208〜218)によって記載されているように単離した。実質的に実施例4に記載のごとくDNAを制限したが、但し制限酵素としてTaqI及びApaIを選択した。アダプターの連結は、下記のアダプターを使用して実施例4に記載のごとく実施した:
TaqIアダプター:5-GACGATGAGTCCTGAC-3
3-TACTCAGGACTGGC-5
ApaIアダプター:5-bio-TCGTAGACTGCGTACAGGCC-3
3-CATCTGACGCATGT-5
【0109】
制限フラグメントの増幅
実施例2に記載のごとく制限フラグメントを増幅した。SRFAに選択したプライマーは、TaqIプライマー 5-CGATGAGTCCTGACCGAg-3(小文字で示した1つの選択的ヌクレオチドを有する)と、ApaIプライマー 5-GACTGCGTACAGGCCCg-3(小文字で示した1つの選択的ヌクレオチドを有する)であった。実施例2に記載のごとく増幅フラグメントを検出するために、ApaIプライマーの5’末端を標識した。
【0110】
上述のプライマーを使用して26種のDNAの各々を増幅した。増幅条件は実施例2に記載の通りであったが、但し、実施例2、3及び4の植物DNAと比較してDNAの複雑性がより低いため、PCRの最後の9サイクルは省略した。
【0111】
この実施例に記載の細菌DNAを用いて得られたDNAフィンガープリントを図17に示す。レーン1〜26は細菌DNA1〜26を示す。ヌクレオチド内のマーカーDNAのサイズ(ゲル上には見えない)はゲルの右側に示されている。この図は、細菌株の類縁関係がバンドパターンの類似性に反映されることを明らかに示している。
【0112】
実施例6
2種の制限酵素を用いた種々の動物のDNAの選択的制限フラグメント増幅
先の実施例では種々の資源の植物DNAにおいて、選択的制限フラグメント増幅(SRFA)を例証した。ここでは、種々の家畜から得られるランダムなDNA試料を使用し、本発明方法の有効性を説明する。試験した動物種は:Gallus domesticus(ニワトリ);Susscrofa domestica L.(ブタ);Bos taurus(ウシ);Equus caballus(ウマ)であった。使用した制限酵素はSse8387I及びMseIであった。
【0113】
DNAの単離及び修飾
Maniatisら(1989)によって記載された方法に従い、血液試料からDNAを単離した。DNA試料1〜3(ニワトリ)、4〜7(ブタ)、8〜11(ウシ)及び12〜15(ウマ)を制限酵素Sse8387I及びMseIによって消化した。実施例2に記載のごとく、DNAフラグメントをアダプターに連結した。制限酵素Sse8387I及びPstIは互換性のある3’突出部を生成するので、本発明者らは実施例2に記載のごときPstI及びMseIアダプターを使用し得た。
【0114】
制限フラグメントの増幅
上記のごとく番号を与え、実施例2に記載のごとく調製した鋳型DNAはSRFA反応における鋳型として作用した。使用したプライマーの組合せは、1つのMseIプライマーと異なるSseIプライマーからなった:
MseIプライマー: 5-GATGAGTCCTGAGTAAtac-3
Sse8387Iプライマー1:5-GACTGCGTACATGCAGGaa-3
Sse8387Iプライマー2:5-GACTGCGTACATGCAGGag-3
【0115】
上述のプライマー対を使用したSse8387I−MseIフラグメントの増幅は、実施例2に記載の方法を使用して実施した。反応生成物を、実施例2に記載の変性ポリアクリルアミドゲルにおいて試験した。上記試料のフィンガープリントを示すオートラジオグラフを図18に示す。レーン1〜15は、Sse8387Iプライマー1と組合わせたMseIプライマーを用いて増幅したDNA1〜15のフィンガープリントを示し、レーン16〜30は、Sse8387Iプライマー2と組合わせたMseIプライマーを用いて得られた同様のパターンを示している。同一種の動物間のフィンガープリントの差異は動物集団内の不均一性(heterogeneity)を反映しているが、総合的なパターンは特定の種に特徴的である。
【0116】
特定の実施態様において、本発明は、所定の特定の(特異的)制限エンドヌクレアーゼに対する制限部位を複数含むと共に、その核酸配列の少なくとも一部分が未知である出発DNAの少なくとも一部分を制御増幅する方法であって、
(a)前記出発DNAを前記特定の制限エンドヌクレアーゼを用いて消化し、それを、それぞれ5’末端及び3’末端を含む対応する一連の制限フラグメントに断片化し、
(b)後述する5’及び3’アダプターがまだ独立した形態になっていないならば、前記特定のエンドヌクレアーゼに対するただ1つの部位をそのヌクレオチド配列内に含む所定の二重鎖オリゴヌクレオチドリンカーを、前記特定のエンドヌクレアーゼを用いて消化し、それぞれかかる5’及び3’アダプターに切断し、
(c)出発DNAから得られた制限フラグメントを、それらの5’及び3’末端で、それぞれ前記3’及び5’アダプターに連結し、それによって、タグをそれぞれ5’及び3’末端に有する、出発DNAのタグ付き制限フラグメント〔そのタグのヌクレオチド配列は、前記特定の制限部位内に含まれるヌクレオチドを含む3’及び5’アダプター配列を含有する〕を生成し、
(d)プライマーに適した鋳型を与えるのに適当であれば、先の連結に先立って前記5’及び3’アダプターが、所定の一定配列のオリゴヌクレオチドセグメントをそれぞれ5’及び3’末端に付加することにより伸長されていない場合、同じ目的で適当であれば、前記タグ付き制限フラグメントの対応末端を前記オリゴヌクレオチドを用いて伸長させ、それによって、前記一定配列によって両末端が伸長されたタグ付き制限フラグメントを得、
(e)前記タグ付きまたは適当であれば伸長された制限フラグメントをハイブリダイジング条件下で2種のオリゴヌクレオチドプライマーと接触させ、
(f)前記プライマーが、前記タグ付きまたは適当であれば伸長された制限フラグメントの5’及び3’末端の鎖〔それ自体は前記プライマーの鋳型として作用する鎖と相補的である〕の末端部分と同じヌクレオチド配列を有する配列を含む場合、前記プライマーはそれぞれ、鋳型鎖の前記所定の特定の制限エンドヌクレアーゼに対する部位の形成に関与するものと相補的なヌクレオチドを含んでおり、
(g)前記プライマーにハイブリダイズした伸長制限フラグメントを、要求されるヌクレオチド及びポリメラーゼの存在下にPCRまたは同様の方法により増幅し、ハイブリダイズしたプライマーを、該プライマーが最初にハイブリダイズした出発DNAの制限フラグメントに沿ってそれらの全長にわたって伸長させ、
(h)前記最後に述べた制限フラグメントを同定または回収する
ことからなる。
【0117】
本発明方法の特定の実施態様においては、少なくとも1つの前記プライマーの求められる伸長方向で最後のヌクレオチドが、前記特定のエンドヌクレアーゼに対する制限部位内に含まれる最後のヌクレオチドに対応しており、該方法は、増幅された出発DNAの制限フラグメントを同定または回収することを含む。
【0118】
本発明の別の特定の実施態様においては、少なくとも1つの前記プライマーは、増幅ステップの間、対応制限フラグメント内で、前記特定のエンドヌクレアーゼに対する制限部位内に含まれる最後のヌクレオチドを超えて、それ自身の伸長の方向に伸びる所定数(1つまたは幾つか)のヌクレオチドからなる選択された配列を含む。
【0119】
上述の方法の特定の実施態様においては、二重鎖DNAリンカーは、全てが相互に異なる特定のエンドヌクレアーゼに対する部位を幾つか含んでおり、該方法は、同じ出発DNAにおいて、かかる制限エンドヌクレアーゼの1種と、もう1種の特定のエンドヌクレアーゼとを使用し、他の特定のエンドヌクレアーゼについては、上記説明において定義したようにヌクレオチド配列が選択されたプライマーを使用し、上述のステップを繰り返すことからなる。
【0120】
上述の方法または本発明のオリゴヌクレオチドは、同じ生体種由来の所定のDNA、例えば細菌、植物、もしくはヒトを含む動物のゲノムDNAまたはそのフラグメントにおいて、それらのなかでのまたは対応する所定の標準DNAに対しての多型現象の同定に適している。これは、調査対象DNAに該方法を実施する、即ち増幅または伸長反応し得る条件下に該オリゴヌクレオチドと接触させ、前記各DNA及び必要によっては前記標準DNAから出発して得られた制限パターンを比較し、DNA多型現象の存在及び適当であればその位置を、種々のDNAの制限フラグメントのサイズ間に認められ差異と相関させることからなる。
【0121】
本発明は更に、その種々のフラグメントが、未切断出発DNAの初期消化物〔これは、同じ所定の特定のエンドヌクレアーゼを用いて当該DNAから生成される〕に対応する配列を有するフラグメント化DNAであって、前記フラグメントの全てが5’及び3’末端にそれぞれ、所定アダプター〔つまり、前記特定のエンドヌクレアーゼに対するただ1つの制限部位を当初含んでいた同じ出発DNAリンカーの切断部分に対応し、更に必要によっては所定の一定配列を用いて伸長された、所定の3’及び5’アダプター〕によってタグが付けられていることを特徴とするフラグメント化DNAに係わる。フラグメント化DNAは、フラグメントを電場の作用下に最初に移動させた適当な支持体、例えばゲル支持体上の移動バンドパターンの形態であり得る。
【0122】
フラグメント化DNAは更に、5’末端から出発して下記の組成を特徴とするオリゴヌクレオチドを含む末端部分を含み得る:
(i)アダプターとして使用される所定のDNA配列と相補的な、10塩基以上且つ30塩基以下のヌクレオチド配列(一定配列)、この直ぐ後に、
(ii)ヌクレオチド配列またはその一部が(ii)に含まれない限り、ステップ(a)で使用された特定の制限エンドヌクレアーゼのターゲット部位と相補的なヌクレオチド配列、この直ぐ後に、
(iii)選択された1以上且つ10未満、例えば長さ1〜5のヌクレオチド配列。
【0123】
本発明は、少なくとも1つの特定の制限エンドヌクレアーゼによって所定のDNAをフラグメントに断片化し、かかるフラグメントを分析するためのキットであって、次のものを含む。
【0124】
−特定の制限エンドヌクレアーゼ;
−前記特定のエンドヌクレアーゼに対するただ1つの部位をそのヌクレオチド配列内に含み、それによって、それぞれ対応する5’及び3’アダプターに切断される二重鎖DNAオリゴヌクレオチドリンカーであって、あとでこのキットのPCRプライマー用鋳型を与え得る5’及び3’部分を与えるに十分なサイズを有する二重鎖DNAオリゴヌクレオチドリンカー;
−一方では、あとで該プライマーの鋳型として作用する鎖と相補的な5’及び3’アダプターの鎖と同じ配列をそれぞれ有するPCRプライマーであって、更に、鋳型鎖内の前記所定の特定の制限部位に対する部位の形成に関与するものと相補的なヌクレオチドを含むプライマー;
−前記リンカーを前記特定の制限エンドヌクレアーゼによって消化してそれぞれ前記5’及び3’アダプターを作製する前に、前記5’アダプターの5’末端もしくは前記3’アダプターの3’末端または両方を伸長するために、或いは、前記5’及び3’アダプターを出発DNAのフラグメントの末端に連結してその後得られたタグ付きフラグメントの伸長のため、前記プライマーを用いたハイブリダイゼーションに十分な長さの部位を生成するための所定(一定)配列のオリゴヌクレオチドセグメント(適当な場合);
−前記特定のエンドヌクレアーゼを用いて消化することにより得られる、断片化試験を実施する所定のDNAに対応するフラグメント化標準DNA(必要による)。
【0125】
上記キットの特定の実施態様は、前記5’及び3’アダプターまたはタグ付きDNAフラグメントの5’及び3’末端の伸長のための前記オリゴヌクレオチドセグメントが同一のヌクレオチド配列を有するものである。
【0126】
別の実施態様においては、該キットのリンカーは、全てが相互に異なる特定のエンドヌクレアーゼに対する固有の部位を幾つか含んでおり、該キットは更に、前記種々の特定のエンドヌクレアーゼを用いて前記リンカーを切断することによりそれぞれ形成される3’及び5アダプターの各々に対応するプライマーを含んでいる。これらのプライマーは、前記リンカーにおいて、前記特定のエンドヌクレアーゼの各々により切断することにより生成される3’及び5’アダプターに関して、請求項8に記載のようなものである。
【0127】
特定の実施態様においては、キットは対応する特定の制限エンドヌクレアーゼに関して上述のごときフラグメント化標準DNAを含み得、各フラグメント化標準DNAは、所定の特定の制限酵素の各々に関係している。
【図面の簡単な説明】
【0128】
【図1】ゲノムDNA分子を制限酵素で切断して得られた標識制限断片の一般的なPCR増幅法の概略図である。
【図2】制限断片の異なる末端、すなわちフラッシュ末端およびスタッガー末端に対するアダプターの結合を示す。
【図3】標識制限断片のPCR増幅を示し、ボックス領域は制限断片に結合するアダプターとPCR増幅に用いるプライマーとを示し、矢印はDNA合成の方向を示す。
【図4】標識制限断片のPCR増幅に関する概略図を示す。
【図5】標識制限断片のPCR増幅に用いる選択的プライマーの一般的設計を示し、矢印は制限断片の各末端における逆反復配列を示し、プライマーの選択性についてはそれぞれ選択的塩基配列と制限断片鋳型DNAの配列との間に完全な整合および全体的な不整合が存在する2つの例で示す。
【図6】アダプター配列に隣接してトリヌクレオチド配列を有する鋳型DNA分子を選択するPCRプライマーを用いた選択的PCR増幅の原理を示す。
【図7】標識制限断片の選択的PCR増幅を示す。
【図8】それぞれ6個の選択的塩基からなる2個のPCRプライマーの組合せ物を用いた断片特異的増幅の原理を示し、各プライマーは制限断片の異なる鎖中に二本鎖構造を形成し、これによってDNA合成を開始させうるプライマー/鋳型複合体を形成する(矢印で示す)。
【図9】選択的制限断片増幅の方法により認識される一般的な配列要素を示し、図はさらに、認識される2つのヌクレオチド配列およびこれら2つの配列を分離する間隔を含む。
【図10】増幅された断片長さ多形性の確認方法で検出されるヌクレオチド配列変化の種類を示す。
【図11】選択性を増大するプライマーを用いるPstIで制限したトマトDNAの増幅結果の分析を含む1.0%アガロースゲルを示す。
【図12】図12は、断片特異的プライマーを用いるトマトDNAの3種の異なるPstI断片に関する特異的増幅結果の分析を含む1.0%アガロースゲルを示す。
【図13】2種のトマト品種の選択的制限断片増幅により得られるDNAフィンガプリントを有する2.5%ポリアクリルアミド/1%アガロースゲルを示す。
【図14】酵素組合せPstI/MseIと共にSRFAを用いる4種のトマト品種のDNAフィンガプリントを有する4.5%変性ポリアクリルアミドゲル部分を示す。
【図15】酵素組合せPstI/MseIと共にSRFAを用いる10種のLactuca品種のDNAフィンガプリントを有する4.5%変性ポリアクリルアミドゲル部分を示す。
【図16】酵素組合せPstI/TaqIおよびEcoRI/TaqIと共にSRFAを用いる2種のコーン品種のDNAフィンガプリントを有する4.5%変性ポリアクリルアミドゲル部分を示す。
【図17】酵素組合せApaI/TaqIと共にSRFAを用いる26種のキサントモナス・キャンペストリス(Xanthomonas campestris)株のDNAフィンガプリントを有する4.5%変性ポリアクリルアミドゲル部分を示す。
【図18】酵素組合せSseI/MseIと共にSRFAを用いる4種の家畜動物、すなわちニワトリ、ブタ、ウシおよびウマの種々異なる個体のDNAフィンガプリントを有する4.5%変性ポリアクリルアミドゲル部分を示す。
【特許請求の範囲】
【請求項1】
標的DNAから得られる制限フラグメントのサブセットを増幅するためのオリゴヌクレオチドプライマーであって、ただし該制限フラグメントはアダプター配列と連結されており、該オリゴヌクレオチドプライマーが前記アダプター配列に相補的な配列、制限エンドヌクレアーゼで消化した後に残る制限部位の部分に相補的な配列、および該標的配列の3’末端に直ぐ隣接した、制限フラグメントのサブセットの選択的増幅を可能にする1〜10個の選択性ヌクレオチドから成り、下記:
・PstIアダプターと連結した制限フラグメントの増幅を可能にするプライマー:
【化1】

・MseIアダプターと連結した制限フラグメントの増幅を可能にするプライマー:
【化2】
・TaqIアダプターと連結した制限フラグメントの増幅を可能にするプライマー:
【化3】
・EcoRIアダプターと連結した制限フラグメントの増幅を可能にするプライマー:
【化4】
・AseIアダプターと連結した制限フラグメントの増幅を可能にするプライマー:
【化5】
・Sse8387Iアダプターと連結した制限フラグメントの増幅を可能にするプライマー:
【化6】
・ApaIアダプターと連結した制限フラグメントの増幅を可能にするプライマー:
【化7】
から選択されるヌクレオチド配列を有することを特徴とする、前記オリゴヌクレオチドプライマー。
【請求項2】
標的DNAから得られる制限フラグメントのサブセットを増幅するためのプライマーのセットであって、該制限フラグメントはアダプター配列と連結されており、該プライマーのセットは請求項1に記載の少なくとも2つのプライマーを含む、前記プライマーのセット。
【請求項3】
標的DNAから得られる制限フラグメントのサブセットを増幅するためのアダプターおよびプライマーのセットであって、該アダプターおよびプライマーのセットは制限フラグメントに連結する少なくとも1つのアダプター並びに10〜50ヌクレオチドを有する少なくとも1つのプライマーを含み、該アダプターおよびプライマーは、下記の群(i)〜(vii):
(i)PstI−アダプター
【化8】
(ii)PstI−アダプター
【化9】
(iii)MseI−アダプター
【化10】
(iv)MseI−アダプター
【化11】
(v)TaqI−アダプター
【化12】
(vi)TaqI−アダプター
【化13】
(vii)PstI−アダプター
【化14】
の中から選択されることを特徴とする、前記アダプターおよびプライマーのセット。
【請求項4】
標的DNAから得られる制限フラグメントのサブセットを増幅するためのキットであって、該制限フラグメントはアダプター配列と連結されており、該キットは請求項1に記載の少なくとも1つのプライマーを含むことを特徴とする前記キット。
【請求項5】
請求項2に記載のプライマーセットを含む請求項4に記載のキット。
【請求項6】
更に、1種または複数種の制限エンドヌクレアーゼを含む請求項5に記載のキット。
【請求項7】
更に、DNAポリメラーゼおよび/またはdNTP、および/または緩衝液溶液を含む請求項6に記載のキット。
【請求項8】
DNAポリメラーゼがTaqポリメラーゼである請求項7に記載のキット。
【請求項9】
遺伝子分析のための請求項4〜8に記載の診断用キット。
【請求項10】
ヒトの法医学的分類のための請求項9に記載のキット。
【請求項11】
動物または植物の形質の遺伝を検出するための請求項9に記載のキット。
【請求項12】
増幅された制限フラグメントのセットであって、
(a) 出発DNAを少なくとも1種の制限エンドヌクレアーゼを用いて消化して、制限フラグメントに断片化し、
(b) 得られた制限フラグメントを、該制限フラグメントの一端または両端に連結するのに適合した一端を有する少なくとも1種の2本鎖合成オリゴヌクレオチドアダプターに連結し、それによって標識付き制限フラグメントを生成し、
(c) 前記標識付き制限フラグメントを、ハイブリダイゼーション条件下に、少なくとも1種のオリゴヌクレオチドプライマーと接触させ、ここで前記プライマーおよびアダプターは、請求項2に記載のものの中から選択され、
(d) 前記1種または複数種のプライマーにハイブリダイズした前記標識付き制限フラグメントを必要なヌクレオチドおよびDNAポリメラーゼの存在下に増幅するか、またはハイブリダイズしたプライマーを前記標識付き制限フラグメントに沿って伸長させ、
(e) ステップ(d)で作製した増幅または伸長したDNAフラグメントを同定または回収する、
ことを含む方法により得られる、増幅された制限フラグメントのセット。
【請求項13】
ゲル、膜またはフィルム上に存在する請求項12に記載の増幅された制限フラグメントのセット。
【請求項14】
少なくとも1種の制限エンドヌクレアーゼを用いて標的DNAから得られる制限フラグメントのサブセットを増幅するためのオリゴヌクレオチドプライマーであって、該オリゴヌクレオチドプライマーが、下記:
・PstIで消化した後に得られる制限フラグメントの増幅を可能にするプライマー:
【化15】
・MseIで消化した後に得られる制限フラグメントの増幅を可能にするプライマー:
【化16】
・TaqIで消化した後に得られる制限フラグメントの増幅を可能にするプライマー:
【化17】
・EcoRIで消化した後に得られる制限フラグメントの増幅を可能にするプライマー:
【化18】
・AseIで消化した後に得られる制限フラグメントの増幅を可能にするプライマー:
【化19】
・Sse8387Iで消化した後に得られる制限フラグメントの増幅を可能にするプライマー:
【化20】
・ApaIで消化した後に得られる制限フラグメントの増幅を可能にするプライマー:
【化21】
から選択されるヌクレオチド配列を有することを特徴とする、前記オリゴヌクレオチドプライマー。
【請求項1】
標的DNAから得られる制限フラグメントのサブセットを増幅するためのオリゴヌクレオチドプライマーであって、ただし該制限フラグメントはアダプター配列と連結されており、該オリゴヌクレオチドプライマーが前記アダプター配列に相補的な配列、制限エンドヌクレアーゼで消化した後に残る制限部位の部分に相補的な配列、および該標的配列の3’末端に直ぐ隣接した、制限フラグメントのサブセットの選択的増幅を可能にする1〜10個の選択性ヌクレオチドから成り、下記:
・PstIアダプターと連結した制限フラグメントの増幅を可能にするプライマー:
【化1】
・MseIアダプターと連結した制限フラグメントの増幅を可能にするプライマー:
【化2】
・TaqIアダプターと連結した制限フラグメントの増幅を可能にするプライマー:
【化3】
・EcoRIアダプターと連結した制限フラグメントの増幅を可能にするプライマー:
【化4】
・AseIアダプターと連結した制限フラグメントの増幅を可能にするプライマー:
【化5】
・Sse8387Iアダプターと連結した制限フラグメントの増幅を可能にするプライマー:
【化6】
・ApaIアダプターと連結した制限フラグメントの増幅を可能にするプライマー:
【化7】
から選択されるヌクレオチド配列を有することを特徴とする、前記オリゴヌクレオチドプライマー。
【請求項2】
標的DNAから得られる制限フラグメントのサブセットを増幅するためのプライマーのセットであって、該制限フラグメントはアダプター配列と連結されており、該プライマーのセットは請求項1に記載の少なくとも2つのプライマーを含む、前記プライマーのセット。
【請求項3】
標的DNAから得られる制限フラグメントのサブセットを増幅するためのアダプターおよびプライマーのセットであって、該アダプターおよびプライマーのセットは制限フラグメントに連結する少なくとも1つのアダプター並びに10〜50ヌクレオチドを有する少なくとも1つのプライマーを含み、該アダプターおよびプライマーは、下記の群(i)〜(vii):
(i)PstI−アダプター
【化8】
(ii)PstI−アダプター
【化9】
(iii)MseI−アダプター
【化10】
(iv)MseI−アダプター
【化11】
(v)TaqI−アダプター
【化12】
(vi)TaqI−アダプター
【化13】
(vii)PstI−アダプター
【化14】
の中から選択されることを特徴とする、前記アダプターおよびプライマーのセット。
【請求項4】
標的DNAから得られる制限フラグメントのサブセットを増幅するためのキットであって、該制限フラグメントはアダプター配列と連結されており、該キットは請求項1に記載の少なくとも1つのプライマーを含むことを特徴とする前記キット。
【請求項5】
請求項2に記載のプライマーセットを含む請求項4に記載のキット。
【請求項6】
更に、1種または複数種の制限エンドヌクレアーゼを含む請求項5に記載のキット。
【請求項7】
更に、DNAポリメラーゼおよび/またはdNTP、および/または緩衝液溶液を含む請求項6に記載のキット。
【請求項8】
DNAポリメラーゼがTaqポリメラーゼである請求項7に記載のキット。
【請求項9】
遺伝子分析のための請求項4〜8に記載の診断用キット。
【請求項10】
ヒトの法医学的分類のための請求項9に記載のキット。
【請求項11】
動物または植物の形質の遺伝を検出するための請求項9に記載のキット。
【請求項12】
増幅された制限フラグメントのセットであって、
(a) 出発DNAを少なくとも1種の制限エンドヌクレアーゼを用いて消化して、制限フラグメントに断片化し、
(b) 得られた制限フラグメントを、該制限フラグメントの一端または両端に連結するのに適合した一端を有する少なくとも1種の2本鎖合成オリゴヌクレオチドアダプターに連結し、それによって標識付き制限フラグメントを生成し、
(c) 前記標識付き制限フラグメントを、ハイブリダイゼーション条件下に、少なくとも1種のオリゴヌクレオチドプライマーと接触させ、ここで前記プライマーおよびアダプターは、請求項2に記載のものの中から選択され、
(d) 前記1種または複数種のプライマーにハイブリダイズした前記標識付き制限フラグメントを必要なヌクレオチドおよびDNAポリメラーゼの存在下に増幅するか、またはハイブリダイズしたプライマーを前記標識付き制限フラグメントに沿って伸長させ、
(e) ステップ(d)で作製した増幅または伸長したDNAフラグメントを同定または回収する、
ことを含む方法により得られる、増幅された制限フラグメントのセット。
【請求項13】
ゲル、膜またはフィルム上に存在する請求項12に記載の増幅された制限フラグメントのセット。
【請求項14】
少なくとも1種の制限エンドヌクレアーゼを用いて標的DNAから得られる制限フラグメントのサブセットを増幅するためのオリゴヌクレオチドプライマーであって、該オリゴヌクレオチドプライマーが、下記:
・PstIで消化した後に得られる制限フラグメントの増幅を可能にするプライマー:
【化15】
・MseIで消化した後に得られる制限フラグメントの増幅を可能にするプライマー:
【化16】
・TaqIで消化した後に得られる制限フラグメントの増幅を可能にするプライマー:
【化17】
・EcoRIで消化した後に得られる制限フラグメントの増幅を可能にするプライマー:
【化18】
・AseIで消化した後に得られる制限フラグメントの増幅を可能にするプライマー:
【化19】
・Sse8387Iで消化した後に得られる制限フラグメントの増幅を可能にするプライマー:
【化20】
・ApaIで消化した後に得られる制限フラグメントの増幅を可能にするプライマー:
【化21】
から選択されるヌクレオチド配列を有することを特徴とする、前記オリゴヌクレオチドプライマー。
【図1】

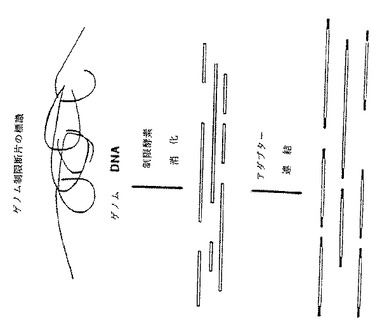
【図2】
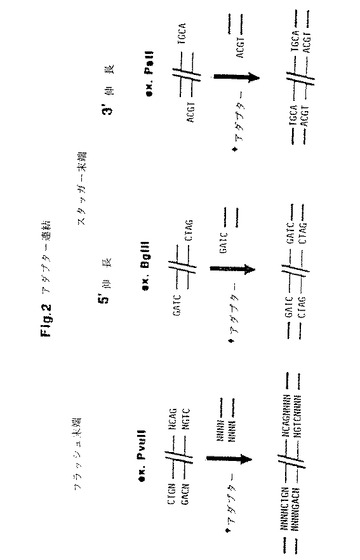
【図3】

【図4】
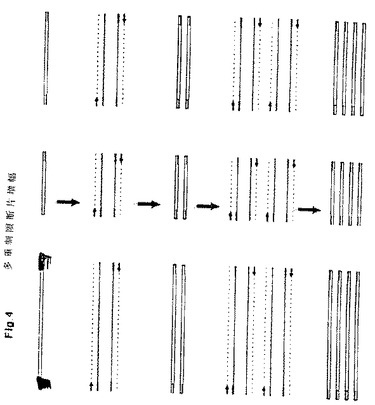
【図5】

【図6】
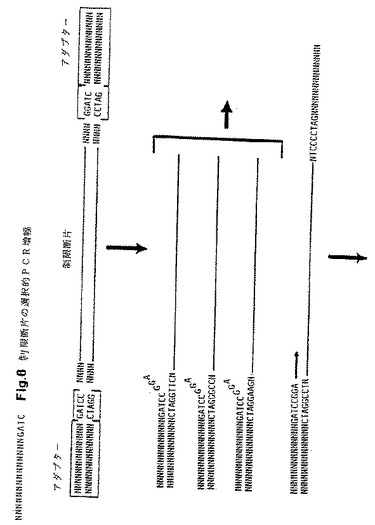
【図7】
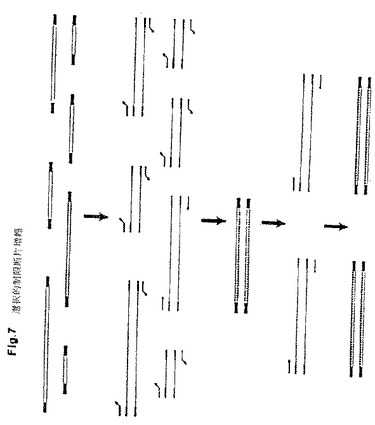
【図8】
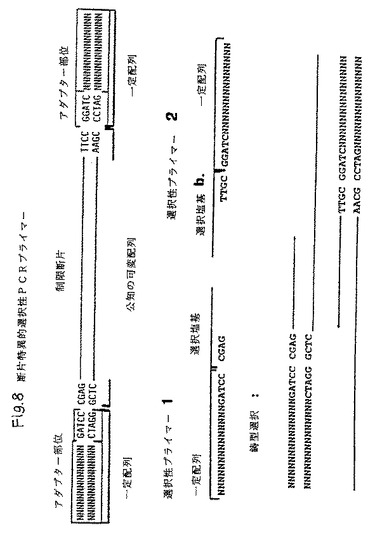
【図9】
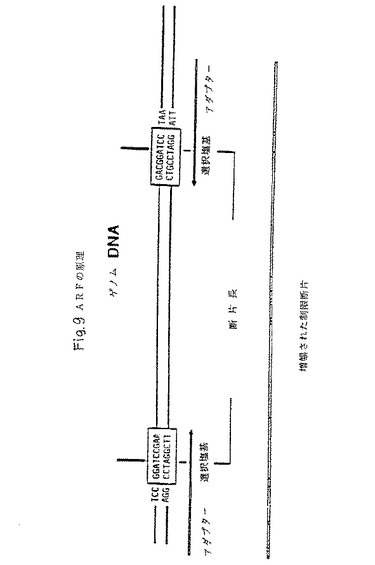
【図10】

【図11】
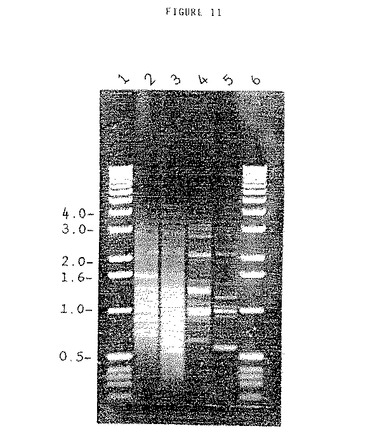
【図12】
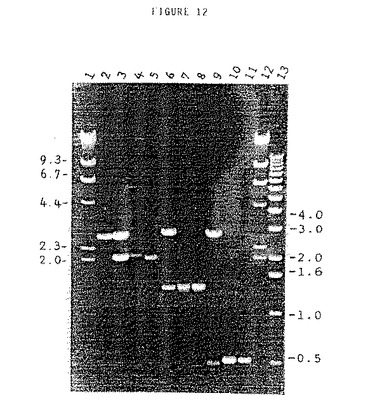
【図13】
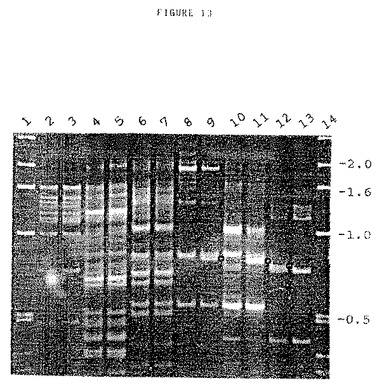
【図14】

【図15】
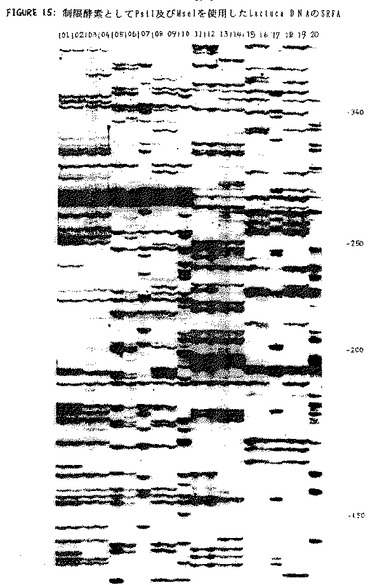
【図16】

【図17】

【図18】
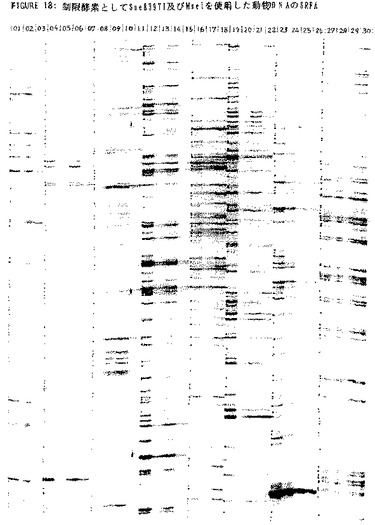
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【公開番号】特開2008−131947(P2008−131947A)
【公開日】平成20年6月12日(2008.6.12)
【国際特許分類】
【出願番号】特願2007−329833(P2007−329833)
【出願日】平成19年12月21日(2007.12.21)
【分割の表示】特願2000−224187(P2000−224187)の分割
【原出願日】平成4年9月24日(1992.9.24)
【出願人】(500345180)
【Fターム(参考)】
【公開日】平成20年6月12日(2008.6.12)
【国際特許分類】
【出願日】平成19年12月21日(2007.12.21)
【分割の表示】特願2000−224187(P2000−224187)の分割
【原出願日】平成4年9月24日(1992.9.24)
【出願人】(500345180)
【Fターム(参考)】
[ Back to top ]