
選択的逐次ポリアシル化法とその装置
【課題】無触媒条件下、カルボン酸無水物とポリヘテロ水素化物からポリアシル化合物を短時間、連続的に高収率・高選択率で合成する方法及びその反応組成物を提供する。
【解決手段】発熱反応の場合、常温流体、吸熱反応の場合、温度100〜400℃、圧力0.1〜40MPaの亜臨界流体、超臨界流体を反応溶媒として使用し、無触媒条件で、発熱反応の場合、流通式常温高圧反応装置、吸熱反応の場合、流通式高温高圧装置に、基質及び反応溶媒を導入し、温度、カルボン酸無水物量の諸条件を変化させることにより、カルボン酸無水物及びヘテロ水素化物からN−アシル化後、O−アシル化し、選択的に逐次ポリアシル化合物を高収率、高選択率、高速・連続的に合成するポリアシル化合物の製造方法、その反応組成物、及びその装置。
【解決手段】発熱反応の場合、常温流体、吸熱反応の場合、温度100〜400℃、圧力0.1〜40MPaの亜臨界流体、超臨界流体を反応溶媒として使用し、無触媒条件で、発熱反応の場合、流通式常温高圧反応装置、吸熱反応の場合、流通式高温高圧装置に、基質及び反応溶媒を導入し、温度、カルボン酸無水物量の諸条件を変化させることにより、カルボン酸無水物及びヘテロ水素化物からN−アシル化後、O−アシル化し、選択的に逐次ポリアシル化合物を高収率、高選択率、高速・連続的に合成するポリアシル化合物の製造方法、その反応組成物、及びその装置。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ポリアシル化合物、その製造方法と装置に関するものであり、更に詳しくは、発熱反応の場合に、常温水、吸熱反応の場合に、高温高圧状態の水あるいは酢酸それらの混合溶媒を反応溶媒とし、無触媒かつ一段階でポリアシル化合物を製造する方法に関するものである。本発明は、常温水あるいは温度100〜400℃、圧力0.1〜40MPaの水あるいは酢酸、それらの混合溶媒を反応溶媒として、触媒無添加で無水カルボン酸とポリヘテロ水素化物からポリアシル化合物を一段階かつ短時間、連続的に合成する方法及びその反応組成物を提供するものである。ここで、ポリヘテロ水素化物におけるヘテロ原子としては、酸素、窒素、硫黄が挙げられ、それぞれポリオール、ポリアミン、ポリチオールに対応し、更に、複数のヘテロ原子が組合わされた化合物も含む。
【0002】
ポリアシル化合物は、原料、基質の機能性を改質向上し、更に、付加価値を付与するため、香料、医薬品、食品分野において有用である。通常、ポリアシル化合物を合成する場合、従来法では、非プロトン性有機溶媒に加えて、酸・塩基触媒が必要であり、食品、医薬品に利用される場合、残存する有機溶媒、触媒の除去は大きな労力を必要とし、環境に影響を与えるのみならず、生体に有害である等の問題点を有していた。本発明は、無水カルボン酸とポリオール、ポリアミン、ポリチオール等ないしは複数種類のヘテロ原子が組み合わされたポリヘテロ水素化物から、無触媒で、水を用いるプロセスのみで、ポリアシル化合物を合成する方法とその反応組成物を提供するものであり、香料、医薬品や食品のみならず、化成品合成にも応用可能であり、ポリアシル化合物を効率良く、短時間で、大量に生産し、提供することを可能にするものである。
【背景技術】
【0003】
従来、無水カルボン酸とポリヘテロ水素化物、ポリオール、ポリアミン、ポリチオールないし複数種類のヘテロ原子が組み合わされたポリヘテロ水素化物からポリアシル化合物を合成する方法が種々報告されている(例えば、非特許文献1参照)。ここで、ポリオールと無水カルボン酸からポリアシル化合物を合成する技術を完成すれば、通常は、ポリアミンやポリチオール等ないしは複数種類のヘテロ原子が組み合わされたポリヘテロ水素化物からポリアシル化合物を合成することが可能となるため、特に、ポリアルコールからポリアシル化合物を合成する技術の報告例は非常に多い。ところが、従来法では、すべてのヘテロ水素をポリアシル化する方法のみであり、アシル化数を調整しながら選択的にポリアシル化する方法は報告されていない(図1)。図1で、R、Rn、Rn+1はアルキル基及びアルキル基以外のヘテロ原子を含む置換基、Qiはヘテロ原子又は置換ヘテロ原子であり、具体的には、酸素(O)、硫黄(S)、窒化水素(NH)、アルキル置換窒素(NR’)、であり、これらの原子の組み合わせも含む。
【0004】
先行技術文献によれば、無溶媒あるいは非プロトン性有機溶媒中、触媒として、CoCl3(非特許文献4)、Me3SiCl(非特許文献5)、塩基であるBu3P(非特許文献6)、ピリジン(非特許文献8)等が使用されてきた。また、安定なアシル中間体形成を経由することで、アシル基を活性化するDMAPの発見は、革新的な技術とされた(特許文献9)。ところが、DMAPは、1等量以上のアミンを利用することから、ルイス酸である金属トリフラートが提案され、Me3SiOTf(非特許文献10)、Sc(OTf)3(特許文献1、2及び非特許文献11)、In(OTf)3(非特許文献12)、Bi(OTf)3(非特許文献13)、Sc(NTf)3(非特許文献14)、が高収率でポリアシル化合物を与えることが示された。更に、V(OTf)3が触媒活性を示さないが、そのオキソ化合物であるV(O)(OTf)2が触媒活性があることが見出され、V=Oの触媒活性化が注目された(特許文献3、非特許文献15)。これらの触媒により、1、2級ポリアルコールから95%以上の収率で、また、3級ポリアルコールから80%以上の収率で、すべてアシル化されたポリアシル化合物が得られると報告されている(図1)。
【0005】
ここで、上記の先行技術文献では、有機塩基、ルイス酸、固体酸のような触媒に加えて、有機溶媒がポリアシル化にとって必要不可欠である。また、高温条件では、不純物が生成し、選択率を低下させるという理由から、アシル化は、常温で行うのが最適であり、高温条件は不適であるとされている(特許文献1、2)。一方、アシル化における溶媒としての水の可能性に関しては、水を除去した粗生成物にアシル化剤を添加し、アシル化する方法が一般的であり、水は負の効果をもたらすとされており(特許文献4)、ある文献では、溶媒として水を列挙しているが、実際には使用されていない(特許文献1、2参照)。ところが、アルドール反応に対する触媒活性と水中でのルイス酸の安定性との相関を元素ごと系統的に比較検討し、他の反応への適用可能性を示唆した例も存在する(非特許文献16)。更に、Bi(OTf)3が触媒の場合には、脱水処理をしていない水を含有する、湿った有機溶媒が反応を促進し、収率向上が観察された事例も存在する(非特許文献13)。したがって、アシル化に対する溶媒としての水の有効性は、これまで明確ではなく、実施もされなかった。
【0006】
他方、Bi(OTf)3を触媒とする場合の無溶媒条件では、収率が低下し、有機溶媒が必要であると報告されている(非特許文献13)。ところが、N−メトキシアセトアミドを触媒として、アミン・アニリン誘導体から有機溶媒又は常温水中、66%以上の収率でN−アシル化する方法が報告され、非ルイス酸触媒を用いた場合には、水が溶媒として利用可能なことが初めて示された。しかし、アシル化剤であるN−メトキシアセトアミドは、無水カルボン酸と比較して、非常に高価であるという問題がある(非特許文献17)。
【0007】
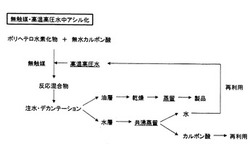
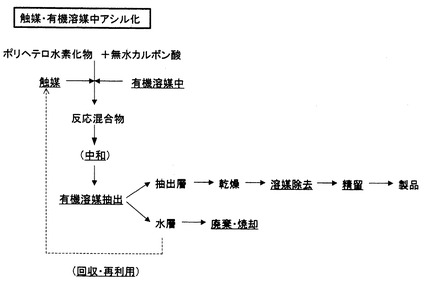
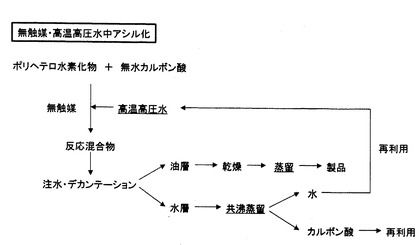
反応後における後処理は、通常の触媒・有機溶媒中アシル化では、反応混合物に中和剤を添加して中和後、抽出溶媒と水あるいは飽和食塩水を加え、分液し、溶媒層は、その後、乾燥、溶媒除去、蒸留あるいは精留のプロセスを得て、目的物を得るが、水層には、水の他に、触媒、有機溶媒、酢酸、基質、生成物、副生成物、無機物の複雑な混合物が含有される。ここで、水層からの触媒の分離が容易である場合には、回収再生され、再使用されるが、分離が困難である場合には、そのまま廃棄・処分される(図2)。無触媒・高温高圧水中アシル化の場合のように、水層に触媒、有機溶媒が含有されず、水、酢酸、生成物のみが含有されるならば、生成物をデカンテーションにより分離後、水層に対して共沸混合物を形成する物質を添加した共沸蒸留を行うことで、水と氷酢酸とに分離することが可能である(特許文献5)。このことは、水の再生を可能にし、通常法に比べて、環境低減型のプロセスであることを意味する(図3)。
【0008】
このように、従来法では、ポリアシル化の場合、触媒及び有機溶媒が必要であるため、製品の品質上、反応後の分離操作において、触媒、有機溶媒やカルボン酸の除去が必要であり、分離操作後の水層は廃棄物となりやすく、廃液の問題を生じる。更に、環境に対する影響や生体への有害性への配慮から、また、ヒトが経口する食品・医薬品の安全性から、触媒・有機溶媒のより高度分離が要求される。高度分離に必要なコストは、合成操作と同程度であり、望ましくは触媒と有機溶媒を使用しない方が良い。以上のことから、当該技術分野においては、簡単、低コスト、環境低減型の合成プロセスで、分離操作が容易かつ高度分離が可能で、触媒や有機溶媒の残存しないアシル化合物の連続的合成を可能とする合成手法が強く要請されていた。
【0009】
【特許文献1】特開平9−169690号公報
【特許文献2】特開平9−176081号公報
【特許文献3】米国特許第6,541,659号明細書
【特許文献4】米国特許第6,005,122号明細書
【特許文献5】米国特許第5,980,696号明細書
【非特許文献1】W.Green,P.G.Wuts,Protective Groups in Organic Synthesis,3rd ed.,Wiley, New York,1999,p150
【非特許文献4】S.Ahmad,J.Iqbal,Chem.Commun.,1987,114
【非特許文献5】N.C.Braus,R.P.Sharma,J.N.Baruah,Tetrahedron Lett.,1983,24,1189;J.Org. Chem.,1987,52,503
【非特許文献6】E.Vedejs,N.S.Bannet,L.M.Conn, S.T.Diver,J.Org.Chem.,1993,58,7286
【非特許文献8】Joseph B.Lambert,Gen Tai Wang, Rodney B.Finzel,and Douglas H.Teramura, J.Am.Chem.Soc.,1987,109.7838
【非特許文献9】G.Hofle,W.Steglich,H.Vorbruggen., Angrew.Chem.Int.Ed.,1978,17,569. b) A.Hassner,L.R.Krepski,V.Alexanian,Tetrahedron,1978,34,2069
【非特許文献10】P.A.Procopiu,S.P.D.Baugh,S.S. Flack,G.G.A.Inglis,J.Org.,Chem.,1998,63, 2342
【非特許文献11】a)K.Ishihara,M.Kubota,H.Kurihara,H.Yamamoto,J.Org.Chem.,1996,61,4560. b)K.Ishihara,M.Kubota,H.Kurihara,H.Yamamoto,J.Am.Chem.Soc.,1995,117,4413
【非特許文献12】K.K.Chauhan,C.G.Frost,I.Love, D.Waite,Synlett,1999,1743
【非特許文献13】J.Otera,A.Orita,C.Tanahashi, A.Kakuta,Angrew.Chem.Int.Ed.,2000,39,2877
【非特許文献14】K.Ishihara,M.Kubota,Synlett, 1996,265
【非特許文献15】C−T.Chen,J−H.Kuo,C−H.Li,N.B. Barhate,S−W.Hon,T−W.Li,S−D.Chao,C−C.Liu, Y−C.Li,I−H.Chang,J−S.Lin,C−J.Liu and Y−C.Chou,Org.Lett.,2001,3,3729
【非特許文献16】S.Kobayashi,S.Nagayama,T.Busujima,J.Am.Chem.Soc.,1998,120,8287
【非特許文献17】Y.Kikugawa,K.Mitsui,T.Sakamoto,M.Kawase,and H.Tamiya,Tetrahedron Lett,1990,31,2,243
【発明の開示】
【発明が解決しようとする課題】
【0010】
このような状況のなかで、本発明者らは、上記従来技術に鑑みて、低コストで、環境に優しい簡便な高速合成プロセスで、上記ポリアシル化合物を連続的かつ選択的に合成することができる新しい合成方法を開発することを目標として鋭意研究を積み重ねた結果、高温高圧水、又は亜臨界水又は超臨界水を反応溶媒とすることで、無触媒で無水カルボン酸とポリヘテロ水素化物からポリアシル化合物を選択的に合成できることを見出し、本発明を完成するに至った。本発明は、無水カルボン酸とポリヘテロ水素化物からポリアシル化合物を無触媒で、短時間の反応条件下で連続的に合成する方法及びその反応組成物を提供することを目的とするものである。
【課題を解決するための手段】
【0011】
上記課題を解決するための本発明は、以下の技術的手段から構成される。
(1)無水カルボン酸とポリヘテロ水素化物との反応組成物であって、触媒及び有機溶媒の残存がないことを特徴とするポリアシル化合物組成物。
(2)無水カルボン酸とポリヘテロ水素化物からポリアシル化合物を合成する方法であって、発熱反応の場合には、常温流体、吸熱反応の場合には、高温高圧状態の亜臨界流体ないしは超臨界流体を反応溶媒として使用し、触媒を用いることなく、無水カルボン酸とポリヘテロ水素化物から一段階の合成反応でポリアシル化合物を選択的に合成することを特徴とするポリアシル化合物の製造方法。
(3)高温高圧状態の亜臨界ないしは超臨界水を反応溶媒として使用する、前記(2)記載の方法。
(4)ポリヘテロ水素化物におけるヘテロ原子又は置換ヘテロ原子が、酸素(O)、硫黄(S)、窒化水素(NH)、アルキル置換窒素(NR’)、又はこれらの原子の組合せである、前記(1)又は(2)記載の方法。
(5)基質におけるヘテロ水素基及びヘテロ水素基に隣接するα位の置換数に対応して、温度と無水カルボン酸の量を調整することにより、N−アシル化後、O−アシル化を実施して、それぞれのアシル化において、無置換(1級)、一置換(2級)、二置換(3級)の順にアシル化しつつアシル化数を調節し、ポリアシル化合物を選択的に合成する、前記(1)又は(2)の方法。
(6)温度100〜400℃、圧力0.1〜40MPaの亜臨界流体ないし超臨界流体を反応溶媒として使用する、前記(2)記載の方法。
(7)常温流体ないし亜臨界流体ないし超臨界流体として、水、酢酸、それ以外の無機溶媒、もしくは有機溶媒もしくは無機溶媒と有機溶媒の混合溶媒を用いる、前記(2)記載の方法。
(8)流通式高温高圧装置に、基質及び反応溶媒を導入し、反応時間を3〜60秒の範囲で変化させることで合成反応を実施する、前記(2)記載の方法。
(9)発熱反応の場合に、流通式常温高圧装置に、基質及び反応溶媒を導入し、反応時間を1〜60秒の範囲で変化させることで合成反応を実施する、前記(2)記載の方法。
(10)水を送液する水送液ポンプ、水加熱用コイル、高温高圧フローセル、基質を送液する反応物送液ポンプ、炉体、反応物を炉体に導入する反応物導入管、反応溶液を排出する排出液ライン、冷却フランジ及び圧力を設定する背圧弁を具備していることを特徴とするポリアシル化合物合成装置。
(11)吸熱反応の場合において、水加熱用コイルが配設されている、前記(10)記載のN−アシル化合物合成装置。
(12)前記(2)記載の方法において、アシル化後、回収水溶液に水を注入してデカンテーションし、油/水二層溶液に分離後、ポリアシル化合物を含む油層を分液回収する一方、水層からは酢酸と水を共沸蒸留によって分離し、回収する簡易な連続分離法。
【0012】
次に、本発明について更に詳細に説明する。
本発明は、化1のカルボン酸無水物と化2のポリヘテロ水素化物から化3のポリアシル化合物を、一段階の反応プロセスで、触媒無添加、短時間の反応条件下で、選択的かつ連続的に合成することを特徴とするものである。本発明では、上記反応溶媒として、発熱反応の場合、常温流体が用いられ、吸熱反応の場合、温度100〜400℃、圧力0.1〜40MPaの亜臨界流体、超臨界流体が用いられ、好適には発熱反応の場合、水、吸熱反応の場合、亜臨界水が用いられる。また、反応条件として、好適には、発熱反応の場合、常温常圧、反応時間が1秒程度に調整され、吸熱反応の場合、温度200〜250℃、圧力5MPa、反応時間が3〜60秒の範囲、好適には10秒程度に調整される。
【0013】
化1の式中、Rはアルキル基又はアルキル基以外のヘテロ原子を含む置換基であり、化2の式中、Rn、Rn+1はアルキル基又はアルキル基以外のヘテロ原子を含む置換基、Qiはヘテロ原子又は置換ヘテロ原子であり、具体的には、酸素(O)、硫黄(S)、窒化水素(NH)、アルキル置換窒素(NR’)、であり、これら原子の組み合わせも含み、また、x、y、zはそれぞれα位が1級、2級、3級であるヘテロ原子の個数を示し、1級、2級、3級の結合順序は限定するものではないため、点線で結合を示している。化3はα位が1級のみであってx個のポリアシル化を示し、化4はα位が1、2級の場合の(x+y)個のポリアシル化を示し、化5はα位が1、2、3級の場合に対する(x+y+z)個のポリアシル化を示す。
【0014】
【化1】

【0015】
【化2】
【0016】
【化3】
【0017】
【化4】
【0018】
【化5】
【0019】
本発明においては、上記基質及び反応溶媒を反応容器に導入して所定の反応時間で合成反応を実施するものである。したがって、上記反応器としては、例えば、バッチ式の常温高圧装置又は高温高圧反応容器、及び連続型の流通式常温高圧装置又は流通式高温高圧反応装置を使用することができるが、本発明は、これらの反応装置型式に特に制限されるものでない。
【0020】
本発明の方法では、反応溶媒として、上記常温流体又は高温高圧状態にある亜臨界流体、超臨界流体が用いられるが、具体的には、亜臨界二酸化炭素(常温以上、0.1MPa以上)、亜臨界水(100℃以上、0.1MPa以上)、亜臨界メタノール(100℃以上、0.1MPa以上)、亜臨界エタノール(100℃以上、0.1MPa以上)、超臨界二酸化炭素(34℃以上、7.38MPa以上)、超臨界水(375℃以上、22MPa以上)、超臨界メタノール(239℃以上、8.1MPa以上)、超臨界エタノール(241℃以上、6.1MPa以上)、同じ状態の混合溶媒が例示され、好適には、亜臨界水(200−250℃、5MPa以上)が用いられる。反応溶媒としては、上記以外の有機溶媒や無機溶媒を任意の割合で含むことができ、具体的には、有機溶媒として、アセトン、アセトニトリル、テトラヒドロフラン等、無機溶媒として酢酸、アンモニア等を含む反応溶液に代替することも可能である。
【0021】
本発明では、上記常温流体、亜臨界流体、超臨界流体の反応溶媒の組成、温度及び圧力条件、基質の種類及びその使用量、反応時間を調整することにより、短時間で、効率良く、反応生成物を合成することができる。また、本発明では、例えば、基質及び反応溶媒を発熱反応の場合に、流通式常温高圧装置に導入し、それらの反応時間を1〜60秒の範囲で変えることにより、あるいは吸熱反応の場合に、流通式高温高圧装置に導入し、それらの反応時間を3〜60秒の範囲で変えることにより、所定の反応生成物を合成することができる。上記反応条件は、使用する出発原料、目的とする反応生成物の種類等により適宜設定することができる。
【0022】
本発明の方法では、従来、触媒存在下で行われていた、カルボン酸無水物とポリヘテロ水素化物からのポリアシル化合物の合成を、高速で連続的に、しかも、無触媒で実施できるため、長時間を要するプロセスを効率化することができる。また、本発明の方法では、従来用いられた触媒を全く使用しないので、反応後の溶液の中和処理、無害化処理等の後処理・処分の必要がなく、環境負荷低減を達成可能である。更に、反応後は、静置分離操作のみであるため、触媒や有機溶媒の分離回収の必要性はなく、生成物分離が容易になる。本発明によれば、触媒無添加で、1〜10秒程度の短時間で、基質が窒素、酸素を含むアミノカルビノール類、アニリノカルビノール類の場合、まず初めに、常温高圧反応装置ないしは高温高圧反応装置で選択的にN−アシル化を実施して、N−アシル化合物を転化率85%以上、選択率99%以上で得た後、引き続き高温高圧反応装置でO−アシル化により、O−アシル化合物を転化率93%以上、選択率87%以上で行い、対応するポリアシル化合物を合成可能である。本発明の合成方法は、香料、医薬品、食品に利用可能な、ポリアシル化合物を効率良く、大量に高速で連続的に生産することを可能にするものとして有用である。
【0023】
従来、二酸化炭素等の亜臨界流体、超臨界流体を利用して、リパーゼや触媒を用いたポリアシル化を実施した例が報告されている。しかし、常温流体、亜臨界水を溶媒とした無触媒条件下で基質の複数の反応点の原子の種類によって、更に、基質の複数の反応点に隣接する1級、2級、3級の骨格に依存して、温度と無水カルボン酸の量を調整することにより、1級、2級、3級の順にアシル化し、アシル化数を調節し、ポリアシル化合物を選択的に合成できることを実証した例はなく、本発明の対象とするポリアシル化合物の選択的合成反応法は、本発明者らによって初めてその有効性が実証されたものである。しかも、従来法でカルボン酸無水物とポリヘテロ水素化物から合成されるポリアシル化合物は、触媒及び有機溶媒の残存が問題とされていたが、本発明で、カルボン酸無水物とポリヘテロ水素化物から合成される反応組成物は、触媒及び有機溶媒の残存がなく、本発明のポリアシル化合物組成物は、従来製品にない利点を有している。
【0024】
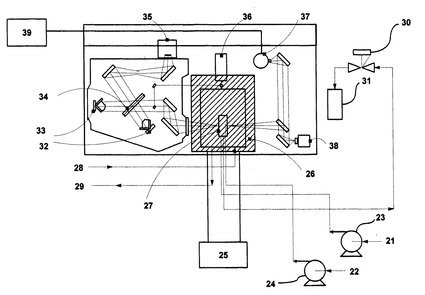
本発明では、無触媒条件で無水カルボン酸とポリヘテロ水素化物の合成反応を実現するために、例えば、基質をあらかじめ溶媒に溶解した溶液を送液し、常温流体、亜臨界流体、超臨界流体中の反応経過を高温高圧赤外フローセル(図4)により赤外分光分析によって観察する流通型高温高圧赤外分光その場測定装置(図5)を用いることも可能である。しかしながら、高温高圧赤外フローセルを窓なし高温高圧フローセル(図6)に交換し、超臨界流体の流れに対して直接反応物の流れを接触反応するように配管配置した方が、高温高圧赤外フローセルにおけるセル窓付近におけるリーク等の問題が発生せず、より高流量で短時間に合成を実施することが可能である。これらのことから、この窓なし高温高圧フローセルを装着した装置を後述する実施例で用いた。
【0025】
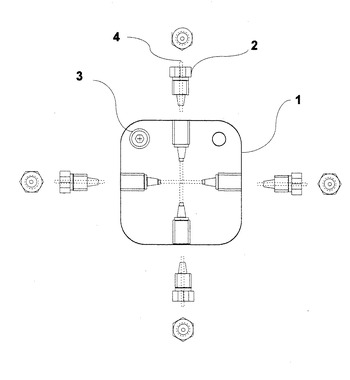
ここで、窓なし高温高圧フローセル本体(図6)については、例えば、市販のSUS316製のクロス1にネジを切り、次に説明する温度センサーシース(図7の12)に固定できるようにする。炉体雰囲気の温度を測定せずに、セル温度を示すように温度センサー位置を調節し、シース固定ネジとオネジ3でネジ止めする。SUS316の配管4はクロス1にワンリングフェラル付きのテーパーネジ2でクロス1に接続される。もちろん、クロス1は、エンドネジで一つの流路を塞ぐことによってティーとしても使用可能である。
【0026】
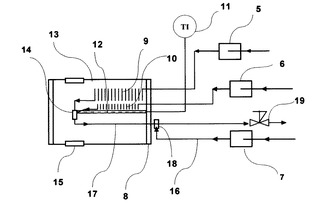
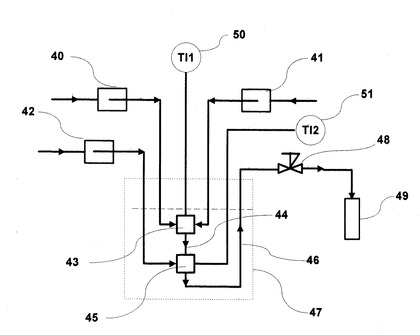
図7は、窓なし高温高圧フローセルを装着した流通式高温高圧反応装置の炉体部分であり、反応装置本体である。これを、図5の流通型高温高圧流体その場赤外分光測定装置の斜線位置に設置すれば、赤外分光は測定できないものの、温度、圧力、流量が可変な亜臨界・超臨界流体接触型の合成反応装置として利用可能となる。また、図8は、発熱反応の場合の流通式常温高圧反応装置であり、反応装置本体である。この場合も、常温付近の温度、圧力、流量が可変な常温流体接触合成反応装置として利用可能となる。更に、図8の流通式常温高圧反応装置と図7の流通式高温高圧反応装置を接続し、選択的に逐次アシル化を行って、目的のポリアシル化合物を得ることも可能である。なお、この場合における反応観察は、排出後の水溶液を採取し、GC−FIDにより、生成物の純品を用いた検量線から定量を実施し、GC/MSにより定性分析を実施した。また、NMRにより定量・定性分析を実施した。
【0027】
以下、図7について説明すると、水送液ポンプ5から水が送液され、冷却フランジ8を通過後、炉体13へ送液される。管コイル9を通過後、高温高圧状態で温度センサー11が挿入された温度センサーシース12に支持固定された高温高圧フローセル14に導入される。一方、反応物が反応物送液ポンプ6から送液され、冷却フランジ8を通過後、炉体13へ送液される。コイル状反応物導入管10を通過後、温度センサーシース12に固定された高温高圧フローセル14に導入される。また、洗浄水がポンプ7により送液され、配管16を通過後、ティー18に導入され、洗浄用に用いられる。高温高圧フローセルを通過した溶液は、配管17を通過後、冷却フランジ8を通過して、炉体外を空冷されながら通過する。その後、圧力を設定している背圧弁19からの排出液を採取し、サンプルとする。ここで、反応物や生成物を含む排出液の加熱による影響を排除する場合には、急速昇温を実施し、反応物導入ライン10と排出液ライン17の配管をできるだけ短く、水加熱用コイル9をできるだけ長くすることが望ましい。本発明は、これらに限らず、これらと同効の反応装置であれば同様に使用することができる。
【0028】
他方、常温反応装置である図8について説明すると、反応物送液ポンプ40と基質送液ポンプ41からそれぞれカルボン酸無水物とアミン又はアニリン誘導体が送液され、温度センサ50が装着された反応ティー43で混合され、反応し、配管44を反応溶液が通過する。その後、反応溶液は、発生する反応熱を抑制するため、温度センサ51が装着された混合ティー45で水送液ポンプ42から送液した水と混合冷却され、排出配管46、背圧弁48を通過後、回収容器49に回収される。この場合、発生する反応熱を抑制するために、反応ティー43、配管44、混合ティー45、排出配管46を冷却器に浸漬することもできる。また、水総液ポンプ42から水を送液せずに、発生する反応熱を抑制するために、反応ティー43、配管44、混合ティー45、排出配管46を冷却器に浸漬し、無溶媒条件で反応させることも可能である。また、図8の配管46と図7の背圧弁19を接続することで、発熱反応を常温流体中で実施後、吸熱反応を亜臨界流体、超臨界流体で連続的に実施することも可能である。本発明は、これらに限らず、これらと同効の反応装置であれば同様に使用することができる。
【発明の効果】
【0029】
本発明により、次のような効果が奏される。
(1)カルボン酸無水物とポリヘテロ水素化物から高速で連続的にポリアシル化合物を合成することができる。
(2)N−アシル化後、O−アシル化を行いつつアシル化数を調節し、ポリアシル化合物を選択的に合成することができる。
(3)触媒及び有機溶媒を用いない合成プロセスを実現できる。
(4)そのため、触媒及び有機溶媒の残存がなく、生体に対して有害性のない安全性の高いポリアシル化合物組成物を提供できる。
(5)生成物が水に溶解しない場合には、排出された油水分散水溶液に対して更に水を注入することで、洗浄しつつ油水二層に分液し、高純度の生成物を容易に回収できる。
(6)香料、医薬品、食品として有用なポリアシル化合物の新しい大量生産プロセスとして、既存の生産プロセスに代替し得る新しい生産技術を提供できる。
【発明を実施するための最良の形態】
【0030】
次に、実施例に基づいて本発明を具体的に説明するが、本発明は、以下の実施例によって何ら限定されるものではない。
【0031】
まず、実施方法を示した後、実施例を示す。
(1)流通式高温高圧反応装置を用いる方法(表1〜2の方法SF)
本実施例では、図7の流通式高温高圧反応装置を用いて、合成条件を、無触媒、温度50〜200℃、圧力5MPa、滞留時間9.9秒で実施した。図7の流通式高温高圧反応装置の本体(主要部分)を、図5の流通型高温高圧流体その場赤外分光測定装置に設置した装置に、まず、温度200℃、圧力5MPaに設定し、窓なしセル(ティー1)の配管コイル9との接続穴をエンドで塞ぎ、ポンプ5から純水を流量5.0ml/minで、ポンプ6から洗浄水を流量0.05ml/minで炉体外のティー18へ送液した。その後、トルエンを内標準として添加した(基質の5mol%)、カルボン酸無水物/アミン又はアニリン誘導体(モル比:1.1/1)混合溶液0.5ml/minをポンプで送液した(混合後の水溶液濃度:0.53mol/kg)。
【0032】
基質送液後、40分後の背圧弁からの排出水溶液を1ml採取した。加熱炉から背圧弁出口までの配管内容積を反応体積とした場合、反応時間は9.9秒であった。回収された1mlの水溶液に1mlのアセトンを加え、振とうし、組成分析をGC/MS分析計(Hewlett Packard社製HP6890、カラム HP−5、注入口温度150℃、初期カラム温度60℃(保持時間2分)、昇温速度10℃/分、最終カラム温度250℃(保持時間2分))で実施し、得られたマススペクトルは、Willey データベースで一致度90%以上で確認した。また、定量分析及び市販試薬がある場合の定性分析は、トルエンを内標準としてGC−FID(Agilent社製GC6890、カラム DB−WAX、注入口温度230℃、スプリット比5.61、初期カラム温度50℃(保持時間0.5分)、昇温速度20℃/分、最終カラム温度230℃(保持時間3分))で実施した。
【0033】
(2)流通式常温高圧反応装置を用いる方法(表1〜2の方法AF1、AF2)
一方、反応が発熱反応であり、上記流通型高温高圧反応装置で合成困難な場合は、図7の流通式高温高圧反応装置では合成が困難な場合、図8の流通式常温高圧反応装置を用いてN−アシル化を実施した。合成条件を、無触媒、圧力0.1MPa、滞留時間1.1秒一定として行った。図8の流通式常温高圧反応装置の本体に、カルボン酸無水物及びトルエンを内標準として添加した(基質の5mol%)アミン又はアニリン誘導体をそれぞれ送液し、更に純水を流量5.0ml/minで送液した。基質送液後、40分後の背圧弁からの排出水溶液を1ml採取した。反応ティーから背圧弁出口までの配管内容積を反応体積とした場合、反応時間は1.1秒であった。
【0034】
回収された1mlの水溶液に1mlのアセトンを加え、振とうし、組成分析をGC/MS分析計(Hewlett Packard社製HP6890、カラム HP−5、注入口温度150℃、初期カラム温度60℃(保持時間2分)、昇温速度10℃/分、最終カラム温度250℃(保持時間2分))で実施し、得られたマススペクトルは、Willey データベースで一致度90%以上で確認した。また、定量分析及び市販試薬がある場合の定性分析は、トルエンを内標準としてGC−FID(Agilent社製GC6890、カラム DB−WAX、注入口温度230℃、スプリット比5.61、初期カラム温度50℃(保持時間0.5分)、昇温速度20℃/分、最終カラム温度230℃(保持時間3分))で実施した。なお、反応が終了して背圧弁から排出時、固体が析出して閉塞する場合には、カルボン酸無水物に酢酸を添加した場合を方法AF1、アミン・アニリン誘導体に酢酸を添加した場合を方法AF2とした。
【0035】
(3)簡易型バッチ方法(表1〜2の方法B)
更に、溶解度及び反応を確認するため、30mlのバイヤル瓶にカルボン酸無水物及びアミン又はアニリン誘導体を滴下後、トルエンを内標準(基質の5mol%)として加えた。ここで、高粘性もしくは固体で溶解しにくい場合には、適量の酢酸を加え、温水で加温振とうし、酢酸に対する溶解度を確認した。更に、この混合溶液から採取された1mlの水溶液に1mlのアセトンを加え、振とうし、組成分析をGC/MS分析計(Hewlett Packard社製HP6890、カラム HP−5、注入口温度150℃、初期カラム温度60℃(保持時間2分)、昇温速度10℃/分、最終カラム温度250℃(保持時間2分))で実施し、得られたマススペクトルは、Willey データベースで一致度90%以上で確認した。また、定量分析及び市販試薬がある場合の定性分析は、トルエンを内標準としてGC−FID(Agilent社製GC6890、カラム DB−WAX、注入口温度230℃、スプリット比5.61、初期カラム温度50℃(保持時間0.5分)、昇温速度20℃/分、最終カラム温度230℃(保持時間3分))で実施した。
【0036】
また、得られた生成物水溶液が油水分散状態で白濁している場合には、水を20ml/minで3分注入し、デカンテーションすると油水2層溶液となり、下(上)層の油層に酢酸を含まないポリアシル化合物を、上(下)層の水相に酢酸水溶液を得た(GCにより確認)。このことは、生成物が水に溶解しない場合、反応終了後の油水分散水溶液に、水を更に注入することで、油水二層に変化してポリアシル化合物と酢酸水溶液を分液することができることを示す。酢酸水溶液は、触媒や有機溶媒を含まないため、酢酸と共沸化合物を作る化合物(例えば、酢酸ターシャリーブチル等)を添加することにより、共沸蒸留により水と氷酢酸に分留することができるため、膨大なエネルギーを必要とする精留を実施しなくても良い。
【0037】
(実施例1、2)
エチレンジアミン1及び1.1モル等量の無水酢酸を基質とし、方法AF1で実施した場合、転化率30%、選択率100%でN−アセチル体1aが得られず、N,N−ジアセチル体1bが得られた。一方、2.2モル等量の無水酢酸を基質とし、方法AF1で実施した場合、転化率99%、選択率100%で1bが得られた。
【0038】
(実施例3−6)
2−アミノエタノール2及び1.1モル等量の無水酢酸を基質とし、方法AF1で行った場合、転化率100%、選択率100%でN−アセチル体2aが得られた。方法AF1後、方法SFで温度200℃、圧力5MPa、滞留時間9.9秒で行った場合、転化率100%で、選択率35%で2aが得られ、選択率65%でN,O−ジアセチル体2bが得られた。2.2モル等量の無水酢酸を基質とし、方法SFにより温度200℃、圧力5MPa、滞留時間9.9秒で行った場合、転化率100%、選択率80%で2bが得られ、選択率20%で環化体2cが得られた。そこで、無水酢酸を減らし、2.0モル等量とし、方法SFにより温度200℃、圧力5MPa、滞留時間9.9秒で行ったところ、転化率100%、選択率87%で2bが得られ、選択率13%で2cが得られた。
【0039】
(実施例7−10)
3−アミノエタノール3及び1.1モル等量の無水酢酸を基質とし、方法AF1で行った場合、転化率100%、選択率100%でN−アセチル体3aが得られた。方法AF1後、方法SFで温度200℃、圧力5MPa、滞留時間9.9秒で行った場合、転化率100%で、選択率59%で3aが得られ、選択率41%でN,O―ジアセチル体3bが得られた。2.2モル等量の無水酢酸を基質とし、方法SFにより温度200℃、圧力5MPa、滞留時間9.9秒で行った場合、転化率100%、選択率85%で3bが得られ、環化体3cが選択率15%で得られた。そこで、無水酢酸を減らし、2.0モル等量とし、方法SFにより温度200℃、圧力5MPa、滞留時間9.9秒で行った場合、転化率100%、選択率90%で3bが得られ、選択率10%で3cが得られた。
【0040】
(実施例11−16)
4−アミノフェノール4及び1.1モル等量の無水酢酸を基質とし、方法AF1で行った場合、転化率7%、選択率2%でN−アシル体4aが得られた。方法SFで、温度100℃、圧力5MPa、滞留時間9.9秒で行った場合には、転化率9%、選択率14%で4aが得られた。また、温度150℃、圧力5MPa、滞留時間9.9秒で行った場合には、転化率78%、選択率65%で4aが得られた。4及び2.2モル等量の無水酢酸を基質として、方法Bで行った場合には、転化率99.7%、選択率100%で4aが選択的に得られた。また、方法SFで、温度50℃、圧力5MPa、滞留時間9.9秒で行った場合、転化率100%、選択率99.5%で4aが得られた。ところが、方法SFで、温度250℃、圧力5MPa、滞留時間9.9秒で行った場合、転化率93%、選択率100%でN,O−ジアセチル体4bが選択的に得られた。
【0041】
以上の実施例から、高温高圧水を反応溶媒として、無触媒でポリアシル化合物が高収率で合成可能であることが明らかとなった。また、ポリアシル化後、回収水溶液に水を注入してデカンテーションし、油/水二層溶液に分離後、ポリアシル化合物を含む油層を分液回収する一方、水層からは酢酸と水を共沸蒸留によって分離し、回収する簡易な連続分離法も構築できることが明らかとなった。
【0042】
【表1】
【0043】
【表2】
【産業上の利用可能性】
【0044】
以上詳述したように、本発明は、カルボン酸無水物及びポリヘテロ水素化物から有機溶媒を用いることなく、高温高圧流体を反応溶媒として、無触媒でポリアシル化合物を合成する方法、及び温度と無水カルボン酸の量を調整することにより、N−アシル化後、O−アシル化し、1級、2級、3級の順にアシル化してアシル化数を調節し、ポリアシル化合物を選択的に合成する方法、及びその反応組成物に係るものであり、従来法では、ポリヘテロ水素化物とカルボン酸無水物からポリアシル化合物の合成は、有機溶媒に触媒を添加し、数時間の反応を行い、すべてをアシル化するため、アシル化数を調整できなかったが、本発明で示した常温流体、亜臨界流体・超臨界流体を用いることにより、触媒無添加で、有機溶媒を使用することなく、高速で連続的に選択的にポリアシル化合物を合成することが可能となった。
【0045】
このことは、香料、医薬品、食品として有用なポリアシル化合物を短時間で、大量に連続的に生産できるというメリットをもたらす。また、アシル化後、回収水溶液に水を注入してデカンテーションし、油/水二層溶液に分離後、ポリアシル化合物を含む油層を分液回収する一方、水層からは酢酸と水を共沸蒸留によって分離し、回収する簡易な連続分離法により、氷酢酸と水を分離し、水をリサイクルすることが可能である。これらのことから、合成・分離プロセスを単純化させることで、プロセスの初期コスト及びランニングコストを圧縮することが可能である。更に、中和処理の後処理も不必要であり、環境調和型生産が可能となる。本発明は、香料、医薬品、食品として有用なポリアシル化合物の新しい大量生産プロセスとして、既存の生産プロセスに代替し得るものである。
【図面の簡単な説明】
【0046】
【図1】触媒・有機溶媒用いるポリヘテロ水素化物のアシル化を示す。
【図2】触媒・有機溶媒を用いるアシル化の後処理フローチャートを示す。
【図3】無触媒・水溶媒を用いるアシル化の後処理フローチャートを示す。
【図4】高温高圧赤外フローセルを示す。
【図5】実施例で用いた流通型高温高圧流体その場赤外分光測定装置を示す。
【図6】窓なし高温高圧フローセルを示す。
【図7】実施例で用いた流通式高温高圧反応装置の主要部分を示す。
【図8】実施例で用いた流通式常温高圧反応装置を示す。
【符号の説明】
【0047】
1 ティー又はクロス(片側口φ4mmネジ切り)
2 φ4mm×5.0mmL六角ネジ
3 ワンリングフェラル付オネジ
4 SUS316チューブ
5 水送液ポンプ
6 反応物送液ポンプ
7 洗浄水送液ポンプ
8 冷却フランジ(冷却水が循環する)
9 水加熱コイル
10 反応物導入管
11 温度センサ
12 温度センサーシース
13 炉体
14 高温高圧フローセル(通常昇温ではティー型、急速昇温ではクロス型)
15 ZnSe窓
16 溶媒導入管
17 排出配管
18 ティー
19 背圧弁
21 水溶液
22 洗浄水
23 水溶液ポンプ
24 洗浄用純水送液ポンプ
25 炉体加熱システム
26 炉体
27 高温高圧赤外フローセル
28 冷却水(入口)
29 冷却水(出口)
30 背圧弁
31 排出水溶液受器
32 可動鏡
33 可動鏡
34 干渉計
35 光源
36 赤外レーザー
37 MCT受光器
38 TGS受光器
39 解析モニター
40 反応物送液ポンプ
41 基質送液ポンプ
42 水送液ポンプ
43 反応ティー
44 配管
45 混合ティー
46 排出配管
47 冷却器
48 背圧弁
49 回収容器
50 温度センサ
51 温度センサ
【技術分野】
【0001】
本発明は、ポリアシル化合物、その製造方法と装置に関するものであり、更に詳しくは、発熱反応の場合に、常温水、吸熱反応の場合に、高温高圧状態の水あるいは酢酸それらの混合溶媒を反応溶媒とし、無触媒かつ一段階でポリアシル化合物を製造する方法に関するものである。本発明は、常温水あるいは温度100〜400℃、圧力0.1〜40MPaの水あるいは酢酸、それらの混合溶媒を反応溶媒として、触媒無添加で無水カルボン酸とポリヘテロ水素化物からポリアシル化合物を一段階かつ短時間、連続的に合成する方法及びその反応組成物を提供するものである。ここで、ポリヘテロ水素化物におけるヘテロ原子としては、酸素、窒素、硫黄が挙げられ、それぞれポリオール、ポリアミン、ポリチオールに対応し、更に、複数のヘテロ原子が組合わされた化合物も含む。
【0002】
ポリアシル化合物は、原料、基質の機能性を改質向上し、更に、付加価値を付与するため、香料、医薬品、食品分野において有用である。通常、ポリアシル化合物を合成する場合、従来法では、非プロトン性有機溶媒に加えて、酸・塩基触媒が必要であり、食品、医薬品に利用される場合、残存する有機溶媒、触媒の除去は大きな労力を必要とし、環境に影響を与えるのみならず、生体に有害である等の問題点を有していた。本発明は、無水カルボン酸とポリオール、ポリアミン、ポリチオール等ないしは複数種類のヘテロ原子が組み合わされたポリヘテロ水素化物から、無触媒で、水を用いるプロセスのみで、ポリアシル化合物を合成する方法とその反応組成物を提供するものであり、香料、医薬品や食品のみならず、化成品合成にも応用可能であり、ポリアシル化合物を効率良く、短時間で、大量に生産し、提供することを可能にするものである。
【背景技術】
【0003】
従来、無水カルボン酸とポリヘテロ水素化物、ポリオール、ポリアミン、ポリチオールないし複数種類のヘテロ原子が組み合わされたポリヘテロ水素化物からポリアシル化合物を合成する方法が種々報告されている(例えば、非特許文献1参照)。ここで、ポリオールと無水カルボン酸からポリアシル化合物を合成する技術を完成すれば、通常は、ポリアミンやポリチオール等ないしは複数種類のヘテロ原子が組み合わされたポリヘテロ水素化物からポリアシル化合物を合成することが可能となるため、特に、ポリアルコールからポリアシル化合物を合成する技術の報告例は非常に多い。ところが、従来法では、すべてのヘテロ水素をポリアシル化する方法のみであり、アシル化数を調整しながら選択的にポリアシル化する方法は報告されていない(図1)。図1で、R、Rn、Rn+1はアルキル基及びアルキル基以外のヘテロ原子を含む置換基、Qiはヘテロ原子又は置換ヘテロ原子であり、具体的には、酸素(O)、硫黄(S)、窒化水素(NH)、アルキル置換窒素(NR’)、であり、これらの原子の組み合わせも含む。
【0004】
先行技術文献によれば、無溶媒あるいは非プロトン性有機溶媒中、触媒として、CoCl3(非特許文献4)、Me3SiCl(非特許文献5)、塩基であるBu3P(非特許文献6)、ピリジン(非特許文献8)等が使用されてきた。また、安定なアシル中間体形成を経由することで、アシル基を活性化するDMAPの発見は、革新的な技術とされた(特許文献9)。ところが、DMAPは、1等量以上のアミンを利用することから、ルイス酸である金属トリフラートが提案され、Me3SiOTf(非特許文献10)、Sc(OTf)3(特許文献1、2及び非特許文献11)、In(OTf)3(非特許文献12)、Bi(OTf)3(非特許文献13)、Sc(NTf)3(非特許文献14)、が高収率でポリアシル化合物を与えることが示された。更に、V(OTf)3が触媒活性を示さないが、そのオキソ化合物であるV(O)(OTf)2が触媒活性があることが見出され、V=Oの触媒活性化が注目された(特許文献3、非特許文献15)。これらの触媒により、1、2級ポリアルコールから95%以上の収率で、また、3級ポリアルコールから80%以上の収率で、すべてアシル化されたポリアシル化合物が得られると報告されている(図1)。
【0005】
ここで、上記の先行技術文献では、有機塩基、ルイス酸、固体酸のような触媒に加えて、有機溶媒がポリアシル化にとって必要不可欠である。また、高温条件では、不純物が生成し、選択率を低下させるという理由から、アシル化は、常温で行うのが最適であり、高温条件は不適であるとされている(特許文献1、2)。一方、アシル化における溶媒としての水の可能性に関しては、水を除去した粗生成物にアシル化剤を添加し、アシル化する方法が一般的であり、水は負の効果をもたらすとされており(特許文献4)、ある文献では、溶媒として水を列挙しているが、実際には使用されていない(特許文献1、2参照)。ところが、アルドール反応に対する触媒活性と水中でのルイス酸の安定性との相関を元素ごと系統的に比較検討し、他の反応への適用可能性を示唆した例も存在する(非特許文献16)。更に、Bi(OTf)3が触媒の場合には、脱水処理をしていない水を含有する、湿った有機溶媒が反応を促進し、収率向上が観察された事例も存在する(非特許文献13)。したがって、アシル化に対する溶媒としての水の有効性は、これまで明確ではなく、実施もされなかった。
【0006】
他方、Bi(OTf)3を触媒とする場合の無溶媒条件では、収率が低下し、有機溶媒が必要であると報告されている(非特許文献13)。ところが、N−メトキシアセトアミドを触媒として、アミン・アニリン誘導体から有機溶媒又は常温水中、66%以上の収率でN−アシル化する方法が報告され、非ルイス酸触媒を用いた場合には、水が溶媒として利用可能なことが初めて示された。しかし、アシル化剤であるN−メトキシアセトアミドは、無水カルボン酸と比較して、非常に高価であるという問題がある(非特許文献17)。
【0007】
反応後における後処理は、通常の触媒・有機溶媒中アシル化では、反応混合物に中和剤を添加して中和後、抽出溶媒と水あるいは飽和食塩水を加え、分液し、溶媒層は、その後、乾燥、溶媒除去、蒸留あるいは精留のプロセスを得て、目的物を得るが、水層には、水の他に、触媒、有機溶媒、酢酸、基質、生成物、副生成物、無機物の複雑な混合物が含有される。ここで、水層からの触媒の分離が容易である場合には、回収再生され、再使用されるが、分離が困難である場合には、そのまま廃棄・処分される(図2)。無触媒・高温高圧水中アシル化の場合のように、水層に触媒、有機溶媒が含有されず、水、酢酸、生成物のみが含有されるならば、生成物をデカンテーションにより分離後、水層に対して共沸混合物を形成する物質を添加した共沸蒸留を行うことで、水と氷酢酸とに分離することが可能である(特許文献5)。このことは、水の再生を可能にし、通常法に比べて、環境低減型のプロセスであることを意味する(図3)。
【0008】
このように、従来法では、ポリアシル化の場合、触媒及び有機溶媒が必要であるため、製品の品質上、反応後の分離操作において、触媒、有機溶媒やカルボン酸の除去が必要であり、分離操作後の水層は廃棄物となりやすく、廃液の問題を生じる。更に、環境に対する影響や生体への有害性への配慮から、また、ヒトが経口する食品・医薬品の安全性から、触媒・有機溶媒のより高度分離が要求される。高度分離に必要なコストは、合成操作と同程度であり、望ましくは触媒と有機溶媒を使用しない方が良い。以上のことから、当該技術分野においては、簡単、低コスト、環境低減型の合成プロセスで、分離操作が容易かつ高度分離が可能で、触媒や有機溶媒の残存しないアシル化合物の連続的合成を可能とする合成手法が強く要請されていた。
【0009】
【特許文献1】特開平9−169690号公報
【特許文献2】特開平9−176081号公報
【特許文献3】米国特許第6,541,659号明細書
【特許文献4】米国特許第6,005,122号明細書
【特許文献5】米国特許第5,980,696号明細書
【非特許文献1】W.Green,P.G.Wuts,Protective Groups in Organic Synthesis,3rd ed.,Wiley, New York,1999,p150
【非特許文献4】S.Ahmad,J.Iqbal,Chem.Commun.,1987,114
【非特許文献5】N.C.Braus,R.P.Sharma,J.N.Baruah,Tetrahedron Lett.,1983,24,1189;J.Org. Chem.,1987,52,503
【非特許文献6】E.Vedejs,N.S.Bannet,L.M.Conn, S.T.Diver,J.Org.Chem.,1993,58,7286
【非特許文献8】Joseph B.Lambert,Gen Tai Wang, Rodney B.Finzel,and Douglas H.Teramura, J.Am.Chem.Soc.,1987,109.7838
【非特許文献9】G.Hofle,W.Steglich,H.Vorbruggen., Angrew.Chem.Int.Ed.,1978,17,569. b) A.Hassner,L.R.Krepski,V.Alexanian,Tetrahedron,1978,34,2069
【非特許文献10】P.A.Procopiu,S.P.D.Baugh,S.S. Flack,G.G.A.Inglis,J.Org.,Chem.,1998,63, 2342
【非特許文献11】a)K.Ishihara,M.Kubota,H.Kurihara,H.Yamamoto,J.Org.Chem.,1996,61,4560. b)K.Ishihara,M.Kubota,H.Kurihara,H.Yamamoto,J.Am.Chem.Soc.,1995,117,4413
【非特許文献12】K.K.Chauhan,C.G.Frost,I.Love, D.Waite,Synlett,1999,1743
【非特許文献13】J.Otera,A.Orita,C.Tanahashi, A.Kakuta,Angrew.Chem.Int.Ed.,2000,39,2877
【非特許文献14】K.Ishihara,M.Kubota,Synlett, 1996,265
【非特許文献15】C−T.Chen,J−H.Kuo,C−H.Li,N.B. Barhate,S−W.Hon,T−W.Li,S−D.Chao,C−C.Liu, Y−C.Li,I−H.Chang,J−S.Lin,C−J.Liu and Y−C.Chou,Org.Lett.,2001,3,3729
【非特許文献16】S.Kobayashi,S.Nagayama,T.Busujima,J.Am.Chem.Soc.,1998,120,8287
【非特許文献17】Y.Kikugawa,K.Mitsui,T.Sakamoto,M.Kawase,and H.Tamiya,Tetrahedron Lett,1990,31,2,243
【発明の開示】
【発明が解決しようとする課題】
【0010】
このような状況のなかで、本発明者らは、上記従来技術に鑑みて、低コストで、環境に優しい簡便な高速合成プロセスで、上記ポリアシル化合物を連続的かつ選択的に合成することができる新しい合成方法を開発することを目標として鋭意研究を積み重ねた結果、高温高圧水、又は亜臨界水又は超臨界水を反応溶媒とすることで、無触媒で無水カルボン酸とポリヘテロ水素化物からポリアシル化合物を選択的に合成できることを見出し、本発明を完成するに至った。本発明は、無水カルボン酸とポリヘテロ水素化物からポリアシル化合物を無触媒で、短時間の反応条件下で連続的に合成する方法及びその反応組成物を提供することを目的とするものである。
【課題を解決するための手段】
【0011】
上記課題を解決するための本発明は、以下の技術的手段から構成される。
(1)無水カルボン酸とポリヘテロ水素化物との反応組成物であって、触媒及び有機溶媒の残存がないことを特徴とするポリアシル化合物組成物。
(2)無水カルボン酸とポリヘテロ水素化物からポリアシル化合物を合成する方法であって、発熱反応の場合には、常温流体、吸熱反応の場合には、高温高圧状態の亜臨界流体ないしは超臨界流体を反応溶媒として使用し、触媒を用いることなく、無水カルボン酸とポリヘテロ水素化物から一段階の合成反応でポリアシル化合物を選択的に合成することを特徴とするポリアシル化合物の製造方法。
(3)高温高圧状態の亜臨界ないしは超臨界水を反応溶媒として使用する、前記(2)記載の方法。
(4)ポリヘテロ水素化物におけるヘテロ原子又は置換ヘテロ原子が、酸素(O)、硫黄(S)、窒化水素(NH)、アルキル置換窒素(NR’)、又はこれらの原子の組合せである、前記(1)又は(2)記載の方法。
(5)基質におけるヘテロ水素基及びヘテロ水素基に隣接するα位の置換数に対応して、温度と無水カルボン酸の量を調整することにより、N−アシル化後、O−アシル化を実施して、それぞれのアシル化において、無置換(1級)、一置換(2級)、二置換(3級)の順にアシル化しつつアシル化数を調節し、ポリアシル化合物を選択的に合成する、前記(1)又は(2)の方法。
(6)温度100〜400℃、圧力0.1〜40MPaの亜臨界流体ないし超臨界流体を反応溶媒として使用する、前記(2)記載の方法。
(7)常温流体ないし亜臨界流体ないし超臨界流体として、水、酢酸、それ以外の無機溶媒、もしくは有機溶媒もしくは無機溶媒と有機溶媒の混合溶媒を用いる、前記(2)記載の方法。
(8)流通式高温高圧装置に、基質及び反応溶媒を導入し、反応時間を3〜60秒の範囲で変化させることで合成反応を実施する、前記(2)記載の方法。
(9)発熱反応の場合に、流通式常温高圧装置に、基質及び反応溶媒を導入し、反応時間を1〜60秒の範囲で変化させることで合成反応を実施する、前記(2)記載の方法。
(10)水を送液する水送液ポンプ、水加熱用コイル、高温高圧フローセル、基質を送液する反応物送液ポンプ、炉体、反応物を炉体に導入する反応物導入管、反応溶液を排出する排出液ライン、冷却フランジ及び圧力を設定する背圧弁を具備していることを特徴とするポリアシル化合物合成装置。
(11)吸熱反応の場合において、水加熱用コイルが配設されている、前記(10)記載のN−アシル化合物合成装置。
(12)前記(2)記載の方法において、アシル化後、回収水溶液に水を注入してデカンテーションし、油/水二層溶液に分離後、ポリアシル化合物を含む油層を分液回収する一方、水層からは酢酸と水を共沸蒸留によって分離し、回収する簡易な連続分離法。
【0012】
次に、本発明について更に詳細に説明する。
本発明は、化1のカルボン酸無水物と化2のポリヘテロ水素化物から化3のポリアシル化合物を、一段階の反応プロセスで、触媒無添加、短時間の反応条件下で、選択的かつ連続的に合成することを特徴とするものである。本発明では、上記反応溶媒として、発熱反応の場合、常温流体が用いられ、吸熱反応の場合、温度100〜400℃、圧力0.1〜40MPaの亜臨界流体、超臨界流体が用いられ、好適には発熱反応の場合、水、吸熱反応の場合、亜臨界水が用いられる。また、反応条件として、好適には、発熱反応の場合、常温常圧、反応時間が1秒程度に調整され、吸熱反応の場合、温度200〜250℃、圧力5MPa、反応時間が3〜60秒の範囲、好適には10秒程度に調整される。
【0013】
化1の式中、Rはアルキル基又はアルキル基以外のヘテロ原子を含む置換基であり、化2の式中、Rn、Rn+1はアルキル基又はアルキル基以外のヘテロ原子を含む置換基、Qiはヘテロ原子又は置換ヘテロ原子であり、具体的には、酸素(O)、硫黄(S)、窒化水素(NH)、アルキル置換窒素(NR’)、であり、これら原子の組み合わせも含み、また、x、y、zはそれぞれα位が1級、2級、3級であるヘテロ原子の個数を示し、1級、2級、3級の結合順序は限定するものではないため、点線で結合を示している。化3はα位が1級のみであってx個のポリアシル化を示し、化4はα位が1、2級の場合の(x+y)個のポリアシル化を示し、化5はα位が1、2、3級の場合に対する(x+y+z)個のポリアシル化を示す。
【0014】
【化1】
【0015】
【化2】
【0016】
【化3】
【0017】
【化4】
【0018】
【化5】
【0019】
本発明においては、上記基質及び反応溶媒を反応容器に導入して所定の反応時間で合成反応を実施するものである。したがって、上記反応器としては、例えば、バッチ式の常温高圧装置又は高温高圧反応容器、及び連続型の流通式常温高圧装置又は流通式高温高圧反応装置を使用することができるが、本発明は、これらの反応装置型式に特に制限されるものでない。
【0020】
本発明の方法では、反応溶媒として、上記常温流体又は高温高圧状態にある亜臨界流体、超臨界流体が用いられるが、具体的には、亜臨界二酸化炭素(常温以上、0.1MPa以上)、亜臨界水(100℃以上、0.1MPa以上)、亜臨界メタノール(100℃以上、0.1MPa以上)、亜臨界エタノール(100℃以上、0.1MPa以上)、超臨界二酸化炭素(34℃以上、7.38MPa以上)、超臨界水(375℃以上、22MPa以上)、超臨界メタノール(239℃以上、8.1MPa以上)、超臨界エタノール(241℃以上、6.1MPa以上)、同じ状態の混合溶媒が例示され、好適には、亜臨界水(200−250℃、5MPa以上)が用いられる。反応溶媒としては、上記以外の有機溶媒や無機溶媒を任意の割合で含むことができ、具体的には、有機溶媒として、アセトン、アセトニトリル、テトラヒドロフラン等、無機溶媒として酢酸、アンモニア等を含む反応溶液に代替することも可能である。
【0021】
本発明では、上記常温流体、亜臨界流体、超臨界流体の反応溶媒の組成、温度及び圧力条件、基質の種類及びその使用量、反応時間を調整することにより、短時間で、効率良く、反応生成物を合成することができる。また、本発明では、例えば、基質及び反応溶媒を発熱反応の場合に、流通式常温高圧装置に導入し、それらの反応時間を1〜60秒の範囲で変えることにより、あるいは吸熱反応の場合に、流通式高温高圧装置に導入し、それらの反応時間を3〜60秒の範囲で変えることにより、所定の反応生成物を合成することができる。上記反応条件は、使用する出発原料、目的とする反応生成物の種類等により適宜設定することができる。
【0022】
本発明の方法では、従来、触媒存在下で行われていた、カルボン酸無水物とポリヘテロ水素化物からのポリアシル化合物の合成を、高速で連続的に、しかも、無触媒で実施できるため、長時間を要するプロセスを効率化することができる。また、本発明の方法では、従来用いられた触媒を全く使用しないので、反応後の溶液の中和処理、無害化処理等の後処理・処分の必要がなく、環境負荷低減を達成可能である。更に、反応後は、静置分離操作のみであるため、触媒や有機溶媒の分離回収の必要性はなく、生成物分離が容易になる。本発明によれば、触媒無添加で、1〜10秒程度の短時間で、基質が窒素、酸素を含むアミノカルビノール類、アニリノカルビノール類の場合、まず初めに、常温高圧反応装置ないしは高温高圧反応装置で選択的にN−アシル化を実施して、N−アシル化合物を転化率85%以上、選択率99%以上で得た後、引き続き高温高圧反応装置でO−アシル化により、O−アシル化合物を転化率93%以上、選択率87%以上で行い、対応するポリアシル化合物を合成可能である。本発明の合成方法は、香料、医薬品、食品に利用可能な、ポリアシル化合物を効率良く、大量に高速で連続的に生産することを可能にするものとして有用である。
【0023】
従来、二酸化炭素等の亜臨界流体、超臨界流体を利用して、リパーゼや触媒を用いたポリアシル化を実施した例が報告されている。しかし、常温流体、亜臨界水を溶媒とした無触媒条件下で基質の複数の反応点の原子の種類によって、更に、基質の複数の反応点に隣接する1級、2級、3級の骨格に依存して、温度と無水カルボン酸の量を調整することにより、1級、2級、3級の順にアシル化し、アシル化数を調節し、ポリアシル化合物を選択的に合成できることを実証した例はなく、本発明の対象とするポリアシル化合物の選択的合成反応法は、本発明者らによって初めてその有効性が実証されたものである。しかも、従来法でカルボン酸無水物とポリヘテロ水素化物から合成されるポリアシル化合物は、触媒及び有機溶媒の残存が問題とされていたが、本発明で、カルボン酸無水物とポリヘテロ水素化物から合成される反応組成物は、触媒及び有機溶媒の残存がなく、本発明のポリアシル化合物組成物は、従来製品にない利点を有している。
【0024】
本発明では、無触媒条件で無水カルボン酸とポリヘテロ水素化物の合成反応を実現するために、例えば、基質をあらかじめ溶媒に溶解した溶液を送液し、常温流体、亜臨界流体、超臨界流体中の反応経過を高温高圧赤外フローセル(図4)により赤外分光分析によって観察する流通型高温高圧赤外分光その場測定装置(図5)を用いることも可能である。しかしながら、高温高圧赤外フローセルを窓なし高温高圧フローセル(図6)に交換し、超臨界流体の流れに対して直接反応物の流れを接触反応するように配管配置した方が、高温高圧赤外フローセルにおけるセル窓付近におけるリーク等の問題が発生せず、より高流量で短時間に合成を実施することが可能である。これらのことから、この窓なし高温高圧フローセルを装着した装置を後述する実施例で用いた。
【0025】
ここで、窓なし高温高圧フローセル本体(図6)については、例えば、市販のSUS316製のクロス1にネジを切り、次に説明する温度センサーシース(図7の12)に固定できるようにする。炉体雰囲気の温度を測定せずに、セル温度を示すように温度センサー位置を調節し、シース固定ネジとオネジ3でネジ止めする。SUS316の配管4はクロス1にワンリングフェラル付きのテーパーネジ2でクロス1に接続される。もちろん、クロス1は、エンドネジで一つの流路を塞ぐことによってティーとしても使用可能である。
【0026】
図7は、窓なし高温高圧フローセルを装着した流通式高温高圧反応装置の炉体部分であり、反応装置本体である。これを、図5の流通型高温高圧流体その場赤外分光測定装置の斜線位置に設置すれば、赤外分光は測定できないものの、温度、圧力、流量が可変な亜臨界・超臨界流体接触型の合成反応装置として利用可能となる。また、図8は、発熱反応の場合の流通式常温高圧反応装置であり、反応装置本体である。この場合も、常温付近の温度、圧力、流量が可変な常温流体接触合成反応装置として利用可能となる。更に、図8の流通式常温高圧反応装置と図7の流通式高温高圧反応装置を接続し、選択的に逐次アシル化を行って、目的のポリアシル化合物を得ることも可能である。なお、この場合における反応観察は、排出後の水溶液を採取し、GC−FIDにより、生成物の純品を用いた検量線から定量を実施し、GC/MSにより定性分析を実施した。また、NMRにより定量・定性分析を実施した。
【0027】
以下、図7について説明すると、水送液ポンプ5から水が送液され、冷却フランジ8を通過後、炉体13へ送液される。管コイル9を通過後、高温高圧状態で温度センサー11が挿入された温度センサーシース12に支持固定された高温高圧フローセル14に導入される。一方、反応物が反応物送液ポンプ6から送液され、冷却フランジ8を通過後、炉体13へ送液される。コイル状反応物導入管10を通過後、温度センサーシース12に固定された高温高圧フローセル14に導入される。また、洗浄水がポンプ7により送液され、配管16を通過後、ティー18に導入され、洗浄用に用いられる。高温高圧フローセルを通過した溶液は、配管17を通過後、冷却フランジ8を通過して、炉体外を空冷されながら通過する。その後、圧力を設定している背圧弁19からの排出液を採取し、サンプルとする。ここで、反応物や生成物を含む排出液の加熱による影響を排除する場合には、急速昇温を実施し、反応物導入ライン10と排出液ライン17の配管をできるだけ短く、水加熱用コイル9をできるだけ長くすることが望ましい。本発明は、これらに限らず、これらと同効の反応装置であれば同様に使用することができる。
【0028】
他方、常温反応装置である図8について説明すると、反応物送液ポンプ40と基質送液ポンプ41からそれぞれカルボン酸無水物とアミン又はアニリン誘導体が送液され、温度センサ50が装着された反応ティー43で混合され、反応し、配管44を反応溶液が通過する。その後、反応溶液は、発生する反応熱を抑制するため、温度センサ51が装着された混合ティー45で水送液ポンプ42から送液した水と混合冷却され、排出配管46、背圧弁48を通過後、回収容器49に回収される。この場合、発生する反応熱を抑制するために、反応ティー43、配管44、混合ティー45、排出配管46を冷却器に浸漬することもできる。また、水総液ポンプ42から水を送液せずに、発生する反応熱を抑制するために、反応ティー43、配管44、混合ティー45、排出配管46を冷却器に浸漬し、無溶媒条件で反応させることも可能である。また、図8の配管46と図7の背圧弁19を接続することで、発熱反応を常温流体中で実施後、吸熱反応を亜臨界流体、超臨界流体で連続的に実施することも可能である。本発明は、これらに限らず、これらと同効の反応装置であれば同様に使用することができる。
【発明の効果】
【0029】
本発明により、次のような効果が奏される。
(1)カルボン酸無水物とポリヘテロ水素化物から高速で連続的にポリアシル化合物を合成することができる。
(2)N−アシル化後、O−アシル化を行いつつアシル化数を調節し、ポリアシル化合物を選択的に合成することができる。
(3)触媒及び有機溶媒を用いない合成プロセスを実現できる。
(4)そのため、触媒及び有機溶媒の残存がなく、生体に対して有害性のない安全性の高いポリアシル化合物組成物を提供できる。
(5)生成物が水に溶解しない場合には、排出された油水分散水溶液に対して更に水を注入することで、洗浄しつつ油水二層に分液し、高純度の生成物を容易に回収できる。
(6)香料、医薬品、食品として有用なポリアシル化合物の新しい大量生産プロセスとして、既存の生産プロセスに代替し得る新しい生産技術を提供できる。
【発明を実施するための最良の形態】
【0030】
次に、実施例に基づいて本発明を具体的に説明するが、本発明は、以下の実施例によって何ら限定されるものではない。
【0031】
まず、実施方法を示した後、実施例を示す。
(1)流通式高温高圧反応装置を用いる方法(表1〜2の方法SF)
本実施例では、図7の流通式高温高圧反応装置を用いて、合成条件を、無触媒、温度50〜200℃、圧力5MPa、滞留時間9.9秒で実施した。図7の流通式高温高圧反応装置の本体(主要部分)を、図5の流通型高温高圧流体その場赤外分光測定装置に設置した装置に、まず、温度200℃、圧力5MPaに設定し、窓なしセル(ティー1)の配管コイル9との接続穴をエンドで塞ぎ、ポンプ5から純水を流量5.0ml/minで、ポンプ6から洗浄水を流量0.05ml/minで炉体外のティー18へ送液した。その後、トルエンを内標準として添加した(基質の5mol%)、カルボン酸無水物/アミン又はアニリン誘導体(モル比:1.1/1)混合溶液0.5ml/minをポンプで送液した(混合後の水溶液濃度:0.53mol/kg)。
【0032】
基質送液後、40分後の背圧弁からの排出水溶液を1ml採取した。加熱炉から背圧弁出口までの配管内容積を反応体積とした場合、反応時間は9.9秒であった。回収された1mlの水溶液に1mlのアセトンを加え、振とうし、組成分析をGC/MS分析計(Hewlett Packard社製HP6890、カラム HP−5、注入口温度150℃、初期カラム温度60℃(保持時間2分)、昇温速度10℃/分、最終カラム温度250℃(保持時間2分))で実施し、得られたマススペクトルは、Willey データベースで一致度90%以上で確認した。また、定量分析及び市販試薬がある場合の定性分析は、トルエンを内標準としてGC−FID(Agilent社製GC6890、カラム DB−WAX、注入口温度230℃、スプリット比5.61、初期カラム温度50℃(保持時間0.5分)、昇温速度20℃/分、最終カラム温度230℃(保持時間3分))で実施した。
【0033】
(2)流通式常温高圧反応装置を用いる方法(表1〜2の方法AF1、AF2)
一方、反応が発熱反応であり、上記流通型高温高圧反応装置で合成困難な場合は、図7の流通式高温高圧反応装置では合成が困難な場合、図8の流通式常温高圧反応装置を用いてN−アシル化を実施した。合成条件を、無触媒、圧力0.1MPa、滞留時間1.1秒一定として行った。図8の流通式常温高圧反応装置の本体に、カルボン酸無水物及びトルエンを内標準として添加した(基質の5mol%)アミン又はアニリン誘導体をそれぞれ送液し、更に純水を流量5.0ml/minで送液した。基質送液後、40分後の背圧弁からの排出水溶液を1ml採取した。反応ティーから背圧弁出口までの配管内容積を反応体積とした場合、反応時間は1.1秒であった。
【0034】
回収された1mlの水溶液に1mlのアセトンを加え、振とうし、組成分析をGC/MS分析計(Hewlett Packard社製HP6890、カラム HP−5、注入口温度150℃、初期カラム温度60℃(保持時間2分)、昇温速度10℃/分、最終カラム温度250℃(保持時間2分))で実施し、得られたマススペクトルは、Willey データベースで一致度90%以上で確認した。また、定量分析及び市販試薬がある場合の定性分析は、トルエンを内標準としてGC−FID(Agilent社製GC6890、カラム DB−WAX、注入口温度230℃、スプリット比5.61、初期カラム温度50℃(保持時間0.5分)、昇温速度20℃/分、最終カラム温度230℃(保持時間3分))で実施した。なお、反応が終了して背圧弁から排出時、固体が析出して閉塞する場合には、カルボン酸無水物に酢酸を添加した場合を方法AF1、アミン・アニリン誘導体に酢酸を添加した場合を方法AF2とした。
【0035】
(3)簡易型バッチ方法(表1〜2の方法B)
更に、溶解度及び反応を確認するため、30mlのバイヤル瓶にカルボン酸無水物及びアミン又はアニリン誘導体を滴下後、トルエンを内標準(基質の5mol%)として加えた。ここで、高粘性もしくは固体で溶解しにくい場合には、適量の酢酸を加え、温水で加温振とうし、酢酸に対する溶解度を確認した。更に、この混合溶液から採取された1mlの水溶液に1mlのアセトンを加え、振とうし、組成分析をGC/MS分析計(Hewlett Packard社製HP6890、カラム HP−5、注入口温度150℃、初期カラム温度60℃(保持時間2分)、昇温速度10℃/分、最終カラム温度250℃(保持時間2分))で実施し、得られたマススペクトルは、Willey データベースで一致度90%以上で確認した。また、定量分析及び市販試薬がある場合の定性分析は、トルエンを内標準としてGC−FID(Agilent社製GC6890、カラム DB−WAX、注入口温度230℃、スプリット比5.61、初期カラム温度50℃(保持時間0.5分)、昇温速度20℃/分、最終カラム温度230℃(保持時間3分))で実施した。
【0036】
また、得られた生成物水溶液が油水分散状態で白濁している場合には、水を20ml/minで3分注入し、デカンテーションすると油水2層溶液となり、下(上)層の油層に酢酸を含まないポリアシル化合物を、上(下)層の水相に酢酸水溶液を得た(GCにより確認)。このことは、生成物が水に溶解しない場合、反応終了後の油水分散水溶液に、水を更に注入することで、油水二層に変化してポリアシル化合物と酢酸水溶液を分液することができることを示す。酢酸水溶液は、触媒や有機溶媒を含まないため、酢酸と共沸化合物を作る化合物(例えば、酢酸ターシャリーブチル等)を添加することにより、共沸蒸留により水と氷酢酸に分留することができるため、膨大なエネルギーを必要とする精留を実施しなくても良い。
【0037】
(実施例1、2)
エチレンジアミン1及び1.1モル等量の無水酢酸を基質とし、方法AF1で実施した場合、転化率30%、選択率100%でN−アセチル体1aが得られず、N,N−ジアセチル体1bが得られた。一方、2.2モル等量の無水酢酸を基質とし、方法AF1で実施した場合、転化率99%、選択率100%で1bが得られた。
【0038】
(実施例3−6)
2−アミノエタノール2及び1.1モル等量の無水酢酸を基質とし、方法AF1で行った場合、転化率100%、選択率100%でN−アセチル体2aが得られた。方法AF1後、方法SFで温度200℃、圧力5MPa、滞留時間9.9秒で行った場合、転化率100%で、選択率35%で2aが得られ、選択率65%でN,O−ジアセチル体2bが得られた。2.2モル等量の無水酢酸を基質とし、方法SFにより温度200℃、圧力5MPa、滞留時間9.9秒で行った場合、転化率100%、選択率80%で2bが得られ、選択率20%で環化体2cが得られた。そこで、無水酢酸を減らし、2.0モル等量とし、方法SFにより温度200℃、圧力5MPa、滞留時間9.9秒で行ったところ、転化率100%、選択率87%で2bが得られ、選択率13%で2cが得られた。
【0039】
(実施例7−10)
3−アミノエタノール3及び1.1モル等量の無水酢酸を基質とし、方法AF1で行った場合、転化率100%、選択率100%でN−アセチル体3aが得られた。方法AF1後、方法SFで温度200℃、圧力5MPa、滞留時間9.9秒で行った場合、転化率100%で、選択率59%で3aが得られ、選択率41%でN,O―ジアセチル体3bが得られた。2.2モル等量の無水酢酸を基質とし、方法SFにより温度200℃、圧力5MPa、滞留時間9.9秒で行った場合、転化率100%、選択率85%で3bが得られ、環化体3cが選択率15%で得られた。そこで、無水酢酸を減らし、2.0モル等量とし、方法SFにより温度200℃、圧力5MPa、滞留時間9.9秒で行った場合、転化率100%、選択率90%で3bが得られ、選択率10%で3cが得られた。
【0040】
(実施例11−16)
4−アミノフェノール4及び1.1モル等量の無水酢酸を基質とし、方法AF1で行った場合、転化率7%、選択率2%でN−アシル体4aが得られた。方法SFで、温度100℃、圧力5MPa、滞留時間9.9秒で行った場合には、転化率9%、選択率14%で4aが得られた。また、温度150℃、圧力5MPa、滞留時間9.9秒で行った場合には、転化率78%、選択率65%で4aが得られた。4及び2.2モル等量の無水酢酸を基質として、方法Bで行った場合には、転化率99.7%、選択率100%で4aが選択的に得られた。また、方法SFで、温度50℃、圧力5MPa、滞留時間9.9秒で行った場合、転化率100%、選択率99.5%で4aが得られた。ところが、方法SFで、温度250℃、圧力5MPa、滞留時間9.9秒で行った場合、転化率93%、選択率100%でN,O−ジアセチル体4bが選択的に得られた。
【0041】
以上の実施例から、高温高圧水を反応溶媒として、無触媒でポリアシル化合物が高収率で合成可能であることが明らかとなった。また、ポリアシル化後、回収水溶液に水を注入してデカンテーションし、油/水二層溶液に分離後、ポリアシル化合物を含む油層を分液回収する一方、水層からは酢酸と水を共沸蒸留によって分離し、回収する簡易な連続分離法も構築できることが明らかとなった。
【0042】
【表1】
【0043】
【表2】
【産業上の利用可能性】
【0044】
以上詳述したように、本発明は、カルボン酸無水物及びポリヘテロ水素化物から有機溶媒を用いることなく、高温高圧流体を反応溶媒として、無触媒でポリアシル化合物を合成する方法、及び温度と無水カルボン酸の量を調整することにより、N−アシル化後、O−アシル化し、1級、2級、3級の順にアシル化してアシル化数を調節し、ポリアシル化合物を選択的に合成する方法、及びその反応組成物に係るものであり、従来法では、ポリヘテロ水素化物とカルボン酸無水物からポリアシル化合物の合成は、有機溶媒に触媒を添加し、数時間の反応を行い、すべてをアシル化するため、アシル化数を調整できなかったが、本発明で示した常温流体、亜臨界流体・超臨界流体を用いることにより、触媒無添加で、有機溶媒を使用することなく、高速で連続的に選択的にポリアシル化合物を合成することが可能となった。
【0045】
このことは、香料、医薬品、食品として有用なポリアシル化合物を短時間で、大量に連続的に生産できるというメリットをもたらす。また、アシル化後、回収水溶液に水を注入してデカンテーションし、油/水二層溶液に分離後、ポリアシル化合物を含む油層を分液回収する一方、水層からは酢酸と水を共沸蒸留によって分離し、回収する簡易な連続分離法により、氷酢酸と水を分離し、水をリサイクルすることが可能である。これらのことから、合成・分離プロセスを単純化させることで、プロセスの初期コスト及びランニングコストを圧縮することが可能である。更に、中和処理の後処理も不必要であり、環境調和型生産が可能となる。本発明は、香料、医薬品、食品として有用なポリアシル化合物の新しい大量生産プロセスとして、既存の生産プロセスに代替し得るものである。
【図面の簡単な説明】
【0046】
【図1】触媒・有機溶媒用いるポリヘテロ水素化物のアシル化を示す。
【図2】触媒・有機溶媒を用いるアシル化の後処理フローチャートを示す。
【図3】無触媒・水溶媒を用いるアシル化の後処理フローチャートを示す。
【図4】高温高圧赤外フローセルを示す。
【図5】実施例で用いた流通型高温高圧流体その場赤外分光測定装置を示す。
【図6】窓なし高温高圧フローセルを示す。
【図7】実施例で用いた流通式高温高圧反応装置の主要部分を示す。
【図8】実施例で用いた流通式常温高圧反応装置を示す。
【符号の説明】
【0047】
1 ティー又はクロス(片側口φ4mmネジ切り)
2 φ4mm×5.0mmL六角ネジ
3 ワンリングフェラル付オネジ
4 SUS316チューブ
5 水送液ポンプ
6 反応物送液ポンプ
7 洗浄水送液ポンプ
8 冷却フランジ(冷却水が循環する)
9 水加熱コイル
10 反応物導入管
11 温度センサ
12 温度センサーシース
13 炉体
14 高温高圧フローセル(通常昇温ではティー型、急速昇温ではクロス型)
15 ZnSe窓
16 溶媒導入管
17 排出配管
18 ティー
19 背圧弁
21 水溶液
22 洗浄水
23 水溶液ポンプ
24 洗浄用純水送液ポンプ
25 炉体加熱システム
26 炉体
27 高温高圧赤外フローセル
28 冷却水(入口)
29 冷却水(出口)
30 背圧弁
31 排出水溶液受器
32 可動鏡
33 可動鏡
34 干渉計
35 光源
36 赤外レーザー
37 MCT受光器
38 TGS受光器
39 解析モニター
40 反応物送液ポンプ
41 基質送液ポンプ
42 水送液ポンプ
43 反応ティー
44 配管
45 混合ティー
46 排出配管
47 冷却器
48 背圧弁
49 回収容器
50 温度センサ
51 温度センサ
【特許請求の範囲】
【請求項1】
無水カルボン酸とポリヘテロ水素化物との反応組成物であって、触媒及び有機溶媒の残存がないことを特徴とするポリアシル化合物組成物。
【請求項2】
無水カルボン酸とポリヘテロ水素化物からポリアシル化合物を合成する方法であって、発熱反応の場合には、常温流体、吸熱反応の場合には、高温高圧状態の亜臨界流体ないしは超臨界流体を反応溶媒として使用し、触媒を用いることなく、無水カルボン酸とポリヘテロ水素化物から一段階の合成反応でポリアシル化合物を選択的に合成することを特徴とするポリアシル化合物の製造方法。
【請求項3】
高温高圧状態の亜臨界ないしは超臨界水を反応溶媒として使用する、請求項2記載の方法。
【請求項4】
ポリヘテロ水素化物におけるヘテロ原子又は置換ヘテロ原子が、酸素(O)、硫黄(S)、窒化水素(NH)、アルキル置換窒素(NR’)、又はこれらの原子の組合せである、請求項1又は請求項2記載の方法。
【請求項5】
基質におけるヘテロ水素基及びヘテロ水素基に隣接するα位の置換数に対応して、温度と無水カルボン酸の量を調整することにより、N−アシル化後、O−アシル化を実施して、それぞれのアシル化において、無置換(1級)、一置換(2級)、二置換(3級)の順にアシル化しつつアシル化数を調節し、ポリアシル化合物を選択的に合成する、請求項1又は請求項2の方法。
【請求項6】
温度100〜400℃、圧力0.1〜40MPaの亜臨界流体ないし超臨界流体を反応溶媒として使用する、請求項2記載の方法。
【請求項7】
常温流体ないし亜臨界流体ないし超臨界流体として、水、酢酸、それ以外の無機溶媒、もしくは有機溶媒もしくは無機溶媒と有機溶媒の混合溶媒を用いる、請求項2記載の方法。
【請求項8】
流通式高温高圧装置に、基質及び反応溶媒を導入し、反応時間を3〜60秒の範囲で変化させることで合成反応を実施する、請求項2記載の方法。
【請求項9】
発熱反応の場合に、流通式常温高圧装置に、基質及び反応溶媒を導入し、反応時間を1〜60秒の範囲で変化させることで合成反応を実施する、請求項2記載の方法。
【請求項10】
水を送液する水送液ポンプ、水加熱用コイル、高温高圧フローセル、基質を送液する反応物送液ポンプ、炉体、反応物を炉体に導入する反応物導入管、反応溶液を排出する排出液ライン、冷却フランジ及び圧力を設定する背圧弁を具備していることを特徴とするポリアシル化合物合成装置。
【請求項11】
吸熱反応の場合において、水加熱用コイルが配設されている、請求項10記載のN−アシル化合物合成装置。
【請求項12】
請求項2記載の方法において、アシル化後、回収水溶液に水を注入してデカンテーションし、油/水二層溶液に分離後、ポリアシル化合物を含む油層を分液回収する一方、水層からは酢酸と水を共沸蒸留によって分離し、回収する簡易な連続分離法。
【請求項1】
無水カルボン酸とポリヘテロ水素化物との反応組成物であって、触媒及び有機溶媒の残存がないことを特徴とするポリアシル化合物組成物。
【請求項2】
無水カルボン酸とポリヘテロ水素化物からポリアシル化合物を合成する方法であって、発熱反応の場合には、常温流体、吸熱反応の場合には、高温高圧状態の亜臨界流体ないしは超臨界流体を反応溶媒として使用し、触媒を用いることなく、無水カルボン酸とポリヘテロ水素化物から一段階の合成反応でポリアシル化合物を選択的に合成することを特徴とするポリアシル化合物の製造方法。
【請求項3】
高温高圧状態の亜臨界ないしは超臨界水を反応溶媒として使用する、請求項2記載の方法。
【請求項4】
ポリヘテロ水素化物におけるヘテロ原子又は置換ヘテロ原子が、酸素(O)、硫黄(S)、窒化水素(NH)、アルキル置換窒素(NR’)、又はこれらの原子の組合せである、請求項1又は請求項2記載の方法。
【請求項5】
基質におけるヘテロ水素基及びヘテロ水素基に隣接するα位の置換数に対応して、温度と無水カルボン酸の量を調整することにより、N−アシル化後、O−アシル化を実施して、それぞれのアシル化において、無置換(1級)、一置換(2級)、二置換(3級)の順にアシル化しつつアシル化数を調節し、ポリアシル化合物を選択的に合成する、請求項1又は請求項2の方法。
【請求項6】
温度100〜400℃、圧力0.1〜40MPaの亜臨界流体ないし超臨界流体を反応溶媒として使用する、請求項2記載の方法。
【請求項7】
常温流体ないし亜臨界流体ないし超臨界流体として、水、酢酸、それ以外の無機溶媒、もしくは有機溶媒もしくは無機溶媒と有機溶媒の混合溶媒を用いる、請求項2記載の方法。
【請求項8】
流通式高温高圧装置に、基質及び反応溶媒を導入し、反応時間を3〜60秒の範囲で変化させることで合成反応を実施する、請求項2記載の方法。
【請求項9】
発熱反応の場合に、流通式常温高圧装置に、基質及び反応溶媒を導入し、反応時間を1〜60秒の範囲で変化させることで合成反応を実施する、請求項2記載の方法。
【請求項10】
水を送液する水送液ポンプ、水加熱用コイル、高温高圧フローセル、基質を送液する反応物送液ポンプ、炉体、反応物を炉体に導入する反応物導入管、反応溶液を排出する排出液ライン、冷却フランジ及び圧力を設定する背圧弁を具備していることを特徴とするポリアシル化合物合成装置。
【請求項11】
吸熱反応の場合において、水加熱用コイルが配設されている、請求項10記載のN−アシル化合物合成装置。
【請求項12】
請求項2記載の方法において、アシル化後、回収水溶液に水を注入してデカンテーションし、油/水二層溶液に分離後、ポリアシル化合物を含む油層を分液回収する一方、水層からは酢酸と水を共沸蒸留によって分離し、回収する簡易な連続分離法。
【図1】


【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公開番号】特開2007−291096(P2007−291096A)
【公開日】平成19年11月8日(2007.11.8)
【国際特許分類】
【出願番号】特願2007−96927(P2007−96927)
【出願日】平成19年4月2日(2007.4.2)
【出願人】(301021533)独立行政法人産業技術総合研究所 (6,529)
【Fターム(参考)】
【公開日】平成19年11月8日(2007.11.8)
【国際特許分類】
【出願日】平成19年4月2日(2007.4.2)
【出願人】(301021533)独立行政法人産業技術総合研究所 (6,529)
【Fターム(参考)】
[ Back to top ]