
遺伝子導入造血細胞から血液細胞を再構築させる方法
【課題】 本発明は、染色体非組込型ウイルスベクターを用いて、遺伝子導入造血細胞を含む造血細胞移植組成物を製造する方法を提供する。また本発明は、染色体非組込型ウイルスベクターを用いて遺伝子導入した造血細胞から、血液細胞を再構築させる方法を提供する。
【解決手段】 マイナス鎖RNAウイルスベクターを造血細胞に導入し、この細胞を造血細胞移植することにより、導入遺伝子を発現する血液細胞を再構築することができる。遺伝子導入造血細胞は、移植された体内において白血球、赤血球、血小板に分化することができた。本発明は、造血細胞移植における遺伝子治療における、宿主染色体損傷の危険がない安全な遺伝子導入システムを提供する。
【解決手段】 マイナス鎖RNAウイルスベクターを造血細胞に導入し、この細胞を造血細胞移植することにより、導入遺伝子を発現する血液細胞を再構築することができる。遺伝子導入造血細胞は、移植された体内において白血球、赤血球、血小板に分化することができた。本発明は、造血細胞移植における遺伝子治療における、宿主染色体損傷の危険がない安全な遺伝子導入システムを提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、遺伝子導入造血細胞を含む造血細胞移植のための組成物に関する。また本発明は、遺伝子導入造血細胞から血液細胞を再構築させる方法に関する。本発明の方法は、造血細胞移植における細胞染色体非損傷型の遺伝子治療に適用され得る。
【背景技術】
【0002】
X連鎖重症複合免疫不全症(X-linked SCID)など、単一遺伝子異常による難治性疾患に対する新たな治療法として遺伝子治療が期待されている。2000年にはフランスにおいて、CD34陽性造血幹細胞に対し原因遺伝子である共通ガンマ鎖(common γ-chain)遺伝子をレトロウイルスベクターにて導入し、11例中9例で有効であったことが報告された(Cavazzana-Calvo M. et al., Science 2000;288:669-672)。しかしその後、2例の患児にT細胞性白血病が発生し、同様なベクターや治療系に対する警鐘が成された(Kaiser J., Science 2003;299:495; Hacein-Bey-Abina S. et al., Science 2003;302:415-419)。その後の解析によりこの2例にはT細胞の発生に必要、かつ胸腺で発現が消失すべき遺伝子であるLMO2の恒常的活性化を認めることが報告されている(Hacein-Bey-Abina S. et al., Science 2003;302:415-419; Fischer A. et al., N Eng Journal Med 2004;350:2526-2527)。現在、このLMO2の恒常的活性化はこの臨床研究プロトコールに特異的なものではないかと考えられているが、レトロウイルスベクターのプロウイルスゲノムの宿主染色体内への取り込み自体が、癌抑制遺伝子の不活化や癌遺伝子の活性化の原因となり得る可能性は否定できず、根本的な問題点と考えられている。この潜在的危険性は同様に染色体へ遺伝子を組み込むアデノ随伴ウイルスベクター(AAV)、レンチウイルスベクターでも想定されている。先天性免疫不全性疾患には骨髄移植が有効性を示さない症例が存在するため、これらの患児には遺伝子治療のみが残されたオプションとなる。従ってこれまでの染色体組込型ベクターと異なる概念に基づく新しいかつ安全なベクターシステムの構築が望まれている。
【非特許文献1】Cavazzana-Calvo M. et al., Science 2000;288:669-672
【非特許文献2】Kaiser J., Science 2003;299:495
【非特許文献3】Hacein-Bey-Abina S. et al., Science 2003;302:415-419
【非特許文献4】Hacein-Bey-Abina S. et al., Science 2003;302:415-419
【非特許文献5】Fischer A. et al., N Eng Journal Med 2004;350:2526-2527
【発明の開示】
【発明が解決しようとする課題】
【0003】
本発明は、染色体非組込型ウイルスベクターを用いて、遺伝子導入造血細胞を含む造血細胞移植組成物を製造する方法を提供する。また本発明は、染色体非組込型ウイルスベクターを用いて遺伝子導入した造血細胞から、血液細胞を再構築させる方法を提供する。
【課題を解決するための手段】
【0004】
本発明者らは、造血細胞移植において、造血細胞の染色体を損傷させることのない安全な遺伝子導入を可能にするため、染色体非組込型ウイルスを用いる方法の開発を行った。このために、染色体非組込型ウイルスの1つであるマイナス鎖RNAウイルスを用いて造血細胞に遺伝子を導入し、この造血細胞からの血球系の再構築を試みた。まず、マウス大腿骨より赤血球・T細胞を除去した骨髄細胞を調製し、外来遺伝子を組み込んだマイナス鎖RNAウイルスを感染させ遺伝子導入骨髄細胞を作製した。そして、放射線照射により骨髄破壊(myeloablation)を施行したマウスに、この遺伝子導入骨髄細胞を注入する実験を行った。その後の生体内における移入骨髄細胞の生着動態と免疫学的反応を検討したところ、マイナス鎖RNAウイルスベクターが導入された骨髄細胞はレシピエント内で生着し、T細胞を含む血球系細胞のrepopulationが確認された。本発明は、染色体非傷害型細胞質型ベクターを用いた遺伝子導入造血細胞による血球系再構築を初めて開示するものであり、マイナス鎖RNAウイルスベクターを用いることにより、細胞の染色体を組み換えることなく遺伝子導入した移植造血細胞から血球系統のrepopulationが可能であることを実証するものである。特にエンベロープ構成蛋白質をコードする遺伝子に変異または欠損を有するマイナス鎖RNAウイルスベクターを用いれば、長期間安定して血球系細胞の再構築が行われることが判明した。本発明により、染色体損傷を懸念することなく、安全に造血細胞移植における遺伝子治療を実施することが可能となる。
【0005】
すなわち本発明は、造血細胞移植に適した遺伝子導入造血細胞組成物およびその製造方法、およびマイナス鎖RNAウイルスベクターを用いて遺伝子導入した造血細胞から血液細胞を再構築させる方法等に関し、より具体的には、請求項の各項に記載の発明に関する。なお同一の請求項を引用する請求項に記載の発明の1つまたは複数の組み合わせからなる発明は、それらの請求項に記載の発現に既に意図されている。すなわち本発明は、
〔1〕造血細胞移植組成物の製造方法であって、マイナス鎖RNAウイルスベクターを造血細胞に導入し、該造血細胞および薬学的に許容される媒体を含む組成物を調製することを含む方法、
〔2〕マイナス鎖RNAウイルスベクターが、エンベロープ構成蛋白質をコードする1つまたは複数の遺伝子が変異または欠損している、〔1〕に記載の方法、
〔3〕エンベロープ構成蛋白質をコードするすべての遺伝子が変異または欠損している、〔2〕に記載の方法、
〔4〕エンベロープ構成蛋白質をコードする少なくとも1つの遺伝子が欠損している、〔2〕または〔3〕に記載の方法、
〔5〕マイナス鎖RNAウイルスがパラミクソウイルス科ウイルスである、〔1〕から〔4〕のいずれかに記載の方法、
〔6〕パラミクソウイルス科ウイルスがセンダイウイルスである、〔5〕に記載の方法、
〔7〕〔1〕から〔6〕のいずれかに記載の方法により得られる造血細胞移植組成物、
〔8〕遺伝子導入された血液細胞を造血細胞から生成させる方法であって、
(a)マイナス鎖RNAウイルスベクターを造血細胞に接触させる工程、および
(b)工程(a)の造血細胞を動物に注入する工程、を含む方法、
〔9〕工程(b)の注入前に該動物を免疫抑制する工程をさらに含む、〔8〕に記載の方法、
〔10〕マイナス鎖RNAウイルスベクターにおいて、エンベロープ構成蛋白質をコードする1つまたは複数の遺伝子が変異または欠損している、〔8〕または〔9〕に記載の方法、
〔11〕エンベロープ構成蛋白質をコードするすべての遺伝子が変異または欠損している、〔10〕に記載の方法、
〔12〕エンベロープ構成蛋白質をコードする少なくとも1つの遺伝子が欠損している、〔10〕または〔11〕に記載の方法、
〔13〕マイナス鎖RNAウイルスがパラミクソウイルス科ウイルスである、〔8〕から〔12〕のいずれかに記載の方法、
〔14〕パラミクソウイルス科ウイルスがセンダイウイルスである、〔13〕に記載の方法、に関する。
【発明の効果】
【0006】
本発明において、染色体非組み込み型の細胞質型ベクターにより遺伝子導入された造血細胞が、生体内でrepopulationが可能であることが初めて実証された。レシピエントにおいて、外来遺伝子を発現するドナー由来のT細胞が検出されたことから、マイナス鎖RNAウイルスベクターが導入された造血幹細胞からrepopulationが起こったことが支持される。マイナス鎖RNAウイルスは、細胞質で転写・複製し、細胞核の染色体遺伝子に相互作用しない。本発明は、例えば先天性免疫不全症などの造血系疾患に対する宿主染色体非傷害遺伝子治療を可能とする。
【発明を実施するための最良の形態】
【0007】
本発明は、マイナス鎖RNAウイルスベクターが導入された造血細胞および薬学的に許容される媒体を含む造血細胞移植組成物およびその製造方法を提供する。本発明においてマイナス鎖RNAウイルスとは、マイナス鎖(ウイルス蛋白質をセンスにコードする鎖と相補的なアンチセンス鎖)のRNAをゲノムとして含むウイルスのことである。マイナス鎖RNAはネガティブ鎖RNAとも呼ばれる。
【0008】
また、マイナス鎖RNAウイルスベクターとは、遺伝子を細胞に導入するため担体(ベクター)としてのマイナス鎖RNAウイルスを言う。マイナス鎖RNAウイルスベクターは外来遺伝子を持っていても持たなくてもよい。
【0009】
組み換えウイルスとは、組み換えポリヌクレオチドを介して生成したウイルス、またはそのウイルスの増幅産物を言う。組み換えポリヌクレオチドとは、両端または片端が自然の状態と同じようには結合していないポリヌクレオチドを言う。具体的には、組み換えポリヌクレオチドは、人為的にポリヌクレオチド鎖の結合が改変(切断および/または結合)されたポリヌクレオチドである。組み換えポリヌクレオチドは、ポリヌクレオチド合成、ヌクレアーゼ処理、リガーゼ処理等を組み合わせて、公知の遺伝子組み換え方法により生成させることができる。組み換えウイルスは、遺伝子操作により構築されたウイルスゲノムをコードするポリヌクレオチドを発現させ、ウイルスを再構築することによって生成することができる。例えば、ウイルスゲノムをコードするcDNAから、ウイルス再構成する方法が知られている(Y. Nagai, A. Kato. Microbiol. Immunol. 1999: 43; 613-624 )。
【0010】
本発明において遺伝子とは遺伝物質を指し、転写単位をコードする核酸を言う。遺伝子はRNAであってもDNAであってもよい。本発明において蛋白質をコードする核酸は、該蛋白質の遺伝子と呼ぶ。また一般に、遺伝子は蛋白質をコードしていなくてもよく、例えば遺伝子はリボザイムまたはアンチセンスRNAなどの機能的RNAをコードするものであってもよい。一般に、遺伝子は天然由来または人為的に設計された配列であり得る。また、本発明において「DNA」とは、一本鎖DNAおよび二本鎖DNAを含む。また蛋白質をコードするとは、ポリヌクレオチドが該蛋白質を適当な条件下で発現できるように、該蛋白質のアミノ酸配列をコードするORFをセンスまたはアンチセンスに含むことを言う。
【0011】
本発明において得に好適に用いられるマイナス鎖RNAウイルスとしては、例えばパラミクソウイルス科 (Paramyxoviridae) ウイルスのセンダイウイルス(Sendai virus)、ニューカッスル病ウイルス(Newcastle disease virus)、おたふくかぜウイルス(Mumps virus)、麻疹ウイルス(Measles virus)、RSウイルス(Respiratory syncytial virus)、牛疫ウイルス(rinderpest virus)、ジステンパーウイルス(distemper virus)、サルパラインフルエンザウイルス(SV5)、ヒトパラインフルエンザウイルス1,2,3型、オルトミクソウイルス科(Orthomyxoviridae)のインフルエンザウイルス(Influenza virus)、ラブドウイルス科(Rhabdoviridae)の水疱性口内炎ウイルス(Vesicular stomatitis virus)、狂犬病ウイルス(Rabies virus)等が挙げられ、具体的には、Sendai virus (SeV)、human parainfluenza virus-1 (HPIV-1)、human parainfluenza virus-3 (HPIV-3)、phocine distemper virus (PDV)、canine distemper virus (CDV)、dolphin molbillivirus (DMV)、peste-des-petits-ruminants virus (PDPR)、measles virus (MV)、rinderpest virus (RPV)、Hendra virus (Hendra)、Nipah virus (Nipah)、human parainfluenza virus-2 (HPIV-2)、simian parainfluenza virus 5 (SV5)、human parainfluenza virus-4a (HPIV-4a)、human parainfluenza virus-4b (HPIV-4b)、mumps virus (Mumps)、およびNewcastle disease virus (NDV) などが含まれる。より好ましくは、Sendai virus (SeV)、human parainfluenza virus-1 (HPIV-1)、human parainfluenza virus-3 (HPIV-3)、phocine distemper virus (PDV)、canine distemper virus (CDV)、dolphin molbillivirus (DMV)、peste-des-petits-ruminants virus (PDPR)、measles virus (MV)、rinderpest virus (RPV)、Hendra virus (Hendra)、Nipah virus (Nipah)、Respiratory syncytial virus (RSV) からなる群より選択されるウイルス等が挙げられる。
【0012】
本発明において用いられるマイナス鎖RNAウイルスとしては、特に一本鎖マイナス鎖RNAウイルス(非分節型(non-segmented)マイナス鎖RNAウイルスとも言う)が好ましい。「一本鎖ネガティブ鎖RNAウイルス」とは、一本鎖ネガティブ鎖[すなわちマイナス鎖]RNAをゲノムに有するウイルスを言う。このようなウイルスとしては、パラミクソウイルス(Paramyxoviridae; Paramyxovirus, Morbillivirus, Rubulavirus, および Pneumovirus属等を含む)、ラブドウイルス(Rhabdoviridae; Vesiculovirus, Lyssavirus, および Ephemerovirus属等を含む)、フィロウイルス(Filoviridae)、オルトミクソウイルス(Orthomyxoviridae; Infuluenza virus A, B, C, および Thogoto-like viruses 等を含む)、ブニヤウイルス(Bunyaviridae; Bunyavirus, Hantavirus, Nairovirus, および Phlebovirus属等を含む)、アレナウイルス(Arenaviridae)などの科に属するウイルスが含まれる。
【0013】
本発明において用いられるマイナス鎖RNAウイルスは、より好ましくは、パラミクソウイルス亜科(レスピロウイルス属、ルブラウイルス属、およびモルビリウイルス属を含む)に属するウイルスまたはその誘導体であり、より好ましくはレスピロウィルス属(genus Respirovirus)(パラミクソウィルス属(Paramyxovirus)とも言う)に属するウィルスまたはその誘導体である。誘導体には、ウイルスによる遺伝子導入能を損なわないように、ウイルス遺伝子が改変されたウイルス、および化学修飾されたウイルス等が含まれる。本発明を適用可能なレスピロウィルス属ウィルスとしては、例えばヒトパラインフルエンザウィルス1型(HPIV-1)、ヒトパラインフルエンザウィルス3型(HPIV-3)、ウシパラインフルエンザウィルス3型(BPIV-3)、センダイウィルス(Sendai virus; マウスパラインフルエンザウィルス1型とも呼ばれる)、およびサルパラインフルエンザウィルス10型(SPIV-10)などが含まれる。本発明においてパラミクソウィルスは、最も好ましくはセンダイウィルスである。これらのウィルスは、天然株、野生株、変異株、ラボ継代株、および人為的に構築された株などに由来してもよい。
【0014】
マイナス鎖RNAウイルスベクターはウイルスゲノムRNAに搭載遺伝子をアンチセンスにコードしている。ウイルスゲノムRNAとは、マイナス鎖RNAウイルスのウイルス蛋白質と共にリボヌクレオプロテイン(RNP)を形成し、該蛋白質によりゲノム中の遺伝子が発現し、このRNAが複製されて娘RNPが形成される機能を持つRNAである。ゲノムRNAは、ウイルス遺伝子に変異および/または欠損があるものや、外来遺伝子が組み込まれるなどして改変されたものであってもよい。一般にマイナス鎖RNAウイルスのゲノムは、3'リーダー領域と5'トレイラー領域の間に、ウイルス遺伝子がアンチセンス配列として並んだ構成をしている。各遺伝子のORFの間には、転写終結配列(E配列) - 介在配列(I配列) - 転写開始配列(S配列) が存在し、これにより各遺伝子のORFをコードするRNAが別々のシストロンとして転写される。本発明のウイルスに含まれるゲノムRNAは、該RNAとRNPを構成するマイナス鎖RNAウイルス蛋白質をコードしている。但し、マイナス鎖RNAウイルスのゲノムRNAはマイナス鎖であるから、これらの蛋白質はアンチセンスとしてコードされている。ゲノムRNAとRNPを構成するマイナス鎖RNAウイルス蛋白質とは、マイナス鎖RNAウイルスのゲノムRNAと複合体を形成し、ゲノムRNAの複製およびゲノムにコードされている遺伝子の発現およびRNA自身の自律的な複製に必要とされるウイルス蛋白質群を言う。これらの蛋白質は、ウイルスのエンベロープを除くコアを形成する蛋白質であり、典型的には、N(ヌクレオキャプシド)、P(ホスホ)、およびL(ラージ)蛋白質である。ウイルス種によっては、表記は異なることもあるが、対応する蛋白質は当業者にとっては自明である (Anjeanette Robert et al., Virology. 1998: 247; 1-6 )。例えばNはNPと表記されることもある。また該RNAは、ウイルス粒子の形成に必要なM(マトリックス)蛋白質をコードしていてもよい。さらに該RNAは、ウイルス粒子の感染に必要なエンベロープ蛋白質をコードしていてもよい。マイナス鎖RNAウイルスのエンベロープ蛋白質としては、細胞膜融合を起こす蛋白質であるF(フュージョン)蛋白質および細胞への接着に必要なHN(ヘマグルチニン-ノイラミニダーゼ)蛋白質(または H (ヘマグルチニン))が挙げられる。また、F蛋白質および/またはHN (またはH) 蛋白質以外のウイルスエンベロープ蛋白質をコードさせてもよい。但し、後述のように、本発明において用いるマイナス鎖RNAウイルスは、エンベロープ構成蛋白質をコードする遺伝子が変異および/または欠損していることが好ましい。
【0015】
例えばパラミクソウイルス亜科に属する各ウィルスにおける各遺伝子は、一般に次のように表記される。一般に、NP遺伝子は"N"とも表記される。また、HNはノイラミニダーゼ活性を有さない場合にはH (ヘマグルチニン) と表記される。
レスピロウイルス属 NP P/C/V M F HN - L
ルブラウイルス属 NP P/V M F HN (SH) L
モービリウイルス属 NP P/C/V M F H - L
【0016】
例えばセンダイウイルスの各遺伝子の塩基配列のデータベースのアクセッション番号は、NP遺伝子については M29343、M30202, M30203, M30204, M51331, M55565, M69046, X17218、P遺伝子については M30202, M30203, M30204, M55565, M69046, X00583, X17007, X17008、M遺伝子については D11446, K02742, M30202, M30203, M30204, M69046, U31956, X00584, X53056、F遺伝子については D00152, D11446, D17334, D17335, M30202, M30203, M30204, M69046, X00152, X02131、HN遺伝子については D26475, M12397, M30202, M30203, M30204, M69046, X00586, X02808, X56131、L遺伝子については D00053, M30202, M30203, M30204, M69040, X00587, X58886を参照のこと。またその他のウイルスがコードするウイルス遺伝子を例示すれば、N遺伝子については、CDV, AF014953; DMV, X75961; HPIV-1, D01070; HPIV-2, M55320; HPIV-3, D10025; Mapuera, X85128; Mumps, D86172; MV, K01711; NDV, AF064091; PDPR, X74443; PDV, X75717; RPV, X68311; SeV, X00087; SV5, M81442; および Tupaia, AF079780、P遺伝子については、CDV, X51869; DMV, Z47758; HPIV-l, M74081; HPIV-3, X04721; HPIV-4a, M55975; HPIV-4b, M55976; Mumps, D86173; MV, M89920; NDV, M20302; PDV, X75960; RPV, X68311; SeV, M30202; SV5, AF052755; および Tupaia, AF079780、C遺伝子については CDV, AF014953; DMV, Z47758; HPIV-1. M74081; HPIV-3, D00047; MV, ABO16162; RPV, X68311; SeV, AB005796; および Tupaia, AF079780、M遺伝子については CDV, M12669; DMV Z30087; HPIV-1, S38067; HPIV-2, M62734; HPIV-3, D00130; HPIV-4a, D10241; HPIV-4b, D10242; Mumps, D86171; MV, AB012948; NDV, AF089819; PDPR, Z47977; PDV, X75717; RPV, M34018; SeV, U31956; および SV5, M32248、F遺伝子については CDV, M21849; DMV, AJ224704; HPN-1. M22347; HPIV-2, M60182; HPIV-3. X05303, HPIV-4a, D49821; HPIV-4b, D49822; Mumps, D86169; MV, AB003178; NDV, AF048763; PDPR, Z37017; PDV, AJ224706; RPV, M21514; SeV, D17334; および SV5, AB021962、HN(HまたはG)遺伝子については CDV, AF112189; DMV, AJ224705; HPIV-1, U709498; HPIV-2. D000865; HPIV-3, AB012132; HPIV-4A, M34033; HPIV-4B, AB006954; Mumps, X99040; MV, K01711; NDV, AF204872; PDPR, Z81358; PDV, Z36979; RPV, AF132934; SeV, U06433; および SV-5, S76876 が例示できる。但し、各ウイルスは複数の株が知られており、株の違いにより上記に例示した以外の配列からなる遺伝子も存在する。
【0017】
これらのウイルス蛋白質をコードするORFおよび外来遺伝子のORFは、ゲノムRNAにおいて上記のE-I-S配列を介してアンチセンスに配置される。ゲノムRNAにおいて最も3'に近いORFは、3'リーダー領域と該ORFとの間にS配列のみが必要であり、EおよびI配列は必要ない。またゲノムRNAにおいて最も5'に近いORFは、5'トレイラー領域と該ORFとの間にE配列のみが必要であり、IおよびS配列は必要ない。また2つのORFは、例えばIRES等の配列を用いて同一シストロンとして転写させることも可能である。このような場合は、これら2つのORFの間にはE-I-S配列は必要ない。例えば、野生型のパラミクソウイルスの場合、典型的なRNAゲノムは、3'リーダー領域に続き、N、P、M、F、HN (H)、およびL蛋白質をアンチセンスにコードする6つのORFが順に並んでおり、それに続いて5'トレイラー領域を他端に有する。本発明においてゲノムRNAは、ウイルス遺伝子の配置はこれに限定されるものではないが、好ましくは、野生型ウイルスと同様に、3'リーダー領域に続き、N、P、M、F、HN (またはH)、およびL蛋白質をコードするORFが順に並び、それに続いて5'トレイラー領域が配置されることが好ましい。ある種のウイルスにおいては、ウイルス遺伝子が異なっているが、そのような場合でも上記と同様に各ウイルス遺伝子を野生型と同様の配置とすることが好ましい。一般に N、P、およびL遺伝子を保持しているベクターは、細胞内で自律的にRNAゲノムから遺伝子が発現し、ゲノムRNAが複製される。さらにFおよびHN (またはH)遺伝子等のエンベロープのスパイク蛋白質をコードする遺伝子、およびM遺伝子の働きにより、感染性のウイルス粒子が形成され、細胞外に放出される。従って、このようなベクターは伝播能を有するウイルスベクターとなる。ベクターに外来遺伝子を搭載させる場合は、後述するように、このゲノム中の蛋白質非コード領域に挿入すればよい。
【0018】
本発明において用いるマイナス鎖RNAウイルスベクターは、1つまたは複数のエンベロープ構成蛋白質の遺伝子に変異および/または欠損を有するものが好ましい。このような変異型マイナス鎖RNAウイルスを用いることにより、血液細胞のリポピュレーションの効率が有意に上昇することが判明した。ここで変異とは、野生型遺伝子を持つウイルスに比べ、変異遺伝子に置換されたウイルスの感染性ウイルス産生能が有意に低下するような変異である。また欠損とは、野生型遺伝子の機能が実質的に失われることであり、機能的蛋白質を発現しないように改変されること、および遺伝子が欠失していることを含む。野生型遺伝子の機能が実質的に失われるとは、当該遺伝子の欠損により、37℃におけるウイルス産生が野生型の1/10以下、好ましくは1/20以下、より好ましくは1/50以下に減少することである。エンベロープ構成蛋白質とは、ウイルスのエンベロープの成分となるウイルス蛋白質を言い、エンベロープ表面に露出し細胞への接着または感染に機能するスパイク蛋白質およびエンベロープの形成等に機能する裏打ち蛋白質が含まれる。典型的には、エンベロープ構成蛋白質の遺伝子としては F、HN、およびMが挙げられ、ウイルス種によっては H、M1、および G 等の遺伝子が含まれる。これらのエンベロープ構成蛋白質の遺伝子の1つまたは複数を変異および/または欠失させたウイルスは、感染細胞における感染性ウイルス粒子の形成能が低下するため安全性が高い。また細胞傷害性が有意に低下する。ゲノムにおいて変異および/または欠損させる遺伝子としては、例えばF遺伝子、HN (またはH) 遺伝子、M遺伝子、またはその任意の組み合わせが挙げられる。特に好ましくは、少なくともF遺伝子が変異または欠損、より好ましくは欠損している。さらに好ましくは、F遺伝子およびHN (またはH) 遺伝子が変異または欠損、より好ましくは欠損している。さらに好ましくは、F遺伝子、HN (またはH) 遺伝子およびM遺伝子が変異または欠損している。
【0019】
特に、少なくともF遺伝子が欠失し、さらにHN (またはH) 遺伝子が変異または欠失するベクターを用いることが好ましい。このようなウイルスベクターは、一度細胞内に入り込んだ後は、F蛋白質は発現せず、HN (またはH) 蛋白質も極低レベル発現または無発現となる。ベクター感染細胞においてHN蛋白質が多数表出すると、細胞同士の吸着やaggregationを来たす可能性があり、事実このような現象がin vitroで起こり得ることを本発明者らは確認した。従って、HN (またはH) 遺伝子に温度感受性変異を有するベクター、さらに好ましくはHN (またはH) 遺伝子を欠損するベクターを用いることにより、造血細胞移植においてより良好な結果を得ることが可能となる。さらに、M遺伝子を変異または欠損させることにより、より良好な生着動態を得ることができる。
【0020】
エンベロープ構成蛋白質において多数の温度感受性変異が知られている。これらの温度感受性変異蛋白質遺伝子を有するウイルスを本発明において好適に用いることができる。温度感受性変異とは、低温 (例えば32℃) に比べ、ウイルス宿主の通常の温度(例えば37℃)において有意に活性が低下する変異のことである。このような、温度感受性変異を持つ蛋白質は、野生型蛋白質などで相補しなくても許容条件(低温)下でウイルスを作製することができるので有用である。
【0021】
例えば、M遺伝子の温度感受性変異としては、特に限定されるものではないが、センダイウィルスのM蛋白質におけるG69、T116、およびA183からなる群より選択される少なくとも1つ、好ましくは任意に選択される2つ、さらに好ましくは3つすべてのアミノ酸部位、あるいは他のマイナス鎖RNAウィルスM蛋白質のそれらと相同な部位が変異しているものを好適に用いることができる。ここでG69とはM蛋白質の69番目のアミノ酸Gly、T116とはM蛋白質の116番目のアミノ酸Thr、A183とはM蛋白質の183番目のアミノ酸Alaを指す。
【0022】
M蛋白質をコードする遺伝子(M遺伝子)は、マイナス鎖RNAウイルスで広く保存されており、ウイルスのヌクレオカプシッドとエンベロープの両者に相互作用する機能を有することが知られている(Garoff, H. et al., Microbiol. Mol. Biol. Rev. 1998: 62; 117-190)。また、SeV M蛋白質においてamphiphilic α-helixと予想されている104-119(104-KACTDLRITVRRTVRA-119/配列番号:1)は粒子形成に重要な領域として同定されている(Genevieve Mottet et al., J. Gen. Virol. 1999: 80; 2977-2986)が、当該領域はマイナス鎖RNAウイルス間で良く保存されている。M蛋白質のアミノ酸配列はマイナス鎖RNAウイルスで類似しており、特にパラミクソウイルス亜科においては既知のM蛋白質は共通して全長約330〜380アミノ酸からなる塩基性蛋白質であり、全領域にわたって類似性があるが、特にC端側半分での類似性が高い(Gould, A. R. Virus Res. 1996: 43; 17-31、Harcourt, B.H. et al., Virology. 2000: 271; 334-349)。従って、例えば SeV M蛋白質のG69、T116、及びA183と相同なアミノ酸は容易に同定することが可能である。
【0023】
SeV M蛋白質のG69、T116、及びA183と対応する他のマイナス鎖RNAウィルスM蛋白質の相同な部位のアミノ酸は、当業者であれば、例えばBLASTなどのアミノ酸配列のホモロジー検索プログラム(アライメント作成機能を持つもの)またはCLUSTAL Wなどのアライメント作成プログラムを用いてSeV M蛋白質のアミノ酸と整列化することにより同定することができる。例えばSeV M蛋白質のG69に相当する各M蛋白質の相同部位としては、human parainfluenza virus-1 (HPIV-1)(括弧は略称)であればG69、human parainfluenza virus-3 (HPIV-3) であればG73、phocine distemper virus (PDV)およびcanine distemper virus (CDV)であればG70、dolphin molbillivirus (DMV)であればG71、peste-des-petits-ruminants virus (PDPR)、measles virus (MV)、およびrinderpest virus (RPV)であればG70、Hendra virus (Hendra)およびNipah virus (Nipah)であればG81、human parainfluenza virus-2 (HPIV-2)であればG70、human parainfluenza virus-4a (HPIV-4a)およびhuman parainfluenza virus-4b (HPIV-4b)であればE47、mumps virus (Mumps)であればE72が挙げられる(文字と番号はアミノ酸とその位置を表す)。また、SeV M蛋白質のT116に相当する各M蛋白質の相同部位としては、human parainfluenza virus-1 (HPIV-1)であればT116、human parainfluenza virus-3 (HPIV-3)でればT120、phocine distemper virus (PDV)およびcanine distemper virus (CDV)であればT104、dolphin molbillivirus (DMV)であれはT105、peste-des-petits-ruminants virus (PDPR)、measles virus (MV)およびrinderpest virus (RPV)であればT104、Hendra virus (Hendra)およびNipah virus (Nipah)であればT120、human parainfluenza virus-2 (HPIV-2)およびsimian parainfluenza virus 5 (SV5)であればT117、human parainfluenza virus-4a (HPIV-4a)およびhuman parainfluenza virus-4b (HPIV-4b)であればT121、mumps virus (Mumps)であればT119、Newcastle disease virus (NDV)であればS120が挙げられる。SeV M蛋白質のA183に相当する各M蛋白質の相同部位としては、human parainfluenza virus-1 (HPIV-1)であればA183、human parainfluenza virus-3 (HPIV-3)であれなF187、phocine distemper virus (PDV)およびcanine distemper virus (CDV)であればY171、dolphin molbillivirus (DMV)であればY172、peste-des-petits-ruminants virus (PDPR)、measles virus (MV)およびrinderpest virus (RPV)であれはY171、Hendra virus (Hendra)およびNipah virus (Nipah)であればY187、human parainfluenza virus-2 (HPIV-2)であれはY184、simian parainfluenza virus 5 (SV5)であればF184、human parainfluenza virus-4a (HPIV-4a)およびhuman parainfluenza virus-4b (HPIV-4b)であれはF188、mumps virus (Mumps)であればF186、Newcastle disease virus (NDV)であればY187が挙げられる。ここに挙げたウイルスにおいて、それぞれのM蛋白質に上記の3つの部位のいずれか、好ましくは任意の2部位の組み合わせ、さらに好ましくは3つの部位全てのアミノ酸が他のアミノ酸に置換された変異M蛋白質をコードするゲノムを有するウイルスは、本発明において好適に用いられる。
【0024】
アミノ酸変異は、所望の他のアミノ酸への置換であってよいが、好ましくは、側鎖の化学的性質の異なるアミノ酸への置換である。例えばアミノ酸は、塩基性アミノ酸(例えばリジン、アルギニン、ヒスチジン)、酸性アミノ酸 (例えばアスパラギン酸、グルタミン酸)、非荷電極性アミノ酸 (例えばグリシン、アスパラギン、グルタミン、セリン、スレオニン、チロシン、システイン)、非極性アミノ酸 (例えばアラニン、バリン、ロイシン、イソロイシン、プロリン、フェニルアラニン、メチオニン、トリプトファン)、β分岐アミノ酸 (例えばスレオニン、バリン、イソロイシン)、および芳香族アミノ酸 (例えばチロシン、フェニルアラニン、トリプトファン、ヒスチジン)などのグループに分類することができるが、あるアミノ酸について、そのアミノ酸が属するグループのアミノ酸以外のアミノ酸に置換することなどが挙げられる。具体的には、塩基性アミノ酸であれは、酸性または中性アミノ酸への置換、極性アミノ酸であれは非極性アミノ酸への置換、20種の天然のアミノ酸の平均分子量より大きい分子量を持つアミノ酸であれば、その平均分子量より小さいアミノ酸への置換、逆にその平均分子量より小さいアミノ酸であれば、それより大きいアミノ酸への置換などが挙げられるが、それに限定されない。
【0025】
具体的に例示すれば、センダイウィルスM蛋白質における G69E、T116A、およびA183Sからなる群より選択される変異あるいはそれらと相同な位置に変異を含む他のパラミクソウイルスのM蛋白質を用いることができる。ここでG69Eとは、M蛋白質の69番目のアミノ酸GlyがGluに置換された変異、T116Aとは、M蛋白質の116番目のアミノ酸ThrがAlaに置換された変異、A183Sとは、M蛋白質の183番目のアミノ酸AlaがSerに置換された変異を言う。すなわち、センダイウィルス M蛋白質のG69、T116、およびA183あるいは他のウイルスM蛋白質の相同部位を、それぞれGlu (E)、Ala (A)、およびSer (S) へ置換することができる。これらの変異は組み合わせて有していることが好ましく、特に上記3変異の全てを保持していることがより好ましい。M遺伝子への変異の導入は、公知の変異導入方法に従って実施することができる。例えば実施例に記載のように目的の変異を入れたオリゴヌクレオチドを用いて導入することが可能である。
【0026】
また、例えば麻疹ウィルスにおいては、M蛋白のモノクローナル抗体に対するエピトープが変化している温度感受性株のP253-505(Morikawa, Y. et al., Kitasato Arch. Exp. Med. 1991: 64; 15-30)のM遺伝子配列を用いてもよい。また、SeV M蛋白質の116番目のThrに対応する麻疹ウィルスM蛋白質の104番目のThr、またはムンプスウィルスのM蛋白質の119番目のThrを他のアミノ酸(例えばAla)に置換してもよい。
【0027】
本発明において用いられるベクターは、さらに好ましい態様においてはM遺伝子を欠損している。M遺伝子の欠損とは、野生型M遺伝子の機能が実質的に失われていることを言い、機能欠失型の変異を有するM遺伝子を持つ場合およびM遺伝子を欠失する場合を含む。M遺伝子の機能欠失型変異は、例えばM遺伝子の蛋白質コード配列を欠失させたり、他の配列を挿入することにより作製することができる。例えば、M蛋白質コード配列の途中に停止コドンを設計することができる(WO00/09700)。本発明のベクターは、最も好ましくはM蛋白質のコード配列を完全に欠失している。M蛋白質のORFを欠失したベクターは、温度感受性変異M蛋白質をコードするベクターとは違い、任意の条件においてウイルス粒子を形成する能力を失っている。
【0028】
HN遺伝子の温度感受性変異としては、特に限定されるものではないが、例えばセンダイウィルスのHN蛋白質のA262、G264、およびK461からなる群より選択される少なくとも1つ、好ましくは任意に選択される2つ、さらに好ましくは3つすべてのアミノ酸部位、あるいは他のマイナス鎖RNAウィルスM蛋白質のそれらと相同な部位に変異を含むものを好適に用いることができる。ここでA262とはHN蛋白質の262番目のアミノ酸Ala、G264とはHN蛋白質の264番目のアミノ酸Gly、K461とはHN蛋白質の461番目のアミノ酸Lysを指す。アミノ酸変異は、所望の他のアミノ酸への置換であってよいが、好ましくは、上記のM蛋白質の変異と同様、側鎖の化学的性質の異なるアミノ酸への置換である。例えば、上記したように異なるグループのアミノ酸に置換することなどが挙げられる。具体的には、例えばセンダイウィルスHN蛋白質における A262T、G264R、およびK461Gからなる群より選択される変異あるいはそれらと相同な変異を含むものを好適に用いることができる。ここでA262Tとは、HN蛋白質の262番目のアミノ酸AlaがThrに置換された変異、G264Rとは、HN蛋白質の264番目のアミノ酸GlyがArgに置換された変異、K461Gとは、HN蛋白質の461番目のアミノ酸LysがGlyに置換された変異を言う。すなわち、センダイウィルス HN蛋白質のA262、G264、およびK461あるいは他のウィルスHN蛋白質の相同部位を、それぞれThr (T)、Arg (R)、およびGly (G) へ置換することができる。これらの変異は組み合わせて有していることが好ましく、特にこれらの3つの変異の全てを保持していることがより好ましい。
【0029】
また、例えば、ムンプスウィルスにおいては、温度感受性の性質を示しワクチンとして使用されているUrabe AM9株(Wright, K. E. et al., Virus Res. 2000: 67; 49-57)を参考に、HN蛋白の464及び468番目のアミノ酸に変異導入することは本発明において好ましい。これと相同な位置のアミノ酸の変異は、他のマイナス鎖RNAウィルスにも適用することができる。
【0030】
またマイナス鎖RNAウィルスは、P遺伝子またはL遺伝子に変異を有していてもよい。このような変異としては、具体的には、SeV P蛋白質の86番目のGlu(E86)の変異、SeV P蛋白質の511番目のLeu(L511)の他のアミノ酸への置換、または他のマイナス鎖RNAウィルスP蛋白質の相同部位の置換が挙げられる。アミノ酸変異は、所望の他のアミノ酸への置換であってよいが、好ましくは、上記と同様、側鎖の化学的性質の異なるアミノ酸への置換である。例えば、上記したように異なるグループのアミノ酸に置換することなどが挙げられる。具体的には、E86のLysへの置換(E86K)、L511のPheへの置換(L511F)などが例示できる。またL蛋白質においては、1197番目のAsn(N1197)および/または1795番目のLys(K1795)の他のアミノ酸への置換、または他のマイナス鎖RNAウィルスL蛋白質の相同部位の置換が挙げられる。L蛋白質のこれら2つの変異の両方を有するL蛋白質遺伝子は特に好ましい。アミノ酸変異は、やはり所望の他のアミノ酸への置換であってよいが、好ましくは、上記と同様、側鎖の化学的性質の異なるアミノ酸への置換である。例えば、上記したように異なるグループのアミノ酸に置換することなどが挙げられる。具体的には、N1197のSerへの置換(N1197S)、K1795のGluへの置換(K1795E)などが例示できる。P遺伝子とL遺伝子の変異は、両方持っていることで、持続感染性、2次粒子放出の抑制、または細胞傷害性の抑制の効果を顕著に高めることができる。さらに、エンベロープ蛋白質遺伝子の変異および/または欠損を組み合わせることで、これらの効果を劇的に上昇させることができる。
【0031】
またマイナス鎖RNAウイルスベクターは、アクセサリー遺伝子が欠損したものであってよい。例えばSeVのアクセサリー遺伝子の1つであるV遺伝子をノックアウトすることにより、培養細胞における遺伝子発現および複製は障害されることなく、マウス等の宿主に対するSeVの病原性が顕著に減少する(Kato, A. et al. J. Virol. 1997: 71; 7266-7272, Kato, A. et al. EMBO J. 1997: 16; 578-587, Curran, J. et al., WO01/04272, EP1067179)。このような弱毒化ベクターは、より毒性の低い遺伝子導入用ウイルスベクターとして特に有用である。
【0032】
マイナス鎖RNAウイルスは宿主細胞の細胞質でのみ転写・複製を行い、DNAフェーズを持たないため染色体への組み込み(integration)は起こらない(Lamb, R.A. and Kolakofsky, D., Paramyxoviridae: The viruses and their replication. In: Fields BN, Knipe DM, Howley PM, (eds). Fields of virology. Lippincott - Raven Publishers: Philadelphia, 1996: 2; 1177-1204)。このため染色体異常による癌化および不死化などの安全面における問題が生じない。マイナス鎖RNAウイルスのこの特徴は、ベクター化した時の安全性に大きく寄与するものと考えられる。異種遺伝子発現の結果では、例えばセンダイウイルス(SeV)を連続多代継代しても殆ど塩基の変異が認められず、ゲノムの安定性が高く、挿入異種遺伝子を長期間に渡って安定に発現する事が示されている(Yu, D. et al., Genes Cells. 1997: 2: 457-466)。また、カプシド構造蛋白質を持たないことによる導入遺伝子のサイズまたはパッケージングの柔軟性(flexibility)など性質上のメリットがある。SeVベクターは、外来遺伝子を少なくとも5kbまで導入可能であり、転写ユニットを付加することによって2種類以上の遺伝子を同時に発現する事も可能である。
【0033】
特にセンダイウイルスは、齧歯類にとっては病原性で肺炎を生じることが知られているが、人に対しては病原性がない。これはまた、野生型センダイウイルスの経鼻的投与によって非ヒト霊長類において重篤な有害作用を示さないというこれまでの報告によっても支持されている(Hurwitz, J.L. et al., Vaccine. 1997: 15; 533-540, Bitzer, M. et al., J. Gene Med. 2003: 5; 543-553, Slobod, K.S. et al., Vaccine. 2004 : 22; 3182-3186)。また、種々の細胞・臓器へ、既存のベクターでは得られない極めて高い遺伝子導入・発現効率を示す(Yonemitsu Y. et al., Nature Biotechnol 2000;18:970-973; Masaki I. et al., FASEB J 2001;15:1294-1296; Yamashita A. et al., J Immunol 2002;168:450-457; Masaki I. et al., Circ Res 2002;90:966-973; Onimaru M. et al., Circ Res 2002;91:723-730; Shoji F. et al., Gene Ther 2003;10:213-218; Okano S. et al., Gene Ther 2003;10:1381-1391)。ヒト造血幹細胞へのin vitro導入においても、3系統への分化能を維持したまま高効率遺伝子導入が可能である(Jin CH. et al., Gene Ther. 2003 ;10:272-277)。センダイウイルスのこれらの特徴は、センダイウイルスベクターが人への造血細胞移植における遺伝子治療ベクターとしての利用を支持するものである。
【0034】
マイナス鎖RNAウイルスベクターは、ゲノムRNA中に所望の外来遺伝子をコードし得る。外来遺伝子を含む組換えウイルスベクターは、上記のウイルスベクターのゲノムに外来遺伝子を挿入することによって得られる。外来遺伝子の挿入位置は、例えばウイルスゲノムの蛋白質非コード領域の所望の部位を選択することができ、例えばゲノムRNAの3'リーダー領域と3'端に最も近いウイルス蛋白質ORFとの間、各ウイルス蛋白質ORFの間、および/または5'端に最も近いウイルス蛋白質ORFと5'トレイラー領域の間に挿入することができる。また、M、FまたはHN遺伝子などのエンベロープ構成蛋白質遺伝子を欠失するゲノムでは、その欠失領域に外来遺伝子をコードする核酸を挿入することができる。パラミクソウイルスに外来遺伝子を導入する場合は、ゲノムへの挿入断片のポリヌクレオチドの鎖長が6の倍数となるように挿入することが望ましい(Kolakofski, D. et al., J. Virol. 1998: 72; 891-899; Calain, P. and Roux, L. J. Virol. 1993: 67; 4822-4830)。挿入した外来遺伝子とウイルスORFとの間には、E-I-S配列が構成されるようにする。E-I-S配列を介して2またはそれ以上の外来遺伝子をタンデムに並べて挿入することができる。
【0035】
マイナス鎖RNAウイルスにより導入する外来遺伝子としては、特に制限はないが、天然の蛋白質としては、例えばサイトカイン、その他の液性因子、増殖因子、受容体、細胞内シグナル分子、酵素、ペプチドなどが挙げられる。蛋白質は分泌蛋白質、膜蛋白質、細胞質蛋白質、核蛋白質などであり得る。人工的な蛋白質としては、例えば、キメラ毒素などの融合蛋白質、ドミナントネガティブ蛋白質(受容体の可溶性分子または膜結合型ドミナントネガティブ受容体を含む)、欠失型の細胞接着分子および細胞表面分子などが挙げられる。また、分泌シグナル、膜局在化シグナル、または核移行シグナル等を付加した蛋白質であってもよい。導入遺伝子としてアンチセンスRNA分子またはRNA切断型リボザイムなどを発現させて、特定の遺伝子の機能を抑制することもできる。外来遺伝子として疾患の治療用遺伝子を用いてウイルスベクターを調製すれば、このベクターを投与して遺伝子治療を行うことが可能となる。本発明のウイルスベクターの遺伝子治療への応用としては、直接投与による遺伝子発現、間接(ex vivo)投与による遺伝子発現のいずれの方法によっても、治療効果を期待できる外来遺伝子もしくは患者の体内で供給が不足している内在遺伝子等を樹状細胞から発現させることが可能である。また本発明の方法は、再生医療における遺伝子治療ベクターとしても利用できる。
【0036】
以下に搭載遺伝子を例示する。
CD45: SCIDの原因1遺伝子、染色体1q31-32に位置。共通リンパ球CD抗原でSrc kinaseを調節するフォスファターゼでこの欠損によりT、NK欠損型のSCIDとなる(Hermiston. et al., Annu. Rev. Immunol. 2003: 21; 107-137; Buckley RH., Annu. Rev. Immunol. 2004: 22; 625-655)
IL-7 receptor alpha: SCIDの原因1遺伝子、染色体5p13に位置。リンパ球特にT細胞の分化、ホメオスターシスに重要なIL-7のレセプターのサブユニット、この欠損によりT欠損型のSCIDとなる(Peschon JJ., J. Exp. Med. 1994: 180; 1955-1960; Buckley RH., Annu. Rev. Immunol. 2004: 22; 625-655)
RAG1/2: SCIDの原因1遺伝子、染色体11p13に位置。T細胞レセプター及び抗体の生成において重要なリコンビナーゼで、この欠損によりT、B欠損型のSCIDとなる(schwarz. et al., Science. 1996: 274; 97-99; Buckley RH., Annu. Rev. Immunol. 2004: 22; 625-655)
Adenosine Deaminase (ADA): SCIDの原因1遺伝子、染色体20q13.11に位置。プリン代謝の再利用経路における酵素で、この遺伝子欠損によりプリン代謝産物の蓄積によりリンパ球がアポトーシスとなりT、NK欠損型のSCIDとなる (Noguchi M., Cell, 1999: 73; 147-157; Puck JM.,Hum. Mol. Gemet., 1993: 1099-1104; Buckley RH., Annu. Rev. Immunol. 2004: 22; 625-655)
common gamma chain (γc) : SCIDの原因1遺伝子、染色体Xq13.1に位置。IL-2, IL-4, IL7, IL-7, IL-15, IL-21のレセプターに共通するサブユニットで、T、B、NK欠損型のSCID(いわゆるX-linked SCID)となる (Buckley RH. et al., J. Pediatr. 1997: 130; 378-387; Buckley RH. et al., N, Engl. J. Med. 1999: 340; 508-516, Buckley RH., Annu. Rev. Immunol. 2004: 22; 625-655)
DAF及びCD59:Glycosil phosphatidylinositol linked proteinで補体活性を抑制する蛋白で両者の欠損は、赤血球の自己の補体活性による溶血を惹起し、重症の夜間血色素尿症を発症する(Johnson RJ. et al., J. Clinic. Pathol. , 2002: 55; 145-152)
copper transporting P-type ATPase (ATP7B, Wilson disease protein):ウイルソン病の原因遺伝子。染色体13に位置し、胆管での銅代謝に重要な蛋白で、この欠損により肝臓、角膜、脳に銅が沈着して発症する (Ferenci P., Clin liver Dis. 1998: 2; 31-49)
GFP(オワンクラゲ蛍光発色体)、luciferase:これらは遺伝子導入細胞のマーカーとして生体内での動態の解析や遺伝子導入効率の判定に利用できる。
FGF-2:(WO2002/042481)
その他、Btk(伴性無ガンマグロブリン血症で欠損)、CD40/CD40 ligand(高IgM症候群で欠損)、(Cooper MD. et al., Hematology, 2003: 313-330)、WASP(Wiskott-Aldrich症候群で欠損;Snapper SB. et al., Annu. Rev. immunol. 1999: 17; 905-929)、などが挙げられるが、これらの限定されない。
【0037】
ベクターに搭載する外来遺伝子の発現レベルは、その遺伝子の上流(マイナス鎖(ネガティブ鎖)の3'側)に付加する転写開始配列の種類により調節することができる(WO01/18223)。また、ゲノム上の外来遺伝子の挿入位置によって制御することができ、マイナス鎖の3'の近くに挿入するほど発現レベルが高く、5'の近くに挿入するほど発現レベルが低くなる。このように、外来遺伝子の挿入位置は、該遺伝子の所望の発現量を得るために、また前後のウイルス蛋白質をコードする遺伝子との組み合わせが最適となる様に適宜調節することができる。一般に、外来遺伝子の高い発現が得られることが有利と考えられるため、外来遺伝子は、効率の高い転写開始配列に連結し、マイナス鎖ゲノムの3'端近くに挿入することが好ましい。具体的には、3'リーダー領域と3'に最も近いウイルス蛋白質ORFとの間に挿入される。あるいは、3'に一番近いウイルス蛋白質遺伝子と2番目のウイルス蛋白質遺伝子のORFの間、または3'から2番目と3番目のウイルス蛋白質遺伝子の間に挿入してもよい。野生型パラミクソウイルスにおいては、ゲノムの3'に最も近いウイルス蛋白質遺伝子はN遺伝子であり、2番目の遺伝子はP遺伝子、3番目の遺伝子はM遺伝子である。逆に、導入遺伝子の高発現が望ましくない場合は、例えば外来遺伝子の挿入位置をマイナス鎖ゲノムのなるべく5'側に設定したり、転写開始配列を効率の低いものにするなどして、ウイルスベクターからの発現レベルを低く抑えることで適切な効果が得られるようにすることも可能である。
【0038】
外来遺伝子をコードする核酸をゲノムに挿入するときに付加するS配列としては、例えばマイナス鎖RNAウイルスの所望のS配列を用いることができるが、センダイウイルスであれば、3'-UCCCWVUUWC-5'(W= AまたはC; V= A, C, またはG)(配列番号:2)の配列を好適に用いることができる。特に 3'-UCCCAGUUUC-5'(配列番号:3)、3'-UCCCACUUAC-5'(配列番号:4)、および 3'-UCCCACUUUC-5'(配列番号:5)が好ましい。これらの配列は、プラス鎖をコードするDNA配列で表すとそれぞれ 5'-AGGGTCAAAG-3'(配列番号:6)、5'-AGGGTGAATG-3'(配列番号:7)、および 5'-AGGGTGAAAG-3'(配列番号:8)である。センダイウイルスベクターのE配列としては、例えば 3'-AUUCUUUUU-5'(配列番号:9)(プラス鎖をコードするDNAでは 5'-TAAGAAAAA-3'(配列番号:10))が好ましい。I配列は、例えば任意の3塩基であってよく、具体的には 3'-GAA-5'(プラス鎖DNAでは 5'-CTT-3')を用いればよい。
【0039】
マイナス鎖RNAウイルスベクターを製造するには、(a)哺乳動物細胞において、マイナス鎖RNAウイルスのゲノムRNAを含むRNPの再構成に必要なウイルス蛋白質の存在下、マイナス鎖RNAウイルスのゲノムRNAをコードするcDNAを転写させる工程、(b)生成したマイナス鎖RNAウイルスを回収する工程、により製造することができる。上記のRNPの再構成に必要なウイルス蛋白質とは、典型的には N、P、およびL蛋白質を言う。転写によりマイナス鎖ゲノム(すなわちウイルスゲノムと同じアンチセンス鎖)を生成させてもよく、あるいはプラス鎖(アンチゲノム。ゲノムRNAの相補鎖。)を生成させても、ウイルスRNPを再構成することができる。ベクターの再構成効率を高めるには、好ましくはプラス鎖を生成させる。RNA末端は、天然のウイルスゲノムと同様に3'リーダー配列と5'トレイラー配列の末端をなるべく正確に反映させることが好ましい。このためには、例えば転写産物の5'端に自己切断型のリボザイムを付加しておき、リボザイムによりマイナス鎖RNAウイルスゲノムの末端を正確に切り出させることにより実現させることができる。あるいは、転写産物の5'端を正確に制御するために、転写開始部位としてバクテリオファージのRNAポリメラーゼ認識配列を利用し、該RNAポリメラーゼを細胞内で発現させる。転写産物の3'端を制御するには、例えば転写産物の3'端に自己切断型リボザイムをコードさせておき、このリボザイムにより正確に3'端が切り出されるようにすることができる(Hasan, M. K. et al., J. Gen. Virol. 1997: 78; 2813-2820、Kato, A. et al., EMBO J. 1997,: 16; 578-587 及び Yu, D. et al., Genes Cells. 1997: 2; 457-466)。リボザイムとしては、デルタ肝炎ウイルスのアンチゲノム鎖(antigenomic strand)由来の自己開裂リボザイムが使用できる。
【0040】
より具体的な組み換えウイルスの再構成は、例えば以下の文献を参照することができる(WO97/16539; WO97/16538; WO00/70055; WO00/70070; WO01/18223; WO03/025570; Durbin, A. P. et al. Virology, 1997: 235; 323-332; Whelan, S. P. et al. Proc. Natl. Acad. Sci. USA, 1995: 92; 8388-8392; Schnell. M. J. et al., EMBO J. 1994: 13; 4195-4203; Radecke, F. et al., EMBO J. 1995: 14; 5773-5784; Lawson, N. D. et al., Proc. Natl. Acad. Sci. USA 92: 4477-4481; Garcin, D. et al. EMBO J. , 1995: 14; 6087-6094; Kato, A. et al., Genes Cells. 1996: 1; 569-579; Baron, M. D. and Barrett, T., J. Virol. 1997: 71; 1265-1271; Bridgen, A. and Elliott, R. M., Proc. Natl. Acad. Sci. USA. 1996: 93; 15400-15404; Hasan, M. K. et al., J. Gen. Virol., 1997: 78; 2813-2820、Kato, A. et al., EMBO J. 1997: 16; 578-587、Yu, D. et al., Genes Cells, 1997: 2; 457-466、Tokusumi, T. et al. Virus Res. 2002: 86; 33-38、Li, H.-O. et al., J. Virol. 2000: 74; 6564-6569)。これらの方法により、パラインフルエンザ、水疱性口内炎ウイルス、狂犬病ウイルス、麻疹ウイルス、リンダーペストウイルス、センダイウイルスなどを含むマイナス鎖RNAウイルスをDNAから再構成させることができる。
【0041】
ウイルス生産細胞でウイルス蛋白質やウイルスゲノムRNAを発現させるためにプラスミドベクターをトランスフェクトする場合、例えばリン酸カルシウム法(Graham, F. L. and Van Der Eb, J., Virology. 1973: 52; 456; Wigler, M. and Silverstein, S., Cell. 1977: 11; 223; Chen, C. and Okayama, H. Mol. Cell. Biol., 1987: 7; 2745)、種々のトランスフェクション試薬を用いた方法、あるいは電気穿孔法等を用いることができる。トランスフェクション試薬については、DEAE-デキストラン(Sigma #D-9885 M.W. 5×105 )、DOTMA(Roche)、Superfect(QIAGEN #301305)、DOTAP、DOPE、DOSPER(Roche #1811169)、TransIT-LT1 (Mirus, Product No. MIR 2300) などを用いることができる。トランスフェクション試薬とDNAとの複合体がエンドソーム中で分解されてしまうのを防ぐため、クロロキンを加えることができる(Calos, M. P., Proc. Natl. Acad. Sci. USA 1983: 80; 3015)。
【0042】
温度感受性変異を有する蛋白質を用いる場合は、組み換えウイルスの産生を許容温度(例えば35℃以下、より好ましくは34℃以下、さらに好ましくは33℃以下、最も好ましくは32℃またはそれ以下)で行う。また、野生型蛋白質を用いる場合でも、ウイルス産生を低温で行うことで効率的な粒子形成を行わせることが可能である。エンベロープ構成蛋白質遺伝子を欠損する組み換えウイルスを製造する場合は、ウイルス産生細胞において、これら欠損させた遺伝子および/または他のウイルスのエンベロープ蛋白質をコードする遺伝子などを細胞に導入して発現させることにより、感染性のウイルス粒子を形成させることができる(Hirata, T. et al., J. Virol. Methods, 2002: 104; 125-133; Inoue, M. et al., J. Virol. 2003 : 77; 6419-6429; Inoue M. et al., J Gene Med. 2004;6:1069-1081)。
【0043】
例えば、ウイルス産生において、水疱性口内炎ウイルス(Vesicular stomatitis virus; VSV)のG蛋白質(VSV-G)やレトロウイルスのアンフォトロピックエンベロープ蛋白質などのヒト細胞に感染するウイルスに由来するエンベロープ蛋白質を用いることができる。VSV-G蛋白質は、任意のVSV株に由来するものであってよい。例えば Indiana血清型株(J. Virology, 1981: 39; 519-528)由来のVSV-G蛋白を用いることができるが、これに限定されない。このようにマイナス鎖RNAウイルスベクターは、他のウイルス由来のエンベロープ蛋白質を任意に組み合わせて製造されたものであってよい。アンフォトロピックエンベロープ蛋白質としては、例えばマウス白血病ウイルス(MuLV)4070A株由来のエンベロープ蛋白質を用い得る。また、MuMLV 10A1由来のエンベロープ蛋白質を用いることもできる(例えばpCL-10A1(Imgenex)(Naviaux, R. K. et al., J. Virol. 1996: 70; 5701-5705)。また、ヘルペスウイルス科の蛋白質としては、例えば単純ヘルペスウイルスのgB、gD、gH、gp85蛋白質、EBウイルスのgp350、gp220蛋白質などが挙げられる。ヘパドナウイルス科の蛋白質としては、B型肝炎ウイルスのS蛋白質などが挙げられる。これらの蛋白質は、細胞外ドメインをF蛋白質またはHN蛋白質の細胞内ドメインと結合させた融合蛋白質として用いてもよい。このように本発明において用いられるウイルスベクターには、VSV-G蛋白質などのように、ゲノムが由来するウイルス以外のウイルスに由来するエンベロープ蛋白質を含むシュードタイプウイルスベクターが含まれる。ウイルスのゲノムRNAにはこれらのエンベロープ蛋白質をゲノムにコードされないように設計すれば、ウイルス粒子が細胞に感染した後は、ウイルスベクターからこの蛋白質が発現されることはない。
【0044】
また、エンベロープ表面に特定の細胞に接着しうるような接着因子、リガンド、受容体等の蛋白質、抗体またはその断片、あるいはこれらの蛋白質を細胞外領域に有し、マイナス鎖RNAウイルスのエンベロープ蛋白質由来のポリペプチドを細胞内領域に有するキメラ蛋白質などを含むウイルスを製造することもできる。これにより、ウイルスベクターの感染の特異性を制御し得る。これらはウイルスゲノムにコードされていてもよいし、ウイルスの再構成時に、ウイルスゲノム以外の遺伝子(例えば別の発現ベクターまたは宿主染色体上などにある遺伝子)からの発現により供給されてもよい。
【0045】
エンベロープ構成蛋白質を発現するヘルパー細胞は、例えば欠損させたエンベロープ構成蛋白質あるいは別のエンベロープ蛋白質をコードする遺伝子をトランスフェクションすることにより作製することができる(WO00/70055 および WO00/70070;Hasan, M. K. et al., J. General Virology. 1997: 78; 2813-2820参照)。誘導発現を可能にするために、例えばCre/loxP誘導型発現プラスミドpCALNdlw(Arai, T. et al., J. Virology, 1998: 72; 1115-1121)等の組み換え酵素標的配列を持つベクターにエンベロープ蛋白質遺伝子を組み込むことが好ましい。細胞は、例えばサル腎臓由来細胞株LLC-MK2細胞(ATCC CCL-7)を用いることができる。LLC-MK2細胞は、10%の熱処理した非動化ウシ胎児血清(FBS)、ペニシリンGナトリウム 50単位/ml、およびストレプトマイシン 50 micro-g/mlを添加したMEMで37℃、5% CO2で培養する。SeV-F遺伝子産物は細胞傷害性を有するため、Cre DNAリコンビナーゼによりエンベロープ蛋白質遺伝子産物を誘導発現されるように設計された上記プラスミドpCALNdLw/Fを、リン酸カルシウム法(mammalian transfection kit (Stratagene))により、周知のプロトコールに従ってLLC-MK2細胞に遺伝子導入を行う。限界希釈により細胞をクローニングした後、細胞を拡大培養し、導入遺伝子の高発現細胞株の選別を行う。このためには、例えばアデノウイルスAxCANCreを斉藤らの方法(Saito et al., Nucl. Acids Res. 1995: 23; 3816-3821; Arai, T.et al., J. Virol. 1998: 72; 1115-1121)により例えば moi=3〜5 で感染させ、ウェスタンブロッティングまたはウイルス生産により、細胞を選択する。
【0046】
回収されたウイルスの力価は、例えばCIU(Cell Infecting Unit)測定または赤血球凝集活性(HA)の測定することにより決定することができる(WO00/70070; Kato, A. et al., Genes Cells. 1996: 1; 569-579; Yonemitsu, Y. & Kaneda, Y., Hemaggulutinating virus of Japan-liposome-mediated gene delivery to vascular cells. Ed. by Baker AH. Molecular Biology of Vascular Diseases. Method in Molecular Medicine: Humana Press. 1999: 295-306; Kiyotani, K. et al., Virology. 1990: 177; 65-74)。また、GFP(緑色蛍光蛋白質)などのマーカー遺伝子を搭載したベクターについては、マーカーを指標に直接的に感染細胞をカウントすることにより力価を定量することができる(例えばGFP-CIUとして)。このようにして測定した力価は、CIUと同等に扱うことができる(WO00/70070)。
【0047】
ウイルスが再構成する限り、再構成に用いる宿主細胞は特に制限されない。例えば、サル腎由来のLLC-MK2細胞およびCV-1細胞 (例えばATCC CCL-70)、ハムスター腎由来のBHK細胞 (例えばATCC CCL-10) などの培養細胞、ヒト由来細胞等を使うことができる。これらの細胞に適当なエンベロープ蛋白質を発現させることで、その蛋白質をエンベロープに有する感染性ウイルス粒子を得ることもできる。また、大量にウイルスベクターを得るために、上記の宿主から得られたウイルスベクターを発育鶏卵に感染させ、ベクターを増幅することができる。鶏卵を使ったウイルスベクターの製造方法は既に開発されている(中西ら編,(1993),「神経科学研究の先端技術プロトコールIII, 分子神経細胞生理学」, 厚生社, 大阪, pp.153-172)。具体的には、例えば、受精卵を培養器に入れ9〜12日間 37〜38℃で培養し、胚を成長させる。ウイルスベクターを尿膜腔へ接種し、数日間(例えば3日間)卵を培養してウイルスベクターを増殖させる。培養期間等の条件は、使用する組み換えセンダイウイルスにより変わり得る。その後、ウイルスを含んだ尿液を回収する。尿液からのセンダイウイルスベクターの分離・精製は常法に従って行うことができる(田代眞人,「ウイルス実験プロトコール」, 永井、石浜監修, メジカルビュー社, pp.68-73,(1995))。
【0048】
回収したウイルスベクターは実質的に純粋になるよう精製することができる。精製方法はフィルトレーション(濾過)、遠心分離、吸着、およびカラム精製等を含む公知の精製・分離方法またはその任意の組み合わせにより行うことができる。「実質的に純粋」とは、ウイルスベクターを含む溶液中で該ウイルスの成分が主要な割合を占めることを言う。例えば実質的に純粋なウイルスベクター組成物は、溶液中に含まれる全蛋白質(但しキャリアーや安定剤として加えた蛋白質は除く)のうち、ウイルスベクターの成分として含まれる蛋白質の割合が10% (重量/重量) 以上、好ましくは20%以上、より好ましくは50%以上、好ましくは70%以上、より好ましくは80%以上、さらに好ましくは90%以上を占めることにより確認することができる。例えばパラミクソウィルスベクターであれば、具体的な精製方法としては、セルロース硫酸エステルまたは架橋ポリサッカライド硫酸エステルを用いる方法(特公昭62-30752号公報、特公昭62-33879号公報、および特公昭62-30753号公報)、およびフコース硫酸含有多糖および/またはその分解物に吸着させる方法(WO97/32010)等を例示することができるが、これらに制限されない。
【0049】
上記のマイナス鎖RNAウイルスベクターを造血細胞に感染させ、造血細胞移植組成物を製造することができる。本発明は、マイナス鎖RNAウイルスベクターを用いた造血細胞移植組成物の製造方法、および造血細胞移植組成物の製造におけるマイナス鎖RNAウイルスベクターの使用に関する。
【0050】
造血細胞とは、白血球、赤血球、または血小板の少なくともいずれかの血液細胞に分化し得る細胞を言う。造血細胞には、骨髄細胞、造血前駆細胞、造血幹細胞(hematopoietic stem cell;HSC)などが含まれる。本発明において用いる造血細胞は、好ましくは白血球、赤血球、および血小板の少なくとも3系統に分化する能力を持つ細胞である。造血前駆細胞とは、血液細胞の少なくともいずれかの系統に分化できる細胞であり、血液細胞の各系統へ分化が進み、多分化能を消失した細胞(リンパ球系、顆粒球系、赤血球系、血小板系へ分化する細胞)も含まれる。造血幹細胞とは、自己複製能と多分化能を有する細胞で、少なくとも白血球、赤血球、および血小板の3系統に分化する能力を持つ細胞である。造血細胞は、体性細胞(神経系、肝臓、心臓など)へ分化することのできるものであってもよい(Weissman IL. et al., Science, 2000: 287; 1442-1446; Lagasse E. et al., Immunity, 2001: 14; 425-436; Anderson DJ. et al. Nat Med, 2001: 7; 393-395)。
【0051】
各血液細胞は、その形態や染色の特異性から同定することが可能である。染色法としては、ギムザ染色、ライト染色、メイ−ギムザ染色、メイ−グリュンワルド−ギムザ染色、ペルオキシダーゼ染色、エラスターゼ染色、PAS染色等を用いることができる(新生化学実験講座8, 血液(上), 1987, 8-15ページ参照)。
【0052】
造血幹細胞は、骨髄のみならず末梢血および臍帯血中にも存在する。本発明において造血細胞移植とは、造血細胞を含む細胞組成物を移植することを言い、骨髄移植(BMT)、末梢血幹細胞移植(PBSCT)、および臍帯血幹細胞移植(CBSCT)などが含まれる。すなわち本発明は、骨髄細胞移植のみならず、末梢血造血幹細胞移植および臍帯血移植においても適用することができる。HSCの同定は、顆粒球、単球の前駆細胞をコロニーアッセイ法で算定したり、HSCに特徴的と考えられるCD34(抗原)陽性細胞をフローサイトメーターで測定することにより実施することができる。具体的には、造血細胞としては、マウスであれば c-kit+、Thy1.1low、lin-、Sca-1+(Osawa M, Hanada K, Hamada H, Nakauchi H. Science. 1996; 273: 242-245.)、ヒトではCD34+、Thy+、Lin-、c-kitlow(Bhatia M, Wang JC, Kapp U, Bonnet D, Dick JE. Proc Natl Acad Sci U S A. 1997; 94:5320-5325)の表現型を有する細胞が挙げられる。lin- (lineage negative) の表現型は、適宜市販のキット等を用いて同定および選択することができるが、例えば GPA、CD3、CD2、CD56、CD24、CD19、CD14、CD16、および CD99b が全てnegativeのものである。
【0053】
ヒト造血幹細胞の分離方法は、以下の文献も参照することができる(Leary AG, Blood 69: 953, 1987; Sutherland HJ, Blood 74: 1563, 1989; Andrews RG, J Exp Med 169: 1721. 1989; Terstappen LWMM, Blood 77: 1218, 1991; Lansdorp PM, J Exp Med 175: 1501, 1992; Briddell RA, Blood 79: 3159, 1992; Gunji Y, Blood 80: 429, 1992; Craig W, J Exp Med 177: 1331, 1993; Gunji Y, Blood 82: 3283, 1993; Traycoff CN, Exp Hematol 22: 215, 1994; Huang S, Blood 83: 1515, 1994; DiGinsto D, Blood 84: 421, 1994; Murray L, Blood 85: 368, 1995; Hao QL, Blood 86: 3745, 1995; Laver JH, Exp Hematol 23: 1515, 1995; Berardi AC, Science 267: 104, 1995; Kawashima I, Blood 87: 4136, 1996; Leemhuis T, Exp Hematol 24: 1215, 1996; Civin CI, Blood 88: 4102, 1996; Larochelle A, Nature Med 2: 1329, 1996; Tajima S, J Exp Med 184: 1357, 1996; Sakabe H, Stem Cells 15: 73, 1997; Sakabe H, Leukemia 12: 728, 1998; 原田実根他編, 新しい造血幹細胞移植, 南江堂, 1998, 9-23ページ)。
【0054】
例えばマウスの骨髄細胞は、大腿骨、及び頸骨より骨髄細胞をフラッシュにて採取し、Lysing buffer(0.38% NH4Cl in Tris-HCl, pH7.65)にて赤血球除去後、抗Thy1.2抗体およびウサギ補体を用いた抗体補体反応による成熟T細胞の除去後に得られる(Tomita Y, Mayumi H, Eto M, Nomoto K. J Immunol. 1990; 144: 463-473.)。または、採取骨髄細胞を抗B220(B細胞)抗体、抗CD3(T細胞)抗体、抗Gr-1(顆粒球)抗体、抗Mac-1(マクロファージ)抗体、抗DX-5(NK細胞)抗体等の抗体でビーズあるいは遠心法にてlineageを除去して得られる(Sato T, Laver JH, Ogawa M. Blood. 1999; 94: 2548-2554.)。
【0055】
ヒト造血細胞を骨髄より採取する場合は、全身麻酔下、腹臥位で、腸骨の左右それぞれ3〜5ヶ所から、1回に数ml程度の骨髄液を採取する(末梢血混入を避けるため)。採取量は同種移植の場合では細胞数は3(2〜5)×108/kg(患者体重あたり)、自家移植の場合で1〜3×108/kg を目標として骨髄採取する。骨組織などの混入があるためメッシュを通した後,バッグにつめる。移植の際には粗いフィルターを用いて輸注する。同種移植の場合は、ヒト白血球抗原(human lymphocyte antigen;HLA)適合ドナーを選択することが好ましい。但し、GVHD予防法の確立、新しい免疫抑制剤の開発、およびCD34陽性細胞の純化技術などにより、HLA不一致のドナーでも移植は可能になってきている。ドナーとしては、HLA-A、-B、および-DRの3組6抗原のうち、4種類以上、より好ましくは5種類以上、より好ましくは全てがレシピエントと一致することが好ましい。ABOmajor不適合移植の場合、遠心分離により赤血球を除去したものを用いる。ABOminor不適合の場合、遠心分離にて血漿を除去したものを用いる。(森下ら編、『造血幹細胞移植マニュアル』第2版、小寺、斎藤監修、日本医学館、東京、(1999年)p260-277、参照 )。
【0056】
末梢血より末梢血幹細胞を採取する場合は、G-CSF 1 日400μg/m2(又は10μg/kg)を1 回、または2 回に分割し、5 日間または採取終了時まで連日皮下注射し、投与開始4日目から6日目までの期間に1から3 回、末梢血(静脈)から血球分離装置を用いて造血幹細胞を採取することによって得られる(日本造血細胞移植学会のウェブページにおける造血幹細胞移植に関するガイドラインの説明資料を(2003年4月21日改訂第3版)を参照)。末梢血幹細胞(PBSC)の動員については、Harada M et al., J. Hematother 5: 63-71, 1996、Waller CF et al., Bone Marrow Transplant 18: 279-283, 1996、Anderlini P et al., Blood 90: 903-908, 1997、原田実根他編, 新しい造血幹細胞移植, 南江堂, 1998, 67-72ページ、名古屋BMTグループ編, 造血細胞移植マニュアル改訂三版, 2004, 237-240ページも参照のこと。動員に用いるG-CSFとしては、野生型蛋白質の他、N末端を改変した誘導体(nartograstinなど)、糖鎖が付加された修飾蛋白質(lenograstinなど)を用いることができる。またG-CSFと他の造血因子を併用してもよい。例えば、GM-CSF、IL-3、またはSCFなどを併用することができる(Huhn RD et al., Exp Hematol 24: 839-847, 1996; Begley CG et al., Blood 90: 3378-3389, 1997; Lane TA et al., Blood 85: 275-282, 1995)。G-CSF投与中は、腰痛、骨痛および発熱などの全身症状の軽減や、血小板凝集能亢進の防止のため、aspirinを投与することができる。
【0057】
ヒト末梢血造血幹細胞(CD34+細胞)を得るには、上記で得られた末梢血幹細胞からCD34陽性細胞をCD34抗体磁気ビーズでの濃縮法Isolex system(Baxter社など)、CliniMACS(AmCell社)、アビジンカラムを通しての濃縮法(CEPRATE、Cellpro社)、CD34抗体をフラスコに固定化し、反応する細胞をフラスコに吸着させ、残りを洗い流す(CELLECTOR、AIS社)等の方法でCD34陽性細胞を分離採取することで得られる(森下ら編、『造血幹細胞移植マニュアル』第2版、小寺、斎藤監修、日本医学館、東京、(1999年)p260-277、参照 )。
【0058】
ヒト臍帯血細胞移植では、採取した臍帯血に、赤血球沈降剤血球を加え、重量によって各血液画分に分け、移植時には不要となる血漿画分、赤血球画分を除き、必要な造血幹細胞を得る。更にCD34陽性細胞をIsolex system(Baxter社)、CliniMACS(AmCell社)、アビジンカラムを通しての濃縮法(CEPRATE、Cellpro社)、CD34抗体をフラスコに固定化し、反応する細胞をフラスコに吸着させ、残りを洗い流す(CELLECTOR、AIS社)等の方法によって純化することで臍帯血CD34陽性造血幹細胞を得ることができる(森下ら編、『造血幹細胞移植マニュアル』第2版、小寺、斎藤監修、日本医学館、東京、(1999年)、p661-680、参照 )。
【0059】
単離した造血細胞は、例えばメチルセスロース法により、SCF、IL-3、GM-CSF、G-CSF、Epoを添加した培養することができる(Sonoda Y. et al., Blood 84: 4099-4106, 1994; Kimura T. et al., Blood 90: 4767-4778, 1997)。造血細胞は1×102から1×104/ml程度で添加し、例えば 37℃、5% CO2、5% O2 下で培養する。但し、培養条件は適宜調整してよい。
【0060】
自家移植においては、簡便性の点から末梢血幹細胞移植が選択されることが多いが、自家骨髄移植も適用される。その場合、通常は、化学療法の影響のない造血回復期に、骨髄の正常造血を確認後、全身麻酔下に腸骨および胸骨から採取する。目標細胞数は、骨髄有核細胞3×108から5×108/kg程度である。連続式血液成分分離装置で得られた細胞浮遊液は、顆粒球や赤血球の混入が非常に少ないため、単核球分離操作を省略することができる。顆粒球や赤血球が多く含まれる場合には、Ficoll比重遠心法により単核球を分離することができる。骨髄液の場合は、顆粒球や赤血球の除去と濃縮のため、Ficoll処理または血液分離装置による単核球分離を行うことができる。
【0061】
混入した血小板をそのまま輸注すると塞栓などの原因となる場合があるため、採取後に除去することが好ましい。例えばダブルバックに集めて弱遠心(200g、15分)するか、遠心管に分注して1600回転、10分遠心して血小板を除去し、RPMI1640培地に交換することができる。採取した細胞を凍結保存する場合は、10%自己血清、10%DMSOを含むRPMI1640培地に浮遊させて凍結させ、programmed freezerで凍結、液体窒素中で保存する。細胞濃度は2×107から6×107/mlとすることができる。比較的短期間の細胞保存のためには、細胞浮遊液(1×108/ml以下の濃度)に終濃度 6% Hydrodyethyl Starch (HES)、5% DMSO、4% albumin となるように、氷冷した等量の保存液を混合し、-80℃のdeep freezerで凍結保存することができる(Knudsen LM et al., J. Hematother. 5: 399-406, 1996)。
【0062】
安全な自家移植に必要なCD34陽性細胞数は、およそ2×106/kgである(Schots R et al., Bone Marrow Transplant. 17: 509-515, 1996; Zimmerman TM et al., Bone Marrow Transplant. 15: 439-444, 1995)。CD34陽性細胞数は、通常の2 color flowcytometryで計測することができる。具体的には、骨髄細胞や末梢血アフェレーシス細胞を溶血処理により赤血球を除去した後、forward scatterと抗CD45抗体で展開し、gateを決めて赤血球および血小板を除く。次に、このgateをside scatterと抗CD34抗体で展開し、好中球などの非特異反応部分を除いた分画中の細胞数の全細胞数に対する割合で算定する。この値と、予め血球計測機で測定した血球数から、全CD34陽性細胞数が算出できる。
凍結保存した細胞に含まれるviableな幹細胞の数は、コロニー形成法によりCFU-GMを数える方法で知ることができる。移植に必要なCFU-GMの基準は、一般的には 1×105〜2×105/kgである。
【0063】
上記のように調整した造血細胞をマイナス鎖RNAウイルスベクターと接触させることにより、該マイナス鎖RNAウイルスベクターを造血細胞に導入することができる。マイナス鎖RNAウイルスベクターと造血細胞との接触は、体内(in vivo)または体外(in vitroまたはex vivo) で行うことができ、例えば培養液、生理食塩水、血液、血漿、血清、体液など所望の生理的水溶液中で実施すればよい。
【0064】
ベクターと造血細胞との接触においては、MOI(多重感染度; 細胞1つあたりの感染ウイルス数)は1〜500の間にすることが好ましく、より好ましくは2〜300、さらに好ましくは3〜200、さらに好ましくは5〜100、さらに好ましくは7〜70である。ベクターと造血細胞との接触は短い時間でも十分であり、例えば1分以上、好ましくは3分以上、5分以上、10分以上、または20分以上接触させればよく、例えば1〜60分程度、より特定すれば5分〜30分程度であってよい。もちろん、それ以上の時間接触させてもよく、例えば数日間またはそれ以上接触させてもよい。
【0065】
ベクターを導入した造血細胞は、薬理学的に許容される所望の担体または媒体と組み合わせて造血細胞移植組成物とすることができる。「薬学的に許容される担体または媒体」とは、生細胞を懸濁できる溶液であれば制限はない。例えばリン酸緩衝生理食塩水(PBS)、塩化ナトリウム溶液、リンゲル溶液、培養液等が挙げられる
【0066】
また造血細胞移植組成物は、ベクターを導入していない造血細胞が含まれていてもよい。ベクターを導入した造血細胞と、ベクターを導入していない造血細胞を混合することにより、造血細胞の生着効率は上昇し得る。混合比率は、それに限定されないが、例えばベクター導入造血細胞:非導入造血細胞を1:10〜10:1の間で適宜調整してよい。
【0067】
また本発明は、マイナス鎖RNAウイルスベクターが導入された造血細胞から血液細胞を再構築させる方法に関する。この方法は、(a)マイナス鎖RNAウイルスベクターを造血細胞に接触させる工程、および(b)工程(a)の造血細胞を動物に注入する工程、を含む。マイナス鎖RNAウイルスベクターは細胞質ベクターであり、造血細胞の染色体にインテグレートすることはないため、レトロウイルスのような染色体損傷のリスクがない。しかも、本発明において、マイナス鎖RNAウイルスは造血細胞の細胞質中で複製し、造血細胞が分裂しても娘細胞にベクターが分配され、再構築された血液細胞中でも導入遺伝子の発現が持続できることが証明された。
【0068】
血液細胞とは、血液中の細胞成分を言い、具体的には白血球、赤血球、および血小板が含まれる。血液細胞を生成させるとは、これらの血液細胞のいずれか、好ましくは白血球、赤血球、および血小板の全てを生成させることである。また白血球は、顆粒球、単球、リンパ球が含まれ、本発明において白血球とは、それらのいずれかの細胞を意味する。本発明の方法により、マイナス鎖RNAウイルスベクターが導入された造血細胞は3系統への分化が可能である。すなわち造血細胞移植により、マイナス鎖RNAウイルスベクターが導入された造血細胞から白血球、赤血球、および血小板が生成する。
【0069】
造血細胞の移植は、適宜周知の移植プロトコルに従って実施することができる。造血細胞移植の対象となる動物に特に制限はないが、例えば所望の哺乳動物が挙げられ、例えばマウス、ラット、イヌ、ブタ、ネコ、ウシ、ウサギ、ヒツジ、ヤギ、サルなどの非ヒト哺乳動物およびヒトが含まれる。例えばマウスにおいては、投与細胞数は1匹あたり、5.0 x 106 から2.0 x 107、好ましくは約1.0x107、好ましくは約2.0x107を尾静脈、陰茎静脈、または眼底静脈よりone shot静脈注射にて投与することができる(Tomita Y, Mayumi H, Eto M, Nomoto K. J Immunol. 1990; 144: 463-473.)。ヒトの骨髄細胞移植では、個体あたり、2 x 108/kgから 3 x 108/kg、好ましくは約3 x 108/kgを経静脈的に点滴投与する。ヒトの末梢血造血幹細胞移植では、個体あたり自家造血幹細胞移植では2 x 106/kg以上、同種移植では5 x 106/kg以上、好ましくは、自家造血幹細胞移植では約5 x 106/kg、同種移植では約10 x 106/kgを経静脈的に点滴投与する。ヒトの臍帯血造血幹細胞移植では、個体あたり、移植必要数は2 x 107/kg以上、好ましくは約4 x 107/kgを経静脈的に点滴投与する。(森下ら編、『造血幹細胞移植マニュアル』第2版、小寺、斎藤監修、日本医学館、東京、(1999年)p260-277、p661-680、参照 )。
【0070】
輸液への血小板の混入による血栓や塞栓を防止するため、抗ヒスタミン剤(hydroxyzine 25mg または chlorphenirmine 5mgなど)または副腎皮質ステロイド(hydrocortisone 100mg)などの前投与を行うことができる。移植に凍結細胞を用いる場合、移植細胞溶液中の赤血球の溶血が問題となる場合には、ハプトグロビン製剤4,000単位の点滴などで尿細管障害を予防することができる。末梢血幹細胞移植の場合は、2日間に分けて輸注することも好ましい。
【0071】
ドナー造血細胞の生着を促進するため、造血細胞の注入前に、レシピエントを免疫抑制しておくことが好ましい。例えば白血病における骨髄移植では、白血病細胞の死滅と宿主の免疫担当細胞の不活化のため、造血細胞の移植前に全身照射(total body irradiation; TBI)を行うことができる。このような処置は、一般に、前処理(Preparative regimen)またはコンディショニング(Conditioning)とも呼ばれる。TBIは急性・慢性のいずれの白血病でも対象となる。このとき、メルファラン、シクロホスファミド(CPA)などの抗癌剤と併用することができる。しかしTBIはさまざまな障害の原因にもなるため、これらを避けるためにブスルファンとCPAのBUCY法など抗癌剤だけの前処置を選択することもできる(Inoue, T. et al., Strahlenther Onkol 169: 250-255, 1993; Blaise, D. et al., Blood. 1992: 79; 2578-2582; Blume, KG. et al., Blood. 1993: 81; 2187-2193; Hartman, A. et al., Bone Marrow Transplant. 1998: 22; 439-443)。また、骨髄移植と同時にDonor Lymphocyte Infusion(DLI)を行うことで、前処置の線量を軽減することが可能である。
【0072】
放射線照射を行う場合は、長SAD(Source axis distance)(またはSSD (Source skin distance)法、スイープビーム法、ビーム移動法、治療寝台移動法等の照射方法を適用することができる。このうち、長SAD(SSD)法は、特殊な装置を必要としない点で簡便であり、良く用いられている。照射は、1回照射または分割照射であってよいが、分割照射の方が正常細胞の回復が期待できる。1回照射は、例えばヒトであれば一回あたり10Gy(6cGy/min)、分割照射であれば、12Gy(6cGy/min)を6回/6日、1.8Gy/回を2〜3回/日(総線量15Gy程度)、または2〜3Gy/回で1〜2回/日(総線量12Gy程度)などが例示できるが、線量は適宜調整されてよい。
【0073】
また、前処置を行わなくても、ドナー造血細胞を多数注入すれば、造血細胞が骨髄に生着することが動物実験により証明されている。これは、ドナー造血細胞の生着には必ずしも骨髄破壊的前処置は必要がないことを示している。これを基に、宿主の免疫抑制のみを実施する骨髄非破壊的前処置を用いる造血幹細胞移植、いわゆるミニ移植(Mini-transplant: ミニトランスプラント)が開発されており、これに本発明を適用することも可能である。ミニ移植は骨髄破壊的前処置を伴う移植とは異なり合併症も軽いため、高齢者や臓器障害のある患者にまで適応が拡大される。本発明において免疫抑制には、TBIなどの骨髄破壊的処置のみならず、免疫抑制剤などによる骨髄非破壊的処置が含まれる。前処置に用いる免疫抑制剤としては、フルダラビン、クラドリビンなどの抗癌剤や抗胸腺グロブリンが用いられる。このような前処置後に造血幹細胞移植を行うことで、患者とドナーの造血細胞が混在する骨髄の混合キメラ、もしくはすべてドナーの造血細胞に入れ替わった骨髄となる完全キメラを作り出すことが可能である。再生不良性貧血のような非悪性疾患では、骨髄非破壊的前処置による混合キメラを適用できるが、白血病などの腫瘍では大量のリンパ球を注入して完全キメラ化する。骨髄破壊を伴わない造血細胞移植が有効と考えられる疾患としては、例えば腎癌、悪性黒色腫、低悪性度リンパ腫、再生不良性貧血などがある。再発リスクの高い腫瘍に対しては骨髄破壊の方が好ましい。ミニ移植の前処置で照射を行う場合は、例えば1〜2Gy/1回程度の小線量TBIが適用される。また、従来の前処置とミニ移植との中間的な前処置である、reduced conditioning療法も開発されている。これらの前処理を本発明において用いることもできる。
【0074】
移植において抗癌剤の大量投与や全身照射を行った場合は、全身の造血が抑えられ、感染に対する抵抗力が著しく低下する。従って、移植期間中は無菌室で過ごし、外界からの感染を予防することが好ましい。但し、HSCのソースとして末梢血を用いる同種末梢血幹細胞移植(allogeneic peripheral blood stem cell transplantation;allo-PBSCT)を行うことで早期の造血能回復が得られるため、完全無菌室管理を行わなくとも移植は可能になっている。移植が成功すれば、約20日程度で白血球数は増加する。一般に、末梢血幹細胞移植のほうが骨髄移植より回復が早い。血球の増加が少ない場合は、さらに造血幹細胞移植を実施することができる。
【0075】
自家移植の場合は、移植した造血幹細胞の数が十分あれば生着に問題はないが、同種移植の場合、ドナーとレシピエントが遺伝学的に同一でない限り、移植片が持つ抗原に対しレシピエントのT細胞が反応し生着不全を起こす。そのため、移植前の前治療には、ドナーのHSCが生着するための免疫抑制効果が必要となる。また逆にHSCが生着してドナー由来の造血能が回復すると、ドナー由来のT細胞がレシピエントの細胞を攻撃し移植片対宿主病(GVHD)が発症する。GVHDは同種移植後の生命予後を決定する重要な因子となるため、発症予防のために免疫抑制療法が行われる。急性GVHDは通常同種移植後2〜6週間以内に発症する。また、サイトメガロウィルスなどの感染症や免疫抑制剤によるmicroangiopathyも併発することもある。診断には、皮膚や胃、十二指腸、大腸、直腸などからの生検が有用である。中等症以上の場合は、副腎皮質ステロイドによる治療を行う。ステロイドに反応しない場合は、抗胸腺細胞グロブリンやタクロリムスなどの免疫抑制剤を用いる。血縁者間の移植では、シクロスポリンとメトトレキサート、非血縁者間ではタクロリムスとメトトレキサートが予防薬として用いられることが多い。慢性GVHDは、典型的には移植後100日前後に、同種移植を受けた長期生存者中に一定の割合で発症する。臨床所見としては、強皮症、扁平苔癬、Sjogren症候群、原発性胆汁性肝硬変などの自己免疫疾患によく似ており、口唇や皮膚の生検、肝生検などによって診断が可能である。治療としてはシクロスポリンと副腎皮質ステロイドが主体で、皮膚病変に対してはPUVA(Psoralens with Ultraviolet A)などの光線療法を実施することもできる。
【0076】
本発明における造血細胞移植は、例えば再生不良性貧血、白血病(急性骨髄性白血病および急性リンパ性白血病などの急性白血病、並びに慢性骨髄性白血病など)、骨髄異形成症候群、リンパ腫(ホジキン病、非ホジキンリンパ腫などを含む)、骨髄腫などの血液疾患、化学療法が有効な悪性腫瘍(乳がん、胚細胞腫などを含む)、先天性疾患などの治療として実施することができる。適応疾患については、単一遺伝子欠損あるいは発現系異常(プロモーターの欠損、あるいは制御異常によるものなど)、または2つ以上の遺伝子欠損あるいは発現系異常による疾患においても適応可能である。また、血球系の異常のみならず、骨髄幹細胞より分化可能とされる神経系細胞、肝細胞などの体細胞の遺伝子異常を要因とする疾患についても適応となる。また、遺伝子異常疾患のみならず、分泌蛋白などの供給細胞として用いた場合に治療効果が期待できる疾患にも応用可能である。例えば、単一遺伝子欠損症としては、X-linked SCID、ADA欠損症、血友病、無免疫グロブリン血症、高IgM血症、慢性肉芽腫症など、複数遺伝子欠損症に関しては、発作性夜間血色素尿症などが挙げられ、それぞれの原因遺伝子を造血幹細胞に遺伝子導入後、細胞移植を行うことで治療効果が期待できる。造血細胞移植における遺伝子治療が有効と考えられる疾患およびその遺伝子を例示すれば、例えばHurler病、Scheie病、およびHurler-Scheie病(α-L-iduronidaseまたはiduronate sulfatase)、Sanfilippo病(N-sulfatase、N-acetyl-α-D-glucosaminidase、α-D-flucosaminidase-N-acetyltransferase)、Morquio病(galactosamine-β-sulfate sulfataseまたはβ-galactosidase)、Maroteaux-Lamy症候群(N-acetylgalactosamine-4-sulphatase)、Sly病(β-glucuronidase)、Gaucher病(β-glucocerebrosidase)、Farber病(acid ceramidase)、Niemann-Pick病(acid sphingomyekinase)、Krabbe病(galactocerebroside β-galactosidase)、Metachromatic leucodystrophy(arylsufatase A)、Fabry病(α-galactosidase A)、SCID(adenosine deaminase)、X-linked SCID(gamma-c receptor)、X連鎖無ガンマグロブリン血症(X-linked agammaglobulinemia;XLA)(Bruton's tyrosine kinase)、JAK-3欠損症(Jak-3)、Aspartyglycoaminuria(aspartyglycosaminidase)、Fucosidoses(α-L-fucosidase)、Guibaud-Vainsel症候群(carbonic anhydrase II)、Thallassemias(α-、β-globin)、G6PD欠損症(glucose-6-phosphate dehydrogenase)、PK欠損症(pyruvate kinase L型)、Erythropoietic porphyia(uroporphyrinogen III synthase)などが挙げられる。また、プリンヌクレオシドフォスフォリラーゼ欠損症については原因遺伝子であるpurine nucleoside phosphorylase (PNP)、X連鎖重症複合免疫不全症については IL-2レセプターγ鎖 (γC)、X染色体連鎖性慢性肉芽腫症については NADPHオキシダーゼを構成するサブユニットの1つであるgp91phoxなどが例示できる。また、固形悪性腫瘍や血液悪性腫瘍の造血幹細胞移植の際に、腫瘍特異的な分子を標的とするキメラ毒素の遺伝子導入などによって、抗腫瘍効果の増強などを惹起することが可能と考えられる。また、癌の化学療法の副作用の軽減のために、多剤耐性遺伝子 MDR-1 を導入することも考えられる。さらに、後天性免疫不全症候群(AIDS)の治療のために抗HIV遺伝子(例えばRRE decoy、envアンチセンス)の遺伝子治療が可能である。
【実施例】
【0077】
以下、実施例により本発明をさらに詳細に説明するが、本発明はこれら実施例に制限されるものではない。なお、本明細書中に引用された文献は、すべて本明細書の一部として組み込まれる。
【0078】
使用動物
7週齢雌性C57BL/6Jマウス及び同Balb/cマウス(KBTオリエンタル)、GFPトランスジェニックマウスC57BL/6-TgN(act-EGFP)OsbC14-Y01-FM131(Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y. FEBS Lett. 1997; 407:313-319)をドナーとして用い、同C57BL/6、Balb/c nu/nuをレシピエントとして使用した。
【0079】
ベクター(SeV18+/dF-GFP、SeV18+/dFMtsHNtsPLmut-GFP)の構築
SeV18+/dF-GFP(以下SeV/dF-GFP)のベクターテンプレートとなるプラスミドpSeV18+/dF-GFPは、センダイウイルスZ株の全長cDNAを挿入、リーダー配列と NP 遺伝子間に挿入されたスペーサー配列に人為的に構築したNotI切断部位に供与核酸であるGFP cDNAを挿入し、さらにF 遺伝子領域を削除することにより作成した。SeV18+/dFMtsHNtsPLmut-GFP(以下tsSeV/dF-GFP)については、同様のプラスミドにさらにM遺伝子(G69E, T116A, A183S)、 HN遺伝子(A262T, G264R, K461G)、および P, L遺伝子(P遺伝子: L511F, L遺伝子: N1197S, K1795E)に対し変異を挿入した(Inoue M, et al. J Virol 2003;77:3238-3246; Inoue M, et al. Mol. Ther. 2003; 7(5):S37)。
双方のベクター構築にはマイナス鎖RNAウイルスをcDNAより作製させるためのリバースジェネティクス法を用いた(Inoue M, et. al. J Virol 2003; 77:3238-3246)。具体的には pSeV/dF-GFPあるいはpSeV18+/dFMtsHNtsPLmut-GFPと、N、P、LおよびF蛋白質を発現するプラスミドを同時にサル腎臓由来 LLC-MK2 細胞にトランスフェクションすることにより作成した。F遺伝子産物はベクターの再構成に必要であるため、F蛋白質を継続的に発現しているLLC-MK2細胞を使ってトランスに供給することにより増幅させた。
【0080】
造血細胞への遺伝子導入とマウスへの移植
以下の実験は、全て無菌操作にて施行した。
ドナーマウスの大腿骨より採取した骨髄細胞は、10%FBS + RPMI1640(Gibco BRL)中に23ゲージ針付き注射器でフラッシュした。その後、赤血球をLysing buffer(0.38% NH4Cl in Tris-HCl, pH7.65)にて除去、さらに成熟T細胞を完全に取り除くために抗Thy1.2抗体(マウス腹水IgMモノクローナル抗体、明治乳業株式会社ヘルスサイエンス研究所、東京)とウサギ補体(CL3051, CEDARLANE, Ontario CANADA)を骨髄細胞に反応させた。
T細胞除去骨髄細胞を1 x 107/mlに調節後、重複感染度(multiplicity of infections: MOI)=10でベクターに感染させ、37℃恒温槽で約1時間インキュベートした。その後一部をプレートに播き48時間後フローサイトメトリー(FACSCaliber, Becton Dickinson, CA)にてGFP遺伝子の発現を確認した。採取された骨髄細胞は3回洗浄、遠心後、serum freeのRPMIで希釈調節した。それぞれレシピエントマウス:C57BL/6は 2 x 107/200μl、nu/nu-BALB/Cは 4 x 107/200μlそれぞれ尾静脈から30ゲージ注射器で注入した。このレシピエントマウスは移植4〜6時間前にγ線をそれぞれC57BL/6は致死量の10 Gry、nu/nu-BALB/Cは亜致死量の7 Gry照射した。なお、細胞性免疫抑制のためのFK506投与群は骨髄移植2週間後からFK506 (tacrolimus, 藤沢薬品工業株式会社, 大阪) を5 mg/kg 連日腹腔内投与した。また、no-spleenマウスは移植2日前に開腹し脾臓を除去した。
【0081】
フローサイトメトリー
骨髄移植から1,4,8,12週間後の末梢血を尾静脈から採血し、GFP陽性細胞についてFACSにて解析した。まず、末梢血は赤血球をLysing bufferで除去し、2回HANKS液で洗浄の後約1x105〜1x106/mlに調節した。解析前に、PI (propidium iodide) 染色を行い、死細胞を除外した。また、それぞれのマウスの脾臓、骨髄、胸腺でのGFP陽性細胞分画の確認のため、Fcブロックの後、樹状細胞(DC)と単球/マクロファージ(MФ)の判定のために、anti-CD11bPE (1:4, Biosciences Pharmingen CA)とanti-CD11cAPC (1:5, Biosciences Pharmingen CA)、B細胞(B-cell)判定にはanti-B220APC (1:13, Becton Dickinson CA)とanti-IgMPE (1:10, Becton Dickinson CA)、helper T-cell(hT-cell)とcytotoxic T-cell(cT-cell)の判定のためanti-CD3APC (1:5, Biosciences Pharmingen CA)、anti-CD8PE (1:10,Becton Dickinson CA)もしくはanti-CD4PE (1:10, Becton Dickinson CA)、そしてナチュラルキラー(NK)細胞、NKT細胞と、T細胞(T-cell)の判定のためanti-CD3APCとanti-DX5PE (1:5, Biosciences Pharmingen CA) 抗体を用いて30分4℃下で染色し,FACS Caliber(Becton Dickinson CA) にて解析を行った。データはFlowJo(TreeStar Inc., San Carlos, CA)もしくはCellQuestTM software(Becton Dickinson CA)を用いて評価した。
【0082】
血清中抗SeV抗体価の測定
骨髄移植から1, 4, 8, 12週間後、マウスの尾静脈から末梢血を採血管で採取し、常温で30分間静置後、10,000rpmで5分間遠心分離した。その上清をストックし-80℃で保管した。検査時に室温で解凍し、血清中の抗SeV抗体価をMONILISA (R) HVJ Enzyme-linked Immunosorbent Assay kit (Wakamoto Pharmaceutical Co. Ltd., 東京) を用いて測定した。
【0083】
血液検査
骨髄移植から1, 4, 8, 12週間後、マウスの尾静脈から末梢血を採血管で採取し、末梢血20μlに対して、血球計算用希釈液50μlで希釈し、ジダ血モードで全自動血球計数器(CelltacαMEK-6158, 日本光電, 東京)を用いて検査した。
【0084】
CTL活性の測定
レシピエントマウスのspleenをスライドガラスにてすりつぶし、lysing後10%FCS入りRPMI + ペニシリン + DM添加培地にて5 x 106/ml希釈し、SeVペプチド 1 mMと混合した後、24穴プレートに播種、5日間培養した。培養3日目では培地交換とともにIL-2を培地1mlにつき3μl(30U/ml 以上)添加し、IL-2と48時間共培養した。
標的細胞(EL4: American Type Culture Collections)は1 x 106/200μlに調整し、SeVペプチドを50μM加えた。さらに、0.1mCiのNa251CrO3を細胞に加え、よく振った後に10%CO2条件下で、15分ごとに振りながら1.5時間インキュベーションした。
5日間培養した細胞を回収し3穴ずつ1 x 106、5 x 105、2.5 x 105、1.25 x 105でU底96穴プレートに播種した。これに標的細胞を100μlずついれ、4時間インキュベーションし、2回洗い後それぞれをγカウンターにかけ、以下の計算式にてSeV特異的Na251CrO3リリースを算出した。
%specific release = ((experimental cpm - spontaneous cpm)/(maximum cpm - spontaneous cpm)) × 100
最大遊離量は1% triton X-100 (Wako Pure Chemical Industries Ltd., Osaka, Japan)を使用した細胞溶解によって得た。自然遊離量は細胞を0.2mlの培養液のみで培養することで得た。自然遊離量は最大遊離量の15% から20% の範囲であった。それぞれの個体は3検体ずつ作成した。
SeVペプチドとしては、既に報告されているCTLの標的配列となり、クラスI抗原に提示されることが明らかとなっているNP蛋白のアミノ酸配列NH2-FAPGNYPAL-COOH(配列番号:11)を合成して用いた。なお、tsSeV/dF-GFPのNP蛋白には、同部位の変異は存在しない。
【0085】
統計解析
統計分析値はすべて平均値±標準誤差として表現した。データはMann Whitney U-testを用いて解析し、P<0.05を統計学的有意差とした。
【0086】
〔実施例1〕マウス造血細胞に対するSeVの遺伝子導入効率と同細胞の生着効率
第1に、C57BL6マウス骨髄細胞(T細胞除去後)に対するSeV/dF-GFPならびにtsSeV/dF-GFPによる遺伝子導入効率を検討した(図1a)。MOI=10にて各々のベクター液を骨髄細胞培養液へ添加、1時間後に新鮮培養液にて洗浄後、48時間後のGFP陽性率をFACSにて解析した。図1に典型的解析図を示す。SeV/dF-GFPによるGFP陽性率は80.7±2.9%(n=3)、tsSeV/dF-GFPによるそれは88.8±1.9%(n=18)であり、いずれも80%以上の高い遺伝子導入効率を示した。
次にSeVにてGFP遺伝子を導入した骨髄細胞の放射線照射同種マウスに対する生着効率を、末梢血細胞におけるGFP陽性率を指標として検討した(図1b)。陽性対照であるGFPトランスジェニックマウス(GFP-TG)をドナーとした場合、生着率は経過中常に98%以上であり、この効率は移植後4週目〜8週目も維持されていた(n=3)。SeV/dF-GFPにて処置した骨髄細胞を移植した場合、移植早期(1週目)より末梢血におけるGFP陽性率は陽性対照に比べ低く(0.1-2.3%, n=9)、以後GFP陽性細胞は消失した。一方でtsSeV/dF-GFPで処理した骨髄細胞を移植した場合、移植早期には比較的高いGFP陽性率を示した(45-85%, n=10)。その後GFP陽性率は次第に漸減し、移植後4週目では、個体差はあるものの(2.0-26%)、依然平均で10%以上の末梢血細胞がGFP陽性であった。
【0087】
〔実施例2〕tsSeV/dF-GFPにて遺伝子導入された造血細胞のレシピエントにおける生体内分布ならびに各細胞分画におけるGFP陽性率
図1において比較的生着効率のよいマウス6頭を用いて、GFP発現細胞が造血系のいずれに分布するかを検討した(図2)。この6頭の末梢血における5週目のGFP陽性率は55.9±5.9%であり、脾臓、胸腺で比較的高かったが、下肢大腿骨骨髄では17.1±8.8%であった。
次に脾臓と骨髄において各細胞分画におけるGFP陽性細胞率をFACSにて解析した(図3)。各細胞分画の同定には、NK cells=CD3-/DX5+、NKT cells= CD3+/DX5+、whole T-cells= CD3+/DX5-、helper T-cells= CD3+/CD4+、cytotoxic T-cells= CD3+/CD8+、dendritic cells (DCs)= CD11b+/CD11c+、monocytes/macrophages(MΦ)= CD11b+/CD11c-、B-cells= B220+/IgM+を用いた。脾臓・骨髄共にNK、DC、MΦ、Bは比較的陽性率が高かった。NKT、Tについては陽性率は比較的低いものの、移植細胞のT細胞分画はほぼ完全に除去されていることより、レシピエントの脾臓・骨髄で検出されたGFP陽性NKT/T細胞は、移入された分化したNKT/T細胞ではなく、むしろ造血幹細胞由来のものと考えられた。
【0088】
〔実施例3〕tsSeV/dF-GFPにて遺伝子導入された造血細胞の5週目以降のレシピエントにおける生着動態
以上の結果から、tsSeV/dF-GFPによって遺伝子導入された骨髄細胞は、同種レシピエントにおいてrepopulation可能であることが示された。しかしGFP陽性率は次第に減少することが明らかになった。このGFP陽性細胞数の減少をより詳細に検討するため、より長期の生着動態について検討した。
計53頭のC57BL6マウスへtsSeV/dF-GFPにて処置した骨髄細胞を同様の方法で移植したところ、約40%のマウスが移植後5〜7週で死亡した(図4a)。これらのマウスについて、移植後1週目及び4週目で末梢血を採取しGFP陽性細胞を確認したが、死亡したマウスの4週目の採血において、全例が比較的良好なGFP陽性率(10%以上)に加え血液の希釈が観察された。そこで移植後4週目における生着率のよいマウスでの血球数を検討した。
図4bに示すように、無処置マウス(n=6)、及びGFP-TGマウス骨髄を移植されたレシピエントマウス(n=6)と比較して、tsSeV/dF-GFPにて処置した骨髄細胞を移植されたレシピエントマウス(全6頭中4週目の末梢血中GFP陽性細胞10%以上:n=3)では移植後4週目における白血球数(WBC)、赤血球数(RBC)、血小板(PLT)全てが著明に減少していた。
【0089】
〔実施例4〕tsSeV/dF-GFPにて遺伝子導入された移植造血細胞に対するレシピエントにおける免疫学的反応の検討
tsSeV/dF-GFP遺伝子導入骨髄を移植されたレシピエントマウスにおける汎血球減少の原因を検討するために、同レシピエントマウスにおけるSeVに対する免疫反応について検索を行った。
4-1.細胞性免疫反応
tsSeV/dF-GFP遺伝子導入骨髄を移植されたレシピエントマウスにおける4週目の末梢血GFP陽性細胞率と、同個体におけるセンダイウイルス感染細胞に対する特異的細胞障害性T 細胞活性(CTL活性)を検討した。
図5aに示すごとく、末梢血GFP陽性細胞率が高い個体(64.9%)では有意なCTL活性を認めるものの、そのレベルは低値に留まっていた。
図5aで得られたCTL活性の軽度の上昇がtsSeV/dF-GFP由来GFP陽性細胞数の減少に有意に関与するか否かを検討するため、主に細胞性免疫を抑制するFK506の連日投与(移植後2週目より、5 mg/kg/day)、及びヌードマウスにおける細胞生着動態を検討した。図5bに示すごとく、双方において細胞生着率の上昇は認められなかったことから、tsSeV/dF-GFP遺伝子導入骨髄を移植されたレシピエントマウスにおける遺伝子導入細胞の排除へのCTLの関与は、比較的低いものと考えられた。
【0090】
4-2.液性免疫反応
次に、tsSeV/dF-GFP遺伝子導入骨髄を移植されたレシピエントマウス(n=34)における液性免疫反応について、4週目の末梢血GFP陽性細胞率と抗SeV抗体の関係について検討した。
図6aに示すごとく、34頭中11頭は5週目にて死亡し、23頭は長期生存した。これらから移植後4週目に採取した末梢血GFP陽性細胞数と同時期の抗SeV抗体価を検討すると、GFP陽性率が高い個体では抗SeV抗体価が低く、またGFP陽性率が低い個体では抗SeV抗体価が高い傾向を認めた(図6aおよび6b)。本発明で使用した抗SeV抗体検出用ELISAシステムはSeVの膜タンパクを認識するものであるため、tsSeV/dF-GFPが感染した骨髄細胞に発現したわずかな膜タンパクに対し、レシピエントが抗体を産生したものと考えられた。
【0091】
〔実施例5〕遺伝子導入ならびに非導入細胞混合造血細胞の移植後動態に関する検討
最後に、tsSeV/dF-GFP遺伝子導入骨髄細胞のレシピエント骨髄中あるいは脾臓の血球細胞の中で、少なくとも一部に除去したはずであるドナー由来のGFP陽性T細胞が存在することから、これらGFP陽性T細胞はtsSeV/dF-GFPにより遺伝子されたドナー細胞中の造血幹細胞に由来する可能性があると考えられる。一方でtsSeV/dF-GFP遺伝子導入骨髄細胞のレシピエントには、比較的高いrepopulationのまま死亡する群と、4週以降速やかに排除されていく群が存在することから、前者は造血幹細胞の多くへ遺伝子導入され、後者では遺伝子導入効率が低いのではないか、と仮説を立てた。
これを検討するため、tsSeV/dF-GFPによる遺伝子導入骨髄細胞と非導入骨髄細胞を等量混合し、レシピエントマウスへ移植した(図7)。本実験では遺伝子導入のみの細胞を移植された群では全てのレシピエントマウスが8週目までに死亡したが、混合移植群では5頭全例においてGFP陽性末梢血細胞率が4週以降に速やかに低下して行った。この結果は上記の仮説に矛盾しない結果であると考えられた。
【産業上の利用可能性】
【0092】
本発明により、染色体非傷害型の細胞質型ベクターを用いた遺伝子導入造血細胞による造血細胞移植が可能になった。本発明の方法は、宿主染色体内へのインテグレーションを介さないため、癌抑制遺伝子の不活化や癌遺伝子の活性化などの危険を伴わずに遺伝子を導入することができるため、安全なベクターシステムを構築することが可能である。先天性免疫不全性疾患には造血細胞移植が有効性を示さない症例が存在するため、これらの治療には遺伝子治療のみが残されたオプションとなる。本発明の方法を、これらの難治性疾患に対する新たな遺伝子治療へ適用することが期待される。
【図面の簡単な説明】
【0093】
【図1】a. フローサイトメトリーによるT細胞除去ドナー(C57BL6マウス)骨髄細胞へのSeV/dF-GFP及びtsSeV/dF-GFPによる遺伝子導入効率解析。代表的プロファイルを示す図である。SeV/dF-GFPの遺伝子導入効率は80.7±2.9%(n=3)、tsSeV/dF-GFPの場合は88.8±1.9%(n=18)であった。b. C57BL6系統GFPトランスジェニックマウス(GFP-TG)より採取した骨髄、あるいはSeV/dF-GFPもしくはtsSeV/dF-GFPで遺伝子導入したC57BL6マウス骨髄を同種レシピエントへ移植した場合の、末梢血血球細胞におけるGFP陽性率の経時的変化を示す図である。縦軸は対数表示である。
【図2】フローサイトメトリーによる、tsSeV/dF-GFPで遺伝子導入したC57BL6マウス骨髄を同種レシピエントへ移植した場合の、各種臓器におけるGFP陽性率(移植後5週、計6頭)を示す図である。図1で解析した個体のうち、4週目の末梢血にて生着率のよいものを選択して解析した。代表的プロファイルを示す。括弧内の数字は、平均値±標準誤差%(n=6)を示す。
【図3】フローサイトメトリーによる、tsSeV/dF-GFPで遺伝子導入したC57BL6マウス骨髄を同種レシピエントへ移植した場合の、脾臓・骨髄における各血球細胞分画におけるGFP陽性率(移植後5週、計6頭)を示す図である。
【図4】a. tsSeV/dF-GFPで遺伝子導入したC57BL6マウス骨髄を同種レシピエントへ移植した場合の、生存率の経時的変化(計53頭)を示す図である。b. 非処理C57BL6マウス、tsSeV/dF-GFPで遺伝子導入したあるいはGFPトランスジェニックマウス骨髄を同種レシピエントへ移植した場合の、末梢血における白血球数(WBC)、赤血球数(RBC)、血小板数(PLT)(移植後4週)を示す図である。tsSeV/dF-GFPのみに著明な汎血球減少を認める。
【図5】a. tsSeV/dF-GFPで遺伝子導入したC57BL6マウス骨髄を同種レシピエントへ移植した場合の、SeV特異的CTL活性(移植後4週)を示す図である。括弧内の数字は、各個体における同時期の末梢血中GFP陽性細胞率を示す。各個体につき、3つ培養ウェルに標的細胞を播種、各データは3ウェルの平均値、エラーバーは標準誤差を示す。b. tsSeV/dF-GFPで遺伝子導入したC57BL6マウス骨髄を同種レシピエントへ移植した場合(n=10)、また同様のマウスに移植後2週目よりFK506 (5 mg/kg/day)を腹腔内連日投与(n=7)、そしてtsSeV/dF-GFPで遺伝子導入したbalb/cマウス骨髄を同種ヌードマウスへ移植した場合の末梢血GFP陽性細胞の経時的変化を示す図である。いずれの操作においても、GFP陽性率の低下を阻止できなかった。
【図6】a. tsSeV/dF-GFPで遺伝子導入したC57BL6マウス骨髄を同種レシピエントへ移植した場合(総個体数n=34)の、5週以内死亡群(n=11、白三角)及び長期生存群(n=23、黒四角)における末梢血GFP陽性細胞率(縦軸)と血清中抗SeV抗体価(横軸)の相関(移植後4週)を示す図である。死亡個体群では、末梢血GFP陽性率が高く、さらに抗SeV抗体価が低いことに注意。b. 上記a.に示したデータを2群(死亡群、生存群)に分けてそれぞれGFP陽性率(左)、抗SeV抗体価(右)を比較したバーグラフである。エラーバーは標準誤差を示す。
【図7】tsSeV/dF-GFPで遺伝子導入したC57BL6マウス骨髄を、単独で同種レシピエントへ移植した場合(ts-BMT, n=3)及び同骨髄を同量の無処置骨髄と混合して移植した場合(ts-Mix-BMT, n=5)における、末梢血GFP陽性細胞率の経時的変化を示す図である。ts-BMT群では、比較的GFP陽性率は保たれたものの、全例が8週以内に死亡した。一方、混合細胞移植群では、速やかにGFP陽性細胞率が減少し、全例が長期生存した。縦軸は対数表示である。
【技術分野】
【0001】
本発明は、遺伝子導入造血細胞を含む造血細胞移植のための組成物に関する。また本発明は、遺伝子導入造血細胞から血液細胞を再構築させる方法に関する。本発明の方法は、造血細胞移植における細胞染色体非損傷型の遺伝子治療に適用され得る。
【背景技術】
【0002】
X連鎖重症複合免疫不全症(X-linked SCID)など、単一遺伝子異常による難治性疾患に対する新たな治療法として遺伝子治療が期待されている。2000年にはフランスにおいて、CD34陽性造血幹細胞に対し原因遺伝子である共通ガンマ鎖(common γ-chain)遺伝子をレトロウイルスベクターにて導入し、11例中9例で有効であったことが報告された(Cavazzana-Calvo M. et al., Science 2000;288:669-672)。しかしその後、2例の患児にT細胞性白血病が発生し、同様なベクターや治療系に対する警鐘が成された(Kaiser J., Science 2003;299:495; Hacein-Bey-Abina S. et al., Science 2003;302:415-419)。その後の解析によりこの2例にはT細胞の発生に必要、かつ胸腺で発現が消失すべき遺伝子であるLMO2の恒常的活性化を認めることが報告されている(Hacein-Bey-Abina S. et al., Science 2003;302:415-419; Fischer A. et al., N Eng Journal Med 2004;350:2526-2527)。現在、このLMO2の恒常的活性化はこの臨床研究プロトコールに特異的なものではないかと考えられているが、レトロウイルスベクターのプロウイルスゲノムの宿主染色体内への取り込み自体が、癌抑制遺伝子の不活化や癌遺伝子の活性化の原因となり得る可能性は否定できず、根本的な問題点と考えられている。この潜在的危険性は同様に染色体へ遺伝子を組み込むアデノ随伴ウイルスベクター(AAV)、レンチウイルスベクターでも想定されている。先天性免疫不全性疾患には骨髄移植が有効性を示さない症例が存在するため、これらの患児には遺伝子治療のみが残されたオプションとなる。従ってこれまでの染色体組込型ベクターと異なる概念に基づく新しいかつ安全なベクターシステムの構築が望まれている。
【非特許文献1】Cavazzana-Calvo M. et al., Science 2000;288:669-672
【非特許文献2】Kaiser J., Science 2003;299:495
【非特許文献3】Hacein-Bey-Abina S. et al., Science 2003;302:415-419
【非特許文献4】Hacein-Bey-Abina S. et al., Science 2003;302:415-419
【非特許文献5】Fischer A. et al., N Eng Journal Med 2004;350:2526-2527
【発明の開示】
【発明が解決しようとする課題】
【0003】
本発明は、染色体非組込型ウイルスベクターを用いて、遺伝子導入造血細胞を含む造血細胞移植組成物を製造する方法を提供する。また本発明は、染色体非組込型ウイルスベクターを用いて遺伝子導入した造血細胞から、血液細胞を再構築させる方法を提供する。
【課題を解決するための手段】
【0004】
本発明者らは、造血細胞移植において、造血細胞の染色体を損傷させることのない安全な遺伝子導入を可能にするため、染色体非組込型ウイルスを用いる方法の開発を行った。このために、染色体非組込型ウイルスの1つであるマイナス鎖RNAウイルスを用いて造血細胞に遺伝子を導入し、この造血細胞からの血球系の再構築を試みた。まず、マウス大腿骨より赤血球・T細胞を除去した骨髄細胞を調製し、外来遺伝子を組み込んだマイナス鎖RNAウイルスを感染させ遺伝子導入骨髄細胞を作製した。そして、放射線照射により骨髄破壊(myeloablation)を施行したマウスに、この遺伝子導入骨髄細胞を注入する実験を行った。その後の生体内における移入骨髄細胞の生着動態と免疫学的反応を検討したところ、マイナス鎖RNAウイルスベクターが導入された骨髄細胞はレシピエント内で生着し、T細胞を含む血球系細胞のrepopulationが確認された。本発明は、染色体非傷害型細胞質型ベクターを用いた遺伝子導入造血細胞による血球系再構築を初めて開示するものであり、マイナス鎖RNAウイルスベクターを用いることにより、細胞の染色体を組み換えることなく遺伝子導入した移植造血細胞から血球系統のrepopulationが可能であることを実証するものである。特にエンベロープ構成蛋白質をコードする遺伝子に変異または欠損を有するマイナス鎖RNAウイルスベクターを用いれば、長期間安定して血球系細胞の再構築が行われることが判明した。本発明により、染色体損傷を懸念することなく、安全に造血細胞移植における遺伝子治療を実施することが可能となる。
【0005】
すなわち本発明は、造血細胞移植に適した遺伝子導入造血細胞組成物およびその製造方法、およびマイナス鎖RNAウイルスベクターを用いて遺伝子導入した造血細胞から血液細胞を再構築させる方法等に関し、より具体的には、請求項の各項に記載の発明に関する。なお同一の請求項を引用する請求項に記載の発明の1つまたは複数の組み合わせからなる発明は、それらの請求項に記載の発現に既に意図されている。すなわち本発明は、
〔1〕造血細胞移植組成物の製造方法であって、マイナス鎖RNAウイルスベクターを造血細胞に導入し、該造血細胞および薬学的に許容される媒体を含む組成物を調製することを含む方法、
〔2〕マイナス鎖RNAウイルスベクターが、エンベロープ構成蛋白質をコードする1つまたは複数の遺伝子が変異または欠損している、〔1〕に記載の方法、
〔3〕エンベロープ構成蛋白質をコードするすべての遺伝子が変異または欠損している、〔2〕に記載の方法、
〔4〕エンベロープ構成蛋白質をコードする少なくとも1つの遺伝子が欠損している、〔2〕または〔3〕に記載の方法、
〔5〕マイナス鎖RNAウイルスがパラミクソウイルス科ウイルスである、〔1〕から〔4〕のいずれかに記載の方法、
〔6〕パラミクソウイルス科ウイルスがセンダイウイルスである、〔5〕に記載の方法、
〔7〕〔1〕から〔6〕のいずれかに記載の方法により得られる造血細胞移植組成物、
〔8〕遺伝子導入された血液細胞を造血細胞から生成させる方法であって、
(a)マイナス鎖RNAウイルスベクターを造血細胞に接触させる工程、および
(b)工程(a)の造血細胞を動物に注入する工程、を含む方法、
〔9〕工程(b)の注入前に該動物を免疫抑制する工程をさらに含む、〔8〕に記載の方法、
〔10〕マイナス鎖RNAウイルスベクターにおいて、エンベロープ構成蛋白質をコードする1つまたは複数の遺伝子が変異または欠損している、〔8〕または〔9〕に記載の方法、
〔11〕エンベロープ構成蛋白質をコードするすべての遺伝子が変異または欠損している、〔10〕に記載の方法、
〔12〕エンベロープ構成蛋白質をコードする少なくとも1つの遺伝子が欠損している、〔10〕または〔11〕に記載の方法、
〔13〕マイナス鎖RNAウイルスがパラミクソウイルス科ウイルスである、〔8〕から〔12〕のいずれかに記載の方法、
〔14〕パラミクソウイルス科ウイルスがセンダイウイルスである、〔13〕に記載の方法、に関する。
【発明の効果】
【0006】
本発明において、染色体非組み込み型の細胞質型ベクターにより遺伝子導入された造血細胞が、生体内でrepopulationが可能であることが初めて実証された。レシピエントにおいて、外来遺伝子を発現するドナー由来のT細胞が検出されたことから、マイナス鎖RNAウイルスベクターが導入された造血幹細胞からrepopulationが起こったことが支持される。マイナス鎖RNAウイルスは、細胞質で転写・複製し、細胞核の染色体遺伝子に相互作用しない。本発明は、例えば先天性免疫不全症などの造血系疾患に対する宿主染色体非傷害遺伝子治療を可能とする。
【発明を実施するための最良の形態】
【0007】
本発明は、マイナス鎖RNAウイルスベクターが導入された造血細胞および薬学的に許容される媒体を含む造血細胞移植組成物およびその製造方法を提供する。本発明においてマイナス鎖RNAウイルスとは、マイナス鎖(ウイルス蛋白質をセンスにコードする鎖と相補的なアンチセンス鎖)のRNAをゲノムとして含むウイルスのことである。マイナス鎖RNAはネガティブ鎖RNAとも呼ばれる。
【0008】
また、マイナス鎖RNAウイルスベクターとは、遺伝子を細胞に導入するため担体(ベクター)としてのマイナス鎖RNAウイルスを言う。マイナス鎖RNAウイルスベクターは外来遺伝子を持っていても持たなくてもよい。
【0009】
組み換えウイルスとは、組み換えポリヌクレオチドを介して生成したウイルス、またはそのウイルスの増幅産物を言う。組み換えポリヌクレオチドとは、両端または片端が自然の状態と同じようには結合していないポリヌクレオチドを言う。具体的には、組み換えポリヌクレオチドは、人為的にポリヌクレオチド鎖の結合が改変(切断および/または結合)されたポリヌクレオチドである。組み換えポリヌクレオチドは、ポリヌクレオチド合成、ヌクレアーゼ処理、リガーゼ処理等を組み合わせて、公知の遺伝子組み換え方法により生成させることができる。組み換えウイルスは、遺伝子操作により構築されたウイルスゲノムをコードするポリヌクレオチドを発現させ、ウイルスを再構築することによって生成することができる。例えば、ウイルスゲノムをコードするcDNAから、ウイルス再構成する方法が知られている(Y. Nagai, A. Kato. Microbiol. Immunol. 1999: 43; 613-624 )。
【0010】
本発明において遺伝子とは遺伝物質を指し、転写単位をコードする核酸を言う。遺伝子はRNAであってもDNAであってもよい。本発明において蛋白質をコードする核酸は、該蛋白質の遺伝子と呼ぶ。また一般に、遺伝子は蛋白質をコードしていなくてもよく、例えば遺伝子はリボザイムまたはアンチセンスRNAなどの機能的RNAをコードするものであってもよい。一般に、遺伝子は天然由来または人為的に設計された配列であり得る。また、本発明において「DNA」とは、一本鎖DNAおよび二本鎖DNAを含む。また蛋白質をコードするとは、ポリヌクレオチドが該蛋白質を適当な条件下で発現できるように、該蛋白質のアミノ酸配列をコードするORFをセンスまたはアンチセンスに含むことを言う。
【0011】
本発明において得に好適に用いられるマイナス鎖RNAウイルスとしては、例えばパラミクソウイルス科 (Paramyxoviridae) ウイルスのセンダイウイルス(Sendai virus)、ニューカッスル病ウイルス(Newcastle disease virus)、おたふくかぜウイルス(Mumps virus)、麻疹ウイルス(Measles virus)、RSウイルス(Respiratory syncytial virus)、牛疫ウイルス(rinderpest virus)、ジステンパーウイルス(distemper virus)、サルパラインフルエンザウイルス(SV5)、ヒトパラインフルエンザウイルス1,2,3型、オルトミクソウイルス科(Orthomyxoviridae)のインフルエンザウイルス(Influenza virus)、ラブドウイルス科(Rhabdoviridae)の水疱性口内炎ウイルス(Vesicular stomatitis virus)、狂犬病ウイルス(Rabies virus)等が挙げられ、具体的には、Sendai virus (SeV)、human parainfluenza virus-1 (HPIV-1)、human parainfluenza virus-3 (HPIV-3)、phocine distemper virus (PDV)、canine distemper virus (CDV)、dolphin molbillivirus (DMV)、peste-des-petits-ruminants virus (PDPR)、measles virus (MV)、rinderpest virus (RPV)、Hendra virus (Hendra)、Nipah virus (Nipah)、human parainfluenza virus-2 (HPIV-2)、simian parainfluenza virus 5 (SV5)、human parainfluenza virus-4a (HPIV-4a)、human parainfluenza virus-4b (HPIV-4b)、mumps virus (Mumps)、およびNewcastle disease virus (NDV) などが含まれる。より好ましくは、Sendai virus (SeV)、human parainfluenza virus-1 (HPIV-1)、human parainfluenza virus-3 (HPIV-3)、phocine distemper virus (PDV)、canine distemper virus (CDV)、dolphin molbillivirus (DMV)、peste-des-petits-ruminants virus (PDPR)、measles virus (MV)、rinderpest virus (RPV)、Hendra virus (Hendra)、Nipah virus (Nipah)、Respiratory syncytial virus (RSV) からなる群より選択されるウイルス等が挙げられる。
【0012】
本発明において用いられるマイナス鎖RNAウイルスとしては、特に一本鎖マイナス鎖RNAウイルス(非分節型(non-segmented)マイナス鎖RNAウイルスとも言う)が好ましい。「一本鎖ネガティブ鎖RNAウイルス」とは、一本鎖ネガティブ鎖[すなわちマイナス鎖]RNAをゲノムに有するウイルスを言う。このようなウイルスとしては、パラミクソウイルス(Paramyxoviridae; Paramyxovirus, Morbillivirus, Rubulavirus, および Pneumovirus属等を含む)、ラブドウイルス(Rhabdoviridae; Vesiculovirus, Lyssavirus, および Ephemerovirus属等を含む)、フィロウイルス(Filoviridae)、オルトミクソウイルス(Orthomyxoviridae; Infuluenza virus A, B, C, および Thogoto-like viruses 等を含む)、ブニヤウイルス(Bunyaviridae; Bunyavirus, Hantavirus, Nairovirus, および Phlebovirus属等を含む)、アレナウイルス(Arenaviridae)などの科に属するウイルスが含まれる。
【0013】
本発明において用いられるマイナス鎖RNAウイルスは、より好ましくは、パラミクソウイルス亜科(レスピロウイルス属、ルブラウイルス属、およびモルビリウイルス属を含む)に属するウイルスまたはその誘導体であり、より好ましくはレスピロウィルス属(genus Respirovirus)(パラミクソウィルス属(Paramyxovirus)とも言う)に属するウィルスまたはその誘導体である。誘導体には、ウイルスによる遺伝子導入能を損なわないように、ウイルス遺伝子が改変されたウイルス、および化学修飾されたウイルス等が含まれる。本発明を適用可能なレスピロウィルス属ウィルスとしては、例えばヒトパラインフルエンザウィルス1型(HPIV-1)、ヒトパラインフルエンザウィルス3型(HPIV-3)、ウシパラインフルエンザウィルス3型(BPIV-3)、センダイウィルス(Sendai virus; マウスパラインフルエンザウィルス1型とも呼ばれる)、およびサルパラインフルエンザウィルス10型(SPIV-10)などが含まれる。本発明においてパラミクソウィルスは、最も好ましくはセンダイウィルスである。これらのウィルスは、天然株、野生株、変異株、ラボ継代株、および人為的に構築された株などに由来してもよい。
【0014】
マイナス鎖RNAウイルスベクターはウイルスゲノムRNAに搭載遺伝子をアンチセンスにコードしている。ウイルスゲノムRNAとは、マイナス鎖RNAウイルスのウイルス蛋白質と共にリボヌクレオプロテイン(RNP)を形成し、該蛋白質によりゲノム中の遺伝子が発現し、このRNAが複製されて娘RNPが形成される機能を持つRNAである。ゲノムRNAは、ウイルス遺伝子に変異および/または欠損があるものや、外来遺伝子が組み込まれるなどして改変されたものであってもよい。一般にマイナス鎖RNAウイルスのゲノムは、3'リーダー領域と5'トレイラー領域の間に、ウイルス遺伝子がアンチセンス配列として並んだ構成をしている。各遺伝子のORFの間には、転写終結配列(E配列) - 介在配列(I配列) - 転写開始配列(S配列) が存在し、これにより各遺伝子のORFをコードするRNAが別々のシストロンとして転写される。本発明のウイルスに含まれるゲノムRNAは、該RNAとRNPを構成するマイナス鎖RNAウイルス蛋白質をコードしている。但し、マイナス鎖RNAウイルスのゲノムRNAはマイナス鎖であるから、これらの蛋白質はアンチセンスとしてコードされている。ゲノムRNAとRNPを構成するマイナス鎖RNAウイルス蛋白質とは、マイナス鎖RNAウイルスのゲノムRNAと複合体を形成し、ゲノムRNAの複製およびゲノムにコードされている遺伝子の発現およびRNA自身の自律的な複製に必要とされるウイルス蛋白質群を言う。これらの蛋白質は、ウイルスのエンベロープを除くコアを形成する蛋白質であり、典型的には、N(ヌクレオキャプシド)、P(ホスホ)、およびL(ラージ)蛋白質である。ウイルス種によっては、表記は異なることもあるが、対応する蛋白質は当業者にとっては自明である (Anjeanette Robert et al., Virology. 1998: 247; 1-6 )。例えばNはNPと表記されることもある。また該RNAは、ウイルス粒子の形成に必要なM(マトリックス)蛋白質をコードしていてもよい。さらに該RNAは、ウイルス粒子の感染に必要なエンベロープ蛋白質をコードしていてもよい。マイナス鎖RNAウイルスのエンベロープ蛋白質としては、細胞膜融合を起こす蛋白質であるF(フュージョン)蛋白質および細胞への接着に必要なHN(ヘマグルチニン-ノイラミニダーゼ)蛋白質(または H (ヘマグルチニン))が挙げられる。また、F蛋白質および/またはHN (またはH) 蛋白質以外のウイルスエンベロープ蛋白質をコードさせてもよい。但し、後述のように、本発明において用いるマイナス鎖RNAウイルスは、エンベロープ構成蛋白質をコードする遺伝子が変異および/または欠損していることが好ましい。
【0015】
例えばパラミクソウイルス亜科に属する各ウィルスにおける各遺伝子は、一般に次のように表記される。一般に、NP遺伝子は"N"とも表記される。また、HNはノイラミニダーゼ活性を有さない場合にはH (ヘマグルチニン) と表記される。
レスピロウイルス属 NP P/C/V M F HN - L
ルブラウイルス属 NP P/V M F HN (SH) L
モービリウイルス属 NP P/C/V M F H - L
【0016】
例えばセンダイウイルスの各遺伝子の塩基配列のデータベースのアクセッション番号は、NP遺伝子については M29343、M30202, M30203, M30204, M51331, M55565, M69046, X17218、P遺伝子については M30202, M30203, M30204, M55565, M69046, X00583, X17007, X17008、M遺伝子については D11446, K02742, M30202, M30203, M30204, M69046, U31956, X00584, X53056、F遺伝子については D00152, D11446, D17334, D17335, M30202, M30203, M30204, M69046, X00152, X02131、HN遺伝子については D26475, M12397, M30202, M30203, M30204, M69046, X00586, X02808, X56131、L遺伝子については D00053, M30202, M30203, M30204, M69040, X00587, X58886を参照のこと。またその他のウイルスがコードするウイルス遺伝子を例示すれば、N遺伝子については、CDV, AF014953; DMV, X75961; HPIV-1, D01070; HPIV-2, M55320; HPIV-3, D10025; Mapuera, X85128; Mumps, D86172; MV, K01711; NDV, AF064091; PDPR, X74443; PDV, X75717; RPV, X68311; SeV, X00087; SV5, M81442; および Tupaia, AF079780、P遺伝子については、CDV, X51869; DMV, Z47758; HPIV-l, M74081; HPIV-3, X04721; HPIV-4a, M55975; HPIV-4b, M55976; Mumps, D86173; MV, M89920; NDV, M20302; PDV, X75960; RPV, X68311; SeV, M30202; SV5, AF052755; および Tupaia, AF079780、C遺伝子については CDV, AF014953; DMV, Z47758; HPIV-1. M74081; HPIV-3, D00047; MV, ABO16162; RPV, X68311; SeV, AB005796; および Tupaia, AF079780、M遺伝子については CDV, M12669; DMV Z30087; HPIV-1, S38067; HPIV-2, M62734; HPIV-3, D00130; HPIV-4a, D10241; HPIV-4b, D10242; Mumps, D86171; MV, AB012948; NDV, AF089819; PDPR, Z47977; PDV, X75717; RPV, M34018; SeV, U31956; および SV5, M32248、F遺伝子については CDV, M21849; DMV, AJ224704; HPN-1. M22347; HPIV-2, M60182; HPIV-3. X05303, HPIV-4a, D49821; HPIV-4b, D49822; Mumps, D86169; MV, AB003178; NDV, AF048763; PDPR, Z37017; PDV, AJ224706; RPV, M21514; SeV, D17334; および SV5, AB021962、HN(HまたはG)遺伝子については CDV, AF112189; DMV, AJ224705; HPIV-1, U709498; HPIV-2. D000865; HPIV-3, AB012132; HPIV-4A, M34033; HPIV-4B, AB006954; Mumps, X99040; MV, K01711; NDV, AF204872; PDPR, Z81358; PDV, Z36979; RPV, AF132934; SeV, U06433; および SV-5, S76876 が例示できる。但し、各ウイルスは複数の株が知られており、株の違いにより上記に例示した以外の配列からなる遺伝子も存在する。
【0017】
これらのウイルス蛋白質をコードするORFおよび外来遺伝子のORFは、ゲノムRNAにおいて上記のE-I-S配列を介してアンチセンスに配置される。ゲノムRNAにおいて最も3'に近いORFは、3'リーダー領域と該ORFとの間にS配列のみが必要であり、EおよびI配列は必要ない。またゲノムRNAにおいて最も5'に近いORFは、5'トレイラー領域と該ORFとの間にE配列のみが必要であり、IおよびS配列は必要ない。また2つのORFは、例えばIRES等の配列を用いて同一シストロンとして転写させることも可能である。このような場合は、これら2つのORFの間にはE-I-S配列は必要ない。例えば、野生型のパラミクソウイルスの場合、典型的なRNAゲノムは、3'リーダー領域に続き、N、P、M、F、HN (H)、およびL蛋白質をアンチセンスにコードする6つのORFが順に並んでおり、それに続いて5'トレイラー領域を他端に有する。本発明においてゲノムRNAは、ウイルス遺伝子の配置はこれに限定されるものではないが、好ましくは、野生型ウイルスと同様に、3'リーダー領域に続き、N、P、M、F、HN (またはH)、およびL蛋白質をコードするORFが順に並び、それに続いて5'トレイラー領域が配置されることが好ましい。ある種のウイルスにおいては、ウイルス遺伝子が異なっているが、そのような場合でも上記と同様に各ウイルス遺伝子を野生型と同様の配置とすることが好ましい。一般に N、P、およびL遺伝子を保持しているベクターは、細胞内で自律的にRNAゲノムから遺伝子が発現し、ゲノムRNAが複製される。さらにFおよびHN (またはH)遺伝子等のエンベロープのスパイク蛋白質をコードする遺伝子、およびM遺伝子の働きにより、感染性のウイルス粒子が形成され、細胞外に放出される。従って、このようなベクターは伝播能を有するウイルスベクターとなる。ベクターに外来遺伝子を搭載させる場合は、後述するように、このゲノム中の蛋白質非コード領域に挿入すればよい。
【0018】
本発明において用いるマイナス鎖RNAウイルスベクターは、1つまたは複数のエンベロープ構成蛋白質の遺伝子に変異および/または欠損を有するものが好ましい。このような変異型マイナス鎖RNAウイルスを用いることにより、血液細胞のリポピュレーションの効率が有意に上昇することが判明した。ここで変異とは、野生型遺伝子を持つウイルスに比べ、変異遺伝子に置換されたウイルスの感染性ウイルス産生能が有意に低下するような変異である。また欠損とは、野生型遺伝子の機能が実質的に失われることであり、機能的蛋白質を発現しないように改変されること、および遺伝子が欠失していることを含む。野生型遺伝子の機能が実質的に失われるとは、当該遺伝子の欠損により、37℃におけるウイルス産生が野生型の1/10以下、好ましくは1/20以下、より好ましくは1/50以下に減少することである。エンベロープ構成蛋白質とは、ウイルスのエンベロープの成分となるウイルス蛋白質を言い、エンベロープ表面に露出し細胞への接着または感染に機能するスパイク蛋白質およびエンベロープの形成等に機能する裏打ち蛋白質が含まれる。典型的には、エンベロープ構成蛋白質の遺伝子としては F、HN、およびMが挙げられ、ウイルス種によっては H、M1、および G 等の遺伝子が含まれる。これらのエンベロープ構成蛋白質の遺伝子の1つまたは複数を変異および/または欠失させたウイルスは、感染細胞における感染性ウイルス粒子の形成能が低下するため安全性が高い。また細胞傷害性が有意に低下する。ゲノムにおいて変異および/または欠損させる遺伝子としては、例えばF遺伝子、HN (またはH) 遺伝子、M遺伝子、またはその任意の組み合わせが挙げられる。特に好ましくは、少なくともF遺伝子が変異または欠損、より好ましくは欠損している。さらに好ましくは、F遺伝子およびHN (またはH) 遺伝子が変異または欠損、より好ましくは欠損している。さらに好ましくは、F遺伝子、HN (またはH) 遺伝子およびM遺伝子が変異または欠損している。
【0019】
特に、少なくともF遺伝子が欠失し、さらにHN (またはH) 遺伝子が変異または欠失するベクターを用いることが好ましい。このようなウイルスベクターは、一度細胞内に入り込んだ後は、F蛋白質は発現せず、HN (またはH) 蛋白質も極低レベル発現または無発現となる。ベクター感染細胞においてHN蛋白質が多数表出すると、細胞同士の吸着やaggregationを来たす可能性があり、事実このような現象がin vitroで起こり得ることを本発明者らは確認した。従って、HN (またはH) 遺伝子に温度感受性変異を有するベクター、さらに好ましくはHN (またはH) 遺伝子を欠損するベクターを用いることにより、造血細胞移植においてより良好な結果を得ることが可能となる。さらに、M遺伝子を変異または欠損させることにより、より良好な生着動態を得ることができる。
【0020】
エンベロープ構成蛋白質において多数の温度感受性変異が知られている。これらの温度感受性変異蛋白質遺伝子を有するウイルスを本発明において好適に用いることができる。温度感受性変異とは、低温 (例えば32℃) に比べ、ウイルス宿主の通常の温度(例えば37℃)において有意に活性が低下する変異のことである。このような、温度感受性変異を持つ蛋白質は、野生型蛋白質などで相補しなくても許容条件(低温)下でウイルスを作製することができるので有用である。
【0021】
例えば、M遺伝子の温度感受性変異としては、特に限定されるものではないが、センダイウィルスのM蛋白質におけるG69、T116、およびA183からなる群より選択される少なくとも1つ、好ましくは任意に選択される2つ、さらに好ましくは3つすべてのアミノ酸部位、あるいは他のマイナス鎖RNAウィルスM蛋白質のそれらと相同な部位が変異しているものを好適に用いることができる。ここでG69とはM蛋白質の69番目のアミノ酸Gly、T116とはM蛋白質の116番目のアミノ酸Thr、A183とはM蛋白質の183番目のアミノ酸Alaを指す。
【0022】
M蛋白質をコードする遺伝子(M遺伝子)は、マイナス鎖RNAウイルスで広く保存されており、ウイルスのヌクレオカプシッドとエンベロープの両者に相互作用する機能を有することが知られている(Garoff, H. et al., Microbiol. Mol. Biol. Rev. 1998: 62; 117-190)。また、SeV M蛋白質においてamphiphilic α-helixと予想されている104-119(104-KACTDLRITVRRTVRA-119/配列番号:1)は粒子形成に重要な領域として同定されている(Genevieve Mottet et al., J. Gen. Virol. 1999: 80; 2977-2986)が、当該領域はマイナス鎖RNAウイルス間で良く保存されている。M蛋白質のアミノ酸配列はマイナス鎖RNAウイルスで類似しており、特にパラミクソウイルス亜科においては既知のM蛋白質は共通して全長約330〜380アミノ酸からなる塩基性蛋白質であり、全領域にわたって類似性があるが、特にC端側半分での類似性が高い(Gould, A. R. Virus Res. 1996: 43; 17-31、Harcourt, B.H. et al., Virology. 2000: 271; 334-349)。従って、例えば SeV M蛋白質のG69、T116、及びA183と相同なアミノ酸は容易に同定することが可能である。
【0023】
SeV M蛋白質のG69、T116、及びA183と対応する他のマイナス鎖RNAウィルスM蛋白質の相同な部位のアミノ酸は、当業者であれば、例えばBLASTなどのアミノ酸配列のホモロジー検索プログラム(アライメント作成機能を持つもの)またはCLUSTAL Wなどのアライメント作成プログラムを用いてSeV M蛋白質のアミノ酸と整列化することにより同定することができる。例えばSeV M蛋白質のG69に相当する各M蛋白質の相同部位としては、human parainfluenza virus-1 (HPIV-1)(括弧は略称)であればG69、human parainfluenza virus-3 (HPIV-3) であればG73、phocine distemper virus (PDV)およびcanine distemper virus (CDV)であればG70、dolphin molbillivirus (DMV)であればG71、peste-des-petits-ruminants virus (PDPR)、measles virus (MV)、およびrinderpest virus (RPV)であればG70、Hendra virus (Hendra)およびNipah virus (Nipah)であればG81、human parainfluenza virus-2 (HPIV-2)であればG70、human parainfluenza virus-4a (HPIV-4a)およびhuman parainfluenza virus-4b (HPIV-4b)であればE47、mumps virus (Mumps)であればE72が挙げられる(文字と番号はアミノ酸とその位置を表す)。また、SeV M蛋白質のT116に相当する各M蛋白質の相同部位としては、human parainfluenza virus-1 (HPIV-1)であればT116、human parainfluenza virus-3 (HPIV-3)でればT120、phocine distemper virus (PDV)およびcanine distemper virus (CDV)であればT104、dolphin molbillivirus (DMV)であれはT105、peste-des-petits-ruminants virus (PDPR)、measles virus (MV)およびrinderpest virus (RPV)であればT104、Hendra virus (Hendra)およびNipah virus (Nipah)であればT120、human parainfluenza virus-2 (HPIV-2)およびsimian parainfluenza virus 5 (SV5)であればT117、human parainfluenza virus-4a (HPIV-4a)およびhuman parainfluenza virus-4b (HPIV-4b)であればT121、mumps virus (Mumps)であればT119、Newcastle disease virus (NDV)であればS120が挙げられる。SeV M蛋白質のA183に相当する各M蛋白質の相同部位としては、human parainfluenza virus-1 (HPIV-1)であればA183、human parainfluenza virus-3 (HPIV-3)であれなF187、phocine distemper virus (PDV)およびcanine distemper virus (CDV)であればY171、dolphin molbillivirus (DMV)であればY172、peste-des-petits-ruminants virus (PDPR)、measles virus (MV)およびrinderpest virus (RPV)であれはY171、Hendra virus (Hendra)およびNipah virus (Nipah)であればY187、human parainfluenza virus-2 (HPIV-2)であれはY184、simian parainfluenza virus 5 (SV5)であればF184、human parainfluenza virus-4a (HPIV-4a)およびhuman parainfluenza virus-4b (HPIV-4b)であれはF188、mumps virus (Mumps)であればF186、Newcastle disease virus (NDV)であればY187が挙げられる。ここに挙げたウイルスにおいて、それぞれのM蛋白質に上記の3つの部位のいずれか、好ましくは任意の2部位の組み合わせ、さらに好ましくは3つの部位全てのアミノ酸が他のアミノ酸に置換された変異M蛋白質をコードするゲノムを有するウイルスは、本発明において好適に用いられる。
【0024】
アミノ酸変異は、所望の他のアミノ酸への置換であってよいが、好ましくは、側鎖の化学的性質の異なるアミノ酸への置換である。例えばアミノ酸は、塩基性アミノ酸(例えばリジン、アルギニン、ヒスチジン)、酸性アミノ酸 (例えばアスパラギン酸、グルタミン酸)、非荷電極性アミノ酸 (例えばグリシン、アスパラギン、グルタミン、セリン、スレオニン、チロシン、システイン)、非極性アミノ酸 (例えばアラニン、バリン、ロイシン、イソロイシン、プロリン、フェニルアラニン、メチオニン、トリプトファン)、β分岐アミノ酸 (例えばスレオニン、バリン、イソロイシン)、および芳香族アミノ酸 (例えばチロシン、フェニルアラニン、トリプトファン、ヒスチジン)などのグループに分類することができるが、あるアミノ酸について、そのアミノ酸が属するグループのアミノ酸以外のアミノ酸に置換することなどが挙げられる。具体的には、塩基性アミノ酸であれは、酸性または中性アミノ酸への置換、極性アミノ酸であれは非極性アミノ酸への置換、20種の天然のアミノ酸の平均分子量より大きい分子量を持つアミノ酸であれば、その平均分子量より小さいアミノ酸への置換、逆にその平均分子量より小さいアミノ酸であれば、それより大きいアミノ酸への置換などが挙げられるが、それに限定されない。
【0025】
具体的に例示すれば、センダイウィルスM蛋白質における G69E、T116A、およびA183Sからなる群より選択される変異あるいはそれらと相同な位置に変異を含む他のパラミクソウイルスのM蛋白質を用いることができる。ここでG69Eとは、M蛋白質の69番目のアミノ酸GlyがGluに置換された変異、T116Aとは、M蛋白質の116番目のアミノ酸ThrがAlaに置換された変異、A183Sとは、M蛋白質の183番目のアミノ酸AlaがSerに置換された変異を言う。すなわち、センダイウィルス M蛋白質のG69、T116、およびA183あるいは他のウイルスM蛋白質の相同部位を、それぞれGlu (E)、Ala (A)、およびSer (S) へ置換することができる。これらの変異は組み合わせて有していることが好ましく、特に上記3変異の全てを保持していることがより好ましい。M遺伝子への変異の導入は、公知の変異導入方法に従って実施することができる。例えば実施例に記載のように目的の変異を入れたオリゴヌクレオチドを用いて導入することが可能である。
【0026】
また、例えば麻疹ウィルスにおいては、M蛋白のモノクローナル抗体に対するエピトープが変化している温度感受性株のP253-505(Morikawa, Y. et al., Kitasato Arch. Exp. Med. 1991: 64; 15-30)のM遺伝子配列を用いてもよい。また、SeV M蛋白質の116番目のThrに対応する麻疹ウィルスM蛋白質の104番目のThr、またはムンプスウィルスのM蛋白質の119番目のThrを他のアミノ酸(例えばAla)に置換してもよい。
【0027】
本発明において用いられるベクターは、さらに好ましい態様においてはM遺伝子を欠損している。M遺伝子の欠損とは、野生型M遺伝子の機能が実質的に失われていることを言い、機能欠失型の変異を有するM遺伝子を持つ場合およびM遺伝子を欠失する場合を含む。M遺伝子の機能欠失型変異は、例えばM遺伝子の蛋白質コード配列を欠失させたり、他の配列を挿入することにより作製することができる。例えば、M蛋白質コード配列の途中に停止コドンを設計することができる(WO00/09700)。本発明のベクターは、最も好ましくはM蛋白質のコード配列を完全に欠失している。M蛋白質のORFを欠失したベクターは、温度感受性変異M蛋白質をコードするベクターとは違い、任意の条件においてウイルス粒子を形成する能力を失っている。
【0028】
HN遺伝子の温度感受性変異としては、特に限定されるものではないが、例えばセンダイウィルスのHN蛋白質のA262、G264、およびK461からなる群より選択される少なくとも1つ、好ましくは任意に選択される2つ、さらに好ましくは3つすべてのアミノ酸部位、あるいは他のマイナス鎖RNAウィルスM蛋白質のそれらと相同な部位に変異を含むものを好適に用いることができる。ここでA262とはHN蛋白質の262番目のアミノ酸Ala、G264とはHN蛋白質の264番目のアミノ酸Gly、K461とはHN蛋白質の461番目のアミノ酸Lysを指す。アミノ酸変異は、所望の他のアミノ酸への置換であってよいが、好ましくは、上記のM蛋白質の変異と同様、側鎖の化学的性質の異なるアミノ酸への置換である。例えば、上記したように異なるグループのアミノ酸に置換することなどが挙げられる。具体的には、例えばセンダイウィルスHN蛋白質における A262T、G264R、およびK461Gからなる群より選択される変異あるいはそれらと相同な変異を含むものを好適に用いることができる。ここでA262Tとは、HN蛋白質の262番目のアミノ酸AlaがThrに置換された変異、G264Rとは、HN蛋白質の264番目のアミノ酸GlyがArgに置換された変異、K461Gとは、HN蛋白質の461番目のアミノ酸LysがGlyに置換された変異を言う。すなわち、センダイウィルス HN蛋白質のA262、G264、およびK461あるいは他のウィルスHN蛋白質の相同部位を、それぞれThr (T)、Arg (R)、およびGly (G) へ置換することができる。これらの変異は組み合わせて有していることが好ましく、特にこれらの3つの変異の全てを保持していることがより好ましい。
【0029】
また、例えば、ムンプスウィルスにおいては、温度感受性の性質を示しワクチンとして使用されているUrabe AM9株(Wright, K. E. et al., Virus Res. 2000: 67; 49-57)を参考に、HN蛋白の464及び468番目のアミノ酸に変異導入することは本発明において好ましい。これと相同な位置のアミノ酸の変異は、他のマイナス鎖RNAウィルスにも適用することができる。
【0030】
またマイナス鎖RNAウィルスは、P遺伝子またはL遺伝子に変異を有していてもよい。このような変異としては、具体的には、SeV P蛋白質の86番目のGlu(E86)の変異、SeV P蛋白質の511番目のLeu(L511)の他のアミノ酸への置換、または他のマイナス鎖RNAウィルスP蛋白質の相同部位の置換が挙げられる。アミノ酸変異は、所望の他のアミノ酸への置換であってよいが、好ましくは、上記と同様、側鎖の化学的性質の異なるアミノ酸への置換である。例えば、上記したように異なるグループのアミノ酸に置換することなどが挙げられる。具体的には、E86のLysへの置換(E86K)、L511のPheへの置換(L511F)などが例示できる。またL蛋白質においては、1197番目のAsn(N1197)および/または1795番目のLys(K1795)の他のアミノ酸への置換、または他のマイナス鎖RNAウィルスL蛋白質の相同部位の置換が挙げられる。L蛋白質のこれら2つの変異の両方を有するL蛋白質遺伝子は特に好ましい。アミノ酸変異は、やはり所望の他のアミノ酸への置換であってよいが、好ましくは、上記と同様、側鎖の化学的性質の異なるアミノ酸への置換である。例えば、上記したように異なるグループのアミノ酸に置換することなどが挙げられる。具体的には、N1197のSerへの置換(N1197S)、K1795のGluへの置換(K1795E)などが例示できる。P遺伝子とL遺伝子の変異は、両方持っていることで、持続感染性、2次粒子放出の抑制、または細胞傷害性の抑制の効果を顕著に高めることができる。さらに、エンベロープ蛋白質遺伝子の変異および/または欠損を組み合わせることで、これらの効果を劇的に上昇させることができる。
【0031】
またマイナス鎖RNAウイルスベクターは、アクセサリー遺伝子が欠損したものであってよい。例えばSeVのアクセサリー遺伝子の1つであるV遺伝子をノックアウトすることにより、培養細胞における遺伝子発現および複製は障害されることなく、マウス等の宿主に対するSeVの病原性が顕著に減少する(Kato, A. et al. J. Virol. 1997: 71; 7266-7272, Kato, A. et al. EMBO J. 1997: 16; 578-587, Curran, J. et al., WO01/04272, EP1067179)。このような弱毒化ベクターは、より毒性の低い遺伝子導入用ウイルスベクターとして特に有用である。
【0032】
マイナス鎖RNAウイルスは宿主細胞の細胞質でのみ転写・複製を行い、DNAフェーズを持たないため染色体への組み込み(integration)は起こらない(Lamb, R.A. and Kolakofsky, D., Paramyxoviridae: The viruses and their replication. In: Fields BN, Knipe DM, Howley PM, (eds). Fields of virology. Lippincott - Raven Publishers: Philadelphia, 1996: 2; 1177-1204)。このため染色体異常による癌化および不死化などの安全面における問題が生じない。マイナス鎖RNAウイルスのこの特徴は、ベクター化した時の安全性に大きく寄与するものと考えられる。異種遺伝子発現の結果では、例えばセンダイウイルス(SeV)を連続多代継代しても殆ど塩基の変異が認められず、ゲノムの安定性が高く、挿入異種遺伝子を長期間に渡って安定に発現する事が示されている(Yu, D. et al., Genes Cells. 1997: 2: 457-466)。また、カプシド構造蛋白質を持たないことによる導入遺伝子のサイズまたはパッケージングの柔軟性(flexibility)など性質上のメリットがある。SeVベクターは、外来遺伝子を少なくとも5kbまで導入可能であり、転写ユニットを付加することによって2種類以上の遺伝子を同時に発現する事も可能である。
【0033】
特にセンダイウイルスは、齧歯類にとっては病原性で肺炎を生じることが知られているが、人に対しては病原性がない。これはまた、野生型センダイウイルスの経鼻的投与によって非ヒト霊長類において重篤な有害作用を示さないというこれまでの報告によっても支持されている(Hurwitz, J.L. et al., Vaccine. 1997: 15; 533-540, Bitzer, M. et al., J. Gene Med. 2003: 5; 543-553, Slobod, K.S. et al., Vaccine. 2004 : 22; 3182-3186)。また、種々の細胞・臓器へ、既存のベクターでは得られない極めて高い遺伝子導入・発現効率を示す(Yonemitsu Y. et al., Nature Biotechnol 2000;18:970-973; Masaki I. et al., FASEB J 2001;15:1294-1296; Yamashita A. et al., J Immunol 2002;168:450-457; Masaki I. et al., Circ Res 2002;90:966-973; Onimaru M. et al., Circ Res 2002;91:723-730; Shoji F. et al., Gene Ther 2003;10:213-218; Okano S. et al., Gene Ther 2003;10:1381-1391)。ヒト造血幹細胞へのin vitro導入においても、3系統への分化能を維持したまま高効率遺伝子導入が可能である(Jin CH. et al., Gene Ther. 2003 ;10:272-277)。センダイウイルスのこれらの特徴は、センダイウイルスベクターが人への造血細胞移植における遺伝子治療ベクターとしての利用を支持するものである。
【0034】
マイナス鎖RNAウイルスベクターは、ゲノムRNA中に所望の外来遺伝子をコードし得る。外来遺伝子を含む組換えウイルスベクターは、上記のウイルスベクターのゲノムに外来遺伝子を挿入することによって得られる。外来遺伝子の挿入位置は、例えばウイルスゲノムの蛋白質非コード領域の所望の部位を選択することができ、例えばゲノムRNAの3'リーダー領域と3'端に最も近いウイルス蛋白質ORFとの間、各ウイルス蛋白質ORFの間、および/または5'端に最も近いウイルス蛋白質ORFと5'トレイラー領域の間に挿入することができる。また、M、FまたはHN遺伝子などのエンベロープ構成蛋白質遺伝子を欠失するゲノムでは、その欠失領域に外来遺伝子をコードする核酸を挿入することができる。パラミクソウイルスに外来遺伝子を導入する場合は、ゲノムへの挿入断片のポリヌクレオチドの鎖長が6の倍数となるように挿入することが望ましい(Kolakofski, D. et al., J. Virol. 1998: 72; 891-899; Calain, P. and Roux, L. J. Virol. 1993: 67; 4822-4830)。挿入した外来遺伝子とウイルスORFとの間には、E-I-S配列が構成されるようにする。E-I-S配列を介して2またはそれ以上の外来遺伝子をタンデムに並べて挿入することができる。
【0035】
マイナス鎖RNAウイルスにより導入する外来遺伝子としては、特に制限はないが、天然の蛋白質としては、例えばサイトカイン、その他の液性因子、増殖因子、受容体、細胞内シグナル分子、酵素、ペプチドなどが挙げられる。蛋白質は分泌蛋白質、膜蛋白質、細胞質蛋白質、核蛋白質などであり得る。人工的な蛋白質としては、例えば、キメラ毒素などの融合蛋白質、ドミナントネガティブ蛋白質(受容体の可溶性分子または膜結合型ドミナントネガティブ受容体を含む)、欠失型の細胞接着分子および細胞表面分子などが挙げられる。また、分泌シグナル、膜局在化シグナル、または核移行シグナル等を付加した蛋白質であってもよい。導入遺伝子としてアンチセンスRNA分子またはRNA切断型リボザイムなどを発現させて、特定の遺伝子の機能を抑制することもできる。外来遺伝子として疾患の治療用遺伝子を用いてウイルスベクターを調製すれば、このベクターを投与して遺伝子治療を行うことが可能となる。本発明のウイルスベクターの遺伝子治療への応用としては、直接投与による遺伝子発現、間接(ex vivo)投与による遺伝子発現のいずれの方法によっても、治療効果を期待できる外来遺伝子もしくは患者の体内で供給が不足している内在遺伝子等を樹状細胞から発現させることが可能である。また本発明の方法は、再生医療における遺伝子治療ベクターとしても利用できる。
【0036】
以下に搭載遺伝子を例示する。
CD45: SCIDの原因1遺伝子、染色体1q31-32に位置。共通リンパ球CD抗原でSrc kinaseを調節するフォスファターゼでこの欠損によりT、NK欠損型のSCIDとなる(Hermiston. et al., Annu. Rev. Immunol. 2003: 21; 107-137; Buckley RH., Annu. Rev. Immunol. 2004: 22; 625-655)
IL-7 receptor alpha: SCIDの原因1遺伝子、染色体5p13に位置。リンパ球特にT細胞の分化、ホメオスターシスに重要なIL-7のレセプターのサブユニット、この欠損によりT欠損型のSCIDとなる(Peschon JJ., J. Exp. Med. 1994: 180; 1955-1960; Buckley RH., Annu. Rev. Immunol. 2004: 22; 625-655)
RAG1/2: SCIDの原因1遺伝子、染色体11p13に位置。T細胞レセプター及び抗体の生成において重要なリコンビナーゼで、この欠損によりT、B欠損型のSCIDとなる(schwarz. et al., Science. 1996: 274; 97-99; Buckley RH., Annu. Rev. Immunol. 2004: 22; 625-655)
Adenosine Deaminase (ADA): SCIDの原因1遺伝子、染色体20q13.11に位置。プリン代謝の再利用経路における酵素で、この遺伝子欠損によりプリン代謝産物の蓄積によりリンパ球がアポトーシスとなりT、NK欠損型のSCIDとなる (Noguchi M., Cell, 1999: 73; 147-157; Puck JM.,Hum. Mol. Gemet., 1993: 1099-1104; Buckley RH., Annu. Rev. Immunol. 2004: 22; 625-655)
common gamma chain (γc) : SCIDの原因1遺伝子、染色体Xq13.1に位置。IL-2, IL-4, IL7, IL-7, IL-15, IL-21のレセプターに共通するサブユニットで、T、B、NK欠損型のSCID(いわゆるX-linked SCID)となる (Buckley RH. et al., J. Pediatr. 1997: 130; 378-387; Buckley RH. et al., N, Engl. J. Med. 1999: 340; 508-516, Buckley RH., Annu. Rev. Immunol. 2004: 22; 625-655)
DAF及びCD59:Glycosil phosphatidylinositol linked proteinで補体活性を抑制する蛋白で両者の欠損は、赤血球の自己の補体活性による溶血を惹起し、重症の夜間血色素尿症を発症する(Johnson RJ. et al., J. Clinic. Pathol. , 2002: 55; 145-152)
copper transporting P-type ATPase (ATP7B, Wilson disease protein):ウイルソン病の原因遺伝子。染色体13に位置し、胆管での銅代謝に重要な蛋白で、この欠損により肝臓、角膜、脳に銅が沈着して発症する (Ferenci P., Clin liver Dis. 1998: 2; 31-49)
GFP(オワンクラゲ蛍光発色体)、luciferase:これらは遺伝子導入細胞のマーカーとして生体内での動態の解析や遺伝子導入効率の判定に利用できる。
FGF-2:(WO2002/042481)
その他、Btk(伴性無ガンマグロブリン血症で欠損)、CD40/CD40 ligand(高IgM症候群で欠損)、(Cooper MD. et al., Hematology, 2003: 313-330)、WASP(Wiskott-Aldrich症候群で欠損;Snapper SB. et al., Annu. Rev. immunol. 1999: 17; 905-929)、などが挙げられるが、これらの限定されない。
【0037】
ベクターに搭載する外来遺伝子の発現レベルは、その遺伝子の上流(マイナス鎖(ネガティブ鎖)の3'側)に付加する転写開始配列の種類により調節することができる(WO01/18223)。また、ゲノム上の外来遺伝子の挿入位置によって制御することができ、マイナス鎖の3'の近くに挿入するほど発現レベルが高く、5'の近くに挿入するほど発現レベルが低くなる。このように、外来遺伝子の挿入位置は、該遺伝子の所望の発現量を得るために、また前後のウイルス蛋白質をコードする遺伝子との組み合わせが最適となる様に適宜調節することができる。一般に、外来遺伝子の高い発現が得られることが有利と考えられるため、外来遺伝子は、効率の高い転写開始配列に連結し、マイナス鎖ゲノムの3'端近くに挿入することが好ましい。具体的には、3'リーダー領域と3'に最も近いウイルス蛋白質ORFとの間に挿入される。あるいは、3'に一番近いウイルス蛋白質遺伝子と2番目のウイルス蛋白質遺伝子のORFの間、または3'から2番目と3番目のウイルス蛋白質遺伝子の間に挿入してもよい。野生型パラミクソウイルスにおいては、ゲノムの3'に最も近いウイルス蛋白質遺伝子はN遺伝子であり、2番目の遺伝子はP遺伝子、3番目の遺伝子はM遺伝子である。逆に、導入遺伝子の高発現が望ましくない場合は、例えば外来遺伝子の挿入位置をマイナス鎖ゲノムのなるべく5'側に設定したり、転写開始配列を効率の低いものにするなどして、ウイルスベクターからの発現レベルを低く抑えることで適切な効果が得られるようにすることも可能である。
【0038】
外来遺伝子をコードする核酸をゲノムに挿入するときに付加するS配列としては、例えばマイナス鎖RNAウイルスの所望のS配列を用いることができるが、センダイウイルスであれば、3'-UCCCWVUUWC-5'(W= AまたはC; V= A, C, またはG)(配列番号:2)の配列を好適に用いることができる。特に 3'-UCCCAGUUUC-5'(配列番号:3)、3'-UCCCACUUAC-5'(配列番号:4)、および 3'-UCCCACUUUC-5'(配列番号:5)が好ましい。これらの配列は、プラス鎖をコードするDNA配列で表すとそれぞれ 5'-AGGGTCAAAG-3'(配列番号:6)、5'-AGGGTGAATG-3'(配列番号:7)、および 5'-AGGGTGAAAG-3'(配列番号:8)である。センダイウイルスベクターのE配列としては、例えば 3'-AUUCUUUUU-5'(配列番号:9)(プラス鎖をコードするDNAでは 5'-TAAGAAAAA-3'(配列番号:10))が好ましい。I配列は、例えば任意の3塩基であってよく、具体的には 3'-GAA-5'(プラス鎖DNAでは 5'-CTT-3')を用いればよい。
【0039】
マイナス鎖RNAウイルスベクターを製造するには、(a)哺乳動物細胞において、マイナス鎖RNAウイルスのゲノムRNAを含むRNPの再構成に必要なウイルス蛋白質の存在下、マイナス鎖RNAウイルスのゲノムRNAをコードするcDNAを転写させる工程、(b)生成したマイナス鎖RNAウイルスを回収する工程、により製造することができる。上記のRNPの再構成に必要なウイルス蛋白質とは、典型的には N、P、およびL蛋白質を言う。転写によりマイナス鎖ゲノム(すなわちウイルスゲノムと同じアンチセンス鎖)を生成させてもよく、あるいはプラス鎖(アンチゲノム。ゲノムRNAの相補鎖。)を生成させても、ウイルスRNPを再構成することができる。ベクターの再構成効率を高めるには、好ましくはプラス鎖を生成させる。RNA末端は、天然のウイルスゲノムと同様に3'リーダー配列と5'トレイラー配列の末端をなるべく正確に反映させることが好ましい。このためには、例えば転写産物の5'端に自己切断型のリボザイムを付加しておき、リボザイムによりマイナス鎖RNAウイルスゲノムの末端を正確に切り出させることにより実現させることができる。あるいは、転写産物の5'端を正確に制御するために、転写開始部位としてバクテリオファージのRNAポリメラーゼ認識配列を利用し、該RNAポリメラーゼを細胞内で発現させる。転写産物の3'端を制御するには、例えば転写産物の3'端に自己切断型リボザイムをコードさせておき、このリボザイムにより正確に3'端が切り出されるようにすることができる(Hasan, M. K. et al., J. Gen. Virol. 1997: 78; 2813-2820、Kato, A. et al., EMBO J. 1997,: 16; 578-587 及び Yu, D. et al., Genes Cells. 1997: 2; 457-466)。リボザイムとしては、デルタ肝炎ウイルスのアンチゲノム鎖(antigenomic strand)由来の自己開裂リボザイムが使用できる。
【0040】
より具体的な組み換えウイルスの再構成は、例えば以下の文献を参照することができる(WO97/16539; WO97/16538; WO00/70055; WO00/70070; WO01/18223; WO03/025570; Durbin, A. P. et al. Virology, 1997: 235; 323-332; Whelan, S. P. et al. Proc. Natl. Acad. Sci. USA, 1995: 92; 8388-8392; Schnell. M. J. et al., EMBO J. 1994: 13; 4195-4203; Radecke, F. et al., EMBO J. 1995: 14; 5773-5784; Lawson, N. D. et al., Proc. Natl. Acad. Sci. USA 92: 4477-4481; Garcin, D. et al. EMBO J. , 1995: 14; 6087-6094; Kato, A. et al., Genes Cells. 1996: 1; 569-579; Baron, M. D. and Barrett, T., J. Virol. 1997: 71; 1265-1271; Bridgen, A. and Elliott, R. M., Proc. Natl. Acad. Sci. USA. 1996: 93; 15400-15404; Hasan, M. K. et al., J. Gen. Virol., 1997: 78; 2813-2820、Kato, A. et al., EMBO J. 1997: 16; 578-587、Yu, D. et al., Genes Cells, 1997: 2; 457-466、Tokusumi, T. et al. Virus Res. 2002: 86; 33-38、Li, H.-O. et al., J. Virol. 2000: 74; 6564-6569)。これらの方法により、パラインフルエンザ、水疱性口内炎ウイルス、狂犬病ウイルス、麻疹ウイルス、リンダーペストウイルス、センダイウイルスなどを含むマイナス鎖RNAウイルスをDNAから再構成させることができる。
【0041】
ウイルス生産細胞でウイルス蛋白質やウイルスゲノムRNAを発現させるためにプラスミドベクターをトランスフェクトする場合、例えばリン酸カルシウム法(Graham, F. L. and Van Der Eb, J., Virology. 1973: 52; 456; Wigler, M. and Silverstein, S., Cell. 1977: 11; 223; Chen, C. and Okayama, H. Mol. Cell. Biol., 1987: 7; 2745)、種々のトランスフェクション試薬を用いた方法、あるいは電気穿孔法等を用いることができる。トランスフェクション試薬については、DEAE-デキストラン(Sigma #D-9885 M.W. 5×105 )、DOTMA(Roche)、Superfect(QIAGEN #301305)、DOTAP、DOPE、DOSPER(Roche #1811169)、TransIT-LT1 (Mirus, Product No. MIR 2300) などを用いることができる。トランスフェクション試薬とDNAとの複合体がエンドソーム中で分解されてしまうのを防ぐため、クロロキンを加えることができる(Calos, M. P., Proc. Natl. Acad. Sci. USA 1983: 80; 3015)。
【0042】
温度感受性変異を有する蛋白質を用いる場合は、組み換えウイルスの産生を許容温度(例えば35℃以下、より好ましくは34℃以下、さらに好ましくは33℃以下、最も好ましくは32℃またはそれ以下)で行う。また、野生型蛋白質を用いる場合でも、ウイルス産生を低温で行うことで効率的な粒子形成を行わせることが可能である。エンベロープ構成蛋白質遺伝子を欠損する組み換えウイルスを製造する場合は、ウイルス産生細胞において、これら欠損させた遺伝子および/または他のウイルスのエンベロープ蛋白質をコードする遺伝子などを細胞に導入して発現させることにより、感染性のウイルス粒子を形成させることができる(Hirata, T. et al., J. Virol. Methods, 2002: 104; 125-133; Inoue, M. et al., J. Virol. 2003 : 77; 6419-6429; Inoue M. et al., J Gene Med. 2004;6:1069-1081)。
【0043】
例えば、ウイルス産生において、水疱性口内炎ウイルス(Vesicular stomatitis virus; VSV)のG蛋白質(VSV-G)やレトロウイルスのアンフォトロピックエンベロープ蛋白質などのヒト細胞に感染するウイルスに由来するエンベロープ蛋白質を用いることができる。VSV-G蛋白質は、任意のVSV株に由来するものであってよい。例えば Indiana血清型株(J. Virology, 1981: 39; 519-528)由来のVSV-G蛋白を用いることができるが、これに限定されない。このようにマイナス鎖RNAウイルスベクターは、他のウイルス由来のエンベロープ蛋白質を任意に組み合わせて製造されたものであってよい。アンフォトロピックエンベロープ蛋白質としては、例えばマウス白血病ウイルス(MuLV)4070A株由来のエンベロープ蛋白質を用い得る。また、MuMLV 10A1由来のエンベロープ蛋白質を用いることもできる(例えばpCL-10A1(Imgenex)(Naviaux, R. K. et al., J. Virol. 1996: 70; 5701-5705)。また、ヘルペスウイルス科の蛋白質としては、例えば単純ヘルペスウイルスのgB、gD、gH、gp85蛋白質、EBウイルスのgp350、gp220蛋白質などが挙げられる。ヘパドナウイルス科の蛋白質としては、B型肝炎ウイルスのS蛋白質などが挙げられる。これらの蛋白質は、細胞外ドメインをF蛋白質またはHN蛋白質の細胞内ドメインと結合させた融合蛋白質として用いてもよい。このように本発明において用いられるウイルスベクターには、VSV-G蛋白質などのように、ゲノムが由来するウイルス以外のウイルスに由来するエンベロープ蛋白質を含むシュードタイプウイルスベクターが含まれる。ウイルスのゲノムRNAにはこれらのエンベロープ蛋白質をゲノムにコードされないように設計すれば、ウイルス粒子が細胞に感染した後は、ウイルスベクターからこの蛋白質が発現されることはない。
【0044】
また、エンベロープ表面に特定の細胞に接着しうるような接着因子、リガンド、受容体等の蛋白質、抗体またはその断片、あるいはこれらの蛋白質を細胞外領域に有し、マイナス鎖RNAウイルスのエンベロープ蛋白質由来のポリペプチドを細胞内領域に有するキメラ蛋白質などを含むウイルスを製造することもできる。これにより、ウイルスベクターの感染の特異性を制御し得る。これらはウイルスゲノムにコードされていてもよいし、ウイルスの再構成時に、ウイルスゲノム以外の遺伝子(例えば別の発現ベクターまたは宿主染色体上などにある遺伝子)からの発現により供給されてもよい。
【0045】
エンベロープ構成蛋白質を発現するヘルパー細胞は、例えば欠損させたエンベロープ構成蛋白質あるいは別のエンベロープ蛋白質をコードする遺伝子をトランスフェクションすることにより作製することができる(WO00/70055 および WO00/70070;Hasan, M. K. et al., J. General Virology. 1997: 78; 2813-2820参照)。誘導発現を可能にするために、例えばCre/loxP誘導型発現プラスミドpCALNdlw(Arai, T. et al., J. Virology, 1998: 72; 1115-1121)等の組み換え酵素標的配列を持つベクターにエンベロープ蛋白質遺伝子を組み込むことが好ましい。細胞は、例えばサル腎臓由来細胞株LLC-MK2細胞(ATCC CCL-7)を用いることができる。LLC-MK2細胞は、10%の熱処理した非動化ウシ胎児血清(FBS)、ペニシリンGナトリウム 50単位/ml、およびストレプトマイシン 50 micro-g/mlを添加したMEMで37℃、5% CO2で培養する。SeV-F遺伝子産物は細胞傷害性を有するため、Cre DNAリコンビナーゼによりエンベロープ蛋白質遺伝子産物を誘導発現されるように設計された上記プラスミドpCALNdLw/Fを、リン酸カルシウム法(mammalian transfection kit (Stratagene))により、周知のプロトコールに従ってLLC-MK2細胞に遺伝子導入を行う。限界希釈により細胞をクローニングした後、細胞を拡大培養し、導入遺伝子の高発現細胞株の選別を行う。このためには、例えばアデノウイルスAxCANCreを斉藤らの方法(Saito et al., Nucl. Acids Res. 1995: 23; 3816-3821; Arai, T.et al., J. Virol. 1998: 72; 1115-1121)により例えば moi=3〜5 で感染させ、ウェスタンブロッティングまたはウイルス生産により、細胞を選択する。
【0046】
回収されたウイルスの力価は、例えばCIU(Cell Infecting Unit)測定または赤血球凝集活性(HA)の測定することにより決定することができる(WO00/70070; Kato, A. et al., Genes Cells. 1996: 1; 569-579; Yonemitsu, Y. & Kaneda, Y., Hemaggulutinating virus of Japan-liposome-mediated gene delivery to vascular cells. Ed. by Baker AH. Molecular Biology of Vascular Diseases. Method in Molecular Medicine: Humana Press. 1999: 295-306; Kiyotani, K. et al., Virology. 1990: 177; 65-74)。また、GFP(緑色蛍光蛋白質)などのマーカー遺伝子を搭載したベクターについては、マーカーを指標に直接的に感染細胞をカウントすることにより力価を定量することができる(例えばGFP-CIUとして)。このようにして測定した力価は、CIUと同等に扱うことができる(WO00/70070)。
【0047】
ウイルスが再構成する限り、再構成に用いる宿主細胞は特に制限されない。例えば、サル腎由来のLLC-MK2細胞およびCV-1細胞 (例えばATCC CCL-70)、ハムスター腎由来のBHK細胞 (例えばATCC CCL-10) などの培養細胞、ヒト由来細胞等を使うことができる。これらの細胞に適当なエンベロープ蛋白質を発現させることで、その蛋白質をエンベロープに有する感染性ウイルス粒子を得ることもできる。また、大量にウイルスベクターを得るために、上記の宿主から得られたウイルスベクターを発育鶏卵に感染させ、ベクターを増幅することができる。鶏卵を使ったウイルスベクターの製造方法は既に開発されている(中西ら編,(1993),「神経科学研究の先端技術プロトコールIII, 分子神経細胞生理学」, 厚生社, 大阪, pp.153-172)。具体的には、例えば、受精卵を培養器に入れ9〜12日間 37〜38℃で培養し、胚を成長させる。ウイルスベクターを尿膜腔へ接種し、数日間(例えば3日間)卵を培養してウイルスベクターを増殖させる。培養期間等の条件は、使用する組み換えセンダイウイルスにより変わり得る。その後、ウイルスを含んだ尿液を回収する。尿液からのセンダイウイルスベクターの分離・精製は常法に従って行うことができる(田代眞人,「ウイルス実験プロトコール」, 永井、石浜監修, メジカルビュー社, pp.68-73,(1995))。
【0048】
回収したウイルスベクターは実質的に純粋になるよう精製することができる。精製方法はフィルトレーション(濾過)、遠心分離、吸着、およびカラム精製等を含む公知の精製・分離方法またはその任意の組み合わせにより行うことができる。「実質的に純粋」とは、ウイルスベクターを含む溶液中で該ウイルスの成分が主要な割合を占めることを言う。例えば実質的に純粋なウイルスベクター組成物は、溶液中に含まれる全蛋白質(但しキャリアーや安定剤として加えた蛋白質は除く)のうち、ウイルスベクターの成分として含まれる蛋白質の割合が10% (重量/重量) 以上、好ましくは20%以上、より好ましくは50%以上、好ましくは70%以上、より好ましくは80%以上、さらに好ましくは90%以上を占めることにより確認することができる。例えばパラミクソウィルスベクターであれば、具体的な精製方法としては、セルロース硫酸エステルまたは架橋ポリサッカライド硫酸エステルを用いる方法(特公昭62-30752号公報、特公昭62-33879号公報、および特公昭62-30753号公報)、およびフコース硫酸含有多糖および/またはその分解物に吸着させる方法(WO97/32010)等を例示することができるが、これらに制限されない。
【0049】
上記のマイナス鎖RNAウイルスベクターを造血細胞に感染させ、造血細胞移植組成物を製造することができる。本発明は、マイナス鎖RNAウイルスベクターを用いた造血細胞移植組成物の製造方法、および造血細胞移植組成物の製造におけるマイナス鎖RNAウイルスベクターの使用に関する。
【0050】
造血細胞とは、白血球、赤血球、または血小板の少なくともいずれかの血液細胞に分化し得る細胞を言う。造血細胞には、骨髄細胞、造血前駆細胞、造血幹細胞(hematopoietic stem cell;HSC)などが含まれる。本発明において用いる造血細胞は、好ましくは白血球、赤血球、および血小板の少なくとも3系統に分化する能力を持つ細胞である。造血前駆細胞とは、血液細胞の少なくともいずれかの系統に分化できる細胞であり、血液細胞の各系統へ分化が進み、多分化能を消失した細胞(リンパ球系、顆粒球系、赤血球系、血小板系へ分化する細胞)も含まれる。造血幹細胞とは、自己複製能と多分化能を有する細胞で、少なくとも白血球、赤血球、および血小板の3系統に分化する能力を持つ細胞である。造血細胞は、体性細胞(神経系、肝臓、心臓など)へ分化することのできるものであってもよい(Weissman IL. et al., Science, 2000: 287; 1442-1446; Lagasse E. et al., Immunity, 2001: 14; 425-436; Anderson DJ. et al. Nat Med, 2001: 7; 393-395)。
【0051】
各血液細胞は、その形態や染色の特異性から同定することが可能である。染色法としては、ギムザ染色、ライト染色、メイ−ギムザ染色、メイ−グリュンワルド−ギムザ染色、ペルオキシダーゼ染色、エラスターゼ染色、PAS染色等を用いることができる(新生化学実験講座8, 血液(上), 1987, 8-15ページ参照)。
【0052】
造血幹細胞は、骨髄のみならず末梢血および臍帯血中にも存在する。本発明において造血細胞移植とは、造血細胞を含む細胞組成物を移植することを言い、骨髄移植(BMT)、末梢血幹細胞移植(PBSCT)、および臍帯血幹細胞移植(CBSCT)などが含まれる。すなわち本発明は、骨髄細胞移植のみならず、末梢血造血幹細胞移植および臍帯血移植においても適用することができる。HSCの同定は、顆粒球、単球の前駆細胞をコロニーアッセイ法で算定したり、HSCに特徴的と考えられるCD34(抗原)陽性細胞をフローサイトメーターで測定することにより実施することができる。具体的には、造血細胞としては、マウスであれば c-kit+、Thy1.1low、lin-、Sca-1+(Osawa M, Hanada K, Hamada H, Nakauchi H. Science. 1996; 273: 242-245.)、ヒトではCD34+、Thy+、Lin-、c-kitlow(Bhatia M, Wang JC, Kapp U, Bonnet D, Dick JE. Proc Natl Acad Sci U S A. 1997; 94:5320-5325)の表現型を有する細胞が挙げられる。lin- (lineage negative) の表現型は、適宜市販のキット等を用いて同定および選択することができるが、例えば GPA、CD3、CD2、CD56、CD24、CD19、CD14、CD16、および CD99b が全てnegativeのものである。
【0053】
ヒト造血幹細胞の分離方法は、以下の文献も参照することができる(Leary AG, Blood 69: 953, 1987; Sutherland HJ, Blood 74: 1563, 1989; Andrews RG, J Exp Med 169: 1721. 1989; Terstappen LWMM, Blood 77: 1218, 1991; Lansdorp PM, J Exp Med 175: 1501, 1992; Briddell RA, Blood 79: 3159, 1992; Gunji Y, Blood 80: 429, 1992; Craig W, J Exp Med 177: 1331, 1993; Gunji Y, Blood 82: 3283, 1993; Traycoff CN, Exp Hematol 22: 215, 1994; Huang S, Blood 83: 1515, 1994; DiGinsto D, Blood 84: 421, 1994; Murray L, Blood 85: 368, 1995; Hao QL, Blood 86: 3745, 1995; Laver JH, Exp Hematol 23: 1515, 1995; Berardi AC, Science 267: 104, 1995; Kawashima I, Blood 87: 4136, 1996; Leemhuis T, Exp Hematol 24: 1215, 1996; Civin CI, Blood 88: 4102, 1996; Larochelle A, Nature Med 2: 1329, 1996; Tajima S, J Exp Med 184: 1357, 1996; Sakabe H, Stem Cells 15: 73, 1997; Sakabe H, Leukemia 12: 728, 1998; 原田実根他編, 新しい造血幹細胞移植, 南江堂, 1998, 9-23ページ)。
【0054】
例えばマウスの骨髄細胞は、大腿骨、及び頸骨より骨髄細胞をフラッシュにて採取し、Lysing buffer(0.38% NH4Cl in Tris-HCl, pH7.65)にて赤血球除去後、抗Thy1.2抗体およびウサギ補体を用いた抗体補体反応による成熟T細胞の除去後に得られる(Tomita Y, Mayumi H, Eto M, Nomoto K. J Immunol. 1990; 144: 463-473.)。または、採取骨髄細胞を抗B220(B細胞)抗体、抗CD3(T細胞)抗体、抗Gr-1(顆粒球)抗体、抗Mac-1(マクロファージ)抗体、抗DX-5(NK細胞)抗体等の抗体でビーズあるいは遠心法にてlineageを除去して得られる(Sato T, Laver JH, Ogawa M. Blood. 1999; 94: 2548-2554.)。
【0055】
ヒト造血細胞を骨髄より採取する場合は、全身麻酔下、腹臥位で、腸骨の左右それぞれ3〜5ヶ所から、1回に数ml程度の骨髄液を採取する(末梢血混入を避けるため)。採取量は同種移植の場合では細胞数は3(2〜5)×108/kg(患者体重あたり)、自家移植の場合で1〜3×108/kg を目標として骨髄採取する。骨組織などの混入があるためメッシュを通した後,バッグにつめる。移植の際には粗いフィルターを用いて輸注する。同種移植の場合は、ヒト白血球抗原(human lymphocyte antigen;HLA)適合ドナーを選択することが好ましい。但し、GVHD予防法の確立、新しい免疫抑制剤の開発、およびCD34陽性細胞の純化技術などにより、HLA不一致のドナーでも移植は可能になってきている。ドナーとしては、HLA-A、-B、および-DRの3組6抗原のうち、4種類以上、より好ましくは5種類以上、より好ましくは全てがレシピエントと一致することが好ましい。ABOmajor不適合移植の場合、遠心分離により赤血球を除去したものを用いる。ABOminor不適合の場合、遠心分離にて血漿を除去したものを用いる。(森下ら編、『造血幹細胞移植マニュアル』第2版、小寺、斎藤監修、日本医学館、東京、(1999年)p260-277、参照 )。
【0056】
末梢血より末梢血幹細胞を採取する場合は、G-CSF 1 日400μg/m2(又は10μg/kg)を1 回、または2 回に分割し、5 日間または採取終了時まで連日皮下注射し、投与開始4日目から6日目までの期間に1から3 回、末梢血(静脈)から血球分離装置を用いて造血幹細胞を採取することによって得られる(日本造血細胞移植学会のウェブページにおける造血幹細胞移植に関するガイドラインの説明資料を(2003年4月21日改訂第3版)を参照)。末梢血幹細胞(PBSC)の動員については、Harada M et al., J. Hematother 5: 63-71, 1996、Waller CF et al., Bone Marrow Transplant 18: 279-283, 1996、Anderlini P et al., Blood 90: 903-908, 1997、原田実根他編, 新しい造血幹細胞移植, 南江堂, 1998, 67-72ページ、名古屋BMTグループ編, 造血細胞移植マニュアル改訂三版, 2004, 237-240ページも参照のこと。動員に用いるG-CSFとしては、野生型蛋白質の他、N末端を改変した誘導体(nartograstinなど)、糖鎖が付加された修飾蛋白質(lenograstinなど)を用いることができる。またG-CSFと他の造血因子を併用してもよい。例えば、GM-CSF、IL-3、またはSCFなどを併用することができる(Huhn RD et al., Exp Hematol 24: 839-847, 1996; Begley CG et al., Blood 90: 3378-3389, 1997; Lane TA et al., Blood 85: 275-282, 1995)。G-CSF投与中は、腰痛、骨痛および発熱などの全身症状の軽減や、血小板凝集能亢進の防止のため、aspirinを投与することができる。
【0057】
ヒト末梢血造血幹細胞(CD34+細胞)を得るには、上記で得られた末梢血幹細胞からCD34陽性細胞をCD34抗体磁気ビーズでの濃縮法Isolex system(Baxter社など)、CliniMACS(AmCell社)、アビジンカラムを通しての濃縮法(CEPRATE、Cellpro社)、CD34抗体をフラスコに固定化し、反応する細胞をフラスコに吸着させ、残りを洗い流す(CELLECTOR、AIS社)等の方法でCD34陽性細胞を分離採取することで得られる(森下ら編、『造血幹細胞移植マニュアル』第2版、小寺、斎藤監修、日本医学館、東京、(1999年)p260-277、参照 )。
【0058】
ヒト臍帯血細胞移植では、採取した臍帯血に、赤血球沈降剤血球を加え、重量によって各血液画分に分け、移植時には不要となる血漿画分、赤血球画分を除き、必要な造血幹細胞を得る。更にCD34陽性細胞をIsolex system(Baxter社)、CliniMACS(AmCell社)、アビジンカラムを通しての濃縮法(CEPRATE、Cellpro社)、CD34抗体をフラスコに固定化し、反応する細胞をフラスコに吸着させ、残りを洗い流す(CELLECTOR、AIS社)等の方法によって純化することで臍帯血CD34陽性造血幹細胞を得ることができる(森下ら編、『造血幹細胞移植マニュアル』第2版、小寺、斎藤監修、日本医学館、東京、(1999年)、p661-680、参照 )。
【0059】
単離した造血細胞は、例えばメチルセスロース法により、SCF、IL-3、GM-CSF、G-CSF、Epoを添加した培養することができる(Sonoda Y. et al., Blood 84: 4099-4106, 1994; Kimura T. et al., Blood 90: 4767-4778, 1997)。造血細胞は1×102から1×104/ml程度で添加し、例えば 37℃、5% CO2、5% O2 下で培養する。但し、培養条件は適宜調整してよい。
【0060】
自家移植においては、簡便性の点から末梢血幹細胞移植が選択されることが多いが、自家骨髄移植も適用される。その場合、通常は、化学療法の影響のない造血回復期に、骨髄の正常造血を確認後、全身麻酔下に腸骨および胸骨から採取する。目標細胞数は、骨髄有核細胞3×108から5×108/kg程度である。連続式血液成分分離装置で得られた細胞浮遊液は、顆粒球や赤血球の混入が非常に少ないため、単核球分離操作を省略することができる。顆粒球や赤血球が多く含まれる場合には、Ficoll比重遠心法により単核球を分離することができる。骨髄液の場合は、顆粒球や赤血球の除去と濃縮のため、Ficoll処理または血液分離装置による単核球分離を行うことができる。
【0061】
混入した血小板をそのまま輸注すると塞栓などの原因となる場合があるため、採取後に除去することが好ましい。例えばダブルバックに集めて弱遠心(200g、15分)するか、遠心管に分注して1600回転、10分遠心して血小板を除去し、RPMI1640培地に交換することができる。採取した細胞を凍結保存する場合は、10%自己血清、10%DMSOを含むRPMI1640培地に浮遊させて凍結させ、programmed freezerで凍結、液体窒素中で保存する。細胞濃度は2×107から6×107/mlとすることができる。比較的短期間の細胞保存のためには、細胞浮遊液(1×108/ml以下の濃度)に終濃度 6% Hydrodyethyl Starch (HES)、5% DMSO、4% albumin となるように、氷冷した等量の保存液を混合し、-80℃のdeep freezerで凍結保存することができる(Knudsen LM et al., J. Hematother. 5: 399-406, 1996)。
【0062】
安全な自家移植に必要なCD34陽性細胞数は、およそ2×106/kgである(Schots R et al., Bone Marrow Transplant. 17: 509-515, 1996; Zimmerman TM et al., Bone Marrow Transplant. 15: 439-444, 1995)。CD34陽性細胞数は、通常の2 color flowcytometryで計測することができる。具体的には、骨髄細胞や末梢血アフェレーシス細胞を溶血処理により赤血球を除去した後、forward scatterと抗CD45抗体で展開し、gateを決めて赤血球および血小板を除く。次に、このgateをside scatterと抗CD34抗体で展開し、好中球などの非特異反応部分を除いた分画中の細胞数の全細胞数に対する割合で算定する。この値と、予め血球計測機で測定した血球数から、全CD34陽性細胞数が算出できる。
凍結保存した細胞に含まれるviableな幹細胞の数は、コロニー形成法によりCFU-GMを数える方法で知ることができる。移植に必要なCFU-GMの基準は、一般的には 1×105〜2×105/kgである。
【0063】
上記のように調整した造血細胞をマイナス鎖RNAウイルスベクターと接触させることにより、該マイナス鎖RNAウイルスベクターを造血細胞に導入することができる。マイナス鎖RNAウイルスベクターと造血細胞との接触は、体内(in vivo)または体外(in vitroまたはex vivo) で行うことができ、例えば培養液、生理食塩水、血液、血漿、血清、体液など所望の生理的水溶液中で実施すればよい。
【0064】
ベクターと造血細胞との接触においては、MOI(多重感染度; 細胞1つあたりの感染ウイルス数)は1〜500の間にすることが好ましく、より好ましくは2〜300、さらに好ましくは3〜200、さらに好ましくは5〜100、さらに好ましくは7〜70である。ベクターと造血細胞との接触は短い時間でも十分であり、例えば1分以上、好ましくは3分以上、5分以上、10分以上、または20分以上接触させればよく、例えば1〜60分程度、より特定すれば5分〜30分程度であってよい。もちろん、それ以上の時間接触させてもよく、例えば数日間またはそれ以上接触させてもよい。
【0065】
ベクターを導入した造血細胞は、薬理学的に許容される所望の担体または媒体と組み合わせて造血細胞移植組成物とすることができる。「薬学的に許容される担体または媒体」とは、生細胞を懸濁できる溶液であれば制限はない。例えばリン酸緩衝生理食塩水(PBS)、塩化ナトリウム溶液、リンゲル溶液、培養液等が挙げられる
【0066】
また造血細胞移植組成物は、ベクターを導入していない造血細胞が含まれていてもよい。ベクターを導入した造血細胞と、ベクターを導入していない造血細胞を混合することにより、造血細胞の生着効率は上昇し得る。混合比率は、それに限定されないが、例えばベクター導入造血細胞:非導入造血細胞を1:10〜10:1の間で適宜調整してよい。
【0067】
また本発明は、マイナス鎖RNAウイルスベクターが導入された造血細胞から血液細胞を再構築させる方法に関する。この方法は、(a)マイナス鎖RNAウイルスベクターを造血細胞に接触させる工程、および(b)工程(a)の造血細胞を動物に注入する工程、を含む。マイナス鎖RNAウイルスベクターは細胞質ベクターであり、造血細胞の染色体にインテグレートすることはないため、レトロウイルスのような染色体損傷のリスクがない。しかも、本発明において、マイナス鎖RNAウイルスは造血細胞の細胞質中で複製し、造血細胞が分裂しても娘細胞にベクターが分配され、再構築された血液細胞中でも導入遺伝子の発現が持続できることが証明された。
【0068】
血液細胞とは、血液中の細胞成分を言い、具体的には白血球、赤血球、および血小板が含まれる。血液細胞を生成させるとは、これらの血液細胞のいずれか、好ましくは白血球、赤血球、および血小板の全てを生成させることである。また白血球は、顆粒球、単球、リンパ球が含まれ、本発明において白血球とは、それらのいずれかの細胞を意味する。本発明の方法により、マイナス鎖RNAウイルスベクターが導入された造血細胞は3系統への分化が可能である。すなわち造血細胞移植により、マイナス鎖RNAウイルスベクターが導入された造血細胞から白血球、赤血球、および血小板が生成する。
【0069】
造血細胞の移植は、適宜周知の移植プロトコルに従って実施することができる。造血細胞移植の対象となる動物に特に制限はないが、例えば所望の哺乳動物が挙げられ、例えばマウス、ラット、イヌ、ブタ、ネコ、ウシ、ウサギ、ヒツジ、ヤギ、サルなどの非ヒト哺乳動物およびヒトが含まれる。例えばマウスにおいては、投与細胞数は1匹あたり、5.0 x 106 から2.0 x 107、好ましくは約1.0x107、好ましくは約2.0x107を尾静脈、陰茎静脈、または眼底静脈よりone shot静脈注射にて投与することができる(Tomita Y, Mayumi H, Eto M, Nomoto K. J Immunol. 1990; 144: 463-473.)。ヒトの骨髄細胞移植では、個体あたり、2 x 108/kgから 3 x 108/kg、好ましくは約3 x 108/kgを経静脈的に点滴投与する。ヒトの末梢血造血幹細胞移植では、個体あたり自家造血幹細胞移植では2 x 106/kg以上、同種移植では5 x 106/kg以上、好ましくは、自家造血幹細胞移植では約5 x 106/kg、同種移植では約10 x 106/kgを経静脈的に点滴投与する。ヒトの臍帯血造血幹細胞移植では、個体あたり、移植必要数は2 x 107/kg以上、好ましくは約4 x 107/kgを経静脈的に点滴投与する。(森下ら編、『造血幹細胞移植マニュアル』第2版、小寺、斎藤監修、日本医学館、東京、(1999年)p260-277、p661-680、参照 )。
【0070】
輸液への血小板の混入による血栓や塞栓を防止するため、抗ヒスタミン剤(hydroxyzine 25mg または chlorphenirmine 5mgなど)または副腎皮質ステロイド(hydrocortisone 100mg)などの前投与を行うことができる。移植に凍結細胞を用いる場合、移植細胞溶液中の赤血球の溶血が問題となる場合には、ハプトグロビン製剤4,000単位の点滴などで尿細管障害を予防することができる。末梢血幹細胞移植の場合は、2日間に分けて輸注することも好ましい。
【0071】
ドナー造血細胞の生着を促進するため、造血細胞の注入前に、レシピエントを免疫抑制しておくことが好ましい。例えば白血病における骨髄移植では、白血病細胞の死滅と宿主の免疫担当細胞の不活化のため、造血細胞の移植前に全身照射(total body irradiation; TBI)を行うことができる。このような処置は、一般に、前処理(Preparative regimen)またはコンディショニング(Conditioning)とも呼ばれる。TBIは急性・慢性のいずれの白血病でも対象となる。このとき、メルファラン、シクロホスファミド(CPA)などの抗癌剤と併用することができる。しかしTBIはさまざまな障害の原因にもなるため、これらを避けるためにブスルファンとCPAのBUCY法など抗癌剤だけの前処置を選択することもできる(Inoue, T. et al., Strahlenther Onkol 169: 250-255, 1993; Blaise, D. et al., Blood. 1992: 79; 2578-2582; Blume, KG. et al., Blood. 1993: 81; 2187-2193; Hartman, A. et al., Bone Marrow Transplant. 1998: 22; 439-443)。また、骨髄移植と同時にDonor Lymphocyte Infusion(DLI)を行うことで、前処置の線量を軽減することが可能である。
【0072】
放射線照射を行う場合は、長SAD(Source axis distance)(またはSSD (Source skin distance)法、スイープビーム法、ビーム移動法、治療寝台移動法等の照射方法を適用することができる。このうち、長SAD(SSD)法は、特殊な装置を必要としない点で簡便であり、良く用いられている。照射は、1回照射または分割照射であってよいが、分割照射の方が正常細胞の回復が期待できる。1回照射は、例えばヒトであれば一回あたり10Gy(6cGy/min)、分割照射であれば、12Gy(6cGy/min)を6回/6日、1.8Gy/回を2〜3回/日(総線量15Gy程度)、または2〜3Gy/回で1〜2回/日(総線量12Gy程度)などが例示できるが、線量は適宜調整されてよい。
【0073】
また、前処置を行わなくても、ドナー造血細胞を多数注入すれば、造血細胞が骨髄に生着することが動物実験により証明されている。これは、ドナー造血細胞の生着には必ずしも骨髄破壊的前処置は必要がないことを示している。これを基に、宿主の免疫抑制のみを実施する骨髄非破壊的前処置を用いる造血幹細胞移植、いわゆるミニ移植(Mini-transplant: ミニトランスプラント)が開発されており、これに本発明を適用することも可能である。ミニ移植は骨髄破壊的前処置を伴う移植とは異なり合併症も軽いため、高齢者や臓器障害のある患者にまで適応が拡大される。本発明において免疫抑制には、TBIなどの骨髄破壊的処置のみならず、免疫抑制剤などによる骨髄非破壊的処置が含まれる。前処置に用いる免疫抑制剤としては、フルダラビン、クラドリビンなどの抗癌剤や抗胸腺グロブリンが用いられる。このような前処置後に造血幹細胞移植を行うことで、患者とドナーの造血細胞が混在する骨髄の混合キメラ、もしくはすべてドナーの造血細胞に入れ替わった骨髄となる完全キメラを作り出すことが可能である。再生不良性貧血のような非悪性疾患では、骨髄非破壊的前処置による混合キメラを適用できるが、白血病などの腫瘍では大量のリンパ球を注入して完全キメラ化する。骨髄破壊を伴わない造血細胞移植が有効と考えられる疾患としては、例えば腎癌、悪性黒色腫、低悪性度リンパ腫、再生不良性貧血などがある。再発リスクの高い腫瘍に対しては骨髄破壊の方が好ましい。ミニ移植の前処置で照射を行う場合は、例えば1〜2Gy/1回程度の小線量TBIが適用される。また、従来の前処置とミニ移植との中間的な前処置である、reduced conditioning療法も開発されている。これらの前処理を本発明において用いることもできる。
【0074】
移植において抗癌剤の大量投与や全身照射を行った場合は、全身の造血が抑えられ、感染に対する抵抗力が著しく低下する。従って、移植期間中は無菌室で過ごし、外界からの感染を予防することが好ましい。但し、HSCのソースとして末梢血を用いる同種末梢血幹細胞移植(allogeneic peripheral blood stem cell transplantation;allo-PBSCT)を行うことで早期の造血能回復が得られるため、完全無菌室管理を行わなくとも移植は可能になっている。移植が成功すれば、約20日程度で白血球数は増加する。一般に、末梢血幹細胞移植のほうが骨髄移植より回復が早い。血球の増加が少ない場合は、さらに造血幹細胞移植を実施することができる。
【0075】
自家移植の場合は、移植した造血幹細胞の数が十分あれば生着に問題はないが、同種移植の場合、ドナーとレシピエントが遺伝学的に同一でない限り、移植片が持つ抗原に対しレシピエントのT細胞が反応し生着不全を起こす。そのため、移植前の前治療には、ドナーのHSCが生着するための免疫抑制効果が必要となる。また逆にHSCが生着してドナー由来の造血能が回復すると、ドナー由来のT細胞がレシピエントの細胞を攻撃し移植片対宿主病(GVHD)が発症する。GVHDは同種移植後の生命予後を決定する重要な因子となるため、発症予防のために免疫抑制療法が行われる。急性GVHDは通常同種移植後2〜6週間以内に発症する。また、サイトメガロウィルスなどの感染症や免疫抑制剤によるmicroangiopathyも併発することもある。診断には、皮膚や胃、十二指腸、大腸、直腸などからの生検が有用である。中等症以上の場合は、副腎皮質ステロイドによる治療を行う。ステロイドに反応しない場合は、抗胸腺細胞グロブリンやタクロリムスなどの免疫抑制剤を用いる。血縁者間の移植では、シクロスポリンとメトトレキサート、非血縁者間ではタクロリムスとメトトレキサートが予防薬として用いられることが多い。慢性GVHDは、典型的には移植後100日前後に、同種移植を受けた長期生存者中に一定の割合で発症する。臨床所見としては、強皮症、扁平苔癬、Sjogren症候群、原発性胆汁性肝硬変などの自己免疫疾患によく似ており、口唇や皮膚の生検、肝生検などによって診断が可能である。治療としてはシクロスポリンと副腎皮質ステロイドが主体で、皮膚病変に対してはPUVA(Psoralens with Ultraviolet A)などの光線療法を実施することもできる。
【0076】
本発明における造血細胞移植は、例えば再生不良性貧血、白血病(急性骨髄性白血病および急性リンパ性白血病などの急性白血病、並びに慢性骨髄性白血病など)、骨髄異形成症候群、リンパ腫(ホジキン病、非ホジキンリンパ腫などを含む)、骨髄腫などの血液疾患、化学療法が有効な悪性腫瘍(乳がん、胚細胞腫などを含む)、先天性疾患などの治療として実施することができる。適応疾患については、単一遺伝子欠損あるいは発現系異常(プロモーターの欠損、あるいは制御異常によるものなど)、または2つ以上の遺伝子欠損あるいは発現系異常による疾患においても適応可能である。また、血球系の異常のみならず、骨髄幹細胞より分化可能とされる神経系細胞、肝細胞などの体細胞の遺伝子異常を要因とする疾患についても適応となる。また、遺伝子異常疾患のみならず、分泌蛋白などの供給細胞として用いた場合に治療効果が期待できる疾患にも応用可能である。例えば、単一遺伝子欠損症としては、X-linked SCID、ADA欠損症、血友病、無免疫グロブリン血症、高IgM血症、慢性肉芽腫症など、複数遺伝子欠損症に関しては、発作性夜間血色素尿症などが挙げられ、それぞれの原因遺伝子を造血幹細胞に遺伝子導入後、細胞移植を行うことで治療効果が期待できる。造血細胞移植における遺伝子治療が有効と考えられる疾患およびその遺伝子を例示すれば、例えばHurler病、Scheie病、およびHurler-Scheie病(α-L-iduronidaseまたはiduronate sulfatase)、Sanfilippo病(N-sulfatase、N-acetyl-α-D-glucosaminidase、α-D-flucosaminidase-N-acetyltransferase)、Morquio病(galactosamine-β-sulfate sulfataseまたはβ-galactosidase)、Maroteaux-Lamy症候群(N-acetylgalactosamine-4-sulphatase)、Sly病(β-glucuronidase)、Gaucher病(β-glucocerebrosidase)、Farber病(acid ceramidase)、Niemann-Pick病(acid sphingomyekinase)、Krabbe病(galactocerebroside β-galactosidase)、Metachromatic leucodystrophy(arylsufatase A)、Fabry病(α-galactosidase A)、SCID(adenosine deaminase)、X-linked SCID(gamma-c receptor)、X連鎖無ガンマグロブリン血症(X-linked agammaglobulinemia;XLA)(Bruton's tyrosine kinase)、JAK-3欠損症(Jak-3)、Aspartyglycoaminuria(aspartyglycosaminidase)、Fucosidoses(α-L-fucosidase)、Guibaud-Vainsel症候群(carbonic anhydrase II)、Thallassemias(α-、β-globin)、G6PD欠損症(glucose-6-phosphate dehydrogenase)、PK欠損症(pyruvate kinase L型)、Erythropoietic porphyia(uroporphyrinogen III synthase)などが挙げられる。また、プリンヌクレオシドフォスフォリラーゼ欠損症については原因遺伝子であるpurine nucleoside phosphorylase (PNP)、X連鎖重症複合免疫不全症については IL-2レセプターγ鎖 (γC)、X染色体連鎖性慢性肉芽腫症については NADPHオキシダーゼを構成するサブユニットの1つであるgp91phoxなどが例示できる。また、固形悪性腫瘍や血液悪性腫瘍の造血幹細胞移植の際に、腫瘍特異的な分子を標的とするキメラ毒素の遺伝子導入などによって、抗腫瘍効果の増強などを惹起することが可能と考えられる。また、癌の化学療法の副作用の軽減のために、多剤耐性遺伝子 MDR-1 を導入することも考えられる。さらに、後天性免疫不全症候群(AIDS)の治療のために抗HIV遺伝子(例えばRRE decoy、envアンチセンス)の遺伝子治療が可能である。
【実施例】
【0077】
以下、実施例により本発明をさらに詳細に説明するが、本発明はこれら実施例に制限されるものではない。なお、本明細書中に引用された文献は、すべて本明細書の一部として組み込まれる。
【0078】
使用動物
7週齢雌性C57BL/6Jマウス及び同Balb/cマウス(KBTオリエンタル)、GFPトランスジェニックマウスC57BL/6-TgN(act-EGFP)OsbC14-Y01-FM131(Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y. FEBS Lett. 1997; 407:313-319)をドナーとして用い、同C57BL/6、Balb/c nu/nuをレシピエントとして使用した。
【0079】
ベクター(SeV18+/dF-GFP、SeV18+/dFMtsHNtsPLmut-GFP)の構築
SeV18+/dF-GFP(以下SeV/dF-GFP)のベクターテンプレートとなるプラスミドpSeV18+/dF-GFPは、センダイウイルスZ株の全長cDNAを挿入、リーダー配列と NP 遺伝子間に挿入されたスペーサー配列に人為的に構築したNotI切断部位に供与核酸であるGFP cDNAを挿入し、さらにF 遺伝子領域を削除することにより作成した。SeV18+/dFMtsHNtsPLmut-GFP(以下tsSeV/dF-GFP)については、同様のプラスミドにさらにM遺伝子(G69E, T116A, A183S)、 HN遺伝子(A262T, G264R, K461G)、および P, L遺伝子(P遺伝子: L511F, L遺伝子: N1197S, K1795E)に対し変異を挿入した(Inoue M, et al. J Virol 2003;77:3238-3246; Inoue M, et al. Mol. Ther. 2003; 7(5):S37)。
双方のベクター構築にはマイナス鎖RNAウイルスをcDNAより作製させるためのリバースジェネティクス法を用いた(Inoue M, et. al. J Virol 2003; 77:3238-3246)。具体的には pSeV/dF-GFPあるいはpSeV18+/dFMtsHNtsPLmut-GFPと、N、P、LおよびF蛋白質を発現するプラスミドを同時にサル腎臓由来 LLC-MK2 細胞にトランスフェクションすることにより作成した。F遺伝子産物はベクターの再構成に必要であるため、F蛋白質を継続的に発現しているLLC-MK2細胞を使ってトランスに供給することにより増幅させた。
【0080】
造血細胞への遺伝子導入とマウスへの移植
以下の実験は、全て無菌操作にて施行した。
ドナーマウスの大腿骨より採取した骨髄細胞は、10%FBS + RPMI1640(Gibco BRL)中に23ゲージ針付き注射器でフラッシュした。その後、赤血球をLysing buffer(0.38% NH4Cl in Tris-HCl, pH7.65)にて除去、さらに成熟T細胞を完全に取り除くために抗Thy1.2抗体(マウス腹水IgMモノクローナル抗体、明治乳業株式会社ヘルスサイエンス研究所、東京)とウサギ補体(CL3051, CEDARLANE, Ontario CANADA)を骨髄細胞に反応させた。
T細胞除去骨髄細胞を1 x 107/mlに調節後、重複感染度(multiplicity of infections: MOI)=10でベクターに感染させ、37℃恒温槽で約1時間インキュベートした。その後一部をプレートに播き48時間後フローサイトメトリー(FACSCaliber, Becton Dickinson, CA)にてGFP遺伝子の発現を確認した。採取された骨髄細胞は3回洗浄、遠心後、serum freeのRPMIで希釈調節した。それぞれレシピエントマウス:C57BL/6は 2 x 107/200μl、nu/nu-BALB/Cは 4 x 107/200μlそれぞれ尾静脈から30ゲージ注射器で注入した。このレシピエントマウスは移植4〜6時間前にγ線をそれぞれC57BL/6は致死量の10 Gry、nu/nu-BALB/Cは亜致死量の7 Gry照射した。なお、細胞性免疫抑制のためのFK506投与群は骨髄移植2週間後からFK506 (tacrolimus, 藤沢薬品工業株式会社, 大阪) を5 mg/kg 連日腹腔内投与した。また、no-spleenマウスは移植2日前に開腹し脾臓を除去した。
【0081】
フローサイトメトリー
骨髄移植から1,4,8,12週間後の末梢血を尾静脈から採血し、GFP陽性細胞についてFACSにて解析した。まず、末梢血は赤血球をLysing bufferで除去し、2回HANKS液で洗浄の後約1x105〜1x106/mlに調節した。解析前に、PI (propidium iodide) 染色を行い、死細胞を除外した。また、それぞれのマウスの脾臓、骨髄、胸腺でのGFP陽性細胞分画の確認のため、Fcブロックの後、樹状細胞(DC)と単球/マクロファージ(MФ)の判定のために、anti-CD11bPE (1:4, Biosciences Pharmingen CA)とanti-CD11cAPC (1:5, Biosciences Pharmingen CA)、B細胞(B-cell)判定にはanti-B220APC (1:13, Becton Dickinson CA)とanti-IgMPE (1:10, Becton Dickinson CA)、helper T-cell(hT-cell)とcytotoxic T-cell(cT-cell)の判定のためanti-CD3APC (1:5, Biosciences Pharmingen CA)、anti-CD8PE (1:10,Becton Dickinson CA)もしくはanti-CD4PE (1:10, Becton Dickinson CA)、そしてナチュラルキラー(NK)細胞、NKT細胞と、T細胞(T-cell)の判定のためanti-CD3APCとanti-DX5PE (1:5, Biosciences Pharmingen CA) 抗体を用いて30分4℃下で染色し,FACS Caliber(Becton Dickinson CA) にて解析を行った。データはFlowJo(TreeStar Inc., San Carlos, CA)もしくはCellQuestTM software(Becton Dickinson CA)を用いて評価した。
【0082】
血清中抗SeV抗体価の測定
骨髄移植から1, 4, 8, 12週間後、マウスの尾静脈から末梢血を採血管で採取し、常温で30分間静置後、10,000rpmで5分間遠心分離した。その上清をストックし-80℃で保管した。検査時に室温で解凍し、血清中の抗SeV抗体価をMONILISA (R) HVJ Enzyme-linked Immunosorbent Assay kit (Wakamoto Pharmaceutical Co. Ltd., 東京) を用いて測定した。
【0083】
血液検査
骨髄移植から1, 4, 8, 12週間後、マウスの尾静脈から末梢血を採血管で採取し、末梢血20μlに対して、血球計算用希釈液50μlで希釈し、ジダ血モードで全自動血球計数器(CelltacαMEK-6158, 日本光電, 東京)を用いて検査した。
【0084】
CTL活性の測定
レシピエントマウスのspleenをスライドガラスにてすりつぶし、lysing後10%FCS入りRPMI + ペニシリン + DM添加培地にて5 x 106/ml希釈し、SeVペプチド 1 mMと混合した後、24穴プレートに播種、5日間培養した。培養3日目では培地交換とともにIL-2を培地1mlにつき3μl(30U/ml 以上)添加し、IL-2と48時間共培養した。
標的細胞(EL4: American Type Culture Collections)は1 x 106/200μlに調整し、SeVペプチドを50μM加えた。さらに、0.1mCiのNa251CrO3を細胞に加え、よく振った後に10%CO2条件下で、15分ごとに振りながら1.5時間インキュベーションした。
5日間培養した細胞を回収し3穴ずつ1 x 106、5 x 105、2.5 x 105、1.25 x 105でU底96穴プレートに播種した。これに標的細胞を100μlずついれ、4時間インキュベーションし、2回洗い後それぞれをγカウンターにかけ、以下の計算式にてSeV特異的Na251CrO3リリースを算出した。
%specific release = ((experimental cpm - spontaneous cpm)/(maximum cpm - spontaneous cpm)) × 100
最大遊離量は1% triton X-100 (Wako Pure Chemical Industries Ltd., Osaka, Japan)を使用した細胞溶解によって得た。自然遊離量は細胞を0.2mlの培養液のみで培養することで得た。自然遊離量は最大遊離量の15% から20% の範囲であった。それぞれの個体は3検体ずつ作成した。
SeVペプチドとしては、既に報告されているCTLの標的配列となり、クラスI抗原に提示されることが明らかとなっているNP蛋白のアミノ酸配列NH2-FAPGNYPAL-COOH(配列番号:11)を合成して用いた。なお、tsSeV/dF-GFPのNP蛋白には、同部位の変異は存在しない。
【0085】
統計解析
統計分析値はすべて平均値±標準誤差として表現した。データはMann Whitney U-testを用いて解析し、P<0.05を統計学的有意差とした。
【0086】
〔実施例1〕マウス造血細胞に対するSeVの遺伝子導入効率と同細胞の生着効率
第1に、C57BL6マウス骨髄細胞(T細胞除去後)に対するSeV/dF-GFPならびにtsSeV/dF-GFPによる遺伝子導入効率を検討した(図1a)。MOI=10にて各々のベクター液を骨髄細胞培養液へ添加、1時間後に新鮮培養液にて洗浄後、48時間後のGFP陽性率をFACSにて解析した。図1に典型的解析図を示す。SeV/dF-GFPによるGFP陽性率は80.7±2.9%(n=3)、tsSeV/dF-GFPによるそれは88.8±1.9%(n=18)であり、いずれも80%以上の高い遺伝子導入効率を示した。
次にSeVにてGFP遺伝子を導入した骨髄細胞の放射線照射同種マウスに対する生着効率を、末梢血細胞におけるGFP陽性率を指標として検討した(図1b)。陽性対照であるGFPトランスジェニックマウス(GFP-TG)をドナーとした場合、生着率は経過中常に98%以上であり、この効率は移植後4週目〜8週目も維持されていた(n=3)。SeV/dF-GFPにて処置した骨髄細胞を移植した場合、移植早期(1週目)より末梢血におけるGFP陽性率は陽性対照に比べ低く(0.1-2.3%, n=9)、以後GFP陽性細胞は消失した。一方でtsSeV/dF-GFPで処理した骨髄細胞を移植した場合、移植早期には比較的高いGFP陽性率を示した(45-85%, n=10)。その後GFP陽性率は次第に漸減し、移植後4週目では、個体差はあるものの(2.0-26%)、依然平均で10%以上の末梢血細胞がGFP陽性であった。
【0087】
〔実施例2〕tsSeV/dF-GFPにて遺伝子導入された造血細胞のレシピエントにおける生体内分布ならびに各細胞分画におけるGFP陽性率
図1において比較的生着効率のよいマウス6頭を用いて、GFP発現細胞が造血系のいずれに分布するかを検討した(図2)。この6頭の末梢血における5週目のGFP陽性率は55.9±5.9%であり、脾臓、胸腺で比較的高かったが、下肢大腿骨骨髄では17.1±8.8%であった。
次に脾臓と骨髄において各細胞分画におけるGFP陽性細胞率をFACSにて解析した(図3)。各細胞分画の同定には、NK cells=CD3-/DX5+、NKT cells= CD3+/DX5+、whole T-cells= CD3+/DX5-、helper T-cells= CD3+/CD4+、cytotoxic T-cells= CD3+/CD8+、dendritic cells (DCs)= CD11b+/CD11c+、monocytes/macrophages(MΦ)= CD11b+/CD11c-、B-cells= B220+/IgM+を用いた。脾臓・骨髄共にNK、DC、MΦ、Bは比較的陽性率が高かった。NKT、Tについては陽性率は比較的低いものの、移植細胞のT細胞分画はほぼ完全に除去されていることより、レシピエントの脾臓・骨髄で検出されたGFP陽性NKT/T細胞は、移入された分化したNKT/T細胞ではなく、むしろ造血幹細胞由来のものと考えられた。
【0088】
〔実施例3〕tsSeV/dF-GFPにて遺伝子導入された造血細胞の5週目以降のレシピエントにおける生着動態
以上の結果から、tsSeV/dF-GFPによって遺伝子導入された骨髄細胞は、同種レシピエントにおいてrepopulation可能であることが示された。しかしGFP陽性率は次第に減少することが明らかになった。このGFP陽性細胞数の減少をより詳細に検討するため、より長期の生着動態について検討した。
計53頭のC57BL6マウスへtsSeV/dF-GFPにて処置した骨髄細胞を同様の方法で移植したところ、約40%のマウスが移植後5〜7週で死亡した(図4a)。これらのマウスについて、移植後1週目及び4週目で末梢血を採取しGFP陽性細胞を確認したが、死亡したマウスの4週目の採血において、全例が比較的良好なGFP陽性率(10%以上)に加え血液の希釈が観察された。そこで移植後4週目における生着率のよいマウスでの血球数を検討した。
図4bに示すように、無処置マウス(n=6)、及びGFP-TGマウス骨髄を移植されたレシピエントマウス(n=6)と比較して、tsSeV/dF-GFPにて処置した骨髄細胞を移植されたレシピエントマウス(全6頭中4週目の末梢血中GFP陽性細胞10%以上:n=3)では移植後4週目における白血球数(WBC)、赤血球数(RBC)、血小板(PLT)全てが著明に減少していた。
【0089】
〔実施例4〕tsSeV/dF-GFPにて遺伝子導入された移植造血細胞に対するレシピエントにおける免疫学的反応の検討
tsSeV/dF-GFP遺伝子導入骨髄を移植されたレシピエントマウスにおける汎血球減少の原因を検討するために、同レシピエントマウスにおけるSeVに対する免疫反応について検索を行った。
4-1.細胞性免疫反応
tsSeV/dF-GFP遺伝子導入骨髄を移植されたレシピエントマウスにおける4週目の末梢血GFP陽性細胞率と、同個体におけるセンダイウイルス感染細胞に対する特異的細胞障害性T 細胞活性(CTL活性)を検討した。
図5aに示すごとく、末梢血GFP陽性細胞率が高い個体(64.9%)では有意なCTL活性を認めるものの、そのレベルは低値に留まっていた。
図5aで得られたCTL活性の軽度の上昇がtsSeV/dF-GFP由来GFP陽性細胞数の減少に有意に関与するか否かを検討するため、主に細胞性免疫を抑制するFK506の連日投与(移植後2週目より、5 mg/kg/day)、及びヌードマウスにおける細胞生着動態を検討した。図5bに示すごとく、双方において細胞生着率の上昇は認められなかったことから、tsSeV/dF-GFP遺伝子導入骨髄を移植されたレシピエントマウスにおける遺伝子導入細胞の排除へのCTLの関与は、比較的低いものと考えられた。
【0090】
4-2.液性免疫反応
次に、tsSeV/dF-GFP遺伝子導入骨髄を移植されたレシピエントマウス(n=34)における液性免疫反応について、4週目の末梢血GFP陽性細胞率と抗SeV抗体の関係について検討した。
図6aに示すごとく、34頭中11頭は5週目にて死亡し、23頭は長期生存した。これらから移植後4週目に採取した末梢血GFP陽性細胞数と同時期の抗SeV抗体価を検討すると、GFP陽性率が高い個体では抗SeV抗体価が低く、またGFP陽性率が低い個体では抗SeV抗体価が高い傾向を認めた(図6aおよび6b)。本発明で使用した抗SeV抗体検出用ELISAシステムはSeVの膜タンパクを認識するものであるため、tsSeV/dF-GFPが感染した骨髄細胞に発現したわずかな膜タンパクに対し、レシピエントが抗体を産生したものと考えられた。
【0091】
〔実施例5〕遺伝子導入ならびに非導入細胞混合造血細胞の移植後動態に関する検討
最後に、tsSeV/dF-GFP遺伝子導入骨髄細胞のレシピエント骨髄中あるいは脾臓の血球細胞の中で、少なくとも一部に除去したはずであるドナー由来のGFP陽性T細胞が存在することから、これらGFP陽性T細胞はtsSeV/dF-GFPにより遺伝子されたドナー細胞中の造血幹細胞に由来する可能性があると考えられる。一方でtsSeV/dF-GFP遺伝子導入骨髄細胞のレシピエントには、比較的高いrepopulationのまま死亡する群と、4週以降速やかに排除されていく群が存在することから、前者は造血幹細胞の多くへ遺伝子導入され、後者では遺伝子導入効率が低いのではないか、と仮説を立てた。
これを検討するため、tsSeV/dF-GFPによる遺伝子導入骨髄細胞と非導入骨髄細胞を等量混合し、レシピエントマウスへ移植した(図7)。本実験では遺伝子導入のみの細胞を移植された群では全てのレシピエントマウスが8週目までに死亡したが、混合移植群では5頭全例においてGFP陽性末梢血細胞率が4週以降に速やかに低下して行った。この結果は上記の仮説に矛盾しない結果であると考えられた。
【産業上の利用可能性】
【0092】
本発明により、染色体非傷害型の細胞質型ベクターを用いた遺伝子導入造血細胞による造血細胞移植が可能になった。本発明の方法は、宿主染色体内へのインテグレーションを介さないため、癌抑制遺伝子の不活化や癌遺伝子の活性化などの危険を伴わずに遺伝子を導入することができるため、安全なベクターシステムを構築することが可能である。先天性免疫不全性疾患には造血細胞移植が有効性を示さない症例が存在するため、これらの治療には遺伝子治療のみが残されたオプションとなる。本発明の方法を、これらの難治性疾患に対する新たな遺伝子治療へ適用することが期待される。
【図面の簡単な説明】
【0093】
【図1】a. フローサイトメトリーによるT細胞除去ドナー(C57BL6マウス)骨髄細胞へのSeV/dF-GFP及びtsSeV/dF-GFPによる遺伝子導入効率解析。代表的プロファイルを示す図である。SeV/dF-GFPの遺伝子導入効率は80.7±2.9%(n=3)、tsSeV/dF-GFPの場合は88.8±1.9%(n=18)であった。b. C57BL6系統GFPトランスジェニックマウス(GFP-TG)より採取した骨髄、あるいはSeV/dF-GFPもしくはtsSeV/dF-GFPで遺伝子導入したC57BL6マウス骨髄を同種レシピエントへ移植した場合の、末梢血血球細胞におけるGFP陽性率の経時的変化を示す図である。縦軸は対数表示である。
【図2】フローサイトメトリーによる、tsSeV/dF-GFPで遺伝子導入したC57BL6マウス骨髄を同種レシピエントへ移植した場合の、各種臓器におけるGFP陽性率(移植後5週、計6頭)を示す図である。図1で解析した個体のうち、4週目の末梢血にて生着率のよいものを選択して解析した。代表的プロファイルを示す。括弧内の数字は、平均値±標準誤差%(n=6)を示す。
【図3】フローサイトメトリーによる、tsSeV/dF-GFPで遺伝子導入したC57BL6マウス骨髄を同種レシピエントへ移植した場合の、脾臓・骨髄における各血球細胞分画におけるGFP陽性率(移植後5週、計6頭)を示す図である。
【図4】a. tsSeV/dF-GFPで遺伝子導入したC57BL6マウス骨髄を同種レシピエントへ移植した場合の、生存率の経時的変化(計53頭)を示す図である。b. 非処理C57BL6マウス、tsSeV/dF-GFPで遺伝子導入したあるいはGFPトランスジェニックマウス骨髄を同種レシピエントへ移植した場合の、末梢血における白血球数(WBC)、赤血球数(RBC)、血小板数(PLT)(移植後4週)を示す図である。tsSeV/dF-GFPのみに著明な汎血球減少を認める。
【図5】a. tsSeV/dF-GFPで遺伝子導入したC57BL6マウス骨髄を同種レシピエントへ移植した場合の、SeV特異的CTL活性(移植後4週)を示す図である。括弧内の数字は、各個体における同時期の末梢血中GFP陽性細胞率を示す。各個体につき、3つ培養ウェルに標的細胞を播種、各データは3ウェルの平均値、エラーバーは標準誤差を示す。b. tsSeV/dF-GFPで遺伝子導入したC57BL6マウス骨髄を同種レシピエントへ移植した場合(n=10)、また同様のマウスに移植後2週目よりFK506 (5 mg/kg/day)を腹腔内連日投与(n=7)、そしてtsSeV/dF-GFPで遺伝子導入したbalb/cマウス骨髄を同種ヌードマウスへ移植した場合の末梢血GFP陽性細胞の経時的変化を示す図である。いずれの操作においても、GFP陽性率の低下を阻止できなかった。
【図6】a. tsSeV/dF-GFPで遺伝子導入したC57BL6マウス骨髄を同種レシピエントへ移植した場合(総個体数n=34)の、5週以内死亡群(n=11、白三角)及び長期生存群(n=23、黒四角)における末梢血GFP陽性細胞率(縦軸)と血清中抗SeV抗体価(横軸)の相関(移植後4週)を示す図である。死亡個体群では、末梢血GFP陽性率が高く、さらに抗SeV抗体価が低いことに注意。b. 上記a.に示したデータを2群(死亡群、生存群)に分けてそれぞれGFP陽性率(左)、抗SeV抗体価(右)を比較したバーグラフである。エラーバーは標準誤差を示す。
【図7】tsSeV/dF-GFPで遺伝子導入したC57BL6マウス骨髄を、単独で同種レシピエントへ移植した場合(ts-BMT, n=3)及び同骨髄を同量の無処置骨髄と混合して移植した場合(ts-Mix-BMT, n=5)における、末梢血GFP陽性細胞率の経時的変化を示す図である。ts-BMT群では、比較的GFP陽性率は保たれたものの、全例が8週以内に死亡した。一方、混合細胞移植群では、速やかにGFP陽性細胞率が減少し、全例が長期生存した。縦軸は対数表示である。
【特許請求の範囲】
【請求項1】
造血細胞移植組成物の製造方法であって、マイナス鎖RNAウイルスベクターを造血細胞に導入し、該造血細胞および薬学的に許容される媒体を含む組成物を調製することを含む方法。
【請求項2】
マイナス鎖RNAウイルスベクターが、エンベロープ構成蛋白質をコードする1つまたは複数の遺伝子が変異または欠損している、請求項1に記載の方法。
【請求項3】
エンベロープ構成蛋白質をコードするすべての遺伝子が変異または欠損している、請求項2に記載の方法。
【請求項4】
エンベロープ構成蛋白質をコードする少なくとも1つの遺伝子が欠損している、請求項2に記載の方法。
【請求項5】
マイナス鎖RNAウイルスがパラミクソウイルス科ウイルスである、請求項1に記載の方法。
【請求項6】
パラミクソウイルス科ウイルスがセンダイウイルスである、請求項5に記載の方法。
【請求項7】
請求項1から6のいずれかに記載の方法により得られる造血細胞移植組成物。
【請求項8】
遺伝子導入された血液細胞を造血細胞から生成させる方法であって、
(a)マイナス鎖RNAウイルスベクターを造血細胞に接触させる工程、および
(b)工程(a)の造血細胞を動物に注入する工程、を含む方法。
【請求項9】
工程(b)の注入前に該動物を免疫抑制する工程をさらに含む、請求項8に記載の方法。
【請求項10】
マイナス鎖RNAウイルスベクターにおいて、エンベロープ構成蛋白質をコードする1つまたは複数の遺伝子が変異または欠損している、請求項8に記載の方法。
【請求項11】
エンベロープ構成蛋白質をコードするすべての遺伝子が変異または欠損している、請求項10に記載の方法。
【請求項12】
エンベロープ構成蛋白質をコードする少なくとも1つの遺伝子が欠損している、請求項10に記載の方法。
【請求項13】
マイナス鎖RNAウイルスがパラミクソウイルス科ウイルスである、請求項8に記載の方法。
【請求項14】
パラミクソウイルス科ウイルスがセンダイウイルスである、請求項13に記載の方法。
【請求項1】
造血細胞移植組成物の製造方法であって、マイナス鎖RNAウイルスベクターを造血細胞に導入し、該造血細胞および薬学的に許容される媒体を含む組成物を調製することを含む方法。
【請求項2】
マイナス鎖RNAウイルスベクターが、エンベロープ構成蛋白質をコードする1つまたは複数の遺伝子が変異または欠損している、請求項1に記載の方法。
【請求項3】
エンベロープ構成蛋白質をコードするすべての遺伝子が変異または欠損している、請求項2に記載の方法。
【請求項4】
エンベロープ構成蛋白質をコードする少なくとも1つの遺伝子が欠損している、請求項2に記載の方法。
【請求項5】
マイナス鎖RNAウイルスがパラミクソウイルス科ウイルスである、請求項1に記載の方法。
【請求項6】
パラミクソウイルス科ウイルスがセンダイウイルスである、請求項5に記載の方法。
【請求項7】
請求項1から6のいずれかに記載の方法により得られる造血細胞移植組成物。
【請求項8】
遺伝子導入された血液細胞を造血細胞から生成させる方法であって、
(a)マイナス鎖RNAウイルスベクターを造血細胞に接触させる工程、および
(b)工程(a)の造血細胞を動物に注入する工程、を含む方法。
【請求項9】
工程(b)の注入前に該動物を免疫抑制する工程をさらに含む、請求項8に記載の方法。
【請求項10】
マイナス鎖RNAウイルスベクターにおいて、エンベロープ構成蛋白質をコードする1つまたは複数の遺伝子が変異または欠損している、請求項8に記載の方法。
【請求項11】
エンベロープ構成蛋白質をコードするすべての遺伝子が変異または欠損している、請求項10に記載の方法。
【請求項12】
エンベロープ構成蛋白質をコードする少なくとも1つの遺伝子が欠損している、請求項10に記載の方法。
【請求項13】
マイナス鎖RNAウイルスがパラミクソウイルス科ウイルスである、請求項8に記載の方法。
【請求項14】
パラミクソウイルス科ウイルスがセンダイウイルスである、請求項13に記載の方法。
【図3】


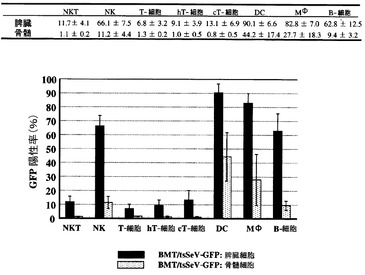
【図4】
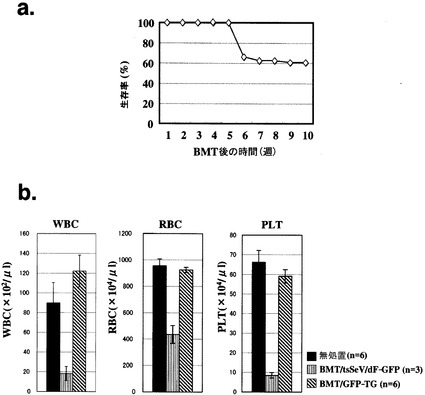
【図5】
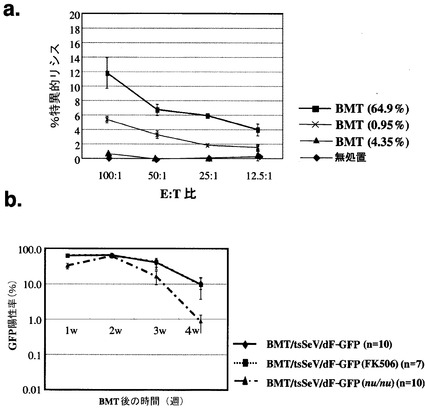
【図6】
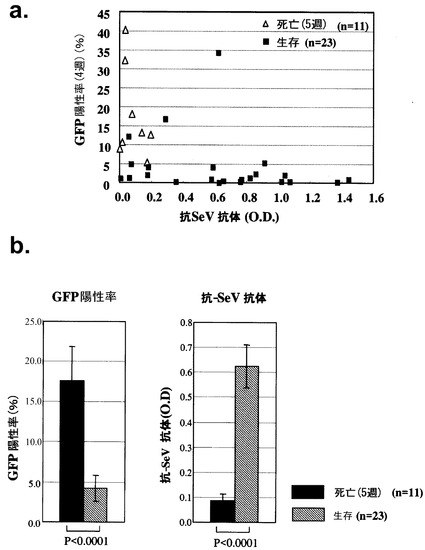
【図7】
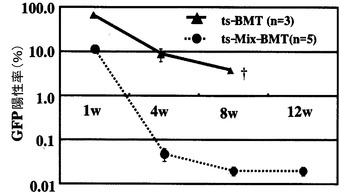
【図1】
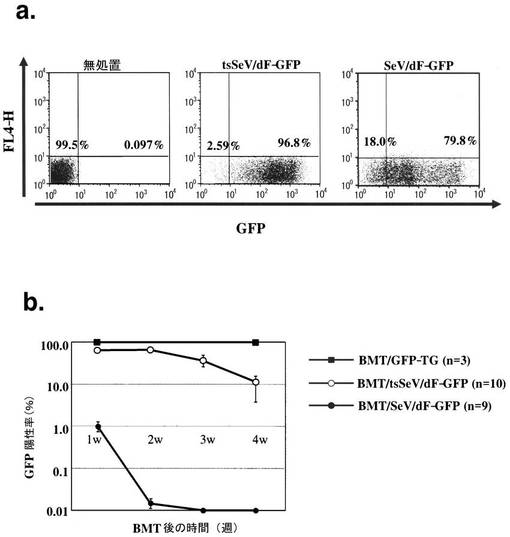
【図2】
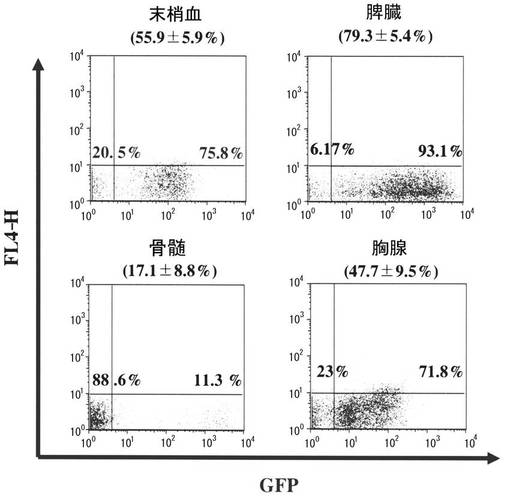
【図4】
【図5】
【図6】
【図7】
【図1】
【図2】
【公開番号】特開2008−105946(P2008−105946A)
【公開日】平成20年5月8日(2008.5.8)
【国際特許分類】
【出願番号】特願2005−32205(P2005−32205)
【出願日】平成17年2月8日(2005.2.8)
【出願人】(505048482)ディナベック株式会社 (5)
【Fターム(参考)】
【公開日】平成20年5月8日(2008.5.8)
【国際特許分類】
【出願日】平成17年2月8日(2005.2.8)
【出願人】(505048482)ディナベック株式会社 (5)
【Fターム(参考)】
[ Back to top ]