
遺伝子欠損株及びそれを用いたタンパク質の製造方法
【課題】培養中の溶菌の減少と、培養液への目的タンパク質又はポリペプチドの分泌促進との両立を可能にする枯草菌変異株、並びに、当該枯草菌変異株を用いた目的タンパク質又はポリペプチドの製造方法の提供。
【解決手段】特定の遺伝子欠失を組み合わせた枯草菌変異株。
【解決手段】特定の遺伝子欠失を組み合わせた枯草菌変異株。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規な遺伝子欠損株、及びそれを用いたタンパク質の製造方法に関する。
【背景技術】
【0002】
微生物による有用物質の工業的生産は、アルコール飲料や味噌、醤油等の食品類、あるいはアミノ酸、有機酸、核酸関連物質、抗生物質、糖質、脂質、タンパク質等、その種類は多岐にわたっており、またそれらの用途も、食品、医薬品、洗剤、化粧品等の日用品、あるいは各種化成品原料等、幅広い分野にわたっている。
【0003】
微生物を用いた有用物質の工業的生産における一つの重要課題として、当該有用物質の生産性向上が挙げられる。従来、当該課題を解決するため、突然変異等の遺伝学的手法による高生産菌の育種が行われてきた。特に最近では、微生物遺伝学、バイオテクノロジーの発展により、遺伝子組換え技術等を用いた、より効率的な高生産菌の育種が行われている。更に、近年のゲノム解析技術の急速な発展を受け、対象とする微生物のゲノム情報を解読し、これらを積極的に産業に応用する試みもなされている。
【0004】
微生物は元来、自然界における環境変化に対応するための多種多様な遺伝子群を有しており、限定された生産培地が使用されるタンパク質等の工業的生産においては、必ずしも生産効率が高いとは言えない状況であった。
【0005】
しかるところ、バチルス属細菌において、胞子形成初期に関わる遺伝子を欠失又は不活性化した菌株が構築され、タンパク質やポリペプチドの生産性向上効果が得られている。例えば、枯草菌のsigE、sigF、spoIIE、spoIISB、sigG、又は、spoIVCBからspoIIICまでの領域に含まれる遺伝子群を欠失した宿主菌株を用いることによって、セルラーゼなどの分泌生産性が向上することが開示されている(特許文献1)。
【0006】
しかしながら、産業的なタンパク質やポリペプチド生産の場においては生産コストをできるだけ低減化する必要があり、このためには更に高い生産性が求められている。この点、上記の各遺伝子は胞子形成の第II期以降に発現し機能する遺伝子であるため、当該遺伝子を欠失や不活性化した場合に於いても細胞は胞子形成の初期段階に入っており、ゆえにタンパク質やポリペプチド生産の観点から改善の余地がある。また、胞子形成期の初期段階を制御するspo0A遺伝子(BG10765)を欠失又は不活性化することによって胞子形成が初期段階で停止することが知られているが、同時に激しい溶菌現象が生じるという問題が存在する。
【0007】
近年、枯草菌ゲノムに存在する約4100種類の遺伝子の破壊株が網羅的に研究され、271個の遺伝子が成育に必須であることが報告され(非特許文献1)、また本出願人は遺伝子組換えのための宿主微生物として、枯草菌(Bacillus subtilis) Marburg No.168系統株の遺伝子破壊株を網羅的に解析し、枯草菌ゲノムの大量域を欠失させ、各種酵素の生産性に優れた宿主変異株の創出に成功している(特許文献2)。
【0008】
また、ある種の微生物では、幾つかの細胞外プロテアーゼ遺伝子を欠失させた菌株が構築されており、例えば、nprE遺伝子の欠失により溶菌の指標となる細胞内酵素イソクエン酸デヒドロゲナーゼの若干の溶出が見られ、更にaprE遺伝子の欠失を組み合わせることにより、その溶出が顕著になることが示されている(特許文献3)。すなわち、複数の細胞外プロテアーゼの欠失を組み合わせることにより枯草菌の溶菌が生じるという問題が存在する。
【0009】
この点、枯草菌の主要な細胞壁溶解酵素CwlB(N−アセチルムラモイル−アラニンアミダーゼ、以前はLytCと呼ばれていた)をコードするcwlB遺伝子(BG10407、以前はlytC遺伝子)を不活性化することによる溶菌現象の抑制が報告されている(非特許文献3)。しかしながら、細胞壁溶解酵素群は細胞分裂や運動性など細胞の増殖にとり重要な役割を果たすといわれ、その欠失や不活性化によって細胞の生育に大きな変化が生じ、タンパク質又はポリペプチドの生産にも影響することが懸念される。例えば、予備検討データではあるが、細胞壁溶解酵素をコードするcwlB(lytC)遺伝子又はcwlG(lytD)遺伝子の不活性化によってタンパク質の分泌が抑制されたとの報告も存在する(非特許文献4)。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】特開2003−47490号公報
【特許文献2】特開2007−130013号公報
【特許文献3】特開2009−38985号公報
【非特許文献】
【0011】
【非特許文献1】K.Kobayashiら、Proc.Natl.Acad.Sci.USA,100,4678−4683,2003
【非特許文献2】Letters in Applied Microbiology,29,141−145(1999)
【非特許文献3】J.Bacteriol.,173,7304−7312(1991)
【非特許文献4】Microbiology,146,249−262(2000)
【発明の概要】
【発明が解決しようとする課題】
【0012】
本発明は、細胞外プロテアーゼ遺伝子の欠失株であっても目的タンパク質又はポリペプチドの培養液からの回収を容易にし、その生産性向上を可能とする枯草菌変異株の提供、並びに、当該枯草菌変異株を用いた目的タンパク質又はポリペプチドの製造方法の提供に関する。
【課題を解決するための手段】
【0013】
本発明者らは、枯草菌の各種遺伝子欠失株について検討したところ、細胞外プロテアーゼ遺伝子の欠失と溶菌関連遺伝子又は遺伝子群の欠失とを組み合わせた枯草菌変異株を構築することにより、細胞外プロテアーゼ遺伝子の欠失株と同等以上の目的タンパク質又はポリペプチドの分泌生産性が得られ、且つ当該枯草菌変異株の溶菌が起こらないため、、その結果、目的タンパク質又はポリペプチドの培養液からの回収が容易になることを見出した。
【0014】
すなわち本発明は、以下の(1)〜(7)に係るものである。
(1) epr、wprA、mpr、nprB、bpr、nprE、vpr、aprE及びaprXから選択される細胞外プロテアーゼ遺伝子が8個以上欠失又は不活性化し、かつ、cwlB、cwlF、PBSX、spoIIIC及びsigDから選択される溶菌酵素関連遺伝子又は遺伝子群が1個以上欠失又は不活性化している枯草菌変異株。
(2) 前記細胞外プロテアーゼ遺伝子が9個欠失又は不活性化している、前記(1)記載の枯草菌変異株。
(3) 細胞外プロテアーゼ遺伝子epr、wprA、mpr、nprB、bpr、nprE、vpr、aprE及びaprXと、溶菌酵素関連遺伝子又は遺伝子群cwlB、cwlF、PBSX、spoIIIC及びsigDと、が全て欠失又は不活性化している枯草菌変異株。
(4) 更に、異種タンパク質又はポリペプチドをコードする遺伝子を有する、前記(1)から(3)のいずれか1つに記載の枯草菌変異株。
(5) 前記異種タンパク質又はポリペプチドをコードする遺伝子の上流に転写開始制御領域、翻訳開始制御領域、又は分泌用シグナル領域が結合している、前記(4)記載の枯草菌変異株。
(6) 前記転写開始制御領域、翻訳開始制御領域又は分泌シグナル領域が、バチルス属細菌のセルラーゼ遺伝子と当該セルラーゼ遺伝子の上流0.6〜1kb領域に由来するものである、前記(5)記載の枯草菌変異株。
(7) 転写開始制御領域、翻訳開始制御領域又は分泌シグナル領域が、配列番号86で示される塩基配列からなるセルラーゼ遺伝子の塩基番号1〜659の塩基配列、配列番号88で示される塩基配列からなるセルラーゼ遺伝子の塩基番号1〜696の塩基配列、又は当該塩基配列のいずれかと70%以上の同一性を有する塩基配列からなるDNA断片、又は、当該塩基配列の一部が欠失した塩基配列からなるDNA断片である、前記(5)記載の枯草菌変異株。
(8) 前記(1)から(7)のいずれか1つに記載の枯草菌変異株を用いる、異種タンパク質又はポリペプチドの製造方法。
【発明の効果】
【0015】
本発明のプロテアーゼ/溶菌酵素欠失枯草菌変異株を用いることにより、(1)培養上清中の不要なタンパク質を低減させることが可能となり、(2)目的タンパク質の比率(比活性)を向上させることが可能となり、(3)細胞内から溶出するプロテアーゼの影響を抑制(目的タンパク質の更なる分解を抑制)することが可能となり、(4)枯草菌の形状がフィラメント状となる結果、膜透過性が向上し、目的タンパク質の生産後の処理工程において除菌が容易になる。
【図面の簡単な説明】
【0016】
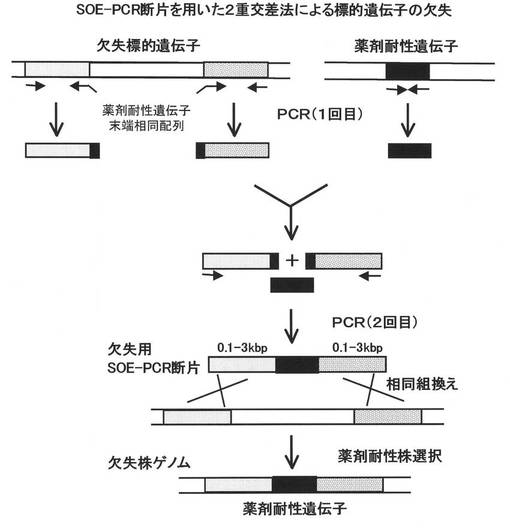
【図1】SOE−PCRによる遺伝子欠失用DNA断片の調製、及び当該DNA断片を用いて標的遺伝子を欠失(薬剤耐性遺伝子と置換)する方法を模式的に示したものである。
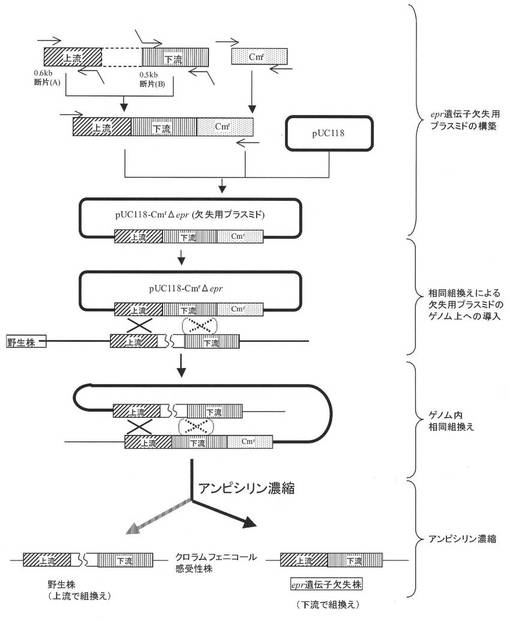
【図2】遺伝子欠失用プラスミドを用いて標的遺伝子を欠失(薬剤耐性遺伝子と置換)する方法を模式的に示したものである。
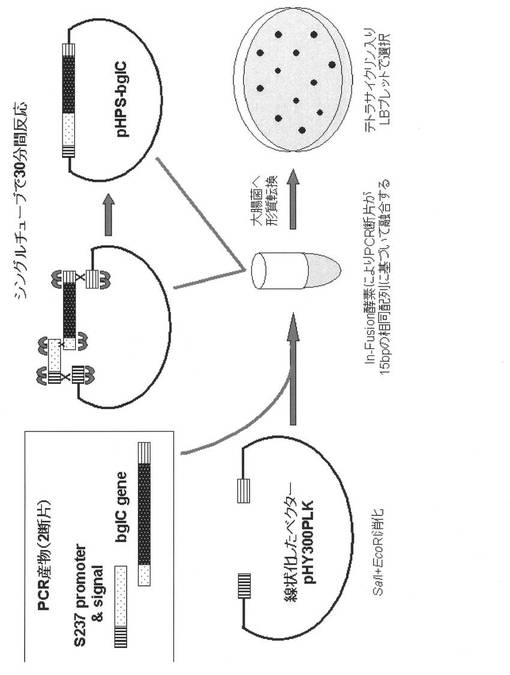
【図3】bglC過剰発現プラスミドの構築方法を模式的に示したものである。
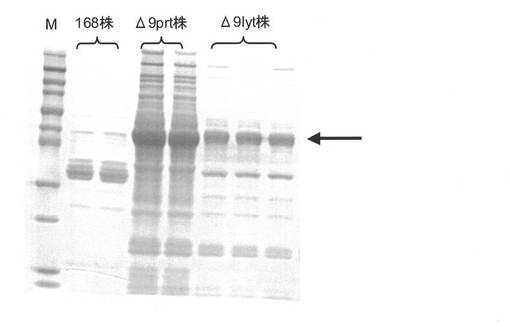
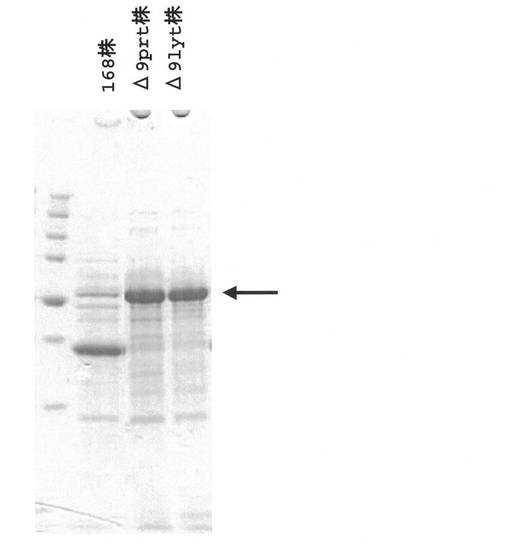
【図4】枯草菌168株、Δ9prt株、Δ9lyt株の培養液上清のタンパク質(セルラーゼBglC)分析を示したものである。
【図5】枯草菌168株、Δ9prt株、Δ9lyt株の培養液上清のタンパク質(セルラーゼN252)分析を示したものである。
【発明を実施するための形態】
【0017】
本発明の宿主微生物である枯草菌は、野生型のものでも変異を施したものでもよいが、全ゲノム情報が明らかにされ、遺伝子工学、ゲノム工学技術が確立され、またタンパク質を菌体外に分泌生産させる能力を有する枯草菌が好ましく、例えば枯草菌168株を元株として用い、更にその元株から所定の遺伝子を欠失させて作製した枯草菌変異株が挙げられる。
【0018】
ここで、枯草菌168株とは、枯草菌Bacillus subtilis Marburg No.168として公知の枯草菌株であり、通常、遺伝子組換えタンパク質生産の際の野生株として用いられている。また枯草菌168株の全塩基配列及び遺伝子は既に報告されており、またインターネット公開されている(Nature,390,249−256,1997及びBSORF Bacillus subtilis Genome Database[http://bacillus.genome.jp/];GenBank:AL009126.2[http://www.ncbi.nlm.nih.gov/nuccore/38680335])。当業者は、これらの情報源から得た枯草菌168株のゲノム情報に基づいて、各種遺伝子操作を行うことができる。
【0019】
本発明において、細胞外プロテアーゼ遺伝子とは、細胞内で産生された後分泌された目的タンパク質又はペプチドの他、溶菌(細胞壁分解)酵素の分解を触媒する酵素タンパク質をコードする遺伝子を意味する。また、溶菌酵素関連遺伝子又は遺伝子群とは、細胞壁の分解を触媒する酵素タンパク質、並びにそれに関連して間接的に細胞壁の分解を促進する他のタンパク質をコードする遺伝子又は遺伝子群を意味する。
【0020】
本発明において欠失又は不活性化の対象となる、細胞外プロテアーゼ遺伝子及び溶菌酵素関連遺伝子の具体例を表1に示す。なお、表中の各遺伝子の名称、番号及び機能等は、Nature,390,249−256(1997)で報告され、JAFAN:Japan Functional Analysis Network for Bacillus subtilis(BSORF DB)でインターネット公開(http://bacillus.genome.ad.jp/、2003年6月17日更新)された枯草菌ゲノムデータに基づき記載されている。
【0021】
【表1】

【0022】
参考文献1:Mol Gen Genet.1990 221(3):486−90
参考文献11:Microbiology.1996 142(Pt12):3437−44
参考文献3:J.Bacteriol.1990 172(2):1019−23
参考文献9:J.Bacteriol.1990 172(2):1024−9
参考文献10:J.Bacteriol.1991 173(20):6364−72
参考文献4:J.Bacteriol.1990 172(3):1470−7
参考文献5:J.Biol Chem.1990 265(12):6845−50
参考文献6:J.Bacteriol.1979 139(2):583−90
参考文献8:J.Bacteriol.1984 160(1):15−21
参考文献2:J.Bacteriol.1991 173(21):6889−95
参考文献7:J.Bacteriol.1984 158(2):411−8
参考文献12:J.Biosci.Bioeng.104,135−143(2007)
参考文献13:特開2003−47490号公報
参考文献14:J.Bacteriol.,173,7304−7312(1991)
参考文献15:J.Bacteriol.180,2549−2555(1998)
参考文献16:Microbiology.140,1855−1867(1994)
参考文献17:J.Bacteriol.180,2110−2117(1998)
参考文献18:Genes Dev.4,525−535(1990)
参考文献19:Bacillus subtilis Closest Relatives:From Genes to Cells,Edited by A.L.Sonenshein,American Society Microbiology,PP.289,(2002)
【0023】
すなわち、表1中のeprからaprXまでの9個の遺伝子が上記の細胞外プロテアーゼ遺伝子に該当し、lytC(cwlB)からsigD遺伝子までの5種類の遺伝子又は遺伝子群が上記の溶菌関連遺伝子又は遺伝子群に該当する。
【0024】
本発明では、細胞外に分泌される目的タンパク質又はポリペプチドの細胞外における分解を効果的に防止して目的タンパク質又はポリペプチドの生産性を向上させるため、上記の9個の細胞外プロテアーゼ遺伝子のうち、8個以上の遺伝子を欠失又は不活性化させることを特徴とし、9個欠失又は不活性化させるのがより好ましい。より具体的には、本願発明に係る枯草菌変異株は、aprE、nprB、nprE、bpr、vpr、mpr、epr及びwprAの8個の遺伝子か、又は、aprX、aprE、nprB、nprE、bpr、vpr、mpr、epr及びwprAの9個の遺伝子を欠失又は不活性化させるのが好ましい。なお、これらのプロテアーゼ遺伝子を欠失又は不活性化した枯草菌変異株は、特開2006−174707号公報に記載の方法により作製することができる。
【0025】
また、本発明では、細胞外プロテアーゼ欠失株と同等以上の目的タンパク質又はポリペプチドを生産し、且つ本発明に係る組換え枯草菌の溶菌を効果的に防止するため、上記の5個の溶菌関連遺伝子又は遺伝子群のうち、1個以上の遺伝子又は遺伝子群を欠失又は不活性化させることを特徴とし、2個以上欠失又は不活性化させるのがより好ましい。なお、本願における5個の溶菌関連遺伝子又は遺伝子群とは、cwlB、cwlF、PBSX、spoIIIC及びsigDを意味するが、そのうちのPBSXとは、以下の38種類の遺伝子群を含む領域のことを意味し、本願ではかかる領域を、欠失又は不活性化させる対象の1個とするものである。
【0026】
【表2】
【0027】
本発明におけるPBSXの欠失又は不活性化は、これらの遺伝子群の一部又は全部の欠失又は不活性化により行うことができ、全部を欠失又は不活性化させることがより好ましい。
【0028】
ここで、sigD及びspoIIICは転写因子の遺伝子である。すなわち本発明によれば、溶菌に直接関与しないが間接的に溶菌酵素に関連する遺伝子を欠失させた場合でも、溶菌を顕著に抑制することができる。
【0029】
表1及び表2に記載の遺伝子は、例えば、当該遺伝子の塩基配列において1若しくは数個の塩基配列が欠失、置換、若しくは付加された塩基配列からなる、当該遺伝子と同じ機能を有する遺伝子であってもよい。更に、表1及び表2に記載の遺伝子と同じ機能を有する、及び/又は、表1及び表2の各遺伝子と塩基配列において70%以上、好ましくは80%以上、より好ましくは90%以上、更に好ましくは95%以上、特に好ましくは98%以上の同一性を有する遺伝子も、表1及び表2に記載の遺伝子に相当する遺伝子と考えられ、本発明において欠失/不活性化しうる遺伝子に含まれる。
【0030】
これらの遺伝子又は遺伝子群は、その内部に他のDNA断片を挿入するか、あるいは当該遺伝子の転写・翻訳開始領域に変異を与える等の方法によっても不活性化できるが、好適には、標的遺伝子を物理的に削除する方がより望ましい。更に本発明の組換え枯草菌の構築において、上記以外の遺伝子の欠失又は不活性化と組み合わせてもよい。
【0031】
遺伝子又は遺伝子群の欠失又は不活性化の手順としては、表1に示す標的遺伝子を計画的に欠失又は不活性化する方法のほか、ランダムな遺伝子の欠失又は不活性化変異を与えた後、適当な方法によりタンパク質生産性の評価及び遺伝子解析を行う方法が挙げられる。
【0032】
標的遺伝子を欠失又は不活性化するには、例えば相同組換えによる方法を用いればよい。すなわち、標的遺伝子の一部を含むDNA断片を適当なプラスミドベクターにクローニングして得られる環状の組換えプラスミドを親枯草菌細胞内に取り込ませ、標的遺伝子の一部領域における相同組換えによって親枯草菌ゲノム上の標的遺伝子を分断して不活性化することが可能である。あるいは、塩基置換や塩基挿入等による不活性化変異を導入した標的遺伝子、又は標的遺伝子の外側領域を含むが標的遺伝子を含まない直鎖状のDNA断片等をPCR等の方法によって構築し、これを親枯草菌細胞内に取り込ませて親枯草菌ゲノムの標的遺伝子内の変異箇所の外側2ヶ所、又は標的遺伝子外側の2ヶ所の領域で2回交差の相同組換えを起こさせることにより、ゲノム上の標的遺伝子が欠失又は不活性化した遺伝子断片と置換することが可能となる。
【0033】
特に、相同組換えにより枯草菌の標的遺伝子を欠失又は不活性化する方法については、既にいくつかの報告例があり(Mol.Gen.Genet.,223,268(1990)等)、かかる方法を2回又はそれ以上繰り返すことにより、本発明の組換え枯草菌を得ることができる。
【0034】
また、ランダムな遺伝子の欠失又は不活性化を行う方法としては、例えばランダムにクローニングしたDNA断片を用いて上述の方法と同様な相同組換えを起こさせる方法や、親枯草菌にγ線等を照射する方法等が挙げられる。
【0035】
以下、より具体的な例として、SOE(splicing by overlap extension)−PCR法(Gene,77,61(1989))によって調製される欠失用DNA断片を用いた二重交差法による欠失方法について説明するが、本発明における遺伝子欠失方法は下記に限定されるものではない。
【0036】
本方法で用いる欠失用DNA断片は、標的遺伝子の上流に隣接する約0.2〜3kb断片(A)と、同じく下流に隣接する約0.2〜3kb断片(B)の間に、薬剤耐性マーカー遺伝子断片(C)を挿入した断片である。まず、1回目のPCRによって、標的遺伝子の上流断片(A)及び下流断片(B)、並びに薬剤耐性マーカー遺伝子断片(C)の3断片を調製するが、この際、例えば、上流断片(A)の下流末端に薬剤耐性マーカー遺伝子断片(C)の上流側10〜30塩基対配列、逆に下流断片(B)の上流末端には薬剤耐性マーカー遺伝子断片(C)の下流側10〜30塩基対配列が付加される様にデザインしたプライマーを用いる(図1)。
【0037】
次いで、1回目に調製した3種類のPCR断片(A)、(B)、(C)を鋳型とし、上流断片の上流側プライマーと下流断片の下流側プライマーを用いて2回目のPCRを行うことによって、上流断片の下流末端及び下流断片の上流末端に付加した薬剤耐性マーカー遺伝子配列に於いて、薬剤耐性マーカー遺伝子断片とのアニールが生じ、PCR増幅の結果、上流側断片と下流側断片の間に、薬剤耐性マーカー遺伝子が挿入し、(A)(C)(B)の順に結合したDNA断片を得ることができる(図1)。
【0038】
本発明では2つ又はそれ以上の遺伝子を欠失又は不活性化することが必要であるため、2種類又はそれ以上の薬剤耐性マーカー遺伝子を用いると簡便に目的の微生物菌株を分離することができる。薬剤耐性マーカー遺伝子の組合せについては特に限定されないが、例えば、カナマイシン耐性遺伝子、スペクチノマイシン耐性遺伝子、クロラムフェニコール耐性遺伝子、エリスロマイシン耐性遺伝子、テトラサイクリン耐性遺伝子などが挙げられる。
【0039】
PCRの方法についても特に限定されず、例えば実施例中に示したプライマーセットを用い、Pyrobest DNAポリメーラーゼ(宝酒造)などの一般のPCR用酵素キット等を用いて、成書(PCR Protocols.Current Methods and Applications,Edited by B.A.White,Humana Press,pp251(1993)、Gene,77,61,(1989)等)に示される通常の条件によりSOE−PCRを行うことによって、各遺伝子の欠失用DNA断片が得られる。
【0040】
また、上記の(A)、(B)及び(C)の3断片を、適当なプラスミドベクター上で、(A)(C)(B)の順になる様にクローニングし、元のプラスミドベクター部分のみを1ヶ所で切断する制限酵素で処理することによっても、同様の欠失用DNA断片を得ることができる(図2)。
【0041】
かくして得られた欠失用DNA断片を、コンピテント法等によって細胞内に導入すると、同一性のある標的遺伝子の上流及び下流の相同領域において相同組換えが生じ、標的遺伝子が薬剤耐性遺伝子で置換される。それにより得られた細胞を、クロラムフェニコールなどの薬剤を含む寒天培地上において生育させ、生じたコロニーを分離することにより、標的遺伝子が欠失した組換え枯草菌を得ることができる。また、標的遺伝子がクロラムフェニコール耐性遺伝子と置換されて欠失していることを、ゲノムを鋳型としたPCR法などによって確認することができる。
【0042】
このようなステップを繰り返し、表1及び表2に示される枯草菌の遺伝子が欠失又は不活性化された枯草菌変異株に、目的タンパク質又はポリペプチドをコードする遺伝子を導入することによって、本発明の組換え微生物を得ることができる。
【0043】
本発明の微生物を用いて生産する目的タンパク質又はポリペプチドは特に限定されないが、例えば食品用、医薬品用、化粧品用、洗浄剤用、繊維処理用、医療検査薬用等として有用な酵素や生理活性因子等のタンパク質やポリペプチドが挙げられる。産業用に用いられる酵素としては、酸化還元酵素(オキシドレダクターゼ)、転移酵素(トランスフェラーゼ)、加水分解酵素(ヒドロラーゼ)、脱離酵素(リアーゼ)、異性化酵素(イソメラーゼ)、合成酵素(リガーゼ/シンセターゼ)等が含まれるが、好適にはセルラーゼ、α−アミラーゼ、プロテアーゼ等の加水分解酵素の遺伝子が挙げられる。具体的には、多糖加水分解酵素の分類(Biochem.J.,280,309(1991))中でファミリー5に属するセルラーゼ(エンドグルカナーゼ)が挙げられ、中でも微生物由来、特にバチルス属細菌由来のセルラーゼ(エンドグルカナーゼ)が挙げられる。
【0044】
より具体的な例としては、配列番号83で示されるアミノ酸配列からなる枯草菌由来エンドグルカナーゼBglC、又は当該アミノ酸配列と70%以上、好ましくは80%以上、より好ましくは90%以上、更に好ましくは95%以上、より更に好ましくは98%以上、最も好ましくは99%以上の同一性を有するアミノ酸配列からなるエンドグルカナーゼ、又は当該アミノ酸配列において1若しくは数個のアミノ酸が置換、欠失若しくは付加されたアミノ酸配列からなるエンドグルカナーゼ;
【0045】
より具体的な例としては、配列番号85で示されるアミノ酸配列からなるバチルス属細菌KSM−N252(FERM P−17474)株由来のアルカリエンドグルカナーゼ、又は当該アミノ酸配列と70%以上、好ましくは80%以上、より好ましくは90%以上、更に好ましくは95%以上、より更に好ましくは98%以上、最も好ましくは99%以上の同一性を有するアミノ酸配列からなるアルカリエンドグルカナーゼ、又は当該アミノ酸配列において1若しくは数個のアミノ酸が置換、欠失若しくは付加されたアミノ酸配列からなるアルカリエンドグルカナーゼ
【0046】
配列番号87で示されるアミノ酸配列からなるバチルス属細菌KSM−S237株(FERM BP−7875)由来のアルカリセルラーゼ、又は当該アミノ酸配列と70%、好ましくは80%、より好ましくは90%以上、更に好ましくは95%以上、より更に好ましくは98%以上、もっとも好ましくは99%以上の同一性を有するアミノ酸配列からなるアルカリセルラーゼ、又は当該アミノ酸配列において1若しくは数個のアミノ酸が置換、欠失若しくは付加されたアミノ酸配列からなるアルカリセルラーゼ;
【0047】
配列番号89で示されるアミノ酸配列からなるバチルス属細菌KSM−64株(FERM BP−2886)由来のアルカリセルラーゼ、又は当該アミノ酸配列と70%、好ましくは80%、より好ましくは90%以上、更に好ましくは95%以上、より更に好ましくは98%以上、もっとも好ましくは99%以上の同一性を有するアミノ酸配列からなるアルカリセルラーゼ、又は当該アミノ酸配列において1若しくは数個のアミノ酸が置換、欠失若しくは付加されたアミノ酸配列からなるアルカリセルラーゼが挙げられる。
【0048】
その他の目的タンパク質又はポリペプチドの具体例としては、バチルス属細菌KSM−K38株(FERM BP−6946、特開2000-184882号)由来のアルカリアミラーゼ、又はそれと70%、好ましくは80%、より好ましくは90%以上、更に好ましくは95%以上、特に好ましくは98%以上のアミノ酸配列同一性を有するアミノ酸配列からなるアミラーゼ、又は当該アミノ酸配列において1若しくは数個のアミノ酸が置換、欠失若しくは付加されたアミノ酸配列からなるアミラーゼ;
【0049】
バチルス属細菌(Bacillus sp.)KSM−KP43株(FERM BP−6532、WO99/18218)由来のアルカリプロテアーゼ、又はそれと70%、好ましくは80%、より好ましくは90%以上、更に好ましくは95%以上、特に好ましくは98%以上のアミノ酸配列同一性を有するアミノ酸配列からなるプロテアーゼ、又は当該アミノ酸配列において1若しくは数個のアミノ酸が置換、欠失若しくは付加されたアミノ酸配列からなるアルカリプロテアーゼが挙げられる。
【0050】
宿主枯草菌に導入される上記目的タンパク質又はポリペプチドの遺伝子は、その上流に当該遺伝子の転写、翻訳、分泌に関わる制御領域、即ち、プロモーター及び転写開始点を含む転写開始制御領域、リボソーム結合部位及び開始コドンを含む翻訳開始領域並びに分泌シグナルペプチド領域から選ばれる1以上の領域が適正な形で結合されていることが望ましい。特に、転写開始制御領域、翻訳開始制御領域及び分泌シグナル領域からなる3領域が結合されていることが好ましく、更に分泌シグナルペプチド領域がバチルス(Bacillus)属細菌のセルラーゼ遺伝子由来のものであり、転写開始制御領域及び翻訳開始制御領域が当該セルラーゼ遺伝子の開始コドンから始まる長さ0.6〜1kbの上流領域であるものが、目的のタンパク質又はポリペプチド遺伝子と適正な形で結合されていることが望ましい。例えば、特開2000−210081号公報や特開平4−190793号公報等に記載されているバチルス(Bacillus)属細菌、すなわちKSM−S237株(FERM BP−7875)、KSM−64株(FERM BP−2886)由来のセルラーゼ遺伝子の転写開始制御領域、翻訳開始領域及び分泌シグナルペプチド領域が目的のタンパク質又はポリペプチドの構造遺伝子と適正に結合されていることが望ましい。より具体的には配列番号86で示される塩基配列の塩基番号1〜659の塩基配列、配列番号88で示される塩基配列からなるセルラーゼ遺伝子の塩基番号1〜696の塩基配列、また当該塩基配列に対して70%以上、好ましくは80%以上、より好ましくは90%以上、更に好ましくは95%以上、特に好ましくは98%以上の同一性を有する塩基配列からなるDNA断片、又は上記いずれかの塩基配列からなるDNAとストリンジェントの条件でハイブリダイズし且つ遺伝子の転写、翻訳、分泌に関わる機能を有するDNA、或いは上記いずれかの塩基配列の一部が欠失した塩基配列からなるDNA断片が、目的のタンパク質又はポリペプチドの構造遺伝子と適正に結合されていることが望ましい。尚、ここで、上記塩基配列の一部が欠失した塩基配列からなるDNA断片とは、上記塩基配列の一部を欠失しているが、遺伝子の転写、翻訳、分泌に関わる機能を保持しているDNA断片を意味する。またここで言うストリンジェントな条件とは、例えば[Molecular cloning−a Laboratory manual 2nd edition(Sambrookら、1989)]に記載の条件等が挙げられる。例えば、6×SSC(1×SSCの組成:0.15M塩化ナトリウム、0.015Mクエン酸ナトリウム、pH7.0)、0.5%SDS、5×デンハート及び100mg/mLニシン精子DNAを含む溶液にプローブとともに65℃で8〜16時間恒温し、ハイブリダイズさせる条件が挙げられる。
【0051】
なお、本発明におけるアミノ酸配列および塩基配列の同一性はLipman−Pearson法(Science,227,1435,(1985))によって計算される。具体的には、遺伝情報処理ソフトウェアGenetyx−Win(ソフトウェア開発)のホモロジー解析(Search homology)プログラムを用いて、パラメータであるUnit size to compare(ktup)を2として解析を行うことにより算出される。
【0052】
本発明の方法による目的タンパク質又はポリペプチドの生産は、上記組換え微生物を同化性の炭素源、窒素源、その他の必須成分を含む培地に接種し、通常の微生物培養法にて培養し、培養終了後、当該目的のタンパク質又はポリペプチドを精製することにより行えばよい。培養に使用される培地の組成及び培養条件については、使用する微生物の種類や目的タンパク質又はポリペプチドの種類等にしたがって、当業者が適宜選択することができる。目的タンパク質又はポリペプチドの精製は、培養終了後の菌体を材料として行っても良いが、本発明の組換え微生物は多量の目的タンパク質又はポリペプチドを細胞外に分泌する能力を有するため、培養終了後の培養上清を材料として行うのが有利である。
【0053】
具体的には、上記の適切なプロモーターが作動可能に連結した目的タンパク質又はポリペプチドの遺伝子を、枯草菌細胞内で自立複製可能なベクターに挿入して枯草菌細胞内に導入することにより、枯草菌形質転換体を得ることが可能であり、当該ベクターの例としてはpHY300PLK等が挙げられる。
【0054】
更に、上記で得られた枯草菌形質転換体を、適切な培地において培養し、目的タンパク質又はポリペプチドの遺伝子を発現させ、更に当該目的タンパク質又はポリペプチドを細胞外へ分泌させる。当該遺伝子を発現させる際、当該遺伝子に連結したプロモーターの性質に応じて、当該培地の組成を適宜調節するのが好ましい。特に発現誘導型プロモーターを用いる場合には、そのプロモーター活性を向上させる誘導物質を適切なタイミングで培地中に添加するのが好ましい。
【0055】
上記の枯草菌変異株は、例えば同化性の炭素源、窒素源、その他の必須成分を含む培地に接種して培養して行うことができる。培養方法は、原則的には一般的な微生物の培養方法であってよく、通常、液体培養による振盪培養、通気撹拌培養等の好気的条件下で実施するのが好ましい。
【0056】
培養終了後、培養液を遠心分離し、得られる上清又は菌体から、硫安沈殿やクロマトグラフィなどを適宜組み合わせ、常法に従い、目的タンパク質又はポリペプチドを抽出・精製することにより得ることができる。
【実施例】
【0057】
以下の実施例におけるDNA断片増幅のためのポリメラーゼ連鎖反応(PCR)は、Pyrobest DNAポリメラーゼ(タカラバイオ社製)及び付属の試薬類を用いた、GeneAmp PCR System(アプライドバイオシステムズ社製)によるDNA増幅により行った。PCR反応液は、適宜希釈した鋳型DNAを1μL、センス及びアンチセンスプライマーを各々20pmol、及びPyrobest DNAポリメラーゼを2.5U添加し、更に総反応液量を50μLとすることにより調製した。PCR反応条件は、98℃で10秒間、55℃で30秒間及び72℃で1〜5分間(目的増幅産物に応じて調整、目安は1kbあたり1分間)の3段階の温度変化を30サイクル繰り返した後、72℃で5分間反応、とした。
【0058】
また、以下の実施例において、遺伝子の上流及び下流とは、複製開始点からの位置を指すのではなく、上流とは各操作・工程において対象となる遺伝子の開始コドンの5’側に続く領域を指し、一方下流とは各操作・工程において対象となる遺伝子の終止コドンの3’側に続く領域を指すものとする。
【0059】
枯草菌の形質転換は、コンピテントセル法(J.Bacteriol.93,1925(1967))により行った。すなわち、枯草菌株をSPI培地(0.20%硫酸アンモニウム、1.40%リン酸水素二カリウム、0.60%リン酸二水素カリウム、0.10%クエン酸三ナトリウム二水和物、0.50%グルコース、0.02%カザミノ酸(Difco社製)、5mM硫酸マグネシウム、0.25μM塩化マンガン、50μg/mLトリプトファン)中で、37℃で、生育度(OD600)の値が約1となるまで振盪培養し、振盪培養後、培養液の一部を9倍量のSPII培地(0.20%硫酸アンモニウム、1.40%リン酸水素二カリウム、0.60%リン酸二水素カリウム、0.10%クエン酸三ナトリウム二水和物、0.50%グルコース、0.01%カザミノ酸(Difco社製)、5mM硫酸マグネシウム、0.40μM塩化マンガン、5μg/mLトリプトファン)に接種し、更に生育度(OD600)の値が約0.4となるまで振盪培養することにより、枯草菌株のコンピテントセルを調製した。
【0060】
次いで調製したコンピテントセル懸濁液(SPII培地における培養液)100μLに、各種DNA断片を含む溶液(SOE−PCRの反応液等)を5μL添加し、37℃で1時間振盪培養後、適切な薬剤を含むLB寒天培地(1%トリプトン、0.5%酵母エキス、1%NaCl、1.5%寒天)に全量を塗沫した。37℃での静置培養の後、生育したコロニーを形質転換体として分離した。得られた形質転換体のゲノムを抽出し、これを鋳型とするPCRを行い、目的とするゲノム構造の改変がなされたことを確認した。
【0061】
目的のタンパク質又はポリペプチドをコードする遺伝子の宿主微生物への導入は、コンピテントセル形質転換法(J.Bacteriol.93,1925(1967))、エレクトロポレーション法(FEMS Microbiol.Lett.55,135(1990))、プロトプラスト形質転換法(Mol.Gen.Genet.168,111(1979))のいずれかによって行った。
【0062】
組換え微生物によるタンパク質生産用の培養には、LB培地(1%トリプトン、0.5%酵母エキス、1%NaCl)、2×YT培地(1.6%トリプトン、1%酵母エキス、0.5%NaCl)、2×L−マルトース培地(2%トリプトン、1%酵母エキス、1%NaCl、7.5%マルトース、7.5ppm硫酸マンガン4−5水和物)、あるいはCSL発酵培地(2%酵母エキス、0.5%コーンスティープリカー(CSL)、0.05%塩化マグネシウム七水和物、0.6%尿素、0.2%L−トリプトファン、10%グルコース、0.15%リン酸二水素ナトリウム、0.35%リン酸水素二ナトリウム、pH7.2)を用いた。
【0063】
実施例1:epr遺伝子欠失用プラスミドの構築:
枯草菌168株から抽出したゲノムDNAを鋳型とし、表3−1、表3−2に示すeprfw1とeprUpr、及びeprDNfとeprrv−repの各プライマーセットを用いて、ゲノム上のepr遺伝子の上流に隣接する0.6kb断片(A)、及び下流に隣接する0.5kb断片(B)をそれぞれ調製した。また別途プラスミドpC194(J.Bacteriol.150(2),815(1982))由来のクロラムフェニコール耐性遺伝子の上流にプラスミドpUB110(Plasmid 15,93(1986))由来のrepU遺伝子のプロモーター領域(Nucleic Acids Res.17,4410(1989))を連結した1.2kb断片(C)を調製した。次に、得られた(A)(B)(C)3断片を混合して鋳型とし、表3−1、表3−2のプライマーeprfw2とCmrv2を用いたSOE−PCRを行うことによって、3断片を(A)(B)(C)の順になる様に結合させ、2.2kbのDNA断片を得た(図2)。このDNA断片の末端を平滑化及び5’−リン酸化し、プラスミドpUC118(Methods Enzymol.153,3(1987))のSmaI制限酵素部位に挿入してepr遺伝子欠失用プラスミドpUC118−CmrΔeprを構築した。尚、上記1.2kb断片(C)は、repUfwとrepUr−Cmのプライマーセット(表3−1、表3−2)及び鋳型としてプラスミドpUB110を用いて調製したrepU遺伝子プロモーター領域を含む0.4kb断片(D)と、CmUf−repとCmrv1のプライマーセット(表3−1、表3−2)及び鋳型としてプラスミドpC194を用いて調製したクロラムフェニコール耐性遺伝子を含む0.8kb断片(E)とを混合して鋳型とし、表3−1、表3−2に示すプライマーrepUfwとCmrv1を用いたSOE−PCRを行なうことによって調製した。
【0064】
【表3−1】
【0065】
【表3−2】
【0066】
実施例2:欠失用プラスミドを用いたepr遺伝子欠失株の構築:
実施例1にて構築したepr遺伝子欠失用プラスミドpUC118−CmrΔeprをコンピテントセル形質転換法(J.Bacteriol.93,1925(1967))によって枯草菌168株に導入し、epr遺伝子上流領域、あるいは下流領域の相当する領域間での1重交差の相同組換えによりゲノムDNAと融合した形質転換株を、クロラムフェニコール耐性を指標に取得した。得られた形質転換株をLB培地に接種し、37℃にて2時間培養後、再度、コンピテンス誘導操作を行うことにより、ゲノム上で重複して存在するepr遺伝子上流領域、あるいは下流領域の間におけるゲノム内相同組換えを誘導した。図2に示す様に、プラスミド導入の際と異なる領域で相同組換えが生じた場合、プラスミドに由来するクロラムフェニコール耐性遺伝子及びpUC118ベクター領域の脱落に伴ってepr遺伝子が欠失する。次に、クロラムフェニコール感受性となった株の存在比率を高める為、以下の要領でアンピシリン濃縮操作を行なった。コンピテントセル誘導後の培養液を終濃度5ppmのクロラムフェニコール及び終濃度100ppmのアンピシリンナトリウムを含むLB培地1mLに、600nmにおける濁度(OD600)が0.003となるように接種した。37℃にて5時間培養後、10,000ppmのアンピシリンナトリウム水溶液を10μL添加し、更に3時間培養した。培養終了後、2%塩化ナトリウム水溶液にて菌体を遠心洗浄した後、1mLの2%塩化ナトリウム水溶液に懸濁し、懸濁液100μLをLB寒天培地に塗沫した。37℃にて約15時間インキュベートし、生育した菌株のうち、プラスミド領域の脱落に伴ってクロラムフェニコール感受性となった株を選抜した。選抜した菌株のゲノムDNAを鋳型とし、表3−1、表3−2に示すプライマーeprfw2とeprrv−repを用いたPCRを行なってepr遺伝子欠失の確認を行ない、epr遺伝子欠失株を取得した。
【0067】
実施例3:プロテアーゼ遺伝子8重欠失株及びプロテアーゼ9重欠失株の構築:
epr遺伝子欠失株に対し、次の欠失としてwprA遺伝子の欠失をepr遺伝子欠失と同様に行なった。即ち、実施例1と同様にしてwprA遺伝子欠失用プラスミドpUC118−CmrΔwprAを構築し、構築したプラスミドのゲノムDNAへの導入とそれに続くゲノム内相同組換えによるwprA遺伝子の欠失によりepr遺伝子とwprA遺伝子の2重欠失株を取得した。以降同様の操作を繰り返すことにより、mpr、nprB、bpr、nprE、vpr、aprEの各遺伝子を順次欠失させ、8種類のプロテアーゼ遺伝子が欠失したプロテアーゼ8重欠失株を構築し、Δ8prt株と命名した。更に同様の操作を繰り返すことによりaprX遺伝子を欠失させたプロテアーゼ9重欠失株を構築し、Δ9prt株と命名した。各欠失を行う際に用いたプライマーの配列を表3−1、表3−2に示し、また各プライマーと実施例1で示したepr遺伝子欠失に用いたプライマーとの対応について表4に示す。
【0068】
【表4】
【0069】
実施例4:プロテアーゼ9重欠失株からのcwlB遺伝子、PBSX遺伝子群、cwlF遺伝子の欠失:
実施例3と同様にして、Δ9prt株より表3−1、表3−2及び表4に示すcwlB遺伝子欠失用のプライマーセットを用いてcwlB遺伝子の欠失を行い、9種類のプロテアーゼ遺伝子とcwlB遺伝子が欠失したΔ9B株を構築した。続いてPBSX遺伝子群の欠失を行うこととした。尚、本発明においてPBSX遺伝子群とは、枯草菌ゲノム上に連続して存在するxlyB遺伝子〜spoIISA遺伝子の38遺伝子群を指すものとする。Δ9B株より表3−1、表3−2及び表4に示すPBSX遺伝子群欠失用のプライマーセットを用いてPBSX遺伝子群の欠失を行い、Δ9BP株を構築した。またΔ9B株より表3−1、表3−2及び表4に示すcwlF遺伝子欠失用のプライマーセットを用いてcwlF遺伝子の欠失を行い、Δ9BF株を構築した。更にΔ9BP株より表3−1、表3−2及び表4に示すcwlF遺伝子欠失用のプライマーセットを用いてcwlF遺伝子の欠失を行い、Δ9BPF株を構築した。
【0070】
実施例5:Δ9BPF株からのspoIIIC遺伝子の欠失:
図1に基づいて、ゲノム中spoIIIC遺伝子の薬剤耐性遺伝子による欠失方法を説明する。尚、spoIIIC遺伝子は、spoIVCB遺伝子と共に枯草菌の胞子形成期に特異的に機能するシグマ因子SigKをコードする遺伝子である。
【0071】
枯草菌168株から抽出したゲノムDNAを鋳型とし、表3−1、表3−2に示すspoIIIC−FW及びspoIIIC/Em−Rのプライマーセットを用いて、ゲノム中のspoIIIC遺伝子の上流に隣接する1.0kb断片(A)をPCRにより増幅した。また、上記ゲノムDNAを鋳型とし、spoIIIC/Em−F及びspoIIIC−RVのプライマーセットを用いて、ゲノム中のspoIIIC遺伝子の下流に隣接する1.0kb断片(B)をPCRにより増幅した。
【0072】
更に、プラスミドpMUTIN4(Microbiology.144,3097(1998))を鋳型として、表3−1、表3−2に示すemf2及びemr2のプライマーセットを用いて、1.3kbのエリスロマイシン(Em)耐性遺伝子領域(C)をPCRにより調製した。
【0073】
次に、図1に示すように、得られた1.0kb断片(A)、1.0kb断片(B)及びCm耐性遺伝子領域(C)の3断片を混合して鋳型として、表1に示したspoIIIC−FW2及びspoIIIC−RV2のプライマーセットを用いたSOE−PCR法によって、3断片が1.0kb断片(A)、Em耐性遺伝子領域(C)、1.0kb断片(B)の順に含まれる3.3kbのDNA断片(D)を得た。
【0074】
更に、コンピテントセル形質転換法によって、得られたDNA断片(D)を用いて、Δ9BPF株の形質転換を行った。形質転換後、エリスロマイシン(1μg/mL)とリンコマイシン(25μg/mL)を含むLB寒天培地上に生育したコロニーを形質転換体として分離した。
【0075】
得られた形質転換体のゲノムDNAを抽出し、PCRによってspoIIIC遺伝子が欠失し、Em耐性遺伝子に置換していることを確認した。以上のようにして、Δ9BPFC株を構築した。
【0076】
実施例6:Δ9BPFC株からのsigD遺伝子の欠失:
図1に基づいて、ゲノム中sigD遺伝子の薬剤耐性遺伝子による欠失方法を説明する。尚、sigD遺伝子は枯草菌の細胞壁溶解、鞭毛形成、あるいは走化性等に関与する遺伝子の発現を制御するシグマ因子SigDをコードする遺伝子である。
【0077】
枯草菌168株から抽出したゲノムDNAを鋳型とし、表3−1、表3−2に示すsigD−FW及びsigD/Cm−Rのプライマーセットを用いて、ゲノム中のsigD遺伝子の上流に隣接する1.0kb断片(A)をPCRにより増幅した。また、上記ゲノムDNAを鋳型とし、sigD/Cm−F及びsigD−RVのプライマーセットを用いて、ゲノム中のsigD遺伝子の下流に隣接する1.0kb断片(B)をPCRにより増幅した。
【0078】
更に、プラスミドDNA pC194を鋳型とし、表3−1、表3−2に示すcatf及びcatrのプライマーセットを用いて、0.85kbのクロラムフェニコール(Cm)耐性遺伝子領域(C)をPCRにより調製した。
【0079】
次に、図1に示すように、得られた1.0kb断片(A)、1.0kb断片(B)及びCm耐性遺伝子領域(C)の3断片を混合して鋳型として、表1に示したsigD−FW2及びsigD−RV2のプライマーセットを用いたSOE−PCR法によって、3断片が1.0kb断片(A)、Cm耐性遺伝子領域(C)、1.0kb断片(B)の順に含まれる2.8kbのDNA断片(D)を得た。
【0080】
更に、コンピテントセル形質転換法によって、得られたDNA断片(D)を用いて、168株の形質転換を行った。形質転換後、クロラムフェニコール(10μg/mL)を含むLB寒天培地上に生育したコロニーを形質転換体として分離した。
【0081】
得られた形質転換体のゲノムDNAを抽出し、PCRによってsigD遺伝子が欠失し、Cm耐性遺伝子に置換していることを確認した。以上のようにして、Δ9BPFCD株(Δ9lyt株)を構築した。
【0082】
実施例7:bglC過剰発現プラスミドの構築:
プラスミドの構築は、In−FusionTM Advantage PCR Cloning Kit(Clontech社)を用いて行なった(図3)。プロトコールに従って設計したプライマー(表3−1、表3−2)を用いて、バチルス エスピー(Bacillus sp.)KSM−S237株(FERM BP−7875)由来のアルカリセルラーゼ遺伝子(特開2000−210081号)のプロモーターとシグナル配列の断片を、pHYS237(Biosci.Biotechnol.Biochem.,64(11):2281−9,2000)を鋳型にして、並びに、配列番号82で示される枯草菌セルラーゼ遺伝子bglCを、枯草菌ゲノムDNAを鋳型にして、それぞれPSF_infu(EcI)とPS237Rのプライマーセット、及びbglC−sig/PSFとbglC_infu(SaI)のプライマーセットを用いて増幅した。得られた増幅産物を、制限酵素EcoRI及びSalI消化により線状化したpHY300PLK及びIn−Fusion酵素を適当量で混合して30分間反応を行ない、S237セルラーゼのプロモーター配列及びシグナル配列にbglCの構造遺伝子が連結したプラスミドを構築し、それをpHPS−bglCと命名した。更に、当該プラスミドを大腸菌HB101コンピテントセル(TaKaRa)に導入し、形質転換した。
【0083】
CMC及びトリパンブルーを添加したLB寒天培地を用い、テトラサイクリン耐性により大腸菌形質転換体を選抜した。また、当該形質転換体がハロー形成をすることが判明した。形質転換体より精製したプラスミドを用いて制限酵素処理あるいはPCRを行い、形質転換が適切に行われていることを確認した。
【0084】
実施例8:枯草菌宿主へのプラスミド導入及び得られた形質転換体の培養:
実施例7にて構築したプラスミドpHPS−bglCを、プロトプラスト形質転換法によって実施例3及び実施例6にて構築した枯草菌変異株Δ9prt株及びΔ9lyt株に導入した。同様に枯草菌野生株168株にも導入した。これにより得られた組換え菌株を、10mLのLB培地で30℃で一晩振盪培養し、更にこの培養液0.05mLを50mLの2×L−マルトース培地(2%トリプトン、1%酵母エキス、1%NaCl、7.5%マルトース、7.5ppm硫酸マンガン4−5水和物、15ppmテトラサイクリン)に接種し、30℃にて3日間振盪培養を行った。遠心分離によって菌体を除くことにより培養液上清を得た。
【0085】
実施例9:枯草菌形質転換体培養液上清のタンパク質分析:
実施例8にて得られた培養液上清5μL分をSDS−ポリアクリルアミドゲル電気泳動(SDS−PAGE;濃縮ゲル4%、分離ゲル12.5%)に供し、タンパク質バンドの分離を行った(168株組換え菌株の2サンプル、Δ9prt株組換え菌株の2サンプル、Δ9lyt株組換え菌株の3サンプル)。その結果を図4に示す。168株組換え菌の培養上清には目的の枯草菌セルラーゼ遺伝子bglCがコードするセルラーゼBglCの分子量に相当する位置にバンドは認められなかった。一方でΔ9prt株組換え菌株とΔ9lyt株組換え菌株の培養上清にはBglCの分子量に相当する位置にバンドが認められたことから、両株において欠失されているプロテアーゼ遺伝子がコードするプロテアーゼがBglCの分解に関与しており、これらのプロテアーゼ遺伝子の欠失によりBglCの分泌生産が可能になったと考えられた。またΔ9prt株組換え菌株の培養上清には目的とするBglC以外のタンパク質が著量認められるのに対し、Δ9lyt株組換え菌株ではこれら夾雑タンパク質は大幅に減少していた。即ち、Δ9prt株では溶菌酵素関連遺伝子群の機能性産物の作用により細胞の溶解、及び細胞内タンパク質の漏出が起こっており、Δ9lyt株ではこの様な溶菌現象に伴うタンパク質の漏出が抑制された結果、培養上清中の夾雑タンパク質が減少したものと考えられた。
【0086】
実施例10:培養上清のセルロース糖化活性:
セルロース(粉砕パルプ)を用い、セルロース糖化反応を行った。なお粉砕パルプは、粉末状パルプ(日本製紙ケミカル社製「W−400G」、400メッシュパス90以上、セルロース含有量99質量%、水分含量1質量%)を媒体撹拌式ミル(アトライタ、三井鉱山(株)製、「MA1D−X」、容器全容量:5.5L)に500g投入し、媒体として、直径10mm、材質ジルコニア、ジルコニアボール:11kgをアトライタに充填(充填率59%)して、回転数307rpmの条件で、2時間粉砕処理することにより粉砕パルプを調製した。
【0087】
<糖化反応>:
150mgの上記粉砕パルプを、3mLの酵素反応液(最終濃度0.1M酢酸緩衝液(pH5)、蓋つきスクリュー管(No.5、φ27×55mm;マルエム製))に懸濁し、Δ9prt株組換え菌株、あるいはΔ9lyt株組換え菌株の培養上清を500μL添加し、50℃で振盪撹拌(150rpm、タイテック製恒温振盪機「BR−15CF」)しながら所定の時間反応させた。反応終了後、遠心分離(4℃、12,000rpm、5分間)によって沈殿物と上清液を分離し、上清液中に遊離した還元糖をDNS法によって定量した。
【0088】
<糖化率測定(DNS法)>:
DNS溶液200μLに上清液2μLを添加し、100℃で5分間熱処理した。冷却後、反応液100μLの吸光度(535nm)をマイクロプレートリーダー(Molecular Devices社製)で測定した。上清液に遊離した還元糖量をDNS法(「生物化学実験法」還元糖の定量法、学会出版センター)によってグルコース換算により定量した。糖化率(%)は、「遊離還元糖量×0.9÷反応前のホロセルロース量×100」で算出した。各培養液上清の粉砕パルプに対する糖化活性を表5に示す。
【0089】
またPROTEIN ASSAY RAPID KIT(和光純薬工業製)を用い、キット添付の牛血アルブミンを標準として各培養上清のタンパク質濃度を測定した。糖化活性とタンパク質濃度より算出される比活性において、Δ9lyt株組換え菌株の培養上清はΔ9prt株組換え菌株を5倍程度上回っており、実施例9の結果と共に、プロテアーゼ遺伝子欠失株からの溶菌酵素関連遺伝子群の欠失が高純度な目的タンパク質の生産に極めて有効であることが示された。
【0090】
【表5】
【0091】
実施例11:N252セルラーゼ分泌生産評価:
N252セルラーゼ分泌生産用プラスミドを以下の様に構築した。
【0092】
バチルス エスピー(Bacillus sp.)KSM−N252株(FERM P−17474)より抽出したゲノムDNAを鋳型として、表3−1、表3−2に示されるN252matu/EGNTR FWと252RBaのプライマーセットを用いてPCRを行い、N252セルラーゼ(特開2001−340074号公報、Appl.Microbiol.Biotechnol.57,10−116,2001)をコードする配列番号84で示される塩基配列の1.5kbのDNA断片(H)を増幅した。またバチルス エスピー(Bacillus sp.)KSM−S237株(FERM BP−7875)より抽出したゲノムDNAを鋳型として、表3−1、表3−2に示されるS237ppp−F2(BamHI)とEGNTR RVのプライマーセットを用いてPCRを行い、S237セルラーゼ遺伝子(特開2000−210081号公報)の転写開始制御領域、翻訳開始制御領域、及び分泌シグナル配列をコードする領域を含む0.6kbのDNA断片(I)を増幅した。次いで、得られた(H)(I)の2断片を混合して鋳型とし、表3−1、表3−2に示されるS237ppp−F2(BamHI)と252RBaのプライマーセットを用いたSOE−PCRを行うことによって、S237セルラーゼ遺伝子の転写開始制御領域、翻訳開始制御領域、及び分泌シグナル配列をコードする領域の下流にN252セルラーゼ遺伝子が連結した2.1kbのDNA断片(J)を得た。得られた2.2kbのDNA断片(J)をシャトルベクターpHY300PLK(ヤクルト)のBamHI−XbaI制限酵素切断点に挿入し、N252セルラーゼ分泌生産評価用プラスミドpHYN252(S237ps)を構築した。
【0093】
つづいてN252セルラーゼ分泌生産評価は以下の様に行った。分泌生産評価用プラスミドpHYN252(S237ps)を、プロトプラスト形質転換法によってΔ9prt株及びΔ9lyt株に導入した。同様に枯草菌野生株168株にも導入した。これにより得られた組換え菌株を、10mLのLB培地で30℃で一晩振盪培養し、更にこの培養液0.05mLを50mLの2×L−マルトース培地(2%トリプトン、1%酵母エキス、1%NaCl、7.5%マルトース、7.5ppm硫酸マンガン4−5水和物、15ppmテトラサイクリン)に接種し、30℃にて3日間振盪培養を行った。遠心分離によって菌体を除くことにより培養液上清を得た。
【0094】
得られた培養液上清5μL分をSDS−ポリアクリルアミドゲル電気泳動(SDS−PAGE;濃縮ゲル4%、分離ゲル12.5%)に供し、タンパク質バンドの分離を行った。その結果を図5に示す。168株組換え菌の培養上清には目的のN252セルラーゼの分子量に相当する位置にバンドは認められなかった。一方でΔ9prt株組換え菌株とΔ9lyt株組換え菌株の培養上清にはN252セルラーゼの分子量に相当する位置にバンドが認められたことから、両株において欠失されているプロテアーゼ遺伝子がコードするプロテアーゼがN252セルラーゼの分解に関与しており、これらのプロテアーゼ遺伝子の欠失によりN252セルラーゼの分泌生産が可能になったと考えられた。またΔ9prt株組換え菌株の培養上清には目的とするN252セルラーゼ以外のタンパク質が著量認められるのに対し、Δ9lyt株組換え菌株ではこれら夾雑タンパク質は大幅に減少していた。即ち、Δ9prt株では溶菌酵素関連遺伝子群の機能性産物の作用により細胞の溶解、及び細胞内タンパク質の漏出が起こっており、Δ9lyt株ではこの様な溶菌現象に伴うタンパク質の漏出が抑制された結果、培養上清中の夾雑タンパク質が減少したものと考えられた。
【0095】
<アルカリセルラーゼ分泌生産評価>
アルカリセルラーゼ分泌生産性評価は以下の様に行った。即ち、バチルス エスピー(Bacillus sp.)KSM−S237株(FERM BP−7875)由来のアルカリセルラーゼ遺伝子(特開2000−210081号公報)断片(3.1kb)がシャトルベクターpHY300PLKのBamHI制限酵素切断点に挿入された組換えプラスミドpHY−S237を、プロトプラスト形質転換法により実施例3及び4にて構築した枯草菌変異株Δ9prt株、Δ9BP株及びΔ9BF株、更に野生株168株に導入した。これによって得られた各組換え菌株を10mLのLB培地で一夜37℃で振盪培養を行い、更にこの培養液0.05mLを50mLの2×L−マルトース培地(2%トリプトン、1%酵母エキス、1% NaCl、7.5%マルトース、7.5ppm硫酸マンガン4−5水和物、15ppmテトラサイクリン)に接種し、30℃にて3日間振盪培養を行った。遠心分離によって菌体を除いた培養液上清のアルカリセルラーゼ活性を測定し、培養によって菌体外に分泌生産されたアルカリセルラーゼの量を求めた。セルラーゼ活性測定については、1/7.5Mリン酸緩衝液(pH7.4、和光純薬)で適宜希釈したサンプル溶液50μLに0.4mM p−nitrophenyl−β−D−cellotrioside(生化学工業)を50μL加えて混和し、30℃にて反応を行った際に遊離するp−ニトロフェノール量を420nmにおける吸光度(OD420nm)変化により定量した。1分間に1μmolのp−ニトロフェノールを遊離させる酵素量を1Uとした。また遠心分離を行う前の培養液を1%塩化ナトリウム水溶液にて適宜希釈した後、分光光度計DU650(ベックマンコールター)を用いて測定した600nmの濁度(OD600)を生育度とした。
【0096】
評価の結果、Δ9prt株、Δ9BP株及びΔ9BF株はいずれも野生株より高いアルカリセルラーゼ生産性を示したが、Δ9prt株では著しい生育度の低下が認められた(表6)。一方でΔ9BP株及びΔ9BF株は高いアルカリセルラーゼ生産性を維持しつつ生育度の改善が認められており、cwlB遺伝子、PBSX遺伝子群、あるいはcwlF遺伝子の欠失によりΔ9prt株で見られる溶菌現象が抑制されたものと考えられた。
【0097】
【表6】
【技術分野】
【0001】
本発明は、新規な遺伝子欠損株、及びそれを用いたタンパク質の製造方法に関する。
【背景技術】
【0002】
微生物による有用物質の工業的生産は、アルコール飲料や味噌、醤油等の食品類、あるいはアミノ酸、有機酸、核酸関連物質、抗生物質、糖質、脂質、タンパク質等、その種類は多岐にわたっており、またそれらの用途も、食品、医薬品、洗剤、化粧品等の日用品、あるいは各種化成品原料等、幅広い分野にわたっている。
【0003】
微生物を用いた有用物質の工業的生産における一つの重要課題として、当該有用物質の生産性向上が挙げられる。従来、当該課題を解決するため、突然変異等の遺伝学的手法による高生産菌の育種が行われてきた。特に最近では、微生物遺伝学、バイオテクノロジーの発展により、遺伝子組換え技術等を用いた、より効率的な高生産菌の育種が行われている。更に、近年のゲノム解析技術の急速な発展を受け、対象とする微生物のゲノム情報を解読し、これらを積極的に産業に応用する試みもなされている。
【0004】
微生物は元来、自然界における環境変化に対応するための多種多様な遺伝子群を有しており、限定された生産培地が使用されるタンパク質等の工業的生産においては、必ずしも生産効率が高いとは言えない状況であった。
【0005】
しかるところ、バチルス属細菌において、胞子形成初期に関わる遺伝子を欠失又は不活性化した菌株が構築され、タンパク質やポリペプチドの生産性向上効果が得られている。例えば、枯草菌のsigE、sigF、spoIIE、spoIISB、sigG、又は、spoIVCBからspoIIICまでの領域に含まれる遺伝子群を欠失した宿主菌株を用いることによって、セルラーゼなどの分泌生産性が向上することが開示されている(特許文献1)。
【0006】
しかしながら、産業的なタンパク質やポリペプチド生産の場においては生産コストをできるだけ低減化する必要があり、このためには更に高い生産性が求められている。この点、上記の各遺伝子は胞子形成の第II期以降に発現し機能する遺伝子であるため、当該遺伝子を欠失や不活性化した場合に於いても細胞は胞子形成の初期段階に入っており、ゆえにタンパク質やポリペプチド生産の観点から改善の余地がある。また、胞子形成期の初期段階を制御するspo0A遺伝子(BG10765)を欠失又は不活性化することによって胞子形成が初期段階で停止することが知られているが、同時に激しい溶菌現象が生じるという問題が存在する。
【0007】
近年、枯草菌ゲノムに存在する約4100種類の遺伝子の破壊株が網羅的に研究され、271個の遺伝子が成育に必須であることが報告され(非特許文献1)、また本出願人は遺伝子組換えのための宿主微生物として、枯草菌(Bacillus subtilis) Marburg No.168系統株の遺伝子破壊株を網羅的に解析し、枯草菌ゲノムの大量域を欠失させ、各種酵素の生産性に優れた宿主変異株の創出に成功している(特許文献2)。
【0008】
また、ある種の微生物では、幾つかの細胞外プロテアーゼ遺伝子を欠失させた菌株が構築されており、例えば、nprE遺伝子の欠失により溶菌の指標となる細胞内酵素イソクエン酸デヒドロゲナーゼの若干の溶出が見られ、更にaprE遺伝子の欠失を組み合わせることにより、その溶出が顕著になることが示されている(特許文献3)。すなわち、複数の細胞外プロテアーゼの欠失を組み合わせることにより枯草菌の溶菌が生じるという問題が存在する。
【0009】
この点、枯草菌の主要な細胞壁溶解酵素CwlB(N−アセチルムラモイル−アラニンアミダーゼ、以前はLytCと呼ばれていた)をコードするcwlB遺伝子(BG10407、以前はlytC遺伝子)を不活性化することによる溶菌現象の抑制が報告されている(非特許文献3)。しかしながら、細胞壁溶解酵素群は細胞分裂や運動性など細胞の増殖にとり重要な役割を果たすといわれ、その欠失や不活性化によって細胞の生育に大きな変化が生じ、タンパク質又はポリペプチドの生産にも影響することが懸念される。例えば、予備検討データではあるが、細胞壁溶解酵素をコードするcwlB(lytC)遺伝子又はcwlG(lytD)遺伝子の不活性化によってタンパク質の分泌が抑制されたとの報告も存在する(非特許文献4)。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】特開2003−47490号公報
【特許文献2】特開2007−130013号公報
【特許文献3】特開2009−38985号公報
【非特許文献】
【0011】
【非特許文献1】K.Kobayashiら、Proc.Natl.Acad.Sci.USA,100,4678−4683,2003
【非特許文献2】Letters in Applied Microbiology,29,141−145(1999)
【非特許文献3】J.Bacteriol.,173,7304−7312(1991)
【非特許文献4】Microbiology,146,249−262(2000)
【発明の概要】
【発明が解決しようとする課題】
【0012】
本発明は、細胞外プロテアーゼ遺伝子の欠失株であっても目的タンパク質又はポリペプチドの培養液からの回収を容易にし、その生産性向上を可能とする枯草菌変異株の提供、並びに、当該枯草菌変異株を用いた目的タンパク質又はポリペプチドの製造方法の提供に関する。
【課題を解決するための手段】
【0013】
本発明者らは、枯草菌の各種遺伝子欠失株について検討したところ、細胞外プロテアーゼ遺伝子の欠失と溶菌関連遺伝子又は遺伝子群の欠失とを組み合わせた枯草菌変異株を構築することにより、細胞外プロテアーゼ遺伝子の欠失株と同等以上の目的タンパク質又はポリペプチドの分泌生産性が得られ、且つ当該枯草菌変異株の溶菌が起こらないため、、その結果、目的タンパク質又はポリペプチドの培養液からの回収が容易になることを見出した。
【0014】
すなわち本発明は、以下の(1)〜(7)に係るものである。
(1) epr、wprA、mpr、nprB、bpr、nprE、vpr、aprE及びaprXから選択される細胞外プロテアーゼ遺伝子が8個以上欠失又は不活性化し、かつ、cwlB、cwlF、PBSX、spoIIIC及びsigDから選択される溶菌酵素関連遺伝子又は遺伝子群が1個以上欠失又は不活性化している枯草菌変異株。
(2) 前記細胞外プロテアーゼ遺伝子が9個欠失又は不活性化している、前記(1)記載の枯草菌変異株。
(3) 細胞外プロテアーゼ遺伝子epr、wprA、mpr、nprB、bpr、nprE、vpr、aprE及びaprXと、溶菌酵素関連遺伝子又は遺伝子群cwlB、cwlF、PBSX、spoIIIC及びsigDと、が全て欠失又は不活性化している枯草菌変異株。
(4) 更に、異種タンパク質又はポリペプチドをコードする遺伝子を有する、前記(1)から(3)のいずれか1つに記載の枯草菌変異株。
(5) 前記異種タンパク質又はポリペプチドをコードする遺伝子の上流に転写開始制御領域、翻訳開始制御領域、又は分泌用シグナル領域が結合している、前記(4)記載の枯草菌変異株。
(6) 前記転写開始制御領域、翻訳開始制御領域又は分泌シグナル領域が、バチルス属細菌のセルラーゼ遺伝子と当該セルラーゼ遺伝子の上流0.6〜1kb領域に由来するものである、前記(5)記載の枯草菌変異株。
(7) 転写開始制御領域、翻訳開始制御領域又は分泌シグナル領域が、配列番号86で示される塩基配列からなるセルラーゼ遺伝子の塩基番号1〜659の塩基配列、配列番号88で示される塩基配列からなるセルラーゼ遺伝子の塩基番号1〜696の塩基配列、又は当該塩基配列のいずれかと70%以上の同一性を有する塩基配列からなるDNA断片、又は、当該塩基配列の一部が欠失した塩基配列からなるDNA断片である、前記(5)記載の枯草菌変異株。
(8) 前記(1)から(7)のいずれか1つに記載の枯草菌変異株を用いる、異種タンパク質又はポリペプチドの製造方法。
【発明の効果】
【0015】
本発明のプロテアーゼ/溶菌酵素欠失枯草菌変異株を用いることにより、(1)培養上清中の不要なタンパク質を低減させることが可能となり、(2)目的タンパク質の比率(比活性)を向上させることが可能となり、(3)細胞内から溶出するプロテアーゼの影響を抑制(目的タンパク質の更なる分解を抑制)することが可能となり、(4)枯草菌の形状がフィラメント状となる結果、膜透過性が向上し、目的タンパク質の生産後の処理工程において除菌が容易になる。
【図面の簡単な説明】
【0016】
【図1】SOE−PCRによる遺伝子欠失用DNA断片の調製、及び当該DNA断片を用いて標的遺伝子を欠失(薬剤耐性遺伝子と置換)する方法を模式的に示したものである。
【図2】遺伝子欠失用プラスミドを用いて標的遺伝子を欠失(薬剤耐性遺伝子と置換)する方法を模式的に示したものである。
【図3】bglC過剰発現プラスミドの構築方法を模式的に示したものである。
【図4】枯草菌168株、Δ9prt株、Δ9lyt株の培養液上清のタンパク質(セルラーゼBglC)分析を示したものである。
【図5】枯草菌168株、Δ9prt株、Δ9lyt株の培養液上清のタンパク質(セルラーゼN252)分析を示したものである。
【発明を実施するための形態】
【0017】
本発明の宿主微生物である枯草菌は、野生型のものでも変異を施したものでもよいが、全ゲノム情報が明らかにされ、遺伝子工学、ゲノム工学技術が確立され、またタンパク質を菌体外に分泌生産させる能力を有する枯草菌が好ましく、例えば枯草菌168株を元株として用い、更にその元株から所定の遺伝子を欠失させて作製した枯草菌変異株が挙げられる。
【0018】
ここで、枯草菌168株とは、枯草菌Bacillus subtilis Marburg No.168として公知の枯草菌株であり、通常、遺伝子組換えタンパク質生産の際の野生株として用いられている。また枯草菌168株の全塩基配列及び遺伝子は既に報告されており、またインターネット公開されている(Nature,390,249−256,1997及びBSORF Bacillus subtilis Genome Database[http://bacillus.genome.jp/];GenBank:AL009126.2[http://www.ncbi.nlm.nih.gov/nuccore/38680335])。当業者は、これらの情報源から得た枯草菌168株のゲノム情報に基づいて、各種遺伝子操作を行うことができる。
【0019】
本発明において、細胞外プロテアーゼ遺伝子とは、細胞内で産生された後分泌された目的タンパク質又はペプチドの他、溶菌(細胞壁分解)酵素の分解を触媒する酵素タンパク質をコードする遺伝子を意味する。また、溶菌酵素関連遺伝子又は遺伝子群とは、細胞壁の分解を触媒する酵素タンパク質、並びにそれに関連して間接的に細胞壁の分解を促進する他のタンパク質をコードする遺伝子又は遺伝子群を意味する。
【0020】
本発明において欠失又は不活性化の対象となる、細胞外プロテアーゼ遺伝子及び溶菌酵素関連遺伝子の具体例を表1に示す。なお、表中の各遺伝子の名称、番号及び機能等は、Nature,390,249−256(1997)で報告され、JAFAN:Japan Functional Analysis Network for Bacillus subtilis(BSORF DB)でインターネット公開(http://bacillus.genome.ad.jp/、2003年6月17日更新)された枯草菌ゲノムデータに基づき記載されている。
【0021】
【表1】
【0022】
参考文献1:Mol Gen Genet.1990 221(3):486−90
参考文献11:Microbiology.1996 142(Pt12):3437−44
参考文献3:J.Bacteriol.1990 172(2):1019−23
参考文献9:J.Bacteriol.1990 172(2):1024−9
参考文献10:J.Bacteriol.1991 173(20):6364−72
参考文献4:J.Bacteriol.1990 172(3):1470−7
参考文献5:J.Biol Chem.1990 265(12):6845−50
参考文献6:J.Bacteriol.1979 139(2):583−90
参考文献8:J.Bacteriol.1984 160(1):15−21
参考文献2:J.Bacteriol.1991 173(21):6889−95
参考文献7:J.Bacteriol.1984 158(2):411−8
参考文献12:J.Biosci.Bioeng.104,135−143(2007)
参考文献13:特開2003−47490号公報
参考文献14:J.Bacteriol.,173,7304−7312(1991)
参考文献15:J.Bacteriol.180,2549−2555(1998)
参考文献16:Microbiology.140,1855−1867(1994)
参考文献17:J.Bacteriol.180,2110−2117(1998)
参考文献18:Genes Dev.4,525−535(1990)
参考文献19:Bacillus subtilis Closest Relatives:From Genes to Cells,Edited by A.L.Sonenshein,American Society Microbiology,PP.289,(2002)
【0023】
すなわち、表1中のeprからaprXまでの9個の遺伝子が上記の細胞外プロテアーゼ遺伝子に該当し、lytC(cwlB)からsigD遺伝子までの5種類の遺伝子又は遺伝子群が上記の溶菌関連遺伝子又は遺伝子群に該当する。
【0024】
本発明では、細胞外に分泌される目的タンパク質又はポリペプチドの細胞外における分解を効果的に防止して目的タンパク質又はポリペプチドの生産性を向上させるため、上記の9個の細胞外プロテアーゼ遺伝子のうち、8個以上の遺伝子を欠失又は不活性化させることを特徴とし、9個欠失又は不活性化させるのがより好ましい。より具体的には、本願発明に係る枯草菌変異株は、aprE、nprB、nprE、bpr、vpr、mpr、epr及びwprAの8個の遺伝子か、又は、aprX、aprE、nprB、nprE、bpr、vpr、mpr、epr及びwprAの9個の遺伝子を欠失又は不活性化させるのが好ましい。なお、これらのプロテアーゼ遺伝子を欠失又は不活性化した枯草菌変異株は、特開2006−174707号公報に記載の方法により作製することができる。
【0025】
また、本発明では、細胞外プロテアーゼ欠失株と同等以上の目的タンパク質又はポリペプチドを生産し、且つ本発明に係る組換え枯草菌の溶菌を効果的に防止するため、上記の5個の溶菌関連遺伝子又は遺伝子群のうち、1個以上の遺伝子又は遺伝子群を欠失又は不活性化させることを特徴とし、2個以上欠失又は不活性化させるのがより好ましい。なお、本願における5個の溶菌関連遺伝子又は遺伝子群とは、cwlB、cwlF、PBSX、spoIIIC及びsigDを意味するが、そのうちのPBSXとは、以下の38種類の遺伝子群を含む領域のことを意味し、本願ではかかる領域を、欠失又は不活性化させる対象の1個とするものである。
【0026】
【表2】
【0027】
本発明におけるPBSXの欠失又は不活性化は、これらの遺伝子群の一部又は全部の欠失又は不活性化により行うことができ、全部を欠失又は不活性化させることがより好ましい。
【0028】
ここで、sigD及びspoIIICは転写因子の遺伝子である。すなわち本発明によれば、溶菌に直接関与しないが間接的に溶菌酵素に関連する遺伝子を欠失させた場合でも、溶菌を顕著に抑制することができる。
【0029】
表1及び表2に記載の遺伝子は、例えば、当該遺伝子の塩基配列において1若しくは数個の塩基配列が欠失、置換、若しくは付加された塩基配列からなる、当該遺伝子と同じ機能を有する遺伝子であってもよい。更に、表1及び表2に記載の遺伝子と同じ機能を有する、及び/又は、表1及び表2の各遺伝子と塩基配列において70%以上、好ましくは80%以上、より好ましくは90%以上、更に好ましくは95%以上、特に好ましくは98%以上の同一性を有する遺伝子も、表1及び表2に記載の遺伝子に相当する遺伝子と考えられ、本発明において欠失/不活性化しうる遺伝子に含まれる。
【0030】
これらの遺伝子又は遺伝子群は、その内部に他のDNA断片を挿入するか、あるいは当該遺伝子の転写・翻訳開始領域に変異を与える等の方法によっても不活性化できるが、好適には、標的遺伝子を物理的に削除する方がより望ましい。更に本発明の組換え枯草菌の構築において、上記以外の遺伝子の欠失又は不活性化と組み合わせてもよい。
【0031】
遺伝子又は遺伝子群の欠失又は不活性化の手順としては、表1に示す標的遺伝子を計画的に欠失又は不活性化する方法のほか、ランダムな遺伝子の欠失又は不活性化変異を与えた後、適当な方法によりタンパク質生産性の評価及び遺伝子解析を行う方法が挙げられる。
【0032】
標的遺伝子を欠失又は不活性化するには、例えば相同組換えによる方法を用いればよい。すなわち、標的遺伝子の一部を含むDNA断片を適当なプラスミドベクターにクローニングして得られる環状の組換えプラスミドを親枯草菌細胞内に取り込ませ、標的遺伝子の一部領域における相同組換えによって親枯草菌ゲノム上の標的遺伝子を分断して不活性化することが可能である。あるいは、塩基置換や塩基挿入等による不活性化変異を導入した標的遺伝子、又は標的遺伝子の外側領域を含むが標的遺伝子を含まない直鎖状のDNA断片等をPCR等の方法によって構築し、これを親枯草菌細胞内に取り込ませて親枯草菌ゲノムの標的遺伝子内の変異箇所の外側2ヶ所、又は標的遺伝子外側の2ヶ所の領域で2回交差の相同組換えを起こさせることにより、ゲノム上の標的遺伝子が欠失又は不活性化した遺伝子断片と置換することが可能となる。
【0033】
特に、相同組換えにより枯草菌の標的遺伝子を欠失又は不活性化する方法については、既にいくつかの報告例があり(Mol.Gen.Genet.,223,268(1990)等)、かかる方法を2回又はそれ以上繰り返すことにより、本発明の組換え枯草菌を得ることができる。
【0034】
また、ランダムな遺伝子の欠失又は不活性化を行う方法としては、例えばランダムにクローニングしたDNA断片を用いて上述の方法と同様な相同組換えを起こさせる方法や、親枯草菌にγ線等を照射する方法等が挙げられる。
【0035】
以下、より具体的な例として、SOE(splicing by overlap extension)−PCR法(Gene,77,61(1989))によって調製される欠失用DNA断片を用いた二重交差法による欠失方法について説明するが、本発明における遺伝子欠失方法は下記に限定されるものではない。
【0036】
本方法で用いる欠失用DNA断片は、標的遺伝子の上流に隣接する約0.2〜3kb断片(A)と、同じく下流に隣接する約0.2〜3kb断片(B)の間に、薬剤耐性マーカー遺伝子断片(C)を挿入した断片である。まず、1回目のPCRによって、標的遺伝子の上流断片(A)及び下流断片(B)、並びに薬剤耐性マーカー遺伝子断片(C)の3断片を調製するが、この際、例えば、上流断片(A)の下流末端に薬剤耐性マーカー遺伝子断片(C)の上流側10〜30塩基対配列、逆に下流断片(B)の上流末端には薬剤耐性マーカー遺伝子断片(C)の下流側10〜30塩基対配列が付加される様にデザインしたプライマーを用いる(図1)。
【0037】
次いで、1回目に調製した3種類のPCR断片(A)、(B)、(C)を鋳型とし、上流断片の上流側プライマーと下流断片の下流側プライマーを用いて2回目のPCRを行うことによって、上流断片の下流末端及び下流断片の上流末端に付加した薬剤耐性マーカー遺伝子配列に於いて、薬剤耐性マーカー遺伝子断片とのアニールが生じ、PCR増幅の結果、上流側断片と下流側断片の間に、薬剤耐性マーカー遺伝子が挿入し、(A)(C)(B)の順に結合したDNA断片を得ることができる(図1)。
【0038】
本発明では2つ又はそれ以上の遺伝子を欠失又は不活性化することが必要であるため、2種類又はそれ以上の薬剤耐性マーカー遺伝子を用いると簡便に目的の微生物菌株を分離することができる。薬剤耐性マーカー遺伝子の組合せについては特に限定されないが、例えば、カナマイシン耐性遺伝子、スペクチノマイシン耐性遺伝子、クロラムフェニコール耐性遺伝子、エリスロマイシン耐性遺伝子、テトラサイクリン耐性遺伝子などが挙げられる。
【0039】
PCRの方法についても特に限定されず、例えば実施例中に示したプライマーセットを用い、Pyrobest DNAポリメーラーゼ(宝酒造)などの一般のPCR用酵素キット等を用いて、成書(PCR Protocols.Current Methods and Applications,Edited by B.A.White,Humana Press,pp251(1993)、Gene,77,61,(1989)等)に示される通常の条件によりSOE−PCRを行うことによって、各遺伝子の欠失用DNA断片が得られる。
【0040】
また、上記の(A)、(B)及び(C)の3断片を、適当なプラスミドベクター上で、(A)(C)(B)の順になる様にクローニングし、元のプラスミドベクター部分のみを1ヶ所で切断する制限酵素で処理することによっても、同様の欠失用DNA断片を得ることができる(図2)。
【0041】
かくして得られた欠失用DNA断片を、コンピテント法等によって細胞内に導入すると、同一性のある標的遺伝子の上流及び下流の相同領域において相同組換えが生じ、標的遺伝子が薬剤耐性遺伝子で置換される。それにより得られた細胞を、クロラムフェニコールなどの薬剤を含む寒天培地上において生育させ、生じたコロニーを分離することにより、標的遺伝子が欠失した組換え枯草菌を得ることができる。また、標的遺伝子がクロラムフェニコール耐性遺伝子と置換されて欠失していることを、ゲノムを鋳型としたPCR法などによって確認することができる。
【0042】
このようなステップを繰り返し、表1及び表2に示される枯草菌の遺伝子が欠失又は不活性化された枯草菌変異株に、目的タンパク質又はポリペプチドをコードする遺伝子を導入することによって、本発明の組換え微生物を得ることができる。
【0043】
本発明の微生物を用いて生産する目的タンパク質又はポリペプチドは特に限定されないが、例えば食品用、医薬品用、化粧品用、洗浄剤用、繊維処理用、医療検査薬用等として有用な酵素や生理活性因子等のタンパク質やポリペプチドが挙げられる。産業用に用いられる酵素としては、酸化還元酵素(オキシドレダクターゼ)、転移酵素(トランスフェラーゼ)、加水分解酵素(ヒドロラーゼ)、脱離酵素(リアーゼ)、異性化酵素(イソメラーゼ)、合成酵素(リガーゼ/シンセターゼ)等が含まれるが、好適にはセルラーゼ、α−アミラーゼ、プロテアーゼ等の加水分解酵素の遺伝子が挙げられる。具体的には、多糖加水分解酵素の分類(Biochem.J.,280,309(1991))中でファミリー5に属するセルラーゼ(エンドグルカナーゼ)が挙げられ、中でも微生物由来、特にバチルス属細菌由来のセルラーゼ(エンドグルカナーゼ)が挙げられる。
【0044】
より具体的な例としては、配列番号83で示されるアミノ酸配列からなる枯草菌由来エンドグルカナーゼBglC、又は当該アミノ酸配列と70%以上、好ましくは80%以上、より好ましくは90%以上、更に好ましくは95%以上、より更に好ましくは98%以上、最も好ましくは99%以上の同一性を有するアミノ酸配列からなるエンドグルカナーゼ、又は当該アミノ酸配列において1若しくは数個のアミノ酸が置換、欠失若しくは付加されたアミノ酸配列からなるエンドグルカナーゼ;
【0045】
より具体的な例としては、配列番号85で示されるアミノ酸配列からなるバチルス属細菌KSM−N252(FERM P−17474)株由来のアルカリエンドグルカナーゼ、又は当該アミノ酸配列と70%以上、好ましくは80%以上、より好ましくは90%以上、更に好ましくは95%以上、より更に好ましくは98%以上、最も好ましくは99%以上の同一性を有するアミノ酸配列からなるアルカリエンドグルカナーゼ、又は当該アミノ酸配列において1若しくは数個のアミノ酸が置換、欠失若しくは付加されたアミノ酸配列からなるアルカリエンドグルカナーゼ
【0046】
配列番号87で示されるアミノ酸配列からなるバチルス属細菌KSM−S237株(FERM BP−7875)由来のアルカリセルラーゼ、又は当該アミノ酸配列と70%、好ましくは80%、より好ましくは90%以上、更に好ましくは95%以上、より更に好ましくは98%以上、もっとも好ましくは99%以上の同一性を有するアミノ酸配列からなるアルカリセルラーゼ、又は当該アミノ酸配列において1若しくは数個のアミノ酸が置換、欠失若しくは付加されたアミノ酸配列からなるアルカリセルラーゼ;
【0047】
配列番号89で示されるアミノ酸配列からなるバチルス属細菌KSM−64株(FERM BP−2886)由来のアルカリセルラーゼ、又は当該アミノ酸配列と70%、好ましくは80%、より好ましくは90%以上、更に好ましくは95%以上、より更に好ましくは98%以上、もっとも好ましくは99%以上の同一性を有するアミノ酸配列からなるアルカリセルラーゼ、又は当該アミノ酸配列において1若しくは数個のアミノ酸が置換、欠失若しくは付加されたアミノ酸配列からなるアルカリセルラーゼが挙げられる。
【0048】
その他の目的タンパク質又はポリペプチドの具体例としては、バチルス属細菌KSM−K38株(FERM BP−6946、特開2000-184882号)由来のアルカリアミラーゼ、又はそれと70%、好ましくは80%、より好ましくは90%以上、更に好ましくは95%以上、特に好ましくは98%以上のアミノ酸配列同一性を有するアミノ酸配列からなるアミラーゼ、又は当該アミノ酸配列において1若しくは数個のアミノ酸が置換、欠失若しくは付加されたアミノ酸配列からなるアミラーゼ;
【0049】
バチルス属細菌(Bacillus sp.)KSM−KP43株(FERM BP−6532、WO99/18218)由来のアルカリプロテアーゼ、又はそれと70%、好ましくは80%、より好ましくは90%以上、更に好ましくは95%以上、特に好ましくは98%以上のアミノ酸配列同一性を有するアミノ酸配列からなるプロテアーゼ、又は当該アミノ酸配列において1若しくは数個のアミノ酸が置換、欠失若しくは付加されたアミノ酸配列からなるアルカリプロテアーゼが挙げられる。
【0050】
宿主枯草菌に導入される上記目的タンパク質又はポリペプチドの遺伝子は、その上流に当該遺伝子の転写、翻訳、分泌に関わる制御領域、即ち、プロモーター及び転写開始点を含む転写開始制御領域、リボソーム結合部位及び開始コドンを含む翻訳開始領域並びに分泌シグナルペプチド領域から選ばれる1以上の領域が適正な形で結合されていることが望ましい。特に、転写開始制御領域、翻訳開始制御領域及び分泌シグナル領域からなる3領域が結合されていることが好ましく、更に分泌シグナルペプチド領域がバチルス(Bacillus)属細菌のセルラーゼ遺伝子由来のものであり、転写開始制御領域及び翻訳開始制御領域が当該セルラーゼ遺伝子の開始コドンから始まる長さ0.6〜1kbの上流領域であるものが、目的のタンパク質又はポリペプチド遺伝子と適正な形で結合されていることが望ましい。例えば、特開2000−210081号公報や特開平4−190793号公報等に記載されているバチルス(Bacillus)属細菌、すなわちKSM−S237株(FERM BP−7875)、KSM−64株(FERM BP−2886)由来のセルラーゼ遺伝子の転写開始制御領域、翻訳開始領域及び分泌シグナルペプチド領域が目的のタンパク質又はポリペプチドの構造遺伝子と適正に結合されていることが望ましい。より具体的には配列番号86で示される塩基配列の塩基番号1〜659の塩基配列、配列番号88で示される塩基配列からなるセルラーゼ遺伝子の塩基番号1〜696の塩基配列、また当該塩基配列に対して70%以上、好ましくは80%以上、より好ましくは90%以上、更に好ましくは95%以上、特に好ましくは98%以上の同一性を有する塩基配列からなるDNA断片、又は上記いずれかの塩基配列からなるDNAとストリンジェントの条件でハイブリダイズし且つ遺伝子の転写、翻訳、分泌に関わる機能を有するDNA、或いは上記いずれかの塩基配列の一部が欠失した塩基配列からなるDNA断片が、目的のタンパク質又はポリペプチドの構造遺伝子と適正に結合されていることが望ましい。尚、ここで、上記塩基配列の一部が欠失した塩基配列からなるDNA断片とは、上記塩基配列の一部を欠失しているが、遺伝子の転写、翻訳、分泌に関わる機能を保持しているDNA断片を意味する。またここで言うストリンジェントな条件とは、例えば[Molecular cloning−a Laboratory manual 2nd edition(Sambrookら、1989)]に記載の条件等が挙げられる。例えば、6×SSC(1×SSCの組成:0.15M塩化ナトリウム、0.015Mクエン酸ナトリウム、pH7.0)、0.5%SDS、5×デンハート及び100mg/mLニシン精子DNAを含む溶液にプローブとともに65℃で8〜16時間恒温し、ハイブリダイズさせる条件が挙げられる。
【0051】
なお、本発明におけるアミノ酸配列および塩基配列の同一性はLipman−Pearson法(Science,227,1435,(1985))によって計算される。具体的には、遺伝情報処理ソフトウェアGenetyx−Win(ソフトウェア開発)のホモロジー解析(Search homology)プログラムを用いて、パラメータであるUnit size to compare(ktup)を2として解析を行うことにより算出される。
【0052】
本発明の方法による目的タンパク質又はポリペプチドの生産は、上記組換え微生物を同化性の炭素源、窒素源、その他の必須成分を含む培地に接種し、通常の微生物培養法にて培養し、培養終了後、当該目的のタンパク質又はポリペプチドを精製することにより行えばよい。培養に使用される培地の組成及び培養条件については、使用する微生物の種類や目的タンパク質又はポリペプチドの種類等にしたがって、当業者が適宜選択することができる。目的タンパク質又はポリペプチドの精製は、培養終了後の菌体を材料として行っても良いが、本発明の組換え微生物は多量の目的タンパク質又はポリペプチドを細胞外に分泌する能力を有するため、培養終了後の培養上清を材料として行うのが有利である。
【0053】
具体的には、上記の適切なプロモーターが作動可能に連結した目的タンパク質又はポリペプチドの遺伝子を、枯草菌細胞内で自立複製可能なベクターに挿入して枯草菌細胞内に導入することにより、枯草菌形質転換体を得ることが可能であり、当該ベクターの例としてはpHY300PLK等が挙げられる。
【0054】
更に、上記で得られた枯草菌形質転換体を、適切な培地において培養し、目的タンパク質又はポリペプチドの遺伝子を発現させ、更に当該目的タンパク質又はポリペプチドを細胞外へ分泌させる。当該遺伝子を発現させる際、当該遺伝子に連結したプロモーターの性質に応じて、当該培地の組成を適宜調節するのが好ましい。特に発現誘導型プロモーターを用いる場合には、そのプロモーター活性を向上させる誘導物質を適切なタイミングで培地中に添加するのが好ましい。
【0055】
上記の枯草菌変異株は、例えば同化性の炭素源、窒素源、その他の必須成分を含む培地に接種して培養して行うことができる。培養方法は、原則的には一般的な微生物の培養方法であってよく、通常、液体培養による振盪培養、通気撹拌培養等の好気的条件下で実施するのが好ましい。
【0056】
培養終了後、培養液を遠心分離し、得られる上清又は菌体から、硫安沈殿やクロマトグラフィなどを適宜組み合わせ、常法に従い、目的タンパク質又はポリペプチドを抽出・精製することにより得ることができる。
【実施例】
【0057】
以下の実施例におけるDNA断片増幅のためのポリメラーゼ連鎖反応(PCR)は、Pyrobest DNAポリメラーゼ(タカラバイオ社製)及び付属の試薬類を用いた、GeneAmp PCR System(アプライドバイオシステムズ社製)によるDNA増幅により行った。PCR反応液は、適宜希釈した鋳型DNAを1μL、センス及びアンチセンスプライマーを各々20pmol、及びPyrobest DNAポリメラーゼを2.5U添加し、更に総反応液量を50μLとすることにより調製した。PCR反応条件は、98℃で10秒間、55℃で30秒間及び72℃で1〜5分間(目的増幅産物に応じて調整、目安は1kbあたり1分間)の3段階の温度変化を30サイクル繰り返した後、72℃で5分間反応、とした。
【0058】
また、以下の実施例において、遺伝子の上流及び下流とは、複製開始点からの位置を指すのではなく、上流とは各操作・工程において対象となる遺伝子の開始コドンの5’側に続く領域を指し、一方下流とは各操作・工程において対象となる遺伝子の終止コドンの3’側に続く領域を指すものとする。
【0059】
枯草菌の形質転換は、コンピテントセル法(J.Bacteriol.93,1925(1967))により行った。すなわち、枯草菌株をSPI培地(0.20%硫酸アンモニウム、1.40%リン酸水素二カリウム、0.60%リン酸二水素カリウム、0.10%クエン酸三ナトリウム二水和物、0.50%グルコース、0.02%カザミノ酸(Difco社製)、5mM硫酸マグネシウム、0.25μM塩化マンガン、50μg/mLトリプトファン)中で、37℃で、生育度(OD600)の値が約1となるまで振盪培養し、振盪培養後、培養液の一部を9倍量のSPII培地(0.20%硫酸アンモニウム、1.40%リン酸水素二カリウム、0.60%リン酸二水素カリウム、0.10%クエン酸三ナトリウム二水和物、0.50%グルコース、0.01%カザミノ酸(Difco社製)、5mM硫酸マグネシウム、0.40μM塩化マンガン、5μg/mLトリプトファン)に接種し、更に生育度(OD600)の値が約0.4となるまで振盪培養することにより、枯草菌株のコンピテントセルを調製した。
【0060】
次いで調製したコンピテントセル懸濁液(SPII培地における培養液)100μLに、各種DNA断片を含む溶液(SOE−PCRの反応液等)を5μL添加し、37℃で1時間振盪培養後、適切な薬剤を含むLB寒天培地(1%トリプトン、0.5%酵母エキス、1%NaCl、1.5%寒天)に全量を塗沫した。37℃での静置培養の後、生育したコロニーを形質転換体として分離した。得られた形質転換体のゲノムを抽出し、これを鋳型とするPCRを行い、目的とするゲノム構造の改変がなされたことを確認した。
【0061】
目的のタンパク質又はポリペプチドをコードする遺伝子の宿主微生物への導入は、コンピテントセル形質転換法(J.Bacteriol.93,1925(1967))、エレクトロポレーション法(FEMS Microbiol.Lett.55,135(1990))、プロトプラスト形質転換法(Mol.Gen.Genet.168,111(1979))のいずれかによって行った。
【0062】
組換え微生物によるタンパク質生産用の培養には、LB培地(1%トリプトン、0.5%酵母エキス、1%NaCl)、2×YT培地(1.6%トリプトン、1%酵母エキス、0.5%NaCl)、2×L−マルトース培地(2%トリプトン、1%酵母エキス、1%NaCl、7.5%マルトース、7.5ppm硫酸マンガン4−5水和物)、あるいはCSL発酵培地(2%酵母エキス、0.5%コーンスティープリカー(CSL)、0.05%塩化マグネシウム七水和物、0.6%尿素、0.2%L−トリプトファン、10%グルコース、0.15%リン酸二水素ナトリウム、0.35%リン酸水素二ナトリウム、pH7.2)を用いた。
【0063】
実施例1:epr遺伝子欠失用プラスミドの構築:
枯草菌168株から抽出したゲノムDNAを鋳型とし、表3−1、表3−2に示すeprfw1とeprUpr、及びeprDNfとeprrv−repの各プライマーセットを用いて、ゲノム上のepr遺伝子の上流に隣接する0.6kb断片(A)、及び下流に隣接する0.5kb断片(B)をそれぞれ調製した。また別途プラスミドpC194(J.Bacteriol.150(2),815(1982))由来のクロラムフェニコール耐性遺伝子の上流にプラスミドpUB110(Plasmid 15,93(1986))由来のrepU遺伝子のプロモーター領域(Nucleic Acids Res.17,4410(1989))を連結した1.2kb断片(C)を調製した。次に、得られた(A)(B)(C)3断片を混合して鋳型とし、表3−1、表3−2のプライマーeprfw2とCmrv2を用いたSOE−PCRを行うことによって、3断片を(A)(B)(C)の順になる様に結合させ、2.2kbのDNA断片を得た(図2)。このDNA断片の末端を平滑化及び5’−リン酸化し、プラスミドpUC118(Methods Enzymol.153,3(1987))のSmaI制限酵素部位に挿入してepr遺伝子欠失用プラスミドpUC118−CmrΔeprを構築した。尚、上記1.2kb断片(C)は、repUfwとrepUr−Cmのプライマーセット(表3−1、表3−2)及び鋳型としてプラスミドpUB110を用いて調製したrepU遺伝子プロモーター領域を含む0.4kb断片(D)と、CmUf−repとCmrv1のプライマーセット(表3−1、表3−2)及び鋳型としてプラスミドpC194を用いて調製したクロラムフェニコール耐性遺伝子を含む0.8kb断片(E)とを混合して鋳型とし、表3−1、表3−2に示すプライマーrepUfwとCmrv1を用いたSOE−PCRを行なうことによって調製した。
【0064】
【表3−1】
【0065】
【表3−2】
【0066】
実施例2:欠失用プラスミドを用いたepr遺伝子欠失株の構築:
実施例1にて構築したepr遺伝子欠失用プラスミドpUC118−CmrΔeprをコンピテントセル形質転換法(J.Bacteriol.93,1925(1967))によって枯草菌168株に導入し、epr遺伝子上流領域、あるいは下流領域の相当する領域間での1重交差の相同組換えによりゲノムDNAと融合した形質転換株を、クロラムフェニコール耐性を指標に取得した。得られた形質転換株をLB培地に接種し、37℃にて2時間培養後、再度、コンピテンス誘導操作を行うことにより、ゲノム上で重複して存在するepr遺伝子上流領域、あるいは下流領域の間におけるゲノム内相同組換えを誘導した。図2に示す様に、プラスミド導入の際と異なる領域で相同組換えが生じた場合、プラスミドに由来するクロラムフェニコール耐性遺伝子及びpUC118ベクター領域の脱落に伴ってepr遺伝子が欠失する。次に、クロラムフェニコール感受性となった株の存在比率を高める為、以下の要領でアンピシリン濃縮操作を行なった。コンピテントセル誘導後の培養液を終濃度5ppmのクロラムフェニコール及び終濃度100ppmのアンピシリンナトリウムを含むLB培地1mLに、600nmにおける濁度(OD600)が0.003となるように接種した。37℃にて5時間培養後、10,000ppmのアンピシリンナトリウム水溶液を10μL添加し、更に3時間培養した。培養終了後、2%塩化ナトリウム水溶液にて菌体を遠心洗浄した後、1mLの2%塩化ナトリウム水溶液に懸濁し、懸濁液100μLをLB寒天培地に塗沫した。37℃にて約15時間インキュベートし、生育した菌株のうち、プラスミド領域の脱落に伴ってクロラムフェニコール感受性となった株を選抜した。選抜した菌株のゲノムDNAを鋳型とし、表3−1、表3−2に示すプライマーeprfw2とeprrv−repを用いたPCRを行なってepr遺伝子欠失の確認を行ない、epr遺伝子欠失株を取得した。
【0067】
実施例3:プロテアーゼ遺伝子8重欠失株及びプロテアーゼ9重欠失株の構築:
epr遺伝子欠失株に対し、次の欠失としてwprA遺伝子の欠失をepr遺伝子欠失と同様に行なった。即ち、実施例1と同様にしてwprA遺伝子欠失用プラスミドpUC118−CmrΔwprAを構築し、構築したプラスミドのゲノムDNAへの導入とそれに続くゲノム内相同組換えによるwprA遺伝子の欠失によりepr遺伝子とwprA遺伝子の2重欠失株を取得した。以降同様の操作を繰り返すことにより、mpr、nprB、bpr、nprE、vpr、aprEの各遺伝子を順次欠失させ、8種類のプロテアーゼ遺伝子が欠失したプロテアーゼ8重欠失株を構築し、Δ8prt株と命名した。更に同様の操作を繰り返すことによりaprX遺伝子を欠失させたプロテアーゼ9重欠失株を構築し、Δ9prt株と命名した。各欠失を行う際に用いたプライマーの配列を表3−1、表3−2に示し、また各プライマーと実施例1で示したepr遺伝子欠失に用いたプライマーとの対応について表4に示す。
【0068】
【表4】
【0069】
実施例4:プロテアーゼ9重欠失株からのcwlB遺伝子、PBSX遺伝子群、cwlF遺伝子の欠失:
実施例3と同様にして、Δ9prt株より表3−1、表3−2及び表4に示すcwlB遺伝子欠失用のプライマーセットを用いてcwlB遺伝子の欠失を行い、9種類のプロテアーゼ遺伝子とcwlB遺伝子が欠失したΔ9B株を構築した。続いてPBSX遺伝子群の欠失を行うこととした。尚、本発明においてPBSX遺伝子群とは、枯草菌ゲノム上に連続して存在するxlyB遺伝子〜spoIISA遺伝子の38遺伝子群を指すものとする。Δ9B株より表3−1、表3−2及び表4に示すPBSX遺伝子群欠失用のプライマーセットを用いてPBSX遺伝子群の欠失を行い、Δ9BP株を構築した。またΔ9B株より表3−1、表3−2及び表4に示すcwlF遺伝子欠失用のプライマーセットを用いてcwlF遺伝子の欠失を行い、Δ9BF株を構築した。更にΔ9BP株より表3−1、表3−2及び表4に示すcwlF遺伝子欠失用のプライマーセットを用いてcwlF遺伝子の欠失を行い、Δ9BPF株を構築した。
【0070】
実施例5:Δ9BPF株からのspoIIIC遺伝子の欠失:
図1に基づいて、ゲノム中spoIIIC遺伝子の薬剤耐性遺伝子による欠失方法を説明する。尚、spoIIIC遺伝子は、spoIVCB遺伝子と共に枯草菌の胞子形成期に特異的に機能するシグマ因子SigKをコードする遺伝子である。
【0071】
枯草菌168株から抽出したゲノムDNAを鋳型とし、表3−1、表3−2に示すspoIIIC−FW及びspoIIIC/Em−Rのプライマーセットを用いて、ゲノム中のspoIIIC遺伝子の上流に隣接する1.0kb断片(A)をPCRにより増幅した。また、上記ゲノムDNAを鋳型とし、spoIIIC/Em−F及びspoIIIC−RVのプライマーセットを用いて、ゲノム中のspoIIIC遺伝子の下流に隣接する1.0kb断片(B)をPCRにより増幅した。
【0072】
更に、プラスミドpMUTIN4(Microbiology.144,3097(1998))を鋳型として、表3−1、表3−2に示すemf2及びemr2のプライマーセットを用いて、1.3kbのエリスロマイシン(Em)耐性遺伝子領域(C)をPCRにより調製した。
【0073】
次に、図1に示すように、得られた1.0kb断片(A)、1.0kb断片(B)及びCm耐性遺伝子領域(C)の3断片を混合して鋳型として、表1に示したspoIIIC−FW2及びspoIIIC−RV2のプライマーセットを用いたSOE−PCR法によって、3断片が1.0kb断片(A)、Em耐性遺伝子領域(C)、1.0kb断片(B)の順に含まれる3.3kbのDNA断片(D)を得た。
【0074】
更に、コンピテントセル形質転換法によって、得られたDNA断片(D)を用いて、Δ9BPF株の形質転換を行った。形質転換後、エリスロマイシン(1μg/mL)とリンコマイシン(25μg/mL)を含むLB寒天培地上に生育したコロニーを形質転換体として分離した。
【0075】
得られた形質転換体のゲノムDNAを抽出し、PCRによってspoIIIC遺伝子が欠失し、Em耐性遺伝子に置換していることを確認した。以上のようにして、Δ9BPFC株を構築した。
【0076】
実施例6:Δ9BPFC株からのsigD遺伝子の欠失:
図1に基づいて、ゲノム中sigD遺伝子の薬剤耐性遺伝子による欠失方法を説明する。尚、sigD遺伝子は枯草菌の細胞壁溶解、鞭毛形成、あるいは走化性等に関与する遺伝子の発現を制御するシグマ因子SigDをコードする遺伝子である。
【0077】
枯草菌168株から抽出したゲノムDNAを鋳型とし、表3−1、表3−2に示すsigD−FW及びsigD/Cm−Rのプライマーセットを用いて、ゲノム中のsigD遺伝子の上流に隣接する1.0kb断片(A)をPCRにより増幅した。また、上記ゲノムDNAを鋳型とし、sigD/Cm−F及びsigD−RVのプライマーセットを用いて、ゲノム中のsigD遺伝子の下流に隣接する1.0kb断片(B)をPCRにより増幅した。
【0078】
更に、プラスミドDNA pC194を鋳型とし、表3−1、表3−2に示すcatf及びcatrのプライマーセットを用いて、0.85kbのクロラムフェニコール(Cm)耐性遺伝子領域(C)をPCRにより調製した。
【0079】
次に、図1に示すように、得られた1.0kb断片(A)、1.0kb断片(B)及びCm耐性遺伝子領域(C)の3断片を混合して鋳型として、表1に示したsigD−FW2及びsigD−RV2のプライマーセットを用いたSOE−PCR法によって、3断片が1.0kb断片(A)、Cm耐性遺伝子領域(C)、1.0kb断片(B)の順に含まれる2.8kbのDNA断片(D)を得た。
【0080】
更に、コンピテントセル形質転換法によって、得られたDNA断片(D)を用いて、168株の形質転換を行った。形質転換後、クロラムフェニコール(10μg/mL)を含むLB寒天培地上に生育したコロニーを形質転換体として分離した。
【0081】
得られた形質転換体のゲノムDNAを抽出し、PCRによってsigD遺伝子が欠失し、Cm耐性遺伝子に置換していることを確認した。以上のようにして、Δ9BPFCD株(Δ9lyt株)を構築した。
【0082】
実施例7:bglC過剰発現プラスミドの構築:
プラスミドの構築は、In−FusionTM Advantage PCR Cloning Kit(Clontech社)を用いて行なった(図3)。プロトコールに従って設計したプライマー(表3−1、表3−2)を用いて、バチルス エスピー(Bacillus sp.)KSM−S237株(FERM BP−7875)由来のアルカリセルラーゼ遺伝子(特開2000−210081号)のプロモーターとシグナル配列の断片を、pHYS237(Biosci.Biotechnol.Biochem.,64(11):2281−9,2000)を鋳型にして、並びに、配列番号82で示される枯草菌セルラーゼ遺伝子bglCを、枯草菌ゲノムDNAを鋳型にして、それぞれPSF_infu(EcI)とPS237Rのプライマーセット、及びbglC−sig/PSFとbglC_infu(SaI)のプライマーセットを用いて増幅した。得られた増幅産物を、制限酵素EcoRI及びSalI消化により線状化したpHY300PLK及びIn−Fusion酵素を適当量で混合して30分間反応を行ない、S237セルラーゼのプロモーター配列及びシグナル配列にbglCの構造遺伝子が連結したプラスミドを構築し、それをpHPS−bglCと命名した。更に、当該プラスミドを大腸菌HB101コンピテントセル(TaKaRa)に導入し、形質転換した。
【0083】
CMC及びトリパンブルーを添加したLB寒天培地を用い、テトラサイクリン耐性により大腸菌形質転換体を選抜した。また、当該形質転換体がハロー形成をすることが判明した。形質転換体より精製したプラスミドを用いて制限酵素処理あるいはPCRを行い、形質転換が適切に行われていることを確認した。
【0084】
実施例8:枯草菌宿主へのプラスミド導入及び得られた形質転換体の培養:
実施例7にて構築したプラスミドpHPS−bglCを、プロトプラスト形質転換法によって実施例3及び実施例6にて構築した枯草菌変異株Δ9prt株及びΔ9lyt株に導入した。同様に枯草菌野生株168株にも導入した。これにより得られた組換え菌株を、10mLのLB培地で30℃で一晩振盪培養し、更にこの培養液0.05mLを50mLの2×L−マルトース培地(2%トリプトン、1%酵母エキス、1%NaCl、7.5%マルトース、7.5ppm硫酸マンガン4−5水和物、15ppmテトラサイクリン)に接種し、30℃にて3日間振盪培養を行った。遠心分離によって菌体を除くことにより培養液上清を得た。
【0085】
実施例9:枯草菌形質転換体培養液上清のタンパク質分析:
実施例8にて得られた培養液上清5μL分をSDS−ポリアクリルアミドゲル電気泳動(SDS−PAGE;濃縮ゲル4%、分離ゲル12.5%)に供し、タンパク質バンドの分離を行った(168株組換え菌株の2サンプル、Δ9prt株組換え菌株の2サンプル、Δ9lyt株組換え菌株の3サンプル)。その結果を図4に示す。168株組換え菌の培養上清には目的の枯草菌セルラーゼ遺伝子bglCがコードするセルラーゼBglCの分子量に相当する位置にバンドは認められなかった。一方でΔ9prt株組換え菌株とΔ9lyt株組換え菌株の培養上清にはBglCの分子量に相当する位置にバンドが認められたことから、両株において欠失されているプロテアーゼ遺伝子がコードするプロテアーゼがBglCの分解に関与しており、これらのプロテアーゼ遺伝子の欠失によりBglCの分泌生産が可能になったと考えられた。またΔ9prt株組換え菌株の培養上清には目的とするBglC以外のタンパク質が著量認められるのに対し、Δ9lyt株組換え菌株ではこれら夾雑タンパク質は大幅に減少していた。即ち、Δ9prt株では溶菌酵素関連遺伝子群の機能性産物の作用により細胞の溶解、及び細胞内タンパク質の漏出が起こっており、Δ9lyt株ではこの様な溶菌現象に伴うタンパク質の漏出が抑制された結果、培養上清中の夾雑タンパク質が減少したものと考えられた。
【0086】
実施例10:培養上清のセルロース糖化活性:
セルロース(粉砕パルプ)を用い、セルロース糖化反応を行った。なお粉砕パルプは、粉末状パルプ(日本製紙ケミカル社製「W−400G」、400メッシュパス90以上、セルロース含有量99質量%、水分含量1質量%)を媒体撹拌式ミル(アトライタ、三井鉱山(株)製、「MA1D−X」、容器全容量:5.5L)に500g投入し、媒体として、直径10mm、材質ジルコニア、ジルコニアボール:11kgをアトライタに充填(充填率59%)して、回転数307rpmの条件で、2時間粉砕処理することにより粉砕パルプを調製した。
【0087】
<糖化反応>:
150mgの上記粉砕パルプを、3mLの酵素反応液(最終濃度0.1M酢酸緩衝液(pH5)、蓋つきスクリュー管(No.5、φ27×55mm;マルエム製))に懸濁し、Δ9prt株組換え菌株、あるいはΔ9lyt株組換え菌株の培養上清を500μL添加し、50℃で振盪撹拌(150rpm、タイテック製恒温振盪機「BR−15CF」)しながら所定の時間反応させた。反応終了後、遠心分離(4℃、12,000rpm、5分間)によって沈殿物と上清液を分離し、上清液中に遊離した還元糖をDNS法によって定量した。
【0088】
<糖化率測定(DNS法)>:
DNS溶液200μLに上清液2μLを添加し、100℃で5分間熱処理した。冷却後、反応液100μLの吸光度(535nm)をマイクロプレートリーダー(Molecular Devices社製)で測定した。上清液に遊離した還元糖量をDNS法(「生物化学実験法」還元糖の定量法、学会出版センター)によってグルコース換算により定量した。糖化率(%)は、「遊離還元糖量×0.9÷反応前のホロセルロース量×100」で算出した。各培養液上清の粉砕パルプに対する糖化活性を表5に示す。
【0089】
またPROTEIN ASSAY RAPID KIT(和光純薬工業製)を用い、キット添付の牛血アルブミンを標準として各培養上清のタンパク質濃度を測定した。糖化活性とタンパク質濃度より算出される比活性において、Δ9lyt株組換え菌株の培養上清はΔ9prt株組換え菌株を5倍程度上回っており、実施例9の結果と共に、プロテアーゼ遺伝子欠失株からの溶菌酵素関連遺伝子群の欠失が高純度な目的タンパク質の生産に極めて有効であることが示された。
【0090】
【表5】
【0091】
実施例11:N252セルラーゼ分泌生産評価:
N252セルラーゼ分泌生産用プラスミドを以下の様に構築した。
【0092】
バチルス エスピー(Bacillus sp.)KSM−N252株(FERM P−17474)より抽出したゲノムDNAを鋳型として、表3−1、表3−2に示されるN252matu/EGNTR FWと252RBaのプライマーセットを用いてPCRを行い、N252セルラーゼ(特開2001−340074号公報、Appl.Microbiol.Biotechnol.57,10−116,2001)をコードする配列番号84で示される塩基配列の1.5kbのDNA断片(H)を増幅した。またバチルス エスピー(Bacillus sp.)KSM−S237株(FERM BP−7875)より抽出したゲノムDNAを鋳型として、表3−1、表3−2に示されるS237ppp−F2(BamHI)とEGNTR RVのプライマーセットを用いてPCRを行い、S237セルラーゼ遺伝子(特開2000−210081号公報)の転写開始制御領域、翻訳開始制御領域、及び分泌シグナル配列をコードする領域を含む0.6kbのDNA断片(I)を増幅した。次いで、得られた(H)(I)の2断片を混合して鋳型とし、表3−1、表3−2に示されるS237ppp−F2(BamHI)と252RBaのプライマーセットを用いたSOE−PCRを行うことによって、S237セルラーゼ遺伝子の転写開始制御領域、翻訳開始制御領域、及び分泌シグナル配列をコードする領域の下流にN252セルラーゼ遺伝子が連結した2.1kbのDNA断片(J)を得た。得られた2.2kbのDNA断片(J)をシャトルベクターpHY300PLK(ヤクルト)のBamHI−XbaI制限酵素切断点に挿入し、N252セルラーゼ分泌生産評価用プラスミドpHYN252(S237ps)を構築した。
【0093】
つづいてN252セルラーゼ分泌生産評価は以下の様に行った。分泌生産評価用プラスミドpHYN252(S237ps)を、プロトプラスト形質転換法によってΔ9prt株及びΔ9lyt株に導入した。同様に枯草菌野生株168株にも導入した。これにより得られた組換え菌株を、10mLのLB培地で30℃で一晩振盪培養し、更にこの培養液0.05mLを50mLの2×L−マルトース培地(2%トリプトン、1%酵母エキス、1%NaCl、7.5%マルトース、7.5ppm硫酸マンガン4−5水和物、15ppmテトラサイクリン)に接種し、30℃にて3日間振盪培養を行った。遠心分離によって菌体を除くことにより培養液上清を得た。
【0094】
得られた培養液上清5μL分をSDS−ポリアクリルアミドゲル電気泳動(SDS−PAGE;濃縮ゲル4%、分離ゲル12.5%)に供し、タンパク質バンドの分離を行った。その結果を図5に示す。168株組換え菌の培養上清には目的のN252セルラーゼの分子量に相当する位置にバンドは認められなかった。一方でΔ9prt株組換え菌株とΔ9lyt株組換え菌株の培養上清にはN252セルラーゼの分子量に相当する位置にバンドが認められたことから、両株において欠失されているプロテアーゼ遺伝子がコードするプロテアーゼがN252セルラーゼの分解に関与しており、これらのプロテアーゼ遺伝子の欠失によりN252セルラーゼの分泌生産が可能になったと考えられた。またΔ9prt株組換え菌株の培養上清には目的とするN252セルラーゼ以外のタンパク質が著量認められるのに対し、Δ9lyt株組換え菌株ではこれら夾雑タンパク質は大幅に減少していた。即ち、Δ9prt株では溶菌酵素関連遺伝子群の機能性産物の作用により細胞の溶解、及び細胞内タンパク質の漏出が起こっており、Δ9lyt株ではこの様な溶菌現象に伴うタンパク質の漏出が抑制された結果、培養上清中の夾雑タンパク質が減少したものと考えられた。
【0095】
<アルカリセルラーゼ分泌生産評価>
アルカリセルラーゼ分泌生産性評価は以下の様に行った。即ち、バチルス エスピー(Bacillus sp.)KSM−S237株(FERM BP−7875)由来のアルカリセルラーゼ遺伝子(特開2000−210081号公報)断片(3.1kb)がシャトルベクターpHY300PLKのBamHI制限酵素切断点に挿入された組換えプラスミドpHY−S237を、プロトプラスト形質転換法により実施例3及び4にて構築した枯草菌変異株Δ9prt株、Δ9BP株及びΔ9BF株、更に野生株168株に導入した。これによって得られた各組換え菌株を10mLのLB培地で一夜37℃で振盪培養を行い、更にこの培養液0.05mLを50mLの2×L−マルトース培地(2%トリプトン、1%酵母エキス、1% NaCl、7.5%マルトース、7.5ppm硫酸マンガン4−5水和物、15ppmテトラサイクリン)に接種し、30℃にて3日間振盪培養を行った。遠心分離によって菌体を除いた培養液上清のアルカリセルラーゼ活性を測定し、培養によって菌体外に分泌生産されたアルカリセルラーゼの量を求めた。セルラーゼ活性測定については、1/7.5Mリン酸緩衝液(pH7.4、和光純薬)で適宜希釈したサンプル溶液50μLに0.4mM p−nitrophenyl−β−D−cellotrioside(生化学工業)を50μL加えて混和し、30℃にて反応を行った際に遊離するp−ニトロフェノール量を420nmにおける吸光度(OD420nm)変化により定量した。1分間に1μmolのp−ニトロフェノールを遊離させる酵素量を1Uとした。また遠心分離を行う前の培養液を1%塩化ナトリウム水溶液にて適宜希釈した後、分光光度計DU650(ベックマンコールター)を用いて測定した600nmの濁度(OD600)を生育度とした。
【0096】
評価の結果、Δ9prt株、Δ9BP株及びΔ9BF株はいずれも野生株より高いアルカリセルラーゼ生産性を示したが、Δ9prt株では著しい生育度の低下が認められた(表6)。一方でΔ9BP株及びΔ9BF株は高いアルカリセルラーゼ生産性を維持しつつ生育度の改善が認められており、cwlB遺伝子、PBSX遺伝子群、あるいはcwlF遺伝子の欠失によりΔ9prt株で見られる溶菌現象が抑制されたものと考えられた。
【0097】
【表6】
【特許請求の範囲】
【請求項1】
epr、wprA、mpr、nprB、bpr、nprE、vpr、aprE及びaprXから選択される細胞外プロテアーゼ遺伝子が8個以上欠失又は不活性化し、かつ、cwlB、cwlF、PBSX、spoIIIC及びsigDから選択される溶菌酵素関連遺伝子又は遺伝子群が1個以上欠失又は不活性化している枯草菌変異株。
【請求項2】
前記細胞外プロテアーゼ遺伝子が9個欠失又は不活性化している、請求項1記載の枯草菌変異株。
【請求項3】
細胞外プロテアーゼ遺伝子epr、wprA、mpr、nprB、bpr、nprE、vpr、aprE及びaprXと、
溶菌酵素関連遺伝子又は遺伝子群cwlB、cwlF、PBSX、spoIIIC及びsigDと、が全て欠失又は不活性化している枯草菌変異株。
【請求項4】
更に、異種タンパク質又はポリペプチドをコードする遺伝子を有する、請求項1から3のいずれか1項記載の枯草菌変異株。
【請求項5】
前記異種タンパク質又はポリペプチドをコードする遺伝子の上流に転写開始制御領域、翻訳開始制御領域、又は分泌用シグナル領域が結合している、請求項4記載の枯草菌変異株。
【請求項6】
前記転写開始制御領域、翻訳開始制御領域又は分泌シグナル領域が、バチルス属細菌のセルラーゼ遺伝子と当該セルラーゼ遺伝子の上流0.6〜1kb領域に由来するものである、請求項5記載の枯草菌変異株。
【請求項7】
転写開始制御領域、翻訳開始制御領域又は分泌シグナル領域が、配列番号86で示される塩基配列からなるセルラーゼ遺伝子の塩基番号1〜659の塩基配列、配列番号88で示される塩基配列からなるセルラーゼ遺伝子の塩基番号1〜696の塩基配列、又は当該塩基配列のいずれかと70%以上の同一性を有する塩基配列からなるDNA断片、又は、当該塩基配列の一部が欠失した塩基配列からなるDNA断片である、請求項5記載の枯草菌変異株。
【請求項8】
請求項1から7のいずれか1項記載の枯草菌変異株を用いる、異種タンパク質又はポリペプチドの製造方法。
【請求項1】
epr、wprA、mpr、nprB、bpr、nprE、vpr、aprE及びaprXから選択される細胞外プロテアーゼ遺伝子が8個以上欠失又は不活性化し、かつ、cwlB、cwlF、PBSX、spoIIIC及びsigDから選択される溶菌酵素関連遺伝子又は遺伝子群が1個以上欠失又は不活性化している枯草菌変異株。
【請求項2】
前記細胞外プロテアーゼ遺伝子が9個欠失又は不活性化している、請求項1記載の枯草菌変異株。
【請求項3】
細胞外プロテアーゼ遺伝子epr、wprA、mpr、nprB、bpr、nprE、vpr、aprE及びaprXと、
溶菌酵素関連遺伝子又は遺伝子群cwlB、cwlF、PBSX、spoIIIC及びsigDと、が全て欠失又は不活性化している枯草菌変異株。
【請求項4】
更に、異種タンパク質又はポリペプチドをコードする遺伝子を有する、請求項1から3のいずれか1項記載の枯草菌変異株。
【請求項5】
前記異種タンパク質又はポリペプチドをコードする遺伝子の上流に転写開始制御領域、翻訳開始制御領域、又は分泌用シグナル領域が結合している、請求項4記載の枯草菌変異株。
【請求項6】
前記転写開始制御領域、翻訳開始制御領域又は分泌シグナル領域が、バチルス属細菌のセルラーゼ遺伝子と当該セルラーゼ遺伝子の上流0.6〜1kb領域に由来するものである、請求項5記載の枯草菌変異株。
【請求項7】
転写開始制御領域、翻訳開始制御領域又は分泌シグナル領域が、配列番号86で示される塩基配列からなるセルラーゼ遺伝子の塩基番号1〜659の塩基配列、配列番号88で示される塩基配列からなるセルラーゼ遺伝子の塩基番号1〜696の塩基配列、又は当該塩基配列のいずれかと70%以上の同一性を有する塩基配列からなるDNA断片、又は、当該塩基配列の一部が欠失した塩基配列からなるDNA断片である、請求項5記載の枯草菌変異株。
【請求項8】
請求項1から7のいずれか1項記載の枯草菌変異株を用いる、異種タンパク質又はポリペプチドの製造方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図2】
【図3】
【図4】
【図5】
【公開番号】特開2012−200168(P2012−200168A)
【公開日】平成24年10月22日(2012.10.22)
【国際特許分類】
【出願番号】特願2011−65907(P2011−65907)
【出願日】平成23年3月24日(2011.3.24)
【出願人】(000000918)花王株式会社 (8,290)
【Fターム(参考)】
【公開日】平成24年10月22日(2012.10.22)
【国際特許分類】
【出願日】平成23年3月24日(2011.3.24)
【出願人】(000000918)花王株式会社 (8,290)
【Fターム(参考)】
[ Back to top ]