
遺伝子治療に使用されるウィルスベクターに対するT細胞介在型免疫反応を検出及び調節する組成物及び方法
【解決手段】 ウィルス性形質導入された導入遺伝子含有細胞のT細胞介在型破壊を抑制する組成物及び方法が提供される。
【発明の詳細な説明】
【技術分野】
【0001】
本願は2006年5月31日付で出願された米国特許仮出願第60/809,956号に優先権を主張するものであり、その全ての内容がこの参照により本願明細書に取り込まれる。
【0002】
米国特許法第202条(35U.S.C.§202(c))に従って、米国国立衛生研究所(National Institutes of Health)の研究助成金番号PO1 HL078810からの資金で一部なされた本願に記載される本発明の特定権利は、米国政府が有するものであることが認められる。
【背景技術】
【0003】
本発明は遺伝子治療及び免疫学の分野に関する。より具体的には、本発明はウィルスベクター抗原の存在を検出する組成物及び方法を提供し、ウィルスベクター抗原に対する免疫反応を抑制及び回避する組成物及び方法を含むものである。
【0004】
複数の出版物と特許文献が本発明に関連する最新の従来技術を説明するために本願明細書の中で参照されている。これらの引用文献の各々は参照により本願明細書に完全に取り込まれる。
【0005】
野生型アデノウィルス随伴ウィルス(adenovirus−associated virus:AAV)は〜4.7kbの一本鎖DNA遺伝子を持つパルボウィルスである。前記ウィルスは複製機能を自然欠損しており、複製にはアデノウィルス及びヘルペスウィルスのようなヘルパーウィルスを必要とする。前記ウィルスはいかなる疾患にも関連が見出されていないが、その代わりにアデノウィルス分離株の混入物質として最初に分離された(4)。これまでに6個の血清型が記述されており、それらは非常に良く保存された配列を持つ(保存率は62%〜99%と異なる)。前記ウィルスの遺伝子は2個の逆方向末端反復配列(inverted terminal repeats:ITRs)によって挟まれており、3個のカプシド(capsid)遺伝子(VP1、2、3)及び4個のレップ(rep)タンパク質をコードするものであり、このレップタンパク質はDNA複製及びAAV生活環の制御に関わる。最近、さらなる血清型(AAV−7、−8、−9)がアカゲザル及びヒトから分離され、AAV−1〜6と比較してこれらの血清型もまた60%よりも高い率で保存されている(5)。
【0006】
野生型AAVが遺伝子導入媒体としての使用のために遺伝子操作された。すなわち、レップ(rep)及びキャップ(cap)遺伝子を欠損させ、ウィルスDNA翻訳領域がないように、目的とする治療遺伝子を2個のITRsの間に挿入する。1990年代の半ばに複数の研究グループ(6〜10)によって、組み換えAAVが骨格筋、肝臓、中枢神経系(CNS)、及び呼吸器官を含む多数の非分裂細胞型に感染し、この組み換えAAVにより免疫学的にコンピテントな動物中で導入遺伝子長期発現が誘導されることが示された。この興味深い知見は多数の研究グループによって利用され、組み換えAAVの投与により小型及び大型動物モデルにおいて遺伝子性疾患が治癒した(11〜17)という目覚ましい結果のポートフォリオが現在集まっている。ヒトにおける経験はより限定されるが(18〜24)、安全性及び遺伝子導入とその発現が証明されているという観点で有望とされている。しかしながら、ほとんどの例で表現型の修正が起こるほど十分に発現レベルが十分に高くない。
【0007】
我々の研究の主要目的は、血友病及び他の血液凝固疾患の治療のための安全且つ有効なアデノ随伴ウィルス(adeno−associated virus:AAV)を介した遺伝子導入システムを確立することである。血友病イヌモデルの長期間にわたる血友病の治癒(1)に基づき臨床試験が計画され、その中で重度のB型血友病の患者にAAV−F.IX(Factor IX:F.IX)経肝動脈注入を行った。1人の患者はベクター注入後第2週目までに11.8%(治療域レベル)の循環ファクターIXを得た。このレベルは約4週間続き、その後徐々に低下し始め、結局患者の基準値<1%に戻った。F.IXレベルの減少に一致して、血中肝アミノ基転移酵素が上昇し始め、注入後5週で最高値に達し、その後数週間で正常値にまで減少した。このように前記患者は、マウス、ラット、ウサギ、血友病イヌ、及び非ヒト霊長類を含む実験動物で観察されたものとはかなり異なる経過をたどった。実験動物とは対照的に、前記ヒト患者は、AAVに対する低い中和抗体適定値の存在、並びにIgG抗体が存在することから推定すればわかるように、既にAAV−2に対する免疫を持っており、前記患者はリンパ球様分画にAAVに特異的な記憶T細胞の一群を持っているようであった(2)。同様の所見が前記臨床試験の別の患者でも観察され、この患者での免疫学的研究によりAAVカプシド中の特異的なペプチドに対するT細胞反応が立証された。明らかに、前記反応はベクターを注入して数週後、末梢血で検出できたが、注入前には検出できなかった。
【0008】
これらの知見から遺伝子治療アプローチを有効にするために、一部の例で、導入遺伝子発現細胞のT細胞介在型破壊を防ぐために免疫反応を調節することが必要であるのは明らかである。
【先行技術文献】
【非特許文献】
【0009】
【非特許文献1】Mount JD,Herzog RW,Tillson DM,et al.Sustained phenotypic correction of hemophilia B dogs with a factor IX null mutation by liver−directed gene therapy.Blood.2002;99:2670−2676.
【非特許文献2】Appay V,Rowland−Jones SL.Lessons from the study of T−cell differentiation in persistent human virus infection.Semin Immunol.2004;16:205−212.
【非特許文献3】Manno CS,Arruda VR,Pierce GF,et al.Successful transduction of liver in hemophilia by AAV−Factor IX and limitations imposed by the host immune response.Nat Med.2006;12:342−347.
【非特許文献4】Blacklow NR,Hoggan MD,Sereno MS,et al.A seroepidemiologic study of adenovirus−associated virus infection in infants and children.Am J Epidemiol.1971;94.
【非特許文献5】Gao GP,Alvira MR,Wang L,Calcedo R,Johnston J,Wilson JM.Novel adeno−associated viruses from rhesus monkeys as vectors for human gene therapy.Proc Natl Acad Sci USA.2002;99:11854−11859.
【非特許文献6】Xiao X,Li J,Samulski RJ.Efficient long−term gene transfer into muscle tissue of immunocompetent mice by adeno−associated virus vector.j Virol.1996;70:8098−8108.
【非特許文献7】Kessler PD,Podsakoff GM,Chen X,et al.Gene delivery to skeletal muscle results in sustained expression and systemic delivery of a therapeutic protein.Proc Natl Acad Sci USA.1996;93:14082−14087.
【非特許文献8】Flotte TR, Afione SA, Conrad C,et al.Stable in vivo expression of the cystic fibrosis transmembrane conductance regulator with an adeno−associated virus vector.Proc Natl Acad Sci USA.1993;90:10613−10617.
【非特許文献9】Kaplitt MG,Leone P,Samulski RJ,et al.Long−term gene expression and phenotypic correction using adeno−associated virus vectors in the mammalian brain.Nat Genet.1994;8:148−154.
【非特許文献10】Flotte TR,Carter BJ.Adeno−associated virus vectors for gene therapy.Gene Ther.1995;2:357−362.
【非特許文献11】Sanchez−Pernaute R,Harvey−White J,Cunningham J,Bankiewicz KS.Functional effect of adeno−associated virus mediated gene transfer of aromatic L−amino acid decarboxylase into the striatum of 6−OHDA−lesioned rats.Mol Ther.2001;4:324−330.
【非特許文献12】Acland GM,Aguirre GD,Ray J,et al.Gene therapy restores vision in a canine model of childhood blindness.Nat Genet.2001;28:92−95.
【非特許文献13】Ho TT,Maguire AM,Aguirre GD,et al.Phenotypic rescue after adeno−associated virus−mediated delivery of 4−sulfatase to the retinal pigment epithelium of feline mucopolysaccharidosis VI.J Gene Med.2002;4:613−621.
【非特許文献14】Mochizuki H,Hayakawa H,Migita M,et al.An AAV−derived Apaf−1 dominant negative inhibitor prevents MPTP toxicity as antiapoptotic gene therapy for Parkinson’s disease.Proc Natl Acad Sci USA.2001;98:10918−10923.
【非特許文献15】Yue Y,Li Z,Harper SQ,Davisson RL,Chamberlain JS,Duan D.Microdystrophin gene therapy of cardiomyopathy restores dystrophin−glycoprotein complex and improves sarcolemma integrity in the mdx mouse heart.Circulation.2003;108:1626−1632.
【非特許文献16】Friedrich O,Both M,Gillis JM,Chamberlain JS,Fink RH.Mini−dystrophin restores l−type calcium currents in skeletal muscle of transgenic mdx mice.J Physiol.2003.
【非特許文献17】Watchko J,O’Day T,Wang B,et al.Adeno−associated virus vector−mediated minidystrophin gene therapy improves dystrophic muscle contractile function in mdx mice.Hum Gene Ther.2002;13:1451−1460.
【非特許文献18】Flotte T,Carter B,Conrad C,et al.A phase I study of an adeno−associated virus−CFTR gene vector in adult CF patients with mild lung disease.Hum Gene Ther.1996;7:1145−1159.
【非特許文献19】Wagner JA,Messner AH,Moran ML,et al.Safety and biological efficacy of an adeno−associated virus vector−cystic fibrosis transmembrane regulator(AAV−CFTR)in the cystic fibrosis maxillary sinus.Laryngoscope.1999;102(2 Pt 1):266−274.
【非特許文献20】Wagner JA,Nepomuceno IB,Messner AH,et al.A phase II, double−blind, randomized, placebo−controlled clinical trial of tgAAVCF using maxillary sinus delivery in patients with cystic fibrosis with antrostomies.Hum Gene Ther.2002;13:1349−1359.
【非特許文献21】Wagner JA,Reynolds T,Moran ML,et al.Efficient and persistent gene transfer of AAV−CFTR in maxillary sinus.Lancet.1998;351(9117):1702−1703.
【非特許文献22】Wagner JA,Moran ML,Messner AH,et al.A phase I/II study of tgAAV−CF for the treatment of chronic sinusitis in patients with cystic fibrosis.Hum Gene Ther.1998;9:889−909.
【非特許文献23】Kay MA,Manno CS,Ragni MV,et al.Evidence for gene transfer and expression of factor IX in haemophilia B patients treated with an AAV vector.Nat Genet.2000;24:257−261.
【非特許文献24】Manno CS,Chew AJ,Hutchison S,et al.AAV−mediated factor IX gene transfer to skeletal muscle in patients with severe hemophilia B.Blood.2003;101:2963−2972.
【非特許文献25】Chirmule N,Propert K,Magosin S,Qian Y,Qian R,Wilson J.Immune responses to adenovirus and adeno−associated virus in humans.Gene Ther.1999;6:1574−1583.
【非特許文献26】Holler PD,Chlewicki LK,Kranz DM.TCRs with high affinity for foreign pMHC show self−reactivity.Nat Immunol.2003;4:55−62.
【非特許文献27】Laugel B,Boulter JM,Lissin N,et al.Design of soluble recombinant T cell receptors for antigen targeting and T cell inhibition.J Biol Chem.2005;280:1882−1892.
【非特許文献28】O’Herrin SM,Lebowitz MS,Bieler JG,et al.Analysis of the expression of peptide−major histocompatibility complexes using high affinity soluble divalent T cell receptors.J Exp Med.1997;186:1333−1345.
【非特許文献29】Peng KW,Holler PD,Orr BA,Kranz DM,Russell SJ.Targeting virus entry and membrane fusion through specific peptide/MHC complexes using a high−affinity T−cell receptor.Gene Ther.2004;11:1234−1239.
【非特許文献30】Subbramanian RA,Moriya C,Martin KL,et al.Engineered T−cell receptor tetramers bind MHC−peptide complexes with high affinity.Nat Biotechnol.2004;22:1429−1434.
【非特許文献31】Zhu X,Belmont HJ,Price−Schiavi S,et al.Visualization of p53(264−272)/HLA−A*0201 complexes naturally presented on tumor cell surface by a multimeric soluble single−chain T cell receptor.J Immunol.2006;176:3223−3232.
【非特許文献32】Yee C,Savage PA,Lee PP,Davis MM,Greenberg PD.Isolation of high avidity melanoma−reactive CTL from heterogeneous populations using peptide−MHC tetramers.J Immunol.1999;162:2227−34.
【発明の概要】
【課題を解決するための手段】
【0010】
本発明ではヒトMHCクラスI分子上のアデノ随伴ウィルス(AAV)に存在するペプチド配列に対して免疫学的に特異的な可溶性T細胞受容体(soluble T cell receptors:sTCR)が開示される。望ましい実施形態において、前記アデノウィルスペプチド配列は、AAV−1、AAV-2、AAV−5、AAV−8及び他の自然発生血清型から成る群から選択される血清型から得られる。特定の望ましい実施形態において、前記ペプチドは表1に示される配列を持ち、前記ヒトMHCクラスI分子はHLA−A1、HLA−A2,HLA−A3、HLA−B7、HLA−B8、HLA−B15、HLA−B44、及びHLA−B51から成る群から選択される。
【0011】
また本発明は、導入遺伝子含有アデノ随伴ウィルスベクター投与前、投与中、投与後のウィルスカプシド抗原に対するT細胞介在型免疫反応の存在を検出する方法を含む。典型的な方法は、患者からT細胞を含む生物学的試料を得る工程と、MHCクラスI分子上の前記カプシドペプチドエピトープを含む五量体又は四量体に前記細胞を接触させる工程と、及び未処理のコントロール細胞と比較して前記接触が前記T細胞を刺激するかどうかを決定する工程とを含み、細胞は前記接触によって刺激され前記ウィルスカプシドの前記ペプチドエピトープに対する特異性を有しており、この特異性がカプシド及び導入遺伝子含有細胞のT細胞介在型破壊に関連する。前記方法はまた刺激を受けた前記T細胞からのmRNAの分離工程と、cDNAを調製する工程と、前記ウィルスカプシド抗原に対して免疫学的特異性のある可溶性T細胞受容体をクローン化する工程とを含む。
【0012】
前記方法によって調製される可溶性T細胞受容体もまた本発明に含まれる。
【0013】
もう1つの態様において、アデノウィルス随伴ベクター投与後のウィルス性形質導入細胞のT細胞介在型破壊の抑制方法が開示される。典型的な方法は、AAVエピトープ/MHC複合体に特異的なsTCRの有効量を提供する工程を含み、問題となるカプシドペプチドへの自然発生T細胞の結合阻害を介して、前記sTCRは前記導入遺伝子含有細胞のT細胞介在型破壊を防ぐ。そのような結合阻害はCTL活性化を防ぐ。
【0014】
また、ウィルス性形質導入細胞のT細胞介在型破壊を回避する方法を提供し、この方法は、上述したようにAAVペプチドエピトープに対する特異性を検出する工程と、前記AAVベクターを改変し同定したペプチドエピトープを除去する工程とを含む。改変後修飾されたAAVベクターが前記患者に投与され、その改変はウィルス性形質導入細胞のT細胞介在型破壊を抑止するものである。そのような改変AAVベクターはまた本発明のさらなる態様を含む。望ましい実施形態において、そのようなAAVベクターは表1で示されるAAVエピトープを欠失するように改変される。
【図面の簡単な説明】
【0015】
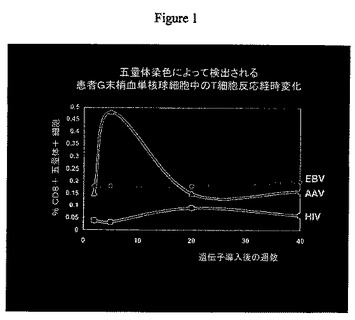
【図1】図1は導入遺伝子含有AAVベクターで処理した患者からの分離末梢血単核細胞(PBMCs)中のT細胞反応の時間的経過を示したグラフを表したものである。
【図2A】図2AはAAVカプシドIFN−γELISpotの結果を示したグラフである。AAV由来ペプチドによってインビトロで再刺激されたヒトリンパ球はIFN−γを産生する。
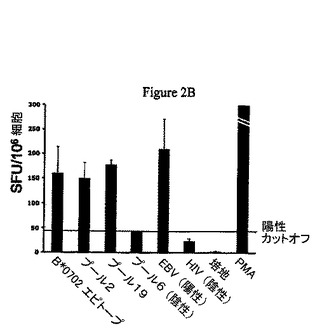
【図2B】図2BはAAVカプシド由来免疫優性エピトープを用いた数回のインビトロ刺激(IVS)を通して正常なドナーの末梢血単核細胞(PBMCs)からAAV−特異的なCD8+T細胞がインビトロで増殖することを表す一連の散布図を示したものである。
【図3】AAV血清型1〜8で非常に良く保存されている、ELISPOTアッセイによって同定された(下線)異なるT細胞エピトープを示したものである。配列ID番号1〜18を示す。
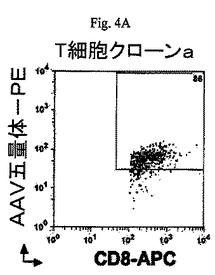
【図4A】図4A及び4Bは流動選別し、続いてインビトロで増加させた2つのCD8+T細胞クローンの一対の散布図である。これらのクローンはmRNA源となり、本願明細書で記載される可溶性T細胞受容体を合成するのに使用される。
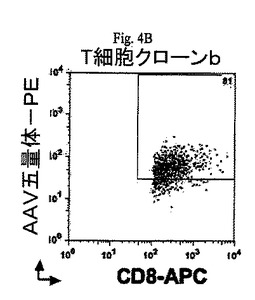
【図4B】図4A及び4Bは流動選別し、続いてインビトロで増加した2つのCD8+T細胞クローンの一対の散布図である。これらのクローンはmRNA源となり、本願明細書で記載される可溶性T細胞受容体を合成するのに使用される。
【図5A】図5A〜5Eは下記に説明されるCTLクローンに対するTCRの配列を示したものである。
【図5B】図5A〜5Eは下記に説明されるCTLクローンに対するTCRの配列を示したものである。
【図5C】図5A〜5Eは下記に説明されるCTLクローンに対するTCRの配列を示したものである。
【図5D】図5A〜5Eは下記に説明されるCTLクローンに対するTCRの配列を示したものである。
【図5E】図5A〜5Eは下記に説明されるCTLクローンに対するTCRの配列を示したものである。
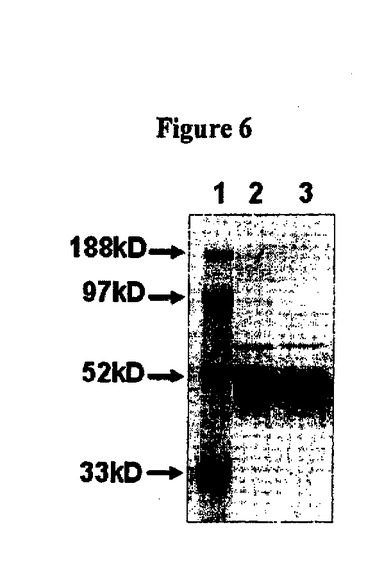
【図6】図6はAAV−a−scTCR−BirA及びAAV−b−scTCR−BirAのSDS−PAGEである。レーン1、標準分子量、レーン2、AAV−a−scTCR−BirA、レーン3、AAV−b−scTCR−BirA。
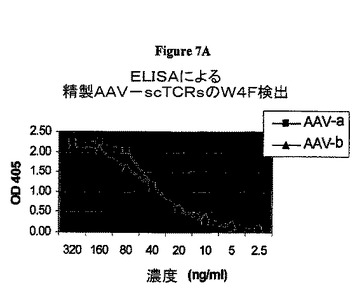
【図7A】図7AはELISAによる精製AAV−scTCRsのW4F検出を示すグラフである。
【図7B】図7BはELISAによる精製AAV−scTCRsの五量体検出を示すグラフである。
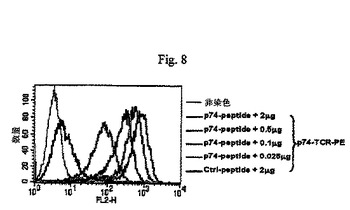
【図8】図8はAAV−scTCR多量体のp74/HLA−B7結合活性を示すプロットである。
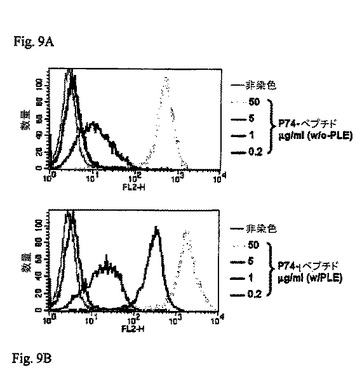
【図9】図9A及び9BはAAV−scTCR多量体のp74/HLA−B7結合のヒストグラムを示すものである。
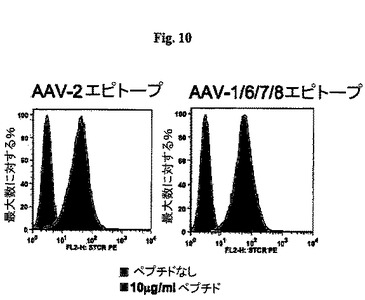
【図10】図10A及び10Bは1対のヒストグラムであり、AAV−scTCR多量体を使用したペプチド負荷した線維芽細胞のインビトロ染色を示したものである。
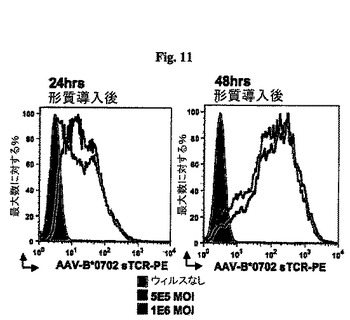
【図11】図11A及び11Bは一対のヒストグラムであり、AAV−scTCR多量体を使用したAAV−形質導入HLA−B*0702ヒト線維芽細胞のインビトロ染色を示したものである。
【図12】図12A及び12Bは一対のヒストグラムで、AAVの1E11ベクター遺伝子(vector genomes:vg)静注後一週間のHLA8 B*0702マウスから集めたCD11c+CD19−細胞のAAV−scTCR多量体染色を示したものである。
【発明を実施するための形態】
【0016】
ウィルスベクターを遺伝子導入媒体として使用するため遺伝子操作し、遺伝的欠陥を修正した。しかしながら、本発明に従ってウィルスベクターの導入遺伝子投与後、前記修正を受けた肝臓細胞が内部にベクターカプシドタンパク質を持つことが明らかになった。これらのタンパク質はT細胞により検出され、前記修正を受けた細胞は次々に破壊される。従って、本発明の一つの態様は、遺伝子導入の標的組織中のウィルスベクター抗原の存在を検出するための新しい試薬として、可溶性T細胞受容体(sTCR)の使用を伴う。ウィルス性ベクター配列又はカプシドタンパク質を検出する従来の方法は、PCR又は免疫特異性のある抗体を使用していた。しかしながら、臨床的により関連のある形態は宿主T細胞と接触するベクターカプシド、すなわち宿主MHCクラスI上に提示されているカプシド由来ペプチドである。可溶性TCRが自然発生T細胞で観察されるものと類似の様式でカプシド抗原のこの形態を認識することが開示される。さらに、このような試薬がベクター由来抗原の存在に関し形質導入組織を直接的且つ定量的に判定するために使用されることが可能である。この情報は免疫抑制療法を中止するかどうか又は中止する時を決定するために必要な情報を臨床医に提供する。
【0017】
本発明のもう1つの態様では、可溶性TCR試薬は、宿主T細胞が同種ベクター抗原に接触するのを特異的に阻害する特異的免疫調節治療法として使用される可能性がある。従って、本願明細書で記載される分子は診断及び治療薬の双方で使用される。
【0018】
様々なAAVの血清型が導入遺伝子の分野において使用され、またヒト集団におけるHLA型にも大きなばらつきがある。従って、本発明は、1)多形HLA拘束性エピトープ及び2)遺伝子療法ベクターで使用される特定のAAV血清型に特異性を持つように設計された可溶性TCRsが含まれる。
【0019】
代替AAVベクター血清型及びそれらのT細胞拘束性エピトープ
様々なAAV血清型、特にAAV−1、2、5及び8の類似した配列にも関わらず、ある自然発生AAVウィルスのエピトープに対して反応するT細胞が、遺伝子治療ベクターとして使用される代替血清型ウィルスに交差反応しないという可能性がある。なぜならばT細胞受容体はMHCクラスIに拘束された小さなペプチド(9〜11アミノ酸)のみを認識し、更にT細胞受容体は非常に高い特異性を持つため、AAV−2エピトープに反応するT細胞が、例えばアミノ酸1個のみが異なるAAV−8のエピトープを認識するかもしれないし認識しないかもしれない。さらに、いかなるAAVウィルスからのペプチドエピトープであっても個々のHLA型(ヒトのMHC)によって拘束される。このように、幾つかのT細胞拘束性AAVエピトープがAAV血清型中に保存される可能性に基づき、我々は、集団において通常見られるHLA−A1、A2、A3、B7、B8、B15、B44及びB51を含むHLA型に拘束される複数の血清型から多様なAAVエピトープを同定した。実際、我々はAAV−1、AAV−2及びAAV−8からの数個の候補エピトープのライブラリーを機能アッセイにおいて発達させた(表1)。可溶性TCRsのベクターカプシド配列検出に対する応用は、我々が既に有するHLA−B7拘束性の1つのAAVエピトープ(AAV−2VPQYGYLTL)から、推定遺伝子治療ベクターとして現在使用中のあらゆるAAV血清型の多数のエピトープへと容易に広げられる可能性がある。
【0020】
MHCクラスI五量体及び可溶性TCRsの産生だけでなく、免疫優性エピトープを同定することは、AAVカプシドタンパク質を遺伝子操作するのに有用であり、不要なエピトープが除去される。一旦エピトープが分かると、AAVカプシド配列中の一致する配列は標準的な分子生物学/組み換えDNA技術を用いて除去される。このように本発明のもう1つの態様はカプシドを再遺伝子操作する工程を含み、同定されたT細胞エピトープを改変する。これによって上に記載された特異的T細胞反応を避けるベクターが得られる。ここで留意するべき点はこれによって一連のベクターが生じ、それぞれが任意のMHCハプロタイプ特異的であり、これらのベクターは従来の臨床試験で見られたT細胞反応を回避するだろうということである(3)。本発明は表1のデータによって示されるように全てのAAV血清型に適用されるだろう。
【0021】
【表1】

【0022】
可溶性TCR技術の使用
sTCRsは免疫調節薬として利用することが可能である。例えば、可溶性TCRはT細胞によるエピトープ−MHCクラスI複合体認識を阻害するのに使用することができ、従って、遺伝子導入目的のAAVベクター導入に続く有害なT細胞反応を防ぐ。この目的では、可溶性TCRはAAVベクター導入直前又は導入時に投与されるだろう。
【0023】
sTCRsはAAV注入組織の表面上にあるAAVカプシドエピトープの染色のための試薬として使用されてもよい。遺伝子治療の分野におけるもっとも重要な疑問の一つはどのくらいの期間細胞内AAVカプシド分解に由来する前記ペプチドがMHC分子複合体の膜表面に提示され、免疫系によって検出可能かどうかである。本願明細書において記載される可溶性TCR試薬は生検組織のAAVエピトープ−MHC複合体を染色する利点を活かして使用することも可能であり、AAVカプシド抗原提示の持続時間を決定するのに役立つ。遺伝子治療を受けている患者がAAVカプシドに対する有害な免疫反応の高まる危険性に晒されている時間の範囲を決定することは有効な免疫抑制レジメンを設定するのに必須である。
【0024】
下記に記載の定義は本発明の理解を助けるために提供される。
【0025】
本願明細書で使用される「可溶性T細胞受容体又はsTCR」という語句は可溶性T細胞抗原受容体(単量体又は多量体)を指し、ウィルス感染細胞上のMHC分子に提示されるペプチド抗原を判定するのに使用される。可溶性TCRはフローサイトメトリー及び免疫組織化学での使用のため異なる標識分子に随意に結合されてもよい。T細胞受容体を生成する方法が米国特許第6,080,840号明細書、国際公開第WO/2005/116646号パンフレット及びBoulter et al.(2003)Protein Engineering 16:707−711に開示される。
【0026】
五量体はペプチドエピトープに結合した5個のMHCクラスI分子の複合体である。全5個の複合体が平面構造に保持されておりT細胞受容体に結合するのに利用されている。五量体は随意に蛍光色素に結合されフローサイトメトリーによって同種抗原特異的なT細胞を検出する。ここで留意すべき点は四量体技術もまた利用可能であり4個のMHCクラスI分子の複合体がペプチドエピトープに結合する。
【0027】
「五量体又は四量体染色」はT細胞を五量体又は四量体と混合し、4度で30分インキュベートし、1%FBS含有PBSで洗浄し、フローサイトメトリーで検出する過程を指す。染色アッセイを行うのに適した試薬はProImmuneから商業的に入手可能である。
【0028】
「細胞傷害性T細胞反応」という語句はエフェクターCD8+T細胞が標的細胞を殺すT細胞介在型破壊過程を指し、その標的細胞はMHCクラスI分子に結合するエピトープを外側表面に提示する。
【0029】
次の実施例は本発明のある実施形態を説明するために提供される。それは本発明をどんな意味においても本発明を限定するものではない。
【実施例1】
【0030】
上に言及したように、血友病治療のための従来の遺伝子治療において、導入遺伝子の発現は初期には高かったが、時間の経過につれて肝酵素が僅かに上昇し導入遺伝子発現は消失した。ベクター導入前及び後の患者からの末梢血単核球細胞(PBMC)を使用し、我々はMHCクラスI拘束性AAVカプシドエピトープを定義し(3)、AAVに特異的なCD8+T細胞を検出する五量体性HLA−B*0702/AAVペプチド(五量体)の可溶型を合成することができた。これらの五量体を使用して、我々は特異的なAAVエピトープに対する前記患者末梢血の特定のT細胞増殖の経時的変化を検出することができ、それは肝アミノ基転移酵素の上昇及び導入遺伝子発現の低下に一致していた。図1を参照。観察された前記T細胞増殖はMHC/ペプチド抗原の存在している時のみに観察された。このように本願明細書において提示される証拠は細胞傷害性T細胞が形質導入肝細胞によって提示されたベクターカプシドタンパク質に反応するということを明示する。ベクターカプシドタンパク質がベクター導入後限られた時間内に存在することを考慮して、本願明細書で開示される方法は免疫抑制の必要がある患者の4ヶ月の免疫抑制治療を含み、ベクター導入時前後にT細胞機能を一時的に停止する。
【0031】
T細胞反応刺激には樹状細胞のような専門の抗原提示細胞(antigen presenting cells:APCs)による抗原提示が必要であると考えられている。専門のAPCsは末梢に存在し、それらは直接感染又はウィルス感染細胞の貪食のいずれかによってウィルス抗原に遭遇する(交差提示)。樹状細胞は抗原を処理し、末梢のリンパ節に移動し、MHCクラスI及びII上の抗原及び適正な共刺激を未感作T細胞に提示する。最初の急速な増殖後、AAVに特異的なCD8+T細胞の発生頻度は減少し少数の貯蔵された記憶T細胞を後に残すことが予想され、この記憶T細胞は恒常的な維持増殖を通して各個人の一生涯の間保持される(2)。一旦適応した免疫システムが刺激されると、専門のAPCsによる共刺激及び抗原提示の必要性は厳密ではなくなり、全ての有核細胞が持つMHCクラスI上にのみウィルス抗原を提示する細胞を記憶T細胞は認識し殺す。AAV−2はT細胞反応刺激に必要な炎症反応を自分自身では誘導しないが、自然感染がヘルパーウィルスと一緒に起こるため、その時点でヘルパーウィルス及びAAV双方の抗原に対して誘導されるCD8+T細胞が刺激される。
【0032】
T細胞刺激に必要な炎症反応は抗体に再遭遇する記憶T細胞を新たに動員し活性化するために必須ではない。実験動物とは異なり、ヒトは幼少時にAAV−2に自然感染する。一般的なヒト集団におけるAAVに対するT細胞反応分析報告はほとんどない。Chirmuleらは正常コントロールの5%が組み換えAAVとともにPBMCをインキュベーション後>2の刺激係数を示したと報告した(25)。それ以来、T細胞分析のより感受性の高く定量的なアッセイが発達し、ヒト患者での抗ウィルスT細胞反応の詳細な特性解析が行われた。我々は数人の健常成人ドナーのAAVに対するT細胞反応を記録することができ、ウィルスカプシドに対する記憶T細胞反応はAAVベクターを使用する遺伝子導入研究における蔓延した問題であることを示した。ヒトリンパ球はAAVカプシドタンパク質配列由来ペプチドエピトープとともにインビトロで増殖させることができる。増殖した細胞はIFN−γを生成することによってエピトープに反応し(図2A)及びAAV特異性MHCクラスI五量体で染色することができる(図2B)。図3はELISPOTアッセイによって同定された一連の異なるT細胞エピトープ(下線を参照)を説明し、それらはAAV血清型1−8で非常に良く保存されている。
【0033】
遺伝子治療ベクターにはウィルスDNAがないため、MHCクラスIによってT細胞に提示される唯一のウィルス抗原は導入ベクターのカプシドタンパク質であり、それは限定された時間内に存在するべきである。しかしながら免疫学的に検出可能な形でカプシドが存在する時間の長さを決定する直接的な方法はない。我々は臨床試験から継続した4ヶ月の免疫抑制治療を選択したが、この時間枠はカプシド分解の動力学の確かな根拠に基づいていない。カプシドが存在する時間を決定する1つの理由は免疫抑制を中止し遺伝的修正を受けた細胞を保つ時期を決定するためである。
【0034】
複数の研究が行われ、それぞれ異なる間接的方法でどれくらいの期間ベクターカプシドが存在するかを決定した。臨床試験において、我々は生体内分布研究にPCRを使用し時間の経過とともにベクター遺伝子の存在を検出した。しかしながら、PCRはベクターDNAを検出するものであり、ベクターDNAは問題となっているT細胞によって認識される形のものではない。同様にベクターカプシドに対する抗体もT細胞によって認識される形のベクターを検出しない。なぜならこのT細胞はMHC+ペプチド複合体の形の中でも抗原部分だけを認識するからである。我々の研究室はまた同じ疑問に対処するため動物モデルの研究も行っている。しかしながら、我々は最終的にヒト患者で、遺伝子導入の標的となった特定の組織における、及び我々が使用する異なるいずれのAAV血清型に対するカプシドの持続性を決定する必要があるだろう、(我々の研究室はAAV−2に焦点をあてているが、他の遺伝子治療研究室はAAV−1、AAV−5、及び/又はAAV−8又はこれらのキメラに焦点を当てている)。このようにベクター形質導入組織に対するT細胞反応を基にした機能アッセイが本願明細書で開示されるが、理想的にはこれらのデータは記載されている可溶性TCRsを使用したデータと組み合わせて解釈される。これらのsTCRsによってカプシドの存在を直接定量的に決定でき、もし生検組織試料が利用できるのであればベクターで治療された個人のヒト患者中においても決定できる。
【0035】
上記のように、ヒトリンパ球をAAVカプシドタンパク質配列由来のペプチドエピトープとともにインビトロで増殖させた。増殖した細胞はIFN−γを生成することによってエピトープに反応し、AAV特異的なMHCクラスI五量体で染色される(図2A及び2B)。
【0036】
AAV特異的MHCクラスI五量体を使用して、増殖したカプシド特異性のあるCD8+T細胞の流動選別を行いCD8+T細胞のクローンを選択する可能性がある(図4A及び4B)。
【0037】
2つの異なるヒトAAV−p74ペプチド特異性のあるCTLクローンをT細胞受容体(T cell receptor:TCR)に特異的プライマーを使用するSMART−RACE法によってTCRα及びβ鎖cDNAを生成するために使用した。cDNA生成物をクローン化して配列を解読した。2個のTCRα鎖遺伝子(AV17/TRAJ43;配列ID番号19、及びAV17/AJ31;配列ID番号20)及び1個のTCRβ鎖遺伝子(BV6−2/BJ1−1;配列ID番号21)をCTLクローンaからクローン化した。1個のTCRα鎖遺伝子(AV27/AJ20;配列ID番号22)及び1個のTCRβ鎖遺伝子(BV4−3/BJ2−7;配列ID番号23)をCTLクローンbからクローン化した。2個のCTLクローンに対するTCRの配列が図5A〜5Eに提供される。
【0038】
同定された2個のα鎖を基に、細菌ビオチンリガーゼに融合した1個のscTCR(scTCR−BirAコンストラクト)をクローンbに生成し、2個の異なるscTCR−BirAコンストラクトをクローンaに生成した。3個の発現ベクターを生成し、CHO細胞に形質移入し、特性解析のための可溶性scTCR−BirA融合タンパク質を生成した。3個の全てのscTCRをAAV−scTCR形質移入CHO細胞に発現させ、ATCCから入手可能な抗ヒトTCR−β鎖抗体、(BF1−)8A3.31及びW4F.5Bを使用したサンドウィッチELISAによって細胞培養上清中に検出した。クローンb scTCR及びクローンa scTCRs(AV17/AJ31:BV6−2/BJ1−1)含有細胞培養上清はELISAによって検出されるAAVp74五量体活性を示し、さらに上清を特性解析した。AAV−scTCR−BirA融合タンパク質をAAV−scTCR−BirA形質移入体の培養上清からBF1−アフィニティークロマトグラフィーによって精製した。精製融合タンパク質をSDS−PAGEに示した(図6)。AAV−a及びAAV−b培養上清からの融合タンパク質精製量は各々4mg/L及び0.2mg/Lであった。精製scTCR−BirA融合タンパク質の特性解析をELISAによって行った。図7A及び7Bを参照。精製AAV−p74−scTCR−BirA融合タンパク質を抗TCR抗体が認識するか否かを決定するために、融合タンパク質の連続希釈したものを抗TCR BF1 mAb塗布プレートでインキュベートし、その後ビオチン標識抗TCR W4F mAb及びSA−HRPで検出した。結果は図7Aに示す。可溶性AAV−p74−scTCRsの機能的結合親和性を試験するため、融合タンパク質の連続希釈したものが抗TCR BF1 mAb塗布プレートにインキュベートされ、その後ビオチン化したAAV−p74/HLA−B*0702五量体及びSA−HRPで検出した。図7Bを参照。
【0039】
精製AAV−b融合タンパク質はAAV−a−AJ31よりもかなり低い五量体結合活性を示すため、AAV−a−AJ31融合タンパク質のみをビオチン標識し可溶性p74−scTCR−PE多量体の生成に使用した。AAV−a−scTCRが細胞表面のAAV−p74ペプチド/HLA−B7複合体に結合できるか否かを決定するためにAAV−p74ペプチド負荷HLA−B7陽性ヒトリンパ芽球様細胞株(JyA2B7)を使用した。50μg/mlのAAV−p74ペプチドを負荷したJyA2B7細胞(不死化B細胞)の特異的な染色を1検査当たり0.025μgのPE結合p74−scTCR多量体とともに観察した(図8)。
【0040】
一方、典型的な状態では最少5μg/mlAAV−p74ペプチドを負荷した(図9A)、又は、負荷中PLEを加えた場合最少1μg/mlAAV−p74ペプチドを負荷したJyA2B7細胞(図7B)を0.5μgのp74−scTCR多量体で染色することができる。(PLEはAltor Bioscience Corp.(Miramar、フロリダ州)の独自の試薬混合物であり、抗原提示細胞に負荷したペプチドを強化する)。
【0041】
AAV−scTCR多量体をHLA−B*0702陽性の正常ヒト線維芽細胞株を使用しインビトロで試験した(ATCC在庫からMalme−3が入手可能である)。細胞に37℃2時間10μg/mlの濃度でAAVカプシドエピトープをペプチド負荷し、その後AAV−scTCR多量体で染色した(図10)。両方の実験においてAAV−scTCR多量体の陽性染色が観察され、多量体がAAVペプチドエピトープを提示するMHC分子に良い親和性で結合することを示した。
【0042】
同様に、AAVベクター静注投与後のHLA−B*0702遺伝子組み換えマウスから採集したリンパ節細胞及び脾臓細胞はAAV−scTCR多量体の陽性染色を示す(図12)。
【0043】
結論
ヒトMHCクラスI分子HLA−B*0702上のAAV2ペプチド(配列VPQYGYLTL)に特異的な典型的な可溶性TCRが本願明細書に記載される。我々はHLA−B*0702ハプロタイプを持つ匿名の健常ドナーからこのペプチドに対して特異性のあるT細胞を増殖させた。このペプチドに特異的なT細胞をクローン化し、T細胞受容体のDNAを分離し発現させた。
【0044】
複数の研究グループがウィルス及び/又は癌抗原発現レベルを研究及び定量するため可溶性TCRsを発達させた(26〜31)。可溶性TCRsの遺伝子操作は様々な理由で困難であり、その理由には自然発生T細胞受容体のMHC/ペプチドに対する低い親和性及び特定の細胞上の特定のペプチド/MHC複合体の低い発現量が含まれる。TCRsの公開された研究において、ペプチド/MHC複合体に対するTCRsの親和性が判定された(30)。MHC/ペプチド抗原レベルの検査はマウス同種異系抗体(28)及び神経学的疾患の原因抗原であるHTLV−1抗原に焦点を当てた(感染(28)及び腫瘍随伴疾患(27))。T細胞レパートリーの発達をよく理解するため可溶性TCRsを使用した研究もあれば、ウィルスの侵入を捕らえるために可溶性TCRを使用した研究もある。最後にZhuら(2006)は多量体一本鎖可溶性TCRを作成し癌細胞上のヒトMHCクラスIに提示された癌抗原由来ペプチドを可視化した。我々が知る限りではヒト、動物、又はインビトロモデルのいずれかにおいてウィルスベクター由来抗原の存在を調べた公開されている研究はない。我々が見い出したHLAB*0702拘束性エピトープを記載した最近の論文(3)以外はAAV由来抗原に関する公開された研究もない。
【0045】
下記に記載された方法は本発明の実践を促進するために提供される。
【0046】
AAVカプシドエピトープの同定
アデノ随伴ウィルス(AAV)はパルボウィルス科に属し、一生のうちの通常早い時期にヒトに自然に感染する。ヒト集団において最も一般的なHLAに対する新しいCD8T細胞エピトープを同定するため、2つのIRB承認プロトコールを開始しthe Children’s Hospital of Philadelphia及びペンシルバニア大学のthe Cooperative Human Tissue Networkと提携してヒト脾臓を収集した。脾臓は他の臓器に比べエピトープ探究のための2つの主要な利点を提供する。1つ目はT細胞記憶保持に関連するリンパ様臓器であること、2つ目は1グラムの組織から5億個の細胞を分離することが可能であり、この数は全血のような他の供給源からは簡単に得られることはできない。
【0047】
T細胞分離及びHLAタイピング
T細胞を組織採取から24時間以内で分離する。脾臓を最初外科用メスによって小片に処理しその後均質化する。赤血球溶解後脾細胞をPBSで2回洗浄し、各々約1000万個の一定分量で10%DMSOとともにヒト血清中に凍結した。
【0048】
2つ若しくはそれ以上の細胞を高分解HLAタイピングのためペンシルバニア大学病院のHLA−タイピング研究室に送付した。
【0049】
インビトロT細胞増殖
CD8T細胞エピトープの同定はAAVカプシドタンパク質に反応する記憶CD8T細胞の低い発生頻度によって妨げられる。この限界を乗り越えるため、脾臓組織からのリンパ球をVP1と呼ばれるAAVカプシドタンパク質に由来する一連のペプチドとともにインビトロで増殖させた。前記VP1ペプチドライブラリーは各々が10アミノ酸ずつ重複しているタンパク質配列に由来する15アミノ酸長のペプチド145個でできている(Mimotopes)。
【0050】
簡潔に説明すると、脾臓からのリンパ球を3%熱不活化ヒト血清を有するAIM−V(Gibco)培地中で1ウェル当たり100万個の細胞を96ウェルプレートに播種し、前記ウェルの細胞の半分を3000radで放射線照射し支持細胞層として機能させる。VP1ライブラリーからの各ペプチドを10μg/mlの最終濃度で1つのウェルに添加する。増殖0日目及び2〜3日毎にIL−2(Roche)を最終濃度10U/mlで培養に添加する。
【0051】
刺激の1巡目は7〜10日間継続するが、AAVカプシドに反応するCD8T細胞の予想される少ない数のため、通常増殖には2−3巡必要である。増殖の各巡の追加は単純にウェルに新しいペプチド及び3000radで放射線照射した500,000同種脾臓細胞を添加することによって行う。
【0052】
IFN−γELISpotによるエピトープ探索
ELISpotは特異的な抗原に反応するT細胞の数を同定するため使用される強力な技術である、すなわちELISpotはペプチドに反応してIFN−γを分泌する細胞の能力を測定する。50,000個の増殖したT細胞を抗ヒトIFN−γ抗体(Mab−Tech)を先に塗布した96ウェルELISpotプレート(Millipore)に増殖のため使用されるペプチドの存在下で播種した。37℃、5%CO2でインキュベーション24時間後細胞を洗浄し、抗ヒトIFN−γビオチン標識2次抗体(Mab−Tech)をウェルに添加する。ストレプトアビジン−アルカリフォスファターゼを特異的基質の存在下で検出試薬として使用する。
【0053】
陽性のウェルを最初に添加された100万個の細胞当たりのスポットを形成する細胞(spot forming cells:SFC)の数を基に判定する。もしSFCの数が陰性対照コントロールウェル(培地のみ)のSFCの数よりも3倍高いならば、前記ウェルは陽性と判定された。陽性ペプチドは上記に記載されたプロトコールを繰り返すことによって通常少なくとも2回確認された。
【0054】
その後この方法は興味のある全てのHLA対立遺伝子に対して繰り返された。
【0055】
オンライン予想アルゴリズム
2つのオンラインエピトープ予想アルゴリズムプログラムをELISpotアッセイに使用される15アミノ酸長ペプチドの中の9アミノ酸配列を同定するのに使用し、その配列はHLA分子に対する結合配列を表す。これらのプログラムはRankpep:http://bio.dfci.harvard.edu/Tools/rankpep.html及びSYFPEITHI:www.syfpeithi.de/のインターネット上で見ることができる。
【0056】
同定された9アミノ酸を合成しELISpot及び細胞内サイトカイン染色によって確認する。陽性のペプチドエピトープ配列をその後HLA−ペプチド五量体検出薬を合成するために利用する(Proimmune)。
【0057】
AAV−特異的T細胞選別及びクローニングのための五量体の使用
特異的なAAVペプチドとともに一度又は二度増殖させた末梢血単核細胞(PBMC)を適切なHLAペプチド五量体とともに滅菌状態で染色する。分析のみに通常使用された五量体染色に関しては、染色を4℃20分間2%ヒト血清を含む滅菌PBS中で実行する。培養を抗CD8抗体とともに共染色する。PBS−2%hAB血清で2回洗浄後、前記細胞を蛍光活性細胞選別器に通し、96ウェル丸底プレートの1ウェル当たり1個の五量体+CD8+T細胞を含むように選別する。細胞を5個から10個のプレートに選別する。各プレートの各ウェルは同じ日に調製し、支持細胞として放射線照射された同種PBMCを含め、EBV形質転換B細胞株からの放射線照射細胞及びT細胞成長因子として50IU/mlの組み換えヒトIL−2を加えた商業的に入手可能な抗CD3T細胞刺激抗体(OKT3)を伴う。選別された(クローン化された)前記細胞をその後2週間37℃の湿潤恒温器でインキュベートする。
【0058】
2週間後T細胞のクローン成長を目視検査によって判定する。成長細胞を五量体染色によるAAVに対する特異性で判定する(上記に記載)。ペプチド特異性を得るクローンを最初の刺激のように同種PBMC、放射線照射EBV形質転換B細胞、OKT3及びIL−2でさらに刺激するが、この作業以外に細胞集団の成長速度によって成長細胞を24ウェルプレート又はT25フラスコに移動させる。Dr.Cassian Yee及びその同僚の記載のようにクローンを2週毎に再刺激をする(32)。
【0059】
一旦少なくとも100万個のT細胞クローンが成長すると、一定分量を標準プロトコールに従って凍結する。この一定分量のT細胞クローンを可溶性TCR生成のためAltor Bioscience Corporationに提供する。もう1つの方法として我々は成長T細胞クローンからRNAを分離しAltor BioscienceにRNAを提供することもある。この時点のRNAをcDNAを生成するのに使用し、この材料によって可溶性T細胞受容体を生成する基礎を作る、以上の作業はこれらの分子を生成する経験を持つグループであるAltor Bioscienceによる(31)。
【0060】
我々はAAVエピトープに特異的な可溶性TCR受容体をクローン化し特性解析するのに成功し、このAAVエピトープは患者への遺伝子導入を妨げる細胞傷害性T細胞反応の発生に関連する。本願明細書において記載される前記材料及び方法は診断及び治療双方に使用され、必要とする患者への治療用異種タンパク質の導入を促進する。
【0061】
本発明の一部の望ましい実施形態を記載し及び特定の実施例を上記に提示したが、本発明はそれらの実施形態に限定されるものではない。様々な改変は次の請求項に記載されるように本発明の要旨に逸脱することなく行われる。
【技術分野】
【0001】
本願は2006年5月31日付で出願された米国特許仮出願第60/809,956号に優先権を主張するものであり、その全ての内容がこの参照により本願明細書に取り込まれる。
【0002】
米国特許法第202条(35U.S.C.§202(c))に従って、米国国立衛生研究所(National Institutes of Health)の研究助成金番号PO1 HL078810からの資金で一部なされた本願に記載される本発明の特定権利は、米国政府が有するものであることが認められる。
【背景技術】
【0003】
本発明は遺伝子治療及び免疫学の分野に関する。より具体的には、本発明はウィルスベクター抗原の存在を検出する組成物及び方法を提供し、ウィルスベクター抗原に対する免疫反応を抑制及び回避する組成物及び方法を含むものである。
【0004】
複数の出版物と特許文献が本発明に関連する最新の従来技術を説明するために本願明細書の中で参照されている。これらの引用文献の各々は参照により本願明細書に完全に取り込まれる。
【0005】
野生型アデノウィルス随伴ウィルス(adenovirus−associated virus:AAV)は〜4.7kbの一本鎖DNA遺伝子を持つパルボウィルスである。前記ウィルスは複製機能を自然欠損しており、複製にはアデノウィルス及びヘルペスウィルスのようなヘルパーウィルスを必要とする。前記ウィルスはいかなる疾患にも関連が見出されていないが、その代わりにアデノウィルス分離株の混入物質として最初に分離された(4)。これまでに6個の血清型が記述されており、それらは非常に良く保存された配列を持つ(保存率は62%〜99%と異なる)。前記ウィルスの遺伝子は2個の逆方向末端反復配列(inverted terminal repeats:ITRs)によって挟まれており、3個のカプシド(capsid)遺伝子(VP1、2、3)及び4個のレップ(rep)タンパク質をコードするものであり、このレップタンパク質はDNA複製及びAAV生活環の制御に関わる。最近、さらなる血清型(AAV−7、−8、−9)がアカゲザル及びヒトから分離され、AAV−1〜6と比較してこれらの血清型もまた60%よりも高い率で保存されている(5)。
【0006】
野生型AAVが遺伝子導入媒体としての使用のために遺伝子操作された。すなわち、レップ(rep)及びキャップ(cap)遺伝子を欠損させ、ウィルスDNA翻訳領域がないように、目的とする治療遺伝子を2個のITRsの間に挿入する。1990年代の半ばに複数の研究グループ(6〜10)によって、組み換えAAVが骨格筋、肝臓、中枢神経系(CNS)、及び呼吸器官を含む多数の非分裂細胞型に感染し、この組み換えAAVにより免疫学的にコンピテントな動物中で導入遺伝子長期発現が誘導されることが示された。この興味深い知見は多数の研究グループによって利用され、組み換えAAVの投与により小型及び大型動物モデルにおいて遺伝子性疾患が治癒した(11〜17)という目覚ましい結果のポートフォリオが現在集まっている。ヒトにおける経験はより限定されるが(18〜24)、安全性及び遺伝子導入とその発現が証明されているという観点で有望とされている。しかしながら、ほとんどの例で表現型の修正が起こるほど十分に発現レベルが十分に高くない。
【0007】
我々の研究の主要目的は、血友病及び他の血液凝固疾患の治療のための安全且つ有効なアデノ随伴ウィルス(adeno−associated virus:AAV)を介した遺伝子導入システムを確立することである。血友病イヌモデルの長期間にわたる血友病の治癒(1)に基づき臨床試験が計画され、その中で重度のB型血友病の患者にAAV−F.IX(Factor IX:F.IX)経肝動脈注入を行った。1人の患者はベクター注入後第2週目までに11.8%(治療域レベル)の循環ファクターIXを得た。このレベルは約4週間続き、その後徐々に低下し始め、結局患者の基準値<1%に戻った。F.IXレベルの減少に一致して、血中肝アミノ基転移酵素が上昇し始め、注入後5週で最高値に達し、その後数週間で正常値にまで減少した。このように前記患者は、マウス、ラット、ウサギ、血友病イヌ、及び非ヒト霊長類を含む実験動物で観察されたものとはかなり異なる経過をたどった。実験動物とは対照的に、前記ヒト患者は、AAVに対する低い中和抗体適定値の存在、並びにIgG抗体が存在することから推定すればわかるように、既にAAV−2に対する免疫を持っており、前記患者はリンパ球様分画にAAVに特異的な記憶T細胞の一群を持っているようであった(2)。同様の所見が前記臨床試験の別の患者でも観察され、この患者での免疫学的研究によりAAVカプシド中の特異的なペプチドに対するT細胞反応が立証された。明らかに、前記反応はベクターを注入して数週後、末梢血で検出できたが、注入前には検出できなかった。
【0008】
これらの知見から遺伝子治療アプローチを有効にするために、一部の例で、導入遺伝子発現細胞のT細胞介在型破壊を防ぐために免疫反応を調節することが必要であるのは明らかである。
【先行技術文献】
【非特許文献】
【0009】
【非特許文献1】Mount JD,Herzog RW,Tillson DM,et al.Sustained phenotypic correction of hemophilia B dogs with a factor IX null mutation by liver−directed gene therapy.Blood.2002;99:2670−2676.
【非特許文献2】Appay V,Rowland−Jones SL.Lessons from the study of T−cell differentiation in persistent human virus infection.Semin Immunol.2004;16:205−212.
【非特許文献3】Manno CS,Arruda VR,Pierce GF,et al.Successful transduction of liver in hemophilia by AAV−Factor IX and limitations imposed by the host immune response.Nat Med.2006;12:342−347.
【非特許文献4】Blacklow NR,Hoggan MD,Sereno MS,et al.A seroepidemiologic study of adenovirus−associated virus infection in infants and children.Am J Epidemiol.1971;94.
【非特許文献5】Gao GP,Alvira MR,Wang L,Calcedo R,Johnston J,Wilson JM.Novel adeno−associated viruses from rhesus monkeys as vectors for human gene therapy.Proc Natl Acad Sci USA.2002;99:11854−11859.
【非特許文献6】Xiao X,Li J,Samulski RJ.Efficient long−term gene transfer into muscle tissue of immunocompetent mice by adeno−associated virus vector.j Virol.1996;70:8098−8108.
【非特許文献7】Kessler PD,Podsakoff GM,Chen X,et al.Gene delivery to skeletal muscle results in sustained expression and systemic delivery of a therapeutic protein.Proc Natl Acad Sci USA.1996;93:14082−14087.
【非特許文献8】Flotte TR, Afione SA, Conrad C,et al.Stable in vivo expression of the cystic fibrosis transmembrane conductance regulator with an adeno−associated virus vector.Proc Natl Acad Sci USA.1993;90:10613−10617.
【非特許文献9】Kaplitt MG,Leone P,Samulski RJ,et al.Long−term gene expression and phenotypic correction using adeno−associated virus vectors in the mammalian brain.Nat Genet.1994;8:148−154.
【非特許文献10】Flotte TR,Carter BJ.Adeno−associated virus vectors for gene therapy.Gene Ther.1995;2:357−362.
【非特許文献11】Sanchez−Pernaute R,Harvey−White J,Cunningham J,Bankiewicz KS.Functional effect of adeno−associated virus mediated gene transfer of aromatic L−amino acid decarboxylase into the striatum of 6−OHDA−lesioned rats.Mol Ther.2001;4:324−330.
【非特許文献12】Acland GM,Aguirre GD,Ray J,et al.Gene therapy restores vision in a canine model of childhood blindness.Nat Genet.2001;28:92−95.
【非特許文献13】Ho TT,Maguire AM,Aguirre GD,et al.Phenotypic rescue after adeno−associated virus−mediated delivery of 4−sulfatase to the retinal pigment epithelium of feline mucopolysaccharidosis VI.J Gene Med.2002;4:613−621.
【非特許文献14】Mochizuki H,Hayakawa H,Migita M,et al.An AAV−derived Apaf−1 dominant negative inhibitor prevents MPTP toxicity as antiapoptotic gene therapy for Parkinson’s disease.Proc Natl Acad Sci USA.2001;98:10918−10923.
【非特許文献15】Yue Y,Li Z,Harper SQ,Davisson RL,Chamberlain JS,Duan D.Microdystrophin gene therapy of cardiomyopathy restores dystrophin−glycoprotein complex and improves sarcolemma integrity in the mdx mouse heart.Circulation.2003;108:1626−1632.
【非特許文献16】Friedrich O,Both M,Gillis JM,Chamberlain JS,Fink RH.Mini−dystrophin restores l−type calcium currents in skeletal muscle of transgenic mdx mice.J Physiol.2003.
【非特許文献17】Watchko J,O’Day T,Wang B,et al.Adeno−associated virus vector−mediated minidystrophin gene therapy improves dystrophic muscle contractile function in mdx mice.Hum Gene Ther.2002;13:1451−1460.
【非特許文献18】Flotte T,Carter B,Conrad C,et al.A phase I study of an adeno−associated virus−CFTR gene vector in adult CF patients with mild lung disease.Hum Gene Ther.1996;7:1145−1159.
【非特許文献19】Wagner JA,Messner AH,Moran ML,et al.Safety and biological efficacy of an adeno−associated virus vector−cystic fibrosis transmembrane regulator(AAV−CFTR)in the cystic fibrosis maxillary sinus.Laryngoscope.1999;102(2 Pt 1):266−274.
【非特許文献20】Wagner JA,Nepomuceno IB,Messner AH,et al.A phase II, double−blind, randomized, placebo−controlled clinical trial of tgAAVCF using maxillary sinus delivery in patients with cystic fibrosis with antrostomies.Hum Gene Ther.2002;13:1349−1359.
【非特許文献21】Wagner JA,Reynolds T,Moran ML,et al.Efficient and persistent gene transfer of AAV−CFTR in maxillary sinus.Lancet.1998;351(9117):1702−1703.
【非特許文献22】Wagner JA,Moran ML,Messner AH,et al.A phase I/II study of tgAAV−CF for the treatment of chronic sinusitis in patients with cystic fibrosis.Hum Gene Ther.1998;9:889−909.
【非特許文献23】Kay MA,Manno CS,Ragni MV,et al.Evidence for gene transfer and expression of factor IX in haemophilia B patients treated with an AAV vector.Nat Genet.2000;24:257−261.
【非特許文献24】Manno CS,Chew AJ,Hutchison S,et al.AAV−mediated factor IX gene transfer to skeletal muscle in patients with severe hemophilia B.Blood.2003;101:2963−2972.
【非特許文献25】Chirmule N,Propert K,Magosin S,Qian Y,Qian R,Wilson J.Immune responses to adenovirus and adeno−associated virus in humans.Gene Ther.1999;6:1574−1583.
【非特許文献26】Holler PD,Chlewicki LK,Kranz DM.TCRs with high affinity for foreign pMHC show self−reactivity.Nat Immunol.2003;4:55−62.
【非特許文献27】Laugel B,Boulter JM,Lissin N,et al.Design of soluble recombinant T cell receptors for antigen targeting and T cell inhibition.J Biol Chem.2005;280:1882−1892.
【非特許文献28】O’Herrin SM,Lebowitz MS,Bieler JG,et al.Analysis of the expression of peptide−major histocompatibility complexes using high affinity soluble divalent T cell receptors.J Exp Med.1997;186:1333−1345.
【非特許文献29】Peng KW,Holler PD,Orr BA,Kranz DM,Russell SJ.Targeting virus entry and membrane fusion through specific peptide/MHC complexes using a high−affinity T−cell receptor.Gene Ther.2004;11:1234−1239.
【非特許文献30】Subbramanian RA,Moriya C,Martin KL,et al.Engineered T−cell receptor tetramers bind MHC−peptide complexes with high affinity.Nat Biotechnol.2004;22:1429−1434.
【非特許文献31】Zhu X,Belmont HJ,Price−Schiavi S,et al.Visualization of p53(264−272)/HLA−A*0201 complexes naturally presented on tumor cell surface by a multimeric soluble single−chain T cell receptor.J Immunol.2006;176:3223−3232.
【非特許文献32】Yee C,Savage PA,Lee PP,Davis MM,Greenberg PD.Isolation of high avidity melanoma−reactive CTL from heterogeneous populations using peptide−MHC tetramers.J Immunol.1999;162:2227−34.
【発明の概要】
【課題を解決するための手段】
【0010】
本発明ではヒトMHCクラスI分子上のアデノ随伴ウィルス(AAV)に存在するペプチド配列に対して免疫学的に特異的な可溶性T細胞受容体(soluble T cell receptors:sTCR)が開示される。望ましい実施形態において、前記アデノウィルスペプチド配列は、AAV−1、AAV-2、AAV−5、AAV−8及び他の自然発生血清型から成る群から選択される血清型から得られる。特定の望ましい実施形態において、前記ペプチドは表1に示される配列を持ち、前記ヒトMHCクラスI分子はHLA−A1、HLA−A2,HLA−A3、HLA−B7、HLA−B8、HLA−B15、HLA−B44、及びHLA−B51から成る群から選択される。
【0011】
また本発明は、導入遺伝子含有アデノ随伴ウィルスベクター投与前、投与中、投与後のウィルスカプシド抗原に対するT細胞介在型免疫反応の存在を検出する方法を含む。典型的な方法は、患者からT細胞を含む生物学的試料を得る工程と、MHCクラスI分子上の前記カプシドペプチドエピトープを含む五量体又は四量体に前記細胞を接触させる工程と、及び未処理のコントロール細胞と比較して前記接触が前記T細胞を刺激するかどうかを決定する工程とを含み、細胞は前記接触によって刺激され前記ウィルスカプシドの前記ペプチドエピトープに対する特異性を有しており、この特異性がカプシド及び導入遺伝子含有細胞のT細胞介在型破壊に関連する。前記方法はまた刺激を受けた前記T細胞からのmRNAの分離工程と、cDNAを調製する工程と、前記ウィルスカプシド抗原に対して免疫学的特異性のある可溶性T細胞受容体をクローン化する工程とを含む。
【0012】
前記方法によって調製される可溶性T細胞受容体もまた本発明に含まれる。
【0013】
もう1つの態様において、アデノウィルス随伴ベクター投与後のウィルス性形質導入細胞のT細胞介在型破壊の抑制方法が開示される。典型的な方法は、AAVエピトープ/MHC複合体に特異的なsTCRの有効量を提供する工程を含み、問題となるカプシドペプチドへの自然発生T細胞の結合阻害を介して、前記sTCRは前記導入遺伝子含有細胞のT細胞介在型破壊を防ぐ。そのような結合阻害はCTL活性化を防ぐ。
【0014】
また、ウィルス性形質導入細胞のT細胞介在型破壊を回避する方法を提供し、この方法は、上述したようにAAVペプチドエピトープに対する特異性を検出する工程と、前記AAVベクターを改変し同定したペプチドエピトープを除去する工程とを含む。改変後修飾されたAAVベクターが前記患者に投与され、その改変はウィルス性形質導入細胞のT細胞介在型破壊を抑止するものである。そのような改変AAVベクターはまた本発明のさらなる態様を含む。望ましい実施形態において、そのようなAAVベクターは表1で示されるAAVエピトープを欠失するように改変される。
【図面の簡単な説明】
【0015】
【図1】図1は導入遺伝子含有AAVベクターで処理した患者からの分離末梢血単核細胞(PBMCs)中のT細胞反応の時間的経過を示したグラフを表したものである。
【図2A】図2AはAAVカプシドIFN−γELISpotの結果を示したグラフである。AAV由来ペプチドによってインビトロで再刺激されたヒトリンパ球はIFN−γを産生する。
【図2B】図2BはAAVカプシド由来免疫優性エピトープを用いた数回のインビトロ刺激(IVS)を通して正常なドナーの末梢血単核細胞(PBMCs)からAAV−特異的なCD8+T細胞がインビトロで増殖することを表す一連の散布図を示したものである。
【図3】AAV血清型1〜8で非常に良く保存されている、ELISPOTアッセイによって同定された(下線)異なるT細胞エピトープを示したものである。配列ID番号1〜18を示す。
【図4A】図4A及び4Bは流動選別し、続いてインビトロで増加させた2つのCD8+T細胞クローンの一対の散布図である。これらのクローンはmRNA源となり、本願明細書で記載される可溶性T細胞受容体を合成するのに使用される。
【図4B】図4A及び4Bは流動選別し、続いてインビトロで増加した2つのCD8+T細胞クローンの一対の散布図である。これらのクローンはmRNA源となり、本願明細書で記載される可溶性T細胞受容体を合成するのに使用される。
【図5A】図5A〜5Eは下記に説明されるCTLクローンに対するTCRの配列を示したものである。
【図5B】図5A〜5Eは下記に説明されるCTLクローンに対するTCRの配列を示したものである。
【図5C】図5A〜5Eは下記に説明されるCTLクローンに対するTCRの配列を示したものである。
【図5D】図5A〜5Eは下記に説明されるCTLクローンに対するTCRの配列を示したものである。
【図5E】図5A〜5Eは下記に説明されるCTLクローンに対するTCRの配列を示したものである。
【図6】図6はAAV−a−scTCR−BirA及びAAV−b−scTCR−BirAのSDS−PAGEである。レーン1、標準分子量、レーン2、AAV−a−scTCR−BirA、レーン3、AAV−b−scTCR−BirA。
【図7A】図7AはELISAによる精製AAV−scTCRsのW4F検出を示すグラフである。
【図7B】図7BはELISAによる精製AAV−scTCRsの五量体検出を示すグラフである。
【図8】図8はAAV−scTCR多量体のp74/HLA−B7結合活性を示すプロットである。
【図9】図9A及び9BはAAV−scTCR多量体のp74/HLA−B7結合のヒストグラムを示すものである。
【図10】図10A及び10Bは1対のヒストグラムであり、AAV−scTCR多量体を使用したペプチド負荷した線維芽細胞のインビトロ染色を示したものである。
【図11】図11A及び11Bは一対のヒストグラムであり、AAV−scTCR多量体を使用したAAV−形質導入HLA−B*0702ヒト線維芽細胞のインビトロ染色を示したものである。
【図12】図12A及び12Bは一対のヒストグラムで、AAVの1E11ベクター遺伝子(vector genomes:vg)静注後一週間のHLA8 B*0702マウスから集めたCD11c+CD19−細胞のAAV−scTCR多量体染色を示したものである。
【発明を実施するための形態】
【0016】
ウィルスベクターを遺伝子導入媒体として使用するため遺伝子操作し、遺伝的欠陥を修正した。しかしながら、本発明に従ってウィルスベクターの導入遺伝子投与後、前記修正を受けた肝臓細胞が内部にベクターカプシドタンパク質を持つことが明らかになった。これらのタンパク質はT細胞により検出され、前記修正を受けた細胞は次々に破壊される。従って、本発明の一つの態様は、遺伝子導入の標的組織中のウィルスベクター抗原の存在を検出するための新しい試薬として、可溶性T細胞受容体(sTCR)の使用を伴う。ウィルス性ベクター配列又はカプシドタンパク質を検出する従来の方法は、PCR又は免疫特異性のある抗体を使用していた。しかしながら、臨床的により関連のある形態は宿主T細胞と接触するベクターカプシド、すなわち宿主MHCクラスI上に提示されているカプシド由来ペプチドである。可溶性TCRが自然発生T細胞で観察されるものと類似の様式でカプシド抗原のこの形態を認識することが開示される。さらに、このような試薬がベクター由来抗原の存在に関し形質導入組織を直接的且つ定量的に判定するために使用されることが可能である。この情報は免疫抑制療法を中止するかどうか又は中止する時を決定するために必要な情報を臨床医に提供する。
【0017】
本発明のもう1つの態様では、可溶性TCR試薬は、宿主T細胞が同種ベクター抗原に接触するのを特異的に阻害する特異的免疫調節治療法として使用される可能性がある。従って、本願明細書で記載される分子は診断及び治療薬の双方で使用される。
【0018】
様々なAAVの血清型が導入遺伝子の分野において使用され、またヒト集団におけるHLA型にも大きなばらつきがある。従って、本発明は、1)多形HLA拘束性エピトープ及び2)遺伝子療法ベクターで使用される特定のAAV血清型に特異性を持つように設計された可溶性TCRsが含まれる。
【0019】
代替AAVベクター血清型及びそれらのT細胞拘束性エピトープ
様々なAAV血清型、特にAAV−1、2、5及び8の類似した配列にも関わらず、ある自然発生AAVウィルスのエピトープに対して反応するT細胞が、遺伝子治療ベクターとして使用される代替血清型ウィルスに交差反応しないという可能性がある。なぜならばT細胞受容体はMHCクラスIに拘束された小さなペプチド(9〜11アミノ酸)のみを認識し、更にT細胞受容体は非常に高い特異性を持つため、AAV−2エピトープに反応するT細胞が、例えばアミノ酸1個のみが異なるAAV−8のエピトープを認識するかもしれないし認識しないかもしれない。さらに、いかなるAAVウィルスからのペプチドエピトープであっても個々のHLA型(ヒトのMHC)によって拘束される。このように、幾つかのT細胞拘束性AAVエピトープがAAV血清型中に保存される可能性に基づき、我々は、集団において通常見られるHLA−A1、A2、A3、B7、B8、B15、B44及びB51を含むHLA型に拘束される複数の血清型から多様なAAVエピトープを同定した。実際、我々はAAV−1、AAV−2及びAAV−8からの数個の候補エピトープのライブラリーを機能アッセイにおいて発達させた(表1)。可溶性TCRsのベクターカプシド配列検出に対する応用は、我々が既に有するHLA−B7拘束性の1つのAAVエピトープ(AAV−2VPQYGYLTL)から、推定遺伝子治療ベクターとして現在使用中のあらゆるAAV血清型の多数のエピトープへと容易に広げられる可能性がある。
【0020】
MHCクラスI五量体及び可溶性TCRsの産生だけでなく、免疫優性エピトープを同定することは、AAVカプシドタンパク質を遺伝子操作するのに有用であり、不要なエピトープが除去される。一旦エピトープが分かると、AAVカプシド配列中の一致する配列は標準的な分子生物学/組み換えDNA技術を用いて除去される。このように本発明のもう1つの態様はカプシドを再遺伝子操作する工程を含み、同定されたT細胞エピトープを改変する。これによって上に記載された特異的T細胞反応を避けるベクターが得られる。ここで留意するべき点はこれによって一連のベクターが生じ、それぞれが任意のMHCハプロタイプ特異的であり、これらのベクターは従来の臨床試験で見られたT細胞反応を回避するだろうということである(3)。本発明は表1のデータによって示されるように全てのAAV血清型に適用されるだろう。
【0021】
【表1】
【0022】
可溶性TCR技術の使用
sTCRsは免疫調節薬として利用することが可能である。例えば、可溶性TCRはT細胞によるエピトープ−MHCクラスI複合体認識を阻害するのに使用することができ、従って、遺伝子導入目的のAAVベクター導入に続く有害なT細胞反応を防ぐ。この目的では、可溶性TCRはAAVベクター導入直前又は導入時に投与されるだろう。
【0023】
sTCRsはAAV注入組織の表面上にあるAAVカプシドエピトープの染色のための試薬として使用されてもよい。遺伝子治療の分野におけるもっとも重要な疑問の一つはどのくらいの期間細胞内AAVカプシド分解に由来する前記ペプチドがMHC分子複合体の膜表面に提示され、免疫系によって検出可能かどうかである。本願明細書において記載される可溶性TCR試薬は生検組織のAAVエピトープ−MHC複合体を染色する利点を活かして使用することも可能であり、AAVカプシド抗原提示の持続時間を決定するのに役立つ。遺伝子治療を受けている患者がAAVカプシドに対する有害な免疫反応の高まる危険性に晒されている時間の範囲を決定することは有効な免疫抑制レジメンを設定するのに必須である。
【0024】
下記に記載の定義は本発明の理解を助けるために提供される。
【0025】
本願明細書で使用される「可溶性T細胞受容体又はsTCR」という語句は可溶性T細胞抗原受容体(単量体又は多量体)を指し、ウィルス感染細胞上のMHC分子に提示されるペプチド抗原を判定するのに使用される。可溶性TCRはフローサイトメトリー及び免疫組織化学での使用のため異なる標識分子に随意に結合されてもよい。T細胞受容体を生成する方法が米国特許第6,080,840号明細書、国際公開第WO/2005/116646号パンフレット及びBoulter et al.(2003)Protein Engineering 16:707−711に開示される。
【0026】
五量体はペプチドエピトープに結合した5個のMHCクラスI分子の複合体である。全5個の複合体が平面構造に保持されておりT細胞受容体に結合するのに利用されている。五量体は随意に蛍光色素に結合されフローサイトメトリーによって同種抗原特異的なT細胞を検出する。ここで留意すべき点は四量体技術もまた利用可能であり4個のMHCクラスI分子の複合体がペプチドエピトープに結合する。
【0027】
「五量体又は四量体染色」はT細胞を五量体又は四量体と混合し、4度で30分インキュベートし、1%FBS含有PBSで洗浄し、フローサイトメトリーで検出する過程を指す。染色アッセイを行うのに適した試薬はProImmuneから商業的に入手可能である。
【0028】
「細胞傷害性T細胞反応」という語句はエフェクターCD8+T細胞が標的細胞を殺すT細胞介在型破壊過程を指し、その標的細胞はMHCクラスI分子に結合するエピトープを外側表面に提示する。
【0029】
次の実施例は本発明のある実施形態を説明するために提供される。それは本発明をどんな意味においても本発明を限定するものではない。
【実施例1】
【0030】
上に言及したように、血友病治療のための従来の遺伝子治療において、導入遺伝子の発現は初期には高かったが、時間の経過につれて肝酵素が僅かに上昇し導入遺伝子発現は消失した。ベクター導入前及び後の患者からの末梢血単核球細胞(PBMC)を使用し、我々はMHCクラスI拘束性AAVカプシドエピトープを定義し(3)、AAVに特異的なCD8+T細胞を検出する五量体性HLA−B*0702/AAVペプチド(五量体)の可溶型を合成することができた。これらの五量体を使用して、我々は特異的なAAVエピトープに対する前記患者末梢血の特定のT細胞増殖の経時的変化を検出することができ、それは肝アミノ基転移酵素の上昇及び導入遺伝子発現の低下に一致していた。図1を参照。観察された前記T細胞増殖はMHC/ペプチド抗原の存在している時のみに観察された。このように本願明細書において提示される証拠は細胞傷害性T細胞が形質導入肝細胞によって提示されたベクターカプシドタンパク質に反応するということを明示する。ベクターカプシドタンパク質がベクター導入後限られた時間内に存在することを考慮して、本願明細書で開示される方法は免疫抑制の必要がある患者の4ヶ月の免疫抑制治療を含み、ベクター導入時前後にT細胞機能を一時的に停止する。
【0031】
T細胞反応刺激には樹状細胞のような専門の抗原提示細胞(antigen presenting cells:APCs)による抗原提示が必要であると考えられている。専門のAPCsは末梢に存在し、それらは直接感染又はウィルス感染細胞の貪食のいずれかによってウィルス抗原に遭遇する(交差提示)。樹状細胞は抗原を処理し、末梢のリンパ節に移動し、MHCクラスI及びII上の抗原及び適正な共刺激を未感作T細胞に提示する。最初の急速な増殖後、AAVに特異的なCD8+T細胞の発生頻度は減少し少数の貯蔵された記憶T細胞を後に残すことが予想され、この記憶T細胞は恒常的な維持増殖を通して各個人の一生涯の間保持される(2)。一旦適応した免疫システムが刺激されると、専門のAPCsによる共刺激及び抗原提示の必要性は厳密ではなくなり、全ての有核細胞が持つMHCクラスI上にのみウィルス抗原を提示する細胞を記憶T細胞は認識し殺す。AAV−2はT細胞反応刺激に必要な炎症反応を自分自身では誘導しないが、自然感染がヘルパーウィルスと一緒に起こるため、その時点でヘルパーウィルス及びAAV双方の抗原に対して誘導されるCD8+T細胞が刺激される。
【0032】
T細胞刺激に必要な炎症反応は抗体に再遭遇する記憶T細胞を新たに動員し活性化するために必須ではない。実験動物とは異なり、ヒトは幼少時にAAV−2に自然感染する。一般的なヒト集団におけるAAVに対するT細胞反応分析報告はほとんどない。Chirmuleらは正常コントロールの5%が組み換えAAVとともにPBMCをインキュベーション後>2の刺激係数を示したと報告した(25)。それ以来、T細胞分析のより感受性の高く定量的なアッセイが発達し、ヒト患者での抗ウィルスT細胞反応の詳細な特性解析が行われた。我々は数人の健常成人ドナーのAAVに対するT細胞反応を記録することができ、ウィルスカプシドに対する記憶T細胞反応はAAVベクターを使用する遺伝子導入研究における蔓延した問題であることを示した。ヒトリンパ球はAAVカプシドタンパク質配列由来ペプチドエピトープとともにインビトロで増殖させることができる。増殖した細胞はIFN−γを生成することによってエピトープに反応し(図2A)及びAAV特異性MHCクラスI五量体で染色することができる(図2B)。図3はELISPOTアッセイによって同定された一連の異なるT細胞エピトープ(下線を参照)を説明し、それらはAAV血清型1−8で非常に良く保存されている。
【0033】
遺伝子治療ベクターにはウィルスDNAがないため、MHCクラスIによってT細胞に提示される唯一のウィルス抗原は導入ベクターのカプシドタンパク質であり、それは限定された時間内に存在するべきである。しかしながら免疫学的に検出可能な形でカプシドが存在する時間の長さを決定する直接的な方法はない。我々は臨床試験から継続した4ヶ月の免疫抑制治療を選択したが、この時間枠はカプシド分解の動力学の確かな根拠に基づいていない。カプシドが存在する時間を決定する1つの理由は免疫抑制を中止し遺伝的修正を受けた細胞を保つ時期を決定するためである。
【0034】
複数の研究が行われ、それぞれ異なる間接的方法でどれくらいの期間ベクターカプシドが存在するかを決定した。臨床試験において、我々は生体内分布研究にPCRを使用し時間の経過とともにベクター遺伝子の存在を検出した。しかしながら、PCRはベクターDNAを検出するものであり、ベクターDNAは問題となっているT細胞によって認識される形のものではない。同様にベクターカプシドに対する抗体もT細胞によって認識される形のベクターを検出しない。なぜならこのT細胞はMHC+ペプチド複合体の形の中でも抗原部分だけを認識するからである。我々の研究室はまた同じ疑問に対処するため動物モデルの研究も行っている。しかしながら、我々は最終的にヒト患者で、遺伝子導入の標的となった特定の組織における、及び我々が使用する異なるいずれのAAV血清型に対するカプシドの持続性を決定する必要があるだろう、(我々の研究室はAAV−2に焦点をあてているが、他の遺伝子治療研究室はAAV−1、AAV−5、及び/又はAAV−8又はこれらのキメラに焦点を当てている)。このようにベクター形質導入組織に対するT細胞反応を基にした機能アッセイが本願明細書で開示されるが、理想的にはこれらのデータは記載されている可溶性TCRsを使用したデータと組み合わせて解釈される。これらのsTCRsによってカプシドの存在を直接定量的に決定でき、もし生検組織試料が利用できるのであればベクターで治療された個人のヒト患者中においても決定できる。
【0035】
上記のように、ヒトリンパ球をAAVカプシドタンパク質配列由来のペプチドエピトープとともにインビトロで増殖させた。増殖した細胞はIFN−γを生成することによってエピトープに反応し、AAV特異的なMHCクラスI五量体で染色される(図2A及び2B)。
【0036】
AAV特異的MHCクラスI五量体を使用して、増殖したカプシド特異性のあるCD8+T細胞の流動選別を行いCD8+T細胞のクローンを選択する可能性がある(図4A及び4B)。
【0037】
2つの異なるヒトAAV−p74ペプチド特異性のあるCTLクローンをT細胞受容体(T cell receptor:TCR)に特異的プライマーを使用するSMART−RACE法によってTCRα及びβ鎖cDNAを生成するために使用した。cDNA生成物をクローン化して配列を解読した。2個のTCRα鎖遺伝子(AV17/TRAJ43;配列ID番号19、及びAV17/AJ31;配列ID番号20)及び1個のTCRβ鎖遺伝子(BV6−2/BJ1−1;配列ID番号21)をCTLクローンaからクローン化した。1個のTCRα鎖遺伝子(AV27/AJ20;配列ID番号22)及び1個のTCRβ鎖遺伝子(BV4−3/BJ2−7;配列ID番号23)をCTLクローンbからクローン化した。2個のCTLクローンに対するTCRの配列が図5A〜5Eに提供される。
【0038】
同定された2個のα鎖を基に、細菌ビオチンリガーゼに融合した1個のscTCR(scTCR−BirAコンストラクト)をクローンbに生成し、2個の異なるscTCR−BirAコンストラクトをクローンaに生成した。3個の発現ベクターを生成し、CHO細胞に形質移入し、特性解析のための可溶性scTCR−BirA融合タンパク質を生成した。3個の全てのscTCRをAAV−scTCR形質移入CHO細胞に発現させ、ATCCから入手可能な抗ヒトTCR−β鎖抗体、(BF1−)8A3.31及びW4F.5Bを使用したサンドウィッチELISAによって細胞培養上清中に検出した。クローンb scTCR及びクローンa scTCRs(AV17/AJ31:BV6−2/BJ1−1)含有細胞培養上清はELISAによって検出されるAAVp74五量体活性を示し、さらに上清を特性解析した。AAV−scTCR−BirA融合タンパク質をAAV−scTCR−BirA形質移入体の培養上清からBF1−アフィニティークロマトグラフィーによって精製した。精製融合タンパク質をSDS−PAGEに示した(図6)。AAV−a及びAAV−b培養上清からの融合タンパク質精製量は各々4mg/L及び0.2mg/Lであった。精製scTCR−BirA融合タンパク質の特性解析をELISAによって行った。図7A及び7Bを参照。精製AAV−p74−scTCR−BirA融合タンパク質を抗TCR抗体が認識するか否かを決定するために、融合タンパク質の連続希釈したものを抗TCR BF1 mAb塗布プレートでインキュベートし、その後ビオチン標識抗TCR W4F mAb及びSA−HRPで検出した。結果は図7Aに示す。可溶性AAV−p74−scTCRsの機能的結合親和性を試験するため、融合タンパク質の連続希釈したものが抗TCR BF1 mAb塗布プレートにインキュベートされ、その後ビオチン化したAAV−p74/HLA−B*0702五量体及びSA−HRPで検出した。図7Bを参照。
【0039】
精製AAV−b融合タンパク質はAAV−a−AJ31よりもかなり低い五量体結合活性を示すため、AAV−a−AJ31融合タンパク質のみをビオチン標識し可溶性p74−scTCR−PE多量体の生成に使用した。AAV−a−scTCRが細胞表面のAAV−p74ペプチド/HLA−B7複合体に結合できるか否かを決定するためにAAV−p74ペプチド負荷HLA−B7陽性ヒトリンパ芽球様細胞株(JyA2B7)を使用した。50μg/mlのAAV−p74ペプチドを負荷したJyA2B7細胞(不死化B細胞)の特異的な染色を1検査当たり0.025μgのPE結合p74−scTCR多量体とともに観察した(図8)。
【0040】
一方、典型的な状態では最少5μg/mlAAV−p74ペプチドを負荷した(図9A)、又は、負荷中PLEを加えた場合最少1μg/mlAAV−p74ペプチドを負荷したJyA2B7細胞(図7B)を0.5μgのp74−scTCR多量体で染色することができる。(PLEはAltor Bioscience Corp.(Miramar、フロリダ州)の独自の試薬混合物であり、抗原提示細胞に負荷したペプチドを強化する)。
【0041】
AAV−scTCR多量体をHLA−B*0702陽性の正常ヒト線維芽細胞株を使用しインビトロで試験した(ATCC在庫からMalme−3が入手可能である)。細胞に37℃2時間10μg/mlの濃度でAAVカプシドエピトープをペプチド負荷し、その後AAV−scTCR多量体で染色した(図10)。両方の実験においてAAV−scTCR多量体の陽性染色が観察され、多量体がAAVペプチドエピトープを提示するMHC分子に良い親和性で結合することを示した。
【0042】
同様に、AAVベクター静注投与後のHLA−B*0702遺伝子組み換えマウスから採集したリンパ節細胞及び脾臓細胞はAAV−scTCR多量体の陽性染色を示す(図12)。
【0043】
結論
ヒトMHCクラスI分子HLA−B*0702上のAAV2ペプチド(配列VPQYGYLTL)に特異的な典型的な可溶性TCRが本願明細書に記載される。我々はHLA−B*0702ハプロタイプを持つ匿名の健常ドナーからこのペプチドに対して特異性のあるT細胞を増殖させた。このペプチドに特異的なT細胞をクローン化し、T細胞受容体のDNAを分離し発現させた。
【0044】
複数の研究グループがウィルス及び/又は癌抗原発現レベルを研究及び定量するため可溶性TCRsを発達させた(26〜31)。可溶性TCRsの遺伝子操作は様々な理由で困難であり、その理由には自然発生T細胞受容体のMHC/ペプチドに対する低い親和性及び特定の細胞上の特定のペプチド/MHC複合体の低い発現量が含まれる。TCRsの公開された研究において、ペプチド/MHC複合体に対するTCRsの親和性が判定された(30)。MHC/ペプチド抗原レベルの検査はマウス同種異系抗体(28)及び神経学的疾患の原因抗原であるHTLV−1抗原に焦点を当てた(感染(28)及び腫瘍随伴疾患(27))。T細胞レパートリーの発達をよく理解するため可溶性TCRsを使用した研究もあれば、ウィルスの侵入を捕らえるために可溶性TCRを使用した研究もある。最後にZhuら(2006)は多量体一本鎖可溶性TCRを作成し癌細胞上のヒトMHCクラスIに提示された癌抗原由来ペプチドを可視化した。我々が知る限りではヒト、動物、又はインビトロモデルのいずれかにおいてウィルスベクター由来抗原の存在を調べた公開されている研究はない。我々が見い出したHLAB*0702拘束性エピトープを記載した最近の論文(3)以外はAAV由来抗原に関する公開された研究もない。
【0045】
下記に記載された方法は本発明の実践を促進するために提供される。
【0046】
AAVカプシドエピトープの同定
アデノ随伴ウィルス(AAV)はパルボウィルス科に属し、一生のうちの通常早い時期にヒトに自然に感染する。ヒト集団において最も一般的なHLAに対する新しいCD8T細胞エピトープを同定するため、2つのIRB承認プロトコールを開始しthe Children’s Hospital of Philadelphia及びペンシルバニア大学のthe Cooperative Human Tissue Networkと提携してヒト脾臓を収集した。脾臓は他の臓器に比べエピトープ探究のための2つの主要な利点を提供する。1つ目はT細胞記憶保持に関連するリンパ様臓器であること、2つ目は1グラムの組織から5億個の細胞を分離することが可能であり、この数は全血のような他の供給源からは簡単に得られることはできない。
【0047】
T細胞分離及びHLAタイピング
T細胞を組織採取から24時間以内で分離する。脾臓を最初外科用メスによって小片に処理しその後均質化する。赤血球溶解後脾細胞をPBSで2回洗浄し、各々約1000万個の一定分量で10%DMSOとともにヒト血清中に凍結した。
【0048】
2つ若しくはそれ以上の細胞を高分解HLAタイピングのためペンシルバニア大学病院のHLA−タイピング研究室に送付した。
【0049】
インビトロT細胞増殖
CD8T細胞エピトープの同定はAAVカプシドタンパク質に反応する記憶CD8T細胞の低い発生頻度によって妨げられる。この限界を乗り越えるため、脾臓組織からのリンパ球をVP1と呼ばれるAAVカプシドタンパク質に由来する一連のペプチドとともにインビトロで増殖させた。前記VP1ペプチドライブラリーは各々が10アミノ酸ずつ重複しているタンパク質配列に由来する15アミノ酸長のペプチド145個でできている(Mimotopes)。
【0050】
簡潔に説明すると、脾臓からのリンパ球を3%熱不活化ヒト血清を有するAIM−V(Gibco)培地中で1ウェル当たり100万個の細胞を96ウェルプレートに播種し、前記ウェルの細胞の半分を3000radで放射線照射し支持細胞層として機能させる。VP1ライブラリーからの各ペプチドを10μg/mlの最終濃度で1つのウェルに添加する。増殖0日目及び2〜3日毎にIL−2(Roche)を最終濃度10U/mlで培養に添加する。
【0051】
刺激の1巡目は7〜10日間継続するが、AAVカプシドに反応するCD8T細胞の予想される少ない数のため、通常増殖には2−3巡必要である。増殖の各巡の追加は単純にウェルに新しいペプチド及び3000radで放射線照射した500,000同種脾臓細胞を添加することによって行う。
【0052】
IFN−γELISpotによるエピトープ探索
ELISpotは特異的な抗原に反応するT細胞の数を同定するため使用される強力な技術である、すなわちELISpotはペプチドに反応してIFN−γを分泌する細胞の能力を測定する。50,000個の増殖したT細胞を抗ヒトIFN−γ抗体(Mab−Tech)を先に塗布した96ウェルELISpotプレート(Millipore)に増殖のため使用されるペプチドの存在下で播種した。37℃、5%CO2でインキュベーション24時間後細胞を洗浄し、抗ヒトIFN−γビオチン標識2次抗体(Mab−Tech)をウェルに添加する。ストレプトアビジン−アルカリフォスファターゼを特異的基質の存在下で検出試薬として使用する。
【0053】
陽性のウェルを最初に添加された100万個の細胞当たりのスポットを形成する細胞(spot forming cells:SFC)の数を基に判定する。もしSFCの数が陰性対照コントロールウェル(培地のみ)のSFCの数よりも3倍高いならば、前記ウェルは陽性と判定された。陽性ペプチドは上記に記載されたプロトコールを繰り返すことによって通常少なくとも2回確認された。
【0054】
その後この方法は興味のある全てのHLA対立遺伝子に対して繰り返された。
【0055】
オンライン予想アルゴリズム
2つのオンラインエピトープ予想アルゴリズムプログラムをELISpotアッセイに使用される15アミノ酸長ペプチドの中の9アミノ酸配列を同定するのに使用し、その配列はHLA分子に対する結合配列を表す。これらのプログラムはRankpep:http://bio.dfci.harvard.edu/Tools/rankpep.html及びSYFPEITHI:www.syfpeithi.de/のインターネット上で見ることができる。
【0056】
同定された9アミノ酸を合成しELISpot及び細胞内サイトカイン染色によって確認する。陽性のペプチドエピトープ配列をその後HLA−ペプチド五量体検出薬を合成するために利用する(Proimmune)。
【0057】
AAV−特異的T細胞選別及びクローニングのための五量体の使用
特異的なAAVペプチドとともに一度又は二度増殖させた末梢血単核細胞(PBMC)を適切なHLAペプチド五量体とともに滅菌状態で染色する。分析のみに通常使用された五量体染色に関しては、染色を4℃20分間2%ヒト血清を含む滅菌PBS中で実行する。培養を抗CD8抗体とともに共染色する。PBS−2%hAB血清で2回洗浄後、前記細胞を蛍光活性細胞選別器に通し、96ウェル丸底プレートの1ウェル当たり1個の五量体+CD8+T細胞を含むように選別する。細胞を5個から10個のプレートに選別する。各プレートの各ウェルは同じ日に調製し、支持細胞として放射線照射された同種PBMCを含め、EBV形質転換B細胞株からの放射線照射細胞及びT細胞成長因子として50IU/mlの組み換えヒトIL−2を加えた商業的に入手可能な抗CD3T細胞刺激抗体(OKT3)を伴う。選別された(クローン化された)前記細胞をその後2週間37℃の湿潤恒温器でインキュベートする。
【0058】
2週間後T細胞のクローン成長を目視検査によって判定する。成長細胞を五量体染色によるAAVに対する特異性で判定する(上記に記載)。ペプチド特異性を得るクローンを最初の刺激のように同種PBMC、放射線照射EBV形質転換B細胞、OKT3及びIL−2でさらに刺激するが、この作業以外に細胞集団の成長速度によって成長細胞を24ウェルプレート又はT25フラスコに移動させる。Dr.Cassian Yee及びその同僚の記載のようにクローンを2週毎に再刺激をする(32)。
【0059】
一旦少なくとも100万個のT細胞クローンが成長すると、一定分量を標準プロトコールに従って凍結する。この一定分量のT細胞クローンを可溶性TCR生成のためAltor Bioscience Corporationに提供する。もう1つの方法として我々は成長T細胞クローンからRNAを分離しAltor BioscienceにRNAを提供することもある。この時点のRNAをcDNAを生成するのに使用し、この材料によって可溶性T細胞受容体を生成する基礎を作る、以上の作業はこれらの分子を生成する経験を持つグループであるAltor Bioscienceによる(31)。
【0060】
我々はAAVエピトープに特異的な可溶性TCR受容体をクローン化し特性解析するのに成功し、このAAVエピトープは患者への遺伝子導入を妨げる細胞傷害性T細胞反応の発生に関連する。本願明細書において記載される前記材料及び方法は診断及び治療双方に使用され、必要とする患者への治療用異種タンパク質の導入を促進する。
【0061】
本発明の一部の望ましい実施形態を記載し及び特定の実施例を上記に提示したが、本発明はそれらの実施形態に限定されるものではない。様々な改変は次の請求項に記載されるように本発明の要旨に逸脱することなく行われる。
【特許請求の範囲】
【請求項1】
ヒトMHCクラスI分子上におけるアデノウィルス随伴ウィルス(adenovirus−associated virus:AAV)に存在するペプチド配列に対して免疫学的に特異的な可溶性T細胞受容体(soluble T cell receptor:sTCR)。
【請求項2】
請求項1記載のsTCRにおいて、前記アデノウィルスペプチド配列は、AAV−1、AAV−2、AAV−5、AAV−8及び他の自然発生血清型から成る群から選択される血清型から得られるものである。
【請求項3】
請求項1記載のsTCRにおいて、前記ヒトMHCクラスI分子は、HLA−A1、HLA−A2、HLA−A3、HLA−B7、HLA−B8、HLA−B15、HLA−B44及びHLA−B51から成る群から選択されるものである。
【請求項4】
導入遺伝子含有アデノ随伴ウィルスベクター投与前、投与中、投与後のウィルスカプシド抗原に対するT細胞介在型免疫反応の存在を検出する方法であって、
a)患者からの生物学的試料、すなわちT細胞を含む前記試料を得る工程と、
b)前記細胞をMHCクラスI分子上の前記カプシド中のペプチドエピトープを含む五量体に接触させる工程と
c)無処理のコントロール細胞と比較して、工程b)の前記接触が前記細胞を刺激するかを決定し、前記ウィルスカプシドの前記ペプチドエピトープに対して特異的な前記接触によって細胞が刺激され、前記細胞がカプシド及び導入遺伝子含有細胞のT細胞介在型破壊を促進する工程と
を有する方法。
【請求項5】
請求項4記載の方法において、前記生物学的試料は、導入遺伝子を含む細胞、末梢血単核球細胞(PBMCs)、肝細胞、上皮細胞、及び筋細胞から成る群から選択される細胞を有するものである。
【請求項6】
請求項4記載の方法において、この方法は、さらに、前記刺激細胞からmRNAを分離する工程と、cDNAを調製する工程と、前記ウィルスカプシド抗原に対して免疫学的に特異的な可溶性T細胞受容体をクローン化する工程とを有するものである。
【請求項7】
請求項6記載の方法よって調製される可溶性T細胞受容体。
【請求項8】
アデノウィルス随伴ウィルスベクター投与後のウィルス性形質導入細胞のT細胞介在型破壊を抑制するための方法であって、
a)AAVエピトープ/MHC複合体に特異的なsTCRの有効量を提供する工程であって、前記sTCRが前記導入遺伝子含有細胞のT細胞介在型破壊を妨げるものである、前記提供する工程
を有する方法。
【請求項9】
ウィルス性形質導入細胞のT細胞介在型破壊を回避するための方法であって、
d)請求項4記載のペプチドエピトープに対する特異性を検出する工程と、
e)前記AAVベクターを改変する工程であって、工程d)において同定されるペプチドエピトープを除去するものである、前記改変する工程と、
f)前記改変AAVベクターの導入遺伝子を投与する工程であって、前記改変は前記ウィルス性形質導入細胞のT細胞介在型破壊を抑止するものである、前記投与する工程と
を有する方法。
【請求項10】
請求項1記載の可溶性T細胞受容体であって、この可溶性T細胞受容体は、表1で示されるAAV血清型のエピトープを有するものである。
【請求項11】
請求項9記載の方法において、前記AAVベクターは表1に記載されるAAVペプチドを除去するように改変されるものである。
【請求項12】
請求項11記載の方法によって調製される改変AAVベクター。
【請求項1】
ヒトMHCクラスI分子上におけるアデノウィルス随伴ウィルス(adenovirus−associated virus:AAV)に存在するペプチド配列に対して免疫学的に特異的な可溶性T細胞受容体(soluble T cell receptor:sTCR)。
【請求項2】
請求項1記載のsTCRにおいて、前記アデノウィルスペプチド配列は、AAV−1、AAV−2、AAV−5、AAV−8及び他の自然発生血清型から成る群から選択される血清型から得られるものである。
【請求項3】
請求項1記載のsTCRにおいて、前記ヒトMHCクラスI分子は、HLA−A1、HLA−A2、HLA−A3、HLA−B7、HLA−B8、HLA−B15、HLA−B44及びHLA−B51から成る群から選択されるものである。
【請求項4】
導入遺伝子含有アデノ随伴ウィルスベクター投与前、投与中、投与後のウィルスカプシド抗原に対するT細胞介在型免疫反応の存在を検出する方法であって、
a)患者からの生物学的試料、すなわちT細胞を含む前記試料を得る工程と、
b)前記細胞をMHCクラスI分子上の前記カプシド中のペプチドエピトープを含む五量体に接触させる工程と
c)無処理のコントロール細胞と比較して、工程b)の前記接触が前記細胞を刺激するかを決定し、前記ウィルスカプシドの前記ペプチドエピトープに対して特異的な前記接触によって細胞が刺激され、前記細胞がカプシド及び導入遺伝子含有細胞のT細胞介在型破壊を促進する工程と
を有する方法。
【請求項5】
請求項4記載の方法において、前記生物学的試料は、導入遺伝子を含む細胞、末梢血単核球細胞(PBMCs)、肝細胞、上皮細胞、及び筋細胞から成る群から選択される細胞を有するものである。
【請求項6】
請求項4記載の方法において、この方法は、さらに、前記刺激細胞からmRNAを分離する工程と、cDNAを調製する工程と、前記ウィルスカプシド抗原に対して免疫学的に特異的な可溶性T細胞受容体をクローン化する工程とを有するものである。
【請求項7】
請求項6記載の方法よって調製される可溶性T細胞受容体。
【請求項8】
アデノウィルス随伴ウィルスベクター投与後のウィルス性形質導入細胞のT細胞介在型破壊を抑制するための方法であって、
a)AAVエピトープ/MHC複合体に特異的なsTCRの有効量を提供する工程であって、前記sTCRが前記導入遺伝子含有細胞のT細胞介在型破壊を妨げるものである、前記提供する工程
を有する方法。
【請求項9】
ウィルス性形質導入細胞のT細胞介在型破壊を回避するための方法であって、
d)請求項4記載のペプチドエピトープに対する特異性を検出する工程と、
e)前記AAVベクターを改変する工程であって、工程d)において同定されるペプチドエピトープを除去するものである、前記改変する工程と、
f)前記改変AAVベクターの導入遺伝子を投与する工程であって、前記改変は前記ウィルス性形質導入細胞のT細胞介在型破壊を抑止するものである、前記投与する工程と
を有する方法。
【請求項10】
請求項1記載の可溶性T細胞受容体であって、この可溶性T細胞受容体は、表1で示されるAAV血清型のエピトープを有するものである。
【請求項11】
請求項9記載の方法において、前記AAVベクターは表1に記載されるAAVペプチドを除去するように改変されるものである。
【請求項12】
請求項11記載の方法によって調製される改変AAVベクター。
【図1】

【図2A】

【図2B】
【図3】

【図4A】
【図4B】
【図5A】

【図5B】

【図5C】

【図5D】

【図5E】

【図6】
【図7A】
【図7B】

【図8】
【図9】
【図10】
【図11】
【図12】

【図2A】
【図2B】
【図3】
【図4A】
【図4B】
【図5A】
【図5B】
【図5C】
【図5D】
【図5E】
【図6】
【図7A】
【図7B】
【図8】
【図9】
【図10】
【図11】
【図12】
【公表番号】特表2009−538633(P2009−538633A)
【公表日】平成21年11月12日(2009.11.12)
【国際特許分類】
【出願番号】特願2009−513463(P2009−513463)
【出願日】平成19年5月31日(2007.5.31)
【国際出願番号】PCT/US2007/070147
【国際公開番号】WO2007/140474
【国際公開日】平成19年12月6日(2007.12.6)
【出願人】(508354153)
【Fターム(参考)】
【公表日】平成21年11月12日(2009.11.12)
【国際特許分類】
【出願日】平成19年5月31日(2007.5.31)
【国際出願番号】PCT/US2007/070147
【国際公開番号】WO2007/140474
【国際公開日】平成19年12月6日(2007.12.6)
【出願人】(508354153)
【Fターム(参考)】
[ Back to top ]