
遺伝子発現アッセイ
RNAの二次構造を実質的に除去する重硫酸塩などの作用物質でRNAを処理することと、処理したRNAの存在または量を測定して遺伝子発現の指標を得ることとを含む遺伝子発現のアッセイ。本発明は、そのアッセイにおけるオリゴヌクレオチド、PNA、LNAまたはINAプローブの使用も含む。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、DNAへのRNAの転換を必ずしも必要としない遺伝子発現のアッセイに関する。
【背景技術】
【0002】
チップを使用するマイクロアレイ発現プロファイリング(Schena et al、1995、Science270:467-470;Chee et al、1996、Science274:610-614)などの細胞集団中のRNA産出量の測定によって、または遺伝子発現の連続分析SAGE(Velculescu et al、1995、Science270:484-487)によって、または全遺伝子発現分析TOGA(Sutcliffe et al、2000、Proc NatlAcad Sci USA97:1976-1981)によって、またはランダムに並んだ位置特定可能な高密度光学センサーアレイ(Michael et al、1998、Anal Chem70:1242-1248)によって、またはマイクロビーズアレイ上での超並列シグネチャー配列決定MPSS(Brenner et al、2000、Nature Biotechnology18:630-634)によって遺伝子発現を推測するための現在使用されている方法は、遺伝子発現の真の程度または量に関する正確な情報を必ずしも与えることができるわけではない。使用されている方法の多くは、まず対応するcDNA分子へのRNAの逆転写仲介転換、次いでcDNA集団の増幅および標識を必要とするので間接的である。良くて、このような現在の方法は遺伝子発現の指標のみをもたらすが、対象の特定遺伝子の発現の正確な測定値はもたらさない。
【0003】
これらの難点は今や非常に明らかである。例えば、SAGE技術(Stollberg et al、2000、Genome Research10:1241-1248)、チップ(Chudin et al、2001、Gene Biology3:0005.1-0005.10;Kothapalli et al、2002、BMC Bioinformatics3:1-10;Workman et al、2002、Genome Biology3:0048.1-0048.16)における偏り(bias)が記載されている。より一般的に、現在の方法における問題点が記載されている(Martin and Pardee、2000、Proc Natl Acad Sci USA 97:3789-3791;Wang et al、2000、Proc Natl Acad Sci USA97:4162-4167)。
【0004】
以前の変性RNA分子に自然かつ迅速に形成する安定した二次構造のために、適切な特異的プローブとの直接的なハイブリダイゼーションによってRNAをアッセイすることは困難である。
【発明の概要】
【発明が解決しようとする課題】
【0005】
本発明者は現在、生物、細胞集団または組織サンプル中の遺伝子発現のより正確な推測を与えることができる改善されたアッセイを開発した。
【課題を解決するための手段】
【0006】
第一の態様において、本発明は、
(a)RNAの二次構造を実質的に除去する作用物質でRNAを処理することと、
(b)処理したRNAの存在または量を測定して遺伝子発現の指標を得ることと
を含む遺伝子発現のアッセイを提供する。
【0007】
第二の態様において、本発明は、遺伝子発現を推測または測定するアッセイにおける、RNAの二次構造を実質的に除去しRNAを安定化させる作用物質の使用を提供する。
【0008】
第三の態様において、本発明は、RNA検出により遺伝子発現をアッセイするための、選択した化学組成を有するプローブの使用を提供する。
【0009】
プローブは塩基A(アデニン)、T(チミン)およびC(シトシン)から実質的に構成され、相当量のG(グアニン)は含まないことが好ましい。プローブは、G(グアニン)を実質的に含まないことが好ましい。
【0010】
本発明は、RNAの二次構造を実質的に除去する作用物質を利用する遺伝子発現のアッセイにおけるオリゴヌクレオチド、PNA、LNAまたはINAプローブの使用も含む。
【0011】
第四の態様において、本発明は、
(a)RNAの二次構造を実質的に除去する作用物質でRNAを処理することと、
(b)RNAの相補配列と結合することができるプライマーを使用してRNAを逆転写および増幅することと、
(c)処理および増幅したRNAの存在または量を測定して遺伝子発現の指標を得ることと
を含む遺伝子発現のアッセイを提供する。
【0012】
好ましい形では、RNAは、微生物、細胞、細胞群または細胞集団を含む、真核生物または原核生物由来である。
【0013】
好ましい形では、RNAはmRNAである。
【0014】
好ましい形では、RNAは微生物由来である。
【0015】
微生物、細胞または細胞集団または他の組織または生物源からRNAを単離するのに適した任意の方法によって、RNAを得ることができる。このような方法は当技術分野でよく知られており、例えば、Sambrook et al、「Molecular Cloning、A Laboratory Manual」second ed.、CSH Press、Cold Spring Harbor、1989を参照。例として、オリゴ−dTコーティング磁気ビーズまたは樹脂があるが、これらだけには限られない。RNA結合樹脂の具体例には、以下のRNeasy(商標)およびOligotex(商標)(Qiagen)、StrataPrep(商標)トータル(Stratagene)、Nucleobond(商標)(Clontech)、RNAgents(商標)およびPolyATract(商標)システム(Promega)などがある。密度勾配遠心分離技法を使用してRNAを単離することもできる。
【0016】
RNAは、真核生物由来でもよく、または細菌などの原核生物およびウイルス由来でもよい。薬物治療、ウイルス量、サンプル中の様々なウイルスの発現アレイなどをモニタリングするためにアッセイを使用することができる。
【0017】
RNAはシトシン塩基を修飾することができる作用物質で処理して、RNAの相補領域間の結合強度を弱めることが好ましいが、これは、シトシンの除去がC:G塩基対形成の消失をもたらすからである。生じる修飾は二次構造を除去し、一本鎖実体としてRNAを実質的に安定させる。作用物質は、亜硫酸水素塩、ヒドロキシルアミン、酢酸塩またはクエン酸塩から選択されることが好ましい。作用物質は、亜硫酸水素塩または酢酸塩試薬であることがより好ましい。作用物質は、水の存在下でシトシンをウラシルに修飾する試薬である亜硫酸水素ナトリウムであることが最も好ましい。
【0018】
亜硫酸水素ナトリウム(NaHSO3)はシトシンの5,6−二重結合と容易に反応して、脱アミノ化の影響を受けやすく水の存在下で亜硫酸ウラシルを生成するスルホン化シトシンの反応中間体を形成する。必要な場合、亜硫酸基を適度なアルカリ条件下で除去して、ウラシルの形成をもたらすことができる。したがって、潜在的には全てのシトシンがウラシルに転換される。ウラシル塩基は、シトシンが形成し得る3つの水素結合ではなく、任意の相補塩基との2つの水素結合のみを形成することができるので、複雑な二次構造を再形成するRNAの傾向は大幅に低下する。次いで、こうして処理した修飾型RNAは、妨げられることなく特定の相補的プローブとの相互作用に利用可能である。
【0019】
重要なことに、特定の実施形態では、標的配列のその含有量に関してサンプルをアッセイする前に、現在実施されているようなRNAを対応する相補的DNA(cDNA)に転換する必要がない。逆転写酵素またはポリメラーゼ連鎖反応(PCR)増幅工程のいずれも必要とされないので、本発明による方法は簡潔でより直接的であり、したがって、標準的手順で用いられる酵素の配列コピーの偏りによって引き起こされる誤差が起こりにくい。
【0020】
存在する標的(修飾型)RNAの量は、任意の適切な手段によって測定することができる。例えば、標的RNAを対象とする特異的プローブは、対象の対応する転写単位の一部または全部から誘導することができる。あるいは、プローブは、適切な配列の適切な抗体または抗体断片または単一ドメイン抗体、オリゴヌクレオチド、またはペプチド核酸(PNA)、固定化核酸(locked nucleic acid)(LNA)または挿入核酸(intercalating nucleic acid)(INA)プローブなどの塩基−配列特異性を示す任意の他の実体から誘導することができる。
【0021】
本発明のプローブは、試験するRNAと「実質的に」相補的であるように設計することができる。プローブが事実上PNA、LNA、オリゴヌクレオチドまたはINAであるとき、それらはA(アデニン)、T(チミン)、またはC(シトシン)塩基のみを含み得るが、これは、修飾型RNAが非修飾C残基を実質的に含まないからである。
【0022】
プローブは、オリゴヌクレオチドプローブまたはPNA、LNAまたはINAプローブなどの任意の適切なリガンドであってよい。例えば、全ての処理済みRNAと結合し得るポリ−TDNAまたはポリ−TPNAまたはLNAプローブまたはポリTINAプローブを使用することができ、これらはいずれも細胞由来のポリA「テイル」を有し、細胞、細胞集団または組織中の全遺伝子発現の測定を可能にする。あるいは、対象のRNAを対象とする特異的プローブを使用して、所与の細胞または組織中の特異的遺伝子発現を測定することが可能である。
【0023】
RNAにおけるランダムな二次構造形成を不安定にするための、シトシンとウラシル、またはその亜硫酸塩付加物の置換も、特異的オリゴ−、PNA、LNA、またはINAプローブと修飾型RNAの結合の強度を有意に低下させ得る。INA分子は、末端位置に限り挿入基(intercalating group)を用いて適切に設計するとき、相補的配列構造のRNAに対して高い結合性を有する。これをさらに補うために、プローブ中のアデニン塩基の代わりに、任意の相補的RNA鎖中の3つの水素結合を形成する2,6−ジアミノプリン(AP)を(アデニンが形成し得る2つに対する)チミンと置換し、これによってプローブとRNAの間の結合を強化することが好ましい。
【0024】
INAプローブは、その塩基配列において相補性を示すDNAまたはRNAの隣接塩基間に挿入することができる挿入分子を、「正常」または「修飾型」ヌクレオチドの配列中の様々な場所に結合させることによって構築する。このようなDNA分子によって、このような挿入部分の存在は、挿入基をINAプローブ中のどこに結合させようとも、プローブと標的核酸分子の間の相互作用を大幅に安定化させる。INAの注目すべき性質は以下に記載する。
【0025】
RNA分子と結合するように設計したINAプローブの場合、挿入基をINAの末端または末端付近に配置して、結合を高めることが好ましい。驚くべきことに、挿入基を内部に配置することはRNAと相補的DNAのハイブリダイゼーションに悪影響を与える可能性があり、かつハイブリッド構造を安定化させずに不安定化し得る。INAプローブを構築するための方法は以下に記載する。
【0026】
本発明の重要性は、処理して全てのそのシトシン残基がウラシル残基に転換された標的RNA種と非常に特異的かつ非常に強く結合する、個々の構築体のINAプローブの驚くべき能力に関する。結果として、本発明は、現在使用されている方法の多くの間接工程によって導かれる誤りまたは偏りを回避することができる方法に関する。
【0027】
示すように、幾つかの他の特異的プローブを本アッセイ中で使用することができるが、それらの使用の詳細な説明から明らかとなる理由で、INAプローブを使用することが好ましい。
【0028】
RNAの増幅は、RNAの相補配列と結合することができるINAプライマーを使用して実施することが好ましい。増幅は、典型的には逆転写酵素PCRベースの方法を使用して実施される。
【0029】
高ストリンジェンシー条件下でハイブリダイズすることができるか本発明中で利用する核酸分子と高い配列類似性を有することができる同等な配列に関して、「高ストリンジェンシー条件下でハイブリダイズする」は、当技術分野でよく知られている用語である「ストリンジェントなハイブリダイゼーション条件」と同義であり得る。例えば、Sambrook、「Molecular Cloning、A Laboratory Manual」second ed.、CSH Press、Cold Spring Harbor、1989;「Nucleic Acid Hybridisation、A Practical Approach」、Hames and Higgins eds.、IRL Press、Oxford、1985を参照。
【0030】
本発明の利点は、cDNAへのRNAの転換を必要とせずに、RNAの直接測定を実施することができることである。本発明のアッセイは、cDNAへのRNAの転換または増幅を必要とする現在の方法による潜在的な誤りを導かずに、細胞集団中の遺伝子活性の真の測定を可能にする。
【0031】
PNAまたはオリゴヌクレオチドプローブは、当業者に知られている任意の適切な方法を使用して調製することができる。INAプローブは、当業者に知られている任意の適切な方法によって調製することができる。
【0032】
適切なプローブを使用するハイブリダイゼーションアッセイ前にINAプライマーを使用して、処理したRNAを少量から増幅することもできる。
【0033】
本発明は、チップなどの現在のアレイ技術中、またはランダムに位置特定可能な高密度光学アレイ中で使用するのに適しており、したがって多数の遺伝子を迅速にアッセイすることができる。この形では、何万もの遺伝子の活性を1回の試験で測定またはアッセイすることができる。本発明は、例えばビーズ技術を使用する、少数の遺伝子を対象とするアッセイにも適応可能である。修飾RNA種はアレイの形で適切な支持体にスポットするまたはつけることが可能であり、アレイは様々なプローブによって測定することができる。
【0034】
1つの好ましい形では、本発明は、PNA分子は正味の電荷を有していないが、一方RNA分子は、それらのリン酸骨格のために、強く負に帯電しているという事実を特に利用する。結合PNAプローブの検出は、正に帯電した蛍光色素などの単分子、その長さに比例して核酸と特異的に結合し直接検出することができる複数の分子を利用することができる。多くのこのような適切な蛍光色素が知られている。
【0035】
検出系は、核酸と選択的に結合し酵素アッセイを使用して検出することができる正に帯電した領域を有する酵素、または捕捉された核酸分子と選択的に結合する正に帯電した放射性分子であってもよい。ナノ結晶も使用することができることは理解されよう。
【0036】
他の適切な検出系は量子ドットバイオコンジュゲートの使用である(Chan and Nie 1998 Science 282:2016-2018)。
【0037】
あるいは、配列特異的プローブおよび幾つかの蛍光色素分子が結合するミクロスフェアを利用することができる。ミクロスフェアは、特定のRNA種を標的化するプローブに直接、またはそのポリアデニンテイルなどのRNAの第二の非特異的構成部分を介して結合させることが可能である。この後者の場合、ミクロスフェアシグナル検出系の結合は、INA、PNA、LNAまたはオリゴヌクレオチド部分としてのポリT配列を介した結合であり得る。
【0038】
蛍光色素マーカーを有するミクロスフェアには様々な色またはスペクトルがあるので、1回の実験で、単細胞サンプル中に存在する幾つかの異なるRNA種のそれぞれの量を測定することが可能である。さらに、そのように標識した一種のミクロスフェアを、容易に目に見える状態にして計数することができ、したがって異なるRNA種間の発現のわずかな違いを相当な精度で決定することができる。
【0039】
基質と反応して着色産物を形成することができる適切な放射性化合物または酵素を用いた標識などの、標的修飾RNAと結合するリガンドを検出するための他の方法を、捕捉RNAまたはプローブまたは基質のいずれかに直接結合させる個々の適用例に使用することもできる。
【0040】
この手順中でのリガンドの1つとしてのINAまたはPNAまたは他のオリゴヌクレオチドプローブの使用には、オリゴヌクレオチドプローブの使用に優る非常に有意な利点がある。INAまたはPNAの結合はより速く平衡に達し、より高い配列特異性を示す。PNA分子は非帯電性であり、従来のオリゴヌクレオチドプローブより高い結合係数で標的修飾RNA分子と結合することができる。特に、INAプローブはA−T−とA−U塩基間の結合を高める。処理してシトシン塩基がウラシル塩基に転換された二次構造を除去したRNAの場合、これは重要である。RNA処理の結果として、少数のG−C塩基間相互作用および対応するA−TおよびA−U塩基間相互作用の数が増大する。
【0041】
本発明は直接的な検出法を使用することができるので、アッセイはサンプル中の標的RNAの量の真の正確な測定値を与えることができる。本発明のアッセイは、PCRなどのプロセスにおけるシグナル増幅に依存する方法に固有の潜在的偏りによって混乱することはなく、この場合、このような手順中で一般的に使用される酵素は、異なる配列の増幅率の差によって組織的偏りを導き得る。
【0042】
本発明は、疾患状態の検出、幹細胞および派生細胞集団の分化状態、遺伝子発現または細胞機能に対する薬剤の影響の検出または測定、およびC型肝炎ウイルス(HCV)およびヒト免疫不全ウイルス(HIV)などのウイルスに感染した患者のための正確な薬物計画の決定に役立つウイルス量モニタリングなどの、遺伝子発現の正確な指標が有用である任意の他の状況に特に適している。
【0043】
本明細書を通じて、文脈で他に必要とならない限り、語句「comprise」、または「comprises」もしくは「comprising」などの変形は、言及する要素、整数または工程、または要素群、複数の整数または工程の包含を意味するが、任意の他の要素、整数または工程、または要素群、複数の整数または工程の除外は意味しないことは理解されよう。
【0044】
本明細書中に含まれている文献、作用、物質、装置、物品などの任意の論述は、本発明の文脈を与える目的のものに過ぎない。これらの事項のいずれかまたは全てが従来技術の土台の一部を形成する、または本発明と関係がある分野の共通の一般知識であったことは当然のこととして解釈すべきでないが、これは、それが本発明の優先日前にオーストラリアに存在したからである。
【0045】
本発明がより明確に理解できるように、好ましい形態を以下の図面および実施例を参照しながら説明する。
【図面の簡単な説明】
【0046】
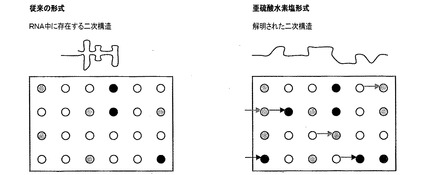
【図1A】典型的なマイクロアレイベースのアッセイの説明を示す図であり、この場合、暗色点は特定のRNA集団中でアップレギュレーションされる遺伝子を示し、かつ淡色点は同じ集団中でダウンレギュレーションされる遺伝子を示す。暗色および淡色の矢印は、従来系中の検出分子がそれらの標的と結合するのを妨げる二次構造のため、従来の方法ではなく亜硫酸水素塩処理RNAにおいて、それぞれアップレギュレーションおよびダウンレギュレーションされるとして検出される遺伝子を示す。
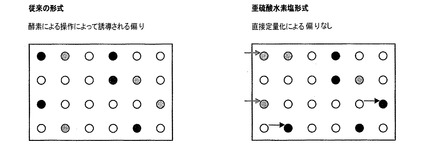
【図1B】図1Aと同様のマイクロアレイベースのアッセイを示す図であるが、暗色および淡色の矢印は、発現分析前の酵素によるRNAの操作中に生じRNA発現レベルの不正確な決定をもたらす偏りのため、従来の方法ではなく亜硫酸水素塩処理RNAにおいて、それぞれアップレギュレーションおよびダウンレギュレーションされるとして検出される遺伝子を示す。酵素による操作は誤解を与える結果を引き起こし、特定の遺伝子は、それらが実際そのように制御されないときにアップまたはダウンレギュレーションされることを示し得る。
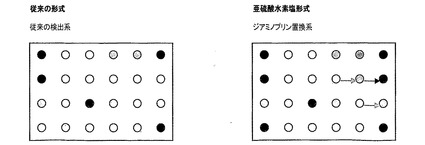
【図1C】図1Aと同様のマイクロアレイベースのアッセイを示す図であるが、暗色および淡色の矢印は、検出分子の改善された特異性のため、従来の方法ではなく亜硫酸水素塩処理RNAにおいて、それぞれアップレギュレーションおよびダウンレギュレーションされるとして検出される遺伝子を示す。検出分子の特異的結合強度の増大は、特異性の欠如のため従来の方法を使用して検出することができないRNA種の検出をもたらす。
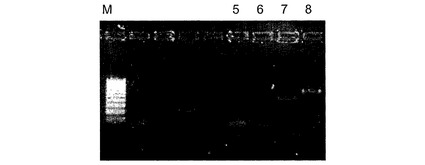
【図2】亜硫酸水素塩処理RNAからのPCR増幅のゲル分離の結果を示す図である。分離ウエル:M、100〜1000bpマーカー;レーン5野生型アクチンプライマーエクソン3a〜3b;レーン6野生型アクチンプライマーエクソン3a〜4;レーン7亜硫酸水素塩転換型アクチンプライマーエクソン3a〜3b;レーン8野生型アクチンプライマーエクソン3a〜4。
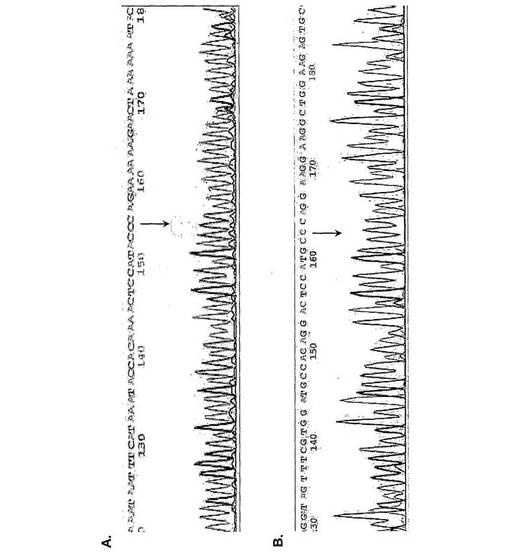
【図3】(図3A)亜硫酸水素塩処理転換型アクチンRNA(配列番号1)の配列分析を示す図である。 (図3B)野生型アクチンRNA(配列番号2)の配列分析を示す図である。
【図4】C型肝炎ウイルス(HCV)に関する直線性のゲルベースの読み出し値を示す図である。
【図5】リアルタイムPCR定量化の報告−C型肝炎ウイルス(HCV)RNAに関する直線性パネルを示す図である。
【図6】リアルタイムPCR定量化の報告−C型肝炎ウイルス(HCV)に関するダイナミックレンジを示す図である。
【発明を実施するための形態】
【0047】
定義
核酸
用語「核酸」は、天然に存在する核酸、DNAおよびRNAを包含する。用語「核酸類似体」は、天然に存在する核酸、DNAおよびRNAの誘導体、および天然に存在する核酸の合成類似体を包含する。合成類似体は1つまたは複数のヌクレオチド類似体を含む。用語ヌクレオチド類似体は、天然に存在するヌクレオチドとほぼ同様の、核酸骨格に取り込むことができ特異的な塩基対形成することができる(以下参照)全てのヌクレオチド類似体を含む。
【0048】
したがって用語「核酸」または「核酸類似体」は、複数のヌクレオチドおよび/またはヌクレオチド類似体および/またはインターカレーター擬ヌクレオチドから本質的になる任意の分子を指す。本発明に有用な核酸または核酸類似体は、異なる骨格のモノマー単位を有する幾つかの異なるヌクレオチドを含むことができる。
【0049】
核酸または核酸類似体の一本鎖は、実質的に相補的な一本鎖核酸および/または核酸類似体とハイブリダイズして、二本鎖核酸または核酸類似体を形成することができることが好ましい。このような二本鎖類似体は、二重らせんを形成することができることがより好ましい。二重らせんは水素結合によって形成されることが好ましく、二重らせんは、A型、B型、Z型およびそれらの中間体の二重らせんからなる群から選択される二重らせんであることがより好ましい。
【0050】
したがって、本発明に有用な核酸および核酸類似体には、それだけに限らないが、DNA、RNA、LNA、PNA、MNA、ANA、HNAおよびそれらの混合物およびそれらのハイブリッド、ならびにそれだけに限らないが、ホスホロチオエート、メチルホスホレート、ホスホラミデート、ホスホロジチエート、ホスホロセレノエート、ホスホトリエステルおよびホスホボラノエートなどのそれらのリン原子修飾体がある。さらに、それだけに限らないが、メチルイミノメチル、ホルムアセテート、チオホルムアセテートおよびアミド含有結合基などの非リン含有化合物をヌクレオチドとの結合に使用することができる。特に、核酸および核酸類似体は、1つまたは複数のインターカレーター擬ヌクレオチドを含むことができる。
【0051】
本文脈内では「混合物」は、異なる種類のヌクレオチドまたはヌクレオチド類似体を含む核酸および核酸類似体鎖を包含することを意味する。さらに、本文脈内では「ハイブリッド」は、1つまたは複数の種類の骨格を有するヌクレオチドまたはヌクレオチド類似体を含む1本の鎖、および異なる種類の骨格を有するヌクレオチドまたはヌクレオチド類似体を含む他の鎖を含む、核酸または核酸類似体を包含することを意味する。
【0052】
HNAによって、例えばVan Aetschot et al.、1995によって記載されたのと同様の核酸を意味する。MNAによって、Hossain et al、1998によって記載されたのと同様の核酸を意味する。ANAはAllert et al、1999によって記載された核酸を指す。LNAはWO99/14226(Exiqon)中に記載されたのと同様の任意のLNA分子であってよく、LNAはWO99/14226の要約書中で示された分子から選択されることが好ましい。LNAは、Singh et al、1998、Koshkin et al、1998またはObika et al、1997中に記載されたのと同様の核酸であることがより好ましい。PNAは例えばNielsen et al、1991によって記載されたのと同様のペプチド核酸を指す。
【0053】
用語ヌクレオチドは核酸または核酸類似体の構成単位を指し、かつ用語ヌクレオチドは、天然に存在するヌクレオチドおよびそれらの誘導体、ならびに天然に存在するヌクレオチドおよびそれらの誘導体とほぼ同じ機能を果たすことができるヌクレオチドを包含する。天然に存在するヌクレオチドは、4つの主な核酸塩基アデニン(A)、チミン(T)、グアニン(G)またはシトシン(C)の1つを含むデオキシリボヌクレオチド、および4つの核酸塩基アデニン(A)、ウラシル(U)、グアニン(G)またはシトシン(C)の1つを含むリボヌクレオチドを含む。
【0054】
ヌクレオチド類似体は、核酸骨格に取り込むことができ特異的な塩基対形成することができる、任意のヌクレオチド様分子であってよい。
【0055】
天然に存在しないヌクレオチドには、DNA、RNA、PNA、HNA、MNA、ANA、LNA、CNA、CeNA、TNA、(2’−NH)−TNA、(3’−NH)−TNA、α−L−リボ−LNA、α−L−キシロ−LNA、β−D−キシロ−LNA、α−D−リボ−LNA、[3.2.1]−LNA、バイシクロ−DNA、6−アミノ−バイシクロ−DNA、5−エピ−バイシクロ−DNA、α−バイシクロ−DNA、トリシクロ−DNA、バイシクロ[4.3.0]−DNA、バイシクロ[3.2.1]−DNA、バイシクロ[4.3.0]アミド−DNA、β−D−リボピラノシル−NA、α−L−リキシオピラノシル−NA、2’−R−RNA、α−L−RNAまたはα−D−RNA、β−D−RNA内に含まれるヌクレオチドがあるが、これらだけには限られない。
【0056】
ヌクレオチドおよびヌクレオチド類似体の機能は、相補的ヌクレオチドの核酸塩基の水素結合を介して相補的ヌクレオチドと特異的に相互作用することができること、および核酸または核酸類似体に取り込まれることができることである。天然に存在するヌクレオチド、および幾つかのヌクレオチド類似体は、酵素によって、例えばRNAまたはDNAポリメラーゼによって、核酸または核酸類似体に取り込まれることができる。しかしながら、ヌクレオチドまたはヌクレオチド類似体は、化学的に核酸または核酸類似体に取り込まれることもできる。
【0057】
さらに核酸または核酸類似体は、2つの小さな核酸または核酸類似体を互いに結合させることによって調製することができ、例えばこれはリガーゼによって酵素的に行うことができ、またはそれは化学的に行うことができる。
【0058】
ヌクレオチドまたはヌクレオチド類似体は、骨格モノマー単位および核酸塩基を含む。核酸塩基は、天然に存在する核酸塩基、またはほぼ同じ機能を果たすことができるその誘導体もしくはその類似体であってよい。核酸塩基の機能は、水素結合を介して1つまたは複数の他の核酸塩基と特異的に結合することができることである。したがって、それは1つまたは少数の他の核酸塩基のみと安定した水素結合を結合することができるが、それは通常それ自体に含まれる大部分の他の核酸塩基とは安定した水素結合を結合することができないことは、核酸塩基の1つの重要な特徴である。1つの核酸塩基と他の核酸塩基の特異的相互作用は、一般に「塩基対形成」と呼ばれる。
【0059】
塩基対形成は、所定のヌクレオチドと相補的ヌクレオチドの間の特異的ハイブリダイゼーションをもたらす。相補的ヌクレオチドは、塩基対形成することができる核酸塩基を含むヌクレオチドである。
【0060】
一般的な天然に存在する核酸塩基の中で、アデニン(A)はチミン(T)またはウラシル(U)と塩基対形成し、かつグアニン(G)はシトシン(C)と塩基対形成する。したがって、Aを含むヌクレオチドはTまたはUのいずれかを含むヌクレオチドと相補的であり、かつGを含むヌクレオチドはCを含むヌクレオチドと相補的である。
【0061】
追加の分子体を含むようにヌクレオチドをさらに誘導体化することができる。ヌクレオチドは、核酸塩基または骨格モノマー単位で誘導体化することができる。塩基上の好ましい誘導体化部位には、アデニンの8−位置、ウラシルの5−位置、シトシンの5−または6−位置、およびグアニンの7−位置がある。複素環修飾は3つの構造クラス、強化型塩基スタッキング(enhanced base stacking)、追加の水素結合、およびこれらのクラスの組合せに分けることができる。平面系のπ−電子雲を拡大することによって塩基スタッキングを強化する修飾は、ピリミジンの5−位置および7−デアザ−プリンの7−位置における共役した親油性修飾によって表される。ピリミジン修飾の5−位置における置換にはプロピン、ヘキシン、チアゾールおよび単にメチル基があり、かつ7−デアザ−プリンの7−位置における置換にはヨード、プロピニル、およびシアノ基がある。プロピンから5員複素環および三環系縮合系までシトシンの5−位置を修飾することもでき、これらは4−および5−位置(シトシンクランプ)から生じる。第二の型の複素環修飾は2−アミノ−アデニンによって表され、この場合、追加のアミノ基が、G−C塩基対における3個の水素結合と類似した他の水素結合をA−T塩基対において与える。組合せの影響を与える複素環修飾は、ヘテロ二本鎖のエトキシアミノ官能基を有する2−アミノ−7−デアザ−7−修飾アデニンおよび三環系シトシン類似体によって表される。さらに、N2−修飾2−アミノアデニン修飾オリゴヌクレオチドは特に一般的な修飾体である。リボースまたはデオキシリボース部分上の誘導体化の好ましい部位は、非結合炭素位置C−2’およびC−4’の修飾、結合炭素C−1’、C−3’およびC−5’の修飾、糖の酸素、O−4’の置換、(立体配座が固定された)無水糖の修飾、(立体配座が固定された)環糖の修飾、リボフラノシル環の大きさの変化、結合部位−糖と糖、(C−3’とC−5’/C−2’とC−5’)、ヘテロ原子環−修飾糖、および前述の修飾の組合せである。しかしながら、核酸または核酸類似体の全体的な塩基対形成の特異性が妨害されない限り、他の部位を誘導体化することができる。最後に、骨格モノマー単位がリン酸基を含むとき、幾つかの骨格モノマー単位のリン酸を誘導体化することができる。
【0062】
本明細書で使用するオリゴヌクレオチドまたはオリゴヌクレオチド類似体は、ヌクレオチドおよび/またはヌクレオチド類似体および/またはインターカレーター擬ヌクレオチドの配列から本質的になる分子である。オリゴヌクレオチドまたはオリゴヌクレオチド類似体は、5〜100個の個別のヌクレオチドを含むことが好ましい。オリゴヌクレオチドまたはオリゴヌクレオチド類似体は、DNA、RNA、LNA、2’−O−メチルRNA、PNA、ANA、HNAおよびこれらの混合物、ならびに任意の他のヌクレオチドおよび/またはヌクレオチド類似体および/またはインターカレーター擬ヌクレオチドを含むことができる。
【0063】
RNA
本明細書で使用するRNAは、細胞などの任意の供給源由来のメッセンジャーRNA(mRNA)未熟mRNA、トランスファーRNA(tRNA)、リボソームRNA(rRNA)およびミクロRNA(miRNA)、ウイルスまたは他の微生物由来のゲノムRNA、DNAから転写されたRNA、対応するDNAのRNAコピーなどを含む。
【0064】
対応する核酸
核酸、核酸類似体、オリゴヌクレオチドまたはオリゴヌクレオチド類似体は、それらがハイブリダイズすることができるとき対応すると考えられる。対応する核酸、核酸類似体、オリゴヌクレオチドまたはオリゴヌクレオチド類似体は、低ストリンジェンシー条件下でハイブリダイズすることができることが好ましく、対応する核酸、核酸類似体、オリゴヌクレオチドまたはオリゴヌクレオチド類似体は、中ストリンジェンシー条件下でハイブリダイズすることができることがより好ましく、対応する核酸、核酸類似体、オリゴヌクレオチドまたはオリゴヌクレオチド類似体は、高ストリンジェンシー条件下でハイブリダイズすることができることがより好ましい。
【0065】
本明細書で使用する高ストリンジェンシー条件は、例えばSouthern E.M.、1975、J.Mol.Biol.98:503−517によって記載されたサザンブロッティングおよびのハイブリダイゼーションに関して通常適用されるストリンジェンシーを示すものとする。このような目的のために、プレハイブリダイゼーションおよびハイブリダイゼーションの工程を含めることは通常業務である。このような工程は通常、Sambrook et al.、1989によって「Molecular Cloning/A Laboratory Manual」、Cold Spring Harbor)中に記載されたように、6×SSPE、5%のDenhardt’s、0.5%のSDS、50%のホルムアミド、100μg/mlの変性したサケ精子DNAを含む溶液を使用し(42℃で18時間インキュベーション)、次に2×SSCおよび0.5%のSDSで洗浄し(室温および37℃)、0.1×SSCおよび0.5%のSDSで洗浄して(30分間68℃でインキュベーション)実施する。
【0066】
本明細書で使用する中ストリンジェンシー条件は、pH7.0において1mMのEDTA,10mMのNa2HPO4H2O、140mMのNaClを含むバッファー中でのハイブリダイゼーションを示すものとする。約1.5μMのそれぞれの核酸または核酸類似体鎖を与えることが好ましい。あるいは中ストリンジェンシーは、50mMのKCl、10mMのTRIS−HCl(pH9,0)、0.1%のTritonX−100、2mMのMgCl2を含むバッファー中でのハイブリダイゼーションを示すことができる。
【0067】
低ストリンジェンシー条件は、pH7.0において1MのNaCl、10mMのNa3PO4で構成されるバッファー中でのハイブリダイゼーションを示す。
【0068】
あるいは、対応する核酸、核酸類似体、オリゴヌクレオチドまたはオリゴヌクレオチド、核酸類似体、オリゴヌクレオチドまたはオリゴヌクレオチドは、所与の配列で互いに実質的に相補的、70%を超えて相補的など、例えば75%を超えて相補的、80%を超えて相補的など、例えば85%を超えて相補的、90%を超えて相補的など、例えば92%を超えて相補的、94%を超えて相補的など、例えば95%を超えて相補的、96%を超えて相補的など、例えば97%を超えて相補的である。
【0069】
所与の配列は少なくとも10ヌクレオチド長、少なくとも15ヌクレオチドなど、例えば少なくとも20ヌクレオチド、少なくとも25ヌクレオチドなど、例えば少なくとも30ヌクレオチド、10ヌクレオチドと500ヌクレオチドの間など、例えば10ヌクレオチド長と100ヌクレオチド長の間、10ヌクレオチド長と50ヌクレオチド長の間であることが好ましい。対応するオリゴヌクレオチドまたはオリゴヌクレオチド類似体は、それらの全長で実質的に相補的であることがより好ましい。
【0070】
クロスハイブリダイゼーション
クロスハイブリダイゼーションとの用語は、少なくとも2つの核酸または核酸類似体間の意図しないハイブリダイゼーションを包含する。したがって、クロスハイブリダイゼーションとの用語を使用して、例えば核酸プローブまたは核酸類似体プローブ配列と、その意図する標的配列以外の他の核酸配列または核酸類似体配列のハイブリダイゼーションを記載することができる。
【0071】
クロスハイブリダイゼーションはプローブと1つまたは複数の対応する非標的配列の間で起こることが多いが、しかしながらこれらは、プローブおよびその対応する標的配列より低度の相補性を有する。この望ましくない影響は、標的に対して大幅に過剰なプローブおよび/または急速なアニーリング動態が原因であり得る。クロスハイブリダイゼーションは、数個の核酸塩基対間、例えばPCR反応中のプライマー間の水素結合によっても起こり、プライマー二量体形成および/または非特異的PCR産物の形成をもたらす。
【0072】
同じ型のヌクレオチド類似体に対して高い親和性を有する1つまたは複数のヌクレオチド類似体を含む核酸は、塩基対形成に基づいて二量体またはさらに高次の複合体を形成する傾向がある。それだけに限らないが、LNA、2’−O−メチルRNAおよびPNAなどのヌクレオチド類似体を含むプローブは、同じ型の骨格モノマー単位を含む他のオリゴヌクレオチド類似体とのハイブリダイズに対する高い親和性を一般に有する。したがって、個々のプローブ分子は低度の相補性のみを有するが、それらはハイブリダイズする傾向がある。
【0073】
自己ハイブリダイゼーション(self-hybridisation)
自己ハイブリダイゼーションとの用語は、核酸または核酸類似体分子が自身で折り畳まることにより自身にアニーリングし、例えばヘアピン構造のような二次構造を生成するプロセスを包含する。大部分の適用例において、自己ハイブリダイゼーションを避けることは重要である。二次構造の生成は、望ましい核酸標的配列とのハイブリダイゼーションを阻害し得る。例えば核酸または核酸類似体をPCR反応におけるプライマーとして、またはエクソヌクレアーゼアッセイ用のフルオロフォア/消光標識プローブとして使用するとき、大部分のアッセイにおいてこれは望ましくない。両方のアッセイにおいて、自己ハイブリダイゼーションは標的核酸配列とのハイブリダイゼーションを阻害し、さらにエクソヌクレアーゼアッセイにおけるフルオロフォアの消光の程度が低下する。
【0074】
同じ型のヌクレオチド類似体に対して高い親和性を有する1つまたは複数のヌクレオチド類似体を含む核酸は、自己ハイブリダイズする傾向がある。それだけに限らないが、LNA、2’−O−メチルRNAおよびPNAなどのヌクレオチド類似体を含むプローブは、自己ハイブリダイズに対する高い親和性を一般に有する。したがって、個々のプローブ分子は低度の相補性のみを有するが、それらは自己ハイブリダイズする傾向がある。
【0075】
融解温度
核酸の融解は、二本鎖核酸分子の2本の鎖の分離を指す。融解温度(Tm)は、50%のらせん(ハイブリダイズ)対コイル(非ハイブリダイズ)形が存在する摂氏温度を示す。
【0076】
高い融解温度は安定した複合体、したがって個々の鎖の間の高い親和性を示す。同様に、低い融解温度は個々の鎖の間の比較的低い親和性を示す。したがって、通常2本の鎖の間の強い水素結合は高い融解温度をもたらす。
【0077】
さらに、二本鎖核酸の核酸塩基間へのインターカレーターの挿入も二本鎖核酸を安定化させ、したがって高い融解温度をもたらす可能性がある。
【0078】
さらに、融解温度は周囲の物理的/化学的状態に依存する。例えば、融解温度は塩濃度およびpHに依存する。
【0079】
融解温度は幾つかのアッセイにより決定することができ、例えばUVスペクトルを使用してハイブリダイゼーションの形成および分解(融解)を決定することによって、融解温度を決定することができる。
【0080】
INA/IPNの定義
挿入核酸(INA)は特有なクラスのDNA結合分子である。INAは、ヌクレオチドおよび/またはヌクレオチド類似体およびインターカレーター擬ヌクレオチド(IPN)モノマーからなる。INAは、内部に位置するIPNに関して10℃までおよび末端位置IPNに関して11℃まで安定状態である相補的DNAに対して、非常に高い親和性を有する。正確に設計した場合、INA自体が、相補的RNAよりDNAとのハイブリダイズを好む選択的分子となり得る。IPNを分子内部に配置した場合、INAはオリゴヌクレオチドプライマーの約25分の1の効率でRNAに結合することが示されている。一方、従来のオリゴヌクレオチド、オリゴヌクレオチド類似体およびPNAは、RNAとDNAの両方に対して等しい親和性を有する。したがってINAは、第一の真に選択的なDNA結合剤である。さらにINAは、他の天然DNA分子より相補的DNAに対して高い特異性および親和性を有する。
【0081】
さらに、IPNはATに富む環境においてDNAを最も安定化させ、これによってIPNはエピゲノミクス研究の分野で特に有用となる。IPNは、典型的には隆起または末端挿入物としてINA分子内に置く。IPNは本質的に、核酸二本鎖中の核酸塩基と共にスタッキングすることができる平面状(ヘテロ)多環芳香族化合物である。
【0082】
INA分子は、エクソヌクレアーゼによる攻撃に耐性があることも示されている。これによって、これらの分子は、phi29などの酵素を使用する増幅用のプライマーとして特に有用となる。phi29は固有のエクソヌクレアーゼ活性を有するので、増幅用の鋳型として使用するプライマーは、特にそれらの3’末端において修飾して酵素による分解を妨げなければならない。しかしながらINA分子は、さらなる修飾なしで加えることができる。
【0083】
INAは従来のPCR増幅反応において使用することができ、従来型プライマーとして挙動する。しかしながら、INAは、DNAまたはRNA鋳型に対して高い特異性を有し、鋳型が制限的であり反応の感度が重要である状況における使用にINAは理想的となる。INAはATに富む環境においてDNAを最も安定化させ、これによってINAは亜硫酸水素塩処理DNA配列の増幅に特に有用となる。これは、亜硫酸水素塩による転換後、全てのシトシン残基はウラシルに、次にPCRまたは他の増幅後チミンに転換される事実に原因がある。したがって亜硫酸水素塩処理DNAは非常にTに富む。INA中のIPN分子の数の増大は、INA/DNA二本鎖の増大した安定性をもたらす。INA中のIPNが増大するほど、DNA/INA二本鎖の融解温度は高くなる。
【0084】
本出願人は、オリゴヌクレオチドまたはオリゴヌクレオチド類似体に取り込まれた際に、挿入核酸(INA)を形成し(WO03/051901、WO03/052132、WO03/052133およびWO03/052134)、オリゴヌクレオチドのサプリメントまたは置換体として新規かつ有用な性質を有するクラスのインターカレーター擬ヌクレオチドを以前に開発している。
【0085】
インターカレーター擬ヌクレオチドは、1−(4,4’−ジメトキシトリフェニルメチルオキシ)−3−ピレンメチルオキシ−2−プロパノールの複数のホスホラミダイトから選択されることが好ましい。インターカレーター擬ヌクレオチドは、(S)−1−(4,4’−ジメトキシトリフェニルメチルオキシ)−3−ピレンメチルオキシ−2−プロパノールのホスホラミダイトまたは(R)−1−(4,4’−ジメトキシトリフェニルメチルオキシ)−3−ピレンメチルオキシ−2−プロパノールのホスホラミダイトから選択されることが好ましい。
【0086】
オリゴヌクレオチドまたはオリゴヌクレオチド類似体は、DNA、RNA、固定核酸(LNA)、ペプチド核酸(PNA)、MNA、アルトリトール核酸(ANA)、ヘキシトール核酸(HNA)、挿入核酸(INA)、シクロヘキサニル核酸(CNA)、およびそれらの混合物およびそれらのハイブリッド、ならびにそれだけに限らないが、ホスホロチオエート、メチルホスホレート、ホスホラミデート、ホスホロジチエート、ホスホロセレノエート、ホスホトリエステルおよびホスホボラノエートなどのそれらのリン原子修飾体から選択することができる。天然に存在しないヌクレオチドには、DNA、RNA、PNA、INA、HNA、MNA、ANA、LNA、CNA、CeNA、TNA、(2’−NH)−TNA、(3’−NH)−TNA、α−L−リボ−LNA、α−L−キシロ−LNA、β−D−キシロ−LNA、α−D−リボ−LNA、[3.2.1]−LNA、バイシクロ−DNA、6−アミノ−バイシクロ−DNA、5−エピ−バイシクロ−DNA、α−バイシクロ−DNA、トリシクロ−DNA、バイシクロ[4.3.0]−DNA、バイシクロ[3.2.1]−DNA、バイシクロ[4.3.0]アミド−DNA、β−D−リボピラノシル−NA、α−L−リキシオピラノシル−NA、2’−R−RNA、α−L−RNAまたはα−D−RNA、β−D−RNA内に含まれるヌクレオチドがあるが、これらだけには限られない。さらに、それだけに限らないが、メチルイミノメチル、ホルムアセテート、チオホルムアセテートおよびアミド含有結合基などの非リン含有化合物をヌクレオチドとの結合に使用することができる。特に、核酸および核酸類似体は、1つまたは複数のインターカレーター擬ヌクレオチドを含むことができる。
【0087】
IPNがメチル化部位の特異的検出用にINA分子内に位置するとき、本発明者は、潜在的CpG部位間にIPNを位置させることは避けることが有用であることを見出している。これは、IPNを使用してCpG部位を隔てると、生成するINAの特異性が低下するという事実によるものである。
【0088】
ペプチド核酸(PNA)
ペプチド核酸は、配列特異性を有する核酸(DNAおよびRNA)とハイブリダイズすることができる天然に存在しないポリアミドである(米国特許第5,539,082号およびEgholm et al.、Nature(1993)365、566-568参照)。PNAはプローブベースのハイブリダイゼーションアッセイにおける核酸プローブに対する代替/代用としての候補であるが、これは、PNAが幾つかの望ましい性質を示すからである。PNAは、核酸とハイブリダイズして、対応する核酸/核酸複合体より熱力学的に安定したハイブリッドを形成するアキラルポリマーである。天然に存在しない分子であるので、それらはペプチドまたは核酸を分解することが知られている酵素の基質であることは知られていない。したがって、PNAは生物サンプル中で安定状態であり、かつ長期の保存寿命を有するはずである。イオン強度に非常に依存する核酸ハイブリダイゼーションとは異なり、PNAと核酸のハイブリダイゼーションはイオン強度にほとんど依存せず、核酸と核酸のハイブリダイゼーションを強く嫌う条件下において低いイオン強度を好む。PNA複合体の安定性および立体配座に対するイオン強度の影響は、広範囲で調べられている。配列識別は、DNA認識DNAよりPNA認識DNAまたはRNAに有効である。しかしながら、ハイブリダイゼーションアッセイにおける、DNAプローブと比較した1塩基の変化、indel、またはPNAプローブを用いた多型識別の利点は、幾分配列依存的であるようである。追加の利点として、PNAは平行方向と逆平行方向の両方で核酸とハイブリダイズするが、逆平行方向が好ましい。
【0089】
PNAは、現在市販されている形式での標準的なペプチド合成手順の適合によって合成する(PNAモノマーおよびオリゴマーの調製の一般評論に関しては、Dueholm et al.、New J.Chem.(1997)、21、19-31またはHyrup et. al.、Bioorganic & Med.Chem.(1996)4、5-23を参照頂きたい)。標識および非標識PNAオリゴマーは購入することができ(PerSeptive Biosystems Promotional Literature:BioConcepts、Publication No.NL612、Practical PNA、Review and Practical PNA、Vol.1、Iss.2を参照)、または市販の製品を使用して調製することができる。
【0090】
実際、PNAプローブと標準的な核酸プローブの間に多くの差異が存在する。これらの差異は、生物学的、構造的、および物理化学的差異に都合良く分けることができる。上記および以下で論じるように、これらの生物学的、構造的、および物理化学的差異は、核酸が典型的に利用されている適用例においてPNAプローブの使用を試みるとき、予期せぬ結果をもたらし得る。異なる組成物のこの非同等性は、化学分野においてしばしば観察される。
【0091】
生物学的差異に関して、核酸は、遺伝的伝達および発現の作用物質として、生存種の寿命において中心的役割を果たす生物学的物質である。それらのin vivoでの性質はほぼ十分に理解されている。しかしながらPNAは、近年開発された完全に人工的な分子であり、化学者の発想で考え出され、合成有機化学を使用して作製される。それは知られている生物学的機能は有していない。
【0092】
構造上、PNAは核酸とも劇的に異なる。両者は一般的な核酸塩基(A、C、G、T、およびU)を利用することができるが、これらの分子の骨格は構造上多様である。RNAおよびDNAの骨格は、反復リン酸ジエステルリボースおよび2−デオキシリボース単位からなる。対照的に、PNAの骨格はN−(2−アミノエチル)グリシン単位からなる。さらにPNA中では、核酸塩基は他のメチレンカルボニル単位によって骨格と結合している。
【0093】
その名称にもかかわらず、PNAは酸ではなく、かつDNAおよびRNA中に存在する基などの帯電した酸性基は含まない。PNAは形式電荷を欠くので、PNAは一般にそれらの同等な核酸分子より疎水性が高い。PNAの疎水特性は、核酸では観察されない非特異的(疎水性/疎水性相互作用)相互作用の可能性を与える。さらに、それがその同等な立体配座がRNA/DNA領域に存在しない構造立体配座をとる能力を有するという条件で、PNAはアキラルである。
【0094】
PNAとDNAまたはRNAの間の物理的/化学的差異も相当にある。PNAは、同じ標的配列と結合する核酸プローブより速く、その相補的核酸と結合する。この挙動は、少なくとも一部分は、PNAはその骨格に電荷を欠くという事実が原因であると考えられる。さらに、近年の刊行物は、PNA中への正に帯電した基の取り込みは、ハイブリダイゼーションの動態を改善することを実証する。それは骨格に電荷を欠くので、PNA/核酸複合体の安定性は、類似のDNA/DNAまたはRNA/DNA複合体の安定性より高い。特定の状況において、PNAは非常に安定した三重らせん複合体を形成するか、または「鎖置換」と呼ばれるプロセスによって小さなループを形成し得る。同等な鎖置換プロセスまたは構造は、DNA/RNA分野では知られていない。
【0095】
要約すると、PNAは配列特異性を有する核酸とハイブリダイズするので、PNAはプローブベースのアッセイを開発するのに有用な候補である。重要なことに、PNAプローブは核酸プローブと同等なプローブではない。それにもかかわらず、最もストリンジェントな条件下でさえ、正確な標的配列と密接に関連した配列(例えば、単一点突然変異(一塩基対のミスマッチ)を有する非標的配列)の両方が、標識核酸または標識PNAプローブとの検出可能な相互作用をしばしば示し得る。密接に関連した非標的配列との任意のハイブリダイゼーションが、望ましくないバックグラウンドシグナルの発生をもたらし得る。配列は非常に密接に関連しているので、複数の点突然変異は、プローブベースのアッセイを使用して検出するのが最も難しい全核酸修飾の一部である。鎌状赤血球貧血および嚢胞性線維症などの多数の疾患は、ゲノム核酸の単一点突然変異によって時折引き起こされる。したがって、プローブベースのアッセイの特異性、感度および信頼性を改善し得る任意の方法、キットまたは組成物が、DNA含有サンプルの検出、分析および定量化において有用であろう。
【0096】
亜硫酸水素ナトリウム
亜硫酸水素ナトリウムで核酸を処理するための方法は、Frommer et al 1992、Proc Natl Acad Sci89:1827-1831;Grigg and Clark 1994 BioAssays 16:431-436;Shapiro et al 1970、J Amer Chem Soc 92:422-423;Wataya and Hayatsu 1972、Biochemistry 11:3583-3588を含む幾つかの参照文献中で見ることができる。
【0097】
核酸の亜硫酸水素塩処理の成功の改善または向上のための複数の方法は、本出願人によっても開発されている。
【0098】
核酸の有効な亜硫酸水素塩処理のための例示的なプロトコルを以下に述べる。このプロトコルによって、実質的に全てのDNAが処理された状態となる。この方法は、本明細書ではヒト遺伝子シグネチャー(HGS)方法とも呼ぶ。サンプルまたは試薬の体積または量は変えることができることは理解されよう。
【0099】
亜硫酸水素塩処理に好ましい方法は、US 10/428310またはPCT/AU2004/000549中で見ることができる。
【0100】
そう望む場合適切な制限酵素で予め消化することができる2μgのDNAに、2μl(1/10体積)の3MのNaOH(6g、50ml水中、新たに作製)を、20μlの最終体積で加えた。この工程は二本鎖DNA分子を一本鎖形に変性させるが、これは、亜硫酸水素塩試薬が一本鎖分子と優先的に反応するからである。混合物は37℃で15分間インキュベートした。室温を超える温度でのインキュベーションを使用して、変性の効率を改善することができる。
【0101】
インキュベーション後、208μlの2Mメタ重亜硫酸ナトリウム(7.6g、20ml水および416ml10NNaOH中;BDH AnalaR#10356.4D;新たに作製)および12μlの10mMキノール(0.055g、50ml水中、BDH AnaIR #103122E;新たに作製)を連続して加えた。キノールは還元剤であり、試薬の酸化の還元を手助けする。他の還元剤、例えばジチオスレイトール(DTT)、メルカプトエタノール、キノン(ヒドロキノン)、または他の適切な還元剤を使用することもできる。サンプルには200μlのミネラルオイルを重層した。ミネラルオイルの重層は試薬の蒸発および酸化を防ぐが、必須ではない。次いでサンプルを55℃で一晩インキュベートした。あるいはサンプルは、サーマルサイクラーにおいて以下のサイクルにかけることが可能である:約4時間または一晩以下のようにインキュベートする:工程1、55℃/2時間サイクル、PCR機器中;工程2、95℃/2分間。工程1は約37℃〜約90℃の任意の温度で実施することができ、かつ5分〜8時間まで長さを変えることができる。工程2は約70℃〜約99℃の任意の温度で実施することができ、かつ約1秒〜60分、またはそれより長くまで長さを変えることができる。
【0102】
メタ重亜硫酸ナトリウムでの処理後、オイルを除去し、DNA濃度が低かった場合、1μlのtRNA(20mg/ml)または2μlのグリコーゲンを加えた。これらの添加剤は任意選択であり、特にDNAが低い濃度で存在するとき、これらを使用して標的DNAとの共沈によって得られるDNAの収率を改善することができる。核酸のより有効な沈殿のための担体としての添加剤の使用は、核酸の量が<0.5μgであるとき一般に望ましい。
【0103】
イソプロパノール浄化処理を以下のように実施した:800μlの水をサンプルに加え、混合し、次いで1mlのイソプロパノールを加えた。水またはバッファーは、塩が対象の標的核酸と共に沈殿しないレベルまで、反応容器中の亜硫酸水素塩の濃度を低下させる。本明細書で開示する所望の範囲以下に塩濃度を希釈する限り、希釈は一般に約1/4〜1/1000である。
【0104】
サンプルを再度混合し、少なくとも5分間4℃に放置した。サンプルは微量遠心管中で10〜15分間回転させ、ペレットは70%ETOHで2回洗浄し、毎回ボルテックスした。この洗浄処理によって、核酸と共に沈殿した任意の残留塩を除去する。
【0105】
ペレットは放置乾燥させ、次いで50μlなどの適切な体積のT/E(10mMトリス/0.1mMEDTA)pH7.0〜12.5中に再懸濁した。pH10.5でのバッファーは特に有効であることが分かっている。核酸を懸濁することが必要であったとき、サンプルは37℃〜95℃で1分間〜96時間インキュベートした。
【0106】
RNAの二次構造を実質的に除去する作用物質
本発明に適した作用物質は、亜硫酸水素塩、ヒドロキシルアミン、酢酸塩またはクエン酸塩を含む。WO2005054502中に記載されたように、亜硫酸水素塩試薬が好ましく、亜硫酸水素ナトリウム、メタ重亜硫酸ナトリウム、および亜硫酸水素グアニジウムを含む。この関連で、グアニジウムイオンおよび亜硫酸イオンを含む溶液の調製および後のRNA処理に亜硫酸水素グアニジウムを使用する、RNA処理を実施することができる。
【0107】
細胞系
【0108】
【表1】
【0109】
細胞からのRNA抽出
I.培地の除去後、1mlのトリゾールを細胞(90%融合)に直接加えた。
II.サンプルをよく混合し、室温で5分間放置して核タンパク質複合体を解離させた。
III.0.5mlをクリーンなRNaseを含まない1.5mlの遠心管に除去した。
IV.次いでサンプルを12,000×gで10分間4℃において回転させて、高分子量DNAおよび他の混入物を除去した。
V.上清をクリーンチューブに除去し、100μlの100%クロロホルムを加え、サンプルは15秒間手作業によって激しく混合し、次いで2〜3分間室温でインキュベートした。
VI.次いでサンプルを12,000×gで10分間4℃において回転させて相を分離した。
VII.上部水相をクリーンチューブに除去し、ピペットの先端が界面から離れた状態に保たれることを確実にし、1μlの20mg/mlのグリコーゲンを加え、サンプルをボルテックスした。
VIII.等体積の100%イソプロパノール(0.25ml)を加え、チューブをボルテックスし、次いで室温で10分間放置した。
IX.次いでサンプルを12,000×gで10分間4℃において回転させてRNAをペレット状にした。
X.上清を除去し、ペレットを0.75mlの80%エタノールで洗浄して、cDNA合成反応の阻害剤を除去し、軽くボルテックスし、次いで7,500×gで5分間4℃において回転させてRNAをペレット状にした。
XI.工程Xをさらに1回繰り返した。
XII.次いでペレットを微量遠心管中で10秒間回転させ、残留エタノールを除去し、ペレットは即座に25μlのRNaseを含まない水中に再懸濁した。NB。ペレットが乾燥する場合、したがってRNAを再懸濁することは非常に困難であり、260/280の比は1.6未満であり得る。
XIII.次いでOD260/280/310を記録し、必要となるまでRNAは−70℃で保存した。
【0110】
RNAの調製
望ましい細胞または組織からの抽出後、20μlのヌクレアーゼを含まない水中にRNAサンプルを再懸濁した。
【0111】
サンプルは60〜100℃で2〜3分間加熱し二次構造を分解して、亜硫酸水素塩の反応において即座に使用した。
【0112】
亜硫酸水素塩処理
本発明によるRNAの亜硫酸水素塩処理の有効性を実証する例示的なプロトコルを以下に述べる。このプロトコルによって首尾よく、実質的に全てのRNAが処理された状態となった。本発明のこの方法は、本明細書ではヒト遺伝子シグネチャー(HGS)方法とも呼ぶ。サンプルまたは試薬の体積または量は変えることができることは理解されよう。
【0113】
2μgのRNAを合計20μlのRNaseを含まない水中に再懸濁する。次いでサンプルを65℃で2分間インキュベートして、二次構造を除去した。インキュベーション後、208μlの2Mメタ重亜硫酸ナトリウムpH5.0(7.6g、20ml水または10mMトリス/1mMEDTAと416ml10NNaOH中;BDH AnalaR#10356.4D;新たに作製)を連続して加えた。製造者の説明書に従い、RNaseOUT(invitrogenカタログ番号10777−019)などのRNase阻害剤もこの時点で加えることができる。サンプルには200μlのミネラルオイルを重層した。ミネラルオイルの重層は試薬の蒸発および酸化を防ぐが、必須ではない。次いでサンプルは55℃で一晩インキュベートした。このインキュベーションは約37℃〜約90℃の任意の温度で実施することができ、かつ5分〜16時間まで長さを変えることができる。
【0114】
メタ重亜硫酸ナトリウムでの処理後、オイルを除去し、特にDNA濃度が低かった場合、1μlのグリコーゲン(20mg/ml)を加えた。この添加剤は任意選択であり、特にRNAが低い濃度で存在するとき、これらを使用して標的RNAとの共沈によって得られるRNAの収率を改善することができる。核酸のより有効な沈殿のための担体としての添加剤の使用は、核酸の量が<0.5μgであるとき一般に望ましい。
【0115】
イソプロパノール浄化処理を以下のように実施した:800μlのRNaseを含まない水をサンプルに加え、混合し、次いで1mlのイソプロパノールを加えた。水またはバッファーは、塩が対象の標的核酸と共に沈殿しないレベルまで、反応容器中の亜硫酸水素塩の濃度を低下させる。本明細書で開示する所望の範囲以下に塩濃度を希釈する限り、希釈は一般に約1/4〜1/1000である。
【0116】
サンプルは再度混合し、60分まで可能であるが、最短で5分間4℃に放置した。サンプルは微量遠心管中で10〜15分間回転させ、ペレットは80%ETOHで2回洗浄した。この洗浄処理によって、核酸と共に沈殿した任意の残留塩を除去する。
【0117】
ペレットは軽く放置乾燥させて残留エタノールを除去した、ただしこれによって最終的なRNA収率が低下する可能性があるので、ペレットが完全には乾燥しないことを確実にし、次いで50μlなどの適切な体積のT/E(10mMトリス/0.1mMEDTA)pH7.0〜12.5中に再懸濁した。製造者の説明書に従い、RNaseOUT(invitrogenカタログ番号10777−019)などのRNase阻害剤もこの時点で加えることができる。pH10.5でのバッファーは特に有効であることが分かっている。核酸を懸濁することが必要であったとき、サンプルは37℃〜95℃で1分間〜96時間インキュベートした。
【0118】
cDNA合成
以下の試薬を、薄壁0.5mlのRNaseを含まないチューブ中でのそれぞれのcDNA合成反応用に調製した。
RNA(1μg) 3.5μl
ランダムヘキサマー(10μM) 1μl
脱イオン水 2.5μl
【0119】
含有物を混合し、微量遠心管中で軽く回転させた。
【0120】
サンプルは70℃で3分間インキュベートして、RNAを変性させた。
【0121】
RNAを変性させていた一方で、以下のマスター混合物を調製した:
Per rxn
5×第一鎖用バッファー 2μl
DTT(20mM) 1μl
50×dNTP混合物 1μl
合計体積 4μl
【0122】
チューブをPCR機器から除去し、氷上で2分間冷却し、次いで軽く回転させて含有物を回収した。
【0123】
次いでサンプルを42℃で2分間インキュベートした。
【0124】
0.5μlのPowerscript逆転写酵素を反応混合物(1.75μl)当たりに加え、マスター混合物はピペッティングによってよく混合した。
【0125】
4.5μlの完全なマスター混合物をそれぞれのサンプルおよび対照チューブに加え、次いでサンプルを42℃で60分間インキュベートし、次いでサンプルを氷上に移した。
【0126】
40μlの10mMのトリス/1mMのEDTA pH7.6をそれぞれのサンプルに加えた。
【0127】
チューブは72℃で7分間加熱し、次いで必要となるまで−70℃で保存した。
【0128】
PCR増幅
PCR増幅は1μlの亜硫酸水素塩処理RNAで実施し、PCR増幅は、Promega PCRマスター混合物、6ng/μlのそれぞれのプライマーを使用して、1μlの亜硫酸水素塩処理ゲノムDNAを含む25μlの反応混合物で実施した。
【0129】
第1ラウンドの増幅の1μlを第2ラウンドの増幅の反応混合物に移した。PCR産物のサンプルは、Clarke et al中に記載された条件下において、ThermoHybaid PX2サーマルサイクラーで増幅した。
【0130】
【表2】
【0131】
アガロースゲル(2%)は、50mlのアガロース当たり1滴の臭化エチジウムを含む1%TAE(CLP#5450)で調製した。5μlのPCR由来産物を1μlの5×アガロースローディングバッファーと混合し、水中水平型電気泳動用タンクを使用して×1TAE中で125mAにおいて電気泳動にかけた。マーカーは低100〜1000bp型であった。Kodak UVldoc EDAS290系を使用してUV照射下で、ゲルを目に見える状態にした。
【0132】
ビーズを使用する検出系
磁気ビーズのコーティング
磁気ビーズとの結合に使用するINAは、幾つかの方法で修飾することができる。この例では、EDCなどのヘテロ二官能性リンカーを使用するビーズとINAの共有結合用に5’または3’アミノ基のいずれかをINAは含んでいた。しかしながら、INAはビオチンなどの5’基で修飾することも可能であり、次いでこれはアビジンまたはステプトアビジン(Steptavidin)基で修飾された磁気ビーズと受動的に結合し得る。
【0133】
10μlのカルボキシレート修飾Magnabind(商標)ビーズ(Pierce)または100μlのDynabeads(商標)ストレプトアビジン(Dynal)をクリーンな1.5mlのチューブに移し、90μlのPBS溶液を加えた。
【0134】
ビーズは混合し次いで磁化し、上清は捨てた。ビーズは洗浄1回当たり100μlのPBSで2回洗浄し、最後に90μlの50mMMESバッファーpH4.5、または製造者の仕様書によって決定した他のバッファー中に再懸濁した。
【0135】
1μlの250μMINA(オリゴハイブリダイゼーションの実験により決定した選択INAの特異的活性に依存する濃度)をサンプルに加え、チューブをボルテックスし室温で10〜20分間放置した。
【0136】
10μlの新たに調製した10mg/mlEDC溶液(Pierce/Sigma)を次いで加え、サンプルをボルテックスし、室温または4℃のいずれかにおいて60分までインキュベートする。
【0137】
次いでサンプルを磁化し上清を捨て、10分間0.25MのNaOHまたは0.5MのトリスpH8.0のいずれかを100μl加えることによってビーズを阻害することができる。
【0138】
次いでビーズをPBS溶液で2回洗浄し、最後に100μlのPBS溶液中に再懸濁した。
【0139】
磁気ビーズを使用するハイブリダイゼーション
10μlのINAコーティングMagnabind(商標)ビーズをクリーンチューブに移し、40μlの非希釈または蒸留水に1:1希釈のExpressHyb(商標)バッファー(Clontech)、または非希釈または蒸留水に1:1/1:2または1:4希釈のUltrahyb(商標)バッファー(Ambion)のいずれか、または室内用ハイブリダイゼーションバッファーを加えた。バッファーは、知られている濃度のカチオン性/アニオン性または両性洗浄剤のいずれか、またはヘパリンおよびポリアミノ酸などの他の添加剤を含むこともできる。
【0140】
次いでサンプルRNA1〜5μlを前述の溶液に加え、チューブをボルテックスし、20〜60分間選択したINA/RNAハイブリッドの融解温度に応じて、55℃または他の温度で次いでインキュベートした。
【0141】
サンプルを磁化し、上清は捨て、ビーズは洗浄1回当たり5分間の初期工程から、ハイブリダイゼーション温度において0.1×SSC/0.1%SDSで2回洗浄し、洗浄の間にサンプルを磁化した。
【0142】
放射標識した検出用球体の調製
INAまたはオリゴ分子は、アミン基、チオール基またはビオチンなどの分子で3’または5’のいずれかを標識することができる。
【0143】
標識分子は、第1標識に対して分子の逆末端に取り込まれたP32またはI125などの第2標識を有することもできる。
【0144】
この二重標識検出用分子は現在、EDCなどのヘテロ二官能性リンカーを使用して、知られている大きさのカルボキシレートまたは修飾ラテックスビーズと共有結合させることが可能である。
【0145】
次いで非結合分子を洗浄によって除去することができ、多数の特異的検出用/シグナル増幅分子でコーティングされたビーズを切り離すことができる。
【0146】
次いでこれらのビーズを対象の核酸サンプルとハイブリダイズさせて、シグナル増幅をもたらすことが可能である。
【0147】
蛍光標識した検出用球体の調製
INAまたはオリゴ分子は、アミン基、チオール基またはビオチンなどの分子で3’または5’のいずれかを標識することができる。
【0148】
標識分子は、第1標識に対して分子の逆末端に取り込まれたCy−3、Cy−5、FAM、HEX、TET、TAMRAまたは任意の他の適切な蛍光分子などの第2標識を有することもできる。
【0149】
この二重標識検出用分子は現在、EDCなどのヘテロ二官能性リンカーを使用して、知られている大きさのカルボキシレートまたは修飾ラテックスビーズと共有結合させることが可能である。
【0150】
次いで非結合分子を洗浄によって除去することができ、多数の特異的検出用/シグナル増幅分子でコーティングされたビーズを切り離すことができる。
【0151】
次いでこれらのビーズを対象のRNAサンプルとハイブリダイズさせて、シグナル増幅をもたらすことが可能である。
【0152】
酵素標識した検出用球体の調製
INAまたはオリゴ分子は、アミン基またはチオール基などの分子で3’または5’のいずれかを標識することができる。
【0153】
標識分子は、第1標識に対して分子の逆末端に、ヘテロ二官能性リンカーによって結合したビオチンまたはホースラディッシュペルオキシダーゼまたはアルカリホスファターゼなどの他の分子などの第2標識を有することもできる。
【0154】
この二重標識検出用分子は現在、EDCなどのヘテロ二官能性リンカーを使用して、知られている大きさのカルボキシレートまたは修飾ラテックスビーズと共有結合させることが可能である。
【0155】
次いで非結合分子を洗浄によって除去することができ、多数の特異的検出用/シグナル増幅分子でコーティングされたビーズを切り離すことができる。
【0156】
次いでこれらのビーズを対象の核酸サンプルとハイブリダイズさせて、シグナル増幅をもたらすことが可能である。
【0157】
次いで、シグナル増幅は、ストレプトアビジンなどの分子の結合または発色基質が関与する酵素反応によって実施することができる。
【0158】
INAオリゴマーの組合せ
初期ハイブリダイゼーション事象では、対象のRNAと相補的なINAでコーティングされた磁気ビーズを使用することが好ましい。
【0159】
必要とされる場合、第二のハイブリダイゼーション事象は、前述の検出法のいずれかを含むことができる。
【0160】
このハイブリダイゼーション反応は、対象の核酸と相補的な第二のINA、または対象のRNAと相補的なオリゴもしくは修飾オリゴのいずれかを用いて行うことができる。
【0161】
デンドリマーおよびアプタマー
デンドリマーは、特異的分子で標識した多層を生成することができるように調節した方法で化学的に合成することができる、枝分かれした樹状の分子である。デンドリマーは中心から末端に、またはその逆に段階的に合成した。
【0162】
デンドリマーの構造およびその生成に影響を与える最も重要なパラメーターの1つは、それぞれの工程で生成する枝の数であり、これが所望の分子を構築するのに必要とされる反復工程の数を決定する。
【0163】
I125またはP32などの放射標識、Cy−3、Cy−5、FAM、HEX、TET、TAMRAまたは任意の他の適切な蛍光分子などの蛍光標識を含むデンドリマーを合成して、シグナル増幅を促進することができる。
【0164】
あるいは、修飾INAまたはDNA分子と結合させるために使用することができるカルボキシレート基または任意の他の反応基を含む、デンドリマーを合成することができる。
【0165】
アレイを使用する検出系
処理したRNAを任意の適切な支持体につけて、対象の遺伝子または発現単位の活性に関してスクリーニングすることができるマイクロアレイなどのアレイを形成することができる。当業者は、適切なアレイを作製するのに適した技法に精通しているはずである。
【実施例】
【0166】
図1A、図1Bおよび図1Cは、本発明(亜硫酸水素塩処理RNA)と典型的なマイクロアレイベースのアッセイを使用する従来技術の比較を示す。図1Aは、典型的なマイクロアレイベースのアッセイの説明を示すが、この場合暗色点は特定のRNA集団中でアップレギュレーションされる遺伝子を示し、かつ淡色点は同じ集団中でダウンレギュレーションされる遺伝子を示す。暗色および淡色の矢印は、従来系中の検出分子がそれらの標的と結合するのを妨げる二次構造のため、従来の方法ではなく亜硫酸水素塩処理RNAにおいて、それぞれアップレギュレーションおよびダウンレギュレーションされるとして検出される遺伝子を示す。
【0167】
図1Bは、図1Aと同様のマイクロアレイベースのアッセイを示すが、暗色および淡色の矢印は、発現分析前の酵素によるRNAの操作中に生じRNA発現レベルの不正確な決定をもたらす偏りのため、従来の方法ではなく亜硫酸水素塩処理RNAにおいて、それぞれアップレギュレーションおよびダウンレギュレーションされるとして検出される遺伝子を示す。酵素による操作は誤解を与える結果を引き起こし、特定の遺伝子は、それらが実際そのように制御されないときにアップまたはダウンレギュレーションされることを示し得る。
【0168】
図1Cは、図1Aと同様のマイクロアレイベースのアッセイを示すが、暗色および淡色の矢印は、検出分子の改善された特異性のため、従来の方法ではなく亜硫酸水素塩処理RNAにおいて、それぞれアップレギュレーションおよびダウンレギュレーションされるとして検出される遺伝子を示す。検出分子の特異的結合強度の増大は、特異性の欠如のため従来の方法を使用して検出することができないRNA種の検出をもたらす。
【0169】
アクチン
既に記載したように、RNAを抽出および精製し、次いで亜硫酸水素塩処理および増幅した。増幅後、製造者によって指示されたようにMarligen PCRクリーンアップキットを使用してPCR産物を精製し、20μlの水中に再懸濁した。100ngの逆方向プライマーを10μlのPCR産物に加え、これらのサンプルはDNA塩基配列決定用にSupermac(Camperdown、Sydney)に送った。
【0170】
図2は、細胞系物質由来の亜硫酸水素塩修飾した全RNAを使用する逆転写酵素のPCRを示す。この図から見ることができるように、アクチン遺伝子のエクソン3a〜4またはエクソン3a〜3bを対象とする野生型プライマー(非亜硫酸水素塩転換型)を使用すると、いずれの場合も増幅産物は得られない。これはRNAの転換は非常に有効であることを示すが、これは、野生型配列を検出することができないからである。逆に、亜硫酸水素塩転換型プライマーを同じサンプルでエクソン3a〜4またはエクソン3a〜3bに使用する場合、異なるPCRバンドが生成し、亜硫酸水素塩転換型物質から特定のmRNAを増幅することが可能であることを示す。
【0171】
図3は、亜硫酸水素塩転換型RNAから生成したPCR産物の直接的な塩基配列決定を示す。塩基配列決定プロファイルから見ることができるように、PCR産物は混入DNAではなくRNAに由来するが、これは、これらの産物の配列がエクソン3と4の間のスプライス部位にわたって続くからである。さらに、図3から見ることができるように、PCR産物は完全に転換されているが、これは、サンプル中の元のC残基がここではT残基に転換されるからである。
【0172】
C型肝炎ウイルス
C型肝炎ウイルス(HCV)のRNAサンプルは、Acrometrix(OptiQual HCV高陽性対照)またはBBI diagnostics(HCV RNA直線性パネル)から得て、製造者の説明に従いUltrasensウイルス精製キットを用いて精製した。サンプルは亜硫酸水素ナトリウムで処理し、転換型HCVのRNAサンプルは以下のようにSuperscriptIII逆転写酵素(Invitrogen)を用いて逆転写した:
11μlの転換型RNA鋳型
1μlのランダムプライマー(300ng/μl)
1μlのdNTP(10mM)
【0173】
サンプルは65℃で5分間加熱し、次いで少なくとも1分間氷上に即座に置き、その後以下の試薬を加えた:
4μlの5×第一鎖用バッファー
1μlのRNaseOUT(40U/μl)
1μlのDTT(100mM)
1μlのSuperscriptIII(200U/μl)
【0174】
サンプルは以下の条件を使用して逆転写した:
25℃、12分間
27℃、2分間
29℃、2分間
31℃、2分間
33℃、2分間
35℃、2分間
37℃、30分間
45℃、15分間
50℃、5分間
75℃、5分間
【0175】
次いで2μlのcDNAを、HCVの5’NTRに特異的なプライマーおよびプローブを用いてPCRで増幅した:
(順方向プライマー−ttatgtagaaagtgtttagttatggtgt(配列番号9);
逆方向プライマー−acccaaatytccaaayattaaacaaat(配列番号10);
プローブ−tcCacAaaCcaCtaTaaCtcTcc(配列番号11)、
(前式で大文字はLNAの存在を示す)、Corbett 6000 Rotor Geneにおいて以下の試薬およびサイクル条件を使用した:
Sigma Jumpstart 2×マスター混合物 12.5μl
順方向プライマー 50ng
逆方向プライマー 50ng
25mMのMgCl2 3.5μl
400nMのプローブ(最終濃度) ×μl
23μlまでの水 ×μl
95℃、10分間
95℃、10秒間(50×)
53℃、90秒間(50×)
60℃、30秒間
【0176】
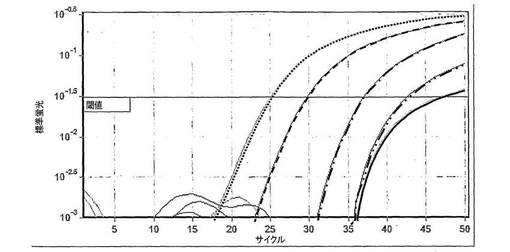
結果は図4中に示し、この場合図4から見ることができるアッセイに関する検出限界は約2.5IUのウイルスである。
【0177】
【表3】
【0178】
図4中に示したゲルの結果は、亜硫酸水素塩の方法を使用して、2.5IUのHCVまで低い広範囲の標的濃度におけるHCVの発現レベルをモニタリングすることができることを示す。
nc=陰性対照
【0179】
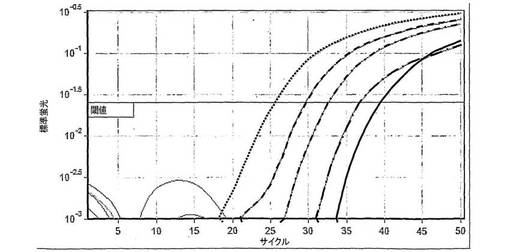
HCVに関する直線性パネルの滴定のリアルタイムqPCRによる結果は図5中に示す。このプロットに関する標準曲線は、0.99947のR値および0.9989のR2値を有する直線であった。
【0180】
【表4】
【0181】
HCVに関するダイナミックレンジの滴定に関する定量化の報告のリアルタイムqPCRによる結果は図6中に示す。このプロットに関する標準曲線は、0.99856のR値および0.99713のR2値を有する直線であった。
【0182】
【表5】
【0183】
直線性パネルおよびダイナミックレンジのサンプルからの結果は、リアルタイムPCR中に作成した定量曲線を示す。曲線のラインが閾値と交差する地点はCt値として知られ、サンプルの定量化に使用する。サンプルのそれぞれのセットに関して3桁を超える、一連の知られている濃度のウイルスを精製し、亜硫酸水素塩転換および増幅し、作成した標準曲線は、1に近いR2値によって例示されるように、反応効率は調べた濃度範囲で一定で直線的であることを示す。これらの結果および図4中に示した結果は、156250IUから1.5IUまでの範囲で、ウイルス特異的プローブを使用する終点PCRおよびリアルタイムPCRを使用すると、HCVウイルスRNAの検出に関して優れた感度および特異性が存在することを実証し、このアッセイは、非常に広範囲の濃度でウイルス遺伝子の発現を検出することができることを示す。
【0184】
広く記載する本発明の精神または範囲から逸脱せずに、具体的な実施形態中に示すように本発明に対して多数の変形および/または変更をなすことができることは、当業者によって理解されよう。したがって、本発明の実施形態は全ての点で例示的であり、かつ制限的ではないとみなすべきである。
【技術分野】
【0001】
本発明は、DNAへのRNAの転換を必ずしも必要としない遺伝子発現のアッセイに関する。
【背景技術】
【0002】
チップを使用するマイクロアレイ発現プロファイリング(Schena et al、1995、Science270:467-470;Chee et al、1996、Science274:610-614)などの細胞集団中のRNA産出量の測定によって、または遺伝子発現の連続分析SAGE(Velculescu et al、1995、Science270:484-487)によって、または全遺伝子発現分析TOGA(Sutcliffe et al、2000、Proc NatlAcad Sci USA97:1976-1981)によって、またはランダムに並んだ位置特定可能な高密度光学センサーアレイ(Michael et al、1998、Anal Chem70:1242-1248)によって、またはマイクロビーズアレイ上での超並列シグネチャー配列決定MPSS(Brenner et al、2000、Nature Biotechnology18:630-634)によって遺伝子発現を推測するための現在使用されている方法は、遺伝子発現の真の程度または量に関する正確な情報を必ずしも与えることができるわけではない。使用されている方法の多くは、まず対応するcDNA分子へのRNAの逆転写仲介転換、次いでcDNA集団の増幅および標識を必要とするので間接的である。良くて、このような現在の方法は遺伝子発現の指標のみをもたらすが、対象の特定遺伝子の発現の正確な測定値はもたらさない。
【0003】
これらの難点は今や非常に明らかである。例えば、SAGE技術(Stollberg et al、2000、Genome Research10:1241-1248)、チップ(Chudin et al、2001、Gene Biology3:0005.1-0005.10;Kothapalli et al、2002、BMC Bioinformatics3:1-10;Workman et al、2002、Genome Biology3:0048.1-0048.16)における偏り(bias)が記載されている。より一般的に、現在の方法における問題点が記載されている(Martin and Pardee、2000、Proc Natl Acad Sci USA 97:3789-3791;Wang et al、2000、Proc Natl Acad Sci USA97:4162-4167)。
【0004】
以前の変性RNA分子に自然かつ迅速に形成する安定した二次構造のために、適切な特異的プローブとの直接的なハイブリダイゼーションによってRNAをアッセイすることは困難である。
【発明の概要】
【発明が解決しようとする課題】
【0005】
本発明者は現在、生物、細胞集団または組織サンプル中の遺伝子発現のより正確な推測を与えることができる改善されたアッセイを開発した。
【課題を解決するための手段】
【0006】
第一の態様において、本発明は、
(a)RNAの二次構造を実質的に除去する作用物質でRNAを処理することと、
(b)処理したRNAの存在または量を測定して遺伝子発現の指標を得ることと
を含む遺伝子発現のアッセイを提供する。
【0007】
第二の態様において、本発明は、遺伝子発現を推測または測定するアッセイにおける、RNAの二次構造を実質的に除去しRNAを安定化させる作用物質の使用を提供する。
【0008】
第三の態様において、本発明は、RNA検出により遺伝子発現をアッセイするための、選択した化学組成を有するプローブの使用を提供する。
【0009】
プローブは塩基A(アデニン)、T(チミン)およびC(シトシン)から実質的に構成され、相当量のG(グアニン)は含まないことが好ましい。プローブは、G(グアニン)を実質的に含まないことが好ましい。
【0010】
本発明は、RNAの二次構造を実質的に除去する作用物質を利用する遺伝子発現のアッセイにおけるオリゴヌクレオチド、PNA、LNAまたはINAプローブの使用も含む。
【0011】
第四の態様において、本発明は、
(a)RNAの二次構造を実質的に除去する作用物質でRNAを処理することと、
(b)RNAの相補配列と結合することができるプライマーを使用してRNAを逆転写および増幅することと、
(c)処理および増幅したRNAの存在または量を測定して遺伝子発現の指標を得ることと
を含む遺伝子発現のアッセイを提供する。
【0012】
好ましい形では、RNAは、微生物、細胞、細胞群または細胞集団を含む、真核生物または原核生物由来である。
【0013】
好ましい形では、RNAはmRNAである。
【0014】
好ましい形では、RNAは微生物由来である。
【0015】
微生物、細胞または細胞集団または他の組織または生物源からRNAを単離するのに適した任意の方法によって、RNAを得ることができる。このような方法は当技術分野でよく知られており、例えば、Sambrook et al、「Molecular Cloning、A Laboratory Manual」second ed.、CSH Press、Cold Spring Harbor、1989を参照。例として、オリゴ−dTコーティング磁気ビーズまたは樹脂があるが、これらだけには限られない。RNA結合樹脂の具体例には、以下のRNeasy(商標)およびOligotex(商標)(Qiagen)、StrataPrep(商標)トータル(Stratagene)、Nucleobond(商標)(Clontech)、RNAgents(商標)およびPolyATract(商標)システム(Promega)などがある。密度勾配遠心分離技法を使用してRNAを単離することもできる。
【0016】
RNAは、真核生物由来でもよく、または細菌などの原核生物およびウイルス由来でもよい。薬物治療、ウイルス量、サンプル中の様々なウイルスの発現アレイなどをモニタリングするためにアッセイを使用することができる。
【0017】
RNAはシトシン塩基を修飾することができる作用物質で処理して、RNAの相補領域間の結合強度を弱めることが好ましいが、これは、シトシンの除去がC:G塩基対形成の消失をもたらすからである。生じる修飾は二次構造を除去し、一本鎖実体としてRNAを実質的に安定させる。作用物質は、亜硫酸水素塩、ヒドロキシルアミン、酢酸塩またはクエン酸塩から選択されることが好ましい。作用物質は、亜硫酸水素塩または酢酸塩試薬であることがより好ましい。作用物質は、水の存在下でシトシンをウラシルに修飾する試薬である亜硫酸水素ナトリウムであることが最も好ましい。
【0018】
亜硫酸水素ナトリウム(NaHSO3)はシトシンの5,6−二重結合と容易に反応して、脱アミノ化の影響を受けやすく水の存在下で亜硫酸ウラシルを生成するスルホン化シトシンの反応中間体を形成する。必要な場合、亜硫酸基を適度なアルカリ条件下で除去して、ウラシルの形成をもたらすことができる。したがって、潜在的には全てのシトシンがウラシルに転換される。ウラシル塩基は、シトシンが形成し得る3つの水素結合ではなく、任意の相補塩基との2つの水素結合のみを形成することができるので、複雑な二次構造を再形成するRNAの傾向は大幅に低下する。次いで、こうして処理した修飾型RNAは、妨げられることなく特定の相補的プローブとの相互作用に利用可能である。
【0019】
重要なことに、特定の実施形態では、標的配列のその含有量に関してサンプルをアッセイする前に、現在実施されているようなRNAを対応する相補的DNA(cDNA)に転換する必要がない。逆転写酵素またはポリメラーゼ連鎖反応(PCR)増幅工程のいずれも必要とされないので、本発明による方法は簡潔でより直接的であり、したがって、標準的手順で用いられる酵素の配列コピーの偏りによって引き起こされる誤差が起こりにくい。
【0020】
存在する標的(修飾型)RNAの量は、任意の適切な手段によって測定することができる。例えば、標的RNAを対象とする特異的プローブは、対象の対応する転写単位の一部または全部から誘導することができる。あるいは、プローブは、適切な配列の適切な抗体または抗体断片または単一ドメイン抗体、オリゴヌクレオチド、またはペプチド核酸(PNA)、固定化核酸(locked nucleic acid)(LNA)または挿入核酸(intercalating nucleic acid)(INA)プローブなどの塩基−配列特異性を示す任意の他の実体から誘導することができる。
【0021】
本発明のプローブは、試験するRNAと「実質的に」相補的であるように設計することができる。プローブが事実上PNA、LNA、オリゴヌクレオチドまたはINAであるとき、それらはA(アデニン)、T(チミン)、またはC(シトシン)塩基のみを含み得るが、これは、修飾型RNAが非修飾C残基を実質的に含まないからである。
【0022】
プローブは、オリゴヌクレオチドプローブまたはPNA、LNAまたはINAプローブなどの任意の適切なリガンドであってよい。例えば、全ての処理済みRNAと結合し得るポリ−TDNAまたはポリ−TPNAまたはLNAプローブまたはポリTINAプローブを使用することができ、これらはいずれも細胞由来のポリA「テイル」を有し、細胞、細胞集団または組織中の全遺伝子発現の測定を可能にする。あるいは、対象のRNAを対象とする特異的プローブを使用して、所与の細胞または組織中の特異的遺伝子発現を測定することが可能である。
【0023】
RNAにおけるランダムな二次構造形成を不安定にするための、シトシンとウラシル、またはその亜硫酸塩付加物の置換も、特異的オリゴ−、PNA、LNA、またはINAプローブと修飾型RNAの結合の強度を有意に低下させ得る。INA分子は、末端位置に限り挿入基(intercalating group)を用いて適切に設計するとき、相補的配列構造のRNAに対して高い結合性を有する。これをさらに補うために、プローブ中のアデニン塩基の代わりに、任意の相補的RNA鎖中の3つの水素結合を形成する2,6−ジアミノプリン(AP)を(アデニンが形成し得る2つに対する)チミンと置換し、これによってプローブとRNAの間の結合を強化することが好ましい。
【0024】
INAプローブは、その塩基配列において相補性を示すDNAまたはRNAの隣接塩基間に挿入することができる挿入分子を、「正常」または「修飾型」ヌクレオチドの配列中の様々な場所に結合させることによって構築する。このようなDNA分子によって、このような挿入部分の存在は、挿入基をINAプローブ中のどこに結合させようとも、プローブと標的核酸分子の間の相互作用を大幅に安定化させる。INAの注目すべき性質は以下に記載する。
【0025】
RNA分子と結合するように設計したINAプローブの場合、挿入基をINAの末端または末端付近に配置して、結合を高めることが好ましい。驚くべきことに、挿入基を内部に配置することはRNAと相補的DNAのハイブリダイゼーションに悪影響を与える可能性があり、かつハイブリッド構造を安定化させずに不安定化し得る。INAプローブを構築するための方法は以下に記載する。
【0026】
本発明の重要性は、処理して全てのそのシトシン残基がウラシル残基に転換された標的RNA種と非常に特異的かつ非常に強く結合する、個々の構築体のINAプローブの驚くべき能力に関する。結果として、本発明は、現在使用されている方法の多くの間接工程によって導かれる誤りまたは偏りを回避することができる方法に関する。
【0027】
示すように、幾つかの他の特異的プローブを本アッセイ中で使用することができるが、それらの使用の詳細な説明から明らかとなる理由で、INAプローブを使用することが好ましい。
【0028】
RNAの増幅は、RNAの相補配列と結合することができるINAプライマーを使用して実施することが好ましい。増幅は、典型的には逆転写酵素PCRベースの方法を使用して実施される。
【0029】
高ストリンジェンシー条件下でハイブリダイズすることができるか本発明中で利用する核酸分子と高い配列類似性を有することができる同等な配列に関して、「高ストリンジェンシー条件下でハイブリダイズする」は、当技術分野でよく知られている用語である「ストリンジェントなハイブリダイゼーション条件」と同義であり得る。例えば、Sambrook、「Molecular Cloning、A Laboratory Manual」second ed.、CSH Press、Cold Spring Harbor、1989;「Nucleic Acid Hybridisation、A Practical Approach」、Hames and Higgins eds.、IRL Press、Oxford、1985を参照。
【0030】
本発明の利点は、cDNAへのRNAの転換を必要とせずに、RNAの直接測定を実施することができることである。本発明のアッセイは、cDNAへのRNAの転換または増幅を必要とする現在の方法による潜在的な誤りを導かずに、細胞集団中の遺伝子活性の真の測定を可能にする。
【0031】
PNAまたはオリゴヌクレオチドプローブは、当業者に知られている任意の適切な方法を使用して調製することができる。INAプローブは、当業者に知られている任意の適切な方法によって調製することができる。
【0032】
適切なプローブを使用するハイブリダイゼーションアッセイ前にINAプライマーを使用して、処理したRNAを少量から増幅することもできる。
【0033】
本発明は、チップなどの現在のアレイ技術中、またはランダムに位置特定可能な高密度光学アレイ中で使用するのに適しており、したがって多数の遺伝子を迅速にアッセイすることができる。この形では、何万もの遺伝子の活性を1回の試験で測定またはアッセイすることができる。本発明は、例えばビーズ技術を使用する、少数の遺伝子を対象とするアッセイにも適応可能である。修飾RNA種はアレイの形で適切な支持体にスポットするまたはつけることが可能であり、アレイは様々なプローブによって測定することができる。
【0034】
1つの好ましい形では、本発明は、PNA分子は正味の電荷を有していないが、一方RNA分子は、それらのリン酸骨格のために、強く負に帯電しているという事実を特に利用する。結合PNAプローブの検出は、正に帯電した蛍光色素などの単分子、その長さに比例して核酸と特異的に結合し直接検出することができる複数の分子を利用することができる。多くのこのような適切な蛍光色素が知られている。
【0035】
検出系は、核酸と選択的に結合し酵素アッセイを使用して検出することができる正に帯電した領域を有する酵素、または捕捉された核酸分子と選択的に結合する正に帯電した放射性分子であってもよい。ナノ結晶も使用することができることは理解されよう。
【0036】
他の適切な検出系は量子ドットバイオコンジュゲートの使用である(Chan and Nie 1998 Science 282:2016-2018)。
【0037】
あるいは、配列特異的プローブおよび幾つかの蛍光色素分子が結合するミクロスフェアを利用することができる。ミクロスフェアは、特定のRNA種を標的化するプローブに直接、またはそのポリアデニンテイルなどのRNAの第二の非特異的構成部分を介して結合させることが可能である。この後者の場合、ミクロスフェアシグナル検出系の結合は、INA、PNA、LNAまたはオリゴヌクレオチド部分としてのポリT配列を介した結合であり得る。
【0038】
蛍光色素マーカーを有するミクロスフェアには様々な色またはスペクトルがあるので、1回の実験で、単細胞サンプル中に存在する幾つかの異なるRNA種のそれぞれの量を測定することが可能である。さらに、そのように標識した一種のミクロスフェアを、容易に目に見える状態にして計数することができ、したがって異なるRNA種間の発現のわずかな違いを相当な精度で決定することができる。
【0039】
基質と反応して着色産物を形成することができる適切な放射性化合物または酵素を用いた標識などの、標的修飾RNAと結合するリガンドを検出するための他の方法を、捕捉RNAまたはプローブまたは基質のいずれかに直接結合させる個々の適用例に使用することもできる。
【0040】
この手順中でのリガンドの1つとしてのINAまたはPNAまたは他のオリゴヌクレオチドプローブの使用には、オリゴヌクレオチドプローブの使用に優る非常に有意な利点がある。INAまたはPNAの結合はより速く平衡に達し、より高い配列特異性を示す。PNA分子は非帯電性であり、従来のオリゴヌクレオチドプローブより高い結合係数で標的修飾RNA分子と結合することができる。特に、INAプローブはA−T−とA−U塩基間の結合を高める。処理してシトシン塩基がウラシル塩基に転換された二次構造を除去したRNAの場合、これは重要である。RNA処理の結果として、少数のG−C塩基間相互作用および対応するA−TおよびA−U塩基間相互作用の数が増大する。
【0041】
本発明は直接的な検出法を使用することができるので、アッセイはサンプル中の標的RNAの量の真の正確な測定値を与えることができる。本発明のアッセイは、PCRなどのプロセスにおけるシグナル増幅に依存する方法に固有の潜在的偏りによって混乱することはなく、この場合、このような手順中で一般的に使用される酵素は、異なる配列の増幅率の差によって組織的偏りを導き得る。
【0042】
本発明は、疾患状態の検出、幹細胞および派生細胞集団の分化状態、遺伝子発現または細胞機能に対する薬剤の影響の検出または測定、およびC型肝炎ウイルス(HCV)およびヒト免疫不全ウイルス(HIV)などのウイルスに感染した患者のための正確な薬物計画の決定に役立つウイルス量モニタリングなどの、遺伝子発現の正確な指標が有用である任意の他の状況に特に適している。
【0043】
本明細書を通じて、文脈で他に必要とならない限り、語句「comprise」、または「comprises」もしくは「comprising」などの変形は、言及する要素、整数または工程、または要素群、複数の整数または工程の包含を意味するが、任意の他の要素、整数または工程、または要素群、複数の整数または工程の除外は意味しないことは理解されよう。
【0044】
本明細書中に含まれている文献、作用、物質、装置、物品などの任意の論述は、本発明の文脈を与える目的のものに過ぎない。これらの事項のいずれかまたは全てが従来技術の土台の一部を形成する、または本発明と関係がある分野の共通の一般知識であったことは当然のこととして解釈すべきでないが、これは、それが本発明の優先日前にオーストラリアに存在したからである。
【0045】
本発明がより明確に理解できるように、好ましい形態を以下の図面および実施例を参照しながら説明する。
【図面の簡単な説明】
【0046】
【図1A】典型的なマイクロアレイベースのアッセイの説明を示す図であり、この場合、暗色点は特定のRNA集団中でアップレギュレーションされる遺伝子を示し、かつ淡色点は同じ集団中でダウンレギュレーションされる遺伝子を示す。暗色および淡色の矢印は、従来系中の検出分子がそれらの標的と結合するのを妨げる二次構造のため、従来の方法ではなく亜硫酸水素塩処理RNAにおいて、それぞれアップレギュレーションおよびダウンレギュレーションされるとして検出される遺伝子を示す。
【図1B】図1Aと同様のマイクロアレイベースのアッセイを示す図であるが、暗色および淡色の矢印は、発現分析前の酵素によるRNAの操作中に生じRNA発現レベルの不正確な決定をもたらす偏りのため、従来の方法ではなく亜硫酸水素塩処理RNAにおいて、それぞれアップレギュレーションおよびダウンレギュレーションされるとして検出される遺伝子を示す。酵素による操作は誤解を与える結果を引き起こし、特定の遺伝子は、それらが実際そのように制御されないときにアップまたはダウンレギュレーションされることを示し得る。
【図1C】図1Aと同様のマイクロアレイベースのアッセイを示す図であるが、暗色および淡色の矢印は、検出分子の改善された特異性のため、従来の方法ではなく亜硫酸水素塩処理RNAにおいて、それぞれアップレギュレーションおよびダウンレギュレーションされるとして検出される遺伝子を示す。検出分子の特異的結合強度の増大は、特異性の欠如のため従来の方法を使用して検出することができないRNA種の検出をもたらす。
【図2】亜硫酸水素塩処理RNAからのPCR増幅のゲル分離の結果を示す図である。分離ウエル:M、100〜1000bpマーカー;レーン5野生型アクチンプライマーエクソン3a〜3b;レーン6野生型アクチンプライマーエクソン3a〜4;レーン7亜硫酸水素塩転換型アクチンプライマーエクソン3a〜3b;レーン8野生型アクチンプライマーエクソン3a〜4。
【図3】(図3A)亜硫酸水素塩処理転換型アクチンRNA(配列番号1)の配列分析を示す図である。 (図3B)野生型アクチンRNA(配列番号2)の配列分析を示す図である。
【図4】C型肝炎ウイルス(HCV)に関する直線性のゲルベースの読み出し値を示す図である。
【図5】リアルタイムPCR定量化の報告−C型肝炎ウイルス(HCV)RNAに関する直線性パネルを示す図である。
【図6】リアルタイムPCR定量化の報告−C型肝炎ウイルス(HCV)に関するダイナミックレンジを示す図である。
【発明を実施するための形態】
【0047】
定義
核酸
用語「核酸」は、天然に存在する核酸、DNAおよびRNAを包含する。用語「核酸類似体」は、天然に存在する核酸、DNAおよびRNAの誘導体、および天然に存在する核酸の合成類似体を包含する。合成類似体は1つまたは複数のヌクレオチド類似体を含む。用語ヌクレオチド類似体は、天然に存在するヌクレオチドとほぼ同様の、核酸骨格に取り込むことができ特異的な塩基対形成することができる(以下参照)全てのヌクレオチド類似体を含む。
【0048】
したがって用語「核酸」または「核酸類似体」は、複数のヌクレオチドおよび/またはヌクレオチド類似体および/またはインターカレーター擬ヌクレオチドから本質的になる任意の分子を指す。本発明に有用な核酸または核酸類似体は、異なる骨格のモノマー単位を有する幾つかの異なるヌクレオチドを含むことができる。
【0049】
核酸または核酸類似体の一本鎖は、実質的に相補的な一本鎖核酸および/または核酸類似体とハイブリダイズして、二本鎖核酸または核酸類似体を形成することができることが好ましい。このような二本鎖類似体は、二重らせんを形成することができることがより好ましい。二重らせんは水素結合によって形成されることが好ましく、二重らせんは、A型、B型、Z型およびそれらの中間体の二重らせんからなる群から選択される二重らせんであることがより好ましい。
【0050】
したがって、本発明に有用な核酸および核酸類似体には、それだけに限らないが、DNA、RNA、LNA、PNA、MNA、ANA、HNAおよびそれらの混合物およびそれらのハイブリッド、ならびにそれだけに限らないが、ホスホロチオエート、メチルホスホレート、ホスホラミデート、ホスホロジチエート、ホスホロセレノエート、ホスホトリエステルおよびホスホボラノエートなどのそれらのリン原子修飾体がある。さらに、それだけに限らないが、メチルイミノメチル、ホルムアセテート、チオホルムアセテートおよびアミド含有結合基などの非リン含有化合物をヌクレオチドとの結合に使用することができる。特に、核酸および核酸類似体は、1つまたは複数のインターカレーター擬ヌクレオチドを含むことができる。
【0051】
本文脈内では「混合物」は、異なる種類のヌクレオチドまたはヌクレオチド類似体を含む核酸および核酸類似体鎖を包含することを意味する。さらに、本文脈内では「ハイブリッド」は、1つまたは複数の種類の骨格を有するヌクレオチドまたはヌクレオチド類似体を含む1本の鎖、および異なる種類の骨格を有するヌクレオチドまたはヌクレオチド類似体を含む他の鎖を含む、核酸または核酸類似体を包含することを意味する。
【0052】
HNAによって、例えばVan Aetschot et al.、1995によって記載されたのと同様の核酸を意味する。MNAによって、Hossain et al、1998によって記載されたのと同様の核酸を意味する。ANAはAllert et al、1999によって記載された核酸を指す。LNAはWO99/14226(Exiqon)中に記載されたのと同様の任意のLNA分子であってよく、LNAはWO99/14226の要約書中で示された分子から選択されることが好ましい。LNAは、Singh et al、1998、Koshkin et al、1998またはObika et al、1997中に記載されたのと同様の核酸であることがより好ましい。PNAは例えばNielsen et al、1991によって記載されたのと同様のペプチド核酸を指す。
【0053】
用語ヌクレオチドは核酸または核酸類似体の構成単位を指し、かつ用語ヌクレオチドは、天然に存在するヌクレオチドおよびそれらの誘導体、ならびに天然に存在するヌクレオチドおよびそれらの誘導体とほぼ同じ機能を果たすことができるヌクレオチドを包含する。天然に存在するヌクレオチドは、4つの主な核酸塩基アデニン(A)、チミン(T)、グアニン(G)またはシトシン(C)の1つを含むデオキシリボヌクレオチド、および4つの核酸塩基アデニン(A)、ウラシル(U)、グアニン(G)またはシトシン(C)の1つを含むリボヌクレオチドを含む。
【0054】
ヌクレオチド類似体は、核酸骨格に取り込むことができ特異的な塩基対形成することができる、任意のヌクレオチド様分子であってよい。
【0055】
天然に存在しないヌクレオチドには、DNA、RNA、PNA、HNA、MNA、ANA、LNA、CNA、CeNA、TNA、(2’−NH)−TNA、(3’−NH)−TNA、α−L−リボ−LNA、α−L−キシロ−LNA、β−D−キシロ−LNA、α−D−リボ−LNA、[3.2.1]−LNA、バイシクロ−DNA、6−アミノ−バイシクロ−DNA、5−エピ−バイシクロ−DNA、α−バイシクロ−DNA、トリシクロ−DNA、バイシクロ[4.3.0]−DNA、バイシクロ[3.2.1]−DNA、バイシクロ[4.3.0]アミド−DNA、β−D−リボピラノシル−NA、α−L−リキシオピラノシル−NA、2’−R−RNA、α−L−RNAまたはα−D−RNA、β−D−RNA内に含まれるヌクレオチドがあるが、これらだけには限られない。
【0056】
ヌクレオチドおよびヌクレオチド類似体の機能は、相補的ヌクレオチドの核酸塩基の水素結合を介して相補的ヌクレオチドと特異的に相互作用することができること、および核酸または核酸類似体に取り込まれることができることである。天然に存在するヌクレオチド、および幾つかのヌクレオチド類似体は、酵素によって、例えばRNAまたはDNAポリメラーゼによって、核酸または核酸類似体に取り込まれることができる。しかしながら、ヌクレオチドまたはヌクレオチド類似体は、化学的に核酸または核酸類似体に取り込まれることもできる。
【0057】
さらに核酸または核酸類似体は、2つの小さな核酸または核酸類似体を互いに結合させることによって調製することができ、例えばこれはリガーゼによって酵素的に行うことができ、またはそれは化学的に行うことができる。
【0058】
ヌクレオチドまたはヌクレオチド類似体は、骨格モノマー単位および核酸塩基を含む。核酸塩基は、天然に存在する核酸塩基、またはほぼ同じ機能を果たすことができるその誘導体もしくはその類似体であってよい。核酸塩基の機能は、水素結合を介して1つまたは複数の他の核酸塩基と特異的に結合することができることである。したがって、それは1つまたは少数の他の核酸塩基のみと安定した水素結合を結合することができるが、それは通常それ自体に含まれる大部分の他の核酸塩基とは安定した水素結合を結合することができないことは、核酸塩基の1つの重要な特徴である。1つの核酸塩基と他の核酸塩基の特異的相互作用は、一般に「塩基対形成」と呼ばれる。
【0059】
塩基対形成は、所定のヌクレオチドと相補的ヌクレオチドの間の特異的ハイブリダイゼーションをもたらす。相補的ヌクレオチドは、塩基対形成することができる核酸塩基を含むヌクレオチドである。
【0060】
一般的な天然に存在する核酸塩基の中で、アデニン(A)はチミン(T)またはウラシル(U)と塩基対形成し、かつグアニン(G)はシトシン(C)と塩基対形成する。したがって、Aを含むヌクレオチドはTまたはUのいずれかを含むヌクレオチドと相補的であり、かつGを含むヌクレオチドはCを含むヌクレオチドと相補的である。
【0061】
追加の分子体を含むようにヌクレオチドをさらに誘導体化することができる。ヌクレオチドは、核酸塩基または骨格モノマー単位で誘導体化することができる。塩基上の好ましい誘導体化部位には、アデニンの8−位置、ウラシルの5−位置、シトシンの5−または6−位置、およびグアニンの7−位置がある。複素環修飾は3つの構造クラス、強化型塩基スタッキング(enhanced base stacking)、追加の水素結合、およびこれらのクラスの組合せに分けることができる。平面系のπ−電子雲を拡大することによって塩基スタッキングを強化する修飾は、ピリミジンの5−位置および7−デアザ−プリンの7−位置における共役した親油性修飾によって表される。ピリミジン修飾の5−位置における置換にはプロピン、ヘキシン、チアゾールおよび単にメチル基があり、かつ7−デアザ−プリンの7−位置における置換にはヨード、プロピニル、およびシアノ基がある。プロピンから5員複素環および三環系縮合系までシトシンの5−位置を修飾することもでき、これらは4−および5−位置(シトシンクランプ)から生じる。第二の型の複素環修飾は2−アミノ−アデニンによって表され、この場合、追加のアミノ基が、G−C塩基対における3個の水素結合と類似した他の水素結合をA−T塩基対において与える。組合せの影響を与える複素環修飾は、ヘテロ二本鎖のエトキシアミノ官能基を有する2−アミノ−7−デアザ−7−修飾アデニンおよび三環系シトシン類似体によって表される。さらに、N2−修飾2−アミノアデニン修飾オリゴヌクレオチドは特に一般的な修飾体である。リボースまたはデオキシリボース部分上の誘導体化の好ましい部位は、非結合炭素位置C−2’およびC−4’の修飾、結合炭素C−1’、C−3’およびC−5’の修飾、糖の酸素、O−4’の置換、(立体配座が固定された)無水糖の修飾、(立体配座が固定された)環糖の修飾、リボフラノシル環の大きさの変化、結合部位−糖と糖、(C−3’とC−5’/C−2’とC−5’)、ヘテロ原子環−修飾糖、および前述の修飾の組合せである。しかしながら、核酸または核酸類似体の全体的な塩基対形成の特異性が妨害されない限り、他の部位を誘導体化することができる。最後に、骨格モノマー単位がリン酸基を含むとき、幾つかの骨格モノマー単位のリン酸を誘導体化することができる。
【0062】
本明細書で使用するオリゴヌクレオチドまたはオリゴヌクレオチド類似体は、ヌクレオチドおよび/またはヌクレオチド類似体および/またはインターカレーター擬ヌクレオチドの配列から本質的になる分子である。オリゴヌクレオチドまたはオリゴヌクレオチド類似体は、5〜100個の個別のヌクレオチドを含むことが好ましい。オリゴヌクレオチドまたはオリゴヌクレオチド類似体は、DNA、RNA、LNA、2’−O−メチルRNA、PNA、ANA、HNAおよびこれらの混合物、ならびに任意の他のヌクレオチドおよび/またはヌクレオチド類似体および/またはインターカレーター擬ヌクレオチドを含むことができる。
【0063】
RNA
本明細書で使用するRNAは、細胞などの任意の供給源由来のメッセンジャーRNA(mRNA)未熟mRNA、トランスファーRNA(tRNA)、リボソームRNA(rRNA)およびミクロRNA(miRNA)、ウイルスまたは他の微生物由来のゲノムRNA、DNAから転写されたRNA、対応するDNAのRNAコピーなどを含む。
【0064】
対応する核酸
核酸、核酸類似体、オリゴヌクレオチドまたはオリゴヌクレオチド類似体は、それらがハイブリダイズすることができるとき対応すると考えられる。対応する核酸、核酸類似体、オリゴヌクレオチドまたはオリゴヌクレオチド類似体は、低ストリンジェンシー条件下でハイブリダイズすることができることが好ましく、対応する核酸、核酸類似体、オリゴヌクレオチドまたはオリゴヌクレオチド類似体は、中ストリンジェンシー条件下でハイブリダイズすることができることがより好ましく、対応する核酸、核酸類似体、オリゴヌクレオチドまたはオリゴヌクレオチド類似体は、高ストリンジェンシー条件下でハイブリダイズすることができることがより好ましい。
【0065】
本明細書で使用する高ストリンジェンシー条件は、例えばSouthern E.M.、1975、J.Mol.Biol.98:503−517によって記載されたサザンブロッティングおよびのハイブリダイゼーションに関して通常適用されるストリンジェンシーを示すものとする。このような目的のために、プレハイブリダイゼーションおよびハイブリダイゼーションの工程を含めることは通常業務である。このような工程は通常、Sambrook et al.、1989によって「Molecular Cloning/A Laboratory Manual」、Cold Spring Harbor)中に記載されたように、6×SSPE、5%のDenhardt’s、0.5%のSDS、50%のホルムアミド、100μg/mlの変性したサケ精子DNAを含む溶液を使用し(42℃で18時間インキュベーション)、次に2×SSCおよび0.5%のSDSで洗浄し(室温および37℃)、0.1×SSCおよび0.5%のSDSで洗浄して(30分間68℃でインキュベーション)実施する。
【0066】
本明細書で使用する中ストリンジェンシー条件は、pH7.0において1mMのEDTA,10mMのNa2HPO4H2O、140mMのNaClを含むバッファー中でのハイブリダイゼーションを示すものとする。約1.5μMのそれぞれの核酸または核酸類似体鎖を与えることが好ましい。あるいは中ストリンジェンシーは、50mMのKCl、10mMのTRIS−HCl(pH9,0)、0.1%のTritonX−100、2mMのMgCl2を含むバッファー中でのハイブリダイゼーションを示すことができる。
【0067】
低ストリンジェンシー条件は、pH7.0において1MのNaCl、10mMのNa3PO4で構成されるバッファー中でのハイブリダイゼーションを示す。
【0068】
あるいは、対応する核酸、核酸類似体、オリゴヌクレオチドまたはオリゴヌクレオチド、核酸類似体、オリゴヌクレオチドまたはオリゴヌクレオチドは、所与の配列で互いに実質的に相補的、70%を超えて相補的など、例えば75%を超えて相補的、80%を超えて相補的など、例えば85%を超えて相補的、90%を超えて相補的など、例えば92%を超えて相補的、94%を超えて相補的など、例えば95%を超えて相補的、96%を超えて相補的など、例えば97%を超えて相補的である。
【0069】
所与の配列は少なくとも10ヌクレオチド長、少なくとも15ヌクレオチドなど、例えば少なくとも20ヌクレオチド、少なくとも25ヌクレオチドなど、例えば少なくとも30ヌクレオチド、10ヌクレオチドと500ヌクレオチドの間など、例えば10ヌクレオチド長と100ヌクレオチド長の間、10ヌクレオチド長と50ヌクレオチド長の間であることが好ましい。対応するオリゴヌクレオチドまたはオリゴヌクレオチド類似体は、それらの全長で実質的に相補的であることがより好ましい。
【0070】
クロスハイブリダイゼーション
クロスハイブリダイゼーションとの用語は、少なくとも2つの核酸または核酸類似体間の意図しないハイブリダイゼーションを包含する。したがって、クロスハイブリダイゼーションとの用語を使用して、例えば核酸プローブまたは核酸類似体プローブ配列と、その意図する標的配列以外の他の核酸配列または核酸類似体配列のハイブリダイゼーションを記載することができる。
【0071】
クロスハイブリダイゼーションはプローブと1つまたは複数の対応する非標的配列の間で起こることが多いが、しかしながらこれらは、プローブおよびその対応する標的配列より低度の相補性を有する。この望ましくない影響は、標的に対して大幅に過剰なプローブおよび/または急速なアニーリング動態が原因であり得る。クロスハイブリダイゼーションは、数個の核酸塩基対間、例えばPCR反応中のプライマー間の水素結合によっても起こり、プライマー二量体形成および/または非特異的PCR産物の形成をもたらす。
【0072】
同じ型のヌクレオチド類似体に対して高い親和性を有する1つまたは複数のヌクレオチド類似体を含む核酸は、塩基対形成に基づいて二量体またはさらに高次の複合体を形成する傾向がある。それだけに限らないが、LNA、2’−O−メチルRNAおよびPNAなどのヌクレオチド類似体を含むプローブは、同じ型の骨格モノマー単位を含む他のオリゴヌクレオチド類似体とのハイブリダイズに対する高い親和性を一般に有する。したがって、個々のプローブ分子は低度の相補性のみを有するが、それらはハイブリダイズする傾向がある。
【0073】
自己ハイブリダイゼーション(self-hybridisation)
自己ハイブリダイゼーションとの用語は、核酸または核酸類似体分子が自身で折り畳まることにより自身にアニーリングし、例えばヘアピン構造のような二次構造を生成するプロセスを包含する。大部分の適用例において、自己ハイブリダイゼーションを避けることは重要である。二次構造の生成は、望ましい核酸標的配列とのハイブリダイゼーションを阻害し得る。例えば核酸または核酸類似体をPCR反応におけるプライマーとして、またはエクソヌクレアーゼアッセイ用のフルオロフォア/消光標識プローブとして使用するとき、大部分のアッセイにおいてこれは望ましくない。両方のアッセイにおいて、自己ハイブリダイゼーションは標的核酸配列とのハイブリダイゼーションを阻害し、さらにエクソヌクレアーゼアッセイにおけるフルオロフォアの消光の程度が低下する。
【0074】
同じ型のヌクレオチド類似体に対して高い親和性を有する1つまたは複数のヌクレオチド類似体を含む核酸は、自己ハイブリダイズする傾向がある。それだけに限らないが、LNA、2’−O−メチルRNAおよびPNAなどのヌクレオチド類似体を含むプローブは、自己ハイブリダイズに対する高い親和性を一般に有する。したがって、個々のプローブ分子は低度の相補性のみを有するが、それらは自己ハイブリダイズする傾向がある。
【0075】
融解温度
核酸の融解は、二本鎖核酸分子の2本の鎖の分離を指す。融解温度(Tm)は、50%のらせん(ハイブリダイズ)対コイル(非ハイブリダイズ)形が存在する摂氏温度を示す。
【0076】
高い融解温度は安定した複合体、したがって個々の鎖の間の高い親和性を示す。同様に、低い融解温度は個々の鎖の間の比較的低い親和性を示す。したがって、通常2本の鎖の間の強い水素結合は高い融解温度をもたらす。
【0077】
さらに、二本鎖核酸の核酸塩基間へのインターカレーターの挿入も二本鎖核酸を安定化させ、したがって高い融解温度をもたらす可能性がある。
【0078】
さらに、融解温度は周囲の物理的/化学的状態に依存する。例えば、融解温度は塩濃度およびpHに依存する。
【0079】
融解温度は幾つかのアッセイにより決定することができ、例えばUVスペクトルを使用してハイブリダイゼーションの形成および分解(融解)を決定することによって、融解温度を決定することができる。
【0080】
INA/IPNの定義
挿入核酸(INA)は特有なクラスのDNA結合分子である。INAは、ヌクレオチドおよび/またはヌクレオチド類似体およびインターカレーター擬ヌクレオチド(IPN)モノマーからなる。INAは、内部に位置するIPNに関して10℃までおよび末端位置IPNに関して11℃まで安定状態である相補的DNAに対して、非常に高い親和性を有する。正確に設計した場合、INA自体が、相補的RNAよりDNAとのハイブリダイズを好む選択的分子となり得る。IPNを分子内部に配置した場合、INAはオリゴヌクレオチドプライマーの約25分の1の効率でRNAに結合することが示されている。一方、従来のオリゴヌクレオチド、オリゴヌクレオチド類似体およびPNAは、RNAとDNAの両方に対して等しい親和性を有する。したがってINAは、第一の真に選択的なDNA結合剤である。さらにINAは、他の天然DNA分子より相補的DNAに対して高い特異性および親和性を有する。
【0081】
さらに、IPNはATに富む環境においてDNAを最も安定化させ、これによってIPNはエピゲノミクス研究の分野で特に有用となる。IPNは、典型的には隆起または末端挿入物としてINA分子内に置く。IPNは本質的に、核酸二本鎖中の核酸塩基と共にスタッキングすることができる平面状(ヘテロ)多環芳香族化合物である。
【0082】
INA分子は、エクソヌクレアーゼによる攻撃に耐性があることも示されている。これによって、これらの分子は、phi29などの酵素を使用する増幅用のプライマーとして特に有用となる。phi29は固有のエクソヌクレアーゼ活性を有するので、増幅用の鋳型として使用するプライマーは、特にそれらの3’末端において修飾して酵素による分解を妨げなければならない。しかしながらINA分子は、さらなる修飾なしで加えることができる。
【0083】
INAは従来のPCR増幅反応において使用することができ、従来型プライマーとして挙動する。しかしながら、INAは、DNAまたはRNA鋳型に対して高い特異性を有し、鋳型が制限的であり反応の感度が重要である状況における使用にINAは理想的となる。INAはATに富む環境においてDNAを最も安定化させ、これによってINAは亜硫酸水素塩処理DNA配列の増幅に特に有用となる。これは、亜硫酸水素塩による転換後、全てのシトシン残基はウラシルに、次にPCRまたは他の増幅後チミンに転換される事実に原因がある。したがって亜硫酸水素塩処理DNAは非常にTに富む。INA中のIPN分子の数の増大は、INA/DNA二本鎖の増大した安定性をもたらす。INA中のIPNが増大するほど、DNA/INA二本鎖の融解温度は高くなる。
【0084】
本出願人は、オリゴヌクレオチドまたはオリゴヌクレオチド類似体に取り込まれた際に、挿入核酸(INA)を形成し(WO03/051901、WO03/052132、WO03/052133およびWO03/052134)、オリゴヌクレオチドのサプリメントまたは置換体として新規かつ有用な性質を有するクラスのインターカレーター擬ヌクレオチドを以前に開発している。
【0085】
インターカレーター擬ヌクレオチドは、1−(4,4’−ジメトキシトリフェニルメチルオキシ)−3−ピレンメチルオキシ−2−プロパノールの複数のホスホラミダイトから選択されることが好ましい。インターカレーター擬ヌクレオチドは、(S)−1−(4,4’−ジメトキシトリフェニルメチルオキシ)−3−ピレンメチルオキシ−2−プロパノールのホスホラミダイトまたは(R)−1−(4,4’−ジメトキシトリフェニルメチルオキシ)−3−ピレンメチルオキシ−2−プロパノールのホスホラミダイトから選択されることが好ましい。
【0086】
オリゴヌクレオチドまたはオリゴヌクレオチド類似体は、DNA、RNA、固定核酸(LNA)、ペプチド核酸(PNA)、MNA、アルトリトール核酸(ANA)、ヘキシトール核酸(HNA)、挿入核酸(INA)、シクロヘキサニル核酸(CNA)、およびそれらの混合物およびそれらのハイブリッド、ならびにそれだけに限らないが、ホスホロチオエート、メチルホスホレート、ホスホラミデート、ホスホロジチエート、ホスホロセレノエート、ホスホトリエステルおよびホスホボラノエートなどのそれらのリン原子修飾体から選択することができる。天然に存在しないヌクレオチドには、DNA、RNA、PNA、INA、HNA、MNA、ANA、LNA、CNA、CeNA、TNA、(2’−NH)−TNA、(3’−NH)−TNA、α−L−リボ−LNA、α−L−キシロ−LNA、β−D−キシロ−LNA、α−D−リボ−LNA、[3.2.1]−LNA、バイシクロ−DNA、6−アミノ−バイシクロ−DNA、5−エピ−バイシクロ−DNA、α−バイシクロ−DNA、トリシクロ−DNA、バイシクロ[4.3.0]−DNA、バイシクロ[3.2.1]−DNA、バイシクロ[4.3.0]アミド−DNA、β−D−リボピラノシル−NA、α−L−リキシオピラノシル−NA、2’−R−RNA、α−L−RNAまたはα−D−RNA、β−D−RNA内に含まれるヌクレオチドがあるが、これらだけには限られない。さらに、それだけに限らないが、メチルイミノメチル、ホルムアセテート、チオホルムアセテートおよびアミド含有結合基などの非リン含有化合物をヌクレオチドとの結合に使用することができる。特に、核酸および核酸類似体は、1つまたは複数のインターカレーター擬ヌクレオチドを含むことができる。
【0087】
IPNがメチル化部位の特異的検出用にINA分子内に位置するとき、本発明者は、潜在的CpG部位間にIPNを位置させることは避けることが有用であることを見出している。これは、IPNを使用してCpG部位を隔てると、生成するINAの特異性が低下するという事実によるものである。
【0088】
ペプチド核酸(PNA)
ペプチド核酸は、配列特異性を有する核酸(DNAおよびRNA)とハイブリダイズすることができる天然に存在しないポリアミドである(米国特許第5,539,082号およびEgholm et al.、Nature(1993)365、566-568参照)。PNAはプローブベースのハイブリダイゼーションアッセイにおける核酸プローブに対する代替/代用としての候補であるが、これは、PNAが幾つかの望ましい性質を示すからである。PNAは、核酸とハイブリダイズして、対応する核酸/核酸複合体より熱力学的に安定したハイブリッドを形成するアキラルポリマーである。天然に存在しない分子であるので、それらはペプチドまたは核酸を分解することが知られている酵素の基質であることは知られていない。したがって、PNAは生物サンプル中で安定状態であり、かつ長期の保存寿命を有するはずである。イオン強度に非常に依存する核酸ハイブリダイゼーションとは異なり、PNAと核酸のハイブリダイゼーションはイオン強度にほとんど依存せず、核酸と核酸のハイブリダイゼーションを強く嫌う条件下において低いイオン強度を好む。PNA複合体の安定性および立体配座に対するイオン強度の影響は、広範囲で調べられている。配列識別は、DNA認識DNAよりPNA認識DNAまたはRNAに有効である。しかしながら、ハイブリダイゼーションアッセイにおける、DNAプローブと比較した1塩基の変化、indel、またはPNAプローブを用いた多型識別の利点は、幾分配列依存的であるようである。追加の利点として、PNAは平行方向と逆平行方向の両方で核酸とハイブリダイズするが、逆平行方向が好ましい。
【0089】
PNAは、現在市販されている形式での標準的なペプチド合成手順の適合によって合成する(PNAモノマーおよびオリゴマーの調製の一般評論に関しては、Dueholm et al.、New J.Chem.(1997)、21、19-31またはHyrup et. al.、Bioorganic & Med.Chem.(1996)4、5-23を参照頂きたい)。標識および非標識PNAオリゴマーは購入することができ(PerSeptive Biosystems Promotional Literature:BioConcepts、Publication No.NL612、Practical PNA、Review and Practical PNA、Vol.1、Iss.2を参照)、または市販の製品を使用して調製することができる。
【0090】
実際、PNAプローブと標準的な核酸プローブの間に多くの差異が存在する。これらの差異は、生物学的、構造的、および物理化学的差異に都合良く分けることができる。上記および以下で論じるように、これらの生物学的、構造的、および物理化学的差異は、核酸が典型的に利用されている適用例においてPNAプローブの使用を試みるとき、予期せぬ結果をもたらし得る。異なる組成物のこの非同等性は、化学分野においてしばしば観察される。
【0091】
生物学的差異に関して、核酸は、遺伝的伝達および発現の作用物質として、生存種の寿命において中心的役割を果たす生物学的物質である。それらのin vivoでの性質はほぼ十分に理解されている。しかしながらPNAは、近年開発された完全に人工的な分子であり、化学者の発想で考え出され、合成有機化学を使用して作製される。それは知られている生物学的機能は有していない。
【0092】
構造上、PNAは核酸とも劇的に異なる。両者は一般的な核酸塩基(A、C、G、T、およびU)を利用することができるが、これらの分子の骨格は構造上多様である。RNAおよびDNAの骨格は、反復リン酸ジエステルリボースおよび2−デオキシリボース単位からなる。対照的に、PNAの骨格はN−(2−アミノエチル)グリシン単位からなる。さらにPNA中では、核酸塩基は他のメチレンカルボニル単位によって骨格と結合している。
【0093】
その名称にもかかわらず、PNAは酸ではなく、かつDNAおよびRNA中に存在する基などの帯電した酸性基は含まない。PNAは形式電荷を欠くので、PNAは一般にそれらの同等な核酸分子より疎水性が高い。PNAの疎水特性は、核酸では観察されない非特異的(疎水性/疎水性相互作用)相互作用の可能性を与える。さらに、それがその同等な立体配座がRNA/DNA領域に存在しない構造立体配座をとる能力を有するという条件で、PNAはアキラルである。
【0094】
PNAとDNAまたはRNAの間の物理的/化学的差異も相当にある。PNAは、同じ標的配列と結合する核酸プローブより速く、その相補的核酸と結合する。この挙動は、少なくとも一部分は、PNAはその骨格に電荷を欠くという事実が原因であると考えられる。さらに、近年の刊行物は、PNA中への正に帯電した基の取り込みは、ハイブリダイゼーションの動態を改善することを実証する。それは骨格に電荷を欠くので、PNA/核酸複合体の安定性は、類似のDNA/DNAまたはRNA/DNA複合体の安定性より高い。特定の状況において、PNAは非常に安定した三重らせん複合体を形成するか、または「鎖置換」と呼ばれるプロセスによって小さなループを形成し得る。同等な鎖置換プロセスまたは構造は、DNA/RNA分野では知られていない。
【0095】
要約すると、PNAは配列特異性を有する核酸とハイブリダイズするので、PNAはプローブベースのアッセイを開発するのに有用な候補である。重要なことに、PNAプローブは核酸プローブと同等なプローブではない。それにもかかわらず、最もストリンジェントな条件下でさえ、正確な標的配列と密接に関連した配列(例えば、単一点突然変異(一塩基対のミスマッチ)を有する非標的配列)の両方が、標識核酸または標識PNAプローブとの検出可能な相互作用をしばしば示し得る。密接に関連した非標的配列との任意のハイブリダイゼーションが、望ましくないバックグラウンドシグナルの発生をもたらし得る。配列は非常に密接に関連しているので、複数の点突然変異は、プローブベースのアッセイを使用して検出するのが最も難しい全核酸修飾の一部である。鎌状赤血球貧血および嚢胞性線維症などの多数の疾患は、ゲノム核酸の単一点突然変異によって時折引き起こされる。したがって、プローブベースのアッセイの特異性、感度および信頼性を改善し得る任意の方法、キットまたは組成物が、DNA含有サンプルの検出、分析および定量化において有用であろう。
【0096】
亜硫酸水素ナトリウム
亜硫酸水素ナトリウムで核酸を処理するための方法は、Frommer et al 1992、Proc Natl Acad Sci89:1827-1831;Grigg and Clark 1994 BioAssays 16:431-436;Shapiro et al 1970、J Amer Chem Soc 92:422-423;Wataya and Hayatsu 1972、Biochemistry 11:3583-3588を含む幾つかの参照文献中で見ることができる。
【0097】
核酸の亜硫酸水素塩処理の成功の改善または向上のための複数の方法は、本出願人によっても開発されている。
【0098】
核酸の有効な亜硫酸水素塩処理のための例示的なプロトコルを以下に述べる。このプロトコルによって、実質的に全てのDNAが処理された状態となる。この方法は、本明細書ではヒト遺伝子シグネチャー(HGS)方法とも呼ぶ。サンプルまたは試薬の体積または量は変えることができることは理解されよう。
【0099】
亜硫酸水素塩処理に好ましい方法は、US 10/428310またはPCT/AU2004/000549中で見ることができる。
【0100】
そう望む場合適切な制限酵素で予め消化することができる2μgのDNAに、2μl(1/10体積)の3MのNaOH(6g、50ml水中、新たに作製)を、20μlの最終体積で加えた。この工程は二本鎖DNA分子を一本鎖形に変性させるが、これは、亜硫酸水素塩試薬が一本鎖分子と優先的に反応するからである。混合物は37℃で15分間インキュベートした。室温を超える温度でのインキュベーションを使用して、変性の効率を改善することができる。
【0101】
インキュベーション後、208μlの2Mメタ重亜硫酸ナトリウム(7.6g、20ml水および416ml10NNaOH中;BDH AnalaR#10356.4D;新たに作製)および12μlの10mMキノール(0.055g、50ml水中、BDH AnaIR #103122E;新たに作製)を連続して加えた。キノールは還元剤であり、試薬の酸化の還元を手助けする。他の還元剤、例えばジチオスレイトール(DTT)、メルカプトエタノール、キノン(ヒドロキノン)、または他の適切な還元剤を使用することもできる。サンプルには200μlのミネラルオイルを重層した。ミネラルオイルの重層は試薬の蒸発および酸化を防ぐが、必須ではない。次いでサンプルを55℃で一晩インキュベートした。あるいはサンプルは、サーマルサイクラーにおいて以下のサイクルにかけることが可能である:約4時間または一晩以下のようにインキュベートする:工程1、55℃/2時間サイクル、PCR機器中;工程2、95℃/2分間。工程1は約37℃〜約90℃の任意の温度で実施することができ、かつ5分〜8時間まで長さを変えることができる。工程2は約70℃〜約99℃の任意の温度で実施することができ、かつ約1秒〜60分、またはそれより長くまで長さを変えることができる。
【0102】
メタ重亜硫酸ナトリウムでの処理後、オイルを除去し、DNA濃度が低かった場合、1μlのtRNA(20mg/ml)または2μlのグリコーゲンを加えた。これらの添加剤は任意選択であり、特にDNAが低い濃度で存在するとき、これらを使用して標的DNAとの共沈によって得られるDNAの収率を改善することができる。核酸のより有効な沈殿のための担体としての添加剤の使用は、核酸の量が<0.5μgであるとき一般に望ましい。
【0103】
イソプロパノール浄化処理を以下のように実施した:800μlの水をサンプルに加え、混合し、次いで1mlのイソプロパノールを加えた。水またはバッファーは、塩が対象の標的核酸と共に沈殿しないレベルまで、反応容器中の亜硫酸水素塩の濃度を低下させる。本明細書で開示する所望の範囲以下に塩濃度を希釈する限り、希釈は一般に約1/4〜1/1000である。
【0104】
サンプルを再度混合し、少なくとも5分間4℃に放置した。サンプルは微量遠心管中で10〜15分間回転させ、ペレットは70%ETOHで2回洗浄し、毎回ボルテックスした。この洗浄処理によって、核酸と共に沈殿した任意の残留塩を除去する。
【0105】
ペレットは放置乾燥させ、次いで50μlなどの適切な体積のT/E(10mMトリス/0.1mMEDTA)pH7.0〜12.5中に再懸濁した。pH10.5でのバッファーは特に有効であることが分かっている。核酸を懸濁することが必要であったとき、サンプルは37℃〜95℃で1分間〜96時間インキュベートした。
【0106】
RNAの二次構造を実質的に除去する作用物質
本発明に適した作用物質は、亜硫酸水素塩、ヒドロキシルアミン、酢酸塩またはクエン酸塩を含む。WO2005054502中に記載されたように、亜硫酸水素塩試薬が好ましく、亜硫酸水素ナトリウム、メタ重亜硫酸ナトリウム、および亜硫酸水素グアニジウムを含む。この関連で、グアニジウムイオンおよび亜硫酸イオンを含む溶液の調製および後のRNA処理に亜硫酸水素グアニジウムを使用する、RNA処理を実施することができる。
【0107】
細胞系
【0108】
【表1】
【0109】
細胞からのRNA抽出
I.培地の除去後、1mlのトリゾールを細胞(90%融合)に直接加えた。
II.サンプルをよく混合し、室温で5分間放置して核タンパク質複合体を解離させた。
III.0.5mlをクリーンなRNaseを含まない1.5mlの遠心管に除去した。
IV.次いでサンプルを12,000×gで10分間4℃において回転させて、高分子量DNAおよび他の混入物を除去した。
V.上清をクリーンチューブに除去し、100μlの100%クロロホルムを加え、サンプルは15秒間手作業によって激しく混合し、次いで2〜3分間室温でインキュベートした。
VI.次いでサンプルを12,000×gで10分間4℃において回転させて相を分離した。
VII.上部水相をクリーンチューブに除去し、ピペットの先端が界面から離れた状態に保たれることを確実にし、1μlの20mg/mlのグリコーゲンを加え、サンプルをボルテックスした。
VIII.等体積の100%イソプロパノール(0.25ml)を加え、チューブをボルテックスし、次いで室温で10分間放置した。
IX.次いでサンプルを12,000×gで10分間4℃において回転させてRNAをペレット状にした。
X.上清を除去し、ペレットを0.75mlの80%エタノールで洗浄して、cDNA合成反応の阻害剤を除去し、軽くボルテックスし、次いで7,500×gで5分間4℃において回転させてRNAをペレット状にした。
XI.工程Xをさらに1回繰り返した。
XII.次いでペレットを微量遠心管中で10秒間回転させ、残留エタノールを除去し、ペレットは即座に25μlのRNaseを含まない水中に再懸濁した。NB。ペレットが乾燥する場合、したがってRNAを再懸濁することは非常に困難であり、260/280の比は1.6未満であり得る。
XIII.次いでOD260/280/310を記録し、必要となるまでRNAは−70℃で保存した。
【0110】
RNAの調製
望ましい細胞または組織からの抽出後、20μlのヌクレアーゼを含まない水中にRNAサンプルを再懸濁した。
【0111】
サンプルは60〜100℃で2〜3分間加熱し二次構造を分解して、亜硫酸水素塩の反応において即座に使用した。
【0112】
亜硫酸水素塩処理
本発明によるRNAの亜硫酸水素塩処理の有効性を実証する例示的なプロトコルを以下に述べる。このプロトコルによって首尾よく、実質的に全てのRNAが処理された状態となった。本発明のこの方法は、本明細書ではヒト遺伝子シグネチャー(HGS)方法とも呼ぶ。サンプルまたは試薬の体積または量は変えることができることは理解されよう。
【0113】
2μgのRNAを合計20μlのRNaseを含まない水中に再懸濁する。次いでサンプルを65℃で2分間インキュベートして、二次構造を除去した。インキュベーション後、208μlの2Mメタ重亜硫酸ナトリウムpH5.0(7.6g、20ml水または10mMトリス/1mMEDTAと416ml10NNaOH中;BDH AnalaR#10356.4D;新たに作製)を連続して加えた。製造者の説明書に従い、RNaseOUT(invitrogenカタログ番号10777−019)などのRNase阻害剤もこの時点で加えることができる。サンプルには200μlのミネラルオイルを重層した。ミネラルオイルの重層は試薬の蒸発および酸化を防ぐが、必須ではない。次いでサンプルは55℃で一晩インキュベートした。このインキュベーションは約37℃〜約90℃の任意の温度で実施することができ、かつ5分〜16時間まで長さを変えることができる。
【0114】
メタ重亜硫酸ナトリウムでの処理後、オイルを除去し、特にDNA濃度が低かった場合、1μlのグリコーゲン(20mg/ml)を加えた。この添加剤は任意選択であり、特にRNAが低い濃度で存在するとき、これらを使用して標的RNAとの共沈によって得られるRNAの収率を改善することができる。核酸のより有効な沈殿のための担体としての添加剤の使用は、核酸の量が<0.5μgであるとき一般に望ましい。
【0115】
イソプロパノール浄化処理を以下のように実施した:800μlのRNaseを含まない水をサンプルに加え、混合し、次いで1mlのイソプロパノールを加えた。水またはバッファーは、塩が対象の標的核酸と共に沈殿しないレベルまで、反応容器中の亜硫酸水素塩の濃度を低下させる。本明細書で開示する所望の範囲以下に塩濃度を希釈する限り、希釈は一般に約1/4〜1/1000である。
【0116】
サンプルは再度混合し、60分まで可能であるが、最短で5分間4℃に放置した。サンプルは微量遠心管中で10〜15分間回転させ、ペレットは80%ETOHで2回洗浄した。この洗浄処理によって、核酸と共に沈殿した任意の残留塩を除去する。
【0117】
ペレットは軽く放置乾燥させて残留エタノールを除去した、ただしこれによって最終的なRNA収率が低下する可能性があるので、ペレットが完全には乾燥しないことを確実にし、次いで50μlなどの適切な体積のT/E(10mMトリス/0.1mMEDTA)pH7.0〜12.5中に再懸濁した。製造者の説明書に従い、RNaseOUT(invitrogenカタログ番号10777−019)などのRNase阻害剤もこの時点で加えることができる。pH10.5でのバッファーは特に有効であることが分かっている。核酸を懸濁することが必要であったとき、サンプルは37℃〜95℃で1分間〜96時間インキュベートした。
【0118】
cDNA合成
以下の試薬を、薄壁0.5mlのRNaseを含まないチューブ中でのそれぞれのcDNA合成反応用に調製した。
RNA(1μg) 3.5μl
ランダムヘキサマー(10μM) 1μl
脱イオン水 2.5μl
【0119】
含有物を混合し、微量遠心管中で軽く回転させた。
【0120】
サンプルは70℃で3分間インキュベートして、RNAを変性させた。
【0121】
RNAを変性させていた一方で、以下のマスター混合物を調製した:
Per rxn
5×第一鎖用バッファー 2μl
DTT(20mM) 1μl
50×dNTP混合物 1μl
合計体積 4μl
【0122】
チューブをPCR機器から除去し、氷上で2分間冷却し、次いで軽く回転させて含有物を回収した。
【0123】
次いでサンプルを42℃で2分間インキュベートした。
【0124】
0.5μlのPowerscript逆転写酵素を反応混合物(1.75μl)当たりに加え、マスター混合物はピペッティングによってよく混合した。
【0125】
4.5μlの完全なマスター混合物をそれぞれのサンプルおよび対照チューブに加え、次いでサンプルを42℃で60分間インキュベートし、次いでサンプルを氷上に移した。
【0126】
40μlの10mMのトリス/1mMのEDTA pH7.6をそれぞれのサンプルに加えた。
【0127】
チューブは72℃で7分間加熱し、次いで必要となるまで−70℃で保存した。
【0128】
PCR増幅
PCR増幅は1μlの亜硫酸水素塩処理RNAで実施し、PCR増幅は、Promega PCRマスター混合物、6ng/μlのそれぞれのプライマーを使用して、1μlの亜硫酸水素塩処理ゲノムDNAを含む25μlの反応混合物で実施した。
【0129】
第1ラウンドの増幅の1μlを第2ラウンドの増幅の反応混合物に移した。PCR産物のサンプルは、Clarke et al中に記載された条件下において、ThermoHybaid PX2サーマルサイクラーで増幅した。
【0130】
【表2】
【0131】
アガロースゲル(2%)は、50mlのアガロース当たり1滴の臭化エチジウムを含む1%TAE(CLP#5450)で調製した。5μlのPCR由来産物を1μlの5×アガロースローディングバッファーと混合し、水中水平型電気泳動用タンクを使用して×1TAE中で125mAにおいて電気泳動にかけた。マーカーは低100〜1000bp型であった。Kodak UVldoc EDAS290系を使用してUV照射下で、ゲルを目に見える状態にした。
【0132】
ビーズを使用する検出系
磁気ビーズのコーティング
磁気ビーズとの結合に使用するINAは、幾つかの方法で修飾することができる。この例では、EDCなどのヘテロ二官能性リンカーを使用するビーズとINAの共有結合用に5’または3’アミノ基のいずれかをINAは含んでいた。しかしながら、INAはビオチンなどの5’基で修飾することも可能であり、次いでこれはアビジンまたはステプトアビジン(Steptavidin)基で修飾された磁気ビーズと受動的に結合し得る。
【0133】
10μlのカルボキシレート修飾Magnabind(商標)ビーズ(Pierce)または100μlのDynabeads(商標)ストレプトアビジン(Dynal)をクリーンな1.5mlのチューブに移し、90μlのPBS溶液を加えた。
【0134】
ビーズは混合し次いで磁化し、上清は捨てた。ビーズは洗浄1回当たり100μlのPBSで2回洗浄し、最後に90μlの50mMMESバッファーpH4.5、または製造者の仕様書によって決定した他のバッファー中に再懸濁した。
【0135】
1μlの250μMINA(オリゴハイブリダイゼーションの実験により決定した選択INAの特異的活性に依存する濃度)をサンプルに加え、チューブをボルテックスし室温で10〜20分間放置した。
【0136】
10μlの新たに調製した10mg/mlEDC溶液(Pierce/Sigma)を次いで加え、サンプルをボルテックスし、室温または4℃のいずれかにおいて60分までインキュベートする。
【0137】
次いでサンプルを磁化し上清を捨て、10分間0.25MのNaOHまたは0.5MのトリスpH8.0のいずれかを100μl加えることによってビーズを阻害することができる。
【0138】
次いでビーズをPBS溶液で2回洗浄し、最後に100μlのPBS溶液中に再懸濁した。
【0139】
磁気ビーズを使用するハイブリダイゼーション
10μlのINAコーティングMagnabind(商標)ビーズをクリーンチューブに移し、40μlの非希釈または蒸留水に1:1希釈のExpressHyb(商標)バッファー(Clontech)、または非希釈または蒸留水に1:1/1:2または1:4希釈のUltrahyb(商標)バッファー(Ambion)のいずれか、または室内用ハイブリダイゼーションバッファーを加えた。バッファーは、知られている濃度のカチオン性/アニオン性または両性洗浄剤のいずれか、またはヘパリンおよびポリアミノ酸などの他の添加剤を含むこともできる。
【0140】
次いでサンプルRNA1〜5μlを前述の溶液に加え、チューブをボルテックスし、20〜60分間選択したINA/RNAハイブリッドの融解温度に応じて、55℃または他の温度で次いでインキュベートした。
【0141】
サンプルを磁化し、上清は捨て、ビーズは洗浄1回当たり5分間の初期工程から、ハイブリダイゼーション温度において0.1×SSC/0.1%SDSで2回洗浄し、洗浄の間にサンプルを磁化した。
【0142】
放射標識した検出用球体の調製
INAまたはオリゴ分子は、アミン基、チオール基またはビオチンなどの分子で3’または5’のいずれかを標識することができる。
【0143】
標識分子は、第1標識に対して分子の逆末端に取り込まれたP32またはI125などの第2標識を有することもできる。
【0144】
この二重標識検出用分子は現在、EDCなどのヘテロ二官能性リンカーを使用して、知られている大きさのカルボキシレートまたは修飾ラテックスビーズと共有結合させることが可能である。
【0145】
次いで非結合分子を洗浄によって除去することができ、多数の特異的検出用/シグナル増幅分子でコーティングされたビーズを切り離すことができる。
【0146】
次いでこれらのビーズを対象の核酸サンプルとハイブリダイズさせて、シグナル増幅をもたらすことが可能である。
【0147】
蛍光標識した検出用球体の調製
INAまたはオリゴ分子は、アミン基、チオール基またはビオチンなどの分子で3’または5’のいずれかを標識することができる。
【0148】
標識分子は、第1標識に対して分子の逆末端に取り込まれたCy−3、Cy−5、FAM、HEX、TET、TAMRAまたは任意の他の適切な蛍光分子などの第2標識を有することもできる。
【0149】
この二重標識検出用分子は現在、EDCなどのヘテロ二官能性リンカーを使用して、知られている大きさのカルボキシレートまたは修飾ラテックスビーズと共有結合させることが可能である。
【0150】
次いで非結合分子を洗浄によって除去することができ、多数の特異的検出用/シグナル増幅分子でコーティングされたビーズを切り離すことができる。
【0151】
次いでこれらのビーズを対象のRNAサンプルとハイブリダイズさせて、シグナル増幅をもたらすことが可能である。
【0152】
酵素標識した検出用球体の調製
INAまたはオリゴ分子は、アミン基またはチオール基などの分子で3’または5’のいずれかを標識することができる。
【0153】
標識分子は、第1標識に対して分子の逆末端に、ヘテロ二官能性リンカーによって結合したビオチンまたはホースラディッシュペルオキシダーゼまたはアルカリホスファターゼなどの他の分子などの第2標識を有することもできる。
【0154】
この二重標識検出用分子は現在、EDCなどのヘテロ二官能性リンカーを使用して、知られている大きさのカルボキシレートまたは修飾ラテックスビーズと共有結合させることが可能である。
【0155】
次いで非結合分子を洗浄によって除去することができ、多数の特異的検出用/シグナル増幅分子でコーティングされたビーズを切り離すことができる。
【0156】
次いでこれらのビーズを対象の核酸サンプルとハイブリダイズさせて、シグナル増幅をもたらすことが可能である。
【0157】
次いで、シグナル増幅は、ストレプトアビジンなどの分子の結合または発色基質が関与する酵素反応によって実施することができる。
【0158】
INAオリゴマーの組合せ
初期ハイブリダイゼーション事象では、対象のRNAと相補的なINAでコーティングされた磁気ビーズを使用することが好ましい。
【0159】
必要とされる場合、第二のハイブリダイゼーション事象は、前述の検出法のいずれかを含むことができる。
【0160】
このハイブリダイゼーション反応は、対象の核酸と相補的な第二のINA、または対象のRNAと相補的なオリゴもしくは修飾オリゴのいずれかを用いて行うことができる。
【0161】
デンドリマーおよびアプタマー
デンドリマーは、特異的分子で標識した多層を生成することができるように調節した方法で化学的に合成することができる、枝分かれした樹状の分子である。デンドリマーは中心から末端に、またはその逆に段階的に合成した。
【0162】
デンドリマーの構造およびその生成に影響を与える最も重要なパラメーターの1つは、それぞれの工程で生成する枝の数であり、これが所望の分子を構築するのに必要とされる反復工程の数を決定する。
【0163】
I125またはP32などの放射標識、Cy−3、Cy−5、FAM、HEX、TET、TAMRAまたは任意の他の適切な蛍光分子などの蛍光標識を含むデンドリマーを合成して、シグナル増幅を促進することができる。
【0164】
あるいは、修飾INAまたはDNA分子と結合させるために使用することができるカルボキシレート基または任意の他の反応基を含む、デンドリマーを合成することができる。
【0165】
アレイを使用する検出系
処理したRNAを任意の適切な支持体につけて、対象の遺伝子または発現単位の活性に関してスクリーニングすることができるマイクロアレイなどのアレイを形成することができる。当業者は、適切なアレイを作製するのに適した技法に精通しているはずである。
【実施例】
【0166】
図1A、図1Bおよび図1Cは、本発明(亜硫酸水素塩処理RNA)と典型的なマイクロアレイベースのアッセイを使用する従来技術の比較を示す。図1Aは、典型的なマイクロアレイベースのアッセイの説明を示すが、この場合暗色点は特定のRNA集団中でアップレギュレーションされる遺伝子を示し、かつ淡色点は同じ集団中でダウンレギュレーションされる遺伝子を示す。暗色および淡色の矢印は、従来系中の検出分子がそれらの標的と結合するのを妨げる二次構造のため、従来の方法ではなく亜硫酸水素塩処理RNAにおいて、それぞれアップレギュレーションおよびダウンレギュレーションされるとして検出される遺伝子を示す。
【0167】
図1Bは、図1Aと同様のマイクロアレイベースのアッセイを示すが、暗色および淡色の矢印は、発現分析前の酵素によるRNAの操作中に生じRNA発現レベルの不正確な決定をもたらす偏りのため、従来の方法ではなく亜硫酸水素塩処理RNAにおいて、それぞれアップレギュレーションおよびダウンレギュレーションされるとして検出される遺伝子を示す。酵素による操作は誤解を与える結果を引き起こし、特定の遺伝子は、それらが実際そのように制御されないときにアップまたはダウンレギュレーションされることを示し得る。
【0168】
図1Cは、図1Aと同様のマイクロアレイベースのアッセイを示すが、暗色および淡色の矢印は、検出分子の改善された特異性のため、従来の方法ではなく亜硫酸水素塩処理RNAにおいて、それぞれアップレギュレーションおよびダウンレギュレーションされるとして検出される遺伝子を示す。検出分子の特異的結合強度の増大は、特異性の欠如のため従来の方法を使用して検出することができないRNA種の検出をもたらす。
【0169】
アクチン
既に記載したように、RNAを抽出および精製し、次いで亜硫酸水素塩処理および増幅した。増幅後、製造者によって指示されたようにMarligen PCRクリーンアップキットを使用してPCR産物を精製し、20μlの水中に再懸濁した。100ngの逆方向プライマーを10μlのPCR産物に加え、これらのサンプルはDNA塩基配列決定用にSupermac(Camperdown、Sydney)に送った。
【0170】
図2は、細胞系物質由来の亜硫酸水素塩修飾した全RNAを使用する逆転写酵素のPCRを示す。この図から見ることができるように、アクチン遺伝子のエクソン3a〜4またはエクソン3a〜3bを対象とする野生型プライマー(非亜硫酸水素塩転換型)を使用すると、いずれの場合も増幅産物は得られない。これはRNAの転換は非常に有効であることを示すが、これは、野生型配列を検出することができないからである。逆に、亜硫酸水素塩転換型プライマーを同じサンプルでエクソン3a〜4またはエクソン3a〜3bに使用する場合、異なるPCRバンドが生成し、亜硫酸水素塩転換型物質から特定のmRNAを増幅することが可能であることを示す。
【0171】
図3は、亜硫酸水素塩転換型RNAから生成したPCR産物の直接的な塩基配列決定を示す。塩基配列決定プロファイルから見ることができるように、PCR産物は混入DNAではなくRNAに由来するが、これは、これらの産物の配列がエクソン3と4の間のスプライス部位にわたって続くからである。さらに、図3から見ることができるように、PCR産物は完全に転換されているが、これは、サンプル中の元のC残基がここではT残基に転換されるからである。
【0172】
C型肝炎ウイルス
C型肝炎ウイルス(HCV)のRNAサンプルは、Acrometrix(OptiQual HCV高陽性対照)またはBBI diagnostics(HCV RNA直線性パネル)から得て、製造者の説明に従いUltrasensウイルス精製キットを用いて精製した。サンプルは亜硫酸水素ナトリウムで処理し、転換型HCVのRNAサンプルは以下のようにSuperscriptIII逆転写酵素(Invitrogen)を用いて逆転写した:
11μlの転換型RNA鋳型
1μlのランダムプライマー(300ng/μl)
1μlのdNTP(10mM)
【0173】
サンプルは65℃で5分間加熱し、次いで少なくとも1分間氷上に即座に置き、その後以下の試薬を加えた:
4μlの5×第一鎖用バッファー
1μlのRNaseOUT(40U/μl)
1μlのDTT(100mM)
1μlのSuperscriptIII(200U/μl)
【0174】
サンプルは以下の条件を使用して逆転写した:
25℃、12分間
27℃、2分間
29℃、2分間
31℃、2分間
33℃、2分間
35℃、2分間
37℃、30分間
45℃、15分間
50℃、5分間
75℃、5分間
【0175】
次いで2μlのcDNAを、HCVの5’NTRに特異的なプライマーおよびプローブを用いてPCRで増幅した:
(順方向プライマー−ttatgtagaaagtgtttagttatggtgt(配列番号9);
逆方向プライマー−acccaaatytccaaayattaaacaaat(配列番号10);
プローブ−tcCacAaaCcaCtaTaaCtcTcc(配列番号11)、
(前式で大文字はLNAの存在を示す)、Corbett 6000 Rotor Geneにおいて以下の試薬およびサイクル条件を使用した:
Sigma Jumpstart 2×マスター混合物 12.5μl
順方向プライマー 50ng
逆方向プライマー 50ng
25mMのMgCl2 3.5μl
400nMのプローブ(最終濃度) ×μl
23μlまでの水 ×μl
95℃、10分間
95℃、10秒間(50×)
53℃、90秒間(50×)
60℃、30秒間
【0176】
結果は図4中に示し、この場合図4から見ることができるアッセイに関する検出限界は約2.5IUのウイルスである。
【0177】
【表3】
【0178】
図4中に示したゲルの結果は、亜硫酸水素塩の方法を使用して、2.5IUのHCVまで低い広範囲の標的濃度におけるHCVの発現レベルをモニタリングすることができることを示す。
nc=陰性対照
【0179】
HCVに関する直線性パネルの滴定のリアルタイムqPCRによる結果は図5中に示す。このプロットに関する標準曲線は、0.99947のR値および0.9989のR2値を有する直線であった。
【0180】
【表4】
【0181】
HCVに関するダイナミックレンジの滴定に関する定量化の報告のリアルタイムqPCRによる結果は図6中に示す。このプロットに関する標準曲線は、0.99856のR値および0.99713のR2値を有する直線であった。
【0182】
【表5】
【0183】
直線性パネルおよびダイナミックレンジのサンプルからの結果は、リアルタイムPCR中に作成した定量曲線を示す。曲線のラインが閾値と交差する地点はCt値として知られ、サンプルの定量化に使用する。サンプルのそれぞれのセットに関して3桁を超える、一連の知られている濃度のウイルスを精製し、亜硫酸水素塩転換および増幅し、作成した標準曲線は、1に近いR2値によって例示されるように、反応効率は調べた濃度範囲で一定で直線的であることを示す。これらの結果および図4中に示した結果は、156250IUから1.5IUまでの範囲で、ウイルス特異的プローブを使用する終点PCRおよびリアルタイムPCRを使用すると、HCVウイルスRNAの検出に関して優れた感度および特異性が存在することを実証し、このアッセイは、非常に広範囲の濃度でウイルス遺伝子の発現を検出することができることを示す。
【0184】
広く記載する本発明の精神または範囲から逸脱せずに、具体的な実施形態中に示すように本発明に対して多数の変形および/または変更をなすことができることは、当業者によって理解されよう。したがって、本発明の実施形態は全ての点で例示的であり、かつ制限的ではないとみなすべきである。
【特許請求の範囲】
【請求項1】
RNAをRNAの二次構造を実質的に除去する作用物質で処理することと、
処理したRNAの存在または量を測定して遺伝子発現の指標を得ることと
を含む遺伝子発現のアッセイ。
【請求項2】
オリゴヌクレオチド、PNA、LNAまたはINAプローブを使用して前記RNAを測定する、請求項1に記載のアッセイ。
【請求項3】
前記プローブが塩基A(アデニン)、T(チミン)およびC(シトシン)を実質的に含む、請求項2に記載のアッセイ。
【請求項4】
前記プローブがINAプローブである、請求項2または3に記載のアッセイ。
【請求項5】
前記RNAがmRNAである、請求項1から4のいずれか一項に記載のアッセイ。
【請求項6】
前記RNAが動物、植物、微生物、細胞、細胞群または細胞集団由来である、請求項1から4のいずれか一項に記載のアッセイ。
【請求項7】
前記微生物がウイルスまたは細菌である、請求項6に記載のアッセイ。
【請求項8】
前記作用物質が、亜硫酸水素塩、ヒドロキシルアミン、酢酸塩またはクエン酸塩から選択される、請求項1から7のいずれか一項に記載のアッセイ。
【請求項9】
前記作用物質が亜硫酸水素ナトリウムである、請求項8に記載のアッセイ。
【請求項10】
RNAの二次構造を実質的に除去する作用物質でRNAを処理することと、
RNAの相補配列と結合することができるプライマーを使用してRNAを逆転写および増幅することと、
処理および増幅したRNAの存在または量を測定して遺伝子発現の指標を得ることと
を含む遺伝子発現のアッセイ。
【請求項11】
前記RNAがmRNAである、請求項10に記載のアッセイ。
【請求項12】
前記RNAが動物、植物、微生物、細胞、細胞群または細胞集団由来である、請求項11に記載のアッセイ。
【請求項13】
前記微生物がウイルスまたは細菌である、請求項12に記載のアッセイ。
【請求項14】
前記作用物質が、亜硫酸水素塩、ヒドロキシルアミン、酢酸塩またはクエン酸塩から選択される、請求項13に記載のアッセイ。
【請求項15】
前記作用物質が亜硫酸水素ナトリウムである、請求項14に記載のアッセイ。
【請求項16】
RNAの二次構造を実質的に除去する作用物質を用いる遺伝子発現のアッセイにおけるオリゴヌクレオチド、PNA、LNAまたはINAプローブの使用。
【請求項17】
前記プローブが塩基A(アデニン)、T(チミン)およびC(シトシン)を実質的に含む、請求項16に記載の使用。
【請求項18】
前記プローブがG(グアニン)を実質的に含まない、請求項16に記載の使用。
【請求項19】
前記作用物質が、亜硫酸水素塩、ヒドロキシルアミン、酢酸塩またはクエン酸塩から選択される、請求項16から18のいずれか一項に記載の使用。
【請求項20】
前記作用物質が亜硫酸水素ナトリウムである、請求項18に記載の使用。
【請求項1】
RNAをRNAの二次構造を実質的に除去する作用物質で処理することと、
処理したRNAの存在または量を測定して遺伝子発現の指標を得ることと
を含む遺伝子発現のアッセイ。
【請求項2】
オリゴヌクレオチド、PNA、LNAまたはINAプローブを使用して前記RNAを測定する、請求項1に記載のアッセイ。
【請求項3】
前記プローブが塩基A(アデニン)、T(チミン)およびC(シトシン)を実質的に含む、請求項2に記載のアッセイ。
【請求項4】
前記プローブがINAプローブである、請求項2または3に記載のアッセイ。
【請求項5】
前記RNAがmRNAである、請求項1から4のいずれか一項に記載のアッセイ。
【請求項6】
前記RNAが動物、植物、微生物、細胞、細胞群または細胞集団由来である、請求項1から4のいずれか一項に記載のアッセイ。
【請求項7】
前記微生物がウイルスまたは細菌である、請求項6に記載のアッセイ。
【請求項8】
前記作用物質が、亜硫酸水素塩、ヒドロキシルアミン、酢酸塩またはクエン酸塩から選択される、請求項1から7のいずれか一項に記載のアッセイ。
【請求項9】
前記作用物質が亜硫酸水素ナトリウムである、請求項8に記載のアッセイ。
【請求項10】
RNAの二次構造を実質的に除去する作用物質でRNAを処理することと、
RNAの相補配列と結合することができるプライマーを使用してRNAを逆転写および増幅することと、
処理および増幅したRNAの存在または量を測定して遺伝子発現の指標を得ることと
を含む遺伝子発現のアッセイ。
【請求項11】
前記RNAがmRNAである、請求項10に記載のアッセイ。
【請求項12】
前記RNAが動物、植物、微生物、細胞、細胞群または細胞集団由来である、請求項11に記載のアッセイ。
【請求項13】
前記微生物がウイルスまたは細菌である、請求項12に記載のアッセイ。
【請求項14】
前記作用物質が、亜硫酸水素塩、ヒドロキシルアミン、酢酸塩またはクエン酸塩から選択される、請求項13に記載のアッセイ。
【請求項15】
前記作用物質が亜硫酸水素ナトリウムである、請求項14に記載のアッセイ。
【請求項16】
RNAの二次構造を実質的に除去する作用物質を用いる遺伝子発現のアッセイにおけるオリゴヌクレオチド、PNA、LNAまたはINAプローブの使用。
【請求項17】
前記プローブが塩基A(アデニン)、T(チミン)およびC(シトシン)を実質的に含む、請求項16に記載の使用。
【請求項18】
前記プローブがG(グアニン)を実質的に含まない、請求項16に記載の使用。
【請求項19】
前記作用物質が、亜硫酸水素塩、ヒドロキシルアミン、酢酸塩またはクエン酸塩から選択される、請求項16から18のいずれか一項に記載の使用。
【請求項20】
前記作用物質が亜硫酸水素ナトリウムである、請求項18に記載の使用。
【図1A】

【図1B】
【図1C】
【図2】
【図3】
【図4】

【図5】
【図6】
【図1B】
【図1C】
【図2】
【図3】
【図4】
【図5】
【図6】
【公表番号】特表2010−521142(P2010−521142A)
【公表日】平成22年6月24日(2010.6.24)
【国際特許分類】
【出願番号】特願2009−552978(P2009−552978)
【出願日】平成20年3月14日(2008.3.14)
【国際出願番号】PCT/AU2008/000367
【国際公開番号】WO2008/113111
【国際公開日】平成20年9月25日(2008.9.25)
【出願人】(505188906)ヒューマン ジェネティック シグネチャーズ ピーティーワイ リミテッド (15)
【Fターム(参考)】
【公表日】平成22年6月24日(2010.6.24)
【国際特許分類】
【出願日】平成20年3月14日(2008.3.14)
【国際出願番号】PCT/AU2008/000367
【国際公開番号】WO2008/113111
【国際公開日】平成20年9月25日(2008.9.25)
【出願人】(505188906)ヒューマン ジェネティック シグネチャーズ ピーティーワイ リミテッド (15)
【Fターム(参考)】
[ Back to top ]