
遺伝子発現制御方法、それに用いる遺伝子組換え用ベクターおよび遺伝子発現制御キット
【課題】 細胞に対する悪影響の恐れがなく、簡単なシステムで実施可能な遺伝子発現制御方法を提供する。
【解決手段】 細胞内の目的遺伝子の転写を調節することで前記目的遺伝子の発現を制御する遺伝子発現制御方法であって、前記目的遺伝子Gの上流に、プロモーター配列Pを配置し、前記プロモーター配列Pの上流に、5’−TGACGT−3’の結合配列を含む応答配列Bを配置し、前記細胞内において、AUREO1タンパク質を存在させ、前記AUREO1タンパク質は、青色光の照射下では、前記結合配列に結合し、青色光の非照射下では、前記結合配列から遊離し、前記青色光の照射または非照射によるAUREO1タンパク質の前記結合配列への結合または遊離により、前記プロモーター配列Pの活性または不活性を調節することで、前記目的遺伝子Gの転写を調節することを特徴とする。
【解決手段】 細胞内の目的遺伝子の転写を調節することで前記目的遺伝子の発現を制御する遺伝子発現制御方法であって、前記目的遺伝子Gの上流に、プロモーター配列Pを配置し、前記プロモーター配列Pの上流に、5’−TGACGT−3’の結合配列を含む応答配列Bを配置し、前記細胞内において、AUREO1タンパク質を存在させ、前記AUREO1タンパク質は、青色光の照射下では、前記結合配列に結合し、青色光の非照射下では、前記結合配列から遊離し、前記青色光の照射または非照射によるAUREO1タンパク質の前記結合配列への結合または遊離により、前記プロモーター配列Pの活性または不活性を調節することで、前記目的遺伝子Gの転写を調節することを特徴とする。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、遺伝子発現制御方法、それに用いる遺伝子組換え用ベクターおよび遺伝子発現制御キットに関する。
【背景技術】
【0002】
350〜500nmの波長光(以下、青色光という)は、多くの植物種において非常に重要な環境シグナルの1つである。例えば、陸上植物では、光屈性や気孔開口のシグナルとして青色光が利用されている。この現象には、光受容体と呼ばれるタンパク質がはたらいている。このような植物の青色光の光受容体タンパク質には、クリプトクロム、フォトトロピン(phot1・phot2)、ADO/FKF/LKP/ZTLファミリーなどがある。また、これらの青色光の光受容体タンパク質は、光センサードメインと制御ドメインを有することが知られている。例えば、光受容体タンパク質である前記フォトトロピンには、光センサードメインである2つのLOV(Light Oxygen Voltage)ドメインと、制御ドメインである1つのプロテインキナーゼドメインが存在する。このうち、光センサードメインであるLOVドメインは、フラビンモノヌクレオチド(FMN)を色素団として持ち、そのFMNが青色光を受容することで、フォトトロピンの立体構造が変化し、フォトトロピンのプロテインキナーゼが活性化されることにより、シグナルを下流分子に伝えるとされている。
【0003】
これまでに、このような青色光によるタンパク質−タンパク質間の相互作用に基づいた遺伝子発現制御方法として、シロイヌナズナの青色光受容体であるLKP1(LOV kelch protein1)等を用いた青色光バイオスイッチが開発されている(特許文献1および2)。また、化学物質を用いた遺伝子の発現制御方法も開発されている(特許文献3)。化学物質を用いた遺伝子の発現制御方法は、化学物質により細胞への悪影響を及ぼす可能性がある。一方、青色光を用いた遺伝子制御方法は、細胞への悪影響がない、優れた方法であるが、前述のように、タンパク質間の相互作用に基づいているために、システムが複雑で、種々の要因の影響を受ける恐れがある。
【特許文献1】特開2006−94704号公報
【特許文献2】特開2007−75019号公報
【特許文献3】特表平11−506901号公報
【発明の開示】
【発明が解決しようとする課題】
【0004】
そこで、本発明は、簡単なシステムで、細胞に悪影響を及ぼす恐れの無い遺伝子発現制御方法を提供することを目的とする。
【課題を解決するための手段】
【0005】
前記目的を達成するために、本発明の遺伝子発現制御方法は、
細胞内の目的遺伝子の転写を調節することで前記目的遺伝子の発現を制御する遺伝子発現制御方法であって、
前記目的遺伝子の上流に、プロモーター配列を配置し、
前記プロモーター配列の上流に、5’−TGACGT−3’の結合配列を含む応答配列を配置し、
前記細胞内において、AUREO1タンパク質を存在させ、
前記AUREO1タンパク質は、青色光の照射下では、前記結合配列に結合し、青色光の非照射下では、前記結合配列から遊離し、
前記青色光の照射または非照射によるAUREO1タンパク質の前記結合配列への結合または遊離により、前記プロモーター配列の活性または不活性を調節することで、前記目的遺伝子の転写を調節することを特徴とする。
【発明の効果】
【0006】
本発明者らは、前記目的を達成するために一連の研究を重ねた。その過程で、藻類の一種であるフシナシミドロから、青色光の照射により、前記特定の塩基配列に結合し、かつ青色光の非照射により、前記特定の塩基配列から遊離するという性質をもつタンパク質であるAUREO1を発見し、分離した。そして、さらに研究を重ねた結果、前記AUREO1を利用すれば、タンパク質間の相互作用無しで、青色光の照射または非照射により、遺伝子の転写を調節できることを見出し、本発明に到達した。したがって、本発明の遺伝子発現制御方法は、化学物質ではなく、青色光を用いるため、細胞への悪影響の恐れがない。また、本発明の遺伝子発現制御方法は、タンパク質間の相互作用を利用していないため、システムが簡単であり、種々の要因による影響も低減可能である。
【発明を実施するための最良の形態】
【0007】
本発明の遺伝子発現制御方法は、AUREO1の結合により遺伝子の転写を開始させる第一の方法と、AUREO1の遊離により遺伝子の転写を開始させる第二の方法がある。
【0008】
本発明の遺伝子発現制御方法の第一の方法は、
青色光の照射により、前記AUREO1タンパク質を、前記応答配列に結合させることにより前記プロモーター配列を活性化して前記目的遺伝子の転写を実施し、
前記青色光の非照射により、前記AUREO1タンパク質を前記結合配列から遊離させることにより前記目的遺伝子の転写を実施しないという方法である。
前記第一の方法では、AUREO1自身が、プロモーター配列を活性化するアクチベーターとして機能する。
【0009】
本発明の遺伝子発現制御方法の第二の方法は、
前記細胞内に、さらに、アクチベーターを存在させ、
前記アクチベーターは、前記応答配列に結合することにより前記プロモーター配列を活性化し、
青色光の照射により、前記AUREO1タンパク質を前記結合配列に結合させることで、
前記アクチベーターの前記応答配列への結合を阻止して前記目的遺伝子の転写を実施せず、
前記青色光の非照射により、前記AUREO1タンパク質を前記結合配列から遊離させることで、前記アクチベーターを前記応答配列に結合させて前記目的遺伝子の転写を実施するという方法である。
前記第二の方法では、AUREO1が、プロモーターを不活性化するリプレッサーとして機能する。
【0010】
本発明において、前記AUREO1タンパク質は、例えば、下記の(A)、(B)または(C)のタンパク質である。
(A) 配列番号2のアミノ酸配列からなるタンパク質
(B) 配列番号2のアミノ酸配列において、1個または数個のアミノ酸残基が、欠失、置換または付加されたアミノ酸配列からなり、青色光の照射下では、前記結合配列に結合し、青色光の非照射下では、前記結合配列から遊離するタンパク質。
(C) 配列番号2のアミノ酸配列に対し、68%以上の相同性を有するアミノ酸配列からなり、青色光の照射下では、前記結合配列に結合し、青色光の非照射下では、前記結合配列から遊離するタンパク質である。
前記(B)のタンパク質は、配列番号2のアミノ酸配列において、例えば、1〜10個、好ましくは1〜5個、より好ましくは1〜3個のアミノ酸残基が、欠失、置換または付加されたアミノ酸配列からなり、青色光の照射下では、前記結合配列に結合し、青色光の非照射下では、前記結合配列から遊離するタンパク質である。
また、前記(C)のタンパク質は、配列番号2のアミノ酸配列に対し、例えば、68%以上、好ましくは80%以上、より好ましくは90%以上の相同性を有するアミノ酸配列からなり、青色光の照射下では、前記結合配列に結合し、青色光の非照射下では、前記結合配列から遊離するタンパク質である。
【0011】
本発明において、前記応答配列は、5’−TGACGT−3’(配列番号1)の結合配列に加えまたは代えて、5’−TGACGC−3’の結合配列を含むという態様であってもよい。ただし、本発明では、5’−TGACGT−3’の結合配列が好ましい。
【0012】
本発明において、前記青色光が、波長350nmから500nmの範囲の光を含むことが好ましい。本発明において、青色光は、特に制限されず、青色光を含めばよく、例えば、太陽光、人工的光源(例えば、蛍光灯、白熱球、ハロゲンランプ、発光ダイオード、有機EL素子)等の光であってもよい。
【0013】
本発明において、前記細胞は、細菌、酵母、動物細胞および植物細胞からなる群から選択される少なくとも一つの細胞であることが好ましい。
【0014】
つぎに、本発明の遺伝子組換え用ベクターは、本発明の遺伝子発現制御方法に使用する遺伝子組換え用ベクターであって、
前記目的遺伝子のクローニングサイト、前記プロモーター配列および前記応答配列を含み、
前記目的遺伝子のクローニングサイトの上流に、前記プロモーター配列が配置され、
前記プロモーター配列の上流に、前記応答配列が配置されていることを特徴とする。
【0015】
本発明の遺伝子組換え用ベクターにおいて、さらに、AUREO1遺伝子を含むという態様であってもよい。
【0016】
つぎに、本発明の遺伝子発現制御用キットは、本発明の遺伝子発現制御方法に使用するキットであって、本発明の遺伝子組換え用ベクターを含むキットである。
【0017】
本発明の遺伝子発現制御用キットにおいて、さらに、AUREO1遺伝子を発現可能な状態で含む遺伝子組み換え用ベクターを含むという態様であってもよい。
【0018】
つぎに、本発明について、詳細に説明する。
【0019】
(A) AUREO1
前述のように、AUREO1は、藻類の一種であるフシナシミドロの細胞内に発現する光受容体タンパク質である。光受容体タンパク質は、光を受容することにより、その立体構造を変化させ、その変化を細胞内の生化学反応に変換する機能を有するタンパク質である。AUREO1は、そのファミリータンパク質であるAUREO2と共に、本発明者等によりフシナシミドロから発見され、分離された。図2に、AUREO1およびAUREO2の構造の模式図を示す。図示のように、両タンパク質共に、一つのLOVドメインと、一つのbZIPドメインを有する。「LOVドメイン」とは、青色光センサー(Light)、酸素センサー(Oxygen)、電位依存カリウムチャンネル(Voltage)の各センサー部分に特異的な立体構造を持つドメインという意味で命名された青色光の光受容体タンパク質が有する発色団結合ドメインであり、前記発色団として、1分子のFMNを非共有結合により有している。LOVドメインを有する光受容体タンパク質は、AUREO1および2の他に、例えば、phot1、phot2、phy3、ADO/FKF/LKP/ZTLファミリータンパク質、TLP1/PLPなどがある。「bZIPドメイン」は、遺伝子の転写を制御するタンパク質のDNA結合ドメインで、DNA結合および二量体化に関与し、コア配列を含有するDNA配列に共通して結合する、いわゆる塩基性ロイシンジッパードメインをいう。
【0020】
AUREO1タンパク質は、例えば、配列番号2のアミノ酸配列からなるタンパク質であり、その分子量は、38.6kDである。AUREO1タンパク質は、青色光を照射すると、そのLOVドメインが、システインを介してFMNに結合し、bZIPドメインが、5’−TGACGT−3’(配列番号1)の配列に可逆的に結合する。また、青色光の非照射の条件下(例えば、暗黒下)では、前記配列から遊離する。一方、AUREO2は、配列番号3のアミノ酸配列からなるタンパク質であり、その分子量が39.1kDである。AUREO2は、AUREO1と異なり、2箇所にイントロンを有している。AUREO1遺伝子の塩基配列は、アクセッション番号AB252504として、AUREO2遺伝子の塩基配列は、アクセッション番号AB252505(mRNA)およびアクセッション番号AB272981(gene)として、それぞれDDBJに登録されている。
【0021】
(B) 本発明の遺伝子発現制御方法の第一の方法
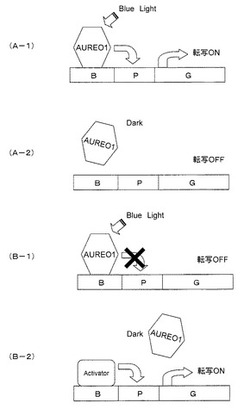
本発明の遺伝子発現制御方法の第一の方法について、図1(A−1)および(A−2)に基づき説明する。前述のように、前記第一の方法では、AUREO1が、プロモーター配列を活性化するアクチベーターとして機能する。図示のように、目的遺伝子Gの上流に、プロモーター配列Pを配置し、前記プロモーター配列Pの上流に、前記応答配列Bを配置する。前記応答配列は、5’−TGACGT−3’の結合配列を含む。前記目的遺伝子G、プロモーター配列Pおよび応答配列Bは、例えば、遺伝子組換え用ベクターを用いて細胞に導入することができる。一方、細胞内に、AUREO1タンパク質を存在させる。AUREO1タンパク質を細胞内に存在させる方法としては、例えば、AUREO1遺伝子を組み込んだベクターを細胞内に導入し、前記遺伝子を発現させるという方法がある。前記AUREO1遺伝子は、前記目的遺伝子Gを組み込んだベクターに組み込んでもよいし、これとは別のベクターに組み込んでもよい。そして、図1(A−1)に示すように、青色光(Blue Light)を照射すると、AUREO1が前記結合配列を特異的に認識して結合し、この結果、前記応答配列Bがプロモーター配列Pを活性化する。プロモーター配列Pの活性化により、目的遺伝子Gの転写が開始される(転写ON)。これとは逆に、青色光の非照射下(例えば、暗黒(Dark)下)では、AUREO1タンパク質は、前記結合配列から遊離しているため、プロモーター配列Pは不活性の状態であり、目的遺伝子は転写されない(転写OFF)。
【0022】
(C) 本発明の遺伝子発現制御方法の第二の方法
本発明の遺伝子発現制御方法の第二の方法について、図1(B−1)および(B−2)に基づき説明する。前述のように、前記第二の方法では、AUREO1が、リプレッサーとして機能し、プロモーター配列を活性化するアクチベーターが応答配列に結合するのを阻止する。したがって、前記アクチベーターは、例えば、AUREO1タンパク質と競合するか、もしくは、青色光の照射下で応答配列から離れる性質を持つ。図示のように、第一の方法と同様に、目的遺伝子Gの上流に、プロモーター配列Pを配置し、前記プロモーター配列Pの上流に、前記応答配列Bを配置する。前記応答配列は、5’−TGACGT−3’の結合配列を含む。前記目的遺伝子G、プロモーター配列Pおよび応答配列Bは、例えば、遺伝子組換え用ベクターを用いて細胞に導入することができる。一方、細胞内に、AUREO1タンパク質およびアクチベーターを存在させる。AUREO1タンパク質およびアクチベーターを細胞内に存在させる方法としては、例えば、AUREO1遺伝子およびアクチベーター遺伝子を組み込んだベクターを細胞内に導入し、前記両遺伝子を発現させるという方法がある。前記AUREO1遺伝子およびアクチベーター遺伝子は、前記目的遺伝子Gを組み込んだベクターに組み込んでもよいし、これとは別のベクターに組み込んでもよい。前記AUREO1遺伝子およびアクチベーター遺伝子は、それぞれ別のベクターに組み込んでもよい。そして、図1(B−1)に示すように、青色光(Blue Light)を照射すると、AUREO1が前記結合配列を特異的に認識して結合し、この結果、アクチベーターが前記応答配列Bに結合できず、プロモーター配列Pは不活性の状態となり、目的遺伝子Gは転写されない(転写OFF)。これとは逆に、青色光の非照射下(例えば、暗黒(Dark)下)では、AUREO1タンパク質は、前記結合配列から遊離しているため、アクチベーターが応答配列Bに結合することができ、この結果、プロモーター配列Pが活性化され、目的遺伝子の転写が開始される(転写ON)。
【0023】
前記プロモーターとしては、例えば、酵母One−hybrid法などに用いられるGAL4遺伝子のミニマムプロモーター領域や、植物で利用されるカリフラワーモザイクウィルス35S遺伝子のミニマムプロモーター領域などが、本発明の前記遺伝子発現制御方法の第一および第二の方法における遺伝子の発現制御において使用可能である。
【実施例】
【0024】
つぎに、本発明の実施例について説明する。
【0025】
(1)DNAおよびRNAの分離
淡水黄緑藻類であるオカフシナシミドロの葉状体を三角フラスコ内の新鮮な培地に移し、白色蛍光ランプ下、20℃で2−3ヶ月間培養した(12時間明条件/12時間暗条件)。培養終了後、培養物のゲノムDNAおよびtotalRNAをセチルトリメチルアンモニウムブロマイド(CTAB)法を用いて単離した。一方、ヒバマタの成熟した生殖器床は、2006年5月下旬に日本の北海道室蘭で採取し、受精後4−6時間後に受精した接合子を回収した。接合子は、直ちにDNA/RNA抽出に用いるか、−80℃で使用時まで保存した。オカフシナシミドロおよびヒバマタのいずれも、DNAおよびRNAは、試料に界面活性剤を含む破砕液を加え、試験管を洗浄するブラシを上下することで細胞を十分に破砕する「ウォッシュブラッシュ」法(Nozaki H, Ito M, Watanabe, MM, Takano H, Kuroiwa T.,“PHYLOGENETIC ANALYSIS OF MORPHOLOGICAL SPECIES OF CARTERIA (VOLVOCALES,CHLOROPHYTA) BASED ON rbcL GENE SEQUENCES”J Phycol 33:864−867(1997))により抽出した。ウォッシュブラッシュ法は、細胞を十分に破砕することでm−RNAやDNAの収量が高まることから、微細な細胞や卵などからm−RNAやDNAを単離する際に用いる方法である。
【0026】
(2)フシナシミドロのLOVドメイン含有タンパク質のクローニング
LOVドメイン含有タンパク質は、縮重プライマー(LOV−F(配列番号4)およびLOV−R(配列番号5))を用いて単離した。PCR生成物は、pGEM-T Easy system(Promega)によりクローニングし、シークエンスを行った。完全長cDNAは、5’および3’RACEにより得た。プライマーには、5’RACE−1−1(配列番号6)、5’RACE−1−2(配列番号7)、5’RACE−1−3(配列番号8)、3’RACE−1−1(配列番号9)、3’RACE−1−2(配列番号10)、5’RACE−2−1(配列番号11)、5’RACE−2−2(配列番号12)、5’RACE−2−3(配列番号13)、3’RACE−2−1(配列番号14)、3’RACE−2−2(配列番号15)を用いた。さらに、特異的プライマー(AUREO1−Full−F(配列番号16)、AUREO1−Full−R(配列番号17)、AUREO2−Full−F(配列番号18)、AUREO2−Full−R(配列番号19))を用いたRT−PCR法により配列を決定した。こうして得た、LOVドメインを有するタンパク質をオーレオクロム(AUREOCHROME)と命名した。図2に示すように、オーレオクロムは、LOVドメインとbZIPドメインを各々1つ有するタンパク質で、AUREO1(配列番号2)とAUREO2(配列番号3)という2つの相同体が存在する。AUREO1において、例えば、配列番号2の第118番から第178番までの配列がbZIPドメインであり、配列番号2の第217番から第320番までの配列がLOVドメインである。また、AUREO2において、配列番号3の第124番から第185番までの配列がbZIPドメインであり、配列番号3の第224番から第327番までの配列がLOVドメインである。また、ヒバマタおよび珪藻においても、高い相同性を有するLOVドメインおよびbZIPドメインの存在が確認された。
【0027】
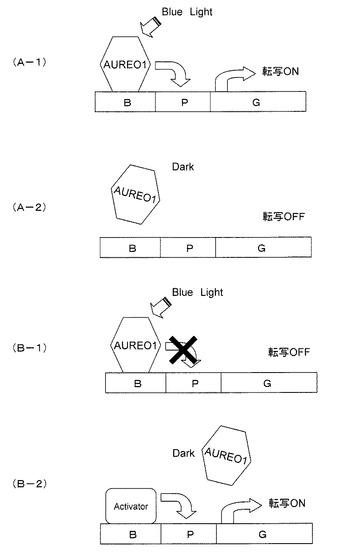
(3)光化学分析
オーレオクロムのフラビン結合能の確認のため、カルモデュリン結合ペプチド(CBP)とAUREO1のLOVドメインとの融合タンパク質と、CBPとAUREO2のLOVドメインとの融合タンパク質を作製し、大腸菌に発現させた。CBP−LOV融合タンパク質の異種発現は、pCAL−n−EK(Stratagene社製)ベクターを用いて大腸菌Rosetta(DE3)pLysS(Novagen社製)を形質転換させて行った。オーレオクロムのLOV(LOVAUREO1およびLOVAUREO2)のcDNA断片は、AUREO1−LOVF−AUREO2−LOVRのプライマーセット(AUREO1−LOVF(配列番号20)、AUREO1−LOVR(配列番号21)、AUREO2−LOVF(配列番号22)、AUREO2−LOVR(配列番号23))を用いてクローニングして得た。得られた形質転換体をLB培地(1.0L)で30℃の暗条件下、3時間培養した。融合タンパク質の発現は、イソプロピル−1−チオ−β−D−galactoside(IPTG)の添加により誘導した。得られた細胞を遠心分離して回収し、使用時まで−20℃で保存した。タンパク質の精製およびクロムフォアの決定は、文献(Kasahara M, Swartz TE, Olney MA, Onodera A, Mochizuki N, Fukuzawa H, Asamizu E, Tabata S, Kanegae H, Takano M et al.,“Photochemical Properties of the Flavin Mononucleotide−Binding Domains of the Phototropins from Arabidopsis, Rice, and Chlamydomonas reinhardtii”Plant Physiol 129:762−773(2002))に従った。概略としては、タンパク質は大腸菌内でつくらせ、タンパク質の精製は、回収した大腸菌のペレットをタンパク質抽出液に懸濁し、超音波洗浄器で大腸菌を破砕後、遠心分離し、市販のカラム(商品名「Colmodulin−affinity−resin」、Stratagene社製)に通し、融合タンパク質を精製した。また、得られた融合タンパク質の吸収スペクトルを測定後、薄層クロマトグラフィによって、結合していると考えられるフラビン類を同定し、クロモフォアを決定した。図3(A)は、AUREO1またはAUREO2のCBP−LOVドメイン(LOVAUREO1およびLOVAUREO2)を発現している大腸菌ペレットの写真である。写真左側のLOVAUREO1において、ペレットが黄緑色に発色し、AUREO1のフラビン結合能が確認された。
【0028】
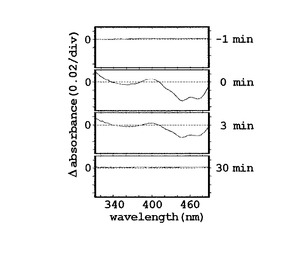
次に、CBP−LOVタンパク質の吸収スペクトルの測定を、スペクトロフォトメーター(Beckman DU−65、Beckman Instruments社製)を用いて行った。なお、青色光による光付加体形成には、LEDイルミネーター(450nm、ISL−150X150BB、CCS株式会社製)を用いた。図3(B)に示す、精製したAUREO1のLOVドメインの吸収スペクトルは、フラボプロテインの典型的な吸収スペクトルを表している。図3(C)は、青色光1分間照射(460nm、500μmole m−2s−1)後のスペクトルの吸収差を、上から下に向かって、青色光照射1分前、照射前0分、照射後3分の暗状態、暗状態30分の順に示している。同図に示すとおり、精製したCBP−LOVタンパク質の吸収スペクトルは、390nm周辺および340nm以下で上昇し、410〜500nm間で著しく低下した。しかし、この吸光度における変化は、20分以内にたちまち消滅した。このことは、青色光による励起状態が、暗条件下でゆっくりと定常状態に戻ったことを示している。また、図3(D)に示すように、有機溶剤を使い、シリカゲル薄層に吸収スペクトルから予想されるフラビン類三種を同時に展開させる薄層クロマトグラフィ法により、FMNがLOVドメインに結合したフラビン種であることが確認された。なお、前記薄層クロマトグラフィ方法は、文献(Iseki M, Matsunaga S, Murakami A, Ohno K, Shiga K, Yoshida K, Sugai M, Takahashi T, Hori T, Watanabe M. “A blue−light−activated adenylyl cyclase mediates photoavoidance in Euglena gracilis”Nature 415(6875):1047−51(2002))に従い、行った。
【0029】
(4) タマネギ表皮細胞における局在
つぎに、前記オーレオクロムの転写制御因子機能を確認するため、タマネギ表皮細胞におけるオーレオクロムの細胞内局在を下記の方法により確認した。
【0030】
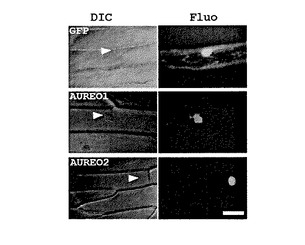
タマネギ表皮細胞におけるオーレオクロムの細胞内局在を観察するために、GFP融合タンパク質であるGFP−AUREO1およびGFP−AUREO2を用いた。融合タンパク質GFP−AUREO1およびGFP−AUREO2を発現する遺伝子コンストタクトを得るために、AUREO1またはAUREO2完全長cDNAをテンプレートとして調整した、BsrGIまたはNotlサイトを有するプライマー(GFPAUREO1F(配列番号24),GFPAUREO1R(配列番号25)、GFPAUREO2F(配列番号26),GFPAUREO2R(配列番号27))を用いてPCR法を実施した。PCR生成物は、BsrGIおよびNotlにより消化し、CAMV35SΩ−sGFP(S65T)プラスミドに挿入した。Bio−Rad PDS−1000/He particle−derlivery systemを用いて、プラスミドをコートした金粒子によりタマネギ表皮細胞に衝撃を付与した。衝撃付与後の表皮を12時間、25℃で培養し、蛍光顕微鏡(Axioskop、Zeiss社製)にて観察した。図4に示した写真は、上段がGFPのみのコントロール区、中段がGFP−AUREO1区、下段がGFP−AUREO2区である。また、左列は位相差顕微鏡(DIC)による位相差観察、右列は蛍光顕微鏡(Fluo)による観察結果である。タマネギ表皮において異種発現したGFP−AUREO融合タンパク質は、AUREO1が核と細胞質の両方に局在したのに対し、AUREO2は核のみに局在した。したがって、核内の局在およびbZIPドメインを有することの双方から、AUREO1およびAUREO2の光感受性転写因子としての役割をサポートしていることが明らかになった。
【0031】
(5) オーレオクロムのDNAへの結合
本発明の遺伝子発現制御方法に用いる前記オーレオクロムのDNAへの直接的な結合を、下記の、野生型プローブ(WT)と6つの単一変異オリゴヌクレオチド(M1−M6)を用いたランダムオリゴヌクレオチドアッセイ方法により確認した。
【0032】
AUREO1−NF(配列番号28)、AUREO1−NR(配列番号29)、AUREO2−NF(配列番号30)およびAUREO2−NR(配列番号31)をプライマーに用いて、AUREO1(1−603bp)およびAUREO2(1−591bp)のN末端部位のDNAインサートをPCR法により生成した。PCR生成物は、pGEM−T Easy Vector(Promega社製)にライゲーションし、シークエンスし、EcoRIおよびBamHIで切断し、さらにpGEX4T2(GE Healthcare Bio−Science社製)のEcoRI−BamHIサイトにライゲーションした。発現したプラスミドにより大腸菌BL2株を形質転換させ、1mM IPTG、37℃、2時間の条件下で、グルタチオン−S−トランスフェラーゼ(GST)融合タンパク質を導入した。細菌ペレットは、2.0mM フェニルメタンスルフォニルフルオライド、15μM ペプスタチン、23μM ロイペプチン、1mM ジチオスレイトールを添加したPBSバッファー(8.1mM NaH2PO4、1.6mM KH2PO4、140mM NaCl)中で超音波処理して溶解させた。GST融合タンパク質は、プロテナーゼ阻害剤存在下で超音波処理により分離し、Glutathione Shepharose4B(GE Healthcare Bio−Sciences社製)を用いたバッチ法により精製した。
【0033】
ランダムオリゴヌクレオチド結合アッセイは、GST融合タンパク質とオリゴヌクレオチド混合物を用いて行った。ランダムオリゴヌクレオチド混合物として、Random1、フォワードプライマー(配列番号32)およびリバースプライマー(配列番号33)を用いた。二本鎖68塩基オリゴヌクレオチド(dsDNA)は、Random1をテンプレートとして用い、PCRにより生成した。Random1プローブ 100ngは、結合バッファー(20mM Tris−HCl pH8.0、50mM KCl、0.5mM EDTA、5mM MgCl2、1mM Dithiothreitol、20μg/ml BSA、2.0mM phenylmethansulfonylfluoride、10μg/ml Polyd[I−C]、10% グリセロール)100μl中でGST−bZIPドメイン(GST−AUREO1−NまたはGST−AUREO2−N)100ngと混合した。10分間穏やかに撹拌した後、glutathione−Sepharose 4Bビーズを加えて、DNA−タンパク質複合体を結合させた。ビーズを遠心分離して回収し、洗浄バッファー(20mM Tris−HCl pH8.0、50mM KCl、0.5mM EDTA、5mM MgCl2、1mM Dithiothreitol、20μg/ml BSA、2.0mM phenylmethansulfonylfluoride、10% グリセロール)200μlにより3回洗浄した。洗浄バッファーで再懸濁したペレットは、3分間、94℃で加温し、遠心分離した。得られた上澄5μlをRandom1のプライマーを用いたPCRのテンプレートとして用いた。PCR反応混合物は、エタノールおよび酢酸ナトリウムで沈殿させ、水に再懸濁した。PCR生成物の一部を次のDNA−タンパク質結合反応に用い、この工程を4回繰り返した。
【0034】
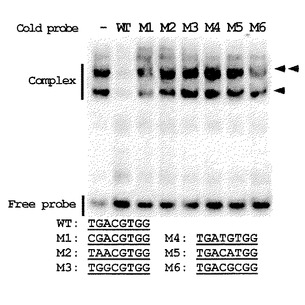
ゲルシフトアッセイは、DIG Gel Shift Kit,2nd Generation(ロシュ社製)を用いて行った。このアッセイで用いたDNAプローブは、7つの各オリゴヌクレオチド対(WT[WT−1(配列番号34)とWT−2(配列番号35)]、M1[M1−1(配列番号36)とM1−2(配列番号37)]、M2[M2−1(配列番号38)とM2−2(配列番号39)]、M3[M3−1(配列番号40)とM3−2(配列番号41)]、M4[M4−1(配列番号42)とM4−2(配列番号43)]、M5[M5−1(配列番号44)とM5−2(配列番号45)]およびM6[M6−1(配列番号46)とM6−2(配列番号47)])のアニーリングにより生成したものを用いた。bZIPタンパク質の混合物および標識したコールドプローブ(25ng GST−AUREO1−N,0.4ng Dig標識プローブおよび50ng コールドプローブ)は、4℃、4%非変性ポリアクリルアミドゲル上で0.5倍Tris−ホウ酸−電気泳動(TBE)バッファーにて電気泳動した。ゲルは、BiodynePLUS(Poll Corporation社製)上でブロットした。Anti−Digoxygenin−APおよびCSPD(ロシュ社製)から化学発光を放出し、Light−Capture及びCSアナライザー Ver.2.0(ATTO Corpotation社製)によって画像化した。図5に、野生型プローブ(WT)と6つの単一変異オリゴヌクレオチド(M1−M6)を用いたGST−AUREO1Nのゲルシフトアッセイを示す。上側のバンド(2つの矢印)はbZIPタンパク質の二量体で、下側のバンド(1つの矢印)はbZIPタンパク質の単量体を示している。WTレーンにおいてのみ、両バンドともに観察できない。すなわち、ランダムオリゴヌクレオチド結合選択アッセイにより、AUREO1(GST−AUREO1N)は、5’−TGACGT−3’(配列番号1)に結合することが示された。
【0035】
(6) AUREO1のDNAへの結合
AUREO1のDNAへの直接的な結合を、下記のゲルシフトアッセイ方法により確認した。
【0036】
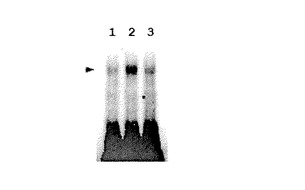
青色光による、AUREO1とTGACGT含有DNAプローブとの結合の活性化は、次のようにして行った。GST−AUREO1タンパク質全長を、完全暗条件化で、ベクターpGEX6P1を用いて大腸菌(Rosetta)に発現させた。得られたタンパク質を弱い赤色光(<0.02μmole m−2s−1)条件下で分離精製し、そのGSTをPreScissionプロテアーゼ(GE Healthcare社製)によって切り取った。AUREO1タンパク質およびDIGで標識したオリゴヌクレオチドプローブを混合し、暗条件下もしくは青色光の短照射条件下(470nm LED、100μmole m−2s−1、5分間)に置いた。30分間の結合反応後、AUREO1標的ヌクレオチド複合体を、暗条件下で電気泳動により分離した(観察のために短時間、安全な弱い赤色光下に置いた)。図6は、ゲルシフトアッセイにおいて、第1列は30分間暗条件下に置いたもの、第2列は25分間暗条件下の後に青色光を5分間短照射したもの、第3列は青色光を5分間短照射後に25分間暗条件化に置いたときの結果を示している。同図に示すように、青色光を照射した第2列および第3列において矢印部にバンドが見られ、AUREO1とターゲットDNA配列との結合が青色光によって活性化されていることを表している。
【0037】
(7) 青色光誘導光形態形成機能
オーレオクロムの青色光誘導光形態形成機能を、下記のRNAi法により確認した。
【0038】
4つのdsRNA断片(LOVAUREO1、bZIPAUREO1、LOVAUREO2およびbZIPAUREO2)はPCR法により合成した。プライマーセットは、bZIPAUREO1F(配列番号48)、bZIPAUREO1R(配列番号49)、LOVAUREO1F(配列番号50),LOVAUREO1R(配列番号51)、bZIPAUREO2F(配列番号52)、bZIPAUREO2R(配列番号53)、LOVAUREO2F(配列番号54),LOVAUREO2R(配列番号55)を用いた。各断片の長さは、おおよそ500塩基対であった。これらのdsRNAをTEバッファー(10mM Tris、1mM EDTA、pH8.0)に5μg/μLになるよう懸濁し、使用時まで冷凍保存した。2または4つのdsRNA混合物は、長さ約10mmのフシナシミドロのセグメントに顕微導入し、導入した個々のセグメントは、数回培地を交換しつつ、6−7ヶ月間、インキュベータ内で別々に培養した(12時間明条件/12時間暗条件、20℃)。導入溶液には、2または4種類のdsRNA混合物(全4種類のdsRNA、LOVAUREO1+bZIPAUREO1またはLOVAUREO2+bZIPAUREO2)7−10μg/μL、20−30mM K2SO4、17−25mM MgSO4、0.07−0.1mM EGTA,40−60mM ソルビトールおよび17−25mM PIPES−K(pH 7.0)を混合したものを用いた。
【0039】
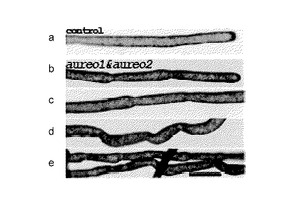
図7(A)は、インジェクション後37日目における、セグメントの形態を示す写真である。写真aは、バッファーのみをインジェクションしたコントロールで、写真b〜eは、dsRNA混合物をマイクロインジェクションしたセグメントから再生した管の異常形態(aureo1&aureo2)を示す写真である。管の狭窄部は日毎の成長制限を表しており、コントロールには見られなかった。
【0040】
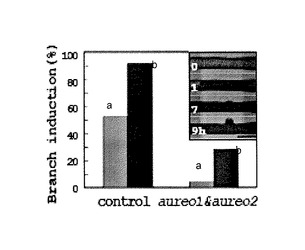
また、図7(B)は、青色光による分枝誘導をAUREO1およびAUREO2のRNAiにより同時阻害したときの分枝率を示すグラフである。5−6ヶ月間培養後、管の一部位(幅200μm)に青色光を照射し、照射した管が分枝した割合(%)を示したもので、グラフの左柱aは青色光を10時間照射し、右柱bは24時間照射したものである。また、コントロール区はバッファーのみをインジェクションした細胞で、aureo1&aureo2区はdsRNA混合物をインジェクションした細胞である。コントロールの分枝率は、10時間照射が52.6%(n=19)、24時間照射が91.7%(n=24)であった。一方、dsRNA混合物をインジェクションした細胞では、10時間照射で4.3%(n=46)、24時間照射で28.6%(n=34)のみが分枝した。グラフ内右上に挿入した写真は、青色光を照射した近接の管の一部位(幅200μm)における、照射後0、1、7および9時間後の、通常の青色光誘導分枝を示している(写真最下部のバーの長さは100μm)。
【0041】
図7(C)は、前記RNAiによりAUREO1又はAUREO2のいずれかがノックアウトされたセグメントの写真である(最下部のバー長さは200μm)。AUREO2のみをノックアウトしたaureo2細胞からは、多くの未成熟な生殖器官が発生した。一方、AUREO1のみをノックアウトしたaureo1細胞は、aureo1&aureo2の表現型(図7(A))に類似していた。
【0042】
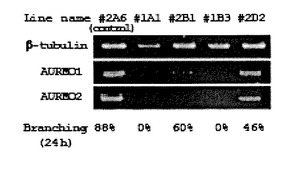
さらに、RT−PCRを用いて、24時間、青色光を照射した管について、細胞内のAUREO1およびAUREO2のmRNAの産生を調べた。図7(D)に示すように、#1A1および#1B3において、オーレオクロムmRNAが全く産生されず、分枝能が完全に欠失していた。これらの結果は、AUREOCHROMEが、青色光による光形態形成における光受容タンパク質であることを示している。
【産業上の利用可能性】
【0043】
以上のように、本発明の遺伝子発現制御方法によれば、細胞に悪影響を及ぼすことなく、簡単なシステムで遺伝子の発現の制御が可能になる。本発明は、遺伝子の発現制御を実施する全ての分野に適用可能であり、その用途は、医学、農学、薬学等の生物に関連する分野全てに適用可能である。
【図面の簡単な説明】
【0044】
【図1】図1(A−1)、(A−2)、(B−1)および(B−2)は、本発明の遺伝子発現制御方法を説明するための模式図である。
【図2】図2は、オーレオクロム(AUREO1およびAUREO2)の構造を示す模式図である。
【図3(A)】図3(A)は、本発明の実施例において、CBP−LOVドメイン(LOVAUREO1、LOVAUREO2)を導入した大腸菌のペレットを示す写真である。
【図3(B)】図3(B)は、本発明の実施例において、精製したCBP−LOVAUREO1の吸収スペクトルを示すグラフである。
【図3(C)】図3(C)は、本発明の実施例において、青色光照射前後のスペクトルの吸収差を示すグラフである。
【図3(D)】図3(D)は、本発明の実施例において、AUREO1の発色団がFMNであることを示す薄層クロマトグラフィの写真である。
【図4】図4は、本発明の実施例において、GFP−AUREO融合タンパク質の細胞内局在を示す顕微鏡写真である。
【図5】図5は、本発明の実施例におけるゲルシフトアッセイ結果を示す写真である。
【図6】図6は、本発明の実施例におけるゲルシフトアッセイ結果を示すその他の写真である。
【図7(A)】図7(A)は、本発明の実施例におけるセグメントから再生した管の形態を示す写真である。
【図7(B)】図7(B)は、本発明の実施例における分枝誘導率の結果を示すグラフである。
【図7(C)】図7(C)は、本発明の実施例におけるRNAiによる表現型の結果を示す写真である。
【図7(D)】図7(D)は、本発明の実施例におけるRT−PCR結果を示す写真である。
【技術分野】
【0001】
本発明は、遺伝子発現制御方法、それに用いる遺伝子組換え用ベクターおよび遺伝子発現制御キットに関する。
【背景技術】
【0002】
350〜500nmの波長光(以下、青色光という)は、多くの植物種において非常に重要な環境シグナルの1つである。例えば、陸上植物では、光屈性や気孔開口のシグナルとして青色光が利用されている。この現象には、光受容体と呼ばれるタンパク質がはたらいている。このような植物の青色光の光受容体タンパク質には、クリプトクロム、フォトトロピン(phot1・phot2)、ADO/FKF/LKP/ZTLファミリーなどがある。また、これらの青色光の光受容体タンパク質は、光センサードメインと制御ドメインを有することが知られている。例えば、光受容体タンパク質である前記フォトトロピンには、光センサードメインである2つのLOV(Light Oxygen Voltage)ドメインと、制御ドメインである1つのプロテインキナーゼドメインが存在する。このうち、光センサードメインであるLOVドメインは、フラビンモノヌクレオチド(FMN)を色素団として持ち、そのFMNが青色光を受容することで、フォトトロピンの立体構造が変化し、フォトトロピンのプロテインキナーゼが活性化されることにより、シグナルを下流分子に伝えるとされている。
【0003】
これまでに、このような青色光によるタンパク質−タンパク質間の相互作用に基づいた遺伝子発現制御方法として、シロイヌナズナの青色光受容体であるLKP1(LOV kelch protein1)等を用いた青色光バイオスイッチが開発されている(特許文献1および2)。また、化学物質を用いた遺伝子の発現制御方法も開発されている(特許文献3)。化学物質を用いた遺伝子の発現制御方法は、化学物質により細胞への悪影響を及ぼす可能性がある。一方、青色光を用いた遺伝子制御方法は、細胞への悪影響がない、優れた方法であるが、前述のように、タンパク質間の相互作用に基づいているために、システムが複雑で、種々の要因の影響を受ける恐れがある。
【特許文献1】特開2006−94704号公報
【特許文献2】特開2007−75019号公報
【特許文献3】特表平11−506901号公報
【発明の開示】
【発明が解決しようとする課題】
【0004】
そこで、本発明は、簡単なシステムで、細胞に悪影響を及ぼす恐れの無い遺伝子発現制御方法を提供することを目的とする。
【課題を解決するための手段】
【0005】
前記目的を達成するために、本発明の遺伝子発現制御方法は、
細胞内の目的遺伝子の転写を調節することで前記目的遺伝子の発現を制御する遺伝子発現制御方法であって、
前記目的遺伝子の上流に、プロモーター配列を配置し、
前記プロモーター配列の上流に、5’−TGACGT−3’の結合配列を含む応答配列を配置し、
前記細胞内において、AUREO1タンパク質を存在させ、
前記AUREO1タンパク質は、青色光の照射下では、前記結合配列に結合し、青色光の非照射下では、前記結合配列から遊離し、
前記青色光の照射または非照射によるAUREO1タンパク質の前記結合配列への結合または遊離により、前記プロモーター配列の活性または不活性を調節することで、前記目的遺伝子の転写を調節することを特徴とする。
【発明の効果】
【0006】
本発明者らは、前記目的を達成するために一連の研究を重ねた。その過程で、藻類の一種であるフシナシミドロから、青色光の照射により、前記特定の塩基配列に結合し、かつ青色光の非照射により、前記特定の塩基配列から遊離するという性質をもつタンパク質であるAUREO1を発見し、分離した。そして、さらに研究を重ねた結果、前記AUREO1を利用すれば、タンパク質間の相互作用無しで、青色光の照射または非照射により、遺伝子の転写を調節できることを見出し、本発明に到達した。したがって、本発明の遺伝子発現制御方法は、化学物質ではなく、青色光を用いるため、細胞への悪影響の恐れがない。また、本発明の遺伝子発現制御方法は、タンパク質間の相互作用を利用していないため、システムが簡単であり、種々の要因による影響も低減可能である。
【発明を実施するための最良の形態】
【0007】
本発明の遺伝子発現制御方法は、AUREO1の結合により遺伝子の転写を開始させる第一の方法と、AUREO1の遊離により遺伝子の転写を開始させる第二の方法がある。
【0008】
本発明の遺伝子発現制御方法の第一の方法は、
青色光の照射により、前記AUREO1タンパク質を、前記応答配列に結合させることにより前記プロモーター配列を活性化して前記目的遺伝子の転写を実施し、
前記青色光の非照射により、前記AUREO1タンパク質を前記結合配列から遊離させることにより前記目的遺伝子の転写を実施しないという方法である。
前記第一の方法では、AUREO1自身が、プロモーター配列を活性化するアクチベーターとして機能する。
【0009】
本発明の遺伝子発現制御方法の第二の方法は、
前記細胞内に、さらに、アクチベーターを存在させ、
前記アクチベーターは、前記応答配列に結合することにより前記プロモーター配列を活性化し、
青色光の照射により、前記AUREO1タンパク質を前記結合配列に結合させることで、
前記アクチベーターの前記応答配列への結合を阻止して前記目的遺伝子の転写を実施せず、
前記青色光の非照射により、前記AUREO1タンパク質を前記結合配列から遊離させることで、前記アクチベーターを前記応答配列に結合させて前記目的遺伝子の転写を実施するという方法である。
前記第二の方法では、AUREO1が、プロモーターを不活性化するリプレッサーとして機能する。
【0010】
本発明において、前記AUREO1タンパク質は、例えば、下記の(A)、(B)または(C)のタンパク質である。
(A) 配列番号2のアミノ酸配列からなるタンパク質
(B) 配列番号2のアミノ酸配列において、1個または数個のアミノ酸残基が、欠失、置換または付加されたアミノ酸配列からなり、青色光の照射下では、前記結合配列に結合し、青色光の非照射下では、前記結合配列から遊離するタンパク質。
(C) 配列番号2のアミノ酸配列に対し、68%以上の相同性を有するアミノ酸配列からなり、青色光の照射下では、前記結合配列に結合し、青色光の非照射下では、前記結合配列から遊離するタンパク質である。
前記(B)のタンパク質は、配列番号2のアミノ酸配列において、例えば、1〜10個、好ましくは1〜5個、より好ましくは1〜3個のアミノ酸残基が、欠失、置換または付加されたアミノ酸配列からなり、青色光の照射下では、前記結合配列に結合し、青色光の非照射下では、前記結合配列から遊離するタンパク質である。
また、前記(C)のタンパク質は、配列番号2のアミノ酸配列に対し、例えば、68%以上、好ましくは80%以上、より好ましくは90%以上の相同性を有するアミノ酸配列からなり、青色光の照射下では、前記結合配列に結合し、青色光の非照射下では、前記結合配列から遊離するタンパク質である。
【0011】
本発明において、前記応答配列は、5’−TGACGT−3’(配列番号1)の結合配列に加えまたは代えて、5’−TGACGC−3’の結合配列を含むという態様であってもよい。ただし、本発明では、5’−TGACGT−3’の結合配列が好ましい。
【0012】
本発明において、前記青色光が、波長350nmから500nmの範囲の光を含むことが好ましい。本発明において、青色光は、特に制限されず、青色光を含めばよく、例えば、太陽光、人工的光源(例えば、蛍光灯、白熱球、ハロゲンランプ、発光ダイオード、有機EL素子)等の光であってもよい。
【0013】
本発明において、前記細胞は、細菌、酵母、動物細胞および植物細胞からなる群から選択される少なくとも一つの細胞であることが好ましい。
【0014】
つぎに、本発明の遺伝子組換え用ベクターは、本発明の遺伝子発現制御方法に使用する遺伝子組換え用ベクターであって、
前記目的遺伝子のクローニングサイト、前記プロモーター配列および前記応答配列を含み、
前記目的遺伝子のクローニングサイトの上流に、前記プロモーター配列が配置され、
前記プロモーター配列の上流に、前記応答配列が配置されていることを特徴とする。
【0015】
本発明の遺伝子組換え用ベクターにおいて、さらに、AUREO1遺伝子を含むという態様であってもよい。
【0016】
つぎに、本発明の遺伝子発現制御用キットは、本発明の遺伝子発現制御方法に使用するキットであって、本発明の遺伝子組換え用ベクターを含むキットである。
【0017】
本発明の遺伝子発現制御用キットにおいて、さらに、AUREO1遺伝子を発現可能な状態で含む遺伝子組み換え用ベクターを含むという態様であってもよい。
【0018】
つぎに、本発明について、詳細に説明する。
【0019】
(A) AUREO1
前述のように、AUREO1は、藻類の一種であるフシナシミドロの細胞内に発現する光受容体タンパク質である。光受容体タンパク質は、光を受容することにより、その立体構造を変化させ、その変化を細胞内の生化学反応に変換する機能を有するタンパク質である。AUREO1は、そのファミリータンパク質であるAUREO2と共に、本発明者等によりフシナシミドロから発見され、分離された。図2に、AUREO1およびAUREO2の構造の模式図を示す。図示のように、両タンパク質共に、一つのLOVドメインと、一つのbZIPドメインを有する。「LOVドメイン」とは、青色光センサー(Light)、酸素センサー(Oxygen)、電位依存カリウムチャンネル(Voltage)の各センサー部分に特異的な立体構造を持つドメインという意味で命名された青色光の光受容体タンパク質が有する発色団結合ドメインであり、前記発色団として、1分子のFMNを非共有結合により有している。LOVドメインを有する光受容体タンパク質は、AUREO1および2の他に、例えば、phot1、phot2、phy3、ADO/FKF/LKP/ZTLファミリータンパク質、TLP1/PLPなどがある。「bZIPドメイン」は、遺伝子の転写を制御するタンパク質のDNA結合ドメインで、DNA結合および二量体化に関与し、コア配列を含有するDNA配列に共通して結合する、いわゆる塩基性ロイシンジッパードメインをいう。
【0020】
AUREO1タンパク質は、例えば、配列番号2のアミノ酸配列からなるタンパク質であり、その分子量は、38.6kDである。AUREO1タンパク質は、青色光を照射すると、そのLOVドメインが、システインを介してFMNに結合し、bZIPドメインが、5’−TGACGT−3’(配列番号1)の配列に可逆的に結合する。また、青色光の非照射の条件下(例えば、暗黒下)では、前記配列から遊離する。一方、AUREO2は、配列番号3のアミノ酸配列からなるタンパク質であり、その分子量が39.1kDである。AUREO2は、AUREO1と異なり、2箇所にイントロンを有している。AUREO1遺伝子の塩基配列は、アクセッション番号AB252504として、AUREO2遺伝子の塩基配列は、アクセッション番号AB252505(mRNA)およびアクセッション番号AB272981(gene)として、それぞれDDBJに登録されている。
【0021】
(B) 本発明の遺伝子発現制御方法の第一の方法
本発明の遺伝子発現制御方法の第一の方法について、図1(A−1)および(A−2)に基づき説明する。前述のように、前記第一の方法では、AUREO1が、プロモーター配列を活性化するアクチベーターとして機能する。図示のように、目的遺伝子Gの上流に、プロモーター配列Pを配置し、前記プロモーター配列Pの上流に、前記応答配列Bを配置する。前記応答配列は、5’−TGACGT−3’の結合配列を含む。前記目的遺伝子G、プロモーター配列Pおよび応答配列Bは、例えば、遺伝子組換え用ベクターを用いて細胞に導入することができる。一方、細胞内に、AUREO1タンパク質を存在させる。AUREO1タンパク質を細胞内に存在させる方法としては、例えば、AUREO1遺伝子を組み込んだベクターを細胞内に導入し、前記遺伝子を発現させるという方法がある。前記AUREO1遺伝子は、前記目的遺伝子Gを組み込んだベクターに組み込んでもよいし、これとは別のベクターに組み込んでもよい。そして、図1(A−1)に示すように、青色光(Blue Light)を照射すると、AUREO1が前記結合配列を特異的に認識して結合し、この結果、前記応答配列Bがプロモーター配列Pを活性化する。プロモーター配列Pの活性化により、目的遺伝子Gの転写が開始される(転写ON)。これとは逆に、青色光の非照射下(例えば、暗黒(Dark)下)では、AUREO1タンパク質は、前記結合配列から遊離しているため、プロモーター配列Pは不活性の状態であり、目的遺伝子は転写されない(転写OFF)。
【0022】
(C) 本発明の遺伝子発現制御方法の第二の方法
本発明の遺伝子発現制御方法の第二の方法について、図1(B−1)および(B−2)に基づき説明する。前述のように、前記第二の方法では、AUREO1が、リプレッサーとして機能し、プロモーター配列を活性化するアクチベーターが応答配列に結合するのを阻止する。したがって、前記アクチベーターは、例えば、AUREO1タンパク質と競合するか、もしくは、青色光の照射下で応答配列から離れる性質を持つ。図示のように、第一の方法と同様に、目的遺伝子Gの上流に、プロモーター配列Pを配置し、前記プロモーター配列Pの上流に、前記応答配列Bを配置する。前記応答配列は、5’−TGACGT−3’の結合配列を含む。前記目的遺伝子G、プロモーター配列Pおよび応答配列Bは、例えば、遺伝子組換え用ベクターを用いて細胞に導入することができる。一方、細胞内に、AUREO1タンパク質およびアクチベーターを存在させる。AUREO1タンパク質およびアクチベーターを細胞内に存在させる方法としては、例えば、AUREO1遺伝子およびアクチベーター遺伝子を組み込んだベクターを細胞内に導入し、前記両遺伝子を発現させるという方法がある。前記AUREO1遺伝子およびアクチベーター遺伝子は、前記目的遺伝子Gを組み込んだベクターに組み込んでもよいし、これとは別のベクターに組み込んでもよい。前記AUREO1遺伝子およびアクチベーター遺伝子は、それぞれ別のベクターに組み込んでもよい。そして、図1(B−1)に示すように、青色光(Blue Light)を照射すると、AUREO1が前記結合配列を特異的に認識して結合し、この結果、アクチベーターが前記応答配列Bに結合できず、プロモーター配列Pは不活性の状態となり、目的遺伝子Gは転写されない(転写OFF)。これとは逆に、青色光の非照射下(例えば、暗黒(Dark)下)では、AUREO1タンパク質は、前記結合配列から遊離しているため、アクチベーターが応答配列Bに結合することができ、この結果、プロモーター配列Pが活性化され、目的遺伝子の転写が開始される(転写ON)。
【0023】
前記プロモーターとしては、例えば、酵母One−hybrid法などに用いられるGAL4遺伝子のミニマムプロモーター領域や、植物で利用されるカリフラワーモザイクウィルス35S遺伝子のミニマムプロモーター領域などが、本発明の前記遺伝子発現制御方法の第一および第二の方法における遺伝子の発現制御において使用可能である。
【実施例】
【0024】
つぎに、本発明の実施例について説明する。
【0025】
(1)DNAおよびRNAの分離
淡水黄緑藻類であるオカフシナシミドロの葉状体を三角フラスコ内の新鮮な培地に移し、白色蛍光ランプ下、20℃で2−3ヶ月間培養した(12時間明条件/12時間暗条件)。培養終了後、培養物のゲノムDNAおよびtotalRNAをセチルトリメチルアンモニウムブロマイド(CTAB)法を用いて単離した。一方、ヒバマタの成熟した生殖器床は、2006年5月下旬に日本の北海道室蘭で採取し、受精後4−6時間後に受精した接合子を回収した。接合子は、直ちにDNA/RNA抽出に用いるか、−80℃で使用時まで保存した。オカフシナシミドロおよびヒバマタのいずれも、DNAおよびRNAは、試料に界面活性剤を含む破砕液を加え、試験管を洗浄するブラシを上下することで細胞を十分に破砕する「ウォッシュブラッシュ」法(Nozaki H, Ito M, Watanabe, MM, Takano H, Kuroiwa T.,“PHYLOGENETIC ANALYSIS OF MORPHOLOGICAL SPECIES OF CARTERIA (VOLVOCALES,CHLOROPHYTA) BASED ON rbcL GENE SEQUENCES”J Phycol 33:864−867(1997))により抽出した。ウォッシュブラッシュ法は、細胞を十分に破砕することでm−RNAやDNAの収量が高まることから、微細な細胞や卵などからm−RNAやDNAを単離する際に用いる方法である。
【0026】
(2)フシナシミドロのLOVドメイン含有タンパク質のクローニング
LOVドメイン含有タンパク質は、縮重プライマー(LOV−F(配列番号4)およびLOV−R(配列番号5))を用いて単離した。PCR生成物は、pGEM-T Easy system(Promega)によりクローニングし、シークエンスを行った。完全長cDNAは、5’および3’RACEにより得た。プライマーには、5’RACE−1−1(配列番号6)、5’RACE−1−2(配列番号7)、5’RACE−1−3(配列番号8)、3’RACE−1−1(配列番号9)、3’RACE−1−2(配列番号10)、5’RACE−2−1(配列番号11)、5’RACE−2−2(配列番号12)、5’RACE−2−3(配列番号13)、3’RACE−2−1(配列番号14)、3’RACE−2−2(配列番号15)を用いた。さらに、特異的プライマー(AUREO1−Full−F(配列番号16)、AUREO1−Full−R(配列番号17)、AUREO2−Full−F(配列番号18)、AUREO2−Full−R(配列番号19))を用いたRT−PCR法により配列を決定した。こうして得た、LOVドメインを有するタンパク質をオーレオクロム(AUREOCHROME)と命名した。図2に示すように、オーレオクロムは、LOVドメインとbZIPドメインを各々1つ有するタンパク質で、AUREO1(配列番号2)とAUREO2(配列番号3)という2つの相同体が存在する。AUREO1において、例えば、配列番号2の第118番から第178番までの配列がbZIPドメインであり、配列番号2の第217番から第320番までの配列がLOVドメインである。また、AUREO2において、配列番号3の第124番から第185番までの配列がbZIPドメインであり、配列番号3の第224番から第327番までの配列がLOVドメインである。また、ヒバマタおよび珪藻においても、高い相同性を有するLOVドメインおよびbZIPドメインの存在が確認された。
【0027】
(3)光化学分析
オーレオクロムのフラビン結合能の確認のため、カルモデュリン結合ペプチド(CBP)とAUREO1のLOVドメインとの融合タンパク質と、CBPとAUREO2のLOVドメインとの融合タンパク質を作製し、大腸菌に発現させた。CBP−LOV融合タンパク質の異種発現は、pCAL−n−EK(Stratagene社製)ベクターを用いて大腸菌Rosetta(DE3)pLysS(Novagen社製)を形質転換させて行った。オーレオクロムのLOV(LOVAUREO1およびLOVAUREO2)のcDNA断片は、AUREO1−LOVF−AUREO2−LOVRのプライマーセット(AUREO1−LOVF(配列番号20)、AUREO1−LOVR(配列番号21)、AUREO2−LOVF(配列番号22)、AUREO2−LOVR(配列番号23))を用いてクローニングして得た。得られた形質転換体をLB培地(1.0L)で30℃の暗条件下、3時間培養した。融合タンパク質の発現は、イソプロピル−1−チオ−β−D−galactoside(IPTG)の添加により誘導した。得られた細胞を遠心分離して回収し、使用時まで−20℃で保存した。タンパク質の精製およびクロムフォアの決定は、文献(Kasahara M, Swartz TE, Olney MA, Onodera A, Mochizuki N, Fukuzawa H, Asamizu E, Tabata S, Kanegae H, Takano M et al.,“Photochemical Properties of the Flavin Mononucleotide−Binding Domains of the Phototropins from Arabidopsis, Rice, and Chlamydomonas reinhardtii”Plant Physiol 129:762−773(2002))に従った。概略としては、タンパク質は大腸菌内でつくらせ、タンパク質の精製は、回収した大腸菌のペレットをタンパク質抽出液に懸濁し、超音波洗浄器で大腸菌を破砕後、遠心分離し、市販のカラム(商品名「Colmodulin−affinity−resin」、Stratagene社製)に通し、融合タンパク質を精製した。また、得られた融合タンパク質の吸収スペクトルを測定後、薄層クロマトグラフィによって、結合していると考えられるフラビン類を同定し、クロモフォアを決定した。図3(A)は、AUREO1またはAUREO2のCBP−LOVドメイン(LOVAUREO1およびLOVAUREO2)を発現している大腸菌ペレットの写真である。写真左側のLOVAUREO1において、ペレットが黄緑色に発色し、AUREO1のフラビン結合能が確認された。
【0028】
次に、CBP−LOVタンパク質の吸収スペクトルの測定を、スペクトロフォトメーター(Beckman DU−65、Beckman Instruments社製)を用いて行った。なお、青色光による光付加体形成には、LEDイルミネーター(450nm、ISL−150X150BB、CCS株式会社製)を用いた。図3(B)に示す、精製したAUREO1のLOVドメインの吸収スペクトルは、フラボプロテインの典型的な吸収スペクトルを表している。図3(C)は、青色光1分間照射(460nm、500μmole m−2s−1)後のスペクトルの吸収差を、上から下に向かって、青色光照射1分前、照射前0分、照射後3分の暗状態、暗状態30分の順に示している。同図に示すとおり、精製したCBP−LOVタンパク質の吸収スペクトルは、390nm周辺および340nm以下で上昇し、410〜500nm間で著しく低下した。しかし、この吸光度における変化は、20分以内にたちまち消滅した。このことは、青色光による励起状態が、暗条件下でゆっくりと定常状態に戻ったことを示している。また、図3(D)に示すように、有機溶剤を使い、シリカゲル薄層に吸収スペクトルから予想されるフラビン類三種を同時に展開させる薄層クロマトグラフィ法により、FMNがLOVドメインに結合したフラビン種であることが確認された。なお、前記薄層クロマトグラフィ方法は、文献(Iseki M, Matsunaga S, Murakami A, Ohno K, Shiga K, Yoshida K, Sugai M, Takahashi T, Hori T, Watanabe M. “A blue−light−activated adenylyl cyclase mediates photoavoidance in Euglena gracilis”Nature 415(6875):1047−51(2002))に従い、行った。
【0029】
(4) タマネギ表皮細胞における局在
つぎに、前記オーレオクロムの転写制御因子機能を確認するため、タマネギ表皮細胞におけるオーレオクロムの細胞内局在を下記の方法により確認した。
【0030】
タマネギ表皮細胞におけるオーレオクロムの細胞内局在を観察するために、GFP融合タンパク質であるGFP−AUREO1およびGFP−AUREO2を用いた。融合タンパク質GFP−AUREO1およびGFP−AUREO2を発現する遺伝子コンストタクトを得るために、AUREO1またはAUREO2完全長cDNAをテンプレートとして調整した、BsrGIまたはNotlサイトを有するプライマー(GFPAUREO1F(配列番号24),GFPAUREO1R(配列番号25)、GFPAUREO2F(配列番号26),GFPAUREO2R(配列番号27))を用いてPCR法を実施した。PCR生成物は、BsrGIおよびNotlにより消化し、CAMV35SΩ−sGFP(S65T)プラスミドに挿入した。Bio−Rad PDS−1000/He particle−derlivery systemを用いて、プラスミドをコートした金粒子によりタマネギ表皮細胞に衝撃を付与した。衝撃付与後の表皮を12時間、25℃で培養し、蛍光顕微鏡(Axioskop、Zeiss社製)にて観察した。図4に示した写真は、上段がGFPのみのコントロール区、中段がGFP−AUREO1区、下段がGFP−AUREO2区である。また、左列は位相差顕微鏡(DIC)による位相差観察、右列は蛍光顕微鏡(Fluo)による観察結果である。タマネギ表皮において異種発現したGFP−AUREO融合タンパク質は、AUREO1が核と細胞質の両方に局在したのに対し、AUREO2は核のみに局在した。したがって、核内の局在およびbZIPドメインを有することの双方から、AUREO1およびAUREO2の光感受性転写因子としての役割をサポートしていることが明らかになった。
【0031】
(5) オーレオクロムのDNAへの結合
本発明の遺伝子発現制御方法に用いる前記オーレオクロムのDNAへの直接的な結合を、下記の、野生型プローブ(WT)と6つの単一変異オリゴヌクレオチド(M1−M6)を用いたランダムオリゴヌクレオチドアッセイ方法により確認した。
【0032】
AUREO1−NF(配列番号28)、AUREO1−NR(配列番号29)、AUREO2−NF(配列番号30)およびAUREO2−NR(配列番号31)をプライマーに用いて、AUREO1(1−603bp)およびAUREO2(1−591bp)のN末端部位のDNAインサートをPCR法により生成した。PCR生成物は、pGEM−T Easy Vector(Promega社製)にライゲーションし、シークエンスし、EcoRIおよびBamHIで切断し、さらにpGEX4T2(GE Healthcare Bio−Science社製)のEcoRI−BamHIサイトにライゲーションした。発現したプラスミドにより大腸菌BL2株を形質転換させ、1mM IPTG、37℃、2時間の条件下で、グルタチオン−S−トランスフェラーゼ(GST)融合タンパク質を導入した。細菌ペレットは、2.0mM フェニルメタンスルフォニルフルオライド、15μM ペプスタチン、23μM ロイペプチン、1mM ジチオスレイトールを添加したPBSバッファー(8.1mM NaH2PO4、1.6mM KH2PO4、140mM NaCl)中で超音波処理して溶解させた。GST融合タンパク質は、プロテナーゼ阻害剤存在下で超音波処理により分離し、Glutathione Shepharose4B(GE Healthcare Bio−Sciences社製)を用いたバッチ法により精製した。
【0033】
ランダムオリゴヌクレオチド結合アッセイは、GST融合タンパク質とオリゴヌクレオチド混合物を用いて行った。ランダムオリゴヌクレオチド混合物として、Random1、フォワードプライマー(配列番号32)およびリバースプライマー(配列番号33)を用いた。二本鎖68塩基オリゴヌクレオチド(dsDNA)は、Random1をテンプレートとして用い、PCRにより生成した。Random1プローブ 100ngは、結合バッファー(20mM Tris−HCl pH8.0、50mM KCl、0.5mM EDTA、5mM MgCl2、1mM Dithiothreitol、20μg/ml BSA、2.0mM phenylmethansulfonylfluoride、10μg/ml Polyd[I−C]、10% グリセロール)100μl中でGST−bZIPドメイン(GST−AUREO1−NまたはGST−AUREO2−N)100ngと混合した。10分間穏やかに撹拌した後、glutathione−Sepharose 4Bビーズを加えて、DNA−タンパク質複合体を結合させた。ビーズを遠心分離して回収し、洗浄バッファー(20mM Tris−HCl pH8.0、50mM KCl、0.5mM EDTA、5mM MgCl2、1mM Dithiothreitol、20μg/ml BSA、2.0mM phenylmethansulfonylfluoride、10% グリセロール)200μlにより3回洗浄した。洗浄バッファーで再懸濁したペレットは、3分間、94℃で加温し、遠心分離した。得られた上澄5μlをRandom1のプライマーを用いたPCRのテンプレートとして用いた。PCR反応混合物は、エタノールおよび酢酸ナトリウムで沈殿させ、水に再懸濁した。PCR生成物の一部を次のDNA−タンパク質結合反応に用い、この工程を4回繰り返した。
【0034】
ゲルシフトアッセイは、DIG Gel Shift Kit,2nd Generation(ロシュ社製)を用いて行った。このアッセイで用いたDNAプローブは、7つの各オリゴヌクレオチド対(WT[WT−1(配列番号34)とWT−2(配列番号35)]、M1[M1−1(配列番号36)とM1−2(配列番号37)]、M2[M2−1(配列番号38)とM2−2(配列番号39)]、M3[M3−1(配列番号40)とM3−2(配列番号41)]、M4[M4−1(配列番号42)とM4−2(配列番号43)]、M5[M5−1(配列番号44)とM5−2(配列番号45)]およびM6[M6−1(配列番号46)とM6−2(配列番号47)])のアニーリングにより生成したものを用いた。bZIPタンパク質の混合物および標識したコールドプローブ(25ng GST−AUREO1−N,0.4ng Dig標識プローブおよび50ng コールドプローブ)は、4℃、4%非変性ポリアクリルアミドゲル上で0.5倍Tris−ホウ酸−電気泳動(TBE)バッファーにて電気泳動した。ゲルは、BiodynePLUS(Poll Corporation社製)上でブロットした。Anti−Digoxygenin−APおよびCSPD(ロシュ社製)から化学発光を放出し、Light−Capture及びCSアナライザー Ver.2.0(ATTO Corpotation社製)によって画像化した。図5に、野生型プローブ(WT)と6つの単一変異オリゴヌクレオチド(M1−M6)を用いたGST−AUREO1Nのゲルシフトアッセイを示す。上側のバンド(2つの矢印)はbZIPタンパク質の二量体で、下側のバンド(1つの矢印)はbZIPタンパク質の単量体を示している。WTレーンにおいてのみ、両バンドともに観察できない。すなわち、ランダムオリゴヌクレオチド結合選択アッセイにより、AUREO1(GST−AUREO1N)は、5’−TGACGT−3’(配列番号1)に結合することが示された。
【0035】
(6) AUREO1のDNAへの結合
AUREO1のDNAへの直接的な結合を、下記のゲルシフトアッセイ方法により確認した。
【0036】
青色光による、AUREO1とTGACGT含有DNAプローブとの結合の活性化は、次のようにして行った。GST−AUREO1タンパク質全長を、完全暗条件化で、ベクターpGEX6P1を用いて大腸菌(Rosetta)に発現させた。得られたタンパク質を弱い赤色光(<0.02μmole m−2s−1)条件下で分離精製し、そのGSTをPreScissionプロテアーゼ(GE Healthcare社製)によって切り取った。AUREO1タンパク質およびDIGで標識したオリゴヌクレオチドプローブを混合し、暗条件下もしくは青色光の短照射条件下(470nm LED、100μmole m−2s−1、5分間)に置いた。30分間の結合反応後、AUREO1標的ヌクレオチド複合体を、暗条件下で電気泳動により分離した(観察のために短時間、安全な弱い赤色光下に置いた)。図6は、ゲルシフトアッセイにおいて、第1列は30分間暗条件下に置いたもの、第2列は25分間暗条件下の後に青色光を5分間短照射したもの、第3列は青色光を5分間短照射後に25分間暗条件化に置いたときの結果を示している。同図に示すように、青色光を照射した第2列および第3列において矢印部にバンドが見られ、AUREO1とターゲットDNA配列との結合が青色光によって活性化されていることを表している。
【0037】
(7) 青色光誘導光形態形成機能
オーレオクロムの青色光誘導光形態形成機能を、下記のRNAi法により確認した。
【0038】
4つのdsRNA断片(LOVAUREO1、bZIPAUREO1、LOVAUREO2およびbZIPAUREO2)はPCR法により合成した。プライマーセットは、bZIPAUREO1F(配列番号48)、bZIPAUREO1R(配列番号49)、LOVAUREO1F(配列番号50),LOVAUREO1R(配列番号51)、bZIPAUREO2F(配列番号52)、bZIPAUREO2R(配列番号53)、LOVAUREO2F(配列番号54),LOVAUREO2R(配列番号55)を用いた。各断片の長さは、おおよそ500塩基対であった。これらのdsRNAをTEバッファー(10mM Tris、1mM EDTA、pH8.0)に5μg/μLになるよう懸濁し、使用時まで冷凍保存した。2または4つのdsRNA混合物は、長さ約10mmのフシナシミドロのセグメントに顕微導入し、導入した個々のセグメントは、数回培地を交換しつつ、6−7ヶ月間、インキュベータ内で別々に培養した(12時間明条件/12時間暗条件、20℃)。導入溶液には、2または4種類のdsRNA混合物(全4種類のdsRNA、LOVAUREO1+bZIPAUREO1またはLOVAUREO2+bZIPAUREO2)7−10μg/μL、20−30mM K2SO4、17−25mM MgSO4、0.07−0.1mM EGTA,40−60mM ソルビトールおよび17−25mM PIPES−K(pH 7.0)を混合したものを用いた。
【0039】
図7(A)は、インジェクション後37日目における、セグメントの形態を示す写真である。写真aは、バッファーのみをインジェクションしたコントロールで、写真b〜eは、dsRNA混合物をマイクロインジェクションしたセグメントから再生した管の異常形態(aureo1&aureo2)を示す写真である。管の狭窄部は日毎の成長制限を表しており、コントロールには見られなかった。
【0040】
また、図7(B)は、青色光による分枝誘導をAUREO1およびAUREO2のRNAiにより同時阻害したときの分枝率を示すグラフである。5−6ヶ月間培養後、管の一部位(幅200μm)に青色光を照射し、照射した管が分枝した割合(%)を示したもので、グラフの左柱aは青色光を10時間照射し、右柱bは24時間照射したものである。また、コントロール区はバッファーのみをインジェクションした細胞で、aureo1&aureo2区はdsRNA混合物をインジェクションした細胞である。コントロールの分枝率は、10時間照射が52.6%(n=19)、24時間照射が91.7%(n=24)であった。一方、dsRNA混合物をインジェクションした細胞では、10時間照射で4.3%(n=46)、24時間照射で28.6%(n=34)のみが分枝した。グラフ内右上に挿入した写真は、青色光を照射した近接の管の一部位(幅200μm)における、照射後0、1、7および9時間後の、通常の青色光誘導分枝を示している(写真最下部のバーの長さは100μm)。
【0041】
図7(C)は、前記RNAiによりAUREO1又はAUREO2のいずれかがノックアウトされたセグメントの写真である(最下部のバー長さは200μm)。AUREO2のみをノックアウトしたaureo2細胞からは、多くの未成熟な生殖器官が発生した。一方、AUREO1のみをノックアウトしたaureo1細胞は、aureo1&aureo2の表現型(図7(A))に類似していた。
【0042】
さらに、RT−PCRを用いて、24時間、青色光を照射した管について、細胞内のAUREO1およびAUREO2のmRNAの産生を調べた。図7(D)に示すように、#1A1および#1B3において、オーレオクロムmRNAが全く産生されず、分枝能が完全に欠失していた。これらの結果は、AUREOCHROMEが、青色光による光形態形成における光受容タンパク質であることを示している。
【産業上の利用可能性】
【0043】
以上のように、本発明の遺伝子発現制御方法によれば、細胞に悪影響を及ぼすことなく、簡単なシステムで遺伝子の発現の制御が可能になる。本発明は、遺伝子の発現制御を実施する全ての分野に適用可能であり、その用途は、医学、農学、薬学等の生物に関連する分野全てに適用可能である。
【図面の簡単な説明】
【0044】
【図1】図1(A−1)、(A−2)、(B−1)および(B−2)は、本発明の遺伝子発現制御方法を説明するための模式図である。
【図2】図2は、オーレオクロム(AUREO1およびAUREO2)の構造を示す模式図である。
【図3(A)】図3(A)は、本発明の実施例において、CBP−LOVドメイン(LOVAUREO1、LOVAUREO2)を導入した大腸菌のペレットを示す写真である。
【図3(B)】図3(B)は、本発明の実施例において、精製したCBP−LOVAUREO1の吸収スペクトルを示すグラフである。
【図3(C)】図3(C)は、本発明の実施例において、青色光照射前後のスペクトルの吸収差を示すグラフである。
【図3(D)】図3(D)は、本発明の実施例において、AUREO1の発色団がFMNであることを示す薄層クロマトグラフィの写真である。
【図4】図4は、本発明の実施例において、GFP−AUREO融合タンパク質の細胞内局在を示す顕微鏡写真である。
【図5】図5は、本発明の実施例におけるゲルシフトアッセイ結果を示す写真である。
【図6】図6は、本発明の実施例におけるゲルシフトアッセイ結果を示すその他の写真である。
【図7(A)】図7(A)は、本発明の実施例におけるセグメントから再生した管の形態を示す写真である。
【図7(B)】図7(B)は、本発明の実施例における分枝誘導率の結果を示すグラフである。
【図7(C)】図7(C)は、本発明の実施例におけるRNAiによる表現型の結果を示す写真である。
【図7(D)】図7(D)は、本発明の実施例におけるRT−PCR結果を示す写真である。
【特許請求の範囲】
【請求項1】
細胞内の目的遺伝子の転写を調節することで前記目的遺伝子の発現を制御する遺伝子発現制御方法であって、
前記目的遺伝子の上流に、プロモーター配列を配置し、
前記プロモーター配列の上流に、5’−TGACGT−3’の結合配列を含む応答配列を配置し、
前記細胞内において、AUREO1タンパク質を存在させ、
前記AUREO1タンパク質は、青色光の照射下では、前記結合配列に結合し、青色光の非照射下では、前記結合配列から遊離し、
前記青色光の照射または非照射によるAUREO1タンパク質の前記結合配列への結合または遊離により、前記プロモーター配列の活性または不活性を調節することで、前記目的遺伝子の転写を調節することを特徴とする遺伝子発現制御方法。
【請求項2】
青色光の照射により、前記AUREO1タンパク質を、前記応答配列に結合させることにより前記プロモーター配列を活性化して前記目的遺伝子の転写を実施し、
前記青色光の非照射により、前記AUREO1タンパク質を前記結合配列から遊離させることにより前記目的遺伝子の転写を実施しない請求項1記載の遺伝子発現制御方法。
【請求項3】
前記細胞内に、さらに、アクチベーターを存在させ、
前記アクチベーターは、前記応答配列に結合することにより前記プロモーター配列を活性化し、
青色光の照射により、前記AUREO1タンパク質を前記結合配列に結合させることで、
前記アクチベーターの前記応答配列への結合を阻止して前記目的遺伝子の転写を実施せず、
前記青色光の非照射により、前記AUREO1タンパク質を前記結合配列から遊離させることで、前記アクチベーターを前記応答配列に結合させて前記目的遺伝子の転写を実施する請求項1記載の遺伝子発現制御方法。
【請求項4】
前記AUREO1タンパク質が、下記の(A)、(B)または(C)のタンパク質である請求項1から3のいずれか一項に記載の遺伝子発現制御方法。
(A) 配列番号2のアミノ酸配列からなるタンパク質
(B) 配列番号2のアミノ酸配列において、1個または数個のアミノ酸残基が、欠失、置換または付加されたアミノ酸配列からなり、青色光の照射下では、前記結合配列に結合し、青色光の非照射下では、前記結合配列から遊離するタンパク質。
(C) 配列番号2のアミノ酸配列に対し、68%以上の相同性を有するアミノ酸配列からなり、青色光の照射下では、前記結合配列に結合し、青色光の非照射下では、前記結合配列から遊離するタンパク質。
【請求項5】
前記応答配列が、5’−TGACGT−3’の結合配列に加えまたは代えて、
5’−TGACGC−3’の結合配列を含む請求項1から4のいずれか一項に記載の遺伝子発現制御方法。
【請求項6】
前記青色光が、波長350nmから500nmの範囲の光を含む請求項1から5のいずれか一項に記載の遺伝子発現制御方法。
【請求項7】
前記細胞が、細菌、酵母、動物細胞および植物細胞からなる群から選択される少なくとも一つの細胞である請求項1から6のいずれか一項に記載の遺伝子発現制御方法。
【請求項8】
請求項1から3のいずれか一項に記載の遺伝子発現制御方法に使用する遺伝子組換え用ベクターであって、
前記目的遺伝子のクローニングサイト、前記プロモーター配列および前記応答配列を含み、
前記目的遺伝子のクローニングサイトの上流に、前記プロモーター配列が配置され、
前記プロモーター配列の上流に、前記応答配列が配置されている遺伝子組換え用ベクター。
【請求項9】
さらに、AUREO1遺伝子を含む請求項8記載の遺伝子組換え用ベクター。
【請求項10】
請求項1から3のいずれか一項に記載の遺伝子発現制御方法に使用する遺伝子発現制御用キットであって、請求項8または9記載のベクターを含むキット。
【請求項11】
請求項1から3のいずれか一項に記載の遺伝子発現制御方法に使用する遺伝子発現制御用キットであって、
請求項8記載のベクターと、
AUREO1遺伝子を発現可能な状態で含む遺伝子組み換え用ベクターと
を含むキット。
【請求項1】
細胞内の目的遺伝子の転写を調節することで前記目的遺伝子の発現を制御する遺伝子発現制御方法であって、
前記目的遺伝子の上流に、プロモーター配列を配置し、
前記プロモーター配列の上流に、5’−TGACGT−3’の結合配列を含む応答配列を配置し、
前記細胞内において、AUREO1タンパク質を存在させ、
前記AUREO1タンパク質は、青色光の照射下では、前記結合配列に結合し、青色光の非照射下では、前記結合配列から遊離し、
前記青色光の照射または非照射によるAUREO1タンパク質の前記結合配列への結合または遊離により、前記プロモーター配列の活性または不活性を調節することで、前記目的遺伝子の転写を調節することを特徴とする遺伝子発現制御方法。
【請求項2】
青色光の照射により、前記AUREO1タンパク質を、前記応答配列に結合させることにより前記プロモーター配列を活性化して前記目的遺伝子の転写を実施し、
前記青色光の非照射により、前記AUREO1タンパク質を前記結合配列から遊離させることにより前記目的遺伝子の転写を実施しない請求項1記載の遺伝子発現制御方法。
【請求項3】
前記細胞内に、さらに、アクチベーターを存在させ、
前記アクチベーターは、前記応答配列に結合することにより前記プロモーター配列を活性化し、
青色光の照射により、前記AUREO1タンパク質を前記結合配列に結合させることで、
前記アクチベーターの前記応答配列への結合を阻止して前記目的遺伝子の転写を実施せず、
前記青色光の非照射により、前記AUREO1タンパク質を前記結合配列から遊離させることで、前記アクチベーターを前記応答配列に結合させて前記目的遺伝子の転写を実施する請求項1記載の遺伝子発現制御方法。
【請求項4】
前記AUREO1タンパク質が、下記の(A)、(B)または(C)のタンパク質である請求項1から3のいずれか一項に記載の遺伝子発現制御方法。
(A) 配列番号2のアミノ酸配列からなるタンパク質
(B) 配列番号2のアミノ酸配列において、1個または数個のアミノ酸残基が、欠失、置換または付加されたアミノ酸配列からなり、青色光の照射下では、前記結合配列に結合し、青色光の非照射下では、前記結合配列から遊離するタンパク質。
(C) 配列番号2のアミノ酸配列に対し、68%以上の相同性を有するアミノ酸配列からなり、青色光の照射下では、前記結合配列に結合し、青色光の非照射下では、前記結合配列から遊離するタンパク質。
【請求項5】
前記応答配列が、5’−TGACGT−3’の結合配列に加えまたは代えて、
5’−TGACGC−3’の結合配列を含む請求項1から4のいずれか一項に記載の遺伝子発現制御方法。
【請求項6】
前記青色光が、波長350nmから500nmの範囲の光を含む請求項1から5のいずれか一項に記載の遺伝子発現制御方法。
【請求項7】
前記細胞が、細菌、酵母、動物細胞および植物細胞からなる群から選択される少なくとも一つの細胞である請求項1から6のいずれか一項に記載の遺伝子発現制御方法。
【請求項8】
請求項1から3のいずれか一項に記載の遺伝子発現制御方法に使用する遺伝子組換え用ベクターであって、
前記目的遺伝子のクローニングサイト、前記プロモーター配列および前記応答配列を含み、
前記目的遺伝子のクローニングサイトの上流に、前記プロモーター配列が配置され、
前記プロモーター配列の上流に、前記応答配列が配置されている遺伝子組換え用ベクター。
【請求項9】
さらに、AUREO1遺伝子を含む請求項8記載の遺伝子組換え用ベクター。
【請求項10】
請求項1から3のいずれか一項に記載の遺伝子発現制御方法に使用する遺伝子発現制御用キットであって、請求項8または9記載のベクターを含むキット。
【請求項11】
請求項1から3のいずれか一項に記載の遺伝子発現制御方法に使用する遺伝子発現制御用キットであって、
請求項8記載のベクターと、
AUREO1遺伝子を発現可能な状態で含む遺伝子組み換え用ベクターと
を含むキット。
【図1】


【図2】

【図3(A)】

【図3(B)】

【図3(C)】
【図3(D)】
【図4】
【図5】
【図6】
【図7(A)】
【図7(B)】
【図7(C)】

【図7(D)】
【図2】
【図3(A)】
【図3(B)】
【図3(C)】
【図3(D)】
【図4】
【図5】
【図6】
【図7(A)】
【図7(B)】
【図7(C)】
【図7(D)】
【公開番号】特開2009−50175(P2009−50175A)
【公開日】平成21年3月12日(2009.3.12)
【国際特許分類】
【出願番号】特願2007−217666(P2007−217666)
【出願日】平成19年8月23日(2007.8.23)
【出願人】(304028346)国立大学法人 香川大学 (285)
【出願人】(504157024)国立大学法人東北大学 (2,297)
【Fターム(参考)】
【公開日】平成21年3月12日(2009.3.12)
【国際特許分類】
【出願日】平成19年8月23日(2007.8.23)
【出願人】(304028346)国立大学法人 香川大学 (285)
【出願人】(504157024)国立大学法人東北大学 (2,297)
【Fターム(参考)】
[ Back to top ]