
部位特異的タンパク質結合体を調製する方法
【課題】タンパク質−ポリマー結合体を迅速にかつ効率的に調製する方法を提供すること。
【解決手段】本発明は、インスリン−ポリマー結合体を含む、タンパク質−ポリマー結合体を迅速にかつ効率的に調製する単一工程の方法に関する。本発明の方法によると、タンパク質と親水性のポリマーを、少なくとも1種の有機溶媒および少なくとも1種の金属キレート剤の存在下で、上記タンパク質とポリマーの結合体の形成が促進される条件下で、接触させる。そのように、本発明は、インスリンのような選択されたタンパク質とポリ(エチレングリコール)との残基PheB1での位置特異的改変に関する。本発明はまた、生分解性ポリマーの結合体をカプセル化する薬学的処方物を提供する。
【解決手段】本発明は、インスリン−ポリマー結合体を含む、タンパク質−ポリマー結合体を迅速にかつ効率的に調製する単一工程の方法に関する。本発明の方法によると、タンパク質と親水性のポリマーを、少なくとも1種の有機溶媒および少なくとも1種の金属キレート剤の存在下で、上記タンパク質とポリマーの結合体の形成が促進される条件下で、接触させる。そのように、本発明は、インスリンのような選択されたタンパク質とポリ(エチレングリコール)との残基PheB1での位置特異的改変に関する。本発明はまた、生分解性ポリマーの結合体をカプセル化する薬学的処方物を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
(関連出願)
本出願は、本明細書に全体の内容を参考として援用している、2003年4月11日出願の米国特許仮出願番号第60/462,364号に基づく優先権を主張する。
【0002】
(発明の分野)
本発明は、非結合体タンパク質の性質より優れた性質を有する化学的に改変されたタンパク質結合体に関する。詳細には、本発明は、非常に簡素化され、費用有効的であり、量産可能な、親水性ポリマーによりタンパク質を改変する方法に関する。より詳細には、本発明は、インスリンのような、選択されたタンパク質のポリエチレングリコールによる部位特異的改変に関する。本発明は、親水性タンパク質との部位特異的改変を有するタンパク質を包含する、生分解性ポリマーベースの薬剤送達処方物に関する。
【背景技術】
【0003】
(関連技術の説明)
PEG化インスリン誘導体を産生する種々の方法は、公知である。Davisら(米国特許第4,179,337号)は、PEGとタンパク質とのリンカーとしてトリクロロ−s−トリアジン(塩化シアヌル)を用いたPEG−インスリン構築物の合成を、記述した。彼等は、大過剰(50×)の塩化シアヌル活性化PEG(2000Da)をホウ酸緩衝液(pH9.2)中のインスリンと2時間反応させる合成スキームに従った。本発明者らは、部分的に活性な(〜50%)PEG−インスリン結合体(それは非免疫原性であり非抗原性である)を、産生することができた。Obermeierら(カナダ特許番号第1,156,217)は、上に引用したDavis特許の実施例Xに従ったPEG−インスリン結合体の調製は、約50%のトリ−PEG−インスリンを含有する結合体の不均質な混合物を産生し、他の可能なPEG−インスリン誘導体の組合せ(モノ−PEGインスリンおよびジ−PEG−インスリン)は残基PheB1で置換されていなかったことを見出した。
【0004】
Obermeierら(カナダ特許番号第1,156,217)は、残基PheB1で特異的に改変されたPEG−インスリン結合体の合成を記述している。彼等の発明の基本は、残基GlyA1およびLysB29の上記反応性アミンを、アルカリ条件下で、有機溶媒(例えば、DMF、DMSO、ピリジンなど)中で、tert−ブチルオキシカルボニル(t−boc)基またはメチルスルホニルエチルオキシカルボニル(Msc)基により、保護する工程を含んでいる。(モノ−、ジ−およびトリ−)保護されたインスリンの複雑な混合物から、NαA1,NεB29−ビス−保護されたインスリン種は、従来のクロマトグラフィー技術で単離された。単離に続いて、上記純粋なNαA1,NεB29−ビス−保護されたインスリンを、活性化(例えば、酸クロライドまたはイソシアネート)PEG誘導体と反応させ、引き続き、ペプチド化学で通例の技術を用いて保護基を除去した。本発明者らは、GlyA1およびLysB29のアミノ基が、アルカリ反応条件下では、PheB1のアミノ基より、より反応性であったことを観察した。彼等は、それらの部位特異的mPEG(1500)−B1−インスリン結合体が、ウサギの血糖レベルの低下に関して100%インスリン効果(モル基準で計算した)を有することを決定した。
【0005】
Geigerら(D.Branderburg,and A.Wollmer(eds.)Insulin:Chemistry、Structure、and Function of Insulin and Related Hormones、Walter de Gruyter & Co.,New York、pp.409〜415、1980)およびEhratら(Biopolymers、22、569〜573、1983)は、Obermeierらにより記載された多段工程方法に類似する、保護/結合/脱保護スキームを用いて、調製された残基PheB1での特異的な改変PEG−インスリン付加物について記述している。GeigerらおよびEhratらは、上記PEG(1500)−B1−インスリン結合体が天然のインスリンより抗原性がずっと低くそしてずっとより安定(肝臓の酵素に対して)であるということを観察した。他のPEG−インスリン調製物(Calicetiら、STP Pharma Sci、9、107〜113、1999;Uchioら、Advanced Drug Delivery Reviews、35、289〜306、1999;Hindsら、Bioconj.Chem.11、195〜201、2000;Hindsら、Advanced Drug Delivery Reviews、54:505〜530、2002)は以下のいずれかである:1)上記に概説されている基礎的3工程の保護/結合/脱保護のスキームを中心としている、2)結果が上記インスリン分子の非特異的改変になるもの、または3)最も効果的な結合体、即ち,PEG−B1−インスリンを産生しない。
【0006】
Liuら(米国特許番号第6,323,311B1号)は、PEG−B1−インスリン結合体の合成の有用な方法を記述している。この方法は、Obermeierの3工程の保護/結合/脱保護スキームの延長であるが、工程間の反応中間物の単離を必要としない(すなわち一回分式合成)。このように、上記インスリンは、残基GlyA1およびLysB29で保護され、直ちにPEGと反応し、引き続きいずれの種の単離の前には、脱保護される。本発明は、それらの一回分式反応で50%までもの正しい位置異性体(即ち,PEG−B1−インスリン)の収率を得、そして次の誘導体化にリサイクルできる未反応インスリンを30%回収し得ることを請求している。これらの構築物の調製が、てきぱきと実行できたと仮定して、完了するまでには、少なくとも5日間はかかる。さらに本発明は、受容可能な結果を達成するために大過剰のPEG試薬を必要とする。本発明の生成物は、効果的であるが、それらの調製は、上記タンパク質が、長時間、タンパク質にとって有害な環境(高pHおよび低pH)で三つの反応工程を経なければならないことを必要としている。
【発明の概要】
【課題を解決するための手段】
【0007】
本発明は、単一工程で、インスリンB鎖のN末端(PheB1)に特異的にPEG化された高純度のインスリン誘導体の単純な調製方法を提供することにより、インスリンをPEG化する方法の従来技術の欠点を取り扱うものである。PheB1でのPEG化が、もっとも可能性の少ない反応性生物であることを示している先行経験(例えば、Calicetiら、1999,上記)とは対照的に、本発明の方法は、PEG化の優勢な部位になるようPheB1アミノ末端の相対的反応性を高めるために、pHの特定の制御条件、金属イオンのキレート剤の使用および有機溶媒の添加を用いている。インスリンに与えられている多くの有益な性質(例えば、低減した免疫原性/抗原性;増加したタンパク質分解に対する安定性、化学的安定性および物理的安定性;増加した循環半減期;増加した水溶性溶解度/有機的溶解度;全生物活性)を考慮すると、残基PheB1への部位特異的PEG化を通じて、この結果を達成するために、単純であり、費用有効的であり、容易にスケールアップできる工程は当該分野では顕著な進展である。
【0008】
(発明の概要)
本発明は、タンパク質−ポリマー結合体を調製する単一工程の方法の発見に基づいている。本発明はまた、親水性タンパク質との部位特異的な改変を有する生分解性ポリマーベースの薬剤送達処方物に関する。特定の実施形態では、本発明は、置換の部位が優勢的に残基PheB1(上記B鎖のN末端)であるPEG化インスリン誘導体の単一工程の合成のための方法を提供する。そのような誘導体が、天然のインスリンより物理的に酵素的により安定であることは当該分野では周知である。さらに、上記誘導体は、インスリンより水系/有機系でより溶解性である。さらにこれらの誘導体は免疫原性がより低く、長時間の循環半減期を有していることが分かっている。
【0009】
本発明の顕著な利点は、上記反応が望ましくない副反応(例えば、脱アミド化、トランスアミド化、酸化等)が無視し得る中性に近い条件で起きることである。別の利点は、比較的少量のポリマー(例えば,PEG試薬)の使用、従って、コストの低減化である。上記の結果できたタンパク質−ポリマー結合体(例えば、PEG化インスリン)は、例えばヒトの治療において、それ自身使用し得るかまたは、米国特許出願第2002/0155158号に開示されているような徐放性送達媒体にカプセル化し得る。
【0010】
従って、一つの実施形態では、本発明は、少なくとも1種の有機溶媒および少なくとも1種の金属キレート剤の存在下で、上記タンパク質とポリマーの結合体が生成する条件下で、インスリンタンパク質と親水性ポリマーとを接触させ、タンパク質−ポリマーの結合体を調製する方法を提供する。そこで、上記結合体は、カラムクロマトグラフィーのような種々の標準的技術で単離され得る。
【0011】
本発明の特定の実施形態では、親水性ポリマーは、ポリエチレングリコール、ポリエチレングリコール/ポリプロピレングリコール共重合体、ポリオキシエチル化グリセリンおよびそれらの線状誘導体、分岐誘導体およびアミノ反応性誘導体からなる群より選択される。適切なアミノ反応性誘導体としては、例えば、アルデヒド、PEGカルボン酸のN−ヒドロキシコハク酸イミドエステル、PNP炭酸塩およびベンゾトリアゾール末端親水性ポリマー誘導体が挙げられる。典型的には、親水性ポリマーおよびインスリンタンパク質は、モル濃度比、約10:1〜1:1で存在する。
【0012】
本発明で使用される適切な有機溶媒としては、限定しないが、例えば、エタノール、メタノール,DMSO、ジオキサン、DMFおよびNMPなどの水と混和可能な有機溶媒を含む、種々の公知の溶媒が挙げられる。典型的には、上記有機溶媒は、濃度が約0.1〜10%で存在する。
【0013】
本発明で使用される適切な金属キレート剤としては、限定されないが、多価(例えば2価)金属イオンキレート剤、例えばEDTA、デフェロキサミン(DEF)、ジエチレントリアミン五酢酸(DTPA)、エチレングリコールビス(2−アミノエチルエーテル)N,N,N’,N’−四酢酸(EGTA)を含む種々の公知の化合物が挙げられる。一般的に、上記キレート剤は、濃度が約0.1〜10mMで存在する。
【0014】
本発明の特定の実施形態では、上記インスリンタンパク質と親水性ポリマー(例えば,PEG)を水溶液中で、タンパク質濃度が約0.1〜5重量%で、接触させる(即ち反応させまたは、結合させる)。本発明の別の実施形態では、上記インスリンタンパク質および親水性ポリマーを、水溶液中pHが約5.0〜7.5、好ましくは約7.0で接触させる。これは、インスリン−ポリマー結合体の上記正しい位置異性体の50%までの収率という結果を生じる。別の特定の実施形態では、上記親水性ポリマーとインスリンタンパク質を約4℃〜50℃、好ましくは約15℃〜25℃の温度で接触させる。
【0015】
一度生成されると、上記タンパク質−ポリマー結合体は、望ましくない副反応性生物および未反応インスリンタンパク質から分離される。これは、クロマトグラフィーのような種々の公知の技術を用いて達成され得る。特定の実施形態では、イオン交換クロマトグラフィーが用いられる。
【0016】
さらに別の実施形態では、本発明の方法には、上記結合生成物を単離する工程に先立ってインスリンタンパク質と親水性のポリマーとの反応(即ち結合)をクエンチする工程が、さらに含まれる。特定の実施形態では、これは、上記反応のpHを約1〜4までに低下させることにより達成される。
【0017】
本発明の方法により産生される特定のタンパク質−ポリマー結合体は、例えば、インスリン−ポリマー結合体、好ましくは、インスリン−PEG結合体(PEG化インスリン)を包含する。これは、配列番号1で示される上記配列を有するヒトインスリンおよび関連ファミリーメンバーのような任意のインスリンもしくはインスリン関連タンパク質を包含し得る。特定の実施形態では、上記インスリンは、残基GlyA1もしくはLysB29での反応がそれ程なく、残基PheB1で特異的に反応(PEG化)する。結果としてできるPEG化インスリンは、当該分野で周知の任意の適切な処方物で治療的に投与され得る。特定の実施形態では、上記結合体は、例えば、投与の前に生分解性ポリマーで上記結合体をカプセル化することによる徐放性処方物で投与される。
【0018】
本発明は、さらに以下を提供する。
(項目1)
タンパク質−ポリマー結合体を調製する方法であって、以下の工程:
(a)インスリンタンパク質を、少なくとも1種の有機溶媒および少なくとも1種の金属キレート剤の存在下、該タンパク質と該ポリマーの結合体の形成を促進する条件下で、親水性ポリマーと接触させる工程;および
(b)該結合体を単離する工程
を包含する、方法。
(項目2)
前記インスリンタンパク質が、ヒトインスリンを含む、項目1に記載の方法。
(項目3)
前記親水性ポリマーが、ポリエチレングリコール、ポリエチレングリコール/ポリプロピレングリコール共重合体、ポリオキシエチル化グリセリンならびにそれらの線状、分岐およびアミノ反応性誘導体からなる群より選択される、項目1または2に記載の方法。
(項目4)
前記アミノ反応性誘導体が、アルデヒド、N−ヒドロキシコハク酸イミド、PNP炭酸塩およびベンゾトリアゾール(benzotrizole)末端親水性ポリマーからなる群より選択される、項目3に記載の方法。
(項目5)
前記親水性ポリマーおよびインスリンタンパク質が、モル比で約10:1〜1:1で接触させられる、項目1〜4のいずれか1項に記載の方法。
(項目6)
前記有機溶媒が、エタノール、メタノール,DMSO、ジオキサン、DMFおよびNMPからなる群より選択される、項目1〜5のいずれか1項に記載の方法。
(項目7)
前記有機溶媒が、約0.1〜10%の濃度で存在する、項目1〜6のいずれか1項に記載の方法。
(項目8)
前記インスリンタンパク質および親水性ポリマーが、約0.1〜5.0%のタンパク質濃度で接触させられる、項目1〜7のいずれか1項に記載の方法。
(項目9)
前記インスリンタンパク質および親水性ポリマーが、pHが約5.0〜7.5で接触させられる、項目1〜8のいずれか1項に記載の方法。
(項目10)
前記キレート剤が、多価金属イオンキレート剤、EDTA、デフェロキサミン(DEF)、ジエチレントリアミン五酢酸(DTPA)およびエチレングリコールビス(2−アミノエチルエーテル)N,N,N’,N’−四酢酸(EGTA)からなる群より選択される、項目1〜9のいずれか1項に記載の方法。
(項目11)
前記キレート剤が、約0.1〜10mMの濃度で存在する、項目1〜10のいずれか1項に記載の方法。
(項目12)
前記インスリンタンパク質および親水性ポリマーが、約4〜50℃の温度で接触させられる、項目1〜11のいずれか1項に記載の方法。
(項目13)
前記方法が、前記結合体を単離する前に、該結合体の形成をクエンチする工程をさらに含む、項目1〜12のいずれか1項に記載の方法。
(項目14)
前記クエンチングが、該pHを約1〜4に低下させることにより達成される、項目13に記載の方法。
(項目15)
前記単離が、クロマトグラフィーにより達成される、項目1〜14のいずれか1項に記載の方法。
(項目16)
前記クロマトグラフィーが、イオン交換クロマトグラフィーを含む、項目15に記載の方法。
(項目17)
前記結合体を、生分解性ポリマーでカプセル化する工程をさらに含む、項目1〜16のいずれか1項に記載の方法。
(項目18)
インスリン−PEG結合体の調製方法であって、以下の工程:
(a)インスリンを、少なくとも1種の有機溶媒および少なくとも1種の金属キレート剤の存在下、該インスリンとPEGとの結合体の形成を促進する条件下で、PEGと接触させる工程;および
(b)該結合体を単離する工程
を包含する、方法。
(項目19)
前記インスリンが、ヒトインスリンを含む、項目18に記載の方法。
(項目20)
前記PEGが、アルデヒド、N−ヒドロキシコハク酸イミド、PNP炭酸塩およびベンゾトリアゾール末端親水性ポリマーからなる群より選択されるアミノ反応性PEG誘導体を含む、項目18または19に記載の方法。
(項目21)
前記PEGおよびインスリンが、モル比で約10:1〜1:1で接触させられる、項目18〜20のいずれか1項に記載の方法。
(項目22)
前記PEGおよびインスリンが、約0.1〜5.0%のタンパク質濃度で接触させられる、項目18〜21のいずれか1項に記載の方法。
(項目23)
前記PEGおよびインスリンが、pHが約5.0〜7.5で接触させられる、項目18〜22のいずれか1項に記載の方法。
(項目24)
前記方法が、前記結合体を単離する前に、該結合体の形成をクエンチする工程をさらに含む、項目18〜23のいずれか1項に記載の方法。
(項目25)
前記クエンチングが、該pHを約1〜4に低下させることにより達成される、項目24に記載の方法。
(項目26)
前記単離が、イオン交換クロマトグラフィーにより達成される、項目18〜25のいずれか1項に記載の方法。
(項目27)
前記結合体を、生分解性ポリマーでカプセル化する工程をさらに含む、項目18〜26のいずれか1項に記載の方法。
本発明の他の実施形態は、以下の詳細な説明および実施例から明らかになる。
【図面の簡単な説明】
【0019】
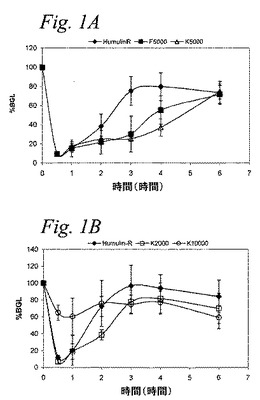
【図1】図1Aおよび1Bは、正常ラットに静脈内注入を行なった後のPEG−インスリン結合体のインビボ薬力学に対する部位置換の効果(図1A)またはポリマー分子量の効果(図1B)を示すグラフである。
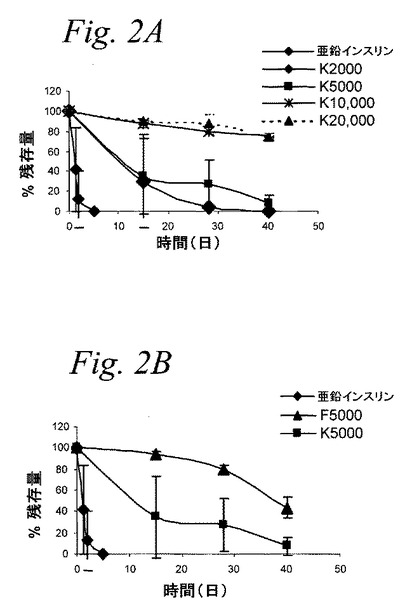
【図2】図2Aおよび2Bは、ポリマー連結の同じ部位で異なる分子量のインスリン−PEG結合体(図2A)および同分子量のPEGFでポリマー連結の部位が異なるインスリン−PEG結合体(図2B)の、物理的凝集に起因する可溶性インスリンの損失を示すグラフである。
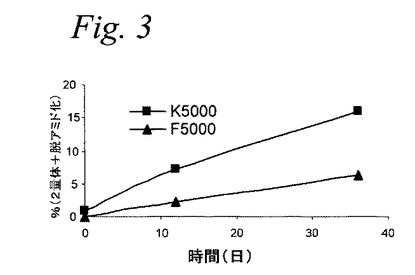
【図3】図3は、2種のmPEG(5000Da)−インスリン位置異性体の化学的安定性を示すグラフである。
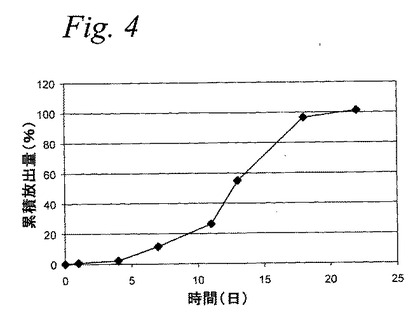
【図4】図4は、PGLAミクロスフェアからのF5000(PEG−インスリン)のインビトロ累積放出を示すグラフである。
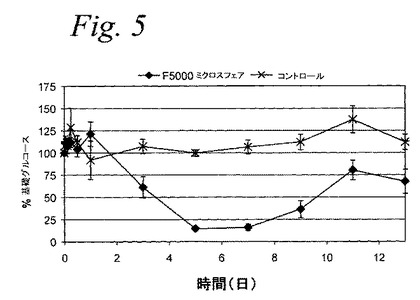
【図5】図5は、糖尿病ラットへ皮下投与後のF5000(PEG−インスリン)ミクロスフェアのインビボ薬力学を示すグラフである。
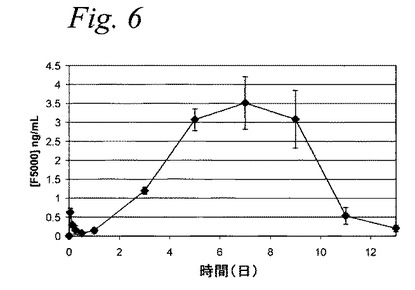
【図6】図6は、糖尿病ラットへ皮下投与後のF5000(PEG−インスリン)ミクロスフェアのインビボ薬物動態を示すグラフである。
【発明を実施するための形態】
【0020】
(詳細な説明)
本発明は、タンパク質−ポリマー結合体を迅速におよび効率的に調製する単一工程の方法に関する。本方法は、少なくとも1種の有機溶媒および少なくとも1種の金属キレート剤の存在下で、中性近辺の条件下で、タンパク質と親水性ポリマーを反応させる工程を包含する。本発明の特定のタンパク質−ポリマー結合体は、最小量のPEG試薬および温和な条件を用いて、アミノ末端PheB1にPEG化されたインスリンを包含する。先行技術は、PheB1が、立体的に嵩高い高分子試薬に対して、通常、インスリンの最も反応性が低いアミノ基であることを示している(例えば、Calicetiら、1999、上記)にもかかわらず、驚くべきことに、本発明の方法は、PheB1が、親水性ポリマーによる改変で利用可能な最も反応性の高い基になるような条件を作り出す。これは、PheB1でPEG化されたインスリンが最高収率の生成物であり、かつ他の結合体および未反応インスリンから分離され得る単純な単一工程の反応を可能にする。後者は、望まれる場合、さらなる結合のためにリサイクルされ得る。PheB1でPEG化されたインスリン結合体は、血糖制御で測定されるように、完全な活性を保持し、かつ上記タンパク質の天然構造が保存されている。
【0021】
(親水性ポリマー)
用語「親水性ポリマー」は、限定しないが、ポリエチレングリコール、およびポリエチレングリコール/ポリプロピレングリコール共重合体、ポリオキシエチル化グリセリンならびに類似のポリマーを含む、如何なる水溶性線状ポリマー、分岐ポリマー、グラフト(forked)ポリマー、分岐−グラフトポリマー、デンドリマー状ポリマー、多枝(multi−armed)ポリマーまたは星型ポリマーをいう。好ましくは、上記ポリマーの分子量は、約500ダルトン〜約50000ダルトンの範囲である。本発明で使用する親水性ポリマーは、典型的には、アミノ、カルボキシル、スルフヒドリル、リン酸またはヒドロキシル基を介して、関心の生理活性分子との連結に組込まれる、少なくとも一つの反応性基を有する。ポリエチレングリコールのような本発明で使用される親水性ポリマーは、メトキシ基で一端が保護され、そして他の一端が生理活性分子の活性基への容易な結合のために活性化される標準的プロトコールに従って、調製され得る。例えば、米国特許番号第6,113,906号は、タンパク質のアミノ基との反応のために、「U型」(即ち、分岐)型形態のポリエチレングリコールにコハク酸コハク酸イミド反応性基または炭酸コハク酸イミド反応性基の使用を記載している。米国特許番号5,650,234号は、安定なウレタン結合を形成するためのタンパク質のアミノ基との反応に、ポリエチレングリコールのN−ヒドロキシベンゾトリアゾール炭酸塩誘導体、2−ヒドロキシピリミジン炭酸塩誘導体およびN−ヒドロキシ−2−ピロリジノン炭酸塩誘導体の使用を記載している。米国特許番号第5,672,662号は、安定なアミド結合を形成するためのタンパク質のアミノ基との反応のために、プロピオン酸および酪酸置換ポリエチレングリコールのコハク酸イミドエステルの使用を記載している。米国特許番号第5,446,090号は、タンパク質のスルフヒドリル基と安定なチオエーテル結合を形成するために、ポリエチレングリコールのビニル−スルホン誘導体の使用を記載している。米国特許番号第5,880,255号は、単純で、安定な2級アミン結合を形成するために、タンパク質のアミノ基との反応のためにポリエチレングリコールのトレシル(2,2,2,−トリフルオロエタンスルホニル誘導体の使用を記載している。米国特許番号第5,252,714号は、安定な2級アミン結合を生じるタンパク質のアミノ基との反応のため、ポリエチレングリコールのプロピオンアルデヒド誘導体の使用を記載している。そのような親水性ポリマーの生理活性分子への連結から生じる結合は、安定または不安定(即ち可逆的)に意図的に設計され得る。さらに本発明で使用される親水性ポリマーは、生理活性分子の活性基への結合を容易にする利用可能な二つの類似な(例えば、ホモ二官能性)、または類似しない(ヘテロ官能性)官能基を用いる標準的なプロトコールに従って調製され得る。例えば、WO126692A1は、タンパク質の改変のために、ヘテロ二官能性のポリエチレングリコール誘導体の使用を記載している。これらの特許の全体的な内容は、本明細書に参考として援用されている。
【0022】
一つの実施形態では、上記親水性ポリマーは、ポリ(エチレングリコール)(PEG)である。PEGは、化学式HO−(CH2CH2O)n−Hを有する線状または分岐の中性ポリエーテルをいう。PEGは水および多くの有機溶媒(例えば、ジクロロメタン、エタノール、トルエン、アセトンおよびクロロホルム)に非常に溶解性があり、種々のサイズ(分子量)および機能的構築物(例えば、アミノ−、カルボキシル−およびスルフヒドリル−末端)で容易に入手できる。PEGは、毒性がないことが分かっており、そして薬物(非経口剤、局所剤、坐薬、鼻スプレー)、食品および化粧品としてFDAにより認可されている。溶液中では,PEGは高度に水和されたポリマーで、各モノマー(エチレンオキシド単位)は、3分子の水と結合し得る。さらに、PEGは、水和水分子とより緩やかに結合している、7つの分子層の構造に影響を与える能力を有すると考えられている。水中での単一表面結合鎖の動きの分子シミュレーションは、上記ポリマーが、大きな程度の分節の自由度を提示することを示している。このように上記ポリマーは、水性環境中では、大きな流体力学的な容量を占めていると考えられている。これらの結果は、何故PEGがその存在から他のポリマー(天然および合成)を排除するのに著しく効果的であるかを説明するのに役立っている。上記の他のポリマーの排除は、PEGがタンパク質を拒絶し、他の合成ポリマーと二相系を形成する主たる推進力であり、このポリマーを非免疫原性および非抗原性にする。PEGが、タンパク質に共有結合的に連結する場合、それは典型的に、ポリマーの有利な特徴の多くを、結果として生じる結合体に、伝達する。上記の多くの有利な性質により、PEGはタンパク質の改変に非常に適している。
【0023】
本明細書で用いられているように、用語「PEG」は、PEG(「PEG試薬」)のアミノ反応性誘導体を含む、如何なるPEGポリマーをも包含する。タンパク質結合のための種々のPEG試薬は公知である。典型的なPEG試薬は、一つの末端がエーテル結合(例えばO−メチル)を有し、かつ他の末端が反応性基で官能基化されている線状PEGポリマーである。他のPEG試薬は、タンパク質への結合が、再び非反応性末端および反応性官能基の組合せによる、分岐またはデンドリマー状である。あるいは、類似もしくは非類似の反応性官能基の組合せを有するホモ−もしくはヘテロ−二官能性PEG試薬は、タンパク質へ結合するために使用され得る。PEG試薬の例としては、限定されないが、アルデヒド、N−ヒドロキコハク酸イミジルカルボネート、N−ヒドロキシコハク酸イミジルプロピオネート、p−ニトロフェニルカルボネート、またはベンゾトリアゾールカルボネート末端種またはPEGの他のアミノ反応性活性化種が挙げられる。
【0024】
上記PEGポリマーは、例えば、500ダルトンから50,000ダルトンの範囲の分子量を有し得る。上記反応性官能基は、リンカー基によりPEG鎖から分離され得る。必要に応じて、上記ポリマーは、上記PEGとリンカーとの間に分解性の内部結合を有する。従って、本発明の一つの実施形態では、PEGポリマーの反応性基は、タンパク質求核剤との反応のために親電子的に活性化され得る。親電子基の例としては、n−ヒドロキシコハク酸イミジルカルボネート、トレシルおよびアルデヒド官能基である。これらの官能基を有するPEG試薬は、タンパク質のアミノ基への共有的に結合を形成するように反応する。タンパク質アミノ基へのPEG結合のための好ましいPEG試薬は、プロピオン酸リンカーmPEG−SPAのmPEGコハク酸イミド活性エステルである。別の好ましいPEG試薬は、モノメトキシPEG−アルデヒド(mPEG−Ald)である。
【0025】
(インスリン−ポリマー結合体)
インスリン処方物の非経口的投与は、75年以上前のインスリンの発見以来、依然としてインスリン依存性糖尿病(IDDM)の処置に用いられる第一治療法である。現在の治療法を不適切にしている要因の多くは、上記インスリン分子に固有の本質的な欠点である。詳細には、インスリンは、弱い物理的安定性および化学的安定性、タンパク質分解の受け易さ、免疫原性および抗原性ならびに比較的短い血漿中半減期を含む、タンパク質製剤に典型的な多くの問題に直面する。
【0026】
タンパク質PEG化は組換えヒトタンパク質の治療効率を改善するのに使用されてきている。ほとんどの非経口的に投与されるタンパク質は、細網内皮系(RES)、腎臓、脾臓および肝臓により、身体から早く消失する。さらに、ろ過率は分子サイズ、電荷および問題のタンパク質の特異的なレセプターの存在に依存する。PEGのタンパク質への連結は、その分子サイズ、電荷およびレセプター結合能に影響を及ぼし、結合体の全体としての消失率を減少させるのに役立つ。
【0027】
さらに、酵素によるタンパク質の代謝は、治療用タンパク質の生物学的活性の早い損失につながる。タンパク質分解攻撃を受けやすい上記タンパク質のドメインを、立体的に遮蔽することにより、PEGは、生物学的に不活性にするタンパク質の分解を、減少させる。
【0028】
さらに、組換えヒトタンパク質でさえも、繰返し使用の後に免疫反応を、誘発する。上記治療用タンパク質の免疫原性決定基/抗原性決定基を立体的に覆い隠すことにより、PEG−連結は、通常非免疫原性および非抗原性の結合体になる。従って、全体として、PEG化による非経口的タンパク質の特質の変化の結果、血漿中の半減期およびタンパク質分解の抵抗性を増加させ、生じたPEG−タンパク質構築物の免疫原性および抗原性を減少させる。
【0029】
(インスリンタンパク質)
本明細書で使用される用語「インスリンタンパク質」は、任意の天然に存在するもしくは組換えインスリンタンパク質または、例えば、残基PheB1で結合され得る関連タンパク質をいう。従って、本発明に使用されるインスリンタンパク質としては、例えば、インスリンアナログ、インスリンホモログおよびインスリン誘導体が挙げられる。任意の適切な種、例えば、ヒト、ブタ、ウシ、イヌ、ラット、マウス、ヒツジまたはサルからのインスリンが、用いられ得る。好ましい実施形態において、インスリンはヒトインスリンである。
【0030】
ヒトインスリンは周知のタンパク質であり、限定されないが、Sigma Chemical CompanyおよびNovo Nordiskを含む、多くの業者から市販品で容易に入手可能である。天然に存在するヒトインスリンは、分子量およそ5,500ダルトンを有し、およそ51アミノ酸を含むタンパク質である。製造業者により、上記インスリンは、重量ベースではわずかに異なる活性を有し得るが、単位で規定されたインスリンの活性は標準である。種々の程度の生物学的活性を有するインスリンは、市販で入手可能である。例えば、低い作用形態、中程度の作用形態および即効性の作用形態のインスリンを購入することは可能である。インスリンと類似した作用を有するかまたはインスリンレセプターを増加させる非インスリン血糖低下剤としては、限定されないが、スルホニル尿素(例えば、グリベンクラミド、グリクラジド、グリピジド、グリブリド(glyburide)、クロルプロパミド、トリブタミド、トラザミド、アセトヘキサミドおよびグリモプリド(glimopride));チアゾリジンジオン(例えば、トログリタゾン(troglitazone)およびプログリタゾン(ploglitazone);αーグルコシダーゼインヒビター(例えば、アカルボースおよびミグリトール);並びに第3世代インスリン放出剤(例えば、KAD 1220、エトキソミール(etoxomir)およびレパグリニド(repaglinide))が挙げられる。
【0031】
上記インスリン分子は、二つのポリペプチド鎖、A鎖およびB鎖からなる。ヒトインスリンの上記アミノ酸配列は、本明細書中の配列番号1として提供される。上記A鎖は、21個のアミノ酸(A1〜A21と表示する)から構成され、そしてより長いB鎖は、30個のアミノ酸(B1〜B30)から構成される。これらの二つの鎖は、残基A7とB7との間ならびにA20とB19との間の二つの鎖間のジスルフィド結合により一緒に保持されているが、別の鎖内ジスルフィド結合は、残基A6およびA11を結合している。インスリンの3次元構造への安定化をさせるのに役立つ、上記の二つの鎖のアミノ酸残基間でまた、多数の非共有結合性相互作用が存在する。
【0032】
改変(例えばPEGによる)のための利用可能な3つの反応性アミノ基があり、即ち、それらは、A鎖N末端およびB鎖N末端(それぞれA1およびB1)に位置し、ならびにB鎖C末端(B29)に隣接するリジンに位置する。
【0033】
インスリンタンパク質はまた、インスリン様成長因子(IおよびII)のような関連タンパク質および成長ホルモンおよびプロラクチンファミリー由来のタンパク質も含む。
【0034】
(インスリンタンパク質の親水性ポリマーへの結合)
本発明に従って、上記インスリンタンパク質および親水性ポリマーを少なくとも1種の有機溶媒および少なくとも1種の金属キレート剤の存在下で、上記タンパク質とポリマーの結合体の形成が促進される条件下で、接触させる(即ち、反応または結合させる)。特定の実施形態では、上記インスリンタンパク質は、最小量のPEG試薬とゆるやかな条件を用いて、PheB1アミノ末端でPEG化される。PheB1アミノ基は、通常インスリンの3つの利用可能なアミノ官能基の中で反応性が最も低い(Calicetiら、1999、上記)。本発明において、条件はPheB1アミノ基をPEG試薬に対して最も反応性にすること見出した。これらの反応条件は、このようにPheB1での単一PEG化を優性の反応生成物として産生する。
【0035】
本発明の特定の実施形態では、上記インスリンタンパク質および親水性ポリマーを、水溶液中で約0.1〜5%(w/w)、好ましくは0.5〜1.5%でのタンパク質濃度で、pHを5.0〜7.5の範囲、好ましくはpH6.5〜7.2に調整して、接触させる。上記pHは、緩衝塩の包含、有機酸/塩基の添加または通常の無機酸/塩基の添加により制御され得る。上記水溶液は、少なくとも1種の水に混和可能な有機溶媒をさらに包含し、それはエタノール、メタノール、DMSO、ジオキサン、DMF、NMPなどを含む群から選択され得る。別の局面では、上記有機溶媒、好ましくはジオキサンは、濃度(v/v)が0〜25%、好ましくは2〜20%、さらに好ましくは、5〜15%で含まれる。
本発明で用いられる適切な金属キレート剤としては、種々の公知のキレート剤、例えばエチレンジアミン四酢酸(EDTA)、ジエチレントリアミン五酢酸(DTPA)、ニトリロ三酢酸(NTA)、N−2−アセトアミド−2−イミノ二酢酸(ADA)、エチレングリコールビス(2−アミノエチルエーテル)N,N,N’,N’−四酢酸(EGTA)、trans−ジアミノシクロヘキサン四酢酸(DCTA)、グルタミン酸およびアスパラギン酸のようなアミノポリカルボン酸;ならびに、例えば、N−ヒドロキシエチルイミノ二酢酸(HIMDA)、N,N−ビス−ヒドロキシエチルグリシン(bicine)、およびN−(トリヒドロキシメチルメチルグリシン(tricine)のようなヒドロキシアミノカルボン酸;ならびにグリシルグリシンのようなN−置換グリシンが挙げられる。他の適切なキレート剤としては、2−(2−アミノ−2−オキソエチル(oxocthyl)アミノエタンスルホン酸(BES)およびデフェロキサミン(DEF)が挙げられる。本発明の方法で用いられる適切なキレート剤としては、例えば、溶液中で利用可能な酸素と反応することを不可能にする金属イオンへ結合し、それにより上記タンパク質と自由に反応および分解するOHラジカルの発生を、最小限にするまたは防止するキレート剤が挙げられる。そのようなキレート剤は、キレート剤の保護なしで処方されたタンパク質の分解を減少させ得、または防止し得る。
【0036】
本発明で用いられるキレート剤は、塩の形態で、例えば前述のキレート剤のカルボキシル基または他の酸性の官能基で存在し得る。そのような塩の例としては、ナトリウム、カリウム、カルシウムおよび他の弱く結合する金属イオンで形成される塩が挙げられる。当該分野で公知のように、上記塩の性質および中和されるべき電荷数は、存在するカルボキシル基の数および安定化キレート剤が供給されるpHに依存する。当該分野で公知のように、キレート剤は、特定の標的イオンと結合する強度は変化する。一般に、重金属イオンは、類似した電荷をもつ低い分子量の対応物よりも、より強く結合する。
本発明の方法で用いられるキレート剤は、EDTA、EGTAおよび当該分野で公知の他の多価カチオンキレート剤から選択され得る。本発明の方法に従って、金属キレート剤、好ましくはEDTAが、濃度が0.1〜10mM、好ましくは1〜5mM、より好ましくは、1〜3mMで存在する。
【0037】
本発明で使用される適切な親水性ポリマーとしては種々の公知のポリマー、例えばポリエチレングリコール、ポリプロピレングリコールおよびそれらの線状、分岐およびアミノ反応性誘導体が挙げられる。本発明の一つの局面では、上記アミノ反応性誘導体は、アルデヒド、N−ヒドロキシコハク酸イミド、PNP炭酸塩およびベンゾトリアゾール末端親水性ポリマー誘導体からなる群より選択される。本発明の特定の実施形態では、親水性ポリマー、例えばPEG試薬、好ましくはPEGのコハク酸イミドエステル、より好ましくは、mPEG−SPAを、インスリンとモル比(PEG:インスリン)を約10:1〜1:1、好ましくは3:1〜1.2:1、より好ましくは1.7:1〜1.5:1で、接触させる。別の特定の実施形態では、親水性ポリマーとインスリンタンパク質を温度が約4℃〜50℃、好ましくは約15℃〜25℃で接触させる。
【0038】
別の実施形態では、本発明は、上記結合体を単離する前に、上記結合反応をクエンチする工程を、さらに含む。これは、例えばpHを約1〜4、好ましくは約2〜3、より好ましくは2.4〜2.6に低下させることにより達成され得る。上記結合体の単離はイオン交換(例えば、カチオン交換)クロマトグラフィーなどの標準的な技術を用いて達成され得、所望の結合体は収集され、濃縮され、脱塩されそして乾燥され得る。
【0039】
(徐放性送達処方物での生理活性結合体薬剤の利用)
PEG化インスリンタンパク質のような生理活性結合体薬剤は、生分解性ポリマーベースの薬物送達処方物で、有利にカプセル化され得る。PEG化生理活性剤は、対応する非PEG化生理活性剤より高濃度で、薬物送達処方物にカプセル化される。生分解性ポリマー薬物送達処方物からのPEG化生理活性剤の放出は、対応する非PEG化の生理活性剤より急速ではないことを示している。生分解性ポリマーの薬物送達処方物の中でのPEG化生理活性剤の物理的安定性および化学的安定性はより大きく、かつ抗原性および免疫原性は対応する非PEG化生理活性剤よりも低い。
【0040】
この応用のための生分解性ポリマーとしては、限定されないが、ポリ(乳酸)、ポリ(グリコール酸)、ポリ(d,l−乳酸−グリコール酸共重合体)、ポリ(カプロラクトン)、ポリ(オルトエステル)、ポリ(エステル)とポリ(エーテル)の共重合体、ポリ(乳酸)とポリ(エチレングリコール)の共重合体などが挙げられる。
【0041】
従って、本発明のタンパク質−ポリマー(例えば、PEG化インスリン)の結合体は、生分解性ポリマーの薬物送達処方物、例えばポリ(d,l−乳酸−グリコール酸共重合体)(PLGA)ミクロスフェアに有利に取り込まれ得る。これは、非結合タンパク質と比較して、上記タンパク質結合体の高いカプセル化を達成し、かつまたバーストを減少させる(最初の24時間にわたる放出)。さらに、PEGのような親水性のポリマーとの結合は、その結合体を特定の有機溶媒に溶解させ、PLGAミクロスフェアの形成を単純化する。
【実施例】
【0042】
I.一工程法による部位特異的PEGインスリンの調製および性質決定
(実施例1)
NαB1−メトキシポリ(エチレングリコール)−インスリン結合体の調製
1グラム(172μモル)のインスリン(Zn2+−インスリン、Intergen Co.)を、室温で100mLの2mM EDTA水溶液に溶解し、希塩酸を用いて、その溶液のpHappを7に調整した。別の容器に、1.4g(インスリンに対して1.6モル当量)の活性化PEG誘導体[モノメトキシポリ(エチレングリコール)プロピオン酸コハク酸イミド、mPEG−SPA、Shearwater Corp.]を、室温で10mLのジオキサンに溶解した。上記mPEG−SPA溶液を、上記インスリン溶液に直接注入で加え、そしてその反応を室温で2時間進行させた。上記反応を、HCl(pHapp2.5)で酸性化してクエンチし、そしてその混合物を0.02%重炭酸アンモニウムに対して限外ろ過[YM3(3000MWCO)膜を取り付けたAmicon 8200 限外ろ過装置]した。その後、その反応混合物を、1M 酢酸/7M 尿素/0.01M NaClに対して限外ろ過し、精製前に10mLまでに濃縮した。上記mPEG−PheB1−インスリン誘導体を、他の反応副生成物(mPEG−GlyA1−インスリン、mPEG−LysB29−インスリン、ジ−mPEG−インスリン、およびトリ−mPEG−インスリン)から、SP セファロース(Amersham Biosciences)カチオン交換カラムを取り付けたFPLCシステムを用いて、単離した。上記カラムを、5mL/分の流速で、1M 酢酸/0.04M NaCl含有7M 尿素で平衡化し、上記結合タンパク質をNaCl−グラジェント(0.04M〜0.12M)を用いて、80分にわたって溶出した。280nmで検出された主要なピークに対応する上記溶出液を収集し、そしていかなる低分子量の不純物をも除去するために0.02% NH4HCO3に対して限外ろ過し、その後凍結乾燥をして性質決定の前に−20℃で保存した。
【0043】
PheB1のアミノ基に連結した、より低分子量の線状ポリマー(即ちmPEG−SPA、Mr=2000Da)を有する1種の結合体、および2種のより高分子量の分岐ポリマー(即ち、mPEG2−SPA、Mr=10,000Da)を有する結合体を調製するのにもこの方法を用い、そしてこれらの2種の異なる結合体の性質決定もまた、以下に概説する技術に従って行なった。この結果は、この方法が、PheB1に連結されたポリマー構造のみ(即ち、線状もしくは分岐)またはサイズのみ(即ち、分子量)が異なる種々様々のPEG−PheB1−インスリン結合体を首尾良く調製するのに用い得る。
【0044】
(実施例2)
結合体純度のFPLC/HPLCによる評価
mPEG−PheB1−インスリンの純度を、流速1.0mL/分を用いた以外は、上述の単離手順で用いた条件と同一の条件下で、分析用カチオン交換カラム(MonoS 5/5、Amersham Biosciences)を用いて、分析した。直交技術(逆相HPLC)もまた、上記結合体の最終純度を証明するのに用いた。Waters Alliance HPLC システムに、Waters 996光ダイオード配列検出器(PDA)およびSymmetry300(C18、5μm 粒子サイズ、4.6×250mm)逆相カラムを取り付けた。流動層Aは、0.1%TFA(トリフルオロ酢酸)MilliQ 品質水からなり、そして流動層Bはまた、0.1%TFAを含有する、95/5ACN(アセトニトリル)/H2Oからなった。15分にわたって、30〜60%Bの直線グラジェントを用いて、化合物の溶出を276nmの検出で、進めた。上記mPEG−インスリンの純度は、95%を超えた。
【0045】
(実施例3)
結合体同一性のN末端タンパク質配列決定(エドマン分解)による確認
上記エドマン分解反応は、PEGに共有結合したN末端アミノ基では進まないということを承知の上で、PEG結合部位を決定するために、N末端タンパク質配列分析を用いた。全ての試料を、Applied Biosystems 477Aタンパク質配列分析器(Pasadena、Ca)を用いて、3回の分解サイクルで分析した。N末端アミノ酸モル比が[GlyA1/PheB1]≒1の場合は、残基LysB29(または無し)への結合を暗示し、[GlyA1/PheB1]≒0の場合は、残基GlyA1への結合を暗示し、[GlyA1/PheB1]≒30の場合はPheB1への結合を暗示する。この結果は、置換の部位が約95%PheB1であることを確証した。
【0046】
(実施例4)
マトリックス支援レーザー脱離イオン化(MALDI)による結合体分子量の同定
この分析の性質決定技術を、分析中にPEG−インスリン結合体が分解する原因にならないような意味を持つ「軟イオン化」であるので、選択した。上記機器の出力は、各試料の質量/電荷比の定量的な尺度を提供する;従って、インスリンに連結しているPEG鎖の数を、上記結合体の分子量と未改変のインスリンの分子量との全体的な差から容易に決定することができる。全ての試料を、直線モードで操作したPerceptive Biosystems model DERP MALDI/TOF質量分析計にかけ、陽イオンをモニターした。全ての試料の上記マトリックスは、α−シアノ−4−ヒドロキシ桂皮酸であり、窒素レーザの337nm線を最終スペクトルのために平均して少なくとも64ショットで用いた。モノマーのインスリンは5807.2Daの計算分子量を有し、結合反応に用いたmPEG(5000)−SPAの数平均分子量は、5129Daであった。上記mPEG(5000)−インスリンの質量スペクトルは、一つのみのmPEG鎖がインスリンに連結しているという結論と一致した。さらに、個々のイオンピークは、一貫して44Da(エチレンオキシドモノマー単位の分子量)ごとに異なっていた。これらの結果、調製した結合体の全てにおいて、一つのみのmPEG鎖がインスリンに連結して、そしてそれらの多分散性が、もっぱらPEG固有の多分散性に、起因するものであることを確証した。
【0047】
(実施例5)
インスリンB1アミノ末端へのPEGの結合における2級アミンの形成
F5000PEG−インスリンを上記タンパク質と末端にアルデヒド基を有する活性化mPEGとを反応させて調製した。この反応は、シフ塩基中間体を経由して進行し、引き続いて上記ポリマーとタンパク質間の安定な2級アミン結合を形成するようシアノトリヒドロホウ酸ナトリウムで還元する。上記反応を以下のようにして行なった:2mM EDTA/25mM リン酸緩衝液を作り、pHをリン酸で6.0に調整した。5.5mg/mL(全容量2mL)のインスリンを440μlのジオキサンを加えたリン酸緩衝液に溶解した。一度、インスリンを2mLの10mM NaCNBH3水溶液に加え、その後5倍モルの過剰のmPEG(5000)−アルデヒド(乾燥粉末として、Shearwater Corporation、Huntsville、Alabama)を加える。結局、上記反応混合物は、およそ12.5mM リン酸(pH6)、1mM EDTA、10%ジオキサン、5mM NaCNBH3、2.5mg/mL インスリンおよび10mg/mL mPEG−アルデヒドを含有していた。上記反応を一晩進行させ、翌日、そのpHが約5.5であることが分かった。上記反応を、酢酸の添加により、約pH2にクエンチした。小アリコートをRP−HPLCを用いて分析した。主要な反応種を以下のように決定した:約70%の上記反応生成物は、モノ−PEG化物(室温で12.5分)、約10%の未反応インスリン(室温で9.8分)および9%のジ−PEG化インスリン(室温13.8分)が残っていた。上記モノ−PEG化分画を溜めて0.02%NH4HCO3に対して透析し、そして凍結乾燥した。MALDI−TOF分析は、インスリンへ1つのPEG−5000鎖の付加に対応する単一分子質量を示した。エドマン分解分析では、約95%のモノ−PEG化種が残基PheB1で置換され、残りのモノ−PEG化種(〜5%)は殆ど残基GlyA1で置換されている。何故なら、ここで用いた反応条件下ではLysB29のアミノ基は99.99%プロトン化されている(従って反応性無い)からである。
【0048】
II.部位特異的PEG−インスリン結合体の代替的調製法
(実施例6)
従来の多工程法を用いて、B鎖アミノ末端(F5000)へ結合したPEG−5000
組換えヒトインスリン(Intergen Co.)をジ−boc保護化中間体を用いて、PheB1位にPEG化した。ジ−NαA1、NεB29−t−boc−インスリン(ジ−boc−インスリン)を、Liuら。1997(Liuら、Bioconj.Chem.8(5):664〜672、1997)に従って合成した。上記mPEG(5000)−PheB1−インスリン結合体をHindsら、2000、上記のプロトコールに従って調製した。
【0049】
FPLCで単離された上記所望の分画は、逆相HPLCおよびイオン交換FPLCのクロマトグラフのピーク面積を基礎にして98%を越える純度であった。上記精製した生成物をMALDI−TOF質量分析およびアミノ酸配列分析でさらに性質決定を行ない、上記B鎖アミノ末端PheB1で、モノ置換PEGであることが分かった。この方法で調製および精製された上記mPEG(5000)−PheB1−インスリン結合体(F5000)は、本発明のより単純で、より時間のかからない方法により作られた同じ結合体(実施例1)と等価であった。
【0050】
(実施例7)
B鎖Lys29に結合したPEG−5000(K5000)
組換えヒトインスリン(Intergen Co.)を、Hindsら、2000、上記の方法により、B鎖のLys29に特異的にPEG化した。上記所望のFPLC分画は、逆相HPLCおよびイオン交換FPLCのクロマトグラフのピーク面積を基礎にして、純度が98%を超えた。その精製された生成物を、MALDI−TOF質量分析およびアミノ酸配列分析でさらに性質決定を行い、そしてB鎖末端から2番目のアミノ酸LysB29で、モノ置換されていることを示した。この同じ方法をまた使用して、LysB29に連結した1種の低分子量の線状ポリマー(即ち、mPEG−SPA、Mr=2000Da)および2種のより高分子量の分岐ポリマー(即ち、mPEG2−SPA、Mr=10,000または20,000Da)を調製し、そして上記に概説した技術に従って、これらの3種の異なる結合体の性質決定を行なった。この結果は、この方法が、LysB29に連結されたポリマー構造のみ(即ち、線状もしくは分岐)またはサイズのみ(即ち、分子量)が異なる種々様々のPEG−LysB29−インスリン結合体を、首尾良く調製するのに用い得る。
【0051】
III.部位特異的PEG−インスリンの性質決定
コンホメーション、活性および安定性
(実施例8)
部位特異的PEG化後のインスリンコンホメーション保持性の評価
遠紫外範囲の円偏光二色性分光法を用いてインスリンのコンホメーションを調べた。通常、以下の二つの負の極小の強度を評価する:水性環境中でのインスリンのコンホメーションの分析において208nm(α−ヘリックス)および223nm(β−シーツ)。208nmの遠紫外のCD−帯は、主として残基B10〜B19、A2〜A6およびA13〜A19から形成されるα−ヘリックスに起因し、一方、β−構造は、223nmの遠紫外CD−帯の主たる成分である。上記試料に特徴的なCDスペクトルは、残基PheB1またはLysB29のいずれかへのインスリンへのmPEGの連結は、Zn2+−インスリンと比較して、上記結合体の全体的なコンホメーション(2次)を変化させない事を確認した。
【0052】
(実施例9)
PEG−インスリン結合体の薬力学
インスリン薬力学に対するPEG化の効果を調査した。これらの研究は、市販で入手可能なインスリン処方物(Humulin R、Lilly)と比較して、置換部位の異なるPEG−インスリン(位置異性体)またはPEGの分子量が異なるPEG−インスリンを含有する結合体処方物のインビボの生物学的活性(血清グルコースを低下させる能力)を評価するために実施した。絶食したSprague−DawleyラットにF5000、K2000、K5000、K10000またはHumulinR(登録商標)を静脈内に投与した後、血中グルコースレベルを測定した。0.3IU/kg(タンパク質濃度を基礎にし、そしてPEGの重量を適切に補正して;25IU/mgタンパク質と仮定する)を通常の生理食塩水に溶解し、尾静脈注射により投与した;1群N=6。試験試料の注射前および注射後6時間の期間、間隔をおいて採血を行なった。血清を標準的な操作方法で単離し、グルコースレベルをAccucheck Advantage(Boeringer Ingelheim)グルコースメータを用いて測定した。
【0053】
図1Aおよび図1Bに示す結果は、F5000、K2000およびK5000は、正常ヒトインスリンと等価の用量で、正常ラットのグルコースレベルを抑制するのに、全て効果的である。興味深いことには、上記K10000結合体は、他の試験試料と同じ程度にはグルコースレベルを減少しなかったが、結合体の作用の持続時間は、6時間を越えて観察された。このように、上記K10000結合体は、糖尿病患者に長時間のグルコース抑制を提供し、従前の基本のインスリン懸濁剤の可溶性代替物として、開発され得る。
【0054】
(実施例10)
代表的なPEG−インスリン結合体の物理的安定性
加速振動試験法を用いて7種のPEG−インスリン結合体(F2000、F5000、F10000、K2000、K5000、K10000およびK20000)および亜鉛インスリンの物理的安定性を調査した。この試験は加速法でインスリン調製物の物理的安定性の正確な測定を提供する文献に、通常記載されている。このアッセイのために、上記タンパク質(結合体)の水溶液を4部(1mg/mL、リン酸緩衝生理食塩水、pH7.3,0.02%アジ化ナトリウム)調製し、37℃で水平振動(100/分の振動数)させた。試料を所定の時間に取り出し、ろ過し(不溶性凝集体の除去のため)、そして溶液中に残っているタンパク質(結合体)分画を、定量化するためにRP−HPLCにより分析した。図2は、各インスリン誘導体に関して、時間の関数として可溶性タンパク質の減少を示している。
【0055】
これらのデータは著しく高い(36〜40倍)物理的安定性を有するPheB1アミノ基で置換されたインスリン誘導体およびLysB29−インスリンが、天然のペプチドより、いくらかより安定(4〜8倍)であった以前の報告を補強するものである。以前の研究では、PheB1−インスリンの繊維形成に対する増加した抵抗性は二つの相補的な効果によるものであることを示している。第一の効果は、mPEG結合によりB鎖N末端を立体特異的に遮断することであり、それがこの表面がインスリンの繊維形成に必要とされる疎水性相互作用に関与するのを防止する。PheB1−インスリンの増加した物理的安定性に貢献する第二の効果は、元々非特異的であり、立体的であり、そしてポリマー分子量に比例的に増加する。上記LysB29−インスリンの全てが、天然のペプチドに比較して、増加した物理的安定性を示すが、ポリマー分子量が10kDaを超えるまでは、PheB1で置換された上記結合体と同じ程度ではない。これは、繊維形成反応におけるLysB29の関与の欠如により説明され得、繊維形成に関与する分子間相互作用の非特異的立体障害に起因する安定化効果を伴う。
【0056】
(実施例11)
代表的PEG−インスリン結合体の化学的安定性
インスリン(またはインスリンアナログ)は、水溶液中で多くの化学分解反応を受けることはよく実証されている。例えば、インスリンA鎖のC末端のアスパラギンは、酸性条件下では、環化中間体により促進される脱アミド化機構により分解する。この高い反応性環化中間体はまた、アミノ転移反応機構を介して、異なるインスリン分子からのN末端アミノ基の一つと反応して分解し得る。
【0057】
上記インスリン結合体、F5000およびK5000を37℃で、0.02%アジ化ナトリウムPBS中で水平振動でインキュベートした。所定の時間に(0、6、12、28および36日)、個々の試料を取り出し、その後、脱アミド化の程度を決定するためにRP−HPLCにより、そして共有結合の2量化の程度を決定するためにサイズ排除(size exclusion)クロマトグラフィーで分析した。
IV.PEG−インスリン生分解性ポリマー徐放送達処方物の調製、性質決定および薬物動態
(実施例12)
PLGAミクロスフェアでのPEG−インスリンのカプセル化
PEG−インスリンF5000(50mg)を、1mlの塩化メチレンに溶解した。上記溶液を150mgのPLGA45:55(lac:gly)、酸末端基を持つ0.15dl/gIVを有する1ml容量の塩化メチレンに添加した。上記塩化メチレン溶液を、15mlの遠心チューブ中の1%ポリ(ビニルアルコール)を含む5mlの水に添加し、そしてボルテックスミキサーでエマルジョンを作った。上記エマルジョンを0.3%のポリ(ビニルアルコール)を含む100mlの水に添加し、塩化メチレンを蒸発させるために3時間攪拌した。上記硬化ミクロスフェアを、Whatman1番のろ紙で真空ろ過し、そして乾燥した。
【0058】
(実施例13)
PEG−インスリンPLGAミクロスフェアの性質決定
上記PEG−インスリンミクロスフェアの表面形態および粒子サイズ分布を、それぞれ、走査型電子顕微鏡およびレーザ光散乱粒子サイズ分析で調査した。ポリマーミクロスフェア内にカプセル化されたPEG−インスリン結合体の量を定量化するために、分析用逆相HPLCを用いた。HPLC分析に先立って、測定した量のミクロスフェアを、ある容量のアセトニトリルに溶解し、そしてポリマーを沈殿させ、水溶液中へ上記結合体を抽出するために、等容量の0.1%TFA水溶液を添加した。K2000、K5000、K10000、K20000およびF5000−Aもまたこの方法を用いてカプセル化した。これらの試験の結果を表1に集めたが、そこには全ミクロスフェア中のPEG−インスリン含量%(w/w)、および(カプセル化されたPEGインスリン重量/最初に添加されたPEG−インスリン重量)として定義されたカプセル化効率を挙げている。28.3%と相対的に高い薬物含量および100%までのカプセル化効率を達成し、上記生成物を、必要とされる減少した全用量により、臨床的に有用になり、かつ出発物質の低い損失で商業的にも魅力的にした。さらに、ミクロスフェアは、乳酸とグリコール酸の比率(50:50および72:25)、分子量(6.5〜24kDa)、固有粘度(0.09〜0.27dL/g)、または末端エステル基(メチルおよびラウリル)を変えたポリマーと広い範囲の薬物負荷(5〜35重量%)を用いて調製した。
【0059】
【表1】

(実施例14)
動物実験で用いられたPEG−インスリンミクロスフェア処方物のインビトロ放出
15mgのF5000PEG−インスリンミクロスフェア(14.1%薬物含量、PLGA 45:55;0.15dl/gIV、酸末端基)を1.5mlのリン酸緩衝生理食塩水(pH7.4、0.02%アジ化ナトリウムおよび0.02%Tween20)に懸濁し、37℃でインキュベートした。上記上清を、間隔的に取り出し、放出されるPEG−インスリンをRP HPLCで分析した。上記緩衝液を、新鮮なPBSで置換し、そのインキュベーションを続けた。データをインキュベーション時間の関数として累積放出量を分析した(図4)。最初の日は、1.0%未満の上記PEG−インスリンの放出で、18日以内で、95%を超える量が放出される。約2週間を越える低い「バースト」放出、高い全放出量および持続期間は、徐放インスリン処方物の非常に所望の特徴である。
異なる生分解性ポリマーおよびF5000とを用い、ならびにK5000PEG−インスリンおよびF5000−A結合体もまた、用いる実施例12の方法で作った他の調製物もまた、1日放出値が0〜7.5%の間で、持続放出期間も60日までもあった。
【0060】
(実施例15)
F5000PEG−インスリンPLGAミクロスフェアのインビボ薬力学および薬物動態PLGA45:55、0.15dl/gIV、酸末端基(14.1%核負荷)から構成されるミクロスフェア中のF5000PEG−インスリンを、糖尿病ラットでグルコース抑制およびF5000薬物動態の試験をした。雄性SDラット(〜250g)に、等張クエン酸緩衝液(10mM、pH4.5)に溶解した40mg/kgのストレプトゾトシン(Sigma、St.Louis、MO)を、試験試料投与する1日前に皮下投与をして糖尿病にした(Junod,Aら、J.Clin.Invest.、48:2129〜2139、1969)。動物を2.5%滅菌カルボキシメチルセルロース(CMC)または生理食塩水(陰性コントロール)に懸濁したPEG−インスリンミクロスフェア(〜11mg/ラット、体重1kgあたり3mgインスリン当量に相当する)で処置し、そして血清単離、引き続くグルコース(PD)およびF5000(PK)レベルの分析のために所定の間隔で採血した。図5および6は、13日の期間の血清グルコースレベルおよび血清PEG−インスリンレベルを示す(データは平均値±標準誤差)。図5は、予想したとおり、実験の間中、血中グルコースレベルがコントロールの糖尿病ラットに対して100%以上(測定誤差内)を示す。PEG−インスリンミクロスフェア処置糖尿病ラットに関しては、血中グルコ−スレベルは、3日後未処置レベルの60%より低いレベルに低下し、さらに7日間、抑制が持続している。
【0061】
図6は、PEG−インスリンの無視し得る初期バースト放出(Cmax=0.62ng/ml)が、ミクロスフェア注入後すぐに(1時間)検出されたことを示している。それから注入後24時間、処置群のPEG−インスリンレベルは、2日を超えて、着実に治療レベル(約1〜3.5ng/ml)まで上がり、約7日間持続し、一方、上記陰性コントロール群のF5000血清レベルは、ずっと検出限界未満であった。これらのデータは、一緒に考えると、結合体の生物学的活性は、ミクロスフェア製造工程で保持され、そしてミクロスフェア注入に続く一週間の放出の間、維持されたことを示している。重要なことは、類似した量の初期PEG−インスリンバースト放出が、インビトロ(図4、第1日<0.5%放出)およびインビボ(図6、AUC0〜1d/AUC0〜13d=0.7%実験)で見出されたことである。さらに、図5および6に示されているデータの評価は、薬物動態/薬力学(PK/PD)相関がこの実施例で存在することを示唆している。
【0062】
(等価物)
当業者は、本明細書に記載している本発明の特定の実施形態に対する多くの等価物を単なる慣用的実験を用いて、認識または確認することができる。そのような等価物は、以下の特許請求の範囲に包含され、網羅されることが意図されている。
【技術分野】
【0001】
(関連出願)
本出願は、本明細書に全体の内容を参考として援用している、2003年4月11日出願の米国特許仮出願番号第60/462,364号に基づく優先権を主張する。
【0002】
(発明の分野)
本発明は、非結合体タンパク質の性質より優れた性質を有する化学的に改変されたタンパク質結合体に関する。詳細には、本発明は、非常に簡素化され、費用有効的であり、量産可能な、親水性ポリマーによりタンパク質を改変する方法に関する。より詳細には、本発明は、インスリンのような、選択されたタンパク質のポリエチレングリコールによる部位特異的改変に関する。本発明は、親水性タンパク質との部位特異的改変を有するタンパク質を包含する、生分解性ポリマーベースの薬剤送達処方物に関する。
【背景技術】
【0003】
(関連技術の説明)
PEG化インスリン誘導体を産生する種々の方法は、公知である。Davisら(米国特許第4,179,337号)は、PEGとタンパク質とのリンカーとしてトリクロロ−s−トリアジン(塩化シアヌル)を用いたPEG−インスリン構築物の合成を、記述した。彼等は、大過剰(50×)の塩化シアヌル活性化PEG(2000Da)をホウ酸緩衝液(pH9.2)中のインスリンと2時間反応させる合成スキームに従った。本発明者らは、部分的に活性な(〜50%)PEG−インスリン結合体(それは非免疫原性であり非抗原性である)を、産生することができた。Obermeierら(カナダ特許番号第1,156,217)は、上に引用したDavis特許の実施例Xに従ったPEG−インスリン結合体の調製は、約50%のトリ−PEG−インスリンを含有する結合体の不均質な混合物を産生し、他の可能なPEG−インスリン誘導体の組合せ(モノ−PEGインスリンおよびジ−PEG−インスリン)は残基PheB1で置換されていなかったことを見出した。
【0004】
Obermeierら(カナダ特許番号第1,156,217)は、残基PheB1で特異的に改変されたPEG−インスリン結合体の合成を記述している。彼等の発明の基本は、残基GlyA1およびLysB29の上記反応性アミンを、アルカリ条件下で、有機溶媒(例えば、DMF、DMSO、ピリジンなど)中で、tert−ブチルオキシカルボニル(t−boc)基またはメチルスルホニルエチルオキシカルボニル(Msc)基により、保護する工程を含んでいる。(モノ−、ジ−およびトリ−)保護されたインスリンの複雑な混合物から、NαA1,NεB29−ビス−保護されたインスリン種は、従来のクロマトグラフィー技術で単離された。単離に続いて、上記純粋なNαA1,NεB29−ビス−保護されたインスリンを、活性化(例えば、酸クロライドまたはイソシアネート)PEG誘導体と反応させ、引き続き、ペプチド化学で通例の技術を用いて保護基を除去した。本発明者らは、GlyA1およびLysB29のアミノ基が、アルカリ反応条件下では、PheB1のアミノ基より、より反応性であったことを観察した。彼等は、それらの部位特異的mPEG(1500)−B1−インスリン結合体が、ウサギの血糖レベルの低下に関して100%インスリン効果(モル基準で計算した)を有することを決定した。
【0005】
Geigerら(D.Branderburg,and A.Wollmer(eds.)Insulin:Chemistry、Structure、and Function of Insulin and Related Hormones、Walter de Gruyter & Co.,New York、pp.409〜415、1980)およびEhratら(Biopolymers、22、569〜573、1983)は、Obermeierらにより記載された多段工程方法に類似する、保護/結合/脱保護スキームを用いて、調製された残基PheB1での特異的な改変PEG−インスリン付加物について記述している。GeigerらおよびEhratらは、上記PEG(1500)−B1−インスリン結合体が天然のインスリンより抗原性がずっと低くそしてずっとより安定(肝臓の酵素に対して)であるということを観察した。他のPEG−インスリン調製物(Calicetiら、STP Pharma Sci、9、107〜113、1999;Uchioら、Advanced Drug Delivery Reviews、35、289〜306、1999;Hindsら、Bioconj.Chem.11、195〜201、2000;Hindsら、Advanced Drug Delivery Reviews、54:505〜530、2002)は以下のいずれかである:1)上記に概説されている基礎的3工程の保護/結合/脱保護のスキームを中心としている、2)結果が上記インスリン分子の非特異的改変になるもの、または3)最も効果的な結合体、即ち,PEG−B1−インスリンを産生しない。
【0006】
Liuら(米国特許番号第6,323,311B1号)は、PEG−B1−インスリン結合体の合成の有用な方法を記述している。この方法は、Obermeierの3工程の保護/結合/脱保護スキームの延長であるが、工程間の反応中間物の単離を必要としない(すなわち一回分式合成)。このように、上記インスリンは、残基GlyA1およびLysB29で保護され、直ちにPEGと反応し、引き続きいずれの種の単離の前には、脱保護される。本発明は、それらの一回分式反応で50%までもの正しい位置異性体(即ち,PEG−B1−インスリン)の収率を得、そして次の誘導体化にリサイクルできる未反応インスリンを30%回収し得ることを請求している。これらの構築物の調製が、てきぱきと実行できたと仮定して、完了するまでには、少なくとも5日間はかかる。さらに本発明は、受容可能な結果を達成するために大過剰のPEG試薬を必要とする。本発明の生成物は、効果的であるが、それらの調製は、上記タンパク質が、長時間、タンパク質にとって有害な環境(高pHおよび低pH)で三つの反応工程を経なければならないことを必要としている。
【発明の概要】
【課題を解決するための手段】
【0007】
本発明は、単一工程で、インスリンB鎖のN末端(PheB1)に特異的にPEG化された高純度のインスリン誘導体の単純な調製方法を提供することにより、インスリンをPEG化する方法の従来技術の欠点を取り扱うものである。PheB1でのPEG化が、もっとも可能性の少ない反応性生物であることを示している先行経験(例えば、Calicetiら、1999,上記)とは対照的に、本発明の方法は、PEG化の優勢な部位になるようPheB1アミノ末端の相対的反応性を高めるために、pHの特定の制御条件、金属イオンのキレート剤の使用および有機溶媒の添加を用いている。インスリンに与えられている多くの有益な性質(例えば、低減した免疫原性/抗原性;増加したタンパク質分解に対する安定性、化学的安定性および物理的安定性;増加した循環半減期;増加した水溶性溶解度/有機的溶解度;全生物活性)を考慮すると、残基PheB1への部位特異的PEG化を通じて、この結果を達成するために、単純であり、費用有効的であり、容易にスケールアップできる工程は当該分野では顕著な進展である。
【0008】
(発明の概要)
本発明は、タンパク質−ポリマー結合体を調製する単一工程の方法の発見に基づいている。本発明はまた、親水性タンパク質との部位特異的な改変を有する生分解性ポリマーベースの薬剤送達処方物に関する。特定の実施形態では、本発明は、置換の部位が優勢的に残基PheB1(上記B鎖のN末端)であるPEG化インスリン誘導体の単一工程の合成のための方法を提供する。そのような誘導体が、天然のインスリンより物理的に酵素的により安定であることは当該分野では周知である。さらに、上記誘導体は、インスリンより水系/有機系でより溶解性である。さらにこれらの誘導体は免疫原性がより低く、長時間の循環半減期を有していることが分かっている。
【0009】
本発明の顕著な利点は、上記反応が望ましくない副反応(例えば、脱アミド化、トランスアミド化、酸化等)が無視し得る中性に近い条件で起きることである。別の利点は、比較的少量のポリマー(例えば,PEG試薬)の使用、従って、コストの低減化である。上記の結果できたタンパク質−ポリマー結合体(例えば、PEG化インスリン)は、例えばヒトの治療において、それ自身使用し得るかまたは、米国特許出願第2002/0155158号に開示されているような徐放性送達媒体にカプセル化し得る。
【0010】
従って、一つの実施形態では、本発明は、少なくとも1種の有機溶媒および少なくとも1種の金属キレート剤の存在下で、上記タンパク質とポリマーの結合体が生成する条件下で、インスリンタンパク質と親水性ポリマーとを接触させ、タンパク質−ポリマーの結合体を調製する方法を提供する。そこで、上記結合体は、カラムクロマトグラフィーのような種々の標準的技術で単離され得る。
【0011】
本発明の特定の実施形態では、親水性ポリマーは、ポリエチレングリコール、ポリエチレングリコール/ポリプロピレングリコール共重合体、ポリオキシエチル化グリセリンおよびそれらの線状誘導体、分岐誘導体およびアミノ反応性誘導体からなる群より選択される。適切なアミノ反応性誘導体としては、例えば、アルデヒド、PEGカルボン酸のN−ヒドロキシコハク酸イミドエステル、PNP炭酸塩およびベンゾトリアゾール末端親水性ポリマー誘導体が挙げられる。典型的には、親水性ポリマーおよびインスリンタンパク質は、モル濃度比、約10:1〜1:1で存在する。
【0012】
本発明で使用される適切な有機溶媒としては、限定しないが、例えば、エタノール、メタノール,DMSO、ジオキサン、DMFおよびNMPなどの水と混和可能な有機溶媒を含む、種々の公知の溶媒が挙げられる。典型的には、上記有機溶媒は、濃度が約0.1〜10%で存在する。
【0013】
本発明で使用される適切な金属キレート剤としては、限定されないが、多価(例えば2価)金属イオンキレート剤、例えばEDTA、デフェロキサミン(DEF)、ジエチレントリアミン五酢酸(DTPA)、エチレングリコールビス(2−アミノエチルエーテル)N,N,N’,N’−四酢酸(EGTA)を含む種々の公知の化合物が挙げられる。一般的に、上記キレート剤は、濃度が約0.1〜10mMで存在する。
【0014】
本発明の特定の実施形態では、上記インスリンタンパク質と親水性ポリマー(例えば,PEG)を水溶液中で、タンパク質濃度が約0.1〜5重量%で、接触させる(即ち反応させまたは、結合させる)。本発明の別の実施形態では、上記インスリンタンパク質および親水性ポリマーを、水溶液中pHが約5.0〜7.5、好ましくは約7.0で接触させる。これは、インスリン−ポリマー結合体の上記正しい位置異性体の50%までの収率という結果を生じる。別の特定の実施形態では、上記親水性ポリマーとインスリンタンパク質を約4℃〜50℃、好ましくは約15℃〜25℃の温度で接触させる。
【0015】
一度生成されると、上記タンパク質−ポリマー結合体は、望ましくない副反応性生物および未反応インスリンタンパク質から分離される。これは、クロマトグラフィーのような種々の公知の技術を用いて達成され得る。特定の実施形態では、イオン交換クロマトグラフィーが用いられる。
【0016】
さらに別の実施形態では、本発明の方法には、上記結合生成物を単離する工程に先立ってインスリンタンパク質と親水性のポリマーとの反応(即ち結合)をクエンチする工程が、さらに含まれる。特定の実施形態では、これは、上記反応のpHを約1〜4までに低下させることにより達成される。
【0017】
本発明の方法により産生される特定のタンパク質−ポリマー結合体は、例えば、インスリン−ポリマー結合体、好ましくは、インスリン−PEG結合体(PEG化インスリン)を包含する。これは、配列番号1で示される上記配列を有するヒトインスリンおよび関連ファミリーメンバーのような任意のインスリンもしくはインスリン関連タンパク質を包含し得る。特定の実施形態では、上記インスリンは、残基GlyA1もしくはLysB29での反応がそれ程なく、残基PheB1で特異的に反応(PEG化)する。結果としてできるPEG化インスリンは、当該分野で周知の任意の適切な処方物で治療的に投与され得る。特定の実施形態では、上記結合体は、例えば、投与の前に生分解性ポリマーで上記結合体をカプセル化することによる徐放性処方物で投与される。
【0018】
本発明は、さらに以下を提供する。
(項目1)
タンパク質−ポリマー結合体を調製する方法であって、以下の工程:
(a)インスリンタンパク質を、少なくとも1種の有機溶媒および少なくとも1種の金属キレート剤の存在下、該タンパク質と該ポリマーの結合体の形成を促進する条件下で、親水性ポリマーと接触させる工程;および
(b)該結合体を単離する工程
を包含する、方法。
(項目2)
前記インスリンタンパク質が、ヒトインスリンを含む、項目1に記載の方法。
(項目3)
前記親水性ポリマーが、ポリエチレングリコール、ポリエチレングリコール/ポリプロピレングリコール共重合体、ポリオキシエチル化グリセリンならびにそれらの線状、分岐およびアミノ反応性誘導体からなる群より選択される、項目1または2に記載の方法。
(項目4)
前記アミノ反応性誘導体が、アルデヒド、N−ヒドロキシコハク酸イミド、PNP炭酸塩およびベンゾトリアゾール(benzotrizole)末端親水性ポリマーからなる群より選択される、項目3に記載の方法。
(項目5)
前記親水性ポリマーおよびインスリンタンパク質が、モル比で約10:1〜1:1で接触させられる、項目1〜4のいずれか1項に記載の方法。
(項目6)
前記有機溶媒が、エタノール、メタノール,DMSO、ジオキサン、DMFおよびNMPからなる群より選択される、項目1〜5のいずれか1項に記載の方法。
(項目7)
前記有機溶媒が、約0.1〜10%の濃度で存在する、項目1〜6のいずれか1項に記載の方法。
(項目8)
前記インスリンタンパク質および親水性ポリマーが、約0.1〜5.0%のタンパク質濃度で接触させられる、項目1〜7のいずれか1項に記載の方法。
(項目9)
前記インスリンタンパク質および親水性ポリマーが、pHが約5.0〜7.5で接触させられる、項目1〜8のいずれか1項に記載の方法。
(項目10)
前記キレート剤が、多価金属イオンキレート剤、EDTA、デフェロキサミン(DEF)、ジエチレントリアミン五酢酸(DTPA)およびエチレングリコールビス(2−アミノエチルエーテル)N,N,N’,N’−四酢酸(EGTA)からなる群より選択される、項目1〜9のいずれか1項に記載の方法。
(項目11)
前記キレート剤が、約0.1〜10mMの濃度で存在する、項目1〜10のいずれか1項に記載の方法。
(項目12)
前記インスリンタンパク質および親水性ポリマーが、約4〜50℃の温度で接触させられる、項目1〜11のいずれか1項に記載の方法。
(項目13)
前記方法が、前記結合体を単離する前に、該結合体の形成をクエンチする工程をさらに含む、項目1〜12のいずれか1項に記載の方法。
(項目14)
前記クエンチングが、該pHを約1〜4に低下させることにより達成される、項目13に記載の方法。
(項目15)
前記単離が、クロマトグラフィーにより達成される、項目1〜14のいずれか1項に記載の方法。
(項目16)
前記クロマトグラフィーが、イオン交換クロマトグラフィーを含む、項目15に記載の方法。
(項目17)
前記結合体を、生分解性ポリマーでカプセル化する工程をさらに含む、項目1〜16のいずれか1項に記載の方法。
(項目18)
インスリン−PEG結合体の調製方法であって、以下の工程:
(a)インスリンを、少なくとも1種の有機溶媒および少なくとも1種の金属キレート剤の存在下、該インスリンとPEGとの結合体の形成を促進する条件下で、PEGと接触させる工程;および
(b)該結合体を単離する工程
を包含する、方法。
(項目19)
前記インスリンが、ヒトインスリンを含む、項目18に記載の方法。
(項目20)
前記PEGが、アルデヒド、N−ヒドロキシコハク酸イミド、PNP炭酸塩およびベンゾトリアゾール末端親水性ポリマーからなる群より選択されるアミノ反応性PEG誘導体を含む、項目18または19に記載の方法。
(項目21)
前記PEGおよびインスリンが、モル比で約10:1〜1:1で接触させられる、項目18〜20のいずれか1項に記載の方法。
(項目22)
前記PEGおよびインスリンが、約0.1〜5.0%のタンパク質濃度で接触させられる、項目18〜21のいずれか1項に記載の方法。
(項目23)
前記PEGおよびインスリンが、pHが約5.0〜7.5で接触させられる、項目18〜22のいずれか1項に記載の方法。
(項目24)
前記方法が、前記結合体を単離する前に、該結合体の形成をクエンチする工程をさらに含む、項目18〜23のいずれか1項に記載の方法。
(項目25)
前記クエンチングが、該pHを約1〜4に低下させることにより達成される、項目24に記載の方法。
(項目26)
前記単離が、イオン交換クロマトグラフィーにより達成される、項目18〜25のいずれか1項に記載の方法。
(項目27)
前記結合体を、生分解性ポリマーでカプセル化する工程をさらに含む、項目18〜26のいずれか1項に記載の方法。
本発明の他の実施形態は、以下の詳細な説明および実施例から明らかになる。
【図面の簡単な説明】
【0019】
【図1】図1Aおよび1Bは、正常ラットに静脈内注入を行なった後のPEG−インスリン結合体のインビボ薬力学に対する部位置換の効果(図1A)またはポリマー分子量の効果(図1B)を示すグラフである。
【図2】図2Aおよび2Bは、ポリマー連結の同じ部位で異なる分子量のインスリン−PEG結合体(図2A)および同分子量のPEGFでポリマー連結の部位が異なるインスリン−PEG結合体(図2B)の、物理的凝集に起因する可溶性インスリンの損失を示すグラフである。
【図3】図3は、2種のmPEG(5000Da)−インスリン位置異性体の化学的安定性を示すグラフである。
【図4】図4は、PGLAミクロスフェアからのF5000(PEG−インスリン)のインビトロ累積放出を示すグラフである。
【図5】図5は、糖尿病ラットへ皮下投与後のF5000(PEG−インスリン)ミクロスフェアのインビボ薬力学を示すグラフである。
【図6】図6は、糖尿病ラットへ皮下投与後のF5000(PEG−インスリン)ミクロスフェアのインビボ薬物動態を示すグラフである。
【発明を実施するための形態】
【0020】
(詳細な説明)
本発明は、タンパク質−ポリマー結合体を迅速におよび効率的に調製する単一工程の方法に関する。本方法は、少なくとも1種の有機溶媒および少なくとも1種の金属キレート剤の存在下で、中性近辺の条件下で、タンパク質と親水性ポリマーを反応させる工程を包含する。本発明の特定のタンパク質−ポリマー結合体は、最小量のPEG試薬および温和な条件を用いて、アミノ末端PheB1にPEG化されたインスリンを包含する。先行技術は、PheB1が、立体的に嵩高い高分子試薬に対して、通常、インスリンの最も反応性が低いアミノ基であることを示している(例えば、Calicetiら、1999、上記)にもかかわらず、驚くべきことに、本発明の方法は、PheB1が、親水性ポリマーによる改変で利用可能な最も反応性の高い基になるような条件を作り出す。これは、PheB1でPEG化されたインスリンが最高収率の生成物であり、かつ他の結合体および未反応インスリンから分離され得る単純な単一工程の反応を可能にする。後者は、望まれる場合、さらなる結合のためにリサイクルされ得る。PheB1でPEG化されたインスリン結合体は、血糖制御で測定されるように、完全な活性を保持し、かつ上記タンパク質の天然構造が保存されている。
【0021】
(親水性ポリマー)
用語「親水性ポリマー」は、限定しないが、ポリエチレングリコール、およびポリエチレングリコール/ポリプロピレングリコール共重合体、ポリオキシエチル化グリセリンならびに類似のポリマーを含む、如何なる水溶性線状ポリマー、分岐ポリマー、グラフト(forked)ポリマー、分岐−グラフトポリマー、デンドリマー状ポリマー、多枝(multi−armed)ポリマーまたは星型ポリマーをいう。好ましくは、上記ポリマーの分子量は、約500ダルトン〜約50000ダルトンの範囲である。本発明で使用する親水性ポリマーは、典型的には、アミノ、カルボキシル、スルフヒドリル、リン酸またはヒドロキシル基を介して、関心の生理活性分子との連結に組込まれる、少なくとも一つの反応性基を有する。ポリエチレングリコールのような本発明で使用される親水性ポリマーは、メトキシ基で一端が保護され、そして他の一端が生理活性分子の活性基への容易な結合のために活性化される標準的プロトコールに従って、調製され得る。例えば、米国特許番号第6,113,906号は、タンパク質のアミノ基との反応のために、「U型」(即ち、分岐)型形態のポリエチレングリコールにコハク酸コハク酸イミド反応性基または炭酸コハク酸イミド反応性基の使用を記載している。米国特許番号5,650,234号は、安定なウレタン結合を形成するためのタンパク質のアミノ基との反応に、ポリエチレングリコールのN−ヒドロキシベンゾトリアゾール炭酸塩誘導体、2−ヒドロキシピリミジン炭酸塩誘導体およびN−ヒドロキシ−2−ピロリジノン炭酸塩誘導体の使用を記載している。米国特許番号第5,672,662号は、安定なアミド結合を形成するためのタンパク質のアミノ基との反応のために、プロピオン酸および酪酸置換ポリエチレングリコールのコハク酸イミドエステルの使用を記載している。米国特許番号第5,446,090号は、タンパク質のスルフヒドリル基と安定なチオエーテル結合を形成するために、ポリエチレングリコールのビニル−スルホン誘導体の使用を記載している。米国特許番号第5,880,255号は、単純で、安定な2級アミン結合を形成するために、タンパク質のアミノ基との反応のためにポリエチレングリコールのトレシル(2,2,2,−トリフルオロエタンスルホニル誘導体の使用を記載している。米国特許番号第5,252,714号は、安定な2級アミン結合を生じるタンパク質のアミノ基との反応のため、ポリエチレングリコールのプロピオンアルデヒド誘導体の使用を記載している。そのような親水性ポリマーの生理活性分子への連結から生じる結合は、安定または不安定(即ち可逆的)に意図的に設計され得る。さらに本発明で使用される親水性ポリマーは、生理活性分子の活性基への結合を容易にする利用可能な二つの類似な(例えば、ホモ二官能性)、または類似しない(ヘテロ官能性)官能基を用いる標準的なプロトコールに従って調製され得る。例えば、WO126692A1は、タンパク質の改変のために、ヘテロ二官能性のポリエチレングリコール誘導体の使用を記載している。これらの特許の全体的な内容は、本明細書に参考として援用されている。
【0022】
一つの実施形態では、上記親水性ポリマーは、ポリ(エチレングリコール)(PEG)である。PEGは、化学式HO−(CH2CH2O)n−Hを有する線状または分岐の中性ポリエーテルをいう。PEGは水および多くの有機溶媒(例えば、ジクロロメタン、エタノール、トルエン、アセトンおよびクロロホルム)に非常に溶解性があり、種々のサイズ(分子量)および機能的構築物(例えば、アミノ−、カルボキシル−およびスルフヒドリル−末端)で容易に入手できる。PEGは、毒性がないことが分かっており、そして薬物(非経口剤、局所剤、坐薬、鼻スプレー)、食品および化粧品としてFDAにより認可されている。溶液中では,PEGは高度に水和されたポリマーで、各モノマー(エチレンオキシド単位)は、3分子の水と結合し得る。さらに、PEGは、水和水分子とより緩やかに結合している、7つの分子層の構造に影響を与える能力を有すると考えられている。水中での単一表面結合鎖の動きの分子シミュレーションは、上記ポリマーが、大きな程度の分節の自由度を提示することを示している。このように上記ポリマーは、水性環境中では、大きな流体力学的な容量を占めていると考えられている。これらの結果は、何故PEGがその存在から他のポリマー(天然および合成)を排除するのに著しく効果的であるかを説明するのに役立っている。上記の他のポリマーの排除は、PEGがタンパク質を拒絶し、他の合成ポリマーと二相系を形成する主たる推進力であり、このポリマーを非免疫原性および非抗原性にする。PEGが、タンパク質に共有結合的に連結する場合、それは典型的に、ポリマーの有利な特徴の多くを、結果として生じる結合体に、伝達する。上記の多くの有利な性質により、PEGはタンパク質の改変に非常に適している。
【0023】
本明細書で用いられているように、用語「PEG」は、PEG(「PEG試薬」)のアミノ反応性誘導体を含む、如何なるPEGポリマーをも包含する。タンパク質結合のための種々のPEG試薬は公知である。典型的なPEG試薬は、一つの末端がエーテル結合(例えばO−メチル)を有し、かつ他の末端が反応性基で官能基化されている線状PEGポリマーである。他のPEG試薬は、タンパク質への結合が、再び非反応性末端および反応性官能基の組合せによる、分岐またはデンドリマー状である。あるいは、類似もしくは非類似の反応性官能基の組合せを有するホモ−もしくはヘテロ−二官能性PEG試薬は、タンパク質へ結合するために使用され得る。PEG試薬の例としては、限定されないが、アルデヒド、N−ヒドロキコハク酸イミジルカルボネート、N−ヒドロキシコハク酸イミジルプロピオネート、p−ニトロフェニルカルボネート、またはベンゾトリアゾールカルボネート末端種またはPEGの他のアミノ反応性活性化種が挙げられる。
【0024】
上記PEGポリマーは、例えば、500ダルトンから50,000ダルトンの範囲の分子量を有し得る。上記反応性官能基は、リンカー基によりPEG鎖から分離され得る。必要に応じて、上記ポリマーは、上記PEGとリンカーとの間に分解性の内部結合を有する。従って、本発明の一つの実施形態では、PEGポリマーの反応性基は、タンパク質求核剤との反応のために親電子的に活性化され得る。親電子基の例としては、n−ヒドロキシコハク酸イミジルカルボネート、トレシルおよびアルデヒド官能基である。これらの官能基を有するPEG試薬は、タンパク質のアミノ基への共有的に結合を形成するように反応する。タンパク質アミノ基へのPEG結合のための好ましいPEG試薬は、プロピオン酸リンカーmPEG−SPAのmPEGコハク酸イミド活性エステルである。別の好ましいPEG試薬は、モノメトキシPEG−アルデヒド(mPEG−Ald)である。
【0025】
(インスリン−ポリマー結合体)
インスリン処方物の非経口的投与は、75年以上前のインスリンの発見以来、依然としてインスリン依存性糖尿病(IDDM)の処置に用いられる第一治療法である。現在の治療法を不適切にしている要因の多くは、上記インスリン分子に固有の本質的な欠点である。詳細には、インスリンは、弱い物理的安定性および化学的安定性、タンパク質分解の受け易さ、免疫原性および抗原性ならびに比較的短い血漿中半減期を含む、タンパク質製剤に典型的な多くの問題に直面する。
【0026】
タンパク質PEG化は組換えヒトタンパク質の治療効率を改善するのに使用されてきている。ほとんどの非経口的に投与されるタンパク質は、細網内皮系(RES)、腎臓、脾臓および肝臓により、身体から早く消失する。さらに、ろ過率は分子サイズ、電荷および問題のタンパク質の特異的なレセプターの存在に依存する。PEGのタンパク質への連結は、その分子サイズ、電荷およびレセプター結合能に影響を及ぼし、結合体の全体としての消失率を減少させるのに役立つ。
【0027】
さらに、酵素によるタンパク質の代謝は、治療用タンパク質の生物学的活性の早い損失につながる。タンパク質分解攻撃を受けやすい上記タンパク質のドメインを、立体的に遮蔽することにより、PEGは、生物学的に不活性にするタンパク質の分解を、減少させる。
【0028】
さらに、組換えヒトタンパク質でさえも、繰返し使用の後に免疫反応を、誘発する。上記治療用タンパク質の免疫原性決定基/抗原性決定基を立体的に覆い隠すことにより、PEG−連結は、通常非免疫原性および非抗原性の結合体になる。従って、全体として、PEG化による非経口的タンパク質の特質の変化の結果、血漿中の半減期およびタンパク質分解の抵抗性を増加させ、生じたPEG−タンパク質構築物の免疫原性および抗原性を減少させる。
【0029】
(インスリンタンパク質)
本明細書で使用される用語「インスリンタンパク質」は、任意の天然に存在するもしくは組換えインスリンタンパク質または、例えば、残基PheB1で結合され得る関連タンパク質をいう。従って、本発明に使用されるインスリンタンパク質としては、例えば、インスリンアナログ、インスリンホモログおよびインスリン誘導体が挙げられる。任意の適切な種、例えば、ヒト、ブタ、ウシ、イヌ、ラット、マウス、ヒツジまたはサルからのインスリンが、用いられ得る。好ましい実施形態において、インスリンはヒトインスリンである。
【0030】
ヒトインスリンは周知のタンパク質であり、限定されないが、Sigma Chemical CompanyおよびNovo Nordiskを含む、多くの業者から市販品で容易に入手可能である。天然に存在するヒトインスリンは、分子量およそ5,500ダルトンを有し、およそ51アミノ酸を含むタンパク質である。製造業者により、上記インスリンは、重量ベースではわずかに異なる活性を有し得るが、単位で規定されたインスリンの活性は標準である。種々の程度の生物学的活性を有するインスリンは、市販で入手可能である。例えば、低い作用形態、中程度の作用形態および即効性の作用形態のインスリンを購入することは可能である。インスリンと類似した作用を有するかまたはインスリンレセプターを増加させる非インスリン血糖低下剤としては、限定されないが、スルホニル尿素(例えば、グリベンクラミド、グリクラジド、グリピジド、グリブリド(glyburide)、クロルプロパミド、トリブタミド、トラザミド、アセトヘキサミドおよびグリモプリド(glimopride));チアゾリジンジオン(例えば、トログリタゾン(troglitazone)およびプログリタゾン(ploglitazone);αーグルコシダーゼインヒビター(例えば、アカルボースおよびミグリトール);並びに第3世代インスリン放出剤(例えば、KAD 1220、エトキソミール(etoxomir)およびレパグリニド(repaglinide))が挙げられる。
【0031】
上記インスリン分子は、二つのポリペプチド鎖、A鎖およびB鎖からなる。ヒトインスリンの上記アミノ酸配列は、本明細書中の配列番号1として提供される。上記A鎖は、21個のアミノ酸(A1〜A21と表示する)から構成され、そしてより長いB鎖は、30個のアミノ酸(B1〜B30)から構成される。これらの二つの鎖は、残基A7とB7との間ならびにA20とB19との間の二つの鎖間のジスルフィド結合により一緒に保持されているが、別の鎖内ジスルフィド結合は、残基A6およびA11を結合している。インスリンの3次元構造への安定化をさせるのに役立つ、上記の二つの鎖のアミノ酸残基間でまた、多数の非共有結合性相互作用が存在する。
【0032】
改変(例えばPEGによる)のための利用可能な3つの反応性アミノ基があり、即ち、それらは、A鎖N末端およびB鎖N末端(それぞれA1およびB1)に位置し、ならびにB鎖C末端(B29)に隣接するリジンに位置する。
【0033】
インスリンタンパク質はまた、インスリン様成長因子(IおよびII)のような関連タンパク質および成長ホルモンおよびプロラクチンファミリー由来のタンパク質も含む。
【0034】
(インスリンタンパク質の親水性ポリマーへの結合)
本発明に従って、上記インスリンタンパク質および親水性ポリマーを少なくとも1種の有機溶媒および少なくとも1種の金属キレート剤の存在下で、上記タンパク質とポリマーの結合体の形成が促進される条件下で、接触させる(即ち、反応または結合させる)。特定の実施形態では、上記インスリンタンパク質は、最小量のPEG試薬とゆるやかな条件を用いて、PheB1アミノ末端でPEG化される。PheB1アミノ基は、通常インスリンの3つの利用可能なアミノ官能基の中で反応性が最も低い(Calicetiら、1999、上記)。本発明において、条件はPheB1アミノ基をPEG試薬に対して最も反応性にすること見出した。これらの反応条件は、このようにPheB1での単一PEG化を優性の反応生成物として産生する。
【0035】
本発明の特定の実施形態では、上記インスリンタンパク質および親水性ポリマーを、水溶液中で約0.1〜5%(w/w)、好ましくは0.5〜1.5%でのタンパク質濃度で、pHを5.0〜7.5の範囲、好ましくはpH6.5〜7.2に調整して、接触させる。上記pHは、緩衝塩の包含、有機酸/塩基の添加または通常の無機酸/塩基の添加により制御され得る。上記水溶液は、少なくとも1種の水に混和可能な有機溶媒をさらに包含し、それはエタノール、メタノール、DMSO、ジオキサン、DMF、NMPなどを含む群から選択され得る。別の局面では、上記有機溶媒、好ましくはジオキサンは、濃度(v/v)が0〜25%、好ましくは2〜20%、さらに好ましくは、5〜15%で含まれる。
本発明で用いられる適切な金属キレート剤としては、種々の公知のキレート剤、例えばエチレンジアミン四酢酸(EDTA)、ジエチレントリアミン五酢酸(DTPA)、ニトリロ三酢酸(NTA)、N−2−アセトアミド−2−イミノ二酢酸(ADA)、エチレングリコールビス(2−アミノエチルエーテル)N,N,N’,N’−四酢酸(EGTA)、trans−ジアミノシクロヘキサン四酢酸(DCTA)、グルタミン酸およびアスパラギン酸のようなアミノポリカルボン酸;ならびに、例えば、N−ヒドロキシエチルイミノ二酢酸(HIMDA)、N,N−ビス−ヒドロキシエチルグリシン(bicine)、およびN−(トリヒドロキシメチルメチルグリシン(tricine)のようなヒドロキシアミノカルボン酸;ならびにグリシルグリシンのようなN−置換グリシンが挙げられる。他の適切なキレート剤としては、2−(2−アミノ−2−オキソエチル(oxocthyl)アミノエタンスルホン酸(BES)およびデフェロキサミン(DEF)が挙げられる。本発明の方法で用いられる適切なキレート剤としては、例えば、溶液中で利用可能な酸素と反応することを不可能にする金属イオンへ結合し、それにより上記タンパク質と自由に反応および分解するOHラジカルの発生を、最小限にするまたは防止するキレート剤が挙げられる。そのようなキレート剤は、キレート剤の保護なしで処方されたタンパク質の分解を減少させ得、または防止し得る。
【0036】
本発明で用いられるキレート剤は、塩の形態で、例えば前述のキレート剤のカルボキシル基または他の酸性の官能基で存在し得る。そのような塩の例としては、ナトリウム、カリウム、カルシウムおよび他の弱く結合する金属イオンで形成される塩が挙げられる。当該分野で公知のように、上記塩の性質および中和されるべき電荷数は、存在するカルボキシル基の数および安定化キレート剤が供給されるpHに依存する。当該分野で公知のように、キレート剤は、特定の標的イオンと結合する強度は変化する。一般に、重金属イオンは、類似した電荷をもつ低い分子量の対応物よりも、より強く結合する。
本発明の方法で用いられるキレート剤は、EDTA、EGTAおよび当該分野で公知の他の多価カチオンキレート剤から選択され得る。本発明の方法に従って、金属キレート剤、好ましくはEDTAが、濃度が0.1〜10mM、好ましくは1〜5mM、より好ましくは、1〜3mMで存在する。
【0037】
本発明で使用される適切な親水性ポリマーとしては種々の公知のポリマー、例えばポリエチレングリコール、ポリプロピレングリコールおよびそれらの線状、分岐およびアミノ反応性誘導体が挙げられる。本発明の一つの局面では、上記アミノ反応性誘導体は、アルデヒド、N−ヒドロキシコハク酸イミド、PNP炭酸塩およびベンゾトリアゾール末端親水性ポリマー誘導体からなる群より選択される。本発明の特定の実施形態では、親水性ポリマー、例えばPEG試薬、好ましくはPEGのコハク酸イミドエステル、より好ましくは、mPEG−SPAを、インスリンとモル比(PEG:インスリン)を約10:1〜1:1、好ましくは3:1〜1.2:1、より好ましくは1.7:1〜1.5:1で、接触させる。別の特定の実施形態では、親水性ポリマーとインスリンタンパク質を温度が約4℃〜50℃、好ましくは約15℃〜25℃で接触させる。
【0038】
別の実施形態では、本発明は、上記結合体を単離する前に、上記結合反応をクエンチする工程を、さらに含む。これは、例えばpHを約1〜4、好ましくは約2〜3、より好ましくは2.4〜2.6に低下させることにより達成され得る。上記結合体の単離はイオン交換(例えば、カチオン交換)クロマトグラフィーなどの標準的な技術を用いて達成され得、所望の結合体は収集され、濃縮され、脱塩されそして乾燥され得る。
【0039】
(徐放性送達処方物での生理活性結合体薬剤の利用)
PEG化インスリンタンパク質のような生理活性結合体薬剤は、生分解性ポリマーベースの薬物送達処方物で、有利にカプセル化され得る。PEG化生理活性剤は、対応する非PEG化生理活性剤より高濃度で、薬物送達処方物にカプセル化される。生分解性ポリマー薬物送達処方物からのPEG化生理活性剤の放出は、対応する非PEG化の生理活性剤より急速ではないことを示している。生分解性ポリマーの薬物送達処方物の中でのPEG化生理活性剤の物理的安定性および化学的安定性はより大きく、かつ抗原性および免疫原性は対応する非PEG化生理活性剤よりも低い。
【0040】
この応用のための生分解性ポリマーとしては、限定されないが、ポリ(乳酸)、ポリ(グリコール酸)、ポリ(d,l−乳酸−グリコール酸共重合体)、ポリ(カプロラクトン)、ポリ(オルトエステル)、ポリ(エステル)とポリ(エーテル)の共重合体、ポリ(乳酸)とポリ(エチレングリコール)の共重合体などが挙げられる。
【0041】
従って、本発明のタンパク質−ポリマー(例えば、PEG化インスリン)の結合体は、生分解性ポリマーの薬物送達処方物、例えばポリ(d,l−乳酸−グリコール酸共重合体)(PLGA)ミクロスフェアに有利に取り込まれ得る。これは、非結合タンパク質と比較して、上記タンパク質結合体の高いカプセル化を達成し、かつまたバーストを減少させる(最初の24時間にわたる放出)。さらに、PEGのような親水性のポリマーとの結合は、その結合体を特定の有機溶媒に溶解させ、PLGAミクロスフェアの形成を単純化する。
【実施例】
【0042】
I.一工程法による部位特異的PEGインスリンの調製および性質決定
(実施例1)
NαB1−メトキシポリ(エチレングリコール)−インスリン結合体の調製
1グラム(172μモル)のインスリン(Zn2+−インスリン、Intergen Co.)を、室温で100mLの2mM EDTA水溶液に溶解し、希塩酸を用いて、その溶液のpHappを7に調整した。別の容器に、1.4g(インスリンに対して1.6モル当量)の活性化PEG誘導体[モノメトキシポリ(エチレングリコール)プロピオン酸コハク酸イミド、mPEG−SPA、Shearwater Corp.]を、室温で10mLのジオキサンに溶解した。上記mPEG−SPA溶液を、上記インスリン溶液に直接注入で加え、そしてその反応を室温で2時間進行させた。上記反応を、HCl(pHapp2.5)で酸性化してクエンチし、そしてその混合物を0.02%重炭酸アンモニウムに対して限外ろ過[YM3(3000MWCO)膜を取り付けたAmicon 8200 限外ろ過装置]した。その後、その反応混合物を、1M 酢酸/7M 尿素/0.01M NaClに対して限外ろ過し、精製前に10mLまでに濃縮した。上記mPEG−PheB1−インスリン誘導体を、他の反応副生成物(mPEG−GlyA1−インスリン、mPEG−LysB29−インスリン、ジ−mPEG−インスリン、およびトリ−mPEG−インスリン)から、SP セファロース(Amersham Biosciences)カチオン交換カラムを取り付けたFPLCシステムを用いて、単離した。上記カラムを、5mL/分の流速で、1M 酢酸/0.04M NaCl含有7M 尿素で平衡化し、上記結合タンパク質をNaCl−グラジェント(0.04M〜0.12M)を用いて、80分にわたって溶出した。280nmで検出された主要なピークに対応する上記溶出液を収集し、そしていかなる低分子量の不純物をも除去するために0.02% NH4HCO3に対して限外ろ過し、その後凍結乾燥をして性質決定の前に−20℃で保存した。
【0043】
PheB1のアミノ基に連結した、より低分子量の線状ポリマー(即ちmPEG−SPA、Mr=2000Da)を有する1種の結合体、および2種のより高分子量の分岐ポリマー(即ち、mPEG2−SPA、Mr=10,000Da)を有する結合体を調製するのにもこの方法を用い、そしてこれらの2種の異なる結合体の性質決定もまた、以下に概説する技術に従って行なった。この結果は、この方法が、PheB1に連結されたポリマー構造のみ(即ち、線状もしくは分岐)またはサイズのみ(即ち、分子量)が異なる種々様々のPEG−PheB1−インスリン結合体を首尾良く調製するのに用い得る。
【0044】
(実施例2)
結合体純度のFPLC/HPLCによる評価
mPEG−PheB1−インスリンの純度を、流速1.0mL/分を用いた以外は、上述の単離手順で用いた条件と同一の条件下で、分析用カチオン交換カラム(MonoS 5/5、Amersham Biosciences)を用いて、分析した。直交技術(逆相HPLC)もまた、上記結合体の最終純度を証明するのに用いた。Waters Alliance HPLC システムに、Waters 996光ダイオード配列検出器(PDA)およびSymmetry300(C18、5μm 粒子サイズ、4.6×250mm)逆相カラムを取り付けた。流動層Aは、0.1%TFA(トリフルオロ酢酸)MilliQ 品質水からなり、そして流動層Bはまた、0.1%TFAを含有する、95/5ACN(アセトニトリル)/H2Oからなった。15分にわたって、30〜60%Bの直線グラジェントを用いて、化合物の溶出を276nmの検出で、進めた。上記mPEG−インスリンの純度は、95%を超えた。
【0045】
(実施例3)
結合体同一性のN末端タンパク質配列決定(エドマン分解)による確認
上記エドマン分解反応は、PEGに共有結合したN末端アミノ基では進まないということを承知の上で、PEG結合部位を決定するために、N末端タンパク質配列分析を用いた。全ての試料を、Applied Biosystems 477Aタンパク質配列分析器(Pasadena、Ca)を用いて、3回の分解サイクルで分析した。N末端アミノ酸モル比が[GlyA1/PheB1]≒1の場合は、残基LysB29(または無し)への結合を暗示し、[GlyA1/PheB1]≒0の場合は、残基GlyA1への結合を暗示し、[GlyA1/PheB1]≒30の場合はPheB1への結合を暗示する。この結果は、置換の部位が約95%PheB1であることを確証した。
【0046】
(実施例4)
マトリックス支援レーザー脱離イオン化(MALDI)による結合体分子量の同定
この分析の性質決定技術を、分析中にPEG−インスリン結合体が分解する原因にならないような意味を持つ「軟イオン化」であるので、選択した。上記機器の出力は、各試料の質量/電荷比の定量的な尺度を提供する;従って、インスリンに連結しているPEG鎖の数を、上記結合体の分子量と未改変のインスリンの分子量との全体的な差から容易に決定することができる。全ての試料を、直線モードで操作したPerceptive Biosystems model DERP MALDI/TOF質量分析計にかけ、陽イオンをモニターした。全ての試料の上記マトリックスは、α−シアノ−4−ヒドロキシ桂皮酸であり、窒素レーザの337nm線を最終スペクトルのために平均して少なくとも64ショットで用いた。モノマーのインスリンは5807.2Daの計算分子量を有し、結合反応に用いたmPEG(5000)−SPAの数平均分子量は、5129Daであった。上記mPEG(5000)−インスリンの質量スペクトルは、一つのみのmPEG鎖がインスリンに連結しているという結論と一致した。さらに、個々のイオンピークは、一貫して44Da(エチレンオキシドモノマー単位の分子量)ごとに異なっていた。これらの結果、調製した結合体の全てにおいて、一つのみのmPEG鎖がインスリンに連結して、そしてそれらの多分散性が、もっぱらPEG固有の多分散性に、起因するものであることを確証した。
【0047】
(実施例5)
インスリンB1アミノ末端へのPEGの結合における2級アミンの形成
F5000PEG−インスリンを上記タンパク質と末端にアルデヒド基を有する活性化mPEGとを反応させて調製した。この反応は、シフ塩基中間体を経由して進行し、引き続いて上記ポリマーとタンパク質間の安定な2級アミン結合を形成するようシアノトリヒドロホウ酸ナトリウムで還元する。上記反応を以下のようにして行なった:2mM EDTA/25mM リン酸緩衝液を作り、pHをリン酸で6.0に調整した。5.5mg/mL(全容量2mL)のインスリンを440μlのジオキサンを加えたリン酸緩衝液に溶解した。一度、インスリンを2mLの10mM NaCNBH3水溶液に加え、その後5倍モルの過剰のmPEG(5000)−アルデヒド(乾燥粉末として、Shearwater Corporation、Huntsville、Alabama)を加える。結局、上記反応混合物は、およそ12.5mM リン酸(pH6)、1mM EDTA、10%ジオキサン、5mM NaCNBH3、2.5mg/mL インスリンおよび10mg/mL mPEG−アルデヒドを含有していた。上記反応を一晩進行させ、翌日、そのpHが約5.5であることが分かった。上記反応を、酢酸の添加により、約pH2にクエンチした。小アリコートをRP−HPLCを用いて分析した。主要な反応種を以下のように決定した:約70%の上記反応生成物は、モノ−PEG化物(室温で12.5分)、約10%の未反応インスリン(室温で9.8分)および9%のジ−PEG化インスリン(室温13.8分)が残っていた。上記モノ−PEG化分画を溜めて0.02%NH4HCO3に対して透析し、そして凍結乾燥した。MALDI−TOF分析は、インスリンへ1つのPEG−5000鎖の付加に対応する単一分子質量を示した。エドマン分解分析では、約95%のモノ−PEG化種が残基PheB1で置換され、残りのモノ−PEG化種(〜5%)は殆ど残基GlyA1で置換されている。何故なら、ここで用いた反応条件下ではLysB29のアミノ基は99.99%プロトン化されている(従って反応性無い)からである。
【0048】
II.部位特異的PEG−インスリン結合体の代替的調製法
(実施例6)
従来の多工程法を用いて、B鎖アミノ末端(F5000)へ結合したPEG−5000
組換えヒトインスリン(Intergen Co.)をジ−boc保護化中間体を用いて、PheB1位にPEG化した。ジ−NαA1、NεB29−t−boc−インスリン(ジ−boc−インスリン)を、Liuら。1997(Liuら、Bioconj.Chem.8(5):664〜672、1997)に従って合成した。上記mPEG(5000)−PheB1−インスリン結合体をHindsら、2000、上記のプロトコールに従って調製した。
【0049】
FPLCで単離された上記所望の分画は、逆相HPLCおよびイオン交換FPLCのクロマトグラフのピーク面積を基礎にして98%を越える純度であった。上記精製した生成物をMALDI−TOF質量分析およびアミノ酸配列分析でさらに性質決定を行ない、上記B鎖アミノ末端PheB1で、モノ置換PEGであることが分かった。この方法で調製および精製された上記mPEG(5000)−PheB1−インスリン結合体(F5000)は、本発明のより単純で、より時間のかからない方法により作られた同じ結合体(実施例1)と等価であった。
【0050】
(実施例7)
B鎖Lys29に結合したPEG−5000(K5000)
組換えヒトインスリン(Intergen Co.)を、Hindsら、2000、上記の方法により、B鎖のLys29に特異的にPEG化した。上記所望のFPLC分画は、逆相HPLCおよびイオン交換FPLCのクロマトグラフのピーク面積を基礎にして、純度が98%を超えた。その精製された生成物を、MALDI−TOF質量分析およびアミノ酸配列分析でさらに性質決定を行い、そしてB鎖末端から2番目のアミノ酸LysB29で、モノ置換されていることを示した。この同じ方法をまた使用して、LysB29に連結した1種の低分子量の線状ポリマー(即ち、mPEG−SPA、Mr=2000Da)および2種のより高分子量の分岐ポリマー(即ち、mPEG2−SPA、Mr=10,000または20,000Da)を調製し、そして上記に概説した技術に従って、これらの3種の異なる結合体の性質決定を行なった。この結果は、この方法が、LysB29に連結されたポリマー構造のみ(即ち、線状もしくは分岐)またはサイズのみ(即ち、分子量)が異なる種々様々のPEG−LysB29−インスリン結合体を、首尾良く調製するのに用い得る。
【0051】
III.部位特異的PEG−インスリンの性質決定
コンホメーション、活性および安定性
(実施例8)
部位特異的PEG化後のインスリンコンホメーション保持性の評価
遠紫外範囲の円偏光二色性分光法を用いてインスリンのコンホメーションを調べた。通常、以下の二つの負の極小の強度を評価する:水性環境中でのインスリンのコンホメーションの分析において208nm(α−ヘリックス)および223nm(β−シーツ)。208nmの遠紫外のCD−帯は、主として残基B10〜B19、A2〜A6およびA13〜A19から形成されるα−ヘリックスに起因し、一方、β−構造は、223nmの遠紫外CD−帯の主たる成分である。上記試料に特徴的なCDスペクトルは、残基PheB1またはLysB29のいずれかへのインスリンへのmPEGの連結は、Zn2+−インスリンと比較して、上記結合体の全体的なコンホメーション(2次)を変化させない事を確認した。
【0052】
(実施例9)
PEG−インスリン結合体の薬力学
インスリン薬力学に対するPEG化の効果を調査した。これらの研究は、市販で入手可能なインスリン処方物(Humulin R、Lilly)と比較して、置換部位の異なるPEG−インスリン(位置異性体)またはPEGの分子量が異なるPEG−インスリンを含有する結合体処方物のインビボの生物学的活性(血清グルコースを低下させる能力)を評価するために実施した。絶食したSprague−DawleyラットにF5000、K2000、K5000、K10000またはHumulinR(登録商標)を静脈内に投与した後、血中グルコースレベルを測定した。0.3IU/kg(タンパク質濃度を基礎にし、そしてPEGの重量を適切に補正して;25IU/mgタンパク質と仮定する)を通常の生理食塩水に溶解し、尾静脈注射により投与した;1群N=6。試験試料の注射前および注射後6時間の期間、間隔をおいて採血を行なった。血清を標準的な操作方法で単離し、グルコースレベルをAccucheck Advantage(Boeringer Ingelheim)グルコースメータを用いて測定した。
【0053】
図1Aおよび図1Bに示す結果は、F5000、K2000およびK5000は、正常ヒトインスリンと等価の用量で、正常ラットのグルコースレベルを抑制するのに、全て効果的である。興味深いことには、上記K10000結合体は、他の試験試料と同じ程度にはグルコースレベルを減少しなかったが、結合体の作用の持続時間は、6時間を越えて観察された。このように、上記K10000結合体は、糖尿病患者に長時間のグルコース抑制を提供し、従前の基本のインスリン懸濁剤の可溶性代替物として、開発され得る。
【0054】
(実施例10)
代表的なPEG−インスリン結合体の物理的安定性
加速振動試験法を用いて7種のPEG−インスリン結合体(F2000、F5000、F10000、K2000、K5000、K10000およびK20000)および亜鉛インスリンの物理的安定性を調査した。この試験は加速法でインスリン調製物の物理的安定性の正確な測定を提供する文献に、通常記載されている。このアッセイのために、上記タンパク質(結合体)の水溶液を4部(1mg/mL、リン酸緩衝生理食塩水、pH7.3,0.02%アジ化ナトリウム)調製し、37℃で水平振動(100/分の振動数)させた。試料を所定の時間に取り出し、ろ過し(不溶性凝集体の除去のため)、そして溶液中に残っているタンパク質(結合体)分画を、定量化するためにRP−HPLCにより分析した。図2は、各インスリン誘導体に関して、時間の関数として可溶性タンパク質の減少を示している。
【0055】
これらのデータは著しく高い(36〜40倍)物理的安定性を有するPheB1アミノ基で置換されたインスリン誘導体およびLysB29−インスリンが、天然のペプチドより、いくらかより安定(4〜8倍)であった以前の報告を補強するものである。以前の研究では、PheB1−インスリンの繊維形成に対する増加した抵抗性は二つの相補的な効果によるものであることを示している。第一の効果は、mPEG結合によりB鎖N末端を立体特異的に遮断することであり、それがこの表面がインスリンの繊維形成に必要とされる疎水性相互作用に関与するのを防止する。PheB1−インスリンの増加した物理的安定性に貢献する第二の効果は、元々非特異的であり、立体的であり、そしてポリマー分子量に比例的に増加する。上記LysB29−インスリンの全てが、天然のペプチドに比較して、増加した物理的安定性を示すが、ポリマー分子量が10kDaを超えるまでは、PheB1で置換された上記結合体と同じ程度ではない。これは、繊維形成反応におけるLysB29の関与の欠如により説明され得、繊維形成に関与する分子間相互作用の非特異的立体障害に起因する安定化効果を伴う。
【0056】
(実施例11)
代表的PEG−インスリン結合体の化学的安定性
インスリン(またはインスリンアナログ)は、水溶液中で多くの化学分解反応を受けることはよく実証されている。例えば、インスリンA鎖のC末端のアスパラギンは、酸性条件下では、環化中間体により促進される脱アミド化機構により分解する。この高い反応性環化中間体はまた、アミノ転移反応機構を介して、異なるインスリン分子からのN末端アミノ基の一つと反応して分解し得る。
【0057】
上記インスリン結合体、F5000およびK5000を37℃で、0.02%アジ化ナトリウムPBS中で水平振動でインキュベートした。所定の時間に(0、6、12、28および36日)、個々の試料を取り出し、その後、脱アミド化の程度を決定するためにRP−HPLCにより、そして共有結合の2量化の程度を決定するためにサイズ排除(size exclusion)クロマトグラフィーで分析した。
IV.PEG−インスリン生分解性ポリマー徐放送達処方物の調製、性質決定および薬物動態
(実施例12)
PLGAミクロスフェアでのPEG−インスリンのカプセル化
PEG−インスリンF5000(50mg)を、1mlの塩化メチレンに溶解した。上記溶液を150mgのPLGA45:55(lac:gly)、酸末端基を持つ0.15dl/gIVを有する1ml容量の塩化メチレンに添加した。上記塩化メチレン溶液を、15mlの遠心チューブ中の1%ポリ(ビニルアルコール)を含む5mlの水に添加し、そしてボルテックスミキサーでエマルジョンを作った。上記エマルジョンを0.3%のポリ(ビニルアルコール)を含む100mlの水に添加し、塩化メチレンを蒸発させるために3時間攪拌した。上記硬化ミクロスフェアを、Whatman1番のろ紙で真空ろ過し、そして乾燥した。
【0058】
(実施例13)
PEG−インスリンPLGAミクロスフェアの性質決定
上記PEG−インスリンミクロスフェアの表面形態および粒子サイズ分布を、それぞれ、走査型電子顕微鏡およびレーザ光散乱粒子サイズ分析で調査した。ポリマーミクロスフェア内にカプセル化されたPEG−インスリン結合体の量を定量化するために、分析用逆相HPLCを用いた。HPLC分析に先立って、測定した量のミクロスフェアを、ある容量のアセトニトリルに溶解し、そしてポリマーを沈殿させ、水溶液中へ上記結合体を抽出するために、等容量の0.1%TFA水溶液を添加した。K2000、K5000、K10000、K20000およびF5000−Aもまたこの方法を用いてカプセル化した。これらの試験の結果を表1に集めたが、そこには全ミクロスフェア中のPEG−インスリン含量%(w/w)、および(カプセル化されたPEGインスリン重量/最初に添加されたPEG−インスリン重量)として定義されたカプセル化効率を挙げている。28.3%と相対的に高い薬物含量および100%までのカプセル化効率を達成し、上記生成物を、必要とされる減少した全用量により、臨床的に有用になり、かつ出発物質の低い損失で商業的にも魅力的にした。さらに、ミクロスフェアは、乳酸とグリコール酸の比率(50:50および72:25)、分子量(6.5〜24kDa)、固有粘度(0.09〜0.27dL/g)、または末端エステル基(メチルおよびラウリル)を変えたポリマーと広い範囲の薬物負荷(5〜35重量%)を用いて調製した。
【0059】
【表1】
(実施例14)
動物実験で用いられたPEG−インスリンミクロスフェア処方物のインビトロ放出
15mgのF5000PEG−インスリンミクロスフェア(14.1%薬物含量、PLGA 45:55;0.15dl/gIV、酸末端基)を1.5mlのリン酸緩衝生理食塩水(pH7.4、0.02%アジ化ナトリウムおよび0.02%Tween20)に懸濁し、37℃でインキュベートした。上記上清を、間隔的に取り出し、放出されるPEG−インスリンをRP HPLCで分析した。上記緩衝液を、新鮮なPBSで置換し、そのインキュベーションを続けた。データをインキュベーション時間の関数として累積放出量を分析した(図4)。最初の日は、1.0%未満の上記PEG−インスリンの放出で、18日以内で、95%を超える量が放出される。約2週間を越える低い「バースト」放出、高い全放出量および持続期間は、徐放インスリン処方物の非常に所望の特徴である。
異なる生分解性ポリマーおよびF5000とを用い、ならびにK5000PEG−インスリンおよびF5000−A結合体もまた、用いる実施例12の方法で作った他の調製物もまた、1日放出値が0〜7.5%の間で、持続放出期間も60日までもあった。
【0060】
(実施例15)
F5000PEG−インスリンPLGAミクロスフェアのインビボ薬力学および薬物動態PLGA45:55、0.15dl/gIV、酸末端基(14.1%核負荷)から構成されるミクロスフェア中のF5000PEG−インスリンを、糖尿病ラットでグルコース抑制およびF5000薬物動態の試験をした。雄性SDラット(〜250g)に、等張クエン酸緩衝液(10mM、pH4.5)に溶解した40mg/kgのストレプトゾトシン(Sigma、St.Louis、MO)を、試験試料投与する1日前に皮下投与をして糖尿病にした(Junod,Aら、J.Clin.Invest.、48:2129〜2139、1969)。動物を2.5%滅菌カルボキシメチルセルロース(CMC)または生理食塩水(陰性コントロール)に懸濁したPEG−インスリンミクロスフェア(〜11mg/ラット、体重1kgあたり3mgインスリン当量に相当する)で処置し、そして血清単離、引き続くグルコース(PD)およびF5000(PK)レベルの分析のために所定の間隔で採血した。図5および6は、13日の期間の血清グルコースレベルおよび血清PEG−インスリンレベルを示す(データは平均値±標準誤差)。図5は、予想したとおり、実験の間中、血中グルコースレベルがコントロールの糖尿病ラットに対して100%以上(測定誤差内)を示す。PEG−インスリンミクロスフェア処置糖尿病ラットに関しては、血中グルコ−スレベルは、3日後未処置レベルの60%より低いレベルに低下し、さらに7日間、抑制が持続している。
【0061】
図6は、PEG−インスリンの無視し得る初期バースト放出(Cmax=0.62ng/ml)が、ミクロスフェア注入後すぐに(1時間)検出されたことを示している。それから注入後24時間、処置群のPEG−インスリンレベルは、2日を超えて、着実に治療レベル(約1〜3.5ng/ml)まで上がり、約7日間持続し、一方、上記陰性コントロール群のF5000血清レベルは、ずっと検出限界未満であった。これらのデータは、一緒に考えると、結合体の生物学的活性は、ミクロスフェア製造工程で保持され、そしてミクロスフェア注入に続く一週間の放出の間、維持されたことを示している。重要なことは、類似した量の初期PEG−インスリンバースト放出が、インビトロ(図4、第1日<0.5%放出)およびインビボ(図6、AUC0〜1d/AUC0〜13d=0.7%実験)で見出されたことである。さらに、図5および6に示されているデータの評価は、薬物動態/薬力学(PK/PD)相関がこの実施例で存在することを示唆している。
【0062】
(等価物)
当業者は、本明細書に記載している本発明の特定の実施形態に対する多くの等価物を単なる慣用的実験を用いて、認識または確認することができる。そのような等価物は、以下の特許請求の範囲に包含され、網羅されることが意図されている。
【特許請求の範囲】
【請求項1】
明細書中に記載の発明。
【請求項1】
明細書中に記載の発明。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2010−248241(P2010−248241A)
【公開日】平成22年11月4日(2010.11.4)
【国際特許分類】
【出願番号】特願2010−160151(P2010−160151)
【出願日】平成22年7月14日(2010.7.14)
【分割の表示】特願2006−509863(P2006−509863)の分割
【原出願日】平成16年4月8日(2004.4.8)
【出願人】(504405497)ピーアール ファーマシューティカルズ, インコーポレイテッド (6)
【Fターム(参考)】
【公開日】平成22年11月4日(2010.11.4)
【国際特許分類】
【出願日】平成22年7月14日(2010.7.14)
【分割の表示】特願2006−509863(P2006−509863)の分割
【原出願日】平成16年4月8日(2004.4.8)
【出願人】(504405497)ピーアール ファーマシューティカルズ, インコーポレイテッド (6)
【Fターム(参考)】
[ Back to top ]