
部分的に付加された抗体およびそれらの結合体化方法
本発明は、タンパク質−薬物結合体ならびにタンパク質−薬物およびタンパク質標識結合体を作成する方法を提供する。本発明の開示により、1個以上の活性化基(例えば、抗体)を含有するタンパク質に、1個以上のそのような結合の部分還元または完全還元を受けさせて、反応性基を形成させる方法が提供される。本発明の方法において、生じたタンパク質は、反応性基の一部と反応する薬物(例えば特定の放射性金属、キレート剤、および毒素)と反応されて、例えばインビトロ診断、インビボ撮像、および治療において有用な結合体を形成する。

【発明の詳細な説明】
【技術分野】
【0001】
(継続)
本願は、2004年3月2日に出願された米国仮特許出願第60/549,476号の優先権を主張し、この開示はその全体が参考として本明細書中に援用される。
【0002】
本発明は、例えばタンパク質−薬物結合体の特定異性体を生じる薬物との少なくとも1個の結合箇所を有する修飾タンパク質と、特定異性体を生じるそのような結合の方法とに関する。本発明はさらに、細胞毒性薬および/または細胞増殖抑制薬が結合可能であり、特定異性体を生じる抗体と、その結合の方法に関する。
【背景技術】
【0003】
(背景)
本発明は、例えばタンパク質−薬物結合体の特定異性体を生じる薬物との少なくとも1個の結合箇所を有する修飾タンパク質と、特定異性体を生じるそのような結合体化の方法とに関する。本発明はさらに、細胞毒性薬および/または細胞増殖抑制薬が結合体化可能であり、特定異性体を生じる抗体と、その結合体化の方法に関する。
【0004】
モノクローナル抗体(mAb)は、癌との闘いにおいて価値のある武器である。mAbは免疫障害の処置でも使用される。癌および免疫障害に対するmAbベースの療法の使用をさらに促進するために、多数の新規な手法が検討されてきた。1つの手法は、殺細胞ペイロードを結合することによって、腫瘍細胞に対するmAbの細胞毒性ポテンシャルを向上させることである。タンパク質毒素、放射性核種、および抗癌剤などの分子は、あるmAbに結合体化されて免疫毒素、放射免疫結合体、および抗体−薬物結合体(ADC)をそれぞれ生成してきた。
【0005】
ADCの開発で以前に考慮された因子は、抗体の選択、および薬物成分の効力の最適化、リンカーの安定性、および薬物がmAbに共有結合される方法を含んでいた。ジスルフィド結合によって結合体化されたADCを生成するための一般的な慣例は、抗体のすべての鎖間ジスルフィド結合を還元することと、還元されたmAbチオールすべてに還元チオールすべてとの相互作用が可能である化合物と反応させて、ある結合部位に対する特異性を得る能力を持たない、薬物8個/mAbで均一に置換された、すなわち「完全付加された」ADCを形成することによるものであった。
【0006】
例えば抗原CD30は、ホジキン病(HD)未分化大細胞リンパ腫(ALCL)などの癌で高度に発現される。CD30のこの発現は、正常細胞での制限された発現と相まって、それをADC療法の魅力的な標的としている。CD30を指向するキメラmAbであるcAC10は、インビトロと、皮下および播種性SCIDマウス異種移植モデルとの両方でHDに対する抗腫瘍活性を有する。cAC10の抗腫瘍活性は、8個すべての鎖間チオールが細胞毒性薬オーリスタチンEの誘導体に薬物成分として結合された完全付加ADCを産生することによって向上された。これらのADCは、マウス異種移植モデル内で耐容性に優れた用量にて非常に有効であった。
【0007】
ADCの産生における慣習は、それらに薬物を完全付加することであったため、部分付加ADCが同じまたはそれ以上の治療有効性を持ちうることは、以前は認識されていなかった。さらに抗体での他の置換パターンが同じまたはそれより少ない毒性で同じまたはそれ以上の治療有効性を発生しうることを考慮できる方法が存在しなかった。これらおよび他の過去の制限および問題は、本発明によって解決される。
【発明の開示】
【課題を解決するための手段】
【0008】
(要旨)
本発明は、タンパク質−薬物結合体ならびにタンパク質−薬物およびタンパク質標識結合体を作成する方法を提供する。薬物または標識を受容するための結合箇所を有するタンパク質も提供される。結合体は、生体内撮像に、または他の用途に治療的に、診断的に(例えばインビトロまたはインビボ)で使用され得る。
【0009】
一般に、割当て可能な結合箇所を有する部分付加された修飾タンパク質が供給される。修飾タンパク質は一般に、結合パートナーとの相互作用のための結合領域と、少なくとも2個の結合箇所とを含み、各結合箇所は薬物または標識と共有結合している。通例、同様の接近性または活性化性を有する、考えられるすべての結合箇所のより少数が薬物または標識に結合される。修飾タンパク質は例えば、抗体、レセプター、レセプターリガンド、ホルモン、サイトカインなどであり得る。結合箇所は例えば、アミノ基、隣接ヒドロキシル基、ヒドロキシル基、カルボキシル基、またはチオール基であり得る。タンパク質は例えば、レセプター、レセプターリガンド、ホルモン、サイトカインなどであり得る。
【0010】
さらなる実施形態において、1個以上のジスルフィド結合を有するタンパク質と、遊離チオールと反応性の薬物との結合体を調製する方法が提供される。該方法は一般に、タンパク質を還元剤によって部分的に還元することと;遊離チオールと反応性の薬物を部分還元タンパク質に結合体化することとを含む。なお別の実施形態において、1個以上のジスルフィド結合を有するタンパク質と、遊離チオールと反応性の薬物との結合体を調製する方法が提供される。該方法は一般に、タンパク質を還元剤によって完全に還元することと;タンパク質を再酸化剤によって部分的に再酸化することと;遊離チオールと反応性の薬物を抗体に結合体化させることとを含む。
【0011】
一部の実施形態において、部分付加抗体が提供される。抗体は、抗原結合領域と、少なくとも1つの鎖間ジスルフィド結合と、それぞれ鎖間チオールに結合体化された、少なくとも2つの薬物または標識を含む。薬物または標識の結合箇所は必要に応じて、容易に割当て可能である。1つの例では、抗体は少なくとも4個の細胞毒性薬または細胞増殖抑制薬を有することが可能であり、各薬物は鎖間チオールに結合体化される。ある実施形態において、抗体は、種4A、種4B、種4C、種4D、種4Eまたは種4Fの配置を有する。
【0012】
部分付加抗体は例えば、マウス抗体、ヒト化抗体、ヒト抗体またはキメラ抗体であり得る。薬物は例えば、MMAF、MMAEまたはAFPなどの細胞毒性薬または細胞増殖抑制薬であり得る。部分付加抗体を含む薬学的組成物も提供される。
【0013】
別の実施形態において、抗体は、細胞毒性薬または細胞増殖抑制薬に対する少なくとも1つの結合箇所を有し、抗体上の結合箇所を細胞毒性薬または細胞増殖抑制薬へ容易に割当てることができるように提供される。抗体では、考えられるすべての結合箇所より少数が細胞毒性薬または細胞増殖抑制薬への結合体化に利用できる。結合箇所は例えば、鎖間チオールであり得る。結合箇所は例えば、種4A〜4Fのうちの少なくとも1個であり得る。
【0014】
割当て可能な結合箇所を有する修飾抗体の組成物も提供される。該組成物は、例えば少なくとも2個の、少なくとも4個の、少なくとも6個の、少なくとも7個の、少なくとも10個以上の種の修飾抗体を有することができる。1つの例において、各種は2個の鎖間チオール、および少なくとも1個の鎖間ジスルフィド結合を有する、少なくとも1つの特定結合対を持ちうる。抗体種は例えば4A、4B、4C、4D、4Eおよび/または4Fを有することができる。さらなる例では、特定結合対は、定常軽鎖−定常重鎖間ジスルフィド結合および/または定常重鎖−定常重鎖間ジスルフィド結合に存在しうる。特定結合対は、ヒンジ領域のN末端より近位におよび/またはヒンジ領域のC末端より近位にあり得る。別の例では、特定結合対は、定常軽鎖−定常重鎖間スルフィド結合に、および修飾抗体のN末端付近に位置するヒンジ領域に、または定常軽鎖−定常重鎖間ジスルフィド結合に、および修飾抗体のC末端付近に位置するヒンジ領域に存在する。
【0015】
抗体の各種は必要に応じて、定常軽鎖−定常重鎖間ジスルフィド結合における少なくとも2個の特定結合対およびヒンジ領域鎖間ジスルフィド結合における少なくとも2個の特定結合対を含むことができる。該組成物は必要に応じて、薬学的に受容可能なキャリアをさらに含むことができる。
【0016】
なお別の実施形態において、部分付加抗体が提供される。該抗体は、少なくとも1個の抗原結合ドメインと、抗体上の少なくとも2個の反応性基と、少なくとも2個の薬物または標識を含み、各薬物または標識は、結合箇所を形成するためにそれぞれ反応性基に結合される。薬物または標識に対する結合箇所は容易に割当てられる。
【0017】
なお他の実施形態において、薬物を還元して、薬物配置の選択性をもたらす抗体に結合させる方法が提供される。該方法は一般に、抗体を還元剤によって完全に還元することと、少なくとも2個の鎖間チオールが残存するように抗体の少なくとも1個の鎖間ジスルフィド結合を再形成するために、完全に還元された抗体を再酸化剤の制限量で処理することと;薬物を鎖間チオールに結合体化することとを含む。再酸化剤は例えば、5,5’−ジチオ−ビス−2−ニトロ安息香酸、4,4’−ジチオジピリジン、2,2’−ジチオジピリジン、テトラチオン酸ナトリウムまたはヨードソ安息香酸であり得る。薬物は例えば、細胞毒性薬または細胞増殖抑制薬あるいは免疫抑制剤であり得る。一部の例では、薬物は副溝バインダ、AEB、AEVB、MMAF、MMAEまたはAFPであり得る。還元剤は例えば、DTTまたはTCEPであり得る。
【0018】
関連する実施形態において、抗体鎖間ジスルフィド結合を還元して、薬物を生じた鎖間チオールに結合させて、抗体上に薬物配置の選択性を生じさせる方法が提供される。該方法は一般に、鎖間チオールを形成するために抗体を還元剤によって完全に還元することと;少なくとも1個の鎖間ジスルフィド結合を再形成するために、抗体を再酸化剤で部分的に再酸化することと;薬物を鎖間チオールに結合させることとを含む。再酸化剤は例えば、5,5’−ジチオ−ビス−2−ニトロ安息香酸、4,4’−ジチオジピリジン、2,2’−ジチオジピリジン、テトラチオン酸ナトリウム、またはヨードソ安息香酸であり得る。還元剤例えば、DTTまたはTCEPであり得る。薬物は例えばMMAF、MMAE、またはAFPであり得る。部分的に再酸化された抗体は必要に応じて、結合体化前に精製され得る。
【0019】
なお他の関連する実施形態において、抗体鎖間ジスルフィド結合を還元して、薬物を生じた鎖間チオールに結合させて、薬物を抗体上に選択的に配置する方法が提供される。該方法は一般に、少なくとも2個の鎖間チオールを形成するために、抗体を還元剤によって部分還元することと;部分的に還元された抗体の鎖間チオールに結合体化させることとを含む。1つの例では、抗体は制限濃度の還元剤によって、キレート剤を含む緩衝液中で部分的に還元される。該薬物は、例えば抗体溶液を冷却して、薬物を冷溶媒に溶解させ、抗体溶液と混合することによって結合体化できる。抗体および薬物溶液は、1個または複数の部分的に付加された抗体−薬物結合体を形成するのに十分な期間に渡ってインキュベートされる。反応は、過剰な薬物をチオール含有試薬によってクエンチさせることによってクエンチできる。結合体はさらに精製できる。具体的な例において、抗体は約37℃にて約1時間、部分的に還元される。還元された抗体は例えば約0℃まで冷却することができる。抗体および薬物溶液は例えば、約0℃にて30分間インキュベートできる。
【0020】
チオール含有試薬は例えば、システインまたはN−アセチルシステインであり得る。還元剤は例えばDTTまたはTCEPであり得る。緩衝液は例えばホウ酸ナトリウム溶液でもよく、キレート剤はジエチレントリアミン五酢酸である。キレート剤例えば、エチレントリアミン五酢酸またはEDTAでもあり得る。溶媒は例えば、アセトニトリル、アルコールまたはDMSOであり得る。薬物は例えば、細胞毒性薬または細胞増殖抑制薬であり得る。
【0021】
一部の実施形態において、還元された抗体は結合体化前に例えばカラムクロマトグラフィー、透析、またはダイアフィルトレーションを使用して精製できる。カラムクロマトグラフィーで使用されるカラムは例えば、PD−10カラムなどの脱塩カラムであり得る。あるいは還元抗体は、部分的還元後および結合体化前に精製されない。
【0022】
結合体は例えばカラムクロマトグラフィー、透析、またはダイアフィルトレーションを使用して精製できる。カラムクロマトグラフィーで使用されるカラムは、例えば脱塩カラム、例えばPD−10カラムであり得る。
【0023】
なお別の実施形態において、薬物の選択的結合体化によって抗体を産生する方法が提供される。該方法は一般に、例えばDTNB滴定によって決定されるように、大幅に過剰の還元剤を添加して、溶液を約37℃にて約30分間インキュベートすることによって、鎖間チオールを産生するのに十分な時間抗体を完全に還元することと;抗体を精製することと;酸化剤を使用して抗体を部分的に再酸化し、還元された抗体を0℃に冷却することによって、少なくとも1個の鎖間ジスルフィド結合を形成することと;還元および冷却された抗体を酸化剤1.5〜2.5モル当量で処理することと;反転により溶液を混合することと;約0℃にて約10分間インキュベートすることによって、抗体を部分的に再酸化することと;部分的に再酸化された抗体を精製することと;結合体化抗体を作成するために、部分的に再酸化された抗体の鎖間チオールに薬物を結合させることと;結合体化抗体を精製することとを含む。
【0024】
還元剤は例えば、DTTまたはTCEPであり得る。部分還元抗体は必要に応じて、例えばカラムクロマトグラフィー、透析、またはダイアフィルトレーションを使用して精製できる。カラムクロマトグラフィーで使用されるカラムは例えば、PD−10カラムなどの脱塩カラムであり得る。結合体化抗体は例えば、カラムクロマトグラフィー、透析、またはダイアフィルトレーションを使用して精製できる。カラムクロマトグラフィーで使用されるカラムは例えば、PD−10カラムなどの脱塩カラムであり得る。再酸化剤は例えば、5,5’−ジチオ−ビス−2−ニトロ安息香酸、4,4’−ジチオジピリジン、2,2’−ジチオジピリジン、テトラチオン酸ナトリウム、またはヨードソ安息香酸であり得る。
【0025】
本発明は、添付図面と併せて、以下の特定の実施形態の詳細な説明を参照することによっても最もよく理解されるで。以下の議論は説明的、例証的、例示的であり、添付の特許請求の範囲によって規定される範囲を限定するものとして解釈すべきではない。
【発明を実施するための最良の形態】
【0026】
(詳細な説明)
開示を明瞭にするために、そして制限するためでなく、本発明の詳細な説明は、以下に続く小節に分割する。
【0027】
(I.定義)
そうでないと定義しない限り、本明細書で使用するすべての技術用語および化学用語は、記載する方法および組成物に関係する当業者によって一般に理解されるのと同じ意味を有する。本明細書で使用される場合、以下の用語および表現は、そうでないと規定しない限り、それらに与えられた意味を有する。
【0028】
「薬物」という用語は本明細書で使用される場合、例えば薬学的化合物、治療化合物、または薬理化合物を含む、元素、化合物、薬剤、または分子実体を意味する。薬物は、天然または合成あるいはその組合せであり得る。「治療薬」は、治療(例えば有益な)効果を癌細胞または免疫細胞(例えば活性化免疫細胞)に単独で、または他の因子との組合せ(例えばプロドラッグと組合されたプロドラッグ変換酵素)のどちらかで作用させる薬剤である。通例、本明細書で述べるおよび方法および組成物によって有用な治療薬は、細胞毒性、細胞増殖抑制、または免疫抑制効果を作用させる薬物である。ある実施形態において、薬物は放射性元素ではない。
【0029】
「細胞毒性薬」は薬剤の細胞に対する効果に関して、細胞を殺すことを意味する。
【0030】
「細胞増殖抑制薬」は、細胞増殖の阻害を意味する。
【0031】
「ポリペプチド」という用語は、アミノ酸のポリマーおよびその同等物を指し、特定の長さの生成物を指すものではない;それゆえ「ペプチド」および「タンパク質」は、ポリペプチドの定義の中に含まれる。ポリペプチドの定義の中には、本明細書で定義された「抗体」も含まれる。「ポリペプチド領域」はポリペプチドのセグメントを指し、そのセグメントは例えば、1個以上のドメインまたはモチーフを含有できる(例えば抗体のポリペプチド領域は、例えば1個以上のCDRを含有できる)。「フラグメント」という用語は、通例、ポリペプチドの少なくとも20個の連続または少なくとも50個の連続アミノ酸を有するポリペプチドの部分を指す。「誘導体」は、第2のポリペプチドに対して保存的アミノ酸置換を有するポリペプチドまたはそのフラグメント;あるいは第2の分子の共有付加によって、例えば非相同ポリペプチドの付加によって、またはグリコシル化、アセチル化、ホスホリル化などによって修飾されたポリペプチドまたはそのフラグメントを含む。さらに、例えばアミノ酸(例えば非天然アミノ酸など)の1個以上のアナログを含有するポリペプチドアナログ、非置換連鎖を有するポリペプチド、ならびに天然および非天然の両方の、当該分野で公知の他の修飾を含むポリペプチドも含まれる。ポリペプチドアナログは例えば、タンパク質模倣物およびボンベシンを含む。
【0032】
「抗体」という用語は本明細書で使用される場合(a)免疫グロブリンポリペプチドおよび免疫グロブリンポリペプチドの免疫活性部分、すなわち特異的抗原へ免疫特異的に結合する抗原結合部位を含有する、免疫グロブリンファミリーのポリペプチド、またはそのフラグメント、あるいは(b)抗原へ免疫特異的に結合する、そのような免疫グロブリンポリペプチドまたはフラグメントの保存的に置換された誘導体を指す。抗体は一般に、例えばHarlow & Lane,Antibodies:A Laboratory Manual(Cold Spring Harbor Laboratory Press,1988)に記載されている。
【0033】
「抗体誘導体」という用語は本明細書で使用される場合、非相同分子の共有付加によって、例えば非相同ポリペプチドの付加によって、あるいは通常は抗体と関連付けられないグリコシル化、アセチル化またはホスホリル化などによって修飾された、上で定義したような抗体を意味する。
【0034】
「モノクローナル抗体」という用語は、それが産生される方法ではなく、いずれかの真核または原核細胞クローン、あるいはファージクローンを含む単細胞クローンに由来する抗体を指す。それゆえ「モノクローナル抗体」という用語は本明細書で使用される場合、ハイブリドーマ技術によって産生される抗体に限定されない。
【0035】
「鎖間ジスルフィド結合」という用語は抗体の文脈において、2個の重鎖間の、または重鎖と軽鎖との間のジスルフィド結合を指す。
【0036】
「鎖間チオール」という用語は、鎖間ジスルフィド結合の形成に関与し得る抗体重鎖または軽鎖のチオール基を指す。
【0037】
タンパク質は、特定の種類のおよび/または同様の反応性の結合箇所すべてが薬物に結合され、タンパク質−薬物結合体の均質な個体群を生じる場合に、「完全付加」と呼ばれる。タンパク質は、特定の種類および/または同様の反応性の考えられる結合箇所の一部のみが薬物に結合され、タンパク質−薬物結合体のある異性体または複数の異性体の生成をもたらす場合に、「部分付加」と呼ばれる。
【0038】
「単離された」という用語は、分子または高分子(例えば抗体)の文脈において、その自然環境の成分から同定ならびに分離および/または回収されたものである。その自然環境の汚染成分は、分子の所望の用途(例えば診断または治療)を妨害する物質であり、酵素、ホルモン、および他のタンパク性または非タンパク性溶質を含み得る。一部の実施形態において、単離された分子または高分子は、(1)例えばLowryまたはBradford法によって決定されたような分子または高分子の重量の95%超まで、または99%超まで、(2)スピニングカップ・シークエネータの使用により、N末端または内部アミノ酸配列の少なくとも15残基を得るのに十分な程度まで、または(3)クマシーブルーまたは好ましくは、銀染色を使用する還元または非還元条件下で、SDS−PAGEによって均質となるまで精製される。単離された分子および高分子が組換え細胞内に分子および高分子をインサイチュで含むのは、抗体の自然環境の少なくとも1つの成分が存在しないからであろう。しかし通常は、単離抗体は少なくとも1つの精製ステップによって調製される。
【0039】
略語「AFP」は、すぐ下に示す一般式を有するジメチルバリン−バリン−ドライソロイシン−ドラプロイン−フェニルアラニン−p−フェニレンジアミンを指す:
【0040】
【化1】
。
【0041】
略語「MMAE」は、すぐ下に示す一般式を有するモノメチルオーリスタチンEを指す:
【0042】
【化2】
。
【0043】
略語「MMAF」は、すぐ下に示す一般式を有するドバリン−バリン−ドライソロイニン−ドラプロイン−フェニルアラニンを指す:
【0044】
【化3】
。
【0045】
略語「AEB」は、オーリスタチンEにパラアセチル安息香酸を反応させることによって生成されるエステルを指す。略語「AEVB」は、オーリスタチンEにベンゾイル吉草酸を反応させることによって生成されるエステルを指す。
【0046】
「薬学的に受容可能な塩」は本明細書で使用されるように、分子または高分子の薬学的に受容可能な有機または無機塩を指す。酸付加塩は、アミノ基によって形成され得る。塩の例としては、これに限定されるわけではないが、サルフェート、シトレート、アセテート、オキサラート、クロライド、ブロミド、ヨージド、ニトレート、ビスルフェート、ホスフェート、酸ホスフェート、イソニコチナート、ラクテート、サリチラート、酸シトレート、タートレート、オレアート、タンナート、パントテナート、バイタートレート、アスコルベート、スクシナート、マレアート、ゲンチシナート、フマレート、グルコナート、グルクロナート、サッカラート、ホルメート、ベンゾアート、グルタメート、メタンスルホナート、エタンスルホナート、ベンゼンスルホナート、p−トルエンスルホナート、およびパモアート(すなわち1,1’メチレンビス−(2−ヒドロキシ3−ナフトアート))塩が挙げられる。薬学的に受容可能な塩は、アセテートイオン、スクシナートイオンまたは他の対イオンなどの、別のイオンの包含も含む。対イオンは、親化合物上に電荷を安定させるいずれかの有機または無機部分でもよい。さらに薬学的に受容可能な塩は、その構造内に1個を超える荷電原子を有してもよい。複数の荷電原子が薬学的に受容可能な塩の一部である例は、複数の対イオンを有することができる。それゆえ薬学的に受容可能な塩は、1個以上の荷電原子および/または1個以上の対イオンを有することができる。
【0047】
「薬学的に受容可能な溶媒和物」または「溶媒和物」は、1個以上の溶媒分子および分子または高分子の会合を指す。薬学的に受容可能な溶媒和物を形成する溶媒の例としては、これに限定されるわけではないが、水、イソプロパノール、エタノール、メタノール、DMSO、エチルアセテート、酢酸、およびエタノールアミンが挙げられる。
【0048】
(II.ポリペプチド、タンパク質、抗体)
本発明は、タンパク質−薬物結合体およびタンパク質−薬物結合体を作成する方法を提供する。薬物を受容するための結合箇所を有するタンパク質も提供される。タンパク質−薬物結合体は治療に、診断に(例えばインビトロまたはインビボで)、インビボ撮像に、そして他の用途に使用され得る。
【0049】
抗体、酵素、グリコシル化タンパク質、レクチン、種々の生物学的レセプター、タンパク質ホルモン、および結合パートナーに対する結合剤として作用できる他のタンパク質を含む、タンパク質の種々のクラスが結合体化され得る。タンパク質は、少なくとも1個の反応部位、例えばジスルフィド結合、アミノ基、ヒドロキシル基またはカルボキシル基を含み、そこで薬物のタンパク質への結合体化が起こり得る。
【0050】
反応部位は、化学反応または手段によって接近可能であり、活性化可能である。一部の実施形態において、結合体化目的で化学的に活性化されるタンパク質は、タンパク質の所期の用途に必須ではないジスルフィド結合を含有するタンパク質、および/またはタンパク質を妨害しないタンパク質(例えばこれに限定されるわけではないが、タンパク質の分解を引き起こす、または結合や他の機能(例えばエフェクタ機能)を妨害する)である。そのようなタンパク質では、ジスルフィド結合は、2個のシステイン残基のチオール(−−SH)側基の酸化の結果として存在し得る。これらの残基は、別のポリペプチド鎖上に、または同じポリペプチド鎖上に存在する。酸化の結果として、ジスルフィド結合(−−S−−S−−)は、元のシステイン残基のベータ炭素間に形成される。還元後、残基は互換的にハーフシステインおよびシステインと呼ばれることが多い。ジスルフィド結合の還元剤による処理は、ジスルフィド結合の還元的開裂を引き起こして、遊離チオール基を残す。ジスルフィド結合を含有するタンパク質の例としては、抗体、多くの酵素、特定のホルモン、および特定のレセプターが挙げられる。
【0051】
一部の実施形態において、ジスルフィド結合は自然発生的であり得る。一部の実施形態において、スルフヒドリル基もタンパク質(例えば抗体)内に化学的に導入され得る。スルフヒドリル基を導入するための適切な方法は、化学的手段(例えば2−ITなどのチオール化剤を使用して)、または組換えDNA技術を使用することを含む。例えばシステイン残基は、タンパク質をコードする核酸の突然変異誘発によって、タンパク質中に導入できる。一般に、参考として本明細書に援用される、Sambrook et al.,Molecular Cloning,A Laboratory Manual,3rd ed.,Cold Spring Harbor Publish.,Cold Spring Harbor, New York(2001);Ausubel et al.,Current Protocols in Molecular Biology,4th ed.,John Wiley and Sons,New York(1999)を参照のこと。スルフヒドリル基は、タンパク質中に、例えばポリペプチド内に、またはカルボキシ末端に導入され得る。
【0052】
一部の実施形態において、タンパク質は抗体である。そのような抗体は、インビトロまたはインビボ診断、インビボ撮像、あるいは特有の抗原に関する疾患または状態の治療に使用できる。抗体構造の基本単位は、非共有結合と、ジスルフィド結合との両方によって共に結合された、4個のポリペプチド−−2個の同じ低分子量(「軽」)鎖および2個の同じ高分子量(「重」)鎖の複合体である。異なる抗体が、これらの基本単位の1〜5個のいずれかに位置するであろう。抗体は、図式的に「Y」として表される。「Y」の各分岐は、重鎖のアミノ末端部と結合した軽鎖によって形成される。「Y」の基部は、2つの重鎖のカルボキシ末端部によって形成される。「Y」の節はヒンジ領域と呼ばれる。
【0053】
5つのヒト抗体クラス(IgG、IgA、IgM、IgDおよびIgE)、およびこれらのクラス内の種々のサブクラス(IgG1、IgG2、IgG3、IgG4、IgA1およびIgA2)または免疫グロブリン分子のサブクラスは、構造の差異、例えば1個の抗体分子中の免疫グロブリン単位の数、個々の単位のジスルフィドブリッジ構造、ならびに鎖長および配列の差異に基づいて認識される。抗体のクラスおよびサブクラスは、そのアイソタイプである。
【0054】
抗体は、インタクトな抗体または抗原結合抗体フラグメント、例えばFab、F(ab’)、F(ab’)2、Fd鎖、単鎖Fv(scFv)、単鎖抗体、ジスルフィド結合Fv(sdFv)、VLまたはVHドメインのどちらかを含むフラグメント、あるいはFab発現ライブラリによって産生されたフラグメントであり得る。単鎖抗体を含む抗原結合抗体フラグメントは、可変領域を単独で、あるいは以下の実体または部分と組合せて含むことができる:ヒンジ領域、CH1、CH2、CH3、CH4およびCLドメイン。また抗原結合フラグメントは、可変領域とヒンジ領域、CH1、CH2、CH3、CH4およびCLドメインとのいずれの組合せも含むことができる。一部の実施形態において、抗体フラグメントは、鎖間ジスルフィド結合を含む少なくとも1つのドメイン、またはドメインの部分を含む。
【0055】
通例、抗体はヒト、げっ歯類(例えばマウスおよびラット)、ロバ、ヒツジ、ウサギ、ヤギ、モルモット、ラクダ科の哺乳類、ウマ、またはニワトリの抗体である。本明細書で使用される場合「ヒト」抗体は、ヒト免疫グロブリンのアミノ酸配列を有する抗体を含み、以下に、そして例えば米国特許第5,939,598号および同第6,111,166号に述べるように、ヒト免疫グロブリンライブラリから、ヒトB細胞から、または1個以上のヒト免疫グロブリンについて遺伝子組換えされた動物から単離された抗体を含む。抗体は単一特異性、二重特異性、三重特異性、またはより多くの多重特異性であり得る。
【0056】
一部の実施形態において、定常ドメインはエフェクター機能を有する。「抗体エフェクター機能」またはAECという用語は本明細書で使用される場合、IgのFcドメインが寄与する機能を指す。そのような機能は例えばFcエフェクタードメインの、食細胞または溶解活性を備えた免疫細胞上のFcレセプターへの結合によって、またはFcエフェクタードメインの補体系の成分への結合によって影響され得る。通例、Fc結合細胞または補体成分によって仲介される効果は、CD70標的細胞の阻害および/または消耗を引き起こす。エフェクター機能は例えば、「抗体依存性細胞毒性」またはADCC、「抗体依存性細胞食作用」またはADCP、「補体依存性細胞毒性」またはCDCであり得る。他の実施形態において、定常ドメインは1つ以上のエフェクター機能を欠いている。
【0057】
抗体は、目的の抗原、例えば医療的および/または治療的目的の抗原に対して産生され得る。例えば抗原は、病原体(これに限定されるわけではないが、ウィルス、細菌、真菌、および原虫など)、寄生虫、腫瘍細胞、または特定の医学的状態と関連付けられる抗原であり得る。腫瘍関連抗原(TAA)の場合、癌は、免疫系、肺、結腸、直腸、乳房、卵巣、前立腺、頭部、頸部、骨、または他のいずれかの解剖学的位置の癌であり得る。目的の抗原はこれに限定されるわけではないが、CD30、CD40、Lewis Y、およびCD70を含む。一部の実施形態において、抗原はCD2、D20、CD22、CD33、CD38、CD40、CD52、HER2、EGFR、VEGF、CEA、HLA−DR、HLA−Dr10、CA125、CA15−3、CA19−9、L6、Lewis X、アルファフェトタンパク質、CA242、胎盤アルカリフォスファターゼ、前立腺特異的抗原、前立腺酸ホスファターゼ、上皮増殖因子、MAGE−1、MAGE−2、MAGE−3、MAGE−4、抗トランスフェリンレセプター、p97、MUC1−KLH、gp100、MART1、IL−2レセプター、ヒト絨毛性ゴナドトロピン、ムチン、P21、MPG、およびNeu癌遺伝子産物である。
【0058】
一部の有用な特異的抗体は、これに限定されるわけではないが、BR96 mAb(Trail et al.(1993),Science 261:212−215)、BR64(Trail et al.(1997),Cancer Research 57:100−105)、CD40抗原に対するmAb、例えばS2C6 mAb(Francisco et al.(2000)Cancer Res.60:3225−3231)、およびCD30抗原に対するmAb、例えばAC10(Bowen et al.(1993)J.Immunol.151:5896−5906)を含む。腫瘍特異的抗原に結合する他の多くの内部移行抗体が使用され得、概説されてきた(例えばFranke et al.(2000),Cancer Biother Radiopharm.15:459−76;Murray(2000),Semin Oncol.27:64−70;Breitling et al.,Recombinant Antibodies,John Wiley,and Sons,New York,1998を参照)。これらの参考文献の開示は、参照により本明細書に援用されている。
【0059】
「腫瘍特異的抗原」という用語は本明細書で使用される場合、特定の腫瘍に特有の、またはそのような腫瘍に強い相関関係のある抗原を示すことが理解されるであろう。しかしながら、腫瘍特異的抗原は腫瘍組織に必ずしも特有でなく、しかしながら、すなわち、それらに対するその抗体は、正常組織の抗原と交差反応することがある。腫瘍特異的抗原が腫瘍細胞にとって特有でない場合、実際問題として、腫瘍特異的抗原に結合する抗体が交差反応による不当なリスクまたは妨害なしに所望の手順を実施するために、腫瘍細胞にとって十分に特異的であることが高い頻度で起こる。多くの因子がこの実際的な特異性に寄与している。例えば腫瘍細胞上の抗原の量は、正常細胞上で見出される交差反応抗原の量を大きく超えるか、または腫瘍細胞上の抗原は、より効果的に提示されうる。したがって「腫瘍特異的抗原」という用語は本明細書では、実際的に有用な特異性に関し、絶対的な特異性を表したり、または抗原が腫瘍に特有であることを示したりするものではない。
【0060】
抗体は、ポリクローナル抗体またはモノクローナル抗体であり得る。被験体がヒト被験体である場合、抗体は抗原に対する有用な免疫反応を開始できるいずれかの動物を免疫化することによって得られ得る。動物はマウス、ラット、ヤギ、ヒツジ、ウサギまたは他の適切な実験動物であり得る。抗原は、天然の免疫原、またはハプテンおよび免疫原性キャリアの合成免疫原性結合体の形で提示できる。モノクローナル抗体の場合、免疫化動物の抗体産生細胞は、抗体を産生するハイブリドーマを得るために、「不死」または「不死化」ヒトまたは動物細胞と融合することができる。所望ならば、免疫グロブリン鎖の1個以上をコードする遺伝子は、抗体が種々の宿主細胞で産生できるようにクローニングすることができ、所望ならば、産生される抗体の配列、およびそれゆえ免疫的特性を変化させるために、遺伝子を突然変異させることができる。ヒトモノクローナル抗体は、当該分野で公知の多数の技術のいずれかによって産生できる(例えばTeng et al.(1983),Proc.Natl.Acad.Sci.USA.80,7308−7312;Kozbor et al.(1983)Immunology Today 4,72−79;およびOlsson et al.(1982),Meth.Enzymol.92,3−16)。
【0061】
抗体は、例えば当業者に周知の技術で産生されたネズミ抗体、キメラ抗体、ヒト化抗体、または完全ヒト抗体であり得る。標準組換えDNA技術を使用して産生できる、ヒトおよび非ヒト部分の両方を含む組換え抗体、例えばキメラ抗体およびヒト化モノクローナル抗体は、有用な抗体である。キメラ抗体は、異なる部分が異なる動物種に由来する分子、例えばマウスモノクローナルに由来する可変領域およびヒト免疫グロブリン定常領域を有する分子である(例えば参考としてその全体が本明細書に援用される、Cabilly et al.,米国特許第4,816,567号;およびBoss et al.,米国特許第4,816,397号を参照のこと)。ヒト化抗体は、非ヒト種からの1個以上の相補性決定領域(CDR)およびヒト免疫グロブリン分子からのフレームワーク領域を有する非ヒト種からの抗体分子である(例えば参考としてその全体が本明細書に援用される、Queen,米国特許第5,585,089号を参照のこと)。そのようなキメラ抗体およびヒト化モノクローナル抗体は、当該分野で公知の組換えDNA技術、例えば国際公開第87/02671号;欧州特許出願公開第184,187号;欧州特許出願公開第171496号;欧州特許出願公開第173494号;国際公開第86/01533号;欧州特許出願公開第12,023号;Berter et al.(1988),Science 240:1041−1043;Liu et al.(1987),Proc.Natl.Acad.Sci.USA 84:3439−3443;Liu et al.(1987),J.Immunol.139:3521−3526;Sun et al.(1987),Proc.Natl.Acad.Sci.USA 84:214−218;Nishimura et al.(1987),Cancer.Res.47:999−1005;Wood et al.(1985),Nature 314:446−449;およびShaw et al.(1988),J.Natl.Cancer Inst.80:1553−1559;Morrison(1985),Science 229:1202−1207;Oi et al.(1986),BioTechniques 4:214;U.S.Patent No.5,225,539;Jones et al.(1986),Nature 321:552−525;Verhoeyan et al.(1988),Science 239:1534;およびBeidler et al.(1988),J.Immunol.141:4053−4060に述べられている方法を使用して産生可能であり、そのそれぞれが参考として全体が本明細書に援用されている。
【0062】
完全ヒト抗体は例えば、内在性免疫グロブリン重鎖および軽鎖遺伝子を発現できないが、ヒト重鎖および軽鎖遺伝子を発現できるトランスジェニックマウスを使用して産生できる。トランスジェニックマウスは、選択した抗原またはその部分を用いて通常の方法で免疫化される。抗原に対して産生されたモノクローナル抗体は、従来のハイブリドーマ技術を使用して得ることができる。トランスジェニックマウスに含まれるヒト免疫グロブリントランス遺伝子は、B細胞分化の間に再配列し、続いてクラス転換および体細胞突然変異を受ける。それゆえそのような技術を使用して、治療的に有用なIgG、IgA、IgMおよびIgE抗体を産生することができる。ヒト抗体を産生するこの技術の概要については、LonbergおよびHuszar(1995,Int.Rev.Immunol.13:65−93)を参照。ヒト抗体およびヒトモノクローナル抗体を産生するためのこの技術ならびにそのような抗体を産生するためのプロトコルの詳細な議論については、例えば米国特許第5,625,126号;同第5,633,425号;同第5,569,825号;同第5,661,016号;同第5,545,806号を参照のこと(そのそれぞれは参考としてその全体が本明細書に援用される)。他のヒト抗体は、例えばAbgenix,Inc.(フリーモント、カリフォルニア州)およびGenpharm(サンノゼ、カリフォルニア州)から市販され得る。
【0063】
選択したエピトープを認識する完全ヒト抗体も、「誘導選択」と呼ばれる技術を使用して産生できる。本手法において、同じエピトープを認識する完全ヒト抗体の選択を誘導するために、選択した非ヒトモノクローナル抗体、例えばマウス抗体が使用される。例えばJespers et al.(1994),Biotechnology 12:899−903を参照のこと。ヒト抗体は、ファージ提示ライブラリ(HoogenboomおよびWinter(1991),J.Mol.Biol.227:381;Marks et al.(1991),J.Mol.Biol.222:581;QuanおよびCarter(2002),”The rise of monoclonal antibodies as therapeutics.”Anti−IgE and Allergic Disease,Jardieu,P.M.およびFick Jr.,R.B,eds.,Marcel Dekker,New York,NY,Chapter 20,pp.427−469を含む、当該分野で公知の種々の技術を使用しても産生できる。
【0064】
抗体は二重特異性抗体でもよい。二重特異性抗体を産生する方法は、当該分野で公知である。全長二重特異性抗体の伝統的な産生は、2つの鎖が異なる特異性を有する、2つの免疫グロブリン重鎖軽鎖対の同時発現に基づいている(Milstein et al.,1983,Nature 305:537−539)。免疫グロブリン重鎖および軽鎖のランダムな組合せのため、これらのハイブリドーマ(クアドローマ)は、1個のみが正しい二重特異性構造を有する、10個の異なる抗体分子の潜在的な混合物を生成する。同様の手順が国際公開第93/08829号、およびTraunecker et al.(1991),EMBO J.10:3655−3659に開示されている。
【0065】
異なる手法に従って、所望の結合特異性(抗体−抗原結合部位)を備えた抗体可変ドメインは、免疫グロブリン定常ドメイン配列に融合される。融合は好ましくは、ヒンジCH2、およびCH3領域の少なくとも一部を含む免疫グロブリン重鎖定常ドメインによってである。融合物の少なくとも1個に存在する、軽鎖結合に必要な部位を含有する第1重鎖定常領域(CH1)を有することが好ましい。免疫グロブリン重鎖融合をコードする配列を含む核酸、そして所望ならば、免疫グロブリン軽鎖は、独立した発現ベクター内に挿入され、適切な宿主生物内に同時形質移入される。これは、構築で使用された3個のポリペプチド鎖の不等比が最適な収率を与えるときに、実施形態において3個のポリペプチドフラグメントの相互比を調整するために高い柔軟性を提供する。しかしながら、等しい比での少なくとも2個のポリペプチド鎖の発現が高い収率を生じるときに、または比が特に重要でないときに、2個または3個すべてのポリペプチド鎖のコード配列を1個の発現ベクターに挿入することが可能である。
【0066】
この手法の実施形態において、二重特異性抗体は、一方のアームに第1の結合特異性を備えたハイブリッド免疫グロブリン重鎖と、他方のアームにハイブリッド免疫グロブリン重鎖−軽鎖対(第2の結合特異性を提供する)を有する。二重特異性分子の半分のみにおける免疫グロブリン軽鎖の存在が、容易な分離方法を提供するため(国際公開第94/04690号(参照としてその全体が本明細書に援用される))、この非対称構造は、所望の二重特異性化合物の、望ましくない免疫グロブリン鎖の組合せからの分離を促進する。
【0067】
二重特異性抗体を産生するためのさらなる詳細については、例えばSuresh et al.(1986),Methods in Enzymology 121:210;Rodrigues et al.(1993),J.Immunology 151:6954−6961;Carter et al.(1992),Bio/Technology 10:163−167;Carter et al.(1995),J.of Hematotherapy 4:463−470;Merchant et al.(1998),Nature Biotechnology 16:677−681を参照のこと。そのような技術を使用して、二重特異性抗体は、疾患の処置または防止への使用のために調製できる。
【0068】
二重特異性抗体は、欧州特許第0 105 360号にも開示されている。この参考文献に開示されているように、ハイブリッドまたは二重特異性抗体は、生物的に、すなわち細胞融合技術によって、または化学的に、特に架橋剤またはジスルフィドブリッジ形成試薬を用いてのどちらかで誘導可能であり、抗体全体またはそのフラグメントを含みうる。そのようなハイブリッド抗体を得るための方法は例えば、国際公開第83/03679号、および欧州特許第0 217 577号に開示されており、その両方とも参考としてその全体が本明細書中に援用される。
【0069】
他の実施形態において、抗体は、抗体の融合タンパク質、またはその機能活性フラグメントであり、例えばその中で抗体は共有結合(例えばペプチド結合)を介して、N末端またはC末端のどちらかにて抗体でない別のタンパク質(またはその部分、好ましくはタンパク質の少なくとも10、20または50アミノ酸部分)のアミノ酸配列に融合される。好ましくは、抗体またはそのフラグメントは、他のタンパク質に定常ドメインのN末端にて共有結合される。
【0070】
なお他の実施形態において、タンパク質は、必要に応じてヒンジ領域を含む抗体重鎖および/または軽鎖定常領域ドメインへ共有結合を介して融合された非抗体分子の結合部分の融合タンパク質であり得る。そのような融合タンパク質は必要に応じて、少なくとも1つの、通例少なくとも2個の鎖間ジスルフィド結合を含むことができる。例えば融合タンパク質は、CH1およびCL領域、ならびにヒンジ領域を含むことができる。
【0071】
(III.活性化方法)
一般に、薬物は活性化部位においてタンパク質または他の適切な分子へ結合され得る。適切な活性化部位としては、チオール基、アミノ基(例えばリジン残基のイプシロンアミノ基またはタンパク質のN末端にて)、隣接ヒドロキシル基(1,2−ジオール)(例えば酸化炭水化物)およびカルボキシル基(例えばタンパク質のC末端、アスパラギン酸およびグルタミン酸残基、および炭水化物、例えばシアル酸)などの結合箇所が挙げられる。
【0072】
薬物は結合箇所に直接結合され得る。例えば薬物は、抗体リジンのε−アミノ基のアルキル化、酸化炭水化物の還元的アミノ化またはヒドラジドとの反応、ヒドロキシル基およびカルボキシル基間のトランスエステル化、アミノ基またはカルボキシル基におけるアミド化、チオール(例えば鎖間チオール)または導入されたチオールへの結合体化、例えばリジンの2−イミノチオラン(Traut試薬)によるアルキル化によって結合体化することができる。薬物を結合箇所に結合体化する適切な方法は、例えばCurrent Protocols in Protein Science (John Wiley & Sons, Inc.),Chapter 15(Chemical Modifications of Proteins)(その開示は、参考としてその全体が本明細書に援用される)に開示されている。
【0073】
薬物は、別の分子、例えばリンカーを介して間接的に結合させることもできる。例えば薬物は、例えばこれに限定されるわけではないが、抗体のヒンジ領域内においてスルフヒドリル基に結合されたマレイミド基を介して結合体化させることもできる。抗体結合体は、薬物のマレイミド誘導形態に抗体を反応させることによって生成できる。さらに詳細には、抗体結合体は、還元された抗体を産生するために抗体を還元して、アミン薬物を生成し、マレイミド誘導体化薬物を生成するためにアミン薬物をマレイミドによって誘導体化して、マレイミド誘導体化薬物に抗体を反応させることによって生成できる。
【0074】
例示的な実施形態において、IgG1、例えばcAC10は多数のジスルフィド結合を所有し、そのわずか4個が鎖間である。4個の鎖間ジスルフィド結合が高度に柔軟性のヒンジ領域に密集し、他の(鎖内)ジスルフィド結合よりはるかに溶媒に接近しやすいため、過剰の例えば還元剤、例えばこれに限定されるわけではないがジチオトレイトール(DTT)、トリス(2−カルボキシエチル)ホスフィン(TCEP)、または2−メルカプトエタノールによる還元が4個すべての結合を切断して、8個のシステインを生成する(すなわち遊離チオール基を含有する)。8個すべてのシステインと薬物−リンカーとの結合は、図7に示すように、抗体に付き約8個の薬物を持つ完全付加結合体を生成する。
【0075】
本発明は驚くべきことに、ADCの生物特性を抗体当たり平均2、2.5、4または6個の薬物を持つ抗体によって改善可能であり、このことが完全付加結合体、すなわち抗体当たり8個の薬物を持つ結合体の有効性を維持しながら、より低い毒性を生じることを実証する。部分薬物付加結合体の治療濃度域(最低有効用量によって分けられた、毒性が最初に見られる薬物−抗体結合体の濃度)は、8個の薬剤を持つ抗体よりも大きい。8個のシステインに4個の薬物を結合体化させて、多数の潜在的な薬物付加種を生じさせる方法が多数ある(合計9種類、抗体当たり0〜8個の薬物、図1〜7を参照のこと)。4個の薬物を持つ抗体では、4個の薬物を配置するための考えられる方法が6つあり、6個の異性体が生じる(図1および7を参照のこと)。抗体当たり4個の薬物が所望されるときには、8個の薬物が付加された種の均質性は失われる。抗体当たり2または6個の薬物では、薬物を分子に配置するために考えられる方法が3つある。
【0076】
部分的に付加されたADCを産生する方法(例えば抗体当たり8個の薬物ではなく4個で(E4))としては、以下が挙げられる:方法1(「部分還元」)これに限定されるわけではないがDTTまたはTCEPなどの還元剤による抗体の部分還元と、それに続く結合体化、および方法2(「完全還元および再酸化」)これに限定されるわけではないがDTTまたはTCEPなどの還元剤におる抗体の完全還元と、それに続く、再酸化剤(例えばこれに限定されるわけではないが5,5’−ジチオ−ビス−2−ニトロ安息香酸(DTNB)、4,4’−ジチオジピリジン、2,2’−ジチオジピリジン、テトラチオン酸ナトリウム、またはヨードソ安息香酸)による抗体の部分再酸化、そして最終的な結合体化。完全還元および再酸化方法では、2つの局面がある:2a、DTNB再酸化後の精製、および2b、DTNB再酸化後の精製なし(ワンポット再酸化および薬物結合)。これらの方法は、異なる種の異なる割合(例えばE4では、25〜40%)を生じ、考えられる種の異なる異性体混合物も生じる。開示には、当業者に公知である上の方法のハイブリッドおよびバリエーションが含まれる。
【0077】
抗体薬物結合体の一例として、図1は、結合反応中に産生できる6つの考えられるE4種(4A〜4F種と呼ばれる)を示す。種4A〜Dは、特定の分析方法によって個別に区別できない;しかしながらそれらは、4Eおよび4Fから区別できる。
【0078】
部分還元の方法1の1つの実施形態において、例えば抗体当たり4個の薬物を持つ結合体は、例えばこれに限定されるわけではないがDTTによる抗体の完全還元によって産生可能であり、8個の抗体システインを生じて、4個の薬物同等物への結合体化が続く。これは、抗体が0〜8個の薬物を有する混合物を生じさせる。あるいは4個のジスルフィドのうち平均2個のみが還元されるように(4個のシステインを遊離する)抗体が例えば制限量のDTTによって還元され、完全薬物結合体化が続く場合、偶数の薬物が付加された種(抗体当たり0、2、4、6、および8個の薬物)のみが形成される。このことにより、混合物の複雑さが低減され、これは異なる薬物付加種を単離するための精製によってさらに低減することができる。
【0079】
一部の実施形態において、タンパク質上のある潜在的な結合箇所は選択的に活性化され得る。この選択的活性化は、タンパク質への薬物の結合部位の即時の割当てを可能にする。例えば制限量の強力な還元剤DTTまたはTCEPによる抗体(例えばcAC10)の処理は、重鎖−軽鎖ジスルフィドの選択的還元を生じさせる。別の例では、抗体の完全DTT還元と、それに続くDTNBなどの強力なチオール酸化剤を使用する部分再酸化は、重鎖−軽鎖ヒンジジスルフィドの選択的再酸化を引き起こして、主に、ヒンジ領域の重鎖に結合した薬剤を生じさせる。これらの方法の両方によって産生されたE2およびE6の異性体個体群は、90%の異性体均質性に到達し得る。
【0080】
薬物の抗体への結合体化後、結合体化された薬物−抗体種を分離され得る。一部の実施形態において、結合した抗体種は、抗体、薬物および/または結合体の特徴に基づいて分離できる。例えば疎水性相互作用クロマトグラフ(HIC)は、抗体当たり0、2、4、6、および8個の薬物に相当する種を単離および分離することに成功している。方法1によるこれらの薬物付加異性体それぞれの収率は、統計的分布によって予想される収率に近い。4個の薬物が付加された種は通例、全物質の30%である。
【0081】
薬物付加および抗体上の薬物の位置を判定するための分析方法が開発されてきた(以下も参照のこと)。部分DTT還元によって調製した純粋な4個の薬物を付加した結合体の、バイオアナライザ(キャピラリー電気泳動)および架橋ジビニルベンゼンカラム(PLRP)でのHPLCによるキャラクタリゼーションは、薬物は主に、抗体の重鎖と軽鎖との間に最初にジスルフィドを生成したシステイン上に位置することを明らかにした。従来とは異なり、1つの異性体が他の5つの異性体よりも有利である薬物位置の特異性は、予想外である。
【0082】
別の実施形態の、方法2、例えば4個の薬物を持つ薬物−抗体結合体を調製するための部分再酸化による完全還元において、抗体は、例えばこれに限定されるわけではないが、DTTによって完全に還元され、次に4個の抗体システインチオールが残るようにジスルフィドの一部を再形成するために、制限量の、例えばこれに限定されるわけではないがDTNBによって処理された。これらのシステインは薬物に結合体化され、本明細書で述べる方法によって分析された。混合物中の4つの薬物が付加された抗体の収率は40%にも上昇し、いったん精製すると、薬物の位置は、ヒンジ領域において重鎖間で最初にジスルフィドを生成したシステイン上への配置が好都合であった。一般的な慣例と比較した、4個の薬物を付加した抗体の収率および選択性はどちらも、ある部分還元方法による異なる異性体に有利であり、予想外であった。種々の化学的方法を使用すると、抗体内の薬物位置は、異なる異性体の産生のために容易に割当てることができる。
【0083】
還元が制限量の還元剤の添加によって制御され、抗体当たり平均して4個未満の鎖間ジスルフィド結合が破壊される部分還元が起こる。4個の鎖間ジスルフィド結合すべてが高度に露出されているため、還元は種々の経路によって進行し、0、2、4、6、または8個のシステインを持つ種の混合物からなる部分還元抗体を生成する。したがって部分還元抗体の結合は、図1および7に示すように、抗体当たり0、2、4、6、または8個の薬物を持つ結合体の混合物を生成できる。部分還元の程度に応じて、薬物付加分布(すなわち0、2、4、6、または8薬物付加種のパーセント)が変化する。
【0084】
部分還元は、抗体当たり可変数の薬物を持つ種を含有する混合物を生成するだけでなく、複数の場所の薬物付着の結果として、さらなる不均一性も生じさせる。図7は、2、4、および6薬物付加種が考えられる異性体が2個以上あることを示している。
【0085】
薬物のタンパク質への結合後、結合された薬物−タンパク質種は分離され得る。例えば一部の実施形態において、結合した抗体種は、抗体、薬物および/または結合体の特徴に基づいて分離できる。例えば疎水性相互作用クロマトグラフ(HIC)は、抗体当たり0、2、4、6、および8薬物に相当する種を単離および分離することに成功している。
【0086】
(IV.分析方法)
結合体の収率および異性体混合物を決定するために、種々の分析方法が使用され得る。例えば1つの実施形態において、HICは、生じた結合体(例えばE4結合体について)からの収率および異性体混合物を決定するために使用される分析方法である。この技術は、可変数の薬物を付加した抗体を分離することができる。薬物付加レベルは、例えば250nmおよび280nmにおける吸光度の比に基づいて決定することができる。例えば薬物が250nmで吸収し、一方抗体が280mmで吸収し得る場合。したがって250/280比は、薬物付加と共に上昇する。本明細書で述べた結合体化方法を使用して、一般に偶数の薬物を持つ抗体は、ジスルフィドの還元が偶数の遊離システインチオールを生じるため、抗体に結合されることが観察された。図2および3は、方法2aおよび2bによってそれぞれ生成されたcAC10−vcMMAEのHIC分離を示す。図4は、方法1からと同様に、これらのクロマトグラムからの種々の置換についてのパーセント組成を示す。方法1は約30%のE4を生じるが、方法2bは約40%のE4を生じる。
【0087】
HICは、置換レベルの混合物からE4を精製するために、ミリグラム〜グラムレベルにて分取的に使用することもできる。図3による純E4(表示された34〜38分の収集時間)を得て、2つの方法によって分析して、異性体E4混合物を決定した。最初に、非共有相互作用を変性させ、タンパク質質量に基づいて分離するAgilentバイオアナライザを使用して、溶出順に以下の抗体成分を得た:軽鎖(L)、重鎖(H)、重鎖−軽鎖(HL)、重鎖−重鎖(HH)、重鎖−重鎖−軽鎖(HHL)、および重鎖−重鎖−軽鎖−軽鎖(HHLL)。ジスルフィドが還元され、遊離チオールがvcMMAEに結合するときに、より小さい種が形成される。
【0088】
図1は、種々のE4異性体の変性から観察される抗体成分も記載する。図5からわかるように、方法2bによって調製された純E4は、HLによって支配され、少量のLおよびHHLを含む。この結果は、多少の種4A−D(HHLおよびLを生じる)を含む、主に種4F(もっぱらHLを生じる)の存在によって説明できる。興味深いことに、方法1によって生成されたcAC10−vcMMAEの同じ分析は、ほぼ等しい量のL、HL、およびHHを生じ、このことは主に種4Eの、多少の種4Fとの混合物と一致する。
【0089】
分析ツールの別の実施形態は、逆相PLRPカラムによるクロマトグラフィーである;カラム支持体は、非特異的にタンパク質を保持できるシリカ支持体で作成された典型的な逆相カラムよりはむしろ、架橋ジビニルベンゼンからなる。この変性および還元技術は、0および1個の薬物を含む軽鎖(L0およびL1)および0〜3個の薬物を含む重鎖(H0〜H3)からなる6種を明確に分離する。図1は、種々のE4種で観察できる薬物付加レベルを示す。方法2bによる純E4は、図6でPLRPによって分離された。未修飾軽鎖(L0)および2個の薬物を含む重鎖(H2)は、4Fから予想された種であるのに対して、L1およびH1は4A〜Dから予想される。バイオアナライザを共に用いて、これらのデータは4E対4Fが約2:1の混合物を生成する方法1と一致するが、方法2bは、4F対4A〜Dを2:1で生成する。それゆえ異なる化学的条件を使用しても、E4収率およびE4異性体の部分はどちらも、方法1と2bでは著しく異なっている。
【0090】
(V.タンパク質への結合が可能である化合物)
タンパク質は、細胞増殖抑制薬または細胞毒性薬、免疫抑制剤、毒素、キレート、化合物、分子、放射性核種などの任意の目的の薬物と結合体化できる。
【0091】
(細胞毒性薬および細胞増殖抑制薬)
細胞毒性薬および細胞増殖抑制薬としては、抗生剤(例えばアドリアマイシン)、抗腫瘍剤、例えばオーリスタチンおよびオーリスタチン誘導体、メトトレキサート、ミトマイシンC、ダウノルビシン、ドキソルビシン、およびビンブラスチン、5−フルオロウラシルDNA副溝バインダ、DNA複製阻害剤、アルキル化剤(例えばシスプラチン、モノ(プラチナ)、ビス(プラチナ)おおび3核プラチナ錯体およびカルボプラチンなどのプラチナ錯体)、駆虫剤(例えばペンタミジンイセチオネート)、アントラサイクリン、葉酸拮抗剤、代謝拮抗物質、化学療法増感剤、デュオカルマイシン、エトポシド、フッ化ピリミジン、イオノフォア、レキシトロプシン、ニトロソウレア、プラチノール、プレフォーミング化合物、プリン代謝拮抗物質、ピューロマイシン、放射線増感剤、ステロイド、タキサン、トポイソメラーゼ阻害剤、ビンカアルカロイド、抗菌剤、抗微小管剤などを含む。抗体がそのような薬物に結合されると、抗体は対応する抗原が発生する部位に薬物を方向付ける。抗体に結合できる他の薬剤および薬物は、当業者に公知であるか、または容易に想到され得る。
【0092】
個々の細胞毒性薬または細胞増殖抑制薬としては、例えばアンドロゲン、アントラマイシン(AMC)、アスパラギナーゼ、5−アザシチジン、アザチオプリン、ブレオマイシン、ブスルファン、ブチオニンスルホキシミン、カンプトセシン、カルボプラチン、カルムスチン(BSNU)、CC−1065、クロラムブシル、シスプラチン、コルヒチン、シクロホスファミド、シタラビン、シチジンアラビノシド、サイトカラシンB、ダカルバジン、ダクチノマイシン(以前はアクチノマイシン)、ダウノルビシン、ダカルバジン、ドセタキセル、ドキソルビシン、エストロゲン、5−フルオロデオキシウリジン、5−フルオロウラシル、グラミシジンD、ヒドロキシウレア、イダルビシン、イフォスファミド、イリノテカン、ロムスチン(CCNU)、メクロレタミン、メルファラン、6−メルカプトプリン、メトトレキサート、ミトラマイシン、ミトマイシンC、ミトキサントロン、ニトロイミダゾール、パクリタキセル、プリカマイシン、プロカルバジン、ストレプトゾトシン、テノポシド、6−チオグアニン、チオTEPA、トポテカン、ビンブラスチン、ビンクリスチン、ビノレルビン、VP−16およびVM−26が挙げられる。
【0093】
他の細胞毒性薬としては例えば、ドラスタチン(以下を参照)、エンジイン(例えばカリケアミシン)およびレキシトロプシン(米国特許第6,130,237号も参照のこと)などのDNA副溝バインダ、デュオカルマイシン、タキサン(例えばパクリタキセルおよびドセタキセル)、ピューロマイシン、CC−1065、SN−38、トポテカン、モルホリノ−ドキソルビシン、リゾキシン、シアノモルホリノ−ドキソルビシン、エキノマイシン、コンブレタスタチン、ネトロプシン、エポチロンA、BまたはD、エストラムスチン、クリプトフィシン、セマドチン、メイタンシノイド、ディスコデルモライド、エレウテロビン(eleutherobin)、およびミトキサントロンが挙げられる。
【0094】
ある実施形態において、細胞毒性薬は例えばドキソルビシン、メルファラン、ビンカアルカロイド、メトトレキサート、ミトマイシンCまたはエトポシドなどの化学療法薬である。加えて、CC−1065アナログ、カリケアミシン、メイタンシン、ドラスタチン10のアナログ、リゾキシン、およびパリトキシンなどの強力な薬剤は、タンパク質に結合することができる。
【0095】
具体的な実施形態において、細胞毒性薬または細胞増殖抑制薬は、オーリスタチンE(当該分野でドラスタチン−10としても公知)またはその誘導体である。通例、オーリスタチンE誘導体は例えばオーリスタチンEとケト酸との間で生成されたエステルである。例えばオーリスタチンEは、パラアセチル安息香酸またはベンゾイル吉草酸と反応させて、それぞれAEBおよびAEVBを生成させることができる。他の代表的なオーリスタチン誘導体としてはAFP、MMAF、およびMMAEが挙げられる。オーリスタチンEおよびその誘導体の合成および構造は、米国特許出願番号第09/845,786号(米国特許出願公開番号第20030083263号)および同第10/001,191号;国際特許出願PCT/US03/24209:国際特許出願PCT/US02/13435:および米国特許第6,323,315号;同第6,239,104号;同第6,034,065号;同第5,780,588号;同第5,665,860号;同第5,663,149号;同第5,635,483号;同第5,599,902号;同第5,554,725号;同第5,530,097号;同第5,521,284号;同第5,504,191号;同第5,410,024号;同第5,138,036号;同第5,076,973号;同第4,986,988号;同第4,978,744号;同第4,879,278号;同第4,816,444号;および同第4,486,414号に述べられている。
【0096】
ある実施形態において、細胞毒性薬または細胞増殖抑制薬は、抗チューブリン剤である。抗チューブリン剤の例としては、これに限定されるわけではないが、タキサン(例えばタキソール(登録商標)(パクリタキセル)、テキソテール(登録商標)(ドセタキセル))、T67(Tularik)、ビンカアルカロイド(例えばビンクリスチン、ビンブラスチン、ビンデシン、およびビノレルビン)、およびドラスタチン(例えばオーリスタチンE、AFP、MMAF、MMAE、AEB、AEVB)が挙げられる。他の抗チューブリン剤としては例えばバッカチン誘導体、タキサンアナログ(例えばエポチロンAおよびB)、ノコダゾール、コルヒチンおよびコルシミド(colcimid)、エストラムスチン、クリプトフィシン、セマドチン、メイタンシノイド、コンブレタスタチン、ディスコデルモライド、およびエレウテロビン(eleutherobin)が挙げられる。
【0097】
ある実施形態において、細胞毒性薬は抗チューブリン剤の別の群である、メイタンシノイドである。例えば具体的な実施形態において、メイタンシノイドはメイタンシンまたはDM−1である(ImmunoGen,Inc.;Chari et al.(1992),Cancer Res.52:127−131も参照のこと)。
【0098】
一部の実施形態において、治療剤は放射性同位体ではない。
【0099】
一部の実施形態において、細胞毒性薬または免疫抑制剤は、代謝拮抗物質である。代謝拮抗物質は、例えばプリンアンタゴニスト(例えばアゾチオプリンまたはミコフェノール酸モフェチル)、ジヒドロ葉酸レダクターゼ阻害剤(例えばメトトレキサート)、アシクロビル、ガンシクロビル、ジドブジン、ビダラビン、リバビリン、アジドチミジン、シチジンアラビノシド、アマンタジン、ジデオキシウリジン、ヨードデオキシウリジン、ポスカルネト(poscarnet)、またはトリフルリジンであり得る。
【0100】
他の実施形態において、細胞毒性薬または免疫抑制剤は、タクロリムス、シクロスポリンまたはラパマイシンである。さらなる実施形態において、細胞毒性薬は、アルデスロイキン、アレムツズマブ、アリトレチノイン、アロプリノール、アルトレタミン、アミフォスチン、アナストロゾール、三酸化ヒ素、ベキサロテン、ベキサロテン、カルステロン、カペシタビン、セレコキシブ、クラドリビン、ダルベポエチンアルファ、デニロイキンディフチトクス、デクスラゾキサン、プロピオン酸ドロモスタノロン、エピルビシン、エポエチンアルファ、エストラムスチン、エキセメスタン、フィルグラスチム、フロクスウリジン、フルダラビン、フルベストラント、ゲムシタビン、ゴセレリン、イダルビシン、イフォスファミド、メシル酸イマチニブ、インターフェロンアルファ−2a、イリノテカン、レトロゾール、ロイコボリン、レバミゾール、メクロレタミンまたは窒素マスタード、メゲストロール、メンサ、メトトレキサート、メトキサレン、ミトマイシンC、ミトタン、ナンドロロンフェンプロピオナート、オプレルベキン、オキサリプラチン、パミドロナート、ペグアデマーゼ、ペグアスパルガーゼ、ペグフィルグラスチム、ペントスタチン、ピポブロマン、プリカマイシン、ポリフィマーナトリウム、プロカルバジン、キナクリン、ラスブリカーゼ、サルグラモスチム、ストレプトゾシン、タモキシフェン、テモゾロマイド、テニポシド、テストラクトン、チオグアニン、トレミフェン、トレチノイン、ウラシルマスタード、バルルビシン、ビンブラスチン、ビンクリスチン、ビノレルビンおよびソレドロナートである。
【0101】
一部の実施形態において、薬剤は免疫抑制剤である。免疫抑制剤は例えばガンシクロビル、タクロリムス、シクロスポリン、ラパマイシン、シクロホスファミド、アザチオプリン、ミコフェノール酸モフェチルまたはメトトレキサートであり得る。あるいは免疫抑制剤は例えば、グルココルチコイド(例えばコルチゾールまたはアルドステロン)またはグルココルチコイドアナログ(例えばプレドニゾンまたはデキサメタゾン)であり得る。
【0102】
一部の実施形態において、免疫抑制剤は抗炎症剤、例えばアリールカルボン酸誘導体、ピラゾール含有誘導体、オキシカム誘導体およびニコチン酸誘導体である。抗炎症剤のクラスとしては例えばシクロオキシゲナーゼ阻害剤、5−リポオキシゲナーゼ阻害剤、およびロイコトリエンレセプターアンタゴニストが挙げられる。
【0103】
適切なシクロオキシゲナーゼ阻害剤としては、メクロフェナム酸、メフェナム酸、カルプロフェン、ジクロフェナク、ジフルニサル、フェンブフェン、フェノプロフェン、イブプロフェン、インドメタシン、ケトプロフェン、ナブメトン、ナプロキセン、スリンダク、テノキシカム、トルメチン、およびアセチルサリチル酸が挙げられる。
【0104】
適切なリポオキシゲナーゼ阻害剤としては、酸化還元阻害剤(例えばカテコールブタン誘導体、ノルジヒドログアヤレチック酸(NDGA)、マソプロコール、フェニドン、イアノパレン(Ianopalen)、インダゾリノン、ナファザトロム、ベンゾフラノール、アルキルヒドロキシアミン)、および非酸化還元阻害剤(例えばヒドロキシチアゾール、メトキシアルキルチアゾール、ベンゾピランおよびその誘導体、メトキシテトラヒドロピラン、ボスウェリア酸およびボスウェリア酸のアセチル化誘導体、ならびにシクロアルキルラジカルによって置換されたキノリンメトキシフェニル酢酸)、および酸化還元阻害剤の前駆物質が挙げられる。
【0105】
他の適切なリポオキシゲナーゼ阻害剤としては、抗酸化剤(例えばフェノール、没食子酸プロピル、フラボノイドおよび/またはフラボノイドを含む天然基質、フラボンのヒドロキシル化誘導体、フラボノール、ジヒドロケルセチン、ルテオリン、ガランギン、オロボル(orobol)、カルコンの誘導体、4,2’,4’−トリヒドロキシカルコン、オルト−アミノフェノール、N−ヒドロキシウレア、ベンゾフラノール、エブセレンおよび還元セレン含有酵素の活性を上昇させる種)、鉄キレート剤(例えばヒドロキサム酸およびその誘導体、N−ヒドロキシウレア、2−ベンジル−1−ナフトール、カテコール、ヒドロキシルアミン、カルノソールトロロックスC、カテコール、ナフトール、スルファサラジン、ジロイトン(zyleuton)、5−アントラニル酸および4−(オメガ−アリールアルキル)フェニルアルカン酸)、イミダゾール含有化合物(例えばケトコナゾールおよびイトラコナゾール)、フェノチアジン、およびベンゾピラン誘導体が挙げられる。
【0106】
なお他の適切なリポオキシゲナーゼ阻害剤としては、エイコサノイド(例えばオクタデカテトラエン酸、エイコサテトラエン酸、ドコサペンタエン酸、エイコサヘキサエン酸およびドコサヘキサエン酸ならびにそのエステル、PGE1(プロスタグランジンE1)、PGA2(プロスタグランジンA2)、ビプロストール、15−モノヒドロキシエイコサテトラエン酸、15−モノヒドロキシ−エイコサトリエン酸、および15−モノヒドロキシエイコサペンタエン酸、およびロイコトリエンB5、C5およびD5)の阻害剤、カルシウム流を妨害する化合物、フェノチアジン、ジフェニルブチルアミン、ベラパミル、フスコシド、クルクミン、クロロゲン酸、コーヒー酸、5,8,11,14−エイコサテトライエン酸(ETYA)、ヒドロキシフェニルレチンアミド、イオナパレン、エスクリン、ジエチルカルバマジン、フェナントリン、バイカレイン、プロキシクロミル、チオエーテル、ジアリルスルフィドおよびジ−(1−プロペニル)スルフィドが挙げられる。
【0107】
ロイコトリエンレセプターアンタゴニストとしては、カルシトリオール、オンタゾラスト、Bayer Bay−x−1005、Ciba−Geigy CGS−25019C、エブセレン、Leo Denmark ETH−615、Lilly LY−293111、Ono ONO−4057、Terumo TMK−688、Boehringer Ingleheim BI−RM−270、Lilly LY 213024, Lilly LY 264086、Lilly LY 292728、Ono ONO LB457、Pfizer 105696、Perdue Frederick PF 10042、Rhone−Poulenc Rorer RP 66153、SmithKline Beecham SB−201146、SmithKline Beecham SB−201993、SmithKline Beecham SB−209247、Searle SC−53228、Sumitamo SM 15178、American Home Products WAY 121006、Bayer Bay−o−8276、Warner−Lambert CI−987、Warner−Lambert CI−987BPC−15LY 223982,Lilly LY 233569、Lilly LY−255283, MacroNex MNX−160、Merck and Co.MK−591、Merck and Co.MK−886、Ono ONO−LB−448、Purdue Frederick PF−5901、Rhone−Poulenc Rorer RG 14893、Rhone−Poulenc Rorer RP 66364、Rhone−Poulenc Rorer RP 69698、Shionoogi S−2474、Searle SC−41930、Searle SC−50505、Searle SC−51146、Searle SC−52798、SmithKline Beecham SK&F−104493、Leo Denmark SR−2566、Tanabe T−757およびTeijin TEI−1338が挙げられる。
【0108】
(毒素)
毒素は、腫瘍、寄生虫または微生物細胞に関連する抗原に特異的な抗体へ有用に結合体化される。毒素は例えば植物(例えばリシンまたはアブリン)、動物(例えばヘビ毒)、または微生物(例えばジフテリアまたは破傷風毒素)に由来する。
【0109】
抗体以外に、薬物または毒素は、他のキャリアタンパク質、例えばアルブミンに結合体化させることができる。
【0110】
(タンパク質への薬物の結合体化)
薬物は、タンパク質の結合箇所と反応性である基を有するか、それを含むように修飾される。例えば薬物は、アルキル化(例えば抗体リジンのε−アミノ基またはタンパク質のN末端にて)、酸化炭水化物の還元的アミノ化、ヒドロキシ基とカルボキシル基との間のトランスエステル化、アミノ基またはカルボキシル基におけるアミド化、チオールへの結合によって付着させることができる。結合に使用できる化学作用の例については、例えばCurrent Protocols in Protein Science(John Wiley & Sons,Inc.),Chapter 15(Chemical Modifications of Proteins)(その開示は、参照によりその全体が本明細書に組み入れられている)を参照。
【0111】
例えばタンパク質の化学活性化が遊離チオール基の形成を生じるとき、タンパク質はスルフヒドリル活性剤と結合体化され得る。1つの局面において、薬剤は、遊離チオール基に実質的に特異的な薬剤である。そのような薬剤としては例えば、マレイミド、ハロアセトアミド(例えばヨード、ブロモまたはクロロ)、ハロエステル(例えばヨード、ブロモまたはクロロ)、ハロメチルケトン(例えばヨード、ブロモまたはクロロ)、ベンジル型ハライド(例えばヨージド、ブロミドまたはクロライド)、ビニルスルホンおよびピリジルチオが挙げられる。
【0112】
(スルフヒドリル反応剤)
スルフヒドリル反応剤としては、ヨードアセトアミドなどのアルファ−ハロアセチル化合物、N−エチルマレイミドなどのマレイミド、アセテート、クロライドまたはニトレートの対イオンを含む3,6−ビス−(水銀メチル)ジオキサンなどの水銀誘導体、ジスルフィドジオキシド誘導体などのジスルフィド誘導体、ポリメチレンビスメタンチオスルホン酸塩試薬およびクラベセイン(還元抗体のジスルフィド結合に添加されることが示されている、2個の遊離スルフヒドリル基を含有するフルオレセインの蛍光誘導体)が挙げられる。
【0113】
ヨードアセテートなどのアルファ−ハロアセチル化合物は、スルフヒドリル基と容易に反応してアミドを生成する。これらの化合物は、遊離チオールをカルボキシメチル化するために使用された。それらは厳密にSH特異性でなく、アミンと反応する。反応は、ハライドの置換を生じる、チオラートイオンの求核攻撃を包含する。反応性ハロアセチル部分X−−CH2CO−−は、種々の目的で化合物に組み込まれている。例えばブロモトリフルオロアセトンは、F−19包含に使用され、N−クロロアセチルヨードチラミンは、放射性ヨウ素のタンパク質への導入に利用されている。
【0114】
N−エチルマレイミドなどのマレイミドは、特に他の基がプロトン化される7以下のpH値では、スルフヒドリル基にかなり特異性であると見なされている。チオールは、マレイミドによるMichael反応を受けて、二重結合への付加体を独占的に生じる。生じたチオエーテル結合は非常に安定である。それらは、はるかに低速にてアミノ基およびイミダゾリル基とも反応する。例えばpH7では、単純なチオールとの反応は、対応するアミンとの反応よりも約1,000倍高速である。反応に関連する300nm領域における特徴的な吸光度の変化は、反応を監視するための好都合な方法を提供する。これらの化合物は低いpHにおいて安定であるが、高いpHにおいて加水分解を受けやすい。一般にWong, Chemistry of Protein Conjugation and Cross−linking;CRC Press,Inc.,Boca Raton,1991:Chapters 2および4)を参照のこと。
【0115】
スルフヒドリルと本質的に反応性でない分子(例えば薬物)は、化学的に活性化されたタンパク質に、目的の分子と反応性の基とスルフヒドリル反応性基の両方を持つ二官能性架橋剤によってなお結合体化させることができる。この薬剤は、目的の分子(例えばアミノ基、カルボキシル基またはヒドロキシ基)および化学的に活性されたタンパク質の両方と同時に反応できるか、または目的の分子を誘導体化して、薬剤に由来する部分のために次にスルフヒドリル反応性であるパートナー分子を形成するために使用できるか、または化学的に活性されたタンパク質を誘導体化して、目的の分子と反応性にするために使用できる。
【0116】
(リンカー)
薬物はリンカーによってタンパク質に結合され得る。適切なリンカーとしては、例えば開裂性および非開裂性リンカーが挙げられる。開裂性リンカーは通例、細胞内条件下で開裂を受けやすい。適切な開裂性リンカーとしては、例えば、細胞内プロテアーゼ、例えばリソソームプロテアーゼまたはエンドソームプロテアーゼによって開裂性であるペプチドリンカーが挙げられる。例示的な実施形態では、リンカーはジペプチドリンカー、例えばバリン−シトルリン(val−cit)またはフェニルアラニン−リジン(phe−lys)リンカーであり得る。他の適切なリンカーとしては、5.5未満のpHで加水分解可能なリンカー、例えばヒドラゾンリンカーが挙げられる。さらなる適切な開裂性リンカーとしては、ジスルフィドリンカーが挙げられる。
【0117】
リンカーは、タンパク質への結合のための基を含むことができる。例えばリンカーとしては、アミノ、ヒドロキシル、カルボキシルまたはスルフヒドリル反応性基(例えばマレイミド、ハロアセトアミド(例えばヨード、ブロモまたはクロロ)、ハロエステル(例えばヨード、ブロモまたはクロロ)、ハロメチルケトン(例えばヨード、ブロモまたはクロロ)、ベンジル型ハライド(例えばヨージド、ブロミドまたはクロライド)、ビニルスルホンおよびピリジルチオ)が挙げられ得る。一般に、Wong,Chemistry of Protein Conjugation and Cross−linking;CRC Press,Inc.,Boca Raton,1991を参照のこと。
【0118】
特定の実施形態において、抗体またはタンパク質薬物結合体は、以下の式:
【0119】
【化4】
の結合体あるいはその薬学的に許容される塩または溶媒和物であり得る。
式中:
Abは、抗体または他のタンパク質であり、
Aは、ストレッチャー単位であり、
aは、0または1であり、
各Wは独立して、リンカー単位であり、
wは、0〜12の範囲の整数であり、
Yは、スペーサー単位であり、
yは、0、1または2であり、
pは、1〜約20の範囲であり、
Dは、薬物、標識または他の分子であり、
zは、タンパク質上の潜在的な結合部位の数であり、p<zである。
他の実施形態において、pは例えば、2、4、8、10、12、16、25またはそれ以上であり得る。
【0120】
ストレッチャー単位は、リンカー単位を抗体または他のタンパク質に結合させることができる。ストレッチャー単位は、抗体または他のタンパク質の官能基と結合を形成できる官能基を有する。有用な官能基としては、これに限定されるわけではないが、スルフヒドリル(−SH)、アミノ、ヒドロキシル、カルボキシ、炭水化物のアノマーヒドロキシル基、およびカルボキシルが挙げられる。
【0121】
リンカー単位は通例、アミノ酸単位、例えばジペプチド、トリペプチド、テトラペプチド、ペンタペプチド、ヘキサペプチド、ヘプタペプチド、オクタペプチド、ノナペプチド、デカペプチド、ウンデカペプチドまたはドデカペプチド単位である。リンカー単位は、細胞内で開裂性または非開裂性であり得る。
【0122】
スペーサー単位は存在する場合、リンカー単位を薬物に結合させる。あるいはスペーサー単位は、リンカー単位が非存在のときに、ストレッチャー単位を薬物部分に結合させることができる。スペーサー単位は、リンカー単位およびストレッチャー単位が非存在のときに、抗体またはタンパク質に結合することもできる。
【0123】
(VI.結合体およびその使用)
(インビトロ免疫診断)
ある実施形態において、タンパク質(例えば抗体)は、インビトロ免疫診断で使用するために、検出可能な標識に結合される。標識は、化学的に活性された抗体の結合箇所(例えば遊離チオール基)に直接または間接的に結合可能である、放射性標識、フルオロフォア、または酵素であり得る。サンプルは、臨床性(例えば血液、尿、精液、または脳脊髄液、あるいは固形組織または臓器)または非臨床性(例えば土壌、水、食物)でよい。アッセイは、定性的または非定性的であり、サンドイッチおよび比較形式を含むいずれの所望の形式でもよい。多数の免疫アッセイ形式、標識、固定化技術などは、参考として本明細書に援用される、以下の刊行物に開示されている:O’Sullivan(1976),Annals Clin.Biochem.16:221−240;McLaren(1981),Med.Lab.Sci.38:245−51;Ollerich(1984),J.Clin.Chem.Clin.Biochem.22:895−904;Ngo and Lenhoff(1982),Mol.Cell.Biochem.,44:3−12。
【0124】
(免疫造影)
免疫結合体は、インビボ免疫造影にも使用され得る。この目的のために、タンパク質(例えば抗体)は、被験体またはその一部、例えば臓器の中のその位置または場所の外部描出を可能にする手段によって標識される。通例、免疫造影剤は、適切な放射性同位体によって直接(テクネチウムと同様に)または間接的に(キレート化インジウムと同様に)標識された抗体である。患者への注射後に、結合体の場所は、放射性標識によって放出された粒子に感受性である検出器によって、例えばガンマエミッタの場合にはガンマ−シンチレーションカメラによって追跡できる。
【0125】
(免疫療法)
免疫療法では、タンパク質は適切な薬物、例えば細胞毒性薬または細胞増殖抑制薬、免疫抑制剤、放射性同位体、毒素などに結合体化することができる。結合体は、腫瘍細胞または癌細胞の増殖を阻害するために、腫瘍または癌細胞においてアポトーシスを発生させるために、または患者の癌を処置するために使用できる。結合体はしたがって、動物の癌の処置に核種の状況において使用できる。結合体は、薬物を腫瘍細胞または癌細胞へ送達するために使用できる。理論に縛られることなく、一部の実施形態において、結合体は癌細胞または腫瘍関連抗原と結合または会合して、結合体および/または薬物は、レセプター仲介エンドサイトーシスを通じて腫瘍細胞または癌細胞内に取り込むことができる。抗原は腫瘍細胞または癌細胞に付着できるか、あるいは腫瘍細胞または癌細胞に関連する細胞外マトリクスタンパク質であり得る。細胞内に入ると、結合体内の(例えばリンカーにおける)1個以上の特異性ペプチド配列は、1個以上の腫瘍細胞または癌細胞関連プロテアーゼによって加水分解によって開裂され、薬物の放出を引き起こす。放出された薬物は次に、細胞内で自由に移動して、細胞毒性または細胞増殖抑制あるいは他の活性を誘起する。一部の実施形態において、薬物は腫瘍細胞または癌細胞の外側の抗体から開裂して、次に薬物は細胞に浸透するか、または細胞表面にて作用する。
【0126】
それゆえ、一部の実施形態において、結合体または他のタンパク質は、腫瘍細胞または癌細胞に結合する。一部の実施形態において、結合体は、腫瘍細胞または癌細胞の表面上にある腫瘍細胞または癌細胞抗原に結合する。他の実施形態において、結合体は、腫瘍細胞または癌細胞に関連する細胞外マトリクスタンパク質である腫瘍細胞または癌細胞抗原に結合する。
【0127】
特定の腫瘍細胞または癌細胞に対するタンパク質の特異性は、最も効果的に処置される腫瘍または癌を判定するために重要であり得る。例えば抗CD30を有する抗体または抗CD40抗体あるいは他の結合タンパク質は、血液悪性腫瘍を処置するのに有用であり得る。
【0128】
タンパク質−薬物結合体によって処置できる他の特定の種類の癌としては、これに限定されるわけではないが:線維肉腫、粘液肉腫、脂肪肉腫、軟骨肉腫、骨原性肉腫、脊索腫、血管肉腫、内皮肉腫、リンパ管肉腫、リンパ管内皮肉腫、滑膜腫、中皮腫、ユーイング腫瘍、平滑筋肉腫、横紋筋肉腫、結腸癌、結腸直腸癌、腎臓癌、膵臓癌、骨肉種、乳癌、卵巣癌、前立腺癌、食道癌、胃癌、口腔癌、鼻癌、咽喉癌、へん平上皮細胞癌、基底細胞癌、腺癌汗腺癌、脂腺癌、乳頭状癌、乳頭状腺癌、嚢胞腺癌、髄様癌、気管支原性癌、腎細胞癌、肝臓癌、胆管癌、絨毛癌、精上皮腫、胎生期癌、ウィルムス腫瘍、子宮頚癌、子宮癌、睾丸癌、小細胞肺癌、膀胱癌、肺癌、上皮癌、神経膠腫、多形性膠芽腫、星状細胞腫、髄芽細胞腫、頭蓋咽頭腫、上衣細胞腫、松果体腫、血管芽細胞腫、聴神経腫、乏突起膠腫、髄膜腫、皮膚癌、メラノーマ、神経芽細胞腫、網膜芽細胞腫を含む固形腫瘍、血液媒介癌(これに限定されるわけではないが:急性リンパ芽球性白血病“ALL”、急性リンパ芽球B細胞白血病、急性リンパ芽球性T細胞白血病、急性骨髄芽球性白血病“AML”、急性骨髄球性白血病“APL”、急性単芽球性白血病、急性赤白血病、急性巨核芽球性白血病、急性骨髄単球性白血病、急性非リンパ球性白血病、急性未分化白血病、慢性骨髄球性白血病“CML”、慢性リンパ球性白血病“CLL”、毛様細胞白血病、多発性骨髄腫を含む)、急性および慢性白血病(例えばリンパ芽球性、骨髄性、リンパ球性、および骨髄球性白血病)、およびリンパ腫(例えばホジキン病、非ホジキンリンパ腫、多発性骨髄腫、ワルデンシュトレームマクログロブリン血症、重鎖病、および真性赤血球増加)が挙げられる。タンパク質は、結合特異性腫瘍または癌ターゲティングを提供する。
【0129】
(癌のマルチモード療法)
上述のように、これに限定されるわけではないが、腫瘍、転移を含む癌、あるいは制御できない細胞増殖を特徴とする他の疾患または障害は、タンパク質−薬物結合体の投与によって処置または予防できる。
【0130】
他の実施形態において、結合体の有効量および化学療法剤をその必要がある患者に投与することを含む、癌を処置または防止するための方法が提供される。一部の実施形態において、化学療法剤は、それによって癌の処置が抗療性であることが見出されていないということである。一部の実施形態において、化学療法剤は、それによって癌の処置が抗療性であることが見出されているということである。結合体は、処置、例えば癌の処置のための外科手術も受けた患者に投与できる。別の実施形態において、処置の追加の方法は放射線療法である。
【0131】
例示的な実施形態において、タンパク質−薬物結合体は、化学療法剤と、または放射線療法と同時に投与される。別の例示的な実施形態において、化学療法剤または放射線療法は、タンパク質−薬物結合体の投与の、1つの局面において、少なくとも1時間、5時間、12時間、1日、1週間、1ヶ月前または後に、さらなる局面において、結合体の投与の、数ヶ月(例えば3ヶ月まで)前または後に投与される。
【0132】
化学療法剤は、一連のセッションに渡って投与できる。以下に挙げる化学療法剤の1つまたは組合せを投与できる。放射線に関しては、処置される癌の種類に応じていずれかの放射線療法プロトコルが使用できる。例えば制限するわけではないが、X線照射を投与できる;特に高エネルギーメガボルテージ(1MeVエネルギーを超える照射)が深い腫瘍に使用可能であり、電子ビームおよびオルトボルテージX線照射は皮膚癌に使用できる。ガンマ線放出放射性同位体、例えばラジウム、コバルトおよび他の元素の放射性同位体も投与できる。
【0133】
加えて、化学療法または放射線療法の毒性が強すぎることが判明した、または判明しうる場合に、例えば処置される対象によって許容されないまたは耐えられない副作用を引き起こす場合に、タンパク質−薬物結合体による癌の処置方法が化学療法または放射線療法の代替手段として提供される。処置される動物は必要に応じて、どの処置が許容されるまたは耐えられることが判明するかによって、外科手術、放射線療法または化学療法などの別の癌処置によって処置できる。
【0134】
タンパク質−薬物結合体は、インビトロまたは生体外方式で、例えばこれに限定されるわけではないが、白血病およびリンパ腫を含むある癌の処置に使用することも可能であり、そのような処置は自己幹細胞移植を包含する。これは動物の自己造血幹細胞が収穫され、癌細胞すべてが一掃される多ステッププロセスを含むことができ、動物の残存する骨髄細胞個体群は次に、付随する高用量放射線療法を伴って、または伴わずに高用量の結合体の投与によって全滅され、そして幹細胞グラフトは動物へ輸液によって戻される。次に対症療法が与えられて、その間に骨髄機能が修復され、動物が回復する。
【0135】
(癌の多剤療法)
タンパク質−薬物結合体の有効量および抗癌剤である別の治療剤をその必要がある患者に投与することを含む癌を処置する方法が開示される。適切な抗癌剤としては、これに限定されるわけではないが、メトトレキサート、タキソール、L−アスパラギナーゼ、メルカプトプリン、チオグアニン、ヒドロキシウレア、シタラビン、シクロホスファミド、イフォスファミド、ニトロソウレア、シスプラチン、カルボプラチン、ミトマイシン、ダカルバジン、プロカルバジン、トポテカン、窒素マスタード、シトキサン、エトポシド、5−フルオロウラシル、BCNU、イリノテカン、カンプトセシン、ブレオマイシン、ドキソルビシン、イダルビシン、ダウノルビシン、ダクチノマイシン、プリカマイシン、ミトキサントロン、アスパラギナーゼ、ビンブラスチン、ビンクリスチン、ビノレルビン、パクリタキセル、およびドセタキセルが挙げられる。
【0136】
抗癌剤としては、これに限定されるわけではないが、アルキル化剤、例えば窒素マスタード(例えばシクロホスファミド,イフォスファミド、トロホスファミド、クロラムブシル、メルファラン)、ニトロソウレア(例えばカルムスチン(BCNU)、ロムスチン(CCNU))、アルキルスルホナート(例えばブスルファン、トレオスルファン)、トリアゼン(例えばデカルバジン)、プラチナ含有化合物(例えばシスプラチン、カルボプラチン);植物アルカロイド、例えばビンカアルカロイド(例えばビンクリスチン、ビンブラスチン、ビンデシン、ビノレルビン)、タキソイド(例えばパクリタキセル、ドセタキソール);DNAトポイソメラーゼ阻害剤、例えばエピポドフィリン(例えばエトポシド、テニポシド、トポテカン、9−アミノカンプトセシン、カンプトセシン、クリスナトール、ミトマイシン(例えばミトマイシンC);代謝拮抗物質、例えば葉酸拮抗剤、例えばDHFR阻害剤(例えばメトトレキサート、トリメトレキサート)、IMPデヒドロゲナーゼ阻害剤(ミコフェノール酸、チアゾフリン、リバビリン、EICAR)およびリボヌクレオチドレダクターゼ阻害剤(例えばヒドロキシウレア、デフェロキサミン)、ピリミジンアナログ、例えばウラシルアナログ(5−フルオロウラシル、フロクスウリジン、ドキシフルリジン、ratitrexed)、シトシンアナログ(例えばシタラビン(ara C)、シトシンアラビノシド、フルダラビン)、およびプリンアナログ(例えばメルカプトプリン、チオグアニン);ホルモン療法、例えばレセプターアンタゴニスト、例えば抗エストロゲン(例えばタモキシフェン、ラロキシフェン、メゲストロール)、LHRHアゴニスト(例えばgoscrclin、酢酸ロイプロリド)、および抗アンドロゲン(例えばフルタミド、ビカルタミド;レチノイド/デルトイド、例えばビタミンD3アナログ(例えばEB 1089、CB 1093、KH 1060)、光線力学療法(例えばベルトポルフィン(BPD−MA)、フタロシアニン、光増感剤Pc4、デメトキシ−ヒポクレリンA(2BA−2−DMHA))、サイトカイン(例えばインターフェロン−α、インターフェロン−γ、腫瘍壊死因子)などの薬物はもちろんのこと、ゲムシタビン、ベルケイド、ラバミド、サラミド、イソプレニル化阻害剤(例えばラバスタチン)、ドーパミン神経毒(例えば1−メチル−4−フェニルピリミジンイオン)、細胞サイクル阻害剤(例えばスタウロスポリン)、アクチノマイシン(例えばアクチノマイシンD、ダクチノマイシン)、ブレオマイシン、ブレオマイシンA2、ブレオマイシンB2、ペプロマイシン)、アントラサイクリン(ダウノルビシン、ドキソルビシン(アドリアマイシン)、イダルビシン、エピルビシン、ピラルビシン、ゾルビシン、ミトキサントロン)、MDR阻害剤(例えばベラパミル)、およびCa2+ATPase阻害剤(例えばタプシガーギン)などの他の薬物が挙げられる。
【0137】
(自己免疫疾患の処置)
タンパク質−薬物結合体は、自己免疫疾患を引き起こす細胞を殺す、またはその複製を阻害するために、あるいは自己免疫疾患を処置するために有用である。したがって結合体は、患者の自己免疫疾患の処置のために種々の状況で使用できる。結合体は、薬物を標的細胞に送達するために使用できる。理論に縛られることなく、1つの実施形態において、結合体は標的細胞の表面上の抗原と会合して、次に結合体はレセプター仲介エンドサイトーシスを通じて標的細胞内に取り込まれる。細胞内に入ると、(例えばリンカー内の)1個以上の特異性ペプチド配列は、酵素または加水分解によって開裂され、薬物の放出を引き起こす。放出された薬物は次に、サイトソルにおいて自由に移動して、細胞毒性または細胞増殖抑制活性を誘起する。代わりの実施形態において、薬物は標的細胞の外側の結合体から開裂して、次に薬物は細胞に浸透する。
【0138】
一部の実施形態において、タンパク質−薬物結合体は自己免疫抗原に結合する。1つの局面において、抗原は自己免疫状態に関与する細胞の表面上にある。一部の実施形態において、抗体は細胞の表面上にある自己免疫抗原に結合する。例示的な実施形態において、抗体は自己免疫疾患状態に関連する活性化リンパ球に結合する。さらなる実施形態において、結合体は、特定の自己免疫疾患に関連する自己免疫抗体を産生する細胞を殺す、またはその増殖を阻害する。
【0139】
タンパク質−薬物結合体によって処置できる特定の種類の自己免疫疾患としては、これに限定されるわけではないが、Th2リンパ球関連障害(例えばアトピー性皮膚炎、アトピー性喘息、鼻結膜炎、アレルギー性鼻炎、Omenn症候群、全身性硬化症および移植片対宿主疾患);Th1リンパ球関連障害(例えば関節リウマチ、多発性硬化症、乾癬、シェーグレン症候群、橋本甲状腺炎、グレーブス病、原発性胆汁性肝硬変症、ウェゲナー肉芽腫症および結核);および活性化Bリンパ球関連障害(例えば全身性エリテマトーデス、グッドパスチャー症候群、関節リウマチおよびI型糖尿病)が挙げられる。他の自己免疫疾患としては、これに限定されるわけではないが、慢性活動性肝炎、アジソン病、アレルギー性肺胞炎、アレルギー反応、アレルギー性鼻炎、アルポート症候群、アナフィラキシー、強直性脊椎炎、抗リン脂質症候群、関節炎、回虫症、アスペルギルス症、アトピー性アレルギー、アトピー性皮膚炎、アトピー性鼻炎、ベーチェット症候群、愛鳥家肺、気管支喘息、カプラン症候群、心筋症、セリアック病、シャーガス病、慢性糸球体腎炎、コーガン症候群、寒冷凝集素症、先天性風疹感染、CREST症候群、クーロン病、クリオグロブリン血症、クッシング症候群、皮膚筋炎、円板状狼瘡、ドレッサー症候群、イートン・ランバート症候群、エコーウィルス感染、脳脊髄炎、内分泌眼疾患、エプスタイン−バーウィルス感染、ウマ肺気腫、エリマトーデス、エヴァンス症候群、フェルティ症候群、線維筋痛、フックス毛様体炎、胃萎縮症、消化管アレルギー、巨細胞性動脈炎、糸球体腎炎、グッドパスチャー症候群、移植片対宿主疾患、グレーブス病、ギラン−バレー病、橋本甲状腺炎、溶血性貧血、シェンライン−ヘノッホ紫斑、特発性成人胆管減少症、特発性肺線維症、IgAネフロパシー、炎症性大腸炎、インシュリン依存性真性糖尿病、若年性関節炎、若年性真性糖尿病(I型)、ランバート−イートン症候群、蹄葉炎、扁平苔癬、ルポイド肝炎、狼瘡、リンパ球減少症、メニエール病、混合結合組織病、多発性硬化症、重症筋無力症、悪性貧血、多腺性症候群、初老期認知症、原発性無ガンマグロブリン血症、原発性胆汁性肝硬変、乾癬、乾癬性関節炎、レイノー現象、反復流産、ライター症候群、リウマチ熱、関節リウマチ、サンプター症候群、住血吸虫症、シュミット症候群、強皮症、シャルマン症候群、シェーグレン症候群、スティフマン症候群、交感性眼炎、全身性エリマトーデス、高安動脈炎、側頭動脈炎、甲状腺炎、血小板減少症、甲状腺中毒症、中毒性表皮壊死症、B型インスリン抵抗性、I型真性糖尿病、潰瘍性大腸炎、ブドウ膜炎、白斑、ワルデンシュトレーム型マクログロブリン血症、およびウェゲナー肉芽腫症が挙げられる。
【0140】
(自己免疫疾患の多剤療法)
タンパク質−薬物結合体の有効量を単独で、または自己免疫疾患の処置で公知の別の治療剤と組合せて、その必要がある患者に投与することを含む、自己免疫疾患を処置する方法も開示されている。抗自己免疫疾患剤としては、これに限定されるわけではないが、以下が挙げられ得る:シクロスポリン、シクロスポリンA、ミコフェノール酸モフェチル、シロリムス、タクロリムス、エナネルセプト(enanercept)、プレドニゾン、アザチオプリン、メトトレキサート、シクロホスファミド、アミノカプロン酸、クロロキン、ヒドロキシクロロキン、ヒドロコルチゾン、デキサメタゾン、クロラムブシル、DHEA、ダナゾール、ブロモクリプチン、メロキシカムおよびインフリキシマブ。
【0141】
(感染性疾患の処置)
タンパク質−薬物結合体は、感染性疾患を引き起こす細胞を殺す、またはその複製を阻害するために、あるいは感染性疾患を処置するために有用である。したがって結合体は、患者の感染性疾患の処置のために種々の状況で使用できる。ADCは、薬物を標的細胞に送達するために使用できる。1つの実施形態において、抗体は感染性疾患細胞に結合する。一部の実施形態において、結合体は、特定の感染性疾患を引き起こす細胞を殺すか、またはその増殖を阻害する。結合体によって処置できる特定の種類の感染性疾患としては、これに限定されるわけではないが以下が挙げられる:細菌性疾患、例えばジフテリア、百日咳、オカルト菌血症、尿路感染症、胃腸炎、蜂巣炎、喉頭蓋炎、気管炎、アデノイド肥大、咽後膿瘍、膿痂疹、膿瘡、肺炎、心内膜炎、敗血症性関節炎、肺炎球菌性腹膜炎、細菌性髄膜炎、急性化膿性髄膜炎、尿道炎、子宮頚管炎、直腸炎、咽頭炎、卵管炎、精巣上体炎、淋病、梅毒、リステリア症、炭疽病、ノカルジア症、サルモネラ、腸チフス、赤痢、結膜炎、副鼻腔炎、ブルセラ症、ツラレミア、コレラ、腺ペスト、破傷風、壊死性腸炎、および放線菌症;混合嫌気性感染症、例えば梅毒、回帰熱、レプトスピラ症、ライム病、ネズミ咬熱、結核、リンパ節炎、ハンセン病、クラミジア症、クラミジア肺炎、トラコーマおよび封入体性結膜炎;全身性真菌疾患、例えばヒストプラスマ症、コクシジオイデス症、ブラストミセス症、スポロトリクム症、クリプトコッカス症、全身性カンジダ症、アスペルギルス症、ムコール菌症、足菌腫、およびクロモミコーシス;リケッチア症、例えばチフス、ロッキ−山紅斑熱、エーリキア症、東半球ダニ媒介性リケッチア症、リケッチア痘症、Q熱およびバルトネラ症;寄生虫症、例えばマラリア、バベシア症、アフリカ睡眠病、シャーガス病、リーシュマニア症、ダムダム熱、トキソプラズマ症、髄膜脳炎、角膜炎、アメーバ症、ランブル鞭毛虫症、クリプトスポリジウム症、イソスポーラ症、サイクロスポリン、微胞子虫症、回虫症、鞭虫感染、鉤虫感染、線虫感染、眼幼虫移行症、旋毛虫病、糸状虫症、リンパ管フィラリア症、ロア糸状虫症、河川盲目症、犬糸状虫感染、住血吸虫症、水泳者かゆみ症、肺吸虫症、肝吸虫症、肝蛭症、肥大吸虫症、オピストルキス症、条虫感染、包虫症、および多包虫症;ウィルス性疾患、例えば麻疹、亜急性硬化性全汎脳炎、風邪、流行性耳下腺炎、風疹、バラ疹、第5病、水痘、呼吸器合胞体ウィルス感染、クループ、気管支梢炎、感染性単核球症、ポリオ、ヘルパンギーナ、手足口病、ボーンホルム病、陰部ヘルペス、陰部疣贅、無菌性髄膜炎、心筋炎、心膜炎、胃腸炎、後天性免疫不全症候群(AIDS)、ヒト免疫不全ウィルス(HIV)、ライ症候群、川崎症候群、インフルエンザ、気管支炎、ウィルス性「歩く」肺炎、急性発熱性呼吸疾患、急性咽頭結膜熱、流行性角結膜炎、単純ヘルペスウイルス1(HSV−1)、単純ヘルペスウイルス2(HSV−2)、帯状疱疹、巨細胞封入体症、狂犬病、進行性多病巣性白質脳障害、クル、致死性家族性不眠症、クロイツフェルト−ヤコブ病、ゲルストマン−ストロイスラー−シャインカー症候群、熱帯性痙性不全対麻痺、西部ウマ脳脊髄炎、カリフォルニア脳炎ウィルス、セントルイス脳炎、黄熱病、デング熱、リンパ球性脈絡髄膜炎、ラッサ熱、出血熱、繁多ウィルス肺症候群、マールブルグウィルス感染、エボラウィルス感染および天然痘。
【0142】
(感染性疾患の多剤療法)
タンパク質−薬物結合体を単独で、または抗感染性疾患剤である別の治療剤と組合せてその必要がある患者に投与することを含む、感染性疾患を処置する方法も開示されている。抗感染性疾患としては、これに限定されるわけではないが、以下が挙げられ得る:β−ラクタム抗生剤、例えばペニシリンG、ペニシリンV、クロキサシリン、ジクロキサシリン、メチシリン、ナフシリン、オキサシリン、アンピシリン、アモキシシリン、バカンピシリン、アズロシリン、カルベニシリン、メズロシリン、ピペラシリンおよびチカルシリン;アミノグリコシド、例えばアミカシン、ゲンタマイシン、カナマイシン、ネオマイシン、ネチルマイシン、ストレプトマイシンおよびトブラマイシン;マクロリド、例えばアジスロマイシン、クラリスロマイシン、エリスロマイシン、リンコマイシンおよびクリンダマイシン;テトラサイクリン、例えばデメクロサイクリン、ドキシサイクリン、ミノサイクリン、オキシテトラサイクリンおよびテトラサイクリン;キノロン、例えばシノキサシン、およびナリジクス酸;フルオロキノロン、例えばシプロフロキサシン、エノキサシン、グレパフロキサシン、レボフロキサシン、ロメフロキサシン、ノルフロキサシン、オフロキサシン、スパルフロキサシンおよびトロバフロキサシン;ポリペプチド、例えばバシトラシン、コリスチンおよびポリミキシンB;スルホンアミド、例えばスルフイソキサゾール、スルファメトキサゾール、スルファジアジン、スルファメチゾールおよびスルファセタミド;ならびに他の抗菌剤、例えばトリメトプリム、スルファメチゾール、クロラムフェニコール、バンコマイシン、メトロニダゾール、キヌプリスチン、ダルフォプリスチン、リファンピン、スペクチノマイシンおよびニトロフラントイン;ならびに抗ウィルス剤、例えば汎用抗ウィルス剤、例えばイドクスウリジン、ビダラビン、トリフルリジン、アシクロビル、ファンシクロビル、ペンシクロビル、バラシクロビル、ガンシクロビル、フォスカルネット、リバビリン、アマンタジン、リマンタジン、シドフォビル;アンチセンスオリゴヌクレオチド、免疫グロブリンおよびインターフェロン;ならびにHIV感染用薬物、例えばテノフォビル、エムトリシタビン、ジドブジン、ディダノシン、ザルシタビン、スタブジン、ラミブジン、ネビラピン、デラビルジン、サクイナビル、リトナビル、インディナビルおよびネルフィナビル。
【0143】
(VII.薬学的組成物)
インビボでの使用において、一般に、免疫造影(immunoimaging)、免疫療法、または他の使用にかかわらず、結合体は被験体に導入される。組成物は、結合体の単一の異性体、または1個以上の部分付加異性体を含むことができる。例えばタンパク質が抗体である場合、組成物は、単一のE2、E4またはE6異性体、選択されたE2、E4またはE6異性体の混合物、すべて単独のE2、E4またはE6異性体、あるいはE2、E4およびE6異性体の混合物を含み得る。一部の実施形態において、ある異性体を含有する組成物は、実質的に他の異性体を含まなくてもよい。この文脈において、「実質的に含まない」とは、約20%未満、約10%未満、約5%未満、約2%未満、または約1%未満の他の異性体を含有する組成物を意味する。
【0144】
組成物は、その組成物が患者に投与され得るいずれの形態でもよい。例えば組成物は、固体、液体または気体(エアゾール)の形態であり得る。代表的な投与経路としては、経口、局所、非経口、舌下、直腸、膣、眼、腫瘍内、および鼻腔内が挙げられるが、これらに限定されない。非経口投与として、皮下注射、静脈内、筋肉内、胸骨内の注射または注入技法が挙げられる。1つの局面において、組成物は非経口投与される。なお別の局面において、結合体または組成物は、静脈内投与される。
【0145】
結合体は、注射によって導入され得る。代表的に、結合体は血管内に(静脈内または動脈内に)または髄腔内(intrathetically)に、しばしば注入によって投与される。さらに、適切な場合には、結合体は、皮下的、粘膜下、筋肉内、頭蓋内、または薬物投与の他の受容される経路によって導入され得る。
【0146】
他の実施形態において、組成物は有効量の結合体および薬学的に受容可能なキャリアまたはビヒクルを含む。そのような組成物は、動物またはヒトへの投与に適切である。
【0147】
薬学的組成物は、患者への組成物の投与時に結合体が生体利用可能であり得るように処方され得る。組成物は、1種以上の投薬単位の形態を取ることができ、ここで例えば錠剤は単一投薬単位であることが可能であり、注射用形態において結合体の容器は、複数回の投薬単位を保持することができる。
【0148】
薬学的組成物を調製する際に使用される物質は、使用される量では非毒性であり得る。薬学的組成物中の活性成分の最適投薬量が種々の要因に依存して変わることは、当業者に明らかである。関連する要因としては、動物の種類(例えばヒト)、結合体の特定の形態、投与様式、使用される組成物が挙げられるが、これらに限定されない。
【0149】
薬学的に受容可能なキャリアまたはビヒクルは、組成物が例えば錠剤または粉末形態であるように、粒子状でもよい。キャリアは液体でもよく、組成物は例えば経口シロップまたは注射用液剤である。さらにキャリアは、例えば吸入投与で有用なエアゾール組成物を提供するために、気体状または粒子状でもよい。
【0150】
経口投与を目的とする場合、組成物は好ましくは固体形態または液体形態であり、半固体形態、半液体形態、懸濁物形態およびゲル形態は、本明細書で固体または液体のどちらかと見なされる形態に含まれる。
【0151】
経口投与用の固体組成物として、組成物は粉剤、顆粒剤、圧縮錠剤、丸剤、カプセル剤、チューイングガム、カシェ剤などで処方され得る。そのような固体組成物は代表的に、1種以上の不活性希釈剤を含有する。さらに、以下の1種以上が存在できる:結合剤(例えばカルボキシメチルセルロース、エチルセルロース、微結晶性セルロース、またはゼラチン);賦形剤(例えばデンプン、ラクトース、またはデキストリン);崩壊剤(例えばアルギン酸、アルギン酸ナトリウム、プリモゲル(Primogel)、コーンスターチなど);滑沢剤(例えばステアリン酸マグネシウムまたはステロテックス);流動促進剤(例えばコロイド状二酸化ケイ素);甘味料(例えばスクロースまたはサッカリン)、矯味矯臭剤(例えばペパーミント、サリチル酸メチルまたはオレンジ香味料)、ならびに着色剤。
【0152】
組成物がカプセル剤、例えばゼラチンカプセル剤の形態である場合、組成物は上の種類の物質に加えて、液体キャリア(例えばポリエチレングリコール、シクロデキストリン、または脂肪油)を含有し得る。
【0153】
組成物は液体(例えばエリキシル剤、シロップ剤、液剤、乳剤または懸濁剤)の形態でもよい。液体は経口投与に、または注射による送達に有用であり得る。経口投与を目的とする場合、組成物は甘味料、保存料、染料/着色料および調味料の1種以上を含むことができる。注射による投与のための組成物では、界面活性剤、保存料、湿潤剤、分散剤、懸濁剤、緩衝剤、安定剤および等張剤の1種以上を含むこともできる。
【0154】
液体組成物は、それらが液剤、懸濁剤または他の同様の形態であるかどうかにかかわらず、以下の1種以上も含むことができる:滅菌希釈剤(例えば注射用水、食塩水、好ましくは生理的食塩水、リンガー液、等張性塩化ナトリウム、不揮発性油(例えば溶媒または懸濁媒体として作用し得る合成モノグリセリドまたはジグリセリド、ポリエチレングリコール、グリセリン、シクロデキストリン、プロピレングリコールまたは他の溶媒);抗菌剤(例えばベンジルアルコールまたはメチルパラベン);抗酸化剤(例えば、アスコルビン酸または亜硫酸水素ナトリウム);キレート剤(例えばエチレンジアミン四酢酸);緩衝剤(例えばアセテート、シトレートまたはホスフェート)および張度の調整のための薬剤(例えば塩化ナトリウムまたはデキストロース)。非経口組成物は、ガラス、プラスチックまたは他の材料からなるアンプル、使い捨て注射器または複数回用量バイアルに封入され得る。生理的食塩水は例示的なアジュバントである。注射用組成物は好ましくは滅菌されている。
【0155】
非経口投与用の調製物は、滅菌水または非水性液剤、懸濁剤、および乳剤を含む。非水性溶媒の例は、プロピレングリコール、ポリエチレングリコール、植物油(例えばオリーブ油)および注射用有機エステル(例えばオレイン酸エチル)である。水性キャリアは、水、アルコール性/水性溶液、乳剤または懸濁物(食塩水および緩衝媒体)を含む。非経口ビヒクルは、塩化ナトリウム溶液、リンガーデキストロース、デキストロースおよび塩化ナトリウム、乳酸加リンガー液、または非揮発性油を含む。静脈内ビヒクルは、流体および栄養素補充薬、電解質補充薬(例えばリンガーデキストロースをベースとするもの)などを含む。保存料および他の添加剤、例えば抗菌薬、抗酸化剤、キレート剤、および不活性ガスなども存在し得る。
【0156】
特定の障害または状態の処置において有効な結合体の量は、障害または状態の性質によって変わり、標準臨床技法によって決定され得る。開示されたタンパク質−薬物結合体の投与のためのこれらの投薬量範囲は、状態または障害の症状が軽減される所望の効果を引き起こすのに十分大きいものである。投薬量は、副作用、例えば望ましくない交差反応、アナフィラキシー反応などを引き起こすほど多くてはならない。加えて、最適投薬量範囲を決定するために、インビトロまたはインビボでのアッセイが必要に応じて利用される。
【0157】
組成物において利用される正確な用量は、患者の年齢、状態、性別および疾患の程度、投与経路、および疾患または症状の重篤度によっても変わり、医師の判断および各患者の状況に従って決定されるべきである。
【0158】
組成物は適切な投薬量が得られるような、有効量の結合体を含む。代表的に、この量は、組成物の重量で少なくとも約0.01%の結合体である。経口投与を目的とする場合、この量は組成物の重量で約0.1%〜約80%の範囲で変化し得る。1つの局面において、経口組成物は組成物の重量で約4%〜約50%の結合体を含むことができる。なお別の局面において、本組成物は、非経口投薬量単位が結合体の重量で約0.01%〜約2%を含有するように調製される。
【0159】
静脈内投与では、組成物は、動物の体重1kg当たり約0.01〜約100mgの結合体を含むことができる。1つの局面において、組成物は動物の体重1kg当たり約1〜約100mgの結合体を含むことができる。別の局面において、投与される量は、約0.1〜約25mg/kg体重の結合体の範囲で変化する。
【0160】
一般に、患者に投与される結合体の投薬量は代表的に、約0.01mg/kg〜約2000mg/kg動物体重である。1つの局面において、患者に投与される投薬量は、約0.01mg/kg〜約10mg/kg動物体重である。別の局面において、患者に投与される投薬量は、約0.1mg/kg〜約250mg/kg動物体重である。なお別の局面において、患者に投与される投薬量は、約0.1mg/kg〜約20mg/kg動物体重である。なお別の局面において、投与される投薬量は、約0.1mg/kg〜約10mg/kg動物体重である。なお別の局面において、投与される投薬量は、約1mg/kg〜約10mg/kg動物体重である。
【0161】
結合体は、いずれかの好都合な経路によって、例えば注入またはボーラス注射によって、上皮または粘膜皮膚内層(例えば口腔粘膜、直腸および腸粘膜など)を介した吸収によって投与され得る。投与は、全身的または局所的であり得る。種々の送達システム、例えばリポソーム、微粒子、マイクロカプセル、カプセルへのカプセル化などが公知であり、結合体または組成物を投与するために使用され得る。ある実施形態において、2つ以上の結合体または組成物が患者に投与される。
【0162】
特定の実施形態において、1種以上の結合体または組成物を処置が必要な領域に局所的に投与することが望ましい。これは例えば、手術中の局所注入によって;例えば手術後の創傷包帯と併せた局所塗布によって;注射によって;カテーテルによって;坐剤によって;膜(例えばシラスティック膜(sialastic))、または繊維を含む、多孔性、非多孔性、あるいはゼラチン性材料の移植)によって達成される。1つの実施形態において、投与は、癌の部位(または形成部位)、腫瘍あるいは新生物または新生物発生前組織への直接注射によって可能である。別の実施形態において、投与は、自己免疫疾患の徴候の部位(または形成部位)への直接注射によって可能である。
【0163】
ある実施形態において、1種以上の結合体または組成物を、脳室内および髄腔内注射を含む任意の適切な経路によって中枢神経系に導入することが望まれ得る。脳室内注射は、例えばオンマヤ(Ommaya)リザーバーなどのリザーバーに取り付けられた脳室内カテーテルによって容易にされ得る。
【0164】
例えば吸入器またはネブライザの使用、およびエアゾール化剤を用いた処方によって、あるいはフルオロカーボンまたは合成肺界面活性剤での潅流を介して、肺投与も利用され得る。
【0165】
なお別の実施形態において、結合体は制御放出システムで送達可能であり、例えばこれに限定されるわけではないが、ポンプまたは種々のポリマー材料が使用され得る。なお別の実施形態において、制御放出システムは結合体の標的の付近、例えば脳に配置可能であり、それゆえ全身用量の一部のみを必要とする(例えばGoodson,in Medical Applications of Controlled Release,supra,vol.2,pp.115−138(1984)を参照)。Langer(Science 249:1527−1533(1990))による概説で述べられている他の制御放出システムが使用され得る。
【0166】
一部の実施形態において、タンパク質結合体をキャリアに結合させて組成物を形成することができる。「キャリア」という用語は、希釈剤、アジュバントまたは賦形剤をいい、それと共に結合体が投与される。そのような薬学的キャリアは、液体(例えば水および石油、動物、植物または合成起源のものを含む油(例えばラッカセイ油、ダイズ油、鉱油、ゴマ油など)であり得る。キャリアは、食塩水、アラビアゴム、ゼラチン、デンプンペースト、タルク、ケラチン、コロイド状シリカ、尿素などであり得る。さらに、補助剤、安定剤、増粘剤、滑剤および着色剤を使用できる。1つの実施形態において、患者に投与される場合、結合体または組成物および薬学的に受容可能なキャリアは滅菌されている。水は、結合体を静脈内投与する場合の例示的なキャリアである。食塩水および水性デキストロースおよびグリセロール溶液は、特に注射用液剤のための液体キャリアとしても利用され得る。適切な薬学的キャリアとしてはまた、デンプン、グルコース、ラクトース、スクロース、ゼラチン、麦芽、コメ、小麦粉、チョーク、シリカゲル、ステアリン酸ナトリウム、モノステアリン酸グリセロール、タルク、塩化ナトリウム、乾燥スキムミルク、グリセロール、プロピレン、グリコール、水、エタノールなどの賦形剤が挙げられる。所望の場合、組成物は少量の湿潤剤または乳化剤、あるいはpH緩衝剤も含むことができる。
【0167】
組成物は、液剤、懸濁剤、乳剤、錠剤、丸剤、ペレット剤、カプセル剤、液体を含有するカプセル剤、粉剤、持続放出製剤、坐剤、乳剤、エアゾール、スプレー、懸濁剤の形態、または使用に適した他の任意の形態を取ることができる。適切な薬学的キャリアの他の例は、E.W.Martinによる”Remington’s Pharmaceutical Sciences”に述べられている。
【0168】
例示的な実施形態において、結合体は通常の手順に従って、動物、特にヒトへの静脈内投与に適した薬学的組成物として処方される。代表的に、静脈内投与のためのキャリアまたはビヒクルは、滅菌等張性水性緩衝溶液である。必要な場合、組成物は可溶化剤も含むことができる。静脈内投与のための組成物は必要に応じて、注射部位の疼痛を緩和するために、リグノカインなどの局所麻酔薬を含むことができる。一般に成分は、例えば活性剤の量を表示したアンプルまたはサシェ(sachette)などの密閉した容器内の乾燥した凍結乾燥粉末または無水濃縮物として、単位投薬形で個別に、または共に混合して提供される。結合体が注入によって投与される場合、例えば滅菌された薬学的用水または食塩水を含有する注入ボトルを用いて投薬できる。結合体が注射によって投与される場合、投与前に成分を混合できるように、注射用の滅菌水または食塩水のアンプルを提供することができる。
【0169】
経口送達用の組成物は、例えば、錠剤、口内錠、水性懸濁剤または油性懸濁剤、顆粒剤、粉末剤、乳剤、カプセル剤、シロップ剤、またはエリキシル剤の形態が可能である。必要に応じて、経口投与組成物は、例えば、薬学的に口当たりのよい調製物を提供するために、必要に応じて1種以上の薬剤(例えばフルクトース、アスパルテームまたはサッカリンなどの甘味料;ペパーミント、ウィンターグリーンオイル、またはチェリーなどの矯味矯臭剤;着色料;および保存料)を含有できる。さらに、錠剤形態または丸剤形態では、組成物は消化管での崩壊および吸収を遅延させて、それにより長期間に渡って持続作用を提供するために、コーティングすることができる。浸透圧的に活性な駆動化合物を包囲する選択的透過性膜も、経口投与化合物に適切である。このような遅延されたプラットフォームでは、カプセル剤を包囲する環境からの流体は、駆動化合物により吸収されて膨潤し、開口部を通じて薬剤または薬剤組成物を放出する。これらの送達プラットフォームは、即時放出処方物の急上昇特性とは対照的に、本質的にゼロ次送達特性を提供することができる。モノステアリン酸グリセロールまたはステアリン酸グリセロールなどの時間遅延材料も使用できる。
【0170】
組成物は局所投与を目的とすることも可能であり、その場合、キャリアは液剤、乳剤、軟膏またはゲルベースの形態であり得る。経皮投与を目的とする場合は、組成物は経皮パッチまたはイオン導入器具の形態を取ることができる。局所処方物は、約0.05%〜約50%w/v(組成物の単位体積当たりの重量)、別の局面では0.1%〜10%w/vの結合体の濃度を含むことができる。
【0171】
組成物は、例えば直腸で溶解して、結合体を放出する坐剤の形態で、直腸投与を目的とすることができる。
【0172】
組成物は、固体または液体投薬単位の物理形態を改良する種々の物質を含むことができる。例えば組成物は、活性成分周囲にコーティングシェルを形成する物質を含むことができる。コーティングシェルを形成する物質は代表的に、不活性であり、例えば糖、シェラック、および他の腸溶コーティング剤から選択できる。あるいは活性成分をゼラチンカプセルに入れることができる。
【0173】
組成物は、ガス状投薬単位から成ることがあり、例えばエアゾールの形態を取ることができる。エアゾールという用語は、コロイド状性質のものから加圧パッケージから成る系に渡る種々の系を示すために使用される。送達は、液化ガスまたは圧縮ガスによって、あるいは活性成分を分配する適切なポンプシステムによって可能である。
【0174】
本組成物は固体、液体またはガス状の形態であるか否かにかかわらず、癌、自己免疫疾患または感染性疾患の処置に使用される薬剤を含むことができる。
【0175】
(VIII.薬物動態学)
マウスにおいて、精製したcAC10−vcMMAEの4個の薬物の結合体の毒性および効果のインビトロでの特徴付けを実施し、実施例でさらに詳細に議論する(例えば実施例8および9を参照のこと)。簡潔には、これらの試験は、抗体当たり平均4個の薬物を含む混合物は、8個の薬物を含む結合体と等しく有効であるが(どちらも1mg/Kgの単回用量)、より毒性が低い(MTDは、抗体当たり4個の薬物では100mg/kgであり、抗体当たり8個の薬物では50mg/kg)ことを示している。DTT方法による抗体当たり4個の薬物を含む精製物質は効果および毒性が同様であるのに対して、DTNB方法による抗体当たり4個の薬物を含む精製物質は、やや高い120mg/KgのMTDを有するが、他の結合体の半分の用量(0.5mg/Kg)で有効である。
【0176】
cAC10に抗体当たり2個、4個、または8個の薬物を付加することは、標的抗原CD30への結合に何の影響も及ぼさなかった。cAC10−ADCのインビトロ有効性は薬物付加に直接依存し、それゆえ全MMAE暴露に直接依存する。
【0177】
cAC10−E4は、Karpas−299異種移植モデルにて、MMAE用量の半分であり、同じ用量の抗体で、cAC10−E8に匹敵する抗腫瘍活性を示した。有効性が薬物付加に直接関連するというインビトロでの発見結果に基づいて、cAC10−E4とcAC10−E8との同等のインビボ抗腫瘍活性は予想されなかった。ADCの薬物動態学の調査は、クリアランスがADCの薬物付加に直接関連し、暴露(曲線下面積−AUC)が薬物付加に反比例することを明らかにした。cAC10−E4のAUCはcAC10−E8よりも3倍高かった。cAC10−E8と比較したcAC10−E4のより高いAUCは、低下した有効性を補償するのに明らかに十分であり、同等の有効性をもたらした。AUCを増大させるために血漿消失半減期を減速させることによって有効性を改善する試みは、インターフェロン−αのためのアルブミン融合タンパク質の構築および抗癌剤ルルトテカン(Lurtotecan)のリポソーム送達を含む方法によって達成されてきた。目的が血漿半減期を延長させることであるこれらの例とは異なり、cAC10−E4の暴露向上は、MMAE付加減少の貴重な結果であった。
【0178】
実施例8に開示するように、cAC10−E2を1.0mg/kg/用量のq4dx4と共に投薬することは、10症例中10の治癒をもたらした。cAC10−E2は同じmAb用量ではcAC10−E4と比べて同等の抗腫瘍活性を示さなかったが、cAC10−E4と比べて同等の抗腫瘍活性を達成するためのcAC10−E2の用量は、インビボ有効性実験に基づいておそらく2分の1未満である。cAC10−E4と同様に、cAC10−E2の暴露の改善は、より低いインビトロでの有効性を解決するのに重要な役割を演じ得る。
【0179】
cAC10−Val−Cit−MMAE ADCの治療可能性を最大限にするためには、高い治療指数が必要である。mAb当たりのMMAE分子の量を8から4に減少させることは、治療指数を100から200へ向上させた。化学療法試薬の急勾配の用量−反応曲線を考えると、治療指数の2倍の差は、毒性に関する臨床上の意義全体の点から見て重要であり得る。
【0180】
MMAEの量をmAb当たり8分子から4分子に減少させることにより、インビトロでの活性の低下があるが、インビボでの同等の抗腫瘍活性がなお示された。抗体当たり2個のMMAE分子への薬物付加でのさらなる減少は、インビトロ活性をさらに低下させ、cAC10−E2は、複数回投薬の状況の2倍の用量で、cAC10−E4およびcAC10−E8と同等またはより優れた有効性を有していた。治療濃度域は、薬物付加を8MMAE分子から4に減少させることにより2倍に上昇し、抗体当たり薬物2個までのさらなる減少により最低にて維持された。ADCの薬物置換の最適化には、注目に値する価値がある。
【実施例】
【0181】
(方法1の実施例1)
cAC10を制限された濃度のDTTによって以下のように部分還元した:cAC10(8mg/mLまたは53.8μM)を0.05Mホウ酸ナトリウム、pH8、0.05M NaCl、および1mMジエチレン−トリアミン五酢酸(DTPA;Aldrich)中のDTT 3.5モル当量(188.4μM;Sigma)によって、37℃にて1時間処理した。次に還元された抗体をPD−10カラム(Amersham Biosciences)で脱塩することによって精製した。PD−10カラムを1mM DTPA(PBSD)を含むリン酸緩衝食塩水(PBS)pH7.4(GIBCO)25mLで平衡にして、上の溶液1mLをカラムに添加し、カラムをPBSD 1.8mLで洗浄して、PBSD 1.4mLを用いてカラムを溶出させた。1.0mg/mL溶液での280nmにおける吸収値1.58を使用してタンパク質濃度を定量して、モル濃度を分子量150,000g/molを用いて決定した。生成された抗体−システインチオールの濃度を5,5’−ジチオ−ビス−(2−ニトロ安息香酸)(DTNB;Pierce)を用いた滴定によって決定すると、概して、この方法を用いた抗体当たり4個の抗体−システインチオールよりもやや高くなった。
【0182】
次に以下のように、薬物vcMMAEを還元cAC10に結合させた:還元cAC10(代表的に、最終濃度が30μM抗体および120μM抗体−システインチオール)を最初に0℃まで冷却した。vcMMAEを冷アセトニトリルに溶解させ、抗体溶液と迅速に混合した。最終アセトニトリル濃度は20%であるのに対して、最終薬物濃度は135〜150μMであった(4.5〜5モル当量、これは抗体−システインチオールよりやや過剰である)。この溶液を0℃にて30分間インキュベートして、過剰なvcMMAEをシステイン(1mM最終濃度)によってクエンチさせ、結合体を上述のようにPD−10カラムを用いて精製した。
【0183】
(方法1の実施例2)
抗体当たり4個のvcMMAEを含むcAC10(E4ミックス)は、制限量のDTTを用いて以下のように調製した:cAC10を0.025Mホウ酸ナトリウム、pH8、0.025M NaCl、1mM DTPA中のDTT 3.25モル当量によって、37℃にて2時間処理した。この混合物を水で5倍に希釈し、ヒドロキシアパタイトカラム(MacroprepセラミックタイプI 40μm、BioRad、ハーキュリーズ、カリフォルニア州)に流速10mL/分で加えた。カラムサイズは、cAC10 10mg当たり1mLであった。カラムは事前に、5カラム容量の0.5Mリン酸ナトリウム、pH7、10mM NaClおよび5カラム容量の10mMリン酸ナトリウム、pH7、10mM NaClで平衡にしておいた。添加後、カラムを5カラム容量の10mMリン酸ナトリウム、pH7、10mM NaClで洗浄して、次に100mMリン酸ナトリウム、pH7、10mM NaClで溶出させた。溶出後にDTPAを1mMまで添加した。1.0mg/mL溶液での280nmにおける吸光度値1.58を使用してタンパク質濃度を定量し、モル濃度を分子量148,449g/molを用いて決定した。生成された抗体−システインチオールの濃度を、DTNBを用いた滴定によって決定すると、代表的に、抗体当たり4.0〜4.5個のチオールが生じた。
【0184】
還元cAC10を抗体−システインチオールに対してわずかに過剰なvcMMAE(1.1モル当量)によってアルキル化した。vcMMAEの溶解性を維持するために、最終反応混合物中に10% DMSOが存在していた。あるいはvcMMAEは、5体積%のアルコール、例えばエタノールおよびイソプロピルアルコールを含む溶液中に保持することが可能であった。アルキル化反応を0℃にて30分間実施した。システイン(最終1mM)を使用して、すべての未反応vcMMAEをクエンチさせた。cAC10−vcMMAEは、上述のようにヒドロキシアパタイトクロマトグラフィーを用いて精製した。溶出後に、緩衝液をAmicon(Millipore、ベッドフォード、マサチューセッツ州)Ultrafree 30Kカットオフスピン濃度デバイスを使用して、リン酸緩衝食塩水(Invitrogen、カールスバッド、カリフォルニア州)に交換した。タンパク質濃度は、1.0mg/mL溶液での280nmにおける吸光度値1.62を使用して定量した。
【0185】
(方法2aの実施例3)
大幅に過剰のDTTを添加することによって、cAC10を完全に還元した。最終反応濃度は、8mg/mL cAC10、0.05Mホウ酸ナトリウム、pH8、0.05M NaCl、10mM DTT、および1mM DTPAであった。この溶液を37℃にて30分間インキュベートして、上述のようにPD−10カラムで脱塩することによって抗体を精製した。DTNB滴定によって決定されたように、これらの条件を使用して8よりもやや多い抗体−システインチオールが生成された。
【0186】
部分再酸化は、DTNBを酸化剤として使用することによって達成された。還元cAC10(代表的に30μM)を0℃まで冷却して、次にDTNB 1.5〜2.5モル当量(最終濃度45〜75μM;E4の最高収率は、2.0当量を使用して得られた)で処理した。溶液を反転によって迅速に混合し、0℃にて10〜20分間インキュベートした。放出されたTNB−が黄色であるため、反応の程度が観察できる。代表的に、反応は数秒以内で完了したことが示される。システインを添加して(最終濃度1mM)、抗体システインとの混合ジスルフィド中ではなく、TNB−としてすべてのTNBが存在することを確認した。次に抗体を上述のようにPD−10カラムまたはヒドロキシアパタイトカラムで精製した。代表的に、4種の抗体−システインチオールは、この部分再酸化手順の後にDTNB滴定によって観察された。vcMMAE薬物は最終的にこれらの抗体−システインチオールに結合され、上の方法1で述べたようにPD−10によって精製した。
【0187】
(方法2bの実施例4)
完全還元cAC10は、上の方法2aで述べたように調製した。完全還元cAC10(代表的に30μM)を0℃まで冷却し、次にDTNB 1.5〜2.5当量(最終濃度45〜75μM)によって処理した。溶液を反転によって迅速に混合し、0℃にて10分間インキュベートした。さらに精製せずに、部分的に再酸化させたcAC10を次に冷アセトニトリルに溶解させたvcMMAE 5当量と迅速に混合した。方法1と同様に、アセトニトリルの最終濃度は20%であった。結合反応では、cAC10最終濃度は、24μMであり(96μM抗体−システインチオールまたは抗体当たり4個)、最終vcMMAE濃度は120μMであった(5モル当量)。この溶液を0℃にて30分間インキュベートしてから、システインによってクエンチさせ、上述のようにPD−10によって精製した。
【0188】
E4ミックスのヒドロキシアパタイトによる分取精製。数個の15mL Amicon Ultrafree 30Kカットオフスピン濃度デバイスを使用して、cAC10の緩衝液(25mM クエン酸ナトリウム、pH6.5、250mM NaCl、および0.02% Tween−80)をPBSに交換した。PBS中のcAC10 1.09gを、最終体積89mLのDTTによって、以下のように完全に還元した:cAC10(82.3μM)を0.025Mホウ酸ナトリウム、pH8、0.025M NaCl中の10mM DTTによって37℃にて1時間処理した。この混合物を水で250mLまで希釈し、70mLヒドロキシアパタイトカラム(MacroprepセラミックタイプI 40μm、BioRad)に流速10mL/分で加えた。カラムは事前に、5カラム容量の0.5Mリン酸ナトリウム、pH7、10mM NaClおよび5カラム容量の10mMリン酸ナトリウム、pH7、10mM NaClで平衡にしておいた。添加後、カラムを5カラム容量の10mMリン酸ナトリウム、pH7、10mM NaClで洗浄して、次に100mMリン酸ナトリウム、pH7、10mM NaClで溶出させた。
【0189】
完全還元cAC10をDTNBによって以下のように再酸化した:上記から溶出された物質(6.02mg/mLまたは40.2μM、170mL中1.02g)を0℃まで冷却し、次にDTNB 2.0当量(10mMストック)で20分間処理した。さらに精製せずに、再酸化させたcAC10をvcMMAEに結合させた。冷cAC10(最終31.9μM)を、最終体積214mLのDMSO(最終20%)中に溶解させたvcMMAE 5当量(最終159.5μM)で処理した。0℃にて40分後、100mMシステイン 1.07mLを添加して、すべての未反応vcMMAEをクエンチさせ、混合物を水で750mLまで希釈した。結合体をDTT還元のために上述のようにヒドロキシアパタイトカラムで精製した。回収したcAC10−vcMMAE E4ミックス(0.99g、またはcAC10に基づいて91%の全収率)を濃縮して、数個の15mL Amicon Ultrafree 30Kカットオフスピン濃度デバイスを使用して緩衝液をPBSに交換した。
【0190】
HICによる純E4の分取精製は、45mL Toyopearlフェニル650M HICカラムによって、流速10mL/分で周囲温度にて実施した。溶媒Aは2M NaClおよび50mMリン酸ナトリウム、pH7であった。溶媒Bは80%v/v 50mMリン酸ナトリウム、pH7および20%v/vアセトニトリルであった。カラムは事前に5カラム容量の溶媒Aで平衡にした。ヒドロキシアパタイト(上記)によって精製したcAC10−vcMMAE E4ミックス最大400mgを1容量の4M NaClおよび50mMリン酸ナトリウム、pH7と混合してカラムに添加した。E0はカラムによって保持されなかった。連続段階勾配によって、異なる薬物付加種を溶出した:E2を35%溶媒Bによって溶出させ、E4を70%溶媒Bによって溶出させ、E6を95%溶媒Bによって溶出させ、E8を100%溶媒Bによって溶出させた。精製したE4を濃縮して、数個の15mL Amicon Ultrafree 30Kカットオフスピン濃度デバイスを使用して緩衝液をPBSに交換し、400mg精製物2回分から純E4 235mgを得た。分析HICによる純度分析(以下)は、90%を超えるE4純度を示した。
【0191】
(方法1の実施例5)
TCEP制限還元と、それに続く中間精製なしのアルキル化は、cAC10を0.025Mホウ酸ナトリウムpH8、0.025M NaCl、1mM DTPA中のTCEP 2.75モル当量により37℃にて2時間処理することによって実施した。図9も参照のこと。次に混合物を0℃まで冷却して、部分還元cAC10を上述のようにvcMMAEによってアルキル化した。cAC10−vcMMAEは、リン酸緩衝食塩水で平衡にしたPD−10カラム(Amersham Biosciences,ピスカタウェイ、ニュージャージー州)を使用して脱塩した(部分還元は、図8に示すように、部分還元抗体の中間精製ステップを用いても実施できる)。
【0192】
異性体分布の動力学を決定するために使用したサンプルは、以下のように調製した:cAC10を50mMリン酸ナトリウム、pH7.5および5mM EDTA中のDTT 3.0当量によって37℃にて還元した。表示した時点でサンプルを取り出して、同量の200mM クエン酸ナトリウム、pH5によってクエンチさせ、5mM EDTAを含有するリン酸緩衝食塩水によって平衡にしたPD−10カラムを使用して精製した。還元cAC10は上述のようにvcMMAEによって処理し、リン酸緩衝食塩水によって平衡にしたPD−10カラムを使用して精製した。
【0193】
HICによる純E2、純E4、および純E6の精製は、Toyopearlフェニル650M HICカラム(Tosoh Biosciences,モンゴメリービル、ペンシルベニア州)によって、流速10mL/分で周囲温度にて実施した。カラムサイズはcAC10−vcMMAE 7.5mg当たり1mLであった。溶媒Aは2.0M NaClおよび50mMリン酸ナトリウム、pH7であった。溶媒Bは80%v/v 50mMリン酸ナトリウム、pH7および20%v/vアセトニトリルであった。カラムは事前に5カラム容量の溶媒Aで平衡にした。cAC10−vcMMAEを0.67容量の5M NaCl(最終2.0M)と混合して、カラムに添加した。E0はカラムによって保持されなかった。連続段階勾配によって、異なる薬物付加種を溶出した:E2を35%溶媒Bによって溶出させ、E4を70%溶媒Bによって溶出させ、E6を95%溶媒Bによって溶出させ、E8を100%溶媒Bによって溶出させた。精製したE4を濃縮して、Amicon Ultrafree 30Kカットオフスピン濃度デバイスを使用して緩衝液をリン酸緩衝食塩水に交換した。
【0194】
(分析方法の実施例6)
薬物付加は、250および280nmにおける吸光度の比(A250/280)を測定することによって決定した。cAC10当たりのvcMMAEの数は実験的に(A250/280−0.36)/0.0686と決定されている。
【0195】
結合体は、Tosoh Biosceinceエーテル−5PWカラム(part08641)を流速1mL/分およびカラム温度30℃にて使用する疎水性相互作用クロマトグラフィー(HIC)によって、E4純度のパーセントについて分析した。溶媒Aは50mMリン酸ナトリウム、pH7および2M NaClであった。溶媒Bは、80% 50mMリン酸ナトリウム、pH7、10% 2−プロパノール、および10%アセトニトリルであった。15分間のアイソクラチック0%B、50分間の0〜100%Bの線形勾配、0.1分間の100〜0%Bの線形勾配、そして14.9分間のアイソクラチック0%B。注入(代表的に90〜100μL)は、1容積の精製vcMMAE−cAC10結合体(少なくとも3mg/mLの濃度)および1容積の50mMリン酸ナトリウム、pH7および4M NaClであった。
【0196】
HICクロマトグラフィーからの純E4を含むADCをAgilentバイオアナライザ(Bioanalyzer)を用いて変性および非還元条件下で分析した。タンパク質200チップを変性条件下であるが、製造業者によって記載されるように使用した。簡潔には、1mg/mL cAC10−vcMMAE 4μLを非還元性付加緩衝液2μLと混合して、100℃まで5分間加熱した。水(84μL)を添加し、この混合物6μLをチップの各ウェルに付加した。
【0197】
最後に純E4をPLRP−Sカラム(Polymer Laboratories)で分析した。流速は1mL/分であり、カラム温度は65℃であった。溶媒Aは0.05%トリフルオロ酢酸水溶液であり、溶媒Bは0.04%トリフルオロ酢酸アセトニトリル溶液であった。3分間のアイソクラチック25%B、15分間の50%Bまでの線形勾配、2分間の95%Bまでの線形勾配、1分間の25%Bまでの線形勾配、そして2分間のアイソクラチック25%B。注入は、以前に20mM DTTによって37℃にて20分間還元して、鎖間ジスルフィドを開裂させたcAC10−vcMMAE 10μLであった。
【0198】
ADCもまた、変性および還元条件下でPLRP−Sカラム(Polymer Laboratories)(2.1x150mm、8μ、1000Å)によって分析した。流速は1mL/分であり、カラム温度は80℃であった。溶媒Aは0.05%トリフルオロ酢酸水溶液であり、溶媒Bは0.04%トリフルオロ酢酸アセトニトリル溶液であった。3分間のアイソクラチック25%B、25分間の50%Bまでの線形勾配、2分間の95%Bまでの線形勾配、1分間の25%Bまでの線形勾配、そして2分間のアイソクラチック25%B。注入は、以前に20mM DTTによって37℃にて15分間還元して、残りの鎖間ジスルフィドを開裂させた1mg/mL cAC10−vcMMAE 10〜20μLであった。各鎖のモル分率は、以下のモル吸光係数を使用して決定した:0vcMMAEを含む軽鎖:30,160M−1cm−1;1vcMMAEを含む軽鎖:31,660M−1cm−1;0vcMMAEを含む重鎖:86,915M−1cm−1;1vcMMAEを含む重鎖:88,415M−1cm−1;2vcMMAEを含む重鎖:89,915M−1cm−1;3vcMMAEを含む重鎖:91,415M−1cm−1。
【0199】
E2およびE6の異性体分布は、PLRP−S HPLCデータのみを使用して決定した。E2異性体Aでは(これらの分析では、「異性体A」は図7の異性体2Aおよび2Bの両方を指す)、0vcMMAEを含む軽鎖(L0)のモル分率は1vcMMAEを含む重鎖(H1)のモル分率と等しいが、E2異性体Cでは、0および1vcMMAEを含む軽鎖のモル分率および0および1vcMMAEを含む重鎖のモル分率はすべて等しい。1vcMMAEを含む軽鎖(L1)および0vcMMAEを含む重鎖(H0)のみが異性体Cのパーセンテージに寄与するため、異性体Cのパーセントは、以下のように表される:
%C=2L1+2H0 (1)。
【0200】
異性体Aのパーセントは100−%Cであると仮定される。2または3vcMMAEを含む少量(合計3%未満)の重鎖がPLRP−S HPLCデータに見られることが多い。これらはおそらく、E2サンプル中のE4汚染またはE6汚染によるものである。E2異性体AおよびCのパーセントを計算するために、L0、L1、H1、およびH2のモルパーセントの合計を100%に設定した。
【0201】
同様に、E6異性体A(これらの分析では「異性体A」は図7の異性体6Aおよび6Bの両方を指す)では、H2のモル分率はL1のモル分率に等しいが、E6異性体Cでは、L0、L1、H2、およびH3のモル分率は等しい。L0およびH3のみが異性体Cのパーセンテージに寄与するため、異性体Cのパーセントは以下のように表すことができる:
%C=2L0+2H3 (2)。
異性体Aのパーセントは100−%Cであると仮定される。E2と同様に、L0、L1、H2、およびH3のモルパーセントの合計を100%に設定した。
【0202】
E4異性体のパーセンテージは、独自の溶液がないため、PLRP−S HPLCデータのみからでは得ることができない。他の2つの異性体についてPLRPデータを使用して解き得る前に、少なくとも1つの異性体を確定する必要がある。HHL、HH、およびHLのモルパーセントは、以下の分子量を使用してバイオアナライザから決定した:124,720.8(HHL)、100,992.6(HH)、および74,224.5(HL)g/mol。バイオアナライザは機器の読み出しのために結合染料の蛍光を使用し、そしてHHL、HH、およびHLが単位分子量当たり染料を等しく結合することが仮定されるが、この仮定が真実である可能性は低い。この仮定から生じる誤差を最小限にするために、以下のようにHHL、HH、およびHLピーク面積を使用してバイオアナライザデータから異性体E4Aのみを計算した(重鎖−重鎖ジスルフィドが開裂される場合に、各抗体は2HLを生成するため、HLピーク面積を2で割る):
【0203】
【化5】
次にPLRP−S HPLCデータを使用して、E4異性体EおよびFの残りの寄与について、以下の式を使用して解く:
%E=H1+L1−0.5%A (4)
%F=H2+LO−0.5%A (5)。
【0204】
E4異性体Aは、L0、L1、H1、およびH2の集団に等しく寄与し、異性体AのH1およびL1による寄与(その全体の寄与の半分)をH1およびL1の観察された総量から引いて、異性体Eの存在に起因するはずであるH1およびL1の残りの量を得る必要がある。異性体Aに対するH2およびL0の寄与に関する同様の引算は、異性体Fによって存在する量を与える。E2およびE6と同様に、L0、L1、H1、およびH2のモルパーセントの合計は100%に設定した。
【0205】
(タンパク質の部分付加のための方法についての実施例7)
部分薬物付加ADCを調製するために、2つの異なる方法を使用した。第1に、限定量のDTTまたはTCEPによるcAC10の部分還元は、8抗体システイン未満で生じた。DTTおよびTCEP約3.25および2.75当量はそれぞれ、2個の鎖間ジスルフィド結合を開裂させて、抗体当たり平均4cAC10システインを生じる(0、2、4、6、および8抗体−システインの混合物)。還元剤の量は経験的に決定できる:cBR96は4抗体システインを生じるためにDTTまたはTCEP 2.1当量のみを必要とするが、マウスIgG1抗体は、還元に対する極度の耐性のためにしばしば使用可能である(データを示さず)。DTTではなくTCEPを使用する利点は、ホスフィンはマレイミドとごくわずかしか反応していないので、vcMMAEを添加する前に残りの還元剤を除去する必要がないことである。過剰なDTTは容易にvcMMAEと反応して、薬物に関して抗体−システインと競合する。抗体還元の後、わずかにモル過剰のvcMMAEによる抗体システインの処理(システイン当たり1.1モル当量)は、抗体当たり4MMAEの平均薬物付加されたcAC10を生じる(E4ミックス)。
【0206】
あるいはcAC10は、10mM DTTによって完全に還元して、次にDTNBによって部分的に再酸化することができる。この再酸化プロセスは非常に効率的であり、8個の抗体システインを4個まで再酸化するために、DTNB 2.0当量を必要とする。この再酸化抗体のシステインなどのチオールによる処理は、結合されたチオニトロ安息香酸を遊離させず、このことは、再酸化システインが混合TNB−システインジスルフィドではなく、むしろ抗体ジスルフィドの形であることを示唆している。以下に述べる分析方法も、抗体ジスルフィドの存在を示している。残りの抗体システインは、上述のようにvcMMAEに結合体化してE4ミックスを生じ得る。
【0207】
薬物付加種の異性体集団を決定するために、E2、E4、およびE6を分離および単離して、純E2、純E4、および純E6を得る。図13Aは、DTT部分還元によって生成されたE4ミックスの疎水性相互作用(HIC)HPLCトレースを示す。偶数の薬物が付加された種はすべて相互に分離可能であり得、奇数の薬物が付加された種の少量が偶数種の間の谷間に見られる。これらの種の薬物付加は、ピークのUVスペクトルの検査によって割当てられ得る。薬物リンカー内のPABA基は、248nm付近に最大吸収を有するが、抗体は同じ波長に最小吸収を有する。248および280nmにおける薬物および抗体吸光係数を使用すると、抗体当たりの薬物数は、開始ADC混合物および観察された各ピークに対して割当てられ得る(Hamblett et al.(2004),Clin Cancer Res 10:7063−70)。
【0208】
表1は、DTT部分還元、TCEP部分還元、および部分DTNB再酸化によって調製された偶数薬物付加種のパーセンテージを示す。DTNB部分還元方法は、部分還元方法(DTTでは30%、TCEPでは33%)よりもわずかに高いE4のパーセンテージ(38%)を生じる。これは主にE6およびE8を犠牲にして得られるものであり、E6およびE8はDTT部分還元では約34%、TCEP部分還元では31%に達するが、DTNB部分再酸化ではわずかに24%である。奇数の薬物が付加された種は表に示しておらず、全物質の7〜10%に相当する。
【0209】
(表1.E4混合物の組成パーセントa)
【0210】
【表1】
aHIC−HPLCクロマトグラムは、組成パーセントに統合した。値は4(DTT部分還元)、3(TCEP部分還元)、または6(DTNB部分再酸化)の独立バッチについての±標準偏差である。奇数種からの寄与分は示されていないため、合計は100%未満となっている。
【0211】
このHIC−HPLC方法は、2、3ミリグラムの純E2、純E4、および純E6を単離するために使用できる。あるいは段階的勾配を使用する分取HICは、図13B、C、Dに示すように、数百ミリグラムの純E2、純E4、および純E6を単離するために使用できる。これらの物質の純度は、薬物付加レベルに関して少なくとも95%である。
【0212】
抗体上での薬物の分布を決定するために、これらの精製物質は、2つの分析方法を受け供された(実施例6を参照)。第1に、PLRP−Sカラムを用いた還元および変性HPLC使用して、抗体鎖当たりの薬物数を決定した。過剰なDTTを用いたADCの予備処理は、残存する鎖間ジスルフィドを切断して、0または1個の薬物を含む軽鎖(L0およびL1)の、0、1、2、または3個の薬物を含む重鎖(H0、H1、H2、およびH3)からの分離を可能にする(図4)。第2に、非還元および変性キャピラリー電気泳動は、残存鎖間ジスルフィドがインタクトのままでの抗体鎖の分離を可能にし、6つの潜在的な種:L、H、HL、HH、HHL、およびHHLLを生じさせる(図15)。
【0213】
PLRP−S HPLCおよびキャピラリー電気泳動によって観察された種の定量化は、異性体集団への分類を可能にする。図1および7は、抗体フラグメントおよび異性体それぞれに結合された薬物の数を示す。E2およびE6の異性体集団は、各異性体が独自のパターンを生じるため、PLRP−S HPLC単独で、またはキャピラリー電気泳動単独で容易に決定できる。たとえば変性および還元条件下では、異性体E2CのみがL1およびH0を生じるのに対して、E2AのみがL0およびH1を生じ、変性および非還元条件では、異性体E2AがHHLLを生じるのに対して、E2CはLおよびHHLを生じる。E4では、PLRP−S HPLCもキャピラリー電気泳動も単独では異性体集団を計算するのに十分ではないため、組成を決定するために2つの方法を組合せて使用する必要がある。表2は、これらの各異性体の組成パーセントを示す。PLRP−S HPLCデータを独占的に使用して、式1および2を用いてE2およびE6異性体の異性体組成を計算した(実施例6を参照)。キャピラリー電気泳動を使用して、式3を用いてE4Aの量を計算し、PLRP−S HPLCを使用して、式4および5を用いて、E4Aの寄与を引いて、E4BおよびE4Cの量を計算した(実施例6を参照)。
【0214】
(表2.精製されたE2、E4およびE6の異性体集団の組成)
【0215】
【表2】
a式1を用いてPLRP−S HPLCデータから決定。b式3を用いてバイオアナライザデータから決定。c式4および5を用いてPLRP−S HPLCデータから決定。d式2を用いてPLRP−S HPLCデータから決定。
【0216】
表2のデータは、産生方法が薬物の配置(location)に著しく影響を及ぼすということから、特筆すべきものであり、抗体ジスルフィドを選択的に還元できることを示唆している。部分DTT還元は、重鎖−軽鎖ジスルフィドの1つの還元から生じる92%の異性体E2Cと、両方の重鎖−軽鎖ジスルフィドの還元から生じる59%の異性体E4Eと、両方の重鎖−重鎖ジスルフィドおよび1つの重鎖−軽鎖ジスルフィドの還元から生じる98%の異性体E6Cを生じる。1つの重鎖−重鎖ジスルフィドが還元された異性体は、極めて少量である。これに対して、部分DTNB再酸化は、E2およびE4異性体については77%の異性体E2Aおよび75%のE4A(1つの重鎖−重鎖ジスルフィドがインタクトである)という、ほぼ反対の結果、そしてE6については96%のE6Cという同じ結果を生じる。AETを用いた酸性還元は、DTT部分還元と非常に類似している異性体集団を生じ、重鎖−軽鎖ジスルフィドの開裂に有利に働く。
【0217】
DTT部分還元の異性体分布の反応動態を表3に示す。cAC10を、3.0当量のDTTによって還元して、サンプルを定期的に取り出し、vcMMAEによってアルキル化した。純E2、純E4、および純E6をHIC−HPLCによって得て、異性体分布をPLRP−S HPLCおよびバイオアナライザによって決定した。異性体組成は、還元時間10〜120分および抗体当たり薬物1.3〜3.9個の全薬物付加の範囲での実験の過程にわたって同一である。これらの結果は、限定量のDTTによってcAC10を2時間還元することによって調製された、表2に示すDTT部分還元の異性体集団が、還元反応の全過程にわたる異性体集団を代表することを示している。
【0218】
(表3.DTT部分還元の異性体分布の反応動態)
【0219】
【表3】
a還元時間。いったん還元された場合、すべての抗体をvcMMAEによって同じ時間処理した。bHIC−HPLCによって決定。c式1を用いてPLRP−S HPLCデータから決定。d式3を用いてバイオアナライザデータから決定。e式4および5を用いてPLRP−S HPLCデータから決定。f式2を用いてPLRP−S HPLCデータから決定。N/D、決定されず。この時点では、ごく少量のE6が産生され、この物質は異性体集団を決定するのには不十分であった。
【0220】
(表4.cAC10−vcMMAEのインビトロ結合および細胞毒性)
【0221】
【表4】
a4〜7回の独立した測定から決定された、抗体成分のμg/mLでのKarpas299への結合±標準偏差、N/D、決定されず。
b3回の独立した測定から決定された抗体成分のng/mLでのインビトロ細胞毒性±標準偏差。N/D、決定されず。cAC10は単独でKarpas299に対して有効性が乏しい。
【0222】
表4に、ADCについて複数の薬物付加レベルで実施したインビトロ結合および細胞毒性実験の結果を挙げる。E2およびE4ミックスは、DTT部分還元およびDTNB部分再酸化からの純E2および純E4と同様に試験した。抗体当たり8個の薬物を持つ完全付加結合体は最も細胞毒性であり、CD30陽性Karpas299細胞株に対して2.7ng/mLのIC50値を有する(抗体の重量に基づいて計算)。抗体当たり4個の薬物を持つADCはわずかに細胞毒性が低く、IC50値は3.4ng/mLと5.0ng/mLとの間であり、抗体当たり2個の薬物を持つADCは最も細胞毒性が低く、IC50値は11.4〜13.8ng/mLである。ADCを産生するために使用した化学作用は、細胞毒性に著しい相違を示さず、混合物とHIC精製ADCとの間にも著しい相違がなかった。インビトロ細胞毒性は、薬物の全用量のみに依存すると考えられる。CD30陽性細胞への結合はE0、E2、およびE4では非常に似ており、E8はわずかに損なわれ、結合体化が抗原結合を妨害しないことを示す。ADCのインビトロ細胞毒性(抗体成分の測定)は、予想された傾向:薬物の数が多くなればなるほど、IC50値が低くなることを示している。実験誤差の範囲内で、薬物の配置はインビトロ細胞毒性に影響を及ぼさないとみられる。
【0223】
(モノクローナル抗体薬物結合体の抗腫瘍活性に対する薬物付加効果についての実施例8)
細胞および試薬。CD30陽性ALCL株Karpas−299をDeutsche Sammlung von Mikroorganism und Zellkulturen GmbH(Braunschweig,ドイツ)より入手した。異種移植片増殖に適したHD株L540の誘導体である、L540cyは、Dr.Harald Stein(Institut fur Pathologie,Univ.Veinikum Benjamin Franklin,Hindenburgdamm30,12200 ベルリン、ドイツ)から快く提供していただいた。細胞株は、10%ウシ胎仔血清を添加したRPMI−1640培地(Life Technologies Inc.,ゲイサーズバーグ、メリーランド州)で培養した。
【0224】
cAC10−Val−Cit−MMAE ADCの構築および精製。簡潔には、抗体当たり8個の薬物を持つcAC10をジチオトレイトール(DTT)と37℃にて30分混合することによって産生して、1mMジエチレントリアミン五酢酸(DTPA)を含有するPBSを用いたSephadex G−25樹脂を介した溶出によって緩衝液を交換した。1mM DTPA(PBS/D)を含有するPBSを還元mAbに添加した(最終濃度2.5mg/mL)。Val−Cit−MMAEと呼ばれる9.5モル過剰のマレイミドカプロイル−Val−Cit−MMAEを還元抗体に4℃にて1時間にわたって添加し、20倍過剰のシステインを添加することによって結合体化反応をクエンチした。反応混合物を遠心分離限外濾過によって濃縮し、4℃にてPBS中で平衡化したSephadex G25を介して緩衝液を交換した。次に結合体を0.2ミクロンフィルタで滅菌条件下にて濾過した。
【0225】
抗体当たり2および4MMAE分子を持つcAC10 ADCの産生は、部分還元と、それに続くVal−Cit−MMAEによる反応を包含した。抗体cAC10(10mg/mL)は、最終DTT:mAbモル比3.0までのDTTの添加と、それに続く37℃での約2時間のインキュベーションにより部分還元された。次に還元反応を約10℃まで冷却して、還元cAC10を過剰DTTからダイアフィルトレーションによって精製した。ダイアフィルトレーション後に、部分還元cAC10のチオール濃度をDTNB(Ellmanの)アッセイによって決定した;この方法では、平均約2個のジスルフィド結合が還元され、それゆえ約4の還元Cys:mAbを露出させた。還元Cysすべてを結合体化するために、最終Val−Cit−MMAE:還元Cysモル比 約1.15までVal−Cit−cMMAEを添加した。15%v/v DMSOの存在下で結合反応を実施して、約10℃にて約30分間進行させた。結合体化反応の後、過剰な遊離Cys(Val−Cit−MMAE 1モル当たり2モルのCys)を添加して、未反応Val−Cit−MMAEをクエンチして、Cys−Val−Cit−MMAE付加体を産生させた。Cysクエンチング反応を約10℃にて約30分間進行させた。Cysクエンチング反応混合物を精製し、ダイアフィルトレーションにより緩衝液をPBSに交換して、部分付加cAC10−Val−Cit−MMAEを得た。
【0226】
分取HIC分画化。すべてのクロマトグラフ工程を室温にて実施した。1.6x25cmカラム(約50mL)にToyopearl Phenyl−650M HIC樹脂(Tosoh Bioscience,モンゴメリービル、ペンシルベニア州)を充填して、>5カラム容積の緩衝液A(50mMリン酸ナトリウム、2M NaCl、pH7.0)により流速15mL/分にて平衡化した。カラムへ充填するためのサンプルを調製するために、部分付加cAC10−vcMMAE 39mL(12.9mg/mL)を緩衝液A’39mL(50mMリン酸ナトリウム、4M NaCl、pH7.0)とブレンドした。サンプルをカラムに10mL/分で充填した;後の工程は、すべて10mL/分の流速にて実施した。サンプル充填の後に、A280ベースラインに到達するまでカラムを緩衝液Aで洗浄した。cAC10−E2は、65%緩衝液A/35%緩衝液Bからなる段階的勾配を用いて溶出および収集した(80%v/v 50mMリン酸ナトリウム、pH7.0、20%v/vアセトニトリル)。ベースラインに再度到達した後、cAC10−E4を30%緩衝液A/70%緩衝液Bからなる段階的勾配によって溶出および収集した。cAC10−E2およびcAC10−E4ピークを、どちらもその各ピーク高さの約20%まで収集した。目的の画分は、30kDaの分子量カットオフを用いたUltrafree−15遠心分離フィルタデバイス(Millipore,ビルリカ、マサチューセッツ州)を使用して、緩衝液をPBSに交換した。
【0227】
結合体の分析。結合体の分析は、エーテル−5PWカラム(Tosoh Bioscience,モンゴメリービル、ペンシルベニア州)を使用してHIC−HPLCによって実施した。方法は、50分間の100%緩衝液Aから100%緩衝液C(80%v/v 50mMリン酸ナトリウム、pH7.0、10%v/vアセトニトリル、10%v/vイソプロパノール)までの線形勾配により構成されていた。流速は1mL/分に設定され、温度は30℃に設定され、検出は248nmおよび280nmの両方にて追跡した。未改変(modify)cAC10およびcAC10−E8ピークの同定は、cAC10およびcAC10−E8標準の注入により確認した。抗体および薬物は別個の吸収極大(それぞれλmax=280nmおよび248nm)を有するため、ピークスペクトルを重ねることによって、抗体当たり2、4、6個の薬物を持つcAC10結合体に相当するピークを同定することが可能であった。
【0228】
cAC10−Val−Cit−MMAE ADCのインビトロ特徴づけ。結合体化または薬物の存在が抗原結合に影響を及ぼすかどうかを判定するために、ADCに対する競合結合を実施した。mAbおよびADCの飽和結合を比較するために、5x105Karpas−299細胞を、氷上の染色培地中で30分間、Alexa Fluor488(Molecular Probes,ユージーン、オレゴン州)によって標識された1μg/mL cAC10の存在下で、cAC10、cAC10−E2、cAC10−E4、またはcAC10−E8の連続希釈と混合し、氷冷染色培地で2回洗浄した。標識細胞をFusionマイクロプレートリーダー(Perkin−Elmer,ボストン、マサチューセッツ州)によって検査した。サンプルデータをバックグラウンド補正して、1μg/mL cAC10−Alexa Fluor(登録商標)488単独で染色された細胞の蛍光で割ったサンプル蛍光によって算出された最大蛍光パーセントとして報告した
DNA合成を測定することによってcAC10結合体の増殖阻害活性を決定した。結合体をCD30+Karpas−299またはL540cy細胞またはCD30−WSU−NHL細胞によってインキュベートした。cAC10またはcAC10を用いた92時間のインキュベーションの後、ADC細胞を0.5μCi/ウェルの[3H]−チミジンによって37℃にて4時間に渡って標識した。ハーベスタを使用して細胞をフィルタ上に収集し、シンチレーション流体と混合して、放射能をTopcountシンチレーションカウンタ(Packard Instruments,メリデン、コネチカット州)を用いて測定した。各分子について未処理のパーセントを濃度に対してプロットして、IC50を決定した(DNA合成の50%阻害を与えるmAb濃度として定義される)。
【0229】
ヒトALCLの異種移植モデル。ALCLの皮下疾患モデルを確立するために、5x106Karpas−299細胞をCB−17 SCIDマウス(Harlan,インディアナポリス、インディアナ州)の右側腹に移植した。各群6〜10匹の動物の腫瘍サイズが平均して約50〜100mm3になったときに、ADCを用いた療法を開始した。処置は、1回の注射、または注射4回について4日ごとに注射1回のスケジュール(q4dx4)を使用した複数回の静脈内注射のどちらかで構成されていた。腫瘍体積は、式(長さx幅2)/2を使用して計算した。触診できないほどにサイズが縮小した腫瘍は、完全後退(CR)として定義した。10腫瘍倍増時間以上続いた完全後退を治癒として定義した。対照グループの腫瘍のサイズが750〜1000mm3に達したときに腫瘍増殖阻害(TGI)を以下のように計算した:
【0230】
【化6】
最大耐容用量。3匹のBALB/cマウス(Harlan,インディアナポリス、インディアナ州)の群に、cAC10−E8 30〜60mg/kg、cAC10−E4 60〜120mg/kg、またはcAC10−vcMMAE2 200〜250mg/kgを尾静脈に注射して、単回投与最大許容用量(MTD)を決定した。マウスを毎日14日間監視し、重量および臨床観察の両方を記録した。著しい苦痛の徴候を発現したマウスをACUCガイドラインに従って殺処分した。最大許容用量は、いずれの動物においても注射の2週間以内に深刻で明白な毒性または20パーセントを超える体重減少を引き起こさない最大用量として定義した。
【0231】
薬物動態学。cAC10、cAC10−E2、cAC10−E4、およびcAC10−E8の薬物動態学をSCIDマウスで評価した。SCIDマウス(n=3)に試験物質10mg/kg(抗体成分に基づいて)を尾静脈注射によって投与した。血液サンプルは、伏在静脈への注射後1時間、4時間、1日、2日、4日、7日、14日、21日、28日、35日、42日、および49日に各マウスから収集した。ヘパリンをコーティングしたチューブに血液を収集して、続いて遠心分離(14,000xg、3分間)にかけて血漿を単離した。cAC10およびADCの血漿濃度をELISAによって測定した。
【0232】
簡潔には、ELISAは以下の工程より構成されていた:プレートコート、ブロック、サンプル結合、二次mAb、TMB、および酸停止。各工程後に、ウェルを洗浄緩衝液(PBS、0.05% Tween−20、pH=7.4)で3回洗浄した。プレートコート工程では、96ウェルプレートへ炭酸塩緩衝液(0.1M炭酸塩/重炭酸塩、pH=9.6)中の2μg/mLの抗cAC10mAbを4℃にて一晩コーティングした。プレートコートの後、ブロッキング緩衝液(PBS、1% BSA、0.05% Tween−20)を添加して、室温にて1時間インキュベートした。次に標準または希釈血漿サンプル100μLを3つのウェルにて室温で1時間添加した。二次工程は、1時間インキュベートされたマウス抗ヒトIgG−HRP結合体(Southern Biotech,バーミンガム、アラバマ)より構成されていた。次に3,3’,5,5’−テトラメチルベンジジン(Sigma,セントルイス、ミズーリ州)100μLを各ウェルに添加して、発色時に1N硫酸100μLによって反応を停止させた。VMax Kinetic Microplateリーダー(Molecular Devices,サニーベール、カリフォルニア州)によって、光学密度を450nmにて、ブランクを630nmにて測定した。非コンパートメント(non−compartment)薬物動態パラメータは、WinNonlin(Pharsight,マウンテンビュー、カリフォルニア州)を用いて計算した。
【0233】
インビトロ特徴づけ。MMAEのcAC10への結合体化がADCのCD30結合能力を阻害したかどうかを評価するために、競合結合実験を実施した。CD30+Karpas−299細胞を、未標識抗体、cAC10−E2、cAC10−E4、またはcAC10−E8の連続希釈と結合させた1μg/mL 蛍光標識cAC10を用いてインキュベートした。図10に示すように、ADC改変体は、それぞれ未標識cAC10と同等に、の蛍光標識cAC10と効果的に競合した。それゆえMMAEとの結合体化は、抗原結合を減少させなかった。
【0234】
ADCのインビトロ細胞毒性活性を、CD30+Karpas−299細胞およびL540cy細胞およびCD30−WSU−NHL細胞を用いた[3H]−TdR包含アッセイによって評価した。cAC10−E8は、1.0ng/mLのIC50が、Karpas−299細胞に対する著しい活性を示した(図11a)。mAb当たり4個のMMAE分子(cAC10−E4)に対する薬物の量を半分に減少させると、IC50が2.9ng/mLへと上昇した。薬物付加を再度半分にすると、cAC10−E2のIC50が6.2ng/mLにさらに上昇した。HD株L540cyに対して、ADCは同様の傾向を有し、cAC10−E8、cAC10−E4、およびcAC10−E2のIC50値はそれぞれ1.4ng/mL、4.5ng/mL、および9.2ng/mLであった(図11b)。ADCの選択性を、CD30−WSU−NHL細胞株を用いて評価した。この細胞株は、すべてのcAC10−ADCに対して不感受性であり、IC50値>1000ng/mLであった(データは示さず)。
【0235】
ヒトALCLの異種移植モデル。インビボ抗腫瘍活性に対する薬物付加の効果をKarpas−299皮下異種移植モデルで評価した。療法は、腫瘍体積が50〜100mm3に達したときに開始し、4日ごとに合計4回の注射を投与した(q4dx4)。このスケジュールを使用した場合、1mg/kgのcAC10−E8は、100% CRを生じさせ、同じ用量でcAC10−E4が100% CRを得たことが以前に見出された(データは示さず)。ADCの活性を比較する目的で、cAC10−E4およびcAC10−E8についてより少ない用量を使用した。皮下Karpas−299異種移植片を保有するマウス集団をcAC10−E2(0.5mg/kg/用量または1mg/kg/用量)、cAC10−E4(0.25または0.5mg/kg/用量)、またはcAC10−E8(0.25または0.5mg/kg/用量)の複数回の投与で処置した。表5は、有効性結果のまとめを示す。
【0236】
(表5)
【0237】
【表5】
表5:皮下Karpas−299異種移植モデルにおけるcAC10−E2、cAC10−E4、cAC10−E4混合物、およびcAC10−E8の抗腫瘍活性。腫瘍体積が50〜100mm3に達したら、動物をADCで処置した。投与は、1回(x1)または4日おきに4回(q4dx4)与えた。完全後退、治癒および腫瘍増殖阻害(TGI)の数を報告する。治癒した1匹のマウスが腫瘍塊の徴候なく第72日に死亡して発見された。
【0238】
cAC10−E8はMMAE量の2倍を同じmAb用量のcAC10−E4として有していたが、それらは両方の用量レベルで等しく有効であった(図12A)。0.5mg/Kg/用量にて、cAC10−E4によって処置した10匹の動物のうち、5匹が完全後退(CR)を達成し、cAC10−E8は10匹中6匹でCRを誘起した。未処置腫瘍は、移植19日後に986mm3の平均腫瘍体積に達した。cAC10−E4の腫瘍増殖阻害は、cAC10−E8の90%に対して91%であった。0.25mg/kg/用量にて、これらのADCの両方が未処置対照動物と比較して腫瘍増殖で同様の遅延を誘発したが、完全後退はなかった。0.5mg/kg/用量のcAC10−E4および0.25mg/kgのcAC10−E8として同じ量のMMAEを含有する用量である、1.0mg/kg/用量でのcAC10−E2は、10/10の治癒を誘起した。0.5mg/kg/用量のcAC10−E2による腫瘍増殖に対する効果は、68%のTGIを生じる0.25mg/kg/用量のcAC10−E4およびcAC10−E8で見られた効果に匹敵していた。薬物MMAEとcAC10との物理的混合物は、0.5mg/kg用量のcAC10−E8と同等であり、未処置と比較して腫瘍増殖のわずかな遅延のみを引き起こし、薬物の抗体への結合が抗腫瘍活性を達成するために重要であることを強調した。
【0239】
次にこの同じモデルでのcAC10−E2、cAC10−E4、およびcAC10−E8の単回投与処置を1.0mg/kgにて比較した(図12B)。cAC10−E8で処置した動物6匹のうち5匹が治癒を達成した。cAC10−E4で処置した動物6匹のうち、すべてが完全後退を達成して、5匹が試験終了の108日までに治癒し、動物1匹が腫瘍塊の徴候なく第72日に死亡して発見された。1.0mg/kgのcAC10−E2は、cAC10−E4の半分のMMAEを含有するが、マウス6匹のうち4匹がCRを達成した。cAC10 1mg/kgと、cAC10−E8 1mg/kgに含有されている薬物の量に相当する遊離MMAE 0.037mg/kgからなる対照グループは、未処置マウスと比較して腫瘍増殖に対する影響をほとんど有していなかった。
【0240】
部分付加ADCの初期結合は、0〜8個の薬剤/mAbを含有する種の混合物を生じた。この混合物(cAC10−E4混合物)の活性を評価するために、cAC10−E4混合物の単回投与抗腫瘍活性を1.0mg/kgの精製cAC10−E4と比較した。前の単回投与実験と同様に、cAC10−E4で処置したマウス10匹中9匹がCRを達成した。平均モル比4.0のcAC10−E4混合物で処置したマウス10匹中8匹で完全後退が生じた。部分付加cAC10−E4混合物は、種々に薬物付加されたADCの集団を含有しているが、精製cAC10−E4と同等の抗腫瘍活性を示した。
【0241】
最大許容用量および治療濃度域。cAC10−E2、cAC10−E4、およびcAC10−E8の単回投与許容性を1群あたり3匹のBALB/cマウスで評価した。最大許容用量(MTD)は、いずれの動物においても20パーセントを超える体重減少あるいは重篤な苦痛の徴候または明白な毒性を誘発しない最高用量として定義した。cAC10−E8では、30〜60mg/kgから10mg/kg間隔でマウスに投薬した。50mg/kgの用量では、マウスは注射6日後に14%の最大体重減少を有し、その後、体重減少は回復した。1匹の動物において、60mg/kgの用量は注射6日後に、23%の体重減少を誘発した。100mg/kgのcAC10−E4では、マウスは約10%の最大体重減少に達した。120mg/kgのcAC10−E4にて、動物1匹が著しい苦痛の徴候および17%の体重減少を示し、この動物を安楽死させた。試験を行った最高用量の250mg/kgまでの用量のcAC10−E2で処置したマウスは、注射6日後に10.5%の最大体重減少を経験し、苦痛の徴候はなかった。我々の観察に基づいて、cAC10−E2のMTDは少なくとも250mg/kgであり、cAC10−E4は100mg/kgであり、cAC10−E8は50mg/kgであった。治療指数は、単回投与MTDの複数回投与有効用量に対する比として定義され、cAC10−E8では100、cAC10−E4では200、cAC10−E2では少なくとも250を得た。
【0242】
薬物動態学。SCIDマウスにcAC10、cAC10−E2、cAC10−E4、およびcAC10−E8を投与して、薬物付加が薬物動態学にどのような影響を及ぼすかを判定した。表6は、非コンパートメント分析によって確立された薬物動態パラメータを示す。
【0243】
【表6】
表6.10mg/kgの用量でのSCIDマウスにおけるcAC10およびcAC10 ADCの薬物動態パラメータ。半減期(t1/2)、曲線下面積(AUC)、クリアランス(CL)、分布体積(Vz)、および注射から第14日までの曲線下面積(AUCt(0−14d))は、非コンパートメント分析を使用して計算した。
【0244】
cAC10、cAC10−E2、cAC10−E4、およびcAC10−E8の時間−濃度曲線は、バイエクスポネンシャルな下降に従った。末期半減期はそれぞれ16.7日、16.9日、14.0日および14.7日であり、それゆえ薬物付加とは直接相関していなかった。しかしながらAUCによって決定されたADCの暴露は薬物付加が減少するにつれ上昇し、未改変cAC10の2638μg日/mLからcAC10−E8の520μg日/mLの範囲であった。反対に、クリアランス値はcAC10の3.8mL/日/kgからcAC10−E2、cAC10−E4、およびcAC10−E8それぞれの4.4、6.0および19.2mL/日/kgまで上昇した。同様に、分布体積は薬物付加に直接相関することが見出された。
【0245】
(インビボ有効性のさらなる試験についての実施例9)
Karpas299 CD30陽性細胞株を使用してインビボ有効性実験を実施し、表7に示す。皮下Karpas−299腫瘍をC.B.−17 SCIDマウスで増殖させ、腫瘍が約100mm3(長さx幅2)に達したときに、試験品を投与した。動物を5〜10匹の群に分けて、各群にADCを静脈内注射した。用量は2倍連続希釈(0.5、1.0、2.0mg/Kg)で作製して、測定できないサイズまで退縮した腫瘍を完全寛解として定義した。数回(3〜8回、動物4匹の一群であるE6Pを除いて、用量当たり動物5〜15匹)の実験で≧80%の完全寛解を生じた用量を有効用量として割当てた。試験したすべてのADCで、注射した薬物成分の量が薬物付加によって変化したという事実にかかわらず、有効用量は1mg/Kgであった。実験(2倍連続希釈)の精度範囲内で、薬物の異性体分布は有効性に影響を及ぼさなかった。
【0246】
ADCの許容性を評価するために、BALB/cマウス(C.B.−17SCIDの親株)にADCを投与した。動物の体重を測定して、14日の期間に渡って臨床観察を記録した。MTDは、>20%の体重減少または苦痛の徴候を生じなかったBALB/cマウスに投与した最高単回用量とした。用量は、E8では、40、50および60mg/Kgであり、用量は、E4では、80、100および120mg/Kgであり、用量は、E2では、200および250mg/Kgである。以前に観察されたように(実施例8を参照)、絶対薬物付加レベルはMTDに影響を及ぼし、より高い薬物付加レベルはより低いMTD値を有する。DTNB部分再酸化によって産生された純E4では、MTDは、DTT部分還元によって産生された純E4よりもわずかに高かった(120対100mg/Kg)。
【0247】
(表7.CD30+Karpas299細胞に対するcAC10−vcMMAEのマウスインビボ有効性およびMTD)
【0248】
【表7】
aインビボ用量は、体重Kg当たりの抗体成分mgに基づいていた。
【0249】
本明細書で言及した、または本明細書に援用された特許または特許出願に対して、明示または黙示を問わず実施権は与えられていない。上の議論は説明的、例証的、かつ例示的であり、添付された請求項のいずれかによって定義された範囲を限定するものとして解釈すべきではない。
【0250】
特許出願、特許および科学の刊行物を含む種々の参考文献が本明細書で引用され、そのそれぞれの開示は、その全体が本明細書に参考として援用される。
【図面の簡単な説明】
【0251】
【図1】図1は、概略図の抗体の「E4」異性体(抗体当たり4個の薬物が結合された異性体)を示す。鎖間ジスルフィド結合は、抗体の重鎖−重鎖間および重鎖−軽鎖間の実線として示されている。薬物および抗体へのその結合箇所を円として示す。非還元(「Non−red」)および還元(「Red」)条件の下で産生されたフラグメントを各異性体の下に示す(カッコ内はフラグメント当たりの薬物の数)。
【図2】図2は、方法2a:部分DTNB再酸化、PD−10精製、およびvcMMAE結合体化の1つの局面によって調製されたvcMMAE−cAC10結合体の、疎水性相互作用クロマトグラフィー(HIC)分析におけるクロマトグラムの溶出プロフィールを示す。「E0」、「E2」、「E4」、「E6」および「E8」は、抗体当たり0、2、4、6および8個のMMAF分子がそれぞれ結合したcAC10抗体の異性体を指す。
【図3】図3は、方法2b:ワンポットDTNB再酸化およびvcMMAE結合体化の別の局面のHICクロマトグラムを示す。「E0」、「E2」、「E4」、「E6」および「E8」は、抗体当たり0、2、4、6および8個のMMAF分子がそれぞれ結合したcAC10抗体の異性体を指す。純粋なE4は、約34〜38分より収集した(矢印で示す)。
【図4】図4は、方法1、2a、および2bからの一様な薬物付加種の抗体−薬物結合体の組成パーセントの棒グラフでの比較を示す。各種では、抗体−薬物結合体をDTT部分還元(左棒)、DTNB再酸化(中央棒)またはワンポットDTNB再酸化(右棒)結合体化方法によって調製した。
【図5】図5は、図3(方法2b)でHICクロマトグラムに示したように収集したE4材料のバイオアナライザによるトレースを示す。「L」は遊離軽鎖を示す。「H」は遊離重鎖を示す。「HL」は結合された重鎖−軽鎖を示す。「HH」は結合された重鎖−重鎖を示す。「HHL」は結合された重鎖−重鎖および重鎖−軽鎖を示す。
【図6】図6は、図3(方法2b)のHICクロマトグラムから収集した材料のPLRP分析を示す。「L0」および「L1」はそれぞれ、薬物分子が結合していない、または薬物分子が1個結合した軽鎖を示す。「H0」、「H1」「H2」および「H3」は、0、1、2または3個の薬物分子が結合した重鎖を示す。
【図7】図7は、鎖間ジスルフィド結合の完全または部分還元と、それに続く薬物の結合体化によって得られた主な結合体種の概略表現を示す。完全還元および結合は主に、抗体当たり8つの薬物を持つ完全付加種を生成するのに対して、部分還元および結合は、図示した種すべての産生を引き起こすことができる。0および8薬物付加種にはそれぞれ異性体が1つのみあるが、2、4および6薬物付加種は3つ、4つおよび3つの異性体をそれぞれ含有する(図1を参照して、種4Aが種4Cの鏡像であり、種4Bが種4Dの鏡像であることに注目する。図7では、種4Aおよび種4Cは種4Aと呼ばれ、種4Bおよび種4Dは種4Bと呼ばれる)。鎖間ジスルフィド結合は、抗体の重鎖−重鎖間または重鎖−軽鎖間の実線として示す。薬物および抗体へのその結合箇所を円として示す。
【図8】図8は、E4混合異性体(E4M)を産生するためにDTTを使用する「部分還元」結合プロセスの1つの局面のプロセスフローダイアグラムである。cAC10抗体のpHは、リン酸水素二ナトリウムによって7.5に調製し、EDTAを5mMの最終濃度まで添加する。次に抗体溶液を37℃に加熱する。抗体を部分還元するために、DTT 2.95モル当量を抗体溶液に添加して、37℃にて105分間還元する。還元後、抗体溶液を2〜8℃に冷却して、過剰なDTTを定容限外濾過/ダイアフィルトレーション(UF/DF)によって濾過し、還元された精製cAC10を得る。還元された精製cAC10のサンプルを取り、A280およびDTNB試験によって、チオール濃度、抗体濃度、およびチオール対抗体モル比を決定する。次にやや過剰の薬物−リンカーvcMMAE(通例、DMSO溶液の形で2〜15%過剰)を抗体溶液に添加して、結合反応を開始させる。結合反応を2〜8℃にて30分間進行させて、粗E4Mを得る。結合反応の終わりに、過剰のvcMMAE薬物−リンカーを、大幅に過剰のシステインと2〜8℃にて15分間反応させることによってクエンチし、クエンチされた粗E4Mを得る。緩衝液交換ならびに遊離薬物および他の小型分子種の除去は、定容量UF/DF(代表的には、6〜10透析体積)によって実施し、E4M薬物物質を得る。
【図9】図9は、E4混合異性体(E4M)を産生するための、中間精製ステップが使用されないTCEPを使用する「部分還元」結合プロセスの別の局面のプロセスフローダイアグラムである。cAC10抗体のpHは、リン酸水素二ナトリウム7.5に調製し、EDTAを5mMの最終濃度まで添加する。次に抗体溶液を37℃に加熱する。抗体を部分還元するために、TCEP 2.20モル当量を抗体溶液に添加して、37℃にて105分間還元する。還元反応のサンプルを取り、A280およびDTNB試験によって、チオール濃度、抗体濃度、およびチオール対抗体モル比を決定する。還元後、抗体溶液を2〜8℃に冷却する。次にやや過剰の薬物−リンカーvcMMAE(通例、DMSO溶液の形で2〜15%過剰)を抗体溶液に添加して、結合反応を開始させる。結合反応を2〜8℃にて20分間進行させて、粗E4Mを得る。結合反応の終わりに、過剰のvcMMAE薬物−リンカーを、大幅に過剰のN−アセチルシステインと2〜8℃にて20分間反応させることによってクエンチし、クエンチされた粗E4Mを得る。緩衝液交換ならびに遊離薬物および他の小型分子種の除去は、定容UF/DF(通例6〜10透析体積)によって実施し、E4M薬物物質を得る。
【図10】図10は、CD30+Karpas−299細胞によるcAC10結合体化抗体の内部移行のグラフを示す。細胞を蛍光標識cAC10 1g/mLおよびcAC10、cAC10−E2、cAC10−E4、またはcAC10−E8のいずれかの20μg/mL〜9ng/mLの連続希釈物と合せた。抗体による細胞のインキュベーションの後、標識細胞を染色溶媒で洗浄し、蛍光を測定した。実施例8で述べるように、正規化蛍光強度をmAb濃度に対してプロットした。
【図11】図11は、CD30+細胞によるcAC10およびcAC10結合体化抗体の内部移行のグラフを示す:A)Karpas−299およびB)L540cy細胞をcAC10ならびにcAC10 ADCのE2、E4およびE8種の連続希釈物によってインキュベートした。サンプルを用いた96時間のインキュベーションの後、[3H]−TdRを添加して、その包含を測定した。処置サンプルの放射能を未処理対照へ正規化して、濃度に対してプロットした。
【図12】図12は、皮下異種移植を持つSCIDマウスにおけるcAC10およびcAC10結合抗体のインビボ有効性を示す。図12Aは、cAC10−E2を0.5mg/kgまたは1.0mg/kgにて注射4回を4日おきに注射された、Karpas−299皮下腫瘍を持つSCIDマウスによる結果を示す。cAC10−E4およびcAC10−8は、0.25または0.5mg/kgのどちらかで、注射4回を4日おきに投薬された。図12Bは、E2、E4またはE8の1.0mg/kgでの単回投与によって処置された、Karpas−299皮下腫瘍を持つSCIDマウスの結果を示した。
【図13】図13は、(A)E4ミックス;(B)分取HICによって作成した純E2;(C)分取HICによって作成した純E4;および(D)分取HICによって作成した純E6のそれぞれの疎水性相互作用クロマトグラフィーHPLCトレースを示す。サンプルは、DTT部分還元と、それに続くMMAE結合によって作成した。クロマトグラムは、各クロマトグラムの最高ピークの高さへ正規化した。注射は、2.0M NaClおよび50mMリン酸ナトリウム、pH7 50μLと混合した、PBS中の5〜10mg/mL cAC10−vcMMAE 50μLであった。分離は30℃にて実施した。
【図14】図14は、(A)部分DTT還元と、それに続くMMAE結合によって作成したE4ミックス(上トレース);(B)DTNB部分再酸化と、それに続くMMAE結合によって作成したE4ミックス(上から2番目のトレース);(C)部分DTT還元と、それに続くMMAE結合によって作成し、分取HICによって精製した純E4(下から2番目のトレース);および(D)部分DTNB再酸化と、それに続くMMAE結合によって作成し、分取HICによって精製した純E4(下トレース)のPLRP−S HPLCトレースを示す。注射は、20mM DTTで37℃にて15分間処理した1mg/mL cAC10−vcMMAE 20μLであった。分離は80℃にて実施した。
【図15】図15は、(A)部分DTT還元と、それに続くMMAE結合によって作成したE4ミックス(上トレース);(B)DTNB部分再酸化と、それに続くMMAE結合によって作成したE4ミックス(上から2番目のトレース);(C)部分DTT還元と、それに続くMMAE結合によって作成し、分取HICによって精製した純E4(下から2番目のトレース);および(D)部分DTNB再酸化と、それに続くMMAE結合によって作成し、分取HICによって精製した純E4(下トレース)のバイオアナライザ(キャピラリー電気泳動)トレースを示す。サンプルは、メーカーが指示した通りに非還元条件下で調製した。
【技術分野】
【0001】
(継続)
本願は、2004年3月2日に出願された米国仮特許出願第60/549,476号の優先権を主張し、この開示はその全体が参考として本明細書中に援用される。
【0002】
本発明は、例えばタンパク質−薬物結合体の特定異性体を生じる薬物との少なくとも1個の結合箇所を有する修飾タンパク質と、特定異性体を生じるそのような結合の方法とに関する。本発明はさらに、細胞毒性薬および/または細胞増殖抑制薬が結合可能であり、特定異性体を生じる抗体と、その結合の方法に関する。
【背景技術】
【0003】
(背景)
本発明は、例えばタンパク質−薬物結合体の特定異性体を生じる薬物との少なくとも1個の結合箇所を有する修飾タンパク質と、特定異性体を生じるそのような結合体化の方法とに関する。本発明はさらに、細胞毒性薬および/または細胞増殖抑制薬が結合体化可能であり、特定異性体を生じる抗体と、その結合体化の方法に関する。
【0004】
モノクローナル抗体(mAb)は、癌との闘いにおいて価値のある武器である。mAbは免疫障害の処置でも使用される。癌および免疫障害に対するmAbベースの療法の使用をさらに促進するために、多数の新規な手法が検討されてきた。1つの手法は、殺細胞ペイロードを結合することによって、腫瘍細胞に対するmAbの細胞毒性ポテンシャルを向上させることである。タンパク質毒素、放射性核種、および抗癌剤などの分子は、あるmAbに結合体化されて免疫毒素、放射免疫結合体、および抗体−薬物結合体(ADC)をそれぞれ生成してきた。
【0005】
ADCの開発で以前に考慮された因子は、抗体の選択、および薬物成分の効力の最適化、リンカーの安定性、および薬物がmAbに共有結合される方法を含んでいた。ジスルフィド結合によって結合体化されたADCを生成するための一般的な慣例は、抗体のすべての鎖間ジスルフィド結合を還元することと、還元されたmAbチオールすべてに還元チオールすべてとの相互作用が可能である化合物と反応させて、ある結合部位に対する特異性を得る能力を持たない、薬物8個/mAbで均一に置換された、すなわち「完全付加された」ADCを形成することによるものであった。
【0006】
例えば抗原CD30は、ホジキン病(HD)未分化大細胞リンパ腫(ALCL)などの癌で高度に発現される。CD30のこの発現は、正常細胞での制限された発現と相まって、それをADC療法の魅力的な標的としている。CD30を指向するキメラmAbであるcAC10は、インビトロと、皮下および播種性SCIDマウス異種移植モデルとの両方でHDに対する抗腫瘍活性を有する。cAC10の抗腫瘍活性は、8個すべての鎖間チオールが細胞毒性薬オーリスタチンEの誘導体に薬物成分として結合された完全付加ADCを産生することによって向上された。これらのADCは、マウス異種移植モデル内で耐容性に優れた用量にて非常に有効であった。
【0007】
ADCの産生における慣習は、それらに薬物を完全付加することであったため、部分付加ADCが同じまたはそれ以上の治療有効性を持ちうることは、以前は認識されていなかった。さらに抗体での他の置換パターンが同じまたはそれより少ない毒性で同じまたはそれ以上の治療有効性を発生しうることを考慮できる方法が存在しなかった。これらおよび他の過去の制限および問題は、本発明によって解決される。
【発明の開示】
【課題を解決するための手段】
【0008】
(要旨)
本発明は、タンパク質−薬物結合体ならびにタンパク質−薬物およびタンパク質標識結合体を作成する方法を提供する。薬物または標識を受容するための結合箇所を有するタンパク質も提供される。結合体は、生体内撮像に、または他の用途に治療的に、診断的に(例えばインビトロまたはインビボ)で使用され得る。
【0009】
一般に、割当て可能な結合箇所を有する部分付加された修飾タンパク質が供給される。修飾タンパク質は一般に、結合パートナーとの相互作用のための結合領域と、少なくとも2個の結合箇所とを含み、各結合箇所は薬物または標識と共有結合している。通例、同様の接近性または活性化性を有する、考えられるすべての結合箇所のより少数が薬物または標識に結合される。修飾タンパク質は例えば、抗体、レセプター、レセプターリガンド、ホルモン、サイトカインなどであり得る。結合箇所は例えば、アミノ基、隣接ヒドロキシル基、ヒドロキシル基、カルボキシル基、またはチオール基であり得る。タンパク質は例えば、レセプター、レセプターリガンド、ホルモン、サイトカインなどであり得る。
【0010】
さらなる実施形態において、1個以上のジスルフィド結合を有するタンパク質と、遊離チオールと反応性の薬物との結合体を調製する方法が提供される。該方法は一般に、タンパク質を還元剤によって部分的に還元することと;遊離チオールと反応性の薬物を部分還元タンパク質に結合体化することとを含む。なお別の実施形態において、1個以上のジスルフィド結合を有するタンパク質と、遊離チオールと反応性の薬物との結合体を調製する方法が提供される。該方法は一般に、タンパク質を還元剤によって完全に還元することと;タンパク質を再酸化剤によって部分的に再酸化することと;遊離チオールと反応性の薬物を抗体に結合体化させることとを含む。
【0011】
一部の実施形態において、部分付加抗体が提供される。抗体は、抗原結合領域と、少なくとも1つの鎖間ジスルフィド結合と、それぞれ鎖間チオールに結合体化された、少なくとも2つの薬物または標識を含む。薬物または標識の結合箇所は必要に応じて、容易に割当て可能である。1つの例では、抗体は少なくとも4個の細胞毒性薬または細胞増殖抑制薬を有することが可能であり、各薬物は鎖間チオールに結合体化される。ある実施形態において、抗体は、種4A、種4B、種4C、種4D、種4Eまたは種4Fの配置を有する。
【0012】
部分付加抗体は例えば、マウス抗体、ヒト化抗体、ヒト抗体またはキメラ抗体であり得る。薬物は例えば、MMAF、MMAEまたはAFPなどの細胞毒性薬または細胞増殖抑制薬であり得る。部分付加抗体を含む薬学的組成物も提供される。
【0013】
別の実施形態において、抗体は、細胞毒性薬または細胞増殖抑制薬に対する少なくとも1つの結合箇所を有し、抗体上の結合箇所を細胞毒性薬または細胞増殖抑制薬へ容易に割当てることができるように提供される。抗体では、考えられるすべての結合箇所より少数が細胞毒性薬または細胞増殖抑制薬への結合体化に利用できる。結合箇所は例えば、鎖間チオールであり得る。結合箇所は例えば、種4A〜4Fのうちの少なくとも1個であり得る。
【0014】
割当て可能な結合箇所を有する修飾抗体の組成物も提供される。該組成物は、例えば少なくとも2個の、少なくとも4個の、少なくとも6個の、少なくとも7個の、少なくとも10個以上の種の修飾抗体を有することができる。1つの例において、各種は2個の鎖間チオール、および少なくとも1個の鎖間ジスルフィド結合を有する、少なくとも1つの特定結合対を持ちうる。抗体種は例えば4A、4B、4C、4D、4Eおよび/または4Fを有することができる。さらなる例では、特定結合対は、定常軽鎖−定常重鎖間ジスルフィド結合および/または定常重鎖−定常重鎖間ジスルフィド結合に存在しうる。特定結合対は、ヒンジ領域のN末端より近位におよび/またはヒンジ領域のC末端より近位にあり得る。別の例では、特定結合対は、定常軽鎖−定常重鎖間スルフィド結合に、および修飾抗体のN末端付近に位置するヒンジ領域に、または定常軽鎖−定常重鎖間ジスルフィド結合に、および修飾抗体のC末端付近に位置するヒンジ領域に存在する。
【0015】
抗体の各種は必要に応じて、定常軽鎖−定常重鎖間ジスルフィド結合における少なくとも2個の特定結合対およびヒンジ領域鎖間ジスルフィド結合における少なくとも2個の特定結合対を含むことができる。該組成物は必要に応じて、薬学的に受容可能なキャリアをさらに含むことができる。
【0016】
なお別の実施形態において、部分付加抗体が提供される。該抗体は、少なくとも1個の抗原結合ドメインと、抗体上の少なくとも2個の反応性基と、少なくとも2個の薬物または標識を含み、各薬物または標識は、結合箇所を形成するためにそれぞれ反応性基に結合される。薬物または標識に対する結合箇所は容易に割当てられる。
【0017】
なお他の実施形態において、薬物を還元して、薬物配置の選択性をもたらす抗体に結合させる方法が提供される。該方法は一般に、抗体を還元剤によって完全に還元することと、少なくとも2個の鎖間チオールが残存するように抗体の少なくとも1個の鎖間ジスルフィド結合を再形成するために、完全に還元された抗体を再酸化剤の制限量で処理することと;薬物を鎖間チオールに結合体化することとを含む。再酸化剤は例えば、5,5’−ジチオ−ビス−2−ニトロ安息香酸、4,4’−ジチオジピリジン、2,2’−ジチオジピリジン、テトラチオン酸ナトリウムまたはヨードソ安息香酸であり得る。薬物は例えば、細胞毒性薬または細胞増殖抑制薬あるいは免疫抑制剤であり得る。一部の例では、薬物は副溝バインダ、AEB、AEVB、MMAF、MMAEまたはAFPであり得る。還元剤は例えば、DTTまたはTCEPであり得る。
【0018】
関連する実施形態において、抗体鎖間ジスルフィド結合を還元して、薬物を生じた鎖間チオールに結合させて、抗体上に薬物配置の選択性を生じさせる方法が提供される。該方法は一般に、鎖間チオールを形成するために抗体を還元剤によって完全に還元することと;少なくとも1個の鎖間ジスルフィド結合を再形成するために、抗体を再酸化剤で部分的に再酸化することと;薬物を鎖間チオールに結合させることとを含む。再酸化剤は例えば、5,5’−ジチオ−ビス−2−ニトロ安息香酸、4,4’−ジチオジピリジン、2,2’−ジチオジピリジン、テトラチオン酸ナトリウム、またはヨードソ安息香酸であり得る。還元剤例えば、DTTまたはTCEPであり得る。薬物は例えばMMAF、MMAE、またはAFPであり得る。部分的に再酸化された抗体は必要に応じて、結合体化前に精製され得る。
【0019】
なお他の関連する実施形態において、抗体鎖間ジスルフィド結合を還元して、薬物を生じた鎖間チオールに結合させて、薬物を抗体上に選択的に配置する方法が提供される。該方法は一般に、少なくとも2個の鎖間チオールを形成するために、抗体を還元剤によって部分還元することと;部分的に還元された抗体の鎖間チオールに結合体化させることとを含む。1つの例では、抗体は制限濃度の還元剤によって、キレート剤を含む緩衝液中で部分的に還元される。該薬物は、例えば抗体溶液を冷却して、薬物を冷溶媒に溶解させ、抗体溶液と混合することによって結合体化できる。抗体および薬物溶液は、1個または複数の部分的に付加された抗体−薬物結合体を形成するのに十分な期間に渡ってインキュベートされる。反応は、過剰な薬物をチオール含有試薬によってクエンチさせることによってクエンチできる。結合体はさらに精製できる。具体的な例において、抗体は約37℃にて約1時間、部分的に還元される。還元された抗体は例えば約0℃まで冷却することができる。抗体および薬物溶液は例えば、約0℃にて30分間インキュベートできる。
【0020】
チオール含有試薬は例えば、システインまたはN−アセチルシステインであり得る。還元剤は例えばDTTまたはTCEPであり得る。緩衝液は例えばホウ酸ナトリウム溶液でもよく、キレート剤はジエチレントリアミン五酢酸である。キレート剤例えば、エチレントリアミン五酢酸またはEDTAでもあり得る。溶媒は例えば、アセトニトリル、アルコールまたはDMSOであり得る。薬物は例えば、細胞毒性薬または細胞増殖抑制薬であり得る。
【0021】
一部の実施形態において、還元された抗体は結合体化前に例えばカラムクロマトグラフィー、透析、またはダイアフィルトレーションを使用して精製できる。カラムクロマトグラフィーで使用されるカラムは例えば、PD−10カラムなどの脱塩カラムであり得る。あるいは還元抗体は、部分的還元後および結合体化前に精製されない。
【0022】
結合体は例えばカラムクロマトグラフィー、透析、またはダイアフィルトレーションを使用して精製できる。カラムクロマトグラフィーで使用されるカラムは、例えば脱塩カラム、例えばPD−10カラムであり得る。
【0023】
なお別の実施形態において、薬物の選択的結合体化によって抗体を産生する方法が提供される。該方法は一般に、例えばDTNB滴定によって決定されるように、大幅に過剰の還元剤を添加して、溶液を約37℃にて約30分間インキュベートすることによって、鎖間チオールを産生するのに十分な時間抗体を完全に還元することと;抗体を精製することと;酸化剤を使用して抗体を部分的に再酸化し、還元された抗体を0℃に冷却することによって、少なくとも1個の鎖間ジスルフィド結合を形成することと;還元および冷却された抗体を酸化剤1.5〜2.5モル当量で処理することと;反転により溶液を混合することと;約0℃にて約10分間インキュベートすることによって、抗体を部分的に再酸化することと;部分的に再酸化された抗体を精製することと;結合体化抗体を作成するために、部分的に再酸化された抗体の鎖間チオールに薬物を結合させることと;結合体化抗体を精製することとを含む。
【0024】
還元剤は例えば、DTTまたはTCEPであり得る。部分還元抗体は必要に応じて、例えばカラムクロマトグラフィー、透析、またはダイアフィルトレーションを使用して精製できる。カラムクロマトグラフィーで使用されるカラムは例えば、PD−10カラムなどの脱塩カラムであり得る。結合体化抗体は例えば、カラムクロマトグラフィー、透析、またはダイアフィルトレーションを使用して精製できる。カラムクロマトグラフィーで使用されるカラムは例えば、PD−10カラムなどの脱塩カラムであり得る。再酸化剤は例えば、5,5’−ジチオ−ビス−2−ニトロ安息香酸、4,4’−ジチオジピリジン、2,2’−ジチオジピリジン、テトラチオン酸ナトリウム、またはヨードソ安息香酸であり得る。
【0025】
本発明は、添付図面と併せて、以下の特定の実施形態の詳細な説明を参照することによっても最もよく理解されるで。以下の議論は説明的、例証的、例示的であり、添付の特許請求の範囲によって規定される範囲を限定するものとして解釈すべきではない。
【発明を実施するための最良の形態】
【0026】
(詳細な説明)
開示を明瞭にするために、そして制限するためでなく、本発明の詳細な説明は、以下に続く小節に分割する。
【0027】
(I.定義)
そうでないと定義しない限り、本明細書で使用するすべての技術用語および化学用語は、記載する方法および組成物に関係する当業者によって一般に理解されるのと同じ意味を有する。本明細書で使用される場合、以下の用語および表現は、そうでないと規定しない限り、それらに与えられた意味を有する。
【0028】
「薬物」という用語は本明細書で使用される場合、例えば薬学的化合物、治療化合物、または薬理化合物を含む、元素、化合物、薬剤、または分子実体を意味する。薬物は、天然または合成あるいはその組合せであり得る。「治療薬」は、治療(例えば有益な)効果を癌細胞または免疫細胞(例えば活性化免疫細胞)に単独で、または他の因子との組合せ(例えばプロドラッグと組合されたプロドラッグ変換酵素)のどちらかで作用させる薬剤である。通例、本明細書で述べるおよび方法および組成物によって有用な治療薬は、細胞毒性、細胞増殖抑制、または免疫抑制効果を作用させる薬物である。ある実施形態において、薬物は放射性元素ではない。
【0029】
「細胞毒性薬」は薬剤の細胞に対する効果に関して、細胞を殺すことを意味する。
【0030】
「細胞増殖抑制薬」は、細胞増殖の阻害を意味する。
【0031】
「ポリペプチド」という用語は、アミノ酸のポリマーおよびその同等物を指し、特定の長さの生成物を指すものではない;それゆえ「ペプチド」および「タンパク質」は、ポリペプチドの定義の中に含まれる。ポリペプチドの定義の中には、本明細書で定義された「抗体」も含まれる。「ポリペプチド領域」はポリペプチドのセグメントを指し、そのセグメントは例えば、1個以上のドメインまたはモチーフを含有できる(例えば抗体のポリペプチド領域は、例えば1個以上のCDRを含有できる)。「フラグメント」という用語は、通例、ポリペプチドの少なくとも20個の連続または少なくとも50個の連続アミノ酸を有するポリペプチドの部分を指す。「誘導体」は、第2のポリペプチドに対して保存的アミノ酸置換を有するポリペプチドまたはそのフラグメント;あるいは第2の分子の共有付加によって、例えば非相同ポリペプチドの付加によって、またはグリコシル化、アセチル化、ホスホリル化などによって修飾されたポリペプチドまたはそのフラグメントを含む。さらに、例えばアミノ酸(例えば非天然アミノ酸など)の1個以上のアナログを含有するポリペプチドアナログ、非置換連鎖を有するポリペプチド、ならびに天然および非天然の両方の、当該分野で公知の他の修飾を含むポリペプチドも含まれる。ポリペプチドアナログは例えば、タンパク質模倣物およびボンベシンを含む。
【0032】
「抗体」という用語は本明細書で使用される場合(a)免疫グロブリンポリペプチドおよび免疫グロブリンポリペプチドの免疫活性部分、すなわち特異的抗原へ免疫特異的に結合する抗原結合部位を含有する、免疫グロブリンファミリーのポリペプチド、またはそのフラグメント、あるいは(b)抗原へ免疫特異的に結合する、そのような免疫グロブリンポリペプチドまたはフラグメントの保存的に置換された誘導体を指す。抗体は一般に、例えばHarlow & Lane,Antibodies:A Laboratory Manual(Cold Spring Harbor Laboratory Press,1988)に記載されている。
【0033】
「抗体誘導体」という用語は本明細書で使用される場合、非相同分子の共有付加によって、例えば非相同ポリペプチドの付加によって、あるいは通常は抗体と関連付けられないグリコシル化、アセチル化またはホスホリル化などによって修飾された、上で定義したような抗体を意味する。
【0034】
「モノクローナル抗体」という用語は、それが産生される方法ではなく、いずれかの真核または原核細胞クローン、あるいはファージクローンを含む単細胞クローンに由来する抗体を指す。それゆえ「モノクローナル抗体」という用語は本明細書で使用される場合、ハイブリドーマ技術によって産生される抗体に限定されない。
【0035】
「鎖間ジスルフィド結合」という用語は抗体の文脈において、2個の重鎖間の、または重鎖と軽鎖との間のジスルフィド結合を指す。
【0036】
「鎖間チオール」という用語は、鎖間ジスルフィド結合の形成に関与し得る抗体重鎖または軽鎖のチオール基を指す。
【0037】
タンパク質は、特定の種類のおよび/または同様の反応性の結合箇所すべてが薬物に結合され、タンパク質−薬物結合体の均質な個体群を生じる場合に、「完全付加」と呼ばれる。タンパク質は、特定の種類および/または同様の反応性の考えられる結合箇所の一部のみが薬物に結合され、タンパク質−薬物結合体のある異性体または複数の異性体の生成をもたらす場合に、「部分付加」と呼ばれる。
【0038】
「単離された」という用語は、分子または高分子(例えば抗体)の文脈において、その自然環境の成分から同定ならびに分離および/または回収されたものである。その自然環境の汚染成分は、分子の所望の用途(例えば診断または治療)を妨害する物質であり、酵素、ホルモン、および他のタンパク性または非タンパク性溶質を含み得る。一部の実施形態において、単離された分子または高分子は、(1)例えばLowryまたはBradford法によって決定されたような分子または高分子の重量の95%超まで、または99%超まで、(2)スピニングカップ・シークエネータの使用により、N末端または内部アミノ酸配列の少なくとも15残基を得るのに十分な程度まで、または(3)クマシーブルーまたは好ましくは、銀染色を使用する還元または非還元条件下で、SDS−PAGEによって均質となるまで精製される。単離された分子および高分子が組換え細胞内に分子および高分子をインサイチュで含むのは、抗体の自然環境の少なくとも1つの成分が存在しないからであろう。しかし通常は、単離抗体は少なくとも1つの精製ステップによって調製される。
【0039】
略語「AFP」は、すぐ下に示す一般式を有するジメチルバリン−バリン−ドライソロイシン−ドラプロイン−フェニルアラニン−p−フェニレンジアミンを指す:
【0040】
【化1】
。
【0041】
略語「MMAE」は、すぐ下に示す一般式を有するモノメチルオーリスタチンEを指す:
【0042】
【化2】
。
【0043】
略語「MMAF」は、すぐ下に示す一般式を有するドバリン−バリン−ドライソロイニン−ドラプロイン−フェニルアラニンを指す:
【0044】
【化3】
。
【0045】
略語「AEB」は、オーリスタチンEにパラアセチル安息香酸を反応させることによって生成されるエステルを指す。略語「AEVB」は、オーリスタチンEにベンゾイル吉草酸を反応させることによって生成されるエステルを指す。
【0046】
「薬学的に受容可能な塩」は本明細書で使用されるように、分子または高分子の薬学的に受容可能な有機または無機塩を指す。酸付加塩は、アミノ基によって形成され得る。塩の例としては、これに限定されるわけではないが、サルフェート、シトレート、アセテート、オキサラート、クロライド、ブロミド、ヨージド、ニトレート、ビスルフェート、ホスフェート、酸ホスフェート、イソニコチナート、ラクテート、サリチラート、酸シトレート、タートレート、オレアート、タンナート、パントテナート、バイタートレート、アスコルベート、スクシナート、マレアート、ゲンチシナート、フマレート、グルコナート、グルクロナート、サッカラート、ホルメート、ベンゾアート、グルタメート、メタンスルホナート、エタンスルホナート、ベンゼンスルホナート、p−トルエンスルホナート、およびパモアート(すなわち1,1’メチレンビス−(2−ヒドロキシ3−ナフトアート))塩が挙げられる。薬学的に受容可能な塩は、アセテートイオン、スクシナートイオンまたは他の対イオンなどの、別のイオンの包含も含む。対イオンは、親化合物上に電荷を安定させるいずれかの有機または無機部分でもよい。さらに薬学的に受容可能な塩は、その構造内に1個を超える荷電原子を有してもよい。複数の荷電原子が薬学的に受容可能な塩の一部である例は、複数の対イオンを有することができる。それゆえ薬学的に受容可能な塩は、1個以上の荷電原子および/または1個以上の対イオンを有することができる。
【0047】
「薬学的に受容可能な溶媒和物」または「溶媒和物」は、1個以上の溶媒分子および分子または高分子の会合を指す。薬学的に受容可能な溶媒和物を形成する溶媒の例としては、これに限定されるわけではないが、水、イソプロパノール、エタノール、メタノール、DMSO、エチルアセテート、酢酸、およびエタノールアミンが挙げられる。
【0048】
(II.ポリペプチド、タンパク質、抗体)
本発明は、タンパク質−薬物結合体およびタンパク質−薬物結合体を作成する方法を提供する。薬物を受容するための結合箇所を有するタンパク質も提供される。タンパク質−薬物結合体は治療に、診断に(例えばインビトロまたはインビボで)、インビボ撮像に、そして他の用途に使用され得る。
【0049】
抗体、酵素、グリコシル化タンパク質、レクチン、種々の生物学的レセプター、タンパク質ホルモン、および結合パートナーに対する結合剤として作用できる他のタンパク質を含む、タンパク質の種々のクラスが結合体化され得る。タンパク質は、少なくとも1個の反応部位、例えばジスルフィド結合、アミノ基、ヒドロキシル基またはカルボキシル基を含み、そこで薬物のタンパク質への結合体化が起こり得る。
【0050】
反応部位は、化学反応または手段によって接近可能であり、活性化可能である。一部の実施形態において、結合体化目的で化学的に活性化されるタンパク質は、タンパク質の所期の用途に必須ではないジスルフィド結合を含有するタンパク質、および/またはタンパク質を妨害しないタンパク質(例えばこれに限定されるわけではないが、タンパク質の分解を引き起こす、または結合や他の機能(例えばエフェクタ機能)を妨害する)である。そのようなタンパク質では、ジスルフィド結合は、2個のシステイン残基のチオール(−−SH)側基の酸化の結果として存在し得る。これらの残基は、別のポリペプチド鎖上に、または同じポリペプチド鎖上に存在する。酸化の結果として、ジスルフィド結合(−−S−−S−−)は、元のシステイン残基のベータ炭素間に形成される。還元後、残基は互換的にハーフシステインおよびシステインと呼ばれることが多い。ジスルフィド結合の還元剤による処理は、ジスルフィド結合の還元的開裂を引き起こして、遊離チオール基を残す。ジスルフィド結合を含有するタンパク質の例としては、抗体、多くの酵素、特定のホルモン、および特定のレセプターが挙げられる。
【0051】
一部の実施形態において、ジスルフィド結合は自然発生的であり得る。一部の実施形態において、スルフヒドリル基もタンパク質(例えば抗体)内に化学的に導入され得る。スルフヒドリル基を導入するための適切な方法は、化学的手段(例えば2−ITなどのチオール化剤を使用して)、または組換えDNA技術を使用することを含む。例えばシステイン残基は、タンパク質をコードする核酸の突然変異誘発によって、タンパク質中に導入できる。一般に、参考として本明細書に援用される、Sambrook et al.,Molecular Cloning,A Laboratory Manual,3rd ed.,Cold Spring Harbor Publish.,Cold Spring Harbor, New York(2001);Ausubel et al.,Current Protocols in Molecular Biology,4th ed.,John Wiley and Sons,New York(1999)を参照のこと。スルフヒドリル基は、タンパク質中に、例えばポリペプチド内に、またはカルボキシ末端に導入され得る。
【0052】
一部の実施形態において、タンパク質は抗体である。そのような抗体は、インビトロまたはインビボ診断、インビボ撮像、あるいは特有の抗原に関する疾患または状態の治療に使用できる。抗体構造の基本単位は、非共有結合と、ジスルフィド結合との両方によって共に結合された、4個のポリペプチド−−2個の同じ低分子量(「軽」)鎖および2個の同じ高分子量(「重」)鎖の複合体である。異なる抗体が、これらの基本単位の1〜5個のいずれかに位置するであろう。抗体は、図式的に「Y」として表される。「Y」の各分岐は、重鎖のアミノ末端部と結合した軽鎖によって形成される。「Y」の基部は、2つの重鎖のカルボキシ末端部によって形成される。「Y」の節はヒンジ領域と呼ばれる。
【0053】
5つのヒト抗体クラス(IgG、IgA、IgM、IgDおよびIgE)、およびこれらのクラス内の種々のサブクラス(IgG1、IgG2、IgG3、IgG4、IgA1およびIgA2)または免疫グロブリン分子のサブクラスは、構造の差異、例えば1個の抗体分子中の免疫グロブリン単位の数、個々の単位のジスルフィドブリッジ構造、ならびに鎖長および配列の差異に基づいて認識される。抗体のクラスおよびサブクラスは、そのアイソタイプである。
【0054】
抗体は、インタクトな抗体または抗原結合抗体フラグメント、例えばFab、F(ab’)、F(ab’)2、Fd鎖、単鎖Fv(scFv)、単鎖抗体、ジスルフィド結合Fv(sdFv)、VLまたはVHドメインのどちらかを含むフラグメント、あるいはFab発現ライブラリによって産生されたフラグメントであり得る。単鎖抗体を含む抗原結合抗体フラグメントは、可変領域を単独で、あるいは以下の実体または部分と組合せて含むことができる:ヒンジ領域、CH1、CH2、CH3、CH4およびCLドメイン。また抗原結合フラグメントは、可変領域とヒンジ領域、CH1、CH2、CH3、CH4およびCLドメインとのいずれの組合せも含むことができる。一部の実施形態において、抗体フラグメントは、鎖間ジスルフィド結合を含む少なくとも1つのドメイン、またはドメインの部分を含む。
【0055】
通例、抗体はヒト、げっ歯類(例えばマウスおよびラット)、ロバ、ヒツジ、ウサギ、ヤギ、モルモット、ラクダ科の哺乳類、ウマ、またはニワトリの抗体である。本明細書で使用される場合「ヒト」抗体は、ヒト免疫グロブリンのアミノ酸配列を有する抗体を含み、以下に、そして例えば米国特許第5,939,598号および同第6,111,166号に述べるように、ヒト免疫グロブリンライブラリから、ヒトB細胞から、または1個以上のヒト免疫グロブリンについて遺伝子組換えされた動物から単離された抗体を含む。抗体は単一特異性、二重特異性、三重特異性、またはより多くの多重特異性であり得る。
【0056】
一部の実施形態において、定常ドメインはエフェクター機能を有する。「抗体エフェクター機能」またはAECという用語は本明細書で使用される場合、IgのFcドメインが寄与する機能を指す。そのような機能は例えばFcエフェクタードメインの、食細胞または溶解活性を備えた免疫細胞上のFcレセプターへの結合によって、またはFcエフェクタードメインの補体系の成分への結合によって影響され得る。通例、Fc結合細胞または補体成分によって仲介される効果は、CD70標的細胞の阻害および/または消耗を引き起こす。エフェクター機能は例えば、「抗体依存性細胞毒性」またはADCC、「抗体依存性細胞食作用」またはADCP、「補体依存性細胞毒性」またはCDCであり得る。他の実施形態において、定常ドメインは1つ以上のエフェクター機能を欠いている。
【0057】
抗体は、目的の抗原、例えば医療的および/または治療的目的の抗原に対して産生され得る。例えば抗原は、病原体(これに限定されるわけではないが、ウィルス、細菌、真菌、および原虫など)、寄生虫、腫瘍細胞、または特定の医学的状態と関連付けられる抗原であり得る。腫瘍関連抗原(TAA)の場合、癌は、免疫系、肺、結腸、直腸、乳房、卵巣、前立腺、頭部、頸部、骨、または他のいずれかの解剖学的位置の癌であり得る。目的の抗原はこれに限定されるわけではないが、CD30、CD40、Lewis Y、およびCD70を含む。一部の実施形態において、抗原はCD2、D20、CD22、CD33、CD38、CD40、CD52、HER2、EGFR、VEGF、CEA、HLA−DR、HLA−Dr10、CA125、CA15−3、CA19−9、L6、Lewis X、アルファフェトタンパク質、CA242、胎盤アルカリフォスファターゼ、前立腺特異的抗原、前立腺酸ホスファターゼ、上皮増殖因子、MAGE−1、MAGE−2、MAGE−3、MAGE−4、抗トランスフェリンレセプター、p97、MUC1−KLH、gp100、MART1、IL−2レセプター、ヒト絨毛性ゴナドトロピン、ムチン、P21、MPG、およびNeu癌遺伝子産物である。
【0058】
一部の有用な特異的抗体は、これに限定されるわけではないが、BR96 mAb(Trail et al.(1993),Science 261:212−215)、BR64(Trail et al.(1997),Cancer Research 57:100−105)、CD40抗原に対するmAb、例えばS2C6 mAb(Francisco et al.(2000)Cancer Res.60:3225−3231)、およびCD30抗原に対するmAb、例えばAC10(Bowen et al.(1993)J.Immunol.151:5896−5906)を含む。腫瘍特異的抗原に結合する他の多くの内部移行抗体が使用され得、概説されてきた(例えばFranke et al.(2000),Cancer Biother Radiopharm.15:459−76;Murray(2000),Semin Oncol.27:64−70;Breitling et al.,Recombinant Antibodies,John Wiley,and Sons,New York,1998を参照)。これらの参考文献の開示は、参照により本明細書に援用されている。
【0059】
「腫瘍特異的抗原」という用語は本明細書で使用される場合、特定の腫瘍に特有の、またはそのような腫瘍に強い相関関係のある抗原を示すことが理解されるであろう。しかしながら、腫瘍特異的抗原は腫瘍組織に必ずしも特有でなく、しかしながら、すなわち、それらに対するその抗体は、正常組織の抗原と交差反応することがある。腫瘍特異的抗原が腫瘍細胞にとって特有でない場合、実際問題として、腫瘍特異的抗原に結合する抗体が交差反応による不当なリスクまたは妨害なしに所望の手順を実施するために、腫瘍細胞にとって十分に特異的であることが高い頻度で起こる。多くの因子がこの実際的な特異性に寄与している。例えば腫瘍細胞上の抗原の量は、正常細胞上で見出される交差反応抗原の量を大きく超えるか、または腫瘍細胞上の抗原は、より効果的に提示されうる。したがって「腫瘍特異的抗原」という用語は本明細書では、実際的に有用な特異性に関し、絶対的な特異性を表したり、または抗原が腫瘍に特有であることを示したりするものではない。
【0060】
抗体は、ポリクローナル抗体またはモノクローナル抗体であり得る。被験体がヒト被験体である場合、抗体は抗原に対する有用な免疫反応を開始できるいずれかの動物を免疫化することによって得られ得る。動物はマウス、ラット、ヤギ、ヒツジ、ウサギまたは他の適切な実験動物であり得る。抗原は、天然の免疫原、またはハプテンおよび免疫原性キャリアの合成免疫原性結合体の形で提示できる。モノクローナル抗体の場合、免疫化動物の抗体産生細胞は、抗体を産生するハイブリドーマを得るために、「不死」または「不死化」ヒトまたは動物細胞と融合することができる。所望ならば、免疫グロブリン鎖の1個以上をコードする遺伝子は、抗体が種々の宿主細胞で産生できるようにクローニングすることができ、所望ならば、産生される抗体の配列、およびそれゆえ免疫的特性を変化させるために、遺伝子を突然変異させることができる。ヒトモノクローナル抗体は、当該分野で公知の多数の技術のいずれかによって産生できる(例えばTeng et al.(1983),Proc.Natl.Acad.Sci.USA.80,7308−7312;Kozbor et al.(1983)Immunology Today 4,72−79;およびOlsson et al.(1982),Meth.Enzymol.92,3−16)。
【0061】
抗体は、例えば当業者に周知の技術で産生されたネズミ抗体、キメラ抗体、ヒト化抗体、または完全ヒト抗体であり得る。標準組換えDNA技術を使用して産生できる、ヒトおよび非ヒト部分の両方を含む組換え抗体、例えばキメラ抗体およびヒト化モノクローナル抗体は、有用な抗体である。キメラ抗体は、異なる部分が異なる動物種に由来する分子、例えばマウスモノクローナルに由来する可変領域およびヒト免疫グロブリン定常領域を有する分子である(例えば参考としてその全体が本明細書に援用される、Cabilly et al.,米国特許第4,816,567号;およびBoss et al.,米国特許第4,816,397号を参照のこと)。ヒト化抗体は、非ヒト種からの1個以上の相補性決定領域(CDR)およびヒト免疫グロブリン分子からのフレームワーク領域を有する非ヒト種からの抗体分子である(例えば参考としてその全体が本明細書に援用される、Queen,米国特許第5,585,089号を参照のこと)。そのようなキメラ抗体およびヒト化モノクローナル抗体は、当該分野で公知の組換えDNA技術、例えば国際公開第87/02671号;欧州特許出願公開第184,187号;欧州特許出願公開第171496号;欧州特許出願公開第173494号;国際公開第86/01533号;欧州特許出願公開第12,023号;Berter et al.(1988),Science 240:1041−1043;Liu et al.(1987),Proc.Natl.Acad.Sci.USA 84:3439−3443;Liu et al.(1987),J.Immunol.139:3521−3526;Sun et al.(1987),Proc.Natl.Acad.Sci.USA 84:214−218;Nishimura et al.(1987),Cancer.Res.47:999−1005;Wood et al.(1985),Nature 314:446−449;およびShaw et al.(1988),J.Natl.Cancer Inst.80:1553−1559;Morrison(1985),Science 229:1202−1207;Oi et al.(1986),BioTechniques 4:214;U.S.Patent No.5,225,539;Jones et al.(1986),Nature 321:552−525;Verhoeyan et al.(1988),Science 239:1534;およびBeidler et al.(1988),J.Immunol.141:4053−4060に述べられている方法を使用して産生可能であり、そのそれぞれが参考として全体が本明細書に援用されている。
【0062】
完全ヒト抗体は例えば、内在性免疫グロブリン重鎖および軽鎖遺伝子を発現できないが、ヒト重鎖および軽鎖遺伝子を発現できるトランスジェニックマウスを使用して産生できる。トランスジェニックマウスは、選択した抗原またはその部分を用いて通常の方法で免疫化される。抗原に対して産生されたモノクローナル抗体は、従来のハイブリドーマ技術を使用して得ることができる。トランスジェニックマウスに含まれるヒト免疫グロブリントランス遺伝子は、B細胞分化の間に再配列し、続いてクラス転換および体細胞突然変異を受ける。それゆえそのような技術を使用して、治療的に有用なIgG、IgA、IgMおよびIgE抗体を産生することができる。ヒト抗体を産生するこの技術の概要については、LonbergおよびHuszar(1995,Int.Rev.Immunol.13:65−93)を参照。ヒト抗体およびヒトモノクローナル抗体を産生するためのこの技術ならびにそのような抗体を産生するためのプロトコルの詳細な議論については、例えば米国特許第5,625,126号;同第5,633,425号;同第5,569,825号;同第5,661,016号;同第5,545,806号を参照のこと(そのそれぞれは参考としてその全体が本明細書に援用される)。他のヒト抗体は、例えばAbgenix,Inc.(フリーモント、カリフォルニア州)およびGenpharm(サンノゼ、カリフォルニア州)から市販され得る。
【0063】
選択したエピトープを認識する完全ヒト抗体も、「誘導選択」と呼ばれる技術を使用して産生できる。本手法において、同じエピトープを認識する完全ヒト抗体の選択を誘導するために、選択した非ヒトモノクローナル抗体、例えばマウス抗体が使用される。例えばJespers et al.(1994),Biotechnology 12:899−903を参照のこと。ヒト抗体は、ファージ提示ライブラリ(HoogenboomおよびWinter(1991),J.Mol.Biol.227:381;Marks et al.(1991),J.Mol.Biol.222:581;QuanおよびCarter(2002),”The rise of monoclonal antibodies as therapeutics.”Anti−IgE and Allergic Disease,Jardieu,P.M.およびFick Jr.,R.B,eds.,Marcel Dekker,New York,NY,Chapter 20,pp.427−469を含む、当該分野で公知の種々の技術を使用しても産生できる。
【0064】
抗体は二重特異性抗体でもよい。二重特異性抗体を産生する方法は、当該分野で公知である。全長二重特異性抗体の伝統的な産生は、2つの鎖が異なる特異性を有する、2つの免疫グロブリン重鎖軽鎖対の同時発現に基づいている(Milstein et al.,1983,Nature 305:537−539)。免疫グロブリン重鎖および軽鎖のランダムな組合せのため、これらのハイブリドーマ(クアドローマ)は、1個のみが正しい二重特異性構造を有する、10個の異なる抗体分子の潜在的な混合物を生成する。同様の手順が国際公開第93/08829号、およびTraunecker et al.(1991),EMBO J.10:3655−3659に開示されている。
【0065】
異なる手法に従って、所望の結合特異性(抗体−抗原結合部位)を備えた抗体可変ドメインは、免疫グロブリン定常ドメイン配列に融合される。融合は好ましくは、ヒンジCH2、およびCH3領域の少なくとも一部を含む免疫グロブリン重鎖定常ドメインによってである。融合物の少なくとも1個に存在する、軽鎖結合に必要な部位を含有する第1重鎖定常領域(CH1)を有することが好ましい。免疫グロブリン重鎖融合をコードする配列を含む核酸、そして所望ならば、免疫グロブリン軽鎖は、独立した発現ベクター内に挿入され、適切な宿主生物内に同時形質移入される。これは、構築で使用された3個のポリペプチド鎖の不等比が最適な収率を与えるときに、実施形態において3個のポリペプチドフラグメントの相互比を調整するために高い柔軟性を提供する。しかしながら、等しい比での少なくとも2個のポリペプチド鎖の発現が高い収率を生じるときに、または比が特に重要でないときに、2個または3個すべてのポリペプチド鎖のコード配列を1個の発現ベクターに挿入することが可能である。
【0066】
この手法の実施形態において、二重特異性抗体は、一方のアームに第1の結合特異性を備えたハイブリッド免疫グロブリン重鎖と、他方のアームにハイブリッド免疫グロブリン重鎖−軽鎖対(第2の結合特異性を提供する)を有する。二重特異性分子の半分のみにおける免疫グロブリン軽鎖の存在が、容易な分離方法を提供するため(国際公開第94/04690号(参照としてその全体が本明細書に援用される))、この非対称構造は、所望の二重特異性化合物の、望ましくない免疫グロブリン鎖の組合せからの分離を促進する。
【0067】
二重特異性抗体を産生するためのさらなる詳細については、例えばSuresh et al.(1986),Methods in Enzymology 121:210;Rodrigues et al.(1993),J.Immunology 151:6954−6961;Carter et al.(1992),Bio/Technology 10:163−167;Carter et al.(1995),J.of Hematotherapy 4:463−470;Merchant et al.(1998),Nature Biotechnology 16:677−681を参照のこと。そのような技術を使用して、二重特異性抗体は、疾患の処置または防止への使用のために調製できる。
【0068】
二重特異性抗体は、欧州特許第0 105 360号にも開示されている。この参考文献に開示されているように、ハイブリッドまたは二重特異性抗体は、生物的に、すなわち細胞融合技術によって、または化学的に、特に架橋剤またはジスルフィドブリッジ形成試薬を用いてのどちらかで誘導可能であり、抗体全体またはそのフラグメントを含みうる。そのようなハイブリッド抗体を得るための方法は例えば、国際公開第83/03679号、および欧州特許第0 217 577号に開示されており、その両方とも参考としてその全体が本明細書中に援用される。
【0069】
他の実施形態において、抗体は、抗体の融合タンパク質、またはその機能活性フラグメントであり、例えばその中で抗体は共有結合(例えばペプチド結合)を介して、N末端またはC末端のどちらかにて抗体でない別のタンパク質(またはその部分、好ましくはタンパク質の少なくとも10、20または50アミノ酸部分)のアミノ酸配列に融合される。好ましくは、抗体またはそのフラグメントは、他のタンパク質に定常ドメインのN末端にて共有結合される。
【0070】
なお他の実施形態において、タンパク質は、必要に応じてヒンジ領域を含む抗体重鎖および/または軽鎖定常領域ドメインへ共有結合を介して融合された非抗体分子の結合部分の融合タンパク質であり得る。そのような融合タンパク質は必要に応じて、少なくとも1つの、通例少なくとも2個の鎖間ジスルフィド結合を含むことができる。例えば融合タンパク質は、CH1およびCL領域、ならびにヒンジ領域を含むことができる。
【0071】
(III.活性化方法)
一般に、薬物は活性化部位においてタンパク質または他の適切な分子へ結合され得る。適切な活性化部位としては、チオール基、アミノ基(例えばリジン残基のイプシロンアミノ基またはタンパク質のN末端にて)、隣接ヒドロキシル基(1,2−ジオール)(例えば酸化炭水化物)およびカルボキシル基(例えばタンパク質のC末端、アスパラギン酸およびグルタミン酸残基、および炭水化物、例えばシアル酸)などの結合箇所が挙げられる。
【0072】
薬物は結合箇所に直接結合され得る。例えば薬物は、抗体リジンのε−アミノ基のアルキル化、酸化炭水化物の還元的アミノ化またはヒドラジドとの反応、ヒドロキシル基およびカルボキシル基間のトランスエステル化、アミノ基またはカルボキシル基におけるアミド化、チオール(例えば鎖間チオール)または導入されたチオールへの結合体化、例えばリジンの2−イミノチオラン(Traut試薬)によるアルキル化によって結合体化することができる。薬物を結合箇所に結合体化する適切な方法は、例えばCurrent Protocols in Protein Science (John Wiley & Sons, Inc.),Chapter 15(Chemical Modifications of Proteins)(その開示は、参考としてその全体が本明細書に援用される)に開示されている。
【0073】
薬物は、別の分子、例えばリンカーを介して間接的に結合させることもできる。例えば薬物は、例えばこれに限定されるわけではないが、抗体のヒンジ領域内においてスルフヒドリル基に結合されたマレイミド基を介して結合体化させることもできる。抗体結合体は、薬物のマレイミド誘導形態に抗体を反応させることによって生成できる。さらに詳細には、抗体結合体は、還元された抗体を産生するために抗体を還元して、アミン薬物を生成し、マレイミド誘導体化薬物を生成するためにアミン薬物をマレイミドによって誘導体化して、マレイミド誘導体化薬物に抗体を反応させることによって生成できる。
【0074】
例示的な実施形態において、IgG1、例えばcAC10は多数のジスルフィド結合を所有し、そのわずか4個が鎖間である。4個の鎖間ジスルフィド結合が高度に柔軟性のヒンジ領域に密集し、他の(鎖内)ジスルフィド結合よりはるかに溶媒に接近しやすいため、過剰の例えば還元剤、例えばこれに限定されるわけではないがジチオトレイトール(DTT)、トリス(2−カルボキシエチル)ホスフィン(TCEP)、または2−メルカプトエタノールによる還元が4個すべての結合を切断して、8個のシステインを生成する(すなわち遊離チオール基を含有する)。8個すべてのシステインと薬物−リンカーとの結合は、図7に示すように、抗体に付き約8個の薬物を持つ完全付加結合体を生成する。
【0075】
本発明は驚くべきことに、ADCの生物特性を抗体当たり平均2、2.5、4または6個の薬物を持つ抗体によって改善可能であり、このことが完全付加結合体、すなわち抗体当たり8個の薬物を持つ結合体の有効性を維持しながら、より低い毒性を生じることを実証する。部分薬物付加結合体の治療濃度域(最低有効用量によって分けられた、毒性が最初に見られる薬物−抗体結合体の濃度)は、8個の薬剤を持つ抗体よりも大きい。8個のシステインに4個の薬物を結合体化させて、多数の潜在的な薬物付加種を生じさせる方法が多数ある(合計9種類、抗体当たり0〜8個の薬物、図1〜7を参照のこと)。4個の薬物を持つ抗体では、4個の薬物を配置するための考えられる方法が6つあり、6個の異性体が生じる(図1および7を参照のこと)。抗体当たり4個の薬物が所望されるときには、8個の薬物が付加された種の均質性は失われる。抗体当たり2または6個の薬物では、薬物を分子に配置するために考えられる方法が3つある。
【0076】
部分的に付加されたADCを産生する方法(例えば抗体当たり8個の薬物ではなく4個で(E4))としては、以下が挙げられる:方法1(「部分還元」)これに限定されるわけではないがDTTまたはTCEPなどの還元剤による抗体の部分還元と、それに続く結合体化、および方法2(「完全還元および再酸化」)これに限定されるわけではないがDTTまたはTCEPなどの還元剤におる抗体の完全還元と、それに続く、再酸化剤(例えばこれに限定されるわけではないが5,5’−ジチオ−ビス−2−ニトロ安息香酸(DTNB)、4,4’−ジチオジピリジン、2,2’−ジチオジピリジン、テトラチオン酸ナトリウム、またはヨードソ安息香酸)による抗体の部分再酸化、そして最終的な結合体化。完全還元および再酸化方法では、2つの局面がある:2a、DTNB再酸化後の精製、および2b、DTNB再酸化後の精製なし(ワンポット再酸化および薬物結合)。これらの方法は、異なる種の異なる割合(例えばE4では、25〜40%)を生じ、考えられる種の異なる異性体混合物も生じる。開示には、当業者に公知である上の方法のハイブリッドおよびバリエーションが含まれる。
【0077】
抗体薬物結合体の一例として、図1は、結合反応中に産生できる6つの考えられるE4種(4A〜4F種と呼ばれる)を示す。種4A〜Dは、特定の分析方法によって個別に区別できない;しかしながらそれらは、4Eおよび4Fから区別できる。
【0078】
部分還元の方法1の1つの実施形態において、例えば抗体当たり4個の薬物を持つ結合体は、例えばこれに限定されるわけではないがDTTによる抗体の完全還元によって産生可能であり、8個の抗体システインを生じて、4個の薬物同等物への結合体化が続く。これは、抗体が0〜8個の薬物を有する混合物を生じさせる。あるいは4個のジスルフィドのうち平均2個のみが還元されるように(4個のシステインを遊離する)抗体が例えば制限量のDTTによって還元され、完全薬物結合体化が続く場合、偶数の薬物が付加された種(抗体当たり0、2、4、6、および8個の薬物)のみが形成される。このことにより、混合物の複雑さが低減され、これは異なる薬物付加種を単離するための精製によってさらに低減することができる。
【0079】
一部の実施形態において、タンパク質上のある潜在的な結合箇所は選択的に活性化され得る。この選択的活性化は、タンパク質への薬物の結合部位の即時の割当てを可能にする。例えば制限量の強力な還元剤DTTまたはTCEPによる抗体(例えばcAC10)の処理は、重鎖−軽鎖ジスルフィドの選択的還元を生じさせる。別の例では、抗体の完全DTT還元と、それに続くDTNBなどの強力なチオール酸化剤を使用する部分再酸化は、重鎖−軽鎖ヒンジジスルフィドの選択的再酸化を引き起こして、主に、ヒンジ領域の重鎖に結合した薬剤を生じさせる。これらの方法の両方によって産生されたE2およびE6の異性体個体群は、90%の異性体均質性に到達し得る。
【0080】
薬物の抗体への結合体化後、結合体化された薬物−抗体種を分離され得る。一部の実施形態において、結合した抗体種は、抗体、薬物および/または結合体の特徴に基づいて分離できる。例えば疎水性相互作用クロマトグラフ(HIC)は、抗体当たり0、2、4、6、および8個の薬物に相当する種を単離および分離することに成功している。方法1によるこれらの薬物付加異性体それぞれの収率は、統計的分布によって予想される収率に近い。4個の薬物が付加された種は通例、全物質の30%である。
【0081】
薬物付加および抗体上の薬物の位置を判定するための分析方法が開発されてきた(以下も参照のこと)。部分DTT還元によって調製した純粋な4個の薬物を付加した結合体の、バイオアナライザ(キャピラリー電気泳動)および架橋ジビニルベンゼンカラム(PLRP)でのHPLCによるキャラクタリゼーションは、薬物は主に、抗体の重鎖と軽鎖との間に最初にジスルフィドを生成したシステイン上に位置することを明らかにした。従来とは異なり、1つの異性体が他の5つの異性体よりも有利である薬物位置の特異性は、予想外である。
【0082】
別の実施形態の、方法2、例えば4個の薬物を持つ薬物−抗体結合体を調製するための部分再酸化による完全還元において、抗体は、例えばこれに限定されるわけではないが、DTTによって完全に還元され、次に4個の抗体システインチオールが残るようにジスルフィドの一部を再形成するために、制限量の、例えばこれに限定されるわけではないがDTNBによって処理された。これらのシステインは薬物に結合体化され、本明細書で述べる方法によって分析された。混合物中の4つの薬物が付加された抗体の収率は40%にも上昇し、いったん精製すると、薬物の位置は、ヒンジ領域において重鎖間で最初にジスルフィドを生成したシステイン上への配置が好都合であった。一般的な慣例と比較した、4個の薬物を付加した抗体の収率および選択性はどちらも、ある部分還元方法による異なる異性体に有利であり、予想外であった。種々の化学的方法を使用すると、抗体内の薬物位置は、異なる異性体の産生のために容易に割当てることができる。
【0083】
還元が制限量の還元剤の添加によって制御され、抗体当たり平均して4個未満の鎖間ジスルフィド結合が破壊される部分還元が起こる。4個の鎖間ジスルフィド結合すべてが高度に露出されているため、還元は種々の経路によって進行し、0、2、4、6、または8個のシステインを持つ種の混合物からなる部分還元抗体を生成する。したがって部分還元抗体の結合は、図1および7に示すように、抗体当たり0、2、4、6、または8個の薬物を持つ結合体の混合物を生成できる。部分還元の程度に応じて、薬物付加分布(すなわち0、2、4、6、または8薬物付加種のパーセント)が変化する。
【0084】
部分還元は、抗体当たり可変数の薬物を持つ種を含有する混合物を生成するだけでなく、複数の場所の薬物付着の結果として、さらなる不均一性も生じさせる。図7は、2、4、および6薬物付加種が考えられる異性体が2個以上あることを示している。
【0085】
薬物のタンパク質への結合後、結合された薬物−タンパク質種は分離され得る。例えば一部の実施形態において、結合した抗体種は、抗体、薬物および/または結合体の特徴に基づいて分離できる。例えば疎水性相互作用クロマトグラフ(HIC)は、抗体当たり0、2、4、6、および8薬物に相当する種を単離および分離することに成功している。
【0086】
(IV.分析方法)
結合体の収率および異性体混合物を決定するために、種々の分析方法が使用され得る。例えば1つの実施形態において、HICは、生じた結合体(例えばE4結合体について)からの収率および異性体混合物を決定するために使用される分析方法である。この技術は、可変数の薬物を付加した抗体を分離することができる。薬物付加レベルは、例えば250nmおよび280nmにおける吸光度の比に基づいて決定することができる。例えば薬物が250nmで吸収し、一方抗体が280mmで吸収し得る場合。したがって250/280比は、薬物付加と共に上昇する。本明細書で述べた結合体化方法を使用して、一般に偶数の薬物を持つ抗体は、ジスルフィドの還元が偶数の遊離システインチオールを生じるため、抗体に結合されることが観察された。図2および3は、方法2aおよび2bによってそれぞれ生成されたcAC10−vcMMAEのHIC分離を示す。図4は、方法1からと同様に、これらのクロマトグラムからの種々の置換についてのパーセント組成を示す。方法1は約30%のE4を生じるが、方法2bは約40%のE4を生じる。
【0087】
HICは、置換レベルの混合物からE4を精製するために、ミリグラム〜グラムレベルにて分取的に使用することもできる。図3による純E4(表示された34〜38分の収集時間)を得て、2つの方法によって分析して、異性体E4混合物を決定した。最初に、非共有相互作用を変性させ、タンパク質質量に基づいて分離するAgilentバイオアナライザを使用して、溶出順に以下の抗体成分を得た:軽鎖(L)、重鎖(H)、重鎖−軽鎖(HL)、重鎖−重鎖(HH)、重鎖−重鎖−軽鎖(HHL)、および重鎖−重鎖−軽鎖−軽鎖(HHLL)。ジスルフィドが還元され、遊離チオールがvcMMAEに結合するときに、より小さい種が形成される。
【0088】
図1は、種々のE4異性体の変性から観察される抗体成分も記載する。図5からわかるように、方法2bによって調製された純E4は、HLによって支配され、少量のLおよびHHLを含む。この結果は、多少の種4A−D(HHLおよびLを生じる)を含む、主に種4F(もっぱらHLを生じる)の存在によって説明できる。興味深いことに、方法1によって生成されたcAC10−vcMMAEの同じ分析は、ほぼ等しい量のL、HL、およびHHを生じ、このことは主に種4Eの、多少の種4Fとの混合物と一致する。
【0089】
分析ツールの別の実施形態は、逆相PLRPカラムによるクロマトグラフィーである;カラム支持体は、非特異的にタンパク質を保持できるシリカ支持体で作成された典型的な逆相カラムよりはむしろ、架橋ジビニルベンゼンからなる。この変性および還元技術は、0および1個の薬物を含む軽鎖(L0およびL1)および0〜3個の薬物を含む重鎖(H0〜H3)からなる6種を明確に分離する。図1は、種々のE4種で観察できる薬物付加レベルを示す。方法2bによる純E4は、図6でPLRPによって分離された。未修飾軽鎖(L0)および2個の薬物を含む重鎖(H2)は、4Fから予想された種であるのに対して、L1およびH1は4A〜Dから予想される。バイオアナライザを共に用いて、これらのデータは4E対4Fが約2:1の混合物を生成する方法1と一致するが、方法2bは、4F対4A〜Dを2:1で生成する。それゆえ異なる化学的条件を使用しても、E4収率およびE4異性体の部分はどちらも、方法1と2bでは著しく異なっている。
【0090】
(V.タンパク質への結合が可能である化合物)
タンパク質は、細胞増殖抑制薬または細胞毒性薬、免疫抑制剤、毒素、キレート、化合物、分子、放射性核種などの任意の目的の薬物と結合体化できる。
【0091】
(細胞毒性薬および細胞増殖抑制薬)
細胞毒性薬および細胞増殖抑制薬としては、抗生剤(例えばアドリアマイシン)、抗腫瘍剤、例えばオーリスタチンおよびオーリスタチン誘導体、メトトレキサート、ミトマイシンC、ダウノルビシン、ドキソルビシン、およびビンブラスチン、5−フルオロウラシルDNA副溝バインダ、DNA複製阻害剤、アルキル化剤(例えばシスプラチン、モノ(プラチナ)、ビス(プラチナ)おおび3核プラチナ錯体およびカルボプラチンなどのプラチナ錯体)、駆虫剤(例えばペンタミジンイセチオネート)、アントラサイクリン、葉酸拮抗剤、代謝拮抗物質、化学療法増感剤、デュオカルマイシン、エトポシド、フッ化ピリミジン、イオノフォア、レキシトロプシン、ニトロソウレア、プラチノール、プレフォーミング化合物、プリン代謝拮抗物質、ピューロマイシン、放射線増感剤、ステロイド、タキサン、トポイソメラーゼ阻害剤、ビンカアルカロイド、抗菌剤、抗微小管剤などを含む。抗体がそのような薬物に結合されると、抗体は対応する抗原が発生する部位に薬物を方向付ける。抗体に結合できる他の薬剤および薬物は、当業者に公知であるか、または容易に想到され得る。
【0092】
個々の細胞毒性薬または細胞増殖抑制薬としては、例えばアンドロゲン、アントラマイシン(AMC)、アスパラギナーゼ、5−アザシチジン、アザチオプリン、ブレオマイシン、ブスルファン、ブチオニンスルホキシミン、カンプトセシン、カルボプラチン、カルムスチン(BSNU)、CC−1065、クロラムブシル、シスプラチン、コルヒチン、シクロホスファミド、シタラビン、シチジンアラビノシド、サイトカラシンB、ダカルバジン、ダクチノマイシン(以前はアクチノマイシン)、ダウノルビシン、ダカルバジン、ドセタキセル、ドキソルビシン、エストロゲン、5−フルオロデオキシウリジン、5−フルオロウラシル、グラミシジンD、ヒドロキシウレア、イダルビシン、イフォスファミド、イリノテカン、ロムスチン(CCNU)、メクロレタミン、メルファラン、6−メルカプトプリン、メトトレキサート、ミトラマイシン、ミトマイシンC、ミトキサントロン、ニトロイミダゾール、パクリタキセル、プリカマイシン、プロカルバジン、ストレプトゾトシン、テノポシド、6−チオグアニン、チオTEPA、トポテカン、ビンブラスチン、ビンクリスチン、ビノレルビン、VP−16およびVM−26が挙げられる。
【0093】
他の細胞毒性薬としては例えば、ドラスタチン(以下を参照)、エンジイン(例えばカリケアミシン)およびレキシトロプシン(米国特許第6,130,237号も参照のこと)などのDNA副溝バインダ、デュオカルマイシン、タキサン(例えばパクリタキセルおよびドセタキセル)、ピューロマイシン、CC−1065、SN−38、トポテカン、モルホリノ−ドキソルビシン、リゾキシン、シアノモルホリノ−ドキソルビシン、エキノマイシン、コンブレタスタチン、ネトロプシン、エポチロンA、BまたはD、エストラムスチン、クリプトフィシン、セマドチン、メイタンシノイド、ディスコデルモライド、エレウテロビン(eleutherobin)、およびミトキサントロンが挙げられる。
【0094】
ある実施形態において、細胞毒性薬は例えばドキソルビシン、メルファラン、ビンカアルカロイド、メトトレキサート、ミトマイシンCまたはエトポシドなどの化学療法薬である。加えて、CC−1065アナログ、カリケアミシン、メイタンシン、ドラスタチン10のアナログ、リゾキシン、およびパリトキシンなどの強力な薬剤は、タンパク質に結合することができる。
【0095】
具体的な実施形態において、細胞毒性薬または細胞増殖抑制薬は、オーリスタチンE(当該分野でドラスタチン−10としても公知)またはその誘導体である。通例、オーリスタチンE誘導体は例えばオーリスタチンEとケト酸との間で生成されたエステルである。例えばオーリスタチンEは、パラアセチル安息香酸またはベンゾイル吉草酸と反応させて、それぞれAEBおよびAEVBを生成させることができる。他の代表的なオーリスタチン誘導体としてはAFP、MMAF、およびMMAEが挙げられる。オーリスタチンEおよびその誘導体の合成および構造は、米国特許出願番号第09/845,786号(米国特許出願公開番号第20030083263号)および同第10/001,191号;国際特許出願PCT/US03/24209:国際特許出願PCT/US02/13435:および米国特許第6,323,315号;同第6,239,104号;同第6,034,065号;同第5,780,588号;同第5,665,860号;同第5,663,149号;同第5,635,483号;同第5,599,902号;同第5,554,725号;同第5,530,097号;同第5,521,284号;同第5,504,191号;同第5,410,024号;同第5,138,036号;同第5,076,973号;同第4,986,988号;同第4,978,744号;同第4,879,278号;同第4,816,444号;および同第4,486,414号に述べられている。
【0096】
ある実施形態において、細胞毒性薬または細胞増殖抑制薬は、抗チューブリン剤である。抗チューブリン剤の例としては、これに限定されるわけではないが、タキサン(例えばタキソール(登録商標)(パクリタキセル)、テキソテール(登録商標)(ドセタキセル))、T67(Tularik)、ビンカアルカロイド(例えばビンクリスチン、ビンブラスチン、ビンデシン、およびビノレルビン)、およびドラスタチン(例えばオーリスタチンE、AFP、MMAF、MMAE、AEB、AEVB)が挙げられる。他の抗チューブリン剤としては例えばバッカチン誘導体、タキサンアナログ(例えばエポチロンAおよびB)、ノコダゾール、コルヒチンおよびコルシミド(colcimid)、エストラムスチン、クリプトフィシン、セマドチン、メイタンシノイド、コンブレタスタチン、ディスコデルモライド、およびエレウテロビン(eleutherobin)が挙げられる。
【0097】
ある実施形態において、細胞毒性薬は抗チューブリン剤の別の群である、メイタンシノイドである。例えば具体的な実施形態において、メイタンシノイドはメイタンシンまたはDM−1である(ImmunoGen,Inc.;Chari et al.(1992),Cancer Res.52:127−131も参照のこと)。
【0098】
一部の実施形態において、治療剤は放射性同位体ではない。
【0099】
一部の実施形態において、細胞毒性薬または免疫抑制剤は、代謝拮抗物質である。代謝拮抗物質は、例えばプリンアンタゴニスト(例えばアゾチオプリンまたはミコフェノール酸モフェチル)、ジヒドロ葉酸レダクターゼ阻害剤(例えばメトトレキサート)、アシクロビル、ガンシクロビル、ジドブジン、ビダラビン、リバビリン、アジドチミジン、シチジンアラビノシド、アマンタジン、ジデオキシウリジン、ヨードデオキシウリジン、ポスカルネト(poscarnet)、またはトリフルリジンであり得る。
【0100】
他の実施形態において、細胞毒性薬または免疫抑制剤は、タクロリムス、シクロスポリンまたはラパマイシンである。さらなる実施形態において、細胞毒性薬は、アルデスロイキン、アレムツズマブ、アリトレチノイン、アロプリノール、アルトレタミン、アミフォスチン、アナストロゾール、三酸化ヒ素、ベキサロテン、ベキサロテン、カルステロン、カペシタビン、セレコキシブ、クラドリビン、ダルベポエチンアルファ、デニロイキンディフチトクス、デクスラゾキサン、プロピオン酸ドロモスタノロン、エピルビシン、エポエチンアルファ、エストラムスチン、エキセメスタン、フィルグラスチム、フロクスウリジン、フルダラビン、フルベストラント、ゲムシタビン、ゴセレリン、イダルビシン、イフォスファミド、メシル酸イマチニブ、インターフェロンアルファ−2a、イリノテカン、レトロゾール、ロイコボリン、レバミゾール、メクロレタミンまたは窒素マスタード、メゲストロール、メンサ、メトトレキサート、メトキサレン、ミトマイシンC、ミトタン、ナンドロロンフェンプロピオナート、オプレルベキン、オキサリプラチン、パミドロナート、ペグアデマーゼ、ペグアスパルガーゼ、ペグフィルグラスチム、ペントスタチン、ピポブロマン、プリカマイシン、ポリフィマーナトリウム、プロカルバジン、キナクリン、ラスブリカーゼ、サルグラモスチム、ストレプトゾシン、タモキシフェン、テモゾロマイド、テニポシド、テストラクトン、チオグアニン、トレミフェン、トレチノイン、ウラシルマスタード、バルルビシン、ビンブラスチン、ビンクリスチン、ビノレルビンおよびソレドロナートである。
【0101】
一部の実施形態において、薬剤は免疫抑制剤である。免疫抑制剤は例えばガンシクロビル、タクロリムス、シクロスポリン、ラパマイシン、シクロホスファミド、アザチオプリン、ミコフェノール酸モフェチルまたはメトトレキサートであり得る。あるいは免疫抑制剤は例えば、グルココルチコイド(例えばコルチゾールまたはアルドステロン)またはグルココルチコイドアナログ(例えばプレドニゾンまたはデキサメタゾン)であり得る。
【0102】
一部の実施形態において、免疫抑制剤は抗炎症剤、例えばアリールカルボン酸誘導体、ピラゾール含有誘導体、オキシカム誘導体およびニコチン酸誘導体である。抗炎症剤のクラスとしては例えばシクロオキシゲナーゼ阻害剤、5−リポオキシゲナーゼ阻害剤、およびロイコトリエンレセプターアンタゴニストが挙げられる。
【0103】
適切なシクロオキシゲナーゼ阻害剤としては、メクロフェナム酸、メフェナム酸、カルプロフェン、ジクロフェナク、ジフルニサル、フェンブフェン、フェノプロフェン、イブプロフェン、インドメタシン、ケトプロフェン、ナブメトン、ナプロキセン、スリンダク、テノキシカム、トルメチン、およびアセチルサリチル酸が挙げられる。
【0104】
適切なリポオキシゲナーゼ阻害剤としては、酸化還元阻害剤(例えばカテコールブタン誘導体、ノルジヒドログアヤレチック酸(NDGA)、マソプロコール、フェニドン、イアノパレン(Ianopalen)、インダゾリノン、ナファザトロム、ベンゾフラノール、アルキルヒドロキシアミン)、および非酸化還元阻害剤(例えばヒドロキシチアゾール、メトキシアルキルチアゾール、ベンゾピランおよびその誘導体、メトキシテトラヒドロピラン、ボスウェリア酸およびボスウェリア酸のアセチル化誘導体、ならびにシクロアルキルラジカルによって置換されたキノリンメトキシフェニル酢酸)、および酸化還元阻害剤の前駆物質が挙げられる。
【0105】
他の適切なリポオキシゲナーゼ阻害剤としては、抗酸化剤(例えばフェノール、没食子酸プロピル、フラボノイドおよび/またはフラボノイドを含む天然基質、フラボンのヒドロキシル化誘導体、フラボノール、ジヒドロケルセチン、ルテオリン、ガランギン、オロボル(orobol)、カルコンの誘導体、4,2’,4’−トリヒドロキシカルコン、オルト−アミノフェノール、N−ヒドロキシウレア、ベンゾフラノール、エブセレンおよび還元セレン含有酵素の活性を上昇させる種)、鉄キレート剤(例えばヒドロキサム酸およびその誘導体、N−ヒドロキシウレア、2−ベンジル−1−ナフトール、カテコール、ヒドロキシルアミン、カルノソールトロロックスC、カテコール、ナフトール、スルファサラジン、ジロイトン(zyleuton)、5−アントラニル酸および4−(オメガ−アリールアルキル)フェニルアルカン酸)、イミダゾール含有化合物(例えばケトコナゾールおよびイトラコナゾール)、フェノチアジン、およびベンゾピラン誘導体が挙げられる。
【0106】
なお他の適切なリポオキシゲナーゼ阻害剤としては、エイコサノイド(例えばオクタデカテトラエン酸、エイコサテトラエン酸、ドコサペンタエン酸、エイコサヘキサエン酸およびドコサヘキサエン酸ならびにそのエステル、PGE1(プロスタグランジンE1)、PGA2(プロスタグランジンA2)、ビプロストール、15−モノヒドロキシエイコサテトラエン酸、15−モノヒドロキシ−エイコサトリエン酸、および15−モノヒドロキシエイコサペンタエン酸、およびロイコトリエンB5、C5およびD5)の阻害剤、カルシウム流を妨害する化合物、フェノチアジン、ジフェニルブチルアミン、ベラパミル、フスコシド、クルクミン、クロロゲン酸、コーヒー酸、5,8,11,14−エイコサテトライエン酸(ETYA)、ヒドロキシフェニルレチンアミド、イオナパレン、エスクリン、ジエチルカルバマジン、フェナントリン、バイカレイン、プロキシクロミル、チオエーテル、ジアリルスルフィドおよびジ−(1−プロペニル)スルフィドが挙げられる。
【0107】
ロイコトリエンレセプターアンタゴニストとしては、カルシトリオール、オンタゾラスト、Bayer Bay−x−1005、Ciba−Geigy CGS−25019C、エブセレン、Leo Denmark ETH−615、Lilly LY−293111、Ono ONO−4057、Terumo TMK−688、Boehringer Ingleheim BI−RM−270、Lilly LY 213024, Lilly LY 264086、Lilly LY 292728、Ono ONO LB457、Pfizer 105696、Perdue Frederick PF 10042、Rhone−Poulenc Rorer RP 66153、SmithKline Beecham SB−201146、SmithKline Beecham SB−201993、SmithKline Beecham SB−209247、Searle SC−53228、Sumitamo SM 15178、American Home Products WAY 121006、Bayer Bay−o−8276、Warner−Lambert CI−987、Warner−Lambert CI−987BPC−15LY 223982,Lilly LY 233569、Lilly LY−255283, MacroNex MNX−160、Merck and Co.MK−591、Merck and Co.MK−886、Ono ONO−LB−448、Purdue Frederick PF−5901、Rhone−Poulenc Rorer RG 14893、Rhone−Poulenc Rorer RP 66364、Rhone−Poulenc Rorer RP 69698、Shionoogi S−2474、Searle SC−41930、Searle SC−50505、Searle SC−51146、Searle SC−52798、SmithKline Beecham SK&F−104493、Leo Denmark SR−2566、Tanabe T−757およびTeijin TEI−1338が挙げられる。
【0108】
(毒素)
毒素は、腫瘍、寄生虫または微生物細胞に関連する抗原に特異的な抗体へ有用に結合体化される。毒素は例えば植物(例えばリシンまたはアブリン)、動物(例えばヘビ毒)、または微生物(例えばジフテリアまたは破傷風毒素)に由来する。
【0109】
抗体以外に、薬物または毒素は、他のキャリアタンパク質、例えばアルブミンに結合体化させることができる。
【0110】
(タンパク質への薬物の結合体化)
薬物は、タンパク質の結合箇所と反応性である基を有するか、それを含むように修飾される。例えば薬物は、アルキル化(例えば抗体リジンのε−アミノ基またはタンパク質のN末端にて)、酸化炭水化物の還元的アミノ化、ヒドロキシ基とカルボキシル基との間のトランスエステル化、アミノ基またはカルボキシル基におけるアミド化、チオールへの結合によって付着させることができる。結合に使用できる化学作用の例については、例えばCurrent Protocols in Protein Science(John Wiley & Sons,Inc.),Chapter 15(Chemical Modifications of Proteins)(その開示は、参照によりその全体が本明細書に組み入れられている)を参照。
【0111】
例えばタンパク質の化学活性化が遊離チオール基の形成を生じるとき、タンパク質はスルフヒドリル活性剤と結合体化され得る。1つの局面において、薬剤は、遊離チオール基に実質的に特異的な薬剤である。そのような薬剤としては例えば、マレイミド、ハロアセトアミド(例えばヨード、ブロモまたはクロロ)、ハロエステル(例えばヨード、ブロモまたはクロロ)、ハロメチルケトン(例えばヨード、ブロモまたはクロロ)、ベンジル型ハライド(例えばヨージド、ブロミドまたはクロライド)、ビニルスルホンおよびピリジルチオが挙げられる。
【0112】
(スルフヒドリル反応剤)
スルフヒドリル反応剤としては、ヨードアセトアミドなどのアルファ−ハロアセチル化合物、N−エチルマレイミドなどのマレイミド、アセテート、クロライドまたはニトレートの対イオンを含む3,6−ビス−(水銀メチル)ジオキサンなどの水銀誘導体、ジスルフィドジオキシド誘導体などのジスルフィド誘導体、ポリメチレンビスメタンチオスルホン酸塩試薬およびクラベセイン(還元抗体のジスルフィド結合に添加されることが示されている、2個の遊離スルフヒドリル基を含有するフルオレセインの蛍光誘導体)が挙げられる。
【0113】
ヨードアセテートなどのアルファ−ハロアセチル化合物は、スルフヒドリル基と容易に反応してアミドを生成する。これらの化合物は、遊離チオールをカルボキシメチル化するために使用された。それらは厳密にSH特異性でなく、アミンと反応する。反応は、ハライドの置換を生じる、チオラートイオンの求核攻撃を包含する。反応性ハロアセチル部分X−−CH2CO−−は、種々の目的で化合物に組み込まれている。例えばブロモトリフルオロアセトンは、F−19包含に使用され、N−クロロアセチルヨードチラミンは、放射性ヨウ素のタンパク質への導入に利用されている。
【0114】
N−エチルマレイミドなどのマレイミドは、特に他の基がプロトン化される7以下のpH値では、スルフヒドリル基にかなり特異性であると見なされている。チオールは、マレイミドによるMichael反応を受けて、二重結合への付加体を独占的に生じる。生じたチオエーテル結合は非常に安定である。それらは、はるかに低速にてアミノ基およびイミダゾリル基とも反応する。例えばpH7では、単純なチオールとの反応は、対応するアミンとの反応よりも約1,000倍高速である。反応に関連する300nm領域における特徴的な吸光度の変化は、反応を監視するための好都合な方法を提供する。これらの化合物は低いpHにおいて安定であるが、高いpHにおいて加水分解を受けやすい。一般にWong, Chemistry of Protein Conjugation and Cross−linking;CRC Press,Inc.,Boca Raton,1991:Chapters 2および4)を参照のこと。
【0115】
スルフヒドリルと本質的に反応性でない分子(例えば薬物)は、化学的に活性化されたタンパク質に、目的の分子と反応性の基とスルフヒドリル反応性基の両方を持つ二官能性架橋剤によってなお結合体化させることができる。この薬剤は、目的の分子(例えばアミノ基、カルボキシル基またはヒドロキシ基)および化学的に活性されたタンパク質の両方と同時に反応できるか、または目的の分子を誘導体化して、薬剤に由来する部分のために次にスルフヒドリル反応性であるパートナー分子を形成するために使用できるか、または化学的に活性されたタンパク質を誘導体化して、目的の分子と反応性にするために使用できる。
【0116】
(リンカー)
薬物はリンカーによってタンパク質に結合され得る。適切なリンカーとしては、例えば開裂性および非開裂性リンカーが挙げられる。開裂性リンカーは通例、細胞内条件下で開裂を受けやすい。適切な開裂性リンカーとしては、例えば、細胞内プロテアーゼ、例えばリソソームプロテアーゼまたはエンドソームプロテアーゼによって開裂性であるペプチドリンカーが挙げられる。例示的な実施形態では、リンカーはジペプチドリンカー、例えばバリン−シトルリン(val−cit)またはフェニルアラニン−リジン(phe−lys)リンカーであり得る。他の適切なリンカーとしては、5.5未満のpHで加水分解可能なリンカー、例えばヒドラゾンリンカーが挙げられる。さらなる適切な開裂性リンカーとしては、ジスルフィドリンカーが挙げられる。
【0117】
リンカーは、タンパク質への結合のための基を含むことができる。例えばリンカーとしては、アミノ、ヒドロキシル、カルボキシルまたはスルフヒドリル反応性基(例えばマレイミド、ハロアセトアミド(例えばヨード、ブロモまたはクロロ)、ハロエステル(例えばヨード、ブロモまたはクロロ)、ハロメチルケトン(例えばヨード、ブロモまたはクロロ)、ベンジル型ハライド(例えばヨージド、ブロミドまたはクロライド)、ビニルスルホンおよびピリジルチオ)が挙げられ得る。一般に、Wong,Chemistry of Protein Conjugation and Cross−linking;CRC Press,Inc.,Boca Raton,1991を参照のこと。
【0118】
特定の実施形態において、抗体またはタンパク質薬物結合体は、以下の式:
【0119】
【化4】
の結合体あるいはその薬学的に許容される塩または溶媒和物であり得る。
式中:
Abは、抗体または他のタンパク質であり、
Aは、ストレッチャー単位であり、
aは、0または1であり、
各Wは独立して、リンカー単位であり、
wは、0〜12の範囲の整数であり、
Yは、スペーサー単位であり、
yは、0、1または2であり、
pは、1〜約20の範囲であり、
Dは、薬物、標識または他の分子であり、
zは、タンパク質上の潜在的な結合部位の数であり、p<zである。
他の実施形態において、pは例えば、2、4、8、10、12、16、25またはそれ以上であり得る。
【0120】
ストレッチャー単位は、リンカー単位を抗体または他のタンパク質に結合させることができる。ストレッチャー単位は、抗体または他のタンパク質の官能基と結合を形成できる官能基を有する。有用な官能基としては、これに限定されるわけではないが、スルフヒドリル(−SH)、アミノ、ヒドロキシル、カルボキシ、炭水化物のアノマーヒドロキシル基、およびカルボキシルが挙げられる。
【0121】
リンカー単位は通例、アミノ酸単位、例えばジペプチド、トリペプチド、テトラペプチド、ペンタペプチド、ヘキサペプチド、ヘプタペプチド、オクタペプチド、ノナペプチド、デカペプチド、ウンデカペプチドまたはドデカペプチド単位である。リンカー単位は、細胞内で開裂性または非開裂性であり得る。
【0122】
スペーサー単位は存在する場合、リンカー単位を薬物に結合させる。あるいはスペーサー単位は、リンカー単位が非存在のときに、ストレッチャー単位を薬物部分に結合させることができる。スペーサー単位は、リンカー単位およびストレッチャー単位が非存在のときに、抗体またはタンパク質に結合することもできる。
【0123】
(VI.結合体およびその使用)
(インビトロ免疫診断)
ある実施形態において、タンパク質(例えば抗体)は、インビトロ免疫診断で使用するために、検出可能な標識に結合される。標識は、化学的に活性された抗体の結合箇所(例えば遊離チオール基)に直接または間接的に結合可能である、放射性標識、フルオロフォア、または酵素であり得る。サンプルは、臨床性(例えば血液、尿、精液、または脳脊髄液、あるいは固形組織または臓器)または非臨床性(例えば土壌、水、食物)でよい。アッセイは、定性的または非定性的であり、サンドイッチおよび比較形式を含むいずれの所望の形式でもよい。多数の免疫アッセイ形式、標識、固定化技術などは、参考として本明細書に援用される、以下の刊行物に開示されている:O’Sullivan(1976),Annals Clin.Biochem.16:221−240;McLaren(1981),Med.Lab.Sci.38:245−51;Ollerich(1984),J.Clin.Chem.Clin.Biochem.22:895−904;Ngo and Lenhoff(1982),Mol.Cell.Biochem.,44:3−12。
【0124】
(免疫造影)
免疫結合体は、インビボ免疫造影にも使用され得る。この目的のために、タンパク質(例えば抗体)は、被験体またはその一部、例えば臓器の中のその位置または場所の外部描出を可能にする手段によって標識される。通例、免疫造影剤は、適切な放射性同位体によって直接(テクネチウムと同様に)または間接的に(キレート化インジウムと同様に)標識された抗体である。患者への注射後に、結合体の場所は、放射性標識によって放出された粒子に感受性である検出器によって、例えばガンマエミッタの場合にはガンマ−シンチレーションカメラによって追跡できる。
【0125】
(免疫療法)
免疫療法では、タンパク質は適切な薬物、例えば細胞毒性薬または細胞増殖抑制薬、免疫抑制剤、放射性同位体、毒素などに結合体化することができる。結合体は、腫瘍細胞または癌細胞の増殖を阻害するために、腫瘍または癌細胞においてアポトーシスを発生させるために、または患者の癌を処置するために使用できる。結合体はしたがって、動物の癌の処置に核種の状況において使用できる。結合体は、薬物を腫瘍細胞または癌細胞へ送達するために使用できる。理論に縛られることなく、一部の実施形態において、結合体は癌細胞または腫瘍関連抗原と結合または会合して、結合体および/または薬物は、レセプター仲介エンドサイトーシスを通じて腫瘍細胞または癌細胞内に取り込むことができる。抗原は腫瘍細胞または癌細胞に付着できるか、あるいは腫瘍細胞または癌細胞に関連する細胞外マトリクスタンパク質であり得る。細胞内に入ると、結合体内の(例えばリンカーにおける)1個以上の特異性ペプチド配列は、1個以上の腫瘍細胞または癌細胞関連プロテアーゼによって加水分解によって開裂され、薬物の放出を引き起こす。放出された薬物は次に、細胞内で自由に移動して、細胞毒性または細胞増殖抑制あるいは他の活性を誘起する。一部の実施形態において、薬物は腫瘍細胞または癌細胞の外側の抗体から開裂して、次に薬物は細胞に浸透するか、または細胞表面にて作用する。
【0126】
それゆえ、一部の実施形態において、結合体または他のタンパク質は、腫瘍細胞または癌細胞に結合する。一部の実施形態において、結合体は、腫瘍細胞または癌細胞の表面上にある腫瘍細胞または癌細胞抗原に結合する。他の実施形態において、結合体は、腫瘍細胞または癌細胞に関連する細胞外マトリクスタンパク質である腫瘍細胞または癌細胞抗原に結合する。
【0127】
特定の腫瘍細胞または癌細胞に対するタンパク質の特異性は、最も効果的に処置される腫瘍または癌を判定するために重要であり得る。例えば抗CD30を有する抗体または抗CD40抗体あるいは他の結合タンパク質は、血液悪性腫瘍を処置するのに有用であり得る。
【0128】
タンパク質−薬物結合体によって処置できる他の特定の種類の癌としては、これに限定されるわけではないが:線維肉腫、粘液肉腫、脂肪肉腫、軟骨肉腫、骨原性肉腫、脊索腫、血管肉腫、内皮肉腫、リンパ管肉腫、リンパ管内皮肉腫、滑膜腫、中皮腫、ユーイング腫瘍、平滑筋肉腫、横紋筋肉腫、結腸癌、結腸直腸癌、腎臓癌、膵臓癌、骨肉種、乳癌、卵巣癌、前立腺癌、食道癌、胃癌、口腔癌、鼻癌、咽喉癌、へん平上皮細胞癌、基底細胞癌、腺癌汗腺癌、脂腺癌、乳頭状癌、乳頭状腺癌、嚢胞腺癌、髄様癌、気管支原性癌、腎細胞癌、肝臓癌、胆管癌、絨毛癌、精上皮腫、胎生期癌、ウィルムス腫瘍、子宮頚癌、子宮癌、睾丸癌、小細胞肺癌、膀胱癌、肺癌、上皮癌、神経膠腫、多形性膠芽腫、星状細胞腫、髄芽細胞腫、頭蓋咽頭腫、上衣細胞腫、松果体腫、血管芽細胞腫、聴神経腫、乏突起膠腫、髄膜腫、皮膚癌、メラノーマ、神経芽細胞腫、網膜芽細胞腫を含む固形腫瘍、血液媒介癌(これに限定されるわけではないが:急性リンパ芽球性白血病“ALL”、急性リンパ芽球B細胞白血病、急性リンパ芽球性T細胞白血病、急性骨髄芽球性白血病“AML”、急性骨髄球性白血病“APL”、急性単芽球性白血病、急性赤白血病、急性巨核芽球性白血病、急性骨髄単球性白血病、急性非リンパ球性白血病、急性未分化白血病、慢性骨髄球性白血病“CML”、慢性リンパ球性白血病“CLL”、毛様細胞白血病、多発性骨髄腫を含む)、急性および慢性白血病(例えばリンパ芽球性、骨髄性、リンパ球性、および骨髄球性白血病)、およびリンパ腫(例えばホジキン病、非ホジキンリンパ腫、多発性骨髄腫、ワルデンシュトレームマクログロブリン血症、重鎖病、および真性赤血球増加)が挙げられる。タンパク質は、結合特異性腫瘍または癌ターゲティングを提供する。
【0129】
(癌のマルチモード療法)
上述のように、これに限定されるわけではないが、腫瘍、転移を含む癌、あるいは制御できない細胞増殖を特徴とする他の疾患または障害は、タンパク質−薬物結合体の投与によって処置または予防できる。
【0130】
他の実施形態において、結合体の有効量および化学療法剤をその必要がある患者に投与することを含む、癌を処置または防止するための方法が提供される。一部の実施形態において、化学療法剤は、それによって癌の処置が抗療性であることが見出されていないということである。一部の実施形態において、化学療法剤は、それによって癌の処置が抗療性であることが見出されているということである。結合体は、処置、例えば癌の処置のための外科手術も受けた患者に投与できる。別の実施形態において、処置の追加の方法は放射線療法である。
【0131】
例示的な実施形態において、タンパク質−薬物結合体は、化学療法剤と、または放射線療法と同時に投与される。別の例示的な実施形態において、化学療法剤または放射線療法は、タンパク質−薬物結合体の投与の、1つの局面において、少なくとも1時間、5時間、12時間、1日、1週間、1ヶ月前または後に、さらなる局面において、結合体の投与の、数ヶ月(例えば3ヶ月まで)前または後に投与される。
【0132】
化学療法剤は、一連のセッションに渡って投与できる。以下に挙げる化学療法剤の1つまたは組合せを投与できる。放射線に関しては、処置される癌の種類に応じていずれかの放射線療法プロトコルが使用できる。例えば制限するわけではないが、X線照射を投与できる;特に高エネルギーメガボルテージ(1MeVエネルギーを超える照射)が深い腫瘍に使用可能であり、電子ビームおよびオルトボルテージX線照射は皮膚癌に使用できる。ガンマ線放出放射性同位体、例えばラジウム、コバルトおよび他の元素の放射性同位体も投与できる。
【0133】
加えて、化学療法または放射線療法の毒性が強すぎることが判明した、または判明しうる場合に、例えば処置される対象によって許容されないまたは耐えられない副作用を引き起こす場合に、タンパク質−薬物結合体による癌の処置方法が化学療法または放射線療法の代替手段として提供される。処置される動物は必要に応じて、どの処置が許容されるまたは耐えられることが判明するかによって、外科手術、放射線療法または化学療法などの別の癌処置によって処置できる。
【0134】
タンパク質−薬物結合体は、インビトロまたは生体外方式で、例えばこれに限定されるわけではないが、白血病およびリンパ腫を含むある癌の処置に使用することも可能であり、そのような処置は自己幹細胞移植を包含する。これは動物の自己造血幹細胞が収穫され、癌細胞すべてが一掃される多ステッププロセスを含むことができ、動物の残存する骨髄細胞個体群は次に、付随する高用量放射線療法を伴って、または伴わずに高用量の結合体の投与によって全滅され、そして幹細胞グラフトは動物へ輸液によって戻される。次に対症療法が与えられて、その間に骨髄機能が修復され、動物が回復する。
【0135】
(癌の多剤療法)
タンパク質−薬物結合体の有効量および抗癌剤である別の治療剤をその必要がある患者に投与することを含む癌を処置する方法が開示される。適切な抗癌剤としては、これに限定されるわけではないが、メトトレキサート、タキソール、L−アスパラギナーゼ、メルカプトプリン、チオグアニン、ヒドロキシウレア、シタラビン、シクロホスファミド、イフォスファミド、ニトロソウレア、シスプラチン、カルボプラチン、ミトマイシン、ダカルバジン、プロカルバジン、トポテカン、窒素マスタード、シトキサン、エトポシド、5−フルオロウラシル、BCNU、イリノテカン、カンプトセシン、ブレオマイシン、ドキソルビシン、イダルビシン、ダウノルビシン、ダクチノマイシン、プリカマイシン、ミトキサントロン、アスパラギナーゼ、ビンブラスチン、ビンクリスチン、ビノレルビン、パクリタキセル、およびドセタキセルが挙げられる。
【0136】
抗癌剤としては、これに限定されるわけではないが、アルキル化剤、例えば窒素マスタード(例えばシクロホスファミド,イフォスファミド、トロホスファミド、クロラムブシル、メルファラン)、ニトロソウレア(例えばカルムスチン(BCNU)、ロムスチン(CCNU))、アルキルスルホナート(例えばブスルファン、トレオスルファン)、トリアゼン(例えばデカルバジン)、プラチナ含有化合物(例えばシスプラチン、カルボプラチン);植物アルカロイド、例えばビンカアルカロイド(例えばビンクリスチン、ビンブラスチン、ビンデシン、ビノレルビン)、タキソイド(例えばパクリタキセル、ドセタキソール);DNAトポイソメラーゼ阻害剤、例えばエピポドフィリン(例えばエトポシド、テニポシド、トポテカン、9−アミノカンプトセシン、カンプトセシン、クリスナトール、ミトマイシン(例えばミトマイシンC);代謝拮抗物質、例えば葉酸拮抗剤、例えばDHFR阻害剤(例えばメトトレキサート、トリメトレキサート)、IMPデヒドロゲナーゼ阻害剤(ミコフェノール酸、チアゾフリン、リバビリン、EICAR)およびリボヌクレオチドレダクターゼ阻害剤(例えばヒドロキシウレア、デフェロキサミン)、ピリミジンアナログ、例えばウラシルアナログ(5−フルオロウラシル、フロクスウリジン、ドキシフルリジン、ratitrexed)、シトシンアナログ(例えばシタラビン(ara C)、シトシンアラビノシド、フルダラビン)、およびプリンアナログ(例えばメルカプトプリン、チオグアニン);ホルモン療法、例えばレセプターアンタゴニスト、例えば抗エストロゲン(例えばタモキシフェン、ラロキシフェン、メゲストロール)、LHRHアゴニスト(例えばgoscrclin、酢酸ロイプロリド)、および抗アンドロゲン(例えばフルタミド、ビカルタミド;レチノイド/デルトイド、例えばビタミンD3アナログ(例えばEB 1089、CB 1093、KH 1060)、光線力学療法(例えばベルトポルフィン(BPD−MA)、フタロシアニン、光増感剤Pc4、デメトキシ−ヒポクレリンA(2BA−2−DMHA))、サイトカイン(例えばインターフェロン−α、インターフェロン−γ、腫瘍壊死因子)などの薬物はもちろんのこと、ゲムシタビン、ベルケイド、ラバミド、サラミド、イソプレニル化阻害剤(例えばラバスタチン)、ドーパミン神経毒(例えば1−メチル−4−フェニルピリミジンイオン)、細胞サイクル阻害剤(例えばスタウロスポリン)、アクチノマイシン(例えばアクチノマイシンD、ダクチノマイシン)、ブレオマイシン、ブレオマイシンA2、ブレオマイシンB2、ペプロマイシン)、アントラサイクリン(ダウノルビシン、ドキソルビシン(アドリアマイシン)、イダルビシン、エピルビシン、ピラルビシン、ゾルビシン、ミトキサントロン)、MDR阻害剤(例えばベラパミル)、およびCa2+ATPase阻害剤(例えばタプシガーギン)などの他の薬物が挙げられる。
【0137】
(自己免疫疾患の処置)
タンパク質−薬物結合体は、自己免疫疾患を引き起こす細胞を殺す、またはその複製を阻害するために、あるいは自己免疫疾患を処置するために有用である。したがって結合体は、患者の自己免疫疾患の処置のために種々の状況で使用できる。結合体は、薬物を標的細胞に送達するために使用できる。理論に縛られることなく、1つの実施形態において、結合体は標的細胞の表面上の抗原と会合して、次に結合体はレセプター仲介エンドサイトーシスを通じて標的細胞内に取り込まれる。細胞内に入ると、(例えばリンカー内の)1個以上の特異性ペプチド配列は、酵素または加水分解によって開裂され、薬物の放出を引き起こす。放出された薬物は次に、サイトソルにおいて自由に移動して、細胞毒性または細胞増殖抑制活性を誘起する。代わりの実施形態において、薬物は標的細胞の外側の結合体から開裂して、次に薬物は細胞に浸透する。
【0138】
一部の実施形態において、タンパク質−薬物結合体は自己免疫抗原に結合する。1つの局面において、抗原は自己免疫状態に関与する細胞の表面上にある。一部の実施形態において、抗体は細胞の表面上にある自己免疫抗原に結合する。例示的な実施形態において、抗体は自己免疫疾患状態に関連する活性化リンパ球に結合する。さらなる実施形態において、結合体は、特定の自己免疫疾患に関連する自己免疫抗体を産生する細胞を殺す、またはその増殖を阻害する。
【0139】
タンパク質−薬物結合体によって処置できる特定の種類の自己免疫疾患としては、これに限定されるわけではないが、Th2リンパ球関連障害(例えばアトピー性皮膚炎、アトピー性喘息、鼻結膜炎、アレルギー性鼻炎、Omenn症候群、全身性硬化症および移植片対宿主疾患);Th1リンパ球関連障害(例えば関節リウマチ、多発性硬化症、乾癬、シェーグレン症候群、橋本甲状腺炎、グレーブス病、原発性胆汁性肝硬変症、ウェゲナー肉芽腫症および結核);および活性化Bリンパ球関連障害(例えば全身性エリテマトーデス、グッドパスチャー症候群、関節リウマチおよびI型糖尿病)が挙げられる。他の自己免疫疾患としては、これに限定されるわけではないが、慢性活動性肝炎、アジソン病、アレルギー性肺胞炎、アレルギー反応、アレルギー性鼻炎、アルポート症候群、アナフィラキシー、強直性脊椎炎、抗リン脂質症候群、関節炎、回虫症、アスペルギルス症、アトピー性アレルギー、アトピー性皮膚炎、アトピー性鼻炎、ベーチェット症候群、愛鳥家肺、気管支喘息、カプラン症候群、心筋症、セリアック病、シャーガス病、慢性糸球体腎炎、コーガン症候群、寒冷凝集素症、先天性風疹感染、CREST症候群、クーロン病、クリオグロブリン血症、クッシング症候群、皮膚筋炎、円板状狼瘡、ドレッサー症候群、イートン・ランバート症候群、エコーウィルス感染、脳脊髄炎、内分泌眼疾患、エプスタイン−バーウィルス感染、ウマ肺気腫、エリマトーデス、エヴァンス症候群、フェルティ症候群、線維筋痛、フックス毛様体炎、胃萎縮症、消化管アレルギー、巨細胞性動脈炎、糸球体腎炎、グッドパスチャー症候群、移植片対宿主疾患、グレーブス病、ギラン−バレー病、橋本甲状腺炎、溶血性貧血、シェンライン−ヘノッホ紫斑、特発性成人胆管減少症、特発性肺線維症、IgAネフロパシー、炎症性大腸炎、インシュリン依存性真性糖尿病、若年性関節炎、若年性真性糖尿病(I型)、ランバート−イートン症候群、蹄葉炎、扁平苔癬、ルポイド肝炎、狼瘡、リンパ球減少症、メニエール病、混合結合組織病、多発性硬化症、重症筋無力症、悪性貧血、多腺性症候群、初老期認知症、原発性無ガンマグロブリン血症、原発性胆汁性肝硬変、乾癬、乾癬性関節炎、レイノー現象、反復流産、ライター症候群、リウマチ熱、関節リウマチ、サンプター症候群、住血吸虫症、シュミット症候群、強皮症、シャルマン症候群、シェーグレン症候群、スティフマン症候群、交感性眼炎、全身性エリマトーデス、高安動脈炎、側頭動脈炎、甲状腺炎、血小板減少症、甲状腺中毒症、中毒性表皮壊死症、B型インスリン抵抗性、I型真性糖尿病、潰瘍性大腸炎、ブドウ膜炎、白斑、ワルデンシュトレーム型マクログロブリン血症、およびウェゲナー肉芽腫症が挙げられる。
【0140】
(自己免疫疾患の多剤療法)
タンパク質−薬物結合体の有効量を単独で、または自己免疫疾患の処置で公知の別の治療剤と組合せて、その必要がある患者に投与することを含む、自己免疫疾患を処置する方法も開示されている。抗自己免疫疾患剤としては、これに限定されるわけではないが、以下が挙げられ得る:シクロスポリン、シクロスポリンA、ミコフェノール酸モフェチル、シロリムス、タクロリムス、エナネルセプト(enanercept)、プレドニゾン、アザチオプリン、メトトレキサート、シクロホスファミド、アミノカプロン酸、クロロキン、ヒドロキシクロロキン、ヒドロコルチゾン、デキサメタゾン、クロラムブシル、DHEA、ダナゾール、ブロモクリプチン、メロキシカムおよびインフリキシマブ。
【0141】
(感染性疾患の処置)
タンパク質−薬物結合体は、感染性疾患を引き起こす細胞を殺す、またはその複製を阻害するために、あるいは感染性疾患を処置するために有用である。したがって結合体は、患者の感染性疾患の処置のために種々の状況で使用できる。ADCは、薬物を標的細胞に送達するために使用できる。1つの実施形態において、抗体は感染性疾患細胞に結合する。一部の実施形態において、結合体は、特定の感染性疾患を引き起こす細胞を殺すか、またはその増殖を阻害する。結合体によって処置できる特定の種類の感染性疾患としては、これに限定されるわけではないが以下が挙げられる:細菌性疾患、例えばジフテリア、百日咳、オカルト菌血症、尿路感染症、胃腸炎、蜂巣炎、喉頭蓋炎、気管炎、アデノイド肥大、咽後膿瘍、膿痂疹、膿瘡、肺炎、心内膜炎、敗血症性関節炎、肺炎球菌性腹膜炎、細菌性髄膜炎、急性化膿性髄膜炎、尿道炎、子宮頚管炎、直腸炎、咽頭炎、卵管炎、精巣上体炎、淋病、梅毒、リステリア症、炭疽病、ノカルジア症、サルモネラ、腸チフス、赤痢、結膜炎、副鼻腔炎、ブルセラ症、ツラレミア、コレラ、腺ペスト、破傷風、壊死性腸炎、および放線菌症;混合嫌気性感染症、例えば梅毒、回帰熱、レプトスピラ症、ライム病、ネズミ咬熱、結核、リンパ節炎、ハンセン病、クラミジア症、クラミジア肺炎、トラコーマおよび封入体性結膜炎;全身性真菌疾患、例えばヒストプラスマ症、コクシジオイデス症、ブラストミセス症、スポロトリクム症、クリプトコッカス症、全身性カンジダ症、アスペルギルス症、ムコール菌症、足菌腫、およびクロモミコーシス;リケッチア症、例えばチフス、ロッキ−山紅斑熱、エーリキア症、東半球ダニ媒介性リケッチア症、リケッチア痘症、Q熱およびバルトネラ症;寄生虫症、例えばマラリア、バベシア症、アフリカ睡眠病、シャーガス病、リーシュマニア症、ダムダム熱、トキソプラズマ症、髄膜脳炎、角膜炎、アメーバ症、ランブル鞭毛虫症、クリプトスポリジウム症、イソスポーラ症、サイクロスポリン、微胞子虫症、回虫症、鞭虫感染、鉤虫感染、線虫感染、眼幼虫移行症、旋毛虫病、糸状虫症、リンパ管フィラリア症、ロア糸状虫症、河川盲目症、犬糸状虫感染、住血吸虫症、水泳者かゆみ症、肺吸虫症、肝吸虫症、肝蛭症、肥大吸虫症、オピストルキス症、条虫感染、包虫症、および多包虫症;ウィルス性疾患、例えば麻疹、亜急性硬化性全汎脳炎、風邪、流行性耳下腺炎、風疹、バラ疹、第5病、水痘、呼吸器合胞体ウィルス感染、クループ、気管支梢炎、感染性単核球症、ポリオ、ヘルパンギーナ、手足口病、ボーンホルム病、陰部ヘルペス、陰部疣贅、無菌性髄膜炎、心筋炎、心膜炎、胃腸炎、後天性免疫不全症候群(AIDS)、ヒト免疫不全ウィルス(HIV)、ライ症候群、川崎症候群、インフルエンザ、気管支炎、ウィルス性「歩く」肺炎、急性発熱性呼吸疾患、急性咽頭結膜熱、流行性角結膜炎、単純ヘルペスウイルス1(HSV−1)、単純ヘルペスウイルス2(HSV−2)、帯状疱疹、巨細胞封入体症、狂犬病、進行性多病巣性白質脳障害、クル、致死性家族性不眠症、クロイツフェルト−ヤコブ病、ゲルストマン−ストロイスラー−シャインカー症候群、熱帯性痙性不全対麻痺、西部ウマ脳脊髄炎、カリフォルニア脳炎ウィルス、セントルイス脳炎、黄熱病、デング熱、リンパ球性脈絡髄膜炎、ラッサ熱、出血熱、繁多ウィルス肺症候群、マールブルグウィルス感染、エボラウィルス感染および天然痘。
【0142】
(感染性疾患の多剤療法)
タンパク質−薬物結合体を単独で、または抗感染性疾患剤である別の治療剤と組合せてその必要がある患者に投与することを含む、感染性疾患を処置する方法も開示されている。抗感染性疾患としては、これに限定されるわけではないが、以下が挙げられ得る:β−ラクタム抗生剤、例えばペニシリンG、ペニシリンV、クロキサシリン、ジクロキサシリン、メチシリン、ナフシリン、オキサシリン、アンピシリン、アモキシシリン、バカンピシリン、アズロシリン、カルベニシリン、メズロシリン、ピペラシリンおよびチカルシリン;アミノグリコシド、例えばアミカシン、ゲンタマイシン、カナマイシン、ネオマイシン、ネチルマイシン、ストレプトマイシンおよびトブラマイシン;マクロリド、例えばアジスロマイシン、クラリスロマイシン、エリスロマイシン、リンコマイシンおよびクリンダマイシン;テトラサイクリン、例えばデメクロサイクリン、ドキシサイクリン、ミノサイクリン、オキシテトラサイクリンおよびテトラサイクリン;キノロン、例えばシノキサシン、およびナリジクス酸;フルオロキノロン、例えばシプロフロキサシン、エノキサシン、グレパフロキサシン、レボフロキサシン、ロメフロキサシン、ノルフロキサシン、オフロキサシン、スパルフロキサシンおよびトロバフロキサシン;ポリペプチド、例えばバシトラシン、コリスチンおよびポリミキシンB;スルホンアミド、例えばスルフイソキサゾール、スルファメトキサゾール、スルファジアジン、スルファメチゾールおよびスルファセタミド;ならびに他の抗菌剤、例えばトリメトプリム、スルファメチゾール、クロラムフェニコール、バンコマイシン、メトロニダゾール、キヌプリスチン、ダルフォプリスチン、リファンピン、スペクチノマイシンおよびニトロフラントイン;ならびに抗ウィルス剤、例えば汎用抗ウィルス剤、例えばイドクスウリジン、ビダラビン、トリフルリジン、アシクロビル、ファンシクロビル、ペンシクロビル、バラシクロビル、ガンシクロビル、フォスカルネット、リバビリン、アマンタジン、リマンタジン、シドフォビル;アンチセンスオリゴヌクレオチド、免疫グロブリンおよびインターフェロン;ならびにHIV感染用薬物、例えばテノフォビル、エムトリシタビン、ジドブジン、ディダノシン、ザルシタビン、スタブジン、ラミブジン、ネビラピン、デラビルジン、サクイナビル、リトナビル、インディナビルおよびネルフィナビル。
【0143】
(VII.薬学的組成物)
インビボでの使用において、一般に、免疫造影(immunoimaging)、免疫療法、または他の使用にかかわらず、結合体は被験体に導入される。組成物は、結合体の単一の異性体、または1個以上の部分付加異性体を含むことができる。例えばタンパク質が抗体である場合、組成物は、単一のE2、E4またはE6異性体、選択されたE2、E4またはE6異性体の混合物、すべて単独のE2、E4またはE6異性体、あるいはE2、E4およびE6異性体の混合物を含み得る。一部の実施形態において、ある異性体を含有する組成物は、実質的に他の異性体を含まなくてもよい。この文脈において、「実質的に含まない」とは、約20%未満、約10%未満、約5%未満、約2%未満、または約1%未満の他の異性体を含有する組成物を意味する。
【0144】
組成物は、その組成物が患者に投与され得るいずれの形態でもよい。例えば組成物は、固体、液体または気体(エアゾール)の形態であり得る。代表的な投与経路としては、経口、局所、非経口、舌下、直腸、膣、眼、腫瘍内、および鼻腔内が挙げられるが、これらに限定されない。非経口投与として、皮下注射、静脈内、筋肉内、胸骨内の注射または注入技法が挙げられる。1つの局面において、組成物は非経口投与される。なお別の局面において、結合体または組成物は、静脈内投与される。
【0145】
結合体は、注射によって導入され得る。代表的に、結合体は血管内に(静脈内または動脈内に)または髄腔内(intrathetically)に、しばしば注入によって投与される。さらに、適切な場合には、結合体は、皮下的、粘膜下、筋肉内、頭蓋内、または薬物投与の他の受容される経路によって導入され得る。
【0146】
他の実施形態において、組成物は有効量の結合体および薬学的に受容可能なキャリアまたはビヒクルを含む。そのような組成物は、動物またはヒトへの投与に適切である。
【0147】
薬学的組成物は、患者への組成物の投与時に結合体が生体利用可能であり得るように処方され得る。組成物は、1種以上の投薬単位の形態を取ることができ、ここで例えば錠剤は単一投薬単位であることが可能であり、注射用形態において結合体の容器は、複数回の投薬単位を保持することができる。
【0148】
薬学的組成物を調製する際に使用される物質は、使用される量では非毒性であり得る。薬学的組成物中の活性成分の最適投薬量が種々の要因に依存して変わることは、当業者に明らかである。関連する要因としては、動物の種類(例えばヒト)、結合体の特定の形態、投与様式、使用される組成物が挙げられるが、これらに限定されない。
【0149】
薬学的に受容可能なキャリアまたはビヒクルは、組成物が例えば錠剤または粉末形態であるように、粒子状でもよい。キャリアは液体でもよく、組成物は例えば経口シロップまたは注射用液剤である。さらにキャリアは、例えば吸入投与で有用なエアゾール組成物を提供するために、気体状または粒子状でもよい。
【0150】
経口投与を目的とする場合、組成物は好ましくは固体形態または液体形態であり、半固体形態、半液体形態、懸濁物形態およびゲル形態は、本明細書で固体または液体のどちらかと見なされる形態に含まれる。
【0151】
経口投与用の固体組成物として、組成物は粉剤、顆粒剤、圧縮錠剤、丸剤、カプセル剤、チューイングガム、カシェ剤などで処方され得る。そのような固体組成物は代表的に、1種以上の不活性希釈剤を含有する。さらに、以下の1種以上が存在できる:結合剤(例えばカルボキシメチルセルロース、エチルセルロース、微結晶性セルロース、またはゼラチン);賦形剤(例えばデンプン、ラクトース、またはデキストリン);崩壊剤(例えばアルギン酸、アルギン酸ナトリウム、プリモゲル(Primogel)、コーンスターチなど);滑沢剤(例えばステアリン酸マグネシウムまたはステロテックス);流動促進剤(例えばコロイド状二酸化ケイ素);甘味料(例えばスクロースまたはサッカリン)、矯味矯臭剤(例えばペパーミント、サリチル酸メチルまたはオレンジ香味料)、ならびに着色剤。
【0152】
組成物がカプセル剤、例えばゼラチンカプセル剤の形態である場合、組成物は上の種類の物質に加えて、液体キャリア(例えばポリエチレングリコール、シクロデキストリン、または脂肪油)を含有し得る。
【0153】
組成物は液体(例えばエリキシル剤、シロップ剤、液剤、乳剤または懸濁剤)の形態でもよい。液体は経口投与に、または注射による送達に有用であり得る。経口投与を目的とする場合、組成物は甘味料、保存料、染料/着色料および調味料の1種以上を含むことができる。注射による投与のための組成物では、界面活性剤、保存料、湿潤剤、分散剤、懸濁剤、緩衝剤、安定剤および等張剤の1種以上を含むこともできる。
【0154】
液体組成物は、それらが液剤、懸濁剤または他の同様の形態であるかどうかにかかわらず、以下の1種以上も含むことができる:滅菌希釈剤(例えば注射用水、食塩水、好ましくは生理的食塩水、リンガー液、等張性塩化ナトリウム、不揮発性油(例えば溶媒または懸濁媒体として作用し得る合成モノグリセリドまたはジグリセリド、ポリエチレングリコール、グリセリン、シクロデキストリン、プロピレングリコールまたは他の溶媒);抗菌剤(例えばベンジルアルコールまたはメチルパラベン);抗酸化剤(例えば、アスコルビン酸または亜硫酸水素ナトリウム);キレート剤(例えばエチレンジアミン四酢酸);緩衝剤(例えばアセテート、シトレートまたはホスフェート)および張度の調整のための薬剤(例えば塩化ナトリウムまたはデキストロース)。非経口組成物は、ガラス、プラスチックまたは他の材料からなるアンプル、使い捨て注射器または複数回用量バイアルに封入され得る。生理的食塩水は例示的なアジュバントである。注射用組成物は好ましくは滅菌されている。
【0155】
非経口投与用の調製物は、滅菌水または非水性液剤、懸濁剤、および乳剤を含む。非水性溶媒の例は、プロピレングリコール、ポリエチレングリコール、植物油(例えばオリーブ油)および注射用有機エステル(例えばオレイン酸エチル)である。水性キャリアは、水、アルコール性/水性溶液、乳剤または懸濁物(食塩水および緩衝媒体)を含む。非経口ビヒクルは、塩化ナトリウム溶液、リンガーデキストロース、デキストロースおよび塩化ナトリウム、乳酸加リンガー液、または非揮発性油を含む。静脈内ビヒクルは、流体および栄養素補充薬、電解質補充薬(例えばリンガーデキストロースをベースとするもの)などを含む。保存料および他の添加剤、例えば抗菌薬、抗酸化剤、キレート剤、および不活性ガスなども存在し得る。
【0156】
特定の障害または状態の処置において有効な結合体の量は、障害または状態の性質によって変わり、標準臨床技法によって決定され得る。開示されたタンパク質−薬物結合体の投与のためのこれらの投薬量範囲は、状態または障害の症状が軽減される所望の効果を引き起こすのに十分大きいものである。投薬量は、副作用、例えば望ましくない交差反応、アナフィラキシー反応などを引き起こすほど多くてはならない。加えて、最適投薬量範囲を決定するために、インビトロまたはインビボでのアッセイが必要に応じて利用される。
【0157】
組成物において利用される正確な用量は、患者の年齢、状態、性別および疾患の程度、投与経路、および疾患または症状の重篤度によっても変わり、医師の判断および各患者の状況に従って決定されるべきである。
【0158】
組成物は適切な投薬量が得られるような、有効量の結合体を含む。代表的に、この量は、組成物の重量で少なくとも約0.01%の結合体である。経口投与を目的とする場合、この量は組成物の重量で約0.1%〜約80%の範囲で変化し得る。1つの局面において、経口組成物は組成物の重量で約4%〜約50%の結合体を含むことができる。なお別の局面において、本組成物は、非経口投薬量単位が結合体の重量で約0.01%〜約2%を含有するように調製される。
【0159】
静脈内投与では、組成物は、動物の体重1kg当たり約0.01〜約100mgの結合体を含むことができる。1つの局面において、組成物は動物の体重1kg当たり約1〜約100mgの結合体を含むことができる。別の局面において、投与される量は、約0.1〜約25mg/kg体重の結合体の範囲で変化する。
【0160】
一般に、患者に投与される結合体の投薬量は代表的に、約0.01mg/kg〜約2000mg/kg動物体重である。1つの局面において、患者に投与される投薬量は、約0.01mg/kg〜約10mg/kg動物体重である。別の局面において、患者に投与される投薬量は、約0.1mg/kg〜約250mg/kg動物体重である。なお別の局面において、患者に投与される投薬量は、約0.1mg/kg〜約20mg/kg動物体重である。なお別の局面において、投与される投薬量は、約0.1mg/kg〜約10mg/kg動物体重である。なお別の局面において、投与される投薬量は、約1mg/kg〜約10mg/kg動物体重である。
【0161】
結合体は、いずれかの好都合な経路によって、例えば注入またはボーラス注射によって、上皮または粘膜皮膚内層(例えば口腔粘膜、直腸および腸粘膜など)を介した吸収によって投与され得る。投与は、全身的または局所的であり得る。種々の送達システム、例えばリポソーム、微粒子、マイクロカプセル、カプセルへのカプセル化などが公知であり、結合体または組成物を投与するために使用され得る。ある実施形態において、2つ以上の結合体または組成物が患者に投与される。
【0162】
特定の実施形態において、1種以上の結合体または組成物を処置が必要な領域に局所的に投与することが望ましい。これは例えば、手術中の局所注入によって;例えば手術後の創傷包帯と併せた局所塗布によって;注射によって;カテーテルによって;坐剤によって;膜(例えばシラスティック膜(sialastic))、または繊維を含む、多孔性、非多孔性、あるいはゼラチン性材料の移植)によって達成される。1つの実施形態において、投与は、癌の部位(または形成部位)、腫瘍あるいは新生物または新生物発生前組織への直接注射によって可能である。別の実施形態において、投与は、自己免疫疾患の徴候の部位(または形成部位)への直接注射によって可能である。
【0163】
ある実施形態において、1種以上の結合体または組成物を、脳室内および髄腔内注射を含む任意の適切な経路によって中枢神経系に導入することが望まれ得る。脳室内注射は、例えばオンマヤ(Ommaya)リザーバーなどのリザーバーに取り付けられた脳室内カテーテルによって容易にされ得る。
【0164】
例えば吸入器またはネブライザの使用、およびエアゾール化剤を用いた処方によって、あるいはフルオロカーボンまたは合成肺界面活性剤での潅流を介して、肺投与も利用され得る。
【0165】
なお別の実施形態において、結合体は制御放出システムで送達可能であり、例えばこれに限定されるわけではないが、ポンプまたは種々のポリマー材料が使用され得る。なお別の実施形態において、制御放出システムは結合体の標的の付近、例えば脳に配置可能であり、それゆえ全身用量の一部のみを必要とする(例えばGoodson,in Medical Applications of Controlled Release,supra,vol.2,pp.115−138(1984)を参照)。Langer(Science 249:1527−1533(1990))による概説で述べられている他の制御放出システムが使用され得る。
【0166】
一部の実施形態において、タンパク質結合体をキャリアに結合させて組成物を形成することができる。「キャリア」という用語は、希釈剤、アジュバントまたは賦形剤をいい、それと共に結合体が投与される。そのような薬学的キャリアは、液体(例えば水および石油、動物、植物または合成起源のものを含む油(例えばラッカセイ油、ダイズ油、鉱油、ゴマ油など)であり得る。キャリアは、食塩水、アラビアゴム、ゼラチン、デンプンペースト、タルク、ケラチン、コロイド状シリカ、尿素などであり得る。さらに、補助剤、安定剤、増粘剤、滑剤および着色剤を使用できる。1つの実施形態において、患者に投与される場合、結合体または組成物および薬学的に受容可能なキャリアは滅菌されている。水は、結合体を静脈内投与する場合の例示的なキャリアである。食塩水および水性デキストロースおよびグリセロール溶液は、特に注射用液剤のための液体キャリアとしても利用され得る。適切な薬学的キャリアとしてはまた、デンプン、グルコース、ラクトース、スクロース、ゼラチン、麦芽、コメ、小麦粉、チョーク、シリカゲル、ステアリン酸ナトリウム、モノステアリン酸グリセロール、タルク、塩化ナトリウム、乾燥スキムミルク、グリセロール、プロピレン、グリコール、水、エタノールなどの賦形剤が挙げられる。所望の場合、組成物は少量の湿潤剤または乳化剤、あるいはpH緩衝剤も含むことができる。
【0167】
組成物は、液剤、懸濁剤、乳剤、錠剤、丸剤、ペレット剤、カプセル剤、液体を含有するカプセル剤、粉剤、持続放出製剤、坐剤、乳剤、エアゾール、スプレー、懸濁剤の形態、または使用に適した他の任意の形態を取ることができる。適切な薬学的キャリアの他の例は、E.W.Martinによる”Remington’s Pharmaceutical Sciences”に述べられている。
【0168】
例示的な実施形態において、結合体は通常の手順に従って、動物、特にヒトへの静脈内投与に適した薬学的組成物として処方される。代表的に、静脈内投与のためのキャリアまたはビヒクルは、滅菌等張性水性緩衝溶液である。必要な場合、組成物は可溶化剤も含むことができる。静脈内投与のための組成物は必要に応じて、注射部位の疼痛を緩和するために、リグノカインなどの局所麻酔薬を含むことができる。一般に成分は、例えば活性剤の量を表示したアンプルまたはサシェ(sachette)などの密閉した容器内の乾燥した凍結乾燥粉末または無水濃縮物として、単位投薬形で個別に、または共に混合して提供される。結合体が注入によって投与される場合、例えば滅菌された薬学的用水または食塩水を含有する注入ボトルを用いて投薬できる。結合体が注射によって投与される場合、投与前に成分を混合できるように、注射用の滅菌水または食塩水のアンプルを提供することができる。
【0169】
経口送達用の組成物は、例えば、錠剤、口内錠、水性懸濁剤または油性懸濁剤、顆粒剤、粉末剤、乳剤、カプセル剤、シロップ剤、またはエリキシル剤の形態が可能である。必要に応じて、経口投与組成物は、例えば、薬学的に口当たりのよい調製物を提供するために、必要に応じて1種以上の薬剤(例えばフルクトース、アスパルテームまたはサッカリンなどの甘味料;ペパーミント、ウィンターグリーンオイル、またはチェリーなどの矯味矯臭剤;着色料;および保存料)を含有できる。さらに、錠剤形態または丸剤形態では、組成物は消化管での崩壊および吸収を遅延させて、それにより長期間に渡って持続作用を提供するために、コーティングすることができる。浸透圧的に活性な駆動化合物を包囲する選択的透過性膜も、経口投与化合物に適切である。このような遅延されたプラットフォームでは、カプセル剤を包囲する環境からの流体は、駆動化合物により吸収されて膨潤し、開口部を通じて薬剤または薬剤組成物を放出する。これらの送達プラットフォームは、即時放出処方物の急上昇特性とは対照的に、本質的にゼロ次送達特性を提供することができる。モノステアリン酸グリセロールまたはステアリン酸グリセロールなどの時間遅延材料も使用できる。
【0170】
組成物は局所投与を目的とすることも可能であり、その場合、キャリアは液剤、乳剤、軟膏またはゲルベースの形態であり得る。経皮投与を目的とする場合は、組成物は経皮パッチまたはイオン導入器具の形態を取ることができる。局所処方物は、約0.05%〜約50%w/v(組成物の単位体積当たりの重量)、別の局面では0.1%〜10%w/vの結合体の濃度を含むことができる。
【0171】
組成物は、例えば直腸で溶解して、結合体を放出する坐剤の形態で、直腸投与を目的とすることができる。
【0172】
組成物は、固体または液体投薬単位の物理形態を改良する種々の物質を含むことができる。例えば組成物は、活性成分周囲にコーティングシェルを形成する物質を含むことができる。コーティングシェルを形成する物質は代表的に、不活性であり、例えば糖、シェラック、および他の腸溶コーティング剤から選択できる。あるいは活性成分をゼラチンカプセルに入れることができる。
【0173】
組成物は、ガス状投薬単位から成ることがあり、例えばエアゾールの形態を取ることができる。エアゾールという用語は、コロイド状性質のものから加圧パッケージから成る系に渡る種々の系を示すために使用される。送達は、液化ガスまたは圧縮ガスによって、あるいは活性成分を分配する適切なポンプシステムによって可能である。
【0174】
本組成物は固体、液体またはガス状の形態であるか否かにかかわらず、癌、自己免疫疾患または感染性疾患の処置に使用される薬剤を含むことができる。
【0175】
(VIII.薬物動態学)
マウスにおいて、精製したcAC10−vcMMAEの4個の薬物の結合体の毒性および効果のインビトロでの特徴付けを実施し、実施例でさらに詳細に議論する(例えば実施例8および9を参照のこと)。簡潔には、これらの試験は、抗体当たり平均4個の薬物を含む混合物は、8個の薬物を含む結合体と等しく有効であるが(どちらも1mg/Kgの単回用量)、より毒性が低い(MTDは、抗体当たり4個の薬物では100mg/kgであり、抗体当たり8個の薬物では50mg/kg)ことを示している。DTT方法による抗体当たり4個の薬物を含む精製物質は効果および毒性が同様であるのに対して、DTNB方法による抗体当たり4個の薬物を含む精製物質は、やや高い120mg/KgのMTDを有するが、他の結合体の半分の用量(0.5mg/Kg)で有効である。
【0176】
cAC10に抗体当たり2個、4個、または8個の薬物を付加することは、標的抗原CD30への結合に何の影響も及ぼさなかった。cAC10−ADCのインビトロ有効性は薬物付加に直接依存し、それゆえ全MMAE暴露に直接依存する。
【0177】
cAC10−E4は、Karpas−299異種移植モデルにて、MMAE用量の半分であり、同じ用量の抗体で、cAC10−E8に匹敵する抗腫瘍活性を示した。有効性が薬物付加に直接関連するというインビトロでの発見結果に基づいて、cAC10−E4とcAC10−E8との同等のインビボ抗腫瘍活性は予想されなかった。ADCの薬物動態学の調査は、クリアランスがADCの薬物付加に直接関連し、暴露(曲線下面積−AUC)が薬物付加に反比例することを明らかにした。cAC10−E4のAUCはcAC10−E8よりも3倍高かった。cAC10−E8と比較したcAC10−E4のより高いAUCは、低下した有効性を補償するのに明らかに十分であり、同等の有効性をもたらした。AUCを増大させるために血漿消失半減期を減速させることによって有効性を改善する試みは、インターフェロン−αのためのアルブミン融合タンパク質の構築および抗癌剤ルルトテカン(Lurtotecan)のリポソーム送達を含む方法によって達成されてきた。目的が血漿半減期を延長させることであるこれらの例とは異なり、cAC10−E4の暴露向上は、MMAE付加減少の貴重な結果であった。
【0178】
実施例8に開示するように、cAC10−E2を1.0mg/kg/用量のq4dx4と共に投薬することは、10症例中10の治癒をもたらした。cAC10−E2は同じmAb用量ではcAC10−E4と比べて同等の抗腫瘍活性を示さなかったが、cAC10−E4と比べて同等の抗腫瘍活性を達成するためのcAC10−E2の用量は、インビボ有効性実験に基づいておそらく2分の1未満である。cAC10−E4と同様に、cAC10−E2の暴露の改善は、より低いインビトロでの有効性を解決するのに重要な役割を演じ得る。
【0179】
cAC10−Val−Cit−MMAE ADCの治療可能性を最大限にするためには、高い治療指数が必要である。mAb当たりのMMAE分子の量を8から4に減少させることは、治療指数を100から200へ向上させた。化学療法試薬の急勾配の用量−反応曲線を考えると、治療指数の2倍の差は、毒性に関する臨床上の意義全体の点から見て重要であり得る。
【0180】
MMAEの量をmAb当たり8分子から4分子に減少させることにより、インビトロでの活性の低下があるが、インビボでの同等の抗腫瘍活性がなお示された。抗体当たり2個のMMAE分子への薬物付加でのさらなる減少は、インビトロ活性をさらに低下させ、cAC10−E2は、複数回投薬の状況の2倍の用量で、cAC10−E4およびcAC10−E8と同等またはより優れた有効性を有していた。治療濃度域は、薬物付加を8MMAE分子から4に減少させることにより2倍に上昇し、抗体当たり薬物2個までのさらなる減少により最低にて維持された。ADCの薬物置換の最適化には、注目に値する価値がある。
【実施例】
【0181】
(方法1の実施例1)
cAC10を制限された濃度のDTTによって以下のように部分還元した:cAC10(8mg/mLまたは53.8μM)を0.05Mホウ酸ナトリウム、pH8、0.05M NaCl、および1mMジエチレン−トリアミン五酢酸(DTPA;Aldrich)中のDTT 3.5モル当量(188.4μM;Sigma)によって、37℃にて1時間処理した。次に還元された抗体をPD−10カラム(Amersham Biosciences)で脱塩することによって精製した。PD−10カラムを1mM DTPA(PBSD)を含むリン酸緩衝食塩水(PBS)pH7.4(GIBCO)25mLで平衡にして、上の溶液1mLをカラムに添加し、カラムをPBSD 1.8mLで洗浄して、PBSD 1.4mLを用いてカラムを溶出させた。1.0mg/mL溶液での280nmにおける吸収値1.58を使用してタンパク質濃度を定量して、モル濃度を分子量150,000g/molを用いて決定した。生成された抗体−システインチオールの濃度を5,5’−ジチオ−ビス−(2−ニトロ安息香酸)(DTNB;Pierce)を用いた滴定によって決定すると、概して、この方法を用いた抗体当たり4個の抗体−システインチオールよりもやや高くなった。
【0182】
次に以下のように、薬物vcMMAEを還元cAC10に結合させた:還元cAC10(代表的に、最終濃度が30μM抗体および120μM抗体−システインチオール)を最初に0℃まで冷却した。vcMMAEを冷アセトニトリルに溶解させ、抗体溶液と迅速に混合した。最終アセトニトリル濃度は20%であるのに対して、最終薬物濃度は135〜150μMであった(4.5〜5モル当量、これは抗体−システインチオールよりやや過剰である)。この溶液を0℃にて30分間インキュベートして、過剰なvcMMAEをシステイン(1mM最終濃度)によってクエンチさせ、結合体を上述のようにPD−10カラムを用いて精製した。
【0183】
(方法1の実施例2)
抗体当たり4個のvcMMAEを含むcAC10(E4ミックス)は、制限量のDTTを用いて以下のように調製した:cAC10を0.025Mホウ酸ナトリウム、pH8、0.025M NaCl、1mM DTPA中のDTT 3.25モル当量によって、37℃にて2時間処理した。この混合物を水で5倍に希釈し、ヒドロキシアパタイトカラム(MacroprepセラミックタイプI 40μm、BioRad、ハーキュリーズ、カリフォルニア州)に流速10mL/分で加えた。カラムサイズは、cAC10 10mg当たり1mLであった。カラムは事前に、5カラム容量の0.5Mリン酸ナトリウム、pH7、10mM NaClおよび5カラム容量の10mMリン酸ナトリウム、pH7、10mM NaClで平衡にしておいた。添加後、カラムを5カラム容量の10mMリン酸ナトリウム、pH7、10mM NaClで洗浄して、次に100mMリン酸ナトリウム、pH7、10mM NaClで溶出させた。溶出後にDTPAを1mMまで添加した。1.0mg/mL溶液での280nmにおける吸光度値1.58を使用してタンパク質濃度を定量し、モル濃度を分子量148,449g/molを用いて決定した。生成された抗体−システインチオールの濃度を、DTNBを用いた滴定によって決定すると、代表的に、抗体当たり4.0〜4.5個のチオールが生じた。
【0184】
還元cAC10を抗体−システインチオールに対してわずかに過剰なvcMMAE(1.1モル当量)によってアルキル化した。vcMMAEの溶解性を維持するために、最終反応混合物中に10% DMSOが存在していた。あるいはvcMMAEは、5体積%のアルコール、例えばエタノールおよびイソプロピルアルコールを含む溶液中に保持することが可能であった。アルキル化反応を0℃にて30分間実施した。システイン(最終1mM)を使用して、すべての未反応vcMMAEをクエンチさせた。cAC10−vcMMAEは、上述のようにヒドロキシアパタイトクロマトグラフィーを用いて精製した。溶出後に、緩衝液をAmicon(Millipore、ベッドフォード、マサチューセッツ州)Ultrafree 30Kカットオフスピン濃度デバイスを使用して、リン酸緩衝食塩水(Invitrogen、カールスバッド、カリフォルニア州)に交換した。タンパク質濃度は、1.0mg/mL溶液での280nmにおける吸光度値1.62を使用して定量した。
【0185】
(方法2aの実施例3)
大幅に過剰のDTTを添加することによって、cAC10を完全に還元した。最終反応濃度は、8mg/mL cAC10、0.05Mホウ酸ナトリウム、pH8、0.05M NaCl、10mM DTT、および1mM DTPAであった。この溶液を37℃にて30分間インキュベートして、上述のようにPD−10カラムで脱塩することによって抗体を精製した。DTNB滴定によって決定されたように、これらの条件を使用して8よりもやや多い抗体−システインチオールが生成された。
【0186】
部分再酸化は、DTNBを酸化剤として使用することによって達成された。還元cAC10(代表的に30μM)を0℃まで冷却して、次にDTNB 1.5〜2.5モル当量(最終濃度45〜75μM;E4の最高収率は、2.0当量を使用して得られた)で処理した。溶液を反転によって迅速に混合し、0℃にて10〜20分間インキュベートした。放出されたTNB−が黄色であるため、反応の程度が観察できる。代表的に、反応は数秒以内で完了したことが示される。システインを添加して(最終濃度1mM)、抗体システインとの混合ジスルフィド中ではなく、TNB−としてすべてのTNBが存在することを確認した。次に抗体を上述のようにPD−10カラムまたはヒドロキシアパタイトカラムで精製した。代表的に、4種の抗体−システインチオールは、この部分再酸化手順の後にDTNB滴定によって観察された。vcMMAE薬物は最終的にこれらの抗体−システインチオールに結合され、上の方法1で述べたようにPD−10によって精製した。
【0187】
(方法2bの実施例4)
完全還元cAC10は、上の方法2aで述べたように調製した。完全還元cAC10(代表的に30μM)を0℃まで冷却し、次にDTNB 1.5〜2.5当量(最終濃度45〜75μM)によって処理した。溶液を反転によって迅速に混合し、0℃にて10分間インキュベートした。さらに精製せずに、部分的に再酸化させたcAC10を次に冷アセトニトリルに溶解させたvcMMAE 5当量と迅速に混合した。方法1と同様に、アセトニトリルの最終濃度は20%であった。結合反応では、cAC10最終濃度は、24μMであり(96μM抗体−システインチオールまたは抗体当たり4個)、最終vcMMAE濃度は120μMであった(5モル当量)。この溶液を0℃にて30分間インキュベートしてから、システインによってクエンチさせ、上述のようにPD−10によって精製した。
【0188】
E4ミックスのヒドロキシアパタイトによる分取精製。数個の15mL Amicon Ultrafree 30Kカットオフスピン濃度デバイスを使用して、cAC10の緩衝液(25mM クエン酸ナトリウム、pH6.5、250mM NaCl、および0.02% Tween−80)をPBSに交換した。PBS中のcAC10 1.09gを、最終体積89mLのDTTによって、以下のように完全に還元した:cAC10(82.3μM)を0.025Mホウ酸ナトリウム、pH8、0.025M NaCl中の10mM DTTによって37℃にて1時間処理した。この混合物を水で250mLまで希釈し、70mLヒドロキシアパタイトカラム(MacroprepセラミックタイプI 40μm、BioRad)に流速10mL/分で加えた。カラムは事前に、5カラム容量の0.5Mリン酸ナトリウム、pH7、10mM NaClおよび5カラム容量の10mMリン酸ナトリウム、pH7、10mM NaClで平衡にしておいた。添加後、カラムを5カラム容量の10mMリン酸ナトリウム、pH7、10mM NaClで洗浄して、次に100mMリン酸ナトリウム、pH7、10mM NaClで溶出させた。
【0189】
完全還元cAC10をDTNBによって以下のように再酸化した:上記から溶出された物質(6.02mg/mLまたは40.2μM、170mL中1.02g)を0℃まで冷却し、次にDTNB 2.0当量(10mMストック)で20分間処理した。さらに精製せずに、再酸化させたcAC10をvcMMAEに結合させた。冷cAC10(最終31.9μM)を、最終体積214mLのDMSO(最終20%)中に溶解させたvcMMAE 5当量(最終159.5μM)で処理した。0℃にて40分後、100mMシステイン 1.07mLを添加して、すべての未反応vcMMAEをクエンチさせ、混合物を水で750mLまで希釈した。結合体をDTT還元のために上述のようにヒドロキシアパタイトカラムで精製した。回収したcAC10−vcMMAE E4ミックス(0.99g、またはcAC10に基づいて91%の全収率)を濃縮して、数個の15mL Amicon Ultrafree 30Kカットオフスピン濃度デバイスを使用して緩衝液をPBSに交換した。
【0190】
HICによる純E4の分取精製は、45mL Toyopearlフェニル650M HICカラムによって、流速10mL/分で周囲温度にて実施した。溶媒Aは2M NaClおよび50mMリン酸ナトリウム、pH7であった。溶媒Bは80%v/v 50mMリン酸ナトリウム、pH7および20%v/vアセトニトリルであった。カラムは事前に5カラム容量の溶媒Aで平衡にした。ヒドロキシアパタイト(上記)によって精製したcAC10−vcMMAE E4ミックス最大400mgを1容量の4M NaClおよび50mMリン酸ナトリウム、pH7と混合してカラムに添加した。E0はカラムによって保持されなかった。連続段階勾配によって、異なる薬物付加種を溶出した:E2を35%溶媒Bによって溶出させ、E4を70%溶媒Bによって溶出させ、E6を95%溶媒Bによって溶出させ、E8を100%溶媒Bによって溶出させた。精製したE4を濃縮して、数個の15mL Amicon Ultrafree 30Kカットオフスピン濃度デバイスを使用して緩衝液をPBSに交換し、400mg精製物2回分から純E4 235mgを得た。分析HICによる純度分析(以下)は、90%を超えるE4純度を示した。
【0191】
(方法1の実施例5)
TCEP制限還元と、それに続く中間精製なしのアルキル化は、cAC10を0.025Mホウ酸ナトリウムpH8、0.025M NaCl、1mM DTPA中のTCEP 2.75モル当量により37℃にて2時間処理することによって実施した。図9も参照のこと。次に混合物を0℃まで冷却して、部分還元cAC10を上述のようにvcMMAEによってアルキル化した。cAC10−vcMMAEは、リン酸緩衝食塩水で平衡にしたPD−10カラム(Amersham Biosciences,ピスカタウェイ、ニュージャージー州)を使用して脱塩した(部分還元は、図8に示すように、部分還元抗体の中間精製ステップを用いても実施できる)。
【0192】
異性体分布の動力学を決定するために使用したサンプルは、以下のように調製した:cAC10を50mMリン酸ナトリウム、pH7.5および5mM EDTA中のDTT 3.0当量によって37℃にて還元した。表示した時点でサンプルを取り出して、同量の200mM クエン酸ナトリウム、pH5によってクエンチさせ、5mM EDTAを含有するリン酸緩衝食塩水によって平衡にしたPD−10カラムを使用して精製した。還元cAC10は上述のようにvcMMAEによって処理し、リン酸緩衝食塩水によって平衡にしたPD−10カラムを使用して精製した。
【0193】
HICによる純E2、純E4、および純E6の精製は、Toyopearlフェニル650M HICカラム(Tosoh Biosciences,モンゴメリービル、ペンシルベニア州)によって、流速10mL/分で周囲温度にて実施した。カラムサイズはcAC10−vcMMAE 7.5mg当たり1mLであった。溶媒Aは2.0M NaClおよび50mMリン酸ナトリウム、pH7であった。溶媒Bは80%v/v 50mMリン酸ナトリウム、pH7および20%v/vアセトニトリルであった。カラムは事前に5カラム容量の溶媒Aで平衡にした。cAC10−vcMMAEを0.67容量の5M NaCl(最終2.0M)と混合して、カラムに添加した。E0はカラムによって保持されなかった。連続段階勾配によって、異なる薬物付加種を溶出した:E2を35%溶媒Bによって溶出させ、E4を70%溶媒Bによって溶出させ、E6を95%溶媒Bによって溶出させ、E8を100%溶媒Bによって溶出させた。精製したE4を濃縮して、Amicon Ultrafree 30Kカットオフスピン濃度デバイスを使用して緩衝液をリン酸緩衝食塩水に交換した。
【0194】
(分析方法の実施例6)
薬物付加は、250および280nmにおける吸光度の比(A250/280)を測定することによって決定した。cAC10当たりのvcMMAEの数は実験的に(A250/280−0.36)/0.0686と決定されている。
【0195】
結合体は、Tosoh Biosceinceエーテル−5PWカラム(part08641)を流速1mL/分およびカラム温度30℃にて使用する疎水性相互作用クロマトグラフィー(HIC)によって、E4純度のパーセントについて分析した。溶媒Aは50mMリン酸ナトリウム、pH7および2M NaClであった。溶媒Bは、80% 50mMリン酸ナトリウム、pH7、10% 2−プロパノール、および10%アセトニトリルであった。15分間のアイソクラチック0%B、50分間の0〜100%Bの線形勾配、0.1分間の100〜0%Bの線形勾配、そして14.9分間のアイソクラチック0%B。注入(代表的に90〜100μL)は、1容積の精製vcMMAE−cAC10結合体(少なくとも3mg/mLの濃度)および1容積の50mMリン酸ナトリウム、pH7および4M NaClであった。
【0196】
HICクロマトグラフィーからの純E4を含むADCをAgilentバイオアナライザ(Bioanalyzer)を用いて変性および非還元条件下で分析した。タンパク質200チップを変性条件下であるが、製造業者によって記載されるように使用した。簡潔には、1mg/mL cAC10−vcMMAE 4μLを非還元性付加緩衝液2μLと混合して、100℃まで5分間加熱した。水(84μL)を添加し、この混合物6μLをチップの各ウェルに付加した。
【0197】
最後に純E4をPLRP−Sカラム(Polymer Laboratories)で分析した。流速は1mL/分であり、カラム温度は65℃であった。溶媒Aは0.05%トリフルオロ酢酸水溶液であり、溶媒Bは0.04%トリフルオロ酢酸アセトニトリル溶液であった。3分間のアイソクラチック25%B、15分間の50%Bまでの線形勾配、2分間の95%Bまでの線形勾配、1分間の25%Bまでの線形勾配、そして2分間のアイソクラチック25%B。注入は、以前に20mM DTTによって37℃にて20分間還元して、鎖間ジスルフィドを開裂させたcAC10−vcMMAE 10μLであった。
【0198】
ADCもまた、変性および還元条件下でPLRP−Sカラム(Polymer Laboratories)(2.1x150mm、8μ、1000Å)によって分析した。流速は1mL/分であり、カラム温度は80℃であった。溶媒Aは0.05%トリフルオロ酢酸水溶液であり、溶媒Bは0.04%トリフルオロ酢酸アセトニトリル溶液であった。3分間のアイソクラチック25%B、25分間の50%Bまでの線形勾配、2分間の95%Bまでの線形勾配、1分間の25%Bまでの線形勾配、そして2分間のアイソクラチック25%B。注入は、以前に20mM DTTによって37℃にて15分間還元して、残りの鎖間ジスルフィドを開裂させた1mg/mL cAC10−vcMMAE 10〜20μLであった。各鎖のモル分率は、以下のモル吸光係数を使用して決定した:0vcMMAEを含む軽鎖:30,160M−1cm−1;1vcMMAEを含む軽鎖:31,660M−1cm−1;0vcMMAEを含む重鎖:86,915M−1cm−1;1vcMMAEを含む重鎖:88,415M−1cm−1;2vcMMAEを含む重鎖:89,915M−1cm−1;3vcMMAEを含む重鎖:91,415M−1cm−1。
【0199】
E2およびE6の異性体分布は、PLRP−S HPLCデータのみを使用して決定した。E2異性体Aでは(これらの分析では、「異性体A」は図7の異性体2Aおよび2Bの両方を指す)、0vcMMAEを含む軽鎖(L0)のモル分率は1vcMMAEを含む重鎖(H1)のモル分率と等しいが、E2異性体Cでは、0および1vcMMAEを含む軽鎖のモル分率および0および1vcMMAEを含む重鎖のモル分率はすべて等しい。1vcMMAEを含む軽鎖(L1)および0vcMMAEを含む重鎖(H0)のみが異性体Cのパーセンテージに寄与するため、異性体Cのパーセントは、以下のように表される:
%C=2L1+2H0 (1)。
【0200】
異性体Aのパーセントは100−%Cであると仮定される。2または3vcMMAEを含む少量(合計3%未満)の重鎖がPLRP−S HPLCデータに見られることが多い。これらはおそらく、E2サンプル中のE4汚染またはE6汚染によるものである。E2異性体AおよびCのパーセントを計算するために、L0、L1、H1、およびH2のモルパーセントの合計を100%に設定した。
【0201】
同様に、E6異性体A(これらの分析では「異性体A」は図7の異性体6Aおよび6Bの両方を指す)では、H2のモル分率はL1のモル分率に等しいが、E6異性体Cでは、L0、L1、H2、およびH3のモル分率は等しい。L0およびH3のみが異性体Cのパーセンテージに寄与するため、異性体Cのパーセントは以下のように表すことができる:
%C=2L0+2H3 (2)。
異性体Aのパーセントは100−%Cであると仮定される。E2と同様に、L0、L1、H2、およびH3のモルパーセントの合計を100%に設定した。
【0202】
E4異性体のパーセンテージは、独自の溶液がないため、PLRP−S HPLCデータのみからでは得ることができない。他の2つの異性体についてPLRPデータを使用して解き得る前に、少なくとも1つの異性体を確定する必要がある。HHL、HH、およびHLのモルパーセントは、以下の分子量を使用してバイオアナライザから決定した:124,720.8(HHL)、100,992.6(HH)、および74,224.5(HL)g/mol。バイオアナライザは機器の読み出しのために結合染料の蛍光を使用し、そしてHHL、HH、およびHLが単位分子量当たり染料を等しく結合することが仮定されるが、この仮定が真実である可能性は低い。この仮定から生じる誤差を最小限にするために、以下のようにHHL、HH、およびHLピーク面積を使用してバイオアナライザデータから異性体E4Aのみを計算した(重鎖−重鎖ジスルフィドが開裂される場合に、各抗体は2HLを生成するため、HLピーク面積を2で割る):
【0203】
【化5】
次にPLRP−S HPLCデータを使用して、E4異性体EおよびFの残りの寄与について、以下の式を使用して解く:
%E=H1+L1−0.5%A (4)
%F=H2+LO−0.5%A (5)。
【0204】
E4異性体Aは、L0、L1、H1、およびH2の集団に等しく寄与し、異性体AのH1およびL1による寄与(その全体の寄与の半分)をH1およびL1の観察された総量から引いて、異性体Eの存在に起因するはずであるH1およびL1の残りの量を得る必要がある。異性体Aに対するH2およびL0の寄与に関する同様の引算は、異性体Fによって存在する量を与える。E2およびE6と同様に、L0、L1、H1、およびH2のモルパーセントの合計は100%に設定した。
【0205】
(タンパク質の部分付加のための方法についての実施例7)
部分薬物付加ADCを調製するために、2つの異なる方法を使用した。第1に、限定量のDTTまたはTCEPによるcAC10の部分還元は、8抗体システイン未満で生じた。DTTおよびTCEP約3.25および2.75当量はそれぞれ、2個の鎖間ジスルフィド結合を開裂させて、抗体当たり平均4cAC10システインを生じる(0、2、4、6、および8抗体−システインの混合物)。還元剤の量は経験的に決定できる:cBR96は4抗体システインを生じるためにDTTまたはTCEP 2.1当量のみを必要とするが、マウスIgG1抗体は、還元に対する極度の耐性のためにしばしば使用可能である(データを示さず)。DTTではなくTCEPを使用する利点は、ホスフィンはマレイミドとごくわずかしか反応していないので、vcMMAEを添加する前に残りの還元剤を除去する必要がないことである。過剰なDTTは容易にvcMMAEと反応して、薬物に関して抗体−システインと競合する。抗体還元の後、わずかにモル過剰のvcMMAEによる抗体システインの処理(システイン当たり1.1モル当量)は、抗体当たり4MMAEの平均薬物付加されたcAC10を生じる(E4ミックス)。
【0206】
あるいはcAC10は、10mM DTTによって完全に還元して、次にDTNBによって部分的に再酸化することができる。この再酸化プロセスは非常に効率的であり、8個の抗体システインを4個まで再酸化するために、DTNB 2.0当量を必要とする。この再酸化抗体のシステインなどのチオールによる処理は、結合されたチオニトロ安息香酸を遊離させず、このことは、再酸化システインが混合TNB−システインジスルフィドではなく、むしろ抗体ジスルフィドの形であることを示唆している。以下に述べる分析方法も、抗体ジスルフィドの存在を示している。残りの抗体システインは、上述のようにvcMMAEに結合体化してE4ミックスを生じ得る。
【0207】
薬物付加種の異性体集団を決定するために、E2、E4、およびE6を分離および単離して、純E2、純E4、および純E6を得る。図13Aは、DTT部分還元によって生成されたE4ミックスの疎水性相互作用(HIC)HPLCトレースを示す。偶数の薬物が付加された種はすべて相互に分離可能であり得、奇数の薬物が付加された種の少量が偶数種の間の谷間に見られる。これらの種の薬物付加は、ピークのUVスペクトルの検査によって割当てられ得る。薬物リンカー内のPABA基は、248nm付近に最大吸収を有するが、抗体は同じ波長に最小吸収を有する。248および280nmにおける薬物および抗体吸光係数を使用すると、抗体当たりの薬物数は、開始ADC混合物および観察された各ピークに対して割当てられ得る(Hamblett et al.(2004),Clin Cancer Res 10:7063−70)。
【0208】
表1は、DTT部分還元、TCEP部分還元、および部分DTNB再酸化によって調製された偶数薬物付加種のパーセンテージを示す。DTNB部分還元方法は、部分還元方法(DTTでは30%、TCEPでは33%)よりもわずかに高いE4のパーセンテージ(38%)を生じる。これは主にE6およびE8を犠牲にして得られるものであり、E6およびE8はDTT部分還元では約34%、TCEP部分還元では31%に達するが、DTNB部分再酸化ではわずかに24%である。奇数の薬物が付加された種は表に示しておらず、全物質の7〜10%に相当する。
【0209】
(表1.E4混合物の組成パーセントa)
【0210】
【表1】
aHIC−HPLCクロマトグラムは、組成パーセントに統合した。値は4(DTT部分還元)、3(TCEP部分還元)、または6(DTNB部分再酸化)の独立バッチについての±標準偏差である。奇数種からの寄与分は示されていないため、合計は100%未満となっている。
【0211】
このHIC−HPLC方法は、2、3ミリグラムの純E2、純E4、および純E6を単離するために使用できる。あるいは段階的勾配を使用する分取HICは、図13B、C、Dに示すように、数百ミリグラムの純E2、純E4、および純E6を単離するために使用できる。これらの物質の純度は、薬物付加レベルに関して少なくとも95%である。
【0212】
抗体上での薬物の分布を決定するために、これらの精製物質は、2つの分析方法を受け供された(実施例6を参照)。第1に、PLRP−Sカラムを用いた還元および変性HPLC使用して、抗体鎖当たりの薬物数を決定した。過剰なDTTを用いたADCの予備処理は、残存する鎖間ジスルフィドを切断して、0または1個の薬物を含む軽鎖(L0およびL1)の、0、1、2、または3個の薬物を含む重鎖(H0、H1、H2、およびH3)からの分離を可能にする(図4)。第2に、非還元および変性キャピラリー電気泳動は、残存鎖間ジスルフィドがインタクトのままでの抗体鎖の分離を可能にし、6つの潜在的な種:L、H、HL、HH、HHL、およびHHLLを生じさせる(図15)。
【0213】
PLRP−S HPLCおよびキャピラリー電気泳動によって観察された種の定量化は、異性体集団への分類を可能にする。図1および7は、抗体フラグメントおよび異性体それぞれに結合された薬物の数を示す。E2およびE6の異性体集団は、各異性体が独自のパターンを生じるため、PLRP−S HPLC単独で、またはキャピラリー電気泳動単独で容易に決定できる。たとえば変性および還元条件下では、異性体E2CのみがL1およびH0を生じるのに対して、E2AのみがL0およびH1を生じ、変性および非還元条件では、異性体E2AがHHLLを生じるのに対して、E2CはLおよびHHLを生じる。E4では、PLRP−S HPLCもキャピラリー電気泳動も単独では異性体集団を計算するのに十分ではないため、組成を決定するために2つの方法を組合せて使用する必要がある。表2は、これらの各異性体の組成パーセントを示す。PLRP−S HPLCデータを独占的に使用して、式1および2を用いてE2およびE6異性体の異性体組成を計算した(実施例6を参照)。キャピラリー電気泳動を使用して、式3を用いてE4Aの量を計算し、PLRP−S HPLCを使用して、式4および5を用いて、E4Aの寄与を引いて、E4BおよびE4Cの量を計算した(実施例6を参照)。
【0214】
(表2.精製されたE2、E4およびE6の異性体集団の組成)
【0215】
【表2】
a式1を用いてPLRP−S HPLCデータから決定。b式3を用いてバイオアナライザデータから決定。c式4および5を用いてPLRP−S HPLCデータから決定。d式2を用いてPLRP−S HPLCデータから決定。
【0216】
表2のデータは、産生方法が薬物の配置(location)に著しく影響を及ぼすということから、特筆すべきものであり、抗体ジスルフィドを選択的に還元できることを示唆している。部分DTT還元は、重鎖−軽鎖ジスルフィドの1つの還元から生じる92%の異性体E2Cと、両方の重鎖−軽鎖ジスルフィドの還元から生じる59%の異性体E4Eと、両方の重鎖−重鎖ジスルフィドおよび1つの重鎖−軽鎖ジスルフィドの還元から生じる98%の異性体E6Cを生じる。1つの重鎖−重鎖ジスルフィドが還元された異性体は、極めて少量である。これに対して、部分DTNB再酸化は、E2およびE4異性体については77%の異性体E2Aおよび75%のE4A(1つの重鎖−重鎖ジスルフィドがインタクトである)という、ほぼ反対の結果、そしてE6については96%のE6Cという同じ結果を生じる。AETを用いた酸性還元は、DTT部分還元と非常に類似している異性体集団を生じ、重鎖−軽鎖ジスルフィドの開裂に有利に働く。
【0217】
DTT部分還元の異性体分布の反応動態を表3に示す。cAC10を、3.0当量のDTTによって還元して、サンプルを定期的に取り出し、vcMMAEによってアルキル化した。純E2、純E4、および純E6をHIC−HPLCによって得て、異性体分布をPLRP−S HPLCおよびバイオアナライザによって決定した。異性体組成は、還元時間10〜120分および抗体当たり薬物1.3〜3.9個の全薬物付加の範囲での実験の過程にわたって同一である。これらの結果は、限定量のDTTによってcAC10を2時間還元することによって調製された、表2に示すDTT部分還元の異性体集団が、還元反応の全過程にわたる異性体集団を代表することを示している。
【0218】
(表3.DTT部分還元の異性体分布の反応動態)
【0219】
【表3】
a還元時間。いったん還元された場合、すべての抗体をvcMMAEによって同じ時間処理した。bHIC−HPLCによって決定。c式1を用いてPLRP−S HPLCデータから決定。d式3を用いてバイオアナライザデータから決定。e式4および5を用いてPLRP−S HPLCデータから決定。f式2を用いてPLRP−S HPLCデータから決定。N/D、決定されず。この時点では、ごく少量のE6が産生され、この物質は異性体集団を決定するのには不十分であった。
【0220】
(表4.cAC10−vcMMAEのインビトロ結合および細胞毒性)
【0221】
【表4】
a4〜7回の独立した測定から決定された、抗体成分のμg/mLでのKarpas299への結合±標準偏差、N/D、決定されず。
b3回の独立した測定から決定された抗体成分のng/mLでのインビトロ細胞毒性±標準偏差。N/D、決定されず。cAC10は単独でKarpas299に対して有効性が乏しい。
【0222】
表4に、ADCについて複数の薬物付加レベルで実施したインビトロ結合および細胞毒性実験の結果を挙げる。E2およびE4ミックスは、DTT部分還元およびDTNB部分再酸化からの純E2および純E4と同様に試験した。抗体当たり8個の薬物を持つ完全付加結合体は最も細胞毒性であり、CD30陽性Karpas299細胞株に対して2.7ng/mLのIC50値を有する(抗体の重量に基づいて計算)。抗体当たり4個の薬物を持つADCはわずかに細胞毒性が低く、IC50値は3.4ng/mLと5.0ng/mLとの間であり、抗体当たり2個の薬物を持つADCは最も細胞毒性が低く、IC50値は11.4〜13.8ng/mLである。ADCを産生するために使用した化学作用は、細胞毒性に著しい相違を示さず、混合物とHIC精製ADCとの間にも著しい相違がなかった。インビトロ細胞毒性は、薬物の全用量のみに依存すると考えられる。CD30陽性細胞への結合はE0、E2、およびE4では非常に似ており、E8はわずかに損なわれ、結合体化が抗原結合を妨害しないことを示す。ADCのインビトロ細胞毒性(抗体成分の測定)は、予想された傾向:薬物の数が多くなればなるほど、IC50値が低くなることを示している。実験誤差の範囲内で、薬物の配置はインビトロ細胞毒性に影響を及ぼさないとみられる。
【0223】
(モノクローナル抗体薬物結合体の抗腫瘍活性に対する薬物付加効果についての実施例8)
細胞および試薬。CD30陽性ALCL株Karpas−299をDeutsche Sammlung von Mikroorganism und Zellkulturen GmbH(Braunschweig,ドイツ)より入手した。異種移植片増殖に適したHD株L540の誘導体である、L540cyは、Dr.Harald Stein(Institut fur Pathologie,Univ.Veinikum Benjamin Franklin,Hindenburgdamm30,12200 ベルリン、ドイツ)から快く提供していただいた。細胞株は、10%ウシ胎仔血清を添加したRPMI−1640培地(Life Technologies Inc.,ゲイサーズバーグ、メリーランド州)で培養した。
【0224】
cAC10−Val−Cit−MMAE ADCの構築および精製。簡潔には、抗体当たり8個の薬物を持つcAC10をジチオトレイトール(DTT)と37℃にて30分混合することによって産生して、1mMジエチレントリアミン五酢酸(DTPA)を含有するPBSを用いたSephadex G−25樹脂を介した溶出によって緩衝液を交換した。1mM DTPA(PBS/D)を含有するPBSを還元mAbに添加した(最終濃度2.5mg/mL)。Val−Cit−MMAEと呼ばれる9.5モル過剰のマレイミドカプロイル−Val−Cit−MMAEを還元抗体に4℃にて1時間にわたって添加し、20倍過剰のシステインを添加することによって結合体化反応をクエンチした。反応混合物を遠心分離限外濾過によって濃縮し、4℃にてPBS中で平衡化したSephadex G25を介して緩衝液を交換した。次に結合体を0.2ミクロンフィルタで滅菌条件下にて濾過した。
【0225】
抗体当たり2および4MMAE分子を持つcAC10 ADCの産生は、部分還元と、それに続くVal−Cit−MMAEによる反応を包含した。抗体cAC10(10mg/mL)は、最終DTT:mAbモル比3.0までのDTTの添加と、それに続く37℃での約2時間のインキュベーションにより部分還元された。次に還元反応を約10℃まで冷却して、還元cAC10を過剰DTTからダイアフィルトレーションによって精製した。ダイアフィルトレーション後に、部分還元cAC10のチオール濃度をDTNB(Ellmanの)アッセイによって決定した;この方法では、平均約2個のジスルフィド結合が還元され、それゆえ約4の還元Cys:mAbを露出させた。還元Cysすべてを結合体化するために、最終Val−Cit−MMAE:還元Cysモル比 約1.15までVal−Cit−cMMAEを添加した。15%v/v DMSOの存在下で結合反応を実施して、約10℃にて約30分間進行させた。結合体化反応の後、過剰な遊離Cys(Val−Cit−MMAE 1モル当たり2モルのCys)を添加して、未反応Val−Cit−MMAEをクエンチして、Cys−Val−Cit−MMAE付加体を産生させた。Cysクエンチング反応を約10℃にて約30分間進行させた。Cysクエンチング反応混合物を精製し、ダイアフィルトレーションにより緩衝液をPBSに交換して、部分付加cAC10−Val−Cit−MMAEを得た。
【0226】
分取HIC分画化。すべてのクロマトグラフ工程を室温にて実施した。1.6x25cmカラム(約50mL)にToyopearl Phenyl−650M HIC樹脂(Tosoh Bioscience,モンゴメリービル、ペンシルベニア州)を充填して、>5カラム容積の緩衝液A(50mMリン酸ナトリウム、2M NaCl、pH7.0)により流速15mL/分にて平衡化した。カラムへ充填するためのサンプルを調製するために、部分付加cAC10−vcMMAE 39mL(12.9mg/mL)を緩衝液A’39mL(50mMリン酸ナトリウム、4M NaCl、pH7.0)とブレンドした。サンプルをカラムに10mL/分で充填した;後の工程は、すべて10mL/分の流速にて実施した。サンプル充填の後に、A280ベースラインに到達するまでカラムを緩衝液Aで洗浄した。cAC10−E2は、65%緩衝液A/35%緩衝液Bからなる段階的勾配を用いて溶出および収集した(80%v/v 50mMリン酸ナトリウム、pH7.0、20%v/vアセトニトリル)。ベースラインに再度到達した後、cAC10−E4を30%緩衝液A/70%緩衝液Bからなる段階的勾配によって溶出および収集した。cAC10−E2およびcAC10−E4ピークを、どちらもその各ピーク高さの約20%まで収集した。目的の画分は、30kDaの分子量カットオフを用いたUltrafree−15遠心分離フィルタデバイス(Millipore,ビルリカ、マサチューセッツ州)を使用して、緩衝液をPBSに交換した。
【0227】
結合体の分析。結合体の分析は、エーテル−5PWカラム(Tosoh Bioscience,モンゴメリービル、ペンシルベニア州)を使用してHIC−HPLCによって実施した。方法は、50分間の100%緩衝液Aから100%緩衝液C(80%v/v 50mMリン酸ナトリウム、pH7.0、10%v/vアセトニトリル、10%v/vイソプロパノール)までの線形勾配により構成されていた。流速は1mL/分に設定され、温度は30℃に設定され、検出は248nmおよび280nmの両方にて追跡した。未改変(modify)cAC10およびcAC10−E8ピークの同定は、cAC10およびcAC10−E8標準の注入により確認した。抗体および薬物は別個の吸収極大(それぞれλmax=280nmおよび248nm)を有するため、ピークスペクトルを重ねることによって、抗体当たり2、4、6個の薬物を持つcAC10結合体に相当するピークを同定することが可能であった。
【0228】
cAC10−Val−Cit−MMAE ADCのインビトロ特徴づけ。結合体化または薬物の存在が抗原結合に影響を及ぼすかどうかを判定するために、ADCに対する競合結合を実施した。mAbおよびADCの飽和結合を比較するために、5x105Karpas−299細胞を、氷上の染色培地中で30分間、Alexa Fluor488(Molecular Probes,ユージーン、オレゴン州)によって標識された1μg/mL cAC10の存在下で、cAC10、cAC10−E2、cAC10−E4、またはcAC10−E8の連続希釈と混合し、氷冷染色培地で2回洗浄した。標識細胞をFusionマイクロプレートリーダー(Perkin−Elmer,ボストン、マサチューセッツ州)によって検査した。サンプルデータをバックグラウンド補正して、1μg/mL cAC10−Alexa Fluor(登録商標)488単独で染色された細胞の蛍光で割ったサンプル蛍光によって算出された最大蛍光パーセントとして報告した
DNA合成を測定することによってcAC10結合体の増殖阻害活性を決定した。結合体をCD30+Karpas−299またはL540cy細胞またはCD30−WSU−NHL細胞によってインキュベートした。cAC10またはcAC10を用いた92時間のインキュベーションの後、ADC細胞を0.5μCi/ウェルの[3H]−チミジンによって37℃にて4時間に渡って標識した。ハーベスタを使用して細胞をフィルタ上に収集し、シンチレーション流体と混合して、放射能をTopcountシンチレーションカウンタ(Packard Instruments,メリデン、コネチカット州)を用いて測定した。各分子について未処理のパーセントを濃度に対してプロットして、IC50を決定した(DNA合成の50%阻害を与えるmAb濃度として定義される)。
【0229】
ヒトALCLの異種移植モデル。ALCLの皮下疾患モデルを確立するために、5x106Karpas−299細胞をCB−17 SCIDマウス(Harlan,インディアナポリス、インディアナ州)の右側腹に移植した。各群6〜10匹の動物の腫瘍サイズが平均して約50〜100mm3になったときに、ADCを用いた療法を開始した。処置は、1回の注射、または注射4回について4日ごとに注射1回のスケジュール(q4dx4)を使用した複数回の静脈内注射のどちらかで構成されていた。腫瘍体積は、式(長さx幅2)/2を使用して計算した。触診できないほどにサイズが縮小した腫瘍は、完全後退(CR)として定義した。10腫瘍倍増時間以上続いた完全後退を治癒として定義した。対照グループの腫瘍のサイズが750〜1000mm3に達したときに腫瘍増殖阻害(TGI)を以下のように計算した:
【0230】
【化6】
最大耐容用量。3匹のBALB/cマウス(Harlan,インディアナポリス、インディアナ州)の群に、cAC10−E8 30〜60mg/kg、cAC10−E4 60〜120mg/kg、またはcAC10−vcMMAE2 200〜250mg/kgを尾静脈に注射して、単回投与最大許容用量(MTD)を決定した。マウスを毎日14日間監視し、重量および臨床観察の両方を記録した。著しい苦痛の徴候を発現したマウスをACUCガイドラインに従って殺処分した。最大許容用量は、いずれの動物においても注射の2週間以内に深刻で明白な毒性または20パーセントを超える体重減少を引き起こさない最大用量として定義した。
【0231】
薬物動態学。cAC10、cAC10−E2、cAC10−E4、およびcAC10−E8の薬物動態学をSCIDマウスで評価した。SCIDマウス(n=3)に試験物質10mg/kg(抗体成分に基づいて)を尾静脈注射によって投与した。血液サンプルは、伏在静脈への注射後1時間、4時間、1日、2日、4日、7日、14日、21日、28日、35日、42日、および49日に各マウスから収集した。ヘパリンをコーティングしたチューブに血液を収集して、続いて遠心分離(14,000xg、3分間)にかけて血漿を単離した。cAC10およびADCの血漿濃度をELISAによって測定した。
【0232】
簡潔には、ELISAは以下の工程より構成されていた:プレートコート、ブロック、サンプル結合、二次mAb、TMB、および酸停止。各工程後に、ウェルを洗浄緩衝液(PBS、0.05% Tween−20、pH=7.4)で3回洗浄した。プレートコート工程では、96ウェルプレートへ炭酸塩緩衝液(0.1M炭酸塩/重炭酸塩、pH=9.6)中の2μg/mLの抗cAC10mAbを4℃にて一晩コーティングした。プレートコートの後、ブロッキング緩衝液(PBS、1% BSA、0.05% Tween−20)を添加して、室温にて1時間インキュベートした。次に標準または希釈血漿サンプル100μLを3つのウェルにて室温で1時間添加した。二次工程は、1時間インキュベートされたマウス抗ヒトIgG−HRP結合体(Southern Biotech,バーミンガム、アラバマ)より構成されていた。次に3,3’,5,5’−テトラメチルベンジジン(Sigma,セントルイス、ミズーリ州)100μLを各ウェルに添加して、発色時に1N硫酸100μLによって反応を停止させた。VMax Kinetic Microplateリーダー(Molecular Devices,サニーベール、カリフォルニア州)によって、光学密度を450nmにて、ブランクを630nmにて測定した。非コンパートメント(non−compartment)薬物動態パラメータは、WinNonlin(Pharsight,マウンテンビュー、カリフォルニア州)を用いて計算した。
【0233】
インビトロ特徴づけ。MMAEのcAC10への結合体化がADCのCD30結合能力を阻害したかどうかを評価するために、競合結合実験を実施した。CD30+Karpas−299細胞を、未標識抗体、cAC10−E2、cAC10−E4、またはcAC10−E8の連続希釈と結合させた1μg/mL 蛍光標識cAC10を用いてインキュベートした。図10に示すように、ADC改変体は、それぞれ未標識cAC10と同等に、の蛍光標識cAC10と効果的に競合した。それゆえMMAEとの結合体化は、抗原結合を減少させなかった。
【0234】
ADCのインビトロ細胞毒性活性を、CD30+Karpas−299細胞およびL540cy細胞およびCD30−WSU−NHL細胞を用いた[3H]−TdR包含アッセイによって評価した。cAC10−E8は、1.0ng/mLのIC50が、Karpas−299細胞に対する著しい活性を示した(図11a)。mAb当たり4個のMMAE分子(cAC10−E4)に対する薬物の量を半分に減少させると、IC50が2.9ng/mLへと上昇した。薬物付加を再度半分にすると、cAC10−E2のIC50が6.2ng/mLにさらに上昇した。HD株L540cyに対して、ADCは同様の傾向を有し、cAC10−E8、cAC10−E4、およびcAC10−E2のIC50値はそれぞれ1.4ng/mL、4.5ng/mL、および9.2ng/mLであった(図11b)。ADCの選択性を、CD30−WSU−NHL細胞株を用いて評価した。この細胞株は、すべてのcAC10−ADCに対して不感受性であり、IC50値>1000ng/mLであった(データは示さず)。
【0235】
ヒトALCLの異種移植モデル。インビボ抗腫瘍活性に対する薬物付加の効果をKarpas−299皮下異種移植モデルで評価した。療法は、腫瘍体積が50〜100mm3に達したときに開始し、4日ごとに合計4回の注射を投与した(q4dx4)。このスケジュールを使用した場合、1mg/kgのcAC10−E8は、100% CRを生じさせ、同じ用量でcAC10−E4が100% CRを得たことが以前に見出された(データは示さず)。ADCの活性を比較する目的で、cAC10−E4およびcAC10−E8についてより少ない用量を使用した。皮下Karpas−299異種移植片を保有するマウス集団をcAC10−E2(0.5mg/kg/用量または1mg/kg/用量)、cAC10−E4(0.25または0.5mg/kg/用量)、またはcAC10−E8(0.25または0.5mg/kg/用量)の複数回の投与で処置した。表5は、有効性結果のまとめを示す。
【0236】
(表5)
【0237】
【表5】
表5:皮下Karpas−299異種移植モデルにおけるcAC10−E2、cAC10−E4、cAC10−E4混合物、およびcAC10−E8の抗腫瘍活性。腫瘍体積が50〜100mm3に達したら、動物をADCで処置した。投与は、1回(x1)または4日おきに4回(q4dx4)与えた。完全後退、治癒および腫瘍増殖阻害(TGI)の数を報告する。治癒した1匹のマウスが腫瘍塊の徴候なく第72日に死亡して発見された。
【0238】
cAC10−E8はMMAE量の2倍を同じmAb用量のcAC10−E4として有していたが、それらは両方の用量レベルで等しく有効であった(図12A)。0.5mg/Kg/用量にて、cAC10−E4によって処置した10匹の動物のうち、5匹が完全後退(CR)を達成し、cAC10−E8は10匹中6匹でCRを誘起した。未処置腫瘍は、移植19日後に986mm3の平均腫瘍体積に達した。cAC10−E4の腫瘍増殖阻害は、cAC10−E8の90%に対して91%であった。0.25mg/kg/用量にて、これらのADCの両方が未処置対照動物と比較して腫瘍増殖で同様の遅延を誘発したが、完全後退はなかった。0.5mg/kg/用量のcAC10−E4および0.25mg/kgのcAC10−E8として同じ量のMMAEを含有する用量である、1.0mg/kg/用量でのcAC10−E2は、10/10の治癒を誘起した。0.5mg/kg/用量のcAC10−E2による腫瘍増殖に対する効果は、68%のTGIを生じる0.25mg/kg/用量のcAC10−E4およびcAC10−E8で見られた効果に匹敵していた。薬物MMAEとcAC10との物理的混合物は、0.5mg/kg用量のcAC10−E8と同等であり、未処置と比較して腫瘍増殖のわずかな遅延のみを引き起こし、薬物の抗体への結合が抗腫瘍活性を達成するために重要であることを強調した。
【0239】
次にこの同じモデルでのcAC10−E2、cAC10−E4、およびcAC10−E8の単回投与処置を1.0mg/kgにて比較した(図12B)。cAC10−E8で処置した動物6匹のうち5匹が治癒を達成した。cAC10−E4で処置した動物6匹のうち、すべてが完全後退を達成して、5匹が試験終了の108日までに治癒し、動物1匹が腫瘍塊の徴候なく第72日に死亡して発見された。1.0mg/kgのcAC10−E2は、cAC10−E4の半分のMMAEを含有するが、マウス6匹のうち4匹がCRを達成した。cAC10 1mg/kgと、cAC10−E8 1mg/kgに含有されている薬物の量に相当する遊離MMAE 0.037mg/kgからなる対照グループは、未処置マウスと比較して腫瘍増殖に対する影響をほとんど有していなかった。
【0240】
部分付加ADCの初期結合は、0〜8個の薬剤/mAbを含有する種の混合物を生じた。この混合物(cAC10−E4混合物)の活性を評価するために、cAC10−E4混合物の単回投与抗腫瘍活性を1.0mg/kgの精製cAC10−E4と比較した。前の単回投与実験と同様に、cAC10−E4で処置したマウス10匹中9匹がCRを達成した。平均モル比4.0のcAC10−E4混合物で処置したマウス10匹中8匹で完全後退が生じた。部分付加cAC10−E4混合物は、種々に薬物付加されたADCの集団を含有しているが、精製cAC10−E4と同等の抗腫瘍活性を示した。
【0241】
最大許容用量および治療濃度域。cAC10−E2、cAC10−E4、およびcAC10−E8の単回投与許容性を1群あたり3匹のBALB/cマウスで評価した。最大許容用量(MTD)は、いずれの動物においても20パーセントを超える体重減少あるいは重篤な苦痛の徴候または明白な毒性を誘発しない最高用量として定義した。cAC10−E8では、30〜60mg/kgから10mg/kg間隔でマウスに投薬した。50mg/kgの用量では、マウスは注射6日後に14%の最大体重減少を有し、その後、体重減少は回復した。1匹の動物において、60mg/kgの用量は注射6日後に、23%の体重減少を誘発した。100mg/kgのcAC10−E4では、マウスは約10%の最大体重減少に達した。120mg/kgのcAC10−E4にて、動物1匹が著しい苦痛の徴候および17%の体重減少を示し、この動物を安楽死させた。試験を行った最高用量の250mg/kgまでの用量のcAC10−E2で処置したマウスは、注射6日後に10.5%の最大体重減少を経験し、苦痛の徴候はなかった。我々の観察に基づいて、cAC10−E2のMTDは少なくとも250mg/kgであり、cAC10−E4は100mg/kgであり、cAC10−E8は50mg/kgであった。治療指数は、単回投与MTDの複数回投与有効用量に対する比として定義され、cAC10−E8では100、cAC10−E4では200、cAC10−E2では少なくとも250を得た。
【0242】
薬物動態学。SCIDマウスにcAC10、cAC10−E2、cAC10−E4、およびcAC10−E8を投与して、薬物付加が薬物動態学にどのような影響を及ぼすかを判定した。表6は、非コンパートメント分析によって確立された薬物動態パラメータを示す。
【0243】
【表6】
表6.10mg/kgの用量でのSCIDマウスにおけるcAC10およびcAC10 ADCの薬物動態パラメータ。半減期(t1/2)、曲線下面積(AUC)、クリアランス(CL)、分布体積(Vz)、および注射から第14日までの曲線下面積(AUCt(0−14d))は、非コンパートメント分析を使用して計算した。
【0244】
cAC10、cAC10−E2、cAC10−E4、およびcAC10−E8の時間−濃度曲線は、バイエクスポネンシャルな下降に従った。末期半減期はそれぞれ16.7日、16.9日、14.0日および14.7日であり、それゆえ薬物付加とは直接相関していなかった。しかしながらAUCによって決定されたADCの暴露は薬物付加が減少するにつれ上昇し、未改変cAC10の2638μg日/mLからcAC10−E8の520μg日/mLの範囲であった。反対に、クリアランス値はcAC10の3.8mL/日/kgからcAC10−E2、cAC10−E4、およびcAC10−E8それぞれの4.4、6.0および19.2mL/日/kgまで上昇した。同様に、分布体積は薬物付加に直接相関することが見出された。
【0245】
(インビボ有効性のさらなる試験についての実施例9)
Karpas299 CD30陽性細胞株を使用してインビボ有効性実験を実施し、表7に示す。皮下Karpas−299腫瘍をC.B.−17 SCIDマウスで増殖させ、腫瘍が約100mm3(長さx幅2)に達したときに、試験品を投与した。動物を5〜10匹の群に分けて、各群にADCを静脈内注射した。用量は2倍連続希釈(0.5、1.0、2.0mg/Kg)で作製して、測定できないサイズまで退縮した腫瘍を完全寛解として定義した。数回(3〜8回、動物4匹の一群であるE6Pを除いて、用量当たり動物5〜15匹)の実験で≧80%の完全寛解を生じた用量を有効用量として割当てた。試験したすべてのADCで、注射した薬物成分の量が薬物付加によって変化したという事実にかかわらず、有効用量は1mg/Kgであった。実験(2倍連続希釈)の精度範囲内で、薬物の異性体分布は有効性に影響を及ぼさなかった。
【0246】
ADCの許容性を評価するために、BALB/cマウス(C.B.−17SCIDの親株)にADCを投与した。動物の体重を測定して、14日の期間に渡って臨床観察を記録した。MTDは、>20%の体重減少または苦痛の徴候を生じなかったBALB/cマウスに投与した最高単回用量とした。用量は、E8では、40、50および60mg/Kgであり、用量は、E4では、80、100および120mg/Kgであり、用量は、E2では、200および250mg/Kgである。以前に観察されたように(実施例8を参照)、絶対薬物付加レベルはMTDに影響を及ぼし、より高い薬物付加レベルはより低いMTD値を有する。DTNB部分再酸化によって産生された純E4では、MTDは、DTT部分還元によって産生された純E4よりもわずかに高かった(120対100mg/Kg)。
【0247】
(表7.CD30+Karpas299細胞に対するcAC10−vcMMAEのマウスインビボ有効性およびMTD)
【0248】
【表7】
aインビボ用量は、体重Kg当たりの抗体成分mgに基づいていた。
【0249】
本明細書で言及した、または本明細書に援用された特許または特許出願に対して、明示または黙示を問わず実施権は与えられていない。上の議論は説明的、例証的、かつ例示的であり、添付された請求項のいずれかによって定義された範囲を限定するものとして解釈すべきではない。
【0250】
特許出願、特許および科学の刊行物を含む種々の参考文献が本明細書で引用され、そのそれぞれの開示は、その全体が本明細書に参考として援用される。
【図面の簡単な説明】
【0251】
【図1】図1は、概略図の抗体の「E4」異性体(抗体当たり4個の薬物が結合された異性体)を示す。鎖間ジスルフィド結合は、抗体の重鎖−重鎖間および重鎖−軽鎖間の実線として示されている。薬物および抗体へのその結合箇所を円として示す。非還元(「Non−red」)および還元(「Red」)条件の下で産生されたフラグメントを各異性体の下に示す(カッコ内はフラグメント当たりの薬物の数)。
【図2】図2は、方法2a:部分DTNB再酸化、PD−10精製、およびvcMMAE結合体化の1つの局面によって調製されたvcMMAE−cAC10結合体の、疎水性相互作用クロマトグラフィー(HIC)分析におけるクロマトグラムの溶出プロフィールを示す。「E0」、「E2」、「E4」、「E6」および「E8」は、抗体当たり0、2、4、6および8個のMMAF分子がそれぞれ結合したcAC10抗体の異性体を指す。
【図3】図3は、方法2b:ワンポットDTNB再酸化およびvcMMAE結合体化の別の局面のHICクロマトグラムを示す。「E0」、「E2」、「E4」、「E6」および「E8」は、抗体当たり0、2、4、6および8個のMMAF分子がそれぞれ結合したcAC10抗体の異性体を指す。純粋なE4は、約34〜38分より収集した(矢印で示す)。
【図4】図4は、方法1、2a、および2bからの一様な薬物付加種の抗体−薬物結合体の組成パーセントの棒グラフでの比較を示す。各種では、抗体−薬物結合体をDTT部分還元(左棒)、DTNB再酸化(中央棒)またはワンポットDTNB再酸化(右棒)結合体化方法によって調製した。
【図5】図5は、図3(方法2b)でHICクロマトグラムに示したように収集したE4材料のバイオアナライザによるトレースを示す。「L」は遊離軽鎖を示す。「H」は遊離重鎖を示す。「HL」は結合された重鎖−軽鎖を示す。「HH」は結合された重鎖−重鎖を示す。「HHL」は結合された重鎖−重鎖および重鎖−軽鎖を示す。
【図6】図6は、図3(方法2b)のHICクロマトグラムから収集した材料のPLRP分析を示す。「L0」および「L1」はそれぞれ、薬物分子が結合していない、または薬物分子が1個結合した軽鎖を示す。「H0」、「H1」「H2」および「H3」は、0、1、2または3個の薬物分子が結合した重鎖を示す。
【図7】図7は、鎖間ジスルフィド結合の完全または部分還元と、それに続く薬物の結合体化によって得られた主な結合体種の概略表現を示す。完全還元および結合は主に、抗体当たり8つの薬物を持つ完全付加種を生成するのに対して、部分還元および結合は、図示した種すべての産生を引き起こすことができる。0および8薬物付加種にはそれぞれ異性体が1つのみあるが、2、4および6薬物付加種は3つ、4つおよび3つの異性体をそれぞれ含有する(図1を参照して、種4Aが種4Cの鏡像であり、種4Bが種4Dの鏡像であることに注目する。図7では、種4Aおよび種4Cは種4Aと呼ばれ、種4Bおよび種4Dは種4Bと呼ばれる)。鎖間ジスルフィド結合は、抗体の重鎖−重鎖間または重鎖−軽鎖間の実線として示す。薬物および抗体へのその結合箇所を円として示す。
【図8】図8は、E4混合異性体(E4M)を産生するためにDTTを使用する「部分還元」結合プロセスの1つの局面のプロセスフローダイアグラムである。cAC10抗体のpHは、リン酸水素二ナトリウムによって7.5に調製し、EDTAを5mMの最終濃度まで添加する。次に抗体溶液を37℃に加熱する。抗体を部分還元するために、DTT 2.95モル当量を抗体溶液に添加して、37℃にて105分間還元する。還元後、抗体溶液を2〜8℃に冷却して、過剰なDTTを定容限外濾過/ダイアフィルトレーション(UF/DF)によって濾過し、還元された精製cAC10を得る。還元された精製cAC10のサンプルを取り、A280およびDTNB試験によって、チオール濃度、抗体濃度、およびチオール対抗体モル比を決定する。次にやや過剰の薬物−リンカーvcMMAE(通例、DMSO溶液の形で2〜15%過剰)を抗体溶液に添加して、結合反応を開始させる。結合反応を2〜8℃にて30分間進行させて、粗E4Mを得る。結合反応の終わりに、過剰のvcMMAE薬物−リンカーを、大幅に過剰のシステインと2〜8℃にて15分間反応させることによってクエンチし、クエンチされた粗E4Mを得る。緩衝液交換ならびに遊離薬物および他の小型分子種の除去は、定容量UF/DF(代表的には、6〜10透析体積)によって実施し、E4M薬物物質を得る。
【図9】図9は、E4混合異性体(E4M)を産生するための、中間精製ステップが使用されないTCEPを使用する「部分還元」結合プロセスの別の局面のプロセスフローダイアグラムである。cAC10抗体のpHは、リン酸水素二ナトリウム7.5に調製し、EDTAを5mMの最終濃度まで添加する。次に抗体溶液を37℃に加熱する。抗体を部分還元するために、TCEP 2.20モル当量を抗体溶液に添加して、37℃にて105分間還元する。還元反応のサンプルを取り、A280およびDTNB試験によって、チオール濃度、抗体濃度、およびチオール対抗体モル比を決定する。還元後、抗体溶液を2〜8℃に冷却する。次にやや過剰の薬物−リンカーvcMMAE(通例、DMSO溶液の形で2〜15%過剰)を抗体溶液に添加して、結合反応を開始させる。結合反応を2〜8℃にて20分間進行させて、粗E4Mを得る。結合反応の終わりに、過剰のvcMMAE薬物−リンカーを、大幅に過剰のN−アセチルシステインと2〜8℃にて20分間反応させることによってクエンチし、クエンチされた粗E4Mを得る。緩衝液交換ならびに遊離薬物および他の小型分子種の除去は、定容UF/DF(通例6〜10透析体積)によって実施し、E4M薬物物質を得る。
【図10】図10は、CD30+Karpas−299細胞によるcAC10結合体化抗体の内部移行のグラフを示す。細胞を蛍光標識cAC10 1g/mLおよびcAC10、cAC10−E2、cAC10−E4、またはcAC10−E8のいずれかの20μg/mL〜9ng/mLの連続希釈物と合せた。抗体による細胞のインキュベーションの後、標識細胞を染色溶媒で洗浄し、蛍光を測定した。実施例8で述べるように、正規化蛍光強度をmAb濃度に対してプロットした。
【図11】図11は、CD30+細胞によるcAC10およびcAC10結合体化抗体の内部移行のグラフを示す:A)Karpas−299およびB)L540cy細胞をcAC10ならびにcAC10 ADCのE2、E4およびE8種の連続希釈物によってインキュベートした。サンプルを用いた96時間のインキュベーションの後、[3H]−TdRを添加して、その包含を測定した。処置サンプルの放射能を未処理対照へ正規化して、濃度に対してプロットした。
【図12】図12は、皮下異種移植を持つSCIDマウスにおけるcAC10およびcAC10結合抗体のインビボ有効性を示す。図12Aは、cAC10−E2を0.5mg/kgまたは1.0mg/kgにて注射4回を4日おきに注射された、Karpas−299皮下腫瘍を持つSCIDマウスによる結果を示す。cAC10−E4およびcAC10−8は、0.25または0.5mg/kgのどちらかで、注射4回を4日おきに投薬された。図12Bは、E2、E4またはE8の1.0mg/kgでの単回投与によって処置された、Karpas−299皮下腫瘍を持つSCIDマウスの結果を示した。
【図13】図13は、(A)E4ミックス;(B)分取HICによって作成した純E2;(C)分取HICによって作成した純E4;および(D)分取HICによって作成した純E6のそれぞれの疎水性相互作用クロマトグラフィーHPLCトレースを示す。サンプルは、DTT部分還元と、それに続くMMAE結合によって作成した。クロマトグラムは、各クロマトグラムの最高ピークの高さへ正規化した。注射は、2.0M NaClおよび50mMリン酸ナトリウム、pH7 50μLと混合した、PBS中の5〜10mg/mL cAC10−vcMMAE 50μLであった。分離は30℃にて実施した。
【図14】図14は、(A)部分DTT還元と、それに続くMMAE結合によって作成したE4ミックス(上トレース);(B)DTNB部分再酸化と、それに続くMMAE結合によって作成したE4ミックス(上から2番目のトレース);(C)部分DTT還元と、それに続くMMAE結合によって作成し、分取HICによって精製した純E4(下から2番目のトレース);および(D)部分DTNB再酸化と、それに続くMMAE結合によって作成し、分取HICによって精製した純E4(下トレース)のPLRP−S HPLCトレースを示す。注射は、20mM DTTで37℃にて15分間処理した1mg/mL cAC10−vcMMAE 20μLであった。分離は80℃にて実施した。
【図15】図15は、(A)部分DTT還元と、それに続くMMAE結合によって作成したE4ミックス(上トレース);(B)DTNB部分再酸化と、それに続くMMAE結合によって作成したE4ミックス(上から2番目のトレース);(C)部分DTT還元と、それに続くMMAE結合によって作成し、分取HICによって精製した純E4(下から2番目のトレース);および(D)部分DTNB再酸化と、それに続くMMAE結合によって作成し、分取HICによって精製した純E4(下トレース)のバイオアナライザ(キャピラリー電気泳動)トレースを示す。サンプルは、メーカーが指示した通りに非還元条件下で調製した。
【特許請求の範囲】
【請求項1】
部分付加抗体であって、該抗体は以下:
少なくとも1個の鎖間ジスルフィド結合、および
少なくとも2個の細胞毒性薬または細胞増殖抑制薬または標識であって、各薬物または標識は、鎖間チオールに結合体化された、薬物または標識;
を含み、ここで該薬物または標識の結合箇所が容易に割当て可能である、
部分付加抗体。
【請求項2】
前記抗体が少なくとも4個の細胞毒性薬または細胞増殖抑制薬を有し、各薬物が鎖間チオールに結合されている、請求項1に記載の部分付加抗体。
【請求項3】
種4Aまたは種4Bの配置を有する、請求項1に記載の部分付加抗体。
【請求項4】
種4Cまたは種4Dの配置を有する、請求項1に記載の部分付加抗体。
【請求項5】
種4Eの配置を有する、請求項1に記載の部分付加抗体。
【請求項6】
種4Fの配置を有する、請求項1に記載の部分付加抗体。
【請求項7】
前記抗体がヒト化またはキメラ抗体である、請求項1に記載の部分付加抗体。
【請求項8】
前記細胞毒性薬または細胞増殖抑制薬がMMAF、MMAE、またはAFPである、請求項1に記載の部分付加抗体。
【請求項9】
請求項1に記載の抗体および薬学的に受容可能なキャリアを含む、薬学的組成物。
【請求項10】
抗体であって、該抗体は、
細胞毒性薬または細胞増殖抑制薬のための少なくとも結合箇所を含み、ここで、該抗体上の細胞毒性薬または細胞増殖抑制薬のための該結合箇所が容易に割当て可能であり、考えられるすべての結合箇所より少数が該細胞毒性薬または細胞増殖抑制薬への結合体化に利用可能である、
抗体。
【請求項11】
前記結合箇所が鎖間チオールである、請求項10に記載の抗体。
【請求項12】
前記結合箇所が4Aから4Fの少なくとも1個である、請求項10に記載の抗体。
【請求項13】
前記結合箇所が4Aから4Fからなる群の少なくとも1個から選択される、請求項10に記載の抗体。
【請求項14】
割当て可能な結合箇所を有する修飾抗体の組成物であって、該組成物は:
修飾抗体の少なくとも4種を含み、それぞれの種が(a)2個の鎖間チオールを有する少なくとも1個の特定結合対および(b)少なくとも1個の鎖間ジスルフィド結合、
を含む、組成物。
【請求項15】
種4Aおよび種4Bを含む、請求項14に記載の組成物。
【請求項16】
種4Cおよび種4Dを含む、請求項14に記載の組成物。
【請求項17】
種4Eを含む、請求項14に記載の組成物。
【請求項18】
種4Fを含む、請求項14に記載の組成物。
【請求項19】
前記特定結合対が定常軽鎖−定常重鎖間ジスルフィド結合にある、請求項14に記載の組成物。
【請求項20】
前記特定結合対が定常重鎖−定常重鎖間ジスルフィド結合にある、請求項14に記載の組成物。
【請求項21】
前記特定結合対がヒンジ領域のN末端より近位である、請求項14に記載の組成物。
【請求項22】
前記特定結合対がヒンジ領域のC末端より近位である、請求項14に記載の組成物。
【請求項23】
前記特定結合対が定常軽鎖−定常重鎖間スルフィド結合に、および前記修飾抗体のN末端付近に位置するヒンジ領域にある、請求項14に記載の組成物。
【請求項24】
前記特定結合対が定常軽鎖−定常重鎖間ジスルフィド結合に、および前記修飾抗体のC末端付近に位置するヒンジ領域にある、請求項14に記載の組成物。
【請求項25】
定常軽鎖−定常重鎖間ジスルフィド結合における少なくとも2個の特定結合対を含む、請求項14に記載の組成物。
【請求項26】
ヒンジ領域鎖間ジスルフィド結合における少なくとも2個の特定結合対を含む、請求項14に記載の組成物。
【請求項27】
薬学的に受容可能なキャリアをさらに含む、請求項14に記載の組成物。
【請求項28】
部分付加抗体であって、該抗体は、以下:
少なくとも1個の抗原結合ドメイン、
該抗体上の少なくとも2個の反応性基、および
少なくとも2個の薬物または標識であって、各薬物または標識は、反応性基に結合体化されて、結合箇所を形成する、薬物または標識
を含み、
ここで、該薬物または標識の結合箇所が容易に割当可能である、
部分付加抗体。
【請求項29】
割当て可能な結合箇所を有する部分的に付加された修飾タンパク質であって、該タンパク質は、以下:
結合パートナーとの相互作用のための結合領域、
それぞれ薬物または標識に共有結合される少なくとも2個の結合箇所
を含み、
ここで、同様の接近性および活性化性を有する、考えられるすべての結合箇所のより少数が薬物または標識に結合される、
修飾タンパク質。
【請求項30】
前記タンパク質が抗体である、請求項29に記載の修飾タンパク質。
【請求項31】
前記結合箇所がアミノ基、隣接ヒドロキシル基、ヒドロキシル基、カルボキシル基、またはチオール基である、請求項29に記載の修飾タンパク質。
【請求項32】
前記タンパク質がレセプター、レセプターリガンド、ホルモン、またはサイトカインである、請求項29に記載の修飾タンパク質。
【請求項33】
癌、免疫疾患、自己免疫疾患または感染性疾患の処置のための医薬品の調製における部分付加抗体の使用であって、該部分付加抗体は、少なくとも1個の鎖間ジスルフィド結合、およびそれぞれ鎖間チオールに結合された少なくとも2個の細胞毒性薬または細胞増殖抑制薬あるいは標識を含み;ここで、該薬物または標識の結合箇所が容易に割当て可能である、部分付加抗体の使用。
【請求項34】
薬物を還元して、薬物配置の選択性をもたらす抗体に結合させる方法であって:
該抗体を還元剤によって完全に還元する工程;
該完全に還元された抗体を再酸化剤の制限量で処理し、少なくとも2個の鎖間チオールが残存するように、該抗体の少なくとも1個の鎖間ジスルフィド結合を再形成する工程;
該薬物を該鎖間チオールに結合体化する工程;
を包含する、方法。
【請求項35】
前記再酸化剤が、5,5’−ジチオ−ビス−2−ニトロ安息香酸、4,4’−ジチオジピリジン、2,2’−ジチオジピリジン、テトラチオン酸ナトリウムまたはヨードソ安息香酸である、請求項34に記載の方法。
【請求項36】
前記薬物が、細胞毒性薬または細胞増殖抑制薬あるいは免疫抑制剤である、請求項34に記載の方法。
【請求項37】
前記細胞毒性薬または細胞増殖抑制薬が、副溝バインダ、AEB、またはAEVBである、請求項34に記載の方法。
【請求項38】
前記薬物が、MMAF、MMAEまたはAFPである、請求項34に記載の方法。
【請求項39】
前記還元剤がDTTまたはTCEPである、請求項34に記載の方法。
【請求項40】
抗体鎖間ジスルフィド結合を還元して、生じた鎖間チオールに薬物を結合させて、該抗体に対する該薬物配置の選択性を生じさせる方法であって、該方法は:
該抗体を還元剤によって完全に還元し、鎖間チオールを形成する工程;
該抗体を再酸化剤で部分的に再酸化し、少なくとも1個の鎖間ジスルフィド結合を再生成する工程;
該薬物を該鎖間チオールに結合させる工程、
を包含する、方法。
【請求項41】
前記再酸化剤が、5,5’−ジチオ−ビス−2−ニトロ安息香酸、4,4’−ジチオジピリジン、2,2’−ジチオジピリジン、テトラチオン酸ナトリウム、またはヨードソ安息香酸である、請求項40に記載の方法。
【請求項42】
前記還元剤が、DTTまたはTCEPである、請求項40に記載の方法。
【請求項43】
前記部分的に再酸化された抗体を精製する工程をさらに包含する、請求項40に記載の方法。
【請求項44】
前記薬物が、MMAF、MMAE、またはAFPである、請求項40に記載の方法。
【請求項45】
抗体鎖間ジスルフィド結合を還元して、薬物を生じた鎖間チオールに結合させて、薬物を該抗体上に選択的に配置する方法であって、該方法は:
該抗体を還元剤によって部分的に還元し、少なくとも2個の鎖間チオールを形成する工程;および
該薬物を、該部分的に還元された抗体の鎖間チオールに結合体化させる工程、
を包含する、方法。
【請求項46】
請求項45に記載の方法であって、
ここで、前記抗体は、制限濃度の還元剤によって、キレート剤を含む緩衝液中で部分的に還元され;そして
ここで、該抗体溶液を冷却して、前記薬物を冷溶媒に溶解させ、該抗体溶液と混合することによって、該薬物が結合体化され;
該抗体および該薬物溶液を、抗体−薬物結合体を形成するのに十分な時間インキュベートさせ;
過剰の薬物をチオール含有試薬によってクエンチさせ;そして
生じた結合体を精製する、
方法。
【請求項47】
前記抗体が約37℃にて約1時間、部分的に還元される、請求項46に記載の方法。
【請求項48】
前記還元された抗体が約0℃まで冷却される、請求項46に記載の方法。
【請求項49】
前記抗体および薬剤溶液が、約0℃にて約30分間インキュベートされる、請求項46に記載の方法。
【請求項50】
前記チオール含有試薬が、システインまたはN−アセチルシステインである、請求項45に記載の方法。
【請求項51】
前記還元剤が、DTTまたはTCEPである、請求項45に記載の方法。
【請求項52】
前記緩衝液がホウ酸ナトリウム溶液であり、前記キレート剤がジエチレントリアミン五酢酸である、請求項46に記載の方法。
【請求項53】
前記キレート剤が、エチレントリアミン五酢酸またはEDTAである、請求項46に記載の方法。
【請求項54】
前記還元された抗体を精製する工程をさらに包含する、請求項45に記載の方法。
【請求項55】
前記還元された抗体が、カラムクロマトグラフィー、透析、またはダイアフィルトレーションを使用して精製される、請求項54に記載の方法。
【請求項56】
前記カラムが脱塩カラムである、請求項55に記載の方法。
【請求項57】
前記脱塩カラムがPD−10カラムである、請求項57に記載の方法。
【請求項58】
前記還元された抗体が、部分的還元後および結合体化前に精製されない、請求項45に記載の方法。
【請求項59】
前記結合体がカラムクロマトグラフィー、透析、またはダイアフィルトレーションを使用して精製される、請求項45に記載の方法。
【請求項60】
前記カラムが脱塩カラムである、請求項59に記載の方法。
【請求項61】
前記脱塩カラムがPD−10カラムである、請求項61に記載の方法。
【請求項62】
前記溶媒がアセトニトリル、アルコールまたはDMSOである、請求項45に記載の方法。
【請求項63】
前記薬物が細胞毒性薬または細胞増殖抑制薬である、請求項45に記載の方法。
【請求項64】
薬物の選択的結合を有する抗体を産生する方法であって、該方法は:
DTNB滴定によって決定されるように、大幅に過剰の還元剤を添加して、溶液を約37℃にて約30分間インキュベートすることによって、鎖間チオールを生成するのに十分な時間、該抗体を完全に還元する工程;
該抗体を精製する工程;
酸化剤を使用して該抗体を部分的に再酸化し、少なくとも1個の鎖間ジスルフィド結合を、以下、
該還元された抗体を0℃に冷却する工程;
該還元および冷却された抗体を該酸化剤1.5〜2.5モル当量で処理する工程;
反転により該溶液を混合する工程;
約0℃にて約10分間インキュベートする工程
によって形成する工程;
該部分的に再酸化された抗体を精製する工程;
該薬物を該部分的に再酸化された抗体の鎖間チオールに結合体化させ、結合体化抗体を作成する工程;ならびに
該結合体化抗体を精製する工程
を包含する、方法。
【請求項65】
前記還元剤が、DTTまたはTCEPである、請求項64に記載の方法。
【請求項66】
前記抗体が、カラムクロマトグラフィー、透析、またはダイアフィルトレーションを使用して精製される、請求項64に記載の方法。
【請求項67】
前記カラムが脱塩カラムである、請求項66に記載の方法。
【請求項68】
前記脱塩カラムがPD−10カラムである、請求項67に記載の方法。
【請求項69】
前記結合体化抗体が、カラムクロマトグラフィー、透析、またはダイアフィルトレーションを使用して精製される、請求項64に記載の方法。
【請求項70】
前記カラムが脱塩カラムである、請求項69に記載の方法。
【請求項71】
前記脱塩カラムがPD−10カラムである、請求項70に記載の方法。
【請求項72】
前記再酸化剤が、5,5’−ジチオ−ビス−2−ニトロ安息香酸、4,4’−ジチオジピリジン、2,2’−ジチオジピリジン、テトラチオン酸ナトリウム、またはヨードソ安息香酸である、請求項64に記載の方法。
【請求項73】
1個以上のジスルフィド結合を有するタンパク質と、遊離チオールと反応性である薬物との結合体を調製する方法であって、該方法は:
該タンパク質を還元剤によって部分的に還元する工程;
該遊離チオールと反応性である薬物を該部分的に還元されたタンパク質に結合体化する工程
を包含する、方法。
【請求項74】
1個以上のジスルフィド結合を有するタンパク質と、遊離チオールと反応性である薬物との結合体を調製する方法であって、該方法は:
該タンパク質を還元剤によって完全に還元する工程;
該タンパク質を再酸化剤によって部分的に再酸化する工程;
該遊離チオールと反応性である薬物を抗体に結合体化する工程;
を包含する、方法。
【請求項75】
部分的に付加された抗体を形成する方法であって、該方法は:
抗体含有溶液を提供する工程、
該抗体溶液のpHを約pH7.5に調整して、キレート剤を添加する工程;
該抗体溶液を約37℃まで加熱する工程;
モル過剰のTCEPを該抗体溶液に添加し、約37℃にて十分な時間反応させて、該抗体の鎖間ジスルフィド基を部分的に還元して鎖間チオールを形成する工程;
該抗体溶液を約2〜8℃まで冷却することと;
わずかにモル過剰の薬物を該抗体溶液に添加することによって、該部分的に還元された抗体の鎖間チオールに該薬物を結合体化させ、そして該部分的に付加された抗体を形成するために十分な時間反応させる工程;ならびに
部分的に付加された抗体を精製する工程、
を包含する、方法。
【請求項1】
部分付加抗体であって、該抗体は以下:
少なくとも1個の鎖間ジスルフィド結合、および
少なくとも2個の細胞毒性薬または細胞増殖抑制薬または標識であって、各薬物または標識は、鎖間チオールに結合体化された、薬物または標識;
を含み、ここで該薬物または標識の結合箇所が容易に割当て可能である、
部分付加抗体。
【請求項2】
前記抗体が少なくとも4個の細胞毒性薬または細胞増殖抑制薬を有し、各薬物が鎖間チオールに結合されている、請求項1に記載の部分付加抗体。
【請求項3】
種4Aまたは種4Bの配置を有する、請求項1に記載の部分付加抗体。
【請求項4】
種4Cまたは種4Dの配置を有する、請求項1に記載の部分付加抗体。
【請求項5】
種4Eの配置を有する、請求項1に記載の部分付加抗体。
【請求項6】
種4Fの配置を有する、請求項1に記載の部分付加抗体。
【請求項7】
前記抗体がヒト化またはキメラ抗体である、請求項1に記載の部分付加抗体。
【請求項8】
前記細胞毒性薬または細胞増殖抑制薬がMMAF、MMAE、またはAFPである、請求項1に記載の部分付加抗体。
【請求項9】
請求項1に記載の抗体および薬学的に受容可能なキャリアを含む、薬学的組成物。
【請求項10】
抗体であって、該抗体は、
細胞毒性薬または細胞増殖抑制薬のための少なくとも結合箇所を含み、ここで、該抗体上の細胞毒性薬または細胞増殖抑制薬のための該結合箇所が容易に割当て可能であり、考えられるすべての結合箇所より少数が該細胞毒性薬または細胞増殖抑制薬への結合体化に利用可能である、
抗体。
【請求項11】
前記結合箇所が鎖間チオールである、請求項10に記載の抗体。
【請求項12】
前記結合箇所が4Aから4Fの少なくとも1個である、請求項10に記載の抗体。
【請求項13】
前記結合箇所が4Aから4Fからなる群の少なくとも1個から選択される、請求項10に記載の抗体。
【請求項14】
割当て可能な結合箇所を有する修飾抗体の組成物であって、該組成物は:
修飾抗体の少なくとも4種を含み、それぞれの種が(a)2個の鎖間チオールを有する少なくとも1個の特定結合対および(b)少なくとも1個の鎖間ジスルフィド結合、
を含む、組成物。
【請求項15】
種4Aおよび種4Bを含む、請求項14に記載の組成物。
【請求項16】
種4Cおよび種4Dを含む、請求項14に記載の組成物。
【請求項17】
種4Eを含む、請求項14に記載の組成物。
【請求項18】
種4Fを含む、請求項14に記載の組成物。
【請求項19】
前記特定結合対が定常軽鎖−定常重鎖間ジスルフィド結合にある、請求項14に記載の組成物。
【請求項20】
前記特定結合対が定常重鎖−定常重鎖間ジスルフィド結合にある、請求項14に記載の組成物。
【請求項21】
前記特定結合対がヒンジ領域のN末端より近位である、請求項14に記載の組成物。
【請求項22】
前記特定結合対がヒンジ領域のC末端より近位である、請求項14に記載の組成物。
【請求項23】
前記特定結合対が定常軽鎖−定常重鎖間スルフィド結合に、および前記修飾抗体のN末端付近に位置するヒンジ領域にある、請求項14に記載の組成物。
【請求項24】
前記特定結合対が定常軽鎖−定常重鎖間ジスルフィド結合に、および前記修飾抗体のC末端付近に位置するヒンジ領域にある、請求項14に記載の組成物。
【請求項25】
定常軽鎖−定常重鎖間ジスルフィド結合における少なくとも2個の特定結合対を含む、請求項14に記載の組成物。
【請求項26】
ヒンジ領域鎖間ジスルフィド結合における少なくとも2個の特定結合対を含む、請求項14に記載の組成物。
【請求項27】
薬学的に受容可能なキャリアをさらに含む、請求項14に記載の組成物。
【請求項28】
部分付加抗体であって、該抗体は、以下:
少なくとも1個の抗原結合ドメイン、
該抗体上の少なくとも2個の反応性基、および
少なくとも2個の薬物または標識であって、各薬物または標識は、反応性基に結合体化されて、結合箇所を形成する、薬物または標識
を含み、
ここで、該薬物または標識の結合箇所が容易に割当可能である、
部分付加抗体。
【請求項29】
割当て可能な結合箇所を有する部分的に付加された修飾タンパク質であって、該タンパク質は、以下:
結合パートナーとの相互作用のための結合領域、
それぞれ薬物または標識に共有結合される少なくとも2個の結合箇所
を含み、
ここで、同様の接近性および活性化性を有する、考えられるすべての結合箇所のより少数が薬物または標識に結合される、
修飾タンパク質。
【請求項30】
前記タンパク質が抗体である、請求項29に記載の修飾タンパク質。
【請求項31】
前記結合箇所がアミノ基、隣接ヒドロキシル基、ヒドロキシル基、カルボキシル基、またはチオール基である、請求項29に記載の修飾タンパク質。
【請求項32】
前記タンパク質がレセプター、レセプターリガンド、ホルモン、またはサイトカインである、請求項29に記載の修飾タンパク質。
【請求項33】
癌、免疫疾患、自己免疫疾患または感染性疾患の処置のための医薬品の調製における部分付加抗体の使用であって、該部分付加抗体は、少なくとも1個の鎖間ジスルフィド結合、およびそれぞれ鎖間チオールに結合された少なくとも2個の細胞毒性薬または細胞増殖抑制薬あるいは標識を含み;ここで、該薬物または標識の結合箇所が容易に割当て可能である、部分付加抗体の使用。
【請求項34】
薬物を還元して、薬物配置の選択性をもたらす抗体に結合させる方法であって:
該抗体を還元剤によって完全に還元する工程;
該完全に還元された抗体を再酸化剤の制限量で処理し、少なくとも2個の鎖間チオールが残存するように、該抗体の少なくとも1個の鎖間ジスルフィド結合を再形成する工程;
該薬物を該鎖間チオールに結合体化する工程;
を包含する、方法。
【請求項35】
前記再酸化剤が、5,5’−ジチオ−ビス−2−ニトロ安息香酸、4,4’−ジチオジピリジン、2,2’−ジチオジピリジン、テトラチオン酸ナトリウムまたはヨードソ安息香酸である、請求項34に記載の方法。
【請求項36】
前記薬物が、細胞毒性薬または細胞増殖抑制薬あるいは免疫抑制剤である、請求項34に記載の方法。
【請求項37】
前記細胞毒性薬または細胞増殖抑制薬が、副溝バインダ、AEB、またはAEVBである、請求項34に記載の方法。
【請求項38】
前記薬物が、MMAF、MMAEまたはAFPである、請求項34に記載の方法。
【請求項39】
前記還元剤がDTTまたはTCEPである、請求項34に記載の方法。
【請求項40】
抗体鎖間ジスルフィド結合を還元して、生じた鎖間チオールに薬物を結合させて、該抗体に対する該薬物配置の選択性を生じさせる方法であって、該方法は:
該抗体を還元剤によって完全に還元し、鎖間チオールを形成する工程;
該抗体を再酸化剤で部分的に再酸化し、少なくとも1個の鎖間ジスルフィド結合を再生成する工程;
該薬物を該鎖間チオールに結合させる工程、
を包含する、方法。
【請求項41】
前記再酸化剤が、5,5’−ジチオ−ビス−2−ニトロ安息香酸、4,4’−ジチオジピリジン、2,2’−ジチオジピリジン、テトラチオン酸ナトリウム、またはヨードソ安息香酸である、請求項40に記載の方法。
【請求項42】
前記還元剤が、DTTまたはTCEPである、請求項40に記載の方法。
【請求項43】
前記部分的に再酸化された抗体を精製する工程をさらに包含する、請求項40に記載の方法。
【請求項44】
前記薬物が、MMAF、MMAE、またはAFPである、請求項40に記載の方法。
【請求項45】
抗体鎖間ジスルフィド結合を還元して、薬物を生じた鎖間チオールに結合させて、薬物を該抗体上に選択的に配置する方法であって、該方法は:
該抗体を還元剤によって部分的に還元し、少なくとも2個の鎖間チオールを形成する工程;および
該薬物を、該部分的に還元された抗体の鎖間チオールに結合体化させる工程、
を包含する、方法。
【請求項46】
請求項45に記載の方法であって、
ここで、前記抗体は、制限濃度の還元剤によって、キレート剤を含む緩衝液中で部分的に還元され;そして
ここで、該抗体溶液を冷却して、前記薬物を冷溶媒に溶解させ、該抗体溶液と混合することによって、該薬物が結合体化され;
該抗体および該薬物溶液を、抗体−薬物結合体を形成するのに十分な時間インキュベートさせ;
過剰の薬物をチオール含有試薬によってクエンチさせ;そして
生じた結合体を精製する、
方法。
【請求項47】
前記抗体が約37℃にて約1時間、部分的に還元される、請求項46に記載の方法。
【請求項48】
前記還元された抗体が約0℃まで冷却される、請求項46に記載の方法。
【請求項49】
前記抗体および薬剤溶液が、約0℃にて約30分間インキュベートされる、請求項46に記載の方法。
【請求項50】
前記チオール含有試薬が、システインまたはN−アセチルシステインである、請求項45に記載の方法。
【請求項51】
前記還元剤が、DTTまたはTCEPである、請求項45に記載の方法。
【請求項52】
前記緩衝液がホウ酸ナトリウム溶液であり、前記キレート剤がジエチレントリアミン五酢酸である、請求項46に記載の方法。
【請求項53】
前記キレート剤が、エチレントリアミン五酢酸またはEDTAである、請求項46に記載の方法。
【請求項54】
前記還元された抗体を精製する工程をさらに包含する、請求項45に記載の方法。
【請求項55】
前記還元された抗体が、カラムクロマトグラフィー、透析、またはダイアフィルトレーションを使用して精製される、請求項54に記載の方法。
【請求項56】
前記カラムが脱塩カラムである、請求項55に記載の方法。
【請求項57】
前記脱塩カラムがPD−10カラムである、請求項57に記載の方法。
【請求項58】
前記還元された抗体が、部分的還元後および結合体化前に精製されない、請求項45に記載の方法。
【請求項59】
前記結合体がカラムクロマトグラフィー、透析、またはダイアフィルトレーションを使用して精製される、請求項45に記載の方法。
【請求項60】
前記カラムが脱塩カラムである、請求項59に記載の方法。
【請求項61】
前記脱塩カラムがPD−10カラムである、請求項61に記載の方法。
【請求項62】
前記溶媒がアセトニトリル、アルコールまたはDMSOである、請求項45に記載の方法。
【請求項63】
前記薬物が細胞毒性薬または細胞増殖抑制薬である、請求項45に記載の方法。
【請求項64】
薬物の選択的結合を有する抗体を産生する方法であって、該方法は:
DTNB滴定によって決定されるように、大幅に過剰の還元剤を添加して、溶液を約37℃にて約30分間インキュベートすることによって、鎖間チオールを生成するのに十分な時間、該抗体を完全に還元する工程;
該抗体を精製する工程;
酸化剤を使用して該抗体を部分的に再酸化し、少なくとも1個の鎖間ジスルフィド結合を、以下、
該還元された抗体を0℃に冷却する工程;
該還元および冷却された抗体を該酸化剤1.5〜2.5モル当量で処理する工程;
反転により該溶液を混合する工程;
約0℃にて約10分間インキュベートする工程
によって形成する工程;
該部分的に再酸化された抗体を精製する工程;
該薬物を該部分的に再酸化された抗体の鎖間チオールに結合体化させ、結合体化抗体を作成する工程;ならびに
該結合体化抗体を精製する工程
を包含する、方法。
【請求項65】
前記還元剤が、DTTまたはTCEPである、請求項64に記載の方法。
【請求項66】
前記抗体が、カラムクロマトグラフィー、透析、またはダイアフィルトレーションを使用して精製される、請求項64に記載の方法。
【請求項67】
前記カラムが脱塩カラムである、請求項66に記載の方法。
【請求項68】
前記脱塩カラムがPD−10カラムである、請求項67に記載の方法。
【請求項69】
前記結合体化抗体が、カラムクロマトグラフィー、透析、またはダイアフィルトレーションを使用して精製される、請求項64に記載の方法。
【請求項70】
前記カラムが脱塩カラムである、請求項69に記載の方法。
【請求項71】
前記脱塩カラムがPD−10カラムである、請求項70に記載の方法。
【請求項72】
前記再酸化剤が、5,5’−ジチオ−ビス−2−ニトロ安息香酸、4,4’−ジチオジピリジン、2,2’−ジチオジピリジン、テトラチオン酸ナトリウム、またはヨードソ安息香酸である、請求項64に記載の方法。
【請求項73】
1個以上のジスルフィド結合を有するタンパク質と、遊離チオールと反応性である薬物との結合体を調製する方法であって、該方法は:
該タンパク質を還元剤によって部分的に還元する工程;
該遊離チオールと反応性である薬物を該部分的に還元されたタンパク質に結合体化する工程
を包含する、方法。
【請求項74】
1個以上のジスルフィド結合を有するタンパク質と、遊離チオールと反応性である薬物との結合体を調製する方法であって、該方法は:
該タンパク質を還元剤によって完全に還元する工程;
該タンパク質を再酸化剤によって部分的に再酸化する工程;
該遊離チオールと反応性である薬物を抗体に結合体化する工程;
を包含する、方法。
【請求項75】
部分的に付加された抗体を形成する方法であって、該方法は:
抗体含有溶液を提供する工程、
該抗体溶液のpHを約pH7.5に調整して、キレート剤を添加する工程;
該抗体溶液を約37℃まで加熱する工程;
モル過剰のTCEPを該抗体溶液に添加し、約37℃にて十分な時間反応させて、該抗体の鎖間ジスルフィド基を部分的に還元して鎖間チオールを形成する工程;
該抗体溶液を約2〜8℃まで冷却することと;
わずかにモル過剰の薬物を該抗体溶液に添加することによって、該部分的に還元された抗体の鎖間チオールに該薬物を結合体化させ、そして該部分的に付加された抗体を形成するために十分な時間反応させる工程;ならびに
部分的に付加された抗体を精製する工程、
を包含する、方法。
【図1】

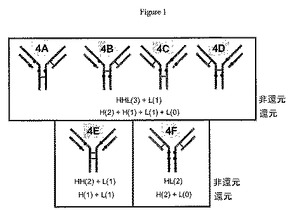
【図2】
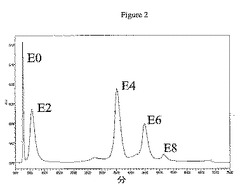
【図3】
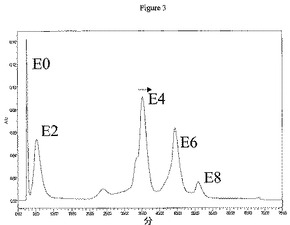
【図4】
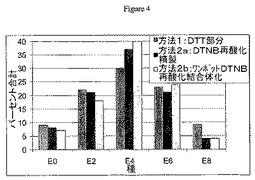
【図5】
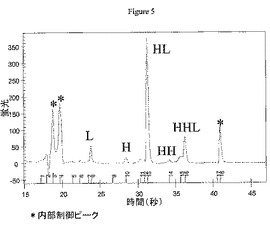
【図6】
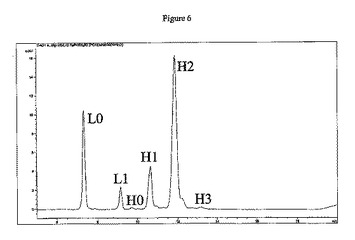
【図7】
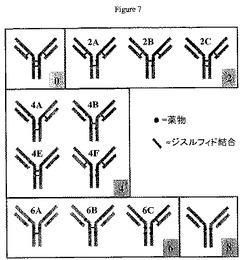
【図8】
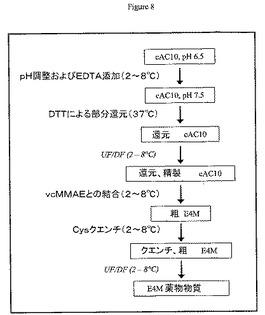
【図9】
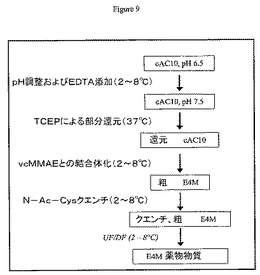
【図10】
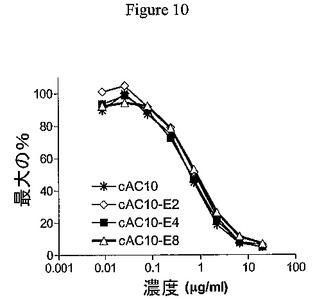
【図11】
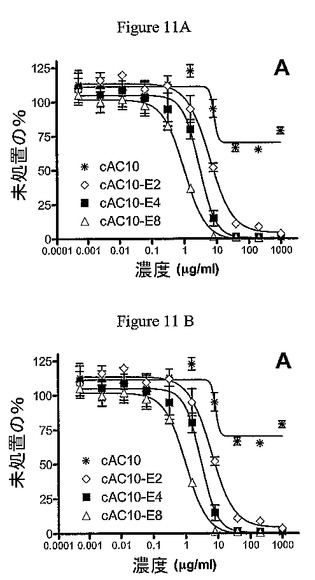
【図12】
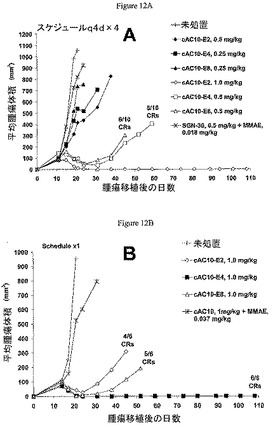
【図13】
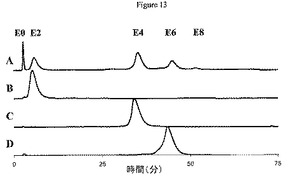
【図14】
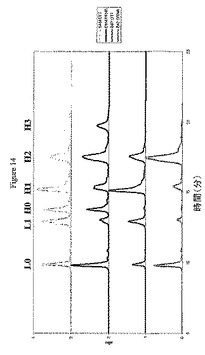
【図15】
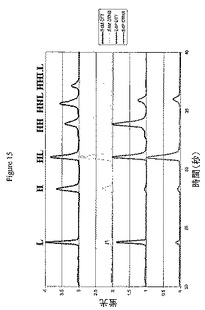
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【公表番号】特表2008−500961(P2008−500961A)
【公表日】平成20年1月17日(2008.1.17)
【国際特許分類】
【出願番号】特願2007−502057(P2007−502057)
【出願日】平成17年3月2日(2005.3.2)
【国際出願番号】PCT/US2005/007239
【国際公開番号】WO2005/084390
【国際公開日】平成17年9月15日(2005.9.15)
【出願人】(505314468)シアトル ジェネティックス, インコーポレイテッド (19)
【Fターム(参考)】
【公表日】平成20年1月17日(2008.1.17)
【国際特許分類】
【出願日】平成17年3月2日(2005.3.2)
【国際出願番号】PCT/US2005/007239
【国際公開番号】WO2005/084390
【国際公開日】平成17年9月15日(2005.9.15)
【出願人】(505314468)シアトル ジェネティックス, インコーポレイテッド (19)
【Fターム(参考)】
[ Back to top ]