
酵母における乳酸の生産方法及びその材料

第1の態様において、ゲノムに組み込まれた複数の外因性LDH遺伝子を有し、かつ、野生型のPDC遺伝子が完全なままである組み換え酵母を提供する。第2の態様において、ゲノムの野生型PDC遺伝子の遺伝子座に組み込まれた外因性LDH遺伝子を有し、かつ、PDC遺伝子が削除された組み換え酵母を提供する。本発明の組み換え酵母は、乳酸を生産するための発酵法に有用である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、酵母におけるL-乳酸の生産方法及びその材料に関する。
【背景技術】
【0002】
本出願は、2002年5月30日に出願された米国仮出願No.60/384,333に基づく優先権を主張する。
【0003】
本発明は、エネルギー省により付与された契約No.DE-FC-36-00GO10598に基づく米国政府の援助によりなされた。米国政府が、本発明について特定の権利を有する。
【0004】
乳酸は、化学処理及び合成、化粧品、医薬品、プラスチック、並びに食糧生産における利用を含む広い産業上の利用可能性を有する。乳酸を作るためのほとんどの工業規模の製法は、発酵法(fermentaion process)である。これらの発酵法においてさまざまな乳酸産生バクテリアが使用されている。
【0005】
最近の研究は、乳酸発酵法における組み換え酵母株の利用について行われている。組み換え酵母は、バクテリアの発酵に対して幾つかの有利性を提供できる可能性を秘めている。酵母の中には、より優れた高温耐性を示す株がある。これにより、より高温の発酵の可能性ができ、この高温発酵が、より速い発酵速度を生み出しうる。より優れた高温耐性は、発酵培地から汚染微生物を一掃することを容易とする。前記培地を、望ましい種が許容でき、望ましくない種が死滅する温度まで、単に加熱すればよいからである。ラクトバシリ(Lactobacilli)等の乳酸産生バクテリアは、効率よく生産するために、複雑な発酵培地を必要とする。発酵培地の複雑さは、原料費を増やし、乳酸を培地から分離することをより困難かつ高価にする。組み換え酵母の使用は、単純化された発酵培地を使用することによるコスト削減の可能性を提供する。
【0006】
加えて、酵母の中には、乳酸産生バクテリアの低いpH(reduced pH)条件により耐性を示す株がある。このことは、潜在的に、非常に重要な特性である。なぜなら、発酵培地のpHは、乳酸が生産されるとともに、自然に下がるからである。従来の製法では、前記培地を、例えば、水酸化カルシウムや炭酸カルシウム等の塩基を用いて、pH約5〜8にバッファーする必要がある。このことが、酸を中和して乳酸塩を形成させる。この乳酸塩は、引き続く工程において、望ましい酸の形で乳酸を回収するために、分離しなければならない。それゆえ、発酵培地のバッファリングの必要性は、結果として、追加される原材料(バッファー剤及び乳酸塩を分離するために通常使用される硫酸)、追加の処理工程(遊離酸の再生のため)、及び、廃棄物(ほとんどの場合、塩分離工程において発生する炭酸カルシウム)処理のための著しい追加費用をもたらす。これらの費用は、発酵が低いpHで実施できるならば、著しく削減できる。それゆえ、低いpHに耐えうる乳酸産生株の開発の成功が、非常に望ましい。
【0007】
Porro とその同僚は、外因性のLDH(乳酸脱水素酵素)遺伝子を S. cerevisiae、 K. lactic 、 T. delbrueckii 及び Z. bailii 由来の酵母細胞に組み込み、前記細胞本来のピルビン酸経路を破壊することにより、乳酸産生酵母を設計する試みをしてきた(非特許文献1〜3参照)。Porro は、乳酸を産生する組み換え酵母を作り出すことができた。しかし、その株は、商業上の製法として実施できるほどの十分な性能とは、到底いえるものではなかった。産業環境での使用にふさわしい株というには、その株は、高い乳酸収率(つまり、基質から乳酸への高い変換)及び高い生産性(つまり、基質から乳酸への迅速な代謝)をもたらすものでなければならない。その酵母は、高い力価の乳酸を含む培地に耐え得ることが好ましい。
【0008】
つい最近、Rajgarhia とその同僚が、Porro のものよりも高い収率及び生産性を示す組み換え酵母を作り出した(例えば、特許文献1〜3参照)。
【特許文献1】WO 00/71738
【特許文献2】WO 02/42471
【特許文献3】PCT/US02/16223
【非特許文献1】ポロら、「乳酸産生のために代謝的に改良した Saccharomyces cerevisiae 細胞」(Porro et al., "Development of metabolically engineered Saccharomyces cerevisiae cells for the production of lactic acid")、Biotechnol. Prog. 1995 May-Jun; 11(3): 294-8
【非特許文献2】ポロら、「乳酸の大規模生産のための改良酵母における代謝経路の置換」(Porro et al., "Replacement of a metabolic pathway for large-scale production of lactic acid from engineered yeasts")、App. Environ. Microbiol. 1999 Sep:65(9):4211-5
【非特許文献3】ビアンチら、「ピルビン酸利用を欠損し、異種由来LDH遺伝子で形質転換された Kluyveromyces lactis 株による効率的なホモ乳酸発酵」(Bianchi et al., "Efficient homolactic fermentation by Kluyveromyces lactis strains defective in pyruvate utilization and transformed with the heterologous LDH gene")、App. Environ. Microbiol. 2001 Dec; 67(12)5621-5.
【発明の開示】
【発明が解決しようとする課題】
【0009】
しかしながら、収率及び/又は生産性がさらにもっと改良された組み換え酵母を提供することが望ましい。特に、乳酸産生に有利な嫌気性及び/又は微好気性条件下で高い収率及び生産性で乳酸を産生する組み換え酵母株を提供することが望ましい。
【課題を解決するための手段】
【0010】
第1の態様として、本発明は、酵母細胞に外因性の乳酸脱水素酵素遺伝子を組み込む方法であって、前記組み込みに先んじて前記細胞は、そのゲノム中の遺伝子座に標的遺伝子を有し、下記(a)〜(d)の工程を含む方法である。(a)乳酸脱水素酵素(LDH)遺伝子、前記LDH遺伝子の上流(つまり、5’)側及び下流(つまり、3’)側の側面配列(flanking sequences)、並びに、少なくとも1つの選択マーカー遺伝子を含む組み換え核酸を用いて、前記細胞を形質転換し、前記LDH遺伝子を前記標的遺伝子の遺伝子座に隣接した前記細胞のゲノムに挿入し、(b)前記形質転換がされた細胞を選択物質存在下で培養することで、前記LDH遺伝子及び前記選択マーカー遺伝子を含む第1の形質転換細胞を得て、(c)前記第1の形質転換細胞を、非選択培地で培養することで、(d)前記第1の形質転換細胞から、前記LDH遺伝子を有するが前記選択マーカー遺伝子及び前記標的遺伝子が削除された第2の形質転換細胞を得る。酵母細胞において、前記標的遺伝子は、ピルビン酸デカルボキシラーゼ(PDC)、アルコール脱水素酵素(ADH)、オロチジン-5'-リン酸デカルボキシラーゼ(ura3)、及び3-イソプロピルリンゴ酸脱水素酵素(leu2)であることが、有利であり、好ましい。
【0011】
本発明の前記態様において、前記選択マーカー遺伝子及び標的遺伝子の欠失は、ある割合の前記第1形質転換細胞において自然発生的に起こる。その結果、挿入された前記LDH遺伝子は、前記標的遺伝子のプロモーター及びターミネーターと相同である機能的なプロモーター及びターミネーターと動作可能に連結する。
【0012】
第2の態様として、本発明は、酵母種細胞の形質転換用組み換え核酸であって、少なくとも1つの選択マーカー及びLDH遺伝子を含み、前記LDH遺伝子が、前記酵母種に対し外因性であって、前記LDH遺伝子の上流(5’)及び下流(3’)側面配列に連結しており、前記側面配列が、それぞれ、前記酵母種の野生型遺伝子の側面配列の上流及び下流に相同である、組み換え核酸である。
【0013】
第3の態様として、本発明は、酵母種の組み換え細胞であって、前記酵母種が、そのゲノム中の遺伝子座に野生型の標的遺伝子を含む組み換え細胞である。前記組み換え細胞は、そのゲノムの前記標的遺伝子の部位に組み込まれた外因性の乳酸脱水素酵素(LDH)遺伝子を有しており、そして、前記標的遺伝子は、欠失している。前記組み込まれたLDH遺伝子は、前記標的遺伝子のものと相同な機能的プロモーター及びターミネーターに動作可能に連結している。このような組み換え酵母細胞は、発酵法に使用した場合、高い生産性とともに、炭水化物基質から予想外に高い乳酸の収率を示す。前記組み換え細胞は、また、高い力価まで乳酸を産生できる。本発明は、あらゆる理論に限定されるものではないが、前記LDH遺伝子が、野生型遺伝子の遺伝子座に、本明細書で述べるように、前記野生型のプロモーター及びターミネーターを利用するように挿入されることで、細胞が、前記挿入されたLDH遺伝子の機能発現に、既存の遺伝子制御システムを利用できるのだと考えられる。このことは、前記細胞が嫌気性又は微好気性の発酵条件に直面する場合に、乳酸への代謝経路に強く有利に働くと考えられる。従って、第4の態様として、本発明は、炭水化物を乳酸に発酵する方法であって、結果として得られる前記細胞を、同細胞が発酵できる炭水化物を含む培地中、発酵条件下で培養することを含む方法である。
【0014】
第5の態様として、本発明は、 K. marxianus 種の細胞であって、複数の外因性の乳酸脱水素酵素が、そのゲノムに組み込まれ、それぞれが、機能的なプロモーター及びターミネーター配列の制御下にあり、さらに、前記 K. marxianus 細胞ゲノムが、機能的なピルビン酸デカルボキシラーゼ遺伝子を含む細胞である。驚くべきことに、低pHの発酵条件かであっても、この細胞により、驚くほど少ないエタノールの収率とともに、非常に優れた乳酸の生産性及び収率が得られた。前述のとおり、本発明は、あらゆる理論に限定されるものではないが、PDC経路を無傷のままにしておくことにより、細胞が、代謝過程のバランスを保つために自身の既存の代謝経路をうまく利用することができ、前記細胞の健康状態及び生命力が、総合的に向上するのだと考えられる。従って、第6の態様として、本発明は、炭水化物を乳酸へ発酵する方法であって、結果として得られる前記細胞を、同細胞が発酵できる炭水化物を含む培地中、発酵条件下で培養することを含む方法である。
【0015】
本発明の具体的な好ましい実施形態は、後に続く、特定の好ましい実施形態のより詳細な説明及び特許請求の範囲から明らかになる。
【発明を実施するための最良の形態】
【0016】
本発明の最初の2つの態様では、外因性のLDH遺伝子が、酵母細胞のゲノム中の標的野生型遺伝子の遺伝子座に組み込まれ、そして、前記標的野生型遺伝子は削除される。前記外因性LDH遺伝子は、前記標的遺伝子のプロモーター配列及びターミネーター配列と、それぞれ、相同なプロモーター配列及びターミネーター配列と動作可能に連結される。
【0017】
本発明において、遺伝子、プロモーター又はターミネーターは、(1)変更されていない細胞のゲノム中に見出されない場合、及び、(2)変更されていない細胞のゲノムに存在する遺伝物質に相同ではない場合、「外因性である(exogenous)」とみなす。ここで用いられる場合、遺伝子、ターミネーター又はプロモーターは、(その機能に影響しない自己から自己の変異を除き)酵母種の変更されていない細胞のゲノム中に見出される場合、その酵母種に対して「野生型(native)」である。
【0018】
遺伝子、プロモーター、ターミネーター又はその他の遺伝物質は、同一である場合、すなわち、ヌクレオチド配列において、その他の遺伝物質と、少なくとも、80%、85%、90%、91%、92%、93%、94%、95%、96%、97%、98%若しくは約99%の同一性を有している場合、又は、その機能を維持できるほど十分に類似している場合、その他の遺伝物質と「相同である(homologous)」とみなす。それゆえ、遺伝物質は、例えば、塩基対の点変異、欠失又は付加による相違があるとしても、これらの点変異、欠失又は付加が、前記遺伝物質の機能に影響を与えないならば、「相同である」とみなされる。側面配列の場合は、その配列が野生型遺伝子の側面配列と十分に類似しており、前記野生型遺伝子の側面配列との単一交差の発生に従事することができる場合、相同性が確立される。
【0019】
「同一性」の用語は、当該技術分野で知られるように、2以上のポリペプチド分子又は2以上の核酸分子の配列間で、これらの配列を比較して決定される関係を参照する。当該技術分野において、「同一性」は、また、場合に応じて、2以上の核酸若しくは2以上のアミノ酸配列の文字列間のマッチで決定される、核酸分子又はポリペプチド間の配列の類縁性の度合いを意味する。「同一性」は、特定の数学的モデル若しくはコンピュータープログラム(つまり、「アルゴリズム」)によるギャップアライメント(もしあれば)の2以上の配列のより小さなものの間の同一マッチのパーセントを評価する。
【0020】
「類似性」の用語は、当該技術分野において、関連する概念に関して使用されている。しかし、「同一性」とは対照的に、「類似性」は、同一マッチおよび同類置換マッチの両方を含む類縁性の指標を参照する。例えば、2つのポリペプチド配列が、10/20の同一アミノ酸を有している例において、残りのものが、すべて非同類置換である場合、同一%及び類似%は、ともに50%となる。同じ例において、5つの位置において同類置換がある場合、同一%は、50%のままであるが、類似%は、75%となる(15/20)。それゆえ、同類置換がある場合には、2つのポリペプチド間の類似%は、前記ポリペプチド間の同一%よりも高くなる。
【0021】
関連した核酸及びポリペプチドの同一性及び類似性は、公知の手段によって容易に計算できる。これらの方法としては、特に限定されないが、例えば、以下の文献に記載される。COMPUTATIONAL MOLECULAR BIOLOGY, (Lesk, A.M., ed.), 1988, Oxford University Press, New York; BIOCOMPUTING: INFORMATICS AND GENOME PROJECTS, (Smith, D.W., ed.), 1993, Academic Press, New York; COMPUTER ANALYSIS OF SEQUENCE DATA, Part 1, (Griffin, A.M., and Griffin, H.G., eds.), 1994, Humana Press, New Jersey; von Heinje, G., SEQUENCE ANALYSIS IN MOLECULAR BIOLOGY, 1987, Academic Press; SEQUENCE ANALYSIS PRIMER, (Gribskov, M. and Devereux, J., eds.), 1991, M. Stockton Press, New York; Carillo et al., 1988, SIAM J. Applied Math., 48:1073; and Durbin et al., 1998, BIOLOGICAL SEQUENCE ANALYSIS, Cambridge University Press.
同一性を決定するために好ましい方法は、テストされる配列間の最大のマッチを与えるように意図される。同一性を決定する方法は、公的に入手可能なコンピュータープログラムに記載されている。2つの配列間の同一性を決定するための好ましいコンピュータープログラム手段は、特に制限されないが、GCGプログラムパッケージを含む。前記GCGプログラムパッケージには、GAP(Devereux et al., 1984, Nucl. Acid. Res., 12:387; Genetics Computer Group, University of Wisconsin, Madison, WI)、BLASTP、BLASTN及びFASTA(Altschul et al., 1990, J. Mol. Biol., 215:403-410)が含まれる。BLASTXプログラムは、NCBI(National Center for Biotechnology Information)及びその他の情報源(BLAST Manual, Altschul et al. NCB/NLM/NIH Bethesda, MD 20894; Altschul et al., 1990,同上)から公的に入手得可能である。周知のSmith Watermanアルゴリズムも、同様に、同一性決定に使用される。
【0022】
2つのアミノ酸又はポリペプチド配列をアライメントするためのアライメントスキームには、2つの配列の短い領域のみのマッチングとなるものがある。そして、2つの全体配列間では有意な関係がないにも関わらず、この小さな領域においては、高い配列同一性を有する。従って、ある態様においては、選択されたアライメント手段(GAPプログラム)は、少なくとも50の連続した標的ポリペプチドのアミノ酸に及ぶアライメントをすることになる。またある態様においては、アライメントは、少なくとも60、70、80、90、100、110又は120の標的ポリペプチドのアミノ酸を含むことができる。ポリヌクレオチドがGAPを利用してアライメントされた場合、そのアライメントは、少なくとも100、150又は200ヌクレオチドに及び、それらは、連続することができる。
【0023】
例えば、コンピューターアルゴリズムGAP(Genetics Computer Group, University of Wisconsin, Madison, WI)を使用すると、配列同一%を決定したい2つのポリペプチドは、それぞれのアミノ酸の最適なマッチング(前記アルゴリズムにより決定される「マッチ範囲」)となるようにアライメントされる。ある態様においては、ギャップ開始ペナルティー(平均対角要素(diagonal)の3倍で計算される。ここで、前記「平均対角要素」とは、使用される比較行列の対角要素の平均値であり、前記「対角要素(diagonal)」とは、特定の比較行列により完全アミノ酸マッチにそれぞれ割り当てられるスコア若しくは数値である。)及びギャップ伸長ペナルティー(通常、ギャップ開始ペナルティーの1割)が、例えば、PAM250若しくはBLOSUM62等の比較行列とともに、前記アルゴリズムと併せて使用される。また、ある態様においては、標準比較行列(PAM250比較行列に関して、Dayhoff et al., 1978, Atlas of Protein Sequence and Structure, 5:345-352参照。BLOSUM62比較行列に関して、Henikoff et al., 1992, Proc. Natl. Acad. Sci USA, 89:10915-10919参照)もまた、前記アルゴリズムにより使用される。
【0024】
ある態様において、ポリペプチド配列の比較のためのパラメータは、以下のものを含む。
アルゴリズム:Needleman et al., 1970, J. Mol. Biol., 48:443-453
比較行列:BLOSUM62 Henikoff et al., 1992 同上
ギャップペナルティー:12
ギャップ長ペナルティー:4
類似性の閾値:0
GAPプログラムには、上記パラメータが有用である。ヌクレオチド配列に対しては、パラメータは、ギャップペナルティー50、及び、各ギャップにおける各シンボルごとにペナルティーが3となるギャップ長ペナルティー3を含む。また、ある態様においては、前述のパラメータは、GAPアルゴリズムを利用した(エンドギャップのペナルティー0を加えた)ポリペプチド比較のためのデフォルトパラメータである。
【0025】
「側面配列」は、遺伝子の上流(すなわち、5’)又は下流(すなわち、3’)の塩基対の配列である。側面配列は、前記遺伝子にすぐ隣接していてもよく、例えば、1〜1000塩基対、好ましくは、1〜100塩基対といった中間配列の塩基対により前記遺伝子と分離していてもよい。本発明の組み換え核酸に用いられる側面配列は、対応する標的遺伝子の側面配列と相同である。前記側面配列の長さは、対応する野生型遺伝子の側面配列との単一交差の発生に従事できるに足る長さである。有用な側面配列の長さは、約50〜約4000塩基対であって、好ましくは、約100〜約2000塩基対であって、とりわけ、約1200塩基対以下である。上流側面配列は、好ましくは、前記標的遺伝子のプロモーターを含み、下流側面配列は、好ましくは、前記標的遺伝子のターミネーター配列を含む。
【0026】
好ましい態様において、前記側面配列は、野生型の酵母のプロモーター及びターミネーター配列と、それぞれ、相同である前記プロモーター及びターミネーター配列を含む。最も好ましくは、組み換え核酸が、酵母ゲノムの染色体DNAにおける標的遺伝子座へ組み込まれる結果、そこにコードされる外因性LDH遺伝子が、前記側面配列を含む野生型遺伝子の発現制御配列の転写コントロール下におかれる。
【0027】
ここで用いられる場合、「プロモーター」の用語は、構造遺伝子の転写開始コドンの上流(すなわち、5’)(通常、約1〜1000bp、好ましくは、1〜500bp、とりわけ、1〜100bp以内)に位置する転写されない配列であって、構造遺伝子の転写開始をコントロールしたり、あるいはそうでなければ、前記遺伝子の転写をコントロールする配列を参照する。
【0028】
同様に、「ターミネーター」の用語は、構造遺伝子の転写ストップコドンの下流(すなわち、3’)(通常、約1〜1000bp、好ましくは、1〜500bp、とりわけ、1〜100bp以内)に位置する転写されない配列であって、構造遺伝子の転写終結をコントロールしたり、あるいはそうでなければ、前記遺伝子の転写をコントロールする配列を参照する。
【0029】
プロモーター又はターミネーターが、必要に応じて、酵母ゲノムに組み込まれた後、構造遺伝子の転写をコントロールするよう機能する場合、前記構造遺伝子(例えば、LDH遺伝子若しくは選択マーカー遺伝子)は、前記プロモーター又はターミネーターと「動作可能に連結」する。
【0030】
「形質転換」の用語は、ここで用いる場合、細胞における遺伝的特徴の変化を参照する。そして、細胞は、新たな核酸を含むように変更された場合、形質転換される。よって、細胞が、野生型の状態から遺伝的に変更された場合、細胞は、形質転換される。例えば、以下のトランスフェクションにおいて、形質転換DNAは、細胞の染色体に物理的に組み込まれることによって、細胞ゲノムDNAと組み換えを起こすことが好ましい。あるいは、少なくとも一時的に(すなわち、細胞形質転換から48〜96時間内に)、前記核酸は、複製されないエピソーム因子として一時的に維持されてもよく、又は、プラスミドとして独立に複製されてもよい。前記DNAが、染色体に組み込まれ、細胞の分裂とともに複製される場合、細胞は、安定して形質転換されたと見なされる。
【0031】
「トランスフェクション」の用語は、細胞による外部又は外因性のDNAの取込みを参照する。外因性DNAが、細胞膜の内側に導入された場合、前記細胞は、「トランスフェクション」される。多くのトランスフェクション技術が当該技術分野において周知である。例えば、Graham et al., 1973, Virology 52:456; Sambrook et al., 2001, MOLECULAR CLONING, A LABORATORY MANUAL, Cold Spring Harbor Laboratories; Davis et al., 1986, BASIC METHODS IN MOLECULAR BIOLOGY, Elsevier; Chu et al., 1981, Gene 13:197.等が、参照できる。これらの技術は、1つ以上の外因性DNA種を、適した宿主細胞に導入する場合に利用できる。
【0032】
本発明の第1の態様によれば、組み換え核酸は、側面配列に連結した外因性LDH遺伝子及び少なくとも1つの選択マーカーを含んで提供される。「組み換え核酸」の用語は、ここでは、宿主細胞にタンパク質をコードする情報を移動するために使用されるあらゆる分子(例えば、核酸断片、プラスミド又はウイルス等)を参照するために使用される。適切な側面配列は、以下のようにして得ることができる。まず、意図する組み込み部位(例えば、酵母細胞ゲノム中の標的遺伝子等)を同定し、その部位に隣接する配列を(任意の都合のよい手段、特に限定されないが、例えば、化学合成、組み換え遺伝学的技術若しくはインビトロ増幅等を含む手段により)得て、そして、これらの配列を、組み換え核酸中の望ましい位置に(例えば、5’及び3’の側面配列を、それぞれ、LDHをコードする遺伝子に共有結合させることで)取り込ませる。前記酵母細胞は、その後、前記組み換え核酸で、任意の適切な形質転換技術により、形質転換される。その結果、LDH遺伝子を含む組み換え核酸の少なくとも1つの断片が前記酵母細胞のゲノムに組み込まれた細胞が得られる。
【0033】
前記組み換え核酸は、1つ以上の選択マーカー遺伝子を含み、前記選択マーカー遺伝子は、それら自身のプロモーター及びターミネーター遺伝子による転写コントロール下にあることが、より好ましい。マーカー遺伝子のプロモーター及びターミネーターの配列は、前記標的遺伝子のプロモーター及びターミネーター配列と相同でないことが好ましい。前記選択マーカー遺伝子並びにそれぞれのプロモーター及びターミネーターは、上流側面配列-LDH遺伝子-下流側面配列の配列を中断しないことが好ましい。前記選択マーカー並びにそれぞれのプロモーター及びターミネーターは、ベクター上で、LDHプロモーター配列(もしあれば、さらなる側面配列)の上流(5’)に位置することが好ましい。
【0034】
本発明の組み換え核酸及び細胞の製造、及び、本発明にかかる方法の実践に有用なLDH遺伝子を有する酵母種としては、 Lactobacillus helveticus, Pediococcus acidolactici, Lactobacillus casei, Kluyveromyces thermotolerans, Torulaspora delbrueckii, Schizosaccharomyces pombii, Rhizopus oryzae 及び B. megaterium を含むことが好ましい。特に、これらの株の中でも、単離でき、本発明の組み換え核酸の製造に使用できる適したL-乳酸脱水素酵素を有する株が好ましい。2つの好ましいL-乳酸脱水素酵素遺伝子は、 L. helveticus 及び B. megaterium のL-乳酸脱水素酵素である。
【0035】
典型的な選択マーカー遺伝子は、(a)抗生物質又はその他の毒に対して耐性を付与するタンパク質をコードする遺伝子、例えば、宿主細胞に対する、ゼオシン( Streptoalloteichus hindustanus ble ブレオマイシン耐性遺伝子)、G418(Tn903由来のカナマイシン耐性遺伝子)、ハイグロマイシン( E. coli 由来のアミノグルコシド抗生物質耐性遺伝子)、アンピシリン、テトラサイクリン、若しくはカナマイシン等、(b)細胞の栄養要求性欠乏症を補完するタンパク質をコードする遺伝子、例えば、アミノ酸ロイシン欠乏症、又は(c)単純培地から得ることができない必須栄養素を補給するタンパク質をコードする遺伝子、例えば、ura3等である。好ましく選択できるマーカーは、特に制限されないが、例えば、ゼオシン耐性遺伝子、G418耐性遺伝子、及びハイグロマイシン耐性遺伝子である。
【0036】
また、本発明の組み換え核酸は、酵母細胞のゲノムに挿入されるために、前記分子が切断され、前記LDH遺伝子、そのプロモーター及び側面配列、マーカー遺伝子並びに付随するプロモーター及びターミネーター等を含む直線状の断片を形成できる制限酵素認識部位を1つ以上有してもよい。
【0037】
さらに、本発明の組み換え核酸は、バックボーン部を含んでもよい。前記バックボーン部は、オリジン、複製(酵母若しくはバクテリアで動作可能であり、有効量の核酸を製造できる)及びその他の有用な特徴部を有利に含むことができ、また、市販の酵母ベクターから都合よく得ることができる。
【0038】
酵母細胞は、本発明の組み換え核酸で形質転換される。細胞を形質転換する方法は、当該技術分野で周知であり、特に制限されないが、例えば、エレクトロポレーション、塩化カルシウム法、又は、酢酸リチウム法等を含む。形質転換に使用する本発明の組み換え核酸は、使用の前に特定の制限酵素で処理してもよく、処理しなくてもよい。LDH遺伝子の側面配列は、標的遺伝子の側面配列と高い相同性を示すので、上流側面配列/LDH遺伝子/下流側面配列の断片の挿入は、標的遺伝子の遺伝子座で起こる傾向がある。
【0039】
標的遺伝子は、LDH遺伝子と入れ替わることを希望する任意の遺伝子である。好ましい標的遺伝子は、ピルビン酸デカルボキシラーゼ遺伝子である。なぜなら、この遺伝子を取り替えることで、エタノールを産生する競合経路を破壊することができるからである。加えて、ピルビン酸遺伝子は、酵母種において活性化する傾向にある。したがって、PCDプロモーター及びターミネーターのコントロール下のゲノム中へのLDH遺伝子の挿入は、LDHをよく発現する変異体を産生する傾向がある。さらなる好ましい標的遺伝子は、ADH、Leu2及びUra3である。
【0040】
酵母細胞を本発明の組み換え核酸で形質転換し、選択マーカーに対する選択培地で生育することにより組み換え細胞を選択し、さらに、選択した形質転換株を非選択条件で生育させると、その結果、外因性LDH遺伝子を有し、前記酵母染色体の標的遺伝子の遺伝子座に組み込まれた組み換え核酸を含む組み換え酵母細胞が得られる。本発明の方法に従い得られた場合、これらの細胞は、前記標的遺伝子が欠失し、組み込まれたLDH遺伝子は、前記標的遺伝子の遺伝子座に動作可能に連結し、前記標的遺伝子の発現コントロール配列(例えば、プロモーター及びターミネーター配列)の転写コントロール下に置かれるように、挿入される。標的遺伝子が、PDC遺伝子の場合、PDC欠失が起きた細胞は、嫌気性条件下では、生育がよくない。よって、同定され、選択された細胞のコロニーは、これらを嫌気性条件下にさらすことで、選択できる。生育しないコロニーが、PDC欠失が起きたものであると同定できる。同様に、任意のその他の標的遺伝子を標的とした組み込みは、各標的遺伝子の欠失に伴う表現型により同定することができる。
【0041】
この結果をもたらす遺伝学的な出来事の説明図を、図17A及びBに示す。図17Bに示すとおり、選択マーカー遺伝子は、自然発生的に、標的遺伝子の欠失とともに、削除できる。選択マーカー遺伝子が欠失した形質転換株の選択をすることが好ましい。なぜなら、遺伝子組み換え操作された酵母細胞は、例えば、薬剤耐性の向上等の特定の性質を有し、望ましくない環境リスクを生じるおそれがあるからである。
【0042】
結果として得られる酵母細胞は、標的遺伝子を失い、そして、酵母細胞のゲノムの前記標的遺伝子の遺伝子座に組み込まれた外因性LDH遺伝子を有する。前記LDH遺伝子は、前記標的遺伝子のプロモーター及びターミネーター配列と相同なプロモーター配列及びターミネーター配列の転写コントロール下に置かれる。
【0043】
図17Bに示すとおり、前記LDHプロモーター及びターミネーター配列は、細胞の形質転換に使用された組み換え核酸に含まれる側面配列に存在したものであってもよく、又は、細胞ゲノムの組み込み部位にもともと存在したものであってもよい。同様に、標的遺伝子のターミネーターも、組み込まれたベクターに存在したターミネーター配列が欠失することにより、維持されることも可能である。
【0044】
本発明の第1及び第2の態様における形質転換に適した酵母細胞は、 Candida 、Saccharomyces 、Kluyveromyces 、 Pichia 及び Hansenula 属由来のものを含む。ピルビン酸を蓄積しない、すなわち、天然に、ピルビン酸をエタノール又はその他の代謝産物へ代謝する酵母細胞が好ましい。 Candida 及び Kluyveromyces 属由来の細胞が特に好ましく、とりわけ好ましくは、 C. sonorensis 及び K. marxianus の細胞である。
【0045】
本発明の第5の態様における細胞は、 K. marxianus 種の組み換え細胞である。これらの細胞は、一般的に、いくらか異なる形質転換方法により調製される。ただし、標的遺伝子が、PDC遺伝子以外であれば、上述の手段が適している。この場合、組み換え核酸は、酵母細胞の野生型のPDC遺伝子の遺伝子座を標的として挿入するようにはデザインされない。それゆえ、細胞の形質転換に使用されるベクターは、一般的に、前記酵母細胞の野生型PDC遺伝子の側面配列と高い相同性を示す側面配列を含まない。
【0046】
第5の態様における細胞は、一般的に、1回につきLDH遺伝子を1コピー導入する形質転換を、2回以上することにより調製される。しかしながら、複数のLDH遺伝子を含む組み換え核酸をコンストラクトし、単一ステップで複数のLDH遺伝子を挿入することもできる。そして、細胞の形質転換に使用するベクターは、それぞれプロモーター及びターミネーターに動作可能に連結した1つ以上のLDH遺伝子を含む。既に述べたとおり、組み換え核酸は、また、様々なマーカー遺伝子を有してよい。これらの遺伝子は、それぞれ、酵母細胞の野生型のPDCプロモーター及びターミネーター配列ではないプロモーター及びターミネーター配列のコントロール下にあることが好ましい。
【0047】
適したLDH遺伝子としては、本発明の最初の3態様に関して上述したものを含む。 L. helveticus L-LDH及び B. megaterium L-LDH遺伝子が好ましい。細胞は、複数のLDH遺伝子、すなわち、少なくとも2以上の前記遺伝子、好ましくは、約2〜10の前記遺伝子、さらに好ましくは、約2〜5の前記遺伝子で形質転換される。挿入されるLDH遺伝子は、全て同じであってもよく、又は、2以上の異なる型のLDH遺伝子(すなわち、2以上の種から得た遺伝子)を含んでもよい。2コピー以上の L. helveticus L-LDH遺伝子を含む組み換え酵母細胞、2コピー以上の B. megaterium L-LDH遺伝子を含む組み換え酵母細胞、並びに、少なくとも1コピー以上の L. helveticus 及び B. megaterium L-LDH遺伝子を含む組み換え酵母細胞が好ましい。
【0048】
第5の態様における細胞の形成の際に、LDHに使用する適したプロモーターとしては、酵母遺伝子であるホスホグリセリン酸キナーゼ(PGK)、グリセルアルデヒド3リン酸デヒドロゲナーゼ(TDH)、ピルビン酸デカルボキシラーゼ(PDC)(K. marxianus 以外の種由来のもの)、トリオースリン酸イソメラーゼ(TPI)、転写エンハンサー因子(TEF)、プリン-シトシンパーミアーゼ(PCPL3)及びアルコールデヒドロゲナーゼ(ADH)のプロモーターを含む。本発明の好ましいプロモーターとしては、 S. cerevisiae PGKプロモーター及び S. cerevisiae PDC1プロモーターをふくむ。
【0049】
適したターミネーターとしては 、S. cerevisiae 又はその他の種由来のGAL10及びCYC-1ターミネーターを含む。
【0050】
好ましい選択マーカーは、本発明の最初の3態様に関して記載したとおりである。
【0051】
形質転換を複数のステップで行う場合、細胞は、まず、少なくとも1つのLDH遺伝子を含む第1のベクターで形質転換される。首尾よく形質転換された細胞が、その後、通常、選択マーカーの存在による特性を利用して、選択される。首尾よく得られた形質転換株は、その後、最終的に、望ましい数及び型の外因性LDH遺伝子を有する細胞となるまで、1以上のさらなる形質転換がされる。
【0052】
本発明の組み換え酵母細胞は、発酵法において、炭水化物から乳酸を生産することに有用である。発酵は、任意の使用しやすい発酵法を用いることができる。一般的には、細胞は、細胞がピルビン酸に代謝できる炭水化物を含む発酵培地とともに使用され、発酵が起こる条件下に置かれる。前記発酵培地は、また、前記細胞の生命力を高める栄養素(例えば、窒素源、リン源、硫黄源、微量元素源等)を含む。
【0053】
使用できる特定の炭水化物は、特定の宿主、及び、その宿主が任意の特定の炭水化物をピルビン酸に代謝できるように遺伝子組み換えされたか否かに依存する。グルコース及びフルクトース等のヘキソースの糖、マルトース、イソマルトース、マルトトリオース、スターチ及びスクロース等のグルコースのオリゴマー、マルトデキストリン並びにキシロース(ペントースの糖)が好ましい。また、より好ましくない炭水化物としては、ガラクトース、マンノース及びアラビノースが挙げられる。
【0054】
発酵の間の温度は、例えば、ほぼ室温以上であって、好ましくは、約30℃以上であり、より好ましくは、約35℃〜約55℃であり、より好ましくは、約50℃以下であって、さらにより好ましくは、約45℃以下である。最高温度は、幾分は、その特定の宿主細胞に依存する。宿主細胞が、例えば、 K. marxianus である場合、(本発明の任意の態様の)組み換え細胞は、比較的高い温度を許容できる(例えば、約40℃〜50℃、とりわけ45℃以下)。もう1つの好ましい宿主である C. sonorensis は、約40℃の温度まで許容できる。この温度範囲は、生産性が著しく低下することなく、より高い温度で発酵できる(それにより、冷却コストを削減できる)可能性を提供する。より高い温度に対する優れた耐性により提供されるその他の利点は、望ましくない微生物に発酵が汚染された場合、多くの場合、発酵培地を40℃以上、とりわけ45℃以上に加熱することにより、本発明の組み換え細胞に大きな害を及ぼすことなく、前記望ましくない微生物を選択的に死滅させることができることである。
【0055】
発酵の間、発酵培地中の細胞濃度は、一般的には、発酵培地1リットルあたりの細胞の乾燥重量で、約1〜150gの範囲であって、好ましくは、約3〜10gの範囲であり、より好ましくは、3〜6gの範囲である。
【0056】
発酵の産生期間は、厳密に嫌気性よりはむしろ微好気性で実施したほうが好ましい場合がある。最適な曝気条件は、各微生物について、特有の酸素摂取速度(OUR)を測定し、これらの速度と、収率、基質消費速度及び望ましい発酵生成物が産生される速度とを関連付けることにより、確立できる。多くの場合、収率及び速度は、特定のOURの範囲内で最適化できる。PDCが破壊された酵母では、最適なOUR値は、約0.8〜約3.5mmolO2/細胞乾燥重量/hrの範囲内である。OURは、発酵中の細胞により酸素が消費される速度を参照し、例えば、mmolO2/細胞乾燥重量/hrのように、単位時間、細胞の乾燥重量あたりの酸素の単位(mmol又はg)で表される。酸素の消費は、発酵中に送り込んだ酸素及び発酵から回収された酸素を測定することで、便利よく決定できる。OURの測定は、特定の生物に最適な範囲にOURを維持するために、発酵の産生期間の曝気条件(とりわけ、ガス導入速度、アジテーション、曝気ガス中の酸素の割合等)をコントロールするための基礎として使用できる。培養液に溶解した酸素濃度は、同時に、飽和の1%未満、とりわけ、10mmolO2/L未満に維持される。特に好ましい製法においては、発酵の増殖期の培養を、培養液に溶解した酸素濃度を、飽和の1%未満、とりわけ、10mmolO2/L未満に減らして、産生期の開始(すなわち、増殖期の好気性条件から産生期の微好気性条件への切り替え)前の一定時間、例えば、約15分〜90分行う。
【0057】
乳酸が産生されると、発酵培地のpHは、形成される酸の全部又は一部を中和するために塩基が添加されない限り、下がる傾向にある。発酵法の一態様において、中和剤、例えば、炭酸カルシウム、水酸化カルシウム、炭酸ナトリウム、水酸化ナトリウム、アンモニア、水酸化アンモニウム等が発酵培養液に添加され、pHが望ましい範囲、一般的には、約5.0〜約8.0、とりわけ、約5.5〜約7.5に維持される。そのような塩が添加された場合、対応する乳酸塩が形成される。それゆえ、乳酸の回収は、遊離酸の再生を含む。これは、一般的に、細胞を取り除き、例えば、硫酸等の強酸で発酵培地を酸性化することにより行われる。塩副産物が形成されるが(カルシウム塩が中和剤で、硫酸が酸性化剤の場合、石膏)、これらは、乳酸から分離される。その後、乳酸は、例えば液-液抽出、蒸留、吸着等の技術により回収される。これらは、以下の文献に開示される。T.B. Vickroy, Vol. 3, Chapter 38 of Comprehensive Biotechnology, (ed. M. Moo-Young), Pergamon, Oxford, 1985; R. Datta, et al., FEMS Microbiol. Rev., 1995; 16:221-231; U.S. Patent Nos. 4,275,234, 4,771,001, 5,132,456, 5,420,304, 5,510,526, 5,641,406, and 5,831,122, 及び国際出願番号 WO 93/00440。
【0058】
あるいは、発酵のpHは、乳酸が細胞により産生されるにつれて下がってもよい。この場合、発酵培養液のpHは、乳酸の産生により、約1.5〜約5.0の範囲、好ましくは、約1.5〜約4.2、より好ましくは、約1.5〜約3.86(乳酸のpKaである)、とりわけ、約2.0以上3.86未満の範囲になってもよい。このような方法で発酵を行うことは、許容できる生産性と収率を達成できるのであれば、いくつかの利点がある。中和剤のコストが、削減又は排除される。発酵pHが(発酵終了時に)乳酸のpKaを下回れば、乳酸は、主に酸の形態で存在することとなる。これは、酸性化ステップを排除することができ、余計な製法ステップ、酸性化コスト、及び塩副産物の廃棄コストを節約できる。よって、とりわけ好ましい製法は、発酵培養液のpHが、3.86を下回るまで発酵を続けることを含む。乳酸は、例えば、WO 99/19290に開示される手段を用いて結果として得られる発酵培養液から分離できる。
【0059】
低pH環境に抵抗できる細胞の能力は、望ましくない微生物による汚染を排除するもう一つのメカニズムを提供する。本発明の細胞を含む培養液を、十分な時間、低pH条件、例えば、約1.5〜4.2、好ましくは、約2.0〜3.86に置くと、酸耐性ではないあらゆる汚染微生物を死滅させることとなる。
【0060】
商業的に有用なため、本発明の組み換え酵母は、いくつかの特徴を有する。前記酵母は、炭水化物のかなりの部分を乳酸に変換する(つまり、生成物の高収率を実現する)。酵母細胞は、高い比生産性(specific productivity)を示す。つまり、単位時間、細胞重量あたりの高い乳酸の生産量を実現する。酵母細胞は、約5.0を下回るpH、好ましくは、約1.5〜4.2、とりわけ、2.0〜3.86の発酵に耐え、これらの条件下でも優れた収率と生産性を発揮することが好ましい。前記細胞は、また、高い乳酸濃度、pH5.0〜8.0、好ましくは、pH1.5〜5.0、より好ましくは、1.5〜4.2、とりわけ、2.0〜3.86に耐え得ることが好ましい。この最後の特性により、開始時の炭水化物濃度が高い場合の発酵法が可能となる。
【0061】
概して、本発明の組み換え細胞を利用した発酵法は。次の特色のいくつか又は全てを提供することが望ましい。
A.乳酸の収率が、炭水化物のグラムあたり、少なくとも30、好ましくは、少なくとも40、より好ましくは、少なくとも60、さらにより好ましくは、少なくとも75g。理論的な望ましい収率は、100%であるが、実際は、約95%の収率が限度である。
B.乳酸の比生産性が、時間、細胞グラムあたり、少なくとも0.1、好ましくは、少なくとも0.3、より好ましくは、少なくとも約0.4、とりわけ、少なくとも0.5g。比生産性は、高ければ高いほど望ましい。
C.力価(つまり、乳酸の最大濃度)が、発酵培地1リッターあたり、少なくとも15g、好ましくは、少なくとも20g、より好ましくは、少なくとも40g、さらにより好ましくは、少なくとも80g、150g以下、好ましくは120g以下。培養培地の温度は、容易に達成できる力価の最大範囲にいくらかの影響を及ぼす。なぜなら、高濃度の乳酸溶液(つまり、約150g/L以上)は、約35℃未満の温度では、非常に粘性を帯びやすいか又はゲル化しやすいからである。高い発酵温度、例えば、約35〜50℃を用いれば、ゲル化や過度の粘性の集積をすることなく、高い力価が可能となる。
【0062】
本発明の第3の態様の細胞は、グルコースの中性発酵(pH5.0〜8.0)に使用すると、収率:85〜95%、比生産性:0.5〜2g/g/hr、力価:80〜120g/Lを提供することがわかっている。pHが約2.8〜3.0に低下する低pH発酵では、本発明の第3の態様の細胞は、収率:75〜81%、比生産性:0.1〜0.4g/g/hr、力価:14〜40g/Lを提供することがわかっている。全ての場合において、これらの結果は、発酵条件の最適化をすることなく得ることができた。
【0063】
本発明の第5の態様の細胞(複数コピーの外因性LDH遺伝子を含み、完全な野生型PDC遺伝子を含む)は、グルコースの中性発酵(pH5.0〜8.0)に使用すると、収率:40%以上、比生産性:0.4〜0.9g/g/hr、力価:40〜75g/Lを提供することがわかっている。グルコースの低pH発酵(最終pH2.8〜3.0)では、前記第5の態様の細胞は、収率:30%以上、比生産性:0.3〜0.5g/g/hr、力価:20〜35g/Lを提供する。このように、前記第5の態様の細胞は、低pH条件下でよく発酵する能力を保持した。前述のとおり、これらの結果は、発酵条件の最適化をすることなく得ることができた。
【0064】
さらに、本発明の発酵法は、高い容積生産性を達成することが好ましい。「容積生産性」は、単位時間、発酵培地の単位容積あたりの生産される生成物量として表され、一般的に、生成物のグラム/培地のリットル/時間(hr)で表される。望ましい容積生産性としては、少なくとも1.5g/L/hrであって、好ましくは、2.0g/L/hrであり、より好ましくは、2.5g/L/hrである。発酵培地リットルあたり3〜6g以下の細胞という好ましい細胞濃度において、最大生産性は、約5.0g/L/hr、より一般的には、約4.0g/L/hr以下となる傾向がある。培地pH、温度、若しくは両方ともが、前節に記載した範囲内である場合に、これらの容積生産性が達成できるように発酵を実施することが、非常に好ましい。
【0065】
本発明により生産された乳酸は、2つの乳酸分子の環状無水物であるラクチドの製造に有用である。乳酸の立体異性体に応じて、ラクチドは、D-ラクチド(2つのD-乳酸分子から作られる)、L-ラクチド(2つのL-乳酸分子から作られる)、または、D-L-ラクチド(各1分子のD-乳酸分子及びL-乳酸分子から作られる)であってもよい。乳酸からラクチドを製造する便利な方法は、USP 5,142,023 to Gruber et al.に開示される重合/解重合法による方法である。
【0066】
次に、ラクチドは、ポリ乳酸ポリマー(PLA)及びコポリマーの製造のためのモノマーとして特に有用である。これらのポリマーを調製する方法は、同様に、USP 5,142,023 to Gruber et al.に開示される。好ましいPLA製品は、USP 5,142,023 to Gruber et al.に開示されるように、安定な溶融ポリマー(melt-stable polymer)である。PLAは、半結晶若しくはアモルファスであってもよい。
【0067】
以下の実施例は、本発明の特定の実施態様を説明するのに役立つものであって、本発明の範囲や精神を限定するものでない。
【実施例1】
【0068】
(実施例1A)
S. cerevisiae ScPGK1プロモーター型発現ベクターpNC2のコンストラクションと、 S. cerevisiae PDC1プロモーター型発現ベクターpNC4のコンストラクション
発現ベクターpNC2(図1)は、 S. cerevisiae ScPGK1とpGEM5Z(+)バックボーンベクター(Promega、Wisconsin)上の S. cerevisiae GAL10ターミネーターとを組み合して作られた。 S. cerevisiae ScPGK1と S. cerevisiae GAL10ターミネーターは、前記酵母プロモーターとターミネーターの間に特定の遺伝子を挿入して発現させるための、XbaI、EcoRI及びBamHIの制限酵素認識部位を有するポリリンカー領域により分離されている。
【0069】
使用した S. cerevisiae ScPGK1プロモーターは、以下の配列(配列番号1)を有する。
5'-GCGGCCGCGG ATCGCTCTTC CGCTATCGAT TAATTTTTTT TTCTTTCCTC TTTTTATTAA CCTTAATTTT TATTTTAGAT TCCTGACTTC AACTCAAGAC GCACAGATAT TATAACATCT GCACAATAGG CATTTGCAAG AATTACTCGT GAGTAAGGAA AGAGTGAGGA ACTATCGCAT ACCTGCATTT AAAGATGCCG ATTTGGGCGC GAATCCTTTA TTTTGGCTTC ACCCTCATAC TATTATCAGG GCCAGAAAAA GGAAGTGTTT CCCTCCTTCT TGAATTGATG TTACCCTCAT AAAGCACGTG GCCTCTTATC GAGAAAGAAA TTACCGTCGC TCGTGATTTG TTTGCAAAAA GAACAAAACT GAAAAAACCC AGACACGCTC GACTTCCTGT CTTCCTATTG ATTGCAGCTT CCAATTTCGT CACACAACAA GGTCCTAGCG ACGGCTCACA GGTTTTGTAA CAAGCAATCG AAGGTTCTGG AATGGCGGGA AAGGGTTTAG TACCACATGC TATGATGCCC ACTGTGATCT CCAGAGCAAA GTTCGTTCGA TCGTACTGTT ACTCTCTCTC TTTCAAACAG AATTGTCCGA ATCGTGTGAC AACAACAGCC TGTTCTCACA CACTCTTTTC TTCTAACCAA GGGGGTGGTT TAGTTTAGTA GAACCTCGTG AAACTTACAT TTACATATAT ATAAACTTGC ATAAATTGGT CAATGCAAGA AATACATATT TGGTCTTTTC TAATTCGTAG TTTTTCAAGT TCTTAGATGC TTTCTTTTTC TCTTTTTTAC AGATCATCAA GGAAGTAATT ATCTACTTTT TACAACAAAT CTAGAATT-3'
この配列は、特許プラスミドであるpBFY004の制限酵素処理断片として得ることができた。あるいは、鋳型として S. cerevisiae 染色体DNAを使用し、配列番号1に基づき設計したプライマーを使用したPCR増幅により得ることができる。
【0070】
使用した S. cerevisiae GAL10ターミネーターは、以下の配列(配列番号2)を有する。
5'-GTAGATACAT TGATGCTATC AATCCAGAGA ACTGGAAAGA TTGTGTAGCC TTGAAAAACG GTGAAACTTA CGGGTCCAAG ATTGTCTACA GATTTTCCTG ATTTGCCAGC TTACTATCCT TCTTGAAAAT ATGCACTCTA TATCTTTTAG TTCTTAATTG CAACACATAG ATTTGCTGTA TAACGAATTT TATGCTATTT TTTAAATTTG GAGTTCAGTG ATAAAAGTGT CACAGCGAAT TTCCTCACAT GTAGGGACCG AATTGTTTAC AAGTTCTCTG TACCACCATG GAGACATCAA AAATTGAAAA TCTATGGAAA GATATGGACG GTAGCAACAA GAATATAGCA CGAGCCGCGG ATTTATTTCG TTACGC-3'
この配列は、特許プラスミドであるpBFY004の制限酵素処理断片として得ることができた。あるいは、鋳型としてS. cerevisiae 染色体DNAを使用し、配列番号2に基づき設計したプライマーを使用したPCR増幅により得ることができる。
【0071】
S. cerevisiae PDC1プロモーター及びScGAL10ターミネーター型発現カセットを含むベクターpNC4をコンストラクトし、一般的な発現ベクターとして使用した。pNC4ベクターを図2に示す。
【0072】
pNC4のベクターバックボーンは、pGEM5Z(Promega社、Madison、WI)である。 S. cerevisiae PDC1プロモーターは、プライマーとして、PSPDCS1(5'-CCA TCG ATA ACA AGC TCA TGC AAA GAG-3':配列番号3)及びPSPDCAS2(5'-GCT CTA GAT TTG ACT GTG TTA TTT TGCG-3':配列番号4)を使用し、鋳型として、 S. cerevisiae GY5098株(ATCC 4005098)の染色体DNAを使用し、PCRにより増幅した。温度サイクルは、PfuTurboDNAポリメラーゼ(Stratagene)を使用して、94℃-1分、56℃-1分、72℃-1分を30サイクル行い、最後に72℃-7分間のインキュベーションを続けた。
【0073】
S. cerevisiae GAL10ターミネーターは、上述のようにして得られた。図2A(配列番号37)及び図2b(配列番号38)は、それぞれ、ScPGK1プロモーター、ScGAL10ターミネーター及びマルチクローニングサイトを含む断片、並びに、ScPDC1プロモーター、ScGAL10ターミネーター及びマルチクローニングサイトを含む断片を示す。
【0074】
(実施例1B)
S. cerevisiae ScPGK1プロモーター及びScGAL10ターミネーターに動作可能に連結したG418耐性遺伝子を有するpVR22ベクターのコンストラクション
G418耐性マーカー(バクテリアneoR遺伝子)は、クローニングされ、S. cerevisiae ScPGK1プロモーターの転写コントロール下に置かれた。このようにコンストラクションされたベクターをpVR22とした(図3)。G418耐性遺伝子は、Pfuポリメラーゼ(Stratagene、Madison、WI)を使用し、プライマーとして、5'-GCT CTA GAT GAG CCA TAT TCA ACG GGA AAC(5'G断片:配列番号5)及び5'-ATG GAT CCT TAG AAA AAC TCA TCG AGC ATC(3'G断片:配列番号6)を使用し、鋳型として、プラスミドpPIC9K(Invitrogen、Carlsbad、CA)を使用し、PCRにより増幅した。温度サイクルは、95℃-30秒、49℃-30秒、72℃-2分を35サイクル行い、最後に72℃-10分のインキュベーションを続けた。PCR産物を、BamHI及びXbaIで制限酵素処理し、821bp断片を単離し、pNC2(実施例1A)の4303bpのBamHI-XbaI断片とライゲーションした。その結果得られたプラスミド(pVR22;図3)は、G418耐性遺伝子に動作可能に連結したScPGK1プロモーター及びScGAL10ターミネーターを有する。
【0075】
(実施例1C)
S. cerevisiae PDC1プロモーター及びScGAL10ターミネーターに機能的に連結したG418耐性遺伝子を有するpVR29プラスミドのコンストラクション(pVR29)
G418耐性マーカー(pVR22;実施例1B)をpNC4(実施例1A)にクローニングしてコンストラクションしたものをpVR29(図4)とした。 S. cerevisiae ScPGK1と S. cerevisiae GAL10ターミネーターとを、G418耐性遺伝子を発現するために使用した。G418耐性遺伝子は、Pfuポリメラーゼ(Stratagene、Madison、WI)を使用し、プライマーとして、5'-GCT CTA GAT GAG CCA TAT TCA ACG GGA AAC(5'G断片:配列番号5)及び5'-ATG GAT CCT TAG AAA AAC TCA TCG AGC ATC(3'G断片:配列番号6)を使用し、鋳型として、プラスミドpVR22(実施例1B)を使用し、PCRにより増幅した。あるいは、鋳型として、プラスミドpPIC9K(Invitrogen、Carlsbad、CA)も使用できる。温度サイクルは、最初に95℃-5分で反応混合物をインキュベートした後、95℃-30秒、49℃-30秒、72℃-2分を35サイクル行い、最後に72℃-10分のインキュベーションを続けた。PCR産物を、BamHI及びXbaIで制限酵素処理し、821bp断片を単離し、pNC4の4303bpのBamHI-XbaI断片とライゲーションした。その結果得られたプラスミドpVR29(図4)は、G418耐性遺伝子に動作可能に連結したPCD1プロモーター及びScGAL10ターミネーターを有する。
【0076】
(実施例1D)
プラスミドpPS1(ハイグロマイシン耐性カセット)、pNC14(Lh-L-LDH発現カセット)及びpPS9(ハイグロマイシンを選択マーカーとして使用し、L-乳酸産生のためにゲノムにLh-L-LDHを組み込むためのベクター)
形質転換した酵母細胞にハイグロマイシン耐性を与え、それにより、乳酸合成に有用なタンパク質をコードする組み換え核酸コンストラクトを含む酵母細胞形質転換株を選択可能とする組み換え核酸を調製した。ベクターpPS9(図5)は、ハイグロマイシン耐性選択による L. helveticus L-LDH遺伝子の酵母ゲノムへの組み込みに使用できる。
【0077】
ハイグロマイシン耐性マーカー( E. coli hph)を、 S. cerevisiae PDC1(ピルビン酸デカルボキシラーゼ)プロモーターの転写コントロール下にクローニングした。ハイグロマイシンBに対して耐性を与える E. coli hph遺伝子は、プライマーとして、5'HYGXBA1(5'-AAG CTC TAG ATG AAA AAG CCT GAA CTC AC-3';配列番号7)及び3'HYGBAMH1(5'-CGC GGA TCC CTA TTC CTT TGC CCT CGG AC-3';配列番号8)を使用し、鋳型として、プラスミドpRLMex30(Mach et al. 1994, Curr. Genet. 25, 567-570; Rajgarhia et al. U.S. Patent Application Serial No: 10/154.360, filed May 23, 2002、その全体を本願に引用して援用する)を使用して、PCRにより増幅した。hph遺伝子は、また、鋳型として働く E. coli 染色体DNAと、前記のプライマーとを使用して得ることもできる。温度サイクルは、PfuTurboDNAポリメラーゼ(Stratagene、Madison、WI)を使用して、94℃-1分、56℃-1分及び72℃-3分を30サイクル行い、最後に72℃-7分のインキュベーションを続けた。PCR産物を0.8%アガロースゲルの電気泳動により分離し、1026bp産物を単離した。次に、この1026bp断片をXbaI及びBamHIで制限酵素処理し、ScPDC1プロモーター及びScGAL10ターミネーターを含むpNC4(実施例1A)のXbaI-BamHI断片とライゲーションして、プラスミドpPS1(図5)を得た。
【0078】
L-乳酸脱水素酵素をコードする L. helveticus L-LDH遺伝子を、S. cerevisiae ScPGK1プロモーターの転写コントロール下にクローニングした。L-乳酸脱水素酵素をコードする L. helveticus L-LDH遺伝子は、プライマーとして、PS15S(5'-GCT CTA GAA TTA TGG CAA GAG AGG AAA AAC-3';配列番号9)及びPS16AS(5'-CGG GAT CCT CAT TGA CGA ACC TTA ACG-3';配列番号10)を使用し、鋳型として、プラスミドpSO20(実施例1H;図10)を使用し、PCRにより増幅した。あるいは、L-LDH遺伝子は、前記のプライマーとともに、 L. helveticus 染色体DNAを鋳型として使用して得ることができる。温度サイクルは、PfuTurboDNAポリメラーゼ(Stratagene、Madison、WI)を使用して、94℃-1分、56℃-1分及び72℃-3分を30サイクル行い、最後に72℃-7分のインキュベーションを続けた。PCR産物を、0.8%アガロースゲルの電気泳動により分離し、990bp産物を単離した。このPCR産物をXbaI及びBamHIで制限酵素処理し、XbaI及びBamHIで制限酵素処理したベクターpNC2(実施例1A)とライゲーションして、プラスミドpPS14(図5)を得た。pNC14をSalI及びBsaHIで制限酵素処理すると、LhLDH発現カセットを得ることができる。このLhLDH発現カセットを単離し、pPS1のSalI-ClaI断片とライゲーションして、プラスミドpPS9を得た(図5)。
【0079】
(実施例1E)
L. helveticus L-LDH遺伝子及びG418マーカーを有するpVR39プラスミドのコンストラクション
pPS9(実施例1H;図5)を、SphIで制限酵素処理し、シュリンプアルカリホスファターゼ(Roche Diagnostics、Indianapolis、IN)を用いてメーカーのマニュアルに従い脱リン酸化し、 L. helveticus L-LDH発現カセットを含む断片を得た。pVR29(実施例1C;図4)をSphIで制限酵素処理し、G418耐性遺伝子カセットを含む2048bp断片を、0.8%アガロースゲル上で分離した。これらの断片をライゲーションし、結果として、互いに隣接する L. helveticus L-LDH遺伝子及びG418耐性遺伝子マーカーを含むプラスミドpVR39(図6)を得た。
【0080】
(実施例1F)
pHES及びpSEHプラスミドのコンストラクション
pHES及びpSEHプラスミドを、PCT出願PCT/US/44041に従い、0.5μgのプラスミドpGAD424(Chien et al., 1991, Proc. Natl Acad. Sci. USA 88:9578-9582に記載される)を使用してコンストラクションし、HindIIIで制限酵素処理した。処理混合物を、TBEバッファー(Biorad、USA)を用いた0.8%アガロースゲル電気泳動により分離した。5.9kbp断片を、Sambrook et al. (1989, Molecular Cloning, second edition, Cold Spring Harbor Laboratory, Plainview, NY)の記載のように、前記ゲルから単離した。複数の制限酵素認識部位を有する、相補的な1対の92bpの合成オリゴマーを設計した。第1のオリゴは、「fwdHES」オリゴであって、以下の配列(配列番号11)を有する。
5'-CCCAAGCTTG AATTCCCCGG GGGATCCCTG CAGGGTACCA CGCGTAGATC TACTAGTGCG GCCGCCTCGA GTCTAGAGGG CCCAAGCTTG GG-3'
第2のオリゴは、「comp(SO1)(CMA2)hes」オリゴであって、以下の配列(配列番号12)を有する。
5'-CCAAGCTTGG GCCCTCTAGA CTCGAGGCGG CCGCACTAGT AGATCTACGC GTGGTACCCT GCAGGGATCC CCCGGGGAA TTCAAGCTTG GG-3'
500nmolの2つの相補的なオリゴマーを、ともに水槽中で10分間(100℃)でボイルした後、室温で徐々に冷ますことにより、互いにアニールさせた。92bpの二重鎖DNAをHindIIIで制限酵素処理し、pGAD424(Clontech、USA)をHindIIIで処理した5.9kbp断片とライゲーションした。ライゲーション混合物を、Sambrook et al.(同上)に記載のエレクトロポレーションによる E. coli DH10B(electromax cells、Life Technologies、Rockville、MD)の形質転換に使用した。組み換え E. coliを、Luria-Bertani ブロースプレート(Difco, USA)にプレーティングし、プラスミドを有する細胞を、100μg/mlの抗生物質アンピシリン(Sigma Chemicals、USA)を使用して選択した。アンピシリン耐性 E. coli クローンから得られたプラスミドDNAをスクリーニングし、2つのプラスミドpHES及びpSEHを得た。この2つのプラスミドは、ベクター上のアルコール脱水素酵素(ADH1)に対する合成二重鎖マルチクローニングサイトの方向が異なる。
【0081】
(実施例1G)
pSO20及びpPS9のさらなるコンストラクションのための L. helveticus L-LDH遺伝子を含むプラスミド:pVR1のコンストラクション
L. helveticus L-LDH遺伝子は、以下のように単離された。 L. helveticus 細胞は、American Type Culture Collection (ATCC Accession #10797)から得て、標準的な条件で生育させた。そして、ゲノムDNAを、以下のプロトコールを使用して精製した。
【0082】
それぞれ50mlの無菌MRSブロース(Difco、USA)が入った2つの250ml無菌フラスコに、バクテリアのグリセロールストックから単一コロニー(又は5ml)を植菌した。その培養液を、37℃で、170rpmのアジテーションを加えながら48時間インキュベートした。この培養液を、50ml無菌青キャップチューブに移し、3000rpmで10分間遠心した。このペレットを、50mlの12.5%w/vスクロース溶液で懸濁し、再度、3000rpmで10分間遠心した。新たに形成されたペレットを、50ml無菌青キャップチューブ中で、5mlの12.5%w/vスクロース溶液に再度懸濁した。5mlのTES溶液を、前記チューブに加えた。(200mlのTES: 2 mlの1.0 M Tris (pH 8.0) [10 mM Tris (pH 8.0)]、20 mlの500 mM EDTA (pH 8.0) [50 mM EDTA (pH 8.0)]、0.584 gのNaCl [50 mM NaCl]を含み、使用前にフィルター滅菌する。)12.5%w/vスクロース溶液を前記チューブに加え、混合した。300mgのリゾチームパウダー(Sigma Chemicals、USA)を添加し、その混合物を2000rpmで1分間ボルテックスした。25μlのムタノリジン(mutanolysin)溶液(Sigma)(濃度2.2mg/ml)を前記混合物に添加し、その混合物を、37℃で一晩(10〜12時間)インキュベートした。
【0083】
2.5mlの20%SDS溶液(Biorad、USA)及び168μlのプロテナーゼK溶液(濃度:28mg/1.4ml)(Sigma)を、前記混合物に添加し、その結果の混合物を上下逆にしながら混合し、50℃-1時間でインキュベーションした。細胞膜物質は、よく破壊され、溶液は、半透明になった。その混合物を、NaClで稀釈し、前記チューブに0.15Mの濃度を得た。
【0084】
前記混合物を、50mlのオークリッジ(Oakridge)チューブに移し、等量のフェノール:クロロホルム:イソアミルアルコール(25:24:1)溶液で処理した。この混合物をよくシェイクし、5000rpmで10分間遠心した。水層を、新しいチューブに吸出し、25μlのRNAse(100mg/ml)を添加し、上下逆にしながら混合した。その結果の混合物を、37℃-15分でインキュベートした。
【0085】
2.5倍量のエタノールを、前記チューブの側面に沿って前記混合物に添加し、混合しなかった。境界面に形成されたDNAを、糸巻きのようにして回収し、70%エタノール中で洗浄し、その混合物を10000rpmで10分間遠心した。底に形成されたペレットDNAを空気乾燥し、微遠心(microfuge)チューブ中で、10mM Tris-HCl、pH8.5に再度懸濁した。その混合物を、DNAが溶液となるまで(2〜3時間)、前記チューブ内で50℃でインキュベートした。
【0086】
プライマーは、 L. helveticus のL-LDHとしてGenbankから入手可能な配列(Genbank accession # Z81318)に基づき設計した。PCR増幅は、Pfuポリメラーゼ(Stratagene、Madison、WI)を使用して行った。各反応は、500ngの濃度の L. helveticus ゲノムDNA、各0.2mMの4種のdNTP、並びに、各1μMの増幅プライマーSO22:5'-CCG GGA TCC ATG GCA AGA GAG GAA AAA CCTC(配列番号13)及びSO23:5'-CCA AGA TCT TTA TTG ACG AAC CTT AAC GCC AG(配列番号14)を含む。温度サイクルは、最初に95℃-10分でインキュベートした後に、95℃-30秒、41℃-30秒及び72℃-60秒を35サイクル行った。この反応で生成された990(bp)の断片を、都合のよい方法を使用してゲルから精製し、BamHI及びBglIIで制限酵素処理し、そして、BamHI及びBglIIで処理したpHES(実施例1F)にクローニングし、プラスミドpLhLDH-HESを得た。結果として得られた配列は、Genbankで公知の L. helveticus L-LDHをコードする遺伝子配列(Accession # Z81318)と極めて相同なポリペプチドに翻訳されうる配列である。
【0087】
L. helveticus L-LDH遺伝子の5'端及び3'端に、それぞれ、NcoI及びDraIIIの制限酵素認識部位を導入するため、さらに、プライマーを設計した。PCR増幅は、Pfuポリメラーゼ(Stratagene、Madison、WI)を使用して行った。各反応は、5ngのpLH-LDH/HES、各0.2mMの4種のdNTP、並びに、各1μMの増幅プライマーVR1:5'-ATC CAT GGC AAG TAT TAC GGA TAA GGA TCA CCAA(配列番号15)及びVR2:5'-ATC ACG AAG TGT CAC GTA CGG GTT TCG ATG TC(配列番号16)を含む。温度サイクルは、最初に95℃-10分でインキュベートした後に、95℃-30秒、55℃-30秒及び72℃-60秒を35サイクル行った。この反応で生成された982(bp)の断片を、都合のよい方法を使用してゲルから精製し、NcoI及びDraIIIで制限酵素処理し、そして、NcoI及びDraIIIで処理したpTEF1/Zeo(Invitrogen、Carlsbad、CA)にクローニングした。結果として得たプラスミドpVR1(図7)は、 L. helveticus L-LDHをコードする遺伝子配列が、 Saccharomyces cerevisiae pTEF1プロモーターのコントロール下にあることが確認された。 E. coli EM7プロモーターが存在するが、これは、本発明の組み込みベクターに必要な構成要素ではない。
【0088】
(実施例1H)
K. marxianus において L. helveticus L-LDHプラスミドを発現するためのプラスミドpSO20のコンストラクション
プラスミドpSO20は、BamHI(NEB)で制限酵素処理され、シュリンプアルカリホスファターゼ(Roche Diagnostics、Indianapolis、IN)処理されたpUC19プラスミド(Life Technologies、USA)と、pVR1(実施例1F)を処理して得られた L. helveticus L-LDH遺伝子とそれに隣接するプロモーター及びターミネーター配列を有するBamHI断片とを、ライゲーションしてコンストラクションした。結果として得られたプラスミドは、pSO18(図8)である。pSO18を、次に、SphIで制限酵素処理し、pKD1のバックボーンを含むpSPH1コンストラクト(Chen et al., 1986, "Sequence organization of the circular plasmid pKD1 from the yeast Kluyveromyces drosophilarum," Nucleic acids Res. 14: 4471-4481)由来のSphI断片とライゲーションすることで、結果として、プラスミドpSO19(図9)を得た。
【0089】
pTEF1/Zeo(Invitorogen, Inc)を、XhoI/XbaIで制限処理して、 S. cerevisiase pTEF1プロモーター、ゼオシン耐性遺伝子、及び、 S. cerevisiase GAL10ターミネーターを放出させた。この1195bp断片の末端を、PfuDNAポリメラーゼ(Stratagene、Madison、WI)を用いて平滑末端とした。コンストラクトpSO19を、AatIIで制限酵素処理し、PfuDNAポリメラーゼで平滑末端を作り出し、結果として得られた断片をシュリンプアルカリホスファターゼで処理した。そして、この9.3kbpを、 S. cerevisiase pTEF1プロモーター、ゼオシン耐性遺伝子、及び、 S. cerevisiase GAL10ターミネーターを含む1195bpのpTEF/Zeo断片とライゲーションした。この結果として、コンストラクトpSO20が得られた(図10)。
【0090】
(実施例1I)
形質転換DNAのランダムな組み込みを介する L. helveticus L-LDH遺伝子の K. marxianus ゲノムへの導入
pVR39をSstI及びApaIで制限酵素処理することで、L. helveticus L-LDH:G418耐性遺伝子カセットを含む4.28kbpの断片を単離した。この断片は、0.8%のアガロースゲルで分離され、下記エレクトロポレーションプロトコールを使用した K. marxianus (CD21)の形質転換に使用される。
【0091】
K. marxianus の単一コロニーを、50mLのYPD培地への植菌(250 mLのバッフル型フラスコ中に、10 g/L 酵母抽出物, 20 g/L ペプトン及び20 g/L グルコースを含む。OD600を0.1まで。)に使用した。この培養液を、30℃-250rpmで16時間培養した。最終OD600は、10となった。10mLの培養液から細胞を、遠心により回収し、エレクトロポレーションバッファー(10 mM Tris-Cl, 270 mM スクロース, 1 mM MgCl2, pH 7.5)で1回洗浄した。その後、この細胞を、インキュベーションバッファー(YPD + 25 mM DTT, 20mM HEPES, pH 8.0)に再度懸濁し、30℃-250rpmで30分インキュベートした。この細胞を、遠心により回収し、エレクトロポレーションバッファーで1回洗浄し、1mLのエレクトロポレーションバッファーに再度懸濁した。そして、400μLの細胞を、0.4cmのエレクトロポレーションキュベット(BioRad;USA)に移した。
【0092】
pVR39を、SstI及びApaIで制限酵素処理し、結果として得られる4.3kbp断片の2μgを、キュベットに添加した(BioRad;USA)。次いで、細胞を、1.8kV、1000Ω、25μFの条件で、エレクトロポレーションした。この細胞を、50mLスクリューキャップ付きファルコンチューブ中の1mLのYPDに移し、30℃-250rpmで4時間インキュベートした後、300μg/mLのG418を含むYPDプレートにプレーティングして選択した。形質転換株は、37℃で3日間生育させた。G418耐性形質転換株は、300μg/mLのG418を含む新しい選択プレートに、再度、ストリークした。
【0093】
K. marxianus のゲノムに断片が組み込まれた確認は、 L. helveticus L-LDH遺伝子に相同なように設計されたPCRプライマー、及び、G418耐性遺伝子と相同なリバースプライマーを使用して達成できる。
【0094】
VR146:5'-GCT GAC TAC CCA GAT TGT AAG GATG-3’(配列番号17)及びVR143:5'-CTG CCA GCG CAT CAA CAA TAT TTT CAC-3’(配列番号18)は、前記カセットを有する株において、 L. helveticus L-LDH遺伝子とG418耐性遺伝子との間の2.3kbpの産物を増幅するように設計した。このプライマーは、前記カセットを有しない株では、少しも断片を増幅しない。
【0095】
形質転換コロニーに対する温度サイクルは、TaqDNAポリメラーゼ(Qiagen、USA)を用いて、最初に94℃-2分で反応混合物をインキュベーションした後、94℃-30秒、53℃-30秒及び72℃-3分を35サイクル行い、最後に72℃-7分のインキュベーションを続けた。分析した10個の形質転換株が、予想される2.3kbpのPCR産物を産出した。前記10個の形質転換株から得たゲノムDNAについての L. helveticus L-LDH遺伝子をプローブとしたサザンブロットを行ったところ、分析した前記10個の形質転換株のうち、2個が、pVR39に由来する L. helveticus L-LDH遺伝子の1コピーの組み込みと一致する適切なバンドパターンを示した。この2つのうち1つを、CD484と認定した。
【0096】
(実施例1J)
形質転換DNAのランダムな組み込みを介する L. helveticus L-LDH遺伝子のさらなるコピーの K. marxianus ゲノムへの導入
pPS9をSstI及びApaIで制限酵素処理することで、L. helveticus L-LDH:HPH耐性遺伝子カセットを含む4.7kbpの断片を単離した。この断片は、0.8%のアガロースゲルで分離され、下記エレクトロポレーションによる K. marxianus CD484の形質転換に使用される。
【0097】
K. marxianus CD484の単一コロニーを、50mLのYPD培地への植菌(250 mLのバッフル型フラスコ中に、10 g/L 酵母抽出物、20 g/L ペプトン、20 g/L グルコース及び2%アガーを含む。OD600を0.1まで。)に使用した。この培養液を、30℃-250rpmで16時間培養した。最終OD600は、10となった。10mLの培養液から細胞を、遠心により回収し、エレクトロポレーションバッファー(10 mM Tris-Cl, 270 mM スクロース, 1 mM MgCl2, pH 7.5)で1回洗浄した。その後、細胞を、インキュベーションバッファー(YPD + 25 mM DTT, 20mM HEPES, pH 8.0)に再度懸濁し、30℃250rpmで30分インキュベートした。この細胞を、遠心により回収し、エレクトロポレーションバッファーで1回洗浄し、1mLのエレクトロポレーションバッファーに再度懸濁した。そして、400μLの細胞を、0.4cmのエレクトロポレーションキュベット(BioRad;USA)に移した。
【0098】
pPS9を、SstI及びApaIで制限酵素処理し、結果として得られる4.7kbp断片の5μgを、キュベットに添加した。次いで、細胞を、1.8kV、1000Ω、25μFの条件で、エレクトロポレーションした。細胞を、50mLスクリューキャップ付きファルコンチューブ中の1mLのYPDに移し、30℃-250rpmで4時間インキュベートした後、200μg/mLのハイグロマイシン(Invitrogen、Carlsbad、CA)を含むYPDプレートにプレーティングして選択した。ハイグロマイシン耐性形質転換株を、37℃で3日間、生育させた。耐性形質転換株が、両方の耐性遺伝子を有することを確認するため、300μg/mLのG418を含む新しい選択プレートに、再度、ストリークした。
【0099】
L. helveticus L-LDH:G418耐性遺伝子カセットの新たな4.7kbp断片が組み込まれた確認は、L. helveticus L-LDH遺伝子に相同となるように設計されたPCRプライマー、及び、ハイグロマイシン耐性遺伝子と相同なリバースプライマーを使用して達成できる。VR146(配列番号17)及びVR142:5'-GTG ACA CCC TGT GCA CGG CGG GAG AT-3’(配列番号19)は、前記カセットを有する株において、 L. helveticus L-LDH遺伝子とハイグロマイシン耐性遺伝子との間の2.3kbpの産物を増幅するように設計した。このプライマーは、前記カセットを有しない株では、少しも断片を増幅しない。
【0100】
温度サイクルは、TaqDNAポリメラーゼ(Qiagen、USA)を用いて、最初に94℃-2分でインキュベーションした後、94℃-30秒、53℃-30秒及び72℃-3分を35サイクル行い、最後に72℃-7分のインキュベーションを続けた。分析した4個の形質転換株が、予想される1.76kbpのPCR産物を産出した。
【0101】
K. marxianus CDC484由来の4288kbpの L. helveticus L-LDH:G418耐性遺伝子の存在確認は、L. helveticus L-LDH遺伝子に相同となるように設計されたPCRプライマー、及び、G418耐性遺伝子と相同なリバースプライマーを使用して達成できる。VR146(配列番号17)及びVR143(配列番号18)は、前記カセットを有する株において、L. helveticus L-LDH遺伝子とG418耐性遺伝子との間の2.3kbpの産物を増幅するように設計した。このプライマーは、前記カセットを有しない株では、少しも断片を増幅しない。
【0102】
形質転換コロニーに対する温度サイクルは、TaqDNAポリメラーゼ(Qiagen、USA)を用いて、最初に94℃-2分でインキュベーションした後、94℃30秒、53℃30秒及び72℃3分を35サイクル行い、最後に72℃で7分のインキュベーションを続けた。分析した4個の形質転換株が、予想される2.3kbpのPCR産物を産出した。
【0103】
ゲノムDNAについて、 L. helveticus L-LDH遺伝子をプローブとしたサザンブロットを行ったところ、PCRでG418及びハイグロマイシンカセットについて陽性であった前記4個の形質転換株のうち、1個が、ゲノム中に組み込まれた L. helveticus L-LDH遺伝子を2コピー有していた。この株を、 K. marxianus CDC492と認定し、さらに、L-乳酸の産生について分析した。
【0104】
(実施例1K)
振とうフラスコ培養のYPD+グルコース培地中におけるL-乳酸の産生
組み換え株CD492を、100g/Lのグルコースを補充した50mLのYPD培地を含む250mLのバッフル型振とうフラスコで培養した。細胞は、YPDアガープレートからOD600が0.1まで、30℃-250rpmで16時間培養した。細胞が対数増殖期であることを確認するため、YSI分析を使用して残存グルコース濃度を測定した。4g/L細胞乾燥重量(「gcdw」)当量を遠心により回収し、250mLのバッフル型振とうフラスコ中の、約100g/Lのグルコース及び55g/LのCaCO3を補充した50mLのYPD培地に再度懸濁した。その培養液を、30℃-70rpmでインキュベートした。0、12、24時間の間隔後にサンプルを回収した。細胞をフィルターによりサンプルから取り除き、その上清を、HPLCにより、グルコース、乳酸塩、ピルビン酸塩及びエタノールについて、分析した。これらの条件下で、CD492株は、鏡像異性体的に99%を越える純度のL-乳酸を産生した。最終力価は、69g/Lであった。容積生産性は、3.6g/L/hrであった。
【0105】
比較のために、CD484(実施例1I)株を同様の条件下で培養した。CD492と比較して、L-乳酸の産生は、低く、エタノールの産生は、高かった。
【0106】
(実施例1L)
振とうフラスコ培養のYPD+グルコース培地中におけるL-乳酸の産生
492株を、100g/Lのグルコースを補充した50mLのYPD培地を含む250mLのバッフル型振とうフラスコで培養した。細胞を、振とうフラスコ中に、OD600が0.1となるように植菌し、前記フラスコを、30℃-250rpmで、16時間インキュベートした。最終pHは、3.1未満であった。細胞が対数増殖期であることを確認するため、YSI分析を使用して、前記フラスコの残存グルコース濃度を測定した。4g/L細胞乾燥重量当量を遠心により回収し、250mLのバッフル型振とうフラスコ中の、約25g/Lのグルコースを補充した50mLのYPD培地に再度懸濁した。その培養液を、30℃-70rpmでインキュベートした。様々な時間間隔後にサンプルを回収し、細胞をフィルターによりサンプルから取り除いた。培養上清を、HPLCにより、グルコース、乳酸塩、ピルビン酸塩及びエタノールについて、分析した。
【0107】
L-乳酸は、鏡像異性体的に99%を越える純度で産生された。乳酸力価は、15g/Lであり、容積生産性は、2g/L/hrであった。CD484株を同様の条件でテストした場合、L-乳酸の産生は、低く、エタノールの産生は、高かった。
【0108】
別個の実験として、492株を、100g/Lのグルコースを補充した100mLのYPD培地を含む500mLのバッフル型振とうフラスコに、OD600が0.1となるように植菌した。前記振とうフラスコを、37℃、250rpmで、約16時間インキュベートした。細胞が対数増殖期であることを確認した後、4g/L細胞乾燥重量当量を遠心により回収した。細胞ペレットを、約40g/Lのグルコースを補充した50mLのYPD培地に再度懸濁し、250mLのバッフル型振とうフラスコに移した。前記振とうフラスコを、37℃-70rpmに置いた。バイオマスの生育終了時及び産生期の0、15、18.5及び23時間後に、グルコースが消費されるまで、HPLC解析用のサンプルを回収した。pHは、産生の開始と終了の時点で測定した。最終pHは、3.00+/-0.06であった。
【0109】
CD492株は、グルコースを、15〜23時間で消費した。遊離酸分析用のサンプルを、産生終了時(グルコースが完全に消費された時)に採取し、乳酸、酢酸塩、エタノール及びピルビン酸塩の総合量を分析した。CD492株は、32g/Lの遊離乳酸、32g/Lのエタノール、並びに、1g/L未満の酢酸塩及びピルビン酸塩を産生した。遊離乳酸は、32g/Lであった。全ての場合で、プロトン化型の酸のパーセントは、発酵終了時で、少なくとも97%であった。乳酸のグルコースに対する収率は、59%であった。
【0110】
CD484株を、同様の条件で評価した。CD484株は、26〜28g/Lの遊離乳酸を産生し、グルコースに対する収率は、46〜58%であった。
【実施例2】
【0111】
(実施例2A)
S. cerevisiase ScPGK1プロモーターのコントロール下でL-LDHを発現するための B. megaterium L-LDHを有するpVR24プラスミドのコンストラクション
LDH遺伝子をコードする B. megaterium DNAは、下記のように単離した。 B. megaterium は、American Type Culture Collection (ATCC Accession #6458)から得て、標準的な条件で生育させた。ゲノムDNAは、これらの細胞から“Easy-DNA"キット(Invitorogen)を使用して、メーカーのプロトコールに従い、精製した。プライマーは、Genbankの B. megaterium のL-LDHとして入手可能な配列(Genbank accession # M22305)に基づき設計した。PCR増幅反応は、buffer II (1.5mM MgCl2) 及び AmpliTaq Gold ポリメラーゼ(ともに、Perkin Elmer社製)を使用して行った。各反応は、6ng/μLの濃度の B. megaterium ゲノムDNA、各0.2mMの4種のdNTP、並びに、各1μMの増幅プライマーBM1270:5'-CCT GAG TCC ACG TCA TTA TTC-3'(配列番号20)及びBM179:5'-TGA AGC TAT TTA TTC TTG TTAC-3'(配列番号21)を含む。
【0112】
温度サイクルは、最初に95℃-10分でインキュベートした後に、95℃-30秒、50℃-30秒及び72℃-60秒を35サイクル行った。この反応で生成された1100bpの断片を、都合のよい方法を使用してゲルから精製し、クローニングし、塩基配列決定をした。結果として得られた配列は、Genbankで公知の L-LDHをコードする遺伝子配列と相同性を示すポリペプチドに翻訳されうる配列である。ここで、前記相同性の程度は、2つの配列間における、少なくとも75%、80%、85%、90%、91%、92%、93%、94%、95%、96%、97%、98%若しくは約99%の同一性である。
【0113】
B. megaterium L-LDHをコードする配列を、pNC2(実施例1A)由来の S. cerevisiae ScPGK1プロモーター及び S. cerevisiae GAL10転写ターミネーターと、動作可能に連結した。2つのオリゴヌクレオチドプライマーBmeg5’:5'-GCT CTA GAT GAA AAC ACA ATT TAC ACC-3'(配列番号22)及びBmeg3’:5'-ATG GAT CCT TAC ACA AAA GCT CTG TCGC-3'(配列番号23)を、前記遺伝子をコードする配列の末端に制限酵素認識部位を導入するように設計した。
【0114】
増幅反応は、PfuTurboDNAポリメラーゼ(Stratagene、Madison、WI)を使用して、上述したようなdNTP及びプライマー濃度で、メーカーが供給するバッファーを使用して行った。温度サイクルは、最初に反応混合物を95℃で3分インキュベートした後、95℃30秒、50℃30秒、72℃60秒を20サイクル行い、最後に72℃9分のインキュベーションを続けた。PCR産物を、XbaI及びBamHIで制限酵素処理し、pNC2(実施例1A)のXbaI-BamHI断片とライゲーションした。このライゲーションの結果、ScPGK1プロモーター及びScGAL10ターミネーターが、 B. megaterium L-LDHをコードする配列に動作可能に連結したプラスミドpVR24(図12)が得られた。
【0115】
(実施例2B)
B. megaterium L-LDHをコードするG418耐性マーカープラスミド(pCA3)
Invitrogen(Carlsbad、CA)から得たG418抗生選択マーカーを変更し、酵母 S. cerevisiae 由来のピルビン酸デカルボキシラーゼ遺伝子プロモーター及び転写ターミネーターに動作可能に連結させた。このコンストラクトを作製する際、下記オリゴヌクレオチドを調製し、G418耐性遺伝子が挿入されたプラスミドからコード配列を増幅するために使用した。2つのオリゴヌクレオチドプライマーG5’:5'-AAA TCT AGA TGA GCC ATA TTC AAC GGGA-3'(配列番号24)及びG3’:5'-CCG GAT CCT TAG AAA AAC TCA TCG AGC AT-3'(配列番号25)は、前記遺伝子をコードする配列の末端に制限酵素認識部位を導入するように設計した。
【0116】
pPIC9Kベクター(Invitrogen、Carlsbad、CA)のG418耐性遺伝子を、G5'及びG3'プライマー、4種のdNTP、並びにPfuTurboポリメラーゼを使用してPCRにより増幅した。増幅反応は、PfuTurboDNAポリメラーゼ(Stratagene、Madison、WI)を使用して、上述したようなdNTP及びプライマー濃度で、メーカーが供給するバッファーを使用して行った。温度サイクルは、最初に95℃-3分で反応混合物をインキュベートした後、95℃-30秒、50℃-30秒、72℃-60秒を20サイクル行い、最後に72℃-9分のインキュベーションを続けた。PCR産物を、XbaI及びBamHIで制限酵素処理し、pNC4(実施例1A)のXbaI-BamHI断片とライゲーションした。
【0117】
このベクターのG418遺伝子のScGAL10ターミネーターの3’末端であるSphI部位に、 S. cerevisiaeのScPGK1プロモーター及びScGAL10ターミネーターに動作可能に連結した B. megaterium L-LDH遺伝子を導入し、その結果、プラスミドpCA3(図13)を得た。
【0118】
(実施例2C)
B. megaterium L-LDH遺伝子及びハイグロマイシン耐性遺伝子を含むpVR38プラスミドのコンストラクション
pVR24(実施例2A)を、SphIで制限酵素処理し、シュリンプアルカリホスファターゼ(Roche Diagnostics、Indianapolis、IN)を用いてメーカーのプロトコールに従い脱リン酸化し、 B. megaterium L-LDH発現カセットを含む断片を得た。pPS1(実施例1D)をSphIで制限酵素処理し、ハイグロマイシン耐性遺伝子カセットを含む2259bp断片を、0.8%アガロースゲル上で分離し、脱リン酸化したpVR24とライゲーションした。その結果、互いに隣接する B. megaterium L-LDH遺伝子及びハイグロマイシン耐性遺伝子マーカーを含むプラスミドpVR38(図14)を得た。
【0119】
(実施例2D)
形質転換DNAのランダムな組み込みを介する B. megaterium L-LDH遺伝子の K. marxianus ゲノムへの導入
pCA3をSstI及びApaIで制限酵素処理することで、 B. megaterium L-LDH:G418耐性遺伝子カセットを含む4.2kbpの断片を単離した。この断片は、0.8%のアガロースゲルで分離され、下記エレクトロポレーションプロトコールを使用した K. marxianus(CD21)の形質転換に使用される。
【0120】
K. marxianus の単一コロニーを、50mLのYPD培地への植菌(250 mLのバッフル型フラスコ中に、10 g/L 酵母抽出物, 20 g/L ペプトン、20 g/L グルコース及び2%アガーを含む。OD600を0.1まで。)に使用した。培養液を、30℃-250rpmで16時間培養した。最終OD600は、10となった。10mLの培養液から細胞を、遠心により回収し、エレクトロポレーションバッファー(10 mM Tris-Cl, 270 mM スクロース, 1 mM MgCl2, pH 7.5)で1回洗浄した。細胞を、インキュベーションバッファー(YPD + 25 mM DTT, 20mM HEPES, pH 8.0)に再度懸濁し、30℃-250rpmで30分インキュベートした。細胞を、遠心により回収し、エレクトロポレーションバッファーで1回洗浄し、1mLのエレクトロポレーションバッファーに再度懸濁した。そして、400μLの細胞を、0.4cmのエレクトロポレーションキュベット(BioRad;USA)に移した。
【0121】
2μgのpCA3由来の4.3kbpSstI/ApaI断片を、キュベットに添加し、細胞を、1.8kV、1000Ω、25μFの条件で、エレクトロポレーションした。細胞を、50mLスクリューキャップ付きファルコンチューブ中の1mLのYPDに移し、30℃250rpmで4時間インキュベートした後、300μg/mLのG418を含むYPDプレートにプレーティングして選択した。形質転換株を、37℃で3日間、生育させた。G418耐性形質転換株を、300μg/mLのG418を含む新しい選択プレートに、再度、ストリークした。
【0122】
K. marxianus のゲノムに断片が組み込まれた確認は、 B. megaterium L-LDH遺伝子に相同となるように設計されたPCRプライマー、及び、G418耐性遺伝子と相同なリバースプライマーを使用して達成できる。BmLDH3:5'-GTA CGC ATT ACC AAG GCT ATT TTA GAT-3’(配列番号26)及びVR143(配列番号18)は、前記カセットを有する株において、 B. megaterium L-LDH遺伝子とG418耐性遺伝子との間の1.8kbpの産物を増幅するように設計した。このプライマーは、前記カセットを有しない株では、少しも断片を増幅しない。
【0123】
形質転換コロニーに対する温度サイクルは、TaqDNAポリメラーゼ(Qiagen、USA)を用いて、最初に94℃-2分で反応混合物をインキュベーションした後、94℃-30秒、53℃-30秒及び72℃-3分を35サイクル行い、最後に72℃-7分のインキュベーションを続けた。分析した10個の形質転換株のうち7個が、予想される1.8kbpのPCR産物を産出した。前記7個の形質転換株から得たゲノムDNAについての B. megaterium L-LDH遺伝子をプローブとしたサザンブロットを行ったところ、分析した前記7個の形質転換株のうち、2個が、pCA3に由来する B. megaterium L-LDH遺伝子の1コピーの組み込みと一致する適切なバンドパターンを示した。この2つのうち1つを、CD162と認定した。
【0124】
(実施例2E)
形質転換DNAのランダムな組み込みを介する B. megaterium L-LDH遺伝子のさらなるコピーの K. marxianus ゲノムへの導入
pVR38をSstI及びApaIで制限酵素処理することで、 B. megaterium L-LDH:HPH耐性遺伝子カセットを含む4.5kbpの断片を単離した。この断片は、0.8%のアガロースゲルで分離し、エレクトロポレーションによる K. marxianus CD162株の形質転換に使用した。
【0125】
K. marxianus CD162の単一コロニーを、50mLのYPD培地への植菌(250 mLのバッフル型フラスコ中に、10 g/L 酵母抽出物、20 g/L ペプトン、20 g/L グルコース及び2%アガーを含む。OD600を0.1まで。)に使用した。培養液を、30℃-250rpmで16時間培養した。最終OD600は、10となった。10mLの培養液から細胞を、遠心により回収し、エレクトロポレーションバッファー(10 mM Tris-Cl, 270 mM スクロース, 1 mM MgCl2, pH 7.5)で1回洗浄した。その後、細胞を、インキュベーションバッファー(YPD + 25 mM DTT, 20mM HEPES, pH 8.0)に再度懸濁し、30℃-250rpmで30分インキュベートした。細胞を、遠心により回収し、エレクトロポレーションバッファーで1回洗浄し、1mLのエレクトロポレーションバッファーに再度懸濁した。そして、一部(400μL)の細胞を、0.4cmのエレクトロポレーションキュベット(BioRad;USA)に移した。
【0126】
pVR38を、SstI及びApaIで制限酵素処理し、結果として得られる4.5kbp断片の5μgを、エレクトロポレーションキュベットに添加した。次いで、細胞を、1.8kV、1000Ω、25μFの条件で、エレクトロポレーションした。細胞を、50mLスクリューキャップ付きファルコンチューブ中の1mLのYPDに移し、30℃-250rpmで4時間インキュベートした後、200μg/mLのハイグロマイシン(Invitrogen、Carlsbad、CA)を含むYPDプレートにプレーティングして選択した。ハイグロマイシン耐性形質転換株を、37℃で3日間生育させた。耐性形質転換株が、両方の耐性遺伝子を有することを確認するため、300μg/mLのG418を含む新しい選択プレートに、再度、ストリークした。
【0127】
B. megaterium L-LDH:HPHカセットの新たな4.5kbp断片が組み込まれた確認は、 L. helveticus L-LDH遺伝子に相同となるように設計されたPCRプライマー及びハイグロマイシン耐性遺伝子と相同なリバースプライマーを使用して達成できる。BmLDH3(配列番号26)及びVR142(配列番号19)は、前記カセットを有する株において B. megaterium L-LDH遺伝子とハイグロマイシン耐性遺伝子との間の1.76kbpの産物を増幅するように設計した。このプライマーは、前記カセットを有しない株では、少しも断片を増幅しない。
【0128】
温度サイクルは、TaqDNAポリメラーゼ(Qiagen、USA)を用いて、最初に94℃-2分で反応混合物をインキュベーションした後、94℃-30秒、53℃-30秒及び72℃-3分を35サイクル行い、最後に72℃-7分のインキュベーションを続けた。上記PCR方法論により分析した9個の形質転換株が、予想される1.76kbpのPCR産物を産出した。
【0129】
K. marxianus CDC162に既に存在した4.2kbpの B. megaterium L-LDH:G418耐性遺伝子の存在確認は、 B. megaterium L-LDH遺伝子に相同となるように設計されたPCRプライマー及びG418耐性遺伝子と相同なリバースプライマーを使用して達成できる。BmLDH3(配列番号26)及びVR143(配列番号18)は、前記カセットを有する株において、 B. megaterium L-LDH遺伝子とG418耐性遺伝子との間1.8kbpの産物を増幅するように設計した。このプライマーは、前記カセットを有しない株では、少しも断片を増幅しない。
【0130】
形質転換コロニーに対する温度サイクルは、TaqDNAポリメラーゼ(Qiagen、USA)を用いて、最初に94℃-2分で反応混合物をインキュベーションした後、94℃-30秒、53℃-30秒及び72℃-3分を35サイクル行い、最後に72℃-7分のインキュベーションを続けた。分析した9個の形質転換株が、予想される1.8kbpのPCR産物を産出した。これらの株のゲノムDNAについて、 B. megaterium L-LDH遺伝子をプローブとしたサザンブロットを行ったところ、PCRでG418及びハイグロマイシンカセットについて陽性であった前記9個の形質転換株のうち、7個が、ゲノム中に組み込まれた B. megaterium L-LDH遺伝子を2コピー有していた。この株のうち、1個を、CD355と認定した。
【0131】
(実施例2F)
振とうフラスコ培養のYPD+グルコース培地中におけるL-乳酸の産生
組み換え株CD355を、実施例Kで記載した一般的な方法で培養した。これらの条件下で、CD355株は、鏡像異性体的に99%を越える純度のL-乳酸を産生した。最終力価は、52g/Lであった。容積生産性は、1.5g/L/hrであった。
【0132】
比較のために、CD162株を同様の条件下で培養した。L-乳酸の産生は、低く、エタノールの産生は、高かった。
【0133】
(実施例2G)
振とうフラスコ培養のYPD+グルコース培地中におけるL-乳酸の産生
355株を、100g/Lのグルコースを補充した50mLのYPD培地を含む250mLのバッフル型振とうフラスコで培養した。細胞を、振とうフラスコ中に、OD600が0.1となるように植菌し、前記フラスコを、30℃-250rpmで、16時間インキュベートした。最終pHは、3.1未満であった。細胞が対数増殖期であることを確認するため、YSI分析を使用して、前記フラスコの残存グルコース濃度を測定した。4g/L細胞乾燥重量当量を遠心により回収し、250mLのバッフル型振とうフラスコ中の、約25g/Lのグルコースを補充した50mLのYPD培地に再度懸濁した。その培養液を、30℃-70rpmでインキュベートした。様々な時間間隔後にサンプルを回収し、細胞をフィルターによりサンプルから取り除いた。培養上清を、HPLCにより、グルコース、乳酸塩、ピルビン酸塩及びエタノールについて、分析した。
【0134】
L-乳酸は、鏡像異性体的に99%を越える純度で産生された。乳酸の力価は、12g/Lであり、容積生産性は、1.9グラム/L/hrであった。CD162株を同様の条件でテストした場合、L-乳酸の産生は、低く、エタノールの産生は、高かった。
【実施例3】
【0135】
(実施例3A)
K. marxianus のPDC1遺伝子座を標的としたDNAの組み込みのためのpBH5a/bのコンストラクション
K. marxianus のピルビン酸デカルボキシラーゼ(KmPDC1)の側面DNAをベクターpVR29(実施例1C)にクローニングし、KmPDC1遺伝子座にDNAを組み込むためのベクターを作製した。最終的に得られたコンストラクトが、pBH5b(図15)である。pBH5bのSbfI制限酵素認識部位にクローニングされた遺伝子は、 K. marxianus のプロモーター及びターミネーターと動作可能に連結される。
【0136】
K. marxianus のPDC1のすぐ上流の1254bpのDNA断片を、プライマーとして、5'-Flank5’:5 -CAA GAA GGT ACC CCT CTC TAA ACT TGA ACA-3'(配列番号27)及び5'-Flank3’:5'-GTA ATT CCT GCA GGT GCA ATT ATT TGG TTT GG-3'(配列番号28)を使用し、鋳型として、プラスミドpSO21を使用して、PCRにより増幅した。温度サイクルは、最初に94℃-2分で反応混合物をインキュベーションした後、94℃-30秒、53℃-30秒及び72℃-1.5分を35サイクル行い、最後に72℃-7分のインキュベーションを続けた。1254bpのPCR産物を、0.8%アガロースゲルの電気泳動により分離して単離した。このPCR産物及びpVR29プラスミド(実施例1C)をともに、KpnI及びSbfIで制限酵素処理した。処理したPCR産物を、5067kbのpVR29断片とライゲーションし、6315bpのpBH5a(図15)を得た。pBH5aは、 S. cerevisiaeのScPDC1プロモーター及びScGAL10ターミネーターに動作可能に連結したG418耐性遺伝子、並びに、K. marxianus のPDC1のすぐ上流の1254bpのDNA断片を有する。
【0137】
K. marxianus のPDC1のすぐ下流の535bpのDNA断片を、プライマーとして、3'-Flank5’:5'-CCA AGC CCT GCA GGA GAG GGA GAG GAT AAA GA-3'(配列番号29)及び3'-Flank3’:5'-CTC GTA ACG CGT GTA CAA GTT GTG GAA CAA-3'(配列番号30)を使用し、鋳型として、プラスミドpSO21を使用して、PCRにより増幅した。温度サイクルは、最初に94℃-2分で反応混合物をインキュベーションした後、94℃-30秒、55℃-30秒及び72℃-45秒を35サイクル行い、最後に72℃-4分のインキュベーションを続けた。PCR産物を、0.8%アガロースゲルの電気泳動により分離し、535bpの産物を単離した。529bpのPCR産物を、SbfI及びMulIで制限酵素処理し、その断片を、pBH5aのSbfI-MulI断片とライゲーションして、プラスミドpBH5b(図15)を得た。pBH5bは、 S. cerevisiaeのScPDC1プロモーター及びScGAL10ターミネーターに動作可能に連結したG418耐性マーカーとともに、K. marxianus のPDC1のすぐ上流の1.2kbpのDNA断片及びK. marxianus のPDC1のすぐ下流の0.5kbpのDNA断片、そしてこれらのPDC1側面配列の間に存在するユニークな制限酵素認識部位SbfIを有する。
【0138】
(実施例3B)
K. marxianus のKmPDC1遺伝子座を標的として L. helveticus L-LDH遺伝子を組み込むためのpBH8のコンストラクション
L. helveticus 由来のL-乳酸脱水素酵素遺伝子をpBH5b(実施例3A)のSbfI部位にクローニングし、 L. helveticus L-LDH遺伝子を、 K. marxianus のKmPDC1遺伝子座に組み込み、外因性の K. marxianus のKmPDC1プロモーター及びターミネーターを使用して L. helveticus L-LDH遺伝子を発現できるベクターを作製した。
【0139】
L. helveticus L-LDH遺伝子を、プライマーとして、L-LDH5’:5'-ACA AAT CCT GCA GGA TGG CAA GAG AGG AAA AA-3'(配列番号31)及びL-LDH3’:5'-TAT CTA CCT GCA GGT CAT TGA CGA ACC TTA AC-3'(配列番号32)を使用し、鋳型として、プラスミドpPS9(実施例1D)を使用して、PCRにより増幅した。温度サイクルは、Failsafe PCR System (Epicentre, Madison, WI)を使用して、94℃-30秒、55℃-30秒及び72℃-1.5分を30サイクル行い、最後に72℃-4分のインキュベーションを続けた。971bpのPCR産物を、0.8%アガロースゲルの電気泳動により分離して単離した。このPCR産物をSbfIで制限酵素処理し、6844bpのSbfIで処理したpBH5b断片とライゲーションし、7824bpのpBH8(図16)を得た。pBH8は、 S. cerevisiaeのScPDC1プロモーター及びScGAL10ターミネーターに動作可能に連結したG418耐性遺伝子とともに、 K. marxianus KmPDC1プロモーター及びターミネーターに動作可能に連結した L. helveticus L-LDH遺伝子を有する。
【0140】
(実施例3C)
pBH8をKmPDC1遺伝子座に組み込むことによる K. marxianus CD606株のコンストラクション
野生型 K. marxianus を、pBH8プラスミド(実施例3B)で形質転換した。CD21のKmPDC1遺伝子座に、pBH8プラスミド全体が、pBH8の L. helveticus L-LDH遺伝子のすぐ上流のKmPDC1の側面DNAと、 K. marxianus 染色体のKmPDC1遺伝子のすぐ上流のDNAとの間で、組み込まれた。結果として得られた株CD606は、KmPDC1の野生型コピーとともに、KmPDC1遺伝子座に組み込まれた単一コピーのpBH8を有する。
【0141】
CD21を、250mLのバッフル型フラスコ中の100g/Lのグルコースを補充した50mLのYPD培地(10 g/L 酵母抽出物, 20 g/L ペプトン、20 g/L グルコース及び2%アガーを含む)に、OD600が0.1となるように植菌した。培養液を、30℃-250rpmで16時間培養した。最終OD600は、12となった。10mLの培養液から細胞を、遠心により回収し、エレクトロポレーションバッファー(10 mM Tris-Cl, 270 mM スクロース, 1 mM MgCl2, pH 7.5)で1回洗浄した。この細胞を、インキュベーションバッファー(YPD + 25 mM DTT, 20mM HEPES, pH 8.0)に再度懸濁し、30℃-250rpmで30分インキュベートした。この細胞を、遠心により回収し、エレクトロポレーションバッファーで1回洗浄し、1mLのエレクトロポレーションバッファーに再度懸濁した。そして、一部(400μL)の細胞を、0.4cmのエレクトロポレーションキュベットに移した。切断していない12μgのpBH8を、合計50μlとして、キュベットに添加し、細胞を、1.8kV、1000Ω、25μFの条件で、エレクトロポレーションした。細胞を、50mLスクリューキャップ付きファルコンチューブ中の1mLのYPDに移し、30℃-250rpmで4時間インキュベートした後、300μg/mLのG418を含むYPDプレートにプレーティングして選択した。形質転換株を、37℃で2日間生育させ、新しい選択プレートに、再度、ストリークした。
【0142】
pBH8の L. helveticus L-LDH遺伝子のすぐ上流のKmPDC1の側面DNAと、 K. marxianus 染色体のKmPDC1遺伝子のすぐ上流のDNAとの間の、KmPDC1遺伝子座へのpBH8の適切な組み込みは、プライマーPDC1Chrmosome5’:5'-AAG CAC CAA GGC CTT CAA CAG-3'(配列番号33)及びL-LDH3’:5'-TCG ATA TCG CTA AGG AAC GCG-3'(配列番号34)を使用して確認した。このプライマーは、KmPDC1遺伝子の上流の染色体DNAであって、pBH8に取り込まれた相同性部分の外側のDNAと、 L. helveticus L-LDH遺伝子との間で、2.7kbpの産物を増幅するように設計した。温度サイクルは、最初に94℃-2分で反応混合物をインキュベーションした後、94℃-30秒、55℃-30秒及び72℃-3分を35サイクル行い、最後に72℃-7分のインキュベーションを続けた。PCRで分析した10個のうち、4個の形質転換株が、予想される2.7kbpのPCR産物を産出した。
【0143】
KmPDC1遺伝子座を増幅するために設計したプライマーPDC15’:5'-CGC AAA GAA AAG CTC CAC ACC-3'(配列番号35)及びPDC13’:5'-CCC ATA CGC TTA TAA TCC CCC-3'(配列番号36)を使用したPCRは、2つの産物を生み出す。第1の産物は、 K. marxianus PDC1に対応する1.7kbpの断片であり、第2の産物は、 L. helveticus L-LDHに対応する1.0kbpの断片である。温度サイクルは、最初に94℃-2分で反応混合物をインキュベーションした後、94℃-30秒、55℃-30秒及び72℃-2分を35サイクル行い、最後に72℃-5分のインキュベーションを続けた。さらに単一コピーの組み込みを確認するために、G418耐性遺伝子をコードする領域をプローブとしたサザンブロット解析を行った。分析した3個の形質転換株のうち、1個が、pHB8の1コピーの組み込みと一致する適切なバンドパターンを示した。1コピーのpBH8がKmPDC1遺伝子座に位置していることを、PCR及びサザンブロット解析により確認した株を、CD606とした。
【0144】
これらの結果は、CD21のKmPDC1遺伝子座を標的とした形質転換されたpBH8DNAの組み込みが、KmPDC1プロモーター配列の間で起こっていたことを示す。
【0145】
(実施例3D)
CD606に由来する K. marxianus 株CD607及びCD608のコンストラクション
標的PDC1遺伝子の欠失を促すため、非選択培地で、CD606を、数回の増殖期間にわたり、増殖させた。 L. helveticus L-LDH遺伝子が、KmPDC1遺伝子と入れ替わった株を単離した。この株は、G418耐性マーカー遺伝子も失っており、G418存在下で生育阻害を受けることで確認される、G418感受性の表現型を示した。neoR遺伝子の欠失は、サザンブロット解析により確認された。さらに、この株は、嫌気性条件で生育することができなかった。
【0146】
CD606を、YPDアガープレート上にプレーティングし、37℃で一晩、約16時間インキュベートした。コロニーのスワッブ(swab)を、新しいYPDアガープレートに移した。この手順を、4回繰り返した。4ラウンドのYPDアガープレート上での非選択増殖に続いて、6つの250mLのバッフル型振とうフラスコ中の100g/Lのグルコース及び42g/LのCaCO3を補充した50mLのYPD培地に、4ラウンドの非選択YPDアガープレート増殖後のプレートから、OD600が0.1となるように植菌した。振とうフラスコを30℃、250rpmで、16時間インキュベートした。次に、前記培養液を、豆粒大のコロニーのスワッブの連続稀釈法によって、PBS(リン酸緩衝生理食塩水)で稀釈した。豆粒大のコロニーのスワッブを1mlのPBSに懸濁した初代懸濁液の、10-5、10-6及び10-7稀釈物を、YPDアガープレートに使用した。
【0147】
さらに、4ラウンドの非選択YPDアガープレート増殖後のコロニーのスワッブを、1mLのPBS(リン酸緩衝生理食塩水;137 mN NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4 pH 7.4)に懸濁し、引き続き希釈し、YPDアガープレート上に単一コロニーを形成するようにプレーティングした。単一コロニーをYPDアガー上にプレーティングした後、メーカーの指示に従って使用した嫌気性チャンバー(Becton Dickinson, "GasPak System"3つのガスパックを使用)に置いた。引き続き、37℃で2日間培養した後、嫌気性チャンバーを開き、嫌気性条件化で生育したコロニーに印をつけた。そのプレートは、もう一日37℃でインキュベートした。好気性条件で生育したが、嫌気性条件で生育しなかったコロニーを、3つにプレーティングしてスクリーニングした。採用したスクリーニングの条件は、好気性YPDプレート、嫌気性YPDプレート、および300μg/mLのG418が補充されたYPDプレートであった。2個の形質転換株が、G418感受性と嫌気性条件化での生育能力の欠落という望ましい表現型を示すことが判明し、これらを、CD607及びCD608とした。
【0148】
KmPDC1と L. helveticus L-LDHとの入れ替わりの確認は、プライマーPDC15’(配列番号35)及びPDC13’(配列番号36)を用いて行った。プライマーは、CD607及びCD608の染色体DNAを鋳型にして、PDC1遺伝子座を増幅するように設計した。温度サイクルは、最初に94℃-2分で反応混合物をインキュベーションした後、94℃-30秒、55℃-30秒及び72℃-2分を35サイクル行い、最後に72℃-5分のインキュベーションを続けた。どちらの株から得た染色体DNAも、 L. helveticus L-LDHに対応する単一の1.0kbpPCR産物を産出した。G418遺伝子とハイブリダイズするように設計されたG418遺伝子の全体のコード領域をプローブとしたサザンブロット解析は、前記PCR産物にG418遺伝子が存在しないことを示した。KmPDC1のコード領域のプローブを用いたサザンブロット解析では、バンドが現れなかった。このことは、KmPDC1のすぐ上流及び下流にハイブリダイズするように設計されたKmPDC1のコード配列プローブが存在しないことを示す。(使用されたプローブは、PDC1遺伝子のすぐ上流及び下流の染色体領域に相同性のあるDNA断片であった。プローブは、オリゴマー5’プローブ=CA110及びCA111;3’プローブ=CA112及びCA113を使用してPCRで増幅した。)KmPDC1がL. helveticus L-LDHとの入れ替ったことと一致するサイズのバンドが生じた。
【0149】
これらの結果は、KmPDC1遺伝子が、 S. cerevisiaeのScPDC1プロモーター及びScGAL10ターミネーターに動作可能に連結したG418耐性遺伝子を含むpBH8プラスミドのバックボーンとともに、CD606の染色体から失われ、その結果、KmPDC1遺伝子が、SbfI部位に隣接する L. helveticus L-LDH遺伝子と、ちょうど入れ替わった株が得られたことを示す。
【0150】
(実施例3E)
CaCO3緩衝複合培地におけるCD607によるL-乳酸の産生
CD607株を、YPDアガープレートから、OD600が0.1となるように植菌し、100g/Lのグルコース及び50g/LのCaCO3を補充した50mLのYPD培地を含む250mLのバッフル型振とうフラスコを、30℃-250rpmで、16時間インキュベートした。フラスコ内に残存グルコースが残っていること、及び、細胞が対数増殖期であることを確認した後、4g/L細胞乾燥重量当量を遠心により回収し、250mLのバッフル型振とうフラスコ中の100g/Lのグルコース及び50g/LのCaCO3を補充した50mLのYPD培地に再度懸濁した。この培養液を、30℃-70rpmで(48時間)培養した。様々な時間(0,8,24及び48時間の間隔)にサンプルを回収し、細胞をフィルターによりサンプルから取り除いた。培養上清を、HPLCにより、グルコース、乳酸塩、ピルビン酸塩及びエタノールについて、分析した。
【0151】
24時間後、CD607株は、59〜61g/Lのグルコースを消費し、58〜60g/Lの乳酸塩及び0.3g/Lのピルビン酸塩を産生した。48時間後、CD607株は、101〜104g/Lのグルコースを消費し、94〜99g/Lの乳酸塩及び0.4g/Lのピルビン酸塩を産生した。これらの乳酸塩の力価は、グルコースに対して90〜98%の乳酸塩の収率を示しており、よって、微好気性産生期における容積生産性は、2.1g/Lhrを超えた。48時間後、培養液中で産生された乳酸塩の濃度が高くなったため、不溶性のカルシウム乳酸塩の沈殿の形成(「ケーキ化」)がおこり、それ以上の解析が妨げられた。酵素を用いた乳酸解析から、前記乳酸塩は、鏡像異性体的に99%を越える純度のL-乳酸塩であることが示された。CD607株の培養液ではエタノールが検出されなかった。このことは、唯一のPCD1コピーが、L−LDH1コピーに置き換わったことを示す。
【0152】
対照的に、CD606株(PDC遺伝子及び L. helveticus L-LDH遺伝子を含む)は、同じ発酵条件において、124g/Lのグルコースを消費し、83g/Lの乳酸塩、20g/Lのエタノール及び0.2g/Lのピルビン酸塩を産生した。
【0153】
(実施例3F)
バッファー剤を補充しない複合培地における遊離L-乳酸の産生
CD607株を、YPDアガープレートから、OD600が0.1となるように植菌し、100g/Lのグルコースを補充した50mLのYPD培地を含む250mLのバッフル型振とうフラスコを、30℃-250rpmで、16時間インキュベートした。フラスコ内に残存グルコースが残っていること、及び、細胞が対数増殖期であることを確認した後、2g/L細胞乾燥重量当量を遠心により回収し、250mLのバッフル型振とうフラスコ中の追加の40g/Lのグルコースを補充した50mLのYPD培地に再度懸濁した。前記培養液を、30℃-70rpmで(72時間)培養した。様々な時間(産生開始から0,24,47及び72時間後)にサンプルを回収した。培養液サンプルを、グルコース、乳酸塩(総計及び遊離酸)、ピルビン酸塩、グリセロール、酢酸塩及びエタノールについて、HPLCにより分析した。pHは、産生の開始と終了の時点で測定した。
【0154】
72時間後、CD607株は、10.8g/Lのグルコースを消費し、9.1g/Lの乳酸塩、0.5g/Lの酢酸塩及び1.2g/Lのピルビン酸を産生した。これらの乳酸塩の力価は、グルコースに対して84%の乳酸の収率を示しており、よって、微好気性産生期における容積生産性は、0.1g/Lhrを超えた。酵素を用いた乳酸塩解析から、前記乳酸塩は、鏡像異性体的に99%を越える純度のL-乳酸塩であることが示された。CD607株の培養液ではエタノールが検出されなかった。さらに、グリセロールも検出されなかった。培養液のpHは、有機酸の産生により、産生期中に、6.50から3.05に減少した。HPLCによる遊離乳酸(プロトン化乳酸)を測定したところ、9.1g/Lの遊離乳酸であった。このことは、基本的には、CD607により産生される全ての乳酸が、そのプロトン化型であることを示す。
【0155】
以上の開示は、本発明の特定の具体的実施態様であって、これらの修正又は代替であって均等なものは、すべて、添付の特許請求の範囲に示される本発明の精神と範囲に含まれるものである。ここで引用した全ての参照文献は、その全体を本願に引用して援用する。
【図面の簡単な説明】
【0156】
【図1】図1は、pNC2プラスミドを描いたダイヤグラムである。
【図2】図2は、pNC4プラスミドを描いたダイヤグラムである。
【図2a】図2aは、遺伝子発現のための Saccharomyces cerevisiae ScPGK1プロモーター、マルチクローニングサイト及びScGAL10ターミネーターを含む1.235kbpのNotI処理断片の配列である。
【図2b】図2bは、 Saccharomyces cerevisiae PDC1プロモーター、ScGAL10ターミネーター及び前記ターミネーターとプロモーターと間のマルチクローニングサイトを含む1.3kbpのNotI処理断片の配列である。
【図3】図3は、pVR22プラスミドを描いたダイヤグラムである。
【図4】図4は、pVR29プラスミドを描いたダイヤグラムである。
【図5】図5は、pPS1プラスミド及びpNC14プラスミドに基づくpPS9のコンストラクションを描いたダイヤグラムである。
【図6】図6は、 L. helveticus L-LDH遺伝子及びG418耐性マーカーを含むpVR39プラスミドを描いたダイヤグラムである。
【図7】図7は、 L. helveticus のL-LDHをコードする遺伝子、 S. cerevisiase pTEF1プロモーター及びE. coli EM7プロモーターを含むpVR1プラスミドを描いたダイヤグラムである。
【図8】図8は、pSO18プラスミドを描いたダイヤグラムである。
【図9】図9は、pSO19プラスミドを描いたダイヤグラムである。
【図10】図10は、pSO20プラスミドを描いたダイヤグラムである。
【図11】図11は、 L. helveticus L-LDH遺伝子及びG418耐性マーカーを含むpVR41プラスミドを描いたダイヤグラムである。
【図12】図12は、pVR24プラスミドを描いたダイヤグラムである。
【図13】図13は、 B. megaterium L-LDH遺伝子及びG418耐性マーカーを含むpCA3プラスミドを描いたダイヤグラムである。
【図14】図14は、 B. megaterium L-LDH遺伝子及びハイグロマイシン耐性遺伝子を含むpVR38プラスミドを描いたダイヤグラムである。
【図15】図15aは、pBH5aのコンストラクションを描いたダイヤグラムである。図15bは、pVR29及びpBH5aに基づくpBH5bのコンストラクションを描いたダイヤグラムである。
【図16】図16は、pBH9プラスミドを描いたダイヤグラムである。
【図17】図17A及び17Bは、本発明の態様に従った、外因性LDH遺伝子の酵母染色体への標的組み込みに関する遺伝学的出来事の説明図である。
【技術分野】
【0001】
本発明は、酵母におけるL-乳酸の生産方法及びその材料に関する。
【背景技術】
【0002】
本出願は、2002年5月30日に出願された米国仮出願No.60/384,333に基づく優先権を主張する。
【0003】
本発明は、エネルギー省により付与された契約No.DE-FC-36-00GO10598に基づく米国政府の援助によりなされた。米国政府が、本発明について特定の権利を有する。
【0004】
乳酸は、化学処理及び合成、化粧品、医薬品、プラスチック、並びに食糧生産における利用を含む広い産業上の利用可能性を有する。乳酸を作るためのほとんどの工業規模の製法は、発酵法(fermentaion process)である。これらの発酵法においてさまざまな乳酸産生バクテリアが使用されている。
【0005】
最近の研究は、乳酸発酵法における組み換え酵母株の利用について行われている。組み換え酵母は、バクテリアの発酵に対して幾つかの有利性を提供できる可能性を秘めている。酵母の中には、より優れた高温耐性を示す株がある。これにより、より高温の発酵の可能性ができ、この高温発酵が、より速い発酵速度を生み出しうる。より優れた高温耐性は、発酵培地から汚染微生物を一掃することを容易とする。前記培地を、望ましい種が許容でき、望ましくない種が死滅する温度まで、単に加熱すればよいからである。ラクトバシリ(Lactobacilli)等の乳酸産生バクテリアは、効率よく生産するために、複雑な発酵培地を必要とする。発酵培地の複雑さは、原料費を増やし、乳酸を培地から分離することをより困難かつ高価にする。組み換え酵母の使用は、単純化された発酵培地を使用することによるコスト削減の可能性を提供する。
【0006】
加えて、酵母の中には、乳酸産生バクテリアの低いpH(reduced pH)条件により耐性を示す株がある。このことは、潜在的に、非常に重要な特性である。なぜなら、発酵培地のpHは、乳酸が生産されるとともに、自然に下がるからである。従来の製法では、前記培地を、例えば、水酸化カルシウムや炭酸カルシウム等の塩基を用いて、pH約5〜8にバッファーする必要がある。このことが、酸を中和して乳酸塩を形成させる。この乳酸塩は、引き続く工程において、望ましい酸の形で乳酸を回収するために、分離しなければならない。それゆえ、発酵培地のバッファリングの必要性は、結果として、追加される原材料(バッファー剤及び乳酸塩を分離するために通常使用される硫酸)、追加の処理工程(遊離酸の再生のため)、及び、廃棄物(ほとんどの場合、塩分離工程において発生する炭酸カルシウム)処理のための著しい追加費用をもたらす。これらの費用は、発酵が低いpHで実施できるならば、著しく削減できる。それゆえ、低いpHに耐えうる乳酸産生株の開発の成功が、非常に望ましい。
【0007】
Porro とその同僚は、外因性のLDH(乳酸脱水素酵素)遺伝子を S. cerevisiae、 K. lactic 、 T. delbrueckii 及び Z. bailii 由来の酵母細胞に組み込み、前記細胞本来のピルビン酸経路を破壊することにより、乳酸産生酵母を設計する試みをしてきた(非特許文献1〜3参照)。Porro は、乳酸を産生する組み換え酵母を作り出すことができた。しかし、その株は、商業上の製法として実施できるほどの十分な性能とは、到底いえるものではなかった。産業環境での使用にふさわしい株というには、その株は、高い乳酸収率(つまり、基質から乳酸への高い変換)及び高い生産性(つまり、基質から乳酸への迅速な代謝)をもたらすものでなければならない。その酵母は、高い力価の乳酸を含む培地に耐え得ることが好ましい。
【0008】
つい最近、Rajgarhia とその同僚が、Porro のものよりも高い収率及び生産性を示す組み換え酵母を作り出した(例えば、特許文献1〜3参照)。
【特許文献1】WO 00/71738
【特許文献2】WO 02/42471
【特許文献3】PCT/US02/16223
【非特許文献1】ポロら、「乳酸産生のために代謝的に改良した Saccharomyces cerevisiae 細胞」(Porro et al., "Development of metabolically engineered Saccharomyces cerevisiae cells for the production of lactic acid")、Biotechnol. Prog. 1995 May-Jun; 11(3): 294-8
【非特許文献2】ポロら、「乳酸の大規模生産のための改良酵母における代謝経路の置換」(Porro et al., "Replacement of a metabolic pathway for large-scale production of lactic acid from engineered yeasts")、App. Environ. Microbiol. 1999 Sep:65(9):4211-5
【非特許文献3】ビアンチら、「ピルビン酸利用を欠損し、異種由来LDH遺伝子で形質転換された Kluyveromyces lactis 株による効率的なホモ乳酸発酵」(Bianchi et al., "Efficient homolactic fermentation by Kluyveromyces lactis strains defective in pyruvate utilization and transformed with the heterologous LDH gene")、App. Environ. Microbiol. 2001 Dec; 67(12)5621-5.
【発明の開示】
【発明が解決しようとする課題】
【0009】
しかしながら、収率及び/又は生産性がさらにもっと改良された組み換え酵母を提供することが望ましい。特に、乳酸産生に有利な嫌気性及び/又は微好気性条件下で高い収率及び生産性で乳酸を産生する組み換え酵母株を提供することが望ましい。
【課題を解決するための手段】
【0010】
第1の態様として、本発明は、酵母細胞に外因性の乳酸脱水素酵素遺伝子を組み込む方法であって、前記組み込みに先んじて前記細胞は、そのゲノム中の遺伝子座に標的遺伝子を有し、下記(a)〜(d)の工程を含む方法である。(a)乳酸脱水素酵素(LDH)遺伝子、前記LDH遺伝子の上流(つまり、5’)側及び下流(つまり、3’)側の側面配列(flanking sequences)、並びに、少なくとも1つの選択マーカー遺伝子を含む組み換え核酸を用いて、前記細胞を形質転換し、前記LDH遺伝子を前記標的遺伝子の遺伝子座に隣接した前記細胞のゲノムに挿入し、(b)前記形質転換がされた細胞を選択物質存在下で培養することで、前記LDH遺伝子及び前記選択マーカー遺伝子を含む第1の形質転換細胞を得て、(c)前記第1の形質転換細胞を、非選択培地で培養することで、(d)前記第1の形質転換細胞から、前記LDH遺伝子を有するが前記選択マーカー遺伝子及び前記標的遺伝子が削除された第2の形質転換細胞を得る。酵母細胞において、前記標的遺伝子は、ピルビン酸デカルボキシラーゼ(PDC)、アルコール脱水素酵素(ADH)、オロチジン-5'-リン酸デカルボキシラーゼ(ura3)、及び3-イソプロピルリンゴ酸脱水素酵素(leu2)であることが、有利であり、好ましい。
【0011】
本発明の前記態様において、前記選択マーカー遺伝子及び標的遺伝子の欠失は、ある割合の前記第1形質転換細胞において自然発生的に起こる。その結果、挿入された前記LDH遺伝子は、前記標的遺伝子のプロモーター及びターミネーターと相同である機能的なプロモーター及びターミネーターと動作可能に連結する。
【0012】
第2の態様として、本発明は、酵母種細胞の形質転換用組み換え核酸であって、少なくとも1つの選択マーカー及びLDH遺伝子を含み、前記LDH遺伝子が、前記酵母種に対し外因性であって、前記LDH遺伝子の上流(5’)及び下流(3’)側面配列に連結しており、前記側面配列が、それぞれ、前記酵母種の野生型遺伝子の側面配列の上流及び下流に相同である、組み換え核酸である。
【0013】
第3の態様として、本発明は、酵母種の組み換え細胞であって、前記酵母種が、そのゲノム中の遺伝子座に野生型の標的遺伝子を含む組み換え細胞である。前記組み換え細胞は、そのゲノムの前記標的遺伝子の部位に組み込まれた外因性の乳酸脱水素酵素(LDH)遺伝子を有しており、そして、前記標的遺伝子は、欠失している。前記組み込まれたLDH遺伝子は、前記標的遺伝子のものと相同な機能的プロモーター及びターミネーターに動作可能に連結している。このような組み換え酵母細胞は、発酵法に使用した場合、高い生産性とともに、炭水化物基質から予想外に高い乳酸の収率を示す。前記組み換え細胞は、また、高い力価まで乳酸を産生できる。本発明は、あらゆる理論に限定されるものではないが、前記LDH遺伝子が、野生型遺伝子の遺伝子座に、本明細書で述べるように、前記野生型のプロモーター及びターミネーターを利用するように挿入されることで、細胞が、前記挿入されたLDH遺伝子の機能発現に、既存の遺伝子制御システムを利用できるのだと考えられる。このことは、前記細胞が嫌気性又は微好気性の発酵条件に直面する場合に、乳酸への代謝経路に強く有利に働くと考えられる。従って、第4の態様として、本発明は、炭水化物を乳酸に発酵する方法であって、結果として得られる前記細胞を、同細胞が発酵できる炭水化物を含む培地中、発酵条件下で培養することを含む方法である。
【0014】
第5の態様として、本発明は、 K. marxianus 種の細胞であって、複数の外因性の乳酸脱水素酵素が、そのゲノムに組み込まれ、それぞれが、機能的なプロモーター及びターミネーター配列の制御下にあり、さらに、前記 K. marxianus 細胞ゲノムが、機能的なピルビン酸デカルボキシラーゼ遺伝子を含む細胞である。驚くべきことに、低pHの発酵条件かであっても、この細胞により、驚くほど少ないエタノールの収率とともに、非常に優れた乳酸の生産性及び収率が得られた。前述のとおり、本発明は、あらゆる理論に限定されるものではないが、PDC経路を無傷のままにしておくことにより、細胞が、代謝過程のバランスを保つために自身の既存の代謝経路をうまく利用することができ、前記細胞の健康状態及び生命力が、総合的に向上するのだと考えられる。従って、第6の態様として、本発明は、炭水化物を乳酸へ発酵する方法であって、結果として得られる前記細胞を、同細胞が発酵できる炭水化物を含む培地中、発酵条件下で培養することを含む方法である。
【0015】
本発明の具体的な好ましい実施形態は、後に続く、特定の好ましい実施形態のより詳細な説明及び特許請求の範囲から明らかになる。
【発明を実施するための最良の形態】
【0016】
本発明の最初の2つの態様では、外因性のLDH遺伝子が、酵母細胞のゲノム中の標的野生型遺伝子の遺伝子座に組み込まれ、そして、前記標的野生型遺伝子は削除される。前記外因性LDH遺伝子は、前記標的遺伝子のプロモーター配列及びターミネーター配列と、それぞれ、相同なプロモーター配列及びターミネーター配列と動作可能に連結される。
【0017】
本発明において、遺伝子、プロモーター又はターミネーターは、(1)変更されていない細胞のゲノム中に見出されない場合、及び、(2)変更されていない細胞のゲノムに存在する遺伝物質に相同ではない場合、「外因性である(exogenous)」とみなす。ここで用いられる場合、遺伝子、ターミネーター又はプロモーターは、(その機能に影響しない自己から自己の変異を除き)酵母種の変更されていない細胞のゲノム中に見出される場合、その酵母種に対して「野生型(native)」である。
【0018】
遺伝子、プロモーター、ターミネーター又はその他の遺伝物質は、同一である場合、すなわち、ヌクレオチド配列において、その他の遺伝物質と、少なくとも、80%、85%、90%、91%、92%、93%、94%、95%、96%、97%、98%若しくは約99%の同一性を有している場合、又は、その機能を維持できるほど十分に類似している場合、その他の遺伝物質と「相同である(homologous)」とみなす。それゆえ、遺伝物質は、例えば、塩基対の点変異、欠失又は付加による相違があるとしても、これらの点変異、欠失又は付加が、前記遺伝物質の機能に影響を与えないならば、「相同である」とみなされる。側面配列の場合は、その配列が野生型遺伝子の側面配列と十分に類似しており、前記野生型遺伝子の側面配列との単一交差の発生に従事することができる場合、相同性が確立される。
【0019】
「同一性」の用語は、当該技術分野で知られるように、2以上のポリペプチド分子又は2以上の核酸分子の配列間で、これらの配列を比較して決定される関係を参照する。当該技術分野において、「同一性」は、また、場合に応じて、2以上の核酸若しくは2以上のアミノ酸配列の文字列間のマッチで決定される、核酸分子又はポリペプチド間の配列の類縁性の度合いを意味する。「同一性」は、特定の数学的モデル若しくはコンピュータープログラム(つまり、「アルゴリズム」)によるギャップアライメント(もしあれば)の2以上の配列のより小さなものの間の同一マッチのパーセントを評価する。
【0020】
「類似性」の用語は、当該技術分野において、関連する概念に関して使用されている。しかし、「同一性」とは対照的に、「類似性」は、同一マッチおよび同類置換マッチの両方を含む類縁性の指標を参照する。例えば、2つのポリペプチド配列が、10/20の同一アミノ酸を有している例において、残りのものが、すべて非同類置換である場合、同一%及び類似%は、ともに50%となる。同じ例において、5つの位置において同類置換がある場合、同一%は、50%のままであるが、類似%は、75%となる(15/20)。それゆえ、同類置換がある場合には、2つのポリペプチド間の類似%は、前記ポリペプチド間の同一%よりも高くなる。
【0021】
関連した核酸及びポリペプチドの同一性及び類似性は、公知の手段によって容易に計算できる。これらの方法としては、特に限定されないが、例えば、以下の文献に記載される。COMPUTATIONAL MOLECULAR BIOLOGY, (Lesk, A.M., ed.), 1988, Oxford University Press, New York; BIOCOMPUTING: INFORMATICS AND GENOME PROJECTS, (Smith, D.W., ed.), 1993, Academic Press, New York; COMPUTER ANALYSIS OF SEQUENCE DATA, Part 1, (Griffin, A.M., and Griffin, H.G., eds.), 1994, Humana Press, New Jersey; von Heinje, G., SEQUENCE ANALYSIS IN MOLECULAR BIOLOGY, 1987, Academic Press; SEQUENCE ANALYSIS PRIMER, (Gribskov, M. and Devereux, J., eds.), 1991, M. Stockton Press, New York; Carillo et al., 1988, SIAM J. Applied Math., 48:1073; and Durbin et al., 1998, BIOLOGICAL SEQUENCE ANALYSIS, Cambridge University Press.
同一性を決定するために好ましい方法は、テストされる配列間の最大のマッチを与えるように意図される。同一性を決定する方法は、公的に入手可能なコンピュータープログラムに記載されている。2つの配列間の同一性を決定するための好ましいコンピュータープログラム手段は、特に制限されないが、GCGプログラムパッケージを含む。前記GCGプログラムパッケージには、GAP(Devereux et al., 1984, Nucl. Acid. Res., 12:387; Genetics Computer Group, University of Wisconsin, Madison, WI)、BLASTP、BLASTN及びFASTA(Altschul et al., 1990, J. Mol. Biol., 215:403-410)が含まれる。BLASTXプログラムは、NCBI(National Center for Biotechnology Information)及びその他の情報源(BLAST Manual, Altschul et al. NCB/NLM/NIH Bethesda, MD 20894; Altschul et al., 1990,同上)から公的に入手得可能である。周知のSmith Watermanアルゴリズムも、同様に、同一性決定に使用される。
【0022】
2つのアミノ酸又はポリペプチド配列をアライメントするためのアライメントスキームには、2つの配列の短い領域のみのマッチングとなるものがある。そして、2つの全体配列間では有意な関係がないにも関わらず、この小さな領域においては、高い配列同一性を有する。従って、ある態様においては、選択されたアライメント手段(GAPプログラム)は、少なくとも50の連続した標的ポリペプチドのアミノ酸に及ぶアライメントをすることになる。またある態様においては、アライメントは、少なくとも60、70、80、90、100、110又は120の標的ポリペプチドのアミノ酸を含むことができる。ポリヌクレオチドがGAPを利用してアライメントされた場合、そのアライメントは、少なくとも100、150又は200ヌクレオチドに及び、それらは、連続することができる。
【0023】
例えば、コンピューターアルゴリズムGAP(Genetics Computer Group, University of Wisconsin, Madison, WI)を使用すると、配列同一%を決定したい2つのポリペプチドは、それぞれのアミノ酸の最適なマッチング(前記アルゴリズムにより決定される「マッチ範囲」)となるようにアライメントされる。ある態様においては、ギャップ開始ペナルティー(平均対角要素(diagonal)の3倍で計算される。ここで、前記「平均対角要素」とは、使用される比較行列の対角要素の平均値であり、前記「対角要素(diagonal)」とは、特定の比較行列により完全アミノ酸マッチにそれぞれ割り当てられるスコア若しくは数値である。)及びギャップ伸長ペナルティー(通常、ギャップ開始ペナルティーの1割)が、例えば、PAM250若しくはBLOSUM62等の比較行列とともに、前記アルゴリズムと併せて使用される。また、ある態様においては、標準比較行列(PAM250比較行列に関して、Dayhoff et al., 1978, Atlas of Protein Sequence and Structure, 5:345-352参照。BLOSUM62比較行列に関して、Henikoff et al., 1992, Proc. Natl. Acad. Sci USA, 89:10915-10919参照)もまた、前記アルゴリズムにより使用される。
【0024】
ある態様において、ポリペプチド配列の比較のためのパラメータは、以下のものを含む。
アルゴリズム:Needleman et al., 1970, J. Mol. Biol., 48:443-453
比較行列:BLOSUM62 Henikoff et al., 1992 同上
ギャップペナルティー:12
ギャップ長ペナルティー:4
類似性の閾値:0
GAPプログラムには、上記パラメータが有用である。ヌクレオチド配列に対しては、パラメータは、ギャップペナルティー50、及び、各ギャップにおける各シンボルごとにペナルティーが3となるギャップ長ペナルティー3を含む。また、ある態様においては、前述のパラメータは、GAPアルゴリズムを利用した(エンドギャップのペナルティー0を加えた)ポリペプチド比較のためのデフォルトパラメータである。
【0025】
「側面配列」は、遺伝子の上流(すなわち、5’)又は下流(すなわち、3’)の塩基対の配列である。側面配列は、前記遺伝子にすぐ隣接していてもよく、例えば、1〜1000塩基対、好ましくは、1〜100塩基対といった中間配列の塩基対により前記遺伝子と分離していてもよい。本発明の組み換え核酸に用いられる側面配列は、対応する標的遺伝子の側面配列と相同である。前記側面配列の長さは、対応する野生型遺伝子の側面配列との単一交差の発生に従事できるに足る長さである。有用な側面配列の長さは、約50〜約4000塩基対であって、好ましくは、約100〜約2000塩基対であって、とりわけ、約1200塩基対以下である。上流側面配列は、好ましくは、前記標的遺伝子のプロモーターを含み、下流側面配列は、好ましくは、前記標的遺伝子のターミネーター配列を含む。
【0026】
好ましい態様において、前記側面配列は、野生型の酵母のプロモーター及びターミネーター配列と、それぞれ、相同である前記プロモーター及びターミネーター配列を含む。最も好ましくは、組み換え核酸が、酵母ゲノムの染色体DNAにおける標的遺伝子座へ組み込まれる結果、そこにコードされる外因性LDH遺伝子が、前記側面配列を含む野生型遺伝子の発現制御配列の転写コントロール下におかれる。
【0027】
ここで用いられる場合、「プロモーター」の用語は、構造遺伝子の転写開始コドンの上流(すなわち、5’)(通常、約1〜1000bp、好ましくは、1〜500bp、とりわけ、1〜100bp以内)に位置する転写されない配列であって、構造遺伝子の転写開始をコントロールしたり、あるいはそうでなければ、前記遺伝子の転写をコントロールする配列を参照する。
【0028】
同様に、「ターミネーター」の用語は、構造遺伝子の転写ストップコドンの下流(すなわち、3’)(通常、約1〜1000bp、好ましくは、1〜500bp、とりわけ、1〜100bp以内)に位置する転写されない配列であって、構造遺伝子の転写終結をコントロールしたり、あるいはそうでなければ、前記遺伝子の転写をコントロールする配列を参照する。
【0029】
プロモーター又はターミネーターが、必要に応じて、酵母ゲノムに組み込まれた後、構造遺伝子の転写をコントロールするよう機能する場合、前記構造遺伝子(例えば、LDH遺伝子若しくは選択マーカー遺伝子)は、前記プロモーター又はターミネーターと「動作可能に連結」する。
【0030】
「形質転換」の用語は、ここで用いる場合、細胞における遺伝的特徴の変化を参照する。そして、細胞は、新たな核酸を含むように変更された場合、形質転換される。よって、細胞が、野生型の状態から遺伝的に変更された場合、細胞は、形質転換される。例えば、以下のトランスフェクションにおいて、形質転換DNAは、細胞の染色体に物理的に組み込まれることによって、細胞ゲノムDNAと組み換えを起こすことが好ましい。あるいは、少なくとも一時的に(すなわち、細胞形質転換から48〜96時間内に)、前記核酸は、複製されないエピソーム因子として一時的に維持されてもよく、又は、プラスミドとして独立に複製されてもよい。前記DNAが、染色体に組み込まれ、細胞の分裂とともに複製される場合、細胞は、安定して形質転換されたと見なされる。
【0031】
「トランスフェクション」の用語は、細胞による外部又は外因性のDNAの取込みを参照する。外因性DNAが、細胞膜の内側に導入された場合、前記細胞は、「トランスフェクション」される。多くのトランスフェクション技術が当該技術分野において周知である。例えば、Graham et al., 1973, Virology 52:456; Sambrook et al., 2001, MOLECULAR CLONING, A LABORATORY MANUAL, Cold Spring Harbor Laboratories; Davis et al., 1986, BASIC METHODS IN MOLECULAR BIOLOGY, Elsevier; Chu et al., 1981, Gene 13:197.等が、参照できる。これらの技術は、1つ以上の外因性DNA種を、適した宿主細胞に導入する場合に利用できる。
【0032】
本発明の第1の態様によれば、組み換え核酸は、側面配列に連結した外因性LDH遺伝子及び少なくとも1つの選択マーカーを含んで提供される。「組み換え核酸」の用語は、ここでは、宿主細胞にタンパク質をコードする情報を移動するために使用されるあらゆる分子(例えば、核酸断片、プラスミド又はウイルス等)を参照するために使用される。適切な側面配列は、以下のようにして得ることができる。まず、意図する組み込み部位(例えば、酵母細胞ゲノム中の標的遺伝子等)を同定し、その部位に隣接する配列を(任意の都合のよい手段、特に限定されないが、例えば、化学合成、組み換え遺伝学的技術若しくはインビトロ増幅等を含む手段により)得て、そして、これらの配列を、組み換え核酸中の望ましい位置に(例えば、5’及び3’の側面配列を、それぞれ、LDHをコードする遺伝子に共有結合させることで)取り込ませる。前記酵母細胞は、その後、前記組み換え核酸で、任意の適切な形質転換技術により、形質転換される。その結果、LDH遺伝子を含む組み換え核酸の少なくとも1つの断片が前記酵母細胞のゲノムに組み込まれた細胞が得られる。
【0033】
前記組み換え核酸は、1つ以上の選択マーカー遺伝子を含み、前記選択マーカー遺伝子は、それら自身のプロモーター及びターミネーター遺伝子による転写コントロール下にあることが、より好ましい。マーカー遺伝子のプロモーター及びターミネーターの配列は、前記標的遺伝子のプロモーター及びターミネーター配列と相同でないことが好ましい。前記選択マーカー遺伝子並びにそれぞれのプロモーター及びターミネーターは、上流側面配列-LDH遺伝子-下流側面配列の配列を中断しないことが好ましい。前記選択マーカー並びにそれぞれのプロモーター及びターミネーターは、ベクター上で、LDHプロモーター配列(もしあれば、さらなる側面配列)の上流(5’)に位置することが好ましい。
【0034】
本発明の組み換え核酸及び細胞の製造、及び、本発明にかかる方法の実践に有用なLDH遺伝子を有する酵母種としては、 Lactobacillus helveticus, Pediococcus acidolactici, Lactobacillus casei, Kluyveromyces thermotolerans, Torulaspora delbrueckii, Schizosaccharomyces pombii, Rhizopus oryzae 及び B. megaterium を含むことが好ましい。特に、これらの株の中でも、単離でき、本発明の組み換え核酸の製造に使用できる適したL-乳酸脱水素酵素を有する株が好ましい。2つの好ましいL-乳酸脱水素酵素遺伝子は、 L. helveticus 及び B. megaterium のL-乳酸脱水素酵素である。
【0035】
典型的な選択マーカー遺伝子は、(a)抗生物質又はその他の毒に対して耐性を付与するタンパク質をコードする遺伝子、例えば、宿主細胞に対する、ゼオシン( Streptoalloteichus hindustanus ble ブレオマイシン耐性遺伝子)、G418(Tn903由来のカナマイシン耐性遺伝子)、ハイグロマイシン( E. coli 由来のアミノグルコシド抗生物質耐性遺伝子)、アンピシリン、テトラサイクリン、若しくはカナマイシン等、(b)細胞の栄養要求性欠乏症を補完するタンパク質をコードする遺伝子、例えば、アミノ酸ロイシン欠乏症、又は(c)単純培地から得ることができない必須栄養素を補給するタンパク質をコードする遺伝子、例えば、ura3等である。好ましく選択できるマーカーは、特に制限されないが、例えば、ゼオシン耐性遺伝子、G418耐性遺伝子、及びハイグロマイシン耐性遺伝子である。
【0036】
また、本発明の組み換え核酸は、酵母細胞のゲノムに挿入されるために、前記分子が切断され、前記LDH遺伝子、そのプロモーター及び側面配列、マーカー遺伝子並びに付随するプロモーター及びターミネーター等を含む直線状の断片を形成できる制限酵素認識部位を1つ以上有してもよい。
【0037】
さらに、本発明の組み換え核酸は、バックボーン部を含んでもよい。前記バックボーン部は、オリジン、複製(酵母若しくはバクテリアで動作可能であり、有効量の核酸を製造できる)及びその他の有用な特徴部を有利に含むことができ、また、市販の酵母ベクターから都合よく得ることができる。
【0038】
酵母細胞は、本発明の組み換え核酸で形質転換される。細胞を形質転換する方法は、当該技術分野で周知であり、特に制限されないが、例えば、エレクトロポレーション、塩化カルシウム法、又は、酢酸リチウム法等を含む。形質転換に使用する本発明の組み換え核酸は、使用の前に特定の制限酵素で処理してもよく、処理しなくてもよい。LDH遺伝子の側面配列は、標的遺伝子の側面配列と高い相同性を示すので、上流側面配列/LDH遺伝子/下流側面配列の断片の挿入は、標的遺伝子の遺伝子座で起こる傾向がある。
【0039】
標的遺伝子は、LDH遺伝子と入れ替わることを希望する任意の遺伝子である。好ましい標的遺伝子は、ピルビン酸デカルボキシラーゼ遺伝子である。なぜなら、この遺伝子を取り替えることで、エタノールを産生する競合経路を破壊することができるからである。加えて、ピルビン酸遺伝子は、酵母種において活性化する傾向にある。したがって、PCDプロモーター及びターミネーターのコントロール下のゲノム中へのLDH遺伝子の挿入は、LDHをよく発現する変異体を産生する傾向がある。さらなる好ましい標的遺伝子は、ADH、Leu2及びUra3である。
【0040】
酵母細胞を本発明の組み換え核酸で形質転換し、選択マーカーに対する選択培地で生育することにより組み換え細胞を選択し、さらに、選択した形質転換株を非選択条件で生育させると、その結果、外因性LDH遺伝子を有し、前記酵母染色体の標的遺伝子の遺伝子座に組み込まれた組み換え核酸を含む組み換え酵母細胞が得られる。本発明の方法に従い得られた場合、これらの細胞は、前記標的遺伝子が欠失し、組み込まれたLDH遺伝子は、前記標的遺伝子の遺伝子座に動作可能に連結し、前記標的遺伝子の発現コントロール配列(例えば、プロモーター及びターミネーター配列)の転写コントロール下に置かれるように、挿入される。標的遺伝子が、PDC遺伝子の場合、PDC欠失が起きた細胞は、嫌気性条件下では、生育がよくない。よって、同定され、選択された細胞のコロニーは、これらを嫌気性条件下にさらすことで、選択できる。生育しないコロニーが、PDC欠失が起きたものであると同定できる。同様に、任意のその他の標的遺伝子を標的とした組み込みは、各標的遺伝子の欠失に伴う表現型により同定することができる。
【0041】
この結果をもたらす遺伝学的な出来事の説明図を、図17A及びBに示す。図17Bに示すとおり、選択マーカー遺伝子は、自然発生的に、標的遺伝子の欠失とともに、削除できる。選択マーカー遺伝子が欠失した形質転換株の選択をすることが好ましい。なぜなら、遺伝子組み換え操作された酵母細胞は、例えば、薬剤耐性の向上等の特定の性質を有し、望ましくない環境リスクを生じるおそれがあるからである。
【0042】
結果として得られる酵母細胞は、標的遺伝子を失い、そして、酵母細胞のゲノムの前記標的遺伝子の遺伝子座に組み込まれた外因性LDH遺伝子を有する。前記LDH遺伝子は、前記標的遺伝子のプロモーター及びターミネーター配列と相同なプロモーター配列及びターミネーター配列の転写コントロール下に置かれる。
【0043】
図17Bに示すとおり、前記LDHプロモーター及びターミネーター配列は、細胞の形質転換に使用された組み換え核酸に含まれる側面配列に存在したものであってもよく、又は、細胞ゲノムの組み込み部位にもともと存在したものであってもよい。同様に、標的遺伝子のターミネーターも、組み込まれたベクターに存在したターミネーター配列が欠失することにより、維持されることも可能である。
【0044】
本発明の第1及び第2の態様における形質転換に適した酵母細胞は、 Candida 、Saccharomyces 、Kluyveromyces 、 Pichia 及び Hansenula 属由来のものを含む。ピルビン酸を蓄積しない、すなわち、天然に、ピルビン酸をエタノール又はその他の代謝産物へ代謝する酵母細胞が好ましい。 Candida 及び Kluyveromyces 属由来の細胞が特に好ましく、とりわけ好ましくは、 C. sonorensis 及び K. marxianus の細胞である。
【0045】
本発明の第5の態様における細胞は、 K. marxianus 種の組み換え細胞である。これらの細胞は、一般的に、いくらか異なる形質転換方法により調製される。ただし、標的遺伝子が、PDC遺伝子以外であれば、上述の手段が適している。この場合、組み換え核酸は、酵母細胞の野生型のPDC遺伝子の遺伝子座を標的として挿入するようにはデザインされない。それゆえ、細胞の形質転換に使用されるベクターは、一般的に、前記酵母細胞の野生型PDC遺伝子の側面配列と高い相同性を示す側面配列を含まない。
【0046】
第5の態様における細胞は、一般的に、1回につきLDH遺伝子を1コピー導入する形質転換を、2回以上することにより調製される。しかしながら、複数のLDH遺伝子を含む組み換え核酸をコンストラクトし、単一ステップで複数のLDH遺伝子を挿入することもできる。そして、細胞の形質転換に使用するベクターは、それぞれプロモーター及びターミネーターに動作可能に連結した1つ以上のLDH遺伝子を含む。既に述べたとおり、組み換え核酸は、また、様々なマーカー遺伝子を有してよい。これらの遺伝子は、それぞれ、酵母細胞の野生型のPDCプロモーター及びターミネーター配列ではないプロモーター及びターミネーター配列のコントロール下にあることが好ましい。
【0047】
適したLDH遺伝子としては、本発明の最初の3態様に関して上述したものを含む。 L. helveticus L-LDH及び B. megaterium L-LDH遺伝子が好ましい。細胞は、複数のLDH遺伝子、すなわち、少なくとも2以上の前記遺伝子、好ましくは、約2〜10の前記遺伝子、さらに好ましくは、約2〜5の前記遺伝子で形質転換される。挿入されるLDH遺伝子は、全て同じであってもよく、又は、2以上の異なる型のLDH遺伝子(すなわち、2以上の種から得た遺伝子)を含んでもよい。2コピー以上の L. helveticus L-LDH遺伝子を含む組み換え酵母細胞、2コピー以上の B. megaterium L-LDH遺伝子を含む組み換え酵母細胞、並びに、少なくとも1コピー以上の L. helveticus 及び B. megaterium L-LDH遺伝子を含む組み換え酵母細胞が好ましい。
【0048】
第5の態様における細胞の形成の際に、LDHに使用する適したプロモーターとしては、酵母遺伝子であるホスホグリセリン酸キナーゼ(PGK)、グリセルアルデヒド3リン酸デヒドロゲナーゼ(TDH)、ピルビン酸デカルボキシラーゼ(PDC)(K. marxianus 以外の種由来のもの)、トリオースリン酸イソメラーゼ(TPI)、転写エンハンサー因子(TEF)、プリン-シトシンパーミアーゼ(PCPL3)及びアルコールデヒドロゲナーゼ(ADH)のプロモーターを含む。本発明の好ましいプロモーターとしては、 S. cerevisiae PGKプロモーター及び S. cerevisiae PDC1プロモーターをふくむ。
【0049】
適したターミネーターとしては 、S. cerevisiae 又はその他の種由来のGAL10及びCYC-1ターミネーターを含む。
【0050】
好ましい選択マーカーは、本発明の最初の3態様に関して記載したとおりである。
【0051】
形質転換を複数のステップで行う場合、細胞は、まず、少なくとも1つのLDH遺伝子を含む第1のベクターで形質転換される。首尾よく形質転換された細胞が、その後、通常、選択マーカーの存在による特性を利用して、選択される。首尾よく得られた形質転換株は、その後、最終的に、望ましい数及び型の外因性LDH遺伝子を有する細胞となるまで、1以上のさらなる形質転換がされる。
【0052】
本発明の組み換え酵母細胞は、発酵法において、炭水化物から乳酸を生産することに有用である。発酵は、任意の使用しやすい発酵法を用いることができる。一般的には、細胞は、細胞がピルビン酸に代謝できる炭水化物を含む発酵培地とともに使用され、発酵が起こる条件下に置かれる。前記発酵培地は、また、前記細胞の生命力を高める栄養素(例えば、窒素源、リン源、硫黄源、微量元素源等)を含む。
【0053】
使用できる特定の炭水化物は、特定の宿主、及び、その宿主が任意の特定の炭水化物をピルビン酸に代謝できるように遺伝子組み換えされたか否かに依存する。グルコース及びフルクトース等のヘキソースの糖、マルトース、イソマルトース、マルトトリオース、スターチ及びスクロース等のグルコースのオリゴマー、マルトデキストリン並びにキシロース(ペントースの糖)が好ましい。また、より好ましくない炭水化物としては、ガラクトース、マンノース及びアラビノースが挙げられる。
【0054】
発酵の間の温度は、例えば、ほぼ室温以上であって、好ましくは、約30℃以上であり、より好ましくは、約35℃〜約55℃であり、より好ましくは、約50℃以下であって、さらにより好ましくは、約45℃以下である。最高温度は、幾分は、その特定の宿主細胞に依存する。宿主細胞が、例えば、 K. marxianus である場合、(本発明の任意の態様の)組み換え細胞は、比較的高い温度を許容できる(例えば、約40℃〜50℃、とりわけ45℃以下)。もう1つの好ましい宿主である C. sonorensis は、約40℃の温度まで許容できる。この温度範囲は、生産性が著しく低下することなく、より高い温度で発酵できる(それにより、冷却コストを削減できる)可能性を提供する。より高い温度に対する優れた耐性により提供されるその他の利点は、望ましくない微生物に発酵が汚染された場合、多くの場合、発酵培地を40℃以上、とりわけ45℃以上に加熱することにより、本発明の組み換え細胞に大きな害を及ぼすことなく、前記望ましくない微生物を選択的に死滅させることができることである。
【0055】
発酵の間、発酵培地中の細胞濃度は、一般的には、発酵培地1リットルあたりの細胞の乾燥重量で、約1〜150gの範囲であって、好ましくは、約3〜10gの範囲であり、より好ましくは、3〜6gの範囲である。
【0056】
発酵の産生期間は、厳密に嫌気性よりはむしろ微好気性で実施したほうが好ましい場合がある。最適な曝気条件は、各微生物について、特有の酸素摂取速度(OUR)を測定し、これらの速度と、収率、基質消費速度及び望ましい発酵生成物が産生される速度とを関連付けることにより、確立できる。多くの場合、収率及び速度は、特定のOURの範囲内で最適化できる。PDCが破壊された酵母では、最適なOUR値は、約0.8〜約3.5mmolO2/細胞乾燥重量/hrの範囲内である。OURは、発酵中の細胞により酸素が消費される速度を参照し、例えば、mmolO2/細胞乾燥重量/hrのように、単位時間、細胞の乾燥重量あたりの酸素の単位(mmol又はg)で表される。酸素の消費は、発酵中に送り込んだ酸素及び発酵から回収された酸素を測定することで、便利よく決定できる。OURの測定は、特定の生物に最適な範囲にOURを維持するために、発酵の産生期間の曝気条件(とりわけ、ガス導入速度、アジテーション、曝気ガス中の酸素の割合等)をコントロールするための基礎として使用できる。培養液に溶解した酸素濃度は、同時に、飽和の1%未満、とりわけ、10mmolO2/L未満に維持される。特に好ましい製法においては、発酵の増殖期の培養を、培養液に溶解した酸素濃度を、飽和の1%未満、とりわけ、10mmolO2/L未満に減らして、産生期の開始(すなわち、増殖期の好気性条件から産生期の微好気性条件への切り替え)前の一定時間、例えば、約15分〜90分行う。
【0057】
乳酸が産生されると、発酵培地のpHは、形成される酸の全部又は一部を中和するために塩基が添加されない限り、下がる傾向にある。発酵法の一態様において、中和剤、例えば、炭酸カルシウム、水酸化カルシウム、炭酸ナトリウム、水酸化ナトリウム、アンモニア、水酸化アンモニウム等が発酵培養液に添加され、pHが望ましい範囲、一般的には、約5.0〜約8.0、とりわけ、約5.5〜約7.5に維持される。そのような塩が添加された場合、対応する乳酸塩が形成される。それゆえ、乳酸の回収は、遊離酸の再生を含む。これは、一般的に、細胞を取り除き、例えば、硫酸等の強酸で発酵培地を酸性化することにより行われる。塩副産物が形成されるが(カルシウム塩が中和剤で、硫酸が酸性化剤の場合、石膏)、これらは、乳酸から分離される。その後、乳酸は、例えば液-液抽出、蒸留、吸着等の技術により回収される。これらは、以下の文献に開示される。T.B. Vickroy, Vol. 3, Chapter 38 of Comprehensive Biotechnology, (ed. M. Moo-Young), Pergamon, Oxford, 1985; R. Datta, et al., FEMS Microbiol. Rev., 1995; 16:221-231; U.S. Patent Nos. 4,275,234, 4,771,001, 5,132,456, 5,420,304, 5,510,526, 5,641,406, and 5,831,122, 及び国際出願番号 WO 93/00440。
【0058】
あるいは、発酵のpHは、乳酸が細胞により産生されるにつれて下がってもよい。この場合、発酵培養液のpHは、乳酸の産生により、約1.5〜約5.0の範囲、好ましくは、約1.5〜約4.2、より好ましくは、約1.5〜約3.86(乳酸のpKaである)、とりわけ、約2.0以上3.86未満の範囲になってもよい。このような方法で発酵を行うことは、許容できる生産性と収率を達成できるのであれば、いくつかの利点がある。中和剤のコストが、削減又は排除される。発酵pHが(発酵終了時に)乳酸のpKaを下回れば、乳酸は、主に酸の形態で存在することとなる。これは、酸性化ステップを排除することができ、余計な製法ステップ、酸性化コスト、及び塩副産物の廃棄コストを節約できる。よって、とりわけ好ましい製法は、発酵培養液のpHが、3.86を下回るまで発酵を続けることを含む。乳酸は、例えば、WO 99/19290に開示される手段を用いて結果として得られる発酵培養液から分離できる。
【0059】
低pH環境に抵抗できる細胞の能力は、望ましくない微生物による汚染を排除するもう一つのメカニズムを提供する。本発明の細胞を含む培養液を、十分な時間、低pH条件、例えば、約1.5〜4.2、好ましくは、約2.0〜3.86に置くと、酸耐性ではないあらゆる汚染微生物を死滅させることとなる。
【0060】
商業的に有用なため、本発明の組み換え酵母は、いくつかの特徴を有する。前記酵母は、炭水化物のかなりの部分を乳酸に変換する(つまり、生成物の高収率を実現する)。酵母細胞は、高い比生産性(specific productivity)を示す。つまり、単位時間、細胞重量あたりの高い乳酸の生産量を実現する。酵母細胞は、約5.0を下回るpH、好ましくは、約1.5〜4.2、とりわけ、2.0〜3.86の発酵に耐え、これらの条件下でも優れた収率と生産性を発揮することが好ましい。前記細胞は、また、高い乳酸濃度、pH5.0〜8.0、好ましくは、pH1.5〜5.0、より好ましくは、1.5〜4.2、とりわけ、2.0〜3.86に耐え得ることが好ましい。この最後の特性により、開始時の炭水化物濃度が高い場合の発酵法が可能となる。
【0061】
概して、本発明の組み換え細胞を利用した発酵法は。次の特色のいくつか又は全てを提供することが望ましい。
A.乳酸の収率が、炭水化物のグラムあたり、少なくとも30、好ましくは、少なくとも40、より好ましくは、少なくとも60、さらにより好ましくは、少なくとも75g。理論的な望ましい収率は、100%であるが、実際は、約95%の収率が限度である。
B.乳酸の比生産性が、時間、細胞グラムあたり、少なくとも0.1、好ましくは、少なくとも0.3、より好ましくは、少なくとも約0.4、とりわけ、少なくとも0.5g。比生産性は、高ければ高いほど望ましい。
C.力価(つまり、乳酸の最大濃度)が、発酵培地1リッターあたり、少なくとも15g、好ましくは、少なくとも20g、より好ましくは、少なくとも40g、さらにより好ましくは、少なくとも80g、150g以下、好ましくは120g以下。培養培地の温度は、容易に達成できる力価の最大範囲にいくらかの影響を及ぼす。なぜなら、高濃度の乳酸溶液(つまり、約150g/L以上)は、約35℃未満の温度では、非常に粘性を帯びやすいか又はゲル化しやすいからである。高い発酵温度、例えば、約35〜50℃を用いれば、ゲル化や過度の粘性の集積をすることなく、高い力価が可能となる。
【0062】
本発明の第3の態様の細胞は、グルコースの中性発酵(pH5.0〜8.0)に使用すると、収率:85〜95%、比生産性:0.5〜2g/g/hr、力価:80〜120g/Lを提供することがわかっている。pHが約2.8〜3.0に低下する低pH発酵では、本発明の第3の態様の細胞は、収率:75〜81%、比生産性:0.1〜0.4g/g/hr、力価:14〜40g/Lを提供することがわかっている。全ての場合において、これらの結果は、発酵条件の最適化をすることなく得ることができた。
【0063】
本発明の第5の態様の細胞(複数コピーの外因性LDH遺伝子を含み、完全な野生型PDC遺伝子を含む)は、グルコースの中性発酵(pH5.0〜8.0)に使用すると、収率:40%以上、比生産性:0.4〜0.9g/g/hr、力価:40〜75g/Lを提供することがわかっている。グルコースの低pH発酵(最終pH2.8〜3.0)では、前記第5の態様の細胞は、収率:30%以上、比生産性:0.3〜0.5g/g/hr、力価:20〜35g/Lを提供する。このように、前記第5の態様の細胞は、低pH条件下でよく発酵する能力を保持した。前述のとおり、これらの結果は、発酵条件の最適化をすることなく得ることができた。
【0064】
さらに、本発明の発酵法は、高い容積生産性を達成することが好ましい。「容積生産性」は、単位時間、発酵培地の単位容積あたりの生産される生成物量として表され、一般的に、生成物のグラム/培地のリットル/時間(hr)で表される。望ましい容積生産性としては、少なくとも1.5g/L/hrであって、好ましくは、2.0g/L/hrであり、より好ましくは、2.5g/L/hrである。発酵培地リットルあたり3〜6g以下の細胞という好ましい細胞濃度において、最大生産性は、約5.0g/L/hr、より一般的には、約4.0g/L/hr以下となる傾向がある。培地pH、温度、若しくは両方ともが、前節に記載した範囲内である場合に、これらの容積生産性が達成できるように発酵を実施することが、非常に好ましい。
【0065】
本発明により生産された乳酸は、2つの乳酸分子の環状無水物であるラクチドの製造に有用である。乳酸の立体異性体に応じて、ラクチドは、D-ラクチド(2つのD-乳酸分子から作られる)、L-ラクチド(2つのL-乳酸分子から作られる)、または、D-L-ラクチド(各1分子のD-乳酸分子及びL-乳酸分子から作られる)であってもよい。乳酸からラクチドを製造する便利な方法は、USP 5,142,023 to Gruber et al.に開示される重合/解重合法による方法である。
【0066】
次に、ラクチドは、ポリ乳酸ポリマー(PLA)及びコポリマーの製造のためのモノマーとして特に有用である。これらのポリマーを調製する方法は、同様に、USP 5,142,023 to Gruber et al.に開示される。好ましいPLA製品は、USP 5,142,023 to Gruber et al.に開示されるように、安定な溶融ポリマー(melt-stable polymer)である。PLAは、半結晶若しくはアモルファスであってもよい。
【0067】
以下の実施例は、本発明の特定の実施態様を説明するのに役立つものであって、本発明の範囲や精神を限定するものでない。
【実施例1】
【0068】
(実施例1A)
S. cerevisiae ScPGK1プロモーター型発現ベクターpNC2のコンストラクションと、 S. cerevisiae PDC1プロモーター型発現ベクターpNC4のコンストラクション
発現ベクターpNC2(図1)は、 S. cerevisiae ScPGK1とpGEM5Z(+)バックボーンベクター(Promega、Wisconsin)上の S. cerevisiae GAL10ターミネーターとを組み合して作られた。 S. cerevisiae ScPGK1と S. cerevisiae GAL10ターミネーターは、前記酵母プロモーターとターミネーターの間に特定の遺伝子を挿入して発現させるための、XbaI、EcoRI及びBamHIの制限酵素認識部位を有するポリリンカー領域により分離されている。
【0069】
使用した S. cerevisiae ScPGK1プロモーターは、以下の配列(配列番号1)を有する。
5'-GCGGCCGCGG ATCGCTCTTC CGCTATCGAT TAATTTTTTT TTCTTTCCTC TTTTTATTAA CCTTAATTTT TATTTTAGAT TCCTGACTTC AACTCAAGAC GCACAGATAT TATAACATCT GCACAATAGG CATTTGCAAG AATTACTCGT GAGTAAGGAA AGAGTGAGGA ACTATCGCAT ACCTGCATTT AAAGATGCCG ATTTGGGCGC GAATCCTTTA TTTTGGCTTC ACCCTCATAC TATTATCAGG GCCAGAAAAA GGAAGTGTTT CCCTCCTTCT TGAATTGATG TTACCCTCAT AAAGCACGTG GCCTCTTATC GAGAAAGAAA TTACCGTCGC TCGTGATTTG TTTGCAAAAA GAACAAAACT GAAAAAACCC AGACACGCTC GACTTCCTGT CTTCCTATTG ATTGCAGCTT CCAATTTCGT CACACAACAA GGTCCTAGCG ACGGCTCACA GGTTTTGTAA CAAGCAATCG AAGGTTCTGG AATGGCGGGA AAGGGTTTAG TACCACATGC TATGATGCCC ACTGTGATCT CCAGAGCAAA GTTCGTTCGA TCGTACTGTT ACTCTCTCTC TTTCAAACAG AATTGTCCGA ATCGTGTGAC AACAACAGCC TGTTCTCACA CACTCTTTTC TTCTAACCAA GGGGGTGGTT TAGTTTAGTA GAACCTCGTG AAACTTACAT TTACATATAT ATAAACTTGC ATAAATTGGT CAATGCAAGA AATACATATT TGGTCTTTTC TAATTCGTAG TTTTTCAAGT TCTTAGATGC TTTCTTTTTC TCTTTTTTAC AGATCATCAA GGAAGTAATT ATCTACTTTT TACAACAAAT CTAGAATT-3'
この配列は、特許プラスミドであるpBFY004の制限酵素処理断片として得ることができた。あるいは、鋳型として S. cerevisiae 染色体DNAを使用し、配列番号1に基づき設計したプライマーを使用したPCR増幅により得ることができる。
【0070】
使用した S. cerevisiae GAL10ターミネーターは、以下の配列(配列番号2)を有する。
5'-GTAGATACAT TGATGCTATC AATCCAGAGA ACTGGAAAGA TTGTGTAGCC TTGAAAAACG GTGAAACTTA CGGGTCCAAG ATTGTCTACA GATTTTCCTG ATTTGCCAGC TTACTATCCT TCTTGAAAAT ATGCACTCTA TATCTTTTAG TTCTTAATTG CAACACATAG ATTTGCTGTA TAACGAATTT TATGCTATTT TTTAAATTTG GAGTTCAGTG ATAAAAGTGT CACAGCGAAT TTCCTCACAT GTAGGGACCG AATTGTTTAC AAGTTCTCTG TACCACCATG GAGACATCAA AAATTGAAAA TCTATGGAAA GATATGGACG GTAGCAACAA GAATATAGCA CGAGCCGCGG ATTTATTTCG TTACGC-3'
この配列は、特許プラスミドであるpBFY004の制限酵素処理断片として得ることができた。あるいは、鋳型としてS. cerevisiae 染色体DNAを使用し、配列番号2に基づき設計したプライマーを使用したPCR増幅により得ることができる。
【0071】
S. cerevisiae PDC1プロモーター及びScGAL10ターミネーター型発現カセットを含むベクターpNC4をコンストラクトし、一般的な発現ベクターとして使用した。pNC4ベクターを図2に示す。
【0072】
pNC4のベクターバックボーンは、pGEM5Z(Promega社、Madison、WI)である。 S. cerevisiae PDC1プロモーターは、プライマーとして、PSPDCS1(5'-CCA TCG ATA ACA AGC TCA TGC AAA GAG-3':配列番号3)及びPSPDCAS2(5'-GCT CTA GAT TTG ACT GTG TTA TTT TGCG-3':配列番号4)を使用し、鋳型として、 S. cerevisiae GY5098株(ATCC 4005098)の染色体DNAを使用し、PCRにより増幅した。温度サイクルは、PfuTurboDNAポリメラーゼ(Stratagene)を使用して、94℃-1分、56℃-1分、72℃-1分を30サイクル行い、最後に72℃-7分間のインキュベーションを続けた。
【0073】
S. cerevisiae GAL10ターミネーターは、上述のようにして得られた。図2A(配列番号37)及び図2b(配列番号38)は、それぞれ、ScPGK1プロモーター、ScGAL10ターミネーター及びマルチクローニングサイトを含む断片、並びに、ScPDC1プロモーター、ScGAL10ターミネーター及びマルチクローニングサイトを含む断片を示す。
【0074】
(実施例1B)
S. cerevisiae ScPGK1プロモーター及びScGAL10ターミネーターに動作可能に連結したG418耐性遺伝子を有するpVR22ベクターのコンストラクション
G418耐性マーカー(バクテリアneoR遺伝子)は、クローニングされ、S. cerevisiae ScPGK1プロモーターの転写コントロール下に置かれた。このようにコンストラクションされたベクターをpVR22とした(図3)。G418耐性遺伝子は、Pfuポリメラーゼ(Stratagene、Madison、WI)を使用し、プライマーとして、5'-GCT CTA GAT GAG CCA TAT TCA ACG GGA AAC(5'G断片:配列番号5)及び5'-ATG GAT CCT TAG AAA AAC TCA TCG AGC ATC(3'G断片:配列番号6)を使用し、鋳型として、プラスミドpPIC9K(Invitrogen、Carlsbad、CA)を使用し、PCRにより増幅した。温度サイクルは、95℃-30秒、49℃-30秒、72℃-2分を35サイクル行い、最後に72℃-10分のインキュベーションを続けた。PCR産物を、BamHI及びXbaIで制限酵素処理し、821bp断片を単離し、pNC2(実施例1A)の4303bpのBamHI-XbaI断片とライゲーションした。その結果得られたプラスミド(pVR22;図3)は、G418耐性遺伝子に動作可能に連結したScPGK1プロモーター及びScGAL10ターミネーターを有する。
【0075】
(実施例1C)
S. cerevisiae PDC1プロモーター及びScGAL10ターミネーターに機能的に連結したG418耐性遺伝子を有するpVR29プラスミドのコンストラクション(pVR29)
G418耐性マーカー(pVR22;実施例1B)をpNC4(実施例1A)にクローニングしてコンストラクションしたものをpVR29(図4)とした。 S. cerevisiae ScPGK1と S. cerevisiae GAL10ターミネーターとを、G418耐性遺伝子を発現するために使用した。G418耐性遺伝子は、Pfuポリメラーゼ(Stratagene、Madison、WI)を使用し、プライマーとして、5'-GCT CTA GAT GAG CCA TAT TCA ACG GGA AAC(5'G断片:配列番号5)及び5'-ATG GAT CCT TAG AAA AAC TCA TCG AGC ATC(3'G断片:配列番号6)を使用し、鋳型として、プラスミドpVR22(実施例1B)を使用し、PCRにより増幅した。あるいは、鋳型として、プラスミドpPIC9K(Invitrogen、Carlsbad、CA)も使用できる。温度サイクルは、最初に95℃-5分で反応混合物をインキュベートした後、95℃-30秒、49℃-30秒、72℃-2分を35サイクル行い、最後に72℃-10分のインキュベーションを続けた。PCR産物を、BamHI及びXbaIで制限酵素処理し、821bp断片を単離し、pNC4の4303bpのBamHI-XbaI断片とライゲーションした。その結果得られたプラスミドpVR29(図4)は、G418耐性遺伝子に動作可能に連結したPCD1プロモーター及びScGAL10ターミネーターを有する。
【0076】
(実施例1D)
プラスミドpPS1(ハイグロマイシン耐性カセット)、pNC14(Lh-L-LDH発現カセット)及びpPS9(ハイグロマイシンを選択マーカーとして使用し、L-乳酸産生のためにゲノムにLh-L-LDHを組み込むためのベクター)
形質転換した酵母細胞にハイグロマイシン耐性を与え、それにより、乳酸合成に有用なタンパク質をコードする組み換え核酸コンストラクトを含む酵母細胞形質転換株を選択可能とする組み換え核酸を調製した。ベクターpPS9(図5)は、ハイグロマイシン耐性選択による L. helveticus L-LDH遺伝子の酵母ゲノムへの組み込みに使用できる。
【0077】
ハイグロマイシン耐性マーカー( E. coli hph)を、 S. cerevisiae PDC1(ピルビン酸デカルボキシラーゼ)プロモーターの転写コントロール下にクローニングした。ハイグロマイシンBに対して耐性を与える E. coli hph遺伝子は、プライマーとして、5'HYGXBA1(5'-AAG CTC TAG ATG AAA AAG CCT GAA CTC AC-3';配列番号7)及び3'HYGBAMH1(5'-CGC GGA TCC CTA TTC CTT TGC CCT CGG AC-3';配列番号8)を使用し、鋳型として、プラスミドpRLMex30(Mach et al. 1994, Curr. Genet. 25, 567-570; Rajgarhia et al. U.S. Patent Application Serial No: 10/154.360, filed May 23, 2002、その全体を本願に引用して援用する)を使用して、PCRにより増幅した。hph遺伝子は、また、鋳型として働く E. coli 染色体DNAと、前記のプライマーとを使用して得ることもできる。温度サイクルは、PfuTurboDNAポリメラーゼ(Stratagene、Madison、WI)を使用して、94℃-1分、56℃-1分及び72℃-3分を30サイクル行い、最後に72℃-7分のインキュベーションを続けた。PCR産物を0.8%アガロースゲルの電気泳動により分離し、1026bp産物を単離した。次に、この1026bp断片をXbaI及びBamHIで制限酵素処理し、ScPDC1プロモーター及びScGAL10ターミネーターを含むpNC4(実施例1A)のXbaI-BamHI断片とライゲーションして、プラスミドpPS1(図5)を得た。
【0078】
L-乳酸脱水素酵素をコードする L. helveticus L-LDH遺伝子を、S. cerevisiae ScPGK1プロモーターの転写コントロール下にクローニングした。L-乳酸脱水素酵素をコードする L. helveticus L-LDH遺伝子は、プライマーとして、PS15S(5'-GCT CTA GAA TTA TGG CAA GAG AGG AAA AAC-3';配列番号9)及びPS16AS(5'-CGG GAT CCT CAT TGA CGA ACC TTA ACG-3';配列番号10)を使用し、鋳型として、プラスミドpSO20(実施例1H;図10)を使用し、PCRにより増幅した。あるいは、L-LDH遺伝子は、前記のプライマーとともに、 L. helveticus 染色体DNAを鋳型として使用して得ることができる。温度サイクルは、PfuTurboDNAポリメラーゼ(Stratagene、Madison、WI)を使用して、94℃-1分、56℃-1分及び72℃-3分を30サイクル行い、最後に72℃-7分のインキュベーションを続けた。PCR産物を、0.8%アガロースゲルの電気泳動により分離し、990bp産物を単離した。このPCR産物をXbaI及びBamHIで制限酵素処理し、XbaI及びBamHIで制限酵素処理したベクターpNC2(実施例1A)とライゲーションして、プラスミドpPS14(図5)を得た。pNC14をSalI及びBsaHIで制限酵素処理すると、LhLDH発現カセットを得ることができる。このLhLDH発現カセットを単離し、pPS1のSalI-ClaI断片とライゲーションして、プラスミドpPS9を得た(図5)。
【0079】
(実施例1E)
L. helveticus L-LDH遺伝子及びG418マーカーを有するpVR39プラスミドのコンストラクション
pPS9(実施例1H;図5)を、SphIで制限酵素処理し、シュリンプアルカリホスファターゼ(Roche Diagnostics、Indianapolis、IN)を用いてメーカーのマニュアルに従い脱リン酸化し、 L. helveticus L-LDH発現カセットを含む断片を得た。pVR29(実施例1C;図4)をSphIで制限酵素処理し、G418耐性遺伝子カセットを含む2048bp断片を、0.8%アガロースゲル上で分離した。これらの断片をライゲーションし、結果として、互いに隣接する L. helveticus L-LDH遺伝子及びG418耐性遺伝子マーカーを含むプラスミドpVR39(図6)を得た。
【0080】
(実施例1F)
pHES及びpSEHプラスミドのコンストラクション
pHES及びpSEHプラスミドを、PCT出願PCT/US/44041に従い、0.5μgのプラスミドpGAD424(Chien et al., 1991, Proc. Natl Acad. Sci. USA 88:9578-9582に記載される)を使用してコンストラクションし、HindIIIで制限酵素処理した。処理混合物を、TBEバッファー(Biorad、USA)を用いた0.8%アガロースゲル電気泳動により分離した。5.9kbp断片を、Sambrook et al. (1989, Molecular Cloning, second edition, Cold Spring Harbor Laboratory, Plainview, NY)の記載のように、前記ゲルから単離した。複数の制限酵素認識部位を有する、相補的な1対の92bpの合成オリゴマーを設計した。第1のオリゴは、「fwdHES」オリゴであって、以下の配列(配列番号11)を有する。
5'-CCCAAGCTTG AATTCCCCGG GGGATCCCTG CAGGGTACCA CGCGTAGATC TACTAGTGCG GCCGCCTCGA GTCTAGAGGG CCCAAGCTTG GG-3'
第2のオリゴは、「comp(SO1)(CMA2)hes」オリゴであって、以下の配列(配列番号12)を有する。
5'-CCAAGCTTGG GCCCTCTAGA CTCGAGGCGG CCGCACTAGT AGATCTACGC GTGGTACCCT GCAGGGATCC CCCGGGGAA TTCAAGCTTG GG-3'
500nmolの2つの相補的なオリゴマーを、ともに水槽中で10分間(100℃)でボイルした後、室温で徐々に冷ますことにより、互いにアニールさせた。92bpの二重鎖DNAをHindIIIで制限酵素処理し、pGAD424(Clontech、USA)をHindIIIで処理した5.9kbp断片とライゲーションした。ライゲーション混合物を、Sambrook et al.(同上)に記載のエレクトロポレーションによる E. coli DH10B(electromax cells、Life Technologies、Rockville、MD)の形質転換に使用した。組み換え E. coliを、Luria-Bertani ブロースプレート(Difco, USA)にプレーティングし、プラスミドを有する細胞を、100μg/mlの抗生物質アンピシリン(Sigma Chemicals、USA)を使用して選択した。アンピシリン耐性 E. coli クローンから得られたプラスミドDNAをスクリーニングし、2つのプラスミドpHES及びpSEHを得た。この2つのプラスミドは、ベクター上のアルコール脱水素酵素(ADH1)に対する合成二重鎖マルチクローニングサイトの方向が異なる。
【0081】
(実施例1G)
pSO20及びpPS9のさらなるコンストラクションのための L. helveticus L-LDH遺伝子を含むプラスミド:pVR1のコンストラクション
L. helveticus L-LDH遺伝子は、以下のように単離された。 L. helveticus 細胞は、American Type Culture Collection (ATCC Accession #10797)から得て、標準的な条件で生育させた。そして、ゲノムDNAを、以下のプロトコールを使用して精製した。
【0082】
それぞれ50mlの無菌MRSブロース(Difco、USA)が入った2つの250ml無菌フラスコに、バクテリアのグリセロールストックから単一コロニー(又は5ml)を植菌した。その培養液を、37℃で、170rpmのアジテーションを加えながら48時間インキュベートした。この培養液を、50ml無菌青キャップチューブに移し、3000rpmで10分間遠心した。このペレットを、50mlの12.5%w/vスクロース溶液で懸濁し、再度、3000rpmで10分間遠心した。新たに形成されたペレットを、50ml無菌青キャップチューブ中で、5mlの12.5%w/vスクロース溶液に再度懸濁した。5mlのTES溶液を、前記チューブに加えた。(200mlのTES: 2 mlの1.0 M Tris (pH 8.0) [10 mM Tris (pH 8.0)]、20 mlの500 mM EDTA (pH 8.0) [50 mM EDTA (pH 8.0)]、0.584 gのNaCl [50 mM NaCl]を含み、使用前にフィルター滅菌する。)12.5%w/vスクロース溶液を前記チューブに加え、混合した。300mgのリゾチームパウダー(Sigma Chemicals、USA)を添加し、その混合物を2000rpmで1分間ボルテックスした。25μlのムタノリジン(mutanolysin)溶液(Sigma)(濃度2.2mg/ml)を前記混合物に添加し、その混合物を、37℃で一晩(10〜12時間)インキュベートした。
【0083】
2.5mlの20%SDS溶液(Biorad、USA)及び168μlのプロテナーゼK溶液(濃度:28mg/1.4ml)(Sigma)を、前記混合物に添加し、その結果の混合物を上下逆にしながら混合し、50℃-1時間でインキュベーションした。細胞膜物質は、よく破壊され、溶液は、半透明になった。その混合物を、NaClで稀釈し、前記チューブに0.15Mの濃度を得た。
【0084】
前記混合物を、50mlのオークリッジ(Oakridge)チューブに移し、等量のフェノール:クロロホルム:イソアミルアルコール(25:24:1)溶液で処理した。この混合物をよくシェイクし、5000rpmで10分間遠心した。水層を、新しいチューブに吸出し、25μlのRNAse(100mg/ml)を添加し、上下逆にしながら混合した。その結果の混合物を、37℃-15分でインキュベートした。
【0085】
2.5倍量のエタノールを、前記チューブの側面に沿って前記混合物に添加し、混合しなかった。境界面に形成されたDNAを、糸巻きのようにして回収し、70%エタノール中で洗浄し、その混合物を10000rpmで10分間遠心した。底に形成されたペレットDNAを空気乾燥し、微遠心(microfuge)チューブ中で、10mM Tris-HCl、pH8.5に再度懸濁した。その混合物を、DNAが溶液となるまで(2〜3時間)、前記チューブ内で50℃でインキュベートした。
【0086】
プライマーは、 L. helveticus のL-LDHとしてGenbankから入手可能な配列(Genbank accession # Z81318)に基づき設計した。PCR増幅は、Pfuポリメラーゼ(Stratagene、Madison、WI)を使用して行った。各反応は、500ngの濃度の L. helveticus ゲノムDNA、各0.2mMの4種のdNTP、並びに、各1μMの増幅プライマーSO22:5'-CCG GGA TCC ATG GCA AGA GAG GAA AAA CCTC(配列番号13)及びSO23:5'-CCA AGA TCT TTA TTG ACG AAC CTT AAC GCC AG(配列番号14)を含む。温度サイクルは、最初に95℃-10分でインキュベートした後に、95℃-30秒、41℃-30秒及び72℃-60秒を35サイクル行った。この反応で生成された990(bp)の断片を、都合のよい方法を使用してゲルから精製し、BamHI及びBglIIで制限酵素処理し、そして、BamHI及びBglIIで処理したpHES(実施例1F)にクローニングし、プラスミドpLhLDH-HESを得た。結果として得られた配列は、Genbankで公知の L. helveticus L-LDHをコードする遺伝子配列(Accession # Z81318)と極めて相同なポリペプチドに翻訳されうる配列である。
【0087】
L. helveticus L-LDH遺伝子の5'端及び3'端に、それぞれ、NcoI及びDraIIIの制限酵素認識部位を導入するため、さらに、プライマーを設計した。PCR増幅は、Pfuポリメラーゼ(Stratagene、Madison、WI)を使用して行った。各反応は、5ngのpLH-LDH/HES、各0.2mMの4種のdNTP、並びに、各1μMの増幅プライマーVR1:5'-ATC CAT GGC AAG TAT TAC GGA TAA GGA TCA CCAA(配列番号15)及びVR2:5'-ATC ACG AAG TGT CAC GTA CGG GTT TCG ATG TC(配列番号16)を含む。温度サイクルは、最初に95℃-10分でインキュベートした後に、95℃-30秒、55℃-30秒及び72℃-60秒を35サイクル行った。この反応で生成された982(bp)の断片を、都合のよい方法を使用してゲルから精製し、NcoI及びDraIIIで制限酵素処理し、そして、NcoI及びDraIIIで処理したpTEF1/Zeo(Invitrogen、Carlsbad、CA)にクローニングした。結果として得たプラスミドpVR1(図7)は、 L. helveticus L-LDHをコードする遺伝子配列が、 Saccharomyces cerevisiae pTEF1プロモーターのコントロール下にあることが確認された。 E. coli EM7プロモーターが存在するが、これは、本発明の組み込みベクターに必要な構成要素ではない。
【0088】
(実施例1H)
K. marxianus において L. helveticus L-LDHプラスミドを発現するためのプラスミドpSO20のコンストラクション
プラスミドpSO20は、BamHI(NEB)で制限酵素処理され、シュリンプアルカリホスファターゼ(Roche Diagnostics、Indianapolis、IN)処理されたpUC19プラスミド(Life Technologies、USA)と、pVR1(実施例1F)を処理して得られた L. helveticus L-LDH遺伝子とそれに隣接するプロモーター及びターミネーター配列を有するBamHI断片とを、ライゲーションしてコンストラクションした。結果として得られたプラスミドは、pSO18(図8)である。pSO18を、次に、SphIで制限酵素処理し、pKD1のバックボーンを含むpSPH1コンストラクト(Chen et al., 1986, "Sequence organization of the circular plasmid pKD1 from the yeast Kluyveromyces drosophilarum," Nucleic acids Res. 14: 4471-4481)由来のSphI断片とライゲーションすることで、結果として、プラスミドpSO19(図9)を得た。
【0089】
pTEF1/Zeo(Invitorogen, Inc)を、XhoI/XbaIで制限処理して、 S. cerevisiase pTEF1プロモーター、ゼオシン耐性遺伝子、及び、 S. cerevisiase GAL10ターミネーターを放出させた。この1195bp断片の末端を、PfuDNAポリメラーゼ(Stratagene、Madison、WI)を用いて平滑末端とした。コンストラクトpSO19を、AatIIで制限酵素処理し、PfuDNAポリメラーゼで平滑末端を作り出し、結果として得られた断片をシュリンプアルカリホスファターゼで処理した。そして、この9.3kbpを、 S. cerevisiase pTEF1プロモーター、ゼオシン耐性遺伝子、及び、 S. cerevisiase GAL10ターミネーターを含む1195bpのpTEF/Zeo断片とライゲーションした。この結果として、コンストラクトpSO20が得られた(図10)。
【0090】
(実施例1I)
形質転換DNAのランダムな組み込みを介する L. helveticus L-LDH遺伝子の K. marxianus ゲノムへの導入
pVR39をSstI及びApaIで制限酵素処理することで、L. helveticus L-LDH:G418耐性遺伝子カセットを含む4.28kbpの断片を単離した。この断片は、0.8%のアガロースゲルで分離され、下記エレクトロポレーションプロトコールを使用した K. marxianus (CD21)の形質転換に使用される。
【0091】
K. marxianus の単一コロニーを、50mLのYPD培地への植菌(250 mLのバッフル型フラスコ中に、10 g/L 酵母抽出物, 20 g/L ペプトン及び20 g/L グルコースを含む。OD600を0.1まで。)に使用した。この培養液を、30℃-250rpmで16時間培養した。最終OD600は、10となった。10mLの培養液から細胞を、遠心により回収し、エレクトロポレーションバッファー(10 mM Tris-Cl, 270 mM スクロース, 1 mM MgCl2, pH 7.5)で1回洗浄した。その後、この細胞を、インキュベーションバッファー(YPD + 25 mM DTT, 20mM HEPES, pH 8.0)に再度懸濁し、30℃-250rpmで30分インキュベートした。この細胞を、遠心により回収し、エレクトロポレーションバッファーで1回洗浄し、1mLのエレクトロポレーションバッファーに再度懸濁した。そして、400μLの細胞を、0.4cmのエレクトロポレーションキュベット(BioRad;USA)に移した。
【0092】
pVR39を、SstI及びApaIで制限酵素処理し、結果として得られる4.3kbp断片の2μgを、キュベットに添加した(BioRad;USA)。次いで、細胞を、1.8kV、1000Ω、25μFの条件で、エレクトロポレーションした。この細胞を、50mLスクリューキャップ付きファルコンチューブ中の1mLのYPDに移し、30℃-250rpmで4時間インキュベートした後、300μg/mLのG418を含むYPDプレートにプレーティングして選択した。形質転換株は、37℃で3日間生育させた。G418耐性形質転換株は、300μg/mLのG418を含む新しい選択プレートに、再度、ストリークした。
【0093】
K. marxianus のゲノムに断片が組み込まれた確認は、 L. helveticus L-LDH遺伝子に相同なように設計されたPCRプライマー、及び、G418耐性遺伝子と相同なリバースプライマーを使用して達成できる。
【0094】
VR146:5'-GCT GAC TAC CCA GAT TGT AAG GATG-3’(配列番号17)及びVR143:5'-CTG CCA GCG CAT CAA CAA TAT TTT CAC-3’(配列番号18)は、前記カセットを有する株において、 L. helveticus L-LDH遺伝子とG418耐性遺伝子との間の2.3kbpの産物を増幅するように設計した。このプライマーは、前記カセットを有しない株では、少しも断片を増幅しない。
【0095】
形質転換コロニーに対する温度サイクルは、TaqDNAポリメラーゼ(Qiagen、USA)を用いて、最初に94℃-2分で反応混合物をインキュベーションした後、94℃-30秒、53℃-30秒及び72℃-3分を35サイクル行い、最後に72℃-7分のインキュベーションを続けた。分析した10個の形質転換株が、予想される2.3kbpのPCR産物を産出した。前記10個の形質転換株から得たゲノムDNAについての L. helveticus L-LDH遺伝子をプローブとしたサザンブロットを行ったところ、分析した前記10個の形質転換株のうち、2個が、pVR39に由来する L. helveticus L-LDH遺伝子の1コピーの組み込みと一致する適切なバンドパターンを示した。この2つのうち1つを、CD484と認定した。
【0096】
(実施例1J)
形質転換DNAのランダムな組み込みを介する L. helveticus L-LDH遺伝子のさらなるコピーの K. marxianus ゲノムへの導入
pPS9をSstI及びApaIで制限酵素処理することで、L. helveticus L-LDH:HPH耐性遺伝子カセットを含む4.7kbpの断片を単離した。この断片は、0.8%のアガロースゲルで分離され、下記エレクトロポレーションによる K. marxianus CD484の形質転換に使用される。
【0097】
K. marxianus CD484の単一コロニーを、50mLのYPD培地への植菌(250 mLのバッフル型フラスコ中に、10 g/L 酵母抽出物、20 g/L ペプトン、20 g/L グルコース及び2%アガーを含む。OD600を0.1まで。)に使用した。この培養液を、30℃-250rpmで16時間培養した。最終OD600は、10となった。10mLの培養液から細胞を、遠心により回収し、エレクトロポレーションバッファー(10 mM Tris-Cl, 270 mM スクロース, 1 mM MgCl2, pH 7.5)で1回洗浄した。その後、細胞を、インキュベーションバッファー(YPD + 25 mM DTT, 20mM HEPES, pH 8.0)に再度懸濁し、30℃250rpmで30分インキュベートした。この細胞を、遠心により回収し、エレクトロポレーションバッファーで1回洗浄し、1mLのエレクトロポレーションバッファーに再度懸濁した。そして、400μLの細胞を、0.4cmのエレクトロポレーションキュベット(BioRad;USA)に移した。
【0098】
pPS9を、SstI及びApaIで制限酵素処理し、結果として得られる4.7kbp断片の5μgを、キュベットに添加した。次いで、細胞を、1.8kV、1000Ω、25μFの条件で、エレクトロポレーションした。細胞を、50mLスクリューキャップ付きファルコンチューブ中の1mLのYPDに移し、30℃-250rpmで4時間インキュベートした後、200μg/mLのハイグロマイシン(Invitrogen、Carlsbad、CA)を含むYPDプレートにプレーティングして選択した。ハイグロマイシン耐性形質転換株を、37℃で3日間、生育させた。耐性形質転換株が、両方の耐性遺伝子を有することを確認するため、300μg/mLのG418を含む新しい選択プレートに、再度、ストリークした。
【0099】
L. helveticus L-LDH:G418耐性遺伝子カセットの新たな4.7kbp断片が組み込まれた確認は、L. helveticus L-LDH遺伝子に相同となるように設計されたPCRプライマー、及び、ハイグロマイシン耐性遺伝子と相同なリバースプライマーを使用して達成できる。VR146(配列番号17)及びVR142:5'-GTG ACA CCC TGT GCA CGG CGG GAG AT-3’(配列番号19)は、前記カセットを有する株において、 L. helveticus L-LDH遺伝子とハイグロマイシン耐性遺伝子との間の2.3kbpの産物を増幅するように設計した。このプライマーは、前記カセットを有しない株では、少しも断片を増幅しない。
【0100】
温度サイクルは、TaqDNAポリメラーゼ(Qiagen、USA)を用いて、最初に94℃-2分でインキュベーションした後、94℃-30秒、53℃-30秒及び72℃-3分を35サイクル行い、最後に72℃-7分のインキュベーションを続けた。分析した4個の形質転換株が、予想される1.76kbpのPCR産物を産出した。
【0101】
K. marxianus CDC484由来の4288kbpの L. helveticus L-LDH:G418耐性遺伝子の存在確認は、L. helveticus L-LDH遺伝子に相同となるように設計されたPCRプライマー、及び、G418耐性遺伝子と相同なリバースプライマーを使用して達成できる。VR146(配列番号17)及びVR143(配列番号18)は、前記カセットを有する株において、L. helveticus L-LDH遺伝子とG418耐性遺伝子との間の2.3kbpの産物を増幅するように設計した。このプライマーは、前記カセットを有しない株では、少しも断片を増幅しない。
【0102】
形質転換コロニーに対する温度サイクルは、TaqDNAポリメラーゼ(Qiagen、USA)を用いて、最初に94℃-2分でインキュベーションした後、94℃30秒、53℃30秒及び72℃3分を35サイクル行い、最後に72℃で7分のインキュベーションを続けた。分析した4個の形質転換株が、予想される2.3kbpのPCR産物を産出した。
【0103】
ゲノムDNAについて、 L. helveticus L-LDH遺伝子をプローブとしたサザンブロットを行ったところ、PCRでG418及びハイグロマイシンカセットについて陽性であった前記4個の形質転換株のうち、1個が、ゲノム中に組み込まれた L. helveticus L-LDH遺伝子を2コピー有していた。この株を、 K. marxianus CDC492と認定し、さらに、L-乳酸の産生について分析した。
【0104】
(実施例1K)
振とうフラスコ培養のYPD+グルコース培地中におけるL-乳酸の産生
組み換え株CD492を、100g/Lのグルコースを補充した50mLのYPD培地を含む250mLのバッフル型振とうフラスコで培養した。細胞は、YPDアガープレートからOD600が0.1まで、30℃-250rpmで16時間培養した。細胞が対数増殖期であることを確認するため、YSI分析を使用して残存グルコース濃度を測定した。4g/L細胞乾燥重量(「gcdw」)当量を遠心により回収し、250mLのバッフル型振とうフラスコ中の、約100g/Lのグルコース及び55g/LのCaCO3を補充した50mLのYPD培地に再度懸濁した。その培養液を、30℃-70rpmでインキュベートした。0、12、24時間の間隔後にサンプルを回収した。細胞をフィルターによりサンプルから取り除き、その上清を、HPLCにより、グルコース、乳酸塩、ピルビン酸塩及びエタノールについて、分析した。これらの条件下で、CD492株は、鏡像異性体的に99%を越える純度のL-乳酸を産生した。最終力価は、69g/Lであった。容積生産性は、3.6g/L/hrであった。
【0105】
比較のために、CD484(実施例1I)株を同様の条件下で培養した。CD492と比較して、L-乳酸の産生は、低く、エタノールの産生は、高かった。
【0106】
(実施例1L)
振とうフラスコ培養のYPD+グルコース培地中におけるL-乳酸の産生
492株を、100g/Lのグルコースを補充した50mLのYPD培地を含む250mLのバッフル型振とうフラスコで培養した。細胞を、振とうフラスコ中に、OD600が0.1となるように植菌し、前記フラスコを、30℃-250rpmで、16時間インキュベートした。最終pHは、3.1未満であった。細胞が対数増殖期であることを確認するため、YSI分析を使用して、前記フラスコの残存グルコース濃度を測定した。4g/L細胞乾燥重量当量を遠心により回収し、250mLのバッフル型振とうフラスコ中の、約25g/Lのグルコースを補充した50mLのYPD培地に再度懸濁した。その培養液を、30℃-70rpmでインキュベートした。様々な時間間隔後にサンプルを回収し、細胞をフィルターによりサンプルから取り除いた。培養上清を、HPLCにより、グルコース、乳酸塩、ピルビン酸塩及びエタノールについて、分析した。
【0107】
L-乳酸は、鏡像異性体的に99%を越える純度で産生された。乳酸力価は、15g/Lであり、容積生産性は、2g/L/hrであった。CD484株を同様の条件でテストした場合、L-乳酸の産生は、低く、エタノールの産生は、高かった。
【0108】
別個の実験として、492株を、100g/Lのグルコースを補充した100mLのYPD培地を含む500mLのバッフル型振とうフラスコに、OD600が0.1となるように植菌した。前記振とうフラスコを、37℃、250rpmで、約16時間インキュベートした。細胞が対数増殖期であることを確認した後、4g/L細胞乾燥重量当量を遠心により回収した。細胞ペレットを、約40g/Lのグルコースを補充した50mLのYPD培地に再度懸濁し、250mLのバッフル型振とうフラスコに移した。前記振とうフラスコを、37℃-70rpmに置いた。バイオマスの生育終了時及び産生期の0、15、18.5及び23時間後に、グルコースが消費されるまで、HPLC解析用のサンプルを回収した。pHは、産生の開始と終了の時点で測定した。最終pHは、3.00+/-0.06であった。
【0109】
CD492株は、グルコースを、15〜23時間で消費した。遊離酸分析用のサンプルを、産生終了時(グルコースが完全に消費された時)に採取し、乳酸、酢酸塩、エタノール及びピルビン酸塩の総合量を分析した。CD492株は、32g/Lの遊離乳酸、32g/Lのエタノール、並びに、1g/L未満の酢酸塩及びピルビン酸塩を産生した。遊離乳酸は、32g/Lであった。全ての場合で、プロトン化型の酸のパーセントは、発酵終了時で、少なくとも97%であった。乳酸のグルコースに対する収率は、59%であった。
【0110】
CD484株を、同様の条件で評価した。CD484株は、26〜28g/Lの遊離乳酸を産生し、グルコースに対する収率は、46〜58%であった。
【実施例2】
【0111】
(実施例2A)
S. cerevisiase ScPGK1プロモーターのコントロール下でL-LDHを発現するための B. megaterium L-LDHを有するpVR24プラスミドのコンストラクション
LDH遺伝子をコードする B. megaterium DNAは、下記のように単離した。 B. megaterium は、American Type Culture Collection (ATCC Accession #6458)から得て、標準的な条件で生育させた。ゲノムDNAは、これらの細胞から“Easy-DNA"キット(Invitorogen)を使用して、メーカーのプロトコールに従い、精製した。プライマーは、Genbankの B. megaterium のL-LDHとして入手可能な配列(Genbank accession # M22305)に基づき設計した。PCR増幅反応は、buffer II (1.5mM MgCl2) 及び AmpliTaq Gold ポリメラーゼ(ともに、Perkin Elmer社製)を使用して行った。各反応は、6ng/μLの濃度の B. megaterium ゲノムDNA、各0.2mMの4種のdNTP、並びに、各1μMの増幅プライマーBM1270:5'-CCT GAG TCC ACG TCA TTA TTC-3'(配列番号20)及びBM179:5'-TGA AGC TAT TTA TTC TTG TTAC-3'(配列番号21)を含む。
【0112】
温度サイクルは、最初に95℃-10分でインキュベートした後に、95℃-30秒、50℃-30秒及び72℃-60秒を35サイクル行った。この反応で生成された1100bpの断片を、都合のよい方法を使用してゲルから精製し、クローニングし、塩基配列決定をした。結果として得られた配列は、Genbankで公知の L-LDHをコードする遺伝子配列と相同性を示すポリペプチドに翻訳されうる配列である。ここで、前記相同性の程度は、2つの配列間における、少なくとも75%、80%、85%、90%、91%、92%、93%、94%、95%、96%、97%、98%若しくは約99%の同一性である。
【0113】
B. megaterium L-LDHをコードする配列を、pNC2(実施例1A)由来の S. cerevisiae ScPGK1プロモーター及び S. cerevisiae GAL10転写ターミネーターと、動作可能に連結した。2つのオリゴヌクレオチドプライマーBmeg5’:5'-GCT CTA GAT GAA AAC ACA ATT TAC ACC-3'(配列番号22)及びBmeg3’:5'-ATG GAT CCT TAC ACA AAA GCT CTG TCGC-3'(配列番号23)を、前記遺伝子をコードする配列の末端に制限酵素認識部位を導入するように設計した。
【0114】
増幅反応は、PfuTurboDNAポリメラーゼ(Stratagene、Madison、WI)を使用して、上述したようなdNTP及びプライマー濃度で、メーカーが供給するバッファーを使用して行った。温度サイクルは、最初に反応混合物を95℃で3分インキュベートした後、95℃30秒、50℃30秒、72℃60秒を20サイクル行い、最後に72℃9分のインキュベーションを続けた。PCR産物を、XbaI及びBamHIで制限酵素処理し、pNC2(実施例1A)のXbaI-BamHI断片とライゲーションした。このライゲーションの結果、ScPGK1プロモーター及びScGAL10ターミネーターが、 B. megaterium L-LDHをコードする配列に動作可能に連結したプラスミドpVR24(図12)が得られた。
【0115】
(実施例2B)
B. megaterium L-LDHをコードするG418耐性マーカープラスミド(pCA3)
Invitrogen(Carlsbad、CA)から得たG418抗生選択マーカーを変更し、酵母 S. cerevisiae 由来のピルビン酸デカルボキシラーゼ遺伝子プロモーター及び転写ターミネーターに動作可能に連結させた。このコンストラクトを作製する際、下記オリゴヌクレオチドを調製し、G418耐性遺伝子が挿入されたプラスミドからコード配列を増幅するために使用した。2つのオリゴヌクレオチドプライマーG5’:5'-AAA TCT AGA TGA GCC ATA TTC AAC GGGA-3'(配列番号24)及びG3’:5'-CCG GAT CCT TAG AAA AAC TCA TCG AGC AT-3'(配列番号25)は、前記遺伝子をコードする配列の末端に制限酵素認識部位を導入するように設計した。
【0116】
pPIC9Kベクター(Invitrogen、Carlsbad、CA)のG418耐性遺伝子を、G5'及びG3'プライマー、4種のdNTP、並びにPfuTurboポリメラーゼを使用してPCRにより増幅した。増幅反応は、PfuTurboDNAポリメラーゼ(Stratagene、Madison、WI)を使用して、上述したようなdNTP及びプライマー濃度で、メーカーが供給するバッファーを使用して行った。温度サイクルは、最初に95℃-3分で反応混合物をインキュベートした後、95℃-30秒、50℃-30秒、72℃-60秒を20サイクル行い、最後に72℃-9分のインキュベーションを続けた。PCR産物を、XbaI及びBamHIで制限酵素処理し、pNC4(実施例1A)のXbaI-BamHI断片とライゲーションした。
【0117】
このベクターのG418遺伝子のScGAL10ターミネーターの3’末端であるSphI部位に、 S. cerevisiaeのScPGK1プロモーター及びScGAL10ターミネーターに動作可能に連結した B. megaterium L-LDH遺伝子を導入し、その結果、プラスミドpCA3(図13)を得た。
【0118】
(実施例2C)
B. megaterium L-LDH遺伝子及びハイグロマイシン耐性遺伝子を含むpVR38プラスミドのコンストラクション
pVR24(実施例2A)を、SphIで制限酵素処理し、シュリンプアルカリホスファターゼ(Roche Diagnostics、Indianapolis、IN)を用いてメーカーのプロトコールに従い脱リン酸化し、 B. megaterium L-LDH発現カセットを含む断片を得た。pPS1(実施例1D)をSphIで制限酵素処理し、ハイグロマイシン耐性遺伝子カセットを含む2259bp断片を、0.8%アガロースゲル上で分離し、脱リン酸化したpVR24とライゲーションした。その結果、互いに隣接する B. megaterium L-LDH遺伝子及びハイグロマイシン耐性遺伝子マーカーを含むプラスミドpVR38(図14)を得た。
【0119】
(実施例2D)
形質転換DNAのランダムな組み込みを介する B. megaterium L-LDH遺伝子の K. marxianus ゲノムへの導入
pCA3をSstI及びApaIで制限酵素処理することで、 B. megaterium L-LDH:G418耐性遺伝子カセットを含む4.2kbpの断片を単離した。この断片は、0.8%のアガロースゲルで分離され、下記エレクトロポレーションプロトコールを使用した K. marxianus(CD21)の形質転換に使用される。
【0120】
K. marxianus の単一コロニーを、50mLのYPD培地への植菌(250 mLのバッフル型フラスコ中に、10 g/L 酵母抽出物, 20 g/L ペプトン、20 g/L グルコース及び2%アガーを含む。OD600を0.1まで。)に使用した。培養液を、30℃-250rpmで16時間培養した。最終OD600は、10となった。10mLの培養液から細胞を、遠心により回収し、エレクトロポレーションバッファー(10 mM Tris-Cl, 270 mM スクロース, 1 mM MgCl2, pH 7.5)で1回洗浄した。細胞を、インキュベーションバッファー(YPD + 25 mM DTT, 20mM HEPES, pH 8.0)に再度懸濁し、30℃-250rpmで30分インキュベートした。細胞を、遠心により回収し、エレクトロポレーションバッファーで1回洗浄し、1mLのエレクトロポレーションバッファーに再度懸濁した。そして、400μLの細胞を、0.4cmのエレクトロポレーションキュベット(BioRad;USA)に移した。
【0121】
2μgのpCA3由来の4.3kbpSstI/ApaI断片を、キュベットに添加し、細胞を、1.8kV、1000Ω、25μFの条件で、エレクトロポレーションした。細胞を、50mLスクリューキャップ付きファルコンチューブ中の1mLのYPDに移し、30℃250rpmで4時間インキュベートした後、300μg/mLのG418を含むYPDプレートにプレーティングして選択した。形質転換株を、37℃で3日間、生育させた。G418耐性形質転換株を、300μg/mLのG418を含む新しい選択プレートに、再度、ストリークした。
【0122】
K. marxianus のゲノムに断片が組み込まれた確認は、 B. megaterium L-LDH遺伝子に相同となるように設計されたPCRプライマー、及び、G418耐性遺伝子と相同なリバースプライマーを使用して達成できる。BmLDH3:5'-GTA CGC ATT ACC AAG GCT ATT TTA GAT-3’(配列番号26)及びVR143(配列番号18)は、前記カセットを有する株において、 B. megaterium L-LDH遺伝子とG418耐性遺伝子との間の1.8kbpの産物を増幅するように設計した。このプライマーは、前記カセットを有しない株では、少しも断片を増幅しない。
【0123】
形質転換コロニーに対する温度サイクルは、TaqDNAポリメラーゼ(Qiagen、USA)を用いて、最初に94℃-2分で反応混合物をインキュベーションした後、94℃-30秒、53℃-30秒及び72℃-3分を35サイクル行い、最後に72℃-7分のインキュベーションを続けた。分析した10個の形質転換株のうち7個が、予想される1.8kbpのPCR産物を産出した。前記7個の形質転換株から得たゲノムDNAについての B. megaterium L-LDH遺伝子をプローブとしたサザンブロットを行ったところ、分析した前記7個の形質転換株のうち、2個が、pCA3に由来する B. megaterium L-LDH遺伝子の1コピーの組み込みと一致する適切なバンドパターンを示した。この2つのうち1つを、CD162と認定した。
【0124】
(実施例2E)
形質転換DNAのランダムな組み込みを介する B. megaterium L-LDH遺伝子のさらなるコピーの K. marxianus ゲノムへの導入
pVR38をSstI及びApaIで制限酵素処理することで、 B. megaterium L-LDH:HPH耐性遺伝子カセットを含む4.5kbpの断片を単離した。この断片は、0.8%のアガロースゲルで分離し、エレクトロポレーションによる K. marxianus CD162株の形質転換に使用した。
【0125】
K. marxianus CD162の単一コロニーを、50mLのYPD培地への植菌(250 mLのバッフル型フラスコ中に、10 g/L 酵母抽出物、20 g/L ペプトン、20 g/L グルコース及び2%アガーを含む。OD600を0.1まで。)に使用した。培養液を、30℃-250rpmで16時間培養した。最終OD600は、10となった。10mLの培養液から細胞を、遠心により回収し、エレクトロポレーションバッファー(10 mM Tris-Cl, 270 mM スクロース, 1 mM MgCl2, pH 7.5)で1回洗浄した。その後、細胞を、インキュベーションバッファー(YPD + 25 mM DTT, 20mM HEPES, pH 8.0)に再度懸濁し、30℃-250rpmで30分インキュベートした。細胞を、遠心により回収し、エレクトロポレーションバッファーで1回洗浄し、1mLのエレクトロポレーションバッファーに再度懸濁した。そして、一部(400μL)の細胞を、0.4cmのエレクトロポレーションキュベット(BioRad;USA)に移した。
【0126】
pVR38を、SstI及びApaIで制限酵素処理し、結果として得られる4.5kbp断片の5μgを、エレクトロポレーションキュベットに添加した。次いで、細胞を、1.8kV、1000Ω、25μFの条件で、エレクトロポレーションした。細胞を、50mLスクリューキャップ付きファルコンチューブ中の1mLのYPDに移し、30℃-250rpmで4時間インキュベートした後、200μg/mLのハイグロマイシン(Invitrogen、Carlsbad、CA)を含むYPDプレートにプレーティングして選択した。ハイグロマイシン耐性形質転換株を、37℃で3日間生育させた。耐性形質転換株が、両方の耐性遺伝子を有することを確認するため、300μg/mLのG418を含む新しい選択プレートに、再度、ストリークした。
【0127】
B. megaterium L-LDH:HPHカセットの新たな4.5kbp断片が組み込まれた確認は、 L. helveticus L-LDH遺伝子に相同となるように設計されたPCRプライマー及びハイグロマイシン耐性遺伝子と相同なリバースプライマーを使用して達成できる。BmLDH3(配列番号26)及びVR142(配列番号19)は、前記カセットを有する株において B. megaterium L-LDH遺伝子とハイグロマイシン耐性遺伝子との間の1.76kbpの産物を増幅するように設計した。このプライマーは、前記カセットを有しない株では、少しも断片を増幅しない。
【0128】
温度サイクルは、TaqDNAポリメラーゼ(Qiagen、USA)を用いて、最初に94℃-2分で反応混合物をインキュベーションした後、94℃-30秒、53℃-30秒及び72℃-3分を35サイクル行い、最後に72℃-7分のインキュベーションを続けた。上記PCR方法論により分析した9個の形質転換株が、予想される1.76kbpのPCR産物を産出した。
【0129】
K. marxianus CDC162に既に存在した4.2kbpの B. megaterium L-LDH:G418耐性遺伝子の存在確認は、 B. megaterium L-LDH遺伝子に相同となるように設計されたPCRプライマー及びG418耐性遺伝子と相同なリバースプライマーを使用して達成できる。BmLDH3(配列番号26)及びVR143(配列番号18)は、前記カセットを有する株において、 B. megaterium L-LDH遺伝子とG418耐性遺伝子との間1.8kbpの産物を増幅するように設計した。このプライマーは、前記カセットを有しない株では、少しも断片を増幅しない。
【0130】
形質転換コロニーに対する温度サイクルは、TaqDNAポリメラーゼ(Qiagen、USA)を用いて、最初に94℃-2分で反応混合物をインキュベーションした後、94℃-30秒、53℃-30秒及び72℃-3分を35サイクル行い、最後に72℃-7分のインキュベーションを続けた。分析した9個の形質転換株が、予想される1.8kbpのPCR産物を産出した。これらの株のゲノムDNAについて、 B. megaterium L-LDH遺伝子をプローブとしたサザンブロットを行ったところ、PCRでG418及びハイグロマイシンカセットについて陽性であった前記9個の形質転換株のうち、7個が、ゲノム中に組み込まれた B. megaterium L-LDH遺伝子を2コピー有していた。この株のうち、1個を、CD355と認定した。
【0131】
(実施例2F)
振とうフラスコ培養のYPD+グルコース培地中におけるL-乳酸の産生
組み換え株CD355を、実施例Kで記載した一般的な方法で培養した。これらの条件下で、CD355株は、鏡像異性体的に99%を越える純度のL-乳酸を産生した。最終力価は、52g/Lであった。容積生産性は、1.5g/L/hrであった。
【0132】
比較のために、CD162株を同様の条件下で培養した。L-乳酸の産生は、低く、エタノールの産生は、高かった。
【0133】
(実施例2G)
振とうフラスコ培養のYPD+グルコース培地中におけるL-乳酸の産生
355株を、100g/Lのグルコースを補充した50mLのYPD培地を含む250mLのバッフル型振とうフラスコで培養した。細胞を、振とうフラスコ中に、OD600が0.1となるように植菌し、前記フラスコを、30℃-250rpmで、16時間インキュベートした。最終pHは、3.1未満であった。細胞が対数増殖期であることを確認するため、YSI分析を使用して、前記フラスコの残存グルコース濃度を測定した。4g/L細胞乾燥重量当量を遠心により回収し、250mLのバッフル型振とうフラスコ中の、約25g/Lのグルコースを補充した50mLのYPD培地に再度懸濁した。その培養液を、30℃-70rpmでインキュベートした。様々な時間間隔後にサンプルを回収し、細胞をフィルターによりサンプルから取り除いた。培養上清を、HPLCにより、グルコース、乳酸塩、ピルビン酸塩及びエタノールについて、分析した。
【0134】
L-乳酸は、鏡像異性体的に99%を越える純度で産生された。乳酸の力価は、12g/Lであり、容積生産性は、1.9グラム/L/hrであった。CD162株を同様の条件でテストした場合、L-乳酸の産生は、低く、エタノールの産生は、高かった。
【実施例3】
【0135】
(実施例3A)
K. marxianus のPDC1遺伝子座を標的としたDNAの組み込みのためのpBH5a/bのコンストラクション
K. marxianus のピルビン酸デカルボキシラーゼ(KmPDC1)の側面DNAをベクターpVR29(実施例1C)にクローニングし、KmPDC1遺伝子座にDNAを組み込むためのベクターを作製した。最終的に得られたコンストラクトが、pBH5b(図15)である。pBH5bのSbfI制限酵素認識部位にクローニングされた遺伝子は、 K. marxianus のプロモーター及びターミネーターと動作可能に連結される。
【0136】
K. marxianus のPDC1のすぐ上流の1254bpのDNA断片を、プライマーとして、5'-Flank5’:5 -CAA GAA GGT ACC CCT CTC TAA ACT TGA ACA-3'(配列番号27)及び5'-Flank3’:5'-GTA ATT CCT GCA GGT GCA ATT ATT TGG TTT GG-3'(配列番号28)を使用し、鋳型として、プラスミドpSO21を使用して、PCRにより増幅した。温度サイクルは、最初に94℃-2分で反応混合物をインキュベーションした後、94℃-30秒、53℃-30秒及び72℃-1.5分を35サイクル行い、最後に72℃-7分のインキュベーションを続けた。1254bpのPCR産物を、0.8%アガロースゲルの電気泳動により分離して単離した。このPCR産物及びpVR29プラスミド(実施例1C)をともに、KpnI及びSbfIで制限酵素処理した。処理したPCR産物を、5067kbのpVR29断片とライゲーションし、6315bpのpBH5a(図15)を得た。pBH5aは、 S. cerevisiaeのScPDC1プロモーター及びScGAL10ターミネーターに動作可能に連結したG418耐性遺伝子、並びに、K. marxianus のPDC1のすぐ上流の1254bpのDNA断片を有する。
【0137】
K. marxianus のPDC1のすぐ下流の535bpのDNA断片を、プライマーとして、3'-Flank5’:5'-CCA AGC CCT GCA GGA GAG GGA GAG GAT AAA GA-3'(配列番号29)及び3'-Flank3’:5'-CTC GTA ACG CGT GTA CAA GTT GTG GAA CAA-3'(配列番号30)を使用し、鋳型として、プラスミドpSO21を使用して、PCRにより増幅した。温度サイクルは、最初に94℃-2分で反応混合物をインキュベーションした後、94℃-30秒、55℃-30秒及び72℃-45秒を35サイクル行い、最後に72℃-4分のインキュベーションを続けた。PCR産物を、0.8%アガロースゲルの電気泳動により分離し、535bpの産物を単離した。529bpのPCR産物を、SbfI及びMulIで制限酵素処理し、その断片を、pBH5aのSbfI-MulI断片とライゲーションして、プラスミドpBH5b(図15)を得た。pBH5bは、 S. cerevisiaeのScPDC1プロモーター及びScGAL10ターミネーターに動作可能に連結したG418耐性マーカーとともに、K. marxianus のPDC1のすぐ上流の1.2kbpのDNA断片及びK. marxianus のPDC1のすぐ下流の0.5kbpのDNA断片、そしてこれらのPDC1側面配列の間に存在するユニークな制限酵素認識部位SbfIを有する。
【0138】
(実施例3B)
K. marxianus のKmPDC1遺伝子座を標的として L. helveticus L-LDH遺伝子を組み込むためのpBH8のコンストラクション
L. helveticus 由来のL-乳酸脱水素酵素遺伝子をpBH5b(実施例3A)のSbfI部位にクローニングし、 L. helveticus L-LDH遺伝子を、 K. marxianus のKmPDC1遺伝子座に組み込み、外因性の K. marxianus のKmPDC1プロモーター及びターミネーターを使用して L. helveticus L-LDH遺伝子を発現できるベクターを作製した。
【0139】
L. helveticus L-LDH遺伝子を、プライマーとして、L-LDH5’:5'-ACA AAT CCT GCA GGA TGG CAA GAG AGG AAA AA-3'(配列番号31)及びL-LDH3’:5'-TAT CTA CCT GCA GGT CAT TGA CGA ACC TTA AC-3'(配列番号32)を使用し、鋳型として、プラスミドpPS9(実施例1D)を使用して、PCRにより増幅した。温度サイクルは、Failsafe PCR System (Epicentre, Madison, WI)を使用して、94℃-30秒、55℃-30秒及び72℃-1.5分を30サイクル行い、最後に72℃-4分のインキュベーションを続けた。971bpのPCR産物を、0.8%アガロースゲルの電気泳動により分離して単離した。このPCR産物をSbfIで制限酵素処理し、6844bpのSbfIで処理したpBH5b断片とライゲーションし、7824bpのpBH8(図16)を得た。pBH8は、 S. cerevisiaeのScPDC1プロモーター及びScGAL10ターミネーターに動作可能に連結したG418耐性遺伝子とともに、 K. marxianus KmPDC1プロモーター及びターミネーターに動作可能に連結した L. helveticus L-LDH遺伝子を有する。
【0140】
(実施例3C)
pBH8をKmPDC1遺伝子座に組み込むことによる K. marxianus CD606株のコンストラクション
野生型 K. marxianus を、pBH8プラスミド(実施例3B)で形質転換した。CD21のKmPDC1遺伝子座に、pBH8プラスミド全体が、pBH8の L. helveticus L-LDH遺伝子のすぐ上流のKmPDC1の側面DNAと、 K. marxianus 染色体のKmPDC1遺伝子のすぐ上流のDNAとの間で、組み込まれた。結果として得られた株CD606は、KmPDC1の野生型コピーとともに、KmPDC1遺伝子座に組み込まれた単一コピーのpBH8を有する。
【0141】
CD21を、250mLのバッフル型フラスコ中の100g/Lのグルコースを補充した50mLのYPD培地(10 g/L 酵母抽出物, 20 g/L ペプトン、20 g/L グルコース及び2%アガーを含む)に、OD600が0.1となるように植菌した。培養液を、30℃-250rpmで16時間培養した。最終OD600は、12となった。10mLの培養液から細胞を、遠心により回収し、エレクトロポレーションバッファー(10 mM Tris-Cl, 270 mM スクロース, 1 mM MgCl2, pH 7.5)で1回洗浄した。この細胞を、インキュベーションバッファー(YPD + 25 mM DTT, 20mM HEPES, pH 8.0)に再度懸濁し、30℃-250rpmで30分インキュベートした。この細胞を、遠心により回収し、エレクトロポレーションバッファーで1回洗浄し、1mLのエレクトロポレーションバッファーに再度懸濁した。そして、一部(400μL)の細胞を、0.4cmのエレクトロポレーションキュベットに移した。切断していない12μgのpBH8を、合計50μlとして、キュベットに添加し、細胞を、1.8kV、1000Ω、25μFの条件で、エレクトロポレーションした。細胞を、50mLスクリューキャップ付きファルコンチューブ中の1mLのYPDに移し、30℃-250rpmで4時間インキュベートした後、300μg/mLのG418を含むYPDプレートにプレーティングして選択した。形質転換株を、37℃で2日間生育させ、新しい選択プレートに、再度、ストリークした。
【0142】
pBH8の L. helveticus L-LDH遺伝子のすぐ上流のKmPDC1の側面DNAと、 K. marxianus 染色体のKmPDC1遺伝子のすぐ上流のDNAとの間の、KmPDC1遺伝子座へのpBH8の適切な組み込みは、プライマーPDC1Chrmosome5’:5'-AAG CAC CAA GGC CTT CAA CAG-3'(配列番号33)及びL-LDH3’:5'-TCG ATA TCG CTA AGG AAC GCG-3'(配列番号34)を使用して確認した。このプライマーは、KmPDC1遺伝子の上流の染色体DNAであって、pBH8に取り込まれた相同性部分の外側のDNAと、 L. helveticus L-LDH遺伝子との間で、2.7kbpの産物を増幅するように設計した。温度サイクルは、最初に94℃-2分で反応混合物をインキュベーションした後、94℃-30秒、55℃-30秒及び72℃-3分を35サイクル行い、最後に72℃-7分のインキュベーションを続けた。PCRで分析した10個のうち、4個の形質転換株が、予想される2.7kbpのPCR産物を産出した。
【0143】
KmPDC1遺伝子座を増幅するために設計したプライマーPDC15’:5'-CGC AAA GAA AAG CTC CAC ACC-3'(配列番号35)及びPDC13’:5'-CCC ATA CGC TTA TAA TCC CCC-3'(配列番号36)を使用したPCRは、2つの産物を生み出す。第1の産物は、 K. marxianus PDC1に対応する1.7kbpの断片であり、第2の産物は、 L. helveticus L-LDHに対応する1.0kbpの断片である。温度サイクルは、最初に94℃-2分で反応混合物をインキュベーションした後、94℃-30秒、55℃-30秒及び72℃-2分を35サイクル行い、最後に72℃-5分のインキュベーションを続けた。さらに単一コピーの組み込みを確認するために、G418耐性遺伝子をコードする領域をプローブとしたサザンブロット解析を行った。分析した3個の形質転換株のうち、1個が、pHB8の1コピーの組み込みと一致する適切なバンドパターンを示した。1コピーのpBH8がKmPDC1遺伝子座に位置していることを、PCR及びサザンブロット解析により確認した株を、CD606とした。
【0144】
これらの結果は、CD21のKmPDC1遺伝子座を標的とした形質転換されたpBH8DNAの組み込みが、KmPDC1プロモーター配列の間で起こっていたことを示す。
【0145】
(実施例3D)
CD606に由来する K. marxianus 株CD607及びCD608のコンストラクション
標的PDC1遺伝子の欠失を促すため、非選択培地で、CD606を、数回の増殖期間にわたり、増殖させた。 L. helveticus L-LDH遺伝子が、KmPDC1遺伝子と入れ替わった株を単離した。この株は、G418耐性マーカー遺伝子も失っており、G418存在下で生育阻害を受けることで確認される、G418感受性の表現型を示した。neoR遺伝子の欠失は、サザンブロット解析により確認された。さらに、この株は、嫌気性条件で生育することができなかった。
【0146】
CD606を、YPDアガープレート上にプレーティングし、37℃で一晩、約16時間インキュベートした。コロニーのスワッブ(swab)を、新しいYPDアガープレートに移した。この手順を、4回繰り返した。4ラウンドのYPDアガープレート上での非選択増殖に続いて、6つの250mLのバッフル型振とうフラスコ中の100g/Lのグルコース及び42g/LのCaCO3を補充した50mLのYPD培地に、4ラウンドの非選択YPDアガープレート増殖後のプレートから、OD600が0.1となるように植菌した。振とうフラスコを30℃、250rpmで、16時間インキュベートした。次に、前記培養液を、豆粒大のコロニーのスワッブの連続稀釈法によって、PBS(リン酸緩衝生理食塩水)で稀釈した。豆粒大のコロニーのスワッブを1mlのPBSに懸濁した初代懸濁液の、10-5、10-6及び10-7稀釈物を、YPDアガープレートに使用した。
【0147】
さらに、4ラウンドの非選択YPDアガープレート増殖後のコロニーのスワッブを、1mLのPBS(リン酸緩衝生理食塩水;137 mN NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4 pH 7.4)に懸濁し、引き続き希釈し、YPDアガープレート上に単一コロニーを形成するようにプレーティングした。単一コロニーをYPDアガー上にプレーティングした後、メーカーの指示に従って使用した嫌気性チャンバー(Becton Dickinson, "GasPak System"3つのガスパックを使用)に置いた。引き続き、37℃で2日間培養した後、嫌気性チャンバーを開き、嫌気性条件化で生育したコロニーに印をつけた。そのプレートは、もう一日37℃でインキュベートした。好気性条件で生育したが、嫌気性条件で生育しなかったコロニーを、3つにプレーティングしてスクリーニングした。採用したスクリーニングの条件は、好気性YPDプレート、嫌気性YPDプレート、および300μg/mLのG418が補充されたYPDプレートであった。2個の形質転換株が、G418感受性と嫌気性条件化での生育能力の欠落という望ましい表現型を示すことが判明し、これらを、CD607及びCD608とした。
【0148】
KmPDC1と L. helveticus L-LDHとの入れ替わりの確認は、プライマーPDC15’(配列番号35)及びPDC13’(配列番号36)を用いて行った。プライマーは、CD607及びCD608の染色体DNAを鋳型にして、PDC1遺伝子座を増幅するように設計した。温度サイクルは、最初に94℃-2分で反応混合物をインキュベーションした後、94℃-30秒、55℃-30秒及び72℃-2分を35サイクル行い、最後に72℃-5分のインキュベーションを続けた。どちらの株から得た染色体DNAも、 L. helveticus L-LDHに対応する単一の1.0kbpPCR産物を産出した。G418遺伝子とハイブリダイズするように設計されたG418遺伝子の全体のコード領域をプローブとしたサザンブロット解析は、前記PCR産物にG418遺伝子が存在しないことを示した。KmPDC1のコード領域のプローブを用いたサザンブロット解析では、バンドが現れなかった。このことは、KmPDC1のすぐ上流及び下流にハイブリダイズするように設計されたKmPDC1のコード配列プローブが存在しないことを示す。(使用されたプローブは、PDC1遺伝子のすぐ上流及び下流の染色体領域に相同性のあるDNA断片であった。プローブは、オリゴマー5’プローブ=CA110及びCA111;3’プローブ=CA112及びCA113を使用してPCRで増幅した。)KmPDC1がL. helveticus L-LDHとの入れ替ったことと一致するサイズのバンドが生じた。
【0149】
これらの結果は、KmPDC1遺伝子が、 S. cerevisiaeのScPDC1プロモーター及びScGAL10ターミネーターに動作可能に連結したG418耐性遺伝子を含むpBH8プラスミドのバックボーンとともに、CD606の染色体から失われ、その結果、KmPDC1遺伝子が、SbfI部位に隣接する L. helveticus L-LDH遺伝子と、ちょうど入れ替わった株が得られたことを示す。
【0150】
(実施例3E)
CaCO3緩衝複合培地におけるCD607によるL-乳酸の産生
CD607株を、YPDアガープレートから、OD600が0.1となるように植菌し、100g/Lのグルコース及び50g/LのCaCO3を補充した50mLのYPD培地を含む250mLのバッフル型振とうフラスコを、30℃-250rpmで、16時間インキュベートした。フラスコ内に残存グルコースが残っていること、及び、細胞が対数増殖期であることを確認した後、4g/L細胞乾燥重量当量を遠心により回収し、250mLのバッフル型振とうフラスコ中の100g/Lのグルコース及び50g/LのCaCO3を補充した50mLのYPD培地に再度懸濁した。この培養液を、30℃-70rpmで(48時間)培養した。様々な時間(0,8,24及び48時間の間隔)にサンプルを回収し、細胞をフィルターによりサンプルから取り除いた。培養上清を、HPLCにより、グルコース、乳酸塩、ピルビン酸塩及びエタノールについて、分析した。
【0151】
24時間後、CD607株は、59〜61g/Lのグルコースを消費し、58〜60g/Lの乳酸塩及び0.3g/Lのピルビン酸塩を産生した。48時間後、CD607株は、101〜104g/Lのグルコースを消費し、94〜99g/Lの乳酸塩及び0.4g/Lのピルビン酸塩を産生した。これらの乳酸塩の力価は、グルコースに対して90〜98%の乳酸塩の収率を示しており、よって、微好気性産生期における容積生産性は、2.1g/Lhrを超えた。48時間後、培養液中で産生された乳酸塩の濃度が高くなったため、不溶性のカルシウム乳酸塩の沈殿の形成(「ケーキ化」)がおこり、それ以上の解析が妨げられた。酵素を用いた乳酸解析から、前記乳酸塩は、鏡像異性体的に99%を越える純度のL-乳酸塩であることが示された。CD607株の培養液ではエタノールが検出されなかった。このことは、唯一のPCD1コピーが、L−LDH1コピーに置き換わったことを示す。
【0152】
対照的に、CD606株(PDC遺伝子及び L. helveticus L-LDH遺伝子を含む)は、同じ発酵条件において、124g/Lのグルコースを消費し、83g/Lの乳酸塩、20g/Lのエタノール及び0.2g/Lのピルビン酸塩を産生した。
【0153】
(実施例3F)
バッファー剤を補充しない複合培地における遊離L-乳酸の産生
CD607株を、YPDアガープレートから、OD600が0.1となるように植菌し、100g/Lのグルコースを補充した50mLのYPD培地を含む250mLのバッフル型振とうフラスコを、30℃-250rpmで、16時間インキュベートした。フラスコ内に残存グルコースが残っていること、及び、細胞が対数増殖期であることを確認した後、2g/L細胞乾燥重量当量を遠心により回収し、250mLのバッフル型振とうフラスコ中の追加の40g/Lのグルコースを補充した50mLのYPD培地に再度懸濁した。前記培養液を、30℃-70rpmで(72時間)培養した。様々な時間(産生開始から0,24,47及び72時間後)にサンプルを回収した。培養液サンプルを、グルコース、乳酸塩(総計及び遊離酸)、ピルビン酸塩、グリセロール、酢酸塩及びエタノールについて、HPLCにより分析した。pHは、産生の開始と終了の時点で測定した。
【0154】
72時間後、CD607株は、10.8g/Lのグルコースを消費し、9.1g/Lの乳酸塩、0.5g/Lの酢酸塩及び1.2g/Lのピルビン酸を産生した。これらの乳酸塩の力価は、グルコースに対して84%の乳酸の収率を示しており、よって、微好気性産生期における容積生産性は、0.1g/Lhrを超えた。酵素を用いた乳酸塩解析から、前記乳酸塩は、鏡像異性体的に99%を越える純度のL-乳酸塩であることが示された。CD607株の培養液ではエタノールが検出されなかった。さらに、グリセロールも検出されなかった。培養液のpHは、有機酸の産生により、産生期中に、6.50から3.05に減少した。HPLCによる遊離乳酸(プロトン化乳酸)を測定したところ、9.1g/Lの遊離乳酸であった。このことは、基本的には、CD607により産生される全ての乳酸が、そのプロトン化型であることを示す。
【0155】
以上の開示は、本発明の特定の具体的実施態様であって、これらの修正又は代替であって均等なものは、すべて、添付の特許請求の範囲に示される本発明の精神と範囲に含まれるものである。ここで引用した全ての参照文献は、その全体を本願に引用して援用する。
【図面の簡単な説明】
【0156】
【図1】図1は、pNC2プラスミドを描いたダイヤグラムである。
【図2】図2は、pNC4プラスミドを描いたダイヤグラムである。
【図2a】図2aは、遺伝子発現のための Saccharomyces cerevisiae ScPGK1プロモーター、マルチクローニングサイト及びScGAL10ターミネーターを含む1.235kbpのNotI処理断片の配列である。
【図2b】図2bは、 Saccharomyces cerevisiae PDC1プロモーター、ScGAL10ターミネーター及び前記ターミネーターとプロモーターと間のマルチクローニングサイトを含む1.3kbpのNotI処理断片の配列である。
【図3】図3は、pVR22プラスミドを描いたダイヤグラムである。
【図4】図4は、pVR29プラスミドを描いたダイヤグラムである。
【図5】図5は、pPS1プラスミド及びpNC14プラスミドに基づくpPS9のコンストラクションを描いたダイヤグラムである。
【図6】図6は、 L. helveticus L-LDH遺伝子及びG418耐性マーカーを含むpVR39プラスミドを描いたダイヤグラムである。
【図7】図7は、 L. helveticus のL-LDHをコードする遺伝子、 S. cerevisiase pTEF1プロモーター及びE. coli EM7プロモーターを含むpVR1プラスミドを描いたダイヤグラムである。
【図8】図8は、pSO18プラスミドを描いたダイヤグラムである。
【図9】図9は、pSO19プラスミドを描いたダイヤグラムである。
【図10】図10は、pSO20プラスミドを描いたダイヤグラムである。
【図11】図11は、 L. helveticus L-LDH遺伝子及びG418耐性マーカーを含むpVR41プラスミドを描いたダイヤグラムである。
【図12】図12は、pVR24プラスミドを描いたダイヤグラムである。
【図13】図13は、 B. megaterium L-LDH遺伝子及びG418耐性マーカーを含むpCA3プラスミドを描いたダイヤグラムである。
【図14】図14は、 B. megaterium L-LDH遺伝子及びハイグロマイシン耐性遺伝子を含むpVR38プラスミドを描いたダイヤグラムである。
【図15】図15aは、pBH5aのコンストラクションを描いたダイヤグラムである。図15bは、pVR29及びpBH5aに基づくpBH5bのコンストラクションを描いたダイヤグラムである。
【図16】図16は、pBH9プラスミドを描いたダイヤグラムである。
【図17】図17A及び17Bは、本発明の態様に従った、外因性LDH遺伝子の酵母染色体への標的組み込みに関する遺伝学的出来事の説明図である。
【特許請求の範囲】
【請求項1】
酵母種の細胞を形質転換するための組み換え核酸であって、前記酵母種に対して外因性であるLDH(乳酸脱水素酵素)遺伝子、及び、少なくとも1つの選択マーカーを含み、前記LDH遺伝子が、上流及び下流の側面配列と共有結合しており、前記側面配列が、それぞれ、前記酵母種に対して野生型である標的遺伝子の上流及び下流の側面配列と相同である組み換え核酸。
【請求項2】
請求項1に記載の組み換え核酸であって、前記LDH遺伝子の上流の側面配列が、前記標的遺伝子のプロモーター配列と相同なプロモーター配列を含み、前記LDH遺伝子の下流の側面配列が、前記標的遺伝子のターミネーター配列と相同なターミネーター配列を含む組み換え核酸。
【請求項3】
請求項2に記載の組み換え核酸であって、前記選択マーカーが、プロモーター及びターミネーター配列に動作可能に連結する組み換え核酸。
【請求項4】
請求項2に記載の組み換え核酸であって、前記標的遺伝子が、PDC(ピルビン酸デカルボキシラーゼ)遺伝子である組み換え核酸。
【請求項5】
請求項3に記載の組み換え核酸であって、前記選択マーカーが、上流側面配列、LDH遺伝子及び下流側面配列の配列を中断しない組み換え核酸。
【請求項6】
請求項1に記載の組み換え核酸であって、前記外因性乳酸脱水素酵素遺伝子が、 L. helveticus 又は B. megaterium に由来する組み換え核酸。
【請求項7】
酵母種の組み換え細胞であって、前記酵母種は、そのゲノムの遺伝子座に標的遺伝子を有し、前記細胞は、そのゲノムに組み込まれて前記標的遺伝子のプロモーター及びターミネーター配列の機能部分の転写コントロール下となった外因性乳酸脱水素酵素(LDH)遺伝子を有し、前記LDH遺伝子が、前記標的遺伝子の遺伝子座に組み込まれ、前記標的遺伝子が、前記細胞のゲノムから削除されている細胞。
【請求項8】
請求項7に記載の細胞であって、前記標的遺伝子が、ピルビン酸デカルボキシラーゼ遺伝子である細胞。
【請求項9】
請求項7に記載の細胞であって、 Saccharomyces 属、 Kluyveromyces 属、 Candida 属、 Pichia 属、Hansenula 属、 Torulopsis 属又は Yamadazyma 属の種である細胞。
【請求項10】
請求項9に記載の細胞であって、前記酵母種が、 K. marxianus 、 K. thermotolerans 、 K. lactis 又は C. sonorensis である細胞。
【請求項11】
請求項8に記載の細胞であって、前記乳酸脱水素酵素遺伝子が、 L. helveticus 又は B. megaterium に由来する細胞。
【請求項12】
細胞に外因性乳酸脱水素酵素遺伝子を組み込む方法であって、前記組み込み以前の前記細胞が、そのゲノムの遺伝子座に標的遺伝子を有し、(a)乳酸脱水素酵素(LDH)遺伝子、前記LDH遺伝子の上流及び下流の側面配列、並びに少なくとも1つの選択マーカー遺伝子を含む組み換え核酸で、前記細胞を形質転換するステップであって、前記側面配列が前記標的遺伝子の上流及び下流の側面配列と相同であり、前記LDH遺伝子が、単一交差が起こる際に、前記標的遺伝子の遺伝子座に近接した前記細胞のゲノムに挿入されるステップ、(b)前記LDH遺伝子及び選択マーカー遺伝子を含む第1の形質転換株を、選択するステップ、(c)前記第1の形質転換株を、非選択条件で培養するステップ、並びに、(d)前記第1の形質転換株の中から、前記LDH遺伝子を有し、かつ、前記選択マーカー遺伝子及び前記標的遺伝子が削除された第2の形質転換細胞を選択するステップ、を含む方法。
【請求項13】
請求項12に記載の方法であって、前記LDH遺伝子の上流の側面配列が、前記標的遺伝子のプロモーター配列と相同なプロモーター配列を含み、前記LDH遺伝子の下流の側面配列が、前記標的遺伝子のターミネーター配列と相同なターミネーター配列を含む方法。
【請求項14】
請求項13に記載の方法であって、前記標的遺伝子が、ピルビン酸デヒドロゲナーゼ遺伝子である方法。
【請求項15】
請求項12に記載の方法であって、前記組み換え核酸が、さらに、プロモーター及びターミネーター配列に動作可能に連結しているマーカー遺伝子を含む方法。
【請求項16】
請求項15に記載の方法であって、前記マーカー遺伝子に動作可能に連結しているプロモーター及びターミネーター配列が、前記酵母種に対して野生型ではない方法。
【請求項17】
K. marxianus 種の細胞であって、そのゲノムに挿入された複数の外因性乳酸脱水素酵素遺伝子を有し、それぞれが、機能的なプロモーター及びターミネーター配列のコントロール下に置かれ、前記 K. marxianus 細胞のゲノムが、さらに、機能的なピルビン酸デカルボキシラーゼ遺伝子を含む細胞。
【請求項18】
請求項17に記載の細胞であって、前記機能的なピルビン酸デカルボキシラーゼ遺伝子が、 K. marxianus に対して野生型である細胞。
【請求項19】
請求項18に記載の細胞であって、前記機能的なピルビン酸デカルボキシラーゼ遺伝子が、PDC1遺伝子である細胞。
【請求項20】
請求項19に記載の細胞であって、前記外因性乳酸脱水素酵素遺伝子が、 L. helveticus 又は B. megaterium に由来する細胞。
【請求項21】
請求項20に記載の細胞であって、 L. helveticus に由来する乳酸脱水素酵素遺伝子が、複数コピー含まれる細胞。
【請求項22】
請求項20に記載の細胞であって、 B. megaterium に由来する乳酸脱水素酵素遺伝子が、複数コピー含まれる細胞。
【請求項23】
請求項20に記載の細胞であって、少なくとも1コピーの L. helveticus に由来する乳酸脱水素酵素遺伝子、及び、少なくとも1コピーの B. megaterium に由来する乳酸脱水素酵素遺伝子が含まれる細胞。
【請求項24】
請求項21に記載の細胞であって、少なくとも1コピーの乳酸脱水素酵素遺伝子が、TEF1プロモーターの転写コントロール下に置かれる細胞。
【請求項25】
請求項22に記載の細胞であって、少なくとも1コピーの乳酸脱水素酵素遺伝子が、TEF1プロモーターの転写コントロール下に置かれる細胞。
【請求項26】
請求項21に記載の細胞であって、少なくとも1コピーの乳酸脱水素酵素遺伝子が、PGKプロモーターの転写コントロール下に置かれる細胞。
【請求項27】
請求項22に記載の細胞であって、少なくとも1コピーの乳酸脱水素酵素遺伝子が、PGKプロモーターの転写コントロール下に置かれる細胞。
【請求項28】
請求項21に記載の細胞であって、少なくとも1コピーの乳酸脱水素酵素遺伝子が、GAL10ターミネーターの転写コントロール下に置かれる細胞。
【請求項29】
請求項22に記載の細胞であって、少なくとも1コピーの乳酸脱水素酵素遺伝子が、GAL10ターミネーターの転写コントロール下に置かれる細胞。
【請求項30】
請求項23に記載の細胞であって、少なくとも1つの乳酸脱水素酵素遺伝子が、TEF1プロモーターの転写コントロール下に置かれる細胞。
【請求項31】
請求項23に記載の細胞であって、少なくとも1つの乳酸脱水素酵素遺伝子が、PGKプロモーターの転写コントロール下に置かれる細胞。
【請求項32】
請求項23に記載の細胞であって、少なくとも1つの乳酸脱水素酵素遺伝子が、GAL10ターミネーターの転写コントロール下に置かれる細胞。
【請求項33】
糖を乳酸に発酵する方法であって、請求項7に記載の細胞を、前記細胞が発酵可能な糖を含む培地中、発酵条件下で培養することを含む方法。
【請求項34】
請求項33に記載の方法であって、前記培地のpHが、発酵の間中、約pH5〜約pH8の範囲で維持される方法。
【請求項35】
請求項33に記載の方法であって、前記細胞により産生される乳酸に起因して、前記培地のpHが、発酵の間に、約2.5〜約5.0に低下する方法。
【請求項36】
請求項34に記載の方法であって、前記細胞により産生される乳酸に起因して、前記培地のpHが、発酵の間に、約2.5〜約3.8に低下する方法。
【請求項37】
請求項33に記載の方法であって、さらに、乳酸の少なくとも一部をラクチドに変換するステップを含む方法。
【請求項38】
請求項37に記載の方法であって、さらに、前記ラクチドを重合し、ポリ乳酸ポリマー又はコポリマーを形成するステップを含む方法。
【請求項39】
乳酸の生産方法であって、請求項7に記載の細胞を、前記細胞が発酵可能な糖を含む培地中、発酵条件下で培養することを含む生産方法。
【請求項40】
請求項39に記載の生産方法であって、前記培地のpHが、発酵の間中、約pH5〜約pH8の範囲で維持される生産方法。
【請求項41】
請求項39に記載の生産方法であって、前記培地のpHが、前記細胞により産生される乳酸に起因して、発酵の間に、約2.5〜約5.0に低下する生産方法。
【請求項42】
請求項41に記載の生産方法であって、前記培地のpHが、前記細胞により産生される乳酸に起因して、発酵の間に、約2.5〜約3.8に低下する生産方法。
【請求項43】
請求項39に記載の生産方法であって、さらに、乳酸の少なくとも一部をラクチドに変換するステップを含む生産方法。
【請求項44】
請求項43に記載の生産方法であって、さらに、前記ラクチドを重合し、ポリ乳酸ポリマー又はコポリマーを形成するステップを含む生産方法。
【請求項45】
糖を乳酸に発酵する方法であって、請求項17に記載の細胞を、前記細胞が発酵可能な糖を含む培地中、発酵条件下で培養することを含む方法。
【請求項46】
請求項45に記載の方法であって、前記培地のpHが、発酵の間中、約pH5〜約pH8の範囲で維持される方法。
【請求項47】
請求項45に記載の方法であって、前記培地のpHが、前記細胞により産生される乳酸に起因して、発酵の間に、約2.5〜約5.0に低下する方法。
【請求項48】
請求項47に記載の方法であって、前記培地のpHが、前記細胞により産生される乳酸に起因して、発酵の間に、約2.5〜約3.8に低下する方法。
【請求項49】
乳酸の生産方法であって、請求項17に記載の細胞を、前記細胞が発酵可能な糖を含む培地中、発酵条件下で培養することを含む生産方法。
【請求項50】
請求項45に記載の方法であって、さらに、乳酸の少なくとも一部をラクチドに変換するステップを含む方法。
【請求項51】
請求項50に記載の方法であって、さらに、前記ラクチドを重合し、ポリ乳酸ポリマー又はコポリマーを形成するステップを含む方法。
【請求項52】
請求項49に記載の生産方法であって、前記培地のpHが、発酵の間中、約pH5〜約pH8の範囲で維持される生産方法。
【請求項53】
請求項49に記載の生産方法であって、前記細胞により産生される乳酸に起因して、前記培地のpHが、発酵の間に、約2.5〜約5.0に低下する生産方法。
【請求項54】
請求項53に記載の生産方法であって、前記細胞により産生される乳酸に起因して、前記培地のpHが、発酵の間に、約2.5〜約3.8に低下する生産方法。
【請求項55】
請求項49に記載の生産方法であって、さらに、乳酸の少なくとも一部をラクチドに変換するステップを含む生産方法。
【請求項56】
請求項55に記載の生産方法であって、さらに、前記ラクチドを重合し、ポリ乳酸ポリマー又はコポリマーを形成するステップを含む生産方法。
【請求項1】
酵母種の細胞を形質転換するための組み換え核酸であって、前記酵母種に対して外因性であるLDH(乳酸脱水素酵素)遺伝子、及び、少なくとも1つの選択マーカーを含み、前記LDH遺伝子が、上流及び下流の側面配列と共有結合しており、前記側面配列が、それぞれ、前記酵母種に対して野生型である標的遺伝子の上流及び下流の側面配列と相同である組み換え核酸。
【請求項2】
請求項1に記載の組み換え核酸であって、前記LDH遺伝子の上流の側面配列が、前記標的遺伝子のプロモーター配列と相同なプロモーター配列を含み、前記LDH遺伝子の下流の側面配列が、前記標的遺伝子のターミネーター配列と相同なターミネーター配列を含む組み換え核酸。
【請求項3】
請求項2に記載の組み換え核酸であって、前記選択マーカーが、プロモーター及びターミネーター配列に動作可能に連結する組み換え核酸。
【請求項4】
請求項2に記載の組み換え核酸であって、前記標的遺伝子が、PDC(ピルビン酸デカルボキシラーゼ)遺伝子である組み換え核酸。
【請求項5】
請求項3に記載の組み換え核酸であって、前記選択マーカーが、上流側面配列、LDH遺伝子及び下流側面配列の配列を中断しない組み換え核酸。
【請求項6】
請求項1に記載の組み換え核酸であって、前記外因性乳酸脱水素酵素遺伝子が、 L. helveticus 又は B. megaterium に由来する組み換え核酸。
【請求項7】
酵母種の組み換え細胞であって、前記酵母種は、そのゲノムの遺伝子座に標的遺伝子を有し、前記細胞は、そのゲノムに組み込まれて前記標的遺伝子のプロモーター及びターミネーター配列の機能部分の転写コントロール下となった外因性乳酸脱水素酵素(LDH)遺伝子を有し、前記LDH遺伝子が、前記標的遺伝子の遺伝子座に組み込まれ、前記標的遺伝子が、前記細胞のゲノムから削除されている細胞。
【請求項8】
請求項7に記載の細胞であって、前記標的遺伝子が、ピルビン酸デカルボキシラーゼ遺伝子である細胞。
【請求項9】
請求項7に記載の細胞であって、 Saccharomyces 属、 Kluyveromyces 属、 Candida 属、 Pichia 属、Hansenula 属、 Torulopsis 属又は Yamadazyma 属の種である細胞。
【請求項10】
請求項9に記載の細胞であって、前記酵母種が、 K. marxianus 、 K. thermotolerans 、 K. lactis 又は C. sonorensis である細胞。
【請求項11】
請求項8に記載の細胞であって、前記乳酸脱水素酵素遺伝子が、 L. helveticus 又は B. megaterium に由来する細胞。
【請求項12】
細胞に外因性乳酸脱水素酵素遺伝子を組み込む方法であって、前記組み込み以前の前記細胞が、そのゲノムの遺伝子座に標的遺伝子を有し、(a)乳酸脱水素酵素(LDH)遺伝子、前記LDH遺伝子の上流及び下流の側面配列、並びに少なくとも1つの選択マーカー遺伝子を含む組み換え核酸で、前記細胞を形質転換するステップであって、前記側面配列が前記標的遺伝子の上流及び下流の側面配列と相同であり、前記LDH遺伝子が、単一交差が起こる際に、前記標的遺伝子の遺伝子座に近接した前記細胞のゲノムに挿入されるステップ、(b)前記LDH遺伝子及び選択マーカー遺伝子を含む第1の形質転換株を、選択するステップ、(c)前記第1の形質転換株を、非選択条件で培養するステップ、並びに、(d)前記第1の形質転換株の中から、前記LDH遺伝子を有し、かつ、前記選択マーカー遺伝子及び前記標的遺伝子が削除された第2の形質転換細胞を選択するステップ、を含む方法。
【請求項13】
請求項12に記載の方法であって、前記LDH遺伝子の上流の側面配列が、前記標的遺伝子のプロモーター配列と相同なプロモーター配列を含み、前記LDH遺伝子の下流の側面配列が、前記標的遺伝子のターミネーター配列と相同なターミネーター配列を含む方法。
【請求項14】
請求項13に記載の方法であって、前記標的遺伝子が、ピルビン酸デヒドロゲナーゼ遺伝子である方法。
【請求項15】
請求項12に記載の方法であって、前記組み換え核酸が、さらに、プロモーター及びターミネーター配列に動作可能に連結しているマーカー遺伝子を含む方法。
【請求項16】
請求項15に記載の方法であって、前記マーカー遺伝子に動作可能に連結しているプロモーター及びターミネーター配列が、前記酵母種に対して野生型ではない方法。
【請求項17】
K. marxianus 種の細胞であって、そのゲノムに挿入された複数の外因性乳酸脱水素酵素遺伝子を有し、それぞれが、機能的なプロモーター及びターミネーター配列のコントロール下に置かれ、前記 K. marxianus 細胞のゲノムが、さらに、機能的なピルビン酸デカルボキシラーゼ遺伝子を含む細胞。
【請求項18】
請求項17に記載の細胞であって、前記機能的なピルビン酸デカルボキシラーゼ遺伝子が、 K. marxianus に対して野生型である細胞。
【請求項19】
請求項18に記載の細胞であって、前記機能的なピルビン酸デカルボキシラーゼ遺伝子が、PDC1遺伝子である細胞。
【請求項20】
請求項19に記載の細胞であって、前記外因性乳酸脱水素酵素遺伝子が、 L. helveticus 又は B. megaterium に由来する細胞。
【請求項21】
請求項20に記載の細胞であって、 L. helveticus に由来する乳酸脱水素酵素遺伝子が、複数コピー含まれる細胞。
【請求項22】
請求項20に記載の細胞であって、 B. megaterium に由来する乳酸脱水素酵素遺伝子が、複数コピー含まれる細胞。
【請求項23】
請求項20に記載の細胞であって、少なくとも1コピーの L. helveticus に由来する乳酸脱水素酵素遺伝子、及び、少なくとも1コピーの B. megaterium に由来する乳酸脱水素酵素遺伝子が含まれる細胞。
【請求項24】
請求項21に記載の細胞であって、少なくとも1コピーの乳酸脱水素酵素遺伝子が、TEF1プロモーターの転写コントロール下に置かれる細胞。
【請求項25】
請求項22に記載の細胞であって、少なくとも1コピーの乳酸脱水素酵素遺伝子が、TEF1プロモーターの転写コントロール下に置かれる細胞。
【請求項26】
請求項21に記載の細胞であって、少なくとも1コピーの乳酸脱水素酵素遺伝子が、PGKプロモーターの転写コントロール下に置かれる細胞。
【請求項27】
請求項22に記載の細胞であって、少なくとも1コピーの乳酸脱水素酵素遺伝子が、PGKプロモーターの転写コントロール下に置かれる細胞。
【請求項28】
請求項21に記載の細胞であって、少なくとも1コピーの乳酸脱水素酵素遺伝子が、GAL10ターミネーターの転写コントロール下に置かれる細胞。
【請求項29】
請求項22に記載の細胞であって、少なくとも1コピーの乳酸脱水素酵素遺伝子が、GAL10ターミネーターの転写コントロール下に置かれる細胞。
【請求項30】
請求項23に記載の細胞であって、少なくとも1つの乳酸脱水素酵素遺伝子が、TEF1プロモーターの転写コントロール下に置かれる細胞。
【請求項31】
請求項23に記載の細胞であって、少なくとも1つの乳酸脱水素酵素遺伝子が、PGKプロモーターの転写コントロール下に置かれる細胞。
【請求項32】
請求項23に記載の細胞であって、少なくとも1つの乳酸脱水素酵素遺伝子が、GAL10ターミネーターの転写コントロール下に置かれる細胞。
【請求項33】
糖を乳酸に発酵する方法であって、請求項7に記載の細胞を、前記細胞が発酵可能な糖を含む培地中、発酵条件下で培養することを含む方法。
【請求項34】
請求項33に記載の方法であって、前記培地のpHが、発酵の間中、約pH5〜約pH8の範囲で維持される方法。
【請求項35】
請求項33に記載の方法であって、前記細胞により産生される乳酸に起因して、前記培地のpHが、発酵の間に、約2.5〜約5.0に低下する方法。
【請求項36】
請求項34に記載の方法であって、前記細胞により産生される乳酸に起因して、前記培地のpHが、発酵の間に、約2.5〜約3.8に低下する方法。
【請求項37】
請求項33に記載の方法であって、さらに、乳酸の少なくとも一部をラクチドに変換するステップを含む方法。
【請求項38】
請求項37に記載の方法であって、さらに、前記ラクチドを重合し、ポリ乳酸ポリマー又はコポリマーを形成するステップを含む方法。
【請求項39】
乳酸の生産方法であって、請求項7に記載の細胞を、前記細胞が発酵可能な糖を含む培地中、発酵条件下で培養することを含む生産方法。
【請求項40】
請求項39に記載の生産方法であって、前記培地のpHが、発酵の間中、約pH5〜約pH8の範囲で維持される生産方法。
【請求項41】
請求項39に記載の生産方法であって、前記培地のpHが、前記細胞により産生される乳酸に起因して、発酵の間に、約2.5〜約5.0に低下する生産方法。
【請求項42】
請求項41に記載の生産方法であって、前記培地のpHが、前記細胞により産生される乳酸に起因して、発酵の間に、約2.5〜約3.8に低下する生産方法。
【請求項43】
請求項39に記載の生産方法であって、さらに、乳酸の少なくとも一部をラクチドに変換するステップを含む生産方法。
【請求項44】
請求項43に記載の生産方法であって、さらに、前記ラクチドを重合し、ポリ乳酸ポリマー又はコポリマーを形成するステップを含む生産方法。
【請求項45】
糖を乳酸に発酵する方法であって、請求項17に記載の細胞を、前記細胞が発酵可能な糖を含む培地中、発酵条件下で培養することを含む方法。
【請求項46】
請求項45に記載の方法であって、前記培地のpHが、発酵の間中、約pH5〜約pH8の範囲で維持される方法。
【請求項47】
請求項45に記載の方法であって、前記培地のpHが、前記細胞により産生される乳酸に起因して、発酵の間に、約2.5〜約5.0に低下する方法。
【請求項48】
請求項47に記載の方法であって、前記培地のpHが、前記細胞により産生される乳酸に起因して、発酵の間に、約2.5〜約3.8に低下する方法。
【請求項49】
乳酸の生産方法であって、請求項17に記載の細胞を、前記細胞が発酵可能な糖を含む培地中、発酵条件下で培養することを含む生産方法。
【請求項50】
請求項45に記載の方法であって、さらに、乳酸の少なくとも一部をラクチドに変換するステップを含む方法。
【請求項51】
請求項50に記載の方法であって、さらに、前記ラクチドを重合し、ポリ乳酸ポリマー又はコポリマーを形成するステップを含む方法。
【請求項52】
請求項49に記載の生産方法であって、前記培地のpHが、発酵の間中、約pH5〜約pH8の範囲で維持される生産方法。
【請求項53】
請求項49に記載の生産方法であって、前記細胞により産生される乳酸に起因して、前記培地のpHが、発酵の間に、約2.5〜約5.0に低下する生産方法。
【請求項54】
請求項53に記載の生産方法であって、前記細胞により産生される乳酸に起因して、前記培地のpHが、発酵の間に、約2.5〜約3.8に低下する生産方法。
【請求項55】
請求項49に記載の生産方法であって、さらに、乳酸の少なくとも一部をラクチドに変換するステップを含む生産方法。
【請求項56】
請求項55に記載の生産方法であって、さらに、前記ラクチドを重合し、ポリ乳酸ポリマー又はコポリマーを形成するステップを含む生産方法。
【図1】



【図2】
【図】
【図】

【図3】
【図4】

【図5】
【図6】
【図】
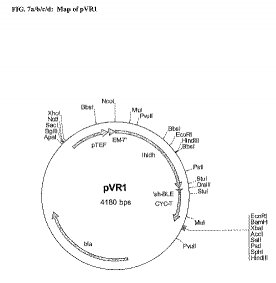
【図8】
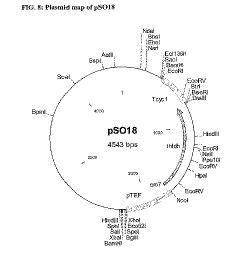
【図9】

【図10】

【図11】

【図12】
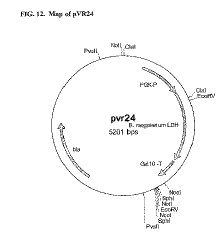
【図13】
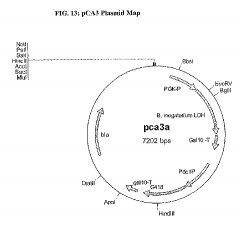
【図14】
【図】

【図16】

【図17】
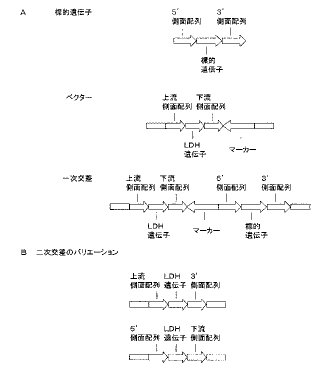
【図2】
【図】
【図】
【図3】
【図4】
【図5】
【図6】
【図】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図】
【図16】
【図17】
【公表番号】特表2005−528106(P2005−528106A)
【公表日】平成17年9月22日(2005.9.22)
【国際特許分類】
【出願番号】特願2004−510394(P2004−510394)
【出願日】平成15年5月30日(2003.5.30)
【国際出願番号】PCT/US2003/017329
【国際公開番号】WO2003/102152
【国際公開日】平成15年12月11日(2003.12.11)
【出願人】(504438060)カーギル ダウ エルエルシー (4)
【Fターム(参考)】
【公表日】平成17年9月22日(2005.9.22)
【国際特許分類】
【出願日】平成15年5月30日(2003.5.30)
【国際出願番号】PCT/US2003/017329
【国際公開番号】WO2003/102152
【国際公開日】平成15年12月11日(2003.12.11)
【出願人】(504438060)カーギル ダウ エルエルシー (4)
【Fターム(参考)】
[ Back to top ]