
酵母及びその育種方法
【課題】酵母に凝集性を付与する活性を有するポリペプチド、酵母に凝集性を付与する活性を有するアミノ酸配列をコードする遺伝子DNAを利用して、凝集性が付与または強化された酵母の育種方法を提供する。
【解決手段】特定な配列からなるアミノ酸配列において、配列の繰り返しを4回以上有するアミノ酸配列を、FLOタンパク質に組み込む遺伝子改変によって、凝集性が付与又は強化されたことを特徴とする酵母、及び、特定な配列からなるアミノ酸配列において、配列の繰り返しを4回以上有するアミノ酸配列を、FLOタンパク質に組み込む遺伝子改変によって、凝集性を付与又は強化することを特徴とする。
【解決手段】特定な配列からなるアミノ酸配列において、配列の繰り返しを4回以上有するアミノ酸配列を、FLOタンパク質に組み込む遺伝子改変によって、凝集性が付与又は強化されたことを特徴とする酵母、及び、特定な配列からなるアミノ酸配列において、配列の繰り返しを4回以上有するアミノ酸配列を、FLOタンパク質に組み込む遺伝子改変によって、凝集性を付与又は強化することを特徴とする。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は酵母に特異的なアミノ酸配列、そのDNA及び形質転換等による遺伝子改変による酵母の育種方法に関する。
【背景技術】
【0002】
細胞間の凝集は、a型細胞とα型細胞間の性的凝集、出芽娘細胞の母細胞からの未分離、非性的凝集などに起因することが知られているが、本発明は、これらのうちの非性的凝集の制御を目的とする。非性的凝集の機構を説明するモデルとしては、凝集性酵母の細胞表層にあるレクチン様タンパク質と糖鎖の結合で隣り合う酵母が結合しているとするレクチン仮説(例えば非特許文献1参照。)が有力であるが、レクチン様タンパク質の同定は成されていない。このことが、酵母凝集の制御が未だ困難である要因でもある。
【0003】
酵母の凝集性に関与する既知の遺伝子としては、FLO1、FLO5、FLO9、FLO10の特異的なレクチン様タンパク質をコードする遺伝子のファミリーがあり、染色体末端領域(テロメア周辺領域)に存在する。一方、凝集性、産膜、浸潤性増殖、基質付着に関連するタンパク質FLO11/MUC1をコードする遺伝子は、非染色体末端領域に存在することが報告されている。
【非特許文献1】J. Bacteriol.,150,878(1982)
【発明の開示】
【発明が解決しようとする課題】
【0004】
既知のFLOタンパク質(FLO1、5、9)は、配列番号1〜5記載の9アミノ酸の配列のいずれか、あるいは組み合わせた配列を1〜3回有する。
【0005】
この配列部位を構成する繰り返し配列は、FLO1では配列番号4、配列番号1、配列番号5を各1回からなる3回、FLO9では配列番号2、配列番号3を各1回からなる2回、YAL065Cでは配列番号2、配列番号3を各1回からなる2回の繰り返しがあり、FLO5では配列番号2のみ1回で繰り返し無しによって構成されている。
【0006】
また、FLO8、FLO10、MUC1(FLO11)、YAR061W、YAR062W、YHR213Wには上記の配列はなかった。
【0007】
上記の従来の酵母を用いたアルコールの発酵生産法は、酵母と生成物とを分離することが困難であるため、非効率的であり、アルコール生産性が不十分である欠点があった。この原因は酵母の凝集性の限界にあり、我々はこの問題を解明した結果、アミノ酸配列の何れかを4回以上繰り返す配列を含むことが凝集性向上に寄与することを見出し、本発明に至った。
【0008】
そこで、本発明の課題は、以下の通りである。
(1) 酵母に凝集性を付与する活性を有するポリペプチドを提供すること;
(2) 酵母に凝集性を付与する活性を有するアミノ酸配列をコードする遺伝子DNAを提供すること;
(3) 上記の遺伝子DNAを利用して、凝集性が付与または強化された酵母の育種方法を提供すること;
【0009】
本発明の他の課題は、以下の記載によって明らかとなる。
【課題を解決するための手段】
【0010】
上記課題は以下の各発明によって解決される。
【0011】
請求項1記載の発明は、配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列を、FLOタンパク質に組み込む遺伝子改変によって、凝集性が付与又は強化されたことを特徴とする酵母である。
請求項2記載の発明は、前記遺伝子改変が、前記FLOタンパク質に存在する配列番号1〜5で示されるアミノ酸配列の何れか又はこれら配列を3回以下の範囲で繰り返してなるアミノ酸配列を、前記配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列に置き換えるものであることを特徴とする請求項1記載の酵母である。
【0012】
請求項3記載の発明は、配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列をコードするDNAを、FLO遺伝子に組み込む遺伝子改変によって、凝集性が付与又は強化されたことを特徴とする酵母である。
請求項4記載の発明は、前記遺伝子改変が、前記FLO遺伝子に存在する配列番号1〜5で示されるアミノ酸配列の何れかをコードするDNA又はこれら配列を3回以下の範囲で繰り返してなるアミノ酸配列をコードするDNAを、前記配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列をコードするDNAに置き換えるものであることを特徴とする請求項3記載の酵母である。
請求項5記載の発明は、前記遺伝子改変が、前記配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列をコードするDNAが組み込まれたベクターによって成されることを特徴とする請求項1〜4の何れかに記載の酵母である。
請求項6記載の発明は、宿主である非凝集性酵母に凝集性を付与してなることを特徴とする請求項1〜5の何れかに記載の酵母である。
請求項7記載の発明は、宿主である凝集性酵母の凝集性を強化してなることを特徴とする請求項1〜5の何れかに記載の酵母である。
【0013】
請求項8記載の発明は、配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列を、FLOタンパク質に組み込む遺伝子改変によって、凝集性を付与又は強化することを特徴とする酵母の育種方法である。
請求項9記載の発明は、前記遺伝子改変が、前記FLOタンパク質に存在する配列番号1〜5で示されるアミノ酸配列の何れか又はこれら配列を3回以下の範囲で繰り返してなるアミノ酸配列を、前記配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列に置き換えるものであることを特徴とする請求項8記載の酵母の育種方法である。
【0014】
請求項10記載の発明は、配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列をコードするDNAを、FLO遺伝子に組み込む遺伝子改変によって、凝集性を付与又は強化することを特徴とする酵母の育種方法である。
請求項11記載の発明は、前記遺伝子改変が、前記FLO遺伝子に存在する配列番号1〜5で示されるアミノ酸配列の何れかをコードするDNA又はこれら配列を3回以下の範囲で繰り返してなるアミノ酸配列をコードするDNAを、前記配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列をコードするDNAに置き換えるものであることを特徴とする請求項10記載の酵母の育種方である。
請求項12記載の発明は、前記遺伝子改変が、前記配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列をコードするDNAが組み込まれたベクターによって成されることを特徴とする請求項8〜11の何れかに記載の酵母の育種方法である。
請求項13記載の発明は、宿主である非凝集性酵母に凝集性を付与することを特徴とする請求項8〜12の何れかに記載の酵母の育種方法である。
請求項14記載の発明は、宿主である凝集性酵母の凝集性を強化することを特徴とする請求項8〜12の何れかに記載の酵母の育種方法である。
【発明の効果】
【0015】
本発明によれば、酵母に凝集性を付与する活性を有するポリペプチドを提供することができ、酵母に凝集性を付与する活性を有するアミノ酸配列をコードする遺伝子DNAを利用して、凝集性が付与または強化された酵母の育種方法を提供することができる。
【図面の簡単な説明】
【0016】
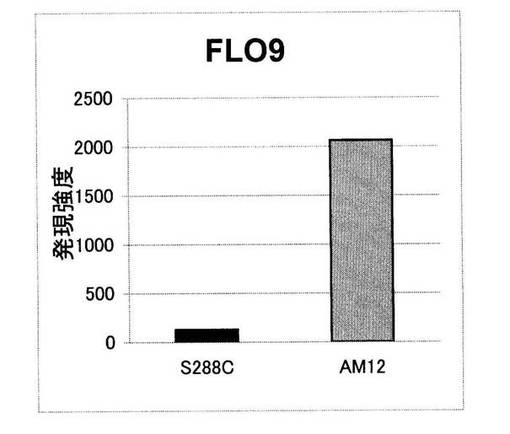
【図1】酵母AM12菌株、及び非凝集性酵母S288C菌株におけるFLO9遺伝子の発現強度の測定結果を示した図
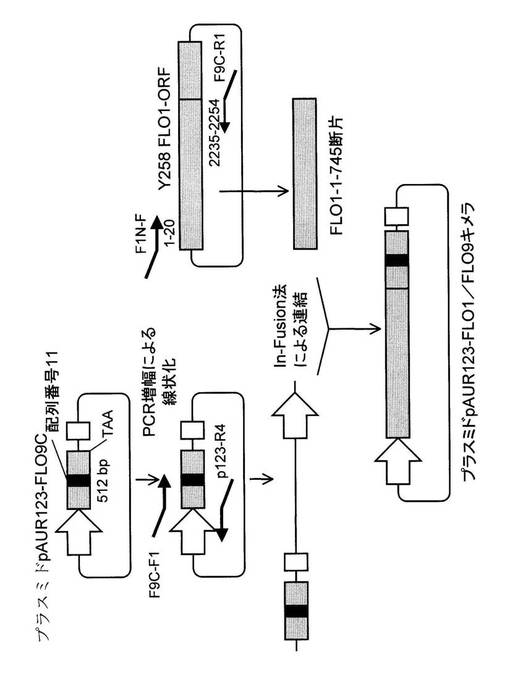
【図2】In−Fusion法によるプラスミドpAUR123−FLO1/FLO9キメラの作製手順とプライマーの位置を示す図
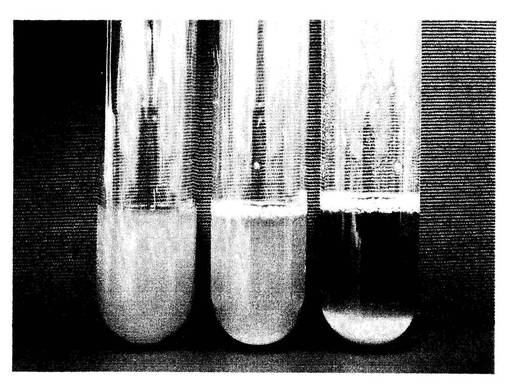
【図3】pAUR123−FLO1、pAUR123−FLO1/FLO9キメラ、FLO1遺伝子を持たないpAUR123のプラスミドをそれぞれ導入した酵母株3種類について、1分間静置後の沈降性を示す写真。
【発明を実施するための最良の形態】
【0017】
はじめに、本発明に用いるアミノ酸配列の繰り返し配列について説明する。
【0018】
サッカロマイセス属セレビシエ菌の凝集性は、細胞表層のマンナン分子のマンノース残基を隣り合った細胞間で直接結合できる特殊な細胞表層のレクチン様タンパク質(または、FLOcculinタンパク質)の関与が報告されている。この細胞間の相互作用によって、多くの細胞が集合し、最終的に酵母の沈降性が生じる。また、サッカロマイセス属セレビシエ菌の凝集性では糖に対する感受性が異なる2つの表現型が知られており、FLO1はマンノース感受性を示し、New FLO(FLONS)はマンノースとグルコー
スに感受性を示す。
【0019】
酵母の凝集性に関与する遺伝子としては、FLO1、FLO5、FLO9、FLO10の特異的なレクチン様タンパク質をコードする遺伝子のファミリーが提案されており、染色体末端領域(テロメア周辺領域)に存在する。一方、凝集性、産膜、浸潤性増殖、基質付着に関連するタンパク質FLO11/MUC1をコードする遺伝子は、非染色体末端領域に存在することが報告されている。
【0020】
これら全てのFLOタンパク質は、グリコシルフォスファチジルイノシトール(GPI)という糖脂質の一種によって修飾を受け、GPIによって細胞膜に繋ぎ留められている。GPIがあたかも錨のように作用することから、このようなタンパク質は GPIアンカー型タンパク質と呼ばれる。
【0021】
FLOタンパク質は、共通の3つのドメイン領域から成り、N末端側のレクチンドメイン、セリン・スレオニン残基を多く含む繰り返し配列を含む中央ドメイン、C末端側のグリコシルフォスファシジルイノシトールとのアンカー配列を含むドメインである。
【0022】
一部の繰り返し配列の配列については、マンノース感受性のFLO1とマンノースとグルコースに感受性のNew FLO(FLONS)の間の糖に対する感受性の違いに関与していることが報告されているが、繰り返し配列が有する機能についてはほとんど分かっていない。
【0023】
これらの酵母の凝集性に関与する遺伝子の分子レベルの研究としては、FLO1遺伝子の単離とその解析がなされている(YEAST,9,423(1993)およびYEAST,10,211(1994))。FLO1タンパク質は、細胞表層に局在するタンパク質で、酵母細胞の凝集性に係わる因子である(Bony et al.,J.Bacteriol.,179:4929−4936(1997))。そのN末端側から、分泌シグナル領域、酵母細胞の凝集性に係わる領域、そしてC末端領域にGPIアンカー結合領域を有している。また、このアミノ酸配列中には13個所の糖鎖結合部位がある。
【0024】
自然界に存在するFLOタンパク質(FLO1、5、9)は、配列番号1〜3の9アミノ酸配列のいずれか、あるいは組み合わせた配列を1〜3回有する。
【0025】
今回本発明者らは、高い凝集性を有する酵母AM12菌由来のFLOタンパク質(AM12 FLO9)中に、9アミノ酸配列の繰り返しを4回有する部位があることを見出した。
【0026】
9アミノ酸の繰り返し配列を4回有するFLOはこれまでに報告がなく、AM12において初めて単離された。
【0027】
AM12 FLOタンパク質のペプチド鎖のC末端側領域に、配列番号1、配列番号2、配列番号3、配列番号3を4回繰り返す配列部位がある。
【0028】
この配列部位を構成する繰り返し配列は、前述のようにFLO1では配列番号4、配列番号1、配列番号5を各1回からなる3回、FLO9では配列番号2、配列番号3を各1回からなる2回、YAL065Cでは配列番号2、配列番号3を各1回からなる2回の繰り返しがあり、FLO5では配列番号2のみ1回で繰り返し無しによって構成されている。
【0029】
また、FLO8、FLO10、MUC1(FLO11)、YAR061W、YAR06
2W、YHR213Wには上記の配列はなかった。
【0030】
これらの配列を解析した結果、配列番号1〜3を任意に選択し4回以上繰り返した配列を、FLOタンパク質のC末端付近に組み込むことで、酵母の凝集性向上が可能であることを見出した。
【0031】
4回以上繰り返した配列の例を挙げると、配列番号11を例示できる。
【0032】
本発明の繰り返し遺伝子DNAを導入することにより、凝集性が付与あるいは強化された酵母を得ることができる。繰り返し遺伝子DNAを導入する方法としては、遺伝子工学の分野において慣用されているものを用いればよく、それを慣用基準(ANALYTICAL BIOCHEMISTRY 163.391(1987)等)に準じて実施すればよい。具体的には、所望のDNAをベクターに組み込んでこれを酵母に導入する方法、ベクターに組み込まずに直接酵母に導入する方法などを挙げることができる。産業に用いる上では、ベクター保持株よりも染色体導入株が望ましい。
【0033】
上記のDNAをベクターに組み込んでこれを酵母に導入する方法において、使用可能なベクターとしては、たとえば、YRp系(酵母染色体のARS配列を複製起点とする酵母用マルチコピーベクター)、YEp系(酵母の2μm DNAの複製起点を持つ酵母用マルチコピーベクター)、YCp系(酵母染色体のARS配列を複製起点として持ち、かつ酵母染色体のセントロメアのDNA配列を持つ酵母用シングルコピーベクター)、YIp系(酵母の複製起点を持たない酵母染色体組み込み用ベクター)等、知られているもの全てのものを用いることができる。これらのベクターは文献に記載されており(医学出版センター刊、「酵母のニューバイオテクノロジー」、p.284)、容易に作製することができる。
【0034】
ベクターに組み込まずに直接酵母にDNAを導入する手法の代表的なものとしては、薬剤耐性遺伝子等のマーカー遺伝子を持つプラスミドと導入するDNA配列とで同時に酵母を形質転換する共形質転換法を挙げることができる(特公平5−60918号公報)。上記のような方法において、導入した遺伝子DNAを酵母中で発現させるために、あるいは発現を増加させるためには、転写および翻訳を制御するユニットであるプロモーターを本発明DNA鎖の5’−上流域に、ターミネーターを3’−下流域にそれぞれ組み込めば良い。このプロモーターおよびターミネーターとしては、繰り返し遺伝子それ自身に由来するものの他、アルコールデヒドロゲナーゼ遺伝子(J.Biol.Chem.,257,3018(1982))、ホスホグリセレートキナーゼ遺伝子(Nucleic Acids Res.,10,7791(1982))、グリセロールアルデヒド−3−燐酸デヒドロゲナーゼ遺伝子(J.Biol.Chem.,254,9839(1979))等既に知られている遺伝子由来のもの、もしくは、人工的にそれを改良したものの使用が可能である。より具体的には、ADH(別名ADC)、GAPDH(別名GPD)、PHO、GAL、PGK、ENO、TRP、HIP等のプロモーターやターミネーターを使用することができる。
【0035】
さらに、適当なプロモーターを選択することにより、本発明DNA鎖の遺伝子を酵母中で制御して発現させることも可能である。例えば、ガラクトキナーゼ遺伝子のプロモーターを使用すれば、培地の糖源をたとえばグルコースからガラクトースに変えることにより発現を増加させることができる。
【0036】
本発明は、5回以上繰り返し遺伝子DNAをAM12菌の4回繰り返し遺伝子DNAなどと入れ替えることによる、元来凝集性を有する酵母の凝集性をさらに向上する方法をも包含する。
【0037】
また、この逆の転換も本発明により提供された繰り返し遺伝子DNAにより可能である。つまり、本発明の繰り返し遺伝子DNAを破壊することによって、繰り返し遺伝子蛋白を発現させる能力を欠失または減少させたDNAを導入することにより、凝集性が欠失または減少した酵母を得ることができる。繰り返し遺伝子DNAの破壊は、繰り返し遺伝子の繰り返し遺伝子蛋白発現に関与する領域、たとえば、プロモーター領域やコード領域の内部へ単一あるいは複数の塩基を付加あるいは欠失させたり、これらの領域全体を欠失させることにより行うことができる。このようにして繰り返し遺伝子を破壊することによって、繰り返し遺伝子蛋白を発現させる能力を欠失または減少させたDNAは、上記したDNA導入法と同じ手法で酵母に導入することができる。その導入によって、ホスト酵母の染色体DNA中の繰り返し遺伝子と導入したDNAとの間で相同組換えが起こり、ホスト酵母の繰り返し遺伝子が分断されて繰り返し遺伝子蛋白を発現する能力が欠失または減少し、その結果、ホスト酵母の凝集性が欠失または減少すると考えられる。
【0038】
本発明は、上記の繰り返し遺伝子DNAの発現を抑制することによって、酵母の凝集性を欠失または減少させる方法をも包含する。このような方法の例としては、繰り返し遺伝子DNAを破壊することによって、繰り返し遺伝子蛋白を発現させる能力を欠失または減少させたDNAを導入する方法、アンチセンスRNA法等を挙げることができる。
【0039】
本発明において形質転換すべき酵母、すなわちホスト酵母、は分類学上、酵母の範疇に入りうる任意のものであり得る。本発明によって高い凝集性を付与された酵母は、バイオエタノール製造の過程において有用である。また本発明の酵母を培養することを含む醸造製品は、ビール、清酒、焼酎、ワイン、ウイスキー、ブランデーを含むアルコール飲料、また醤油、味噌、みりんなどの調味料、さらには、燃料用アルコールなどを包含する。本発明における醸造製品の製造法としては、前記醸造製品に係わる醸造過程を包含する。
【0040】
本発明タンパク質を細胞凝集剤として非凝集性酵母を含む溶液に単純混合することにより酵母凝集効果を得ることもできる。また、細胞間凝集に限らず、例えば細胞を基盤上に固定するような場合(バイオセンサー等)においても応用できる。
【実施例】
【0041】
以下に本発明の実施例を説明するが、本発明はかかる実施例によって限定されない。
【0042】
実施例1(酵母AM12菌株由来RNAからのFLO遺伝子断片の単離)
(1−1) Total RNAの抽出
サッカロマイセス属セレビシエ(Saccharomyces cerevisiae)AM12菌株をYPD培地(1% Yeast extract、2% Polypeptone、2% D−glucose)で30℃、8時間培養し、菌体を回収後、Roche Diagnostics社製 High Pure RNA Isolation kitを用いてTotal RNAの抽出を行った。
【0043】
(1−2) 3’−RACE法によるPCR産物の取得
抽出したTotal RNAを鋳型に、タカラバイオ社製 3’−Full RACE
Core Setを用いて、逆転写反応を、続いてPCR反応を行った。Total RNAは65℃で10分間加熱後、氷上で急冷し、1μgを逆転写反応(反応液量20μlの系)に用いた。逆転写反応液組成は、キットのプロトコールに従って行った。PCRサーマルサイクラーの反応条件は、30℃10分間、42℃60分間、95℃5分間、5℃5分間を1サイクル行った。
【0044】
次に、得られた逆転写反応液1μlを用い、以下の反応液組成、および反応条件でPC
R反応を行った。PCR用酵素としては、東洋紡社製の高正確性ポリメラーゼKOD−Plus−Ver.2を用いた。その結果、約0.6kbpのPCR断片が得られた。PCR反応条件として94℃2分間の後、98℃10秒間、60℃30秒間、68℃5分間を1サイクルとして30サイクル行い、反応終了後は4℃にした。
【0045】
Forwardプライマーとして、c00258−F1を用いた(配列番号6)。
5’−CTGCTCGAGCTCGGCTACTGTGAATGATGTTG−3’
【0046】
Reverseプライマーはタカラバイオ社製 3’−Full RACE Core
Setの3sites Adaptor Primerを用いた。
【0047】
(1−3) PCR産物の回収
PCR産物を1%のTAEアガロースゲルにて電気泳動を行った結果、約0.6kbpの位置にバンドが見られた。このバンドをゲルから切り出し、キアゲン社製のQiagen quick Gel extraction kitを用いて抽出精製を行った。
【0048】
(1−4) TAクローニング
東洋紡社製TArget Clone(登録商標)−Plus−キットを用いて、PCR産物をpTA2ベクター(アンピシリン耐性遺伝子をマーカーとして持つ)に組み込み、大腸菌DH5αに形質転換した。次に、得られたアンピシリン耐性を示す大腸菌から東洋紡社製のMagExtractor−Plasmid−を用いてプラスミド抽出を行った。抽出したプラスミドを制限酵素EcoRIで処理し、1%のTAEアガロース電気泳動を行った結果、pTA2ベクターにPCR産物が組み込まれていることが確認された。
【0049】
(1−5) 塩基配列決定
PCR産物を組み込まれたpTA2ベクターを鋳型に、Dye Terminatorサイクルシーケンス反応を行った。次に、BECKMAN COULTER社製のCEQ8000 DNA Analysis Systemを用い、塩基配列の決定を行った。PCR産物の両側は、ユニバーサルプライマーのM13 Primer M3とM13 Primer RVを用いて解読を行った。次に、得られた塩基配列を基に設計した以下のプライマーを用いてPCR産物の内側の配列を決定した。最終的に、二つの独立した実験から得られたPCR産物を組み込まれたpTA2ベクターについて、遺伝子断片の全長配列を解読し、二つが一致していることを確認した。
【0050】
<配列決定に用いたプライマー>
M13 Primer M3 (配列番号7)
5’−GTAAAACGACGGCCAGT−3’
M13 Primer RV (配列番号8)
5’−CAGGAAACAGCTATGAC−3’
c00258−F3 (配列番号9)
5’−CTGCTCGAGCTCATGAACAGTGCTACCAGTGAG−3’c00258−F4 (配列番号10)
5’−ACAGTAGTCACCTCTTCGCT−3’
【0051】
(1−6) 塩基配列からアミノ酸への変換、比較解析
得られた塩基配列からアミノ酸への変換は、株式会社ゼネティックス製の遺伝子情報処理ソフトウェア GENETYX(登録商標)Ver.9 Windows(登録商標)版を用いて行った。
【0052】
得られた酵母AM12菌株由来RNAからのFLO遺伝子断片の配列を配列番号12に
示す。
【0053】
実施例2
<既知配列との比較>
既知配列との比較は、インターネットを介した同ソフトウェアの塩基配列対塩基配列データベースのBLAST検索(BLASTN)、タンパク質配列対タンパク質配列データベースのBLAST検索(BLASTP)、および付属のホモロジー解析機能、マルチプルアライメント機能を用いて行った。
【0054】
既知の遺伝子と比較した結果、FLO1と最も相同性が高く、他にFLO9、YAL065C、FLO5とも相同性が見られた。
【0055】
また、遺伝子断片は、FLO1(1537アミノ酸)のC末側の1376〜1554に位置する。FLO9、5でもC末端側に位置し、YAL065Cでは全長に相当することが分かった。
【0056】
更に比較をすることにより、分離したアミノ酸配列には、配列番号1、配列番号2、配列番号3、配列番号3の4回繰り返し配列が存在することを見出した。
【0057】
他の遺伝子の相当する部位では、同様のアミノ酸の配列が、FLO1で3回、FLO9とYAL065Cで2回、FLO5で1回見つかった。
【0058】
実施例3(酵母AM12菌由来ゲノムDNAからのFLO9遺伝子の単離、及び部分配列の決定)
(3−1)フォスミドゲノムライブラリーの作製
AM12菌株をYPD培地で30℃、8時間培養し、菌体を回収後、Genomic DNA Buffer Set & Genomic−tip (キアゲン製)を用いてゲノムDNAを抽出した。ゲノムDNAをDNA断片化装置によりランダムに断片化し、Mighty Cloning Blunt End(タカラバイオ製)を使用してDNA断片の末端を平滑処理し、パルスフィールド電気泳動による33〜48kb付近のサイズのDNAをゲルから分画した。切り出したゲル断片からDNAを精製し、インサートDNAとした。調整したインサートDNA断片とベクターpCC1FOS(EPICENTER製)をT4DNAリガーゼ(タカラバイオ製)を用いて4℃で終夜ライゲーション反応を行なった。ベクターライゲーション後、MaxPlax Lambda packaging Extract(EP ICENTER製)を用いて、in vitro packagingを行った。ライブラリーの一部を用いて宿主大腸菌EPI300に形質転換し、タイターを測定した。また、ランダムに選択した16個の白コロニーよりFosmid DNAを抽出し、Not Iで制限酵素処理した後、パルスフィールド電気泳動でインサートサイズが30Kb以上であることを確認した。
【0059】
(3−2)クローンプールグリセロールストック作製
約1万クローン分のライブラリー溶液を宿主大腸菌EPI300に導入し、形質転換体を作製した。方法は、10mM MgSO4と0.2%(w/v)のマルトースを添加したLB培地に宿主大腸菌EPI300のシングルコロニーを植菌し、37℃で3〜5時間振とう培養を行った。遠心して集菌し、上清を廃棄した後、上清と等量の10mM MgSO4に、菌体を懸濁した。
【0060】
SM buffer(50mM Tris−HCl pH7.5、100mM NaCl、10mM MgSO4、0.01% gelatin)を加えて希釈し100μlに合わせたライブラリー溶液を菌体懸濁液100μlに加えて混合し、37℃で1時間静置培
養した。終濃度が12.5μg/mlになるように、クロラムフェニコールを添加したLB寒天培地(100μmol/ml IPTG、40μg/ml X−Gal)に播種し、37℃で一晩培養した。クロラムフェニコール耐性寒天培地を入れた96穴プレートの1ウェルに、100クローンになるように形質転換体を添加し、終夜37℃で、静置培養をした。実際に含まれるクローン数は、これと同時に1ウェルに添加したのと同じ量をシャーレ寒天培地にプレーティングし計測した。
【0061】
培養後の96穴プレートの各ウェルに、20%グリセロール含有クロラムフェニコールLB液体培地を添加した。コロニーを懸濁後に新しい96穴プレートに移し変え、クローンプールグリセロールストックプレートとした。作製したグリセロールストック溶液についてはタイターの確認を行った。
【0062】
(3−3)クローンプールDNA調整
作製したクローンプールグリセロールストックプレートから一部をLB培地に植菌し、培養後にアルカリ−SDS法を用いてフォスミドDNAを調整し、20μL/wellの滅菌蒸留水に溶解した。
【0063】
(3−4)一次スクリーニング
作製した各ウェルのクローンプールDNAを鋳型に、LA Taq polymerase(タカラバイオ製)、FLO9−3Fプライマー(配列番号14)、FLO9−2Rプライマー(配列番号15)を用いて、一次スクリーニングPCR(アニーリング温度55℃、30サイクル)を行った。PCR産物の電気泳動を行い、バンドが見られたウェルを一次陽性クローンとした。
【0064】
FLO9−3F(配列番号14)
5’−TGGTCAAGCAGTTATAGTGTA−3’
FLO9−2R(配列番号15)
5’−AGTTATCAAAGCATTCGCCAA−3’
【0065】
(3−5)配列の決定
得られた一次陽性クローンのPCR産物から直接、プライマーウォーキング法によってシーケンスを行い、獲得したクローンの塩基配列の一部を決定した。
【0066】
酵母AM12菌由来FLO9遺伝子配列の一部、及び推定される翻訳物の配列は配列番号16に示す。参照配列(Accession No.U12980 Saccharomyces cerevisiae chromosome I left arm sequence)との相同性から推定ORFを決定した。また、3’側の非翻訳領域の相同性から当該のFLO遺伝子は、FLO9遺伝子であった。さらに、ORFの3’側の配列は、実施例1で酵母抽出RNAより逆転写によって得られた配列と一致していることが分かった。
【0067】
実施例4(酵母AM12菌株由来FLO9遺伝子の発現解析)
酵母AM12菌株においてFLO9遺伝子が発現しているかどうか、また、その発現強度を調べるために、DNAマイクロアレイ解析を実施した。AM12菌株、及び非凝集性酵母としてS288C菌株をYPD培地で30℃、3時間培養し、菌体を回収後、実施例1と同様に、High Pure RNA Isolation kit(Roche Diagnostics製)を用いてTotal RNAの抽出を行った。抽出したRNAを用いてDNAマイクロアレイ実験を行った。DNAチップは、Yeast Oligo Microarray(V2)(Agilent Order Number 251507210525)(アジレント製)を用い、一色のラベリング方法で行った。このDNA
チップには標準株S288C株の全遺伝子6000個に対応したオリゴDNAがのっており、酵母より抽出したRNAとのハイブリダイゼーションにより各遺伝子の発現強度を測定することができる。FLO9に関しては、A_06_P1087のプローブを用いた。
【0068】
解析結果を図1に示す。AM12菌株ではFLO9遺伝子が強く発現していた。一方、凝集性を持たない株であるS288C菌株では、FLO9遺伝子の発現が低い値に留まっていた。
【0069】
実施例5(FLO遺伝子導入酵母の凝集沈降性試験)
(5−1)FLO1遺伝子の酵母発現ベクター(pAUR123−FLO1)の作製
サッカロマイセス属セレビシエY258菌株由来のYAR050W(FLO1)のORF領域(開始コドンから終止コドンを含む)を挿入したBG1805−ampベクターを保持する大腸菌DH5α株をOpen Biosystems社のYeast ORF collectionより入手した(カタログNo.YSC3867−9520537)。Y258菌株由来FLO1遺伝子の配列、及び推定される翻訳物の配列を配列番号18に示す。常法に従って大腸菌よりプラスミドを抽出し、それを鋳型にc02553−F2プライマー(配列番号20)及びc00258−R3プライマー(配列番号21)を用いてPCRを行い、FLO1 ORF領域を増幅した。得られたPCR産物を制限酵素Xho Iで処理し、電気泳動後、ゲルから目的サイズのバンドを切り出し、MagExtractor−PCR&Gel Clean up−キット(東洋紡製)を用いて抽出し、FLO1断片とした。一方、pAUR123ベクター(タカラバイオ製)を制限酵素Xho Iで処理後に、CIAP処理し、電気泳動後、ゲルから目的バンドを切り出し、MagExtractor−PCR&Gel Clean up−キット(東洋紡製)を用いて抽出し、pAUR123−Xho I断片とした。pAUR123−Xho I断片とFLO1断片はT4リガーゼを用いて連結し、連結物を大腸菌HST08Premium(タカラバイオ製)に形質転換し、アンピシリン耐性を示すコロニーを獲得した。コロニーを培養後、常法に従ってプラスミドを抽出精製し、プラスミドpAUR123−FLO1とした。シーケンス解析により、FLO1断片の挿入の向きが正しいことを確認した。
【0070】
c02553−F2プライマー(配列番号20)
5’−CTGCTCGAGCTCATGACAATGCCTCATCGCTA−3’
c00258−R3プライマー(配列番号21)
5’−CGACTCGAGTTAAATAATTGCCAGCAATAAGGACGC−3’
【0071】
(5−2)FLO1/FLO9キメラ遺伝子の酵母発現ベクター(pAUR123−FLO1/FLO9)の作製
実施例1で取得した酵母AM12菌株由来FLO9C末端の断片(171アミノ酸)は、Y258菌株由来FLO1(907アミノ酸、配列番号18)上の746番目アミノ酸から終止コドンまでの位置に該当する。そこで、Y258菌株由来のFLO1の746番目から終始コドンまでの領域とAM12菌株由来FLO9C末端の断片(171アミノ酸)とを置き換えたFLO1/FLO9キメラタンパク質を発現できるプラスミドの作製を行った。
【0072】
Y258菌株FLO1遺伝子の翻訳産物(配列番号18)新たに作製したFLO1/FLO9キメラ遺伝子の翻訳産物(配列番号22)では、1番目から745番目までのアミノ酸は全く同じである。746番目のアミノ酸以降では、778番目以降、前者が、配列番号4、配列番号1、配列番号5の3回繰り返し配列が存在し、後者では、配列番号1、配列番号2、配列番号3、配列番号3の4回繰り返し配列が存在する。
【0073】
その他の部位では、3アミノ酸の違いがあるが、新たに作製したFLO1/FLO9キメラ遺伝子の翻訳産物(配列番号22)は、実質的に、Y258菌株FLO1遺伝子の翻訳産物(配列番号18)の778番目以降の3回繰り返し配列が、配列番号1、配列番号2、配列番号3、配列番号3からなる4回繰り返し配列(配列番号11)に置き換えられているものである。
【0074】
具体的な作製方法は、次の通りである。実施例1で作製したAM12菌株由来FLO9のC末端の遺伝子断片(FLO9C)を有するpTAベクターを制限酵素Xho Iで処理し、電気泳動にてFLO9C断片部分とベクター部分を分離後、ゲルから目的サイズのバンドを切り出し、MagExtractor−PCR&Gel Clean up−キット(東洋紡社製)を用いて抽出し、FLO9C断片とした。上記で作製したpAUR123−Xho I断片を用い、FLO9C断片とT4リガーゼを用いて連結し、連結物を大腸菌HST08 Premium(タカラバイオ社製)に形質転換し、アンピシリン耐性を示すコロニーを獲得した。コロニーを培養後、常法に従ってプラスミドを調製し、プラスミドpAUR123−FLO9Cとした。シーケンス解析により、FLO9C断片の挿入の向きが正しいことを確認した。
【0075】
次に、図2に示すようにプラスミドpAUR123−FLO1/FLO9キメラを作製した。プラスミドpAUR123−FLO9Cを鋳型に、F9C−F1プライマー(配列番号24)及びp123−R4プライマー(配列番号25)を用いてPCRを行い、ベクター部分を含むプラスミド全長領域を増幅した。得られたPCR産物を電気泳動後、ゲルから目的サイズのバンドを切り出し、MagExtractor−PCR&Gel Clean up−キット(東洋紡製)を用いて抽出し、pAUR123−FLO9C断片とした。一方、前述したOpen Biosystems社のYeast ORF collectionのFLO1 ORF領域(カタログNo.YSC3867−9520537)のプラスミド抽出物を鋳型に、F1N−Fプライマー(配列番号26)及びF9C−R1プライマー(配列番号27)を用いてPCRを行い、Y258FLO1 ORFの1番目から745番目のアミノ酸までの領域を増幅した。得られたPCR産物を電気泳動後、ゲルから目的サイズのバンドを切り出し、MagExtractor−PCR&Gel
Clean up−キット(東洋紡製)を用いて抽出し、FLO1−1−745断片とした。pAUR123−FLO9C断片とFLO1−1−745断片はIn−Fusionキット(タカラバイオ社製)を用いて連結し、連結物を大腸菌HST08 Premium(タカラバイオ社製)に形質転換し、アンピシリン耐性を示すコロニーを獲得した。コロニーを培養後、常法に従ってプラスミドを調製し、プラスミドpAUR123−FLO1/FLO9キメラとした。シーケンス解析により、FLO1/FLO9キメラ断片の配列、及び挿入の向きが正しいことを確認し、次の実験に用いた。
【0076】
F9C−F1プライマー(配列番号24)
5’−CTGTGAATGATGTTGTTACG−3’
p123−R4プライマー(配列番号25)
5’−CAGTTGATTGTATGCTTGGT−3’
F1N−Fプライマー(配列番号26)
5’−GCATACAATCAACTGATGACAATGCCTCATCGCTA−3’
F9C−R1プライマー(配列番号27)
5’−CAACATCATTCACAGTAGCC−3’
【0077】
(5−3)非凝集性酵母への各発現ベクターの導入
文部科学省NBRP「酵母」から提供された非凝集性のサッカロマイセス属セレビシエBY1994株(DKD−5D、SH1994)を宿主として用いた。また、上述のpA
UR123−FLO1、pAUR123−FLO1/FLO9キメラ、pAUR123の3種類のプラスミドをそれぞれ酵母への導入に用いた。
【0078】
具体的な実験は次の通りである。BY1994株をYPD液体培地で30℃、一晩培養したものを前培養液とした。前培養液を40mlのYPD液体培地に植菌して、菌体濁度がOD600で約1になるまで30℃で培養した。培養後の菌体を遠心して集菌し、20mlの0.1M酢酸リチウム溶液(0.1M 酢酸リチウム、0.1M DTT、10mM Tris−HCl、1mM EDTA、pH7.5)に懸濁し、室温で1時間放置した。再度遠心して、集菌後に、滅菌水で2回洗菌し、さらに、1Mソルビトールで1回洗菌した。最終的に、50μlの1Mソルビトールを加えて、菌を懸濁した。100μlの酵母懸濁液と0.1μg〜1μgのプラスミドDNAを混合して、0.2cmギャップのキュベットに分注して、氷中で10分間静置した。キュベットをジーンパルサーシステム(バイオラッド社製)にセットし、1.5kV、25μF、200Ω、パルス1回の条件で、エレクトロポレーションを行った。エレクトロポレーション後の菌液を1Mソルビトールで希釈し、オーレオバシジン1μg/mlを添加したYPD寒天平板培地上に塗末した。30℃で3〜4日間培養後に、オーレオバシジン耐性を示すコロニーを獲得した。オーレオバシジン1μg/mlの終濃度で添加したYPD液体培地でコロニーを培養後、常法に従ってゲノムDNAを調製し、それを鋳型に、pAUR123Fプライマー(タカラバイオ社製)及びpAUR123Rプライマー(タカラバイオ社製)を用いてPCR(アニーリング温度60℃、30サイクル)を行い、増幅が確認できたものをプラスミド保持株とした。
【0079】
(5−4)各ベクター導入酵母の凝集沈降性の比較
前述のpAUR123−FLO1、pAUR123−FLO1/FLO9キメラ、FLO1遺伝子を持たないpAUR123のプラスミド(タカラバイオ社製)をそれぞれ導入した酵母株3種類について、オーレオバシジンの終濃度が1μg/mlになるように添加したYPD液体培地5mlに植菌し、30℃で2日間の振とう培養を行った。培養後に試験管を静置し、目視によって各株の凝集沈降性を比較した。
【0080】
図3は、1分間静置後の試験管の写真である。図3中、左がpAUR123ベクターを導入した酵母、中央がpAUR123−FLO1を導入した酵母、右がpAUR123−FLO1/FLO9キメラを導入した酵母である。pAUR123ベクターを導入した酵母では、その後10分間以上静置しても菌体の沈降は見られなかった。pAUR123−FLO1を導入した酵母についても、目視でわずかに判別できる沈降性が見られる程度であった。一方、pAUR123−FLO1/FLO9キメラを導入した酵母では、1分間の静置で、試験管の底面への沈降が見られた。このことより、遺伝子に配列番号1〜3で示される繰り返し配列の何れかを4回繰り返すように遺伝子改変することで、FLO遺伝子が機能して生じる酵母の凝集沈降性が強化されることが示された。
【技術分野】
【0001】
本発明は酵母に特異的なアミノ酸配列、そのDNA及び形質転換等による遺伝子改変による酵母の育種方法に関する。
【背景技術】
【0002】
細胞間の凝集は、a型細胞とα型細胞間の性的凝集、出芽娘細胞の母細胞からの未分離、非性的凝集などに起因することが知られているが、本発明は、これらのうちの非性的凝集の制御を目的とする。非性的凝集の機構を説明するモデルとしては、凝集性酵母の細胞表層にあるレクチン様タンパク質と糖鎖の結合で隣り合う酵母が結合しているとするレクチン仮説(例えば非特許文献1参照。)が有力であるが、レクチン様タンパク質の同定は成されていない。このことが、酵母凝集の制御が未だ困難である要因でもある。
【0003】
酵母の凝集性に関与する既知の遺伝子としては、FLO1、FLO5、FLO9、FLO10の特異的なレクチン様タンパク質をコードする遺伝子のファミリーがあり、染色体末端領域(テロメア周辺領域)に存在する。一方、凝集性、産膜、浸潤性増殖、基質付着に関連するタンパク質FLO11/MUC1をコードする遺伝子は、非染色体末端領域に存在することが報告されている。
【非特許文献1】J. Bacteriol.,150,878(1982)
【発明の開示】
【発明が解決しようとする課題】
【0004】
既知のFLOタンパク質(FLO1、5、9)は、配列番号1〜5記載の9アミノ酸の配列のいずれか、あるいは組み合わせた配列を1〜3回有する。
【0005】
この配列部位を構成する繰り返し配列は、FLO1では配列番号4、配列番号1、配列番号5を各1回からなる3回、FLO9では配列番号2、配列番号3を各1回からなる2回、YAL065Cでは配列番号2、配列番号3を各1回からなる2回の繰り返しがあり、FLO5では配列番号2のみ1回で繰り返し無しによって構成されている。
【0006】
また、FLO8、FLO10、MUC1(FLO11)、YAR061W、YAR062W、YHR213Wには上記の配列はなかった。
【0007】
上記の従来の酵母を用いたアルコールの発酵生産法は、酵母と生成物とを分離することが困難であるため、非効率的であり、アルコール生産性が不十分である欠点があった。この原因は酵母の凝集性の限界にあり、我々はこの問題を解明した結果、アミノ酸配列の何れかを4回以上繰り返す配列を含むことが凝集性向上に寄与することを見出し、本発明に至った。
【0008】
そこで、本発明の課題は、以下の通りである。
(1) 酵母に凝集性を付与する活性を有するポリペプチドを提供すること;
(2) 酵母に凝集性を付与する活性を有するアミノ酸配列をコードする遺伝子DNAを提供すること;
(3) 上記の遺伝子DNAを利用して、凝集性が付与または強化された酵母の育種方法を提供すること;
【0009】
本発明の他の課題は、以下の記載によって明らかとなる。
【課題を解決するための手段】
【0010】
上記課題は以下の各発明によって解決される。
【0011】
請求項1記載の発明は、配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列を、FLOタンパク質に組み込む遺伝子改変によって、凝集性が付与又は強化されたことを特徴とする酵母である。
請求項2記載の発明は、前記遺伝子改変が、前記FLOタンパク質に存在する配列番号1〜5で示されるアミノ酸配列の何れか又はこれら配列を3回以下の範囲で繰り返してなるアミノ酸配列を、前記配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列に置き換えるものであることを特徴とする請求項1記載の酵母である。
【0012】
請求項3記載の発明は、配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列をコードするDNAを、FLO遺伝子に組み込む遺伝子改変によって、凝集性が付与又は強化されたことを特徴とする酵母である。
請求項4記載の発明は、前記遺伝子改変が、前記FLO遺伝子に存在する配列番号1〜5で示されるアミノ酸配列の何れかをコードするDNA又はこれら配列を3回以下の範囲で繰り返してなるアミノ酸配列をコードするDNAを、前記配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列をコードするDNAに置き換えるものであることを特徴とする請求項3記載の酵母である。
請求項5記載の発明は、前記遺伝子改変が、前記配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列をコードするDNAが組み込まれたベクターによって成されることを特徴とする請求項1〜4の何れかに記載の酵母である。
請求項6記載の発明は、宿主である非凝集性酵母に凝集性を付与してなることを特徴とする請求項1〜5の何れかに記載の酵母である。
請求項7記載の発明は、宿主である凝集性酵母の凝集性を強化してなることを特徴とする請求項1〜5の何れかに記載の酵母である。
【0013】
請求項8記載の発明は、配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列を、FLOタンパク質に組み込む遺伝子改変によって、凝集性を付与又は強化することを特徴とする酵母の育種方法である。
請求項9記載の発明は、前記遺伝子改変が、前記FLOタンパク質に存在する配列番号1〜5で示されるアミノ酸配列の何れか又はこれら配列を3回以下の範囲で繰り返してなるアミノ酸配列を、前記配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列に置き換えるものであることを特徴とする請求項8記載の酵母の育種方法である。
【0014】
請求項10記載の発明は、配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列をコードするDNAを、FLO遺伝子に組み込む遺伝子改変によって、凝集性を付与又は強化することを特徴とする酵母の育種方法である。
請求項11記載の発明は、前記遺伝子改変が、前記FLO遺伝子に存在する配列番号1〜5で示されるアミノ酸配列の何れかをコードするDNA又はこれら配列を3回以下の範囲で繰り返してなるアミノ酸配列をコードするDNAを、前記配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列をコードするDNAに置き換えるものであることを特徴とする請求項10記載の酵母の育種方である。
請求項12記載の発明は、前記遺伝子改変が、前記配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列をコードするDNAが組み込まれたベクターによって成されることを特徴とする請求項8〜11の何れかに記載の酵母の育種方法である。
請求項13記載の発明は、宿主である非凝集性酵母に凝集性を付与することを特徴とする請求項8〜12の何れかに記載の酵母の育種方法である。
請求項14記載の発明は、宿主である凝集性酵母の凝集性を強化することを特徴とする請求項8〜12の何れかに記載の酵母の育種方法である。
【発明の効果】
【0015】
本発明によれば、酵母に凝集性を付与する活性を有するポリペプチドを提供することができ、酵母に凝集性を付与する活性を有するアミノ酸配列をコードする遺伝子DNAを利用して、凝集性が付与または強化された酵母の育種方法を提供することができる。
【図面の簡単な説明】
【0016】
【図1】酵母AM12菌株、及び非凝集性酵母S288C菌株におけるFLO9遺伝子の発現強度の測定結果を示した図
【図2】In−Fusion法によるプラスミドpAUR123−FLO1/FLO9キメラの作製手順とプライマーの位置を示す図
【図3】pAUR123−FLO1、pAUR123−FLO1/FLO9キメラ、FLO1遺伝子を持たないpAUR123のプラスミドをそれぞれ導入した酵母株3種類について、1分間静置後の沈降性を示す写真。
【発明を実施するための最良の形態】
【0017】
はじめに、本発明に用いるアミノ酸配列の繰り返し配列について説明する。
【0018】
サッカロマイセス属セレビシエ菌の凝集性は、細胞表層のマンナン分子のマンノース残基を隣り合った細胞間で直接結合できる特殊な細胞表層のレクチン様タンパク質(または、FLOcculinタンパク質)の関与が報告されている。この細胞間の相互作用によって、多くの細胞が集合し、最終的に酵母の沈降性が生じる。また、サッカロマイセス属セレビシエ菌の凝集性では糖に対する感受性が異なる2つの表現型が知られており、FLO1はマンノース感受性を示し、New FLO(FLONS)はマンノースとグルコー
スに感受性を示す。
【0019】
酵母の凝集性に関与する遺伝子としては、FLO1、FLO5、FLO9、FLO10の特異的なレクチン様タンパク質をコードする遺伝子のファミリーが提案されており、染色体末端領域(テロメア周辺領域)に存在する。一方、凝集性、産膜、浸潤性増殖、基質付着に関連するタンパク質FLO11/MUC1をコードする遺伝子は、非染色体末端領域に存在することが報告されている。
【0020】
これら全てのFLOタンパク質は、グリコシルフォスファチジルイノシトール(GPI)という糖脂質の一種によって修飾を受け、GPIによって細胞膜に繋ぎ留められている。GPIがあたかも錨のように作用することから、このようなタンパク質は GPIアンカー型タンパク質と呼ばれる。
【0021】
FLOタンパク質は、共通の3つのドメイン領域から成り、N末端側のレクチンドメイン、セリン・スレオニン残基を多く含む繰り返し配列を含む中央ドメイン、C末端側のグリコシルフォスファシジルイノシトールとのアンカー配列を含むドメインである。
【0022】
一部の繰り返し配列の配列については、マンノース感受性のFLO1とマンノースとグルコースに感受性のNew FLO(FLONS)の間の糖に対する感受性の違いに関与していることが報告されているが、繰り返し配列が有する機能についてはほとんど分かっていない。
【0023】
これらの酵母の凝集性に関与する遺伝子の分子レベルの研究としては、FLO1遺伝子の単離とその解析がなされている(YEAST,9,423(1993)およびYEAST,10,211(1994))。FLO1タンパク質は、細胞表層に局在するタンパク質で、酵母細胞の凝集性に係わる因子である(Bony et al.,J.Bacteriol.,179:4929−4936(1997))。そのN末端側から、分泌シグナル領域、酵母細胞の凝集性に係わる領域、そしてC末端領域にGPIアンカー結合領域を有している。また、このアミノ酸配列中には13個所の糖鎖結合部位がある。
【0024】
自然界に存在するFLOタンパク質(FLO1、5、9)は、配列番号1〜3の9アミノ酸配列のいずれか、あるいは組み合わせた配列を1〜3回有する。
【0025】
今回本発明者らは、高い凝集性を有する酵母AM12菌由来のFLOタンパク質(AM12 FLO9)中に、9アミノ酸配列の繰り返しを4回有する部位があることを見出した。
【0026】
9アミノ酸の繰り返し配列を4回有するFLOはこれまでに報告がなく、AM12において初めて単離された。
【0027】
AM12 FLOタンパク質のペプチド鎖のC末端側領域に、配列番号1、配列番号2、配列番号3、配列番号3を4回繰り返す配列部位がある。
【0028】
この配列部位を構成する繰り返し配列は、前述のようにFLO1では配列番号4、配列番号1、配列番号5を各1回からなる3回、FLO9では配列番号2、配列番号3を各1回からなる2回、YAL065Cでは配列番号2、配列番号3を各1回からなる2回の繰り返しがあり、FLO5では配列番号2のみ1回で繰り返し無しによって構成されている。
【0029】
また、FLO8、FLO10、MUC1(FLO11)、YAR061W、YAR06
2W、YHR213Wには上記の配列はなかった。
【0030】
これらの配列を解析した結果、配列番号1〜3を任意に選択し4回以上繰り返した配列を、FLOタンパク質のC末端付近に組み込むことで、酵母の凝集性向上が可能であることを見出した。
【0031】
4回以上繰り返した配列の例を挙げると、配列番号11を例示できる。
【0032】
本発明の繰り返し遺伝子DNAを導入することにより、凝集性が付与あるいは強化された酵母を得ることができる。繰り返し遺伝子DNAを導入する方法としては、遺伝子工学の分野において慣用されているものを用いればよく、それを慣用基準(ANALYTICAL BIOCHEMISTRY 163.391(1987)等)に準じて実施すればよい。具体的には、所望のDNAをベクターに組み込んでこれを酵母に導入する方法、ベクターに組み込まずに直接酵母に導入する方法などを挙げることができる。産業に用いる上では、ベクター保持株よりも染色体導入株が望ましい。
【0033】
上記のDNAをベクターに組み込んでこれを酵母に導入する方法において、使用可能なベクターとしては、たとえば、YRp系(酵母染色体のARS配列を複製起点とする酵母用マルチコピーベクター)、YEp系(酵母の2μm DNAの複製起点を持つ酵母用マルチコピーベクター)、YCp系(酵母染色体のARS配列を複製起点として持ち、かつ酵母染色体のセントロメアのDNA配列を持つ酵母用シングルコピーベクター)、YIp系(酵母の複製起点を持たない酵母染色体組み込み用ベクター)等、知られているもの全てのものを用いることができる。これらのベクターは文献に記載されており(医学出版センター刊、「酵母のニューバイオテクノロジー」、p.284)、容易に作製することができる。
【0034】
ベクターに組み込まずに直接酵母にDNAを導入する手法の代表的なものとしては、薬剤耐性遺伝子等のマーカー遺伝子を持つプラスミドと導入するDNA配列とで同時に酵母を形質転換する共形質転換法を挙げることができる(特公平5−60918号公報)。上記のような方法において、導入した遺伝子DNAを酵母中で発現させるために、あるいは発現を増加させるためには、転写および翻訳を制御するユニットであるプロモーターを本発明DNA鎖の5’−上流域に、ターミネーターを3’−下流域にそれぞれ組み込めば良い。このプロモーターおよびターミネーターとしては、繰り返し遺伝子それ自身に由来するものの他、アルコールデヒドロゲナーゼ遺伝子(J.Biol.Chem.,257,3018(1982))、ホスホグリセレートキナーゼ遺伝子(Nucleic Acids Res.,10,7791(1982))、グリセロールアルデヒド−3−燐酸デヒドロゲナーゼ遺伝子(J.Biol.Chem.,254,9839(1979))等既に知られている遺伝子由来のもの、もしくは、人工的にそれを改良したものの使用が可能である。より具体的には、ADH(別名ADC)、GAPDH(別名GPD)、PHO、GAL、PGK、ENO、TRP、HIP等のプロモーターやターミネーターを使用することができる。
【0035】
さらに、適当なプロモーターを選択することにより、本発明DNA鎖の遺伝子を酵母中で制御して発現させることも可能である。例えば、ガラクトキナーゼ遺伝子のプロモーターを使用すれば、培地の糖源をたとえばグルコースからガラクトースに変えることにより発現を増加させることができる。
【0036】
本発明は、5回以上繰り返し遺伝子DNAをAM12菌の4回繰り返し遺伝子DNAなどと入れ替えることによる、元来凝集性を有する酵母の凝集性をさらに向上する方法をも包含する。
【0037】
また、この逆の転換も本発明により提供された繰り返し遺伝子DNAにより可能である。つまり、本発明の繰り返し遺伝子DNAを破壊することによって、繰り返し遺伝子蛋白を発現させる能力を欠失または減少させたDNAを導入することにより、凝集性が欠失または減少した酵母を得ることができる。繰り返し遺伝子DNAの破壊は、繰り返し遺伝子の繰り返し遺伝子蛋白発現に関与する領域、たとえば、プロモーター領域やコード領域の内部へ単一あるいは複数の塩基を付加あるいは欠失させたり、これらの領域全体を欠失させることにより行うことができる。このようにして繰り返し遺伝子を破壊することによって、繰り返し遺伝子蛋白を発現させる能力を欠失または減少させたDNAは、上記したDNA導入法と同じ手法で酵母に導入することができる。その導入によって、ホスト酵母の染色体DNA中の繰り返し遺伝子と導入したDNAとの間で相同組換えが起こり、ホスト酵母の繰り返し遺伝子が分断されて繰り返し遺伝子蛋白を発現する能力が欠失または減少し、その結果、ホスト酵母の凝集性が欠失または減少すると考えられる。
【0038】
本発明は、上記の繰り返し遺伝子DNAの発現を抑制することによって、酵母の凝集性を欠失または減少させる方法をも包含する。このような方法の例としては、繰り返し遺伝子DNAを破壊することによって、繰り返し遺伝子蛋白を発現させる能力を欠失または減少させたDNAを導入する方法、アンチセンスRNA法等を挙げることができる。
【0039】
本発明において形質転換すべき酵母、すなわちホスト酵母、は分類学上、酵母の範疇に入りうる任意のものであり得る。本発明によって高い凝集性を付与された酵母は、バイオエタノール製造の過程において有用である。また本発明の酵母を培養することを含む醸造製品は、ビール、清酒、焼酎、ワイン、ウイスキー、ブランデーを含むアルコール飲料、また醤油、味噌、みりんなどの調味料、さらには、燃料用アルコールなどを包含する。本発明における醸造製品の製造法としては、前記醸造製品に係わる醸造過程を包含する。
【0040】
本発明タンパク質を細胞凝集剤として非凝集性酵母を含む溶液に単純混合することにより酵母凝集効果を得ることもできる。また、細胞間凝集に限らず、例えば細胞を基盤上に固定するような場合(バイオセンサー等)においても応用できる。
【実施例】
【0041】
以下に本発明の実施例を説明するが、本発明はかかる実施例によって限定されない。
【0042】
実施例1(酵母AM12菌株由来RNAからのFLO遺伝子断片の単離)
(1−1) Total RNAの抽出
サッカロマイセス属セレビシエ(Saccharomyces cerevisiae)AM12菌株をYPD培地(1% Yeast extract、2% Polypeptone、2% D−glucose)で30℃、8時間培養し、菌体を回収後、Roche Diagnostics社製 High Pure RNA Isolation kitを用いてTotal RNAの抽出を行った。
【0043】
(1−2) 3’−RACE法によるPCR産物の取得
抽出したTotal RNAを鋳型に、タカラバイオ社製 3’−Full RACE
Core Setを用いて、逆転写反応を、続いてPCR反応を行った。Total RNAは65℃で10分間加熱後、氷上で急冷し、1μgを逆転写反応(反応液量20μlの系)に用いた。逆転写反応液組成は、キットのプロトコールに従って行った。PCRサーマルサイクラーの反応条件は、30℃10分間、42℃60分間、95℃5分間、5℃5分間を1サイクル行った。
【0044】
次に、得られた逆転写反応液1μlを用い、以下の反応液組成、および反応条件でPC
R反応を行った。PCR用酵素としては、東洋紡社製の高正確性ポリメラーゼKOD−Plus−Ver.2を用いた。その結果、約0.6kbpのPCR断片が得られた。PCR反応条件として94℃2分間の後、98℃10秒間、60℃30秒間、68℃5分間を1サイクルとして30サイクル行い、反応終了後は4℃にした。
【0045】
Forwardプライマーとして、c00258−F1を用いた(配列番号6)。
5’−CTGCTCGAGCTCGGCTACTGTGAATGATGTTG−3’
【0046】
Reverseプライマーはタカラバイオ社製 3’−Full RACE Core
Setの3sites Adaptor Primerを用いた。
【0047】
(1−3) PCR産物の回収
PCR産物を1%のTAEアガロースゲルにて電気泳動を行った結果、約0.6kbpの位置にバンドが見られた。このバンドをゲルから切り出し、キアゲン社製のQiagen quick Gel extraction kitを用いて抽出精製を行った。
【0048】
(1−4) TAクローニング
東洋紡社製TArget Clone(登録商標)−Plus−キットを用いて、PCR産物をpTA2ベクター(アンピシリン耐性遺伝子をマーカーとして持つ)に組み込み、大腸菌DH5αに形質転換した。次に、得られたアンピシリン耐性を示す大腸菌から東洋紡社製のMagExtractor−Plasmid−を用いてプラスミド抽出を行った。抽出したプラスミドを制限酵素EcoRIで処理し、1%のTAEアガロース電気泳動を行った結果、pTA2ベクターにPCR産物が組み込まれていることが確認された。
【0049】
(1−5) 塩基配列決定
PCR産物を組み込まれたpTA2ベクターを鋳型に、Dye Terminatorサイクルシーケンス反応を行った。次に、BECKMAN COULTER社製のCEQ8000 DNA Analysis Systemを用い、塩基配列の決定を行った。PCR産物の両側は、ユニバーサルプライマーのM13 Primer M3とM13 Primer RVを用いて解読を行った。次に、得られた塩基配列を基に設計した以下のプライマーを用いてPCR産物の内側の配列を決定した。最終的に、二つの独立した実験から得られたPCR産物を組み込まれたpTA2ベクターについて、遺伝子断片の全長配列を解読し、二つが一致していることを確認した。
【0050】
<配列決定に用いたプライマー>
M13 Primer M3 (配列番号7)
5’−GTAAAACGACGGCCAGT−3’
M13 Primer RV (配列番号8)
5’−CAGGAAACAGCTATGAC−3’
c00258−F3 (配列番号9)
5’−CTGCTCGAGCTCATGAACAGTGCTACCAGTGAG−3’c00258−F4 (配列番号10)
5’−ACAGTAGTCACCTCTTCGCT−3’
【0051】
(1−6) 塩基配列からアミノ酸への変換、比較解析
得られた塩基配列からアミノ酸への変換は、株式会社ゼネティックス製の遺伝子情報処理ソフトウェア GENETYX(登録商標)Ver.9 Windows(登録商標)版を用いて行った。
【0052】
得られた酵母AM12菌株由来RNAからのFLO遺伝子断片の配列を配列番号12に
示す。
【0053】
実施例2
<既知配列との比較>
既知配列との比較は、インターネットを介した同ソフトウェアの塩基配列対塩基配列データベースのBLAST検索(BLASTN)、タンパク質配列対タンパク質配列データベースのBLAST検索(BLASTP)、および付属のホモロジー解析機能、マルチプルアライメント機能を用いて行った。
【0054】
既知の遺伝子と比較した結果、FLO1と最も相同性が高く、他にFLO9、YAL065C、FLO5とも相同性が見られた。
【0055】
また、遺伝子断片は、FLO1(1537アミノ酸)のC末側の1376〜1554に位置する。FLO9、5でもC末端側に位置し、YAL065Cでは全長に相当することが分かった。
【0056】
更に比較をすることにより、分離したアミノ酸配列には、配列番号1、配列番号2、配列番号3、配列番号3の4回繰り返し配列が存在することを見出した。
【0057】
他の遺伝子の相当する部位では、同様のアミノ酸の配列が、FLO1で3回、FLO9とYAL065Cで2回、FLO5で1回見つかった。
【0058】
実施例3(酵母AM12菌由来ゲノムDNAからのFLO9遺伝子の単離、及び部分配列の決定)
(3−1)フォスミドゲノムライブラリーの作製
AM12菌株をYPD培地で30℃、8時間培養し、菌体を回収後、Genomic DNA Buffer Set & Genomic−tip (キアゲン製)を用いてゲノムDNAを抽出した。ゲノムDNAをDNA断片化装置によりランダムに断片化し、Mighty Cloning Blunt End(タカラバイオ製)を使用してDNA断片の末端を平滑処理し、パルスフィールド電気泳動による33〜48kb付近のサイズのDNAをゲルから分画した。切り出したゲル断片からDNAを精製し、インサートDNAとした。調整したインサートDNA断片とベクターpCC1FOS(EPICENTER製)をT4DNAリガーゼ(タカラバイオ製)を用いて4℃で終夜ライゲーション反応を行なった。ベクターライゲーション後、MaxPlax Lambda packaging Extract(EP ICENTER製)を用いて、in vitro packagingを行った。ライブラリーの一部を用いて宿主大腸菌EPI300に形質転換し、タイターを測定した。また、ランダムに選択した16個の白コロニーよりFosmid DNAを抽出し、Not Iで制限酵素処理した後、パルスフィールド電気泳動でインサートサイズが30Kb以上であることを確認した。
【0059】
(3−2)クローンプールグリセロールストック作製
約1万クローン分のライブラリー溶液を宿主大腸菌EPI300に導入し、形質転換体を作製した。方法は、10mM MgSO4と0.2%(w/v)のマルトースを添加したLB培地に宿主大腸菌EPI300のシングルコロニーを植菌し、37℃で3〜5時間振とう培養を行った。遠心して集菌し、上清を廃棄した後、上清と等量の10mM MgSO4に、菌体を懸濁した。
【0060】
SM buffer(50mM Tris−HCl pH7.5、100mM NaCl、10mM MgSO4、0.01% gelatin)を加えて希釈し100μlに合わせたライブラリー溶液を菌体懸濁液100μlに加えて混合し、37℃で1時間静置培
養した。終濃度が12.5μg/mlになるように、クロラムフェニコールを添加したLB寒天培地(100μmol/ml IPTG、40μg/ml X−Gal)に播種し、37℃で一晩培養した。クロラムフェニコール耐性寒天培地を入れた96穴プレートの1ウェルに、100クローンになるように形質転換体を添加し、終夜37℃で、静置培養をした。実際に含まれるクローン数は、これと同時に1ウェルに添加したのと同じ量をシャーレ寒天培地にプレーティングし計測した。
【0061】
培養後の96穴プレートの各ウェルに、20%グリセロール含有クロラムフェニコールLB液体培地を添加した。コロニーを懸濁後に新しい96穴プレートに移し変え、クローンプールグリセロールストックプレートとした。作製したグリセロールストック溶液についてはタイターの確認を行った。
【0062】
(3−3)クローンプールDNA調整
作製したクローンプールグリセロールストックプレートから一部をLB培地に植菌し、培養後にアルカリ−SDS法を用いてフォスミドDNAを調整し、20μL/wellの滅菌蒸留水に溶解した。
【0063】
(3−4)一次スクリーニング
作製した各ウェルのクローンプールDNAを鋳型に、LA Taq polymerase(タカラバイオ製)、FLO9−3Fプライマー(配列番号14)、FLO9−2Rプライマー(配列番号15)を用いて、一次スクリーニングPCR(アニーリング温度55℃、30サイクル)を行った。PCR産物の電気泳動を行い、バンドが見られたウェルを一次陽性クローンとした。
【0064】
FLO9−3F(配列番号14)
5’−TGGTCAAGCAGTTATAGTGTA−3’
FLO9−2R(配列番号15)
5’−AGTTATCAAAGCATTCGCCAA−3’
【0065】
(3−5)配列の決定
得られた一次陽性クローンのPCR産物から直接、プライマーウォーキング法によってシーケンスを行い、獲得したクローンの塩基配列の一部を決定した。
【0066】
酵母AM12菌由来FLO9遺伝子配列の一部、及び推定される翻訳物の配列は配列番号16に示す。参照配列(Accession No.U12980 Saccharomyces cerevisiae chromosome I left arm sequence)との相同性から推定ORFを決定した。また、3’側の非翻訳領域の相同性から当該のFLO遺伝子は、FLO9遺伝子であった。さらに、ORFの3’側の配列は、実施例1で酵母抽出RNAより逆転写によって得られた配列と一致していることが分かった。
【0067】
実施例4(酵母AM12菌株由来FLO9遺伝子の発現解析)
酵母AM12菌株においてFLO9遺伝子が発現しているかどうか、また、その発現強度を調べるために、DNAマイクロアレイ解析を実施した。AM12菌株、及び非凝集性酵母としてS288C菌株をYPD培地で30℃、3時間培養し、菌体を回収後、実施例1と同様に、High Pure RNA Isolation kit(Roche Diagnostics製)を用いてTotal RNAの抽出を行った。抽出したRNAを用いてDNAマイクロアレイ実験を行った。DNAチップは、Yeast Oligo Microarray(V2)(Agilent Order Number 251507210525)(アジレント製)を用い、一色のラベリング方法で行った。このDNA
チップには標準株S288C株の全遺伝子6000個に対応したオリゴDNAがのっており、酵母より抽出したRNAとのハイブリダイゼーションにより各遺伝子の発現強度を測定することができる。FLO9に関しては、A_06_P1087のプローブを用いた。
【0068】
解析結果を図1に示す。AM12菌株ではFLO9遺伝子が強く発現していた。一方、凝集性を持たない株であるS288C菌株では、FLO9遺伝子の発現が低い値に留まっていた。
【0069】
実施例5(FLO遺伝子導入酵母の凝集沈降性試験)
(5−1)FLO1遺伝子の酵母発現ベクター(pAUR123−FLO1)の作製
サッカロマイセス属セレビシエY258菌株由来のYAR050W(FLO1)のORF領域(開始コドンから終止コドンを含む)を挿入したBG1805−ampベクターを保持する大腸菌DH5α株をOpen Biosystems社のYeast ORF collectionより入手した(カタログNo.YSC3867−9520537)。Y258菌株由来FLO1遺伝子の配列、及び推定される翻訳物の配列を配列番号18に示す。常法に従って大腸菌よりプラスミドを抽出し、それを鋳型にc02553−F2プライマー(配列番号20)及びc00258−R3プライマー(配列番号21)を用いてPCRを行い、FLO1 ORF領域を増幅した。得られたPCR産物を制限酵素Xho Iで処理し、電気泳動後、ゲルから目的サイズのバンドを切り出し、MagExtractor−PCR&Gel Clean up−キット(東洋紡製)を用いて抽出し、FLO1断片とした。一方、pAUR123ベクター(タカラバイオ製)を制限酵素Xho Iで処理後に、CIAP処理し、電気泳動後、ゲルから目的バンドを切り出し、MagExtractor−PCR&Gel Clean up−キット(東洋紡製)を用いて抽出し、pAUR123−Xho I断片とした。pAUR123−Xho I断片とFLO1断片はT4リガーゼを用いて連結し、連結物を大腸菌HST08Premium(タカラバイオ製)に形質転換し、アンピシリン耐性を示すコロニーを獲得した。コロニーを培養後、常法に従ってプラスミドを抽出精製し、プラスミドpAUR123−FLO1とした。シーケンス解析により、FLO1断片の挿入の向きが正しいことを確認した。
【0070】
c02553−F2プライマー(配列番号20)
5’−CTGCTCGAGCTCATGACAATGCCTCATCGCTA−3’
c00258−R3プライマー(配列番号21)
5’−CGACTCGAGTTAAATAATTGCCAGCAATAAGGACGC−3’
【0071】
(5−2)FLO1/FLO9キメラ遺伝子の酵母発現ベクター(pAUR123−FLO1/FLO9)の作製
実施例1で取得した酵母AM12菌株由来FLO9C末端の断片(171アミノ酸)は、Y258菌株由来FLO1(907アミノ酸、配列番号18)上の746番目アミノ酸から終止コドンまでの位置に該当する。そこで、Y258菌株由来のFLO1の746番目から終始コドンまでの領域とAM12菌株由来FLO9C末端の断片(171アミノ酸)とを置き換えたFLO1/FLO9キメラタンパク質を発現できるプラスミドの作製を行った。
【0072】
Y258菌株FLO1遺伝子の翻訳産物(配列番号18)新たに作製したFLO1/FLO9キメラ遺伝子の翻訳産物(配列番号22)では、1番目から745番目までのアミノ酸は全く同じである。746番目のアミノ酸以降では、778番目以降、前者が、配列番号4、配列番号1、配列番号5の3回繰り返し配列が存在し、後者では、配列番号1、配列番号2、配列番号3、配列番号3の4回繰り返し配列が存在する。
【0073】
その他の部位では、3アミノ酸の違いがあるが、新たに作製したFLO1/FLO9キメラ遺伝子の翻訳産物(配列番号22)は、実質的に、Y258菌株FLO1遺伝子の翻訳産物(配列番号18)の778番目以降の3回繰り返し配列が、配列番号1、配列番号2、配列番号3、配列番号3からなる4回繰り返し配列(配列番号11)に置き換えられているものである。
【0074】
具体的な作製方法は、次の通りである。実施例1で作製したAM12菌株由来FLO9のC末端の遺伝子断片(FLO9C)を有するpTAベクターを制限酵素Xho Iで処理し、電気泳動にてFLO9C断片部分とベクター部分を分離後、ゲルから目的サイズのバンドを切り出し、MagExtractor−PCR&Gel Clean up−キット(東洋紡社製)を用いて抽出し、FLO9C断片とした。上記で作製したpAUR123−Xho I断片を用い、FLO9C断片とT4リガーゼを用いて連結し、連結物を大腸菌HST08 Premium(タカラバイオ社製)に形質転換し、アンピシリン耐性を示すコロニーを獲得した。コロニーを培養後、常法に従ってプラスミドを調製し、プラスミドpAUR123−FLO9Cとした。シーケンス解析により、FLO9C断片の挿入の向きが正しいことを確認した。
【0075】
次に、図2に示すようにプラスミドpAUR123−FLO1/FLO9キメラを作製した。プラスミドpAUR123−FLO9Cを鋳型に、F9C−F1プライマー(配列番号24)及びp123−R4プライマー(配列番号25)を用いてPCRを行い、ベクター部分を含むプラスミド全長領域を増幅した。得られたPCR産物を電気泳動後、ゲルから目的サイズのバンドを切り出し、MagExtractor−PCR&Gel Clean up−キット(東洋紡製)を用いて抽出し、pAUR123−FLO9C断片とした。一方、前述したOpen Biosystems社のYeast ORF collectionのFLO1 ORF領域(カタログNo.YSC3867−9520537)のプラスミド抽出物を鋳型に、F1N−Fプライマー(配列番号26)及びF9C−R1プライマー(配列番号27)を用いてPCRを行い、Y258FLO1 ORFの1番目から745番目のアミノ酸までの領域を増幅した。得られたPCR産物を電気泳動後、ゲルから目的サイズのバンドを切り出し、MagExtractor−PCR&Gel
Clean up−キット(東洋紡製)を用いて抽出し、FLO1−1−745断片とした。pAUR123−FLO9C断片とFLO1−1−745断片はIn−Fusionキット(タカラバイオ社製)を用いて連結し、連結物を大腸菌HST08 Premium(タカラバイオ社製)に形質転換し、アンピシリン耐性を示すコロニーを獲得した。コロニーを培養後、常法に従ってプラスミドを調製し、プラスミドpAUR123−FLO1/FLO9キメラとした。シーケンス解析により、FLO1/FLO9キメラ断片の配列、及び挿入の向きが正しいことを確認し、次の実験に用いた。
【0076】
F9C−F1プライマー(配列番号24)
5’−CTGTGAATGATGTTGTTACG−3’
p123−R4プライマー(配列番号25)
5’−CAGTTGATTGTATGCTTGGT−3’
F1N−Fプライマー(配列番号26)
5’−GCATACAATCAACTGATGACAATGCCTCATCGCTA−3’
F9C−R1プライマー(配列番号27)
5’−CAACATCATTCACAGTAGCC−3’
【0077】
(5−3)非凝集性酵母への各発現ベクターの導入
文部科学省NBRP「酵母」から提供された非凝集性のサッカロマイセス属セレビシエBY1994株(DKD−5D、SH1994)を宿主として用いた。また、上述のpA
UR123−FLO1、pAUR123−FLO1/FLO9キメラ、pAUR123の3種類のプラスミドをそれぞれ酵母への導入に用いた。
【0078】
具体的な実験は次の通りである。BY1994株をYPD液体培地で30℃、一晩培養したものを前培養液とした。前培養液を40mlのYPD液体培地に植菌して、菌体濁度がOD600で約1になるまで30℃で培養した。培養後の菌体を遠心して集菌し、20mlの0.1M酢酸リチウム溶液(0.1M 酢酸リチウム、0.1M DTT、10mM Tris−HCl、1mM EDTA、pH7.5)に懸濁し、室温で1時間放置した。再度遠心して、集菌後に、滅菌水で2回洗菌し、さらに、1Mソルビトールで1回洗菌した。最終的に、50μlの1Mソルビトールを加えて、菌を懸濁した。100μlの酵母懸濁液と0.1μg〜1μgのプラスミドDNAを混合して、0.2cmギャップのキュベットに分注して、氷中で10分間静置した。キュベットをジーンパルサーシステム(バイオラッド社製)にセットし、1.5kV、25μF、200Ω、パルス1回の条件で、エレクトロポレーションを行った。エレクトロポレーション後の菌液を1Mソルビトールで希釈し、オーレオバシジン1μg/mlを添加したYPD寒天平板培地上に塗末した。30℃で3〜4日間培養後に、オーレオバシジン耐性を示すコロニーを獲得した。オーレオバシジン1μg/mlの終濃度で添加したYPD液体培地でコロニーを培養後、常法に従ってゲノムDNAを調製し、それを鋳型に、pAUR123Fプライマー(タカラバイオ社製)及びpAUR123Rプライマー(タカラバイオ社製)を用いてPCR(アニーリング温度60℃、30サイクル)を行い、増幅が確認できたものをプラスミド保持株とした。
【0079】
(5−4)各ベクター導入酵母の凝集沈降性の比較
前述のpAUR123−FLO1、pAUR123−FLO1/FLO9キメラ、FLO1遺伝子を持たないpAUR123のプラスミド(タカラバイオ社製)をそれぞれ導入した酵母株3種類について、オーレオバシジンの終濃度が1μg/mlになるように添加したYPD液体培地5mlに植菌し、30℃で2日間の振とう培養を行った。培養後に試験管を静置し、目視によって各株の凝集沈降性を比較した。
【0080】
図3は、1分間静置後の試験管の写真である。図3中、左がpAUR123ベクターを導入した酵母、中央がpAUR123−FLO1を導入した酵母、右がpAUR123−FLO1/FLO9キメラを導入した酵母である。pAUR123ベクターを導入した酵母では、その後10分間以上静置しても菌体の沈降は見られなかった。pAUR123−FLO1を導入した酵母についても、目視でわずかに判別できる沈降性が見られる程度であった。一方、pAUR123−FLO1/FLO9キメラを導入した酵母では、1分間の静置で、試験管の底面への沈降が見られた。このことより、遺伝子に配列番号1〜3で示される繰り返し配列の何れかを4回繰り返すように遺伝子改変することで、FLO遺伝子が機能して生じる酵母の凝集沈降性が強化されることが示された。
【特許請求の範囲】
【請求項1】
配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列を、FLOタンパク質に組み込む遺伝子改変によって、凝集性が付与又は強化されたことを特徴とする酵母。
【請求項2】
前記遺伝子改変が、前記FLOタンパク質に存在する配列番号1〜5で示されるアミノ酸配列の何れか又はこれら配列を3回以下の範囲で繰り返してなるアミノ酸配列を、前記配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列に置き換えるものであることを特徴とする請求項1記載の酵母。
【請求項3】
配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列をコードするDNAを、FLO遺伝子に組み込む遺伝子改変によって、凝集性が付与又は強化されたことを特徴とする酵母。
【請求項4】
前記遺伝子改変が、前記FLO遺伝子に存在する配列番号1〜5で示されるアミノ酸配列の何れかをコードするDNA又はこれら配列を3回以下の範囲で繰り返してなるアミノ酸配列をコードするDNAを、前記配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列をコードするDNAに置き換えるものであることを特徴とする請求項3記載の酵母。
【請求項5】
前記遺伝子改変が、前記配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列をコードするDNAが組み込まれたベクターによって成されることを特徴とする請求項1〜4の何れかに記載の酵母。
【請求項6】
宿主である非凝集性酵母に凝集性を付与してなることを特徴とする請求項1〜5の何れかに記載の酵母。
【請求項7】
宿主である凝集性酵母の凝集性を強化してなることを特徴とする請求項1〜5の何れかに記載の酵母。
【請求項8】
配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列を、FLOタンパク質に組み込む遺伝子改変によって、凝集性を付与又は強化することを特徴とする酵母の育種方法。
【請求項9】
前記遺伝子改変が、前記FLOタンパク質に存在する配列番号1〜5で示されるアミノ酸配列の何れか又はこれら配列を3回以下の範囲で繰り返してなるアミノ酸配列を、前記配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列に置き換えるものであることを特徴とする請求項8記載の酵母の育種方法。
【請求項10】
配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列をコードするDNAを、FLO遺伝子に組み込む遺伝子改変によって、凝集性を付与又は強化することを特徴とする酵母の育種方法。
【請求項11】
前記遺伝子改変が、前記FLO遺伝子に存在する配列番号1〜5で示されるアミノ酸配列の何れかをコードするDNA又はこれら配列を3回以下の範囲で繰り返してなるアミノ酸配列をコードするDNAを、前記配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列をコードするDNAに置き換えるものであることを特徴とする請求項10記載の酵母の育種方法。
【請求項12】
前記遺伝子改変が、前記配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列をコードするDNAが組み込まれたベクターによって成されることを特徴とする請求項8〜11の何れかに記載の酵母の育種方法。
【請求項13】
宿主である非凝集性酵母に凝集性を付与することを特徴とする請求項8〜12の何れかに記載の酵母の育種方法。
【請求項14】
宿主である凝集性酵母の凝集性を強化することを特徴とする請求項8〜12の何れかに記載の酵母の育種方法。
【請求項1】
配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列を、FLOタンパク質に組み込む遺伝子改変によって、凝集性が付与又は強化されたことを特徴とする酵母。
【請求項2】
前記遺伝子改変が、前記FLOタンパク質に存在する配列番号1〜5で示されるアミノ酸配列の何れか又はこれら配列を3回以下の範囲で繰り返してなるアミノ酸配列を、前記配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列に置き換えるものであることを特徴とする請求項1記載の酵母。
【請求項3】
配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列をコードするDNAを、FLO遺伝子に組み込む遺伝子改変によって、凝集性が付与又は強化されたことを特徴とする酵母。
【請求項4】
前記遺伝子改変が、前記FLO遺伝子に存在する配列番号1〜5で示されるアミノ酸配列の何れかをコードするDNA又はこれら配列を3回以下の範囲で繰り返してなるアミノ酸配列をコードするDNAを、前記配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列をコードするDNAに置き換えるものであることを特徴とする請求項3記載の酵母。
【請求項5】
前記遺伝子改変が、前記配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列をコードするDNAが組み込まれたベクターによって成されることを特徴とする請求項1〜4の何れかに記載の酵母。
【請求項6】
宿主である非凝集性酵母に凝集性を付与してなることを特徴とする請求項1〜5の何れかに記載の酵母。
【請求項7】
宿主である凝集性酵母の凝集性を強化してなることを特徴とする請求項1〜5の何れかに記載の酵母。
【請求項8】
配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列を、FLOタンパク質に組み込む遺伝子改変によって、凝集性を付与又は強化することを特徴とする酵母の育種方法。
【請求項9】
前記遺伝子改変が、前記FLOタンパク質に存在する配列番号1〜5で示されるアミノ酸配列の何れか又はこれら配列を3回以下の範囲で繰り返してなるアミノ酸配列を、前記配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列に置き換えるものであることを特徴とする請求項8記載の酵母の育種方法。
【請求項10】
配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列をコードするDNAを、FLO遺伝子に組み込む遺伝子改変によって、凝集性を付与又は強化することを特徴とする酵母の育種方法。
【請求項11】
前記遺伝子改変が、前記FLO遺伝子に存在する配列番号1〜5で示されるアミノ酸配列の何れかをコードするDNA又はこれら配列を3回以下の範囲で繰り返してなるアミノ酸配列をコードするDNAを、前記配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列をコードするDNAに置き換えるものであることを特徴とする請求項10記載の酵母の育種方法。
【請求項12】
前記遺伝子改変が、前記配列番号1〜3で示される配列の繰り返しを4回以上有するアミノ酸配列をコードするDNAが組み込まれたベクターによって成されることを特徴とする請求項8〜11の何れかに記載の酵母の育種方法。
【請求項13】
宿主である非凝集性酵母に凝集性を付与することを特徴とする請求項8〜12の何れかに記載の酵母の育種方法。
【請求項14】
宿主である凝集性酵母の凝集性を強化することを特徴とする請求項8〜12の何れかに記載の酵母の育種方法。
【図1】


【図2】
【図3】
【図2】
【図3】
【公開番号】特開2011−254827(P2011−254827A)
【公開日】平成23年12月22日(2011.12.22)
【国際特許分類】
【出願番号】特願2011−161849(P2011−161849)
【出願日】平成23年7月25日(2011.7.25)
【分割の表示】特願2010−540528(P2010−540528)の分割
【原出願日】平成21年11月27日(2009.11.27)
【出願人】(000005902)三井造船株式会社 (1,723)
【Fターム(参考)】
【公開日】平成23年12月22日(2011.12.22)
【国際特許分類】
【出願日】平成23年7月25日(2011.7.25)
【分割の表示】特願2010−540528(P2010−540528)の分割
【原出願日】平成21年11月27日(2009.11.27)
【出願人】(000005902)三井造船株式会社 (1,723)
【Fターム(参考)】
[ Back to top ]