
酵素基質修飾ヌクレオシド三リン酸、核酸プローブ、マルチラベル化核酸プローブ、マルチラベル化核酸プローブの製造方法および標的核酸の検出方法
【課題】新規なヌクレオシド三リン酸誘導体、核酸プローブ、および簡便かつ高感度に標的核酸を検出することができるマルチラベル化核酸プローブ、そのマルチラベル化核酸プローブの製造方法、そのマルチラベル化核酸プローブまたは核酸プローブを用いた標的核酸の検出方法を提供する。
【解決手段】トランスグルタミナーゼ(TGase)を用いて、予め共有結合的に複数の標識部分を導入したマルチラベル化核酸プローブを使用することにより、または、標的核酸にハイブリダイズさせた核酸プローブに、共有結合的に複数の標識部分を導入することにより、簡便かつ高感度に標的核酸を検出することができる。
【解決手段】トランスグルタミナーゼ(TGase)を用いて、予め共有結合的に複数の標識部分を導入したマルチラベル化核酸プローブを使用することにより、または、標的核酸にハイブリダイズさせた核酸プローブに、共有結合的に複数の標識部分を導入することにより、簡便かつ高感度に標的核酸を検出することができる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ヌクレオシド三リン酸誘導体、核酸プローブ、マルチラベル化核酸プローブ、マルチラベル化核酸プローブの製造方法および標的核酸の検出方法に関する。
【背景技術】
【0002】
何らかの標識が施されたRNAプローブなどの核酸プローブを用い、細胞レベルにおけるDNAやRNAの発現パターンを検出、可視化することによって、生命現象に関する数多くの疑問点を解明することが可能となる。細胞レベルでの遺伝子発現パターンを可視化するこのような手法をin situ ハイブリダイゼーション(in situ hybridization:ISH)と言うが、この際に使用されるプローブの標識法は大別して、「放射性同位体標識法」、「蛍光抗体標識法」、「酵素抗体標識法」に分類される。歴史的には、放射性同位体を取り込ませた核酸プローブが先に確立したが、近年その取り扱いが制限されてきたこともあって、放射性同位体元素を用いない蛍光抗体標識法や酵素抗体標識法が注目されている。
【0003】
これらの手法は、核酸プローブ作製時に抗原やビオチンでラベル化しておき、それらを標的核酸にハイブリダイズした後、酵素もしくは蛍光物質によって標識された抗体やアビジンを用いて免疫染色法により検出するといった手法である。感度という観点から見ると、酵素反応によるシグナル増幅効果が得られる酵素抗体標識法が優れており、現在最も広く使用されている。
【0004】
酵素抗体標識法を利用した核酸プローブとしては、例えば、ジゴキシゲニン(DIG)などの抗体認識部位で修飾したヌクレオチド誘導体を複数個ランダムに導入した抗原マルチラベル化核酸プローブが知られている。この抗原マルチラベル化核酸プローブと標的核酸とのin situ ハイブリダイゼーションの後、抗体認識部位を認識する酵素標識化抗体との抗原−抗体反応を行い、酵素アルカリホスファターゼとのハイブリッドによる発色反応を利用して検出を行う。しかしながら、酵素標識された抗体が非常に高価であること、抗原−抗体反応に伴う操作の煩雑化や非特異吸着などによるバックグラウンドの増加など、幾つかの問題を有している。
【0005】
一方、トランスグルタミナーゼ(TGase)を用いて、TGaseが認識可能なリシン(Lys)残基または第一級アミンを有するペプチドまたはタンパク質へ、アニオン性であり、かつTGaseが認識可能なグルタミン(Gln)残基を有する外来分子を部位特異的に連結する方法が知られている(例えば、特許文献1参照)。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特開2008−54658号公報
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明は、新規なヌクレオシド三リン酸誘導体、核酸プローブ、および簡便かつ高感度に標的核酸を検出することができるマルチラベル化核酸プローブ、そのマルチラベル化核酸プローブの製造方法、そのマルチラベル化核酸プローブまたは核酸プローブを用いた標的核酸の検出方法である。
【課題を解決するための手段】
【0008】
本発明は、グルタミン(Gln)残基を有するヌクレオシド三リン酸誘導体である。
【0009】
また、前記ヌクレオシド三リン酸誘導体が、下記式(1)で示されるものであることが好ましい。
【化1】

(式(1)中、Aは、グルタミン(Gln)残基を有する置換基、Bは水素原子またはヒドロキシル基を表す。)
【0010】
また、前記ヌクレオシド三リン酸誘導体が、下記式(2)で示されるものであることが好ましい。
【化2】
(式(2)中、A1およびA2のうち少なくとも1つは、グルタミン(Gln)残基を有する置換基で残りは水素原子を表し、Bは水素原子またはヒドロキシル基を表す。)
【0011】
また、前記ヌクレオシド三リン酸誘導体が、下記式(3)で示されるものであることが好ましい。
【化3】
(式(3)中、Aは、グルタミン(Gln)残基を有する置換基、Bは水素原子またはヒドロキシル基を表す。)
【0012】
また、前記ヌクレオシド三リン酸誘導体が、下記式(4)で示されるものであることが好ましい。
【化4】
(式(4)中、Aは、グルタミン(Gln)残基を有する置換基、Bは水素原子またはヒドロキシル基を表す。)
【0013】
また、前記ヌクレオシド三リン酸誘導体が、下記式(5)で示されるものであることが好ましい。
【化5】
(式(5)中、XおよびYは、それぞれ独立して2価の連結基を表し、Zは、置換基を表す。)
【0014】
また、前記ヌクレオシド三リン酸誘導体において、前記XおよびYは、それぞれ独立して炭素数1〜48のアルキレン基または炭素数2〜48のアルケニレン基であることが好ましく、Zは、炭素数1〜48のアルキル基、炭素数1〜48のアルコキシ基、炭素数6〜48のアリール基、炭素数6〜48のアリールオキシ基、炭素数7〜48のアリールアルキル基または炭素数7〜48のアリールアルキルオキシ基であることが好ましい。また、Yは、炭素数2〜48のオキシアルキレン基(例えば、オキシエチレン基、オキシプロピレン基)また、Y,Zは独立してLys以外のアミノ酸により少なくとも一方が置換されていてもよい。
【0015】
また、前記ヌクレオシド三リン酸誘導体において、前記Xはエテニレン基、Yはメチレン基であり、Zはベンジルオキシ基であることが好ましい。
【0016】
また、本発明は、構成単位として、前記ヌクレオシド三リン酸誘導体が複数個導入されている核酸である核酸プローブである。
【0017】
また、本発明は、前記核酸プローブにおけるグルタミン(Gln)残基のうち少なくとも2つに、リシン(Lys)残基または第一級アミンを有し標識部分を含む標識化合物が結合されて構成されているマルチラベル化核酸プローブである。
【0018】
また、前記マルチラベル化核酸プローブにおいて、前記標識部分が、酵素および蛍光色素のうち少なくとも1つであることが好ましい。
【0019】
また、前記マルチラベル化核酸プローブにおいて、前記酵素が、超好熱菌由来の酵素であることが好ましい。
【0020】
また、前記マルチラベル化核酸プローブにおいて、前記酵素が、有機溶媒や熱に対して安定な酵素であることが好ましい。
【0021】
また、本発明は、マルチラベル化核酸プローブの製造方法であって、トランスグルタミナーゼ(TGase)を用いて、前記核酸プローブにおけるグルタミン(Gln)残基のうち少なくとも2つに、リシン(Lys)残基または第一級アミンを有し標識部分を含む標識化合物を結合するマルチラベル化核酸プローブの製造方法である。
【0022】
また、前記マルチラベル化核酸プローブの製造方法において、前記標識部分が、酵素および蛍光色素のうち少なくとも1つであることが好ましい。
【0023】
また、前記マルチラベル化核酸プローブの製造方法において、前記酵素が、超好熱菌由来の酵素であることが好ましい。
【0024】
また、本発明は、標的核酸の検出方法であって、前記マルチラベル化核酸プローブと、対象物中に存在する標的核酸とを核酸部分により特異的に結合させ、結合している前記マルチラベル化核酸プローブを、前記標識部分により検出する標的核酸の検出方法である。
【0025】
また、本発明は、標的核酸の検出方法であって、前記核酸プローブと、対象物中に存在する標的核酸とを核酸部分により特異的に結合させた後、トランスグルタミナーゼ(TGase)を用いて、前記核酸プローブにおけるグルタミン(Gln)残基と、リシン(Lys)残基または第一級アミンを有し標識部分を含む標識化合物のリシン(Lys)残基または第一級アミンとを反応させて複数の標識部分を導入し、結合している前記核酸プローブを、前記標識部分により検出する標的核酸の検出方法である。
【発明の効果】
【0026】
本発明では、共有結合的に複数の標識部分を導入したマルチラベル化核酸プローブを得るための新規なヌクレオシド三リン酸誘導体、核酸プローブを提供することができる。
【0027】
また、本発明では、ターゲットとなる標的核酸にハイブリダイズする前に、予め共有結合的に複数の標識部分を核酸プローブに導入することにより、簡便かつ高感度に標的核酸を検出することができるマルチラベル化核酸プローブを提供することができる。
【0028】
また、本発明では、トランスグルタミナーゼ(TGase)を用いて、共有結合的に複数の標識部分を核酸プローブに導入することにより、簡便かつ高感度に標的核酸を検出することができるマルチラベル化核酸プローブの製造方法を提供することができる。
【0029】
また、本発明では、予め共有結合的に複数の酵素を導入したマルチラベル化核酸プローブを用いることにより、簡便かつ高感度に標的核酸を検出することができる標的核酸の検出方法を提供することができる。
【0030】
また、本発明では、核酸プローブをターゲットとなる標的核酸にハイブリダイズさせた後、核酸プローブにおけるグルタミン(Gln)残基と標識化合物のリシン(Lys)残基または第一級アミンとの簡易な反応によって、共有結合的に複数の標識部分を導入することにより、簡便かつ高感度に標的核酸を検出することが可能な標的核酸の検出方法を提供することができる。
【図面の簡単な説明】
【0031】
【図1】本発明の実施形態に係るヌクレオチド誘導体の構造の一例(Z−QG−UTP)を示す図である。
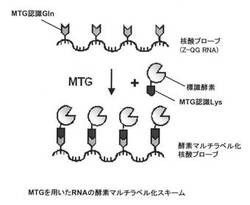
【図2】本発明の実施形態に係る酵素マルチラベル化核酸プローブの調製方法を示す概略図である。
【図3】本発明の実施形態に係るヌクレオチド誘導体の一例であるZ−QG−UTPの合成方法の一例を示す図である。
【図4】本発明の実施例1において、Z−QG−UTPを合成した後、逆相HPLC(表1のHPLC測定条件)を行った際の結果を示す図である。
【図5】本発明の実施例1において、Z−QG−UTPを合成した後、逆相HPLC(表2のHPLC測定条件)を行った際の結果を示す図である。
【図6】本発明の実施例1において合成したZ−QG−UTPのMALDI TOF−MS分析の結果を示す図である。
【図7】実施例1における実験1の標準プローブを用いたドットブロットの結果を示す図である。
【図8】実施例1における実験2の酵素マルチラベル化核酸プローブを用いてAlkphos Directのプロトコールに準じて検出した結果を示す図である。
【図9】UTPとZ−QG−UTPの割合を変化させてRNAを調製した場合の分子量変化について、(A)アガロースゲル電気泳動装置、(B)マイクロチップ型電気泳動装置にて電気泳動を行った結果を示す図である。
【図10】MTGを用いてPfuAPラベル化を行った際のアガロースゲル電気泳動装置による電気泳動の結果を示す図である。
【図11】PfuAPマルチラベル化RNAプローブの核酸プローブを用いた室温におけるドットブロットの結果を示す図である。
【図12】Alkphos Directキットにより調製された酵素ラベル化核酸プローブによるISHの検出感度評価を示す図である。
【図13】PfuAPマルチラベル化RNAプローブの核酸プローブを用いた50℃におけるドットブロットの結果を示す図である。
【図14】aminoallyl−UTP in TE Buffer(pH8.0)におけるaminoallyl−UTPの濃度に対する吸収スペクトル測定結果を示す図である。
【図15】スペクトルより290nmの吸光度を算出し検量線を示す図である。
【図16】本発明の実施形態に係る標的核酸の検出方法の一例を示す概略図である。
【図17】本発明の実施例3において合成したZ−QG−dUTPのMALDI TOF−MS分析の結果を示す図である。
【図18】本発明の実施例3において、Z−QG−dUTPを合成した後、逆相HPLC(表13のHPLC測定条件)を行った際の結果を示す図である。
【図19】本発明の実施例3において、aminoallyl−dUTPの逆相HPLC(表13のHPLC測定条件)を行った際の結果を示す図である。
【図20】本発明の実施例3のZ−QG DNAの調製において、Z−QG−dUTPの割合を変化させてPCRした際の電気泳動の結果を示す図である。
【図21】本発明の実施例3のZ−QG DNAへのPfuAPのラベル化の検討において、MTG反応後のアガロース電気泳動の結果を示す図である。
【図22】本発明の実施例3のPfuAPマルチラベル化DNAプローブの調製において、MTG反応後のアガロース電気泳動の結果を示す図である。
【図23】本発明の実施例3において、各種PfuAPラベル化DNAプローブのドットブロットの結果を示す図である。
【図24】本発明の実施例4のPfuAPマルチラベル化DNAプローブの調製において、MTG反応後のアガロース電気泳動の結果を示す図である。
【図25】本発明の実施例4において、各種PfuAPラベル化DNAプローブのドットブロットの結果を示す図である。
【図26】本発明の実施例5において、MTGを用いてBAPラベル化を行った際のアガロースゲル電気泳動装置による電気泳動の結果を示す図である。
【図27】本発明の実施例5において、BAPマルチラベル化RNAプローブの核酸プローブを用いた室温におけるドットブロットの結果を示す図である。
【図28】本発明の実施例6において、MTGを用いて、Z−QG RNAに対してAlexa Fluor 555 cadaverineラベル化を行った際の結果を示す図である。
【図29】本発明の実施例6において、Alexa Fluor 555マルチラベル化RNAの核酸プローブを用いた室温におけるドットブロットの結果を示す図である。
【発明を実施するための形態】
【0032】
本発明の実施の形態について以下説明する。本実施形態は本発明を実施する一例であって、本発明は本実施形態に限定されるものではない。
【0033】
本発明者らは、核酸プローブに複数の酵素を共有結合的に導入する手法として、微生物由来トランスグルタミナーゼ(MTG)などのトランスグルタミナーゼ(TGase)が有する部位特異的なタンパク質修飾能に着目した。TGaseはアシル転移反応を触媒する酵素であり、例えば、タンパク質中の特定のGln残基(Q)のγ−カルボキシアミド基と、Lys残基(K)のε−アミノ基や各種一級アミンとの共有結合を触媒する酵素である。このTGaseを用いて、複数の標識酵素などの標識部分を導入したマルチラベル化核酸プローブの創製を行う。
【0034】
具体的には、例えば、図1に示す、ウリジン三リン酸(uridine triphosphate:UTP)に、TGaseが認識するGln(MTG認識Gln)を有するZ−QGを結合させたヌクレオチド誘導体であるZ−QG−UTPを合成し、図2に示すように、核酸プローブとなるRNAを調製する際にZ−QG−UTPを取り込ませることによって、TGase認識Glnが複数箇所導入されたZ−QG RNAを調製する。その後、MTGなどのTGaseを用いて、MTG認識LysなどのTGase認識Lysを導入した標識酵素などの標識化合物を結合させることによって、RNAと標識酵素などの標識部分が1:n(nは2以上の整数)であるマルチラベル化核酸プローブを創製することができる。
【0035】
このマルチラベル化核酸プローブは、ターゲットとなる標的核酸にハイブリダイズした後、直ちに検出反応を行うことができるため、既存の手法と比較して、操作の大幅な簡素化、バックグラウンドの低下、バルク酵素である微生物由来トランスグルタミナーゼ(MTG)を用いることによるコストの抑制などが見込まれる。
【0036】
なお、図2において、核酸プローブにおけるGln残基と、標識化合物におけるLys残基とは逆であってもよい。すなわち、TGaseが認識するLys(MTG認識Lys)を有するヌクレオシド三リン酸誘導体を合成し、核酸プローブとなるRNAを調製する際に取り込ませることによって、TGase認識Lysが複数箇所導入された核酸プローブを調製する。その後、MTGなどのTGaseを用いて、MTG認識GlnなどのTGase認識Glnを導入した標識化合物を結合させることによって、RNAと標識部分が1:n(nは2以上の整数)であるマルチラベル化核酸プローブを創製してもよい。
【0037】
また、図16に示すように、Z−QG RNAなどの核酸プローブをターゲットとなる標的核酸にハイブリダイズした後、MTGなどのTGaseを用いて、MTG認識LysなどのTGase認識Lysを導入した標識酵素などの標識化合物を結合させてもよい。RNAと標識酵素などの標識部分が1:n(nは2以上の整数)となり、このように導入した標識部分の検出反応を行うことにより、既存の手法と比較して、操作の大幅な簡素化、バックグラウンドの低下、バルク酵素である微生物由来トランスグルタミナーゼ(MTG)を用いることによるコストの抑制などが見込まれる。
【0038】
なお、図16において、核酸プローブにおけるGln残基と、標識化合物におけるLys残基とは逆であってもよい。すなわち、TGaseが認識するLys(MTG認識Lys)を有するヌクレオシド三リン酸誘導体を合成し、核酸プローブとなるRNAを調製する際に取り込ませることによって、TGase認識Lysが複数箇所導入された核酸プローブを調製する。その後、核酸プローブをターゲットとなる標的核酸にハイブリダイズした後、MTGなどのTGaseを用いて、MTG認識GlnなどのTGase認識Glnを導入した標識化合物を結合させてもよい。
【0039】
<ヌクレオシド三リン酸誘導体>
本実施形態に係るヌクレオシド三リン酸誘導体は、グルタミン(Gln)残基を有する。グルタミン(Gln)残基を有するヌクレオシド三リン酸誘導体としては、グルタミン(Gln)残基を有する、ウリジン三リン酸(uridine triphosphate:UTP)誘導体、アデノシン三リン酸(adenosine triphosphate:ATP)誘導体、グアノシン三リン酸(guanosine triphosphate:GTP)誘導体、シチジン三リン酸(cytidine triphosphate:CTP)誘導体、デオキシウリジン三リン酸(deoxyuridine triphosphate:dUTP)誘導体、デオキシアデノシン三リン酸(deoxyadenosine triphosphate:dATP)誘導体、デオキシグアノシン三リン酸(deoxyguanosine triphosphate:dGTP)誘導体、デオキシシチジン三リン酸(deoxycytidine triphosphate:dCTP)誘導体などが挙げられる。本実施形態に係るヌクレオシド三リン酸誘導体において、グルタミン(Gln)残基は、例えば、ウラシル、アデニン、グアニン、シトシンの部分に直接または置換基を介して結合されている。
【0040】
これらのヌクレオシド三リン酸誘導体は、UTP、ATP、GTP、CTP、dUTP、dATP、dGTP、dCTPまたはそれらの各種誘導体から得ることができる。
【0041】
また、これらのヌクレオシド三リン酸誘導体は、ウリジン、ウリジンの一リン酸(UMP)および二リン酸(UDP)、アデノシン、アデノシンの一リン酸(AMP)および二リン酸(ADP)、グアノシン、グアノシンの一リン酸(GMP)および二リン酸(GDP)、シチジン、シチジンの一リン酸(CMP)および二リン酸(CDP)、デオキシウリジン、デオキシウリジンの一リン酸(dUMP)および二リン酸(dUDP)、デオキシアデノシン、デオキシアデノシンの一リン酸(dAMP)および二リン酸(dADP)、デオキシグアノシン、デオキシグアノシンの一リン酸(dGMP)および二リン酸(dGDP)、デオキシシチジン、デオキシシチジンの一リン酸(dCMP)および二リン酸(dCDP)ならびにそれらの各種誘導体から得てもよい。
【0042】
例えば、ウリジン、アデノシン、グアノシン、シチジン、デオキシウリジン、デオキシアデノシン、デオキシグアノシン、デオキシシチジンのリン酸化酵素などによるリン酸化(例えば、生物工学会誌,85(9),p397−399(2007)、Journal of Bioscience and Bioengineering,87(6),p.732−738(1999)など参照)や、プロトンスポンジ存在下でのオキシ塩化リンなどによるリン酸化(例えば、Tetrahedron Letters,29(36),p.4525−4528(1988)など参照)などによって、それらの三リン酸体を得ることができる。
【0043】
本実施形態に係るヌクレオシド三リン酸誘導体は、例えば、下記式(1)で示され、TGaseが認識可能なGln残基を有するウリジン三リン酸誘導体である。
【化6】
(式(1)中、Aは、グルタミン(Gln)残基を有する置換基、Bは水素原子またはヒドロキシル基を表す。)
【0044】
本実施形態に係るヌクレオシド三リン酸誘導体は、例えば、下記式(2)で示され、TGaseが認識可能なGln残基を有するアデノシン三リン酸誘導体である。
【化7】
(式(2)中、A1およびA2のうち少なくとも1つは、グルタミン(Gln)残基を有する置換基で残りは水素原子を表し、Bは水素原子またはヒドロキシル基を表す。)
【0045】
本実施形態に係るヌクレオシド三リン酸誘導体は、例えば、下記式(3)で示され、TGaseが認識可能なGln残基を有するシチジン三リン酸誘導体である。
【化8】
(式(3)中、Aは、グルタミン(Gln)残基を有する置換基、Bは水素原子またはヒドロキシル基を表す。)
【0046】
本実施形態に係るヌクレオシド三リン酸誘導体は、例えば、下記式(4)で示され、TGaseが認識可能なGln残基を有するグアノシン三リン酸誘導体である。
【化9】
(式(4)中、Aは、グルタミン(Gln)残基を有する置換基、Bは水素原子またはヒドロキシル基を表す。)
【0047】
Aで表されるグルタミン(Gln)残基を有する置換基としては、特に制限はないが、例えば、グルタミン(Gln)残基を有する直鎖、分岐、環状の飽和または不飽和のアルキル基、アミノアルキル基、アリール基、ヘテロアリール基を含む置換基が挙げられ、合成のし易さなどを考慮して決めればよい。
【0048】
本実施形態に係るヌクレオシド三リン酸誘導体は、例えば、下記式(5)で示され、TGaseが認識可能なGln残基を有するTGase基質修飾ヌクレオチド誘導体である。
【化10】
(式(5)中、XおよびYは、それぞれ独立して2価の連結基を表し、Zは、置換基を表す。)
【0049】
XおよびYで表される2価の連結基としては、それぞれ独立して、メチレン基、エチレン基、プロピレン基、ブチレン基などの炭素数1〜48のアルキレン基、エテニレン基、プロペニレン基、ブテニレン基などの炭素数2〜48のアルケニレン基などが挙げられる。これらのうち、X,Yは、それぞれ独立して炭素数1〜48のアルキレン基または炭素数2〜48のアルケニレン基、炭素数1〜48のアルコキシ基であることが好ましく、Xはエテニレン基、Yはメチレン基であることがより好ましい。X,Yはさらにエテニレン基、オキシアルキレン基、例えば−(C2H4O)n−または−(C3H6O)n−(nは繰り返し数でありn=2,4,8,12,24)基などで置換されていてもよい。
【0050】
Zで表される置換基としては、メチル基、エチル基、プロピル基などの炭素数1〜48のアルキル基、メトキシ基、エトキシ基、プロピオキシ基などの炭素数1〜48のアルコキシ基、フェニル基、ナフチル基などの炭素数6〜48のアリール基、フェニルオキシ基などの炭素数6〜48のアリールオキシ基、ベンジル基などの炭素数7〜48のアリールアルキル基、ベンジルオキシ基などの炭素数7〜48のアリールアルキルオキシ基などが挙げられる。これらのうち、Zは、炭素数1〜48のアルキル基、炭素数1〜48のアルコキシ基、炭素数6〜48のアリール基、炭素数6〜48のアリールオキシ基、炭素数7〜48のアリールアルキル基または炭素数7〜48のアリールアルキルオキシ基であることが好ましく、Zはベンジルオキシ基であることがより好ましい。Zはさらにジニトロフェニル基、L−3,4−ジヒドロキシフェニル基などで置換されていてもよい。また、上述したYで表される置換との組み合わせで、Y,Zは独立してLys以外のアミノ酸により少なくとも一方が置換されていてもよい。
【0051】
Xを適宜選択することにより、Z−QGとUTPとを連結するリンカー部位の構造を最適化し、例えば柔軟なリンカー部位を導入することで、酵素などのアクセスを向上することができる。また、Y,Zを適宜選択することにより、基質ペプチド配列を最適化し、例えば酵素などの親和性を向上することができる。
【0052】
微生物由来TGase(MTG)を用いる場合、MTGが認識可能なGln残基は、ベンジルオキシカルボニル−L−グルタミルグリシン(Z−QG)として存在することが好ましい。Z−QGは、ジゴキシゲニン(DIG)などよりも分子サイズが小さいため好ましい。式(5)で示されるヌクレオシド三リン酸誘導体において、Xがエテニレン基、Yがメチレン基、Zがベンジルオキシ基であるヌクレオチド誘導体が、UTPにZ−QGを結合させたヌクレオチド誘導体Z−QG−UTPである。また、ヌクレオシド三リン酸誘導体中には、TGaseが認識可能なLys残基または第一級アミンが共存しないようなものを選択することが好ましい。共存する場合には、TGaseにより、自己架橋する可能性があり、目的のマルチラベル化核酸プローブの収率に好ましくない影響を与える場合があるからである。
【0053】
また、微生物由来TGaseの良基質として、LLQG(配列番号:1)、LAQG(配列番号:2)、LGQG(配列番号:3)、PLAQSH(配列番号:4)、FERQHMDS(配列番号:5)、もしくはTEQKLISEEDL(配列番号:6)のアミノ酸配列からなるペプチド、またはGLGQGGG(配列番号:7)、GFGQGGG(配列番号:8)、GVGQGGG(配列番号:9)、もしくはGGLQGGG(配列番号:10)のアミノ酸配列からなるペプチドが知られている。また、guinea pig liver由来のTGaseの良基質として、ベンジルオキシカルボニル−L−グルタミルフェニルアラニン(Z−QF)、またはEAQQIVM(配列番号:11)のアミノ酸配列からなるペプチド、またはGGGQLGG(配列番号:12)、GGGQVGG(配列番号:13)、GGGQRGG(配列番号:14)、GQQQLG(配列番号:15)、PNPQLPF(配列番号:16)もしくはPKPQQFM(配列番号:17)のアミノ酸配列からなるペプチドが知られている。TGaseが認識可能なGln残基は、用いるTGaseの種類に応じ、このようなペプチドとして存在してもよい。
【0054】
なお、N末端がグリシン(G)である基質ペプチドは、N末端アミノ基がTGaseの基質になりうるため、自己架橋による副産物が生じうる。したがって、N末端がグリシン(G)である基質ペプチドについては、N末端アミノ基の水素を適切な基で置換することによりTGaseの基質とはならないように保護して、所望の連結を行うことができるようにするとよい。なお、本明細書において「N末端保護」というときは、特別な場合を除き、このような意味で用いている。そして、N末端保護の手段により、反応性が異なることが知られている。詳細には、ほ乳類由来TGaseに関して、GQQQLGのN末端アセチル化による保護(すなわち、Ac−GQQQLG)、またN末端アミノ酸をDOPA(L−3,4−dihydroxyphenylalanine)にする(すなわち、DOPA−GQQQLG)と反応性が向上することが知られている。このような保護の例を、本実施形態においても利用することができる。
【0055】
Z−QG−UTPの調製方法を図3に示す。これは、本実施形態に係るヌクレオシド三リン酸誘導体の調製方法の一例であって、これに限定されるものではない。
【0056】
まず、ベンジルオキシカルボニル−L−グルタミルグリシン(Z−QG)にN−ヒドロキシスクシンイミド(N−hydroxysuccinimide:NHS)基などを導入し、活性化しておく(NHS化Z−QG)。そして、アミノアリルUTPなどの末端をアミノ化した置換基を有するUTPと、NHS化Z−QGとを縮合することにより、Z−QG−UTPを得ることができる。
【0057】
また、C末端のカルボキシル基を活性エステル化する上述の方法に加えて、TGaseが認識可能なGln残基を有するペプチドをUTPに導入する方法として、アミノ基と反応性の高い官能基をペプチドに導入する方法がある。例えば、アルデヒド化、アシルアジド化、スルフォニルクロライド化、エポキシ化、イソシアネート化、またはイソチオシアネート化した基質ペプチドを調製できれば、これをアミノ化UTPと反応させることにより、TGaseが認識可能なGln残基を有するUTPを調製することができる。ただし、これらの反応性官能基は、基質ペプチドにおいてTGase認識に影響がない部分に導入する必要がある。したがって、上述のように、Gln残基とは離れたC末端のカルボキシル基を活性化する方法は、この目的において最も優れたものの一つである。
【0058】
Z−QG−UTPの精製は、高速液体クロマトグラフィ(HPLC)、ゲル濾過クロマトグラフィ(GPC)などにより行うことができる。また、Z−QG−UTPの同定は、MALDI TOF−MS、NMR、IRなどにより行うことができる。また、HPLCにより、生成物の確認および収率を求めることができる。
【0059】
<TGase基質マルチラベル化核酸>
本実施形態に係るTGase基質マルチラベル化核酸は、構成単位として、例えば式(1)〜(5)で示されるTGase基質ラベル化ヌクレオシド三リン酸誘導体を調製したのち、このTGase基質ラベル化ヌクレオシド三リン酸誘導体が複数個導入されているTGase基質マルチラベル化核酸であり、核酸部分が、検出対象である標的核酸の標的分子特異的配列の全部または一部に相補的な配列を有するものである。
【0060】
核酸には、DNA、PNAおよびRNAが含まれる。核酸の配列および長さには特に制限はない。長さに関しては、例えば少なくとも20mer程度であればよい。
【0061】
例えば、Z−QG−UTPを基質として、RNAポリメラーゼ活性のある酵素を用いて複数のZ−QG−UTPを取り込ませて、TGase認識Glnが複数箇所に導入され、所望の配列を有するTGase基質マルチラベル化核酸プローブZ−QG RNA(図2)を調製することができる。
【0062】
<マルチラベル化核酸プローブ>
本実施形態に係るマルチラベル化核酸プローブは、上記核酸プローブにおける構成単位であるヌクレオシド三リン酸誘導体のグルタミン(Gln)残基のうち少なくとも2つに、リシン(Lys)残基または第一級アミンを有し標識部分を含む標識化合物が結合されて構成されたものである。マルチラベル化核酸プローブは、例えば、MTGなどのTGaseを用いて、MTG認識GlnなどのTGase認識Glnが複数箇所に導入された核酸プローブZ−QG RNAに、TGase認識Lysを導入した標識酵素などを結合させることによって、RNAと標識酵素などの標識部分が1:nであるマルチラベル化核酸プローブを創製することができる(図2)。
【0063】
RNAと標識酵素などの標識部分との比率nは、2以上であれば特に制限はなく、適宜調整することができるが、nが大きいほど検出感度が高くなり好ましい。ただし、nが大きすぎると、標的核酸とのハイブリダイゼーションの効率が低下する場合がある。
【0064】
また、例えば、以下の方法により、異なる標識酵素、異なる蛍光色素などの異なる標識部分を有する複数のマルチラベル化核酸プローブを調製することができる。
(1)TGaseの由来を変える。
(2)TGaseの基質特異性を変える。
【0065】
(1)の方法では、例えば、用いるTGaseの種類に応じて、異なる基質ペプチドで修飾されたUTPなどを調製すればよい。
【0066】
(2)の方法では、例えば、TGaseに、タンパク質工学的にアミノ酸変異を導入して基質特異性を変えればよい。例えば、MTGを大腸菌で調製し(例えば、Christian K. Marx,Thomas C. Hertel and Markus Pietzsch,Enzyme and Microbial Technology,Volume 40,Issue 6,2 May 2007,p.1543−1550,”Soluble expression of a pro−transglutaminase from Streptomyces mobaraensis in Escherichia coli”参照)、さらに変異体ライブラリーを作って耐熱性の向上したMTGを取得することができる(例えば、Christian K. Marx,Thomas C. Hertel and Markus Pietzsch,Journal of Biotechnology,Volume 136,Issues 3−4,10 September 2008,p.156−162,”Random mutagenesis of a recombinant microbial transglutaminase for the generation of thermostable and heat−sensitive variants”参照)。
【0067】
本明細書において、「TGaseにより結合する」というときは、特別な場合を除き、得られる連結部は、Lys残基とGln残基とが、ε(γ−グルタミル)リシン結合を形成することにより構成されている。
【0068】
本実施形態においては、TGaseが認識可能なLys残基は、第一級アミンであってもよい。本明細書では、Lys残基を例に説明するが、その説明は、特別な場合を除き、第一級アミンにも当てはまる。
【0069】
リシン(Lys)残基または第一級アミンを有し標識部分を含む標識化合物としては、特に制限はない。
【0070】
標識部分としては、酵素、蛍光色素、放射性同位体を含む化合物、磁気的に検出可能な標識(例えば、磁性ナノ粒子)、熱的に検出可能な標識(例えば、温度応答性高分子)、電気的に検出可能な標識(例えば、フェロセン部位を有する高分子)などが挙げられ、検出感度、取り扱い性などの点から、酵素および蛍光色素のうち少なくとも1つが好ましい。
【0071】
蛍光色素としては、選択された波長の紫外光、可視光などの放射線による照射に応答して蛍光または燐光を発する物質であればよく、特に制限はないが、例えば、蛍光色素としてフルオレセイン、ローダミン、ダンシル、カルボシアニン誘導体など、あるいは蛍光タンパク質として緑色蛍光タンパク質とその変異体などが挙げられる。
【0072】
放射性同位体としては、例えば、重水素(2H)、三重水素(3H)、10B、11B、13C、15N、18Oなどが挙げられる。
【0073】
TGaseに対し、Lys残基(または第一級アミン)供与体となる基質は、Gln残基供与体となる基質に比較して構造的な制約が少ないと考えられる。したがって、修飾しようとする標識酵素が、TGaseが認識可能なLys残基を元来有している場合もあり、TGaseが認識可能なLys残基を含むタグを酵素に付加する場合もある。
【0074】
TGaseが認識可能なLys残基(K)は、MKHKGS(配列番号:18)、MRHKGS(配列番号:35)、MRRKGS(配列番号:36)、MHRKGS(配列番号:37)のアミノ酸配列を有するペプチドとして存在してもよい。このようなTGaseが認識可能なLys残基を含むペプチドによるタグ化は、標識酵素を、タンパク質の所望の部位、例えばC末端またはN末端に連結する目的で用いることができる。TGaseが認識可能なLys残基を含む他のペプチドまたはそのアミノ酸配列の例としては、改変型S−peptide(GSGMKETAAARFERAHMDSGS(配列番号:19))、MGGSTKHKIPGGS(配列番号:20)、N末端グリシン(N−terminal GGG、N−terminal GGGGG(配列番号:21))、N末端MKHKGSと対象タンパク質間のリンカー部位を伸ばしたMKHKGGGSGGGSGS(配列番号:22)などが挙げられる。
【0075】
C末端またはN末端にTGaseが認識可能なLys残基を含むペプチドを付加した標識酵素は、遺伝子工学的な手法を用いて、組換えタンパク質として調製することができる。C末端またはN末端にTGaseの基質ペプチドタグが導入された当該組換えタンパク質の精製は、それぞれN末端またはC末端に付加した精製用ペプチドタグ(例えば、(His)6−tag(ヘキサヒスチジンタグ))を利用し(TGaseの反応性の低下を回避するために、基質ペプチドタグを入れた末端とは異なる末端に精製用ペプチドタグを入れるようにデザインするとよい。)、ゲル濾過クロマトグラフィなどにより行うことができ、またアミノ酸配列の確認は当該タンパク質をコードするプラスミドベクターの遺伝子配列をDNAシーケンサにて確認するか、N末端に導入された基質ペプチドについてはN末端分析により直接同定することができる。タンパク質の精製の確認は、SDS−PAGEなどで行うことができる。
【0076】
標識酵素としては、発色反応などを利用して検出を行うことができる性質を有するものであればよく特に制限はない。例えば、アルカリホスファターゼ(AP)、グルタチオン−S−トランスフェラーゼ(GST)、ルシフェラーゼ、ペルオキシダーゼなどが挙げられる。これらのうち、高い触媒活性と安定性の観点から、アルカリホスファターゼあるいはペルオキシダーゼが好ましい。ペプチドタグが容易に導入可能との観点からは、遺伝子工学的に製造可能なタンパク質が好ましい。
【0077】
酵素マルチラベル化核酸プローブと標的核酸の間でより厳密に塩基配列特異的な二本鎖形成を行うためには、比較的高温(例えば、70℃以上)の条件下で行うことがあるため、常温菌由来の酵素を利用すると活性の損失が懸念される。そこで、標的酵素としては、超好熱菌Pyrococcus furiosus由来アルカリホスファターゼ(PfuAP)が好ましい。
【0078】
超好熱菌由来の酵素は、一般的に有機溶媒や熱に対して高い安定性を示すことが知られているため好ましく(例えば、H.Atomi,Current Opinion in Chemical Biology,9,p.166−173(2005)参照)、さらに、大腸菌を宿主とした大量調製が比較的容易に行うことができる点においても好ましい。大腸菌を宿主として耐熱性酵素を調製する場合、細胞破砕液を高温処理(例えば、80℃で30分温置)することで、大腸菌由来の共雑タンパク質のほとんどを沈殿させ、粗精製を容易に行うことができる。
【0079】
超好熱菌は一般の生物がほとんど生育できない極限環境で生育することができる微生物であるため、超好熱菌由来のタンパク質は非常に高い耐熱性を有している。さらに、熱に対する耐性だけでなく、一般的に、変性剤、有機溶媒、pHなどに対する耐性も常温菌由来酵素に比べて極めて高いことから、PfuAPを使用することによって、酵素の失活を伴わない厳密な二本鎖形成を達成することができると考えられる。
【0080】
本実施形態に係る酵素マルチラベル化核酸プローブの好ましい態様の一つは、PfuAPとZ−QG RNAとの複合体である。このような複合体は、安定な酵素であるPfuAPと、安定な分子であるRNAが、アミド結合という安定な共有結合で連結されているため、複合体全体としても安定性であるというメリットがある。
【0081】
また、酵素マルチラベル化核酸プローブにおいて、酵素が有機溶媒や熱に対して安定な酵素であってもよい。このような安定性の高い酵素は、自然界からのスクリーニング(例えば、化学と工業,vol.61(No.6),p.571−575(2008)、内山拓,宮崎健太郎,バイオサイエンスとインダストリー,vol.66(No.5),p.234−239(2008)、道久則之,バイオサイエンスとインダストリー,vol.66(No.12),p.667−670(2008))や、タンパク質工学的手法により安定性を高める技術(例えば、荻野博康,BIO INDUSTRY,vol.25(No.7),p.16−23(2008)、宮崎健太郎,BIO INDUSTRY,vol.25(No.7),p.52−58(2008))により得ることができる。これらの手法により、常温菌由来の酵素であっても、有機溶媒耐性や耐熱性を有する酵素へと変換することができる。
【0082】
蛍光色素部分を導入した、リシン(Lys)残基または第一級アミンを有する標識化合物は、例えば、カルボキシル基にジアミンを導入する方法で調製することができる(例えば、G.T.Hermanson(1996),Bioconjugate Techniques,Chapter 1,Section 4.3,p.100−104,Academic Press,San Diego.を参照)。
【0083】
トランスグルタミナーゼ(TGase)としては、種々のものを用いることができる。現在、TGaseとして、哺乳類(guinea pig、ヒト)、無脊椎動物(昆虫、カブトガニ、ウニ)、植物、菌類、原生生物(粘菌)由来のものが知られており、またヒトの場合については、8種類のアイソザイムが見つかっている。本実施形態において用いることのできるTGaseの好ましい例は、安定性、ハンドリングの容易さ、バルク生産が可能などの点から微生物由来微生物由来トランスグルタミナーゼ(MTG)である。
【0084】
本実施形態においてMTGを用いた場合、予想されているMTGの触媒反応から、Lys残基を有する標識酵素などの標識部分を含む標識化合物とZ−QG RNAとの連結反応は、MTG活性中心であるシステイン(Cys)残基の、Z−QG RNAのGlnへの求核置換反応によるアシル−酵素複合体の形成と、続いて起こる標識化合物のLysによるアシル−酵素複合体への求核置換反応によるMTGの脱離、の2段階で進行すると予想される。
【0085】
本実施形態の好ましい態様においては、TGaseが認識可能なGln残基を有するZ−QG RNAに対する、TGaseが認識可能なLys残基または第一級アミンを有する標識化合物のモル濃度比は、好ましくは2以上であり、より好ましくは5以上である。なお、本明細書で単に「濃度比」というときは、特別な場合を除き、モル濃度による比を指す。例えば、Z−QG RNAに対するNK14−PfuAPのモル濃度比は、[NK14−PfuAP]/[Z−QG RNA]と表すこともできる。
【0086】
[NK14−PfuAPの調製]
NK14−PfuAPとは、MKHKGGGSGGGSGSの配列を有するアミノ酸14残基からなる付加配列を遺伝子工学的にPfuAPのN末端に、精製用タグをC末端に導入したものである。PfuAPの発現ベクターは香川大学櫻庭春彦教授より委譲頂いた。PCRによりPfuAPをコードする領域を増幅する際、それぞれのタグが導入されるようにタンパク質発現用ベクターpET22に組換え、大腸菌BL21株を形質転換した。アンピシリンを含むLB培地にて前培養、本培養を経て、得られた形質転換体を遠心分離により収菌し、25mM TBSで2回洗浄した。得られた菌体を凍結・融解させた後、超音波処理により細胞を粉砕し、遠心分離により可溶性画分を回収した。超高熱菌由来のPfuAPは高温条件下でも安定であるので、得られた無細胞抽出液を80℃で30分処理し、他のタンパク質を沈殿させることによって粗精製を行った。粗精製後、遠心分離・フィルター濾過によって上澄みを回収し、His−tagカラムによって精製した。精製後、限外濾過による濃縮を行い、PD−10カラムを用いて溶媒を10mM Tris−HCl(pH8.0)へと置換し、実験に供するまで凍結保存した。
【0087】
また、NK14−PfuAPの大腸菌での発現量の向上のため、大腸菌のコドン使用頻度に合わせて塩基配列が改変されたNK14−PfuAPの発現ベクター(アクセッション番号:AB479383、配列番号:26)を用いてもよい。この発現ベクターは、Codon Devices社(http://www.codondevices.com)に受託合成して得た。NK14−PfuAPをコードする遺伝子領域の両端に適切な制限酵素サイトを導入しておき、これらを利用することでタンパク質発現用ベクターpET22に組換え、得られたNK14−PfuAP発現ベクターにより大腸菌BL21株を形質転換した。アンピシリンを含むLB培地にて前培養、本培養を経て、得られた形質転換体を遠心分離により収菌し、25mM TBSで2回洗浄した。得られた菌体を凍結・融解させた後、超音波処理により細胞を粉砕し、遠心分離により可溶性画分を回収した。超高熱菌由来のPfuAPは高温条件下でも安定であるので、得られた無細胞抽出液を80℃で30分処理し、他のタンパク質を沈殿させることによって粗精製を行った。粗精製後、遠心分離・フィルター濾過によって上澄みを回収し、His−tagカラムによって精製した。精製後、限外濾過による濃縮を行い、PD−10カラムを用いて溶媒を10mM Tris−HCl(pH8.0)へと置換し、実験に供するまで凍結保存した。
【0088】
TGaseとしてMTGを用いて連結反応を行う場合には、上述のようにモル濃度比が適切な範囲となるようにすることに加えて、pH5.5〜8.0、温度4〜50℃(例えば、室温(例えば、18℃〜22℃))の条件で行うことが好ましい。このようにすれば、12時間以内、好ましくは6時間以内、より好ましくは3時間以内に、充分に高い反応率が達成可能である。
【0089】
このような高い反応率が達成できる方法により得られたマルチラベル化核酸プローブ溶液には、未反応の核酸プローブ(例えば、遊離のZ−QG RNA)がほとんど存在せず、以下で詳述する、標的核酸の検出にそのまま用いたとしても、マルチラベル化核酸プローブと未反応分子との競合が実質的に生じないか、生じたとしても目的とする検出の結果には実質的な影響を与えないほど少ないと考えられる。したがって、マルチラベル化核酸プローブ溶液を精製することなく、直接検出へと利用できるメリットがある。
【0090】
本実施形態以前に、TGaseが認識可能なLys残基または第一級アミンを有する標識酵素などの標識化合物と、TGaseが認識可能なGln残基を有するZ−QG RNAとをTGaseを用いて連結することにより、マルチラベル化核酸プローブを得る例は存在しなかった。したがって、本実施形態の方法により連結されたマルチラベル化核酸プローブは、物質として新規なものということができる。
【0091】
本実施形態における酵素マルチラベル化核酸プローブの形成方法は、従来法に比較して、以下の特徴およびメリットを有する。
・対象となる標識酵素は、TGaseが認識可能なLys残基または第一級アミンを有する酵素、TGaseが認識可能なLys残基または第一級アミンを導入することができるあらゆる酵素を包含する。また、インテインのような大きなタンパク質性タグを必要としない。
・酵素の修飾部位は、C末端に限定されない。TGase活性なLys残基が存在するか、そのようなLys残基を有するタグを導入することができる部位であれば、修飾可能である。
・C末端、N末端に加え、loop領域のようなタンパク質構造中の揺らぎの大きな部位も修飾対象となりうる。
・結合する核酸の塩基配列および長さには、特に制限がない。
【0092】
<標的核酸の検出方法>
本実施形態に係る標的核酸の検出方法は、構成単位として例えば上記式(1)〜(5)で示されるヌクレオシド三リン酸誘導体が複数個導入されている核酸である核酸プローブにおけるグルタミン(Gln)残基のうち少なくとも2つに、リシン(Lys)残基または第一級アミンを有する標識酵素などの標識化合物が結合されて構成されているマルチラベル化核酸プローブであって、標識部分が容易に検出可能な性質を有し、かつ核酸部分が標的核酸に特異的に結合可能な塩基配列を有するものであるマルチラベル化核酸プローブを準備し、マルチラベル化核酸プローブと対象物中に存在する標的核酸とを核酸部分により特異的に結合させ、結合しているマルチラベル化核酸プローブを、酵素部分などの標識部分により検出する工程を含む。
【0093】
本実施形態に係る標的核酸の検出方法は、標的核酸の定性、定量、識別、染色、局在化の調査などの目的で用いることができる。
【0094】
この方法においては、上記マルチラベル化核酸プローブが用いられるが、核酸部分は、標的核酸に特異的に結合可能な核酸配列を有するものとする。酵素部分などの標識部分は、容易に検出可能な性質を有するものである。この方法において、標的核酸を含む標的分子は、核酸、比較的低分子の有機化合物(ATP)、タンパク質、ペプチド、金属イオン、複雑な構造を持つ多量体、ウイルスなどでありうる。検出対象は、(i)DNA転写膜、または(ii)細胞もしくは個体組織切片などである。
【0095】
検出対象が(i)の場合、標的核酸は、PCRにより増幅されたDNAまたはゲノム断片DNAなどであり、(ii)の場合、標的核酸は、細胞または個体組織中に含まれる核酸(mRNAまたはDNA)などである。本方法は、従来法、例えばジゴキシゲニン(DIG)標識化プローブを用いる方法に比較して、種々の点で優れている。例えば、DIG法では、核酸プローブのDIG修飾および標識化された抗DIG抗体が必要であり、それに伴う煩雑な洗浄操作が必要となるが、本実施形態に係るマルチラベル化核酸プローブを用いれば、DIG標識化プローブおよび標識化抗DIG抗体が不要になるため、試薬、手間および時間を大幅に削減することが可能となる。また、1つの認識部位(核酸)に対して複数のシグナル増幅部位(酵素など)を配置しているため、検出感度の向上が可能になる。
【0096】
この方法においては、標的核酸に、マルチラベル化核酸プローブを供し、標的核酸とマルチラベル化核酸プローブの標的核酸相補的配列を有する部分とをハイブリダイズさせるが、このハイブリダイズのための条件は、当業者であれば、用いる核酸部分の長さ、塩基配列などに応じて、適宜設計することができる。
【0097】
本実施形態に係る標的核酸の検出方法において、異なる標識酵素、異なる蛍光色素などの異なる標識部分を有する複数のマルチラベル化核酸プローブを準備して、同時に複数の標的核酸を検出してもよい。
【0098】
また、本実施形態において、標的核酸の検出方法は、
(a)基材表面に固定化された核酸、
(b)固定化された固定化核酸の全部または一部に相補的な配列(固定化核酸相補的配列)、および標的核酸に特異的に結合可能な配列(標的核酸特異的配列)を有するアプタマ核酸、
(c)構成単位として上記式(1)で示されるヌクレオチド誘導体が複数個導入されている核酸である核酸プローブにおけるグルタミン(Gln)残基のうち少なくとも2つに、リシン(Lys)残基または第一級アミンを有する標識酵素などの標識化合物が結合されて構成されているマルチラベル化核酸プローブであって、標識部分が容易に検出可能な性質を有し、及び/又は核酸部分がアプタマ核酸の標的核酸特異的配列の全部または一部に相補的な配列を有するものであるマルチラベル化核酸プローブ、
を準備し、
(A)固定化核酸に、アプタマ核酸を供し、固定化核酸とアプタマ核酸の固定化核酸相補的配列を有する部分とをハイブリダイズさせることにより、アプタマ核酸を固定化し、
(B)固定化された固定化アプタマ核酸に、標的核酸を含む標的分子を含む可能性のある試料を供し、標的分子が存在する場合には標的核酸とアプタマ核酸の標的核酸分子特異的配列を有する部分とを結合させ、かつ上記マルチラベル化核酸プローブを供し、標的分子が存在しない場合にはマルチラベル化核酸プローブの核酸部分とアプタマ核酸とをハイブリダイズさせることにより、タンパク質を固定化し、
(C)固定化された酵素部分の有無またはその量を酵素の性質に基づいて検出することにより、試料中の標的分子を検出する工程を含む。
【0099】
この方法における基材表面への核酸の固定化のためには、従来技術、例えばDNAマイクロアレイなどを調製する際に用いられる技術を適用することができる。「基材」は、ガラス、シリコンなどのプラスチック製の、チップ、ビーズ、ウェル、プレートなどの形態とすることができ、核酸は、従来技術を用いて、基材表面に非共有結合(静電結合)的に、または共有結合で固定することができる。あらかじめ調製した核酸を基材に固定化してもよく、基材上で直接核酸を合成してもよい。簡便には、アビジンなどで被覆された市販のプレートを用い、ビオチン化した所望のDNAを固定することができる。
【0100】
この方法ではさらに、固定化核酸の全部または一部に相補的な配列(固定化核酸相補的配列)、および標的分子を含まれる標的核酸に特異性的に結合可能な配列(標的核酸特異的配列)を有する核酸(アプタマ核酸)が用いられる。標的分子は、核酸、比較的低分子の有機化合物、タンパク質、ペプチド、金属イオン、複雑な構造を持つ多量体、ウイルスなどでありうる。標的核酸特異的配列を有する部分は、従来技術、例えばSELEX(試験管内人工進化法)工程を用いることにより産生することができ、また標的分子に対して非常に高い標的親和性及び特異性を有するものとすることができる。標的核酸特異的配列を有する部分は、修飾ヌクレオチドで構成されていてもよい。
【0101】
この方法においては、上記マルチラベル化核酸プローブが用いられるが、核酸部分は、アプタマ核酸の標的核酸特異的配列の全部または一部に相補的な配列を有するものとする。酵素部分などの標識部分は、容易に検出可能な性質を有するものである。この方法において、標的核酸を含む標的分子は、核酸、比較的低分子の有機化合物(ATP)、タンパク質、ペプチド、金属イオン、複雑な構造を持つ多量体、ウイルスなどでありうる。検出対象は、(i)DNA転写膜、または(ii)細胞もしくは個体組織切片などである。
【0102】
この方法においては、固定化核酸に、アプタマ核酸を供し、固定化核酸とアプタマ核酸の固定化核酸相補的配列を有する部分とをハイブリダイズさせることにより、アプタマ核酸を固定化するが、このハイブリダイズのための条件は、当業者であれば、用いる核酸部分の長さ、塩基配列などに応じて、適宜設計することができる。
【0103】
また、この方法においては、次いで固定化アプタマ核酸に、標的分子を含む可能性のある試料を供し、標的分子が存在する場合には標的分子とアプタマ核酸の標的核酸特異的配列を有する部分とを結合させ、さらにマルチラベル化核酸プローブを供し、標的分子が存在しない場合にはマルチラベル化核酸プローブの核酸部分とアプタマ核酸とをハイブリダイズさせる。試料は、細胞もしくは組織抽出液、体液などでありうる。
【0104】
さらにこの方法においては、固定化された酵素部分の有無またはその量を酵素の性質に基づいて検出することにより、試料中の標的分子を検出することができる。
【0105】
アプタマ核酸においては、固定化核酸相補的配列を有する部分と、標的核酸特異的配列を有する部分とは、重複してもよく、連続してもよく、また適当なスペーサを介して両者が存在するように設計することができる。標的核酸特異的配列を有する部分(アプタマ領域)が分子認識のためにある立体構造をとることを考慮すると、該部分は、固定化核酸相補的配列を有する部分とは少なくとも重複しないようにするのがよい。
【0106】
アプタマ核酸は、固定化核酸相補的配列および標的核酸特異的配列以外に、所望の場合にはマルチラベル化核酸プローブと適切にハイブリダイズするための配列をさらに含んでいてもよい。マルチラベル化核酸プローブの核酸部分は、アプタマ核酸の標的分子特異的配列の全部または一部に相補的な配列を有するが、この特異的配列の全部または一部に相補的な配列からなる領域が長い(標的核酸特異的配列と重複が多い)と、かえってアプタマ核酸とはハイブリダイズできない場合が生じうる。また短い(重複が少ない)と、標的分子が存在し、アプタマ核酸と結合している場合にも、マルチラベル化核酸プローブとアプタマ核酸とがハイブリダイズしてしまう場合が生じうる。当業者であれば、このようなことを考慮して、固定化核酸、アプタマ核酸、マルチラベル化核酸プローブの核酸部分を、適宜設計することができる。
【0107】
また、本実施形態に係る標的核酸の検出方法は、核酸部分が標的核酸に特異的に結合可能な塩基配列を有するものである、構成単位として、例えば上記式(1)〜(5)で示されるヌクレオシド三リン酸誘導体が複数個導入されている核酸である核酸プローブと、対象物中に存在する標的核酸とを核酸部分により特異的に結合させた後、トランスグルタミナーゼ(TGase)を用いて、核酸プローブにおけるグルタミン(Gln)残基と、リシン(Lys)残基または第一級アミンを有し標識部分を含む標識化合物のリシン(Lys)残基または第一級アミンとを反応させて、容易に検出可能な性質を有する複数の標識部分を導入し、結合している核酸プローブを、標識部分により検出する工程を含む(図16参照)。
【0108】
この方法においては、上記核酸プローブが用いられるが、核酸部分は、標的核酸に特異的に結合可能な核酸配列を有するものとする。また、標識部分は、容易に検出可能な性質を有するものである。
【0109】
本方法は、従来法、例えばジゴキシゲニン(DIG)標識化プローブを用いる方法に比較して、種々の点で優れている。例えば、DIG法では、核酸プローブのDIG修飾および標識化された抗DIG抗体が必要であり、それに伴う煩雑な洗浄操作が必要となるが、本実施形態に係る核酸プローブを用いれば、DIG標識化プローブおよび標識化抗DIG抗体が不要になるため、試薬、手間および時間を大幅に削減することが可能となる。また、1つの認識部位(核酸)に対して複数のシグナル増幅部位(標識部分)を配置することにより、検出感度の向上が可能になる。
【0110】
例えば、MTGなどのTGaseを用いて、MTG認識GlnなどのTGase認識Glnが複数箇所に導入された核酸プローブZ−QG RNAに、TGase認識Lysを導入した酵素、蛍光色素などの標識化合物を結合させる。
【実施例】
【0111】
以下、実施例および比較例を挙げ、本発明をより具体的に詳細に説明するが、本発明は、以下の実施例に限定されるものではない。
【0112】
本実施例では、In situ ハイブリダイゼーション(ISH)法への応用を目指して、超好熱菌由来酵素マルチラベル化RNAの開発を行った。酵素ラベル化の手法としては、微生物由来トランスグルタミナーゼ(MTG)のタンパク質修飾能を利用した。MTGの良基質であるZ−QGを導入したZ−QG−UTPを合成し、RNA polymeraseによる転写反応によってZ−QG RNAを調製した。さらに、MTGを用いて超好熱菌由来酵素マルチラベル化核酸プローブを調製し、その性能をドットブロットによって評価した。
【0113】
(実施例1)
<Z−QG−UTPの合成・精製>
Z−QG−UTPの合成スキームは、図3に示す通りである。
【0114】
まず100mM N,N’−ジイソプロピルカルボジイミド(DIC)、100mM N-ヒドロキシスクシンイミド(NHS)、50mM Z−QGを、室温(調製した日は27.0℃)でN,N−ジメチルホルムアミド(DMF)4mL中で20時間反応させることにより、NHS化Z−QG(50mM)を調製した(この段階で500μLずつに分注し、−80℃で保存)。その後、25mMのNHS化Z−QGと5mMの5−(3−aminoallyl)−UTP(以下、aminoallyl−UTPと略記、SIGMA社製)とを、100mM ホウ酸緩衝液(pH8.8)とDMFの混合溶媒(v/v=1/1)0.32mL中において25℃で12時間反応させた。反応終了後、サンプルをMilli−Qで10倍希釈し、HPLC(日本分光製)(高速液体クロマトグラフ・ポンプ:TRI ROTAR−V型、バリアブル・ループ・インジェクタ:VL−613型、紫外可視分光検出器:UVIDEC−100−IV型)によって精製を行った。HPLCの測定条件は表1の通りとした。生成物の同定は、レーザーイオン化飛行時間型質量分析装置であるMALDI TOF−MS(島津製作所製 AXIMA(登録商標)−CFR Plus)によって行った。なお、サンプル調製手順は、まず、試料1μLをMALDI用試料プレートの上に滴下し、次にその上から、マトリックス溶液;2,5−ジヒドロキ安息香酸(DHB)の10mg/ml溶液(超純水)を滴下し、その後、風乾して、得られた試料プレートをMALDI TOF−MS装置のイオン源内に導入して計測した。
【0115】
Z−QG−UTPを合成した後、表1に示す条件で逆相HPLCを行った際の結果を図4に示す。aminoallyl−UTPの場合(図4(A))と比較して、疎水側に新たなピーク(22min)が出現しており(図4(B))、このピークがZ−QG−UTPであると推測される。しかし、生成物ピークの分離が十分でなかったので、HPLC条件を表2に示すように変えて検討を行った。結果を図5に示す。
【0116】
【表1】
【0117】
【表2】
【0118】
そこで、保持時間18.8分のピークを回収してMALDI TOF−MS分析を行った(図6参照)。その結果、856.89のピークが確認され、理論分子量の857.13と良く一致した結果が得られたため、Z−QG−UTPの合成が示された。
【0119】
[実験1:Alkphos Directキットの検出感度の評価]
<センス鎖RNA、アンチセンス鎖RNAの調製>
RNA polymeraseにより転写を行うことによって、センス鎖RNA、アンチセンス鎖RNAを調製した。標的核酸(ターゲット)としては、マウス中にて発現パターンが既知であるSonic hedgehog(SHH)というタンパク質をコードするmRNA(shh)をモデルとした。そこでまず、SHHをコードした遺伝子を含むプラスミドをテンプレートとしてPCRを行い、長さ約1000bpのDNA断片を得た。この際、上流にはT3プロモータ、下流にはT7プロモータが含まれるようにプライマー設計を施した。次に得られたPCR産物を基に、T3 polymerase(センス鎖(S) 調製用)を用いてRNA合成を行い、T3 polymeraseで転写反応を行ってセンス鎖配列を調製した(配列番号:24)。同様にT7 polymerase(アンチセンス鎖(AS) 調製用)を用いてRNA合成を行い、T7 polymeraseにで転写反応を行ってアンチセンス鎖配列を合成した(配列番号:25)。条件は以下の表3に示す。反応は37℃で2時間行い、得られたセンス鎖RNA、アンチセンス鎖はエタノール沈殿によって回収し、TEバッファー20μLで縣濁した。RNase inhibitorとDTTを添加し、RNaseによる分解を抑えた。
【0120】
【表3】
【0121】
<RNA濃度の測定>
転写反応後、Nano DropにてRNA濃度を測定した。その結果、センス鎖は1019ng/μL、アンチセンス鎖は815ng/μLであった。
【0122】
<標識プローブの作製>
Alkphos Direct(商標、GEヘルスケアバイオサイエンス社製)のキットに付随する試薬にて標識プローブを調製した。まず、上述したアンチセンス鎖RNAを滅菌水を用いて10ng/μLに希釈したもの60μL(総量600ng)を99.9℃で熱変性後、急冷した。キットに添付されているReaction buffer 60μL、Labeling reagent 12μL、Cross−linker溶液60μLの順に熱変性後のRNAに添加し、37℃で30分間インキュベートし、標識プローブを作製した。
【0123】
<検出用メンブレンの作製>
まず、ターゲットとなるセンス鎖RNA、アンチセンス鎖RNAを1pg/μL〜50ng/μlの6段階の濃度に滅菌水を用いて希釈したものを、それぞれ1μLずつプラスのチャージを持つメンブレン(Hybond N+、GEヘルスケアバイオサイエンス社製)にアプライした。その後、80℃で2時間ベーキングした。
【0124】
<ハイブリダイゼーション>
上記検出用メンブレンの作製で作製したメンブレンをキットに添付されているハイブリダイゼーションバッファーに移し、55℃で15分間プレハイブリダイゼーションを行った。その後、上述した標準プローブを濃度が50ng/mLとなるように添加して55℃で一晩(14時間〜16時間程度)ハイブリダイゼーションを行った。
【0125】
<メンブレンの洗浄>
ハイブリダイゼーション後、メンブレンをWashIバッファー(表4)に移し、55℃で振とうしながら15分洗浄した。この操作を2回繰り返した。その後、WashIIバッファー(表5)にて洗浄した。この操作を2回繰り返した。
【0126】
【表4】
【0127】
【表5】
【0128】
<発色法による検出>
NTMTxバッファー(表6)で室温(実験した日は18〜22℃程度)、5分間洗浄したメンブレンをStaining溶液(375μg/mL NBT+188μg/mL BCIP in NTMTx)に移し、室温で2時間、遮光下で発色させた。なお、NBTとは、Nitro blue tetrazolium chlorideの略称であり、BCIPは、5-Bromo-4-chloro-3-indolyl phosphate, toluidine saltのことである。
【0129】
【表6】
【0130】
ドットブロットの結果を図7に示す。プローブと相補的なセンス鎖のメンブレンでは、100pg/μLのドットまで検出できた。また、アンチセンス鎖でも50,10ng/μLのドットに若干のシグナルがみられた。なお、この結果は、ターゲットRNAの調製時に使用した鋳型のDNAがDNaseIで処理できず残っていた可能性が考えられる。また、洗浄操作の際に温度が低下し、ストリンジェンシーの低下による非特異的吸着も考えられる。
【0131】
[実験2:Alkphos Directのプロトコールに準じた系におけるTGase基質マルチラベル化核酸プローブを用いた場合の検出感度の評価]
<TGase基質マルチラベル化核酸の調製>
上記実験1のセンス鎖RNA、アンチセンス鎖RNAの調製に加え、表7、表8に準じてTGase基質マルチラベル化核酸もT7 polymeraseを用いて同様に調製した。調製後、DNaseIで処理し、エタノール沈殿でRNAを濃縮・精製してTEバッファー20μLにて縣濁した。また、RNase inhibitorとDTTも添加し、新規酵素マルチラベル化核酸プローブを作製した。
【0132】
【表7】
【0133】
【表8】
【0134】
<RNA濃度の測定>
転写反応後、Nano DropにてRNA濃度を測定した。以下に測定結果を示す。なお、表8中の○数字を以下括弧数字で表示する。
(1)Z−QG−UTP 0%:410ng/μL
(2)Z−QG−UTP 20%:424ng/μL
(3)Z−QG−UTP 40%:378ng/μL
(4)Z−QG−UTP 60%:368ng/μL
(5)Z−QG−UTP 80%:311ng/μL
(6)Z−QG−UTP 100%:256ng/μL
【0135】
<標識プローブの作製>
Z−QG−UTPを異なる割合で取り込んだTGase基質マルチラベル化核酸を99.9℃で5分間熱変性後、急冷した。熱変性した核酸を用いて表9の条件となるように反応溶液を調製し、4℃で6時間反応させて酵素マルチラベル化プローブを作製した(配列番号:23)。
【0136】
【表9】
【0137】
以下、上述した実験1の検出用メンブレンの作製、ハイブリダイゼーション、メンブレン洗浄を行った。
【0138】
<発色法による検出>
検出は発色時の温度を室温(実験した日は18〜22℃程度)と50℃に設定し、それ以外の操作は実験1の発色法による検出操作に準じて行った。その結果、酵素マルチラベル化核酸プローブを用いてAlkphos Directのプロトコールに準じて検出した結果を図8に示した。室温で発色反応を行った場合には、Z−QG−UTPの割合が20〜40%のプローブでは10ng/μLまで、Z−QG−UTPの割合が60〜100%のプローブでは1ng/μLのドットまで検出することができた。また、50℃で発色反応を行った場合では、Z−QG−UTPの割合が20%では10ng/μL、40〜60%では1ng/μL、80〜100%では100pg/μLのドットまで検出できた。
【0139】
以上より、Z−QG導入割合の増加に伴って、検出感度は向上することが分かった。また、室温での発色条件下ではZ−QG−UTP60%以上の時、1ng/μLのターゲット核酸の検出に成功した。また、発色時の温度を新規酵素(アルカリホスファターゼ:PfuAP)の至適温度に近づけると、Z−QG−UTP80%以上で市販のAlkphos Directの検出限界(100pg/μL)と同等の検出感度で、コントラストの高い像を得ることができた。
【0140】
(実施例2:ドットブロットによるISHプロトコールに準じた検出感度の評価)
[PfuAPマルチラベル化RNAプローブの調製および機能評価]
細胞組織内での遺伝子発現パターンが既知であるソニックヘッジホッグ遺伝子(shh)をターゲットのmRNAとする。まず、PCRによってshhをコードするDNA断片(約1000bp)を得た。この際、遺伝子の上流側にはT3プロモーター、下流側にはT7プロモーターが含まれるようプライマー設計を施した。次に、得られたPCR産物を基にT7Polymeraseを用いて転写反応を行った(表10)。この際、UTPの代わりに、実験1で合成したZ−QG−UTPを様々な割合(0〜100%)にて加えることで、導入率の異なるZ−QG RNAを調製した。得られたZ−QG RNAはエタノール沈殿によって回収し、アガロースゲル電気泳動およびマイクロチップ型電気泳道装置(Bio−Rad,Experion)により評価した。
【0141】
【表10】
【0142】
<PfuAPマルチラベル化RNAの調製>
遺伝子工学的手法により、N末端にMTGが認識するKを含んだペプチドタグ(MKHK(GGGS)2GS)を導入したNK14−PfuAPが、MTGによってZ−QG RNAと結合するか検討するために、まず、様々な割合のZ−QG−UTPを用いて調製したZ−QG RNAを、99.9℃まで加熱した後、急冷することによって、変性処理を行った。続いて、100mM Tris−HCl(pH8.0)緩衝液中にて、各種Z−QG RNA50μg/mL、NK14−PfuAP 0.4mg/mL、MTG5.0U/mL、RNase inhibtor 1.0U/μLの条件下にて、6時間反応を行った。コントロールとして、MTG未添加の場合にも同様の操作を行い、反応はアガロースゲル電気泳動によって評価した。
【0143】
<ドットブロットによるプローブ能評価>
shh S(ターゲット)をメンブレン上に固定化し、PfuAPマルチラベル化核酸プローブ(shh アンチセンス、AS)を用いて検出を試みた。まず、未標識shh S(T3 Polymeraseにより調製)の希釈系列(50ng/μL、10ng/μL、1ng/μL、0.1mg/μL)を調製し、メンブレンに1μLずつスポットし、オーブンにて固定した(80℃、2時間)。その後、表11に示す操作を行い、PfuAPマルチラベル化プローブを用いてshh Sの検出を行った。コントロールとして、shh AS鎖を同様にして固定化したメンブレンを調製し、これを用いて行った。尚、溶液の組成は表12に記す。
【0144】
【表11】
【0145】
【表12】
【0146】
<Z−QGマルチラベル化RNAの調製>
UTPとZ−QG−UTPの割合を変化させてRNAを調製した際の結果を、図9の(A)、(B)に示す。結果より、いずれの条件においてもRNAの合成に成功していることから、合成したZ−QG−UTPは、比較的良好な基質として、T7 Polymeraseに認識されることが分かる。また、Z−QG−UTPの割合増加とともに高分子量側へとシフトしていることから、Z−QG−UTPの濃度を変化させることによって、Z−QG RNAへのZ−QG−UTP導入量が調節可能である。
【0147】
<PfuAPマルチラベル化核酸プローブの調製>
それぞれ導入量の異なる、Z−QG RNAに対して、MTGを用いてPfuAPラベル化を行った際の結果を図10に示す。Lane2はMTG未添加時、Lane3はMTG添加時の結果である。20%〜100%の条件では、MTG添加時において高分子量側へのシフトが見られるが、Z−QG−UTPが0%の際には確認されないために、導入したZ−QG特異的にZ−QG−UTPがラベル化されたことが分かる。また、Z−QG導入量の増加とともに高分子量側へのシフトも大きくなっていることからZ−QG−UTPの割合を変化させることによって、プローブのPfuAPのラベル化量が制御可能である。
【0148】
<ドットブロットによるプローブ能評価>
調製したPfuAPマルチラベル化RNAプローブの核酸プローブとしての機能について検討するためにドットブロットを行った(図11)。AS(アンチセンス)鎖をメンブレンに固定化した際にはスポットが確認されないことから、塩基配列特異的な検出ができている。20〜100%導入プローブにおいて1ng/μLのセンス鎖の検出が可能であった。以上のことから、転写反応時のZ−QG−UTP濃度を変化させることによって、得られる修飾RNA中のZ−QG導入量、その後のMTGによるPfuAPラベル化量が制御可能なことが示された。なお、実施例1とは異なり、Z−QG導入量増加に伴う検出感度向上は確認されなかった。この理由としては、上記表11,12に示す様に、ハイブリダイズ条件および洗浄条件が、Alkphos Direct条件に比べるとかなり厳しい条件であることが挙げられる。図10に示すようにいずれの場合においても、PfuAPラベル化した際のバンドはブロード化しており、PfuAPが均一に導入されているとは言えない。そのためPfuAPの導入数が少ないプローブ、あるいは未修飾のプローブの方がより強固にターゲット(AS鎖)とハイブリダイズするため、いずれの場合においても同等の感度となったのではないかと推察される。また、PfuAPの導入に伴うプローブの二本鎖形成能低下の可能性も、その一因として挙げられる。
【0149】
一方、超好熱菌由来酵素であるPfuAPを活性化させるために、染色時の温度を室温から50℃程度まで上昇させれば、現段階においても更なる感度向上が見込まれる。
【0150】
<高温条件での発色を行う場合の酵素ラベル化核酸プローブによるISHの検出感度評価>
本発明における核酸プローブにラベル化されたPfuAPは、室温よりも高温条件でより高い触媒活性を発現する。そこで、表11の酵素発色条件時の反応温度を、室温から50℃に上げた場合の検出感度を評価した。酵素ラベル化核酸プローブとしての性能を評価するため、まず、Alkphos Directキットにより得られる酵素標識核酸プローブと、本発明のPfuAPラベル化核酸プローブの性能比較を、ISH条件において検討した。実験は、酵素標識核酸プローブをキットにより調製する以外は、上述同様の操作によりドットブロットを行った。その結果、1ng/μLのセンス鎖の検出が可能であった(図12)。このことから、今回新たに調製されたPfuAPマルチラベル化核酸プローブは、ISH条件下において、市販のAlkphos Directにより調製されたプローブと同等の検出感度を有するプローブとして利用できることが示唆された。そこでは、最終的に核酸を検出するステップ、すなわち、酵素(アルカリホスファターゼ)部位による発色反応における温度を室温で行っていた。一方、本発明の核酸プローブにラベル化されたPfuAPは、室温よりも高温条件でより高い触媒活性を発現する。そこで、表10の酵素発色条件時の反応温度を、室温から50℃に上げた場合の検出感度を評価した。その結果、図13に示すように、検出限界はZ−QG導入割合により異なり、Z−QG80%、100%で100pg/μLから1ng/μL、Z−QG60%においては10pg/μLの核酸を検出することが可能であり、上述した実験1のAlkphos Directプローブを用いた場合(1ng/μL)より、少なくとも2桁程度、検出感度の向上が可能なことが明らかとなった。
【0151】
上述した実施例では、以下に示すような濃度決定したサンプルを用いて実験を行った。
【0152】
[Z−QG−UTPの濃度算出]
<aminoallyl−UTPの吸光係数算出(@290nm)>
aminoallyl−UTPをTE Buffer(pH8.0)に溶解した後、0、0.02、0.04、0.06、0.08、0.1mMとなるように調製し、吸収スペクトル測定を行った(図14)。スペクトルより290nmの吸光度を算出し検量線を作成した(図15)。尚、図14のスペクトルは、スペクトルチャートの吸収感度の一番高いのが0.1mMであり、以下順に0.08mM、0.06mM、0.04mM、0.02mM、0mMと吸収感度が下がっている。
【0153】
図15より、290nmの吸光係数が算出される(ε290=5760M−1cm−1)が、市販のaminoallyl−UTP(SIGMA社製)の純度が84%であることを考慮すると、ε290=6860M−1cm−1となる。なお、290nmにおけるZ−QGの吸収はほとんどないため、Z−QG−UTPサンプル中に未反応のZ−QGが混入していたとしても、濃度決定への影響は無視できる。
【0154】
<Z−QG−UTPストックサンプルの調製>
HPLCによる精製後、凍結乾燥によって溶媒を除去し、滅菌水(2mL)に溶解した。その後、Speed vacを用いて濃縮した(この際、析出物が見られたため、未反応のZ−QGが析出していると考えられる。濃縮に伴いaminoallyl−UTPと同形のスペクトルは増加したため、Z−QG−UTPの析出ではないと考える)。濃縮後、上清を回収し、TE bufferによって10倍希釈した後、Nano Drop(光路長:1mm)を用いて吸光度測定を行った所、吸収度ABS=1.061であった。したがって、希釈前のZ−QG−UTP濃度Cは次式により算出される。
【0155】
(数1)
1.061=6860[M−1cm−1]×0.1[cm]×C/10
したがって、C=約15.5[mM]である。
【0156】
(実施例3)
[Z−QG DNAを用いたPfuAPマルチラベル化DNAプローブの調製とドットブロットによる検出感度の評価]
Z−QG DNAを用いたPfuAPマルチラベル化DNAプローブの調製とドットブロットによる検出感度の評価を行った。
【0157】
<Z−QG−dUTPの合成・精製>
まず、N,N−ジメチルホルムアミド(DMF)4mL中にて、100mM N,N’−ジイソプロピルカルボジイミド(DIC)、100mM N−ヒドロキシスクシンイミド(NHS)、50mM Z−QGを、室温(調製した日は18〜22℃程度)で20時間反応させることによって、NHS化Z−QG(50mM)を調製した。一方、50mM 5−(3−aminoallyl)−dUTP(以下、aminoallyl−dUTPと略記、SIGMA社製)を含む10mM Tris−HCl(pH7.5)溶液(Ambion製)16μLと200mM ホウ酸緩衝液(pH8.8)40μL、滅菌水16μLとを混合し、10mM aminoallyl−dUTP溶液を80μL調製した。この溶液に対して、上記で調製したNHS化Z−QG溶液を80μL添加し、25℃で一晩反応させた。反応終了後、サンプルをMilli−Qで10倍希釈し、HPLC(日本分光製、高速液体クロマトグラフ・ポンプ:TRI ROTAR−V型、バリアブル・ループ・インジェクタ:VL−613型、紫外可視分光検出器:UVIDEC−100−IV型)によって、表13に示す条件で精製を行った。生成物の同定はMALDI TOF−MS(BRUKER DALTONICS製、autoflex III)によって行った。結果を図17に示す。この際、3−ヒドロキシピコリン酸(3−HPA)をマトリックスとして使用した。
【0158】
【表13】
【0159】
Z−QG−dUTPを合成した後、逆相HPLCを行った際の結果を図18に示す。aminoallyl−dUTPの場合(図19参照)と比較して、疎水側に新たなピークが出現しており、このピークがZ−QG−dUTPであると推測された。そこで、保持時間19.1分のピークを回収してMALDI TOF−MS分析を行った。その結果、図17に示すように、841.46のピークが確認され、理論分子量の842.13と良く一致した結果が得られたため、Z−QG−dUTPの合成が示された。
【0160】
<PCRによるZ−QG DNAの調製>
まず、マウス由来のSonic hedgehog(SHH)をコードする遺伝子中の約300bp(配列番号:27)をPCR増幅するプライマーを設計した(表14、配列番号:28,29)。PCRのテンプレートにはマウス由来のShhをコードする遺伝子の全配列(約990bp)を組み込んだプラスミドDNAを用い、DNA polymeraseとしてKOD−Fx(TOYOBO社製)を使用して反応液を調製した(表15、表16)。PCR条件は94℃×2分を1サイクル、94℃×20秒、52℃×20秒、72℃×15秒を30サイクルとした。PCR後、アガロース電気泳動で増幅断片を確認した。得られた増幅断片は、MinElute PCR Purification kit(QIAGEN社製)で精製し、Nano DropにてZ−QG DNAの濃度を測定した。
【0161】
【表14】
【0162】
【表15】
【0163】
【表16】
【0164】
Z−QG−dUTPの割合を変化させてPCRした際の電気泳動の結果を図20に示す。Z−QG−dUTPの割合を50%、100%としてPCRした場合でも増幅断片のバンドが確認されたことから、Z−QG−dUTPはDNA polymeraseの基質として認識されていることが示された。また、Z−QG−dUTPの割合の増加に伴ってバンドが高分子側にシフトしていることから、PCRを行う際のZ−QG−dUTPの量を変化させることで、Z−QGの導入量を調節することが可能であることがわかった。精製後にNano DropでそれぞれのZ−QG DNAの濃度を計った結果、Z−QG−dUTP 0%で調製したZ−QG DNAは73.8ng/μL、Z−QG−dUTP 50%で調製したZ−QG DNAは111.6ng/μL、Z−QG−dUTP 100%で調製したZ−QG DNAは59.1ng/μLであった。
【0165】
<Z−QG DNAへのPfuAPのラベル化の検討>
遺伝子工学的にMTGが認識可能なリジン残基(K)を含むペプチドダグを導入したNK14−PfuAPがMTGによってZ−QG DNAに結合するかを、Z−QGの導入量が異なるZ−QG DNAを用いて検討した。まず、100mM Tris−HCl(pH8.0)緩衝液中にて、各種Z−QG DNA 15ng/μL、NK14−PfuAP 0.4mg/mL、MTG 5.0Unit/mLの条件下で4℃、6時間反応させた。MTG未添加の場合も同様に検討した。PfuAPのラベル化の確認はアガロース電気泳動で行った。
【0166】
MTG反応後のアガロース電気泳動の結果を図21に示す。Z−QG−dUTP 50%、100%の条件で調製したZ−QG DNAを用いた場合はMTG添加時に高分子側へシフトしているが、Z−QG未ラベルDNA(Z−QG−dUTP 0%の条件で調製した陰性対照DNA)では分子量シフトが確認されなかった。また、Z−QG DNAに対して、MTG未添加時にも高分子量化は確認されなかった。この結果より、NK14−PfuAPはMTGによってZ−QG DNAのZ−QG部位へ特異的に結合していることがわかった。また、Z−QG−dUTP 50%の場合よりZ−QG−dUTP 100%の条件で調製したZ−QG DNAを用いたほうが、より明確に高分子量化が進んでいることから、PCRによる増幅の際のZ−QG−dUTPの割合を変えることによって、PfuAPの結合量を調節することが可能なことが示された。
【0167】
<PfuAPマルチラベル化DNAプローブの調製>
Z−QG−dUTP 50%、100%の条件で調製したZ−QG DNAとNK14−PfuAPをMTGにて結合し、PfuAPマルチラベル化DNAプローブを調製した。まず、各種Z−QG DNAを90℃で5分間熱処理してDNAを一本鎖に変性させた後、100mM Tris−HCl(pH8.0)緩衝液中にて、各種Z−QG DNA 25ng/μL、NK14−PfuAP 0.4mg/mL、MTG 5.0Unit/mLの条件下で4℃、6時間反応させた(MTG反応前熱処理プローブ)。DNAプローブは熱処理で一本鎖にすることがプローブの検出能に大きく影響することから、熱処理していないZ−QG DNAについても同様の条件でPfuAPのラベル化を行い、その後、90℃で5分間熱処理を施したプローブも調製した(MTG反応後熱処理プローブ)。DNAプローブのPfuAPラベル化の確認はアガロース電気泳動で行った。
【0168】
MTG反応後のアガロース電気泳動の結果を図22に示した。MTG反応前に熱処理したZ−QG DNAおよびMTG反応後に熱処理したZ−QG DNAとも高分子側へシフトしていることから、Z−QG DNAへのPfuAPのラベル化が確認された。
【0169】
<ドットブロットによる検出感度の評価>
それぞれ調製したPfuAPマルチラベル化プローブの検出感度の評価をドットブロットで行った。なお、ハイブリダイゼーション時の各種組成およびプロトコールは、市販の検出キットAlkphos Direct(GEヘルスケアバイオサイエンス社製)に準拠した。ターゲットDNAとして、プローブ配列を含む990bpのShhのDNA断片(配列番号:30、プライマー配列は表17参照(配列番号:31))、およびプローブ配列と同じ300bpのShhのDNA断片を用いた。また、ネガティブコントロールとして、PfuAPをコードする遺伝子約1.7kbのDNA断片(配列番号:32、プライマー配列は表18参照(配列番号:33,34))を用いた。
【0170】
【表17】
【0171】
【表18】
【0172】
それぞれのターゲットDNAをTE Bufferを用いて50ng/μLと50pg/μLに希釈し(ネガティブコントロールは50ng/μLのみ)、それぞれを1μLずつプラスチャージのメンブラン(Hybond N+、GEヘルスケアバイオサイエンス社製)にスポットし、80℃で2時間熱処理することでDNAをメンブランに固定した。その後、検出キットに添付されているハイブリダイゼーションバッファーにメンブランを移し、55℃で30分間、プレハイブリダイゼーションを行った。そして、上記方法で調製した各種PfuAPマルチラベル化DNAプローブを50ng/mLとなるようにハイブリダイゼーションバッファーに添加して、55℃で一晩(14時間〜16時間程度)ハイブリダイゼーションを行った。
【0173】
ハイブリダイゼーション後、メンブレンをWash Iバッファー(表19)に移し、55℃で震盪しながら15分間洗浄した。この操作を2回繰り返した。その後、室温(実験した日は18〜22℃程度)でWash IIバッファー(表20)にて5分間洗浄した。この操作を2回繰り返した。
【0174】
【表19】
【0175】
【表20】
【0176】
NTMTxバッファー(表21)で室温、5分間洗浄したメンブランをStaining溶液(375μg/mL NBT + 188μg/mL BCIP in NTMTx)に移し、50℃に2時間、遮光下で静置して発色させた。
【0177】
【表21】
【0178】
各種PfuAPラベル化DNAプローブのドットブロットの結果を図23に示した。どのDNAプローブを用いた場合もドットのシグナルが検出されていることから、MTGを用いてZ−QG DNAにPfuAPでラベルしたDNAプローブは、核酸プローブとしての検出能を有していることが示された。また、MTG反応後に熱処理する場合(MTG反応後熱処理プローブ)よりも、MTG反応前にZ−QG DNAを熱処理する場合(MTG反応前熱処理プローブ)の方が、検出感度を向上できることがわかった。これはMTG反応後熱処理プローブでは、PfuAPがラベル化されたDNAプローブを熱処理することになるため、熱処理時の反応容量の増加により熱処理が不十分となりDNAが完全に熱変性されなかったことや、ラベル化された状態での熱処理によるPfuAPの失活が考えられる。
【0179】
MTG反応前熱処理プローブでは、Z−QGの導入量が50%および100%のプローブのどちらも50pg/μLのドットまで検出することができた。ネガティブコントロールにおけるターゲット核酸濃度が50ng/μLと高い場合には、非特異的なシグナルも若干検出されたが、相補的なDNAを明確に判別することができた。また、ターゲットDNAの鎖長の違いは、検出感度に大きな影響を与えなかった。
【0180】
(実施例4)
[Z−QG DNAを用いたPfuAPマルチラベル化DNAプローブのISHプロトコールに準じたRNA検出]
実施例3では、検出対象核酸はDNAであった。本実施例4では、検出対象核酸をRNAとした場合の、PfuAPマルチラベル化DNAプローブの核酸プローブとしての機能について評価した。
【0181】
<PCRによるZ−QG DNAの調製>
実施例3と同様にして、PCRによるZ−QG DNAの調製を行った。
【0182】
<PfuAPマルチラベル化DNAプローブの調製>
Z−QG−dUTP 50%、100%の条件で調製したZ−QG DNAとNK14−PfuAPをMTGにて結合し、PfuAPマルチラベル化DNAプローブを調製した。まず、各種Z−QG DNAを90℃で5分間熱処理してDNAを一本鎖に変性させた後、100mM Tris−HCl(pH8.0)緩衝液中にて、各種Z−QG DNA 25ng/μL、NK14−PfuAP 0.4mg/mL、MTG 5.0Unit/mLの条件下で4℃、6時間反応させた。DNAプローブのPfuAPラベル化の確認はアガロース電気泳動で行った。
【0183】
MTG反応後のアガロース電気泳動の結果を図24に示した。Z−QG−dUTPを50%、100%導入したZ−QG DNAはどちらもMTG反応後に高分子側にバンドがシフトしていた。この結果よりZ−QG DNAへのPfuAPのラベル化が確認されたため、これらをドットブロットのDNAプローブとして使用した。
【0184】
<ISHプロトコールに準じたドットブロットによる検出感度の評価>
実施例3と同様にして、PCRによってプローブ配列を含む990bpのShhのDNA断片(配列番号:30)を得た。この際、Shh遺伝子の下流側にT7プロモーターが含まれるようプライマー設計(配列番号:31、表17参照)を施した。次に、得られたPCR産物を基にT7 Polymeraseを用いて転写反応を行った(表22)。転写反応後、ゲル濾過カラム(Mini Quick Spin Columns、Roche社製)で未反応のNTP等を精製したものをターゲットRNAとして用いた。
【0185】
【表22】
【0186】
得られたターゲットRNAをTE Bufferを用いて50ng/μL、1ng/μL、50pg/μLの三段階に希釈し、それぞれを1μLずつプラスチャージのメンブラン(Hybond N+、GEヘルスケアバイオサイエンス社製)にスポットし、80℃で2時間熱処理することでRNAをメンブランに固定した。ネガティブコントロールとして、市販の酵母RNA(Ribonucleic torula yeast RNA、シグマ・アルドリッチ製)を50ng/μLに希釈したものを同様にメンブランに1μLスポットし、固定化した。その後、ハイブリダイゼーションバッファー(表23)中にメンブランを移し、60℃で30分間、プレハイブリダイゼーションを行った。そして、上記方法で調製した各種DNAプローブを50ng/μLとなるように添加し、60℃で一晩(14時間〜16時間程度)ハイブリダイゼーションを行った。
【0187】
【表23】
【0188】
ハイブリダイゼーション後、メンブレンの洗浄操作を行った。各種洗浄溶液を表24に示す。メンブランをWash Iバッファーに移し、60℃で震盪しながら20分間洗浄した。この操作を3回繰り返した。そして、55℃でWash IIバッファーにて20分間洗浄した。この操作を3回繰り返した。続いてNTMTxバッファーを用いて室温(実験した日は18〜22℃程度)で5分間、2回洗浄し、洗浄したメンブランをStaining溶液(375μg/mL NBT + 188μg/mL BCIP in NTMTx)に移し、50℃で2時間、遮光下に静置して発色させた。
【0189】
【表24】
【0190】
各種PfuAPラベル化DNAプローブのISHプロトコールに準じたドットブロットの結果を図25に示した。Z−QGの導入量が50%および100%のどちらのDNAプローブも1ng/μLのドットまで検出することができ、シグナルの発色強度もどちらのプローブを用いた場合もほぼ同等であった。また、ネガティブコントロールの非特異的なシグナルは検出されなかった。このことより、MTGを用いてZ−QG DNAにPfuAPをラベルしたDNAプローブは、RNAを検出するプローブとして利用可能であることが明らかとなった。
【0191】
(実施例5)
実施例2と同様にして、常温菌であるEscherichia coli由来のAP(BAP)を用いてマルチラベル化したマルチラベル化核酸プローブについても、プローブ能評価を行った。
【0192】
<BAPマルチラベル化RNAの調製>
実施例2と同様にして、RNAの変性処理を行った後に、続いて、100mM Tris−HCl(pH8.0)緩衝液中にて、各種Z−QG RNA 50μg/mL、NK14−BAP 0.4mg/mL、MTG 5.0U/mL、RNase inhibtor 1.0U/μLの条件下にて、6時間反応を行った。コントロールとして、MTG未添加の場合にも同様の操作を行い、反応はアガロースゲル電気泳動によって評価した。
【0193】
<ドットブロットによるプローブ能評価>
実施例2と同様にして、ドットブロットによりプローブ能について評価した。
【0194】
ラベル化した際の分子量変化を図26に示す。結果より、Z−QG導入量に比例して、顕著な分子量増加が生じた。また、Z−QG RNA(100%)を用いた際には、分子量分布が小さくなっていることが確認された。RNA調製時にZ−QG−UTPのみを使用した場合には、Z−QGが導入される数や位置が単分散となるため(理論上全てのU)、ほぼ均一にラベル化が進行したと考えられる。PfuAP(図10)に比べて著しい分子量増加が見られた点から、Z−QG RNAとの反応性はBAPの方が優れていることが示唆される結果となった。
【0195】
一方で、得られたBAPマルチラベル化RNAを用い、ドットブロットによってプローブ能を評価した(図27参照)。結果より、Z−QG導入量が60%、80%、100%のmRNAに対してBAPラベル化を行った際において、1ng程度のRNAが検出可能であった。20%、40%においてほとんど検出されなかった理由としては、BAP未修飾のRNAが残存しているためであると予想される(図26参照)。PfuAPをラベル化した際のプローブ(検出限界:10pg)と比較すると、本系における標識酵素としてはPfuAPの方が適していることが示された。
【0196】
(実施例6:MTGによるZ−QG RNAの蛍光色素ラベル化およびドットブロットによる標的核酸の検出)
<Alexa Fluor 555マルチラベル化RNAの調製>
蛍光色素として、一級アミン誘導化されたAlexa Fluor(登録商標) 555 cadaverine(invitrogen社製)が、MTGによってZ−QG RNAと結合するかについて検討した。まず、Z−QG−UTP使用率100%の条件で調製したZ−QG RNAを、99.0℃まで加熱した後、急冷することによって、変性処理を行った。続いて、100mM Tris−HCl(pH8.0)緩衝液中にて、Z−QG RNA 50μg/mL、Alexa Fluor 555 cadaverine 20μM、MTG 5.0units/mL、RNase inhibitor 1.0U/μLの条件下にて、6時間反応を行った。コントロールとして、MTG未添加の場合およびZ−QGを導入していない未修飾RNAに対しても同様の操作を行い、反応はアガロースゲル電気泳動によって評価した。
【0197】
MTGを用いて、Z−QG RNAに対してAlexa Fluor 555 cadaverineラベル化を行った際の結果を図28に示す。ここで、Lane2は未修飾のRNAとAlexa Fluor 555 cadaverineを、Lane3はLane2の条件にMTGを加えたものを、Lane4にはZ−QG RNAとAlexa Fluor 555 cadaverineを、Lane5にはLane4にMTGを加えたものを示す。Lane4,5において未修飾RNAとほぼ同等の位置に見られるシグナルは未反応のAlexa Fluor 555 cadaverineに由来するものと考えられる。このことは、エチジウムブロミド染色前にLane2〜4のいずれのLaneにおいても同様のバンドが確認されたことからも支持される。Lane2とLane3では変化が見られないのに対して、Lane4とLane5とを比較すると、MTG添加時にはZ−QG RNA由来のバンドが消失し、低分子量側へのシフトが確認された。このことは、負電荷を帯びたAlexa Fluor 555 cadaverineがZ−QG RNAに結合したことを示しており、Alexa Fluor 555マルチラベル化RNAの生成が示唆された。
【0198】
<ドットブロットによるプローブ能評価>
shh S(ターゲット)をメンブレン上に固定化し、Alexa Fluor 555マルチラベル化核酸プローブを用いて検出を試みた。まず、500ng/μLの未標識shh Sを調製し、メンブレンに1μL添加し、乾燥させた後、再度1μL添加し(計1,000ng)、オーブンにて固定化した(80℃、2時間)。その後、表25に示す操作を行い、蛍光イメージャ(BIO−RAD製、Molecular Imager FX Pro、532nm excitation、555nm long pass)を用いて、Alexa Fluor 555マルチラベル化核酸プローブによるshh Sの検出を行った。コントロールとして、shh AS鎖を同様にして固定化したメンブレンに対しても同様の操作を行った。なお、溶液の組成は表26に示す。
【0199】
【表25】
【0200】
【表26】
【0201】
調製したAlexa Fluor 555マルチラベル化RNAの核酸プローブとしての応用性について検討するためにドットブロットを行った(図29参照)。AS鎖を固定化した際にはシグナルが得られなかったのに対して、S鎖を固定化した場合には蛍光が確認されたことから、Alexa Fluor 555マルチラベル化RNAはプローブとして応用可能であることが示された。また、塩基配列特異性を保持しているため、今後、ハイブリダイゼーション過程の温度やホルムアミド濃度を最適化することによって、より高感度な検出が期待される。
【技術分野】
【0001】
本発明は、ヌクレオシド三リン酸誘導体、核酸プローブ、マルチラベル化核酸プローブ、マルチラベル化核酸プローブの製造方法および標的核酸の検出方法に関する。
【背景技術】
【0002】
何らかの標識が施されたRNAプローブなどの核酸プローブを用い、細胞レベルにおけるDNAやRNAの発現パターンを検出、可視化することによって、生命現象に関する数多くの疑問点を解明することが可能となる。細胞レベルでの遺伝子発現パターンを可視化するこのような手法をin situ ハイブリダイゼーション(in situ hybridization:ISH)と言うが、この際に使用されるプローブの標識法は大別して、「放射性同位体標識法」、「蛍光抗体標識法」、「酵素抗体標識法」に分類される。歴史的には、放射性同位体を取り込ませた核酸プローブが先に確立したが、近年その取り扱いが制限されてきたこともあって、放射性同位体元素を用いない蛍光抗体標識法や酵素抗体標識法が注目されている。
【0003】
これらの手法は、核酸プローブ作製時に抗原やビオチンでラベル化しておき、それらを標的核酸にハイブリダイズした後、酵素もしくは蛍光物質によって標識された抗体やアビジンを用いて免疫染色法により検出するといった手法である。感度という観点から見ると、酵素反応によるシグナル増幅効果が得られる酵素抗体標識法が優れており、現在最も広く使用されている。
【0004】
酵素抗体標識法を利用した核酸プローブとしては、例えば、ジゴキシゲニン(DIG)などの抗体認識部位で修飾したヌクレオチド誘導体を複数個ランダムに導入した抗原マルチラベル化核酸プローブが知られている。この抗原マルチラベル化核酸プローブと標的核酸とのin situ ハイブリダイゼーションの後、抗体認識部位を認識する酵素標識化抗体との抗原−抗体反応を行い、酵素アルカリホスファターゼとのハイブリッドによる発色反応を利用して検出を行う。しかしながら、酵素標識された抗体が非常に高価であること、抗原−抗体反応に伴う操作の煩雑化や非特異吸着などによるバックグラウンドの増加など、幾つかの問題を有している。
【0005】
一方、トランスグルタミナーゼ(TGase)を用いて、TGaseが認識可能なリシン(Lys)残基または第一級アミンを有するペプチドまたはタンパク質へ、アニオン性であり、かつTGaseが認識可能なグルタミン(Gln)残基を有する外来分子を部位特異的に連結する方法が知られている(例えば、特許文献1参照)。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特開2008−54658号公報
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明は、新規なヌクレオシド三リン酸誘導体、核酸プローブ、および簡便かつ高感度に標的核酸を検出することができるマルチラベル化核酸プローブ、そのマルチラベル化核酸プローブの製造方法、そのマルチラベル化核酸プローブまたは核酸プローブを用いた標的核酸の検出方法である。
【課題を解決するための手段】
【0008】
本発明は、グルタミン(Gln)残基を有するヌクレオシド三リン酸誘導体である。
【0009】
また、前記ヌクレオシド三リン酸誘導体が、下記式(1)で示されるものであることが好ましい。
【化1】
(式(1)中、Aは、グルタミン(Gln)残基を有する置換基、Bは水素原子またはヒドロキシル基を表す。)
【0010】
また、前記ヌクレオシド三リン酸誘導体が、下記式(2)で示されるものであることが好ましい。
【化2】
(式(2)中、A1およびA2のうち少なくとも1つは、グルタミン(Gln)残基を有する置換基で残りは水素原子を表し、Bは水素原子またはヒドロキシル基を表す。)
【0011】
また、前記ヌクレオシド三リン酸誘導体が、下記式(3)で示されるものであることが好ましい。
【化3】
(式(3)中、Aは、グルタミン(Gln)残基を有する置換基、Bは水素原子またはヒドロキシル基を表す。)
【0012】
また、前記ヌクレオシド三リン酸誘導体が、下記式(4)で示されるものであることが好ましい。
【化4】
(式(4)中、Aは、グルタミン(Gln)残基を有する置換基、Bは水素原子またはヒドロキシル基を表す。)
【0013】
また、前記ヌクレオシド三リン酸誘導体が、下記式(5)で示されるものであることが好ましい。
【化5】
(式(5)中、XおよびYは、それぞれ独立して2価の連結基を表し、Zは、置換基を表す。)
【0014】
また、前記ヌクレオシド三リン酸誘導体において、前記XおよびYは、それぞれ独立して炭素数1〜48のアルキレン基または炭素数2〜48のアルケニレン基であることが好ましく、Zは、炭素数1〜48のアルキル基、炭素数1〜48のアルコキシ基、炭素数6〜48のアリール基、炭素数6〜48のアリールオキシ基、炭素数7〜48のアリールアルキル基または炭素数7〜48のアリールアルキルオキシ基であることが好ましい。また、Yは、炭素数2〜48のオキシアルキレン基(例えば、オキシエチレン基、オキシプロピレン基)また、Y,Zは独立してLys以外のアミノ酸により少なくとも一方が置換されていてもよい。
【0015】
また、前記ヌクレオシド三リン酸誘導体において、前記Xはエテニレン基、Yはメチレン基であり、Zはベンジルオキシ基であることが好ましい。
【0016】
また、本発明は、構成単位として、前記ヌクレオシド三リン酸誘導体が複数個導入されている核酸である核酸プローブである。
【0017】
また、本発明は、前記核酸プローブにおけるグルタミン(Gln)残基のうち少なくとも2つに、リシン(Lys)残基または第一級アミンを有し標識部分を含む標識化合物が結合されて構成されているマルチラベル化核酸プローブである。
【0018】
また、前記マルチラベル化核酸プローブにおいて、前記標識部分が、酵素および蛍光色素のうち少なくとも1つであることが好ましい。
【0019】
また、前記マルチラベル化核酸プローブにおいて、前記酵素が、超好熱菌由来の酵素であることが好ましい。
【0020】
また、前記マルチラベル化核酸プローブにおいて、前記酵素が、有機溶媒や熱に対して安定な酵素であることが好ましい。
【0021】
また、本発明は、マルチラベル化核酸プローブの製造方法であって、トランスグルタミナーゼ(TGase)を用いて、前記核酸プローブにおけるグルタミン(Gln)残基のうち少なくとも2つに、リシン(Lys)残基または第一級アミンを有し標識部分を含む標識化合物を結合するマルチラベル化核酸プローブの製造方法である。
【0022】
また、前記マルチラベル化核酸プローブの製造方法において、前記標識部分が、酵素および蛍光色素のうち少なくとも1つであることが好ましい。
【0023】
また、前記マルチラベル化核酸プローブの製造方法において、前記酵素が、超好熱菌由来の酵素であることが好ましい。
【0024】
また、本発明は、標的核酸の検出方法であって、前記マルチラベル化核酸プローブと、対象物中に存在する標的核酸とを核酸部分により特異的に結合させ、結合している前記マルチラベル化核酸プローブを、前記標識部分により検出する標的核酸の検出方法である。
【0025】
また、本発明は、標的核酸の検出方法であって、前記核酸プローブと、対象物中に存在する標的核酸とを核酸部分により特異的に結合させた後、トランスグルタミナーゼ(TGase)を用いて、前記核酸プローブにおけるグルタミン(Gln)残基と、リシン(Lys)残基または第一級アミンを有し標識部分を含む標識化合物のリシン(Lys)残基または第一級アミンとを反応させて複数の標識部分を導入し、結合している前記核酸プローブを、前記標識部分により検出する標的核酸の検出方法である。
【発明の効果】
【0026】
本発明では、共有結合的に複数の標識部分を導入したマルチラベル化核酸プローブを得るための新規なヌクレオシド三リン酸誘導体、核酸プローブを提供することができる。
【0027】
また、本発明では、ターゲットとなる標的核酸にハイブリダイズする前に、予め共有結合的に複数の標識部分を核酸プローブに導入することにより、簡便かつ高感度に標的核酸を検出することができるマルチラベル化核酸プローブを提供することができる。
【0028】
また、本発明では、トランスグルタミナーゼ(TGase)を用いて、共有結合的に複数の標識部分を核酸プローブに導入することにより、簡便かつ高感度に標的核酸を検出することができるマルチラベル化核酸プローブの製造方法を提供することができる。
【0029】
また、本発明では、予め共有結合的に複数の酵素を導入したマルチラベル化核酸プローブを用いることにより、簡便かつ高感度に標的核酸を検出することができる標的核酸の検出方法を提供することができる。
【0030】
また、本発明では、核酸プローブをターゲットとなる標的核酸にハイブリダイズさせた後、核酸プローブにおけるグルタミン(Gln)残基と標識化合物のリシン(Lys)残基または第一級アミンとの簡易な反応によって、共有結合的に複数の標識部分を導入することにより、簡便かつ高感度に標的核酸を検出することが可能な標的核酸の検出方法を提供することができる。
【図面の簡単な説明】
【0031】
【図1】本発明の実施形態に係るヌクレオチド誘導体の構造の一例(Z−QG−UTP)を示す図である。
【図2】本発明の実施形態に係る酵素マルチラベル化核酸プローブの調製方法を示す概略図である。
【図3】本発明の実施形態に係るヌクレオチド誘導体の一例であるZ−QG−UTPの合成方法の一例を示す図である。
【図4】本発明の実施例1において、Z−QG−UTPを合成した後、逆相HPLC(表1のHPLC測定条件)を行った際の結果を示す図である。
【図5】本発明の実施例1において、Z−QG−UTPを合成した後、逆相HPLC(表2のHPLC測定条件)を行った際の結果を示す図である。
【図6】本発明の実施例1において合成したZ−QG−UTPのMALDI TOF−MS分析の結果を示す図である。
【図7】実施例1における実験1の標準プローブを用いたドットブロットの結果を示す図である。
【図8】実施例1における実験2の酵素マルチラベル化核酸プローブを用いてAlkphos Directのプロトコールに準じて検出した結果を示す図である。
【図9】UTPとZ−QG−UTPの割合を変化させてRNAを調製した場合の分子量変化について、(A)アガロースゲル電気泳動装置、(B)マイクロチップ型電気泳動装置にて電気泳動を行った結果を示す図である。
【図10】MTGを用いてPfuAPラベル化を行った際のアガロースゲル電気泳動装置による電気泳動の結果を示す図である。
【図11】PfuAPマルチラベル化RNAプローブの核酸プローブを用いた室温におけるドットブロットの結果を示す図である。
【図12】Alkphos Directキットにより調製された酵素ラベル化核酸プローブによるISHの検出感度評価を示す図である。
【図13】PfuAPマルチラベル化RNAプローブの核酸プローブを用いた50℃におけるドットブロットの結果を示す図である。
【図14】aminoallyl−UTP in TE Buffer(pH8.0)におけるaminoallyl−UTPの濃度に対する吸収スペクトル測定結果を示す図である。
【図15】スペクトルより290nmの吸光度を算出し検量線を示す図である。
【図16】本発明の実施形態に係る標的核酸の検出方法の一例を示す概略図である。
【図17】本発明の実施例3において合成したZ−QG−dUTPのMALDI TOF−MS分析の結果を示す図である。
【図18】本発明の実施例3において、Z−QG−dUTPを合成した後、逆相HPLC(表13のHPLC測定条件)を行った際の結果を示す図である。
【図19】本発明の実施例3において、aminoallyl−dUTPの逆相HPLC(表13のHPLC測定条件)を行った際の結果を示す図である。
【図20】本発明の実施例3のZ−QG DNAの調製において、Z−QG−dUTPの割合を変化させてPCRした際の電気泳動の結果を示す図である。
【図21】本発明の実施例3のZ−QG DNAへのPfuAPのラベル化の検討において、MTG反応後のアガロース電気泳動の結果を示す図である。
【図22】本発明の実施例3のPfuAPマルチラベル化DNAプローブの調製において、MTG反応後のアガロース電気泳動の結果を示す図である。
【図23】本発明の実施例3において、各種PfuAPラベル化DNAプローブのドットブロットの結果を示す図である。
【図24】本発明の実施例4のPfuAPマルチラベル化DNAプローブの調製において、MTG反応後のアガロース電気泳動の結果を示す図である。
【図25】本発明の実施例4において、各種PfuAPラベル化DNAプローブのドットブロットの結果を示す図である。
【図26】本発明の実施例5において、MTGを用いてBAPラベル化を行った際のアガロースゲル電気泳動装置による電気泳動の結果を示す図である。
【図27】本発明の実施例5において、BAPマルチラベル化RNAプローブの核酸プローブを用いた室温におけるドットブロットの結果を示す図である。
【図28】本発明の実施例6において、MTGを用いて、Z−QG RNAに対してAlexa Fluor 555 cadaverineラベル化を行った際の結果を示す図である。
【図29】本発明の実施例6において、Alexa Fluor 555マルチラベル化RNAの核酸プローブを用いた室温におけるドットブロットの結果を示す図である。
【発明を実施するための形態】
【0032】
本発明の実施の形態について以下説明する。本実施形態は本発明を実施する一例であって、本発明は本実施形態に限定されるものではない。
【0033】
本発明者らは、核酸プローブに複数の酵素を共有結合的に導入する手法として、微生物由来トランスグルタミナーゼ(MTG)などのトランスグルタミナーゼ(TGase)が有する部位特異的なタンパク質修飾能に着目した。TGaseはアシル転移反応を触媒する酵素であり、例えば、タンパク質中の特定のGln残基(Q)のγ−カルボキシアミド基と、Lys残基(K)のε−アミノ基や各種一級アミンとの共有結合を触媒する酵素である。このTGaseを用いて、複数の標識酵素などの標識部分を導入したマルチラベル化核酸プローブの創製を行う。
【0034】
具体的には、例えば、図1に示す、ウリジン三リン酸(uridine triphosphate:UTP)に、TGaseが認識するGln(MTG認識Gln)を有するZ−QGを結合させたヌクレオチド誘導体であるZ−QG−UTPを合成し、図2に示すように、核酸プローブとなるRNAを調製する際にZ−QG−UTPを取り込ませることによって、TGase認識Glnが複数箇所導入されたZ−QG RNAを調製する。その後、MTGなどのTGaseを用いて、MTG認識LysなどのTGase認識Lysを導入した標識酵素などの標識化合物を結合させることによって、RNAと標識酵素などの標識部分が1:n(nは2以上の整数)であるマルチラベル化核酸プローブを創製することができる。
【0035】
このマルチラベル化核酸プローブは、ターゲットとなる標的核酸にハイブリダイズした後、直ちに検出反応を行うことができるため、既存の手法と比較して、操作の大幅な簡素化、バックグラウンドの低下、バルク酵素である微生物由来トランスグルタミナーゼ(MTG)を用いることによるコストの抑制などが見込まれる。
【0036】
なお、図2において、核酸プローブにおけるGln残基と、標識化合物におけるLys残基とは逆であってもよい。すなわち、TGaseが認識するLys(MTG認識Lys)を有するヌクレオシド三リン酸誘導体を合成し、核酸プローブとなるRNAを調製する際に取り込ませることによって、TGase認識Lysが複数箇所導入された核酸プローブを調製する。その後、MTGなどのTGaseを用いて、MTG認識GlnなどのTGase認識Glnを導入した標識化合物を結合させることによって、RNAと標識部分が1:n(nは2以上の整数)であるマルチラベル化核酸プローブを創製してもよい。
【0037】
また、図16に示すように、Z−QG RNAなどの核酸プローブをターゲットとなる標的核酸にハイブリダイズした後、MTGなどのTGaseを用いて、MTG認識LysなどのTGase認識Lysを導入した標識酵素などの標識化合物を結合させてもよい。RNAと標識酵素などの標識部分が1:n(nは2以上の整数)となり、このように導入した標識部分の検出反応を行うことにより、既存の手法と比較して、操作の大幅な簡素化、バックグラウンドの低下、バルク酵素である微生物由来トランスグルタミナーゼ(MTG)を用いることによるコストの抑制などが見込まれる。
【0038】
なお、図16において、核酸プローブにおけるGln残基と、標識化合物におけるLys残基とは逆であってもよい。すなわち、TGaseが認識するLys(MTG認識Lys)を有するヌクレオシド三リン酸誘導体を合成し、核酸プローブとなるRNAを調製する際に取り込ませることによって、TGase認識Lysが複数箇所導入された核酸プローブを調製する。その後、核酸プローブをターゲットとなる標的核酸にハイブリダイズした後、MTGなどのTGaseを用いて、MTG認識GlnなどのTGase認識Glnを導入した標識化合物を結合させてもよい。
【0039】
<ヌクレオシド三リン酸誘導体>
本実施形態に係るヌクレオシド三リン酸誘導体は、グルタミン(Gln)残基を有する。グルタミン(Gln)残基を有するヌクレオシド三リン酸誘導体としては、グルタミン(Gln)残基を有する、ウリジン三リン酸(uridine triphosphate:UTP)誘導体、アデノシン三リン酸(adenosine triphosphate:ATP)誘導体、グアノシン三リン酸(guanosine triphosphate:GTP)誘導体、シチジン三リン酸(cytidine triphosphate:CTP)誘導体、デオキシウリジン三リン酸(deoxyuridine triphosphate:dUTP)誘導体、デオキシアデノシン三リン酸(deoxyadenosine triphosphate:dATP)誘導体、デオキシグアノシン三リン酸(deoxyguanosine triphosphate:dGTP)誘導体、デオキシシチジン三リン酸(deoxycytidine triphosphate:dCTP)誘導体などが挙げられる。本実施形態に係るヌクレオシド三リン酸誘導体において、グルタミン(Gln)残基は、例えば、ウラシル、アデニン、グアニン、シトシンの部分に直接または置換基を介して結合されている。
【0040】
これらのヌクレオシド三リン酸誘導体は、UTP、ATP、GTP、CTP、dUTP、dATP、dGTP、dCTPまたはそれらの各種誘導体から得ることができる。
【0041】
また、これらのヌクレオシド三リン酸誘導体は、ウリジン、ウリジンの一リン酸(UMP)および二リン酸(UDP)、アデノシン、アデノシンの一リン酸(AMP)および二リン酸(ADP)、グアノシン、グアノシンの一リン酸(GMP)および二リン酸(GDP)、シチジン、シチジンの一リン酸(CMP)および二リン酸(CDP)、デオキシウリジン、デオキシウリジンの一リン酸(dUMP)および二リン酸(dUDP)、デオキシアデノシン、デオキシアデノシンの一リン酸(dAMP)および二リン酸(dADP)、デオキシグアノシン、デオキシグアノシンの一リン酸(dGMP)および二リン酸(dGDP)、デオキシシチジン、デオキシシチジンの一リン酸(dCMP)および二リン酸(dCDP)ならびにそれらの各種誘導体から得てもよい。
【0042】
例えば、ウリジン、アデノシン、グアノシン、シチジン、デオキシウリジン、デオキシアデノシン、デオキシグアノシン、デオキシシチジンのリン酸化酵素などによるリン酸化(例えば、生物工学会誌,85(9),p397−399(2007)、Journal of Bioscience and Bioengineering,87(6),p.732−738(1999)など参照)や、プロトンスポンジ存在下でのオキシ塩化リンなどによるリン酸化(例えば、Tetrahedron Letters,29(36),p.4525−4528(1988)など参照)などによって、それらの三リン酸体を得ることができる。
【0043】
本実施形態に係るヌクレオシド三リン酸誘導体は、例えば、下記式(1)で示され、TGaseが認識可能なGln残基を有するウリジン三リン酸誘導体である。
【化6】
(式(1)中、Aは、グルタミン(Gln)残基を有する置換基、Bは水素原子またはヒドロキシル基を表す。)
【0044】
本実施形態に係るヌクレオシド三リン酸誘導体は、例えば、下記式(2)で示され、TGaseが認識可能なGln残基を有するアデノシン三リン酸誘導体である。
【化7】
(式(2)中、A1およびA2のうち少なくとも1つは、グルタミン(Gln)残基を有する置換基で残りは水素原子を表し、Bは水素原子またはヒドロキシル基を表す。)
【0045】
本実施形態に係るヌクレオシド三リン酸誘導体は、例えば、下記式(3)で示され、TGaseが認識可能なGln残基を有するシチジン三リン酸誘導体である。
【化8】
(式(3)中、Aは、グルタミン(Gln)残基を有する置換基、Bは水素原子またはヒドロキシル基を表す。)
【0046】
本実施形態に係るヌクレオシド三リン酸誘導体は、例えば、下記式(4)で示され、TGaseが認識可能なGln残基を有するグアノシン三リン酸誘導体である。
【化9】
(式(4)中、Aは、グルタミン(Gln)残基を有する置換基、Bは水素原子またはヒドロキシル基を表す。)
【0047】
Aで表されるグルタミン(Gln)残基を有する置換基としては、特に制限はないが、例えば、グルタミン(Gln)残基を有する直鎖、分岐、環状の飽和または不飽和のアルキル基、アミノアルキル基、アリール基、ヘテロアリール基を含む置換基が挙げられ、合成のし易さなどを考慮して決めればよい。
【0048】
本実施形態に係るヌクレオシド三リン酸誘導体は、例えば、下記式(5)で示され、TGaseが認識可能なGln残基を有するTGase基質修飾ヌクレオチド誘導体である。
【化10】
(式(5)中、XおよびYは、それぞれ独立して2価の連結基を表し、Zは、置換基を表す。)
【0049】
XおよびYで表される2価の連結基としては、それぞれ独立して、メチレン基、エチレン基、プロピレン基、ブチレン基などの炭素数1〜48のアルキレン基、エテニレン基、プロペニレン基、ブテニレン基などの炭素数2〜48のアルケニレン基などが挙げられる。これらのうち、X,Yは、それぞれ独立して炭素数1〜48のアルキレン基または炭素数2〜48のアルケニレン基、炭素数1〜48のアルコキシ基であることが好ましく、Xはエテニレン基、Yはメチレン基であることがより好ましい。X,Yはさらにエテニレン基、オキシアルキレン基、例えば−(C2H4O)n−または−(C3H6O)n−(nは繰り返し数でありn=2,4,8,12,24)基などで置換されていてもよい。
【0050】
Zで表される置換基としては、メチル基、エチル基、プロピル基などの炭素数1〜48のアルキル基、メトキシ基、エトキシ基、プロピオキシ基などの炭素数1〜48のアルコキシ基、フェニル基、ナフチル基などの炭素数6〜48のアリール基、フェニルオキシ基などの炭素数6〜48のアリールオキシ基、ベンジル基などの炭素数7〜48のアリールアルキル基、ベンジルオキシ基などの炭素数7〜48のアリールアルキルオキシ基などが挙げられる。これらのうち、Zは、炭素数1〜48のアルキル基、炭素数1〜48のアルコキシ基、炭素数6〜48のアリール基、炭素数6〜48のアリールオキシ基、炭素数7〜48のアリールアルキル基または炭素数7〜48のアリールアルキルオキシ基であることが好ましく、Zはベンジルオキシ基であることがより好ましい。Zはさらにジニトロフェニル基、L−3,4−ジヒドロキシフェニル基などで置換されていてもよい。また、上述したYで表される置換との組み合わせで、Y,Zは独立してLys以外のアミノ酸により少なくとも一方が置換されていてもよい。
【0051】
Xを適宜選択することにより、Z−QGとUTPとを連結するリンカー部位の構造を最適化し、例えば柔軟なリンカー部位を導入することで、酵素などのアクセスを向上することができる。また、Y,Zを適宜選択することにより、基質ペプチド配列を最適化し、例えば酵素などの親和性を向上することができる。
【0052】
微生物由来TGase(MTG)を用いる場合、MTGが認識可能なGln残基は、ベンジルオキシカルボニル−L−グルタミルグリシン(Z−QG)として存在することが好ましい。Z−QGは、ジゴキシゲニン(DIG)などよりも分子サイズが小さいため好ましい。式(5)で示されるヌクレオシド三リン酸誘導体において、Xがエテニレン基、Yがメチレン基、Zがベンジルオキシ基であるヌクレオチド誘導体が、UTPにZ−QGを結合させたヌクレオチド誘導体Z−QG−UTPである。また、ヌクレオシド三リン酸誘導体中には、TGaseが認識可能なLys残基または第一級アミンが共存しないようなものを選択することが好ましい。共存する場合には、TGaseにより、自己架橋する可能性があり、目的のマルチラベル化核酸プローブの収率に好ましくない影響を与える場合があるからである。
【0053】
また、微生物由来TGaseの良基質として、LLQG(配列番号:1)、LAQG(配列番号:2)、LGQG(配列番号:3)、PLAQSH(配列番号:4)、FERQHMDS(配列番号:5)、もしくはTEQKLISEEDL(配列番号:6)のアミノ酸配列からなるペプチド、またはGLGQGGG(配列番号:7)、GFGQGGG(配列番号:8)、GVGQGGG(配列番号:9)、もしくはGGLQGGG(配列番号:10)のアミノ酸配列からなるペプチドが知られている。また、guinea pig liver由来のTGaseの良基質として、ベンジルオキシカルボニル−L−グルタミルフェニルアラニン(Z−QF)、またはEAQQIVM(配列番号:11)のアミノ酸配列からなるペプチド、またはGGGQLGG(配列番号:12)、GGGQVGG(配列番号:13)、GGGQRGG(配列番号:14)、GQQQLG(配列番号:15)、PNPQLPF(配列番号:16)もしくはPKPQQFM(配列番号:17)のアミノ酸配列からなるペプチドが知られている。TGaseが認識可能なGln残基は、用いるTGaseの種類に応じ、このようなペプチドとして存在してもよい。
【0054】
なお、N末端がグリシン(G)である基質ペプチドは、N末端アミノ基がTGaseの基質になりうるため、自己架橋による副産物が生じうる。したがって、N末端がグリシン(G)である基質ペプチドについては、N末端アミノ基の水素を適切な基で置換することによりTGaseの基質とはならないように保護して、所望の連結を行うことができるようにするとよい。なお、本明細書において「N末端保護」というときは、特別な場合を除き、このような意味で用いている。そして、N末端保護の手段により、反応性が異なることが知られている。詳細には、ほ乳類由来TGaseに関して、GQQQLGのN末端アセチル化による保護(すなわち、Ac−GQQQLG)、またN末端アミノ酸をDOPA(L−3,4−dihydroxyphenylalanine)にする(すなわち、DOPA−GQQQLG)と反応性が向上することが知られている。このような保護の例を、本実施形態においても利用することができる。
【0055】
Z−QG−UTPの調製方法を図3に示す。これは、本実施形態に係るヌクレオシド三リン酸誘導体の調製方法の一例であって、これに限定されるものではない。
【0056】
まず、ベンジルオキシカルボニル−L−グルタミルグリシン(Z−QG)にN−ヒドロキシスクシンイミド(N−hydroxysuccinimide:NHS)基などを導入し、活性化しておく(NHS化Z−QG)。そして、アミノアリルUTPなどの末端をアミノ化した置換基を有するUTPと、NHS化Z−QGとを縮合することにより、Z−QG−UTPを得ることができる。
【0057】
また、C末端のカルボキシル基を活性エステル化する上述の方法に加えて、TGaseが認識可能なGln残基を有するペプチドをUTPに導入する方法として、アミノ基と反応性の高い官能基をペプチドに導入する方法がある。例えば、アルデヒド化、アシルアジド化、スルフォニルクロライド化、エポキシ化、イソシアネート化、またはイソチオシアネート化した基質ペプチドを調製できれば、これをアミノ化UTPと反応させることにより、TGaseが認識可能なGln残基を有するUTPを調製することができる。ただし、これらの反応性官能基は、基質ペプチドにおいてTGase認識に影響がない部分に導入する必要がある。したがって、上述のように、Gln残基とは離れたC末端のカルボキシル基を活性化する方法は、この目的において最も優れたものの一つである。
【0058】
Z−QG−UTPの精製は、高速液体クロマトグラフィ(HPLC)、ゲル濾過クロマトグラフィ(GPC)などにより行うことができる。また、Z−QG−UTPの同定は、MALDI TOF−MS、NMR、IRなどにより行うことができる。また、HPLCにより、生成物の確認および収率を求めることができる。
【0059】
<TGase基質マルチラベル化核酸>
本実施形態に係るTGase基質マルチラベル化核酸は、構成単位として、例えば式(1)〜(5)で示されるTGase基質ラベル化ヌクレオシド三リン酸誘導体を調製したのち、このTGase基質ラベル化ヌクレオシド三リン酸誘導体が複数個導入されているTGase基質マルチラベル化核酸であり、核酸部分が、検出対象である標的核酸の標的分子特異的配列の全部または一部に相補的な配列を有するものである。
【0060】
核酸には、DNA、PNAおよびRNAが含まれる。核酸の配列および長さには特に制限はない。長さに関しては、例えば少なくとも20mer程度であればよい。
【0061】
例えば、Z−QG−UTPを基質として、RNAポリメラーゼ活性のある酵素を用いて複数のZ−QG−UTPを取り込ませて、TGase認識Glnが複数箇所に導入され、所望の配列を有するTGase基質マルチラベル化核酸プローブZ−QG RNA(図2)を調製することができる。
【0062】
<マルチラベル化核酸プローブ>
本実施形態に係るマルチラベル化核酸プローブは、上記核酸プローブにおける構成単位であるヌクレオシド三リン酸誘導体のグルタミン(Gln)残基のうち少なくとも2つに、リシン(Lys)残基または第一級アミンを有し標識部分を含む標識化合物が結合されて構成されたものである。マルチラベル化核酸プローブは、例えば、MTGなどのTGaseを用いて、MTG認識GlnなどのTGase認識Glnが複数箇所に導入された核酸プローブZ−QG RNAに、TGase認識Lysを導入した標識酵素などを結合させることによって、RNAと標識酵素などの標識部分が1:nであるマルチラベル化核酸プローブを創製することができる(図2)。
【0063】
RNAと標識酵素などの標識部分との比率nは、2以上であれば特に制限はなく、適宜調整することができるが、nが大きいほど検出感度が高くなり好ましい。ただし、nが大きすぎると、標的核酸とのハイブリダイゼーションの効率が低下する場合がある。
【0064】
また、例えば、以下の方法により、異なる標識酵素、異なる蛍光色素などの異なる標識部分を有する複数のマルチラベル化核酸プローブを調製することができる。
(1)TGaseの由来を変える。
(2)TGaseの基質特異性を変える。
【0065】
(1)の方法では、例えば、用いるTGaseの種類に応じて、異なる基質ペプチドで修飾されたUTPなどを調製すればよい。
【0066】
(2)の方法では、例えば、TGaseに、タンパク質工学的にアミノ酸変異を導入して基質特異性を変えればよい。例えば、MTGを大腸菌で調製し(例えば、Christian K. Marx,Thomas C. Hertel and Markus Pietzsch,Enzyme and Microbial Technology,Volume 40,Issue 6,2 May 2007,p.1543−1550,”Soluble expression of a pro−transglutaminase from Streptomyces mobaraensis in Escherichia coli”参照)、さらに変異体ライブラリーを作って耐熱性の向上したMTGを取得することができる(例えば、Christian K. Marx,Thomas C. Hertel and Markus Pietzsch,Journal of Biotechnology,Volume 136,Issues 3−4,10 September 2008,p.156−162,”Random mutagenesis of a recombinant microbial transglutaminase for the generation of thermostable and heat−sensitive variants”参照)。
【0067】
本明細書において、「TGaseにより結合する」というときは、特別な場合を除き、得られる連結部は、Lys残基とGln残基とが、ε(γ−グルタミル)リシン結合を形成することにより構成されている。
【0068】
本実施形態においては、TGaseが認識可能なLys残基は、第一級アミンであってもよい。本明細書では、Lys残基を例に説明するが、その説明は、特別な場合を除き、第一級アミンにも当てはまる。
【0069】
リシン(Lys)残基または第一級アミンを有し標識部分を含む標識化合物としては、特に制限はない。
【0070】
標識部分としては、酵素、蛍光色素、放射性同位体を含む化合物、磁気的に検出可能な標識(例えば、磁性ナノ粒子)、熱的に検出可能な標識(例えば、温度応答性高分子)、電気的に検出可能な標識(例えば、フェロセン部位を有する高分子)などが挙げられ、検出感度、取り扱い性などの点から、酵素および蛍光色素のうち少なくとも1つが好ましい。
【0071】
蛍光色素としては、選択された波長の紫外光、可視光などの放射線による照射に応答して蛍光または燐光を発する物質であればよく、特に制限はないが、例えば、蛍光色素としてフルオレセイン、ローダミン、ダンシル、カルボシアニン誘導体など、あるいは蛍光タンパク質として緑色蛍光タンパク質とその変異体などが挙げられる。
【0072】
放射性同位体としては、例えば、重水素(2H)、三重水素(3H)、10B、11B、13C、15N、18Oなどが挙げられる。
【0073】
TGaseに対し、Lys残基(または第一級アミン)供与体となる基質は、Gln残基供与体となる基質に比較して構造的な制約が少ないと考えられる。したがって、修飾しようとする標識酵素が、TGaseが認識可能なLys残基を元来有している場合もあり、TGaseが認識可能なLys残基を含むタグを酵素に付加する場合もある。
【0074】
TGaseが認識可能なLys残基(K)は、MKHKGS(配列番号:18)、MRHKGS(配列番号:35)、MRRKGS(配列番号:36)、MHRKGS(配列番号:37)のアミノ酸配列を有するペプチドとして存在してもよい。このようなTGaseが認識可能なLys残基を含むペプチドによるタグ化は、標識酵素を、タンパク質の所望の部位、例えばC末端またはN末端に連結する目的で用いることができる。TGaseが認識可能なLys残基を含む他のペプチドまたはそのアミノ酸配列の例としては、改変型S−peptide(GSGMKETAAARFERAHMDSGS(配列番号:19))、MGGSTKHKIPGGS(配列番号:20)、N末端グリシン(N−terminal GGG、N−terminal GGGGG(配列番号:21))、N末端MKHKGSと対象タンパク質間のリンカー部位を伸ばしたMKHKGGGSGGGSGS(配列番号:22)などが挙げられる。
【0075】
C末端またはN末端にTGaseが認識可能なLys残基を含むペプチドを付加した標識酵素は、遺伝子工学的な手法を用いて、組換えタンパク質として調製することができる。C末端またはN末端にTGaseの基質ペプチドタグが導入された当該組換えタンパク質の精製は、それぞれN末端またはC末端に付加した精製用ペプチドタグ(例えば、(His)6−tag(ヘキサヒスチジンタグ))を利用し(TGaseの反応性の低下を回避するために、基質ペプチドタグを入れた末端とは異なる末端に精製用ペプチドタグを入れるようにデザインするとよい。)、ゲル濾過クロマトグラフィなどにより行うことができ、またアミノ酸配列の確認は当該タンパク質をコードするプラスミドベクターの遺伝子配列をDNAシーケンサにて確認するか、N末端に導入された基質ペプチドについてはN末端分析により直接同定することができる。タンパク質の精製の確認は、SDS−PAGEなどで行うことができる。
【0076】
標識酵素としては、発色反応などを利用して検出を行うことができる性質を有するものであればよく特に制限はない。例えば、アルカリホスファターゼ(AP)、グルタチオン−S−トランスフェラーゼ(GST)、ルシフェラーゼ、ペルオキシダーゼなどが挙げられる。これらのうち、高い触媒活性と安定性の観点から、アルカリホスファターゼあるいはペルオキシダーゼが好ましい。ペプチドタグが容易に導入可能との観点からは、遺伝子工学的に製造可能なタンパク質が好ましい。
【0077】
酵素マルチラベル化核酸プローブと標的核酸の間でより厳密に塩基配列特異的な二本鎖形成を行うためには、比較的高温(例えば、70℃以上)の条件下で行うことがあるため、常温菌由来の酵素を利用すると活性の損失が懸念される。そこで、標的酵素としては、超好熱菌Pyrococcus furiosus由来アルカリホスファターゼ(PfuAP)が好ましい。
【0078】
超好熱菌由来の酵素は、一般的に有機溶媒や熱に対して高い安定性を示すことが知られているため好ましく(例えば、H.Atomi,Current Opinion in Chemical Biology,9,p.166−173(2005)参照)、さらに、大腸菌を宿主とした大量調製が比較的容易に行うことができる点においても好ましい。大腸菌を宿主として耐熱性酵素を調製する場合、細胞破砕液を高温処理(例えば、80℃で30分温置)することで、大腸菌由来の共雑タンパク質のほとんどを沈殿させ、粗精製を容易に行うことができる。
【0079】
超好熱菌は一般の生物がほとんど生育できない極限環境で生育することができる微生物であるため、超好熱菌由来のタンパク質は非常に高い耐熱性を有している。さらに、熱に対する耐性だけでなく、一般的に、変性剤、有機溶媒、pHなどに対する耐性も常温菌由来酵素に比べて極めて高いことから、PfuAPを使用することによって、酵素の失活を伴わない厳密な二本鎖形成を達成することができると考えられる。
【0080】
本実施形態に係る酵素マルチラベル化核酸プローブの好ましい態様の一つは、PfuAPとZ−QG RNAとの複合体である。このような複合体は、安定な酵素であるPfuAPと、安定な分子であるRNAが、アミド結合という安定な共有結合で連結されているため、複合体全体としても安定性であるというメリットがある。
【0081】
また、酵素マルチラベル化核酸プローブにおいて、酵素が有機溶媒や熱に対して安定な酵素であってもよい。このような安定性の高い酵素は、自然界からのスクリーニング(例えば、化学と工業,vol.61(No.6),p.571−575(2008)、内山拓,宮崎健太郎,バイオサイエンスとインダストリー,vol.66(No.5),p.234−239(2008)、道久則之,バイオサイエンスとインダストリー,vol.66(No.12),p.667−670(2008))や、タンパク質工学的手法により安定性を高める技術(例えば、荻野博康,BIO INDUSTRY,vol.25(No.7),p.16−23(2008)、宮崎健太郎,BIO INDUSTRY,vol.25(No.7),p.52−58(2008))により得ることができる。これらの手法により、常温菌由来の酵素であっても、有機溶媒耐性や耐熱性を有する酵素へと変換することができる。
【0082】
蛍光色素部分を導入した、リシン(Lys)残基または第一級アミンを有する標識化合物は、例えば、カルボキシル基にジアミンを導入する方法で調製することができる(例えば、G.T.Hermanson(1996),Bioconjugate Techniques,Chapter 1,Section 4.3,p.100−104,Academic Press,San Diego.を参照)。
【0083】
トランスグルタミナーゼ(TGase)としては、種々のものを用いることができる。現在、TGaseとして、哺乳類(guinea pig、ヒト)、無脊椎動物(昆虫、カブトガニ、ウニ)、植物、菌類、原生生物(粘菌)由来のものが知られており、またヒトの場合については、8種類のアイソザイムが見つかっている。本実施形態において用いることのできるTGaseの好ましい例は、安定性、ハンドリングの容易さ、バルク生産が可能などの点から微生物由来微生物由来トランスグルタミナーゼ(MTG)である。
【0084】
本実施形態においてMTGを用いた場合、予想されているMTGの触媒反応から、Lys残基を有する標識酵素などの標識部分を含む標識化合物とZ−QG RNAとの連結反応は、MTG活性中心であるシステイン(Cys)残基の、Z−QG RNAのGlnへの求核置換反応によるアシル−酵素複合体の形成と、続いて起こる標識化合物のLysによるアシル−酵素複合体への求核置換反応によるMTGの脱離、の2段階で進行すると予想される。
【0085】
本実施形態の好ましい態様においては、TGaseが認識可能なGln残基を有するZ−QG RNAに対する、TGaseが認識可能なLys残基または第一級アミンを有する標識化合物のモル濃度比は、好ましくは2以上であり、より好ましくは5以上である。なお、本明細書で単に「濃度比」というときは、特別な場合を除き、モル濃度による比を指す。例えば、Z−QG RNAに対するNK14−PfuAPのモル濃度比は、[NK14−PfuAP]/[Z−QG RNA]と表すこともできる。
【0086】
[NK14−PfuAPの調製]
NK14−PfuAPとは、MKHKGGGSGGGSGSの配列を有するアミノ酸14残基からなる付加配列を遺伝子工学的にPfuAPのN末端に、精製用タグをC末端に導入したものである。PfuAPの発現ベクターは香川大学櫻庭春彦教授より委譲頂いた。PCRによりPfuAPをコードする領域を増幅する際、それぞれのタグが導入されるようにタンパク質発現用ベクターpET22に組換え、大腸菌BL21株を形質転換した。アンピシリンを含むLB培地にて前培養、本培養を経て、得られた形質転換体を遠心分離により収菌し、25mM TBSで2回洗浄した。得られた菌体を凍結・融解させた後、超音波処理により細胞を粉砕し、遠心分離により可溶性画分を回収した。超高熱菌由来のPfuAPは高温条件下でも安定であるので、得られた無細胞抽出液を80℃で30分処理し、他のタンパク質を沈殿させることによって粗精製を行った。粗精製後、遠心分離・フィルター濾過によって上澄みを回収し、His−tagカラムによって精製した。精製後、限外濾過による濃縮を行い、PD−10カラムを用いて溶媒を10mM Tris−HCl(pH8.0)へと置換し、実験に供するまで凍結保存した。
【0087】
また、NK14−PfuAPの大腸菌での発現量の向上のため、大腸菌のコドン使用頻度に合わせて塩基配列が改変されたNK14−PfuAPの発現ベクター(アクセッション番号:AB479383、配列番号:26)を用いてもよい。この発現ベクターは、Codon Devices社(http://www.codondevices.com)に受託合成して得た。NK14−PfuAPをコードする遺伝子領域の両端に適切な制限酵素サイトを導入しておき、これらを利用することでタンパク質発現用ベクターpET22に組換え、得られたNK14−PfuAP発現ベクターにより大腸菌BL21株を形質転換した。アンピシリンを含むLB培地にて前培養、本培養を経て、得られた形質転換体を遠心分離により収菌し、25mM TBSで2回洗浄した。得られた菌体を凍結・融解させた後、超音波処理により細胞を粉砕し、遠心分離により可溶性画分を回収した。超高熱菌由来のPfuAPは高温条件下でも安定であるので、得られた無細胞抽出液を80℃で30分処理し、他のタンパク質を沈殿させることによって粗精製を行った。粗精製後、遠心分離・フィルター濾過によって上澄みを回収し、His−tagカラムによって精製した。精製後、限外濾過による濃縮を行い、PD−10カラムを用いて溶媒を10mM Tris−HCl(pH8.0)へと置換し、実験に供するまで凍結保存した。
【0088】
TGaseとしてMTGを用いて連結反応を行う場合には、上述のようにモル濃度比が適切な範囲となるようにすることに加えて、pH5.5〜8.0、温度4〜50℃(例えば、室温(例えば、18℃〜22℃))の条件で行うことが好ましい。このようにすれば、12時間以内、好ましくは6時間以内、より好ましくは3時間以内に、充分に高い反応率が達成可能である。
【0089】
このような高い反応率が達成できる方法により得られたマルチラベル化核酸プローブ溶液には、未反応の核酸プローブ(例えば、遊離のZ−QG RNA)がほとんど存在せず、以下で詳述する、標的核酸の検出にそのまま用いたとしても、マルチラベル化核酸プローブと未反応分子との競合が実質的に生じないか、生じたとしても目的とする検出の結果には実質的な影響を与えないほど少ないと考えられる。したがって、マルチラベル化核酸プローブ溶液を精製することなく、直接検出へと利用できるメリットがある。
【0090】
本実施形態以前に、TGaseが認識可能なLys残基または第一級アミンを有する標識酵素などの標識化合物と、TGaseが認識可能なGln残基を有するZ−QG RNAとをTGaseを用いて連結することにより、マルチラベル化核酸プローブを得る例は存在しなかった。したがって、本実施形態の方法により連結されたマルチラベル化核酸プローブは、物質として新規なものということができる。
【0091】
本実施形態における酵素マルチラベル化核酸プローブの形成方法は、従来法に比較して、以下の特徴およびメリットを有する。
・対象となる標識酵素は、TGaseが認識可能なLys残基または第一級アミンを有する酵素、TGaseが認識可能なLys残基または第一級アミンを導入することができるあらゆる酵素を包含する。また、インテインのような大きなタンパク質性タグを必要としない。
・酵素の修飾部位は、C末端に限定されない。TGase活性なLys残基が存在するか、そのようなLys残基を有するタグを導入することができる部位であれば、修飾可能である。
・C末端、N末端に加え、loop領域のようなタンパク質構造中の揺らぎの大きな部位も修飾対象となりうる。
・結合する核酸の塩基配列および長さには、特に制限がない。
【0092】
<標的核酸の検出方法>
本実施形態に係る標的核酸の検出方法は、構成単位として例えば上記式(1)〜(5)で示されるヌクレオシド三リン酸誘導体が複数個導入されている核酸である核酸プローブにおけるグルタミン(Gln)残基のうち少なくとも2つに、リシン(Lys)残基または第一級アミンを有する標識酵素などの標識化合物が結合されて構成されているマルチラベル化核酸プローブであって、標識部分が容易に検出可能な性質を有し、かつ核酸部分が標的核酸に特異的に結合可能な塩基配列を有するものであるマルチラベル化核酸プローブを準備し、マルチラベル化核酸プローブと対象物中に存在する標的核酸とを核酸部分により特異的に結合させ、結合しているマルチラベル化核酸プローブを、酵素部分などの標識部分により検出する工程を含む。
【0093】
本実施形態に係る標的核酸の検出方法は、標的核酸の定性、定量、識別、染色、局在化の調査などの目的で用いることができる。
【0094】
この方法においては、上記マルチラベル化核酸プローブが用いられるが、核酸部分は、標的核酸に特異的に結合可能な核酸配列を有するものとする。酵素部分などの標識部分は、容易に検出可能な性質を有するものである。この方法において、標的核酸を含む標的分子は、核酸、比較的低分子の有機化合物(ATP)、タンパク質、ペプチド、金属イオン、複雑な構造を持つ多量体、ウイルスなどでありうる。検出対象は、(i)DNA転写膜、または(ii)細胞もしくは個体組織切片などである。
【0095】
検出対象が(i)の場合、標的核酸は、PCRにより増幅されたDNAまたはゲノム断片DNAなどであり、(ii)の場合、標的核酸は、細胞または個体組織中に含まれる核酸(mRNAまたはDNA)などである。本方法は、従来法、例えばジゴキシゲニン(DIG)標識化プローブを用いる方法に比較して、種々の点で優れている。例えば、DIG法では、核酸プローブのDIG修飾および標識化された抗DIG抗体が必要であり、それに伴う煩雑な洗浄操作が必要となるが、本実施形態に係るマルチラベル化核酸プローブを用いれば、DIG標識化プローブおよび標識化抗DIG抗体が不要になるため、試薬、手間および時間を大幅に削減することが可能となる。また、1つの認識部位(核酸)に対して複数のシグナル増幅部位(酵素など)を配置しているため、検出感度の向上が可能になる。
【0096】
この方法においては、標的核酸に、マルチラベル化核酸プローブを供し、標的核酸とマルチラベル化核酸プローブの標的核酸相補的配列を有する部分とをハイブリダイズさせるが、このハイブリダイズのための条件は、当業者であれば、用いる核酸部分の長さ、塩基配列などに応じて、適宜設計することができる。
【0097】
本実施形態に係る標的核酸の検出方法において、異なる標識酵素、異なる蛍光色素などの異なる標識部分を有する複数のマルチラベル化核酸プローブを準備して、同時に複数の標的核酸を検出してもよい。
【0098】
また、本実施形態において、標的核酸の検出方法は、
(a)基材表面に固定化された核酸、
(b)固定化された固定化核酸の全部または一部に相補的な配列(固定化核酸相補的配列)、および標的核酸に特異的に結合可能な配列(標的核酸特異的配列)を有するアプタマ核酸、
(c)構成単位として上記式(1)で示されるヌクレオチド誘導体が複数個導入されている核酸である核酸プローブにおけるグルタミン(Gln)残基のうち少なくとも2つに、リシン(Lys)残基または第一級アミンを有する標識酵素などの標識化合物が結合されて構成されているマルチラベル化核酸プローブであって、標識部分が容易に検出可能な性質を有し、及び/又は核酸部分がアプタマ核酸の標的核酸特異的配列の全部または一部に相補的な配列を有するものであるマルチラベル化核酸プローブ、
を準備し、
(A)固定化核酸に、アプタマ核酸を供し、固定化核酸とアプタマ核酸の固定化核酸相補的配列を有する部分とをハイブリダイズさせることにより、アプタマ核酸を固定化し、
(B)固定化された固定化アプタマ核酸に、標的核酸を含む標的分子を含む可能性のある試料を供し、標的分子が存在する場合には標的核酸とアプタマ核酸の標的核酸分子特異的配列を有する部分とを結合させ、かつ上記マルチラベル化核酸プローブを供し、標的分子が存在しない場合にはマルチラベル化核酸プローブの核酸部分とアプタマ核酸とをハイブリダイズさせることにより、タンパク質を固定化し、
(C)固定化された酵素部分の有無またはその量を酵素の性質に基づいて検出することにより、試料中の標的分子を検出する工程を含む。
【0099】
この方法における基材表面への核酸の固定化のためには、従来技術、例えばDNAマイクロアレイなどを調製する際に用いられる技術を適用することができる。「基材」は、ガラス、シリコンなどのプラスチック製の、チップ、ビーズ、ウェル、プレートなどの形態とすることができ、核酸は、従来技術を用いて、基材表面に非共有結合(静電結合)的に、または共有結合で固定することができる。あらかじめ調製した核酸を基材に固定化してもよく、基材上で直接核酸を合成してもよい。簡便には、アビジンなどで被覆された市販のプレートを用い、ビオチン化した所望のDNAを固定することができる。
【0100】
この方法ではさらに、固定化核酸の全部または一部に相補的な配列(固定化核酸相補的配列)、および標的分子を含まれる標的核酸に特異性的に結合可能な配列(標的核酸特異的配列)を有する核酸(アプタマ核酸)が用いられる。標的分子は、核酸、比較的低分子の有機化合物、タンパク質、ペプチド、金属イオン、複雑な構造を持つ多量体、ウイルスなどでありうる。標的核酸特異的配列を有する部分は、従来技術、例えばSELEX(試験管内人工進化法)工程を用いることにより産生することができ、また標的分子に対して非常に高い標的親和性及び特異性を有するものとすることができる。標的核酸特異的配列を有する部分は、修飾ヌクレオチドで構成されていてもよい。
【0101】
この方法においては、上記マルチラベル化核酸プローブが用いられるが、核酸部分は、アプタマ核酸の標的核酸特異的配列の全部または一部に相補的な配列を有するものとする。酵素部分などの標識部分は、容易に検出可能な性質を有するものである。この方法において、標的核酸を含む標的分子は、核酸、比較的低分子の有機化合物(ATP)、タンパク質、ペプチド、金属イオン、複雑な構造を持つ多量体、ウイルスなどでありうる。検出対象は、(i)DNA転写膜、または(ii)細胞もしくは個体組織切片などである。
【0102】
この方法においては、固定化核酸に、アプタマ核酸を供し、固定化核酸とアプタマ核酸の固定化核酸相補的配列を有する部分とをハイブリダイズさせることにより、アプタマ核酸を固定化するが、このハイブリダイズのための条件は、当業者であれば、用いる核酸部分の長さ、塩基配列などに応じて、適宜設計することができる。
【0103】
また、この方法においては、次いで固定化アプタマ核酸に、標的分子を含む可能性のある試料を供し、標的分子が存在する場合には標的分子とアプタマ核酸の標的核酸特異的配列を有する部分とを結合させ、さらにマルチラベル化核酸プローブを供し、標的分子が存在しない場合にはマルチラベル化核酸プローブの核酸部分とアプタマ核酸とをハイブリダイズさせる。試料は、細胞もしくは組織抽出液、体液などでありうる。
【0104】
さらにこの方法においては、固定化された酵素部分の有無またはその量を酵素の性質に基づいて検出することにより、試料中の標的分子を検出することができる。
【0105】
アプタマ核酸においては、固定化核酸相補的配列を有する部分と、標的核酸特異的配列を有する部分とは、重複してもよく、連続してもよく、また適当なスペーサを介して両者が存在するように設計することができる。標的核酸特異的配列を有する部分(アプタマ領域)が分子認識のためにある立体構造をとることを考慮すると、該部分は、固定化核酸相補的配列を有する部分とは少なくとも重複しないようにするのがよい。
【0106】
アプタマ核酸は、固定化核酸相補的配列および標的核酸特異的配列以外に、所望の場合にはマルチラベル化核酸プローブと適切にハイブリダイズするための配列をさらに含んでいてもよい。マルチラベル化核酸プローブの核酸部分は、アプタマ核酸の標的分子特異的配列の全部または一部に相補的な配列を有するが、この特異的配列の全部または一部に相補的な配列からなる領域が長い(標的核酸特異的配列と重複が多い)と、かえってアプタマ核酸とはハイブリダイズできない場合が生じうる。また短い(重複が少ない)と、標的分子が存在し、アプタマ核酸と結合している場合にも、マルチラベル化核酸プローブとアプタマ核酸とがハイブリダイズしてしまう場合が生じうる。当業者であれば、このようなことを考慮して、固定化核酸、アプタマ核酸、マルチラベル化核酸プローブの核酸部分を、適宜設計することができる。
【0107】
また、本実施形態に係る標的核酸の検出方法は、核酸部分が標的核酸に特異的に結合可能な塩基配列を有するものである、構成単位として、例えば上記式(1)〜(5)で示されるヌクレオシド三リン酸誘導体が複数個導入されている核酸である核酸プローブと、対象物中に存在する標的核酸とを核酸部分により特異的に結合させた後、トランスグルタミナーゼ(TGase)を用いて、核酸プローブにおけるグルタミン(Gln)残基と、リシン(Lys)残基または第一級アミンを有し標識部分を含む標識化合物のリシン(Lys)残基または第一級アミンとを反応させて、容易に検出可能な性質を有する複数の標識部分を導入し、結合している核酸プローブを、標識部分により検出する工程を含む(図16参照)。
【0108】
この方法においては、上記核酸プローブが用いられるが、核酸部分は、標的核酸に特異的に結合可能な核酸配列を有するものとする。また、標識部分は、容易に検出可能な性質を有するものである。
【0109】
本方法は、従来法、例えばジゴキシゲニン(DIG)標識化プローブを用いる方法に比較して、種々の点で優れている。例えば、DIG法では、核酸プローブのDIG修飾および標識化された抗DIG抗体が必要であり、それに伴う煩雑な洗浄操作が必要となるが、本実施形態に係る核酸プローブを用いれば、DIG標識化プローブおよび標識化抗DIG抗体が不要になるため、試薬、手間および時間を大幅に削減することが可能となる。また、1つの認識部位(核酸)に対して複数のシグナル増幅部位(標識部分)を配置することにより、検出感度の向上が可能になる。
【0110】
例えば、MTGなどのTGaseを用いて、MTG認識GlnなどのTGase認識Glnが複数箇所に導入された核酸プローブZ−QG RNAに、TGase認識Lysを導入した酵素、蛍光色素などの標識化合物を結合させる。
【実施例】
【0111】
以下、実施例および比較例を挙げ、本発明をより具体的に詳細に説明するが、本発明は、以下の実施例に限定されるものではない。
【0112】
本実施例では、In situ ハイブリダイゼーション(ISH)法への応用を目指して、超好熱菌由来酵素マルチラベル化RNAの開発を行った。酵素ラベル化の手法としては、微生物由来トランスグルタミナーゼ(MTG)のタンパク質修飾能を利用した。MTGの良基質であるZ−QGを導入したZ−QG−UTPを合成し、RNA polymeraseによる転写反応によってZ−QG RNAを調製した。さらに、MTGを用いて超好熱菌由来酵素マルチラベル化核酸プローブを調製し、その性能をドットブロットによって評価した。
【0113】
(実施例1)
<Z−QG−UTPの合成・精製>
Z−QG−UTPの合成スキームは、図3に示す通りである。
【0114】
まず100mM N,N’−ジイソプロピルカルボジイミド(DIC)、100mM N-ヒドロキシスクシンイミド(NHS)、50mM Z−QGを、室温(調製した日は27.0℃)でN,N−ジメチルホルムアミド(DMF)4mL中で20時間反応させることにより、NHS化Z−QG(50mM)を調製した(この段階で500μLずつに分注し、−80℃で保存)。その後、25mMのNHS化Z−QGと5mMの5−(3−aminoallyl)−UTP(以下、aminoallyl−UTPと略記、SIGMA社製)とを、100mM ホウ酸緩衝液(pH8.8)とDMFの混合溶媒(v/v=1/1)0.32mL中において25℃で12時間反応させた。反応終了後、サンプルをMilli−Qで10倍希釈し、HPLC(日本分光製)(高速液体クロマトグラフ・ポンプ:TRI ROTAR−V型、バリアブル・ループ・インジェクタ:VL−613型、紫外可視分光検出器:UVIDEC−100−IV型)によって精製を行った。HPLCの測定条件は表1の通りとした。生成物の同定は、レーザーイオン化飛行時間型質量分析装置であるMALDI TOF−MS(島津製作所製 AXIMA(登録商標)−CFR Plus)によって行った。なお、サンプル調製手順は、まず、試料1μLをMALDI用試料プレートの上に滴下し、次にその上から、マトリックス溶液;2,5−ジヒドロキ安息香酸(DHB)の10mg/ml溶液(超純水)を滴下し、その後、風乾して、得られた試料プレートをMALDI TOF−MS装置のイオン源内に導入して計測した。
【0115】
Z−QG−UTPを合成した後、表1に示す条件で逆相HPLCを行った際の結果を図4に示す。aminoallyl−UTPの場合(図4(A))と比較して、疎水側に新たなピーク(22min)が出現しており(図4(B))、このピークがZ−QG−UTPであると推測される。しかし、生成物ピークの分離が十分でなかったので、HPLC条件を表2に示すように変えて検討を行った。結果を図5に示す。
【0116】
【表1】
【0117】
【表2】
【0118】
そこで、保持時間18.8分のピークを回収してMALDI TOF−MS分析を行った(図6参照)。その結果、856.89のピークが確認され、理論分子量の857.13と良く一致した結果が得られたため、Z−QG−UTPの合成が示された。
【0119】
[実験1:Alkphos Directキットの検出感度の評価]
<センス鎖RNA、アンチセンス鎖RNAの調製>
RNA polymeraseにより転写を行うことによって、センス鎖RNA、アンチセンス鎖RNAを調製した。標的核酸(ターゲット)としては、マウス中にて発現パターンが既知であるSonic hedgehog(SHH)というタンパク質をコードするmRNA(shh)をモデルとした。そこでまず、SHHをコードした遺伝子を含むプラスミドをテンプレートとしてPCRを行い、長さ約1000bpのDNA断片を得た。この際、上流にはT3プロモータ、下流にはT7プロモータが含まれるようにプライマー設計を施した。次に得られたPCR産物を基に、T3 polymerase(センス鎖(S) 調製用)を用いてRNA合成を行い、T3 polymeraseで転写反応を行ってセンス鎖配列を調製した(配列番号:24)。同様にT7 polymerase(アンチセンス鎖(AS) 調製用)を用いてRNA合成を行い、T7 polymeraseにで転写反応を行ってアンチセンス鎖配列を合成した(配列番号:25)。条件は以下の表3に示す。反応は37℃で2時間行い、得られたセンス鎖RNA、アンチセンス鎖はエタノール沈殿によって回収し、TEバッファー20μLで縣濁した。RNase inhibitorとDTTを添加し、RNaseによる分解を抑えた。
【0120】
【表3】
【0121】
<RNA濃度の測定>
転写反応後、Nano DropにてRNA濃度を測定した。その結果、センス鎖は1019ng/μL、アンチセンス鎖は815ng/μLであった。
【0122】
<標識プローブの作製>
Alkphos Direct(商標、GEヘルスケアバイオサイエンス社製)のキットに付随する試薬にて標識プローブを調製した。まず、上述したアンチセンス鎖RNAを滅菌水を用いて10ng/μLに希釈したもの60μL(総量600ng)を99.9℃で熱変性後、急冷した。キットに添付されているReaction buffer 60μL、Labeling reagent 12μL、Cross−linker溶液60μLの順に熱変性後のRNAに添加し、37℃で30分間インキュベートし、標識プローブを作製した。
【0123】
<検出用メンブレンの作製>
まず、ターゲットとなるセンス鎖RNA、アンチセンス鎖RNAを1pg/μL〜50ng/μlの6段階の濃度に滅菌水を用いて希釈したものを、それぞれ1μLずつプラスのチャージを持つメンブレン(Hybond N+、GEヘルスケアバイオサイエンス社製)にアプライした。その後、80℃で2時間ベーキングした。
【0124】
<ハイブリダイゼーション>
上記検出用メンブレンの作製で作製したメンブレンをキットに添付されているハイブリダイゼーションバッファーに移し、55℃で15分間プレハイブリダイゼーションを行った。その後、上述した標準プローブを濃度が50ng/mLとなるように添加して55℃で一晩(14時間〜16時間程度)ハイブリダイゼーションを行った。
【0125】
<メンブレンの洗浄>
ハイブリダイゼーション後、メンブレンをWashIバッファー(表4)に移し、55℃で振とうしながら15分洗浄した。この操作を2回繰り返した。その後、WashIIバッファー(表5)にて洗浄した。この操作を2回繰り返した。
【0126】
【表4】
【0127】
【表5】
【0128】
<発色法による検出>
NTMTxバッファー(表6)で室温(実験した日は18〜22℃程度)、5分間洗浄したメンブレンをStaining溶液(375μg/mL NBT+188μg/mL BCIP in NTMTx)に移し、室温で2時間、遮光下で発色させた。なお、NBTとは、Nitro blue tetrazolium chlorideの略称であり、BCIPは、5-Bromo-4-chloro-3-indolyl phosphate, toluidine saltのことである。
【0129】
【表6】
【0130】
ドットブロットの結果を図7に示す。プローブと相補的なセンス鎖のメンブレンでは、100pg/μLのドットまで検出できた。また、アンチセンス鎖でも50,10ng/μLのドットに若干のシグナルがみられた。なお、この結果は、ターゲットRNAの調製時に使用した鋳型のDNAがDNaseIで処理できず残っていた可能性が考えられる。また、洗浄操作の際に温度が低下し、ストリンジェンシーの低下による非特異的吸着も考えられる。
【0131】
[実験2:Alkphos Directのプロトコールに準じた系におけるTGase基質マルチラベル化核酸プローブを用いた場合の検出感度の評価]
<TGase基質マルチラベル化核酸の調製>
上記実験1のセンス鎖RNA、アンチセンス鎖RNAの調製に加え、表7、表8に準じてTGase基質マルチラベル化核酸もT7 polymeraseを用いて同様に調製した。調製後、DNaseIで処理し、エタノール沈殿でRNAを濃縮・精製してTEバッファー20μLにて縣濁した。また、RNase inhibitorとDTTも添加し、新規酵素マルチラベル化核酸プローブを作製した。
【0132】
【表7】
【0133】
【表8】
【0134】
<RNA濃度の測定>
転写反応後、Nano DropにてRNA濃度を測定した。以下に測定結果を示す。なお、表8中の○数字を以下括弧数字で表示する。
(1)Z−QG−UTP 0%:410ng/μL
(2)Z−QG−UTP 20%:424ng/μL
(3)Z−QG−UTP 40%:378ng/μL
(4)Z−QG−UTP 60%:368ng/μL
(5)Z−QG−UTP 80%:311ng/μL
(6)Z−QG−UTP 100%:256ng/μL
【0135】
<標識プローブの作製>
Z−QG−UTPを異なる割合で取り込んだTGase基質マルチラベル化核酸を99.9℃で5分間熱変性後、急冷した。熱変性した核酸を用いて表9の条件となるように反応溶液を調製し、4℃で6時間反応させて酵素マルチラベル化プローブを作製した(配列番号:23)。
【0136】
【表9】
【0137】
以下、上述した実験1の検出用メンブレンの作製、ハイブリダイゼーション、メンブレン洗浄を行った。
【0138】
<発色法による検出>
検出は発色時の温度を室温(実験した日は18〜22℃程度)と50℃に設定し、それ以外の操作は実験1の発色法による検出操作に準じて行った。その結果、酵素マルチラベル化核酸プローブを用いてAlkphos Directのプロトコールに準じて検出した結果を図8に示した。室温で発色反応を行った場合には、Z−QG−UTPの割合が20〜40%のプローブでは10ng/μLまで、Z−QG−UTPの割合が60〜100%のプローブでは1ng/μLのドットまで検出することができた。また、50℃で発色反応を行った場合では、Z−QG−UTPの割合が20%では10ng/μL、40〜60%では1ng/μL、80〜100%では100pg/μLのドットまで検出できた。
【0139】
以上より、Z−QG導入割合の増加に伴って、検出感度は向上することが分かった。また、室温での発色条件下ではZ−QG−UTP60%以上の時、1ng/μLのターゲット核酸の検出に成功した。また、発色時の温度を新規酵素(アルカリホスファターゼ:PfuAP)の至適温度に近づけると、Z−QG−UTP80%以上で市販のAlkphos Directの検出限界(100pg/μL)と同等の検出感度で、コントラストの高い像を得ることができた。
【0140】
(実施例2:ドットブロットによるISHプロトコールに準じた検出感度の評価)
[PfuAPマルチラベル化RNAプローブの調製および機能評価]
細胞組織内での遺伝子発現パターンが既知であるソニックヘッジホッグ遺伝子(shh)をターゲットのmRNAとする。まず、PCRによってshhをコードするDNA断片(約1000bp)を得た。この際、遺伝子の上流側にはT3プロモーター、下流側にはT7プロモーターが含まれるようプライマー設計を施した。次に、得られたPCR産物を基にT7Polymeraseを用いて転写反応を行った(表10)。この際、UTPの代わりに、実験1で合成したZ−QG−UTPを様々な割合(0〜100%)にて加えることで、導入率の異なるZ−QG RNAを調製した。得られたZ−QG RNAはエタノール沈殿によって回収し、アガロースゲル電気泳動およびマイクロチップ型電気泳道装置(Bio−Rad,Experion)により評価した。
【0141】
【表10】
【0142】
<PfuAPマルチラベル化RNAの調製>
遺伝子工学的手法により、N末端にMTGが認識するKを含んだペプチドタグ(MKHK(GGGS)2GS)を導入したNK14−PfuAPが、MTGによってZ−QG RNAと結合するか検討するために、まず、様々な割合のZ−QG−UTPを用いて調製したZ−QG RNAを、99.9℃まで加熱した後、急冷することによって、変性処理を行った。続いて、100mM Tris−HCl(pH8.0)緩衝液中にて、各種Z−QG RNA50μg/mL、NK14−PfuAP 0.4mg/mL、MTG5.0U/mL、RNase inhibtor 1.0U/μLの条件下にて、6時間反応を行った。コントロールとして、MTG未添加の場合にも同様の操作を行い、反応はアガロースゲル電気泳動によって評価した。
【0143】
<ドットブロットによるプローブ能評価>
shh S(ターゲット)をメンブレン上に固定化し、PfuAPマルチラベル化核酸プローブ(shh アンチセンス、AS)を用いて検出を試みた。まず、未標識shh S(T3 Polymeraseにより調製)の希釈系列(50ng/μL、10ng/μL、1ng/μL、0.1mg/μL)を調製し、メンブレンに1μLずつスポットし、オーブンにて固定した(80℃、2時間)。その後、表11に示す操作を行い、PfuAPマルチラベル化プローブを用いてshh Sの検出を行った。コントロールとして、shh AS鎖を同様にして固定化したメンブレンを調製し、これを用いて行った。尚、溶液の組成は表12に記す。
【0144】
【表11】
【0145】
【表12】
【0146】
<Z−QGマルチラベル化RNAの調製>
UTPとZ−QG−UTPの割合を変化させてRNAを調製した際の結果を、図9の(A)、(B)に示す。結果より、いずれの条件においてもRNAの合成に成功していることから、合成したZ−QG−UTPは、比較的良好な基質として、T7 Polymeraseに認識されることが分かる。また、Z−QG−UTPの割合増加とともに高分子量側へとシフトしていることから、Z−QG−UTPの濃度を変化させることによって、Z−QG RNAへのZ−QG−UTP導入量が調節可能である。
【0147】
<PfuAPマルチラベル化核酸プローブの調製>
それぞれ導入量の異なる、Z−QG RNAに対して、MTGを用いてPfuAPラベル化を行った際の結果を図10に示す。Lane2はMTG未添加時、Lane3はMTG添加時の結果である。20%〜100%の条件では、MTG添加時において高分子量側へのシフトが見られるが、Z−QG−UTPが0%の際には確認されないために、導入したZ−QG特異的にZ−QG−UTPがラベル化されたことが分かる。また、Z−QG導入量の増加とともに高分子量側へのシフトも大きくなっていることからZ−QG−UTPの割合を変化させることによって、プローブのPfuAPのラベル化量が制御可能である。
【0148】
<ドットブロットによるプローブ能評価>
調製したPfuAPマルチラベル化RNAプローブの核酸プローブとしての機能について検討するためにドットブロットを行った(図11)。AS(アンチセンス)鎖をメンブレンに固定化した際にはスポットが確認されないことから、塩基配列特異的な検出ができている。20〜100%導入プローブにおいて1ng/μLのセンス鎖の検出が可能であった。以上のことから、転写反応時のZ−QG−UTP濃度を変化させることによって、得られる修飾RNA中のZ−QG導入量、その後のMTGによるPfuAPラベル化量が制御可能なことが示された。なお、実施例1とは異なり、Z−QG導入量増加に伴う検出感度向上は確認されなかった。この理由としては、上記表11,12に示す様に、ハイブリダイズ条件および洗浄条件が、Alkphos Direct条件に比べるとかなり厳しい条件であることが挙げられる。図10に示すようにいずれの場合においても、PfuAPラベル化した際のバンドはブロード化しており、PfuAPが均一に導入されているとは言えない。そのためPfuAPの導入数が少ないプローブ、あるいは未修飾のプローブの方がより強固にターゲット(AS鎖)とハイブリダイズするため、いずれの場合においても同等の感度となったのではないかと推察される。また、PfuAPの導入に伴うプローブの二本鎖形成能低下の可能性も、その一因として挙げられる。
【0149】
一方、超好熱菌由来酵素であるPfuAPを活性化させるために、染色時の温度を室温から50℃程度まで上昇させれば、現段階においても更なる感度向上が見込まれる。
【0150】
<高温条件での発色を行う場合の酵素ラベル化核酸プローブによるISHの検出感度評価>
本発明における核酸プローブにラベル化されたPfuAPは、室温よりも高温条件でより高い触媒活性を発現する。そこで、表11の酵素発色条件時の反応温度を、室温から50℃に上げた場合の検出感度を評価した。酵素ラベル化核酸プローブとしての性能を評価するため、まず、Alkphos Directキットにより得られる酵素標識核酸プローブと、本発明のPfuAPラベル化核酸プローブの性能比較を、ISH条件において検討した。実験は、酵素標識核酸プローブをキットにより調製する以外は、上述同様の操作によりドットブロットを行った。その結果、1ng/μLのセンス鎖の検出が可能であった(図12)。このことから、今回新たに調製されたPfuAPマルチラベル化核酸プローブは、ISH条件下において、市販のAlkphos Directにより調製されたプローブと同等の検出感度を有するプローブとして利用できることが示唆された。そこでは、最終的に核酸を検出するステップ、すなわち、酵素(アルカリホスファターゼ)部位による発色反応における温度を室温で行っていた。一方、本発明の核酸プローブにラベル化されたPfuAPは、室温よりも高温条件でより高い触媒活性を発現する。そこで、表10の酵素発色条件時の反応温度を、室温から50℃に上げた場合の検出感度を評価した。その結果、図13に示すように、検出限界はZ−QG導入割合により異なり、Z−QG80%、100%で100pg/μLから1ng/μL、Z−QG60%においては10pg/μLの核酸を検出することが可能であり、上述した実験1のAlkphos Directプローブを用いた場合(1ng/μL)より、少なくとも2桁程度、検出感度の向上が可能なことが明らかとなった。
【0151】
上述した実施例では、以下に示すような濃度決定したサンプルを用いて実験を行った。
【0152】
[Z−QG−UTPの濃度算出]
<aminoallyl−UTPの吸光係数算出(@290nm)>
aminoallyl−UTPをTE Buffer(pH8.0)に溶解した後、0、0.02、0.04、0.06、0.08、0.1mMとなるように調製し、吸収スペクトル測定を行った(図14)。スペクトルより290nmの吸光度を算出し検量線を作成した(図15)。尚、図14のスペクトルは、スペクトルチャートの吸収感度の一番高いのが0.1mMであり、以下順に0.08mM、0.06mM、0.04mM、0.02mM、0mMと吸収感度が下がっている。
【0153】
図15より、290nmの吸光係数が算出される(ε290=5760M−1cm−1)が、市販のaminoallyl−UTP(SIGMA社製)の純度が84%であることを考慮すると、ε290=6860M−1cm−1となる。なお、290nmにおけるZ−QGの吸収はほとんどないため、Z−QG−UTPサンプル中に未反応のZ−QGが混入していたとしても、濃度決定への影響は無視できる。
【0154】
<Z−QG−UTPストックサンプルの調製>
HPLCによる精製後、凍結乾燥によって溶媒を除去し、滅菌水(2mL)に溶解した。その後、Speed vacを用いて濃縮した(この際、析出物が見られたため、未反応のZ−QGが析出していると考えられる。濃縮に伴いaminoallyl−UTPと同形のスペクトルは増加したため、Z−QG−UTPの析出ではないと考える)。濃縮後、上清を回収し、TE bufferによって10倍希釈した後、Nano Drop(光路長:1mm)を用いて吸光度測定を行った所、吸収度ABS=1.061であった。したがって、希釈前のZ−QG−UTP濃度Cは次式により算出される。
【0155】
(数1)
1.061=6860[M−1cm−1]×0.1[cm]×C/10
したがって、C=約15.5[mM]である。
【0156】
(実施例3)
[Z−QG DNAを用いたPfuAPマルチラベル化DNAプローブの調製とドットブロットによる検出感度の評価]
Z−QG DNAを用いたPfuAPマルチラベル化DNAプローブの調製とドットブロットによる検出感度の評価を行った。
【0157】
<Z−QG−dUTPの合成・精製>
まず、N,N−ジメチルホルムアミド(DMF)4mL中にて、100mM N,N’−ジイソプロピルカルボジイミド(DIC)、100mM N−ヒドロキシスクシンイミド(NHS)、50mM Z−QGを、室温(調製した日は18〜22℃程度)で20時間反応させることによって、NHS化Z−QG(50mM)を調製した。一方、50mM 5−(3−aminoallyl)−dUTP(以下、aminoallyl−dUTPと略記、SIGMA社製)を含む10mM Tris−HCl(pH7.5)溶液(Ambion製)16μLと200mM ホウ酸緩衝液(pH8.8)40μL、滅菌水16μLとを混合し、10mM aminoallyl−dUTP溶液を80μL調製した。この溶液に対して、上記で調製したNHS化Z−QG溶液を80μL添加し、25℃で一晩反応させた。反応終了後、サンプルをMilli−Qで10倍希釈し、HPLC(日本分光製、高速液体クロマトグラフ・ポンプ:TRI ROTAR−V型、バリアブル・ループ・インジェクタ:VL−613型、紫外可視分光検出器:UVIDEC−100−IV型)によって、表13に示す条件で精製を行った。生成物の同定はMALDI TOF−MS(BRUKER DALTONICS製、autoflex III)によって行った。結果を図17に示す。この際、3−ヒドロキシピコリン酸(3−HPA)をマトリックスとして使用した。
【0158】
【表13】
【0159】
Z−QG−dUTPを合成した後、逆相HPLCを行った際の結果を図18に示す。aminoallyl−dUTPの場合(図19参照)と比較して、疎水側に新たなピークが出現しており、このピークがZ−QG−dUTPであると推測された。そこで、保持時間19.1分のピークを回収してMALDI TOF−MS分析を行った。その結果、図17に示すように、841.46のピークが確認され、理論分子量の842.13と良く一致した結果が得られたため、Z−QG−dUTPの合成が示された。
【0160】
<PCRによるZ−QG DNAの調製>
まず、マウス由来のSonic hedgehog(SHH)をコードする遺伝子中の約300bp(配列番号:27)をPCR増幅するプライマーを設計した(表14、配列番号:28,29)。PCRのテンプレートにはマウス由来のShhをコードする遺伝子の全配列(約990bp)を組み込んだプラスミドDNAを用い、DNA polymeraseとしてKOD−Fx(TOYOBO社製)を使用して反応液を調製した(表15、表16)。PCR条件は94℃×2分を1サイクル、94℃×20秒、52℃×20秒、72℃×15秒を30サイクルとした。PCR後、アガロース電気泳動で増幅断片を確認した。得られた増幅断片は、MinElute PCR Purification kit(QIAGEN社製)で精製し、Nano DropにてZ−QG DNAの濃度を測定した。
【0161】
【表14】
【0162】
【表15】
【0163】
【表16】
【0164】
Z−QG−dUTPの割合を変化させてPCRした際の電気泳動の結果を図20に示す。Z−QG−dUTPの割合を50%、100%としてPCRした場合でも増幅断片のバンドが確認されたことから、Z−QG−dUTPはDNA polymeraseの基質として認識されていることが示された。また、Z−QG−dUTPの割合の増加に伴ってバンドが高分子側にシフトしていることから、PCRを行う際のZ−QG−dUTPの量を変化させることで、Z−QGの導入量を調節することが可能であることがわかった。精製後にNano DropでそれぞれのZ−QG DNAの濃度を計った結果、Z−QG−dUTP 0%で調製したZ−QG DNAは73.8ng/μL、Z−QG−dUTP 50%で調製したZ−QG DNAは111.6ng/μL、Z−QG−dUTP 100%で調製したZ−QG DNAは59.1ng/μLであった。
【0165】
<Z−QG DNAへのPfuAPのラベル化の検討>
遺伝子工学的にMTGが認識可能なリジン残基(K)を含むペプチドダグを導入したNK14−PfuAPがMTGによってZ−QG DNAに結合するかを、Z−QGの導入量が異なるZ−QG DNAを用いて検討した。まず、100mM Tris−HCl(pH8.0)緩衝液中にて、各種Z−QG DNA 15ng/μL、NK14−PfuAP 0.4mg/mL、MTG 5.0Unit/mLの条件下で4℃、6時間反応させた。MTG未添加の場合も同様に検討した。PfuAPのラベル化の確認はアガロース電気泳動で行った。
【0166】
MTG反応後のアガロース電気泳動の結果を図21に示す。Z−QG−dUTP 50%、100%の条件で調製したZ−QG DNAを用いた場合はMTG添加時に高分子側へシフトしているが、Z−QG未ラベルDNA(Z−QG−dUTP 0%の条件で調製した陰性対照DNA)では分子量シフトが確認されなかった。また、Z−QG DNAに対して、MTG未添加時にも高分子量化は確認されなかった。この結果より、NK14−PfuAPはMTGによってZ−QG DNAのZ−QG部位へ特異的に結合していることがわかった。また、Z−QG−dUTP 50%の場合よりZ−QG−dUTP 100%の条件で調製したZ−QG DNAを用いたほうが、より明確に高分子量化が進んでいることから、PCRによる増幅の際のZ−QG−dUTPの割合を変えることによって、PfuAPの結合量を調節することが可能なことが示された。
【0167】
<PfuAPマルチラベル化DNAプローブの調製>
Z−QG−dUTP 50%、100%の条件で調製したZ−QG DNAとNK14−PfuAPをMTGにて結合し、PfuAPマルチラベル化DNAプローブを調製した。まず、各種Z−QG DNAを90℃で5分間熱処理してDNAを一本鎖に変性させた後、100mM Tris−HCl(pH8.0)緩衝液中にて、各種Z−QG DNA 25ng/μL、NK14−PfuAP 0.4mg/mL、MTG 5.0Unit/mLの条件下で4℃、6時間反応させた(MTG反応前熱処理プローブ)。DNAプローブは熱処理で一本鎖にすることがプローブの検出能に大きく影響することから、熱処理していないZ−QG DNAについても同様の条件でPfuAPのラベル化を行い、その後、90℃で5分間熱処理を施したプローブも調製した(MTG反応後熱処理プローブ)。DNAプローブのPfuAPラベル化の確認はアガロース電気泳動で行った。
【0168】
MTG反応後のアガロース電気泳動の結果を図22に示した。MTG反応前に熱処理したZ−QG DNAおよびMTG反応後に熱処理したZ−QG DNAとも高分子側へシフトしていることから、Z−QG DNAへのPfuAPのラベル化が確認された。
【0169】
<ドットブロットによる検出感度の評価>
それぞれ調製したPfuAPマルチラベル化プローブの検出感度の評価をドットブロットで行った。なお、ハイブリダイゼーション時の各種組成およびプロトコールは、市販の検出キットAlkphos Direct(GEヘルスケアバイオサイエンス社製)に準拠した。ターゲットDNAとして、プローブ配列を含む990bpのShhのDNA断片(配列番号:30、プライマー配列は表17参照(配列番号:31))、およびプローブ配列と同じ300bpのShhのDNA断片を用いた。また、ネガティブコントロールとして、PfuAPをコードする遺伝子約1.7kbのDNA断片(配列番号:32、プライマー配列は表18参照(配列番号:33,34))を用いた。
【0170】
【表17】
【0171】
【表18】
【0172】
それぞれのターゲットDNAをTE Bufferを用いて50ng/μLと50pg/μLに希釈し(ネガティブコントロールは50ng/μLのみ)、それぞれを1μLずつプラスチャージのメンブラン(Hybond N+、GEヘルスケアバイオサイエンス社製)にスポットし、80℃で2時間熱処理することでDNAをメンブランに固定した。その後、検出キットに添付されているハイブリダイゼーションバッファーにメンブランを移し、55℃で30分間、プレハイブリダイゼーションを行った。そして、上記方法で調製した各種PfuAPマルチラベル化DNAプローブを50ng/mLとなるようにハイブリダイゼーションバッファーに添加して、55℃で一晩(14時間〜16時間程度)ハイブリダイゼーションを行った。
【0173】
ハイブリダイゼーション後、メンブレンをWash Iバッファー(表19)に移し、55℃で震盪しながら15分間洗浄した。この操作を2回繰り返した。その後、室温(実験した日は18〜22℃程度)でWash IIバッファー(表20)にて5分間洗浄した。この操作を2回繰り返した。
【0174】
【表19】
【0175】
【表20】
【0176】
NTMTxバッファー(表21)で室温、5分間洗浄したメンブランをStaining溶液(375μg/mL NBT + 188μg/mL BCIP in NTMTx)に移し、50℃に2時間、遮光下で静置して発色させた。
【0177】
【表21】
【0178】
各種PfuAPラベル化DNAプローブのドットブロットの結果を図23に示した。どのDNAプローブを用いた場合もドットのシグナルが検出されていることから、MTGを用いてZ−QG DNAにPfuAPでラベルしたDNAプローブは、核酸プローブとしての検出能を有していることが示された。また、MTG反応後に熱処理する場合(MTG反応後熱処理プローブ)よりも、MTG反応前にZ−QG DNAを熱処理する場合(MTG反応前熱処理プローブ)の方が、検出感度を向上できることがわかった。これはMTG反応後熱処理プローブでは、PfuAPがラベル化されたDNAプローブを熱処理することになるため、熱処理時の反応容量の増加により熱処理が不十分となりDNAが完全に熱変性されなかったことや、ラベル化された状態での熱処理によるPfuAPの失活が考えられる。
【0179】
MTG反応前熱処理プローブでは、Z−QGの導入量が50%および100%のプローブのどちらも50pg/μLのドットまで検出することができた。ネガティブコントロールにおけるターゲット核酸濃度が50ng/μLと高い場合には、非特異的なシグナルも若干検出されたが、相補的なDNAを明確に判別することができた。また、ターゲットDNAの鎖長の違いは、検出感度に大きな影響を与えなかった。
【0180】
(実施例4)
[Z−QG DNAを用いたPfuAPマルチラベル化DNAプローブのISHプロトコールに準じたRNA検出]
実施例3では、検出対象核酸はDNAであった。本実施例4では、検出対象核酸をRNAとした場合の、PfuAPマルチラベル化DNAプローブの核酸プローブとしての機能について評価した。
【0181】
<PCRによるZ−QG DNAの調製>
実施例3と同様にして、PCRによるZ−QG DNAの調製を行った。
【0182】
<PfuAPマルチラベル化DNAプローブの調製>
Z−QG−dUTP 50%、100%の条件で調製したZ−QG DNAとNK14−PfuAPをMTGにて結合し、PfuAPマルチラベル化DNAプローブを調製した。まず、各種Z−QG DNAを90℃で5分間熱処理してDNAを一本鎖に変性させた後、100mM Tris−HCl(pH8.0)緩衝液中にて、各種Z−QG DNA 25ng/μL、NK14−PfuAP 0.4mg/mL、MTG 5.0Unit/mLの条件下で4℃、6時間反応させた。DNAプローブのPfuAPラベル化の確認はアガロース電気泳動で行った。
【0183】
MTG反応後のアガロース電気泳動の結果を図24に示した。Z−QG−dUTPを50%、100%導入したZ−QG DNAはどちらもMTG反応後に高分子側にバンドがシフトしていた。この結果よりZ−QG DNAへのPfuAPのラベル化が確認されたため、これらをドットブロットのDNAプローブとして使用した。
【0184】
<ISHプロトコールに準じたドットブロットによる検出感度の評価>
実施例3と同様にして、PCRによってプローブ配列を含む990bpのShhのDNA断片(配列番号:30)を得た。この際、Shh遺伝子の下流側にT7プロモーターが含まれるようプライマー設計(配列番号:31、表17参照)を施した。次に、得られたPCR産物を基にT7 Polymeraseを用いて転写反応を行った(表22)。転写反応後、ゲル濾過カラム(Mini Quick Spin Columns、Roche社製)で未反応のNTP等を精製したものをターゲットRNAとして用いた。
【0185】
【表22】
【0186】
得られたターゲットRNAをTE Bufferを用いて50ng/μL、1ng/μL、50pg/μLの三段階に希釈し、それぞれを1μLずつプラスチャージのメンブラン(Hybond N+、GEヘルスケアバイオサイエンス社製)にスポットし、80℃で2時間熱処理することでRNAをメンブランに固定した。ネガティブコントロールとして、市販の酵母RNA(Ribonucleic torula yeast RNA、シグマ・アルドリッチ製)を50ng/μLに希釈したものを同様にメンブランに1μLスポットし、固定化した。その後、ハイブリダイゼーションバッファー(表23)中にメンブランを移し、60℃で30分間、プレハイブリダイゼーションを行った。そして、上記方法で調製した各種DNAプローブを50ng/μLとなるように添加し、60℃で一晩(14時間〜16時間程度)ハイブリダイゼーションを行った。
【0187】
【表23】
【0188】
ハイブリダイゼーション後、メンブレンの洗浄操作を行った。各種洗浄溶液を表24に示す。メンブランをWash Iバッファーに移し、60℃で震盪しながら20分間洗浄した。この操作を3回繰り返した。そして、55℃でWash IIバッファーにて20分間洗浄した。この操作を3回繰り返した。続いてNTMTxバッファーを用いて室温(実験した日は18〜22℃程度)で5分間、2回洗浄し、洗浄したメンブランをStaining溶液(375μg/mL NBT + 188μg/mL BCIP in NTMTx)に移し、50℃で2時間、遮光下に静置して発色させた。
【0189】
【表24】
【0190】
各種PfuAPラベル化DNAプローブのISHプロトコールに準じたドットブロットの結果を図25に示した。Z−QGの導入量が50%および100%のどちらのDNAプローブも1ng/μLのドットまで検出することができ、シグナルの発色強度もどちらのプローブを用いた場合もほぼ同等であった。また、ネガティブコントロールの非特異的なシグナルは検出されなかった。このことより、MTGを用いてZ−QG DNAにPfuAPをラベルしたDNAプローブは、RNAを検出するプローブとして利用可能であることが明らかとなった。
【0191】
(実施例5)
実施例2と同様にして、常温菌であるEscherichia coli由来のAP(BAP)を用いてマルチラベル化したマルチラベル化核酸プローブについても、プローブ能評価を行った。
【0192】
<BAPマルチラベル化RNAの調製>
実施例2と同様にして、RNAの変性処理を行った後に、続いて、100mM Tris−HCl(pH8.0)緩衝液中にて、各種Z−QG RNA 50μg/mL、NK14−BAP 0.4mg/mL、MTG 5.0U/mL、RNase inhibtor 1.0U/μLの条件下にて、6時間反応を行った。コントロールとして、MTG未添加の場合にも同様の操作を行い、反応はアガロースゲル電気泳動によって評価した。
【0193】
<ドットブロットによるプローブ能評価>
実施例2と同様にして、ドットブロットによりプローブ能について評価した。
【0194】
ラベル化した際の分子量変化を図26に示す。結果より、Z−QG導入量に比例して、顕著な分子量増加が生じた。また、Z−QG RNA(100%)を用いた際には、分子量分布が小さくなっていることが確認された。RNA調製時にZ−QG−UTPのみを使用した場合には、Z−QGが導入される数や位置が単分散となるため(理論上全てのU)、ほぼ均一にラベル化が進行したと考えられる。PfuAP(図10)に比べて著しい分子量増加が見られた点から、Z−QG RNAとの反応性はBAPの方が優れていることが示唆される結果となった。
【0195】
一方で、得られたBAPマルチラベル化RNAを用い、ドットブロットによってプローブ能を評価した(図27参照)。結果より、Z−QG導入量が60%、80%、100%のmRNAに対してBAPラベル化を行った際において、1ng程度のRNAが検出可能であった。20%、40%においてほとんど検出されなかった理由としては、BAP未修飾のRNAが残存しているためであると予想される(図26参照)。PfuAPをラベル化した際のプローブ(検出限界:10pg)と比較すると、本系における標識酵素としてはPfuAPの方が適していることが示された。
【0196】
(実施例6:MTGによるZ−QG RNAの蛍光色素ラベル化およびドットブロットによる標的核酸の検出)
<Alexa Fluor 555マルチラベル化RNAの調製>
蛍光色素として、一級アミン誘導化されたAlexa Fluor(登録商標) 555 cadaverine(invitrogen社製)が、MTGによってZ−QG RNAと結合するかについて検討した。まず、Z−QG−UTP使用率100%の条件で調製したZ−QG RNAを、99.0℃まで加熱した後、急冷することによって、変性処理を行った。続いて、100mM Tris−HCl(pH8.0)緩衝液中にて、Z−QG RNA 50μg/mL、Alexa Fluor 555 cadaverine 20μM、MTG 5.0units/mL、RNase inhibitor 1.0U/μLの条件下にて、6時間反応を行った。コントロールとして、MTG未添加の場合およびZ−QGを導入していない未修飾RNAに対しても同様の操作を行い、反応はアガロースゲル電気泳動によって評価した。
【0197】
MTGを用いて、Z−QG RNAに対してAlexa Fluor 555 cadaverineラベル化を行った際の結果を図28に示す。ここで、Lane2は未修飾のRNAとAlexa Fluor 555 cadaverineを、Lane3はLane2の条件にMTGを加えたものを、Lane4にはZ−QG RNAとAlexa Fluor 555 cadaverineを、Lane5にはLane4にMTGを加えたものを示す。Lane4,5において未修飾RNAとほぼ同等の位置に見られるシグナルは未反応のAlexa Fluor 555 cadaverineに由来するものと考えられる。このことは、エチジウムブロミド染色前にLane2〜4のいずれのLaneにおいても同様のバンドが確認されたことからも支持される。Lane2とLane3では変化が見られないのに対して、Lane4とLane5とを比較すると、MTG添加時にはZ−QG RNA由来のバンドが消失し、低分子量側へのシフトが確認された。このことは、負電荷を帯びたAlexa Fluor 555 cadaverineがZ−QG RNAに結合したことを示しており、Alexa Fluor 555マルチラベル化RNAの生成が示唆された。
【0198】
<ドットブロットによるプローブ能評価>
shh S(ターゲット)をメンブレン上に固定化し、Alexa Fluor 555マルチラベル化核酸プローブを用いて検出を試みた。まず、500ng/μLの未標識shh Sを調製し、メンブレンに1μL添加し、乾燥させた後、再度1μL添加し(計1,000ng)、オーブンにて固定化した(80℃、2時間)。その後、表25に示す操作を行い、蛍光イメージャ(BIO−RAD製、Molecular Imager FX Pro、532nm excitation、555nm long pass)を用いて、Alexa Fluor 555マルチラベル化核酸プローブによるshh Sの検出を行った。コントロールとして、shh AS鎖を同様にして固定化したメンブレンに対しても同様の操作を行った。なお、溶液の組成は表26に示す。
【0199】
【表25】
【0200】
【表26】
【0201】
調製したAlexa Fluor 555マルチラベル化RNAの核酸プローブとしての応用性について検討するためにドットブロットを行った(図29参照)。AS鎖を固定化した際にはシグナルが得られなかったのに対して、S鎖を固定化した場合には蛍光が確認されたことから、Alexa Fluor 555マルチラベル化RNAはプローブとして応用可能であることが示された。また、塩基配列特異性を保持しているため、今後、ハイブリダイゼーション過程の温度やホルムアミド濃度を最適化することによって、より高感度な検出が期待される。
【特許請求の範囲】
【請求項1】
グルタミン(Gln)残基を有することを特徴とするヌクレオシド三リン酸誘導体。
【請求項2】
請求項1に記載のヌクレオシド三リン酸誘導体であって、
下記式(1)で示されることを特徴とするヌクレオシド三リン酸誘導体。
【化1】
(式(1)中、Aは、グルタミン(Gln)残基を有する置換基、Bは水素原子またはヒドロキシル基を表す。)
【請求項3】
請求項1に記載のヌクレオシド三リン酸誘導体であって、
下記式(2)で示されることを特徴とするヌクレオシド三リン酸誘導体。
【化2】
(式(2)中、A1およびA2のうち少なくとも1つは、グルタミン(Gln)残基を有する置換基で残りは水素原子を表し、Bは水素原子またはヒドロキシル基を表す。)
【請求項4】
請求項1に記載のヌクレオシド三リン酸誘導体であって、
下記式(3)で示されることを特徴とするヌクレオシド三リン酸誘導体。
【化3】
(式(3)中、Aは、グルタミン(Gln)残基を有する置換基、Bは水素原子またはヒドロキシル基を表す。)
【請求項5】
請求項1に記載のヌクレオシド三リン酸誘導体であって、
下記式(4)で示されることを特徴とするヌクレオシド三リン酸誘導体。
【化4】
(式(4)中、Aは、グルタミン(Gln)残基を有する置換基、Bは水素原子またはヒドロキシル基を表す。)
【請求項6】
請求項1に記載のヌクレオシド三リン酸誘導体であって、
下記式(5)で示されることを特徴とするヌクレオシド三リン酸誘導体。
【化5】
(式(5)中、XおよびYは、それぞれ独立して2価の連結基を表し、Zは、置換基を表す。)
【請求項7】
請求項6に記載のヌクレオシド三リン酸誘導体であって、
前記XおよびYは、それぞれ独立して炭素数1〜48のアルキレン基または炭素数2〜48のアルケニレン基であり、Zは、炭素数1〜48のアルキル基、炭素数1〜48のアルコキシ基、炭素数6〜48のアリール基、炭素数6〜48のアリールオキシ基、炭素数7〜48のアリールアルキル基または炭素数7〜48のアリールアルキルオキシ基であることを特徴とするヌクレオシド三リン酸誘導体。
【請求項8】
請求項7に記載のヌクレオシド三リン酸誘導体であって、
前記Xはエテニレン基、Yはメチレン基であり、Zはベンジルオキシ基であることを特徴とするヌクレオシド三リン酸誘導体。
【請求項9】
構成単位として、請求項1〜8のいずれか1項に記載のヌクレオシド三リン酸誘導体が複数個導入されている核酸であることを特徴とする核酸プローブ。
【請求項10】
請求項9に記載の核酸プローブにおけるグルタミン(Gln)残基のうち少なくとも2つに、リシン(Lys)残基または第一級アミンを有し標識部分を含む標識化合物が結合されて構成されていることを特徴とするマルチラベル化核酸プローブ。
【請求項11】
請求項10に記載のマルチラベル化核酸プローブであって、
前記標識部分が、酵素および蛍光色素のうち少なくとも1つであることを特徴とするマルチラベル化核酸プローブ。
【請求項12】
請求項11に記載のマルチラベル化核酸プローブであって、
前記酵素が、超好熱菌由来の酵素であることを特徴とするマルチラベル化核酸プローブ。
【請求項13】
マルチラベル化核酸プローブの製造方法であって、
トランスグルタミナーゼ(TGase)を用いて、請求項9に記載の核酸プローブにおけるグルタミン(Gln)残基のうち少なくとも2つに、リシン(Lys)残基または第一級アミンを有し標識部分を含む標識化合物を結合することを特徴とするマルチラベル化核酸プローブの製造方法。
【請求項14】
請求項13に記載のマルチラベル化核酸プローブの製造方法であって、
前記標識部分が、酵素および蛍光色素のうち少なくとも1つであることを特徴とするマルチラベル化核酸プローブの製造方法。
【請求項15】
請求項14に記載のマルチラベル化核酸プローブの製造方法であって、
前記酵素が、超好熱菌由来の酵素であることを特徴とするマルチラベル化核酸プローブの製造方法。
【請求項16】
標的核酸の検出方法であって、
請求項10〜12のいずれか1項に記載のマルチラベル化核酸プローブと、対象物中に存在する標的核酸とを核酸部分により特異的に結合させ、結合している前記マルチラベル化核酸プローブを、前記標識部分により検出することを特徴とする標的核酸の検出方法。
【請求項17】
標的核酸の検出方法であって、
請求項9に記載の核酸プローブと、対象物中に存在する標的核酸とを核酸部分により特異的に結合させた後、
トランスグルタミナーゼ(TGase)を用いて、前記核酸プローブにおけるグルタミン(Gln)残基と、リシン(Lys)残基または第一級アミンを有し標識部分を含む標識化合物のリシン(Lys)残基または第一級アミンとを反応させて複数の標識部分を導入し、結合している前記核酸プローブを、前記標識部分により検出することを特徴とする標的核酸の検出方法。
【請求項1】
グルタミン(Gln)残基を有することを特徴とするヌクレオシド三リン酸誘導体。
【請求項2】
請求項1に記載のヌクレオシド三リン酸誘導体であって、
下記式(1)で示されることを特徴とするヌクレオシド三リン酸誘導体。
【化1】
(式(1)中、Aは、グルタミン(Gln)残基を有する置換基、Bは水素原子またはヒドロキシル基を表す。)
【請求項3】
請求項1に記載のヌクレオシド三リン酸誘導体であって、
下記式(2)で示されることを特徴とするヌクレオシド三リン酸誘導体。
【化2】
(式(2)中、A1およびA2のうち少なくとも1つは、グルタミン(Gln)残基を有する置換基で残りは水素原子を表し、Bは水素原子またはヒドロキシル基を表す。)
【請求項4】
請求項1に記載のヌクレオシド三リン酸誘導体であって、
下記式(3)で示されることを特徴とするヌクレオシド三リン酸誘導体。
【化3】
(式(3)中、Aは、グルタミン(Gln)残基を有する置換基、Bは水素原子またはヒドロキシル基を表す。)
【請求項5】
請求項1に記載のヌクレオシド三リン酸誘導体であって、
下記式(4)で示されることを特徴とするヌクレオシド三リン酸誘導体。
【化4】
(式(4)中、Aは、グルタミン(Gln)残基を有する置換基、Bは水素原子またはヒドロキシル基を表す。)
【請求項6】
請求項1に記載のヌクレオシド三リン酸誘導体であって、
下記式(5)で示されることを特徴とするヌクレオシド三リン酸誘導体。
【化5】
(式(5)中、XおよびYは、それぞれ独立して2価の連結基を表し、Zは、置換基を表す。)
【請求項7】
請求項6に記載のヌクレオシド三リン酸誘導体であって、
前記XおよびYは、それぞれ独立して炭素数1〜48のアルキレン基または炭素数2〜48のアルケニレン基であり、Zは、炭素数1〜48のアルキル基、炭素数1〜48のアルコキシ基、炭素数6〜48のアリール基、炭素数6〜48のアリールオキシ基、炭素数7〜48のアリールアルキル基または炭素数7〜48のアリールアルキルオキシ基であることを特徴とするヌクレオシド三リン酸誘導体。
【請求項8】
請求項7に記載のヌクレオシド三リン酸誘導体であって、
前記Xはエテニレン基、Yはメチレン基であり、Zはベンジルオキシ基であることを特徴とするヌクレオシド三リン酸誘導体。
【請求項9】
構成単位として、請求項1〜8のいずれか1項に記載のヌクレオシド三リン酸誘導体が複数個導入されている核酸であることを特徴とする核酸プローブ。
【請求項10】
請求項9に記載の核酸プローブにおけるグルタミン(Gln)残基のうち少なくとも2つに、リシン(Lys)残基または第一級アミンを有し標識部分を含む標識化合物が結合されて構成されていることを特徴とするマルチラベル化核酸プローブ。
【請求項11】
請求項10に記載のマルチラベル化核酸プローブであって、
前記標識部分が、酵素および蛍光色素のうち少なくとも1つであることを特徴とするマルチラベル化核酸プローブ。
【請求項12】
請求項11に記載のマルチラベル化核酸プローブであって、
前記酵素が、超好熱菌由来の酵素であることを特徴とするマルチラベル化核酸プローブ。
【請求項13】
マルチラベル化核酸プローブの製造方法であって、
トランスグルタミナーゼ(TGase)を用いて、請求項9に記載の核酸プローブにおけるグルタミン(Gln)残基のうち少なくとも2つに、リシン(Lys)残基または第一級アミンを有し標識部分を含む標識化合物を結合することを特徴とするマルチラベル化核酸プローブの製造方法。
【請求項14】
請求項13に記載のマルチラベル化核酸プローブの製造方法であって、
前記標識部分が、酵素および蛍光色素のうち少なくとも1つであることを特徴とするマルチラベル化核酸プローブの製造方法。
【請求項15】
請求項14に記載のマルチラベル化核酸プローブの製造方法であって、
前記酵素が、超好熱菌由来の酵素であることを特徴とするマルチラベル化核酸プローブの製造方法。
【請求項16】
標的核酸の検出方法であって、
請求項10〜12のいずれか1項に記載のマルチラベル化核酸プローブと、対象物中に存在する標的核酸とを核酸部分により特異的に結合させ、結合している前記マルチラベル化核酸プローブを、前記標識部分により検出することを特徴とする標的核酸の検出方法。
【請求項17】
標的核酸の検出方法であって、
請求項9に記載の核酸プローブと、対象物中に存在する標的核酸とを核酸部分により特異的に結合させた後、
トランスグルタミナーゼ(TGase)を用いて、前記核酸プローブにおけるグルタミン(Gln)残基と、リシン(Lys)残基または第一級アミンを有し標識部分を含む標識化合物のリシン(Lys)残基または第一級アミンとを反応させて複数の標識部分を導入し、結合している前記核酸プローブを、前記標識部分により検出することを特徴とする標的核酸の検出方法。
【図1】

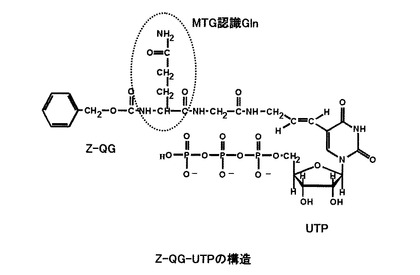
【図3】
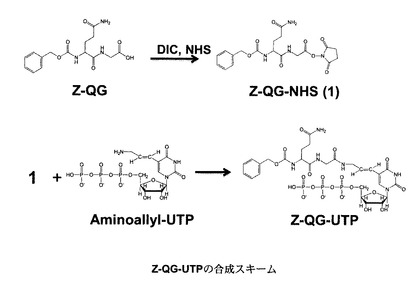
【図4】
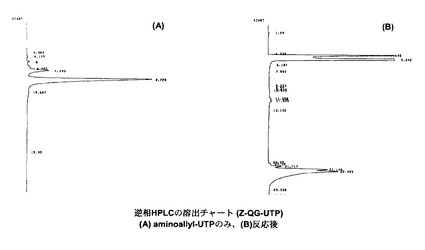
【図5】
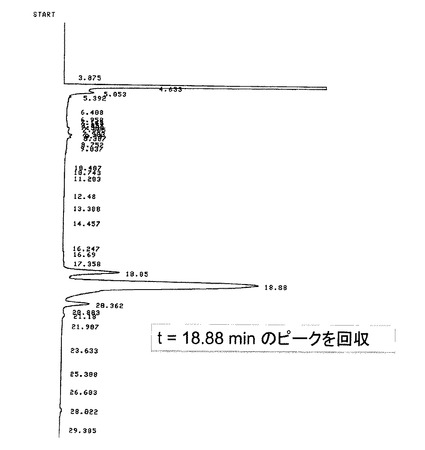
【図18】
【図19】
【図2】
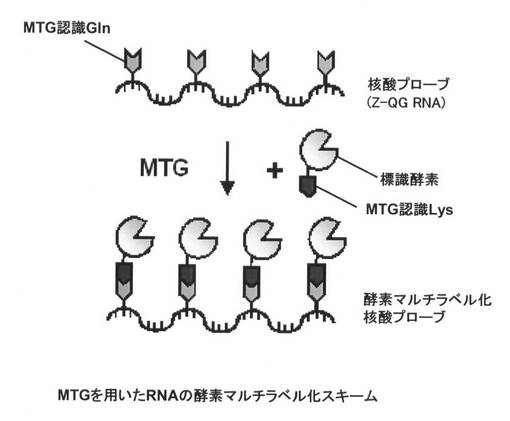
【図6】
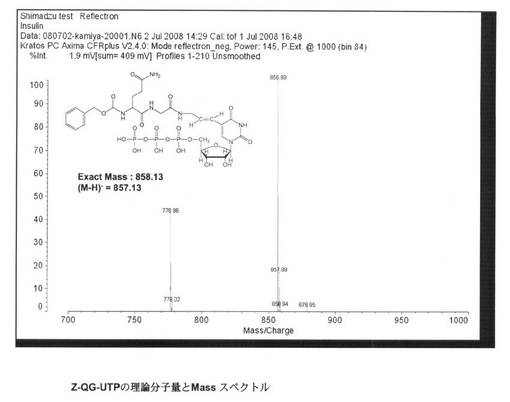
【図7】
【図8】
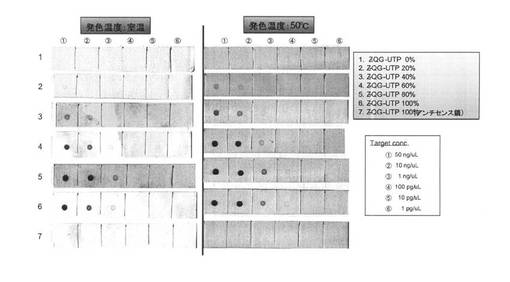
【図9】
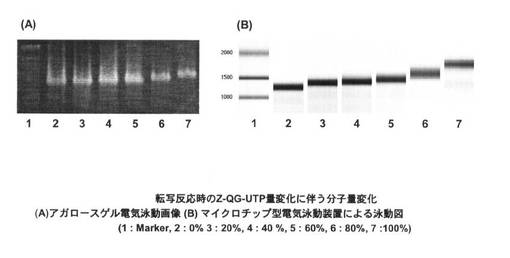
【図10】
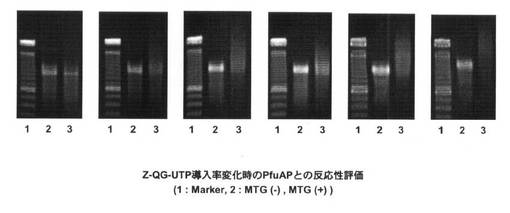
【図11】
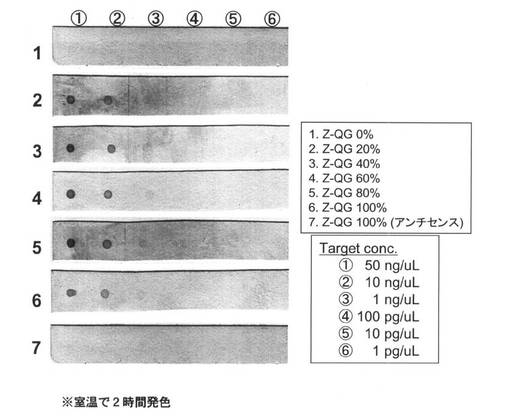
【図12】
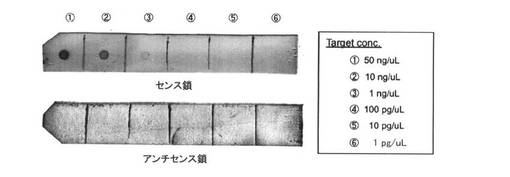
【図13】
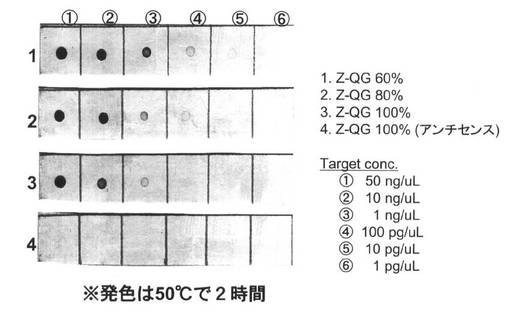
【図14】
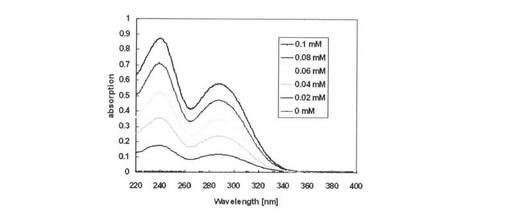
【図15】
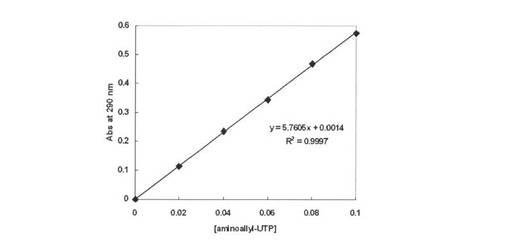
【図16】
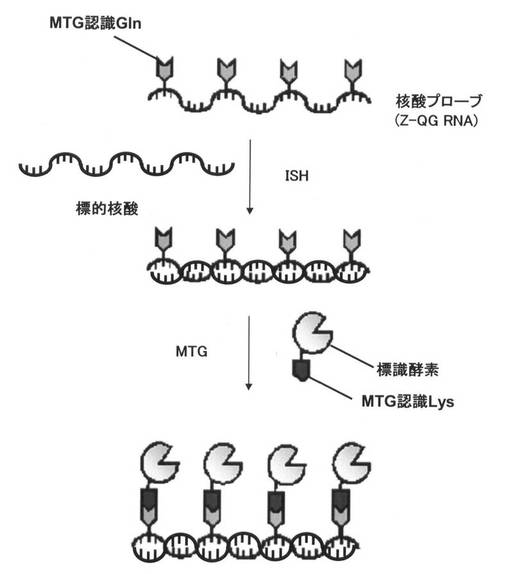
【図17】
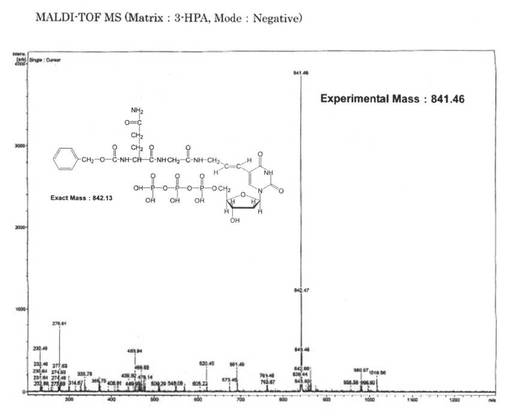
【図20】
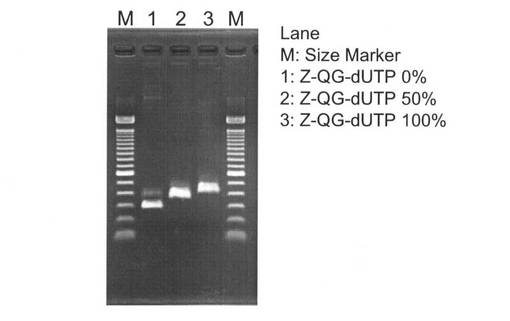
【図21】
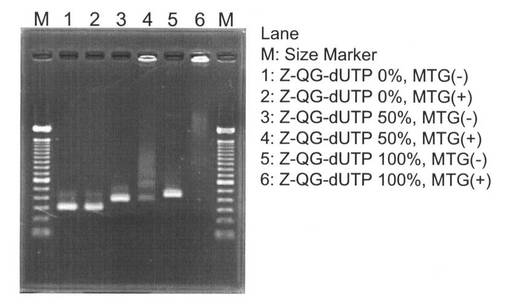
【図22】
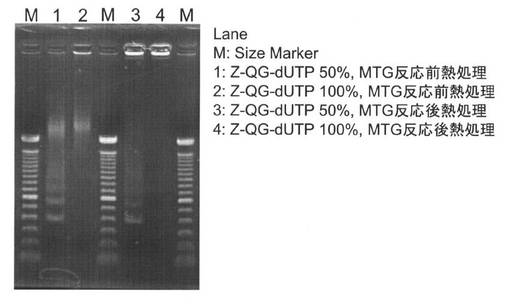
【図23】
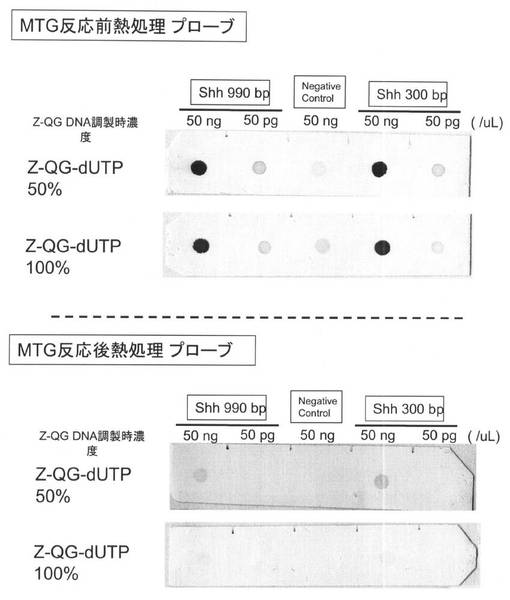
【図24】
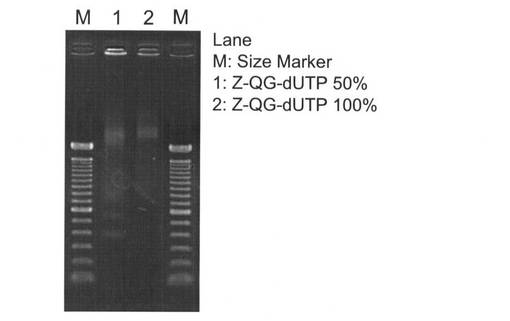
【図25】
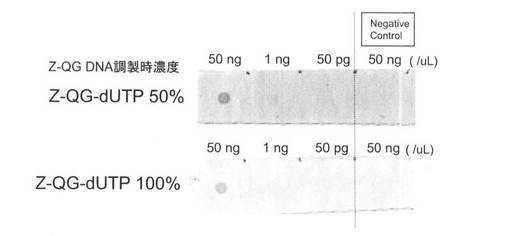
【図26】
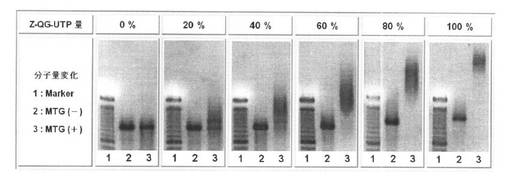
【図27】
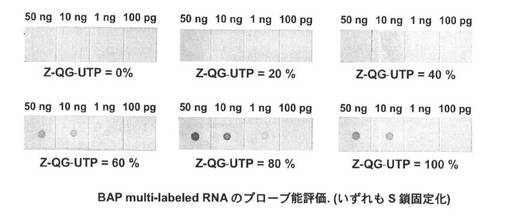
【図28】
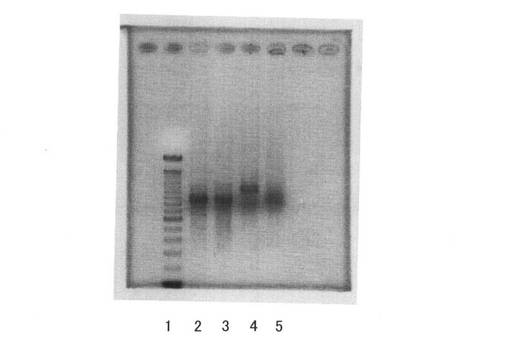
【図29】

【図3】
【図4】
【図5】
【図18】
【図19】
【図2】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【公開番号】特開2010−47559(P2010−47559A)
【公開日】平成22年3月4日(2010.3.4)
【国際特許分類】
【出願番号】特願2009−32763(P2009−32763)
【出願日】平成21年2月16日(2009.2.16)
【国等の委託研究の成果に係る記載事項】(出願人による申告)国等の委託研究の成果に係る特許出願(独立行政法人科学技術振興機構 地域イノベーション創出総合支援事業、重点地域研究開発推進プログラム(育成研究)、産業技術力強化法第19条の適用を受けるもの)
【出願人】(504145342)国立大学法人九州大学 (960)
【出願人】(304020292)国立大学法人徳島大学 (307)
【出願人】(390029791)アロカ株式会社 (899)
【Fターム(参考)】
【公開日】平成22年3月4日(2010.3.4)
【国際特許分類】
【出願日】平成21年2月16日(2009.2.16)
【国等の委託研究の成果に係る記載事項】(出願人による申告)国等の委託研究の成果に係る特許出願(独立行政法人科学技術振興機構 地域イノベーション創出総合支援事業、重点地域研究開発推進プログラム(育成研究)、産業技術力強化法第19条の適用を受けるもの)
【出願人】(504145342)国立大学法人九州大学 (960)
【出願人】(304020292)国立大学法人徳島大学 (307)
【出願人】(390029791)アロカ株式会社 (899)
【Fターム(参考)】
[ Back to top ]