
酸化チタン膜および酸化チタン膜形成方法
【課題】酸化チタン膜および酸化チタン膜形成方法を提供すること。
【解決手段】本発明の酸化チタン膜は、表面被膜として使用され、1μm以下のミクロポア構造と、1μmを超えるマクロポア構造を有し、マクロポア構造の空孔率が、酸化チタン膜の表面に対する面積率で0.02以上とされている。また、本発明では、マクロポア構造の空孔率は、0.06〜0.4とされることが好ましい。さらに本発明は、4級アンモニウム化合物を含む電解液にチタンを含むアノードを浸漬し、アノードをプラズマ電解酸化してアノード表面に酸化チタン膜を形成する工程を含む、酸化チタン膜形成方法を低供する。
【解決手段】本発明の酸化チタン膜は、表面被膜として使用され、1μm以下のミクロポア構造と、1μmを超えるマクロポア構造を有し、マクロポア構造の空孔率が、酸化チタン膜の表面に対する面積率で0.02以上とされている。また、本発明では、マクロポア構造の空孔率は、0.06〜0.4とされることが好ましい。さらに本発明は、4級アンモニウム化合物を含む電解液にチタンを含むアノードを浸漬し、アノードをプラズマ電解酸化してアノード表面に酸化チタン膜を形成する工程を含む、酸化チタン膜形成方法を低供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、表面改質技術に関し、より詳細には、本発明は、アパタイト形成能を有する酸化チタン膜および酸化チタン膜形成方法に関する。
【背景技術】
【0002】
チタンおよびチタン合金は、軽量で生体適合性を有していること、および酸化物が光触媒活性を有することなどの点から注目されている金属材料である。チタンの性質のうちでも生体適合性は、歯科医療、整形外科医療などの医療分野で実用化している。しかしながらチタンおよびチタン合金は、その表面が生体不活性であるため、生体組織に適合するのに長い時間を要してしまうという問題点があることが知られている。
【0003】
このため、チタン及びチタン合金をインプラントとして、そのまま治療に利用すると、チタンと骨組織との間の密着強度が増大するまでに長期間を要し、この結果、埋設したインプラントがゆるんでしまうといった問題が発生する場合もあった。チタンおよびチタン合金の生体活性を向上させるため、チタン表面の改質が行われており、従来、生体適合性を付与するため、チタンまたはチタン合金の表面の改質が行われている。
【0004】
チタンの生体活性を付与する表面改質方法としては、アパタイトまたは金属酸化物などの酸化物材料をチタン表面に付着させる方法を挙げることができる。アパタイトをチタンに付着させるため、従来では、プラズマ溶射法、フレーム溶射法などの溶射法、ゾルゲルコーティング法などが使用されている。溶射法は、被覆材料粉末を高温ガス流中に存在させ、高温ガス流とともにチタン表面に衝突させて付着させることで、生体活性が付与されているが、それぞれ次のような問題点があることが指摘されている。
【0005】
プラズマ溶射法およびフレーム溶射法では、アパタイトの高温処理を伴うことからアパタイトが部分的に熱分解してしまい、形成されたアパタイト膜を水熱処理などにより後処理し、アパタイトを再結晶化する必要がある。また、溶射法を使用する場合、チタン基材と形成されたアパタイト膜との間の密着性が低いという別の問題点もある。
【0006】
酸化チタンのゾルゲルコーティング法は、チタンアルコキシドなどの酸化チタン前駆体を使用し、ゾルゲル法を利用して酸化チタンをチタン基材コーティングする方法である。形成さる酸化チタン膜は、形成段階から湿式で被膜化され、初期から高い生体活性を示すことが知られている。しかしながら、ゾルゲル法で生成された酸化チタン膜は、チタン基板との密着性が非常に低いという問題点があることが知られている。
【0007】
さらに、生体活性を付与する目的では使用されていないが、酸化チタン粉末を各種の有機バインダで調整した塗料とし、塗料でチタン基材を塗布する方法も知られている。塗布法は、基材に対し、光触媒活性を付与する方法として汎用されている。塗布法で形成される被膜は、酸化チタンが有機バインダに埋もれた構造になるため、光触媒活性点を表面に露出させる効率が悪い、光触媒活性によって有機バインダが分解する、などの問題点があり、また酸化チタン粉末の人体に対する影響も無視できないものとなる。
【0008】
チタンおよびチタン合金に対して表面コーティングする技術として特許文献1〜3および非特許文献1に示すように各種の検討が行われている。例えば、特開2008−80102号公報(特許文献1)では、インプラント材の表面に光触媒活性および生体緩和性を有する金属酸化物層を有するインプラントが記載されている。金属酸化物層は、陽極酸化法またはプラズマソースイオン注入成膜法で形成されており、短期間での生体適合性を可能とするインプラントが提供されている。また、特開2005−240139号公報(特許文献2)では、陽極電界酸化処理を使用してアナターゼ型酸化チタン被膜の形成が記載されている。さらに、特開2003−129290号公報(特許文献3)では、陽極酸化法を使用してルチルを含む光触媒用チタン陽極酸化被膜の形成が開示されている。そして、Cuiら(非特許文献1)では、陽極酸化処理によってチタン基板の表面に酸化チタンを形成する方法を記載している。また、生体活性セラミックス表面での骨類似水酸アパタイト層の形成を、in vitroで再現する方法としてKokubo等(非特許文献2)には、SBFが知られている。
【0009】
特許文献1および非特許文献1では、基材の生体活性を改善するために酸化チタンが陽極酸化法を使用して形成されているものの、さらに、基材との密着性に優れ、アパタイト形成能の高い酸化チタン被膜および当該酸化チタン被膜の効率的な形成方法が必要とされていた。また、特許文献2および特許文献3は、酸化チタン被膜を陽極酸化法を使用して形成する点を開示するものの、密着性およびアパタイト形成能の改善および製造効率の改善を課題とするものではない。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】特開2008−80102号公報
【特許文献2】特開2005−240139号公報
【特許文献3】特開2003−129290号公報
【非特許文献】
【0011】
【非特許文献1】DENTAL MATERIALS, Vol. 25 (2009), pp.80-86
【非特許文献2】Kokubo,T., Kushimatu, H., Sakka, S., Kitsugi, T. and Yamamuro, T. J. Biomed. Res., 24,721-734 (1990)
【発明の概要】
【発明が解決しようとする課題】
【0012】
上述したように、チタンおよびチタン合金は生体材料として実用化しているが、その表面は生体不活性であるため、各種の問題があった。また、従来の湿式成膜法ではチタンおよびチタン合金の基材と酸化物被膜との間の密着性が充分ではないという問題もあった。
【0013】
本発明は、上記従来技術の問題点に鑑みてなされたものであり、チタンおよびチタン合金の表面に対し、高いアパタイト形成能および触媒活性を有する新奇な酸化チタン膜および酸化チタン膜形成方法を提供することを目的とする。
【課題を解決するための手段】
【0014】
本発明では、酸化チタン被膜を形成するために、プラズマ電解酸化法(以下、PEO法として参照する。)を使用して酸化チタン基材の表面に酸化チタン被膜を形成する。本発明では、酸化皮膜に絶縁破壊電圧以上の電圧を印加してスパークを発生させ、生成するスパークにより基板表面を瞬間的に高温で加熱して改質しながら陽極酸化することにより早い成膜速度で、かつ強い密着強度を持った酸化チタン皮膜を形成する。
【0015】
さらに、陽極酸化における電解質として、酢酸アンモニウムを使用する場合、触媒活性を有する他、高いアパタイト形成能を示す酸化チタン被膜を形成することができ、生体活性の高い酸化チタン被膜を含むチタンまたはチタン合金材料が形成できる。
【0016】
すなわち、本発明によれば、表面被膜として使用される酸化チタン膜であって、前記酸化チタン膜は、1μm以下のミクロポア構造と、1μmを超えるマクロポア構造を有し、前記マクロポア構造の空孔率が、前記酸化チタン膜の表面に対する面積率で0.02以上である、酸化チタン膜が提供される。
【0017】
本発明では、前記マクロポア構造の空孔率が0.06〜0.4でることが好ましい。さらに本発明では、前記酸化チタン膜の膜厚が、0.6μm〜5μmであり、質量比でアナターゼをルチルよりも多く含有することが好ましい。また、本発明では、前記酸化チタン膜は、電解質として下記構造式(1)で与えられる4級アンモニウム化合物を使用するプラズマ電解酸化法により形成されることが好ましい。
【0018】
【化1】

(上記式(1)中、上記式(1)中、R1〜R3は、同一でも異なっていてもよい水素原子、アルキル基であり、R4は、水酸基または炭素数1個から4個のカルボン酸からなる群から選択される基である。)
また、本発明では、前記4級アンモニウム化合物は、酢酸アンモニウム塩であることが好ましい。さらに、本発明では、前記酸化チタン膜のアパタイト形成能が、前記酸化チタン膜の表面被覆速度として、0.018/day以上であることが好ましい。また、本発明では、前記酸化チタン膜は、インプラントの表面被膜または光触媒膜とすることが好ましい。
【0019】
本発明では、酸化チタン膜を陽極酸化により形成する酸化チタン膜形成方法であって、
4級アンモニウム化合物を含む電解液にチタンを含むアノードを浸漬し、前記アノードをプラズマ電解酸化して前記アノード表面に質量比でアナターゼをルチルよりも多く含有する酸化チタン膜を形成する工程を含む、酸化チタン膜形成方法が提供される。
【0020】
本発明では、前記プラズマ電解酸化は、電解電圧を、150V〜250Vの繰り返しパルスを印加して行うことができる。また、本発明では、インプラントの表面被膜または光触媒膜として使用される前記酸化チタン膜を製造することが好ましい。
【図面の簡単な説明】
【0021】
【図1】本実施形態の酸化チタン膜を形成するためのプラズマ電気分解装置を示した図。
【図2】本発明の酸化チタン膜のサブミクロンオーダーのミクロポア構造の電界効果電子顕微鏡写真。
【図3】図2に示したミクロポア構造について白黒2値化前後の画像イメージを示した図。
【図4】本発明の酸化チタン膜のマクロポア構造のFE−SEM写真。
【図5】図4に示したマクロポア構造の与えるモルフォロジーを画像解析し、マクロポア構造の空孔率を、酸化チタン膜厚に対するプロット。
【図6】代表的な本発明の酸化チタン膜および比較例(電解質:リン酸ナトリウム)のモルフォロジーを示したFE−SEM写真。
【図7】図6に示したマクロポア構造のポア分布を、画素ビットの白黒ハーフトーンの輝度レベル分布として示した図。
【図8】本発明の酸化チタン膜のXRDパターンを示した図。
【図9】本発明による酸化チタン膜のアパタイト形成能を示した図。
【図10】本発明により得られた図9に示した酸化チタン膜に対するアパタイトの沈着性を、アパタイトによる酸化チタン膜の被覆率としてプロットした図。
【図11】本発明による酸化チタン膜の光触媒能を示した図。
【発明を実施するための形態】
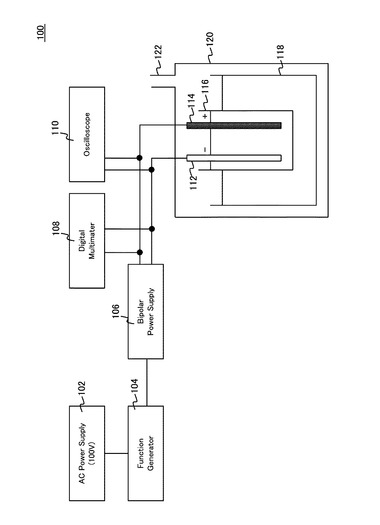
【0022】
以下、本発明を実施形態をもって説明するが、本発明は、後述する実施形態に限定されるものではない。図1は、本実施形態の酸化チタン膜を形成するためのプラズマ電気分解装置の概略図である。図1に示した製造装置は、いわゆるプラズマ電解酸化(以下、PEO:Plasma Electrolysis Oxidizationとして参照する。)法により、陽極酸化を行って、アノード電極表面に陽極酸化被膜を形成する。図1のプラズマ電気分解装置100は、電解セル116内に電解液を貯留しており、電解液内にカソード112と、アノード114とが浸漬されていて、アノード114上で水の電解によって生じた酸素(原子状酸素または酸素分子)により、アノード114を構成する材料の表面を酸化する。
【0023】
カソード112を形成する材料としては、当方性黒鉛などを使用することができる。また、アノード114を形成する材料としては、金属チタンまたはチタン合金を使用することができる。アノード114を構成する材料は、例えばインプラントなどの形状に予め形成しておき、電解液中に浸漬してPEO法を適用することにより、高速で陽極酸化膜として酸化チタン膜を形成することができる。電解液としては、本実施形態で高いアパタイト形成能を得るためには、下記一般式(1)で与えられる4級アンモニウム化合物を使用する。
【0024】
【化2】
上記式(1)中、R1〜R3は、同一でも異なっていてもよい水素原子、アルキル基であり、R4は、水酸基または炭素数2個から4個のカルボン酸から得られる陰イオン基、またはこれらのいかなる混合物からなる群から選択することができる。
【0025】
4級アンモニウム化合物は、水溶液を形成するために充分な溶解性を有し、溶解時の水素イオン濃度(pH)が、7.0以上となることが、チタンまたはチタン合金を陽極酸化するためには好ましい。本実施形態で使用することができる4級アンモニウム化合物としては、アンモニア水、酢酸アンモニウム、プロピオン酸アンモニウム、酪酸アンモニウムを挙げることができ、溶解度およびpHの観点から4級アンモニウム化合物は、4級アンモニウム塩とすることが好ましく、溶解度および取扱性の点から酢酸アンモニウムを最適な実施例として挙げることができる。また、本発明では、アパタイト形成能を調整するために、上述した4級アンモニウム塩をその溶解度の範囲で適宜混合して使用することができる。
【0026】
電解液の濃度は、0.1mol/L〜飽和濃度とすることができるが、本実施形態で、酸化チタン膜の形成効率および電解の安定性などを考慮すると、0.3mol/L〜2mol/L程度が好ましく、さらに、本発明で、生体内にインプラントする点を考慮し、ルチル型の結晶構造を有する酸化チタンの形成を抑制する点では、0.3mol/L〜1.5mol/Lの範囲とすることが好ましく、アパタイト沈着性を考慮する場合、0.3mol/L〜0.8mol/Lの範囲が最も好ましい。
【0027】
電解セル116は、冷却システム118内に配置され、電解液の温度が、概ね室温となるように制御されている。冷却システム118は、さらに電解容器120に収容され、電解容器120には、発生する気体をパージするパージ配管122が接続されていて、酸素ガス、水素ガスなどの電気分解生成ガスを処理系に送っている。カソード112およびアノード114には、パルス電源システムを介して直流パルス電流が供給されている。パルス電源システムは、交流電源102と、パルスを発生するためのファンクションジェネレーター104と、2極電圧発生装置106とを含んで構成されている。ファンクションジェネレーター104は、本発明においてアノード近傍にスパークを発生させることが可能なパルス波形を生成している。パルス波形は、本発明では、周波数、0.1〜3kHz、デューティ比5%〜50%程度とすることができ、より好ましい実施形態では、PEO法の制御および成膜速度の点で、周波数を約1kHzとし、デューティ比を約10%とすることができる。
【0028】
高圧バイポーラ電圧発生装置106の出力は、それぞれカソード112およびアノード114に接続され、電気分解のための正負電圧が印加される。カソード112およびアノード114に印加される電圧および電流値は、デジタルマルチメーター108によりモニタされて、電圧および電流値の制御にフィードバックされる。また、パルス波形は、オシロスコープ110によりモニタされる。
【0029】
アノード114で電気分解が発生すると、初期には、フレッシュな金属表面が電解液に露出しているため、過電圧は加えられることないので電圧が高くともスパークが発生することなく電気分解が進行する。電気分解の進行につれ電気分解により発生した酸素原子、酸素イオン過酸化物などによりアノード114の酸化が進行して行き、表面に酸化チタン膜が生成される。純粋な酸化チタンは、誘電体なので、パルス電源システムから供給される電荷は、誘電体によりアノード114の表面に蓄積し、絶縁破壊電圧を超えると、付近のアノード114の表面に向けて電子雪崩を生じさせ、これがアノード上で火花放電を生じさせる。高エネルギーの火花放電が、アノード114の表面に生成したチタン表面または酸化チタン層を攻撃し、酸化チタン膜を改質しながら電解酸化膜として酸化チタンを形成する。
【0030】
以上のメカニズムによりPEO法は、高速にアノード114の表面酸化処理を行うことができ、また、通常の電解酸化膜よりも強固な膜質を電解酸化膜に対して与えることができる。本発明で好ましい電気分解条件としては、電気分解電圧が250Vの場合、電解酸化処理の時間を、約5min〜約20min程度とすることができる。本発明の好ましい電解酸化においては、電気分解電圧を250V、パルス繰り返し周波数を、1kHzとした場合、約15minの電気分解処理で、膜厚を、約0.6μm〜約5μmとする膜形成速度とすることができる。さらに、充分なアパタイト形成能および光触媒能、並びに被膜強度を得る目的でいえば約1μm〜約5μmの電気分解速度とすることが、PEOの制御および良好な特性の酸化チタン膜を形成できるのでより好ましい。また、電気分解電圧は、本発明では、50V〜300Vの範囲で可変とすることができ、酸化チタン膜の形成効率の点から言えば、100〜300Vの範囲でPEOを行うことができ、最も好ましくは、酸化チタン膜の形成速度およびPEO処理プロセスの制御性の観点から、150V〜250Vの範囲とすることができる。
【0031】
本発明の酸化チタン膜は、酸化チタンがアナターゼとルチルの結晶系の混合した形態として形成される。本発明のPEO処理では、アナターゼとルチルとの結晶の生成割合は、電解質の濃度により変化することが見出された。本発明により形成される酸化チタン膜のアナターゼ/ルチルの比は、X線散乱(以下、XRDとして参照する。)の回折パターンのピーク比で表現した場合、電解質濃度が、0.1mol/L〜1.5mol/Lの範囲で、0.6以上のアナターゼ/ルチル質量比とすることができる。一方、生体適合性に関していえば、ルチルをできるだけ低く抑制することが好ましいので、本発明による酸化チタン膜は、アナターゼ/ルチル質量比を1〜1.5の範囲とすることが最も好ましい。
【0032】
また、本発明では、基材上に形成される酸化チタン膜は、酸化チタン膜の膜厚に対応してサブミクロンオーダーのミクロポア構造と、数ミクロンオーダーのマクロポア構造とを有する。本発明の酸化チタン膜のサブミクロンオーダーのミクロポア構造の電界効果型電子顕微鏡(以下、FE−SEMとして参照する。)写真を図2に示す。図2(a)が、FE−SEMによる、倍率5000で撮影したミクロポア構造を示し、図2(b)が、倍率15000とした場合のより詳細なミクロポア構造を示す。本発明のミクロポア構造は、図2に示すように、数100nmオーダーのポアが、比較的緊密な酸化チタンブリッジにより相互連結されて形成されている。
【0033】
図2に示したミクロポア構造を画像解析して、図2に示したミクロポアの空孔率を見積もることができる。図3は、図2のミクロポア構造を画像解析した場合の画像イメージである。画像解析は、画像解析ソフトウェアであるPhotoshop(Photoshopは、Adobe Co.Ltd.の登録商標である。)を使用し、画像を8ビットダイナミックレンジの白黒2値化処理を行い、白黒2値化処理された後の画像のうち、レベル=0(ブラック)の属性を有する画素ビットの数を、全体画素ビット数に対する比として得た。
【0034】
図3に示した画像イメージは、白黒2値化前後の画像である。図3(a)が、図2(a)に対応する画像であり、図3(b)が、図2(a)の画像を白黒2値化したときの白黒2値化画像である。図3に示されるようにミクロポア構造は、サブミクロンオーダーで形成され、画像面積比での空孔率は、約20.3%程度であることがわかった。なお、空孔率について、白黒2値化処理のしきい値ビットを変化させることで、オーバーエスティメートとアンダーエスティメートの場合について平均した値である。また、2値化レベルのオーバーエスティメートとアンダーエスティメートとの間では、面積率は、±3.5%変動したに過ぎず、空孔率として充分ロバストな結果として得られた。
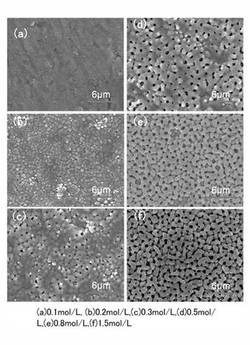
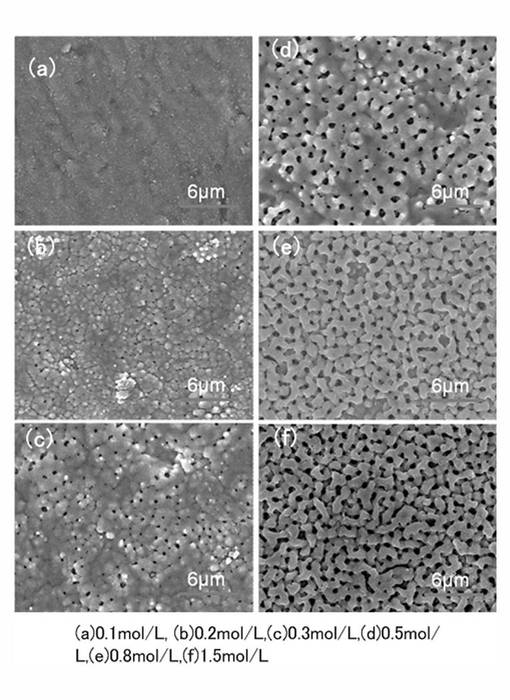
【0035】
さらに、本発明の酸化チタン膜は、図2および図3に示したミクロポア構造に加え、マクロポア構造を有する。図4は、本発明の酸化チタン膜のマクロポア構造のFE−SEM写真を示す。図4に示すマクロポア構造(a)〜(f)は、電解質を酢酸アンモニウムとし、電解質濃度を、0.1mol/L(a)、0.2mol/L(b)、0.3mol/L(c)、0.5mol/L(d)、0.8mol/L(e)、1.5mol/L(f)とし、陽極酸化電圧250Vで、15minの陽極酸化を行って形成されたものである。図4に示されるように、本発明では、電解液濃度が増加するにつれて、マクロポア構造が順次発達し、明確に形成されて行く。
【0036】
また、マクロポア構造のポア特性は、膜厚が薄い場合には、概ね円形として形成されるが、膜厚の増加にともなって、アスペクト比の大きなポア構造が成長する、このマクロポア構造のポアを、長径と短径とを有するポアとして形成される。この理由は明確ではないが、例えば、電解質の濃度により電解速度が増加しその結果、酸化チタンの形成速度が増加することで、絶縁破壊による火花放電の発生頻度が変化し、ミクロポア構造に対して施される放電処理の程度が高まるPEO法の特性によるものと推定される。
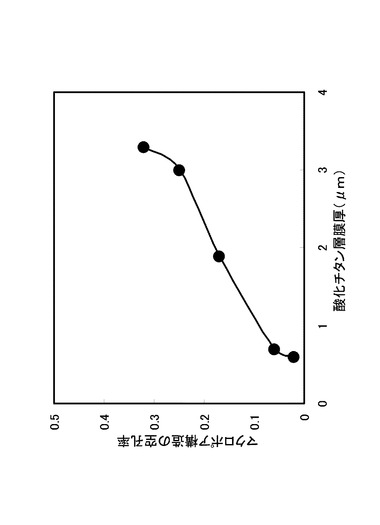
【0037】
図4に示したマクロポア構造の与えるモルフォロジーを、図3で説明したと同一の方法で画像解析し、マクロポアの存在割合(以下、マクロポア構造の空孔率(マクロポア構造の空孔率は、全面がマクロポアである場合にマクロポア率=1がとなる値である。)として参照する。)を、そのときの酸化チタン膜厚に対して行ったプロットを図5に示す。図5に示すプロットは、マクロポア構造の空孔率を縦軸とし、酸化チタン層膜厚(μm)を横軸として示したプロットである。図5に示すように、酸化チタン層の膜厚が増加するにつれて、マクロポア構造の空孔率が増加していることが示されている。また、本発明では、アパタイト形成能および光触媒能の両方について、酸化チタン膜の膜厚、マクロポア構造の空孔率への依存性がみられ、酸化チタン膜の膜厚およびマクロポア構造の発達に伴い、アパタイト形成能および触媒活性が向上する傾向が見出された。
【0038】
なお、本発明の酸化チタン膜のアパタイト形成能は、SBF法を使用することにより判断することができる。SBF(Simulated Body Fluid: SBF)法は、SBFを使用して、基材の生体活性を判断する方法である。SBFは、細胞やタンパク質のような有機物を含まず無機イオン濃度だけをヒトの細胞外液のそれにほぼ等しくして、pHをトリス緩衝液で7.25または7.4に調整した水溶液である。なお、SBFの詳細については、例えば、非特許文献2に詳細が記載されており、SBF法により体内における生体活性セラミックス表面での骨類似水酸アパタイト層の形成を、in vitroで再現する方法である。本発明では、SBF法を使用してアパタイト沈着性を検討するにあたり、SBF濃度の調整および酸化チタン膜のSBF溶液に対する浸漬時間を変化させてアパタイト形成能を評価することができる。
【0039】
本発明の酸化チタン膜について、SBF法により評価したところ、本発明により形成された酸化チタン膜は、6日間で充分な被覆率で表面にアパタイトを沈着させる。本発明の酸化チタン膜のアパタイト形成能は、FE−SEMによる目視観測の他、FE−SEMの画像を使用し、酸化チタン膜がアパタイトで被覆される速度を、例えば酸化チタン膜の被覆速度として評価することができる。本発明の酸化チタン膜は、1.0SBFモードで試験を使用した評価で、酸化チタン膜を生成する際の電解質濃度に依存し、約0.018/day〜0.122/dayの範囲の析出速度を示す。なお、本発明では、アパタイトの析出は、酸化チタン膜が完全にアパタイトで被覆された後も、発生し、SBFへの浸漬期間とともにアパタイトの生成が継続する。このため、本発明の酸化チタン膜は、インプラント表面の表面コーティングに適用した場合、迅速に生体に対して適合し良好な表面コートを与えることができる。
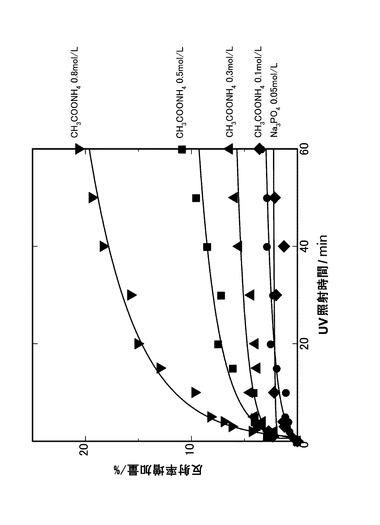
【0040】
また、本発明の酸化チタン膜の光触媒活性は、光照射下での、色素の分解速度により評価することができる。本発明の光触媒活性の評価は、酸化チタン膜上を酸化状態により光吸収性を変化させる染料で着色し、着色した酸化チタン膜をUV照射して色素の退色を評価することにより、酸化チタン膜間の相対的な光触媒活性として評価する。
【0041】
本発明の酸化チタン膜は、良好な結晶性を有しており、良好な光触媒活性を示すことが見出された。本発明では、アパタイト形成能および触媒活性は、酸化チタン膜の膜厚に伴って増加する傾向が観測された。本発明で、良好なアパタイト形成速度を得る目的からは、酸化チタン膜の膜厚は、0.6μm〜10μmとすることができ、より好ましいアパタイト形成速度を得る点では、約1μm〜5μmとすることができ、製造プロセスの制御性の観点では、約1μm〜5μmとすることが好ましい。
【0042】
本発明の酸化チタン膜の膜厚は、電解質として使用する酢酸アンモニウムの濃度上昇に従って増加する。酢酸アンモニウムを電解質として形成した酸化チタン膜は、XRD測定の結果から、皮膜内に電解質成分がほとんど取り込まれておらず、高い結晶性を有することが見出された。一方、酢酸アンモニウム溶液の濃度とともに酸化チタン膜のルチル形成量が増加し、相対的にアナターゼが減少する。この理由は、種々想定できるが、1つの要因としては、PEO処理の進行に伴って火花放電によってアナターゼからルチルへの熱転位温度915±15℃以上に被膜が局所加熱されることも関連するものと考えられる。
【0043】
また、本発明により得られる酸化チタン膜は、電解質としてリン酸ナトリウムを使用した場合に比較して早期に火花放電が停止する現象が観測された。この理由は、本発明の酸化チタン膜は、高純度の酸化チタンとして形成され、この結果抵抗が高い良質の酸化チタン膜が形成され、早期の絶縁膜形成を促進する結果と推定される。
【0044】
以下、本発明について、詳細な実施例をもって説明するが、本発明は、以下に説明する実施例に限定されるものではない。
【実施例】
【0045】
[材料]
電極材料としては、陽極として3×10×40〜80mm3に切り出した純チタン板(株式会社ニラコ製、99.5%、3×200×400mm)を使用した。陰極を構成する電極材料としては、等方性黒鉛板(東海カーボン株式会社製、3×10×80mm)を用いた。等方性黒鉛板は、耐水研磨紙(#2000)で研磨し、アセトン洗浄した後乾燥させた。当該等方性黒鉛板を絶縁塗料(古藤産業株式会社製、フロンマスクMA−1)でマスキングし、10mm×10mmの開口を形成して電解液に露出させてカソードとした。
【0046】
電解液としては、実施例では、酢酸アンモニウム水溶液(和光純薬工業株式会社製;特級)を使用した。また、比較例の電解質として、リン酸ナトリウム(和光純薬工業株式会社製;CAS#7601-54-9)を使用した。各電解液を、0.05〜1.5mol/Lの濃度に調整し、実施例および比較例の電解液とした。
【0047】
[PEO装置およびPEO条件]
PEO装置は、図1の構成として、電解セルとしては、陽極酸化装置本発明のPEO処理に適するように設計された陽極酸化装置を用いた。交流安定化電源として、EC100S(エヌエフ回路設計ブロック株式会社製)、ファンクションジェネレーターとして、SG−4105(岩崎通信機株式会社製)を使用し、火花放電を発生させる電圧を生成させるため、高速バイポーラ電源(エヌエフ回路設計ブロック製:HSA4052)を高圧電源として使用した。
【0048】
ファンクションジェネレーターは、任意のパルス波形を生成し、初期電圧2.5Vまでの可変電圧のパルスを出力する。パルス周波数は、1kHzとした。高速バイポーラ電源は、ファンクションジェネレーターの直流出力パルスを電圧増幅する。電極間の実効電圧値は、オシロスコープ(岩通計測株式会社製、DS−8822P)を使用してモニタし、電極間を流れる電流の経時変化をデジタルマルチメーター(岩通計測株式会社製、VOAC21)により記録した。電解セル内はPEOによって発生する火花放電によって加熱されるため、電解セルを恒温水槽(アズワン株式会社製、TR−3A)および冷却水循環装置(旭テクノグラス株式会社製、CLUMINI2型を用いて冷却した。
【0049】
また、電極を電解セル内の電解液に浸る様に固定し、電解セルを排気口を除き密閉して電圧を印加することにより、PEO処理を施した。電解処理時間を3sec〜15minとし、PEO処理を介した後15min後、電極を取り出し、アセトン洗浄超音波洗浄して、デシケーター中に保存した。電気分解電圧は、150V〜250V、電解液濃度(0.05〜1.5mol/L)の範囲に設定した。
【0050】
結晶相・結晶性の同定は、X線回折により行った。X線回折測定は、X線回折装置(理学電機株式会社製、RINT2000)を用いた。使用したX線源は、CuKα線とし、X線発生条件は、管球電圧:40kV、走査速度:1deg・min、管球電流:200mAであった。
【0051】
生成膜の微構造の観測は、FE−SEMにより行った。FE−SEMとしては、株式会社日立製作所製S4500を使用し、観察条件を、加速電圧15kV、ワーキングディスタンス(W.D.)を15mmとした。また、観察試料は、イオンスパッタ装置(株式会社日立製作所製、E−1030)を用いて、白金−パラジウムコーティングを、30sとした。生成した酸化チタン膜の膜断面観察は、試料を、FIB(株式会社日立ハイテクノロジーズ、FB−2100)を使用し断面観察用試料を作製し、FE−SEMを使用して観測した。断面観察資料は、酸化チタン膜を部分的に生成したチタン基板を、酸化チタン膜を切断する方向にGaイオンビーム照射し、酸化チタン膜およびチタン基板をイオンビームエッチングすることで、矩形ノッチを生成させて作成した。その後、形成した矩形ノッチに隣接する酸化チタン膜の断面を、FE−SEMで観測した。各観察結果は、デジタルカメラにより画像イメージとして取得した。
【0052】
[SBFの調整および評価方法]
疑似体液(SBF)は、ヒトの血漿の無機成分とほぼ等しい濃度となるように調整し、その組成を下記表1示す。
【0053】
【表1】
【0054】
上記組成のSBFは、1000mLのポリエチレンビーカーに約700mLの蒸留水と攪拌子とを入れ、それをマグネティックスターラー上に置いた恒温槽中に設置し、37℃でに攪拌した。この蒸留水に上記表の試薬を順次加えた。試薬は一度に複数投入せず、一つの試薬が完全に溶解した後で投入した。溶液は、37℃でpH7.4になるように塩酸を加えて調整し、最後に1000mLメスフラスコに移して蒸留水を加え、1Lとなるように調整した。
【0055】
生体活性は、作製した試料の疑似体液(SBF)浸漬によるアパタイト沈着をFE−SEMによる観測により評価した。試験条件は、作製した酸化チタン膜を37℃で、30mLのSBFに6、9、12日間浸漬し、試験終了後、資料を蒸留水でよくすすぎ、室温で乾燥後、FE−SEMおよびラマン分光測定によってアパタイトの形成を検討した。
【0056】
ラマン分光は、愛宕物産株式会社/Jovin Yvon社製(T64000)のトリプルモノクロメーターとCCDディテクターとを用い、測定範囲をレーザ波長の波長から10000m−1〜120000m−1として測定を行った。なお、レーザラマン散乱を生じさせるためのレーザ光源としては、アルゴンイオンレーザの波長514.5nmを使用した。
【0057】
[光触媒能]
光触媒能を、酸化チタン膜による染料の光学特性の変化を用いて評価し、酸化チタン膜を、酢酸アンモニウム溶液を電解液とし、その濃度を0.1〜0.8mol/Lの範囲で変化させて調整した。染料としてメチレンブルーを使用し、生成した酸化チタン膜をメチレンブルーで着色し、試料表面をUV照射し、光触媒反応によるメチレンブルーの分解で回復する反射率の変化を分光測色計(株式会社コニカミノルタセンシング製CM−2500d)で観測して、光触媒活性を評価した。
【0058】
[結果]
上述した条件を使用し、実施例として酢酸アンモニウムを電解液として使用し、比較例として、リン酸ナトリウムを電解質とした場合について、PEO法を適用して酸化チタン膜を生成し、酸化チタン膜のモルフォロジー、アパタイト形成能、および光触媒能について検討を加えた。
【0059】
I.膜モルフォロジーの評価
本発明により形成された酸化チタン膜の膜モルフォロジーを、電解液を酢酸アンモニウムとした場合(実施例)と、電解液をリン酸ナトリウムとした場合(比較例)について評価した。PEO条件は、酢酸アンモニウムを電解質とした場合、電解質濃度を、0.1mol/L、0.3mol/L、0.8mol/L(3条件)とし、電解電圧=250V、パルス周波数=1kHz、PEO処理時間=15minとした。また、比較例では電解質をリン酸ナトリウムとし、電解質濃度を、見かけ上の酸化チタン膜の膜厚が同じオーダーとなる濃度の0.2mol/Lとし、電解電圧=250V、パルス周波数=1kHz、PEO処理時間15minとしてそれぞれ酸化チタン膜を形成し、FE−SEMにより膜モルフォロジーの比較を行った。
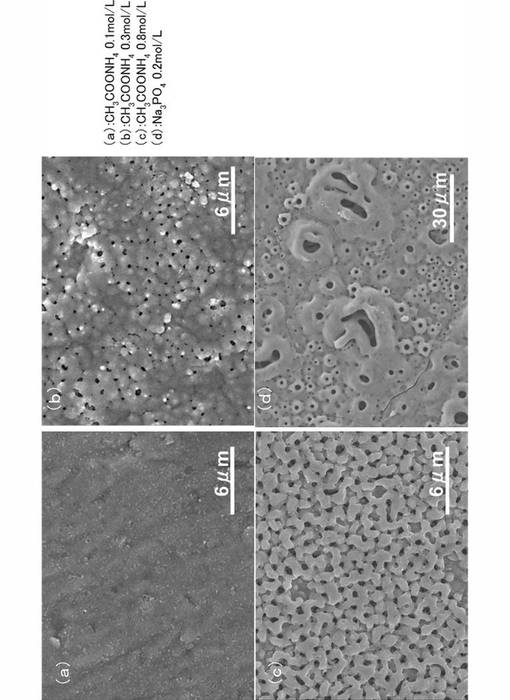
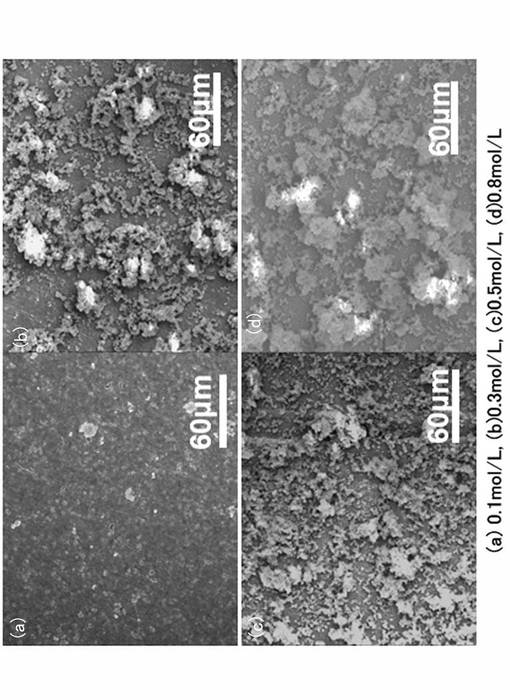
【0060】
図6に代表的な酸化チタン膜のモルフォロジーを示す。図6に示したFE−SEM写真は、2500倍である。図6(a)〜図6(c)が実施例であり、図6(d)が比較例である。図6に示すように、本発明により形成された酸化チタン膜は、電解質の濃度増加に伴いマクロポア構造が順次発達しており、膜厚は、それぞれ、図6(a)で0.6μm、図6(b)で、0.7μm、図6(c)で3.0μmである。なお、図6(a)の実施形態で、PEO処理時間=15minの酸化チタン膜では、明確に目視観測できるマクロポア構造が未だ充分に発達していない。一方、比較例である図6(d)は、形成される酸化チタン膜のマクロポア構造が粗いため、平均値として約7μmと見積もられた。
【0061】
図6に示すように、本発明により形成された酸化チタン膜(図6(a)〜図6(c)は、電解質濃度の増加に伴い、良好なマクロポア構造が形成されている。一方、比較例である図6(d)の酸化チタン膜は、マクロポア構造が不均一であった。図6に示した酸化チタン膜は、PEO処理時間を15minに統一していることから、本発明では、被膜形成速度は比較例に比較して遅いということができるものの、本実施例で形成される酸化チタン膜は、良好なマクロポア構造を示した。
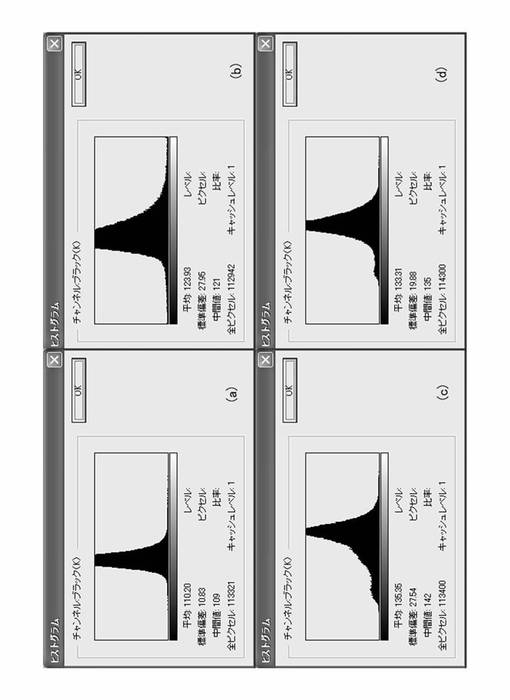
【0062】
図7は、図6に示したマクロポア構造のポア分布を、画素ビットの白黒ハーフトーン(400dpi、ダイナミックレンジ8ビット)の輝度レベル分布として示した図であり、図7(a)〜図7(d)は、それぞれ図6(a)〜図6(d)のマクロポア構造の輝度レベル分布に対応する。図7に示すように、本実施形態で明確なマクロポア構造が観測された酸化チタン膜(図6(b)、図6(c))については、輝度レベル分布の標準偏差が、それぞれ27.96および27.54という値が得られている。一方、比較例である図6(d)に対応する酸化チタン膜では、標準偏差が19.88として得られ、マクロポア構造の均質なランダムさという観点から見ると、本実施形態の酸化チタン膜の方が比較例に比してよりランダム、すなわち均一な分布のマクロポア構造を与えていることが示される。
【0063】
なお、図7(d)に示す比較例の場合、所々大きなポアが認められるものの、標準偏差が19.88として得られている。この理由は、図6(d)に示されるように、大きなポアを除けば比較的均一なポアが形成されていることから、マクロポア分布としては、本発明により形成されるマクロポア構造に比較して比較的均一であることを示すものと考えられる。
【0064】
II.膜の結晶性の評価
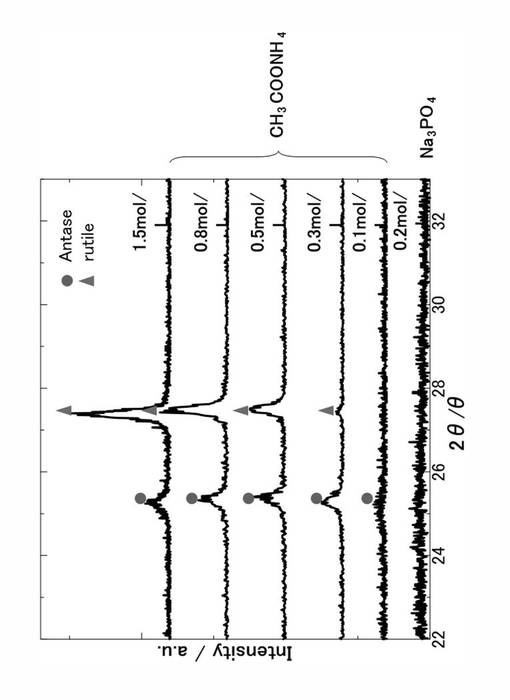
図8は、本発明の酸化チタン膜のXRDパターンを示す、電解質として酢酸アンモニウムを使用し、その濃度を、0.1mol/L〜1.5mol/Lまで変化させて得られた酸化チタン膜が実施例についてのXRDパターンであり、電解質としてリン酸ナトリウム0.2mol/Lとして形成された酸化チタン膜が比較例についてのXRDパターンである。図8に示すように、本実施形態の酸化チタン膜については、0.1mol/Lの電解質濃度であっても酸化チタンのアナターゼのピークがトレース程度であるが形成されているのが示された。
【0065】
また、電解質濃度が、0.1mol/L〜1.5mol/Lに増加するにつれて、結晶構造の割合がアナターゼからルチルへと増加しているのが観測された。アナターゼ/ルチル質量比は、電解質濃度が0.1mol/Lで約1.5であり、電解質濃度が1.5mol/Lで約0.6という結果が得られた。一方、比較例であるリン酸ナトリウムを電解質として使用して形成された酸化チタン膜では、図6で説明したように充分な膜厚の酸化チタン膜が形成されているにもかかわらず、アナターゼおよびルチルの結晶構造に由来するピークが観測されなかった。
【0066】
このため、本発明により形成される酸化チタン膜は、比較例により形成される酸化チタン膜に比較してより高結晶性の酸化チタン被膜が形成されていることが示された。
【0067】
III.アパタイト形成能の評価
図9は、酸化チタン膜のアパタイト形成能を示した図である。図9(a)〜図9(d)は、いずれも本発明により形成された酸化チタン膜について、1.0SBFを使用し、6日間、9日間、12日間の浸漬を行い、アパタイト形成能を評価した。図9に示した実施例では、いずれもSBF浸漬6日間の結果を示す。図9に示された酸化チタン膜へのアパタイト形成については、レーザラマン散乱を使用して確認し、全試料について、アパタイトが沈着していることが確認できた。
【0068】
一方、比較例として、電解液としてリン酸アンモニウム(0.2mol/L)を使用して形成した酸化チタン膜については、12日間浸漬してもアパタイトの酸化チタン膜表面への沈着は観測されなかった。
【0069】
以上のことから、本発明では、電解質としてリン酸ナトリウムを使用して形成された酸化チタン膜に比較してはるかに高いアパタイト形成能を有する酸化チタン膜が得られることがわかった。なお、図9に示す実施例について、さらに12日間までアパタイト沈着性をトレースしたが、アパタイト沈着が阻害されることなく、継続的な沈着を示し、良好なアパタイト層の形成が認められた。
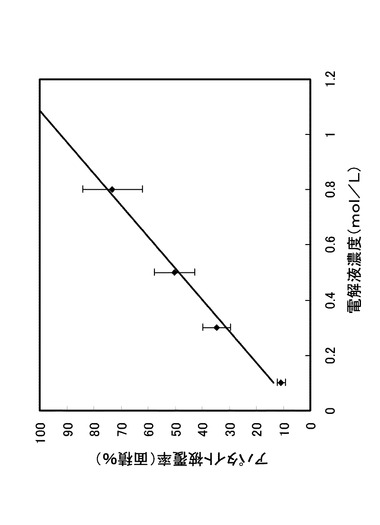
【0070】
図10は、本発明により得られた図9に示す酸化チタン膜に対するアパタイトの沈着性を、アパタイトによる酸化チタン膜の被覆率をもって示した図である。横軸は、電解液濃度(mol/L)であり、縦軸は、アパタイト被覆率(面積%)である。なお、アパタイト被覆率は、画像解析により行い、画像解析は、画像解析ソフトウェアであるPhotoshop(Photoshopは、Adobe Co.Ltd.の登録商標である。)を使用し、画像を8ビットダイナミックレンジの白黒2値化処理を行なった後、アパタイト粒子が酸化チタン膜から立体的に上側に成長し輝度の高いポイントとして観測されることから、白黒2値化処理された後の画像のうち、レベル=255(ホワイト)の属性を有する画素ビットの数を、全体画素ビット数に対する比として得た。なお、図10のプロットの誤差バーは、白黒2値化処理の相対誤差の3倍(約10%)を示す。
【0071】
図10に示すようにアパタイト沈着速度は、電解液濃度(mol/L)に対して依存性を示しており、電解液濃度が0.1mol/L〜0.8mol/Lの各ポイントについて、約0.018/day〜0.122/dayの沈着速度が得られた。
【0072】
IV.光触媒能の評価
図11には、酸化チタン膜の光触媒能についての結果を示す。図11に示すように、本発明により形成される酸化チタン膜は、電解液濃度0.1mol/Lであっても、光触媒活性を有しており、比較例である、リン酸ナトリウムを電解質として形成された酸化チタン膜に比較して相対的に高い光触媒活性を示すことがわかった。これは、本発明により形成される酸化チタン膜がより結晶性が高いことに起因するものと考えられる。また、本発明の酸化チタン膜は、良好な光触媒活性も併せ持っているので、生体内に導入する前に紫外線で簡単にかつ効率よく滅菌できる利点があり、実用上製造プロセスのプロセス工程を削減でき、製造を効率化できる。
【産業上の利用可能性】
【0073】
以上説明したように、本発明によれば、プラズマ電界酸化法により新奇なモルフォロジーを有する酸化チタン膜を形成することができ、当該酸化チタン膜は、良好なアパタイト沈着性および光触媒活性を示す。さらに本発明により形成される酸化チタン膜は、火花放電による表面処理を受け、基材に対して酸化チタン層が偏在せず、よりランダムかつ強固に形成され、より高い強度の酸化チタン陽極酸化膜を形成することができる。このため、本発明の酸化チタン膜は、インプラント材の表面被覆として高い生体活性を提供するばかりではなく、良好な光触媒層を提供することができる。
【0074】
これまで本実施形態につき説明してきたが、本発明は、上述した実施形態に限定されるものではなく、他の実施形態、追加、変更、削除など、当業者が想到することができる範囲内で変更することができ、いずれの態様においても本発明の作用・効果を奏する限り、本発明の範囲に含まれるものである。
【符号の説明】
【0075】
100…プラズマ電解酸化装置、102…AC電源、104…ファンクションジェネレーター、106…高圧バイポーラ電源、108…デジタルマルチメーター、110オシロスコープ、112…カソード、114…アノード、116…電界セル、118…冷却システム、120…電解容器、122…パージ配管
【技術分野】
【0001】
本発明は、表面改質技術に関し、より詳細には、本発明は、アパタイト形成能を有する酸化チタン膜および酸化チタン膜形成方法に関する。
【背景技術】
【0002】
チタンおよびチタン合金は、軽量で生体適合性を有していること、および酸化物が光触媒活性を有することなどの点から注目されている金属材料である。チタンの性質のうちでも生体適合性は、歯科医療、整形外科医療などの医療分野で実用化している。しかしながらチタンおよびチタン合金は、その表面が生体不活性であるため、生体組織に適合するのに長い時間を要してしまうという問題点があることが知られている。
【0003】
このため、チタン及びチタン合金をインプラントとして、そのまま治療に利用すると、チタンと骨組織との間の密着強度が増大するまでに長期間を要し、この結果、埋設したインプラントがゆるんでしまうといった問題が発生する場合もあった。チタンおよびチタン合金の生体活性を向上させるため、チタン表面の改質が行われており、従来、生体適合性を付与するため、チタンまたはチタン合金の表面の改質が行われている。
【0004】
チタンの生体活性を付与する表面改質方法としては、アパタイトまたは金属酸化物などの酸化物材料をチタン表面に付着させる方法を挙げることができる。アパタイトをチタンに付着させるため、従来では、プラズマ溶射法、フレーム溶射法などの溶射法、ゾルゲルコーティング法などが使用されている。溶射法は、被覆材料粉末を高温ガス流中に存在させ、高温ガス流とともにチタン表面に衝突させて付着させることで、生体活性が付与されているが、それぞれ次のような問題点があることが指摘されている。
【0005】
プラズマ溶射法およびフレーム溶射法では、アパタイトの高温処理を伴うことからアパタイトが部分的に熱分解してしまい、形成されたアパタイト膜を水熱処理などにより後処理し、アパタイトを再結晶化する必要がある。また、溶射法を使用する場合、チタン基材と形成されたアパタイト膜との間の密着性が低いという別の問題点もある。
【0006】
酸化チタンのゾルゲルコーティング法は、チタンアルコキシドなどの酸化チタン前駆体を使用し、ゾルゲル法を利用して酸化チタンをチタン基材コーティングする方法である。形成さる酸化チタン膜は、形成段階から湿式で被膜化され、初期から高い生体活性を示すことが知られている。しかしながら、ゾルゲル法で生成された酸化チタン膜は、チタン基板との密着性が非常に低いという問題点があることが知られている。
【0007】
さらに、生体活性を付与する目的では使用されていないが、酸化チタン粉末を各種の有機バインダで調整した塗料とし、塗料でチタン基材を塗布する方法も知られている。塗布法は、基材に対し、光触媒活性を付与する方法として汎用されている。塗布法で形成される被膜は、酸化チタンが有機バインダに埋もれた構造になるため、光触媒活性点を表面に露出させる効率が悪い、光触媒活性によって有機バインダが分解する、などの問題点があり、また酸化チタン粉末の人体に対する影響も無視できないものとなる。
【0008】
チタンおよびチタン合金に対して表面コーティングする技術として特許文献1〜3および非特許文献1に示すように各種の検討が行われている。例えば、特開2008−80102号公報(特許文献1)では、インプラント材の表面に光触媒活性および生体緩和性を有する金属酸化物層を有するインプラントが記載されている。金属酸化物層は、陽極酸化法またはプラズマソースイオン注入成膜法で形成されており、短期間での生体適合性を可能とするインプラントが提供されている。また、特開2005−240139号公報(特許文献2)では、陽極電界酸化処理を使用してアナターゼ型酸化チタン被膜の形成が記載されている。さらに、特開2003−129290号公報(特許文献3)では、陽極酸化法を使用してルチルを含む光触媒用チタン陽極酸化被膜の形成が開示されている。そして、Cuiら(非特許文献1)では、陽極酸化処理によってチタン基板の表面に酸化チタンを形成する方法を記載している。また、生体活性セラミックス表面での骨類似水酸アパタイト層の形成を、in vitroで再現する方法としてKokubo等(非特許文献2)には、SBFが知られている。
【0009】
特許文献1および非特許文献1では、基材の生体活性を改善するために酸化チタンが陽極酸化法を使用して形成されているものの、さらに、基材との密着性に優れ、アパタイト形成能の高い酸化チタン被膜および当該酸化チタン被膜の効率的な形成方法が必要とされていた。また、特許文献2および特許文献3は、酸化チタン被膜を陽極酸化法を使用して形成する点を開示するものの、密着性およびアパタイト形成能の改善および製造効率の改善を課題とするものではない。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】特開2008−80102号公報
【特許文献2】特開2005−240139号公報
【特許文献3】特開2003−129290号公報
【非特許文献】
【0011】
【非特許文献1】DENTAL MATERIALS, Vol. 25 (2009), pp.80-86
【非特許文献2】Kokubo,T., Kushimatu, H., Sakka, S., Kitsugi, T. and Yamamuro, T. J. Biomed. Res., 24,721-734 (1990)
【発明の概要】
【発明が解決しようとする課題】
【0012】
上述したように、チタンおよびチタン合金は生体材料として実用化しているが、その表面は生体不活性であるため、各種の問題があった。また、従来の湿式成膜法ではチタンおよびチタン合金の基材と酸化物被膜との間の密着性が充分ではないという問題もあった。
【0013】
本発明は、上記従来技術の問題点に鑑みてなされたものであり、チタンおよびチタン合金の表面に対し、高いアパタイト形成能および触媒活性を有する新奇な酸化チタン膜および酸化チタン膜形成方法を提供することを目的とする。
【課題を解決するための手段】
【0014】
本発明では、酸化チタン被膜を形成するために、プラズマ電解酸化法(以下、PEO法として参照する。)を使用して酸化チタン基材の表面に酸化チタン被膜を形成する。本発明では、酸化皮膜に絶縁破壊電圧以上の電圧を印加してスパークを発生させ、生成するスパークにより基板表面を瞬間的に高温で加熱して改質しながら陽極酸化することにより早い成膜速度で、かつ強い密着強度を持った酸化チタン皮膜を形成する。
【0015】
さらに、陽極酸化における電解質として、酢酸アンモニウムを使用する場合、触媒活性を有する他、高いアパタイト形成能を示す酸化チタン被膜を形成することができ、生体活性の高い酸化チタン被膜を含むチタンまたはチタン合金材料が形成できる。
【0016】
すなわち、本発明によれば、表面被膜として使用される酸化チタン膜であって、前記酸化チタン膜は、1μm以下のミクロポア構造と、1μmを超えるマクロポア構造を有し、前記マクロポア構造の空孔率が、前記酸化チタン膜の表面に対する面積率で0.02以上である、酸化チタン膜が提供される。
【0017】
本発明では、前記マクロポア構造の空孔率が0.06〜0.4でることが好ましい。さらに本発明では、前記酸化チタン膜の膜厚が、0.6μm〜5μmであり、質量比でアナターゼをルチルよりも多く含有することが好ましい。また、本発明では、前記酸化チタン膜は、電解質として下記構造式(1)で与えられる4級アンモニウム化合物を使用するプラズマ電解酸化法により形成されることが好ましい。
【0018】
【化1】
(上記式(1)中、上記式(1)中、R1〜R3は、同一でも異なっていてもよい水素原子、アルキル基であり、R4は、水酸基または炭素数1個から4個のカルボン酸からなる群から選択される基である。)
また、本発明では、前記4級アンモニウム化合物は、酢酸アンモニウム塩であることが好ましい。さらに、本発明では、前記酸化チタン膜のアパタイト形成能が、前記酸化チタン膜の表面被覆速度として、0.018/day以上であることが好ましい。また、本発明では、前記酸化チタン膜は、インプラントの表面被膜または光触媒膜とすることが好ましい。
【0019】
本発明では、酸化チタン膜を陽極酸化により形成する酸化チタン膜形成方法であって、
4級アンモニウム化合物を含む電解液にチタンを含むアノードを浸漬し、前記アノードをプラズマ電解酸化して前記アノード表面に質量比でアナターゼをルチルよりも多く含有する酸化チタン膜を形成する工程を含む、酸化チタン膜形成方法が提供される。
【0020】
本発明では、前記プラズマ電解酸化は、電解電圧を、150V〜250Vの繰り返しパルスを印加して行うことができる。また、本発明では、インプラントの表面被膜または光触媒膜として使用される前記酸化チタン膜を製造することが好ましい。
【図面の簡単な説明】
【0021】
【図1】本実施形態の酸化チタン膜を形成するためのプラズマ電気分解装置を示した図。
【図2】本発明の酸化チタン膜のサブミクロンオーダーのミクロポア構造の電界効果電子顕微鏡写真。
【図3】図2に示したミクロポア構造について白黒2値化前後の画像イメージを示した図。
【図4】本発明の酸化チタン膜のマクロポア構造のFE−SEM写真。
【図5】図4に示したマクロポア構造の与えるモルフォロジーを画像解析し、マクロポア構造の空孔率を、酸化チタン膜厚に対するプロット。
【図6】代表的な本発明の酸化チタン膜および比較例(電解質:リン酸ナトリウム)のモルフォロジーを示したFE−SEM写真。
【図7】図6に示したマクロポア構造のポア分布を、画素ビットの白黒ハーフトーンの輝度レベル分布として示した図。
【図8】本発明の酸化チタン膜のXRDパターンを示した図。
【図9】本発明による酸化チタン膜のアパタイト形成能を示した図。
【図10】本発明により得られた図9に示した酸化チタン膜に対するアパタイトの沈着性を、アパタイトによる酸化チタン膜の被覆率としてプロットした図。
【図11】本発明による酸化チタン膜の光触媒能を示した図。
【発明を実施するための形態】
【0022】
以下、本発明を実施形態をもって説明するが、本発明は、後述する実施形態に限定されるものではない。図1は、本実施形態の酸化チタン膜を形成するためのプラズマ電気分解装置の概略図である。図1に示した製造装置は、いわゆるプラズマ電解酸化(以下、PEO:Plasma Electrolysis Oxidizationとして参照する。)法により、陽極酸化を行って、アノード電極表面に陽極酸化被膜を形成する。図1のプラズマ電気分解装置100は、電解セル116内に電解液を貯留しており、電解液内にカソード112と、アノード114とが浸漬されていて、アノード114上で水の電解によって生じた酸素(原子状酸素または酸素分子)により、アノード114を構成する材料の表面を酸化する。
【0023】
カソード112を形成する材料としては、当方性黒鉛などを使用することができる。また、アノード114を形成する材料としては、金属チタンまたはチタン合金を使用することができる。アノード114を構成する材料は、例えばインプラントなどの形状に予め形成しておき、電解液中に浸漬してPEO法を適用することにより、高速で陽極酸化膜として酸化チタン膜を形成することができる。電解液としては、本実施形態で高いアパタイト形成能を得るためには、下記一般式(1)で与えられる4級アンモニウム化合物を使用する。
【0024】
【化2】
上記式(1)中、R1〜R3は、同一でも異なっていてもよい水素原子、アルキル基であり、R4は、水酸基または炭素数2個から4個のカルボン酸から得られる陰イオン基、またはこれらのいかなる混合物からなる群から選択することができる。
【0025】
4級アンモニウム化合物は、水溶液を形成するために充分な溶解性を有し、溶解時の水素イオン濃度(pH)が、7.0以上となることが、チタンまたはチタン合金を陽極酸化するためには好ましい。本実施形態で使用することができる4級アンモニウム化合物としては、アンモニア水、酢酸アンモニウム、プロピオン酸アンモニウム、酪酸アンモニウムを挙げることができ、溶解度およびpHの観点から4級アンモニウム化合物は、4級アンモニウム塩とすることが好ましく、溶解度および取扱性の点から酢酸アンモニウムを最適な実施例として挙げることができる。また、本発明では、アパタイト形成能を調整するために、上述した4級アンモニウム塩をその溶解度の範囲で適宜混合して使用することができる。
【0026】
電解液の濃度は、0.1mol/L〜飽和濃度とすることができるが、本実施形態で、酸化チタン膜の形成効率および電解の安定性などを考慮すると、0.3mol/L〜2mol/L程度が好ましく、さらに、本発明で、生体内にインプラントする点を考慮し、ルチル型の結晶構造を有する酸化チタンの形成を抑制する点では、0.3mol/L〜1.5mol/Lの範囲とすることが好ましく、アパタイト沈着性を考慮する場合、0.3mol/L〜0.8mol/Lの範囲が最も好ましい。
【0027】
電解セル116は、冷却システム118内に配置され、電解液の温度が、概ね室温となるように制御されている。冷却システム118は、さらに電解容器120に収容され、電解容器120には、発生する気体をパージするパージ配管122が接続されていて、酸素ガス、水素ガスなどの電気分解生成ガスを処理系に送っている。カソード112およびアノード114には、パルス電源システムを介して直流パルス電流が供給されている。パルス電源システムは、交流電源102と、パルスを発生するためのファンクションジェネレーター104と、2極電圧発生装置106とを含んで構成されている。ファンクションジェネレーター104は、本発明においてアノード近傍にスパークを発生させることが可能なパルス波形を生成している。パルス波形は、本発明では、周波数、0.1〜3kHz、デューティ比5%〜50%程度とすることができ、より好ましい実施形態では、PEO法の制御および成膜速度の点で、周波数を約1kHzとし、デューティ比を約10%とすることができる。
【0028】
高圧バイポーラ電圧発生装置106の出力は、それぞれカソード112およびアノード114に接続され、電気分解のための正負電圧が印加される。カソード112およびアノード114に印加される電圧および電流値は、デジタルマルチメーター108によりモニタされて、電圧および電流値の制御にフィードバックされる。また、パルス波形は、オシロスコープ110によりモニタされる。
【0029】
アノード114で電気分解が発生すると、初期には、フレッシュな金属表面が電解液に露出しているため、過電圧は加えられることないので電圧が高くともスパークが発生することなく電気分解が進行する。電気分解の進行につれ電気分解により発生した酸素原子、酸素イオン過酸化物などによりアノード114の酸化が進行して行き、表面に酸化チタン膜が生成される。純粋な酸化チタンは、誘電体なので、パルス電源システムから供給される電荷は、誘電体によりアノード114の表面に蓄積し、絶縁破壊電圧を超えると、付近のアノード114の表面に向けて電子雪崩を生じさせ、これがアノード上で火花放電を生じさせる。高エネルギーの火花放電が、アノード114の表面に生成したチタン表面または酸化チタン層を攻撃し、酸化チタン膜を改質しながら電解酸化膜として酸化チタンを形成する。
【0030】
以上のメカニズムによりPEO法は、高速にアノード114の表面酸化処理を行うことができ、また、通常の電解酸化膜よりも強固な膜質を電解酸化膜に対して与えることができる。本発明で好ましい電気分解条件としては、電気分解電圧が250Vの場合、電解酸化処理の時間を、約5min〜約20min程度とすることができる。本発明の好ましい電解酸化においては、電気分解電圧を250V、パルス繰り返し周波数を、1kHzとした場合、約15minの電気分解処理で、膜厚を、約0.6μm〜約5μmとする膜形成速度とすることができる。さらに、充分なアパタイト形成能および光触媒能、並びに被膜強度を得る目的でいえば約1μm〜約5μmの電気分解速度とすることが、PEOの制御および良好な特性の酸化チタン膜を形成できるのでより好ましい。また、電気分解電圧は、本発明では、50V〜300Vの範囲で可変とすることができ、酸化チタン膜の形成効率の点から言えば、100〜300Vの範囲でPEOを行うことができ、最も好ましくは、酸化チタン膜の形成速度およびPEO処理プロセスの制御性の観点から、150V〜250Vの範囲とすることができる。
【0031】
本発明の酸化チタン膜は、酸化チタンがアナターゼとルチルの結晶系の混合した形態として形成される。本発明のPEO処理では、アナターゼとルチルとの結晶の生成割合は、電解質の濃度により変化することが見出された。本発明により形成される酸化チタン膜のアナターゼ/ルチルの比は、X線散乱(以下、XRDとして参照する。)の回折パターンのピーク比で表現した場合、電解質濃度が、0.1mol/L〜1.5mol/Lの範囲で、0.6以上のアナターゼ/ルチル質量比とすることができる。一方、生体適合性に関していえば、ルチルをできるだけ低く抑制することが好ましいので、本発明による酸化チタン膜は、アナターゼ/ルチル質量比を1〜1.5の範囲とすることが最も好ましい。
【0032】
また、本発明では、基材上に形成される酸化チタン膜は、酸化チタン膜の膜厚に対応してサブミクロンオーダーのミクロポア構造と、数ミクロンオーダーのマクロポア構造とを有する。本発明の酸化チタン膜のサブミクロンオーダーのミクロポア構造の電界効果型電子顕微鏡(以下、FE−SEMとして参照する。)写真を図2に示す。図2(a)が、FE−SEMによる、倍率5000で撮影したミクロポア構造を示し、図2(b)が、倍率15000とした場合のより詳細なミクロポア構造を示す。本発明のミクロポア構造は、図2に示すように、数100nmオーダーのポアが、比較的緊密な酸化チタンブリッジにより相互連結されて形成されている。
【0033】
図2に示したミクロポア構造を画像解析して、図2に示したミクロポアの空孔率を見積もることができる。図3は、図2のミクロポア構造を画像解析した場合の画像イメージである。画像解析は、画像解析ソフトウェアであるPhotoshop(Photoshopは、Adobe Co.Ltd.の登録商標である。)を使用し、画像を8ビットダイナミックレンジの白黒2値化処理を行い、白黒2値化処理された後の画像のうち、レベル=0(ブラック)の属性を有する画素ビットの数を、全体画素ビット数に対する比として得た。
【0034】
図3に示した画像イメージは、白黒2値化前後の画像である。図3(a)が、図2(a)に対応する画像であり、図3(b)が、図2(a)の画像を白黒2値化したときの白黒2値化画像である。図3に示されるようにミクロポア構造は、サブミクロンオーダーで形成され、画像面積比での空孔率は、約20.3%程度であることがわかった。なお、空孔率について、白黒2値化処理のしきい値ビットを変化させることで、オーバーエスティメートとアンダーエスティメートの場合について平均した値である。また、2値化レベルのオーバーエスティメートとアンダーエスティメートとの間では、面積率は、±3.5%変動したに過ぎず、空孔率として充分ロバストな結果として得られた。
【0035】
さらに、本発明の酸化チタン膜は、図2および図3に示したミクロポア構造に加え、マクロポア構造を有する。図4は、本発明の酸化チタン膜のマクロポア構造のFE−SEM写真を示す。図4に示すマクロポア構造(a)〜(f)は、電解質を酢酸アンモニウムとし、電解質濃度を、0.1mol/L(a)、0.2mol/L(b)、0.3mol/L(c)、0.5mol/L(d)、0.8mol/L(e)、1.5mol/L(f)とし、陽極酸化電圧250Vで、15minの陽極酸化を行って形成されたものである。図4に示されるように、本発明では、電解液濃度が増加するにつれて、マクロポア構造が順次発達し、明確に形成されて行く。
【0036】
また、マクロポア構造のポア特性は、膜厚が薄い場合には、概ね円形として形成されるが、膜厚の増加にともなって、アスペクト比の大きなポア構造が成長する、このマクロポア構造のポアを、長径と短径とを有するポアとして形成される。この理由は明確ではないが、例えば、電解質の濃度により電解速度が増加しその結果、酸化チタンの形成速度が増加することで、絶縁破壊による火花放電の発生頻度が変化し、ミクロポア構造に対して施される放電処理の程度が高まるPEO法の特性によるものと推定される。
【0037】
図4に示したマクロポア構造の与えるモルフォロジーを、図3で説明したと同一の方法で画像解析し、マクロポアの存在割合(以下、マクロポア構造の空孔率(マクロポア構造の空孔率は、全面がマクロポアである場合にマクロポア率=1がとなる値である。)として参照する。)を、そのときの酸化チタン膜厚に対して行ったプロットを図5に示す。図5に示すプロットは、マクロポア構造の空孔率を縦軸とし、酸化チタン層膜厚(μm)を横軸として示したプロットである。図5に示すように、酸化チタン層の膜厚が増加するにつれて、マクロポア構造の空孔率が増加していることが示されている。また、本発明では、アパタイト形成能および光触媒能の両方について、酸化チタン膜の膜厚、マクロポア構造の空孔率への依存性がみられ、酸化チタン膜の膜厚およびマクロポア構造の発達に伴い、アパタイト形成能および触媒活性が向上する傾向が見出された。
【0038】
なお、本発明の酸化チタン膜のアパタイト形成能は、SBF法を使用することにより判断することができる。SBF(Simulated Body Fluid: SBF)法は、SBFを使用して、基材の生体活性を判断する方法である。SBFは、細胞やタンパク質のような有機物を含まず無機イオン濃度だけをヒトの細胞外液のそれにほぼ等しくして、pHをトリス緩衝液で7.25または7.4に調整した水溶液である。なお、SBFの詳細については、例えば、非特許文献2に詳細が記載されており、SBF法により体内における生体活性セラミックス表面での骨類似水酸アパタイト層の形成を、in vitroで再現する方法である。本発明では、SBF法を使用してアパタイト沈着性を検討するにあたり、SBF濃度の調整および酸化チタン膜のSBF溶液に対する浸漬時間を変化させてアパタイト形成能を評価することができる。
【0039】
本発明の酸化チタン膜について、SBF法により評価したところ、本発明により形成された酸化チタン膜は、6日間で充分な被覆率で表面にアパタイトを沈着させる。本発明の酸化チタン膜のアパタイト形成能は、FE−SEMによる目視観測の他、FE−SEMの画像を使用し、酸化チタン膜がアパタイトで被覆される速度を、例えば酸化チタン膜の被覆速度として評価することができる。本発明の酸化チタン膜は、1.0SBFモードで試験を使用した評価で、酸化チタン膜を生成する際の電解質濃度に依存し、約0.018/day〜0.122/dayの範囲の析出速度を示す。なお、本発明では、アパタイトの析出は、酸化チタン膜が完全にアパタイトで被覆された後も、発生し、SBFへの浸漬期間とともにアパタイトの生成が継続する。このため、本発明の酸化チタン膜は、インプラント表面の表面コーティングに適用した場合、迅速に生体に対して適合し良好な表面コートを与えることができる。
【0040】
また、本発明の酸化チタン膜の光触媒活性は、光照射下での、色素の分解速度により評価することができる。本発明の光触媒活性の評価は、酸化チタン膜上を酸化状態により光吸収性を変化させる染料で着色し、着色した酸化チタン膜をUV照射して色素の退色を評価することにより、酸化チタン膜間の相対的な光触媒活性として評価する。
【0041】
本発明の酸化チタン膜は、良好な結晶性を有しており、良好な光触媒活性を示すことが見出された。本発明では、アパタイト形成能および触媒活性は、酸化チタン膜の膜厚に伴って増加する傾向が観測された。本発明で、良好なアパタイト形成速度を得る目的からは、酸化チタン膜の膜厚は、0.6μm〜10μmとすることができ、より好ましいアパタイト形成速度を得る点では、約1μm〜5μmとすることができ、製造プロセスの制御性の観点では、約1μm〜5μmとすることが好ましい。
【0042】
本発明の酸化チタン膜の膜厚は、電解質として使用する酢酸アンモニウムの濃度上昇に従って増加する。酢酸アンモニウムを電解質として形成した酸化チタン膜は、XRD測定の結果から、皮膜内に電解質成分がほとんど取り込まれておらず、高い結晶性を有することが見出された。一方、酢酸アンモニウム溶液の濃度とともに酸化チタン膜のルチル形成量が増加し、相対的にアナターゼが減少する。この理由は、種々想定できるが、1つの要因としては、PEO処理の進行に伴って火花放電によってアナターゼからルチルへの熱転位温度915±15℃以上に被膜が局所加熱されることも関連するものと考えられる。
【0043】
また、本発明により得られる酸化チタン膜は、電解質としてリン酸ナトリウムを使用した場合に比較して早期に火花放電が停止する現象が観測された。この理由は、本発明の酸化チタン膜は、高純度の酸化チタンとして形成され、この結果抵抗が高い良質の酸化チタン膜が形成され、早期の絶縁膜形成を促進する結果と推定される。
【0044】
以下、本発明について、詳細な実施例をもって説明するが、本発明は、以下に説明する実施例に限定されるものではない。
【実施例】
【0045】
[材料]
電極材料としては、陽極として3×10×40〜80mm3に切り出した純チタン板(株式会社ニラコ製、99.5%、3×200×400mm)を使用した。陰極を構成する電極材料としては、等方性黒鉛板(東海カーボン株式会社製、3×10×80mm)を用いた。等方性黒鉛板は、耐水研磨紙(#2000)で研磨し、アセトン洗浄した後乾燥させた。当該等方性黒鉛板を絶縁塗料(古藤産業株式会社製、フロンマスクMA−1)でマスキングし、10mm×10mmの開口を形成して電解液に露出させてカソードとした。
【0046】
電解液としては、実施例では、酢酸アンモニウム水溶液(和光純薬工業株式会社製;特級)を使用した。また、比較例の電解質として、リン酸ナトリウム(和光純薬工業株式会社製;CAS#7601-54-9)を使用した。各電解液を、0.05〜1.5mol/Lの濃度に調整し、実施例および比較例の電解液とした。
【0047】
[PEO装置およびPEO条件]
PEO装置は、図1の構成として、電解セルとしては、陽極酸化装置本発明のPEO処理に適するように設計された陽極酸化装置を用いた。交流安定化電源として、EC100S(エヌエフ回路設計ブロック株式会社製)、ファンクションジェネレーターとして、SG−4105(岩崎通信機株式会社製)を使用し、火花放電を発生させる電圧を生成させるため、高速バイポーラ電源(エヌエフ回路設計ブロック製:HSA4052)を高圧電源として使用した。
【0048】
ファンクションジェネレーターは、任意のパルス波形を生成し、初期電圧2.5Vまでの可変電圧のパルスを出力する。パルス周波数は、1kHzとした。高速バイポーラ電源は、ファンクションジェネレーターの直流出力パルスを電圧増幅する。電極間の実効電圧値は、オシロスコープ(岩通計測株式会社製、DS−8822P)を使用してモニタし、電極間を流れる電流の経時変化をデジタルマルチメーター(岩通計測株式会社製、VOAC21)により記録した。電解セル内はPEOによって発生する火花放電によって加熱されるため、電解セルを恒温水槽(アズワン株式会社製、TR−3A)および冷却水循環装置(旭テクノグラス株式会社製、CLUMINI2型を用いて冷却した。
【0049】
また、電極を電解セル内の電解液に浸る様に固定し、電解セルを排気口を除き密閉して電圧を印加することにより、PEO処理を施した。電解処理時間を3sec〜15minとし、PEO処理を介した後15min後、電極を取り出し、アセトン洗浄超音波洗浄して、デシケーター中に保存した。電気分解電圧は、150V〜250V、電解液濃度(0.05〜1.5mol/L)の範囲に設定した。
【0050】
結晶相・結晶性の同定は、X線回折により行った。X線回折測定は、X線回折装置(理学電機株式会社製、RINT2000)を用いた。使用したX線源は、CuKα線とし、X線発生条件は、管球電圧:40kV、走査速度:1deg・min、管球電流:200mAであった。
【0051】
生成膜の微構造の観測は、FE−SEMにより行った。FE−SEMとしては、株式会社日立製作所製S4500を使用し、観察条件を、加速電圧15kV、ワーキングディスタンス(W.D.)を15mmとした。また、観察試料は、イオンスパッタ装置(株式会社日立製作所製、E−1030)を用いて、白金−パラジウムコーティングを、30sとした。生成した酸化チタン膜の膜断面観察は、試料を、FIB(株式会社日立ハイテクノロジーズ、FB−2100)を使用し断面観察用試料を作製し、FE−SEMを使用して観測した。断面観察資料は、酸化チタン膜を部分的に生成したチタン基板を、酸化チタン膜を切断する方向にGaイオンビーム照射し、酸化チタン膜およびチタン基板をイオンビームエッチングすることで、矩形ノッチを生成させて作成した。その後、形成した矩形ノッチに隣接する酸化チタン膜の断面を、FE−SEMで観測した。各観察結果は、デジタルカメラにより画像イメージとして取得した。
【0052】
[SBFの調整および評価方法]
疑似体液(SBF)は、ヒトの血漿の無機成分とほぼ等しい濃度となるように調整し、その組成を下記表1示す。
【0053】
【表1】
【0054】
上記組成のSBFは、1000mLのポリエチレンビーカーに約700mLの蒸留水と攪拌子とを入れ、それをマグネティックスターラー上に置いた恒温槽中に設置し、37℃でに攪拌した。この蒸留水に上記表の試薬を順次加えた。試薬は一度に複数投入せず、一つの試薬が完全に溶解した後で投入した。溶液は、37℃でpH7.4になるように塩酸を加えて調整し、最後に1000mLメスフラスコに移して蒸留水を加え、1Lとなるように調整した。
【0055】
生体活性は、作製した試料の疑似体液(SBF)浸漬によるアパタイト沈着をFE−SEMによる観測により評価した。試験条件は、作製した酸化チタン膜を37℃で、30mLのSBFに6、9、12日間浸漬し、試験終了後、資料を蒸留水でよくすすぎ、室温で乾燥後、FE−SEMおよびラマン分光測定によってアパタイトの形成を検討した。
【0056】
ラマン分光は、愛宕物産株式会社/Jovin Yvon社製(T64000)のトリプルモノクロメーターとCCDディテクターとを用い、測定範囲をレーザ波長の波長から10000m−1〜120000m−1として測定を行った。なお、レーザラマン散乱を生じさせるためのレーザ光源としては、アルゴンイオンレーザの波長514.5nmを使用した。
【0057】
[光触媒能]
光触媒能を、酸化チタン膜による染料の光学特性の変化を用いて評価し、酸化チタン膜を、酢酸アンモニウム溶液を電解液とし、その濃度を0.1〜0.8mol/Lの範囲で変化させて調整した。染料としてメチレンブルーを使用し、生成した酸化チタン膜をメチレンブルーで着色し、試料表面をUV照射し、光触媒反応によるメチレンブルーの分解で回復する反射率の変化を分光測色計(株式会社コニカミノルタセンシング製CM−2500d)で観測して、光触媒活性を評価した。
【0058】
[結果]
上述した条件を使用し、実施例として酢酸アンモニウムを電解液として使用し、比較例として、リン酸ナトリウムを電解質とした場合について、PEO法を適用して酸化チタン膜を生成し、酸化チタン膜のモルフォロジー、アパタイト形成能、および光触媒能について検討を加えた。
【0059】
I.膜モルフォロジーの評価
本発明により形成された酸化チタン膜の膜モルフォロジーを、電解液を酢酸アンモニウムとした場合(実施例)と、電解液をリン酸ナトリウムとした場合(比較例)について評価した。PEO条件は、酢酸アンモニウムを電解質とした場合、電解質濃度を、0.1mol/L、0.3mol/L、0.8mol/L(3条件)とし、電解電圧=250V、パルス周波数=1kHz、PEO処理時間=15minとした。また、比較例では電解質をリン酸ナトリウムとし、電解質濃度を、見かけ上の酸化チタン膜の膜厚が同じオーダーとなる濃度の0.2mol/Lとし、電解電圧=250V、パルス周波数=1kHz、PEO処理時間15minとしてそれぞれ酸化チタン膜を形成し、FE−SEMにより膜モルフォロジーの比較を行った。
【0060】
図6に代表的な酸化チタン膜のモルフォロジーを示す。図6に示したFE−SEM写真は、2500倍である。図6(a)〜図6(c)が実施例であり、図6(d)が比較例である。図6に示すように、本発明により形成された酸化チタン膜は、電解質の濃度増加に伴いマクロポア構造が順次発達しており、膜厚は、それぞれ、図6(a)で0.6μm、図6(b)で、0.7μm、図6(c)で3.0μmである。なお、図6(a)の実施形態で、PEO処理時間=15minの酸化チタン膜では、明確に目視観測できるマクロポア構造が未だ充分に発達していない。一方、比較例である図6(d)は、形成される酸化チタン膜のマクロポア構造が粗いため、平均値として約7μmと見積もられた。
【0061】
図6に示すように、本発明により形成された酸化チタン膜(図6(a)〜図6(c)は、電解質濃度の増加に伴い、良好なマクロポア構造が形成されている。一方、比較例である図6(d)の酸化チタン膜は、マクロポア構造が不均一であった。図6に示した酸化チタン膜は、PEO処理時間を15minに統一していることから、本発明では、被膜形成速度は比較例に比較して遅いということができるものの、本実施例で形成される酸化チタン膜は、良好なマクロポア構造を示した。
【0062】
図7は、図6に示したマクロポア構造のポア分布を、画素ビットの白黒ハーフトーン(400dpi、ダイナミックレンジ8ビット)の輝度レベル分布として示した図であり、図7(a)〜図7(d)は、それぞれ図6(a)〜図6(d)のマクロポア構造の輝度レベル分布に対応する。図7に示すように、本実施形態で明確なマクロポア構造が観測された酸化チタン膜(図6(b)、図6(c))については、輝度レベル分布の標準偏差が、それぞれ27.96および27.54という値が得られている。一方、比較例である図6(d)に対応する酸化チタン膜では、標準偏差が19.88として得られ、マクロポア構造の均質なランダムさという観点から見ると、本実施形態の酸化チタン膜の方が比較例に比してよりランダム、すなわち均一な分布のマクロポア構造を与えていることが示される。
【0063】
なお、図7(d)に示す比較例の場合、所々大きなポアが認められるものの、標準偏差が19.88として得られている。この理由は、図6(d)に示されるように、大きなポアを除けば比較的均一なポアが形成されていることから、マクロポア分布としては、本発明により形成されるマクロポア構造に比較して比較的均一であることを示すものと考えられる。
【0064】
II.膜の結晶性の評価
図8は、本発明の酸化チタン膜のXRDパターンを示す、電解質として酢酸アンモニウムを使用し、その濃度を、0.1mol/L〜1.5mol/Lまで変化させて得られた酸化チタン膜が実施例についてのXRDパターンであり、電解質としてリン酸ナトリウム0.2mol/Lとして形成された酸化チタン膜が比較例についてのXRDパターンである。図8に示すように、本実施形態の酸化チタン膜については、0.1mol/Lの電解質濃度であっても酸化チタンのアナターゼのピークがトレース程度であるが形成されているのが示された。
【0065】
また、電解質濃度が、0.1mol/L〜1.5mol/Lに増加するにつれて、結晶構造の割合がアナターゼからルチルへと増加しているのが観測された。アナターゼ/ルチル質量比は、電解質濃度が0.1mol/Lで約1.5であり、電解質濃度が1.5mol/Lで約0.6という結果が得られた。一方、比較例であるリン酸ナトリウムを電解質として使用して形成された酸化チタン膜では、図6で説明したように充分な膜厚の酸化チタン膜が形成されているにもかかわらず、アナターゼおよびルチルの結晶構造に由来するピークが観測されなかった。
【0066】
このため、本発明により形成される酸化チタン膜は、比較例により形成される酸化チタン膜に比較してより高結晶性の酸化チタン被膜が形成されていることが示された。
【0067】
III.アパタイト形成能の評価
図9は、酸化チタン膜のアパタイト形成能を示した図である。図9(a)〜図9(d)は、いずれも本発明により形成された酸化チタン膜について、1.0SBFを使用し、6日間、9日間、12日間の浸漬を行い、アパタイト形成能を評価した。図9に示した実施例では、いずれもSBF浸漬6日間の結果を示す。図9に示された酸化チタン膜へのアパタイト形成については、レーザラマン散乱を使用して確認し、全試料について、アパタイトが沈着していることが確認できた。
【0068】
一方、比較例として、電解液としてリン酸アンモニウム(0.2mol/L)を使用して形成した酸化チタン膜については、12日間浸漬してもアパタイトの酸化チタン膜表面への沈着は観測されなかった。
【0069】
以上のことから、本発明では、電解質としてリン酸ナトリウムを使用して形成された酸化チタン膜に比較してはるかに高いアパタイト形成能を有する酸化チタン膜が得られることがわかった。なお、図9に示す実施例について、さらに12日間までアパタイト沈着性をトレースしたが、アパタイト沈着が阻害されることなく、継続的な沈着を示し、良好なアパタイト層の形成が認められた。
【0070】
図10は、本発明により得られた図9に示す酸化チタン膜に対するアパタイトの沈着性を、アパタイトによる酸化チタン膜の被覆率をもって示した図である。横軸は、電解液濃度(mol/L)であり、縦軸は、アパタイト被覆率(面積%)である。なお、アパタイト被覆率は、画像解析により行い、画像解析は、画像解析ソフトウェアであるPhotoshop(Photoshopは、Adobe Co.Ltd.の登録商標である。)を使用し、画像を8ビットダイナミックレンジの白黒2値化処理を行なった後、アパタイト粒子が酸化チタン膜から立体的に上側に成長し輝度の高いポイントとして観測されることから、白黒2値化処理された後の画像のうち、レベル=255(ホワイト)の属性を有する画素ビットの数を、全体画素ビット数に対する比として得た。なお、図10のプロットの誤差バーは、白黒2値化処理の相対誤差の3倍(約10%)を示す。
【0071】
図10に示すようにアパタイト沈着速度は、電解液濃度(mol/L)に対して依存性を示しており、電解液濃度が0.1mol/L〜0.8mol/Lの各ポイントについて、約0.018/day〜0.122/dayの沈着速度が得られた。
【0072】
IV.光触媒能の評価
図11には、酸化チタン膜の光触媒能についての結果を示す。図11に示すように、本発明により形成される酸化チタン膜は、電解液濃度0.1mol/Lであっても、光触媒活性を有しており、比較例である、リン酸ナトリウムを電解質として形成された酸化チタン膜に比較して相対的に高い光触媒活性を示すことがわかった。これは、本発明により形成される酸化チタン膜がより結晶性が高いことに起因するものと考えられる。また、本発明の酸化チタン膜は、良好な光触媒活性も併せ持っているので、生体内に導入する前に紫外線で簡単にかつ効率よく滅菌できる利点があり、実用上製造プロセスのプロセス工程を削減でき、製造を効率化できる。
【産業上の利用可能性】
【0073】
以上説明したように、本発明によれば、プラズマ電界酸化法により新奇なモルフォロジーを有する酸化チタン膜を形成することができ、当該酸化チタン膜は、良好なアパタイト沈着性および光触媒活性を示す。さらに本発明により形成される酸化チタン膜は、火花放電による表面処理を受け、基材に対して酸化チタン層が偏在せず、よりランダムかつ強固に形成され、より高い強度の酸化チタン陽極酸化膜を形成することができる。このため、本発明の酸化チタン膜は、インプラント材の表面被覆として高い生体活性を提供するばかりではなく、良好な光触媒層を提供することができる。
【0074】
これまで本実施形態につき説明してきたが、本発明は、上述した実施形態に限定されるものではなく、他の実施形態、追加、変更、削除など、当業者が想到することができる範囲内で変更することができ、いずれの態様においても本発明の作用・効果を奏する限り、本発明の範囲に含まれるものである。
【符号の説明】
【0075】
100…プラズマ電解酸化装置、102…AC電源、104…ファンクションジェネレーター、106…高圧バイポーラ電源、108…デジタルマルチメーター、110オシロスコープ、112…カソード、114…アノード、116…電界セル、118…冷却システム、120…電解容器、122…パージ配管
【特許請求の範囲】
【請求項1】
表面被膜として使用される酸化チタン膜であって、前記酸化チタン膜は、
1μm以下のミクロポア構造と、1μmを超えるマクロポア構造を有し、前記マクロポア構造の空孔率が、前記酸化チタン膜の表面に対する面積率で0.02以上である、酸化チタン膜。
【請求項2】
前記マクロポア構造の空孔率が0.06〜0.4である請求項1に記載の酸化チタン膜。
【請求項3】
前記酸化チタン膜の膜厚が、0.6μm〜5μmであり、質量比でアナターゼをルチルよりも多く含有する、請求項1または2の記載の酸化チタン膜。
【請求項4】
前記酸化チタン膜は、電解質として下記構造式(1)で与えられる4級アンモニウム化合物を使用するプラズマ電解酸化法により形成される、請求項1〜3のいずれか1項に記載の酸化チタン膜。
【化1】
(上記式(1)中、上記式(1)中、R1〜R3は、同一でも異なっていてもよい水素原子、アルキル基であり、R4は、水酸基または炭素数1個から4個のカルボン酸から得られる陰イオン基からなる群から選択される基である。)
【請求項5】
前記4級アンモニウム化合物は、酢酸アンモニウム塩である、請求項4に記載の酸化チタン膜。
【請求項6】
前記酸化チタン膜のアパタイト形成能が、前記酸化チタン膜の表面被覆速度として、0.018/day以上である、請求項5に記載の酸化チタン膜。
【請求項7】
前記酸化チタン膜は、インプラントの表面被膜または光触媒膜である、請求項1〜6のいずれか1項に記載の酸化チタン膜。
【請求項8】
酸化チタン膜を陽極酸化により形成する酸化チタン膜形成方法であって、
4級アンモニウム化合物を含む電解液にチタンを含むアノードを浸漬し、前記アノードをプラズマ電解酸化して前記アノード表面に質量比でアナターゼをルチルよりも多く含有する酸化チタン膜を形成する工程を含む、酸化チタン膜形成方法。
【請求項9】
前記プラズマ電解酸化は、電解電圧を、150V〜250Vの繰り返しパルスを印加して行う、請求項8に記載の酸化チタン膜形成方法。
【請求項10】
インプラントの表面被膜または光触媒膜として使用される前記酸化チタン膜を製造する請求項8または9に記載の酸化チタン膜形成方法。
【請求項1】
表面被膜として使用される酸化チタン膜であって、前記酸化チタン膜は、
1μm以下のミクロポア構造と、1μmを超えるマクロポア構造を有し、前記マクロポア構造の空孔率が、前記酸化チタン膜の表面に対する面積率で0.02以上である、酸化チタン膜。
【請求項2】
前記マクロポア構造の空孔率が0.06〜0.4である請求項1に記載の酸化チタン膜。
【請求項3】
前記酸化チタン膜の膜厚が、0.6μm〜5μmであり、質量比でアナターゼをルチルよりも多く含有する、請求項1または2の記載の酸化チタン膜。
【請求項4】
前記酸化チタン膜は、電解質として下記構造式(1)で与えられる4級アンモニウム化合物を使用するプラズマ電解酸化法により形成される、請求項1〜3のいずれか1項に記載の酸化チタン膜。
【化1】
(上記式(1)中、上記式(1)中、R1〜R3は、同一でも異なっていてもよい水素原子、アルキル基であり、R4は、水酸基または炭素数1個から4個のカルボン酸から得られる陰イオン基からなる群から選択される基である。)
【請求項5】
前記4級アンモニウム化合物は、酢酸アンモニウム塩である、請求項4に記載の酸化チタン膜。
【請求項6】
前記酸化チタン膜のアパタイト形成能が、前記酸化チタン膜の表面被覆速度として、0.018/day以上である、請求項5に記載の酸化チタン膜。
【請求項7】
前記酸化チタン膜は、インプラントの表面被膜または光触媒膜である、請求項1〜6のいずれか1項に記載の酸化チタン膜。
【請求項8】
酸化チタン膜を陽極酸化により形成する酸化チタン膜形成方法であって、
4級アンモニウム化合物を含む電解液にチタンを含むアノードを浸漬し、前記アノードをプラズマ電解酸化して前記アノード表面に質量比でアナターゼをルチルよりも多く含有する酸化チタン膜を形成する工程を含む、酸化チタン膜形成方法。
【請求項9】
前記プラズマ電解酸化は、電解電圧を、150V〜250Vの繰り返しパルスを印加して行う、請求項8に記載の酸化チタン膜形成方法。
【請求項10】
インプラントの表面被膜または光触媒膜として使用される前記酸化チタン膜を製造する請求項8または9に記載の酸化チタン膜形成方法。
【図1】

【図5】
【図10】
【図11】
【図2】

【図3】

【図4】
【図6】
【図7】
【図8】
【図9】
【図5】
【図10】
【図11】
【図2】
【図3】
【図4】
【図6】
【図7】
【図8】
【図9】
【公開番号】特開2010−215438(P2010−215438A)
【公開日】平成22年9月30日(2010.9.30)
【国際特許分類】
【出願番号】特願2009−62153(P2009−62153)
【出願日】平成21年3月15日(2009.3.15)
【出願人】(304021417)国立大学法人東京工業大学 (1,821)
【Fターム(参考)】
【公開日】平成22年9月30日(2010.9.30)
【国際特許分類】
【出願日】平成21年3月15日(2009.3.15)
【出願人】(304021417)国立大学法人東京工業大学 (1,821)
【Fターム(参考)】
[ Back to top ]