
酸化処理を用いて固体支持体の表面に有機膜を調製するための方法
本発明は、(コ)ポリマーから作られている固体支持体の表面の一部の上に有機膜を調製するための方法に関し、それが、(i)前記表面部分を酸化処理にかける工程、および(ii)ラジカル化学的グラフト化によって前記表面部分に有機膜をグラフト化させる工程、からなる連続的な工程を含むことを特徴とする。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、有機表面コーティングの分野に関し、前記コーティングは、有機膜の形態にある。
【0002】
さらに詳しくは、本発明は、導電性または非導電性表面、有利にはプラスチックまたはポリマーから作られている表面の上に、それらの表面を酸化処理するという予備工程を用い、コーティングすることによってそのような有機コーティングを調製するための方法に関する。
【背景技術】
【0003】
現在のところ、基材の上に有機薄膜を調製することを可能とするいくつかの技術が存在している。
【0004】
いかなるタイプの表面にでも適用することが可能な、いくつかの技術では、その膜とコーティングされる表面との間の親和性を必要とせず、それら二つの構成成分の間の物理吸着のみが含まれている。これらの技術は、遠心力によるコーティングの形成(「スピンコーティング」)、浸漬によるコーティングの形成(「ディップコーティング」)、または蒸発による析出(「スプレーコーティング」)の方法を含む。したがって、膜とコーティングされる表面との間には実際のグラフトは存在せず、特に最も薄い析出層(20nm未満)では、得られる厚みを調節することは容易にはできない。
【0005】
支持体の表面に有機コーティングを形成させる他の方法、たとえばプラズマ蒸着または光化学的活性化(すなわち光活性化)は、同一の原理に基づいていて、被覆する表面の近くに不安定な前駆体の形態を発生させ、その不安定な形態が変化して、その基材の上に膜を形成させる。これらの方法は一般的には、付着性の膜を形成させることになるが、その付着が、対象物にトポロジー的に密接した膜の架橋によるものなのか、あるいはその界面に結合が形成されたことによるものかを識別することは通常不可能である。さらに、これらの方法では以下のようなことが必要とされる:特定の前駆体、特に光活性化の場合であれば感光性前駆体、比較的に複雑でコストのかかる前処理、プラズマ法の場合であれば真空下の真空装置の使用、電気化学的方法では照射および/またはポテンシオスタットの使用(これらの場合、接続についてのいくつもの問題が伴う)。
【0006】
電気泳動もまた、有機膜を用いて導電性表面をコーティングするための一つの方法である。電気泳動またはカチオン電着プロセスによって、(析出させるより前に合成しておいた)荷電ポリマーを使用して金属部品を覆うことが可能となり、導電性表面の上に均質な膜を与える方法となる。しかしながら、電着条件を厳密に調節することに加えて、その処理では、電着膜に物理的強度または耐薬品性を付与するためには、さらに焼付け工程が必要となる。
【0007】
単分子層を自己組織化させることは、実施するには極めて単純な方法ではあるが、コーティングの対象となる表面に対して十分な親和性を有する、一般的には分子状の前駆体を必要とする。したがって、ここで使用した用語は、たとえば金または銀に対して親和性を有する硫黄化合物特にチオール官能基を有するもの、酸化物たとえばシリカまたはアルミナに対してはトリハロシラン、あるいはグラファイトまたはカーボンナノチューブに対してはポリ芳香族化合物といった「前駆体と表面のペア」となるであろう。いずれの場合においても、分子の厚み(10nm未満)の膜を形成させることは、前駆体分子の一部と、その表面のある種の「受容体」部位との間の特定の化学反応に基づいている。化学吸着反応によって、付着が確実になる。しかしながら、表面と単分子膜との間の界面結合は、ある種の組合せでは弱い可能性もあるし、あるいは、特定の環境条件、特に湿潤状態では弱くなる可能性がある。
【0008】
ポリマーの電気グラフト化(electrografting)は、電極としておよび重合開始剤としての両方の作用を有する対象物の表面上で電気活性モノマーを、電気的に誘導された連鎖生長反応によって、重合を開始させ、次いで重合させることに基づいた方法である[1]。電気グラフト化では、還元によって開始され、連鎖生長するメカニズムに適合した前駆体を使用することが必要である。電気グラフト化された膜への付着は、炭素−金属共有結合によって与えられる[2]。上で思い起こした各種の方法の中で、電気グラフト化は、界面結合性を特定的に調節したグラフト膜を製造することを可能とする唯一の方法である。具体的には、活性化されたビニルモノマーから得られるポリマーの膜を表面(必然的に導電性表面である)にグラフトさせることを可能とする、その唯一の方法は、ポテンシオスタットを介して、表面から始まる重合反応を電気的に開始させること、それに続けて、次々とモノマーを加えていってその鎖を生長させることからなり、カソードおよびアノードを有する電気化学的セルを使用し、後者の端子に電圧を印加することが必要である。したがって、国際公開第03/018212号パンフレットには、導電性表面の上に導電性有機膜をグラフトさせ、生長させるためのプロセスが特に記載されており、前記有機膜の前駆体のジアゾニウム塩を電解還元させることによって、そのグラフト化と生長とを同時に行わせている[3]。
【0009】
最後に、非電気化学的条件下でグラフト化有機コーティングを作成することを可能とし、いかなるタイプの表面にも容易に実施されるプロセスが、最近報告された。国際公開第2008/078052号パンフレットに記載されているこのプロセスには、簡単な工程で共有結合的なグラフト化を可能とする、ラジカル反応的グラフト化が含まれている[4]。
【0010】
この新規な方法では、特に、不対電子を有する、たとえばジアゾニウム塩のような開裂可能なアリール塩から得られる分子的エンティティ(molecular entities)を採用して、グラフトさせる対象物の表面と共有結合タイプの結合を形成しているが、前記分子的エンティティは、それらがグラフト化される対象の表面からは独立して発生させる。これらの分子的エンティティは、その表面にグラフトされ、特にエチレン性モノマーを含むラジカル反応を開始させる。
【発明の概要】
【発明が解決しようとする課題】
【0011】
本願発明者らは、国際公開第2008/078052号パンフレットに記載されたプロセスをさらに改良することを可能とするための、各種の手段および/または各種の工程を確認するための研究を続けてきた。
【課題を解決するための手段】
【0012】
したがって、本発明は、非電気化学的条件下で、固体支持体の表面の少なくとも一部に有機膜をグラフト化させるための改良された方法に関する。
【0013】
その理由は、本願発明者らの研究によって、国際公開第2008/078052号パンフレット[4]に記載のプロセスにおいて、固体支持体の表面に酸化的前処理を行うことによって、有機膜のグラフト化を改良すること、および/または前記部分にグラフト化される有機膜の厚み、従って量を増やすことが可能となることを見出したためである。そのような酸化処理は、さらに詳しくは、固体支持体の表面に対して、および/またはポリマー、たとえばプラスチックから作られている固体支持体に対して適用される。
【0014】
なにか一つの理論に拘束されることを望むものではないが、酸化的前処理を使用することによって、国際公開第2008/078052号パンフレット[4]に記載のプロセスに、より良好な寄与が可能となる。さらに、グラフト化された表面の反応性がより高いために、酸化的前処理を行うことによって、国際公開第2008/078052号パンフレット[4]に記載のプロセスの通常の反応時間を下げることも可能となる。
【0015】
したがって、本発明は、(コ)ポリマーから作られている固体支持体の表面の一部の上に有機膜を調製するための方法に関し、以下のものからなる連続的な工程を含むことを特徴としている:
i)前記表面部分を酸化処理にかける工程;
ii)ラジカル化学的グラフト化によって前記表面部分に有機膜をグラフト化させる工程。
【0016】
「(コ)ポリマーから作られている固体支持体の表面」という用語は、(コ)ポリマーから作られている固体支持体、または(コ)ポリマーから作られている表面を正に有している固体支持体の両方を意味しているが、その支持体の残りの部分は、いかなる材料から作られていてもよいと理解されたい。本発明における固体支持体は、いかなるサイズ、いかなる形状であってもよい。
【0017】
「(コ)ポリマーから作られている」という用語は、本発明の文脈においては、正に1種の(コ)ポリマーかまたは複数の異なった(コ)ポリマーから実質的になる支持体または表面を意味していると理解されたい。
【0018】
「実質的になる(essentially composed)」という用語は、本発明の文脈においては、重量で表して、その構成成分の少なくとも50%、少なくとも60%、少なくとも70%、少なくとも80%、少なくとも90%、少なくとも95%、および/または少なくとも98%が、1種(または複数)の(コ)ポリマーである、支持体または表面を意味している。
【0019】
その支持体またはその支持体の表面が、1種(または複数)の(コ)ポリマー(1種または複数)によってのみ構成されているのが有利である。
【0020】
また別な形態においては、その支持体またはその支持体の表面が、1種(または複数)の(コ)ポリマーに加えて、充填剤、可塑剤および添加剤からなる群から選択された少なくとも1種の成分を含んでいる。この(これらの)追加の成分は、ポリマー材料の中に組み入れられるかおよび/または分散されているのが有利である。
【0021】
念のために言えば、プラスチックは、有利には3000を超える重合度を有する少なくとも1種の(コ)ポリマーと、少なくとも1種の添加剤とから形成されている。したがって、本発明の文脈において実施されるポリマーから作られている支持体またはその支持体の表面には、プラスチックから作られた支持体または支持体表面が含まれている。
【0022】
無機充填剤たとえば、シリカ、タルク、ガラス繊維もしくはガラスビーズ、または有機充填剤たとえば、穀物粉もしくはセルロースパルプが、コストを下げるためおよびポリマー材料のある種の性質たとえば機械的性質を改良するために、一般的に使用されている。添加剤は、ポリマー材料の特定の性質を改良するために主として使用されるが、前記性質としては、架橋性、スリップ性、耐分解性、難燃性および/または細菌および真菌の攻撃に対する抵抗性などが挙げられる。
【0023】
各種の天然(たとえばラテックスもしくはゴム)、人工、合成の熱可塑性、熱硬化性、耐熱性、エラストマー性、直鎖状(すなわち、直鎖状もしくは分岐状、一次元)および/または三次元のポリマーが、本発明の文脈において使用することができる。
【0024】
本発明の文脈において実現される(コ)ポリマーが、以下のものからなる群より選択される熱可塑性(コ)ポリマーであるのが有利である:
−ポリオレフィン、たとえば、ポリエチレン、ポリプロピレン、エチレン/プロピレンコポリマー、ポリブチレン、ポリメチルペンテン、エチレン/酢酸ビニルコポリマー、エチレン/ビニルアルコールコポリマー、それらの誘導体の1種、それらのコポリマーの1種、それらのブレンド物の1種、およびそれらの組合せの1種;
−ポリエステル、たとえば、(場合によってはグリコールによって変性された)ポリエチレンテレフタレート(PET)、ポリブチレンテレフタレート、ポリラクチド、ポリカーボネート、それらのコポリマーの1種、それらのブレンド物の1種、およびそれらの組合せの1種;
−ポリエーテル、たとえば、ポリ(オキシメチレン)、ポリ(オキシエチレン)、ポリ(オキシプロピレン)、ポリ(フェニレンエーテル)、それらのコポリマーの1種、それらのブレンド物の1種およびそれらの組合せの1種;
−ビニルポリマー、たとえば、(場合によっては塩素化された)ポリ塩化ビニル、ポリビニルアルコール、ポリ(酢酸ビニル)、ポリ(ビニルアセタール)、ポリ(ビニルホルマール)、ポリフッ化ビニル、ポリ(塩化ビニル/酢酸ビニル)、それらのコポリマーの1種、それらのブレンド物の1種およびそれらの組合せの1種;
−ビニリデンポリマー、たとえば、ポリ(塩化ビニリデン)、ポリフッ化ビニリデン、それらのコポリマーの1種、それらのブレンド物の1種およびそれらの組合せの1種;
−スチレンポリマー、たとえば、ポリスチレン、ポリ(スチレン/ブタジエン)、ポリ(アクリロニトリル/ブタジエン/スチレン)(ABS)、アクリロニトリル/ブタジエン/スチレン−ポリカーボネート(ABS/PC)、ポリ(アクリロニトリル/スチレン)、ポリ(アクリロニトリル/エチレン/プロピレン/スチレン)、ポリ(アクリロニトリル/スチレン/アクリレート)、それらのコポリマーの1種、それらのブレンド物の1種およびそれらの組合せの1種;
−(メタ)アクリル系ポリマー、たとえば、ポリアクリロニトリル、ポリ(アクリル酸メチル)、ポリ(メタクリル酸メチル)、それらの誘導体の1種、それらのコポリマーの1種、それらのブレンド物の1種およびそれらの組合せの1種;
−ポリアミド、たとえば、ポリ(カプロラクタム)、ポリ(ヘキサメチレンアジパミド)、ポリ(ラウロアミド)、ポリエーテル−ブロック−アミド、ポリ(メタキシリレンアジパミド)、ポリ(メタフェニレンイソフタルアミド)、それらのコポリマーの1種、それらのブレンド物の1種およびそれらの組合せの1種;
−フルオロポリマー(またはポリフルオロエテン)、たとえば、ポリテトラフルオロエチレン、ポリクロロトリフルオロエチレン、ペルフルオロ化ポリ(エチレン/プロピレン)、ポリフッ化ビニリデン(PVDF)、それらのコポリマーの1種[たとえば、テトラフルオロエチレンとテトラフルオロプロピレンとのコポリマー(FEP)、エチレンとテトラフルオロエチレンとのコポリマー(ETFE)、ヘキサフルオロプロペンとフッ化ビニリデンとのコポリマー(HFP−co−VDF)、フッ化ビニリデンとトリフルオロエチレンとのコポリマー(VDF−co−TrFE)、およびフッ化ビニリデンとトリフルオロエチレンとモノクロロトリフルオロエチレンとのコポリマー(VDF−co−TrFE−co−chloro−TrFE)]、それらのブレンド物の1種およびそれらの組合せの1種;
−セルロースポリマー、たとえば、酢酸セルロース、硝酸セルロース、メチルセルロース、カルボキシメチルセルロース、それらのコポリマーの1種、それらのブレンド物の1種およびそれらの組合せの1種;
−ポリ(アリーレンスルホン)、たとえば、ポリスルホン、ポリエーテルスルホン、ポリアリールスルホン、それらのコポリマーの1種、それらのブレンド物の1種およびそれらの組合せの1種;
−ポリスルフィド、たとえば、ポリ(フェニレンスルフィド);
−ポリ(アリールエーテル)ケトン、たとえば、ポリ(エーテルケトン)、ポリ(エーテルエーテルケトン)、ポリ(エーテルケトンケトン)、それらのコポリマーの1種、それらのブレンド物の1種およびそれらの組合せの1種;
−ポリアミド−イミド;
−ポリ(エーテル)イミド;
−ポリベンズイミダゾール;
−ポリ(インデン/クマロン);
−ポリ(パラキシリレン);
−それらのコポリマーの1種、それらのブレンド物の1種およびそれらの組合せの1種。
【0025】
また別な形態においては、本発明の文脈において実現される(コ)ポリマーが、以下のものからなる群より選択される熱硬化性(コ)ポリマーである:アミノプラスト、たとえば、尿素/ホルムアルデヒド、メラミン/ホルムアルデヒド、メラミン/ホルムアルデヒド−ポリエステル、それらのコポリマーの1種、それらのブレンド物の1種およびそれらの組合せの1種;ポリウレタン;不飽和ポリエステル;ポリシロキサン;フェノール/ホルムアルデヒド、エポキシド、アリルもしくはビニルエステル樹脂;アルキド;ポリウレア;ポリイソシアヌレート;ポリ(ビスマレイミド);ポリベンズイミダゾール;ポリジシクロペンタジエン;それらのコポリマーの1種、それらのブレンド物の1種およびそれらの組合せの1種。
【0026】
同様にして、塩基性の基たとえば三級もしくは二級アミンたとえばピリジン類を担持している(コ)ポリマー、たとえばポリ(4−ビニルピリジン)およびポリ(2−ビニルピリジン)(P4VPおよびP2VP)を使用するか、または、より一般的には芳香族およびニトロ芳香族基を担持するポリマーを使用して本発明を実現することができる。
【0027】
本発明の文脈において使用することが可能なポリマーについてのさらなる情報は、ナウジン(Naudin)による論文(1995)[5]に見ることができる。
【0028】
「酸化処理」という用語は、本発明の文脈においては、採用された固体支持体の表面を酸化すること、および/またはラジカルを形成させることによってさらなる酸化のための表面を調製することを目的とした、処理(または前処理)を意味していると理解されたい。酸化は、特に、固体支持体の表面を、酸素リッチな基たとえば、カルボキシル(−COOH)、ヒドロキシル(−OH)、アルコキシル(−OR,Rについては以下において定義する)、カルボニル(−C=O)、過炭酸(−C−O−OH)、および場合によってはアミド(−CONH)タイプのような基をそれに付着させるかおよび/またはその中に導入することによって、変性させる。
【0029】
この処理がベースとしているのは、固体支持体の表面および/または固体支持体を構成するポリマーの表面で、表面酸化を起こさせるため、および/またはラジカルを形成させることによって表面酸化のための表面を形成させるために、各種の反応剤を使用することである。そのようにして得られた酸化によって、国際公開第2008/078052号パンフレット[4]に記載のプロセスによるグラフト化よりは、良好な付着性および/またはより大量のポリマーが得られるようになる。この処理は、物理的処理または化学的処理をベースとする、主として2種のタイプの表面変性をベースとしている。
【0030】
本発明の第一の実施態様においては、使用される酸化処理が化学的酸化処理である。そのような化学的酸化処理は、フェントン(Fenton)化学反応、アルコール性水酸化カリウムを用いた処理、強酸を用いた処理、水酸化ナトリウムを用いた処理、強酸化剤を用いた処理、オゾンを用いた処理、およびそれらの組合せからなる群より選択するのが有利である。
【0031】
「組合せ」という用語は、その化学的酸化処理が、上に列記した処理の少なくとも2種を採用していることを意味していると考えられたい。例を挙げれば、そのような組合せが、強酸化剤を用いた処理と、それに続くフェントン化学反応からなっていてもよい。
【0032】
「フェントン化学反応」という用語は、1894年にフェントン(Fenton)によって報告された反応を意味していると理解されたい。この反応では、次の反応スキームで表されるように、過酸化水素水溶液を鉄(II)と反応させることによってヒドロキシルラジカルを生成させることが可能となる:
Fe2++H2O2 → Fe3++・OH+OH−
【0033】
本発明の文脈においては、フェントン化学反応は、固体支持体の表面および/または固体支持体を、第一鉄(Fe2+)イオンと式ROORの化合物とを含む溶液と接触させることからなっているが、ここでRは、水素、1〜15個の炭素原子を含むアルキル基、アシル基−COR’(ここでR’は、1〜15個の炭素原子を含むアルキル基を表す)、またはアロイル基−COAr(ここでArは、6〜15個の炭素原子を含む芳香族基を表す)を表している。タイプ・OR(Rは先に定義されたもの)のラジカルは、Fe2+イオンを用いてペルオキシドROORを開裂させることによって得られる。
【0034】
「1〜15個の炭素原子を含むアルキル基」という用語は、直鎖状、分岐状、または環状、そして場合によっては置換された、1〜15個の炭素原子、とりわけ1〜10個の炭素原子、特には2〜6個の炭素原子と、場合によってはヘテロ原子たとえば、N、O、F、Cl、P、Si、Br、またはSを含むアルキル基を意味していると理解されたい。
【0035】
「6〜15個の炭素原子を含む芳香族基」という用語は、本発明の文脈においては、それぞれ3〜10個の原子を含む1個または複数の芳香族環またはヘテロ芳香族環(その1個または複数のヘテロ原子が、N、O、PまたはSであってよい)からなる、場合によっては置換された芳香族基またはヘテロ芳香族基を意味していると理解されたい。
【0036】
「置換された(substituted)」という用語は、本発明の文脈においては、1〜4個の炭素原子を含む直鎖状または分岐状のアルキル基によるか、アミン基によるか、カルボキシル基によるか、および/またはニトロ基によって単置換または多置換されたアルキル基または芳香族基を意味していると理解されたい。
【0037】
第一鉄(Fe2+)イオンは、第一鉄(Fe2+)イオンおよび式ROORの化合物を、有利には0.05M〜5Mの間、とりわけ0.1M〜3Mの間、特には0.25M〜2Mの間の濃度で含む溶液の中に存在している。第一鉄(Fe2+)イオンおよび式ROORの化合物を含む溶液にはさらに、対イオンたとえば、テトラフルオロホウ酸イオン、硫酸イオン、または塩素イオンが含まれる。
【0038】
式ROORの化合物は、0.1M〜5Mの間、とりわけ0.5M〜3Mの間、特には1M〜2.5Mの間の濃度で第一鉄(Fe2+)イオンおよび式ROORの化合物を含む溶液の中に存在している。
【0039】
第一鉄(Fe2+)イオンおよび式ROORの化合物を含む溶液は、酸性溶液であるのが有利である。「酸性溶液」という用語は、7未満、とりわけ2〜4の間、特には3のオーダー(すなわち、3±0.5)のpHを有する溶液を意味していると理解されたいこの溶液にはさらに、とりわけ0.05mM〜50mMの間、特に0.1mM〜10mMの間、さらに特には1mMのオーダー(すなわち、1mM±0.25mM)の濃度で硫酸を含んでいる。
【0040】
フェントン化学反応の処理時間は変化させることができる。限定する訳ではないが例を挙げれば、この時間は、有利には5分〜5時間の間、とりわけ10分〜3時間の間、特に15分〜2時間の間、さらに特には25分のオーダー(すなわち、25±5分)である。
【0041】
アルコール性水酸化カリウムを用いた処理は、固体支持体の表面および/または固体支持体を、アルコール性水酸化カリウム溶液と接触させることからなっている。
【0042】
「アルコール性水酸化カリウム溶液」という用語は、溶媒としてアルコールを含む溶液(以後アルコール性溶液と呼ぶ)で希釈された水酸化カリウムを意味していると理解されたい。このアルコールは、メタノール、エタノール、およびプロパノールからなる群より選択するのが有利である。このアルコールがエタノールであるのが好ましい。
【0043】
アルコール性溶液中のKOHの濃度は、0.1M〜10Mの間、とりわけ0.5M〜5Mの間、特には3.5Mのオーダー(すなわち、3.5M±0.5M)である。
【0044】
アルコール性水酸化カリウムを用いて処理する時間は、変化させることができる。限定する訳ではないが例を挙げれば、この時間は有利には5分〜5時間の間、とりわけ10分〜3時間の間、特には20分〜2時間の間である。
【0045】
アルコール性水酸化カリウムを用いた処理は、フルオロポリマーたとえばPVDFの場合に、より特に適している。その理由は、アルコール性水酸化カリウム溶液によるそれらの酸化が、ポリマーの表面の脱フッ素を起こして、−CH=CF−結合、およびそれらの酸化された同等物を形成するからである。これらの結合は、フッ素原子が存在しているために反応性が高い。
【0046】
強酸を用いた処理は、固体支持体の表面および/または固体支持体を、強酸または強酸混合物の溶液と接触させることからなっている。そのような処理によって、表面酸化により、酸素含有基の数が増える。
【0047】
本発明による強酸を用いた酸化処理の文脈においては、いかなる強酸も使用することができる。限定する訳ではないが例を挙げれば、その強酸は、HCl、H2SO4、HNO3、HClO4、およびそれらの混合物からなる群より選択するのが有利である。
【0048】
強酸(1種または複数)の溶液の強酸の重量比は、変化させることができる。限定する訳ではないが例を挙げれば、この濃度は、有利には10〜100重量%の間、とりわけ50〜95重量%の間、特には70〜90重量%の間である。
【0049】
強酸を用いた処理の時間は、変化させることができる。限定する訳ではないが例を挙げれば、この時間は、有利には1分〜5時間の間、とりわけ1分〜3時間の間、特には1分〜1時間の間である。
【0050】
水酸化ナトリウムを用いた処理は、固体支持体の表面および/または固体支持体を、水酸化ナトリウム溶液と接触させることからなっている。
【0051】
その溶液の水酸化ナトリウムの重量比は、変化させることができる。限定する訳ではないが例を挙げれば、この濃度は、有利には10〜100重量%の間、とりわけ15〜70重量%の間、特には20〜50重量%の間である。
【0052】
水酸化ナトリウムを用いて処理する時間は、変化させることができる。限定する訳ではないが例を挙げれば、この時間は、有利には1分〜5時間の間、とりわけ1分〜3時間の間、特には1分〜1時間の間である。
【0053】
強酸化剤を用いた処理は、固体支持体の表面および/または固体支持体を、強酸化剤の溶液と接触させることからなっている。この強酸化剤の溶液は、中性、酸性、塩基性のいずれであってもよい。
【0054】
この溶液が酸性であるのが、有利である。より具体的には、その強酸化剤は、塩酸、硫酸または硝酸中のKMnO4、K2Cr2O7、KClO3、CrO3、およびそれらの混合物からなる群より選択する。したがって、以下のものが強酸化剤の溶液として考えられる:KMnO4/HCl、K2Cr2O7/HCl、KClO3/HCl、CrO3/HCl、KMnO4/H2SO4、K2Cr2O7/H2SO4、KClO3/H2SO4、CrO3/H2SO4、KMnO4/HNO3、K2Cr2O7/HNO3、KClO3/HNO3、およびCrO3/HNO3。
【0055】
塩酸中、硫酸中、または硝酸中のKMnO4、K2Cr2O7、KClO3またはCrO3の濃度は、有利には10mM〜1Mの間、とりわけ0.1M〜0.5Mの間、特には0.2Mのオーダー(すなわち、0.2M±50mM)である。
【0056】
強酸化剤の溶液の中の塩酸、硫酸、または硝酸の濃度は、有利には0.1M〜10Mの間、とりわけ0.5M〜5Mの間、特には3.5Mのオーダー(すなわち、3.5M±0.5M)である。
【0057】
強酸化剤を用いた処理の時間は、変化させることができる。限定する訳ではないが例を挙げれば、この時間は、有利には1分〜3時間の間、とりわけ5分〜1時間の間、特には10分〜30分の間、さらに特には15分のオーダー(すなわち、15分±5分)である。
【0058】
オゾンを用いた処理は、固体支持体の表面および/または固体支持体を、オゾンに曝露させることからなる。この曝露には、固体支持体の表面および/または固体支持体をオゾンの流れと接触させるか、または固体支持体の表面および/または固体支持体を、オゾンを含む雰囲気の中に置くかの、いずれかを含むことができる。
【0059】
必要なオゾンは、オゾン発生器たとえば、低圧水銀蒸気ランプ(28mW/cm2、254nm)を備えたUVO−クリーナー(UVO−Cleaner)モデル42−200を介して、酸素を多く含むガスたとえば空気、酸素、酸素富化空気または酸素富化ガスから得ることができる。
【0060】
オゾンを用いて処理する時間は、変化させることができる。限定する訳ではないが例を挙げれば、この時間は、有利には30秒〜3時間の間、とりわけ1分〜1時間の間、特には5分〜30分間の間、さらに特には10分のオーダー(10分±3分)である。
【0061】
化学的酸化処理は、電気化学的前処理からなっていてよい。そのような処理は、酸化させる基材を電気化学的に発生させた酸化性溶液の中に浸漬させることからなっているが、これについては、ブルーズ(Brewis)およびダーム(Dahm)の論文(2001)[6]に具体的な記載がある。国際公開第2007/042659号パンフレットにも、その文献ではエレクトロフェントン(ElectroFenton)と呼ばれている、そのような電気化学的前処理についての記載がある[7]。
【0062】
本発明の第二の実施態様においては、使用される酸化処理が物理的酸化処理である。この物理的酸化処理は、乾式ルートによる処理からなっていて、それが目的としているのは、表面を直接酸化させるか、またはラジカルを発生させることによってその表面を後ほど酸化させるための準備をすることである。乾式ルートによるいくつかの処理の場合においては、表面の酸化数を直接的に増加させる必要はない。その理由は、酸化数の増加が、その処理した表面を水溶液の中に浸漬させた後でのみ起きてもよいからである。そのような物理的酸化処理は、火炎処理、コロナ作用による処理、プラズマ処理、UV照射線を用いた処理、X線またはγ線を用いた処理、電子を用いるかまたは重イオンを用いた照射処理、およびそれらの組合せからなる群より選択するのが有利である。
【0063】
「組合せ」という用語は、その物理的酸化処理が、上に列記した処理の少なくとも2種を採用していることを意味していると考えられたい。
【0064】
さらに、本発明の文脈においては、先に定義された化学的酸化処理の少なくとも一つと、先に定義された物理的酸化処理の少なくとも一つとを組み合わせて、固体支持体の表面を処理することができる。
【0065】
火炎処理(flame treatment)または「火炎吹付(flaming)」は、固体支持体の表面および/または固体支持体を火炎に曝露させることからなっている。その火炎は特に、固体支持体の表面からおよび/または固体支持体から、0.1cm〜20cmの間、とりわけ0.3cm〜10cmの間、さらに特には0.5cm〜5cmの間の距離に位置させる。
【0066】
この火炎は、少なくとも2種のガスの混合物から発生させるのが有利であり、その第一のガスおよび第二のガスは、それぞれ、水素、メタン、エタン、およびプロパンからなる群より、および空気、オゾン、および酸素からなる群より選択する。そのようにして得られる火炎の温度は、500℃〜1600℃の間、とりわけ800℃〜1400℃の間、特には1200℃のオーダー(すなわち、1200℃±100℃)である。
【0067】
この高温処理により、活性なエンティティが発生するが、それは、ラジカル、イオン、または励起分子に当てはめることが可能である。それらは、ヒドロキシル、カルボニル、またはカルボキシル官能基の性質を有することができる。
【0068】
火炎処理の時間は、0.01秒〜10秒の間、とりわけ0.015秒〜1秒の間、特には0.02秒〜0.1秒の間である。
【0069】
コロナ作用による処理は、「コロナ放電処理」とも呼ばれているが、数mm、特には1mm〜2mmの距離で離した二つの電極の間に高電圧交流を印加することによって作り出されたイオン化場に、固体支持体の表面および/または固体支持体を曝露させることからなっている。したがって、電位が臨界値を超えるが、アークの形成にはいたらない条件である場合に、導電体を取り囲む媒体のイオン化によって放電がもたらされる。
【0070】
このイオン化の際に、放出された電子が沈殿(precipitate)し、固体支持体の表面および/または固体支持体の周りの媒体の分子にそれらのエネルギーを伝えるが、その媒体が、場合によっては酸素を富化させた空気または不活性ガスであるのが有利である。このことが、固体支持体のポリマー(1種または複数)の鎖を切断し、その媒体の中に存在している化学的エンティティと同時反応をもたらすことになる。そのコロナ作用による処理が、酸素の寄与がない不活性な雰囲気下で実施されたとしても、不純物に基づく酸化が、表面酸化をもたらす。
【0071】
コロナ放電の密度は、有利には10W・min/m2〜500W・min/m2の間、とりわけ20W・min/m2〜400W・min/m2の間、特には30W・min/m2〜300W・min/m2の間である。
【0072】
コロナ作用による処理の時間は、0.1秒〜600秒の間、とりわけ1秒〜120秒の間、特には10秒〜50秒の間である。
【0073】
プラズマ処理は、固体支持体の表面および/または固体支持体をプラズマに曝露させることからなっている。
【0074】
念のために言えば、プラズマはイオン化状態にあるガスであって、便宜的には、物質の第四状態とみなされている。ガスをイオン化させるのに必要なエネルギーは、電磁波(無線周波数またはマイクロ波)の手段によって与えられる。プラズマは、中性分子、イオン、電子、ラジカルエンティティ(化学的に高活性)、および励起エンティティから構成されているが、それらは、物質の表面と反応することになるであろう。
【0075】
「コールド」プラズマと「ホット」プラズマとの間では区別がされていて、プラズマの中に存在しているエンティティのイオン化度に関連して、相互に異なっている。「コールド」プラズマの場合には、反応性エンティティのイオン化度が、10−4未満であるのに対して、「ホット」プラズマでは、10−4よりも高い。「ホット」および「コールド」の用語は、「ホット」プラズマの方が、「コールド」プラズマよりも、はるかに高エネルギーであるという事実に基づいている。本発明における酸化的前処理の場合においては、コールドプラズマの方が、より適している。しかしながら、採用したプラズマがコールドであっても、あるいはホットであっても、いずれに場合においても、その処理は、固体支持体の表面に、酸素リッチな官能基の出現をもたらすこととなる。
【0076】
プラズマは、2種のガスの混合物によって発生させるのが有利であるが、その第一のガスおよび第二のガスはそれぞれ、不活性ガスからなる群から、および空気および酸素からなる群から選択される。
【0077】
プラズマ処理の時間は、1秒〜5分の間、とりわけ10秒〜60秒の間、特には20秒〜40秒の間である。
【0078】
UV照射線を用いた処理は、固体支持体の表面および/または固体支持体をUV光にあてることからなる。
【0079】
採用されるUV光が、10nm〜400nmの間、とりわけ100nm〜380nmの間、特には180nm〜360nmの間の波長を示しているのが有利である。
【0080】
そのようなUV光を発生させるためには、各種のUV光源を使用することができる。例として、以下のものをあげることができる:UVランプ、低圧水銀ランプ、中圧水銀ランプ、高圧水銀ランプ、超高圧水銀ランプ、アーク灯、ハライドランプ、キセノンランプ、レーザー、ArFエキシマレーザー、KrFエキシマレーザー、エキシマランプ、またはシンクロトロン放射。
【0081】
本発明の文脈においては、UV照射線を用いた処理は、5℃〜120℃の間、とりわけ10℃〜80℃の間、特には15℃〜40℃の間の温度で実施することができる。さらに詳しくは、本発明におけるUV照射線を用いた処理は、周囲温度で実施される。「周囲温度」という用語は、20℃±5℃の温度を意味していると理解されたい。
【0082】
このUV照射線を用いた処理は、ガスの存在下、特に酸素および/またはオゾンを多く含むガス、たとえば空気、酸素、オゾン、酸素富化および/またはオゾン富化空気、酸素富化および/またはオゾン富化ガスの存在下に、実施するのが有利である。その理由は、固体支持体の表面の光酸化、すなわち、酸素および/またはオゾンの存在下にUV照射すると、前記固体支持体を構成しているポリマー材料の表面に酸素を導入することが可能となるからである。
【0083】
本発明の文脈においては、UV照射線を用いた処理は、1分〜5時間、とりわけ5分〜1時間、特には10分〜45分続ける。照射は、1回だけ実施してもよいし、あるいは数回、とりわけ2〜20回、特には3〜10回繰り返して実施することもできる。
【0084】
X線またはγ線を用いた処理は、固体支持体の表面および/または固体支持体を、X線またはγ線(すなわち、電磁線)にあてることからなっている。
【0085】
採用するX線が、5pm未満の波長を示し、採用するγ線が、5pm〜10nmの間の波長を示すのが有利である。
【0086】
X線またはγ線は発生させることを可能とし、それを本発明における固体支持体の表面に向かわせることが可能であり、そして当業者には公知の、各種の線源を、本発明の文脈においては、使用することができる。例を挙げれば、γ線は、放射性線源たとえば、ガンマ線装置の中のコバルト−60またはセシウム−137から放出させることができる。
【0087】
本発明の文脈においては、X線またはγ線を用いた処理は、5℃〜120℃の間、とりわけ10℃〜80℃の間、特には15℃〜40℃の間の温度で実施することができる。さらに詳しくは、本発明におけるX線またはγ線を用いた処理は、周囲温度で実施される。「周囲温度」という用語は、20℃±5℃の温度を意味していると理解されたい。
【0088】
このX線またはγ線を用いた処理は、酸素を多く含むガス、たとえば空気、酸素、酸素富化空気、または酸素富化ガスの存在下に実施するのが有利である。その理由は、放出されるフォトンのエネルギーが極めて高いために、フォトンが材料を通過してしまうことが可能ではあるが、その照射の際に酸素が存在していることで、ポリマーの表面に(すなわち固体支持体の表面に)ラジカルが形成されるからである。それらのラジカルが次いで、周辺の酸素と反応して、固体支持体の表面に酸素リッチな官能基を形成する。
【0089】
X線またはγ線を用いた処理の時間は、1分〜2時間の間、とりわけ5分〜1時間の間、特には10分〜30分間の間である。
【0090】
電子を用いるかまたは重イオンを用いた照射処理は、固体支持体の表面および/または固体支持体を、電子ビームにあてるか(「電子照射」とも呼ばれている)、または重イオンを衝突させるか、からなっている。
【0091】
固体支持体の表面および/または固体支持体を電子ビームにあてることからなる工程は、固体支持体の表面および/または固体支持体を、加速された電子のビームを用いて掃引することからなっていてよく、このビームは、電子加速器(たとえば、ファン・デ・グラーフ(Van de Graaf)加速器、2.5MeV)によって放出させることが可能である。電子線の線量は、5kGy〜1000kGy、とりわけ10kGy〜500kGy、特には50kGy〜150kGyの範囲で変化させることができる。
【0092】
「重イオン」という用語は、炭素の質量よりも大きい質量を有するイオンを意味していると理解されたい。一般的には、それらは、クリプトン、鉛、およびキセノンから選択されるイオンである。この工程は、重イオンのビーム、たとえばPbのビームまたはKrイオンのビームを用いて、固体支持体の表面および/または固体支持体に衝突させることからなっていてよい。イオンビームの強度は、有利には0.1MeV/amu〜100MeV/amuの間、とりわけ1MeV/amu〜50MeV/amuの間、特には4MeV/amu〜20MeV/amuの間である。
【0093】
本発明の文脈においては、電子を用いるかまたは重イオンを用いた照射処理は、5℃〜120℃の間、とりわけ10℃〜80℃の間、特には15℃〜40℃の間の温度で実施することができる。さらに詳しくは、本発明における電子を用いるかまたは重イオンを用いた照射処理は、周囲温度で実施される。「周囲温度」という用語は、20℃±5℃の温度を意味していると理解されたい。
【0094】
この電子を用いるかまたは重イオンを用いた照射処理は、酸素を多く含むガス、たとえば空気、酸素、酸素富化空気もしくは酸素富化ガスの存在下、またはそのようなガスに曝露させた後に実施するのが有利である。先にも述べたように、酸素リッチな雰囲気下またはそのような雰囲気に曝露させた後に、電子を用いるかまたは重イオンを用いた照射をすると、ポリマーの表面の上に酸素を導入することが可能となる[8]。
【0095】
電子を用いるかまたは重イオンを用いた照射処理の時間は、1分〜2時間の間、とりわけ5分〜1時間の間、特には10分〜30分間の間である。
【0096】
本発明によるプロセスの一つの特定の実施態様においては、物理的および化学的両方の酸化処理が、マスクの存在下に実施される。この酸化処理の際にマスクを使用することによって、「ブランクの」表面と、「酸化された」(すなわち、処理された)表面との間で反応性に差をつけることが可能となる。この反応性における差は、パターン化された表面を作り出すのに利用することが可能であり、その理由は、国際公開第2008/078052号パンフレット[4]に記載のプロセスは、「ブランクの」表面に比較して「酸化された」表面において、より効果的となるであろうからである。
【0097】
したがって、その上に有機膜を形成させねばならない固体支持体の表面には、それを少なくとも部分的に覆い、酸化処理の際にそれを保護するマスクが備わっている。そのマスクによって、酸化処理に関連して、その表面の化学的な反応性を局所的に「マスクする」ことが可能となる。
【0098】
そのマスクは典型的には、物理的なエンティティに相当し、表面にグラフト化されてもいないし、また後者に共有結合的に結合されていることもない。具体的には、それは、バルクの物質であっても、あるいは表面の上に析出された無機または有機物質の、典型的には数オングストロームから数ミクロンまでの薄層であってもよい。そのマスクは、穏やかな条件下で容易に剥がすことが可能な、凝集力の低い層として機能する薄層からなっているのが有利である。典型的には、その穏やかな条件とは、一般的にそのマスクが溶解可能な溶媒を用いて実施される単純な化学的洗浄、そのマスクが溶解可能な溶媒の中における超音波を用いた処理、または温度を上げることなどに相当する。
【0099】
したがって、そのマスクを構成する物質は、広い範囲から選択することができる。それは、一般的には、その固体支持体の性質に従って選択されることになるであろう。そのマスクは、アルカンチオール、特には長鎖アルカンチオール、多くの場合C15〜C20、典型的にはC18アルカンチオールから構成されているのが有利である。
【0100】
マスクをした析出方法は、当業者には公知である。具体的には、それらは、コーティング法、スプレー法、または浸漬法とすることができる。したがって、材料の薄層の形態のマスクは、たとえば、選択された材料を含ませたフェルトペンからはじめて直接書き込むか、「パッド(pad)」プロセスによるか、および/または慣用されるリソグラフィー法たとえばスピンコーティング法のいずれかによって析出させ、それに続けて物理的マスクを通すか、または誘導することが可能な光もしくは粒子のビームを介して照射し、次いで現像させることができる。
【0101】
固体支持体の表面および/または固体支持体を先に定義されたような酸化処理にかけたら、ラジカル反応的グラフト化の工程の前に、その固体支持体の表面および/または固体支持体を、洗い流すか、洗浄するか、および/または乾燥させることができる。
【0102】
その固体支持体の表面および/または固体支持体を、特に水中、たとえばミリQ(Milli−Q)水中での1回または複数回の洗い流し操作にかけるのが有利である。次いで、その固体支持体の表面および/または固体支持体を、5分〜30分、とりわけ10分のオーダー(すなわち、10分±2分)の時間、超音波を用いた処理にかけてから、乾燥させる。
【0103】
工程(ii)の際の「ラジカル反応的グラフト化」という用語は、具体的には、固体支持体の表面と共有結合タイプの結合を形成させるために、不対電子を有する分子的エンティティを使用することを指しているが、前記分子的エンティティは、それらをグラフト化させるつもりの表面とは独立して発生させる。したがって、ラジカル反応によって、固体支持体の表面と有機膜との間に共有結合が生成することとなる。
【0104】
固体支持体またはこの支持体の少なくとも表面を構成しているポリマーにグラフト化される有機膜は、50nm未満、有利には40nm未満、とりわけ30nm未満、特には20nm未満の長さを示す。
【0105】
固体支持体またはこの支持体の少なくとも表面を構成しているポリマーにグラフト化される有機膜は、いくつかの同一および/または異なったモノマー単位から主として得られる(コ)ポリマーである。
【0106】
本発明の文脈において採用されるモノマー単位の全部または一部が、ラジカル経路によって重合可能なモノマーであるのが有利である。「ラジカル経路によって重合可能なモノマー」という用語は、ラジカル性化学的エンティティによって開始された後に、ラジカル条件下で重合可能なモノマーを意味していると理解されたい。典型的には、それらは、エチレン性のタイプの少なくとも一つの結合を含むモノマーである、すなわちそれらは、エチレン性のタイプの分子である。
【0107】
ビニルモノマー、具体的には、国際公開第2005/033378号パンフレットおよび国際公開第2006/097611号パンフレットに記載されているモノマーが、特に関連する[9、10]。
【0108】
本発明の特に有利な実施態様においては、その1種または複数のビニルモノマーが、次の式(II)のモノマーから選択される:
【0109】
【化1】
【0110】
[式中、R1〜R4基(同一であっても、異なっていてもよい)は、1価の非金属原子たとえば、ハロゲン原子もしくは水素原子、または飽和もしくは不飽和の化学基たとえば、アルキルもしくはアリール基、ニトリル、カルボニル、アミン、アミド、または−COOR5基(ここで、R5は、水素原子またはC1〜C12、好ましくはC1〜C6のアルキル基を表す)を表す。]
【0111】
上述の式(II)のモノマーは、具体的には、以下のものからなる群より選択される:アクリル酸、酢酸ビニル、アクリロニトリル、メタクリロニトリル、メタクリル酸メチル、メタクリル酸エチル、メタクリル酸ブチル、メタクリル酸プロピル、メタクリル酸ヒドロキシエチル(HEMA)、メタクリル酸ヒドロキシプロピル、メタクリル酸グリシジルおよびそれらの誘導体;エチル−、プロピル−、ブチル−、ペンチル−、およびヘキシル−アクリルアミド、特に−メタクリルアミド、シアノアクリレート、ジアクリレート、およびジメタクリレート、トリアクリレートおよびトリメタクリレート、テトラアクリレートおよびテトラメタクリレート(たとえば、ペンタエリスリトールテトラメタクリレート)、スチレンおよびその誘導体、パラ−クロロスチレン、ペンタフルオロスチレン、N−ビニルピロリドン、4−ビニル−ピリジン、2−ビニルピリジン、ビニル、アクリロイルまたはメタクリロイルハライド、ジビニルベンゼン(DVB)、ならびにより一般的には、ビニル架橋剤または、アクリレート、メタクリレート、およびそれらの誘導体をベースとする架橋剤。
【0112】
有機膜は、固体支持体の表面に間接的にグラフト化される。「間接的なグラフト化」という用語は、分子的エンティティが、有機膜を構成している(コ)ポリマーを固体支持体の表面から分離しているケースを意味していると理解されたい。したがって、この分子的エンティティは、一つの共有結合によって固体支持体の表面に結合され、また別な共有結合によって有機膜に結合されている。
【0113】
そのような分子的エンティティは、先に定義された1種または複数のモノマーとは別の接着プライマーから得られたものであるのが有利である。より詳しくは、そのような接着プライマーは、まず第一に、ラジカル反応的グラフト化によって固体支持体の表面と反応し、次いで、その接着プライマーから得られたグラフト化誘導体が、ラジカル反応によって、先に定義されたモノマーと反応する。この代替えの形態においては、有機膜を形成する(コ)ポリマーが、場合によっては、それを構成するいくつかのモノマー単位の間に、前記ポリマーを固体支持体の表面にグラフト化させるために使用される接着プライマーから生じる一つまたは複数の単位を含むことができるということは明らかである。
【0114】
「接着プライマー」という用語は、本発明の文脈においては、ある種の条件下においては、ラジカル反応、たとえばラジカル反応的グラフト化によって固体支持体の表面に化学吸着される、各種の有機分子を意味していると理解されたい。そのような分子には、ラジカルと反応することが可能な少なくとも一つの官能基と、さらに化学吸着の後に他のラジカルに対して反応性である官能基とを含んでいる。
【0115】
その接着プライマーが、アリールジアゾニウム塩、アリールアンモニウム塩、アリールホスホニウム塩、およびアリールスルホニウム塩からなる群より選択される開裂可能なアリール塩であるのが有利である。これらの塩では、そのアリール基は、以下において定義されるR6で表すことが可能なアリール基である。
【0116】
開裂可能なアリール塩の中でも、次の式(I)の化合物を特に挙げることができる:
R6−N2+A− (I)
[式中、
−Aは、1価のアニオンを表し、そして
−R6は、アリール基を表す。]
【0117】
開裂可能なアリール塩、特に上述の式(I)の化合物のアリール基としては、有利には、芳香族炭素またはヘテロ芳香族構造を挙げることができるが、それらは場合によっては一置換または多置換されており、それぞれ3〜8個の原子を含み、その1個または複数のヘテロ原子がN、O、PまたはSであることが可能な一つまたは複数の芳香族またはヘテロ芳香族環からなっている。その1個または複数の置換基には、1個または複数のヘテロ原子たとえば、N、O、P、Cl、P、Si、BrまたはS,および特にC1〜C6アルキル基を含むことができる。
【0118】
開裂可能なアリール塩、特に上述の式(I)の化合物の中では、R6を、電子求引性基、たとえばNO2、COH、ケトン、CN、CO2H、NH2(NH3+の形で)、エステル、およびハロゲンによって置換されたアリール基から選択するのが好ましい。特に好ましいタイプの、アリールのR6基は、カルボキシフェニル、アミノフェニル、ニトロフェニル、およびフェニル基である。
【0119】
上述の式(I)の化合物の中では、Aは特に、以下のものから選択することができる:無機アニオン、たとえばハライド、たとえばI−、Br−、およびCl−、ハロホウ酸塩、たとえばテトラフルオロホウ酸塩、過塩素酸塩およびスルホン酸塩、ならびに有機アニオン、たとえばアルコキシドおよびカルボキシレート。
【0120】
式(I)の化合物としては、以下のものからなる群より選択される化合物を使用するのが特に有利である:フェニルジアゾニウムテトラフルオロボレート、4−ニトロフェニルジアゾニウムテトラフルオロボレート、4−ブロモ−フェニル−ジアゾニウムテトラフルオロボレート、4−アミノフェニル−ジアゾニウムクロリド、4−アミノメチルフェニルジアゾニウムクロリド、2−メチル−4−クロロフェニルジアゾニウムクロリド、4−ベンゾイルベンゼンジアゾニウムテトラフルオロボレート、4−シアノ−フェニルジアゾニウムテトラフルオロボレート、4−カルボキシフェニル−ジアゾニウムテトラフルオロボレート、4−アセトアミドフェニルジアゾニウムテトラフルオロボレート、4−カルボキシメチルフェニルジアゾニウムテトラフルオロボレート、2−メチル−4−[(2−メチルフェニル)ジアゼニル]ベンゼンジアゾニウムスルフェート、9,10−ジオキソ−9,10−ジヒドロ−1−アントラセンジアゾニウムクロリド、4−ニトロナフタレンジアゾニウムテトラフルオロボレート、およびナフタレンジアゾニウムテトラフルオロボレート。
【0121】
第一の代替えの形態においては、本発明によるプロセスの工程(ii)には以下のものからなる工程を含むことができる:
a)少なくとも1種の溶媒の存在下に、少なくとも1種のモノマー(特に先に定義されたもの)を、前記モノマー(特に先に定義されたもの)とは異なる少なくとも1種の接着プライマーを含む溶液に添加する工程、
b)工程(a)において得られた溶液を、その接着プライマーからラジカルエンティティを生成させることを可能とする、非電気化学的条件下に置く工程、
c)固体支持体の表面を、工程(b)の溶液と接触させる工程。
【0122】
第二の代替えの形態においては、本発明によるプロセスの工程(ii)には以下のものからなる工程を含むことができる:
a’)少なくとも1種の溶媒の存在下に、固体支持体の表面を、少なくとも1種の接着プライマー(特に先に定義されたもの)と、場合によっては前記接着プライマー(特に先に定義されたもの)とは異なる少なくとも1種のモノマーとを含む溶液と接触させる工程、
b’)その固体支持体の表面を、前記接着プライマーからラジカルエンティティを生成させることを可能とする非電気化学的条件下に、工程(a’)の溶液と接触状態に置く工程、
c’)場合によっては、前記接着プライマー(特に先に定義されたもの)とは異なる少なくとも1種のモノマーを工程(b’)で得られた溶液に添加する工程。
【0123】
先に定義されたプロセスの第二の代替えの形態において、以下の三つのシナリオを描くことが可能である。
【0124】
シナリオ1.
工程(a’)の溶液の中にはモノマーは存在せず、工程(c’)においてのみ添加する。このシナリオは、使用するモノマーが、特に水溶液中において、極めて溶解性が高い訳ではないという場合;そのモノマーが水に不溶性であり、接着プライマーがジアゾニウム塩である場合には、特に有利である。したがって、工程(c’)においては、そのモノマーを、溶液中、特に工程(a’)において採用されたのと同じ溶媒の中に、そして有利には超音波または界面活性剤を使用して予め製造しておいたエマルションまたは分散体の形態で添加することができる。
【0125】
シナリオ2.
工程(a’)の溶液の中にモノマーが存在し、そのプロセスには、工程(c’)がない。このシナリオは、そのプライマーがジアゾニウム塩である場合、そしてそのモノマーが水溶性である場合に特にあてはまる。第一の代替えの形態に従ったプロセスも、このシナリオで使用することもできる。
【0126】
シナリオ3.
モノマーの一部が、工程(a’)の溶液の中に存在し、そして(本質的に同一であったも、異なっていてもよい)モノマーの他の部分が、工程(c’)の中にのみ添加される。
【0127】
接着プライマーは、溶液(1種または複数)の中に導入してもよいし、あるいは後者の中でインサイチューで調製してもよい。
【0128】
接着プライマーをインサイチューで調製する場合には、たとえば接着プライマーの前駆体を使用するのが有利である。「接着プライマーの前駆体」という用語は、本発明の文脈においては、実施することが容易な単一の操作工程によって、前記接着プライマーから分離される分子を意味していると理解されたい、
【0129】
一般的には、それらの前駆体は、同一の環境条件下では、接着プライマーよりも高い安定性を示す。たとえば、アリールアミンは、アリールジアゾニウム塩の前駆体である。この理由は、たとえば酸性水性媒体中でのNaNO2との、あるいは有機媒体中でのNOBF4との単純な反応によって、対応するアリールジアゾニウム塩を生成させることが可能であるからである。
【0130】
本発明の文脈において採用される好適な前駆体は、次の式(III)のアリールジアゾニウム塩の前駆体である:
R6−NH2 (III)
[R6は、先に定義されたものである。]
【0131】
限定する訳ではないが例を挙げれば、本発明の文脈において採用することが可能な前駆体は、特に、4−アミノフェニルアミン(すなわち、p−フェニレンジアミン、または1,4−ジアミノフェニレン)、4−アミノ安息香酸、および4−アミノメチルフェニルアミンからなる群より選択される。
【0132】
工程(a)および(a’)の溶液中での接着プライマーまたは接着プライマー前駆体の量は、実験担当者の望むように変化させることができる。この量は、有利にはほぼ10−6M〜5Mの間、好ましくは、5×10−2M〜10−1Mの間である。
【0133】
工程(a)、(a’)および/または(c’)の溶液の中の重合性モノマーの量は、実験担当者の望むように変化させることができる。この量は、採用されるこれらの溶液の溶媒(すなわち、反応溶媒)中への、考慮対象のモノマーの溶解度よりも高くすることも可能であり、所定の温度、一般的には周囲温度または反応温度における、その溶液中での前記モノマーの溶解度のたとえば18〜40倍とすることができる。それらの条件下では、モノマーの分子を溶液の中に分散させることを可能とする手段、たとえば界面活性剤または超音波を採用するのが有利である。
【0134】
その溶媒が、本発明の特定の実施態様に従って、プロトン性溶媒である場合には、そのモノマーが5×10−2Mよりも低い溶解度を示すときには、界面活性剤を使用することが推奨される。使用可能な界面活性剤は、具体的には、アニオン性界面活性剤、カチオン性界面活性剤、両性イオン性界面活性剤、両性界面活性剤、中性(ノニオン性)界面活性剤、および特には、国際公開第2008/078052号パンフレット[4]に記載されている界面活性剤である。界面活性剤の濃度は、典型的にはほぼ0.5mM〜5Mの間、好ましくはほぼ0.1mM〜150mMの間となるであろう。推奨される界面活性剤の濃度は、通常、10mMである。
【0135】
プロセスの工程(ii)は、一般的には、その上に膜をグラフト化することを望んでいるサンプルの表面に対しても、採用したモノマーに対しても、破壊的ではない穏やかな条件下で実施する。したがって、モノマーが分解しないような条件下で操作するのが望ましい。さらに、溶液の温度は、反応溶媒によって制限され、反応溶媒を液状に維持できるのが好ましい。典型的には、そのプロセスは、0〜100℃の間、一般的には標準温度圧力条件(STPC)下、利用者が見出した場所に従って、多くの場合ほぼ25℃、1気圧前後で実施する。
【0136】
本発明によるプロセスの工程(a)、(a’)、(b)、(b’)、(c)、または(c’)の際に採用される溶液には、溶媒として、以下に記載の溶媒を使用することができる:
−プロトン性溶媒、すなわち、プロトンの形態で放出されることが可能な少なくとも1個の水素原子を含む溶媒であって、有利には、水、脱イオン水、蒸留水(それらの水は酸性化されていても、塩基性であってもよい)、酢酸、ヒドロキシル化溶媒たとえばメタノールおよびエタノール、低分子量液状グリコールたとえばエチレングリコール、およびそれらの混合物からなる群より選択される溶媒か;
−または、非プロトン性溶媒、すなわち、プロトンを放出することができないか、または極端な条件以外ではそれらの一つを受け入れることができない溶媒であって、有利には、ジメチルホルムアミド(DMF)、アセトン、アセトニトリル、およびジメチルスルホキシド(DMSO)から選択される溶媒;
−または、少なくとも1種のプロトン性溶媒と少なくとも1種の非プロトン性溶媒との混合物。
【0137】
本発明のプロセスの工程(b)または(b’)において、少なくとも1種のラジカルエンティティの形成を可能とする条件は、工程(b)もしくは(b’)の溶液または固体支持体に、電圧をまったく印加しなくても、ラジカルエンティティの形成が可能となるような条件である。
【0138】
それらの条件としては、たとえば、温度、溶媒の性質、特定の添加剤の存在、撹拌、または圧力などのパラメーターが挙げられるが、その一方で、ラジカルエンティティの形成の際には電流は含まれない。ラジカルエンティティの形成を可能とする条件は多く存在しているが、このタイプの反応は公知であり、従来技術において詳しく検討されている。
【0139】
したがって、たとえば、ラジカルエンティティを形成させるために、接着プライマーを不安定化させる目的で、熱的、動力学的、化学的、光化学的、または放射化学的環境を作用させることが可能である。言うまでもないことであるが、いくつかのそれらのパラメーターを同時に作用させることも可能である。
【0140】
本発明の文脈においては、本発明におけるグラフト化工程の際にラジカルエンティティの生成を可能とする条件は、その接着プライマーが置かれる、典型的には、熱的条件、動力学的条件、化学的条件、光化学的条件、放射化学的条件、およびそれらの組合せからなる群より選択される。本発明によるプロセスのグラフト化工程の文脈において採用される条件が、熱的条件、化学的条件、光化学的条件、放射化学的条件、およびそれらの相互の組合せ、および/または動力学的条件との組合せ、からなる群より選択されるのが有利である。本発明によるプロセスのグラフト化工程の文脈において採用される条件は、より具体的には、化学的条件である。
【0141】
熱的環境は温度に依存する。それは、当業者によって通常採用される加熱手段を使用すれば、容易に調節される。サーモスタットで調節する環境を使用するのが、特に有利であるが、その理由は、反応条件を正確に調節することが可能となるからである。
【0142】
動力学的環境は、実質的には、系の撹拌および摩擦力に対応する。この場合、それは、本来的な分子の撹拌(結合の伸びなど)ではなく、分子全体の動きの撹拌である。
【0143】
したがって、前記グラフト化工程の際に、工程(b)または(b’)の溶液を、機械的撹拌または超音波を用いた処理にかける。第一の代替えの形態においては、工程(b)または(b’)の際に採用される溶液を、マグネチックスターラーおよびマグネチックバーを介して、5分〜24時間の間、とりわけ10分〜12時間の間、特には15分〜6時間の間の撹拌時間で、高回転速度にかける。第二の代替えの形態においては、工程(b)または(b’)の際に採用される溶液を、具体的には、典型的には吸収性能(absorption capacity)が500W、周波数が25kHzまたは45kHzの超音波浴を使用し、1分〜24時間の間、とりわけ15分〜12時間の間、特には30分〜6時間の間の撹拌時間で、超音波を用いた処理にかける。
【0144】
最後に、たとえば電磁線、γ線、UV線、電子ビームまたはイオンビームのような、各種のタイプの放射線の作用もまた、ラジカルを形成させるに十分なほど接着プライマーを不安定化させることができる。使用する接着プライマーに合わせて、採用する波長を選択することになるが、その選択は、発明に値するようなことではない。
【0145】
化学的条件の文脈においては、1種または複数の化学的重合開始剤が、工程(b)または(b’)の際に使用される溶液において採用される。化学的重合開始剤を存在させることが、たとえば先に挙げたような非化学的環境条件と組み合わされることも多い。典型的には、化学的重合開始剤(その安定性が、選択された環境条件下においては、採用された接着プライマーの安定性よりも低い)が、不安定な形態に変化し、それが後者に作用して、後者から始まる、ラジカルエンティティの生成をもたらすことになるであろう。環境条件に本質的にはつながらない作用を有する化学的重合開始剤を採用することもまた可能であり、それらの重合開始剤は、熱的条件またはさらには動力学的条件の広い範囲にわたって作用を有することができる。重合開始剤は、反応の環境、たとえば採用される溶媒に適したものであるのが好ましいであろう。
【0146】
多くの化学的重合開始剤が存在している。一般的には、採用される環境条件に従って、それらは三つのタイプに分類される:
−熱重合開始剤、その最も一般的なものは、ペルオキシド化合物またはアゾ化合物である。熱の作用が加わると、これらの化合物が分解して、フリーラジカルとなる。この場合においては、その反応は、重合開始剤から生ずるラジカルの発生のために必要とされる温度に相当する、最低温度で実施される。このタイプの化学的重合開始剤は、一般的には、それらの分解の動力学に従って、ある種の温度範囲内に限定して使用される;
−放射線によって(一般的にはUV照射線によるが、γ線または電子ビームによっても)引き起こされる照射によって励起される、光化学的重合開始剤または放射化学的重合開始剤は、多少なりとも複雑なメカニズムによって、ラジカルの発生を可能とする。光化学的重合開始剤または放射化学的重合開始剤としては、Bu3SnHおよびI2が挙げられる;
−本質的に化学的重合開始剤、このタイプの重合開始剤は、迅速かつ標準温度圧力条件で接着プライマーに作用して、それからラジカルを生成させる。そのような重合開始剤は、一般的には、反応条件下では使用される接着プライマーの還元電位よりも低い酸化/還元電位を有している。したがって、接着プライマーの性質に従って、それらは、接着プライマーの不安定化を起こさせるに十分な割合の、たとえば、還元性金属、たとえば鉄、亜鉛、またはニッケル;メタロセン;有機還元剤、たとえば次亜リン酸(H3PO2)またはアスコルビン酸;または有機もしくは無機塩基であってよい。
【0147】
化学的重合開始剤として使用される還元性金属は、微細な形態、たとえば金属ウールまたは金属の削り屑(metal filings)として加えるのが有利である。一般的には、化学的重合開始剤として有機または無機の塩基を使用する場合には、4以上のpHであれば一般的には十分である。ラジカルリザーバータイプの構造、たとえば、電子ビームを用いるか、または重イオンのビームを用いるか、および/または上述の照射手段を組み合わせて用いることによって予め照射しておいたポリマーマトリックスを、接着プライマーを不安定化させ、そしてその結果このプライマーからラジカルエンティティを生成させるための化学的重合開始剤として採用することもまた可能である。
【0148】
本発明によるプロセスの工程(ii)は、先に定義されたグラフト化工程に相当する。それは、10分〜6時間、とりわけ30分〜4時間、特には1時間〜2時間、さらに特にはほぼ90分(すなわち、90分±10分)のあいだ、続けることができる。
【0149】
ラジカル経路によって重合することが可能な接着プライマーおよびモノマーが、工程(c)または(c’)の溶液の中には大量に存在しているので、それらの分子が固体支持体に結合されるより前にグラフト化工程を停止させることができる。当業者ならば、グラフト化工程を停止させることを可能とする別な方法を知っているし、採用した接着プライマーおよび重合性モノマーに対して最も適した方法の決め方も知っていることであろう。そのような方法の例として、以下のようなものをあげることができる:工程(c)または(c’)の溶液のpHを、特にそれに塩基性の溶液(たとえば、pHが10よりも高い塩基性の水)を添加することによって変化させる方法、工程(c)または(c’)の溶液から(たとえば濾過によるか沈殿によるか、または錯化によって)接着プライマーおよび重合性モノマーを除去する方法、または工程(c)または(c’)の溶液から固体支持体を抜き出す方法。
【0150】
本発明はさらに、表面の一部にグラフト化させた、本発明によるプロセスによって調製可能な、先に定義されたような有機膜を有する固体支持体にも関する。本明細書で定義されたような酸化的前処理を用いると、グラフト化された有機膜がより厚く、そのためにそのようにグラフト化された支持体の物理化学的性質が極めて大きく改質される。さらには、ポリマーの接触角、したがって親水性の改質の観点からは、このことは、本特許出願において広く確認される。接触角を変化させることは、その物質の化学的な性質を変えることになる。
【0151】
本発明はさらに、先に定義された有機膜を固体支持体の表面の全部または一部にラジカル反応的にグラフト化させるためのプロセスを改良するために、酸化処理を使用することにも関する。
【0152】
この改良は、酸化処理の後に先に定義された有機膜を用いて、ラジカル反応的グラフト化によってグラフト化された固体支持体の上に位置する液滴の接触角を、酸化処理なしの有機膜を用いてラジカル反応的グラフト化によってグラフト化された固体支持体の上に位置する液滴の接触角と比較することによって、証明することができる。
【0153】
酸化処理の後でグラフト化された固体支持体の上の液滴の接触角は、酸化処理なしでグラフト化された固体支持体の上の液滴の接触角に比較して、とりわけ5〜70%、特には20〜50%小さくなる。
【0154】
この改良はさらに、酸化処理の後でラジカル反応的グラフト化によって固体支持体にグラフト化された先に定義されたような有機膜の厚みを、酸化処理なしで固体支持体に同一の条件下(同一の溶媒、同一の接着プライマー、および同一のモノマー、同一のグラフト化時間)でグラフト化された有機膜の厚みと比較することによって証明することもできる。この厚みにおける増加は、IR分析によって可視化することができる。これは、IR分析によると、グラフト化された有機膜に帰属された吸収帯が、膜の厚みが増加したと言えるほど、よりはっきりと見えるようになっているからである。
【0155】
添付した図面を参照し、以下の実施例(説明のためであって、限定を意図するものではない)を読めば、本発明のその他の特徴および利点は、当業者に、より明らかとなるであろう。
【図面の簡単な説明】
【0156】
【図1】以下の表面の上に載せた水の液滴について測定した接触角を示す図である:ABS、ABS/PC、PVDFまたはPETから作られている未処理かつ未グラフト化表面(「ブランク」);国際公開第2008/078052号パンフレット[4]に記載のプロセスに従ってPAAを用いてグラフト化されたABS、ABS/PC、PVDFまたはPETから作られている表面(「グラフトファスト(GraftFast)(著作権)」);およびフェントン酸化によって前処理され、次いで国際公開第2008/078052号パンフレット[4]に記載のプロセスに従ってPAAを用いてグラフト化されたABS、ABS/PC、PVDFまたはPETから作られている表面(「前処理+グラフトファスト(GraftFast)(著作権)」)。
【図2】以下の表面の上に載せた水の液滴について測定した接触角を示す図である:PVDFから作られている未処理かつ未グラフト化の表面(「ブランク」);国際公開第2008/078052号パンフレット[4]に記載のプロセスに従ってPAAを用いてグラフト化されたPVDFから作られている表面(「グラフトファスト(GraftFast)(著作権)」);および、アルコール性水酸化カリウムを用いて前処理され、次いで国際公開第2008/078052号パンフレット[4]に記載のプロセスに従ってPAAを用いてグラフト化されたPVDFから作られている表面(「前処理+フラフトファスト(GraftFast)(著作権)」)。
【図3】以下の表面の上に載せた水の液滴について測定した接触角を示す図である:PP、PET、PVDFまたはPCから作られている未処理かつ未グラフト化の表面(「ブランク」);国際公開第2008/078052号パンフレット[4]に記載のプロセスに従ってPAAを用いてグラフト化されたPP、PET、PVDFまたはPCから作られている表面(「グラフトファスト(GraftFast)(著作権)」);および、オゾン用いて前処理され、次いで国際公開第2008/078052号パンフレット[4]に記載のプロセスに従ってPAAを用いてグラフト化されたPP、PET、PVDFまたはPCから作られている表面(「前処理+フラフトファスト(GraftFast)(著作権)」)。
【図4】以下の表面の上に載せた水の液滴について測定した接触角を示す図である:ABS、ABS/PC、またはPPから作られている未処理かつ未グラフト化表面(「ブランク」);国際公開第2008/078052号パンフレット[4]に記載のプロセスに従ってpHEMAを用いてグラフト化されたABS、ABS/PC、またはPPから作られている表面(「グラフトファスト(GraftFast)(著作権)」);およびフェントン酸化によって前処理され、次いで国際公開第2008/078052号パンフレット[4]に記載のプロセスに従ってpHEMAを用いてグラフト化されたABS、ABS/PC、またはPPから作られている表面(「前処理+フラフトファスト(GraftFast)(著作権)」)。
【図5】以下の表面の上に載せた水の液滴について測定した接触角を示す図である:PPから作られている未処理かつ未グラフト化表面(「ブランク」);国際公開第2008/078052号パンフレット[4]に記載のプロセスに従ってp(4−VP)を用いてグラフト化されたPPから作られている表面(「グラフトファスト(GraftFast)(著作権)」);および、フェントン酸化によって前処理され、次いで国際公開第2008/078052号パンフレット[4]に記載のプロセスに従ってp(4−VP)を用いてグラフト化されたPPから作られている表面(「前処理+フラフトファスト(GraftFast)(著作権)」)。
【発明を実施するための形態】
【0157】
以下の実施例は、ガラス容器の中で実施した。
【0158】
特に断らない限り、それらは、大気中、標準温度圧力条件下(ほぼ25℃、ほぼ1気圧)で実施した。特に断らない限り、採用した反応剤は市場で直接購入したものであって、さらなる精製は行わなかった。
【0159】
金の細片およびプラスチックまたはポリマーのサンプルは、1×5cm2の表面積を有していた。試験をしたプラスチックのサンプルには、アクリロニトリル/ブタジエン/スチレン(ABS)、アクリロニトリル/ブタジエン/スチレン−ポリカーボネート(ABS/PC)、ポリプロピレン(PP)、およびポリアミド(PA)が含まれる。試験したポリマーのサンプルには、ポリフッ化ビニリデン(PVDF)、ポリカーボネート(PC)、およびポリエチレンテレフタレート(PET)が含まれる。
【0160】
雰囲気の組成には何の注意も払わなかったし、溶液の脱気も行わなかった。プラスチックまたはポリマーのサンプルはあらかじめ、超音波の下で10分間、工業用洗剤(TDF4)の溶液を使用して清浄化させ、次いでミリQ(Milli−Q)水中で10分間超音波にかけてから、乾燥させた。
【0161】
各種のサンプルは、IR分光測定法によるか、および/またはそれらのサンプルの表面上に載せた一定容積(2μL)の液滴との間の接触角を測定することによって分析した。
【0162】
1:プラスチックおよびポリマーの酸化処理
実施例1.1:鉄(II)テトラフルオロボレートから出発するフェントン反応による酸化
鉄(II)テトラフルオロボレート(1.69g、5×10−2mol)を、水中0.001M硫酸100mLの中に溶解させた。プラスチックおよび/またはポリマーのサンプルをこの溶液の中に浸漬させた。次いで、pH3一定に保ちながら、12mL(0.125mol)の35%過酸化水素水溶液を添加した。25分後に、ミリQ(Milli−Q)水を用いてサンプルを洗い流し、水中で10分間超音波をかけてから、乾燥させた。
【0163】
実施例1.2:硫酸鉄(II)から出発するフェントン反応による酸化
硫酸鉄(II)(3.475g、5×10−2mol)を、0.001M硫酸水溶液25mLの中に溶解させた。プラスチックのサンプルおよび/またはポリマーをこの溶液の中に浸漬させた。次いで、pH3一定に保ちながら、5mL(0.062mol)の35%過酸化水素水溶液を添加した。25分後に、ミリQ(Milli−Q)水を用いてサンプルを洗い流し、水中で10分間超音波をかけてから、乾燥させた。
【0164】
実施例1.3:アルコール性水酸化カリウムを用いた処理による酸化
4g(7.1×10−2mol)の水酸化カリウムを、20mLの95%エタノールの中に溶解させた。この溶液の中に、ポリマーのサンプルを、典型的には20分〜2時間の間で変化させた時間のあいだ、浸漬させた。ミリQ(Milli−Q)水を用いてサンプルを洗い流し、水中で10分間超音波をかけてから、乾燥させた。
【0165】
実施例1.4:オゾンを用いた処理による酸化
各種のプラスチックおよびポリマーを、10分間、オゾンの流れに置いた(オゾン雰囲気に曝露させた)。
【0166】
そのオゾン発生器は、低圧水銀蒸気ランプ(28mW/cm2、254nm)を有する、UVO−クリーナー(UVO−Cleaner)モデル42−200である。
【0167】
実施例1.5:KMnO4を用いた処理による酸化
0.75gの過マンガン酸カリウム(5×10−3mol)を、25mLの3.3M硫酸溶液に添加した。プラスチックおよび/またはポリマーのサンプルをこの溶液の中に15分間浸漬させた。各種のサンプルを、順次、ミリQ(Milli−Q)水を用いて洗い流し、ミリQ(Milli−Q)水中で超音波をかけてから、乾燥させた。
【0168】
ABSまたはABS−PCに関しては、ABSおよびABS−PCの中に存在しているポリブタジエンの小塊を、KMnO4が酸化し、破壊する。ポリアクリロニトリルもまた酸化して、特にポリアミドとすることができる。
【0169】
本発明の文脈において実施される試験では、これらの酸化的改質は、当然のことながらIRスペクトルでは見分けられない。その一方で、それらのポリマーをKMnO4溶液の中に6時間浸漬させることによって、そのような酸化的改質を実証することが可能であった。
【0170】
実施例1.6:KMnO4(実施例1.5)に続けてフェントン反応(実施例1.2)を用いた処理による酸化
実施例1.5に記載された形態に従って、この実施例を実施した。そうして調製されたサンプルを、次いで、フェントン反応のための実施例1.2に記載された反応媒体の中に導入すると、その結果として、それらが清浄化されたので、上述のようにして乾燥させた。
【0171】
実施例1.1〜1.6で得られた結果を、接触角の測定については表1に、そしてIR分光測定法については表2に示す。
【0172】
【表1】
【0173】
【表2】
【0174】
2:国際公開第2008/078052号パンフレット[4]に記載のプロトコールに従った、プラスチックのサンプルおよび/またはポリマーへの膜のグラフト化
手順:
フェントン反応に従った処理をしていないプラスチック(ABS、ABS/PC、PP、およびPA)を、超音波下に石けん水を用いて10分間、次いで、MQ水を用いて10分間かけて清浄化した。
【0175】
実施例2.1:4−アミノ安息香酸から出発する、国際公開第2008/078052号パンフレット[4]に記載のプロセスによるポリアクリル酸膜のグラフト化
4−アミノ安息香酸(2.7g、2×10−2mol)を、塩酸溶液(100mL、0.5M)の中に溶解させた。100mLのNaNO2の水溶液(1.38g、2×10−2mol)を、その溶液に添加した。
【0176】
このジアゾニウム塩溶液に、6.8mLのアクリル酸AA(10−1mol)および2gの鉄の削り屑を添加した。
【0177】
次いで、プラスチックのサンプルおよび/またはポリマーおよび金のストリップを、反応媒体の中に導入し、90分間置いた。ミリQ(Milli−Q)水を用いて各種のサンプルを連続的に洗い流し、0.01M水酸化ナトリウム溶液中で超音波にかけ、次いでミリQ(Milli−Q)水を用いて洗浄してから、乾燥させた。
【0178】
金のストリップのIR分光測定法による分析から、そのグラフト化浴が活性であったことが確認される。3356cm−1(COOH変角振動)、1710cm−1(C=O変角振動)、および1265cm−1(C−O変角振動)に特性吸収帯が存在している。
【0179】
実施例2.2:1,4−ジアミノフェニレンから出発する、国際公開第2008/078052号パンフレット[4]に記載のプロセスによるポリアクリル酸膜のグラフト化
手順1:
この手順は、実施例2.1の場合と同じであるが、ただし、4−アミノ安息香酸を、1,4−ジアミノフェニレン(2.13g、2×10−2mol)に置き換えた。
【0180】
次いで、プラスチックのサンプルおよび/またはポリマーおよび金のストリップを、反応媒体の中に導入し、90分間おいた。ミリQ(Milli−Q)水を用いて各種のサンプルを連続的に洗い流し、0.01M水酸化ナトリウム溶液中で超音波にかけ、次いでミリQ(Milli−Q)水を用いて洗浄してから、乾燥させた。
【0181】
手順2:
4−アミノ安息香酸(1.07g、1×10−2mol)を、塩酸溶液(100mL、0.5M)の中に溶解させた。100mLのNaNO2の水溶液(0.69g、1×10−2mol)を、この溶液に添加した。100mLのアクリル酸AA(1.46mol)および次いで15g(0.27mol)の鉄の削り屑を、このジアゾニウム塩の溶液に添加した。
【0182】
次いで、プラスチックのサンプルおよび/またはポリマーおよび金のストリップを、反応媒体の中に導入し、周囲温度で30分間、38℃のオーブン中に90分間置いた。ミリQ(Milli−Q)水を用いて各種のサンプルを連続的に洗い流し、0.1M水酸化ナトリウム溶液中で超音波にかけ、次いでミリQ(Milli−Q)水を用いて洗浄してから、乾燥させた。
【0183】
金のストリップのIR分光測定法による分析から、その両方の手順において、そのグラフト化浴が活性であったことが確認される。3207cm−1(COOH変角振動)、1720cm−1(C=O変角振動)、および1262cm−1(C−O変角振動)に特性吸収帯が存在している。
【0184】
実施例2.3:4−ニトロベンゼンジアゾニウムテトラフルオロボレートから出発する、国際公開第2008/078052号パンフレット[4]に記載のプロセスによるポリアクリル酸膜のグラフト化
その手順は、実施例2.1の場合と同じである。4−アミノ安息香酸を4−ニトロベンゼンジアゾニウムテトラフルオロボレート(4.7g、2×10−2mol)に置き換えたが、NaNO2の存在下にジアゾニウム塩前駆体をジアゾニウム塩に転換させる工程はもはや必要ない。
【0185】
次いで、プラスチックおよび/またはポリマーのサンプルならびに金のストリップを、反応媒体の中に導入し、90分間置いた。ミリQ(Milli−Q)水を用いて各種のサンプルを連続的に洗い流し、0.01M水酸化ナトリウム溶液中で超音波にかけ、次いでミリQ(Milli−Q)水を用いて洗浄してから、乾燥させた。
【0186】
金のストリップのIR分光測定法による分析から、そのグラフト化浴が活性であったことが確認される。3405cm−1(COOH変角振動)、1695cm−1(C=O変角振動)、1597cm−1(C=C変角振動)、1518cm−1(N=O変角振動)、1345cm−1(N=O変角振動)、および1162cm−1(C−O変角振動)に特性吸収帯が存在している。
【0187】
実施例2.4:4−アミノ安息香酸から出発する、国際公開第2008/078052号パンフレット[4]に記載のプロセスによるポリ(HEMA)膜のグラフト化
その手順は、実施例2.1の場合と同じである。アクリル酸AAを、12.5mLのメタクリル酸2−ヒドロキシエチル(10−1mol)に置き換えた。
【0188】
ミリQ(Milli−Q)水を用いて各種のサンプルを連続的に洗い流し、次いで加熱条件下でエタノールの浴の中に30分間浸漬させてから、乾燥させた。
【0189】
金のストリップのIR分光測定法による分析から、そのグラフト化浴が活性であったことが確認される。3387cm−1(OH変角振動)、1726cm−1(C=O変角振動)、および1160cm−1(C−O変角振動)に特性吸収帯が存在している。
【0190】
実施例2.5:4−アミノ安息香酸から出発する、国際公開第2008/078052号パンフレット[4]に記載のプロセスによるポリ(4−ビニルピリジン)膜のグラフト化
その手順は、実施例2.1の場合と同じである。アクリル酸AAを、100mLの水中0.1Mの4−ビニルピリジン(4−VP)および1MのH2SO4の溶液に置き換えた。
【0191】
ミリQ(Milli−Q)水を用いて各種のサンプルを連続的に洗い流し、次いで加熱条件下でエタノールの浴の中に30分間浸漬させてから、乾燥させた。
【0192】
金のストリップのIR分光測定法による分析から、そのグラフト化浴が活性であったことが確認される。3371cm−1(COOH変角振動)、1600cm−1(C=N変角振動)、および1556cm−1(C=C変角振動)に特性吸収帯が存在している。
【0193】
実施例2.1〜2.5についての接触角の測定から得られた結果を表3に示す。
【0194】
【表3】
【0195】
3:国際公開第2008/078052号パンフレット[4]に記載のプロセスによる膜の表面酸化+グラフト化
実施例3.1:フェントン酸化(実施例1.1)とそれに続く、4−アミノ安息香酸から出発する、国際公開第2008/078052号パンフレット[4]に記載のプロセスによるPAA膜のグラフト化(実施例2.1)
実施例1.1に記載された形態に従って、実施例を実施した。調製されたサンプルを、次いで、グラフト化にかけるために、実施例2.1に記載の反応媒体の中に導入すると、その結果として、それらが清浄化されたので、上述のようにして乾燥させた。
【0196】
実施例3.2:フェントン酸化(実施例1.1)と、それに続く、1,4−ジアミノフェニレンから出発する、国際公開第2008/078052号パンフレット[4]に記載のプロセスによるPAA膜のグラフト化(実施例2.2)
実施例1.1に記載された形態に従って、実施例を実施した。調製されたサンプルを、次いで、グラフト化にかけるために、実施例2.2に記載の反応媒体の中に導入すると、その結果として、それらが清浄化されたので、上述のようにして乾燥させた。
【0197】
実施例3.3:フェントン酸化(実施例1.1)と、それに続く、4−ニトロベンゼンジアゾニウムテトラフルオロボレートから出発する、国際公開第2008/078052号パンフレット[4]に記載のプロセスによるPAA膜のグラフト化(実施例2.3)
実施例1.1に記載された形態に従って、実施例を実施した。調製されたサンプルを、次いで、グラフト化にかけるために、実施例2.3に記載の反応媒体の中に導入すると、その結果として、それらが清浄化されたので、上述のようにして乾燥させた。
【0198】
実施例3.4:アルコール性水酸化カリウムを用いた処理による酸化(実施例1.3)と、それに続く、4−アミノ安息香酸CBDから出発する、国際公開第2008/078052号パンフレット[4]に記載のプロセスによるPAA膜のグラフト化(実施例2.1)
実施例1.3に記載された形態に従って、実施例を実施した。調製されたサンプルを、次いで、グラフト化にかけるために、実施例2.1に記載の反応媒体の中に導入すると、その結果として、それらが清浄化されたので、上述のようにして乾燥させた。
【0199】
実施例3.5:KMnO4を用いた処理による酸化(実施例1.5)と、それに続く、1,4−ジアミノフェニレンから出発する、国際公開第2008/078052号パンフレット[4]に記載のプロセスによるPAA膜のグラフト化(実施例2.2)
実施例1.5に記載された形態に従って、実施例を実施した。調製されたサンプルを、次いで、グラフト化にかけるために、実施例2.2(第二の手順)に記載の反応媒体の中に導入すると、その結果として、それらが清浄化されたので、上述のようにして乾燥させた。
【0200】
実施例3.6:オゾンを用いた処理による酸化(実施例1.4)と、それに続く、4−アミノ安息香酸から出発する、国際公開第2008/078052号パンフレット[4]に記載のプロセスによるPAA膜のグラフト化(実施例2.1)
実施例1.4に記載された形態に従って、実施例を実施した。調製されたサンプルを、次いで、グラフト化にかけるために、実施例2.1に記載の反応媒体の中に導入すると、その結果として、それらが清浄化されたので、上述のようにして乾燥させた。
【0201】
実施例3.7:フェントン酸化(実施例1.1)と、それに続く、4−アミノ安息香酸から出発する、国際公開第2008/078052号パンフレット[4]に記載のプロセスによるpHEMA膜のグラフト化(実施例2.4)
実施例1.1に記載された形態に従って、実施例を実施した。調製されたサンプルを、次いで、グラフト化にかけるために、実施例2.4に記載の反応媒体の中に導入すると、その結果として、それらが清浄化されたので、上述のようにして乾燥させた。
【0202】
実施例3.8:フェントン酸化(実施例1.1)と、それに続く、4−アミノ安息香酸から出発する、国際公開第2008/078052号パンフレット[4]に記載のプロセスによるp(4−VP)膜のグラフト化(実施例2.5)
実施例1.1に記載された形態に従って、実施例を実施した。調製されたサンプルを、次いで、グラフト化にかけるために、実施例2.5に記載の反応媒体の中に導入すると、その結果として、それらが清浄化されたので、上述のようにして乾燥させた。
【0203】
実施例3.9:KMnO4/フェントン酸化(実施例1.5)と、それに続く、1,4−ジアミノフェニレンから出発する、国際公開第2008/078052号パンフレット[4]に記載のプロセスによるPAA膜のグラフト化(実施例2.2)
実施例1.5に記載された形態に従って、実施例を実施した。調製されたサンプルを、次いで、グラフト化にかけるために、実施例2.2(第二の手順)に記載の反応媒体の中に導入すると、その結果として、それらが清浄化されたので、上述のようにして乾燥させた。
【0204】
実施例3.1〜3.9についての接触角の測定から得られた結果を表4に示す。
【0205】
【表4】
【0206】
4:国際公開第2008/078052号パンフレット[4]に記載のプロセスによるポリマー膜の作成におよぼす表面酸化の影響
4.1:国際公開第2008/078052号パンフレット[4]に記載のプロセスによるPAA膜のグラフト化に対する酸化的前処理の改良
フェントン酸化を介しての酸化的前処理をすることによって、国際公開第2008/078052号パンフレット[4]に記載の方法によるポリアクリル酸のグラフト化を向上させることが可能となる。この向上効果は、ブランクのABS、ABS/PC、PVDFまたはPETから作られている支持体、または国際公開第2008/078052号パンフレット[4]に記載のプロセスによってグラフト化されたABS、ABS/PC、PVDFまたはPETから作られている支持体の上に載せた液滴に比較して、前処理をされ、次いで国際公開第2008/078052号パンフレット[4]に記載のプロセスによってグラフト化されたABS、ABS/PC、PVDFまたはPETから作られている支持体の上に載せた液滴について測定された接触角が顕著に小さくなるということに反映されている(図1)。
【0207】
下記の表5は、IR分析によるポリアクリル酸のC=O特性吸収帯の強度を示している。この吸収帯は、本発明におけるフェントン酸化を介しての酸化的前処理に続けてグラフト化させた後のみに観察できる。
【0208】
【表5】
【0209】
アルコール性水酸化カリウムを用いた処理を介しての酸化的前処理をすることによって、国際公開第2008/078052号パンフレット[4]に記載の方法によるポリアクリル酸のグラフト化を向上させることが可能となる。この向上効果は、ブランクのPVDFから作られている支持体、または国際公開第2008/078052号パンフレット[4]に記載のプロセスによってグラフト化されたPVDFから作られている支持体に比較して、前処理をされ、次いで国際公開第2008/078052号パンフレット[4]に記載のプロセスによってグラフト化されたPVDFから作られている支持体の上に載せた液滴について測定された接触角が顕著に小さくなるということに反映されている(図2)。
【0210】
下記の表6は、IR分析によるポリアクリル酸のC=O特性吸収帯の強度を示している。この吸収帯は、本発明におけるアルコール性水酸化カリウムを用いた処理を介しての酸化的前処理に続けてグラフト化させた後にのみに観察できる。
【0211】
【表6】
【0212】
オゾンを用いた処理を介しての酸化的前処理をすることによって、国際公開第2008/078052号パンフレット[4]に記載の方法によるポリアクリル酸のグラフト化を向上させることが可能となる。この向上効果は、ブランクのPVDFから作られている支持体、または国際公開第2008/078052号パンフレット[4]に記載のプロセスによってグラフト化されたPVDFから作られている支持体に比較して、前処理をされ、次いで国際公開第2008/078052号パンフレット[4]に記載のプロセスによってグラフト化されたPVDFから作られている支持体の上に載せた液滴について測定された接触角が顕著に小さくなるということに反映されている(図3)。
【0213】
PETおよびPCの構造には基本的に、酸素含有基が含まれている。したがって、これらのポリマーでは、オゾンを用いた前処理の効果はPPおよびPVDFの場合よりは弱い。下記の表7は、IR分析によるポリアクリル酸のC=O特性吸収帯の強度を示している。この吸収帯は、本発明におけるオゾンを用いた処理を介しての酸化的前処理に続けてグラフト化させた後にのみに観察できる。
【0214】
【表7】
【0215】
4.2:国際公開第2008/078052号パンフレット[4]に記載のプロセスによるpHEMA膜のグラフト化に対する酸化的前処理の改良
フェントン酸化を介しての酸化的前処理をすることによって、国際公開第2008/078052号パンフレット[4]に記載の方法によるpHEMAのグラフト化を向上させることが可能となる。この向上効果は、ブランクのABS、ABS/PC、またはPPから作られている支持体、または国際公開第2008/078052号パンフレット[4]に記載のプロセスによってグラフト化されたABS、ABS/PC、またはPPから作られている支持体の上に載せた液滴に比較して、前処理をされ、次いで国際公開第2008/078052号パンフレット[4]に記載のプロセスによってグラフト化されたABS、ABS/PC、またはPPから作られている支持体の上に載せた液滴について測定された接触角が顕著に小さくなるということに反映されている(図4)。
【0216】
4.3:国際公開第2008/078052号パンフレット[4]に記載のプロセスによるp(4−VP)膜のグラフト化に対する酸化的前処理の改良
フェントン酸化を介しての酸化的前処理をすることによって、国際公開第2008/078052号パンフレット[4]に記載の方法によるp(4−VP)のグラフト化を向上させることが可能となる。この向上効果は、ブランクのPPから作られている支持体、または国際公開第2008/078052号パンフレット[4]に記載のプロセスによってグラフト化されたPPから作られている支持体に比較して、前処理をされ、次いで国際公開第2008/078052号パンフレット[4]に記載のプロセスによってグラフト化されたPPから作られている支持体の上に載せた液滴について測定された接触角が顕著に小さくなるということに反映されている(図5)。
【0217】
引用文献
[1]パラシン(Palacin)ら、「分子−金属結合:導電性表面上のポリマーの電気グラフト化(Molecule−to−metal bonds:Electrografting polymers on conducting surfaces)」、ケミカル・フィジックス・ケミストリー(Chem.Phys.Chem.)、2004、5(10)、1469〜1481。
[2]デニアウ(Deniau)ら、「炭素−金属結合:2−ブテンニトリルの電解還元」、サーフェス・サイエンス(Surf.Sci.)、2006、600(3)、675〜684。
[3]国際公開第03/018212号パンフレット(出願人:CEA,公開日:2003年3月6日)。
[4]国際公開第2008/078052号パンフレット(出願人:CEA、公開日:2008年7月3日)。
[5]ナウディン(Naudin)、「ポリマーの命名、分類および化学式(Nomenclature,classificaiton et formules chimques des polymeres[Nomenclature,Classification and Chemical Formulae of Polymers])」、テクニークス・ド・リンゲイヌール(Techniques de l’Ingenieur)、1995、A3035。
[6]ブルーズ(Brewis)およびダーム(Dahm)、「総説:ポリマーの電気化学的前処理(A review of electrochemical pretreatments of polymes)」、インターナショナル・ジャーナル・オブ・アドヘージョン・アンド・アドヘーシブズ(Intl.J.of Adhesion & Adhesives)、2001、21、397〜409。
[7]国際公開第2007/042659号パンフレット(出願人:Alchimer,公開日:2007年4月19日)。
[8]ゼンキエビッチ(Zenkiewicz)ら、「ポリマー複合材料の表面酸化に及ぼす電子ビーム照射の影響(Effects of electron−beam irradiation on surface oxidation of polymer composites)」、アプライド・サーフェス・サイエンス(Applied Surface Science)、2007、253(22)、8992〜8999。
[9]国際公開第2005/033378号パンフレット(出願人:CEA、公開日:2005年4月14日)。
[10]国際公開第2006/097611号パンフレット(出願人:CEA、公開日:2006年9月21日)。
【技術分野】
【0001】
本発明は、有機表面コーティングの分野に関し、前記コーティングは、有機膜の形態にある。
【0002】
さらに詳しくは、本発明は、導電性または非導電性表面、有利にはプラスチックまたはポリマーから作られている表面の上に、それらの表面を酸化処理するという予備工程を用い、コーティングすることによってそのような有機コーティングを調製するための方法に関する。
【背景技術】
【0003】
現在のところ、基材の上に有機薄膜を調製することを可能とするいくつかの技術が存在している。
【0004】
いかなるタイプの表面にでも適用することが可能な、いくつかの技術では、その膜とコーティングされる表面との間の親和性を必要とせず、それら二つの構成成分の間の物理吸着のみが含まれている。これらの技術は、遠心力によるコーティングの形成(「スピンコーティング」)、浸漬によるコーティングの形成(「ディップコーティング」)、または蒸発による析出(「スプレーコーティング」)の方法を含む。したがって、膜とコーティングされる表面との間には実際のグラフトは存在せず、特に最も薄い析出層(20nm未満)では、得られる厚みを調節することは容易にはできない。
【0005】
支持体の表面に有機コーティングを形成させる他の方法、たとえばプラズマ蒸着または光化学的活性化(すなわち光活性化)は、同一の原理に基づいていて、被覆する表面の近くに不安定な前駆体の形態を発生させ、その不安定な形態が変化して、その基材の上に膜を形成させる。これらの方法は一般的には、付着性の膜を形成させることになるが、その付着が、対象物にトポロジー的に密接した膜の架橋によるものなのか、あるいはその界面に結合が形成されたことによるものかを識別することは通常不可能である。さらに、これらの方法では以下のようなことが必要とされる:特定の前駆体、特に光活性化の場合であれば感光性前駆体、比較的に複雑でコストのかかる前処理、プラズマ法の場合であれば真空下の真空装置の使用、電気化学的方法では照射および/またはポテンシオスタットの使用(これらの場合、接続についてのいくつもの問題が伴う)。
【0006】
電気泳動もまた、有機膜を用いて導電性表面をコーティングするための一つの方法である。電気泳動またはカチオン電着プロセスによって、(析出させるより前に合成しておいた)荷電ポリマーを使用して金属部品を覆うことが可能となり、導電性表面の上に均質な膜を与える方法となる。しかしながら、電着条件を厳密に調節することに加えて、その処理では、電着膜に物理的強度または耐薬品性を付与するためには、さらに焼付け工程が必要となる。
【0007】
単分子層を自己組織化させることは、実施するには極めて単純な方法ではあるが、コーティングの対象となる表面に対して十分な親和性を有する、一般的には分子状の前駆体を必要とする。したがって、ここで使用した用語は、たとえば金または銀に対して親和性を有する硫黄化合物特にチオール官能基を有するもの、酸化物たとえばシリカまたはアルミナに対してはトリハロシラン、あるいはグラファイトまたはカーボンナノチューブに対してはポリ芳香族化合物といった「前駆体と表面のペア」となるであろう。いずれの場合においても、分子の厚み(10nm未満)の膜を形成させることは、前駆体分子の一部と、その表面のある種の「受容体」部位との間の特定の化学反応に基づいている。化学吸着反応によって、付着が確実になる。しかしながら、表面と単分子膜との間の界面結合は、ある種の組合せでは弱い可能性もあるし、あるいは、特定の環境条件、特に湿潤状態では弱くなる可能性がある。
【0008】
ポリマーの電気グラフト化(electrografting)は、電極としておよび重合開始剤としての両方の作用を有する対象物の表面上で電気活性モノマーを、電気的に誘導された連鎖生長反応によって、重合を開始させ、次いで重合させることに基づいた方法である[1]。電気グラフト化では、還元によって開始され、連鎖生長するメカニズムに適合した前駆体を使用することが必要である。電気グラフト化された膜への付着は、炭素−金属共有結合によって与えられる[2]。上で思い起こした各種の方法の中で、電気グラフト化は、界面結合性を特定的に調節したグラフト膜を製造することを可能とする唯一の方法である。具体的には、活性化されたビニルモノマーから得られるポリマーの膜を表面(必然的に導電性表面である)にグラフトさせることを可能とする、その唯一の方法は、ポテンシオスタットを介して、表面から始まる重合反応を電気的に開始させること、それに続けて、次々とモノマーを加えていってその鎖を生長させることからなり、カソードおよびアノードを有する電気化学的セルを使用し、後者の端子に電圧を印加することが必要である。したがって、国際公開第03/018212号パンフレットには、導電性表面の上に導電性有機膜をグラフトさせ、生長させるためのプロセスが特に記載されており、前記有機膜の前駆体のジアゾニウム塩を電解還元させることによって、そのグラフト化と生長とを同時に行わせている[3]。
【0009】
最後に、非電気化学的条件下でグラフト化有機コーティングを作成することを可能とし、いかなるタイプの表面にも容易に実施されるプロセスが、最近報告された。国際公開第2008/078052号パンフレットに記載されているこのプロセスには、簡単な工程で共有結合的なグラフト化を可能とする、ラジカル反応的グラフト化が含まれている[4]。
【0010】
この新規な方法では、特に、不対電子を有する、たとえばジアゾニウム塩のような開裂可能なアリール塩から得られる分子的エンティティ(molecular entities)を採用して、グラフトさせる対象物の表面と共有結合タイプの結合を形成しているが、前記分子的エンティティは、それらがグラフト化される対象の表面からは独立して発生させる。これらの分子的エンティティは、その表面にグラフトされ、特にエチレン性モノマーを含むラジカル反応を開始させる。
【発明の概要】
【発明が解決しようとする課題】
【0011】
本願発明者らは、国際公開第2008/078052号パンフレットに記載されたプロセスをさらに改良することを可能とするための、各種の手段および/または各種の工程を確認するための研究を続けてきた。
【課題を解決するための手段】
【0012】
したがって、本発明は、非電気化学的条件下で、固体支持体の表面の少なくとも一部に有機膜をグラフト化させるための改良された方法に関する。
【0013】
その理由は、本願発明者らの研究によって、国際公開第2008/078052号パンフレット[4]に記載のプロセスにおいて、固体支持体の表面に酸化的前処理を行うことによって、有機膜のグラフト化を改良すること、および/または前記部分にグラフト化される有機膜の厚み、従って量を増やすことが可能となることを見出したためである。そのような酸化処理は、さらに詳しくは、固体支持体の表面に対して、および/またはポリマー、たとえばプラスチックから作られている固体支持体に対して適用される。
【0014】
なにか一つの理論に拘束されることを望むものではないが、酸化的前処理を使用することによって、国際公開第2008/078052号パンフレット[4]に記載のプロセスに、より良好な寄与が可能となる。さらに、グラフト化された表面の反応性がより高いために、酸化的前処理を行うことによって、国際公開第2008/078052号パンフレット[4]に記載のプロセスの通常の反応時間を下げることも可能となる。
【0015】
したがって、本発明は、(コ)ポリマーから作られている固体支持体の表面の一部の上に有機膜を調製するための方法に関し、以下のものからなる連続的な工程を含むことを特徴としている:
i)前記表面部分を酸化処理にかける工程;
ii)ラジカル化学的グラフト化によって前記表面部分に有機膜をグラフト化させる工程。
【0016】
「(コ)ポリマーから作られている固体支持体の表面」という用語は、(コ)ポリマーから作られている固体支持体、または(コ)ポリマーから作られている表面を正に有している固体支持体の両方を意味しているが、その支持体の残りの部分は、いかなる材料から作られていてもよいと理解されたい。本発明における固体支持体は、いかなるサイズ、いかなる形状であってもよい。
【0017】
「(コ)ポリマーから作られている」という用語は、本発明の文脈においては、正に1種の(コ)ポリマーかまたは複数の異なった(コ)ポリマーから実質的になる支持体または表面を意味していると理解されたい。
【0018】
「実質的になる(essentially composed)」という用語は、本発明の文脈においては、重量で表して、その構成成分の少なくとも50%、少なくとも60%、少なくとも70%、少なくとも80%、少なくとも90%、少なくとも95%、および/または少なくとも98%が、1種(または複数)の(コ)ポリマーである、支持体または表面を意味している。
【0019】
その支持体またはその支持体の表面が、1種(または複数)の(コ)ポリマー(1種または複数)によってのみ構成されているのが有利である。
【0020】
また別な形態においては、その支持体またはその支持体の表面が、1種(または複数)の(コ)ポリマーに加えて、充填剤、可塑剤および添加剤からなる群から選択された少なくとも1種の成分を含んでいる。この(これらの)追加の成分は、ポリマー材料の中に組み入れられるかおよび/または分散されているのが有利である。
【0021】
念のために言えば、プラスチックは、有利には3000を超える重合度を有する少なくとも1種の(コ)ポリマーと、少なくとも1種の添加剤とから形成されている。したがって、本発明の文脈において実施されるポリマーから作られている支持体またはその支持体の表面には、プラスチックから作られた支持体または支持体表面が含まれている。
【0022】
無機充填剤たとえば、シリカ、タルク、ガラス繊維もしくはガラスビーズ、または有機充填剤たとえば、穀物粉もしくはセルロースパルプが、コストを下げるためおよびポリマー材料のある種の性質たとえば機械的性質を改良するために、一般的に使用されている。添加剤は、ポリマー材料の特定の性質を改良するために主として使用されるが、前記性質としては、架橋性、スリップ性、耐分解性、難燃性および/または細菌および真菌の攻撃に対する抵抗性などが挙げられる。
【0023】
各種の天然(たとえばラテックスもしくはゴム)、人工、合成の熱可塑性、熱硬化性、耐熱性、エラストマー性、直鎖状(すなわち、直鎖状もしくは分岐状、一次元)および/または三次元のポリマーが、本発明の文脈において使用することができる。
【0024】
本発明の文脈において実現される(コ)ポリマーが、以下のものからなる群より選択される熱可塑性(コ)ポリマーであるのが有利である:
−ポリオレフィン、たとえば、ポリエチレン、ポリプロピレン、エチレン/プロピレンコポリマー、ポリブチレン、ポリメチルペンテン、エチレン/酢酸ビニルコポリマー、エチレン/ビニルアルコールコポリマー、それらの誘導体の1種、それらのコポリマーの1種、それらのブレンド物の1種、およびそれらの組合せの1種;
−ポリエステル、たとえば、(場合によってはグリコールによって変性された)ポリエチレンテレフタレート(PET)、ポリブチレンテレフタレート、ポリラクチド、ポリカーボネート、それらのコポリマーの1種、それらのブレンド物の1種、およびそれらの組合せの1種;
−ポリエーテル、たとえば、ポリ(オキシメチレン)、ポリ(オキシエチレン)、ポリ(オキシプロピレン)、ポリ(フェニレンエーテル)、それらのコポリマーの1種、それらのブレンド物の1種およびそれらの組合せの1種;
−ビニルポリマー、たとえば、(場合によっては塩素化された)ポリ塩化ビニル、ポリビニルアルコール、ポリ(酢酸ビニル)、ポリ(ビニルアセタール)、ポリ(ビニルホルマール)、ポリフッ化ビニル、ポリ(塩化ビニル/酢酸ビニル)、それらのコポリマーの1種、それらのブレンド物の1種およびそれらの組合せの1種;
−ビニリデンポリマー、たとえば、ポリ(塩化ビニリデン)、ポリフッ化ビニリデン、それらのコポリマーの1種、それらのブレンド物の1種およびそれらの組合せの1種;
−スチレンポリマー、たとえば、ポリスチレン、ポリ(スチレン/ブタジエン)、ポリ(アクリロニトリル/ブタジエン/スチレン)(ABS)、アクリロニトリル/ブタジエン/スチレン−ポリカーボネート(ABS/PC)、ポリ(アクリロニトリル/スチレン)、ポリ(アクリロニトリル/エチレン/プロピレン/スチレン)、ポリ(アクリロニトリル/スチレン/アクリレート)、それらのコポリマーの1種、それらのブレンド物の1種およびそれらの組合せの1種;
−(メタ)アクリル系ポリマー、たとえば、ポリアクリロニトリル、ポリ(アクリル酸メチル)、ポリ(メタクリル酸メチル)、それらの誘導体の1種、それらのコポリマーの1種、それらのブレンド物の1種およびそれらの組合せの1種;
−ポリアミド、たとえば、ポリ(カプロラクタム)、ポリ(ヘキサメチレンアジパミド)、ポリ(ラウロアミド)、ポリエーテル−ブロック−アミド、ポリ(メタキシリレンアジパミド)、ポリ(メタフェニレンイソフタルアミド)、それらのコポリマーの1種、それらのブレンド物の1種およびそれらの組合せの1種;
−フルオロポリマー(またはポリフルオロエテン)、たとえば、ポリテトラフルオロエチレン、ポリクロロトリフルオロエチレン、ペルフルオロ化ポリ(エチレン/プロピレン)、ポリフッ化ビニリデン(PVDF)、それらのコポリマーの1種[たとえば、テトラフルオロエチレンとテトラフルオロプロピレンとのコポリマー(FEP)、エチレンとテトラフルオロエチレンとのコポリマー(ETFE)、ヘキサフルオロプロペンとフッ化ビニリデンとのコポリマー(HFP−co−VDF)、フッ化ビニリデンとトリフルオロエチレンとのコポリマー(VDF−co−TrFE)、およびフッ化ビニリデンとトリフルオロエチレンとモノクロロトリフルオロエチレンとのコポリマー(VDF−co−TrFE−co−chloro−TrFE)]、それらのブレンド物の1種およびそれらの組合せの1種;
−セルロースポリマー、たとえば、酢酸セルロース、硝酸セルロース、メチルセルロース、カルボキシメチルセルロース、それらのコポリマーの1種、それらのブレンド物の1種およびそれらの組合せの1種;
−ポリ(アリーレンスルホン)、たとえば、ポリスルホン、ポリエーテルスルホン、ポリアリールスルホン、それらのコポリマーの1種、それらのブレンド物の1種およびそれらの組合せの1種;
−ポリスルフィド、たとえば、ポリ(フェニレンスルフィド);
−ポリ(アリールエーテル)ケトン、たとえば、ポリ(エーテルケトン)、ポリ(エーテルエーテルケトン)、ポリ(エーテルケトンケトン)、それらのコポリマーの1種、それらのブレンド物の1種およびそれらの組合せの1種;
−ポリアミド−イミド;
−ポリ(エーテル)イミド;
−ポリベンズイミダゾール;
−ポリ(インデン/クマロン);
−ポリ(パラキシリレン);
−それらのコポリマーの1種、それらのブレンド物の1種およびそれらの組合せの1種。
【0025】
また別な形態においては、本発明の文脈において実現される(コ)ポリマーが、以下のものからなる群より選択される熱硬化性(コ)ポリマーである:アミノプラスト、たとえば、尿素/ホルムアルデヒド、メラミン/ホルムアルデヒド、メラミン/ホルムアルデヒド−ポリエステル、それらのコポリマーの1種、それらのブレンド物の1種およびそれらの組合せの1種;ポリウレタン;不飽和ポリエステル;ポリシロキサン;フェノール/ホルムアルデヒド、エポキシド、アリルもしくはビニルエステル樹脂;アルキド;ポリウレア;ポリイソシアヌレート;ポリ(ビスマレイミド);ポリベンズイミダゾール;ポリジシクロペンタジエン;それらのコポリマーの1種、それらのブレンド物の1種およびそれらの組合せの1種。
【0026】
同様にして、塩基性の基たとえば三級もしくは二級アミンたとえばピリジン類を担持している(コ)ポリマー、たとえばポリ(4−ビニルピリジン)およびポリ(2−ビニルピリジン)(P4VPおよびP2VP)を使用するか、または、より一般的には芳香族およびニトロ芳香族基を担持するポリマーを使用して本発明を実現することができる。
【0027】
本発明の文脈において使用することが可能なポリマーについてのさらなる情報は、ナウジン(Naudin)による論文(1995)[5]に見ることができる。
【0028】
「酸化処理」という用語は、本発明の文脈においては、採用された固体支持体の表面を酸化すること、および/またはラジカルを形成させることによってさらなる酸化のための表面を調製することを目的とした、処理(または前処理)を意味していると理解されたい。酸化は、特に、固体支持体の表面を、酸素リッチな基たとえば、カルボキシル(−COOH)、ヒドロキシル(−OH)、アルコキシル(−OR,Rについては以下において定義する)、カルボニル(−C=O)、過炭酸(−C−O−OH)、および場合によってはアミド(−CONH)タイプのような基をそれに付着させるかおよび/またはその中に導入することによって、変性させる。
【0029】
この処理がベースとしているのは、固体支持体の表面および/または固体支持体を構成するポリマーの表面で、表面酸化を起こさせるため、および/またはラジカルを形成させることによって表面酸化のための表面を形成させるために、各種の反応剤を使用することである。そのようにして得られた酸化によって、国際公開第2008/078052号パンフレット[4]に記載のプロセスによるグラフト化よりは、良好な付着性および/またはより大量のポリマーが得られるようになる。この処理は、物理的処理または化学的処理をベースとする、主として2種のタイプの表面変性をベースとしている。
【0030】
本発明の第一の実施態様においては、使用される酸化処理が化学的酸化処理である。そのような化学的酸化処理は、フェントン(Fenton)化学反応、アルコール性水酸化カリウムを用いた処理、強酸を用いた処理、水酸化ナトリウムを用いた処理、強酸化剤を用いた処理、オゾンを用いた処理、およびそれらの組合せからなる群より選択するのが有利である。
【0031】
「組合せ」という用語は、その化学的酸化処理が、上に列記した処理の少なくとも2種を採用していることを意味していると考えられたい。例を挙げれば、そのような組合せが、強酸化剤を用いた処理と、それに続くフェントン化学反応からなっていてもよい。
【0032】
「フェントン化学反応」という用語は、1894年にフェントン(Fenton)によって報告された反応を意味していると理解されたい。この反応では、次の反応スキームで表されるように、過酸化水素水溶液を鉄(II)と反応させることによってヒドロキシルラジカルを生成させることが可能となる:
Fe2++H2O2 → Fe3++・OH+OH−
【0033】
本発明の文脈においては、フェントン化学反応は、固体支持体の表面および/または固体支持体を、第一鉄(Fe2+)イオンと式ROORの化合物とを含む溶液と接触させることからなっているが、ここでRは、水素、1〜15個の炭素原子を含むアルキル基、アシル基−COR’(ここでR’は、1〜15個の炭素原子を含むアルキル基を表す)、またはアロイル基−COAr(ここでArは、6〜15個の炭素原子を含む芳香族基を表す)を表している。タイプ・OR(Rは先に定義されたもの)のラジカルは、Fe2+イオンを用いてペルオキシドROORを開裂させることによって得られる。
【0034】
「1〜15個の炭素原子を含むアルキル基」という用語は、直鎖状、分岐状、または環状、そして場合によっては置換された、1〜15個の炭素原子、とりわけ1〜10個の炭素原子、特には2〜6個の炭素原子と、場合によってはヘテロ原子たとえば、N、O、F、Cl、P、Si、Br、またはSを含むアルキル基を意味していると理解されたい。
【0035】
「6〜15個の炭素原子を含む芳香族基」という用語は、本発明の文脈においては、それぞれ3〜10個の原子を含む1個または複数の芳香族環またはヘテロ芳香族環(その1個または複数のヘテロ原子が、N、O、PまたはSであってよい)からなる、場合によっては置換された芳香族基またはヘテロ芳香族基を意味していると理解されたい。
【0036】
「置換された(substituted)」という用語は、本発明の文脈においては、1〜4個の炭素原子を含む直鎖状または分岐状のアルキル基によるか、アミン基によるか、カルボキシル基によるか、および/またはニトロ基によって単置換または多置換されたアルキル基または芳香族基を意味していると理解されたい。
【0037】
第一鉄(Fe2+)イオンは、第一鉄(Fe2+)イオンおよび式ROORの化合物を、有利には0.05M〜5Mの間、とりわけ0.1M〜3Mの間、特には0.25M〜2Mの間の濃度で含む溶液の中に存在している。第一鉄(Fe2+)イオンおよび式ROORの化合物を含む溶液にはさらに、対イオンたとえば、テトラフルオロホウ酸イオン、硫酸イオン、または塩素イオンが含まれる。
【0038】
式ROORの化合物は、0.1M〜5Mの間、とりわけ0.5M〜3Mの間、特には1M〜2.5Mの間の濃度で第一鉄(Fe2+)イオンおよび式ROORの化合物を含む溶液の中に存在している。
【0039】
第一鉄(Fe2+)イオンおよび式ROORの化合物を含む溶液は、酸性溶液であるのが有利である。「酸性溶液」という用語は、7未満、とりわけ2〜4の間、特には3のオーダー(すなわち、3±0.5)のpHを有する溶液を意味していると理解されたいこの溶液にはさらに、とりわけ0.05mM〜50mMの間、特に0.1mM〜10mMの間、さらに特には1mMのオーダー(すなわち、1mM±0.25mM)の濃度で硫酸を含んでいる。
【0040】
フェントン化学反応の処理時間は変化させることができる。限定する訳ではないが例を挙げれば、この時間は、有利には5分〜5時間の間、とりわけ10分〜3時間の間、特に15分〜2時間の間、さらに特には25分のオーダー(すなわち、25±5分)である。
【0041】
アルコール性水酸化カリウムを用いた処理は、固体支持体の表面および/または固体支持体を、アルコール性水酸化カリウム溶液と接触させることからなっている。
【0042】
「アルコール性水酸化カリウム溶液」という用語は、溶媒としてアルコールを含む溶液(以後アルコール性溶液と呼ぶ)で希釈された水酸化カリウムを意味していると理解されたい。このアルコールは、メタノール、エタノール、およびプロパノールからなる群より選択するのが有利である。このアルコールがエタノールであるのが好ましい。
【0043】
アルコール性溶液中のKOHの濃度は、0.1M〜10Mの間、とりわけ0.5M〜5Mの間、特には3.5Mのオーダー(すなわち、3.5M±0.5M)である。
【0044】
アルコール性水酸化カリウムを用いて処理する時間は、変化させることができる。限定する訳ではないが例を挙げれば、この時間は有利には5分〜5時間の間、とりわけ10分〜3時間の間、特には20分〜2時間の間である。
【0045】
アルコール性水酸化カリウムを用いた処理は、フルオロポリマーたとえばPVDFの場合に、より特に適している。その理由は、アルコール性水酸化カリウム溶液によるそれらの酸化が、ポリマーの表面の脱フッ素を起こして、−CH=CF−結合、およびそれらの酸化された同等物を形成するからである。これらの結合は、フッ素原子が存在しているために反応性が高い。
【0046】
強酸を用いた処理は、固体支持体の表面および/または固体支持体を、強酸または強酸混合物の溶液と接触させることからなっている。そのような処理によって、表面酸化により、酸素含有基の数が増える。
【0047】
本発明による強酸を用いた酸化処理の文脈においては、いかなる強酸も使用することができる。限定する訳ではないが例を挙げれば、その強酸は、HCl、H2SO4、HNO3、HClO4、およびそれらの混合物からなる群より選択するのが有利である。
【0048】
強酸(1種または複数)の溶液の強酸の重量比は、変化させることができる。限定する訳ではないが例を挙げれば、この濃度は、有利には10〜100重量%の間、とりわけ50〜95重量%の間、特には70〜90重量%の間である。
【0049】
強酸を用いた処理の時間は、変化させることができる。限定する訳ではないが例を挙げれば、この時間は、有利には1分〜5時間の間、とりわけ1分〜3時間の間、特には1分〜1時間の間である。
【0050】
水酸化ナトリウムを用いた処理は、固体支持体の表面および/または固体支持体を、水酸化ナトリウム溶液と接触させることからなっている。
【0051】
その溶液の水酸化ナトリウムの重量比は、変化させることができる。限定する訳ではないが例を挙げれば、この濃度は、有利には10〜100重量%の間、とりわけ15〜70重量%の間、特には20〜50重量%の間である。
【0052】
水酸化ナトリウムを用いて処理する時間は、変化させることができる。限定する訳ではないが例を挙げれば、この時間は、有利には1分〜5時間の間、とりわけ1分〜3時間の間、特には1分〜1時間の間である。
【0053】
強酸化剤を用いた処理は、固体支持体の表面および/または固体支持体を、強酸化剤の溶液と接触させることからなっている。この強酸化剤の溶液は、中性、酸性、塩基性のいずれであってもよい。
【0054】
この溶液が酸性であるのが、有利である。より具体的には、その強酸化剤は、塩酸、硫酸または硝酸中のKMnO4、K2Cr2O7、KClO3、CrO3、およびそれらの混合物からなる群より選択する。したがって、以下のものが強酸化剤の溶液として考えられる:KMnO4/HCl、K2Cr2O7/HCl、KClO3/HCl、CrO3/HCl、KMnO4/H2SO4、K2Cr2O7/H2SO4、KClO3/H2SO4、CrO3/H2SO4、KMnO4/HNO3、K2Cr2O7/HNO3、KClO3/HNO3、およびCrO3/HNO3。
【0055】
塩酸中、硫酸中、または硝酸中のKMnO4、K2Cr2O7、KClO3またはCrO3の濃度は、有利には10mM〜1Mの間、とりわけ0.1M〜0.5Mの間、特には0.2Mのオーダー(すなわち、0.2M±50mM)である。
【0056】
強酸化剤の溶液の中の塩酸、硫酸、または硝酸の濃度は、有利には0.1M〜10Mの間、とりわけ0.5M〜5Mの間、特には3.5Mのオーダー(すなわち、3.5M±0.5M)である。
【0057】
強酸化剤を用いた処理の時間は、変化させることができる。限定する訳ではないが例を挙げれば、この時間は、有利には1分〜3時間の間、とりわけ5分〜1時間の間、特には10分〜30分の間、さらに特には15分のオーダー(すなわち、15分±5分)である。
【0058】
オゾンを用いた処理は、固体支持体の表面および/または固体支持体を、オゾンに曝露させることからなる。この曝露には、固体支持体の表面および/または固体支持体をオゾンの流れと接触させるか、または固体支持体の表面および/または固体支持体を、オゾンを含む雰囲気の中に置くかの、いずれかを含むことができる。
【0059】
必要なオゾンは、オゾン発生器たとえば、低圧水銀蒸気ランプ(28mW/cm2、254nm)を備えたUVO−クリーナー(UVO−Cleaner)モデル42−200を介して、酸素を多く含むガスたとえば空気、酸素、酸素富化空気または酸素富化ガスから得ることができる。
【0060】
オゾンを用いて処理する時間は、変化させることができる。限定する訳ではないが例を挙げれば、この時間は、有利には30秒〜3時間の間、とりわけ1分〜1時間の間、特には5分〜30分間の間、さらに特には10分のオーダー(10分±3分)である。
【0061】
化学的酸化処理は、電気化学的前処理からなっていてよい。そのような処理は、酸化させる基材を電気化学的に発生させた酸化性溶液の中に浸漬させることからなっているが、これについては、ブルーズ(Brewis)およびダーム(Dahm)の論文(2001)[6]に具体的な記載がある。国際公開第2007/042659号パンフレットにも、その文献ではエレクトロフェントン(ElectroFenton)と呼ばれている、そのような電気化学的前処理についての記載がある[7]。
【0062】
本発明の第二の実施態様においては、使用される酸化処理が物理的酸化処理である。この物理的酸化処理は、乾式ルートによる処理からなっていて、それが目的としているのは、表面を直接酸化させるか、またはラジカルを発生させることによってその表面を後ほど酸化させるための準備をすることである。乾式ルートによるいくつかの処理の場合においては、表面の酸化数を直接的に増加させる必要はない。その理由は、酸化数の増加が、その処理した表面を水溶液の中に浸漬させた後でのみ起きてもよいからである。そのような物理的酸化処理は、火炎処理、コロナ作用による処理、プラズマ処理、UV照射線を用いた処理、X線またはγ線を用いた処理、電子を用いるかまたは重イオンを用いた照射処理、およびそれらの組合せからなる群より選択するのが有利である。
【0063】
「組合せ」という用語は、その物理的酸化処理が、上に列記した処理の少なくとも2種を採用していることを意味していると考えられたい。
【0064】
さらに、本発明の文脈においては、先に定義された化学的酸化処理の少なくとも一つと、先に定義された物理的酸化処理の少なくとも一つとを組み合わせて、固体支持体の表面を処理することができる。
【0065】
火炎処理(flame treatment)または「火炎吹付(flaming)」は、固体支持体の表面および/または固体支持体を火炎に曝露させることからなっている。その火炎は特に、固体支持体の表面からおよび/または固体支持体から、0.1cm〜20cmの間、とりわけ0.3cm〜10cmの間、さらに特には0.5cm〜5cmの間の距離に位置させる。
【0066】
この火炎は、少なくとも2種のガスの混合物から発生させるのが有利であり、その第一のガスおよび第二のガスは、それぞれ、水素、メタン、エタン、およびプロパンからなる群より、および空気、オゾン、および酸素からなる群より選択する。そのようにして得られる火炎の温度は、500℃〜1600℃の間、とりわけ800℃〜1400℃の間、特には1200℃のオーダー(すなわち、1200℃±100℃)である。
【0067】
この高温処理により、活性なエンティティが発生するが、それは、ラジカル、イオン、または励起分子に当てはめることが可能である。それらは、ヒドロキシル、カルボニル、またはカルボキシル官能基の性質を有することができる。
【0068】
火炎処理の時間は、0.01秒〜10秒の間、とりわけ0.015秒〜1秒の間、特には0.02秒〜0.1秒の間である。
【0069】
コロナ作用による処理は、「コロナ放電処理」とも呼ばれているが、数mm、特には1mm〜2mmの距離で離した二つの電極の間に高電圧交流を印加することによって作り出されたイオン化場に、固体支持体の表面および/または固体支持体を曝露させることからなっている。したがって、電位が臨界値を超えるが、アークの形成にはいたらない条件である場合に、導電体を取り囲む媒体のイオン化によって放電がもたらされる。
【0070】
このイオン化の際に、放出された電子が沈殿(precipitate)し、固体支持体の表面および/または固体支持体の周りの媒体の分子にそれらのエネルギーを伝えるが、その媒体が、場合によっては酸素を富化させた空気または不活性ガスであるのが有利である。このことが、固体支持体のポリマー(1種または複数)の鎖を切断し、その媒体の中に存在している化学的エンティティと同時反応をもたらすことになる。そのコロナ作用による処理が、酸素の寄与がない不活性な雰囲気下で実施されたとしても、不純物に基づく酸化が、表面酸化をもたらす。
【0071】
コロナ放電の密度は、有利には10W・min/m2〜500W・min/m2の間、とりわけ20W・min/m2〜400W・min/m2の間、特には30W・min/m2〜300W・min/m2の間である。
【0072】
コロナ作用による処理の時間は、0.1秒〜600秒の間、とりわけ1秒〜120秒の間、特には10秒〜50秒の間である。
【0073】
プラズマ処理は、固体支持体の表面および/または固体支持体をプラズマに曝露させることからなっている。
【0074】
念のために言えば、プラズマはイオン化状態にあるガスであって、便宜的には、物質の第四状態とみなされている。ガスをイオン化させるのに必要なエネルギーは、電磁波(無線周波数またはマイクロ波)の手段によって与えられる。プラズマは、中性分子、イオン、電子、ラジカルエンティティ(化学的に高活性)、および励起エンティティから構成されているが、それらは、物質の表面と反応することになるであろう。
【0075】
「コールド」プラズマと「ホット」プラズマとの間では区別がされていて、プラズマの中に存在しているエンティティのイオン化度に関連して、相互に異なっている。「コールド」プラズマの場合には、反応性エンティティのイオン化度が、10−4未満であるのに対して、「ホット」プラズマでは、10−4よりも高い。「ホット」および「コールド」の用語は、「ホット」プラズマの方が、「コールド」プラズマよりも、はるかに高エネルギーであるという事実に基づいている。本発明における酸化的前処理の場合においては、コールドプラズマの方が、より適している。しかしながら、採用したプラズマがコールドであっても、あるいはホットであっても、いずれに場合においても、その処理は、固体支持体の表面に、酸素リッチな官能基の出現をもたらすこととなる。
【0076】
プラズマは、2種のガスの混合物によって発生させるのが有利であるが、その第一のガスおよび第二のガスはそれぞれ、不活性ガスからなる群から、および空気および酸素からなる群から選択される。
【0077】
プラズマ処理の時間は、1秒〜5分の間、とりわけ10秒〜60秒の間、特には20秒〜40秒の間である。
【0078】
UV照射線を用いた処理は、固体支持体の表面および/または固体支持体をUV光にあてることからなる。
【0079】
採用されるUV光が、10nm〜400nmの間、とりわけ100nm〜380nmの間、特には180nm〜360nmの間の波長を示しているのが有利である。
【0080】
そのようなUV光を発生させるためには、各種のUV光源を使用することができる。例として、以下のものをあげることができる:UVランプ、低圧水銀ランプ、中圧水銀ランプ、高圧水銀ランプ、超高圧水銀ランプ、アーク灯、ハライドランプ、キセノンランプ、レーザー、ArFエキシマレーザー、KrFエキシマレーザー、エキシマランプ、またはシンクロトロン放射。
【0081】
本発明の文脈においては、UV照射線を用いた処理は、5℃〜120℃の間、とりわけ10℃〜80℃の間、特には15℃〜40℃の間の温度で実施することができる。さらに詳しくは、本発明におけるUV照射線を用いた処理は、周囲温度で実施される。「周囲温度」という用語は、20℃±5℃の温度を意味していると理解されたい。
【0082】
このUV照射線を用いた処理は、ガスの存在下、特に酸素および/またはオゾンを多く含むガス、たとえば空気、酸素、オゾン、酸素富化および/またはオゾン富化空気、酸素富化および/またはオゾン富化ガスの存在下に、実施するのが有利である。その理由は、固体支持体の表面の光酸化、すなわち、酸素および/またはオゾンの存在下にUV照射すると、前記固体支持体を構成しているポリマー材料の表面に酸素を導入することが可能となるからである。
【0083】
本発明の文脈においては、UV照射線を用いた処理は、1分〜5時間、とりわけ5分〜1時間、特には10分〜45分続ける。照射は、1回だけ実施してもよいし、あるいは数回、とりわけ2〜20回、特には3〜10回繰り返して実施することもできる。
【0084】
X線またはγ線を用いた処理は、固体支持体の表面および/または固体支持体を、X線またはγ線(すなわち、電磁線)にあてることからなっている。
【0085】
採用するX線が、5pm未満の波長を示し、採用するγ線が、5pm〜10nmの間の波長を示すのが有利である。
【0086】
X線またはγ線は発生させることを可能とし、それを本発明における固体支持体の表面に向かわせることが可能であり、そして当業者には公知の、各種の線源を、本発明の文脈においては、使用することができる。例を挙げれば、γ線は、放射性線源たとえば、ガンマ線装置の中のコバルト−60またはセシウム−137から放出させることができる。
【0087】
本発明の文脈においては、X線またはγ線を用いた処理は、5℃〜120℃の間、とりわけ10℃〜80℃の間、特には15℃〜40℃の間の温度で実施することができる。さらに詳しくは、本発明におけるX線またはγ線を用いた処理は、周囲温度で実施される。「周囲温度」という用語は、20℃±5℃の温度を意味していると理解されたい。
【0088】
このX線またはγ線を用いた処理は、酸素を多く含むガス、たとえば空気、酸素、酸素富化空気、または酸素富化ガスの存在下に実施するのが有利である。その理由は、放出されるフォトンのエネルギーが極めて高いために、フォトンが材料を通過してしまうことが可能ではあるが、その照射の際に酸素が存在していることで、ポリマーの表面に(すなわち固体支持体の表面に)ラジカルが形成されるからである。それらのラジカルが次いで、周辺の酸素と反応して、固体支持体の表面に酸素リッチな官能基を形成する。
【0089】
X線またはγ線を用いた処理の時間は、1分〜2時間の間、とりわけ5分〜1時間の間、特には10分〜30分間の間である。
【0090】
電子を用いるかまたは重イオンを用いた照射処理は、固体支持体の表面および/または固体支持体を、電子ビームにあてるか(「電子照射」とも呼ばれている)、または重イオンを衝突させるか、からなっている。
【0091】
固体支持体の表面および/または固体支持体を電子ビームにあてることからなる工程は、固体支持体の表面および/または固体支持体を、加速された電子のビームを用いて掃引することからなっていてよく、このビームは、電子加速器(たとえば、ファン・デ・グラーフ(Van de Graaf)加速器、2.5MeV)によって放出させることが可能である。電子線の線量は、5kGy〜1000kGy、とりわけ10kGy〜500kGy、特には50kGy〜150kGyの範囲で変化させることができる。
【0092】
「重イオン」という用語は、炭素の質量よりも大きい質量を有するイオンを意味していると理解されたい。一般的には、それらは、クリプトン、鉛、およびキセノンから選択されるイオンである。この工程は、重イオンのビーム、たとえばPbのビームまたはKrイオンのビームを用いて、固体支持体の表面および/または固体支持体に衝突させることからなっていてよい。イオンビームの強度は、有利には0.1MeV/amu〜100MeV/amuの間、とりわけ1MeV/amu〜50MeV/amuの間、特には4MeV/amu〜20MeV/amuの間である。
【0093】
本発明の文脈においては、電子を用いるかまたは重イオンを用いた照射処理は、5℃〜120℃の間、とりわけ10℃〜80℃の間、特には15℃〜40℃の間の温度で実施することができる。さらに詳しくは、本発明における電子を用いるかまたは重イオンを用いた照射処理は、周囲温度で実施される。「周囲温度」という用語は、20℃±5℃の温度を意味していると理解されたい。
【0094】
この電子を用いるかまたは重イオンを用いた照射処理は、酸素を多く含むガス、たとえば空気、酸素、酸素富化空気もしくは酸素富化ガスの存在下、またはそのようなガスに曝露させた後に実施するのが有利である。先にも述べたように、酸素リッチな雰囲気下またはそのような雰囲気に曝露させた後に、電子を用いるかまたは重イオンを用いた照射をすると、ポリマーの表面の上に酸素を導入することが可能となる[8]。
【0095】
電子を用いるかまたは重イオンを用いた照射処理の時間は、1分〜2時間の間、とりわけ5分〜1時間の間、特には10分〜30分間の間である。
【0096】
本発明によるプロセスの一つの特定の実施態様においては、物理的および化学的両方の酸化処理が、マスクの存在下に実施される。この酸化処理の際にマスクを使用することによって、「ブランクの」表面と、「酸化された」(すなわち、処理された)表面との間で反応性に差をつけることが可能となる。この反応性における差は、パターン化された表面を作り出すのに利用することが可能であり、その理由は、国際公開第2008/078052号パンフレット[4]に記載のプロセスは、「ブランクの」表面に比較して「酸化された」表面において、より効果的となるであろうからである。
【0097】
したがって、その上に有機膜を形成させねばならない固体支持体の表面には、それを少なくとも部分的に覆い、酸化処理の際にそれを保護するマスクが備わっている。そのマスクによって、酸化処理に関連して、その表面の化学的な反応性を局所的に「マスクする」ことが可能となる。
【0098】
そのマスクは典型的には、物理的なエンティティに相当し、表面にグラフト化されてもいないし、また後者に共有結合的に結合されていることもない。具体的には、それは、バルクの物質であっても、あるいは表面の上に析出された無機または有機物質の、典型的には数オングストロームから数ミクロンまでの薄層であってもよい。そのマスクは、穏やかな条件下で容易に剥がすことが可能な、凝集力の低い層として機能する薄層からなっているのが有利である。典型的には、その穏やかな条件とは、一般的にそのマスクが溶解可能な溶媒を用いて実施される単純な化学的洗浄、そのマスクが溶解可能な溶媒の中における超音波を用いた処理、または温度を上げることなどに相当する。
【0099】
したがって、そのマスクを構成する物質は、広い範囲から選択することができる。それは、一般的には、その固体支持体の性質に従って選択されることになるであろう。そのマスクは、アルカンチオール、特には長鎖アルカンチオール、多くの場合C15〜C20、典型的にはC18アルカンチオールから構成されているのが有利である。
【0100】
マスクをした析出方法は、当業者には公知である。具体的には、それらは、コーティング法、スプレー法、または浸漬法とすることができる。したがって、材料の薄層の形態のマスクは、たとえば、選択された材料を含ませたフェルトペンからはじめて直接書き込むか、「パッド(pad)」プロセスによるか、および/または慣用されるリソグラフィー法たとえばスピンコーティング法のいずれかによって析出させ、それに続けて物理的マスクを通すか、または誘導することが可能な光もしくは粒子のビームを介して照射し、次いで現像させることができる。
【0101】
固体支持体の表面および/または固体支持体を先に定義されたような酸化処理にかけたら、ラジカル反応的グラフト化の工程の前に、その固体支持体の表面および/または固体支持体を、洗い流すか、洗浄するか、および/または乾燥させることができる。
【0102】
その固体支持体の表面および/または固体支持体を、特に水中、たとえばミリQ(Milli−Q)水中での1回または複数回の洗い流し操作にかけるのが有利である。次いで、その固体支持体の表面および/または固体支持体を、5分〜30分、とりわけ10分のオーダー(すなわち、10分±2分)の時間、超音波を用いた処理にかけてから、乾燥させる。
【0103】
工程(ii)の際の「ラジカル反応的グラフト化」という用語は、具体的には、固体支持体の表面と共有結合タイプの結合を形成させるために、不対電子を有する分子的エンティティを使用することを指しているが、前記分子的エンティティは、それらをグラフト化させるつもりの表面とは独立して発生させる。したがって、ラジカル反応によって、固体支持体の表面と有機膜との間に共有結合が生成することとなる。
【0104】
固体支持体またはこの支持体の少なくとも表面を構成しているポリマーにグラフト化される有機膜は、50nm未満、有利には40nm未満、とりわけ30nm未満、特には20nm未満の長さを示す。
【0105】
固体支持体またはこの支持体の少なくとも表面を構成しているポリマーにグラフト化される有機膜は、いくつかの同一および/または異なったモノマー単位から主として得られる(コ)ポリマーである。
【0106】
本発明の文脈において採用されるモノマー単位の全部または一部が、ラジカル経路によって重合可能なモノマーであるのが有利である。「ラジカル経路によって重合可能なモノマー」という用語は、ラジカル性化学的エンティティによって開始された後に、ラジカル条件下で重合可能なモノマーを意味していると理解されたい。典型的には、それらは、エチレン性のタイプの少なくとも一つの結合を含むモノマーである、すなわちそれらは、エチレン性のタイプの分子である。
【0107】
ビニルモノマー、具体的には、国際公開第2005/033378号パンフレットおよび国際公開第2006/097611号パンフレットに記載されているモノマーが、特に関連する[9、10]。
【0108】
本発明の特に有利な実施態様においては、その1種または複数のビニルモノマーが、次の式(II)のモノマーから選択される:
【0109】
【化1】
【0110】
[式中、R1〜R4基(同一であっても、異なっていてもよい)は、1価の非金属原子たとえば、ハロゲン原子もしくは水素原子、または飽和もしくは不飽和の化学基たとえば、アルキルもしくはアリール基、ニトリル、カルボニル、アミン、アミド、または−COOR5基(ここで、R5は、水素原子またはC1〜C12、好ましくはC1〜C6のアルキル基を表す)を表す。]
【0111】
上述の式(II)のモノマーは、具体的には、以下のものからなる群より選択される:アクリル酸、酢酸ビニル、アクリロニトリル、メタクリロニトリル、メタクリル酸メチル、メタクリル酸エチル、メタクリル酸ブチル、メタクリル酸プロピル、メタクリル酸ヒドロキシエチル(HEMA)、メタクリル酸ヒドロキシプロピル、メタクリル酸グリシジルおよびそれらの誘導体;エチル−、プロピル−、ブチル−、ペンチル−、およびヘキシル−アクリルアミド、特に−メタクリルアミド、シアノアクリレート、ジアクリレート、およびジメタクリレート、トリアクリレートおよびトリメタクリレート、テトラアクリレートおよびテトラメタクリレート(たとえば、ペンタエリスリトールテトラメタクリレート)、スチレンおよびその誘導体、パラ−クロロスチレン、ペンタフルオロスチレン、N−ビニルピロリドン、4−ビニル−ピリジン、2−ビニルピリジン、ビニル、アクリロイルまたはメタクリロイルハライド、ジビニルベンゼン(DVB)、ならびにより一般的には、ビニル架橋剤または、アクリレート、メタクリレート、およびそれらの誘導体をベースとする架橋剤。
【0112】
有機膜は、固体支持体の表面に間接的にグラフト化される。「間接的なグラフト化」という用語は、分子的エンティティが、有機膜を構成している(コ)ポリマーを固体支持体の表面から分離しているケースを意味していると理解されたい。したがって、この分子的エンティティは、一つの共有結合によって固体支持体の表面に結合され、また別な共有結合によって有機膜に結合されている。
【0113】
そのような分子的エンティティは、先に定義された1種または複数のモノマーとは別の接着プライマーから得られたものであるのが有利である。より詳しくは、そのような接着プライマーは、まず第一に、ラジカル反応的グラフト化によって固体支持体の表面と反応し、次いで、その接着プライマーから得られたグラフト化誘導体が、ラジカル反応によって、先に定義されたモノマーと反応する。この代替えの形態においては、有機膜を形成する(コ)ポリマーが、場合によっては、それを構成するいくつかのモノマー単位の間に、前記ポリマーを固体支持体の表面にグラフト化させるために使用される接着プライマーから生じる一つまたは複数の単位を含むことができるということは明らかである。
【0114】
「接着プライマー」という用語は、本発明の文脈においては、ある種の条件下においては、ラジカル反応、たとえばラジカル反応的グラフト化によって固体支持体の表面に化学吸着される、各種の有機分子を意味していると理解されたい。そのような分子には、ラジカルと反応することが可能な少なくとも一つの官能基と、さらに化学吸着の後に他のラジカルに対して反応性である官能基とを含んでいる。
【0115】
その接着プライマーが、アリールジアゾニウム塩、アリールアンモニウム塩、アリールホスホニウム塩、およびアリールスルホニウム塩からなる群より選択される開裂可能なアリール塩であるのが有利である。これらの塩では、そのアリール基は、以下において定義されるR6で表すことが可能なアリール基である。
【0116】
開裂可能なアリール塩の中でも、次の式(I)の化合物を特に挙げることができる:
R6−N2+A− (I)
[式中、
−Aは、1価のアニオンを表し、そして
−R6は、アリール基を表す。]
【0117】
開裂可能なアリール塩、特に上述の式(I)の化合物のアリール基としては、有利には、芳香族炭素またはヘテロ芳香族構造を挙げることができるが、それらは場合によっては一置換または多置換されており、それぞれ3〜8個の原子を含み、その1個または複数のヘテロ原子がN、O、PまたはSであることが可能な一つまたは複数の芳香族またはヘテロ芳香族環からなっている。その1個または複数の置換基には、1個または複数のヘテロ原子たとえば、N、O、P、Cl、P、Si、BrまたはS,および特にC1〜C6アルキル基を含むことができる。
【0118】
開裂可能なアリール塩、特に上述の式(I)の化合物の中では、R6を、電子求引性基、たとえばNO2、COH、ケトン、CN、CO2H、NH2(NH3+の形で)、エステル、およびハロゲンによって置換されたアリール基から選択するのが好ましい。特に好ましいタイプの、アリールのR6基は、カルボキシフェニル、アミノフェニル、ニトロフェニル、およびフェニル基である。
【0119】
上述の式(I)の化合物の中では、Aは特に、以下のものから選択することができる:無機アニオン、たとえばハライド、たとえばI−、Br−、およびCl−、ハロホウ酸塩、たとえばテトラフルオロホウ酸塩、過塩素酸塩およびスルホン酸塩、ならびに有機アニオン、たとえばアルコキシドおよびカルボキシレート。
【0120】
式(I)の化合物としては、以下のものからなる群より選択される化合物を使用するのが特に有利である:フェニルジアゾニウムテトラフルオロボレート、4−ニトロフェニルジアゾニウムテトラフルオロボレート、4−ブロモ−フェニル−ジアゾニウムテトラフルオロボレート、4−アミノフェニル−ジアゾニウムクロリド、4−アミノメチルフェニルジアゾニウムクロリド、2−メチル−4−クロロフェニルジアゾニウムクロリド、4−ベンゾイルベンゼンジアゾニウムテトラフルオロボレート、4−シアノ−フェニルジアゾニウムテトラフルオロボレート、4−カルボキシフェニル−ジアゾニウムテトラフルオロボレート、4−アセトアミドフェニルジアゾニウムテトラフルオロボレート、4−カルボキシメチルフェニルジアゾニウムテトラフルオロボレート、2−メチル−4−[(2−メチルフェニル)ジアゼニル]ベンゼンジアゾニウムスルフェート、9,10−ジオキソ−9,10−ジヒドロ−1−アントラセンジアゾニウムクロリド、4−ニトロナフタレンジアゾニウムテトラフルオロボレート、およびナフタレンジアゾニウムテトラフルオロボレート。
【0121】
第一の代替えの形態においては、本発明によるプロセスの工程(ii)には以下のものからなる工程を含むことができる:
a)少なくとも1種の溶媒の存在下に、少なくとも1種のモノマー(特に先に定義されたもの)を、前記モノマー(特に先に定義されたもの)とは異なる少なくとも1種の接着プライマーを含む溶液に添加する工程、
b)工程(a)において得られた溶液を、その接着プライマーからラジカルエンティティを生成させることを可能とする、非電気化学的条件下に置く工程、
c)固体支持体の表面を、工程(b)の溶液と接触させる工程。
【0122】
第二の代替えの形態においては、本発明によるプロセスの工程(ii)には以下のものからなる工程を含むことができる:
a’)少なくとも1種の溶媒の存在下に、固体支持体の表面を、少なくとも1種の接着プライマー(特に先に定義されたもの)と、場合によっては前記接着プライマー(特に先に定義されたもの)とは異なる少なくとも1種のモノマーとを含む溶液と接触させる工程、
b’)その固体支持体の表面を、前記接着プライマーからラジカルエンティティを生成させることを可能とする非電気化学的条件下に、工程(a’)の溶液と接触状態に置く工程、
c’)場合によっては、前記接着プライマー(特に先に定義されたもの)とは異なる少なくとも1種のモノマーを工程(b’)で得られた溶液に添加する工程。
【0123】
先に定義されたプロセスの第二の代替えの形態において、以下の三つのシナリオを描くことが可能である。
【0124】
シナリオ1.
工程(a’)の溶液の中にはモノマーは存在せず、工程(c’)においてのみ添加する。このシナリオは、使用するモノマーが、特に水溶液中において、極めて溶解性が高い訳ではないという場合;そのモノマーが水に不溶性であり、接着プライマーがジアゾニウム塩である場合には、特に有利である。したがって、工程(c’)においては、そのモノマーを、溶液中、特に工程(a’)において採用されたのと同じ溶媒の中に、そして有利には超音波または界面活性剤を使用して予め製造しておいたエマルションまたは分散体の形態で添加することができる。
【0125】
シナリオ2.
工程(a’)の溶液の中にモノマーが存在し、そのプロセスには、工程(c’)がない。このシナリオは、そのプライマーがジアゾニウム塩である場合、そしてそのモノマーが水溶性である場合に特にあてはまる。第一の代替えの形態に従ったプロセスも、このシナリオで使用することもできる。
【0126】
シナリオ3.
モノマーの一部が、工程(a’)の溶液の中に存在し、そして(本質的に同一であったも、異なっていてもよい)モノマーの他の部分が、工程(c’)の中にのみ添加される。
【0127】
接着プライマーは、溶液(1種または複数)の中に導入してもよいし、あるいは後者の中でインサイチューで調製してもよい。
【0128】
接着プライマーをインサイチューで調製する場合には、たとえば接着プライマーの前駆体を使用するのが有利である。「接着プライマーの前駆体」という用語は、本発明の文脈においては、実施することが容易な単一の操作工程によって、前記接着プライマーから分離される分子を意味していると理解されたい、
【0129】
一般的には、それらの前駆体は、同一の環境条件下では、接着プライマーよりも高い安定性を示す。たとえば、アリールアミンは、アリールジアゾニウム塩の前駆体である。この理由は、たとえば酸性水性媒体中でのNaNO2との、あるいは有機媒体中でのNOBF4との単純な反応によって、対応するアリールジアゾニウム塩を生成させることが可能であるからである。
【0130】
本発明の文脈において採用される好適な前駆体は、次の式(III)のアリールジアゾニウム塩の前駆体である:
R6−NH2 (III)
[R6は、先に定義されたものである。]
【0131】
限定する訳ではないが例を挙げれば、本発明の文脈において採用することが可能な前駆体は、特に、4−アミノフェニルアミン(すなわち、p−フェニレンジアミン、または1,4−ジアミノフェニレン)、4−アミノ安息香酸、および4−アミノメチルフェニルアミンからなる群より選択される。
【0132】
工程(a)および(a’)の溶液中での接着プライマーまたは接着プライマー前駆体の量は、実験担当者の望むように変化させることができる。この量は、有利にはほぼ10−6M〜5Mの間、好ましくは、5×10−2M〜10−1Mの間である。
【0133】
工程(a)、(a’)および/または(c’)の溶液の中の重合性モノマーの量は、実験担当者の望むように変化させることができる。この量は、採用されるこれらの溶液の溶媒(すなわち、反応溶媒)中への、考慮対象のモノマーの溶解度よりも高くすることも可能であり、所定の温度、一般的には周囲温度または反応温度における、その溶液中での前記モノマーの溶解度のたとえば18〜40倍とすることができる。それらの条件下では、モノマーの分子を溶液の中に分散させることを可能とする手段、たとえば界面活性剤または超音波を採用するのが有利である。
【0134】
その溶媒が、本発明の特定の実施態様に従って、プロトン性溶媒である場合には、そのモノマーが5×10−2Mよりも低い溶解度を示すときには、界面活性剤を使用することが推奨される。使用可能な界面活性剤は、具体的には、アニオン性界面活性剤、カチオン性界面活性剤、両性イオン性界面活性剤、両性界面活性剤、中性(ノニオン性)界面活性剤、および特には、国際公開第2008/078052号パンフレット[4]に記載されている界面活性剤である。界面活性剤の濃度は、典型的にはほぼ0.5mM〜5Mの間、好ましくはほぼ0.1mM〜150mMの間となるであろう。推奨される界面活性剤の濃度は、通常、10mMである。
【0135】
プロセスの工程(ii)は、一般的には、その上に膜をグラフト化することを望んでいるサンプルの表面に対しても、採用したモノマーに対しても、破壊的ではない穏やかな条件下で実施する。したがって、モノマーが分解しないような条件下で操作するのが望ましい。さらに、溶液の温度は、反応溶媒によって制限され、反応溶媒を液状に維持できるのが好ましい。典型的には、そのプロセスは、0〜100℃の間、一般的には標準温度圧力条件(STPC)下、利用者が見出した場所に従って、多くの場合ほぼ25℃、1気圧前後で実施する。
【0136】
本発明によるプロセスの工程(a)、(a’)、(b)、(b’)、(c)、または(c’)の際に採用される溶液には、溶媒として、以下に記載の溶媒を使用することができる:
−プロトン性溶媒、すなわち、プロトンの形態で放出されることが可能な少なくとも1個の水素原子を含む溶媒であって、有利には、水、脱イオン水、蒸留水(それらの水は酸性化されていても、塩基性であってもよい)、酢酸、ヒドロキシル化溶媒たとえばメタノールおよびエタノール、低分子量液状グリコールたとえばエチレングリコール、およびそれらの混合物からなる群より選択される溶媒か;
−または、非プロトン性溶媒、すなわち、プロトンを放出することができないか、または極端な条件以外ではそれらの一つを受け入れることができない溶媒であって、有利には、ジメチルホルムアミド(DMF)、アセトン、アセトニトリル、およびジメチルスルホキシド(DMSO)から選択される溶媒;
−または、少なくとも1種のプロトン性溶媒と少なくとも1種の非プロトン性溶媒との混合物。
【0137】
本発明のプロセスの工程(b)または(b’)において、少なくとも1種のラジカルエンティティの形成を可能とする条件は、工程(b)もしくは(b’)の溶液または固体支持体に、電圧をまったく印加しなくても、ラジカルエンティティの形成が可能となるような条件である。
【0138】
それらの条件としては、たとえば、温度、溶媒の性質、特定の添加剤の存在、撹拌、または圧力などのパラメーターが挙げられるが、その一方で、ラジカルエンティティの形成の際には電流は含まれない。ラジカルエンティティの形成を可能とする条件は多く存在しているが、このタイプの反応は公知であり、従来技術において詳しく検討されている。
【0139】
したがって、たとえば、ラジカルエンティティを形成させるために、接着プライマーを不安定化させる目的で、熱的、動力学的、化学的、光化学的、または放射化学的環境を作用させることが可能である。言うまでもないことであるが、いくつかのそれらのパラメーターを同時に作用させることも可能である。
【0140】
本発明の文脈においては、本発明におけるグラフト化工程の際にラジカルエンティティの生成を可能とする条件は、その接着プライマーが置かれる、典型的には、熱的条件、動力学的条件、化学的条件、光化学的条件、放射化学的条件、およびそれらの組合せからなる群より選択される。本発明によるプロセスのグラフト化工程の文脈において採用される条件が、熱的条件、化学的条件、光化学的条件、放射化学的条件、およびそれらの相互の組合せ、および/または動力学的条件との組合せ、からなる群より選択されるのが有利である。本発明によるプロセスのグラフト化工程の文脈において採用される条件は、より具体的には、化学的条件である。
【0141】
熱的環境は温度に依存する。それは、当業者によって通常採用される加熱手段を使用すれば、容易に調節される。サーモスタットで調節する環境を使用するのが、特に有利であるが、その理由は、反応条件を正確に調節することが可能となるからである。
【0142】
動力学的環境は、実質的には、系の撹拌および摩擦力に対応する。この場合、それは、本来的な分子の撹拌(結合の伸びなど)ではなく、分子全体の動きの撹拌である。
【0143】
したがって、前記グラフト化工程の際に、工程(b)または(b’)の溶液を、機械的撹拌または超音波を用いた処理にかける。第一の代替えの形態においては、工程(b)または(b’)の際に採用される溶液を、マグネチックスターラーおよびマグネチックバーを介して、5分〜24時間の間、とりわけ10分〜12時間の間、特には15分〜6時間の間の撹拌時間で、高回転速度にかける。第二の代替えの形態においては、工程(b)または(b’)の際に採用される溶液を、具体的には、典型的には吸収性能(absorption capacity)が500W、周波数が25kHzまたは45kHzの超音波浴を使用し、1分〜24時間の間、とりわけ15分〜12時間の間、特には30分〜6時間の間の撹拌時間で、超音波を用いた処理にかける。
【0144】
最後に、たとえば電磁線、γ線、UV線、電子ビームまたはイオンビームのような、各種のタイプの放射線の作用もまた、ラジカルを形成させるに十分なほど接着プライマーを不安定化させることができる。使用する接着プライマーに合わせて、採用する波長を選択することになるが、その選択は、発明に値するようなことではない。
【0145】
化学的条件の文脈においては、1種または複数の化学的重合開始剤が、工程(b)または(b’)の際に使用される溶液において採用される。化学的重合開始剤を存在させることが、たとえば先に挙げたような非化学的環境条件と組み合わされることも多い。典型的には、化学的重合開始剤(その安定性が、選択された環境条件下においては、採用された接着プライマーの安定性よりも低い)が、不安定な形態に変化し、それが後者に作用して、後者から始まる、ラジカルエンティティの生成をもたらすことになるであろう。環境条件に本質的にはつながらない作用を有する化学的重合開始剤を採用することもまた可能であり、それらの重合開始剤は、熱的条件またはさらには動力学的条件の広い範囲にわたって作用を有することができる。重合開始剤は、反応の環境、たとえば採用される溶媒に適したものであるのが好ましいであろう。
【0146】
多くの化学的重合開始剤が存在している。一般的には、採用される環境条件に従って、それらは三つのタイプに分類される:
−熱重合開始剤、その最も一般的なものは、ペルオキシド化合物またはアゾ化合物である。熱の作用が加わると、これらの化合物が分解して、フリーラジカルとなる。この場合においては、その反応は、重合開始剤から生ずるラジカルの発生のために必要とされる温度に相当する、最低温度で実施される。このタイプの化学的重合開始剤は、一般的には、それらの分解の動力学に従って、ある種の温度範囲内に限定して使用される;
−放射線によって(一般的にはUV照射線によるが、γ線または電子ビームによっても)引き起こされる照射によって励起される、光化学的重合開始剤または放射化学的重合開始剤は、多少なりとも複雑なメカニズムによって、ラジカルの発生を可能とする。光化学的重合開始剤または放射化学的重合開始剤としては、Bu3SnHおよびI2が挙げられる;
−本質的に化学的重合開始剤、このタイプの重合開始剤は、迅速かつ標準温度圧力条件で接着プライマーに作用して、それからラジカルを生成させる。そのような重合開始剤は、一般的には、反応条件下では使用される接着プライマーの還元電位よりも低い酸化/還元電位を有している。したがって、接着プライマーの性質に従って、それらは、接着プライマーの不安定化を起こさせるに十分な割合の、たとえば、還元性金属、たとえば鉄、亜鉛、またはニッケル;メタロセン;有機還元剤、たとえば次亜リン酸(H3PO2)またはアスコルビン酸;または有機もしくは無機塩基であってよい。
【0147】
化学的重合開始剤として使用される還元性金属は、微細な形態、たとえば金属ウールまたは金属の削り屑(metal filings)として加えるのが有利である。一般的には、化学的重合開始剤として有機または無機の塩基を使用する場合には、4以上のpHであれば一般的には十分である。ラジカルリザーバータイプの構造、たとえば、電子ビームを用いるか、または重イオンのビームを用いるか、および/または上述の照射手段を組み合わせて用いることによって予め照射しておいたポリマーマトリックスを、接着プライマーを不安定化させ、そしてその結果このプライマーからラジカルエンティティを生成させるための化学的重合開始剤として採用することもまた可能である。
【0148】
本発明によるプロセスの工程(ii)は、先に定義されたグラフト化工程に相当する。それは、10分〜6時間、とりわけ30分〜4時間、特には1時間〜2時間、さらに特にはほぼ90分(すなわち、90分±10分)のあいだ、続けることができる。
【0149】
ラジカル経路によって重合することが可能な接着プライマーおよびモノマーが、工程(c)または(c’)の溶液の中には大量に存在しているので、それらの分子が固体支持体に結合されるより前にグラフト化工程を停止させることができる。当業者ならば、グラフト化工程を停止させることを可能とする別な方法を知っているし、採用した接着プライマーおよび重合性モノマーに対して最も適した方法の決め方も知っていることであろう。そのような方法の例として、以下のようなものをあげることができる:工程(c)または(c’)の溶液のpHを、特にそれに塩基性の溶液(たとえば、pHが10よりも高い塩基性の水)を添加することによって変化させる方法、工程(c)または(c’)の溶液から(たとえば濾過によるか沈殿によるか、または錯化によって)接着プライマーおよび重合性モノマーを除去する方法、または工程(c)または(c’)の溶液から固体支持体を抜き出す方法。
【0150】
本発明はさらに、表面の一部にグラフト化させた、本発明によるプロセスによって調製可能な、先に定義されたような有機膜を有する固体支持体にも関する。本明細書で定義されたような酸化的前処理を用いると、グラフト化された有機膜がより厚く、そのためにそのようにグラフト化された支持体の物理化学的性質が極めて大きく改質される。さらには、ポリマーの接触角、したがって親水性の改質の観点からは、このことは、本特許出願において広く確認される。接触角を変化させることは、その物質の化学的な性質を変えることになる。
【0151】
本発明はさらに、先に定義された有機膜を固体支持体の表面の全部または一部にラジカル反応的にグラフト化させるためのプロセスを改良するために、酸化処理を使用することにも関する。
【0152】
この改良は、酸化処理の後に先に定義された有機膜を用いて、ラジカル反応的グラフト化によってグラフト化された固体支持体の上に位置する液滴の接触角を、酸化処理なしの有機膜を用いてラジカル反応的グラフト化によってグラフト化された固体支持体の上に位置する液滴の接触角と比較することによって、証明することができる。
【0153】
酸化処理の後でグラフト化された固体支持体の上の液滴の接触角は、酸化処理なしでグラフト化された固体支持体の上の液滴の接触角に比較して、とりわけ5〜70%、特には20〜50%小さくなる。
【0154】
この改良はさらに、酸化処理の後でラジカル反応的グラフト化によって固体支持体にグラフト化された先に定義されたような有機膜の厚みを、酸化処理なしで固体支持体に同一の条件下(同一の溶媒、同一の接着プライマー、および同一のモノマー、同一のグラフト化時間)でグラフト化された有機膜の厚みと比較することによって証明することもできる。この厚みにおける増加は、IR分析によって可視化することができる。これは、IR分析によると、グラフト化された有機膜に帰属された吸収帯が、膜の厚みが増加したと言えるほど、よりはっきりと見えるようになっているからである。
【0155】
添付した図面を参照し、以下の実施例(説明のためであって、限定を意図するものではない)を読めば、本発明のその他の特徴および利点は、当業者に、より明らかとなるであろう。
【図面の簡単な説明】
【0156】
【図1】以下の表面の上に載せた水の液滴について測定した接触角を示す図である:ABS、ABS/PC、PVDFまたはPETから作られている未処理かつ未グラフト化表面(「ブランク」);国際公開第2008/078052号パンフレット[4]に記載のプロセスに従ってPAAを用いてグラフト化されたABS、ABS/PC、PVDFまたはPETから作られている表面(「グラフトファスト(GraftFast)(著作権)」);およびフェントン酸化によって前処理され、次いで国際公開第2008/078052号パンフレット[4]に記載のプロセスに従ってPAAを用いてグラフト化されたABS、ABS/PC、PVDFまたはPETから作られている表面(「前処理+グラフトファスト(GraftFast)(著作権)」)。
【図2】以下の表面の上に載せた水の液滴について測定した接触角を示す図である:PVDFから作られている未処理かつ未グラフト化の表面(「ブランク」);国際公開第2008/078052号パンフレット[4]に記載のプロセスに従ってPAAを用いてグラフト化されたPVDFから作られている表面(「グラフトファスト(GraftFast)(著作権)」);および、アルコール性水酸化カリウムを用いて前処理され、次いで国際公開第2008/078052号パンフレット[4]に記載のプロセスに従ってPAAを用いてグラフト化されたPVDFから作られている表面(「前処理+フラフトファスト(GraftFast)(著作権)」)。
【図3】以下の表面の上に載せた水の液滴について測定した接触角を示す図である:PP、PET、PVDFまたはPCから作られている未処理かつ未グラフト化の表面(「ブランク」);国際公開第2008/078052号パンフレット[4]に記載のプロセスに従ってPAAを用いてグラフト化されたPP、PET、PVDFまたはPCから作られている表面(「グラフトファスト(GraftFast)(著作権)」);および、オゾン用いて前処理され、次いで国際公開第2008/078052号パンフレット[4]に記載のプロセスに従ってPAAを用いてグラフト化されたPP、PET、PVDFまたはPCから作られている表面(「前処理+フラフトファスト(GraftFast)(著作権)」)。
【図4】以下の表面の上に載せた水の液滴について測定した接触角を示す図である:ABS、ABS/PC、またはPPから作られている未処理かつ未グラフト化表面(「ブランク」);国際公開第2008/078052号パンフレット[4]に記載のプロセスに従ってpHEMAを用いてグラフト化されたABS、ABS/PC、またはPPから作られている表面(「グラフトファスト(GraftFast)(著作権)」);およびフェントン酸化によって前処理され、次いで国際公開第2008/078052号パンフレット[4]に記載のプロセスに従ってpHEMAを用いてグラフト化されたABS、ABS/PC、またはPPから作られている表面(「前処理+フラフトファスト(GraftFast)(著作権)」)。
【図5】以下の表面の上に載せた水の液滴について測定した接触角を示す図である:PPから作られている未処理かつ未グラフト化表面(「ブランク」);国際公開第2008/078052号パンフレット[4]に記載のプロセスに従ってp(4−VP)を用いてグラフト化されたPPから作られている表面(「グラフトファスト(GraftFast)(著作権)」);および、フェントン酸化によって前処理され、次いで国際公開第2008/078052号パンフレット[4]に記載のプロセスに従ってp(4−VP)を用いてグラフト化されたPPから作られている表面(「前処理+フラフトファスト(GraftFast)(著作権)」)。
【発明を実施するための形態】
【0157】
以下の実施例は、ガラス容器の中で実施した。
【0158】
特に断らない限り、それらは、大気中、標準温度圧力条件下(ほぼ25℃、ほぼ1気圧)で実施した。特に断らない限り、採用した反応剤は市場で直接購入したものであって、さらなる精製は行わなかった。
【0159】
金の細片およびプラスチックまたはポリマーのサンプルは、1×5cm2の表面積を有していた。試験をしたプラスチックのサンプルには、アクリロニトリル/ブタジエン/スチレン(ABS)、アクリロニトリル/ブタジエン/スチレン−ポリカーボネート(ABS/PC)、ポリプロピレン(PP)、およびポリアミド(PA)が含まれる。試験したポリマーのサンプルには、ポリフッ化ビニリデン(PVDF)、ポリカーボネート(PC)、およびポリエチレンテレフタレート(PET)が含まれる。
【0160】
雰囲気の組成には何の注意も払わなかったし、溶液の脱気も行わなかった。プラスチックまたはポリマーのサンプルはあらかじめ、超音波の下で10分間、工業用洗剤(TDF4)の溶液を使用して清浄化させ、次いでミリQ(Milli−Q)水中で10分間超音波にかけてから、乾燥させた。
【0161】
各種のサンプルは、IR分光測定法によるか、および/またはそれらのサンプルの表面上に載せた一定容積(2μL)の液滴との間の接触角を測定することによって分析した。
【0162】
1:プラスチックおよびポリマーの酸化処理
実施例1.1:鉄(II)テトラフルオロボレートから出発するフェントン反応による酸化
鉄(II)テトラフルオロボレート(1.69g、5×10−2mol)を、水中0.001M硫酸100mLの中に溶解させた。プラスチックおよび/またはポリマーのサンプルをこの溶液の中に浸漬させた。次いで、pH3一定に保ちながら、12mL(0.125mol)の35%過酸化水素水溶液を添加した。25分後に、ミリQ(Milli−Q)水を用いてサンプルを洗い流し、水中で10分間超音波をかけてから、乾燥させた。
【0163】
実施例1.2:硫酸鉄(II)から出発するフェントン反応による酸化
硫酸鉄(II)(3.475g、5×10−2mol)を、0.001M硫酸水溶液25mLの中に溶解させた。プラスチックのサンプルおよび/またはポリマーをこの溶液の中に浸漬させた。次いで、pH3一定に保ちながら、5mL(0.062mol)の35%過酸化水素水溶液を添加した。25分後に、ミリQ(Milli−Q)水を用いてサンプルを洗い流し、水中で10分間超音波をかけてから、乾燥させた。
【0164】
実施例1.3:アルコール性水酸化カリウムを用いた処理による酸化
4g(7.1×10−2mol)の水酸化カリウムを、20mLの95%エタノールの中に溶解させた。この溶液の中に、ポリマーのサンプルを、典型的には20分〜2時間の間で変化させた時間のあいだ、浸漬させた。ミリQ(Milli−Q)水を用いてサンプルを洗い流し、水中で10分間超音波をかけてから、乾燥させた。
【0165】
実施例1.4:オゾンを用いた処理による酸化
各種のプラスチックおよびポリマーを、10分間、オゾンの流れに置いた(オゾン雰囲気に曝露させた)。
【0166】
そのオゾン発生器は、低圧水銀蒸気ランプ(28mW/cm2、254nm)を有する、UVO−クリーナー(UVO−Cleaner)モデル42−200である。
【0167】
実施例1.5:KMnO4を用いた処理による酸化
0.75gの過マンガン酸カリウム(5×10−3mol)を、25mLの3.3M硫酸溶液に添加した。プラスチックおよび/またはポリマーのサンプルをこの溶液の中に15分間浸漬させた。各種のサンプルを、順次、ミリQ(Milli−Q)水を用いて洗い流し、ミリQ(Milli−Q)水中で超音波をかけてから、乾燥させた。
【0168】
ABSまたはABS−PCに関しては、ABSおよびABS−PCの中に存在しているポリブタジエンの小塊を、KMnO4が酸化し、破壊する。ポリアクリロニトリルもまた酸化して、特にポリアミドとすることができる。
【0169】
本発明の文脈において実施される試験では、これらの酸化的改質は、当然のことながらIRスペクトルでは見分けられない。その一方で、それらのポリマーをKMnO4溶液の中に6時間浸漬させることによって、そのような酸化的改質を実証することが可能であった。
【0170】
実施例1.6:KMnO4(実施例1.5)に続けてフェントン反応(実施例1.2)を用いた処理による酸化
実施例1.5に記載された形態に従って、この実施例を実施した。そうして調製されたサンプルを、次いで、フェントン反応のための実施例1.2に記載された反応媒体の中に導入すると、その結果として、それらが清浄化されたので、上述のようにして乾燥させた。
【0171】
実施例1.1〜1.6で得られた結果を、接触角の測定については表1に、そしてIR分光測定法については表2に示す。
【0172】
【表1】
【0173】
【表2】
【0174】
2:国際公開第2008/078052号パンフレット[4]に記載のプロトコールに従った、プラスチックのサンプルおよび/またはポリマーへの膜のグラフト化
手順:
フェントン反応に従った処理をしていないプラスチック(ABS、ABS/PC、PP、およびPA)を、超音波下に石けん水を用いて10分間、次いで、MQ水を用いて10分間かけて清浄化した。
【0175】
実施例2.1:4−アミノ安息香酸から出発する、国際公開第2008/078052号パンフレット[4]に記載のプロセスによるポリアクリル酸膜のグラフト化
4−アミノ安息香酸(2.7g、2×10−2mol)を、塩酸溶液(100mL、0.5M)の中に溶解させた。100mLのNaNO2の水溶液(1.38g、2×10−2mol)を、その溶液に添加した。
【0176】
このジアゾニウム塩溶液に、6.8mLのアクリル酸AA(10−1mol)および2gの鉄の削り屑を添加した。
【0177】
次いで、プラスチックのサンプルおよび/またはポリマーおよび金のストリップを、反応媒体の中に導入し、90分間置いた。ミリQ(Milli−Q)水を用いて各種のサンプルを連続的に洗い流し、0.01M水酸化ナトリウム溶液中で超音波にかけ、次いでミリQ(Milli−Q)水を用いて洗浄してから、乾燥させた。
【0178】
金のストリップのIR分光測定法による分析から、そのグラフト化浴が活性であったことが確認される。3356cm−1(COOH変角振動)、1710cm−1(C=O変角振動)、および1265cm−1(C−O変角振動)に特性吸収帯が存在している。
【0179】
実施例2.2:1,4−ジアミノフェニレンから出発する、国際公開第2008/078052号パンフレット[4]に記載のプロセスによるポリアクリル酸膜のグラフト化
手順1:
この手順は、実施例2.1の場合と同じであるが、ただし、4−アミノ安息香酸を、1,4−ジアミノフェニレン(2.13g、2×10−2mol)に置き換えた。
【0180】
次いで、プラスチックのサンプルおよび/またはポリマーおよび金のストリップを、反応媒体の中に導入し、90分間おいた。ミリQ(Milli−Q)水を用いて各種のサンプルを連続的に洗い流し、0.01M水酸化ナトリウム溶液中で超音波にかけ、次いでミリQ(Milli−Q)水を用いて洗浄してから、乾燥させた。
【0181】
手順2:
4−アミノ安息香酸(1.07g、1×10−2mol)を、塩酸溶液(100mL、0.5M)の中に溶解させた。100mLのNaNO2の水溶液(0.69g、1×10−2mol)を、この溶液に添加した。100mLのアクリル酸AA(1.46mol)および次いで15g(0.27mol)の鉄の削り屑を、このジアゾニウム塩の溶液に添加した。
【0182】
次いで、プラスチックのサンプルおよび/またはポリマーおよび金のストリップを、反応媒体の中に導入し、周囲温度で30分間、38℃のオーブン中に90分間置いた。ミリQ(Milli−Q)水を用いて各種のサンプルを連続的に洗い流し、0.1M水酸化ナトリウム溶液中で超音波にかけ、次いでミリQ(Milli−Q)水を用いて洗浄してから、乾燥させた。
【0183】
金のストリップのIR分光測定法による分析から、その両方の手順において、そのグラフト化浴が活性であったことが確認される。3207cm−1(COOH変角振動)、1720cm−1(C=O変角振動)、および1262cm−1(C−O変角振動)に特性吸収帯が存在している。
【0184】
実施例2.3:4−ニトロベンゼンジアゾニウムテトラフルオロボレートから出発する、国際公開第2008/078052号パンフレット[4]に記載のプロセスによるポリアクリル酸膜のグラフト化
その手順は、実施例2.1の場合と同じである。4−アミノ安息香酸を4−ニトロベンゼンジアゾニウムテトラフルオロボレート(4.7g、2×10−2mol)に置き換えたが、NaNO2の存在下にジアゾニウム塩前駆体をジアゾニウム塩に転換させる工程はもはや必要ない。
【0185】
次いで、プラスチックおよび/またはポリマーのサンプルならびに金のストリップを、反応媒体の中に導入し、90分間置いた。ミリQ(Milli−Q)水を用いて各種のサンプルを連続的に洗い流し、0.01M水酸化ナトリウム溶液中で超音波にかけ、次いでミリQ(Milli−Q)水を用いて洗浄してから、乾燥させた。
【0186】
金のストリップのIR分光測定法による分析から、そのグラフト化浴が活性であったことが確認される。3405cm−1(COOH変角振動)、1695cm−1(C=O変角振動)、1597cm−1(C=C変角振動)、1518cm−1(N=O変角振動)、1345cm−1(N=O変角振動)、および1162cm−1(C−O変角振動)に特性吸収帯が存在している。
【0187】
実施例2.4:4−アミノ安息香酸から出発する、国際公開第2008/078052号パンフレット[4]に記載のプロセスによるポリ(HEMA)膜のグラフト化
その手順は、実施例2.1の場合と同じである。アクリル酸AAを、12.5mLのメタクリル酸2−ヒドロキシエチル(10−1mol)に置き換えた。
【0188】
ミリQ(Milli−Q)水を用いて各種のサンプルを連続的に洗い流し、次いで加熱条件下でエタノールの浴の中に30分間浸漬させてから、乾燥させた。
【0189】
金のストリップのIR分光測定法による分析から、そのグラフト化浴が活性であったことが確認される。3387cm−1(OH変角振動)、1726cm−1(C=O変角振動)、および1160cm−1(C−O変角振動)に特性吸収帯が存在している。
【0190】
実施例2.5:4−アミノ安息香酸から出発する、国際公開第2008/078052号パンフレット[4]に記載のプロセスによるポリ(4−ビニルピリジン)膜のグラフト化
その手順は、実施例2.1の場合と同じである。アクリル酸AAを、100mLの水中0.1Mの4−ビニルピリジン(4−VP)および1MのH2SO4の溶液に置き換えた。
【0191】
ミリQ(Milli−Q)水を用いて各種のサンプルを連続的に洗い流し、次いで加熱条件下でエタノールの浴の中に30分間浸漬させてから、乾燥させた。
【0192】
金のストリップのIR分光測定法による分析から、そのグラフト化浴が活性であったことが確認される。3371cm−1(COOH変角振動)、1600cm−1(C=N変角振動)、および1556cm−1(C=C変角振動)に特性吸収帯が存在している。
【0193】
実施例2.1〜2.5についての接触角の測定から得られた結果を表3に示す。
【0194】
【表3】
【0195】
3:国際公開第2008/078052号パンフレット[4]に記載のプロセスによる膜の表面酸化+グラフト化
実施例3.1:フェントン酸化(実施例1.1)とそれに続く、4−アミノ安息香酸から出発する、国際公開第2008/078052号パンフレット[4]に記載のプロセスによるPAA膜のグラフト化(実施例2.1)
実施例1.1に記載された形態に従って、実施例を実施した。調製されたサンプルを、次いで、グラフト化にかけるために、実施例2.1に記載の反応媒体の中に導入すると、その結果として、それらが清浄化されたので、上述のようにして乾燥させた。
【0196】
実施例3.2:フェントン酸化(実施例1.1)と、それに続く、1,4−ジアミノフェニレンから出発する、国際公開第2008/078052号パンフレット[4]に記載のプロセスによるPAA膜のグラフト化(実施例2.2)
実施例1.1に記載された形態に従って、実施例を実施した。調製されたサンプルを、次いで、グラフト化にかけるために、実施例2.2に記載の反応媒体の中に導入すると、その結果として、それらが清浄化されたので、上述のようにして乾燥させた。
【0197】
実施例3.3:フェントン酸化(実施例1.1)と、それに続く、4−ニトロベンゼンジアゾニウムテトラフルオロボレートから出発する、国際公開第2008/078052号パンフレット[4]に記載のプロセスによるPAA膜のグラフト化(実施例2.3)
実施例1.1に記載された形態に従って、実施例を実施した。調製されたサンプルを、次いで、グラフト化にかけるために、実施例2.3に記載の反応媒体の中に導入すると、その結果として、それらが清浄化されたので、上述のようにして乾燥させた。
【0198】
実施例3.4:アルコール性水酸化カリウムを用いた処理による酸化(実施例1.3)と、それに続く、4−アミノ安息香酸CBDから出発する、国際公開第2008/078052号パンフレット[4]に記載のプロセスによるPAA膜のグラフト化(実施例2.1)
実施例1.3に記載された形態に従って、実施例を実施した。調製されたサンプルを、次いで、グラフト化にかけるために、実施例2.1に記載の反応媒体の中に導入すると、その結果として、それらが清浄化されたので、上述のようにして乾燥させた。
【0199】
実施例3.5:KMnO4を用いた処理による酸化(実施例1.5)と、それに続く、1,4−ジアミノフェニレンから出発する、国際公開第2008/078052号パンフレット[4]に記載のプロセスによるPAA膜のグラフト化(実施例2.2)
実施例1.5に記載された形態に従って、実施例を実施した。調製されたサンプルを、次いで、グラフト化にかけるために、実施例2.2(第二の手順)に記載の反応媒体の中に導入すると、その結果として、それらが清浄化されたので、上述のようにして乾燥させた。
【0200】
実施例3.6:オゾンを用いた処理による酸化(実施例1.4)と、それに続く、4−アミノ安息香酸から出発する、国際公開第2008/078052号パンフレット[4]に記載のプロセスによるPAA膜のグラフト化(実施例2.1)
実施例1.4に記載された形態に従って、実施例を実施した。調製されたサンプルを、次いで、グラフト化にかけるために、実施例2.1に記載の反応媒体の中に導入すると、その結果として、それらが清浄化されたので、上述のようにして乾燥させた。
【0201】
実施例3.7:フェントン酸化(実施例1.1)と、それに続く、4−アミノ安息香酸から出発する、国際公開第2008/078052号パンフレット[4]に記載のプロセスによるpHEMA膜のグラフト化(実施例2.4)
実施例1.1に記載された形態に従って、実施例を実施した。調製されたサンプルを、次いで、グラフト化にかけるために、実施例2.4に記載の反応媒体の中に導入すると、その結果として、それらが清浄化されたので、上述のようにして乾燥させた。
【0202】
実施例3.8:フェントン酸化(実施例1.1)と、それに続く、4−アミノ安息香酸から出発する、国際公開第2008/078052号パンフレット[4]に記載のプロセスによるp(4−VP)膜のグラフト化(実施例2.5)
実施例1.1に記載された形態に従って、実施例を実施した。調製されたサンプルを、次いで、グラフト化にかけるために、実施例2.5に記載の反応媒体の中に導入すると、その結果として、それらが清浄化されたので、上述のようにして乾燥させた。
【0203】
実施例3.9:KMnO4/フェントン酸化(実施例1.5)と、それに続く、1,4−ジアミノフェニレンから出発する、国際公開第2008/078052号パンフレット[4]に記載のプロセスによるPAA膜のグラフト化(実施例2.2)
実施例1.5に記載された形態に従って、実施例を実施した。調製されたサンプルを、次いで、グラフト化にかけるために、実施例2.2(第二の手順)に記載の反応媒体の中に導入すると、その結果として、それらが清浄化されたので、上述のようにして乾燥させた。
【0204】
実施例3.1〜3.9についての接触角の測定から得られた結果を表4に示す。
【0205】
【表4】
【0206】
4:国際公開第2008/078052号パンフレット[4]に記載のプロセスによるポリマー膜の作成におよぼす表面酸化の影響
4.1:国際公開第2008/078052号パンフレット[4]に記載のプロセスによるPAA膜のグラフト化に対する酸化的前処理の改良
フェントン酸化を介しての酸化的前処理をすることによって、国際公開第2008/078052号パンフレット[4]に記載の方法によるポリアクリル酸のグラフト化を向上させることが可能となる。この向上効果は、ブランクのABS、ABS/PC、PVDFまたはPETから作られている支持体、または国際公開第2008/078052号パンフレット[4]に記載のプロセスによってグラフト化されたABS、ABS/PC、PVDFまたはPETから作られている支持体の上に載せた液滴に比較して、前処理をされ、次いで国際公開第2008/078052号パンフレット[4]に記載のプロセスによってグラフト化されたABS、ABS/PC、PVDFまたはPETから作られている支持体の上に載せた液滴について測定された接触角が顕著に小さくなるということに反映されている(図1)。
【0207】
下記の表5は、IR分析によるポリアクリル酸のC=O特性吸収帯の強度を示している。この吸収帯は、本発明におけるフェントン酸化を介しての酸化的前処理に続けてグラフト化させた後のみに観察できる。
【0208】
【表5】
【0209】
アルコール性水酸化カリウムを用いた処理を介しての酸化的前処理をすることによって、国際公開第2008/078052号パンフレット[4]に記載の方法によるポリアクリル酸のグラフト化を向上させることが可能となる。この向上効果は、ブランクのPVDFから作られている支持体、または国際公開第2008/078052号パンフレット[4]に記載のプロセスによってグラフト化されたPVDFから作られている支持体に比較して、前処理をされ、次いで国際公開第2008/078052号パンフレット[4]に記載のプロセスによってグラフト化されたPVDFから作られている支持体の上に載せた液滴について測定された接触角が顕著に小さくなるということに反映されている(図2)。
【0210】
下記の表6は、IR分析によるポリアクリル酸のC=O特性吸収帯の強度を示している。この吸収帯は、本発明におけるアルコール性水酸化カリウムを用いた処理を介しての酸化的前処理に続けてグラフト化させた後にのみに観察できる。
【0211】
【表6】
【0212】
オゾンを用いた処理を介しての酸化的前処理をすることによって、国際公開第2008/078052号パンフレット[4]に記載の方法によるポリアクリル酸のグラフト化を向上させることが可能となる。この向上効果は、ブランクのPVDFから作られている支持体、または国際公開第2008/078052号パンフレット[4]に記載のプロセスによってグラフト化されたPVDFから作られている支持体に比較して、前処理をされ、次いで国際公開第2008/078052号パンフレット[4]に記載のプロセスによってグラフト化されたPVDFから作られている支持体の上に載せた液滴について測定された接触角が顕著に小さくなるということに反映されている(図3)。
【0213】
PETおよびPCの構造には基本的に、酸素含有基が含まれている。したがって、これらのポリマーでは、オゾンを用いた前処理の効果はPPおよびPVDFの場合よりは弱い。下記の表7は、IR分析によるポリアクリル酸のC=O特性吸収帯の強度を示している。この吸収帯は、本発明におけるオゾンを用いた処理を介しての酸化的前処理に続けてグラフト化させた後にのみに観察できる。
【0214】
【表7】
【0215】
4.2:国際公開第2008/078052号パンフレット[4]に記載のプロセスによるpHEMA膜のグラフト化に対する酸化的前処理の改良
フェントン酸化を介しての酸化的前処理をすることによって、国際公開第2008/078052号パンフレット[4]に記載の方法によるpHEMAのグラフト化を向上させることが可能となる。この向上効果は、ブランクのABS、ABS/PC、またはPPから作られている支持体、または国際公開第2008/078052号パンフレット[4]に記載のプロセスによってグラフト化されたABS、ABS/PC、またはPPから作られている支持体の上に載せた液滴に比較して、前処理をされ、次いで国際公開第2008/078052号パンフレット[4]に記載のプロセスによってグラフト化されたABS、ABS/PC、またはPPから作られている支持体の上に載せた液滴について測定された接触角が顕著に小さくなるということに反映されている(図4)。
【0216】
4.3:国際公開第2008/078052号パンフレット[4]に記載のプロセスによるp(4−VP)膜のグラフト化に対する酸化的前処理の改良
フェントン酸化を介しての酸化的前処理をすることによって、国際公開第2008/078052号パンフレット[4]に記載の方法によるp(4−VP)のグラフト化を向上させることが可能となる。この向上効果は、ブランクのPPから作られている支持体、または国際公開第2008/078052号パンフレット[4]に記載のプロセスによってグラフト化されたPPから作られている支持体に比較して、前処理をされ、次いで国際公開第2008/078052号パンフレット[4]に記載のプロセスによってグラフト化されたPPから作られている支持体の上に載せた液滴について測定された接触角が顕著に小さくなるということに反映されている(図5)。
【0217】
引用文献
[1]パラシン(Palacin)ら、「分子−金属結合:導電性表面上のポリマーの電気グラフト化(Molecule−to−metal bonds:Electrografting polymers on conducting surfaces)」、ケミカル・フィジックス・ケミストリー(Chem.Phys.Chem.)、2004、5(10)、1469〜1481。
[2]デニアウ(Deniau)ら、「炭素−金属結合:2−ブテンニトリルの電解還元」、サーフェス・サイエンス(Surf.Sci.)、2006、600(3)、675〜684。
[3]国際公開第03/018212号パンフレット(出願人:CEA,公開日:2003年3月6日)。
[4]国際公開第2008/078052号パンフレット(出願人:CEA、公開日:2008年7月3日)。
[5]ナウディン(Naudin)、「ポリマーの命名、分類および化学式(Nomenclature,classificaiton et formules chimques des polymeres[Nomenclature,Classification and Chemical Formulae of Polymers])」、テクニークス・ド・リンゲイヌール(Techniques de l’Ingenieur)、1995、A3035。
[6]ブルーズ(Brewis)およびダーム(Dahm)、「総説:ポリマーの電気化学的前処理(A review of electrochemical pretreatments of polymes)」、インターナショナル・ジャーナル・オブ・アドヘージョン・アンド・アドヘーシブズ(Intl.J.of Adhesion & Adhesives)、2001、21、397〜409。
[7]国際公開第2007/042659号パンフレット(出願人:Alchimer,公開日:2007年4月19日)。
[8]ゼンキエビッチ(Zenkiewicz)ら、「ポリマー複合材料の表面酸化に及ぼす電子ビーム照射の影響(Effects of electron−beam irradiation on surface oxidation of polymer composites)」、アプライド・サーフェス・サイエンス(Applied Surface Science)、2007、253(22)、8992〜8999。
[9]国際公開第2005/033378号パンフレット(出願人:CEA、公開日:2005年4月14日)。
[10]国際公開第2006/097611号パンフレット(出願人:CEA、公開日:2006年9月21日)。
【特許請求の範囲】
【請求項1】
(コ)ポリマーから作られている固体支持体の表面の一部の上に有機膜を調製するための方法であって、それが、
i)前記表面部分を酸化処理にかける工程、
ii)ラジカル化学的グラフト化によって前記表面部分に有機膜をグラフト化させる工程、
からなる連続的な工程を含むことを特徴とする、有機膜の調製方法。
【請求項2】
前記酸化処理が、化学的酸化処理であることを特徴とする、請求項1に記載の有機膜の調製方法。
【請求項3】
前記化学的酸化処理が、フェントン化学反応、アルコール性水酸化カリウムを用いた処理、強酸を用いた処理、水酸化ナトリウムを用いた処理、強酸化剤を用いた処理、オゾンを用いた処理、およびそれらの組合せからなる群より選択されることを特徴とする、請求項2に記載の有機膜の調製方法。
【請求項4】
前記強酸化剤が、塩酸、硫酸または硝酸中のKMnO4、K2Cr2O7、KClO3、CrO3、およびそれらの混合物からなる群より選択されることを特徴とする、請求項3に記載の有機膜の調製方法。
【請求項5】
前記酸化処理が、物理的酸化処理であることを特徴とする、請求項1〜4のいずれか1項に記載の有機膜の調製方法。
【請求項6】
前記物理的酸化処理が、火炎処理、コロナ作用による処理、プラズマ処理、UV照射線を用いた処理、X線またはγ線を用いた処理、電子を用いるかまたは重イオンを用いた照射処理、およびそれらの組合せからなる群より選択されることを特徴とする、請求項5に記載の有機膜の調製方法。
【請求項7】
前記固体支持体の前記表面が、マスクを備えていることを特徴とする、請求項1〜6のいずれか1項に記載の有機膜の調製方法。
【請求項8】
前記有機膜が、ラジカル経路によって重合可能な、いくつかの同一および/または異なったモノマー単位から主として得られた(コ)ポリマーであることを特徴とする、請求項1〜7のいずれか1項に記載の有機膜の調製方法。
【請求項9】
前記1種または複数のモノマーが、次の式(II)のモノマーから選択されることを特徴とする、請求項8に記載の有機膜の調製方法。
【化1】
[式中、R1〜R4基は、同一でも異なっていてもよく、1価の非金属原子たとえば、ハロゲン原子もしくは水素原子、または飽和もしくは不飽和の化学基たとえば、アルキルもしくはアリール基、ニトリル、カルボニル、アミン、アミド、または−COOR5基を表し、R5は、水素原子またはC1〜C12、好ましくはC1〜C6のアルキル基を表す。]
【請求項10】
請求項8または請求項9において定義された1種または複数のモノマーとは異なる接着プライマーから得られた分子実体が、前記固体支持体の前記表面に対して共有結合により結合され、そして前記有機膜に対してはまた別の共有結合によって結合されていることを特徴とする、請求項1〜9のいずれか1項に記載の有機膜の調製方法。
【請求項11】
前記接着プライマーが、アリールジアゾニウム塩、アリールアンモニウム塩、アリールホスホニウム塩、およびアリールスルホニウム塩からなる群より選択される開裂可能なアリール塩であることを特徴とする、請求項10に記載の有機膜の調製方法。
【請求項12】
前記工程(ii)が、以下の:
a)少なくとも1種の溶媒の存在下に、請求項8または請求項9において定義されたような少なくとも1種のモノマーを、前記モノマーとは異なる、請求項11において定義されたような少なくとも1種の接着プライマーを含む溶液に添加する工程、
b)工程(a)において得られた溶液を、前記接着プライマーからラジカル実体を生成させることを可能とする、非電気化学的条件下に置く工程、
c)前記固体支持体の表面を、工程(b)の溶液と接触させる工程、
からなる工程を含むことを特徴とする、請求項1〜11のいずれか1項に記載の有機膜の調製方法。
【請求項13】
前記工程(ii)が、以下の:
a’)前記固体支持体の表面を、少なくとも1種の請求項11において定義されたような接着プライマーを含む溶液を、少なくとも1種の溶媒、および、場合によっては、前記接着プライマーとは異なる請求項8または請求項9に定義されたような少なくとも1種のモノマーの存在下に、接触させる工程、
b’)前記固体支持体の表面を、前記接着プライマーからラジカル実体を生成させることを可能とする非電気化学的条件下に、工程(a’)の前記溶液と接触状態に置く工程、
c’)場合によっては、前記接着プライマーと異なる、少なくとも1種の請求項8または請求項9において定義されたようなモノマーを、工程(b’)において得られた前記溶液に添加する工程、
からなる工程を含むことを特徴とする、請求項1〜11のいずれか1項に記載の有機膜の調製方法。
【請求項14】
工程(b)または(b’)の際に、1種(または複数)の化学的重合開始剤、たとえば金属ウールまたは金属の削り屑を使用することを特徴とする、請求項12または請求項13に記載の有機膜の調製方法。
【請求項1】
(コ)ポリマーから作られている固体支持体の表面の一部の上に有機膜を調製するための方法であって、それが、
i)前記表面部分を酸化処理にかける工程、
ii)ラジカル化学的グラフト化によって前記表面部分に有機膜をグラフト化させる工程、
からなる連続的な工程を含むことを特徴とする、有機膜の調製方法。
【請求項2】
前記酸化処理が、化学的酸化処理であることを特徴とする、請求項1に記載の有機膜の調製方法。
【請求項3】
前記化学的酸化処理が、フェントン化学反応、アルコール性水酸化カリウムを用いた処理、強酸を用いた処理、水酸化ナトリウムを用いた処理、強酸化剤を用いた処理、オゾンを用いた処理、およびそれらの組合せからなる群より選択されることを特徴とする、請求項2に記載の有機膜の調製方法。
【請求項4】
前記強酸化剤が、塩酸、硫酸または硝酸中のKMnO4、K2Cr2O7、KClO3、CrO3、およびそれらの混合物からなる群より選択されることを特徴とする、請求項3に記載の有機膜の調製方法。
【請求項5】
前記酸化処理が、物理的酸化処理であることを特徴とする、請求項1〜4のいずれか1項に記載の有機膜の調製方法。
【請求項6】
前記物理的酸化処理が、火炎処理、コロナ作用による処理、プラズマ処理、UV照射線を用いた処理、X線またはγ線を用いた処理、電子を用いるかまたは重イオンを用いた照射処理、およびそれらの組合せからなる群より選択されることを特徴とする、請求項5に記載の有機膜の調製方法。
【請求項7】
前記固体支持体の前記表面が、マスクを備えていることを特徴とする、請求項1〜6のいずれか1項に記載の有機膜の調製方法。
【請求項8】
前記有機膜が、ラジカル経路によって重合可能な、いくつかの同一および/または異なったモノマー単位から主として得られた(コ)ポリマーであることを特徴とする、請求項1〜7のいずれか1項に記載の有機膜の調製方法。
【請求項9】
前記1種または複数のモノマーが、次の式(II)のモノマーから選択されることを特徴とする、請求項8に記載の有機膜の調製方法。
【化1】
[式中、R1〜R4基は、同一でも異なっていてもよく、1価の非金属原子たとえば、ハロゲン原子もしくは水素原子、または飽和もしくは不飽和の化学基たとえば、アルキルもしくはアリール基、ニトリル、カルボニル、アミン、アミド、または−COOR5基を表し、R5は、水素原子またはC1〜C12、好ましくはC1〜C6のアルキル基を表す。]
【請求項10】
請求項8または請求項9において定義された1種または複数のモノマーとは異なる接着プライマーから得られた分子実体が、前記固体支持体の前記表面に対して共有結合により結合され、そして前記有機膜に対してはまた別の共有結合によって結合されていることを特徴とする、請求項1〜9のいずれか1項に記載の有機膜の調製方法。
【請求項11】
前記接着プライマーが、アリールジアゾニウム塩、アリールアンモニウム塩、アリールホスホニウム塩、およびアリールスルホニウム塩からなる群より選択される開裂可能なアリール塩であることを特徴とする、請求項10に記載の有機膜の調製方法。
【請求項12】
前記工程(ii)が、以下の:
a)少なくとも1種の溶媒の存在下に、請求項8または請求項9において定義されたような少なくとも1種のモノマーを、前記モノマーとは異なる、請求項11において定義されたような少なくとも1種の接着プライマーを含む溶液に添加する工程、
b)工程(a)において得られた溶液を、前記接着プライマーからラジカル実体を生成させることを可能とする、非電気化学的条件下に置く工程、
c)前記固体支持体の表面を、工程(b)の溶液と接触させる工程、
からなる工程を含むことを特徴とする、請求項1〜11のいずれか1項に記載の有機膜の調製方法。
【請求項13】
前記工程(ii)が、以下の:
a’)前記固体支持体の表面を、少なくとも1種の請求項11において定義されたような接着プライマーを含む溶液を、少なくとも1種の溶媒、および、場合によっては、前記接着プライマーとは異なる請求項8または請求項9に定義されたような少なくとも1種のモノマーの存在下に、接触させる工程、
b’)前記固体支持体の表面を、前記接着プライマーからラジカル実体を生成させることを可能とする非電気化学的条件下に、工程(a’)の前記溶液と接触状態に置く工程、
c’)場合によっては、前記接着プライマーと異なる、少なくとも1種の請求項8または請求項9において定義されたようなモノマーを、工程(b’)において得られた前記溶液に添加する工程、
からなる工程を含むことを特徴とする、請求項1〜11のいずれか1項に記載の有機膜の調製方法。
【請求項14】
工程(b)または(b’)の際に、1種(または複数)の化学的重合開始剤、たとえば金属ウールまたは金属の削り屑を使用することを特徴とする、請求項12または請求項13に記載の有機膜の調製方法。
【図1】

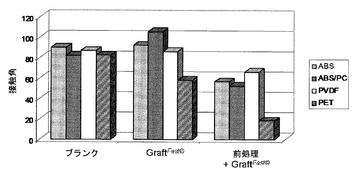
【図2】

【図3】
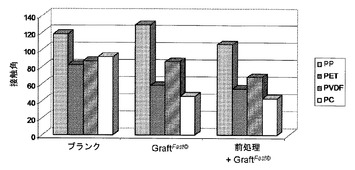
【図4】
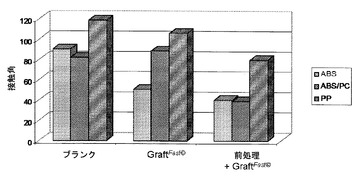
【図5】
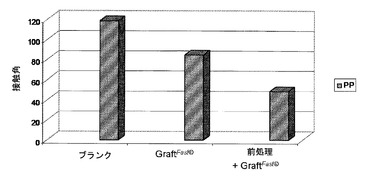
【図2】
【図3】
【図4】
【図5】
【公表番号】特表2012−525462(P2012−525462A)
【公表日】平成24年10月22日(2012.10.22)
【国際特許分類】
【出願番号】特願2012−507782(P2012−507782)
【出願日】平成22年4月30日(2010.4.30)
【国際出願番号】PCT/EP2010/055921
【国際公開番号】WO2010/125190
【国際公開日】平成22年11月4日(2010.11.4)
【出願人】(502124444)コミッサリア ア レネルジー アトミーク エ オ ゼネルジ ザルタナテイヴ (383)
【Fターム(参考)】
【公表日】平成24年10月22日(2012.10.22)
【国際特許分類】
【出願日】平成22年4月30日(2010.4.30)
【国際出願番号】PCT/EP2010/055921
【国際公開番号】WO2010/125190
【国際公開日】平成22年11月4日(2010.11.4)
【出願人】(502124444)コミッサリア ア レネルジー アトミーク エ オ ゼネルジ ザルタナテイヴ (383)
【Fターム(参考)】
[ Back to top ]