
酸化発色化合物の製造方法
【課題】生体成分測定のための酸化発色化合物の製造方法の提供。
【解決手段】下記化学式1:

(式中、R1、R2、R3、R4およびR5は、それぞれ独立して、水素原子またはスルホン酸基であり、この際R1、R2、R3、R4、およびR5の少なくとも一つがスルホン酸基である)で示される化合物またはその塩を、亜硝酸塩を用いてピラゾール環の4位をニトロソ化する工程と;該ニトロソ基を亜鉛を用いてアミノ基に還元する工程と;を含む、酸化発色化合物またはその塩の製造方法。
【解決手段】下記化学式1:
(式中、R1、R2、R3、R4およびR5は、それぞれ独立して、水素原子またはスルホン酸基であり、この際R1、R2、R3、R4、およびR5の少なくとも一つがスルホン酸基である)で示される化合物またはその塩を、亜硝酸塩を用いてピラゾール環の4位をニトロソ化する工程と;該ニトロソ基を亜鉛を用いてアミノ基に還元する工程と;を含む、酸化発色化合物またはその塩の製造方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、酸化発色化合物の製造方法ならびに当該方法によって製造された化合物を用いた試薬組成物および試験具に関する。
【背景技術】
【0002】
今日、血液や尿をはじめとする体液中の生体成分の測定法の一種として、酵素を用いた比色分析法が広く用いられている。これらの方法は、測定キット、自動分析機、ドライケミストリー試験具等に組み込まれ、日常的な臨床検査に数多く用いられている(例えば、特許文献1を参照)。
【0003】
このような測定法の一種として、被測定物質に特異的な酸化酵素を作用させて発生する過酸化水素をさらにペルオキシダーゼの作用で活性酸素とし、これで発色試薬を酸化して生成する色素を比色定量する方法がある。
【0004】
ここで使用される酵素としては、例えばグルコースの測定ではグルコースオキシダーゼ、コレステロールの測定ではコレステロールオキシダーゼ、尿酸の測定にはウリカーゼ、ピルビン酸の測定にはピルベートオキシダーゼ等が用いられる。これらの酵素は検体中の基質のみを特異的に酸化するために、色々な成分を含む検体からそれぞれの測定対象物のみを限定して定量することができる。
【0005】
また、発色試薬としては、活性酸素で酸化されることによって、その吸収波長特性や吸収強度等が変化する色素もしくは色素の前駆体(色原体)が用いられる。すなわち、活性酸素の量に応じて変化する色を測定することによって、被測定物質の量を定量することができる。これらの色素や色原体には、1分子だけで機能するものや、異なる2分子がカップリングして機能するものがある。
【0006】
1分子で機能するものには、ロイコ型色素等が挙げられる。具体的な化合物としては、ベンチジン、o−トリジン、o−ジアニシジン、2,2’−アミノ−ビス(3−エチルベンゾチアゾリノン−6−スルホン酸(ABTS)、ビス−(4−ジエチルアミノ)−2−スルホフェニルメタン(BSPM)、ビス[3−ビス(4−クロロフェニル)メチル−4−ジメチルアミノフェニル]アミン(BCMA)、10−N−メチルカルバモイル−3,7−ジメチルアミノ−10H−フェノチアジン(MCDP)、3,3’,5,5’−テトラメチルベンチジン(TMBZ)、ビス[4−(N−アルキル−N−スルホプロピル)−2,6−ジメチルフェニル]メタン(Bis−MAPS)、N,N,N’,N’,N’’,N’’−ヘキサ(3−スルホプロピル)−4,4’,4’’’−トリアミノトリフェニルメタン(TMP)などが挙げられる。
【0007】
2分子で機能する代表的なものには、カプラーとトリンダー試薬を酸化的にカップリングさせたものが挙げられる(例えば、非特許文献1および2を参照)。
【0008】
カプラーとしては、4−アミノアンチピリン(4−AA)、バニリンジアミンスルホン酸、メチルベンズチアゾリノンヒドラゾン(MBTH)、スルホン化メチルベンズチアゾリノンヒドラゾン(SMBTH)、アミノジフェニルアミンまたはその誘導体などが用いられる。
【0009】
トリンダー試薬としては、フェノール誘導体、アニリン誘導体が用いられる。フェノール誘導体としては、フェノール、4−クロロフェノール、2,4−ジクロロフェノール、2,6−ジクロロフェノール、3,5−ジクロロフェノール、2,4−ジブロモフェノール、2,6,4−トリクロロフェノール、2,6,4−トリブロモフェノール、3,5−ジクロロ−2−ヒドロキシベンゼンスルホン酸、3−ヒドロキシ−2,4,6−トリヨードベンゾイル酸、などが挙げられる。アニリン誘導体としては、N−エチル−N−(3−スルホプロピル)−3−メチルアニリン(TOPS)、N−エチル−N−(3−メチルフェニル)−3−アセチルエチレンジアミン(EMAE)、N−エチル−N−(3−メチルフェニル)−N−サクシニルエチレンジアミン(EMSE)、N−エチル−N−(2−ヒドロキシ−3−スルホプロピル)−3−メチルアニリン(TOOS)、N−(2−カルボキシエチル)−N−エチル−3−メチルアニリン(CEMB)、N,N−ビス−(4−スルホブチル)−3−メチルアニリン(TODB)、N−エチル−N−(2−サクシニルアミノエチル)−3−メチルアニリン(ESET)、N−エチル−N−(3−スルホプロピル)−3−メトキシアニリン(ADPS)、N−(2−ヒドロキシ−3−スルホプロピル)−3,5−ジメトキシアニリン(HSDA)、N−エチル−N−(2−ヒドロキシ−3−スルホプロピル)−3,5−ジメトキシアニリン(DAOS)、N−エチル−N−(2−ヒドロキシ−3−スルホプロピル)−3,5−ジメトキシ−4−フルオロアニリン(FDAOS)、N−エチル−N−(2−ヒドロキシ−3−スルホプロピル)−3,5−ジメチルアニリン(MAOS)などが挙げられる。
【0010】
ここで、保存安定性に鑑みると、1分子で機能する色素より2分子で機能する色素の方が良い傾向にある。また、2分子で機能する色素のうち、カプラーとしては、前記のうち最も安定な4−アミノアンチピリン、メチルベンズチアゾリノンヒドラゾンが多く用いられ、トリンダー試薬としては安定でかつ発色強度や波長の点でより有利なアニリン誘導体が多く用いられている。
【0011】
このような現状の下、前記の発色原理を用いた製品の価値を上げるためには、測定精度をさらに向上させる、あるいは経時的劣化を低減させる必要があり、また、より微量な成分の検出には、現存のものよりさらに感度のよい検出系が必要となり、それらの目的において、より高性能の発色試薬の開発が望まれている。
【0012】
このように、従来のカプラーに変わる新規な発色化合物の開発が進められており、例えば、特許文献2には、酵素を阻害しないとされるアミノアンチピリン誘導体が開示されている。
【0013】
ところで、このようなアミノアンチピリン誘導体のようなアミノ化合物を得るためには、ニトロソ化を行い、さらに還元を行なう方法が一般的に知られており、アミノアンチピリンの製造過程においては、ニトロソ化した後還元し、アミノ化をする手法が開示されている(例えば、特許文献3参照)。なお、特許文献2においても、ニトロソ化、アミノ化を行い、所望の発色化合物を得ている。
【特許文献1】特開2004−223115号公報
【特許文献2】特公平6−99403号公報
【特許文献3】米国特許第1,877,166号明細書
【非特許文献1】Trinder,P.,Ann.Clin.Biochem.,6,24,1969
【非特許文献2】Barham,D.and Trinder,P.,Analyst(London),97,142,1972
【発明の開示】
【発明が解決しようとする課題】
【0014】
上記の通り、より高性能の発色試薬の開発が望まれている一方で、実際に工業化を念頭においた場合、その高性能の発色試薬を、高純度かつ高収率で製造する必要性が生じる。
【0015】
しかしながら、前記したような技術においては、高純度かつ高収率で製造することができず、このような技術手段の開発が、現実問題として希求されている。
【0016】
よって、本発明は、上記実情を鑑みてなされたものであり、従来に比して性能が向上した発色化合物を、高純度かつ高収率で製造するための方法を提供することを目的とする。
【課題を解決するための手段】
【0017】
本発明者らは、上記実情に鑑み、鋭意研究を行った。その過程でまず、前記のような技術によっても、従来に比して性能が向上した発色化合物を、高純度かつ高収率で製造することができない理由を、以下の通り詳細に探索した。
【0018】
まず、所望の発色化合物を得るためには、ニトロソ化、アミノ化を経る必要があるが、高純度かつ高収率で所望の発色化合物を得ることができないのは、ニトロソ化により生じるニトロソ体が非常に不安定で、水溶液中では容易に分解して不純物を生じるのではないかと予測した。
【0019】
さらには、本発明が提供する酸化発色化合物は、従来のアミノアンチピリンと比較して極めて水溶性が高いことも、従来の方法によっては、高純度かつ高収率で目的物を得ることができない一因ではないかと予測した。
【0020】
その予測の下、鋭意研究を行った結果、本発明の製造方法によると、驚くべきことに、非常に高い純度および収率で、目的物を得ることができたのである。本発明は、このようにして完成した。
【0021】
すなわち、本発明は、下記(1)〜(11)で示される、高い性能を持つ新規酸化発色化合物の製造方法を提供する。
【0022】
(1)下記化学式1:
【0023】
【化1】
【0024】
ただし、R1、R2、R3、R4、およびR5は、それぞれ独立して、水素原子またはスルホン酸基であり、この際R1、R2、R3、R4、およびR5の少なくとも一つがスルホン酸基である、
で示される化合物またはその塩を、亜硝酸塩を用いてニトロソ化する、ニトロソ化工程と;
前記ニトロソ化工程の後、
下記化学式2:
【0025】
【化2】
【0026】
ただし、R1、R2、R3、R4およびR5は、化学式1の定義と同様である、
で示される化合物またはその塩を単離する、単離工程と;
前記単離工程により得られた化学式2で示される化合物またはその塩を、亜鉛を用いて還元する、還元工程と;
を含む、
下記化学式3:
【0027】
【化3】
【0028】
ただし、R1、R2、R3、R4およびR5は、化学式1の定義と同様である、
で示される酸化発色化合物またはその塩の製造方法。
【0029】
(2)前記ニトロソ化工程において、亜硝酸塩の量が、化学式1で示される化合物またはその塩に対して1.0〜1.5倍モルである、(1)に記載の製造方法。
【0030】
(3)前記ニトロソ化工程におけるニトロソ化反応を、化学式1で示される化合物またはその塩に対して1.0〜1.5倍モルの酸を用いて行う、(1)または(2)に記載の製造方法。
【0031】
(4)前記ニトロソ化工程において、酸またはその水溶液と、亜硝酸塩の水溶液と、を同時に化学式1で示される化合物またはその塩の水溶液に滴下して添加する、(3)に記載の製造方法。
【0032】
(5)前記還元工程において、亜鉛の量が、前記単離工程により得られた化学式2で示される化合物またはその塩に対して1〜30倍モルである、(1)〜(4)のいずれか1つに記載の製造方法。
【0033】
(6)前記還元工程における還元反応を、前記単離工程により得られた化学式2で示される化合物またはその塩に対して1〜30倍モルの酸をさらに用いて行う、(1)〜(5)のいずれか1つに記載の製造方法。
【0034】
(7)前記還元工程を、反応溶媒を用いて行い、該反応溶媒が、アルコールである、(1)〜(6)のいずれか1つに記載の製造方法。
【0035】
(8)前記還元工程を、反応溶媒を用いて行い、該反応溶媒の量が、前記単離工程により得られた化学式2で示される化合物またはその塩1gに対して20〜100mLである、(1)〜(7)のいずれか1つに記載の製造方法。
【0036】
(9)前記還元工程後、反応混合物中に残留する亜鉛を、水酸化亜鉛として除去する工程をさらに含む、(1)〜(8)のいずれか1つに記載の製造方法。
【0037】
(10)前記還元工程後、反応混合物からアルコールを用いて、化学式3で示される酸化発色化合物またはその塩を抽出精製する、抽出精製工程をさらに含む、(1)〜(9)のいずれか1つに記載の製造方法。
【0038】
(11)(1)〜(10)のいずれか1つに記載の製造方法で得られた、化学式3で示される酸化発色化合物またはその塩。
【発明の効果】
【0039】
本発明の製造方法においては、従来の方法と比較して、下記(ア)〜(ウ)で優れている。
【0040】
(ア)純度の向上
従来法では、50%程度であった純度が、本願発明の製造方法によると、95%以上に向上しうる。
【0041】
(イ)反応収率の向上
従来法では、30%程度であった収率が、本願発明の製造方法によると、80%以上の収率が得られうる。
【0042】
(ウ)吸湿性の改善
従来法で得られるアミノアンチピリン誘導体は、そのアミノ基が、酸(塩酸、酢酸等)との塩を形成しており、非常に吸湿性が高かった。本願発明の製造方法によると、スルホン酸基がアルカリ塩となっているため、吸湿性が有意に低い。さらに、本願発明により製造される発色化合物は、その呈色性能において、下記(エ)〜(キ)の点でこれまでの試薬より優れている。特に担体に保持した試験具として使用する場合、その効果が最も顕著である。
【0043】
(エ)測定時間の短縮
本願発明の製造方法により製造される発色化合物は、水溶性が有意に高いので、特に担体に保持して使用する場合には、血液などの検体にすばやく溶解して均一化し、迅速に呈色反応が起こる。
【0044】
(オ)感度が高い
呈色反応によって生成した色素化合物は、検体への溶解性や親和性が有意に高く、担体内での展開性が良くなるので、担体表面での発色が鮮やかで均一性の高いものとなる。それによって感度が高く安定した測定が可能となる。
【0045】
(カ)精度が高い
本願発明の製造方法により製造される発色化合物は、溶解性が有意に高いので、従来の試薬より高濃度の試薬液が調製可能である。また、担体に担持して使用する場合塗布液濃度を高くしてより多くの試薬を担体に保持させることが可能となる。さらには、塗布液の濃度を高く設定し全体液量を少なくできることで、塗工均一性の高い、グラビア印刷法、ドット印刷法、インクジェット印刷等の精密印刷法が使用できる。塗工液量が少なくなれば、乾燥時間が短縮され乾燥むらを低減させる効果もある。つまり、十分な試薬を高い均一性で塗工できることにより、塗りむらなどに起因するばらつきを低減させることができ、高い精度の試験具を提供することができる。
【0046】
(キ)経時安定性に優れる
本願発明の製造方法により製造された発色化合物は、親水性が有意に向上して、分子量が有意に大きいため、保管中の昇華による試薬の損失を押さえることができる。また、本願発明の製造方法により製造された発色化合物は、試薬濃度の変化における呈色強度への影響が少ないため、発色化合物が経時的に劣化減少した場合でも測定値への影響が少ない。さらには、本発明の製造方法により製造される発色化合物における親水性官能基は、担体への吸着性を高め、担体内での化合物の安定性を向上させる。つまり、経時劣化を低減させ、有効期間を延ばすことが可能となる。
【発明を実施するための最良の形態】
【0047】
以下に本発明をより具体的に説明する。
【0048】
<本発明の第一>
本発明の第一は、
下記化学式1:
【0049】
【化4】
【0050】
ただし、R1、R2、R3、R4、およびR5は、それぞれ独立して、水素原子またはスルホン酸基であり、この際R1、R2、R3、R4、およびR5の少なくとも一つがスルホン酸基である、
で示される化合物またはその塩を、亜硝酸塩を用いてニトロソ化する、ニトロソ化工程と;
前記ニトロソ化工程の後、
下記化学式2:
【0051】
【化5】
【0052】
ただし、R1、R2、R3、R4およびR5は、化学式1の定義と同様である、
で示される化合物またはその塩を単離する、単離工程と;
前記単離工程により得られた化学式2で示される化合物またはその塩を、亜鉛を用いて還元する、還元工程と;
を含む、
下記化学式3:
【0053】
【化6】
【0054】
ただし、R1、R2、R3、R4およびR5は、化学式1の定義と同様である、
で示される酸化発色化合物またはその塩の製造方法である。
【0055】
上記の通り、従来、所望の発色化合物を得ることができないのは、ニトロソ化により生じるニトロソ体が非常に不安定ではないかとの予測の下に鋭意研究を行った。
【0056】
その結果、中間体であるニトロソ体(化学式2で示される化合物またはその塩)は不安定であり、それが、所望の発色化合物の収率減少や純度低下の一因となっていた。
【0057】
さらに研究を進めていく過程で、本発明の製造方法により製造される発色化合物は、従来のアミノアンチピリンと比較して極めて水溶性が高いため、従来の方法によっては、高純度かつ高収率で所望の酸化発色化合物を得ることができないとの知見も見出した。
【0058】
よって、ニトロソ化反応、還元反応の条件および精製法等に、様々な技術的工夫を行うことにより、従来法に比較して、より効率的に高純度の発色化合物を合成する方法を見出し、本発明を完成させたのである。
【0059】
以下、本発明を詳説する。
【0060】
[ニトロソ化工程]
ニトロソ化を行う対象化合物は、
下記化学式1:
【0061】
【化7】
【0062】
ただし、R1、R2、R3、R4、およびR5は、それぞれ独立して、水素原子またはスルホン酸基であり、この際R1、R2、R3、R4、およびR5の少なくとも一つがスルホン酸基である、で示される化合物またはその塩である。
【0063】
R1、R2、R3、R4およびR5のうち1つのみが、スルホン酸基である場合、好ましい部位は、呈色性の観点で、R2、R3またはR4である。また、R1、R2、R3、R4およびR5のうちの2つがスルホン酸基である場合、好ましい部位は、呈色性の観点で、R2、R4である。
【0064】
なお、スルホン酸基は、イオン性官能基であるが、これらの官能基は、遊離の状態であっても、または塩を形成していてもよい、すなわち、R1、R2、R3、R4およびR5は、スルホン酸基の塩であってもよい。
【0065】
この際、これらの酸基と塩を形成する対イオンは、無機イオンでもよいし、有機イオンでもよい。例えば、ナトリウムイオン、カリウムイオン、カルシウムイオン等の無機塩基由来の陽イオン;アンモニウムイオン;アルキルアミン類等の有機塩基由来の陽イオン;およびアミノ酸類等の両性化合物由来のイオンなどが挙げられる。なかでも、塩の形態である場合には、好ましくはナトリウム塩、カリウム塩、またはアンモニウム塩であり、より好ましくはナトリウム塩である。
【0066】
なお、後述する化学式2〜化学式5における「R1、R2、R3、R4およびR5」は、いずれも、上記化学式1において行った「R1、R2、R3、R4およびR5」の定義と同様である。よって、下記ではその説明を割愛する。
【0067】
なお、ここで、化学式1で示される化合物またはその塩を得る方法について下記に説明する。
【0068】
本発明の化学式1で示される化合物またはその塩の製造方法は、特に制限されず、公知の製造方法が同様にしてあるいは適宜修飾してあるいは適宜組み合せて適用できる。以下、本発明の化学式1で示される化合物またはその塩の製造方法の好ましい実施形態を説明するが、本発明は下記好ましい実施形態に限定されるものではない。
【0069】
すなわち、本発明の化学式1で示される化合物またはその塩の製造方法は、下記化学式4:
【0070】
【化8】
【0071】
で示される化合物[以下、「化合物4」とも称する]と、下記化学式5:
【0072】
【化9】
【0073】
で示される化合物[以下、「化合物5」とも称する]とを、反応させる工程[工程(A)]を有することが好ましい。
【0074】
化合物4と化合物5とを反応させる上記工程(A)において、化合物4及び5の混合比は、特に制限されない。好ましくは、化合物4及び5をそれぞれ等モル混合する、あるいは化合物4を化合物5に対してやや多めに混合することが好ましい。後者の場合、化合物4は、化合物5を1モルに対して、0.8〜2モル、より好ましくは1〜1.2モル程度の割合で、混合することが好ましい。また、化合物4と化合物5との反応条件は、これらの反応が進行する条件であれば特に制限されないが、例えば、反応温度は、好ましくは20〜200℃、より好ましくは、80〜150℃の温度であり、反応時間は、好ましくは0.5〜5時間、より好ましくは1〜1.5時間である。また、化合物4と化合物5との反応は、無溶媒下で行なってもよく、あるいは溶媒中で行なってもよい。後者の場合に使用できる溶媒としては、メタノール、エタノール、プロパノール等の、アルコール類;THF、ジエチルエーテル、ジオキサン等の、エーテル類;ピリジン、ジメチルホルムアミド、ジメチルスルホキシド等の溶媒が挙げられる。これらのうち、THFが好ましい。化合物4と化合物5との反応後は、反応混合物を適当な溶媒中に加えることによって生成物を固化させることによって、原料化合物と分離回収することができる。
【0075】
次に、上記工程(A)で得られた反応生成物を、メチル化する[工程(B)]ことによって、前記化学式1で示される化合物が得られる。
【0076】
上記工程(B)において、メチル化は、ヨウ化メチルや硫酸ジメチルなどのメチル化剤を用いて行なうことができる。ここで、メチル化剤としては、上記のものに限定されず、他の公知のメチル化剤も同様にして使用できる。この反応は適当な溶媒や塩基を用いてもよく、室温あるいは加熱下で行なわれる。より具体的には、使用できる溶媒としては、上記工程(A)で列挙したのと同様の溶媒が使用でき、これらのうち、メタノール、THFが好ましい。また、塩基としては、トリエチルアミン(TEA)、ジイソプロピルエチルアミン(DIEA)、4−(ジメチルアミノ)ピリジンなどが使用でき、これらのうち、TEAが好ましい。
【0077】
上記工程(B)において、メチル化剤の使用量は、上記工程(A)で得られた反応生成物を十分メチル化できる量であれば特に制限されないが、上記工程(A)で得られた反応生成物1モルに対して、好ましくは0.5〜5モル、より好ましくは1〜2モルである。反応条件もまた特に制限されないが、上記工程(A)で得られた反応生成物のメチル化反応を、好ましくは40〜200℃、より好ましくは100〜140℃の温度で、好ましくは1〜24時間、より好ましくは10〜20時間、行なう。
【0078】
また、化学式1で示される化合物またはその塩を得る他の方法としては、上記工程(A)で得られた反応生成物を、以下の通りメチル化する[工程(C)]ことによって行う方法が挙げられる。
【0079】
すなわち、上記工程(A)で得られた反応生成物と、硫酸ジメチルと、を混合し、無溶媒下で加熱反応させる工程を含む、下記化学式1で示される化合物またはその塩:
【0080】
【化10】
【0081】
で示される化合物またはその塩の製造方法である。
【0082】
工程(C)の製造方法のように、溶媒を用いずに(無溶媒で)合成できるメリットとしては、反応容量を少量化でき、反応容器容積あたりの仕込み可能量が大きいことがあげられる。すなわち、通常の反応より小型の反応装置でより多くの製造ができるので、大量合成に好都合である。また、塩基を用いずに合成することもできる。
【0083】
硫酸ジメチルは、市販のものを使用しても合成して使用してもよく、特に制限されることはない。
【0084】
上記工程(A)で得られた反応生成物に対する前記硫酸ジメチルの量は、1.0〜2.0倍モルであることが好ましく、1.0〜1.5倍モルがより好ましく、1.0〜1.3倍モルが特に好ましい。上記工程(A)で得られた反応生成物に対する硫酸ジメチルの添加量が、1.0倍モル未満だと、反応効率低下の虞れがあり、2.0倍モルを越えると副生成物の増大の虞れがある。
【0085】
本発明に係る硫酸ジメチルの添加の方法としては、滴下ロート、ピペットなどを用いることができるが、外気中の水分による硫酸ジメチルの加水分解を防ぐために滴下ロートを用いて反応系を密閉して行なうのが好ましい。また、室温、1atmにおいて上記工程(A)で得られた反応生成物と、硫酸ジメチルとを混合する際には、液体である硫酸ジメチルに完全に溶解させてもまたは溶解させなくてもよく、両者がどのような状態であろうが混合していればよい。
【0086】
工程(C)に用いられる反応容器としては、ガラス、ステンレス、ホーロー、テフロン(登録商標)製のフラスコが好ましく、摺合せのガラス製フラスコが特に好ましい。
【0087】
工程(C)に用いられる反応容器としては、なす型フラスコ、丸底フラスコ、平底フラスコ、三角フラスコ、セパラブルフラスコ、多口フラスコを用いることができるが、これらは、仕込み量や加熱効率を考慮して選択される。
【0088】
工程(C)に用いられる冷却器としては、通常の合成反応に用いられる還流冷却器を用いることができ、リービッヒ冷却器、玉入冷却器(アーリン氏タイプ)、ジムロート冷却器、蛇管冷却器(グラハムタイプ)が好ましく、リービッヒ冷却器、玉入冷却器が特に好ましい。
【0089】
また、工程(C)における加熱反応中、外気からの水分の混入を防ぐ為に、塩化カルシウム管、窒素バブラーを取り付けることが好ましい。
【0090】
工程(C)における製造方法における加熱反応の加熱方法としては、マントルヒーター、油浴、砂浴(バーナー直火)が好ましく、マントルヒーターが特に好ましい。必要に応じて、センサー付き温度コントローラーを用いることができる。
【0091】
工程(C)における加熱反応の温度としては、190〜260℃が好ましく、200〜250℃が特に好ましく、220〜240℃がさらに好ましい。
【0092】
上記加熱反応の温度が190℃未満だと、反応効率低下の虞れがあり、260℃超だと、副生成物の増大の虞れがある。
【0093】
工程(C)における加熱反応の反応時間としては、0.5〜5時間が好ましく、0.5〜3時間が特に好ましく、1〜2時間がさらに好ましい。
【0094】
上記加熱反応の反応時間が0.5時間未満だと、反応効率低下の虞れがあり、5時間超だと、副生成物の増大の虞れがある。
【0095】
工程(C)において、上記工程(A)で得られた反応生成物と、硫酸ジメチルとを混合し、無溶媒下で加熱反応させて得られた反応混合物をアルカリ水溶液に溶解させて反応混合溶液を得る工程と、前記反応混合溶液に酸を加えて固体を単離する工程とを含むことが好ましい。
【0096】
本発明に係る化学式(1)で示される化合物は、溶媒抽出による回収が困難であるため酸を添加することで強制的に沈殿するいわゆる“酸沈”により回収することが好ましい。これにより、最終目的物を高い純度で得られることができる。
これにより、化学式1で示される化合物またはその塩を高い純度で得られることができる。
【0097】
本発明に係る製造方法に用いられるアルカリ水溶液のアルカリとしては、水酸化ナトリウム、水酸化カリウム、水酸化リチウムが好ましく、水酸化ナトリウムが特に好ましい。
【0098】
本発明に係る製造方法に用いられるアルカリ水溶液の濃度としては、10〜30w/v%(重量対体積%)が好ましく、15〜25w/v%が特に好ましく、20〜25w/v%がさらに好ましい。
【0099】
また、上記反応混合物をアルカリ水溶液に溶解させて得られる反応混合溶液のpHは、7〜14が好ましい。
【0100】
本発明に係る製造方法に用いられるアルカリ水溶液中のアルカリの量としては、使用する硫酸ジメチルに対して2〜5倍モルが好ましく、2.5〜4倍モルが特に好ましく、3倍モルがさらに好ましい。
【0101】
本発明に係る製造方法に用いられる酸としては、有機酸または無機酸のいずれでもよく、塩酸、硫酸、硝酸、酢酸、クエン酸、コハク酸、ピクリン酸、濃硫酸、濃硝酸などが用いられるが、塩酸、硫酸、硝酸がより好ましく、塩酸がさらに好ましい。
【0102】
本発明に係る製造方法において、上記反応物に添加する酸の濃度としては、なるべく水分量を減らすべく濃度が高いものが好ましい。
【0103】
上記反応物混合溶液に添加する酸の濃度は、5〜100w/v%(重量/体積%)が好ましく、15〜100w/v%がより好ましく、30〜100w/v%が特に好ましい。例えば、添加する酸として塩酸を用いる場合は、15〜36w/v%が好ましく、30〜36w/v%が特に好ましい。
【0104】
上記反応物混合溶液に添加する酸の量は、1〜10倍モルが好ましく、1〜5倍モルがより好ましく、1.5〜3倍モルが特に好ましい。
【0105】
上記反応物に添加する塩酸の量としては、出発物質の使用量に対して、1〜5倍モルが好ましく、1.5〜3倍モルが特に好ましく、1.8〜2.2倍モルがさらに好ましい。
【0106】
また、本発明に係る製造方法において酸を加え沈殿を生成させる工程で用いる攪拌装置としては、磁気スターラー、メカニカルスターラーなどを用いることができる。
【0107】
本発明に係る製造方法において、前記単離工程で得られた固体を水から再結晶する工程をさらに含むことが好ましい。すなわち、本発明の製造方法は、上記工程(A)で得られた反応生成物と、硫酸ジメチルを混合し、無溶媒下で加熱反応させて得られた反応混合物を得る工程と、アルカリ水溶液に溶解させて反応混合溶液を得た後、さらに前記反応混合溶液に酸を加えて固体(上記化学式1に示される化合物を含むもの)を単離する工程と、単離された前記固体を水から再結晶する工程とを含むことが好ましい。
【0108】
本発明に係る製造方法における再結晶で用いる水の量としては、固体100g当り、500ml〜3000mlが好ましく、1000ml〜2000mlが特に好ましく、1000ml〜1500mlがさらに好ましい。なお、当該単離された固体は、0.5〜24時間、室温条件下で減圧乾燥したものが使用される。
【0109】
上記再結晶工程で固体を水に溶解させる際に、加熱を行なってよい。加熱器具としては、水浴、油浴、マントルヒーター、ホットプレートなどが用いられる。加熱する際の加熱器具の温度設定は、50〜150℃が好ましく、70〜130℃が特に好ましく、90〜110℃がさらに好ましい。
【0110】
また上記再結晶のための放置時間は、30分〜3日が好ましく、1時間〜1日が特に好ましく、10時間〜15時間がさらに好ましい。さらに、上記再結晶のための放置時の温度は、0〜40℃が好ましく、0〜25℃が特に好ましく、0〜15℃がさらに好ましい。
【0111】
本発明に係る製造方法において使用される水は、特に制限されるものではなく、水道水、イオン交換水、純水、超純水、または工業用水などの水洗水を使用することができ、純水が特に好ましい。
【0112】
上記により得られた、前記化学式1で示される化合物またはその塩の純度の測定方法は、後述する。
【0113】
(亜硝酸塩)
前記化学式1で示される化合物またはその塩を、亜硝酸塩を用いてニトロソ化する。
【0114】
ニトロソ化工程に用いる亜硝酸塩は、特に制限はないが、亜硝酸のカチオン塩が用いられると好ましい。
【0115】
カチオン塩としても、特に制限はなく、アルカリ金属塩、アルカリ土類金属塩、アンモニウム塩などが好適に挙げられる。具体的には、ナトリウム塩、カリウム塩、リチウム塩等が好ましい。これらは、単独で用いてもよく、混合して用いてもよい。なお、入手のし易さの観点で、ナトリウム塩が特に好ましい。
【0116】
ニトロソ化工程に用いる亜硝酸塩の量は、化学式1で示される化合物またはその塩(本明細書中、単に「出発物質」とも称する。)の量に対して、反応効率および副生成物の観点で、1.0〜2.0倍モルが好ましく、1.0〜1.5倍モルがより好ましく、1.0〜1.2倍モルがさらに好ましい。
【0117】
(酸)
前記ニトロソ化工程におけるニトロソ化反応は、反応性の観点で、酸の存在下において行うとよい。
【0118】
ニトロソ化工程に用いられる酸は、特に制限はないが、反応性の観点で、塩酸、硫酸、硝酸、濃塩酸、濃硫酸、濃硝酸、酢酸、クエン酸、コハク酸、ピクリン酸が好ましい。これらは、単独で用いてもよく、混合して用いてもよい。なお、取り扱いの観点で、塩酸が特に好ましい。
【0119】
ニトロソ化工程に用いられうる酸の量にも、特に制限はないが、出発物質の量に対して、反応効率および副生成物の生成の観点で、0.5〜2.0倍モルが好ましく、1.0〜1.5倍モルがより好ましく、1.0〜1.2倍モルがさらに好ましい。
【0120】
また、添加する酸は、副生成物の生成を抑える観点で、好ましくは0〜15℃、より好ましくは1〜10℃、さらに好ましくは1〜5℃にしておくとよい。なお、添加する酸を、後述する溶媒で希釈した場合も同様の温度に制御しておくとよい。
【0121】
(溶媒)
ニトロソ化工程において、出発物質と上記亜硝酸塩、酸を混合する場合、それぞれを予め溶媒に含有させておいてもよい。
【0122】
つまり、溶媒に、それぞれの原末(出発物質と亜硝酸塩)または原液(酸)を加えていく方法や、出発物質を溶媒に含有させた溶液(本明細書中、「出発物質の溶液」とも称する)に、亜硝酸塩、酸を溶媒に含有させた溶液(それぞれ、「亜硝酸塩の溶液」、「酸の溶液」とも称する。)を添加する方法を用いてもよい。
【0123】
好ましくは、反応効率の観点で、出発物質の溶液に、亜硝酸塩の溶液および酸の溶液を添加する方法である。つまり、酸の溶液と、亜硝酸塩の溶液とを、化学式1で示される化合物またはその塩の溶液に滴下して添加する。
【0124】
この際、添加する亜硝酸塩の溶液は、副生成物の生成を抑える観点で、好ましくは0〜15℃、より好ましくは1〜10℃、さらに好ましくは1〜5℃にしておくとよい。また、添加する亜硝酸塩の溶液の濃度は、反応性の観点で、好ましくは1〜10mol/l、より好ましくは1〜5mol/l、さらに好ましくは3〜5mol/lにしておくとよい。
【0125】
また、添加する酸の溶液の濃度は、反応性の観点で、好ましくは1〜10mol/l、より好ましくは1〜5mol/l、さらに好ましくは3〜5mol/lにしておくとよい。
【0126】
また、出発物質の溶液の濃度は、反応性の観点で、好ましくは0.1〜10mol/l、より好ましくは0.1〜5mol/l、さらに好ましくは0.5〜2mol/lである。
【0127】
また、予め出発物質の溶液を、副生成物を抑える観点で、好ましくは0〜15℃、より好ましくは0〜5℃、さらに好ましくは0〜3℃にしておくとよい。
【0128】
前記溶媒は、特に制限はないが、水、有機溶媒またはこれら混合物(混合溶媒)が用いられうる。なお、本明細書中、溶媒として「水」を主成分(好ましくは、全部)とした溶液の名称を「水溶液」とも称する。なお、本明細書中、「水を主成分とする」とは、全溶液中に水の占める割合が、10〜100v/v%であることを意味する。
【0129】
有機溶媒としては、特に制限はないが、アルコール、エーテル類、ケトン類、ジメチルホルムアミド、ジメチルスルホキシドなどが用いられる。
【0130】
アルコールとしては、メタノール、エタノール、n−プロパノール、イソプロパノールなどが好適に用いられうる。これらは、単独で用いてもよく、混合して用いてもよい。
【0131】
エーテル類としては、ジエチルエーテル、イソプロピルエーテル、テトラヒドロフラン(THF)、ジオキサン、ジフェニルエーテル、ベンジルエーテル及びtert−ブチルエーテルなどが好適に用いられうる。これらは、単独で用いてもよく、混合して用いてもよい。
【0132】
ケトン類としては、アセトン、メチルイソブチルケトン(MIBK)、メチルエチルケトン(MEK)及びシクロヘキサノンなどが好適に用いられうる。これらは、単独で用いてもよく、混合して用いてもよい。
【0133】
これらのうち、反応性の観点で、水単独または水とアルコールとの混合物が好ましく、水単独が特に好ましい。
【0134】
水および有機溶媒の混合溶媒を用いる際は、例えば、好ましくは水/有機溶媒(容量比)=0/100〜100/0、より好ましくは水/有機溶媒(容量比)=10/90〜100/0程度がよい。この際、有機溶媒の含有量が、混合溶媒中、50容量%を超えると、亜硝酸塩の析出の虞れもある。
【0135】
また、上記の通り、出発物質と、亜硝酸塩と酸とを混合させる方法にも特に制限はないが、反応性の観点で、亜硝酸塩の溶液と、酸の溶液とを、攪拌下で、出発物質の溶液に添加する方法が好ましい。さらには、添加する条件は、副生成物を抑える観点で、亜硝酸塩の溶液と酸の溶液を、ほぼ同じ速度で、同時に添加することが好ましい。
【0136】
ここで、「同時に添加する」とは、亜硝酸塩の溶液と、酸の溶液とを添加しているある時点が、一瞬でも互いに重複すればよいが、亜硝酸塩の溶液と、酸の溶液との添加を開始する時点と、亜硝酸塩の溶液と、酸の溶液との添加を終了する時点とが、実質的に同じであると好ましい。
【0137】
このように、「同じ速度で添加する」および「同時に添加する」ことで、亜硝酸を一定で連続的に発生させる効果を奏しうる。
【0138】
なお、この際の、亜硝酸塩の溶液と、酸の溶液の添加の方法にも特に制限はないが、ピペット、シリンジ、滴下ロートなどを用いて行なわれることが好ましい。また、これらの添加に要する時間は、グラムスケールの反応では、生成物の安定性の観点で、1〜10分が好ましく、3〜7分が特に好ましく、4〜6分がさらに好ましい。
【0139】
また、添加は、磁気攪拌子等で、攪拌しながら行うことが、好ましい。
【0140】
さらに、亜硝酸塩の溶液と、酸の溶液と、を添加した後、そのまま静置しておいてもよいが、反応が添加後、時間とともに進行するため、好ましくは、上述の磁気攪拌子等で、好ましくは1〜10分程度、より好ましくは3〜5分程度、激しく攪拌するとよい。この際、「激しく攪拌する」の攪拌速度にも特に制限はないが、好ましくは300〜1200rpm、より好ましくは600〜1000rpmである。
【0141】
(反応時間)
ニトロソ化工程における反応時間にも特に制限はないが、あまりに反応時間が短すぎると、反応が不十分で出発物質の残留が生じる虞れがある。一方で、あまりに反応時間が長過ぎると、生成したニトロソ体の分解が起こる虞れがある。
【0142】
反応時間は、亜硝酸塩の溶液と酸の溶液を添加する時間を含めて、グラムスケールの反応では、1〜20分が好ましく、5〜15分がより好ましく、7〜12分がさらに好ましい。
【0143】
(反応温度)
ニトロソ化工程において設定される反応温度としては、副生成物の生成を抑える観点で、0〜15℃が好ましく、1〜10℃がより好ましく、1〜5℃がさらに好ましい。かような温度条件にするためには、従来公知の知見を適宜参照して制御することができ、例えば、水浴や氷浴等を用いればよい。
【0144】
(反応容器)
ニトロソ化工程に用いられる反応容器の素材としても、特に制限はないが、ガラス製、ステンレス製、ホーロー製、テフロン(登録商標)製などが好適に例示でき、ガラス製が特に好ましい。また、反応容器の形状としても、特に制限はないが、なす型フラスコ、丸底フラスコ、平底フラスコ、三角フラスコ、セパラブルフラスコ、多口フラスコなどが挙げられる。
【0145】
上記ニトロソ化工程を経ることにより、
下記化学式2:
【0146】
【化11】
【0147】
ただし、R1、R2、R3、R4およびR5は、化学式1の定義と同様である、で示される化合物またはその塩を得ることができる。
【0148】
[単離工程]
本発明においては、上記ニトロソ化工程の後、化学式2で示される化合物またはその塩を単離する、単離工程を経る。つまり、本発明においては、ニトロソ化工程終了後、そのままワンポットで、一気に、後述の還元工程を行わず、単離工程を必須の工程として有する点に特徴の一つを有する。
【0149】
上記の通り、従来、所望の発色化合物を、高純度かつ高収率で得ることができないのは、ニトロソ化により生じるニトロソ体が非常に不安定ではないかとの予測の下に鋭意研究を行った。そして、その予測は正しく、中間体であるニトロソ体(化学式2で示される化合物またはその塩)は不安定であり、それが、収率減少や目的物の純度低下の一因となっていることを見出したのである。
【0150】
つまりは、中間体であるニトロソ体(化学式2で示される化合物またはその塩)を適切な精製を行わなければ、結局は、最終生成物たる化学式3で示される酸化発色化合物またはその塩の収率減少や純度低下という結果に直結するのである。
【0151】
一方で、ニトロソ化工程終了後、ワンポット(一気に)で、後述の還元工程に移行する条件の下で、化学式3で示される酸化発色化合物またはその塩の製造を試みると、中間体であるニトロソ体(化学式2で示される化合物またはその塩)の単離をすることが非常に困難なのである。つまりは、中間体であるニトロソ体(化学式2で示される化合物またはその塩)中に存在する副生成物の分離除去を行うことが困難なのである。
【0152】
その観点からして、上記ニトロソ化工程においては、中間体であるニトロソ体(化学式2で示される化合物またはその塩)の単離を容易に行わせしめるべく、最適なニトロソ化反応条件を詳細に検討する点も、非常に有用であると言えるのである。具体的には、本発明の製造方法におけるニトロソ化工程によれば、化学式2で示される化合物またはその塩のみが、沈殿分離するため、容易に単離精製することができる。また、上記にて、亜硝酸塩、酸を、それぞれ化学式1で示させる化合物またはその塩に対して、ほぼ等量で添加するのも、副生成物の生成をより抑え、ひいては化学式2で示される化合物またはその塩を容易に単離精製させるために他ならない。
【0153】
上記の通り、ニトロソ体(化学式2で示される化合物またはその塩)を固体として単離することによって、不純物が除去され、次の還元工程が効率的に進行するため、最終的に得られるアミノ体(化学式3で示される酸化発色化合物またはその塩)の純度が高くなり、また還元工程における収率も向上するのである。
【0154】
以下、単離工程における、ニトロソ体を単離する方法について詳説する。
【0155】
ニトロソ体(化学式2で示される化合物またはその塩)を単離する具体的方法としては、特に制限はないが、ニトロソ化工程終了後に得られた反応混合物を、濾過したり、遠心分離したりすることなどによって行なわれるとよい。なお、かようにして得られたものを、「ニトロソ化物固体」とも称する。
【0156】
この際の濾過方法や条件としては、特に制限ないが、ろ紙を用いた吸引ろ過によって行うことができる。
【0157】
なお、得られたニトロソ化物固体は、溶媒で洗浄することにより、さらに純度が高くなる。
【0158】
この洗浄に用いられうる溶媒は、特に制限はないが、有機溶媒、またはこれらの混合物が用いられる。これらのうち、ニトロソ体の安定性の観点で、有機溶媒は、アルコールが好ましい。アルコールとしても特に制限はなく、上記にて列挙した具体例が同様に妥当であるが、ニトロソ体の安定性・純度の観点で、メタノールが特に好ましい。
【0159】
この際、洗浄に用いるアルコールは、ニトロソ体の安定性の観点から、好ましくは−20℃〜10℃程度、より好ましくは0〜5℃程度に冷却しておくと良い。
【0160】
このように得られたニトロソ体(化学式2で示される化合物またはその塩)の固体は、減圧乾燥などによって乾燥することによって、さらに、保存安定性が向上する。この際の減圧乾燥方法や条件としても、特に制限ないが、真空ライン(好ましくは0〜10mmHg、より好ましくは1〜2mmHg)に接続して行うことができる。
【0161】
かようにして、化学式2で示される化合物またはその塩を単離することができる。
【0162】
[還元工程]
本発明における還元工程では、前記単離工程により得られた化学式2で示される化合物またはその塩を、亜鉛を用いて還元する。
【0163】
かような還元工程を経ることにより、
下記化学式3:
【0164】
【化12】
【0165】
ただし、R1、R2、R3、R4およびR5は、化学式1の定義と同様である、で示される酸化発色化合物またはその塩を製造することができる。
【0166】
上記の通り、ニトロソ化工程終了後、ワンポット(一気に)で、後述の還元工程に移行する条件下で、化学式3で示される酸化発色化合物またはその塩の製造を試みると、生成する無機塩(亜鉛等)の除去が困難である。
【0167】
本発明の製造方法によると、無機塩(亜鉛等)を水酸化物として分離除去できるため、精製が非常に容易であり、最終生成物たる化学式3で示される酸化発色化合物またはその塩の収率が有意に向上する。
【0168】
(亜鉛)
還元工程において、亜鉛を必須成分として用いる。ただし、亜鉛以外でも、本発明の効果を奏する範囲であれば、他の還元剤を用いてもよい。他の還元剤としては、ハイドロサルファイド塩、サルファイド塩などが挙げられる。
【0169】
還元工程において、亜鉛の量は、前記単離工程により得られた化学式2で示される化合物またはその塩に対して1〜30倍モルが好ましく、5〜10倍モルがより好ましく、7〜8倍モルがさらに好ましい。
【0170】
(酸)
還元工程における還元反応は、酸の存在下において行うとよい。
【0171】
還元工程に用いられる酸は、特に制限はなく、どのような種の酸でも用いうるが、反応性の観点で、塩酸、硫酸、硝酸、酢酸が好ましい。これらは、単独で用いてもよく、混合して用いてもよい。なお、取り扱いやすさの観点で、塩酸が好ましい。
【0172】
還元工程に用いられうる酸の量にも、特に制限はないが、前記単離工程により得られた化学式2で示される化合物またはその塩の量に対して、副生成物を抑える観点で、1〜30倍モルが好ましく、5〜15倍モルが特に好ましく、7〜9倍モルがさらに好ましい。
【0173】
(溶媒)
化学式3で示される酸化発色化合物またはその塩の製造のために、前記単離工程により得られた化学式2で示される化合物またはその塩と、亜鉛と、を用い、上記の通り、還元反応は、酸の存在下で行うとよいが、必要に応じ反応溶媒を用いるとよい。
【0174】
反応溶媒としては、特に制限はなく、上記ニトロソ化工程で説明した具体例が、同様に妥当である。
【0175】
これら具体例のうち、化学式2で示される化合物の安定性の観点で、アルコール単独が好ましく、中でもメタノール単独が特に好ましい。
【0176】
還元工程における溶媒の量は、前記単離工程により得られた化学式2で示される化合物またはその塩1gに対して、反応性の観点で、20〜100mLが好ましく、30〜70mLがより好ましく、30〜50mLがさらに好ましい。
【0177】
また、前記単離工程により得られた化学式2で示される化合物またはその塩と、亜鉛と酸とを混合させる方法にも特に制限はないが、反応性の観点で、亜鉛粉末と、酸の溶液とを、攪拌下で、単離工程により得られた化学式2で示される化合物またはその塩を溶媒に含有した溶液(本明細書中、「単離工程により得られた化学式2で示される化合物またはその塩の溶液」とも称する)に添加する方法が好ましい。
【0178】
より具体的には、単離工程により得られた化学式2で示される化合物またはその塩を、予め溶媒(この溶媒は、上記の通り、メタノールが特に好ましい。また、この際、この溶媒を、安定性の観点で、好ましくは−20〜10℃、より好ましくは−10〜5℃に冷却しておくとよい。)に含有させておき、攪拌しながら、酸を添加し、さらに添加すべき亜鉛を一度に添加することが好ましい。「一度に添加する」理由は、ニトロソ体が不安定なため、早く反応を終了させるためである。この際、酸と亜鉛の添加順序にも特に制限はないが、ニトロソ体の安定性の観点で、好ましくは、亜鉛を先に添加する。
【0179】
また、添加は、磁気攪拌子等で、攪拌しながら行うことが、好ましい。
【0180】
また、亜鉛、酸の溶液を添加した後、副生成物を抑える観点で、好ましくは、上述の磁気攪拌子等で、好ましくは5〜20分程度、より好ましくは5〜10分程度、攪拌するとよい。また、攪拌速度にも特に制限はないが、好ましくは100〜1200rpm、より好ましくは300〜700rpmである。
【0181】
(反応温度)
還元工程において設定される反応温度としては、−20〜35℃が好ましく、−15〜25℃が特に好ましい。かような温度条件にするためには、従来公知の知見を適宜参照して制御することができ、例えば、水浴や氷浴、ドライアイス−メタノール浴等を用いればよい。
【0182】
(反応容器)
還元工程に用いられる反応容器の素材としても、特に制限はなく、前記ニトロソ化工程で説明した具体例が同様に妥当である。
【0183】
また、反応容器の形状としても、特に制限はなく、前記ニトロソ化工程で説明した具体例が同様に妥当である。
【0184】
かような還元工程を経ることにより、前記化学式3で示される酸化発色化合物またはその塩を製造することができる。
【0185】
[亜鉛除去工程]
上記還元工程終了後、亜鉛を除去することが好ましい。
【0186】
上記還元工程終了後、亜鉛は、固体状および/または水溶性の亜鉛の形態で残留しうる。
【0187】
固体状で残留している亜鉛は、濾過によって除去することが好ましく、さらに反応溶液中に残留する水溶性の亜鉛の除去を行なうとよい(亜鉛除去工程)。かような亜鉛除去工程を経ることにより、より高純度かつ高収率で、化学式3で示される酸化発色化合物またはその塩を得ることができる。
【0188】
このような水溶性の亜鉛を除去する方法も特に制限されないが、例えば、以下の方法が挙げられる。
【0189】
すなわち、還元工程終了後の反応混合物を、濾過し、固体状で残留している亜鉛を除去した後、水を添加して洗浄を行う。その後、濾液と洗液をあわせ、濃縮する。そして、もとの化学式2で示される化合物の重量に対し、好ましくは5〜1000質量部、より好ましくは10〜100質量部、さらに好ましくは10〜50質量部となる水を添加して、濃縮物の水溶液を準備する。そして、該濃縮物の水溶液に、アルカリ性物質を添加することにより、水酸化亜鉛の形で残留亜鉛を析出させて、濾過によって除去することができる。
【0190】
かようにすることで、亜鉛除去工程を経た反応混合物を得ることができる。
【0191】
(アルカリ性物質)
水酸化亜鉛を生成させるアルカリ性物質としては、特に制限はないが、水酸化塩が好ましく、より具体的には、水酸化ナトリウム、水酸化カリウム、水酸化リチウムなどが挙げられる。これらは、単独で用いてもよく、混合して用いてもよい。なお、中でも水酸化ナトリウムが特に好ましい。
【0192】
水酸化亜鉛を生成させるためのアルカリ性物質は、水溶液の形態(アルカリ性物質水溶液)で添加することができる。添加するアルカリ性物質水溶液の濃度は、残留する亜鉛濃度の観点から、1〜10規定濃度が好ましく、3〜5規定濃度がさらに好ましい。
【0193】
なお、添加するアルカリ性物質の量は、残留亜鉛の除去と、化学式3で示される化合物の安定性の観点から、アルカリ性物質を添加した後、pHが、8.0〜10.0になるよう設定するのが好ましく、8.5〜9.5になるよう設定するのが特に好ましい。
【0194】
[抽出精製工程]
上記亜鉛除去工程を経た後、亜鉛除去工程を経て得られた反応混合物から、さらに残留する無機塩を除くため、抽出精製工程を経ることが好ましい。かような抽出精製工程を経ることにより、より高純度かつ高収率で、化学式3で示される酸化発色化合物またはその塩を得ることができる。
【0195】
抽出精製工程は、亜鉛除去工程を経て得られた反応混合物を固体状態とし、溶媒で抽出する方法が用いられうる。抽出精製工程における抽出方法は、反応混合物を粉末状態として溶媒を加えて攪拌する他、一般に用いられる抽出装置を使用して行なってもよい。
【0196】
亜鉛除去工程を経て得られた反応混合物を固体状態にする方法にも特に制限はないが、例えば、減圧乾燥方法等により蒸発乾固する方法が挙げられる。
【0197】
この際の減圧乾燥方法や条件としても、特に制限ないが、真空ライン(好ましくは0〜30mmHg、より好ましくは1〜2mmHg)に接続して行うことができる。
【0198】
抽出精製工程における溶媒としては、最終目的物(化学式3で示される酸化発色化合物またはその塩)を溶解できるものであれば特に制限されないが、アルコールが好ましく、より具体的には、メタノール、エタノール、n−プロパノール、イソプロパノールなどが好適に使用できる。これらは、単独で用いてもよく、混合して用いてもよい。なお、抽出効率の観点で、エタノールが特に好ましい。
【0199】
また、本発明の製造方法により得られる化学式3で示される酸化発色化合物またはその塩は、両性でありうるため、単離をすること自体も困難である。しかしながら、上記の通り、ニトロソ化物の単離(不純物除去)、残留亜鉛の除去、エタノールによる無機塩(NaCl)の分離などの工夫を行うため、容易に単離を行うことができる。
【0200】
本発明の製造方法によると、従来法では50%程度であった純度が、少なくとも80%以上、実際には95%以上に向上する。
【0201】
この点においても、本発明は非常に優れていると言える。
【0202】
なお、目的物の純度は以下の方法で調べることができる。
【0203】
【表1】
【0204】
標品と同時に展開を行い、目視で反応進行度合いの確認に用いられる。
【0205】
【表2】
【0206】
全ピーク面積の和のうち目的物のピーク面積の%を計算し、純度とした。
【0207】
<本発明の第二>
本実施形態では、本発明の製造方法により製造された酸化発色化合物またはその塩、つまり、化学式3で示される酸化発色化合物またはその塩(本明細書中、「本発明の酸化発色化合物/塩」とも称する)の特に好ましい用途・使用に関する実施形態を説明する。
【0208】
本発明の酸化発色化合物/塩は、血液や尿をはじめとする体液中の生体成分の測定法の一つである、酵素を用いた比色分析法に好適に使用される。特に2分子で機能する酸化型発色試薬のうちのカプラーとして用いられうる。すなわち、本発明の酸化発色化合物/塩は、例えば、前記背景技術の項で記載したトリンダー試薬と共に用いられ、被測定物質の酵素的酸化で生じる過酸化酸素をさらにPOD酸化して生成した活性酸素によって、2分子が酸化縮合することによって呈色する。
【0209】
すなわち、本発明の本発明の第二は、本発明の酸化発色化合物/塩と、トリンダー試薬と、測定対象物に対して選択的に作用するオキシダーゼと、ペルオキシダーゼと、を含む試薬組成物(本明細書中、「本発明の試薬組成物」とも称する)である。
【0210】
つまり、本発明の酸化発色化合物/塩は、カプラーとして使用することが特に好ましく、それ以外については従来公知の知見が適宜参照され、特に限定されるものではない。
【0211】
(トリンダー試薬)
トリンダー試薬としては、特に制限はなく、従来公知の化合物がいずれも好ましく用いられうる。ただ、中でも中でもN−エチル−N−(3−スルホプロピル)−3−メチルアニリン、N−エチル−N−(3−メチルフェニル)−3−アセチルエチレンジアミン、N−エチル−N−(3−メチルフェニル)−N−サクシニルエチレンジアミン、N−エチル−N−(2−ヒドロキシ−3−スルホプロピル)−3−メチルアニリン、N−(2−カルボキシエチル)−N−エチル−3−メチルアニリン、N,N−ビス−(4−スルホブチル)−3−メチルアニリン、N−エチル−N−(2−サクシニルアミノエチル)−3−メチルアニリン、N−エチル−N−(3−スルホプロピル)−3−メトキシアニリン、N−(2−ヒドロキシ−3−スルホプロピル)−3,5−ジメトキシアニリン、N−エチル−N−(2−ヒドロキシ−3−スルホプロピル)−3,5−ジメトキシアニリン、N−エチル−N−(2−ヒドロキシ−3−スルホプロピル)−3,5−ジメトキシ−4−フルオロアニリン、N−エチル−N−(2−ヒドロキシ−3−スルホプロピル)−3,5−ジメチルアニリンが好ましく、N−エチル−N−(2−ヒドロキシ−3−スルホプロピル)−3−メチルアニリン、N−エチル−N−(2−ヒドロキシ−3−スルホプロピル)−3,5−ジメチルアニリンが、発色強度や波長といった観点で、特に好ましい。
【0212】
(測定対象物に対して選択的に作用するオキシダーゼ)
「測定対象物に対して選択的に作用するオキシダーゼ」としては、例えば、測定対象物がグルコースである場合、該オキシダーゼはグルコースオキシダーゼであり、測定対象物がコレステロールである場合、該オキシダーゼはコレステロールオキシダーゼであり、測定対象物が尿酸である場合、該オキシダーゼはウリカーゼであり、測定対象物がピルビン酸である場合、該オキシダーゼはピルベートオキシダーゼである。これらは測定対象物の基質のみを特異的に酸化するために、色々な成分を含む検体からそれぞれの測定対象物のみを限定して定量することができる。
【0213】
本発明に係る試薬組成物において、本発明の酸化発色化合物/塩とトリンダー試薬とのモル比は、1:1〜2:1であることが好ましく、1.3:1がより好ましい。本発明の酸化発色化合物/塩とトリンダー試薬とのモル比が1:1未満である場合、また、本発明の酸化発色化合物/塩とトリンダー試薬とのモル比が2:1を超える場合、発色不良を生じることがある。
【0214】
本発明に係る試薬組成物中の、測定対象物に対して選択的に作用するオキシダーゼの含有量は、カプラー15mmolに対して、433k〜2600kUであることが好ましく、866k〜2600kUであることがより好ましい。測定対象物に対して選択的に作用するオキシダーゼの含有量が、カプラー15mmolに対して433kU未満である場合、発色不良を生じることがある。なお、本発明において、前記測定対象物に対して選択的に作用するオキシダーゼの含有量は、4AA−TOOS法により測定したユニット数を採用するものとする。
【0215】
本発明に係る試薬組成物中のペルオキシダーゼの含有量は、カプラー15mmolに対して166.5k〜1666kUであることが好ましく、333k〜999kUであることがより好ましい。ペルオキシダーゼの含有量が、カプラー15mmolに対して166.5kU未満である場合、発色不良を生じることがある。なお、本発明において、前記ペルオキシダーゼの含有量は、ピロガロール法により測定したユニット数を採用するものとする。
【0216】
本発明に係る試薬組成物は、本発明の酸化発色化合物/塩と、トリンダー試薬と、測定対象物に対して選択的に作用するオキシダーゼと、ペルオキシダーゼと、を含む。すなわち、「本発明の酸化発色化合物/塩と、トリンダー試薬と、測定対象物に対して選択的に作用するオキシダーゼと、ペルオキシダーゼ」以外の他の成分を、試薬組成物としての機能を害さない程度含んでいてもよい。該成分としては、例えば、pH緩衝剤、浸透圧調整剤、界面活性剤、可溶化剤、安定化剤、保護剤等が挙げられる。
【0217】
なお、これら他の成分の含有量は、試薬組成物としての機能を害さなければ特に制限されないが、本発明の酸化発色化合物/塩と、トリンダー試薬と、測定対象物に対して選択的に作用するオキシダーゼと、ペルオキシダーゼとの総質量に対して、好ましくは10〜30質量%、より好ましくは13〜20質量%含まれてもよい。
【0218】
本発明に係る酸化発色化合物/塩は、血液や尿をはじめとする体液中の生体成分の測定法の一種である、酵素を用いた比色分析法に好適に使用されるカプラーとして用いられうる。すなわち、本発明に係る酸化発色化合物/塩は、前記トリンダー試薬と共に用いられ、測定対象物の酵素的酸化で生じる過酸化酸素を、さらにペルオキシダーゼ(POD)酸化して生成した活性酸素によって、本発明に係る酸化発色化合物/塩を含むカプラーと、トリンダー試薬と、が酸化縮合することによって呈色する。そのため本発明の酸化発色化合物/塩は、上記の通り、その化合物自体に特徴を有するものであるが、カプラーとして使用することが特に好ましい。
【0219】
上述の通り、本発明の酸化発色化合物/塩は水溶性が高いため、それを含む試薬組成物(本明細書中、「本発明に係る試薬組成物」とも称する)や、該試薬組成物を担体に保持して試験具として使用する場合には、血液などの検体にすばやく溶解して均一化し、迅速に呈色反応が起こる点、非常に優れたものである。また、本発明に係る試薬組成物を用いた呈色反応(比色分析法)において、それによって生じた色素化合物についても、検体への溶解性や親和性が高く、担体内での展開性が良くなるので、担体表面での発色が鮮やかで均一性の高いものとなり、感度が高く安定した測定が可能となる。さらには、本発明に係る酸化発色化合物/塩は、溶解性が高いので、従来の試薬より高濃度の試薬組成物液(試薬液)が調製可能である。すると、該試薬組成物を担体に担持して、試験具として使用する場合、塗布液濃度を高くしてより多くの試薬を担体に保持させることが可能となる。加えて、本発明の酸化発色化合物/塩は親水性が向上し分子量が高くなっている為、保管中の昇華による試薬組成物の損失を抑えることができる。
【0220】
また、本発明に係る酸化発色化合物/塩は、試薬濃度の変化における呈色強度への影響が少ない為に、化合物が経時的に劣化減少した場合でも測定値への影響が少ない。さらに、本発明に係る試薬組成物に含まれる本発明の酸化発色化合物/塩の親水性官能基は、担体への吸着性を高め、試験具として使用した場合でも、担体内での化合物の安定性を向上させる。このように本発明に係る試薬組成物を提供することにより、ひいては、経時劣化を低減させ有効期間が延びた試験具を提供することが可能となる。
【0221】
<本発明の第三>
上記本発明の第二の通り、本発明の酸化発色化合物/塩は、測定対象物に対して選択的に作用するオキシダーゼと、トリンダー試薬と、を組み合わせて、本発明に係る試薬組成物となる。
【0222】
本発明の酸化発色化合物/塩は、前記トリンダー試薬と、測定対象物に対して選択的に作用するオキシダーゼと、ペルオキシダーゼと、組み合わせて、液体、粉末、錠剤、もしくは担体に保持した状態で分析用組成物として用いられうる。
【0223】
これら分析用組成物には、上記に加えて、pH緩衝剤、浸透圧調整剤、界面活性剤、可溶化剤、安定化剤、保護剤等の成分を含んでいてもよい。
【0224】
なお、これら成分の含有量は、試薬組成物としての機能を害さなければ特に制限されないが、本発明に係る酸化発色化合物またはこれらの塩と、トリンダー試薬と、測定対象物に対して選択的に作用するオキシダーゼと、ペルオキシダーゼとの総重量に対して、好ましくは10〜30質量%、より好ましくは13〜20質量%含まれてもよい。
【0225】
また、液体に保持して用いる場合には、検体と混合して反応させた後、色の変化を目視で判定する他、分光光度計で透過吸光度を測定してもよい。また、本発明に係る試薬組成物は、担体に保持した状態で試験具として用いてもよい。
【0226】
このように、本発明に係る試薬組成物は、種々の物に保持して用いることができるが、本発明に係る試薬組成物は、担体に保持した状態で試験具として用いることが好ましい。
【0227】
よって、本発明の第三は、本発明に係る試薬組成物が、担体に保持されてなる試験具である。
【0228】
かようなドライケミストリーで用いられる試験用具のように、担体に保持した状態で用いる場合には、これらの試薬組成物を含む層の他、これの機能を害さない範囲において、計量層、展開層、濾過層、保持層等を含んでいてもよい。このように担体に保持して用いる場合には、検体を付与した後、色の変化を目視で判定する他、分光光度計で反射吸光度を測定してもよい。測定値は予め作製した検量線を用いて測定対象物の量に換算することができる。
【0229】
(担体)
本発明に係る担体の素材としては、紙、布帛、高分子膜等の多孔質物質を用いることができるが、特に、発色性能といった点で、高分子膜が好ましい。
【0230】
上記高分子膜とは、高分子よりなる水不溶性の多孔質体である。高分子としては、ポリスルホン、ポリエーテルスルホン、セルロース、セルロースアセテート、硝酸セルロース、ポリアクリロニトリル、ポリアミド、芳香族ポリアミド、ポリカーボネート、ポリエチレンテレフタレート、ポリイミド、ポリエチレン、ポリプロピレン、ポリテトラフルオロエチレン、ポリフッ化ビニリデン、ポリ塩化ビニル、ポリビニルアルコールが挙げられる。これらの高分子は一般的に知られている製膜方法を用いて膜を形成することができる。これらの高分子膜の中でもポリスルホンまたはポリエーテルスルホンが、発色性能といった点で、特に好ましい。
【0231】
(担体に保持する方法)
本発明に係る試薬組成物を担体に保持する方法にも特に制限はないが、担体に適当な溶剤に溶解させた試薬組成物溶液を担体にコーティングする他、試薬組成物を含むマトリックス前駆体を成型して試験層を形成させる等の公知の方法が用いられうる。
【0232】
コーティングは、工業用に使用される一般的なコート法を用いることができるが、担体が多孔質の場合には、しばしば塗工直後の液移動や不均一な乾燥に起因するコートむらが問題となる。担体と塗工液の物性や、塗工乾燥の方法、機器、条件がこれらを支配する重要因子となりうる。このため、精密印刷法が有効な場合もある。
【0233】
塗工後の液移動や不均一な乾燥を低減する為には、少量の液を正確に計量して塗工するような精密印刷技術が適するが、この方法では高い試薬濃度の塗工液を作製できることが必須となる。
【0234】
例えば、4−アミノアンチピリン等の既存のカプラーは、測定に用いる種々の試薬の中でも最も溶解性が低く、これが高い濃度の塗工液を作製するときの問題点となっていた。しかしながら、本発明の化合物は溶解性が高く、濃度の高い塗工液を作製するに好都合である。
【0235】
試薬を担体に保持させた試験具では、検体付与後、検体の液体成分によってまず試薬が溶解し、混合して反応が起こり、色素化合物が生成し、通常試験具の検体付与面の反対側にある読み取り面に移動し、その色調変化を測定する、というステップを経て定量が行なわれる。このため、試薬の溶解性が高いことは、検体による均一な溶解、均一化、迅速な反応に有利であるだけではなく、生成する色素も溶解性に優れるために読み取り面への移動がスムースかつ均一性が高いというメリットもある。これらの特徴は、測定時間の短縮化、測定精度の向上、測定値ばらつきの低減に寄与する。したがって、本発明の酸化発色化合物/塩は高い溶解性を有するため、測定時間の短縮化、測定精度の向上、測定値ばらつきの低減に寄与できることが期待される。
【0236】
例えば、従来公知のカプラーである4−アミノアンチピリンを担体に保持させた試験具として使用した場合、経時的に劣化減少し測定値が上昇するという問題が生じる。減少による測定値上昇の原因としては、酵素反応や呈色反応の阻害ではなく化合物の濃度が高いほど他の試薬類の溶解や移動、また生成色素の流動性に何らかの障害を与えるのではないかと考えられている。しかしながら、本発明の化合物は4−アミノアンチピリンと比較して量が増減しても測定値への影響が少なく、4−アミノアンチピリンでみられた問題を改善できた。
4−アミノアンチピリン等の既存のカプラーは、ある担体に保持させた試験具として使用した場合、濃度変化が測定値に影響するという問題点がある。バルク担体にコートする際むらが生じた場合には、それを切り抜いて用いる各試験具間に感度差が生じる、あるいは保存中に劣化分解して量が変化した場合測定値に影響が出るということを示唆している。原因は、酵素反応や呈色反応の阻害ではなく、化合物の濃度が高いほど他の試薬類の溶解や移動、また生成色素の流動性に何らかの障害を与えるのではないかと考えられている。これに対して、本発明の酸化発色化合物/塩は4−アミノアンチピリンと比較して濃度の測定値への影響が少なく、これらの問題を改善できる。
【0237】
本発明の製造方法により得られる化学式3で示される酸化発色化合物またはその塩は、トリンダー試薬と組み合わせて酸化発色させた場合には、4−アミノアンチピリン等の既存のカプラーを用いた場合と比較して吸光度が大きくなり、測定感度が向上することができる。例えば、本発明の4−アミノ−1−(4−スルホフェニル)−2,3−ジメチル−5−ピラゾロン、4−アミノ−1−(3−スルホフェニル)−2,3−ジメチル−5−ピラゾロン等は、N−エチル−N−(2−ヒドロキシ−3−スルホプロピル)−3−メチルアニリン(TOOS)やN−エチル−N−(2−ヒドロキシ−3−スルホプロピル)−3,5−ジメチルアニリン(MAOS)と組み合わせて発色させた場合、その検量線の傾きは改善され、測定感度が向上されうる。
【0238】
上述の通り、本発明の製造方法により得られる化学式3で示される酸化発色化合物またはその塩は水溶性が高いため、それを含む試薬組成物を担体に保持しなる試験具(以下、単に、「本発明に係る試験具」とも称する)を使用する場合、本発明に係る試薬組成物は、血液などの検体にすばやく溶解して均一化し、迅速に呈色反応が起こるため、本発明に係る試験具は、非常に優れたものとして使用されうる。
【0239】
また、本発明に係る試験具を用いた場合、本発明に係る試薬組成物の呈色反応において生じた色素化合物についても、検体への溶解性や親和性が高く、担体内での展開性が良くなるので、担体表面での発色が鮮やかで均一性の高いものとなる。
【0240】
本願発明の製造方法により製造された化合物は、溶解性が有意に高いので、従来の試薬より高濃度の試薬液が調製可能である。また、担体に担持して使用する場合塗布液濃度を高くしてより多くの試薬を担体に保持させることが可能となる。さらには、塗布液の濃度を高く設定し全体液量を少なくできることで、塗工均一性の高い、グラビア印刷法、ドット印刷法、インクジェット印刷等の精密印刷法が使用できる。塗工液量が少なくなれば、乾燥時間が短縮され乾燥むらを低減させる効果もある。以上、十分な試薬を高い均一性で塗工できることにより、塗りむらなどに起因するばらつきを低減させることができ、高い精度の試験具を提供することができる。すなわち、本発明に係る試験具を用いると、感度が高く安定した測定が可能となる。さらには、本発明の酸化発色化合物/塩は、溶解性が高いので、従来の試薬より高濃度の試薬組成物液(試薬液)が調製可能である。すると、該試薬組成物を担体に担持して、試験具として使用する場合、塗布液濃度を高くしてより多くの試薬を担体に保持させることが可能となる。
【0241】
加えて、本発明の製造方法により得られる化学式3で示される酸化発色化合物またはその塩は、親水性が向上し分子量が高くなっている為、保管中の昇華による試薬組成物の損失を抑えることができる。
【0242】
また、本発明の酸化発色化合物/塩は、試薬濃度の変化における呈色強度への影響が少ない為に、化合物が経時的に劣化減少した場合でも測定値への影響が少ない。さらに、本発明に係る試薬組成物に含まれる本発明の酸化発色化合物/塩の親水性官能基は、担体への吸着性を高める。すなわち、本発明に係る試験具は、担体内での化合物の安定性を向上されている。
【0243】
このように本発明に係る試験具は、従来に比べ、経時劣化を低減させ有効期間が有意に延びたものといえる。
【実施例】
【0244】
次に実施例によって本発明を説明する。本発明の化合物は以下の実施例の製法で合成される。
【0245】
<実施例1>
1−(4−スルホフェニル)−2,3−ジメチル−5−ピラゾロンの合成
200gの1−(4−スルホフェニル)−3−メチル−5−ピラゾロンを1Lナスフラスコに入れ、89mLの硫酸ジメチルを滴下ロートから均一にかかるように室温下にて滴下した。硫酸ジメチルの滴下後、ナスフラスコ上部に塩化カルシウム管を接続した玉入冷却器を取り付けた。次いで、温度コントローラーのセンサー部分をマントルヒーター上に置き、ナスフラスコを装着した。設定温度を230℃にして加熱を開始した。1−(4−スルホフェニル)−3−メチル−5−ピラゾロンの粉末が完全に融解し内部が茶色透明の均一状態になるまで、約2時間加熱した。ナスフラスコを室温まで放冷後、氷冷下、水酸化ナトリウム水溶液(水酸化ナトリウム:113g+純水:500 mL)を徐々に添加した後、発熱、発煙が収まってから、70〜80℃の水浴上でガラス棒にて攪拌しながら反応混合物の固形物を溶解した。さらに、溶解後、pH試験紙で内容液が強アルカリ性(pH11以上)であることを確認した。
【0246】
次いで、上記反応混合物の固形物をアルカリ水溶液で溶解した反応物をサンプリングし、TLCでチェックを行なった。ナスフラスコ内容液中の黒色不溶物を吸引濾過し濾別した。氷冷下、得られた濾液に120 mLの塩酸を攪拌しながら添加し、pH試験紙で内容液が強酸性(pH1以下)であることを確認した。氷冷下、沈殿が生成するまで攪拌を続けた。生成した沈殿を吸引濾過し、ロート上で、純水500mL、アセトン500mLにて順次洗浄した。真空ラインに接続して、減圧乾燥を行ない、粗生成物174gを得た。粗生成物を2000mLナスフラスコに入れ、純水から再結晶して、純水、アセトンにて洗浄後、減圧乾燥して目的物119gを得た(収率:56%)。純度95%以上(HPLC)。
【0247】
【表3】
【0248】
<実施例2>
1−(4−スルホフェニル)−2,3−ジメチル−5−ピラゾロンの合成(高圧法)
6.75gの1−(4−スルホフェニル)−3−メチル−5−ピラゾロン(アルドリッチ社製 134163)、250mLの脱水メタノール(ナカライテスク社製 04100−25)、4.2mLのヨウ化メチル(関東化学社製 I0060)を、高圧反応装置〈耐圧硝子社製TPR−1型)シリンダー容器内に入れ、130℃で18時間加熱攪拌した。反応後、反応混合液を300mLのナスフラスコに注ぎロータリーエバポレーターにて40±3℃の水浴上で乾固するまで減圧濃縮を行なった。真空ラインに接続して室温にて30分間減圧乾燥を行なった。乾燥後、純水15mLを加え100〜110℃のオイルバス中で攪拌しながら内容物を完全に溶解させた。すり潰した塩化ナトリウム5.53gを加え、オイルバス上で完全に溶解させた。ナスフラスコをオイルバスから取り出し、室温で1時間攪拌した。茶色の沈殿が生成することを確認し、冷蔵庫に入れ、15時間以上放置した後、生成した沈殿を吸引濾過し、純水、アセトンで洗浄し、減圧乾燥して粗生成物を得た。粗生成物を純水から再結晶して、純水、アセトンにて洗浄後、減圧乾燥して目的物1.5gを得た(収率:21%)。所要時間3日、純度95%以上(HPLC)。
【0249】
【表4】
【0250】
<実施例3>
4−アミノ−1−(4−スルホフェニル)−2,3−ジメチル−5−ピラゾロンの合成
(1)「ニトロソ化工程」
4−ニトロソ−1−(4−スルホフェニル)−2,3−ジメチル−5−ピラゾロンの合成
実施例1または2で合成した1−(4−スルホフェニル)−2,3−ジメチル−5−ピラゾロン30.0g(0.111mol)を乳鉢で細かくすりつぶし、500mL三つ口フラスコに秤量し、磁気攪拌子を入れた。三つ口フラスコを磁気スターラー上に置いた氷浴(0〜3℃)中に浸し、氷冷した純水111mLを加え、10分程度攪拌した。
【0251】
攪拌下、2〜3℃に冷却した亜硝酸ナトリウム水溶液31mL(亜硝酸ナトリウム13.8g/純水50mL)(0.124mol)と、2〜3℃に冷却した4N塩酸31mL(0.124mol)と、を同時に、5分かけて滴下ロートから滴下した。滴下後、磁気スターラーで3分間激しく攪拌した。生成した沈殿を吸引濾過した。フィルターケーキ上に、2〜3℃に冷却した脱水メタノール約150mLをまんべんなくかけて洗浄し、真空ライン(<2mmHg)に接続して、室温にて、減圧乾燥した。
【0252】
その後、目的物(4−ニトロソ−1−(4−スルホフェニル)−2,3−ジメチル−5−ピラゾロンの合成)25gを得た。
【0253】
なお、1−(4−スルホフェニル)−2,3−ジメチル−5−ピラゾロンに対する収率は、75%であった。
【0254】
(2)「還元工程」
4−アミノ−1−(4−スルホフェニル)−2,3−ジメチル−5−ピラゾロンの合成
上記(1)で得られた4−ニトロソ−1−(4−スルホフェニル)−2,3−ジメチル−5−ピラゾロン4.0g(0.0134mol)を、500mLナスフラスコに秤量した。ナスフラスコに、磁気攪拌子を入れ、磁気スターラー上に置いた氷浴中に浸しクランプで固定した。
【0255】
それに、2〜3℃に冷却したメタノール156mLを入れ、3秒間攪拌して分散させた。亜鉛粉末6.6g(0.100mol)、2〜3℃に冷却した2N塩酸54mL(0.108mol)を加えて、5分間攪拌を続けた。反応後、ナスフラスコ中の内容物を吸引濾過して亜鉛残渣を濾別した。亜鉛残渣を、純水を用いてロート上で洗浄して、濾液と洗液をあわせ、ナスフラスコに入れた。ロータリーエバポレーターにて減圧濃縮を行ない、溶媒を留去した。130mLの純水を加えて濃縮物を懸濁させ、ビーカーに移して磁気攪拌子を入れ、磁気スターラー上に置いた氷浴中に浸し攪拌を行った。濃縮物のpHをpHメーターでモニターしながら、pHが9.0±0.5になるまで、攪拌下、4N水酸化ナトリウム水溶液をピペットで滴下した。
【0256】
生成した水酸化亜鉛の沈殿物を吸引濾過して水酸化亜鉛残渣を濾別した。水酸化亜鉛残渣を、純水を用いてロート上で洗浄して濾液と洗液をあわせ、ナスフラスコに入れた。ロータリーエバポレーターにて減圧濃縮を行ない、溶媒を留去して、真空ライン(<2mmHg)に接続して、室温にて減圧乾燥した。ナスフラスコ中の生成物にエタノール800mLを加え、磁気攪拌子を入れ磁気スターラーにて目的物を攪拌抽出した。ナスフラスコ内容物を吸引濾過して塩化ナトリウム残渣を濾別した。塩化ナトリウム残渣を、エタノールを用いてロート上で洗浄して濾液と洗液をあわせ、ナスフラスコに入れた。ロータリーエバポレーターにて減圧濃縮を行ない、溶媒を留去して、真空ライン(<2mmHg)に接続して、室温にて減圧乾燥した。目的物:3.7gを得た。
【0257】
なお、4−ニトロソ−1−(4−スルホフェニル)−2,3−ジメチル−5−ピラゾロン対する収率は、90%であった。
【0258】
2工程(「ニトロソ化工程」および「還元工程」の2工程)の収率は68%((1)×(2))であり、純度は、97%以上(HPLC)であった。
【0259】
【表5】
【0260】
<比較例1>
4−アミノ−1−(4−スルホフェニル)−2,3−ジメチル−5−ピラゾロンの合成
実施例1または2で得られた1−(4−スルホフェニル)−2,3−ジメチル−5−ピラゾロン 0.5gを0.4N塩酸10mLに溶解し、氷冷下40%亜硝酸ナトリウム水溶液0.35mLを加えて1分間攪拌し、亜鉛末500mgを加え、さらに室温で10分攪拌した。反応混合物をろ過後、濾液を濃縮乾固し、目的物0.41gを得た。収率は36%、純度は50%(HPLC測定により)であった。
【0261】
【表6】
【0262】
<実施例4>
1−(3−スルホフェニル)−2,3−ジメチル−5−ピラゾロンの合成
200gの1−(3−スルホフェニル)−3−メチル−5−ピラゾロンを1000mLナスフラスコに入れ、89mLの硫酸ジメチルを滴下ロートから均一にかかるように室温下にて滴下した後、ナスフラスコ上部に塩化カルシウム管を接続した玉入冷却器を取り付けた。温度コントローラーのセンサー部分をマントルヒーター上に置き、ナスフラスコを装着した。設定温度を230℃にして加熱を開始した。1−(3−スルホフェニル)−3−メチル−5−ピラゾロンの粉末が完全に融解し内部が茶色透明の均一状態になるまで、約2時間加熱した。ナスフラスコを室温まで放冷後、氷冷下、水酸化ナトリウム水溶液(水酸化ナトリウム:113g+純水:500 mL)を徐々に添加した後、発熱、発煙が収まってから、70〜80℃の水浴上でガラス棒にて攪拌しながら固形物を溶解した。溶解後、pH試験紙で内容液が強アルカリ性(pH11以上)であることを確認した。
【0263】
反応物をサンプリングし、TLCでチェックを行なった。ナスフラスコ内容液中の黒色不溶物を吸引濾過を行ない濾別した。氷冷下、得られた濾液に120 mLの塩酸を攪拌しながら添加し、pH試験紙で内容液が強酸性(pH1以下)であることを確認した。氷冷下、沈殿が生成するまで攪拌を続けた。生成した沈殿を吸引濾過し、ロート上で、純水500mL、アセトン500mLにて順次洗浄した。真空ラインに接続して、減圧乾燥を行ない、粗生成物180gを得た。粗生成物を2000mLナスフラスコに入れ、純水から再結晶して、純水、アセトンにて洗浄後、減圧乾燥して目的物125gを得た(収率:60%)。所要時間2日、純度95%以上(HPLC測定により)。
【0264】
【表7】
【0265】
以上のことから、比較例に比べて本発明の製造方法で製造された実施例1または実施例4では、同規模の反応装置ではるかに仕込み量が大きく出来ることにより効率化された。また、所要時間は1日、収率は38%程度向上している。
【0266】
<実施例5>
1−(3−スルホフェニル)−2,3−ジメチル−5−ピラゾロンの合成
6.75gの1−(3−スルホフェニル)−3−メチル−5−ピラゾロン(アルドリッチ社製 556890)、250mLの脱水メタノール(ナカライテスク社製 04100−25)、4.2mLのヨウ化メチル(関東化学社製 I0060)を、高圧反応装置〈耐圧硝子社製TPR−1型)シリンダー容器内に入れ、130℃で18時間加熱攪拌した。
【0267】
反応後、反応混合液を300mLのナスフラスコに注ぎロータリーエバポレーターにて40±3℃の水浴上で、乾固するまで減圧濃縮を行なった。さらに、真空ラインに接続して、室温にて30分間減圧乾燥を行なった。乾燥後、純水15mLを加え、100〜110℃のオイルバス中で攪拌しながら内容物を完全に溶解させた。これに、すり潰した塩化ナトリウム5.53gを加え、オイルバス上で完全に溶解させた。ナスフラスコをオイルバスから取り出し、室温で1時間攪拌した。ここで、茶色の沈殿が生成したことを確認し、冷蔵庫に入れ、15時間以上(一昼夜)放置した。
【0268】
放置後、生成した沈殿を吸引濾過し、純水、アセトンで洗浄し、減圧乾燥して粗生成物を得た。粗生成物を純水から再結晶して、純水、アセトンにて洗浄後、減圧乾燥して目的物1.4gを得た(収率:20%)。なお、純度は95%以上(HPLC)であった。
【0269】
【表8】
【0270】
<実施例6>
4−アミノ−1−(3−スルホフェニル)−2,3−ジメチル−5−ピラゾロンの合成
(1)「ニトロソ化工程」
4−ニトロソ−1−(3−スルホフェニル)−2,3−ジメチル−5−ピラゾロンの合成
実施例4または5で合成した1−(3−スルホフェニル)−2,3−ジメチル−5−ピラゾロン30.0g(0.111mol)を、乳鉢で細かくすりつぶし、500mL三つ口フラスコに秤量し、磁気攪拌子を入れた。三つ口フラスコを磁気スターラー上に置いた氷浴(0〜3℃)中に浸し、氷冷した純水110mLを加え、10分攪拌した。
【0271】
攪拌下、2〜3℃に冷却した亜硝酸ナトリウム水溶液31mL(亜硝酸ナトリウム13.8g/純水50mL)(0.124mol)と、2〜3℃に冷却した4N塩酸31mL(0.124mol)と、を同時に、5分かけて滴下ロートから滴下する。滴下後、磁気スターラーで3分間激しく攪拌した。生成した沈殿を吸引濾過した。フィルターケーキ上に、2〜3℃に冷却した脱水メタノール約150mLをまんべんなくかけて洗浄し、真空ライン(<2mmHg)に接続して、室温にて、減圧乾燥した。目的物25gを得た。
【0272】
なお、1−(3−スルホフェニル)−2,3−ジメチル−5−ピラゾロン対しての収率は、75%であった。
【0273】
(2)「還元工程」
4−アミノ−1−(3−スルホフェニル)−2,3−ジメチル−5−ピラゾロンの合成
上記(1)で得られた4−ニトロソ−1−(3−スルホフェニル)−2,3−ジメチル−5−ピラゾロン4.0g(0.01mol)を、500mLナスフラスコに秤量した。ナスフラスコに、磁気攪拌子を入れ、磁気スターラー上に置いた氷浴中に浸しクランプで固定した。
【0274】
それに、2〜3℃に冷却したメタノール160mLを入れ、3秒間攪拌して分散させた。亜鉛粉末6.6g(0.1mol)、2〜3℃に冷却した2N塩酸54mL(0.1mol)を加えて、5分間攪拌を続ける。反応後、ナスフラスコ中の内容物を吸引濾過して亜鉛残渣を濾別した。亜鉛残渣を、純水を用いてロート上で洗浄して、濾液と洗液をあわせ、ナスフラスコに入れた。ロータリーエバポレーターにて減圧濃縮を行ない、溶媒を留去した。130mLの純水を加えて濃縮物を懸濁させ、ビーカーに移して磁気攪拌子を入れ、磁気スターラー上に置いた氷浴中に浸し攪拌を行なった。濃縮物のpHをpHメーターでモニターしながら、pHが9.0±0.5になるまで、攪拌下、4N水酸化ナトリウム水溶液をピペットで滴下した。
【0275】
生成した水酸化亜鉛の沈殿物を吸引濾過して水酸化亜鉛残渣を濾別した。
【0276】
水酸化亜鉛残渣を、純水を用いてロート上で洗浄して濾液と洗液をあわせ、ナスフラスコに入れた。ロータリーエバポレーターにて減圧濃縮を行ない、溶媒を留去して、真空ライン(<2mmHg)に接続して、室温にて減圧乾燥した。
【0277】
ナスフラスコ中の生成物にエタノール800mLを加え、磁気攪拌子を入れ磁気スターラーにて目的物を攪拌抽出した。
【0278】
ナスフラスコ内容物を吸引濾過して塩化ナトリウム残渣を濾別した。
【0279】
塩化ナトリウム残渣を、エタノールを用いてロート上で洗浄して濾液と洗液をあわせ、ナスフラスコに入れた。ロータリーエバポレーターにて減圧濃縮を行ない、溶媒を留去して、真空ライン(<2mmHg)に接続して、室温にて減圧乾燥した。目的物:3.6gを得た(収率87%)。
【0280】
2工程(「ニトロソ化工程」および「還元工程」の2工程)の収率は65%であり、純度は、97%以上(HPLC)であった。
【0281】
【表9】
【0282】
比較例に比べて本発明の製造方法で製造された実施例では、純度は47%以上、収率は32%および29%向上している。また性状は、比較例で得られた化合物は吸湿性が高い粉末であり扱いにくいのに対し、実施例3および実施例6で得られた化合物は吸湿性が低い粉末で取扱いがしやすい。
【0283】
<実施例7>
試験片による性能評価
0.13Mコハク酸ナトリウム緩衝液(pH=5.0)100mL中に、実施例3で合成した4−アミノ−1−(4−スルホフェニル)−2,3−ジメチル−5−ピラゾロン 0.46g 、 N−エチル−N−(2−ヒドロキシ−3−スルホプロピル)−3,5−ジメチルアニリン 0.95g、Triton X−100 3.0g、グルコースオキシダーゼ 150K unit、ペルオキシダーゼ 250K unitを溶解して塗工液を作成した。ポリエーテルスルホン膜(テルモ株式会社製、膜厚130μm)に塗工液を塗布し、35℃で18時間乾燥を行なった。膜を1cm角の正方形に打ち抜き、試験片を得た。試験片を反射分光光度計検出部に固定した後、1μLの検体を試験膜上部より滴下し、10秒後の反射吸光度を測定した。ヘマトクリット値40、種々のグルコース濃度の血液で測定した値から検量線を作成し、定量感度を観察した。(図1)反射吸光度値はそれぞれの試験片に対して最高となる時間の値を用いた。図1中、x軸に反射吸光度、y軸に血糖値をとっているため、傾きがなだらかなほど感度が高いことを意味する。
図2から示されるように、本発明の製造方法により合成した中間体を用いて合成された4−アミノ−1−(4−スルホフェニル)−2,3−ジメチル−5−ピラゾロンを用いた試験片の検量線の傾きは、従来から広く使用されている4−アミノアンチピリン(4−AA)を用いた場合より改善され、測定感度を有意に向上させることが示される。
【図面の簡単な説明】
【0284】
【図1】実施例3における、本発明の酸化発色化合物/塩および4−アミノアンチピリンの反射吸光度値の時間変化を表した図である。
【技術分野】
【0001】
本発明は、酸化発色化合物の製造方法ならびに当該方法によって製造された化合物を用いた試薬組成物および試験具に関する。
【背景技術】
【0002】
今日、血液や尿をはじめとする体液中の生体成分の測定法の一種として、酵素を用いた比色分析法が広く用いられている。これらの方法は、測定キット、自動分析機、ドライケミストリー試験具等に組み込まれ、日常的な臨床検査に数多く用いられている(例えば、特許文献1を参照)。
【0003】
このような測定法の一種として、被測定物質に特異的な酸化酵素を作用させて発生する過酸化水素をさらにペルオキシダーゼの作用で活性酸素とし、これで発色試薬を酸化して生成する色素を比色定量する方法がある。
【0004】
ここで使用される酵素としては、例えばグルコースの測定ではグルコースオキシダーゼ、コレステロールの測定ではコレステロールオキシダーゼ、尿酸の測定にはウリカーゼ、ピルビン酸の測定にはピルベートオキシダーゼ等が用いられる。これらの酵素は検体中の基質のみを特異的に酸化するために、色々な成分を含む検体からそれぞれの測定対象物のみを限定して定量することができる。
【0005】
また、発色試薬としては、活性酸素で酸化されることによって、その吸収波長特性や吸収強度等が変化する色素もしくは色素の前駆体(色原体)が用いられる。すなわち、活性酸素の量に応じて変化する色を測定することによって、被測定物質の量を定量することができる。これらの色素や色原体には、1分子だけで機能するものや、異なる2分子がカップリングして機能するものがある。
【0006】
1分子で機能するものには、ロイコ型色素等が挙げられる。具体的な化合物としては、ベンチジン、o−トリジン、o−ジアニシジン、2,2’−アミノ−ビス(3−エチルベンゾチアゾリノン−6−スルホン酸(ABTS)、ビス−(4−ジエチルアミノ)−2−スルホフェニルメタン(BSPM)、ビス[3−ビス(4−クロロフェニル)メチル−4−ジメチルアミノフェニル]アミン(BCMA)、10−N−メチルカルバモイル−3,7−ジメチルアミノ−10H−フェノチアジン(MCDP)、3,3’,5,5’−テトラメチルベンチジン(TMBZ)、ビス[4−(N−アルキル−N−スルホプロピル)−2,6−ジメチルフェニル]メタン(Bis−MAPS)、N,N,N’,N’,N’’,N’’−ヘキサ(3−スルホプロピル)−4,4’,4’’’−トリアミノトリフェニルメタン(TMP)などが挙げられる。
【0007】
2分子で機能する代表的なものには、カプラーとトリンダー試薬を酸化的にカップリングさせたものが挙げられる(例えば、非特許文献1および2を参照)。
【0008】
カプラーとしては、4−アミノアンチピリン(4−AA)、バニリンジアミンスルホン酸、メチルベンズチアゾリノンヒドラゾン(MBTH)、スルホン化メチルベンズチアゾリノンヒドラゾン(SMBTH)、アミノジフェニルアミンまたはその誘導体などが用いられる。
【0009】
トリンダー試薬としては、フェノール誘導体、アニリン誘導体が用いられる。フェノール誘導体としては、フェノール、4−クロロフェノール、2,4−ジクロロフェノール、2,6−ジクロロフェノール、3,5−ジクロロフェノール、2,4−ジブロモフェノール、2,6,4−トリクロロフェノール、2,6,4−トリブロモフェノール、3,5−ジクロロ−2−ヒドロキシベンゼンスルホン酸、3−ヒドロキシ−2,4,6−トリヨードベンゾイル酸、などが挙げられる。アニリン誘導体としては、N−エチル−N−(3−スルホプロピル)−3−メチルアニリン(TOPS)、N−エチル−N−(3−メチルフェニル)−3−アセチルエチレンジアミン(EMAE)、N−エチル−N−(3−メチルフェニル)−N−サクシニルエチレンジアミン(EMSE)、N−エチル−N−(2−ヒドロキシ−3−スルホプロピル)−3−メチルアニリン(TOOS)、N−(2−カルボキシエチル)−N−エチル−3−メチルアニリン(CEMB)、N,N−ビス−(4−スルホブチル)−3−メチルアニリン(TODB)、N−エチル−N−(2−サクシニルアミノエチル)−3−メチルアニリン(ESET)、N−エチル−N−(3−スルホプロピル)−3−メトキシアニリン(ADPS)、N−(2−ヒドロキシ−3−スルホプロピル)−3,5−ジメトキシアニリン(HSDA)、N−エチル−N−(2−ヒドロキシ−3−スルホプロピル)−3,5−ジメトキシアニリン(DAOS)、N−エチル−N−(2−ヒドロキシ−3−スルホプロピル)−3,5−ジメトキシ−4−フルオロアニリン(FDAOS)、N−エチル−N−(2−ヒドロキシ−3−スルホプロピル)−3,5−ジメチルアニリン(MAOS)などが挙げられる。
【0010】
ここで、保存安定性に鑑みると、1分子で機能する色素より2分子で機能する色素の方が良い傾向にある。また、2分子で機能する色素のうち、カプラーとしては、前記のうち最も安定な4−アミノアンチピリン、メチルベンズチアゾリノンヒドラゾンが多く用いられ、トリンダー試薬としては安定でかつ発色強度や波長の点でより有利なアニリン誘導体が多く用いられている。
【0011】
このような現状の下、前記の発色原理を用いた製品の価値を上げるためには、測定精度をさらに向上させる、あるいは経時的劣化を低減させる必要があり、また、より微量な成分の検出には、現存のものよりさらに感度のよい検出系が必要となり、それらの目的において、より高性能の発色試薬の開発が望まれている。
【0012】
このように、従来のカプラーに変わる新規な発色化合物の開発が進められており、例えば、特許文献2には、酵素を阻害しないとされるアミノアンチピリン誘導体が開示されている。
【0013】
ところで、このようなアミノアンチピリン誘導体のようなアミノ化合物を得るためには、ニトロソ化を行い、さらに還元を行なう方法が一般的に知られており、アミノアンチピリンの製造過程においては、ニトロソ化した後還元し、アミノ化をする手法が開示されている(例えば、特許文献3参照)。なお、特許文献2においても、ニトロソ化、アミノ化を行い、所望の発色化合物を得ている。
【特許文献1】特開2004−223115号公報
【特許文献2】特公平6−99403号公報
【特許文献3】米国特許第1,877,166号明細書
【非特許文献1】Trinder,P.,Ann.Clin.Biochem.,6,24,1969
【非特許文献2】Barham,D.and Trinder,P.,Analyst(London),97,142,1972
【発明の開示】
【発明が解決しようとする課題】
【0014】
上記の通り、より高性能の発色試薬の開発が望まれている一方で、実際に工業化を念頭においた場合、その高性能の発色試薬を、高純度かつ高収率で製造する必要性が生じる。
【0015】
しかしながら、前記したような技術においては、高純度かつ高収率で製造することができず、このような技術手段の開発が、現実問題として希求されている。
【0016】
よって、本発明は、上記実情を鑑みてなされたものであり、従来に比して性能が向上した発色化合物を、高純度かつ高収率で製造するための方法を提供することを目的とする。
【課題を解決するための手段】
【0017】
本発明者らは、上記実情に鑑み、鋭意研究を行った。その過程でまず、前記のような技術によっても、従来に比して性能が向上した発色化合物を、高純度かつ高収率で製造することができない理由を、以下の通り詳細に探索した。
【0018】
まず、所望の発色化合物を得るためには、ニトロソ化、アミノ化を経る必要があるが、高純度かつ高収率で所望の発色化合物を得ることができないのは、ニトロソ化により生じるニトロソ体が非常に不安定で、水溶液中では容易に分解して不純物を生じるのではないかと予測した。
【0019】
さらには、本発明が提供する酸化発色化合物は、従来のアミノアンチピリンと比較して極めて水溶性が高いことも、従来の方法によっては、高純度かつ高収率で目的物を得ることができない一因ではないかと予測した。
【0020】
その予測の下、鋭意研究を行った結果、本発明の製造方法によると、驚くべきことに、非常に高い純度および収率で、目的物を得ることができたのである。本発明は、このようにして完成した。
【0021】
すなわち、本発明は、下記(1)〜(11)で示される、高い性能を持つ新規酸化発色化合物の製造方法を提供する。
【0022】
(1)下記化学式1:
【0023】
【化1】
【0024】
ただし、R1、R2、R3、R4、およびR5は、それぞれ独立して、水素原子またはスルホン酸基であり、この際R1、R2、R3、R4、およびR5の少なくとも一つがスルホン酸基である、
で示される化合物またはその塩を、亜硝酸塩を用いてニトロソ化する、ニトロソ化工程と;
前記ニトロソ化工程の後、
下記化学式2:
【0025】
【化2】
【0026】
ただし、R1、R2、R3、R4およびR5は、化学式1の定義と同様である、
で示される化合物またはその塩を単離する、単離工程と;
前記単離工程により得られた化学式2で示される化合物またはその塩を、亜鉛を用いて還元する、還元工程と;
を含む、
下記化学式3:
【0027】
【化3】
【0028】
ただし、R1、R2、R3、R4およびR5は、化学式1の定義と同様である、
で示される酸化発色化合物またはその塩の製造方法。
【0029】
(2)前記ニトロソ化工程において、亜硝酸塩の量が、化学式1で示される化合物またはその塩に対して1.0〜1.5倍モルである、(1)に記載の製造方法。
【0030】
(3)前記ニトロソ化工程におけるニトロソ化反応を、化学式1で示される化合物またはその塩に対して1.0〜1.5倍モルの酸を用いて行う、(1)または(2)に記載の製造方法。
【0031】
(4)前記ニトロソ化工程において、酸またはその水溶液と、亜硝酸塩の水溶液と、を同時に化学式1で示される化合物またはその塩の水溶液に滴下して添加する、(3)に記載の製造方法。
【0032】
(5)前記還元工程において、亜鉛の量が、前記単離工程により得られた化学式2で示される化合物またはその塩に対して1〜30倍モルである、(1)〜(4)のいずれか1つに記載の製造方法。
【0033】
(6)前記還元工程における還元反応を、前記単離工程により得られた化学式2で示される化合物またはその塩に対して1〜30倍モルの酸をさらに用いて行う、(1)〜(5)のいずれか1つに記載の製造方法。
【0034】
(7)前記還元工程を、反応溶媒を用いて行い、該反応溶媒が、アルコールである、(1)〜(6)のいずれか1つに記載の製造方法。
【0035】
(8)前記還元工程を、反応溶媒を用いて行い、該反応溶媒の量が、前記単離工程により得られた化学式2で示される化合物またはその塩1gに対して20〜100mLである、(1)〜(7)のいずれか1つに記載の製造方法。
【0036】
(9)前記還元工程後、反応混合物中に残留する亜鉛を、水酸化亜鉛として除去する工程をさらに含む、(1)〜(8)のいずれか1つに記載の製造方法。
【0037】
(10)前記還元工程後、反応混合物からアルコールを用いて、化学式3で示される酸化発色化合物またはその塩を抽出精製する、抽出精製工程をさらに含む、(1)〜(9)のいずれか1つに記載の製造方法。
【0038】
(11)(1)〜(10)のいずれか1つに記載の製造方法で得られた、化学式3で示される酸化発色化合物またはその塩。
【発明の効果】
【0039】
本発明の製造方法においては、従来の方法と比較して、下記(ア)〜(ウ)で優れている。
【0040】
(ア)純度の向上
従来法では、50%程度であった純度が、本願発明の製造方法によると、95%以上に向上しうる。
【0041】
(イ)反応収率の向上
従来法では、30%程度であった収率が、本願発明の製造方法によると、80%以上の収率が得られうる。
【0042】
(ウ)吸湿性の改善
従来法で得られるアミノアンチピリン誘導体は、そのアミノ基が、酸(塩酸、酢酸等)との塩を形成しており、非常に吸湿性が高かった。本願発明の製造方法によると、スルホン酸基がアルカリ塩となっているため、吸湿性が有意に低い。さらに、本願発明により製造される発色化合物は、その呈色性能において、下記(エ)〜(キ)の点でこれまでの試薬より優れている。特に担体に保持した試験具として使用する場合、その効果が最も顕著である。
【0043】
(エ)測定時間の短縮
本願発明の製造方法により製造される発色化合物は、水溶性が有意に高いので、特に担体に保持して使用する場合には、血液などの検体にすばやく溶解して均一化し、迅速に呈色反応が起こる。
【0044】
(オ)感度が高い
呈色反応によって生成した色素化合物は、検体への溶解性や親和性が有意に高く、担体内での展開性が良くなるので、担体表面での発色が鮮やかで均一性の高いものとなる。それによって感度が高く安定した測定が可能となる。
【0045】
(カ)精度が高い
本願発明の製造方法により製造される発色化合物は、溶解性が有意に高いので、従来の試薬より高濃度の試薬液が調製可能である。また、担体に担持して使用する場合塗布液濃度を高くしてより多くの試薬を担体に保持させることが可能となる。さらには、塗布液の濃度を高く設定し全体液量を少なくできることで、塗工均一性の高い、グラビア印刷法、ドット印刷法、インクジェット印刷等の精密印刷法が使用できる。塗工液量が少なくなれば、乾燥時間が短縮され乾燥むらを低減させる効果もある。つまり、十分な試薬を高い均一性で塗工できることにより、塗りむらなどに起因するばらつきを低減させることができ、高い精度の試験具を提供することができる。
【0046】
(キ)経時安定性に優れる
本願発明の製造方法により製造された発色化合物は、親水性が有意に向上して、分子量が有意に大きいため、保管中の昇華による試薬の損失を押さえることができる。また、本願発明の製造方法により製造された発色化合物は、試薬濃度の変化における呈色強度への影響が少ないため、発色化合物が経時的に劣化減少した場合でも測定値への影響が少ない。さらには、本発明の製造方法により製造される発色化合物における親水性官能基は、担体への吸着性を高め、担体内での化合物の安定性を向上させる。つまり、経時劣化を低減させ、有効期間を延ばすことが可能となる。
【発明を実施するための最良の形態】
【0047】
以下に本発明をより具体的に説明する。
【0048】
<本発明の第一>
本発明の第一は、
下記化学式1:
【0049】
【化4】
【0050】
ただし、R1、R2、R3、R4、およびR5は、それぞれ独立して、水素原子またはスルホン酸基であり、この際R1、R2、R3、R4、およびR5の少なくとも一つがスルホン酸基である、
で示される化合物またはその塩を、亜硝酸塩を用いてニトロソ化する、ニトロソ化工程と;
前記ニトロソ化工程の後、
下記化学式2:
【0051】
【化5】
【0052】
ただし、R1、R2、R3、R4およびR5は、化学式1の定義と同様である、
で示される化合物またはその塩を単離する、単離工程と;
前記単離工程により得られた化学式2で示される化合物またはその塩を、亜鉛を用いて還元する、還元工程と;
を含む、
下記化学式3:
【0053】
【化6】
【0054】
ただし、R1、R2、R3、R4およびR5は、化学式1の定義と同様である、
で示される酸化発色化合物またはその塩の製造方法である。
【0055】
上記の通り、従来、所望の発色化合物を得ることができないのは、ニトロソ化により生じるニトロソ体が非常に不安定ではないかとの予測の下に鋭意研究を行った。
【0056】
その結果、中間体であるニトロソ体(化学式2で示される化合物またはその塩)は不安定であり、それが、所望の発色化合物の収率減少や純度低下の一因となっていた。
【0057】
さらに研究を進めていく過程で、本発明の製造方法により製造される発色化合物は、従来のアミノアンチピリンと比較して極めて水溶性が高いため、従来の方法によっては、高純度かつ高収率で所望の酸化発色化合物を得ることができないとの知見も見出した。
【0058】
よって、ニトロソ化反応、還元反応の条件および精製法等に、様々な技術的工夫を行うことにより、従来法に比較して、より効率的に高純度の発色化合物を合成する方法を見出し、本発明を完成させたのである。
【0059】
以下、本発明を詳説する。
【0060】
[ニトロソ化工程]
ニトロソ化を行う対象化合物は、
下記化学式1:
【0061】
【化7】
【0062】
ただし、R1、R2、R3、R4、およびR5は、それぞれ独立して、水素原子またはスルホン酸基であり、この際R1、R2、R3、R4、およびR5の少なくとも一つがスルホン酸基である、で示される化合物またはその塩である。
【0063】
R1、R2、R3、R4およびR5のうち1つのみが、スルホン酸基である場合、好ましい部位は、呈色性の観点で、R2、R3またはR4である。また、R1、R2、R3、R4およびR5のうちの2つがスルホン酸基である場合、好ましい部位は、呈色性の観点で、R2、R4である。
【0064】
なお、スルホン酸基は、イオン性官能基であるが、これらの官能基は、遊離の状態であっても、または塩を形成していてもよい、すなわち、R1、R2、R3、R4およびR5は、スルホン酸基の塩であってもよい。
【0065】
この際、これらの酸基と塩を形成する対イオンは、無機イオンでもよいし、有機イオンでもよい。例えば、ナトリウムイオン、カリウムイオン、カルシウムイオン等の無機塩基由来の陽イオン;アンモニウムイオン;アルキルアミン類等の有機塩基由来の陽イオン;およびアミノ酸類等の両性化合物由来のイオンなどが挙げられる。なかでも、塩の形態である場合には、好ましくはナトリウム塩、カリウム塩、またはアンモニウム塩であり、より好ましくはナトリウム塩である。
【0066】
なお、後述する化学式2〜化学式5における「R1、R2、R3、R4およびR5」は、いずれも、上記化学式1において行った「R1、R2、R3、R4およびR5」の定義と同様である。よって、下記ではその説明を割愛する。
【0067】
なお、ここで、化学式1で示される化合物またはその塩を得る方法について下記に説明する。
【0068】
本発明の化学式1で示される化合物またはその塩の製造方法は、特に制限されず、公知の製造方法が同様にしてあるいは適宜修飾してあるいは適宜組み合せて適用できる。以下、本発明の化学式1で示される化合物またはその塩の製造方法の好ましい実施形態を説明するが、本発明は下記好ましい実施形態に限定されるものではない。
【0069】
すなわち、本発明の化学式1で示される化合物またはその塩の製造方法は、下記化学式4:
【0070】
【化8】
【0071】
で示される化合物[以下、「化合物4」とも称する]と、下記化学式5:
【0072】
【化9】
【0073】
で示される化合物[以下、「化合物5」とも称する]とを、反応させる工程[工程(A)]を有することが好ましい。
【0074】
化合物4と化合物5とを反応させる上記工程(A)において、化合物4及び5の混合比は、特に制限されない。好ましくは、化合物4及び5をそれぞれ等モル混合する、あるいは化合物4を化合物5に対してやや多めに混合することが好ましい。後者の場合、化合物4は、化合物5を1モルに対して、0.8〜2モル、より好ましくは1〜1.2モル程度の割合で、混合することが好ましい。また、化合物4と化合物5との反応条件は、これらの反応が進行する条件であれば特に制限されないが、例えば、反応温度は、好ましくは20〜200℃、より好ましくは、80〜150℃の温度であり、反応時間は、好ましくは0.5〜5時間、より好ましくは1〜1.5時間である。また、化合物4と化合物5との反応は、無溶媒下で行なってもよく、あるいは溶媒中で行なってもよい。後者の場合に使用できる溶媒としては、メタノール、エタノール、プロパノール等の、アルコール類;THF、ジエチルエーテル、ジオキサン等の、エーテル類;ピリジン、ジメチルホルムアミド、ジメチルスルホキシド等の溶媒が挙げられる。これらのうち、THFが好ましい。化合物4と化合物5との反応後は、反応混合物を適当な溶媒中に加えることによって生成物を固化させることによって、原料化合物と分離回収することができる。
【0075】
次に、上記工程(A)で得られた反応生成物を、メチル化する[工程(B)]ことによって、前記化学式1で示される化合物が得られる。
【0076】
上記工程(B)において、メチル化は、ヨウ化メチルや硫酸ジメチルなどのメチル化剤を用いて行なうことができる。ここで、メチル化剤としては、上記のものに限定されず、他の公知のメチル化剤も同様にして使用できる。この反応は適当な溶媒や塩基を用いてもよく、室温あるいは加熱下で行なわれる。より具体的には、使用できる溶媒としては、上記工程(A)で列挙したのと同様の溶媒が使用でき、これらのうち、メタノール、THFが好ましい。また、塩基としては、トリエチルアミン(TEA)、ジイソプロピルエチルアミン(DIEA)、4−(ジメチルアミノ)ピリジンなどが使用でき、これらのうち、TEAが好ましい。
【0077】
上記工程(B)において、メチル化剤の使用量は、上記工程(A)で得られた反応生成物を十分メチル化できる量であれば特に制限されないが、上記工程(A)で得られた反応生成物1モルに対して、好ましくは0.5〜5モル、より好ましくは1〜2モルである。反応条件もまた特に制限されないが、上記工程(A)で得られた反応生成物のメチル化反応を、好ましくは40〜200℃、より好ましくは100〜140℃の温度で、好ましくは1〜24時間、より好ましくは10〜20時間、行なう。
【0078】
また、化学式1で示される化合物またはその塩を得る他の方法としては、上記工程(A)で得られた反応生成物を、以下の通りメチル化する[工程(C)]ことによって行う方法が挙げられる。
【0079】
すなわち、上記工程(A)で得られた反応生成物と、硫酸ジメチルと、を混合し、無溶媒下で加熱反応させる工程を含む、下記化学式1で示される化合物またはその塩:
【0080】
【化10】
【0081】
で示される化合物またはその塩の製造方法である。
【0082】
工程(C)の製造方法のように、溶媒を用いずに(無溶媒で)合成できるメリットとしては、反応容量を少量化でき、反応容器容積あたりの仕込み可能量が大きいことがあげられる。すなわち、通常の反応より小型の反応装置でより多くの製造ができるので、大量合成に好都合である。また、塩基を用いずに合成することもできる。
【0083】
硫酸ジメチルは、市販のものを使用しても合成して使用してもよく、特に制限されることはない。
【0084】
上記工程(A)で得られた反応生成物に対する前記硫酸ジメチルの量は、1.0〜2.0倍モルであることが好ましく、1.0〜1.5倍モルがより好ましく、1.0〜1.3倍モルが特に好ましい。上記工程(A)で得られた反応生成物に対する硫酸ジメチルの添加量が、1.0倍モル未満だと、反応効率低下の虞れがあり、2.0倍モルを越えると副生成物の増大の虞れがある。
【0085】
本発明に係る硫酸ジメチルの添加の方法としては、滴下ロート、ピペットなどを用いることができるが、外気中の水分による硫酸ジメチルの加水分解を防ぐために滴下ロートを用いて反応系を密閉して行なうのが好ましい。また、室温、1atmにおいて上記工程(A)で得られた反応生成物と、硫酸ジメチルとを混合する際には、液体である硫酸ジメチルに完全に溶解させてもまたは溶解させなくてもよく、両者がどのような状態であろうが混合していればよい。
【0086】
工程(C)に用いられる反応容器としては、ガラス、ステンレス、ホーロー、テフロン(登録商標)製のフラスコが好ましく、摺合せのガラス製フラスコが特に好ましい。
【0087】
工程(C)に用いられる反応容器としては、なす型フラスコ、丸底フラスコ、平底フラスコ、三角フラスコ、セパラブルフラスコ、多口フラスコを用いることができるが、これらは、仕込み量や加熱効率を考慮して選択される。
【0088】
工程(C)に用いられる冷却器としては、通常の合成反応に用いられる還流冷却器を用いることができ、リービッヒ冷却器、玉入冷却器(アーリン氏タイプ)、ジムロート冷却器、蛇管冷却器(グラハムタイプ)が好ましく、リービッヒ冷却器、玉入冷却器が特に好ましい。
【0089】
また、工程(C)における加熱反応中、外気からの水分の混入を防ぐ為に、塩化カルシウム管、窒素バブラーを取り付けることが好ましい。
【0090】
工程(C)における製造方法における加熱反応の加熱方法としては、マントルヒーター、油浴、砂浴(バーナー直火)が好ましく、マントルヒーターが特に好ましい。必要に応じて、センサー付き温度コントローラーを用いることができる。
【0091】
工程(C)における加熱反応の温度としては、190〜260℃が好ましく、200〜250℃が特に好ましく、220〜240℃がさらに好ましい。
【0092】
上記加熱反応の温度が190℃未満だと、反応効率低下の虞れがあり、260℃超だと、副生成物の増大の虞れがある。
【0093】
工程(C)における加熱反応の反応時間としては、0.5〜5時間が好ましく、0.5〜3時間が特に好ましく、1〜2時間がさらに好ましい。
【0094】
上記加熱反応の反応時間が0.5時間未満だと、反応効率低下の虞れがあり、5時間超だと、副生成物の増大の虞れがある。
【0095】
工程(C)において、上記工程(A)で得られた反応生成物と、硫酸ジメチルとを混合し、無溶媒下で加熱反応させて得られた反応混合物をアルカリ水溶液に溶解させて反応混合溶液を得る工程と、前記反応混合溶液に酸を加えて固体を単離する工程とを含むことが好ましい。
【0096】
本発明に係る化学式(1)で示される化合物は、溶媒抽出による回収が困難であるため酸を添加することで強制的に沈殿するいわゆる“酸沈”により回収することが好ましい。これにより、最終目的物を高い純度で得られることができる。
これにより、化学式1で示される化合物またはその塩を高い純度で得られることができる。
【0097】
本発明に係る製造方法に用いられるアルカリ水溶液のアルカリとしては、水酸化ナトリウム、水酸化カリウム、水酸化リチウムが好ましく、水酸化ナトリウムが特に好ましい。
【0098】
本発明に係る製造方法に用いられるアルカリ水溶液の濃度としては、10〜30w/v%(重量対体積%)が好ましく、15〜25w/v%が特に好ましく、20〜25w/v%がさらに好ましい。
【0099】
また、上記反応混合物をアルカリ水溶液に溶解させて得られる反応混合溶液のpHは、7〜14が好ましい。
【0100】
本発明に係る製造方法に用いられるアルカリ水溶液中のアルカリの量としては、使用する硫酸ジメチルに対して2〜5倍モルが好ましく、2.5〜4倍モルが特に好ましく、3倍モルがさらに好ましい。
【0101】
本発明に係る製造方法に用いられる酸としては、有機酸または無機酸のいずれでもよく、塩酸、硫酸、硝酸、酢酸、クエン酸、コハク酸、ピクリン酸、濃硫酸、濃硝酸などが用いられるが、塩酸、硫酸、硝酸がより好ましく、塩酸がさらに好ましい。
【0102】
本発明に係る製造方法において、上記反応物に添加する酸の濃度としては、なるべく水分量を減らすべく濃度が高いものが好ましい。
【0103】
上記反応物混合溶液に添加する酸の濃度は、5〜100w/v%(重量/体積%)が好ましく、15〜100w/v%がより好ましく、30〜100w/v%が特に好ましい。例えば、添加する酸として塩酸を用いる場合は、15〜36w/v%が好ましく、30〜36w/v%が特に好ましい。
【0104】
上記反応物混合溶液に添加する酸の量は、1〜10倍モルが好ましく、1〜5倍モルがより好ましく、1.5〜3倍モルが特に好ましい。
【0105】
上記反応物に添加する塩酸の量としては、出発物質の使用量に対して、1〜5倍モルが好ましく、1.5〜3倍モルが特に好ましく、1.8〜2.2倍モルがさらに好ましい。
【0106】
また、本発明に係る製造方法において酸を加え沈殿を生成させる工程で用いる攪拌装置としては、磁気スターラー、メカニカルスターラーなどを用いることができる。
【0107】
本発明に係る製造方法において、前記単離工程で得られた固体を水から再結晶する工程をさらに含むことが好ましい。すなわち、本発明の製造方法は、上記工程(A)で得られた反応生成物と、硫酸ジメチルを混合し、無溶媒下で加熱反応させて得られた反応混合物を得る工程と、アルカリ水溶液に溶解させて反応混合溶液を得た後、さらに前記反応混合溶液に酸を加えて固体(上記化学式1に示される化合物を含むもの)を単離する工程と、単離された前記固体を水から再結晶する工程とを含むことが好ましい。
【0108】
本発明に係る製造方法における再結晶で用いる水の量としては、固体100g当り、500ml〜3000mlが好ましく、1000ml〜2000mlが特に好ましく、1000ml〜1500mlがさらに好ましい。なお、当該単離された固体は、0.5〜24時間、室温条件下で減圧乾燥したものが使用される。
【0109】
上記再結晶工程で固体を水に溶解させる際に、加熱を行なってよい。加熱器具としては、水浴、油浴、マントルヒーター、ホットプレートなどが用いられる。加熱する際の加熱器具の温度設定は、50〜150℃が好ましく、70〜130℃が特に好ましく、90〜110℃がさらに好ましい。
【0110】
また上記再結晶のための放置時間は、30分〜3日が好ましく、1時間〜1日が特に好ましく、10時間〜15時間がさらに好ましい。さらに、上記再結晶のための放置時の温度は、0〜40℃が好ましく、0〜25℃が特に好ましく、0〜15℃がさらに好ましい。
【0111】
本発明に係る製造方法において使用される水は、特に制限されるものではなく、水道水、イオン交換水、純水、超純水、または工業用水などの水洗水を使用することができ、純水が特に好ましい。
【0112】
上記により得られた、前記化学式1で示される化合物またはその塩の純度の測定方法は、後述する。
【0113】
(亜硝酸塩)
前記化学式1で示される化合物またはその塩を、亜硝酸塩を用いてニトロソ化する。
【0114】
ニトロソ化工程に用いる亜硝酸塩は、特に制限はないが、亜硝酸のカチオン塩が用いられると好ましい。
【0115】
カチオン塩としても、特に制限はなく、アルカリ金属塩、アルカリ土類金属塩、アンモニウム塩などが好適に挙げられる。具体的には、ナトリウム塩、カリウム塩、リチウム塩等が好ましい。これらは、単独で用いてもよく、混合して用いてもよい。なお、入手のし易さの観点で、ナトリウム塩が特に好ましい。
【0116】
ニトロソ化工程に用いる亜硝酸塩の量は、化学式1で示される化合物またはその塩(本明細書中、単に「出発物質」とも称する。)の量に対して、反応効率および副生成物の観点で、1.0〜2.0倍モルが好ましく、1.0〜1.5倍モルがより好ましく、1.0〜1.2倍モルがさらに好ましい。
【0117】
(酸)
前記ニトロソ化工程におけるニトロソ化反応は、反応性の観点で、酸の存在下において行うとよい。
【0118】
ニトロソ化工程に用いられる酸は、特に制限はないが、反応性の観点で、塩酸、硫酸、硝酸、濃塩酸、濃硫酸、濃硝酸、酢酸、クエン酸、コハク酸、ピクリン酸が好ましい。これらは、単独で用いてもよく、混合して用いてもよい。なお、取り扱いの観点で、塩酸が特に好ましい。
【0119】
ニトロソ化工程に用いられうる酸の量にも、特に制限はないが、出発物質の量に対して、反応効率および副生成物の生成の観点で、0.5〜2.0倍モルが好ましく、1.0〜1.5倍モルがより好ましく、1.0〜1.2倍モルがさらに好ましい。
【0120】
また、添加する酸は、副生成物の生成を抑える観点で、好ましくは0〜15℃、より好ましくは1〜10℃、さらに好ましくは1〜5℃にしておくとよい。なお、添加する酸を、後述する溶媒で希釈した場合も同様の温度に制御しておくとよい。
【0121】
(溶媒)
ニトロソ化工程において、出発物質と上記亜硝酸塩、酸を混合する場合、それぞれを予め溶媒に含有させておいてもよい。
【0122】
つまり、溶媒に、それぞれの原末(出発物質と亜硝酸塩)または原液(酸)を加えていく方法や、出発物質を溶媒に含有させた溶液(本明細書中、「出発物質の溶液」とも称する)に、亜硝酸塩、酸を溶媒に含有させた溶液(それぞれ、「亜硝酸塩の溶液」、「酸の溶液」とも称する。)を添加する方法を用いてもよい。
【0123】
好ましくは、反応効率の観点で、出発物質の溶液に、亜硝酸塩の溶液および酸の溶液を添加する方法である。つまり、酸の溶液と、亜硝酸塩の溶液とを、化学式1で示される化合物またはその塩の溶液に滴下して添加する。
【0124】
この際、添加する亜硝酸塩の溶液は、副生成物の生成を抑える観点で、好ましくは0〜15℃、より好ましくは1〜10℃、さらに好ましくは1〜5℃にしておくとよい。また、添加する亜硝酸塩の溶液の濃度は、反応性の観点で、好ましくは1〜10mol/l、より好ましくは1〜5mol/l、さらに好ましくは3〜5mol/lにしておくとよい。
【0125】
また、添加する酸の溶液の濃度は、反応性の観点で、好ましくは1〜10mol/l、より好ましくは1〜5mol/l、さらに好ましくは3〜5mol/lにしておくとよい。
【0126】
また、出発物質の溶液の濃度は、反応性の観点で、好ましくは0.1〜10mol/l、より好ましくは0.1〜5mol/l、さらに好ましくは0.5〜2mol/lである。
【0127】
また、予め出発物質の溶液を、副生成物を抑える観点で、好ましくは0〜15℃、より好ましくは0〜5℃、さらに好ましくは0〜3℃にしておくとよい。
【0128】
前記溶媒は、特に制限はないが、水、有機溶媒またはこれら混合物(混合溶媒)が用いられうる。なお、本明細書中、溶媒として「水」を主成分(好ましくは、全部)とした溶液の名称を「水溶液」とも称する。なお、本明細書中、「水を主成分とする」とは、全溶液中に水の占める割合が、10〜100v/v%であることを意味する。
【0129】
有機溶媒としては、特に制限はないが、アルコール、エーテル類、ケトン類、ジメチルホルムアミド、ジメチルスルホキシドなどが用いられる。
【0130】
アルコールとしては、メタノール、エタノール、n−プロパノール、イソプロパノールなどが好適に用いられうる。これらは、単独で用いてもよく、混合して用いてもよい。
【0131】
エーテル類としては、ジエチルエーテル、イソプロピルエーテル、テトラヒドロフラン(THF)、ジオキサン、ジフェニルエーテル、ベンジルエーテル及びtert−ブチルエーテルなどが好適に用いられうる。これらは、単独で用いてもよく、混合して用いてもよい。
【0132】
ケトン類としては、アセトン、メチルイソブチルケトン(MIBK)、メチルエチルケトン(MEK)及びシクロヘキサノンなどが好適に用いられうる。これらは、単独で用いてもよく、混合して用いてもよい。
【0133】
これらのうち、反応性の観点で、水単独または水とアルコールとの混合物が好ましく、水単独が特に好ましい。
【0134】
水および有機溶媒の混合溶媒を用いる際は、例えば、好ましくは水/有機溶媒(容量比)=0/100〜100/0、より好ましくは水/有機溶媒(容量比)=10/90〜100/0程度がよい。この際、有機溶媒の含有量が、混合溶媒中、50容量%を超えると、亜硝酸塩の析出の虞れもある。
【0135】
また、上記の通り、出発物質と、亜硝酸塩と酸とを混合させる方法にも特に制限はないが、反応性の観点で、亜硝酸塩の溶液と、酸の溶液とを、攪拌下で、出発物質の溶液に添加する方法が好ましい。さらには、添加する条件は、副生成物を抑える観点で、亜硝酸塩の溶液と酸の溶液を、ほぼ同じ速度で、同時に添加することが好ましい。
【0136】
ここで、「同時に添加する」とは、亜硝酸塩の溶液と、酸の溶液とを添加しているある時点が、一瞬でも互いに重複すればよいが、亜硝酸塩の溶液と、酸の溶液との添加を開始する時点と、亜硝酸塩の溶液と、酸の溶液との添加を終了する時点とが、実質的に同じであると好ましい。
【0137】
このように、「同じ速度で添加する」および「同時に添加する」ことで、亜硝酸を一定で連続的に発生させる効果を奏しうる。
【0138】
なお、この際の、亜硝酸塩の溶液と、酸の溶液の添加の方法にも特に制限はないが、ピペット、シリンジ、滴下ロートなどを用いて行なわれることが好ましい。また、これらの添加に要する時間は、グラムスケールの反応では、生成物の安定性の観点で、1〜10分が好ましく、3〜7分が特に好ましく、4〜6分がさらに好ましい。
【0139】
また、添加は、磁気攪拌子等で、攪拌しながら行うことが、好ましい。
【0140】
さらに、亜硝酸塩の溶液と、酸の溶液と、を添加した後、そのまま静置しておいてもよいが、反応が添加後、時間とともに進行するため、好ましくは、上述の磁気攪拌子等で、好ましくは1〜10分程度、より好ましくは3〜5分程度、激しく攪拌するとよい。この際、「激しく攪拌する」の攪拌速度にも特に制限はないが、好ましくは300〜1200rpm、より好ましくは600〜1000rpmである。
【0141】
(反応時間)
ニトロソ化工程における反応時間にも特に制限はないが、あまりに反応時間が短すぎると、反応が不十分で出発物質の残留が生じる虞れがある。一方で、あまりに反応時間が長過ぎると、生成したニトロソ体の分解が起こる虞れがある。
【0142】
反応時間は、亜硝酸塩の溶液と酸の溶液を添加する時間を含めて、グラムスケールの反応では、1〜20分が好ましく、5〜15分がより好ましく、7〜12分がさらに好ましい。
【0143】
(反応温度)
ニトロソ化工程において設定される反応温度としては、副生成物の生成を抑える観点で、0〜15℃が好ましく、1〜10℃がより好ましく、1〜5℃がさらに好ましい。かような温度条件にするためには、従来公知の知見を適宜参照して制御することができ、例えば、水浴や氷浴等を用いればよい。
【0144】
(反応容器)
ニトロソ化工程に用いられる反応容器の素材としても、特に制限はないが、ガラス製、ステンレス製、ホーロー製、テフロン(登録商標)製などが好適に例示でき、ガラス製が特に好ましい。また、反応容器の形状としても、特に制限はないが、なす型フラスコ、丸底フラスコ、平底フラスコ、三角フラスコ、セパラブルフラスコ、多口フラスコなどが挙げられる。
【0145】
上記ニトロソ化工程を経ることにより、
下記化学式2:
【0146】
【化11】
【0147】
ただし、R1、R2、R3、R4およびR5は、化学式1の定義と同様である、で示される化合物またはその塩を得ることができる。
【0148】
[単離工程]
本発明においては、上記ニトロソ化工程の後、化学式2で示される化合物またはその塩を単離する、単離工程を経る。つまり、本発明においては、ニトロソ化工程終了後、そのままワンポットで、一気に、後述の還元工程を行わず、単離工程を必須の工程として有する点に特徴の一つを有する。
【0149】
上記の通り、従来、所望の発色化合物を、高純度かつ高収率で得ることができないのは、ニトロソ化により生じるニトロソ体が非常に不安定ではないかとの予測の下に鋭意研究を行った。そして、その予測は正しく、中間体であるニトロソ体(化学式2で示される化合物またはその塩)は不安定であり、それが、収率減少や目的物の純度低下の一因となっていることを見出したのである。
【0150】
つまりは、中間体であるニトロソ体(化学式2で示される化合物またはその塩)を適切な精製を行わなければ、結局は、最終生成物たる化学式3で示される酸化発色化合物またはその塩の収率減少や純度低下という結果に直結するのである。
【0151】
一方で、ニトロソ化工程終了後、ワンポット(一気に)で、後述の還元工程に移行する条件の下で、化学式3で示される酸化発色化合物またはその塩の製造を試みると、中間体であるニトロソ体(化学式2で示される化合物またはその塩)の単離をすることが非常に困難なのである。つまりは、中間体であるニトロソ体(化学式2で示される化合物またはその塩)中に存在する副生成物の分離除去を行うことが困難なのである。
【0152】
その観点からして、上記ニトロソ化工程においては、中間体であるニトロソ体(化学式2で示される化合物またはその塩)の単離を容易に行わせしめるべく、最適なニトロソ化反応条件を詳細に検討する点も、非常に有用であると言えるのである。具体的には、本発明の製造方法におけるニトロソ化工程によれば、化学式2で示される化合物またはその塩のみが、沈殿分離するため、容易に単離精製することができる。また、上記にて、亜硝酸塩、酸を、それぞれ化学式1で示させる化合物またはその塩に対して、ほぼ等量で添加するのも、副生成物の生成をより抑え、ひいては化学式2で示される化合物またはその塩を容易に単離精製させるために他ならない。
【0153】
上記の通り、ニトロソ体(化学式2で示される化合物またはその塩)を固体として単離することによって、不純物が除去され、次の還元工程が効率的に進行するため、最終的に得られるアミノ体(化学式3で示される酸化発色化合物またはその塩)の純度が高くなり、また還元工程における収率も向上するのである。
【0154】
以下、単離工程における、ニトロソ体を単離する方法について詳説する。
【0155】
ニトロソ体(化学式2で示される化合物またはその塩)を単離する具体的方法としては、特に制限はないが、ニトロソ化工程終了後に得られた反応混合物を、濾過したり、遠心分離したりすることなどによって行なわれるとよい。なお、かようにして得られたものを、「ニトロソ化物固体」とも称する。
【0156】
この際の濾過方法や条件としては、特に制限ないが、ろ紙を用いた吸引ろ過によって行うことができる。
【0157】
なお、得られたニトロソ化物固体は、溶媒で洗浄することにより、さらに純度が高くなる。
【0158】
この洗浄に用いられうる溶媒は、特に制限はないが、有機溶媒、またはこれらの混合物が用いられる。これらのうち、ニトロソ体の安定性の観点で、有機溶媒は、アルコールが好ましい。アルコールとしても特に制限はなく、上記にて列挙した具体例が同様に妥当であるが、ニトロソ体の安定性・純度の観点で、メタノールが特に好ましい。
【0159】
この際、洗浄に用いるアルコールは、ニトロソ体の安定性の観点から、好ましくは−20℃〜10℃程度、より好ましくは0〜5℃程度に冷却しておくと良い。
【0160】
このように得られたニトロソ体(化学式2で示される化合物またはその塩)の固体は、減圧乾燥などによって乾燥することによって、さらに、保存安定性が向上する。この際の減圧乾燥方法や条件としても、特に制限ないが、真空ライン(好ましくは0〜10mmHg、より好ましくは1〜2mmHg)に接続して行うことができる。
【0161】
かようにして、化学式2で示される化合物またはその塩を単離することができる。
【0162】
[還元工程]
本発明における還元工程では、前記単離工程により得られた化学式2で示される化合物またはその塩を、亜鉛を用いて還元する。
【0163】
かような還元工程を経ることにより、
下記化学式3:
【0164】
【化12】
【0165】
ただし、R1、R2、R3、R4およびR5は、化学式1の定義と同様である、で示される酸化発色化合物またはその塩を製造することができる。
【0166】
上記の通り、ニトロソ化工程終了後、ワンポット(一気に)で、後述の還元工程に移行する条件下で、化学式3で示される酸化発色化合物またはその塩の製造を試みると、生成する無機塩(亜鉛等)の除去が困難である。
【0167】
本発明の製造方法によると、無機塩(亜鉛等)を水酸化物として分離除去できるため、精製が非常に容易であり、最終生成物たる化学式3で示される酸化発色化合物またはその塩の収率が有意に向上する。
【0168】
(亜鉛)
還元工程において、亜鉛を必須成分として用いる。ただし、亜鉛以外でも、本発明の効果を奏する範囲であれば、他の還元剤を用いてもよい。他の還元剤としては、ハイドロサルファイド塩、サルファイド塩などが挙げられる。
【0169】
還元工程において、亜鉛の量は、前記単離工程により得られた化学式2で示される化合物またはその塩に対して1〜30倍モルが好ましく、5〜10倍モルがより好ましく、7〜8倍モルがさらに好ましい。
【0170】
(酸)
還元工程における還元反応は、酸の存在下において行うとよい。
【0171】
還元工程に用いられる酸は、特に制限はなく、どのような種の酸でも用いうるが、反応性の観点で、塩酸、硫酸、硝酸、酢酸が好ましい。これらは、単独で用いてもよく、混合して用いてもよい。なお、取り扱いやすさの観点で、塩酸が好ましい。
【0172】
還元工程に用いられうる酸の量にも、特に制限はないが、前記単離工程により得られた化学式2で示される化合物またはその塩の量に対して、副生成物を抑える観点で、1〜30倍モルが好ましく、5〜15倍モルが特に好ましく、7〜9倍モルがさらに好ましい。
【0173】
(溶媒)
化学式3で示される酸化発色化合物またはその塩の製造のために、前記単離工程により得られた化学式2で示される化合物またはその塩と、亜鉛と、を用い、上記の通り、還元反応は、酸の存在下で行うとよいが、必要に応じ反応溶媒を用いるとよい。
【0174】
反応溶媒としては、特に制限はなく、上記ニトロソ化工程で説明した具体例が、同様に妥当である。
【0175】
これら具体例のうち、化学式2で示される化合物の安定性の観点で、アルコール単独が好ましく、中でもメタノール単独が特に好ましい。
【0176】
還元工程における溶媒の量は、前記単離工程により得られた化学式2で示される化合物またはその塩1gに対して、反応性の観点で、20〜100mLが好ましく、30〜70mLがより好ましく、30〜50mLがさらに好ましい。
【0177】
また、前記単離工程により得られた化学式2で示される化合物またはその塩と、亜鉛と酸とを混合させる方法にも特に制限はないが、反応性の観点で、亜鉛粉末と、酸の溶液とを、攪拌下で、単離工程により得られた化学式2で示される化合物またはその塩を溶媒に含有した溶液(本明細書中、「単離工程により得られた化学式2で示される化合物またはその塩の溶液」とも称する)に添加する方法が好ましい。
【0178】
より具体的には、単離工程により得られた化学式2で示される化合物またはその塩を、予め溶媒(この溶媒は、上記の通り、メタノールが特に好ましい。また、この際、この溶媒を、安定性の観点で、好ましくは−20〜10℃、より好ましくは−10〜5℃に冷却しておくとよい。)に含有させておき、攪拌しながら、酸を添加し、さらに添加すべき亜鉛を一度に添加することが好ましい。「一度に添加する」理由は、ニトロソ体が不安定なため、早く反応を終了させるためである。この際、酸と亜鉛の添加順序にも特に制限はないが、ニトロソ体の安定性の観点で、好ましくは、亜鉛を先に添加する。
【0179】
また、添加は、磁気攪拌子等で、攪拌しながら行うことが、好ましい。
【0180】
また、亜鉛、酸の溶液を添加した後、副生成物を抑える観点で、好ましくは、上述の磁気攪拌子等で、好ましくは5〜20分程度、より好ましくは5〜10分程度、攪拌するとよい。また、攪拌速度にも特に制限はないが、好ましくは100〜1200rpm、より好ましくは300〜700rpmである。
【0181】
(反応温度)
還元工程において設定される反応温度としては、−20〜35℃が好ましく、−15〜25℃が特に好ましい。かような温度条件にするためには、従来公知の知見を適宜参照して制御することができ、例えば、水浴や氷浴、ドライアイス−メタノール浴等を用いればよい。
【0182】
(反応容器)
還元工程に用いられる反応容器の素材としても、特に制限はなく、前記ニトロソ化工程で説明した具体例が同様に妥当である。
【0183】
また、反応容器の形状としても、特に制限はなく、前記ニトロソ化工程で説明した具体例が同様に妥当である。
【0184】
かような還元工程を経ることにより、前記化学式3で示される酸化発色化合物またはその塩を製造することができる。
【0185】
[亜鉛除去工程]
上記還元工程終了後、亜鉛を除去することが好ましい。
【0186】
上記還元工程終了後、亜鉛は、固体状および/または水溶性の亜鉛の形態で残留しうる。
【0187】
固体状で残留している亜鉛は、濾過によって除去することが好ましく、さらに反応溶液中に残留する水溶性の亜鉛の除去を行なうとよい(亜鉛除去工程)。かような亜鉛除去工程を経ることにより、より高純度かつ高収率で、化学式3で示される酸化発色化合物またはその塩を得ることができる。
【0188】
このような水溶性の亜鉛を除去する方法も特に制限されないが、例えば、以下の方法が挙げられる。
【0189】
すなわち、還元工程終了後の反応混合物を、濾過し、固体状で残留している亜鉛を除去した後、水を添加して洗浄を行う。その後、濾液と洗液をあわせ、濃縮する。そして、もとの化学式2で示される化合物の重量に対し、好ましくは5〜1000質量部、より好ましくは10〜100質量部、さらに好ましくは10〜50質量部となる水を添加して、濃縮物の水溶液を準備する。そして、該濃縮物の水溶液に、アルカリ性物質を添加することにより、水酸化亜鉛の形で残留亜鉛を析出させて、濾過によって除去することができる。
【0190】
かようにすることで、亜鉛除去工程を経た反応混合物を得ることができる。
【0191】
(アルカリ性物質)
水酸化亜鉛を生成させるアルカリ性物質としては、特に制限はないが、水酸化塩が好ましく、より具体的には、水酸化ナトリウム、水酸化カリウム、水酸化リチウムなどが挙げられる。これらは、単独で用いてもよく、混合して用いてもよい。なお、中でも水酸化ナトリウムが特に好ましい。
【0192】
水酸化亜鉛を生成させるためのアルカリ性物質は、水溶液の形態(アルカリ性物質水溶液)で添加することができる。添加するアルカリ性物質水溶液の濃度は、残留する亜鉛濃度の観点から、1〜10規定濃度が好ましく、3〜5規定濃度がさらに好ましい。
【0193】
なお、添加するアルカリ性物質の量は、残留亜鉛の除去と、化学式3で示される化合物の安定性の観点から、アルカリ性物質を添加した後、pHが、8.0〜10.0になるよう設定するのが好ましく、8.5〜9.5になるよう設定するのが特に好ましい。
【0194】
[抽出精製工程]
上記亜鉛除去工程を経た後、亜鉛除去工程を経て得られた反応混合物から、さらに残留する無機塩を除くため、抽出精製工程を経ることが好ましい。かような抽出精製工程を経ることにより、より高純度かつ高収率で、化学式3で示される酸化発色化合物またはその塩を得ることができる。
【0195】
抽出精製工程は、亜鉛除去工程を経て得られた反応混合物を固体状態とし、溶媒で抽出する方法が用いられうる。抽出精製工程における抽出方法は、反応混合物を粉末状態として溶媒を加えて攪拌する他、一般に用いられる抽出装置を使用して行なってもよい。
【0196】
亜鉛除去工程を経て得られた反応混合物を固体状態にする方法にも特に制限はないが、例えば、減圧乾燥方法等により蒸発乾固する方法が挙げられる。
【0197】
この際の減圧乾燥方法や条件としても、特に制限ないが、真空ライン(好ましくは0〜30mmHg、より好ましくは1〜2mmHg)に接続して行うことができる。
【0198】
抽出精製工程における溶媒としては、最終目的物(化学式3で示される酸化発色化合物またはその塩)を溶解できるものであれば特に制限されないが、アルコールが好ましく、より具体的には、メタノール、エタノール、n−プロパノール、イソプロパノールなどが好適に使用できる。これらは、単独で用いてもよく、混合して用いてもよい。なお、抽出効率の観点で、エタノールが特に好ましい。
【0199】
また、本発明の製造方法により得られる化学式3で示される酸化発色化合物またはその塩は、両性でありうるため、単離をすること自体も困難である。しかしながら、上記の通り、ニトロソ化物の単離(不純物除去)、残留亜鉛の除去、エタノールによる無機塩(NaCl)の分離などの工夫を行うため、容易に単離を行うことができる。
【0200】
本発明の製造方法によると、従来法では50%程度であった純度が、少なくとも80%以上、実際には95%以上に向上する。
【0201】
この点においても、本発明は非常に優れていると言える。
【0202】
なお、目的物の純度は以下の方法で調べることができる。
【0203】
【表1】
【0204】
標品と同時に展開を行い、目視で反応進行度合いの確認に用いられる。
【0205】
【表2】
【0206】
全ピーク面積の和のうち目的物のピーク面積の%を計算し、純度とした。
【0207】
<本発明の第二>
本実施形態では、本発明の製造方法により製造された酸化発色化合物またはその塩、つまり、化学式3で示される酸化発色化合物またはその塩(本明細書中、「本発明の酸化発色化合物/塩」とも称する)の特に好ましい用途・使用に関する実施形態を説明する。
【0208】
本発明の酸化発色化合物/塩は、血液や尿をはじめとする体液中の生体成分の測定法の一つである、酵素を用いた比色分析法に好適に使用される。特に2分子で機能する酸化型発色試薬のうちのカプラーとして用いられうる。すなわち、本発明の酸化発色化合物/塩は、例えば、前記背景技術の項で記載したトリンダー試薬と共に用いられ、被測定物質の酵素的酸化で生じる過酸化酸素をさらにPOD酸化して生成した活性酸素によって、2分子が酸化縮合することによって呈色する。
【0209】
すなわち、本発明の本発明の第二は、本発明の酸化発色化合物/塩と、トリンダー試薬と、測定対象物に対して選択的に作用するオキシダーゼと、ペルオキシダーゼと、を含む試薬組成物(本明細書中、「本発明の試薬組成物」とも称する)である。
【0210】
つまり、本発明の酸化発色化合物/塩は、カプラーとして使用することが特に好ましく、それ以外については従来公知の知見が適宜参照され、特に限定されるものではない。
【0211】
(トリンダー試薬)
トリンダー試薬としては、特に制限はなく、従来公知の化合物がいずれも好ましく用いられうる。ただ、中でも中でもN−エチル−N−(3−スルホプロピル)−3−メチルアニリン、N−エチル−N−(3−メチルフェニル)−3−アセチルエチレンジアミン、N−エチル−N−(3−メチルフェニル)−N−サクシニルエチレンジアミン、N−エチル−N−(2−ヒドロキシ−3−スルホプロピル)−3−メチルアニリン、N−(2−カルボキシエチル)−N−エチル−3−メチルアニリン、N,N−ビス−(4−スルホブチル)−3−メチルアニリン、N−エチル−N−(2−サクシニルアミノエチル)−3−メチルアニリン、N−エチル−N−(3−スルホプロピル)−3−メトキシアニリン、N−(2−ヒドロキシ−3−スルホプロピル)−3,5−ジメトキシアニリン、N−エチル−N−(2−ヒドロキシ−3−スルホプロピル)−3,5−ジメトキシアニリン、N−エチル−N−(2−ヒドロキシ−3−スルホプロピル)−3,5−ジメトキシ−4−フルオロアニリン、N−エチル−N−(2−ヒドロキシ−3−スルホプロピル)−3,5−ジメチルアニリンが好ましく、N−エチル−N−(2−ヒドロキシ−3−スルホプロピル)−3−メチルアニリン、N−エチル−N−(2−ヒドロキシ−3−スルホプロピル)−3,5−ジメチルアニリンが、発色強度や波長といった観点で、特に好ましい。
【0212】
(測定対象物に対して選択的に作用するオキシダーゼ)
「測定対象物に対して選択的に作用するオキシダーゼ」としては、例えば、測定対象物がグルコースである場合、該オキシダーゼはグルコースオキシダーゼであり、測定対象物がコレステロールである場合、該オキシダーゼはコレステロールオキシダーゼであり、測定対象物が尿酸である場合、該オキシダーゼはウリカーゼであり、測定対象物がピルビン酸である場合、該オキシダーゼはピルベートオキシダーゼである。これらは測定対象物の基質のみを特異的に酸化するために、色々な成分を含む検体からそれぞれの測定対象物のみを限定して定量することができる。
【0213】
本発明に係る試薬組成物において、本発明の酸化発色化合物/塩とトリンダー試薬とのモル比は、1:1〜2:1であることが好ましく、1.3:1がより好ましい。本発明の酸化発色化合物/塩とトリンダー試薬とのモル比が1:1未満である場合、また、本発明の酸化発色化合物/塩とトリンダー試薬とのモル比が2:1を超える場合、発色不良を生じることがある。
【0214】
本発明に係る試薬組成物中の、測定対象物に対して選択的に作用するオキシダーゼの含有量は、カプラー15mmolに対して、433k〜2600kUであることが好ましく、866k〜2600kUであることがより好ましい。測定対象物に対して選択的に作用するオキシダーゼの含有量が、カプラー15mmolに対して433kU未満である場合、発色不良を生じることがある。なお、本発明において、前記測定対象物に対して選択的に作用するオキシダーゼの含有量は、4AA−TOOS法により測定したユニット数を採用するものとする。
【0215】
本発明に係る試薬組成物中のペルオキシダーゼの含有量は、カプラー15mmolに対して166.5k〜1666kUであることが好ましく、333k〜999kUであることがより好ましい。ペルオキシダーゼの含有量が、カプラー15mmolに対して166.5kU未満である場合、発色不良を生じることがある。なお、本発明において、前記ペルオキシダーゼの含有量は、ピロガロール法により測定したユニット数を採用するものとする。
【0216】
本発明に係る試薬組成物は、本発明の酸化発色化合物/塩と、トリンダー試薬と、測定対象物に対して選択的に作用するオキシダーゼと、ペルオキシダーゼと、を含む。すなわち、「本発明の酸化発色化合物/塩と、トリンダー試薬と、測定対象物に対して選択的に作用するオキシダーゼと、ペルオキシダーゼ」以外の他の成分を、試薬組成物としての機能を害さない程度含んでいてもよい。該成分としては、例えば、pH緩衝剤、浸透圧調整剤、界面活性剤、可溶化剤、安定化剤、保護剤等が挙げられる。
【0217】
なお、これら他の成分の含有量は、試薬組成物としての機能を害さなければ特に制限されないが、本発明の酸化発色化合物/塩と、トリンダー試薬と、測定対象物に対して選択的に作用するオキシダーゼと、ペルオキシダーゼとの総質量に対して、好ましくは10〜30質量%、より好ましくは13〜20質量%含まれてもよい。
【0218】
本発明に係る酸化発色化合物/塩は、血液や尿をはじめとする体液中の生体成分の測定法の一種である、酵素を用いた比色分析法に好適に使用されるカプラーとして用いられうる。すなわち、本発明に係る酸化発色化合物/塩は、前記トリンダー試薬と共に用いられ、測定対象物の酵素的酸化で生じる過酸化酸素を、さらにペルオキシダーゼ(POD)酸化して生成した活性酸素によって、本発明に係る酸化発色化合物/塩を含むカプラーと、トリンダー試薬と、が酸化縮合することによって呈色する。そのため本発明の酸化発色化合物/塩は、上記の通り、その化合物自体に特徴を有するものであるが、カプラーとして使用することが特に好ましい。
【0219】
上述の通り、本発明の酸化発色化合物/塩は水溶性が高いため、それを含む試薬組成物(本明細書中、「本発明に係る試薬組成物」とも称する)や、該試薬組成物を担体に保持して試験具として使用する場合には、血液などの検体にすばやく溶解して均一化し、迅速に呈色反応が起こる点、非常に優れたものである。また、本発明に係る試薬組成物を用いた呈色反応(比色分析法)において、それによって生じた色素化合物についても、検体への溶解性や親和性が高く、担体内での展開性が良くなるので、担体表面での発色が鮮やかで均一性の高いものとなり、感度が高く安定した測定が可能となる。さらには、本発明に係る酸化発色化合物/塩は、溶解性が高いので、従来の試薬より高濃度の試薬組成物液(試薬液)が調製可能である。すると、該試薬組成物を担体に担持して、試験具として使用する場合、塗布液濃度を高くしてより多くの試薬を担体に保持させることが可能となる。加えて、本発明の酸化発色化合物/塩は親水性が向上し分子量が高くなっている為、保管中の昇華による試薬組成物の損失を抑えることができる。
【0220】
また、本発明に係る酸化発色化合物/塩は、試薬濃度の変化における呈色強度への影響が少ない為に、化合物が経時的に劣化減少した場合でも測定値への影響が少ない。さらに、本発明に係る試薬組成物に含まれる本発明の酸化発色化合物/塩の親水性官能基は、担体への吸着性を高め、試験具として使用した場合でも、担体内での化合物の安定性を向上させる。このように本発明に係る試薬組成物を提供することにより、ひいては、経時劣化を低減させ有効期間が延びた試験具を提供することが可能となる。
【0221】
<本発明の第三>
上記本発明の第二の通り、本発明の酸化発色化合物/塩は、測定対象物に対して選択的に作用するオキシダーゼと、トリンダー試薬と、を組み合わせて、本発明に係る試薬組成物となる。
【0222】
本発明の酸化発色化合物/塩は、前記トリンダー試薬と、測定対象物に対して選択的に作用するオキシダーゼと、ペルオキシダーゼと、組み合わせて、液体、粉末、錠剤、もしくは担体に保持した状態で分析用組成物として用いられうる。
【0223】
これら分析用組成物には、上記に加えて、pH緩衝剤、浸透圧調整剤、界面活性剤、可溶化剤、安定化剤、保護剤等の成分を含んでいてもよい。
【0224】
なお、これら成分の含有量は、試薬組成物としての機能を害さなければ特に制限されないが、本発明に係る酸化発色化合物またはこれらの塩と、トリンダー試薬と、測定対象物に対して選択的に作用するオキシダーゼと、ペルオキシダーゼとの総重量に対して、好ましくは10〜30質量%、より好ましくは13〜20質量%含まれてもよい。
【0225】
また、液体に保持して用いる場合には、検体と混合して反応させた後、色の変化を目視で判定する他、分光光度計で透過吸光度を測定してもよい。また、本発明に係る試薬組成物は、担体に保持した状態で試験具として用いてもよい。
【0226】
このように、本発明に係る試薬組成物は、種々の物に保持して用いることができるが、本発明に係る試薬組成物は、担体に保持した状態で試験具として用いることが好ましい。
【0227】
よって、本発明の第三は、本発明に係る試薬組成物が、担体に保持されてなる試験具である。
【0228】
かようなドライケミストリーで用いられる試験用具のように、担体に保持した状態で用いる場合には、これらの試薬組成物を含む層の他、これの機能を害さない範囲において、計量層、展開層、濾過層、保持層等を含んでいてもよい。このように担体に保持して用いる場合には、検体を付与した後、色の変化を目視で判定する他、分光光度計で反射吸光度を測定してもよい。測定値は予め作製した検量線を用いて測定対象物の量に換算することができる。
【0229】
(担体)
本発明に係る担体の素材としては、紙、布帛、高分子膜等の多孔質物質を用いることができるが、特に、発色性能といった点で、高分子膜が好ましい。
【0230】
上記高分子膜とは、高分子よりなる水不溶性の多孔質体である。高分子としては、ポリスルホン、ポリエーテルスルホン、セルロース、セルロースアセテート、硝酸セルロース、ポリアクリロニトリル、ポリアミド、芳香族ポリアミド、ポリカーボネート、ポリエチレンテレフタレート、ポリイミド、ポリエチレン、ポリプロピレン、ポリテトラフルオロエチレン、ポリフッ化ビニリデン、ポリ塩化ビニル、ポリビニルアルコールが挙げられる。これらの高分子は一般的に知られている製膜方法を用いて膜を形成することができる。これらの高分子膜の中でもポリスルホンまたはポリエーテルスルホンが、発色性能といった点で、特に好ましい。
【0231】
(担体に保持する方法)
本発明に係る試薬組成物を担体に保持する方法にも特に制限はないが、担体に適当な溶剤に溶解させた試薬組成物溶液を担体にコーティングする他、試薬組成物を含むマトリックス前駆体を成型して試験層を形成させる等の公知の方法が用いられうる。
【0232】
コーティングは、工業用に使用される一般的なコート法を用いることができるが、担体が多孔質の場合には、しばしば塗工直後の液移動や不均一な乾燥に起因するコートむらが問題となる。担体と塗工液の物性や、塗工乾燥の方法、機器、条件がこれらを支配する重要因子となりうる。このため、精密印刷法が有効な場合もある。
【0233】
塗工後の液移動や不均一な乾燥を低減する為には、少量の液を正確に計量して塗工するような精密印刷技術が適するが、この方法では高い試薬濃度の塗工液を作製できることが必須となる。
【0234】
例えば、4−アミノアンチピリン等の既存のカプラーは、測定に用いる種々の試薬の中でも最も溶解性が低く、これが高い濃度の塗工液を作製するときの問題点となっていた。しかしながら、本発明の化合物は溶解性が高く、濃度の高い塗工液を作製するに好都合である。
【0235】
試薬を担体に保持させた試験具では、検体付与後、検体の液体成分によってまず試薬が溶解し、混合して反応が起こり、色素化合物が生成し、通常試験具の検体付与面の反対側にある読み取り面に移動し、その色調変化を測定する、というステップを経て定量が行なわれる。このため、試薬の溶解性が高いことは、検体による均一な溶解、均一化、迅速な反応に有利であるだけではなく、生成する色素も溶解性に優れるために読み取り面への移動がスムースかつ均一性が高いというメリットもある。これらの特徴は、測定時間の短縮化、測定精度の向上、測定値ばらつきの低減に寄与する。したがって、本発明の酸化発色化合物/塩は高い溶解性を有するため、測定時間の短縮化、測定精度の向上、測定値ばらつきの低減に寄与できることが期待される。
【0236】
例えば、従来公知のカプラーである4−アミノアンチピリンを担体に保持させた試験具として使用した場合、経時的に劣化減少し測定値が上昇するという問題が生じる。減少による測定値上昇の原因としては、酵素反応や呈色反応の阻害ではなく化合物の濃度が高いほど他の試薬類の溶解や移動、また生成色素の流動性に何らかの障害を与えるのではないかと考えられている。しかしながら、本発明の化合物は4−アミノアンチピリンと比較して量が増減しても測定値への影響が少なく、4−アミノアンチピリンでみられた問題を改善できた。
4−アミノアンチピリン等の既存のカプラーは、ある担体に保持させた試験具として使用した場合、濃度変化が測定値に影響するという問題点がある。バルク担体にコートする際むらが生じた場合には、それを切り抜いて用いる各試験具間に感度差が生じる、あるいは保存中に劣化分解して量が変化した場合測定値に影響が出るということを示唆している。原因は、酵素反応や呈色反応の阻害ではなく、化合物の濃度が高いほど他の試薬類の溶解や移動、また生成色素の流動性に何らかの障害を与えるのではないかと考えられている。これに対して、本発明の酸化発色化合物/塩は4−アミノアンチピリンと比較して濃度の測定値への影響が少なく、これらの問題を改善できる。
【0237】
本発明の製造方法により得られる化学式3で示される酸化発色化合物またはその塩は、トリンダー試薬と組み合わせて酸化発色させた場合には、4−アミノアンチピリン等の既存のカプラーを用いた場合と比較して吸光度が大きくなり、測定感度が向上することができる。例えば、本発明の4−アミノ−1−(4−スルホフェニル)−2,3−ジメチル−5−ピラゾロン、4−アミノ−1−(3−スルホフェニル)−2,3−ジメチル−5−ピラゾロン等は、N−エチル−N−(2−ヒドロキシ−3−スルホプロピル)−3−メチルアニリン(TOOS)やN−エチル−N−(2−ヒドロキシ−3−スルホプロピル)−3,5−ジメチルアニリン(MAOS)と組み合わせて発色させた場合、その検量線の傾きは改善され、測定感度が向上されうる。
【0238】
上述の通り、本発明の製造方法により得られる化学式3で示される酸化発色化合物またはその塩は水溶性が高いため、それを含む試薬組成物を担体に保持しなる試験具(以下、単に、「本発明に係る試験具」とも称する)を使用する場合、本発明に係る試薬組成物は、血液などの検体にすばやく溶解して均一化し、迅速に呈色反応が起こるため、本発明に係る試験具は、非常に優れたものとして使用されうる。
【0239】
また、本発明に係る試験具を用いた場合、本発明に係る試薬組成物の呈色反応において生じた色素化合物についても、検体への溶解性や親和性が高く、担体内での展開性が良くなるので、担体表面での発色が鮮やかで均一性の高いものとなる。
【0240】
本願発明の製造方法により製造された化合物は、溶解性が有意に高いので、従来の試薬より高濃度の試薬液が調製可能である。また、担体に担持して使用する場合塗布液濃度を高くしてより多くの試薬を担体に保持させることが可能となる。さらには、塗布液の濃度を高く設定し全体液量を少なくできることで、塗工均一性の高い、グラビア印刷法、ドット印刷法、インクジェット印刷等の精密印刷法が使用できる。塗工液量が少なくなれば、乾燥時間が短縮され乾燥むらを低減させる効果もある。以上、十分な試薬を高い均一性で塗工できることにより、塗りむらなどに起因するばらつきを低減させることができ、高い精度の試験具を提供することができる。すなわち、本発明に係る試験具を用いると、感度が高く安定した測定が可能となる。さらには、本発明の酸化発色化合物/塩は、溶解性が高いので、従来の試薬より高濃度の試薬組成物液(試薬液)が調製可能である。すると、該試薬組成物を担体に担持して、試験具として使用する場合、塗布液濃度を高くしてより多くの試薬を担体に保持させることが可能となる。
【0241】
加えて、本発明の製造方法により得られる化学式3で示される酸化発色化合物またはその塩は、親水性が向上し分子量が高くなっている為、保管中の昇華による試薬組成物の損失を抑えることができる。
【0242】
また、本発明の酸化発色化合物/塩は、試薬濃度の変化における呈色強度への影響が少ない為に、化合物が経時的に劣化減少した場合でも測定値への影響が少ない。さらに、本発明に係る試薬組成物に含まれる本発明の酸化発色化合物/塩の親水性官能基は、担体への吸着性を高める。すなわち、本発明に係る試験具は、担体内での化合物の安定性を向上されている。
【0243】
このように本発明に係る試験具は、従来に比べ、経時劣化を低減させ有効期間が有意に延びたものといえる。
【実施例】
【0244】
次に実施例によって本発明を説明する。本発明の化合物は以下の実施例の製法で合成される。
【0245】
<実施例1>
1−(4−スルホフェニル)−2,3−ジメチル−5−ピラゾロンの合成
200gの1−(4−スルホフェニル)−3−メチル−5−ピラゾロンを1Lナスフラスコに入れ、89mLの硫酸ジメチルを滴下ロートから均一にかかるように室温下にて滴下した。硫酸ジメチルの滴下後、ナスフラスコ上部に塩化カルシウム管を接続した玉入冷却器を取り付けた。次いで、温度コントローラーのセンサー部分をマントルヒーター上に置き、ナスフラスコを装着した。設定温度を230℃にして加熱を開始した。1−(4−スルホフェニル)−3−メチル−5−ピラゾロンの粉末が完全に融解し内部が茶色透明の均一状態になるまで、約2時間加熱した。ナスフラスコを室温まで放冷後、氷冷下、水酸化ナトリウム水溶液(水酸化ナトリウム:113g+純水:500 mL)を徐々に添加した後、発熱、発煙が収まってから、70〜80℃の水浴上でガラス棒にて攪拌しながら反応混合物の固形物を溶解した。さらに、溶解後、pH試験紙で内容液が強アルカリ性(pH11以上)であることを確認した。
【0246】
次いで、上記反応混合物の固形物をアルカリ水溶液で溶解した反応物をサンプリングし、TLCでチェックを行なった。ナスフラスコ内容液中の黒色不溶物を吸引濾過し濾別した。氷冷下、得られた濾液に120 mLの塩酸を攪拌しながら添加し、pH試験紙で内容液が強酸性(pH1以下)であることを確認した。氷冷下、沈殿が生成するまで攪拌を続けた。生成した沈殿を吸引濾過し、ロート上で、純水500mL、アセトン500mLにて順次洗浄した。真空ラインに接続して、減圧乾燥を行ない、粗生成物174gを得た。粗生成物を2000mLナスフラスコに入れ、純水から再結晶して、純水、アセトンにて洗浄後、減圧乾燥して目的物119gを得た(収率:56%)。純度95%以上(HPLC)。
【0247】
【表3】
【0248】
<実施例2>
1−(4−スルホフェニル)−2,3−ジメチル−5−ピラゾロンの合成(高圧法)
6.75gの1−(4−スルホフェニル)−3−メチル−5−ピラゾロン(アルドリッチ社製 134163)、250mLの脱水メタノール(ナカライテスク社製 04100−25)、4.2mLのヨウ化メチル(関東化学社製 I0060)を、高圧反応装置〈耐圧硝子社製TPR−1型)シリンダー容器内に入れ、130℃で18時間加熱攪拌した。反応後、反応混合液を300mLのナスフラスコに注ぎロータリーエバポレーターにて40±3℃の水浴上で乾固するまで減圧濃縮を行なった。真空ラインに接続して室温にて30分間減圧乾燥を行なった。乾燥後、純水15mLを加え100〜110℃のオイルバス中で攪拌しながら内容物を完全に溶解させた。すり潰した塩化ナトリウム5.53gを加え、オイルバス上で完全に溶解させた。ナスフラスコをオイルバスから取り出し、室温で1時間攪拌した。茶色の沈殿が生成することを確認し、冷蔵庫に入れ、15時間以上放置した後、生成した沈殿を吸引濾過し、純水、アセトンで洗浄し、減圧乾燥して粗生成物を得た。粗生成物を純水から再結晶して、純水、アセトンにて洗浄後、減圧乾燥して目的物1.5gを得た(収率:21%)。所要時間3日、純度95%以上(HPLC)。
【0249】
【表4】
【0250】
<実施例3>
4−アミノ−1−(4−スルホフェニル)−2,3−ジメチル−5−ピラゾロンの合成
(1)「ニトロソ化工程」
4−ニトロソ−1−(4−スルホフェニル)−2,3−ジメチル−5−ピラゾロンの合成
実施例1または2で合成した1−(4−スルホフェニル)−2,3−ジメチル−5−ピラゾロン30.0g(0.111mol)を乳鉢で細かくすりつぶし、500mL三つ口フラスコに秤量し、磁気攪拌子を入れた。三つ口フラスコを磁気スターラー上に置いた氷浴(0〜3℃)中に浸し、氷冷した純水111mLを加え、10分程度攪拌した。
【0251】
攪拌下、2〜3℃に冷却した亜硝酸ナトリウム水溶液31mL(亜硝酸ナトリウム13.8g/純水50mL)(0.124mol)と、2〜3℃に冷却した4N塩酸31mL(0.124mol)と、を同時に、5分かけて滴下ロートから滴下した。滴下後、磁気スターラーで3分間激しく攪拌した。生成した沈殿を吸引濾過した。フィルターケーキ上に、2〜3℃に冷却した脱水メタノール約150mLをまんべんなくかけて洗浄し、真空ライン(<2mmHg)に接続して、室温にて、減圧乾燥した。
【0252】
その後、目的物(4−ニトロソ−1−(4−スルホフェニル)−2,3−ジメチル−5−ピラゾロンの合成)25gを得た。
【0253】
なお、1−(4−スルホフェニル)−2,3−ジメチル−5−ピラゾロンに対する収率は、75%であった。
【0254】
(2)「還元工程」
4−アミノ−1−(4−スルホフェニル)−2,3−ジメチル−5−ピラゾロンの合成
上記(1)で得られた4−ニトロソ−1−(4−スルホフェニル)−2,3−ジメチル−5−ピラゾロン4.0g(0.0134mol)を、500mLナスフラスコに秤量した。ナスフラスコに、磁気攪拌子を入れ、磁気スターラー上に置いた氷浴中に浸しクランプで固定した。
【0255】
それに、2〜3℃に冷却したメタノール156mLを入れ、3秒間攪拌して分散させた。亜鉛粉末6.6g(0.100mol)、2〜3℃に冷却した2N塩酸54mL(0.108mol)を加えて、5分間攪拌を続けた。反応後、ナスフラスコ中の内容物を吸引濾過して亜鉛残渣を濾別した。亜鉛残渣を、純水を用いてロート上で洗浄して、濾液と洗液をあわせ、ナスフラスコに入れた。ロータリーエバポレーターにて減圧濃縮を行ない、溶媒を留去した。130mLの純水を加えて濃縮物を懸濁させ、ビーカーに移して磁気攪拌子を入れ、磁気スターラー上に置いた氷浴中に浸し攪拌を行った。濃縮物のpHをpHメーターでモニターしながら、pHが9.0±0.5になるまで、攪拌下、4N水酸化ナトリウム水溶液をピペットで滴下した。
【0256】
生成した水酸化亜鉛の沈殿物を吸引濾過して水酸化亜鉛残渣を濾別した。水酸化亜鉛残渣を、純水を用いてロート上で洗浄して濾液と洗液をあわせ、ナスフラスコに入れた。ロータリーエバポレーターにて減圧濃縮を行ない、溶媒を留去して、真空ライン(<2mmHg)に接続して、室温にて減圧乾燥した。ナスフラスコ中の生成物にエタノール800mLを加え、磁気攪拌子を入れ磁気スターラーにて目的物を攪拌抽出した。ナスフラスコ内容物を吸引濾過して塩化ナトリウム残渣を濾別した。塩化ナトリウム残渣を、エタノールを用いてロート上で洗浄して濾液と洗液をあわせ、ナスフラスコに入れた。ロータリーエバポレーターにて減圧濃縮を行ない、溶媒を留去して、真空ライン(<2mmHg)に接続して、室温にて減圧乾燥した。目的物:3.7gを得た。
【0257】
なお、4−ニトロソ−1−(4−スルホフェニル)−2,3−ジメチル−5−ピラゾロン対する収率は、90%であった。
【0258】
2工程(「ニトロソ化工程」および「還元工程」の2工程)の収率は68%((1)×(2))であり、純度は、97%以上(HPLC)であった。
【0259】
【表5】
【0260】
<比較例1>
4−アミノ−1−(4−スルホフェニル)−2,3−ジメチル−5−ピラゾロンの合成
実施例1または2で得られた1−(4−スルホフェニル)−2,3−ジメチル−5−ピラゾロン 0.5gを0.4N塩酸10mLに溶解し、氷冷下40%亜硝酸ナトリウム水溶液0.35mLを加えて1分間攪拌し、亜鉛末500mgを加え、さらに室温で10分攪拌した。反応混合物をろ過後、濾液を濃縮乾固し、目的物0.41gを得た。収率は36%、純度は50%(HPLC測定により)であった。
【0261】
【表6】
【0262】
<実施例4>
1−(3−スルホフェニル)−2,3−ジメチル−5−ピラゾロンの合成
200gの1−(3−スルホフェニル)−3−メチル−5−ピラゾロンを1000mLナスフラスコに入れ、89mLの硫酸ジメチルを滴下ロートから均一にかかるように室温下にて滴下した後、ナスフラスコ上部に塩化カルシウム管を接続した玉入冷却器を取り付けた。温度コントローラーのセンサー部分をマントルヒーター上に置き、ナスフラスコを装着した。設定温度を230℃にして加熱を開始した。1−(3−スルホフェニル)−3−メチル−5−ピラゾロンの粉末が完全に融解し内部が茶色透明の均一状態になるまで、約2時間加熱した。ナスフラスコを室温まで放冷後、氷冷下、水酸化ナトリウム水溶液(水酸化ナトリウム:113g+純水:500 mL)を徐々に添加した後、発熱、発煙が収まってから、70〜80℃の水浴上でガラス棒にて攪拌しながら固形物を溶解した。溶解後、pH試験紙で内容液が強アルカリ性(pH11以上)であることを確認した。
【0263】
反応物をサンプリングし、TLCでチェックを行なった。ナスフラスコ内容液中の黒色不溶物を吸引濾過を行ない濾別した。氷冷下、得られた濾液に120 mLの塩酸を攪拌しながら添加し、pH試験紙で内容液が強酸性(pH1以下)であることを確認した。氷冷下、沈殿が生成するまで攪拌を続けた。生成した沈殿を吸引濾過し、ロート上で、純水500mL、アセトン500mLにて順次洗浄した。真空ラインに接続して、減圧乾燥を行ない、粗生成物180gを得た。粗生成物を2000mLナスフラスコに入れ、純水から再結晶して、純水、アセトンにて洗浄後、減圧乾燥して目的物125gを得た(収率:60%)。所要時間2日、純度95%以上(HPLC測定により)。
【0264】
【表7】
【0265】
以上のことから、比較例に比べて本発明の製造方法で製造された実施例1または実施例4では、同規模の反応装置ではるかに仕込み量が大きく出来ることにより効率化された。また、所要時間は1日、収率は38%程度向上している。
【0266】
<実施例5>
1−(3−スルホフェニル)−2,3−ジメチル−5−ピラゾロンの合成
6.75gの1−(3−スルホフェニル)−3−メチル−5−ピラゾロン(アルドリッチ社製 556890)、250mLの脱水メタノール(ナカライテスク社製 04100−25)、4.2mLのヨウ化メチル(関東化学社製 I0060)を、高圧反応装置〈耐圧硝子社製TPR−1型)シリンダー容器内に入れ、130℃で18時間加熱攪拌した。
【0267】
反応後、反応混合液を300mLのナスフラスコに注ぎロータリーエバポレーターにて40±3℃の水浴上で、乾固するまで減圧濃縮を行なった。さらに、真空ラインに接続して、室温にて30分間減圧乾燥を行なった。乾燥後、純水15mLを加え、100〜110℃のオイルバス中で攪拌しながら内容物を完全に溶解させた。これに、すり潰した塩化ナトリウム5.53gを加え、オイルバス上で完全に溶解させた。ナスフラスコをオイルバスから取り出し、室温で1時間攪拌した。ここで、茶色の沈殿が生成したことを確認し、冷蔵庫に入れ、15時間以上(一昼夜)放置した。
【0268】
放置後、生成した沈殿を吸引濾過し、純水、アセトンで洗浄し、減圧乾燥して粗生成物を得た。粗生成物を純水から再結晶して、純水、アセトンにて洗浄後、減圧乾燥して目的物1.4gを得た(収率:20%)。なお、純度は95%以上(HPLC)であった。
【0269】
【表8】
【0270】
<実施例6>
4−アミノ−1−(3−スルホフェニル)−2,3−ジメチル−5−ピラゾロンの合成
(1)「ニトロソ化工程」
4−ニトロソ−1−(3−スルホフェニル)−2,3−ジメチル−5−ピラゾロンの合成
実施例4または5で合成した1−(3−スルホフェニル)−2,3−ジメチル−5−ピラゾロン30.0g(0.111mol)を、乳鉢で細かくすりつぶし、500mL三つ口フラスコに秤量し、磁気攪拌子を入れた。三つ口フラスコを磁気スターラー上に置いた氷浴(0〜3℃)中に浸し、氷冷した純水110mLを加え、10分攪拌した。
【0271】
攪拌下、2〜3℃に冷却した亜硝酸ナトリウム水溶液31mL(亜硝酸ナトリウム13.8g/純水50mL)(0.124mol)と、2〜3℃に冷却した4N塩酸31mL(0.124mol)と、を同時に、5分かけて滴下ロートから滴下する。滴下後、磁気スターラーで3分間激しく攪拌した。生成した沈殿を吸引濾過した。フィルターケーキ上に、2〜3℃に冷却した脱水メタノール約150mLをまんべんなくかけて洗浄し、真空ライン(<2mmHg)に接続して、室温にて、減圧乾燥した。目的物25gを得た。
【0272】
なお、1−(3−スルホフェニル)−2,3−ジメチル−5−ピラゾロン対しての収率は、75%であった。
【0273】
(2)「還元工程」
4−アミノ−1−(3−スルホフェニル)−2,3−ジメチル−5−ピラゾロンの合成
上記(1)で得られた4−ニトロソ−1−(3−スルホフェニル)−2,3−ジメチル−5−ピラゾロン4.0g(0.01mol)を、500mLナスフラスコに秤量した。ナスフラスコに、磁気攪拌子を入れ、磁気スターラー上に置いた氷浴中に浸しクランプで固定した。
【0274】
それに、2〜3℃に冷却したメタノール160mLを入れ、3秒間攪拌して分散させた。亜鉛粉末6.6g(0.1mol)、2〜3℃に冷却した2N塩酸54mL(0.1mol)を加えて、5分間攪拌を続ける。反応後、ナスフラスコ中の内容物を吸引濾過して亜鉛残渣を濾別した。亜鉛残渣を、純水を用いてロート上で洗浄して、濾液と洗液をあわせ、ナスフラスコに入れた。ロータリーエバポレーターにて減圧濃縮を行ない、溶媒を留去した。130mLの純水を加えて濃縮物を懸濁させ、ビーカーに移して磁気攪拌子を入れ、磁気スターラー上に置いた氷浴中に浸し攪拌を行なった。濃縮物のpHをpHメーターでモニターしながら、pHが9.0±0.5になるまで、攪拌下、4N水酸化ナトリウム水溶液をピペットで滴下した。
【0275】
生成した水酸化亜鉛の沈殿物を吸引濾過して水酸化亜鉛残渣を濾別した。
【0276】
水酸化亜鉛残渣を、純水を用いてロート上で洗浄して濾液と洗液をあわせ、ナスフラスコに入れた。ロータリーエバポレーターにて減圧濃縮を行ない、溶媒を留去して、真空ライン(<2mmHg)に接続して、室温にて減圧乾燥した。
【0277】
ナスフラスコ中の生成物にエタノール800mLを加え、磁気攪拌子を入れ磁気スターラーにて目的物を攪拌抽出した。
【0278】
ナスフラスコ内容物を吸引濾過して塩化ナトリウム残渣を濾別した。
【0279】
塩化ナトリウム残渣を、エタノールを用いてロート上で洗浄して濾液と洗液をあわせ、ナスフラスコに入れた。ロータリーエバポレーターにて減圧濃縮を行ない、溶媒を留去して、真空ライン(<2mmHg)に接続して、室温にて減圧乾燥した。目的物:3.6gを得た(収率87%)。
【0280】
2工程(「ニトロソ化工程」および「還元工程」の2工程)の収率は65%であり、純度は、97%以上(HPLC)であった。
【0281】
【表9】
【0282】
比較例に比べて本発明の製造方法で製造された実施例では、純度は47%以上、収率は32%および29%向上している。また性状は、比較例で得られた化合物は吸湿性が高い粉末であり扱いにくいのに対し、実施例3および実施例6で得られた化合物は吸湿性が低い粉末で取扱いがしやすい。
【0283】
<実施例7>
試験片による性能評価
0.13Mコハク酸ナトリウム緩衝液(pH=5.0)100mL中に、実施例3で合成した4−アミノ−1−(4−スルホフェニル)−2,3−ジメチル−5−ピラゾロン 0.46g 、 N−エチル−N−(2−ヒドロキシ−3−スルホプロピル)−3,5−ジメチルアニリン 0.95g、Triton X−100 3.0g、グルコースオキシダーゼ 150K unit、ペルオキシダーゼ 250K unitを溶解して塗工液を作成した。ポリエーテルスルホン膜(テルモ株式会社製、膜厚130μm)に塗工液を塗布し、35℃で18時間乾燥を行なった。膜を1cm角の正方形に打ち抜き、試験片を得た。試験片を反射分光光度計検出部に固定した後、1μLの検体を試験膜上部より滴下し、10秒後の反射吸光度を測定した。ヘマトクリット値40、種々のグルコース濃度の血液で測定した値から検量線を作成し、定量感度を観察した。(図1)反射吸光度値はそれぞれの試験片に対して最高となる時間の値を用いた。図1中、x軸に反射吸光度、y軸に血糖値をとっているため、傾きがなだらかなほど感度が高いことを意味する。
図2から示されるように、本発明の製造方法により合成した中間体を用いて合成された4−アミノ−1−(4−スルホフェニル)−2,3−ジメチル−5−ピラゾロンを用いた試験片の検量線の傾きは、従来から広く使用されている4−アミノアンチピリン(4−AA)を用いた場合より改善され、測定感度を有意に向上させることが示される。
【図面の簡単な説明】
【0284】
【図1】実施例3における、本発明の酸化発色化合物/塩および4−アミノアンチピリンの反射吸光度値の時間変化を表した図である。
【特許請求の範囲】
【請求項1】
下記化学式1:
【化1】
ただし、R1、R2、R3、R4、およびR5は、それぞれ独立して、水素原子またはスルホン酸基であり、この際R1、R2、R3、R4、およびR5の少なくとも一つがスルホン酸基である、
で示される化合物またはその塩を、亜硝酸塩を用いてニトロソ化する、ニトロソ化工程と;
前記ニトロソ化工程の後、
下記化学式2:
【化2】
ただし、R1、R2、R3、R4およびR5は、化学式1の定義と同様である、
で示される化合物またはその塩を単離する、単離工程と;
前記単離工程により得られた化学式2で示される化合物またはその塩を、亜鉛を用いて還元する、還元工程と;
を含む、
下記化学式3:
【化3】
ただし、R1、R2、R3、R4およびR5は、化学式1の定義と同様である、
で示される酸化発色化合物またはその塩の製造方法。
【請求項2】
前記ニトロソ化工程において、亜硝酸塩の量が、化学式1で示される化合物またはその塩に対して1.0〜1.5倍モルである、請求項1に記載の製造方法。
【請求項3】
前記ニトロソ化工程におけるニトロソ化反応を、化学式1で示される化合物またはその塩に対して1.0〜1.5倍モルの酸を用いて行う、請求項1または2に記載の製造方法。
【請求項4】
前記ニトロソ化工程において、酸またはその水溶液と、亜硝酸塩の水溶液と、を同時に化学式1で示される化合物またはその塩の水溶液に滴下して添加する、請求項3に製造方法。
【請求項5】
前記還元工程において、亜鉛の量が、前記単離工程により得られた化学式2で示される化合物またはその塩に対して1〜30倍モルである、請求項1〜4のいずれか1項に記載の製造方法。
【請求項6】
前記還元工程における還元反応を、前記単離工程により得られた化学式2で示される化合物またはその塩に対して1〜30倍モルの酸を用いて行う、請求項1〜5のいずれか1項に記載の製造方法。
【請求項7】
前記還元工程を、反応溶媒を用いて行い、該反応溶媒が、アルコールである、請求項1〜6のいずれか1項に記載の製造方法。
【請求項8】
前記還元工程を、反応溶媒を用いて行い、該反応溶媒の量が、前記単離工程により得られた化学式2で示される化合物またはその塩1gに対して20〜100mLである、請求項1〜7のいずれか1項に記載の製造方法。
【請求項9】
前記還元工程後、反応混合物中に残留する水溶性の亜鉛を、水酸化亜鉛として除去する、亜鉛除去工程をさらに含む、請求項1〜8のいずれか1項に記載の製造方法。
【請求項10】
前記還元工程後、反応混合物からアルコールを用いて、化学式3で示される酸化発色化合物またはその塩を抽出精製する、抽出精製工程をさらに含む、請求項1〜9のいずれか1項に記載の製造方法。
【請求項11】
請求項1〜10のいずれか1項に製造方法で得られる、化学式3で示される酸化発色化合物またはその塩。
【請求項1】
下記化学式1:
【化1】
ただし、R1、R2、R3、R4、およびR5は、それぞれ独立して、水素原子またはスルホン酸基であり、この際R1、R2、R3、R4、およびR5の少なくとも一つがスルホン酸基である、
で示される化合物またはその塩を、亜硝酸塩を用いてニトロソ化する、ニトロソ化工程と;
前記ニトロソ化工程の後、
下記化学式2:
【化2】
ただし、R1、R2、R3、R4およびR5は、化学式1の定義と同様である、
で示される化合物またはその塩を単離する、単離工程と;
前記単離工程により得られた化学式2で示される化合物またはその塩を、亜鉛を用いて還元する、還元工程と;
を含む、
下記化学式3:
【化3】
ただし、R1、R2、R3、R4およびR5は、化学式1の定義と同様である、
で示される酸化発色化合物またはその塩の製造方法。
【請求項2】
前記ニトロソ化工程において、亜硝酸塩の量が、化学式1で示される化合物またはその塩に対して1.0〜1.5倍モルである、請求項1に記載の製造方法。
【請求項3】
前記ニトロソ化工程におけるニトロソ化反応を、化学式1で示される化合物またはその塩に対して1.0〜1.5倍モルの酸を用いて行う、請求項1または2に記載の製造方法。
【請求項4】
前記ニトロソ化工程において、酸またはその水溶液と、亜硝酸塩の水溶液と、を同時に化学式1で示される化合物またはその塩の水溶液に滴下して添加する、請求項3に製造方法。
【請求項5】
前記還元工程において、亜鉛の量が、前記単離工程により得られた化学式2で示される化合物またはその塩に対して1〜30倍モルである、請求項1〜4のいずれか1項に記載の製造方法。
【請求項6】
前記還元工程における還元反応を、前記単離工程により得られた化学式2で示される化合物またはその塩に対して1〜30倍モルの酸を用いて行う、請求項1〜5のいずれか1項に記載の製造方法。
【請求項7】
前記還元工程を、反応溶媒を用いて行い、該反応溶媒が、アルコールである、請求項1〜6のいずれか1項に記載の製造方法。
【請求項8】
前記還元工程を、反応溶媒を用いて行い、該反応溶媒の量が、前記単離工程により得られた化学式2で示される化合物またはその塩1gに対して20〜100mLである、請求項1〜7のいずれか1項に記載の製造方法。
【請求項9】
前記還元工程後、反応混合物中に残留する水溶性の亜鉛を、水酸化亜鉛として除去する、亜鉛除去工程をさらに含む、請求項1〜8のいずれか1項に記載の製造方法。
【請求項10】
前記還元工程後、反応混合物からアルコールを用いて、化学式3で示される酸化発色化合物またはその塩を抽出精製する、抽出精製工程をさらに含む、請求項1〜9のいずれか1項に記載の製造方法。
【請求項11】
請求項1〜10のいずれか1項に製造方法で得られる、化学式3で示される酸化発色化合物またはその塩。
【図1】

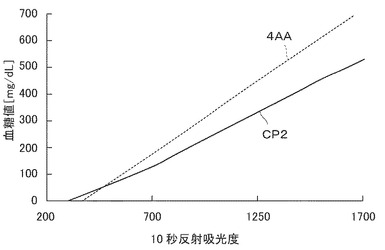
【公開番号】特開2009−242246(P2009−242246A)
【公開日】平成21年10月22日(2009.10.22)
【国際特許分類】
【出願番号】特願2008−87197(P2008−87197)
【出願日】平成20年3月28日(2008.3.28)
【出願人】(000109543)テルモ株式会社 (2,232)
【Fターム(参考)】
【公開日】平成21年10月22日(2009.10.22)
【国際特許分類】
【出願日】平成20年3月28日(2008.3.28)
【出願人】(000109543)テルモ株式会社 (2,232)
【Fターム(参考)】
[ Back to top ]