
金属表面処理用組成物、これを用いた金属表面処理方法及びこれらを用いた金属表面処理皮膜
【課題】金属材料、特に形状が複雑な金属構成体に対し、優れた耐食性及び塗膜外観を付与し得る皮膜を形成せしめることが可能な金属表面処理組成物を提供する。
【解決手段】水系樹脂を5〜30重量%、3価のBiイオンを100〜5000ppm及びBiイオンに対して0.1〜5倍モル濃度のアミノポリカルボン酸を含有し、水系樹脂の少なくとも一部が、下記式(1):

{ここで、R1及びR2は、相互に独立して、−(R)m−X(ここで、Rは、炭素数1〜6のアルキレン基であり、mは1か0であり、Xは、水素、カルボキシル、ヒドロキシル又はアシルである}で示される、Biイオンとキレートを形成可能な窒素含有基をポリマー骨格中に有するノニオン性及び/又はカチオン性樹脂であることを特徴とする金属表面処理用組成物。
【解決手段】水系樹脂を5〜30重量%、3価のBiイオンを100〜5000ppm及びBiイオンに対して0.1〜5倍モル濃度のアミノポリカルボン酸を含有し、水系樹脂の少なくとも一部が、下記式(1):
{ここで、R1及びR2は、相互に独立して、−(R)m−X(ここで、Rは、炭素数1〜6のアルキレン基であり、mは1か0であり、Xは、水素、カルボキシル、ヒドロキシル又はアシルである}で示される、Biイオンとキレートを形成可能な窒素含有基をポリマー骨格中に有するノニオン性及び/又はカチオン性樹脂であることを特徴とする金属表面処理用組成物。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、金属材料、特に形状が複雑な金属構成体に対し、優れた耐食性及び塗膜外観を付与し得る皮膜を形成せしめることが可能な金属表面処理組成物、これを用いた金属表面処理方法及びこれらを用いた金属表面処理皮膜に関するものである。
【背景技術】
【0002】
従来、各種金属材料、特に形状が複雑な金属構成体に対して優れた耐食性を付与するための手法としては、高い付き廻り性を有する電着塗装が一般的に用いられてきた。しかし、電着塗装によって得られる電着塗膜のみでは、所望の耐食性が得られない場合が多いため、電着塗装の前段には標準的にリン酸亜鉛系化成処理等の化成型の塗装下地処理が適用されていた。
【0003】
電着塗装は、アニオン性樹脂エマルジョンを含有する水性塗料中で被塗物をアノード電解することによって塗膜を析出させるアニオン電着塗装と、カチオン樹脂エマルジョンを含有する水性塗料中で被塗物をカソード電解することによって塗膜を析出させるカチオン電着塗装とに大別できるが、鉄系金属材料の耐食性向上に対しては、電解処理中に素地金属が塗料中に溶出する心配の無いカチオン電着塗装が有利であり、鉄系材料を主とする金属構成体である自動車車体、自動車部品、家電製品、建築材料等に対してはカチオン電着塗装が広く適用されている。
【0004】
カチオン電着塗装の市場での歴史は長く、かつてはクロム化合物や鉛化合物を配合することによって防錆性を確保していた。但し、これによっても防錆性は不充分であったため、リン酸亜鉛系化成処理等の下地処理が必須であった。現在では環境規制、特に欧州におけるELV規制によりクロム化合物や鉛化合物が実質使用できなくなったため、代替成分が検討され、ビスマス化合物にその効果が見出されており、具体的には次に挙げる特許文献が開示されている。
【0005】
特許文献1(特開平5−32919)には、ビスマス化合物をコーティングした顔料を少なくとも1種含有することを特徴とする電着塗料用樹脂組成物が開示されている。
【0006】
特許文献2(WO99/31187)には、有機酸変性ビスマス化合物が非水溶性の形態で存在する水性分散液を配合した水性分散ペーストからなることを特徴とするカチオン電着塗料組成物が開示されている。
【0007】
特許文献3(特開2004−137367)には、コロイド状ビスマス金属、及び、スルホニウム基とプロパルギル基とを持つ樹脂組成物からなることを特徴とするカチオン電着塗料が開示されている。
【0008】
特許文献4(特開2007−197688)には、水酸化ビスマス、ジルコニウム化合物及びタングステン化合物から選ばれる少なくとも1種の金属化合物の粒子を含んでなる電着塗料であって、該金属化合物が1〜1000nmであることを特徴とする電着塗料が開示されている。
【0009】
特許文献5(特開平11−80621)には、脂肪族アルコキシカルボン酸ビスマス塩水溶液を含有することを特徴とするカチオン電着塗料組成物が開示されている。
【0010】
特許文献6(特開平11−80622)には、2種以上の有機酸によるビスマス塩の水溶液であって、該有機酸の少なくとも1種が脂肪族ヒドロキシカルボン酸である有機酸ビスマス塩水溶液を含有することを特徴とするカチオン電着塗料組成物が開示されている。
【0011】
特許文献7(特開平11−100533)には、光学異性体のうちのL体が80%以上含まれる乳酸を用いてなる乳酸ビスマスを含有することを特徴とするカチオン電着塗料組成物が開示されている。
【0012】
特許文献8(特開平11−106687)には、2種以上の有機酸によるビスマス塩の水溶液であって、該有機酸の少なくとも1種が脂肪族アルコキシカルボン酸である有機酸ビスマス塩水溶液を含有することを特徴とするカチオン電着塗料組成物が開示されている。
【0013】
これらの特許文献は特許文献1〜4及び特許文献5〜8に大別できる。すなわち、特許文献1〜4は水性塗料に対して不溶性のビスマス化合物又は金属ビスマスを分散させたものであり、特許文献5〜8は少なくともビスマス化合物を固形分の残存が無くなるまで溶解させる、つまりBiイオンの状態にしてから塗料に添加することを特徴としている。
【0014】
しかしながら、これらの特許文献におけるビスマス化合物は、あくまでクロム化合物や鉛化合物の代替として作用するものであり、リン酸亜鉛系化成処理等の下地処理無しには充分な耐食性は得られない。事実、これらの特許文献ではリン酸亜鉛系化成処理との組合せを前提とした実施例のみが開示されている。
【0015】
一方、昨今ビスマス化合物以外の手法により耐食性を更に向上させ、リン酸亜鉛系化成処理等の下地処理を施さなくても、1コートにて充分な耐食性を確保し得る技術が検討されてきている。
【0016】
例えば特許文献9(特開2008−274392)には、金属基材に、皮膜形成剤を少なくとも2段階の多段通電方式で塗装することによって皮膜を形成する方法であって、(i)皮膜形成剤が、ジルコニウム化合物と、必要に応じて、チタン、コバルト、バナジウム、タングステン、モリブデン、銅、亜鉛、インジウム、アルミニウム、ビスマス、イットリウム、ランタノイド金属、アルカリ金属及びアルカリ土類金属から選ばれる少なくとも1種の金属(a)を含有する化合物とを合計金属量(質量換算)で30〜20,000ppmと、樹脂成分1〜40質量%とを含んでなり、(ii)金属基材を陰極として1段目の塗装を1〜50Vの電圧(V1)で10〜360秒間通電することにより行い、次いで、金属基材を陰極として2段目以降の塗装を50〜400Vの電圧(V2)で60〜600秒間通電することにより行い、そして(iii)電圧(V2)と電圧(V1)の差が少なくとも10Vであることを特徴とする表面処理皮膜の形成方法が開示されている。
【0017】
また、特許文献10(特開2008−538383)には、(A)希土類金属化合物、(B)カチオン基を有する基体樹脂、及び(C)硬化剤を含む水性塗料組成物であって、該水性塗料組成物に含まれる(A)希土類金属化合物の量が、塗料固形分に対して、希土類金属に換算して、0.05〜10重量%である水性塗料組成物に、被塗物を浸漬する、浸漬工程、該水性塗料組成物中において、被塗物を陰極として50V未満の電圧を印加する、前処理工程、及び該水性塗料組成物中において、被塗物を陰極として50〜450Vの電圧を印加する、電着塗装工程、を包含する、複層塗膜形成方法が開示されている。
【先行技術文献】
【特許文献】
【0018】
【特許文献1】特開平5−32919号公報
【特許文献2】WO99/31187号公報
【特許文献3】特開2004−137367号公報
【特許文献4】特開2007−197688号公報
【特許文献5】特開平11−80621号公報
【特許文献6】特開平11−80622号公報
【特許文献7】特開平11−100533号公報
【特許文献8】特開平11−106687号公報
【特許文献9】特開2008−274392号公報
【特許文献10】特開2008−538383号公報
【発明の概要】
【発明が解決しようとする課題】
【0019】
本発明者らは、これら従来技術について種々検討した結果、やはりリン酸亜鉛系化成皮膜等の前処理無しに充分な耐食性を付与する皮膜を金属材料の上に形成させるためには、Biの適用が最も効果的であるとの結論に達した。そしてBiの作用効果について再検討することとした。
【0020】
そして、Biの作用効果としては従来から、樹脂の硬化触媒としての機能と、素地金属の防食作用が注目されていたが、従来技術では、硬化触媒としての機能は望めるものの、金属の防食作用については極めて不充分であり、この作用を最大限に発揮させることこそ課題解決につながるものとして検討を進めた。
【0021】
素地金属の防食作用はBiが金属に接触する面、すなわち素地金属表面と皮膜の界面に存在しなくてはならないが、従来技術ではBi成分が皮膜中に均一に分散してしまい、耐食性を発揮するに充分なBiが素地金属表面に存在していないものと推定した。
【0022】
前述の如く特許文献1〜4は水性塗料に対して不溶性のビスマス化合物又は金属ビスマスを分散させたものであるが、このような組成物から皮膜を析出させた場合、他の顔料と同様、皮膜中にBiは均一に分散してしまう。
【0023】
特許文献5〜8は少なくともビスマス化合物を固形分の残存が無くなるまで溶解させる、つまりBiイオンの状態にしてから塗料に添加することを特徴としているが、Biの安定化剤である有機酸のキレート能力が微弱であるため、組成物に投入した際、Biは徐々に加水分解してしまい、酸化物又は水酸化物へと変化してしまうため、Biイオンとしての長期的な安定化は望めない。これによって、やはりBiは皮膜中に均一に分散してしまうのである。これらの特許文献において、やはりリン酸亜鉛系化成処理が下地処理として用いられていたのは、上記の推察を裏付けている。
【0024】
一方、特許文献9及び特許文献10は、素地金属上に無機系の皮膜を析出させた上に樹脂皮膜を積層させる技術であり、素地金属の防食の面で有利であるが、無機系の皮膜も樹脂皮膜もカソード電解による素地金属表面のpH上昇によって析出する機構であるため、積層皮膜の形成が容易でない。
【課題を解決するための手段】
【0025】
本発明者らは、上記の従来技術の課題を解決するために、Biイオンを組成物中でより安定に存在させるために、キレート能力の高いアミノポリカルボン酸を適用し、低電圧カソード電解にてBiを還元析出させ、次いで高電圧カソード電解でBiイオンの拡散が不充分になった段階で、かかるpH上昇によって樹脂が析出する反応機構を見出した。
【0026】
そして、これによって得られた皮膜は、Biの持つ樹脂の硬化触媒能はもちろん、素地金属表面により高濃度で存在するBiにより、素地金属の耐食性をも充分に向上し得ることを確認した。
【0027】
更に、本発明者らは、防錆効果をより高めるため、Bi濃度を高くすることを検討した。この際、Bi濃度を高くするにはキレート剤であるアミノポリカルボン酸濃度を高くすればよいが、この場合、過剰のアミノポリカルボン酸が塗膜に取り込まれると塗膜外観(具体的には光沢度)が低下するという別の問題を招く。以上の問題を解決することができる本発明は、次に示す(1)〜(10)である。
【0028】
本発明(1)は、水系樹脂を5〜30重量%、3価のBiイオンを100〜5000ppm及びBiイオンに対して0.1〜5倍モル濃度のアミノポリカルボン酸を含有し、
水系樹脂の少なくとも一部が、下記式(1):
【化3】
{ここで、R1及びR2は、相互に独立して、−(R)m−X(ここで、Rは、炭素数1〜6のアルキレン基であり、mは1か0であり、Xは、水素、カルボキシル、ヒドロキシル又はアシルである}で示される、Biイオンとキレートを形成可能な窒素含有基をポリマー骨格中に有するノニオン性及び/又はカチオン性樹脂であることを特徴とする金属表面処理用組成物(電解によって有機無機複合皮膜を析出させるための金属表面処理組成物)である。
【0029】
本発明(2)は、式(1)において、R1及びR2は、相互に独立して、水素、アルキル、ヒドロキシアルキル、カルボキシアルキル(−R−COOH:Rはアルキル基)又はアルキルカルボニル(−CO−R:Rはアルキル基)であり、ここで、アルキル、ヒドロキシアルキル、カルボキシアルキル及びアルキルカルボニルのアルキル部分は、炭素数1〜6の直鎖状、分岐鎖状又は環状である、前記発明(1)の金属表面処理用組成物である。
【0030】
本発明(3)は、水系樹脂が、変性エポキシ樹脂である、前記発明(1)又は(2)の金属表面処理用組成物である。
【0031】
本発明(4)は、変性エポキシ樹脂が、ビスフェノールA型であり、前記窒素含有基が、前記変性エポキシ樹脂中のグリシジルエーテル部ではなくフェニレン基のベンゼン環部に直接結合している、前記発明(3)の金属表面処理用組成物である。
【0032】
本発明(5)は、ビスフェノールA型の変性エポキシ樹脂(A)が、原料として、変性樹脂(B)、エポキシ当量180〜2500のエポキシ樹脂(C)及び前記窒素含有基を有するフェノール化合物(D)を用い、或いは更に2級アミノ基含有化合物(F)及び/又はビスフェノールA(E)をも用い、これらを反応させることで得られる変性エポキシ樹脂のアミノ化物である、前記発明(4)の金属表面処理用組成物である。
【0033】
本発明(6)は、化合物(D)が、下記式(2):
(式4)
(式中、R3は、前記窒素含有基又は水素原子であり、R3の少なくとも1個は前記窒素含有基であり;R4は、相互に独立して水素又は炭素数1〜2のアルキル基である)で示されるアミン付加フェノール化合物である、前記発明(5)の金属表面処理用組成物である。
【0034】
本発明(7)は、3価のBiイオンに対して、前記窒素含有基のモル濃度が0.1〜200倍である、前記発明(1)〜(6)のいずれか一つの金属表面処理用組成物である。
【0035】
本発明(8)は、前記発明(1)〜(7)のいずれか一つの組成物を用い、材料を陰極とした電解処理工程にて金属材料表面に塗膜を析出させる工程を含む、金属表面処理方法である。
【0036】
本発明(9)は、表面が清浄化された金属材料に対して電解処理を施す電解処理工程と、電解処理工程後に実行する水洗及び焼付け工程を含む、金属材料上に皮膜を析出させる金属表面処理方法において、
前記電解処理工程は、
前記発明(1)〜(7)のいずれか一つの組成物中に前記金属材料を浸漬させた状態で、電圧0〜15Vにて10〜120秒間電解する第一工程と、
前記発明(1)〜(7)のいずれか一つの組成物中に前記金属材料を浸漬させた状態で、電圧50〜300Vにて30〜300秒間電解する、前記第一工程の後に実施する第二工程と
を含み、ここで、前記第二工程は、前記第一工程に引き続いて同一浴内で実施するか又は前記第一工程とは異なる別浴内で実施することを特徴とする方法である。ここで、第一工程及び第二工程における「電圧X〜Y(V)」は、電圧X〜Yの範囲内で一定電圧を印加する態様でも又は経時的に印加電圧を変化させる態様でもよい。尚、第一工程における「電圧0〜15V」の下限値「0V」は、一定電圧での態様ではなく、経時的に印加電圧を変化させる態様における所定時の電圧を意味する。
【0037】
本発明(10)は、前記発明(8)又は(9)の方法によって形成される、金属Bi及び酸化BiがBiとして20〜500mg/m2付着し、全皮膜厚が5〜40μmであり、かつ皮膜厚の中心から金属材料側のBi付着量(G)が、全Bi付着量(H)に対して55%以上(G/H≧55%)となるBi付着分布であることを特徴とする金属表面処理皮膜である。
【図面の簡単な説明】
【0038】
【図1】第1図は、実施例1における皮膜のEPMA線分析プロファイルである。
【図2】第2図は、Alイオン濃度及びpHの適正範囲を示した図である。
【発明を実施するための最良形態】
【0039】
以下、本最良形態に係る金属表面処理用組成物、当該組成物を使用した金属表面処理方法及び当該方法により形成される金属表面処理皮膜を順に説明することとする。
【0040】
《金属表面処理組成物》
本発明に係る金属表面処理用組成物は、水系樹脂を5〜30重量%、3価のBiイオンを100〜5000ppm及びBiイオンに対して0.1〜5倍モル濃度のアミノポリカルボン酸を含有し、水系樹脂の少なくとも一部が所定の置換基を有するノニオン性及び/又はカチオン性樹脂であることを特徴とする。以下、本組成物を構成する各成分について詳述する。
【0041】
<組成物構成成分:水系樹脂>
本発明に係る水系樹脂は、好適には、ノニオン性樹脂及びカチオン性樹脂である。また、水系樹脂には、ブロック化ポリイソシアネートをはじめとする硬化剤を任意に配合することもできる。ここで、この水系樹脂の少なくとも一部は、下記式(1):
【化5】
{ここで、R1及びR2は、相互に独立して、−(R)m−X(ここで、Rは、炭素数1〜6のアルキレン基であり、mは1か0であり、Xは、水素、カルボキシル、ヒドロキシル又はアシルである}で示される、Biイオンとキレートを形成可能な窒素含有基をポリマー骨格中に有するノニオン性及び/又はカチオン性樹脂である。尚、本発明における水系樹脂とは、水分散するエマルジョンと水溶性樹脂の総称である。ここで、アシルにおけるRは、脂肪族でも芳香族でもよく、炭素数は1〜6であることが好適である。
【0042】
ここで、式(1)において、R1及びR2は、相互に独立して、水素、アルキル、ヒドロキシアルキル、カルボキシアルキル又はアルキルカルボニルであり、ここで、アルキル、ヒドロキシアルキル、カルボキシアルキル及びアルキルカルボニルのアルキル部分は、炭素数1〜6の直鎖状、分岐鎖状又は環状であることが好適である。これらの中で、キレート形成能の観点からO(酸素原子)を含有するものが好適であり、更に合成の容易性に鑑みるとヒドロキシアルキルが最適である。
【0043】
式(1)で示される窒素含有基は、ノニオン性及び/又はカチオン性樹脂のポリマー骨格中に存在する。ここで、ノニオン性樹脂及び/又はカチオン性樹脂は、いずれも特に限定されるものではなく、一例として、エポキシ樹脂、ウレタン樹脂、アクリル樹脂を挙げることができる。これらの中では、エポキシ樹脂が好適であり、変性エポキシ樹脂がより好適であり、ビスフェノールA型の変性エポキシ樹脂が特に好適である。
【0044】
ここで、樹脂にカチオン性を付与するには、典型的にはアミノ基を樹脂骨格中(特に末端)に導入する手法(例えば、エポキシ樹脂では、末端のグリシジル基にアミノ基含有化合物を付加する手法)が採用されている。尚、これについては後で詳述する。
【0045】
また、樹脂にノニオン性を付与するには、例えば、エチレンオキサイド基を含有する化合物も導入させることができる。これを導入することで、このエチレンオキサイド基がノニオン性の活性剤的働きを有することとなり、エマルション状態となったときの乳化安定性向上などといった効果が得られる。具体的には、ポリエチレングリコール、もしくはポリエチレングリコールジグリシジルエーテルを反応させることで、ノニオン性樹脂の性質を示すこととなる。また、ビスフェノールA型の変性エポキシ樹脂を用いる場合には、同じようなビスフェノールA構造を持つ、ビスフェノールAエチレンオキサイド付加物を用いることが好ましい。
【0046】
以下、カチオン性樹脂として特に好適である、ビスフェノールA型の変性エポキシ樹脂のアミノ化物について詳述する。
【0047】
(ビスフェノールA型の変性エポキシ樹脂のアミノ化物)
ここで、特に好適なビスフェノールA型の変性エポキシ樹脂(A)のアミノ化物について説明することとする。ビスフェノールA型の変性エポキシ樹脂は、原料として、変性樹脂(B)、エポキシ当量180〜2500のエポキシ樹脂(C)、前記窒素含有基を有するフェノール化合物(D)及び2級アミノ基含有化合物(F)を用い或いは更にビスフェノールA(E)をも用い、これらを反応させることで得られる変性エポキシ樹脂のアミノ化物である。以下、各成分について説明する。
【0048】
*ビスフェノールA型の変性エポキシ樹脂(A)の原料
まず、変性樹脂(B)としては、通常、ポリオール化合物が用いられる。これらは、エポキシ樹脂の可塑性向上などを目的として適用される。具体的には、ポリエステルポリオール、ポリエーテルポリオール、ポリウレタンポリオール、アクリルポリオール等のポリオール樹脂、末端にフェノールを付加し、水酸基を有する芳香族縮合化合物などが挙げられる。更に具体的には、ポリカプロラクトンジオール、ポリエチレングリコール、ポリプロピレングリコール、フェノール性水酸基を有するキシレンホルムアルデヒド樹脂等が挙げられる。これら化合物により変性を行うことは、これら化合物が有する水酸基とエポキシ樹脂のグリシジルエーテル部が容易に反応し得ることから、従来より用いられてきた技術である。変性エポキシ樹脂中において、これら変性樹脂は5〜30重量%含まれる。
【0049】
次に、エポキシ当量180〜2500のエポキシ樹脂(C)としては、塗膜の防食性等の観点から、特に、ポリフェノール化合物とエピハロヒドリン、例えば、エピクロルヒドリンとの反応により得られるエポキシ樹脂が好適である。中でも、ビスフェノールAとエピクロロヒドリンとの反応により得られるビスフェノールAジグリシジルエーテルが最適である。また、ビスフェノールAを基本構造として重合させたエポキシ樹脂も同様の効果を示し、エポキシ当量として180〜2500、好ましくは180〜2000、更に好ましくは180〜1500のものが最適である。変性エポキシ樹脂中において、これらエポキシ樹脂は5〜30重量%含まれる。
【0050】
次に、前記窒素含有基を有するフェノール化合物(D)としては、下記式(2):
(式6)
(式中、R3は、前記窒素含有基又は水素原子であり、R3の少なくとも1個は前記窒素含有基であり;R4は、相互に独立して水素又は炭素数1〜2のアルキル基である)で示されるアミン付加フェノール化合物であることが好適である。中でも、R3の付加数が1のものが好ましい。また、R4が炭素数1の場合、即ちビスフェノールA型のものが好ましい。変性エポキシ樹脂中において、これら窒素含有基を有するフェノール化合物は5〜30重量%含まれる。これらは、ジフェノール化合物に対し、カルボニル化合物と1級もしくは2級アミンとのマンニッヒ反応により合成させることで、得ることができる。この時のカルボニル化合物としてはホルムアルデヒドを用いることが一般的である。1級、2級のアミンとしては、アンモニア;モノメチルアミン、ジメチルアミン、モノエチルアミン、ジエチルアミン、モノイソプロピルアミン、ジイソプロピルアミン、モノブチルアミン、ジブチルアミン等のモノ−、もしくはジ−アルキルアミン;モノエタノールアミン、ジエタノールアミン、モノ(2−ヒドロキシプロピル)アミン、ジ(2−ヒドロキシプロピル)アミン、トリ(2−ヒドロキシプロピル)アミン、モノメチルアミノエタノール、モノエチルアミノエタノール等のアルカノールアミン;N−ホルミルメチルアミン、N−ホルミルエチルアミン、アセトアミド、N−メチルアセトアミド、N−エチルアセトアミド、プロピオン酸アミド、N−メチルプロピオンアミド、N−エチルプロピレンアミド、グリシン、サルコシン等のカルボキシル基あるいはアシル基を含有するアミン等が挙げられる。
【0051】
次に、ビスフェノールA(E)の含有量(添加量)は、前記窒素含有基を有するフェノール化合物(D)の含有量(添加量)との関係で任意の割合で比率を変えることができる。変性エポキシ樹脂中において、両者の合計量として5〜30重量%含まれる。
【0052】
次に、2級アミノ基含有化合物(F)は、エポキシ樹脂基体にアミノ基を導入して、該エポキシ樹脂をカチオン化するためのカチオン性付与成分であり、前記キレート能を期待するアミンとはその効果が異なる。ここで使用されるアミンはエポキシ基と反応する活性水素を少なくとも1個含有するものが用いられる。そのような目的で使用されるアミノ基含有化合物としては、例えば、モノメチルアミン、ジメチルアミン、モノエチルアミン、ジエチルアミン、モノイソプロピルアミン、ジイソプロピルアミン、モノブチルアミン、ジブチルアミンなどのモノ−、もしくはジ−アルキルアミン;モノエタノールアミン、ジエタノールアミン、モノ(2−ヒドロキシプロピル)アミン、ジ(2−ヒドロキシプロピル)アミン、トリ(2−ヒドロキシプロピル)アミン、モノメチルアミノエタノール、モノエチルアミノエタノールなどのアルカノールアミン;エチレンジアミン、プロピレンジアミン、ブチレンジアミン、ヘキサメチレンジアミン、テトラエチレンペンタミン、ペンタエチレンヘキサミン、ジエチルアミノプロピルアミン、ジエチレントリアミン、トリエチレンテトラミン等のアルキレンポリアミン及びこれらのポリアミンのケチミン化物;エチレンイミン、プロピレンイミンなどのアルキレンイミン;ピペラジン、モルホリン等の環状アミン等が挙げられる。変性エポキシ樹脂中において、2級アミノ基含有化合物(F)は0.5〜20重量%含まれる。
【0053】
*ビスフェノールA型の変性エポキシ樹脂のアミノ化物の製造方法
先ず、変性樹脂(B)、エポキシ樹脂(C)、前記窒素含有基を有するフェノール化合物(D)、ビスフェノールA(E)を所定量混合し、加熱撹拌を行う。加熱温度は70〜100℃が好ましい。各原料が溶解した後、触媒を添加し、加熱温度を上げ、合成を行う。触媒は、通常、ジメチルベンジルアミンのような3級アミンが使用される。合成温度は120℃〜150℃で制御するのが一般的である。
【0054】
合成温度と時間を調整することにより、所定のエポキシ当量を持ったエポキシ樹脂を合成できる。エポキシ当量はJIS K7236に定められるエポキシ当量測定によって算出される。この時のエポキシ当量は800〜10000が好適であり、800〜5000がより好適であり、800〜3000が最も好適である。エポキシ当量が大きくなるほど、エマルション作製時の乳化安定性が低下する傾向がある。
【0055】
次に、この合成した変性エポキシ樹脂に2級アミノ基含有化合物(F)を付加する。変性エポキシ樹脂を70℃〜110℃に保ちながら2級アミノ基含有化合物(F)を添加し、1〜3時間合成を行うことで、変性エポキシ樹脂のアミノ化物が得られる。
【0056】
<組成物構成成分:3価のBiイオン>
本発明でいうBiイオンとは、組成物中で固形化せず、完全に溶解状態になっているBi成分のことを指し、具体的には後述するアミノポリカルボン酸やポリマー中の窒素含有基によってキレートを構成し、安定的に水溶化された状態であることを意味している。
【0057】
<組成物構成成分:アミノポリカルボン酸>
アミノポリカルボン酸とは、分子中にアミノ基と複数のカルボキシル基を有するキレート剤の総称であり、具体的にはEDTA(エチレンジアミン四酢酸)、HEDTA(ヒドロキシエチルエチレンジアミン三酢酸)、NTA(ニトリロ三酢酸)、DTPA(ジエチレントリアミン五酢酸)、TTHA(トリエチレンテトラミン六酢酸)等が該当するが、Biイオンとのキレート安定度の観点からEDTA、HEDTA、NTAがより好ましい。
【0058】
<組成物構成成分:他の成分>
本発明の組成物には、更に必要に応じて顔料、触媒、有機溶剤、顔料分散剤、界面活性剤等、塗料分野で通常使用されている添加剤を適用することもできる。顔料としては、チタン白、カーボンブラック等の着色顔料、クレー、タルク、バリタ等の体質顔料、トリポリリン酸アルミニウム、リン酸亜鉛等の防錆顔料、ジブチル錫オキサイド、ジオクチル錫オキサイド等の有機錫化合物、ジブチル錫ラウレート、ジブチル錫ジベンゾエート等のジアルキル錫の脂肪酸もしくは芳香族カルボン酸塩などの錫化合物が挙げられる。また、後で詳述するように水系樹脂の析出を促進させる観点から、Alイオンを添加してもよい(特にノニオン系樹脂の場合には必須)。更には、本発明の組成物の中には、Biイオン、Alイオンの他に、Feイオン、Znイオン、Ceイオン等の金属イオンを含有しても、本発明の効果を損なうものではない。むしろ、これらの金属イオンには、Alイオンほどではないものの、水系樹脂の析出を促進させる作用を有する。尚、Feイオンは2価よりも3価がより好ましい。
【0059】
<組成物構成成分:液体媒体>
本発明に係る金属表面処理用組成物の液体媒体としては、水性媒体が好適であり、水がより好適である。尚、液体媒体が水である場合、液体媒体として水以外の他の水系溶媒(例えば、水溶性のアルコール類)を含有していてもよい。
【0060】
<組成物の組成>
次に、本発明に係る金属表面処理用組成物の組成について説明することとする。
【0061】
(水系樹脂)
まず、本発明に係る金属表面処理用組成物は、組成物の全重量を基準として水系樹脂を5〜30重量%含み、好適には10〜25重量%、より好適には10〜20重量%含む。ここで、全水系樹脂における式(1)で示されるノニオン性及び/又はカチオン性樹脂の比率(重量比)は、好適には30〜100%であり、より好適には70〜100%である。式(1)で示される樹脂の割合は、少ないほどキレート能が低下し、Biの安定性に寄与できないこととなる。また、全水系樹脂含有量が低過ぎると皮膜析出量が不足し、含有量が高過ぎると経済的に不利である。
【0062】
ここで、本発明に係る金属表面処理用組成物中での、式(1)で示される前記窒素含有基のモル濃度は、3価のBiイオンに対して、0.1〜200倍、0.5〜100倍であることがより好適であり、1.0〜50倍であることが更に好適である。モル濃度が低いほどキレート能が低下し、Biの安定性に寄与できない。モル濃度が高いほど、経済的に不利である。
【0063】
(3価のBiイオン)
次に、本発明に係る金属表面処理用組成物は、3価のBiイオンを100〜5000ppm含有する。500〜4000ppmが更に好ましく、1000〜3000ppmが最も好ましい。Biイオン濃度が低過ぎる場合、耐食性向上に必要な充分なBi付着量が得られず、高過ぎると組成物の電気伝導度が高くなり過ぎ、複雑な形状を有する金属材料への皮膜の付き廻り性が劣化すると共に、Bi付着量過多となり皮膜密着性を損なう恐れがある。組成物中のBiイオン濃度は、超遠心機により組成物を固液分離し、液相を高周波誘導結合プラズマ発光分光分析(ICP)もしくは原子吸光分光分析(AA)を用いて定量することができる。
【0064】
(アミノポリカルボン酸)
本発明に係る金属表面処理用組成物は、Biイオンに対して0.1〜5倍モル濃度でアミノポリカルボン酸を含有する。0.1〜4倍モル濃度が更に好ましく、0.1〜3倍モル濃度であることが最も好ましい。Biイオンに対する濃度比率が低過ぎるとBiイオンが組成物中で加水分解し、酸化物となってしまうため、有効なBiイオン濃度が低下し、結果として充分なBi付着量が得られなくなる。高過ぎると逆にBiイオンが安定化し過ぎ、やはり充分なBi付着量が得られなくなる。後述のように、過剰なアミノポリカルボン酸はカチオン性樹脂のゲル化を招くこともあり、また、電解時に塗膜中に取り込まれることにより、塗膜外観の低下を招く。
【0065】
(Alイオン)
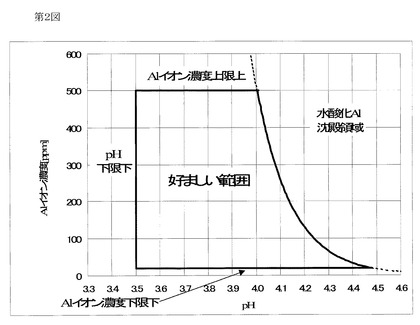
ここで、本発明に係る組成物はアミノポリカルボン酸を含有するが、特にカチオン性の樹脂と組み合わせた場合、過剰なアミノポリカルボン酸の存在により、ときとしてカチオン性樹脂のゲル化を招くことがある。このような場合には、カチオン性樹脂のカチオン基の量を減らすか或いはノニオン性の樹脂とする(或いは、カチオン性樹脂とノニオン性樹脂とを混合し、全体的なカチオン基量を相対的に減少させる)のが好適である。ところで、この場合、pHの上昇によっても樹脂があまり析出しないという別の問題を生じることがある。ここで、当該問題は、Alイオンを含有させることにより解消することが可能となる。この際、Alイオンを20〜500ppm含有することが好ましい。50〜400ppmが更に好ましく、100〜300ppmが最も好ましい。下限を下回るとAlイオンの塗膜析出向上効果が不充分となり、上限を上回ると組成物の電気伝導度が過剰となり、かえって付き廻り性を低下させる。
【0066】
ここで、前述したAlイオンの作用機序は以下の通りである。つまり、イオン状のAlがカソード電解による金属表面pH上昇により微細な水酸化物コロイドになり、それがpH9前後でゼータ電荷を完全に失い急激に凝集を始める際、周りの樹脂をも巻き込んで析出するものと推定される。
【0067】
カソード電解によってAlイオンから水酸化物コロイドの電荷の消失にいたる一連の反応は瞬時に完了する必要がある。あらかじめ水酸化物になっていては、経時で凝集が始まってしまい、pH9前後での凝集能力が極端に減退する。よって、当該態様におけるAl成分は、組成物中ではあくまでイオンでいなければならないのである。
【0068】
また、金属イオンは通常キレート剤の存在によって安定化されるが、Alイオンの場合は、pH上昇に伴う水酸化物コロイドの生成を阻止する程の安定度を有するキレート剤は無い又は稀である。少なくとも、電着塗料組成物に通常配合されている、酢酸、蟻酸、スルファミン酸、乳酸等の有機酸及びアミノポリカルボン酸には、Alイオンを安定化させるほどのキレート能力はない。
【0069】
AlイオンはAl化合物を用いて添加することができる。Al化合物は特に限定されないが、硝酸塩、硫酸塩と言った無機酸塩又は乳酸塩、酢酸塩と言った有機酸塩の形で添加することが可能である。
【0070】
更に、Alイオンを前述の範囲で含有することに加え、当該態様に係る組成物のpHをAlイオン濃度をA[ppm]としたとき次の計算式を満足するようにすることが好ましい。
3.5≦pH≦−Log((A×1.93×10−15)1/3)
下記式であることが更に好ましい。
3.6≦pH≦−Log((A×1.93×10−15)1/3)
下記式であることが最も好ましい。
3.7≦pH≦−Log((A×1.93×10−15)1/3)
pHが下限を下回ると、析出効率が低下し付き廻り性も低下していく。pHが上限を上回ると、Alイオンが加水分解を起こしてしまうため、好ましくない。
【0071】
−Log((A×1.93×10−15)1/3)の項は、25℃における水酸化Alの溶解度積:1.92×10−32から求められる。つまり、このpH以上になるとAlイオンは水酸化物として沈殿析出してしまい、もはやイオンではいられなくなる。ここで、25℃は、組成物の保存時及び使用時の典型的な温度である。
【0072】
また、本発明の組成物の中には、Biイオン、Alイオンの他に、Feイオン、Znイオン、Ceイオン等の金属イオンを含有しても、本発明の効果を損なうものではない。むしろ、これらの金属イオンには、Alイオンほどではないものの、水系樹脂の析出を促進させる作用を有する。なお、Feイオンは2価よりも3価がより好ましい。
【0073】
参考のため、Alイオン濃度及びpHの適正範囲を第2図に示す。
【0074】
<金属表面処理用組成物の物性>
(pH)
本発明に係る金属表面処理用組成物のpHは特に制限されるものではないが、通常2.0〜7.0、好ましくは3.0〜6.5の範囲に調整して使用することができる。
【0075】
(温度)
本発明に係る金属表面処理用組成物の温度についても特に制約は無いが、電解処理によって皮膜を析出させる際は、通常15〜40℃、好ましくは20〜35℃の範囲内で使用することができる。
【0076】
《金属表面処理用組成物の製造方法》
次に、本発明に係る金属表面処理用組成物の製造方法を説明することとする。尚、水系樹脂として変性エポキシ樹脂のアミノ化物を用いた場合を例に採り説明する。先ず、合成した変性エポキシ樹脂のアミノ化物に中和酸を添加し、撹拌混合した後、水で希釈し、所定濃度の樹脂エマルションを作製する。中和酸は、蟻酸、酢酸、乳酸、スルファミン酸などが用いられる。
【0077】
この際、中和酸を添加する前に硬化剤や硬化触媒、有機溶剤などを添加しておくことが好ましい。あらかじめ添加をすることで、均一なエマルションを得ることができる。
【0078】
硬化剤はブロックポリイソシアネートを用いることが一般的である。ブロックポリイソシアネートは、ポリイソシアネート化合物とイソシアネートブロック剤とのほぼ化学理論量での付加反応生成物である。ここで使用されるポリイソシアネート化合物としては、例えば、トリレンジイソシアネート、キシリレンジイソシアネート、フェニレンジイソシアネート、ジフェニルメタン−2,4'−ジイソシアネート、ジフェニルメタン−4,4'−ジイソシアネート(通常「MDI」と呼ばれる)、クルードMDI、ビス(イソシアネートメチル)シクロヘキサン、テトラメチレンジイソシアネート、ヘキサメチレンジイソシアネート、メチレンジイソシアネート、イソホロンジイソシアネートなどの芳香族、脂肪族又は脂環族のポリイソシアネート化合物;これらのポリイシアネート化合物の環化重合体、イソシアネートビゥレット体;これらのイソシアネート化合物の過剰量にエチレングリコール、プロピレングリコール、トリメチロールプロパン、ヘキサントリオール、ヒマシ油などの低分子活性水素含有化合物を反応させて得られる末端イソシアネート含有化合物などを挙げることができる。これらはそれぞれ単独で又は2種以上組合わせて使用することができる。
【0079】
一方、前記イソシアネートブロック剤は、ポリイソシアネート化合物のイソシアネート基に付加してブロックするものであり、そして付加によって生成するブロックポリイソシアネート化合物は常温において安定であるが、塗膜の焼付け温度(通常約100〜約200℃)に加熱した際、ブロック剤が解離して遊離のイソシアネート基を再生しうるものであることが望ましい。
【0080】
このような要件を満たすブロック剤としては、例えば、ε−カプロラクタム、γ−ブチロラクタムなどのラクタム系化合物;メチルエチルケトオキシム、シクロヘキサノンオキシムなどのオキシム系化合物;フェノール、パラ−t−ブチルフェノール、クレゾールなどのフェノール系化合物;n−ブタノール、2−エチルヘキサノールなどの脂肪族アルコール類;フェニルカルビノール、メチルフェニルカルビノールなどの芳香族アルキルアルコール類;エチレングリコールモノブチルエーテル、ジエチレングリコールモノエチルエーテルなどのエーテルアルコール系化合物等を挙げることができる。これらのブロック剤はそれぞれ単独で又は2種以上組み合わせて使用することができる。また、ブロック剤の解離、硬化反応などを効率よく進め、また、意図する硬化反応物を生成させるために、あらかじめ、変性エポキシ樹脂の骨格にイソシアネート基の一部を付加し、かつ、残りのイソシアネート基をブロック剤でブロックする手法もとられる。
【0081】
次に、得られたエマルションに対し、Biイオン水溶液を添加する。Biイオン水溶液は、所定濃度のポリアミノカルボン酸を水に溶解させ、そこに硝酸ビスマス5水和物を添加し、50〜70℃で溶解するまで撹拌することで得ることができる。
【0082】
通常は塗膜性能を向上させたり、着色のため、ここに顔料を添加する。顔料は分散樹脂を用い、あらかじめ分散体(ペースト)としたものを用いる。
【0083】
これらを充分に撹拌したものが金属表面処理用組成物となる。
【0084】
《金属表面処理用組成物の使用方法(用途)》
(適用対象)
本発明に係る金属表面処理用組成物は、各種金属を腐食から防止する目的で使用される。金属材料は、特に限定されるものではないが、冷延鋼板、熱延鋼板、鋳物材、鋼管等の鉄鋼材料、それらの鉄鋼材料の上に亜鉛系めっき処理及び/又はアルミニウム系めっきが施された材料、アルミニウム合金板、アルミニウム系鋳物材、マグネシウム合金版、マグネシウム系鋳物材等が挙げられる。特に形状が複雑な金属構成体、例えば、鉄系材料を主とする金属構成体である自動車車体、自動車部品、家電製品、建築材料等への使用に適している。
【0085】
(使用方法<金属表面処理方法>)
本発明に係る使用方法(金属表面処理方法)は、前述した金属表面処理用組成物を用い、材料を陰極とした電解処理工程にて金属材料表面に塗膜を析出させる工程を含む。より好適には、本発明に係る使用方法(金属表面処理方法)は、金属材料上に皮膜を析出させるべく、表面が清浄化された金属材料に対して電解処理を施す電解処理工程と、電解処理工程後に実行する水洗及び焼付け工程を含む。以下、本方法に特徴的な電解処理工程について詳述する。
【0086】
この電解処理工程(カソード電解)は、金属表面処理用組成物中に前記金属材料を浸漬させた状態で、電圧0〜15Vにて10〜120秒間電解する第一工程と、金属表面処理用組成物中に前記金属材料を浸漬させた状態で、電圧50〜300Vにて30〜300秒間電解する、前記第一工程の後に実施する第二工程とを含み、ここで、前記第二工程は、前記第一工程に引き続いて同一浴内で実施するか又は前記第一工程とは異なる別浴内で実施する。
【0087】
ここで、第一工程は主としてBiを優先的に付着させるために行われる工程であり、第二工程は主として樹脂を優先的に析出させるために行われる工程である。充分な耐食性を得るためには、金属材料に直接接触しているBi、つまり金属材料と皮膜の界面に存在する界面Biの存在が必要であり、そのためには第一工程と第二工程の順番と条件が極めて重要となってくる。
【0088】
第一工程の電圧は0〜15Vであり、10〜120秒間電解することが好ましい。電圧が下限を下回る場合、すなわち金属材料を陽極として電解した場合は、金属材料が組成物中に溶出してしまい、組成物の安定性を低下させるばかりか、耐食性の向上に必要な界面Biが充分付着しなくなる。上限を超える場合も、Biが金属表面に優先的に析出する前に樹脂析出が始まってしまうため、やはり充分な耐食性が得られなくなる。
【0089】
処理時間が下限を下回る場合も充分な界面Biが析出せず、上限を上回る場合は界面Biの付着量過多となり、皮膜の密着性が損なわれる場合がある。
【0090】
第二工程の電圧は50〜300Vであり、30〜300秒間電解することが好ましい。電圧が下限を下回る場合は、樹脂皮膜の析出量が不充分となり、上限を上回る場合は、樹脂皮膜の析出過多により経済的に不利であるばかりか、皮膜の仕上がり外観が損なわれる場合がある。
【0091】
第一工程に次いで第二工程に移行する際、電圧を瞬時に増加させる必要は無く、緩やかに増加させても本発明の効果を損なうものではない。
【0092】
更に、本発明における二段階電解処理は同一槽で行う必要は必ずしもなく、それぞれ異なる槽で実行してもよい。二段階電解処理を二槽で行う場合、第一槽と第二槽とで第一行程、第二行程にあわせたBi濃度、樹脂濃度の調整が可能となり、コストメリットが図れる。例えば、第二槽のBi濃度を低下させることで、持ち出し液中に含まれるBi量を減らすことができ、コスト低減につなげられる。
【0093】
《金属表面処理皮膜》
本発明に係る金属表面処理皮膜は、本発明の金属表面処理用組成物を用い、本発明の処理方法によって得られる。ここで、皮膜中に存在するBiは金属及び酸化物の形態で存在する。カソード電解によって析出するBiは、基本的に還元析出した金属Biであるが、その一部は特に皮膜の焼付け工程で酸化されて酸化物となる。また、第二工程において高電圧がかかった場合、皮膜表面のpH上昇により、アミノポリカルボン酸によるBiの安定化が不充分となるため、特に皮膜表面側では酸化Biとしても析出する。
【0094】
Bi付着量は20〜500mg/m2が好ましく、30〜400mg/m2が更に好ましく、50〜300mg/m2が最も好ましい。Bi付着量が低過ぎると充分な耐食性が得られず、高過ぎるともはや耐食性の向上が望めないばかりか皮膜密着性を損なう場合もある。尚、Bi付着量は蛍光X線分光分析により定量可能である。尚、本特許請求の範囲及び本明細書における「金属Bi付着量」及び「酸化Bi付着量」は、当該蛍光X線分光分析で定量された値とする。尚、その他の形態として水酸化物の存在も否定できないが、当該測定方法で「金属Bi」又は「酸化Bi」として定量された場合には、その数値は「金属Bi付着量」又は「酸化Bi付着量」とすることとする。
【0095】
得られる皮膜の全皮膜厚は5〜40μmが好ましく、5〜30μmが更に好ましく、7〜25μmが最も好ましい。薄過ぎると充分な耐食性が得られず、厚過ぎると経済的に不利なばかりか付き廻り性が低下する場合がある。皮膜厚は、素地金属が磁性金属であれば電磁誘導式膜厚計、素地金属が非磁性金属であれば渦電流式膜厚計により、測定可能である。
【0096】
皮膜中のBiは、皮膜表面よりも素地金属側により多く存在する必要がある。具体的には、皮膜厚の中心から金属材料側のBi付着量:Bが、全Bi付着量:Aに対して55%以上(B/A≧55%)となるBi付着分布であることが好ましい。58%以上が更に好ましく、60%以上が最も好ましい。低過ぎると充分な耐食性が得られない。なお、90%を超えると皮膜表面側のBi濃度が極端に低下し、Biの持つ硬化触媒としての機能を失うので好ましくない。
【0097】
皮膜中のBi付着分布については、EPMAを用いて皮膜断面を線分析することにより測定可能である。同時に撮影した反射電子像によって素地金属と皮膜の界面及び皮膜表面の位置を特定し、EPMA線分析による皮膜中のBi強度の積分値:A及び皮膜厚の中心から素地金属側のみの積分値:Bを求め、B/Aを算出することができる。
【実施例】
【0098】
フェノール化合物(D)の合成
ビスフェノールA(和光純薬):264.8gをブチルセロソルブ(和光純薬):100gとエタノール(純正化学):100gの混合溶媒に完全に溶解させた後、ジエタノールアミン(和光純薬):121.8g、ホルムアルデヒド液(純正化学):96.8gを加え、75℃に加温した。75℃の条件下で4日間撹拌を行い、その後、エバポレーターにて溶剤を除去することでフェノール化合物(D1)を得た。NMR分析より、この時得られた化合物の構造は式3であることが確認できた。
(式7)
【0099】
ビスフェノールA(和光純薬):264.8gをブチルセロソルブ(和光純薬):100gとエタノール(純正化学):100gの混合溶媒に完全に溶解させた後、グリシン(東京化成):87.1g、ホルムアルデヒド液(純正化学):96.8gを加え、75℃に加温した。75℃の条件下で6日間撹拌を行い、その後、エバポレーターにて溶剤を除去することでフェノール化合物(D2)を得た。NMR分析より、この時得られた化合物の構造は式4であることが確認できた。
(式8)
【0100】
ビスフェノールA(和光純薬):264.8gをブチルセロソルブ(和光純薬):100gとエタノール(純正化学):100gの混合溶媒に完全に溶解させた後、アセチルエチルアミン(東京化成):101.1g、ホルムアルデヒド液(純正化学):96.8gを加え、75℃に加温した。75℃の条件下で6日間撹拌を行い、その後、エバポレーターにて溶剤を除去することでフェノール化合物(D3)を得た。NMR分析より、この時得られた化合物の構造は式5であることが確認できた。
(式9)
【0101】
ビスフェノールA(和光純薬):264.8gをブチルセロソルブ(和光純薬):100gとエタノール(純正化学):100gの混合溶媒に完全に溶解させた後、ジエチルアミン(東京化成):84.8g、ホルムアルデヒド液(純正化学):96.8gを加え、75℃に加温した。75℃の条件下で4日間撹拌を行い、その後、エバポレーターにて溶剤を除去することでフェノール化合物(D4)を得た。NMR分析より、この時得られた化合物の構造は式6であることが確認できた。
(式10)
【0102】
ビスフェノールA(和光純薬):264.8gをブチルセロソルブ(和光純薬):100gとエタノール(純正化学):100gの混合溶媒に完全に溶解させた後、エチルアミン(メタノール溶液)(東京化成):149.4g、ホルムアルデヒド液(純正化学):96.8gを加え、75℃に加温した。75℃の条件下で6日間撹拌を行い、その後、エバポレーターにて溶剤を除去することでフェノール化合物(D5)を得た。NMR分析より、この時得られた化合物の構造は式7であることが確認できた。
(式11)
【0103】
ビスフェノールA(和光純薬):264.8gをブチルセロソルブ(和光純薬):100gとエタノール(純正化学):100gの混合溶媒に完全に溶解させた後、アンモニア(エタノール溶液)(東京化成):580.0g、ホルムアルデヒド液(純正化学):96.8gを加え、75℃に加温した。75℃の条件下で6日間撹拌を行い、その後、エバポレーターにて溶剤を除去することでフェノール化合物(D6)を得た。NMR分析より、この時得られた化合物の構造は式8であることが確認できた。
(式12)
【0104】
Biイオンキレート能の確認
Biイオン水溶液(B2、下記参照):100gにあらかじめエタノール50gに溶解させたフェノール化合物(D1):1.7gを加え、60℃に加温しながら撹拌を行った。得られた溶液について、液体クロマトグラフィー質量分析法を用いることで、フェノール化合物(D1)はBiをキレートしていることが確認できた。なお、この場合フェノール化合物(D1)はBiの1.0倍モル濃度となる。
【0105】
Biイオン水溶液(B2、下記参照):100gにあらかじめエタノール50gに溶解させたビスフェノールAの溶媒溶液:1.1gを加え、60℃に加温しながら撹拌を行った。得られた溶液について、液体クロマトグラフィー質量分析法を用いることで、ビスフェノールAはBiをキレートしていないことが確認できた。なお、この場合ビスフェノールAはBiの1.0倍モル濃度となる。
【0106】
Biイオン水溶液(B2、下記参照):100gにあらかじめエタノール50gに溶解させたフェノール化合物(D2):1.5gを加え、60℃に加温しながら撹拌を行った。得られた溶液について、液体クロマトグラフィー質量分析法を用いることで、フェノール化合物(D2)はBiをキレートしていることが確認できた。なお、この場合フェノール化合物(D2)はBiの1.0倍モル濃度となる。
【0107】
Biイオン水溶液(B2、下記参照):100gにあらかじめエタノール50gに溶解させたフェノール化合物(D3):1.6gを加え、60℃に加温しながら撹拌を行った。得られた溶液について、液体クロマトグラフィー質量分析法を用いることで、フェノール化合物(D3)はBiをキレートしていることが確認できた。なお、この場合フェノール化合物(D3)はBiの1.0倍モル濃度となる。
【0108】
Biイオン水溶液(B2、下記参照):100gにあらかじめエタノール50gに溶解させたフェノール化合物(D4):1.5gを加え、60℃に加温しながら撹拌を行った。得られた溶液について、液体クロマトグラフィー質量分析法を用いることで、フェノール化合物(D4)はBiをキレートしていることが確認できた。なお、この場合フェノール化合物(D4)はBiの1.0倍モル濃度となる。
【0109】
Biイオン水溶液(B2、下記参照):100gにあらかじめエタノール50gに溶解させたフェノール化合物(D5):1.4gを加え、60℃に加温しながら撹拌を行った。得られた溶液について、液体クロマトグラフィー質量分析法を用いることで、フェノール化合物(D5)はBiをキレートしていることが確認できた。なお、この場合フェノール化合物(D5)はBiの1.0倍モル濃度となる。
【0110】
Biイオン水溶液(B2、下記参照):100gにあらかじめエタノール50gに溶解させたフェノール化合物(D6):1.2gを加え、60℃に加温しながら撹拌を行った。得られた溶液について、液体クロマトグラフィー質量分析法を用いることで、フェノール化合物(D6)はBiをキレートしていることが確認できた。なお、この場合フェノール化合物(D6)はBiの1.0倍モル濃度となる。
【0111】
ブロック化イソシアネートの作製
コスモネートM200(三井化学株式会社製):678.4gにメチルイソブチルケトン:115.6gを加え、70℃に昇温した後、ジエチレングリコールモノエチルエーテル:706.0gをゆっくり滴下し、滴下終了後、90℃に昇温した。90℃の条件下で12時間反応させ、ブロック化イソシアネートを得た。赤外吸収スペクトル測定を行ったところ、未反応のイソシアネート基由来の吸収が見られず、イソシアネートが完全にブロック化されたことが確認できた。
【0112】
水系樹脂エマルションの作製
製造例1
温度計、コンデンサ、攪拌機を備えた1000mlセパラブルフラスコにエポキシ樹脂・#828(ジャパンエポキシレジン株式会社製、エポキシ当量:180):114.0g、変性樹脂としてポリカプロラクトンジオール・プラクセル208(ダイセル化学株式会社製):41.5g、ビスフェノールA:45.6g、ジメチルベンジルアミン0.1gを加え、130℃でエポキシ当量1000になるまで反応を行った。反応終了後にブチルセロソルブ55.5gを加え、更にジエタノールアミン:12.6g、ジエチレントリアミンのケチミン化物:8.0gを加え、90℃で2時間反応を行った。ここにブロック化イソシアネート:105.5g、ジブチル錫ジアセテート:3.2g、酢酸5.4gを加え、均一になるまで撹拌を行った後、脱イオン水578.1gを強く撹拌しながら約1時間かけて滴下し、固形分濃度33%の水系樹脂エマルション(A1)を得た。
【0113】
製造例2
製造例1において、ビスフェノールAの代わりに、ビスフェノールA:31.9g、フェノール化合物(D1):20.8gを使用し、同様な反応を行うことで、水系樹脂エマルション(A2)を得た。
【0114】
製造例3
製造例1において、ビスフェノールAの代わりに、ビスフェノールA:13.7g、フェノール化合物(D1):48.4gを使用し、同様な反応を行うことで、水系樹脂エマルション(A3)を得た。
【0115】
製造例4
同様に、ビスフェノールAの代わりに、ビスフェノールA:31.9g、D2:19.0gを使用し、同様な反応を行うことで、水系樹脂エマルション(A4)を得た。
【0116】
製造例5
同様に、ビスフェノールAの代わりに、ビスフェノールA:31.9g、D3:19.7gを使用し、同様な反応を行うことで、水系樹脂エマルション(A5)を得た。
【0117】
製造例6
同様に、ビスフェノールAの代わりに、ビスフェノールA:22.8g、D4:18.9gを使用し、同様な反応を行うことで、水系樹脂エマルション(A6)を得た。
【0118】
製造例7
同様に、ビスフェノールAの代わりに、ビスフェノールA:22.8g、D5:17.2gを使用し、同様な反応を行うことで、水系樹脂エマルション(A7)を得た。
【0119】
製造例8
同様に、ビスフェノールAの代わりに、ビスフェノールA:22.8g、D6:15.5gを使用し、同様な反応を行うことで、水系樹脂エマルション(A8)を得た。
【0120】
製造例9
温度計、コンデンサ、攪拌機を備えた2000mlセパラブルフラスコにエポキシ樹脂・#828(ジャパンエポキシレジン株式会社製、エポキシ当量:180):114.0g、変性樹脂としてポリカプロラクトンジオール・プラクセル208(ダイセル化学株式会社製):41.5g、ビスフェノールA:31.9g、フェノール化合物(D1):20.8g、ビスフェノールAエチレンオキサイド付加物・ニューポールBPE−100(三洋化成工業株式会社製):100.2g、ジメチルベンジルアミン0.1gを加え、130℃でエポキシ当量1500になるまで反応を行い、反応終了後にブチルセロソルブ75.3gを加えた。ここにブロック化イソシアネート:143.4g、ジブチル錫ジアセテート:4.3gを加え、均一になるまで撹拌を行った後、脱イオン水792.9gを強く撹拌しながら約1時間かけて滴下し、固形分濃度33%の水系樹脂エマルション(A9)を得た。
【0121】
顔料ペーストの作製
60%の第四級塩化エポキシ樹脂8.3部に対し、精製クレー7.0部、カーボンブラック0.3部、リン酸亜鉛3.0部および脱イオン水を加え、ボールミルにて20時間分散し、固形分50重量%の顔料分散ペーストを得た。
【0122】
Biイオン液の作製
蒸留水:500gにHEDTA:13.3gを溶解させ、60℃に加温した後、硝酸ビスマス5水和物:23.2gを加えて固形分が完全に溶解するまで撹拌した。最終的に全量が1.0Lとなるように更に蒸留水を加え、Biイオン水溶液(B1)を作製した。なお、この場合HEDTAはBiの1.0倍モル濃度となる。
蒸留水:500gにHEDTA:6.65gを溶解させ、60℃に加温した後、硝酸ビスマス5水和物:23.2gを加えて撹拌した。最終的に全量が1.0Lとなるように更に蒸留水を加え、薄く濁ったBiイオン液(B2)を作製した。なお、この場合HEDTAはBiの0.5倍モル濃度となる。
蒸留水:500gにHEDTA:2.66gを溶解させ、60℃に加温した後、硝酸ビスマス5水和物:23.2gを加えて撹拌した。最終的に全量が1.0Lとなるように更に蒸留水を加え、濁ったBiイオン液(B3)を作製した。なお、この場合HEDTAはBiの0.2倍モル濃度となる。
蒸留水:500gにHEDTA:1.33gを溶解させ、60℃に加温した後、硝酸ビスマス5水和物:23.2gを加えて撹拌した。最終的に全量が1.0Lとなるように更に蒸留水を加え、濃く濁ったBiイオン液(B4)を作製した。なお、この場合HEDTAはBiの0.1倍モル濃度となる。
蒸留水:500gにEDTA:6.99gを溶解させ、60℃に加温した後、硝酸ビスマス5水和物:23.2gを加えて固形分が完全に溶解するまで撹拌した。最終的に全量が1.0Lとなるように更に蒸留水を加えBiイオン水溶液(B5)を作製した。なお、この場合、EDTAはBiの0.5倍モル濃度となる。
蒸留水:500gにNTA:45.8gを溶解させ、60℃に加温した後、硝酸ビスマス5水和物:23.2gを加えて固形分が完全に溶解するまで撹拌した。最終的に全量が1.0Lとなるように更に蒸留水を加えBiイオン水溶液(B6)を作製した。なお、この場合NTAはBiの5.0倍モル濃度となる。
【0123】
組成物の作製
第1表に示す組合せの固形分16.0重量%になる量の樹脂エマルジョンに無機固形分4.0重量%になる量の顔料分散ペースト及びBi添加剤を配合し、組成物を作製した。なお、それぞれの濃度は脱イオン水を用いて希釈し調整した。尚、実施例1〜11、比較例1、2については、いずれも処理液のpHは5であった。実施例12については、硝酸アルミニウム・9水和物と硝酸を用い、Al濃度:200ppm、pH4に調整を行った。
【0124】
電解条件
電解工程(1)として8Vにて60秒間電解後、直ちに電解工程(2)として180Vにて180秒間電解処理を行った。
【0125】
試験板の作製
試験板として、冷延鋼板:SPCC(JIS3141)70×150×0.8mm(以下、SPCと略す)を用い、あらかじめその表面を日本パーカライジング社製強アルカリ脱脂剤「FC−E2001」を使用して、120秒間スプレー処理することにより脱脂処理した。脱脂処理後は30秒間スプレー水洗し、実施例及び比較例に示す組成物に浸漬させ、実施例及び比較例に示す電解条件にてカソード電解処理を実施した。電解終了後の試験板は直ちに脱イオン水にて30秒間スプレー水洗し、電気オーブン中で180℃にて20分間焼付けを行った。
【0126】
皮膜特性の調査
試験板の上に析出した皮膜の皮膜特性を以下の方法で調査した。
皮膜厚測定:電磁誘導式膜厚計を用いて測定した。
Bi付着量:蛍光X線分光分析によって定量した。
Bi付着分布:試料断面をEPMAの線分析にて分析した。具体的方法は下記参照。
【0127】
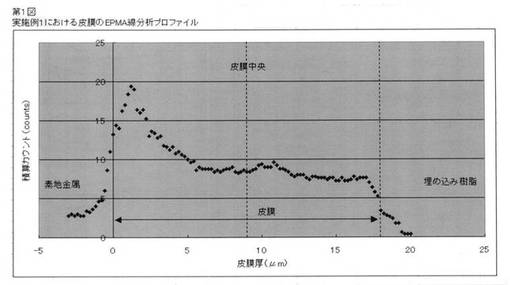
皮膜中のBi付着量分布測定は、EPMAを用いて分析した。皮膜処理後の金属材料を、埋め込み樹脂によって固定し、断面を研磨し、素地金属方向から析出皮膜表面方向にBiの線分析プロファイルを求めた。線分析プロファイルとは、マッピング分析データを基に、分析エリアの1次元方向に任意の幅で特性X線強度の平均値を算出したもので、幅を持った線分析と解することができる。測定条件は以下の通り。
【0128】
測定機器:島津製作所製EPMA−1610型
電子銃:CeB6カソード型
ビーム電流:50nA、ビーム電圧:15kV、ビーム径:1μmφ以下
積算回数:1回、1点あたりのサンプリング時間:100ms
分光結晶:PET(Bi Mα)
【0129】
同時に撮影した反射電子像によって素地金属と皮膜の界面及び皮膜表面の位置を特定し、皮膜中のBi強度の積分値:A及び皮膜厚の中心から素地金属側のみの積分値:Bを求め、B/Aを算出した。
なお、参考のため代表的なプロファイルとして実施例1で得られた皮膜の分析結果を第1図に示す。
【0130】
耐食性試験方法および評価方法
カソード電解処理により作製された樹脂塗装板にクロスカットを施し、塩水噴霧試験(JIS−Z2371)を実施し、1500時間後のクロスカット部の片側最大膨れ幅を測定した。測定結果を基に、2mm未満:◎、2mm以上3mm未満:○、3mm以上4mm未満:△、4mm以上:×にて評価した。結果を第2表に示す。
【0131】
外観性試験方法および評価方法
カソード電解処理により作製された樹脂塗装板の光沢度を測定した。
光沢計:日本電色工業株式会社製 VG2000
測定角度:60°
測定結果を基に、65以上:○、65未満:×にて評価した。結果を第2表に示す。
【表1】
【表2】
【技術分野】
【0001】
本発明は、金属材料、特に形状が複雑な金属構成体に対し、優れた耐食性及び塗膜外観を付与し得る皮膜を形成せしめることが可能な金属表面処理組成物、これを用いた金属表面処理方法及びこれらを用いた金属表面処理皮膜に関するものである。
【背景技術】
【0002】
従来、各種金属材料、特に形状が複雑な金属構成体に対して優れた耐食性を付与するための手法としては、高い付き廻り性を有する電着塗装が一般的に用いられてきた。しかし、電着塗装によって得られる電着塗膜のみでは、所望の耐食性が得られない場合が多いため、電着塗装の前段には標準的にリン酸亜鉛系化成処理等の化成型の塗装下地処理が適用されていた。
【0003】
電着塗装は、アニオン性樹脂エマルジョンを含有する水性塗料中で被塗物をアノード電解することによって塗膜を析出させるアニオン電着塗装と、カチオン樹脂エマルジョンを含有する水性塗料中で被塗物をカソード電解することによって塗膜を析出させるカチオン電着塗装とに大別できるが、鉄系金属材料の耐食性向上に対しては、電解処理中に素地金属が塗料中に溶出する心配の無いカチオン電着塗装が有利であり、鉄系材料を主とする金属構成体である自動車車体、自動車部品、家電製品、建築材料等に対してはカチオン電着塗装が広く適用されている。
【0004】
カチオン電着塗装の市場での歴史は長く、かつてはクロム化合物や鉛化合物を配合することによって防錆性を確保していた。但し、これによっても防錆性は不充分であったため、リン酸亜鉛系化成処理等の下地処理が必須であった。現在では環境規制、特に欧州におけるELV規制によりクロム化合物や鉛化合物が実質使用できなくなったため、代替成分が検討され、ビスマス化合物にその効果が見出されており、具体的には次に挙げる特許文献が開示されている。
【0005】
特許文献1(特開平5−32919)には、ビスマス化合物をコーティングした顔料を少なくとも1種含有することを特徴とする電着塗料用樹脂組成物が開示されている。
【0006】
特許文献2(WO99/31187)には、有機酸変性ビスマス化合物が非水溶性の形態で存在する水性分散液を配合した水性分散ペーストからなることを特徴とするカチオン電着塗料組成物が開示されている。
【0007】
特許文献3(特開2004−137367)には、コロイド状ビスマス金属、及び、スルホニウム基とプロパルギル基とを持つ樹脂組成物からなることを特徴とするカチオン電着塗料が開示されている。
【0008】
特許文献4(特開2007−197688)には、水酸化ビスマス、ジルコニウム化合物及びタングステン化合物から選ばれる少なくとも1種の金属化合物の粒子を含んでなる電着塗料であって、該金属化合物が1〜1000nmであることを特徴とする電着塗料が開示されている。
【0009】
特許文献5(特開平11−80621)には、脂肪族アルコキシカルボン酸ビスマス塩水溶液を含有することを特徴とするカチオン電着塗料組成物が開示されている。
【0010】
特許文献6(特開平11−80622)には、2種以上の有機酸によるビスマス塩の水溶液であって、該有機酸の少なくとも1種が脂肪族ヒドロキシカルボン酸である有機酸ビスマス塩水溶液を含有することを特徴とするカチオン電着塗料組成物が開示されている。
【0011】
特許文献7(特開平11−100533)には、光学異性体のうちのL体が80%以上含まれる乳酸を用いてなる乳酸ビスマスを含有することを特徴とするカチオン電着塗料組成物が開示されている。
【0012】
特許文献8(特開平11−106687)には、2種以上の有機酸によるビスマス塩の水溶液であって、該有機酸の少なくとも1種が脂肪族アルコキシカルボン酸である有機酸ビスマス塩水溶液を含有することを特徴とするカチオン電着塗料組成物が開示されている。
【0013】
これらの特許文献は特許文献1〜4及び特許文献5〜8に大別できる。すなわち、特許文献1〜4は水性塗料に対して不溶性のビスマス化合物又は金属ビスマスを分散させたものであり、特許文献5〜8は少なくともビスマス化合物を固形分の残存が無くなるまで溶解させる、つまりBiイオンの状態にしてから塗料に添加することを特徴としている。
【0014】
しかしながら、これらの特許文献におけるビスマス化合物は、あくまでクロム化合物や鉛化合物の代替として作用するものであり、リン酸亜鉛系化成処理等の下地処理無しには充分な耐食性は得られない。事実、これらの特許文献ではリン酸亜鉛系化成処理との組合せを前提とした実施例のみが開示されている。
【0015】
一方、昨今ビスマス化合物以外の手法により耐食性を更に向上させ、リン酸亜鉛系化成処理等の下地処理を施さなくても、1コートにて充分な耐食性を確保し得る技術が検討されてきている。
【0016】
例えば特許文献9(特開2008−274392)には、金属基材に、皮膜形成剤を少なくとも2段階の多段通電方式で塗装することによって皮膜を形成する方法であって、(i)皮膜形成剤が、ジルコニウム化合物と、必要に応じて、チタン、コバルト、バナジウム、タングステン、モリブデン、銅、亜鉛、インジウム、アルミニウム、ビスマス、イットリウム、ランタノイド金属、アルカリ金属及びアルカリ土類金属から選ばれる少なくとも1種の金属(a)を含有する化合物とを合計金属量(質量換算)で30〜20,000ppmと、樹脂成分1〜40質量%とを含んでなり、(ii)金属基材を陰極として1段目の塗装を1〜50Vの電圧(V1)で10〜360秒間通電することにより行い、次いで、金属基材を陰極として2段目以降の塗装を50〜400Vの電圧(V2)で60〜600秒間通電することにより行い、そして(iii)電圧(V2)と電圧(V1)の差が少なくとも10Vであることを特徴とする表面処理皮膜の形成方法が開示されている。
【0017】
また、特許文献10(特開2008−538383)には、(A)希土類金属化合物、(B)カチオン基を有する基体樹脂、及び(C)硬化剤を含む水性塗料組成物であって、該水性塗料組成物に含まれる(A)希土類金属化合物の量が、塗料固形分に対して、希土類金属に換算して、0.05〜10重量%である水性塗料組成物に、被塗物を浸漬する、浸漬工程、該水性塗料組成物中において、被塗物を陰極として50V未満の電圧を印加する、前処理工程、及び該水性塗料組成物中において、被塗物を陰極として50〜450Vの電圧を印加する、電着塗装工程、を包含する、複層塗膜形成方法が開示されている。
【先行技術文献】
【特許文献】
【0018】
【特許文献1】特開平5−32919号公報
【特許文献2】WO99/31187号公報
【特許文献3】特開2004−137367号公報
【特許文献4】特開2007−197688号公報
【特許文献5】特開平11−80621号公報
【特許文献6】特開平11−80622号公報
【特許文献7】特開平11−100533号公報
【特許文献8】特開平11−106687号公報
【特許文献9】特開2008−274392号公報
【特許文献10】特開2008−538383号公報
【発明の概要】
【発明が解決しようとする課題】
【0019】
本発明者らは、これら従来技術について種々検討した結果、やはりリン酸亜鉛系化成皮膜等の前処理無しに充分な耐食性を付与する皮膜を金属材料の上に形成させるためには、Biの適用が最も効果的であるとの結論に達した。そしてBiの作用効果について再検討することとした。
【0020】
そして、Biの作用効果としては従来から、樹脂の硬化触媒としての機能と、素地金属の防食作用が注目されていたが、従来技術では、硬化触媒としての機能は望めるものの、金属の防食作用については極めて不充分であり、この作用を最大限に発揮させることこそ課題解決につながるものとして検討を進めた。
【0021】
素地金属の防食作用はBiが金属に接触する面、すなわち素地金属表面と皮膜の界面に存在しなくてはならないが、従来技術ではBi成分が皮膜中に均一に分散してしまい、耐食性を発揮するに充分なBiが素地金属表面に存在していないものと推定した。
【0022】
前述の如く特許文献1〜4は水性塗料に対して不溶性のビスマス化合物又は金属ビスマスを分散させたものであるが、このような組成物から皮膜を析出させた場合、他の顔料と同様、皮膜中にBiは均一に分散してしまう。
【0023】
特許文献5〜8は少なくともビスマス化合物を固形分の残存が無くなるまで溶解させる、つまりBiイオンの状態にしてから塗料に添加することを特徴としているが、Biの安定化剤である有機酸のキレート能力が微弱であるため、組成物に投入した際、Biは徐々に加水分解してしまい、酸化物又は水酸化物へと変化してしまうため、Biイオンとしての長期的な安定化は望めない。これによって、やはりBiは皮膜中に均一に分散してしまうのである。これらの特許文献において、やはりリン酸亜鉛系化成処理が下地処理として用いられていたのは、上記の推察を裏付けている。
【0024】
一方、特許文献9及び特許文献10は、素地金属上に無機系の皮膜を析出させた上に樹脂皮膜を積層させる技術であり、素地金属の防食の面で有利であるが、無機系の皮膜も樹脂皮膜もカソード電解による素地金属表面のpH上昇によって析出する機構であるため、積層皮膜の形成が容易でない。
【課題を解決するための手段】
【0025】
本発明者らは、上記の従来技術の課題を解決するために、Biイオンを組成物中でより安定に存在させるために、キレート能力の高いアミノポリカルボン酸を適用し、低電圧カソード電解にてBiを還元析出させ、次いで高電圧カソード電解でBiイオンの拡散が不充分になった段階で、かかるpH上昇によって樹脂が析出する反応機構を見出した。
【0026】
そして、これによって得られた皮膜は、Biの持つ樹脂の硬化触媒能はもちろん、素地金属表面により高濃度で存在するBiにより、素地金属の耐食性をも充分に向上し得ることを確認した。
【0027】
更に、本発明者らは、防錆効果をより高めるため、Bi濃度を高くすることを検討した。この際、Bi濃度を高くするにはキレート剤であるアミノポリカルボン酸濃度を高くすればよいが、この場合、過剰のアミノポリカルボン酸が塗膜に取り込まれると塗膜外観(具体的には光沢度)が低下するという別の問題を招く。以上の問題を解決することができる本発明は、次に示す(1)〜(10)である。
【0028】
本発明(1)は、水系樹脂を5〜30重量%、3価のBiイオンを100〜5000ppm及びBiイオンに対して0.1〜5倍モル濃度のアミノポリカルボン酸を含有し、
水系樹脂の少なくとも一部が、下記式(1):
【化3】
{ここで、R1及びR2は、相互に独立して、−(R)m−X(ここで、Rは、炭素数1〜6のアルキレン基であり、mは1か0であり、Xは、水素、カルボキシル、ヒドロキシル又はアシルである}で示される、Biイオンとキレートを形成可能な窒素含有基をポリマー骨格中に有するノニオン性及び/又はカチオン性樹脂であることを特徴とする金属表面処理用組成物(電解によって有機無機複合皮膜を析出させるための金属表面処理組成物)である。
【0029】
本発明(2)は、式(1)において、R1及びR2は、相互に独立して、水素、アルキル、ヒドロキシアルキル、カルボキシアルキル(−R−COOH:Rはアルキル基)又はアルキルカルボニル(−CO−R:Rはアルキル基)であり、ここで、アルキル、ヒドロキシアルキル、カルボキシアルキル及びアルキルカルボニルのアルキル部分は、炭素数1〜6の直鎖状、分岐鎖状又は環状である、前記発明(1)の金属表面処理用組成物である。
【0030】
本発明(3)は、水系樹脂が、変性エポキシ樹脂である、前記発明(1)又は(2)の金属表面処理用組成物である。
【0031】
本発明(4)は、変性エポキシ樹脂が、ビスフェノールA型であり、前記窒素含有基が、前記変性エポキシ樹脂中のグリシジルエーテル部ではなくフェニレン基のベンゼン環部に直接結合している、前記発明(3)の金属表面処理用組成物である。
【0032】
本発明(5)は、ビスフェノールA型の変性エポキシ樹脂(A)が、原料として、変性樹脂(B)、エポキシ当量180〜2500のエポキシ樹脂(C)及び前記窒素含有基を有するフェノール化合物(D)を用い、或いは更に2級アミノ基含有化合物(F)及び/又はビスフェノールA(E)をも用い、これらを反応させることで得られる変性エポキシ樹脂のアミノ化物である、前記発明(4)の金属表面処理用組成物である。
【0033】
本発明(6)は、化合物(D)が、下記式(2):
(式4)
(式中、R3は、前記窒素含有基又は水素原子であり、R3の少なくとも1個は前記窒素含有基であり;R4は、相互に独立して水素又は炭素数1〜2のアルキル基である)で示されるアミン付加フェノール化合物である、前記発明(5)の金属表面処理用組成物である。
【0034】
本発明(7)は、3価のBiイオンに対して、前記窒素含有基のモル濃度が0.1〜200倍である、前記発明(1)〜(6)のいずれか一つの金属表面処理用組成物である。
【0035】
本発明(8)は、前記発明(1)〜(7)のいずれか一つの組成物を用い、材料を陰極とした電解処理工程にて金属材料表面に塗膜を析出させる工程を含む、金属表面処理方法である。
【0036】
本発明(9)は、表面が清浄化された金属材料に対して電解処理を施す電解処理工程と、電解処理工程後に実行する水洗及び焼付け工程を含む、金属材料上に皮膜を析出させる金属表面処理方法において、
前記電解処理工程は、
前記発明(1)〜(7)のいずれか一つの組成物中に前記金属材料を浸漬させた状態で、電圧0〜15Vにて10〜120秒間電解する第一工程と、
前記発明(1)〜(7)のいずれか一つの組成物中に前記金属材料を浸漬させた状態で、電圧50〜300Vにて30〜300秒間電解する、前記第一工程の後に実施する第二工程と
を含み、ここで、前記第二工程は、前記第一工程に引き続いて同一浴内で実施するか又は前記第一工程とは異なる別浴内で実施することを特徴とする方法である。ここで、第一工程及び第二工程における「電圧X〜Y(V)」は、電圧X〜Yの範囲内で一定電圧を印加する態様でも又は経時的に印加電圧を変化させる態様でもよい。尚、第一工程における「電圧0〜15V」の下限値「0V」は、一定電圧での態様ではなく、経時的に印加電圧を変化させる態様における所定時の電圧を意味する。
【0037】
本発明(10)は、前記発明(8)又は(9)の方法によって形成される、金属Bi及び酸化BiがBiとして20〜500mg/m2付着し、全皮膜厚が5〜40μmであり、かつ皮膜厚の中心から金属材料側のBi付着量(G)が、全Bi付着量(H)に対して55%以上(G/H≧55%)となるBi付着分布であることを特徴とする金属表面処理皮膜である。
【図面の簡単な説明】
【0038】
【図1】第1図は、実施例1における皮膜のEPMA線分析プロファイルである。
【図2】第2図は、Alイオン濃度及びpHの適正範囲を示した図である。
【発明を実施するための最良形態】
【0039】
以下、本最良形態に係る金属表面処理用組成物、当該組成物を使用した金属表面処理方法及び当該方法により形成される金属表面処理皮膜を順に説明することとする。
【0040】
《金属表面処理組成物》
本発明に係る金属表面処理用組成物は、水系樹脂を5〜30重量%、3価のBiイオンを100〜5000ppm及びBiイオンに対して0.1〜5倍モル濃度のアミノポリカルボン酸を含有し、水系樹脂の少なくとも一部が所定の置換基を有するノニオン性及び/又はカチオン性樹脂であることを特徴とする。以下、本組成物を構成する各成分について詳述する。
【0041】
<組成物構成成分:水系樹脂>
本発明に係る水系樹脂は、好適には、ノニオン性樹脂及びカチオン性樹脂である。また、水系樹脂には、ブロック化ポリイソシアネートをはじめとする硬化剤を任意に配合することもできる。ここで、この水系樹脂の少なくとも一部は、下記式(1):
【化5】
{ここで、R1及びR2は、相互に独立して、−(R)m−X(ここで、Rは、炭素数1〜6のアルキレン基であり、mは1か0であり、Xは、水素、カルボキシル、ヒドロキシル又はアシルである}で示される、Biイオンとキレートを形成可能な窒素含有基をポリマー骨格中に有するノニオン性及び/又はカチオン性樹脂である。尚、本発明における水系樹脂とは、水分散するエマルジョンと水溶性樹脂の総称である。ここで、アシルにおけるRは、脂肪族でも芳香族でもよく、炭素数は1〜6であることが好適である。
【0042】
ここで、式(1)において、R1及びR2は、相互に独立して、水素、アルキル、ヒドロキシアルキル、カルボキシアルキル又はアルキルカルボニルであり、ここで、アルキル、ヒドロキシアルキル、カルボキシアルキル及びアルキルカルボニルのアルキル部分は、炭素数1〜6の直鎖状、分岐鎖状又は環状であることが好適である。これらの中で、キレート形成能の観点からO(酸素原子)を含有するものが好適であり、更に合成の容易性に鑑みるとヒドロキシアルキルが最適である。
【0043】
式(1)で示される窒素含有基は、ノニオン性及び/又はカチオン性樹脂のポリマー骨格中に存在する。ここで、ノニオン性樹脂及び/又はカチオン性樹脂は、いずれも特に限定されるものではなく、一例として、エポキシ樹脂、ウレタン樹脂、アクリル樹脂を挙げることができる。これらの中では、エポキシ樹脂が好適であり、変性エポキシ樹脂がより好適であり、ビスフェノールA型の変性エポキシ樹脂が特に好適である。
【0044】
ここで、樹脂にカチオン性を付与するには、典型的にはアミノ基を樹脂骨格中(特に末端)に導入する手法(例えば、エポキシ樹脂では、末端のグリシジル基にアミノ基含有化合物を付加する手法)が採用されている。尚、これについては後で詳述する。
【0045】
また、樹脂にノニオン性を付与するには、例えば、エチレンオキサイド基を含有する化合物も導入させることができる。これを導入することで、このエチレンオキサイド基がノニオン性の活性剤的働きを有することとなり、エマルション状態となったときの乳化安定性向上などといった効果が得られる。具体的には、ポリエチレングリコール、もしくはポリエチレングリコールジグリシジルエーテルを反応させることで、ノニオン性樹脂の性質を示すこととなる。また、ビスフェノールA型の変性エポキシ樹脂を用いる場合には、同じようなビスフェノールA構造を持つ、ビスフェノールAエチレンオキサイド付加物を用いることが好ましい。
【0046】
以下、カチオン性樹脂として特に好適である、ビスフェノールA型の変性エポキシ樹脂のアミノ化物について詳述する。
【0047】
(ビスフェノールA型の変性エポキシ樹脂のアミノ化物)
ここで、特に好適なビスフェノールA型の変性エポキシ樹脂(A)のアミノ化物について説明することとする。ビスフェノールA型の変性エポキシ樹脂は、原料として、変性樹脂(B)、エポキシ当量180〜2500のエポキシ樹脂(C)、前記窒素含有基を有するフェノール化合物(D)及び2級アミノ基含有化合物(F)を用い或いは更にビスフェノールA(E)をも用い、これらを反応させることで得られる変性エポキシ樹脂のアミノ化物である。以下、各成分について説明する。
【0048】
*ビスフェノールA型の変性エポキシ樹脂(A)の原料
まず、変性樹脂(B)としては、通常、ポリオール化合物が用いられる。これらは、エポキシ樹脂の可塑性向上などを目的として適用される。具体的には、ポリエステルポリオール、ポリエーテルポリオール、ポリウレタンポリオール、アクリルポリオール等のポリオール樹脂、末端にフェノールを付加し、水酸基を有する芳香族縮合化合物などが挙げられる。更に具体的には、ポリカプロラクトンジオール、ポリエチレングリコール、ポリプロピレングリコール、フェノール性水酸基を有するキシレンホルムアルデヒド樹脂等が挙げられる。これら化合物により変性を行うことは、これら化合物が有する水酸基とエポキシ樹脂のグリシジルエーテル部が容易に反応し得ることから、従来より用いられてきた技術である。変性エポキシ樹脂中において、これら変性樹脂は5〜30重量%含まれる。
【0049】
次に、エポキシ当量180〜2500のエポキシ樹脂(C)としては、塗膜の防食性等の観点から、特に、ポリフェノール化合物とエピハロヒドリン、例えば、エピクロルヒドリンとの反応により得られるエポキシ樹脂が好適である。中でも、ビスフェノールAとエピクロロヒドリンとの反応により得られるビスフェノールAジグリシジルエーテルが最適である。また、ビスフェノールAを基本構造として重合させたエポキシ樹脂も同様の効果を示し、エポキシ当量として180〜2500、好ましくは180〜2000、更に好ましくは180〜1500のものが最適である。変性エポキシ樹脂中において、これらエポキシ樹脂は5〜30重量%含まれる。
【0050】
次に、前記窒素含有基を有するフェノール化合物(D)としては、下記式(2):
(式6)
(式中、R3は、前記窒素含有基又は水素原子であり、R3の少なくとも1個は前記窒素含有基であり;R4は、相互に独立して水素又は炭素数1〜2のアルキル基である)で示されるアミン付加フェノール化合物であることが好適である。中でも、R3の付加数が1のものが好ましい。また、R4が炭素数1の場合、即ちビスフェノールA型のものが好ましい。変性エポキシ樹脂中において、これら窒素含有基を有するフェノール化合物は5〜30重量%含まれる。これらは、ジフェノール化合物に対し、カルボニル化合物と1級もしくは2級アミンとのマンニッヒ反応により合成させることで、得ることができる。この時のカルボニル化合物としてはホルムアルデヒドを用いることが一般的である。1級、2級のアミンとしては、アンモニア;モノメチルアミン、ジメチルアミン、モノエチルアミン、ジエチルアミン、モノイソプロピルアミン、ジイソプロピルアミン、モノブチルアミン、ジブチルアミン等のモノ−、もしくはジ−アルキルアミン;モノエタノールアミン、ジエタノールアミン、モノ(2−ヒドロキシプロピル)アミン、ジ(2−ヒドロキシプロピル)アミン、トリ(2−ヒドロキシプロピル)アミン、モノメチルアミノエタノール、モノエチルアミノエタノール等のアルカノールアミン;N−ホルミルメチルアミン、N−ホルミルエチルアミン、アセトアミド、N−メチルアセトアミド、N−エチルアセトアミド、プロピオン酸アミド、N−メチルプロピオンアミド、N−エチルプロピレンアミド、グリシン、サルコシン等のカルボキシル基あるいはアシル基を含有するアミン等が挙げられる。
【0051】
次に、ビスフェノールA(E)の含有量(添加量)は、前記窒素含有基を有するフェノール化合物(D)の含有量(添加量)との関係で任意の割合で比率を変えることができる。変性エポキシ樹脂中において、両者の合計量として5〜30重量%含まれる。
【0052】
次に、2級アミノ基含有化合物(F)は、エポキシ樹脂基体にアミノ基を導入して、該エポキシ樹脂をカチオン化するためのカチオン性付与成分であり、前記キレート能を期待するアミンとはその効果が異なる。ここで使用されるアミンはエポキシ基と反応する活性水素を少なくとも1個含有するものが用いられる。そのような目的で使用されるアミノ基含有化合物としては、例えば、モノメチルアミン、ジメチルアミン、モノエチルアミン、ジエチルアミン、モノイソプロピルアミン、ジイソプロピルアミン、モノブチルアミン、ジブチルアミンなどのモノ−、もしくはジ−アルキルアミン;モノエタノールアミン、ジエタノールアミン、モノ(2−ヒドロキシプロピル)アミン、ジ(2−ヒドロキシプロピル)アミン、トリ(2−ヒドロキシプロピル)アミン、モノメチルアミノエタノール、モノエチルアミノエタノールなどのアルカノールアミン;エチレンジアミン、プロピレンジアミン、ブチレンジアミン、ヘキサメチレンジアミン、テトラエチレンペンタミン、ペンタエチレンヘキサミン、ジエチルアミノプロピルアミン、ジエチレントリアミン、トリエチレンテトラミン等のアルキレンポリアミン及びこれらのポリアミンのケチミン化物;エチレンイミン、プロピレンイミンなどのアルキレンイミン;ピペラジン、モルホリン等の環状アミン等が挙げられる。変性エポキシ樹脂中において、2級アミノ基含有化合物(F)は0.5〜20重量%含まれる。
【0053】
*ビスフェノールA型の変性エポキシ樹脂のアミノ化物の製造方法
先ず、変性樹脂(B)、エポキシ樹脂(C)、前記窒素含有基を有するフェノール化合物(D)、ビスフェノールA(E)を所定量混合し、加熱撹拌を行う。加熱温度は70〜100℃が好ましい。各原料が溶解した後、触媒を添加し、加熱温度を上げ、合成を行う。触媒は、通常、ジメチルベンジルアミンのような3級アミンが使用される。合成温度は120℃〜150℃で制御するのが一般的である。
【0054】
合成温度と時間を調整することにより、所定のエポキシ当量を持ったエポキシ樹脂を合成できる。エポキシ当量はJIS K7236に定められるエポキシ当量測定によって算出される。この時のエポキシ当量は800〜10000が好適であり、800〜5000がより好適であり、800〜3000が最も好適である。エポキシ当量が大きくなるほど、エマルション作製時の乳化安定性が低下する傾向がある。
【0055】
次に、この合成した変性エポキシ樹脂に2級アミノ基含有化合物(F)を付加する。変性エポキシ樹脂を70℃〜110℃に保ちながら2級アミノ基含有化合物(F)を添加し、1〜3時間合成を行うことで、変性エポキシ樹脂のアミノ化物が得られる。
【0056】
<組成物構成成分:3価のBiイオン>
本発明でいうBiイオンとは、組成物中で固形化せず、完全に溶解状態になっているBi成分のことを指し、具体的には後述するアミノポリカルボン酸やポリマー中の窒素含有基によってキレートを構成し、安定的に水溶化された状態であることを意味している。
【0057】
<組成物構成成分:アミノポリカルボン酸>
アミノポリカルボン酸とは、分子中にアミノ基と複数のカルボキシル基を有するキレート剤の総称であり、具体的にはEDTA(エチレンジアミン四酢酸)、HEDTA(ヒドロキシエチルエチレンジアミン三酢酸)、NTA(ニトリロ三酢酸)、DTPA(ジエチレントリアミン五酢酸)、TTHA(トリエチレンテトラミン六酢酸)等が該当するが、Biイオンとのキレート安定度の観点からEDTA、HEDTA、NTAがより好ましい。
【0058】
<組成物構成成分:他の成分>
本発明の組成物には、更に必要に応じて顔料、触媒、有機溶剤、顔料分散剤、界面活性剤等、塗料分野で通常使用されている添加剤を適用することもできる。顔料としては、チタン白、カーボンブラック等の着色顔料、クレー、タルク、バリタ等の体質顔料、トリポリリン酸アルミニウム、リン酸亜鉛等の防錆顔料、ジブチル錫オキサイド、ジオクチル錫オキサイド等の有機錫化合物、ジブチル錫ラウレート、ジブチル錫ジベンゾエート等のジアルキル錫の脂肪酸もしくは芳香族カルボン酸塩などの錫化合物が挙げられる。また、後で詳述するように水系樹脂の析出を促進させる観点から、Alイオンを添加してもよい(特にノニオン系樹脂の場合には必須)。更には、本発明の組成物の中には、Biイオン、Alイオンの他に、Feイオン、Znイオン、Ceイオン等の金属イオンを含有しても、本発明の効果を損なうものではない。むしろ、これらの金属イオンには、Alイオンほどではないものの、水系樹脂の析出を促進させる作用を有する。尚、Feイオンは2価よりも3価がより好ましい。
【0059】
<組成物構成成分:液体媒体>
本発明に係る金属表面処理用組成物の液体媒体としては、水性媒体が好適であり、水がより好適である。尚、液体媒体が水である場合、液体媒体として水以外の他の水系溶媒(例えば、水溶性のアルコール類)を含有していてもよい。
【0060】
<組成物の組成>
次に、本発明に係る金属表面処理用組成物の組成について説明することとする。
【0061】
(水系樹脂)
まず、本発明に係る金属表面処理用組成物は、組成物の全重量を基準として水系樹脂を5〜30重量%含み、好適には10〜25重量%、より好適には10〜20重量%含む。ここで、全水系樹脂における式(1)で示されるノニオン性及び/又はカチオン性樹脂の比率(重量比)は、好適には30〜100%であり、より好適には70〜100%である。式(1)で示される樹脂の割合は、少ないほどキレート能が低下し、Biの安定性に寄与できないこととなる。また、全水系樹脂含有量が低過ぎると皮膜析出量が不足し、含有量が高過ぎると経済的に不利である。
【0062】
ここで、本発明に係る金属表面処理用組成物中での、式(1)で示される前記窒素含有基のモル濃度は、3価のBiイオンに対して、0.1〜200倍、0.5〜100倍であることがより好適であり、1.0〜50倍であることが更に好適である。モル濃度が低いほどキレート能が低下し、Biの安定性に寄与できない。モル濃度が高いほど、経済的に不利である。
【0063】
(3価のBiイオン)
次に、本発明に係る金属表面処理用組成物は、3価のBiイオンを100〜5000ppm含有する。500〜4000ppmが更に好ましく、1000〜3000ppmが最も好ましい。Biイオン濃度が低過ぎる場合、耐食性向上に必要な充分なBi付着量が得られず、高過ぎると組成物の電気伝導度が高くなり過ぎ、複雑な形状を有する金属材料への皮膜の付き廻り性が劣化すると共に、Bi付着量過多となり皮膜密着性を損なう恐れがある。組成物中のBiイオン濃度は、超遠心機により組成物を固液分離し、液相を高周波誘導結合プラズマ発光分光分析(ICP)もしくは原子吸光分光分析(AA)を用いて定量することができる。
【0064】
(アミノポリカルボン酸)
本発明に係る金属表面処理用組成物は、Biイオンに対して0.1〜5倍モル濃度でアミノポリカルボン酸を含有する。0.1〜4倍モル濃度が更に好ましく、0.1〜3倍モル濃度であることが最も好ましい。Biイオンに対する濃度比率が低過ぎるとBiイオンが組成物中で加水分解し、酸化物となってしまうため、有効なBiイオン濃度が低下し、結果として充分なBi付着量が得られなくなる。高過ぎると逆にBiイオンが安定化し過ぎ、やはり充分なBi付着量が得られなくなる。後述のように、過剰なアミノポリカルボン酸はカチオン性樹脂のゲル化を招くこともあり、また、電解時に塗膜中に取り込まれることにより、塗膜外観の低下を招く。
【0065】
(Alイオン)
ここで、本発明に係る組成物はアミノポリカルボン酸を含有するが、特にカチオン性の樹脂と組み合わせた場合、過剰なアミノポリカルボン酸の存在により、ときとしてカチオン性樹脂のゲル化を招くことがある。このような場合には、カチオン性樹脂のカチオン基の量を減らすか或いはノニオン性の樹脂とする(或いは、カチオン性樹脂とノニオン性樹脂とを混合し、全体的なカチオン基量を相対的に減少させる)のが好適である。ところで、この場合、pHの上昇によっても樹脂があまり析出しないという別の問題を生じることがある。ここで、当該問題は、Alイオンを含有させることにより解消することが可能となる。この際、Alイオンを20〜500ppm含有することが好ましい。50〜400ppmが更に好ましく、100〜300ppmが最も好ましい。下限を下回るとAlイオンの塗膜析出向上効果が不充分となり、上限を上回ると組成物の電気伝導度が過剰となり、かえって付き廻り性を低下させる。
【0066】
ここで、前述したAlイオンの作用機序は以下の通りである。つまり、イオン状のAlがカソード電解による金属表面pH上昇により微細な水酸化物コロイドになり、それがpH9前後でゼータ電荷を完全に失い急激に凝集を始める際、周りの樹脂をも巻き込んで析出するものと推定される。
【0067】
カソード電解によってAlイオンから水酸化物コロイドの電荷の消失にいたる一連の反応は瞬時に完了する必要がある。あらかじめ水酸化物になっていては、経時で凝集が始まってしまい、pH9前後での凝集能力が極端に減退する。よって、当該態様におけるAl成分は、組成物中ではあくまでイオンでいなければならないのである。
【0068】
また、金属イオンは通常キレート剤の存在によって安定化されるが、Alイオンの場合は、pH上昇に伴う水酸化物コロイドの生成を阻止する程の安定度を有するキレート剤は無い又は稀である。少なくとも、電着塗料組成物に通常配合されている、酢酸、蟻酸、スルファミン酸、乳酸等の有機酸及びアミノポリカルボン酸には、Alイオンを安定化させるほどのキレート能力はない。
【0069】
AlイオンはAl化合物を用いて添加することができる。Al化合物は特に限定されないが、硝酸塩、硫酸塩と言った無機酸塩又は乳酸塩、酢酸塩と言った有機酸塩の形で添加することが可能である。
【0070】
更に、Alイオンを前述の範囲で含有することに加え、当該態様に係る組成物のpHをAlイオン濃度をA[ppm]としたとき次の計算式を満足するようにすることが好ましい。
3.5≦pH≦−Log((A×1.93×10−15)1/3)
下記式であることが更に好ましい。
3.6≦pH≦−Log((A×1.93×10−15)1/3)
下記式であることが最も好ましい。
3.7≦pH≦−Log((A×1.93×10−15)1/3)
pHが下限を下回ると、析出効率が低下し付き廻り性も低下していく。pHが上限を上回ると、Alイオンが加水分解を起こしてしまうため、好ましくない。
【0071】
−Log((A×1.93×10−15)1/3)の項は、25℃における水酸化Alの溶解度積:1.92×10−32から求められる。つまり、このpH以上になるとAlイオンは水酸化物として沈殿析出してしまい、もはやイオンではいられなくなる。ここで、25℃は、組成物の保存時及び使用時の典型的な温度である。
【0072】
また、本発明の組成物の中には、Biイオン、Alイオンの他に、Feイオン、Znイオン、Ceイオン等の金属イオンを含有しても、本発明の効果を損なうものではない。むしろ、これらの金属イオンには、Alイオンほどではないものの、水系樹脂の析出を促進させる作用を有する。なお、Feイオンは2価よりも3価がより好ましい。
【0073】
参考のため、Alイオン濃度及びpHの適正範囲を第2図に示す。
【0074】
<金属表面処理用組成物の物性>
(pH)
本発明に係る金属表面処理用組成物のpHは特に制限されるものではないが、通常2.0〜7.0、好ましくは3.0〜6.5の範囲に調整して使用することができる。
【0075】
(温度)
本発明に係る金属表面処理用組成物の温度についても特に制約は無いが、電解処理によって皮膜を析出させる際は、通常15〜40℃、好ましくは20〜35℃の範囲内で使用することができる。
【0076】
《金属表面処理用組成物の製造方法》
次に、本発明に係る金属表面処理用組成物の製造方法を説明することとする。尚、水系樹脂として変性エポキシ樹脂のアミノ化物を用いた場合を例に採り説明する。先ず、合成した変性エポキシ樹脂のアミノ化物に中和酸を添加し、撹拌混合した後、水で希釈し、所定濃度の樹脂エマルションを作製する。中和酸は、蟻酸、酢酸、乳酸、スルファミン酸などが用いられる。
【0077】
この際、中和酸を添加する前に硬化剤や硬化触媒、有機溶剤などを添加しておくことが好ましい。あらかじめ添加をすることで、均一なエマルションを得ることができる。
【0078】
硬化剤はブロックポリイソシアネートを用いることが一般的である。ブロックポリイソシアネートは、ポリイソシアネート化合物とイソシアネートブロック剤とのほぼ化学理論量での付加反応生成物である。ここで使用されるポリイソシアネート化合物としては、例えば、トリレンジイソシアネート、キシリレンジイソシアネート、フェニレンジイソシアネート、ジフェニルメタン−2,4'−ジイソシアネート、ジフェニルメタン−4,4'−ジイソシアネート(通常「MDI」と呼ばれる)、クルードMDI、ビス(イソシアネートメチル)シクロヘキサン、テトラメチレンジイソシアネート、ヘキサメチレンジイソシアネート、メチレンジイソシアネート、イソホロンジイソシアネートなどの芳香族、脂肪族又は脂環族のポリイソシアネート化合物;これらのポリイシアネート化合物の環化重合体、イソシアネートビゥレット体;これらのイソシアネート化合物の過剰量にエチレングリコール、プロピレングリコール、トリメチロールプロパン、ヘキサントリオール、ヒマシ油などの低分子活性水素含有化合物を反応させて得られる末端イソシアネート含有化合物などを挙げることができる。これらはそれぞれ単独で又は2種以上組合わせて使用することができる。
【0079】
一方、前記イソシアネートブロック剤は、ポリイソシアネート化合物のイソシアネート基に付加してブロックするものであり、そして付加によって生成するブロックポリイソシアネート化合物は常温において安定であるが、塗膜の焼付け温度(通常約100〜約200℃)に加熱した際、ブロック剤が解離して遊離のイソシアネート基を再生しうるものであることが望ましい。
【0080】
このような要件を満たすブロック剤としては、例えば、ε−カプロラクタム、γ−ブチロラクタムなどのラクタム系化合物;メチルエチルケトオキシム、シクロヘキサノンオキシムなどのオキシム系化合物;フェノール、パラ−t−ブチルフェノール、クレゾールなどのフェノール系化合物;n−ブタノール、2−エチルヘキサノールなどの脂肪族アルコール類;フェニルカルビノール、メチルフェニルカルビノールなどの芳香族アルキルアルコール類;エチレングリコールモノブチルエーテル、ジエチレングリコールモノエチルエーテルなどのエーテルアルコール系化合物等を挙げることができる。これらのブロック剤はそれぞれ単独で又は2種以上組み合わせて使用することができる。また、ブロック剤の解離、硬化反応などを効率よく進め、また、意図する硬化反応物を生成させるために、あらかじめ、変性エポキシ樹脂の骨格にイソシアネート基の一部を付加し、かつ、残りのイソシアネート基をブロック剤でブロックする手法もとられる。
【0081】
次に、得られたエマルションに対し、Biイオン水溶液を添加する。Biイオン水溶液は、所定濃度のポリアミノカルボン酸を水に溶解させ、そこに硝酸ビスマス5水和物を添加し、50〜70℃で溶解するまで撹拌することで得ることができる。
【0082】
通常は塗膜性能を向上させたり、着色のため、ここに顔料を添加する。顔料は分散樹脂を用い、あらかじめ分散体(ペースト)としたものを用いる。
【0083】
これらを充分に撹拌したものが金属表面処理用組成物となる。
【0084】
《金属表面処理用組成物の使用方法(用途)》
(適用対象)
本発明に係る金属表面処理用組成物は、各種金属を腐食から防止する目的で使用される。金属材料は、特に限定されるものではないが、冷延鋼板、熱延鋼板、鋳物材、鋼管等の鉄鋼材料、それらの鉄鋼材料の上に亜鉛系めっき処理及び/又はアルミニウム系めっきが施された材料、アルミニウム合金板、アルミニウム系鋳物材、マグネシウム合金版、マグネシウム系鋳物材等が挙げられる。特に形状が複雑な金属構成体、例えば、鉄系材料を主とする金属構成体である自動車車体、自動車部品、家電製品、建築材料等への使用に適している。
【0085】
(使用方法<金属表面処理方法>)
本発明に係る使用方法(金属表面処理方法)は、前述した金属表面処理用組成物を用い、材料を陰極とした電解処理工程にて金属材料表面に塗膜を析出させる工程を含む。より好適には、本発明に係る使用方法(金属表面処理方法)は、金属材料上に皮膜を析出させるべく、表面が清浄化された金属材料に対して電解処理を施す電解処理工程と、電解処理工程後に実行する水洗及び焼付け工程を含む。以下、本方法に特徴的な電解処理工程について詳述する。
【0086】
この電解処理工程(カソード電解)は、金属表面処理用組成物中に前記金属材料を浸漬させた状態で、電圧0〜15Vにて10〜120秒間電解する第一工程と、金属表面処理用組成物中に前記金属材料を浸漬させた状態で、電圧50〜300Vにて30〜300秒間電解する、前記第一工程の後に実施する第二工程とを含み、ここで、前記第二工程は、前記第一工程に引き続いて同一浴内で実施するか又は前記第一工程とは異なる別浴内で実施する。
【0087】
ここで、第一工程は主としてBiを優先的に付着させるために行われる工程であり、第二工程は主として樹脂を優先的に析出させるために行われる工程である。充分な耐食性を得るためには、金属材料に直接接触しているBi、つまり金属材料と皮膜の界面に存在する界面Biの存在が必要であり、そのためには第一工程と第二工程の順番と条件が極めて重要となってくる。
【0088】
第一工程の電圧は0〜15Vであり、10〜120秒間電解することが好ましい。電圧が下限を下回る場合、すなわち金属材料を陽極として電解した場合は、金属材料が組成物中に溶出してしまい、組成物の安定性を低下させるばかりか、耐食性の向上に必要な界面Biが充分付着しなくなる。上限を超える場合も、Biが金属表面に優先的に析出する前に樹脂析出が始まってしまうため、やはり充分な耐食性が得られなくなる。
【0089】
処理時間が下限を下回る場合も充分な界面Biが析出せず、上限を上回る場合は界面Biの付着量過多となり、皮膜の密着性が損なわれる場合がある。
【0090】
第二工程の電圧は50〜300Vであり、30〜300秒間電解することが好ましい。電圧が下限を下回る場合は、樹脂皮膜の析出量が不充分となり、上限を上回る場合は、樹脂皮膜の析出過多により経済的に不利であるばかりか、皮膜の仕上がり外観が損なわれる場合がある。
【0091】
第一工程に次いで第二工程に移行する際、電圧を瞬時に増加させる必要は無く、緩やかに増加させても本発明の効果を損なうものではない。
【0092】
更に、本発明における二段階電解処理は同一槽で行う必要は必ずしもなく、それぞれ異なる槽で実行してもよい。二段階電解処理を二槽で行う場合、第一槽と第二槽とで第一行程、第二行程にあわせたBi濃度、樹脂濃度の調整が可能となり、コストメリットが図れる。例えば、第二槽のBi濃度を低下させることで、持ち出し液中に含まれるBi量を減らすことができ、コスト低減につなげられる。
【0093】
《金属表面処理皮膜》
本発明に係る金属表面処理皮膜は、本発明の金属表面処理用組成物を用い、本発明の処理方法によって得られる。ここで、皮膜中に存在するBiは金属及び酸化物の形態で存在する。カソード電解によって析出するBiは、基本的に還元析出した金属Biであるが、その一部は特に皮膜の焼付け工程で酸化されて酸化物となる。また、第二工程において高電圧がかかった場合、皮膜表面のpH上昇により、アミノポリカルボン酸によるBiの安定化が不充分となるため、特に皮膜表面側では酸化Biとしても析出する。
【0094】
Bi付着量は20〜500mg/m2が好ましく、30〜400mg/m2が更に好ましく、50〜300mg/m2が最も好ましい。Bi付着量が低過ぎると充分な耐食性が得られず、高過ぎるともはや耐食性の向上が望めないばかりか皮膜密着性を損なう場合もある。尚、Bi付着量は蛍光X線分光分析により定量可能である。尚、本特許請求の範囲及び本明細書における「金属Bi付着量」及び「酸化Bi付着量」は、当該蛍光X線分光分析で定量された値とする。尚、その他の形態として水酸化物の存在も否定できないが、当該測定方法で「金属Bi」又は「酸化Bi」として定量された場合には、その数値は「金属Bi付着量」又は「酸化Bi付着量」とすることとする。
【0095】
得られる皮膜の全皮膜厚は5〜40μmが好ましく、5〜30μmが更に好ましく、7〜25μmが最も好ましい。薄過ぎると充分な耐食性が得られず、厚過ぎると経済的に不利なばかりか付き廻り性が低下する場合がある。皮膜厚は、素地金属が磁性金属であれば電磁誘導式膜厚計、素地金属が非磁性金属であれば渦電流式膜厚計により、測定可能である。
【0096】
皮膜中のBiは、皮膜表面よりも素地金属側により多く存在する必要がある。具体的には、皮膜厚の中心から金属材料側のBi付着量:Bが、全Bi付着量:Aに対して55%以上(B/A≧55%)となるBi付着分布であることが好ましい。58%以上が更に好ましく、60%以上が最も好ましい。低過ぎると充分な耐食性が得られない。なお、90%を超えると皮膜表面側のBi濃度が極端に低下し、Biの持つ硬化触媒としての機能を失うので好ましくない。
【0097】
皮膜中のBi付着分布については、EPMAを用いて皮膜断面を線分析することにより測定可能である。同時に撮影した反射電子像によって素地金属と皮膜の界面及び皮膜表面の位置を特定し、EPMA線分析による皮膜中のBi強度の積分値:A及び皮膜厚の中心から素地金属側のみの積分値:Bを求め、B/Aを算出することができる。
【実施例】
【0098】
フェノール化合物(D)の合成
ビスフェノールA(和光純薬):264.8gをブチルセロソルブ(和光純薬):100gとエタノール(純正化学):100gの混合溶媒に完全に溶解させた後、ジエタノールアミン(和光純薬):121.8g、ホルムアルデヒド液(純正化学):96.8gを加え、75℃に加温した。75℃の条件下で4日間撹拌を行い、その後、エバポレーターにて溶剤を除去することでフェノール化合物(D1)を得た。NMR分析より、この時得られた化合物の構造は式3であることが確認できた。
(式7)
【0099】
ビスフェノールA(和光純薬):264.8gをブチルセロソルブ(和光純薬):100gとエタノール(純正化学):100gの混合溶媒に完全に溶解させた後、グリシン(東京化成):87.1g、ホルムアルデヒド液(純正化学):96.8gを加え、75℃に加温した。75℃の条件下で6日間撹拌を行い、その後、エバポレーターにて溶剤を除去することでフェノール化合物(D2)を得た。NMR分析より、この時得られた化合物の構造は式4であることが確認できた。
(式8)
【0100】
ビスフェノールA(和光純薬):264.8gをブチルセロソルブ(和光純薬):100gとエタノール(純正化学):100gの混合溶媒に完全に溶解させた後、アセチルエチルアミン(東京化成):101.1g、ホルムアルデヒド液(純正化学):96.8gを加え、75℃に加温した。75℃の条件下で6日間撹拌を行い、その後、エバポレーターにて溶剤を除去することでフェノール化合物(D3)を得た。NMR分析より、この時得られた化合物の構造は式5であることが確認できた。
(式9)
【0101】
ビスフェノールA(和光純薬):264.8gをブチルセロソルブ(和光純薬):100gとエタノール(純正化学):100gの混合溶媒に完全に溶解させた後、ジエチルアミン(東京化成):84.8g、ホルムアルデヒド液(純正化学):96.8gを加え、75℃に加温した。75℃の条件下で4日間撹拌を行い、その後、エバポレーターにて溶剤を除去することでフェノール化合物(D4)を得た。NMR分析より、この時得られた化合物の構造は式6であることが確認できた。
(式10)
【0102】
ビスフェノールA(和光純薬):264.8gをブチルセロソルブ(和光純薬):100gとエタノール(純正化学):100gの混合溶媒に完全に溶解させた後、エチルアミン(メタノール溶液)(東京化成):149.4g、ホルムアルデヒド液(純正化学):96.8gを加え、75℃に加温した。75℃の条件下で6日間撹拌を行い、その後、エバポレーターにて溶剤を除去することでフェノール化合物(D5)を得た。NMR分析より、この時得られた化合物の構造は式7であることが確認できた。
(式11)
【0103】
ビスフェノールA(和光純薬):264.8gをブチルセロソルブ(和光純薬):100gとエタノール(純正化学):100gの混合溶媒に完全に溶解させた後、アンモニア(エタノール溶液)(東京化成):580.0g、ホルムアルデヒド液(純正化学):96.8gを加え、75℃に加温した。75℃の条件下で6日間撹拌を行い、その後、エバポレーターにて溶剤を除去することでフェノール化合物(D6)を得た。NMR分析より、この時得られた化合物の構造は式8であることが確認できた。
(式12)
【0104】
Biイオンキレート能の確認
Biイオン水溶液(B2、下記参照):100gにあらかじめエタノール50gに溶解させたフェノール化合物(D1):1.7gを加え、60℃に加温しながら撹拌を行った。得られた溶液について、液体クロマトグラフィー質量分析法を用いることで、フェノール化合物(D1)はBiをキレートしていることが確認できた。なお、この場合フェノール化合物(D1)はBiの1.0倍モル濃度となる。
【0105】
Biイオン水溶液(B2、下記参照):100gにあらかじめエタノール50gに溶解させたビスフェノールAの溶媒溶液:1.1gを加え、60℃に加温しながら撹拌を行った。得られた溶液について、液体クロマトグラフィー質量分析法を用いることで、ビスフェノールAはBiをキレートしていないことが確認できた。なお、この場合ビスフェノールAはBiの1.0倍モル濃度となる。
【0106】
Biイオン水溶液(B2、下記参照):100gにあらかじめエタノール50gに溶解させたフェノール化合物(D2):1.5gを加え、60℃に加温しながら撹拌を行った。得られた溶液について、液体クロマトグラフィー質量分析法を用いることで、フェノール化合物(D2)はBiをキレートしていることが確認できた。なお、この場合フェノール化合物(D2)はBiの1.0倍モル濃度となる。
【0107】
Biイオン水溶液(B2、下記参照):100gにあらかじめエタノール50gに溶解させたフェノール化合物(D3):1.6gを加え、60℃に加温しながら撹拌を行った。得られた溶液について、液体クロマトグラフィー質量分析法を用いることで、フェノール化合物(D3)はBiをキレートしていることが確認できた。なお、この場合フェノール化合物(D3)はBiの1.0倍モル濃度となる。
【0108】
Biイオン水溶液(B2、下記参照):100gにあらかじめエタノール50gに溶解させたフェノール化合物(D4):1.5gを加え、60℃に加温しながら撹拌を行った。得られた溶液について、液体クロマトグラフィー質量分析法を用いることで、フェノール化合物(D4)はBiをキレートしていることが確認できた。なお、この場合フェノール化合物(D4)はBiの1.0倍モル濃度となる。
【0109】
Biイオン水溶液(B2、下記参照):100gにあらかじめエタノール50gに溶解させたフェノール化合物(D5):1.4gを加え、60℃に加温しながら撹拌を行った。得られた溶液について、液体クロマトグラフィー質量分析法を用いることで、フェノール化合物(D5)はBiをキレートしていることが確認できた。なお、この場合フェノール化合物(D5)はBiの1.0倍モル濃度となる。
【0110】
Biイオン水溶液(B2、下記参照):100gにあらかじめエタノール50gに溶解させたフェノール化合物(D6):1.2gを加え、60℃に加温しながら撹拌を行った。得られた溶液について、液体クロマトグラフィー質量分析法を用いることで、フェノール化合物(D6)はBiをキレートしていることが確認できた。なお、この場合フェノール化合物(D6)はBiの1.0倍モル濃度となる。
【0111】
ブロック化イソシアネートの作製
コスモネートM200(三井化学株式会社製):678.4gにメチルイソブチルケトン:115.6gを加え、70℃に昇温した後、ジエチレングリコールモノエチルエーテル:706.0gをゆっくり滴下し、滴下終了後、90℃に昇温した。90℃の条件下で12時間反応させ、ブロック化イソシアネートを得た。赤外吸収スペクトル測定を行ったところ、未反応のイソシアネート基由来の吸収が見られず、イソシアネートが完全にブロック化されたことが確認できた。
【0112】
水系樹脂エマルションの作製
製造例1
温度計、コンデンサ、攪拌機を備えた1000mlセパラブルフラスコにエポキシ樹脂・#828(ジャパンエポキシレジン株式会社製、エポキシ当量:180):114.0g、変性樹脂としてポリカプロラクトンジオール・プラクセル208(ダイセル化学株式会社製):41.5g、ビスフェノールA:45.6g、ジメチルベンジルアミン0.1gを加え、130℃でエポキシ当量1000になるまで反応を行った。反応終了後にブチルセロソルブ55.5gを加え、更にジエタノールアミン:12.6g、ジエチレントリアミンのケチミン化物:8.0gを加え、90℃で2時間反応を行った。ここにブロック化イソシアネート:105.5g、ジブチル錫ジアセテート:3.2g、酢酸5.4gを加え、均一になるまで撹拌を行った後、脱イオン水578.1gを強く撹拌しながら約1時間かけて滴下し、固形分濃度33%の水系樹脂エマルション(A1)を得た。
【0113】
製造例2
製造例1において、ビスフェノールAの代わりに、ビスフェノールA:31.9g、フェノール化合物(D1):20.8gを使用し、同様な反応を行うことで、水系樹脂エマルション(A2)を得た。
【0114】
製造例3
製造例1において、ビスフェノールAの代わりに、ビスフェノールA:13.7g、フェノール化合物(D1):48.4gを使用し、同様な反応を行うことで、水系樹脂エマルション(A3)を得た。
【0115】
製造例4
同様に、ビスフェノールAの代わりに、ビスフェノールA:31.9g、D2:19.0gを使用し、同様な反応を行うことで、水系樹脂エマルション(A4)を得た。
【0116】
製造例5
同様に、ビスフェノールAの代わりに、ビスフェノールA:31.9g、D3:19.7gを使用し、同様な反応を行うことで、水系樹脂エマルション(A5)を得た。
【0117】
製造例6
同様に、ビスフェノールAの代わりに、ビスフェノールA:22.8g、D4:18.9gを使用し、同様な反応を行うことで、水系樹脂エマルション(A6)を得た。
【0118】
製造例7
同様に、ビスフェノールAの代わりに、ビスフェノールA:22.8g、D5:17.2gを使用し、同様な反応を行うことで、水系樹脂エマルション(A7)を得た。
【0119】
製造例8
同様に、ビスフェノールAの代わりに、ビスフェノールA:22.8g、D6:15.5gを使用し、同様な反応を行うことで、水系樹脂エマルション(A8)を得た。
【0120】
製造例9
温度計、コンデンサ、攪拌機を備えた2000mlセパラブルフラスコにエポキシ樹脂・#828(ジャパンエポキシレジン株式会社製、エポキシ当量:180):114.0g、変性樹脂としてポリカプロラクトンジオール・プラクセル208(ダイセル化学株式会社製):41.5g、ビスフェノールA:31.9g、フェノール化合物(D1):20.8g、ビスフェノールAエチレンオキサイド付加物・ニューポールBPE−100(三洋化成工業株式会社製):100.2g、ジメチルベンジルアミン0.1gを加え、130℃でエポキシ当量1500になるまで反応を行い、反応終了後にブチルセロソルブ75.3gを加えた。ここにブロック化イソシアネート:143.4g、ジブチル錫ジアセテート:4.3gを加え、均一になるまで撹拌を行った後、脱イオン水792.9gを強く撹拌しながら約1時間かけて滴下し、固形分濃度33%の水系樹脂エマルション(A9)を得た。
【0121】
顔料ペーストの作製
60%の第四級塩化エポキシ樹脂8.3部に対し、精製クレー7.0部、カーボンブラック0.3部、リン酸亜鉛3.0部および脱イオン水を加え、ボールミルにて20時間分散し、固形分50重量%の顔料分散ペーストを得た。
【0122】
Biイオン液の作製
蒸留水:500gにHEDTA:13.3gを溶解させ、60℃に加温した後、硝酸ビスマス5水和物:23.2gを加えて固形分が完全に溶解するまで撹拌した。最終的に全量が1.0Lとなるように更に蒸留水を加え、Biイオン水溶液(B1)を作製した。なお、この場合HEDTAはBiの1.0倍モル濃度となる。
蒸留水:500gにHEDTA:6.65gを溶解させ、60℃に加温した後、硝酸ビスマス5水和物:23.2gを加えて撹拌した。最終的に全量が1.0Lとなるように更に蒸留水を加え、薄く濁ったBiイオン液(B2)を作製した。なお、この場合HEDTAはBiの0.5倍モル濃度となる。
蒸留水:500gにHEDTA:2.66gを溶解させ、60℃に加温した後、硝酸ビスマス5水和物:23.2gを加えて撹拌した。最終的に全量が1.0Lとなるように更に蒸留水を加え、濁ったBiイオン液(B3)を作製した。なお、この場合HEDTAはBiの0.2倍モル濃度となる。
蒸留水:500gにHEDTA:1.33gを溶解させ、60℃に加温した後、硝酸ビスマス5水和物:23.2gを加えて撹拌した。最終的に全量が1.0Lとなるように更に蒸留水を加え、濃く濁ったBiイオン液(B4)を作製した。なお、この場合HEDTAはBiの0.1倍モル濃度となる。
蒸留水:500gにEDTA:6.99gを溶解させ、60℃に加温した後、硝酸ビスマス5水和物:23.2gを加えて固形分が完全に溶解するまで撹拌した。最終的に全量が1.0Lとなるように更に蒸留水を加えBiイオン水溶液(B5)を作製した。なお、この場合、EDTAはBiの0.5倍モル濃度となる。
蒸留水:500gにNTA:45.8gを溶解させ、60℃に加温した後、硝酸ビスマス5水和物:23.2gを加えて固形分が完全に溶解するまで撹拌した。最終的に全量が1.0Lとなるように更に蒸留水を加えBiイオン水溶液(B6)を作製した。なお、この場合NTAはBiの5.0倍モル濃度となる。
【0123】
組成物の作製
第1表に示す組合せの固形分16.0重量%になる量の樹脂エマルジョンに無機固形分4.0重量%になる量の顔料分散ペースト及びBi添加剤を配合し、組成物を作製した。なお、それぞれの濃度は脱イオン水を用いて希釈し調整した。尚、実施例1〜11、比較例1、2については、いずれも処理液のpHは5であった。実施例12については、硝酸アルミニウム・9水和物と硝酸を用い、Al濃度:200ppm、pH4に調整を行った。
【0124】
電解条件
電解工程(1)として8Vにて60秒間電解後、直ちに電解工程(2)として180Vにて180秒間電解処理を行った。
【0125】
試験板の作製
試験板として、冷延鋼板:SPCC(JIS3141)70×150×0.8mm(以下、SPCと略す)を用い、あらかじめその表面を日本パーカライジング社製強アルカリ脱脂剤「FC−E2001」を使用して、120秒間スプレー処理することにより脱脂処理した。脱脂処理後は30秒間スプレー水洗し、実施例及び比較例に示す組成物に浸漬させ、実施例及び比較例に示す電解条件にてカソード電解処理を実施した。電解終了後の試験板は直ちに脱イオン水にて30秒間スプレー水洗し、電気オーブン中で180℃にて20分間焼付けを行った。
【0126】
皮膜特性の調査
試験板の上に析出した皮膜の皮膜特性を以下の方法で調査した。
皮膜厚測定:電磁誘導式膜厚計を用いて測定した。
Bi付着量:蛍光X線分光分析によって定量した。
Bi付着分布:試料断面をEPMAの線分析にて分析した。具体的方法は下記参照。
【0127】
皮膜中のBi付着量分布測定は、EPMAを用いて分析した。皮膜処理後の金属材料を、埋め込み樹脂によって固定し、断面を研磨し、素地金属方向から析出皮膜表面方向にBiの線分析プロファイルを求めた。線分析プロファイルとは、マッピング分析データを基に、分析エリアの1次元方向に任意の幅で特性X線強度の平均値を算出したもので、幅を持った線分析と解することができる。測定条件は以下の通り。
【0128】
測定機器:島津製作所製EPMA−1610型
電子銃:CeB6カソード型
ビーム電流:50nA、ビーム電圧:15kV、ビーム径:1μmφ以下
積算回数:1回、1点あたりのサンプリング時間:100ms
分光結晶:PET(Bi Mα)
【0129】
同時に撮影した反射電子像によって素地金属と皮膜の界面及び皮膜表面の位置を特定し、皮膜中のBi強度の積分値:A及び皮膜厚の中心から素地金属側のみの積分値:Bを求め、B/Aを算出した。
なお、参考のため代表的なプロファイルとして実施例1で得られた皮膜の分析結果を第1図に示す。
【0130】
耐食性試験方法および評価方法
カソード電解処理により作製された樹脂塗装板にクロスカットを施し、塩水噴霧試験(JIS−Z2371)を実施し、1500時間後のクロスカット部の片側最大膨れ幅を測定した。測定結果を基に、2mm未満:◎、2mm以上3mm未満:○、3mm以上4mm未満:△、4mm以上:×にて評価した。結果を第2表に示す。
【0131】
外観性試験方法および評価方法
カソード電解処理により作製された樹脂塗装板の光沢度を測定した。
光沢計:日本電色工業株式会社製 VG2000
測定角度:60°
測定結果を基に、65以上:○、65未満:×にて評価した。結果を第2表に示す。
【表1】
【表2】
【特許請求の範囲】
【請求項1】
水系樹脂を5〜30重量%、3価のBiイオンを100〜5000ppm及びBiイオンに対して0.1〜5倍モル濃度のアミノポリカルボン酸を含有し、
水系樹脂の少なくとも一部が、下記式(1):
【化1】
{ここで、R1及びR2は、相互に独立して、−(R)m−X(ここで、Rは、炭素数1〜6のアルキレン基であり、mは1か0であり、Xは、水素、カルボキシル、ヒドロキシル又はアシルである}で示される、Biイオンとキレートを形成可能な窒素含有基をポリマー骨格中に有するノニオン性及び/又はカチオン性樹脂である
ことを特徴とする金属表面処理用組成物。
【請求項2】
式(1)において、R1及びR2は、相互に独立して、水素、アルキル、ヒドロキシアルキル、カルボキシアルキル又はアルキルカルボニルであり、ここで、アルキル、ヒドロキシアルキル、カルボキシアルキル及びアルキルカルボニルのアルキル部分は、炭素数1〜6の直鎖状、分岐鎖状又は環状である、請求項1記載の金属表面処理用組成物。
【請求項3】
水系樹脂が、変性エポキシ樹脂である、請求項1又は2記載の金属表面処理用組成物。
【請求項4】
変性エポキシ樹脂が、ビスフェノールA型であり、前記窒素含有基が、前記変性エポキシ樹脂中のグリシジルエーテル部ではなくフェニレン基のベンゼン環部に直接結合している、請求項3記載の金属表面処理用組成物。
【請求項5】
ビスフェノールA型の変性エポキシ樹脂(A)が、原料として、変性樹脂(B)、エポキシ当量180〜2500のエポキシ樹脂(C)及び前記窒素含有基を有するフェノール化合物(D)を用い、或いは更に2級アミノ基含有化合物(F)及び/又はビスフェノールA(E)をも用い、これらを反応させることで得られる変性エポキシ樹脂のアミノ化物である、請求項4記載の金属表面処理用組成物。
【請求項6】
化合物(D)が、下記式(2):
(式2)
(式中、R3は、前記窒素含有基又は水素原子であり、R3の少なくとも1個は前記窒素含有基であり;R4は、相互に独立して水素又は炭素数1〜2のアルキル基である)で示されるアミン付加フェノール化合物である、請求項5記載の金属表面処理用組成物。
【請求項7】
3価のBiイオンに対して、前記窒素含有基のモル濃度が0.1〜200倍である、請求項1〜6のいずれか一項記載の金属表面処理用組成物。
【請求項8】
請求項1〜7のいずれか一項に記載の組成物を用い、材料を陰極とした電解処理工程にて金属材料表面に塗膜を析出させる工程を含む、金属表面処理方法。
【請求項9】
表面が清浄化された金属材料に対して電解処理を施す電解処理工程と、電解処理工程後に実行する水洗及び焼付け工程を含む、金属材料上に皮膜を析出させる金属表面処理方法において、
前記電解処理工程は、
請求項1〜7のいずれか一項記載の組成物中に前記金属材料を浸漬させた状態で、電圧0〜15Vにて10〜120秒間電解する第一工程と、
請求項1〜7のいずれか一項記載の組成物中に前記金属材料を浸漬させた状態で、電圧50〜300Vにて30〜300秒間電解する、前記第一工程の後に実施する第二工程と
を含み、ここで、前記第二工程は、前記第一工程に引き続いて同一浴内で実施するか又は前記第一工程とは異なる別浴内で実施することを特徴とする方法。
【請求項10】
請求項8又は9記載の方法によって形成される、金属Bi及び酸化BiがBiとして20〜500mg/m2付着し、全皮膜厚が5〜40μmであり、かつ皮膜厚の中心から金属材料側のBi付着量(G)が、全Bi付着量(H)に対して55%以上(G/H≧55%)となるBi付着分布であることを特徴とする金属表面処理皮膜。
【請求項1】
水系樹脂を5〜30重量%、3価のBiイオンを100〜5000ppm及びBiイオンに対して0.1〜5倍モル濃度のアミノポリカルボン酸を含有し、
水系樹脂の少なくとも一部が、下記式(1):
【化1】
{ここで、R1及びR2は、相互に独立して、−(R)m−X(ここで、Rは、炭素数1〜6のアルキレン基であり、mは1か0であり、Xは、水素、カルボキシル、ヒドロキシル又はアシルである}で示される、Biイオンとキレートを形成可能な窒素含有基をポリマー骨格中に有するノニオン性及び/又はカチオン性樹脂である
ことを特徴とする金属表面処理用組成物。
【請求項2】
式(1)において、R1及びR2は、相互に独立して、水素、アルキル、ヒドロキシアルキル、カルボキシアルキル又はアルキルカルボニルであり、ここで、アルキル、ヒドロキシアルキル、カルボキシアルキル及びアルキルカルボニルのアルキル部分は、炭素数1〜6の直鎖状、分岐鎖状又は環状である、請求項1記載の金属表面処理用組成物。
【請求項3】
水系樹脂が、変性エポキシ樹脂である、請求項1又は2記載の金属表面処理用組成物。
【請求項4】
変性エポキシ樹脂が、ビスフェノールA型であり、前記窒素含有基が、前記変性エポキシ樹脂中のグリシジルエーテル部ではなくフェニレン基のベンゼン環部に直接結合している、請求項3記載の金属表面処理用組成物。
【請求項5】
ビスフェノールA型の変性エポキシ樹脂(A)が、原料として、変性樹脂(B)、エポキシ当量180〜2500のエポキシ樹脂(C)及び前記窒素含有基を有するフェノール化合物(D)を用い、或いは更に2級アミノ基含有化合物(F)及び/又はビスフェノールA(E)をも用い、これらを反応させることで得られる変性エポキシ樹脂のアミノ化物である、請求項4記載の金属表面処理用組成物。
【請求項6】
化合物(D)が、下記式(2):
(式2)
(式中、R3は、前記窒素含有基又は水素原子であり、R3の少なくとも1個は前記窒素含有基であり;R4は、相互に独立して水素又は炭素数1〜2のアルキル基である)で示されるアミン付加フェノール化合物である、請求項5記載の金属表面処理用組成物。
【請求項7】
3価のBiイオンに対して、前記窒素含有基のモル濃度が0.1〜200倍である、請求項1〜6のいずれか一項記載の金属表面処理用組成物。
【請求項8】
請求項1〜7のいずれか一項に記載の組成物を用い、材料を陰極とした電解処理工程にて金属材料表面に塗膜を析出させる工程を含む、金属表面処理方法。
【請求項9】
表面が清浄化された金属材料に対して電解処理を施す電解処理工程と、電解処理工程後に実行する水洗及び焼付け工程を含む、金属材料上に皮膜を析出させる金属表面処理方法において、
前記電解処理工程は、
請求項1〜7のいずれか一項記載の組成物中に前記金属材料を浸漬させた状態で、電圧0〜15Vにて10〜120秒間電解する第一工程と、
請求項1〜7のいずれか一項記載の組成物中に前記金属材料を浸漬させた状態で、電圧50〜300Vにて30〜300秒間電解する、前記第一工程の後に実施する第二工程と
を含み、ここで、前記第二工程は、前記第一工程に引き続いて同一浴内で実施するか又は前記第一工程とは異なる別浴内で実施することを特徴とする方法。
【請求項10】
請求項8又は9記載の方法によって形成される、金属Bi及び酸化BiがBiとして20〜500mg/m2付着し、全皮膜厚が5〜40μmであり、かつ皮膜厚の中心から金属材料側のBi付着量(G)が、全Bi付着量(H)に対して55%以上(G/H≧55%)となるBi付着分布であることを特徴とする金属表面処理皮膜。
【図2】

【図1】
【図1】
【公開番号】特開2011−57944(P2011−57944A)
【公開日】平成23年3月24日(2011.3.24)
【国際特許分類】
【出願番号】特願2009−212211(P2009−212211)
【出願日】平成21年9月14日(2009.9.14)
【出願人】(000229597)日本パーカライジング株式会社 (198)
【Fターム(参考)】
【公開日】平成23年3月24日(2011.3.24)
【国際特許分類】
【出願日】平成21年9月14日(2009.9.14)
【出願人】(000229597)日本パーカライジング株式会社 (198)
【Fターム(参考)】
[ Back to top ]