
金属試料中の着目元素の固溶含有率を求める方法
【課題】金属試料中の着目元素の固溶含有率を簡便、迅速かつ直接的に求める方法を提供する
【解決手段】まず、金属試料を電解する。次いで、電解中および/または電解後に電解液の一部を採取し、採取された電解液を分析する。そして、分析の結果を基に、電解液中における、比較元素に対する着目元素の濃度比を算出し、算出された濃度比に金属試料における比較元素の含有率を乗じることで、金属試料中の着目元素の固溶含有率を求める。例えば、鉄鋼試料中のチタンの固溶含有率を求める場合、上記方法により、分析溶液中のチタン濃度(KTi)及び比較元素として選択した鉄の濃度(KFe)を、それぞれICP質量分析装置で測定する。そして、その濃度比(KTi/KFe)に、比較元素の含有率(鉄の組成値)を乗じて、鉄鋼試料中のチタンの固溶含有率を求めることができる。
【解決手段】まず、金属試料を電解する。次いで、電解中および/または電解後に電解液の一部を採取し、採取された電解液を分析する。そして、分析の結果を基に、電解液中における、比較元素に対する着目元素の濃度比を算出し、算出された濃度比に金属試料における比較元素の含有率を乗じることで、金属試料中の着目元素の固溶含有率を求める。例えば、鉄鋼試料中のチタンの固溶含有率を求める場合、上記方法により、分析溶液中のチタン濃度(KTi)及び比較元素として選択した鉄の濃度(KFe)を、それぞれICP質量分析装置で測定する。そして、その濃度比(KTi/KFe)に、比較元素の含有率(鉄の組成値)を乗じて、鉄鋼試料中のチタンの固溶含有率を求めることができる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は金属試料中の元素分析方法に関し、特に、金属試料中に固溶状態で存在する元素の含有率を迅速かつ正確に求める方法に関するものである。
【背景技術】
【0002】
金属試料中に存在する析出物は、その形態、大きさ、ならびに分布によっては材料の諸特性、例えば、疲労的性質、熱間および冷間加工性、深絞り性、被削性、あるいは電磁気的性質などに著しい影響を及ぼす。
【0003】
鉄鋼を例に説明すると、特に近年は、微細な析出物を利用して鉄鋼製品の特性を向上させる技術が著しく発展し、それに伴って製造工程における析出物制御が厳格化してきた。添加元素の金属試料中に固溶状態で存在する元素の含有率(以下、固溶含有率と称す)と析出状態で存在する元素の含有率(以下、析出含有率と称す)は、鉄鋼製品の各製造工程を通じて、漸次変化している。例えば、溶鋼が凝固する際に添加元素は窒素や炭素と結合して一部が析出し、さらにこの析出物はスラブ加熱段階で固溶部分と析出部分に分かれる。固溶部分はその後の圧延段階で歪誘起析出により一部が析出し、残りの固溶部分が続く変態過程で析出する。このとき、冷却速度に応じて一部は固溶状態のまま製品にもたらされることもある。
【0004】
以上から、優れた最終製品を製造するためには、添加元素の析出量と固溶量の変化を制御することが必要となる。そして、そのためには、鋼中の添加元素の固溶含有率あるいは析出含有率を正確に分析することが必要不可欠である。
【0005】
鉄鋼材料中の析出物および/または介在物(以下、これらをまとめて析出物等と称す)には、ほう化物、りん化物、硫化物、窒化物、炭化物、酸化物などがある。これら析出物等を定量する公知技術として、非特許文献1、非特許文献2には、酸分解法、ハロゲン法、電解法などが開示されている。非特許文献1、非特許文献2に記載されている方法の共通点は、抽出液中でマトリクスを化学的に溶解してから、ろ過によってそれぞれの抽出液から目的の析出物等を回収して分析する点にあり、これらの方法は目的とする析出物等の種類に応じて使い分けられている。例えば、酸溶解法やハロゲン法は炭化物や硫化物を溶解するので酸化物系介在物だけを選択的に抽出する場合に使われることが多い。ほとんどすべての析出物等を鉄鋼材料から損失なく抽出する場合には、特に非水溶媒系電解液を用いた電解法が利用される。
【0006】
一方、添加元素の固溶含有率を直接的に分析する公知技術は無い。類似技術として非特許文献3が挙げられる。非特許文献3は、前記酸分解法におけるろ液部分を分析する方法、すなわち、酸で溶解した鋼中のアルミニウムを分析する方法であり、分析結果は固溶したアルミニウムの含有率と酸可溶性の析出したアルミニウムの含有率の合算値となるため、正確なアルミニウムの固溶含有率を得ることはできない。従って、固溶成分のみを分析するためには、例えば、スパーク放電発光分光分析法(JIS G1253 2002)などで分析した着目成分の含有率から、前記電解法で分析した着目成分の析出含有率を差し引く間接的な(1)式の方法(以下、間接法と称す)が考えられる。
【0007】
【数1】

【0008】
(1)式における各含有率の分析精度は、各含有率の分析結果の標準偏差σで議論できる。固溶含有率[sol.A]の分析結果の標準偏差σsol.Aは、含有率[total A]の分析結果の標準偏差σtotalAと析出含有率[pre.A]の分析結果の標準偏差σpre.Aによって、統計的に(σtotalA2+σpre.A2)1/2で表される。すなわち、含有率の標準偏差σtotalAと析出含有率の標準偏差σpre.Aの少なくとも1つが大きい場合には、間接法の固溶含有率[sol.A]の分析精度は、必然的に悪化する。また、一般に、分析結果の誤差の大きさは分析対象元素の含有率に比例して大きくなるため、間接法は析出割合が大きい場合に、特に分析精度が悪くなる問題がある。
【0009】
ここで、上記間接法においての鋼中析出物等の微細化に伴う問題を述べる。近年の鉄鋼材料製造技術は著しく進展し鋼材中の析出物等は数ナノメートルの大きさにまで微細化した。これらの微細な析出物等を鋼中から抽出してろ過回収操作を行うと、析出物等の捕集漏れが不可避的に発生するため析出含有率には負の誤差が生じ、反対に間接法の固溶含有率には正の誤差が生じる。そこで、微細な析出物等を含む試料の場合には、必然的に分析の対象となる析出物等の大きさよりも小さな孔径のフィルタを用いることになるが、数ナノメートルサイズの析出物粒子を液体から完全に分離することのできるフィルタは存在しない。仮にサブナノメートルの孔径のフィルタが開発されたとしても、ろ過速度の著しい低下が作業性を多大に阻害することは明白である。これらは鉄鋼に限った問題ではなく、非特許文献4では銅合金中の微細析出物等をろ過回収した場合において、着目元素の析出部分と固溶部分とは分離できないと述べられている。つまり、微細な析出物等を含む試料において、間接法を適用することが難しいと考えられる。
【0010】
また、特許文献1には、金属試料をそのまま溶液化し、次いで、その溶液を分析して得られた元素スペクトルに補正式を用いて各元素の含有率を求める溶液発光分析方法が開示されている。この特許文献1の場合、(1)金属試料中に含有されている全ての元素含有率を合計すると100質量%となること、(2)同時に測定した2元素の強度スペクトル比と2元素の含有率比は一定の関係にあること、が前提となっている。即ち、上記(1)と(2)の前提を同時に満たす必要があるため、着目元素以外の溶液試料中のほとんど全ての元素を測定する必要がある。さらに上記(1)の前提から、金属試料の組成と溶液試料の組成がほぼ同一である必要があり、溶液化された試料の組成が金属試料の組成と異なる場合には適用できない。
【0011】
以上述べてきたような問題が想定されるため、金属試料中の固溶成分を直接定量できる具体的な公知技術は存在しなかったものと考えられる。
【特許文献1】特開昭59−58342号公報
【非特許文献1】日本鉄鋼協会 「鉄鋼便覧第四版(CD-ROM)」第四巻 2編 3.5
【非特許文献2】アグネ 「最新の鉄鋼状態分析」40頁 1979
【非特許文献3】JIS G 1257「鉄及び鋼−原子吸光分析方法」8.3.1.3 70頁 1997
【非特許文献4】日本金属学会 「まてりあ」第45巻 第1号 52頁 2006
【非特許文献5】日本鉄鋼協会 「鉄と鋼」第79巻 第6号 628頁 1993
【発明の開示】
【発明が解決しようとする課題】
【0012】
本発明は、かかる事情に鑑みなされたもので、金属試料中の着目元素の固溶含有率を簡便、迅速かつ直接的に求める方法を提供することを目的とするものである。
【課題を解決するための手段】
【0013】
上記課題を解決するために、まず、固溶成分のみを直接分析する方法として、非水溶媒系電解液を用いた電解法でマトリクスを溶解した後、この非水溶媒系電解液を分析することに着目した。
【0014】
しかし、上述したように、微細な析出物等を含有する試料においては、非水溶媒系電解液をろ過して未溶解残渣を除いた後のろ液を用いたとしても、ろ液中には析出物等が混入して固溶含有率の分析値に正の誤差を与える、という問題がある。
【0015】
そこで、発明者等は、改めて、非水溶媒系電解液を用いた電解法における、微細な析出物等を含む試料について検討した。そうしたところ、発明者等は、電解中または電解後の金属試料に析出物等が付着している状態で電解液を採取することで、析出物等は電解後または電解中の金属試料の残部(表面)に基本的には全て付着しており、電解液中には着目元素の固溶分のみが抽出されていることを見出した。そして、上記知見をもとにすれば、電解液を分析することで、析出物等の混入のない着目元素の固溶含有率が得られることになる。
【0016】
上記検討結果に基づき、次に、非水溶媒系電解液中の着目元素の分析方法について検討した。非水溶媒中の金属元素を正確に定量するにあたっては、以下の問題点が考えられる。
【0017】
電解液のまま直接あるいは別の有機溶媒で希釈して測定する方法の場合は、有機溶媒は、気化しやすいため一定量とすることが難しく、火気や人体へ影響のなどの安全面でも配慮しなければならない等、取り扱いが簡単ではない。また、元素分析装置では、直接測定できる有機溶媒が限定され、非水溶媒系電解液の多くはこれに該当しない。また、電解法のような固体を溶液化して分析する手法で得られる結果は単位体積あたりの質量(例えば、g/l)となるため、固体中の質量含有率を求めるには、溶解した着目元素の質量と溶解した固体の質量に換算しなければならない。着目元素の減量分を知るためには、溶解させた液体の体積を正確に測定しなければならない。しかしながら、非水溶媒系電解液の体積を正確に測定するのは簡単ではなく、その分析結果の真度に問題がある。
【0018】
電解液を乾固させて水溶液に置換してから測定する方法の場合は、大量の電解液(例えば、鉄鋼材料の電解作業では数百mlの電解液が用いられる)を乾固させなければならず、蒸発操作の後期において、電解液成分のキレート剤や電解によって溶解した金属成分などが濃化し蒸発速度が低下してしまう点等を考慮すると非常に煩雑である。
【0019】
このように、電解液中の着目元素の分析が行えたとしても、簡便で、かつ迅速に行うという点で問題が残る。
【0020】
そこで、電解液を全量分析するのではなく、電解液から一部を採取し分析することで上記問題を解決できないかと考えた。検討した結果、固体から液体に変化しても、状態変化に対して不変的な元素を指標として、即ち比較元素として着目元素の固溶濃度を相対的に求めれば、液体中の濃度から目的とする着目元素の固溶含有率に帰着することができることを見出した。すなわち、採取された電解液中の着目元素と比較元素との濃度比を、別の方法で求めた固体試料中の比較元素の含有率で補正することで、大量の有機溶媒を扱うことなく着目元素の固溶含有率が得られることに想到した。
【0021】
以上の検討からすると、金属試料(固体)中における着目元素iの固溶含有率sol.Ciは、採取した電解液中の着目元素iの濃度Kiと比較元素mの濃度Kmをそれぞれ測定しておき、両者の比Ki/Kmに金属試料(固体)中の比較元素の含有率Cmを乗じることによって得られることがわかる(下記式(2)参照)。そして、上記の考え方を用いれば、電解液全量を分析し測定せずに、目的とする着目元素の固溶含有率が求められることになる。
【0022】
【数2】
【0023】
本発明は、以上の知見に基づきなされたもので、その要旨は以下のとおりである。
[1]金属試料を電解液中で電解し、電解中および/または電解後に前記電解液の一部を採取し、前記採取された電解液を分析し、該分析の結果を基に、下記(1)〜(2)の方法により着目元素の固溶含有率を得ることを特徴とする金属試料中の着目元素の固溶含有率を求める方法。
(1)電解液中における、比較元素に対する着目元素の濃度比を算出する。
(2)(1)により算出された濃度比に、金属試料における比較元素の含有率を乗じる。
[2]金属試料を電解液中で電解し、電解後、金属試料の残部を電解液から取り除き、次いで該電解液の一部を採取し、前記採取された電解液を分析し、該分析の結果を基に、下記(1)〜(2)の方法により着目元素の固溶含有率を得ることを特徴とする金属試料中の着目元素の固溶含有率を求める方法。
(1)電解液中における、比較元素に対する着目元素の濃度比を算出する。
(2)(1)により算出された濃度比に、金属試料における比較元素の含有率を乗じる。
[3]前記[1]または[2]において、前記電解液の一部を採取するに際し、前記電解液をろ過し、ろ過後の電解液の一部を採取することを特徴とする金属試料中の着目元素の固溶含有率を求める方法。
[4]前記[1]または[2]において、前記採取後の電解液をろ過し、次いで、分析することを特徴とする金属試料中の着目元素の固溶含有率を求める方法。
[5]前記[1]〜[4]のいずれかにおいて、前記採取する電解液の量が、5ml以下であることを特徴とする金属試料中の着目元素の固溶含有率を求める方法。
[6]前記[1]〜[5]のいずれかにおいて、前記採取後の電解液にキレート剤水溶液を添加し、比較元素および着目元素を水溶性キレートとしてから分析することを特徴とする金属試料中の着目元素の固溶含有率を求める方法。
[7]前記[6]において、前記キレート剤水溶液は、エチレンジアミン四酢酸塩水溶液であることを特徴とする金属試料中の着目元素の固溶含有率を求める方法。
【発明の効果】
【0024】
本発明によれば、金属試料中の着目元素の固溶含有率を正確に直接定量することができる。そして、例えば、ナノ・サブナノサイズといった微小な析出物等を含む金属試料に対しても適用でき、析出物等の大きさや量に影響をうけず、あらゆる金属試料に対して適用可能な方法である。
【0025】
そして、本発明では、電解液の一部を採取・分析して電解液中の着目元素と比較元素との濃度比をもとに着目元素の固溶含有率を求めるため、着目元素の固溶含有率を簡便、迅速に求めることができる。また、非水溶媒系電解液を扱う場合に考えられる諸問題(環境に対する問題、揮発性に伴う定量安定性の問題等)も解決される。
【0026】
さらに、金属試料の固溶含有率は、金属製品開発を促進させる重要な評価因子であり、あるいは製品品質を保証する重要な要素となりうるものであることから、金属試料中の着目元素の固溶含有率を直接的、且つ正確に得ることができる本発明は産業上有益な発明といえる。
【発明を実施するための最良の形態】
【0027】
本発明は、電解中および/または電解後に前記電解液の一部を採取し、前記採取された電解液を分析し、分析の結果を基に、下記(1)〜(2)の方法により着目元素の固溶含有率を得ることを特徴とする。なお、着目元素と比較元素は共に、金属試料中に含まれている元素から選択されるものである。
(1)電解液中における、比較元素に対する着目元素の濃度比を算出する。
(2)(1)により算出された濃度比に、金属試料における比較元素の含有率を乗じる。
そして、本発明は、1)電解中または電解後の金属試料に析出物等が付着している状態で電解液を採取することで、電解液中には着目元素の固溶分のみを抽出すること、2)電解液の一部を採取すること、3)電解液中の着目元素と比較元素との濃度比を、別の方法で求めた固体中の比較元素濃度で補正することを基本的な技術思想とする。
【0028】
例えば、金属試料に含まれる元素は、マトリクス中に固溶する部分と、窒素や炭素等と化合物を形成して析出する部分とに分けられる。特に、鉄鋼試料を非水溶媒系電解液(キレート剤+支持電解質+有機溶媒)を用いて電解する場合、固溶部分は比較元素とともに電解液中に溶解し、析出部分は未溶解残渣として試料表面に露出する。露出した析出物等は電気的引力によって陽極である鉄鋼試料表面に付着していると考えられるため、析出物等を試料とともに電解液から取り出すことができ、その結果、固溶部分と析出部分の分離が容易に可能となる。
【0029】
分離後の電解液中に溶解した着目元素の量を測定すれば固溶部分の濃度を得ることができる。しかし、非水溶媒系電解液はメタノールを主体とした有機溶媒で揮発性が高いうえに数百mlもの容量となることから、無機元素の含有量を測定することは容易ではない。そこで、数百mlの電解液から適当量を採取して乾燥した後、硝酸などの鉱酸で溶解して水溶液としてから着目元素と比較元素をそれぞれ適切な溶液分析法で測定し、その濃度比に固体中の比較元素濃度を乗ずることにより、着目元素の鋼中固溶部分の含有率を得る。
【0030】
このように、本発明は特許文献1と異なり、少なくとも1種類の着目元素と少なくとも1種類の比較元素とを測定すれば効果を奏する。さらに、溶液化された試料の組成が析出物を除いた固溶成分と母相成分のみという、金属試料の組成と異なる場合でも適応可能である。
以下、本発明を詳細に説明する。
【0031】
1)金属試料を電解液中で電解する。
【0032】
電解液中に溶出した元素は電解液中のキレート剤と錯体を形成するため、溶媒であるメタノールの蒸発によって得られる物質は、容易に水溶液化が可能であるという利点がある。そのため、非水溶媒系電解液に用いるキレート剤としては、アセチルアセトン、無水マレイン酸、トリエタノールアミン、サリチル酸メチル及びサリチル酸が挙げられる。また、支持電解質としては、テトラメチルアンモニウムクロライドや塩化リチウムなどが好適である。
【0033】
なお、電解を行う際の諸条件については、特に限定されず、キレート剤の着目成分に対する錯形成能や着目成分を含む析出物等の安定性等によって、適宜設計される。
【0034】
2)電解中および/または電解後に前記電解液の一部を採取する。
電解液の一部を採取するに際しては、金属試料に析出物等が付着している状態で電解液を採取すればよく、その採取するタイミングについては、材料や条件等により定義設定される。しかしながら、電気的に析出物等が試料へ付着する引力の強さの点から通電中に電解液を採取するのが好ましい。
【0035】
また、通電停止後に電解液を採取する際には、金属試料の残部は電解液中に浸漬してある状態でもよいが、一般に電解液上層は溶解した金属の濃度が低いため、最終的な元素分析装置の感度などを考慮すると、金属試料の残部を電解液から取り除いた後、攪拌して、あるいは下層から電解液を採取することが好ましい。
【0036】
さらに、電解液中に析出物等が分散したことが懸念される場合には、採取した電解液を適当なフィルタでろ過、あるいは遠心分離処理して析出物等を除去してから分析処理をすることもできる。または、電解液を適当なフィルタでろ過、あるいは遠心分離処理して析出物等を除去した後、ろ過後の電解液を採取し分析することもできる。
【0037】
また、採取する量については、次の工程である電解液の分析において、例えば検出器感度等を考慮した上で分析に必要可能な量であればよく、特に限定しない。中でも、例えば、1mlの電解液は常温環境でもおよそ5分程度で乾燥するため、電解液の採取量としては、5ml以下が好ましく、より好ましくは1ml以下である。
【0038】
3)採取された電解液を分析する。
分析する方法は、特に限定はしないが、中でも、誘導結合プラズマ発光分光分析法(以下、ICP-AESと略すこともある)、誘導結合プラズマ質量分析法(以下、ICP-MSと略すこともある)及び原子吸光分析法が好適である。
なお、採取した電解液を前記元素分析方法で分析する際には、乾燥により非水溶媒を除去し、電解液を水溶液とすることが好ましい。
【0039】
ただし、着目元素の中には非水溶媒の電解液から水溶液に置換されることで沈殿を形成してしまうものがあり、元素測定時に様々な問題(例えば、沈殿となることで、分析装置からの出力が安定し測定可能となるまでに時間がかかるなど)が生じることがある。そこで、電解液を乾燥する前に、含有する着目元素の非水溶媒可溶性錯体を水溶性錯体に変化させておくことが望ましい。具体的には、採取された電解液を分析する前に、上記2)の工程にて採取した後のまたは採取してから析出物等をろ過等により除去した後の電解液に、キレート剤水溶液を添加して混合して、電解液中の着目元素と比較元素を水溶性キレートとして錯体化させてから、乾燥させて非水溶媒を除去し水溶液とした後に分析する。このように着目元素を錯体化させることで、上記のような水溶液化による沈殿の発生を防ぐことができ、迅速に正確な濃度測定が可能となる。錯体化のためのキレート剤としては、金属元素との親和力の強いエチレンジアミン四酢酸塩(以下、EDTAと略すこともある)が最も好適である。この他、1,2-シクロヘキサンジアミン四酢酸(CyDTAと略すこともある)や、1、2−ジヒドロキシ−3、5−ベンゼンジスルホン酸、二ナトリウム塩(Tironと略すこともある)などが、好適である。
【0040】
さらに、上述のように水溶性キレートとして錯体化させた場合、キレート剤を添加して水溶液化後、分析前に、当該水溶液を半透膜を用いて透析処理をしても良い。当該水溶液中に含まれる金属錯体は、半透膜を介して透析液(純水)へ移行するが、粒子である析出物等は移行できないので、採取した電解液に析出物等が含まれている場合には有効となる。
【0041】
また、一般的に、試料の組成・大きさ等から電解液中への析出物等の混入の有無を判断することは難しい。そこで、上記透析処理は、微粒子の混入を判別した後に行うことが、効率的でより望ましい。
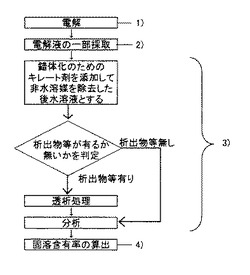
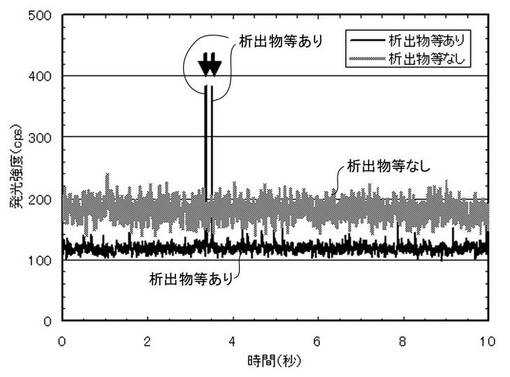
具体的には、以下の通りである。まず、錯体化のためのキレート剤を添加し非水溶媒を除去した後の水溶液を、そのまま、時間分解測定機能を有する分析装置で時間分解測定を行なう。図3はICP-AESで測定した、水溶液中に微細な析出物等が含まれる場合と、水溶液中に微細な析出物等が含まれない場合の光強度の推移を示す図である。図3によると、微細な析出物等が含まれない場合には、溶解成分の検出のみであるため検出される光強度は安定している。これに対して、数10nm以下の微細なものであっても析出物等が含まれる場合には、間欠的に強いピーク(図3中に矢印で示す)が検出される。これは析出物等が分析装置へ取り込まれた際、短時間に大量の測定対象元素が励起、検出されたためであり、固溶成分の定量の観点からは、正の誤差を与えることになる。
このように、まず、時間分解測定を行い微粒子の混入の有無を判別し、図3の矢印のように間欠的に強いピークが散見される場合には透析処理を行なってから分析を行い、ピークがない場合には透析処理を行わずに分析を行う。但し、粒子の存在を示すピークの検出頻度が、分析値に対して無視できるほどに小さい場合には、検出されたとしても透析処理を省略して構わない。具体的な判定の目安は、試料や場合によって詳細を決定すればよい。また、時間分解測定機能を有する分析装置としては、ICP-AESやICP-MSがあげられる。
以上のように、本発明では透析処理を行う前に時間分解測定機能を有する分析装置に採取した電解液の一部(上記では水溶液)を導入し、その結果を検討することで、透析処理など析出物等の除去操作を行なうか否かの判定を行なうことが可能となる。このような判定ステップを入れた場合の分析フローの一例を図4に示す。
【0042】
また、前述したように、本発明では、採取電解液中に含まれる着目元素と比較元素の濃度の相対値を重要な要件とするため、電解液からの採取量は正確である必要はなく、さらに乾燥や溶解工程で溶液の一部が損失することもまったく問題とならない。
【0043】
比較元素としては、鉄鋼の場合は鉄、ステンレス鋼の場合は鉄、クロムもしくはニッケル、銅合金の場合は銅のように、金属試料中の主たる成分で且つ析出物等を形成しない、または形成したとしてもその割合が極僅かである元素を選択することが望ましい。ただし、具体的な比較元素としては上記例に限られず、ある程度以上の量が含まれる元素であれば、比較元素として利用できる可能性がある。さらに、複数元素を比較元素として選択しても良い。例えば、ステンレス鋼の場合、鉄、クロムおよびニッケルの内2種以上の元素を選択し、比較元素の電解液中の濃度は選択した元素の電解液中の濃度の合計値、および比較元素の含有率は選択した元素の含有率の合計値としてもよい。
【0044】
4)上記3)の分析結果を基に、下記(1)〜(2)の方法により着目元素の固溶含有率を得る。
(1)電解液中における、着目元素iの濃度Kiと比較元素mの濃度Kmをそれぞれ測定して、濃度比(Ki/Km)を算出する。
(2)(1)により算出された濃度比に、金属試料における比較元素Cmの含有率を乗じる。上記(1)(2)の結果、固体中の着目元素iの固溶含有率sol.Ciが得られる(式(2)参照)
【0045】
【数2】
【0046】
ここで、固体中の比較元素の含有率を求めるための方法としては、スパーク放電発光分光分析方法(JIS G1253 2002)、蛍光X線分析方法(JIS G1256 1997)、ICP発光分光分析法及びICP質量分析法などにより得られた比較元素以外の元素の合計値を100%から減算する方法が適当である。
【実施例1】
【0047】
以下、鉄鋼試料中のバナジウム(V)の固溶含有率sol.CVとニオブ(Nb)の固溶含有率sol.CNbを分析した例を、具体的に説明する。
【0048】
表1に示す鉄鋼認証標準物質を適切な大きさに切断して、表面を十分研削した後、以下に示す本発明法と比較法により分析を行った。なお、表1の鉄以外の組成値は認証値であり、鉄の組成値は、100%から鉄以外の元素の組成値を合計した値を減算して求めた計算値である。
【0049】
【表1】
【0050】
[本発明法]
まず、約300mlの10%AA系電解液(10vol%アセチルアセトン-1mass%塩化テトラメチルアンモニウム-メタノール)を用いて、前記鉄鋼試料を約0.5g定電位電解する。
【0051】
次いで、通電完了直前に、試料を電解液中に保持したまま約300mlの電解液より約1mlを採取してビーカ内で乾固し、残留物を硝酸で加熱溶解して分析溶液とする(残りの電解液及び取り出した試料に付着した残渣は以降の分析には供しない)。
【0052】
得られた分析溶液中のバナジウムの濃度(KV)およびニオブの濃度(KNb)と、比較元素として選択した鉄の濃度(KFe)を、それぞれICP質量分析装置で測定する。得られたそれぞれの濃度比(KV/KFe、KNb/KFe)に、比較元素(鉄)の含有率(CFe)(表1の鉄の組成値)を乗じて、鉄鋼試料中のバナジウムの固溶含有率(sol.CV)とニオブの固溶含有率(sol.CNb)を得る。
【0053】
[比較法]
従来技術として、鉄鋼試料中の金属元素の固溶含有率を直接分析する方法がないので、ここでは、比較法として、(社)日本鉄鋼協会の推奨する析出物分析法で、前記鉄鋼認証標準物質の析出物等を分析して着目元素ごとの析出含有率を求め、認証値(鋼組成値)から前記析出含有率を減算して固溶含有率を求めることとする。
【0054】
先ず、約300mlの10%AA系電解液を用いて、前記鉄鋼試料を約0.5g定電位電解する。
次いで、通電完了後、試料を電解液中から静かに引き上げて取り出し、約100mlのメタノールを入れた別のビーカに移し変え、超音波振動を与えて試料表面に付着した析出物等を除去する。試料表面が金属光沢を呈したら超音波振動を停止し、試料をビーカから取り出してメタノールで洗浄してから乾燥する。乾燥後、天秤で試料重量を測定して、電解前の試料重量から差し引いて電解重量を計算する。
【0055】
そして、前記電解液及びメタノールを孔径0.2μmのフィルタで吸引ろ過して、残渣をフィルタ上に捕集する。さらに、残渣をフィルタとともに混酸で加熱溶解して溶液化したのち、ICP発光分光分析装置で分析して残渣中のバナジウムの絶対量およびニオブの絶対量を測定する。得られた残渣中のバナジウムの絶対量およびニオブの絶対量を電解前後の試料減量でそれぞれ除して、バナジウムの析出含有率およびニオブの析出含有率を得る。
以上により得られた結果を表2に示す。
【0056】
【表2】
【0057】
表2より、本発明法の固溶含有率は比較法と良く一致していることわかるが、これは、比較法とともに本発明法による固溶含有率が正しい結果であることを示している。なお、実施例1では、発明法の検証のために含有析出物等が十分大きいと思われる鉄鋼認証標準試料を用いたので、比較法でも正確な固溶金属濃度が分析できた。しかし、ナノ・サブナノサイズの析出物等を含むような試料の場合には、比較法では正しい固溶金属濃度を分析することはできない。ナノ・サブナノサイズの析出物等を含むような試料の場合には、現状、本発明法のみが正確な固溶金属含有率定量法となる。
また、上記本発明法と比較法における一試料あたりの分析時間の結果を表3に示す。なお、共通操作である電解時間は控除した。
【0058】
【表3】
【0059】
表3より、本発明法では、ろ過、残渣分解などの煩雑な工程、並びに及び定容・冷却工程のような時間のかかる工程が無い分だけ、比較法に比べて分析時間が短く、簡便かつ迅速な方法であることがわかる。
【実施例2】
【0060】
以下、実施例2として、鉄鋼試料中のチタンの固溶含有率sol.CTiを分析した例を具体的に説明する。
表4に示す組成からなる鋼塊を2つ用意し、それぞれ1250℃×60分間加熱し、仕上げ温度1050℃で圧延した後、表5に示す条件で熱処理した。次いで、放冷後、適切な大きさに切断して表面を十分研削した後、以下に示す発明法と比較法により分析を行った。
【0061】
なお、表4の鉄以外の組成値はスパーク放電発光分光分析法などの一般的な方法で分析した値であり、鉄の組成値は、100%から鉄以外の元素の組成値を合計した値を減算して求めた計算値である。
また、各試料について、発明法および比較法による固溶したチタンの分析は、それぞれ4回繰り返した。
【0062】
【表4】
【0063】
【表5】
【0064】
また、電子顕微鏡観察によって、それぞれの試料で確認された析出物等の大きさの概略を表5に併せて示す。表5より、試料Aは、通常良く見られる大きさの析出物等が有するが、試料Bは、ナノメートルオーダーの微細な析出物等も有しているのがわかる。
【0065】
[本発明法]
まず、約300mlの10%AA系電解液(10vol%アセチルアセトン-1mass%塩化テトラメチルアンモニウム-メタノール)を用いて、前記鉄鋼試料を約0.5g定電位電解する。
次いで、通電完了直前に、試料を電解液中に保持したまま約300mlの電解液より約1mlを採取してビーカ内で乾固し、残留物を硝酸で加熱溶解して分析溶液とする(残りの電解液及び取り出した試料に付着した残渣は以降の分析には供しない)。
【0066】
得られた分析溶液中のチタン濃度(KTi)及び比較元素として選択した鉄の濃度(KFe)を、それぞれICP質量分析装置で測定する。その濃度比(KTi/KFe)に、比較元素の含有率(CFe)(表4の鉄の組成値)を乗じて、鉄鋼試料中のチタンの固溶含有率(sol.CTi)を得る。
【0067】
[比較法]
従来技術として、鉄鋼試料中の金属元素の固溶含有率を直接分析する方法がないので、ここでは、比較法として、(社)日本鉄鋼協会の推奨する析出物分析法で前記試料中のチタンの析出含有率を分析し、表4に示した鋼中におけるチタンの含有率から、析出含有率を減算して固溶含有率を求めることとする。
【0068】
まず、約300mlの10%AA系電解液を用いて、あらかじめ天秤で重量を測定した前記鉄鋼試料を陽極として約0.5gを定電位電解する。
【0069】
次いで、通電完了後、試料を電解液中から静かに引き上げて取り出し、約100mlのメタノールを入れた別の容器に移し変え、超音波振動を与えて試料表面に付着した析出物等を除去する。試料表面が金属光沢を呈したら超音波振動を停止し、試料を容器から取り出してメタノールで洗浄してから乾燥する。乾燥後、天秤で試料重量を測定して、電解前の試料重量から差し引いて電解重量を計算する。
【0070】
そして、電解液及びメタノールを孔径0.2μmのフィルタで吸引ろ過して、残渣をフィルタ上に捕集する。さらに、残渣をフィルタとともに硝酸、過塩素酸並びに硫酸の混合溶液で加熱溶解して溶液化したのち、ICP発光分光分析装置で分析して残渣中のチタン絶対量を測定する。この残渣中のチタン絶対量を、先に求めた電解重量で除してチタンの析出含有率を得る。得られたチタンの析出含有率を、表4のチタンの組成値から減算して、チタンの固溶含有率を算出する。
【0071】
以上、本発明法および比較法により得られた結果を表6に示す。
【0072】
【表6】
【0073】
表6より、まず、本発明法と比較法の真度を比較する。析出物等がマイクロメートルオーダーで通常大の試料Aにおいては、本発明法と比較法の平均分析値(固溶含有率)はほぼ一致した。従って、本発明法で得られる結果は、析出物等を含まない正確なチタンの固溶含有率であることが示された。また、微細な析出物等を有する試料Bにおいては、本発明法の固溶含有率は比較法よりも非常に低い結果を示した。これは、試料Bに含まれる析出物等が非常に微細かつ量が少ないため、比較法では析出物等のろ過回収操作で捕集漏れが発生して、チタンの析出含有率が低値を示し、計算上、固溶含有率が高くなったことが原因である。これより、小さな析出物等を含み、さらにその量が少ない試料の場合には、比較法の固溶含有率よりも本発明法の固溶含有率の方が真度が高いことが明らかとなった。
【0074】
次に、本発明法と比較法の分析精度を比較する。試料Aでは、比較法の標準偏差σが本発明法に比べてやや小さいが、試料Bでは本発明法の標準偏差σが比較法に比べて非常に小さい。
【0075】
試料Aでは、チタン含有率の2割強が析出部分、残りの8割弱の固溶部分である。そのため、本発明法では、8割弱の固溶部分を分析対象とするのに対し、比較法では、2割強の析出部分を分析対象とするため、比較法の標準偏差σが小さくなったと考える。
【0076】
一方、試料Bでは、析出したチタン(析出部分)がチタン含有率の6割程度となっている。そのため、4割程度の固溶部分を直接分析する本発明法のほうが、6割程度の析出部分を分析する比較法に比べて精度が優れている。さらに、比較法では、含有している析出物等の大きさと量の問題から、比較法における析出物等のろ過回収が安定していないために、分析精度が非常に悪くなっているものと考えられる。従って、本発明法は、微細な析出物等を有する試料に対して、また、特に析出割合が高い元素に対して、比較法よりも精度よく固溶含有率を分析可能であるといえる。
【0077】
以上から、本方法は、材料中の着目元素の固溶含有率を直接分析可能で、試料に含有する析出物等の微細化やその量に影響されることなく、真度の高い固溶含有率を提供することが可能であることが明らかになった。
【実施例3】
【0078】
採取した電解液に錯体化のためのキレート剤を分析前に添加することで、沈殿形成を防ぎ、着目元素によっては分析時間を短縮できることを示した例を、具体的に説明する。
【0079】
まず、表1に示した鉄鋼認証標準物質JSS1008-1を陽極として、約300mlの10%AA系電解液中で約0.5gを定電位電解する。
次いで、通電完了後、試料を電解液中から静かに引き上げて取り出した後の電解液を、約1ml採取して、以下に示したキレート剤水溶液を添加した場合とキレート剤水溶液を添加しない場合、それぞれの処理を行った。そして、それぞれの処理を行って得られた分析試料を、分析装置に導入してから出力が安定して測定可能となるまでの経時変化を比較した。
【0080】
[キレート剤水溶液を添加した場合]
採取された約1mlの電解液に、錯体化のためのキレート剤水溶液として0.1mol/lエチレンジアミン四酢酸二アンモニウム(EDTA)水溶液を0.4ml添加して十分攪拌してから、試験管内で乾燥させた。この乾燥後の残留物を、約20mlの純水で加温溶解して分析溶液とした。この分析溶液を、ICP質量分析装置にて、バナジウム、鉄およびモリブデンのイオン強度を、上記分析装置への導入直後から経時的に計測した。
【0081】
[キレート剤水溶液を添加しない場合]
採取された約1mlの電解液を、試験管内で乾燥させた。この乾燥後の残留物に30%硝酸2mlを添加して加温溶解し、純水で約20mlに定容して分析溶液とした。この分析溶液を、ICP質量分析装置にて、バナジウム、鉄およびにモリブデンのイオン強度を、記分析装置への導入直後から経時的に計測した。
【0082】
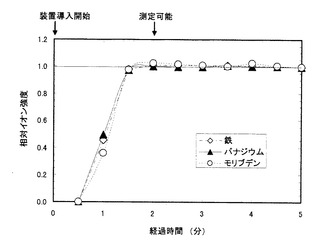
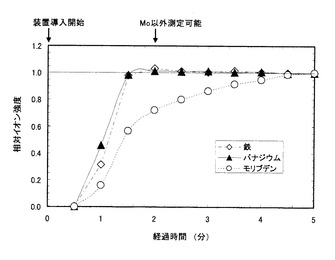
図1にキレート剤水溶液を添加した場合の、図2にキレート剤水溶液を添加しない場合の、計測されたイオン強度の経時変化をそれぞれ示す。図1、図2共に、横軸は計測時間を示し、分析装置への導入直後(=0分)から5分後までを示した。一方、縦軸はイオン強度の計測値を示し、分析装置導入開始から5分後のイオン強度の値を1として、相対化してある。
キレート剤水溶液を添加した場合は、バナジウム、鉄およびモリブデンのいずれのイオン強度も分析装置導入開始から約2分後には安定し、測定可能となった。通常、ICP質量分析装置を用いての測定は、当該分析装置に液体試料を導入した後、出力されるイオン強度値が安定するまで待って行われる。つまりこの場合、約2分で3種類の元素とも測定可能な状態になったことを意味する。
一方、キレート剤水溶液を添加しなかった場合は、バナジウムと鉄は分析装置導入開始から2分間で強度が安定化したが、モリブデンのイオン強度は分析装置導入開始から5分が経過しても安定しなかった。つまり、バナジウムと鉄は、約2分で測定可能となったものの、モリブデンは5分待っても測定可能な状態になっていないことを意味する。
【0083】
一般にICP質量分析装置では、液体試料を毛細チューブで吸い上げてから霧化、イオン化、検出という工程で計測が行なわれる。そのため、試料溶液を装置に導入直後から計測を開始した場合、試料溶液中の元素を検出するまでにはある程度の時間が必要となる。キレート剤水溶液を添加しなかった場合のモリブデンの挙動は、採取した電解液中のモリブデンの一部が硝酸溶液中で不安定になり微細な沈殿を形成したため、完全に溶解しているバナジウムや鉄と比べて、分析装置の検出部への到達に遅れが生じたためと考えられる。一方、キレート剤水溶液を添加した場合は、溶液中のモリブデンイオンは添加されたEDTA水溶液によって水溶性の安定したキレートを形成したため沈殿生成が防止され、モリブデンのイオン強度はバナジウムや鉄と同様に、イオン強度が速やかに安定したものと考えられる。
【0084】
以上の結果から、例えば、モリブデンの固溶含有率を測定する場合には、分析する前に錯体化のためのキレート剤を添加して沈殿形成を防げば、鉄やバナジウムと同等に迅速かつ正確な分析が可能となることが明らかになった。
【図面の簡単な説明】
【0085】
【図1】キレート剤水溶液を添加した場合の、各元素における相対イオン強度の経時変化を示す図である。(実施例3)
【図2】キレート剤水溶液を添加しない場合の、各元素における相対イオン強度の経時変化を示す図である。(実施例3)
【図3】析出物等が電解液に混入した場合のICP-AES装置の出力例を示す図である。
【図4】3)の電解液の分析工程に、キレート剤添加処理、析出物等混入判定処理、および透析処理を追加した場合の、本発明に係る分析フローの例を示す図である。
【技術分野】
【0001】
本発明は金属試料中の元素分析方法に関し、特に、金属試料中に固溶状態で存在する元素の含有率を迅速かつ正確に求める方法に関するものである。
【背景技術】
【0002】
金属試料中に存在する析出物は、その形態、大きさ、ならびに分布によっては材料の諸特性、例えば、疲労的性質、熱間および冷間加工性、深絞り性、被削性、あるいは電磁気的性質などに著しい影響を及ぼす。
【0003】
鉄鋼を例に説明すると、特に近年は、微細な析出物を利用して鉄鋼製品の特性を向上させる技術が著しく発展し、それに伴って製造工程における析出物制御が厳格化してきた。添加元素の金属試料中に固溶状態で存在する元素の含有率(以下、固溶含有率と称す)と析出状態で存在する元素の含有率(以下、析出含有率と称す)は、鉄鋼製品の各製造工程を通じて、漸次変化している。例えば、溶鋼が凝固する際に添加元素は窒素や炭素と結合して一部が析出し、さらにこの析出物はスラブ加熱段階で固溶部分と析出部分に分かれる。固溶部分はその後の圧延段階で歪誘起析出により一部が析出し、残りの固溶部分が続く変態過程で析出する。このとき、冷却速度に応じて一部は固溶状態のまま製品にもたらされることもある。
【0004】
以上から、優れた最終製品を製造するためには、添加元素の析出量と固溶量の変化を制御することが必要となる。そして、そのためには、鋼中の添加元素の固溶含有率あるいは析出含有率を正確に分析することが必要不可欠である。
【0005】
鉄鋼材料中の析出物および/または介在物(以下、これらをまとめて析出物等と称す)には、ほう化物、りん化物、硫化物、窒化物、炭化物、酸化物などがある。これら析出物等を定量する公知技術として、非特許文献1、非特許文献2には、酸分解法、ハロゲン法、電解法などが開示されている。非特許文献1、非特許文献2に記載されている方法の共通点は、抽出液中でマトリクスを化学的に溶解してから、ろ過によってそれぞれの抽出液から目的の析出物等を回収して分析する点にあり、これらの方法は目的とする析出物等の種類に応じて使い分けられている。例えば、酸溶解法やハロゲン法は炭化物や硫化物を溶解するので酸化物系介在物だけを選択的に抽出する場合に使われることが多い。ほとんどすべての析出物等を鉄鋼材料から損失なく抽出する場合には、特に非水溶媒系電解液を用いた電解法が利用される。
【0006】
一方、添加元素の固溶含有率を直接的に分析する公知技術は無い。類似技術として非特許文献3が挙げられる。非特許文献3は、前記酸分解法におけるろ液部分を分析する方法、すなわち、酸で溶解した鋼中のアルミニウムを分析する方法であり、分析結果は固溶したアルミニウムの含有率と酸可溶性の析出したアルミニウムの含有率の合算値となるため、正確なアルミニウムの固溶含有率を得ることはできない。従って、固溶成分のみを分析するためには、例えば、スパーク放電発光分光分析法(JIS G1253 2002)などで分析した着目成分の含有率から、前記電解法で分析した着目成分の析出含有率を差し引く間接的な(1)式の方法(以下、間接法と称す)が考えられる。
【0007】
【数1】
【0008】
(1)式における各含有率の分析精度は、各含有率の分析結果の標準偏差σで議論できる。固溶含有率[sol.A]の分析結果の標準偏差σsol.Aは、含有率[total A]の分析結果の標準偏差σtotalAと析出含有率[pre.A]の分析結果の標準偏差σpre.Aによって、統計的に(σtotalA2+σpre.A2)1/2で表される。すなわち、含有率の標準偏差σtotalAと析出含有率の標準偏差σpre.Aの少なくとも1つが大きい場合には、間接法の固溶含有率[sol.A]の分析精度は、必然的に悪化する。また、一般に、分析結果の誤差の大きさは分析対象元素の含有率に比例して大きくなるため、間接法は析出割合が大きい場合に、特に分析精度が悪くなる問題がある。
【0009】
ここで、上記間接法においての鋼中析出物等の微細化に伴う問題を述べる。近年の鉄鋼材料製造技術は著しく進展し鋼材中の析出物等は数ナノメートルの大きさにまで微細化した。これらの微細な析出物等を鋼中から抽出してろ過回収操作を行うと、析出物等の捕集漏れが不可避的に発生するため析出含有率には負の誤差が生じ、反対に間接法の固溶含有率には正の誤差が生じる。そこで、微細な析出物等を含む試料の場合には、必然的に分析の対象となる析出物等の大きさよりも小さな孔径のフィルタを用いることになるが、数ナノメートルサイズの析出物粒子を液体から完全に分離することのできるフィルタは存在しない。仮にサブナノメートルの孔径のフィルタが開発されたとしても、ろ過速度の著しい低下が作業性を多大に阻害することは明白である。これらは鉄鋼に限った問題ではなく、非特許文献4では銅合金中の微細析出物等をろ過回収した場合において、着目元素の析出部分と固溶部分とは分離できないと述べられている。つまり、微細な析出物等を含む試料において、間接法を適用することが難しいと考えられる。
【0010】
また、特許文献1には、金属試料をそのまま溶液化し、次いで、その溶液を分析して得られた元素スペクトルに補正式を用いて各元素の含有率を求める溶液発光分析方法が開示されている。この特許文献1の場合、(1)金属試料中に含有されている全ての元素含有率を合計すると100質量%となること、(2)同時に測定した2元素の強度スペクトル比と2元素の含有率比は一定の関係にあること、が前提となっている。即ち、上記(1)と(2)の前提を同時に満たす必要があるため、着目元素以外の溶液試料中のほとんど全ての元素を測定する必要がある。さらに上記(1)の前提から、金属試料の組成と溶液試料の組成がほぼ同一である必要があり、溶液化された試料の組成が金属試料の組成と異なる場合には適用できない。
【0011】
以上述べてきたような問題が想定されるため、金属試料中の固溶成分を直接定量できる具体的な公知技術は存在しなかったものと考えられる。
【特許文献1】特開昭59−58342号公報
【非特許文献1】日本鉄鋼協会 「鉄鋼便覧第四版(CD-ROM)」第四巻 2編 3.5
【非特許文献2】アグネ 「最新の鉄鋼状態分析」40頁 1979
【非特許文献3】JIS G 1257「鉄及び鋼−原子吸光分析方法」8.3.1.3 70頁 1997
【非特許文献4】日本金属学会 「まてりあ」第45巻 第1号 52頁 2006
【非特許文献5】日本鉄鋼協会 「鉄と鋼」第79巻 第6号 628頁 1993
【発明の開示】
【発明が解決しようとする課題】
【0012】
本発明は、かかる事情に鑑みなされたもので、金属試料中の着目元素の固溶含有率を簡便、迅速かつ直接的に求める方法を提供することを目的とするものである。
【課題を解決するための手段】
【0013】
上記課題を解決するために、まず、固溶成分のみを直接分析する方法として、非水溶媒系電解液を用いた電解法でマトリクスを溶解した後、この非水溶媒系電解液を分析することに着目した。
【0014】
しかし、上述したように、微細な析出物等を含有する試料においては、非水溶媒系電解液をろ過して未溶解残渣を除いた後のろ液を用いたとしても、ろ液中には析出物等が混入して固溶含有率の分析値に正の誤差を与える、という問題がある。
【0015】
そこで、発明者等は、改めて、非水溶媒系電解液を用いた電解法における、微細な析出物等を含む試料について検討した。そうしたところ、発明者等は、電解中または電解後の金属試料に析出物等が付着している状態で電解液を採取することで、析出物等は電解後または電解中の金属試料の残部(表面)に基本的には全て付着しており、電解液中には着目元素の固溶分のみが抽出されていることを見出した。そして、上記知見をもとにすれば、電解液を分析することで、析出物等の混入のない着目元素の固溶含有率が得られることになる。
【0016】
上記検討結果に基づき、次に、非水溶媒系電解液中の着目元素の分析方法について検討した。非水溶媒中の金属元素を正確に定量するにあたっては、以下の問題点が考えられる。
【0017】
電解液のまま直接あるいは別の有機溶媒で希釈して測定する方法の場合は、有機溶媒は、気化しやすいため一定量とすることが難しく、火気や人体へ影響のなどの安全面でも配慮しなければならない等、取り扱いが簡単ではない。また、元素分析装置では、直接測定できる有機溶媒が限定され、非水溶媒系電解液の多くはこれに該当しない。また、電解法のような固体を溶液化して分析する手法で得られる結果は単位体積あたりの質量(例えば、g/l)となるため、固体中の質量含有率を求めるには、溶解した着目元素の質量と溶解した固体の質量に換算しなければならない。着目元素の減量分を知るためには、溶解させた液体の体積を正確に測定しなければならない。しかしながら、非水溶媒系電解液の体積を正確に測定するのは簡単ではなく、その分析結果の真度に問題がある。
【0018】
電解液を乾固させて水溶液に置換してから測定する方法の場合は、大量の電解液(例えば、鉄鋼材料の電解作業では数百mlの電解液が用いられる)を乾固させなければならず、蒸発操作の後期において、電解液成分のキレート剤や電解によって溶解した金属成分などが濃化し蒸発速度が低下してしまう点等を考慮すると非常に煩雑である。
【0019】
このように、電解液中の着目元素の分析が行えたとしても、簡便で、かつ迅速に行うという点で問題が残る。
【0020】
そこで、電解液を全量分析するのではなく、電解液から一部を採取し分析することで上記問題を解決できないかと考えた。検討した結果、固体から液体に変化しても、状態変化に対して不変的な元素を指標として、即ち比較元素として着目元素の固溶濃度を相対的に求めれば、液体中の濃度から目的とする着目元素の固溶含有率に帰着することができることを見出した。すなわち、採取された電解液中の着目元素と比較元素との濃度比を、別の方法で求めた固体試料中の比較元素の含有率で補正することで、大量の有機溶媒を扱うことなく着目元素の固溶含有率が得られることに想到した。
【0021】
以上の検討からすると、金属試料(固体)中における着目元素iの固溶含有率sol.Ciは、採取した電解液中の着目元素iの濃度Kiと比較元素mの濃度Kmをそれぞれ測定しておき、両者の比Ki/Kmに金属試料(固体)中の比較元素の含有率Cmを乗じることによって得られることがわかる(下記式(2)参照)。そして、上記の考え方を用いれば、電解液全量を分析し測定せずに、目的とする着目元素の固溶含有率が求められることになる。
【0022】
【数2】
【0023】
本発明は、以上の知見に基づきなされたもので、その要旨は以下のとおりである。
[1]金属試料を電解液中で電解し、電解中および/または電解後に前記電解液の一部を採取し、前記採取された電解液を分析し、該分析の結果を基に、下記(1)〜(2)の方法により着目元素の固溶含有率を得ることを特徴とする金属試料中の着目元素の固溶含有率を求める方法。
(1)電解液中における、比較元素に対する着目元素の濃度比を算出する。
(2)(1)により算出された濃度比に、金属試料における比較元素の含有率を乗じる。
[2]金属試料を電解液中で電解し、電解後、金属試料の残部を電解液から取り除き、次いで該電解液の一部を採取し、前記採取された電解液を分析し、該分析の結果を基に、下記(1)〜(2)の方法により着目元素の固溶含有率を得ることを特徴とする金属試料中の着目元素の固溶含有率を求める方法。
(1)電解液中における、比較元素に対する着目元素の濃度比を算出する。
(2)(1)により算出された濃度比に、金属試料における比較元素の含有率を乗じる。
[3]前記[1]または[2]において、前記電解液の一部を採取するに際し、前記電解液をろ過し、ろ過後の電解液の一部を採取することを特徴とする金属試料中の着目元素の固溶含有率を求める方法。
[4]前記[1]または[2]において、前記採取後の電解液をろ過し、次いで、分析することを特徴とする金属試料中の着目元素の固溶含有率を求める方法。
[5]前記[1]〜[4]のいずれかにおいて、前記採取する電解液の量が、5ml以下であることを特徴とする金属試料中の着目元素の固溶含有率を求める方法。
[6]前記[1]〜[5]のいずれかにおいて、前記採取後の電解液にキレート剤水溶液を添加し、比較元素および着目元素を水溶性キレートとしてから分析することを特徴とする金属試料中の着目元素の固溶含有率を求める方法。
[7]前記[6]において、前記キレート剤水溶液は、エチレンジアミン四酢酸塩水溶液であることを特徴とする金属試料中の着目元素の固溶含有率を求める方法。
【発明の効果】
【0024】
本発明によれば、金属試料中の着目元素の固溶含有率を正確に直接定量することができる。そして、例えば、ナノ・サブナノサイズといった微小な析出物等を含む金属試料に対しても適用でき、析出物等の大きさや量に影響をうけず、あらゆる金属試料に対して適用可能な方法である。
【0025】
そして、本発明では、電解液の一部を採取・分析して電解液中の着目元素と比較元素との濃度比をもとに着目元素の固溶含有率を求めるため、着目元素の固溶含有率を簡便、迅速に求めることができる。また、非水溶媒系電解液を扱う場合に考えられる諸問題(環境に対する問題、揮発性に伴う定量安定性の問題等)も解決される。
【0026】
さらに、金属試料の固溶含有率は、金属製品開発を促進させる重要な評価因子であり、あるいは製品品質を保証する重要な要素となりうるものであることから、金属試料中の着目元素の固溶含有率を直接的、且つ正確に得ることができる本発明は産業上有益な発明といえる。
【発明を実施するための最良の形態】
【0027】
本発明は、電解中および/または電解後に前記電解液の一部を採取し、前記採取された電解液を分析し、分析の結果を基に、下記(1)〜(2)の方法により着目元素の固溶含有率を得ることを特徴とする。なお、着目元素と比較元素は共に、金属試料中に含まれている元素から選択されるものである。
(1)電解液中における、比較元素に対する着目元素の濃度比を算出する。
(2)(1)により算出された濃度比に、金属試料における比較元素の含有率を乗じる。
そして、本発明は、1)電解中または電解後の金属試料に析出物等が付着している状態で電解液を採取することで、電解液中には着目元素の固溶分のみを抽出すること、2)電解液の一部を採取すること、3)電解液中の着目元素と比較元素との濃度比を、別の方法で求めた固体中の比較元素濃度で補正することを基本的な技術思想とする。
【0028】
例えば、金属試料に含まれる元素は、マトリクス中に固溶する部分と、窒素や炭素等と化合物を形成して析出する部分とに分けられる。特に、鉄鋼試料を非水溶媒系電解液(キレート剤+支持電解質+有機溶媒)を用いて電解する場合、固溶部分は比較元素とともに電解液中に溶解し、析出部分は未溶解残渣として試料表面に露出する。露出した析出物等は電気的引力によって陽極である鉄鋼試料表面に付着していると考えられるため、析出物等を試料とともに電解液から取り出すことができ、その結果、固溶部分と析出部分の分離が容易に可能となる。
【0029】
分離後の電解液中に溶解した着目元素の量を測定すれば固溶部分の濃度を得ることができる。しかし、非水溶媒系電解液はメタノールを主体とした有機溶媒で揮発性が高いうえに数百mlもの容量となることから、無機元素の含有量を測定することは容易ではない。そこで、数百mlの電解液から適当量を採取して乾燥した後、硝酸などの鉱酸で溶解して水溶液としてから着目元素と比較元素をそれぞれ適切な溶液分析法で測定し、その濃度比に固体中の比較元素濃度を乗ずることにより、着目元素の鋼中固溶部分の含有率を得る。
【0030】
このように、本発明は特許文献1と異なり、少なくとも1種類の着目元素と少なくとも1種類の比較元素とを測定すれば効果を奏する。さらに、溶液化された試料の組成が析出物を除いた固溶成分と母相成分のみという、金属試料の組成と異なる場合でも適応可能である。
以下、本発明を詳細に説明する。
【0031】
1)金属試料を電解液中で電解する。
【0032】
電解液中に溶出した元素は電解液中のキレート剤と錯体を形成するため、溶媒であるメタノールの蒸発によって得られる物質は、容易に水溶液化が可能であるという利点がある。そのため、非水溶媒系電解液に用いるキレート剤としては、アセチルアセトン、無水マレイン酸、トリエタノールアミン、サリチル酸メチル及びサリチル酸が挙げられる。また、支持電解質としては、テトラメチルアンモニウムクロライドや塩化リチウムなどが好適である。
【0033】
なお、電解を行う際の諸条件については、特に限定されず、キレート剤の着目成分に対する錯形成能や着目成分を含む析出物等の安定性等によって、適宜設計される。
【0034】
2)電解中および/または電解後に前記電解液の一部を採取する。
電解液の一部を採取するに際しては、金属試料に析出物等が付着している状態で電解液を採取すればよく、その採取するタイミングについては、材料や条件等により定義設定される。しかしながら、電気的に析出物等が試料へ付着する引力の強さの点から通電中に電解液を採取するのが好ましい。
【0035】
また、通電停止後に電解液を採取する際には、金属試料の残部は電解液中に浸漬してある状態でもよいが、一般に電解液上層は溶解した金属の濃度が低いため、最終的な元素分析装置の感度などを考慮すると、金属試料の残部を電解液から取り除いた後、攪拌して、あるいは下層から電解液を採取することが好ましい。
【0036】
さらに、電解液中に析出物等が分散したことが懸念される場合には、採取した電解液を適当なフィルタでろ過、あるいは遠心分離処理して析出物等を除去してから分析処理をすることもできる。または、電解液を適当なフィルタでろ過、あるいは遠心分離処理して析出物等を除去した後、ろ過後の電解液を採取し分析することもできる。
【0037】
また、採取する量については、次の工程である電解液の分析において、例えば検出器感度等を考慮した上で分析に必要可能な量であればよく、特に限定しない。中でも、例えば、1mlの電解液は常温環境でもおよそ5分程度で乾燥するため、電解液の採取量としては、5ml以下が好ましく、より好ましくは1ml以下である。
【0038】
3)採取された電解液を分析する。
分析する方法は、特に限定はしないが、中でも、誘導結合プラズマ発光分光分析法(以下、ICP-AESと略すこともある)、誘導結合プラズマ質量分析法(以下、ICP-MSと略すこともある)及び原子吸光分析法が好適である。
なお、採取した電解液を前記元素分析方法で分析する際には、乾燥により非水溶媒を除去し、電解液を水溶液とすることが好ましい。
【0039】
ただし、着目元素の中には非水溶媒の電解液から水溶液に置換されることで沈殿を形成してしまうものがあり、元素測定時に様々な問題(例えば、沈殿となることで、分析装置からの出力が安定し測定可能となるまでに時間がかかるなど)が生じることがある。そこで、電解液を乾燥する前に、含有する着目元素の非水溶媒可溶性錯体を水溶性錯体に変化させておくことが望ましい。具体的には、採取された電解液を分析する前に、上記2)の工程にて採取した後のまたは採取してから析出物等をろ過等により除去した後の電解液に、キレート剤水溶液を添加して混合して、電解液中の着目元素と比較元素を水溶性キレートとして錯体化させてから、乾燥させて非水溶媒を除去し水溶液とした後に分析する。このように着目元素を錯体化させることで、上記のような水溶液化による沈殿の発生を防ぐことができ、迅速に正確な濃度測定が可能となる。錯体化のためのキレート剤としては、金属元素との親和力の強いエチレンジアミン四酢酸塩(以下、EDTAと略すこともある)が最も好適である。この他、1,2-シクロヘキサンジアミン四酢酸(CyDTAと略すこともある)や、1、2−ジヒドロキシ−3、5−ベンゼンジスルホン酸、二ナトリウム塩(Tironと略すこともある)などが、好適である。
【0040】
さらに、上述のように水溶性キレートとして錯体化させた場合、キレート剤を添加して水溶液化後、分析前に、当該水溶液を半透膜を用いて透析処理をしても良い。当該水溶液中に含まれる金属錯体は、半透膜を介して透析液(純水)へ移行するが、粒子である析出物等は移行できないので、採取した電解液に析出物等が含まれている場合には有効となる。
【0041】
また、一般的に、試料の組成・大きさ等から電解液中への析出物等の混入の有無を判断することは難しい。そこで、上記透析処理は、微粒子の混入を判別した後に行うことが、効率的でより望ましい。
具体的には、以下の通りである。まず、錯体化のためのキレート剤を添加し非水溶媒を除去した後の水溶液を、そのまま、時間分解測定機能を有する分析装置で時間分解測定を行なう。図3はICP-AESで測定した、水溶液中に微細な析出物等が含まれる場合と、水溶液中に微細な析出物等が含まれない場合の光強度の推移を示す図である。図3によると、微細な析出物等が含まれない場合には、溶解成分の検出のみであるため検出される光強度は安定している。これに対して、数10nm以下の微細なものであっても析出物等が含まれる場合には、間欠的に強いピーク(図3中に矢印で示す)が検出される。これは析出物等が分析装置へ取り込まれた際、短時間に大量の測定対象元素が励起、検出されたためであり、固溶成分の定量の観点からは、正の誤差を与えることになる。
このように、まず、時間分解測定を行い微粒子の混入の有無を判別し、図3の矢印のように間欠的に強いピークが散見される場合には透析処理を行なってから分析を行い、ピークがない場合には透析処理を行わずに分析を行う。但し、粒子の存在を示すピークの検出頻度が、分析値に対して無視できるほどに小さい場合には、検出されたとしても透析処理を省略して構わない。具体的な判定の目安は、試料や場合によって詳細を決定すればよい。また、時間分解測定機能を有する分析装置としては、ICP-AESやICP-MSがあげられる。
以上のように、本発明では透析処理を行う前に時間分解測定機能を有する分析装置に採取した電解液の一部(上記では水溶液)を導入し、その結果を検討することで、透析処理など析出物等の除去操作を行なうか否かの判定を行なうことが可能となる。このような判定ステップを入れた場合の分析フローの一例を図4に示す。
【0042】
また、前述したように、本発明では、採取電解液中に含まれる着目元素と比較元素の濃度の相対値を重要な要件とするため、電解液からの採取量は正確である必要はなく、さらに乾燥や溶解工程で溶液の一部が損失することもまったく問題とならない。
【0043】
比較元素としては、鉄鋼の場合は鉄、ステンレス鋼の場合は鉄、クロムもしくはニッケル、銅合金の場合は銅のように、金属試料中の主たる成分で且つ析出物等を形成しない、または形成したとしてもその割合が極僅かである元素を選択することが望ましい。ただし、具体的な比較元素としては上記例に限られず、ある程度以上の量が含まれる元素であれば、比較元素として利用できる可能性がある。さらに、複数元素を比較元素として選択しても良い。例えば、ステンレス鋼の場合、鉄、クロムおよびニッケルの内2種以上の元素を選択し、比較元素の電解液中の濃度は選択した元素の電解液中の濃度の合計値、および比較元素の含有率は選択した元素の含有率の合計値としてもよい。
【0044】
4)上記3)の分析結果を基に、下記(1)〜(2)の方法により着目元素の固溶含有率を得る。
(1)電解液中における、着目元素iの濃度Kiと比較元素mの濃度Kmをそれぞれ測定して、濃度比(Ki/Km)を算出する。
(2)(1)により算出された濃度比に、金属試料における比較元素Cmの含有率を乗じる。上記(1)(2)の結果、固体中の着目元素iの固溶含有率sol.Ciが得られる(式(2)参照)
【0045】
【数2】
【0046】
ここで、固体中の比較元素の含有率を求めるための方法としては、スパーク放電発光分光分析方法(JIS G1253 2002)、蛍光X線分析方法(JIS G1256 1997)、ICP発光分光分析法及びICP質量分析法などにより得られた比較元素以外の元素の合計値を100%から減算する方法が適当である。
【実施例1】
【0047】
以下、鉄鋼試料中のバナジウム(V)の固溶含有率sol.CVとニオブ(Nb)の固溶含有率sol.CNbを分析した例を、具体的に説明する。
【0048】
表1に示す鉄鋼認証標準物質を適切な大きさに切断して、表面を十分研削した後、以下に示す本発明法と比較法により分析を行った。なお、表1の鉄以外の組成値は認証値であり、鉄の組成値は、100%から鉄以外の元素の組成値を合計した値を減算して求めた計算値である。
【0049】
【表1】
【0050】
[本発明法]
まず、約300mlの10%AA系電解液(10vol%アセチルアセトン-1mass%塩化テトラメチルアンモニウム-メタノール)を用いて、前記鉄鋼試料を約0.5g定電位電解する。
【0051】
次いで、通電完了直前に、試料を電解液中に保持したまま約300mlの電解液より約1mlを採取してビーカ内で乾固し、残留物を硝酸で加熱溶解して分析溶液とする(残りの電解液及び取り出した試料に付着した残渣は以降の分析には供しない)。
【0052】
得られた分析溶液中のバナジウムの濃度(KV)およびニオブの濃度(KNb)と、比較元素として選択した鉄の濃度(KFe)を、それぞれICP質量分析装置で測定する。得られたそれぞれの濃度比(KV/KFe、KNb/KFe)に、比較元素(鉄)の含有率(CFe)(表1の鉄の組成値)を乗じて、鉄鋼試料中のバナジウムの固溶含有率(sol.CV)とニオブの固溶含有率(sol.CNb)を得る。
【0053】
[比較法]
従来技術として、鉄鋼試料中の金属元素の固溶含有率を直接分析する方法がないので、ここでは、比較法として、(社)日本鉄鋼協会の推奨する析出物分析法で、前記鉄鋼認証標準物質の析出物等を分析して着目元素ごとの析出含有率を求め、認証値(鋼組成値)から前記析出含有率を減算して固溶含有率を求めることとする。
【0054】
先ず、約300mlの10%AA系電解液を用いて、前記鉄鋼試料を約0.5g定電位電解する。
次いで、通電完了後、試料を電解液中から静かに引き上げて取り出し、約100mlのメタノールを入れた別のビーカに移し変え、超音波振動を与えて試料表面に付着した析出物等を除去する。試料表面が金属光沢を呈したら超音波振動を停止し、試料をビーカから取り出してメタノールで洗浄してから乾燥する。乾燥後、天秤で試料重量を測定して、電解前の試料重量から差し引いて電解重量を計算する。
【0055】
そして、前記電解液及びメタノールを孔径0.2μmのフィルタで吸引ろ過して、残渣をフィルタ上に捕集する。さらに、残渣をフィルタとともに混酸で加熱溶解して溶液化したのち、ICP発光分光分析装置で分析して残渣中のバナジウムの絶対量およびニオブの絶対量を測定する。得られた残渣中のバナジウムの絶対量およびニオブの絶対量を電解前後の試料減量でそれぞれ除して、バナジウムの析出含有率およびニオブの析出含有率を得る。
以上により得られた結果を表2に示す。
【0056】
【表2】
【0057】
表2より、本発明法の固溶含有率は比較法と良く一致していることわかるが、これは、比較法とともに本発明法による固溶含有率が正しい結果であることを示している。なお、実施例1では、発明法の検証のために含有析出物等が十分大きいと思われる鉄鋼認証標準試料を用いたので、比較法でも正確な固溶金属濃度が分析できた。しかし、ナノ・サブナノサイズの析出物等を含むような試料の場合には、比較法では正しい固溶金属濃度を分析することはできない。ナノ・サブナノサイズの析出物等を含むような試料の場合には、現状、本発明法のみが正確な固溶金属含有率定量法となる。
また、上記本発明法と比較法における一試料あたりの分析時間の結果を表3に示す。なお、共通操作である電解時間は控除した。
【0058】
【表3】
【0059】
表3より、本発明法では、ろ過、残渣分解などの煩雑な工程、並びに及び定容・冷却工程のような時間のかかる工程が無い分だけ、比較法に比べて分析時間が短く、簡便かつ迅速な方法であることがわかる。
【実施例2】
【0060】
以下、実施例2として、鉄鋼試料中のチタンの固溶含有率sol.CTiを分析した例を具体的に説明する。
表4に示す組成からなる鋼塊を2つ用意し、それぞれ1250℃×60分間加熱し、仕上げ温度1050℃で圧延した後、表5に示す条件で熱処理した。次いで、放冷後、適切な大きさに切断して表面を十分研削した後、以下に示す発明法と比較法により分析を行った。
【0061】
なお、表4の鉄以外の組成値はスパーク放電発光分光分析法などの一般的な方法で分析した値であり、鉄の組成値は、100%から鉄以外の元素の組成値を合計した値を減算して求めた計算値である。
また、各試料について、発明法および比較法による固溶したチタンの分析は、それぞれ4回繰り返した。
【0062】
【表4】
【0063】
【表5】
【0064】
また、電子顕微鏡観察によって、それぞれの試料で確認された析出物等の大きさの概略を表5に併せて示す。表5より、試料Aは、通常良く見られる大きさの析出物等が有するが、試料Bは、ナノメートルオーダーの微細な析出物等も有しているのがわかる。
【0065】
[本発明法]
まず、約300mlの10%AA系電解液(10vol%アセチルアセトン-1mass%塩化テトラメチルアンモニウム-メタノール)を用いて、前記鉄鋼試料を約0.5g定電位電解する。
次いで、通電完了直前に、試料を電解液中に保持したまま約300mlの電解液より約1mlを採取してビーカ内で乾固し、残留物を硝酸で加熱溶解して分析溶液とする(残りの電解液及び取り出した試料に付着した残渣は以降の分析には供しない)。
【0066】
得られた分析溶液中のチタン濃度(KTi)及び比較元素として選択した鉄の濃度(KFe)を、それぞれICP質量分析装置で測定する。その濃度比(KTi/KFe)に、比較元素の含有率(CFe)(表4の鉄の組成値)を乗じて、鉄鋼試料中のチタンの固溶含有率(sol.CTi)を得る。
【0067】
[比較法]
従来技術として、鉄鋼試料中の金属元素の固溶含有率を直接分析する方法がないので、ここでは、比較法として、(社)日本鉄鋼協会の推奨する析出物分析法で前記試料中のチタンの析出含有率を分析し、表4に示した鋼中におけるチタンの含有率から、析出含有率を減算して固溶含有率を求めることとする。
【0068】
まず、約300mlの10%AA系電解液を用いて、あらかじめ天秤で重量を測定した前記鉄鋼試料を陽極として約0.5gを定電位電解する。
【0069】
次いで、通電完了後、試料を電解液中から静かに引き上げて取り出し、約100mlのメタノールを入れた別の容器に移し変え、超音波振動を与えて試料表面に付着した析出物等を除去する。試料表面が金属光沢を呈したら超音波振動を停止し、試料を容器から取り出してメタノールで洗浄してから乾燥する。乾燥後、天秤で試料重量を測定して、電解前の試料重量から差し引いて電解重量を計算する。
【0070】
そして、電解液及びメタノールを孔径0.2μmのフィルタで吸引ろ過して、残渣をフィルタ上に捕集する。さらに、残渣をフィルタとともに硝酸、過塩素酸並びに硫酸の混合溶液で加熱溶解して溶液化したのち、ICP発光分光分析装置で分析して残渣中のチタン絶対量を測定する。この残渣中のチタン絶対量を、先に求めた電解重量で除してチタンの析出含有率を得る。得られたチタンの析出含有率を、表4のチタンの組成値から減算して、チタンの固溶含有率を算出する。
【0071】
以上、本発明法および比較法により得られた結果を表6に示す。
【0072】
【表6】
【0073】
表6より、まず、本発明法と比較法の真度を比較する。析出物等がマイクロメートルオーダーで通常大の試料Aにおいては、本発明法と比較法の平均分析値(固溶含有率)はほぼ一致した。従って、本発明法で得られる結果は、析出物等を含まない正確なチタンの固溶含有率であることが示された。また、微細な析出物等を有する試料Bにおいては、本発明法の固溶含有率は比較法よりも非常に低い結果を示した。これは、試料Bに含まれる析出物等が非常に微細かつ量が少ないため、比較法では析出物等のろ過回収操作で捕集漏れが発生して、チタンの析出含有率が低値を示し、計算上、固溶含有率が高くなったことが原因である。これより、小さな析出物等を含み、さらにその量が少ない試料の場合には、比較法の固溶含有率よりも本発明法の固溶含有率の方が真度が高いことが明らかとなった。
【0074】
次に、本発明法と比較法の分析精度を比較する。試料Aでは、比較法の標準偏差σが本発明法に比べてやや小さいが、試料Bでは本発明法の標準偏差σが比較法に比べて非常に小さい。
【0075】
試料Aでは、チタン含有率の2割強が析出部分、残りの8割弱の固溶部分である。そのため、本発明法では、8割弱の固溶部分を分析対象とするのに対し、比較法では、2割強の析出部分を分析対象とするため、比較法の標準偏差σが小さくなったと考える。
【0076】
一方、試料Bでは、析出したチタン(析出部分)がチタン含有率の6割程度となっている。そのため、4割程度の固溶部分を直接分析する本発明法のほうが、6割程度の析出部分を分析する比較法に比べて精度が優れている。さらに、比較法では、含有している析出物等の大きさと量の問題から、比較法における析出物等のろ過回収が安定していないために、分析精度が非常に悪くなっているものと考えられる。従って、本発明法は、微細な析出物等を有する試料に対して、また、特に析出割合が高い元素に対して、比較法よりも精度よく固溶含有率を分析可能であるといえる。
【0077】
以上から、本方法は、材料中の着目元素の固溶含有率を直接分析可能で、試料に含有する析出物等の微細化やその量に影響されることなく、真度の高い固溶含有率を提供することが可能であることが明らかになった。
【実施例3】
【0078】
採取した電解液に錯体化のためのキレート剤を分析前に添加することで、沈殿形成を防ぎ、着目元素によっては分析時間を短縮できることを示した例を、具体的に説明する。
【0079】
まず、表1に示した鉄鋼認証標準物質JSS1008-1を陽極として、約300mlの10%AA系電解液中で約0.5gを定電位電解する。
次いで、通電完了後、試料を電解液中から静かに引き上げて取り出した後の電解液を、約1ml採取して、以下に示したキレート剤水溶液を添加した場合とキレート剤水溶液を添加しない場合、それぞれの処理を行った。そして、それぞれの処理を行って得られた分析試料を、分析装置に導入してから出力が安定して測定可能となるまでの経時変化を比較した。
【0080】
[キレート剤水溶液を添加した場合]
採取された約1mlの電解液に、錯体化のためのキレート剤水溶液として0.1mol/lエチレンジアミン四酢酸二アンモニウム(EDTA)水溶液を0.4ml添加して十分攪拌してから、試験管内で乾燥させた。この乾燥後の残留物を、約20mlの純水で加温溶解して分析溶液とした。この分析溶液を、ICP質量分析装置にて、バナジウム、鉄およびモリブデンのイオン強度を、上記分析装置への導入直後から経時的に計測した。
【0081】
[キレート剤水溶液を添加しない場合]
採取された約1mlの電解液を、試験管内で乾燥させた。この乾燥後の残留物に30%硝酸2mlを添加して加温溶解し、純水で約20mlに定容して分析溶液とした。この分析溶液を、ICP質量分析装置にて、バナジウム、鉄およびにモリブデンのイオン強度を、記分析装置への導入直後から経時的に計測した。
【0082】
図1にキレート剤水溶液を添加した場合の、図2にキレート剤水溶液を添加しない場合の、計測されたイオン強度の経時変化をそれぞれ示す。図1、図2共に、横軸は計測時間を示し、分析装置への導入直後(=0分)から5分後までを示した。一方、縦軸はイオン強度の計測値を示し、分析装置導入開始から5分後のイオン強度の値を1として、相対化してある。
キレート剤水溶液を添加した場合は、バナジウム、鉄およびモリブデンのいずれのイオン強度も分析装置導入開始から約2分後には安定し、測定可能となった。通常、ICP質量分析装置を用いての測定は、当該分析装置に液体試料を導入した後、出力されるイオン強度値が安定するまで待って行われる。つまりこの場合、約2分で3種類の元素とも測定可能な状態になったことを意味する。
一方、キレート剤水溶液を添加しなかった場合は、バナジウムと鉄は分析装置導入開始から2分間で強度が安定化したが、モリブデンのイオン強度は分析装置導入開始から5分が経過しても安定しなかった。つまり、バナジウムと鉄は、約2分で測定可能となったものの、モリブデンは5分待っても測定可能な状態になっていないことを意味する。
【0083】
一般にICP質量分析装置では、液体試料を毛細チューブで吸い上げてから霧化、イオン化、検出という工程で計測が行なわれる。そのため、試料溶液を装置に導入直後から計測を開始した場合、試料溶液中の元素を検出するまでにはある程度の時間が必要となる。キレート剤水溶液を添加しなかった場合のモリブデンの挙動は、採取した電解液中のモリブデンの一部が硝酸溶液中で不安定になり微細な沈殿を形成したため、完全に溶解しているバナジウムや鉄と比べて、分析装置の検出部への到達に遅れが生じたためと考えられる。一方、キレート剤水溶液を添加した場合は、溶液中のモリブデンイオンは添加されたEDTA水溶液によって水溶性の安定したキレートを形成したため沈殿生成が防止され、モリブデンのイオン強度はバナジウムや鉄と同様に、イオン強度が速やかに安定したものと考えられる。
【0084】
以上の結果から、例えば、モリブデンの固溶含有率を測定する場合には、分析する前に錯体化のためのキレート剤を添加して沈殿形成を防げば、鉄やバナジウムと同等に迅速かつ正確な分析が可能となることが明らかになった。
【図面の簡単な説明】
【0085】
【図1】キレート剤水溶液を添加した場合の、各元素における相対イオン強度の経時変化を示す図である。(実施例3)
【図2】キレート剤水溶液を添加しない場合の、各元素における相対イオン強度の経時変化を示す図である。(実施例3)
【図3】析出物等が電解液に混入した場合のICP-AES装置の出力例を示す図である。
【図4】3)の電解液の分析工程に、キレート剤添加処理、析出物等混入判定処理、および透析処理を追加した場合の、本発明に係る分析フローの例を示す図である。
【特許請求の範囲】
【請求項1】
金属試料を電解液中で電解し、電解中および/または電解後に前記電解液の一部を採取し、前記採取された電解液を分析し、該分析の結果を基に、下記(1)〜(2)の方法により着目元素の固溶含有率を得ることを特徴とする金属試料中の着目元素の固溶含有率を求める方法。
(1)電解液中における、比較元素に対する着目元素の濃度比を算出する。
(2)(1)により算出された濃度比に、金属試料における比較元素の含有率を乗じる。
【請求項2】
金属試料を電解液中で電解し、電解後、金属試料の残部を電解液から取り除き、次いで該電解液の一部を採取し、前記採取された電解液を分析し、該分析の結果を基に、下記(1)〜(2)の方法により着目元素の固溶含有率を得ることを特徴とする金属試料中の着目元素の固溶含有率を求める方法。
(1)電解液中における、比較元素に対する着目元素の濃度比を算出する。
(2)(1)により算出された濃度比に、金属試料における比較元素の含有率を乗じる。
【請求項3】
前記電解液の一部を採取するに際し、前記電解液をろ過し、ろ過後の電解液の一部を採取することを特徴とする請求項1または2に記載の金属試料中の着目元素の固溶含有率を求める方法。
【請求項4】
前記採取後の電解液をろ過し、次いで、分析することを特徴とする請求項1または2に記載の金属試料中の着目元素の固溶含有率を求める方法。
【請求項5】
前記採取する電解液の量が、5ml以下であることを特徴とする請求項1ないし4のいずれかに記載の金属試料中の着目元素の固溶含有率を求める方法。
【請求項6】
前記採取後の電解液にキレート剤水溶液を添加し、比較元素および着目元素を水溶性キレートとしてから分析することを特徴とする請求項1ないし5のいずれかに記載の金属試料中の着目元素の固溶含有率を求める方法。
【請求項7】
前記キレート剤水溶液は、エチレンジアミン四酢酸塩水溶液であることを特徴とする請求項6に記載の金属試料中の着目元素の固溶含有率を求める方法。
【請求項1】
金属試料を電解液中で電解し、電解中および/または電解後に前記電解液の一部を採取し、前記採取された電解液を分析し、該分析の結果を基に、下記(1)〜(2)の方法により着目元素の固溶含有率を得ることを特徴とする金属試料中の着目元素の固溶含有率を求める方法。
(1)電解液中における、比較元素に対する着目元素の濃度比を算出する。
(2)(1)により算出された濃度比に、金属試料における比較元素の含有率を乗じる。
【請求項2】
金属試料を電解液中で電解し、電解後、金属試料の残部を電解液から取り除き、次いで該電解液の一部を採取し、前記採取された電解液を分析し、該分析の結果を基に、下記(1)〜(2)の方法により着目元素の固溶含有率を得ることを特徴とする金属試料中の着目元素の固溶含有率を求める方法。
(1)電解液中における、比較元素に対する着目元素の濃度比を算出する。
(2)(1)により算出された濃度比に、金属試料における比較元素の含有率を乗じる。
【請求項3】
前記電解液の一部を採取するに際し、前記電解液をろ過し、ろ過後の電解液の一部を採取することを特徴とする請求項1または2に記載の金属試料中の着目元素の固溶含有率を求める方法。
【請求項4】
前記採取後の電解液をろ過し、次いで、分析することを特徴とする請求項1または2に記載の金属試料中の着目元素の固溶含有率を求める方法。
【請求項5】
前記採取する電解液の量が、5ml以下であることを特徴とする請求項1ないし4のいずれかに記載の金属試料中の着目元素の固溶含有率を求める方法。
【請求項6】
前記採取後の電解液にキレート剤水溶液を添加し、比較元素および着目元素を水溶性キレートとしてから分析することを特徴とする請求項1ないし5のいずれかに記載の金属試料中の着目元素の固溶含有率を求める方法。
【請求項7】
前記キレート剤水溶液は、エチレンジアミン四酢酸塩水溶液であることを特徴とする請求項6に記載の金属試料中の着目元素の固溶含有率を求める方法。
【図1】

【図2】
【図4】
【図3】
【図2】
【図4】
【図3】
【公開番号】特開2009−31269(P2009−31269A)
【公開日】平成21年2月12日(2009.2.12)
【国際特許分類】
【出願番号】特願2008−162651(P2008−162651)
【出願日】平成20年6月23日(2008.6.23)
【出願人】(000001258)JFEスチール株式会社 (8,589)
【Fターム(参考)】
【公開日】平成21年2月12日(2009.2.12)
【国際特許分類】
【出願日】平成20年6月23日(2008.6.23)
【出願人】(000001258)JFEスチール株式会社 (8,589)
【Fターム(参考)】
[ Back to top ]