
金属配位組成物
生物活性部分と金属からなる金属配位錯体が開示されている。この錯体は有効性、安定性、吸収性、標的デリバリー及びそれらの組合せを含む改善された性能を生物活性部分に与える。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規な生物活性分子の金属配位錯体に関する。
【背景技術】
【0002】
公知の生物活性分子の特性をその構造を修飾することにより改善することは望ましい。前記修飾の目的は、分子を有効性、安定性、少ない副作用または標的デリバリーのような幾つかの点で改善することである。この改善を分子の望ましい特性を犠牲にすることなく達成する。上記目的は安易に述べられているが、具体的修飾の効果は極めて予測不可能なことが多いので実際行うと達成することが困難である。
【発明の開示】
【0003】
本発明によれば、公知の生物活性分子の構造を修飾すると、金属配位錯体として知られる新しい分子が生ずる。これらの新しい分子は予期せぬほど優れた特性を有している。本発明の金属配位錯体にはサイロニン、テトラサイクリン系抗生物質、オキシコドン及びヒドロコドンの錯体並びにそれらの誘導体の錯体が含まれる。
【0004】
キレート化は金属配位錯体の安定化における重要な要素である。s−ブロック金属に関して、このことはカルシウム及びマグネシウムに特に当てはまる。例えば、酢酸−マグネシウム錯体のlogKeqは0.47である。分子上に1つのアミノ基を導入すると(すなわち、グリシン)logKeqは1.34に上昇した。マグネシウムは典型的には窒素よりもむしろ酸素とキレート化し、この効果はアデニンのlogKeq(logKeq=2.08)を6−ヒドロキシプリンのlogKeq(logKeq=6.65)と比較することにより認められる。マグネシウムは、アデノシンのlogKeq(logKeq=0.50)とアデノシン−5’−モノリン酸のlogKeq(logKeq=1.80)の比較から明らかにされるように、酸化リン(例えば、ホスフェート)と特に強い結合を形成する。
【0005】
通常、亜鉛錯体は同等のマグネシウム錯体よりも安定である。このことは、配位子が酸素または硫黄を有している場合に特に当てはまる(これは、酸素のみを有している配位子の場合に当てはまらないことがあり、配位子がホスフェートである場合には余りあてはまらない)。上のグリシンの例を用いると、グリシン−亜鉛錯体のlogKeqは4.85である。亜鉛−酸素結合に対する亜鉛−硫黄結合の強度は、ヒドロキシプロパン酸の亜鉛錯体のlogKeq(logKeq=0.86)とメルカプトプロパン酸の亜鉛錯体のlogKeq(logKeq=6.43)の相対logKeq値で明らかである。logKeq値を他の金属や配位子と比較すると、このキレート化安定化は金属配位化学に勝っていることが明らかとなる。
【0006】
固有の共有原子価を有する安定な金属配位錯体を形成するためにキレート化を起こすことが必要でない場合もあり、このことは遷移金属を窒素配位子と組み合わせたときに特にあてはまるが、多くの場合、本発明の好ましい実施形態では活性物質を金属、特にマグネシウムとキレート化する。
【0007】
本発明の実施形態では、マグネシウム及びカルシウムとの錯体形成のための最良の候補になる活性物質はヘテロ原子(すなわち、酸素、窒素または硫黄)上に僅かに水よりも高いかまたは水よりも低いpKaを有するプロトンを有し、金属との結合あるいはキレート化に参加し得るように第1プロトン化ヘテロ原子に非常に近接して追加のヘテロ原子を有するものである。この官能基の配置を有する薬物は金属と結合する可能性が最も高いが、生じた金属配位活性物質は生体系中で十分に安定であり、前記系での加水分解を乗り越え、よって活性物質の性能が十分にモジュレートされる。多座配位子により付与される加水分解安定性は、アミドさえも配位金属の存在下で弱塩基(例えば、トリエチルアミン)を用いて脱プロトン化され得るように配位子のpKaを低下させ得るという事実により裏付けられる。従って、典型的な生物学的pHでは通常イオン化されないヘテロ原子上にプロトンを有する活性物質は共有的に配位された金属で置換されるプロトンを有し得、共有結合性は関与するヘテロ原子からの追加的なキレート化により強化される。本発明の好ましい実施形態では、マグネシウムまたはカルシウムに結合する活性物質上のヘテロ原子の少なくとも1つは酸素または硫黄である。マグネシウムはホスフェート及びホスホネートと著しく強い結合を形成し、よって本発明の追加の実施態様ではマグネシウムと配位する活性物質はオルガノホスフェートまたはオルガノホスホネート化合物である。
【0008】
本発明の実施形態では、亜鉛及びp−ブロック金属と錯体形成するための最良の候補になる活性物質は、活性物質が適切なキレート化配置で2個の窒素、1個の窒素と1個のメルカプタン、または2個のメルカプタンを有しているならば安定な金属配位錯体を形成するためにヘテロ原子上にプロトンが存在していなくてもよいという追加の柔軟性で、s−ブロック金属と錯体形成するものと同じである。本発明の更なる実施形態では、遷移金属は、配位子が少なくとも1個の窒素またはメルカプト基を有しているならば、共有配位錯体のためにキレート化は余り必要でない点で更なる連結反応柔軟性を有している。
【0009】
本発明において具体化される活性物質は表1に示す化学分類に分類され得る(実際には、異種キレート化ポテンシャルを反映すべく化学分類の組み合わせに分類され得る)。表1にリストした薬物は、本発明の実施形態を満たす全薬物の網羅的リストであると意図されず、医薬品中に存在する化学分類の代表例であると意図され、表1にリストした同一分類に入るかまたは本発明の実施形態により満足されない原子の配置を有する他の医薬品も本発明により特許請求される。
【表1】

【0010】
本発明を明確に理解し、容易に実施するために、本発明を図面と関連づけて説明する。
【発明を実施するための最良の形態】
【0011】
化学結合は3つの基本的形態:イオン結合、共有結合及び配位結合、すなわち典型的には無機金属塩よりも大きい所謂ウェルナー錯体で存在している。(ウェルナー錯体は中性配位子を有していると見なされることを指摘しなければならない。)3つの結合タイプの違いは特に溶液中での結合の熱力学的安定性に一部起因し得る。逆に、化合物の安定性は分子の原子が溶液中で分離または解離する傾向で表され得る。
【0012】
化合物の熱力学的安定性は、式1に従ってその形成の自由エネルギーに換算して表される:
ΔG=−RTlnK 式1
ΔGはギブズ自由エネルギーであり、化合物の熱力学的安定性を示す。ΔGがより負の値であるほどに化合物はより安定である。Rはガス定数であり、Tは絶対温度であり、Kは平衡定数である。平衡定数は反応物質に対する生成物の比で表される。反応:
【化1】
【0013】
に対する錯体化合物の場合、Kは式2:
K=[MLx]/[M][L]x 式2
で表される。よって、化合物の熱力学的安定性の増大は平衡定数の増大に直接関連している。
【0014】
ある場合には、平衡定数を金属−配位子結合の解離ポテンシャルに換算して表すことが有利である。反応は、上述のように
【化2】
【0015】
である。解離定数Kdissは式3:
Kdiss=[M][L]x/[MLx] 式3
で示される。イオン結合は溶液中でほぼ完全に解離すると一般的に認められているのに対して、多くの共有結合は全く解離しない。よって、解離定数またはより一般的に表されているパラメータである平衡定数を測定することにより溶液中の結合強度を求めることは結合のタイプを識別する方法である。金属と15〜17族からの配位子間の結合を含む配位化合物について熱力学的安定性はまだ確立されていない。
【0016】
文献の精査から、有機金属結合の共有原子価は分光分析データ(すなわち、NMR及びMS)、アプイニシオ分子力学計算、またはその2つの組み合わせから決定され得る。通常、共有原子価は遷移金属、(酸素よりも)窒素及び硫黄配位子原子、配位子の結合次数またはハプト数(“η”で示す)の増加で生ずる可能性が高い。複数のハプト数を有する配位子を有する有機金属化合物はキレートと記載されている。1及び2族の金属、すなわち所謂s−ブロック主族元素の中で、ベリリウム及びマグネシウムのみが重要なキレート形成元素と見られている。
【0017】
アプイニシオ理論計算を用いる最近の研究から、配位子−金属結合のイオン性対共有結合性の点で配位結合の種類が更に限定された。Pierlootは、これらの有機金属錯体の共有原子価の程度を調べるために一連のウェルナー錯体にCASSCF(完全活性空間自己無撞着場)モデルを適用した。彼女は概括的に、CASSCF計算から得た静的相関エネルギーが金属−配位子結合の共有原子価とうまく相関する傾向が存在すると結論づけた。彼女は更に、同一金属について金属−配位子共有原子価及び関連する相関効果は以下の配位子の順
F−<OH2<NH3<Cl−<Br−<I−
で増加すると結論づけた。これは、Jorgensenが記載したネフェルクス効果(nephelauxetic effect)と一致した。この効果の大きさは配位子場での配位に対する遷移金属の電子間反発の減少に直接相関した。この減少は配位子に依存し、錯体及び遊離金属イオン中のラカーパラメータBの比(β=B錯体/Bイオン)として表された。Bの減少は、金属錯体を形成するために配位子を添加した後の遊離金属イオンの電子−電子反発作用の低下から生じた。Bが大きく減少することは強いネフェルクス効果を示している。よって、例えば、F−のようなイオン性配位子はBを余り減少させず、より大きなβ値を有する。分光測定に基づいて、配位子はネフェルクス系列:
【化3】
【0018】
が生じるβ値の減少する順に従って並べられた。2つの系列の相関は、両技術が記載した効果間の類似性により裏付けられる。前者の場合、CASSCF計算は金属−配位子結合に対する金属d軌道の寄与を評価している。錯体形成によるラカーパラメータ(B錯体)の減少は配位子上の遷移金属d軌道電子雲の非局在化により生じ、これは共有結合が形成されていることを示している。Li+とBe2+のCp配位子との結合は、Cp結合に比して寄与金属結合のエネルギー状態が低いためにほぼイオン性である。加えて、これらの元素のイオン半径は2個以上のCp配位子が結合するには余りに小さい。
【0019】
マグネソセン(図1)Cp2Mgの理論的な計算から、該化合物の構造はCp2Ca、Cp2Sr及びCp2Baの構造に類似しているが、Mgのd軌道密度はCp2Mgでは無視できることが判明した。しかしながら、Cp2MgにおけるMgに対するマリケン電荷は密度汎関数理論(DFT)を用いて0.66と予想された。MgCl2のような高い解離定数を有する化合物に関しては2に近い値が予想される。これはFaegri、Almloef及びLuethiの論文と一致しており、彼らはアプイニシオMO−LCAO計算に従ってマグネソセンの電荷分離は公知の共有配位化合物であるフェロセン(Cp2Fe)の電荷分離よりも僅かしか高くないと結論づけている。これらのデータから、Mg−Cp結合がやや共有結合であることが示唆される。Cp部分は、一部はその負の電荷から、一部は二重結合由来のπ結合からその共有結合に寄与している。こうしたアニオン性及びπ結合と金属の組合せはレチノイン酸及びその類似体でも生ずる。よって、レチノイン酸の金属配位化合物を形成することは本発明の実施形態である。
【0020】
Cp2Mg及び関連するMg−Cp化合物の反応性をWinterらが研究し、彼らはマグネシウムがアミジナート配位子と安定な結合を形成することを知見した。おそらくこれを最も物語っているのは[CpMg(η2−tBuC(N(2,6−iPr2C6H3))2)]の安定性であり、これは未変化で昇華し、180℃/0.05トルで80%回収された(図2)。このように、安定なマグネシウム−アミジナート化合物の例が報告されており、該化合物の共有結合性に対する更なる裏付けを与えている。このことは、プリン及びアルギニン含有化合物のようなアミジナート官能基を含む化合物の場合重要である。
【0021】
遷移メタロセンが強い共有結合性を有していることは一般に認められているが、主族メタロセンが顕著な共有結合性元素を有するという道理にかなった論議もなされている。金属−配位子結合にd軌道が関与しないと化合物の安定性が低下する恐れがあるが、主族金属、特にマグネシウムが配位子とイオン性よりも共有結合性の結合を形成することができるという見解を妨げるものではない。結晶化の際に6−配位マグネシウム錯体が形成されるのはsp3d2混成のためであることは一般的に公知である。よって、ある状況ではマグネシウムのd軌道でも配位錯体の結合に関与し得ると考えられる。
【0022】
ウェルナー錯体の平衡定数を金属ハライドと比較したとき安定性に大きな差を見つけることができる。例えば、logKMg−ピリジン=2.08であり、logKMgCl2≒−1.0である。ピリジンが中性配位子であることを心に留めると、このlogKの差はマグネシウム−クロライド結合のイオン性と相対するマグネシウム−ピリジン結合の共有結合性のみに起因し得る。安定なマグネシウム錯体の別の例はマグネシウム−サリチルアルデヒド(SA)錯体であり、logKMg−SA2=6.80である(図3)。この結合の安定性は、配位子結合原子が通常金属とイオン結合を形成する傾向にある酸素である場合に顕著である。しかしながら、キレート化酸素が存在していると錯体を純粋なイオン結合以上に安定化させた。窒素はカルシウムよりもマグネシウムに対してより強い結合を形成するが、通常酸素は窒素よりもマグネシウムのより強いキレート化剤である。
【0023】
キレートの平衡定数は通常非常に大きく(例えば、マグネシウムエチレンジアミン−N,N’−ジスクシネート錯体のlogKeqは6.09である)、配位子の中性部分と金属間の共有原子価の程度を表していないことがある。しかしながら、平衡定数は配位化合物の安定性を指し、化学物質の性質及び特殊な用途でどのように機能するかを決定するための重要な基準である。錯体内の共有結合の存在及びそのキレートの安定性に対する寄与は非常に高いlogKeqを説明し得、分子構造の剛性にも寄与し得る。多くの場合共有結合性は配位錯体の安定性に対する最も重要な寄与因子であることを指摘しなければならない。
【0024】
マグネシウムポルフィリン錯体またはキレートはおそらく最もよく知られた有機マグネシウム化合物である。クロロフィルはマグネシウムポルフィンである。フタロシアニンはその種の化合物の基本的要素を表すポルフィリンであり、金属−ポルフィリン結合を研究するためのモデルシステムとして広く使用されている。遷移金属がフタロシアニンと非常に簡単に錯体(図4)を形成することは判明しているが、アルカリ及びアルカリ土類塩は水及び他のプロトン性溶媒中にほぼ完全に解離するのでLi+、Na+、K+、Sr2+及びBa2+をこれらの塩の溶液から直接導入するのに適した溶媒は今まで見つかっていない。Mg2+及びBe2+の錯体形成が容易であることから予測されるように、これら2つのs−ブロック元素のみがCa2+と一緒にフタロシアニンに直接導入され得、通常はピリジン中でヨウ化物または過塩素酸塩から導入される。
【0025】
金属−ポルフィリン錯体の合成、構造、安定性及び物理的特性は十分に研究されている。マグネシウムフタロシアニンの構造及び物理的特性は各種技術を用いて、より最近は近IR吸収及びX線結晶学を用いて更に解明されている。マグネシウム−ポルフィリンキレートが金属配位化合物の非常に安定な例に相当するという結論が再び得られた。
【0026】
特定のマグネシウム−配位子錯体は実際共有結合性であり、イオン性ではなく、よって新しい組成物であり、新しい塩の形態ではない。金属−有機配位子化合物はその結合の種類の点で共有結合性である。
【0027】
本発明の実施形態では、配位子が金属(好ましくは、マグネシウム)の内圏に対して直接結合する機会を有しているときには配位錯体の形成が好ましい。これは、無水マグネシウム及び非プロトン性溶媒(または、溶媒がプロトン性ならばその溶媒は嵩高くなければならない)を使用することにより達成される。このコンセプトは、金属イオンの触媒反応性が水和形態では低下するという事実により裏付けられる。水性系での錯体形成は配位子と水間の水素結合ならびに水和及び配位子の錯体形成能による金属上の結合部位に対する競合の微妙なバランスである。よって、当然の結果として配位子と金属の内圏の錯体形成も水性系で低下する。更に、当然の結果として、逆も真なりであり、すなわち非水性系での金属の配位子とのキレート化または錯体形成の速度は水性系に比して促進される。
【0028】
固有の共有原子価を有する安定な金属−配位子配位化合物としての金属に結合した有機活性物質を含む組成は新しい分子である。本発明の別の好ましい実施形態では、金属は主族元素から選択される。本発明の更なる実施形態では、金属はs−ブロック元素から選択される。本発明の好ましい実施形態では、金属はマグネシウムである。
【0029】
更に、本発明の実施形態では、実質的にKeq>1.0である薬物−マグネシウム錯体は解離、吸収、分布、代謝及び排泄の薬物動態をモジュレートするのに十分な固有の安定性を有している。Mg(OH)2の解離定数が−11.5であると仮定すると、多くのマグネシウム錯体が酸条件よりもアルカリ条件でより非常に安定であるとの知見は驚くことでない。よって、小腸での薬物−マグネシウム錯体の安定性は薬物吸収の薬物動態をモジュレートする可能性が高い。酸不安定なそれらの金属−薬物錯体の場合、本発明の実施形態では、錯体は小腸に進入時に錯体を放出するコーティングまたはカプセル化材料により胃の酸性環境から保護される。本発明の更なる実施形態では、カプセル化物質は外配位圏を形成する配位子または配位子群である。
【0030】
本発明の別の重要なコンセプトは、溶液中で金属と配位子を単に組み合わせても常に同一の生成物が生成されないことである。多くはないが幾つかの特許は主題を裏付けることなく従属クレームとして各種塩を特許請求していることを認識されたい。このことは、当業者に公知の方法で有機酸の塩が塩基及び金属塩で処理することにより容易に製造され、予想される生成物は有機酸の金属塩であるので認められる。しかしながら、配位化学が有機酸と金属間の結合に寄与する場合、溶媒、温度、及びおそらく最も重要である金属に結合する配位子のような各種条件が配位錯体の構造及び安定性に影響を及ぼす。
【0031】
薬物以外の別の配位子は金属−薬物錯体を安定化させ得る。例えば、グリシン(G)マグネシウム結合のKeqは1.34である。しかしながら、この錯体にサリチルアルデヒドを添加すると、反応:
【化4】
【0032】
の平衡は4.77である。明らかに、サリチルアルデヒドはマグネシウムグリシン結合に対して安定化効果を加える。本発明の実施形態では、有利な物理化学的特性を付与するためにサリチルアルデヒドのようなアジュバントを薬物:金属錯体に配合する。本発明の更なる実施形態では、アジュバントの効果は水溶液中のような特定の環境下で薬物:金属錯体を安定化させる。
【0033】
遷移金属との配位錯体の非常に少ない例がPhysician’s Desk Reference(“PDR”)に記載されており、その中には1)亜鉛で修飾したインスリン;2)カルボプラチンは白金を含むこと;3)ニフェレクスは多糖−鉄錯体であること;4)ふけ防止シャンプー中の活性成分として使用されているピリチオン亜鉛、が挙げられている。加えて、幾つかの栄養サプリメントが錯体として記載されている。3つのピコリン酸基が八面体で1つのCr+3に結合している(窒素が3つの他の結合部位を与える)ピコリン酸クロムが一例である。これに対し、本発明の実施態様では金属は遷移金属に相当する族から選択され、本発明のより好ましい実施形態では金属は1及び2族のs−ブロック主族元素から選択される。本発明の最も好ましい実施形態では、金属はマグネシウムである。
【0034】
特許文献は幾つかの新規なマグネシウム配位薬物を例示しており、その中には1)Trilisate(登録商標)関節痛を治療するための上記した安定な固体コリンマグネシウムサリチレート組成物;2)高い組織特異性、精製及び薬剤へのコンパウンディングの容易さを与える2−デスカルボキシ−2−(テトラゾル−5−イル)−11−デスオキシ−15−置換−ω−ペンタノルプロスタグランジンのマグネシウム塩;3)インスリン抵抗性症候群の治療において有用なインスリン様特性を有するバナジウム酸マグネシウム;4)血栓または塞栓性卒中を治療するため、子癇前症/子癇及び急性心臓状態を予防的治療するための結晶性マグネシウム−タウリン化合物;5)GERDを治療するための上記したマグネシウムオメプラゾール“塩”誘導体が含まれる。
【0035】
医薬用塩の科学は十分に研究されている分野であり、塩形態の選択は所与の医薬性能に影響を及ぼし得る。塩形態が薬物に対して有し得る効果の例には解離速度、溶解度、官能性、安定性、処方効果、吸収モジュレーション及び薬物動態が含まれる。2003年10月に発表された定期刊行物Drug Deliveryの記事は薬剤を安定化するために、おそらく塩を形成するであろう金属を用いた3つの賦形剤用途を記載している。ヒト成長ホルモンを亜鉛と錯体形成すると親水性が低下し、これにより薬物の放出が遅れる。カプセル化ポリマーの分解により生ずる酸性環境からのタンパク質の安定化は製剤に水酸化マグネシウムを添加することによりなされた。ビンカアルカロイドを酸加水分解から安定化させるために炭酸亜鉛が使用された。これらの生成物は明らかに医薬的に活性な物質を安定化させるために金属を使用しているのに対して、後者の2つは活性物質の構造を修飾させたとは記載していない。
【0036】
更に、塩形態を単に変化させるだけで小腸での吸収の薬物動態が大きくモジュレートされることは十分に確証されている。例えば、テトラサイクリンのリン酸塩及びテトラサイクリン塩酸塩の化学構造はファルマコフォアでない塩形態の部分の点で異なり、各々の相対的物理的特性は相対的バイオアベイラビリティーに対して大きな影響を持たないと予想される。しかし、実際リン酸塩は塩酸塩よりも2倍多く吸収される。逆に、ワルファリンの遊離酸のバイオアベイラビリティーはそのナトリウム塩とほぼ等しい。このことは、ワルファリンナトリウム錠の溶解速度が遊離酸を含有する錠剤よりも350倍速いので予期せぬことである。異なる塩形態が吸収のカイネティクスに変化を与え得るならば、s−ブロック主族元素との錯体は吸収に対してより顕著な効果を有することがあり得る。
【0037】
テトラサイクリン系抗生物質のバイオアベイラビリティーが消化管中で最も形成する可能性の高い金属錯体の物理化学的特性により主に影響されることは公知である。このことは、明らかにインビボでの薬物:金属結合形成を示している。インビボで薬物と金属間に共有結合が形成されることは、薬物が窒素を含有し、金属が10〜12族(例えば、ニッケル、銅、亜鉛)に属するときにはより合理的な予想である。本発明の実施形態では、薬物性能のモジュレーションは薬物と金属間の安定な錯体の形成を促進する薬物と金属の製剤化により付与される。
【0038】
本発明の実施形態では、金属と錯体形成または配位したとき薬物に対して以下の効果が与えられ得る:
1.より良好なバイオアベイラビリティーと同一視し得る改善された水溶性(以下の討論参照);
2.細胞膜を介する吸収を改善するための高い親油性;
3.改善された受容体結合のためのファルマコフォアのコンフォメーションへの閉じ込め;
4.多形に起因する製剤化の問題の改善(以下の討論参照);
5.胃のような酸性環境での分解から保護するための酸吸収性;
6.配位錯体の安定性は活性ファルマコフォアの吸収の遅れを暗示し得る。このことは、薬物の迅速吸収が毒性ポテンシャルを上昇させるリオサイロニンのような薬物にとって重要である;
7.活性ファルマコフォアの持続吸収のための生物接着性;
8.麻酔薬のファルマコフォアに有機金属錯体を介して結合し、摂取されない限り麻酔薬を不活性とすることによる麻酔性鎮痛剤の乱用の防止。
【0039】
自然界は、どのように遷移金属を輸送し、貯蔵し、利用するかの多くの例を提示している。おそらく、最もよく知られている例は鉄ポルフィリンであるヘモグロビンである。上記したように、クロロフィルはマグネシウムを囲むポルフィン構造である。幾つかの酵素は該酵素を活性とするために金属を必要とする。これが、微量金属(例えば、銅、亜鉛、クロム等)が適切な栄養供給のために重要な理由である。幾つかの抗体もこれに関連する遷移金属を有している。金属が配位錯体の形成により酵素のペプチド構造をコンフォメーションに閉じ込めるため、金属が酵素活性のために必要である。
【0040】
薬物のコンピューター支援設計を組み入れるコンセプトは最近評判を得ている。イン・シリコと称されているこの技術は、何の物質も生成していない段階で基質と受容体間のアロストリック、クーロン力及び非共有相互作用を理解して、リード薬物候補をコンピューターモデリングにより同定するまでに開発されている。本発明の実施形態では、コンピューターシミュレートした分子に金属配位を加えることにより新しく改善されたリード化合物を同定することができる。本発明の更なる実施形態では、リード化合物の錯体としての金属の導入を計算に組み入れるとき、イン・シリコに由来するリード化合物は変化した結合熱力学を有している。本発明の更なる実施形態では、以前に不十分なイン・シリコ分析に基づいて考察から除外された化合物は金属錯体を計算に組み入れて再分析したとき重要なリード化合物になるであろう。本発明の好ましい実施形態では、上記したような改正イン・シリコ計算のために使用される金属は主族元素から選択される。本発明のより好ましい実施形態では、金属はs−ブロック主族元素から選択される。本発明の好ましい実施形態では、金属はマグネシウムである。
【0041】
錯体を生体系に直接適用する点で、本発明の実施形態では、配位子−受容体結合を必要とする活性物質には、金属と錯体形成して活性物質のコンフォメーション構造を適所に閉じ込めることによって、高い生物学的活性が付与されている。受容体は膜結合性であるか、細胞質内にあるか、または体内を循環している。本発明の実施形態では、標的受容体との最適相互作用を与えるコンフォメーションで薬物を閉じ込めるために金属を注射薬に配合する。本発明の好ましい実施形態では、金属は注射のために安全であると考えられる。本発明の更なる実施形態では、金属はアルミニウム、ビスマス、マグネシウム、カルシウム、鉄または亜鉛のリストから選択される。本発明の更に好ましい実施形態では、活性物質は注射薬のリストから選択され、前記注射薬にはワクチン、抗悪性腫瘍薬、抗糖尿病薬、及びアンチセンスRNAまたは他の代謝モジュレーターが含まれるが、これらに限定されない。
【0042】
本発明の金属配位技術はワクチン設計における現在の研究を前進させることもできる。例えば、開発中の新しい癌ワクチンはリポタンパク質アジュバント、ペプチド抗原を、癌細胞に対して特異的な炭水化物抗原と組み合わせる。ワクチン構築物の3つの成分をリンカーを介して一緒に共有結合させる。このワクチンの構築方法はバイオコンジュゲート化学では一般的である。金属配位は、組み合わせる成分がアプタマー、ポリエチレングリコール及び脂質であるペガプタニブのようなバイオコンジュゲートの各種成分を結合させるための足場として使用され得る。本発明の実施形態では、バイオコンジュゲートの諸成分は各成分を中心金属に錯体形成することにより単一分子中に組み合わされ得る。本発明の更なる実施形態では、金属配位をバイオコンジュゲート化学における一般的な技術として使用する。
【0043】
マグネシウムが核酸に対して著しい親和力を有していることに特に注目されたい。アンチセンスRNA、干渉RNA及びアプタマーの治療薬としての進歩に伴って、これらの核酸薬に対してデリバリー技術を取り入れることがますます重要であろう。現在研究されている薬物デリバリー技術の幾つかにはペグ化、リポソームまたはアニオン性クレーが含まれる。興味深いことに、Howard Hughes Medical Research Labから発表された最近の研究でモンモリロナイトクレーがRNAの脂質小胞への進入を促進することが示された。アルミナ、シリカ、マグネシア、鉄及びカリウムの量の点で異なる各種クレーがある。よって、RNAのマグネシウム錯体を形成すると、細胞膜の実験室モデルと見なされる小胞へRNAの進入が促進され得る。
【0044】
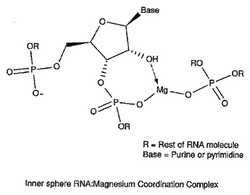
マグネシウム−核酸錯体の有効性を核酸単独と比較して上記したイン・シリコ技術を用いて評価し得る。よって、核酸薬の有効性が金属と配位させることにより高まることが本発明の重要な実施形態となる。本発明の更なる実施形態では、投与前に核酸を金属と組み合わせて配位錯体を形成する。水性系中で金属塩を核酸と単に組み合わせた核酸の重要な部分は外圏配位配位子(図5)であり、受容体結合のための特に膜輸送用途のために最適なコンフォメーションを与えないことがある。本発明の主たる前提は、配位子が外圏配位子の存在下で内圏配位子(図6)となる機会を有しているならば金属−配位子錯体構造が大きく影響されることである。
【0045】
本発明の更なる前提は、内圏配位子の形成が金属−配位子錯体を製造するために無水条件を使用することにより促進されることである。従って、本発明の実施形態では、金属配位子錯体を無水条件下で製造し、前記錯体を水中で再構成すると、水中で製造した錯体に比してより大きい共有結合性、より高い安定性、より優れた細胞透過性及びモジュレートされた生物学的性能を有する配位錯体が生ずる。この系は、トランスフェクション効率を高めるための内配位圏内へのアジュバント(例えば、ポリアルギニン)の導入に適している(図7)。
【0046】
おそらく、最近の遺伝子治療での最も重要な進歩は干渉RNA(“iRNA”)に関する発見及び進歩した研究であった。アンチセンスRNAとは異なり、iRNAはmRNAによりコードされる遺伝子産物を更に抑制させるために細胞の生化学機構により再利用される。こうすると、遺伝子抑制の効率が高まる。iRNAに関連する主要な問題には細胞への透過性及び(特にヌクレアーゼの存在下での)安定性が含まれる。
【0047】
これらの問題は、肺表面活性物質(“SAM”)、脂質またはアミンを主成分とするトランスフェクション剤、エレクトロポレーション、ウイルスベクターまたはプラスミドベクターを導入することにより解決されてきた。後者の技術は、プラスミドベクターによりsiRNAが“ヘアピン”構造を採用し、iRNA変異体に短ヘアピンRNAまたはshRNAの名前が付けられている点で特に興味深い。これらのshRNA分子は高い抑制能力を有している。更に、siRNA分子が細胞に進入するのにトランスフェクション剤が必ずしも必要でないことを示唆する証拠は多数存在する。siRNA及びウイロイドを肺適用するという最近の知見は、裸RNAが細胞に進入し、遺伝子産物を抑制させ得る2つの報告された現象である。実際、当業界の科学者にはRNAの二次構造がその遺伝子抑制効果に影響を及ぼさないようであることが知られている。
【0048】
RNAは複数のホスフェート基を有するオリゴヌクレオチドである。マグネシウムはホスフェート基と非常に強い結合を形成し、よってRNA−Mg錯体ではおそらくマグネシウムはホスフェート基と結合している可能性が高い。無水条件下でマグネシウムをRNAと組み合わせることにより、共有結合が形成され、こうすると理論上RNA分子の当該部分の親油性が高まる。更に、マグネシウム中心は複数のホスフェート基を結合し得、理論上上記したヘアピン構造が形成される。このヘアピン構造により親油性残基が現れるだけでなく、ヌクレアーゼからの攻撃に対してより高い耐性が生じ、これにより高い安定性が得られる。
【0049】
RNAはマグネシウムに結合しているホスフェート基以外に他のホスフェート基を有しているので、RNA分子の当該部分はその水溶性を保持している。この新規な形態のRNAは物質輸送(親水性)及び吸収(親油性)にとって重要な所望の両親媒性を有する。この点の更なる検討については、以下の「改善された溶解性」を参照されたい。
【0050】
典型的な方法は無水溶媒中でRNAをマグネシウムと組み合わせることを伴う。適当な溶媒はDMSO、またはおそらくイオン性液体である。イオン性液体の利点は、アルコールのようなイオン性液体混和性非溶媒(または、幾つかの場合には超臨界CO2が機能することがある)に溶液を添加するだけで所望の生成物が析出し、マグネシウム−RNA錯体が回収されることである。次いで、イオン性液体はアルコールを留去させることにより次の反応のために再利用され得る。
【0051】
上記方法はおそらく水溶性の生物活性物質に適用することが可能である。よって、本発明の実施形態では、生物活性物質は糖、ペプチドまたはヌクレオチドである。本発明の好ましい実施形態では、生物活性物質はヌクレオチドである。本発明のより好ましい実施形態では、生物活性物質はアンチセンスRNA、干渉RNAまたはアプタマーである。本発明の好ましい実施形態では、金属は主族元素から選択される。本発明の更に好ましい実施形態では、金属はs−ブロック主族元素から選択される。マグネシウムがカルシウムよりも核酸に対してよりしっかりと結合することは認められており、よって金属がマグネシウムであることが本発明の最も好ましい実施形態である。
【0052】
改善された溶解性/浸透性
薬物吸収を定量する際、用語「バイオアベイラビリティー」を適用することが有用である。これは体循環に達する用量の分数(F)として定義される。極端な場合、薬物が消化管に全く吸収されない場合にはF=0であり、薬物が完全に吸収される(且つ、初回通過効果により代謝されない)場合にはF=1である。バイオアベイラビリティーは血清レベル対時間プロットの曲線下面積(AUC)から計算され得る。バイオアベイラビリティーは多くの因子に依存し、これらの因子の幾つかは正常な個人の間でも異なる。バイオアベイラビリティーの点で、薬物は下表に従う4つのカテゴリーに分類されている。
【表2】
【0053】
分かるように、更なる開発のための実行可能な候補となるよう、薬物は細胞膜透過性と溶解性の微妙なバランスに適合していなければならない。この理由は、溶解性(すなわち、親水性)を高める物性が通常透過性(すなわち、疎水性または親油性)を高める物性と相反しているからである。
【0054】
金属とテトラサイクリン系抗生物質の相互作用により薬物及び金属の両方のバイオアベイラビリティーが低下することは判明している。上記したように、テトラサイクリン系抗生物質のバイオアベイラビリティーは主に消化管に広がっている金属錯体の物理化学的特性により影響される。中性種は腸管細胞のリン脂質膜に容易に吸収される可能性が高いので、電荷はバイオアベイラビリティーに大きな影響を有している。親油性金属配位錯体は、電荷を有している薬物の金属塩と比較してバイオアベイラビリティーを高めるのに役立たなければならない。よって、本発明の実施形態では、親油性金属−抗生物質共有結合性錯体を投与することにより抗生物質の物理化学的特性がコントロールされ得、更に消化管中の金属が薬物との金属相互作用の動態、最終的には吸収に影響を及ぼすのを防げ得る。本発明の更なる実施形態では、上記した原則が一般的にすべての薬物に適用される。
【0055】
この技術は、高度に水溶性の薬物または所謂クラスIII薬物の親油性を高めるためにも使用され得る。この場合、ホスフェートまたはスルフェート基のようなイオン性中心が共有結合に変換される。この金属と配位子間の結合の変化により配位子の水溶性が低下し、配位子の有機溶媒溶解性または親油性が上昇することは知られている。
【0056】
薬物が余り溶解しないが容易に透過し得るならば、その溶解性を高め得る1つの方法は薬物に対して水溶性物質(例えば、アミノ酸または炭水化物)を共有結合させることによる。或いは、薬物とイオン化金属中心間で金属−配位子錯体を形成することにより、固有の親水性が付与された新しい化学物質が形成される。本発明の実施形態では、活性物質を遷移金属またはアルカリ土類金属に結合させて、その透過性を保持しながら溶解性を改善した新規な組成物が形成される。新しい金属配位子結合は共有結合であるので、新しく形成された共有結合金属中心の親油性を相殺するように金属に追加の配位子(例えば、アミノ酸)が結合されていることが好ましい。
【0057】
共有結合性により、金属−活性物質錯体の安定性は刷子縁膜の水フィルムコーティングへの輸送まで保持されている。錯体が膜に達すると、金属と薬物は、親油性活性物質を受容し親水性金属を拒絶する膜中の脂質により分離される。これは物理化学的作用により行われ、薬物の水溶性を上昇させる従来方法とは対照的に酵素を必要としない。
【0058】
薬物は1)皮膚表面に対する効果、2)角質層内の効果、3)表皮及び真皮へのより高い浸透を必要とする効果、または4)血管系への浸透を介する全身効果を引き出すために皮膚に適用される。この研究の目的は、薬物を表皮を介してまたは血管系に浸透させ得る新しい経皮薬物デリバリー(TDD)システムを設計することである。浸透の所望レベルは薬物に依存する。
【0059】
角質層は有効なバリアを与え、水及び化学物質が表皮に、表皮から浸透するのを防止する。角質層における脂質の構造組織化は水及び化学物質の迅速な輸送を防止するのに重要な因子であることが提案されている。このように脂質が組織化すると、液晶形態が生じ、このマトリックスを介する浸透は、脂質炭化水素鎖が乱され液晶が不安定化することにより生ずる。これが、ハイドロトロープの局所的に適用された薬物の浸透性を高める能力について提案されているメカニズムである。
【0060】
皮膚透過性を高めるために使用されている化学物質の分類の幾つかにはアルコール、アルキルメチルスルホキシド、ピロリドン、界面活性剤(アニオン性、カチオン性及び非イオン性)、脂肪酸及び脂肪アルコールが含まれる。加えて、ラウロカプラム、尿素、カルシウムチオグリクレート、アセトン及びジメチル−m−トルアミドが特定の生物活性物質の皮膚浸透性を高めるために使用されている。これらの薬物のビヒクル効果の多くはその屈水性による。化学的には、これらの多くは大きい双極子モーメントを有している。すなわち、親油性部分及び親水性部分を有している。この大きな双極子モーメントが化学物質が角質層中の脂質を乱す主要な要因である。
【0061】
多くの薬物は本質的に局所的に使用するのに十分な皮膚浸透能を有していない。よって、実質的にすべての局所適用される薬物は所望の効果を得るためにビヒクルまたはTDDエンハンサーを含む製剤を必要とする。すべての医薬品が従わなければならない安全性及び有効性の一般的要件に加えて、局所適用される薬物はビヒクル中に溶解し、安定でなければならず、製剤は含量均一性を有していなければならず、製剤は適正な粘度及び分散特性を有していなければならず、患者のコンプライアンスを最大にしなければならない(このことは、製剤が適用するのに不快であってはならず、不快な臭いを有していたり皮膚刺激を生じさせてはならないことを意味する)。
【0062】
最も注目することは、薬物が表皮に浸透するためのタイムラグはビヒクルから角質層へ分散する薬物の能力に依存するが、これがTDD製剤の開発にあたり大きな障害となっている。以前の報告で、このタイムラグが数分〜数日にも及び得ることを示している。よって、TDDシステムの開発の重大な障害はこの適用に特有の追加の考慮すべき事項であり、歴史的によれば経皮医薬品の開発時間はしばしば途方もなくかかると見られていた。
【0063】
本発明に従う薬物錯体の高い経皮透過性は錯体の安定性と両親媒性の組合せに依存している。よって、実施形態では、共有結合金属−薬物結合を形成させて薬物それ自体のTDDを強化し得る有効なハイドロトロープに薬物を変換させる。本発明の更なる実施形態では、TDDエンハンサーがなお必要とされているならば金属をビヒクルに対するアンカーとして作用させ、全錯体を単一分子として挙動させる。この作用効果は、錯体からの薬物の放出のためにビヒクルと表皮の脂質マトリックス間の分配係数を差別化する必要がないことである。
【0064】
共有結合性のために、金属−活性物質錯体の安定性は角質層を介する輸送の間保持されていなければならない。錯体が表皮中にあるとき、金属と薬物は、親油性活性物質を受容し親水性金属を拒絶する膜中の脂質により分離される。これは物理化学的作用により付与され、薬物の溶解度を高める従来方法とは対照的に酵素を必要としない。
【0065】
薬物を金属配位錯体に変換すると、眼への進入も容易となる。眼圧(IOP)を治療するためにスルホンアミドをその金属配位錯体に変換するとそのIOP降下効果が高まることは判明している。これは、一部は眼中にスルホンアミドが多く存在しているためであり、よって金属配位錯体の親油性と親水性のバランスが適切であるためであると考えられる。眼病を治療するための薬物は本発明に従って金属配位錯体に変換させることにより改善され得る。このことは、現在の治療では薬物を眼の奥部に注射している加齢黄斑変性(AMD)を治療するために非常に重要である。AMDを治療するために薬物を点眼すると患者のコンプライアンスが大きく改善される。これは、AMD薬物を金属と配位させることにより達成される。
【0066】
多形のコントロール
多形は、一部は溶解度の変動のために投与量の変動に大きく寄与している。歴史的に言えば、有機金属化合物の固有の物理的特性は安定な結晶形を製造するのが比較的容易であることである。よって、本発明の更なる実施形態では、活性物質を金属錯体に変換させ、前記錯体を当業者に一般的に公知の方法により再結晶化方法にかけることにより多形を解消する。こうすると、活性部分は所望の多形に閉じ込められる。
【0067】
薬物吸収のモジュレーション
最近では、薬物のデリバリー方法をモジュレートすることにより薬物を改善するための活動が活発である。薬物デリバリー技術は経口から注射、インプラント、皮膚パッチのすべての投与形態に広がっている。これらの技術の大部分は、活性成分をポリマー球体の内部に封入または“トラップ”するカプセル化技術またはビーズ技術を利用している。このポリマー球体はミセルとして、自己組織化分子ロッドまたはボールとして、または活性成分の周りのコーティングとして存在し得る。薬物は、血液中を循環したり消化管を横断するとカプセル化剤から溶媒和または膨潤することにより放出される。薬物のデリバリーをモジュレートする主な利点は、その放出を広げたり、改善された安全性のために血液レベルを調節したり、または改善された有効性のために吸収を高めることである。よって、本発明の実施形態では、薬物−金属錯体放出はインビボで錯体それ自体に対する物理化学的作用によりモジュレートされる。
【0068】
ある場合には、薬物−金属錯体をポルフィン、ペプチドまたはポリマーマトリックス内に封入することにより活性物質−金属錯体の安定性を高めることが有利であり得る。このことは、活性物質が金属と安定な錯体を形成するために必要な要素(例えば、上記した第1級アミンまたはアルコール)を含んでいない場合に特に当てはまる。本発明の好ましい実施形態では、マトリックスは活性物質を金属に結合させるために所要により修飾されているポルフィン誘導体である。本発明の実施形態では、薬物−金属錯体放出はインビボでポルフィン、ペプチドまたはポリマーマトリックスに対する物理化学的作用によりモジュレートされる。本発明の更なる実施形態では、マトリックスは元々小腸に存在する化合物である。本発明の更に好ましい実施形態では、ポルフィンマトリックスはビリルビンまたはその誘導体である。
【0069】
本発明の更に好ましい実施形態では、カプセル化マトリックスはアミノ酸またはジペプチドであり、前記アミノ酸または複数のジペプチドは金属−配位子錯体と配位させるかまたはその周りで自己組織化させるために添加され得る。ヒスチジンは、ヒスチジン中のイミダゾール部分の強い金属結合能のために理想的なアミノ酸である。アルギニンは、ペプチド結合アルギニンのグアニジン部分のアミジナート連結によりマグネシウムと錯体形成するのにうまく適している別のアミノ酸である(図8)。本発明の関連実施形態では、その錯体形成及び酸中和のためにマグネシウムは胃においてアルギニンを安定化させ、その有効性を増加させる。このことは、アルギニンをCOPD及び関連する病的状態を治療するためのNO源として使用する場合に役立つ。
【0070】
金属上の第2配位子としてアミノ酸を使用することは、内配位圏を安定化させ、内圏の周りに疎水性シェルを生成し、よって金属−薬物結合の加水分解を防止するためである。よって、本発明の実施形態では、錯体を特に水性系中で安定化させるためにアミノ酸、ジペプチドまたはオリゴペプチドを金属−薬物錯体上の第2配位子またはアジュバントとして作用させる。本発明の好ましい実施形態では、第2配位子はジペプチドである。本発明の別の好ましい実施形態では、第2配位子はアミノ酸である。本発明の別の好ましい実施形態では、アミノ酸はヒスチジン及びアルギニンの群から選択する。
【0071】
遊離アミノ基を有する(例えば、錯体の一部としてヒスチジンのようなアミノ酸を有する)有機金属錯体はアミノ酸−NCAの重合を開始させてポリペプチドを形成し、有機金属錯体を配座的に保護する。この技術の別の利点は、アミノ酸NCAを有機金属錯体の周りで自己組織化させ、次いで重合の開始時にポリペプチドを自己組織化構造にコアセルベートすることである。
【0072】
本発明の実施形態では、カプセル化技術をコアセルベーション技術と組み合わせて活性物質と遷移金属間に内圏共有結合を形成して新しい組成物を生成し、次いで前記錯体をポリマーマトリックス内に外圏配位でカプセル化して、安定な錯体を提供する。図9は、活性物質(構造簡素化のために、サリチル酸を例として使用している)、内圏でマグネシウムに結合させたポリマー結合アルギニン、及び外圏で錯体を封入しているペプチドを示している。
【0073】
本発明の更なる実施形態では、カプセル化マトリックスを水、油、エマルションまたは生体流体(例えば、胃液)により膨潤または溶解させた場合のみ活性物質が放出する。本発明の実施形態では、活性物質は抗体−抗原錯体で生ずるようなカプセル化剤と活性物質間の強い結合によりカプセル化マトリックスから放出され得ない。幾つかの場合には、カプセル化剤からの薬物の放出を消化酵素によりモジュレートすることが有利である。本発明の好ましい実施形態では、腸中に分泌される、細胞膜内に存在する、または血流中を循環する酵素による化学分解により活性物質をカプセル化剤から放出させる。本発明の好ましい実施形態では、活性物質はアルミニウム、マグネシウム、カルシウム、鉄、ビスマス、ケイ素または亜鉛に結合している。本発明の別の最も好ましい実施形態では、カプセル化剤は金属−配位子錯体に対する抗体である。本発明の更に別の実施形態では、錯体は活性物質−金属錯体からなり、カプセル化剤はアミノ酸、ポルフィン、炭水化物またはそれらの混合物の組合せから自己組織化される。本発明の最も好ましい実施形態では、活性物質−金属−カプセル化剤錯体は医薬品である。
【0074】
本発明の別の実施形態では、配位錯体は該錯体を形成し得るすべての金属から選択される金属であり、薬物はすべての生物活性物質または医薬活性物質の群から選択される。本発明の好ましい実施形態では、医薬活性物質は生物活性のために特殊なコンフォメーションを必要とする。活性は活性物質の細胞膜を横断する能力に依存し得、配位金属は活性物質の膜転移のために適正な構造を与える。好ましい実施形態では、医薬活性物質は小分子、ペプチド、炭水化物、DNAまたはRNAからなる群から選択され、後者の2つは遺伝子治療において、アプタマーとして、またはアンチセンスヌクレオチド治療用途において使用されている。好ましい実施形態では、金属はアルミニウム、ビスマス、カルシウム、マグネシウム、鉄、ケイ素及び亜鉛からなる群から選択される。
【0075】
生体接着性
マグネシウムまたはカルシウムイオンを薬物の分子式に導入して薬物に対する生体接着性を推定する方法は各種ある。例えば、マグネシウム及びカルシウムがインテグリンの接着機能にとって重要であることは公知であり、よって腸管での薬物のマグネシウムまたはカルシウム塩、或いは薬物の錯体が腸壁の刷子縁膜上で発現するインテグリンに対する薬物の生体接着を高めると予想することは合理的である。また、生体接着により腸の通過時間が遅くなるので、これらの錯体は腸における吸収を持続性とする。従って、本発明の実施形態では、薬物の持続吸収は薬物をマグネシウムまたはカルシウムと錯体形成させることにより高められる。本発明の更なる実施形態では、より強い生体接着活性によりマグネシウムまたはカルシウム薬物錯体に対して持続放出が付与される。
【0076】
麻酔薬の常習防止
麻酔薬は非常に有効な鎮痛剤であるが、非常に常習性でもある。最近数年間、アヘン剤常習者及び気晴らし薬の使用者がOxyContinを常習していることを記載している報告書が多数ある。典型的には、薬物常習者は例えば水を添加することにより錠剤マトリックスを機械的または化学的に崩壊して、全12時間用量を1度に服用している。加えて、このタイプの常習は通常経口投与から始めるが、常習者はしばしば濃厚麻酔薬を鼻から吸入したり注入したりするようになることがある。
【0077】
オキシモルホン及びオキシコドンの構造分析から、その分子は金属とのキレート化のための理想的な候補であることが分かる。9位のβ−ヒドロキシ及び窒素は、その2つの間に金属を錯体形成すると非常に熱力学的に好ましい5員環が形成されるように配置されている。9−ヒドロキシを脱プロトン化してアニオン性アルコキシドを形成することが好ましい(図10)。窒素の孤立電子対は金属キレートを安定化するために十分な電子密度に寄与し得る。サリチルアルデヒドがグリシン−マグネシウム錯体を安定化する場合のように錯体に第2配位子またはアジュバントを添加することにより更なる安定化が付与され得る。本発明の更なる実施形態では、金属−麻酔薬錯体を上記したようにマトリックス内に封入する。
【0078】
よって、本発明の実施形態では、麻酔薬を金属と錯体形成させることにより、麻酔薬は物理化学的作用により錯体からゆっくり放出される。このことは、麻酔薬が即座にまたは一度に利用され得ないことを意味する。更に、本発明の実施形態では、金属−麻酔薬錯体は血液脳関門を通過することができず、麻酔薬が錯体から放出されるまで麻酔薬は無効である。放出速度は遅いので、一度に血液脳関門を超えて移動することができる麻酔薬の量は経口投与される用量よりも非常に少なく、陶酔効果は得られない。本発明の更なる実施形態では、第2配位子、カプセル化剤またはその両者の組合せを導入することにより麻酔薬の放出カイネティクスを更に遅くすることができる。好ましい実施形態では、金属はアルミニウム、ビスマス、カルシウム、マグネシウム、鉄、ケイ素及び亜鉛からなる群から選択される。本発明のより好ましい実施形態では、金属は主族元素から選択される。本発明の更に好ましい実施形態では、金属はs−ブロック主族元素から選択される。本発明の最も好ましい実施形態では、金属はマグネシウムである。
【0079】
金属の選択
配位錯体中に使用しようとする好ましい金属について言及する。医薬用途では、本発明の錯体中で使用する金属を選択するときには、全ての金属配位医薬品の安全性を考慮しなければならない。本発明を実施するために多くの金属を使用することができるが、本発明の好ましい実施形態では金属を一般に安全と認められている(GRAS)一覧表から選択する。金属を選択するための1つの基準は、現在市販されているミネラルサプリメントのリストを検討し、表1にリストされている薬物との配位錯体として配合されるであろう用量をはるかに超える投与量を有するものを選択することである。非処方薬物及びダイエタリーサプリメントに関するPDRから、2mg/投与以上の量を有する7つの金属(アルカリ金属、すなわちナトリウム、カリウム等を除く)のリストを表2に示す。
【表3】
【0080】
本発明の好ましい実施形態では、金属はアルミニウム、ビスマス、カルシウム、鉄、マグネシウム、ケイ素及び亜鉛からなる群から選択される。本発明の実施形態では、医薬品とランタニド、アクチニド、遷移金属及び主族金属(s−及びp−ブロック)を含めた任意の金属間で共有結合を形成することにより新しい組成物を形成するが、本発明の好ましい実施形態では金属をs−ブロック主族元素から選択する。この理由は、s−ブロック元素が遷移金属またはp−ブロック主族元素(ランタニドまたはアクチニドはOTC製品中に決して使用されない)よりもGRASである可能性が高く、OTC医薬品やビタミンサプリメント中によりしばしば使用されているからである。他のs−ブロック元素(例えば、カルシウム)よりもマグネシウムを選択するのには幾つかの理由がある:
1.カルシウムは優先順で8>7>6>9という配位数の高い変動性を示す。カルシウムよりも小さいマグネシウムは殆どもっぱら、合成手順を簡素化し、生成物の混合物ではなく単一の生成物をより高い収率で生成する可能性が高い八面体である;
2.マグネシウムは他のs−ブロック元素よりもより容易にキレート化配位子と共有結合を形成し得る;
3.マグネシウムはカルシウムよりもタンパク質及び核酸とより安定な結合を形成し、よって生物学的医薬品の安定性を高める;
4.マグネシウム欠乏は幾つかの病的状態(例えば、心臓血管関連、偏頭痛、ADHD)に関与しており、予防の視点からマグネシウムは重要な効果を有し得る。例えば、トリプタンマグネシウムはこの技術の理想的な候補であり得る;
5.カルシウムは能動輸送メカニズムにより腸に吸収されるのに対して、マグネシウムは受動的に輸送される。マグネシウム(塩として)及びフロセミドの腸管輸送は両者を一緒に経口投与したときに促進された。よって、吸収されにくい薬物であるフロセミドはこの技術の別の注目すべき候補に相当する。
【0081】
本発明の最も好ましい実施形態では、金属はマグネシウムである。
【0082】
溶媒の選択
上記したように、錯体形成反応のための溶媒の選択は金属配位化合物の構造及び安定性に影響を及ぼす。マグネシウムは水と強い結合を形成し、配位圏水和マグネシウムは生成物形成のカイネティクスだけでなく、生成物の構造及び安定性に対して影響を及ぼす。窒素含有配位子を遷移金属(例えば、亜鉛)と反応させる場合には強い窒素−遷移金属結合のために、反応混合物中の水の存在は通常金属配位生成物の構造及び安定性に対して強い影響を持たない。
【0083】
幾つかの場合には、配位子、金属及び所望生成物に応じて水は好ましい溶媒であり得る。生成物の大部分は無水有機溶媒が最良の選択であるとしている。幾つかの適当な溶媒にはアルコール、アセトンまたはTHFが含まれる。実質的にすべての医薬品及び栄養品を溶解し、塩化マグネシウムを含めた大部分のハロゲン化金属をも溶解する優れた万能溶媒であるので、DMSOが最も好ましい溶媒である。これにより、単相反応ができる。加えて、安定な金属配位医薬品はコアセルベーションに類似の方法により単離され得る。コアセルベーションは典型的には反応混合物に非溶媒を単に添加することを含む。
【0084】
DMSOはマグネシウムを含めた金属とその場で錯体を形成し得、DMSO−金属錯体は薬物配位子と反応し始めて金属中心でDMSO配位子を置換させる。その後、DMSOはアジュバントを錯体中に配合しようとする反応では一時保護基として機能し得る。このインプロセス反応スキームは、DMSO配位子間に水素結合が欠けているためにDMSO−金属錯体は水のような外配位圏を形成できないという事実により促進される。こうすると、金属中心は入ってくる配位子に容易に接近し得る。形成された金属配位錯体に応じて、最終生成物はDMSOを配位子として保持していることも、保持していないこともある。DMSOが配位子に結合しているならば、DMSOの用量が毒性レベルに近くまで達するような状況はありそうもない。
【0085】
本発明の前提では、生物活性配位子の水溶液に金属塩を添加するだけでは、形成された配位錯体は無水条件下で反応物質を組み合わせたかのように同じでない。更に、乾燥した配位錯体を同一水性環境において再構成しても、その2つの錯体の構造は異なる。その目的を達するために、幾つかの配位錯体をFDA承認医薬品を用いて製造し、これらの錯体が安定であることを立証することができる。生成物はできるだけ完全にキャラクタライズされ、バイオアベイラビリティ研究が行われるだろう。金属配位錯体は水及び有機溶媒中で製造され得る。多くの場合それぞれの生成物が安定性、構造及び生物活性の点で異なると予想される。
【0086】
薬物の選択
金属と安定な錯体を形成し得る殆どの薬物の錯体が本発明により形成可能である。以下の実施例のために選択した薬物は表2に示すように化学分類及び薬効分類の代表に相当する。
【表4】
【0087】
小分子の検討
合成:
重要であり得る他の薬物−マグネシウム錯体には、頭痛を軽減するためにマグネシウムが重要であるためトリプタン−Mgが、及び乱用耐性麻酔薬が重要であるためオキシコドン−Mgが含まれる。
【0088】
典型的には、tBuOH/DMSO中の薬物とKOtBuの混合物に無水金属ハロゲン化物(ヨウ化物、臭化物及び塩化物)を添加する。或いは、薬物及びDMSO中の第3級アミン(例えば、トリエチルアミン)の溶液に金属ハロゲン化物を添加してもよい。別のオプションでは、金属ハロゲン化物を薬物及びTHF中のKHに添加し得る。好ましい金属はマグネシウム及び亜鉛であり、好ましいハロゲン化物は塩化物である。塩化亜鉛はDMSO、アセトンまたはエタノール中に可溶性であり、これらは亜鉛錯体形成、特に窒素含有配位子との錯体形成のために好ましい溶媒である。
【0089】
生成物を沈澱により単離し、吸引濾過または遠心分離により液体から分離し、洗浄した後、最後の微量の水分を除去するために高真空下で乾燥する。薬物:金属錯体は水和物を形成していることがあり、水のすべてが高真空下で除去されないことがある。或いは、添加した水が残っているその場で形成した金属上のDMSO配位子を置換し得ないことがある。結果として、生成物は薬物:金属:DMSO錯体であり得る。
【0090】
マグネシウムと結合したとき高い解離定数を有する特定の薬物がDMSOとの錯体形成にとって有利である。その場での三元錯体の形成により錯体が更に安定化し、経口投与後の吸収過程中その分子の完全性が保持される。このために、多くの薬物はマグネシウムハロゲン化物と反応させるときDMSOが好ましい溶媒である。
【0091】
実施例(アシクログアノシン−Mg及びT3−Znの例を除く)における錯体形成反応に対する比較として、下記実施例に記載したように反応媒体に水を含める以外は同様に反応を行う。反応物を上記したように後処理し、乾燥させる。
【0092】
T3−Mg錯体形成反応に水を添加すると、明らかに単離された生成物に影響を及ぼした。DMSO単独中で製造したT3−Mg化合物は、その1H NMRスペクトルにおいて脂肪族領域のみに幅広化し、シャープな芳香族ピークを示した(図11)。
【0093】
脂肪族領域にシャープなピークを示すT3の1H NMRを図12に示す。比較すると、水の存在下で製造したT3−Mg生成物の1H NMRは1H NMRスペクトル全体に広範囲にわたる幅広化を示した。更に、水の非存在下で製造したT3−Mg生成物のマグネシウム含量(1.62%)はビス(トリヨードサイロニナト)−ビス(ジメチルスルホキシド)−マグネシウムの含量と非常にうまく整合していた。対照的に、水の存在下で製造したT3−Mg錯体のマグネシウム含量はたった0.96%であった。そのカリウム含量は0.23%であり、ビス(トリヨードサイロニナト)−ビス(ジメチルスルホキシド)−マグネシウム生成物ではカリウムは検出されなかった。
【0094】
テトラサイクリンはβ−ジケトン及びβ−ケトフェノール官能基を有しており、マグネシウムと安定な錯体を形成する。DMSO中の反応に水を添加してもそれぞれの生成物の溶解性、1H NMRスペクトルまたは金属含量に対する影響は殆どない。実際、水単独中で実施した反応から単離した生成物の1H NMRスペクトルはDMSO単独から、または5:1のDMSO:水混合物中で単離した生成物のものとは余り異なっていない。反応溶媒中の水含量が多くなるとマグネシウム含量は少なくなる傾向があるが、これは主に生成物の水和度に起因し得る。
【0095】
亜鉛は窒素含有化合物と非常に安定な結合を形成するので、水は亜鉛と配位子間の錯体形成を干渉しないが、錯体構造に影響を及ぼす恐れがある。無水DMSO中での金属と薬物の反応から生ずる生成物は通常十分にキャラクタライズされた配位錯体であった。構造的にうまく規定されなかった化合物(すなわち、HCTZ−亜鉛)または若干不安定であった化合物(すなわち、ジメチルビグアニド−亜鉛錯体)の場合、亜鉛配位錯体が単離され得、少なくとも部分的にキャラクタライズされた。水の存在下で製造した同一の錯体は極性溶媒中で高い溶解性を有していた。それぞれの製造方法からの生成物の溶解性の差から、生成物それ自体の差が明らかに証明された。無水溶媒系中で製造した亜鉛生成物はイオン性塩、外配位錯体、水和錯体またはその組合せを生じたと考えられる。これは5:1 DMSO:水混合物中で製造したジメチルビグアニド−亜鉛錯体の場合であるようで、1H NMRスペクトルは2.85及び2.80ppmにN−メチル基に相当し、それぞれ遊離(イオン性)及び錯体形成されたジメチルビグアニドを示す共鳴を示した。
【0096】
キャラクタリゼーション
合成した各生成物をNMR、MS(TOFまたはFAB)及びICPによりキャラクタライズした。NMRスペクトルによりサンプルの完全性が確認され、幅広化、ピークシフトまたは多重共鳴の存在により金属が錯体形成されていることが分かる。
【0097】
無水DMSO中で製造した多くの錯体の金属含量は2つの薬物を金属に結合させた錯体と一致した。加えて、金属含量はバッチ毎に一定のままであった。ジメチルビグアニドの錯体は単離方法に応じて異なる金属含量を有しており、決して薬物:金属比は一定でなかった。金属含量をICP分析により測定し、NMR及びMSと共に、そのデータに基づいて薬物対金属の比を計算することができる。
【0098】
T3錯体の1H NMRスペクトルは、分子のアミノ酸部分との錯体形成を示す脂肪族領域の幾つかの幅広化及び高磁場シフト化を示していた。これは、それぞれ図11、12及び13に示すビス(T3)ビス(DMSO)Mg、T3遊離酸及びビス(T3)Znの1H NMRスペクトルの2.5〜3.5ppmの領域を比較することにより見つけることができる。
【0099】
ジメチルビグアニド錯体の1H NMRスペクトルは、窒素原子との錯体形成を示す−NH共鳴の大きな高磁場シフトを示した。加えて、N−ジメチル基の0.05ppm高磁場シフトがジメチルビグアニドのスペクトル(図15)と比したジメチルビグアニド−亜鉛錯体スペクトル(図14)中に見られた。
【0100】
ミノサイクリン及びテトラサイクリン錯体の1H NMRスペクトルは非常な大きい幅広化及びスペクトル全体にわたる多くの新しい共鳴の出現を有するポリマー構造に似ていた。これらのスペクトルが動的異性体混合物に起因し、分解にも重合にも起因していなかったことを立証するために、テトラサイクリン及びそのマグネシウム錯体のNMRサンプルに12N HClを添加し、スペクトルを再びとった。テトラサイクリン、ビス(テトラサイクリナト)マグネシウム及びHClを添加した錯体の1H NMRスペクトルをそれぞれ図16〜18に示す。一連のスペクトルから分かるように、マグネシウム錯体は参照テトラサイクリン化合物に逆戻りした。興味深いことに、HClをテトラサイクリンに添加したときNMRスペクトルの芳香族領域にかなりの量の分解が見られ(図19)、その量はテトラサイクリン−マグネシウム錯体の同等のスペクトルでは見られなかった。これはマグネシウムと錯体形成により与えられる酸保護効果を示し、酸環境(例えば、胃)において不安定である薬物に対するこの技術の重要な属性となり得る。
【0101】
ヒドロクロロチアジド錯体の1H NMRスペクトルも抗生物質−金属錯体のスペクトルで見られるのと同様の幅広化及び新しい特徴のない共鳴を有するポリマー構造に似ていた。ヒドロクロロチアジド、ヒドロクロロチアジド−亜鉛錯体及びHClを添加した錯体の1H NMRスペクトルをそれぞれ図20〜22に示す。これらのスペクトルから分かるように、幅広化した線は参照薬物で観察されるシャープな共鳴に逆戻りした。このことから、各錯体の1H NMRスペクトルで観察された幅広化及び追加の共鳴が溶液中の錯体の複数の立体化学及び幾何異性体に起因したことが分かる。溶液中の異性体間の中程度にゆっくりした相互変換も観察された幅広化に寄与し得る。
【0102】
アシクログアノシン−マグネシウム錯体に関するスペクトルデータから、錯体が形成されたことが判明した。アシクログアノシンの1H NMRスペクトルとそのマグネシウム錯体の1H NMRスペクトル(図23)の比較から、アシクログアノシン上の錯体形成部位がアミド酸素及びイミダゾール窒素であったことが示唆された。錯体のNMRスペクトルで欠けている10.6ppmの共鳴はアミドプロトンに帰属している。
【0103】
配位錯体の有意な存在を表す質量スペクトルは錯体の安定性の重要な指標である。これにより、分子イオンはビス(T3)Mg、ビス(T3)Zn、ビス(ミノサイクリナト)Mg、ビス(テトラサイクリナト)Mg及びビス(アシクログアノシナト)Mgで見られた。亜鉛同位体パターンを有する2つの分子イオンがヒドロクロロチアジド−亜鉛錯体のMALDIスペクトルで観察された。このとき、これらの質量に相当する構造は知られていない。ジメチルビグアニド−亜鉛錯体はそのMALDIスペクトルで亜鉛含有分子イオンを有していなかった。このことは化合物の不安定性に起因すると考えられる。
【0104】
FTIR研究は、観察された特定の錯体について配位子結合原子があるかどうか、錯体がDMSO、水と配位するか、または全く溶媒和しないかを調べるために使用され得る。
【0105】
安定性
製造した配位錯体の平衡定数を類似化合物の文献記載値から推定した。例えば、T3−Znの平衡定数logKeqは別のアミノ酸亜鉛錯体のフェニルアラニン−亜鉛に基づいて4〜5と推定される。また、ジメチルビグアニド−亜鉛のlogKeqは5〜7と推定される。ヒドロクロロチアジド−亜鉛のlogKeqを文献値から推定するのは困難である。テトラサイクリン−マグネシウムの異なるpH値での水中における安定度定数は報告されており、テトラサイクリン−マグネシウムのlogKeqは4〜5と予想される。アシクログアノシン−マグネシウムのlogKeqは1.6と推定される。T3−MgのlogKeqは良好な比較物質がないために推定するのが困難であるが、T3のようにアミノ酸であるグリシンのlogKeqは1.34である。T3はグリシンに比して疎水性が非常に高いために、T3−MgのlogKeqは1.34よりもかなり大きいと予想される。
【0106】
薬物−金属または薬物−金属−アジュバントの安定性の別の指標は、Keqに関連するが更に多座配位子の段階的安定性を示す結合定数である。金属と配位子間の最大結合についての累積結合定数βnは式4により求められる。
【数1】
【0107】
この差はフェニルアラニン−亜鉛の測定logKeqと8.5というその近似β2値で見られ得る。このβ2値はT3−Zn錯体の安定性をも非常にうまく反映し得る。
【0108】
異なる環境での金属−薬物錯体の結合定数を推定し得る方法は幾つかある。その幾つかの方法は光学吸収分光法、NMR分光法、質量分光法、反応カイネティクス、電位差測定法及びクロマトグラフィーである。
【0109】
分配係数(Partition Coefficient)及び分布係数(Distribution Coefficient)
分配係数は一定であり、式5に示すように水性相中の中性化合物の濃度/非混和性有機相中の濃度の比として定義される。
【0110】
分配係数P=[有機]/[水性] 式5
実際、式6に定義するLogPは、測定する条件によって、特にpHによって変動する。なぜならば、低pHでは塩基がイオン化し、高pHでは酸がイオン化するからである。
【0111】
LogP=logl0(分配係数) 式6
よって、LogP=1は10:1 有機:水性を意味し、LogP=0は1:1 有機:水性を意味し、LogP=−1は1:10 有機:水性を意味する。当然、イオン化化合物は水性相に優先的に分配し、よってLogPは低下する。塩基である中性分子の場合pHがそのpKaよりも2単位以上高いならば中性のままであり、中性酸の場合pHがそのpKaよりも2単位低いならば中性のままである。
【0112】
分配溶媒の選択もLogPに影響を及ぼす。多くのLogP測定では、オクタノール:水系を使用している。イオンペアリング作用がLogP測定に影響を及ぼし、特に本発明において具体化されているような金属配位化合物の場合考慮しなければならない。
【0113】
医薬用途の点から、以下のガイドラインが投与方法、製剤及び剤形を決定するために使用される:
・低いLogP(0以下) 注射
・中(0〜3) 経口
・高い(3〜4) 経皮
・非常に高い(4〜7) 脂肪組織に毒物蓄積
また、経口投与薬物の領域ではこれらのガイドラインが使用されている:
1.最適CNS浸透のために、LogP=2±0.7(Hansch rules)
2.最適経口吸収のために、LogP=1.8
3.最適腸内吸収のために、LogP=1.35
4.最適結腸吸収のために、LogP=1.32
5.最適舌下吸収のために、LogP=5.5
6.最適経皮浸透のために、LogP=2.6(及び、低いmw)。
【0114】
分布係数(D)は、式7で示される有機相中の非イオン化化合物/水性相中の化合物の全量の比である。
【0115】
D=[非イオン化](o)/[非イオン化](aq)+[イオン化](aq) 式7
LogDは特定pHでのlog分布係数である(式8)。これは一定でなく、分子のプロトン性によって変動する。pH7.4でのLogDは血漿のpHでの薬物の親油性を示すためにしばしば引用されている。
【0116】
LogD=log10(分布係数) 式8
LogDは以下の式によりLogP及びpKaに関連している:
酸に対して
LogD(pH)=logP−log[1+10(pH−pKa)] 式9
塩基に対して
LogD(pH)=logP−log[1+10(pKa−pKH)] 式10
よって、イオン化が最小であるようにpHを調節すると、logDはlogPにほぼ等しい。これらの条件下で、logDは具体的用途における薬物のバイオアベイラビリティーの信頼できる指標である。金属配位薬物の点から、参照薬物と比した薬物−金属錯体のlogDの上昇は親油性の増加を示すだけでなく、その水中安定性をも示している。テトラサイクリン、ビス(テトラサイクリナト)マグネシウム、トリヨードサイロニン及びビス(トリヨードサイロニナト)亜鉛のpH7.4でのlogDを測定し、pKaと一緒に表4に示す。
【表5】
【0117】
これらの結果は、中性化合物のアニオンを金属塩と組み合わせると低い親油性及び低いlogDを有する化合物を生成するはずであるという従来技術の教示に反している。おそらくより興味深いことには、本発明にこの技術を適用することによりテトラサイクリンは理論上注射薬物(logD<0)から経口薬物(logD>0)に変換され、T3は経口薬物(logD<3)から経皮薬物(logD>3)に変換された。この後者の結果は薬物を徐放性経皮デポ剤で投与することによりT3製品の安全性を高める際に重要な関係を有している。
【0118】
バイオアベイラビリティー研究
概要:参照薬物の吸収に対する金属と薬物の配位が有する影響を観察するためにラットモデルを用いて予備研究を実施した。この特別研究のために選択した参照薬物はトリヨードサイロニン(T3)であり、これはサイトメル(Cytomel)及びサイロラー(Thyrolar)の活性成分である。サイトメル及びサイロラーは共に現在甲状腺機能低下症を治療するために使用されている。サイトメルは特定の精神病の治療においても適応されている。
【0119】
研究計画:T3−亜鉛及びT3−マグネシウムを参照薬物と比して5時間にわたるバイオアベイラビリティーについて試験した。3つの化合物を別々にゼラチンカプセル剤に製剤化し、108±12μg/kgの全用量で投与した。ラット胃における試験化合物の酸分解を避けるために、製剤に金属酸化物を添加した。T3遊離酸に酸化亜鉛を添加した別のT3対照を含めた。製剤化したゼラチンカプセル剤の各々を各ラットの食道に直接経口投与し、血液サンプルを投与前、投与から0.5、1、2、2.5、5時間目に採取した。血清トリヨードサイロニンレベルを業界標準アッセイ方法を用いて独立研究所で分析した。
【0120】
結果:図30に示す結果から、4つすべての薬物製剤がラットにより急速に吸収され、金属配位T3を供給されたラットでは血清T3レベルは2.5時間目まで上昇し、その後通常横ばい状態になる。T3遊離酸を供給されたラットでは血清T3レベルが5時間目になお上昇する可能性があることを示していた。金属錯体形成したT3及び酸化亜鉛を添加したT3を投与されたラットでは血清T3血液レベルが上昇し、そのレベルは5時間目でT3遊離酸よりも約65〜85%高かった。
【0121】
結論:上記データは、金属をT3と錯体形成すると経口投与T3の吸収がT3単独よりもほぼ2倍増加したことを明確に示している。T3及び酸化亜鉛の製剤を供給した動物では、T3遊離酸はT3の吸収前に酸化亜鉛と錯体形成することが分かる。
【0122】
この研究は薬物デリバリー技術の有意な進歩、および金属を医薬品と配位することがバイオアベイラビリティー限界を有する薬物の性能に適用され、これを改善し得ることを表している。
【0123】
大分子プロトコル
合成:
規定サイズを有する二本鎖iRNAを実施例の節に記載されている標準プロトコルに従って作成した。次いで、iRNAを実施例の節に記載されているように無水条件下または水の存在下でマグネシウムまたは亜鉛と反応させた。
【0124】
iRNAの生物活性を各種方法で他の金属(例えば、カルシウム、亜鉛、コバルト及びマンガン)と錯体形成することによりモジュレートし得る。加えて、プリン/ピリミジン基とホスフェート基の結合を促進するために多価金属(例えば、CuまたはAg33)を組み合わせると、iRNAのトランスフェクション効率及び安定性を改善させることができる。
【0125】
キャラクタリゼーション:
金属:RNA生成物を実施例の節に記載されている標準プロトコルに従って等電点電気泳動(IEF)ゲルを用いてイオン性/共有結合性挙動の変化について調べた。IEFゲルの利点は、標的RNA分子上の電荷分布の変化をモニターするための安価なツールを提供することである。IEF研究のデータを製造した各iRNA錯体についてΔpKaとして表した。
【0126】
IEF実験の結論:
異なる等電点(pKa)を有する無水DMSO反応から得られたRNA分子の存在は新しいRNA分子の存在を示している(図31)。これはRNA分解に起因しないので、新しいRNA分子は金属(マグネシウムまたは亜鉛)とRNAの安定な錯体であることを意味している。加えて、そのpKaが天然RNAのpKaよりも低いので、金属とRNA間の共有結合の形成が裏付けられる。
【実施例】
【0127】
DMSO−マグネシウム錯体のFTIR分析
DMSOのいずれの原子がマグネシウムに結合するかを調べるために、DMSO−マグネシウム錯体のFTIRスペクトルを集めた。FTIRスペクトルは954cm−1にS=O−Mg伸縮を示すエキストラ伸縮を示した。T3錯体のFTIRはS=O−Mg、C=O及びN−H伸縮の存在について調べた。
【0128】
ビス(トリヨードサイロニナト)−ビス(ジメチルスルホキシド)マグネシウムの製造
トリヨードサイロニン、すなわちT3(218mg)を無水DMSO(4mL)中に溶解し、その後t−ブタノール中1M カリウムt−ブトキシド(0.34mL)を添加し、溶液を10分間撹拌した。塩化マグネシウム(16mg)を添加し、溶液を一晩撹拌した。溶液を脱イオン水(10mL)中に注いで生成物を沈澱させた。この沈澱を吸引濾過し、風乾させた。高真空下で一晩乾燥した後、164mgの明ベージュ色粉末が生じた。生成物の構造を1H NMR、FAB−MS及びICPによりキャラクタライズした。1H NMR(DMSO):δ 7.83(s),7.05(d),6.82(d),6.62(dd),3.26(bm),3.15(bd),2.71(bm),2.54(s)。側鎖領域に幅広多重項が存在することはマグネシウム結合のための部位を示している(図11参照)。FAB−MS:1325にビス(トリヨードサイロニナト)マグネシウムを示す分子イオン(DMSO配位子のロス後)。FTIR(ニート):cm−1 1596(C=O伸縮),1014(S=O伸縮),949(S=O−Mg伸縮);1633にN−H伸縮の不在。マグネシウム分析(ICP):予測値=1.68%,実測値=1.62%。これは図24に示す構造を確認するものであり、純度は約96%であり、汚染の大部分はおそらく1H NMRに示されている水に起因している。
【0129】
ビス(トリヨードサイロニナト)亜鉛の製造
トリヨードサイロニン、すなわちT3(192mg)を無水DMSO(4mL)中に溶解し、その後t−ブタノール中1M カリウムt−ブトキシド(0.30mL)を添加し、溶液を10分間撹拌した。ジエチルエーテル中塩化亜鉛(0.16mLの1M溶液)を添加し、溶液を一晩撹拌した。溶液を脱イオン水(10mL)中に注いで生成物を沈澱させた。この沈澱を吸引濾過し、風乾した。高真空下で一晩乾燥した後、140mgの明ベージュ色粉末を得た。生成物の構造を1H NMR、FAB−MS及びICPによりキャラクタライズした。1H NMR(DMSO):δ 7.82(s),7.02(d),6.80(d),6.61(dd),3.49(bm),3.22(bm),2.67(bm)。側鎖領域に幅広多重項が存在することは亜鉛結合のための部位を示している(図13)。FAB−MS:1364及び1366に亜鉛と一致する同位体存在度パターンを有するビス(トリヨードサイロニナト)亜鉛を示す分子イオン。FTIR(ニート):cm−1 1582(C=O伸縮);1633にN−H伸縮の不在。亜鉛分析(ICP):予想値=4.8%,実測値=4.3%。これは図25に示す構造を裏付け、純度は約90%であり、汚染の大部分はおそらく水に起因し、1H NMRスペクトルで見られるように水和物であり得る。
【0130】
水の存在下でのマグネシウムトリヨードサイロニン錯体の製造
トリヨードサイロニン、すなわちT3(188mg)を無水DMSO(3.5mL)中に溶解し、その後t−ブタノール中1M カリウムt−ブトキシド(0.29mL)を添加し、溶液を10分間撹拌した。水(0.5mL)中塩化マグネシウム(16mg)を添加し、溶液を一晩撹拌した。溶液を脱イオン水(10mL)中に注いで生成物を沈澱させた。この沈澱を吸引濾過し、風乾した。高真空下で一晩乾燥した後、188mgの明ベージュ色粉末が得られた。生成物構造を1H NMR、FAB−MS及びICPによりキャラクタライズした。1H NMR(DMSO):δ 7.81(bs),7.02(bs),6.81(bd),6.59(dd)。他の共鳴は溶媒ピーク及び水ピークの後に隠れていた。サンプルはDMSO中で曇った懸濁液を形成した。FAB−MS:652にMgが結合していないプロトン化T3を示す分子イオン。FTIR(ニート):cm−1 1633(N−H伸縮),1535(C=O伸縮),1012(S=O伸縮),949(S=O−Mg伸縮)。T3関連伸縮に比したDMSO関連伸縮の強度は(DMSO)xMgカチオン及びT3アニオンの混合物を示していた。マグネシウム分析(ICP):予想値=1.8%,実測値=0.96%。カリウム分析(ICP):実測値=0.23%。ICPデータから、この生成物はマグネシウム塩、カリウム及び双性イオンの混合物であるようである。
【0131】
ビス(ミノサイクリナト)マグネシウムの製造
ミノサイクリン(104mg)を無水DMSO(3mL)中に溶解し、その後t−ブタノール中1M カリウムt−ブトキシド(0.44mL)を添加し、溶液を10分間撹拌した。塩化マグネシウム(11mg)を添加し、溶液を一晩撹拌した。溶液を脱イオン水(10mL)中に注いで生成物を沈澱させた。この沈澱を吸引濾過し、風乾した。高真空下で一晩乾燥して、52mgの深黄色粉末を得た。生成物の構造は、おそらくアニオン性ミノサイクリン及びマグネシウムがとりうる二座錯体形態の各種順列のために1H NMRによりキャラクタライズすることができなかった。その後、生成物をFAB−MS及びICPによりキャラクタライズした。FAB−MS:937.4にビス(ミノサイクリナト)マグネシウムを示す分子イオン。マグネシウム分析(ICP):予想値=2.68%,実測値=2.61%。このことから、図26に示すもっともらしい構造により表されるようにマグネシウム1原子あたり2つのミノサイクリンがあることが確認される(以下のテトラサイクリンに関する検討参照)。生成物の純度は約97%であり、汚染の大部分はおそらく1H NMRスペクトルで見られる水に起因する。
【0132】
ビス(テトラサイクリナト)マグネシウムの製造
テトラサイクリン(89mg)を無水DMSO(0.5mL)中に溶解し、その後t−ブタノール中1M カリウムt−ブトキシド(0.2mL)を添加し、溶液を10分間撹拌した。塩化マグネシウム(11mg)を添加し、溶液を3時間撹拌した。溶液を真空中30℃で濃縮し、その後脱イオン水(0.5mL)を添加し、混合物を摩砕し、2mLマイクロ遠心管に移した。8,000rpmで6分間遠心分離することにより生成物を液体から分離し、上清をデカントした。水(0.5mL)を添加し、渦状に混合し、遠心分離し、上清をデカントすることによりペレットを洗浄した。洗浄手順を繰り返した。高真空下で一晩乾燥した後、72mgの深黄色粉末を得た。1H NMRスペクトル(図20)はポリマー構造に似ており、8.8〜10.1ppm、6.4〜7.8ppm、4.2〜5.0ppm及び1.1〜3.1ppmに幅広多重項を含んでいた。生成物構造は、おそらくアニオン性テトラサイクリン及びマグネシウムがとりうる二座錯体形態の各種順列及びこれらの異性体錯体形態間での適度にゆっくりした平衡のために1H NMRにより正確にキャラクタライズすることができなかった。NMRサンプルに約1当量の12N HClを添加し、1H NMRにより再分析することにより確認が観察された。これは、マグネシウム錯体がテトラサイクリン及びおそらく塩化マグネシウムに逆戻りしたことを示した(図18)。生成物を更にMALDI−ES及びICPによりキャラクタライズした。MALDI−ES:911.3にビス(テトラサイクリナト)マグネシウムを示す分子イオン。マグネシウム分析(ICP):予測値=2.67%,実測値=2.53%。FTIRはDMSO配位子の存在を示さない。これは、マグネシウム1原子あたり2個のテトラサイクリン分子があることを示している。水性系中でのテトラサイクリン−マグネシウム錯体形成の以前に公表された研究からのNMR証拠に従って、ビス(テトラサイクリナト)マグネシウムのとりうる構造を図27に示す。生成物の純度は約95%であり、汚染の大部分はおそらく1H NMRスペクトルで見られる溶媒に起因している。
【0133】
水中でのマグネシウムテトラサイクリン錯体の製造
テトラサイクリン(89mg)を水(1.5mL)中に溶解し、その後t−ブタノール中1M カリウムt−ブトキシド(0.2mL)を添加し、溶液を10分間撹拌した。塩化マグネシウム(0.11mLの1M溶液)を添加し、溶液を3時間撹拌した。8,000rpmで6分間遠心分離することにより生じた沈澱を水から分離し、上清をデカントした。ペレットを水(1mL)を添加し、渦状に混合し、遠心分離し、上清をデカントすることにより洗浄した。高真空中で一晩乾燥した後、65mgの深黄色粉末が得られた。1H NMRスペクトルは無水DMSO中で製造した錯体に非常に似ていた。マグネシウム分析(ICP):予測値=2.67%,実測値=2.42%。水中での錯体形成対無水条件下での錯体形成はテトラサイクリン−マグネシウム錯体の安定性及び構造に対して殆ど影響を持たないようである。
【0134】
ヒドロクロロチアジド亜鉛錯体の製造
ヒドロクロロチアジド(120mg)を無水DMSO(0.5mL)中に溶解し、その後t−ブタノール中1M カリウムt−ブトキシド(0.4mL)を添加し、溶液を10分間撹拌した。ジエチルエーテル中塩化亜鉛(0.2mLの1M溶液)を添加し、溶液を4時間撹拌した。溶液を真空中30℃で濃縮した後、メタノール(0.5mL)を添加し、混合物を摩砕し、2mLマイクロ遠心管に移した。8,000rpmで6分間遠心分離することにより生成物を液体から分離し、上清をデカントした。ペレットをメタノール(0.5mL)を添加し、渦状に混合し、遠心分離し、上清をデカントすることにより洗浄した。洗浄手順を繰り返した。高真空下で一晩乾燥した後、104mgの自由流動性白色粉末を得た。生成物は、周囲空気に数分間さらした後粉末がガム状に変わったために明らかに吸湿性であった。1H NMRスペクトルはポリマー構造に似ており(図21)、6.3〜8.0ppm及び4.4〜5.9ppmの幅広多重項を含んでいた。生成物の構造は、おそらくアニオン性ヒドロクロロチアジド及び亜鉛がとりうる錯体形態の各種順列及び異性体錯体形態間での適度にゆっくりした平衡のために1H NMRにより正確にキャラクタライズできなかった。約1当量の12N HClをNMRサンプルに添加し、1H NMRにより再分析することにより確認を観察した。この分析は亜鉛錯体がヒドロクロロチアジド及びおそらく塩化亜鉛に逆戻りしたことを示す(図22)。生成物を更にMALDI−ES及びICPによりキャラクタライズした。MALDI−ES:705.9及び932.9に典型的な亜鉛同位体存在度を有するが特定のヒドロクロロチアジド亜鉛錯体構造を示さない分子イオン。亜鉛分析(ICP):実測値=10.4%。FTIR(ニート):cm−1 1027(S=O伸縮),953(S=O−Zn伸縮);1646にN−H伸縮の不在。NMR、MALDI及びICPデータは明らかにヒドロクロロチアジド−亜鉛錯体の形成を示している。錯体形成の部位はヒドロクロロチアジドのスルホンアミドの窒素の一方及び両方上にあり得ることは合理的であるようである。FTIRデータはDMSO配位子の存在を示唆しており、1H NMR積分値によればHCTZ:DMSO比は1:1である。0.08ppmのDMSOメチル基の化学シフトから、亜鉛とDMSO間のO−結合が示唆される。
【0135】
水の存在下での亜鉛とヒドロクロロチアジドの反応
この手順は、塩化亜鉛を水(100μL)に添加し、反応混合物に添加する前にエーテルを蒸発除去させた以外は同様の無水製造に従った。全混合物が沈澱ステップ中にメタノール中にゆっくり溶解するために生成物は単離されなかった。これは推定上メタノール可溶性水和錯体が形成されたためであった。
【0136】
ジメチルビグアニド亜鉛錯体の製造
ジメチルビグアニド(66mg)を無水DMSO4(1mL)中に溶解し、その後t−ブタノール1M カリウムt−ブトキシド(0.88mL)を添加し、溶液を10分間撹拌した。ジエチルエーテル中塩化亜鉛(0.22mLの1M溶液)を添加し、溶液を3時間撹拌した。溶液を真空中35℃で濃縮し、その後エタノール(0.5mL)を添加し、混合物を摩砕し、2mLマイクロ遠心管に移した。8,000rpmで6分間遠心分離することにより生成物を液体から分離し、上清をデカントした。ペレットをエタノール(0.5mL)を添加し、渦状に混合し、遠心分離し、上清をデカントすることにより洗浄した。洗浄手順を繰り返した。高真空下で一晩乾燥した後、57mgの自由流動性白色粉末を得た。1H NMR(DMSO):δ 4.90(s),4.66(s),4.50(s),2.80(s)。ジメチルビグアニドに比して−NHプロトンが大きく高磁場シフトしていることは亜鉛との錯体形成を示している。ジメチルビグアニド−亜鉛錯体ではジメチル基中の0.05ppmの小さな高磁場シフトも観察された。ジメチルビグアニド−亜鉛及びジメチルビグアニドのNMRスペクトルをそれぞれ図14及び15に示す。生成物を更にICPによりキャラクタライズした。亜鉛分析(ICP):実測値=8.14%。FTIRデータはDMSO配位子の存在を示さなかった。NMR及びICPデータは、明らかにジメチルビグアニド−亜鉛錯体の形成を示している。図28は製造したビグアニド−金属錯体を表している。MALDI−ES分析は亜鉛含有化合物を表さなかった。
【0137】
水の存在下での亜鉛ジメチルビグアニド錯体の製造
この手順は、塩化亜鉛を水(100μL)に添加し、反応混合物に添加する前にエーテルを蒸発させた以外は類似の無水製造に従った。同一方法で後処理し、乾燥させて、47mgの自由流動性白色粉末を得た。この化合物はDMSO中に僅かしか溶解せず、NMRにおいて低い信号対雑音比が生じた。1H NMR(DMSO):δ 4.90(bs),4.66(bs),4.50(bs),2.85(s),2.80(s)。2.85ppmの一重項は亜鉛と錯体形成していないジメチルビグアニドの存在を示している。2.85及び2.80ppmの2つのピークの積分値から判断して、遊離ジメチルビグアニド/亜鉛錯体の比は約1:1である。
【0138】
水中での亜鉛ジメチルビグアニド錯体の製造
ジメチルビグアニド(33mg)を水(1mL)中に溶解し、その後t−ブタノール中1M カリウムt−ブトキシド(0.44mL)を添加し、溶液を10分間撹拌した。ジエチルエーテル中塩化亜鉛(0.22mLの1M溶液)を添加し、溶液を5時間撹拌した。8,000rpmで6分間遠心分離することにより生じた沈澱を液体から分離し、上清をデカントした。ペレットを水(1mL)を添加し、渦状に混合し、遠心分離し、上清をデカントすることにより洗浄した。高真空下で一晩乾燥した後、20mgの自由流動性白色粉末を得た。この粉末は1H NMRによれば有機物質を含んでいなかった。単離された生成物は各種水和形態の亜鉛塩であった。
【0139】
ビス(アシクログアノシナト)マグネシウムの製造
アシクログアノシン(45mg)を無水DMSO(0.5mL)中に溶解し、その後t−ブタノール中1M カリウムt−ブトキシド(0.2mL)を添加し、溶液を10分間撹拌した。塩化マグネシウム(11mg)を添加し、溶液を4時間撹拌した。溶液を真空中30℃で濃縮し、その後メタノール(0.5mL)を添加し、混合物を摩砕し、2mLマイクロ遠心管に移した。8,000rpmで6分間遠心分離することにより生成物を液体から分離し、上清をデカントした。ペレットをメタノール(0.5mL)を添加し、渦状に混合し、遠心分離し、上清をデカントすることにより洗浄した。洗浄手段を繰り返した。高真空下で一晩乾燥した後、15mgのベージュ色の粗い粉末を得た。生成物の構造を1H NMR、MALDI−ES及びICPによリキャラクタライズした。1H NMR(DMSO):δ 7.70(s),6.75(bs),5.30(s),4.64(bs),3.42(s)。MALDI−ES:473.1の分子イオンはビス(アシクログアノシン)マグネシウムを示唆している。666、702、893及び1065の他の分子イオンは特定の構造に帰属されなかった。マグネシウム分析(ICP):予想値=5.14%,実測値=4.16%。FTIRデータはDMSO配位子の存在を示さなかった。10.6ppmにアミドプロトンが存在しないことと、ICP分析及びMALDI−ES分析を併せて、ビス(アシクログアノシナト)マグネシウムの構造を図29に示す。
【0140】
T3錯体、テトラサイクリン錯体及び参照薬物の分配係数、分布係数及びpKa
pKa及びlogPを電位差測定法及び分光光度法により測定した。電位差測定法はアナライトの簡単な酸及び塩基滴定からpKa及びlogPを計算するために専門ソフトウェアを使用することを含む。pKaをまず約2mgの純粋物質をアッセイバイアルに入れて秤量して測定した。イオン強度を0.15M KClを用いて調節し、化合物を溶解させるために水を添加し、次いでpHを所望の出発値に低下または上昇させるために酸または塩基滴定剤を添加した。次いで、溶液を酸(0.5N HCl)または塩基(0.5N NaOH)を用いて最終pHまで滴定した。概算のpKa値を示し、後に正確なデータに絞り込んだ。
【0141】
水に僅かしか溶解しない(T3)2Znについては、pKaを水とDMSO補助溶媒の混合物中で測定した。水/DMSOの3つの比の最小値を滴定してpsKa(補助溶媒の存在下での見かけpKa)を得た。水性pKaをYasuda-Shedlovsky技術を用いて外挿することにより求めた。
【0142】
logPは、オクタノール(水飽和)の存在下での滴定により求めた。水中のpKa及びオクタノールの存在下での見かけpKa(poKa)を比較し、logPを求めた。別の容量のオクタノールの存在下で更に滴定して、イオンペアリング(オクタノール中への荷電種の分配、logP+またはlogP−と称される)を求めた。実験的に測定したpKa及びlogPを用いて、薬物の親油性プロファイル(logD対pH)を計算した。logD7.4はpH7.4でのこのプロファイルから求めた。
【0143】
分光光度法は、光ファィバーディッププローブ、UV光源(パルス型重水素ランプ)及び酸または塩基溶液の添加中にサンプル溶液の吸収スペクトルを自動的に捕らえるためのフォトダイオードアレイ検出器を使用した。
【0144】
最高10mMのストック溶液を、幾つかの0.5mgサンプルを0.5〜1.0mLの水または補助溶媒中に溶解させることにより調製した。滴定のために十分量のストックをバイアルにピペットで添加する。ディッププローブをアッセイバイアルに入れ、ディッププローブをカバーするために0.15M KCl水溶液を添加する。pHを所望出発値とするために酸または塩基を添加する。選択したpH範囲で、その後の分析のためにイオン化に起因するスペクトルの変化をフォトダイオードアレイ検出器で捕らえた。サンプルのpKa値を推理し、還元種の主要吸収スペクトルを分析するために多変量解析(TFA)を適用した。Yasuda-Shedlovsky技術を用いる外挿により水性pKaを求めた。分光光度実験から得たpKa値は電位差滴定から得た値と非常にうまく一致している。
【0145】
バイオアベイラビリティー研究
表題:Sprague Dawleyラットでのホルモン錯体栄養補充の吸収及び効果の評価
被験体:15匹の若い雌Sprague Dawleyラット(180〜225g)を使用した。これらのラットは業者(バージニア州ダブリンに所在のHarlan Laboratory Animals)から入手し、Litton Reeves Hall(Division of Laboratory Animal Resources)のビバリウムにポリプロピレンシューボックスケージ中に1群3匹で収容した。飲水は自由とした。ラットには自由に認可げっ歯類固形飼料を与えた。到着後、ラットの健康を評価し、動物を最低5日間隔離して置いた。この間に全身健康状態を調べた。隔離後、ラットをアクセス及び研究のために耐久性動物小屋に移動させた。
【0146】
研究計画:これは、ラットに対して食道に直接経口投与した3つのホルモンの吸収及び効果を比較するための研究である。2つの試験化合物であるT3−亜鉛及びT3−マグネシウムを合成した。トリヨードサイロニン遊離酸(T3)はポジティブ対照であった。3つの化合物を別々にゼラチンカプセル剤に製剤化し、108±12μg/kgの全用量で投与した。試験化合物のラットの胃での酸分解を避けるために、製剤に金属酸化物を添加した。T3−マグネシウム化合物に対しては1.08±0.13mgの酸化マグネシウムを添加し、T3−亜鉛化合物に対しては106±13μgの酸化亜鉛を添加した。T3遊離酸に145±50μgの酸化亜鉛を添加した別のT3対照を含めた。
【0147】
投与し、採血するために、反復実験1回あたり3匹のラットを麻酔した。基準血液サンプルを後眼窩サンプリングにより採取した。次いで、化合物を胃管栄養チューブにより経口投与した。投与後、血液サンプル(500μL)を投与から0.5、1、2、2.5、5時間目に採取した。血液サンプルを遠心分離し、血清を除去し、血清を氷上に保持した後、血清トリヨードサイロニンレベルについて分析した。
【0148】
サンプルの分析:血清トリヨードサイロニンレベルをRIAにより測定した。
【0149】
結果:各ラット群からの個々の血清T3レベルの平均を求め、T3濃度(ng/mL)対時のプロットを作成した。このプロットを図30に示す。
【0150】
大分子の例
干渉RNAの製造:
干渉RNAを修飾New England Biolabs Litmus 28i RNAi双方向転写ベクターを用いて製造した。922bpウシ血清アルブミンcDNA断片をLitmus RNAiベクターのBglII部位及びStuI部位に導入した。T7 RNAポリメラーゼを用いるインビトロ転写により標的RNAi転写物を製造して、1mg/mlを得た。次いで、RNAを50ugサンプルに分割し、凍結乾燥した。
【0151】
RNA:マグネシウム内部配位(共有結合性)錯体の製造:
iRNA(約50μg)を50℃で無水DMSO(100μL)中に溶解した。無水塩化マグネシウム(19mg)を無水DMSO中に溶解することにより4mM 塩化マグネシウムのストック溶液を調製した。3つの別々のiRNAのDMSO溶液に15μL、30μL及び60μLの4mM 塩化マグネシウムを添加して、3つの別々の反応を行ったた。溶液を時々渦状に混合しながら室温で90分間放置し、この後7.5M 水性塩化アンモニウム(20μL)、次いでRNAアーゼ非含有エタノール(400μL)を添加し、渦状に混合した。生成物を1時間かけて溶液から沈澱させ、遠心分離し、液体をペレットからデカントした。このペレットをRNAアーゼ非含有エタノール(100μL)で洗浄し、渦状に混合し、遠心分離し、デカントによりエタノール上清をペレットから分離した。生じた無色ペレットを数分間風乾した後、等電点電気泳動ゲルで試験した。
【0152】
マグネシウム:RNA外部配位(イオン性)錯体の製造:
塩化マグネシウムストック溶液を無水DMSOではなくRNAアーゼ非含有水を用いて調製した以外は共有結合性類似体の手順を正確に追跡して、イオン性マグネシウム:RNA錯体を製造した。生じた無色ペレットを数分間風乾した後、等電点電気泳動ゲルで試験した。
【0153】
RNA:亜鉛内部配位(共有結合性)錯体の製造:
塩化マグネシウムストック溶液ではなく塩化亜鉛ストック溶液を調製した以外はマグネシウム類似体の手順を正確に追跡して、共有結合性亜鉛:RNA錯体を製造した。生じた無色ペレットを数分間風乾した後、等電点電気泳動ゲルで試験した。
【0154】
亜鉛:RNA外部配位(イオン性)錯体の製造:
塩化亜鉛ストック溶液を無水DMSOではなくRNAアーゼ非含有水を用いて調製した以外は共有結合性類似体の手順を正確に追跡して、イオン性亜鉛:RNA錯体を製造した。生じた無色ペレットを数分間風乾した後、等電点電気泳動ゲルで試験した。
【0155】
等電点電気泳動ゲル実験:
これは、iRNA標的の予測される修飾をモニターするためにIEFゲルを用いる新規な方法である。図31は初期実験の結果を示している。
【0156】
IEF実験の解釈:
IEF実験から、3種の塩化マグネシウム濃度で無水(A)条件で製造したマグネシウムRNA錯体は約50%の収率で共有結合性錯体を生じたことが分かった。3種の塩化マグネシウム濃度で水性(W)条件下で製造したマグネシウムRNA錯体はイオン性錯体を生じた。塩化亜鉛を用いて無水(A)条件で製造した亜鉛RNA錯体は約50%の収率で共有結合性錯体を生じた。塩化亜鉛を用いて水性(W)条件で製造した亜鉛RNA錯体はイオン性錯体を生じた。
【図面の簡単な説明】
【0157】
【図1】従来技術によるマグネソセンの構造を示す。
【図2】従来技術による(シクロペンタジエニル)−tブチルメチルビス(N,N’−[2,6−ジイソプロピルフェニル]アミジナート)マグネシウムの構造を示す。
【図3】従来技術によるマグネシウム:サリチルアルデヒド錯体の構造を示す。
【図4】従来技術によるマグネシウムフタロシアニンの構造を示す。
【図5】従来技術による外圏RNA:マグネシウム配位錯体を示す。
【図6】本発明による内圏RNA:マグネシウム配位錯体を示す。
【図7】本発明によるRNA:マグネシウム:アルギニン配位錯体を示す。
【図8】本発明による置換アルギニン:マグネシウム錯体を示す。
【図9】本発明による内圏でマグネシウムと錯体形成したサリチル酸とポリマー結合アルギニン及び外圏で配位子:金属錯体を封入したペプチドを示す。
【図10】本発明によるマグネシウム:オキシコドン錯体を示す。
【図11】本発明によるビス(トリヨードサイロニナト)−ビス(ジメチルスルホキシド)マグネシウムのプロトンNMRを示す。
【図12】従来技術によるトリヨードサイロニン(T3)のプロトンNMRを示す。
【図13】本発明によるビス(トリヨードサイロニナト)亜鉛のプロトンNMRを示す。
【図14】本発明によるジメチルビグアニド:亜鉛錯体のプロトンNMRを示す。
【図15】従来技術によるジメチルビグアニドのプロトンNMRを示す。
【図16】従来技術によるテトラサイクリンのプロトンNMRを示す。
【図17】本発明によるビス(テトラサイクリナト)マグネシウムのプロトンNMRを示す。
【図18】本発明による1N HClを添加したテトラサイクリン−マグネシウム錯体のプロトンNMRを示す。
【図19】本発明による1N HClを添加したテトラサイクリンのプロトンNMRを示す。
【図20】従来技術によるヒドロクロロチアジドのプロトンNMRを示す。
【図21】本発明によるヒドロクロロチアジド−亜鉛錯体のプロトンNMRを示す。
【図22】本発明による1N HClを添加したヒドロクロロチアジド−亜鉛錯体のプロトンNMRを示す。
【図23】本発明によるビス(アシクログアノシナト)マグネシウムのプロトンNMRを示す。
【図24】本発明によるビス(トリヨードサイロニナト)−ビス(ジメチルスルホキシド)マグネシウムの構造を示す。
【図25】本発明によるビス(トリヨードサイロニナト)亜鉛の構造を示す。
【図26】本発明によるビス(ミノサイクリナト)マグネシウムの構造を示す。
【図27】本発明によるビス(テトラサイクリナト)マグネシウムの構造を示す。
【図28】本発明によるジメチルビグアニド−亜鉛錯体の構造を示す。
【図29】本発明によるビス(アシクログアノシナト)マグネシウムの構造を示す。
【図30】本発明によるラット動物モデルにおけるT3、T3Mg、T3Znの相対的薬物動態プロファイルを示す。
【図31】本発明によるマグネシウム及び亜鉛iRNA錯体のIEFプロファイルを示す。“A”を付した列は無水条件で製造した錯体を指す。“W”を付した列は水中で製造した錯体を指す。
【技術分野】
【0001】
本発明は、新規な生物活性分子の金属配位錯体に関する。
【背景技術】
【0002】
公知の生物活性分子の特性をその構造を修飾することにより改善することは望ましい。前記修飾の目的は、分子を有効性、安定性、少ない副作用または標的デリバリーのような幾つかの点で改善することである。この改善を分子の望ましい特性を犠牲にすることなく達成する。上記目的は安易に述べられているが、具体的修飾の効果は極めて予測不可能なことが多いので実際行うと達成することが困難である。
【発明の開示】
【0003】
本発明によれば、公知の生物活性分子の構造を修飾すると、金属配位錯体として知られる新しい分子が生ずる。これらの新しい分子は予期せぬほど優れた特性を有している。本発明の金属配位錯体にはサイロニン、テトラサイクリン系抗生物質、オキシコドン及びヒドロコドンの錯体並びにそれらの誘導体の錯体が含まれる。
【0004】
キレート化は金属配位錯体の安定化における重要な要素である。s−ブロック金属に関して、このことはカルシウム及びマグネシウムに特に当てはまる。例えば、酢酸−マグネシウム錯体のlogKeqは0.47である。分子上に1つのアミノ基を導入すると(すなわち、グリシン)logKeqは1.34に上昇した。マグネシウムは典型的には窒素よりもむしろ酸素とキレート化し、この効果はアデニンのlogKeq(logKeq=2.08)を6−ヒドロキシプリンのlogKeq(logKeq=6.65)と比較することにより認められる。マグネシウムは、アデノシンのlogKeq(logKeq=0.50)とアデノシン−5’−モノリン酸のlogKeq(logKeq=1.80)の比較から明らかにされるように、酸化リン(例えば、ホスフェート)と特に強い結合を形成する。
【0005】
通常、亜鉛錯体は同等のマグネシウム錯体よりも安定である。このことは、配位子が酸素または硫黄を有している場合に特に当てはまる(これは、酸素のみを有している配位子の場合に当てはまらないことがあり、配位子がホスフェートである場合には余りあてはまらない)。上のグリシンの例を用いると、グリシン−亜鉛錯体のlogKeqは4.85である。亜鉛−酸素結合に対する亜鉛−硫黄結合の強度は、ヒドロキシプロパン酸の亜鉛錯体のlogKeq(logKeq=0.86)とメルカプトプロパン酸の亜鉛錯体のlogKeq(logKeq=6.43)の相対logKeq値で明らかである。logKeq値を他の金属や配位子と比較すると、このキレート化安定化は金属配位化学に勝っていることが明らかとなる。
【0006】
固有の共有原子価を有する安定な金属配位錯体を形成するためにキレート化を起こすことが必要でない場合もあり、このことは遷移金属を窒素配位子と組み合わせたときに特にあてはまるが、多くの場合、本発明の好ましい実施形態では活性物質を金属、特にマグネシウムとキレート化する。
【0007】
本発明の実施形態では、マグネシウム及びカルシウムとの錯体形成のための最良の候補になる活性物質はヘテロ原子(すなわち、酸素、窒素または硫黄)上に僅かに水よりも高いかまたは水よりも低いpKaを有するプロトンを有し、金属との結合あるいはキレート化に参加し得るように第1プロトン化ヘテロ原子に非常に近接して追加のヘテロ原子を有するものである。この官能基の配置を有する薬物は金属と結合する可能性が最も高いが、生じた金属配位活性物質は生体系中で十分に安定であり、前記系での加水分解を乗り越え、よって活性物質の性能が十分にモジュレートされる。多座配位子により付与される加水分解安定性は、アミドさえも配位金属の存在下で弱塩基(例えば、トリエチルアミン)を用いて脱プロトン化され得るように配位子のpKaを低下させ得るという事実により裏付けられる。従って、典型的な生物学的pHでは通常イオン化されないヘテロ原子上にプロトンを有する活性物質は共有的に配位された金属で置換されるプロトンを有し得、共有結合性は関与するヘテロ原子からの追加的なキレート化により強化される。本発明の好ましい実施形態では、マグネシウムまたはカルシウムに結合する活性物質上のヘテロ原子の少なくとも1つは酸素または硫黄である。マグネシウムはホスフェート及びホスホネートと著しく強い結合を形成し、よって本発明の追加の実施態様ではマグネシウムと配位する活性物質はオルガノホスフェートまたはオルガノホスホネート化合物である。
【0008】
本発明の実施形態では、亜鉛及びp−ブロック金属と錯体形成するための最良の候補になる活性物質は、活性物質が適切なキレート化配置で2個の窒素、1個の窒素と1個のメルカプタン、または2個のメルカプタンを有しているならば安定な金属配位錯体を形成するためにヘテロ原子上にプロトンが存在していなくてもよいという追加の柔軟性で、s−ブロック金属と錯体形成するものと同じである。本発明の更なる実施形態では、遷移金属は、配位子が少なくとも1個の窒素またはメルカプト基を有しているならば、共有配位錯体のためにキレート化は余り必要でない点で更なる連結反応柔軟性を有している。
【0009】
本発明において具体化される活性物質は表1に示す化学分類に分類され得る(実際には、異種キレート化ポテンシャルを反映すべく化学分類の組み合わせに分類され得る)。表1にリストした薬物は、本発明の実施形態を満たす全薬物の網羅的リストであると意図されず、医薬品中に存在する化学分類の代表例であると意図され、表1にリストした同一分類に入るかまたは本発明の実施形態により満足されない原子の配置を有する他の医薬品も本発明により特許請求される。
【表1】
【0010】
本発明を明確に理解し、容易に実施するために、本発明を図面と関連づけて説明する。
【発明を実施するための最良の形態】
【0011】
化学結合は3つの基本的形態:イオン結合、共有結合及び配位結合、すなわち典型的には無機金属塩よりも大きい所謂ウェルナー錯体で存在している。(ウェルナー錯体は中性配位子を有していると見なされることを指摘しなければならない。)3つの結合タイプの違いは特に溶液中での結合の熱力学的安定性に一部起因し得る。逆に、化合物の安定性は分子の原子が溶液中で分離または解離する傾向で表され得る。
【0012】
化合物の熱力学的安定性は、式1に従ってその形成の自由エネルギーに換算して表される:
ΔG=−RTlnK 式1
ΔGはギブズ自由エネルギーであり、化合物の熱力学的安定性を示す。ΔGがより負の値であるほどに化合物はより安定である。Rはガス定数であり、Tは絶対温度であり、Kは平衡定数である。平衡定数は反応物質に対する生成物の比で表される。反応:
【化1】
【0013】
に対する錯体化合物の場合、Kは式2:
K=[MLx]/[M][L]x 式2
で表される。よって、化合物の熱力学的安定性の増大は平衡定数の増大に直接関連している。
【0014】
ある場合には、平衡定数を金属−配位子結合の解離ポテンシャルに換算して表すことが有利である。反応は、上述のように
【化2】
【0015】
である。解離定数Kdissは式3:
Kdiss=[M][L]x/[MLx] 式3
で示される。イオン結合は溶液中でほぼ完全に解離すると一般的に認められているのに対して、多くの共有結合は全く解離しない。よって、解離定数またはより一般的に表されているパラメータである平衡定数を測定することにより溶液中の結合強度を求めることは結合のタイプを識別する方法である。金属と15〜17族からの配位子間の結合を含む配位化合物について熱力学的安定性はまだ確立されていない。
【0016】
文献の精査から、有機金属結合の共有原子価は分光分析データ(すなわち、NMR及びMS)、アプイニシオ分子力学計算、またはその2つの組み合わせから決定され得る。通常、共有原子価は遷移金属、(酸素よりも)窒素及び硫黄配位子原子、配位子の結合次数またはハプト数(“η”で示す)の増加で生ずる可能性が高い。複数のハプト数を有する配位子を有する有機金属化合物はキレートと記載されている。1及び2族の金属、すなわち所謂s−ブロック主族元素の中で、ベリリウム及びマグネシウムのみが重要なキレート形成元素と見られている。
【0017】
アプイニシオ理論計算を用いる最近の研究から、配位子−金属結合のイオン性対共有結合性の点で配位結合の種類が更に限定された。Pierlootは、これらの有機金属錯体の共有原子価の程度を調べるために一連のウェルナー錯体にCASSCF(完全活性空間自己無撞着場)モデルを適用した。彼女は概括的に、CASSCF計算から得た静的相関エネルギーが金属−配位子結合の共有原子価とうまく相関する傾向が存在すると結論づけた。彼女は更に、同一金属について金属−配位子共有原子価及び関連する相関効果は以下の配位子の順
F−<OH2<NH3<Cl−<Br−<I−
で増加すると結論づけた。これは、Jorgensenが記載したネフェルクス効果(nephelauxetic effect)と一致した。この効果の大きさは配位子場での配位に対する遷移金属の電子間反発の減少に直接相関した。この減少は配位子に依存し、錯体及び遊離金属イオン中のラカーパラメータBの比(β=B錯体/Bイオン)として表された。Bの減少は、金属錯体を形成するために配位子を添加した後の遊離金属イオンの電子−電子反発作用の低下から生じた。Bが大きく減少することは強いネフェルクス効果を示している。よって、例えば、F−のようなイオン性配位子はBを余り減少させず、より大きなβ値を有する。分光測定に基づいて、配位子はネフェルクス系列:
【化3】
【0018】
が生じるβ値の減少する順に従って並べられた。2つの系列の相関は、両技術が記載した効果間の類似性により裏付けられる。前者の場合、CASSCF計算は金属−配位子結合に対する金属d軌道の寄与を評価している。錯体形成によるラカーパラメータ(B錯体)の減少は配位子上の遷移金属d軌道電子雲の非局在化により生じ、これは共有結合が形成されていることを示している。Li+とBe2+のCp配位子との結合は、Cp結合に比して寄与金属結合のエネルギー状態が低いためにほぼイオン性である。加えて、これらの元素のイオン半径は2個以上のCp配位子が結合するには余りに小さい。
【0019】
マグネソセン(図1)Cp2Mgの理論的な計算から、該化合物の構造はCp2Ca、Cp2Sr及びCp2Baの構造に類似しているが、Mgのd軌道密度はCp2Mgでは無視できることが判明した。しかしながら、Cp2MgにおけるMgに対するマリケン電荷は密度汎関数理論(DFT)を用いて0.66と予想された。MgCl2のような高い解離定数を有する化合物に関しては2に近い値が予想される。これはFaegri、Almloef及びLuethiの論文と一致しており、彼らはアプイニシオMO−LCAO計算に従ってマグネソセンの電荷分離は公知の共有配位化合物であるフェロセン(Cp2Fe)の電荷分離よりも僅かしか高くないと結論づけている。これらのデータから、Mg−Cp結合がやや共有結合であることが示唆される。Cp部分は、一部はその負の電荷から、一部は二重結合由来のπ結合からその共有結合に寄与している。こうしたアニオン性及びπ結合と金属の組合せはレチノイン酸及びその類似体でも生ずる。よって、レチノイン酸の金属配位化合物を形成することは本発明の実施形態である。
【0020】
Cp2Mg及び関連するMg−Cp化合物の反応性をWinterらが研究し、彼らはマグネシウムがアミジナート配位子と安定な結合を形成することを知見した。おそらくこれを最も物語っているのは[CpMg(η2−tBuC(N(2,6−iPr2C6H3))2)]の安定性であり、これは未変化で昇華し、180℃/0.05トルで80%回収された(図2)。このように、安定なマグネシウム−アミジナート化合物の例が報告されており、該化合物の共有結合性に対する更なる裏付けを与えている。このことは、プリン及びアルギニン含有化合物のようなアミジナート官能基を含む化合物の場合重要である。
【0021】
遷移メタロセンが強い共有結合性を有していることは一般に認められているが、主族メタロセンが顕著な共有結合性元素を有するという道理にかなった論議もなされている。金属−配位子結合にd軌道が関与しないと化合物の安定性が低下する恐れがあるが、主族金属、特にマグネシウムが配位子とイオン性よりも共有結合性の結合を形成することができるという見解を妨げるものではない。結晶化の際に6−配位マグネシウム錯体が形成されるのはsp3d2混成のためであることは一般的に公知である。よって、ある状況ではマグネシウムのd軌道でも配位錯体の結合に関与し得ると考えられる。
【0022】
ウェルナー錯体の平衡定数を金属ハライドと比較したとき安定性に大きな差を見つけることができる。例えば、logKMg−ピリジン=2.08であり、logKMgCl2≒−1.0である。ピリジンが中性配位子であることを心に留めると、このlogKの差はマグネシウム−クロライド結合のイオン性と相対するマグネシウム−ピリジン結合の共有結合性のみに起因し得る。安定なマグネシウム錯体の別の例はマグネシウム−サリチルアルデヒド(SA)錯体であり、logKMg−SA2=6.80である(図3)。この結合の安定性は、配位子結合原子が通常金属とイオン結合を形成する傾向にある酸素である場合に顕著である。しかしながら、キレート化酸素が存在していると錯体を純粋なイオン結合以上に安定化させた。窒素はカルシウムよりもマグネシウムに対してより強い結合を形成するが、通常酸素は窒素よりもマグネシウムのより強いキレート化剤である。
【0023】
キレートの平衡定数は通常非常に大きく(例えば、マグネシウムエチレンジアミン−N,N’−ジスクシネート錯体のlogKeqは6.09である)、配位子の中性部分と金属間の共有原子価の程度を表していないことがある。しかしながら、平衡定数は配位化合物の安定性を指し、化学物質の性質及び特殊な用途でどのように機能するかを決定するための重要な基準である。錯体内の共有結合の存在及びそのキレートの安定性に対する寄与は非常に高いlogKeqを説明し得、分子構造の剛性にも寄与し得る。多くの場合共有結合性は配位錯体の安定性に対する最も重要な寄与因子であることを指摘しなければならない。
【0024】
マグネシウムポルフィリン錯体またはキレートはおそらく最もよく知られた有機マグネシウム化合物である。クロロフィルはマグネシウムポルフィンである。フタロシアニンはその種の化合物の基本的要素を表すポルフィリンであり、金属−ポルフィリン結合を研究するためのモデルシステムとして広く使用されている。遷移金属がフタロシアニンと非常に簡単に錯体(図4)を形成することは判明しているが、アルカリ及びアルカリ土類塩は水及び他のプロトン性溶媒中にほぼ完全に解離するのでLi+、Na+、K+、Sr2+及びBa2+をこれらの塩の溶液から直接導入するのに適した溶媒は今まで見つかっていない。Mg2+及びBe2+の錯体形成が容易であることから予測されるように、これら2つのs−ブロック元素のみがCa2+と一緒にフタロシアニンに直接導入され得、通常はピリジン中でヨウ化物または過塩素酸塩から導入される。
【0025】
金属−ポルフィリン錯体の合成、構造、安定性及び物理的特性は十分に研究されている。マグネシウムフタロシアニンの構造及び物理的特性は各種技術を用いて、より最近は近IR吸収及びX線結晶学を用いて更に解明されている。マグネシウム−ポルフィリンキレートが金属配位化合物の非常に安定な例に相当するという結論が再び得られた。
【0026】
特定のマグネシウム−配位子錯体は実際共有結合性であり、イオン性ではなく、よって新しい組成物であり、新しい塩の形態ではない。金属−有機配位子化合物はその結合の種類の点で共有結合性である。
【0027】
本発明の実施形態では、配位子が金属(好ましくは、マグネシウム)の内圏に対して直接結合する機会を有しているときには配位錯体の形成が好ましい。これは、無水マグネシウム及び非プロトン性溶媒(または、溶媒がプロトン性ならばその溶媒は嵩高くなければならない)を使用することにより達成される。このコンセプトは、金属イオンの触媒反応性が水和形態では低下するという事実により裏付けられる。水性系での錯体形成は配位子と水間の水素結合ならびに水和及び配位子の錯体形成能による金属上の結合部位に対する競合の微妙なバランスである。よって、当然の結果として配位子と金属の内圏の錯体形成も水性系で低下する。更に、当然の結果として、逆も真なりであり、すなわち非水性系での金属の配位子とのキレート化または錯体形成の速度は水性系に比して促進される。
【0028】
固有の共有原子価を有する安定な金属−配位子配位化合物としての金属に結合した有機活性物質を含む組成は新しい分子である。本発明の別の好ましい実施形態では、金属は主族元素から選択される。本発明の更なる実施形態では、金属はs−ブロック元素から選択される。本発明の好ましい実施形態では、金属はマグネシウムである。
【0029】
更に、本発明の実施形態では、実質的にKeq>1.0である薬物−マグネシウム錯体は解離、吸収、分布、代謝及び排泄の薬物動態をモジュレートするのに十分な固有の安定性を有している。Mg(OH)2の解離定数が−11.5であると仮定すると、多くのマグネシウム錯体が酸条件よりもアルカリ条件でより非常に安定であるとの知見は驚くことでない。よって、小腸での薬物−マグネシウム錯体の安定性は薬物吸収の薬物動態をモジュレートする可能性が高い。酸不安定なそれらの金属−薬物錯体の場合、本発明の実施形態では、錯体は小腸に進入時に錯体を放出するコーティングまたはカプセル化材料により胃の酸性環境から保護される。本発明の更なる実施形態では、カプセル化物質は外配位圏を形成する配位子または配位子群である。
【0030】
本発明の別の重要なコンセプトは、溶液中で金属と配位子を単に組み合わせても常に同一の生成物が生成されないことである。多くはないが幾つかの特許は主題を裏付けることなく従属クレームとして各種塩を特許請求していることを認識されたい。このことは、当業者に公知の方法で有機酸の塩が塩基及び金属塩で処理することにより容易に製造され、予想される生成物は有機酸の金属塩であるので認められる。しかしながら、配位化学が有機酸と金属間の結合に寄与する場合、溶媒、温度、及びおそらく最も重要である金属に結合する配位子のような各種条件が配位錯体の構造及び安定性に影響を及ぼす。
【0031】
薬物以外の別の配位子は金属−薬物錯体を安定化させ得る。例えば、グリシン(G)マグネシウム結合のKeqは1.34である。しかしながら、この錯体にサリチルアルデヒドを添加すると、反応:
【化4】
【0032】
の平衡は4.77である。明らかに、サリチルアルデヒドはマグネシウムグリシン結合に対して安定化効果を加える。本発明の実施形態では、有利な物理化学的特性を付与するためにサリチルアルデヒドのようなアジュバントを薬物:金属錯体に配合する。本発明の更なる実施形態では、アジュバントの効果は水溶液中のような特定の環境下で薬物:金属錯体を安定化させる。
【0033】
遷移金属との配位錯体の非常に少ない例がPhysician’s Desk Reference(“PDR”)に記載されており、その中には1)亜鉛で修飾したインスリン;2)カルボプラチンは白金を含むこと;3)ニフェレクスは多糖−鉄錯体であること;4)ふけ防止シャンプー中の活性成分として使用されているピリチオン亜鉛、が挙げられている。加えて、幾つかの栄養サプリメントが錯体として記載されている。3つのピコリン酸基が八面体で1つのCr+3に結合している(窒素が3つの他の結合部位を与える)ピコリン酸クロムが一例である。これに対し、本発明の実施態様では金属は遷移金属に相当する族から選択され、本発明のより好ましい実施形態では金属は1及び2族のs−ブロック主族元素から選択される。本発明の最も好ましい実施形態では、金属はマグネシウムである。
【0034】
特許文献は幾つかの新規なマグネシウム配位薬物を例示しており、その中には1)Trilisate(登録商標)関節痛を治療するための上記した安定な固体コリンマグネシウムサリチレート組成物;2)高い組織特異性、精製及び薬剤へのコンパウンディングの容易さを与える2−デスカルボキシ−2−(テトラゾル−5−イル)−11−デスオキシ−15−置換−ω−ペンタノルプロスタグランジンのマグネシウム塩;3)インスリン抵抗性症候群の治療において有用なインスリン様特性を有するバナジウム酸マグネシウム;4)血栓または塞栓性卒中を治療するため、子癇前症/子癇及び急性心臓状態を予防的治療するための結晶性マグネシウム−タウリン化合物;5)GERDを治療するための上記したマグネシウムオメプラゾール“塩”誘導体が含まれる。
【0035】
医薬用塩の科学は十分に研究されている分野であり、塩形態の選択は所与の医薬性能に影響を及ぼし得る。塩形態が薬物に対して有し得る効果の例には解離速度、溶解度、官能性、安定性、処方効果、吸収モジュレーション及び薬物動態が含まれる。2003年10月に発表された定期刊行物Drug Deliveryの記事は薬剤を安定化するために、おそらく塩を形成するであろう金属を用いた3つの賦形剤用途を記載している。ヒト成長ホルモンを亜鉛と錯体形成すると親水性が低下し、これにより薬物の放出が遅れる。カプセル化ポリマーの分解により生ずる酸性環境からのタンパク質の安定化は製剤に水酸化マグネシウムを添加することによりなされた。ビンカアルカロイドを酸加水分解から安定化させるために炭酸亜鉛が使用された。これらの生成物は明らかに医薬的に活性な物質を安定化させるために金属を使用しているのに対して、後者の2つは活性物質の構造を修飾させたとは記載していない。
【0036】
更に、塩形態を単に変化させるだけで小腸での吸収の薬物動態が大きくモジュレートされることは十分に確証されている。例えば、テトラサイクリンのリン酸塩及びテトラサイクリン塩酸塩の化学構造はファルマコフォアでない塩形態の部分の点で異なり、各々の相対的物理的特性は相対的バイオアベイラビリティーに対して大きな影響を持たないと予想される。しかし、実際リン酸塩は塩酸塩よりも2倍多く吸収される。逆に、ワルファリンの遊離酸のバイオアベイラビリティーはそのナトリウム塩とほぼ等しい。このことは、ワルファリンナトリウム錠の溶解速度が遊離酸を含有する錠剤よりも350倍速いので予期せぬことである。異なる塩形態が吸収のカイネティクスに変化を与え得るならば、s−ブロック主族元素との錯体は吸収に対してより顕著な効果を有することがあり得る。
【0037】
テトラサイクリン系抗生物質のバイオアベイラビリティーが消化管中で最も形成する可能性の高い金属錯体の物理化学的特性により主に影響されることは公知である。このことは、明らかにインビボでの薬物:金属結合形成を示している。インビボで薬物と金属間に共有結合が形成されることは、薬物が窒素を含有し、金属が10〜12族(例えば、ニッケル、銅、亜鉛)に属するときにはより合理的な予想である。本発明の実施形態では、薬物性能のモジュレーションは薬物と金属間の安定な錯体の形成を促進する薬物と金属の製剤化により付与される。
【0038】
本発明の実施形態では、金属と錯体形成または配位したとき薬物に対して以下の効果が与えられ得る:
1.より良好なバイオアベイラビリティーと同一視し得る改善された水溶性(以下の討論参照);
2.細胞膜を介する吸収を改善するための高い親油性;
3.改善された受容体結合のためのファルマコフォアのコンフォメーションへの閉じ込め;
4.多形に起因する製剤化の問題の改善(以下の討論参照);
5.胃のような酸性環境での分解から保護するための酸吸収性;
6.配位錯体の安定性は活性ファルマコフォアの吸収の遅れを暗示し得る。このことは、薬物の迅速吸収が毒性ポテンシャルを上昇させるリオサイロニンのような薬物にとって重要である;
7.活性ファルマコフォアの持続吸収のための生物接着性;
8.麻酔薬のファルマコフォアに有機金属錯体を介して結合し、摂取されない限り麻酔薬を不活性とすることによる麻酔性鎮痛剤の乱用の防止。
【0039】
自然界は、どのように遷移金属を輸送し、貯蔵し、利用するかの多くの例を提示している。おそらく、最もよく知られている例は鉄ポルフィリンであるヘモグロビンである。上記したように、クロロフィルはマグネシウムを囲むポルフィン構造である。幾つかの酵素は該酵素を活性とするために金属を必要とする。これが、微量金属(例えば、銅、亜鉛、クロム等)が適切な栄養供給のために重要な理由である。幾つかの抗体もこれに関連する遷移金属を有している。金属が配位錯体の形成により酵素のペプチド構造をコンフォメーションに閉じ込めるため、金属が酵素活性のために必要である。
【0040】
薬物のコンピューター支援設計を組み入れるコンセプトは最近評判を得ている。イン・シリコと称されているこの技術は、何の物質も生成していない段階で基質と受容体間のアロストリック、クーロン力及び非共有相互作用を理解して、リード薬物候補をコンピューターモデリングにより同定するまでに開発されている。本発明の実施形態では、コンピューターシミュレートした分子に金属配位を加えることにより新しく改善されたリード化合物を同定することができる。本発明の更なる実施形態では、リード化合物の錯体としての金属の導入を計算に組み入れるとき、イン・シリコに由来するリード化合物は変化した結合熱力学を有している。本発明の更なる実施形態では、以前に不十分なイン・シリコ分析に基づいて考察から除外された化合物は金属錯体を計算に組み入れて再分析したとき重要なリード化合物になるであろう。本発明の好ましい実施形態では、上記したような改正イン・シリコ計算のために使用される金属は主族元素から選択される。本発明のより好ましい実施形態では、金属はs−ブロック主族元素から選択される。本発明の好ましい実施形態では、金属はマグネシウムである。
【0041】
錯体を生体系に直接適用する点で、本発明の実施形態では、配位子−受容体結合を必要とする活性物質には、金属と錯体形成して活性物質のコンフォメーション構造を適所に閉じ込めることによって、高い生物学的活性が付与されている。受容体は膜結合性であるか、細胞質内にあるか、または体内を循環している。本発明の実施形態では、標的受容体との最適相互作用を与えるコンフォメーションで薬物を閉じ込めるために金属を注射薬に配合する。本発明の好ましい実施形態では、金属は注射のために安全であると考えられる。本発明の更なる実施形態では、金属はアルミニウム、ビスマス、マグネシウム、カルシウム、鉄または亜鉛のリストから選択される。本発明の更に好ましい実施形態では、活性物質は注射薬のリストから選択され、前記注射薬にはワクチン、抗悪性腫瘍薬、抗糖尿病薬、及びアンチセンスRNAまたは他の代謝モジュレーターが含まれるが、これらに限定されない。
【0042】
本発明の金属配位技術はワクチン設計における現在の研究を前進させることもできる。例えば、開発中の新しい癌ワクチンはリポタンパク質アジュバント、ペプチド抗原を、癌細胞に対して特異的な炭水化物抗原と組み合わせる。ワクチン構築物の3つの成分をリンカーを介して一緒に共有結合させる。このワクチンの構築方法はバイオコンジュゲート化学では一般的である。金属配位は、組み合わせる成分がアプタマー、ポリエチレングリコール及び脂質であるペガプタニブのようなバイオコンジュゲートの各種成分を結合させるための足場として使用され得る。本発明の実施形態では、バイオコンジュゲートの諸成分は各成分を中心金属に錯体形成することにより単一分子中に組み合わされ得る。本発明の更なる実施形態では、金属配位をバイオコンジュゲート化学における一般的な技術として使用する。
【0043】
マグネシウムが核酸に対して著しい親和力を有していることに特に注目されたい。アンチセンスRNA、干渉RNA及びアプタマーの治療薬としての進歩に伴って、これらの核酸薬に対してデリバリー技術を取り入れることがますます重要であろう。現在研究されている薬物デリバリー技術の幾つかにはペグ化、リポソームまたはアニオン性クレーが含まれる。興味深いことに、Howard Hughes Medical Research Labから発表された最近の研究でモンモリロナイトクレーがRNAの脂質小胞への進入を促進することが示された。アルミナ、シリカ、マグネシア、鉄及びカリウムの量の点で異なる各種クレーがある。よって、RNAのマグネシウム錯体を形成すると、細胞膜の実験室モデルと見なされる小胞へRNAの進入が促進され得る。
【0044】
マグネシウム−核酸錯体の有効性を核酸単独と比較して上記したイン・シリコ技術を用いて評価し得る。よって、核酸薬の有効性が金属と配位させることにより高まることが本発明の重要な実施形態となる。本発明の更なる実施形態では、投与前に核酸を金属と組み合わせて配位錯体を形成する。水性系中で金属塩を核酸と単に組み合わせた核酸の重要な部分は外圏配位配位子(図5)であり、受容体結合のための特に膜輸送用途のために最適なコンフォメーションを与えないことがある。本発明の主たる前提は、配位子が外圏配位子の存在下で内圏配位子(図6)となる機会を有しているならば金属−配位子錯体構造が大きく影響されることである。
【0045】
本発明の更なる前提は、内圏配位子の形成が金属−配位子錯体を製造するために無水条件を使用することにより促進されることである。従って、本発明の実施形態では、金属配位子錯体を無水条件下で製造し、前記錯体を水中で再構成すると、水中で製造した錯体に比してより大きい共有結合性、より高い安定性、より優れた細胞透過性及びモジュレートされた生物学的性能を有する配位錯体が生ずる。この系は、トランスフェクション効率を高めるための内配位圏内へのアジュバント(例えば、ポリアルギニン)の導入に適している(図7)。
【0046】
おそらく、最近の遺伝子治療での最も重要な進歩は干渉RNA(“iRNA”)に関する発見及び進歩した研究であった。アンチセンスRNAとは異なり、iRNAはmRNAによりコードされる遺伝子産物を更に抑制させるために細胞の生化学機構により再利用される。こうすると、遺伝子抑制の効率が高まる。iRNAに関連する主要な問題には細胞への透過性及び(特にヌクレアーゼの存在下での)安定性が含まれる。
【0047】
これらの問題は、肺表面活性物質(“SAM”)、脂質またはアミンを主成分とするトランスフェクション剤、エレクトロポレーション、ウイルスベクターまたはプラスミドベクターを導入することにより解決されてきた。後者の技術は、プラスミドベクターによりsiRNAが“ヘアピン”構造を採用し、iRNA変異体に短ヘアピンRNAまたはshRNAの名前が付けられている点で特に興味深い。これらのshRNA分子は高い抑制能力を有している。更に、siRNA分子が細胞に進入するのにトランスフェクション剤が必ずしも必要でないことを示唆する証拠は多数存在する。siRNA及びウイロイドを肺適用するという最近の知見は、裸RNAが細胞に進入し、遺伝子産物を抑制させ得る2つの報告された現象である。実際、当業界の科学者にはRNAの二次構造がその遺伝子抑制効果に影響を及ぼさないようであることが知られている。
【0048】
RNAは複数のホスフェート基を有するオリゴヌクレオチドである。マグネシウムはホスフェート基と非常に強い結合を形成し、よってRNA−Mg錯体ではおそらくマグネシウムはホスフェート基と結合している可能性が高い。無水条件下でマグネシウムをRNAと組み合わせることにより、共有結合が形成され、こうすると理論上RNA分子の当該部分の親油性が高まる。更に、マグネシウム中心は複数のホスフェート基を結合し得、理論上上記したヘアピン構造が形成される。このヘアピン構造により親油性残基が現れるだけでなく、ヌクレアーゼからの攻撃に対してより高い耐性が生じ、これにより高い安定性が得られる。
【0049】
RNAはマグネシウムに結合しているホスフェート基以外に他のホスフェート基を有しているので、RNA分子の当該部分はその水溶性を保持している。この新規な形態のRNAは物質輸送(親水性)及び吸収(親油性)にとって重要な所望の両親媒性を有する。この点の更なる検討については、以下の「改善された溶解性」を参照されたい。
【0050】
典型的な方法は無水溶媒中でRNAをマグネシウムと組み合わせることを伴う。適当な溶媒はDMSO、またはおそらくイオン性液体である。イオン性液体の利点は、アルコールのようなイオン性液体混和性非溶媒(または、幾つかの場合には超臨界CO2が機能することがある)に溶液を添加するだけで所望の生成物が析出し、マグネシウム−RNA錯体が回収されることである。次いで、イオン性液体はアルコールを留去させることにより次の反応のために再利用され得る。
【0051】
上記方法はおそらく水溶性の生物活性物質に適用することが可能である。よって、本発明の実施形態では、生物活性物質は糖、ペプチドまたはヌクレオチドである。本発明の好ましい実施形態では、生物活性物質はヌクレオチドである。本発明のより好ましい実施形態では、生物活性物質はアンチセンスRNA、干渉RNAまたはアプタマーである。本発明の好ましい実施形態では、金属は主族元素から選択される。本発明の更に好ましい実施形態では、金属はs−ブロック主族元素から選択される。マグネシウムがカルシウムよりも核酸に対してよりしっかりと結合することは認められており、よって金属がマグネシウムであることが本発明の最も好ましい実施形態である。
【0052】
改善された溶解性/浸透性
薬物吸収を定量する際、用語「バイオアベイラビリティー」を適用することが有用である。これは体循環に達する用量の分数(F)として定義される。極端な場合、薬物が消化管に全く吸収されない場合にはF=0であり、薬物が完全に吸収される(且つ、初回通過効果により代謝されない)場合にはF=1である。バイオアベイラビリティーは血清レベル対時間プロットの曲線下面積(AUC)から計算され得る。バイオアベイラビリティーは多くの因子に依存し、これらの因子の幾つかは正常な個人の間でも異なる。バイオアベイラビリティーの点で、薬物は下表に従う4つのカテゴリーに分類されている。
【表2】
【0053】
分かるように、更なる開発のための実行可能な候補となるよう、薬物は細胞膜透過性と溶解性の微妙なバランスに適合していなければならない。この理由は、溶解性(すなわち、親水性)を高める物性が通常透過性(すなわち、疎水性または親油性)を高める物性と相反しているからである。
【0054】
金属とテトラサイクリン系抗生物質の相互作用により薬物及び金属の両方のバイオアベイラビリティーが低下することは判明している。上記したように、テトラサイクリン系抗生物質のバイオアベイラビリティーは主に消化管に広がっている金属錯体の物理化学的特性により影響される。中性種は腸管細胞のリン脂質膜に容易に吸収される可能性が高いので、電荷はバイオアベイラビリティーに大きな影響を有している。親油性金属配位錯体は、電荷を有している薬物の金属塩と比較してバイオアベイラビリティーを高めるのに役立たなければならない。よって、本発明の実施形態では、親油性金属−抗生物質共有結合性錯体を投与することにより抗生物質の物理化学的特性がコントロールされ得、更に消化管中の金属が薬物との金属相互作用の動態、最終的には吸収に影響を及ぼすのを防げ得る。本発明の更なる実施形態では、上記した原則が一般的にすべての薬物に適用される。
【0055】
この技術は、高度に水溶性の薬物または所謂クラスIII薬物の親油性を高めるためにも使用され得る。この場合、ホスフェートまたはスルフェート基のようなイオン性中心が共有結合に変換される。この金属と配位子間の結合の変化により配位子の水溶性が低下し、配位子の有機溶媒溶解性または親油性が上昇することは知られている。
【0056】
薬物が余り溶解しないが容易に透過し得るならば、その溶解性を高め得る1つの方法は薬物に対して水溶性物質(例えば、アミノ酸または炭水化物)を共有結合させることによる。或いは、薬物とイオン化金属中心間で金属−配位子錯体を形成することにより、固有の親水性が付与された新しい化学物質が形成される。本発明の実施形態では、活性物質を遷移金属またはアルカリ土類金属に結合させて、その透過性を保持しながら溶解性を改善した新規な組成物が形成される。新しい金属配位子結合は共有結合であるので、新しく形成された共有結合金属中心の親油性を相殺するように金属に追加の配位子(例えば、アミノ酸)が結合されていることが好ましい。
【0057】
共有結合性により、金属−活性物質錯体の安定性は刷子縁膜の水フィルムコーティングへの輸送まで保持されている。錯体が膜に達すると、金属と薬物は、親油性活性物質を受容し親水性金属を拒絶する膜中の脂質により分離される。これは物理化学的作用により行われ、薬物の水溶性を上昇させる従来方法とは対照的に酵素を必要としない。
【0058】
薬物は1)皮膚表面に対する効果、2)角質層内の効果、3)表皮及び真皮へのより高い浸透を必要とする効果、または4)血管系への浸透を介する全身効果を引き出すために皮膚に適用される。この研究の目的は、薬物を表皮を介してまたは血管系に浸透させ得る新しい経皮薬物デリバリー(TDD)システムを設計することである。浸透の所望レベルは薬物に依存する。
【0059】
角質層は有効なバリアを与え、水及び化学物質が表皮に、表皮から浸透するのを防止する。角質層における脂質の構造組織化は水及び化学物質の迅速な輸送を防止するのに重要な因子であることが提案されている。このように脂質が組織化すると、液晶形態が生じ、このマトリックスを介する浸透は、脂質炭化水素鎖が乱され液晶が不安定化することにより生ずる。これが、ハイドロトロープの局所的に適用された薬物の浸透性を高める能力について提案されているメカニズムである。
【0060】
皮膚透過性を高めるために使用されている化学物質の分類の幾つかにはアルコール、アルキルメチルスルホキシド、ピロリドン、界面活性剤(アニオン性、カチオン性及び非イオン性)、脂肪酸及び脂肪アルコールが含まれる。加えて、ラウロカプラム、尿素、カルシウムチオグリクレート、アセトン及びジメチル−m−トルアミドが特定の生物活性物質の皮膚浸透性を高めるために使用されている。これらの薬物のビヒクル効果の多くはその屈水性による。化学的には、これらの多くは大きい双極子モーメントを有している。すなわち、親油性部分及び親水性部分を有している。この大きな双極子モーメントが化学物質が角質層中の脂質を乱す主要な要因である。
【0061】
多くの薬物は本質的に局所的に使用するのに十分な皮膚浸透能を有していない。よって、実質的にすべての局所適用される薬物は所望の効果を得るためにビヒクルまたはTDDエンハンサーを含む製剤を必要とする。すべての医薬品が従わなければならない安全性及び有効性の一般的要件に加えて、局所適用される薬物はビヒクル中に溶解し、安定でなければならず、製剤は含量均一性を有していなければならず、製剤は適正な粘度及び分散特性を有していなければならず、患者のコンプライアンスを最大にしなければならない(このことは、製剤が適用するのに不快であってはならず、不快な臭いを有していたり皮膚刺激を生じさせてはならないことを意味する)。
【0062】
最も注目することは、薬物が表皮に浸透するためのタイムラグはビヒクルから角質層へ分散する薬物の能力に依存するが、これがTDD製剤の開発にあたり大きな障害となっている。以前の報告で、このタイムラグが数分〜数日にも及び得ることを示している。よって、TDDシステムの開発の重大な障害はこの適用に特有の追加の考慮すべき事項であり、歴史的によれば経皮医薬品の開発時間はしばしば途方もなくかかると見られていた。
【0063】
本発明に従う薬物錯体の高い経皮透過性は錯体の安定性と両親媒性の組合せに依存している。よって、実施形態では、共有結合金属−薬物結合を形成させて薬物それ自体のTDDを強化し得る有効なハイドロトロープに薬物を変換させる。本発明の更なる実施形態では、TDDエンハンサーがなお必要とされているならば金属をビヒクルに対するアンカーとして作用させ、全錯体を単一分子として挙動させる。この作用効果は、錯体からの薬物の放出のためにビヒクルと表皮の脂質マトリックス間の分配係数を差別化する必要がないことである。
【0064】
共有結合性のために、金属−活性物質錯体の安定性は角質層を介する輸送の間保持されていなければならない。錯体が表皮中にあるとき、金属と薬物は、親油性活性物質を受容し親水性金属を拒絶する膜中の脂質により分離される。これは物理化学的作用により付与され、薬物の溶解度を高める従来方法とは対照的に酵素を必要としない。
【0065】
薬物を金属配位錯体に変換すると、眼への進入も容易となる。眼圧(IOP)を治療するためにスルホンアミドをその金属配位錯体に変換するとそのIOP降下効果が高まることは判明している。これは、一部は眼中にスルホンアミドが多く存在しているためであり、よって金属配位錯体の親油性と親水性のバランスが適切であるためであると考えられる。眼病を治療するための薬物は本発明に従って金属配位錯体に変換させることにより改善され得る。このことは、現在の治療では薬物を眼の奥部に注射している加齢黄斑変性(AMD)を治療するために非常に重要である。AMDを治療するために薬物を点眼すると患者のコンプライアンスが大きく改善される。これは、AMD薬物を金属と配位させることにより達成される。
【0066】
多形のコントロール
多形は、一部は溶解度の変動のために投与量の変動に大きく寄与している。歴史的に言えば、有機金属化合物の固有の物理的特性は安定な結晶形を製造するのが比較的容易であることである。よって、本発明の更なる実施形態では、活性物質を金属錯体に変換させ、前記錯体を当業者に一般的に公知の方法により再結晶化方法にかけることにより多形を解消する。こうすると、活性部分は所望の多形に閉じ込められる。
【0067】
薬物吸収のモジュレーション
最近では、薬物のデリバリー方法をモジュレートすることにより薬物を改善するための活動が活発である。薬物デリバリー技術は経口から注射、インプラント、皮膚パッチのすべての投与形態に広がっている。これらの技術の大部分は、活性成分をポリマー球体の内部に封入または“トラップ”するカプセル化技術またはビーズ技術を利用している。このポリマー球体はミセルとして、自己組織化分子ロッドまたはボールとして、または活性成分の周りのコーティングとして存在し得る。薬物は、血液中を循環したり消化管を横断するとカプセル化剤から溶媒和または膨潤することにより放出される。薬物のデリバリーをモジュレートする主な利点は、その放出を広げたり、改善された安全性のために血液レベルを調節したり、または改善された有効性のために吸収を高めることである。よって、本発明の実施形態では、薬物−金属錯体放出はインビボで錯体それ自体に対する物理化学的作用によりモジュレートされる。
【0068】
ある場合には、薬物−金属錯体をポルフィン、ペプチドまたはポリマーマトリックス内に封入することにより活性物質−金属錯体の安定性を高めることが有利であり得る。このことは、活性物質が金属と安定な錯体を形成するために必要な要素(例えば、上記した第1級アミンまたはアルコール)を含んでいない場合に特に当てはまる。本発明の好ましい実施形態では、マトリックスは活性物質を金属に結合させるために所要により修飾されているポルフィン誘導体である。本発明の実施形態では、薬物−金属錯体放出はインビボでポルフィン、ペプチドまたはポリマーマトリックスに対する物理化学的作用によりモジュレートされる。本発明の更なる実施形態では、マトリックスは元々小腸に存在する化合物である。本発明の更に好ましい実施形態では、ポルフィンマトリックスはビリルビンまたはその誘導体である。
【0069】
本発明の更に好ましい実施形態では、カプセル化マトリックスはアミノ酸またはジペプチドであり、前記アミノ酸または複数のジペプチドは金属−配位子錯体と配位させるかまたはその周りで自己組織化させるために添加され得る。ヒスチジンは、ヒスチジン中のイミダゾール部分の強い金属結合能のために理想的なアミノ酸である。アルギニンは、ペプチド結合アルギニンのグアニジン部分のアミジナート連結によりマグネシウムと錯体形成するのにうまく適している別のアミノ酸である(図8)。本発明の関連実施形態では、その錯体形成及び酸中和のためにマグネシウムは胃においてアルギニンを安定化させ、その有効性を増加させる。このことは、アルギニンをCOPD及び関連する病的状態を治療するためのNO源として使用する場合に役立つ。
【0070】
金属上の第2配位子としてアミノ酸を使用することは、内配位圏を安定化させ、内圏の周りに疎水性シェルを生成し、よって金属−薬物結合の加水分解を防止するためである。よって、本発明の実施形態では、錯体を特に水性系中で安定化させるためにアミノ酸、ジペプチドまたはオリゴペプチドを金属−薬物錯体上の第2配位子またはアジュバントとして作用させる。本発明の好ましい実施形態では、第2配位子はジペプチドである。本発明の別の好ましい実施形態では、第2配位子はアミノ酸である。本発明の別の好ましい実施形態では、アミノ酸はヒスチジン及びアルギニンの群から選択する。
【0071】
遊離アミノ基を有する(例えば、錯体の一部としてヒスチジンのようなアミノ酸を有する)有機金属錯体はアミノ酸−NCAの重合を開始させてポリペプチドを形成し、有機金属錯体を配座的に保護する。この技術の別の利点は、アミノ酸NCAを有機金属錯体の周りで自己組織化させ、次いで重合の開始時にポリペプチドを自己組織化構造にコアセルベートすることである。
【0072】
本発明の実施形態では、カプセル化技術をコアセルベーション技術と組み合わせて活性物質と遷移金属間に内圏共有結合を形成して新しい組成物を生成し、次いで前記錯体をポリマーマトリックス内に外圏配位でカプセル化して、安定な錯体を提供する。図9は、活性物質(構造簡素化のために、サリチル酸を例として使用している)、内圏でマグネシウムに結合させたポリマー結合アルギニン、及び外圏で錯体を封入しているペプチドを示している。
【0073】
本発明の更なる実施形態では、カプセル化マトリックスを水、油、エマルションまたは生体流体(例えば、胃液)により膨潤または溶解させた場合のみ活性物質が放出する。本発明の実施形態では、活性物質は抗体−抗原錯体で生ずるようなカプセル化剤と活性物質間の強い結合によりカプセル化マトリックスから放出され得ない。幾つかの場合には、カプセル化剤からの薬物の放出を消化酵素によりモジュレートすることが有利である。本発明の好ましい実施形態では、腸中に分泌される、細胞膜内に存在する、または血流中を循環する酵素による化学分解により活性物質をカプセル化剤から放出させる。本発明の好ましい実施形態では、活性物質はアルミニウム、マグネシウム、カルシウム、鉄、ビスマス、ケイ素または亜鉛に結合している。本発明の別の最も好ましい実施形態では、カプセル化剤は金属−配位子錯体に対する抗体である。本発明の更に別の実施形態では、錯体は活性物質−金属錯体からなり、カプセル化剤はアミノ酸、ポルフィン、炭水化物またはそれらの混合物の組合せから自己組織化される。本発明の最も好ましい実施形態では、活性物質−金属−カプセル化剤錯体は医薬品である。
【0074】
本発明の別の実施形態では、配位錯体は該錯体を形成し得るすべての金属から選択される金属であり、薬物はすべての生物活性物質または医薬活性物質の群から選択される。本発明の好ましい実施形態では、医薬活性物質は生物活性のために特殊なコンフォメーションを必要とする。活性は活性物質の細胞膜を横断する能力に依存し得、配位金属は活性物質の膜転移のために適正な構造を与える。好ましい実施形態では、医薬活性物質は小分子、ペプチド、炭水化物、DNAまたはRNAからなる群から選択され、後者の2つは遺伝子治療において、アプタマーとして、またはアンチセンスヌクレオチド治療用途において使用されている。好ましい実施形態では、金属はアルミニウム、ビスマス、カルシウム、マグネシウム、鉄、ケイ素及び亜鉛からなる群から選択される。
【0075】
生体接着性
マグネシウムまたはカルシウムイオンを薬物の分子式に導入して薬物に対する生体接着性を推定する方法は各種ある。例えば、マグネシウム及びカルシウムがインテグリンの接着機能にとって重要であることは公知であり、よって腸管での薬物のマグネシウムまたはカルシウム塩、或いは薬物の錯体が腸壁の刷子縁膜上で発現するインテグリンに対する薬物の生体接着を高めると予想することは合理的である。また、生体接着により腸の通過時間が遅くなるので、これらの錯体は腸における吸収を持続性とする。従って、本発明の実施形態では、薬物の持続吸収は薬物をマグネシウムまたはカルシウムと錯体形成させることにより高められる。本発明の更なる実施形態では、より強い生体接着活性によりマグネシウムまたはカルシウム薬物錯体に対して持続放出が付与される。
【0076】
麻酔薬の常習防止
麻酔薬は非常に有効な鎮痛剤であるが、非常に常習性でもある。最近数年間、アヘン剤常習者及び気晴らし薬の使用者がOxyContinを常習していることを記載している報告書が多数ある。典型的には、薬物常習者は例えば水を添加することにより錠剤マトリックスを機械的または化学的に崩壊して、全12時間用量を1度に服用している。加えて、このタイプの常習は通常経口投与から始めるが、常習者はしばしば濃厚麻酔薬を鼻から吸入したり注入したりするようになることがある。
【0077】
オキシモルホン及びオキシコドンの構造分析から、その分子は金属とのキレート化のための理想的な候補であることが分かる。9位のβ−ヒドロキシ及び窒素は、その2つの間に金属を錯体形成すると非常に熱力学的に好ましい5員環が形成されるように配置されている。9−ヒドロキシを脱プロトン化してアニオン性アルコキシドを形成することが好ましい(図10)。窒素の孤立電子対は金属キレートを安定化するために十分な電子密度に寄与し得る。サリチルアルデヒドがグリシン−マグネシウム錯体を安定化する場合のように錯体に第2配位子またはアジュバントを添加することにより更なる安定化が付与され得る。本発明の更なる実施形態では、金属−麻酔薬錯体を上記したようにマトリックス内に封入する。
【0078】
よって、本発明の実施形態では、麻酔薬を金属と錯体形成させることにより、麻酔薬は物理化学的作用により錯体からゆっくり放出される。このことは、麻酔薬が即座にまたは一度に利用され得ないことを意味する。更に、本発明の実施形態では、金属−麻酔薬錯体は血液脳関門を通過することができず、麻酔薬が錯体から放出されるまで麻酔薬は無効である。放出速度は遅いので、一度に血液脳関門を超えて移動することができる麻酔薬の量は経口投与される用量よりも非常に少なく、陶酔効果は得られない。本発明の更なる実施形態では、第2配位子、カプセル化剤またはその両者の組合せを導入することにより麻酔薬の放出カイネティクスを更に遅くすることができる。好ましい実施形態では、金属はアルミニウム、ビスマス、カルシウム、マグネシウム、鉄、ケイ素及び亜鉛からなる群から選択される。本発明のより好ましい実施形態では、金属は主族元素から選択される。本発明の更に好ましい実施形態では、金属はs−ブロック主族元素から選択される。本発明の最も好ましい実施形態では、金属はマグネシウムである。
【0079】
金属の選択
配位錯体中に使用しようとする好ましい金属について言及する。医薬用途では、本発明の錯体中で使用する金属を選択するときには、全ての金属配位医薬品の安全性を考慮しなければならない。本発明を実施するために多くの金属を使用することができるが、本発明の好ましい実施形態では金属を一般に安全と認められている(GRAS)一覧表から選択する。金属を選択するための1つの基準は、現在市販されているミネラルサプリメントのリストを検討し、表1にリストされている薬物との配位錯体として配合されるであろう用量をはるかに超える投与量を有するものを選択することである。非処方薬物及びダイエタリーサプリメントに関するPDRから、2mg/投与以上の量を有する7つの金属(アルカリ金属、すなわちナトリウム、カリウム等を除く)のリストを表2に示す。
【表3】
【0080】
本発明の好ましい実施形態では、金属はアルミニウム、ビスマス、カルシウム、鉄、マグネシウム、ケイ素及び亜鉛からなる群から選択される。本発明の実施形態では、医薬品とランタニド、アクチニド、遷移金属及び主族金属(s−及びp−ブロック)を含めた任意の金属間で共有結合を形成することにより新しい組成物を形成するが、本発明の好ましい実施形態では金属をs−ブロック主族元素から選択する。この理由は、s−ブロック元素が遷移金属またはp−ブロック主族元素(ランタニドまたはアクチニドはOTC製品中に決して使用されない)よりもGRASである可能性が高く、OTC医薬品やビタミンサプリメント中によりしばしば使用されているからである。他のs−ブロック元素(例えば、カルシウム)よりもマグネシウムを選択するのには幾つかの理由がある:
1.カルシウムは優先順で8>7>6>9という配位数の高い変動性を示す。カルシウムよりも小さいマグネシウムは殆どもっぱら、合成手順を簡素化し、生成物の混合物ではなく単一の生成物をより高い収率で生成する可能性が高い八面体である;
2.マグネシウムは他のs−ブロック元素よりもより容易にキレート化配位子と共有結合を形成し得る;
3.マグネシウムはカルシウムよりもタンパク質及び核酸とより安定な結合を形成し、よって生物学的医薬品の安定性を高める;
4.マグネシウム欠乏は幾つかの病的状態(例えば、心臓血管関連、偏頭痛、ADHD)に関与しており、予防の視点からマグネシウムは重要な効果を有し得る。例えば、トリプタンマグネシウムはこの技術の理想的な候補であり得る;
5.カルシウムは能動輸送メカニズムにより腸に吸収されるのに対して、マグネシウムは受動的に輸送される。マグネシウム(塩として)及びフロセミドの腸管輸送は両者を一緒に経口投与したときに促進された。よって、吸収されにくい薬物であるフロセミドはこの技術の別の注目すべき候補に相当する。
【0081】
本発明の最も好ましい実施形態では、金属はマグネシウムである。
【0082】
溶媒の選択
上記したように、錯体形成反応のための溶媒の選択は金属配位化合物の構造及び安定性に影響を及ぼす。マグネシウムは水と強い結合を形成し、配位圏水和マグネシウムは生成物形成のカイネティクスだけでなく、生成物の構造及び安定性に対して影響を及ぼす。窒素含有配位子を遷移金属(例えば、亜鉛)と反応させる場合には強い窒素−遷移金属結合のために、反応混合物中の水の存在は通常金属配位生成物の構造及び安定性に対して強い影響を持たない。
【0083】
幾つかの場合には、配位子、金属及び所望生成物に応じて水は好ましい溶媒であり得る。生成物の大部分は無水有機溶媒が最良の選択であるとしている。幾つかの適当な溶媒にはアルコール、アセトンまたはTHFが含まれる。実質的にすべての医薬品及び栄養品を溶解し、塩化マグネシウムを含めた大部分のハロゲン化金属をも溶解する優れた万能溶媒であるので、DMSOが最も好ましい溶媒である。これにより、単相反応ができる。加えて、安定な金属配位医薬品はコアセルベーションに類似の方法により単離され得る。コアセルベーションは典型的には反応混合物に非溶媒を単に添加することを含む。
【0084】
DMSOはマグネシウムを含めた金属とその場で錯体を形成し得、DMSO−金属錯体は薬物配位子と反応し始めて金属中心でDMSO配位子を置換させる。その後、DMSOはアジュバントを錯体中に配合しようとする反応では一時保護基として機能し得る。このインプロセス反応スキームは、DMSO配位子間に水素結合が欠けているためにDMSO−金属錯体は水のような外配位圏を形成できないという事実により促進される。こうすると、金属中心は入ってくる配位子に容易に接近し得る。形成された金属配位錯体に応じて、最終生成物はDMSOを配位子として保持していることも、保持していないこともある。DMSOが配位子に結合しているならば、DMSOの用量が毒性レベルに近くまで達するような状況はありそうもない。
【0085】
本発明の前提では、生物活性配位子の水溶液に金属塩を添加するだけでは、形成された配位錯体は無水条件下で反応物質を組み合わせたかのように同じでない。更に、乾燥した配位錯体を同一水性環境において再構成しても、その2つの錯体の構造は異なる。その目的を達するために、幾つかの配位錯体をFDA承認医薬品を用いて製造し、これらの錯体が安定であることを立証することができる。生成物はできるだけ完全にキャラクタライズされ、バイオアベイラビリティ研究が行われるだろう。金属配位錯体は水及び有機溶媒中で製造され得る。多くの場合それぞれの生成物が安定性、構造及び生物活性の点で異なると予想される。
【0086】
薬物の選択
金属と安定な錯体を形成し得る殆どの薬物の錯体が本発明により形成可能である。以下の実施例のために選択した薬物は表2に示すように化学分類及び薬効分類の代表に相当する。
【表4】
【0087】
小分子の検討
合成:
重要であり得る他の薬物−マグネシウム錯体には、頭痛を軽減するためにマグネシウムが重要であるためトリプタン−Mgが、及び乱用耐性麻酔薬が重要であるためオキシコドン−Mgが含まれる。
【0088】
典型的には、tBuOH/DMSO中の薬物とKOtBuの混合物に無水金属ハロゲン化物(ヨウ化物、臭化物及び塩化物)を添加する。或いは、薬物及びDMSO中の第3級アミン(例えば、トリエチルアミン)の溶液に金属ハロゲン化物を添加してもよい。別のオプションでは、金属ハロゲン化物を薬物及びTHF中のKHに添加し得る。好ましい金属はマグネシウム及び亜鉛であり、好ましいハロゲン化物は塩化物である。塩化亜鉛はDMSO、アセトンまたはエタノール中に可溶性であり、これらは亜鉛錯体形成、特に窒素含有配位子との錯体形成のために好ましい溶媒である。
【0089】
生成物を沈澱により単離し、吸引濾過または遠心分離により液体から分離し、洗浄した後、最後の微量の水分を除去するために高真空下で乾燥する。薬物:金属錯体は水和物を形成していることがあり、水のすべてが高真空下で除去されないことがある。或いは、添加した水が残っているその場で形成した金属上のDMSO配位子を置換し得ないことがある。結果として、生成物は薬物:金属:DMSO錯体であり得る。
【0090】
マグネシウムと結合したとき高い解離定数を有する特定の薬物がDMSOとの錯体形成にとって有利である。その場での三元錯体の形成により錯体が更に安定化し、経口投与後の吸収過程中その分子の完全性が保持される。このために、多くの薬物はマグネシウムハロゲン化物と反応させるときDMSOが好ましい溶媒である。
【0091】
実施例(アシクログアノシン−Mg及びT3−Znの例を除く)における錯体形成反応に対する比較として、下記実施例に記載したように反応媒体に水を含める以外は同様に反応を行う。反応物を上記したように後処理し、乾燥させる。
【0092】
T3−Mg錯体形成反応に水を添加すると、明らかに単離された生成物に影響を及ぼした。DMSO単独中で製造したT3−Mg化合物は、その1H NMRスペクトルにおいて脂肪族領域のみに幅広化し、シャープな芳香族ピークを示した(図11)。
【0093】
脂肪族領域にシャープなピークを示すT3の1H NMRを図12に示す。比較すると、水の存在下で製造したT3−Mg生成物の1H NMRは1H NMRスペクトル全体に広範囲にわたる幅広化を示した。更に、水の非存在下で製造したT3−Mg生成物のマグネシウム含量(1.62%)はビス(トリヨードサイロニナト)−ビス(ジメチルスルホキシド)−マグネシウムの含量と非常にうまく整合していた。対照的に、水の存在下で製造したT3−Mg錯体のマグネシウム含量はたった0.96%であった。そのカリウム含量は0.23%であり、ビス(トリヨードサイロニナト)−ビス(ジメチルスルホキシド)−マグネシウム生成物ではカリウムは検出されなかった。
【0094】
テトラサイクリンはβ−ジケトン及びβ−ケトフェノール官能基を有しており、マグネシウムと安定な錯体を形成する。DMSO中の反応に水を添加してもそれぞれの生成物の溶解性、1H NMRスペクトルまたは金属含量に対する影響は殆どない。実際、水単独中で実施した反応から単離した生成物の1H NMRスペクトルはDMSO単独から、または5:1のDMSO:水混合物中で単離した生成物のものとは余り異なっていない。反応溶媒中の水含量が多くなるとマグネシウム含量は少なくなる傾向があるが、これは主に生成物の水和度に起因し得る。
【0095】
亜鉛は窒素含有化合物と非常に安定な結合を形成するので、水は亜鉛と配位子間の錯体形成を干渉しないが、錯体構造に影響を及ぼす恐れがある。無水DMSO中での金属と薬物の反応から生ずる生成物は通常十分にキャラクタライズされた配位錯体であった。構造的にうまく規定されなかった化合物(すなわち、HCTZ−亜鉛)または若干不安定であった化合物(すなわち、ジメチルビグアニド−亜鉛錯体)の場合、亜鉛配位錯体が単離され得、少なくとも部分的にキャラクタライズされた。水の存在下で製造した同一の錯体は極性溶媒中で高い溶解性を有していた。それぞれの製造方法からの生成物の溶解性の差から、生成物それ自体の差が明らかに証明された。無水溶媒系中で製造した亜鉛生成物はイオン性塩、外配位錯体、水和錯体またはその組合せを生じたと考えられる。これは5:1 DMSO:水混合物中で製造したジメチルビグアニド−亜鉛錯体の場合であるようで、1H NMRスペクトルは2.85及び2.80ppmにN−メチル基に相当し、それぞれ遊離(イオン性)及び錯体形成されたジメチルビグアニドを示す共鳴を示した。
【0096】
キャラクタリゼーション
合成した各生成物をNMR、MS(TOFまたはFAB)及びICPによりキャラクタライズした。NMRスペクトルによりサンプルの完全性が確認され、幅広化、ピークシフトまたは多重共鳴の存在により金属が錯体形成されていることが分かる。
【0097】
無水DMSO中で製造した多くの錯体の金属含量は2つの薬物を金属に結合させた錯体と一致した。加えて、金属含量はバッチ毎に一定のままであった。ジメチルビグアニドの錯体は単離方法に応じて異なる金属含量を有しており、決して薬物:金属比は一定でなかった。金属含量をICP分析により測定し、NMR及びMSと共に、そのデータに基づいて薬物対金属の比を計算することができる。
【0098】
T3錯体の1H NMRスペクトルは、分子のアミノ酸部分との錯体形成を示す脂肪族領域の幾つかの幅広化及び高磁場シフト化を示していた。これは、それぞれ図11、12及び13に示すビス(T3)ビス(DMSO)Mg、T3遊離酸及びビス(T3)Znの1H NMRスペクトルの2.5〜3.5ppmの領域を比較することにより見つけることができる。
【0099】
ジメチルビグアニド錯体の1H NMRスペクトルは、窒素原子との錯体形成を示す−NH共鳴の大きな高磁場シフトを示した。加えて、N−ジメチル基の0.05ppm高磁場シフトがジメチルビグアニドのスペクトル(図15)と比したジメチルビグアニド−亜鉛錯体スペクトル(図14)中に見られた。
【0100】
ミノサイクリン及びテトラサイクリン錯体の1H NMRスペクトルは非常な大きい幅広化及びスペクトル全体にわたる多くの新しい共鳴の出現を有するポリマー構造に似ていた。これらのスペクトルが動的異性体混合物に起因し、分解にも重合にも起因していなかったことを立証するために、テトラサイクリン及びそのマグネシウム錯体のNMRサンプルに12N HClを添加し、スペクトルを再びとった。テトラサイクリン、ビス(テトラサイクリナト)マグネシウム及びHClを添加した錯体の1H NMRスペクトルをそれぞれ図16〜18に示す。一連のスペクトルから分かるように、マグネシウム錯体は参照テトラサイクリン化合物に逆戻りした。興味深いことに、HClをテトラサイクリンに添加したときNMRスペクトルの芳香族領域にかなりの量の分解が見られ(図19)、その量はテトラサイクリン−マグネシウム錯体の同等のスペクトルでは見られなかった。これはマグネシウムと錯体形成により与えられる酸保護効果を示し、酸環境(例えば、胃)において不安定である薬物に対するこの技術の重要な属性となり得る。
【0101】
ヒドロクロロチアジド錯体の1H NMRスペクトルも抗生物質−金属錯体のスペクトルで見られるのと同様の幅広化及び新しい特徴のない共鳴を有するポリマー構造に似ていた。ヒドロクロロチアジド、ヒドロクロロチアジド−亜鉛錯体及びHClを添加した錯体の1H NMRスペクトルをそれぞれ図20〜22に示す。これらのスペクトルから分かるように、幅広化した線は参照薬物で観察されるシャープな共鳴に逆戻りした。このことから、各錯体の1H NMRスペクトルで観察された幅広化及び追加の共鳴が溶液中の錯体の複数の立体化学及び幾何異性体に起因したことが分かる。溶液中の異性体間の中程度にゆっくりした相互変換も観察された幅広化に寄与し得る。
【0102】
アシクログアノシン−マグネシウム錯体に関するスペクトルデータから、錯体が形成されたことが判明した。アシクログアノシンの1H NMRスペクトルとそのマグネシウム錯体の1H NMRスペクトル(図23)の比較から、アシクログアノシン上の錯体形成部位がアミド酸素及びイミダゾール窒素であったことが示唆された。錯体のNMRスペクトルで欠けている10.6ppmの共鳴はアミドプロトンに帰属している。
【0103】
配位錯体の有意な存在を表す質量スペクトルは錯体の安定性の重要な指標である。これにより、分子イオンはビス(T3)Mg、ビス(T3)Zn、ビス(ミノサイクリナト)Mg、ビス(テトラサイクリナト)Mg及びビス(アシクログアノシナト)Mgで見られた。亜鉛同位体パターンを有する2つの分子イオンがヒドロクロロチアジド−亜鉛錯体のMALDIスペクトルで観察された。このとき、これらの質量に相当する構造は知られていない。ジメチルビグアニド−亜鉛錯体はそのMALDIスペクトルで亜鉛含有分子イオンを有していなかった。このことは化合物の不安定性に起因すると考えられる。
【0104】
FTIR研究は、観察された特定の錯体について配位子結合原子があるかどうか、錯体がDMSO、水と配位するか、または全く溶媒和しないかを調べるために使用され得る。
【0105】
安定性
製造した配位錯体の平衡定数を類似化合物の文献記載値から推定した。例えば、T3−Znの平衡定数logKeqは別のアミノ酸亜鉛錯体のフェニルアラニン−亜鉛に基づいて4〜5と推定される。また、ジメチルビグアニド−亜鉛のlogKeqは5〜7と推定される。ヒドロクロロチアジド−亜鉛のlogKeqを文献値から推定するのは困難である。テトラサイクリン−マグネシウムの異なるpH値での水中における安定度定数は報告されており、テトラサイクリン−マグネシウムのlogKeqは4〜5と予想される。アシクログアノシン−マグネシウムのlogKeqは1.6と推定される。T3−MgのlogKeqは良好な比較物質がないために推定するのが困難であるが、T3のようにアミノ酸であるグリシンのlogKeqは1.34である。T3はグリシンに比して疎水性が非常に高いために、T3−MgのlogKeqは1.34よりもかなり大きいと予想される。
【0106】
薬物−金属または薬物−金属−アジュバントの安定性の別の指標は、Keqに関連するが更に多座配位子の段階的安定性を示す結合定数である。金属と配位子間の最大結合についての累積結合定数βnは式4により求められる。
【数1】
【0107】
この差はフェニルアラニン−亜鉛の測定logKeqと8.5というその近似β2値で見られ得る。このβ2値はT3−Zn錯体の安定性をも非常にうまく反映し得る。
【0108】
異なる環境での金属−薬物錯体の結合定数を推定し得る方法は幾つかある。その幾つかの方法は光学吸収分光法、NMR分光法、質量分光法、反応カイネティクス、電位差測定法及びクロマトグラフィーである。
【0109】
分配係数(Partition Coefficient)及び分布係数(Distribution Coefficient)
分配係数は一定であり、式5に示すように水性相中の中性化合物の濃度/非混和性有機相中の濃度の比として定義される。
【0110】
分配係数P=[有機]/[水性] 式5
実際、式6に定義するLogPは、測定する条件によって、特にpHによって変動する。なぜならば、低pHでは塩基がイオン化し、高pHでは酸がイオン化するからである。
【0111】
LogP=logl0(分配係数) 式6
よって、LogP=1は10:1 有機:水性を意味し、LogP=0は1:1 有機:水性を意味し、LogP=−1は1:10 有機:水性を意味する。当然、イオン化化合物は水性相に優先的に分配し、よってLogPは低下する。塩基である中性分子の場合pHがそのpKaよりも2単位以上高いならば中性のままであり、中性酸の場合pHがそのpKaよりも2単位低いならば中性のままである。
【0112】
分配溶媒の選択もLogPに影響を及ぼす。多くのLogP測定では、オクタノール:水系を使用している。イオンペアリング作用がLogP測定に影響を及ぼし、特に本発明において具体化されているような金属配位化合物の場合考慮しなければならない。
【0113】
医薬用途の点から、以下のガイドラインが投与方法、製剤及び剤形を決定するために使用される:
・低いLogP(0以下) 注射
・中(0〜3) 経口
・高い(3〜4) 経皮
・非常に高い(4〜7) 脂肪組織に毒物蓄積
また、経口投与薬物の領域ではこれらのガイドラインが使用されている:
1.最適CNS浸透のために、LogP=2±0.7(Hansch rules)
2.最適経口吸収のために、LogP=1.8
3.最適腸内吸収のために、LogP=1.35
4.最適結腸吸収のために、LogP=1.32
5.最適舌下吸収のために、LogP=5.5
6.最適経皮浸透のために、LogP=2.6(及び、低いmw)。
【0114】
分布係数(D)は、式7で示される有機相中の非イオン化化合物/水性相中の化合物の全量の比である。
【0115】
D=[非イオン化](o)/[非イオン化](aq)+[イオン化](aq) 式7
LogDは特定pHでのlog分布係数である(式8)。これは一定でなく、分子のプロトン性によって変動する。pH7.4でのLogDは血漿のpHでの薬物の親油性を示すためにしばしば引用されている。
【0116】
LogD=log10(分布係数) 式8
LogDは以下の式によりLogP及びpKaに関連している:
酸に対して
LogD(pH)=logP−log[1+10(pH−pKa)] 式9
塩基に対して
LogD(pH)=logP−log[1+10(pKa−pKH)] 式10
よって、イオン化が最小であるようにpHを調節すると、logDはlogPにほぼ等しい。これらの条件下で、logDは具体的用途における薬物のバイオアベイラビリティーの信頼できる指標である。金属配位薬物の点から、参照薬物と比した薬物−金属錯体のlogDの上昇は親油性の増加を示すだけでなく、その水中安定性をも示している。テトラサイクリン、ビス(テトラサイクリナト)マグネシウム、トリヨードサイロニン及びビス(トリヨードサイロニナト)亜鉛のpH7.4でのlogDを測定し、pKaと一緒に表4に示す。
【表5】
【0117】
これらの結果は、中性化合物のアニオンを金属塩と組み合わせると低い親油性及び低いlogDを有する化合物を生成するはずであるという従来技術の教示に反している。おそらくより興味深いことには、本発明にこの技術を適用することによりテトラサイクリンは理論上注射薬物(logD<0)から経口薬物(logD>0)に変換され、T3は経口薬物(logD<3)から経皮薬物(logD>3)に変換された。この後者の結果は薬物を徐放性経皮デポ剤で投与することによりT3製品の安全性を高める際に重要な関係を有している。
【0118】
バイオアベイラビリティー研究
概要:参照薬物の吸収に対する金属と薬物の配位が有する影響を観察するためにラットモデルを用いて予備研究を実施した。この特別研究のために選択した参照薬物はトリヨードサイロニン(T3)であり、これはサイトメル(Cytomel)及びサイロラー(Thyrolar)の活性成分である。サイトメル及びサイロラーは共に現在甲状腺機能低下症を治療するために使用されている。サイトメルは特定の精神病の治療においても適応されている。
【0119】
研究計画:T3−亜鉛及びT3−マグネシウムを参照薬物と比して5時間にわたるバイオアベイラビリティーについて試験した。3つの化合物を別々にゼラチンカプセル剤に製剤化し、108±12μg/kgの全用量で投与した。ラット胃における試験化合物の酸分解を避けるために、製剤に金属酸化物を添加した。T3遊離酸に酸化亜鉛を添加した別のT3対照を含めた。製剤化したゼラチンカプセル剤の各々を各ラットの食道に直接経口投与し、血液サンプルを投与前、投与から0.5、1、2、2.5、5時間目に採取した。血清トリヨードサイロニンレベルを業界標準アッセイ方法を用いて独立研究所で分析した。
【0120】
結果:図30に示す結果から、4つすべての薬物製剤がラットにより急速に吸収され、金属配位T3を供給されたラットでは血清T3レベルは2.5時間目まで上昇し、その後通常横ばい状態になる。T3遊離酸を供給されたラットでは血清T3レベルが5時間目になお上昇する可能性があることを示していた。金属錯体形成したT3及び酸化亜鉛を添加したT3を投与されたラットでは血清T3血液レベルが上昇し、そのレベルは5時間目でT3遊離酸よりも約65〜85%高かった。
【0121】
結論:上記データは、金属をT3と錯体形成すると経口投与T3の吸収がT3単独よりもほぼ2倍増加したことを明確に示している。T3及び酸化亜鉛の製剤を供給した動物では、T3遊離酸はT3の吸収前に酸化亜鉛と錯体形成することが分かる。
【0122】
この研究は薬物デリバリー技術の有意な進歩、および金属を医薬品と配位することがバイオアベイラビリティー限界を有する薬物の性能に適用され、これを改善し得ることを表している。
【0123】
大分子プロトコル
合成:
規定サイズを有する二本鎖iRNAを実施例の節に記載されている標準プロトコルに従って作成した。次いで、iRNAを実施例の節に記載されているように無水条件下または水の存在下でマグネシウムまたは亜鉛と反応させた。
【0124】
iRNAの生物活性を各種方法で他の金属(例えば、カルシウム、亜鉛、コバルト及びマンガン)と錯体形成することによりモジュレートし得る。加えて、プリン/ピリミジン基とホスフェート基の結合を促進するために多価金属(例えば、CuまたはAg33)を組み合わせると、iRNAのトランスフェクション効率及び安定性を改善させることができる。
【0125】
キャラクタリゼーション:
金属:RNA生成物を実施例の節に記載されている標準プロトコルに従って等電点電気泳動(IEF)ゲルを用いてイオン性/共有結合性挙動の変化について調べた。IEFゲルの利点は、標的RNA分子上の電荷分布の変化をモニターするための安価なツールを提供することである。IEF研究のデータを製造した各iRNA錯体についてΔpKaとして表した。
【0126】
IEF実験の結論:
異なる等電点(pKa)を有する無水DMSO反応から得られたRNA分子の存在は新しいRNA分子の存在を示している(図31)。これはRNA分解に起因しないので、新しいRNA分子は金属(マグネシウムまたは亜鉛)とRNAの安定な錯体であることを意味している。加えて、そのpKaが天然RNAのpKaよりも低いので、金属とRNA間の共有結合の形成が裏付けられる。
【実施例】
【0127】
DMSO−マグネシウム錯体のFTIR分析
DMSOのいずれの原子がマグネシウムに結合するかを調べるために、DMSO−マグネシウム錯体のFTIRスペクトルを集めた。FTIRスペクトルは954cm−1にS=O−Mg伸縮を示すエキストラ伸縮を示した。T3錯体のFTIRはS=O−Mg、C=O及びN−H伸縮の存在について調べた。
【0128】
ビス(トリヨードサイロニナト)−ビス(ジメチルスルホキシド)マグネシウムの製造
トリヨードサイロニン、すなわちT3(218mg)を無水DMSO(4mL)中に溶解し、その後t−ブタノール中1M カリウムt−ブトキシド(0.34mL)を添加し、溶液を10分間撹拌した。塩化マグネシウム(16mg)を添加し、溶液を一晩撹拌した。溶液を脱イオン水(10mL)中に注いで生成物を沈澱させた。この沈澱を吸引濾過し、風乾させた。高真空下で一晩乾燥した後、164mgの明ベージュ色粉末が生じた。生成物の構造を1H NMR、FAB−MS及びICPによりキャラクタライズした。1H NMR(DMSO):δ 7.83(s),7.05(d),6.82(d),6.62(dd),3.26(bm),3.15(bd),2.71(bm),2.54(s)。側鎖領域に幅広多重項が存在することはマグネシウム結合のための部位を示している(図11参照)。FAB−MS:1325にビス(トリヨードサイロニナト)マグネシウムを示す分子イオン(DMSO配位子のロス後)。FTIR(ニート):cm−1 1596(C=O伸縮),1014(S=O伸縮),949(S=O−Mg伸縮);1633にN−H伸縮の不在。マグネシウム分析(ICP):予測値=1.68%,実測値=1.62%。これは図24に示す構造を確認するものであり、純度は約96%であり、汚染の大部分はおそらく1H NMRに示されている水に起因している。
【0129】
ビス(トリヨードサイロニナト)亜鉛の製造
トリヨードサイロニン、すなわちT3(192mg)を無水DMSO(4mL)中に溶解し、その後t−ブタノール中1M カリウムt−ブトキシド(0.30mL)を添加し、溶液を10分間撹拌した。ジエチルエーテル中塩化亜鉛(0.16mLの1M溶液)を添加し、溶液を一晩撹拌した。溶液を脱イオン水(10mL)中に注いで生成物を沈澱させた。この沈澱を吸引濾過し、風乾した。高真空下で一晩乾燥した後、140mgの明ベージュ色粉末を得た。生成物の構造を1H NMR、FAB−MS及びICPによりキャラクタライズした。1H NMR(DMSO):δ 7.82(s),7.02(d),6.80(d),6.61(dd),3.49(bm),3.22(bm),2.67(bm)。側鎖領域に幅広多重項が存在することは亜鉛結合のための部位を示している(図13)。FAB−MS:1364及び1366に亜鉛と一致する同位体存在度パターンを有するビス(トリヨードサイロニナト)亜鉛を示す分子イオン。FTIR(ニート):cm−1 1582(C=O伸縮);1633にN−H伸縮の不在。亜鉛分析(ICP):予想値=4.8%,実測値=4.3%。これは図25に示す構造を裏付け、純度は約90%であり、汚染の大部分はおそらく水に起因し、1H NMRスペクトルで見られるように水和物であり得る。
【0130】
水の存在下でのマグネシウムトリヨードサイロニン錯体の製造
トリヨードサイロニン、すなわちT3(188mg)を無水DMSO(3.5mL)中に溶解し、その後t−ブタノール中1M カリウムt−ブトキシド(0.29mL)を添加し、溶液を10分間撹拌した。水(0.5mL)中塩化マグネシウム(16mg)を添加し、溶液を一晩撹拌した。溶液を脱イオン水(10mL)中に注いで生成物を沈澱させた。この沈澱を吸引濾過し、風乾した。高真空下で一晩乾燥した後、188mgの明ベージュ色粉末が得られた。生成物構造を1H NMR、FAB−MS及びICPによりキャラクタライズした。1H NMR(DMSO):δ 7.81(bs),7.02(bs),6.81(bd),6.59(dd)。他の共鳴は溶媒ピーク及び水ピークの後に隠れていた。サンプルはDMSO中で曇った懸濁液を形成した。FAB−MS:652にMgが結合していないプロトン化T3を示す分子イオン。FTIR(ニート):cm−1 1633(N−H伸縮),1535(C=O伸縮),1012(S=O伸縮),949(S=O−Mg伸縮)。T3関連伸縮に比したDMSO関連伸縮の強度は(DMSO)xMgカチオン及びT3アニオンの混合物を示していた。マグネシウム分析(ICP):予想値=1.8%,実測値=0.96%。カリウム分析(ICP):実測値=0.23%。ICPデータから、この生成物はマグネシウム塩、カリウム及び双性イオンの混合物であるようである。
【0131】
ビス(ミノサイクリナト)マグネシウムの製造
ミノサイクリン(104mg)を無水DMSO(3mL)中に溶解し、その後t−ブタノール中1M カリウムt−ブトキシド(0.44mL)を添加し、溶液を10分間撹拌した。塩化マグネシウム(11mg)を添加し、溶液を一晩撹拌した。溶液を脱イオン水(10mL)中に注いで生成物を沈澱させた。この沈澱を吸引濾過し、風乾した。高真空下で一晩乾燥して、52mgの深黄色粉末を得た。生成物の構造は、おそらくアニオン性ミノサイクリン及びマグネシウムがとりうる二座錯体形態の各種順列のために1H NMRによりキャラクタライズすることができなかった。その後、生成物をFAB−MS及びICPによりキャラクタライズした。FAB−MS:937.4にビス(ミノサイクリナト)マグネシウムを示す分子イオン。マグネシウム分析(ICP):予想値=2.68%,実測値=2.61%。このことから、図26に示すもっともらしい構造により表されるようにマグネシウム1原子あたり2つのミノサイクリンがあることが確認される(以下のテトラサイクリンに関する検討参照)。生成物の純度は約97%であり、汚染の大部分はおそらく1H NMRスペクトルで見られる水に起因する。
【0132】
ビス(テトラサイクリナト)マグネシウムの製造
テトラサイクリン(89mg)を無水DMSO(0.5mL)中に溶解し、その後t−ブタノール中1M カリウムt−ブトキシド(0.2mL)を添加し、溶液を10分間撹拌した。塩化マグネシウム(11mg)を添加し、溶液を3時間撹拌した。溶液を真空中30℃で濃縮し、その後脱イオン水(0.5mL)を添加し、混合物を摩砕し、2mLマイクロ遠心管に移した。8,000rpmで6分間遠心分離することにより生成物を液体から分離し、上清をデカントした。水(0.5mL)を添加し、渦状に混合し、遠心分離し、上清をデカントすることによりペレットを洗浄した。洗浄手順を繰り返した。高真空下で一晩乾燥した後、72mgの深黄色粉末を得た。1H NMRスペクトル(図20)はポリマー構造に似ており、8.8〜10.1ppm、6.4〜7.8ppm、4.2〜5.0ppm及び1.1〜3.1ppmに幅広多重項を含んでいた。生成物構造は、おそらくアニオン性テトラサイクリン及びマグネシウムがとりうる二座錯体形態の各種順列及びこれらの異性体錯体形態間での適度にゆっくりした平衡のために1H NMRにより正確にキャラクタライズすることができなかった。NMRサンプルに約1当量の12N HClを添加し、1H NMRにより再分析することにより確認が観察された。これは、マグネシウム錯体がテトラサイクリン及びおそらく塩化マグネシウムに逆戻りしたことを示した(図18)。生成物を更にMALDI−ES及びICPによりキャラクタライズした。MALDI−ES:911.3にビス(テトラサイクリナト)マグネシウムを示す分子イオン。マグネシウム分析(ICP):予測値=2.67%,実測値=2.53%。FTIRはDMSO配位子の存在を示さない。これは、マグネシウム1原子あたり2個のテトラサイクリン分子があることを示している。水性系中でのテトラサイクリン−マグネシウム錯体形成の以前に公表された研究からのNMR証拠に従って、ビス(テトラサイクリナト)マグネシウムのとりうる構造を図27に示す。生成物の純度は約95%であり、汚染の大部分はおそらく1H NMRスペクトルで見られる溶媒に起因している。
【0133】
水中でのマグネシウムテトラサイクリン錯体の製造
テトラサイクリン(89mg)を水(1.5mL)中に溶解し、その後t−ブタノール中1M カリウムt−ブトキシド(0.2mL)を添加し、溶液を10分間撹拌した。塩化マグネシウム(0.11mLの1M溶液)を添加し、溶液を3時間撹拌した。8,000rpmで6分間遠心分離することにより生じた沈澱を水から分離し、上清をデカントした。ペレットを水(1mL)を添加し、渦状に混合し、遠心分離し、上清をデカントすることにより洗浄した。高真空中で一晩乾燥した後、65mgの深黄色粉末が得られた。1H NMRスペクトルは無水DMSO中で製造した錯体に非常に似ていた。マグネシウム分析(ICP):予測値=2.67%,実測値=2.42%。水中での錯体形成対無水条件下での錯体形成はテトラサイクリン−マグネシウム錯体の安定性及び構造に対して殆ど影響を持たないようである。
【0134】
ヒドロクロロチアジド亜鉛錯体の製造
ヒドロクロロチアジド(120mg)を無水DMSO(0.5mL)中に溶解し、その後t−ブタノール中1M カリウムt−ブトキシド(0.4mL)を添加し、溶液を10分間撹拌した。ジエチルエーテル中塩化亜鉛(0.2mLの1M溶液)を添加し、溶液を4時間撹拌した。溶液を真空中30℃で濃縮した後、メタノール(0.5mL)を添加し、混合物を摩砕し、2mLマイクロ遠心管に移した。8,000rpmで6分間遠心分離することにより生成物を液体から分離し、上清をデカントした。ペレットをメタノール(0.5mL)を添加し、渦状に混合し、遠心分離し、上清をデカントすることにより洗浄した。洗浄手順を繰り返した。高真空下で一晩乾燥した後、104mgの自由流動性白色粉末を得た。生成物は、周囲空気に数分間さらした後粉末がガム状に変わったために明らかに吸湿性であった。1H NMRスペクトルはポリマー構造に似ており(図21)、6.3〜8.0ppm及び4.4〜5.9ppmの幅広多重項を含んでいた。生成物の構造は、おそらくアニオン性ヒドロクロロチアジド及び亜鉛がとりうる錯体形態の各種順列及び異性体錯体形態間での適度にゆっくりした平衡のために1H NMRにより正確にキャラクタライズできなかった。約1当量の12N HClをNMRサンプルに添加し、1H NMRにより再分析することにより確認を観察した。この分析は亜鉛錯体がヒドロクロロチアジド及びおそらく塩化亜鉛に逆戻りしたことを示す(図22)。生成物を更にMALDI−ES及びICPによりキャラクタライズした。MALDI−ES:705.9及び932.9に典型的な亜鉛同位体存在度を有するが特定のヒドロクロロチアジド亜鉛錯体構造を示さない分子イオン。亜鉛分析(ICP):実測値=10.4%。FTIR(ニート):cm−1 1027(S=O伸縮),953(S=O−Zn伸縮);1646にN−H伸縮の不在。NMR、MALDI及びICPデータは明らかにヒドロクロロチアジド−亜鉛錯体の形成を示している。錯体形成の部位はヒドロクロロチアジドのスルホンアミドの窒素の一方及び両方上にあり得ることは合理的であるようである。FTIRデータはDMSO配位子の存在を示唆しており、1H NMR積分値によればHCTZ:DMSO比は1:1である。0.08ppmのDMSOメチル基の化学シフトから、亜鉛とDMSO間のO−結合が示唆される。
【0135】
水の存在下での亜鉛とヒドロクロロチアジドの反応
この手順は、塩化亜鉛を水(100μL)に添加し、反応混合物に添加する前にエーテルを蒸発除去させた以外は同様の無水製造に従った。全混合物が沈澱ステップ中にメタノール中にゆっくり溶解するために生成物は単離されなかった。これは推定上メタノール可溶性水和錯体が形成されたためであった。
【0136】
ジメチルビグアニド亜鉛錯体の製造
ジメチルビグアニド(66mg)を無水DMSO4(1mL)中に溶解し、その後t−ブタノール1M カリウムt−ブトキシド(0.88mL)を添加し、溶液を10分間撹拌した。ジエチルエーテル中塩化亜鉛(0.22mLの1M溶液)を添加し、溶液を3時間撹拌した。溶液を真空中35℃で濃縮し、その後エタノール(0.5mL)を添加し、混合物を摩砕し、2mLマイクロ遠心管に移した。8,000rpmで6分間遠心分離することにより生成物を液体から分離し、上清をデカントした。ペレットをエタノール(0.5mL)を添加し、渦状に混合し、遠心分離し、上清をデカントすることにより洗浄した。洗浄手順を繰り返した。高真空下で一晩乾燥した後、57mgの自由流動性白色粉末を得た。1H NMR(DMSO):δ 4.90(s),4.66(s),4.50(s),2.80(s)。ジメチルビグアニドに比して−NHプロトンが大きく高磁場シフトしていることは亜鉛との錯体形成を示している。ジメチルビグアニド−亜鉛錯体ではジメチル基中の0.05ppmの小さな高磁場シフトも観察された。ジメチルビグアニド−亜鉛及びジメチルビグアニドのNMRスペクトルをそれぞれ図14及び15に示す。生成物を更にICPによりキャラクタライズした。亜鉛分析(ICP):実測値=8.14%。FTIRデータはDMSO配位子の存在を示さなかった。NMR及びICPデータは、明らかにジメチルビグアニド−亜鉛錯体の形成を示している。図28は製造したビグアニド−金属錯体を表している。MALDI−ES分析は亜鉛含有化合物を表さなかった。
【0137】
水の存在下での亜鉛ジメチルビグアニド錯体の製造
この手順は、塩化亜鉛を水(100μL)に添加し、反応混合物に添加する前にエーテルを蒸発させた以外は類似の無水製造に従った。同一方法で後処理し、乾燥させて、47mgの自由流動性白色粉末を得た。この化合物はDMSO中に僅かしか溶解せず、NMRにおいて低い信号対雑音比が生じた。1H NMR(DMSO):δ 4.90(bs),4.66(bs),4.50(bs),2.85(s),2.80(s)。2.85ppmの一重項は亜鉛と錯体形成していないジメチルビグアニドの存在を示している。2.85及び2.80ppmの2つのピークの積分値から判断して、遊離ジメチルビグアニド/亜鉛錯体の比は約1:1である。
【0138】
水中での亜鉛ジメチルビグアニド錯体の製造
ジメチルビグアニド(33mg)を水(1mL)中に溶解し、その後t−ブタノール中1M カリウムt−ブトキシド(0.44mL)を添加し、溶液を10分間撹拌した。ジエチルエーテル中塩化亜鉛(0.22mLの1M溶液)を添加し、溶液を5時間撹拌した。8,000rpmで6分間遠心分離することにより生じた沈澱を液体から分離し、上清をデカントした。ペレットを水(1mL)を添加し、渦状に混合し、遠心分離し、上清をデカントすることにより洗浄した。高真空下で一晩乾燥した後、20mgの自由流動性白色粉末を得た。この粉末は1H NMRによれば有機物質を含んでいなかった。単離された生成物は各種水和形態の亜鉛塩であった。
【0139】
ビス(アシクログアノシナト)マグネシウムの製造
アシクログアノシン(45mg)を無水DMSO(0.5mL)中に溶解し、その後t−ブタノール中1M カリウムt−ブトキシド(0.2mL)を添加し、溶液を10分間撹拌した。塩化マグネシウム(11mg)を添加し、溶液を4時間撹拌した。溶液を真空中30℃で濃縮し、その後メタノール(0.5mL)を添加し、混合物を摩砕し、2mLマイクロ遠心管に移した。8,000rpmで6分間遠心分離することにより生成物を液体から分離し、上清をデカントした。ペレットをメタノール(0.5mL)を添加し、渦状に混合し、遠心分離し、上清をデカントすることにより洗浄した。洗浄手段を繰り返した。高真空下で一晩乾燥した後、15mgのベージュ色の粗い粉末を得た。生成物の構造を1H NMR、MALDI−ES及びICPによリキャラクタライズした。1H NMR(DMSO):δ 7.70(s),6.75(bs),5.30(s),4.64(bs),3.42(s)。MALDI−ES:473.1の分子イオンはビス(アシクログアノシン)マグネシウムを示唆している。666、702、893及び1065の他の分子イオンは特定の構造に帰属されなかった。マグネシウム分析(ICP):予想値=5.14%,実測値=4.16%。FTIRデータはDMSO配位子の存在を示さなかった。10.6ppmにアミドプロトンが存在しないことと、ICP分析及びMALDI−ES分析を併せて、ビス(アシクログアノシナト)マグネシウムの構造を図29に示す。
【0140】
T3錯体、テトラサイクリン錯体及び参照薬物の分配係数、分布係数及びpKa
pKa及びlogPを電位差測定法及び分光光度法により測定した。電位差測定法はアナライトの簡単な酸及び塩基滴定からpKa及びlogPを計算するために専門ソフトウェアを使用することを含む。pKaをまず約2mgの純粋物質をアッセイバイアルに入れて秤量して測定した。イオン強度を0.15M KClを用いて調節し、化合物を溶解させるために水を添加し、次いでpHを所望の出発値に低下または上昇させるために酸または塩基滴定剤を添加した。次いで、溶液を酸(0.5N HCl)または塩基(0.5N NaOH)を用いて最終pHまで滴定した。概算のpKa値を示し、後に正確なデータに絞り込んだ。
【0141】
水に僅かしか溶解しない(T3)2Znについては、pKaを水とDMSO補助溶媒の混合物中で測定した。水/DMSOの3つの比の最小値を滴定してpsKa(補助溶媒の存在下での見かけpKa)を得た。水性pKaをYasuda-Shedlovsky技術を用いて外挿することにより求めた。
【0142】
logPは、オクタノール(水飽和)の存在下での滴定により求めた。水中のpKa及びオクタノールの存在下での見かけpKa(poKa)を比較し、logPを求めた。別の容量のオクタノールの存在下で更に滴定して、イオンペアリング(オクタノール中への荷電種の分配、logP+またはlogP−と称される)を求めた。実験的に測定したpKa及びlogPを用いて、薬物の親油性プロファイル(logD対pH)を計算した。logD7.4はpH7.4でのこのプロファイルから求めた。
【0143】
分光光度法は、光ファィバーディッププローブ、UV光源(パルス型重水素ランプ)及び酸または塩基溶液の添加中にサンプル溶液の吸収スペクトルを自動的に捕らえるためのフォトダイオードアレイ検出器を使用した。
【0144】
最高10mMのストック溶液を、幾つかの0.5mgサンプルを0.5〜1.0mLの水または補助溶媒中に溶解させることにより調製した。滴定のために十分量のストックをバイアルにピペットで添加する。ディッププローブをアッセイバイアルに入れ、ディッププローブをカバーするために0.15M KCl水溶液を添加する。pHを所望出発値とするために酸または塩基を添加する。選択したpH範囲で、その後の分析のためにイオン化に起因するスペクトルの変化をフォトダイオードアレイ検出器で捕らえた。サンプルのpKa値を推理し、還元種の主要吸収スペクトルを分析するために多変量解析(TFA)を適用した。Yasuda-Shedlovsky技術を用いる外挿により水性pKaを求めた。分光光度実験から得たpKa値は電位差滴定から得た値と非常にうまく一致している。
【0145】
バイオアベイラビリティー研究
表題:Sprague Dawleyラットでのホルモン錯体栄養補充の吸収及び効果の評価
被験体:15匹の若い雌Sprague Dawleyラット(180〜225g)を使用した。これらのラットは業者(バージニア州ダブリンに所在のHarlan Laboratory Animals)から入手し、Litton Reeves Hall(Division of Laboratory Animal Resources)のビバリウムにポリプロピレンシューボックスケージ中に1群3匹で収容した。飲水は自由とした。ラットには自由に認可げっ歯類固形飼料を与えた。到着後、ラットの健康を評価し、動物を最低5日間隔離して置いた。この間に全身健康状態を調べた。隔離後、ラットをアクセス及び研究のために耐久性動物小屋に移動させた。
【0146】
研究計画:これは、ラットに対して食道に直接経口投与した3つのホルモンの吸収及び効果を比較するための研究である。2つの試験化合物であるT3−亜鉛及びT3−マグネシウムを合成した。トリヨードサイロニン遊離酸(T3)はポジティブ対照であった。3つの化合物を別々にゼラチンカプセル剤に製剤化し、108±12μg/kgの全用量で投与した。試験化合物のラットの胃での酸分解を避けるために、製剤に金属酸化物を添加した。T3−マグネシウム化合物に対しては1.08±0.13mgの酸化マグネシウムを添加し、T3−亜鉛化合物に対しては106±13μgの酸化亜鉛を添加した。T3遊離酸に145±50μgの酸化亜鉛を添加した別のT3対照を含めた。
【0147】
投与し、採血するために、反復実験1回あたり3匹のラットを麻酔した。基準血液サンプルを後眼窩サンプリングにより採取した。次いで、化合物を胃管栄養チューブにより経口投与した。投与後、血液サンプル(500μL)を投与から0.5、1、2、2.5、5時間目に採取した。血液サンプルを遠心分離し、血清を除去し、血清を氷上に保持した後、血清トリヨードサイロニンレベルについて分析した。
【0148】
サンプルの分析:血清トリヨードサイロニンレベルをRIAにより測定した。
【0149】
結果:各ラット群からの個々の血清T3レベルの平均を求め、T3濃度(ng/mL)対時のプロットを作成した。このプロットを図30に示す。
【0150】
大分子の例
干渉RNAの製造:
干渉RNAを修飾New England Biolabs Litmus 28i RNAi双方向転写ベクターを用いて製造した。922bpウシ血清アルブミンcDNA断片をLitmus RNAiベクターのBglII部位及びStuI部位に導入した。T7 RNAポリメラーゼを用いるインビトロ転写により標的RNAi転写物を製造して、1mg/mlを得た。次いで、RNAを50ugサンプルに分割し、凍結乾燥した。
【0151】
RNA:マグネシウム内部配位(共有結合性)錯体の製造:
iRNA(約50μg)を50℃で無水DMSO(100μL)中に溶解した。無水塩化マグネシウム(19mg)を無水DMSO中に溶解することにより4mM 塩化マグネシウムのストック溶液を調製した。3つの別々のiRNAのDMSO溶液に15μL、30μL及び60μLの4mM 塩化マグネシウムを添加して、3つの別々の反応を行ったた。溶液を時々渦状に混合しながら室温で90分間放置し、この後7.5M 水性塩化アンモニウム(20μL)、次いでRNAアーゼ非含有エタノール(400μL)を添加し、渦状に混合した。生成物を1時間かけて溶液から沈澱させ、遠心分離し、液体をペレットからデカントした。このペレットをRNAアーゼ非含有エタノール(100μL)で洗浄し、渦状に混合し、遠心分離し、デカントによりエタノール上清をペレットから分離した。生じた無色ペレットを数分間風乾した後、等電点電気泳動ゲルで試験した。
【0152】
マグネシウム:RNA外部配位(イオン性)錯体の製造:
塩化マグネシウムストック溶液を無水DMSOではなくRNAアーゼ非含有水を用いて調製した以外は共有結合性類似体の手順を正確に追跡して、イオン性マグネシウム:RNA錯体を製造した。生じた無色ペレットを数分間風乾した後、等電点電気泳動ゲルで試験した。
【0153】
RNA:亜鉛内部配位(共有結合性)錯体の製造:
塩化マグネシウムストック溶液ではなく塩化亜鉛ストック溶液を調製した以外はマグネシウム類似体の手順を正確に追跡して、共有結合性亜鉛:RNA錯体を製造した。生じた無色ペレットを数分間風乾した後、等電点電気泳動ゲルで試験した。
【0154】
亜鉛:RNA外部配位(イオン性)錯体の製造:
塩化亜鉛ストック溶液を無水DMSOではなくRNAアーゼ非含有水を用いて調製した以外は共有結合性類似体の手順を正確に追跡して、イオン性亜鉛:RNA錯体を製造した。生じた無色ペレットを数分間風乾した後、等電点電気泳動ゲルで試験した。
【0155】
等電点電気泳動ゲル実験:
これは、iRNA標的の予測される修飾をモニターするためにIEFゲルを用いる新規な方法である。図31は初期実験の結果を示している。
【0156】
IEF実験の解釈:
IEF実験から、3種の塩化マグネシウム濃度で無水(A)条件で製造したマグネシウムRNA錯体は約50%の収率で共有結合性錯体を生じたことが分かった。3種の塩化マグネシウム濃度で水性(W)条件下で製造したマグネシウムRNA錯体はイオン性錯体を生じた。塩化亜鉛を用いて無水(A)条件で製造した亜鉛RNA錯体は約50%の収率で共有結合性錯体を生じた。塩化亜鉛を用いて水性(W)条件で製造した亜鉛RNA錯体はイオン性錯体を生じた。
【図面の簡単な説明】
【0157】
【図1】従来技術によるマグネソセンの構造を示す。
【図2】従来技術による(シクロペンタジエニル)−tブチルメチルビス(N,N’−[2,6−ジイソプロピルフェニル]アミジナート)マグネシウムの構造を示す。
【図3】従来技術によるマグネシウム:サリチルアルデヒド錯体の構造を示す。
【図4】従来技術によるマグネシウムフタロシアニンの構造を示す。
【図5】従来技術による外圏RNA:マグネシウム配位錯体を示す。
【図6】本発明による内圏RNA:マグネシウム配位錯体を示す。
【図7】本発明によるRNA:マグネシウム:アルギニン配位錯体を示す。
【図8】本発明による置換アルギニン:マグネシウム錯体を示す。
【図9】本発明による内圏でマグネシウムと錯体形成したサリチル酸とポリマー結合アルギニン及び外圏で配位子:金属錯体を封入したペプチドを示す。
【図10】本発明によるマグネシウム:オキシコドン錯体を示す。
【図11】本発明によるビス(トリヨードサイロニナト)−ビス(ジメチルスルホキシド)マグネシウムのプロトンNMRを示す。
【図12】従来技術によるトリヨードサイロニン(T3)のプロトンNMRを示す。
【図13】本発明によるビス(トリヨードサイロニナト)亜鉛のプロトンNMRを示す。
【図14】本発明によるジメチルビグアニド:亜鉛錯体のプロトンNMRを示す。
【図15】従来技術によるジメチルビグアニドのプロトンNMRを示す。
【図16】従来技術によるテトラサイクリンのプロトンNMRを示す。
【図17】本発明によるビス(テトラサイクリナト)マグネシウムのプロトンNMRを示す。
【図18】本発明による1N HClを添加したテトラサイクリン−マグネシウム錯体のプロトンNMRを示す。
【図19】本発明による1N HClを添加したテトラサイクリンのプロトンNMRを示す。
【図20】従来技術によるヒドロクロロチアジドのプロトンNMRを示す。
【図21】本発明によるヒドロクロロチアジド−亜鉛錯体のプロトンNMRを示す。
【図22】本発明による1N HClを添加したヒドロクロロチアジド−亜鉛錯体のプロトンNMRを示す。
【図23】本発明によるビス(アシクログアノシナト)マグネシウムのプロトンNMRを示す。
【図24】本発明によるビス(トリヨードサイロニナト)−ビス(ジメチルスルホキシド)マグネシウムの構造を示す。
【図25】本発明によるビス(トリヨードサイロニナト)亜鉛の構造を示す。
【図26】本発明によるビス(ミノサイクリナト)マグネシウムの構造を示す。
【図27】本発明によるビス(テトラサイクリナト)マグネシウムの構造を示す。
【図28】本発明によるジメチルビグアニド−亜鉛錯体の構造を示す。
【図29】本発明によるビス(アシクログアノシナト)マグネシウムの構造を示す。
【図30】本発明によるラット動物モデルにおけるT3、T3Mg、T3Znの相対的薬物動態プロファイルを示す。
【図31】本発明によるマグネシウム及び亜鉛iRNA錯体のIEFプロファイルを示す。“A”を付した列は無水条件で製造した錯体を指す。“W”を付した列は水中で製造した錯体を指す。
【特許請求の範囲】
【請求項1】
サイロニン、テトラサイクリン系抗生物質、ジメチルビグアニド、ヒドロクロロチアジド、アシクログアノシン、ヒドロコドン、オキシコドン及びそれらの誘導体からなる群から選択される生物活性部分とアルミニウム、ビスマス、カルシウム、鉄、マグネシウム、ケイ素及び亜鉛からなる群から選択される金属との金属配位錯体。
【請求項2】
生物活性部分がサイロニン及びその誘導体からなる群から選択される請求項1に記載の錯体。
【請求項3】
錯体がビス(トリヨードサイロニナト)亜鉛である請求項2に記載の錯体。
【請求項4】
錯体がビス(トリヨードサイロニナト)マグネシウムである請求項2に記載の錯体。
【請求項5】
生物活性部分がテトラサイクリン系抗生物質及びその誘導体からなる群から選択される請求項1に記載の錯体。
【請求項6】
錯体がビス(テトラサイクリナト)マグネシウムである請求項4に記載の錯体。
【請求項7】
生物活性部分がオキシコドン及びその誘導体からなる群から選択される請求項1に記載の錯体。
【請求項8】
錯体が(オキシコドン)マグネシウムである請求項7に記載の錯体。
【請求項9】
錯体が(オキシコドン)亜鉛である請求項7に記載の錯体。
【請求項10】
生物活性部分がヒドロコドン及びその誘導体からなる群から選択される請求項1に記載の錯体。
【請求項11】
錯体がビス(ヒドロコドナト)マグネシウムである請求項10に記載の錯体。
【請求項12】
錯体がビス(ヒドロコドナト)亜鉛である請求項10に記載の錯体。
【請求項13】
生物活性部分がペニシリン及びその誘導体からなる群から選択される請求項1に記載の錯体。
【請求項14】
錯体がビス(ペニシリナト)亜鉛である請求項13に記載の錯体。
【請求項15】
錯体がビス(ペニシリナト)マグネシウムである請求項13に記載の錯体。
【請求項16】
生物活性部分がジメチルビグアニド及びその誘導体からなる群から選択される請求項1に記載の錯体。
【請求項17】
錯体がビス(ジメチルビグアニド)亜鉛である請求項16に記載の錯体。
【請求項18】
生物活性部分がヒドロクロロチアジド及びその誘導体からなる群から選択される請求項1に記載の錯体。
【請求項19】
錯体がビス(ヒドロクロロチアジド)亜鉛である請求項18に記載の錯体。
【請求項20】
生物活性部分がアシクログアノシン及びその誘導体からなる群から選択される請求項1に記載の錯体。
【請求項21】
錯体がビス(アシクログアノシナト)マグネシウムである請求項19に記載の錯体。
【請求項22】
生物活性部分がフロセミド及びその誘導体からなる群から選択される請求項1に記載の錯体。
【請求項23】
錯体がビス(フロセミダト)亜鉛である請求項22に記載の錯体。
【請求項24】
錯体がビス(フロセミダト)マグネシウムである請求項22に記載の錯体。
【請求項25】
生物活性部分がDOPA及びその誘導体からなる群から選択される請求項1に記載の錯体。
【請求項26】
錯体がビス(DOPA)亜鉛である請求項25に記載の錯体。
【請求項27】
錯体がビス(DOPA)マグネシウムである請求項25に記載の錯体。
【請求項28】
生物活性部分がアレンドロネート及びその誘導体からなる群から選択される請求項1に記載の錯体。
【請求項29】
錯体がビス(アレンドロナト)亜鉛である請求項28に記載の錯体。
【請求項30】
錯体がビス(アレンドロナト)マグネシウムである請求項28に記載の錯体。
【請求項31】
β−ジケトン、ケトフェノールまたはβ−ケトアルコール官能基を含む生物活性部分とアルミニウム、ビスマス、カルシウム、鉄、マグネシウム、ケイ素及び亜鉛からなる群から選択される金属との金属配位錯体。
【請求項32】
アルコール及びアゾール官能基を含む生物活性部分とアルミニウム、ビスマス、カルシウム、鉄、マグネシウム、ケイ素及び亜鉛からなる群から選択される金属との金属配位錯体。
【請求項33】
グアニドまたはジアミン官能基を含む生物活性部分とアルミニウム、ビスマス、カルシウム、鉄、マグネシウム、ケイ素及び亜鉛からなる群から選択される金属との金属配位錯体。
【請求項34】
生物活性部分及び金属を用いて金属配位錯体を形成することを含む生物活性部分の特性のモジュレート方法であって、
前記生物活性部分をサイロニン、テトラサイクリン系抗生物質、ヒドロコドン、オキシコドン及びそれらの誘導体からなる群から選択し、
前記金属をアルミニウム、ビスマス、カルシウム、鉄、マグネシウム、ケイ素及び亜鉛からなる群から選択する前記方法。
【請求項35】
更に、生物活性物質に対して所望の性能パラメータを付与するペプチド、炭水化物または脂質からなる群から選択されるアジュバントを添加することを含む請求項34に記載の方法。
【請求項36】
モジュレートしようとする特性が有効性、安定性、吸収性、標的デリバリー及びそれらの組合せからなる群から選択される請求項34に記載の方法。
【請求項1】
サイロニン、テトラサイクリン系抗生物質、ジメチルビグアニド、ヒドロクロロチアジド、アシクログアノシン、ヒドロコドン、オキシコドン及びそれらの誘導体からなる群から選択される生物活性部分とアルミニウム、ビスマス、カルシウム、鉄、マグネシウム、ケイ素及び亜鉛からなる群から選択される金属との金属配位錯体。
【請求項2】
生物活性部分がサイロニン及びその誘導体からなる群から選択される請求項1に記載の錯体。
【請求項3】
錯体がビス(トリヨードサイロニナト)亜鉛である請求項2に記載の錯体。
【請求項4】
錯体がビス(トリヨードサイロニナト)マグネシウムである請求項2に記載の錯体。
【請求項5】
生物活性部分がテトラサイクリン系抗生物質及びその誘導体からなる群から選択される請求項1に記載の錯体。
【請求項6】
錯体がビス(テトラサイクリナト)マグネシウムである請求項4に記載の錯体。
【請求項7】
生物活性部分がオキシコドン及びその誘導体からなる群から選択される請求項1に記載の錯体。
【請求項8】
錯体が(オキシコドン)マグネシウムである請求項7に記載の錯体。
【請求項9】
錯体が(オキシコドン)亜鉛である請求項7に記載の錯体。
【請求項10】
生物活性部分がヒドロコドン及びその誘導体からなる群から選択される請求項1に記載の錯体。
【請求項11】
錯体がビス(ヒドロコドナト)マグネシウムである請求項10に記載の錯体。
【請求項12】
錯体がビス(ヒドロコドナト)亜鉛である請求項10に記載の錯体。
【請求項13】
生物活性部分がペニシリン及びその誘導体からなる群から選択される請求項1に記載の錯体。
【請求項14】
錯体がビス(ペニシリナト)亜鉛である請求項13に記載の錯体。
【請求項15】
錯体がビス(ペニシリナト)マグネシウムである請求項13に記載の錯体。
【請求項16】
生物活性部分がジメチルビグアニド及びその誘導体からなる群から選択される請求項1に記載の錯体。
【請求項17】
錯体がビス(ジメチルビグアニド)亜鉛である請求項16に記載の錯体。
【請求項18】
生物活性部分がヒドロクロロチアジド及びその誘導体からなる群から選択される請求項1に記載の錯体。
【請求項19】
錯体がビス(ヒドロクロロチアジド)亜鉛である請求項18に記載の錯体。
【請求項20】
生物活性部分がアシクログアノシン及びその誘導体からなる群から選択される請求項1に記載の錯体。
【請求項21】
錯体がビス(アシクログアノシナト)マグネシウムである請求項19に記載の錯体。
【請求項22】
生物活性部分がフロセミド及びその誘導体からなる群から選択される請求項1に記載の錯体。
【請求項23】
錯体がビス(フロセミダト)亜鉛である請求項22に記載の錯体。
【請求項24】
錯体がビス(フロセミダト)マグネシウムである請求項22に記載の錯体。
【請求項25】
生物活性部分がDOPA及びその誘導体からなる群から選択される請求項1に記載の錯体。
【請求項26】
錯体がビス(DOPA)亜鉛である請求項25に記載の錯体。
【請求項27】
錯体がビス(DOPA)マグネシウムである請求項25に記載の錯体。
【請求項28】
生物活性部分がアレンドロネート及びその誘導体からなる群から選択される請求項1に記載の錯体。
【請求項29】
錯体がビス(アレンドロナト)亜鉛である請求項28に記載の錯体。
【請求項30】
錯体がビス(アレンドロナト)マグネシウムである請求項28に記載の錯体。
【請求項31】
β−ジケトン、ケトフェノールまたはβ−ケトアルコール官能基を含む生物活性部分とアルミニウム、ビスマス、カルシウム、鉄、マグネシウム、ケイ素及び亜鉛からなる群から選択される金属との金属配位錯体。
【請求項32】
アルコール及びアゾール官能基を含む生物活性部分とアルミニウム、ビスマス、カルシウム、鉄、マグネシウム、ケイ素及び亜鉛からなる群から選択される金属との金属配位錯体。
【請求項33】
グアニドまたはジアミン官能基を含む生物活性部分とアルミニウム、ビスマス、カルシウム、鉄、マグネシウム、ケイ素及び亜鉛からなる群から選択される金属との金属配位錯体。
【請求項34】
生物活性部分及び金属を用いて金属配位錯体を形成することを含む生物活性部分の特性のモジュレート方法であって、
前記生物活性部分をサイロニン、テトラサイクリン系抗生物質、ヒドロコドン、オキシコドン及びそれらの誘導体からなる群から選択し、
前記金属をアルミニウム、ビスマス、カルシウム、鉄、マグネシウム、ケイ素及び亜鉛からなる群から選択する前記方法。
【請求項35】
更に、生物活性物質に対して所望の性能パラメータを付与するペプチド、炭水化物または脂質からなる群から選択されるアジュバントを添加することを含む請求項34に記載の方法。
【請求項36】
モジュレートしようとする特性が有効性、安定性、吸収性、標的デリバリー及びそれらの組合せからなる群から選択される請求項34に記載の方法。
【図1】

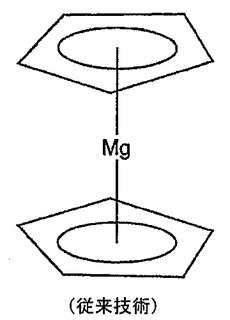
【図2】
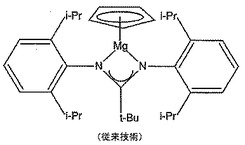
【図3】
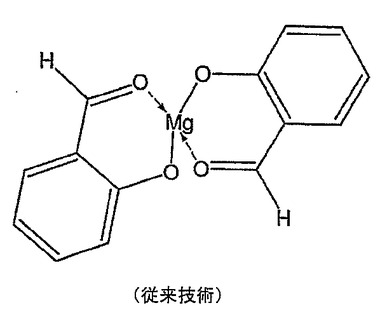
【図4】
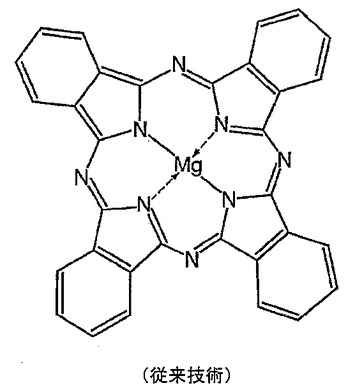
【図5】
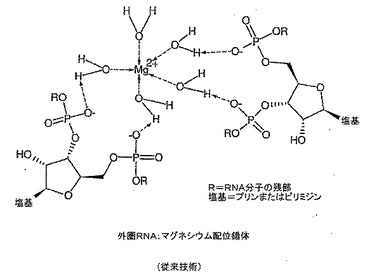
【図6】
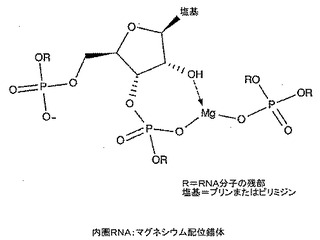
【図7】
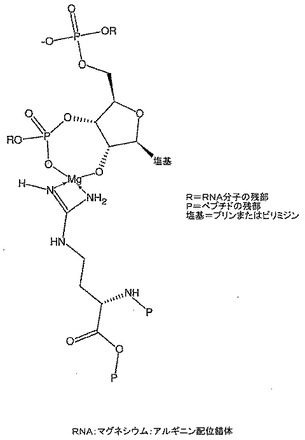
【図8】
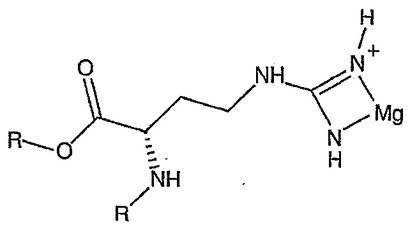
【図9】
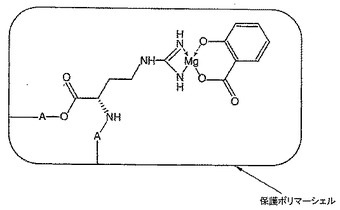
【図10】
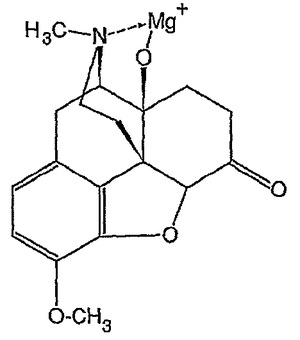
【図11】
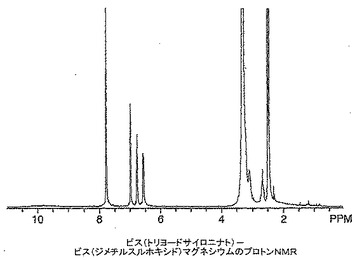
【図12】
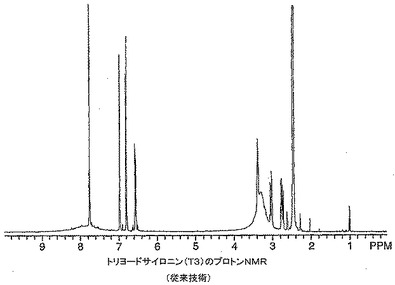
【図13】
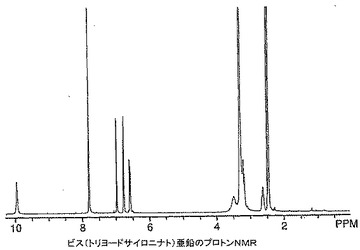
【図14】
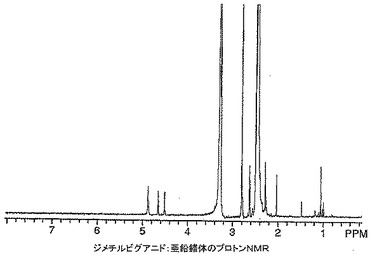
【図15】
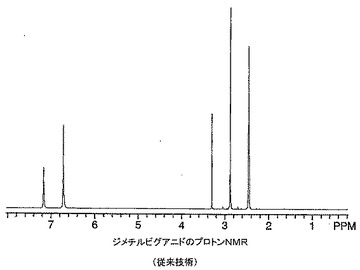
【図16】
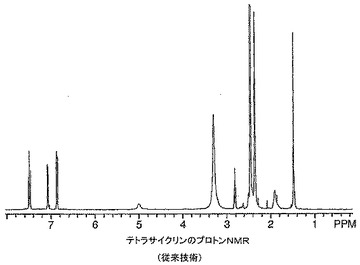
【図17】
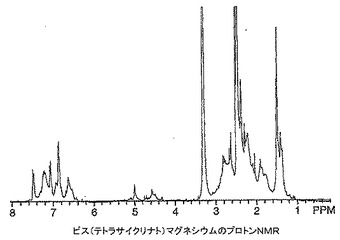
【図18】
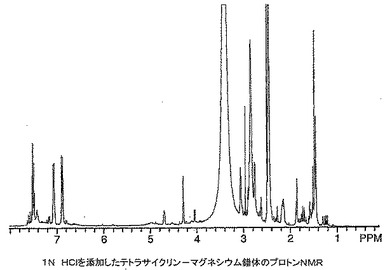
【図19】
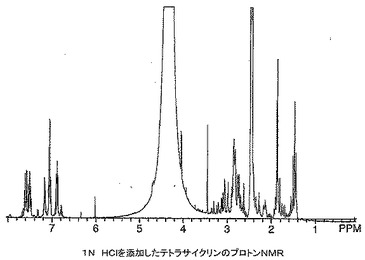
【図20】
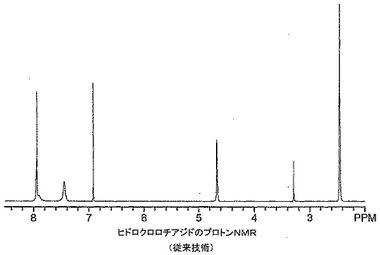
【図21】
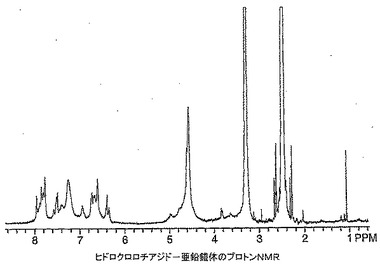
【図22】
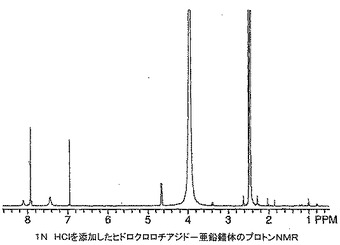
【図23】
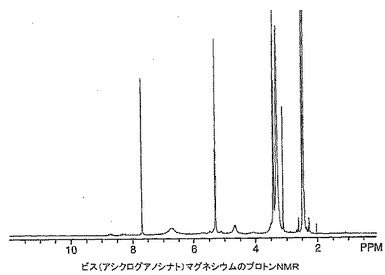
【図24】
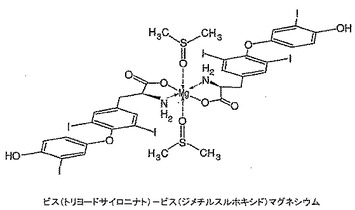
【図25】
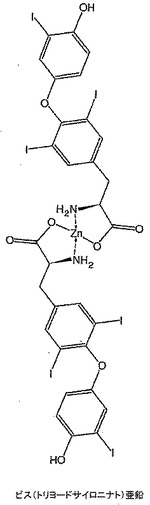
【図26】
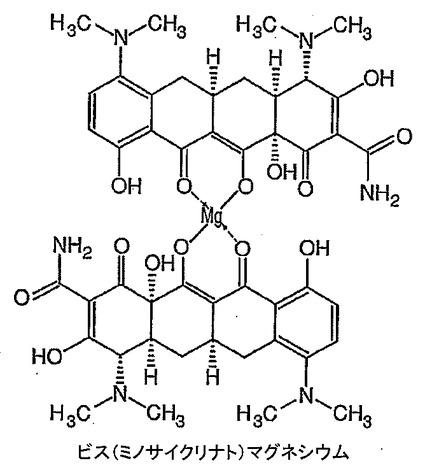
【図27】
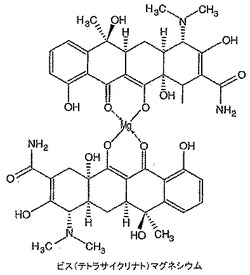
【図28】
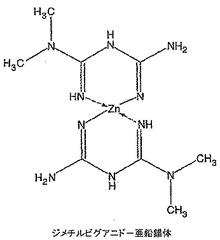
【図29】
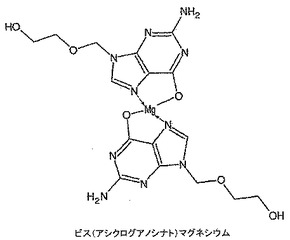
【図30】
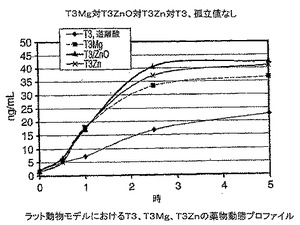
【図31】
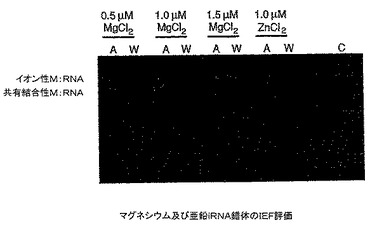
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【図31】
【公表番号】特表2009−512728(P2009−512728A)
【公表日】平成21年3月26日(2009.3.26)
【国際特許分類】
【出願番号】特願2008−537702(P2008−537702)
【出願日】平成18年8月15日(2006.8.15)
【国際出願番号】PCT/US2006/031757
【国際公開番号】WO2007/050181
【国際公開日】平成19年5月3日(2007.5.3)
【出願人】(508125531)シントニクス,インコーポレーテッド (1)
【Fターム(参考)】
【公表日】平成21年3月26日(2009.3.26)
【国際特許分類】
【出願日】平成18年8月15日(2006.8.15)
【国際出願番号】PCT/US2006/031757
【国際公開番号】WO2007/050181
【国際公開日】平成19年5月3日(2007.5.3)
【出願人】(508125531)シントニクス,インコーポレーテッド (1)
【Fターム(参考)】
[ Back to top ]