
針状の活性物質を含む押出物
本発明は、針状医薬活性物質の粒径とストランド径との比が少なくとも1:15である少なくとも1種の針の形の医薬活性物質を含む押出物、および、医薬を製造するための該押出物の使用に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、針状医薬活性物質の粒径とストランド径(strand diameter)との比が少なくとも1:15である少なくとも1種の針状晶の形態の医薬活性物質を含有する押出物、および、医薬の製造のためのこれらの押出物の使用に関する。
【背景技術】
【0002】
苦い薬物物質の味をマスキングすることは、ヒトの医薬、特に小児科の製剤の服薬率の改善にとって重要であるが、獣医学の薬物においてもそうである。最も簡単なタイプの味のマスキングは、芳香の添加であるが、それは、非常に苦く、水溶性の物質では問題となり得る(Bienz, 1996)。活性物質を疎水性担体で顆粒に加工することによる味のマスキングも記載された(Kalbe and Hopkins, 1998)。他の可能性は、剤形の被覆である。このために使用される物質の例は、Eudragit E(Cerea et al., 2004; Lovrecich et al., 1996; Ohta and Buckton, 2004; Petereit and Weisbrod, 1999)、セラック(shellac)(Pearnchob et al., 2003b; Pearnchob et al., 2003a)およびセルロース誘導体(Al-Omran et al., 2002; Li et al., 2002; Shirai et al., 1993)である。Eudragit E の欠点は、味のマスキングが陽イオン性添加物と陰イオン性活性物質のイオン性相互作用に基づく点である。セラックの欠点は、それが天然ポリマーであり、その組成が変動し得る点である。この他に、剤形の被覆は、追加的な費用と時間のかかる工程である。加えて、不溶性セラックマトリックス中のキノロンまたはナフチリドンカルボン酸の固体分散物が記載された(Cabrera, 2002)。
【0003】
イオン交換樹脂および封入複合体(inclusion complexes)も、味のマスキングに使用される。イオン交換樹脂の有用性は、薬物物質がイオン特性を持っていなければならないという事実により限定される(Chun and Choi, 2004; Lu et al., 1991; Prompruk et al., 2005)。封入複合体は、少量の薬物物質しか担持できない(Sohi et al., 2004)。
【0004】
脂質基剤も、味のマスキングに使用できる。味のマスキングのためにレシチンおよび甘味料も含有する、固い脂肪をベースとする一体化した剤形が記載された(Suzuki et al., 2003; Suzuki et al., 2004)。ここでの欠点は、製造工程で脂質を完全に融解しなければならず、物理的な不安定性を導き得ることである。さらに、固い脂肪、ジステアリン酸グリセロールおよびステアリン酸は、冷間押出において親油性結合剤として使用され、味のマスキングを達成するために、被覆としての Eudragit E の使用も必要である(Breitkreutz et al., 2003)。味のマスキングを目的とするものではないが、剤形を製造するための融点より低温での脂質の押出も記載された(Reitz and Kleinebudde, 2007; Windbergs et al., 2008)。
【0005】
味がマスキングされたキノロン型のジャイレース阻害剤の製剤は、活性物質を高級脂肪酸、および、必要であれば他の添加物と混合し、加熱し、冷却の後に造粒または微粉砕することにより得られた(Ahrens et al., 1998)。さらに、蝋をベースとするペレット剤も製造された(Adeyeye and Price, 1991; Adeyeye and Price, 1994; Zhou et al., 1996; Zhou et al., 1998)。これらの研究は、活性物質の放出は、使用した蝋の融点およびペレット中の濃度に依存することを示した。蝋の融点および含有量が高いほど、放出は遅い。味のマスキングのさらなる可能性は、Kim および Choi(2004)により記載され、彼らは、ココアバターの核または固い脂肪および活性物質を調製し、これにアルギン酸ナトリウムまたはカラゲナンの被覆を施した。しかしながら、この方法では、脂肪は完全に融解し、それは安定性の理由で不利であり、さらに、被覆のための製造工程は追加的な工程であった。
【0006】
加えて、Compritol(登録商標)888 ATO は、マトリックス形成成分として記載された。そのペレット剤は、融解したCompritol(登録商標)、活性物質および多糖類の被覆からなった(Mirghani et al., 2000)。他の研究では、マトリックス錠剤は、粉末混合物から直接、または微粉砕された固体分散物から圧縮された。微粉砕された固体分散物に由来する錠剤は、より良好な味のマスキングを示したが、これらの製造のためには、Compritol(登録商標)を完全に融解しなければならなかった(Li et al., 2006)。Barthelemy は、Compritol(登録商標)を、テオフィリンのペレット剤および顆粒剤の被覆に使用した。ここでも、この脂肪は完全に融解された(Barthelemy et al., 1999)。
【0007】
リン脂質の使用は、苦味をマスキングするもう1つの可能性であるが、他の味のタイプのものではない(Katsuragi et al., 1997; Takagi et al., 2001)。加えて、リン脂質の添加は、脂質の結晶化度に影響し、不安定性を導き得る(Schubert, 2005)。粉末の味のマスキングは、小さい添加物の粒子が大きい活性物質粒子に沈着することにより可能である(Barra et al., 1999)。
【0008】
押出により中に活性物質を組み込んだ動物飼料も記載された(Huber et al., 2003)。塩基性薬物物質およびメタクリル酸ポリマーの溶融押出により、Petereit らは、味がマスキングされた押出物を得、次いで、それを顆粒または粉末に破砕し(Petereit et al., 2003)、そして、この2つの成分を中鎖ないし長鎖脂肪酸と融解状態で混合することにより、迅速に崩壊する剤形を得た。凝固後、生成物を破砕し、水溶性マトリックスに包埋した(Petereit et al., 2004)。活性物質をベヘン酸エステルの脂質マトリックスおよび疎水性希釈剤の中に含有する制御放出の医薬も研究された(Nabil et al., 1998)。Thombre は、味をマスキングする添加物を含む多粒子形態のペット用製剤を記載している(Thombre, 2004)。
【0009】
係属中の出願「味のマスキングが改善された押出物」(ドイツ特許出願番号102007026550.8、対応するPCT出願番号PCT/EP2008/004218も参照)では、医薬的に有用なストランド径0.5mm以下の押出物が記載された。これらの押出物は、薬物物質の味のマスキングに適する。
【0010】
しかしながら、活性物質を針状の形態で含有する混合物の押出では、予期せぬ困難が生じる:使用する薬物物質の結晶形が針状であるならば、針状晶の長さがストランド径よりも顕著に小さいときでさえ、安定かつ再現性のある製造方法は実施できない。原則として、針状薬物物質に伴う加工の困難性は他の製造技術で周知であった。粉末および結果的な錠剤の特性は、使用する物質の粒子の形態に依存することが、様々な研究で示された(Alderborn and Nystroem, 1982; Wong and Pilpel, 1990)。例えば、針状結晶相のパラセタモールは、他の結晶形態よりもはるかに不十分に錠剤に圧縮され得、それは錠剤のキャッピングおよび粉末流動性の乏しさに現れる(Wang & Zhang, 1995)。針状結晶の形態では、イブプロフェンも非常に乏しい流動性、粘着性および接着性を示し、圧縮および打錠に高いエネルギーの投入が必要であり、得られた錠剤は機械的に不安定であった。凝集または等軸結晶形態への再結晶により、イブプロフェンの打錠特性を顕著に改善できた(Jbilou et al., 1999; Rasenack and Mueller, 2002)。
【0011】
しかしながら、押出方法に関しては、針状粒子が押出の方向に並ぶことがガラス加工から知られていた。この場合、製造されるガラスにおける異方性を達成するために、整列は望ましかった(Moisescu et al., 1999)。等軸の粒子と比較して、溶融押出中の軟化したガラス塊の粘度は、針状粒子を含有する場合に顕著に高かった(Yue et al., 1999)。
【0012】
当業者は、特に、粒子が押出に都合よく平行に整列すると確かに想定できたので、粒径がストランド径よりもかなりの倍率(例えば5)で小さい限り、問題はないと予想したであろう。
【0013】
しかしながら、驚くべきことに、針状活性物質を含有する混合物の押出には問題が生じる:製造方法の問題は、ノズルプレートの前での蓄積の形で現れ、ノズル器具が詰まり、その結果、押出ノズルプレートの前の圧力が上昇する。それに加えて、粉末混合物の流動が乏しいので、高い加工速度で一定の供給速度を維持することは殆ど不可能である。
【0014】
満足のいく針状活性物質の押出方法に到達するために、様々なアプローチが可能である:製剤の改変、例えば、脂質基剤の変更、さらなる添加物の添加、または、様々な活性物質の負荷量を用いる実験は、十分な改善をもたらさない。さらに、加工の方向にノズル管が段階的に広くなるノズルプレートも使用できるが、この場合、これは、いくつかの状況下で、工程の均一性の悪化を導く。開口部のドリル(aperture drillings)に特に滑らかな表面を有するノズルプレートを介する押出は、有意な変化をもたらさない。さらなる装置の変形として、様々なスクリュー配置を使用できるが、これも何の改善ももたらさない。さらに、温度、供給速度およびスクリュー回転速度の工程パラメーターを変更できる。使用する脂質の融解範囲より20℃低い押出温度から、融解範囲内までが可能である。低すぎる温度では、ノズルはすぐに詰まり、圧力は非常に急速に高まり、高すぎる温度では、脂質は完全に融解し、柔らかいペーストとしてノズルを出る。
【0015】
しかしながら、大きいノズル直径を用いると、一定の改善が達成されることが見出されている。他の改変のいずれも決定的にこの方法を改善しないので、活性物質粉末を破砕する(例えば、エアジェットミル(air jet mill)で);針状結晶構造を、破砕により微粉砕する。十分に小さい粒径では、押出物を何の問題もなく製造できることが見出されている。十分に細かく破砕された活性物質を用いる押出方法は、高い薬物物質の負荷量(例えば、50%または80%まででさえ)および例えば0.3mmまたはわずか0.2mmのノズル直径でも、原則として円滑かつ再現可能に一定の圧力で進行する。
【発明の概要】
【0016】
従って、本発明は、以下のものに関する:
・針状医薬活性物質の粒径のストランド径に対する比が少なくとも1:15であることを特徴とする、少なくとも1種の医薬活性物質を針状晶の形態で含有する押出物。
・医薬を製造するための、上記の押出物の使用。
【0017】
針状医薬活性物質の粒径とストランド径との比は、通常、少なくとも1:15、好ましくは少なくとも1:20、特に好ましくは少なくとも1:25、ことさら特に好ましくは少なくとも1:50、特に少なくとも1:100である。
【0018】
疑念がある場合には、ここでの粒径は、レーザー回折法で測定されるd(0.9)値を意味すると理解されるべきである。本発明の意味では、d(0.9)は、全粒子の90%がこの値以下の寸法(直径)を有する、体積に基づく粒径分布を意味すると理解される(場合により、用語d(90)またはd(v,90)もこのために使用され、後者は、これが体積に基づく粒径分布であることを明確にするためである)。用語d(0.5)、d(0.1)なども、対応して理解されるべきである。ここで述べる粒径は、活性物質粒子の屈折率が不明であるので、Malvern Mastersizer 2000(分散ユニット Hydro 2000G)およびフラウンホーファー回折評価モードを用いるレーザー回折法により測定した。このために、適量のサンプル溶液を分散媒体(例えば、プラジカンテルには0.1%スルホコハク酸ジオクチルナトリウム水溶液、または、メサラジンにはエタノール)2−3mlに、撹拌しながら予め分散させた。次いで、分散物を装置の分散ユニットに撹拌(300rpm)およびポンプ輸送(900rpm)しながら供給し、測定を行った。評価ソフトウェアは、粒径をd(0.9)値(またはd(0.5)値など)として出力した。
【0019】
一般的な溶媒に溶解しすぎる活性物質粒子(例えば、カフェイン)は、適当なユニット(例えば、Scirocco 2000 乾燥粉末供給機)を用いて、0.5barの空気圧の気流を利用して乾燥分散させる。
【0020】
本発明による押出物のストランド径は、好ましくは最大で0.5mm、特に好ましくは最大で0.3mmである。通常、直径0.1mmを超える、好ましくは0.2mmを超える押出物を使用できる。非円筒形の押出物では、最大の縁の長さまたは楕円の長さは、最大で0.5mm、好ましくは最大で0.3mmである。
【0021】
押出物は、熱可塑的に変形できる物質またはいくつかの熱可塑的に変形できる物質の混合物からなる押出に適する基剤、並びに、必要であれば、さらなる医薬的に許容し得る補助物質および添加物を含有する。
【0022】
基剤は、熱可塑的に変形できる物質、例えば、ポリマー、例えば、ポリアクリル酸塩、または、セルロース誘導体、脂質、例えば、アシルグリセリド類、界面活性剤、例えば、モノステアリン酸グリセロールまたはステアリン酸ナトリウム、マクロゴール類、例えば、ポリエチレングリコール6000、糖または糖アルコール、例えば、マンニトールまたはキシリトールからなる。好ましくは、脂質基剤を使用する。脂質基剤として、例えば、脂肪基剤、特に、グリセロールエステル類が適し、これらは、好ましくはC12−C24脂肪酸とのエステルである。グリセロールエステルとして、グリセロールジエステル類、例えば、ジベヘン酸グリセロール、グリセロールトリエステル類、例えば、トリラウリン酸グリセロール、トリミリスチン酸グリセロール、トリパルミチン酸グリセロールまたはトリステアリン酸グリセロール、および、グリセロールモノ−、ジ−およびトリエステルの混合物、例えば、パルミチン酸ステアリン酸グリセロールに言及し得る。ココナッツバター、パーム油および/またはパームナッツ油(例えば、Witocan(登録商標)の名称で購入できる固い脂肪)をベースとするトリグリセリド類にも言及し得る。クエン酸および/または乳酸のモノ−またはジグリセリド類も使用できる。
さらに、蝋、特に、30ないし60個の炭素原子のもの、例えば、パルミチン酸セチルに言及し得る。
そのような脂質は、例えば、Precirol(登録商標)、Compritol(登録商標)および Dynasan(登録商標)の名称で購入できる。
【0023】
グリセロールジエステルの系列で特に好ましい例は、ジベヘン酸グリセロール(例えば、Compritol(登録商標)888 ATO、これは、主にジベヘン酸グリセロールを含有するが、モノベヘン酸グリセロールおよびトリベヘン酸グリセロールも含有する)である。グリセロールトリエステルの系列で特に好ましい例は、トリミリスチン酸グリセロール(例えば、Dynasan(登録商標)114)、トリパルミチン酸グリセロール(例えば、Dynasan(登録商標)116)およびトリステアリン酸グリセロール(例えば、Dynasan(登録商標)118)である。
【0024】
好ましくは、脂肪基剤は、粉末形態である。多くの脂質は多形性であり、いくつかの状況下で、温度および圧力が変化する場合に、準安定な形態を形成できる。いくつかの状況下で保存すると、変態の変換が起こり得、より安定な変態が形成され得る。文献の記載によると [Reitz and Kleinebudde, 2007; Windbergs et al., 2008]、グリセロールトリエステル類(例えば Dynasan(登録商標)として知られる)、特にトリミリスチン酸グリセロール(Dynasan 114(登録商標))またはトリパルミチン酸グリセロール(例えば、Dynasan(登録商標)116)またはトリステアリン酸グリセロール(例えば、Dynasan(登録商標)118)は、そのような変化に対して比較的安定であり、従って、医薬用の脂質基剤として特に適する。
【0025】
特に、脂肪基剤として使用する物質は、しばしば、例えば、モノ−、ジ−および/またはトリグリセリド類の混合物として販売される。本質的に1種の成分のみからなる均質な脂肪基剤は、これらより好ましい。これらの添加物を用いて製造される製剤は、良好な保存安定性を特徴とする。
【0026】
使用する基剤(熱可塑的に変形できる物質からなる)の量は、押出物に含まれる他の物質の量に応じて決まる。通常、15ないし99%[w/w]、好ましくは20ないし99%[w/w]、特に好ましくは25ないし80%[w/w]、ことさら特に好ましくは30ないし70%[w/w]を使用する。
使用する基剤、特に脂質基剤は、概して、その下限が通常少なくとも50℃、好ましくは少なくとも60℃である融点範囲を有する。
【0027】
本発明による押出物は、必要であれば、1種またはそれ以上のさらなる補助物質および添加物を含有できる。これらとして可能なものは、流動調節剤、好ましくは、濃度0.2ないし2%[w/w]のコロイド状二酸化ケイ素、滑沢剤、好ましくは濃度0.2ないし5%[w/w]のステアリン酸マグネシウムまたはジベヘン酸カルシウム、および、界面活性剤、好ましくは濃度0.5ないし10%[w/w]のレシチンである。さらに、抗酸化剤を使用でき、例えば、通常の量、概して0.01ないし0.5%[w/w]、好ましくは0.05ないし0.2%[w/w]で使用されるブチルヒドロキシアニソール(BHA)またはブチルヒドロキシトルエン(BHT)が適する。活性物質の放出は、例えば、いわゆる孔形成物質の添加により制御できる。これらの例は、糖、特にラクトース、ポリオール類、特にマンニトール、または、ポリエチレングリコール類(PEG)、好ましくはPEG1500ないし10000、特に好ましくはPEG1500ないし6000、例えば、PEG1500(Macrogol 1500)である。孔形成物質は、5ないし40%[w/w]の濃度で、好ましくは5ないし20%[w/w]の濃度で使用する。活性物質の放出に影響を与える他の可能性は、崩壊補助剤の添加である。加えて、クロスポビドン、クロスカルメロースナトリウムまたは架橋ナトリウムカルボキシメチルスターチなどの、いわゆる超崩壊剤(super disintegrator)を使用できる。超崩壊剤は、1ないし15%[w/w]の濃度、好ましくは3ないし10%[w/w]の濃度で使用する。これに代わって、酸に溶ける、かつ/または、二酸化炭素を放出する物質、例えば、炭酸マグネシウムまたは炭酸カルシウムを使用できる。二酸化炭素放出物質は、5ないし15%[w/w]の濃度で、好ましくは5ないし10%[w/w]の濃度で使用する。
【0028】
さらに、本発明による押出物は、帯電防止剤を含有できる。これは、特に、帯電が押出に影響する場合に推奨される。帯電は、ノズル開口部の詰まりをもたらし得、これは、帯電防止剤の添加により防止できる。帯電防止剤として、PEGを好ましく使用でき、特に、可能なのはPEG1500ないし6000である。PEGは、帯電防止効果を発揮するために、好ましくは粉末形態であり、押出中に融解するべきである。故に、帯電防止剤としてのPEGの添加に伴い、押出中にPEGが融解するが、使用する脂肪基剤は融解しないように、PEGの融解温度は十分に低いものであるべきである。実際に、帯電は、全ての基剤で起こるわけではない。それは、とりわけ、脂肪酸とのグリセロール脂肪酸トリエステル類、例えば、トリミリスチン酸グリセロール、トリパルミチン酸グリセロールまたはトリステアリン酸グリセロールで観察される。故に、そのような基剤の使用に伴い、問題のない押出を確実にするために、押出される混合物に帯電防止剤を添加することが推奨される。帯電防止剤は、少なくとも5%[w/w]、好ましくは少なくとも10%[w/w]の濃度で使用する。通常、30%[w/w]以下を使用する。
【0029】
医薬活性物質として、薬物の活性物質、特に、不快な味をマスキングしなければならないものを使用できる。
本発明によると、針状の活性物質を使用する。一般に、これらは針状結晶である。
ここで、「針状晶」または「針状」は、長さが直径よりも顕著に大きく、長さ/直径の比が、少なくとも3:1より大きく、好ましくは5:1より大きく、特に好ましくは10:1より大きく、ことさら特に好ましくは20:1より大きい粒子を意味すると理解されるべきである。針状晶は、概して丸くないので、疑念がある場合には、「直径」は、長さに対して垂直な最大の寸法を意味すると理解されるべきである。
【0030】
本質的に、活性物質を融解する必要がないので、針状活性物質の選択に大きい制限はない。押出物の味のマスキング作用のために、それらは、好ましくは、不快な(例えば、苦い)味の活性物質に適する。
活性物質は、例えば、以下の群に属するものであり得る:抗生物質、寄生性原生動物に対する薬物、駆虫剤、代謝刺激剤および炎症阻害性物質。
当然、活性物質の用語には、針状の塩および溶媒和物も含まれる。
【0031】
活性物質の特定の例として、メサラジンに言及し得る。メサラジンは、慢性炎症性腸疾患に対して使用される炎症阻害性薬物物質である。それは水にあまり溶けず、280℃の融点および針状の結晶形を有する。
さらなる例は、結晶性カフェインである。
【0032】
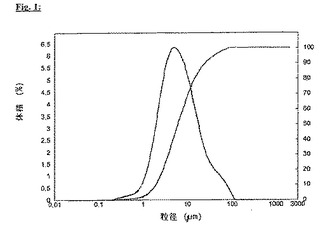
針状活性物質の好ましい特定の例として、条虫および住血吸虫に対して有効な昔から知られてきた駆虫剤であるプラジカンテルに言及し得る。それは水にあまり溶けず、139℃の融点および針状の結晶形を有する。ピンディスクミル(pinned disc mill)で破砕した、レーザー回折法により測定されるd(0.9)として表される粒径25μmの等級(図1)を、通常は使用する。
【0033】
親油性マトリックスに包埋することにより、使用する活性物質の性質に応じて、遅延放出、および、故に、効果の遅延を達成できる。
押出物中で使用する活性物質の量は、作用の強さおよび所望の投与量によって決まる。80%[w/w]まで、好ましくは70%[w/w]まで、特に好ましくは60%[w/w]までの濃度の、高い活性物質濃度の押出物でさえ製造できることが見出されている。通常の濃度範囲の例は、1ないし80%[w/w]、好ましくは5ないし70%[w/w]、そして、特に好ましくは30ないし60%[w/w]である。
【0034】
本発明による押出物は、出発物質(医薬活性物質、基剤および必要であれば補助物質および添加物)を混合し、次いで押し出すことにより製造する。押出は、好ましくは、熱可塑的に変形できる物質の完全な融解に至らない温度で、実際には、通常、室温の範囲の温度で、好ましくは40℃で、熱可塑的に変形できる物質の融解範囲より低温で実施する。実際に、これは、通常、問題の基剤について定められている融解範囲に基づく。概して、押出の温度は、基剤、特に脂肪基剤の融解範囲の下限よりも20℃、好ましくは15℃、特に好ましくは10℃より低くは設定しない。通常、押出の温度は、基剤の融解範囲の下限よりも高くは設定せず、好ましくは1℃低く、特に好ましくは5℃低い。目的は、柔らかいペーストの押出を回避することである。押出工程は、できるだけ一定の物質温度で実施すべきである。この目的で、スクリュー押出機(screw extruder)、特に二軸スクリュー押出機は、特に適する。押し出されたストランドは、好ましくは円形の断面および上記の直径を有する。押し出されたストランドは、押出の際にナイフで直接粉砕できるか、または、通常のミル、例えば遠心ミルで、穏やかにひくことにより、別工程で粉砕できる。得られる生成物の粒度は、使用するノズルの直径によって決まり、粉砕されたストランドは、最大でストランド径の3倍に相当する長さを有する。典型的な粒度は、例えば、300ないし500μmであり、あるいは、小さいノズル直径の場合、200ないし500μmでさえある。好ましい実施態様によると、破砕された生成物を篩にかけることもできる。かくして、微粉の画分を除去できる。
【0035】
本明細書で時折使用される、押出物はそれらの融点より低温で押し出されるという記述は、上述の通り、押出物が、使用する熱可塑性の基剤が完全に融解しない温度で押し出されることを意味すると理解されるべきである。しばしば、他の成分、例えば活性物質は、これより高い融点を有する。そのような押出物は、不快な味の成分の味のマスキングに適する。
【0036】
穏やかにひいた後、本発明による押出物は、必要であれば、さらに適する剤形に加工できる。さらなる添加物の添加が、さらなる加工のために場合により必要である。本発明によると好ましい剤形は、必要であれば所望の適用に適する形状を有していてもよい錠剤である。可能な他の剤形は、ペースト剤、懸濁剤、サシェ剤、カプセル剤などである。
【0037】
本発明による押出物または医薬は、一般的に、ヒトおよび動物における使用に適する。それらは、好ましくは、農業、育種、動物園、研究室および実験用の動物、および、「趣味の動物」、および、特に哺乳動物において、動物の管理および動物の繁殖に使用する。
【0038】
農業用および繁殖用の動物には、哺乳動物、例えば、ウシ、ウマ、ヒツジ、ブタ、ヤギ、ラクダ、スイギュウ、ロバ、ウサギ、ダマジカ、トナカイ、毛皮動物、例えば、ミンク、チンチラ、アライグマ、および、鳥類、例えば、ニワトリ、ガチョウ、シチメンチョウ、アヒル、ハトおよびダチョウのタイプが含まれる。好ましい農業用動物の例は、ウシ、ヒツジ、ブタおよびニワトリである。
【0039】
研究室および実験用の動物には、イヌ、ネコ、ウサギ並びに齧歯類、例えば、マウス、ラット、モルモットおよびハムスターが含まれる。
愛玩動物には、イヌ、ネコ、ウマ、ウサギ、齧歯類、例えば、ハムスター、モルモット、マウス、並びに、家庭および動物園で飼育するための爬虫類、両生類および鳥類が含まれる。
【0040】
押出物は、通常、経腸で、特に経口で、直接または適する製剤の形態(剤形)で使用する。
経腸使用は、例えば、経口で、顆粒剤、錠剤、カプセル剤、ペースト剤、懸濁剤または薬用動物飼料の形態で行う。経口投与の1つの選択肢は、いわゆるトップドレッシング(top dressing)であり、これは、動物飼料に乗せられ、飼料と共に摂取される散剤、顆粒剤またはペースト剤である。
【0041】
適する製剤は以下のものである:
固体製剤、例えば、顆粒剤、ペレット剤、錠剤、巨丸剤および有効成分を含有する成形品(moulded body)。
固体製剤の製造には、粉砕した押出物を、適する担体と、必要であれば添加物を添加して混合し、所望の形態にする。
担体として、全ての生理的に適合する固体の不活性物質に言及し得る。無機および有機物質をそのように使用する。無機物質の例は、食塩、炭酸カルシウムなどの炭酸塩、炭酸水素塩、酸化アルミニウム、ケイ酸、アルミナ、沈殿またはコロイド状二酸化ケイ素およびリン酸塩である。
有機物質の例は、糖、セルロース、食糧および動物飼料、例えば、粉乳、動物食餌、穀粉および穀物、並びに、スターチである。
【0042】
添加物は、防腐剤、抗酸化剤および着色料である。適する添加物および添加する必要量は、本質的に当業者に知られている。防腐剤として、例えばソルビン酸に言及し得る。抗酸化剤として、例えばブチルヒドロキシアニソール(BHA)またはブチルヒドロキシトルエン(BHT)が適する。可能な着色料は、有機および無機着色料、または、医薬の目的に適する色素、例えば、酸化鉄である。
【0043】
さらなる適する添加物は、滑沢剤および離型剤、例えば、ステアリン酸マグネシウム、ステアリン酸、タルク、ベントナイト類、崩壊促進物質、例えば、スターチまたは架橋ポリビニルピロリドン、結合剤、例えば、スターチ、ゼラチンまたは直鎖状ポリビニルピロリドン、および、乾燥結合剤、例えば、結晶セルロースである。
【0044】
さらなる添加物として、油、例えば、植物油(例えば、オリーブ油、大豆油、ヒマワリ油)、または、動物起源の油、例えば、魚油を使用できる。通常量は、0.5ないし20%[w/w]、好ましくは0.5ないし10%[w/w]、特に好ましくは1ないし2%[w/w]である。
【0045】
懸濁剤を経口で使用できる。それらは、粉砕した押出物を、担体の液体に、必要であれば他の添加物、例えば、湿潤剤、着色料、吸収促進物質、防腐剤、抗酸化剤または光安定化剤を添加して、懸濁することにより製造する。
【0046】
可能な担体の液体は、問題の押出物が溶解しない均質な溶媒または溶媒混合物である。例えば、生理的に適合する溶媒、例えば、水、アルコール、例えば、エタノール、ブタノール、グリセリン、プロピレングリコール、ポリエチレングリコールおよびそれらの混合物に言及し得る。
【0047】
湿潤剤(分散剤)として、界面活性剤を使用できる。例えば:
非イオン性界面活性剤、例えば、ポリエトキシル化ヒマシ油、ポリエトキシル化モノオレイン酸ソルビタン、モノステアリン酸ソルビタン、モノステアリン酸グリセリン、ステアリン酸ポリオキシエチル、アルキルフェノールポリグリコールエーテル;
両性界面活性剤、例えば、ジ−NaN−ラウリル−β−イミノジプロピオネートまたはレシチン;
陰イオン性界面活性剤、例えば、ラウリル硫酸Na、脂肪アルコールエーテルサルフェート、モノ/ジアルキルポリグリコールエーテルオルトリン酸エステルモノエタノールアミン塩;および、
陽イオン性界面活性剤、例えば、セチルトリメチル塩化アンモニウムに言及し得る。
【0048】
さらなる添加物として、例えば:
増粘性物質および懸濁安定化物質、例えば、カルボキシメチルセルロース、メチルセルロースおよび他のセルロースおよびスターチ誘導体、ポリアクリル酸塩、アルギン酸塩、ゼラチン、アラビアゴム、ポリビニルピロリドン、ポリビニルアルコール、メチルビニルエーテルおよび無水マレイン酸のコポリマー、ポリエチレングリコール、蝋、コロイド状ケイ酸または列挙した物質の混合物に言及し得る。
【0049】
半固体製剤は、経口投与できる。それらは、それらの高い粘性でのみ、上記の懸濁剤および乳剤と異なる。
活性物質は、共力剤または他の活性物質と組み合わせて使用することもできる。
【実施例】
【0050】
実施例
断りのない限り、百分率の値は、完成した混合物を基準とする重量パーセントである。
I. 比較例:プラジカンテルを含む押出物:
粒径d(0.9)=25μm(図1参照)の活性物質プラジカンテル(50%[w/w])、および、添加物の Compritol(登録商標)888 ATO(49%[w/w])、主成分がジベヘン酸グリセロールの脂肪基剤(モノ−およびトリエステル、および、それより少ない量のC16−C20脂肪酸とのエステルも含有する)、および、Aerosil(登録商標)200(1%[w/w])、その使用が粉末塊の流動性の改善に貢献する発熱性コロイド状二酸化ケイ素(高分散二酸化ケイ素とも記載される)からなる粉末混合物を、押出の前に、実験用ミキサー中、室温で混合し(15分間、40rpm)、粉末混合物を押出機の重量測定による供給機に移した。
【0051】
溶融押出のために、円形の工具およびブラントスクリューアタッチメントを備えた一定速度の二軸スクリュー押出機を使用した。
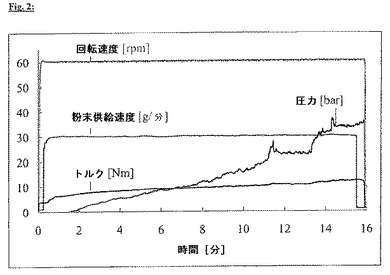
この製剤の押出の過程では、多くのノズル開口部が詰まり(ノズルプレートは、各々直径0.3mmの全部で67個のノズル孔を有する)、結果的に、一定の回転速度および供給速度で、圧力は継続的に上昇した(図2)。未破砕のプラジカンテルを用いる工程の間にノズルプレートの後ろに蓄積した圧力のために、開いたままのノズル開口部から押出物が出てくる速度は非常に顕著に上昇した。
【0052】
II. 実施例:細かく破砕したプラジカンテルを含む押出物
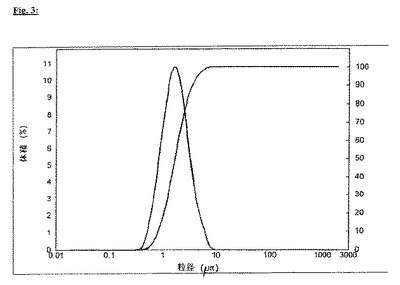
エアジェットミルで2回破砕したプラジカンテル(d(0.5)=1.7μm、d(0.9)=3.6μm、図3参照)を使用して、比較例の実験を繰り返した。
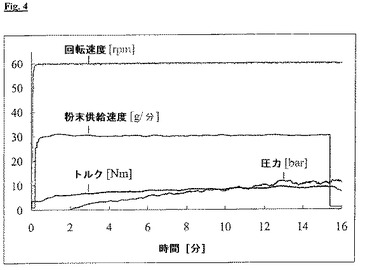
破砕したプラジカンテルを用い、薬物物質充填率50%およびノズル直径0.3mmを用いる押出工程は、一定の圧力で均一かつ再現可能であった。図4に示す工程の過程から、約12分後に一定のレベルで、10barで安定した圧力は、未破砕のプラジカンテルを用いる工程(図2)のもの(15分間の加工時間の後、圧力は約40barまで上昇した)より顕著に低かったことがわかる。押出は、全てのノズル開口部を通して、均一の速度で起こった。
【0053】
比較例Iと本実施例の押出物を比較すると、違いが見られる:かなりの摩擦のために、より多くの脂質が押出物の表面に押しつけられ、それは、走査型電子顕微鏡で非常に明確に見ることができる;押出物の表面は滑らかで平坦である。これと対照的に、破砕したプラジカンテルを含む押出物の表面は平坦ではなく、プラジカンテル粒子は表面下に直接位置し、それらの形状は薄い脂質層を通して明白である。
【0054】
III. 比較例:未破砕のメサラジンを含む押出物
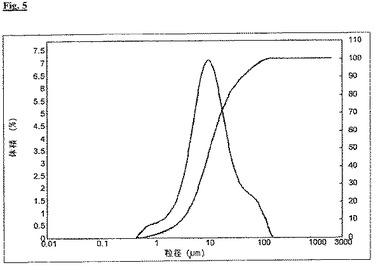
未破砕のメサラジン(d(0.5)=10.7μm、d(0.9)=44.0μm、図5参照)を含む押出物を、比較例Iと同様に製造した。50%[w/w]未破砕メサラジン/49%[w/w]Compritol(登録商標)/1%[w/w]高分散二酸化ケイ素(Aerosil(登録商標))の粉末混合物を使用し、直径0.3mmのノズルプレートを通して押出を実施した。製造工程で、比較例Iと同様の問題が起こった。工程の過程から(図6)、10分後に圧力は約25barで安定したが、依然として本当に強く上下することが明らかである。この実験でも、部分的なノズル開口部の詰まりが観察されたが、これは、プラジカンテルほど急速にも、大規模にも起こらなかった。
【0055】
IV. 実施例:破砕したメサラジンを含む押出物
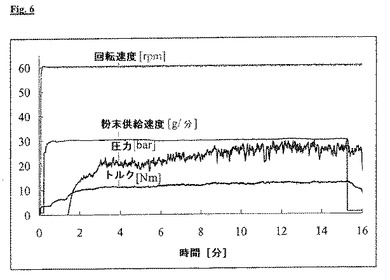
エアジェットミルで2回破砕することにより、より小さい粒径のメサラジンを得た:d(0.5)=4.9μm、d(0.9)=11.9μm、図7参照。破砕の後、粒子は顕著に変化した形状を示した。
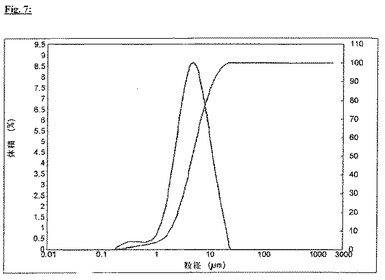
比較例IIIと同様の押出方法におけるこの破砕したメサラジンの使用を通じて、この方法を決定的に改善することが可能であった。図8に、圧力は5分後に20barの一定レベルで既に安定し、押出は全てのノズル開口部を通して均一の速度で起こったことを見ることができる。故に、破砕したメサラジンの使用により、ノズル開口部の詰まりを防止でき、かくして押出方法は改良された。
【0056】
押出物を比較すると、比較例の針状メサラジン結晶は押出中に大きく破壊され、一方、この実施例の破砕したメサラジン粉末は、押出後にもとの粒子の形状および大きさを依然として有し、それは、例えば加熱できるスライドを備えた光学顕微鏡下で明確に見られることが見出された。これと対照的に、針状プラジカンテル結晶は、押出中に破壊されず、もとの形状および大きさで、押出物中に依然として存在した。
【0057】
上記の結果に基づき、当業者は、大きい針状メサラジン結晶は、小さいプラジカンテル針状晶よりも押出において多大な問題を引き起こすと予測するであろう。しかしながら、大きいメサラジン針状晶は、工程中の押出機のスクリューの剪断作用により既に殆ど粉砕されていたので、ノズル開口部の詰まり、および、これに起因する急速な圧力の蓄積は、メサラジンではプラジカンテルほど強く起こらなかった。
【0058】
V. 比較例:帯電防止剤を含まない押出物
破砕したプラジカンテルを用いて、実施例IIと同様に押出物を製造した。50%[w/w]破砕プラジカンテル/49%[w/w]Dynasan 116(登録商標)/1%[w/w]高分散二酸化ケイ素(Aerosil(登録商標))の粉末混合物を使用した;Dynasan 116(登録商標)は、98%がトリパルミチン酸グリセロールからなる脂肪基剤である。押出は、直径0.3mmのノズルプレートを通して実施した。
【0059】
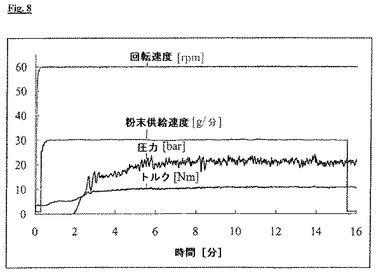
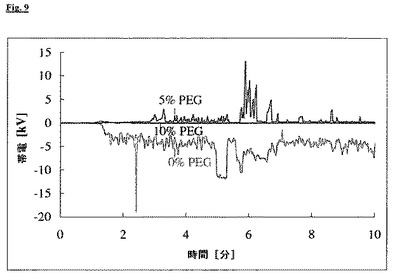
この製剤の押出の過程で、リン脂質塊のノズル内表面に対する摩擦によりかなり強い帯電が押出物に生じ、数分後に、押出物は既に強く押出機自体に静電気的に引き付けられ、ノズルヘッドに付着したままになった。この電荷のために、破砕したプラジカンテルを使用したが、いくつかのノズル開口部は詰まり、ノズルプレートの後ろの圧力は上昇した。電荷の発生は、工程中にノズルプレートの直前に取り付けた静電気検出器IZD 10-510(SMCより)を利用して、継続的に記録できた。この比較例の押出中に、わずか2分後に、帯電は既に−5kVの値に達した(図9)。
【0060】
VI. 実施例:帯電防止剤を含む押出物
比較例Vと同様に、5および10%のPEG1500を帯電防止剤として添加して、押出物を製造した。50%[w/w]破砕プラジカンテル/44%[w/w]Dynasan 116(登録商標)/5%[w/w]PEG1500/1%[w/w]Aerosil(登録商標)または50%[w/w]破砕プラジカンテル/39%[w/w]Dynasan 116(登録商標)/10%[w/w]PEG1500/1%[w/w]高分散二酸化ケイ素(Aerosil(登録商標))の粉末混合物を使用した。シリンダーの温度は約60℃であり、それは、押出工程中のPEG1500の融解を確実にした。
【0061】
5%PEGを用いる押出の過程では、数個のノズル開口部が再度詰まり、3分間の工程時間後に、ノズルプレートの前に1−2kVの帯電が測定された。これと対照的に、10%PEGを用いる押出工程は、ノズル開口部の詰まりおよび測定可能な帯電を伴わず、均一かつ再現可能に進行した(図9)。
帯電防止剤としての10%PEGの添加により、高純度のグリセロールトリエステル類を含有する押出物の帯電を防止し、かくして、この方法を顕著に改良することが可能であった。
【0062】
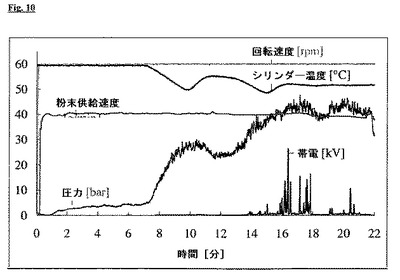
VII. 比較例:未融解の帯電防止剤を含む押出物
実施例VIと同様に、10%PEG6000を帯電防止剤として添加して押出物を製造した。50%[w/w]破砕プラジカンテル/39%[w/w]Dynasan 116(登録商標)/10%[w/w]PEG6000/1%[w/w]高分散二酸化ケイ素(Aerosil(登録商標))の粉末混合物を使用した。シリンダーの温度は、最初に約60℃であり、次いで8分後にまず55℃に、最終的に52℃に冷却した。PEG6000の融解範囲は約55−60℃であり、シリンダー温度の低下後に、PEGはもはや融解形態で存在しなかった。
【0063】
最初に、10%PEG1500を用いた実施例VIと同様に、この方法は均一に帯電せずに行われ、全てのノズル開口部は開いていた(図10)。温度低下後、押出物は帯電し、いくつかのノズル開口部は詰まり、結果として、ノズルプレートの後ろの圧力は上昇した。
PEGは、その帯電防止作用を発揮するために、押出工程中に融解形態で存在するべきであると示すことができた。
【0064】
VIII. 比較例:未破砕のカフェインを含む押出物
比較例Iと同様に、未破砕のカフェインを用いて押出物を製造した。50%[w/w]未破砕カフェイン(粒径d(0.9)=1170μm)/49%[w/w]Compritol(登録商標)/1%[w/w]高分散二酸化ケイ素(Aerosil(登録商標))の粉末混合物を使用した;押出は、直径0.3mmのノズルプレートを通して実施した。製造工程で比較例Iと同様の問題が生じた。8分間の押出の後に、ノズル開口部の完全な詰まりにより圧力は既にかなり上昇したので、工程を止めなければならなかった。
【0065】
IX. 実施例:破砕したカフェインを含む押出物
エアジェットミルで2回破砕することにより、より小さい粒径のカフェインを得た。
この破砕したカフェインを実施例IIと同様の押出工程で使用することにより、この方法を決定的に改良できた。押出中に、3分後に圧力は5barの一定レベルで既に安定し、押出は全てのノズル開口部を通して均一の速度で起こった。故に、破砕カフェインの使用を介して、ノズル開口部の詰まりを防止でき、押出方法を顕著に改良できた。
【0066】
以下の押出物を、上記の実施例と同様に製造した:
X. 実施例
70%[w/w]プラジカンテル(破砕、d(0.9)=5.7μm)
17%[w/w]トリステアリン酸グリセロール(Dynasan(登録商標)118)
12%[w/w]PEG6000
1%[w/w]高分散二酸化ケイ素(Aerosil(登録商標))
ノズル直径:0.3mm(押出中に、100%開いているノズル開口部)、スクリュー速度:60rpm、押出温度:67℃
【0067】
XI. 実施例
50%[w/w]プラジカンテル(破砕、d(0.9)=5.7μm)
29%[w/w]トリステアリン酸グリセロール(Dynasan(登録商標)118)
20%[w/w]PEG6000
1%[w/w]高分散二酸化ケイ素(Aerosil(登録商標))
ノズル直径:0.2mm(180個のノズル開口部、押出中に、100%開いているノズル開口部)、スクリュー速度:60rpm、押出温度:67℃
【0068】
XII. 実施例
50%[w/w]プラジカンテル(破砕、d(0.9)=5.7μm)
29%[w/w]トリステアリン酸グリセロール(Dynasan(登録商標)118)
20%[w/w]PEG6000
1%[w/w]高分散二酸化ケイ素(Aerosil(登録商標))
ノズル直径:0.3mm(押出中に、100%開いているノズル開口部)、スクリュー速度:60rpm、押出温度:67℃
【0069】
XIII. 実施例
50%[w/w]プラジカンテル(破砕、d(0.9)=5.7μm)
49%[w/w]トリステアリン酸グリセロール(Dynasan(登録商標)118)
1%[w/w]高分散二酸化ケイ素(Aerosil(登録商標))
ノズル直径:0.3mm(押出中に、3%開いているノズル開口部)、スクリュー速度:60rpm、押出温度:67℃
【0070】
生物学的実施例:ネコにおける受入試験
40匹のネコを、10匹ずつの4つの群に分けた。以下の方法で、実施例XないしXIIIの各押出物を、それぞれネコ10匹の1つの群で試験した:
プラジカンテル5mg/体重kgの投与量に相当する量の押出物を、乾燥ネコ飼料にトップドレッシングとして添加し、通常の給餌時間でネコに与えた。全てのネコは、飼料を全て食べた。これは、100%の受入の証拠と見なされた。
1週間の休止後、実験を繰り返し、ただし、ここではネコ用の缶詰の飼料(湿っている)を使用した。乾燥飼料と同じ結果が観察され、100%の受入と評価された。
【図面の簡単な説明】
【0071】
図面
【図1】図1:未破砕プラジカンテルの粒径分布、d(0.5)=6.2μm、d(0.9)=25.1μm、湿式分散後にレーザー回折法により測定(Mastersizer 2000 Ver. 5.54, Malvern Instruments, Malvern UK)。
【図2】図2:50%未破砕プラジカンテル/49%Compritol(登録商標)/1%高分散二酸化ケイ素(Aerosil(登録商標))、ノズルプレート直径0.3mmの押出工程の過程
【図3】図3:2回破砕したプラジカンテルの粒径分布、d(0.5)=1.7μm、d(0.9)=3.6μm、湿式分散後にレーザー回折法により測定(Mastersizer 2000 Ver. 5.54, Malvern Instruments, Malvern UK)。
【図4】図4:50%破砕プラジカンテル/49%Compritol(登録商標)/1%高分散二酸化ケイ素(Aerosil(登録商標))、ノズルプレート直径0.3mmの押出工程の過程
【図5】図5:未破砕メサラジンの粒径分布、d(0.5)=10.7μm、d(0.9)=44.0μm、湿式分散後にレーザー回折法により測定(Mastersizer 2000 Ver. 5.54, Malvern Instruments, Malvern UK)。
【図6】図6:50%未破砕メサラジン/49%Compritol(登録商標)/1%高分散二酸化ケイ素(Aerosil(登録商標))、ノズルプレート直径0.3mmの押出工程の過程。
【図7】図7:2回破砕したメサラジンの粒径分布、d(0.5)=4.9μm、d(0.9)=11.9μm、湿式分散後にレーザー回折法により測定(Mastersizer 2000 Ver. 5.54, Malvern Instruments, Malvern UK)。
【図8】図8:50%破砕メサラジン/49%Compritol(登録商標)/1%高分散二酸化ケイ素(Aerosil(登録商標))、ノズルプレート直径0.3mmの押出工程の過程。
【図9】図9:各々0、5および10%のPEG1500含有量の押出工程における、ノズルプレート前の帯電
【図10】図10:50%破砕プラジカンテル/39%Dynasan 116(登録商標)/10%PEG6000/1%高分散二酸化ケイ素(Aerosil(登録商標))の押出工程の過程。
【0072】
文献
Adeyeye C.M., Price J.C., 1991. Development and Evaluation of Sustained-Release Ibuprofen-Wax Microspheres. 1. Effect of Formulation Variables on Physical Characteristics. Pharm. Res. 8, 1377-1383.
Adeyeye C.M., Price J.C., 1994. Development and Evaluation of Sustained-Release Ibuprofen-Wax Microspheres. 2. In-Vitro Dissolution. Pharm. Res. 11, 575-579.
Ahrens G., Mentrup E., Maas J., Radau M., 1998. Production of taste-masked preparations of antibacterially active quinolone derivatives. EP 1998/0855183.
Alderborn G., Nystroem C., 1982. Studies on direct compression of tablets. 3. The effect on tablet strength of changes in particle-shape and texture obtained by milling. Acta Pharm. Suec. 19, 147-156.
Al-Omran M.F., Al-Suwayeh S.A., El-Helw A.M., Saleh S.I., 2002. Taste masking of diclofenac sodium using microencapsulation. J. Microencapsul. 19, 45-52.
Barra J., Lescure F., Doelker E., 1999. Taste masking as a consequence of the organisation of powder mixes. Pharm. Acta Helv. 74, 37-42.
Barthelemy P., Laforet J.P., Farah N., Joachim J., 1999. Compritol(登録商標)888 ATO: an innovative hot-melt coating agent for prolonged-release drug formulations. Eur. J. Pharm. Biopharm. 47, 87-90.
Bienz M., 1996. Taste masking strategies for drug dosage forms. Manufacturing Chemist. 67, 17-20.
Breitkreutz J., El-Saleh F., Kiera C., Kleinebudde P., Wiedey W., 2003. Pediatric drug formulations of sodium benzoate: II. Coated granules with a lipophilic binder. Eur. J. Pharm. Biopharm. 56, 255-260.
Cabrera F.A., 2002. Solid Phase Dispersion of Quinolone-OR Naphthyridonecarboxylic acids. WO 2002/058669.
Cerea M., Zheng W.J., Young C.R., McGinity J.W., 2004. A novel powder coating process for attaining taste masking and moisture protective films applied to tablets. Int. J. Pharm. 279, 127-139.
Chun M.K., Choi H.K., 2004. Preparation and characterization of enrofloxacin/carbopol complex in aqueous solution. Arch. Pharm. Res. 27, 670-675.
Farah N., Barthelemy P., Joachim J., 1998. Method for preparing a pharmaceutical composition with modified release of the active principle, comprising a matrix. WO 1998/14176.
【0073】
Huber G.R., Jones D.R., Kuenzi J.C., Kuenzi K.D., Cabrera F.A., 2003. Animal Feeds including Actives and Methods of using same. WO 2003/030653.
Jbilou M., Ettabia A., Guyot-Hermann A.M., Guyot J.C., 1999. Ibuprofen Agglomerates Preparation by Phase Separation. Drug Dev. Ind. Pharm. 25, 297-305.
Kalbe J., Hopkins T., 1998. Orally administrable granules of hexahydropyrazine derivatives. WO 1998/03157.
Katsuragi Y., Mitsui Y., Umeda T., Otsuji K., Yamasawa S., Kurihara K., 1997. Basic studies for the practical use of bitterness inhibitors: Selective inhibition of bitterness by phospholipids. Pharm. Res. 14, 720-724.
Kim E.-H., Choi H.K., 2004. Preparation of various solid-lipid beads for Drug Delivery of Enrofloxacin. Drug Deliv. 11, 365-370.
Li F.-Q., Hu J.-H., Deng J.-X., Su H., Xu S., Liu J.-Y., 2006. In vitro controlled release of sodium ferulate from Compritol(登録商標)888 ATO-based matrix tablets. Int. J. Pharm. 324, 152-157.
Li S.P., Martellucci S.A., Bruce R.D., Kinyon A.C., Hay M.B., Higgins J.D., 2002. Evaluation of the film-coating properties of a hydroxyethyl cellulose/hydroxypropyl methylcellulose polymer system. Drug Dev. Ind. Pharm. 28, 389-401.
Lovrecich M., Nobile F., Rubessa F., Zingone G., 1996. Effect of ageing on the release of indomethacin from solid dispersions with Eudragits. Int. J. Pharm. 131, 247-255.
Lu M.Y.F., Borodkin S., Woodward L., Li P., Diesner C., Hernandez L., Vadnere M., 1991. A Polymer Carrier System for Taste Masking of Macrolide Antibiotics. Pharm. Res. 8, 706-712.
Moisescu C., Jana C., Habelitz S., Carl G., Ruessel C., 1999. Oriented fluoroapatite glass-ceramics. J. Noncry. Sol. 248, 176-182.
Mirghani A., Idkaidek N.M., Salem M.S., Najib N.M., 2000. Formulation and release behavior of diclofenac sodium in Compritol(登録商標)888 ATO matrix beads encapsulated in alginate. Drug Dev. Ind. Pharm. 26, 791-795.
Nabil, F. et al., 1998. Method for preparing a pharmaceutical composition with modified release of the active principle, comprising a matrix method for preparing a pharmaceutical composition with modified release of the active principle, comprising a matrix. FR2753904(A1).
Ohta M., Buckton G., 2004. The use of inverse gas chromatography to assess the acid-base contributions to surface energies of cefditoren pivoxil and methacrylate copolymers and possible links to instability. Int. J. Pharm. 272, 121-128.
【0074】
Pearnchob N., Dashevsky A., Siepmann J., Bodmeier R., 2003a. Shellac used as coating material for solid pharmaceutical dosage forms: understanding the effects of formulation and processing variables. Stp Pharma Sciences. 13, 387-396.
Pearnchob N., Siepmann J., Bodmeier R., 2003b. Pharmaceutical applications of shellac: Moisture protective and taste masking coatings and extended-release matrix tablets. Drug Dev. Ind. Pharm. 29, 925-938.
Petereit H.U., Weisbrod W., 1999. Formulation and process considerations affecting the stability of solid dosage forms formulated with methacrylate copolymers. Eur. J. Pharm. Biopharm. 47, 15-25.
Petereit H.U., Meier C., Gryczke A., 2003. Melt extrusion of active substance salts. WO 2003/072083.
Petereit H.U., Meier C., Gryczke A., 2004. Process for the production of an oral drug form with direct disintegration and active substance release. WO 2004/066976.
Prompruk K., Govender T., Zhang S., Xiong C.D., Stolnik S., 2005. Synthesis of a novel PEG-blockpoly(aspartic acid-stat-phenylalanine)copolymer shows potential for formation of a micellar drug carrier. Int. J. Pharm. 297, 242-253.
Rasenack N., Mueller B.W., 2002. Crystal habit and tableting behaviour. Int. J. Pharm. 244, 45-57.
Reitz C., Kleinebudde P., 2007. Solid lipid extrusion of sustained release dosage forms. Eur. J. Pharm. Biopharm. 67, 440-448.
Schubert M.A., Schicke B.C., Mueller-Goymann C.C., 2005. Thermal analysis of the crystallization and melting behavior of lipid matrices and lipid nanoparticles containing high amounts of lecithin. Int. J. Pharm. 298, 242-254.
Shirai Y., Sogo K., Yamamoto K., Kojima K., Fujioka H., Makita H., Nakamura Y., 1993. A Novel Fine Granule System for Masking Bitter Taste. Biol. Pharm. Bull. 16, 172-177.
Sohi H., Sultana Y., Khar R.K., 2004. Taste masking technologies in oral pharmaceuticals: Recent developments and approaches. Drug Dev. Ind. Pharm. 30, 429-448.
Suzuki H., Onishi H., Takahashi Y., Iwata M., Machida Y., 2003. Development of oral acetaminophen chewable tablets with inhibited bitter taste. Int. J. Pharm. 251, 123-132.
Suzuki H., Onishi H., Hisamatsu S., Masuda K., Takahashi Y., Iwata M., Machida Y., 2004. Acetaminophen-containing chewable tablets with suppressed bitterness and improved oral feeling. Int. J. Pharm. 278, 51-61.
【0075】
Takagi S., Toko K., Wada K., Ohki T., 2001. Quantification of suppression of bitterness using an electronic tongue. J. Pharm. Sci. 90, 2042-2048.
Thombre A.G., 2004. Palatable controlled-release formulations for companion animals. WO 2004/014346.
Wang H., Zhang R., 1995. Compaction behaviour of paracetamol powders of different crystal shapes. Drug Dev. Ind. Pharm. 21, 863-868.
Windbergs M., Strachan C.J., Kleinebudde P., 2008 Understanding the solid-state behaviour of triglyceride solid lipid extrudates and its influence on dissolution. Eur. J. Pharm. Biopharm. In Press.
Wong L.W., Pilpel N., 1990. The effect of particle shape on the mechanical properties of powders. Int. J. Pharm. 59, 145-154.
Yue Y., Moisescu C., Carl G., Ruessel C., 1999. Influence of suspended iso- and anisometric crystals on the flow behaviour of fluoroapatite glass melts during extrusion. Physics and Chemistry of Glasses. 40, 243-247.
Zhou F., Vervaet C., Remon J.P., 1996. Matrix pellets based on the combination of waxes, starches and maltodextrins. Int. J. Pharm. 133, 155-160.
Zhou F., Vervaet C., Schelkens M., Lefebvre R., Remon J.P., 1998. Bioavailability of ibuprofen from matrix pellets based on the combination of waxes and starch derivatives. Int. J. Pharm. 168, 79-84.
【技術分野】
【0001】
本発明は、針状医薬活性物質の粒径とストランド径(strand diameter)との比が少なくとも1:15である少なくとも1種の針状晶の形態の医薬活性物質を含有する押出物、および、医薬の製造のためのこれらの押出物の使用に関する。
【背景技術】
【0002】
苦い薬物物質の味をマスキングすることは、ヒトの医薬、特に小児科の製剤の服薬率の改善にとって重要であるが、獣医学の薬物においてもそうである。最も簡単なタイプの味のマスキングは、芳香の添加であるが、それは、非常に苦く、水溶性の物質では問題となり得る(Bienz, 1996)。活性物質を疎水性担体で顆粒に加工することによる味のマスキングも記載された(Kalbe and Hopkins, 1998)。他の可能性は、剤形の被覆である。このために使用される物質の例は、Eudragit E(Cerea et al., 2004; Lovrecich et al., 1996; Ohta and Buckton, 2004; Petereit and Weisbrod, 1999)、セラック(shellac)(Pearnchob et al., 2003b; Pearnchob et al., 2003a)およびセルロース誘導体(Al-Omran et al., 2002; Li et al., 2002; Shirai et al., 1993)である。Eudragit E の欠点は、味のマスキングが陽イオン性添加物と陰イオン性活性物質のイオン性相互作用に基づく点である。セラックの欠点は、それが天然ポリマーであり、その組成が変動し得る点である。この他に、剤形の被覆は、追加的な費用と時間のかかる工程である。加えて、不溶性セラックマトリックス中のキノロンまたはナフチリドンカルボン酸の固体分散物が記載された(Cabrera, 2002)。
【0003】
イオン交換樹脂および封入複合体(inclusion complexes)も、味のマスキングに使用される。イオン交換樹脂の有用性は、薬物物質がイオン特性を持っていなければならないという事実により限定される(Chun and Choi, 2004; Lu et al., 1991; Prompruk et al., 2005)。封入複合体は、少量の薬物物質しか担持できない(Sohi et al., 2004)。
【0004】
脂質基剤も、味のマスキングに使用できる。味のマスキングのためにレシチンおよび甘味料も含有する、固い脂肪をベースとする一体化した剤形が記載された(Suzuki et al., 2003; Suzuki et al., 2004)。ここでの欠点は、製造工程で脂質を完全に融解しなければならず、物理的な不安定性を導き得ることである。さらに、固い脂肪、ジステアリン酸グリセロールおよびステアリン酸は、冷間押出において親油性結合剤として使用され、味のマスキングを達成するために、被覆としての Eudragit E の使用も必要である(Breitkreutz et al., 2003)。味のマスキングを目的とするものではないが、剤形を製造するための融点より低温での脂質の押出も記載された(Reitz and Kleinebudde, 2007; Windbergs et al., 2008)。
【0005】
味がマスキングされたキノロン型のジャイレース阻害剤の製剤は、活性物質を高級脂肪酸、および、必要であれば他の添加物と混合し、加熱し、冷却の後に造粒または微粉砕することにより得られた(Ahrens et al., 1998)。さらに、蝋をベースとするペレット剤も製造された(Adeyeye and Price, 1991; Adeyeye and Price, 1994; Zhou et al., 1996; Zhou et al., 1998)。これらの研究は、活性物質の放出は、使用した蝋の融点およびペレット中の濃度に依存することを示した。蝋の融点および含有量が高いほど、放出は遅い。味のマスキングのさらなる可能性は、Kim および Choi(2004)により記載され、彼らは、ココアバターの核または固い脂肪および活性物質を調製し、これにアルギン酸ナトリウムまたはカラゲナンの被覆を施した。しかしながら、この方法では、脂肪は完全に融解し、それは安定性の理由で不利であり、さらに、被覆のための製造工程は追加的な工程であった。
【0006】
加えて、Compritol(登録商標)888 ATO は、マトリックス形成成分として記載された。そのペレット剤は、融解したCompritol(登録商標)、活性物質および多糖類の被覆からなった(Mirghani et al., 2000)。他の研究では、マトリックス錠剤は、粉末混合物から直接、または微粉砕された固体分散物から圧縮された。微粉砕された固体分散物に由来する錠剤は、より良好な味のマスキングを示したが、これらの製造のためには、Compritol(登録商標)を完全に融解しなければならなかった(Li et al., 2006)。Barthelemy は、Compritol(登録商標)を、テオフィリンのペレット剤および顆粒剤の被覆に使用した。ここでも、この脂肪は完全に融解された(Barthelemy et al., 1999)。
【0007】
リン脂質の使用は、苦味をマスキングするもう1つの可能性であるが、他の味のタイプのものではない(Katsuragi et al., 1997; Takagi et al., 2001)。加えて、リン脂質の添加は、脂質の結晶化度に影響し、不安定性を導き得る(Schubert, 2005)。粉末の味のマスキングは、小さい添加物の粒子が大きい活性物質粒子に沈着することにより可能である(Barra et al., 1999)。
【0008】
押出により中に活性物質を組み込んだ動物飼料も記載された(Huber et al., 2003)。塩基性薬物物質およびメタクリル酸ポリマーの溶融押出により、Petereit らは、味がマスキングされた押出物を得、次いで、それを顆粒または粉末に破砕し(Petereit et al., 2003)、そして、この2つの成分を中鎖ないし長鎖脂肪酸と融解状態で混合することにより、迅速に崩壊する剤形を得た。凝固後、生成物を破砕し、水溶性マトリックスに包埋した(Petereit et al., 2004)。活性物質をベヘン酸エステルの脂質マトリックスおよび疎水性希釈剤の中に含有する制御放出の医薬も研究された(Nabil et al., 1998)。Thombre は、味をマスキングする添加物を含む多粒子形態のペット用製剤を記載している(Thombre, 2004)。
【0009】
係属中の出願「味のマスキングが改善された押出物」(ドイツ特許出願番号102007026550.8、対応するPCT出願番号PCT/EP2008/004218も参照)では、医薬的に有用なストランド径0.5mm以下の押出物が記載された。これらの押出物は、薬物物質の味のマスキングに適する。
【0010】
しかしながら、活性物質を針状の形態で含有する混合物の押出では、予期せぬ困難が生じる:使用する薬物物質の結晶形が針状であるならば、針状晶の長さがストランド径よりも顕著に小さいときでさえ、安定かつ再現性のある製造方法は実施できない。原則として、針状薬物物質に伴う加工の困難性は他の製造技術で周知であった。粉末および結果的な錠剤の特性は、使用する物質の粒子の形態に依存することが、様々な研究で示された(Alderborn and Nystroem, 1982; Wong and Pilpel, 1990)。例えば、針状結晶相のパラセタモールは、他の結晶形態よりもはるかに不十分に錠剤に圧縮され得、それは錠剤のキャッピングおよび粉末流動性の乏しさに現れる(Wang & Zhang, 1995)。針状結晶の形態では、イブプロフェンも非常に乏しい流動性、粘着性および接着性を示し、圧縮および打錠に高いエネルギーの投入が必要であり、得られた錠剤は機械的に不安定であった。凝集または等軸結晶形態への再結晶により、イブプロフェンの打錠特性を顕著に改善できた(Jbilou et al., 1999; Rasenack and Mueller, 2002)。
【0011】
しかしながら、押出方法に関しては、針状粒子が押出の方向に並ぶことがガラス加工から知られていた。この場合、製造されるガラスにおける異方性を達成するために、整列は望ましかった(Moisescu et al., 1999)。等軸の粒子と比較して、溶融押出中の軟化したガラス塊の粘度は、針状粒子を含有する場合に顕著に高かった(Yue et al., 1999)。
【0012】
当業者は、特に、粒子が押出に都合よく平行に整列すると確かに想定できたので、粒径がストランド径よりもかなりの倍率(例えば5)で小さい限り、問題はないと予想したであろう。
【0013】
しかしながら、驚くべきことに、針状活性物質を含有する混合物の押出には問題が生じる:製造方法の問題は、ノズルプレートの前での蓄積の形で現れ、ノズル器具が詰まり、その結果、押出ノズルプレートの前の圧力が上昇する。それに加えて、粉末混合物の流動が乏しいので、高い加工速度で一定の供給速度を維持することは殆ど不可能である。
【0014】
満足のいく針状活性物質の押出方法に到達するために、様々なアプローチが可能である:製剤の改変、例えば、脂質基剤の変更、さらなる添加物の添加、または、様々な活性物質の負荷量を用いる実験は、十分な改善をもたらさない。さらに、加工の方向にノズル管が段階的に広くなるノズルプレートも使用できるが、この場合、これは、いくつかの状況下で、工程の均一性の悪化を導く。開口部のドリル(aperture drillings)に特に滑らかな表面を有するノズルプレートを介する押出は、有意な変化をもたらさない。さらなる装置の変形として、様々なスクリュー配置を使用できるが、これも何の改善ももたらさない。さらに、温度、供給速度およびスクリュー回転速度の工程パラメーターを変更できる。使用する脂質の融解範囲より20℃低い押出温度から、融解範囲内までが可能である。低すぎる温度では、ノズルはすぐに詰まり、圧力は非常に急速に高まり、高すぎる温度では、脂質は完全に融解し、柔らかいペーストとしてノズルを出る。
【0015】
しかしながら、大きいノズル直径を用いると、一定の改善が達成されることが見出されている。他の改変のいずれも決定的にこの方法を改善しないので、活性物質粉末を破砕する(例えば、エアジェットミル(air jet mill)で);針状結晶構造を、破砕により微粉砕する。十分に小さい粒径では、押出物を何の問題もなく製造できることが見出されている。十分に細かく破砕された活性物質を用いる押出方法は、高い薬物物質の負荷量(例えば、50%または80%まででさえ)および例えば0.3mmまたはわずか0.2mmのノズル直径でも、原則として円滑かつ再現可能に一定の圧力で進行する。
【発明の概要】
【0016】
従って、本発明は、以下のものに関する:
・針状医薬活性物質の粒径のストランド径に対する比が少なくとも1:15であることを特徴とする、少なくとも1種の医薬活性物質を針状晶の形態で含有する押出物。
・医薬を製造するための、上記の押出物の使用。
【0017】
針状医薬活性物質の粒径とストランド径との比は、通常、少なくとも1:15、好ましくは少なくとも1:20、特に好ましくは少なくとも1:25、ことさら特に好ましくは少なくとも1:50、特に少なくとも1:100である。
【0018】
疑念がある場合には、ここでの粒径は、レーザー回折法で測定されるd(0.9)値を意味すると理解されるべきである。本発明の意味では、d(0.9)は、全粒子の90%がこの値以下の寸法(直径)を有する、体積に基づく粒径分布を意味すると理解される(場合により、用語d(90)またはd(v,90)もこのために使用され、後者は、これが体積に基づく粒径分布であることを明確にするためである)。用語d(0.5)、d(0.1)なども、対応して理解されるべきである。ここで述べる粒径は、活性物質粒子の屈折率が不明であるので、Malvern Mastersizer 2000(分散ユニット Hydro 2000G)およびフラウンホーファー回折評価モードを用いるレーザー回折法により測定した。このために、適量のサンプル溶液を分散媒体(例えば、プラジカンテルには0.1%スルホコハク酸ジオクチルナトリウム水溶液、または、メサラジンにはエタノール)2−3mlに、撹拌しながら予め分散させた。次いで、分散物を装置の分散ユニットに撹拌(300rpm)およびポンプ輸送(900rpm)しながら供給し、測定を行った。評価ソフトウェアは、粒径をd(0.9)値(またはd(0.5)値など)として出力した。
【0019】
一般的な溶媒に溶解しすぎる活性物質粒子(例えば、カフェイン)は、適当なユニット(例えば、Scirocco 2000 乾燥粉末供給機)を用いて、0.5barの空気圧の気流を利用して乾燥分散させる。
【0020】
本発明による押出物のストランド径は、好ましくは最大で0.5mm、特に好ましくは最大で0.3mmである。通常、直径0.1mmを超える、好ましくは0.2mmを超える押出物を使用できる。非円筒形の押出物では、最大の縁の長さまたは楕円の長さは、最大で0.5mm、好ましくは最大で0.3mmである。
【0021】
押出物は、熱可塑的に変形できる物質またはいくつかの熱可塑的に変形できる物質の混合物からなる押出に適する基剤、並びに、必要であれば、さらなる医薬的に許容し得る補助物質および添加物を含有する。
【0022】
基剤は、熱可塑的に変形できる物質、例えば、ポリマー、例えば、ポリアクリル酸塩、または、セルロース誘導体、脂質、例えば、アシルグリセリド類、界面活性剤、例えば、モノステアリン酸グリセロールまたはステアリン酸ナトリウム、マクロゴール類、例えば、ポリエチレングリコール6000、糖または糖アルコール、例えば、マンニトールまたはキシリトールからなる。好ましくは、脂質基剤を使用する。脂質基剤として、例えば、脂肪基剤、特に、グリセロールエステル類が適し、これらは、好ましくはC12−C24脂肪酸とのエステルである。グリセロールエステルとして、グリセロールジエステル類、例えば、ジベヘン酸グリセロール、グリセロールトリエステル類、例えば、トリラウリン酸グリセロール、トリミリスチン酸グリセロール、トリパルミチン酸グリセロールまたはトリステアリン酸グリセロール、および、グリセロールモノ−、ジ−およびトリエステルの混合物、例えば、パルミチン酸ステアリン酸グリセロールに言及し得る。ココナッツバター、パーム油および/またはパームナッツ油(例えば、Witocan(登録商標)の名称で購入できる固い脂肪)をベースとするトリグリセリド類にも言及し得る。クエン酸および/または乳酸のモノ−またはジグリセリド類も使用できる。
さらに、蝋、特に、30ないし60個の炭素原子のもの、例えば、パルミチン酸セチルに言及し得る。
そのような脂質は、例えば、Precirol(登録商標)、Compritol(登録商標)および Dynasan(登録商標)の名称で購入できる。
【0023】
グリセロールジエステルの系列で特に好ましい例は、ジベヘン酸グリセロール(例えば、Compritol(登録商標)888 ATO、これは、主にジベヘン酸グリセロールを含有するが、モノベヘン酸グリセロールおよびトリベヘン酸グリセロールも含有する)である。グリセロールトリエステルの系列で特に好ましい例は、トリミリスチン酸グリセロール(例えば、Dynasan(登録商標)114)、トリパルミチン酸グリセロール(例えば、Dynasan(登録商標)116)およびトリステアリン酸グリセロール(例えば、Dynasan(登録商標)118)である。
【0024】
好ましくは、脂肪基剤は、粉末形態である。多くの脂質は多形性であり、いくつかの状況下で、温度および圧力が変化する場合に、準安定な形態を形成できる。いくつかの状況下で保存すると、変態の変換が起こり得、より安定な変態が形成され得る。文献の記載によると [Reitz and Kleinebudde, 2007; Windbergs et al., 2008]、グリセロールトリエステル類(例えば Dynasan(登録商標)として知られる)、特にトリミリスチン酸グリセロール(Dynasan 114(登録商標))またはトリパルミチン酸グリセロール(例えば、Dynasan(登録商標)116)またはトリステアリン酸グリセロール(例えば、Dynasan(登録商標)118)は、そのような変化に対して比較的安定であり、従って、医薬用の脂質基剤として特に適する。
【0025】
特に、脂肪基剤として使用する物質は、しばしば、例えば、モノ−、ジ−および/またはトリグリセリド類の混合物として販売される。本質的に1種の成分のみからなる均質な脂肪基剤は、これらより好ましい。これらの添加物を用いて製造される製剤は、良好な保存安定性を特徴とする。
【0026】
使用する基剤(熱可塑的に変形できる物質からなる)の量は、押出物に含まれる他の物質の量に応じて決まる。通常、15ないし99%[w/w]、好ましくは20ないし99%[w/w]、特に好ましくは25ないし80%[w/w]、ことさら特に好ましくは30ないし70%[w/w]を使用する。
使用する基剤、特に脂質基剤は、概して、その下限が通常少なくとも50℃、好ましくは少なくとも60℃である融点範囲を有する。
【0027】
本発明による押出物は、必要であれば、1種またはそれ以上のさらなる補助物質および添加物を含有できる。これらとして可能なものは、流動調節剤、好ましくは、濃度0.2ないし2%[w/w]のコロイド状二酸化ケイ素、滑沢剤、好ましくは濃度0.2ないし5%[w/w]のステアリン酸マグネシウムまたはジベヘン酸カルシウム、および、界面活性剤、好ましくは濃度0.5ないし10%[w/w]のレシチンである。さらに、抗酸化剤を使用でき、例えば、通常の量、概して0.01ないし0.5%[w/w]、好ましくは0.05ないし0.2%[w/w]で使用されるブチルヒドロキシアニソール(BHA)またはブチルヒドロキシトルエン(BHT)が適する。活性物質の放出は、例えば、いわゆる孔形成物質の添加により制御できる。これらの例は、糖、特にラクトース、ポリオール類、特にマンニトール、または、ポリエチレングリコール類(PEG)、好ましくはPEG1500ないし10000、特に好ましくはPEG1500ないし6000、例えば、PEG1500(Macrogol 1500)である。孔形成物質は、5ないし40%[w/w]の濃度で、好ましくは5ないし20%[w/w]の濃度で使用する。活性物質の放出に影響を与える他の可能性は、崩壊補助剤の添加である。加えて、クロスポビドン、クロスカルメロースナトリウムまたは架橋ナトリウムカルボキシメチルスターチなどの、いわゆる超崩壊剤(super disintegrator)を使用できる。超崩壊剤は、1ないし15%[w/w]の濃度、好ましくは3ないし10%[w/w]の濃度で使用する。これに代わって、酸に溶ける、かつ/または、二酸化炭素を放出する物質、例えば、炭酸マグネシウムまたは炭酸カルシウムを使用できる。二酸化炭素放出物質は、5ないし15%[w/w]の濃度で、好ましくは5ないし10%[w/w]の濃度で使用する。
【0028】
さらに、本発明による押出物は、帯電防止剤を含有できる。これは、特に、帯電が押出に影響する場合に推奨される。帯電は、ノズル開口部の詰まりをもたらし得、これは、帯電防止剤の添加により防止できる。帯電防止剤として、PEGを好ましく使用でき、特に、可能なのはPEG1500ないし6000である。PEGは、帯電防止効果を発揮するために、好ましくは粉末形態であり、押出中に融解するべきである。故に、帯電防止剤としてのPEGの添加に伴い、押出中にPEGが融解するが、使用する脂肪基剤は融解しないように、PEGの融解温度は十分に低いものであるべきである。実際に、帯電は、全ての基剤で起こるわけではない。それは、とりわけ、脂肪酸とのグリセロール脂肪酸トリエステル類、例えば、トリミリスチン酸グリセロール、トリパルミチン酸グリセロールまたはトリステアリン酸グリセロールで観察される。故に、そのような基剤の使用に伴い、問題のない押出を確実にするために、押出される混合物に帯電防止剤を添加することが推奨される。帯電防止剤は、少なくとも5%[w/w]、好ましくは少なくとも10%[w/w]の濃度で使用する。通常、30%[w/w]以下を使用する。
【0029】
医薬活性物質として、薬物の活性物質、特に、不快な味をマスキングしなければならないものを使用できる。
本発明によると、針状の活性物質を使用する。一般に、これらは針状結晶である。
ここで、「針状晶」または「針状」は、長さが直径よりも顕著に大きく、長さ/直径の比が、少なくとも3:1より大きく、好ましくは5:1より大きく、特に好ましくは10:1より大きく、ことさら特に好ましくは20:1より大きい粒子を意味すると理解されるべきである。針状晶は、概して丸くないので、疑念がある場合には、「直径」は、長さに対して垂直な最大の寸法を意味すると理解されるべきである。
【0030】
本質的に、活性物質を融解する必要がないので、針状活性物質の選択に大きい制限はない。押出物の味のマスキング作用のために、それらは、好ましくは、不快な(例えば、苦い)味の活性物質に適する。
活性物質は、例えば、以下の群に属するものであり得る:抗生物質、寄生性原生動物に対する薬物、駆虫剤、代謝刺激剤および炎症阻害性物質。
当然、活性物質の用語には、針状の塩および溶媒和物も含まれる。
【0031】
活性物質の特定の例として、メサラジンに言及し得る。メサラジンは、慢性炎症性腸疾患に対して使用される炎症阻害性薬物物質である。それは水にあまり溶けず、280℃の融点および針状の結晶形を有する。
さらなる例は、結晶性カフェインである。
【0032】
針状活性物質の好ましい特定の例として、条虫および住血吸虫に対して有効な昔から知られてきた駆虫剤であるプラジカンテルに言及し得る。それは水にあまり溶けず、139℃の融点および針状の結晶形を有する。ピンディスクミル(pinned disc mill)で破砕した、レーザー回折法により測定されるd(0.9)として表される粒径25μmの等級(図1)を、通常は使用する。
【0033】
親油性マトリックスに包埋することにより、使用する活性物質の性質に応じて、遅延放出、および、故に、効果の遅延を達成できる。
押出物中で使用する活性物質の量は、作用の強さおよび所望の投与量によって決まる。80%[w/w]まで、好ましくは70%[w/w]まで、特に好ましくは60%[w/w]までの濃度の、高い活性物質濃度の押出物でさえ製造できることが見出されている。通常の濃度範囲の例は、1ないし80%[w/w]、好ましくは5ないし70%[w/w]、そして、特に好ましくは30ないし60%[w/w]である。
【0034】
本発明による押出物は、出発物質(医薬活性物質、基剤および必要であれば補助物質および添加物)を混合し、次いで押し出すことにより製造する。押出は、好ましくは、熱可塑的に変形できる物質の完全な融解に至らない温度で、実際には、通常、室温の範囲の温度で、好ましくは40℃で、熱可塑的に変形できる物質の融解範囲より低温で実施する。実際に、これは、通常、問題の基剤について定められている融解範囲に基づく。概して、押出の温度は、基剤、特に脂肪基剤の融解範囲の下限よりも20℃、好ましくは15℃、特に好ましくは10℃より低くは設定しない。通常、押出の温度は、基剤の融解範囲の下限よりも高くは設定せず、好ましくは1℃低く、特に好ましくは5℃低い。目的は、柔らかいペーストの押出を回避することである。押出工程は、できるだけ一定の物質温度で実施すべきである。この目的で、スクリュー押出機(screw extruder)、特に二軸スクリュー押出機は、特に適する。押し出されたストランドは、好ましくは円形の断面および上記の直径を有する。押し出されたストランドは、押出の際にナイフで直接粉砕できるか、または、通常のミル、例えば遠心ミルで、穏やかにひくことにより、別工程で粉砕できる。得られる生成物の粒度は、使用するノズルの直径によって決まり、粉砕されたストランドは、最大でストランド径の3倍に相当する長さを有する。典型的な粒度は、例えば、300ないし500μmであり、あるいは、小さいノズル直径の場合、200ないし500μmでさえある。好ましい実施態様によると、破砕された生成物を篩にかけることもできる。かくして、微粉の画分を除去できる。
【0035】
本明細書で時折使用される、押出物はそれらの融点より低温で押し出されるという記述は、上述の通り、押出物が、使用する熱可塑性の基剤が完全に融解しない温度で押し出されることを意味すると理解されるべきである。しばしば、他の成分、例えば活性物質は、これより高い融点を有する。そのような押出物は、不快な味の成分の味のマスキングに適する。
【0036】
穏やかにひいた後、本発明による押出物は、必要であれば、さらに適する剤形に加工できる。さらなる添加物の添加が、さらなる加工のために場合により必要である。本発明によると好ましい剤形は、必要であれば所望の適用に適する形状を有していてもよい錠剤である。可能な他の剤形は、ペースト剤、懸濁剤、サシェ剤、カプセル剤などである。
【0037】
本発明による押出物または医薬は、一般的に、ヒトおよび動物における使用に適する。それらは、好ましくは、農業、育種、動物園、研究室および実験用の動物、および、「趣味の動物」、および、特に哺乳動物において、動物の管理および動物の繁殖に使用する。
【0038】
農業用および繁殖用の動物には、哺乳動物、例えば、ウシ、ウマ、ヒツジ、ブタ、ヤギ、ラクダ、スイギュウ、ロバ、ウサギ、ダマジカ、トナカイ、毛皮動物、例えば、ミンク、チンチラ、アライグマ、および、鳥類、例えば、ニワトリ、ガチョウ、シチメンチョウ、アヒル、ハトおよびダチョウのタイプが含まれる。好ましい農業用動物の例は、ウシ、ヒツジ、ブタおよびニワトリである。
【0039】
研究室および実験用の動物には、イヌ、ネコ、ウサギ並びに齧歯類、例えば、マウス、ラット、モルモットおよびハムスターが含まれる。
愛玩動物には、イヌ、ネコ、ウマ、ウサギ、齧歯類、例えば、ハムスター、モルモット、マウス、並びに、家庭および動物園で飼育するための爬虫類、両生類および鳥類が含まれる。
【0040】
押出物は、通常、経腸で、特に経口で、直接または適する製剤の形態(剤形)で使用する。
経腸使用は、例えば、経口で、顆粒剤、錠剤、カプセル剤、ペースト剤、懸濁剤または薬用動物飼料の形態で行う。経口投与の1つの選択肢は、いわゆるトップドレッシング(top dressing)であり、これは、動物飼料に乗せられ、飼料と共に摂取される散剤、顆粒剤またはペースト剤である。
【0041】
適する製剤は以下のものである:
固体製剤、例えば、顆粒剤、ペレット剤、錠剤、巨丸剤および有効成分を含有する成形品(moulded body)。
固体製剤の製造には、粉砕した押出物を、適する担体と、必要であれば添加物を添加して混合し、所望の形態にする。
担体として、全ての生理的に適合する固体の不活性物質に言及し得る。無機および有機物質をそのように使用する。無機物質の例は、食塩、炭酸カルシウムなどの炭酸塩、炭酸水素塩、酸化アルミニウム、ケイ酸、アルミナ、沈殿またはコロイド状二酸化ケイ素およびリン酸塩である。
有機物質の例は、糖、セルロース、食糧および動物飼料、例えば、粉乳、動物食餌、穀粉および穀物、並びに、スターチである。
【0042】
添加物は、防腐剤、抗酸化剤および着色料である。適する添加物および添加する必要量は、本質的に当業者に知られている。防腐剤として、例えばソルビン酸に言及し得る。抗酸化剤として、例えばブチルヒドロキシアニソール(BHA)またはブチルヒドロキシトルエン(BHT)が適する。可能な着色料は、有機および無機着色料、または、医薬の目的に適する色素、例えば、酸化鉄である。
【0043】
さらなる適する添加物は、滑沢剤および離型剤、例えば、ステアリン酸マグネシウム、ステアリン酸、タルク、ベントナイト類、崩壊促進物質、例えば、スターチまたは架橋ポリビニルピロリドン、結合剤、例えば、スターチ、ゼラチンまたは直鎖状ポリビニルピロリドン、および、乾燥結合剤、例えば、結晶セルロースである。
【0044】
さらなる添加物として、油、例えば、植物油(例えば、オリーブ油、大豆油、ヒマワリ油)、または、動物起源の油、例えば、魚油を使用できる。通常量は、0.5ないし20%[w/w]、好ましくは0.5ないし10%[w/w]、特に好ましくは1ないし2%[w/w]である。
【0045】
懸濁剤を経口で使用できる。それらは、粉砕した押出物を、担体の液体に、必要であれば他の添加物、例えば、湿潤剤、着色料、吸収促進物質、防腐剤、抗酸化剤または光安定化剤を添加して、懸濁することにより製造する。
【0046】
可能な担体の液体は、問題の押出物が溶解しない均質な溶媒または溶媒混合物である。例えば、生理的に適合する溶媒、例えば、水、アルコール、例えば、エタノール、ブタノール、グリセリン、プロピレングリコール、ポリエチレングリコールおよびそれらの混合物に言及し得る。
【0047】
湿潤剤(分散剤)として、界面活性剤を使用できる。例えば:
非イオン性界面活性剤、例えば、ポリエトキシル化ヒマシ油、ポリエトキシル化モノオレイン酸ソルビタン、モノステアリン酸ソルビタン、モノステアリン酸グリセリン、ステアリン酸ポリオキシエチル、アルキルフェノールポリグリコールエーテル;
両性界面活性剤、例えば、ジ−NaN−ラウリル−β−イミノジプロピオネートまたはレシチン;
陰イオン性界面活性剤、例えば、ラウリル硫酸Na、脂肪アルコールエーテルサルフェート、モノ/ジアルキルポリグリコールエーテルオルトリン酸エステルモノエタノールアミン塩;および、
陽イオン性界面活性剤、例えば、セチルトリメチル塩化アンモニウムに言及し得る。
【0048】
さらなる添加物として、例えば:
増粘性物質および懸濁安定化物質、例えば、カルボキシメチルセルロース、メチルセルロースおよび他のセルロースおよびスターチ誘導体、ポリアクリル酸塩、アルギン酸塩、ゼラチン、アラビアゴム、ポリビニルピロリドン、ポリビニルアルコール、メチルビニルエーテルおよび無水マレイン酸のコポリマー、ポリエチレングリコール、蝋、コロイド状ケイ酸または列挙した物質の混合物に言及し得る。
【0049】
半固体製剤は、経口投与できる。それらは、それらの高い粘性でのみ、上記の懸濁剤および乳剤と異なる。
活性物質は、共力剤または他の活性物質と組み合わせて使用することもできる。
【実施例】
【0050】
実施例
断りのない限り、百分率の値は、完成した混合物を基準とする重量パーセントである。
I. 比較例:プラジカンテルを含む押出物:
粒径d(0.9)=25μm(図1参照)の活性物質プラジカンテル(50%[w/w])、および、添加物の Compritol(登録商標)888 ATO(49%[w/w])、主成分がジベヘン酸グリセロールの脂肪基剤(モノ−およびトリエステル、および、それより少ない量のC16−C20脂肪酸とのエステルも含有する)、および、Aerosil(登録商標)200(1%[w/w])、その使用が粉末塊の流動性の改善に貢献する発熱性コロイド状二酸化ケイ素(高分散二酸化ケイ素とも記載される)からなる粉末混合物を、押出の前に、実験用ミキサー中、室温で混合し(15分間、40rpm)、粉末混合物を押出機の重量測定による供給機に移した。
【0051】
溶融押出のために、円形の工具およびブラントスクリューアタッチメントを備えた一定速度の二軸スクリュー押出機を使用した。
この製剤の押出の過程では、多くのノズル開口部が詰まり(ノズルプレートは、各々直径0.3mmの全部で67個のノズル孔を有する)、結果的に、一定の回転速度および供給速度で、圧力は継続的に上昇した(図2)。未破砕のプラジカンテルを用いる工程の間にノズルプレートの後ろに蓄積した圧力のために、開いたままのノズル開口部から押出物が出てくる速度は非常に顕著に上昇した。
【0052】
II. 実施例:細かく破砕したプラジカンテルを含む押出物
エアジェットミルで2回破砕したプラジカンテル(d(0.5)=1.7μm、d(0.9)=3.6μm、図3参照)を使用して、比較例の実験を繰り返した。
破砕したプラジカンテルを用い、薬物物質充填率50%およびノズル直径0.3mmを用いる押出工程は、一定の圧力で均一かつ再現可能であった。図4に示す工程の過程から、約12分後に一定のレベルで、10barで安定した圧力は、未破砕のプラジカンテルを用いる工程(図2)のもの(15分間の加工時間の後、圧力は約40barまで上昇した)より顕著に低かったことがわかる。押出は、全てのノズル開口部を通して、均一の速度で起こった。
【0053】
比較例Iと本実施例の押出物を比較すると、違いが見られる:かなりの摩擦のために、より多くの脂質が押出物の表面に押しつけられ、それは、走査型電子顕微鏡で非常に明確に見ることができる;押出物の表面は滑らかで平坦である。これと対照的に、破砕したプラジカンテルを含む押出物の表面は平坦ではなく、プラジカンテル粒子は表面下に直接位置し、それらの形状は薄い脂質層を通して明白である。
【0054】
III. 比較例:未破砕のメサラジンを含む押出物
未破砕のメサラジン(d(0.5)=10.7μm、d(0.9)=44.0μm、図5参照)を含む押出物を、比較例Iと同様に製造した。50%[w/w]未破砕メサラジン/49%[w/w]Compritol(登録商標)/1%[w/w]高分散二酸化ケイ素(Aerosil(登録商標))の粉末混合物を使用し、直径0.3mmのノズルプレートを通して押出を実施した。製造工程で、比較例Iと同様の問題が起こった。工程の過程から(図6)、10分後に圧力は約25barで安定したが、依然として本当に強く上下することが明らかである。この実験でも、部分的なノズル開口部の詰まりが観察されたが、これは、プラジカンテルほど急速にも、大規模にも起こらなかった。
【0055】
IV. 実施例:破砕したメサラジンを含む押出物
エアジェットミルで2回破砕することにより、より小さい粒径のメサラジンを得た:d(0.5)=4.9μm、d(0.9)=11.9μm、図7参照。破砕の後、粒子は顕著に変化した形状を示した。
比較例IIIと同様の押出方法におけるこの破砕したメサラジンの使用を通じて、この方法を決定的に改善することが可能であった。図8に、圧力は5分後に20barの一定レベルで既に安定し、押出は全てのノズル開口部を通して均一の速度で起こったことを見ることができる。故に、破砕したメサラジンの使用により、ノズル開口部の詰まりを防止でき、かくして押出方法は改良された。
【0056】
押出物を比較すると、比較例の針状メサラジン結晶は押出中に大きく破壊され、一方、この実施例の破砕したメサラジン粉末は、押出後にもとの粒子の形状および大きさを依然として有し、それは、例えば加熱できるスライドを備えた光学顕微鏡下で明確に見られることが見出された。これと対照的に、針状プラジカンテル結晶は、押出中に破壊されず、もとの形状および大きさで、押出物中に依然として存在した。
【0057】
上記の結果に基づき、当業者は、大きい針状メサラジン結晶は、小さいプラジカンテル針状晶よりも押出において多大な問題を引き起こすと予測するであろう。しかしながら、大きいメサラジン針状晶は、工程中の押出機のスクリューの剪断作用により既に殆ど粉砕されていたので、ノズル開口部の詰まり、および、これに起因する急速な圧力の蓄積は、メサラジンではプラジカンテルほど強く起こらなかった。
【0058】
V. 比較例:帯電防止剤を含まない押出物
破砕したプラジカンテルを用いて、実施例IIと同様に押出物を製造した。50%[w/w]破砕プラジカンテル/49%[w/w]Dynasan 116(登録商標)/1%[w/w]高分散二酸化ケイ素(Aerosil(登録商標))の粉末混合物を使用した;Dynasan 116(登録商標)は、98%がトリパルミチン酸グリセロールからなる脂肪基剤である。押出は、直径0.3mmのノズルプレートを通して実施した。
【0059】
この製剤の押出の過程で、リン脂質塊のノズル内表面に対する摩擦によりかなり強い帯電が押出物に生じ、数分後に、押出物は既に強く押出機自体に静電気的に引き付けられ、ノズルヘッドに付着したままになった。この電荷のために、破砕したプラジカンテルを使用したが、いくつかのノズル開口部は詰まり、ノズルプレートの後ろの圧力は上昇した。電荷の発生は、工程中にノズルプレートの直前に取り付けた静電気検出器IZD 10-510(SMCより)を利用して、継続的に記録できた。この比較例の押出中に、わずか2分後に、帯電は既に−5kVの値に達した(図9)。
【0060】
VI. 実施例:帯電防止剤を含む押出物
比較例Vと同様に、5および10%のPEG1500を帯電防止剤として添加して、押出物を製造した。50%[w/w]破砕プラジカンテル/44%[w/w]Dynasan 116(登録商標)/5%[w/w]PEG1500/1%[w/w]Aerosil(登録商標)または50%[w/w]破砕プラジカンテル/39%[w/w]Dynasan 116(登録商標)/10%[w/w]PEG1500/1%[w/w]高分散二酸化ケイ素(Aerosil(登録商標))の粉末混合物を使用した。シリンダーの温度は約60℃であり、それは、押出工程中のPEG1500の融解を確実にした。
【0061】
5%PEGを用いる押出の過程では、数個のノズル開口部が再度詰まり、3分間の工程時間後に、ノズルプレートの前に1−2kVの帯電が測定された。これと対照的に、10%PEGを用いる押出工程は、ノズル開口部の詰まりおよび測定可能な帯電を伴わず、均一かつ再現可能に進行した(図9)。
帯電防止剤としての10%PEGの添加により、高純度のグリセロールトリエステル類を含有する押出物の帯電を防止し、かくして、この方法を顕著に改良することが可能であった。
【0062】
VII. 比較例:未融解の帯電防止剤を含む押出物
実施例VIと同様に、10%PEG6000を帯電防止剤として添加して押出物を製造した。50%[w/w]破砕プラジカンテル/39%[w/w]Dynasan 116(登録商標)/10%[w/w]PEG6000/1%[w/w]高分散二酸化ケイ素(Aerosil(登録商標))の粉末混合物を使用した。シリンダーの温度は、最初に約60℃であり、次いで8分後にまず55℃に、最終的に52℃に冷却した。PEG6000の融解範囲は約55−60℃であり、シリンダー温度の低下後に、PEGはもはや融解形態で存在しなかった。
【0063】
最初に、10%PEG1500を用いた実施例VIと同様に、この方法は均一に帯電せずに行われ、全てのノズル開口部は開いていた(図10)。温度低下後、押出物は帯電し、いくつかのノズル開口部は詰まり、結果として、ノズルプレートの後ろの圧力は上昇した。
PEGは、その帯電防止作用を発揮するために、押出工程中に融解形態で存在するべきであると示すことができた。
【0064】
VIII. 比較例:未破砕のカフェインを含む押出物
比較例Iと同様に、未破砕のカフェインを用いて押出物を製造した。50%[w/w]未破砕カフェイン(粒径d(0.9)=1170μm)/49%[w/w]Compritol(登録商標)/1%[w/w]高分散二酸化ケイ素(Aerosil(登録商標))の粉末混合物を使用した;押出は、直径0.3mmのノズルプレートを通して実施した。製造工程で比較例Iと同様の問題が生じた。8分間の押出の後に、ノズル開口部の完全な詰まりにより圧力は既にかなり上昇したので、工程を止めなければならなかった。
【0065】
IX. 実施例:破砕したカフェインを含む押出物
エアジェットミルで2回破砕することにより、より小さい粒径のカフェインを得た。
この破砕したカフェインを実施例IIと同様の押出工程で使用することにより、この方法を決定的に改良できた。押出中に、3分後に圧力は5barの一定レベルで既に安定し、押出は全てのノズル開口部を通して均一の速度で起こった。故に、破砕カフェインの使用を介して、ノズル開口部の詰まりを防止でき、押出方法を顕著に改良できた。
【0066】
以下の押出物を、上記の実施例と同様に製造した:
X. 実施例
70%[w/w]プラジカンテル(破砕、d(0.9)=5.7μm)
17%[w/w]トリステアリン酸グリセロール(Dynasan(登録商標)118)
12%[w/w]PEG6000
1%[w/w]高分散二酸化ケイ素(Aerosil(登録商標))
ノズル直径:0.3mm(押出中に、100%開いているノズル開口部)、スクリュー速度:60rpm、押出温度:67℃
【0067】
XI. 実施例
50%[w/w]プラジカンテル(破砕、d(0.9)=5.7μm)
29%[w/w]トリステアリン酸グリセロール(Dynasan(登録商標)118)
20%[w/w]PEG6000
1%[w/w]高分散二酸化ケイ素(Aerosil(登録商標))
ノズル直径:0.2mm(180個のノズル開口部、押出中に、100%開いているノズル開口部)、スクリュー速度:60rpm、押出温度:67℃
【0068】
XII. 実施例
50%[w/w]プラジカンテル(破砕、d(0.9)=5.7μm)
29%[w/w]トリステアリン酸グリセロール(Dynasan(登録商標)118)
20%[w/w]PEG6000
1%[w/w]高分散二酸化ケイ素(Aerosil(登録商標))
ノズル直径:0.3mm(押出中に、100%開いているノズル開口部)、スクリュー速度:60rpm、押出温度:67℃
【0069】
XIII. 実施例
50%[w/w]プラジカンテル(破砕、d(0.9)=5.7μm)
49%[w/w]トリステアリン酸グリセロール(Dynasan(登録商標)118)
1%[w/w]高分散二酸化ケイ素(Aerosil(登録商標))
ノズル直径:0.3mm(押出中に、3%開いているノズル開口部)、スクリュー速度:60rpm、押出温度:67℃
【0070】
生物学的実施例:ネコにおける受入試験
40匹のネコを、10匹ずつの4つの群に分けた。以下の方法で、実施例XないしXIIIの各押出物を、それぞれネコ10匹の1つの群で試験した:
プラジカンテル5mg/体重kgの投与量に相当する量の押出物を、乾燥ネコ飼料にトップドレッシングとして添加し、通常の給餌時間でネコに与えた。全てのネコは、飼料を全て食べた。これは、100%の受入の証拠と見なされた。
1週間の休止後、実験を繰り返し、ただし、ここではネコ用の缶詰の飼料(湿っている)を使用した。乾燥飼料と同じ結果が観察され、100%の受入と評価された。
【図面の簡単な説明】
【0071】
図面
【図1】図1:未破砕プラジカンテルの粒径分布、d(0.5)=6.2μm、d(0.9)=25.1μm、湿式分散後にレーザー回折法により測定(Mastersizer 2000 Ver. 5.54, Malvern Instruments, Malvern UK)。
【図2】図2:50%未破砕プラジカンテル/49%Compritol(登録商標)/1%高分散二酸化ケイ素(Aerosil(登録商標))、ノズルプレート直径0.3mmの押出工程の過程
【図3】図3:2回破砕したプラジカンテルの粒径分布、d(0.5)=1.7μm、d(0.9)=3.6μm、湿式分散後にレーザー回折法により測定(Mastersizer 2000 Ver. 5.54, Malvern Instruments, Malvern UK)。
【図4】図4:50%破砕プラジカンテル/49%Compritol(登録商標)/1%高分散二酸化ケイ素(Aerosil(登録商標))、ノズルプレート直径0.3mmの押出工程の過程
【図5】図5:未破砕メサラジンの粒径分布、d(0.5)=10.7μm、d(0.9)=44.0μm、湿式分散後にレーザー回折法により測定(Mastersizer 2000 Ver. 5.54, Malvern Instruments, Malvern UK)。
【図6】図6:50%未破砕メサラジン/49%Compritol(登録商標)/1%高分散二酸化ケイ素(Aerosil(登録商標))、ノズルプレート直径0.3mmの押出工程の過程。
【図7】図7:2回破砕したメサラジンの粒径分布、d(0.5)=4.9μm、d(0.9)=11.9μm、湿式分散後にレーザー回折法により測定(Mastersizer 2000 Ver. 5.54, Malvern Instruments, Malvern UK)。
【図8】図8:50%破砕メサラジン/49%Compritol(登録商標)/1%高分散二酸化ケイ素(Aerosil(登録商標))、ノズルプレート直径0.3mmの押出工程の過程。
【図9】図9:各々0、5および10%のPEG1500含有量の押出工程における、ノズルプレート前の帯電
【図10】図10:50%破砕プラジカンテル/39%Dynasan 116(登録商標)/10%PEG6000/1%高分散二酸化ケイ素(Aerosil(登録商標))の押出工程の過程。
【0072】
文献
Adeyeye C.M., Price J.C., 1991. Development and Evaluation of Sustained-Release Ibuprofen-Wax Microspheres. 1. Effect of Formulation Variables on Physical Characteristics. Pharm. Res. 8, 1377-1383.
Adeyeye C.M., Price J.C., 1994. Development and Evaluation of Sustained-Release Ibuprofen-Wax Microspheres. 2. In-Vitro Dissolution. Pharm. Res. 11, 575-579.
Ahrens G., Mentrup E., Maas J., Radau M., 1998. Production of taste-masked preparations of antibacterially active quinolone derivatives. EP 1998/0855183.
Alderborn G., Nystroem C., 1982. Studies on direct compression of tablets. 3. The effect on tablet strength of changes in particle-shape and texture obtained by milling. Acta Pharm. Suec. 19, 147-156.
Al-Omran M.F., Al-Suwayeh S.A., El-Helw A.M., Saleh S.I., 2002. Taste masking of diclofenac sodium using microencapsulation. J. Microencapsul. 19, 45-52.
Barra J., Lescure F., Doelker E., 1999. Taste masking as a consequence of the organisation of powder mixes. Pharm. Acta Helv. 74, 37-42.
Barthelemy P., Laforet J.P., Farah N., Joachim J., 1999. Compritol(登録商標)888 ATO: an innovative hot-melt coating agent for prolonged-release drug formulations. Eur. J. Pharm. Biopharm. 47, 87-90.
Bienz M., 1996. Taste masking strategies for drug dosage forms. Manufacturing Chemist. 67, 17-20.
Breitkreutz J., El-Saleh F., Kiera C., Kleinebudde P., Wiedey W., 2003. Pediatric drug formulations of sodium benzoate: II. Coated granules with a lipophilic binder. Eur. J. Pharm. Biopharm. 56, 255-260.
Cabrera F.A., 2002. Solid Phase Dispersion of Quinolone-OR Naphthyridonecarboxylic acids. WO 2002/058669.
Cerea M., Zheng W.J., Young C.R., McGinity J.W., 2004. A novel powder coating process for attaining taste masking and moisture protective films applied to tablets. Int. J. Pharm. 279, 127-139.
Chun M.K., Choi H.K., 2004. Preparation and characterization of enrofloxacin/carbopol complex in aqueous solution. Arch. Pharm. Res. 27, 670-675.
Farah N., Barthelemy P., Joachim J., 1998. Method for preparing a pharmaceutical composition with modified release of the active principle, comprising a matrix. WO 1998/14176.
【0073】
Huber G.R., Jones D.R., Kuenzi J.C., Kuenzi K.D., Cabrera F.A., 2003. Animal Feeds including Actives and Methods of using same. WO 2003/030653.
Jbilou M., Ettabia A., Guyot-Hermann A.M., Guyot J.C., 1999. Ibuprofen Agglomerates Preparation by Phase Separation. Drug Dev. Ind. Pharm. 25, 297-305.
Kalbe J., Hopkins T., 1998. Orally administrable granules of hexahydropyrazine derivatives. WO 1998/03157.
Katsuragi Y., Mitsui Y., Umeda T., Otsuji K., Yamasawa S., Kurihara K., 1997. Basic studies for the practical use of bitterness inhibitors: Selective inhibition of bitterness by phospholipids. Pharm. Res. 14, 720-724.
Kim E.-H., Choi H.K., 2004. Preparation of various solid-lipid beads for Drug Delivery of Enrofloxacin. Drug Deliv. 11, 365-370.
Li F.-Q., Hu J.-H., Deng J.-X., Su H., Xu S., Liu J.-Y., 2006. In vitro controlled release of sodium ferulate from Compritol(登録商標)888 ATO-based matrix tablets. Int. J. Pharm. 324, 152-157.
Li S.P., Martellucci S.A., Bruce R.D., Kinyon A.C., Hay M.B., Higgins J.D., 2002. Evaluation of the film-coating properties of a hydroxyethyl cellulose/hydroxypropyl methylcellulose polymer system. Drug Dev. Ind. Pharm. 28, 389-401.
Lovrecich M., Nobile F., Rubessa F., Zingone G., 1996. Effect of ageing on the release of indomethacin from solid dispersions with Eudragits. Int. J. Pharm. 131, 247-255.
Lu M.Y.F., Borodkin S., Woodward L., Li P., Diesner C., Hernandez L., Vadnere M., 1991. A Polymer Carrier System for Taste Masking of Macrolide Antibiotics. Pharm. Res. 8, 706-712.
Moisescu C., Jana C., Habelitz S., Carl G., Ruessel C., 1999. Oriented fluoroapatite glass-ceramics. J. Noncry. Sol. 248, 176-182.
Mirghani A., Idkaidek N.M., Salem M.S., Najib N.M., 2000. Formulation and release behavior of diclofenac sodium in Compritol(登録商標)888 ATO matrix beads encapsulated in alginate. Drug Dev. Ind. Pharm. 26, 791-795.
Nabil, F. et al., 1998. Method for preparing a pharmaceutical composition with modified release of the active principle, comprising a matrix method for preparing a pharmaceutical composition with modified release of the active principle, comprising a matrix. FR2753904(A1).
Ohta M., Buckton G., 2004. The use of inverse gas chromatography to assess the acid-base contributions to surface energies of cefditoren pivoxil and methacrylate copolymers and possible links to instability. Int. J. Pharm. 272, 121-128.
【0074】
Pearnchob N., Dashevsky A., Siepmann J., Bodmeier R., 2003a. Shellac used as coating material for solid pharmaceutical dosage forms: understanding the effects of formulation and processing variables. Stp Pharma Sciences. 13, 387-396.
Pearnchob N., Siepmann J., Bodmeier R., 2003b. Pharmaceutical applications of shellac: Moisture protective and taste masking coatings and extended-release matrix tablets. Drug Dev. Ind. Pharm. 29, 925-938.
Petereit H.U., Weisbrod W., 1999. Formulation and process considerations affecting the stability of solid dosage forms formulated with methacrylate copolymers. Eur. J. Pharm. Biopharm. 47, 15-25.
Petereit H.U., Meier C., Gryczke A., 2003. Melt extrusion of active substance salts. WO 2003/072083.
Petereit H.U., Meier C., Gryczke A., 2004. Process for the production of an oral drug form with direct disintegration and active substance release. WO 2004/066976.
Prompruk K., Govender T., Zhang S., Xiong C.D., Stolnik S., 2005. Synthesis of a novel PEG-blockpoly(aspartic acid-stat-phenylalanine)copolymer shows potential for formation of a micellar drug carrier. Int. J. Pharm. 297, 242-253.
Rasenack N., Mueller B.W., 2002. Crystal habit and tableting behaviour. Int. J. Pharm. 244, 45-57.
Reitz C., Kleinebudde P., 2007. Solid lipid extrusion of sustained release dosage forms. Eur. J. Pharm. Biopharm. 67, 440-448.
Schubert M.A., Schicke B.C., Mueller-Goymann C.C., 2005. Thermal analysis of the crystallization and melting behavior of lipid matrices and lipid nanoparticles containing high amounts of lecithin. Int. J. Pharm. 298, 242-254.
Shirai Y., Sogo K., Yamamoto K., Kojima K., Fujioka H., Makita H., Nakamura Y., 1993. A Novel Fine Granule System for Masking Bitter Taste. Biol. Pharm. Bull. 16, 172-177.
Sohi H., Sultana Y., Khar R.K., 2004. Taste masking technologies in oral pharmaceuticals: Recent developments and approaches. Drug Dev. Ind. Pharm. 30, 429-448.
Suzuki H., Onishi H., Takahashi Y., Iwata M., Machida Y., 2003. Development of oral acetaminophen chewable tablets with inhibited bitter taste. Int. J. Pharm. 251, 123-132.
Suzuki H., Onishi H., Hisamatsu S., Masuda K., Takahashi Y., Iwata M., Machida Y., 2004. Acetaminophen-containing chewable tablets with suppressed bitterness and improved oral feeling. Int. J. Pharm. 278, 51-61.
【0075】
Takagi S., Toko K., Wada K., Ohki T., 2001. Quantification of suppression of bitterness using an electronic tongue. J. Pharm. Sci. 90, 2042-2048.
Thombre A.G., 2004. Palatable controlled-release formulations for companion animals. WO 2004/014346.
Wang H., Zhang R., 1995. Compaction behaviour of paracetamol powders of different crystal shapes. Drug Dev. Ind. Pharm. 21, 863-868.
Windbergs M., Strachan C.J., Kleinebudde P., 2008 Understanding the solid-state behaviour of triglyceride solid lipid extrudates and its influence on dissolution. Eur. J. Pharm. Biopharm. In Press.
Wong L.W., Pilpel N., 1990. The effect of particle shape on the mechanical properties of powders. Int. J. Pharm. 59, 145-154.
Yue Y., Moisescu C., Carl G., Ruessel C., 1999. Influence of suspended iso- and anisometric crystals on the flow behaviour of fluoroapatite glass melts during extrusion. Physics and Chemistry of Glasses. 40, 243-247.
Zhou F., Vervaet C., Remon J.P., 1996. Matrix pellets based on the combination of waxes, starches and maltodextrins. Int. J. Pharm. 133, 155-160.
Zhou F., Vervaet C., Schelkens M., Lefebvre R., Remon J.P., 1998. Bioavailability of ibuprofen from matrix pellets based on the combination of waxes and starch derivatives. Int. J. Pharm. 168, 79-84.
【特許請求の範囲】
【請求項1】
針状医薬活性物質の粒径とストランド径との比が、少なくとも1:15であることを特徴とする、少なくとも1種の針状晶の形態の医薬活性物質を含有する押出物。
【請求項2】
針状医薬活性物質の粒径とストランド径との比が、少なくとも1:20であることを特徴とする、請求項1に記載の押出物。
【請求項3】
針状医薬活性物質の粒径とストランド径との比が、少なくとも1:25であることを特徴とする、請求項1または請求項2に記載の押出物。
【請求項4】
ストランド径が0.5mm以下であることを特徴とする、請求項1ないし請求項3のいずれかに記載の押出物。
【請求項5】
脂質基剤を添加物として含有することを特徴とする、請求項1ないし請求項4のいずれかに記載の押出物。
【請求項6】
C12−C24脂肪酸のグリセロールエステルを脂質基剤として含有することを特徴とする、請求項1ないし請求項5のいずれかに記載の押出物。
【請求項7】
グリセロールジエステルを脂質基剤として含有することを特徴とする、請求項1ないし請求項6のいずれかに記載の押出物。
【請求項8】
ジベヘン酸グリセロールを脂質基剤として含有することを特徴とする、請求項7に記載の押出物。
【請求項9】
グリセロールトリエステルを脂質基剤として含有することを特徴とする、請求項1ないし請求項6のいずれかに記載の押出物。
【請求項10】
トリミリスチン酸グリセロール、トリパルミチン酸グリセロールまたはトリステアリン酸グリセロールを脂質基剤として含有することを特徴とする、請求項9に記載の押出物。
【請求項11】
トリステアリン酸グリセロールを脂質基剤として含有することを特徴とする、請求項10に記載の押出物。
【請求項12】
帯電防止剤、特にポリエチレングリコールを含有することを特徴とする、請求項1ないし請求項11のいずれかに記載の押出物。
【請求項13】
含有される基剤の融解範囲の下限よりも低温で押出されたことを特徴とする、請求項1ないし請求項12のいずれかに記載の押出物。
【請求項14】
医薬を製造するための、請求項1ないし請求項13のいずれかに記載の押出物の使用。
【請求項15】
請求項1ないし請求項13のいずれかに記載の押出物および1種またはそれ以上の医薬的に許容し得る補助物質および/または添加物を含有することを特徴とする、医薬。
【請求項1】
針状医薬活性物質の粒径とストランド径との比が、少なくとも1:15であることを特徴とする、少なくとも1種の針状晶の形態の医薬活性物質を含有する押出物。
【請求項2】
針状医薬活性物質の粒径とストランド径との比が、少なくとも1:20であることを特徴とする、請求項1に記載の押出物。
【請求項3】
針状医薬活性物質の粒径とストランド径との比が、少なくとも1:25であることを特徴とする、請求項1または請求項2に記載の押出物。
【請求項4】
ストランド径が0.5mm以下であることを特徴とする、請求項1ないし請求項3のいずれかに記載の押出物。
【請求項5】
脂質基剤を添加物として含有することを特徴とする、請求項1ないし請求項4のいずれかに記載の押出物。
【請求項6】
C12−C24脂肪酸のグリセロールエステルを脂質基剤として含有することを特徴とする、請求項1ないし請求項5のいずれかに記載の押出物。
【請求項7】
グリセロールジエステルを脂質基剤として含有することを特徴とする、請求項1ないし請求項6のいずれかに記載の押出物。
【請求項8】
ジベヘン酸グリセロールを脂質基剤として含有することを特徴とする、請求項7に記載の押出物。
【請求項9】
グリセロールトリエステルを脂質基剤として含有することを特徴とする、請求項1ないし請求項6のいずれかに記載の押出物。
【請求項10】
トリミリスチン酸グリセロール、トリパルミチン酸グリセロールまたはトリステアリン酸グリセロールを脂質基剤として含有することを特徴とする、請求項9に記載の押出物。
【請求項11】
トリステアリン酸グリセロールを脂質基剤として含有することを特徴とする、請求項10に記載の押出物。
【請求項12】
帯電防止剤、特にポリエチレングリコールを含有することを特徴とする、請求項1ないし請求項11のいずれかに記載の押出物。
【請求項13】
含有される基剤の融解範囲の下限よりも低温で押出されたことを特徴とする、請求項1ないし請求項12のいずれかに記載の押出物。
【請求項14】
医薬を製造するための、請求項1ないし請求項13のいずれかに記載の押出物の使用。
【請求項15】
請求項1ないし請求項13のいずれかに記載の押出物および1種またはそれ以上の医薬的に許容し得る補助物質および/または添加物を含有することを特徴とする、医薬。
【図1】


【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【公表番号】特表2012−510956(P2012−510956A)
【公表日】平成24年5月17日(2012.5.17)
【国際特許分類】
【出願番号】特願2011−538866(P2011−538866)
【出願日】平成21年11月24日(2009.11.24)
【国際出願番号】PCT/EP2009/008341
【国際公開番号】WO2010/063387
【国際公開日】平成22年6月10日(2010.6.10)
【出願人】(508270727)バイエル・アニマル・ヘルス・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツング (32)
【氏名又は名称原語表記】BAYER ANIMAL HEALTH GMBH
【Fターム(参考)】
【公表日】平成24年5月17日(2012.5.17)
【国際特許分類】
【出願日】平成21年11月24日(2009.11.24)
【国際出願番号】PCT/EP2009/008341
【国際公開番号】WO2010/063387
【国際公開日】平成22年6月10日(2010.6.10)
【出願人】(508270727)バイエル・アニマル・ヘルス・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツング (32)
【氏名又は名称原語表記】BAYER ANIMAL HEALTH GMBH
【Fターム(参考)】
[ Back to top ]