
銀及び有機酸アニオン含有アルミニウム硫酸塩水酸物粒子よりなる抗菌剤およびその利用
【課題】抗菌性、分散性、透明性、白色性、水道水接触環境下抗菌力維持特性に優れた銀および有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子からなる抗菌剤を提供すること。および該菌剤の製造方法を提供すること。さらに抗カビ剤、抗菌消臭剤、抗菌紙、農薬及び化粧料を提供すること。
【解決手段】下記式(1)で表される銀および有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子からなる抗菌剤、さらに該化合物粒子を含有する抗カビ剤、抗菌消臭剤、抗菌紙、農薬及び化粧料。および該抗菌剤の製造方法。および該菌剤が樹脂に配合された抗菌性樹脂組成物、および該樹脂組成物から形成された抗菌性樹脂製品。
(AgaBb−a)bAlcAx(SO4)y(OH)z・pH2O (1)
【解決手段】下記式(1)で表される銀および有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子からなる抗菌剤、さらに該化合物粒子を含有する抗カビ剤、抗菌消臭剤、抗菌紙、農薬及び化粧料。および該抗菌剤の製造方法。および該菌剤が樹脂に配合された抗菌性樹脂組成物、および該樹脂組成物から形成された抗菌性樹脂製品。
(AgaBb−a)bAlcAx(SO4)y(OH)z・pH2O (1)
【発明の詳細な説明】
【技術分野】
【0001】
本発明は銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子よりなる抗菌剤に関する。さらに詳しくは特定粒子性状(粒子形状、粒子径均一性、平均2次粒子径、比表面積等)を有する銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子よりなる抗菌剤に関する。また該抗菌剤の製造方法にも関する。
さらには該抗菌剤を樹脂に混練混合する時の性能として混練押出加工時フィルター通過性、分散性に優れた抗菌性樹脂組成物(マスターバッチを含む)にも関する。また該樹脂組成物から形成された、分散性、透明性、白色性、抗菌性(水道水接触環境後抗菌力維持特性も含む)に優れた抗菌性樹脂成形品、抗菌性フィルム、抗菌性繊維、抗菌性塗料、抗菌性不織布、抗菌性コーキング材のような抗菌性樹脂製品にも関する。さらに抗カビ剤、抗菌消臭剤、抗菌紙、農薬及び化粧料にも関する。
【背景技術】
【0002】
一般的に細菌は高温多湿な環境で繁殖が進み、安全衛生上、住環境上重大な問題を引き起こす場合がある。これらの問題を解決するため従来有機系抗菌剤や無機系抗菌剤を抗菌性付与対象物となる樹脂その他の物に配合して細菌からの被害を防止する抗菌性樹脂組成物等の技術が提案されているが、その中では無機系抗菌剤が比較的安全なため最近無機抗菌剤の需要は伸びてきている。
無機系抗菌剤においては、銀が比較的高い抗菌活性と比較的高い安全性を有することから、銀を無機化合物に担持又はイオン交換させた抗菌剤を使用した抗菌性樹脂組成物が多数提案されている。
【0003】
例えば、特許文献1ではリン酸ジルコニウムに銀を担持した抗菌性樹脂組成物の技術が開示されている。しかしながら、該抗菌剤を樹脂に配合した抗菌性樹脂組成物においては白色性は若干改善されているものの抗菌性、分散性、透明性、水道水接触環境後抗菌力維持特性の全ての点において完全ではなく解決すべき問題が残っていた。
なかでも、この従来技術では抗菌性樹脂組成物をしばらく水道水に接触させて使用すると該樹脂組成物の抗菌活性は全くなくなるか、又は著しく低下してしまい長時間の使用に耐えなくなり、細菌からの被害を防止できなくなるという問題があり、この問題を解決することは重要な課題であった。
この問題をさらに具体例で説明すると、台所、風呂場やトイレの周りでは水道水が常に使用され、衣類等は水道水で何回も洗濯して使用される。従って、このような場所であるいは条件で使用する抗菌性樹脂製品は本来水道水に長時間接触しても抗菌性を発揮して細菌からの被害を防止できるものでなければならない。ところが、前記従来の技術による抗菌性樹脂製品は樹脂製品製造直後には一定程度の抗菌性を発現するものの、しばらく水道水と接触して使用すると、該樹脂製品の抗菌活性は全くなくなるか又は著しく低下してしまい、さらに長時間の経過の後は細菌からの被害を全く防止できなくなるという問題があった。
【0004】
特許文献2では、MAl3(SO4)2(OH)6なる式で表されたBET法比表面積が30m2/g以下で、該式中のMをアルカリ金属又はアンモニウム基とした粒子形状が紡錘状乃至球状を呈するアルカリアルミニウム硫酸塩水酸化物が開示され、この文献にはコールターカウンター法で測定された体積基準累積粒子径の25%値の粒子径D25(大粒子側)を75%値の粒子径D75(小粒子側)で除した算式で表される粒子径分布のシャープ度Rs=D25/D75が1.45乃至1.61のものが実施例で具体的に示され、さらにその製造方法及び樹脂配合例が紹介されている。
前記特許文献の、段落番号0035では該化合物のMがAg,Zn、Cu等の抗菌性発現効果のある元素を含むことが可能であり抗菌性粒子を得ることができるとの極一般的な説明はある。しかしこの文献には抗菌剤及び抗菌性樹脂製品及び抗カビ剤、抗菌消臭剤、抗菌紙、農薬、化粧料に関する具体的な説明もまた実施例もない。
【特許文献1】特開平6−212019号公報
【特許文献2】特開2000−7326号公報
【発明の開示】
【発明が解決しようとする課題】
【0005】
かくして本発明の目的は、前記従来技術の無機系抗菌剤の種々の問題点を克服し、抗菌剤の性能として樹脂等の抗菌化対象物に配合使用された時、分散性、透明性、白色性に優れ、抗菌性特に水道水接触環境後の抗菌力維持特性にも優れた銀と有機酸アニオンがアルカリアルミニウム硫酸塩水酸化物粒子に特定範囲で含有したことからなる銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子よりなる抗菌剤を提供すること、さらには該抗菌剤において該諸特性が特に優れるように設計された特定粒子性状(粒子形状、粒子径均一性、平均2次粒子径、比表面積等)を有する銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子よりなる抗菌剤を提供すること、さらには該抗菌剤の製造方法を提供することである。
【0006】
さらに他の目的は、その抗菌剤を樹脂混練押出機を用いて混練混合する時に樹脂混練押出加工時フィルター通過性、分散性に優れた性質を有する抗菌性樹脂組成物(マスターバッチを含む)を提供すること、さらには該樹脂組成物から成形されて分散性、透明性、白色性、抗菌性特に水道水接触環境後抗菌力維持特性に優れた抗菌性樹脂成形品、抗菌性フィルム、抗菌性繊維、抗菌性不織布、抗菌性塗料、抗菌性コーキング材等の抗菌性樹脂製品を提供することである。
従来技術では抗菌性樹脂製品を水道水接触環境下でしばらく使用すると抗菌活性が全くなくなるか、又は短時間で著しく低下するというような問題があったことに鑑み、本発明はその問題を解決し、台所、風呂場やトイレ等で水道水を常に使用する場所、あるいは衣類のような製品において水道水で何回も洗濯して使用する条件下でも抗菌性を長時間維持できる抗菌性樹脂組成物、及びそれからの製品、及び該樹脂組成物に配合する該抗菌剤、及び該抗菌剤の製造方法を提供することが本発明の課題の1つである。
もう1つの重要な課題は、抗菌性樹脂製品を得るための前段として通常良く実施される技術、すなわち一旦樹脂と抗菌剤とを樹脂混練押出機を用いてマスターバッチ(MB)を製造する時に樹脂混練押出加工時フィルター通過性(押出機圧力)が悪くなって機械を長時間運転できずフィルターを短時間で交換しなければならなかった問題を解決することである。機械をより長時間運転できれば、フィルター交換に伴う資源、エネルギー、労力、時間が節減できそれだけ低コストで抗菌性樹脂組成物、及び抗菌性樹脂製品が社会に提供でき工業的な価値は大である。
【0007】
本発明のさらに他の目的は、前記した抗菌性樹脂製品の他に、抗菌剤の特性を利用した別の製品を提供することにある。すなわち、抗カビ剤、抗菌消臭剤、抗菌紙、農薬または化粧料を提供することにある。
【課題を解決するための手段】
【0008】
本発明者等は前記課題の達成のため鋭意研究した結果下記式(1)で表される銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子及び/又は該化合物粒子の150℃〜600℃で乾燥処理又は焼成処理された化合物粒子が抗菌剤の性能として抗菌性が非常に優れ、且つその粒子を樹脂100重量部に対し0.001〜300重量部配合した樹脂組成物も抗菌性が非常に優れていること、しかも樹脂混練押出加工時フィルター通過性、白色性、透明性、水道水接触環境後抗菌力維持特性等の諸特性に優れていること、及び該樹脂組成物から形成された抗菌性樹脂成形品、抗菌性フィルム、抗菌性繊維、抗菌性不織布、抗菌性塗料、抗菌性コーキング材のような抗菌性樹脂製品も同様に白色性、透明性、水道水接触環境後抗菌力維持特性等の諸特性に優れていることを見出し本発明に到達した。
さらに前記粒子は、その抗菌性を利用して、成形品以外にも抗カビ剤、抗菌消臭剤、農薬および化粧料としても有利に利用しうることを見出し本発明に到達した。
【0009】
本発明によれば、下記式(1)で表わされる銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子よりなる抗菌剤が提供される。
(AgaBb−a)bAlcAx(SO4)y(OH)z・pH2O (1)
式(1)中,a,b,c,x,y,z及びpは0.00001≦a<0.5、 0.7≦b≦1.35、2.7<c<3.3、 0.001≦x≦0.5, 1.7<y<2.5、4<z<7及び0≦p≦5を満足し、BはNa+、NH4+、K+及びH3O+の群から選ばれた少なくとも1種の1価陽イオンを表わし、陽イオンの価数×モル数の合計値(1b+3c)の範囲が8<(1b+3c)<12であり、Aは有機酸アニオンを表す。
【発明の効果】
【0010】
本発明の銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子からなる抗菌剤を樹脂に使用すれば、分散性、透明性、白色性、抗菌性特に水道水接触環境後抗菌力維持特性、混練押出加工時フィルター通過性の全ての性質に優れた抗菌性樹脂組成物、及び該樹脂組成物から形成された抗微菌樹脂成形品、抗菌性フィルム、抗菌性繊維、抗菌性不織布、抗菌性塗料、抗菌性コーキング材等の抗菌性樹脂製品を提供することが可能となった。
なかでも、水道水接触環境後抗菌力特性を長時間維持し続けることは、従来技術では到底成し得なかったことであり、それが本発明によって例えば台所、風呂場、トイレ等の水道水を常に使用する場所でも長時間抗菌性を維持できること、つまり細菌からの被害をそのような条件でも長時間維持し続ける抗菌剤、該抗菌剤の製造方法、抗菌性樹脂組成物、及びそれからの抗菌性樹脂製品を提供することに関する新規な技術が提供できるようになったことは本発明の大きな成果である。
【0011】
もう一つの本発明の大きな成果は抗菌性樹脂製品を得るための前段として通常良く実施される技術、すなわち一旦樹脂と抗菌剤とを樹脂混練押出機を用いてマスターバッチ(MB)を製造する時に樹脂混練押出加工時フィルター通過性(押出機圧力)が悪くなって機械を長時間運転できずフィルターを短時間で交換しなければならなかった問題をも解決したことである。
この成果をさらに具体的に説明すると、本発明によって樹脂混練押出加工時フィルター通過性が改善され機械をより長時間運転できるようになり、フィルター交換の頻度が少なくて済むことに伴う樹脂及び樹脂に配合する添加剤・充填剤等の副資材、水、エネルギー、電力、労力、時間が節減できそれだけ低コストで抗菌性樹脂組成物、及び抗菌性樹脂製品が社会に提供できるようになったことによる工業的な価値は非常に大であるということである。
【発明を実施するための最良の形態】
【0012】
本発明について以下さらに詳しく説明する。
【0013】
本発明によれば、前記式(1)で表わされる抗菌剤粒子の性状として、下記(i)、(ii)及び(iii)の特性を、独立して有していることも見出された。
【0014】
(i)レーザー回折散乱法で測定された平均二次粒子径が0.1μm〜12μm、好ましくは0.1〜5μm
(ii)BET法比表面積が0.1〜250m2/g、好ましくは1〜100m2/g
(iii)Dr=D75/D25[D25がレーザー回折散乱法による体積基準累積粒子径分布曲線の25%値(微粒子径側)の粒子径を表し、D75がその75%値(大粒子径側)の粒子径を表す]で定義される粒子径分布のシャープ度が1.0〜1.8、好ましくは1.01〜1.5、さらに好ましくは1.01〜1.3、最も好ましくは1.01〜1.2
【0015】
前記(i)の平均二次粒子径、(ii)のBET法比表面積及び(iii)の粒子径分布のシャープ度(Dr)は、それぞれ独した特性であり、これらの2つの特性が同時に満足するものが、より好ましく、さらにこれら3つの特性を同時に満足する粒子が最も本発明の目的が達成のため好ましい。
さらに本発明の抗菌剤粒子は凝集粒子ではなく単分散状であって以下の粒子形状を有する点にも特徴を有している。
本発明の抗菌剤の粒子は、種々の粒子形状を有しているが、その形状が均一で大きさが揃っていることおよび凝集が少なく単分散状を呈する点にも特徴を有している。粒子形状について説明すると、抗菌剤の粒子は、大きく分けると、球状、円盤状(基石状)、一対状(ハンバーガー状)、米粒状、直方体状、六角板状、円柱状(酒樽状)及び八面体状に分類される。これら種々の粒子形状を図1〜図13により具体的に説明する。
【0016】
図1〜図13は、本発明の実施例により得られた粒子の代表的なSEM写真である。粒子の形状は約1万倍乃至約2万倍に拡大されたSEM写真に基いて観察される。なお、図14は従来知られたアルカリアルミニウム硫酸塩水酸化物粒子のSEM写真である。
球状粒子の例は図1〜図6に示され、これら球状粒子は図1に示す表面が平滑である球状、図2に示す表面に小粒を有する球状、図3に示す表面が荒くさらには球に皺(傷や割れ)を有する球状、図4に示す小穴(凹凸)を有する球状、図5に示す表面が平滑であり且つ直線部分を図1のものより少し多く含む球状、及び図6に示す表面が荒く皺を有する球状に分類できる。
円盤状粒子の例は図7に示され、この形状は表と裏がほぼ対象であり、ドーム形であって、碁石にも似ている。図7の円盤状粒子は表面が平滑である。
一対状粒子の例は図8に示されている。この粒子の特徴は、底面が平板でその反対面がドーム形の円盤粒子の2つが底面を対称面として一対状の形状を有していることであり、その2つの粒子の重なり合う周囲の間隙には空間が存在している。また重なり合う中心部は2つの円盤を接合しているアルミニウム塩水酸化物が存在している。この一対状粒子は、一見ハンバーガーに似ている。
米粒状粒子の例は、図9に示されている。この米粒状粒子は、投影した形が楕円形で長さ方向の直角断面がほぼ円形の形状をしている。図9の粒子は、表面に小さなしわ(皺)を有している。
直方体状粒子の例は図10に示され正六面体に近い直方体であって表面が平滑である。
六角板状粒子の例は図11に示され、この六角板状粒子は六つの辺で形成された六面体の表面を有する板状のものである。この六つの辺は同じ長さであることを要せず、また2つの辺の接点は丸味を有していてもよい。
八面体状粒子の例は図12に示され正方両錐体状乃至偏八面体状の八面体状の形状を有している。
円柱状粒子の例は図13に示されている。この円柱状粒子は、大略酒樽状(またはワイン樽状)のように中間部分が膨らんだものでよく、また断面がほぼ円形の筒状のものでもよい。
図13の粒子は表面に多数の凹凸を有している。
図1〜図13から理解されるように本発明の粒子は各々の図(写真)において、粒子形状が揃っており、その大きさが均一でありかつ分散性がよい点に特徴を有している。前記した各粒子の形状は、それぞれ区分するために分類して表現したものであり、若干の変形や少割合の他の粒子の混合があっても差支えない。また粒子の表面における平滑性、微小凹凸の存在または小さいしわ(皺)の存在は、特に限定されるものではなく、存在してもしなくてもよい。
【0017】
次に本発明の粒子の形状を特定する方法について説明する。
粒子の形状を特定する尺度の一つに、粉体工業分野において従来から用いられてきたWadellの円形度及び球形度がある。
Wadellの球形度sは、s=(粒子と等体積の球の表面積)/(粒子の表面積)
で定義され、sが1に近い程真球に近い。
Wadellの円形度cは、c=(粒子の投影面積と等面積の周長)/(粒子の投影面の周長)で定義され、cが1に近い程真円に近い。
【0018】
本発明において粒子の形状が球状であるとは、図1〜図6に示すようなボール様の形状であれば良く、前記のWadellの球形度sが0.95≦s≦1であることが好ましい。
【0019】
本発明において粒子の形状が円盤状(碁石状)であるとは、図7に示すように短径を回転軸とした回転楕円状の形状である。具体的には、回転軸の方向から見た粒子の投影像に関して、Wadellの円形度cが、0.95≦s≦1であって、断面である楕円の(短径/長径)の比率aが0.05≦a≦0.5であることが好ましい。
【0020】
本発明において粒子の形状が一対状(ハンバーガー状)であるとは、図8に示すように半球状の粒子が2個重なり合うような形状で対を形成した粒子である。そして一対状粒子は、二つの半球状粒子の重なり合う面の周縁に、隙間(溝)が存在している。一対状粒子の(短径/長径)の比率tは0.1<t<0.5であり、(該半球の合わせ目の隙間幅)/(短径)の比率uが0.05<u<0.5であることが好ましい。
【0021】
本発明において粒子の形状が米粒状であるとは、図9に示すように短径を回転軸とした回転楕円状の形状であり、楕円の(短径/長径)の比率aが0.1≦a≦0.5であり、前記のWadellの球形度sが,0.4≦s<0.75であることが好ましい。
【0022】
本発明において粒子の形状が直方体状であるとは、図10に示すような六面体又は正六面体に類似する形状であれば良く、前記のWadellの球形度sが0.5≦s≦0.8であることが好ましい。
【0023】
本発明において粒子の形状が六角板状であるとは、図11に示すような扁平な正六角柱様の形状で、上面又は下面方向から見た粒子の投影像に関してWadellの円形度cが,0.95≦c<0.99であって、厚さ/(正六角形の対角線長さ)の比率bが0.05≦b≦0.5であることが好ましい。
【0024】
本発明において粒子の形状が8面体状であるとは、図12で示されるように正方両錐体状乃至偏八面体状の八面体状の形状を有していると考えられ、前記のWadellの球形度sが0.5≦s≦0.9であることが好ましい。ただし、この八面体状粒子は熟視しないと、SEM写真の分解能不足からくる不鮮明さにより一見六面体状粒子に見える恐れがある。
【0025】
本発明において粒子の形状が円柱状(酒樽状)であるとは、図13に示すように円柱を含み、円柱の高さ方向の中心部の半径が上面及び下面の半径の1.0〜1.2倍までの形状をいい、上面及び下面の投影像に関して、前記のWadellの円形度cが,0.95≦c<0.99であって、(高さ)/(上面又は下面の直径)の値dが1.5≦d≦3であることが好ましい。
【0026】
本発明によれば、上記のように、銀及び有機酸アニオン含有アルミニウム塩水酸化物粒子は、用途や目的に応じて球状、円盤状(碁石状)、一対状、直方体状、六角板状、米粒状、八面体状または円柱状などの種々の形状を提供でき、かつ粒子径をコントロールできる。
【0027】
一方粒子径に関しても、用途および必要な充填率に応じて最適な粒子径の銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子を提供することが可能である。
それらの粒子には粒子同士の凝集がなく樹脂への分散性に優れ、さらに樹脂に配合した時も該抗菌剤の樹脂中での凝集も全くあるいはほとんどないことが該抗菌剤を樹脂に配合した樹脂製品において銀の含有量が極めて少量でも抗菌性を発揮する要因の1つだと考えられ、この単分散技術だけでも従来技術では到底達成できなかった予想外の高抗菌活性を発揮するものである。
【0028】
本発明の該抗菌剤がこのような予想外の高抗菌活性を発揮するメカニズムについてはさらに次の第2の要因も推定される。
本発明の該抗菌剤粒子に高い抗菌性発現効果があるのは、本発明の該抗菌剤の粒子には分子の内部構造にまで有機酸がアニオンとして取り込まれているため本発明の該抗菌剤に光が当たった時、本発明の該抗菌剤粒子からヒドロキシルラジカル(OH−)のようなラジカルが長期間にわたり発生し易くなり、これが本発明の該抗菌剤の抗菌性能が長期間にわたって維持できる要因にもなっているものと推定される。
それは、従来の大部分の銀系無機抗菌剤は抗菌剤から銀イオンを除放できる期間においては抗菌性を発現するが、その期間が終わると抗菌性が発現しなくなるという致命的な欠点があったが、本発明はその問題を根本的に解決するものである。
さらにそればかりでなく、本発明によると該抗菌剤の分子構造中に有機酸をアニオンとして取り込んだことによる抗菌性発現向上効果の第3の要因として、前記の分散効果及びラジカル発生効果のみならず、該抗菌剤粒子の有機酸の炭素部分による樹脂との相溶性向上効果がさらに加わって、つまり本発明ではこれら3つの効果を相乗作用させることに成功しより一層高い抗菌性能を奏することができるようになったものと推定される。
【0029】
本発明では前記式(1)で表される抗菌剤を樹脂100重量部に対し0.001〜300重量部配合することにより混練押出加工時フィルター通過性、抗菌性、分散性、白色性に優れた性質を有しながらしかも尚且つ水道水接触環境後の抗菌力維持特性に優れた抗菌性樹脂組成物及びそれから形成される樹脂製品を提供することができるが、該樹脂製品に高い透明性という付加価値を重視する目的においては0.001〜10重量部、好ましくは0.001重量部〜2重量部配合することが推奨できる。該抗菌剤の配合量が0.001重量部以下であると充分な抗菌性を発揮できない恐れがあり、300重量部以上配合することは銀の含有量にもよるが不経済となり、透明性も低下する傾向がある。
本発明の抗菌剤の屈折率は約1.48〜約1.56であり多くの樹脂と屈折率が重複あるいは接近していため、樹脂にかなり高濃度に本発明の抗菌剤を配合しても透明性をあまり損なわないが、透明性を高度に維持するためには該抗菌剤の配合量は10重量部以下、好ましくは2重量部以下にすることが推奨できる。
【0030】
本発明における式(1)中のaは銀の該抗菌剤粒子へのイオン交換量を示し、aの数値が高ければそれだけ銀が該抗菌剤粒子にイオン交換していることを示し抗菌性が向上するが、あまり高くなりすぎるとイオン交換体(固溶体)から銀が環境中で析出や溶出し酸化銀になる恐れがあり該抗菌剤が配合された樹脂成形品等の色が暗褐色を呈することにもなり、また経済的でもないし、またaは0.5以上イオン交換しにくい。一方aの数値が低過ぎる場合はそれだけ銀が該抗菌剤粒子へのイオン交換量が少ないことを示し抗菌性が発現しないので、抗菌性発現力と色の問題を適度にバランスするためaは一定範囲にすることが望まれる。かかる意味において式(1)におけるaは0.00001〜0.5、好ましくは0.00001〜0.35、さらに好ましくは0.001〜0.3の範囲であることが適している。
【0031】
本発明において、「銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子よりなる抗菌剤」なる記述の中の、含有という言葉は該粒子を粉末X線回折法で測定した時、式(1)以外の化合物のピークが現らわれない程度に式(1)以外の銀及び有機酸からなる化合物の混入量が少ない範囲の物であることを意味している。
従って該抗菌剤粒子にはイオン交換体のみならず、粉末X線回折法の測定ではピークが現らわれない若干の銀イオンがイオン交換体以外の形態で該粒子に担持された形態及び/又は若干の有機酸アニオンが該粒子の表面に吸着している形態も含んでいるものと考えられる。
その場合、該粒子を銀に着目して考えると、銀がイオン交換許容範囲内でイオン交換した固溶体のみからなるものは樹脂製品への着色の影響が少なくなっていると考えられ、そういう意味では完全な銀イオン交換体(固溶体)のみからなる物が好ましい。
【0032】
本発明における式(1)中Bには種々の1価のカチオンであればよいが、実際には銀イオンとイオン半径が比較的近く広い範囲で強くイオン交換体を形成することができるという意味、つまり樹脂に配合した時に銀が該イオン交換体から遊離して酸化銀になることがなく、その結果樹脂製品の白色度の低下(樹脂製品の成形直後の白色から光による作用で時間とともに暗褐色乃至褐色への変色すること)を招き難いということ、さらに安全性、及び経済性を考慮すると、BとしてはNa+,NH4+,K+及びH3O+が適している。さらにその中ではNa+,NH4+及びH3O+が比較的好ましく、その中でもさらにNH4+及びH3O+がさらに好ましく、NH4+が該目的において最も好ましい。この変色防止という観点からは、BとしてNa+及び/又はK+を用いる量はできるだけ少ないことが好ましく、特にK+を用いる量はBなる1価陽イオンの合計モルの1/2より小であることが好ましい。ただしこれらの変色は蛍光増白剤を樹脂中に0.000001%〜0.1%添加することにより防止できる程度のものである。従ってBとしてNa+及びK+を多く用いる場合は変色防止のため蛍光増白剤を用いることは好ましいことである。
【0033】
本発明の抗菌性樹脂組成物の一部分ではあるが、前記比較的好ましい態様すなわち、NH4+及びNa+、H3O+の3種1価陽イオンが用いられる抗菌剤粒子の場合、蛍光増白剤を用いなくとも変色しないあるいは比較的変色が少ない樹脂製品を得るためには、Naの使用量は、Naのモル含有量がこれらBなる1価陽イオンの合計モル含有量の1/2より小であるべきである。そうすることによって本発明では変色のないあるいは少ない樹脂製品を得ることができる。
該蛍光増白剤を用いる場合、その例としてはベンゾヘキサゾール系の2,5−チオフェンジイル(5−tert−ブチル−1,3−ベンゾヘキサゾール、4,4’−ビス(ベンゾヘキサゾール−2−イル)スチルベン、及びピラゾリン系、カーマリン系のものが例示できるが、FDA(米国Food & Drag Administration)やポリオレフィン等衛生協議会に登録されているものを用いることが好ましい。
【0034】
本発明における式(1)中bは0.7〜1.35、好ましくは0.8〜1.2、最も好ましくは0.9〜1.1であれば本発明の該抗菌剤粒子を形成し易く、cは2.7〜3.3、好ましくは2.8〜3.2、最も好ましくは2.9〜3.1であれば本発明の該抗菌剤粒子を形成し易い。
式(1)の陽イオンのイオン価×モル数は(1b+3c)として表される。本発明でのその範囲は8<(1b+3c)<12、好ましくは9<(1b+3c)<11であれば本発明の該抗菌剤粒子を形成し易い。
本発明の抗菌剤では式(1)なる粒子の合成において、粒子形状と粒子サイズを選択して製造すること、粒子径分布を均一に整えること、該粒子を出来る限りに単分散状態で樹脂に分散できるようにするためには、該反応において反応時に有機酸類を添加する必要があり、添加された有機酸類は該抗菌剤粒子の分子構造中に組み込むことができる。
【0035】
かかる有機酸類としては式(1)中の有機酸アニオン(A)が有機カルボン酸又は有機オキシカルボン酸に基づくアニオン群から選ばれる少なくとも1種であるものを例示できるが、有機酸アニオン(A)が、炭素数1〜15、好ましくは1〜10有し、且つカルボキシル基を1〜4個有する有機カルボン酸又は有機オキシカルボン酸に基づくアニオン群から選ばれる少なくとも1種であることも好ましく、例えばジカルボン酸、モノカルボン酸、トリカルボン酸、鎖式カルボン酸、芳香族カルボン酸、ヒドロキシ酸、ケトン酸、アルデヒド酸、フェノール酸、アミノ酸、ハロゲンカルボン酸及びそれらの塩類が例示できるが、その中でも有機酸アニオン(A)が、蓚酸イオン、クエン酸イオン、リンゴ酸イオン、酒石酸イオン、グリセリン酸イオン、没食子酸イオン、及び乳酸イオンの群から選ばれる少なくとも1種であることが最も好ましい。
【0036】
式(1)中の有機酸アニオン(A)の割合xを0.0001≦x≦0.5、好ましくは0.0001≦x≦0.4、さらに好ましくは0.001≦x≦0.2にすれば前記の目的を達成し且つ粒子形状が前記のを形状を有し且つ粒子径均一性の該抗菌剤粒子が得られる。xが0.5を超えると特にこの効果は高くなるものでもなく又経済的でもない。
xが0.0001未満の場合は粒子形状が前記の形状を有し且つ粒子径均一性の該抗菌剤粒子が得られ難くなるし、又前記のヒドロキシラジカルの発生、及び有機酸による樹脂との相溶性向上効果が原因と考えられる抗菌性の向上等本発明の目的は達成し難くなる。
【0037】
本発明の式(1)におけるyは1.7<y<2.5、好ましくは1.8<y<2.2であれば本発明の該抗菌剤粒子を形成し易く、zは4<z<7、好ましくは5<z<7であれば本発明の該抗菌剤粒子をより一層形成し易い。
式(1)中のpは結晶水の量を示し、通常pは0≦p≦5の範囲である。このpを限りなく0に近づけるか、あるいは0にするためには350℃以下の追加の乾燥処理、あるいは焼成処理すれば良い。焼成処理は600℃以下が好ましい。焼成温度が500℃以上、さらに550℃以上、特に600℃以上の温度であると、下記式で表される水溶性のアルミニウム硫酸塩が一部生成する恐れがあり、またそれを添加した樹脂製品は耐水性が低下する恐れがある。ただし添加量が少ない場合には特に耐水性に問題はない。
(AgaBb−a)bAlAx(SO4)y
焼成温度が500℃以下、特に450℃以下であれば前記式で表される水溶性のアルミニウム硫酸塩は生成せず、これを樹脂製品に多量に使用しても耐水性は低下せず何ら問題ない。また、本発明の抗菌剤粒子は600℃以上の温度で焼成すると図1〜図13に示すような、本発明の該抗菌剤粒子の粒子形状が維持できない恐れもある。かかる耐水性及び形状維持の観点から本発明の抗菌剤粒子の焼成温度は350℃〜600℃、好ましくは350℃〜550℃、さらに好ましくは350℃〜500℃、最も好ましくは350℃〜450℃である。
【0038】
前述した乾燥処理又は焼成処理を窒素雰囲気下で実施することは、該抗菌剤粒子及び該抗菌剤粒子が配合された樹脂製品の着色防止の点で好ましいことである。乾燥処理は真空乾燥で実施しても着色防止の点で好まい。
樹脂加工の際、pが0でなくても問題にならない場合、例えば該抗菌剤の配合量が非常に少ない場合あるいは樹脂加工時の水分が特に問題にならない樹脂の場合は、乾燥処理又は焼成処理を実施していないpが0≦p≦5、好ましくは0≦p≦3のものを樹脂に配合して樹脂組成物を製造することができる。
逆に、そのpが0又は0に近づけたものでないと問題になる場合は追加乾燥処理又は焼成処理を加えることによりp=0又は0に限りなく近づけたものを使用すれば良い。
例えばPET,PBTのようなポリエステル系樹脂、ポリアミド系樹脂、ポリウレタン系樹脂、ポリカーボネート樹脂、ポリアセタール樹脂等の樹脂では前記の条件で追加乾燥処理、あるいは焼成処理されたp(水分量)を0又は限りなく0に近づける配慮をした該抗菌剤を使用することは推奨できることである。
【0039】
本発明の粒子は、前記レーザー回折散乱法粒子径分布のシャープ度(Dr)を1.0≦Dr≦1.8、好ましくは1.0≦Dr≦1.5、さらに好ましくは1.01≦Dr≦1.3、最も好ましくは1.01≦Dr≦1.2とすることにより樹脂への分散性という点において凝集がなく完全分散することができ、それが抗菌効果を高める要素であると考えられるが、さらには樹脂の押出加工等の際、特にマスターバッチを製造する際、フィルター(スクリーンメッシュ)を使用しても、該抗菌剤のスクリーンメッシュへの目詰まりがなくなるあるいは少なくなるという利点もある。
【0040】
本発明の該抗菌剤配合抗菌性樹脂製品に高い抗菌性を付与する目的においては粒子の平均二次粒子が小さい程適しているが0.1μm〜12μm、好ましくは0.1〜5μm、さらに好ましくは0.1〜2μm、さらにより好ましくは0.1〜1μm、最も好ましくは0.1μm〜0.5μmの物が用いられる。
ただし該抗菌剤の平均二次粒子径が0.1μm未満のものは製造し難い恐れがあり、12μm以上のものは樹脂に配合しても樹脂組成物等の抗菌性があまり高くならない恐れがある。本発明の樹脂組成物に抗菌性のみならず、高度の透明性を付与する目的においては、本発明の抗菌剤の中でも平均二次粒子径が0.1〜0.5μm、好ましくは0.1〜0.4μm、さらに好ましくは0.1〜0.3μmである超微粒子のものを使用すると、もともと樹脂と屈折率が重複又は接近している本発明の抗菌剤粒子の特性を最大限に発揮でき、樹脂製品に従来技術では到底達成し得なかった尚一層高度な透明性を付与する効果を絶大に発揮するものである。
【0041】
本発明で使用される抗菌剤のBET法比表面積は0.1〜250m2/gの物が用いられる。
樹脂組成物に高い抗菌性を付与するためにはBET法比表面積の高い方が有利ではあるが、BET法比表面積のあまり高い物は一方では樹脂に充填し難い問題が生ずる恐れがあり、一方BET法比表面積が低すぎると樹脂組成物に十分な抗菌性を付与できない恐れがある。
かかる意味において該抗菌剤のBET法比表面積は0.1〜250m2/g、好ましくは1〜100m2/gさらに好ましくは10〜100m2/g、最も好ましくは30〜100m2/gである。
【0042】
本発明の、該抗菌剤粒子が配合された抗菌性樹脂製品は元々高い耐酸性を有しているが、さらに高い耐酸性を樹脂製品に付与する目的、あるいは変色防止の目的においては、本発明の該抗菌剤粒子の表面を、ケイ素化合物、リン化合物、ホウ素化合物、アルミニュウム化合物,ジルコニウム化合物、チタン化合物、亜鉛化合物、錫化合物の群から選ばれた少なくとも1種の耐酸性改質剤で被覆してさらに耐酸性を向上することができる。
かかる耐酸性改質剤を例示するとケイ素化合物としてはメタケイ酸ナトリウム、オルトケイ酸ナトリウム、メタケイ酸カリウム、オルトケイ酸カリウム、水ガラス、ケイ酸、シリコーンオイル;ホウ素化合物としては四ホウ酸ナトリウム、メタホウ酸ナトリウム、四ホウ酸カリウム、メタホウ酸カリウム、ホウ酸;アルミニュウム化合物としてはオルトアルミン酸ナトリウム、メタアルミン酸ナトリウム、オルトアルミン酸カリウム、メタアルミン酸カリウム、塩化アルミニュウム、硝酸アルミニュウム、硫酸アルミニュウム、リン酸アルミニュウム;リン化合物としてはリン酸カリウム、リン酸ナトリウム、リン酸;ジルコニウム化合物としてはリン酸ジルコニウム、ジルコン酸ナトリウム、ジルコン酸カリウム、ジルコン酸;チタン化合物としては塩化チタン、チタン酸ナトリウム、チタン酸カリウム、チタン酸;亜鉛化合物としては塩化亜鉛、硝酸亜鉛、炭酸亜鉛、硫酸亜鉛、亜鉛酸塩;錫化合物としては錫酸ソーダ、錫酸カリウム等の改質剤を一例として挙げることができる。
【0043】
本発明の該抗菌剤粒子は元々単分散粒子であるため樹脂への分散性は極めて優れているが、さらに分散性を向上させる目的、あるいは樹脂製品の変色防止の目的、あるいは本発明の該抗菌剤粒子が樹脂に比較的多量に配合された時の機械的強度の低下を抑制する目的においては、本発明の該抗菌剤粒子の表面を、高級脂肪酸類、シラン系カップリング剤、アルミネート系カップリング剤、アルコールリン酸エステル類、界面活性剤類等の群から選ばれた少なくとも1種で表面処理することもできる。
かかる表面処理剤としては、ステアリン酸、オレイン酸、エルカ酸、パルミチン酸、ラウリン酸、ベヘニン酸等の高級脂肪酸類及びその塩類、ポリエチレンエーテルの硫酸エステル塩、アミド結合硫酸エステル塩、エステル結合硫酸エステル塩、エステル結合スルホネート、アミド結合スルホン酸塩、エーテル結合スルホン酸塩、エーテル結合アルキルアリルスルホン酸塩、アミド結合アルキルアリルスルホン酸塩等の界面活性剤類、ステアリルアルコール、オレイルアルコール等の高級アルコールの硫酸エステル塩、オルトリン酸とステアリルアルコール又はオレイルアルコールなどのモノ又はジエステル又は両者の混合物であって、その酸型又はアルカリ金属塩又はアミン等の燐酸エステル類、ビニルエトキシシラン、ビニル−トリル(2−メトキシ−エトキシ)シラン、ガンマ−メタクリロキシプロピルトリメトキシシラン、ガンマ−アミノプロピルトリメトキシシラン、N−フェニル−ガンマ−アミノプロピルトリメトキシシラン、N−ベータ(アミノエチル)ガンマ−アミノプロピルトリメトキシシラン、N−ベータ(アミノエチル)ガンマ−アミノプロピルトリエトキシシラン、N−フェニル−ガンマ−アミノプロピルトリエトキシシラン、ベータ(3,4−エポキシシクロヘキシル)エチルトリメトキシシラン、ガンマ−メルカプトプロピルとりメトキシシラン等のシランカップリング剤類、イソプロピルトリイソステアロイルチタネート、イソプロピルトリ(アミノエチル)チタネート、イソプロピルトリデシルベンゼンスルホニルチタネート等のチタネートカップリング剤類、アセトアルコキシアルミニュウムジイソプロピレート等のアルミネートカップリング剤を例示できる。
【0044】
本発明の該抗菌剤粒子は下記の方法で製造することができる。
その際、工業的に食品衛生法厚生省告示第20号で規定された、鉛、カドミュム等の重金属含有量が少なく、さらには樹脂製品の熱劣化防止(耐熱劣化性の向上)及び着色防止という意味で鉄、マンガン、クロム、銅、ニッケル等の重金属含有量を1%以下、好ましくは0.1%以下、さらに好ましくは0.01重量%以下、最も好ましくは0.001%以下の高純度の該抗菌剤粒子を得る場合には、先ず原料面で高純度の物を選択すること、さらには化学操作装置の材質面において特に腐食が起き易い水熱処理工程等では装置材料の溶出によって鉄、マンガン、クロム、銅、ニッケル等の重金属化合物が固溶体及び又は夾雑物として本発明の該抗菌剤粒子に混入しないように配慮された耐腐食性に優れたハステロイ鋼、ステンレスSUS−316鋼等を選択することは好ましいことである。
【0045】
本発明の該抗菌剤粒子は、基本的には国際特許出願PCT/JP2005/003831(出願日:2005年3月1日)明細書に記載された方法によって製造できる下記一般式(2)で表される有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子の1価陽イオン(B)の部分を銀とイオン交換する方法であり、その方法を後記1〜4に例示する。
[B]bAlcAx(SO4)y(OH)z・pH2O 式(2)
式中、b、c、x、y、z、p、BおよびAの定義は、前記式(1)と同じものを意味する。
式(2)なる化合物を合成するためには、硫酸アルミニウム、硫酸ナトリウム、硫酸カリウム、硫酸アンモニウム、及び硫酸カルシュウム等のアルミニウム及び硫酸原料と、蓚酸等の有機酸類原料と、水酸化ナトリウム、水酸化カリウム及びアンモニア水溶液等のアルカリ原料とを湿式法又は乾式法で反応させ、Bがナトリウム型、カリウム型またはアンモニウム型の銀を含まない有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子〔粒子形状が球状、円盤状、(碁石状)、一対状(ハンバーガー状)、米粒状、六角板状、六面体状、円柱状のものについては後記1〜3に示す〕を先ず合成しておき、然る後それらの有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子を水等の懸濁液中において銀溶液と接触攪拌すれば、銀が有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子にイオン交換した本発明の該抗菌剤粒子が製造できる。
【0046】
粒子形状が直方体状の銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子の製造方法については後記4に示す。
いずれの際も、銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子を、形状と粒子サイズを選択して製造すること、粒子径分布を均一に整えること、該粒子を完璧なまでに単分散状態で樹脂に分散できるようにするためには、該反応において前記の有機酸類を前記の反応時に添加する必要があり、添加された有機酸類は該抗菌剤粒子の構造中に組み込むことができる。
ただし有機酸類の一部は該抗菌剤粒子の表面に吸着している場合も考えられる。
いずれにしても有機酸類は本発明の目的を達成するために該抗菌剤に含有することができる。
有機酸類を前記の反応時に添加せず、反応が終わった後に添加しても前記粒子形状、粒子径均一性、分散性等の性質を有する本発明の抗菌剤粒子は製造できない。
【0047】
製造された本発明の抗菌剤粒子の粉砕処理に関しては従来技術のように強力な機械力で実施する必要はなく弱い力で簡単に処理してもの凝集のないのものが得られることも本発明の技術の特徴である。
以下に、ナトリウム型、カリウム型、アンモニウム型、水素型の銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子抗菌剤の製造方法について例示する。
【0048】
1、ナトリウム型銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子の製造方法
原料としては硫酸アルミニウム、硫酸ナトリウム等硫酸塩、及び水酸化ナトリウム等のAl原料及びSO4原料及びNa原料、蓚酸等の有機酸原料、及びイオン交換反応で使用する硝酸銀等の可溶性銀塩を銀原料として用い、下記の方法で製造する方法が例示できる。
例えばAl2(SO4)3+Na2SO4(或いはNaNO3)+H2C2O4を水の中に充分溶解した後、攪拌しながら、水酸化ナトリウムを添加する。
添加終了後、よく分散させるために、好ましくは更に20分以上攪拌する。
その後水熱処理を行うことは好ましいことである。
水熱処理の温度は100℃〜250℃が好ましく、処理時間は1時間〜30時間が好ましい。
【0049】
このようにして得られた有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子は濾過、及び必要に応じ水洗する。、
それを水等の液に懸濁し硫酸銀、硝酸銀等の可溶性銀塩の溶液と攪拌すればイオン交換反応処理が実施でき、本発明の銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子が製造できる。
イオン交換反応処理の温度は0℃〜100℃が好ましく、処理時間は0.1時間〜30時間遮光下で実施することが好ましい。
【0050】
イオン交換するときの処理温度が低かったり、処理時間が短か過ぎると銀のイオン交換量が少なくなる恐れがある。一方イオン交換するときの処理温度が高かったり、処理時間が長過ぎるとイオン交換処理物が褐色に着色する傾向がある。
イオン交換反応処理での攪拌の方法は振動及び回転等の方法が例示できる。
イオン交換したものは濾過・遠心分離の方法で濾別した後、必要ならばさらに水洗・表面処理・乾燥・粉砕等の操作を必要に応じ実施し回収すれば良い。濾別が困難な場合、本発明の目的に反しない範囲で凝集剤を使用し濾別操作を改善しても良い。凝集剤としてはポリアクリルアミドのような高分子凝集剤が例示できる。高分子凝集剤の添加量は0.2%以下が好ましい。0.2%以上添加しても特に炉別操作は改善しない。
【0051】
2、カリウム型銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子の製造方法
原料としては硫酸アルミニウム、硫酸カリウム等硫酸塩、及び水酸化カリウム等のAl原料及びSO4原料及びK原料、蓚酸等の有機酸原料、及びイオン交換反応で使用する硝酸銀等の可溶性銀塩を銀原料として用い、下記の方法で製造する方法が例示できる。
例えばAl2(SO4)3+K2SO4(或いはKNO3)+H2C2O4を水の中に充分溶解した後、攪拌しながら、水酸化カリウムを添加する。それ以後の操作方法は上記1に準じて行う。
【0052】
3、アンモニウム型銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子の製造方法
原料としては硫酸アルミニウム、硫酸アンモニウム等硫酸塩、及び硝酸アンモニウム等のAl原料及びSO4原料及びNH4原料、蓚酸等の有機酸原料、及びイオン交換反応で使用する硝酸銀等の可溶性銀塩を銀原料として用い、下記の方法で製造する方法が例示できる。
例えばAl2(SO4)3+K2SO4(或いはKNO3)+H2C2O4を水の中に充分溶解した後、攪拌しながら、アンモニア水溶液を添加する。それ以後の操作方法は上記1に準じて行う。
【0053】
4、水素型{(H3O)+型}直方体状銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子の製造方法
直方体状粒子は前記の球状及び円板状粒子の製造方法とは異なり、硫酸アルミニウム水溶液と水酸化アルミニウム懸濁液、及び蓚酸等の有機酸を混合攪拌したものを100℃〜250℃、好ましくは120℃〜200℃で、0.5時間以上、好ましくは0.5〜30時間、好ましくは2〜20時間水熱処理すれば直方体状で水素型、化学式(H3O)Al3(SO4)2(OH)6で表されるものを得ることができる。
用いる水酸化アルミニウムとしては、結晶性のギプサイト、バイアライト、ベーマイト、擬ベーマイト、ダイアスポア、無定形の水酸化アルミニウムが例示できるが、無定形の水酸化アルミニウムが粒子径の均一性が高くなるという点で好ましい。
無定形の水酸化アルミニウムとしては、協和化学工業(株)製乾燥水酸化アルミニウムゲルS−100,及び同FMが例示できる。
【0054】
この方法において微粒子化と粒子径均一性を向上ためには、硫酸アルミニウム水溶液と水酸化アルミニウム懸濁液を混合攪拌した反応物を直ちに水熱処理するよりは、反応後ある程度時間が経過したもの、例えば0.5時間以上、好ましくは5時間以上、さらに好ましくは16時間以上静置又は攪拌したものを水熱処理すると微粒子で粒子径均一性を有するアルミニウム硫酸塩水酸化物粒子が得られる。それ以後の操作方法は上記1に準じて行う。
【0055】
本発明の銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子は粉末X線回折法により同定することができる。銀が該粒子にイオン交換していればその回折パターンはイオン交換前の該粒子と同じ回折パターンとなるので化学分析値と照らし合わせて回折X線の回折パターンを精査すれば良い。もし銀が該粒子にイオン交換せずに、酸化銀のような不純物で共存する製造物を樹脂に配合すると樹脂製品の色は暗褐色を呈し本発明の重要な目的の一つである白色の樹脂製品を得ることから逸脱するので好ましくない。
本発明においては、式(1)なる該抗菌剤の3価陽イオンであるアルミニウム(Al)の一部をZn2+、Ti4+の群から選ばれた少なくとも1種の陽イオンで、及び/又は1価陽イオンBの一部をCa2+で本発明の目的を害しない範囲で置き換えて含有させても良い。
その場合、該抗菌剤を樹脂に配合した時樹脂製品の透明性を低下させないようにするためZn2+、Ti4+の合計モル含有量はアルミニウムのモル含有量の1/2以下好ましくは1/3以下、Ca2+のモル含有量は1価陽イオン(B)のモル含有量の1/2以下好ましくは1/3以下にして該抗菌剤粒子に含有させれば良い。
【0056】
式(1)なる該抗菌剤の3価陽イオンのアルミニウム(Al)の一部を置き換える方法としては、該金属カチオンを含む硫酸亜鉛、硫酸チタン、硫酸カルシウム等の塩類を前記式(2)なる化合物を合成する時に用いて式(2)に組み込むか、あるいは式(2)なる化合物に該金属カチオンを含む化合物を用いて溶媒中でイオン交換した物に、さらに前記の方法で銀とイオン交換する方法が例示できる。その場合、Zn2+、Ti4+は抗菌性発現効果がある元素であるため式(1)においてAg1+の相対的含有量を減らしても抗菌性を発揮し、その結果これを樹脂に配合した樹脂成形品の着色を減少させる利点がありそういう意味で好ましいことである。
【0057】
本発明においては、式(1)なる該抗菌剤の(SO4)yは(SO4)yの一部、好ましくyモルの1/2以下を他の無機酸イオンで置き換えることもでき、置き換える量が特に1/2以下であれば本発明の粒子形状や粒子径均一性が何の問題もなく維持でき本発明の目的が達成される。該無機酸イオンとしてはSO32−、PO43−、HPO32−、CO32−、NO3−、SiO44−及びBO33−等が例示できる。式(1)なる該抗菌剤の(SO4)yの一部を他の無機酸イオンで置き換える方法としては、該無機酸イオンを含む塩類を式(2)なる化合物を反応する時に硫酸アルミニウム、硫酸ナトリウム、硫酸カリウム等の代りに用いて式(2)に組み込むか、あるいは式(2)なる化合物に該無機酸イオンを含む化合物を用いて溶媒中でイオン交換した物に、さらに前記の方法で銀とイオン交換する方法が例示できる。該粒子はさらに式(1)の該抗菌剤粒子同様、追加乾燥処理、焼成処理、表面処理、耐酸性被覆処理をして抗菌剤として利用することもできるし、樹脂に配合して抗菌性樹脂製品を得ることもできる。
【0058】
本発明においては、式(1)なる該抗菌剤の(OH)zは(OH)zの一部がCl−で置き換わって含有されている場合もあるが、その含有量は着色防止という意味で該抗菌剤式(1)中0.1モル以下、好ましくは0.01モル以下、最も好ましくは0.001モル以下である。
本発明において抗菌化の対象樹脂としては特に限定されるものではなく合成樹脂・ゴム、天然樹脂・ゴム等を原料としそれを加工して通常成形品、繊維、不織布、塗料、コーキング材、フィルム等樹脂製品として使用されるものであれば良く、以下にその一部を例示する。
【0059】
熱可塑性樹脂としては、ポリプロピレン、エチレンプロピレン共重合体、高密度ポリエチレン、低密度ポリエチレン、超高分子量ポリエチレン、直鎖状低密度ポリエチレン、ポリブテン、ポリ・4メチルペンテン−1等の如きC2〜C12のオレフィン(αオレフィン)の重合体もしくは共重合体、これらのオレフィンとジエンとの共重合体、エチレン酢酸ビニル共重合体、エチレン・アクリレート共重合体、TPO(熱可塑性ポリオレフィン)樹脂等のポリオレフィン系樹脂、ポリエチレンオキサイド樹脂、ポリスチレン、ABS(アクリロニトリルブタジエンスチレン)樹脂、AAS(アクリロニトリルアクリルスチレン)樹脂、AS(アクリロニトリルスチレン)樹脂、AES(アクリロニトリルイーピーディーエムスチレン)樹脂、MBS(メチルメタクリレートブタジエンスチレン)樹脂、ポリパラメチルスチレン樹脂等のスチレン系樹脂、ACS(アクリロニトリルクロリネイティドポリエチレンスチレン)樹脂、酢酸ビニル樹脂、プロピオン酸ビニル樹脂、フェノキシ樹脂、アイオノマー樹脂、ポリアセタール樹脂、ナイロン6、ナイロン66等のポリアミド系樹脂、ポリエチレンテレフタレート、ポリブチレンテレフタレート等のポリエステル系樹脂、ポリアミドイミド樹脂、ポリサルホン樹脂、ポリアリレート樹脂、ポリエーテルイミド樹脂、ポリエーテルケトン樹脂、ポリフェニレンエーテル樹脂、ポリフェニレンサルファイド樹脂、メタクリル樹脂、セルロース樹脂、ポリカーボネート樹脂、フッソ樹脂、ポリウレタン樹脂、シリコーン樹脂、ポリビニルエーテル樹脂、ポリビニルホルマール樹脂、ポリビニルブチラール樹脂、ポリビニルアルコール樹脂、イソブチレン−無水マレイン酸共重合樹脂、さらにはポリ塩化ビニル樹脂、エチレン塩ビ共重合樹脂、エチレン酢ビ共重合樹脂、塩素化塩化ビニル樹脂、塩素化ポリエチレン、塩素化ポリプロピレン、クマロン樹脂、ケトン樹脂、ポリ塩化ビニリデン、ポリ2塩化ビニル樹脂、塩素化ポリエーテル等の分子構造中に塩素を有する樹脂、アセテートプラスチック、酢酸セルロース、セルロイド、液晶ポリマー、さらには吸水性樹脂等がそれらの一部として例示できる。
【0060】
熱硬化性樹脂としては、エポキシ樹脂、フェノール樹脂、メラミン樹脂、不飽和ポリエステル樹脂、アルキド樹脂、グアナミン樹脂、ポリイミド樹脂、尿素樹脂、シリコーン樹脂、フェノールホルムアルデヒド樹脂、メラミンホルムアルデヒド樹脂、ポリパラ安息香酸樹脂、ポリウレタン樹脂、マレイン酸樹脂、ユリア樹脂、フラン樹脂、キシレン樹脂、ジアリルフタレート樹脂等の樹脂を例示できる。
ゴムとしてはEPDM(エチレンプロピレンジエン共重合体ゴム),EPM(エチレンプロピレン共重合体ゴム),ブチルゴム、イソプレンゴム、SBR(スチレンブタジエンゴム),NIR(ニトリルイソプレンゴム),NBR(ニトリルブタジエンゴム),ウレタンゴム、クロロプレンゴム、水素化ニトリルゴム、ポリエーテル系ゴム、四フッ化エチレン・プロピレンゴム、クロロスルホン化ゴム、ブタジエンゴム、アクリルゴム、塩素化ポリエチレン、エピクロルヒドリンゴム、プロピレンオキサイドゴム、エチレン・アクリルゴム、ノルボルネンゴム、多硫化ゴム、フッ素ゴム、シリコーンゴム、天然ゴム等が例示できる。
これらの樹脂及びゴムは単独で用いても良く、あるいは複数種同時に用いても良い。複数種同時に用いる場合はポリマーアロイ化して用いても良く、ブレンドして用いても良く、あるいは積層成形して用いても良い。これら合成樹脂類は製造方法によって限定されるものではなく、例えばポリオレフィンの重合触媒としては、チーグラー法、チーグラーナッタ法、フリーデルクラフト法、メタロセン法、フィリップス法等いかなものであっても良い。
【0061】
本発明の抗菌性樹脂組成物には通常使用される添加剤、補強剤、充填剤を本発明の目的を害しない範囲で配合することができる。以下これらの一部を例示する。
酸化防止剤、紫外線吸収剤、光安定化剤、熱安定剤、金属不活性化剤、可塑剤、帯電防止剤、難燃剤、加硫剤、加硫促進剤、老化防止剤、素練り促進剤、粘着付与剤、香料、滑剤、着色剤、造核剤、発砲剤、脱臭剤、ポリマーアロイ相溶化剤等。
本発明の抗菌性樹脂組成物を得る方法には、特別の制約はなく前記の樹脂類、ゴム類と本発明の抗菌剤粒子を、例えば2軸押出機、加圧ニーダー、オープンロール、バンバリーミキサー等の装置を使用しできるだけ均一に混合混練しておき、それをペレッターザー、粉砕機等の装置を使用しペレットや粉末にし樹脂組成物を得る方法や、樹脂類が溶媒を使用して溶解されたものの中に本発明の抗菌剤粒子を混合する方法が例示できる。
【0062】
本発明の抗菌性樹脂組成物の成形品を得る方法には、特別の制約はなく前記の方法で得られた該抗菌剤配合樹脂組成物をそのままの濃度で成形品等の樹脂製品にする方法、あるいはマスターバッチとして一旦高濃度に加工したものからそれを最終的に樹脂製品として使用する濃度に希釈混合し、それを射出成形機、押出成形機、ブロー成形機、カレンダー成形機、圧縮成形機、積層成形機等の装置を用いて抗菌性樹脂製品とする方法、さらには抗菌剤粒子を樹脂と混合後、前記の機械に直接投入し直接射出成形、直接押出成形する方法等を一例として例示できる。
得られた成形品の形状、大きさ、厚さ、太さ、さらには使用方法についても何ら制約はなく例えば板状、ボトル状、球状、円盤状、シート状、電線被覆状、発砲体、積層体等何でも良くその成形品は例えば台所、風呂場、トイレ周りの製品、衛生用品、エアコンの吹き出し口等抗菌性を必要とする分野で好適に使用できる。
【0063】
本発明の抗菌性フィルムを得る方法には、特別の制約はなくフィルム製造に適した樹脂の中から前記の方法で得られた抗菌性樹脂組成物を製造し、それをインフレーション法、Tダイ法、カレンダー法、キャスト法等でフィルムを得ることができ、さらにそのフィルムを延伸しても良く、さらには共押出し法により2層以上の積層フィルムにした物で抗菌性が必要な層にだけ本発明の該抗菌剤粒子を抗菌剤として使用する経済性を追求した利用方法も例示できる。
得られたフィルムの形状や形態、使用方法についても特に制約はなく、例えば野菜果物菓子乾物魚肉等食品包装用フィルムとして鮮度保持を兼ねた抗菌性を必要とする分野を一例として例示できる。
【0064】
本発明の繊維を製造する方法において特別の制約はなく、前記の樹脂組成物のうち繊維として紡糸できる樹脂組成物を使用し従来公知の溶融押し出し法、乾式紡糸法、湿式紡糸法等で紡糸し、必要に応じさらに延伸や撚糸、及び/又は、さらには綿、ウール、麻等の天然繊維との混合等の工程を採用し製品とすれば良い。得られた繊維の利用方法についても特に制約はなく、例えば絨毯、衣服、タオル、ナプキン、ハンカチ、手袋、ソックス、帽子、マフラー等の抗菌性を必要とする分野が一例として例示できる。
繊維としては、ポリプロピレン系、ポリエチレン系、ポリアミド(ナイロン)系、アラミド系、アクリル系、ポリウレタン系、フッソ系、ポリクラール系、ポリエステル系、ポリ塩化ビニル系、ビニロン系、ビニリデン系、アセテート系、トリアセテート系、レーヨン系、キュプラ系、ノボロイド系、プロミックス系、ポリアセタール系、ポリノジック系、プラスチック光ファイバー等の繊維が例示できる。
【0065】
本発明の抗菌性不織布を製造する方法には特に制約はなく従来公知の方法を採用すれば良い。
例えば、抄造ウエブ法、ランダムウエブ法、パラレルウエブ法、クロスレイドウエブ法、糸交錯ウエブ法、トウ拡開ウエブ法、短繊維ウエブ法、フィラメントウエブ法、マイクロファイバーウエブ法、スプリットフィルムウエブ法等の方法が採用し得る。これらの方法で得られた抗菌性不織布は抗菌性を有しながら軽くて通気性、防縮性等の性質に優れるため生理用用品、衣服の芯地や裏地、ビニルレザーや擬革の基布等抗菌性を必要とする分野で利用できることが一例として挙げられる。
【0066】
本発明の抗菌性塗料を工業的に製造する方法において特別の制約はなく、従来公知の方法が採用でき、顔料、展色剤、耐候剤、溶剤、助溶剤、希釈剤、可塑剤等を混合する工程で本発明の銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子抗菌剤を混合し、その後は所望により練り合わせ、フルイ(濾過)等の工程を通して製品とすれば良い。家庭で個人的に使用する場合は市販の塗料に本発明の該抗菌剤粒子を混合して使用することもできる。本発明の抗菌性塗料は例えば、病院内、老人ホーム、学校、レストラン等で使用すれば大腸菌や黄色ブドウ球菌等の細菌からの被害を防止でき本発明の目的が達成される。塗料としては、合成樹脂塗料(アルキド、アミノアルキド、エポキシ、フッ素、ウレタン、酢酸ビニル、アクリル酸エステル、不飽和ポリエステル、フェノール、グアナミン、ブチラール、スチレンブタジエン、スチレン、塩化ビニル、塩化ビニリデン、塩化ゴム系塗料、サビ止めペイント、粉体塗料、電気絶縁塗料)、セルロース塗料、ゴム系塗料、水溶性合成樹脂塗料(水溶性アルキド、水溶性エポキシ、水溶性ポリブタジエン、水溶性メラミン、水溶性尿素、水溶性フェノール、水溶性アクリル等)、酒精塗料、油性塗料等樹脂を使用する塗料を例示できる。
【0067】
本発明の抗菌性コーキング材を得る方法には、特別の制約はなく例えばコーキング材としての基礎成分オルガノポリシロキサンとγアミノプロピルビス(メチルエチルケトキシアミノ)メトキシシランのようなメトキシシラン系の接着促進剤を混合しておき、次に4官能性や3官能性のシラン架橋剤及びシリカエアロジル等の増粘剤、その他必要に応じポリマーと架橋剤の反応を促進する有機スズカルボキシレートのような触媒、さらには酸化防止剤、紫外線吸収剤、可塑剤等とともに本発明の抗菌剤粒子を従来公知の方法で得る方法が例示できる。
【0068】
本発明の前記式(1)で表わされる抗菌剤は、前述したように樹脂に配合して種々の成形品として価値のある製品を提供することができる。一方、本発明の抗菌剤は、その分散性、白色性、粒形の均一性および粒子径の均一性を利用して、樹脂以外の用途にも適用することができる。
すなわち、その抗菌性を利用して、抗カビ剤、抗菌消臭剤(スプレー)、抗菌紙、農薬および化粧料にも適用することができる。本発明の抗菌剤は人体に対して安全であり且つ人体に接触しても刺激を与えないので、人間の生活する住宅内の台所、風呂場やトイレなどの製品に対して抗菌剤および抗カビ剤として利用することができる。また農薬として野菜や果物などにも直接散布することができる。さらに化粧料の中に配合しても有利に利用することができる。
【0069】
以下に本発明の該抗菌剤粒子からなる抗菌剤、及び該抗菌剤粒子が配合された抗菌性樹脂組成物、及び該抗菌性樹脂組成物から形成された成形品等の各物性の測定方法を示す。
銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子抗菌剤の粒子性状の測定方法;
最近の微粒子の平均二次粒子径及び粒子径分布の測定は、レーザー回折散乱法が主流になっており、本発明ではこれらの測定はレーザー回折散乱法で行われた。
【0070】
(1)平均二次粒子径;堀場製作所製粒度分布測定装置LA―910(レーザー回折散乱法)を用いて測定した。
【0071】
(2)粒子径分布のシャープ度 Dr=D75/D25
堀場製作所製粒度分布測定装置LA―910を用いて測定されたレーザー回折散乱法体積基準累積粒子径分布曲線の75%値の粒子径D75(大粒子径側)を25%値の粒子径D25(微粒子径側)で除した算式で表される粒子径分布のシャープ度をDr=D75/D25として定義した。 Drの数値が小さい程粒子径分布のシャープ度が認められ粒子径均一性を意味する。
だだし、球形粒子についてはレーザー回折散乱法で測定されたD75/D25と、下記のSEM写真による粒子径分布測定方法と組み合わせて粒子径を評価した。
その場合、レーザー回折散乱法によるD75/D25の数値はSEM写真によるD75/D25の数値と約−10〜+10%の範囲で異なる程度でほぼ一致したので本発明ではレーザー回折散乱法によるD75/D25を採用した。
電子顕微鏡写真(SEM写真)による粒子径分布測定方法(球形粒子の場合);
1枚のSEM写真で観察される全ての粒子(50個〜数百個)の球形粒子をそれぞれ個別に長径と短径をノギスで1/50mmまで測定し長径と短径の平均値を求めて各球形粒子の粒子径(μmに換算)とし、それから累積粒子径のD75%とD25%に該当する粒子径を認定しDr=D75/D25を算出した。
【0072】
(3)BET法比表面積;湯浅アイオニクス(株)製の12検体全自動表面測定装置マルチソーブ−12で測定した。
【0073】
(4)粒子形状;走査型電子顕微鏡(SEM写真)で観察した。
尚粒子形状としての図(写真)のと実施例、比較例の抗菌剤粒子との関係は以下のとおりである。
図1、表面が平滑である球状粒子、A1粒子
図2、表面に小粒を有する球状粒子、A20粒子
図3、表面が荒くさらには皺(球に傷や割れ)を有する球状粒子、A21粒子
図4、穴(凹凸)を有する球状粒子、A22粒子
図5、表面が平滑であり且つ直線部分を図1のものより少し多く含む球状粒子、A30粒子
図6、表面が荒く皺を有する球状粒子、A31粒子
図7、円盤状粒子粒子、B1−1粒子、
図8、一対状粒子(ハンバーガー状粒子)、C1粒子
図9、米粒状粒子、D1粒子、
図10、直方体粒子、E1粒子
図11、六角板状粒子、F1粒子
図12、8面体状粒子、G1粒子
図13、円柱状(酒樽状)粒子、H1粒子
図14、表面が荒く延伸されたような状態の凝集体粒子、V1粒子
【0074】
(5)イオン交換体形成;粉末X線回折法により銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子の回折パターン及びそれ以外の回折パターンの有無を調査した。
【0075】
(6)有機酸の含有量;銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子を1000℃で焼成しCO2を発生させ回収し、JIS R 9101によりCO2を測定し、それに基づいて炭素の含有量を計算しそれから有機酸の含有量を求めた。
抗菌性樹脂組成物、樹脂成形品、フィルム、繊維、塗料、コーキング材等樹脂製品の測定方法;
【0076】
(7)抗菌性試験方法
ただし下記(a)〜(e)の試験において、大腸菌はE.coli NBRC 3972 黄色ブドウ球菌は S.aureus NBRC 12732を使用した。抗菌試験の結果も示す表−7〜表−17においてcfuはcolony forming unitの略である。cfu/mlは1ml中の生菌数を表わし、その数値が小さい程抗菌性が高いことを意味している。
(a)成形板;JIS−Z 2801(2000年版)
テストピースサイズ;50mm×50mm×2mm
(b)繊維、不織布;JIS−L 1902(2002年版)
(c)塗料;塗料を鉄板サイズ50mm×50mm×2mmのテストピース(材質はJIS G 3101 SS400)の上に約0.1mmになるように塗装し上記(a)の方法で測定した。
(d)フィルム;JIS−Z 2801(2000年版)
(e)コーキング材;コーキング材を鉄板サイズ50mm×50mm×2mmのテストピース(材質はJIS G 3101 SS400)の上に約1mmになるようにコーキングし上記(a)の方法で測定した。
【0077】
(8)透明性;厚さ2mmの板又は50μmのフィルムでヘイズ値HをTOKYO DENSYOKU CO,LTDのAUTOMATIC HAZE METER OPTICAL UNIT TC−HIIIDPで測定し下記の算式により%で表示した。
【0078】
H={h1−(h2−h1)}×100/h1 %
h1=抗菌剤未添加の樹脂成形品又はフィルムのヘイズ値
h2=抗菌剤を添加した樹脂成形品又はフィルムのヘイズ値
Hが100%に近い程抗菌剤未添加品に近い透明性を維持していることを意味している。
【0079】
(9)白色性; 成形品、フィルム、繊維、不織布等のサンプルを直射日光のない北側の室内で60日間静置したものがどの程度変色したかを目視により測定した。
【0080】
(10)水道水接触後の抗菌力維持特性;
上記記載の試験資料を、水道水1000mlの中に90℃又は70℃で120時間浸水させた後水道水で十分に水洗し,前記(7)の(a)〜(e)の方法で測定した。尚試験片の比重が1以下のものについてはステンレス鋼SUS−304で作られた金網を錘に使い水中に強制的に浸水した。水道水を90℃又は70℃に加温した理由は水道水接触後の抗菌力維持特性を短時間で見るためであり、これは常温で水道水接触後の抗菌力維持特性を試験する場合の促進試験となる。
【0081】
(11)混練押出加工時フィルター通過性試験(押出機圧力の測定)
プラスチック工学研究所製の30φ2軸混練押出機(スクリュー径30mm)にフィルター(スクリーンメッシュ)を樹脂の流れ方向から見てブレーカープレートの手前に設置した。フィルターは50メッシュのものをブレーカープレート側に、80メッシュのものを中央に、100メッシュのものをホッパー側にそれぞれ1枚づつ設置した。その状態で、該機械をスクリュー回転数が170rpm,吐出量が10Kg/Hrとなるようにして2時間、及び24時間運転し、その時のフィルター手前に設置してある圧力計により圧力を測定した。
圧力はフィルターを通過できない粗大粒子がフィルターに多く目詰まりする程早く高くなりそれはフィルター通過性が悪いことを意味し、それだけフィルターを入れ替える必要が早く生じ工業的には問題である。
この試験において、圧力が200Kg/cm2以上は機械が破損寸前での運転状態であり、150Kg/cm2以上はフィルター交換しないと機械に負荷をかけ過ぎ機械運転に支障をきたす恐れがあることを意味し、100Kg/cm2になるとフィルター交換の準備が必要なことを意味し、100Kg/cm2以下は何の問題もなく運転できることを意味している。
【0082】
機械を運転開始して24時間後の圧力が150Kg/cm2までであれば、一日に1回程度のフィルター交換で済み工業的にはかろうじて何とか意味のあることである。
尚本測定は樹脂95重量%と抗菌剤5重量%の合計100重量%になる抗菌剤配合濃度において測定したもので、表−7〜表−9に示した縦2重線より左側の抗菌剤配合量濃度とは異なる配合条件で別途実施したものである。混練押出する時のその他の条件は縦2重線より左側と同じにした。
【実施例】
【0083】
以下先ず本発明の抗菌剤粒子の製造方法を実施例に基づいて具体的に説明する。
【0084】
尚、以下の実施例の式に於いて式(1)のbの数値が1の場合は1が省略してある。
銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子抗菌剤の製造;
【0085】
<実施例>I−1〜I−47及び<比較例>I−1〜I−9
(A,B,C,D,E,F,G,H粒子の製造)
球状粒子(A粒子)の製造;実施例I−1〜31
実施例I−1
[Ag0.1(NH4)0.9]Al3(C2O4)0.1(SO4)1.9(OH)6・0.5H2Oの製造
下記の原料を使用し、下記の合成方法によりA1粒子を得た。
使用原料 硫酸アンモニウム 264.28g=2モル
蓚酸(H2C2O4・2H2O) 38g=0.3モル
1.04mol/Lの硫酸アルミニウム×1923ml=2.0モル
9.0mol/Lのアンモニア水溶液×894.5ml=8.05モル
合成方法 264.28gの硫酸アンモニウム(NH4)2SO4を4.0Lのイオン交換水に溶解した。
38gの蓚酸(H2C2O4・2H2O)を1.0Lのイオン交換水に溶解した。
【0086】
攪拌下の該硫酸アンモニウム(NH4)2SO4水溶液に該蓚酸水溶液及び上記硫酸アルミニウムAl2(SO4)3水溶液を添加し混合酸性水溶液を作った。
該混合酸性水溶液は良く攪拌しながら、50℃以下に加熱し(析出した結晶物を溶解させるため)、その後上記のアンモニア水溶液を894.5ml を25分かけて該混合酸性水溶液に添加しアンモニウム型の有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子沈殿物のスラリーを作った。
そのスラリーを更に100℃で1時間水熱処理した。
処理後の沈殿物を濾過・水洗・乾燥・粉砕処理し粉末サンプルを得た。
そのサンプル100gをとって0.025mol/Lの硫酸銀水溶液600mlに懸濁させ攪拌して、30℃でアンモニウムイオンと銀イオンとのイオン交換処理を遮光下で8時間行った。
イオン交換後のサンプルを濾過・水洗・乾燥(105℃×6時間)・粉砕した。
これらの処理過程によって、A1粒子を得た。結果を表−1に示す。
【0087】
実施例I−2
[Ag0.1(NH4)0.9]Al3(C2O4)0.1(SO4)1.9(OH)6の製造)
実施例I−1で得たA1粒子を150℃で2時間乾燥し、A2粒子を得た。
【0088】
実施例I−3
[Ag0.1(NH4)0.9]Al3(C2O4)0.0001(SO4)1.9999(OH)6の合成
実施例I−1において、蓚酸の使用量を0.0005モル(0.063g)に変更したことが実施例I−1と異なるのみで、その他の処理過程は実施例I−1に準じて行い、さらに150℃で2時間乾燥しA3粒子を得た。
【0089】
実施例I−4
[Ag0.1(NH4)0.9]Al3(C2O4)0.01(SO4)1.999(OH)6の製造
実施例I−1において、蓚酸の使用量を0.05モル(6.3g)に変更したことが実施例I−1と異なるのみで、その他の処理過程は実施例I−1に準じて行い、さらに150℃で2時間乾燥しA4粒子を得た。
【0090】
実施例I−5
[Ag0.1(NH4)0.9]Al3(C2O4)0.4(SO4)1.7(OH)5.8の製造
実施例I−1において、蓚酸の使用量を1.5モル(189.1g)に変更したことが実施例I−1と異なるのみで、その他の処理過程は実施例I−1に準じて行い、さらに150℃で2時間乾燥しA5粒子を得た。
【0091】
実施例I−6
[Ag0.1(NH4)0.9]Al2.7(C2O4)0.1(SO4)1.9(OH)5.1の製造
実施例I−1において、硫酸アルミニウムの使用量を1731ml(1.8モル)に変更したことが実施例I−1と異なるのみで、その他の処理過程は実施例I−1に準じて行い、さらに150℃で2時間乾燥しA6粒子を得た。
【0092】
実施例I−7 [Ag0.1(NH4)0.9]Al3.3(C2O4)0.1(SO4)1.9(OH)6.9の製造
実施例I−1において、硫酸アルミニウムの使用量を2115ml(2.2モル)に変更したことが実施例I−1と異なるのみで、その他の処理過程は実施例I−1に準じて行い、さらに150℃で2時間乾燥しA7粒子を得た。
【0093】
実施例I−8
[Ag0.1(NH4)0.9]Al3(C2O4)0.1(SO4)2.4(OH)5の製造)
実施例I−1において、硫酸アンモニウム(NH4)2SO4の使用量を2.3モル(303.9g)に変更したことが実施例I−1と異なるのみで、その他の処理過程は実施例I−1に準じて行い、さらに150℃で2時間乾燥しA8粒子を得た。
【0094】
実施例I−9
[Ag0.1(NH4)1.10]1.2Al3(C2O4)0.1(SO4)1.9(OH)6.02の製造
実施例I−1において、アンモニア水溶液添加後の攪拌時間を2時間に変更したこと、添加したアンモニア水溶液の量を1093ml(9.84モル)に変更したこと、及び水熱処理時間を30分に変更したことが実施例I−1と異なるのみで、その他の処理過程は実施例I−1に準じて行い、さらに150℃で2時間乾燥しA9粒子を得た。
【0095】
実施例I−10
[Ag0.1(NH4)0.9]Al3(C2O4)0.1(SO4)1.9(OH)6の製造
実施例I−1において、100℃での水熱処理時間を45分に変更したことが実施例I−1と異なるのみで、その他の処理過程は実施例I−1に準じて行い、さらに150℃で2時間乾燥しA10粒子を得た。
【0096】
実施例I−11
[Ag0.1(NH4)0.76]0.86Al3(C2O4)0.1(SO4)1.9(OH)5.68の製造
実施例I−1において、アンモニア水溶液添加の時間を40分に変更したこと、及び添加したアンモニア水溶液の量を755ml(6.8モル)に変更したことが実施例I−1と異なるのみで、その他の処理過程は実施例I−1に準じて行い、さらに150℃で2時間乾燥しA11粒子を得た。
【0097】
実施例I−12
[Ag0.3(NH4)0.7]Al3(C2O4)0.1(SO4)1.9(OH)6の合成
実施例I−1において、イオン交換用硫酸銀水溶液の量を1800mlに変更したことが実施例I−1と異なるのみで、その他の処理過程は実施例I−1に準じて行い、さらに150℃で2時間乾燥しA12粒子を得た。
【0098】
実施例I−13
[Ag0.001(NH4)0.999]Al3(C2O4)0.1(SO4)1.9(OH)6の製造
実施例I−1において、イオン交換用硫酸銀水溶液を硝酸銀水溶液に変更し、その濃度を0.001mol/L,液量を300mlに変更し処理温度を15℃に変更したことが実施例I−1と異なるのみで、その他の処理過程は実施例I−1に準じて行い、さらに150℃で2時間乾燥しA13粒子を得た。
【0099】
実施例I−14
[Ag0.00001(NH4)0.99999]Al3(C2O4)0.1(SO4)1.9(OH)6 の製造
実施例I−13において、イオン交換用硝酸銀水溶液の濃度を0.00001mol/Lに変更したことが実施例I−13と異なるのみで、その他の処理過程は実施例I−1に準じて行い、さらに150℃で2時間乾燥しA14粒子を得た。
【0100】
実施例I−15
[Ag0.1(NH4)0.9]Al3(C2O4)0.1(SO4)1.9(OH)6・0.5H2Oの製造
実施例I−1で得たサンプル50gをステアリン酸ソーダ1.5g含む80℃水溶液に懸濁させ、30分間攪拌し表面処理を行った後脱水、水洗、乾燥(105℃×6時間)、粉砕しA15粒子を得た。
【0101】
実施例I−16
[Ag0.1(NH4)0.9]Al3(C2O4)0.1(SO4)1.9(OH)6・2H2Oの製造
実施例I−1で得たサンプルを30℃で相対湿度75%のNaCl雰囲気において、25時間の吸湿処理を行い、A16粒子を得た。
【0102】
実施例I−17 実施例1の400℃ 1時間焼成物
実施例I−1で得たサンプルを400℃1時間窒素雰囲気下で焼成処理を行い、A17粒子を得た。
粉末X線回折法によると全て[Ag0.1(NH4)0.9]Al3(C2O4)0.1(SO4)1.9(OH)6であった。形状は球状であった。
A17粒子10.00gをイオン交換水100mlに入れ20℃で30分攪拌し、それを脱水・水洗120℃で16時間乾燥した後の重量減少はなく10.00gであった。
【0103】
実施例I−18 実施例1の500℃ 1時間焼成物
実施例I−1で得たサンプルを500℃1時間窒素雰囲気下で焼成処理を行い、A18粒子を得た。
粉末X線回折法によると全て[Ag0.1(NH4)0.9]Al3(C2O4)0.1(SO4)1.9(OH)6であった。形状は球状であった。
A18粒子10.00gをイオン交換水100mlに入れ20℃で30分攪拌し、それを脱水・水洗し120℃で16時間乾燥した後の重量減少はなく10.00gであった。
【0104】
実施例I−19
[Ag0.001K0.999]Al3(C2O4)0.1(SO4)1.9(OH)6の製造
下記の原料を使用し、下記の方法によりA19粒子を得た。
使用原料 硫酸カリウム 2モル 348g
蓚酸(H2C2O4・2H2O)0.3モル 38g
1.04mol/Lの硫酸アルミニウム水溶液 2.0モル 1923ml
7.5mol/L KOH水溶液 8.4モル 1120ml
合成方法 348gの硫酸カリウム(K2SO4)は4.0Lのイオン交換水に溶解した。
38gの蓚酸(H2C2O4・2H2O) は1.0Lのイオン交換水に溶解した。
攪拌下の該硫酸カリウム(K2SO4)水溶液に該蓚酸水溶液及び上記硫酸アルミニウムAl2(SO4)3水溶液を添加し混合酸性水溶液を作った。
該混合酸性水溶液を良く攪拌して、そして、上記水酸化カリウム水溶液を1120mlを25分かけて該混合酸性水性液に添加しカリウム型の有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子沈殿物スラリーを作った。。
そのスラリーを更に1時間攪拌後、オートクレーブによって、140℃2時間の水熱処理を行った。
処理後の沈殿物を濾過・水洗・乾燥処理し粉末サンプルを得た。
その100gサンプルをとって0.001mol/Lの硝酸銀水溶液300mlに懸濁し攪拌して、カリウムと銀とのイオン交換処理を25℃で1時間遮光下で行った。
イオン交換後のサンプルを濾過・水洗・乾燥(105℃×6時間)・粉砕した。
これらの処理過程を経てさらに200℃で2時間乾燥し、A19粒子を得た。
【0105】
実施例I−20
[Ag0.001K1.179]1.18Al3(SO4)2.26(NO3)0.04(C2O4)0.07(OH)5.48の製造
実施例I−19において、使用した硫酸カリウムを硝酸カリウムKNO3に変更したこと及びが実施例I−19と異なるのみで、その他の処理過程は実施例I−19に準じて行い、A20粒子を得た。
【0106】
実施例I−21
[Ag0.001K1.179]1.18Al3(SO4)2.26(NO3)0.04(C2O4)0.07(OH)5.48の製造
実施例I−19において、K2SO4の代りにKNO3に変更したこと及び水熱処理条件を160°×2時間に変更した実施例I−19と異なるのみで、その他の処理過程は実施例I−19に準じて行い、A21粒子を得た。
【0107】
実施例I−22
[Ag0.001K0.999]Al3(C2O4)0.1(SO4)1.9(OH)6 の製造
実施例I−19において、水熱処理の温度を140℃から170℃に変更したことが実施例I−19と異なるのみで、その他の処理過程は実施例I−19に準じて行い、A22粒子を得た。
【0108】
実施例I−23 [Ag0.1Na0.9]Al3(C6 H5O7)0.067(SO4)1.9(OH)6の製造
下記の原料を使用し、下記の方法によりA23粒子を得た。
使用原料 硫酸ナトリウム 2モル 284.06g
クエン酸(C6H8O7・H2O) 0.3モル 63g
1.04mol/Lの硫酸アルミニウム水溶液 2.0モル 1923ml
3.36mol/L NaOH水溶液 8.2モル 2440.5ml
合成方法 284.06gの硫酸ナトリウム(Na2SO4)を4.0Lのイオン交換水に溶解した。
63gのクエン酸は1.0Lのイオン交換水に溶解した。
攪拌下の該硫酸ナトリウム(Na2SO4)水溶液に該クエン酸水溶液及び上記硫酸アルミニウムAl2(SO4)3水溶液を添加し混合酸性水溶液を作った。
攪拌下の該混合酸性水溶液に上記の水酸化ナトリウム水溶液2440.5mlを 25分かけて添加しナトリウム型の銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子沈殿物スラリーを作った。
そのスラリーを更に10時間攪拌継続後、オートクレーブによって、170℃2時間の水熱処理を行った。水熱処理後の沈殿物を濾過・水洗・乾燥・粉砕処理し粉末サンプルを得た。
その他の処理過程は実施例I−1に準じて行い150℃で2時間乾燥し、A23粒子を得た。
【0109】
実施例I−24
[Ag0.1Na0.9]Al3(C6 H5O7)0.067(SO4)1.9(OH)6 の製造
実施例I−23において、水酸化ナトリウムの添加時間は40分に、及び水酸化ナトリウム添加後の攪拌時間は1時間に変更したことが実施例I−23と異なるのみで、その他の処理過程は実施例I−23に準じて行い、さらに150℃で2時間乾燥しA24粒子を得た。
【0110】
実施例I−25
[Ag0.1Na0.9]Al3(C4 H4O6)0.1(SO4)1.9(OH)6 の製造
下記の原料を使用し、下記の方法によりA25粒子を得た。
使用原料 硫酸ナトリウム 2モル 284.06g
酒石酸(C4H6O6) 0.3モル 45g
1.04mol/Lの硫酸アルミニウム水溶液 2.0モル 1923ml
3.36mol/L NaOH水溶液 8.2モル 2440.5ml
合成方法 284.06gの硫酸ナトリウム(Na2SO4)は4.0Lのイオン交換水に溶解した。
45gの酒石酸(C4H6O6) は1.0Lのイオン交換水に溶解した。
攪拌下の該硫酸カリウム(K2SO4)水溶液に該蓚酸水溶液及び上記硫酸アルミニウムAl2(SO4)3水溶液を添加し混合酸性水溶液を作った。
該混合酸性水溶液を良く攪拌して、そして、上記水酸化ナトリウム水溶液244.05mlを25分かけて該混合酸性水溶液に添加しナトリウム型の銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子沈殿物スラリーを作った。
そのスラリーを更に5時間攪拌後、オートクレーブによって、170℃2時間の水熱処理を行った。
水熱処理後の懸濁液を濾過・水洗・乾燥・粉砕処理した。
その他の処理過程は実施例I−1に準じて行い150℃で2時間乾燥し、A25粒子を作った。
【0111】
実施例I−26
[Ag0.1Na0.9]Al3(C4 H4O6)0.1(SO4)1.9(OH)6 の製造
実施例I−24において、水酸化ナトリウムの量は8モル(2381ml)に変更し、添加の時間も40分に変更したことが実施例I−25と異なるのみで、その他の処理過程は実施例I−25に準じて行い、A26粒子をった。
【0112】
実施例I−27
[Ag0.1(NH4)0.9] Al2.94Zn0.01 Ti0.01(C2O4)0.1(SO4)1.9(OH)6・0.5H2Oの製造
実施例I−1の混合酸性溶液(ただし該混合酸性溶液の該硫酸アルミニウム水溶液使用量は1885mlに減らした)に硫酸亜鉛2.5g,及び硫酸チタン{Ti2(SO4)3}2.5gを含む硫酸溶液を追加添加したこと及び銀イオン交換処理温度を80℃に処理時間を16時間に変更したことが実施例I−1と異なるだけでその他は実施例I−1に準じて行いA27粒子を得た。
【0113】
実施例I−28
[Ag0.1(NH4)0.9 ]Al2.25Zn0.5 Ti0.25(C2O4)0.1(SO4)1.9(OH)6・0.5H2O の製造
実施例I−1の混合酸性溶液(ただし該混合酸性溶液の該硫酸アルミニウム水溶液使用量は1442mlに減らした)に硫酸亜鉛125g,及び硫酸チタン{Ti2(SO4)3}63gを含む硫酸溶液を追加添加したこと及び銀イオン交換処理温度を80℃に処理時間を30時間に変更したことが実施例I−1と異なるだけでその他は実施例I−1に準じて行いA28粒子を得た。
【0114】
実施例I−29
[Ag0.1(NH4)0.9]Al3(C2O4)0.1(SO4)1.15(PO4)0.5(OH)6 の製造
リン酸アンモニウム74.5gを水500mlに溶解させた水溶液に、銀とイオン交換する直前(水熱処理後)のA−1粒子371gを添加して100℃で1時間攪拌しPO43−を該粒子に組み込んだ。その後の処理過程は実施例I−1に準じて行いさらに150℃で2時間乾燥し銀及び有機酸アニオン含有アルミニウム硫酸塩リン酸塩水酸化物A29粒子を得た。
尚この例において、リン酸ナトリウムの代わりに炭酸ナトリウム、硝酸ナトリウム、珪酸ナトリウム、ホウ酸ナトリウムに置き換えて使用したことだけが異なるように試験したがCO32−、NO3−、SiO42−、BO33−それぞれの無機酸イオンを含むA−29粒子同様の粒子性状を有する銀及び有機酸アニオン含有アルミニウム無機酸塩水酸化物粒子が得られた。
【0115】
実施例I−30
[Ag0.1Na0.9]Al3(SO4)1.9(C4H4O5)0.1(OH)6の製造
実施例I−25において、使用した有機酸を酒石酸からDL−林檎酸に変更したことが実施例I−25と異なるのみで、その他の処理過程は実施例I−25に準じて行いA30粒子を得た。
諸特性を表1に示す。このときの粒子形状は図5に示す球状であった。
【0116】
実施例I−31
[Ag0.1Na0.9]Al3(SO4)1.9[C6H2(OH)3COO]0.067(OH)6の製造
実施例I−23において、使用した有機酸をクエン酸から没食子酸[C6H4(OH)3COOH]に変更したことが実施例I−23と異なるのみで、その他の処理過程は実施例I−23に準じて行いA31粒子を得た。諸特性を表1に示す。
このときの粒子形状は図6に示す球状であった。
【0117】
比較例I−1
[Ag0.1(NH4)0.9]Al3(SO4)2(OH)6 の製造
実施例I−1において、蓚酸を使用しなかったことが実施例I−1と異なるのみで、その他の処理過程は実施例I−1に準じて行い、さらに150℃で2時間乾燥しV1粒子を得た。
【0118】
比較例I−2
NH4Al3(C2O4)0.1(SO4)1.9(OH)6の製造
実施例I−1において、銀溶液でイオン交換を行わなかったことが実施例I−1と異なるのみで、その他の処理過程は実施例I−1に準じて行い、さらに150℃で2時間乾燥しV2粒子を得た。
【0119】
比較例I−3
KAl3(C2O4)0.1(SO4)1.9(OH)6の製造
実施例I−19において、銀溶液でイオン交換を行わなかったことが実施例I−19と異なるのみで、その他の処理過程は実施例I−19に準じて行い、V3粒子を得た。
【0120】
【表1】

【0121】
【表2】
【0122】
円盤状粒子(碁石状粒子;B粒子)の製造;(実施例I−32〜34)
実施例I−32−1
[Ag0.1Na0.9 ]Al3(C2O4)0.1(SO4)1.9(OH)6 の製造
下記の原料を使用し、下記の方法によりB1粒子を得た。
使用原料 硫酸ナトリウム 2モル 284.06g
蓚酸(H2C2O4・2H2O) 0.3モル 38g
1.04mol/Lの硫酸アルミニウム水溶液 2.0モル 1923ml
3.36mol/L NaOH水溶液 8.2モル 2440.5ml
合成方法 284.06gの硫酸ナトリウム(Na2SO4)は4.0Lのイオン交換水に溶解した。
38gの蓚酸は1.0Lのイオン交換水に溶解した。
攪拌下の該硫酸ナトリウム(Na2SO4)水溶液に該蓚酸水溶液及び該硫酸アルミニウムAl2(SO4)3水溶液を添加し混合酸性水溶液を作った。
【0123】
該混合酸性水溶液を良く攪拌して、そして、該水酸化ナトリウム水溶液2440.5mlを20分かけて該混合酸性水溶液に添加しナトリウム型の銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子沈殿物スラリーを作った。
そのスラリーを更に5時間攪拌後、オートクレーブによって、170℃2時間の水熱処理を行った。処理後の沈殿物を濾過・水洗・乾燥処理した。そして、そのサンプル100gをとって0.025mol/Lの硫酸銀水溶液600mlに懸濁し攪拌してナトリウムと銀のイオン交換処理を25℃で5時間行った。交換後のサンプルにポリアクリルアミド系の高分子凝集剤(住友化学製 スミフロック FN−20)を0.02g添加し10分間攪拌後、濾過・水洗・乾燥(105℃×6時間)・粉砕した。これらの処理過程を経てさらに200℃で2時間乾燥しB1−1粒子を得た。
【0124】
実施例I−32−2
80℃のイオン交換水1,000mlに2.6gの硫酸亜鉛7水和物(和光純薬工業試薬1級)と2.2gの硫酸アンモニウム(和光純薬工業試薬1級)を入れ混液を作った。その混液の中にB1−1粒子95gを添加して6時間攪拌した後脱水・水洗・乾燥して亜鉛及びアンモニウムで処理されたB1−2粒子を作った。該粒子のZnの含有量は0.1%、NH4の含有率は0.4%BET法比表面積は60m2/gであったが、その他の粒子性状はB1−1粒子と同じであった。
次にA20粒子、C1粒子、D1粒子、E1粒子、F1粒子、G1粒子、H1粒子それぞれについても該処理同様の処理を行い処理物を得た。
処理物のZn含有量は0.1%、NH4の含有量は0.4%でありどれもほぼ同じであった。
処理物のBET法比表面積はどれも処理前の約6倍に増大していたが、平均2次粒子径、粒子径均一性粒子の形状、その他の粒子形状は処理前の粒子性状とほぼ同じであった。
処理物を樹脂に添加した時の樹脂組成物の白色性は処理前の物を添加した場合により一層改善されていた。
【0125】
実施例I−33
[Ag0.1Na0.9 ]Al3(C2O4)0.1(SO4)1.9(OH)6 の製造
実施例I−32において、使用した硫酸ナトリウムを0.5モル/L硫酸ナトリウムを含む反応母液4.0L使用に変更したこと、又、水酸化ナトリウムの添加時間40分に変更したこと、添加後の撹拌時間1時間に変更したことが実施例I−32と異なり、その他の処理過程は実施例I−32に準じて行い、B2粒子を得た。
【0126】
実施例I−34 [Ag0.001Na0.999] Al3(C2O4)0.1(SO4)1.9(OH)6 の製造
実施例I−32において、硫酸銀水溶液の濃度は0.00025モル/Lに変更したことが実施例I−32と異なるのみで、その他の処理過程は実施例I−32に準じて行い、B3粒子を得た。
【0127】
比較例I−4−(1)
NaAl3(C2O4)0.1(SO4)1.9(OH)6 の製造
実施例I−32において、銀イオンとのイオン交換処理を行わないことが実施例I−32と異なるのみで、その他の処理過程は実施例I−32に準じて行い、W1粒子を得た。
【0128】
比較例I−4−(2)
[Ag0.1Na0.9 ]Al3(SO4)2(OH)6 の製造
実施例I−32において、蓚酸を使用しなかったことが実施例I−32と異なるのみで、その他の処理過程は実施例I−32に準じて行い、W2粒子を得た。
【0129】
【表3】
【0130】
一対状(ハンバーガー状)粒子(C粒子)の製造;実施例I−35〜37
実施例I−35 [Ag0.1Na0.9] Al3(C2O4)0.1(SO4)1.9(OH)6 の製造
実施例I−32において、オートクレーブの処理温度は180℃に、処理時間10時間に変更したことが実施例I−32と異なるのみで、その他の処理過程は実施例I−32に準じて行い、C1粒子を得た。
【0131】
実施例 I−36
[Ag0.1Na0.9] Al3(C2O4)0.1(SO4)1.9(OH)6 の製造
実施例I−32において、オートクレーブの処理温度は180℃、処理時間10時間に変更したこと、使用した硫酸ナトリウムは0.5モル/L硫酸ナトリウムを含む反応母液4.0Lを使用したこと、又、水酸化ナトリウムの添加時間は40分に変更したことが実施例I−32と異なるのみで、その他の処理過程は実施例I−32に準じて行い、C2粒子を得た。
【0132】
実施例 I−37
[Ag0.001Na0.999] Al3(C2O4)0.1(SO4)1.9(OH)6 の製造
実施例I−32において、硫酸銀溶液の濃度は0.00025モル/Lに変更したこと、又、オートクレーブの処理温度180℃、処理時間10時間に変更したことが実施例I−32と異なるのみで、その他の処理過程は実施例I−32に準じて行い、C3粒子を得た。
【0133】
比較例I−5−(1) NaAl3(C2O4)0.1(SO4)1.9(OH)6
実施例I−35において、銀イオンとのイオン交換処理を行わないことが実施例I−35と異なるのみで、その他の処理過程は実施例I−35に準じて行い、X1粒子を得た。
【0134】
比較例I−5−(2)
[Ag0.1Na0.9] Al3(SO4)2(OH)6 の製造
実施例I−35において、蓚酸を使用しなかったことが実施例I−35と異なるのみで、その他の処理過程は実施例I−35に準じて行い、X2粒子を得た。
【0135】
【表4】
【0136】
米粒状粒子(D粒子)の製造;実施例I−38〜40
実施例I−38
[Ag0.1Na0.9]Al3(C2H4O6)0.1(SO4)1.9(OH)6の製造
実施例I−23において、クエン酸の代りに酒石酸に変更したことが実施例I−23と異なるのみで、その他の処理過程は実施例I−23に準じて行い、さらに150℃で2時間乾燥しD1粒子を得た。
【0137】
実施例I−39
[Ag0.1Na0.9]Al3(C2H4O6)0.1(SO4)1.9(OH)6の製造
実施例I−23において、クエン酸の代りに酒石酸に変更したこと、又、NaOH水溶液の添加時間35分に変更したことが実施例I−23と異なるのみで、その他の処理過程は実施例I−23に準じて行い、さらに150℃で2時間乾燥しD2粒子を得た。
【0138】
実施例I−40
[Ag0.001Na0.999]Al3(C2O4)0.1(SO4)1.9(OH)6 の製造
実施例I−23において、クエン酸の代りに酒石酸に変更したこと、又、銀イオンの濃度を0.00025モル/Lに変更したが実施例I−23と異なるのみで、その他の処理過程は実施例I−23に準じて行い、さらに150℃で2時間乾燥しD3粒子を得た。
【0139】
比較例I−6−(1)
NaAl3(C2H4O6)0.1(SO4)1.9(OH)6
実施例I−38において、銀イオンとの交換を行わないことに変更したことが実施例I−38と異なるのみで、その他の処理過程は実施例I−38に準じて行い、Y1粒子を得た。
【0140】
比較例I−6−(2)
[Ag0.1Na0.9]Al3(SO4)2(OH)6 の製造
実施例I−38において、酒石酸を使用しなかったことが実施例I−38と異なるのみで、その他の合成処理過程は実施例I−38に準じて行い、さらに200℃で2時間乾燥しY2粒子を得た。
【0141】
【表5】
【0142】
直方体状粒子(E粒子)の製造;実施例I−41〜44
実施例I−41
[Ag0.1(H3O)0.9 ]Al3(C2O4)0.1(SO4)1.9(OH)6 の製造
下記の原料を使用し、下記の方法によりE1粒子を得た。
使用原料 1.04mol/Lの硫酸アルミニウム 2.0モル 1923ml
水酸化アルミニウムAl(OH)3 2.0モル 156.02g
(水酸化アルミニウム;協和化学工業(株)製乾燥水酸化アルミニウムゲルS−100:無定形)
蓚酸(H2C2O4・2H2O) 0.25モル 31.52g
合成方法 上記硫酸アルミニウム水溶液中を撹拌しながら、上記蓚酸を添加し、さらに攪拌しながら上記水酸化アルミニウムAl(OH)3を添加し水素型の有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子沈殿物スラリーを作った。
該スラリーにイオン交換水を加え該スラリーが7.0Lになるように希釈して、更に室温で15時間攪拌後、オートクレーブによって、170℃5時間の水熱処理を行った。
処理後の溶液を濾過・水洗・乾燥・粉砕処理しサンプルを得た。
その他の処理過程は実施例I−1に準じて行いさらに150℃で2時間乾燥し、E1粒子を得た。
【0143】
実施例I−42
[Ag0.1(H3O)0.9 ]Al3(C2O4)0.1(SO4)1.9(OH)6 の製造
実施例I−41において、オートクレーブ処理前の撹拌時間は30分に変更したこと、又オートクレーブ処理温度150℃、処理時間2時間に変更したことが、実施例I−41と異なり、その他の処理過程は実施例I−41に準じて行い、E2粒子を得た。
【0144】
実施例I−43
[Ag0.001(H3O)0.999 ]Al3(C2O4)0.1(SO4)1.9(OH)6
実施例I−41において、交換用銀イオン濃度は0.00025モル/Lに変更したことが、実施例I−41と異なるのみで、その他の処理過程は実施例I−41に準じて行い、E3粒子を得た。
【0145】
実施例I−44
[Ag0.1(H3O)0.9 ]Al3(C2O4)0.1(SO4)1.9(OH)6 の製造
実施例I−41において、オートクレーブ処理前の攪拌時間を2時間に変更したことが、実施例I−41と異なるのみで、その他の処理過程は実施例I−41に準じて行い、E4粒子を得た。
【0146】
比較例I−7−(1)
(H3O)Al3(C2O4)0.1(SO4)1.9(OH)6 の製造
実施例I−41において、銀イオン交換処理を行わないことが、実施例I−41と異なるのみで、その他の処理過程は実施例I−41に準じて行い、Z1粒子を得た。
【0147】
比較例I−7−(2)
[Ag0.1(H3O)0.9 ]Al3(SO4)2(OH)6 の製造
実施例I−41において、蓚酸を使用しなかったことが実施例I−41と異なるのみで、その他の処理過程は実施例I−41に準じて行い、Z2粒子を得た。
【0148】
比較例I−9
比較例I−9のR1粒子は平均2次粒子径1.0μm、Dr=4.5、BET法比表面積4m2/g、銀含有量3%である銀担持リン酸ジルコニウムであるが、R1粒子の粒子性状は表―5に示す。
【0149】
【表6】
【0150】
六角板状粒子(F粒子)の製造;実施例I−45
実施例I−45
[Ag0.1Na0.9]Al3(SO4)1.9(C2O4)0.1 (OH)6の製造
硫酸アルミニウム2モルと硫酸ナトリウム2モルを6000mlの純水に溶解させ、その中に蓚酸31.52g(0.25モル)を入れた。攪拌中の前記混合液に水酸化ナトリウムを8.8モル(352g)含む水溶液1800mlを添加し、さらに室温で30分攪拌したのち、180℃で20時間の水熱処理を行った後、常温まで冷却し濾過水洗し、95℃で15時間乾燥処理して有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子を得た。
その後の処理過程は実施例I−1に準じて行いさらに150℃で2時間乾燥しF1粒子を得た。
諸特性を表6に示す。
このときの粒子形状は図11に示す六角板状であった。
【0151】
8面体状粒子(G粒子)の製造;実施例I−46
実施例I−46
[Ag0.1Na0.9]Al3(SO4)1.9[CH3CH(OH)COO]0.067(OH)6の製造
実施例I−23において、使用した有機酸をクエン酸からL−乳酸[CH3CH(OH)COOH]に変更したことが実施例I−23と異なるのみで、その他の処理過程は実施例I−23に準じて行いG1粒子を得た。諸特性を表6に示す。
このときの粒子形状は図12に示す八面体状であった。
【0152】
円柱状粒子(酒樽状粒子;H粒子)の製造;実施例I−47
実施例I−47
[Ag0.1Na0.9]Al3(SO4)1.9[HOCH2CH(OH)COO]0.067(OH)6の製造
実施例I−23において、使用した有機酸をクエン酸からDL−グリセリン酸[HOCH2CH(OH)COOH]に変更したことが実施例I−23と異なるのみで、その他の処理過程は実施例I−23に準じて行いH1粒子を得た。諸特性を表6に示す。
このときの粒子形状は図13に示す円柱状(酒樽状粒子)であった。
【0153】
【表7】
【0154】
尚前記の実施例IのA1〜A31、B1〜B3、C1〜C3,D1〜D3,E1〜E4、F1,G1,H1の各粒子は高純度の原料(不純物含有量Pb,Cd、As,Baは全て0.1ppm以下、Feは10ppm以下、Mn,Cu,Cr,Niは1ppm以下に精製されたもの)を使用し、且つ設備面では耐腐食性の前記の材質で構成された装置を使用し合成した。
そのため不純物含有量は天然の明礬石とは異なりPb,Cd、As,Baは全て0.1ppm以下、Feは10ppm以下、Mn,Cu,Cr,Niは1ppm以下,Clは100ppm以下であった。
尚不純物含有量は原子吸光法、又はICP−AES法(Inductively Coupled Plasma −Atomic Emission Spectroscopy)、又は蛍光X線法で測定した。
次に本発明の抗菌剤を樹脂に配合した場合の抗菌性樹脂組成物及び抗菌性樹脂製品の製造方法を実施例に基づいて具体的に説明する。尚配合剤の配合量は樹脂100重量部に対するものである。
蛍光増白剤は2,5−チオフェンジイル(5−tert−ブチル−1,3−ベンゾヘキサゾール)はそれぞれの表に示してある量を添加した。
【0155】
ポリプロピレン成形品;
実施例II−1〜32及び比較例II−1〜4
実施例II−1は透明射出成形用グレードポリプロピレン100重量部、表−1記載のA1粒子0.06重量部、蛍光増白剤は2,5−チオフェンジイル(5−tert−ブチル−1,3−ベンゾヘキサゾール)表−1に示す添加量}を予め混合しておき、それを2軸混練押出機を使用し230℃で混練して混和ペレットを得それを230℃で2mmに射出成形して前記各試験用のテストピースを作成し抗菌性(成形直後及び耐水道水接触環境後抗菌力維持特性)、透明性、白色性を前記の樹脂製品の測定方法(7)〜(10)に従って測定した。その結果を表−7の縦2重線より左側に示す。
混練押出加工時フィルター通過性試験(押出機圧力)の試験は上記(11)の方法で行った。
その結果を表−7の縦2重線より右側に示す。
実施例II−2〜32の試験においては実施例II−1で使用した使用抗菌剤粒子及び配合量を表−7に示すように一部変更したが、それ以外は実施例II−1同様にテストピースを作成し各試験を実施した。
その結果を表−7に示す。
【0156】
比較例II−1は有機酸が含まれていない銀含有アルミニウム硫酸塩水酸化物粒子でしかもDrの幅(粒度分布幅)が大きいV1,W2,X2,Y2,Z2粒子であるところ又は銀担持リン酸ジルコニウムであるR1粒子であるところが、比較例II−2の試験は抗菌剤無添加であるところが、比較例II−3は銀を含まないアルミニウム硫酸塩水酸化物粒子V2であるところが、比較例II−4も銀を含まないアルミニウム硫酸塩水酸化物粒子V3粒子であるところが、実施例II−1と異なり、抗菌剤配合量については一部の抗菌剤が表−1に示すように異なるのみで、その他の条件は実施例II−1同様にテストピースを作成し同じ測定を行った。
その結果を表−7に示す。
【0157】
【表8】
【0158】
【表9】
【0159】
【表10】
【0160】
実施例においては、成形直後の抗菌性、水道水接触環境後抗菌力維持特性、透明性、色、混練押出加工時フィルター通過性において優れた特性を有することが確認されたが、一方比較例においてはそれらの特性の中で少なくとも1つ以上の項目に問題があった。
又本実験においては、銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子抗菌剤の平均二次粒子径が微粒子である程抗菌性と透明性が高くなる、BET法比表面積については高いものほど程抗菌性が高くなる、Drの幅(粒度分布幅)は小さく粒子径が均一で有機酸を一定範囲で含んでいるものは抗菌性が高くなる傾向が認められた。
一方比較例について見てみると、比較例II−1において有機酸が含まれていない銀含有アルミニウム硫酸塩水酸化物粒子でしかもDrの幅(粒度分布幅)が大きいV1,W2,X2,Y2,Z2粒子を使用した場合、前記の有機酸が含まれていないことの特徴が現われ抗菌性が不十分で、混練押出加工時フィルター通過性は機械の運転が困難なまで運転直後から悪くなり、また透明性も若干低下していた。
【0161】
比較例II−1においてはリン酸ジルコニウム系のR1粒子を配合したものは、実施例に比べると、水道水接触環境後抗菌力維持特性、及び透明性が劣っていることが認められた。
比較例II−2では抗菌剤を配合しなかったため透明性と色には問題はなかったが、抗菌効果は全く認めらず本発明の目的を達成していないことはいうまでもなかった。
比較例II−4では有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子を300重量部も配合したにも係らず、銀を含有した有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子ではなかったため抗菌効果は全く認められなかった。
つまり、比較例をまとめるとアルミニウム硫酸塩水酸化物粒子を使用しても、比較例II−3及びII−4から解るようにアルミニウム硫酸塩水酸化物粒子であっても銀を含有していないものでは成形品に抗菌性を付与できないことが判明した。
【0162】
ポリスチレン、アクリル樹透明性樹脂成形品
実施例II−33、34及び比較例II−5、6、7
実施例II−33,34は実施例II−1において抗菌化対象樹脂をポリプロピレンから、ポリスチレン、アクリル樹脂等の透明性樹脂に変更し、抗菌剤及び蛍光増白剤配合量を表−8に示すように変更し、混練時、成形時の温度を210℃に変更しただけでそれ以外は実施例II−1に準じておこなった。尚配合剤の添加量は表−8に示す通りである。
比較例II−5は実施例II−33において抗菌剤を添加しなかったこと、比較例II−6は抗菌剤をV2粒子に,比較例II−7は抗菌剤をY1粒子に変更したのみでそれ以外は実施例II−33に準じておこなった。抗菌性、透明性、色の結果を表−8に示す。
下記表−8に示すとおり、実施例の成形品については透明性が損なわれることなく、色も無色(白)であり且つ抗菌性(成形直後及び水道水接触環境後抗菌力維持特性)に関しても優れていることが確認された。
一方比較例においてはアルミニウム硫酸塩水酸化物粒子であっても銀を含有していないため抗菌性が全く発現しなかった。
【0163】
【表11】
【0164】
ポリカーボネート、ポリエチレンテレフタレート、ナイロン6・6、ポリアセタール樹脂成形品
実施例II−35、36及び比較例II−8、9
実施例II−35、36は実施例II−1において抗菌化対象樹脂をポリプロピレンから、ポリカーボネート、ポリエチレンテレフタレート、ナイロン6・6、ポリアセタール樹脂等透明性を有し且つ加工時に水分が少ないことが要求される樹脂に変更し、抗菌剤及び抗菌剤の配合量及び蛍光増白剤の配合量を表−9に示すように変更し、さらに混練時、成形時の温度をそれぞれの樹脂の常識的加工温度(PC,PET,ナイロン6・6では290℃、ポリアセタールでは190℃)に変更し、その他は実施例II−1に準じて試験を行った。
ただし、実施例II−35、36では抗菌剤は乾燥処理したA2,B1,C1,D1,E1,F1,G1,H1粒子又は焼成処理したA17,A18粒子を0.1重量部使用し、比較例8は抗菌剤無添加であり、比較例9はV2粒子の400℃又は500℃焼成品を0.1重量部使用した。抗菌性、透明性、色、押出機圧力の結果を表−9に示す。
無論実施例II−35、36で得られた成形品にはシルバーストリークは発生しなかった。
【0165】
下記表−9に示すとおり、実施例においては成形品の透明性がほとんど損なわれることなく、色も無色(白)であり且つ抗菌性(成形直後及び水道水接触環境後抗菌力維持特性)に関しても優れていることが確認された。
一方比較例においては抗菌性が全く発現しなかった。
【0166】
【表12】
【0167】
フィルム
実施例II−37及び比較例II−10
LDPE樹脂90重量%と抗菌剤A2,B1,C1,D1,E1,F1,G1,H1粒子それぞれを10重量%の合計100重量%を予め加圧ニーダーを用い120℃で15分間混練したものを押出造粒機でホットカットし120℃で直径約3mmのマスターバッチ・ペレットを得た。
【0168】
そのマスターバッチ・ペレットとマスターバッチにする前のLDPE樹脂とを混合し表−10の組成になるようにしたものをTダイ法及びインフレーション法によりそれぞれ厚さ50μmのフィルムを得た。
尚比較例II−10ではマスターバッチにする前のLDPE樹脂のみで同様にフィルムを得た。
そのフィルムについて抗菌性(成形直後及び水道水接触環境後抗菌力維持特性)、透明性、白色性を前記の方法で測定した。その結果を表−10に示す。比較例II−10では抗菌剤無添加のものについてもフィルムを作成し、実施例II−37と同じ測定を行った。その結果を表−10に示す。
下記表−10に示すとおり、実施例のフィルムについては透明性が損なわれることなく、色も無色(白)であり且つ抗菌性(成形直後及び水道接触用環境後抗菌力維持特性)に関しても優れていることが確認された。
一方比較例においては抗菌性が全く発現しなかった。
【0169】
実施例II−38
ポリプロピレン、LDPE,HDPE,アイオノマー樹脂、ナイロン6/66共重合樹脂、PET樹脂,AS樹脂のそれぞれの樹脂100重量部につき、2軸混練押出機を用いA2粒子0.1重量部の混和ペレットを得た。
そのペレットをTダイ法によりそれぞれ厚さ50μmのフィルムを得た。
そのフィルムについて抗菌性(成形直後及び水道水接触環境下抗菌力維持特性)、透明性、白色性を前記の方法で測定した。その結果を表−10に示す。
実施例で得られた各フィルムについては透明性が損なわれることなく、色も無色(白)であり且つ抗菌性(成形直後及び水道水接触環境後抗菌力維持特性)に関しても優れていることが確認された。
一方比較例においては抗菌性が全く発現しなかった。
【0170】
【表13】
【0171】
PVC成形品
実施例II−39及び比較例II−11
実施例II−39ではポリ塩化ビニル樹脂100重量部、表−11に示す抗菌剤粒子0.1重量部、オクチル錫メルカプト1.2重量部、グリセリンリシノレート0.8重量部、モンタン酸エステル0.4重量部からなる組成物をオープンロールを用い180℃で3分間混練したものを圧縮成形機により180℃で厚さ2mmの成形板を得た。
その成形板について抗菌性(成形直後及び水道水接触環境後抗菌力維持特性)を前記の方法で測定した。その結果を表−11に示す。
比較例II−11では同様に抗菌剤無添加のものについて厚さ2mmの成形板にし、同じ測定を行った。その結果を表−11に示す。
下記表−11に示すとおり、実施例の成形板については抗菌性(成形直後及び水道水接触環境後抗菌力維持特性)及び透明性が優れていることが確認された。一方比較例においては抗菌性が全く発現しなかった。
【0172】
【表14】
【0173】
熱硬化性樹脂成形品
実施例II−40及び比較例II−12、13
実施例II−40では不飽和ポリエステル樹脂100重量部、表−12に示す抗菌剤粒子1重量部、硬化剤(チバスペシャルケミカル社製HY951)3重量部、ステアリン酸1重量部、酸化防止剤(チバスペシャルケミカル社製イルガノックス1010)0.5重量部、蛍光増白剤0.001重量部、平均二次粒子径30μm、BET法比表面積1m2/gの人造代理石用途水酸化アルミニュウム150重量部をニーダーで混練して、それを90℃で15分間硬化させ厚さ2mmの板を得た。
【0174】
その成形板について抗菌性(成形直後及び水道水接触環境後抗菌力維持特性)を前記の方法で測定した。その結果を表−12に示す。
比較例II−12、13では実施例II−40同様に抗菌剤無添加のも及びR1粒子についてもそれぞれ個別に厚さ2mmの成形板にし、同じ測定を行った。その結果を表−12に示す。
実施例の成形板について抗菌性(成形直後及び水道水接触環境後抗菌力維持特性)が優れていることが確認された。一方比較例II−12においては抗菌性が全く発現しなかった。
比較例II−13においては成形直後の抗菌性は認められるものの、水道水接触環境下抗菌力維持特性が全くないことが判明した。
【0175】
実施例II−41
実施例II−41は実施例II−40において抗菌剤使用粒子がE2粒子に変更されたこと、及び配合量が300重量部に変更されたこと、ステアリン酸が3重量部に変更された以外は実施例II−40同様に成形板を作成し、同じ測定を行った。その結果を表−12に示す。
成形板については抗菌性(成形直後及び水道水接触環境後抗菌力維持特性)が優れていることが確認された。
【0176】
実施例II−42
実施例II−40において抗菌化対象樹脂を不飽和ポリエステル樹脂からフェノール樹脂、メラミン樹脂、エポキシ樹脂へ変更、抗菌剤をE4粒子に変更し、その他は実施例II−40に準じて成形板を作成し、同じ測定を行った。その結果を表−12に示す。得られた該成形板については抗菌性(成形直後及び水道水接触環境後抗菌力維持特性)が優れていることが確認された。
【0177】
実施例II−40〜42及び比較例II−12、13をまとめると以下のことが理解できる。
本発明の熱硬化性樹脂抗菌性製品は手洗い、風呂等水場で使用される人造大理石用途においても抗菌性(水道水接触環境後抗菌力維持特性)を長時間失うことがなく好適に使用できることを示している。
一方従来技術の銀担持リン酸ジルコニュムを配合した比較例II−13では成形直後の抗菌性は認められるものの、水道水接触環境下抗菌力維持特性が全くなりそのような用途では意味がないことを示している。
【0178】
【表15】
【0179】
ゴム成形品
実施例II−43及び比較例II−14
EPDM(エチレン/プロピレン比=50/50)100重量部、表−13に示す抗菌剤粒子0.5重量部、ディクミルパーオキサイド3重量部、ポリ(2,2,4−トリメチル−1,2ディヒドロキノリン)0.5重量部、シランカップリング剤(日本ユニカ製A−172)1重量部、ステアリン酸0.5重量部、イオウ1重量部、蛍光増白剤0.001重量部、ルチル型酸化チタン5重量部からなる組成物をオープンロールを用い50℃で混練し、それを1日後に160℃で30分加硫し厚さ2mmの成形板を得た。
【0180】
その成形板について抗菌性(成形直後及び水道水接触環境後抗菌力維持特性)を前記の方法で測定した。その結果を表−13に示す。
比較例II−14では同様に抗菌剤無添加のものについて個別に厚さ2mmの成形板にし、同じ測定を行った。その結果を表−13に示す。
実施例の成形板については抗菌性(成形直後及び水道水接触環境後抗菌力維持特性)が優れていることが確認された。一方比較例II−14においては抗菌性が全く発現しなかった。
【0181】
実施例II−44
実施例II−43において抗菌剤使用粒子がA14粒子に変更されたこと、及び配合量が200重量部に変更されたこと、ステアリン酸が2重量部に変更された以外は実施例II−43同様に成形板を作成し、同じ測定を行った。その結果を表−13に示す。
成形板については抗菌性(成形直後及び水道水接触環境後抗菌力維持特性)が優れていることが確認された。
【0182】
【表16】
【0183】
繊維
実施例II−45、比較例15
繊維用ポリプロピレン100重量部、抗菌剤A2粒子を2重量部を2軸混練押出機を用いて予め混練しておき、それを300メッシュのスクリーンを付設した押出機を用い溶融法により100デニルに紡糸した繊維を得た。その繊維について抗菌性(成形直後及び水道水接触環境後抗菌力維持特性)を前記の方法で測定した。その結果を表−14に示す。
比較例II−15では同様に抗菌剤無添加のものについて100デニルの繊維にし、同じ測定を行った。その結果を表−14に示す。
実施例の繊維については抗菌性(紡糸直後及び水道水接触環境後抗菌力維持特性)が優れていることが確認された。
このことは例えば手袋、ソックス等の繊維製品において数十回、数百回洗濯しても抗菌性を失わないあるいは失い難いことを意味している。
尚前記の押出溶融法において実施例においては該スクリーンに粗大粒子が詰まって紡糸作業に支障をきたすことはなかった。
【0184】
【表17】
【0185】
不織布
実施例II−46、比較例II−16
実施例II−45及び比較例II−15で得られた各ポリプロピレン繊維を抄造ウエブ法及びランダムウエブ法により0.06g/cm3の密度に不織布を作成し前記の抗菌性の試験を行ったが実施例においては優れた抗菌性効果(成形直後及び水道水接触環境後抗菌力維持特性)が得られた。
一方比較例においては抗菌性が全く発現しなかった。結果を表−15に示す。
【0186】
【表18】
【0187】
塗料
実施例II−47、比較例II−17
冷却設備を有する設備の中に、メチルメタクリレート60重量部、2−エチルヘキシルアクリレート40重量部の合計100重量部に対し、トリエチレングリコールジメタクリレート3重量部、ジアルキルフタレート10重量部、ハイドロキノン0.003重量部、融点46℃のパラフィンワックス0.5重量部、融点54℃のパラフィンワックス0.5重量部、N,N−ジ8ヒドロキシプロピル9−Pトルイジン0.7重量部を投入し攪拌しながらメチルメタクリレートとn−ブチルメタクリレートの共重合体(Tg=66℃、Mw=40,000)25重量部を除々に加え60℃で2時間攪拌し30℃まで冷却した。
【0188】
このものに実施例I−2のA2粒子1重量部、着色剤三菱レイヨン製トナーP−400を7重量部、骨材三菱レイヨン製KM17を300重量部、重合開始剤ジアシルパーオキサイド2重量部からなる塗料を20℃で1時間放置し塗幕を作り前記(7)の(c)の方法により抗菌のテストを実施した。その結果を表−16に示す。
比較例II−17では抗菌剤無添加のものについて実施例II−47同様に塗布をし、同じ測定を行った。その結果を表−16に示す。
実施例の塗料については抗菌性((成形直後及び水道水接触環境後抗菌力維持特性)が優れていることが確認された。一方比較例においては抗菌性が全く発現しなかった。
【0189】
【表19】
【0190】
コーキング材
実施例II−48及び比較例II−18
シラノールを末端基とする25℃の粘度が50、000センチポイズのポリジメチルシロキサン100重量部に対し1重量部のγアミノプロピルビス(メチルエチルケトキシアミノ)メトキシシランを真空下で10分間混合し、ついでその混合物に5重量部のメチルトリス(メチルエチルケトキシアミノ)シランを加え真空下15分間混合し、BET法比表面積200m2/gのヒュームドシリカ5重量部、ジブチルスズラウレート0.1重量部、抗菌剤A2粒子1重量部を加え真空下で10分間混合した。
この混合された組成物をポリエチレンシートの上に垂らして厚さ2mmにし抗菌性テスト用のテストピースを得た。それを前記の抗菌性試験法(7)の(e)で塗布し抗菌のテストを実施した。その結果を表−17に示す。比較例では同様に抗菌剤無添加のものについてコーキング材にし、同じ測定を行った。その結果を表−17に示す。
実施例のコーキング材については抗菌性(成形直後及び水道水接触環境後抗菌力維持特性)が優れていることが確認された。一方比較例においては抗菌性が全く発現しなかった。
【0191】
【表20】
【0192】
尚前記の実施例II−1〜II−48の各成形品、フィルム等を一旦900℃で焼成した灰分を硫酸及び硝酸に溶解し溶液とし、それから各実施例の成形品、フィルム等に含まれる重金属を原子吸光法、又はICP−AE法(Inductively Coupled Plasma −Atomic Emission Spectroscopy)、又は蛍光X線法で測定したが、実施例II−1〜II−48の各樹脂成形品、フィルム、繊維等に含まれるPb、Cd、As,Baは全て0.1ppm以下で、且つFeは5ppm以下、Mn,Cu,Cr,Niはそれぞれ1ppm以下であった。
従って本発明の抗菌性樹脂組成物及びそれから形成された製品は、安全性が高いだけだけでなく、さらには耐熱劣化性にも優れた材料であることが判明した。
【0193】
抗カビ剤
実施例II−49、比較例II−19およびII−20
培地が感性MHB培地から日水製薬製ポテトデキストロース寒天培地に変更したことだけが異なる日本化学療法学会標準法(2003年改訂版)において最小発育阻止濃度を本発明の抗カビ剤粒子、及び比較例の粒子について抗カビ性能を測定し最小発育阻止濃度としてppmで示した。
この数値が小さいほど抗カビ性能が高いことを示している。
測定の結果を下記表−18に示す。
試供カビは ※1:Caldosporium Caldosporides NBRC 6348 (黒カワカビ)、
※2:Colletotricum coccodes NBRC 5256 (ナス黒点根腐病原菌)、
※3:Ustilaginoidia virens NBRC 9175 (稲こうじ病原菌)を使用した。
【0194】
【表21】
【0195】
本発明抗カビ剤は比較例の抗カビ剤よりはるかに優れた抗カビ性能を示すことが判明した。
【0196】
化粧料
実施例II−50、比較例II−21
下記表−19に示す実施例と比較例の配合成分を用いた油中水型のクリーム状の乳化化粧料を調整した。表中1〜5は油性材料、6、7は界面活性剤、10抗菌剤とを混合したものをAとし、一方、表中の物質8精製水と,9の保湿剤を混合したものを混合物Bとし、各々の混合物A,Bを70℃に加熱した後、混合物A中に混合物Bを攪拌しながら流し入れ、乳化させその後冷却した。
抗菌性テストは乳化30日後に大腸菌E.coli NBRC 3972 を使用して前記(7)の(c)に準じて実施した。さらに実施例と比較例のクリームを日頃脇に悪臭の有る人の脇下に1g塗って8時間後に悪臭のあるかないかを10人に嗅いでもらった。
測定の結果を下記表−19に示す。
【0197】
【表22】
【0198】
実験の結果実施例では抗菌性に優れ且つ悪臭を抑える効果が高いことが判明した。
また本発明においては化粧料を顔に塗った時の感触としてのざらつき感がいずれもなかったが、特に円盤状粒子(A1粒子)においては非常になめらかな塗り心地であった。
一方、比較例においては実施例の抗菌剤がR1粒子に置き換えられたこと、又は抗菌剤が全く添加されなかったことが異なるのみであるが、抗菌性は無く且つ悪臭防止効果も無かったことが判明した。
【0199】
抗菌消臭スプレー剤
実施例II−51、比較例II−21およびII−22
70℃に加温した容器の中に2.3重量部のジプロピレングリコールを入れ、さらに2.5重量部のラウリン酸と1重量部のミリスチン酸、さらに3.2重量部のトリエタノールアミンを加えて溶液を作成した。
次にこの溶液を70℃に加温した90重量部のイオン交換水に徐々に注加して乳化させ泡基材溶液を調製し、それを25℃まで冷却した。
該泡基材溶液に本発明の抗菌剤粒子1重量部添加して攪拌、混合し抗菌消臭泡沫状エアゾール組成物を調製した。
この組成物180gと噴射剤(LPG 0.34MPa)20gをブリキ缶のスプレー式容器に充填して抗菌消臭泡沫状エアゾールスプレーを作成した。
一方、日頃脇に悪臭の有る人に充分運動してもらった後で15cm×15cmの綿製のハンカチが0.5g、及び5g重くなるように汗を採取した。該ハンカチの表と裏に該抗菌消臭泡沫状エアゾールスプレーをそれぞれ1gづつ噴霧し、恒温槽に30℃で10日間静置した後で10人に悪臭を嗅いでもらった。測定の結果を下記表−20に示す。
【0200】
【表23】
【0201】
本発明抗菌剤は比較例の抗菌剤よりはるかに優れた悪臭防止性能を示すことが判明した。
【0202】
抗菌紙
実施例II−52、比較例II−23およびII−24
さらしケミパルプ82%の中に、本発明の抗菌剤1%、デンプン(乾燥紙力増強剤)5%、尿素−ホルムアルデヒド樹脂(湿潤紙力増強剤)5%、ニ酸化チタン(無機填料)2%、ポリアミド系樹脂を主成分とするインキ(接着性バインダー)5%を混合し、抄紙機を用いて厚さ0.1mmの紙を抄いた。この紙を5cm×5cmに切り取り前記の抗菌性試験方法(7)の(a)同様のテストを実施した。測定の結果を下記表−21に示す。
【0203】
【表24】
【0204】
本発明抗菌剤は比較例の抗菌剤よりはるかに優れた抗菌性能を示すことが判明した。
【0205】
農薬(抗カビ剤)
実施例II−53、比較例II−25およびII−26
実施例では本発明の粒子10重量部、シランカップリング剤表面改質軽質炭酸カルシウム(無機微粉体)30重量部、ポリオキシエチレンアルキルアリルエーテル(界面活性剤)5重量部、エチレングリコール(界面活性剤)10重量部、キサンタンガム(乳化安定剤)0.2重量部、水44.8重量部をホモミキサーで均一に混合した後、ボールミルで均一に湿式粉砕して水性懸濁状農薬組成物をえた。これを水で1/100に希釈して市販のプラスチック製スプレー装置に入れた。
比較例は実施例の粒子が下記の粒子、又はボルドー撒粉に置き換えられたことが異なるだけでその他の実験方法は実施例の要領に準じた。
【0206】
一方、それぞれ約20cmに成長したナス、及び稲を用意した。
ナスには1×106個/mlに調製したColletotricum coccodes NBRC 5256 (ナス黒点根腐病原菌)懸濁液1gづつを葉、茎、根元に噴霧し、その1日後に前記の1/100に希釈された該水性懸濁状農薬組成物1gを同様に噴霧し、さらにその30日後にナス黒点根腐病の発生の程度を観察した。ただしナスの試験は直径33cm、深さ30cmの鉢に土が27cmの高さになるように入れて実施した。
稲には1×106個/mlに調製したUstilaginoidia virens NBRC 9175 (稲こうじ病原菌)懸濁液を1gづつを葉、茎、根元に噴霧し、その1日後に前記の1/100に希釈された該水性懸濁状農薬組成物1gを同様に噴霧し、さらにその30日後に稲こうじ病の発生の程度を観察した。ただし稲の試験は直径33cm、深さ30cmの鉢に土が27cmの高さになるように入れて土壌の水が表面からかろうじて切れるが土壌中には十分水が存在する条件下で実施した。測定の結果を下記表−22に示す。
【0207】
【表25】
【0208】
本発明農薬組成物は比較例の農薬組成物よりはるかに優れた農薬性能を示すことが判明した。
【図面の簡単な説明】
【0209】
【図1】実施例I−1における球状粒子(粒子名:A1)のSEM写真である。
【図2】実施例I−20における球状粒子(粒子名:A20)のSEM写真である。
【図3】実施例I−21における球状粒子(粒子名:A21)のSEM写真である。
【図4】実施例I−22における球状粒子(粒子名:A22)のSEM写真である。
【図5】実施例I−30における球状粒子(粒子名:A30)のSEM写真である。
【図6】実施例I−31における球状粒子(粒子名:A31)のSEM写真である。
【図7】実施例I−32−1における円盤状粒子(粒子名:B1−1)のSEM写真である。
【図8】実施例I−35における一対状粒子(粒子名:C1)のSEM写真である。
【図9】実施例I−38における米粒状粒子(粒子名:D1)のSEM写真である。
【図10】実施例I−41における直方体状粒子(粒子名:E1)のSEM写真である。
【図11】実施例I−45における六角板状粒子(粒子名:F1)のSEM写真である。
【図12】実施例I−46における八面体状粒子(粒子名:G1)のSEM写真である。
【図13】実施例I−47における円柱状粒子(粒子名:H1)のSEM写真である。
【図14】比較例I−1における凝集状粒子(粒子名:V1)のSEM写真である。
【技術分野】
【0001】
本発明は銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子よりなる抗菌剤に関する。さらに詳しくは特定粒子性状(粒子形状、粒子径均一性、平均2次粒子径、比表面積等)を有する銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子よりなる抗菌剤に関する。また該抗菌剤の製造方法にも関する。
さらには該抗菌剤を樹脂に混練混合する時の性能として混練押出加工時フィルター通過性、分散性に優れた抗菌性樹脂組成物(マスターバッチを含む)にも関する。また該樹脂組成物から形成された、分散性、透明性、白色性、抗菌性(水道水接触環境後抗菌力維持特性も含む)に優れた抗菌性樹脂成形品、抗菌性フィルム、抗菌性繊維、抗菌性塗料、抗菌性不織布、抗菌性コーキング材のような抗菌性樹脂製品にも関する。さらに抗カビ剤、抗菌消臭剤、抗菌紙、農薬及び化粧料にも関する。
【背景技術】
【0002】
一般的に細菌は高温多湿な環境で繁殖が進み、安全衛生上、住環境上重大な問題を引き起こす場合がある。これらの問題を解決するため従来有機系抗菌剤や無機系抗菌剤を抗菌性付与対象物となる樹脂その他の物に配合して細菌からの被害を防止する抗菌性樹脂組成物等の技術が提案されているが、その中では無機系抗菌剤が比較的安全なため最近無機抗菌剤の需要は伸びてきている。
無機系抗菌剤においては、銀が比較的高い抗菌活性と比較的高い安全性を有することから、銀を無機化合物に担持又はイオン交換させた抗菌剤を使用した抗菌性樹脂組成物が多数提案されている。
【0003】
例えば、特許文献1ではリン酸ジルコニウムに銀を担持した抗菌性樹脂組成物の技術が開示されている。しかしながら、該抗菌剤を樹脂に配合した抗菌性樹脂組成物においては白色性は若干改善されているものの抗菌性、分散性、透明性、水道水接触環境後抗菌力維持特性の全ての点において完全ではなく解決すべき問題が残っていた。
なかでも、この従来技術では抗菌性樹脂組成物をしばらく水道水に接触させて使用すると該樹脂組成物の抗菌活性は全くなくなるか、又は著しく低下してしまい長時間の使用に耐えなくなり、細菌からの被害を防止できなくなるという問題があり、この問題を解決することは重要な課題であった。
この問題をさらに具体例で説明すると、台所、風呂場やトイレの周りでは水道水が常に使用され、衣類等は水道水で何回も洗濯して使用される。従って、このような場所であるいは条件で使用する抗菌性樹脂製品は本来水道水に長時間接触しても抗菌性を発揮して細菌からの被害を防止できるものでなければならない。ところが、前記従来の技術による抗菌性樹脂製品は樹脂製品製造直後には一定程度の抗菌性を発現するものの、しばらく水道水と接触して使用すると、該樹脂製品の抗菌活性は全くなくなるか又は著しく低下してしまい、さらに長時間の経過の後は細菌からの被害を全く防止できなくなるという問題があった。
【0004】
特許文献2では、MAl3(SO4)2(OH)6なる式で表されたBET法比表面積が30m2/g以下で、該式中のMをアルカリ金属又はアンモニウム基とした粒子形状が紡錘状乃至球状を呈するアルカリアルミニウム硫酸塩水酸化物が開示され、この文献にはコールターカウンター法で測定された体積基準累積粒子径の25%値の粒子径D25(大粒子側)を75%値の粒子径D75(小粒子側)で除した算式で表される粒子径分布のシャープ度Rs=D25/D75が1.45乃至1.61のものが実施例で具体的に示され、さらにその製造方法及び樹脂配合例が紹介されている。
前記特許文献の、段落番号0035では該化合物のMがAg,Zn、Cu等の抗菌性発現効果のある元素を含むことが可能であり抗菌性粒子を得ることができるとの極一般的な説明はある。しかしこの文献には抗菌剤及び抗菌性樹脂製品及び抗カビ剤、抗菌消臭剤、抗菌紙、農薬、化粧料に関する具体的な説明もまた実施例もない。
【特許文献1】特開平6−212019号公報
【特許文献2】特開2000−7326号公報
【発明の開示】
【発明が解決しようとする課題】
【0005】
かくして本発明の目的は、前記従来技術の無機系抗菌剤の種々の問題点を克服し、抗菌剤の性能として樹脂等の抗菌化対象物に配合使用された時、分散性、透明性、白色性に優れ、抗菌性特に水道水接触環境後の抗菌力維持特性にも優れた銀と有機酸アニオンがアルカリアルミニウム硫酸塩水酸化物粒子に特定範囲で含有したことからなる銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子よりなる抗菌剤を提供すること、さらには該抗菌剤において該諸特性が特に優れるように設計された特定粒子性状(粒子形状、粒子径均一性、平均2次粒子径、比表面積等)を有する銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子よりなる抗菌剤を提供すること、さらには該抗菌剤の製造方法を提供することである。
【0006】
さらに他の目的は、その抗菌剤を樹脂混練押出機を用いて混練混合する時に樹脂混練押出加工時フィルター通過性、分散性に優れた性質を有する抗菌性樹脂組成物(マスターバッチを含む)を提供すること、さらには該樹脂組成物から成形されて分散性、透明性、白色性、抗菌性特に水道水接触環境後抗菌力維持特性に優れた抗菌性樹脂成形品、抗菌性フィルム、抗菌性繊維、抗菌性不織布、抗菌性塗料、抗菌性コーキング材等の抗菌性樹脂製品を提供することである。
従来技術では抗菌性樹脂製品を水道水接触環境下でしばらく使用すると抗菌活性が全くなくなるか、又は短時間で著しく低下するというような問題があったことに鑑み、本発明はその問題を解決し、台所、風呂場やトイレ等で水道水を常に使用する場所、あるいは衣類のような製品において水道水で何回も洗濯して使用する条件下でも抗菌性を長時間維持できる抗菌性樹脂組成物、及びそれからの製品、及び該樹脂組成物に配合する該抗菌剤、及び該抗菌剤の製造方法を提供することが本発明の課題の1つである。
もう1つの重要な課題は、抗菌性樹脂製品を得るための前段として通常良く実施される技術、すなわち一旦樹脂と抗菌剤とを樹脂混練押出機を用いてマスターバッチ(MB)を製造する時に樹脂混練押出加工時フィルター通過性(押出機圧力)が悪くなって機械を長時間運転できずフィルターを短時間で交換しなければならなかった問題を解決することである。機械をより長時間運転できれば、フィルター交換に伴う資源、エネルギー、労力、時間が節減できそれだけ低コストで抗菌性樹脂組成物、及び抗菌性樹脂製品が社会に提供でき工業的な価値は大である。
【0007】
本発明のさらに他の目的は、前記した抗菌性樹脂製品の他に、抗菌剤の特性を利用した別の製品を提供することにある。すなわち、抗カビ剤、抗菌消臭剤、抗菌紙、農薬または化粧料を提供することにある。
【課題を解決するための手段】
【0008】
本発明者等は前記課題の達成のため鋭意研究した結果下記式(1)で表される銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子及び/又は該化合物粒子の150℃〜600℃で乾燥処理又は焼成処理された化合物粒子が抗菌剤の性能として抗菌性が非常に優れ、且つその粒子を樹脂100重量部に対し0.001〜300重量部配合した樹脂組成物も抗菌性が非常に優れていること、しかも樹脂混練押出加工時フィルター通過性、白色性、透明性、水道水接触環境後抗菌力維持特性等の諸特性に優れていること、及び該樹脂組成物から形成された抗菌性樹脂成形品、抗菌性フィルム、抗菌性繊維、抗菌性不織布、抗菌性塗料、抗菌性コーキング材のような抗菌性樹脂製品も同様に白色性、透明性、水道水接触環境後抗菌力維持特性等の諸特性に優れていることを見出し本発明に到達した。
さらに前記粒子は、その抗菌性を利用して、成形品以外にも抗カビ剤、抗菌消臭剤、農薬および化粧料としても有利に利用しうることを見出し本発明に到達した。
【0009】
本発明によれば、下記式(1)で表わされる銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子よりなる抗菌剤が提供される。
(AgaBb−a)bAlcAx(SO4)y(OH)z・pH2O (1)
式(1)中,a,b,c,x,y,z及びpは0.00001≦a<0.5、 0.7≦b≦1.35、2.7<c<3.3、 0.001≦x≦0.5, 1.7<y<2.5、4<z<7及び0≦p≦5を満足し、BはNa+、NH4+、K+及びH3O+の群から選ばれた少なくとも1種の1価陽イオンを表わし、陽イオンの価数×モル数の合計値(1b+3c)の範囲が8<(1b+3c)<12であり、Aは有機酸アニオンを表す。
【発明の効果】
【0010】
本発明の銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子からなる抗菌剤を樹脂に使用すれば、分散性、透明性、白色性、抗菌性特に水道水接触環境後抗菌力維持特性、混練押出加工時フィルター通過性の全ての性質に優れた抗菌性樹脂組成物、及び該樹脂組成物から形成された抗微菌樹脂成形品、抗菌性フィルム、抗菌性繊維、抗菌性不織布、抗菌性塗料、抗菌性コーキング材等の抗菌性樹脂製品を提供することが可能となった。
なかでも、水道水接触環境後抗菌力特性を長時間維持し続けることは、従来技術では到底成し得なかったことであり、それが本発明によって例えば台所、風呂場、トイレ等の水道水を常に使用する場所でも長時間抗菌性を維持できること、つまり細菌からの被害をそのような条件でも長時間維持し続ける抗菌剤、該抗菌剤の製造方法、抗菌性樹脂組成物、及びそれからの抗菌性樹脂製品を提供することに関する新規な技術が提供できるようになったことは本発明の大きな成果である。
【0011】
もう一つの本発明の大きな成果は抗菌性樹脂製品を得るための前段として通常良く実施される技術、すなわち一旦樹脂と抗菌剤とを樹脂混練押出機を用いてマスターバッチ(MB)を製造する時に樹脂混練押出加工時フィルター通過性(押出機圧力)が悪くなって機械を長時間運転できずフィルターを短時間で交換しなければならなかった問題をも解決したことである。
この成果をさらに具体的に説明すると、本発明によって樹脂混練押出加工時フィルター通過性が改善され機械をより長時間運転できるようになり、フィルター交換の頻度が少なくて済むことに伴う樹脂及び樹脂に配合する添加剤・充填剤等の副資材、水、エネルギー、電力、労力、時間が節減できそれだけ低コストで抗菌性樹脂組成物、及び抗菌性樹脂製品が社会に提供できるようになったことによる工業的な価値は非常に大であるということである。
【発明を実施するための最良の形態】
【0012】
本発明について以下さらに詳しく説明する。
【0013】
本発明によれば、前記式(1)で表わされる抗菌剤粒子の性状として、下記(i)、(ii)及び(iii)の特性を、独立して有していることも見出された。
【0014】
(i)レーザー回折散乱法で測定された平均二次粒子径が0.1μm〜12μm、好ましくは0.1〜5μm
(ii)BET法比表面積が0.1〜250m2/g、好ましくは1〜100m2/g
(iii)Dr=D75/D25[D25がレーザー回折散乱法による体積基準累積粒子径分布曲線の25%値(微粒子径側)の粒子径を表し、D75がその75%値(大粒子径側)の粒子径を表す]で定義される粒子径分布のシャープ度が1.0〜1.8、好ましくは1.01〜1.5、さらに好ましくは1.01〜1.3、最も好ましくは1.01〜1.2
【0015】
前記(i)の平均二次粒子径、(ii)のBET法比表面積及び(iii)の粒子径分布のシャープ度(Dr)は、それぞれ独した特性であり、これらの2つの特性が同時に満足するものが、より好ましく、さらにこれら3つの特性を同時に満足する粒子が最も本発明の目的が達成のため好ましい。
さらに本発明の抗菌剤粒子は凝集粒子ではなく単分散状であって以下の粒子形状を有する点にも特徴を有している。
本発明の抗菌剤の粒子は、種々の粒子形状を有しているが、その形状が均一で大きさが揃っていることおよび凝集が少なく単分散状を呈する点にも特徴を有している。粒子形状について説明すると、抗菌剤の粒子は、大きく分けると、球状、円盤状(基石状)、一対状(ハンバーガー状)、米粒状、直方体状、六角板状、円柱状(酒樽状)及び八面体状に分類される。これら種々の粒子形状を図1〜図13により具体的に説明する。
【0016】
図1〜図13は、本発明の実施例により得られた粒子の代表的なSEM写真である。粒子の形状は約1万倍乃至約2万倍に拡大されたSEM写真に基いて観察される。なお、図14は従来知られたアルカリアルミニウム硫酸塩水酸化物粒子のSEM写真である。
球状粒子の例は図1〜図6に示され、これら球状粒子は図1に示す表面が平滑である球状、図2に示す表面に小粒を有する球状、図3に示す表面が荒くさらには球に皺(傷や割れ)を有する球状、図4に示す小穴(凹凸)を有する球状、図5に示す表面が平滑であり且つ直線部分を図1のものより少し多く含む球状、及び図6に示す表面が荒く皺を有する球状に分類できる。
円盤状粒子の例は図7に示され、この形状は表と裏がほぼ対象であり、ドーム形であって、碁石にも似ている。図7の円盤状粒子は表面が平滑である。
一対状粒子の例は図8に示されている。この粒子の特徴は、底面が平板でその反対面がドーム形の円盤粒子の2つが底面を対称面として一対状の形状を有していることであり、その2つの粒子の重なり合う周囲の間隙には空間が存在している。また重なり合う中心部は2つの円盤を接合しているアルミニウム塩水酸化物が存在している。この一対状粒子は、一見ハンバーガーに似ている。
米粒状粒子の例は、図9に示されている。この米粒状粒子は、投影した形が楕円形で長さ方向の直角断面がほぼ円形の形状をしている。図9の粒子は、表面に小さなしわ(皺)を有している。
直方体状粒子の例は図10に示され正六面体に近い直方体であって表面が平滑である。
六角板状粒子の例は図11に示され、この六角板状粒子は六つの辺で形成された六面体の表面を有する板状のものである。この六つの辺は同じ長さであることを要せず、また2つの辺の接点は丸味を有していてもよい。
八面体状粒子の例は図12に示され正方両錐体状乃至偏八面体状の八面体状の形状を有している。
円柱状粒子の例は図13に示されている。この円柱状粒子は、大略酒樽状(またはワイン樽状)のように中間部分が膨らんだものでよく、また断面がほぼ円形の筒状のものでもよい。
図13の粒子は表面に多数の凹凸を有している。
図1〜図13から理解されるように本発明の粒子は各々の図(写真)において、粒子形状が揃っており、その大きさが均一でありかつ分散性がよい点に特徴を有している。前記した各粒子の形状は、それぞれ区分するために分類して表現したものであり、若干の変形や少割合の他の粒子の混合があっても差支えない。また粒子の表面における平滑性、微小凹凸の存在または小さいしわ(皺)の存在は、特に限定されるものではなく、存在してもしなくてもよい。
【0017】
次に本発明の粒子の形状を特定する方法について説明する。
粒子の形状を特定する尺度の一つに、粉体工業分野において従来から用いられてきたWadellの円形度及び球形度がある。
Wadellの球形度sは、s=(粒子と等体積の球の表面積)/(粒子の表面積)
で定義され、sが1に近い程真球に近い。
Wadellの円形度cは、c=(粒子の投影面積と等面積の周長)/(粒子の投影面の周長)で定義され、cが1に近い程真円に近い。
【0018】
本発明において粒子の形状が球状であるとは、図1〜図6に示すようなボール様の形状であれば良く、前記のWadellの球形度sが0.95≦s≦1であることが好ましい。
【0019】
本発明において粒子の形状が円盤状(碁石状)であるとは、図7に示すように短径を回転軸とした回転楕円状の形状である。具体的には、回転軸の方向から見た粒子の投影像に関して、Wadellの円形度cが、0.95≦s≦1であって、断面である楕円の(短径/長径)の比率aが0.05≦a≦0.5であることが好ましい。
【0020】
本発明において粒子の形状が一対状(ハンバーガー状)であるとは、図8に示すように半球状の粒子が2個重なり合うような形状で対を形成した粒子である。そして一対状粒子は、二つの半球状粒子の重なり合う面の周縁に、隙間(溝)が存在している。一対状粒子の(短径/長径)の比率tは0.1<t<0.5であり、(該半球の合わせ目の隙間幅)/(短径)の比率uが0.05<u<0.5であることが好ましい。
【0021】
本発明において粒子の形状が米粒状であるとは、図9に示すように短径を回転軸とした回転楕円状の形状であり、楕円の(短径/長径)の比率aが0.1≦a≦0.5であり、前記のWadellの球形度sが,0.4≦s<0.75であることが好ましい。
【0022】
本発明において粒子の形状が直方体状であるとは、図10に示すような六面体又は正六面体に類似する形状であれば良く、前記のWadellの球形度sが0.5≦s≦0.8であることが好ましい。
【0023】
本発明において粒子の形状が六角板状であるとは、図11に示すような扁平な正六角柱様の形状で、上面又は下面方向から見た粒子の投影像に関してWadellの円形度cが,0.95≦c<0.99であって、厚さ/(正六角形の対角線長さ)の比率bが0.05≦b≦0.5であることが好ましい。
【0024】
本発明において粒子の形状が8面体状であるとは、図12で示されるように正方両錐体状乃至偏八面体状の八面体状の形状を有していると考えられ、前記のWadellの球形度sが0.5≦s≦0.9であることが好ましい。ただし、この八面体状粒子は熟視しないと、SEM写真の分解能不足からくる不鮮明さにより一見六面体状粒子に見える恐れがある。
【0025】
本発明において粒子の形状が円柱状(酒樽状)であるとは、図13に示すように円柱を含み、円柱の高さ方向の中心部の半径が上面及び下面の半径の1.0〜1.2倍までの形状をいい、上面及び下面の投影像に関して、前記のWadellの円形度cが,0.95≦c<0.99であって、(高さ)/(上面又は下面の直径)の値dが1.5≦d≦3であることが好ましい。
【0026】
本発明によれば、上記のように、銀及び有機酸アニオン含有アルミニウム塩水酸化物粒子は、用途や目的に応じて球状、円盤状(碁石状)、一対状、直方体状、六角板状、米粒状、八面体状または円柱状などの種々の形状を提供でき、かつ粒子径をコントロールできる。
【0027】
一方粒子径に関しても、用途および必要な充填率に応じて最適な粒子径の銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子を提供することが可能である。
それらの粒子には粒子同士の凝集がなく樹脂への分散性に優れ、さらに樹脂に配合した時も該抗菌剤の樹脂中での凝集も全くあるいはほとんどないことが該抗菌剤を樹脂に配合した樹脂製品において銀の含有量が極めて少量でも抗菌性を発揮する要因の1つだと考えられ、この単分散技術だけでも従来技術では到底達成できなかった予想外の高抗菌活性を発揮するものである。
【0028】
本発明の該抗菌剤がこのような予想外の高抗菌活性を発揮するメカニズムについてはさらに次の第2の要因も推定される。
本発明の該抗菌剤粒子に高い抗菌性発現効果があるのは、本発明の該抗菌剤の粒子には分子の内部構造にまで有機酸がアニオンとして取り込まれているため本発明の該抗菌剤に光が当たった時、本発明の該抗菌剤粒子からヒドロキシルラジカル(OH−)のようなラジカルが長期間にわたり発生し易くなり、これが本発明の該抗菌剤の抗菌性能が長期間にわたって維持できる要因にもなっているものと推定される。
それは、従来の大部分の銀系無機抗菌剤は抗菌剤から銀イオンを除放できる期間においては抗菌性を発現するが、その期間が終わると抗菌性が発現しなくなるという致命的な欠点があったが、本発明はその問題を根本的に解決するものである。
さらにそればかりでなく、本発明によると該抗菌剤の分子構造中に有機酸をアニオンとして取り込んだことによる抗菌性発現向上効果の第3の要因として、前記の分散効果及びラジカル発生効果のみならず、該抗菌剤粒子の有機酸の炭素部分による樹脂との相溶性向上効果がさらに加わって、つまり本発明ではこれら3つの効果を相乗作用させることに成功しより一層高い抗菌性能を奏することができるようになったものと推定される。
【0029】
本発明では前記式(1)で表される抗菌剤を樹脂100重量部に対し0.001〜300重量部配合することにより混練押出加工時フィルター通過性、抗菌性、分散性、白色性に優れた性質を有しながらしかも尚且つ水道水接触環境後の抗菌力維持特性に優れた抗菌性樹脂組成物及びそれから形成される樹脂製品を提供することができるが、該樹脂製品に高い透明性という付加価値を重視する目的においては0.001〜10重量部、好ましくは0.001重量部〜2重量部配合することが推奨できる。該抗菌剤の配合量が0.001重量部以下であると充分な抗菌性を発揮できない恐れがあり、300重量部以上配合することは銀の含有量にもよるが不経済となり、透明性も低下する傾向がある。
本発明の抗菌剤の屈折率は約1.48〜約1.56であり多くの樹脂と屈折率が重複あるいは接近していため、樹脂にかなり高濃度に本発明の抗菌剤を配合しても透明性をあまり損なわないが、透明性を高度に維持するためには該抗菌剤の配合量は10重量部以下、好ましくは2重量部以下にすることが推奨できる。
【0030】
本発明における式(1)中のaは銀の該抗菌剤粒子へのイオン交換量を示し、aの数値が高ければそれだけ銀が該抗菌剤粒子にイオン交換していることを示し抗菌性が向上するが、あまり高くなりすぎるとイオン交換体(固溶体)から銀が環境中で析出や溶出し酸化銀になる恐れがあり該抗菌剤が配合された樹脂成形品等の色が暗褐色を呈することにもなり、また経済的でもないし、またaは0.5以上イオン交換しにくい。一方aの数値が低過ぎる場合はそれだけ銀が該抗菌剤粒子へのイオン交換量が少ないことを示し抗菌性が発現しないので、抗菌性発現力と色の問題を適度にバランスするためaは一定範囲にすることが望まれる。かかる意味において式(1)におけるaは0.00001〜0.5、好ましくは0.00001〜0.35、さらに好ましくは0.001〜0.3の範囲であることが適している。
【0031】
本発明において、「銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子よりなる抗菌剤」なる記述の中の、含有という言葉は該粒子を粉末X線回折法で測定した時、式(1)以外の化合物のピークが現らわれない程度に式(1)以外の銀及び有機酸からなる化合物の混入量が少ない範囲の物であることを意味している。
従って該抗菌剤粒子にはイオン交換体のみならず、粉末X線回折法の測定ではピークが現らわれない若干の銀イオンがイオン交換体以外の形態で該粒子に担持された形態及び/又は若干の有機酸アニオンが該粒子の表面に吸着している形態も含んでいるものと考えられる。
その場合、該粒子を銀に着目して考えると、銀がイオン交換許容範囲内でイオン交換した固溶体のみからなるものは樹脂製品への着色の影響が少なくなっていると考えられ、そういう意味では完全な銀イオン交換体(固溶体)のみからなる物が好ましい。
【0032】
本発明における式(1)中Bには種々の1価のカチオンであればよいが、実際には銀イオンとイオン半径が比較的近く広い範囲で強くイオン交換体を形成することができるという意味、つまり樹脂に配合した時に銀が該イオン交換体から遊離して酸化銀になることがなく、その結果樹脂製品の白色度の低下(樹脂製品の成形直後の白色から光による作用で時間とともに暗褐色乃至褐色への変色すること)を招き難いということ、さらに安全性、及び経済性を考慮すると、BとしてはNa+,NH4+,K+及びH3O+が適している。さらにその中ではNa+,NH4+及びH3O+が比較的好ましく、その中でもさらにNH4+及びH3O+がさらに好ましく、NH4+が該目的において最も好ましい。この変色防止という観点からは、BとしてNa+及び/又はK+を用いる量はできるだけ少ないことが好ましく、特にK+を用いる量はBなる1価陽イオンの合計モルの1/2より小であることが好ましい。ただしこれらの変色は蛍光増白剤を樹脂中に0.000001%〜0.1%添加することにより防止できる程度のものである。従ってBとしてNa+及びK+を多く用いる場合は変色防止のため蛍光増白剤を用いることは好ましいことである。
【0033】
本発明の抗菌性樹脂組成物の一部分ではあるが、前記比較的好ましい態様すなわち、NH4+及びNa+、H3O+の3種1価陽イオンが用いられる抗菌剤粒子の場合、蛍光増白剤を用いなくとも変色しないあるいは比較的変色が少ない樹脂製品を得るためには、Naの使用量は、Naのモル含有量がこれらBなる1価陽イオンの合計モル含有量の1/2より小であるべきである。そうすることによって本発明では変色のないあるいは少ない樹脂製品を得ることができる。
該蛍光増白剤を用いる場合、その例としてはベンゾヘキサゾール系の2,5−チオフェンジイル(5−tert−ブチル−1,3−ベンゾヘキサゾール、4,4’−ビス(ベンゾヘキサゾール−2−イル)スチルベン、及びピラゾリン系、カーマリン系のものが例示できるが、FDA(米国Food & Drag Administration)やポリオレフィン等衛生協議会に登録されているものを用いることが好ましい。
【0034】
本発明における式(1)中bは0.7〜1.35、好ましくは0.8〜1.2、最も好ましくは0.9〜1.1であれば本発明の該抗菌剤粒子を形成し易く、cは2.7〜3.3、好ましくは2.8〜3.2、最も好ましくは2.9〜3.1であれば本発明の該抗菌剤粒子を形成し易い。
式(1)の陽イオンのイオン価×モル数は(1b+3c)として表される。本発明でのその範囲は8<(1b+3c)<12、好ましくは9<(1b+3c)<11であれば本発明の該抗菌剤粒子を形成し易い。
本発明の抗菌剤では式(1)なる粒子の合成において、粒子形状と粒子サイズを選択して製造すること、粒子径分布を均一に整えること、該粒子を出来る限りに単分散状態で樹脂に分散できるようにするためには、該反応において反応時に有機酸類を添加する必要があり、添加された有機酸類は該抗菌剤粒子の分子構造中に組み込むことができる。
【0035】
かかる有機酸類としては式(1)中の有機酸アニオン(A)が有機カルボン酸又は有機オキシカルボン酸に基づくアニオン群から選ばれる少なくとも1種であるものを例示できるが、有機酸アニオン(A)が、炭素数1〜15、好ましくは1〜10有し、且つカルボキシル基を1〜4個有する有機カルボン酸又は有機オキシカルボン酸に基づくアニオン群から選ばれる少なくとも1種であることも好ましく、例えばジカルボン酸、モノカルボン酸、トリカルボン酸、鎖式カルボン酸、芳香族カルボン酸、ヒドロキシ酸、ケトン酸、アルデヒド酸、フェノール酸、アミノ酸、ハロゲンカルボン酸及びそれらの塩類が例示できるが、その中でも有機酸アニオン(A)が、蓚酸イオン、クエン酸イオン、リンゴ酸イオン、酒石酸イオン、グリセリン酸イオン、没食子酸イオン、及び乳酸イオンの群から選ばれる少なくとも1種であることが最も好ましい。
【0036】
式(1)中の有機酸アニオン(A)の割合xを0.0001≦x≦0.5、好ましくは0.0001≦x≦0.4、さらに好ましくは0.001≦x≦0.2にすれば前記の目的を達成し且つ粒子形状が前記のを形状を有し且つ粒子径均一性の該抗菌剤粒子が得られる。xが0.5を超えると特にこの効果は高くなるものでもなく又経済的でもない。
xが0.0001未満の場合は粒子形状が前記の形状を有し且つ粒子径均一性の該抗菌剤粒子が得られ難くなるし、又前記のヒドロキシラジカルの発生、及び有機酸による樹脂との相溶性向上効果が原因と考えられる抗菌性の向上等本発明の目的は達成し難くなる。
【0037】
本発明の式(1)におけるyは1.7<y<2.5、好ましくは1.8<y<2.2であれば本発明の該抗菌剤粒子を形成し易く、zは4<z<7、好ましくは5<z<7であれば本発明の該抗菌剤粒子をより一層形成し易い。
式(1)中のpは結晶水の量を示し、通常pは0≦p≦5の範囲である。このpを限りなく0に近づけるか、あるいは0にするためには350℃以下の追加の乾燥処理、あるいは焼成処理すれば良い。焼成処理は600℃以下が好ましい。焼成温度が500℃以上、さらに550℃以上、特に600℃以上の温度であると、下記式で表される水溶性のアルミニウム硫酸塩が一部生成する恐れがあり、またそれを添加した樹脂製品は耐水性が低下する恐れがある。ただし添加量が少ない場合には特に耐水性に問題はない。
(AgaBb−a)bAlAx(SO4)y
焼成温度が500℃以下、特に450℃以下であれば前記式で表される水溶性のアルミニウム硫酸塩は生成せず、これを樹脂製品に多量に使用しても耐水性は低下せず何ら問題ない。また、本発明の抗菌剤粒子は600℃以上の温度で焼成すると図1〜図13に示すような、本発明の該抗菌剤粒子の粒子形状が維持できない恐れもある。かかる耐水性及び形状維持の観点から本発明の抗菌剤粒子の焼成温度は350℃〜600℃、好ましくは350℃〜550℃、さらに好ましくは350℃〜500℃、最も好ましくは350℃〜450℃である。
【0038】
前述した乾燥処理又は焼成処理を窒素雰囲気下で実施することは、該抗菌剤粒子及び該抗菌剤粒子が配合された樹脂製品の着色防止の点で好ましいことである。乾燥処理は真空乾燥で実施しても着色防止の点で好まい。
樹脂加工の際、pが0でなくても問題にならない場合、例えば該抗菌剤の配合量が非常に少ない場合あるいは樹脂加工時の水分が特に問題にならない樹脂の場合は、乾燥処理又は焼成処理を実施していないpが0≦p≦5、好ましくは0≦p≦3のものを樹脂に配合して樹脂組成物を製造することができる。
逆に、そのpが0又は0に近づけたものでないと問題になる場合は追加乾燥処理又は焼成処理を加えることによりp=0又は0に限りなく近づけたものを使用すれば良い。
例えばPET,PBTのようなポリエステル系樹脂、ポリアミド系樹脂、ポリウレタン系樹脂、ポリカーボネート樹脂、ポリアセタール樹脂等の樹脂では前記の条件で追加乾燥処理、あるいは焼成処理されたp(水分量)を0又は限りなく0に近づける配慮をした該抗菌剤を使用することは推奨できることである。
【0039】
本発明の粒子は、前記レーザー回折散乱法粒子径分布のシャープ度(Dr)を1.0≦Dr≦1.8、好ましくは1.0≦Dr≦1.5、さらに好ましくは1.01≦Dr≦1.3、最も好ましくは1.01≦Dr≦1.2とすることにより樹脂への分散性という点において凝集がなく完全分散することができ、それが抗菌効果を高める要素であると考えられるが、さらには樹脂の押出加工等の際、特にマスターバッチを製造する際、フィルター(スクリーンメッシュ)を使用しても、該抗菌剤のスクリーンメッシュへの目詰まりがなくなるあるいは少なくなるという利点もある。
【0040】
本発明の該抗菌剤配合抗菌性樹脂製品に高い抗菌性を付与する目的においては粒子の平均二次粒子が小さい程適しているが0.1μm〜12μm、好ましくは0.1〜5μm、さらに好ましくは0.1〜2μm、さらにより好ましくは0.1〜1μm、最も好ましくは0.1μm〜0.5μmの物が用いられる。
ただし該抗菌剤の平均二次粒子径が0.1μm未満のものは製造し難い恐れがあり、12μm以上のものは樹脂に配合しても樹脂組成物等の抗菌性があまり高くならない恐れがある。本発明の樹脂組成物に抗菌性のみならず、高度の透明性を付与する目的においては、本発明の抗菌剤の中でも平均二次粒子径が0.1〜0.5μm、好ましくは0.1〜0.4μm、さらに好ましくは0.1〜0.3μmである超微粒子のものを使用すると、もともと樹脂と屈折率が重複又は接近している本発明の抗菌剤粒子の特性を最大限に発揮でき、樹脂製品に従来技術では到底達成し得なかった尚一層高度な透明性を付与する効果を絶大に発揮するものである。
【0041】
本発明で使用される抗菌剤のBET法比表面積は0.1〜250m2/gの物が用いられる。
樹脂組成物に高い抗菌性を付与するためにはBET法比表面積の高い方が有利ではあるが、BET法比表面積のあまり高い物は一方では樹脂に充填し難い問題が生ずる恐れがあり、一方BET法比表面積が低すぎると樹脂組成物に十分な抗菌性を付与できない恐れがある。
かかる意味において該抗菌剤のBET法比表面積は0.1〜250m2/g、好ましくは1〜100m2/gさらに好ましくは10〜100m2/g、最も好ましくは30〜100m2/gである。
【0042】
本発明の、該抗菌剤粒子が配合された抗菌性樹脂製品は元々高い耐酸性を有しているが、さらに高い耐酸性を樹脂製品に付与する目的、あるいは変色防止の目的においては、本発明の該抗菌剤粒子の表面を、ケイ素化合物、リン化合物、ホウ素化合物、アルミニュウム化合物,ジルコニウム化合物、チタン化合物、亜鉛化合物、錫化合物の群から選ばれた少なくとも1種の耐酸性改質剤で被覆してさらに耐酸性を向上することができる。
かかる耐酸性改質剤を例示するとケイ素化合物としてはメタケイ酸ナトリウム、オルトケイ酸ナトリウム、メタケイ酸カリウム、オルトケイ酸カリウム、水ガラス、ケイ酸、シリコーンオイル;ホウ素化合物としては四ホウ酸ナトリウム、メタホウ酸ナトリウム、四ホウ酸カリウム、メタホウ酸カリウム、ホウ酸;アルミニュウム化合物としてはオルトアルミン酸ナトリウム、メタアルミン酸ナトリウム、オルトアルミン酸カリウム、メタアルミン酸カリウム、塩化アルミニュウム、硝酸アルミニュウム、硫酸アルミニュウム、リン酸アルミニュウム;リン化合物としてはリン酸カリウム、リン酸ナトリウム、リン酸;ジルコニウム化合物としてはリン酸ジルコニウム、ジルコン酸ナトリウム、ジルコン酸カリウム、ジルコン酸;チタン化合物としては塩化チタン、チタン酸ナトリウム、チタン酸カリウム、チタン酸;亜鉛化合物としては塩化亜鉛、硝酸亜鉛、炭酸亜鉛、硫酸亜鉛、亜鉛酸塩;錫化合物としては錫酸ソーダ、錫酸カリウム等の改質剤を一例として挙げることができる。
【0043】
本発明の該抗菌剤粒子は元々単分散粒子であるため樹脂への分散性は極めて優れているが、さらに分散性を向上させる目的、あるいは樹脂製品の変色防止の目的、あるいは本発明の該抗菌剤粒子が樹脂に比較的多量に配合された時の機械的強度の低下を抑制する目的においては、本発明の該抗菌剤粒子の表面を、高級脂肪酸類、シラン系カップリング剤、アルミネート系カップリング剤、アルコールリン酸エステル類、界面活性剤類等の群から選ばれた少なくとも1種で表面処理することもできる。
かかる表面処理剤としては、ステアリン酸、オレイン酸、エルカ酸、パルミチン酸、ラウリン酸、ベヘニン酸等の高級脂肪酸類及びその塩類、ポリエチレンエーテルの硫酸エステル塩、アミド結合硫酸エステル塩、エステル結合硫酸エステル塩、エステル結合スルホネート、アミド結合スルホン酸塩、エーテル結合スルホン酸塩、エーテル結合アルキルアリルスルホン酸塩、アミド結合アルキルアリルスルホン酸塩等の界面活性剤類、ステアリルアルコール、オレイルアルコール等の高級アルコールの硫酸エステル塩、オルトリン酸とステアリルアルコール又はオレイルアルコールなどのモノ又はジエステル又は両者の混合物であって、その酸型又はアルカリ金属塩又はアミン等の燐酸エステル類、ビニルエトキシシラン、ビニル−トリル(2−メトキシ−エトキシ)シラン、ガンマ−メタクリロキシプロピルトリメトキシシラン、ガンマ−アミノプロピルトリメトキシシラン、N−フェニル−ガンマ−アミノプロピルトリメトキシシラン、N−ベータ(アミノエチル)ガンマ−アミノプロピルトリメトキシシラン、N−ベータ(アミノエチル)ガンマ−アミノプロピルトリエトキシシラン、N−フェニル−ガンマ−アミノプロピルトリエトキシシラン、ベータ(3,4−エポキシシクロヘキシル)エチルトリメトキシシラン、ガンマ−メルカプトプロピルとりメトキシシラン等のシランカップリング剤類、イソプロピルトリイソステアロイルチタネート、イソプロピルトリ(アミノエチル)チタネート、イソプロピルトリデシルベンゼンスルホニルチタネート等のチタネートカップリング剤類、アセトアルコキシアルミニュウムジイソプロピレート等のアルミネートカップリング剤を例示できる。
【0044】
本発明の該抗菌剤粒子は下記の方法で製造することができる。
その際、工業的に食品衛生法厚生省告示第20号で規定された、鉛、カドミュム等の重金属含有量が少なく、さらには樹脂製品の熱劣化防止(耐熱劣化性の向上)及び着色防止という意味で鉄、マンガン、クロム、銅、ニッケル等の重金属含有量を1%以下、好ましくは0.1%以下、さらに好ましくは0.01重量%以下、最も好ましくは0.001%以下の高純度の該抗菌剤粒子を得る場合には、先ず原料面で高純度の物を選択すること、さらには化学操作装置の材質面において特に腐食が起き易い水熱処理工程等では装置材料の溶出によって鉄、マンガン、クロム、銅、ニッケル等の重金属化合物が固溶体及び又は夾雑物として本発明の該抗菌剤粒子に混入しないように配慮された耐腐食性に優れたハステロイ鋼、ステンレスSUS−316鋼等を選択することは好ましいことである。
【0045】
本発明の該抗菌剤粒子は、基本的には国際特許出願PCT/JP2005/003831(出願日:2005年3月1日)明細書に記載された方法によって製造できる下記一般式(2)で表される有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子の1価陽イオン(B)の部分を銀とイオン交換する方法であり、その方法を後記1〜4に例示する。
[B]bAlcAx(SO4)y(OH)z・pH2O 式(2)
式中、b、c、x、y、z、p、BおよびAの定義は、前記式(1)と同じものを意味する。
式(2)なる化合物を合成するためには、硫酸アルミニウム、硫酸ナトリウム、硫酸カリウム、硫酸アンモニウム、及び硫酸カルシュウム等のアルミニウム及び硫酸原料と、蓚酸等の有機酸類原料と、水酸化ナトリウム、水酸化カリウム及びアンモニア水溶液等のアルカリ原料とを湿式法又は乾式法で反応させ、Bがナトリウム型、カリウム型またはアンモニウム型の銀を含まない有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子〔粒子形状が球状、円盤状、(碁石状)、一対状(ハンバーガー状)、米粒状、六角板状、六面体状、円柱状のものについては後記1〜3に示す〕を先ず合成しておき、然る後それらの有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子を水等の懸濁液中において銀溶液と接触攪拌すれば、銀が有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子にイオン交換した本発明の該抗菌剤粒子が製造できる。
【0046】
粒子形状が直方体状の銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子の製造方法については後記4に示す。
いずれの際も、銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子を、形状と粒子サイズを選択して製造すること、粒子径分布を均一に整えること、該粒子を完璧なまでに単分散状態で樹脂に分散できるようにするためには、該反応において前記の有機酸類を前記の反応時に添加する必要があり、添加された有機酸類は該抗菌剤粒子の構造中に組み込むことができる。
ただし有機酸類の一部は該抗菌剤粒子の表面に吸着している場合も考えられる。
いずれにしても有機酸類は本発明の目的を達成するために該抗菌剤に含有することができる。
有機酸類を前記の反応時に添加せず、反応が終わった後に添加しても前記粒子形状、粒子径均一性、分散性等の性質を有する本発明の抗菌剤粒子は製造できない。
【0047】
製造された本発明の抗菌剤粒子の粉砕処理に関しては従来技術のように強力な機械力で実施する必要はなく弱い力で簡単に処理してもの凝集のないのものが得られることも本発明の技術の特徴である。
以下に、ナトリウム型、カリウム型、アンモニウム型、水素型の銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子抗菌剤の製造方法について例示する。
【0048】
1、ナトリウム型銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子の製造方法
原料としては硫酸アルミニウム、硫酸ナトリウム等硫酸塩、及び水酸化ナトリウム等のAl原料及びSO4原料及びNa原料、蓚酸等の有機酸原料、及びイオン交換反応で使用する硝酸銀等の可溶性銀塩を銀原料として用い、下記の方法で製造する方法が例示できる。
例えばAl2(SO4)3+Na2SO4(或いはNaNO3)+H2C2O4を水の中に充分溶解した後、攪拌しながら、水酸化ナトリウムを添加する。
添加終了後、よく分散させるために、好ましくは更に20分以上攪拌する。
その後水熱処理を行うことは好ましいことである。
水熱処理の温度は100℃〜250℃が好ましく、処理時間は1時間〜30時間が好ましい。
【0049】
このようにして得られた有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子は濾過、及び必要に応じ水洗する。、
それを水等の液に懸濁し硫酸銀、硝酸銀等の可溶性銀塩の溶液と攪拌すればイオン交換反応処理が実施でき、本発明の銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子が製造できる。
イオン交換反応処理の温度は0℃〜100℃が好ましく、処理時間は0.1時間〜30時間遮光下で実施することが好ましい。
【0050】
イオン交換するときの処理温度が低かったり、処理時間が短か過ぎると銀のイオン交換量が少なくなる恐れがある。一方イオン交換するときの処理温度が高かったり、処理時間が長過ぎるとイオン交換処理物が褐色に着色する傾向がある。
イオン交換反応処理での攪拌の方法は振動及び回転等の方法が例示できる。
イオン交換したものは濾過・遠心分離の方法で濾別した後、必要ならばさらに水洗・表面処理・乾燥・粉砕等の操作を必要に応じ実施し回収すれば良い。濾別が困難な場合、本発明の目的に反しない範囲で凝集剤を使用し濾別操作を改善しても良い。凝集剤としてはポリアクリルアミドのような高分子凝集剤が例示できる。高分子凝集剤の添加量は0.2%以下が好ましい。0.2%以上添加しても特に炉別操作は改善しない。
【0051】
2、カリウム型銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子の製造方法
原料としては硫酸アルミニウム、硫酸カリウム等硫酸塩、及び水酸化カリウム等のAl原料及びSO4原料及びK原料、蓚酸等の有機酸原料、及びイオン交換反応で使用する硝酸銀等の可溶性銀塩を銀原料として用い、下記の方法で製造する方法が例示できる。
例えばAl2(SO4)3+K2SO4(或いはKNO3)+H2C2O4を水の中に充分溶解した後、攪拌しながら、水酸化カリウムを添加する。それ以後の操作方法は上記1に準じて行う。
【0052】
3、アンモニウム型銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子の製造方法
原料としては硫酸アルミニウム、硫酸アンモニウム等硫酸塩、及び硝酸アンモニウム等のAl原料及びSO4原料及びNH4原料、蓚酸等の有機酸原料、及びイオン交換反応で使用する硝酸銀等の可溶性銀塩を銀原料として用い、下記の方法で製造する方法が例示できる。
例えばAl2(SO4)3+K2SO4(或いはKNO3)+H2C2O4を水の中に充分溶解した後、攪拌しながら、アンモニア水溶液を添加する。それ以後の操作方法は上記1に準じて行う。
【0053】
4、水素型{(H3O)+型}直方体状銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子の製造方法
直方体状粒子は前記の球状及び円板状粒子の製造方法とは異なり、硫酸アルミニウム水溶液と水酸化アルミニウム懸濁液、及び蓚酸等の有機酸を混合攪拌したものを100℃〜250℃、好ましくは120℃〜200℃で、0.5時間以上、好ましくは0.5〜30時間、好ましくは2〜20時間水熱処理すれば直方体状で水素型、化学式(H3O)Al3(SO4)2(OH)6で表されるものを得ることができる。
用いる水酸化アルミニウムとしては、結晶性のギプサイト、バイアライト、ベーマイト、擬ベーマイト、ダイアスポア、無定形の水酸化アルミニウムが例示できるが、無定形の水酸化アルミニウムが粒子径の均一性が高くなるという点で好ましい。
無定形の水酸化アルミニウムとしては、協和化学工業(株)製乾燥水酸化アルミニウムゲルS−100,及び同FMが例示できる。
【0054】
この方法において微粒子化と粒子径均一性を向上ためには、硫酸アルミニウム水溶液と水酸化アルミニウム懸濁液を混合攪拌した反応物を直ちに水熱処理するよりは、反応後ある程度時間が経過したもの、例えば0.5時間以上、好ましくは5時間以上、さらに好ましくは16時間以上静置又は攪拌したものを水熱処理すると微粒子で粒子径均一性を有するアルミニウム硫酸塩水酸化物粒子が得られる。それ以後の操作方法は上記1に準じて行う。
【0055】
本発明の銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子は粉末X線回折法により同定することができる。銀が該粒子にイオン交換していればその回折パターンはイオン交換前の該粒子と同じ回折パターンとなるので化学分析値と照らし合わせて回折X線の回折パターンを精査すれば良い。もし銀が該粒子にイオン交換せずに、酸化銀のような不純物で共存する製造物を樹脂に配合すると樹脂製品の色は暗褐色を呈し本発明の重要な目的の一つである白色の樹脂製品を得ることから逸脱するので好ましくない。
本発明においては、式(1)なる該抗菌剤の3価陽イオンであるアルミニウム(Al)の一部をZn2+、Ti4+の群から選ばれた少なくとも1種の陽イオンで、及び/又は1価陽イオンBの一部をCa2+で本発明の目的を害しない範囲で置き換えて含有させても良い。
その場合、該抗菌剤を樹脂に配合した時樹脂製品の透明性を低下させないようにするためZn2+、Ti4+の合計モル含有量はアルミニウムのモル含有量の1/2以下好ましくは1/3以下、Ca2+のモル含有量は1価陽イオン(B)のモル含有量の1/2以下好ましくは1/3以下にして該抗菌剤粒子に含有させれば良い。
【0056】
式(1)なる該抗菌剤の3価陽イオンのアルミニウム(Al)の一部を置き換える方法としては、該金属カチオンを含む硫酸亜鉛、硫酸チタン、硫酸カルシウム等の塩類を前記式(2)なる化合物を合成する時に用いて式(2)に組み込むか、あるいは式(2)なる化合物に該金属カチオンを含む化合物を用いて溶媒中でイオン交換した物に、さらに前記の方法で銀とイオン交換する方法が例示できる。その場合、Zn2+、Ti4+は抗菌性発現効果がある元素であるため式(1)においてAg1+の相対的含有量を減らしても抗菌性を発揮し、その結果これを樹脂に配合した樹脂成形品の着色を減少させる利点がありそういう意味で好ましいことである。
【0057】
本発明においては、式(1)なる該抗菌剤の(SO4)yは(SO4)yの一部、好ましくyモルの1/2以下を他の無機酸イオンで置き換えることもでき、置き換える量が特に1/2以下であれば本発明の粒子形状や粒子径均一性が何の問題もなく維持でき本発明の目的が達成される。該無機酸イオンとしてはSO32−、PO43−、HPO32−、CO32−、NO3−、SiO44−及びBO33−等が例示できる。式(1)なる該抗菌剤の(SO4)yの一部を他の無機酸イオンで置き換える方法としては、該無機酸イオンを含む塩類を式(2)なる化合物を反応する時に硫酸アルミニウム、硫酸ナトリウム、硫酸カリウム等の代りに用いて式(2)に組み込むか、あるいは式(2)なる化合物に該無機酸イオンを含む化合物を用いて溶媒中でイオン交換した物に、さらに前記の方法で銀とイオン交換する方法が例示できる。該粒子はさらに式(1)の該抗菌剤粒子同様、追加乾燥処理、焼成処理、表面処理、耐酸性被覆処理をして抗菌剤として利用することもできるし、樹脂に配合して抗菌性樹脂製品を得ることもできる。
【0058】
本発明においては、式(1)なる該抗菌剤の(OH)zは(OH)zの一部がCl−で置き換わって含有されている場合もあるが、その含有量は着色防止という意味で該抗菌剤式(1)中0.1モル以下、好ましくは0.01モル以下、最も好ましくは0.001モル以下である。
本発明において抗菌化の対象樹脂としては特に限定されるものではなく合成樹脂・ゴム、天然樹脂・ゴム等を原料としそれを加工して通常成形品、繊維、不織布、塗料、コーキング材、フィルム等樹脂製品として使用されるものであれば良く、以下にその一部を例示する。
【0059】
熱可塑性樹脂としては、ポリプロピレン、エチレンプロピレン共重合体、高密度ポリエチレン、低密度ポリエチレン、超高分子量ポリエチレン、直鎖状低密度ポリエチレン、ポリブテン、ポリ・4メチルペンテン−1等の如きC2〜C12のオレフィン(αオレフィン)の重合体もしくは共重合体、これらのオレフィンとジエンとの共重合体、エチレン酢酸ビニル共重合体、エチレン・アクリレート共重合体、TPO(熱可塑性ポリオレフィン)樹脂等のポリオレフィン系樹脂、ポリエチレンオキサイド樹脂、ポリスチレン、ABS(アクリロニトリルブタジエンスチレン)樹脂、AAS(アクリロニトリルアクリルスチレン)樹脂、AS(アクリロニトリルスチレン)樹脂、AES(アクリロニトリルイーピーディーエムスチレン)樹脂、MBS(メチルメタクリレートブタジエンスチレン)樹脂、ポリパラメチルスチレン樹脂等のスチレン系樹脂、ACS(アクリロニトリルクロリネイティドポリエチレンスチレン)樹脂、酢酸ビニル樹脂、プロピオン酸ビニル樹脂、フェノキシ樹脂、アイオノマー樹脂、ポリアセタール樹脂、ナイロン6、ナイロン66等のポリアミド系樹脂、ポリエチレンテレフタレート、ポリブチレンテレフタレート等のポリエステル系樹脂、ポリアミドイミド樹脂、ポリサルホン樹脂、ポリアリレート樹脂、ポリエーテルイミド樹脂、ポリエーテルケトン樹脂、ポリフェニレンエーテル樹脂、ポリフェニレンサルファイド樹脂、メタクリル樹脂、セルロース樹脂、ポリカーボネート樹脂、フッソ樹脂、ポリウレタン樹脂、シリコーン樹脂、ポリビニルエーテル樹脂、ポリビニルホルマール樹脂、ポリビニルブチラール樹脂、ポリビニルアルコール樹脂、イソブチレン−無水マレイン酸共重合樹脂、さらにはポリ塩化ビニル樹脂、エチレン塩ビ共重合樹脂、エチレン酢ビ共重合樹脂、塩素化塩化ビニル樹脂、塩素化ポリエチレン、塩素化ポリプロピレン、クマロン樹脂、ケトン樹脂、ポリ塩化ビニリデン、ポリ2塩化ビニル樹脂、塩素化ポリエーテル等の分子構造中に塩素を有する樹脂、アセテートプラスチック、酢酸セルロース、セルロイド、液晶ポリマー、さらには吸水性樹脂等がそれらの一部として例示できる。
【0060】
熱硬化性樹脂としては、エポキシ樹脂、フェノール樹脂、メラミン樹脂、不飽和ポリエステル樹脂、アルキド樹脂、グアナミン樹脂、ポリイミド樹脂、尿素樹脂、シリコーン樹脂、フェノールホルムアルデヒド樹脂、メラミンホルムアルデヒド樹脂、ポリパラ安息香酸樹脂、ポリウレタン樹脂、マレイン酸樹脂、ユリア樹脂、フラン樹脂、キシレン樹脂、ジアリルフタレート樹脂等の樹脂を例示できる。
ゴムとしてはEPDM(エチレンプロピレンジエン共重合体ゴム),EPM(エチレンプロピレン共重合体ゴム),ブチルゴム、イソプレンゴム、SBR(スチレンブタジエンゴム),NIR(ニトリルイソプレンゴム),NBR(ニトリルブタジエンゴム),ウレタンゴム、クロロプレンゴム、水素化ニトリルゴム、ポリエーテル系ゴム、四フッ化エチレン・プロピレンゴム、クロロスルホン化ゴム、ブタジエンゴム、アクリルゴム、塩素化ポリエチレン、エピクロルヒドリンゴム、プロピレンオキサイドゴム、エチレン・アクリルゴム、ノルボルネンゴム、多硫化ゴム、フッ素ゴム、シリコーンゴム、天然ゴム等が例示できる。
これらの樹脂及びゴムは単独で用いても良く、あるいは複数種同時に用いても良い。複数種同時に用いる場合はポリマーアロイ化して用いても良く、ブレンドして用いても良く、あるいは積層成形して用いても良い。これら合成樹脂類は製造方法によって限定されるものではなく、例えばポリオレフィンの重合触媒としては、チーグラー法、チーグラーナッタ法、フリーデルクラフト法、メタロセン法、フィリップス法等いかなものであっても良い。
【0061】
本発明の抗菌性樹脂組成物には通常使用される添加剤、補強剤、充填剤を本発明の目的を害しない範囲で配合することができる。以下これらの一部を例示する。
酸化防止剤、紫外線吸収剤、光安定化剤、熱安定剤、金属不活性化剤、可塑剤、帯電防止剤、難燃剤、加硫剤、加硫促進剤、老化防止剤、素練り促進剤、粘着付与剤、香料、滑剤、着色剤、造核剤、発砲剤、脱臭剤、ポリマーアロイ相溶化剤等。
本発明の抗菌性樹脂組成物を得る方法には、特別の制約はなく前記の樹脂類、ゴム類と本発明の抗菌剤粒子を、例えば2軸押出機、加圧ニーダー、オープンロール、バンバリーミキサー等の装置を使用しできるだけ均一に混合混練しておき、それをペレッターザー、粉砕機等の装置を使用しペレットや粉末にし樹脂組成物を得る方法や、樹脂類が溶媒を使用して溶解されたものの中に本発明の抗菌剤粒子を混合する方法が例示できる。
【0062】
本発明の抗菌性樹脂組成物の成形品を得る方法には、特別の制約はなく前記の方法で得られた該抗菌剤配合樹脂組成物をそのままの濃度で成形品等の樹脂製品にする方法、あるいはマスターバッチとして一旦高濃度に加工したものからそれを最終的に樹脂製品として使用する濃度に希釈混合し、それを射出成形機、押出成形機、ブロー成形機、カレンダー成形機、圧縮成形機、積層成形機等の装置を用いて抗菌性樹脂製品とする方法、さらには抗菌剤粒子を樹脂と混合後、前記の機械に直接投入し直接射出成形、直接押出成形する方法等を一例として例示できる。
得られた成形品の形状、大きさ、厚さ、太さ、さらには使用方法についても何ら制約はなく例えば板状、ボトル状、球状、円盤状、シート状、電線被覆状、発砲体、積層体等何でも良くその成形品は例えば台所、風呂場、トイレ周りの製品、衛生用品、エアコンの吹き出し口等抗菌性を必要とする分野で好適に使用できる。
【0063】
本発明の抗菌性フィルムを得る方法には、特別の制約はなくフィルム製造に適した樹脂の中から前記の方法で得られた抗菌性樹脂組成物を製造し、それをインフレーション法、Tダイ法、カレンダー法、キャスト法等でフィルムを得ることができ、さらにそのフィルムを延伸しても良く、さらには共押出し法により2層以上の積層フィルムにした物で抗菌性が必要な層にだけ本発明の該抗菌剤粒子を抗菌剤として使用する経済性を追求した利用方法も例示できる。
得られたフィルムの形状や形態、使用方法についても特に制約はなく、例えば野菜果物菓子乾物魚肉等食品包装用フィルムとして鮮度保持を兼ねた抗菌性を必要とする分野を一例として例示できる。
【0064】
本発明の繊維を製造する方法において特別の制約はなく、前記の樹脂組成物のうち繊維として紡糸できる樹脂組成物を使用し従来公知の溶融押し出し法、乾式紡糸法、湿式紡糸法等で紡糸し、必要に応じさらに延伸や撚糸、及び/又は、さらには綿、ウール、麻等の天然繊維との混合等の工程を採用し製品とすれば良い。得られた繊維の利用方法についても特に制約はなく、例えば絨毯、衣服、タオル、ナプキン、ハンカチ、手袋、ソックス、帽子、マフラー等の抗菌性を必要とする分野が一例として例示できる。
繊維としては、ポリプロピレン系、ポリエチレン系、ポリアミド(ナイロン)系、アラミド系、アクリル系、ポリウレタン系、フッソ系、ポリクラール系、ポリエステル系、ポリ塩化ビニル系、ビニロン系、ビニリデン系、アセテート系、トリアセテート系、レーヨン系、キュプラ系、ノボロイド系、プロミックス系、ポリアセタール系、ポリノジック系、プラスチック光ファイバー等の繊維が例示できる。
【0065】
本発明の抗菌性不織布を製造する方法には特に制約はなく従来公知の方法を採用すれば良い。
例えば、抄造ウエブ法、ランダムウエブ法、パラレルウエブ法、クロスレイドウエブ法、糸交錯ウエブ法、トウ拡開ウエブ法、短繊維ウエブ法、フィラメントウエブ法、マイクロファイバーウエブ法、スプリットフィルムウエブ法等の方法が採用し得る。これらの方法で得られた抗菌性不織布は抗菌性を有しながら軽くて通気性、防縮性等の性質に優れるため生理用用品、衣服の芯地や裏地、ビニルレザーや擬革の基布等抗菌性を必要とする分野で利用できることが一例として挙げられる。
【0066】
本発明の抗菌性塗料を工業的に製造する方法において特別の制約はなく、従来公知の方法が採用でき、顔料、展色剤、耐候剤、溶剤、助溶剤、希釈剤、可塑剤等を混合する工程で本発明の銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子抗菌剤を混合し、その後は所望により練り合わせ、フルイ(濾過)等の工程を通して製品とすれば良い。家庭で個人的に使用する場合は市販の塗料に本発明の該抗菌剤粒子を混合して使用することもできる。本発明の抗菌性塗料は例えば、病院内、老人ホーム、学校、レストラン等で使用すれば大腸菌や黄色ブドウ球菌等の細菌からの被害を防止でき本発明の目的が達成される。塗料としては、合成樹脂塗料(アルキド、アミノアルキド、エポキシ、フッ素、ウレタン、酢酸ビニル、アクリル酸エステル、不飽和ポリエステル、フェノール、グアナミン、ブチラール、スチレンブタジエン、スチレン、塩化ビニル、塩化ビニリデン、塩化ゴム系塗料、サビ止めペイント、粉体塗料、電気絶縁塗料)、セルロース塗料、ゴム系塗料、水溶性合成樹脂塗料(水溶性アルキド、水溶性エポキシ、水溶性ポリブタジエン、水溶性メラミン、水溶性尿素、水溶性フェノール、水溶性アクリル等)、酒精塗料、油性塗料等樹脂を使用する塗料を例示できる。
【0067】
本発明の抗菌性コーキング材を得る方法には、特別の制約はなく例えばコーキング材としての基礎成分オルガノポリシロキサンとγアミノプロピルビス(メチルエチルケトキシアミノ)メトキシシランのようなメトキシシラン系の接着促進剤を混合しておき、次に4官能性や3官能性のシラン架橋剤及びシリカエアロジル等の増粘剤、その他必要に応じポリマーと架橋剤の反応を促進する有機スズカルボキシレートのような触媒、さらには酸化防止剤、紫外線吸収剤、可塑剤等とともに本発明の抗菌剤粒子を従来公知の方法で得る方法が例示できる。
【0068】
本発明の前記式(1)で表わされる抗菌剤は、前述したように樹脂に配合して種々の成形品として価値のある製品を提供することができる。一方、本発明の抗菌剤は、その分散性、白色性、粒形の均一性および粒子径の均一性を利用して、樹脂以外の用途にも適用することができる。
すなわち、その抗菌性を利用して、抗カビ剤、抗菌消臭剤(スプレー)、抗菌紙、農薬および化粧料にも適用することができる。本発明の抗菌剤は人体に対して安全であり且つ人体に接触しても刺激を与えないので、人間の生活する住宅内の台所、風呂場やトイレなどの製品に対して抗菌剤および抗カビ剤として利用することができる。また農薬として野菜や果物などにも直接散布することができる。さらに化粧料の中に配合しても有利に利用することができる。
【0069】
以下に本発明の該抗菌剤粒子からなる抗菌剤、及び該抗菌剤粒子が配合された抗菌性樹脂組成物、及び該抗菌性樹脂組成物から形成された成形品等の各物性の測定方法を示す。
銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子抗菌剤の粒子性状の測定方法;
最近の微粒子の平均二次粒子径及び粒子径分布の測定は、レーザー回折散乱法が主流になっており、本発明ではこれらの測定はレーザー回折散乱法で行われた。
【0070】
(1)平均二次粒子径;堀場製作所製粒度分布測定装置LA―910(レーザー回折散乱法)を用いて測定した。
【0071】
(2)粒子径分布のシャープ度 Dr=D75/D25
堀場製作所製粒度分布測定装置LA―910を用いて測定されたレーザー回折散乱法体積基準累積粒子径分布曲線の75%値の粒子径D75(大粒子径側)を25%値の粒子径D25(微粒子径側)で除した算式で表される粒子径分布のシャープ度をDr=D75/D25として定義した。 Drの数値が小さい程粒子径分布のシャープ度が認められ粒子径均一性を意味する。
だだし、球形粒子についてはレーザー回折散乱法で測定されたD75/D25と、下記のSEM写真による粒子径分布測定方法と組み合わせて粒子径を評価した。
その場合、レーザー回折散乱法によるD75/D25の数値はSEM写真によるD75/D25の数値と約−10〜+10%の範囲で異なる程度でほぼ一致したので本発明ではレーザー回折散乱法によるD75/D25を採用した。
電子顕微鏡写真(SEM写真)による粒子径分布測定方法(球形粒子の場合);
1枚のSEM写真で観察される全ての粒子(50個〜数百個)の球形粒子をそれぞれ個別に長径と短径をノギスで1/50mmまで測定し長径と短径の平均値を求めて各球形粒子の粒子径(μmに換算)とし、それから累積粒子径のD75%とD25%に該当する粒子径を認定しDr=D75/D25を算出した。
【0072】
(3)BET法比表面積;湯浅アイオニクス(株)製の12検体全自動表面測定装置マルチソーブ−12で測定した。
【0073】
(4)粒子形状;走査型電子顕微鏡(SEM写真)で観察した。
尚粒子形状としての図(写真)のと実施例、比較例の抗菌剤粒子との関係は以下のとおりである。
図1、表面が平滑である球状粒子、A1粒子
図2、表面に小粒を有する球状粒子、A20粒子
図3、表面が荒くさらには皺(球に傷や割れ)を有する球状粒子、A21粒子
図4、穴(凹凸)を有する球状粒子、A22粒子
図5、表面が平滑であり且つ直線部分を図1のものより少し多く含む球状粒子、A30粒子
図6、表面が荒く皺を有する球状粒子、A31粒子
図7、円盤状粒子粒子、B1−1粒子、
図8、一対状粒子(ハンバーガー状粒子)、C1粒子
図9、米粒状粒子、D1粒子、
図10、直方体粒子、E1粒子
図11、六角板状粒子、F1粒子
図12、8面体状粒子、G1粒子
図13、円柱状(酒樽状)粒子、H1粒子
図14、表面が荒く延伸されたような状態の凝集体粒子、V1粒子
【0074】
(5)イオン交換体形成;粉末X線回折法により銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子の回折パターン及びそれ以外の回折パターンの有無を調査した。
【0075】
(6)有機酸の含有量;銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子を1000℃で焼成しCO2を発生させ回収し、JIS R 9101によりCO2を測定し、それに基づいて炭素の含有量を計算しそれから有機酸の含有量を求めた。
抗菌性樹脂組成物、樹脂成形品、フィルム、繊維、塗料、コーキング材等樹脂製品の測定方法;
【0076】
(7)抗菌性試験方法
ただし下記(a)〜(e)の試験において、大腸菌はE.coli NBRC 3972 黄色ブドウ球菌は S.aureus NBRC 12732を使用した。抗菌試験の結果も示す表−7〜表−17においてcfuはcolony forming unitの略である。cfu/mlは1ml中の生菌数を表わし、その数値が小さい程抗菌性が高いことを意味している。
(a)成形板;JIS−Z 2801(2000年版)
テストピースサイズ;50mm×50mm×2mm
(b)繊維、不織布;JIS−L 1902(2002年版)
(c)塗料;塗料を鉄板サイズ50mm×50mm×2mmのテストピース(材質はJIS G 3101 SS400)の上に約0.1mmになるように塗装し上記(a)の方法で測定した。
(d)フィルム;JIS−Z 2801(2000年版)
(e)コーキング材;コーキング材を鉄板サイズ50mm×50mm×2mmのテストピース(材質はJIS G 3101 SS400)の上に約1mmになるようにコーキングし上記(a)の方法で測定した。
【0077】
(8)透明性;厚さ2mmの板又は50μmのフィルムでヘイズ値HをTOKYO DENSYOKU CO,LTDのAUTOMATIC HAZE METER OPTICAL UNIT TC−HIIIDPで測定し下記の算式により%で表示した。
【0078】
H={h1−(h2−h1)}×100/h1 %
h1=抗菌剤未添加の樹脂成形品又はフィルムのヘイズ値
h2=抗菌剤を添加した樹脂成形品又はフィルムのヘイズ値
Hが100%に近い程抗菌剤未添加品に近い透明性を維持していることを意味している。
【0079】
(9)白色性; 成形品、フィルム、繊維、不織布等のサンプルを直射日光のない北側の室内で60日間静置したものがどの程度変色したかを目視により測定した。
【0080】
(10)水道水接触後の抗菌力維持特性;
上記記載の試験資料を、水道水1000mlの中に90℃又は70℃で120時間浸水させた後水道水で十分に水洗し,前記(7)の(a)〜(e)の方法で測定した。尚試験片の比重が1以下のものについてはステンレス鋼SUS−304で作られた金網を錘に使い水中に強制的に浸水した。水道水を90℃又は70℃に加温した理由は水道水接触後の抗菌力維持特性を短時間で見るためであり、これは常温で水道水接触後の抗菌力維持特性を試験する場合の促進試験となる。
【0081】
(11)混練押出加工時フィルター通過性試験(押出機圧力の測定)
プラスチック工学研究所製の30φ2軸混練押出機(スクリュー径30mm)にフィルター(スクリーンメッシュ)を樹脂の流れ方向から見てブレーカープレートの手前に設置した。フィルターは50メッシュのものをブレーカープレート側に、80メッシュのものを中央に、100メッシュのものをホッパー側にそれぞれ1枚づつ設置した。その状態で、該機械をスクリュー回転数が170rpm,吐出量が10Kg/Hrとなるようにして2時間、及び24時間運転し、その時のフィルター手前に設置してある圧力計により圧力を測定した。
圧力はフィルターを通過できない粗大粒子がフィルターに多く目詰まりする程早く高くなりそれはフィルター通過性が悪いことを意味し、それだけフィルターを入れ替える必要が早く生じ工業的には問題である。
この試験において、圧力が200Kg/cm2以上は機械が破損寸前での運転状態であり、150Kg/cm2以上はフィルター交換しないと機械に負荷をかけ過ぎ機械運転に支障をきたす恐れがあることを意味し、100Kg/cm2になるとフィルター交換の準備が必要なことを意味し、100Kg/cm2以下は何の問題もなく運転できることを意味している。
【0082】
機械を運転開始して24時間後の圧力が150Kg/cm2までであれば、一日に1回程度のフィルター交換で済み工業的にはかろうじて何とか意味のあることである。
尚本測定は樹脂95重量%と抗菌剤5重量%の合計100重量%になる抗菌剤配合濃度において測定したもので、表−7〜表−9に示した縦2重線より左側の抗菌剤配合量濃度とは異なる配合条件で別途実施したものである。混練押出する時のその他の条件は縦2重線より左側と同じにした。
【実施例】
【0083】
以下先ず本発明の抗菌剤粒子の製造方法を実施例に基づいて具体的に説明する。
【0084】
尚、以下の実施例の式に於いて式(1)のbの数値が1の場合は1が省略してある。
銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子抗菌剤の製造;
【0085】
<実施例>I−1〜I−47及び<比較例>I−1〜I−9
(A,B,C,D,E,F,G,H粒子の製造)
球状粒子(A粒子)の製造;実施例I−1〜31
実施例I−1
[Ag0.1(NH4)0.9]Al3(C2O4)0.1(SO4)1.9(OH)6・0.5H2Oの製造
下記の原料を使用し、下記の合成方法によりA1粒子を得た。
使用原料 硫酸アンモニウム 264.28g=2モル
蓚酸(H2C2O4・2H2O) 38g=0.3モル
1.04mol/Lの硫酸アルミニウム×1923ml=2.0モル
9.0mol/Lのアンモニア水溶液×894.5ml=8.05モル
合成方法 264.28gの硫酸アンモニウム(NH4)2SO4を4.0Lのイオン交換水に溶解した。
38gの蓚酸(H2C2O4・2H2O)を1.0Lのイオン交換水に溶解した。
【0086】
攪拌下の該硫酸アンモニウム(NH4)2SO4水溶液に該蓚酸水溶液及び上記硫酸アルミニウムAl2(SO4)3水溶液を添加し混合酸性水溶液を作った。
該混合酸性水溶液は良く攪拌しながら、50℃以下に加熱し(析出した結晶物を溶解させるため)、その後上記のアンモニア水溶液を894.5ml を25分かけて該混合酸性水溶液に添加しアンモニウム型の有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子沈殿物のスラリーを作った。
そのスラリーを更に100℃で1時間水熱処理した。
処理後の沈殿物を濾過・水洗・乾燥・粉砕処理し粉末サンプルを得た。
そのサンプル100gをとって0.025mol/Lの硫酸銀水溶液600mlに懸濁させ攪拌して、30℃でアンモニウムイオンと銀イオンとのイオン交換処理を遮光下で8時間行った。
イオン交換後のサンプルを濾過・水洗・乾燥(105℃×6時間)・粉砕した。
これらの処理過程によって、A1粒子を得た。結果を表−1に示す。
【0087】
実施例I−2
[Ag0.1(NH4)0.9]Al3(C2O4)0.1(SO4)1.9(OH)6の製造)
実施例I−1で得たA1粒子を150℃で2時間乾燥し、A2粒子を得た。
【0088】
実施例I−3
[Ag0.1(NH4)0.9]Al3(C2O4)0.0001(SO4)1.9999(OH)6の合成
実施例I−1において、蓚酸の使用量を0.0005モル(0.063g)に変更したことが実施例I−1と異なるのみで、その他の処理過程は実施例I−1に準じて行い、さらに150℃で2時間乾燥しA3粒子を得た。
【0089】
実施例I−4
[Ag0.1(NH4)0.9]Al3(C2O4)0.01(SO4)1.999(OH)6の製造
実施例I−1において、蓚酸の使用量を0.05モル(6.3g)に変更したことが実施例I−1と異なるのみで、その他の処理過程は実施例I−1に準じて行い、さらに150℃で2時間乾燥しA4粒子を得た。
【0090】
実施例I−5
[Ag0.1(NH4)0.9]Al3(C2O4)0.4(SO4)1.7(OH)5.8の製造
実施例I−1において、蓚酸の使用量を1.5モル(189.1g)に変更したことが実施例I−1と異なるのみで、その他の処理過程は実施例I−1に準じて行い、さらに150℃で2時間乾燥しA5粒子を得た。
【0091】
実施例I−6
[Ag0.1(NH4)0.9]Al2.7(C2O4)0.1(SO4)1.9(OH)5.1の製造
実施例I−1において、硫酸アルミニウムの使用量を1731ml(1.8モル)に変更したことが実施例I−1と異なるのみで、その他の処理過程は実施例I−1に準じて行い、さらに150℃で2時間乾燥しA6粒子を得た。
【0092】
実施例I−7 [Ag0.1(NH4)0.9]Al3.3(C2O4)0.1(SO4)1.9(OH)6.9の製造
実施例I−1において、硫酸アルミニウムの使用量を2115ml(2.2モル)に変更したことが実施例I−1と異なるのみで、その他の処理過程は実施例I−1に準じて行い、さらに150℃で2時間乾燥しA7粒子を得た。
【0093】
実施例I−8
[Ag0.1(NH4)0.9]Al3(C2O4)0.1(SO4)2.4(OH)5の製造)
実施例I−1において、硫酸アンモニウム(NH4)2SO4の使用量を2.3モル(303.9g)に変更したことが実施例I−1と異なるのみで、その他の処理過程は実施例I−1に準じて行い、さらに150℃で2時間乾燥しA8粒子を得た。
【0094】
実施例I−9
[Ag0.1(NH4)1.10]1.2Al3(C2O4)0.1(SO4)1.9(OH)6.02の製造
実施例I−1において、アンモニア水溶液添加後の攪拌時間を2時間に変更したこと、添加したアンモニア水溶液の量を1093ml(9.84モル)に変更したこと、及び水熱処理時間を30分に変更したことが実施例I−1と異なるのみで、その他の処理過程は実施例I−1に準じて行い、さらに150℃で2時間乾燥しA9粒子を得た。
【0095】
実施例I−10
[Ag0.1(NH4)0.9]Al3(C2O4)0.1(SO4)1.9(OH)6の製造
実施例I−1において、100℃での水熱処理時間を45分に変更したことが実施例I−1と異なるのみで、その他の処理過程は実施例I−1に準じて行い、さらに150℃で2時間乾燥しA10粒子を得た。
【0096】
実施例I−11
[Ag0.1(NH4)0.76]0.86Al3(C2O4)0.1(SO4)1.9(OH)5.68の製造
実施例I−1において、アンモニア水溶液添加の時間を40分に変更したこと、及び添加したアンモニア水溶液の量を755ml(6.8モル)に変更したことが実施例I−1と異なるのみで、その他の処理過程は実施例I−1に準じて行い、さらに150℃で2時間乾燥しA11粒子を得た。
【0097】
実施例I−12
[Ag0.3(NH4)0.7]Al3(C2O4)0.1(SO4)1.9(OH)6の合成
実施例I−1において、イオン交換用硫酸銀水溶液の量を1800mlに変更したことが実施例I−1と異なるのみで、その他の処理過程は実施例I−1に準じて行い、さらに150℃で2時間乾燥しA12粒子を得た。
【0098】
実施例I−13
[Ag0.001(NH4)0.999]Al3(C2O4)0.1(SO4)1.9(OH)6の製造
実施例I−1において、イオン交換用硫酸銀水溶液を硝酸銀水溶液に変更し、その濃度を0.001mol/L,液量を300mlに変更し処理温度を15℃に変更したことが実施例I−1と異なるのみで、その他の処理過程は実施例I−1に準じて行い、さらに150℃で2時間乾燥しA13粒子を得た。
【0099】
実施例I−14
[Ag0.00001(NH4)0.99999]Al3(C2O4)0.1(SO4)1.9(OH)6 の製造
実施例I−13において、イオン交換用硝酸銀水溶液の濃度を0.00001mol/Lに変更したことが実施例I−13と異なるのみで、その他の処理過程は実施例I−1に準じて行い、さらに150℃で2時間乾燥しA14粒子を得た。
【0100】
実施例I−15
[Ag0.1(NH4)0.9]Al3(C2O4)0.1(SO4)1.9(OH)6・0.5H2Oの製造
実施例I−1で得たサンプル50gをステアリン酸ソーダ1.5g含む80℃水溶液に懸濁させ、30分間攪拌し表面処理を行った後脱水、水洗、乾燥(105℃×6時間)、粉砕しA15粒子を得た。
【0101】
実施例I−16
[Ag0.1(NH4)0.9]Al3(C2O4)0.1(SO4)1.9(OH)6・2H2Oの製造
実施例I−1で得たサンプルを30℃で相対湿度75%のNaCl雰囲気において、25時間の吸湿処理を行い、A16粒子を得た。
【0102】
実施例I−17 実施例1の400℃ 1時間焼成物
実施例I−1で得たサンプルを400℃1時間窒素雰囲気下で焼成処理を行い、A17粒子を得た。
粉末X線回折法によると全て[Ag0.1(NH4)0.9]Al3(C2O4)0.1(SO4)1.9(OH)6であった。形状は球状であった。
A17粒子10.00gをイオン交換水100mlに入れ20℃で30分攪拌し、それを脱水・水洗120℃で16時間乾燥した後の重量減少はなく10.00gであった。
【0103】
実施例I−18 実施例1の500℃ 1時間焼成物
実施例I−1で得たサンプルを500℃1時間窒素雰囲気下で焼成処理を行い、A18粒子を得た。
粉末X線回折法によると全て[Ag0.1(NH4)0.9]Al3(C2O4)0.1(SO4)1.9(OH)6であった。形状は球状であった。
A18粒子10.00gをイオン交換水100mlに入れ20℃で30分攪拌し、それを脱水・水洗し120℃で16時間乾燥した後の重量減少はなく10.00gであった。
【0104】
実施例I−19
[Ag0.001K0.999]Al3(C2O4)0.1(SO4)1.9(OH)6の製造
下記の原料を使用し、下記の方法によりA19粒子を得た。
使用原料 硫酸カリウム 2モル 348g
蓚酸(H2C2O4・2H2O)0.3モル 38g
1.04mol/Lの硫酸アルミニウム水溶液 2.0モル 1923ml
7.5mol/L KOH水溶液 8.4モル 1120ml
合成方法 348gの硫酸カリウム(K2SO4)は4.0Lのイオン交換水に溶解した。
38gの蓚酸(H2C2O4・2H2O) は1.0Lのイオン交換水に溶解した。
攪拌下の該硫酸カリウム(K2SO4)水溶液に該蓚酸水溶液及び上記硫酸アルミニウムAl2(SO4)3水溶液を添加し混合酸性水溶液を作った。
該混合酸性水溶液を良く攪拌して、そして、上記水酸化カリウム水溶液を1120mlを25分かけて該混合酸性水性液に添加しカリウム型の有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子沈殿物スラリーを作った。。
そのスラリーを更に1時間攪拌後、オートクレーブによって、140℃2時間の水熱処理を行った。
処理後の沈殿物を濾過・水洗・乾燥処理し粉末サンプルを得た。
その100gサンプルをとって0.001mol/Lの硝酸銀水溶液300mlに懸濁し攪拌して、カリウムと銀とのイオン交換処理を25℃で1時間遮光下で行った。
イオン交換後のサンプルを濾過・水洗・乾燥(105℃×6時間)・粉砕した。
これらの処理過程を経てさらに200℃で2時間乾燥し、A19粒子を得た。
【0105】
実施例I−20
[Ag0.001K1.179]1.18Al3(SO4)2.26(NO3)0.04(C2O4)0.07(OH)5.48の製造
実施例I−19において、使用した硫酸カリウムを硝酸カリウムKNO3に変更したこと及びが実施例I−19と異なるのみで、その他の処理過程は実施例I−19に準じて行い、A20粒子を得た。
【0106】
実施例I−21
[Ag0.001K1.179]1.18Al3(SO4)2.26(NO3)0.04(C2O4)0.07(OH)5.48の製造
実施例I−19において、K2SO4の代りにKNO3に変更したこと及び水熱処理条件を160°×2時間に変更した実施例I−19と異なるのみで、その他の処理過程は実施例I−19に準じて行い、A21粒子を得た。
【0107】
実施例I−22
[Ag0.001K0.999]Al3(C2O4)0.1(SO4)1.9(OH)6 の製造
実施例I−19において、水熱処理の温度を140℃から170℃に変更したことが実施例I−19と異なるのみで、その他の処理過程は実施例I−19に準じて行い、A22粒子を得た。
【0108】
実施例I−23 [Ag0.1Na0.9]Al3(C6 H5O7)0.067(SO4)1.9(OH)6の製造
下記の原料を使用し、下記の方法によりA23粒子を得た。
使用原料 硫酸ナトリウム 2モル 284.06g
クエン酸(C6H8O7・H2O) 0.3モル 63g
1.04mol/Lの硫酸アルミニウム水溶液 2.0モル 1923ml
3.36mol/L NaOH水溶液 8.2モル 2440.5ml
合成方法 284.06gの硫酸ナトリウム(Na2SO4)を4.0Lのイオン交換水に溶解した。
63gのクエン酸は1.0Lのイオン交換水に溶解した。
攪拌下の該硫酸ナトリウム(Na2SO4)水溶液に該クエン酸水溶液及び上記硫酸アルミニウムAl2(SO4)3水溶液を添加し混合酸性水溶液を作った。
攪拌下の該混合酸性水溶液に上記の水酸化ナトリウム水溶液2440.5mlを 25分かけて添加しナトリウム型の銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子沈殿物スラリーを作った。
そのスラリーを更に10時間攪拌継続後、オートクレーブによって、170℃2時間の水熱処理を行った。水熱処理後の沈殿物を濾過・水洗・乾燥・粉砕処理し粉末サンプルを得た。
その他の処理過程は実施例I−1に準じて行い150℃で2時間乾燥し、A23粒子を得た。
【0109】
実施例I−24
[Ag0.1Na0.9]Al3(C6 H5O7)0.067(SO4)1.9(OH)6 の製造
実施例I−23において、水酸化ナトリウムの添加時間は40分に、及び水酸化ナトリウム添加後の攪拌時間は1時間に変更したことが実施例I−23と異なるのみで、その他の処理過程は実施例I−23に準じて行い、さらに150℃で2時間乾燥しA24粒子を得た。
【0110】
実施例I−25
[Ag0.1Na0.9]Al3(C4 H4O6)0.1(SO4)1.9(OH)6 の製造
下記の原料を使用し、下記の方法によりA25粒子を得た。
使用原料 硫酸ナトリウム 2モル 284.06g
酒石酸(C4H6O6) 0.3モル 45g
1.04mol/Lの硫酸アルミニウム水溶液 2.0モル 1923ml
3.36mol/L NaOH水溶液 8.2モル 2440.5ml
合成方法 284.06gの硫酸ナトリウム(Na2SO4)は4.0Lのイオン交換水に溶解した。
45gの酒石酸(C4H6O6) は1.0Lのイオン交換水に溶解した。
攪拌下の該硫酸カリウム(K2SO4)水溶液に該蓚酸水溶液及び上記硫酸アルミニウムAl2(SO4)3水溶液を添加し混合酸性水溶液を作った。
該混合酸性水溶液を良く攪拌して、そして、上記水酸化ナトリウム水溶液244.05mlを25分かけて該混合酸性水溶液に添加しナトリウム型の銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子沈殿物スラリーを作った。
そのスラリーを更に5時間攪拌後、オートクレーブによって、170℃2時間の水熱処理を行った。
水熱処理後の懸濁液を濾過・水洗・乾燥・粉砕処理した。
その他の処理過程は実施例I−1に準じて行い150℃で2時間乾燥し、A25粒子を作った。
【0111】
実施例I−26
[Ag0.1Na0.9]Al3(C4 H4O6)0.1(SO4)1.9(OH)6 の製造
実施例I−24において、水酸化ナトリウムの量は8モル(2381ml)に変更し、添加の時間も40分に変更したことが実施例I−25と異なるのみで、その他の処理過程は実施例I−25に準じて行い、A26粒子をった。
【0112】
実施例I−27
[Ag0.1(NH4)0.9] Al2.94Zn0.01 Ti0.01(C2O4)0.1(SO4)1.9(OH)6・0.5H2Oの製造
実施例I−1の混合酸性溶液(ただし該混合酸性溶液の該硫酸アルミニウム水溶液使用量は1885mlに減らした)に硫酸亜鉛2.5g,及び硫酸チタン{Ti2(SO4)3}2.5gを含む硫酸溶液を追加添加したこと及び銀イオン交換処理温度を80℃に処理時間を16時間に変更したことが実施例I−1と異なるだけでその他は実施例I−1に準じて行いA27粒子を得た。
【0113】
実施例I−28
[Ag0.1(NH4)0.9 ]Al2.25Zn0.5 Ti0.25(C2O4)0.1(SO4)1.9(OH)6・0.5H2O の製造
実施例I−1の混合酸性溶液(ただし該混合酸性溶液の該硫酸アルミニウム水溶液使用量は1442mlに減らした)に硫酸亜鉛125g,及び硫酸チタン{Ti2(SO4)3}63gを含む硫酸溶液を追加添加したこと及び銀イオン交換処理温度を80℃に処理時間を30時間に変更したことが実施例I−1と異なるだけでその他は実施例I−1に準じて行いA28粒子を得た。
【0114】
実施例I−29
[Ag0.1(NH4)0.9]Al3(C2O4)0.1(SO4)1.15(PO4)0.5(OH)6 の製造
リン酸アンモニウム74.5gを水500mlに溶解させた水溶液に、銀とイオン交換する直前(水熱処理後)のA−1粒子371gを添加して100℃で1時間攪拌しPO43−を該粒子に組み込んだ。その後の処理過程は実施例I−1に準じて行いさらに150℃で2時間乾燥し銀及び有機酸アニオン含有アルミニウム硫酸塩リン酸塩水酸化物A29粒子を得た。
尚この例において、リン酸ナトリウムの代わりに炭酸ナトリウム、硝酸ナトリウム、珪酸ナトリウム、ホウ酸ナトリウムに置き換えて使用したことだけが異なるように試験したがCO32−、NO3−、SiO42−、BO33−それぞれの無機酸イオンを含むA−29粒子同様の粒子性状を有する銀及び有機酸アニオン含有アルミニウム無機酸塩水酸化物粒子が得られた。
【0115】
実施例I−30
[Ag0.1Na0.9]Al3(SO4)1.9(C4H4O5)0.1(OH)6の製造
実施例I−25において、使用した有機酸を酒石酸からDL−林檎酸に変更したことが実施例I−25と異なるのみで、その他の処理過程は実施例I−25に準じて行いA30粒子を得た。
諸特性を表1に示す。このときの粒子形状は図5に示す球状であった。
【0116】
実施例I−31
[Ag0.1Na0.9]Al3(SO4)1.9[C6H2(OH)3COO]0.067(OH)6の製造
実施例I−23において、使用した有機酸をクエン酸から没食子酸[C6H4(OH)3COOH]に変更したことが実施例I−23と異なるのみで、その他の処理過程は実施例I−23に準じて行いA31粒子を得た。諸特性を表1に示す。
このときの粒子形状は図6に示す球状であった。
【0117】
比較例I−1
[Ag0.1(NH4)0.9]Al3(SO4)2(OH)6 の製造
実施例I−1において、蓚酸を使用しなかったことが実施例I−1と異なるのみで、その他の処理過程は実施例I−1に準じて行い、さらに150℃で2時間乾燥しV1粒子を得た。
【0118】
比較例I−2
NH4Al3(C2O4)0.1(SO4)1.9(OH)6の製造
実施例I−1において、銀溶液でイオン交換を行わなかったことが実施例I−1と異なるのみで、その他の処理過程は実施例I−1に準じて行い、さらに150℃で2時間乾燥しV2粒子を得た。
【0119】
比較例I−3
KAl3(C2O4)0.1(SO4)1.9(OH)6の製造
実施例I−19において、銀溶液でイオン交換を行わなかったことが実施例I−19と異なるのみで、その他の処理過程は実施例I−19に準じて行い、V3粒子を得た。
【0120】
【表1】
【0121】
【表2】
【0122】
円盤状粒子(碁石状粒子;B粒子)の製造;(実施例I−32〜34)
実施例I−32−1
[Ag0.1Na0.9 ]Al3(C2O4)0.1(SO4)1.9(OH)6 の製造
下記の原料を使用し、下記の方法によりB1粒子を得た。
使用原料 硫酸ナトリウム 2モル 284.06g
蓚酸(H2C2O4・2H2O) 0.3モル 38g
1.04mol/Lの硫酸アルミニウム水溶液 2.0モル 1923ml
3.36mol/L NaOH水溶液 8.2モル 2440.5ml
合成方法 284.06gの硫酸ナトリウム(Na2SO4)は4.0Lのイオン交換水に溶解した。
38gの蓚酸は1.0Lのイオン交換水に溶解した。
攪拌下の該硫酸ナトリウム(Na2SO4)水溶液に該蓚酸水溶液及び該硫酸アルミニウムAl2(SO4)3水溶液を添加し混合酸性水溶液を作った。
【0123】
該混合酸性水溶液を良く攪拌して、そして、該水酸化ナトリウム水溶液2440.5mlを20分かけて該混合酸性水溶液に添加しナトリウム型の銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子沈殿物スラリーを作った。
そのスラリーを更に5時間攪拌後、オートクレーブによって、170℃2時間の水熱処理を行った。処理後の沈殿物を濾過・水洗・乾燥処理した。そして、そのサンプル100gをとって0.025mol/Lの硫酸銀水溶液600mlに懸濁し攪拌してナトリウムと銀のイオン交換処理を25℃で5時間行った。交換後のサンプルにポリアクリルアミド系の高分子凝集剤(住友化学製 スミフロック FN−20)を0.02g添加し10分間攪拌後、濾過・水洗・乾燥(105℃×6時間)・粉砕した。これらの処理過程を経てさらに200℃で2時間乾燥しB1−1粒子を得た。
【0124】
実施例I−32−2
80℃のイオン交換水1,000mlに2.6gの硫酸亜鉛7水和物(和光純薬工業試薬1級)と2.2gの硫酸アンモニウム(和光純薬工業試薬1級)を入れ混液を作った。その混液の中にB1−1粒子95gを添加して6時間攪拌した後脱水・水洗・乾燥して亜鉛及びアンモニウムで処理されたB1−2粒子を作った。該粒子のZnの含有量は0.1%、NH4の含有率は0.4%BET法比表面積は60m2/gであったが、その他の粒子性状はB1−1粒子と同じであった。
次にA20粒子、C1粒子、D1粒子、E1粒子、F1粒子、G1粒子、H1粒子それぞれについても該処理同様の処理を行い処理物を得た。
処理物のZn含有量は0.1%、NH4の含有量は0.4%でありどれもほぼ同じであった。
処理物のBET法比表面積はどれも処理前の約6倍に増大していたが、平均2次粒子径、粒子径均一性粒子の形状、その他の粒子形状は処理前の粒子性状とほぼ同じであった。
処理物を樹脂に添加した時の樹脂組成物の白色性は処理前の物を添加した場合により一層改善されていた。
【0125】
実施例I−33
[Ag0.1Na0.9 ]Al3(C2O4)0.1(SO4)1.9(OH)6 の製造
実施例I−32において、使用した硫酸ナトリウムを0.5モル/L硫酸ナトリウムを含む反応母液4.0L使用に変更したこと、又、水酸化ナトリウムの添加時間40分に変更したこと、添加後の撹拌時間1時間に変更したことが実施例I−32と異なり、その他の処理過程は実施例I−32に準じて行い、B2粒子を得た。
【0126】
実施例I−34 [Ag0.001Na0.999] Al3(C2O4)0.1(SO4)1.9(OH)6 の製造
実施例I−32において、硫酸銀水溶液の濃度は0.00025モル/Lに変更したことが実施例I−32と異なるのみで、その他の処理過程は実施例I−32に準じて行い、B3粒子を得た。
【0127】
比較例I−4−(1)
NaAl3(C2O4)0.1(SO4)1.9(OH)6 の製造
実施例I−32において、銀イオンとのイオン交換処理を行わないことが実施例I−32と異なるのみで、その他の処理過程は実施例I−32に準じて行い、W1粒子を得た。
【0128】
比較例I−4−(2)
[Ag0.1Na0.9 ]Al3(SO4)2(OH)6 の製造
実施例I−32において、蓚酸を使用しなかったことが実施例I−32と異なるのみで、その他の処理過程は実施例I−32に準じて行い、W2粒子を得た。
【0129】
【表3】
【0130】
一対状(ハンバーガー状)粒子(C粒子)の製造;実施例I−35〜37
実施例I−35 [Ag0.1Na0.9] Al3(C2O4)0.1(SO4)1.9(OH)6 の製造
実施例I−32において、オートクレーブの処理温度は180℃に、処理時間10時間に変更したことが実施例I−32と異なるのみで、その他の処理過程は実施例I−32に準じて行い、C1粒子を得た。
【0131】
実施例 I−36
[Ag0.1Na0.9] Al3(C2O4)0.1(SO4)1.9(OH)6 の製造
実施例I−32において、オートクレーブの処理温度は180℃、処理時間10時間に変更したこと、使用した硫酸ナトリウムは0.5モル/L硫酸ナトリウムを含む反応母液4.0Lを使用したこと、又、水酸化ナトリウムの添加時間は40分に変更したことが実施例I−32と異なるのみで、その他の処理過程は実施例I−32に準じて行い、C2粒子を得た。
【0132】
実施例 I−37
[Ag0.001Na0.999] Al3(C2O4)0.1(SO4)1.9(OH)6 の製造
実施例I−32において、硫酸銀溶液の濃度は0.00025モル/Lに変更したこと、又、オートクレーブの処理温度180℃、処理時間10時間に変更したことが実施例I−32と異なるのみで、その他の処理過程は実施例I−32に準じて行い、C3粒子を得た。
【0133】
比較例I−5−(1) NaAl3(C2O4)0.1(SO4)1.9(OH)6
実施例I−35において、銀イオンとのイオン交換処理を行わないことが実施例I−35と異なるのみで、その他の処理過程は実施例I−35に準じて行い、X1粒子を得た。
【0134】
比較例I−5−(2)
[Ag0.1Na0.9] Al3(SO4)2(OH)6 の製造
実施例I−35において、蓚酸を使用しなかったことが実施例I−35と異なるのみで、その他の処理過程は実施例I−35に準じて行い、X2粒子を得た。
【0135】
【表4】
【0136】
米粒状粒子(D粒子)の製造;実施例I−38〜40
実施例I−38
[Ag0.1Na0.9]Al3(C2H4O6)0.1(SO4)1.9(OH)6の製造
実施例I−23において、クエン酸の代りに酒石酸に変更したことが実施例I−23と異なるのみで、その他の処理過程は実施例I−23に準じて行い、さらに150℃で2時間乾燥しD1粒子を得た。
【0137】
実施例I−39
[Ag0.1Na0.9]Al3(C2H4O6)0.1(SO4)1.9(OH)6の製造
実施例I−23において、クエン酸の代りに酒石酸に変更したこと、又、NaOH水溶液の添加時間35分に変更したことが実施例I−23と異なるのみで、その他の処理過程は実施例I−23に準じて行い、さらに150℃で2時間乾燥しD2粒子を得た。
【0138】
実施例I−40
[Ag0.001Na0.999]Al3(C2O4)0.1(SO4)1.9(OH)6 の製造
実施例I−23において、クエン酸の代りに酒石酸に変更したこと、又、銀イオンの濃度を0.00025モル/Lに変更したが実施例I−23と異なるのみで、その他の処理過程は実施例I−23に準じて行い、さらに150℃で2時間乾燥しD3粒子を得た。
【0139】
比較例I−6−(1)
NaAl3(C2H4O6)0.1(SO4)1.9(OH)6
実施例I−38において、銀イオンとの交換を行わないことに変更したことが実施例I−38と異なるのみで、その他の処理過程は実施例I−38に準じて行い、Y1粒子を得た。
【0140】
比較例I−6−(2)
[Ag0.1Na0.9]Al3(SO4)2(OH)6 の製造
実施例I−38において、酒石酸を使用しなかったことが実施例I−38と異なるのみで、その他の合成処理過程は実施例I−38に準じて行い、さらに200℃で2時間乾燥しY2粒子を得た。
【0141】
【表5】
【0142】
直方体状粒子(E粒子)の製造;実施例I−41〜44
実施例I−41
[Ag0.1(H3O)0.9 ]Al3(C2O4)0.1(SO4)1.9(OH)6 の製造
下記の原料を使用し、下記の方法によりE1粒子を得た。
使用原料 1.04mol/Lの硫酸アルミニウム 2.0モル 1923ml
水酸化アルミニウムAl(OH)3 2.0モル 156.02g
(水酸化アルミニウム;協和化学工業(株)製乾燥水酸化アルミニウムゲルS−100:無定形)
蓚酸(H2C2O4・2H2O) 0.25モル 31.52g
合成方法 上記硫酸アルミニウム水溶液中を撹拌しながら、上記蓚酸を添加し、さらに攪拌しながら上記水酸化アルミニウムAl(OH)3を添加し水素型の有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子沈殿物スラリーを作った。
該スラリーにイオン交換水を加え該スラリーが7.0Lになるように希釈して、更に室温で15時間攪拌後、オートクレーブによって、170℃5時間の水熱処理を行った。
処理後の溶液を濾過・水洗・乾燥・粉砕処理しサンプルを得た。
その他の処理過程は実施例I−1に準じて行いさらに150℃で2時間乾燥し、E1粒子を得た。
【0143】
実施例I−42
[Ag0.1(H3O)0.9 ]Al3(C2O4)0.1(SO4)1.9(OH)6 の製造
実施例I−41において、オートクレーブ処理前の撹拌時間は30分に変更したこと、又オートクレーブ処理温度150℃、処理時間2時間に変更したことが、実施例I−41と異なり、その他の処理過程は実施例I−41に準じて行い、E2粒子を得た。
【0144】
実施例I−43
[Ag0.001(H3O)0.999 ]Al3(C2O4)0.1(SO4)1.9(OH)6
実施例I−41において、交換用銀イオン濃度は0.00025モル/Lに変更したことが、実施例I−41と異なるのみで、その他の処理過程は実施例I−41に準じて行い、E3粒子を得た。
【0145】
実施例I−44
[Ag0.1(H3O)0.9 ]Al3(C2O4)0.1(SO4)1.9(OH)6 の製造
実施例I−41において、オートクレーブ処理前の攪拌時間を2時間に変更したことが、実施例I−41と異なるのみで、その他の処理過程は実施例I−41に準じて行い、E4粒子を得た。
【0146】
比較例I−7−(1)
(H3O)Al3(C2O4)0.1(SO4)1.9(OH)6 の製造
実施例I−41において、銀イオン交換処理を行わないことが、実施例I−41と異なるのみで、その他の処理過程は実施例I−41に準じて行い、Z1粒子を得た。
【0147】
比較例I−7−(2)
[Ag0.1(H3O)0.9 ]Al3(SO4)2(OH)6 の製造
実施例I−41において、蓚酸を使用しなかったことが実施例I−41と異なるのみで、その他の処理過程は実施例I−41に準じて行い、Z2粒子を得た。
【0148】
比較例I−9
比較例I−9のR1粒子は平均2次粒子径1.0μm、Dr=4.5、BET法比表面積4m2/g、銀含有量3%である銀担持リン酸ジルコニウムであるが、R1粒子の粒子性状は表―5に示す。
【0149】
【表6】
【0150】
六角板状粒子(F粒子)の製造;実施例I−45
実施例I−45
[Ag0.1Na0.9]Al3(SO4)1.9(C2O4)0.1 (OH)6の製造
硫酸アルミニウム2モルと硫酸ナトリウム2モルを6000mlの純水に溶解させ、その中に蓚酸31.52g(0.25モル)を入れた。攪拌中の前記混合液に水酸化ナトリウムを8.8モル(352g)含む水溶液1800mlを添加し、さらに室温で30分攪拌したのち、180℃で20時間の水熱処理を行った後、常温まで冷却し濾過水洗し、95℃で15時間乾燥処理して有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子を得た。
その後の処理過程は実施例I−1に準じて行いさらに150℃で2時間乾燥しF1粒子を得た。
諸特性を表6に示す。
このときの粒子形状は図11に示す六角板状であった。
【0151】
8面体状粒子(G粒子)の製造;実施例I−46
実施例I−46
[Ag0.1Na0.9]Al3(SO4)1.9[CH3CH(OH)COO]0.067(OH)6の製造
実施例I−23において、使用した有機酸をクエン酸からL−乳酸[CH3CH(OH)COOH]に変更したことが実施例I−23と異なるのみで、その他の処理過程は実施例I−23に準じて行いG1粒子を得た。諸特性を表6に示す。
このときの粒子形状は図12に示す八面体状であった。
【0152】
円柱状粒子(酒樽状粒子;H粒子)の製造;実施例I−47
実施例I−47
[Ag0.1Na0.9]Al3(SO4)1.9[HOCH2CH(OH)COO]0.067(OH)6の製造
実施例I−23において、使用した有機酸をクエン酸からDL−グリセリン酸[HOCH2CH(OH)COOH]に変更したことが実施例I−23と異なるのみで、その他の処理過程は実施例I−23に準じて行いH1粒子を得た。諸特性を表6に示す。
このときの粒子形状は図13に示す円柱状(酒樽状粒子)であった。
【0153】
【表7】
【0154】
尚前記の実施例IのA1〜A31、B1〜B3、C1〜C3,D1〜D3,E1〜E4、F1,G1,H1の各粒子は高純度の原料(不純物含有量Pb,Cd、As,Baは全て0.1ppm以下、Feは10ppm以下、Mn,Cu,Cr,Niは1ppm以下に精製されたもの)を使用し、且つ設備面では耐腐食性の前記の材質で構成された装置を使用し合成した。
そのため不純物含有量は天然の明礬石とは異なりPb,Cd、As,Baは全て0.1ppm以下、Feは10ppm以下、Mn,Cu,Cr,Niは1ppm以下,Clは100ppm以下であった。
尚不純物含有量は原子吸光法、又はICP−AES法(Inductively Coupled Plasma −Atomic Emission Spectroscopy)、又は蛍光X線法で測定した。
次に本発明の抗菌剤を樹脂に配合した場合の抗菌性樹脂組成物及び抗菌性樹脂製品の製造方法を実施例に基づいて具体的に説明する。尚配合剤の配合量は樹脂100重量部に対するものである。
蛍光増白剤は2,5−チオフェンジイル(5−tert−ブチル−1,3−ベンゾヘキサゾール)はそれぞれの表に示してある量を添加した。
【0155】
ポリプロピレン成形品;
実施例II−1〜32及び比較例II−1〜4
実施例II−1は透明射出成形用グレードポリプロピレン100重量部、表−1記載のA1粒子0.06重量部、蛍光増白剤は2,5−チオフェンジイル(5−tert−ブチル−1,3−ベンゾヘキサゾール)表−1に示す添加量}を予め混合しておき、それを2軸混練押出機を使用し230℃で混練して混和ペレットを得それを230℃で2mmに射出成形して前記各試験用のテストピースを作成し抗菌性(成形直後及び耐水道水接触環境後抗菌力維持特性)、透明性、白色性を前記の樹脂製品の測定方法(7)〜(10)に従って測定した。その結果を表−7の縦2重線より左側に示す。
混練押出加工時フィルター通過性試験(押出機圧力)の試験は上記(11)の方法で行った。
その結果を表−7の縦2重線より右側に示す。
実施例II−2〜32の試験においては実施例II−1で使用した使用抗菌剤粒子及び配合量を表−7に示すように一部変更したが、それ以外は実施例II−1同様にテストピースを作成し各試験を実施した。
その結果を表−7に示す。
【0156】
比較例II−1は有機酸が含まれていない銀含有アルミニウム硫酸塩水酸化物粒子でしかもDrの幅(粒度分布幅)が大きいV1,W2,X2,Y2,Z2粒子であるところ又は銀担持リン酸ジルコニウムであるR1粒子であるところが、比較例II−2の試験は抗菌剤無添加であるところが、比較例II−3は銀を含まないアルミニウム硫酸塩水酸化物粒子V2であるところが、比較例II−4も銀を含まないアルミニウム硫酸塩水酸化物粒子V3粒子であるところが、実施例II−1と異なり、抗菌剤配合量については一部の抗菌剤が表−1に示すように異なるのみで、その他の条件は実施例II−1同様にテストピースを作成し同じ測定を行った。
その結果を表−7に示す。
【0157】
【表8】
【0158】
【表9】
【0159】
【表10】
【0160】
実施例においては、成形直後の抗菌性、水道水接触環境後抗菌力維持特性、透明性、色、混練押出加工時フィルター通過性において優れた特性を有することが確認されたが、一方比較例においてはそれらの特性の中で少なくとも1つ以上の項目に問題があった。
又本実験においては、銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子抗菌剤の平均二次粒子径が微粒子である程抗菌性と透明性が高くなる、BET法比表面積については高いものほど程抗菌性が高くなる、Drの幅(粒度分布幅)は小さく粒子径が均一で有機酸を一定範囲で含んでいるものは抗菌性が高くなる傾向が認められた。
一方比較例について見てみると、比較例II−1において有機酸が含まれていない銀含有アルミニウム硫酸塩水酸化物粒子でしかもDrの幅(粒度分布幅)が大きいV1,W2,X2,Y2,Z2粒子を使用した場合、前記の有機酸が含まれていないことの特徴が現われ抗菌性が不十分で、混練押出加工時フィルター通過性は機械の運転が困難なまで運転直後から悪くなり、また透明性も若干低下していた。
【0161】
比較例II−1においてはリン酸ジルコニウム系のR1粒子を配合したものは、実施例に比べると、水道水接触環境後抗菌力維持特性、及び透明性が劣っていることが認められた。
比較例II−2では抗菌剤を配合しなかったため透明性と色には問題はなかったが、抗菌効果は全く認めらず本発明の目的を達成していないことはいうまでもなかった。
比較例II−4では有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子を300重量部も配合したにも係らず、銀を含有した有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子ではなかったため抗菌効果は全く認められなかった。
つまり、比較例をまとめるとアルミニウム硫酸塩水酸化物粒子を使用しても、比較例II−3及びII−4から解るようにアルミニウム硫酸塩水酸化物粒子であっても銀を含有していないものでは成形品に抗菌性を付与できないことが判明した。
【0162】
ポリスチレン、アクリル樹透明性樹脂成形品
実施例II−33、34及び比較例II−5、6、7
実施例II−33,34は実施例II−1において抗菌化対象樹脂をポリプロピレンから、ポリスチレン、アクリル樹脂等の透明性樹脂に変更し、抗菌剤及び蛍光増白剤配合量を表−8に示すように変更し、混練時、成形時の温度を210℃に変更しただけでそれ以外は実施例II−1に準じておこなった。尚配合剤の添加量は表−8に示す通りである。
比較例II−5は実施例II−33において抗菌剤を添加しなかったこと、比較例II−6は抗菌剤をV2粒子に,比較例II−7は抗菌剤をY1粒子に変更したのみでそれ以外は実施例II−33に準じておこなった。抗菌性、透明性、色の結果を表−8に示す。
下記表−8に示すとおり、実施例の成形品については透明性が損なわれることなく、色も無色(白)であり且つ抗菌性(成形直後及び水道水接触環境後抗菌力維持特性)に関しても優れていることが確認された。
一方比較例においてはアルミニウム硫酸塩水酸化物粒子であっても銀を含有していないため抗菌性が全く発現しなかった。
【0163】
【表11】
【0164】
ポリカーボネート、ポリエチレンテレフタレート、ナイロン6・6、ポリアセタール樹脂成形品
実施例II−35、36及び比較例II−8、9
実施例II−35、36は実施例II−1において抗菌化対象樹脂をポリプロピレンから、ポリカーボネート、ポリエチレンテレフタレート、ナイロン6・6、ポリアセタール樹脂等透明性を有し且つ加工時に水分が少ないことが要求される樹脂に変更し、抗菌剤及び抗菌剤の配合量及び蛍光増白剤の配合量を表−9に示すように変更し、さらに混練時、成形時の温度をそれぞれの樹脂の常識的加工温度(PC,PET,ナイロン6・6では290℃、ポリアセタールでは190℃)に変更し、その他は実施例II−1に準じて試験を行った。
ただし、実施例II−35、36では抗菌剤は乾燥処理したA2,B1,C1,D1,E1,F1,G1,H1粒子又は焼成処理したA17,A18粒子を0.1重量部使用し、比較例8は抗菌剤無添加であり、比較例9はV2粒子の400℃又は500℃焼成品を0.1重量部使用した。抗菌性、透明性、色、押出機圧力の結果を表−9に示す。
無論実施例II−35、36で得られた成形品にはシルバーストリークは発生しなかった。
【0165】
下記表−9に示すとおり、実施例においては成形品の透明性がほとんど損なわれることなく、色も無色(白)であり且つ抗菌性(成形直後及び水道水接触環境後抗菌力維持特性)に関しても優れていることが確認された。
一方比較例においては抗菌性が全く発現しなかった。
【0166】
【表12】
【0167】
フィルム
実施例II−37及び比較例II−10
LDPE樹脂90重量%と抗菌剤A2,B1,C1,D1,E1,F1,G1,H1粒子それぞれを10重量%の合計100重量%を予め加圧ニーダーを用い120℃で15分間混練したものを押出造粒機でホットカットし120℃で直径約3mmのマスターバッチ・ペレットを得た。
【0168】
そのマスターバッチ・ペレットとマスターバッチにする前のLDPE樹脂とを混合し表−10の組成になるようにしたものをTダイ法及びインフレーション法によりそれぞれ厚さ50μmのフィルムを得た。
尚比較例II−10ではマスターバッチにする前のLDPE樹脂のみで同様にフィルムを得た。
そのフィルムについて抗菌性(成形直後及び水道水接触環境後抗菌力維持特性)、透明性、白色性を前記の方法で測定した。その結果を表−10に示す。比較例II−10では抗菌剤無添加のものについてもフィルムを作成し、実施例II−37と同じ測定を行った。その結果を表−10に示す。
下記表−10に示すとおり、実施例のフィルムについては透明性が損なわれることなく、色も無色(白)であり且つ抗菌性(成形直後及び水道接触用環境後抗菌力維持特性)に関しても優れていることが確認された。
一方比較例においては抗菌性が全く発現しなかった。
【0169】
実施例II−38
ポリプロピレン、LDPE,HDPE,アイオノマー樹脂、ナイロン6/66共重合樹脂、PET樹脂,AS樹脂のそれぞれの樹脂100重量部につき、2軸混練押出機を用いA2粒子0.1重量部の混和ペレットを得た。
そのペレットをTダイ法によりそれぞれ厚さ50μmのフィルムを得た。
そのフィルムについて抗菌性(成形直後及び水道水接触環境下抗菌力維持特性)、透明性、白色性を前記の方法で測定した。その結果を表−10に示す。
実施例で得られた各フィルムについては透明性が損なわれることなく、色も無色(白)であり且つ抗菌性(成形直後及び水道水接触環境後抗菌力維持特性)に関しても優れていることが確認された。
一方比較例においては抗菌性が全く発現しなかった。
【0170】
【表13】
【0171】
PVC成形品
実施例II−39及び比較例II−11
実施例II−39ではポリ塩化ビニル樹脂100重量部、表−11に示す抗菌剤粒子0.1重量部、オクチル錫メルカプト1.2重量部、グリセリンリシノレート0.8重量部、モンタン酸エステル0.4重量部からなる組成物をオープンロールを用い180℃で3分間混練したものを圧縮成形機により180℃で厚さ2mmの成形板を得た。
その成形板について抗菌性(成形直後及び水道水接触環境後抗菌力維持特性)を前記の方法で測定した。その結果を表−11に示す。
比較例II−11では同様に抗菌剤無添加のものについて厚さ2mmの成形板にし、同じ測定を行った。その結果を表−11に示す。
下記表−11に示すとおり、実施例の成形板については抗菌性(成形直後及び水道水接触環境後抗菌力維持特性)及び透明性が優れていることが確認された。一方比較例においては抗菌性が全く発現しなかった。
【0172】
【表14】
【0173】
熱硬化性樹脂成形品
実施例II−40及び比較例II−12、13
実施例II−40では不飽和ポリエステル樹脂100重量部、表−12に示す抗菌剤粒子1重量部、硬化剤(チバスペシャルケミカル社製HY951)3重量部、ステアリン酸1重量部、酸化防止剤(チバスペシャルケミカル社製イルガノックス1010)0.5重量部、蛍光増白剤0.001重量部、平均二次粒子径30μm、BET法比表面積1m2/gの人造代理石用途水酸化アルミニュウム150重量部をニーダーで混練して、それを90℃で15分間硬化させ厚さ2mmの板を得た。
【0174】
その成形板について抗菌性(成形直後及び水道水接触環境後抗菌力維持特性)を前記の方法で測定した。その結果を表−12に示す。
比較例II−12、13では実施例II−40同様に抗菌剤無添加のも及びR1粒子についてもそれぞれ個別に厚さ2mmの成形板にし、同じ測定を行った。その結果を表−12に示す。
実施例の成形板について抗菌性(成形直後及び水道水接触環境後抗菌力維持特性)が優れていることが確認された。一方比較例II−12においては抗菌性が全く発現しなかった。
比較例II−13においては成形直後の抗菌性は認められるものの、水道水接触環境下抗菌力維持特性が全くないことが判明した。
【0175】
実施例II−41
実施例II−41は実施例II−40において抗菌剤使用粒子がE2粒子に変更されたこと、及び配合量が300重量部に変更されたこと、ステアリン酸が3重量部に変更された以外は実施例II−40同様に成形板を作成し、同じ測定を行った。その結果を表−12に示す。
成形板については抗菌性(成形直後及び水道水接触環境後抗菌力維持特性)が優れていることが確認された。
【0176】
実施例II−42
実施例II−40において抗菌化対象樹脂を不飽和ポリエステル樹脂からフェノール樹脂、メラミン樹脂、エポキシ樹脂へ変更、抗菌剤をE4粒子に変更し、その他は実施例II−40に準じて成形板を作成し、同じ測定を行った。その結果を表−12に示す。得られた該成形板については抗菌性(成形直後及び水道水接触環境後抗菌力維持特性)が優れていることが確認された。
【0177】
実施例II−40〜42及び比較例II−12、13をまとめると以下のことが理解できる。
本発明の熱硬化性樹脂抗菌性製品は手洗い、風呂等水場で使用される人造大理石用途においても抗菌性(水道水接触環境後抗菌力維持特性)を長時間失うことがなく好適に使用できることを示している。
一方従来技術の銀担持リン酸ジルコニュムを配合した比較例II−13では成形直後の抗菌性は認められるものの、水道水接触環境下抗菌力維持特性が全くなりそのような用途では意味がないことを示している。
【0178】
【表15】
【0179】
ゴム成形品
実施例II−43及び比較例II−14
EPDM(エチレン/プロピレン比=50/50)100重量部、表−13に示す抗菌剤粒子0.5重量部、ディクミルパーオキサイド3重量部、ポリ(2,2,4−トリメチル−1,2ディヒドロキノリン)0.5重量部、シランカップリング剤(日本ユニカ製A−172)1重量部、ステアリン酸0.5重量部、イオウ1重量部、蛍光増白剤0.001重量部、ルチル型酸化チタン5重量部からなる組成物をオープンロールを用い50℃で混練し、それを1日後に160℃で30分加硫し厚さ2mmの成形板を得た。
【0180】
その成形板について抗菌性(成形直後及び水道水接触環境後抗菌力維持特性)を前記の方法で測定した。その結果を表−13に示す。
比較例II−14では同様に抗菌剤無添加のものについて個別に厚さ2mmの成形板にし、同じ測定を行った。その結果を表−13に示す。
実施例の成形板については抗菌性(成形直後及び水道水接触環境後抗菌力維持特性)が優れていることが確認された。一方比較例II−14においては抗菌性が全く発現しなかった。
【0181】
実施例II−44
実施例II−43において抗菌剤使用粒子がA14粒子に変更されたこと、及び配合量が200重量部に変更されたこと、ステアリン酸が2重量部に変更された以外は実施例II−43同様に成形板を作成し、同じ測定を行った。その結果を表−13に示す。
成形板については抗菌性(成形直後及び水道水接触環境後抗菌力維持特性)が優れていることが確認された。
【0182】
【表16】
【0183】
繊維
実施例II−45、比較例15
繊維用ポリプロピレン100重量部、抗菌剤A2粒子を2重量部を2軸混練押出機を用いて予め混練しておき、それを300メッシュのスクリーンを付設した押出機を用い溶融法により100デニルに紡糸した繊維を得た。その繊維について抗菌性(成形直後及び水道水接触環境後抗菌力維持特性)を前記の方法で測定した。その結果を表−14に示す。
比較例II−15では同様に抗菌剤無添加のものについて100デニルの繊維にし、同じ測定を行った。その結果を表−14に示す。
実施例の繊維については抗菌性(紡糸直後及び水道水接触環境後抗菌力維持特性)が優れていることが確認された。
このことは例えば手袋、ソックス等の繊維製品において数十回、数百回洗濯しても抗菌性を失わないあるいは失い難いことを意味している。
尚前記の押出溶融法において実施例においては該スクリーンに粗大粒子が詰まって紡糸作業に支障をきたすことはなかった。
【0184】
【表17】
【0185】
不織布
実施例II−46、比較例II−16
実施例II−45及び比較例II−15で得られた各ポリプロピレン繊維を抄造ウエブ法及びランダムウエブ法により0.06g/cm3の密度に不織布を作成し前記の抗菌性の試験を行ったが実施例においては優れた抗菌性効果(成形直後及び水道水接触環境後抗菌力維持特性)が得られた。
一方比較例においては抗菌性が全く発現しなかった。結果を表−15に示す。
【0186】
【表18】
【0187】
塗料
実施例II−47、比較例II−17
冷却設備を有する設備の中に、メチルメタクリレート60重量部、2−エチルヘキシルアクリレート40重量部の合計100重量部に対し、トリエチレングリコールジメタクリレート3重量部、ジアルキルフタレート10重量部、ハイドロキノン0.003重量部、融点46℃のパラフィンワックス0.5重量部、融点54℃のパラフィンワックス0.5重量部、N,N−ジ8ヒドロキシプロピル9−Pトルイジン0.7重量部を投入し攪拌しながらメチルメタクリレートとn−ブチルメタクリレートの共重合体(Tg=66℃、Mw=40,000)25重量部を除々に加え60℃で2時間攪拌し30℃まで冷却した。
【0188】
このものに実施例I−2のA2粒子1重量部、着色剤三菱レイヨン製トナーP−400を7重量部、骨材三菱レイヨン製KM17を300重量部、重合開始剤ジアシルパーオキサイド2重量部からなる塗料を20℃で1時間放置し塗幕を作り前記(7)の(c)の方法により抗菌のテストを実施した。その結果を表−16に示す。
比較例II−17では抗菌剤無添加のものについて実施例II−47同様に塗布をし、同じ測定を行った。その結果を表−16に示す。
実施例の塗料については抗菌性((成形直後及び水道水接触環境後抗菌力維持特性)が優れていることが確認された。一方比較例においては抗菌性が全く発現しなかった。
【0189】
【表19】
【0190】
コーキング材
実施例II−48及び比較例II−18
シラノールを末端基とする25℃の粘度が50、000センチポイズのポリジメチルシロキサン100重量部に対し1重量部のγアミノプロピルビス(メチルエチルケトキシアミノ)メトキシシランを真空下で10分間混合し、ついでその混合物に5重量部のメチルトリス(メチルエチルケトキシアミノ)シランを加え真空下15分間混合し、BET法比表面積200m2/gのヒュームドシリカ5重量部、ジブチルスズラウレート0.1重量部、抗菌剤A2粒子1重量部を加え真空下で10分間混合した。
この混合された組成物をポリエチレンシートの上に垂らして厚さ2mmにし抗菌性テスト用のテストピースを得た。それを前記の抗菌性試験法(7)の(e)で塗布し抗菌のテストを実施した。その結果を表−17に示す。比較例では同様に抗菌剤無添加のものについてコーキング材にし、同じ測定を行った。その結果を表−17に示す。
実施例のコーキング材については抗菌性(成形直後及び水道水接触環境後抗菌力維持特性)が優れていることが確認された。一方比較例においては抗菌性が全く発現しなかった。
【0191】
【表20】
【0192】
尚前記の実施例II−1〜II−48の各成形品、フィルム等を一旦900℃で焼成した灰分を硫酸及び硝酸に溶解し溶液とし、それから各実施例の成形品、フィルム等に含まれる重金属を原子吸光法、又はICP−AE法(Inductively Coupled Plasma −Atomic Emission Spectroscopy)、又は蛍光X線法で測定したが、実施例II−1〜II−48の各樹脂成形品、フィルム、繊維等に含まれるPb、Cd、As,Baは全て0.1ppm以下で、且つFeは5ppm以下、Mn,Cu,Cr,Niはそれぞれ1ppm以下であった。
従って本発明の抗菌性樹脂組成物及びそれから形成された製品は、安全性が高いだけだけでなく、さらには耐熱劣化性にも優れた材料であることが判明した。
【0193】
抗カビ剤
実施例II−49、比較例II−19およびII−20
培地が感性MHB培地から日水製薬製ポテトデキストロース寒天培地に変更したことだけが異なる日本化学療法学会標準法(2003年改訂版)において最小発育阻止濃度を本発明の抗カビ剤粒子、及び比較例の粒子について抗カビ性能を測定し最小発育阻止濃度としてppmで示した。
この数値が小さいほど抗カビ性能が高いことを示している。
測定の結果を下記表−18に示す。
試供カビは ※1:Caldosporium Caldosporides NBRC 6348 (黒カワカビ)、
※2:Colletotricum coccodes NBRC 5256 (ナス黒点根腐病原菌)、
※3:Ustilaginoidia virens NBRC 9175 (稲こうじ病原菌)を使用した。
【0194】
【表21】
【0195】
本発明抗カビ剤は比較例の抗カビ剤よりはるかに優れた抗カビ性能を示すことが判明した。
【0196】
化粧料
実施例II−50、比較例II−21
下記表−19に示す実施例と比較例の配合成分を用いた油中水型のクリーム状の乳化化粧料を調整した。表中1〜5は油性材料、6、7は界面活性剤、10抗菌剤とを混合したものをAとし、一方、表中の物質8精製水と,9の保湿剤を混合したものを混合物Bとし、各々の混合物A,Bを70℃に加熱した後、混合物A中に混合物Bを攪拌しながら流し入れ、乳化させその後冷却した。
抗菌性テストは乳化30日後に大腸菌E.coli NBRC 3972 を使用して前記(7)の(c)に準じて実施した。さらに実施例と比較例のクリームを日頃脇に悪臭の有る人の脇下に1g塗って8時間後に悪臭のあるかないかを10人に嗅いでもらった。
測定の結果を下記表−19に示す。
【0197】
【表22】
【0198】
実験の結果実施例では抗菌性に優れ且つ悪臭を抑える効果が高いことが判明した。
また本発明においては化粧料を顔に塗った時の感触としてのざらつき感がいずれもなかったが、特に円盤状粒子(A1粒子)においては非常になめらかな塗り心地であった。
一方、比較例においては実施例の抗菌剤がR1粒子に置き換えられたこと、又は抗菌剤が全く添加されなかったことが異なるのみであるが、抗菌性は無く且つ悪臭防止効果も無かったことが判明した。
【0199】
抗菌消臭スプレー剤
実施例II−51、比較例II−21およびII−22
70℃に加温した容器の中に2.3重量部のジプロピレングリコールを入れ、さらに2.5重量部のラウリン酸と1重量部のミリスチン酸、さらに3.2重量部のトリエタノールアミンを加えて溶液を作成した。
次にこの溶液を70℃に加温した90重量部のイオン交換水に徐々に注加して乳化させ泡基材溶液を調製し、それを25℃まで冷却した。
該泡基材溶液に本発明の抗菌剤粒子1重量部添加して攪拌、混合し抗菌消臭泡沫状エアゾール組成物を調製した。
この組成物180gと噴射剤(LPG 0.34MPa)20gをブリキ缶のスプレー式容器に充填して抗菌消臭泡沫状エアゾールスプレーを作成した。
一方、日頃脇に悪臭の有る人に充分運動してもらった後で15cm×15cmの綿製のハンカチが0.5g、及び5g重くなるように汗を採取した。該ハンカチの表と裏に該抗菌消臭泡沫状エアゾールスプレーをそれぞれ1gづつ噴霧し、恒温槽に30℃で10日間静置した後で10人に悪臭を嗅いでもらった。測定の結果を下記表−20に示す。
【0200】
【表23】
【0201】
本発明抗菌剤は比較例の抗菌剤よりはるかに優れた悪臭防止性能を示すことが判明した。
【0202】
抗菌紙
実施例II−52、比較例II−23およびII−24
さらしケミパルプ82%の中に、本発明の抗菌剤1%、デンプン(乾燥紙力増強剤)5%、尿素−ホルムアルデヒド樹脂(湿潤紙力増強剤)5%、ニ酸化チタン(無機填料)2%、ポリアミド系樹脂を主成分とするインキ(接着性バインダー)5%を混合し、抄紙機を用いて厚さ0.1mmの紙を抄いた。この紙を5cm×5cmに切り取り前記の抗菌性試験方法(7)の(a)同様のテストを実施した。測定の結果を下記表−21に示す。
【0203】
【表24】
【0204】
本発明抗菌剤は比較例の抗菌剤よりはるかに優れた抗菌性能を示すことが判明した。
【0205】
農薬(抗カビ剤)
実施例II−53、比較例II−25およびII−26
実施例では本発明の粒子10重量部、シランカップリング剤表面改質軽質炭酸カルシウム(無機微粉体)30重量部、ポリオキシエチレンアルキルアリルエーテル(界面活性剤)5重量部、エチレングリコール(界面活性剤)10重量部、キサンタンガム(乳化安定剤)0.2重量部、水44.8重量部をホモミキサーで均一に混合した後、ボールミルで均一に湿式粉砕して水性懸濁状農薬組成物をえた。これを水で1/100に希釈して市販のプラスチック製スプレー装置に入れた。
比較例は実施例の粒子が下記の粒子、又はボルドー撒粉に置き換えられたことが異なるだけでその他の実験方法は実施例の要領に準じた。
【0206】
一方、それぞれ約20cmに成長したナス、及び稲を用意した。
ナスには1×106個/mlに調製したColletotricum coccodes NBRC 5256 (ナス黒点根腐病原菌)懸濁液1gづつを葉、茎、根元に噴霧し、その1日後に前記の1/100に希釈された該水性懸濁状農薬組成物1gを同様に噴霧し、さらにその30日後にナス黒点根腐病の発生の程度を観察した。ただしナスの試験は直径33cm、深さ30cmの鉢に土が27cmの高さになるように入れて実施した。
稲には1×106個/mlに調製したUstilaginoidia virens NBRC 9175 (稲こうじ病原菌)懸濁液を1gづつを葉、茎、根元に噴霧し、その1日後に前記の1/100に希釈された該水性懸濁状農薬組成物1gを同様に噴霧し、さらにその30日後に稲こうじ病の発生の程度を観察した。ただし稲の試験は直径33cm、深さ30cmの鉢に土が27cmの高さになるように入れて土壌の水が表面からかろうじて切れるが土壌中には十分水が存在する条件下で実施した。測定の結果を下記表−22に示す。
【0207】
【表25】
【0208】
本発明農薬組成物は比較例の農薬組成物よりはるかに優れた農薬性能を示すことが判明した。
【図面の簡単な説明】
【0209】
【図1】実施例I−1における球状粒子(粒子名:A1)のSEM写真である。
【図2】実施例I−20における球状粒子(粒子名:A20)のSEM写真である。
【図3】実施例I−21における球状粒子(粒子名:A21)のSEM写真である。
【図4】実施例I−22における球状粒子(粒子名:A22)のSEM写真である。
【図5】実施例I−30における球状粒子(粒子名:A30)のSEM写真である。
【図6】実施例I−31における球状粒子(粒子名:A31)のSEM写真である。
【図7】実施例I−32−1における円盤状粒子(粒子名:B1−1)のSEM写真である。
【図8】実施例I−35における一対状粒子(粒子名:C1)のSEM写真である。
【図9】実施例I−38における米粒状粒子(粒子名:D1)のSEM写真である。
【図10】実施例I−41における直方体状粒子(粒子名:E1)のSEM写真である。
【図11】実施例I−45における六角板状粒子(粒子名:F1)のSEM写真である。
【図12】実施例I−46における八面体状粒子(粒子名:G1)のSEM写真である。
【図13】実施例I−47における円柱状粒子(粒子名:H1)のSEM写真である。
【図14】比較例I−1における凝集状粒子(粒子名:V1)のSEM写真である。
【特許請求の範囲】
【請求項1】
下記式(1)で表される銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子よりなる抗菌剤。
(AgaBb−a)bAlcAx(SO4)y(OH)z・pH2O (1)
式(1)中,a,b,c,x,y,z及びpは0.00001≦a<0.5、 0.7≦b≦1.35、2.7<c<3.3、 0.001≦x≦0.5, 1.7<y<2.5、4<z<7、0≦p≦5、BはNa+、NH4+、K+及びH3O+の群から選ばれた少なくとも1種の1価陽イオンを表わし、陽イオンの価数×モル数の合計値(1b+3c)の範囲が8<(1b+3c)<12であり、Aは有機酸アニオンを表す。
【請求項2】
請求項1記載の有機酸アニオンAが有機カルボン酸又は有機オキシカルボン酸に基づくアニオン群から選ばれる少なくとも1種であることを特徴とする請求項1記載の抗菌剤。
【請求項3】
請求項1記載の有機酸アニオンAが、炭素数1〜15を有する、有機カルボン酸又は有機オキシカルボン酸に基づくアニオン群から選ばれる少なくとも1種であることを特徴とする請求項1記載の抗菌剤。
【請求項4】
式(1)中BはNa+,H3O+,及びNH4+の群から選ばれた少なくとも1種の1価陽イオンである請求項1記載の抗菌剤。
【請求項5】
式(1)中AlcにおいてAlのcモルをZn2+及び/又はTi4+の合計モル数で計算してアルミニウムcモルの1/2以下をZn2+及び/又はTi4+で置き換えたことからなる請求項1記載の抗菌剤。
【請求項6】
式(1)中(SO4)yのSO42−の一部を、PO43−、CO32−、NO3−、SiO44−及びBO33−の群から選ばれた少なくとも1種の他の無機酸イオンで置き換えた請求項1記載の抗菌剤。
【請求項7】
該粒子は、下記(i)及び(iiの粒子性状を具備する請求項1記載の抗菌剤。
(i)レーザー回折散乱法で測定された平均二次粒子径が0.1μm〜12μm、
(ii)BET法比表面積が0.1〜250m2/g。
【請求項8】
該粒子は、(iii)Dr=D75/D25(D25がレーザー回折散乱法による体積基準累積粒子径分布曲線の25%値の粒子径を表し、D75がその75%値の粒子径を表す)で定義される粒子径分布のシャープ度が1.0≦Dr≦1.8の範囲である請求項1記載の抗菌剤。
【請求項9】
該粒子は、SEM写真で観察された粒子形状が球状、円盤状(碁石状)、一対状(ハンバーガー状)、米粒状、直方体状、六角板状、円柱状(酒樽状)、八面体状のいずれかである請求項1記載の抗菌剤。
【請求項10】
該粒子は、レーザー回折散乱法で測定された平均二次粒子径が0.1〜5μmである請求項1記載の抗菌剤。
【請求項11】
該粒子は、BET法比表面積が1〜100m2/gである請求項1記載の抗菌剤。
【請求項12】
式(1)中、a,b,c,x,y,z及びpは0.001≦a<0.3,0.9≦b≦1.2,2.7<c<3.3、0.001≦x≦0.2,1.7<y<2.3,5<z<7,0≦p<3を満足し、BはNa+、NH4+,H3O+の群から選ばれた少なくとも1種の1価陽イオンを表わし、陽イオンの価数×モル数(1b+3c)の範囲が9<(1b+3c)<11を満足する請求項1記載の抗菌剤。
【請求項13】
式(1)中Aは蓚酸イオン、クエン酸イオン、リンゴ酸イオン、酒石酸イオン、グリセリン酸イオン、没食子酸イオン及び乳酸イオンの群から選ばれる少なくとも1種である有機酸アニオンを表わす請求項1記載の抗菌剤。
【請求項14】
請求項1記載の抗菌剤の表面が、高級脂肪酸類、シラン系カップリング剤、チタネート系カップリング剤、アルミネート系カップリング剤、アルコールリン酸エステル類、界面活性剤類等の群から選ばれた少なくとも1種で表面処理された請求項1記載の抗菌剤。
【請求項15】
硫酸アルミニウムと、1価陽イオン硫酸塩及び/又は硝酸塩との混合水溶液に、1価の陽イオンからなるアルカリ水溶液と、及び有機酸とを添加して加熱反応することにより有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子を生成した後、得られた粒子と銀を含む水溶液とを接触攪拌して該粒子の陽イオンの一部を銀とイオン交換することを特徴とする請求項1記載の銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子抗菌剤の製造方法。
【請求項16】
請求項1記載の抗菌剤が樹脂100重量部に対し0.001〜300重量部配合されて抗菌性を有する樹脂組成物。
【請求項17】
請求項1記載記載の抗菌剤が樹脂100重量部に対し0.001〜2重量部配合されて透明性に優れたことを特徴とする抗菌性樹脂組成物。
【請求項18】
請求項16記載の樹脂組成物から形成された抗菌性樹脂成形品。
【請求項19】
請求項16記載の樹脂組成物から形成された抗菌性フィルム。
【請求項20】
請求項16記載の樹脂組成物から形成された抗菌性繊維。
【請求項21】
請求項16記載の樹脂組成物から形成された抗菌性不織布。
【請求項22】
請求項16記載の樹脂組成物から形成された抗菌性塗料。
【請求項23】
請求項16記載の樹脂組成物から形成された抗菌性コーキング材。
【請求項24】
蛍光増白剤が樹脂100重量部に対して0.000001〜0.1重量部添加されて白色度が改善された請求項16〜23いずれかに記載の抗菌性樹脂製品。
【請求項25】
請求項1記載の抗菌剤を含む抗カビ剤。
【請求項26】
請求項1記載の抗菌剤を含む化粧料。
【請求項27】
請求項1記載の抗菌剤を含む抗菌紙。
【請求項28】
請求項1記載の抗菌剤を含む抗菌消臭スプレー。
【請求項29】
請求項1記載の抗菌剤を含む農薬。
【請求項1】
下記式(1)で表される銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子よりなる抗菌剤。
(AgaBb−a)bAlcAx(SO4)y(OH)z・pH2O (1)
式(1)中,a,b,c,x,y,z及びpは0.00001≦a<0.5、 0.7≦b≦1.35、2.7<c<3.3、 0.001≦x≦0.5, 1.7<y<2.5、4<z<7、0≦p≦5、BはNa+、NH4+、K+及びH3O+の群から選ばれた少なくとも1種の1価陽イオンを表わし、陽イオンの価数×モル数の合計値(1b+3c)の範囲が8<(1b+3c)<12であり、Aは有機酸アニオンを表す。
【請求項2】
請求項1記載の有機酸アニオンAが有機カルボン酸又は有機オキシカルボン酸に基づくアニオン群から選ばれる少なくとも1種であることを特徴とする請求項1記載の抗菌剤。
【請求項3】
請求項1記載の有機酸アニオンAが、炭素数1〜15を有する、有機カルボン酸又は有機オキシカルボン酸に基づくアニオン群から選ばれる少なくとも1種であることを特徴とする請求項1記載の抗菌剤。
【請求項4】
式(1)中BはNa+,H3O+,及びNH4+の群から選ばれた少なくとも1種の1価陽イオンである請求項1記載の抗菌剤。
【請求項5】
式(1)中AlcにおいてAlのcモルをZn2+及び/又はTi4+の合計モル数で計算してアルミニウムcモルの1/2以下をZn2+及び/又はTi4+で置き換えたことからなる請求項1記載の抗菌剤。
【請求項6】
式(1)中(SO4)yのSO42−の一部を、PO43−、CO32−、NO3−、SiO44−及びBO33−の群から選ばれた少なくとも1種の他の無機酸イオンで置き換えた請求項1記載の抗菌剤。
【請求項7】
該粒子は、下記(i)及び(iiの粒子性状を具備する請求項1記載の抗菌剤。
(i)レーザー回折散乱法で測定された平均二次粒子径が0.1μm〜12μm、
(ii)BET法比表面積が0.1〜250m2/g。
【請求項8】
該粒子は、(iii)Dr=D75/D25(D25がレーザー回折散乱法による体積基準累積粒子径分布曲線の25%値の粒子径を表し、D75がその75%値の粒子径を表す)で定義される粒子径分布のシャープ度が1.0≦Dr≦1.8の範囲である請求項1記載の抗菌剤。
【請求項9】
該粒子は、SEM写真で観察された粒子形状が球状、円盤状(碁石状)、一対状(ハンバーガー状)、米粒状、直方体状、六角板状、円柱状(酒樽状)、八面体状のいずれかである請求項1記載の抗菌剤。
【請求項10】
該粒子は、レーザー回折散乱法で測定された平均二次粒子径が0.1〜5μmである請求項1記載の抗菌剤。
【請求項11】
該粒子は、BET法比表面積が1〜100m2/gである請求項1記載の抗菌剤。
【請求項12】
式(1)中、a,b,c,x,y,z及びpは0.001≦a<0.3,0.9≦b≦1.2,2.7<c<3.3、0.001≦x≦0.2,1.7<y<2.3,5<z<7,0≦p<3を満足し、BはNa+、NH4+,H3O+の群から選ばれた少なくとも1種の1価陽イオンを表わし、陽イオンの価数×モル数(1b+3c)の範囲が9<(1b+3c)<11を満足する請求項1記載の抗菌剤。
【請求項13】
式(1)中Aは蓚酸イオン、クエン酸イオン、リンゴ酸イオン、酒石酸イオン、グリセリン酸イオン、没食子酸イオン及び乳酸イオンの群から選ばれる少なくとも1種である有機酸アニオンを表わす請求項1記載の抗菌剤。
【請求項14】
請求項1記載の抗菌剤の表面が、高級脂肪酸類、シラン系カップリング剤、チタネート系カップリング剤、アルミネート系カップリング剤、アルコールリン酸エステル類、界面活性剤類等の群から選ばれた少なくとも1種で表面処理された請求項1記載の抗菌剤。
【請求項15】
硫酸アルミニウムと、1価陽イオン硫酸塩及び/又は硝酸塩との混合水溶液に、1価の陽イオンからなるアルカリ水溶液と、及び有機酸とを添加して加熱反応することにより有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子を生成した後、得られた粒子と銀を含む水溶液とを接触攪拌して該粒子の陽イオンの一部を銀とイオン交換することを特徴とする請求項1記載の銀及び有機酸アニオン含有アルミニウム硫酸塩水酸化物粒子抗菌剤の製造方法。
【請求項16】
請求項1記載の抗菌剤が樹脂100重量部に対し0.001〜300重量部配合されて抗菌性を有する樹脂組成物。
【請求項17】
請求項1記載記載の抗菌剤が樹脂100重量部に対し0.001〜2重量部配合されて透明性に優れたことを特徴とする抗菌性樹脂組成物。
【請求項18】
請求項16記載の樹脂組成物から形成された抗菌性樹脂成形品。
【請求項19】
請求項16記載の樹脂組成物から形成された抗菌性フィルム。
【請求項20】
請求項16記載の樹脂組成物から形成された抗菌性繊維。
【請求項21】
請求項16記載の樹脂組成物から形成された抗菌性不織布。
【請求項22】
請求項16記載の樹脂組成物から形成された抗菌性塗料。
【請求項23】
請求項16記載の樹脂組成物から形成された抗菌性コーキング材。
【請求項24】
蛍光増白剤が樹脂100重量部に対して0.000001〜0.1重量部添加されて白色度が改善された請求項16〜23いずれかに記載の抗菌性樹脂製品。
【請求項25】
請求項1記載の抗菌剤を含む抗カビ剤。
【請求項26】
請求項1記載の抗菌剤を含む化粧料。
【請求項27】
請求項1記載の抗菌剤を含む抗菌紙。
【請求項28】
請求項1記載の抗菌剤を含む抗菌消臭スプレー。
【請求項29】
請求項1記載の抗菌剤を含む農薬。
【図1】


【図2】

【図3】

【図4】

【図5】

【図6】

【図7】

【図8】

【図9】

【図10】

【図11】
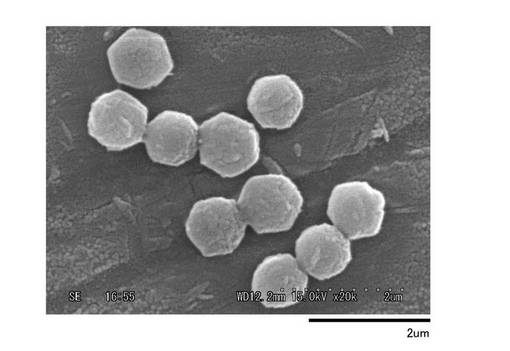
【図12】

【図13】

【図14】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【公開番号】特開2007−39442(P2007−39442A)
【公開日】平成19年2月15日(2007.2.15)
【国際特許分類】
【出願番号】特願2006−179771(P2006−179771)
【出願日】平成18年6月29日(2006.6.29)
【出願人】(000162489)協和化学工業株式会社 (66)
【Fターム(参考)】
【公開日】平成19年2月15日(2007.2.15)
【国際特許分類】
【出願日】平成18年6月29日(2006.6.29)
【出願人】(000162489)協和化学工業株式会社 (66)
【Fターム(参考)】
[ Back to top ]