
錯体混合リガンド開骨格材料
本発明は、異なる官能性リガンドを含む多変量の金属有機骨格を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、混合リガンドを含むセンサとして使用するための、ガス分離、貯蔵用有機骨格を提供する。
【背景技術】
【0002】
結晶拡張構造は、少数の別個の構築単位から構成されている点において「単純」である傾向がある。
【発明の概要】
【発明が解決しようとする課題】
【0003】
拡張結晶固体の構造は、少数の別個の構築単位から通常構築されている点において根本的に「単純」である。このことは、1種類の連結、官能性基及び金属イオン単位から一般に構成されている金属有機骨格(MOF)において確実に当てはまる。
【0004】
本発明は、連結部分上、又は少なくとも2個の異なる連結部分上に複数の異なる官能基を含む金属有機骨格を提供する。多孔質有機骨格は、細孔機能の変異が、少なくとも2個の異なる官能基を有機骨格中に組み込むことによって容易に修飾することができる点において多重変異性である。一実施形態において、連結部分のサブ構造は均一であるが、連結部分上の側鎖基は変異される。
【課題を解決するための手段】
【0005】
本発明は、主鎖に沿った配向、数、相対位置及び比率が連結部分の変化のないサイズ及び主鎖の不変の結合性のために制御可能である異なる官能基であって、骨格における細孔の化学的及び物理的性質を修飾する官能基を有する複数の連結部分を含む多孔質有機骨格を提供する。一実施形態において、該有機骨格は、n≧2であるn個の異なる有機連結から構成されている。別の実施形態において、該官能基は、金属酸化物及びフェニルの単位を含むコアに沿っている。また、別の実施形態において、該有機骨格は、金属−酸化物の結合部及び有機連結部分の反復単位、並びに連結部分に共有結合されている複数の官能基を含み、ここで、該官能基は不均一であるか、及び/又は該官能基は連結に沿って異なった間隔で配置されている。一実施形態において、該有機骨格は、MOF−5骨格と実質的に同一のMOFトポロジーを含む。別の実施形態において、該骨格は、対応する金属塩対アニオンとともに、Li+、Na+、Rb+、Mg2+、Ca2+、Sr2+、Ba2+、Sc3+、Ti4+、Zr4+、Ta3+、Cr3+、Mo3+、W3+、Mn3+、Fe3+、Fe2+、Ru3+、Ru2+、Os3+、Os2+、Co3+、Co2+、Ni2+、Ni+、Pd2+、Pd+、Pt2+、Pt+、Cu2+、Cu+、Au+、Zn2+、Al3+、Ga3+、In3+、Si4+、Si2+、Ge4+、Ge2+、Sn4+、Sn2+、Bi5+、Bi3+からなる群及びこれらの組合せから選択される金属を含む。また、別の実施形態において、該連結部分は、
【化1】

【0006】
(式中、R1〜R4は、−H、−NH2、−BR、−Cl、−NO2、−CH3、−OCH2R5及び−O−CH2R6からなる群から選択され、ここで、R5は約1個〜5個の炭素のアルキルもしくはアルケンであり、R6はアリールもしくは置換アリールである、又はR1〜R2は隣接する場合、環を形成することができる)からなる群から選択される一般構造を有する。さらなる実施形態において、該連結部分は、
【化2】
【0007】
からなる群から選択されるメンバーを含む。また、別の実施形態において、該有機骨格は、第一官能基を含む該複数の連結部分の第一連結部分及び第二官能基を含む該複数の連結部分の第二連結部分を含み、ここで、第二官能基は反応後の基との合成後の反応を受けることで、骨格をさらに官能化することができる。また別の実施形態において、mvMOFは、同じトポロジーであるが均一の連結部分を有する骨格と比較して改善されたガス収着能を有する。
【0008】
本発明は、連結部分が所望の比率で連結部分の特定の組合せを有機骨格に組み込むための所望の比率である複数の化学的官能化連結部分と、金属イオン又は金属硝酸塩とを混合すること、結晶を精製すること、及び溶媒を除去することを含む、mvMOFを作製する方法を提供する。具体的な一実施形態において、該方法は、ベンゼンジカルボン酸を含む有機骨格に所望の比率で連結部分の特定の組合せを組み込むための所望の比率で、複数の化学的官能化連結部分と硝酸亜鉛とをDEF/DMF中にて混合することを含む。
【0009】
本発明は、mvMOFを含むガス分離装置も提供する。本発明は、mvMOFを含むガス貯蔵装置も提供する。
【0010】
本発明は、具体的な実施形態において、多重の官能性基の連結を使用することで多変量の(MTV)構造を生成し、これによって、より複雑なMOFを作製するための戦略を実証する。1,4−ベンゼンジカルボキシレート(BDC、A)及びその官能化誘導体、−NH2、−Br、−(Cl)2、−NO2、−(CH3)2、−C4H4、−(C3H5O)2並びに−(C7H7O)2(それぞれB−I)は、18個のmvMOFを構築するために使用されたが、これらのそれぞれが立方MOF−5型結晶構造を有し、最大8個の異なる官能性基を含有する(2:mvMOF−5−AB、−AC、−AD、−AE、−AF、−AG、−AH、−AI、−EI、3:mvMOF−5−ABC、−AHI、−EHI、4:mvMOF−5−ABCD、−ACEF、5:mvMOF−5−ABCHI、6:mvMOF−5−ABCGHI、7:mvMOF−5−ABCEGHI、8:mvMOF−5−ABCEFGHI)。この系列(mvMOF−5−ACEF)の典型的メンバーの単結晶回折研究は、MOF主鎖(金属酸化物及びフェニルの単位)が規則化されており、官能性基が不可避的に不規則化されていることを確証しており、このことは、これらのMOFが固溶体であるという可能性を排除する。核磁気共鳴分光法を使用して、mvMOF系列の各メンバーの構造内における各官能基の存在、それらの同定及び比率を決定した。これらの測定は、それらのバルク均一性を確証するために固体生成物から選択されるいくつかの結晶に対して、及び結晶全体における同一連結比の存在を確証するために単結晶のセグメントに対しても行われた。後者の観察は結晶内における官能性基のランダム分布を論証することができるが、おそらくそれらは、特異的非障害位置での特異的連結に対する先入観を必然的に作り出す連結−連結相互作用により、特異的配列に配置されている可能性が大きい。これは、それぞれのmvMOFの大部分において最小障害連結(A)の比較的高い比率が観察されたことによって裏付けられている。細孔内における官能基の「複雑な」配置は、同じ連結対応物(MOF−5)と比較して、CO2に対するmvMOF−5−EHIの選択性においてCOを超える最大400%の向上をもたらす。
【0011】
本発明は、n個(n≧2である)の異なる有機連結から構成されている錯体自己集合開骨格材料を提供する。本発明は、多座有機又は無機コアによって架橋されている有機連結から構成されている全ての開骨格材料を包含する。開骨格材料;共有結合性有機骨格(COF)、ゼオライトイミダゾレート骨格(ZIF)及び金属有機骨格(MOF)の全ての部類、並びに網状化学構造リソース(http://rcsr.anu.edu.au/)内に記載されている通りの全ての可能な結果として生じるネットトポロジーを含める。該骨格における2個を超える連結を利用することによって、多種多様の材料を生み出す錯体構成物を合成することができる。こうした材料は、ガス貯蔵及び分離、並びに触媒作用等の応用において様々に使用される。
【0012】
本発明の一つ又は複数の実施形態の詳細を添付の図及び下の記述で説明する。本発明の他の特徴、対象物及び利点は、記述及び図面から、並びに請求項から明らかであろう。
【図面の簡単な説明】
【0013】
【図1】mvMOF−5−ABCDの試料に関して本明細書で示されているmvMOFに対して行われた通常の分析を示す図である。(A)MOF−5に関してシミュレートしたものと比較した結晶粉末のX線回折パターン。(B)各連結に関する固有の共鳴を示す13C CP/MAS NMRスペクトル。(C)連結の比率を決定するために使用された溶液1H NMRスペクトル。(D)それぞれ黒丸及び白丸によって表される吸着点及び脱着点を持つ77KでのN2吸着等温線。(E)セグメントが連結の比率に関して分析され、全体において同一であると判明した大結晶。
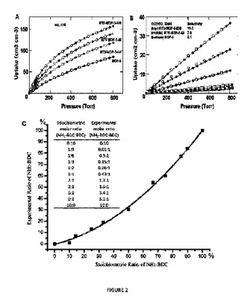
【図2】以下を示す図である。(A)mvMOF−5−AH(緑色)、−AI、−AHI及びMOF−5の77KでのH2吸着等温線。(B)mvMOF−5−EI、−EHI及びMOF−5の298KでのCO2(丸)及びCO(四角)の吸着等温線。吸着及び脱着枝は、それぞれ黒丸(COには四角)及び白丸(COには四角)によって表される。(C)NH2−BDC対BDCのモル比の表形式データと一緒に、溶液1H NMR対合成に使用された化学量論比によって決定されたMOF−5−ABにおけるNH2−BDCのパーセント比率のプロット。
【図3】調製されたままのmvMOF−5−ABの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図5】調製されたままのmvMOF−5−ACの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図6】調製されたままのmvMOF−5−ADの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図7】調製されたままのmvMOF−5−AEの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図8】調製されたままのmvMOF−5−AFの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図9】調製されたままのmvMOF−5−AGの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図10】調製されたままのmvMOF−5−AHの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図11】調製されたままのmvMOF−5−AIの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図12】調製されたままのmvMOF−5−EIの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図13】調製されたままのmvMOF−5−ABCの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図14】調製されたままのmvMOF−5−AHIの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図15】調製されたままのmvMOF−5−EHIの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図16】調製されたままのmvMOF−5−ABCDの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図17】調製されたままのmvMOF−5−ACEFの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図18】調製されたままのmvMOF−5−ABCHIの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図19】調製されたままのmvMOF−5−ABCGHIの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図20】調製されたままのmvMOF−5−ABCEGHIの実験PXRDパターンと、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図21】調製されたままのmvMOF−5−ABCEFGHIの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図22】活性化mvMOF−5−ABのTGAトレースの図である。
【図23】活性化mvMOF−5−ACのTGAトレースの図である。
【図24】活性化mvMOF−5−ADのTGAトレースの図である。
【図25】活性化mvMOF−5−AEのTGAトレースの図である。
【図26】活性化mvMOF−5−AFのTGAトレースの図である。
【図27】活性化MOF−5−AGのTGAトレースの図である。
【図28】活性化mvMOF−5−AHのTGAトレースの図である。
【図29】活性化MOF−5−AIのTGAトレースの図である。
【図30】活性化mvMOF−5−EIのTGAトレースの図である。
【図31】活性化MOF−5−AHIのTGAトレースの図である。
【図32】活性化mvMOF−5−EHIのTGAトレースの図である。
【図33】活性化mvMOF−5−ACEFのTGAトレースの図である。
【図34】活性化mvMOF−5−ABCDのTGAトレースの図である。
【図35】活性化MOF−5−ABCEFGHIのTGAトレースの図である。
【図36】光学的画像の図である。(A及びB):合成されたままのmvMOF−5−AB−lscの光学的画像。(C及びD):合成されたままのmvMOF−5−ABCD−lscの光学的画像。(E及びF):合成されたままのmvMOF−5−ABCEFGHI−lscの光学的画像。
【図37】mvMOF−5−ABの調製されたままの単結晶の実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較の図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図38】mvMOF−5−ABCDの調製されたままの単結晶の実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較の図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図39】mvMOF−5−ABCEFGHIの調製されたままの単結晶の実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較の図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図40】調製されたままのmvMOF−5−AB系列の実験PXRDパターン(a〜k)と、シミュレートされたMOF−5回折パターン(黒色)との比較の図である。該パターン間の非常に高い対応度は、バルク材料全てがMOF−5と同じトポロジーを有することを表す。
【図41】無色から赤色へ、それぞれ左から右へ、mvMOF−5−AB−aからmvMOF−5−AB−k(下部)の色変化を示す結晶の光学的画像と一緒に、溶液1H NMR対合成に使用された化学量論比(上部)によって決定されたmvMOF−5−AB系列におけるBのパーセント比率のプロットの図である。
【図42】調製されたままのmvMOF−5−AI系列の実験PXRDパターン(a〜c)と、シミュレートされたMOF−5回折パターン(黒色)との比較の図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図43】調製されたままのmvMOF−5−ABCD系列の実験PXRDパターン(a〜e)と、シミュレートされたMOF−5回折パターン(黒色)との比較の図である。該パターン間の非常に高い対応度は、バルク材料全てがMOF−5と同じトポロジーを有することを表す。
【図44】水素原子及びBr原子を含めて、不規則化基の両方の成分が示されているmvMOF−5−AC単位のORTEP図を示す。Zn(暗緑色)原子及びO(暗赤色)原子は異方的に精密化されており、一方、C(灰色)原子及びBr(赤色)原子は等方的に精密化されており、H(明緑色)原子は算出位置に置かれていた。全ての楕円体は、15%の確率水準で表示されている。各フェニル環上において、同等の可能性でBrによって占有されている全四つ位置から、一つの位置だけがBrによって占有されていることに留意されたい。
【図45】示されている不規則化基の全ての成分(Br、CH3、H)とのmvMOF−5−ACEF単位のORTEP図を示す。Zn(暗緑色)だけが異方的に精密化されており、一方、O(暗赤色)原子、C(灰色)原子及びBr(赤色)原子は等方的に精密化されており、H(明緑色)原子は算出位置に置かれていた。全ての楕円体は15%の確率水準で表示されている。各フェニル環は四つの位置の一つにBrを、及び四つの位置の二つにメチル基を有することができ、これらは共存することができないことに留意されたい。この図面上及び構造精密化において、非常に少量で存在するNO2の組成は無視されている。
【図46】77Kで測定されたmvMOF−5−AB(青色)、−ABCD(緑色)、及び−ABCEFGHI(赤色)の窒素吸着等温線を示す図である。中黒記号及び中白記号はそれぞれ吸着及び脱着枝を表す。連結線は目線のための案内である。
【図47】87Kで測定されたmvMOF−5(青色)及びmvMOF−5−AI(赤色)に関するアルゴン吸着等温線を示す図である。中黒記号及び中白記号はそれぞれ吸着及び脱着枝を表す。連結線は目線のための案内である。
【図48】NLDFTモデルに基づくmvMOF−5−AI−a及びMOF−5の算出孔径分布を示す図である。
【図49】77Kで測定されたMOF−5−AI−a(赤色)、mvMOF−5−AI−b(青色)及びmvMOF−5−AI−cの窒素吸着等温線を示す図である。中黒記号及び中白記号はそれぞれ吸着及び脱着枝を表す。連結線は目線のための案内である.
【図50】273(青色)、283(緑色)及び298K(赤色)で取得されたMOF−5に関するCO等温線を示す図である。中黒記号及び中白記号は吸着及び脱着枝を表す。連結線は目線のための案内である。
【図51】273(青色)、283(緑色)及び298K(赤色)で取得されたMOF−5に関するCO2等温線を示す図である。中黒記号及び中白記号は吸着及び脱着枝を表す。連結線は目線のための案内である。
【図52】273(青色)、283(緑色)及び298K(赤色)で取得されたmvMOF−5−EIに関するCO等温線を示す図である。中黒記号及び中白記号は吸着及び脱着枝を表す。連結線は目線のための案内である。
【図53】273(青色)、283(緑色)及び298K(赤色)で取得されたmvMOF−5−EIに関するCO2等温線を示す図である。中黒記号及び中白記号は吸着及び脱着枝を表す。連結線は目線のための案内である。
【図54】273(青色)、283(緑色)及び298K(赤色)で取得されたmvMOF−5−EHIに関するCO等温線を示す図である。中黒記号及び中白記号は吸着及び脱着枝を表す。連結線は目線のための案内である。
【図55】273(青色)、283(緑色)及び298K(赤色)で取得されたmvMOF−5−EHIに関するCO2等温線を示す図である。中黒記号及び中白記号は吸着及び脱着枝を表す。連結線は目線のための案内である。
【発明を実施するための形態】
【0014】
本明細書で使用される場合、及び添付の請求項において、単数形「一(a)」「及び」及び「前記」は、文脈が別途明確に指示していない限り、複数の指示対象を含める。したがって、例えば、「一骨格」の言及は複数のこうした骨格を含め、「前記金属」という言及は1種又は複数の金属及び当業者に知られているこれらの同等物等への言及を含める。
【0015】
別途定義されていない限り、本明細書において使用される全ての技術的及び科学的用語は、この開示が属する分野の当業者に通例理解されるのと同じ意味を有する。本明細書に記載されているものと類似又は同等の任意の方法及び試薬は開示の方法及び組成物の実践において使用することができるが、例示的な方法及び材料がここに記載されている。
【0016】
また、「又は」の使用は、別途記載されていない限り、「及び/又は」を意味する。同様に、「含む(comprise)」「含む(comprises)」「含む(comprising)」「含める(include)」、「含める(includes)」及び「含めて」は交換可能であり、限定することを意図していない。
【0017】
各種実施形態の記載が「含む(comprising)」という用語を使用する場合、一部の具体的例において、「から基本的になる」又は「からなる」という言語を使用して、実施形態は別法で記載され得ると当業者が理解すると、さらに理解されるべきである。
【0018】
本明細書に記述されている全ての文献は、本明細書に記載と関連して使用されると思われる文献に記載されている方法論を記載及び開示する目的で、参照により全文を本明細書に組み込む。上記及び本文を通して考察されている文献は、本出願の出願日より前に、単にそれらの開示内容のために提供されている。本発明者らが前の開示のためにこうした開示に先行する権利を与えられていないことを容認すると解釈されるべきものは、本明細書において何もない。
【0019】
結晶拡張構造は、少数の別個の構築単位から構成されているという点において、「単純」である傾向がある。こうした単位の数及び多様性を増加させることは、結晶構造の特性を改善する機会をもたらす。実際に、多数の異なる構築単位の特異的配置が特異的機能をコードする又は新たな現象に至る人工材料を開発することを想像することができる。今日まで、この種類の「複雑性」の結晶材料は、これらの合成が概して混合単位の単相よりむしろ混合相、又は非晶質材料のいずれかを生成するので存在しない。ブロックコポリマーの確立された化学的性質においてさえ、側鎖の官能性におけるわずかな変化が、ポリマー構造における大きな望ましくない変化又はその相分離に至り、したがってそれらの構造及び複雑性に対する制御の発現防止に至る。
【0020】
金属−酸化物単位が有機単位によって連結されている強固な規則化金属−有機骨格(MOF)を構築する方法を開発し、本発明は、MOF主鎖の本来の規則性が、MOFに制御された複雑性を達成する際に有用であり得ることを実証する。本発明は、主鎖に沿った配向、数、相対位置及び比率が連結の変化のないサイズ、及び主鎖の不変の結合性のために制御可能である異なる官能基を持つ連結を導入する戦略を実証する。こうしたMOFは、金属−酸化物及び有機連結単位の「単純な」反復パターンからなる第一次構造、及び該連結に共有結合されている官能基の広範に変動する配置によって形成される「複雑な」第二次構造を有するとみなすことができる。このように、MOF内の細孔のそれぞれは、それらの中心に向いている一連の混合官能性基を有すると思われる。すなわち、こうした官能性基の配列、及びそれらのいくつかが配列に現れる頻度が、元の単一連結のMOFのそれによって保有されるいかなるレベルもはるかに超える新たなレベルの複雑性を細孔に与える。特性に好ましい意味を持つ細孔環境の微調整を可能にする一態様。
【0021】
本発明はしたがって、金属−有機骨格(MOF)の本来の強剛性及びポリマーの機能可撓性を組み合わせることによって、これらの問題を克服し、それぞれが多変量の(MTV)官能性基を有する多数の単相材料を作り出すことができることを実証する。
【0022】
本発明は、主鎖(金属−酸化物及びフェニルの単位)に沿った配向、数、相対位置及び比率を連結の変化のない長さ及びその不変の結合性のために制御することができる異なる官能基を持つ連結からそれらの構造を組み立てることによって、多変量の金属有機骨格(mvMOFs)を作製する方法も提供する。スキーム1は、こうした変異の概略を表す。すなわち、こうした官能性基の配列及び特定の官能基が配列に現れる頻度が、元の同連結MOFのレベルによって保有されるいかなるレベルもはるかに超える新たなレベルの複雑性を細孔に与える。特性に好ましい意味を持つ細孔環境の微調整を可能にする一態様。
【0023】
スキーム1
【化3】
【0024】
本発明は、異なる官能基の2個、3個、4個、5個、6個、7個、8個、9個、10個、11個又は12個の連結のセットを組み合わせる結晶MOF材料を生成するための一般的方法を記載しており、これらのそれぞれは連結の比率が制御されている単一構造に組み込まれており、該材料はバルク純度で生成することができる。本発明は、単相としての多重変異体MOFの単離を実証する。mvMOFの多孔性は多様であり、多変異連結が様々な官能性基を導入することができることを示す。例えば、MOF構造に導入することができる変異連結として、これらに限定されないがNO2−BDC及び(Cl)2−BDCが挙げられ、MOF−5型構造(mvMOF−5−AD及び−AE)に導入され、それ以外単独で使用される場合はこの構造を形成しない。本発明は、この系列(例えば、mvMOF−5−AHI及び−EHI)のメンバーが、単純な同連結類似体と比較して多変異連結MOFのガス吸着及び分離特性の有意な向上によって証明されている通り「全体のほうが部分の和より良好である」ことを示すことも実証する。下記スキーム2は、mvMOFを発生させるために使用される多変異連結、並びにmvMOFの例を実証する。
【0025】
多重変異体金属有機骨格(mvMOF)は、変異官能性基を有する少なくとも2個、3個、4個、5個、6個、7個、8個、9個、10個、11個又は12個の異なる連結部分を含むMOF構造を指す。mvMOFは、連結クラスターを介して連結部分に共役している金属を含む。連結部分のサブ構造は、サブ構造上の側鎖基を変異させることによって起因し得る異なる官能性を含む。例えば、立方構造に含まれているmvMOFは、その角に金属(例えば、4個の金属原子)を含み、各金属原子は3個の連結クラスターに共役しており、各連結クラスターは連結部分サブ構造に共役している。すなわち、立方構造は12個の連結部分を含む。連結部分上の1個又は複数の側基における変異は、生じる立方構造の変異官能性を生じさせる。MOF骨格のサイズに取り入れると、骨格における変異は、ガス貯蔵、分離及び精製の改善及び多様な機能をもたらす。
【0026】
本明細書で使用される場合、「コア」は、骨格にみられる反復単位又は単位(複数)を指す。こうした骨格は、同種の反復コア構造又は異種の反復コア構造を含むことができる。コアは、遷移金属又は遷移金属のクラスター、及び連結部分を含む。一緒に連結されている複数のコアが骨格の特徴となる。
【0027】
「クラスター」という用語は、2個以上の原子の同定可能な会合を指す。こうした会合は通常、ある型の結合−イオン結合、共有結合及びファンデルワール等によって確立される。
【0028】
「連結クラスター」は、連結部分サブ構造と金属基との間、又は連結部分と別の連結部分との間に結合を形成する能力がある原子を含む縮合能力がある1種又は複数の反応種を指す。こうした種の例は、ホウ素原子、酸素原子、炭素原子、窒素原子及びリン原子からなる群から選択される。一部の実施形態において、連結クラスターは、架橋酸素原子と連結を形成する能力がある1種又は複数の異なる反応種を含むことができる。例えば、連結クラスターは、Rが1個から5個の炭素原子を有するアルキル基又は1個から2個のフェニル環を含むアリール基であるCO2H、CS2H、NO2、SO3H、Si(OH)3、Ge(OH)3、Sn(OH)3、Si(SH)4、Ge(SH)4、Sn(SH)4、PO3H、AsO3H、AsO4H、P(SH)3、As(SH)3、CH(RSH)2、C(RSH)3、CH(RNH2)2、C(RNH2)3、CH(ROH)2、C(ROH)3、CH(RCN)2、C(RCN)3、CH(SH)2、C(SH)3、CH(NH2)2、C(NH2)3、CH(OH)2、C(OH)3、CH(CN)2、及びC(CN)3、並びにCH(SH)2、C(SH)3、CH(NH2)2、C(NH2)3、CH(OH)2、C(OH)3、CH(CN)2及びC(CN)3を含むことができる。通常、MOFの発生において金属を結合するための連結クラスターは、カルボン酸官能基を含有する。
【0029】
本発明は、環を構成する全炭素原子、又は炭素原子と窒素原子、酸素原子、硫黄原子、ホウ素原子、リン原子、ケイ素原子及びアルミニウム原子との混合物のいずれかからなる1個から5個の環を含むシクロアルキルサブ構造又はアリールサブ構造を含める。
【0030】
「共有結合性有機多面体」という用語は、非拡張共有結合性有機ネットワークを指す。こうした多面体における重合は、重合を阻害するキャッピングリガンドの存在により一般に生じない。共有結合有機多面体は、多座コアを一緒に連結する複数の連結部分を含む共有結合有機ネットワークであるので、ネットワークの空間構造は多面体である。通常、この変異の多面体は二次元又は三次元構造である。
【0031】
「連結部分」は、遷移金属又は複数の遷移金属をそれぞれ結合する単座又は多座化合物を指す。一般に、連結部分は、1個から20個の炭素原子を含むアルキル基もしくはシクロアルキル基、1個から5個のフェニル環を含むアリール基、又は1個から20個の炭素原子を有するアルキル基もしくはシクロアルキル基、又は1個から5個のフェニル環を含むアリール基を含むアルキルアミンもしくはアリールアミンを含むサブ構造、及び金属との縮合を促進するためサブ構造の一つ又は複数の位置に連結クラスターを含む。シクロアルキル又はアリールのサブ構造は、環を構成する全炭素原子、又は炭素原子と窒素原子、酸素原子、硫黄原子、ホウ素原子、リン原子、ケイ素原子及び/又はアルミニウム原子との混合物のいずれかを含む1個から5個の環を含むことができる。通常、連結部分は、共有結合している1個又は複数のカルボン酸連結クラスターを有するサブ構造を含む。サブ構造は、反応側基で官能化することができる。
【0032】
本明細書で使用される場合、一方の末端上に原子があり、他方の末端上に何もない化学式における線は、該式が、原子が付加されていない末端上で別の実体に結合されている化学フラグメントを指すことを意味する。強調のために時々波線が該線に交差する。
【0033】
一態様において、連結部分サブ構造は、以下のいずれかから選択される。
【化4】
【0034】
式中、R1〜R4は、−H、−NH2、−BR、−Cl、−NO2、−CH3、−OCH2R5及び−O−CH2R6からなる群から選択され、R5は、約1〜5個の炭素のアルキル又はアルケンであり、R6は、アリールもしくは置換アリールであるか、又はR1〜R2は隣接する場合、環を形成することができる。一実施形態において、連結リガンドは以下からなる群から選択されるメンバーを含む。
【化5】
【0035】
本発明のmvMOFは、異なる官能基を有する複数の連結部分を最初に利用することによって生じさせることができ、ここで、少なくとも1個の官能基は、反応基で合成後に修飾することができることがさらに企図される。言い換えると、少なくとも1個の連結部分は、反応後の骨格と合成後に反応させることで有機骨格内における官能基の多様性をさらに増加させることができる官能基を含む。
【0036】
また別の実施形態において、連結部分は、下記に説明される通りの一般構造を有することができる。
【化6】
【0037】
式中、R1〜R5は、H、NH2、COOH、CN、NO2、F、Cl、Br、I、S、O、SH、SO3H、PO3H2、OH、CHO、CS2H、SO3H、Si(OH)3、Ge(OH)3、Sn(OH)3、Si(SH)4、Ge(SH)4、PO3H、AsO3H、AsO4H、P(SH)3、As(SH)3、CH(RSH)2、C(RSH)3、CH(RNH2)2、C(RNH2)3、CH(ROH)2、C(ROH)3、CH(RCN)2、C(RCN)3である。
【化7】
【0038】
式中、X=1、2又は3である。
【0039】
適切な反応性官能性基を保有する全ての前述の連結部分を、骨格合成後の適当な反応剤によって化学的に変換することで、さらなる官能性基を細孔に付加することができる。骨格における有機連結を合成後的に修飾することによって、以前はアクセス不可能であるか又は多大な困難及び/又はコストを介してのみアクセス可能であった官能基へのアクセスが可能及び容易である。後の骨格反応剤として、全ての既知有機変換剤及びそれぞれの反応剤;N、S、O等の原子を含めた官能基との1〜20個の炭素の環が挙げられる。付加された官能基に、又は以前から存在する官能基及び新規に付加された官能基の組合せにキレートすることができる全ての金属。例えば不均一触媒として使用するために有機金属錯体を骨格に繋ぐ結果になる全ての反応物。後の骨格反応剤の一部の例として以下のものが挙げられる。
【化8】
【0040】
式中、R=H、アルキル、アリール、OH、アルコキシ、アルケン、アルキン、フェニル及び前述の置換、硫黄含有基(例えば、チオアルコキシ)、ケイ素含有基、窒素含有基(例えば、アミド)、酸素含有基(例えば、ケトン及びアルデヒド)、ハロゲン、ニトロ、アミノ、シアノ、ホウ素含有基、リン含有基、カルボン酸又はエステルである。
【0041】
さらに、金属、並びに付加された官能基に、又は以前から存在する官能基及び新規に付加された官能基の組合せにキレートすることができる金属含有化合物も有用である。例えば不均一触媒として使用するために有機金属錯体を骨格に繋ぐ結果になる反応物も使用することができる。後の骨格反応剤の例として、これらに限定されないが、複素環化合物が挙げられる。
【0042】
一実施形態において、後の骨格反応剤は、飽和又は不飽和複素環であってよい。単独又は接尾語もしくは接頭語として使用される「複素環」という用語は、環構造の一部としてN、O及びSから独立して選択される1個又は複数の多価ヘテロ原子を有し、環(単数又は複数)内に少なくとも3個及び最大約20個の原子を含める環含有構造又は分子を指す。複素環は1個又は複数の二重結合を含有する飽和又は不飽和であってよく、複素環は1個を超える環を含有することができる。複素環が1個を超える環を含有する場合、該環は縮合又は非縮合であってよい。縮合環は一般に、その間に2個の原子を共有する少なくとも2個の環を指す。複素環は芳香族性を有してよい、又は芳香族性を有していなくてよい。単独又は接尾語もしくは接頭語として使用される「複素環基」、「複素環部分」、「複素環」又は「ヘテロシクロ」という用語は、複素環から1個又は複数の水素を除去することによって誘導される基を指す。単独又は接尾語もしくは接頭語として使用される「ヘテロシクリル」という用語は、複素環から1個の水素を除去することによって誘導される一価の基を指す。単独又は接尾語もしくは接頭語として使用される「ヘテロアリール」という用語は、芳香族性を有するヘテロシクリルを指す。複素環として、例えば、以下等の単環式複素環が挙げられる。アジリジン、オキシラン、チイラン、アゼチジン、オキセタン、チエタン、ピロリジン、ピロリン、イミダゾリジン、ピラゾリジン、ピラゾリン、ジオキソラン、スルホラン2,3−ジヒドロフラン、2,5−ジヒドロフランテトラヒドロフラン、チオファン、ピペリジン、1,2,3,6−テトラヒドロ−ピリジン、ピペラジン、モルホリン、チオモルホリン、ピラン、チオピラン、2,3−ジヒドロピラン、テトラヒドロピラン、1,4−ジヒドロピリジン、1,4−ジオキサン、1,3−ジオキサン、ジオキサン、ホモピペリジン、2,3,4,7−テトラヒドロ−1H−アゼピンホモピペラジン、1,3−ジオキセパン、4,7−ジヒドロ−1,3−ジオキセピン、及びヘキサメチレンオキシド。
【0043】
さらに、複素環として、芳香族複素環(ヘテロアリール基)、例えば、ピリジン、ピラジン、ピリミジン、ピリダジン、チオフェン、フラン、フラザン、ピロール、イミダゾール、チアゾール、オキサゾール、ピラゾール、イソチアゾール、イソオキサゾール、1,2,3−トリアゾール、テトラゾール、1,2,3−チアジアゾール、1,2,3−オキサジアゾール、1,2,4−トリアゾール、1,2,4−チアジアゾール、1,2,4−オキサジアゾール、1,3,4−トリアゾール、1,3,4−チアジアゾール、及び1,3,4−オキサジアゾールが挙げられる。
【0044】
さらに、複素環は、多環式複素環、例えば、インドール、インドリン、イソインドリン、キノリン、テトラヒドロキノリン、イソキノリン、テトラヒドロイソキノリン、1,4−ベンゾジオキサン、クマリン、ジヒドロクマリン、ベンゾフラン、2,3−ジヒドロベンゾフラン、イソベンゾフラン、クロメン、クロマン、イソクロマン、キサンテン、フェノキサチイン、チアントレン、インドリジン、イソインドール、インダゾール、プリン、フタラジン、ナフチリジン、キノキサリン、キナゾリン、シンノリン、プテリジン、フェナントリジン、ペリミジン、フェナントロリン、フェナジン、フェノチアジン、フェノキサジン、1,2−ベンゾイソオキサゾール、ベンゾチオフェン、ベンゾオキサゾール、ベンゾチアゾール、ベンゾイミダゾール、ベンゾトリアゾール、チオキサンチン、カルバゾール、カルボリン、アクリジン、ピロリジジン、及びキノリジジンを包含する。
【0045】
上に記載されている多環式複素環に加えて、複素環として、2個以上の環の間の環縮合が、両方の環に共通する1個を超える結合、及び両方の環に共通する2個を超える原子を含める多環式複素環が挙げられる。こうした架橋複素環の例として、キヌクリジン、ジアザビシクロ[2.2.1]ヘプタン及び7−オキサビシクロ[2.2.1]ヘプタンが挙げられる。
【0046】
ヘテロシクリルとして、例えば、以下等の単環式ヘテロシクリルが挙げられる。アジリジニル、オキシラニル、チイラニル、アゼチジニル、オキセタニル、チエタニル、ピロリジニル、ピロリニル、イミダゾリジニル、ピラゾリジニル、ピラゾリニル、ジオキソラニル、スルホラニル、2,3−ジヒドロフラニル、2,5−ジヒドロフラニル、テトラヒドロフラニル、チオファニル、ピペリジニル、1,2,3,6−テトラヒドロ−ピリジニル、ピペラジニル、モルホリニル、チオモルホリニル、ピラニル、チオピラニル、2,3−ジヒドロピラニル、テトラヒドロピラニル、1,4−ジヒドロピリジニル、1,4−ジオキサニル、1,3−ジオキサニル、ジオキサニル、ホモピペリジニル、2,3,4,7−テトラヒドロ−1H−アゼピニル、ホモピペラジニル、1,3−ジオキセパニル、4,7−ジヒドロ−1,3−ジオキセピニル、及びヘキサメチレンオキシジル。
【0047】
さらに、ヘテロシクリルとして、芳香族ヘテロシクリル又はヘテロアリール、例えば、ピリジニル、ピラジニル、ピリミジニル、ピリダジニル、チエニル、フリル、フラザニル、ピロリル、イミダゾリル、チアゾリル、オキサゾリル、ピラゾリル、イソチアゾリル、イソオキサゾリル、1,2,3−トリアゾリル、テトラゾリル、1,2,3−チアジアゾリル、1,2,3−オキサジアゾリル、1,2,4−トリアゾリル、1,2,4−チアジアゾリル、1,2,4−オキサジアゾリル、1,3,4−トリアゾリル、1,3,4−チアジアゾリル、及び1,3,4オキサジアゾリルが挙げられる。
【0048】
さらに、ヘテロシクリルは、多環式ヘテロシクリル(芳香族又は非芳香族の両方を含める)、例えば、インドリル、インドリニル、イソインドリニル、キノリニル、テトラヒドロキノリニル、イソキノリニル、テトラヒドロイソキノリニル、1,4−ベンゾジオキサニル、クマリニル、ジヒドロクマリニル、ベンゾフラニル、2,3−ジヒドロベンゾフラニル、イソベンゾフラニル、クロメニル、クロマニル、イソクロマニル、キサンテニル、フェノキサチイニル、チアントレニル、インドリジニル、イソインドリル、インダゾリル、プリニル、フタラジニル、ナフチリジニル、キノキサリニル、キナゾリニル、シンノリニル、プテリジニル、フェナンスリジニル、ペリミジニル、フェナントロリニル、フェナジニル、フェノチアジニル、フェノキサジニル、1,2−ベンゾイソオキサゾリル、ベンゾチオフェニル、ベンゾオキサゾリル、ベンゾチアゾリル、ベンゾイミダゾリル、ベンゾトリアゾリル、チオキサンチニル、カルバゾリル、カルボリニル、アクリジニル、ピロリジジニル、及びキノリジジニルを包含する。
【0049】
上に記載されている多環式ヘテロシクリルに加えて、ヘテロシクリルとして、2個以上の環の間の環縮合が、両方の環に共通する1個を超える結合及び両方の環に共通する2個を超える原子を含める多環式ヘテロシクリルが挙げられる。こうした架橋複素環の例として、キヌクリジニル、ジアザビシクロ[2.2.1]ヘプチル;及び7−オキサビシクロ[2.2.1]ヘプチルが挙げられる。
【0050】
本発明の骨格の合成に使用することができる金属イオンとして、Li+、Na+、Rb+、Mg2+、Ca2+、Sr2+、Ba2+、Sc3+、Ti4+、Zr4+、Ta3+、Cr3+、Mo3+、W3+、Mn3+、Fe3+、Fe2+、Ru3+、Ru2+、Os3+、Os2+、Co3+、Co2+、Ni2+、Ni+、Pd2+、Pd+、Pt2+、Pt+、Cu2+、Cu+、Au+、Zn2+、Al3+、Ga3+、In3+、Si4+、Si2+、Ge4+、Ge2+、Sn4+、Sn2+、Bi5+、Bi3+、及びこれらの組合せが、対応する金属塩対アニオンとともに挙げられる。
【0051】
本発明は、本発明のmvMOFを作製する方法も提供する。該方法は、有機骨格に所望の比率で連結部分の特定の組合せを組み込むための所望の比率で、複数の化学的官能化連結部分と、金属イオン又は金属−硝酸塩とを、適切な緩衝液中で混合することを含む。結果として生じる結晶物質を次いで精製し、溶媒を除去する。一実施形態において、該方法は、ベンゼンジカルボン酸を含む有機骨格中に所望の比率で連結部分の特定の組合せを組み込むための所望の比率で、複数の化学的官能化連結部分と硝酸亜鉛とをDEF/DMF中にて85〜100℃で24〜48時間混合することを含む。結果として生じる結晶物質を次いでDMF中に24時間、次いで順次クロロホルム中に、24時間を3周期の間浸漬する。最終的に、この多孔質材料は、溶媒を真空下で24時間室温又は最大120℃までの加熱で除去することによって活性化される。
【0052】
本発明の骨格の調製は、水性系又は非水性系のいずれかにおいて実施することができる。溶媒は各場合によって極性又は非極性であってよい。溶媒は、テンプレート剤又は単座官能基を含有する任意選択のリガンドを含むことができる。非水性溶媒の例として、ペンタン、ヘキサン等のn−アルカン、ベンゼン、トルエン、キシレン、クロロベンゼン、ニトロベンゼン、シアノベンゼン、アニリン、ナフタレン、ナフサ、メタノール等のn−アルコール、エタノール、n−プロパノール、イソプロパノール、アセトン、1,3−ジクロロエタン、塩化メチレン、クロロホルム、四塩化炭素、テトラヒドロフラン、ジメチルホルムアミド、ジメチルスルホキシド、N−メチルピロリドン、ジメチルアセトアミド、ジエチルホルムアミド、チオフェン、ピリジン、エタノールアミン、トリエチルアミン及びエチレンジアミン等が挙げられる。当業者は出発反応剤に基づいて適切な溶媒を容易に決定することができ、溶媒の選択は本発明の材料を入手するのに重大であるとは思われない。
【0053】
本発明の方法にテンプレート剤を使用することができる。本発明に用いられるテンプレート剤は、生じる結晶塩基骨格における細孔を占有する目的で、反応混合物によって付加される。本発明の一部の変異において、空隙充填薬剤、吸着された化学種及びゲスト種は、金属−有機骨格の表面積を増加させる。適当な空隙充填薬剤として、例えば、以下のものからなる群から選択される成分が挙げられる。(i)1個から20個の炭素原子を有する直鎖、分枝又は環状脂肪族基を含有する、アルキルアミン及びそれらの対応するアルキルアンモニウム塩;(ii)1個から5個のフェニル環を有する、アリールアミン及びそれらの対応するアリールアンモニウム塩;(iii)1個から20個の炭素原子を有する直鎖、分枝又は環状脂肪族基を含有するアルキルホスホニウム塩;(iv)1個から5個のフェニル環を有するアリールホスホニウム塩;(v)1個から20個の炭素原子を有する直鎖、分枝又は環状脂肪族基を含有する、アルキル有機酸及びそれらの対応する塩;(vi)1個から5個のフェニル環を有する、アリール有機酸及びそれらの対応する塩;(vii)1個から20個の炭素原子を有する直鎖、分枝又は環状脂肪族基を含有する脂肪族アルコール;又は(viii)1個から5個のフェニル環を有するアリールアルコール。
【0054】
結晶化は、室温又は等温の乾燥器内で最大300℃までの間、溶液を放置すること;希釈塩基を溶液に添加することで結晶化を促進すること;希釈塩基を溶液中に拡散することで結晶化を促進すること;及び/又は溶液を密閉容器に移し、所定温度に加熱することによって実施することができる。
【0055】
さらに提供されているのは、化学種の収着取込み用装置である。該装置は、本明細書において提供されているか又は本発明の方法によって得られる骨格を含む収着剤を含める。該取込みは可逆的又は非可逆的であってよい。一部の実施形態において、収着剤は個別の収着粒子中に含められる。収着粒子は、固体、液体及び/又はガス透過性三次元担持体に埋め込むか、又は固定することができる。一部の実施形態において、収着粒子は液体又はガスの可逆的取込み又は貯蔵のための細孔を有し、ここで、収着粒子は液体又はガスを可逆的に吸着又は吸収することができる。
【0056】
一部の実施形態において、本発明において提供されている装置は、アンモニア、二酸化炭素、一酸化炭素、水素、アミン、メタン、酸素、アルゴン、窒素、アルゴン、有機染料、多環式有機分子、及びそれらの組合せ等の化学種の貯蔵のための貯蔵ユニットを含む。
【0057】
さらに提供されているのは、化学種の収着取込みのための方法である。該方法は、本明細書において提供されている骨格を含む収着剤と化学種を接触させることを含める。化学種の取込みは、化学種の貯蔵を含める。一部の態様において、化学種はエネルギー供給源としての使用に適当な条件下で貯蔵される。
【0058】
天然ガスは重要な燃料ガスであり、石油化学工業及び他の化学プロセス工業における塩基性原料として広範に使用されている。天然ガスの組成は分野により広く変動する。多くの天然ガス貯留層は、比較的低い百分率の炭化水素(例えば40%未満)及び高い百分率の酸性ガス、主に二酸化炭素だが硫化水素、硫化カルボニル、二硫化炭素及び様々なメルカプタンも含有している。遠隔地で生産される天然ガスからの酸性ガスの除去は、パイプラインへの送達、天然ガス液体回収、ヘリウム回収、液化天然ガス(LNG)への変換、又は後続の窒素排除のいずれかのための調節又はスイート乾性天然ガスを供給するのに望ましい。CO2は水の存在下で腐食性であり、ドライアイス、水和物を形成する恐れがあり、パイプライン及び天然ガスを加工する際にしばしば使用される極低温装置における凍結問題を引き起こす恐れがある。その上、発熱量に寄与しないことによって、CO2はガス輸送のコストを増大するだけである。
【0059】
いかなる天然ガス処理プロセスも経済的側面が重要である。天然ガスは通常大量に処理され、プロセス技術の選択における処理ユニットの有意な要因である資本コスト及び操作コストにわずかであるが違いを生ずる。一部の天然ガス資源は、加工コストのため生産するのに不経済である。高い信頼性を有し、操作の簡易性を示す天然ガス処理プロセスの改善が引き続き必要とされている。
【0060】
さらに、現在、人為的二酸化炭素の主要な供給源である発電所の煙道排気からの二酸化炭素の除去は、排気を冷却及び加圧すること、又はアミン水溶液の流動床に煙霧を通過させることによって一般に達成され、これらの両方ともコストがかかり、非効率的である。酸化物表面上への二酸化炭素の化学吸着、又は多孔質シリケート、炭素及び膜内への吸着に基づく他の方法は、二酸化炭素取込みのための手段として追及されてきた。しかし、有効な吸着媒体が二酸化炭素除去において長期生存能を有するために、下記の二つの特徴を組み合わせるべきである。(i)二酸化炭素の取込み及び放出が十分可逆的である周期構造、及び(ii)化学的機能化及び分子レベルの微調整が、最適化された取込み能力のために達成できる可撓性。
【0061】
ガス蒸気からの二酸化炭素の回復又は除去のための多くのプロセスが提案され、工業規模で実践されている。プロセスは広く多様であるが、一般に、溶媒吸収、多孔質吸着剤上の吸着、蒸留、又は半透膜を介する拡散の一部の形態を伴う。
【0062】
以下の実施例は本発明を例示することを意図するが、限定するものではない。使用することができる通常の例であるが、当業者に知られている他の手順も別法として使用することができる。
【実施例】
【0063】
以下の実施例は例示に過ぎず、様々なリガンド及び官能基を含めるために修飾することができる該MOF構造の多様性を限定することを意図するものではない。一実施形態において、立方MOF−5構造を使用し、1,4−ベンゼンジカルボキシレート(BDC)、NH2−BDC、Br−BDC、(Cl)2−BDC、NO2−BDC、(CH3)2−BDC、C4H4−BDC、(C3H5O)2−BDC及び(C7H7O)2−BDCの連結(それぞれ、スキーム2−A−I)の酸形態と組み合わせることで、それぞれが2個以上の異なる官能性基(2:mvMOF−5−AB、−AC、−AD、−AE、−AF、−AG、−AH、−AI、−EI;3:mvMOF−5−ABC、−AHI、−EHI;4:mvMOF−5−ABCD、−ACEF;5:mvMOF−5−ABCHI;6:mvMOF−5−ABCGHI;7:mvMOF−5−ABCEGHI;8:mvMOF−5−ABCEFGHI、スキーム2)を有する18個のmvMOFの対応するセットを形成した。本発明は、単相として単離、MOF−5主鎖の構造及びそれらの多孔性を実証し、この多変異連結合成戦略が、NO2−BDC及び(Cl)2−BDC等の官能性基を、それ以外単独で使用される場合この構造を形成しないMOF−5型構造(mvMOF−5−AD及び−AE)に導入するのに有用であることを示す。該データは、この系列(mvMOF−5−AHI及び−EHI)のメンバーが、それらの単純な同連結類似体と比較して、多変異連結MOFのガス吸着及び分離特性の有意な向上によって証明されている通り「全体のほうが部分の和より良好である」ことも実証する。
【化9】
【0064】
スキーム2
【0065】
mvMOFsの結晶は、MOF−5の合成にすでに使用されている条件下で、選択された有機連結の酸形態のN,N−ジメチルホルムアミド(DMF)溶液混合物に、Zn(NO3)2・4H2Oを添加することによって得られた。全ての化合物は、それぞれ、粉末X線回折(PXRD)、13C交差分極マジック角スピニング(CP/MAS)NMR、それらの結晶の酸蒸解溶液に対する1H NMR、及び熱重量分析(TGA)によって特徴づけることで、それらの結晶化度、連結組成、連結比及び熱安定性を評価した。これらの化合物(全て、2個、3個又は4個の異なる連結、及びmvMOF−5−ABCEFGHIを含有する)のサブセットの多孔性は、窒素ガス吸着測定によって評価した。全ての化合物に対する完全な特性決定手順及びデータ取得を行ったが、mvMOF−5−ABCDの詳細は例示的な例として提供されている。
【0066】
化合物は、連結A、B、C及びDの等モル量から合成された(上記スキーム2において説明されている通り)。その高い結晶化度は、親MOF−5構造のものと一致する鋭い回折線を示す合成されたままの試料のPXRDパターンから証明された(図1A)。mvMOF−5−ABCDにおける四つの型の連結の比率を決定するため、試料を50℃にて真空下で(10mTorr)24時間加熱することによって脱気することで、合成中に閉塞した細孔から任意のゲスト溶媒分子を除去した。この試料に対して行ったTGAは最大400℃まで重量損失を示さず、全てのゲスト分子が細孔から除去されたこと、及び脱気された骨格が熱的に安定であることを確証している。
【0067】
mvMOF−5−ABCDの脱気された試料の13C CP/MAS NMRスペクトルは、それぞれ、NH2−BDC、Br−BDC、(Cl)2−BDC及びBDC連結の固有の炭素原子に特有である150.3ppm、127.0ppm、133.7ppm及び136.3ppmで共鳴を示した(図1B)。これらのスペクトルは、MOF主鎖におけるそれらの存在を明確に表している。さらに、同じ実験をmvMOF−5−ABCDの構成物質フリー連結の混合物に行ったが、2ppmの特有のシフトは、フリー連結のカルボニル炭素と骨格に組み込まれた連結のカルボニル炭素との間で認められ、したがって非結合有機連結はMOF結晶内に存在しないことを確証している。全ての残りのmvMOFに対する同様の分析は同じ結果になった。
【0068】
正確な連結比は、mvMOF−5−ABCD固体のDCl蒸解溶液の1H NMRスペクトルから得た(表1、連結組成)。予測カップリングパターンとの共鳴は、連結の固有のプロトンのそれぞれの予想領域において認められた(図1C)。共鳴ピーク強度を積分することによって、連結はMOFに、それぞれ1.00:0.12:0.56:0.40の比率で存在することが実証されている。これらの比率が結晶においてバルク固体と同じであることを示すために、上記に考察されている溶液1H NMR実験を、mvMOF−5−ABCDバルク試料から無作為に選択される4種の異なる結晶に対して行い、該比率がほぼ同一であることを示した。同じ実験をmvMOF−5−AB及び−ABCEFGHIに対しても行い、mvMOF系列のバルク均一性を再度確証した(表1、バルク均一性)。さらに、本来のMOF−5構造の多孔性及び構成安定性は、mvMOF−5−ABCDに関する図1Dに示されているI型窒素吸着等温線及びその高表面積(2860m2g−1)によって例示されている通り、mvMOF化合物中に保持されている。さらに、様々な連結モル比からmvMOF−ABCDを合成することによって、データは、所定のmvMOFにおいて反応化学量を改変することによって連結比を制御することができることを実証した(表1、連結比の制御)。本質的に、連結比におけるこの型の制御は、それらの保持されているPXRDパターンによって証明されている通り、第一次構造の下部結合度を変更することなく、細孔に向いている官能基の占有数及び多様性の制御につながる(図29)。
【0069】
mvMOF−5−AC及び−ACEFの単結晶に対して行われたX線結晶学的研究は、予想通り、Zn4O(CO2)6頂点によって結合されている強固なフェニル単位からなる規則化立方MOF−5構造を明らかにした。これらの材料におけるフェニル単位上の官能基の非水素原子は全て、非常に低い占有率で存在する。フェニル環の4個の炭素原子のそれらの位置が等確率のため、対称には、各官能基が二つ(連結Fのジメチル基)又は四つ(連結CのBr基、又は連結Eのニトロ基)の位置で不規則化されていることが必要である。mvMOF−5−ACにおけるBrは、その低い占有率及びデータの強度に対する低い寄与にもかかわらず精密化することができる。mvMOF−5−ACEFにおいても、官能基原子の占有率は全く低いが、Br原子(連結C)、N原子(連結E)及びC原子(連結F)の位置が重なっているため、差異ピークはあり得る。フェニル単位原子が全てのmvMOFに存在する前提で、これらのパラメーターの全てを、主鎖非水素原子に関して精密化するのに成功した。これは、mvMOFの構造は固溶体でないが、むしろ規則化された骨格に共有結合的に連結された変異官能基の系を表していることを明確に示している。
【0070】
mvMOFの構成の固有性を考慮すると、浮かび上がる有意な疑問は、結晶が官能性基の巨視的領域からなるのか、又は骨格主鎖全体にわたり反復された官能性単位の特有の配列からなるのかである。これらの二つの可能性を区別するため、それぞれ4.0mm×4.0mm×2.0mm、2.0mm×2.0mm×2.0mm及び2.0mm×2.0mm×1.0mmの寸法のmvMOF−5−AB、−ABCD(図1E)及び−ABCEFGHIの大きな単結晶を調製した。各場合において、各単結晶の構造をそのPXRDパターンによって確認した(図25〜27)。各結晶を3個の等しいセグメントに切断し、次いで、溶液1H NMRスペクトルを、それぞれ各結晶の各セグメントの酸蒸解試料上に回収した。同種連結の巨視的領域がmvMOFの単結晶に存在すれば、異なる連結比がそれぞれの親結晶の3個のセグメントのそれぞれに予想される。しかし、データは、連結分布の比率が研究した3mvMOFの各セグメントに同一であると明確に示しており(表1、単結晶のセグメント)、したがって巨視的領域の非存在を示唆している。この結論を支持するさらなる証拠は、MOF−5又は任意の他の同連結MOFに関して認められる通り、mvMOF−5−AIに関する狭い孔径分布の非存在であり、これは連結Iが細孔全体に分布されていることを示唆する。これは、ナノメートルスケール上の特異的官能基(又は官能性基のサブセット)の優位が予想される微視的領域の存在を排除するものではない。
【0071】
表1.付加比率と比較したmvMOF結晶(太字)に認められる連結の比率。連結Aの数値を各場合において1に正規化した。
【表1】
【0072】
MOF主鎖に沿った官能性基の特有の配列の可能な存在は必然的に錯体細孔環境に至り、珍しい特性を明らかにする機会をもたらす。同連結MOF−5構造が有意な量のガス(例えば、H2、CO2)を吸収することが知られているので、mvMOFをこれらの用途で試験することで、それらの性能がそれらの構成物質の性能より大きいかどうかを決定した。図2Aにおいて、mvMOF−5−AHI、−AH、−HI及びMOF−5のH2貯蔵容量間の比較を示す。驚くべきことに、等温線は、mvMOF−5−AHIの取込み容量がmvMOF−5−AH、−HI及び−A(MOF−5)の取込み容量より最大84%大きいことを明確に実証している。同様に、COを超えるCO2の選択的取込み容量における珍しい増加が認められた。MOF−5と比較してCO2に対するmvMOF−5−EHIの場合、400%より良好な選択性(図2B)。
【0073】
これらの発見は、mvMOFの特性が構成物質の単純な直鎖組合せではないことを実証しており、したがってmvMOF内における官能性基の配列が、特異的特性を向上又は新たな特性を実現するための基準として有用であり得るという考えを支持している。
【0074】
複数グラムスケールの生成物を含めたmvMOFの調製の詳細な合成手順、並びに実験及びシミュレートPXRDパターン。
【0075】
テレフタル酸(ベンゼン−1,4−ジカルボン酸又はBDCH2)、2−アミノテレフタル酸(NH2−BDCH2)、2−ブロモテレフタル酸(Br−BDCH2)、2,5−ジクロロテレフタル酸((Cl)2−BDCH2)、2−ニトロテレフタル酸(NO2−BDCH2)、ナフタレン−1,4−ジカルボン酸(C4H4−BDCH2)をAldrich Chemical Co.から購入した。N,N−ジメチルホルムアミド(DMF)は、Fisher Scientific International Inc.から購入する。硝酸亜鉛四水和物Zn(NO3)2・4H2OをEM Scienceから購入した。2,5−ジメチルテレフタル酸((CH3)2−BDCH2)をTCI Americaから購入した。上記全ての出発原料は、さらに精製することなく使用した。N,N−ジエチルホルムアミド(DEF、BASF)溶媒を、活性炭素(Calgon)及びシリカゲル(EMD、シリカゲル60)を充填したカラムに通す濾過によって精製し、クロロホルム(Fisher、HPLC等級、ペンテンを安定化)を使用前に、新たに活性化したモレキュラーシーブ4A上で乾燥させた。2,5−ビス(アリルオキシ)テレフタル酸((C3H5O)2−BDCH2)、2,5−ビス(ベンジルオキシ)テレフタル酸((C7H7O)2−BDCH2)を合成した。
【0076】
多変量の金属−有機骨格(mvMOF)は、変動する量の化学的官能化ベンゼンジカルボン酸と硝酸亜鉛とをDEF/DMF中にて85〜100℃で24〜48時間混合することによって合成される。結果として生じる結晶物質は、次いで、DMF中で24時間、及び次いで順次クロロホルム中で24時間を3周期の間浸漬する。最終的に、この多孔質材料は、溶媒を真空下で24時間室温又は最大120℃までの加熱で除去することによって活性化する。
【0077】
出発原料の量を正確に制御するため、テラフタル酸連結のDMF/DEF溶液(0.025〜0.10M(1M=1moldm−3))及び硝酸亜鉛四水和物のDMF/DEF溶液(0.10M)を原液として使用した。DMF及びDEFは、それらの高沸点により合成の溶媒として選択されており、ソルボサーマル合成に適当である。より低い沸点の溶媒は、生成物よりむしろ反応剤の析出をもたらす恐れがある。反応後、結晶を光学顕微鏡下で検査し、回収し、粉末X線回折(PXRD)によって特徴づけた。破砕した大きな単立方結晶のPXRDデータを、40kV、40mAのCuKα(λ=1.5406Å)、3°/分の走査速度及び2θで0.050°のステップサイズにて操作するBruker AXS D8 Advance回折計に記録した。シミュレートしたMOF−5 PXRDパターンを、ソフトウェアPowder Cell v.2.2を使用して前に公開した単結晶構造から算出した。
【0078】
蒸解mvMOFの1H NMR:一般に、8mgの乾燥(又はDMFと交換した溶媒)mvMOFを蒸解し、1.0mLの希釈DCl溶液(200μLの20%DCl/D2O溶液(Aldrich)及び10mLのDMSO−d6から調製した)中の超音波処理で溶解した。蒸解溶液を1H−NMRに直接使用した。
【0079】
mvMOFの合成:mvMOF−5−AB、Zn4O(BDC)1.92(NH2−BDC)1.08:0.10mLのBDCH2原液(0.10M、1.0×10−5mol)、0.10mLのNH2−BDCH2原液(0.10M、1.0×10−5mol)、0.20mLのZn(NO3)2・4H2O原液(0.30M、6.0×10−5mol)及び0.60mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに48時間反応させた。生成物は立方形の褐色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5粉末回折パターンに整合する(図5)。
【0080】
蒸解mvMOF−5−AB結晶(400MHz、DMSO−d6)の1H NMR。BDCH2 δ:8.00(s、4H)。NH2−BDCH2 δ:7.03(d、1H)、7.38(s、1H)、7.74(d、1H)。ピークの積分に基づくモル比:BDC:NH2−BDC=1:0.57。
【0081】
mvMOF−5−AC、Zn4O(BDC)1.86(Br−BDC)1.14:0.10mLのBr−BDCH2原液(0.10M、1.0×10−5mol)、0.10mLのBDCH2原液(0.10M、1.0×10−5mol)、0.20mLのZn(NO3)2・4H2O原液(0.30M、6.0×10−5mol)及び0.60mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに24時間反応させた。生成物は立方形の淡黄色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図5)。
【0082】
蒸解mvMOF−5−AC結晶の1H NMR(400MHz、DMSO−d6)。BDCH2 δ:8.00(s、4H)。Br−BDCH2 δ:7.78(d、1H)、7.94(d、1H)、8.10(s、1H)。ピークの積分に基づくモル比:BDC:NH2−BDC=1:0.61。
【0083】
mvMOF−5−AD、Zn4O(BDC)1.83((Cl)2−BDC)1.17:0.10mLの(Cl)2−BDCH2原液(0.10M、1.0×10−5mol)、0.10mLのBDCH2原液(0.10M、1.0×10−5mol)、0.20mLのZn(NO3)2・4H2O原液(0.30M、6.0×10−5mol)及び0.60mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに24時間反応させた。生成物は立方形の淡黄色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図6)。
【0084】
蒸解mvMOF−5−AD結晶の1H NMR(400MHz、DMSO−d6)。BDCH2 δ:8.00(s、4H)。(Cl)2−BDCH2 δ:7.90(s、2H)。ピークの積分に基づくモル比:BDC:(Cl)2−BDC=1:0.63。
【0085】
mvMOF−5−AE、Zn4O(BDC)2.13(NO2−BDC)0.87:0.10mLのNO2−BDCH2原液(0.10M、1.0×10−5mol)、0.10mLのBDCH2原液(0.10M、1.0×10−5mol)、0.20mLのZn(NO3)2・4H2O原液(0.30M、6.0×10−5mol)及び0.60mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに48時間反応させた。生成物は立方形の褐色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図7)。
【0086】
蒸解mvMOF−5−AE結晶の1H NMR(400MHz、DMSO−d6)。BDCH2 δ:8.00(s、4H)。NO2−BDCH2 δ:7.92(d、1H)、8.25(d、1H)、8.35(s、1H)。ピークの積分に基づくモル比:BDC:NO2−BDC=1:0.40。
【0087】
mvMOF−5−AF、Zn4O(BDC)1.35((CH3)2−BDC)1.65:0.10mLの(CH3)2−BDCH2原液(0.10M、1.0×10−5mol)、0.10mLのBDCH2原液(0.10M、1.0×10−5mol)、0.20mLのZn(NO3)2・4H2O原液(0.30M、6.0×10−5mol)及び0.60mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに24時間反応させた。生成物は立方形の淡黄色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図8)。
【0088】
蒸解mvMOF−5−AF結晶の1H NMR(400MHz、DMSO−d6)。BDCH2 δ:8.00(s、4H)。(CH3)2−BDCH2 δ:2.43(d、6H)、7.64(s、2H)。ピークの積分に基づくモル比:BDC:(CH3)2−BDC=1:1.24。
【0089】
mvMOF−5−AG、Zn4O(BDC)1.98(C4H4−BDC)1.02:0.40mLのC4H4−BDCH2原液(0.025M、1.0×10−5mol)、0.10mLのBDCH2原液(0.10M、1.0×10−5mol)、0.20mLのZn(NO3)2・4H2O原液(0.30M、6.0×10−5mol)及び0.30mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに24時間反応させた。生成物は立方形の淡緑色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図9)。
【0090】
蒸解mvMOF−5−AG結晶の1H NMR(400MHz、DMSO−d6)。BDCH2 δ:8.00(s、4H)。C4H4−BDCH2 δ:7.64〜7.68(m、2H)、8.05(s、2H)、8.70〜8.74(m、2H)。ピークの積分に基づくモル比:BDC:C4H4−BDC=1:0.52。
【0091】
mvMOF−5−AH、Zn4O(BDC)2.04((C3H5O)2−BDC)0.96:0.10mLの(C3H5O)2−BDCH2原液(0.10M、1.0×10−5mol)、0.10mLのBDCH2原液(0.10M、1.0×10−5mol)、0.20mLのZn(NO3)2・4H2O原液(0.30M、6.0×10−5mol)及び0.60mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに24時間反応させた。生成物は立方形の褐色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図10)。
【0092】
蒸解mvMOF−5−AH結晶の1H NMR(400MHz、DMSO−d6)。BDCH2 δ:8.00(s、4H)。(C3H5O)2−BDCH2 δ:4.54(d、4H)、5.19(d、2H)、5.40(d、2H)、5.94〜6.01(m、2H)、7.30(s、2H)。ピークの積分に基づくモル比:BDC:(C3H5O)2−BDC=1:0.46。
【0093】
mvMOF−5−AI、Zn4O(BDC)2.13((C7H7O)2−BDC)0.87:0.10mLの(C7H7O)2−BDCH2原液(0.10M、1.0×10−5mol)、0.10mLのBDCH2原液(0.10M、1.0×10−5mol)、0.20mLのZn(NO3)2・4H2O原液(0.30M、6.0×10−5mol)及び0.60mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに24時間反応させた。生成物は立方形の淡黄色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図11)。
【0094】
蒸解mvMOF−5−AI結晶の1H NMR(400MHz、DMSO−d6)。BDCH2 δ:8.00(s、4H)。(C7H7O)2−BDCH2 δ:5.12(s、4H)、7.25〜7.244(m、12H)。ピークの積分に基づくモル比:BDC:(C7H7O)2−BDC=1:0.40。
【0095】
mvMOF−5−EI、Zn4O((C7H7O)2‐BDC)2.49(NIDC) 0.51:0.10mLの(C7H7O)2−BDCH2原液(0.10M、1.0×10−5mol)、0.40mLのNO2‐BDCH2原液(0.025M、1.0×10−5mol)、0.20mLのZn(NO3)2・4H2O原液(0.30M、6.0×10−5mol)及び0.30mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに48時間反応させた。生成物は立方形の褐色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図12)。
【0096】
蒸解mvMOF−5−EI結晶の1H NMR(400MHz、DMSO−d6)。NO2‐BDCH2 δ:7.92(d、1H)、8.25(d、1H)、8.35(s、1H)、(C7H7O)2‐BDCH2 δ:5.12(s、4H)、7.25〜7.44(m、12H)。ピークの積分に基づくモル比:NO2‐BDC:(C7H7O)2−BDC=0.20:1。
【0097】
mvMOF−5−ABC、Zn4O(BDC)1.90(NH2‐BDC)0.11(Br−BDC)0.99:0.133mLのBDCH2原液(0.10M、1.33×10−5mol)、0.133mLのNH2‐BDCH2原液(0.10M、1.33×10−5mol)、0.133mLのBr−BDCH2原液(0.10M、1.33×10−5mol)、0.40mLのZn(NO3)2・4H2O原液(0.30M、1.2×10−4mol)及び1.20mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに48時間反応させた。生成物は立方形の褐色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図13)。
【0098】
蒸解mvMOF−5−ABC結晶の1H NMR(400MHz、DMSO−d6)。BDCH2 δ:8.00(s、4H)。NH2‐BDCH2 δ:7.03(d、1H)、7.40(d、1H)、7.38(s、1H)。Br−BDCH2 δ:7.78(d、1H)、7.94(d、1H)、8.10(s、1H)。ピークの積分に基づくモル比:BDC:NH2−BDC:Br−BDC=1:0.052:0.52。
【0099】
mvMOF−5−AHI、Zn4O(BDC)1.52((C3H5O)2−BDC)0.73((C7H7O)2−BDC)0.75:0.133mLのBr−BDCH2原液(0.10M、1.33×10−5mol)、0.133mLの(C3H5O)2−BDCH2原液(0.10M、1.33×10−5mol)、0.133mLの(C7H7O)2‐BDCH2原液(0.10M、1.33×10−5mol)、0.40mLのZn(NO3)2・4H2O原液(0.30M、1.2×10−4mol)及び1.20mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに24時間反応させた。生成物は立方形の褐色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図14)。
【0100】
蒸解mvMOF−5−AHI結晶の1H NMR(400MHz、DMSO−d6)。BDCH2 δ:8.00(s、4H)。(C3H5O)2−BDCH2 δ:4.55(d、4H)、5.20(d、2H)、5.38(d、2H) 、5.93〜6.00(m、2H)。(C7H7O)2‐BDCH2 δ:5.12(s、4H)、7.26〜7.44(m、12H)。ピークの積分に基づくモル比:BDC:(C3H5O)2−BDC:(C7H7O)2−BDC=1:0.48:0.50。
【0101】
mvMOF−5−EHI、Zn4O(NO2−BDC)1.19((C3H5O)2−BDC)1.07((C7H7O)2−BDC)0.74:0.133mLの(C7H7O)2−BDCH2原液(0.10M、1.33×10−5mol)、0.133mLのNO2−BDCH2原液(0.10M、1.33×10−5mol)、0.133mLの(C3H5O)2−BDCH2原液(0.10M、1.33×10−5mol)、0.40mLのZn(NO3)2・4H2O原液(0.30M、1.2×10−4mol)及び1.20mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに24時間反応させた。生成物は立方形の褐色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図15)。
【0102】
蒸解mvMOF−5‐EHI結晶の1H NMR(400MHz、DMSO−d6)。NO2−BDCH2 δ:7.93(d、1H)、8.25(d、1H)、8.35(s、1H)。(C3H5O)2−BDCH2 δ:4.55(d、4H)、5.20(d、2H)、5.38(d、2H) 、5.93〜6.00(m、2H)。(C7H7O)2−BDCH2 δ:5.12(s、4H)、7.26〜7.44(m、12H)。ピークの積分に基づくモル比:NO2−BDC:(C3H5O)2−BDC:(C7H7O)2−BDC=1:0.89:0.62。
【0103】
mvMOF−5−ABCD、Zn4O(BDC)1.44(NH2−BDC)0.18(Br−BDC)0.81((Cl)2−BDC)0.57:0.10mLのBDCH2原液(0.10M、1.0×10−5mol)、0.10mLのNH2−BDCH2原液(0.10M、1.0×10−5mol)、0.10mLのBr−BDCH2原液(0.10M、1.0×10−5mol)、0.10mLの(Cl)2−BDCH2原液(0.10M、1.0×10−5mol)、0.40mLのZn(NO3)2・4H2O原液(0.30M、1.2×10−4mol)及び1.20mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに48時間反応させた。生成物は立方形の暗褐色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図16)。
【0104】
蒸解mvMOF−5−ABCD結晶の1H NMR(400MHz、DMSO−d6)。BDCH2 δ:8.00(s、4H)。NH2−BDCH2 δ:7.03(d、1H)、7.40(d、1H)、7.38(s、1H)。Br−BDCH2 δ:7.78(d、1H)、7.94(d、1H)、8.10(s、1H)。(Cl)2−BDCH2 δ:7.90(s、2H)。ピークの積分に基づくモル比:BDC:NH2−BDC:Br−BDC:(Cl)2−BDC =1:0.12:0.56:0.40。
【0105】
mvMOF−5−ACEF、Zn4O((BDC)1.29(Br−BDC)0.63(NO2−BDC)0.28((CH3)2−BDC)0.80:0.10mLのBDCH2原液(0.10M、1.0×10−5mol)、0.10mLのBr−BDCH2原液(0.10M、1.0×10−5mol)、0.10mLのNO2−BDCH2原液(0.10M、1.0×10−5mol)、0.10mLの(CH3)2−BDCH2原液(0.10M、1.0×10−5mol)、0.40mLのZn(NO3)2・4H2O原液(0.30M、1.2×10−4mol)及び1.20mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに24時間反応させた。生成物は立方形の暗褐色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図17)。
【0106】
蒸解mvMOF−5−ACEF結晶の1H NMR(400MHz、DMSO−d6)。BDCH2 δ:8.00(s、4H)。Br−BDCH2 δ:7.78(d、1H)、7.94(d、1H)、8.10(s、1H)。NO2−BDCH2 δ:7.92(d、1H)、8.25(d、1H)、8.35(s、1H)。(CH3)2−BDCH2 δ:2.43(s、6H)、7.64(s、2H)。ピークの積分に基づくモル比:BDC:Br−BDC:(CH3)2−BDC:NO2−BDC=1:0.49:0.62:0.22。
【0107】
mvMOF−5−ABCHI、Zn4O(BDC)1.38(NH2−BDC)0.03(Br−BDC)0.30((C3H5O)2−BDC)0.86((C7H7O)2−BDC)0.43:80μLのBDCH2原液(0.10M、8.0×10−6mol)、80μLのNH2−BDCH2原液(0.10M、8.0×10−6mol)、80μLのBr −BDCH2原液(0.10M、8.0×10−6mol)、80μLの(C3H5O)2−BDCH2原液(0.10M、8.0×10−6mol)、80μLの(C7H7O)2−BDCH2原液(0.10M、8.0×10−6mol)、0.40mLのZn(NO3)2・4H2O原液(0.30M、1.2×10−4mol)及び1.20mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに48時間反応させた。生成物は立方形の暗褐色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図18)。
【0108】
蒸解mvMOF−5−ABCHI結晶の1H NMR(400MHz、DMSO−d6)。BDCH2 δ:8.00(s、4H)。NH2−BDCH2 δ:7.03(d、1H)。Br−BDCH2 δ:7.78(d、1H)、7.94(d、1H)、8.10(s、1H)。(C3H5O)2−BDCH2 δ:4.54(d、4H)、5.19(d、2H)、5.40(d、2H)、5.94〜6.01(m、2H)。(C7H7O)2−BDCH2 δ:5.12(s、4H)、7.25〜7.44(m、12H)。ピークの積分に基づくモル比:BDC:NH2−BDC:Br−BDC:(C3H5O)2−BDC :(C7H7O)2−BDC=1:0.017:0.22:0.62:0.32。
【0109】
mvMOF−5−ABCGHI、Zn4O(BDC)0.72(NH2−BDC)0.08(Br−BDC)0.63((C4H4−BDC)0.48((C3H5O)2−BDC)0.52((C7H7O)2−BDC)0.57:67μLのBDCH2原液(0.10M、6.7×10−6mol)、67μLのNH2−BDCH2原液(0.10M、6.7×10−6mol)、67μLのBr −BDCH2原液(0.10M、6.7×10−6mol)、267μLのC4H4−BDCH2原液(0.10M、6.7×10−6mol)、67μLの(C3H5O)2−BDCH2原液(0.10M、6.7×10−6mol)、67μLの(C7H7O)2−BDCH2原液(0.10M、6.7×10−6mol)、0.40mLのZn(NO3)2・4H2O原液(0.30M、1.2×10−4mol)及び1.0mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに48時間反応させた。生成物は立方形の暗褐色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図19)。
【0110】
蒸解mvMOF−5−ABCGHI結晶の1H NMR(400MHz、DMSO−d6)。BDCH2 δ:8.00(s、4H)。NH2−BDCH2 δ:7.03(d、1H)。Br−BDCH2 δ:7.78(d、1H)、7.94(d、1H)、8.10(s、1H)。C4H4−BDCH2 δ:7.64〜7.68(m、2H)、8.05(s、2H)、8.70〜8.74(m、2H)。(C3H5O)2−BDCH2 δ:4.54(d、4H)、5.19(d、2H)、5.40(d、2H)、5.94〜6.01(m、2H)。(C7H7O)2−BDCH2 δ:5.12(s、4H)、7.25〜7.44(m、12H)。ピークの積分に基づくモル比:BDC:NH2−BDC:Br−BDC:C4H4−BDC:(C3H5O)2−BDC:(C7H7O)2−BDC=1:0.093:0.87:0.67:0.73:0.80。
【0111】
mvMOF−5−ABCEGHI、Zn4O(BDC)0.57(NH2−BDC)0.05(Br−BDC)0.57(NO2−BDC)0.39(C4H4−BDC)0.44((C3H5O)2−BDC)0.42((C7H7O)2−BDC)0.56:57μLのBDCH2原液(0.10M、5.7×10−6mol)、57μLのNH2−BDCH2原液(0.10M、5.7×10−6mol)、57μLのBr−BDCH2原液(0.10M、5.7×10−6mol)、57μLのNO2−BDCH2原液(0.10M、5.7×10−6mol)、228μLのC4H4−BDCH2原液(0.10M、5.7×10−6mol)、57μLの(C3H5O)2−BDCH2原液(0.10M、5.7×10−6mol)、57μLの(C7H7O)2−BDCH2原液(0.10M、5.7×10−6mol)、0.40mLのZn(NO3)2・4H2O原液(0.30M、1.2×10−4mol)及び1.03mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに48時間反応させた。生成物は立方形の暗褐色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図20)。
【0112】
蒸解mvMOF−5−ABCEGHI結晶の1H NMR(400MHz、DMSO−d6)。BDCH2 δ:8.00(s、4H)。NH2−BDCH2 δ:7.03(d、1H)。Br−BDCH2 δ:7.78(d、1H)、7.94(d、1H)、8.10(s、1H)。NO2−BDCH2 δ:7.92(d、1H)、8.25(d、1H)、8.35(s、1H)。C4H4−BDCH2 δ:7.64〜7.68(m、2H)、8.05(s、2H)、8.70〜8.74(m、2H)。(C3H5O)2−BDCH2 δ:4.54(d、4H)、5.19(d、2H)、5.40(d、2H)、5.94〜6.01(m、2H)。(C7H7O)2−BDCH2 δ:5.12(s、4H)、7.25〜7.44(m、12H)。ピークの積分に基づくモル比:BDC:NH2−BDC:Br−BDC:NO2−BDC:C4H4−BDC:(C3H5O)2−BDC:(C7H7O)2−BDC=1:0.077:1:0.69:0.77:0.73:0.96。
【0113】
mvMOF−5−ABCEFGHI、Zn4O(BDC)0.70(NH2−BDC)0.011(Br−BDC)0.39(NO2−BDC)0.21((CH3)2−BDC)0.46(C4H4−BDC)0.39((C3H5O)2−BDC)0.35((C7H7O)2−BDC)0.39:50μLのBDCH2原液(0.10M、5.0×10−6mol)、50μLのNH2−BDCH2原液(0.10M、5.0×10−6mol)、50μLのBr−BDCH2原液(0.10M、5.0×10−6mol)、50μLのNO2−BDCH2原液(0.10M、5.0×10−6mol)、50μLの(CH3)2−BDCH2原液(0.10M、5.0×10−6mol)、200μLのC4H4 −BDCH2原液(0.10M、5.0×10−6mol)、50μLの(C3H5O)2−BDCH2原液(0.10M、5.0×10−6mol)、50μLの(C7H7O)2−BDCH2原液(0.10M、5.0×10−6mol)、0.40mLのZn(NO3)2・4H2O原液(0.30M、1.2×10−4mol)及び1.05mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに48時間反応させた。生成物は立方形の暗褐色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図21)。
【0114】
蒸解mvMOF−5−ABCEFGHI結晶の1H NMR(400MHz、DMSO−d6)。BDCH2 δ:8.00(s、4H)。NH2−BDCH2 δ:7.03(d、1H)。Br−BDCH2 δ:7.78(d、1H)、7.94(d、1H)、8.10(s、1H)。NO2−BDCH2 δ:7.92(d、1H)、8.25(d、1H)、8.35(s、1H)。(CH3)2−BDCH2 δ:2.43(s、6H)、7.64(s、2H)。C4H4−BDCH2 δ:7.64−7.68(m、2H)、8.05(s、2H)、8.70−8.74(m、2H)。(C3H5O)2−BDCH2 δ:4.54(d、4H)、5.19(d、2H)、5.40(d、2H)、5.94−6.01(m、2H)。(C7H7O)2−BDCH2 δ:5.12(s、4H)、7.25−7.44(m、12H)。ピークの積分に基づくモル比:BDC:NH2−BDC:Br−BDC:(CH3)2−BDC:NO2−BDC:C4H4−BDC:(C3H5O)2−BDC:(C7H7O)2−BDC=1:0.14:0.56:0.29:0.67:0.56:0.48:0.56。
【0115】
2個、3個又は4個の異なる連結、及びmvMOF−5−ABCEFGHIを含有する全ての化合物を、上記したのと同じ濃度でグラムスケールにスケールアップした。スケールアップした試料のPXRDは、小スケール試料と同一である。さらに、スケールアップした試料のバルク均一性を、無作為に選択した結晶溶液1H NMRによって調べたところ、各化合物において同一の連結比を示した。
【0116】
熱重量分析。全ての試料を、試料を持続的気流雰囲気中の白金受皿に保持するTA Instruments Q−500シリーズ熱重量分析器にかけた。試料を5℃/分の一定速度で全てのTGA実験中に加熱した。
【0117】
連結におけるダングリング二重結合及びベンゼン環の損失により、連結H及び連結Iを含有するmvMOFは、200℃から徐々に重量を失い、続いて400℃で急に低下する。全ての他のmvMOFは400℃まで重量を失わない。
【0118】
活性化mvMOFの固体状態13C MAS NMR。高い分解能の固体状態核磁気共鳴(NMR)スペクトルを周囲温度で、4mm(外径)のジルコニア回転子を持つ標準Brukerマジックアングルスピニング(MAS)プローブを用いるBruker DSX−300分光計上に記録した。MASとともに交差分極(CP/MAS)を使用することで、13Cデータを75.47MHzで獲得した。1H及び13Cの90度パルス幅はともに4μsであった。CP接触時間は1.5分及び5分で変動した。高出力2パルス位相変調(TPPM)1Hデカップリングを、データ獲得中に適用した。デカップリング周波数は72kHzに相当した。MAS試料スピニング速度は10kHzであった。走査間の(CP/MAS)に関するリサイクル遅延は、13C信号強度において一走査から次の走査まで明らかな損失を認められないことによって決定された通りの化合物に依存して、3秒から20秒の間で変動した。第二次参照として37.77ppmに定めたアダマンタンのメチレン炭素信号を使用して較正された、テトラメチルシランをゼロppmとして割り当てた13C化学シフトを、表2に列挙した。
【0119】
mvMOF−5−AB、−ABCD及び−ABCEFGHIにおけるのと同じ比率の連結混合物に対する13C CP/MAS NMRの対照実験を調べた。mvMOFに対する実験と比較する172ppmから174ppmまで、カルボニル炭素の有意なシフトがある。これは、これらの3個のmvMOFにおける全ての連結が亜鉛クラスターに化学的に結合していることを実証している。さらに、静電気試料の13Cスペクトルを、CP/MAS方法を使用して獲得し、移動ゲスト分子を検出するため調べた。この実験において、リサイクル遅延を20秒に設定することで、走査間の十分な緩和を可能にした。mvMOFsの完全な細孔構造を確証する検出可能な炭素信号は発見されなかった。
【表2】
【0120】
大きな単結晶mvMOF−5−AB−lscの合成。4.20gのZn(NO3)2・4H2O(16.0mmol)、450mgのBDCH2(2.7mmol)及び490mgのNH2−BDCH2(2.7mmol)を、混合物を15分間超音波処理することによって、50mLDEF中にガラスビーカー内で溶解した。PTFEフィルター(Whatman、0.45μmの孔径)を備えたプラスチックシリンジを使用することによって、溶液を10シンチレーションバイアル(20mLサイズ)に均等に分配した。バイアルに次いでしっかり蓋をし、等温の乾燥器内に置いた。85℃で72時間加熱した後で反応を止めた。各バイアルにおける母液を温めながらデカントし、生成物を新たなDEF(各バイアルに対して3×5mL)で洗浄した。生成物の大部分は、大きなチャンクの内部成長した立方褐色結晶であった。時折、一部の大きな単一立方体(サイズ1.5〜4.0mm)が認められた。上記した通りの通常のバッチに、8〜13個のこうした結晶を得ることができた。大きな立方結晶はMOF−5トポロジーを有することが、実験PXRDパターンがシミュレートした一つと一致すること、及びこれらの結晶を光学顕微鏡下で検査することによって確認された(図36A、B及び37)。高い当初の濃度が、大きなmvMOF単結晶の形成に有用である。
【0121】
大きな単結晶mvMOF−5−ABCD−lscの合成。6.20gのZn(NO3)2・4H2O(23.5mmol)、332mgのBDCH2(2.0mmol)及び362mgのNH2−BDCH2(2.0mmol)、490mgのBr−BDCH2(2.0mmol) 及び470mgの(Cl)2−BDCH2(2.0mmol)を、混合物を15分間超音波処理することによって、50mLのDEF中にガラスビーカー内で溶解した。PTFEフィルター(Whatman、0.45μmの孔径)を備えたプラスチックシリンジを使用することによって、溶液を10シンチレーションバイアル(20mLサイズ)に均等に分配した。バイアルに次いでしっかり蓋をし、等温の乾燥器内に置いた。85℃で120時間加熱した後で反応を止めた。各バイアルにおける母液を温めながらデカントし、生成物を新たなDEF(各バイアルに対して3×5mL)で洗浄した。生成物の大部分は、大きなチャンクの内部成長した褐色立方結晶であった。時折、一部の大きな単一立方体(サイズ1.0〜2.0mm)が認められた。上記した通りの通常のバッチに、6〜10個のこうした結晶を得ることができた。大きな立方結晶はMOF−5トポロジーを有することが、実験PXRDパターンがシミュレートした一つと一致すること、及びこれらの結晶を光学顕微鏡下で検査することによって確認された(図36C、D及び38)。
【0122】
大きな単結晶mvMOF−5−ABCEFGHI−lscの合成。4.20gのZn(NO3)2・4H2O(23.5mmol)、166mgのBDCH2(1.0mmol)、181mgのNH2−BDCH2(1.0mmol)、245mgのBr−BDCH2(1.0mmol)、211mgのNO2−BDCH2(1.0mmol)、194mgの(CH3)2−BDCH2(1.0mmol)、216mgのC4H4−BDCH2(1.0mmol)、278mgの(C3H5O)2−BDCH2(1.0mmol)及び374mgの(C7H7O)2−BDCH2(1.0mmol)を、混合物を15分間超音波処理することによって、50mLのDEF中にガラスビーカー内で溶解した。PTFEフィルター(Whatman、0.45μmの孔径)を備えたプラスチックシリンジを使用することによって、溶液を10シンチレーションバイアル(20mLサイズ)に均等に分配した。バイアルに次いでしっかり蓋をし、等温の乾燥器内に置いた。85℃で7日間加熱した後で反応を止めた。各バイアルにおける母液を温めながらデカントし、生成物を新たなDEF(各バイアルに対して3×5mL)で洗浄した。生成物の大部分は、大きなチャンクの内部成長した褐色立方結晶であった。時折、一部の大きな単一立方体(サイズ1.0〜2.0mm)が認められた。上記した通りの通常のバッチに、5〜6個のこうした結晶を得ることができた。大きな立方結晶はMOF−5トポロジーを有することが、実験PXRDパターンがシミュレートした一つと一致すること、及びこれらの結晶を光学顕微鏡下で検査することによって確認された(図36E、F及び39)。
【0123】
mvMOFの大きな単結晶の溶媒交換。適当な結晶を20mLシンチレーションバイアルに回収した。ピペットを使用してDEF溶媒を可能な限りきれいに除去した後、この開口バイアルを、クロロホルム蒸気で飽和したデシケーター中に置いたところ、該蒸気はゆっくりバイアル中に凝縮し、3日で約5mmの高さに蓄積した。結晶はこの時点でまだ完全に透明であった。蓄積したクロロホルムの除去後、バイアルに新たなクロロホルムを充填し、蓋をした。溶媒容量をそれぞれ1日目及び2日目の浸漬後に2度交換し、さらに2日間静置した。mvMOFの大きな単結晶のクロロホルム交換の合計時間は、デシケーター中で3日及び寝かせて5日であった。
【0124】
mvMOF内における連結の比率を1H NMRで決定した。結果を表1に列挙した。
【0125】
mvMOF−5−AB系列の合成。0.30MのZn(NO3)2・4H2O原液、0.10MのBDCH2(連結A)及びNH2−BDCH2(連結B)原液を事前に調製した。様々な容量の連結A及び連結B原液を2.0mLの0.30MのZn(NO3)2・4H2O原液中に、続いてDEFを添加することで、20mLのガラスバイアル中における合計容量を10mLにした。バイアルを密封し、等温の乾燥器内にて100℃で加熱し、48時間ソルボサーマルに反応させた。11種のmvMOF−5−AB化合物それぞれa〜kを合成したが、ここで、mvMOF−5−AB−aは連結Aをのみ有し、mvMOF−5−AB−kは連結Bのみを有しているので、それらは実際にそれぞれMOF−5及びIRMOF−3である。全ての生成物は、立方形の単結晶の形態であった。各化合物の結晶化度をPXRDによって確認したが、シミュレートしたMOF−5パターンと対応する非常に高い程度は、これらの化合物が本来不変のMOF−5トポロジー(図40)であることを表している。mvMOF内における連結の比率を、前に記載されているのと同様に1H NMRによって決定する。表3に、各化合物に使用した原液の量、及びそれらの当初の連結比、及びそれらの生成物の連結比を概要した。mvMOF−5−AB系列の結晶生成物におけるBのパーセント比率を、当初の化学量論比に対してプロットした(図41)。より多くの連結Bを出発原料として添加するにつれて、より多くの連結Bが結晶生成物に発見された。これは、mvMOFにおける連結比が、全範囲において、合成中に使用される連結の当初の量を制御することによりたやすく微調整することができることを明確に実証している。NH2−BDC(連結B)はMOF構造においてその特有な赤色を有するが、ここで、さらにmvMOFにおける連結比の正確な制御の能力を表す、合成されたままのmvMOF−5−AB結晶の色における段階的変化が認められた。連結Bの百分率が化合物aからkに増加するにつれて、結晶の色は無色から赤色に、次いで暗赤色に変化する(図41)。
【表3】
【0126】
mvMOF−5−AI系列の合成。0.30MのZn(NO3)2・4H2O原液、0.10MのBDCH2(連結A)及び(C7H7O)2−BDCH2(連結I)原液を事前に調製した。様々な容量の連結A及び連結I原液を2.0mLの0.30M Zn(NO3)2・4H2O原液中に、続いてDEFを添加することで、20mLのガラスバイアルにおける合計容量を10mLにした。バイアルを密封し、等温の乾燥器内にて100℃で加熱し、24時間ソルボサーマルに反応させた。3種のmvMOF−5−AI化合物それぞれa〜cを合成した。全ての生成物は、立方形の単結晶の形態であった。各化合物の結晶化度をPXRDによって確認したが、シミュレートしたMOF−5パターンと対応する非常に高い程度は、これらの化合物が本来不変のMOF−5トポロジー(図42)であることを表している。mvMOF内における連結の比率を、前に記載されているのと同様に1H NMRによって決定する。表4に、各化合物に使用した原液の量、及びそれらの当初の連結比、及びそれらの生成物の連結比を概要した。
【0127】
mvMOF−5−AB系列と同様に、mvMOF−5−AI系列の結晶生成物におけるIのパーセント比率は、より多くの連結Iが出発原料中に使用されたことに伴い増加する。
【表4】
【0128】
mvMOF−5−ABCD系列の合成。0.30MのZn(NO3)2・4H2O原液、0.10MのBDCH2(連結A)及びNH2−BDCH2(連結B)、Br−BDCH2(連結C)、(Cl)2−BDCH2(連結D)原液を事前に調製した。様々な容量の連結A及び連結I原液を2.0mLの0.30M Zn(NO3)2・4H2O原液中に、続いてDEFを添加することで、20mLのガラスバイアルにおける合計容量を10mLにした。バイアル密封し、等温の乾燥器内にて100℃で加熱し、48時間ソルボサーマルに反応させた。5種のmvMOF−5−ABCD化合物それぞれa〜eを合成した。全ての生成物は、立方形の単結晶の形態であった。各化合物の結晶化度をPXRDによって確認したが、シミュレートしたMOF−5パターンと対応する非常に高い程度は、これらの化合物が本来不変のMOF−5トポロジー(図43)であることを表している。mvMOF内における連結の比率は、セクション1に記載されているのと同様に1H NMRによって決定する。表5に、本文における各化合物及び表1(連結比部分の制御)に使用された原液の量を概要した。mvMOF−5−ABCD結晶の連結比を各化合物の合成に使用された連結の化学量論比と比較した場合、出発原料中と同様に使用された特定の連結の量が増加(又は減少)するにつれて、生じるmvMOFにおける比率は対応して増加(又は減少)する。これらは、mvMOFs内における連結の比率が制御可能であることを再び実証している。
【表5】
【0129】
mvMOF−5−AC及び−ACEFに関する単一X線回折データ収集、構造溶液及び精密化の手順。単結晶データ収集、構造溶液及び精密化に関する一般的手順をここに提示する。構造上の不規則を含めて各構造の固有の詳細を、各構造に関する実験及び計量の前に記載する。
【0130】
一般のデータ収集及び精密化の手順:CCDエリア検出器を備え及び1200W出力(40kV、30mA)で操作されるBruker SMART APEXII三軸回折計上にデータを回収して、CuKα放射線(λ=1.5418Å)を発生させた。Bruker Excalibur Gobelミラー光学を使用して入射X線ビームを集中させ、単色にした。少量の母液を含有するフレームシールしたボロシリケート細管に結晶を全て装填することで、データ収集中の脱溶媒和を防止した。
【0131】
各試験片の当初のω−φを走査することで、予備単位格子パラメーターが得られ、結晶のモザイク性(すなわち、骨組み間の箇所の幅)を評価して、データ収集に必要なフレーム幅を選択した。全ての場合に関して、0.5°の骨組み幅が適切であると判断し、異なる検出器(2θ)の設定でφ及びωの走査を重複することを実施するため、Bruker APEX2ソフトウェア一式を使用してデータの完全半球を回収した。データ収集に続いて、データ統合用の単位格子パラメーターを再決定するため、及びCELL_NOWを使用して回転双晶化を調べるため、Ewald球体の全ての領域から反射光をサンプリングした。
【0132】
回収した骨組みの徹底的見直しに続いて、データセットの分解能を判断し、必要に応じて、干渉性散乱が認められない骨組みの領域をデータ統合に対する検討から除いた。Bruker APEX2 V2.1ソフトウェアを使用し、狭いフレームアルゴリズム及び平均強度の少数下限を0.400で、データを統合した。吸収補正はデータの品質を向上させるのに効果的ではなかったが、これは、主に軽原子を含有する低密度材料の小さな結晶には予想外ではない。
【0133】
XPREPを使用して、欠面双晶化に関する空間群決定及び試験を実施した。全ての場合において、最大限の空間群を選択し、欠面双晶化の兆候は認められなかった。全ての構造を直接的方法によって解析し、SHELXTL’97ソフトウェア一式を使用して精密化した。初期モデルの最小二乗精密化に続いて差異Fマップの反復検査から、原子の位置を決定した。完全な収束が達成されるまで、最終モデルを異方的に精密化した(許容されたデータの数及び安定な精密化が達する場合)。部分的占有率を、不規則化基における原子の各対に割り当てた。一部の原子は、高不規則及び低占有率のため、位置を正確に決定できなかった(mvMOF−ACEFにおける連結E上のニトロ基)。水素原子を算出位置に置き、ライディング原子として含めた。骨格の空隙内の電子密度のモデル化は、低減したデータ分解能のため、構造における全てのゲスト実体の同定に至らなかった。溶媒が充填された細孔を含有する多孔質結晶に典型的であるこの問題は、認められた強力な(高強度)散乱がせいぜい約1.0Åに制限される生データにあり、より高い分解能データが存在するが弱い(低強度)。X線データを向上させるための一般戦略のように、結晶をX線に暴露する時間を増加させることは、低角度データからの強度が検出器を飽和し、高角度データにおいて最小限の向上が達成されたため、これらの場合における高角度データの品質を改良しなかった。さらに、MOF主鎖のフェニル単位上の不規則化官能基からの散漫散乱、及び結晶内の空隙空間における溶媒、及び結晶を装填させるのに使用された細管からの溶媒が、背景雑音及び時々高角度データの「洗い出し」に寄与した。これらの拡張骨格構造に関して、構造におけるゲスト実体が自由に移動することが可能になり、したがって認められた構造因子に干渉性散乱期間を与えない場合、より高い範囲の温度(−120から−100℃よりむしろ−50から−15℃)で回収したデータに反してモデル化することも、より妥当であった。
【0134】
骨格における原子位置の精度を証明するため、適用可能な場合、A.SpekのSQUEEZEルーチンの適用が行われてきた。しかし、「非SQUEEZE」構造のための原子配位も提案されており、プログラムSQUEEZEが用いられた場合にCIFも提示された。PLATONのAdsymサブルーチンを使用して全ての構造を検査することで、追加の対称がモデルに適用され得ることがないことを確保した。ORTEP図表における全ての楕円体は、別段の注記がない限り、15%の確率レベルで表示されている。全ての構造に関して、上昇したR値は、上記に表した理由により、MOF結晶学において一般に遭遇する。
【0135】
mvMOF−5−ACに関する実験及び精密化の詳細。mvMOF−5−ACの無色平行六面体結晶(0.41×0.41×0.28mm3)を、0.5mm直径のボロシリケート細管中に少量の母液と一緒に置いた。細管をフレームシールし、CCDエリア検出器を備え、1200W出力(40kV、30mA)で操作されるSMART APEXII三軸回折計上に装填することで、窒素の液体N2冷却流中で258(2)Kに冷却しながらCuKα放射線(λ=1.5418Å)を発生させた。3種の異なる検出器(2θ)設定(2θ=28°、60°、100°)でφ及びωの走査を重複することを実施するため、Bruker APEX2ソフトウェア一式を使用してデータの完全半球を回収した。合計14178の反射光を回収し、これらのうち2951が固有であり、これらの1994が2σ(I)を超えていた。θの範囲は2.97°から41.28°であった。データの分析は回収中の軽視し得る減衰を示した。プログラムScaleを行うことで、対称関連反射光又は繰り返し測定した反射光の間の差異を最小にした。
【0136】
直接的方法を使用してZ=8で単斜C2/m空間群において、構造を解析した。水素原子をそれらの親原子の配位に乗る球体として生成して、骨格の主鎖におけるZn及びO原子を異方的に精密化し、全ての他の非水素原子を等方的に精密化した。モデル溶媒分子にした試みは、ゲスト実体の同定に至らなかった。溶媒が骨格に結合していないので、MOF構造に関して溶媒分子の不正確な位置決定が予想された。さらに、非常に高い置き換えパラメーター、不規則による高いesd及び部分的な占有率が、溶媒分子の正確な位置を決定することを不可能にした。それでもなお、mvMOF−5−ACの主鎖骨格及び−Br基の帰属及び精密化は、生じた結合及び角度の測定基準によって判断した通り明らかであった。
【0137】
骨格における原子位置を向上するため、A.SpekのSQUEEZEルーチンの適用が行われてきた。しかし、「非SQUEEZE」構造に関する原子配位も提案されている。F2に対する最終フルマトリックス最小二乗精密化は、GOF=0.998で、R1=0.1257(F>2σ(F))及びwR2=0.3084(全データ)に収束した。SQUEEZEプログラムが用いられていない場合の構造では、F2に対する最終フルマトリックス最小二乗精密化は、GOF=1.426で、R1=0.1892(F>2σ(F))及びwR2=0.4184(全データ)に収束した。骨格原子のみを後者の構造因子算出に含めた場合、Fマップにおける残留電子密度は、mvMOF−5−ACの細孔内に位置していた。結晶構造精密化のための実験式は、0.683gcm−3の密度で、1H NMRによる結晶における2種の連結の測定比率に基づき、C12 H5.43 Br0.57 O7.25 Zn2である。
【表6】
【表7】
【表8】
【表9】
【表10】
【表11】
【0138】
mvMOF−5−ACに関する実験及び精密化の詳細。mvMOF−5−ACEFの赤色平行六面体結晶(0.30×0.30×0.21mm3)を、0.4mm直径のボロシリケート細管中に少量の母液と一緒に置いた。細管をフレームシールし、CCDエリア検出器を備え、1200W出力(40kV、30mA)で操作されるSMART APEXII三軸回折計上に装填することで、窒素の液体N2冷却流中で233(2)Kに冷却しながらCuKα放射線(λ=1.5418Å)を発生させた。3種の異なる検出器(2θ)設定(2θ=28°、60°、100°)でφ及びωの走査を重複することを実施するため、Bruker APEX2ソフトウェア一式を使用してデータの完全半球を回収した。合計13758の反射光を回収し、これらのうち2855が固有であり、これらの1918が2σ(I)を超えていた。θの範囲は2.99°から41.02°であった。データの分析は回収中の軽視し得る減衰を示した。プログラムScaleを行うことで、対称関連反射光又は繰り返し測定した反射光の間の差異を最小にした。
【0139】
直接的方法を使用してZ=8で単斜C2/m空間群において、構造を解析した。水素原子をそれらの親原子の配位に乗る球体として生成して、骨格の主鎖におけるZn原子を異方的に精密化し、全ての他の非水素原子を等方的に精密化した。モデル溶媒分子にした試みは、ゲスト実体の同定に至らなかった。溶媒が骨格に結合していないので、MOF構造に関して溶媒分子の不正確な位置決定が予想された。さらに、非常に高い置き換えパラメーター、不規則による高いesd及び部分的な占有率が、溶媒分子の正確な位置を決定することを不可能にした。それでもなお、mvMOF−5−ACEFの主鎖骨格及び官能基(−Br及び−CH3)の一般位置の帰属及び精密化は、生じた結合及び角度の測定基準によって判断した通り明らかであった。低い占有率(モルパーセントで9.4%のみ)及び四つの位置上のその不規則を考えると、非常に少量で存在するNO2の組成は、したがって、この精密化において無視されている。
【0140】
骨格における原子位置の精度を向上するため、A.SpekのSQUEEZEルーチンの適用が行われてきた。しかし、「非SQUEEZE」構造に関する原子配位も提案されている。F2に対する最終フルマトリックス最小二乗精密化は、GOF=1.003で、R1=0.1219(F>2σ(F))及びwR2=0.3041(全データ)に収束した。SQUEEZEプログラムが用いられていない場合の構造では、F2に対する最終フルマトリックス最小二乗精密化は、GOF=1.378で、R1=0.1737(F>2σ(F))及びwR2=0.3972(全データ)に収束した。骨格原子のみを後者の構造因子算出に含めた場合、Fマップにおける残留電子密度は、mvMOF−5−ACEFの細孔内に位置していた。結晶構造精密化の実験式は、1H NMRによる結晶における4種の連結の測定比率に基づき、C12.81 H6.65 Br0.31 N0.14 O7.78 Zn2であった。
【表12】
【表13】
【表14】
【表15】
【表16】
【表17】
【0141】
mvMOFの多孔性、H2取込み、及びCO2分離研究。全ての低圧力ガス吸着実験(最大1バール)をQuantachrome Autosorb−1自動容量測定器上で行った。液体窒素槽(77K)及び液体アルゴン槽(87K)をN2、H2及びArの等温線測定に使用し、一方恒温槽(273k、283k及び298K)をCO2及びCOの等温線測定に使用した。超高純度グレードのN2、H2、Ar、CO、He(99.999%純度)及びCO2のガス(99.995%純度)を吸着実験全体で使用した。ガスの非理想度を実験温度で第二ビリアル係数から得た。見かけ表面積の測定のため、N2(Ar)断面積を16.2(14.2)Å2/分子と仮定してN2(Ar)等温線の吸着枝を使用するラングミュア方法を適用した。MOFの合成されたままの試料をクロロホルム中に周囲温度で24時間を3周期の間浸漬し、周囲温度で12時間脱気した。
【0142】
構成剛性、並びにその結果としての脱気したMOF−5、mvMOF−5−AB、−AI−a、−ABCD及び−ABCEFGHIの永久的多孔性を、ガス収着分析によって明解に証明した。これらのMOFに関してI型N2/Ar吸着等温線の挙動が認められ(図46及び47)、微孔性の性質を明らかにした。mvMOF−ABのラングミュア表面積を算出したところ、3640m2/gであった。同様に、MOF−5、mvMOF−5−AI−a、−ABCD及び−ABCEFGHIの見かけ表面積を算出したところ、それぞれ4140m2/g、2680m2/g、2860m2/g及び1860m2/gであった。MVMOF−5−AI系列において、より嵩高い連結Iを結晶生成物に導入するにつれて、算出密度は−aから−cへと増加し、表面積は減少する(図49)。
【表18】
【0143】
mvMOF−5−AI及びMOF−5の孔径分布分析。さらに、嵩高い連結を持つmvMOFの一つであるMOF−5−AI−aの孔径分布を研究した。MOF−5−AI−aに関するAr吸着等温線と非局所密度汎関数理論(NLDFT)モデルとの一致は、孔径分布が12Å未満で大きく集中していることを明らかにしており、これはMOF−5の典型的孔径分布である(図41)。これは、MOF−5−AI−aのほとんど全ての大きなケージが連結Iによって部分的に官能化されており、MOF−5の細孔環境が存在しないことを表している。
【0144】
mvMOF−5−EIに関するCO2及びCOの吸着等温線は、図48〜51にそれぞれ例示されている。確かなヘンリー定数を推定するため、温度非依存性パラメーターai及びbiを含むビリアル型数式を適用した。
【数1】
【0145】
ここで、Pは圧力であり、Nは吸着量であり、Tは温度であり、並びにm及びnは等温線を適切に表示するのに必要な係数の数を表す。これらの結果から、ヘンリー定数(KH)を算出するが、Tは温度である。
【数2】
【0146】
298Kでのガス成分jを超えたiのヘンリー則選択率を等式(3)に基づいて算出する。
【数3】
【0147】
本発明の実施形態が多数記載されている。それでもなお、本発明の趣旨及び範囲から逸脱することなく様々な改変が行われ得ることを理解されよう。すなわち、他の実施形態は、以下の請求項の範囲内である。
【技術分野】
【0001】
本発明は、混合リガンドを含むセンサとして使用するための、ガス分離、貯蔵用有機骨格を提供する。
【背景技術】
【0002】
結晶拡張構造は、少数の別個の構築単位から構成されている点において「単純」である傾向がある。
【発明の概要】
【発明が解決しようとする課題】
【0003】
拡張結晶固体の構造は、少数の別個の構築単位から通常構築されている点において根本的に「単純」である。このことは、1種類の連結、官能性基及び金属イオン単位から一般に構成されている金属有機骨格(MOF)において確実に当てはまる。
【0004】
本発明は、連結部分上、又は少なくとも2個の異なる連結部分上に複数の異なる官能基を含む金属有機骨格を提供する。多孔質有機骨格は、細孔機能の変異が、少なくとも2個の異なる官能基を有機骨格中に組み込むことによって容易に修飾することができる点において多重変異性である。一実施形態において、連結部分のサブ構造は均一であるが、連結部分上の側鎖基は変異される。
【課題を解決するための手段】
【0005】
本発明は、主鎖に沿った配向、数、相対位置及び比率が連結部分の変化のないサイズ及び主鎖の不変の結合性のために制御可能である異なる官能基であって、骨格における細孔の化学的及び物理的性質を修飾する官能基を有する複数の連結部分を含む多孔質有機骨格を提供する。一実施形態において、該有機骨格は、n≧2であるn個の異なる有機連結から構成されている。別の実施形態において、該官能基は、金属酸化物及びフェニルの単位を含むコアに沿っている。また、別の実施形態において、該有機骨格は、金属−酸化物の結合部及び有機連結部分の反復単位、並びに連結部分に共有結合されている複数の官能基を含み、ここで、該官能基は不均一であるか、及び/又は該官能基は連結に沿って異なった間隔で配置されている。一実施形態において、該有機骨格は、MOF−5骨格と実質的に同一のMOFトポロジーを含む。別の実施形態において、該骨格は、対応する金属塩対アニオンとともに、Li+、Na+、Rb+、Mg2+、Ca2+、Sr2+、Ba2+、Sc3+、Ti4+、Zr4+、Ta3+、Cr3+、Mo3+、W3+、Mn3+、Fe3+、Fe2+、Ru3+、Ru2+、Os3+、Os2+、Co3+、Co2+、Ni2+、Ni+、Pd2+、Pd+、Pt2+、Pt+、Cu2+、Cu+、Au+、Zn2+、Al3+、Ga3+、In3+、Si4+、Si2+、Ge4+、Ge2+、Sn4+、Sn2+、Bi5+、Bi3+からなる群及びこれらの組合せから選択される金属を含む。また、別の実施形態において、該連結部分は、
【化1】
【0006】
(式中、R1〜R4は、−H、−NH2、−BR、−Cl、−NO2、−CH3、−OCH2R5及び−O−CH2R6からなる群から選択され、ここで、R5は約1個〜5個の炭素のアルキルもしくはアルケンであり、R6はアリールもしくは置換アリールである、又はR1〜R2は隣接する場合、環を形成することができる)からなる群から選択される一般構造を有する。さらなる実施形態において、該連結部分は、
【化2】
【0007】
からなる群から選択されるメンバーを含む。また、別の実施形態において、該有機骨格は、第一官能基を含む該複数の連結部分の第一連結部分及び第二官能基を含む該複数の連結部分の第二連結部分を含み、ここで、第二官能基は反応後の基との合成後の反応を受けることで、骨格をさらに官能化することができる。また別の実施形態において、mvMOFは、同じトポロジーであるが均一の連結部分を有する骨格と比較して改善されたガス収着能を有する。
【0008】
本発明は、連結部分が所望の比率で連結部分の特定の組合せを有機骨格に組み込むための所望の比率である複数の化学的官能化連結部分と、金属イオン又は金属硝酸塩とを混合すること、結晶を精製すること、及び溶媒を除去することを含む、mvMOFを作製する方法を提供する。具体的な一実施形態において、該方法は、ベンゼンジカルボン酸を含む有機骨格に所望の比率で連結部分の特定の組合せを組み込むための所望の比率で、複数の化学的官能化連結部分と硝酸亜鉛とをDEF/DMF中にて混合することを含む。
【0009】
本発明は、mvMOFを含むガス分離装置も提供する。本発明は、mvMOFを含むガス貯蔵装置も提供する。
【0010】
本発明は、具体的な実施形態において、多重の官能性基の連結を使用することで多変量の(MTV)構造を生成し、これによって、より複雑なMOFを作製するための戦略を実証する。1,4−ベンゼンジカルボキシレート(BDC、A)及びその官能化誘導体、−NH2、−Br、−(Cl)2、−NO2、−(CH3)2、−C4H4、−(C3H5O)2並びに−(C7H7O)2(それぞれB−I)は、18個のmvMOFを構築するために使用されたが、これらのそれぞれが立方MOF−5型結晶構造を有し、最大8個の異なる官能性基を含有する(2:mvMOF−5−AB、−AC、−AD、−AE、−AF、−AG、−AH、−AI、−EI、3:mvMOF−5−ABC、−AHI、−EHI、4:mvMOF−5−ABCD、−ACEF、5:mvMOF−5−ABCHI、6:mvMOF−5−ABCGHI、7:mvMOF−5−ABCEGHI、8:mvMOF−5−ABCEFGHI)。この系列(mvMOF−5−ACEF)の典型的メンバーの単結晶回折研究は、MOF主鎖(金属酸化物及びフェニルの単位)が規則化されており、官能性基が不可避的に不規則化されていることを確証しており、このことは、これらのMOFが固溶体であるという可能性を排除する。核磁気共鳴分光法を使用して、mvMOF系列の各メンバーの構造内における各官能基の存在、それらの同定及び比率を決定した。これらの測定は、それらのバルク均一性を確証するために固体生成物から選択されるいくつかの結晶に対して、及び結晶全体における同一連結比の存在を確証するために単結晶のセグメントに対しても行われた。後者の観察は結晶内における官能性基のランダム分布を論証することができるが、おそらくそれらは、特異的非障害位置での特異的連結に対する先入観を必然的に作り出す連結−連結相互作用により、特異的配列に配置されている可能性が大きい。これは、それぞれのmvMOFの大部分において最小障害連結(A)の比較的高い比率が観察されたことによって裏付けられている。細孔内における官能基の「複雑な」配置は、同じ連結対応物(MOF−5)と比較して、CO2に対するmvMOF−5−EHIの選択性においてCOを超える最大400%の向上をもたらす。
【0011】
本発明は、n個(n≧2である)の異なる有機連結から構成されている錯体自己集合開骨格材料を提供する。本発明は、多座有機又は無機コアによって架橋されている有機連結から構成されている全ての開骨格材料を包含する。開骨格材料;共有結合性有機骨格(COF)、ゼオライトイミダゾレート骨格(ZIF)及び金属有機骨格(MOF)の全ての部類、並びに網状化学構造リソース(http://rcsr.anu.edu.au/)内に記載されている通りの全ての可能な結果として生じるネットトポロジーを含める。該骨格における2個を超える連結を利用することによって、多種多様の材料を生み出す錯体構成物を合成することができる。こうした材料は、ガス貯蔵及び分離、並びに触媒作用等の応用において様々に使用される。
【0012】
本発明の一つ又は複数の実施形態の詳細を添付の図及び下の記述で説明する。本発明の他の特徴、対象物及び利点は、記述及び図面から、並びに請求項から明らかであろう。
【図面の簡単な説明】
【0013】
【図1】mvMOF−5−ABCDの試料に関して本明細書で示されているmvMOFに対して行われた通常の分析を示す図である。(A)MOF−5に関してシミュレートしたものと比較した結晶粉末のX線回折パターン。(B)各連結に関する固有の共鳴を示す13C CP/MAS NMRスペクトル。(C)連結の比率を決定するために使用された溶液1H NMRスペクトル。(D)それぞれ黒丸及び白丸によって表される吸着点及び脱着点を持つ77KでのN2吸着等温線。(E)セグメントが連結の比率に関して分析され、全体において同一であると判明した大結晶。
【図2】以下を示す図である。(A)mvMOF−5−AH(緑色)、−AI、−AHI及びMOF−5の77KでのH2吸着等温線。(B)mvMOF−5−EI、−EHI及びMOF−5の298KでのCO2(丸)及びCO(四角)の吸着等温線。吸着及び脱着枝は、それぞれ黒丸(COには四角)及び白丸(COには四角)によって表される。(C)NH2−BDC対BDCのモル比の表形式データと一緒に、溶液1H NMR対合成に使用された化学量論比によって決定されたMOF−5−ABにおけるNH2−BDCのパーセント比率のプロット。
【図3】調製されたままのmvMOF−5−ABの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図5】調製されたままのmvMOF−5−ACの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図6】調製されたままのmvMOF−5−ADの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図7】調製されたままのmvMOF−5−AEの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図8】調製されたままのmvMOF−5−AFの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図9】調製されたままのmvMOF−5−AGの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図10】調製されたままのmvMOF−5−AHの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図11】調製されたままのmvMOF−5−AIの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図12】調製されたままのmvMOF−5−EIの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図13】調製されたままのmvMOF−5−ABCの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図14】調製されたままのmvMOF−5−AHIの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図15】調製されたままのmvMOF−5−EHIの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図16】調製されたままのmvMOF−5−ABCDの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図17】調製されたままのmvMOF−5−ACEFの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図18】調製されたままのmvMOF−5−ABCHIの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図19】調製されたままのmvMOF−5−ABCGHIの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図20】調製されたままのmvMOF−5−ABCEGHIの実験PXRDパターンと、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図21】調製されたままのmvMOF−5−ABCEFGHIの実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較を示す図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図22】活性化mvMOF−5−ABのTGAトレースの図である。
【図23】活性化mvMOF−5−ACのTGAトレースの図である。
【図24】活性化mvMOF−5−ADのTGAトレースの図である。
【図25】活性化mvMOF−5−AEのTGAトレースの図である。
【図26】活性化mvMOF−5−AFのTGAトレースの図である。
【図27】活性化MOF−5−AGのTGAトレースの図である。
【図28】活性化mvMOF−5−AHのTGAトレースの図である。
【図29】活性化MOF−5−AIのTGAトレースの図である。
【図30】活性化mvMOF−5−EIのTGAトレースの図である。
【図31】活性化MOF−5−AHIのTGAトレースの図である。
【図32】活性化mvMOF−5−EHIのTGAトレースの図である。
【図33】活性化mvMOF−5−ACEFのTGAトレースの図である。
【図34】活性化mvMOF−5−ABCDのTGAトレースの図である。
【図35】活性化MOF−5−ABCEFGHIのTGAトレースの図である。
【図36】光学的画像の図である。(A及びB):合成されたままのmvMOF−5−AB−lscの光学的画像。(C及びD):合成されたままのmvMOF−5−ABCD−lscの光学的画像。(E及びF):合成されたままのmvMOF−5−ABCEFGHI−lscの光学的画像。
【図37】mvMOF−5−ABの調製されたままの単結晶の実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較の図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図38】mvMOF−5−ABCDの調製されたままの単結晶の実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較の図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図39】mvMOF−5−ABCEFGHIの調製されたままの単結晶の実験PXRDパターン(上部)と、シミュレートされたMOF−5回折パターン(下部)との比較の図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図40】調製されたままのmvMOF−5−AB系列の実験PXRDパターン(a〜k)と、シミュレートされたMOF−5回折パターン(黒色)との比較の図である。該パターン間の非常に高い対応度は、バルク材料全てがMOF−5と同じトポロジーを有することを表す。
【図41】無色から赤色へ、それぞれ左から右へ、mvMOF−5−AB−aからmvMOF−5−AB−k(下部)の色変化を示す結晶の光学的画像と一緒に、溶液1H NMR対合成に使用された化学量論比(上部)によって決定されたmvMOF−5−AB系列におけるBのパーセント比率のプロットの図である。
【図42】調製されたままのmvMOF−5−AI系列の実験PXRDパターン(a〜c)と、シミュレートされたMOF−5回折パターン(黒色)との比較の図である。該パターン間の非常に高い対応度は、バルク材料がMOF−5と同じトポロジーを有することを表す。
【図43】調製されたままのmvMOF−5−ABCD系列の実験PXRDパターン(a〜e)と、シミュレートされたMOF−5回折パターン(黒色)との比較の図である。該パターン間の非常に高い対応度は、バルク材料全てがMOF−5と同じトポロジーを有することを表す。
【図44】水素原子及びBr原子を含めて、不規則化基の両方の成分が示されているmvMOF−5−AC単位のORTEP図を示す。Zn(暗緑色)原子及びO(暗赤色)原子は異方的に精密化されており、一方、C(灰色)原子及びBr(赤色)原子は等方的に精密化されており、H(明緑色)原子は算出位置に置かれていた。全ての楕円体は、15%の確率水準で表示されている。各フェニル環上において、同等の可能性でBrによって占有されている全四つ位置から、一つの位置だけがBrによって占有されていることに留意されたい。
【図45】示されている不規則化基の全ての成分(Br、CH3、H)とのmvMOF−5−ACEF単位のORTEP図を示す。Zn(暗緑色)だけが異方的に精密化されており、一方、O(暗赤色)原子、C(灰色)原子及びBr(赤色)原子は等方的に精密化されており、H(明緑色)原子は算出位置に置かれていた。全ての楕円体は15%の確率水準で表示されている。各フェニル環は四つの位置の一つにBrを、及び四つの位置の二つにメチル基を有することができ、これらは共存することができないことに留意されたい。この図面上及び構造精密化において、非常に少量で存在するNO2の組成は無視されている。
【図46】77Kで測定されたmvMOF−5−AB(青色)、−ABCD(緑色)、及び−ABCEFGHI(赤色)の窒素吸着等温線を示す図である。中黒記号及び中白記号はそれぞれ吸着及び脱着枝を表す。連結線は目線のための案内である。
【図47】87Kで測定されたmvMOF−5(青色)及びmvMOF−5−AI(赤色)に関するアルゴン吸着等温線を示す図である。中黒記号及び中白記号はそれぞれ吸着及び脱着枝を表す。連結線は目線のための案内である。
【図48】NLDFTモデルに基づくmvMOF−5−AI−a及びMOF−5の算出孔径分布を示す図である。
【図49】77Kで測定されたMOF−5−AI−a(赤色)、mvMOF−5−AI−b(青色)及びmvMOF−5−AI−cの窒素吸着等温線を示す図である。中黒記号及び中白記号はそれぞれ吸着及び脱着枝を表す。連結線は目線のための案内である.
【図50】273(青色)、283(緑色)及び298K(赤色)で取得されたMOF−5に関するCO等温線を示す図である。中黒記号及び中白記号は吸着及び脱着枝を表す。連結線は目線のための案内である。
【図51】273(青色)、283(緑色)及び298K(赤色)で取得されたMOF−5に関するCO2等温線を示す図である。中黒記号及び中白記号は吸着及び脱着枝を表す。連結線は目線のための案内である。
【図52】273(青色)、283(緑色)及び298K(赤色)で取得されたmvMOF−5−EIに関するCO等温線を示す図である。中黒記号及び中白記号は吸着及び脱着枝を表す。連結線は目線のための案内である。
【図53】273(青色)、283(緑色)及び298K(赤色)で取得されたmvMOF−5−EIに関するCO2等温線を示す図である。中黒記号及び中白記号は吸着及び脱着枝を表す。連結線は目線のための案内である。
【図54】273(青色)、283(緑色)及び298K(赤色)で取得されたmvMOF−5−EHIに関するCO等温線を示す図である。中黒記号及び中白記号は吸着及び脱着枝を表す。連結線は目線のための案内である。
【図55】273(青色)、283(緑色)及び298K(赤色)で取得されたmvMOF−5−EHIに関するCO2等温線を示す図である。中黒記号及び中白記号は吸着及び脱着枝を表す。連結線は目線のための案内である。
【発明を実施するための形態】
【0014】
本明細書で使用される場合、及び添付の請求項において、単数形「一(a)」「及び」及び「前記」は、文脈が別途明確に指示していない限り、複数の指示対象を含める。したがって、例えば、「一骨格」の言及は複数のこうした骨格を含め、「前記金属」という言及は1種又は複数の金属及び当業者に知られているこれらの同等物等への言及を含める。
【0015】
別途定義されていない限り、本明細書において使用される全ての技術的及び科学的用語は、この開示が属する分野の当業者に通例理解されるのと同じ意味を有する。本明細書に記載されているものと類似又は同等の任意の方法及び試薬は開示の方法及び組成物の実践において使用することができるが、例示的な方法及び材料がここに記載されている。
【0016】
また、「又は」の使用は、別途記載されていない限り、「及び/又は」を意味する。同様に、「含む(comprise)」「含む(comprises)」「含む(comprising)」「含める(include)」、「含める(includes)」及び「含めて」は交換可能であり、限定することを意図していない。
【0017】
各種実施形態の記載が「含む(comprising)」という用語を使用する場合、一部の具体的例において、「から基本的になる」又は「からなる」という言語を使用して、実施形態は別法で記載され得ると当業者が理解すると、さらに理解されるべきである。
【0018】
本明細書に記述されている全ての文献は、本明細書に記載と関連して使用されると思われる文献に記載されている方法論を記載及び開示する目的で、参照により全文を本明細書に組み込む。上記及び本文を通して考察されている文献は、本出願の出願日より前に、単にそれらの開示内容のために提供されている。本発明者らが前の開示のためにこうした開示に先行する権利を与えられていないことを容認すると解釈されるべきものは、本明細書において何もない。
【0019】
結晶拡張構造は、少数の別個の構築単位から構成されているという点において、「単純」である傾向がある。こうした単位の数及び多様性を増加させることは、結晶構造の特性を改善する機会をもたらす。実際に、多数の異なる構築単位の特異的配置が特異的機能をコードする又は新たな現象に至る人工材料を開発することを想像することができる。今日まで、この種類の「複雑性」の結晶材料は、これらの合成が概して混合単位の単相よりむしろ混合相、又は非晶質材料のいずれかを生成するので存在しない。ブロックコポリマーの確立された化学的性質においてさえ、側鎖の官能性におけるわずかな変化が、ポリマー構造における大きな望ましくない変化又はその相分離に至り、したがってそれらの構造及び複雑性に対する制御の発現防止に至る。
【0020】
金属−酸化物単位が有機単位によって連結されている強固な規則化金属−有機骨格(MOF)を構築する方法を開発し、本発明は、MOF主鎖の本来の規則性が、MOFに制御された複雑性を達成する際に有用であり得ることを実証する。本発明は、主鎖に沿った配向、数、相対位置及び比率が連結の変化のないサイズ、及び主鎖の不変の結合性のために制御可能である異なる官能基を持つ連結を導入する戦略を実証する。こうしたMOFは、金属−酸化物及び有機連結単位の「単純な」反復パターンからなる第一次構造、及び該連結に共有結合されている官能基の広範に変動する配置によって形成される「複雑な」第二次構造を有するとみなすことができる。このように、MOF内の細孔のそれぞれは、それらの中心に向いている一連の混合官能性基を有すると思われる。すなわち、こうした官能性基の配列、及びそれらのいくつかが配列に現れる頻度が、元の単一連結のMOFのそれによって保有されるいかなるレベルもはるかに超える新たなレベルの複雑性を細孔に与える。特性に好ましい意味を持つ細孔環境の微調整を可能にする一態様。
【0021】
本発明はしたがって、金属−有機骨格(MOF)の本来の強剛性及びポリマーの機能可撓性を組み合わせることによって、これらの問題を克服し、それぞれが多変量の(MTV)官能性基を有する多数の単相材料を作り出すことができることを実証する。
【0022】
本発明は、主鎖(金属−酸化物及びフェニルの単位)に沿った配向、数、相対位置及び比率を連結の変化のない長さ及びその不変の結合性のために制御することができる異なる官能基を持つ連結からそれらの構造を組み立てることによって、多変量の金属有機骨格(mvMOFs)を作製する方法も提供する。スキーム1は、こうした変異の概略を表す。すなわち、こうした官能性基の配列及び特定の官能基が配列に現れる頻度が、元の同連結MOFのレベルによって保有されるいかなるレベルもはるかに超える新たなレベルの複雑性を細孔に与える。特性に好ましい意味を持つ細孔環境の微調整を可能にする一態様。
【0023】
スキーム1
【化3】
【0024】
本発明は、異なる官能基の2個、3個、4個、5個、6個、7個、8個、9個、10個、11個又は12個の連結のセットを組み合わせる結晶MOF材料を生成するための一般的方法を記載しており、これらのそれぞれは連結の比率が制御されている単一構造に組み込まれており、該材料はバルク純度で生成することができる。本発明は、単相としての多重変異体MOFの単離を実証する。mvMOFの多孔性は多様であり、多変異連結が様々な官能性基を導入することができることを示す。例えば、MOF構造に導入することができる変異連結として、これらに限定されないがNO2−BDC及び(Cl)2−BDCが挙げられ、MOF−5型構造(mvMOF−5−AD及び−AE)に導入され、それ以外単独で使用される場合はこの構造を形成しない。本発明は、この系列(例えば、mvMOF−5−AHI及び−EHI)のメンバーが、単純な同連結類似体と比較して多変異連結MOFのガス吸着及び分離特性の有意な向上によって証明されている通り「全体のほうが部分の和より良好である」ことを示すことも実証する。下記スキーム2は、mvMOFを発生させるために使用される多変異連結、並びにmvMOFの例を実証する。
【0025】
多重変異体金属有機骨格(mvMOF)は、変異官能性基を有する少なくとも2個、3個、4個、5個、6個、7個、8個、9個、10個、11個又は12個の異なる連結部分を含むMOF構造を指す。mvMOFは、連結クラスターを介して連結部分に共役している金属を含む。連結部分のサブ構造は、サブ構造上の側鎖基を変異させることによって起因し得る異なる官能性を含む。例えば、立方構造に含まれているmvMOFは、その角に金属(例えば、4個の金属原子)を含み、各金属原子は3個の連結クラスターに共役しており、各連結クラスターは連結部分サブ構造に共役している。すなわち、立方構造は12個の連結部分を含む。連結部分上の1個又は複数の側基における変異は、生じる立方構造の変異官能性を生じさせる。MOF骨格のサイズに取り入れると、骨格における変異は、ガス貯蔵、分離及び精製の改善及び多様な機能をもたらす。
【0026】
本明細書で使用される場合、「コア」は、骨格にみられる反復単位又は単位(複数)を指す。こうした骨格は、同種の反復コア構造又は異種の反復コア構造を含むことができる。コアは、遷移金属又は遷移金属のクラスター、及び連結部分を含む。一緒に連結されている複数のコアが骨格の特徴となる。
【0027】
「クラスター」という用語は、2個以上の原子の同定可能な会合を指す。こうした会合は通常、ある型の結合−イオン結合、共有結合及びファンデルワール等によって確立される。
【0028】
「連結クラスター」は、連結部分サブ構造と金属基との間、又は連結部分と別の連結部分との間に結合を形成する能力がある原子を含む縮合能力がある1種又は複数の反応種を指す。こうした種の例は、ホウ素原子、酸素原子、炭素原子、窒素原子及びリン原子からなる群から選択される。一部の実施形態において、連結クラスターは、架橋酸素原子と連結を形成する能力がある1種又は複数の異なる反応種を含むことができる。例えば、連結クラスターは、Rが1個から5個の炭素原子を有するアルキル基又は1個から2個のフェニル環を含むアリール基であるCO2H、CS2H、NO2、SO3H、Si(OH)3、Ge(OH)3、Sn(OH)3、Si(SH)4、Ge(SH)4、Sn(SH)4、PO3H、AsO3H、AsO4H、P(SH)3、As(SH)3、CH(RSH)2、C(RSH)3、CH(RNH2)2、C(RNH2)3、CH(ROH)2、C(ROH)3、CH(RCN)2、C(RCN)3、CH(SH)2、C(SH)3、CH(NH2)2、C(NH2)3、CH(OH)2、C(OH)3、CH(CN)2、及びC(CN)3、並びにCH(SH)2、C(SH)3、CH(NH2)2、C(NH2)3、CH(OH)2、C(OH)3、CH(CN)2及びC(CN)3を含むことができる。通常、MOFの発生において金属を結合するための連結クラスターは、カルボン酸官能基を含有する。
【0029】
本発明は、環を構成する全炭素原子、又は炭素原子と窒素原子、酸素原子、硫黄原子、ホウ素原子、リン原子、ケイ素原子及びアルミニウム原子との混合物のいずれかからなる1個から5個の環を含むシクロアルキルサブ構造又はアリールサブ構造を含める。
【0030】
「共有結合性有機多面体」という用語は、非拡張共有結合性有機ネットワークを指す。こうした多面体における重合は、重合を阻害するキャッピングリガンドの存在により一般に生じない。共有結合有機多面体は、多座コアを一緒に連結する複数の連結部分を含む共有結合有機ネットワークであるので、ネットワークの空間構造は多面体である。通常、この変異の多面体は二次元又は三次元構造である。
【0031】
「連結部分」は、遷移金属又は複数の遷移金属をそれぞれ結合する単座又は多座化合物を指す。一般に、連結部分は、1個から20個の炭素原子を含むアルキル基もしくはシクロアルキル基、1個から5個のフェニル環を含むアリール基、又は1個から20個の炭素原子を有するアルキル基もしくはシクロアルキル基、又は1個から5個のフェニル環を含むアリール基を含むアルキルアミンもしくはアリールアミンを含むサブ構造、及び金属との縮合を促進するためサブ構造の一つ又は複数の位置に連結クラスターを含む。シクロアルキル又はアリールのサブ構造は、環を構成する全炭素原子、又は炭素原子と窒素原子、酸素原子、硫黄原子、ホウ素原子、リン原子、ケイ素原子及び/又はアルミニウム原子との混合物のいずれかを含む1個から5個の環を含むことができる。通常、連結部分は、共有結合している1個又は複数のカルボン酸連結クラスターを有するサブ構造を含む。サブ構造は、反応側基で官能化することができる。
【0032】
本明細書で使用される場合、一方の末端上に原子があり、他方の末端上に何もない化学式における線は、該式が、原子が付加されていない末端上で別の実体に結合されている化学フラグメントを指すことを意味する。強調のために時々波線が該線に交差する。
【0033】
一態様において、連結部分サブ構造は、以下のいずれかから選択される。
【化4】
【0034】
式中、R1〜R4は、−H、−NH2、−BR、−Cl、−NO2、−CH3、−OCH2R5及び−O−CH2R6からなる群から選択され、R5は、約1〜5個の炭素のアルキル又はアルケンであり、R6は、アリールもしくは置換アリールであるか、又はR1〜R2は隣接する場合、環を形成することができる。一実施形態において、連結リガンドは以下からなる群から選択されるメンバーを含む。
【化5】
【0035】
本発明のmvMOFは、異なる官能基を有する複数の連結部分を最初に利用することによって生じさせることができ、ここで、少なくとも1個の官能基は、反応基で合成後に修飾することができることがさらに企図される。言い換えると、少なくとも1個の連結部分は、反応後の骨格と合成後に反応させることで有機骨格内における官能基の多様性をさらに増加させることができる官能基を含む。
【0036】
また別の実施形態において、連結部分は、下記に説明される通りの一般構造を有することができる。
【化6】
【0037】
式中、R1〜R5は、H、NH2、COOH、CN、NO2、F、Cl、Br、I、S、O、SH、SO3H、PO3H2、OH、CHO、CS2H、SO3H、Si(OH)3、Ge(OH)3、Sn(OH)3、Si(SH)4、Ge(SH)4、PO3H、AsO3H、AsO4H、P(SH)3、As(SH)3、CH(RSH)2、C(RSH)3、CH(RNH2)2、C(RNH2)3、CH(ROH)2、C(ROH)3、CH(RCN)2、C(RCN)3である。
【化7】
【0038】
式中、X=1、2又は3である。
【0039】
適切な反応性官能性基を保有する全ての前述の連結部分を、骨格合成後の適当な反応剤によって化学的に変換することで、さらなる官能性基を細孔に付加することができる。骨格における有機連結を合成後的に修飾することによって、以前はアクセス不可能であるか又は多大な困難及び/又はコストを介してのみアクセス可能であった官能基へのアクセスが可能及び容易である。後の骨格反応剤として、全ての既知有機変換剤及びそれぞれの反応剤;N、S、O等の原子を含めた官能基との1〜20個の炭素の環が挙げられる。付加された官能基に、又は以前から存在する官能基及び新規に付加された官能基の組合せにキレートすることができる全ての金属。例えば不均一触媒として使用するために有機金属錯体を骨格に繋ぐ結果になる全ての反応物。後の骨格反応剤の一部の例として以下のものが挙げられる。
【化8】
【0040】
式中、R=H、アルキル、アリール、OH、アルコキシ、アルケン、アルキン、フェニル及び前述の置換、硫黄含有基(例えば、チオアルコキシ)、ケイ素含有基、窒素含有基(例えば、アミド)、酸素含有基(例えば、ケトン及びアルデヒド)、ハロゲン、ニトロ、アミノ、シアノ、ホウ素含有基、リン含有基、カルボン酸又はエステルである。
【0041】
さらに、金属、並びに付加された官能基に、又は以前から存在する官能基及び新規に付加された官能基の組合せにキレートすることができる金属含有化合物も有用である。例えば不均一触媒として使用するために有機金属錯体を骨格に繋ぐ結果になる反応物も使用することができる。後の骨格反応剤の例として、これらに限定されないが、複素環化合物が挙げられる。
【0042】
一実施形態において、後の骨格反応剤は、飽和又は不飽和複素環であってよい。単独又は接尾語もしくは接頭語として使用される「複素環」という用語は、環構造の一部としてN、O及びSから独立して選択される1個又は複数の多価ヘテロ原子を有し、環(単数又は複数)内に少なくとも3個及び最大約20個の原子を含める環含有構造又は分子を指す。複素環は1個又は複数の二重結合を含有する飽和又は不飽和であってよく、複素環は1個を超える環を含有することができる。複素環が1個を超える環を含有する場合、該環は縮合又は非縮合であってよい。縮合環は一般に、その間に2個の原子を共有する少なくとも2個の環を指す。複素環は芳香族性を有してよい、又は芳香族性を有していなくてよい。単独又は接尾語もしくは接頭語として使用される「複素環基」、「複素環部分」、「複素環」又は「ヘテロシクロ」という用語は、複素環から1個又は複数の水素を除去することによって誘導される基を指す。単独又は接尾語もしくは接頭語として使用される「ヘテロシクリル」という用語は、複素環から1個の水素を除去することによって誘導される一価の基を指す。単独又は接尾語もしくは接頭語として使用される「ヘテロアリール」という用語は、芳香族性を有するヘテロシクリルを指す。複素環として、例えば、以下等の単環式複素環が挙げられる。アジリジン、オキシラン、チイラン、アゼチジン、オキセタン、チエタン、ピロリジン、ピロリン、イミダゾリジン、ピラゾリジン、ピラゾリン、ジオキソラン、スルホラン2,3−ジヒドロフラン、2,5−ジヒドロフランテトラヒドロフラン、チオファン、ピペリジン、1,2,3,6−テトラヒドロ−ピリジン、ピペラジン、モルホリン、チオモルホリン、ピラン、チオピラン、2,3−ジヒドロピラン、テトラヒドロピラン、1,4−ジヒドロピリジン、1,4−ジオキサン、1,3−ジオキサン、ジオキサン、ホモピペリジン、2,3,4,7−テトラヒドロ−1H−アゼピンホモピペラジン、1,3−ジオキセパン、4,7−ジヒドロ−1,3−ジオキセピン、及びヘキサメチレンオキシド。
【0043】
さらに、複素環として、芳香族複素環(ヘテロアリール基)、例えば、ピリジン、ピラジン、ピリミジン、ピリダジン、チオフェン、フラン、フラザン、ピロール、イミダゾール、チアゾール、オキサゾール、ピラゾール、イソチアゾール、イソオキサゾール、1,2,3−トリアゾール、テトラゾール、1,2,3−チアジアゾール、1,2,3−オキサジアゾール、1,2,4−トリアゾール、1,2,4−チアジアゾール、1,2,4−オキサジアゾール、1,3,4−トリアゾール、1,3,4−チアジアゾール、及び1,3,4−オキサジアゾールが挙げられる。
【0044】
さらに、複素環は、多環式複素環、例えば、インドール、インドリン、イソインドリン、キノリン、テトラヒドロキノリン、イソキノリン、テトラヒドロイソキノリン、1,4−ベンゾジオキサン、クマリン、ジヒドロクマリン、ベンゾフラン、2,3−ジヒドロベンゾフラン、イソベンゾフラン、クロメン、クロマン、イソクロマン、キサンテン、フェノキサチイン、チアントレン、インドリジン、イソインドール、インダゾール、プリン、フタラジン、ナフチリジン、キノキサリン、キナゾリン、シンノリン、プテリジン、フェナントリジン、ペリミジン、フェナントロリン、フェナジン、フェノチアジン、フェノキサジン、1,2−ベンゾイソオキサゾール、ベンゾチオフェン、ベンゾオキサゾール、ベンゾチアゾール、ベンゾイミダゾール、ベンゾトリアゾール、チオキサンチン、カルバゾール、カルボリン、アクリジン、ピロリジジン、及びキノリジジンを包含する。
【0045】
上に記載されている多環式複素環に加えて、複素環として、2個以上の環の間の環縮合が、両方の環に共通する1個を超える結合、及び両方の環に共通する2個を超える原子を含める多環式複素環が挙げられる。こうした架橋複素環の例として、キヌクリジン、ジアザビシクロ[2.2.1]ヘプタン及び7−オキサビシクロ[2.2.1]ヘプタンが挙げられる。
【0046】
ヘテロシクリルとして、例えば、以下等の単環式ヘテロシクリルが挙げられる。アジリジニル、オキシラニル、チイラニル、アゼチジニル、オキセタニル、チエタニル、ピロリジニル、ピロリニル、イミダゾリジニル、ピラゾリジニル、ピラゾリニル、ジオキソラニル、スルホラニル、2,3−ジヒドロフラニル、2,5−ジヒドロフラニル、テトラヒドロフラニル、チオファニル、ピペリジニル、1,2,3,6−テトラヒドロ−ピリジニル、ピペラジニル、モルホリニル、チオモルホリニル、ピラニル、チオピラニル、2,3−ジヒドロピラニル、テトラヒドロピラニル、1,4−ジヒドロピリジニル、1,4−ジオキサニル、1,3−ジオキサニル、ジオキサニル、ホモピペリジニル、2,3,4,7−テトラヒドロ−1H−アゼピニル、ホモピペラジニル、1,3−ジオキセパニル、4,7−ジヒドロ−1,3−ジオキセピニル、及びヘキサメチレンオキシジル。
【0047】
さらに、ヘテロシクリルとして、芳香族ヘテロシクリル又はヘテロアリール、例えば、ピリジニル、ピラジニル、ピリミジニル、ピリダジニル、チエニル、フリル、フラザニル、ピロリル、イミダゾリル、チアゾリル、オキサゾリル、ピラゾリル、イソチアゾリル、イソオキサゾリル、1,2,3−トリアゾリル、テトラゾリル、1,2,3−チアジアゾリル、1,2,3−オキサジアゾリル、1,2,4−トリアゾリル、1,2,4−チアジアゾリル、1,2,4−オキサジアゾリル、1,3,4−トリアゾリル、1,3,4−チアジアゾリル、及び1,3,4オキサジアゾリルが挙げられる。
【0048】
さらに、ヘテロシクリルは、多環式ヘテロシクリル(芳香族又は非芳香族の両方を含める)、例えば、インドリル、インドリニル、イソインドリニル、キノリニル、テトラヒドロキノリニル、イソキノリニル、テトラヒドロイソキノリニル、1,4−ベンゾジオキサニル、クマリニル、ジヒドロクマリニル、ベンゾフラニル、2,3−ジヒドロベンゾフラニル、イソベンゾフラニル、クロメニル、クロマニル、イソクロマニル、キサンテニル、フェノキサチイニル、チアントレニル、インドリジニル、イソインドリル、インダゾリル、プリニル、フタラジニル、ナフチリジニル、キノキサリニル、キナゾリニル、シンノリニル、プテリジニル、フェナンスリジニル、ペリミジニル、フェナントロリニル、フェナジニル、フェノチアジニル、フェノキサジニル、1,2−ベンゾイソオキサゾリル、ベンゾチオフェニル、ベンゾオキサゾリル、ベンゾチアゾリル、ベンゾイミダゾリル、ベンゾトリアゾリル、チオキサンチニル、カルバゾリル、カルボリニル、アクリジニル、ピロリジジニル、及びキノリジジニルを包含する。
【0049】
上に記載されている多環式ヘテロシクリルに加えて、ヘテロシクリルとして、2個以上の環の間の環縮合が、両方の環に共通する1個を超える結合及び両方の環に共通する2個を超える原子を含める多環式ヘテロシクリルが挙げられる。こうした架橋複素環の例として、キヌクリジニル、ジアザビシクロ[2.2.1]ヘプチル;及び7−オキサビシクロ[2.2.1]ヘプチルが挙げられる。
【0050】
本発明の骨格の合成に使用することができる金属イオンとして、Li+、Na+、Rb+、Mg2+、Ca2+、Sr2+、Ba2+、Sc3+、Ti4+、Zr4+、Ta3+、Cr3+、Mo3+、W3+、Mn3+、Fe3+、Fe2+、Ru3+、Ru2+、Os3+、Os2+、Co3+、Co2+、Ni2+、Ni+、Pd2+、Pd+、Pt2+、Pt+、Cu2+、Cu+、Au+、Zn2+、Al3+、Ga3+、In3+、Si4+、Si2+、Ge4+、Ge2+、Sn4+、Sn2+、Bi5+、Bi3+、及びこれらの組合せが、対応する金属塩対アニオンとともに挙げられる。
【0051】
本発明は、本発明のmvMOFを作製する方法も提供する。該方法は、有機骨格に所望の比率で連結部分の特定の組合せを組み込むための所望の比率で、複数の化学的官能化連結部分と、金属イオン又は金属−硝酸塩とを、適切な緩衝液中で混合することを含む。結果として生じる結晶物質を次いで精製し、溶媒を除去する。一実施形態において、該方法は、ベンゼンジカルボン酸を含む有機骨格中に所望の比率で連結部分の特定の組合せを組み込むための所望の比率で、複数の化学的官能化連結部分と硝酸亜鉛とをDEF/DMF中にて85〜100℃で24〜48時間混合することを含む。結果として生じる結晶物質を次いでDMF中に24時間、次いで順次クロロホルム中に、24時間を3周期の間浸漬する。最終的に、この多孔質材料は、溶媒を真空下で24時間室温又は最大120℃までの加熱で除去することによって活性化される。
【0052】
本発明の骨格の調製は、水性系又は非水性系のいずれかにおいて実施することができる。溶媒は各場合によって極性又は非極性であってよい。溶媒は、テンプレート剤又は単座官能基を含有する任意選択のリガンドを含むことができる。非水性溶媒の例として、ペンタン、ヘキサン等のn−アルカン、ベンゼン、トルエン、キシレン、クロロベンゼン、ニトロベンゼン、シアノベンゼン、アニリン、ナフタレン、ナフサ、メタノール等のn−アルコール、エタノール、n−プロパノール、イソプロパノール、アセトン、1,3−ジクロロエタン、塩化メチレン、クロロホルム、四塩化炭素、テトラヒドロフラン、ジメチルホルムアミド、ジメチルスルホキシド、N−メチルピロリドン、ジメチルアセトアミド、ジエチルホルムアミド、チオフェン、ピリジン、エタノールアミン、トリエチルアミン及びエチレンジアミン等が挙げられる。当業者は出発反応剤に基づいて適切な溶媒を容易に決定することができ、溶媒の選択は本発明の材料を入手するのに重大であるとは思われない。
【0053】
本発明の方法にテンプレート剤を使用することができる。本発明に用いられるテンプレート剤は、生じる結晶塩基骨格における細孔を占有する目的で、反応混合物によって付加される。本発明の一部の変異において、空隙充填薬剤、吸着された化学種及びゲスト種は、金属−有機骨格の表面積を増加させる。適当な空隙充填薬剤として、例えば、以下のものからなる群から選択される成分が挙げられる。(i)1個から20個の炭素原子を有する直鎖、分枝又は環状脂肪族基を含有する、アルキルアミン及びそれらの対応するアルキルアンモニウム塩;(ii)1個から5個のフェニル環を有する、アリールアミン及びそれらの対応するアリールアンモニウム塩;(iii)1個から20個の炭素原子を有する直鎖、分枝又は環状脂肪族基を含有するアルキルホスホニウム塩;(iv)1個から5個のフェニル環を有するアリールホスホニウム塩;(v)1個から20個の炭素原子を有する直鎖、分枝又は環状脂肪族基を含有する、アルキル有機酸及びそれらの対応する塩;(vi)1個から5個のフェニル環を有する、アリール有機酸及びそれらの対応する塩;(vii)1個から20個の炭素原子を有する直鎖、分枝又は環状脂肪族基を含有する脂肪族アルコール;又は(viii)1個から5個のフェニル環を有するアリールアルコール。
【0054】
結晶化は、室温又は等温の乾燥器内で最大300℃までの間、溶液を放置すること;希釈塩基を溶液に添加することで結晶化を促進すること;希釈塩基を溶液中に拡散することで結晶化を促進すること;及び/又は溶液を密閉容器に移し、所定温度に加熱することによって実施することができる。
【0055】
さらに提供されているのは、化学種の収着取込み用装置である。該装置は、本明細書において提供されているか又は本発明の方法によって得られる骨格を含む収着剤を含める。該取込みは可逆的又は非可逆的であってよい。一部の実施形態において、収着剤は個別の収着粒子中に含められる。収着粒子は、固体、液体及び/又はガス透過性三次元担持体に埋め込むか、又は固定することができる。一部の実施形態において、収着粒子は液体又はガスの可逆的取込み又は貯蔵のための細孔を有し、ここで、収着粒子は液体又はガスを可逆的に吸着又は吸収することができる。
【0056】
一部の実施形態において、本発明において提供されている装置は、アンモニア、二酸化炭素、一酸化炭素、水素、アミン、メタン、酸素、アルゴン、窒素、アルゴン、有機染料、多環式有機分子、及びそれらの組合せ等の化学種の貯蔵のための貯蔵ユニットを含む。
【0057】
さらに提供されているのは、化学種の収着取込みのための方法である。該方法は、本明細書において提供されている骨格を含む収着剤と化学種を接触させることを含める。化学種の取込みは、化学種の貯蔵を含める。一部の態様において、化学種はエネルギー供給源としての使用に適当な条件下で貯蔵される。
【0058】
天然ガスは重要な燃料ガスであり、石油化学工業及び他の化学プロセス工業における塩基性原料として広範に使用されている。天然ガスの組成は分野により広く変動する。多くの天然ガス貯留層は、比較的低い百分率の炭化水素(例えば40%未満)及び高い百分率の酸性ガス、主に二酸化炭素だが硫化水素、硫化カルボニル、二硫化炭素及び様々なメルカプタンも含有している。遠隔地で生産される天然ガスからの酸性ガスの除去は、パイプラインへの送達、天然ガス液体回収、ヘリウム回収、液化天然ガス(LNG)への変換、又は後続の窒素排除のいずれかのための調節又はスイート乾性天然ガスを供給するのに望ましい。CO2は水の存在下で腐食性であり、ドライアイス、水和物を形成する恐れがあり、パイプライン及び天然ガスを加工する際にしばしば使用される極低温装置における凍結問題を引き起こす恐れがある。その上、発熱量に寄与しないことによって、CO2はガス輸送のコストを増大するだけである。
【0059】
いかなる天然ガス処理プロセスも経済的側面が重要である。天然ガスは通常大量に処理され、プロセス技術の選択における処理ユニットの有意な要因である資本コスト及び操作コストにわずかであるが違いを生ずる。一部の天然ガス資源は、加工コストのため生産するのに不経済である。高い信頼性を有し、操作の簡易性を示す天然ガス処理プロセスの改善が引き続き必要とされている。
【0060】
さらに、現在、人為的二酸化炭素の主要な供給源である発電所の煙道排気からの二酸化炭素の除去は、排気を冷却及び加圧すること、又はアミン水溶液の流動床に煙霧を通過させることによって一般に達成され、これらの両方ともコストがかかり、非効率的である。酸化物表面上への二酸化炭素の化学吸着、又は多孔質シリケート、炭素及び膜内への吸着に基づく他の方法は、二酸化炭素取込みのための手段として追及されてきた。しかし、有効な吸着媒体が二酸化炭素除去において長期生存能を有するために、下記の二つの特徴を組み合わせるべきである。(i)二酸化炭素の取込み及び放出が十分可逆的である周期構造、及び(ii)化学的機能化及び分子レベルの微調整が、最適化された取込み能力のために達成できる可撓性。
【0061】
ガス蒸気からの二酸化炭素の回復又は除去のための多くのプロセスが提案され、工業規模で実践されている。プロセスは広く多様であるが、一般に、溶媒吸収、多孔質吸着剤上の吸着、蒸留、又は半透膜を介する拡散の一部の形態を伴う。
【0062】
以下の実施例は本発明を例示することを意図するが、限定するものではない。使用することができる通常の例であるが、当業者に知られている他の手順も別法として使用することができる。
【実施例】
【0063】
以下の実施例は例示に過ぎず、様々なリガンド及び官能基を含めるために修飾することができる該MOF構造の多様性を限定することを意図するものではない。一実施形態において、立方MOF−5構造を使用し、1,4−ベンゼンジカルボキシレート(BDC)、NH2−BDC、Br−BDC、(Cl)2−BDC、NO2−BDC、(CH3)2−BDC、C4H4−BDC、(C3H5O)2−BDC及び(C7H7O)2−BDCの連結(それぞれ、スキーム2−A−I)の酸形態と組み合わせることで、それぞれが2個以上の異なる官能性基(2:mvMOF−5−AB、−AC、−AD、−AE、−AF、−AG、−AH、−AI、−EI;3:mvMOF−5−ABC、−AHI、−EHI;4:mvMOF−5−ABCD、−ACEF;5:mvMOF−5−ABCHI;6:mvMOF−5−ABCGHI;7:mvMOF−5−ABCEGHI;8:mvMOF−5−ABCEFGHI、スキーム2)を有する18個のmvMOFの対応するセットを形成した。本発明は、単相として単離、MOF−5主鎖の構造及びそれらの多孔性を実証し、この多変異連結合成戦略が、NO2−BDC及び(Cl)2−BDC等の官能性基を、それ以外単独で使用される場合この構造を形成しないMOF−5型構造(mvMOF−5−AD及び−AE)に導入するのに有用であることを示す。該データは、この系列(mvMOF−5−AHI及び−EHI)のメンバーが、それらの単純な同連結類似体と比較して、多変異連結MOFのガス吸着及び分離特性の有意な向上によって証明されている通り「全体のほうが部分の和より良好である」ことも実証する。
【化9】
【0064】
スキーム2
【0065】
mvMOFsの結晶は、MOF−5の合成にすでに使用されている条件下で、選択された有機連結の酸形態のN,N−ジメチルホルムアミド(DMF)溶液混合物に、Zn(NO3)2・4H2Oを添加することによって得られた。全ての化合物は、それぞれ、粉末X線回折(PXRD)、13C交差分極マジック角スピニング(CP/MAS)NMR、それらの結晶の酸蒸解溶液に対する1H NMR、及び熱重量分析(TGA)によって特徴づけることで、それらの結晶化度、連結組成、連結比及び熱安定性を評価した。これらの化合物(全て、2個、3個又は4個の異なる連結、及びmvMOF−5−ABCEFGHIを含有する)のサブセットの多孔性は、窒素ガス吸着測定によって評価した。全ての化合物に対する完全な特性決定手順及びデータ取得を行ったが、mvMOF−5−ABCDの詳細は例示的な例として提供されている。
【0066】
化合物は、連結A、B、C及びDの等モル量から合成された(上記スキーム2において説明されている通り)。その高い結晶化度は、親MOF−5構造のものと一致する鋭い回折線を示す合成されたままの試料のPXRDパターンから証明された(図1A)。mvMOF−5−ABCDにおける四つの型の連結の比率を決定するため、試料を50℃にて真空下で(10mTorr)24時間加熱することによって脱気することで、合成中に閉塞した細孔から任意のゲスト溶媒分子を除去した。この試料に対して行ったTGAは最大400℃まで重量損失を示さず、全てのゲスト分子が細孔から除去されたこと、及び脱気された骨格が熱的に安定であることを確証している。
【0067】
mvMOF−5−ABCDの脱気された試料の13C CP/MAS NMRスペクトルは、それぞれ、NH2−BDC、Br−BDC、(Cl)2−BDC及びBDC連結の固有の炭素原子に特有である150.3ppm、127.0ppm、133.7ppm及び136.3ppmで共鳴を示した(図1B)。これらのスペクトルは、MOF主鎖におけるそれらの存在を明確に表している。さらに、同じ実験をmvMOF−5−ABCDの構成物質フリー連結の混合物に行ったが、2ppmの特有のシフトは、フリー連結のカルボニル炭素と骨格に組み込まれた連結のカルボニル炭素との間で認められ、したがって非結合有機連結はMOF結晶内に存在しないことを確証している。全ての残りのmvMOFに対する同様の分析は同じ結果になった。
【0068】
正確な連結比は、mvMOF−5−ABCD固体のDCl蒸解溶液の1H NMRスペクトルから得た(表1、連結組成)。予測カップリングパターンとの共鳴は、連結の固有のプロトンのそれぞれの予想領域において認められた(図1C)。共鳴ピーク強度を積分することによって、連結はMOFに、それぞれ1.00:0.12:0.56:0.40の比率で存在することが実証されている。これらの比率が結晶においてバルク固体と同じであることを示すために、上記に考察されている溶液1H NMR実験を、mvMOF−5−ABCDバルク試料から無作為に選択される4種の異なる結晶に対して行い、該比率がほぼ同一であることを示した。同じ実験をmvMOF−5−AB及び−ABCEFGHIに対しても行い、mvMOF系列のバルク均一性を再度確証した(表1、バルク均一性)。さらに、本来のMOF−5構造の多孔性及び構成安定性は、mvMOF−5−ABCDに関する図1Dに示されているI型窒素吸着等温線及びその高表面積(2860m2g−1)によって例示されている通り、mvMOF化合物中に保持されている。さらに、様々な連結モル比からmvMOF−ABCDを合成することによって、データは、所定のmvMOFにおいて反応化学量を改変することによって連結比を制御することができることを実証した(表1、連結比の制御)。本質的に、連結比におけるこの型の制御は、それらの保持されているPXRDパターンによって証明されている通り、第一次構造の下部結合度を変更することなく、細孔に向いている官能基の占有数及び多様性の制御につながる(図29)。
【0069】
mvMOF−5−AC及び−ACEFの単結晶に対して行われたX線結晶学的研究は、予想通り、Zn4O(CO2)6頂点によって結合されている強固なフェニル単位からなる規則化立方MOF−5構造を明らかにした。これらの材料におけるフェニル単位上の官能基の非水素原子は全て、非常に低い占有率で存在する。フェニル環の4個の炭素原子のそれらの位置が等確率のため、対称には、各官能基が二つ(連結Fのジメチル基)又は四つ(連結CのBr基、又は連結Eのニトロ基)の位置で不規則化されていることが必要である。mvMOF−5−ACにおけるBrは、その低い占有率及びデータの強度に対する低い寄与にもかかわらず精密化することができる。mvMOF−5−ACEFにおいても、官能基原子の占有率は全く低いが、Br原子(連結C)、N原子(連結E)及びC原子(連結F)の位置が重なっているため、差異ピークはあり得る。フェニル単位原子が全てのmvMOFに存在する前提で、これらのパラメーターの全てを、主鎖非水素原子に関して精密化するのに成功した。これは、mvMOFの構造は固溶体でないが、むしろ規則化された骨格に共有結合的に連結された変異官能基の系を表していることを明確に示している。
【0070】
mvMOFの構成の固有性を考慮すると、浮かび上がる有意な疑問は、結晶が官能性基の巨視的領域からなるのか、又は骨格主鎖全体にわたり反復された官能性単位の特有の配列からなるのかである。これらの二つの可能性を区別するため、それぞれ4.0mm×4.0mm×2.0mm、2.0mm×2.0mm×2.0mm及び2.0mm×2.0mm×1.0mmの寸法のmvMOF−5−AB、−ABCD(図1E)及び−ABCEFGHIの大きな単結晶を調製した。各場合において、各単結晶の構造をそのPXRDパターンによって確認した(図25〜27)。各結晶を3個の等しいセグメントに切断し、次いで、溶液1H NMRスペクトルを、それぞれ各結晶の各セグメントの酸蒸解試料上に回収した。同種連結の巨視的領域がmvMOFの単結晶に存在すれば、異なる連結比がそれぞれの親結晶の3個のセグメントのそれぞれに予想される。しかし、データは、連結分布の比率が研究した3mvMOFの各セグメントに同一であると明確に示しており(表1、単結晶のセグメント)、したがって巨視的領域の非存在を示唆している。この結論を支持するさらなる証拠は、MOF−5又は任意の他の同連結MOFに関して認められる通り、mvMOF−5−AIに関する狭い孔径分布の非存在であり、これは連結Iが細孔全体に分布されていることを示唆する。これは、ナノメートルスケール上の特異的官能基(又は官能性基のサブセット)の優位が予想される微視的領域の存在を排除するものではない。
【0071】
表1.付加比率と比較したmvMOF結晶(太字)に認められる連結の比率。連結Aの数値を各場合において1に正規化した。
【表1】
【0072】
MOF主鎖に沿った官能性基の特有の配列の可能な存在は必然的に錯体細孔環境に至り、珍しい特性を明らかにする機会をもたらす。同連結MOF−5構造が有意な量のガス(例えば、H2、CO2)を吸収することが知られているので、mvMOFをこれらの用途で試験することで、それらの性能がそれらの構成物質の性能より大きいかどうかを決定した。図2Aにおいて、mvMOF−5−AHI、−AH、−HI及びMOF−5のH2貯蔵容量間の比較を示す。驚くべきことに、等温線は、mvMOF−5−AHIの取込み容量がmvMOF−5−AH、−HI及び−A(MOF−5)の取込み容量より最大84%大きいことを明確に実証している。同様に、COを超えるCO2の選択的取込み容量における珍しい増加が認められた。MOF−5と比較してCO2に対するmvMOF−5−EHIの場合、400%より良好な選択性(図2B)。
【0073】
これらの発見は、mvMOFの特性が構成物質の単純な直鎖組合せではないことを実証しており、したがってmvMOF内における官能性基の配列が、特異的特性を向上又は新たな特性を実現するための基準として有用であり得るという考えを支持している。
【0074】
複数グラムスケールの生成物を含めたmvMOFの調製の詳細な合成手順、並びに実験及びシミュレートPXRDパターン。
【0075】
テレフタル酸(ベンゼン−1,4−ジカルボン酸又はBDCH2)、2−アミノテレフタル酸(NH2−BDCH2)、2−ブロモテレフタル酸(Br−BDCH2)、2,5−ジクロロテレフタル酸((Cl)2−BDCH2)、2−ニトロテレフタル酸(NO2−BDCH2)、ナフタレン−1,4−ジカルボン酸(C4H4−BDCH2)をAldrich Chemical Co.から購入した。N,N−ジメチルホルムアミド(DMF)は、Fisher Scientific International Inc.から購入する。硝酸亜鉛四水和物Zn(NO3)2・4H2OをEM Scienceから購入した。2,5−ジメチルテレフタル酸((CH3)2−BDCH2)をTCI Americaから購入した。上記全ての出発原料は、さらに精製することなく使用した。N,N−ジエチルホルムアミド(DEF、BASF)溶媒を、活性炭素(Calgon)及びシリカゲル(EMD、シリカゲル60)を充填したカラムに通す濾過によって精製し、クロロホルム(Fisher、HPLC等級、ペンテンを安定化)を使用前に、新たに活性化したモレキュラーシーブ4A上で乾燥させた。2,5−ビス(アリルオキシ)テレフタル酸((C3H5O)2−BDCH2)、2,5−ビス(ベンジルオキシ)テレフタル酸((C7H7O)2−BDCH2)を合成した。
【0076】
多変量の金属−有機骨格(mvMOF)は、変動する量の化学的官能化ベンゼンジカルボン酸と硝酸亜鉛とをDEF/DMF中にて85〜100℃で24〜48時間混合することによって合成される。結果として生じる結晶物質は、次いで、DMF中で24時間、及び次いで順次クロロホルム中で24時間を3周期の間浸漬する。最終的に、この多孔質材料は、溶媒を真空下で24時間室温又は最大120℃までの加熱で除去することによって活性化する。
【0077】
出発原料の量を正確に制御するため、テラフタル酸連結のDMF/DEF溶液(0.025〜0.10M(1M=1moldm−3))及び硝酸亜鉛四水和物のDMF/DEF溶液(0.10M)を原液として使用した。DMF及びDEFは、それらの高沸点により合成の溶媒として選択されており、ソルボサーマル合成に適当である。より低い沸点の溶媒は、生成物よりむしろ反応剤の析出をもたらす恐れがある。反応後、結晶を光学顕微鏡下で検査し、回収し、粉末X線回折(PXRD)によって特徴づけた。破砕した大きな単立方結晶のPXRDデータを、40kV、40mAのCuKα(λ=1.5406Å)、3°/分の走査速度及び2θで0.050°のステップサイズにて操作するBruker AXS D8 Advance回折計に記録した。シミュレートしたMOF−5 PXRDパターンを、ソフトウェアPowder Cell v.2.2を使用して前に公開した単結晶構造から算出した。
【0078】
蒸解mvMOFの1H NMR:一般に、8mgの乾燥(又はDMFと交換した溶媒)mvMOFを蒸解し、1.0mLの希釈DCl溶液(200μLの20%DCl/D2O溶液(Aldrich)及び10mLのDMSO−d6から調製した)中の超音波処理で溶解した。蒸解溶液を1H−NMRに直接使用した。
【0079】
mvMOFの合成:mvMOF−5−AB、Zn4O(BDC)1.92(NH2−BDC)1.08:0.10mLのBDCH2原液(0.10M、1.0×10−5mol)、0.10mLのNH2−BDCH2原液(0.10M、1.0×10−5mol)、0.20mLのZn(NO3)2・4H2O原液(0.30M、6.0×10−5mol)及び0.60mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに48時間反応させた。生成物は立方形の褐色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5粉末回折パターンに整合する(図5)。
【0080】
蒸解mvMOF−5−AB結晶(400MHz、DMSO−d6)の1H NMR。BDCH2 δ:8.00(s、4H)。NH2−BDCH2 δ:7.03(d、1H)、7.38(s、1H)、7.74(d、1H)。ピークの積分に基づくモル比:BDC:NH2−BDC=1:0.57。
【0081】
mvMOF−5−AC、Zn4O(BDC)1.86(Br−BDC)1.14:0.10mLのBr−BDCH2原液(0.10M、1.0×10−5mol)、0.10mLのBDCH2原液(0.10M、1.0×10−5mol)、0.20mLのZn(NO3)2・4H2O原液(0.30M、6.0×10−5mol)及び0.60mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに24時間反応させた。生成物は立方形の淡黄色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図5)。
【0082】
蒸解mvMOF−5−AC結晶の1H NMR(400MHz、DMSO−d6)。BDCH2 δ:8.00(s、4H)。Br−BDCH2 δ:7.78(d、1H)、7.94(d、1H)、8.10(s、1H)。ピークの積分に基づくモル比:BDC:NH2−BDC=1:0.61。
【0083】
mvMOF−5−AD、Zn4O(BDC)1.83((Cl)2−BDC)1.17:0.10mLの(Cl)2−BDCH2原液(0.10M、1.0×10−5mol)、0.10mLのBDCH2原液(0.10M、1.0×10−5mol)、0.20mLのZn(NO3)2・4H2O原液(0.30M、6.0×10−5mol)及び0.60mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに24時間反応させた。生成物は立方形の淡黄色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図6)。
【0084】
蒸解mvMOF−5−AD結晶の1H NMR(400MHz、DMSO−d6)。BDCH2 δ:8.00(s、4H)。(Cl)2−BDCH2 δ:7.90(s、2H)。ピークの積分に基づくモル比:BDC:(Cl)2−BDC=1:0.63。
【0085】
mvMOF−5−AE、Zn4O(BDC)2.13(NO2−BDC)0.87:0.10mLのNO2−BDCH2原液(0.10M、1.0×10−5mol)、0.10mLのBDCH2原液(0.10M、1.0×10−5mol)、0.20mLのZn(NO3)2・4H2O原液(0.30M、6.0×10−5mol)及び0.60mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに48時間反応させた。生成物は立方形の褐色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図7)。
【0086】
蒸解mvMOF−5−AE結晶の1H NMR(400MHz、DMSO−d6)。BDCH2 δ:8.00(s、4H)。NO2−BDCH2 δ:7.92(d、1H)、8.25(d、1H)、8.35(s、1H)。ピークの積分に基づくモル比:BDC:NO2−BDC=1:0.40。
【0087】
mvMOF−5−AF、Zn4O(BDC)1.35((CH3)2−BDC)1.65:0.10mLの(CH3)2−BDCH2原液(0.10M、1.0×10−5mol)、0.10mLのBDCH2原液(0.10M、1.0×10−5mol)、0.20mLのZn(NO3)2・4H2O原液(0.30M、6.0×10−5mol)及び0.60mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに24時間反応させた。生成物は立方形の淡黄色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図8)。
【0088】
蒸解mvMOF−5−AF結晶の1H NMR(400MHz、DMSO−d6)。BDCH2 δ:8.00(s、4H)。(CH3)2−BDCH2 δ:2.43(d、6H)、7.64(s、2H)。ピークの積分に基づくモル比:BDC:(CH3)2−BDC=1:1.24。
【0089】
mvMOF−5−AG、Zn4O(BDC)1.98(C4H4−BDC)1.02:0.40mLのC4H4−BDCH2原液(0.025M、1.0×10−5mol)、0.10mLのBDCH2原液(0.10M、1.0×10−5mol)、0.20mLのZn(NO3)2・4H2O原液(0.30M、6.0×10−5mol)及び0.30mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに24時間反応させた。生成物は立方形の淡緑色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図9)。
【0090】
蒸解mvMOF−5−AG結晶の1H NMR(400MHz、DMSO−d6)。BDCH2 δ:8.00(s、4H)。C4H4−BDCH2 δ:7.64〜7.68(m、2H)、8.05(s、2H)、8.70〜8.74(m、2H)。ピークの積分に基づくモル比:BDC:C4H4−BDC=1:0.52。
【0091】
mvMOF−5−AH、Zn4O(BDC)2.04((C3H5O)2−BDC)0.96:0.10mLの(C3H5O)2−BDCH2原液(0.10M、1.0×10−5mol)、0.10mLのBDCH2原液(0.10M、1.0×10−5mol)、0.20mLのZn(NO3)2・4H2O原液(0.30M、6.0×10−5mol)及び0.60mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに24時間反応させた。生成物は立方形の褐色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図10)。
【0092】
蒸解mvMOF−5−AH結晶の1H NMR(400MHz、DMSO−d6)。BDCH2 δ:8.00(s、4H)。(C3H5O)2−BDCH2 δ:4.54(d、4H)、5.19(d、2H)、5.40(d、2H)、5.94〜6.01(m、2H)、7.30(s、2H)。ピークの積分に基づくモル比:BDC:(C3H5O)2−BDC=1:0.46。
【0093】
mvMOF−5−AI、Zn4O(BDC)2.13((C7H7O)2−BDC)0.87:0.10mLの(C7H7O)2−BDCH2原液(0.10M、1.0×10−5mol)、0.10mLのBDCH2原液(0.10M、1.0×10−5mol)、0.20mLのZn(NO3)2・4H2O原液(0.30M、6.0×10−5mol)及び0.60mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに24時間反応させた。生成物は立方形の淡黄色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図11)。
【0094】
蒸解mvMOF−5−AI結晶の1H NMR(400MHz、DMSO−d6)。BDCH2 δ:8.00(s、4H)。(C7H7O)2−BDCH2 δ:5.12(s、4H)、7.25〜7.244(m、12H)。ピークの積分に基づくモル比:BDC:(C7H7O)2−BDC=1:0.40。
【0095】
mvMOF−5−EI、Zn4O((C7H7O)2‐BDC)2.49(NIDC) 0.51:0.10mLの(C7H7O)2−BDCH2原液(0.10M、1.0×10−5mol)、0.40mLのNO2‐BDCH2原液(0.025M、1.0×10−5mol)、0.20mLのZn(NO3)2・4H2O原液(0.30M、6.0×10−5mol)及び0.30mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに48時間反応させた。生成物は立方形の褐色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図12)。
【0096】
蒸解mvMOF−5−EI結晶の1H NMR(400MHz、DMSO−d6)。NO2‐BDCH2 δ:7.92(d、1H)、8.25(d、1H)、8.35(s、1H)、(C7H7O)2‐BDCH2 δ:5.12(s、4H)、7.25〜7.44(m、12H)。ピークの積分に基づくモル比:NO2‐BDC:(C7H7O)2−BDC=0.20:1。
【0097】
mvMOF−5−ABC、Zn4O(BDC)1.90(NH2‐BDC)0.11(Br−BDC)0.99:0.133mLのBDCH2原液(0.10M、1.33×10−5mol)、0.133mLのNH2‐BDCH2原液(0.10M、1.33×10−5mol)、0.133mLのBr−BDCH2原液(0.10M、1.33×10−5mol)、0.40mLのZn(NO3)2・4H2O原液(0.30M、1.2×10−4mol)及び1.20mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに48時間反応させた。生成物は立方形の褐色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図13)。
【0098】
蒸解mvMOF−5−ABC結晶の1H NMR(400MHz、DMSO−d6)。BDCH2 δ:8.00(s、4H)。NH2‐BDCH2 δ:7.03(d、1H)、7.40(d、1H)、7.38(s、1H)。Br−BDCH2 δ:7.78(d、1H)、7.94(d、1H)、8.10(s、1H)。ピークの積分に基づくモル比:BDC:NH2−BDC:Br−BDC=1:0.052:0.52。
【0099】
mvMOF−5−AHI、Zn4O(BDC)1.52((C3H5O)2−BDC)0.73((C7H7O)2−BDC)0.75:0.133mLのBr−BDCH2原液(0.10M、1.33×10−5mol)、0.133mLの(C3H5O)2−BDCH2原液(0.10M、1.33×10−5mol)、0.133mLの(C7H7O)2‐BDCH2原液(0.10M、1.33×10−5mol)、0.40mLのZn(NO3)2・4H2O原液(0.30M、1.2×10−4mol)及び1.20mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに24時間反応させた。生成物は立方形の褐色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図14)。
【0100】
蒸解mvMOF−5−AHI結晶の1H NMR(400MHz、DMSO−d6)。BDCH2 δ:8.00(s、4H)。(C3H5O)2−BDCH2 δ:4.55(d、4H)、5.20(d、2H)、5.38(d、2H) 、5.93〜6.00(m、2H)。(C7H7O)2‐BDCH2 δ:5.12(s、4H)、7.26〜7.44(m、12H)。ピークの積分に基づくモル比:BDC:(C3H5O)2−BDC:(C7H7O)2−BDC=1:0.48:0.50。
【0101】
mvMOF−5−EHI、Zn4O(NO2−BDC)1.19((C3H5O)2−BDC)1.07((C7H7O)2−BDC)0.74:0.133mLの(C7H7O)2−BDCH2原液(0.10M、1.33×10−5mol)、0.133mLのNO2−BDCH2原液(0.10M、1.33×10−5mol)、0.133mLの(C3H5O)2−BDCH2原液(0.10M、1.33×10−5mol)、0.40mLのZn(NO3)2・4H2O原液(0.30M、1.2×10−4mol)及び1.20mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに24時間反応させた。生成物は立方形の褐色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図15)。
【0102】
蒸解mvMOF−5‐EHI結晶の1H NMR(400MHz、DMSO−d6)。NO2−BDCH2 δ:7.93(d、1H)、8.25(d、1H)、8.35(s、1H)。(C3H5O)2−BDCH2 δ:4.55(d、4H)、5.20(d、2H)、5.38(d、2H) 、5.93〜6.00(m、2H)。(C7H7O)2−BDCH2 δ:5.12(s、4H)、7.26〜7.44(m、12H)。ピークの積分に基づくモル比:NO2−BDC:(C3H5O)2−BDC:(C7H7O)2−BDC=1:0.89:0.62。
【0103】
mvMOF−5−ABCD、Zn4O(BDC)1.44(NH2−BDC)0.18(Br−BDC)0.81((Cl)2−BDC)0.57:0.10mLのBDCH2原液(0.10M、1.0×10−5mol)、0.10mLのNH2−BDCH2原液(0.10M、1.0×10−5mol)、0.10mLのBr−BDCH2原液(0.10M、1.0×10−5mol)、0.10mLの(Cl)2−BDCH2原液(0.10M、1.0×10−5mol)、0.40mLのZn(NO3)2・4H2O原液(0.30M、1.2×10−4mol)及び1.20mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに48時間反応させた。生成物は立方形の暗褐色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図16)。
【0104】
蒸解mvMOF−5−ABCD結晶の1H NMR(400MHz、DMSO−d6)。BDCH2 δ:8.00(s、4H)。NH2−BDCH2 δ:7.03(d、1H)、7.40(d、1H)、7.38(s、1H)。Br−BDCH2 δ:7.78(d、1H)、7.94(d、1H)、8.10(s、1H)。(Cl)2−BDCH2 δ:7.90(s、2H)。ピークの積分に基づくモル比:BDC:NH2−BDC:Br−BDC:(Cl)2−BDC =1:0.12:0.56:0.40。
【0105】
mvMOF−5−ACEF、Zn4O((BDC)1.29(Br−BDC)0.63(NO2−BDC)0.28((CH3)2−BDC)0.80:0.10mLのBDCH2原液(0.10M、1.0×10−5mol)、0.10mLのBr−BDCH2原液(0.10M、1.0×10−5mol)、0.10mLのNO2−BDCH2原液(0.10M、1.0×10−5mol)、0.10mLの(CH3)2−BDCH2原液(0.10M、1.0×10−5mol)、0.40mLのZn(NO3)2・4H2O原液(0.30M、1.2×10−4mol)及び1.20mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに24時間反応させた。生成物は立方形の暗褐色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図17)。
【0106】
蒸解mvMOF−5−ACEF結晶の1H NMR(400MHz、DMSO−d6)。BDCH2 δ:8.00(s、4H)。Br−BDCH2 δ:7.78(d、1H)、7.94(d、1H)、8.10(s、1H)。NO2−BDCH2 δ:7.92(d、1H)、8.25(d、1H)、8.35(s、1H)。(CH3)2−BDCH2 δ:2.43(s、6H)、7.64(s、2H)。ピークの積分に基づくモル比:BDC:Br−BDC:(CH3)2−BDC:NO2−BDC=1:0.49:0.62:0.22。
【0107】
mvMOF−5−ABCHI、Zn4O(BDC)1.38(NH2−BDC)0.03(Br−BDC)0.30((C3H5O)2−BDC)0.86((C7H7O)2−BDC)0.43:80μLのBDCH2原液(0.10M、8.0×10−6mol)、80μLのNH2−BDCH2原液(0.10M、8.0×10−6mol)、80μLのBr −BDCH2原液(0.10M、8.0×10−6mol)、80μLの(C3H5O)2−BDCH2原液(0.10M、8.0×10−6mol)、80μLの(C7H7O)2−BDCH2原液(0.10M、8.0×10−6mol)、0.40mLのZn(NO3)2・4H2O原液(0.30M、1.2×10−4mol)及び1.20mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに48時間反応させた。生成物は立方形の暗褐色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図18)。
【0108】
蒸解mvMOF−5−ABCHI結晶の1H NMR(400MHz、DMSO−d6)。BDCH2 δ:8.00(s、4H)。NH2−BDCH2 δ:7.03(d、1H)。Br−BDCH2 δ:7.78(d、1H)、7.94(d、1H)、8.10(s、1H)。(C3H5O)2−BDCH2 δ:4.54(d、4H)、5.19(d、2H)、5.40(d、2H)、5.94〜6.01(m、2H)。(C7H7O)2−BDCH2 δ:5.12(s、4H)、7.25〜7.44(m、12H)。ピークの積分に基づくモル比:BDC:NH2−BDC:Br−BDC:(C3H5O)2−BDC :(C7H7O)2−BDC=1:0.017:0.22:0.62:0.32。
【0109】
mvMOF−5−ABCGHI、Zn4O(BDC)0.72(NH2−BDC)0.08(Br−BDC)0.63((C4H4−BDC)0.48((C3H5O)2−BDC)0.52((C7H7O)2−BDC)0.57:67μLのBDCH2原液(0.10M、6.7×10−6mol)、67μLのNH2−BDCH2原液(0.10M、6.7×10−6mol)、67μLのBr −BDCH2原液(0.10M、6.7×10−6mol)、267μLのC4H4−BDCH2原液(0.10M、6.7×10−6mol)、67μLの(C3H5O)2−BDCH2原液(0.10M、6.7×10−6mol)、67μLの(C7H7O)2−BDCH2原液(0.10M、6.7×10−6mol)、0.40mLのZn(NO3)2・4H2O原液(0.30M、1.2×10−4mol)及び1.0mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに48時間反応させた。生成物は立方形の暗褐色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図19)。
【0110】
蒸解mvMOF−5−ABCGHI結晶の1H NMR(400MHz、DMSO−d6)。BDCH2 δ:8.00(s、4H)。NH2−BDCH2 δ:7.03(d、1H)。Br−BDCH2 δ:7.78(d、1H)、7.94(d、1H)、8.10(s、1H)。C4H4−BDCH2 δ:7.64〜7.68(m、2H)、8.05(s、2H)、8.70〜8.74(m、2H)。(C3H5O)2−BDCH2 δ:4.54(d、4H)、5.19(d、2H)、5.40(d、2H)、5.94〜6.01(m、2H)。(C7H7O)2−BDCH2 δ:5.12(s、4H)、7.25〜7.44(m、12H)。ピークの積分に基づくモル比:BDC:NH2−BDC:Br−BDC:C4H4−BDC:(C3H5O)2−BDC:(C7H7O)2−BDC=1:0.093:0.87:0.67:0.73:0.80。
【0111】
mvMOF−5−ABCEGHI、Zn4O(BDC)0.57(NH2−BDC)0.05(Br−BDC)0.57(NO2−BDC)0.39(C4H4−BDC)0.44((C3H5O)2−BDC)0.42((C7H7O)2−BDC)0.56:57μLのBDCH2原液(0.10M、5.7×10−6mol)、57μLのNH2−BDCH2原液(0.10M、5.7×10−6mol)、57μLのBr−BDCH2原液(0.10M、5.7×10−6mol)、57μLのNO2−BDCH2原液(0.10M、5.7×10−6mol)、228μLのC4H4−BDCH2原液(0.10M、5.7×10−6mol)、57μLの(C3H5O)2−BDCH2原液(0.10M、5.7×10−6mol)、57μLの(C7H7O)2−BDCH2原液(0.10M、5.7×10−6mol)、0.40mLのZn(NO3)2・4H2O原液(0.30M、1.2×10−4mol)及び1.03mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに48時間反応させた。生成物は立方形の暗褐色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図20)。
【0112】
蒸解mvMOF−5−ABCEGHI結晶の1H NMR(400MHz、DMSO−d6)。BDCH2 δ:8.00(s、4H)。NH2−BDCH2 δ:7.03(d、1H)。Br−BDCH2 δ:7.78(d、1H)、7.94(d、1H)、8.10(s、1H)。NO2−BDCH2 δ:7.92(d、1H)、8.25(d、1H)、8.35(s、1H)。C4H4−BDCH2 δ:7.64〜7.68(m、2H)、8.05(s、2H)、8.70〜8.74(m、2H)。(C3H5O)2−BDCH2 δ:4.54(d、4H)、5.19(d、2H)、5.40(d、2H)、5.94〜6.01(m、2H)。(C7H7O)2−BDCH2 δ:5.12(s、4H)、7.25〜7.44(m、12H)。ピークの積分に基づくモル比:BDC:NH2−BDC:Br−BDC:NO2−BDC:C4H4−BDC:(C3H5O)2−BDC:(C7H7O)2−BDC=1:0.077:1:0.69:0.77:0.73:0.96。
【0113】
mvMOF−5−ABCEFGHI、Zn4O(BDC)0.70(NH2−BDC)0.011(Br−BDC)0.39(NO2−BDC)0.21((CH3)2−BDC)0.46(C4H4−BDC)0.39((C3H5O)2−BDC)0.35((C7H7O)2−BDC)0.39:50μLのBDCH2原液(0.10M、5.0×10−6mol)、50μLのNH2−BDCH2原液(0.10M、5.0×10−6mol)、50μLのBr−BDCH2原液(0.10M、5.0×10−6mol)、50μLのNO2−BDCH2原液(0.10M、5.0×10−6mol)、50μLの(CH3)2−BDCH2原液(0.10M、5.0×10−6mol)、200μLのC4H4 −BDCH2原液(0.10M、5.0×10−6mol)、50μLの(C3H5O)2−BDCH2原液(0.10M、5.0×10−6mol)、50μLの(C7H7O)2−BDCH2原液(0.10M、5.0×10−6mol)、0.40mLのZn(NO3)2・4H2O原液(0.30M、1.2×10−4mol)及び1.05mLのDEFを、4mLガラスバイアルに順次添加した。バイアルを密封し、等温の乾燥器にて100℃で加熱し、ソルボサーマルに48時間反応させた。生成物は立方形の暗褐色単結晶の形態であった。PXRDを調べたところ、シミュレートしたMOF−5構造に整合した(図21)。
【0114】
蒸解mvMOF−5−ABCEFGHI結晶の1H NMR(400MHz、DMSO−d6)。BDCH2 δ:8.00(s、4H)。NH2−BDCH2 δ:7.03(d、1H)。Br−BDCH2 δ:7.78(d、1H)、7.94(d、1H)、8.10(s、1H)。NO2−BDCH2 δ:7.92(d、1H)、8.25(d、1H)、8.35(s、1H)。(CH3)2−BDCH2 δ:2.43(s、6H)、7.64(s、2H)。C4H4−BDCH2 δ:7.64−7.68(m、2H)、8.05(s、2H)、8.70−8.74(m、2H)。(C3H5O)2−BDCH2 δ:4.54(d、4H)、5.19(d、2H)、5.40(d、2H)、5.94−6.01(m、2H)。(C7H7O)2−BDCH2 δ:5.12(s、4H)、7.25−7.44(m、12H)。ピークの積分に基づくモル比:BDC:NH2−BDC:Br−BDC:(CH3)2−BDC:NO2−BDC:C4H4−BDC:(C3H5O)2−BDC:(C7H7O)2−BDC=1:0.14:0.56:0.29:0.67:0.56:0.48:0.56。
【0115】
2個、3個又は4個の異なる連結、及びmvMOF−5−ABCEFGHIを含有する全ての化合物を、上記したのと同じ濃度でグラムスケールにスケールアップした。スケールアップした試料のPXRDは、小スケール試料と同一である。さらに、スケールアップした試料のバルク均一性を、無作為に選択した結晶溶液1H NMRによって調べたところ、各化合物において同一の連結比を示した。
【0116】
熱重量分析。全ての試料を、試料を持続的気流雰囲気中の白金受皿に保持するTA Instruments Q−500シリーズ熱重量分析器にかけた。試料を5℃/分の一定速度で全てのTGA実験中に加熱した。
【0117】
連結におけるダングリング二重結合及びベンゼン環の損失により、連結H及び連結Iを含有するmvMOFは、200℃から徐々に重量を失い、続いて400℃で急に低下する。全ての他のmvMOFは400℃まで重量を失わない。
【0118】
活性化mvMOFの固体状態13C MAS NMR。高い分解能の固体状態核磁気共鳴(NMR)スペクトルを周囲温度で、4mm(外径)のジルコニア回転子を持つ標準Brukerマジックアングルスピニング(MAS)プローブを用いるBruker DSX−300分光計上に記録した。MASとともに交差分極(CP/MAS)を使用することで、13Cデータを75.47MHzで獲得した。1H及び13Cの90度パルス幅はともに4μsであった。CP接触時間は1.5分及び5分で変動した。高出力2パルス位相変調(TPPM)1Hデカップリングを、データ獲得中に適用した。デカップリング周波数は72kHzに相当した。MAS試料スピニング速度は10kHzであった。走査間の(CP/MAS)に関するリサイクル遅延は、13C信号強度において一走査から次の走査まで明らかな損失を認められないことによって決定された通りの化合物に依存して、3秒から20秒の間で変動した。第二次参照として37.77ppmに定めたアダマンタンのメチレン炭素信号を使用して較正された、テトラメチルシランをゼロppmとして割り当てた13C化学シフトを、表2に列挙した。
【0119】
mvMOF−5−AB、−ABCD及び−ABCEFGHIにおけるのと同じ比率の連結混合物に対する13C CP/MAS NMRの対照実験を調べた。mvMOFに対する実験と比較する172ppmから174ppmまで、カルボニル炭素の有意なシフトがある。これは、これらの3個のmvMOFにおける全ての連結が亜鉛クラスターに化学的に結合していることを実証している。さらに、静電気試料の13Cスペクトルを、CP/MAS方法を使用して獲得し、移動ゲスト分子を検出するため調べた。この実験において、リサイクル遅延を20秒に設定することで、走査間の十分な緩和を可能にした。mvMOFsの完全な細孔構造を確証する検出可能な炭素信号は発見されなかった。
【表2】
【0120】
大きな単結晶mvMOF−5−AB−lscの合成。4.20gのZn(NO3)2・4H2O(16.0mmol)、450mgのBDCH2(2.7mmol)及び490mgのNH2−BDCH2(2.7mmol)を、混合物を15分間超音波処理することによって、50mLDEF中にガラスビーカー内で溶解した。PTFEフィルター(Whatman、0.45μmの孔径)を備えたプラスチックシリンジを使用することによって、溶液を10シンチレーションバイアル(20mLサイズ)に均等に分配した。バイアルに次いでしっかり蓋をし、等温の乾燥器内に置いた。85℃で72時間加熱した後で反応を止めた。各バイアルにおける母液を温めながらデカントし、生成物を新たなDEF(各バイアルに対して3×5mL)で洗浄した。生成物の大部分は、大きなチャンクの内部成長した立方褐色結晶であった。時折、一部の大きな単一立方体(サイズ1.5〜4.0mm)が認められた。上記した通りの通常のバッチに、8〜13個のこうした結晶を得ることができた。大きな立方結晶はMOF−5トポロジーを有することが、実験PXRDパターンがシミュレートした一つと一致すること、及びこれらの結晶を光学顕微鏡下で検査することによって確認された(図36A、B及び37)。高い当初の濃度が、大きなmvMOF単結晶の形成に有用である。
【0121】
大きな単結晶mvMOF−5−ABCD−lscの合成。6.20gのZn(NO3)2・4H2O(23.5mmol)、332mgのBDCH2(2.0mmol)及び362mgのNH2−BDCH2(2.0mmol)、490mgのBr−BDCH2(2.0mmol) 及び470mgの(Cl)2−BDCH2(2.0mmol)を、混合物を15分間超音波処理することによって、50mLのDEF中にガラスビーカー内で溶解した。PTFEフィルター(Whatman、0.45μmの孔径)を備えたプラスチックシリンジを使用することによって、溶液を10シンチレーションバイアル(20mLサイズ)に均等に分配した。バイアルに次いでしっかり蓋をし、等温の乾燥器内に置いた。85℃で120時間加熱した後で反応を止めた。各バイアルにおける母液を温めながらデカントし、生成物を新たなDEF(各バイアルに対して3×5mL)で洗浄した。生成物の大部分は、大きなチャンクの内部成長した褐色立方結晶であった。時折、一部の大きな単一立方体(サイズ1.0〜2.0mm)が認められた。上記した通りの通常のバッチに、6〜10個のこうした結晶を得ることができた。大きな立方結晶はMOF−5トポロジーを有することが、実験PXRDパターンがシミュレートした一つと一致すること、及びこれらの結晶を光学顕微鏡下で検査することによって確認された(図36C、D及び38)。
【0122】
大きな単結晶mvMOF−5−ABCEFGHI−lscの合成。4.20gのZn(NO3)2・4H2O(23.5mmol)、166mgのBDCH2(1.0mmol)、181mgのNH2−BDCH2(1.0mmol)、245mgのBr−BDCH2(1.0mmol)、211mgのNO2−BDCH2(1.0mmol)、194mgの(CH3)2−BDCH2(1.0mmol)、216mgのC4H4−BDCH2(1.0mmol)、278mgの(C3H5O)2−BDCH2(1.0mmol)及び374mgの(C7H7O)2−BDCH2(1.0mmol)を、混合物を15分間超音波処理することによって、50mLのDEF中にガラスビーカー内で溶解した。PTFEフィルター(Whatman、0.45μmの孔径)を備えたプラスチックシリンジを使用することによって、溶液を10シンチレーションバイアル(20mLサイズ)に均等に分配した。バイアルに次いでしっかり蓋をし、等温の乾燥器内に置いた。85℃で7日間加熱した後で反応を止めた。各バイアルにおける母液を温めながらデカントし、生成物を新たなDEF(各バイアルに対して3×5mL)で洗浄した。生成物の大部分は、大きなチャンクの内部成長した褐色立方結晶であった。時折、一部の大きな単一立方体(サイズ1.0〜2.0mm)が認められた。上記した通りの通常のバッチに、5〜6個のこうした結晶を得ることができた。大きな立方結晶はMOF−5トポロジーを有することが、実験PXRDパターンがシミュレートした一つと一致すること、及びこれらの結晶を光学顕微鏡下で検査することによって確認された(図36E、F及び39)。
【0123】
mvMOFの大きな単結晶の溶媒交換。適当な結晶を20mLシンチレーションバイアルに回収した。ピペットを使用してDEF溶媒を可能な限りきれいに除去した後、この開口バイアルを、クロロホルム蒸気で飽和したデシケーター中に置いたところ、該蒸気はゆっくりバイアル中に凝縮し、3日で約5mmの高さに蓄積した。結晶はこの時点でまだ完全に透明であった。蓄積したクロロホルムの除去後、バイアルに新たなクロロホルムを充填し、蓋をした。溶媒容量をそれぞれ1日目及び2日目の浸漬後に2度交換し、さらに2日間静置した。mvMOFの大きな単結晶のクロロホルム交換の合計時間は、デシケーター中で3日及び寝かせて5日であった。
【0124】
mvMOF内における連結の比率を1H NMRで決定した。結果を表1に列挙した。
【0125】
mvMOF−5−AB系列の合成。0.30MのZn(NO3)2・4H2O原液、0.10MのBDCH2(連結A)及びNH2−BDCH2(連結B)原液を事前に調製した。様々な容量の連結A及び連結B原液を2.0mLの0.30MのZn(NO3)2・4H2O原液中に、続いてDEFを添加することで、20mLのガラスバイアル中における合計容量を10mLにした。バイアルを密封し、等温の乾燥器内にて100℃で加熱し、48時間ソルボサーマルに反応させた。11種のmvMOF−5−AB化合物それぞれa〜kを合成したが、ここで、mvMOF−5−AB−aは連結Aをのみ有し、mvMOF−5−AB−kは連結Bのみを有しているので、それらは実際にそれぞれMOF−5及びIRMOF−3である。全ての生成物は、立方形の単結晶の形態であった。各化合物の結晶化度をPXRDによって確認したが、シミュレートしたMOF−5パターンと対応する非常に高い程度は、これらの化合物が本来不変のMOF−5トポロジー(図40)であることを表している。mvMOF内における連結の比率を、前に記載されているのと同様に1H NMRによって決定する。表3に、各化合物に使用した原液の量、及びそれらの当初の連結比、及びそれらの生成物の連結比を概要した。mvMOF−5−AB系列の結晶生成物におけるBのパーセント比率を、当初の化学量論比に対してプロットした(図41)。より多くの連結Bを出発原料として添加するにつれて、より多くの連結Bが結晶生成物に発見された。これは、mvMOFにおける連結比が、全範囲において、合成中に使用される連結の当初の量を制御することによりたやすく微調整することができることを明確に実証している。NH2−BDC(連結B)はMOF構造においてその特有な赤色を有するが、ここで、さらにmvMOFにおける連結比の正確な制御の能力を表す、合成されたままのmvMOF−5−AB結晶の色における段階的変化が認められた。連結Bの百分率が化合物aからkに増加するにつれて、結晶の色は無色から赤色に、次いで暗赤色に変化する(図41)。
【表3】
【0126】
mvMOF−5−AI系列の合成。0.30MのZn(NO3)2・4H2O原液、0.10MのBDCH2(連結A)及び(C7H7O)2−BDCH2(連結I)原液を事前に調製した。様々な容量の連結A及び連結I原液を2.0mLの0.30M Zn(NO3)2・4H2O原液中に、続いてDEFを添加することで、20mLのガラスバイアルにおける合計容量を10mLにした。バイアルを密封し、等温の乾燥器内にて100℃で加熱し、24時間ソルボサーマルに反応させた。3種のmvMOF−5−AI化合物それぞれa〜cを合成した。全ての生成物は、立方形の単結晶の形態であった。各化合物の結晶化度をPXRDによって確認したが、シミュレートしたMOF−5パターンと対応する非常に高い程度は、これらの化合物が本来不変のMOF−5トポロジー(図42)であることを表している。mvMOF内における連結の比率を、前に記載されているのと同様に1H NMRによって決定する。表4に、各化合物に使用した原液の量、及びそれらの当初の連結比、及びそれらの生成物の連結比を概要した。
【0127】
mvMOF−5−AB系列と同様に、mvMOF−5−AI系列の結晶生成物におけるIのパーセント比率は、より多くの連結Iが出発原料中に使用されたことに伴い増加する。
【表4】
【0128】
mvMOF−5−ABCD系列の合成。0.30MのZn(NO3)2・4H2O原液、0.10MのBDCH2(連結A)及びNH2−BDCH2(連結B)、Br−BDCH2(連結C)、(Cl)2−BDCH2(連結D)原液を事前に調製した。様々な容量の連結A及び連結I原液を2.0mLの0.30M Zn(NO3)2・4H2O原液中に、続いてDEFを添加することで、20mLのガラスバイアルにおける合計容量を10mLにした。バイアル密封し、等温の乾燥器内にて100℃で加熱し、48時間ソルボサーマルに反応させた。5種のmvMOF−5−ABCD化合物それぞれa〜eを合成した。全ての生成物は、立方形の単結晶の形態であった。各化合物の結晶化度をPXRDによって確認したが、シミュレートしたMOF−5パターンと対応する非常に高い程度は、これらの化合物が本来不変のMOF−5トポロジー(図43)であることを表している。mvMOF内における連結の比率は、セクション1に記載されているのと同様に1H NMRによって決定する。表5に、本文における各化合物及び表1(連結比部分の制御)に使用された原液の量を概要した。mvMOF−5−ABCD結晶の連結比を各化合物の合成に使用された連結の化学量論比と比較した場合、出発原料中と同様に使用された特定の連結の量が増加(又は減少)するにつれて、生じるmvMOFにおける比率は対応して増加(又は減少)する。これらは、mvMOFs内における連結の比率が制御可能であることを再び実証している。
【表5】
【0129】
mvMOF−5−AC及び−ACEFに関する単一X線回折データ収集、構造溶液及び精密化の手順。単結晶データ収集、構造溶液及び精密化に関する一般的手順をここに提示する。構造上の不規則を含めて各構造の固有の詳細を、各構造に関する実験及び計量の前に記載する。
【0130】
一般のデータ収集及び精密化の手順:CCDエリア検出器を備え及び1200W出力(40kV、30mA)で操作されるBruker SMART APEXII三軸回折計上にデータを回収して、CuKα放射線(λ=1.5418Å)を発生させた。Bruker Excalibur Gobelミラー光学を使用して入射X線ビームを集中させ、単色にした。少量の母液を含有するフレームシールしたボロシリケート細管に結晶を全て装填することで、データ収集中の脱溶媒和を防止した。
【0131】
各試験片の当初のω−φを走査することで、予備単位格子パラメーターが得られ、結晶のモザイク性(すなわち、骨組み間の箇所の幅)を評価して、データ収集に必要なフレーム幅を選択した。全ての場合に関して、0.5°の骨組み幅が適切であると判断し、異なる検出器(2θ)の設定でφ及びωの走査を重複することを実施するため、Bruker APEX2ソフトウェア一式を使用してデータの完全半球を回収した。データ収集に続いて、データ統合用の単位格子パラメーターを再決定するため、及びCELL_NOWを使用して回転双晶化を調べるため、Ewald球体の全ての領域から反射光をサンプリングした。
【0132】
回収した骨組みの徹底的見直しに続いて、データセットの分解能を判断し、必要に応じて、干渉性散乱が認められない骨組みの領域をデータ統合に対する検討から除いた。Bruker APEX2 V2.1ソフトウェアを使用し、狭いフレームアルゴリズム及び平均強度の少数下限を0.400で、データを統合した。吸収補正はデータの品質を向上させるのに効果的ではなかったが、これは、主に軽原子を含有する低密度材料の小さな結晶には予想外ではない。
【0133】
XPREPを使用して、欠面双晶化に関する空間群決定及び試験を実施した。全ての場合において、最大限の空間群を選択し、欠面双晶化の兆候は認められなかった。全ての構造を直接的方法によって解析し、SHELXTL’97ソフトウェア一式を使用して精密化した。初期モデルの最小二乗精密化に続いて差異Fマップの反復検査から、原子の位置を決定した。完全な収束が達成されるまで、最終モデルを異方的に精密化した(許容されたデータの数及び安定な精密化が達する場合)。部分的占有率を、不規則化基における原子の各対に割り当てた。一部の原子は、高不規則及び低占有率のため、位置を正確に決定できなかった(mvMOF−ACEFにおける連結E上のニトロ基)。水素原子を算出位置に置き、ライディング原子として含めた。骨格の空隙内の電子密度のモデル化は、低減したデータ分解能のため、構造における全てのゲスト実体の同定に至らなかった。溶媒が充填された細孔を含有する多孔質結晶に典型的であるこの問題は、認められた強力な(高強度)散乱がせいぜい約1.0Åに制限される生データにあり、より高い分解能データが存在するが弱い(低強度)。X線データを向上させるための一般戦略のように、結晶をX線に暴露する時間を増加させることは、低角度データからの強度が検出器を飽和し、高角度データにおいて最小限の向上が達成されたため、これらの場合における高角度データの品質を改良しなかった。さらに、MOF主鎖のフェニル単位上の不規則化官能基からの散漫散乱、及び結晶内の空隙空間における溶媒、及び結晶を装填させるのに使用された細管からの溶媒が、背景雑音及び時々高角度データの「洗い出し」に寄与した。これらの拡張骨格構造に関して、構造におけるゲスト実体が自由に移動することが可能になり、したがって認められた構造因子に干渉性散乱期間を与えない場合、より高い範囲の温度(−120から−100℃よりむしろ−50から−15℃)で回収したデータに反してモデル化することも、より妥当であった。
【0134】
骨格における原子位置の精度を証明するため、適用可能な場合、A.SpekのSQUEEZEルーチンの適用が行われてきた。しかし、「非SQUEEZE」構造のための原子配位も提案されており、プログラムSQUEEZEが用いられた場合にCIFも提示された。PLATONのAdsymサブルーチンを使用して全ての構造を検査することで、追加の対称がモデルに適用され得ることがないことを確保した。ORTEP図表における全ての楕円体は、別段の注記がない限り、15%の確率レベルで表示されている。全ての構造に関して、上昇したR値は、上記に表した理由により、MOF結晶学において一般に遭遇する。
【0135】
mvMOF−5−ACに関する実験及び精密化の詳細。mvMOF−5−ACの無色平行六面体結晶(0.41×0.41×0.28mm3)を、0.5mm直径のボロシリケート細管中に少量の母液と一緒に置いた。細管をフレームシールし、CCDエリア検出器を備え、1200W出力(40kV、30mA)で操作されるSMART APEXII三軸回折計上に装填することで、窒素の液体N2冷却流中で258(2)Kに冷却しながらCuKα放射線(λ=1.5418Å)を発生させた。3種の異なる検出器(2θ)設定(2θ=28°、60°、100°)でφ及びωの走査を重複することを実施するため、Bruker APEX2ソフトウェア一式を使用してデータの完全半球を回収した。合計14178の反射光を回収し、これらのうち2951が固有であり、これらの1994が2σ(I)を超えていた。θの範囲は2.97°から41.28°であった。データの分析は回収中の軽視し得る減衰を示した。プログラムScaleを行うことで、対称関連反射光又は繰り返し測定した反射光の間の差異を最小にした。
【0136】
直接的方法を使用してZ=8で単斜C2/m空間群において、構造を解析した。水素原子をそれらの親原子の配位に乗る球体として生成して、骨格の主鎖におけるZn及びO原子を異方的に精密化し、全ての他の非水素原子を等方的に精密化した。モデル溶媒分子にした試みは、ゲスト実体の同定に至らなかった。溶媒が骨格に結合していないので、MOF構造に関して溶媒分子の不正確な位置決定が予想された。さらに、非常に高い置き換えパラメーター、不規則による高いesd及び部分的な占有率が、溶媒分子の正確な位置を決定することを不可能にした。それでもなお、mvMOF−5−ACの主鎖骨格及び−Br基の帰属及び精密化は、生じた結合及び角度の測定基準によって判断した通り明らかであった。
【0137】
骨格における原子位置を向上するため、A.SpekのSQUEEZEルーチンの適用が行われてきた。しかし、「非SQUEEZE」構造に関する原子配位も提案されている。F2に対する最終フルマトリックス最小二乗精密化は、GOF=0.998で、R1=0.1257(F>2σ(F))及びwR2=0.3084(全データ)に収束した。SQUEEZEプログラムが用いられていない場合の構造では、F2に対する最終フルマトリックス最小二乗精密化は、GOF=1.426で、R1=0.1892(F>2σ(F))及びwR2=0.4184(全データ)に収束した。骨格原子のみを後者の構造因子算出に含めた場合、Fマップにおける残留電子密度は、mvMOF−5−ACの細孔内に位置していた。結晶構造精密化のための実験式は、0.683gcm−3の密度で、1H NMRによる結晶における2種の連結の測定比率に基づき、C12 H5.43 Br0.57 O7.25 Zn2である。
【表6】
【表7】
【表8】
【表9】
【表10】
【表11】
【0138】
mvMOF−5−ACに関する実験及び精密化の詳細。mvMOF−5−ACEFの赤色平行六面体結晶(0.30×0.30×0.21mm3)を、0.4mm直径のボロシリケート細管中に少量の母液と一緒に置いた。細管をフレームシールし、CCDエリア検出器を備え、1200W出力(40kV、30mA)で操作されるSMART APEXII三軸回折計上に装填することで、窒素の液体N2冷却流中で233(2)Kに冷却しながらCuKα放射線(λ=1.5418Å)を発生させた。3種の異なる検出器(2θ)設定(2θ=28°、60°、100°)でφ及びωの走査を重複することを実施するため、Bruker APEX2ソフトウェア一式を使用してデータの完全半球を回収した。合計13758の反射光を回収し、これらのうち2855が固有であり、これらの1918が2σ(I)を超えていた。θの範囲は2.99°から41.02°であった。データの分析は回収中の軽視し得る減衰を示した。プログラムScaleを行うことで、対称関連反射光又は繰り返し測定した反射光の間の差異を最小にした。
【0139】
直接的方法を使用してZ=8で単斜C2/m空間群において、構造を解析した。水素原子をそれらの親原子の配位に乗る球体として生成して、骨格の主鎖におけるZn原子を異方的に精密化し、全ての他の非水素原子を等方的に精密化した。モデル溶媒分子にした試みは、ゲスト実体の同定に至らなかった。溶媒が骨格に結合していないので、MOF構造に関して溶媒分子の不正確な位置決定が予想された。さらに、非常に高い置き換えパラメーター、不規則による高いesd及び部分的な占有率が、溶媒分子の正確な位置を決定することを不可能にした。それでもなお、mvMOF−5−ACEFの主鎖骨格及び官能基(−Br及び−CH3)の一般位置の帰属及び精密化は、生じた結合及び角度の測定基準によって判断した通り明らかであった。低い占有率(モルパーセントで9.4%のみ)及び四つの位置上のその不規則を考えると、非常に少量で存在するNO2の組成は、したがって、この精密化において無視されている。
【0140】
骨格における原子位置の精度を向上するため、A.SpekのSQUEEZEルーチンの適用が行われてきた。しかし、「非SQUEEZE」構造に関する原子配位も提案されている。F2に対する最終フルマトリックス最小二乗精密化は、GOF=1.003で、R1=0.1219(F>2σ(F))及びwR2=0.3041(全データ)に収束した。SQUEEZEプログラムが用いられていない場合の構造では、F2に対する最終フルマトリックス最小二乗精密化は、GOF=1.378で、R1=0.1737(F>2σ(F))及びwR2=0.3972(全データ)に収束した。骨格原子のみを後者の構造因子算出に含めた場合、Fマップにおける残留電子密度は、mvMOF−5−ACEFの細孔内に位置していた。結晶構造精密化の実験式は、1H NMRによる結晶における4種の連結の測定比率に基づき、C12.81 H6.65 Br0.31 N0.14 O7.78 Zn2であった。
【表12】
【表13】
【表14】
【表15】
【表16】
【表17】
【0141】
mvMOFの多孔性、H2取込み、及びCO2分離研究。全ての低圧力ガス吸着実験(最大1バール)をQuantachrome Autosorb−1自動容量測定器上で行った。液体窒素槽(77K)及び液体アルゴン槽(87K)をN2、H2及びArの等温線測定に使用し、一方恒温槽(273k、283k及び298K)をCO2及びCOの等温線測定に使用した。超高純度グレードのN2、H2、Ar、CO、He(99.999%純度)及びCO2のガス(99.995%純度)を吸着実験全体で使用した。ガスの非理想度を実験温度で第二ビリアル係数から得た。見かけ表面積の測定のため、N2(Ar)断面積を16.2(14.2)Å2/分子と仮定してN2(Ar)等温線の吸着枝を使用するラングミュア方法を適用した。MOFの合成されたままの試料をクロロホルム中に周囲温度で24時間を3周期の間浸漬し、周囲温度で12時間脱気した。
【0142】
構成剛性、並びにその結果としての脱気したMOF−5、mvMOF−5−AB、−AI−a、−ABCD及び−ABCEFGHIの永久的多孔性を、ガス収着分析によって明解に証明した。これらのMOFに関してI型N2/Ar吸着等温線の挙動が認められ(図46及び47)、微孔性の性質を明らかにした。mvMOF−ABのラングミュア表面積を算出したところ、3640m2/gであった。同様に、MOF−5、mvMOF−5−AI−a、−ABCD及び−ABCEFGHIの見かけ表面積を算出したところ、それぞれ4140m2/g、2680m2/g、2860m2/g及び1860m2/gであった。MVMOF−5−AI系列において、より嵩高い連結Iを結晶生成物に導入するにつれて、算出密度は−aから−cへと増加し、表面積は減少する(図49)。
【表18】
【0143】
mvMOF−5−AI及びMOF−5の孔径分布分析。さらに、嵩高い連結を持つmvMOFの一つであるMOF−5−AI−aの孔径分布を研究した。MOF−5−AI−aに関するAr吸着等温線と非局所密度汎関数理論(NLDFT)モデルとの一致は、孔径分布が12Å未満で大きく集中していることを明らかにしており、これはMOF−5の典型的孔径分布である(図41)。これは、MOF−5−AI−aのほとんど全ての大きなケージが連結Iによって部分的に官能化されており、MOF−5の細孔環境が存在しないことを表している。
【0144】
mvMOF−5−EIに関するCO2及びCOの吸着等温線は、図48〜51にそれぞれ例示されている。確かなヘンリー定数を推定するため、温度非依存性パラメーターai及びbiを含むビリアル型数式を適用した。
【数1】
【0145】
ここで、Pは圧力であり、Nは吸着量であり、Tは温度であり、並びにm及びnは等温線を適切に表示するのに必要な係数の数を表す。これらの結果から、ヘンリー定数(KH)を算出するが、Tは温度である。
【数2】
【0146】
298Kでのガス成分jを超えたiのヘンリー則選択率を等式(3)に基づいて算出する。
【数3】
【0147】
本発明の実施形態が多数記載されている。それでもなお、本発明の趣旨及び範囲から逸脱することなく様々な改変が行われ得ることを理解されよう。すなわち、他の実施形態は、以下の請求項の範囲内である。
【特許請求の範囲】
【請求項1】
主鎖に沿った配向、数、相対位置及び比率が連結部分の変化のないサイズ及び主鎖の不変の結合性のために制御可能である異なる官能基であって、骨格における細孔の化学的及び物理的性質を修飾する官能基を有する複数の連結部分を含む多孔質有機骨格。
【請求項2】
n≧2であるn個の異なる有機連結から構成されている、請求項1に記載の有機骨格。
【請求項3】
官能基が金属−酸化物及びフェニル単位を含むコアに沿っている、請求項1に記載の有機骨格。
【請求項4】
有機骨格が、金属−酸化物結合部及び有機連結部分の反復単位、並びに連結部分に共有結合している複数の官能基を含み、官能基が不均一であり、及び/又は官能基が連結に沿って異なった間隔で配置されている、請求項1に記載の有機骨格。
【請求項5】
骨格内における細孔のそれぞれが、細孔の中央を向いている複数の異なる官能基を含む、請求項1〜4のいずれかに記載の有機骨格。
【請求項6】
骨格が、MOF−5骨格のトポロジーを含む、請求項1に記載の有機骨格。
【請求項7】
骨格が、Li+、Na+、Rb+、Mg2+、Ca2+、Sr2+、Ba2+、Sc3+、Ti4+、Zr4+、Ta3+、Cr3+、Mo3+、W3+、Mn3+、Fe3+、Fe2+、Ru3+、Ru2+、Os3+、Os2+、Co3+、Co2+、Ni2+、Ni+、Pd2+、Pd+、Pt2+、Pt+、Cu2+、Cu+、Au+、Zn2+、Al3+、Ga3+、In3+、Si4+、Si2+、Ge4+、Ge2+、Sn4+、Sn2+、Bi5+、Bi3+、及びそれらの組合せからなる群から選択される金属イオンを、対応する金属塩対アニオンとともに含む、請求項1に記載の有機骨格。
【請求項8】
連結が、
【化1】
(式中、R1〜R4は−H、−NH2、−BR、−Cl、−NO2、−CH3、−OCH2R5及び−O−CH2R6からなる群から選択され、R5は約1〜5個の炭素のアルキル又はアルケンであり、R6はアリール又は置換アリールであるか、又はR1〜R2は隣接する場合、環を形成することができる)
からなる群から選択される一般構造を有する、請求項1〜4、6又は7のいずれかに記載の有機骨格。
【請求項9】
連結が、
【化2】
からなる群から選択されるメンバーを含む、請求項8に記載の有機骨格。
【請求項10】
複数の連結部分の第一連結部分が第一官能基を含み、複数の連結部分の第二連結部分が第二官能基を含み、第二官能基が反応後の基との合成後反応を受けることで骨格をさらに官能化することができる、請求項1に記載の有機骨格。
【請求項11】
連結部分が所望の比率で連結部分の特定の組合せを有機骨格に組み込むための所望の比率になっている複数の化学的官能化連結部分と、金属イオン又は金属硝酸塩とを混合すること、結晶を精製すること、及び溶媒を除去することを含む方法によって作製される、請求項1に記載の有機骨格。
【請求項12】
方法が、ベンゼンジカルボン酸を含む有機骨格に、所望の比率で連結部分の特定の組合せを組み込むための所望の比率で、複数の化学的官能化連結部分と硝酸亜鉛とをDEF/DMF中にて混合することを含む、請求項11に記載の有機骨格。
【請求項13】
請求項1〜10のいずれかに記載の有機骨格を含むガス分離装置。
【請求項14】
請求項1〜10のいずれかに記載の有機骨格を含むガス貯蔵装置。
【請求項15】
同じトポロジーであるが均一な連結部分を有する骨格と比較して向上したガス収着能を有する、請求項1に記載の有機骨格。
【請求項1】
主鎖に沿った配向、数、相対位置及び比率が連結部分の変化のないサイズ及び主鎖の不変の結合性のために制御可能である異なる官能基であって、骨格における細孔の化学的及び物理的性質を修飾する官能基を有する複数の連結部分を含む多孔質有機骨格。
【請求項2】
n≧2であるn個の異なる有機連結から構成されている、請求項1に記載の有機骨格。
【請求項3】
官能基が金属−酸化物及びフェニル単位を含むコアに沿っている、請求項1に記載の有機骨格。
【請求項4】
有機骨格が、金属−酸化物結合部及び有機連結部分の反復単位、並びに連結部分に共有結合している複数の官能基を含み、官能基が不均一であり、及び/又は官能基が連結に沿って異なった間隔で配置されている、請求項1に記載の有機骨格。
【請求項5】
骨格内における細孔のそれぞれが、細孔の中央を向いている複数の異なる官能基を含む、請求項1〜4のいずれかに記載の有機骨格。
【請求項6】
骨格が、MOF−5骨格のトポロジーを含む、請求項1に記載の有機骨格。
【請求項7】
骨格が、Li+、Na+、Rb+、Mg2+、Ca2+、Sr2+、Ba2+、Sc3+、Ti4+、Zr4+、Ta3+、Cr3+、Mo3+、W3+、Mn3+、Fe3+、Fe2+、Ru3+、Ru2+、Os3+、Os2+、Co3+、Co2+、Ni2+、Ni+、Pd2+、Pd+、Pt2+、Pt+、Cu2+、Cu+、Au+、Zn2+、Al3+、Ga3+、In3+、Si4+、Si2+、Ge4+、Ge2+、Sn4+、Sn2+、Bi5+、Bi3+、及びそれらの組合せからなる群から選択される金属イオンを、対応する金属塩対アニオンとともに含む、請求項1に記載の有機骨格。
【請求項8】
連結が、
【化1】
(式中、R1〜R4は−H、−NH2、−BR、−Cl、−NO2、−CH3、−OCH2R5及び−O−CH2R6からなる群から選択され、R5は約1〜5個の炭素のアルキル又はアルケンであり、R6はアリール又は置換アリールであるか、又はR1〜R2は隣接する場合、環を形成することができる)
からなる群から選択される一般構造を有する、請求項1〜4、6又は7のいずれかに記載の有機骨格。
【請求項9】
連結が、
【化2】
からなる群から選択されるメンバーを含む、請求項8に記載の有機骨格。
【請求項10】
複数の連結部分の第一連結部分が第一官能基を含み、複数の連結部分の第二連結部分が第二官能基を含み、第二官能基が反応後の基との合成後反応を受けることで骨格をさらに官能化することができる、請求項1に記載の有機骨格。
【請求項11】
連結部分が所望の比率で連結部分の特定の組合せを有機骨格に組み込むための所望の比率になっている複数の化学的官能化連結部分と、金属イオン又は金属硝酸塩とを混合すること、結晶を精製すること、及び溶媒を除去することを含む方法によって作製される、請求項1に記載の有機骨格。
【請求項12】
方法が、ベンゼンジカルボン酸を含む有機骨格に、所望の比率で連結部分の特定の組合せを組み込むための所望の比率で、複数の化学的官能化連結部分と硝酸亜鉛とをDEF/DMF中にて混合することを含む、請求項11に記載の有機骨格。
【請求項13】
請求項1〜10のいずれかに記載の有機骨格を含むガス分離装置。
【請求項14】
請求項1〜10のいずれかに記載の有機骨格を含むガス貯蔵装置。
【請求項15】
同じトポロジーであるが均一な連結部分を有する骨格と比較して向上したガス収着能を有する、請求項1に記載の有機骨格。
【図1】

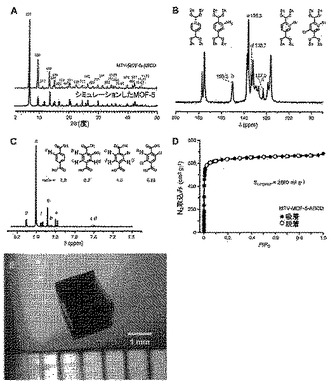
【図2】
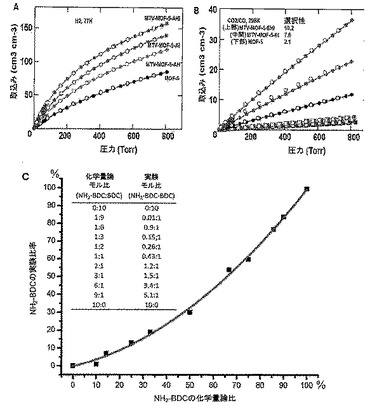
【図3】
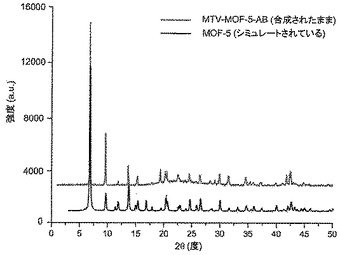
【図5】
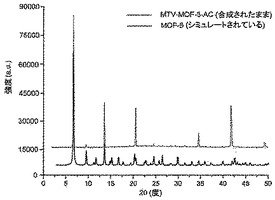
【図6】
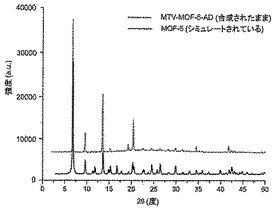
【図7】
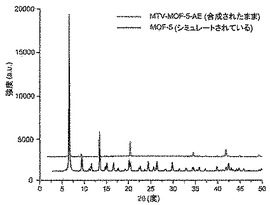
【図8】
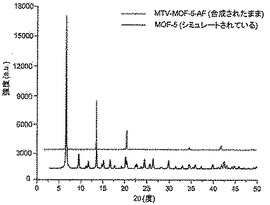
【図9】
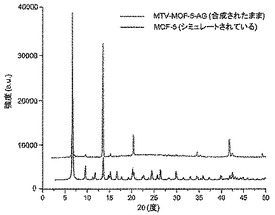
【図10】
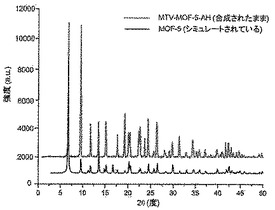
【図11】
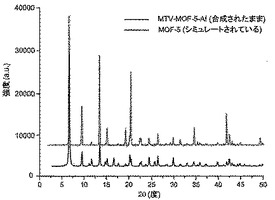
【図12】
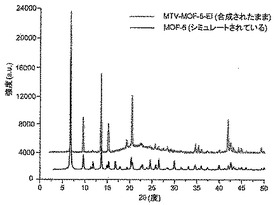
【図13】
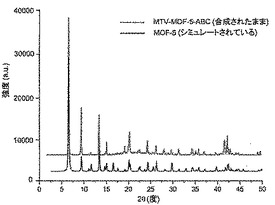
【図14】
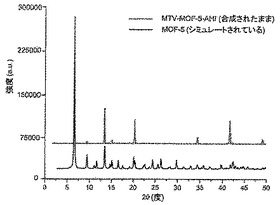
【図15】
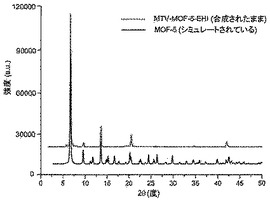
【図16】
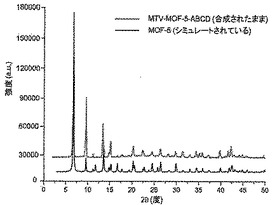
【図17】
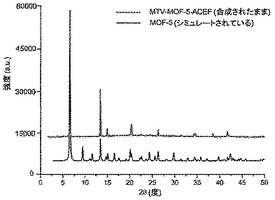
【図18】
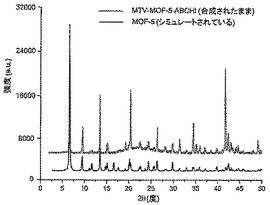
【図19】
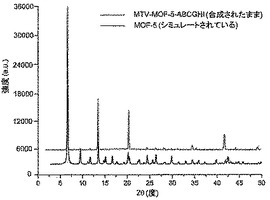
【図20】
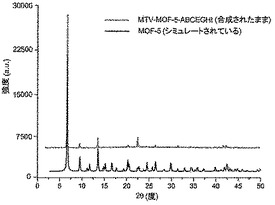
【図21】
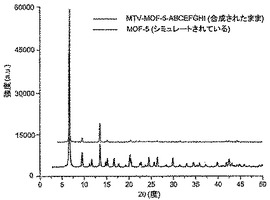
【図22】
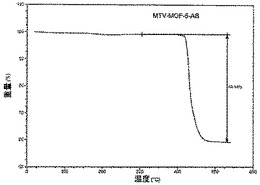
【図23】
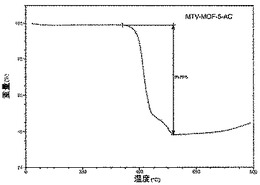
【図24】
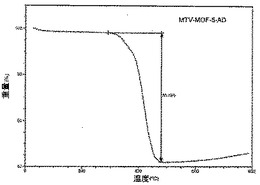
【図25】
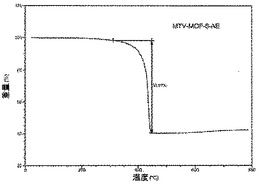
【図26】
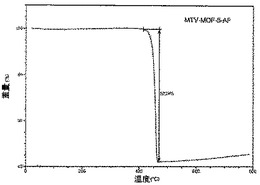
【図27】
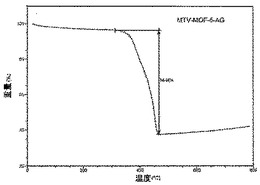
【図28】
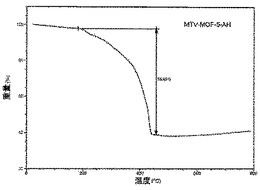
【図29】
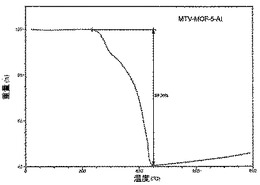
【図30】
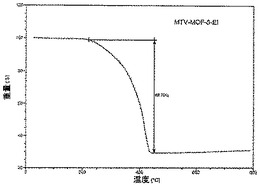
【図31】
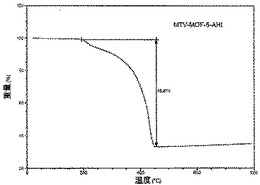
【図32】
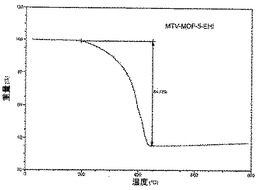
【図33】
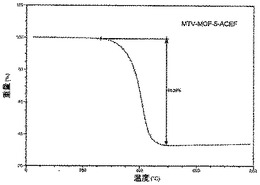
【図34】
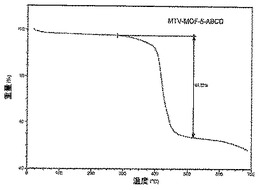
【図35】
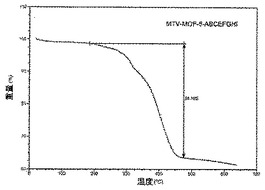
【図36】

【図37】
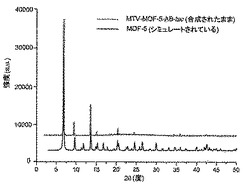
【図38】
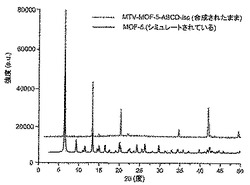
【図39】
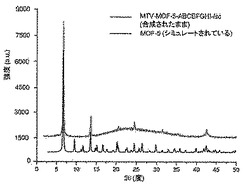
【図40】
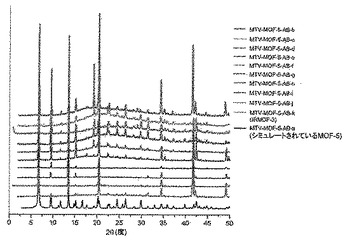
【図41】
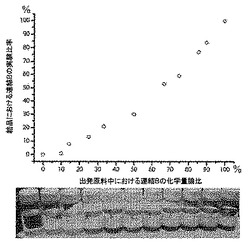
【図42】
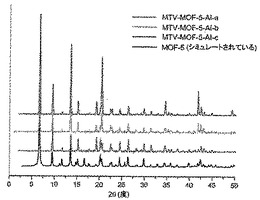
【図43】
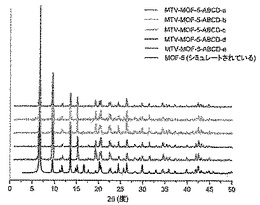
【図44】
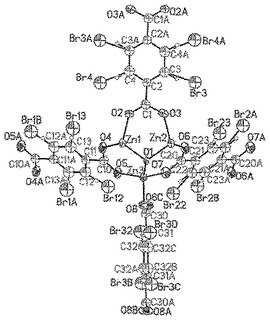
【図45】
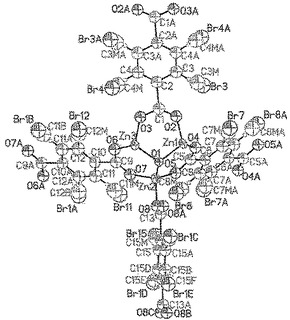
【図46】
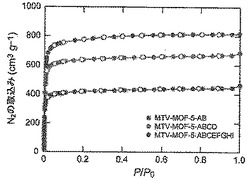
【図47】
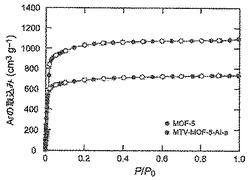
【図48】

【図49】
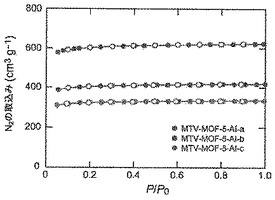
【図50】
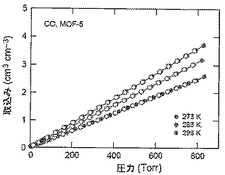
【図51】
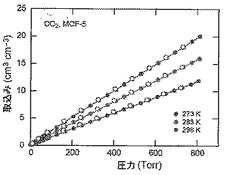
【図52】
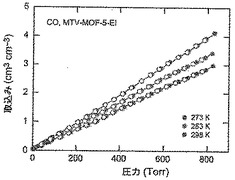
【図53】
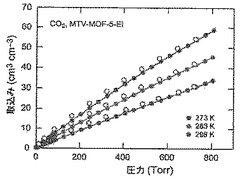
【図54】
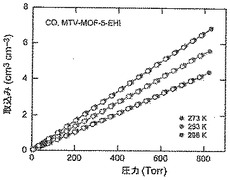
【図55】
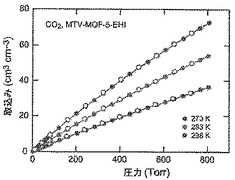
【図2】
【図3】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【図31】
【図32】
【図33】
【図34】
【図35】
【図36】
【図37】
【図38】
【図39】
【図40】
【図41】
【図42】
【図43】
【図44】
【図45】
【図46】
【図47】
【図48】
【図49】
【図50】
【図51】
【図52】
【図53】
【図54】
【図55】
【公表番号】特表2012−530718(P2012−530718A)
【公表日】平成24年12月6日(2012.12.6)
【国際特許分類】
【出願番号】特願2012−516333(P2012−516333)
【出願日】平成22年6月18日(2010.6.18)
【国際出願番号】PCT/US2010/039154
【国際公開番号】WO2010/148296
【国際公開日】平成22年12月23日(2010.12.23)
【出願人】(304056899)ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア (9)
【氏名又は名称原語表記】THE REGENTS OF THE UNIVERSITY OF CALIFORNIA
【住所又は居所原語表記】1111 Franklin Street, Oakland, CA 94607−5200, United States of America
【Fターム(参考)】
【公表日】平成24年12月6日(2012.12.6)
【国際特許分類】
【出願日】平成22年6月18日(2010.6.18)
【国際出願番号】PCT/US2010/039154
【国際公開番号】WO2010/148296
【国際公開日】平成22年12月23日(2010.12.23)
【出願人】(304056899)ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア (9)
【氏名又は名称原語表記】THE REGENTS OF THE UNIVERSITY OF CALIFORNIA
【住所又は居所原語表記】1111 Franklin Street, Oakland, CA 94607−5200, United States of America
【Fターム(参考)】
[ Back to top ]