
鎮痛剤
【課題】グリチルリチンの類縁体を有効成分とする新規な鎮痛剤の提供。
【解決手段】下記式(1)で表される化合物又はその薬学上許容される塩を有効成分とする鎮痛剤。
[化1]

【解決手段】下記式(1)で表される化合物又はその薬学上許容される塩を有効成分とする鎮痛剤。
[化1]
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規の鎮痛剤に関する。
【背景技術】
【0002】
痛みの緩和は、日常生活での不快感の改善から、発病時の病状の軽減、そして終末期医療での深刻な苦痛の軽減に至るまで、極めて広範囲の目的に沿って行われている。
痛みには、炎症性のものと神経因性のものがあると考えられ、その病態に応じて極めて多種類の鎮痛剤がこれまでに開発されており、比較的良く知られているものだけでも、アセトアミノフェン、アセチルサリチル酸、イブプロフェン、エテンザミド、モルヒネ等、枚挙に暇がない。
このような中、目的に即した鎮痛剤を使用することで、副作用を抑えつつ、十分な鎮痛効果を得ようとするのはごく自然なことであり、新規な鎮痛剤の開発が強く望まれている。
【0003】
ところで、甘草の主成分であるグリチルリチンは、抗炎症作用を有することが知られている(非特許文献1参照)。そして、例えば、グリチルリチンのアグリコンであるグリチルレチン酸の誘導体に相当する、オレアン−11,13(18)−ジエン−3β,30−ジオールの、二つの水酸基がフタル酸エステル化された化合物は、抗潰瘍剤の有効成分となることが開示されている(特許文献1参照)。しかし、これに対して、グリチルリチンやその類縁体については、それ自体が鎮痛剤として十分に有効であるか否か、これまでに十分な検討がなされてこなかったのが実情である。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特開昭60−260540号公報
【非特許文献】
【0005】
【非特許文献1】Inoue et al.,1996,Jpn.J.Pharmacol.71,281−289
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明は上記事情に鑑みてなされたものであり、グリチルリチンの類縁体を有効成分とする新規な鎮痛剤を提供することを課題とする。
【課題を解決するための手段】
【0007】
本発明者らは、上記課題を解決するために鋭意研究を行った結果、グリチルリチンのアグリコンであるグリチルレチン酸の特定の誘導体が、疼痛の抑制効果を示すことを見出し、本発明を完成するに至った。
【0008】
すなわち、上記課題を解決するため、
本発明は、下記式(1)で表される化合物又はその薬学上許容される塩を有効成分とする鎮痛剤を提供する。
【0009】
【化1】
【0010】
本発明の鎮痛剤においては、下記式(10−1)で表される化合物又はその薬学上許容される塩を有効成分とすることが好ましい。
【0011】
【化2】
【発明の効果】
【0012】
本発明によれば、グリチルリチンの類縁体を有効成分とする新規な鎮痛剤を提供できる。
【図面の簡単な説明】
【0013】
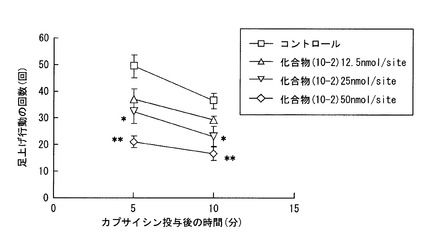
【図1】実施例1におけるラットのカプサイシン投与後の足上げ行動の回数を示すグラフである。
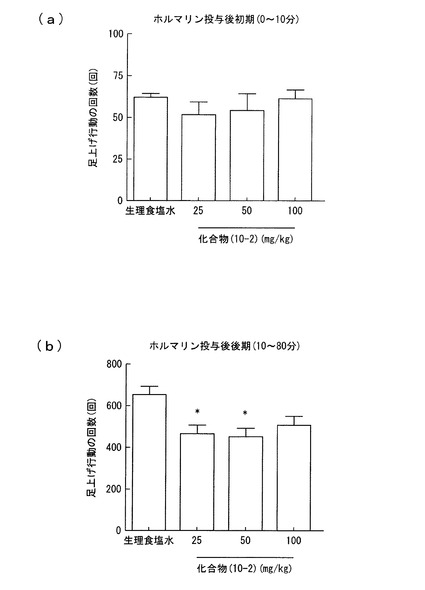
【図2】実施例2におけるラットの足上げ行動の回数を示すグラフであり、(a)はホルマリン投与後初期(0〜10分)における回数を、(b)はホルマリン投与後後期(10〜80分)における回数をそれぞれ示す。
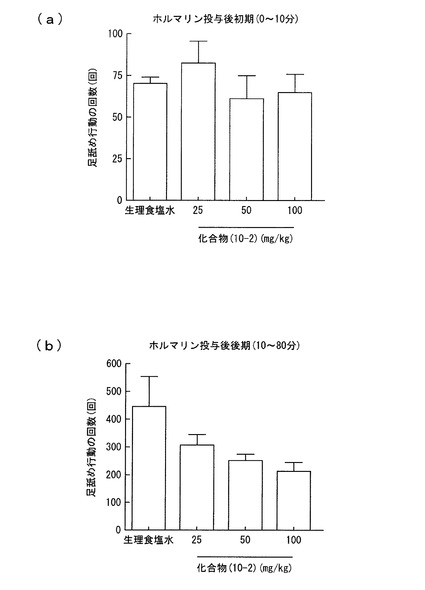
【図3】実施例2におけるラットの足舐め行動の回数を示すグラフであり、(a)はホルマリン投与後初期(0〜10分)における回数を、(b)はホルマリン投与後後期(10〜80分)における回数をそれぞれ示す。
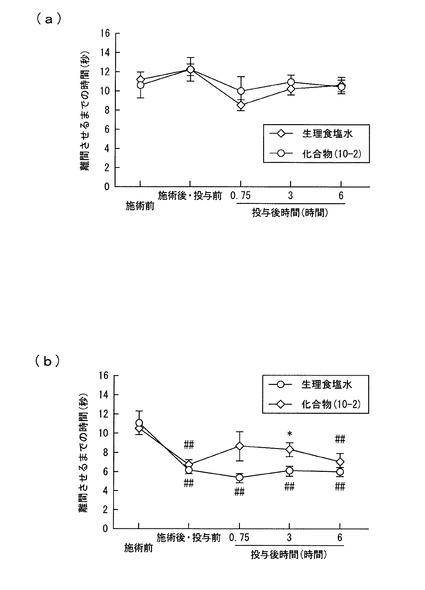
【図4】実施例3において、左坐骨神経をゆるく結紮したラットに熱刺激を加えた時の、ラットが足を熱源から離間させるまでの時間を示すグラフであり、(a)は右足を、(b)は左足を、それぞれ熱源から離間させるまでの時間を示す。
【発明を実施するための形態】
【0014】
本発明の鎮痛剤は、下記式(1)で表される化合物(以下、化合物(1)と略記する)又はその薬学上許容される塩を有効成分とする。
【0015】
【化3】
【0016】
化合物(1)又はその薬学上許容される塩を有効成分とする鎮痛剤は、炎症性の痛み及び神経因性の痛みのいずれにも抑制効果を示す。
【0017】
化合物(1)は、下記式(30−1)で表されるグリチルレチン酸の誘導体であり、これまでに知られている鎮痛剤とは異なる構造を有し、主要骨格も全く異なる。
【0018】
【化4】
【0019】
化合物(1)としては、下記式(10−1)で表される化合物(以下、化合物(10−1)と略記する)が好ましい。すなわち、本発明の鎮痛剤は、化合物(10−1)又はその薬学上許容される塩を有効成分とするものが好ましい。
【0020】
【化5】
【0021】
化合物(1)の薬学上許容される塩の好ましいものとして、化合物(1)モノアンモニウム塩、化合物(1)ジアンモニウム塩等のアンモニウム塩;化合物(1)モノナトリウム塩、化合物(1)ジナトリウム塩、化合物(1)モノカリウム塩、化合物(1)ジカリウム塩等のアルカリ金属塩;化合物(1)カルシウム塩;化合物(1)マグネシウム塩;化合物(1)アルミニウム塩;化合物(1)モノコリン塩、化合物(1)ジコリン塩等の有機アミン塩等が例示できる。
【0022】
化合物(1)の薬学上許容される塩が二塩の場合、塩を構成する二つのカチオン種は、上記のように同じでも良いが、異なっていても良く、異なる場合には、アンモニウムイオン、ナトリウムイオン、カリウムイオン、カルシウムイオン、マグネシウムイオン、アルミニウムイオン及びコリンからなる群から選択される異なる二種のカチオンが例示できる。
【0023】
化合物(1)の薬学上許容される塩としては、化合物(1)アルカリ金属塩が好ましく、化合物(1)ナトリウム塩がより好ましい。
【0024】
化合物(1)又はその薬学上許容される塩は、一種を単独で使用しても良いし、二種以上を併用しても良い。二種以上を併用する場合には、その組み合わせ及び比率は、目的に応じて適宜選択すれば良い。
【0025】
本発明の鎮痛剤の製剤形態は特に限定されず、目的に応じて錠剤、散剤、顆粒剤、カプセル剤、細粒剤、液剤(水薬等)等の経口剤;吸入剤、座剤、注射剤、貼付剤、スプレー剤、軟膏等の非経口剤等から適宜選択すれば良い。これら製剤形態の鎮痛剤は、いずれも公知の方法で製造できる。
【0026】
鎮痛剤を経口剤等の製剤形態とする場合には、これら製剤の製造で通常使用される各種添加剤を配合しても良い。前記添加剤としては、賦形剤、滑沢剤、可塑剤、界面活性剤、結合剤、崩壊剤、湿潤剤、安定剤、矯味剤、着色剤、香料等が例示できる。
前記添加剤は、一種を単独で使用しても良いし、二種以上を併用しても良い。二種以上を併用する場合には、その組み合わせ及び比率は、目的に応じて適宜選択すれば良い。
【0027】
前記賦形剤としては、乳糖、ブドウ糖、D−マンニトール、果糖、デキストリン、デンプン、食塩、炭酸水素ナトリウム、炭酸カルシウム、アルギン酸ナトリウム、エチルセルロース、ナトリウムカルボキシメチルセルロース、ヒドロキシプロピルセルロース、無水ケイ酸、カオリン等が例示できる。
【0028】
前記滑沢剤としては、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸、タルク、トウモロコシデンプン、マクロゴール等が例示できる。
【0029】
前記可塑剤としては、ポリエチレングリコール、プロピレングリコール、グリセリン類、トリアセチン、中鎖脂肪酸トリグリセリド、アセチルグリセリン脂肪酸エステル、クエン酸トリエチル等が例示できる。
【0030】
前記結合剤としては、ゼラチン、アラビアゴム、セルロースエステル、ポリビニルピロリドン、水飴、甘草エキス、トラガント、単シロップ等が例示できる。
前記崩壊剤としては、デンプン、カンテン、カルメロースカルシウム、カルメロース、結晶セルロース等が例示できる。
前記湿潤剤としては、アラビアゴム、ポリビニルピロリドン、メチルセルロース、カルメロースナトリウム、ヒドロキシプロピルセルロース等が挙げられる。
【0031】
前記矯味剤としては、白糖、ハチミツ、サッカリンナトリウム、ハッカ、ユーカリ油、ケイヒ油等が例示できる。
前記着色剤としては、酸化鉄、β−カロチン、クロロフィル、水溶性食用タール色素等が例示できる。
前記香料としては、レモン油、オレンジ油、dl−又はl−メントール等が例示できる。
【0032】
鎮痛剤を吸入剤、注射剤、貼付剤、スプレー剤、軟膏等、非経口剤の製剤形態とする場合には、使用できる溶媒として、注射用蒸留水、無菌の非水性溶媒、懸濁剤等が例示できる。非水性溶媒又は懸濁剤の基剤としては、プロピレングリコール、ポリエチレングリコール、グリセリン、オリーブ油、コーン油、オレイン酸エチル等が好ましいものとして例示できる。
【0033】
さらに、鎮痛剤を貼付剤等、非経口剤の製剤形態とする場合には、有効成分等の各成分と基剤との混合物を、布、紙、プラスチックフィルム等に薄く塗布すれば良い。
【0034】
本発明の鎮痛剤には、本発明の効果を妨げない範囲内で、上記成分以外の薬学上許容される任意成分を、必要に応じて適宜配合しても良い。
前記任意成分としては、緩衝剤、防腐剤、抗酸化剤等が例示できる。
【0035】
本発明の鎮痛剤は、経口投与及び非経口投与のいずれでも投与できる。
本発明の鎮痛剤の投与量は、患者の年齢、症状等により適宜調節することが好ましい。経口投与の場合には、通常、成人一人一日あたり、化合物(1)又はその薬学上許容される塩の量として、1500〜6000mg/人であることが好ましい。非経口投与の場合には、通常、成人一人一日あたり、化合物(1)又はその薬学上許容される塩の量として、0.5〜2mg/人であることが好ましい。
本発明の鎮痛剤は、所定量を一日に一回又は複数回に分けて投与される。
【0036】
化合物(1)は、公知の方法に従って合成することで得られる。
また、化合物(1)の薬学上許容される塩は、例えば、化合物(1)と無機塩基及び/又は有機塩基とを、特定のモル比で作用させることで得られる。また、化合物(1)の特定の塩を塩交換することでも得られる。
化合物(1)又はその薬学上許容される塩は、公知の手法で取り出すことができる。すなわち、反応終了後に適宜必要に応じて、ろ過、洗浄、抽出、pH調整、脱水、濃縮等の後処理を行った後、結晶化やカラムクロマトグラフィー等により、生成物を取り出せば良い。また、取り出した生成物は、さらに結晶化やカラムクロマトグラフィー等を繰り返すことで精製を行っても良い。
化合物(1)又はその薬学上許容される塩としては、市販品を使用しても良い。
【実施例】
【0037】
以下、具体的に実施例を挙げ、本発明についてさらに詳しく説明する。ただし、本発明は以下に示す実施例に何ら限定されるものではない。
【0038】
<炎症性疼痛モデルにおける化合物(10−2)の鎮痛作用>
[実施例1]
(カプサイシン誘発疼痛に対する鎮痛作用)
完全フロインドアジュバント(Complete Freund‘s adjuvant:CFA)をラットの後足板に皮下注射し、二日間経過させることで、炎症ラットを作製した。そして、この炎症ラットに、さらに0.1%カプサイシンを10μL皮下注射することで疼痛を誘発したところ、足上げ行動が認められた。この状態のラットを炎症性疼痛モデルとして、以下の実験に使用した。
下記式(10−2)で表される、化合物(10−1)のジナトリウム塩(以下、化合物(10−2)と略記する)を、0.1%tween80(ポリオキシエチレンソルビタンモノオレアート)に懸濁させ、これを生理的食塩水に溶解させた溶液を調製し、この溶液を前記炎症性疼痛モデルに対して、カプサイシン投与の10分前に、12.5nmol/site、25nmol/site、50nmol/siteの量だけ、それぞれ後足板に皮下注射し、その後のラットの足上げ行動を観察した。ここでは、化合物(10−1)の薬学上許容される塩である化合物(10−2)を選択し、注射に供する溶液の溶媒種に応じて、有効成分の溶解性を向上させることで、投与の形態に適した製剤を選択した。
同様の観察を、前記溶液に代えて生理食塩水を皮下注射して行った(コントロール)。結果を図1に示す。図1のグラフにおける縦軸は、ラットの足上げ行動の回数を、横軸はカプサイシン投与後の時間(分)を示す。
【0039】
【化6】
【0040】
図1から明らかなように、化合物(10−2)を投与した場合には、濃度依存的に足上げ行動の抑制が認められ、そのIC50値は、39.7nmol/siteであった。化合物(10−2)は、特に25nmol/site、50nmol/siteの投与量で、有意に足上げ行動の抑制が認められた。
【0041】
ラットでは、CFAの投与により長期の炎症反応が惹起され、C−ファイバー上にカプサイシンのレセプター(TRPV1)が発現する。そして、カプサイシンの投与により疼痛が起きる。化合物(10−2)は、このようなカプサイシンを投与されたラットに対して、鎮痛作用を発現した。
【0042】
[実施例2]
(ホルマリン誘発疼痛に対する鎮痛作用)
1.85%ホルマリンを50μLラットの後足板に皮下注射して疼痛行動を誘発させ、このラットを疼痛モデルとして、以下の実験に使用した。このラットには、ホルマリンによって誘発された疼痛に伴い、足上げ行動及び足舐め行動が認められた。
この疼痛モデルについて、ホルマリン投与の45分前に、化合物(10−2)を25mg/kg、50mg/kg、100mg/kgの用量でそれぞれ経口投与し、ラットの足上げ行動及び足舐め行動を観察した。同様の観察を、化合物(10−2)に代えて生理食塩水をホルマリン投与の30分前に経口投与して行った。結果を図2及び3に示す。図2(a)はホルマリン投与後初期(0〜10分)における足上げ行動の回数を、(b)はホルマリン投与後後期(10〜80分)における足上げ行動の回数をそれぞれ示す。また、図3(a)はホルマリン投与後初期(0〜10分)における足舐め行動の回数を、(b)はホルマリン投与後後期(10〜80分)における足舐め行動の回数をそれぞれ示す。
【0043】
その結果、図2(a)及び図3(a)から明らかなように、化合物(10−2)を投与した場合には、ホルマリン投与後初期(0〜10分)では、足上げ行動及び足舐め行動の抑制は認められなかった。しかし、ホルマリン投与後後期(10〜80分)では、足上げ行動及び足舐め行動の抑制が認められた。化合物(10−2)は、特に25mg/kg、50mg/kgの投与量で有意に足上げ行動の抑制が認められ、足舐め行動の抑制には、用量依存的な抑制効果が認められた。
【0044】
ラットでは、ホルマリンの投与により、投与後一時間以内に二相性(第一相、第二相)の疼痛反応が起きることが知られている。化合物(10−2)の投与により、第一相の疼痛反応に対しては鎮痛作用がなく、第二相の疼痛反応に対しては鎮痛作用があったと考えられる。
【0045】
<神経因性疼痛モデルにおける化合物(10−2)の鎮痛作用>
[実施例3]
ラットの左坐骨神経をゆるく結紮し、五日後に熱刺激により、右足には疼痛反応が起こらず、左足に疼痛反応が起こることを確認して、このラットを神経因性疼痛モデルとして、以下の実験に使用した。なお、熱刺激は、輻射熱を加えることで行った。
左坐骨神経結紮の施術後七日目に、熱刺激により疼痛反応が起こることを確認した。そして同日に、化合物(10−2)を50mg/kgの用量で経口投与し、投与後45分(0.75時間)、3時間、6時間の段階で、熱刺激による疼痛反応の程度について観察した。同様の観察を、化合物(10−2)に代えて生理食塩水を経口投与して行った。結果を図4に示す。図4(a)は熱刺激を加えた時に右足を熱源から離間させるまでの時間を、(b)は熱刺激を加えた時に左足を熱源から離間させるまでの時間をそれぞれ示す。
【0046】
熱刺激をラットの左足に加えた場合には、右足の場合よりも、刺激に反応する閾値が各時間で低下した。そして、左足の場合、投与後3時間の段階で、化合物(10−2)を投与したものは、生理食塩水を投与したものよりも閾値が有意に上昇していることを確認した。
【0047】
化合物(10−2)は、分子量が800以上であり、経口投与によって、活性成分が中枢神経系にまで到達することは考えにくい。実際に、化合物(10−2)は、投与後三時間の血清からは検出されたが、脳ホモジネートからは検出されなかった。このように、化合物(10−2)は、活性成分の中枢神経系への移行性が低い化合物であることが示唆された。
【産業上の利用可能性】
【0048】
本発明は、医療分野で痛みの緩和等に利用可能である。
【技術分野】
【0001】
本発明は、新規の鎮痛剤に関する。
【背景技術】
【0002】
痛みの緩和は、日常生活での不快感の改善から、発病時の病状の軽減、そして終末期医療での深刻な苦痛の軽減に至るまで、極めて広範囲の目的に沿って行われている。
痛みには、炎症性のものと神経因性のものがあると考えられ、その病態に応じて極めて多種類の鎮痛剤がこれまでに開発されており、比較的良く知られているものだけでも、アセトアミノフェン、アセチルサリチル酸、イブプロフェン、エテンザミド、モルヒネ等、枚挙に暇がない。
このような中、目的に即した鎮痛剤を使用することで、副作用を抑えつつ、十分な鎮痛効果を得ようとするのはごく自然なことであり、新規な鎮痛剤の開発が強く望まれている。
【0003】
ところで、甘草の主成分であるグリチルリチンは、抗炎症作用を有することが知られている(非特許文献1参照)。そして、例えば、グリチルリチンのアグリコンであるグリチルレチン酸の誘導体に相当する、オレアン−11,13(18)−ジエン−3β,30−ジオールの、二つの水酸基がフタル酸エステル化された化合物は、抗潰瘍剤の有効成分となることが開示されている(特許文献1参照)。しかし、これに対して、グリチルリチンやその類縁体については、それ自体が鎮痛剤として十分に有効であるか否か、これまでに十分な検討がなされてこなかったのが実情である。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特開昭60−260540号公報
【非特許文献】
【0005】
【非特許文献1】Inoue et al.,1996,Jpn.J.Pharmacol.71,281−289
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明は上記事情に鑑みてなされたものであり、グリチルリチンの類縁体を有効成分とする新規な鎮痛剤を提供することを課題とする。
【課題を解決するための手段】
【0007】
本発明者らは、上記課題を解決するために鋭意研究を行った結果、グリチルリチンのアグリコンであるグリチルレチン酸の特定の誘導体が、疼痛の抑制効果を示すことを見出し、本発明を完成するに至った。
【0008】
すなわち、上記課題を解決するため、
本発明は、下記式(1)で表される化合物又はその薬学上許容される塩を有効成分とする鎮痛剤を提供する。
【0009】
【化1】
【0010】
本発明の鎮痛剤においては、下記式(10−1)で表される化合物又はその薬学上許容される塩を有効成分とすることが好ましい。
【0011】
【化2】
【発明の効果】
【0012】
本発明によれば、グリチルリチンの類縁体を有効成分とする新規な鎮痛剤を提供できる。
【図面の簡単な説明】
【0013】
【図1】実施例1におけるラットのカプサイシン投与後の足上げ行動の回数を示すグラフである。
【図2】実施例2におけるラットの足上げ行動の回数を示すグラフであり、(a)はホルマリン投与後初期(0〜10分)における回数を、(b)はホルマリン投与後後期(10〜80分)における回数をそれぞれ示す。
【図3】実施例2におけるラットの足舐め行動の回数を示すグラフであり、(a)はホルマリン投与後初期(0〜10分)における回数を、(b)はホルマリン投与後後期(10〜80分)における回数をそれぞれ示す。
【図4】実施例3において、左坐骨神経をゆるく結紮したラットに熱刺激を加えた時の、ラットが足を熱源から離間させるまでの時間を示すグラフであり、(a)は右足を、(b)は左足を、それぞれ熱源から離間させるまでの時間を示す。
【発明を実施するための形態】
【0014】
本発明の鎮痛剤は、下記式(1)で表される化合物(以下、化合物(1)と略記する)又はその薬学上許容される塩を有効成分とする。
【0015】
【化3】
【0016】
化合物(1)又はその薬学上許容される塩を有効成分とする鎮痛剤は、炎症性の痛み及び神経因性の痛みのいずれにも抑制効果を示す。
【0017】
化合物(1)は、下記式(30−1)で表されるグリチルレチン酸の誘導体であり、これまでに知られている鎮痛剤とは異なる構造を有し、主要骨格も全く異なる。
【0018】
【化4】
【0019】
化合物(1)としては、下記式(10−1)で表される化合物(以下、化合物(10−1)と略記する)が好ましい。すなわち、本発明の鎮痛剤は、化合物(10−1)又はその薬学上許容される塩を有効成分とするものが好ましい。
【0020】
【化5】
【0021】
化合物(1)の薬学上許容される塩の好ましいものとして、化合物(1)モノアンモニウム塩、化合物(1)ジアンモニウム塩等のアンモニウム塩;化合物(1)モノナトリウム塩、化合物(1)ジナトリウム塩、化合物(1)モノカリウム塩、化合物(1)ジカリウム塩等のアルカリ金属塩;化合物(1)カルシウム塩;化合物(1)マグネシウム塩;化合物(1)アルミニウム塩;化合物(1)モノコリン塩、化合物(1)ジコリン塩等の有機アミン塩等が例示できる。
【0022】
化合物(1)の薬学上許容される塩が二塩の場合、塩を構成する二つのカチオン種は、上記のように同じでも良いが、異なっていても良く、異なる場合には、アンモニウムイオン、ナトリウムイオン、カリウムイオン、カルシウムイオン、マグネシウムイオン、アルミニウムイオン及びコリンからなる群から選択される異なる二種のカチオンが例示できる。
【0023】
化合物(1)の薬学上許容される塩としては、化合物(1)アルカリ金属塩が好ましく、化合物(1)ナトリウム塩がより好ましい。
【0024】
化合物(1)又はその薬学上許容される塩は、一種を単独で使用しても良いし、二種以上を併用しても良い。二種以上を併用する場合には、その組み合わせ及び比率は、目的に応じて適宜選択すれば良い。
【0025】
本発明の鎮痛剤の製剤形態は特に限定されず、目的に応じて錠剤、散剤、顆粒剤、カプセル剤、細粒剤、液剤(水薬等)等の経口剤;吸入剤、座剤、注射剤、貼付剤、スプレー剤、軟膏等の非経口剤等から適宜選択すれば良い。これら製剤形態の鎮痛剤は、いずれも公知の方法で製造できる。
【0026】
鎮痛剤を経口剤等の製剤形態とする場合には、これら製剤の製造で通常使用される各種添加剤を配合しても良い。前記添加剤としては、賦形剤、滑沢剤、可塑剤、界面活性剤、結合剤、崩壊剤、湿潤剤、安定剤、矯味剤、着色剤、香料等が例示できる。
前記添加剤は、一種を単独で使用しても良いし、二種以上を併用しても良い。二種以上を併用する場合には、その組み合わせ及び比率は、目的に応じて適宜選択すれば良い。
【0027】
前記賦形剤としては、乳糖、ブドウ糖、D−マンニトール、果糖、デキストリン、デンプン、食塩、炭酸水素ナトリウム、炭酸カルシウム、アルギン酸ナトリウム、エチルセルロース、ナトリウムカルボキシメチルセルロース、ヒドロキシプロピルセルロース、無水ケイ酸、カオリン等が例示できる。
【0028】
前記滑沢剤としては、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸、タルク、トウモロコシデンプン、マクロゴール等が例示できる。
【0029】
前記可塑剤としては、ポリエチレングリコール、プロピレングリコール、グリセリン類、トリアセチン、中鎖脂肪酸トリグリセリド、アセチルグリセリン脂肪酸エステル、クエン酸トリエチル等が例示できる。
【0030】
前記結合剤としては、ゼラチン、アラビアゴム、セルロースエステル、ポリビニルピロリドン、水飴、甘草エキス、トラガント、単シロップ等が例示できる。
前記崩壊剤としては、デンプン、カンテン、カルメロースカルシウム、カルメロース、結晶セルロース等が例示できる。
前記湿潤剤としては、アラビアゴム、ポリビニルピロリドン、メチルセルロース、カルメロースナトリウム、ヒドロキシプロピルセルロース等が挙げられる。
【0031】
前記矯味剤としては、白糖、ハチミツ、サッカリンナトリウム、ハッカ、ユーカリ油、ケイヒ油等が例示できる。
前記着色剤としては、酸化鉄、β−カロチン、クロロフィル、水溶性食用タール色素等が例示できる。
前記香料としては、レモン油、オレンジ油、dl−又はl−メントール等が例示できる。
【0032】
鎮痛剤を吸入剤、注射剤、貼付剤、スプレー剤、軟膏等、非経口剤の製剤形態とする場合には、使用できる溶媒として、注射用蒸留水、無菌の非水性溶媒、懸濁剤等が例示できる。非水性溶媒又は懸濁剤の基剤としては、プロピレングリコール、ポリエチレングリコール、グリセリン、オリーブ油、コーン油、オレイン酸エチル等が好ましいものとして例示できる。
【0033】
さらに、鎮痛剤を貼付剤等、非経口剤の製剤形態とする場合には、有効成分等の各成分と基剤との混合物を、布、紙、プラスチックフィルム等に薄く塗布すれば良い。
【0034】
本発明の鎮痛剤には、本発明の効果を妨げない範囲内で、上記成分以外の薬学上許容される任意成分を、必要に応じて適宜配合しても良い。
前記任意成分としては、緩衝剤、防腐剤、抗酸化剤等が例示できる。
【0035】
本発明の鎮痛剤は、経口投与及び非経口投与のいずれでも投与できる。
本発明の鎮痛剤の投与量は、患者の年齢、症状等により適宜調節することが好ましい。経口投与の場合には、通常、成人一人一日あたり、化合物(1)又はその薬学上許容される塩の量として、1500〜6000mg/人であることが好ましい。非経口投与の場合には、通常、成人一人一日あたり、化合物(1)又はその薬学上許容される塩の量として、0.5〜2mg/人であることが好ましい。
本発明の鎮痛剤は、所定量を一日に一回又は複数回に分けて投与される。
【0036】
化合物(1)は、公知の方法に従って合成することで得られる。
また、化合物(1)の薬学上許容される塩は、例えば、化合物(1)と無機塩基及び/又は有機塩基とを、特定のモル比で作用させることで得られる。また、化合物(1)の特定の塩を塩交換することでも得られる。
化合物(1)又はその薬学上許容される塩は、公知の手法で取り出すことができる。すなわち、反応終了後に適宜必要に応じて、ろ過、洗浄、抽出、pH調整、脱水、濃縮等の後処理を行った後、結晶化やカラムクロマトグラフィー等により、生成物を取り出せば良い。また、取り出した生成物は、さらに結晶化やカラムクロマトグラフィー等を繰り返すことで精製を行っても良い。
化合物(1)又はその薬学上許容される塩としては、市販品を使用しても良い。
【実施例】
【0037】
以下、具体的に実施例を挙げ、本発明についてさらに詳しく説明する。ただし、本発明は以下に示す実施例に何ら限定されるものではない。
【0038】
<炎症性疼痛モデルにおける化合物(10−2)の鎮痛作用>
[実施例1]
(カプサイシン誘発疼痛に対する鎮痛作用)
完全フロインドアジュバント(Complete Freund‘s adjuvant:CFA)をラットの後足板に皮下注射し、二日間経過させることで、炎症ラットを作製した。そして、この炎症ラットに、さらに0.1%カプサイシンを10μL皮下注射することで疼痛を誘発したところ、足上げ行動が認められた。この状態のラットを炎症性疼痛モデルとして、以下の実験に使用した。
下記式(10−2)で表される、化合物(10−1)のジナトリウム塩(以下、化合物(10−2)と略記する)を、0.1%tween80(ポリオキシエチレンソルビタンモノオレアート)に懸濁させ、これを生理的食塩水に溶解させた溶液を調製し、この溶液を前記炎症性疼痛モデルに対して、カプサイシン投与の10分前に、12.5nmol/site、25nmol/site、50nmol/siteの量だけ、それぞれ後足板に皮下注射し、その後のラットの足上げ行動を観察した。ここでは、化合物(10−1)の薬学上許容される塩である化合物(10−2)を選択し、注射に供する溶液の溶媒種に応じて、有効成分の溶解性を向上させることで、投与の形態に適した製剤を選択した。
同様の観察を、前記溶液に代えて生理食塩水を皮下注射して行った(コントロール)。結果を図1に示す。図1のグラフにおける縦軸は、ラットの足上げ行動の回数を、横軸はカプサイシン投与後の時間(分)を示す。
【0039】
【化6】
【0040】
図1から明らかなように、化合物(10−2)を投与した場合には、濃度依存的に足上げ行動の抑制が認められ、そのIC50値は、39.7nmol/siteであった。化合物(10−2)は、特に25nmol/site、50nmol/siteの投与量で、有意に足上げ行動の抑制が認められた。
【0041】
ラットでは、CFAの投与により長期の炎症反応が惹起され、C−ファイバー上にカプサイシンのレセプター(TRPV1)が発現する。そして、カプサイシンの投与により疼痛が起きる。化合物(10−2)は、このようなカプサイシンを投与されたラットに対して、鎮痛作用を発現した。
【0042】
[実施例2]
(ホルマリン誘発疼痛に対する鎮痛作用)
1.85%ホルマリンを50μLラットの後足板に皮下注射して疼痛行動を誘発させ、このラットを疼痛モデルとして、以下の実験に使用した。このラットには、ホルマリンによって誘発された疼痛に伴い、足上げ行動及び足舐め行動が認められた。
この疼痛モデルについて、ホルマリン投与の45分前に、化合物(10−2)を25mg/kg、50mg/kg、100mg/kgの用量でそれぞれ経口投与し、ラットの足上げ行動及び足舐め行動を観察した。同様の観察を、化合物(10−2)に代えて生理食塩水をホルマリン投与の30分前に経口投与して行った。結果を図2及び3に示す。図2(a)はホルマリン投与後初期(0〜10分)における足上げ行動の回数を、(b)はホルマリン投与後後期(10〜80分)における足上げ行動の回数をそれぞれ示す。また、図3(a)はホルマリン投与後初期(0〜10分)における足舐め行動の回数を、(b)はホルマリン投与後後期(10〜80分)における足舐め行動の回数をそれぞれ示す。
【0043】
その結果、図2(a)及び図3(a)から明らかなように、化合物(10−2)を投与した場合には、ホルマリン投与後初期(0〜10分)では、足上げ行動及び足舐め行動の抑制は認められなかった。しかし、ホルマリン投与後後期(10〜80分)では、足上げ行動及び足舐め行動の抑制が認められた。化合物(10−2)は、特に25mg/kg、50mg/kgの投与量で有意に足上げ行動の抑制が認められ、足舐め行動の抑制には、用量依存的な抑制効果が認められた。
【0044】
ラットでは、ホルマリンの投与により、投与後一時間以内に二相性(第一相、第二相)の疼痛反応が起きることが知られている。化合物(10−2)の投与により、第一相の疼痛反応に対しては鎮痛作用がなく、第二相の疼痛反応に対しては鎮痛作用があったと考えられる。
【0045】
<神経因性疼痛モデルにおける化合物(10−2)の鎮痛作用>
[実施例3]
ラットの左坐骨神経をゆるく結紮し、五日後に熱刺激により、右足には疼痛反応が起こらず、左足に疼痛反応が起こることを確認して、このラットを神経因性疼痛モデルとして、以下の実験に使用した。なお、熱刺激は、輻射熱を加えることで行った。
左坐骨神経結紮の施術後七日目に、熱刺激により疼痛反応が起こることを確認した。そして同日に、化合物(10−2)を50mg/kgの用量で経口投与し、投与後45分(0.75時間)、3時間、6時間の段階で、熱刺激による疼痛反応の程度について観察した。同様の観察を、化合物(10−2)に代えて生理食塩水を経口投与して行った。結果を図4に示す。図4(a)は熱刺激を加えた時に右足を熱源から離間させるまでの時間を、(b)は熱刺激を加えた時に左足を熱源から離間させるまでの時間をそれぞれ示す。
【0046】
熱刺激をラットの左足に加えた場合には、右足の場合よりも、刺激に反応する閾値が各時間で低下した。そして、左足の場合、投与後3時間の段階で、化合物(10−2)を投与したものは、生理食塩水を投与したものよりも閾値が有意に上昇していることを確認した。
【0047】
化合物(10−2)は、分子量が800以上であり、経口投与によって、活性成分が中枢神経系にまで到達することは考えにくい。実際に、化合物(10−2)は、投与後三時間の血清からは検出されたが、脳ホモジネートからは検出されなかった。このように、化合物(10−2)は、活性成分の中枢神経系への移行性が低い化合物であることが示唆された。
【産業上の利用可能性】
【0048】
本発明は、医療分野で痛みの緩和等に利用可能である。
【特許請求の範囲】
【請求項1】
下記式(1)で表される化合物又はその薬学上許容される塩を有効成分とする鎮痛剤。
【化1】
【請求項2】
下記式(10−1)で表される化合物又はその薬学上許容される塩を有効成分とする請求項1に記載の鎮痛剤。
【化2】
【請求項1】
下記式(1)で表される化合物又はその薬学上許容される塩を有効成分とする鎮痛剤。
【化1】
【請求項2】
下記式(10−1)で表される化合物又はその薬学上許容される塩を有効成分とする請求項1に記載の鎮痛剤。
【化2】
【図1】

【図2】
【図3】
【図4】
【図2】
【図3】
【図4】
【公開番号】特開2012−250936(P2012−250936A)
【公開日】平成24年12月20日(2012.12.20)
【国際特許分類】
【出願番号】特願2011−125089(P2011−125089)
【出願日】平成23年6月3日(2011.6.3)
【出願人】(000170358)株式会社ミノファーゲン製薬 (16)
【出願人】(500557048)学校法人日本医科大学 (20)
【Fターム(参考)】
【公開日】平成24年12月20日(2012.12.20)
【国際特許分類】
【出願日】平成23年6月3日(2011.6.3)
【出願人】(000170358)株式会社ミノファーゲン製薬 (16)
【出願人】(500557048)学校法人日本医科大学 (20)
【Fターム(参考)】
[ Back to top ]