
鎮痛薬の改善された製造方法
【課題】改善された鎮痛作用のあるオキシモルホン塩酸塩の提供。
【解決手段】14−ヒドロキシモルヒノンの含量が10ppm未満であるオキシモルホン塩酸塩を含む医薬製剤。このオキシモルホン塩酸塩は、原料のオキシモルホン塩酸塩を、ガス状水素を用いて、規定の酸性度、溶媒系及び温度条件下還元することにより製造する。水和により、特定のオキシモルホン塩酸塩多形体を得ることができる。
【解決手段】14−ヒドロキシモルヒノンの含量が10ppm未満であるオキシモルホン塩酸塩を含む医薬製剤。このオキシモルホン塩酸塩は、原料のオキシモルホン塩酸塩を、ガス状水素を用いて、規定の酸性度、溶媒系及び温度条件下還元することにより製造する。水和により、特定のオキシモルホン塩酸塩多形体を得ることができる。
【発明の詳細な説明】
【発明の分野】
【0001】
本発明は、鎮痛薬、より詳細にはアヘンオキシモルホンを塩酸塩として製造する改善された方法に関する。
【0002】
一般的に塩酸塩の形態で投与されるオキシモルホンは、中程度〜激しい痛みを軽減するための強力な半合成アヘン系鎮痛薬であり、1959年以来認可されているものである。オキシモルホンは、注射液、坐剤、錠剤又は徐放錠剤として投与されることができる。高純度のオキシモルホン及びその合成方法を開発することが望ましい。
【0003】
ケシから単離した化合物又はそれから得た化合物から、例えば、モルヒネ、テバイン又はオキシコドンを原料として、オキシモルホンを合成するいくつかの方法が知られている。α,β−不飽和ケトンによる汚染度が低いオキシモルホンを形成できる方法が必要とされている。本発明によれば、改善されたオキシモルホン生成物及びこのようなオキシモルホンの製造方法が提供される。
【0004】
米国特許第7,129,248号は、14−ヒドロキシコデイノン含量が100ppm超であるオキシコドンを水素添加することにより、14−ヒドロキシコデイノン含量が25ppm未満であるオキシコドン塩酸塩を製造する方法を開示している。米国特許第7,129,248号に教示されているオキシコドンへの合成経路では、テバインを原料とし、中間生成物として14−ヒドロキシコデイノンを製造し、テバインの過酸化から得られる副生成物として8,14−ジヒドロキシ−7,8−ジヒドロコデイノンが生成する。オキシコドン遊離塩基のその塩酸塩への転化中、副生成物が酸触媒下脱水し、14−ヒドロキシコデイノンに転化することがある。したがって、最終的なオキシコドン塩酸塩は、未反応14−ヒドロキシコデイノン、及び副生成物である8,14−ジヒドロキシ−7,8−ジヒドロコデイノンから得られた14−ヒドロキシコデイノンを含んでいる。水素添加工程により、14−ヒドロキシコデイノン含量が少なくとも100ppmから25ppm未満に減少するとしている。
【0005】
本発明によれば、α,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩生成物が提供される。また、本発明によれば、オキシモルホン塩酸塩を精製してα,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩生成物を得る方法が提供される。この方法は、原料のオキシモルホン塩酸塩を、強酸水及びアルコール溶媒中60〜70℃の温度範囲で、水素ガスにより還元することを含む。還元時間は少なくとも20時間が好適である。しかしながら、別の態様によれば、還元は1〜20時間実施する。
【図面の簡単な説明】
【0006】
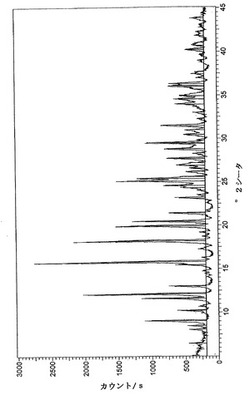
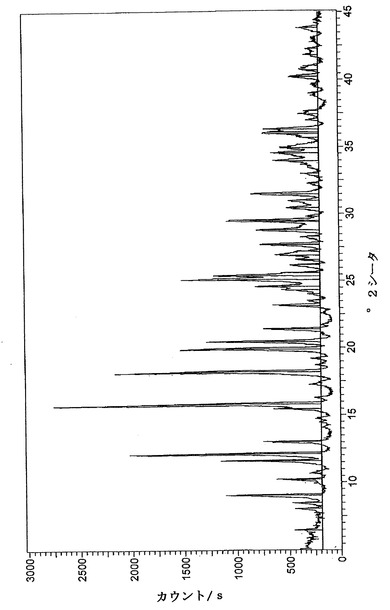
【図1】実施例3.2Dにより得られた水和オキシモルホン塩酸塩生成物についての粉末X線回折パターンである。
【発明を実施するための形態】
【0007】
好ましくは、溶媒は、エタノール/水である。しかしながら、イソプロパノール及びn−プロパノール等の他の水混和性アルコールを使用してもよい。反応媒体は、少なくとも2当量の塩酸を含有させることにより強酸性とすることが好ましい。pHは、1未満が望ましい。
【0008】
反応温度は、約65℃に維持するのが最も好ましい。水素は、反応容器に2.41バールの圧力で供給するのが都合がよい。
【0009】
本発明の方法は、原料であるオキシモルホン塩酸塩の非常に高含量(0.3〜0.5%程度又は3,000〜5,000ppm程度)のα,β−不飽和ケトンを、10ppm未満、そして多くの場合、検出不能なレベル(HPLCにより)まで減少することができた。
【0010】
原料であるオキシモルホン塩酸塩は、単離物質でもよいし、非単離物質でもよい。オキシモルホン塩酸塩は、オキシモルホン遊離塩基を塩酸及びアルコール/水反応媒体の存在下で加熱することにより得られたものが望ましい。好適な温度は、60〜70℃である。反応媒体は本発明の方法の還元に理想的であるので、一般的にオキシモルホン塩酸塩を単離する必要がないことが明らかである。しかしながら、原料であるオキシモルホン塩酸塩は、反応媒体から単離してもよいし、別の源から得てもよい。
【0011】
オキシモルホン遊離塩基自体は、14−ヒドロキシモルヒノンの還元により調製することが好ましい。この還元は、一段又は二段プロセスで実施できる。好ましくは、この還元は、ガス状水素及び炭素に担持されたパラジウム触媒を用いて酢酸中で実施する。好ましい温度は、30℃の程度である。塩基は、アンモニア水(NH4OH)を添加することにより析出する。
【0012】
この還元は、メタノール、フロラシル(Florasil)及びn−プロパノールにジクロロメタンを加えた反応媒体の存在下でおこなってもよい。
【0013】
14−ヒドロキシモルヒノン自体は、過酸化水素を用いて、蟻酸の存在下、オリパビンを水酸化することにより最も好適に調製される。
【0014】
オリパビンは、ケシがら(ケシ滓、ケシ茎根)から抽出できる公知化合物である。テバイン高収量株であるタスマニアで発育した株は、通常よりも高収量のオリパビンも産生する。
【0015】
本発明の方法は、非常に柔軟性があり、数多くの反応工程を中間生成物を単離することなく実施することができる。しかも、この場合、オリパビンからの全収率(50%程度)を高い状態に保持するとともに、純度を著しく高く保持することができる。好条件下では、α,β−不飽和ケトンは、HPLC等の通常の手段によってはその存在が検出できない程度である。しかしながら、当業者は、汚染レベルを10ppm未満に容易にできる。本発明の方法は、キログラムスケールでうまく実施することができた。
【0016】
α,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩は、例えば、α,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩を通常の賦形剤、すなわち、薬学的に許容される有機又は無機担体物質と混ぜて混合剤とすることにより、医薬剤形に含有させることができる。経口製剤の場合、剤形により、活性成分が除放性とすることができる。好適な薬学的に許容される担体には、アルコール、アラビア・ゴム、植物油、ベンジルアルコール、ポリエチレングリコール、ゲル、炭水化物、例えば、ラクトース、アミロース又はでんぷん、ステアリン酸マグネシウム、タルク、ケイ酸、粘性パラフィン、香油、脂肪酸モノグリセリド及びジグリセリド、ペンタエリスリトール脂肪酸エステル、ヒドロキシーメチルセルロース、ポリビニルピロリドンなどがあるが、これらには限定されない。製剤は、殺菌し、必要に応じて補助剤、例えば、滑剤、崩壊剤、防腐剤、安定剤、湿潤剤、乳化剤、浸透圧緩衝液に影響を与える塩、着色物質、香味物質及び/又は芳香物質等と混合できる。経口用組成物は、当該技術分野において公知の方法により調製でき、このような組成物は、錠剤の製造に好適である不活性、非毒性の薬学的に許容される賦形剤からなる群から選択される一種以上の薬剤を含有していてもよい。このような賦形剤には、例えば、不活性希釈剤、例えば、ラクトース;造粒及び崩壊剤、例えば、コーンスターチ;結合剤、例えば、でんぷん;並びに滑剤、例えば、ステアリン酸マグネシウムなどがある。錠剤は、被覆されていないものでもよく、又は外見よくしたり又は有効成分の放出を遅くする公知の方法により被覆されていてもよい。また、経口用製剤は、硬質ゼラチンカプセル剤として提供してもよい。この場合、有効成分は不活性希釈剤と混合する。本発明の経口用剤形は、錠剤(除放性及び/又は即時放出性)、トローチ、ロゼンジ、散剤又は顆粒剤、硬質又は軟質カプセル剤、微粒子(例えば、マイクロカプセル剤、微小球等)、バッカル錠、液剤、懸濁剤等の形態でよい。
【0017】
本発明のある実施態様によれば、ヒト患者に本明細書に記載の剤形を投与することにより痛みを治療する方法が提供される。
【0018】
剤形が経口用であるとき、本発明の剤形は、α,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩約1mg〜約40mgを含有する。特に好ましい投与量は、約5mg、約10mg、約20mg又は約40mgであるが、他の投与量を使用してもよい。また、α,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩は、好適な薬学的に許容される賦形剤を配合して、α,β−不飽和ケトン含量が10ppm未満の徐放性製剤としてもよい。このような製剤は、米国特許第2003/129230A1号、米国特許第2003/129234A1号及び米国特許第2003/157167A1号に準じて調製できる。
【0019】
α,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩は、当業者に知られている好適な錠剤、被覆錠剤又は多粒子製剤における徐放性経口製剤として製剤化でてきる。
【0020】
徐放性剤形は、徐放性物質を、マトリックスにそのオキシモルホン塩とともに含有させて含んでいてもよい。
【0021】
徐放性剤形は、必要に応じて、α,β−不飽和ケトン含量10ppm未満のオキシモルホン塩酸塩を含有する粒子を含んでいてもよい。ある態様によれば、粒子は、直径が約0.1mm〜約2.5mm、好ましくは約0.5mm〜約2mmである。好ましくは、粒子は、活性成分を水媒体中で徐放できる物質の被膜が塗布されているものである。被膜状コートは、他の記載の特性との組み合わせで所望の放出特性が得られるように選択する。本発明の徐放性コーティング製剤は、好ましくは、平滑且つ外観のよい強度のある連続被膜を製造することができ、顔量及び他のコーティング添加剤を担持することができ、非毒性、不活性且つ不粘着性のものである。
【0022】
塗布ビーズ
本発明のある実施態様によれば、nu pariel18/20ビーズなどの不活性医薬ビーズに疎水性物質を塗布する。その後、複数の得られた固体状徐放性ビーズを、摂取され、環境流体、例えば、胃液又は溶解媒体により接触されたときに、有効な徐放投与量となるのに十分な量で、ゼラチンカプセル剤に入れることができる。
【0023】
本発明の徐放性ビーズ製剤は、例えば、摂取され、胃液及び次に腸液に暴露されたときに本発明の活性成分をゆっくりと放出する。本発明の製剤の徐放プロフィールは、例えば、疎水性物質で上塗りする量を変更することにより、可塑剤を疎水性物質に添加する方法を変更することにより、疎水性物質に対する可塑剤の量を変更することにより、追加の成分又は賦形剤を含有させることにより、又は製造方法を変更することにより、変更することができる。最終生成物の溶解プロフィールも、例えば、遅延コーティングの厚さを増加するか又は減少させることにより変更することができる。
【0024】
本発明の薬剤を塗布した球状物又はビーズは、例えば、薬剤を水に溶解し、その後溶液を基材、例えば、nu pariel18/20ビーズ上にウスター(Wuster)インサートを用いて噴霧することにより調製される。必要に応じて、ビーズに塗布する前に、追加の成分も添加して、ビーズへの活性成分の結合の促進及び/又は溶液の着色などをおこなう。例えば、着色剤を添加してあるか又は添加していないヒドロキシプロピルメチルセルロース等を含む生成物(例えば、Colorcon社から市販されているOpadry(商品名))を、溶液に添加し、その溶液を混合(例えば、約1時間)してから、その溶液をビーズ上に適用してもよい。得られた塗布基材(この場合ビーズ)の上に、次に必要に応じてバリア剤を塗布して、活性成分を疎水性徐放性コーティングから分離してもよい。好適なバリア剤の一例として、ヒドロキシプロピルメチルセルロースを含むものがある。しかしながら、当該技術分野において公知のいずれの膜形成剤を用いてもよい。バリア剤は、最終製品の溶解速度に影響しないことが好ましい。
【0025】
次にビーズの上に、疎水性物質の水性分散液を塗布してもよい。疎水性物質の水性分散液は、好ましくはさらに有効量の可塑剤、例えば、クエン酸トリエチルを含む。予め調製したエチルセルロースの水性分散液、例えば、Aquacoat(商品名)又はSurelease(商品名)を使用してもよい。Surelease(商品名)を使用する場合、可塑剤を別途添加する必要はない。別法として、予め調製したアクリル系ポリマーの水性分散液、例えば、Eudragit(商品名)を使用することができる。
【0026】
好ましくは、本発明のコーティング溶液は、膜形成剤、可塑剤及び溶媒系(すなわち、水)の他に、着色剤を含有して、良好な外観及び製品の識別性を付与する。疎水性物質の水性分散液を加える代わりに、又は疎水性物質の水性分散液に加えて、着色剤を治療活性剤の溶液に添加してもよい。例えば、着色剤を、Aquacoat(商品名)に、アルコール又はプロピレングリコール系着色剤分散液、粉砕アルミニウムレーキ及び二酸化チタン等の乳白剤を用いて、着色剤を剪断力により水溶性ポリマー溶液に添加し、その後低剪断力を用いて可塑化したAquacoat(商品名)に添加してもよい。別法として、本発明の製剤を着色するいずれかの好適な方法を用いてもよい。アクリル系ポリマーの水性分散液を使用するとき、製剤を着色するのに好適な成分としては、二酸化チタン、着色顔料、例えば、酸化鉄顔料などがある。しかしながら、顔料を含有させることにより、コーティングの効果を顕著に妨害することがある。
【0027】
可塑化した疎水性物質は、当該技術分野おいて公知のいずれかの好適な噴霧装置を用いて噴霧することにより、薬剤を含む基材上に適用してもよい。好ましい方法では、Wurster流動床装置を使用する。Wurster流動床装置では、下部から注入された空気ジェットにより、コア物質が流動化し、アクリル系ポリマーコーティングをコア物質上に噴霧しながら乾燥をおこなう。塗布された基材が水溶液、例えば、胃液にさらされたときに、薬剤が所定の徐放性を示すのに十分な量の疎水性物質を適用することができる。疎水性物質を塗布した後、必要に応じて、さらにOpadry(商品名)等の膜形成剤をビーズにオーバーコーティングする。このオーバーコートは、ビーズの凝集があるとしてもその凝集を実質的に減少させるために設けられる。
【0028】
本発明の除放性製剤から薬剤を放出させることは、一種以上の放出調整剤の添加、又はコーティングを介して一つ以上の経路を設けることによりさらに所望の速度に変更、すなわち、調整できる。疎水性物質の水溶性物質に対する比は、他の因子のうち、必要放出速度と選択物質の溶解度特性により求める。
【0029】
孔形成剤として機能する放出調整剤は、有機物質でも、無機物質でもよく、使用環境において、コーティングから溶解、抽出又は侵出することができる物質などがある。孔形成剤は、ヒドロキシプロピルメチルセルロース等の一種以上の親水性物質を含むことができる。
【0030】
また、本発明の除放性コーティングは、でんぷん及びゴム等の浸食促進剤を含むことができる。
【0031】
また、本発明の除放性コーティングは、使用環境において微細孔性薄層を形成するのに有効な物質、例えば、ポリマー鎖にカーボネート基が繰り返している炭酸の線状ポリエステルからなるポリカーボネートを含むことができる。
【0032】
また、放出調整剤は、半浸透性ポリマーも含んでいてもよい。
【0033】
ある好ましい実施態様によれば、放出調整剤は、ヒドロキシプロピルメチルセルロース、ラクトース、ステアリン酸金属塩及びこれらの混合物から選択される。
【0034】
本発明の除放性コーティングは、少なくとも一つの経路、オリフィス等を備えた出口手段を備えていてもよい。この経路は、米国特許第3,845,770号、米国特許第3,916,899号、米国特許第4,063,064号及び米国特許第4,088,864号に開示てれているような方法により形成してもよい。
【0035】
マトリックス製剤
本発明の他の実施態様によれば、除放性製剤は、必要に応じて本明細書に記載の除放性コーティングを有するマトリックスにより提供される。除放性マトリックスに含有させるのに好適な物質は、マトリックス形成に使用される方法によって異なることができる。
【0036】
例えば、α,β−不飽和ケトン含量が10ppm未満のオキシモルホン塩酸塩の他にマトリックスは、親水性及び/又は疎水性物質、例えば、ゴム、セルロースエーテル、アクリル樹脂、タンパク質由来物質などを含んでいてもよい。含有される物質は、これらには限定されず、薬剤に除放性を付与させることのでき、且つ溶融する(又は押出に必要とする程度まで軟化する)いずれの薬学的に許容される疎水性物質又は親水性物質も、本発明において使用してもよい。
【0037】
消化性長鎖(C8−C50、とりわけC12−C40)置換又は非置換炭化水素、例えば、脂肪酸、脂肪アルコール、脂肪酸のグリセリルエステル、鉱油、植物油、ろう及びステアリ
ルアルコール;並びにポリアルキレングリコールなどがあげられる。これらのポリマーのうち、アクリル系ポリマー、とりわけEudragit(商品名)、セルロースエーテル、とりわけヒドロキシアルキルセルロース及びカルボキシアルキルセルロースが好ましい。経口剤は、少なくとも一種の親水性又は疎水性物質を1%〜80%(重量)含有してもよい。
【0038】
疎水性物質が炭化水素であるとき、炭化水素の融点は好ましくは25℃〜90℃である。長鎖炭化水素物質のうち、脂肪(脂肪族)アルコールが好ましい。経口剤は、少なくとも一種の消化性長鎖炭化水素を60%(重量)以下を含有してもよい。
【0039】
好ましくは、経口剤は、少なくとも一種のポリアルキレングリコールを60%(重量)以下含む。
【0040】
疎水性物質は、好ましくはアルキルセルロース、アクリル酸ポリマー、メタクリル酸ポリマー及びコポリマー、セラック、ゼイン、水素添加ひまし油、水素添加植物油又はこれらの混合物からなる群から選択される。本発明のある好ましい実施態様によれば、疎水性物質は、アクリル酸コポリマー、メタクリル酸コポリマー、メチルメタクリレート、メチルメタクリレートコポリマー、エトキシエチルメタクリレート、シアノエチルメタクリレート、アミノアルキルメタクリレートコポリマー、ポリ(アクリル酸)、ポリ(メタクリル酸)、メタクリル酸アルキルアミンコポリマー、ポリ(メチルメタクリレート)、ポリ(メタクリル酸)(無水物)、ポリメタクリレート、ポリアクリルアミド、ポリ(メタクリル酸無水物)、及びグリシジルメタクリレートコポリマーなどの薬学的に許容されるアクリル系ポリマーであるが、これらには限定されない。他の実施態様によれば、疎水性物質は、ヒドロキシアルキルセルロース、例えば、ヒドロキシプロピルメチルセルロース及びこれらの混合物などの物質から選択される。
【0041】
好ましい疎水性物質は、水不溶性であって、事実上顕著な親水性及び/又は疎水性を有するものである。好ましくは、本発明に有用な疎水性物質は、融点が約25℃〜約200℃、好ましくは約45℃〜約90℃である。具体的には、疎水性物質は、天然又は合成のろう、脂肪アルコール(例えば、ラウリルアルコール、ミリスチルアルコール、ステアリルアルコール、セチルアルコール又は好ましくはセトステアリルアルコール)、脂肪酸(以下のものをなどがあるが、これらには限定されない)、例えば、脂肪酸エステル、脂肪酸グリセリド(モノ−、ジ−及びトリ−グリセリド)、硬化油脂、炭化水素、通常ろう、ステアリン酸、ステアリルアルコール並びに炭化水素骨格を有する疎水性及び親水性物質を含んでいてもよい。好適なろうには、例えば、ミツロウ、グリコろう、カスターろう及びカルナウバろうなどがある。本発明の目的からして、ろう様物質は、通常室温で固体であり且つ融点が約25℃〜約100℃である物質として定義される。
【0042】
本発明に使用することができる好適な疎水性物質は、消化性長鎖(C8−C50、とりわけC12−C40)置換又は非置換炭化水素、例えば、脂肪酸、脂肪アルコール、脂肪酸のグリセリルエステル、鉱油、植物油及び天然ろう又は合成ろうなどがある。融点が25℃〜90℃の炭化水素が好ましい。長鎖炭化水素物質のうち、脂肪(脂肪族)アルコールが、ある実施態様においては好ましい。経口剤は、少なくとも一種の消化性長鎖炭化水素を60%(重量)以下含有してもよい。
【0043】
好ましくは、二種以上の疎水性物質を組み合わせてマトリックス製剤に含有させる。追加の疎水性物質を含有させる場合、天然又は合成ろう、脂肪酸、脂肪アルコール及びこれらの混合物から選択するのが好ましい。例えば、ミツロウ、カルナウバろう、ステアリン酸及びステアリルアルコールなどがある。しかしながら、これらには限定されない。
【0044】
一つの具体的な好適なマトリックスは、少なくとも一種の水溶性ヒドロキシアルキルセルロース、少なくとも一種のC12〜C36、好ましくはC14−C22の脂肪族アルコール及び必要に応じて少なくとも一種のポリアルキレングリコールを含む。少なくとも一種の ヒドロキシアルキルセルロースは、好ましくはヒドロキシ(C1〜C6)アルキルセルロース、例えば、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース及びとりわけ、ヒドロキシエチルセルロースである。本発明の経口剤中の少なくとも一種のヒドロキシアルキルセルロースの量は、とりわけ必要とされるオキシモルホン塩酸塩の正確な放出速度により決定する。少なくとも一種の脂肪族アルコールは、例えば、ラウリルアルコール、ミリスチルアルコール又はステアリルアルコールでよい。しかしながら、本発明の経口剤の特に好ましい実施態様よれば、少なくとも一種の脂肪族アルコールは、セチルアルコール又はセトステアリルアルコールである。本発明の経口剤中の少なくとも一種の脂肪族アルコールの量は、上記のように、オピオイドキシモルホンの正確な必要とする放出速度により決定される。また、これは、少なくとも一種のポリアルキレングリコールが経口剤に存在するか又は存在しないかにより異なる。少なくとも一種のポリアルキレングリコールが存在しない場合、経口剤は好ましくは少なくとも一種の脂肪族アルコールを20%〜50%(重量)を含有する。少なくとも一種のポリアルキレングリコールが経口剤に存在するときには、少なくとも一種の脂肪族アルコールと少なくとも一種のポリアルキレングリコールの合計重量は、好ましくは総投与量の20%〜50%(重量)である。
【0045】
一実施態様によれば、少なくとも一種のヒドロキシアルキルセルロース又はアクリル樹脂の少なくとも一種の脂肪族アルコール/ポリアルキレングリコールに対する比、例えば、少なくとも一種のヒドロキシアルキルセルロースの少なくとも一種の脂肪族アルコール/ポリアルキレングリコールに対する比(w/w)は、1:2〜1:4が好ましく、特に好ましくは1:3〜1:4である。
【0046】
少なくとも一種のポリアルキレングリコールは、例えば、ポリプロピレングリコールでよく、好ましくはポリエチレングリコールである。少なくとも一種のポリアルキレングリコールの数平均分子量は、好ましくは1,000〜15,000、とりわけ1,500〜12,000である。
【0047】
別の好適な除放性マトリックスは、アルキルセルロース(とりわけエチルセルロース)、 C12〜C36脂肪族アルコール及び必要に応じてポリアルキレングリコールを含む。
【0048】
別の好ましい実施態様によれば、マトリックスは、薬学的に許容される少なくとも二種の疎水性物質の組合わせを含む。
【0049】
除放性マトリックスは、上記成分の他に、医薬分野において一般的に用いられている好適な量の他の物質、例えば、希釈剤、滑剤、結合剤、造粒助剤、着色剤、香味剤、流動促進剤を含有してもよい。
【0050】
マトリックス−微粒子
本発明の固体状除放性経口剤の調製を容易にするために、当業者に公知のマトリックス製剤の調製方法を用いることができる。例えば、(a)少なくとも一種の水溶性ヒドロキシアルキルセルロースとα,β−不飽和ケトン含有量が10ppm未満であるオキシモルホン塩酸塩とを含む顆粒を形成し、(b)ヒドロキシアルキルセルロース含有顆粒を少なくとも一種のC12〜C36脂肪族アルコールと混合し、(c)必要に応じて顆粒を圧縮し、造形することにより、マトリックスに含有させることができる。好ましくは、顆粒は、ヒドロキシアルキルセルロース顆粒を水を用いて湿式造粒する。
【0051】
さらに他の別の実施態様によれば、球形化剤を活性成分とともに球形として、球状体を形成することができる。微結晶性セルロースは、好ましい球状化剤である。好適な微結晶性セルロースは、例えば、Avicel PH101(商品名、FMC社製)として販売されている物質である。このような実施態様によれば、球状体は、有効成分及び球状化剤の他に、結合剤を含んでいてもよい。低粘度水溶性ポリマー等の好適な結合剤が、医薬分野の当業者には周知である。しかしながら、ヒドロキシプロピルセルロース等の水溶性ヒドロシキ低級アルキルセルロースが好ましい。さらに又は別法として、球状体は、水不溶性ポリマー、とりわけアクリル系ポリマー、アクリル系コポリマー、例えば、メタクリル酸−エチルアクリレートコポリマー又はエチルセルロースを含んでいてもよい。このような実施態様では、除放性コーティングは、一般的に疎水性物質、例えば、(a)ろう(単独又は脂肪アルコールとの混合物として);(b)セラック又はゼインを含む。
【0052】
溶融押出マトリックス
除放性マトリックスは、溶融造粒法又は溶融押出法により調製することもできる。一般的に、溶融造粒法は、通常固体である疎水性物質、例えば、ろうを溶融し、粉末状医薬をそこに含有させることを含む。除放性製剤を得るためには、追加の疎水性物質、例えば、エチルセルロース又は水不溶性アクリル系ポリマーを溶融ろう疎水性物質に含有させることが必要なことがある。溶融造粒法により調製される除放性製剤の例が、米国特許第4,861,598号に記載されている。
【0053】
追加の疎水性物質は、一種以上の水不溶性ろう様熱可塑性物質(場合によっては、前記一種以上の水不溶性ろう様物質よりも疎水性度が小さい一種以上のろう様熱可塑性物質との混合物)を含んでいてもよい。一定の放出を維持するためには、製剤中の個々のろう様物質は、初期放出段階中実質的に非分解性であり、胃腸液に不溶でなければならない。有用な水不溶性ろう様物質は、水溶解度が約1:5,000(w/w)より低いものでよい。
【0054】
上記成分の他に、除放性マトリックスは、医薬分野において一般的に用いられている好適な量の他の物質、例えば、希釈剤、滑剤、結合剤、造粒助剤、着色剤、香味剤及び流動促進剤を含有してもよい。これらの追加の物質の量は、所望の製剤に所望の効果を付与するのに十分な量である。
【0055】
溶融押出多微粒子を含有する除放性マトリックスは、上記成分の他に、医薬分野において一般的に用いられている好適な量の他の物質、例えば、希釈剤、滑剤、結合剤、造粒助剤、着色剤、香味剤及び流動促進剤を、必要に応じて微粒子の約50重量%以下の量で含有してもよい。
【0056】
経口剤を調製するのに使用できる薬学的に許容される担体及び賦形剤の具体例が、Handbook of Pharmaceutical Excipients(医薬賦形剤ハンドブック)、 American Pharmaceutical Association (1986)に記載されている。
【0057】
溶融押出多微粒子
本発明による好適な溶融押出マトリックスの調製は、例えば、α,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩を、少なくとも一種の疎水性物質及び好ましくは追加の疎水性物質とともに、配合して均一混合物を得る工程を含んでいてもよい。次に、均一混合物を、混合物を少なくとも軟化し、混合物を十分に押しだせるのに十分な温度に加熱する。次に、得られた均一混合物を、押出してストランドを形成する。押出物は、好ましくは冷却し、当該技術分野において公知のいずれかの手段により切断して多微粒子にする。ストランドは、冷却し、切断して多微粒子にする。次に、多微粒子を、一回量に分ける。好ましくは、押出物は、直径が約0.1mm〜約5mmであり、治療に有効な薬剤を約8時間〜約24時間の間除放するものである。
【0058】
本発明の溶融押出の任意のプロセスには、疎水性物質、α,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩及び必要に応じて結合剤を押出機に直接計量して入れ;得られた均一混合物を加熱し;均一混合物を押出しストランドを形成し;均一混合物を含むストランドを冷却し;得られたストランドを切断して約0.1mm〜約12mmの大きさの粒子とし;そして得られた粒子を一回量に分けることを含む。本発明のこの実施態様では、比較的連続した製造手順が可能となる。
【0059】
押出機の開口又は出口ポートの直径を調整して、押出ストランドの厚さを変化させることもできる。さらに、押出機の出口部分は、丸い必要はなく、長円形、矩形等であってもよい。出てきたストランドは、熱線カッター、ギロチン等を用いてサイズを粒子サイズまで減少させることができる。
【0060】
溶融押出多微粒子系は、押出機の出口オリフィスに応じて、例えば、顆粒、球状体又はペレットの形態であることができる。本発明の目的のために、用語「溶融押出多微粒子」、「溶融押出多微粒子系」及び「溶融押出粒子」は、好ましくは一定範囲の同様なサイズ及び/又は形状内で、且つ一種以上の活性剤及び一種以上の賦形剤(好ましくは本明細書に記載されているような疎水性物質を含む)を含有する複数単位を意味する。この点において、溶融押出多微粒子は、長さが約0.1mm〜約12mmであり、直径が約0.1mm〜約5mmである。さらに、溶融押出多微粒子は、このサイズ範囲内のいずれの幾何学的形状であってもよい。別法として、押出物は、球状化工程の必要なく、単純に所望の長さに切断し、治療効果のある活性剤の一回量に分けてもよい。
【0061】
好ましい一実施態様によれば、経口剤は、カプセル剤内に、有効量の溶融押出多微粒子を含有させて調製する。例えば、複数の溶融押出多微粒子を、ゼラチンカプセルに、摂取され、胃液に接触したときに、有効な除放投与量となるのに十分な量を入れてもよい。
【0062】
別の好ましい実施態様によれば、多微粒子押出物の好適量を、標準的な方法を用いて、通常の打錠装置により圧縮して経口錠剤とする。当業者には、錠剤(圧縮及び成形したもの)、カプセル剤(硬質及び軟質ゼラチン)及びピルを製造するための種々の方法及び組成物が知られている。
【0063】
まだ別の好ましい実施態様によれば、押出物は、上記でより詳細に記載した米国特許第4,957,681号に記載したように、賦形して錠剤とすることができる。
【0064】
必要に応じて、上記で説明した除放性コーティング等の除放性コーティングにより、除放性溶融押出多微粒子系又は錠剤を被覆したり、又は多微粒子を含むゼラチンカプセル剤をさらに被覆することができる。このようなコーティングは、好ましくは十分な量の疎水性物質を含有して、約2%〜約30%の重量増加する。但し、オーバーコートは、とりわけ所望の除放速度に応じて大きく異なることができる。
【0065】
本発明の溶融押出単位剤形は、カプセル化する前に溶融押出粒子の組み合わせをさらに含むことができる。さらに、単位剤形は、即時放出用の即時放出剤もかなりの量で含むことができる。即時放出剤は、例えば、ゼラチンカプセル剤内に別個のペレットとして含有させてもいいし、又は剤形(例えば、除放性コーティング又はマトリックス系)の調製後、多微粒子表面上に塗布してもよい。また、本発明の単位剤形は、除放性ビーズとマトリックス多微粒子との組み合わせを含有させて所望の効果を得ることもできる。
【0066】
本発明の除放性製剤は、好ましくは、例えば、摂取され、胃液にさらされ、その後腸液にさらされたときにゆっくりと薬剤を放出する。本発明の溶融押出製剤の除放性プロフィールは、例えば、抑制剤、すなわち、疎水性物質の量を変化することにより、疎水性物質に対する可塑剤の量を変更することにより、追加の成分又は賦形剤を含有させることにより、製造方法を変更することによる等により変更できる。
【0067】
本発明の他の実施態様によれば、溶融押出物質は、α,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩を含有させることなく調製し、その後オキシモルホン塩酸塩を押出物に添加することができる。このような製剤は、典型的には薬剤を、押出したマトリックス物質とともにブレンドし、その後混合物を錠剤化して除放性製剤を得る。
【0068】
コーティング(被覆)
本発明の剤形は、必要に応じて放出の調節又は製剤の保護に好適な一種以上の物質を被覆してもよい。一実施態様によれば、コーティングを設けて、pH依存放出性又はpH非依存放出性とする。pH依存性コーティングは、少なくとも約8時間以上、好ましくは約12時間以上、約24時間以下、患者に対して鎮痛作用を与えることができる吸収プロフィールが得られるように、活性成分を胃腸(GI)管、例えば、胃又は小腸の所望の領域に放出する。pH非依存コーティングが望ましいときには、環境流体、例えば、GI管におけるpH変化とは無関係に最適に放出するようにコーティングを設計する。また、GI管の一つの所望領域、例えば、胃において投与量の一部分を放出し、投与量の残部をGI管の別の領域、例えば、小腸において放出する組成物を製剤化することもできる。
【0069】
pH依存性コーティングを利用して製剤化した本発明による製剤は、反復作用効果を付与することもできる。それにより、未保護薬剤を腸溶コーティング上に被覆して胃で放出され、一方、腸溶コーティングにより保護されている残部は胃腸管でさらに放出される。本発明で使用できるpH依存性コーティングとしては、セラック、セルロースアセテートフタレート、ポリビニルアセテートフタレート、ヒドロキシプロピルメチルセルロースフタレート及びメタクリル酸エステルコポリマー、ゼイン等がある。
【0070】
ある好ましい実施態様によれば、α,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩を含有する基材(例えば、錠剤コアビーズ、マトリックス粒子)に、(i) アルキルセルロース;(ii)アクリル系ポリマー;又は(iii)これらの混合物から選択された疎水性物質で被覆する。コーティングは、有機物又は水溶液又は分散液の形態で適用することができる。コーティングは、基材の約2%〜約25%の重量増加となるように適用して、所望の除放性プロフィールが得られるようにすることができる。水性分散液から得られるコーティングは、米国特許第5,273,760号、米国特許第5,286,493号、米国特許第5,324,351号、米国特許第5,356,467号及び米国特許第5,472,712号に詳細に記載されている。
【0071】
アルキルセルロースポリマー
アルキルセルロースなどのセルロース系物質及びポリマーにより、本発明によるビーズをコーティングするのに適している疎水性物質が得られる。例えば、一つの好ましいアルキルセルロース系ポリマーとしては、エチルセルロースがあげられる。但し、当業者には、他のセルロース及び/又はアルキルセルロースポリマーが、単一又は組み合わせにおいて、本発明による疎水性コーティングの全体又は一部分として容易に使用可能であることが理解できるであろう。
【0072】
アクリル系ポリマー
本発明の他の好ましい実施態様によれば、除放性コーティングを構成する疎水性物質は、薬学的に許容されるアクリル系ポリマー、例えば、アクリル酸コポリマー及びメタクリル酸コポリマー、メチルメタクリレートコポリマー、エトキシエチルメタクリレート、シアノエチルメタクリレート、ポリ(アクリル酸)、ポリ(メタクリル酸)、メタクリル酸アルキルアミドコポリマー、ポリ(メチルメタクリレート)、ポリメタクリレート、ポリ(メチルメタクリレート)コポリマー、ポリアクリルアミド、アミノアルキルメタクリレートコポリマー、ポリ(メタクリル酸無水物)及びグリシジルメタクリレートコポリマーがあげられる。疎水性物質は、これらには限定されない。
【0073】
ある好ましい実施態様によれば、アクリル系ポリマーは、一種以上のアンモニオメタクリレートコポリマーからなる。アンモニオメタクリレートコポリマーは、当該技術分野において周知であり、第4級アンモニウム基低含量のアクリル酸エステル及びメタクリル酸エステルの完全重合コポリマーとして記載されている。
【0074】
所望の溶解プロフィールを得るために、異なる物性、例えば、第4級アンモニウム基の中性(メタ)アクリル酸エステルに対するモル比が異なる二種以上のアンモニオメタクリレートコポリマーを含有させることが必要なことがある。
【0075】
ある種のメタクリル酸エステル型ポリマーは、本発明に使用することができるpH依存コーティングを形成するのに有用である。例えば、メタクリル酸コポリマー又は高分子メタクリレートとしても知られており、Rohm Tech社からEudragit(商品名)の商品名で市販されている、ジエチルアミノエチルメタクリレート及び他の中性メタアクリル酸エステルから合成されたコポリマー群がある。Eudragit(商品名)には、いくつかの異なる種類のものがある。例えば、Eudragit(商品名)Eは、酸性媒体で膨潤し、そこに溶解するメタクリル酸コポリマーの一例である。Eudragit(商品名)Lは、概略pH<5.7では膨潤せず、概略pH>6で溶解するメタクリル酸コポリマーである。Eudragit(商品名)Sは、概略pH<6.5では膨潤せず、概略pH>7で溶解する。Eudragit(商品名)RL及びEudragit(商品名)RSは、水膨潤性である。これらのポリマーにより吸収される水の量は、pH依存性であるが、Eudragit(商品名)RL及びRSで被覆された剤形は、pH非依存性である。
【0076】
ある好ましい実施態様によれば、アクリル系コーティングは、Rohm Pharma社からEudragit(商品名)RL30D及びEudragit(商品名)RS30Dの商品名で入手できる2種のアクリル樹脂ラッカーの混合物を含む。Eudragit(商品名)RL30D及びEudragit(商品名)RS30Dは、第4級アンモニウム基低含量(アンモニウム基の残りの中性(メタ)アクリル酸エステルに対するモル比は、Eudragit(商品名)RL30Dでは1:20、Eudragit(商品名)RS30Dでは1:40である)のアクリル酸エステル及びメタクリル酸エステルのコポリマーである。平均分子量は、約150,000である。コード表示RL(高浸透性)及びRS(低浸透性)は、これらの薬剤の浸透性を示す。Eudragit(商品名)RL/RS混合物は、水及び消化液に不溶性である。しかしながら、この混合物から形成したコーティングは、水溶液及び消化液において膨潤性及び浸透性である。
【0077】
本発明のEudragit(商品名)RL/RS分散液は、いずれかの所望の比で混合して、最終的に所望の溶解プロフィールを有する除放性製剤を得ることができる。所望の除放性製剤は、例えば、100%Eudragit(商品名)RL、50%Eudragit(商品名)RL及び、50%Eudragit(商品名)RS、又は10%Eudragit(商品名)RL及び90%Eudragit(商品名)RSから得られた遅延コーティングから得ることができる。もちろん、当業者には、例えば、Eudragit(商品名)Lなどの他のアクリル系ポリマーを使用することができるとわかるであろう。
【0078】
可塑剤
コーティングが疎水性物質の水性分散液を含む本発明の実施態様によれば、有効量の可塑剤を疎水性物質の水性分散液に含有させると、除放性コーティングの物性がさらに改善される。例えば、エチルセルロースはガラス転移温度が比較的高く、通常のコーティング条件下では可撓性被膜を形成しないので、可塑剤を、除放性コーティングを含有するエチルセルロースコーティングに、コーティングを塗料として使用する前に含有させることが好ましい。一般的に、コーティング溶液に含有させる可塑剤の量は、膜形成剤の濃度に基づいて決定され、例えば、最も一般的には膜形成剤の約1重量%〜約50重量%である。しかしながら、可塑剤濃度は、特定のコーティング溶液及び適用方法についての慎重な実験をすることによってのみ適切に決定できる。
【0079】
エチルセルロースに好適な可塑剤としては、例えば、水不溶性可塑剤、例えば、セバシン酸ジブチル、ジエチルフタレート、クエン酸トリエチル、クエン酸トリブチル及びトリアセチンなどがある。但し、他の水不溶性可塑剤(例えば、アセチル化モノグリセリド、フタレートエステル、ひまし油等)を使用することもできる。クエン酸トリエチルが、本発明のエチルセルロースの水性分散液にとってとりわけ好ましい可塑剤である。
【0080】
本発明のアクリル系ポリマーに好適な可塑剤としては、例えば、クエン酸エステル、例えば、クエン酸トリエチル、クエン酸トリブチル、ジブチルフタレート及び場合により1,2−プロピレングリコールがあげられるが、これらには限定されない。Eudragit(商品名)RL/RSラッカー溶液等のアクリル被膜から形成される被膜の弾性を高めるのに好適であることが分かった他の可塑剤には、ポリエチレングリコール、プロピレングリコール、ジエチルフタレート、ひまし油及びトリアセチンなどがある。クエン酸トリエチルが、本発明のエチルセルロースの水性分散液にとってとりわけ好ましい可塑剤である。
【0081】
少量のタルクを添加することも、処理中に水性分散液がくっつく傾向を減少するのに役立ち、研磨剤としての役割を果たすことができる。
【0082】
除放性浸透剤
また、本発明の除放性製剤は、浸透剤として調製することもできる。浸透剤は、好ましくは二重層コアを備えている。二重層コアは、薬物層(α,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩)及び送達層又は押圧層を備えている。ここで、二重層コアは、半浸透性壁により包囲されており、必要に応じて少なくとも一個の経路がその中に設けられている。
【0083】
本発明の目的で使用される表現「経路」は、開口、オリフィス、穴、細孔、多孔性要素を備えていて、それにより、α,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩が、繊維、毛細管、多孔性オーバーレイ、多孔性インサート、微細孔部材又は多孔性組成物を介して汲み上げられるか、拡散するか、又は移動する。また、経路は、使用流体環境において壁を浸食したり、また壁から滲出して少なくとも一つの経路を生じる化合物を含んでいてもよい。経路を形成する代表的な化合物には、壁における浸食性ポリグリコール酸又はポリ乳酸;ゼラチン状フィラメント;水除去性ポリ(ビニルアルコール);滲出性化合物、例えば、流体除去性細孔形成多糖類、酸、塩又は酸化物などがある。経路は、ソルビトール、スクロース、ラクトース、マルトース又はフルクトース等の化合物を壁から滲出させて、除放性次元細孔経路を形成することにより形成できる。一つ以上の表面上に間隔をあけて一つ以上の経路を有する剤形を製造できる。経路及び経路形成装置については、米国特許第3,845,770号、米国特許第3,916,899号、米国特許第4,063,064号及び米国特許第4,088,864号に開示されている。水性滲出により形成される放出性細孔としての大きさとし、賦形し、適合させた除放寸法を有して除放速度を有する放出細孔を設けた経路について、米国特許第4,200,098号及び米国特許第4,285,987号に開示されている。
【0084】
ある実施態様によれば、薬物層は、また少なくとも一種のポリマーヒドロゲルを含んでいてもよい。ポリマーヒドロゲルは、平均分子量が約500〜約6,000,000でよい。ポリマーヒドロゲルとしては、例えば、式(C6H12O5)nH2O(式中、nは3〜7,500である)で表されるマルトデキストリンポリマー(マルトデキストリンポリマーの数平均分子量が500〜1,250,000である);ポリ(アルキレンオキシド)、例えば、重量平均分子量が50,000〜750,000であるポリ(エチレンオキシド)及びポリ(プロピレンオキシド)、より具体的には重量平均分子量が100,000、200,000、300,000又は400,000のうちの少なくとも一つであるポリ(エチレンオキシド);アルカリカルボキシアルキルセルロース(但し、アルカリはナトリウム又はカリウムであり、アルキルはメチル、エチル、プロピル又はブチルである)(重量平均分子量:10,000〜175,000);及びメタアクリル酸及びエタアクリル酸を含むエチレン−アクリル酸のコポリマー(数平均分子量:10,000〜500,000)などがあげられるが、これらには限定されない。
【0085】
本発明のある実施態様によれば、送達層又は押圧層は、オスモポリマーを含む。オスモポリマーとしては、例えば、ポリアルキレンオキシド及びカルボキシアルキルセルロースからなる群から選択されるものがあげられるが、これらには限定されない。ポリアルキレンオキシドは、重量平均分子量が1,000,000〜10,000,000である。ポリアルキレンオキシドは、ポリメチレンオキシド、ポリエチレンオキシド、ポリプロピレンオキシド、平均分子量が1,000,000であるポリエチレンオキシド、平均分子量が5,000,000であるポリエチレンオキシド、平均分子量が7,000,000であるポリエチレンオキシド、平均分子量が1,000,000である架橋ポリメチレンオキシド及び平均分子量が1,200,000であるポリプロピレンオキシドからなる群から選択されたものでよい。典型的なオスモポリマーであるカルボキシアルキルセルロースは、アルカリカルボキシアルキルセルロース、カルボキシメチルセルロースナトリウム、カルボキシメチルセルロースカリウム、カルボキシエチルセルロースナトリウム、カルボキシメチルセルロースリチウム、カルボキシエチルセルロースナトリウム、カルボキシアルキルヒドロキシアルキルセルロース、カルボキシメチルヒドロキシエチルセルロース、カルボキシエチルヒドロキシエチルセルロース及びカルボキシメチルヒドロキシプロピルセルロースからなる群から選択されるものを含む。置換層に使用されるオスモポリマーは、半浸透性壁の浸透圧勾配を示す。オスモポリマーにより、流体が剤形に吸収され、それにより浸透ヒドロゲル(オスモゲルとしても知られている)として膨潤且つ膨張し、その結果、浸透ヒドロゲルがα,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩を浸透製剤から押し出す。
【0086】
押圧層は、オスマジェント及び浸透圧的に有効な溶質としても知られている一種以上の浸透圧的に有効な化合物を含んでいてもよい。これらは、環境流体を、例えば、胃腸管から剤形に吸収させ、置換層の送達動態に影響する。浸透圧的に活性な化合物としては、例えば、浸透性塩及び浸透性炭水化物からなる群から選択されるものがあげられる。オスマジェントの具体例としては、塩化ナトリウム、塩化カリウム、硫酸マグネシウム、リン酸リチウム、塩化リチウム、リン酸ナトリウム、硫酸カリウム、硫酸ナトリウム、リン酸カリウム、グルコース、フルクトース及びマルトースなどがあるが、これらには限定されない。
【0087】
押圧層は、必要に応じて、数平均分子量が9,000〜450,000であるヒドロキシプロピルアルキルセルロースを含んでいてもよい。ヒドロキシプロピルアルキルセルロースは、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルエチルセルロース、ヒドロキシプロピルイソプロピルセルロース、ヒドロキシプロピルブチルセルロース及び ヒドロキシプロピルペンチルセルロースからなる群から選択されたものが代表的なものである。
【0088】
押圧層は、必要に応じて非毒性着色剤又は色素を含んでいてもよい。着色剤又は色素としては、例えば、食品医薬品局着色剤(FD&C)、例えば、FD&C No.1青色色素、FD&C No.4赤色色素、赤色酸化第二鉄、黄色酸化第二鉄、二酸化チタン、カーボンブラック及びインジゴなどがあるが、これらには限定されない。
【0089】
また、押圧層は、必要に応じて酸化防止剤を含有して、成分の酸化を抑制するようにしてもよい。アスコルビン酸、パルミチン酸アスコルビル、ブチル化ヒドロキシアニソール、 ジ−tert−ブチル−4−ヒドロキシアニソールとトリ−tert−ブチル−4−ヒドロキシアニソールとの混合物、ブチル化ヒドロキシトルエン、イソアスコルビン酸ナトリウム、ジヒドログアレティック酸、ソルビン酸カリウム、硫酸水素ナトリウム、メタ重亜硫酸ナトリウム、ソルビン酸、アスコルビン酸カリウム、ビタミンE、4−クロロ−2,6−ジ−tert−ブチルフェノール、α−トコフェロール及び没食子酸プロピルからなる群から選択されるものが、酸化防止剤の一例としてあげられるが、これらには限定されない。
【0090】
ある別の実施態様によれば、剤形は、均質なコアを備えている。このコアは、α,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩、薬学的に許容されるポリマー(例えば、ポリエチレンオキシド)、任意の崩壊剤(例えば、ポリビニルピロリドン)、任意の吸収促進剤(例えば、脂肪酸、界面活性剤、キレート剤、胆汁酸塩等)を含む。均質コアは、α,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩を放出するための経路(上記したような)を有する半浸透性壁により包囲されている。
ある実施態様によれば、半浸透性壁は、セルロースエステルポリマー、セルロースエーテルポリマー及びセルロースエステル−エーテルポリマーからなる群から選択されるものを含む。代表的な壁ポリマーは、セルロースアシレート、セルロースジアシレート、セルローストリアシレート、セルロースアセテート、セルロースジアセテート、セルローストリアセテート、モノ−、ジ−及びトリセルロースアルケニレート並びにモノ−、ジ−及びトリセルロースアルキニレートからなる群から選択されるものを含む。本発明に使用されるポリ(セルロース)は、数平均分子量が20,000〜7,500,000である。
【0091】
本発明の目的のための追加の半浸透性ポリマーは、アセトアルデヒドジメチルセルロースアセテート、セルロースアセテートエチルカルバメート、セルロースアセテートメチルカルバメート、セルロースジアセテート、プロピルカルバメート、セルロースアセテートジエチルアミノアセテート;半浸透性ポリアミド;半浸透性ポリウレタン;半浸透性スルホン化ポリスチレン;ポリアニオンとポリカチオンの共沈により形成された半浸透性架橋ポリマー、半浸透性架橋ポリスチレン、半浸透性架橋ポリ(スチレンスルホン酸ナトリウム)、半浸透性架橋ポリ(ビニルベンジルトリメチル塩化アンモニウム)及び半浸透性ポリマー(流体浸透性:半浸透性壁の静水圧又は浸透圧差当りで、2.5x10-8〜2.5x10-2(cm2/hr・atm))を含む。本発明に有用な他のポリマーは、当該技術分野において知られており、例えば、Handbook of Common Polymers(一般ポリマーハンドブック)、Scott、J.R.及びW.J.Roff、1971、オハイオ州クリーブランドにあるCRC Press社発行に記載のものなどがあげられる。
【0092】
ある実施態様によれば、好ましくは半浸透性壁が非毒性、不活性であり、薬物の投薬寿命中は、その物理的及び化学的健全性が維持されるものである。ある実施態様によれば、剤形は、結合剤を含む。結合剤としては、例えば、粘度平均分子量が5,000〜350,000である治療学的に許容されるビニルポリマーがあげられ、代表的には、ビニルアセテート、ビニルアルコール、塩化ビニル、フッ化ビニル、酪酸ビニル、ラウリン酸ビニル及びステアリン酸ビニルからなる群から選択されるものを有する、ポリ−n−ビニルアミド、ポリ−n−ビニルアセトアミド、ポリ(ビニルピロリドン)(ポリ−n−ビニルピロリドンとしても知られている)、ポリ−n−ビニルカプロラクトン、ポリ−n−ビニル−5−メチル−2−ピロリドン及びポリ−n−ビニルピロリドンコポリマーからなる群から選択されるものがあげられるが、これには限定されない。他の結合剤として、例えば、アラビアゴム、でんぷん、ゼラチン及び平均分子量が9,200〜250,000のヒドロキシプロピルアルキルセルロースなどがある。
【0093】
ある実施態様によれば、剤形は、滑剤を含む。滑剤は、剤形の製造中に使用されて、型壁又はパンチ面への粘着を防止することができるものである。滑剤としては、例えば、ステアリン酸マグネシウム、ステアリン酸ナトリウム、ステアリン酸、ステアリン酸カルシウム、オレイン酸マグネシウム、オレイン酸、オレイン酸カリウム、カプリル酸、フマル酸ステアリルナトリウム及びパルミチン酸マグネシウムなどがあるが、これらには限定されない。
【0094】
ある好ましい実施態様によれば、本発明は、オキシモルホン塩酸塩10〜40mgに相当する量のα,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩、平均分子量が150,000〜500,000であるポリ(アルキレンオキシド)25mg〜500mg、平均分子量が40,000であるポリビニルピロリドン1mg〜50mg、滑剤0mg〜約7.5mgを含有する治療組成物を含む。
【0095】
坐剤
本発明の除放製剤は、直腸投与用薬用坐剤として製剤化してもよい。この坐剤は、好適な坐剤基剤及びα,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩を含む。除放性坐剤の調製については、例えば、米国特許第5,215,758号に記載されている。
【0096】
吸収前には、薬物は、溶液状態でなければならない。坐剤の場合、まず坐剤の基剤の溶解又は基剤の溶融が生じて、続いて薬物が坐剤の基剤から直腸液に分配される。薬物の体内への吸収は、坐剤の基剤により変更できる。したがって、薬物の物性を考慮しながら、特定の薬物に応じて、使用される特定の坐剤の基剤を選ぶ必要がある。例えば、脂溶性薬物は直腸液には容易に分配されないが、脂質基剤に微溶性である薬物は直腸液に容易に分配される。
【0097】
薬物の溶解時間(又は放出速度)に影響する異なる因子には、溶解溶媒にさらされる薬物物質の表面積、使用溶液のpH、特定の溶媒への物質の溶解度及び溶媒への溶解物質の飽和濃度の駆動力がある。一般的に、直腸内に投与された坐剤からの薬物の吸収に影響する因子には、坐剤のビヒクル、吸収部位pH、薬物pKa、イオン化度及び脂溶性などがある。
【0098】
選択される坐剤の基剤は、本発明の活性成分と適合するものでなければならない。さらに、坐剤の基剤は、好ましくは非毒性であり、粘膜を刺激せず、直腸液で溶融又は溶解し、そして貯蔵中安定であることが好ましい。
【0099】
本発明のある好ましい実施態様によれば、水溶性薬物及び水不溶性薬物の両方において、坐剤の基剤は、鎖長がC12〜C18である飽和天然脂肪酸のモノ−、ジ−及びトリグリセリドからなる群から選択される脂肪酸ろうを含む。
【0100】
本発明の坐剤を調製する際、他の賦形剤を使用することもできる。例えば、ろうを使用して直腸経路を介した投与のための適切な形状を形成してもよい。この系は、直腸投与及び経口投与用のゼラチンカプセル剤に希釈剤を添加し、ろうなしで使用することもできる。
【0101】
好適な市販のモノ−、ジ−及びトリグリセリドとしては、商品名Novata(商品名) (タイプAB、AB、B、BC、BD、BBC、E、BCF、C、D及び299)(Henkel社製)及びWitepsol(商品名)(タイプH5、H12、H15、H175、H185、H19、H32、H35、H39、H42、W25、W31、W35、W45、S55、S58、E75、E76及びE85)(ダイナマイト・ノーベル社製)の炭素長が12〜18の飽和天然脂肪酸などがある。
【0102】
他の薬学的に許容される坐剤の基剤は、上記したモノ−、ジ−及びトリグリセリドが全体又は部分的に置換されたものでもよい。坐剤中の基剤の量は、剤形のサイズ(すなわち、実重量)、使用される基剤(例えば、アルギン酸塩)、薬物の量により決定される。一般的に、坐剤の基剤の量は、坐剤の総重量の約20重量%〜約90重量%である。好ましくは、坐剤中の坐剤の基剤の量は、坐剤の総重量の約65重量%〜約80重量%である。
【0103】
さらなる実施態様
α,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩は、既存の市販の製品、例えば、Opana(商品名)、 Opana ER(商品名)及びNumorphan(商品名)におけるオキシモルホン塩酸塩の代替品として使用できる。このような製剤は、FDA Orange Book(FDAオレンジブック)に挙げられている。
【0104】
以下、実施例により本発明を説明する。実施例では、オリパビンを原料として用い、添付図面を参照して高純度オキシモルホンの合成を示す。
【0105】
図1は、実施例3.2Dにより得られた水和オキシモルホン塩酸塩生成物についての粉末X線回折パターンである。
【0106】
〔実施例1.1A〕
オリパビンの14−ヒドロキシモルヒノンへの水酸化
オリパビン1kgを、攪拌しながら、ぎ酸2.76kg及び水0.53kgの入った反応容器に添加する。オリパビンが完全に溶解するまで攪拌を継続し、温度が20〜30℃の範囲を維持する。続いて、35重量%過酸化水素溶液0.36kgを添加し、反応混合物を3時間以上攪拌するとともに、温度を20〜35℃の範囲に維持する。反応容器を10℃に冷却し、希薄水酸化アンモニウム7.12リットルをゆっくりと添加するとともに、反応混合物を40℃未満に維持する。必要ならば、反応混合物のpHを、必要に応じてさらに希薄水酸化アンモニウム溶液又は塩酸を添加して8〜10の範囲に調整し、攪拌を3〜5時間継続する。
【0107】
生成物である14−ヒドロキシモルヒノンの沈殿が形成し、その沈殿を濾取する。沈殿を水で無色になるまで洗浄した後、湿気のあるケーキとなるまで乾燥し、採集して次の段階に使用する。
【0108】
〔実施例1.1B〕
オキシモルホン基剤の形成
水素添加容器に、水(kgリットル)と酢酸0.73kgを入れてから、実施例1.1Aと同様にして調製した14−ヒドロキシモルヒノン1kgを添加し、混合物を溶液が透明になるまで攪拌する。炭素に担持された湿った10%パラジウム触媒40gを窒素流下で添加し、水素を35〜40psi(2.41〜2.76バール)で供給する。温度を、水素吸収が停止するまで30±5℃に維持し、次に容器を35〜40psi(2.41〜2.76バール)及び30±5℃に3〜4時間維持する。反応容器を25℃未満に冷却し、試料をHPLC分析して、14−ヒドロキシモルヒノンを確認する。HPLC分析で検出された14−ヒドロキシモルヒノン面積が>0.1%である場合には、水素添加を繰り返す。
【0109】
反応が完了したことを確認したら、触媒を濾去し、濾液のpHを水酸化アンモニウム溶液を用いて9に調製する。生成物が沈殿し、濾過により単離し、真空乾燥する。生成物をジクロロメタン/メタノール(9:1v/v)に溶解し、フロリジルにスラリー化し、濾過し、濾液を蒸留して、n−プロパノールに交換する。n−プロパノール混合物を冷却する。生成物が沈殿し、それを濾取する(収率66%)。生成物試料を、HPLC分析してα,β−不飽和ケトン検出したところ、測定面積で0.51%含有することが判明する。
【0110】
〔実施例1.1C〕
高純度オキシモルホン塩酸塩の形成
反応容器に、実施例1.1Bと同様の方法により調製したオキシモルホン基剤1kgを、無水アルコール2.05kg及び水0.66kgとともに入れる。混合物を60±2℃に加熱し、攪拌してスラリーを形成する。濃塩酸0.66kg、水0.24kg及び無水アルコール0.31kgから調製した塩酸溶液を、オキシモルホン基剤スラリーに添加し、pHが<1.0であることを確認する。炭素に担持された10%パラジウム触媒水湿潤ペースト40gを、窒素流下、反応混合物に添加し、混合物を35±5psi(2.41バール)で20時間水素添加するとともに、この間、温度を65±3℃に維持する。反応混合物を、熱い状態で、セライト及び0.2μmポリッシュフィルターで濾過する。濾液を0〜5℃で2〜3時間かけて冷却し、さらに2時間攪拌して、オキシモルホン塩酸塩沈殿を得る。沈殿を無水アルコールで洗浄後乾燥する。収率は、80%である。
【0111】
生成物の試料を、HPLC分析してα,β−不飽和ケトンを検出すると、6.2ppmである。
【0112】
〔実施例1.2A〕
オリパビンの14−ヒドロキシモルヒノンへの水酸化
オリパビン40gを、攪拌しながら、水30g及びぎ酸85gを入れた反応容器に添加し、オリパビンが完全に溶解するまで攪拌を続ける。温度を20〜30℃の範囲に維持する。続いて、30重量%過酸化水素溶液17.72gを添加し、その反応混合物を3時間以上攪拌し、この間、温度を20〜35℃の範囲に維持する。反応混合物を<20℃に冷却し、希薄水酸化アンモニウム335mLをゆっくりと添加する。この間、反応混合物の温度を32℃未満に維持する。必要ならば、反応混合物のpHを、必要に応じてさらに希薄水酸化アンモニウム溶液又は塩酸を添加して9.0に調整し、攪拌を20℃で2時間継続し、4〜5℃で2時間継続する。
【0113】
14−ヒドロキシモルヒノンの沈殿が形成する。この沈殿を濾取する。濾取した沈殿を水で洗浄した後、乾燥して湿ったケーキを得て、採集して次の段階で使用する。
【0114】
〔実施例1.2B〕
オキシモルホン基剤の形成
水素添加容器に、水148g、酢酸90.6g、実施例1.2Aと同様にして調製した湿った14−ヒドロキシモルヒノン(水分48%)250gを入れる。混合物を、溶液が透明になるまで攪拌し、ペーストの形態の炭素に担持された10%パラジウム触媒(乾燥重量)1.34gを、窒素流下添加する。水素添加容器を窒素及び水素でフラッシュした後、反応混合物を、30℃及び35psi(2.41バール)で5時間水素添加する。製造過程でのHPLC分析から、14−ヒドロキシモルヒノン面積が0.07%であることが分かる。
【0115】
反応が完了したことが確認されたら、触媒をセライトパッドで濾去し、セライトケーキを水25mLで洗浄する。濾液を0〜5℃に冷却し、pHを濃水酸化アンモニウムと水の1:1混合物(V/V)により9.5±0.5に調整する。生じた沈殿を0〜5℃で1時間攪拌し、濾過により単離する。粗生成物を、真空オーブン中50℃で乾燥して淡いベージュ色の固体113g(収率86.9%)を得る。生成物の試料をHPLCで分析してα,β−不飽和ケトンを検出し、測定面積で0.27%含有していることが分かる。
【0116】
粗生成物であるオキシモルホン基剤113gを、ジクロロメタン/メタノール(9:1、 v/v)1.13Lに入れる。フロリジル113gを溶液に添加し、混合物を12時間攪拌する。混合物をフロリジル113gのパッドにより濾過し、フロリジルケーキをジクロロメタン/メタノール120mLですすぐ。溶媒を留去した後、n−プロパノールに切り換える。バッチを0〜5℃に冷却し、1時間攪拌してオキシモルホン基剤を沈殿させる。この沈殿を、濾取し、冷n−プロパノールで洗浄し、真空オーブン中で乾燥して白色固体67.2g(59.47%)を得る。
【0117】
生成物の試料をHPLCで分析してα,β−不飽和ケトンを検出したところ、測定面積で0.027%含有していることが分かる。
【0118】
〔実施例1.2C〕
高純度オキシモルホン塩酸塩の形成
反応容器に、実施例1.2Bと同様の方法により調製したオキシモルホン基剤50.1gを、無水アルコール120gとともに入れる。混合物を60±2℃に加熱し、攪拌してスラリーを形成する。濃塩酸32.7g、水33.6gから調製した塩酸溶液を、オキシモルホン基剤スラリーに添加し、pHが<1.0であることを確認する。炭素に担持された10%パラジウム触媒水湿潤ペースト2.0gを、窒素流下、反応混合物に添加し、混合物を35psi(2.41バール)で20時間水素添加するとともに、この間、温度を65℃に維持する。反応混合物を、熱い状態で、セライトで濾過する。濾液を2〜3時間かけて0〜5℃に冷却し、さらに2時間攪拌して、オキシモルホン塩酸塩沈殿を得る。沈殿を濾取し、無水アルコールで洗浄後乾燥して、白色結晶を収率77%で得る。
【0119】
生成物の試料を、HPLC分析してα,β−不飽和ケトンを検出すると、1.1ppmであることが分かる。
【0120】
上記の方法は、当業者により、生成物であるオキシモルホン塩酸塩の高純度を維持しながら変更することができ、このような変形例の実施例を以下に示す。
【0121】
〔実施例2.1B〕
14−ヒドロキシモルヒノンのオキシモルホン基剤への還元
水素添加容器に水2.5kg及び酢酸0.73kgを入れ、14−ヒドロキシモルヒノン1kgを添加する。透明溶液が得られるまで反応混合物を攪拌し、それから炭素に担持された湿った10%パラジウム触媒40gを窒素流下で添加する。水素を、35〜40psi(2.41〜2.76バール)で供給する。温度を、水素吸収が停止するまで30±5℃に維持した後、容器を35〜40psi(2.41〜2.76バール)及び30±5℃で3〜4時間維持する。反応容器を25℃未満に冷却し、試料をHPLC分析して14−ヒドロキシモルヒノンを検出する。HPLC分析により検出された14−ヒドロキシモルヒノンの面積が>0.1%である場合には、水素添加を繰り返す。
【0122】
反応が完了したことが確認されたら、触媒を濾去し、ジクロロメタン/メタノール(9:1 v/v)を濾液に添加し、混合物に水酸化アンモニウム溶液を添加することによりpH9〜10に調整する。ジクロロメタン/メタノール相を分離し、フロリジルにスラリー化し、濾過し、濾液を蒸溜し、n−プロパノールに交換する。n−プロパノール混合物を冷却する。生成物が沈殿する。この沈殿を、濾取する(収率73%)。生成物の試料をHPLC分析してα,β−不飽和ケトンを検出すると、面積で0.32%含有していることが分かる。
【0123】
〔実施例2.2B〕
14−ヒドロキシモルヒノンのオキシモルホン基剤への還元
水素添加容器に、水35g、酢酸17g及び実施例1.2Aで調製した14−ヒドロキシモルヒノン38.08gを入れる。反応混合物を透明溶液となるまで攪拌した後、炭素に担持された湿った5%パラジウム触媒1.8gを窒素流下添加する。水素を、35〜40psi(2.41〜2.76バール)で供給する。温度を、水素の吸収が停止するまで30±5℃に維持する。次に、容器を、35〜40psi(2.41〜2.76バール)及び30±5℃で4時間維持する。反応容器を25℃未満に冷却し、試料をHPLCで分析して14−ヒドロキシモルヒノンを検出する。その結果、HPLC分析で検出した14−ヒドロキシモルヒノン面積は、0.26%である。
【0124】
反応が完了したことを確認したら、触媒を濾去し、得られたケーキを水15mLで洗浄する。ジクロロメタン/メタノール(9:1、v/v)180mLを濾液に添加する。濃水酸化アンモニウムを添加することにより、pHを9〜10に調整する。ジクロロメタン/メタノール層を分離し、フロリジル約20gでスラリー化することにより精製する。スラリーを濾過し、濾液を蒸溜し、n−プロパノールに交換する。混合物を 0〜5℃に冷却し、1〜2時間攪拌してオキシモルホン基剤沈殿を生じさせ、その沈殿を濾過により単離する。次に、オキシモルホン基剤をn−プロパノールでスラリー化して生成物を得る(収率74%)。生成物の試料をHPLC分析してα,β−不飽和ケトンを検出すると、面積で0.32%含有していることが分かる。
【0125】
〔実施例2.2C〕
高純度オキシモルホン塩酸塩の形成
反応容器に、実施例2.2Bで同様にして調製したオキシモルホン基剤2.5gを、無水アルコール7.5mL、水2.5g及び濃塩酸1.66gとともに入れる。混合物を50〜60℃に加熱して溶液を得る。pHが<1.0であることを確認する。炭素に担持された10%パラジウム触媒水湿潤ペースト0.111gを反応混合物に窒素流下で添加する。混合物を35±5psi(2.41バール)で21時間水素添加する。この間、温度は、65±3℃に維持する。反応混合物を、熱い状態で、0.45μmフィルターにより濾過する。濾液を、2〜3時間かけて、0〜5℃に冷却し、さらに2時間攪拌してオキシモルホン塩酸塩沈殿を形成する。沈殿を濾取し、冷無水アルコールで洗浄し、真空乾燥して白色結晶を得る(収率77%)。
【0126】
生成物の試料をHPLC分析してα,β−不飽和ケトンを検出すると、2.8ppm含有されていることが分かる。
【0127】
〔実施例3.1B〕
14−ヒドロキシモルヒノンのオキシモルホン塩酸塩への還元
オキシモルヒノン遊離塩基の形成後、上記した手順をおこなう。しかしながら、この場合、ジクロロメタン/メタノール溶液から遊離塩基を単離する代わりに、0.35容積当量の3N塩酸を添加(ジクロロメタン/メタノール溶液の容積に対して)し、反応混合物を攪拌し、放置し、水層(生成物を含有)を有機層から分離する。水層を、真空蒸溜して容積の約35%を除去した後、残りの溶液を2時間かけて20〜25℃に冷却し、1〜2時間攪拌し、0〜5℃に冷却し、2〜3時間攪拌する。攪拌中に形成する白色固体を濾取し、冷イソプロパノールで洗浄する。収率は64%であり、生成物はα,β−不飽和ケトンを0.34%含有する。
【0128】
〔実施例3.1C〕
オキシモルホン塩酸塩の精製
実施例3.1Bの生成物を原料として用いること以外は実施例1.1Cと同様の方法により、精製オキシモルホン塩酸塩を、収率92%で得る。この生成物中のα,β−不飽和ケトン含量は、検出できない程度である。
【0129】
〔実施例3.2C〕
高純度オキシモルホン塩酸塩の調製
反応容器に、実施例3.1Bで調製したオキシモルホン塩酸塩5.05gを、無水アルコール13.5mL、水4.5mL及び濃塩酸1.51gとともに入れる。混合物を、50〜60℃に加熱して溶液を得る。pHが<1.0であることを確認する。炭に担持された10%Pd触媒水湿潤ペースト0.21gを、反応混合物に窒素流下で添加する。混合物を、35±5psi(2.41バール)で20時間水素添加する。この間、温度を65±3℃に維持する。反応混合物を、熱い状態で、0.45μmフィルターで濾過する。濾液を、2〜3時間かけて、0〜5℃に冷却し、さらに2時間攪拌して沈殿を形成する。沈殿を濾取し、冷無水アルコールで洗浄した後、乾燥する。収率は、92%である。
【0130】
生成物の試料をHPLC分析すると、α,β−不飽和ケトン含量が検出できない程度あることが分かる。
基本プロセス工程を変更することなく、原料についてのプロセス工程を少し変更すること、例えば、このような原料を単離するか又は単離しないことにより、そして精製オキシモルホン塩酸塩への最終工程についての本発明に不可欠な還元要件を利用することにより、α,β−不飽和ケトン含量レベルが3.8ppm、1.7ppm、6.2ppm、6.9ppm、2.8ppm、3.1ppm、0.9ppm、6.0ppm及び別の例では検出不可能レベル、すなわちゼロである他の生成物が得られた。
【0131】
〔実施例3.2D〕
オキシモルホン塩酸塩の水和
乾燥皿に、エタノールを約5〜13重量%含有する実施例1.1C、1.2C、2.2C、3.1C又は3.2Cで同様に調製したオキシモルホン塩酸塩を入れる。試料を、水100mL入っている皿とともに真空オーブンに入れる。真空24〜29Hgとし、オーブンを20〜40℃で24時間維持する。水分約10〜13重量%のエタノール不含又はエタノール低含量(約0.04重量%)生成物が得られる。試料により吸収された水は、真空オーブン中50〜55℃で除去してよい。生成物のKFが6〜8重量%になったら乾燥プロセスを停止する。最終的な水和オキシモルホン塩酸塩は、一致したX線回折パターンを有する均一な多形体である。
【発明の分野】
【0001】
本発明は、鎮痛薬、より詳細にはアヘンオキシモルホンを塩酸塩として製造する改善された方法に関する。
【0002】
一般的に塩酸塩の形態で投与されるオキシモルホンは、中程度〜激しい痛みを軽減するための強力な半合成アヘン系鎮痛薬であり、1959年以来認可されているものである。オキシモルホンは、注射液、坐剤、錠剤又は徐放錠剤として投与されることができる。高純度のオキシモルホン及びその合成方法を開発することが望ましい。
【0003】
ケシから単離した化合物又はそれから得た化合物から、例えば、モルヒネ、テバイン又はオキシコドンを原料として、オキシモルホンを合成するいくつかの方法が知られている。α,β−不飽和ケトンによる汚染度が低いオキシモルホンを形成できる方法が必要とされている。本発明によれば、改善されたオキシモルホン生成物及びこのようなオキシモルホンの製造方法が提供される。
【0004】
米国特許第7,129,248号は、14−ヒドロキシコデイノン含量が100ppm超であるオキシコドンを水素添加することにより、14−ヒドロキシコデイノン含量が25ppm未満であるオキシコドン塩酸塩を製造する方法を開示している。米国特許第7,129,248号に教示されているオキシコドンへの合成経路では、テバインを原料とし、中間生成物として14−ヒドロキシコデイノンを製造し、テバインの過酸化から得られる副生成物として8,14−ジヒドロキシ−7,8−ジヒドロコデイノンが生成する。オキシコドン遊離塩基のその塩酸塩への転化中、副生成物が酸触媒下脱水し、14−ヒドロキシコデイノンに転化することがある。したがって、最終的なオキシコドン塩酸塩は、未反応14−ヒドロキシコデイノン、及び副生成物である8,14−ジヒドロキシ−7,8−ジヒドロコデイノンから得られた14−ヒドロキシコデイノンを含んでいる。水素添加工程により、14−ヒドロキシコデイノン含量が少なくとも100ppmから25ppm未満に減少するとしている。
【0005】
本発明によれば、α,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩生成物が提供される。また、本発明によれば、オキシモルホン塩酸塩を精製してα,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩生成物を得る方法が提供される。この方法は、原料のオキシモルホン塩酸塩を、強酸水及びアルコール溶媒中60〜70℃の温度範囲で、水素ガスにより還元することを含む。還元時間は少なくとも20時間が好適である。しかしながら、別の態様によれば、還元は1〜20時間実施する。
【図面の簡単な説明】
【0006】
【図1】実施例3.2Dにより得られた水和オキシモルホン塩酸塩生成物についての粉末X線回折パターンである。
【発明を実施するための形態】
【0007】
好ましくは、溶媒は、エタノール/水である。しかしながら、イソプロパノール及びn−プロパノール等の他の水混和性アルコールを使用してもよい。反応媒体は、少なくとも2当量の塩酸を含有させることにより強酸性とすることが好ましい。pHは、1未満が望ましい。
【0008】
反応温度は、約65℃に維持するのが最も好ましい。水素は、反応容器に2.41バールの圧力で供給するのが都合がよい。
【0009】
本発明の方法は、原料であるオキシモルホン塩酸塩の非常に高含量(0.3〜0.5%程度又は3,000〜5,000ppm程度)のα,β−不飽和ケトンを、10ppm未満、そして多くの場合、検出不能なレベル(HPLCにより)まで減少することができた。
【0010】
原料であるオキシモルホン塩酸塩は、単離物質でもよいし、非単離物質でもよい。オキシモルホン塩酸塩は、オキシモルホン遊離塩基を塩酸及びアルコール/水反応媒体の存在下で加熱することにより得られたものが望ましい。好適な温度は、60〜70℃である。反応媒体は本発明の方法の還元に理想的であるので、一般的にオキシモルホン塩酸塩を単離する必要がないことが明らかである。しかしながら、原料であるオキシモルホン塩酸塩は、反応媒体から単離してもよいし、別の源から得てもよい。
【0011】
オキシモルホン遊離塩基自体は、14−ヒドロキシモルヒノンの還元により調製することが好ましい。この還元は、一段又は二段プロセスで実施できる。好ましくは、この還元は、ガス状水素及び炭素に担持されたパラジウム触媒を用いて酢酸中で実施する。好ましい温度は、30℃の程度である。塩基は、アンモニア水(NH4OH)を添加することにより析出する。
【0012】
この還元は、メタノール、フロラシル(Florasil)及びn−プロパノールにジクロロメタンを加えた反応媒体の存在下でおこなってもよい。
【0013】
14−ヒドロキシモルヒノン自体は、過酸化水素を用いて、蟻酸の存在下、オリパビンを水酸化することにより最も好適に調製される。
【0014】
オリパビンは、ケシがら(ケシ滓、ケシ茎根)から抽出できる公知化合物である。テバイン高収量株であるタスマニアで発育した株は、通常よりも高収量のオリパビンも産生する。
【0015】
本発明の方法は、非常に柔軟性があり、数多くの反応工程を中間生成物を単離することなく実施することができる。しかも、この場合、オリパビンからの全収率(50%程度)を高い状態に保持するとともに、純度を著しく高く保持することができる。好条件下では、α,β−不飽和ケトンは、HPLC等の通常の手段によってはその存在が検出できない程度である。しかしながら、当業者は、汚染レベルを10ppm未満に容易にできる。本発明の方法は、キログラムスケールでうまく実施することができた。
【0016】
α,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩は、例えば、α,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩を通常の賦形剤、すなわち、薬学的に許容される有機又は無機担体物質と混ぜて混合剤とすることにより、医薬剤形に含有させることができる。経口製剤の場合、剤形により、活性成分が除放性とすることができる。好適な薬学的に許容される担体には、アルコール、アラビア・ゴム、植物油、ベンジルアルコール、ポリエチレングリコール、ゲル、炭水化物、例えば、ラクトース、アミロース又はでんぷん、ステアリン酸マグネシウム、タルク、ケイ酸、粘性パラフィン、香油、脂肪酸モノグリセリド及びジグリセリド、ペンタエリスリトール脂肪酸エステル、ヒドロキシーメチルセルロース、ポリビニルピロリドンなどがあるが、これらには限定されない。製剤は、殺菌し、必要に応じて補助剤、例えば、滑剤、崩壊剤、防腐剤、安定剤、湿潤剤、乳化剤、浸透圧緩衝液に影響を与える塩、着色物質、香味物質及び/又は芳香物質等と混合できる。経口用組成物は、当該技術分野において公知の方法により調製でき、このような組成物は、錠剤の製造に好適である不活性、非毒性の薬学的に許容される賦形剤からなる群から選択される一種以上の薬剤を含有していてもよい。このような賦形剤には、例えば、不活性希釈剤、例えば、ラクトース;造粒及び崩壊剤、例えば、コーンスターチ;結合剤、例えば、でんぷん;並びに滑剤、例えば、ステアリン酸マグネシウムなどがある。錠剤は、被覆されていないものでもよく、又は外見よくしたり又は有効成分の放出を遅くする公知の方法により被覆されていてもよい。また、経口用製剤は、硬質ゼラチンカプセル剤として提供してもよい。この場合、有効成分は不活性希釈剤と混合する。本発明の経口用剤形は、錠剤(除放性及び/又は即時放出性)、トローチ、ロゼンジ、散剤又は顆粒剤、硬質又は軟質カプセル剤、微粒子(例えば、マイクロカプセル剤、微小球等)、バッカル錠、液剤、懸濁剤等の形態でよい。
【0017】
本発明のある実施態様によれば、ヒト患者に本明細書に記載の剤形を投与することにより痛みを治療する方法が提供される。
【0018】
剤形が経口用であるとき、本発明の剤形は、α,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩約1mg〜約40mgを含有する。特に好ましい投与量は、約5mg、約10mg、約20mg又は約40mgであるが、他の投与量を使用してもよい。また、α,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩は、好適な薬学的に許容される賦形剤を配合して、α,β−不飽和ケトン含量が10ppm未満の徐放性製剤としてもよい。このような製剤は、米国特許第2003/129230A1号、米国特許第2003/129234A1号及び米国特許第2003/157167A1号に準じて調製できる。
【0019】
α,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩は、当業者に知られている好適な錠剤、被覆錠剤又は多粒子製剤における徐放性経口製剤として製剤化でてきる。
【0020】
徐放性剤形は、徐放性物質を、マトリックスにそのオキシモルホン塩とともに含有させて含んでいてもよい。
【0021】
徐放性剤形は、必要に応じて、α,β−不飽和ケトン含量10ppm未満のオキシモルホン塩酸塩を含有する粒子を含んでいてもよい。ある態様によれば、粒子は、直径が約0.1mm〜約2.5mm、好ましくは約0.5mm〜約2mmである。好ましくは、粒子は、活性成分を水媒体中で徐放できる物質の被膜が塗布されているものである。被膜状コートは、他の記載の特性との組み合わせで所望の放出特性が得られるように選択する。本発明の徐放性コーティング製剤は、好ましくは、平滑且つ外観のよい強度のある連続被膜を製造することができ、顔量及び他のコーティング添加剤を担持することができ、非毒性、不活性且つ不粘着性のものである。
【0022】
塗布ビーズ
本発明のある実施態様によれば、nu pariel18/20ビーズなどの不活性医薬ビーズに疎水性物質を塗布する。その後、複数の得られた固体状徐放性ビーズを、摂取され、環境流体、例えば、胃液又は溶解媒体により接触されたときに、有効な徐放投与量となるのに十分な量で、ゼラチンカプセル剤に入れることができる。
【0023】
本発明の徐放性ビーズ製剤は、例えば、摂取され、胃液及び次に腸液に暴露されたときに本発明の活性成分をゆっくりと放出する。本発明の製剤の徐放プロフィールは、例えば、疎水性物質で上塗りする量を変更することにより、可塑剤を疎水性物質に添加する方法を変更することにより、疎水性物質に対する可塑剤の量を変更することにより、追加の成分又は賦形剤を含有させることにより、又は製造方法を変更することにより、変更することができる。最終生成物の溶解プロフィールも、例えば、遅延コーティングの厚さを増加するか又は減少させることにより変更することができる。
【0024】
本発明の薬剤を塗布した球状物又はビーズは、例えば、薬剤を水に溶解し、その後溶液を基材、例えば、nu pariel18/20ビーズ上にウスター(Wuster)インサートを用いて噴霧することにより調製される。必要に応じて、ビーズに塗布する前に、追加の成分も添加して、ビーズへの活性成分の結合の促進及び/又は溶液の着色などをおこなう。例えば、着色剤を添加してあるか又は添加していないヒドロキシプロピルメチルセルロース等を含む生成物(例えば、Colorcon社から市販されているOpadry(商品名))を、溶液に添加し、その溶液を混合(例えば、約1時間)してから、その溶液をビーズ上に適用してもよい。得られた塗布基材(この場合ビーズ)の上に、次に必要に応じてバリア剤を塗布して、活性成分を疎水性徐放性コーティングから分離してもよい。好適なバリア剤の一例として、ヒドロキシプロピルメチルセルロースを含むものがある。しかしながら、当該技術分野において公知のいずれの膜形成剤を用いてもよい。バリア剤は、最終製品の溶解速度に影響しないことが好ましい。
【0025】
次にビーズの上に、疎水性物質の水性分散液を塗布してもよい。疎水性物質の水性分散液は、好ましくはさらに有効量の可塑剤、例えば、クエン酸トリエチルを含む。予め調製したエチルセルロースの水性分散液、例えば、Aquacoat(商品名)又はSurelease(商品名)を使用してもよい。Surelease(商品名)を使用する場合、可塑剤を別途添加する必要はない。別法として、予め調製したアクリル系ポリマーの水性分散液、例えば、Eudragit(商品名)を使用することができる。
【0026】
好ましくは、本発明のコーティング溶液は、膜形成剤、可塑剤及び溶媒系(すなわち、水)の他に、着色剤を含有して、良好な外観及び製品の識別性を付与する。疎水性物質の水性分散液を加える代わりに、又は疎水性物質の水性分散液に加えて、着色剤を治療活性剤の溶液に添加してもよい。例えば、着色剤を、Aquacoat(商品名)に、アルコール又はプロピレングリコール系着色剤分散液、粉砕アルミニウムレーキ及び二酸化チタン等の乳白剤を用いて、着色剤を剪断力により水溶性ポリマー溶液に添加し、その後低剪断力を用いて可塑化したAquacoat(商品名)に添加してもよい。別法として、本発明の製剤を着色するいずれかの好適な方法を用いてもよい。アクリル系ポリマーの水性分散液を使用するとき、製剤を着色するのに好適な成分としては、二酸化チタン、着色顔料、例えば、酸化鉄顔料などがある。しかしながら、顔料を含有させることにより、コーティングの効果を顕著に妨害することがある。
【0027】
可塑化した疎水性物質は、当該技術分野おいて公知のいずれかの好適な噴霧装置を用いて噴霧することにより、薬剤を含む基材上に適用してもよい。好ましい方法では、Wurster流動床装置を使用する。Wurster流動床装置では、下部から注入された空気ジェットにより、コア物質が流動化し、アクリル系ポリマーコーティングをコア物質上に噴霧しながら乾燥をおこなう。塗布された基材が水溶液、例えば、胃液にさらされたときに、薬剤が所定の徐放性を示すのに十分な量の疎水性物質を適用することができる。疎水性物質を塗布した後、必要に応じて、さらにOpadry(商品名)等の膜形成剤をビーズにオーバーコーティングする。このオーバーコートは、ビーズの凝集があるとしてもその凝集を実質的に減少させるために設けられる。
【0028】
本発明の除放性製剤から薬剤を放出させることは、一種以上の放出調整剤の添加、又はコーティングを介して一つ以上の経路を設けることによりさらに所望の速度に変更、すなわち、調整できる。疎水性物質の水溶性物質に対する比は、他の因子のうち、必要放出速度と選択物質の溶解度特性により求める。
【0029】
孔形成剤として機能する放出調整剤は、有機物質でも、無機物質でもよく、使用環境において、コーティングから溶解、抽出又は侵出することができる物質などがある。孔形成剤は、ヒドロキシプロピルメチルセルロース等の一種以上の親水性物質を含むことができる。
【0030】
また、本発明の除放性コーティングは、でんぷん及びゴム等の浸食促進剤を含むことができる。
【0031】
また、本発明の除放性コーティングは、使用環境において微細孔性薄層を形成するのに有効な物質、例えば、ポリマー鎖にカーボネート基が繰り返している炭酸の線状ポリエステルからなるポリカーボネートを含むことができる。
【0032】
また、放出調整剤は、半浸透性ポリマーも含んでいてもよい。
【0033】
ある好ましい実施態様によれば、放出調整剤は、ヒドロキシプロピルメチルセルロース、ラクトース、ステアリン酸金属塩及びこれらの混合物から選択される。
【0034】
本発明の除放性コーティングは、少なくとも一つの経路、オリフィス等を備えた出口手段を備えていてもよい。この経路は、米国特許第3,845,770号、米国特許第3,916,899号、米国特許第4,063,064号及び米国特許第4,088,864号に開示てれているような方法により形成してもよい。
【0035】
マトリックス製剤
本発明の他の実施態様によれば、除放性製剤は、必要に応じて本明細書に記載の除放性コーティングを有するマトリックスにより提供される。除放性マトリックスに含有させるのに好適な物質は、マトリックス形成に使用される方法によって異なることができる。
【0036】
例えば、α,β−不飽和ケトン含量が10ppm未満のオキシモルホン塩酸塩の他にマトリックスは、親水性及び/又は疎水性物質、例えば、ゴム、セルロースエーテル、アクリル樹脂、タンパク質由来物質などを含んでいてもよい。含有される物質は、これらには限定されず、薬剤に除放性を付与させることのでき、且つ溶融する(又は押出に必要とする程度まで軟化する)いずれの薬学的に許容される疎水性物質又は親水性物質も、本発明において使用してもよい。
【0037】
消化性長鎖(C8−C50、とりわけC12−C40)置換又は非置換炭化水素、例えば、脂肪酸、脂肪アルコール、脂肪酸のグリセリルエステル、鉱油、植物油、ろう及びステアリ
ルアルコール;並びにポリアルキレングリコールなどがあげられる。これらのポリマーのうち、アクリル系ポリマー、とりわけEudragit(商品名)、セルロースエーテル、とりわけヒドロキシアルキルセルロース及びカルボキシアルキルセルロースが好ましい。経口剤は、少なくとも一種の親水性又は疎水性物質を1%〜80%(重量)含有してもよい。
【0038】
疎水性物質が炭化水素であるとき、炭化水素の融点は好ましくは25℃〜90℃である。長鎖炭化水素物質のうち、脂肪(脂肪族)アルコールが好ましい。経口剤は、少なくとも一種の消化性長鎖炭化水素を60%(重量)以下を含有してもよい。
【0039】
好ましくは、経口剤は、少なくとも一種のポリアルキレングリコールを60%(重量)以下含む。
【0040】
疎水性物質は、好ましくはアルキルセルロース、アクリル酸ポリマー、メタクリル酸ポリマー及びコポリマー、セラック、ゼイン、水素添加ひまし油、水素添加植物油又はこれらの混合物からなる群から選択される。本発明のある好ましい実施態様によれば、疎水性物質は、アクリル酸コポリマー、メタクリル酸コポリマー、メチルメタクリレート、メチルメタクリレートコポリマー、エトキシエチルメタクリレート、シアノエチルメタクリレート、アミノアルキルメタクリレートコポリマー、ポリ(アクリル酸)、ポリ(メタクリル酸)、メタクリル酸アルキルアミンコポリマー、ポリ(メチルメタクリレート)、ポリ(メタクリル酸)(無水物)、ポリメタクリレート、ポリアクリルアミド、ポリ(メタクリル酸無水物)、及びグリシジルメタクリレートコポリマーなどの薬学的に許容されるアクリル系ポリマーであるが、これらには限定されない。他の実施態様によれば、疎水性物質は、ヒドロキシアルキルセルロース、例えば、ヒドロキシプロピルメチルセルロース及びこれらの混合物などの物質から選択される。
【0041】
好ましい疎水性物質は、水不溶性であって、事実上顕著な親水性及び/又は疎水性を有するものである。好ましくは、本発明に有用な疎水性物質は、融点が約25℃〜約200℃、好ましくは約45℃〜約90℃である。具体的には、疎水性物質は、天然又は合成のろう、脂肪アルコール(例えば、ラウリルアルコール、ミリスチルアルコール、ステアリルアルコール、セチルアルコール又は好ましくはセトステアリルアルコール)、脂肪酸(以下のものをなどがあるが、これらには限定されない)、例えば、脂肪酸エステル、脂肪酸グリセリド(モノ−、ジ−及びトリ−グリセリド)、硬化油脂、炭化水素、通常ろう、ステアリン酸、ステアリルアルコール並びに炭化水素骨格を有する疎水性及び親水性物質を含んでいてもよい。好適なろうには、例えば、ミツロウ、グリコろう、カスターろう及びカルナウバろうなどがある。本発明の目的からして、ろう様物質は、通常室温で固体であり且つ融点が約25℃〜約100℃である物質として定義される。
【0042】
本発明に使用することができる好適な疎水性物質は、消化性長鎖(C8−C50、とりわけC12−C40)置換又は非置換炭化水素、例えば、脂肪酸、脂肪アルコール、脂肪酸のグリセリルエステル、鉱油、植物油及び天然ろう又は合成ろうなどがある。融点が25℃〜90℃の炭化水素が好ましい。長鎖炭化水素物質のうち、脂肪(脂肪族)アルコールが、ある実施態様においては好ましい。経口剤は、少なくとも一種の消化性長鎖炭化水素を60%(重量)以下含有してもよい。
【0043】
好ましくは、二種以上の疎水性物質を組み合わせてマトリックス製剤に含有させる。追加の疎水性物質を含有させる場合、天然又は合成ろう、脂肪酸、脂肪アルコール及びこれらの混合物から選択するのが好ましい。例えば、ミツロウ、カルナウバろう、ステアリン酸及びステアリルアルコールなどがある。しかしながら、これらには限定されない。
【0044】
一つの具体的な好適なマトリックスは、少なくとも一種の水溶性ヒドロキシアルキルセルロース、少なくとも一種のC12〜C36、好ましくはC14−C22の脂肪族アルコール及び必要に応じて少なくとも一種のポリアルキレングリコールを含む。少なくとも一種の ヒドロキシアルキルセルロースは、好ましくはヒドロキシ(C1〜C6)アルキルセルロース、例えば、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース及びとりわけ、ヒドロキシエチルセルロースである。本発明の経口剤中の少なくとも一種のヒドロキシアルキルセルロースの量は、とりわけ必要とされるオキシモルホン塩酸塩の正確な放出速度により決定する。少なくとも一種の脂肪族アルコールは、例えば、ラウリルアルコール、ミリスチルアルコール又はステアリルアルコールでよい。しかしながら、本発明の経口剤の特に好ましい実施態様よれば、少なくとも一種の脂肪族アルコールは、セチルアルコール又はセトステアリルアルコールである。本発明の経口剤中の少なくとも一種の脂肪族アルコールの量は、上記のように、オピオイドキシモルホンの正確な必要とする放出速度により決定される。また、これは、少なくとも一種のポリアルキレングリコールが経口剤に存在するか又は存在しないかにより異なる。少なくとも一種のポリアルキレングリコールが存在しない場合、経口剤は好ましくは少なくとも一種の脂肪族アルコールを20%〜50%(重量)を含有する。少なくとも一種のポリアルキレングリコールが経口剤に存在するときには、少なくとも一種の脂肪族アルコールと少なくとも一種のポリアルキレングリコールの合計重量は、好ましくは総投与量の20%〜50%(重量)である。
【0045】
一実施態様によれば、少なくとも一種のヒドロキシアルキルセルロース又はアクリル樹脂の少なくとも一種の脂肪族アルコール/ポリアルキレングリコールに対する比、例えば、少なくとも一種のヒドロキシアルキルセルロースの少なくとも一種の脂肪族アルコール/ポリアルキレングリコールに対する比(w/w)は、1:2〜1:4が好ましく、特に好ましくは1:3〜1:4である。
【0046】
少なくとも一種のポリアルキレングリコールは、例えば、ポリプロピレングリコールでよく、好ましくはポリエチレングリコールである。少なくとも一種のポリアルキレングリコールの数平均分子量は、好ましくは1,000〜15,000、とりわけ1,500〜12,000である。
【0047】
別の好適な除放性マトリックスは、アルキルセルロース(とりわけエチルセルロース)、 C12〜C36脂肪族アルコール及び必要に応じてポリアルキレングリコールを含む。
【0048】
別の好ましい実施態様によれば、マトリックスは、薬学的に許容される少なくとも二種の疎水性物質の組合わせを含む。
【0049】
除放性マトリックスは、上記成分の他に、医薬分野において一般的に用いられている好適な量の他の物質、例えば、希釈剤、滑剤、結合剤、造粒助剤、着色剤、香味剤、流動促進剤を含有してもよい。
【0050】
マトリックス−微粒子
本発明の固体状除放性経口剤の調製を容易にするために、当業者に公知のマトリックス製剤の調製方法を用いることができる。例えば、(a)少なくとも一種の水溶性ヒドロキシアルキルセルロースとα,β−不飽和ケトン含有量が10ppm未満であるオキシモルホン塩酸塩とを含む顆粒を形成し、(b)ヒドロキシアルキルセルロース含有顆粒を少なくとも一種のC12〜C36脂肪族アルコールと混合し、(c)必要に応じて顆粒を圧縮し、造形することにより、マトリックスに含有させることができる。好ましくは、顆粒は、ヒドロキシアルキルセルロース顆粒を水を用いて湿式造粒する。
【0051】
さらに他の別の実施態様によれば、球形化剤を活性成分とともに球形として、球状体を形成することができる。微結晶性セルロースは、好ましい球状化剤である。好適な微結晶性セルロースは、例えば、Avicel PH101(商品名、FMC社製)として販売されている物質である。このような実施態様によれば、球状体は、有効成分及び球状化剤の他に、結合剤を含んでいてもよい。低粘度水溶性ポリマー等の好適な結合剤が、医薬分野の当業者には周知である。しかしながら、ヒドロキシプロピルセルロース等の水溶性ヒドロシキ低級アルキルセルロースが好ましい。さらに又は別法として、球状体は、水不溶性ポリマー、とりわけアクリル系ポリマー、アクリル系コポリマー、例えば、メタクリル酸−エチルアクリレートコポリマー又はエチルセルロースを含んでいてもよい。このような実施態様では、除放性コーティングは、一般的に疎水性物質、例えば、(a)ろう(単独又は脂肪アルコールとの混合物として);(b)セラック又はゼインを含む。
【0052】
溶融押出マトリックス
除放性マトリックスは、溶融造粒法又は溶融押出法により調製することもできる。一般的に、溶融造粒法は、通常固体である疎水性物質、例えば、ろうを溶融し、粉末状医薬をそこに含有させることを含む。除放性製剤を得るためには、追加の疎水性物質、例えば、エチルセルロース又は水不溶性アクリル系ポリマーを溶融ろう疎水性物質に含有させることが必要なことがある。溶融造粒法により調製される除放性製剤の例が、米国特許第4,861,598号に記載されている。
【0053】
追加の疎水性物質は、一種以上の水不溶性ろう様熱可塑性物質(場合によっては、前記一種以上の水不溶性ろう様物質よりも疎水性度が小さい一種以上のろう様熱可塑性物質との混合物)を含んでいてもよい。一定の放出を維持するためには、製剤中の個々のろう様物質は、初期放出段階中実質的に非分解性であり、胃腸液に不溶でなければならない。有用な水不溶性ろう様物質は、水溶解度が約1:5,000(w/w)より低いものでよい。
【0054】
上記成分の他に、除放性マトリックスは、医薬分野において一般的に用いられている好適な量の他の物質、例えば、希釈剤、滑剤、結合剤、造粒助剤、着色剤、香味剤及び流動促進剤を含有してもよい。これらの追加の物質の量は、所望の製剤に所望の効果を付与するのに十分な量である。
【0055】
溶融押出多微粒子を含有する除放性マトリックスは、上記成分の他に、医薬分野において一般的に用いられている好適な量の他の物質、例えば、希釈剤、滑剤、結合剤、造粒助剤、着色剤、香味剤及び流動促進剤を、必要に応じて微粒子の約50重量%以下の量で含有してもよい。
【0056】
経口剤を調製するのに使用できる薬学的に許容される担体及び賦形剤の具体例が、Handbook of Pharmaceutical Excipients(医薬賦形剤ハンドブック)、 American Pharmaceutical Association (1986)に記載されている。
【0057】
溶融押出多微粒子
本発明による好適な溶融押出マトリックスの調製は、例えば、α,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩を、少なくとも一種の疎水性物質及び好ましくは追加の疎水性物質とともに、配合して均一混合物を得る工程を含んでいてもよい。次に、均一混合物を、混合物を少なくとも軟化し、混合物を十分に押しだせるのに十分な温度に加熱する。次に、得られた均一混合物を、押出してストランドを形成する。押出物は、好ましくは冷却し、当該技術分野において公知のいずれかの手段により切断して多微粒子にする。ストランドは、冷却し、切断して多微粒子にする。次に、多微粒子を、一回量に分ける。好ましくは、押出物は、直径が約0.1mm〜約5mmであり、治療に有効な薬剤を約8時間〜約24時間の間除放するものである。
【0058】
本発明の溶融押出の任意のプロセスには、疎水性物質、α,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩及び必要に応じて結合剤を押出機に直接計量して入れ;得られた均一混合物を加熱し;均一混合物を押出しストランドを形成し;均一混合物を含むストランドを冷却し;得られたストランドを切断して約0.1mm〜約12mmの大きさの粒子とし;そして得られた粒子を一回量に分けることを含む。本発明のこの実施態様では、比較的連続した製造手順が可能となる。
【0059】
押出機の開口又は出口ポートの直径を調整して、押出ストランドの厚さを変化させることもできる。さらに、押出機の出口部分は、丸い必要はなく、長円形、矩形等であってもよい。出てきたストランドは、熱線カッター、ギロチン等を用いてサイズを粒子サイズまで減少させることができる。
【0060】
溶融押出多微粒子系は、押出機の出口オリフィスに応じて、例えば、顆粒、球状体又はペレットの形態であることができる。本発明の目的のために、用語「溶融押出多微粒子」、「溶融押出多微粒子系」及び「溶融押出粒子」は、好ましくは一定範囲の同様なサイズ及び/又は形状内で、且つ一種以上の活性剤及び一種以上の賦形剤(好ましくは本明細書に記載されているような疎水性物質を含む)を含有する複数単位を意味する。この点において、溶融押出多微粒子は、長さが約0.1mm〜約12mmであり、直径が約0.1mm〜約5mmである。さらに、溶融押出多微粒子は、このサイズ範囲内のいずれの幾何学的形状であってもよい。別法として、押出物は、球状化工程の必要なく、単純に所望の長さに切断し、治療効果のある活性剤の一回量に分けてもよい。
【0061】
好ましい一実施態様によれば、経口剤は、カプセル剤内に、有効量の溶融押出多微粒子を含有させて調製する。例えば、複数の溶融押出多微粒子を、ゼラチンカプセルに、摂取され、胃液に接触したときに、有効な除放投与量となるのに十分な量を入れてもよい。
【0062】
別の好ましい実施態様によれば、多微粒子押出物の好適量を、標準的な方法を用いて、通常の打錠装置により圧縮して経口錠剤とする。当業者には、錠剤(圧縮及び成形したもの)、カプセル剤(硬質及び軟質ゼラチン)及びピルを製造するための種々の方法及び組成物が知られている。
【0063】
まだ別の好ましい実施態様によれば、押出物は、上記でより詳細に記載した米国特許第4,957,681号に記載したように、賦形して錠剤とすることができる。
【0064】
必要に応じて、上記で説明した除放性コーティング等の除放性コーティングにより、除放性溶融押出多微粒子系又は錠剤を被覆したり、又は多微粒子を含むゼラチンカプセル剤をさらに被覆することができる。このようなコーティングは、好ましくは十分な量の疎水性物質を含有して、約2%〜約30%の重量増加する。但し、オーバーコートは、とりわけ所望の除放速度に応じて大きく異なることができる。
【0065】
本発明の溶融押出単位剤形は、カプセル化する前に溶融押出粒子の組み合わせをさらに含むことができる。さらに、単位剤形は、即時放出用の即時放出剤もかなりの量で含むことができる。即時放出剤は、例えば、ゼラチンカプセル剤内に別個のペレットとして含有させてもいいし、又は剤形(例えば、除放性コーティング又はマトリックス系)の調製後、多微粒子表面上に塗布してもよい。また、本発明の単位剤形は、除放性ビーズとマトリックス多微粒子との組み合わせを含有させて所望の効果を得ることもできる。
【0066】
本発明の除放性製剤は、好ましくは、例えば、摂取され、胃液にさらされ、その後腸液にさらされたときにゆっくりと薬剤を放出する。本発明の溶融押出製剤の除放性プロフィールは、例えば、抑制剤、すなわち、疎水性物質の量を変化することにより、疎水性物質に対する可塑剤の量を変更することにより、追加の成分又は賦形剤を含有させることにより、製造方法を変更することによる等により変更できる。
【0067】
本発明の他の実施態様によれば、溶融押出物質は、α,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩を含有させることなく調製し、その後オキシモルホン塩酸塩を押出物に添加することができる。このような製剤は、典型的には薬剤を、押出したマトリックス物質とともにブレンドし、その後混合物を錠剤化して除放性製剤を得る。
【0068】
コーティング(被覆)
本発明の剤形は、必要に応じて放出の調節又は製剤の保護に好適な一種以上の物質を被覆してもよい。一実施態様によれば、コーティングを設けて、pH依存放出性又はpH非依存放出性とする。pH依存性コーティングは、少なくとも約8時間以上、好ましくは約12時間以上、約24時間以下、患者に対して鎮痛作用を与えることができる吸収プロフィールが得られるように、活性成分を胃腸(GI)管、例えば、胃又は小腸の所望の領域に放出する。pH非依存コーティングが望ましいときには、環境流体、例えば、GI管におけるpH変化とは無関係に最適に放出するようにコーティングを設計する。また、GI管の一つの所望領域、例えば、胃において投与量の一部分を放出し、投与量の残部をGI管の別の領域、例えば、小腸において放出する組成物を製剤化することもできる。
【0069】
pH依存性コーティングを利用して製剤化した本発明による製剤は、反復作用効果を付与することもできる。それにより、未保護薬剤を腸溶コーティング上に被覆して胃で放出され、一方、腸溶コーティングにより保護されている残部は胃腸管でさらに放出される。本発明で使用できるpH依存性コーティングとしては、セラック、セルロースアセテートフタレート、ポリビニルアセテートフタレート、ヒドロキシプロピルメチルセルロースフタレート及びメタクリル酸エステルコポリマー、ゼイン等がある。
【0070】
ある好ましい実施態様によれば、α,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩を含有する基材(例えば、錠剤コアビーズ、マトリックス粒子)に、(i) アルキルセルロース;(ii)アクリル系ポリマー;又は(iii)これらの混合物から選択された疎水性物質で被覆する。コーティングは、有機物又は水溶液又は分散液の形態で適用することができる。コーティングは、基材の約2%〜約25%の重量増加となるように適用して、所望の除放性プロフィールが得られるようにすることができる。水性分散液から得られるコーティングは、米国特許第5,273,760号、米国特許第5,286,493号、米国特許第5,324,351号、米国特許第5,356,467号及び米国特許第5,472,712号に詳細に記載されている。
【0071】
アルキルセルロースポリマー
アルキルセルロースなどのセルロース系物質及びポリマーにより、本発明によるビーズをコーティングするのに適している疎水性物質が得られる。例えば、一つの好ましいアルキルセルロース系ポリマーとしては、エチルセルロースがあげられる。但し、当業者には、他のセルロース及び/又はアルキルセルロースポリマーが、単一又は組み合わせにおいて、本発明による疎水性コーティングの全体又は一部分として容易に使用可能であることが理解できるであろう。
【0072】
アクリル系ポリマー
本発明の他の好ましい実施態様によれば、除放性コーティングを構成する疎水性物質は、薬学的に許容されるアクリル系ポリマー、例えば、アクリル酸コポリマー及びメタクリル酸コポリマー、メチルメタクリレートコポリマー、エトキシエチルメタクリレート、シアノエチルメタクリレート、ポリ(アクリル酸)、ポリ(メタクリル酸)、メタクリル酸アルキルアミドコポリマー、ポリ(メチルメタクリレート)、ポリメタクリレート、ポリ(メチルメタクリレート)コポリマー、ポリアクリルアミド、アミノアルキルメタクリレートコポリマー、ポリ(メタクリル酸無水物)及びグリシジルメタクリレートコポリマーがあげられる。疎水性物質は、これらには限定されない。
【0073】
ある好ましい実施態様によれば、アクリル系ポリマーは、一種以上のアンモニオメタクリレートコポリマーからなる。アンモニオメタクリレートコポリマーは、当該技術分野において周知であり、第4級アンモニウム基低含量のアクリル酸エステル及びメタクリル酸エステルの完全重合コポリマーとして記載されている。
【0074】
所望の溶解プロフィールを得るために、異なる物性、例えば、第4級アンモニウム基の中性(メタ)アクリル酸エステルに対するモル比が異なる二種以上のアンモニオメタクリレートコポリマーを含有させることが必要なことがある。
【0075】
ある種のメタクリル酸エステル型ポリマーは、本発明に使用することができるpH依存コーティングを形成するのに有用である。例えば、メタクリル酸コポリマー又は高分子メタクリレートとしても知られており、Rohm Tech社からEudragit(商品名)の商品名で市販されている、ジエチルアミノエチルメタクリレート及び他の中性メタアクリル酸エステルから合成されたコポリマー群がある。Eudragit(商品名)には、いくつかの異なる種類のものがある。例えば、Eudragit(商品名)Eは、酸性媒体で膨潤し、そこに溶解するメタクリル酸コポリマーの一例である。Eudragit(商品名)Lは、概略pH<5.7では膨潤せず、概略pH>6で溶解するメタクリル酸コポリマーである。Eudragit(商品名)Sは、概略pH<6.5では膨潤せず、概略pH>7で溶解する。Eudragit(商品名)RL及びEudragit(商品名)RSは、水膨潤性である。これらのポリマーにより吸収される水の量は、pH依存性であるが、Eudragit(商品名)RL及びRSで被覆された剤形は、pH非依存性である。
【0076】
ある好ましい実施態様によれば、アクリル系コーティングは、Rohm Pharma社からEudragit(商品名)RL30D及びEudragit(商品名)RS30Dの商品名で入手できる2種のアクリル樹脂ラッカーの混合物を含む。Eudragit(商品名)RL30D及びEudragit(商品名)RS30Dは、第4級アンモニウム基低含量(アンモニウム基の残りの中性(メタ)アクリル酸エステルに対するモル比は、Eudragit(商品名)RL30Dでは1:20、Eudragit(商品名)RS30Dでは1:40である)のアクリル酸エステル及びメタクリル酸エステルのコポリマーである。平均分子量は、約150,000である。コード表示RL(高浸透性)及びRS(低浸透性)は、これらの薬剤の浸透性を示す。Eudragit(商品名)RL/RS混合物は、水及び消化液に不溶性である。しかしながら、この混合物から形成したコーティングは、水溶液及び消化液において膨潤性及び浸透性である。
【0077】
本発明のEudragit(商品名)RL/RS分散液は、いずれかの所望の比で混合して、最終的に所望の溶解プロフィールを有する除放性製剤を得ることができる。所望の除放性製剤は、例えば、100%Eudragit(商品名)RL、50%Eudragit(商品名)RL及び、50%Eudragit(商品名)RS、又は10%Eudragit(商品名)RL及び90%Eudragit(商品名)RSから得られた遅延コーティングから得ることができる。もちろん、当業者には、例えば、Eudragit(商品名)Lなどの他のアクリル系ポリマーを使用することができるとわかるであろう。
【0078】
可塑剤
コーティングが疎水性物質の水性分散液を含む本発明の実施態様によれば、有効量の可塑剤を疎水性物質の水性分散液に含有させると、除放性コーティングの物性がさらに改善される。例えば、エチルセルロースはガラス転移温度が比較的高く、通常のコーティング条件下では可撓性被膜を形成しないので、可塑剤を、除放性コーティングを含有するエチルセルロースコーティングに、コーティングを塗料として使用する前に含有させることが好ましい。一般的に、コーティング溶液に含有させる可塑剤の量は、膜形成剤の濃度に基づいて決定され、例えば、最も一般的には膜形成剤の約1重量%〜約50重量%である。しかしながら、可塑剤濃度は、特定のコーティング溶液及び適用方法についての慎重な実験をすることによってのみ適切に決定できる。
【0079】
エチルセルロースに好適な可塑剤としては、例えば、水不溶性可塑剤、例えば、セバシン酸ジブチル、ジエチルフタレート、クエン酸トリエチル、クエン酸トリブチル及びトリアセチンなどがある。但し、他の水不溶性可塑剤(例えば、アセチル化モノグリセリド、フタレートエステル、ひまし油等)を使用することもできる。クエン酸トリエチルが、本発明のエチルセルロースの水性分散液にとってとりわけ好ましい可塑剤である。
【0080】
本発明のアクリル系ポリマーに好適な可塑剤としては、例えば、クエン酸エステル、例えば、クエン酸トリエチル、クエン酸トリブチル、ジブチルフタレート及び場合により1,2−プロピレングリコールがあげられるが、これらには限定されない。Eudragit(商品名)RL/RSラッカー溶液等のアクリル被膜から形成される被膜の弾性を高めるのに好適であることが分かった他の可塑剤には、ポリエチレングリコール、プロピレングリコール、ジエチルフタレート、ひまし油及びトリアセチンなどがある。クエン酸トリエチルが、本発明のエチルセルロースの水性分散液にとってとりわけ好ましい可塑剤である。
【0081】
少量のタルクを添加することも、処理中に水性分散液がくっつく傾向を減少するのに役立ち、研磨剤としての役割を果たすことができる。
【0082】
除放性浸透剤
また、本発明の除放性製剤は、浸透剤として調製することもできる。浸透剤は、好ましくは二重層コアを備えている。二重層コアは、薬物層(α,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩)及び送達層又は押圧層を備えている。ここで、二重層コアは、半浸透性壁により包囲されており、必要に応じて少なくとも一個の経路がその中に設けられている。
【0083】
本発明の目的で使用される表現「経路」は、開口、オリフィス、穴、細孔、多孔性要素を備えていて、それにより、α,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩が、繊維、毛細管、多孔性オーバーレイ、多孔性インサート、微細孔部材又は多孔性組成物を介して汲み上げられるか、拡散するか、又は移動する。また、経路は、使用流体環境において壁を浸食したり、また壁から滲出して少なくとも一つの経路を生じる化合物を含んでいてもよい。経路を形成する代表的な化合物には、壁における浸食性ポリグリコール酸又はポリ乳酸;ゼラチン状フィラメント;水除去性ポリ(ビニルアルコール);滲出性化合物、例えば、流体除去性細孔形成多糖類、酸、塩又は酸化物などがある。経路は、ソルビトール、スクロース、ラクトース、マルトース又はフルクトース等の化合物を壁から滲出させて、除放性次元細孔経路を形成することにより形成できる。一つ以上の表面上に間隔をあけて一つ以上の経路を有する剤形を製造できる。経路及び経路形成装置については、米国特許第3,845,770号、米国特許第3,916,899号、米国特許第4,063,064号及び米国特許第4,088,864号に開示されている。水性滲出により形成される放出性細孔としての大きさとし、賦形し、適合させた除放寸法を有して除放速度を有する放出細孔を設けた経路について、米国特許第4,200,098号及び米国特許第4,285,987号に開示されている。
【0084】
ある実施態様によれば、薬物層は、また少なくとも一種のポリマーヒドロゲルを含んでいてもよい。ポリマーヒドロゲルは、平均分子量が約500〜約6,000,000でよい。ポリマーヒドロゲルとしては、例えば、式(C6H12O5)nH2O(式中、nは3〜7,500である)で表されるマルトデキストリンポリマー(マルトデキストリンポリマーの数平均分子量が500〜1,250,000である);ポリ(アルキレンオキシド)、例えば、重量平均分子量が50,000〜750,000であるポリ(エチレンオキシド)及びポリ(プロピレンオキシド)、より具体的には重量平均分子量が100,000、200,000、300,000又は400,000のうちの少なくとも一つであるポリ(エチレンオキシド);アルカリカルボキシアルキルセルロース(但し、アルカリはナトリウム又はカリウムであり、アルキルはメチル、エチル、プロピル又はブチルである)(重量平均分子量:10,000〜175,000);及びメタアクリル酸及びエタアクリル酸を含むエチレン−アクリル酸のコポリマー(数平均分子量:10,000〜500,000)などがあげられるが、これらには限定されない。
【0085】
本発明のある実施態様によれば、送達層又は押圧層は、オスモポリマーを含む。オスモポリマーとしては、例えば、ポリアルキレンオキシド及びカルボキシアルキルセルロースからなる群から選択されるものがあげられるが、これらには限定されない。ポリアルキレンオキシドは、重量平均分子量が1,000,000〜10,000,000である。ポリアルキレンオキシドは、ポリメチレンオキシド、ポリエチレンオキシド、ポリプロピレンオキシド、平均分子量が1,000,000であるポリエチレンオキシド、平均分子量が5,000,000であるポリエチレンオキシド、平均分子量が7,000,000であるポリエチレンオキシド、平均分子量が1,000,000である架橋ポリメチレンオキシド及び平均分子量が1,200,000であるポリプロピレンオキシドからなる群から選択されたものでよい。典型的なオスモポリマーであるカルボキシアルキルセルロースは、アルカリカルボキシアルキルセルロース、カルボキシメチルセルロースナトリウム、カルボキシメチルセルロースカリウム、カルボキシエチルセルロースナトリウム、カルボキシメチルセルロースリチウム、カルボキシエチルセルロースナトリウム、カルボキシアルキルヒドロキシアルキルセルロース、カルボキシメチルヒドロキシエチルセルロース、カルボキシエチルヒドロキシエチルセルロース及びカルボキシメチルヒドロキシプロピルセルロースからなる群から選択されるものを含む。置換層に使用されるオスモポリマーは、半浸透性壁の浸透圧勾配を示す。オスモポリマーにより、流体が剤形に吸収され、それにより浸透ヒドロゲル(オスモゲルとしても知られている)として膨潤且つ膨張し、その結果、浸透ヒドロゲルがα,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩を浸透製剤から押し出す。
【0086】
押圧層は、オスマジェント及び浸透圧的に有効な溶質としても知られている一種以上の浸透圧的に有効な化合物を含んでいてもよい。これらは、環境流体を、例えば、胃腸管から剤形に吸収させ、置換層の送達動態に影響する。浸透圧的に活性な化合物としては、例えば、浸透性塩及び浸透性炭水化物からなる群から選択されるものがあげられる。オスマジェントの具体例としては、塩化ナトリウム、塩化カリウム、硫酸マグネシウム、リン酸リチウム、塩化リチウム、リン酸ナトリウム、硫酸カリウム、硫酸ナトリウム、リン酸カリウム、グルコース、フルクトース及びマルトースなどがあるが、これらには限定されない。
【0087】
押圧層は、必要に応じて、数平均分子量が9,000〜450,000であるヒドロキシプロピルアルキルセルロースを含んでいてもよい。ヒドロキシプロピルアルキルセルロースは、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルエチルセルロース、ヒドロキシプロピルイソプロピルセルロース、ヒドロキシプロピルブチルセルロース及び ヒドロキシプロピルペンチルセルロースからなる群から選択されたものが代表的なものである。
【0088】
押圧層は、必要に応じて非毒性着色剤又は色素を含んでいてもよい。着色剤又は色素としては、例えば、食品医薬品局着色剤(FD&C)、例えば、FD&C No.1青色色素、FD&C No.4赤色色素、赤色酸化第二鉄、黄色酸化第二鉄、二酸化チタン、カーボンブラック及びインジゴなどがあるが、これらには限定されない。
【0089】
また、押圧層は、必要に応じて酸化防止剤を含有して、成分の酸化を抑制するようにしてもよい。アスコルビン酸、パルミチン酸アスコルビル、ブチル化ヒドロキシアニソール、 ジ−tert−ブチル−4−ヒドロキシアニソールとトリ−tert−ブチル−4−ヒドロキシアニソールとの混合物、ブチル化ヒドロキシトルエン、イソアスコルビン酸ナトリウム、ジヒドログアレティック酸、ソルビン酸カリウム、硫酸水素ナトリウム、メタ重亜硫酸ナトリウム、ソルビン酸、アスコルビン酸カリウム、ビタミンE、4−クロロ−2,6−ジ−tert−ブチルフェノール、α−トコフェロール及び没食子酸プロピルからなる群から選択されるものが、酸化防止剤の一例としてあげられるが、これらには限定されない。
【0090】
ある別の実施態様によれば、剤形は、均質なコアを備えている。このコアは、α,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩、薬学的に許容されるポリマー(例えば、ポリエチレンオキシド)、任意の崩壊剤(例えば、ポリビニルピロリドン)、任意の吸収促進剤(例えば、脂肪酸、界面活性剤、キレート剤、胆汁酸塩等)を含む。均質コアは、α,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩を放出するための経路(上記したような)を有する半浸透性壁により包囲されている。
ある実施態様によれば、半浸透性壁は、セルロースエステルポリマー、セルロースエーテルポリマー及びセルロースエステル−エーテルポリマーからなる群から選択されるものを含む。代表的な壁ポリマーは、セルロースアシレート、セルロースジアシレート、セルローストリアシレート、セルロースアセテート、セルロースジアセテート、セルローストリアセテート、モノ−、ジ−及びトリセルロースアルケニレート並びにモノ−、ジ−及びトリセルロースアルキニレートからなる群から選択されるものを含む。本発明に使用されるポリ(セルロース)は、数平均分子量が20,000〜7,500,000である。
【0091】
本発明の目的のための追加の半浸透性ポリマーは、アセトアルデヒドジメチルセルロースアセテート、セルロースアセテートエチルカルバメート、セルロースアセテートメチルカルバメート、セルロースジアセテート、プロピルカルバメート、セルロースアセテートジエチルアミノアセテート;半浸透性ポリアミド;半浸透性ポリウレタン;半浸透性スルホン化ポリスチレン;ポリアニオンとポリカチオンの共沈により形成された半浸透性架橋ポリマー、半浸透性架橋ポリスチレン、半浸透性架橋ポリ(スチレンスルホン酸ナトリウム)、半浸透性架橋ポリ(ビニルベンジルトリメチル塩化アンモニウム)及び半浸透性ポリマー(流体浸透性:半浸透性壁の静水圧又は浸透圧差当りで、2.5x10-8〜2.5x10-2(cm2/hr・atm))を含む。本発明に有用な他のポリマーは、当該技術分野において知られており、例えば、Handbook of Common Polymers(一般ポリマーハンドブック)、Scott、J.R.及びW.J.Roff、1971、オハイオ州クリーブランドにあるCRC Press社発行に記載のものなどがあげられる。
【0092】
ある実施態様によれば、好ましくは半浸透性壁が非毒性、不活性であり、薬物の投薬寿命中は、その物理的及び化学的健全性が維持されるものである。ある実施態様によれば、剤形は、結合剤を含む。結合剤としては、例えば、粘度平均分子量が5,000〜350,000である治療学的に許容されるビニルポリマーがあげられ、代表的には、ビニルアセテート、ビニルアルコール、塩化ビニル、フッ化ビニル、酪酸ビニル、ラウリン酸ビニル及びステアリン酸ビニルからなる群から選択されるものを有する、ポリ−n−ビニルアミド、ポリ−n−ビニルアセトアミド、ポリ(ビニルピロリドン)(ポリ−n−ビニルピロリドンとしても知られている)、ポリ−n−ビニルカプロラクトン、ポリ−n−ビニル−5−メチル−2−ピロリドン及びポリ−n−ビニルピロリドンコポリマーからなる群から選択されるものがあげられるが、これには限定されない。他の結合剤として、例えば、アラビアゴム、でんぷん、ゼラチン及び平均分子量が9,200〜250,000のヒドロキシプロピルアルキルセルロースなどがある。
【0093】
ある実施態様によれば、剤形は、滑剤を含む。滑剤は、剤形の製造中に使用されて、型壁又はパンチ面への粘着を防止することができるものである。滑剤としては、例えば、ステアリン酸マグネシウム、ステアリン酸ナトリウム、ステアリン酸、ステアリン酸カルシウム、オレイン酸マグネシウム、オレイン酸、オレイン酸カリウム、カプリル酸、フマル酸ステアリルナトリウム及びパルミチン酸マグネシウムなどがあるが、これらには限定されない。
【0094】
ある好ましい実施態様によれば、本発明は、オキシモルホン塩酸塩10〜40mgに相当する量のα,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩、平均分子量が150,000〜500,000であるポリ(アルキレンオキシド)25mg〜500mg、平均分子量が40,000であるポリビニルピロリドン1mg〜50mg、滑剤0mg〜約7.5mgを含有する治療組成物を含む。
【0095】
坐剤
本発明の除放製剤は、直腸投与用薬用坐剤として製剤化してもよい。この坐剤は、好適な坐剤基剤及びα,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩を含む。除放性坐剤の調製については、例えば、米国特許第5,215,758号に記載されている。
【0096】
吸収前には、薬物は、溶液状態でなければならない。坐剤の場合、まず坐剤の基剤の溶解又は基剤の溶融が生じて、続いて薬物が坐剤の基剤から直腸液に分配される。薬物の体内への吸収は、坐剤の基剤により変更できる。したがって、薬物の物性を考慮しながら、特定の薬物に応じて、使用される特定の坐剤の基剤を選ぶ必要がある。例えば、脂溶性薬物は直腸液には容易に分配されないが、脂質基剤に微溶性である薬物は直腸液に容易に分配される。
【0097】
薬物の溶解時間(又は放出速度)に影響する異なる因子には、溶解溶媒にさらされる薬物物質の表面積、使用溶液のpH、特定の溶媒への物質の溶解度及び溶媒への溶解物質の飽和濃度の駆動力がある。一般的に、直腸内に投与された坐剤からの薬物の吸収に影響する因子には、坐剤のビヒクル、吸収部位pH、薬物pKa、イオン化度及び脂溶性などがある。
【0098】
選択される坐剤の基剤は、本発明の活性成分と適合するものでなければならない。さらに、坐剤の基剤は、好ましくは非毒性であり、粘膜を刺激せず、直腸液で溶融又は溶解し、そして貯蔵中安定であることが好ましい。
【0099】
本発明のある好ましい実施態様によれば、水溶性薬物及び水不溶性薬物の両方において、坐剤の基剤は、鎖長がC12〜C18である飽和天然脂肪酸のモノ−、ジ−及びトリグリセリドからなる群から選択される脂肪酸ろうを含む。
【0100】
本発明の坐剤を調製する際、他の賦形剤を使用することもできる。例えば、ろうを使用して直腸経路を介した投与のための適切な形状を形成してもよい。この系は、直腸投与及び経口投与用のゼラチンカプセル剤に希釈剤を添加し、ろうなしで使用することもできる。
【0101】
好適な市販のモノ−、ジ−及びトリグリセリドとしては、商品名Novata(商品名) (タイプAB、AB、B、BC、BD、BBC、E、BCF、C、D及び299)(Henkel社製)及びWitepsol(商品名)(タイプH5、H12、H15、H175、H185、H19、H32、H35、H39、H42、W25、W31、W35、W45、S55、S58、E75、E76及びE85)(ダイナマイト・ノーベル社製)の炭素長が12〜18の飽和天然脂肪酸などがある。
【0102】
他の薬学的に許容される坐剤の基剤は、上記したモノ−、ジ−及びトリグリセリドが全体又は部分的に置換されたものでもよい。坐剤中の基剤の量は、剤形のサイズ(すなわち、実重量)、使用される基剤(例えば、アルギン酸塩)、薬物の量により決定される。一般的に、坐剤の基剤の量は、坐剤の総重量の約20重量%〜約90重量%である。好ましくは、坐剤中の坐剤の基剤の量は、坐剤の総重量の約65重量%〜約80重量%である。
【0103】
さらなる実施態様
α,β−不飽和ケトン含量が10ppm未満であるオキシモルホン塩酸塩は、既存の市販の製品、例えば、Opana(商品名)、 Opana ER(商品名)及びNumorphan(商品名)におけるオキシモルホン塩酸塩の代替品として使用できる。このような製剤は、FDA Orange Book(FDAオレンジブック)に挙げられている。
【0104】
以下、実施例により本発明を説明する。実施例では、オリパビンを原料として用い、添付図面を参照して高純度オキシモルホンの合成を示す。
【0105】
図1は、実施例3.2Dにより得られた水和オキシモルホン塩酸塩生成物についての粉末X線回折パターンである。
【0106】
〔実施例1.1A〕
オリパビンの14−ヒドロキシモルヒノンへの水酸化
オリパビン1kgを、攪拌しながら、ぎ酸2.76kg及び水0.53kgの入った反応容器に添加する。オリパビンが完全に溶解するまで攪拌を継続し、温度が20〜30℃の範囲を維持する。続いて、35重量%過酸化水素溶液0.36kgを添加し、反応混合物を3時間以上攪拌するとともに、温度を20〜35℃の範囲に維持する。反応容器を10℃に冷却し、希薄水酸化アンモニウム7.12リットルをゆっくりと添加するとともに、反応混合物を40℃未満に維持する。必要ならば、反応混合物のpHを、必要に応じてさらに希薄水酸化アンモニウム溶液又は塩酸を添加して8〜10の範囲に調整し、攪拌を3〜5時間継続する。
【0107】
生成物である14−ヒドロキシモルヒノンの沈殿が形成し、その沈殿を濾取する。沈殿を水で無色になるまで洗浄した後、湿気のあるケーキとなるまで乾燥し、採集して次の段階に使用する。
【0108】
〔実施例1.1B〕
オキシモルホン基剤の形成
水素添加容器に、水(kgリットル)と酢酸0.73kgを入れてから、実施例1.1Aと同様にして調製した14−ヒドロキシモルヒノン1kgを添加し、混合物を溶液が透明になるまで攪拌する。炭素に担持された湿った10%パラジウム触媒40gを窒素流下で添加し、水素を35〜40psi(2.41〜2.76バール)で供給する。温度を、水素吸収が停止するまで30±5℃に維持し、次に容器を35〜40psi(2.41〜2.76バール)及び30±5℃に3〜4時間維持する。反応容器を25℃未満に冷却し、試料をHPLC分析して、14−ヒドロキシモルヒノンを確認する。HPLC分析で検出された14−ヒドロキシモルヒノン面積が>0.1%である場合には、水素添加を繰り返す。
【0109】
反応が完了したことを確認したら、触媒を濾去し、濾液のpHを水酸化アンモニウム溶液を用いて9に調製する。生成物が沈殿し、濾過により単離し、真空乾燥する。生成物をジクロロメタン/メタノール(9:1v/v)に溶解し、フロリジルにスラリー化し、濾過し、濾液を蒸留して、n−プロパノールに交換する。n−プロパノール混合物を冷却する。生成物が沈殿し、それを濾取する(収率66%)。生成物試料を、HPLC分析してα,β−不飽和ケトン検出したところ、測定面積で0.51%含有することが判明する。
【0110】
〔実施例1.1C〕
高純度オキシモルホン塩酸塩の形成
反応容器に、実施例1.1Bと同様の方法により調製したオキシモルホン基剤1kgを、無水アルコール2.05kg及び水0.66kgとともに入れる。混合物を60±2℃に加熱し、攪拌してスラリーを形成する。濃塩酸0.66kg、水0.24kg及び無水アルコール0.31kgから調製した塩酸溶液を、オキシモルホン基剤スラリーに添加し、pHが<1.0であることを確認する。炭素に担持された10%パラジウム触媒水湿潤ペースト40gを、窒素流下、反応混合物に添加し、混合物を35±5psi(2.41バール)で20時間水素添加するとともに、この間、温度を65±3℃に維持する。反応混合物を、熱い状態で、セライト及び0.2μmポリッシュフィルターで濾過する。濾液を0〜5℃で2〜3時間かけて冷却し、さらに2時間攪拌して、オキシモルホン塩酸塩沈殿を得る。沈殿を無水アルコールで洗浄後乾燥する。収率は、80%である。
【0111】
生成物の試料を、HPLC分析してα,β−不飽和ケトンを検出すると、6.2ppmである。
【0112】
〔実施例1.2A〕
オリパビンの14−ヒドロキシモルヒノンへの水酸化
オリパビン40gを、攪拌しながら、水30g及びぎ酸85gを入れた反応容器に添加し、オリパビンが完全に溶解するまで攪拌を続ける。温度を20〜30℃の範囲に維持する。続いて、30重量%過酸化水素溶液17.72gを添加し、その反応混合物を3時間以上攪拌し、この間、温度を20〜35℃の範囲に維持する。反応混合物を<20℃に冷却し、希薄水酸化アンモニウム335mLをゆっくりと添加する。この間、反応混合物の温度を32℃未満に維持する。必要ならば、反応混合物のpHを、必要に応じてさらに希薄水酸化アンモニウム溶液又は塩酸を添加して9.0に調整し、攪拌を20℃で2時間継続し、4〜5℃で2時間継続する。
【0113】
14−ヒドロキシモルヒノンの沈殿が形成する。この沈殿を濾取する。濾取した沈殿を水で洗浄した後、乾燥して湿ったケーキを得て、採集して次の段階で使用する。
【0114】
〔実施例1.2B〕
オキシモルホン基剤の形成
水素添加容器に、水148g、酢酸90.6g、実施例1.2Aと同様にして調製した湿った14−ヒドロキシモルヒノン(水分48%)250gを入れる。混合物を、溶液が透明になるまで攪拌し、ペーストの形態の炭素に担持された10%パラジウム触媒(乾燥重量)1.34gを、窒素流下添加する。水素添加容器を窒素及び水素でフラッシュした後、反応混合物を、30℃及び35psi(2.41バール)で5時間水素添加する。製造過程でのHPLC分析から、14−ヒドロキシモルヒノン面積が0.07%であることが分かる。
【0115】
反応が完了したことが確認されたら、触媒をセライトパッドで濾去し、セライトケーキを水25mLで洗浄する。濾液を0〜5℃に冷却し、pHを濃水酸化アンモニウムと水の1:1混合物(V/V)により9.5±0.5に調整する。生じた沈殿を0〜5℃で1時間攪拌し、濾過により単離する。粗生成物を、真空オーブン中50℃で乾燥して淡いベージュ色の固体113g(収率86.9%)を得る。生成物の試料をHPLCで分析してα,β−不飽和ケトンを検出し、測定面積で0.27%含有していることが分かる。
【0116】
粗生成物であるオキシモルホン基剤113gを、ジクロロメタン/メタノール(9:1、 v/v)1.13Lに入れる。フロリジル113gを溶液に添加し、混合物を12時間攪拌する。混合物をフロリジル113gのパッドにより濾過し、フロリジルケーキをジクロロメタン/メタノール120mLですすぐ。溶媒を留去した後、n−プロパノールに切り換える。バッチを0〜5℃に冷却し、1時間攪拌してオキシモルホン基剤を沈殿させる。この沈殿を、濾取し、冷n−プロパノールで洗浄し、真空オーブン中で乾燥して白色固体67.2g(59.47%)を得る。
【0117】
生成物の試料をHPLCで分析してα,β−不飽和ケトンを検出したところ、測定面積で0.027%含有していることが分かる。
【0118】
〔実施例1.2C〕
高純度オキシモルホン塩酸塩の形成
反応容器に、実施例1.2Bと同様の方法により調製したオキシモルホン基剤50.1gを、無水アルコール120gとともに入れる。混合物を60±2℃に加熱し、攪拌してスラリーを形成する。濃塩酸32.7g、水33.6gから調製した塩酸溶液を、オキシモルホン基剤スラリーに添加し、pHが<1.0であることを確認する。炭素に担持された10%パラジウム触媒水湿潤ペースト2.0gを、窒素流下、反応混合物に添加し、混合物を35psi(2.41バール)で20時間水素添加するとともに、この間、温度を65℃に維持する。反応混合物を、熱い状態で、セライトで濾過する。濾液を2〜3時間かけて0〜5℃に冷却し、さらに2時間攪拌して、オキシモルホン塩酸塩沈殿を得る。沈殿を濾取し、無水アルコールで洗浄後乾燥して、白色結晶を収率77%で得る。
【0119】
生成物の試料を、HPLC分析してα,β−不飽和ケトンを検出すると、1.1ppmであることが分かる。
【0120】
上記の方法は、当業者により、生成物であるオキシモルホン塩酸塩の高純度を維持しながら変更することができ、このような変形例の実施例を以下に示す。
【0121】
〔実施例2.1B〕
14−ヒドロキシモルヒノンのオキシモルホン基剤への還元
水素添加容器に水2.5kg及び酢酸0.73kgを入れ、14−ヒドロキシモルヒノン1kgを添加する。透明溶液が得られるまで反応混合物を攪拌し、それから炭素に担持された湿った10%パラジウム触媒40gを窒素流下で添加する。水素を、35〜40psi(2.41〜2.76バール)で供給する。温度を、水素吸収が停止するまで30±5℃に維持した後、容器を35〜40psi(2.41〜2.76バール)及び30±5℃で3〜4時間維持する。反応容器を25℃未満に冷却し、試料をHPLC分析して14−ヒドロキシモルヒノンを検出する。HPLC分析により検出された14−ヒドロキシモルヒノンの面積が>0.1%である場合には、水素添加を繰り返す。
【0122】
反応が完了したことが確認されたら、触媒を濾去し、ジクロロメタン/メタノール(9:1 v/v)を濾液に添加し、混合物に水酸化アンモニウム溶液を添加することによりpH9〜10に調整する。ジクロロメタン/メタノール相を分離し、フロリジルにスラリー化し、濾過し、濾液を蒸溜し、n−プロパノールに交換する。n−プロパノール混合物を冷却する。生成物が沈殿する。この沈殿を、濾取する(収率73%)。生成物の試料をHPLC分析してα,β−不飽和ケトンを検出すると、面積で0.32%含有していることが分かる。
【0123】
〔実施例2.2B〕
14−ヒドロキシモルヒノンのオキシモルホン基剤への還元
水素添加容器に、水35g、酢酸17g及び実施例1.2Aで調製した14−ヒドロキシモルヒノン38.08gを入れる。反応混合物を透明溶液となるまで攪拌した後、炭素に担持された湿った5%パラジウム触媒1.8gを窒素流下添加する。水素を、35〜40psi(2.41〜2.76バール)で供給する。温度を、水素の吸収が停止するまで30±5℃に維持する。次に、容器を、35〜40psi(2.41〜2.76バール)及び30±5℃で4時間維持する。反応容器を25℃未満に冷却し、試料をHPLCで分析して14−ヒドロキシモルヒノンを検出する。その結果、HPLC分析で検出した14−ヒドロキシモルヒノン面積は、0.26%である。
【0124】
反応が完了したことを確認したら、触媒を濾去し、得られたケーキを水15mLで洗浄する。ジクロロメタン/メタノール(9:1、v/v)180mLを濾液に添加する。濃水酸化アンモニウムを添加することにより、pHを9〜10に調整する。ジクロロメタン/メタノール層を分離し、フロリジル約20gでスラリー化することにより精製する。スラリーを濾過し、濾液を蒸溜し、n−プロパノールに交換する。混合物を 0〜5℃に冷却し、1〜2時間攪拌してオキシモルホン基剤沈殿を生じさせ、その沈殿を濾過により単離する。次に、オキシモルホン基剤をn−プロパノールでスラリー化して生成物を得る(収率74%)。生成物の試料をHPLC分析してα,β−不飽和ケトンを検出すると、面積で0.32%含有していることが分かる。
【0125】
〔実施例2.2C〕
高純度オキシモルホン塩酸塩の形成
反応容器に、実施例2.2Bで同様にして調製したオキシモルホン基剤2.5gを、無水アルコール7.5mL、水2.5g及び濃塩酸1.66gとともに入れる。混合物を50〜60℃に加熱して溶液を得る。pHが<1.0であることを確認する。炭素に担持された10%パラジウム触媒水湿潤ペースト0.111gを反応混合物に窒素流下で添加する。混合物を35±5psi(2.41バール)で21時間水素添加する。この間、温度は、65±3℃に維持する。反応混合物を、熱い状態で、0.45μmフィルターにより濾過する。濾液を、2〜3時間かけて、0〜5℃に冷却し、さらに2時間攪拌してオキシモルホン塩酸塩沈殿を形成する。沈殿を濾取し、冷無水アルコールで洗浄し、真空乾燥して白色結晶を得る(収率77%)。
【0126】
生成物の試料をHPLC分析してα,β−不飽和ケトンを検出すると、2.8ppm含有されていることが分かる。
【0127】
〔実施例3.1B〕
14−ヒドロキシモルヒノンのオキシモルホン塩酸塩への還元
オキシモルヒノン遊離塩基の形成後、上記した手順をおこなう。しかしながら、この場合、ジクロロメタン/メタノール溶液から遊離塩基を単離する代わりに、0.35容積当量の3N塩酸を添加(ジクロロメタン/メタノール溶液の容積に対して)し、反応混合物を攪拌し、放置し、水層(生成物を含有)を有機層から分離する。水層を、真空蒸溜して容積の約35%を除去した後、残りの溶液を2時間かけて20〜25℃に冷却し、1〜2時間攪拌し、0〜5℃に冷却し、2〜3時間攪拌する。攪拌中に形成する白色固体を濾取し、冷イソプロパノールで洗浄する。収率は64%であり、生成物はα,β−不飽和ケトンを0.34%含有する。
【0128】
〔実施例3.1C〕
オキシモルホン塩酸塩の精製
実施例3.1Bの生成物を原料として用いること以外は実施例1.1Cと同様の方法により、精製オキシモルホン塩酸塩を、収率92%で得る。この生成物中のα,β−不飽和ケトン含量は、検出できない程度である。
【0129】
〔実施例3.2C〕
高純度オキシモルホン塩酸塩の調製
反応容器に、実施例3.1Bで調製したオキシモルホン塩酸塩5.05gを、無水アルコール13.5mL、水4.5mL及び濃塩酸1.51gとともに入れる。混合物を、50〜60℃に加熱して溶液を得る。pHが<1.0であることを確認する。炭に担持された10%Pd触媒水湿潤ペースト0.21gを、反応混合物に窒素流下で添加する。混合物を、35±5psi(2.41バール)で20時間水素添加する。この間、温度を65±3℃に維持する。反応混合物を、熱い状態で、0.45μmフィルターで濾過する。濾液を、2〜3時間かけて、0〜5℃に冷却し、さらに2時間攪拌して沈殿を形成する。沈殿を濾取し、冷無水アルコールで洗浄した後、乾燥する。収率は、92%である。
【0130】
生成物の試料をHPLC分析すると、α,β−不飽和ケトン含量が検出できない程度あることが分かる。
基本プロセス工程を変更することなく、原料についてのプロセス工程を少し変更すること、例えば、このような原料を単離するか又は単離しないことにより、そして精製オキシモルホン塩酸塩への最終工程についての本発明に不可欠な還元要件を利用することにより、α,β−不飽和ケトン含量レベルが3.8ppm、1.7ppm、6.2ppm、6.9ppm、2.8ppm、3.1ppm、0.9ppm、6.0ppm及び別の例では検出不可能レベル、すなわちゼロである他の生成物が得られた。
【0131】
〔実施例3.2D〕
オキシモルホン塩酸塩の水和
乾燥皿に、エタノールを約5〜13重量%含有する実施例1.1C、1.2C、2.2C、3.1C又は3.2Cで同様に調製したオキシモルホン塩酸塩を入れる。試料を、水100mL入っている皿とともに真空オーブンに入れる。真空24〜29Hgとし、オーブンを20〜40℃で24時間維持する。水分約10〜13重量%のエタノール不含又はエタノール低含量(約0.04重量%)生成物が得られる。試料により吸収された水は、真空オーブン中50〜55℃で除去してよい。生成物のKFが6〜8重量%になったら乾燥プロセスを停止する。最終的な水和オキシモルホン塩酸塩は、一致したX線回折パターンを有する均一な多形体である。
【特許請求の範囲】
【請求項1】
HPLCで測定したときに、14−ヒドロキシモルヒノンの含量が10ppm未満である、オキシモルホン塩酸塩。
【請求項2】
14−ヒドロキシモルヒノンの含量が5ppm未満である、請求項1に記載のオキシモルホン塩酸塩。
【請求項3】
医薬として使用するための請求項1又は2に記載のオキシモルホン塩酸塩。
【請求項4】
少なくとも一種の薬学的に許容される賦形剤と、請求項1又は2に記載のオキシモルホン塩酸塩とを含んでなる、医薬製剤。
【請求項5】
HPLCにより測定したときに、14−ヒドロキシモルヒノンの含量が10ppm未満であり、KF含量が6〜8重量%である、水和オキシモルホン塩酸塩。
【請求項6】
HPLCにより測定したときに、14−ヒドロキシモルヒノンの含量が10ppm未満であり、且つ、
粉末X線回折により分析した時に、以下の2θ範囲:
8.5〜9.5、11.0〜12.0、11.5〜12.5、12.4〜13.4、15.2〜16.2、17.6〜18.6、19.3〜20.3、19.9〜20.9、24.6〜25.6、24.9〜25.9、29.0〜30.0及び31.0〜32.0にピークを有する、水和オキシモルホン塩酸塩。
【請求項1】
HPLCで測定したときに、14−ヒドロキシモルヒノンの含量が10ppm未満である、オキシモルホン塩酸塩。
【請求項2】
14−ヒドロキシモルヒノンの含量が5ppm未満である、請求項1に記載のオキシモルホン塩酸塩。
【請求項3】
医薬として使用するための請求項1又は2に記載のオキシモルホン塩酸塩。
【請求項4】
少なくとも一種の薬学的に許容される賦形剤と、請求項1又は2に記載のオキシモルホン塩酸塩とを含んでなる、医薬製剤。
【請求項5】
HPLCにより測定したときに、14−ヒドロキシモルヒノンの含量が10ppm未満であり、KF含量が6〜8重量%である、水和オキシモルホン塩酸塩。
【請求項6】
HPLCにより測定したときに、14−ヒドロキシモルヒノンの含量が10ppm未満であり、且つ、
粉末X線回折により分析した時に、以下の2θ範囲:
8.5〜9.5、11.0〜12.0、11.5〜12.5、12.4〜13.4、15.2〜16.2、17.6〜18.6、19.3〜20.3、19.9〜20.9、24.6〜25.6、24.9〜25.9、29.0〜30.0及び31.0〜32.0にピークを有する、水和オキシモルホン塩酸塩。
【図1】


【公開番号】特開2011−201906(P2011−201906A)
【公開日】平成23年10月13日(2011.10.13)
【国際特許分類】
【出願番号】特願2011−118861(P2011−118861)
【出願日】平成23年5月27日(2011.5.27)
【分割の表示】特願2009−540878(P2009−540878)の分割
【原出願日】平成19年12月14日(2007.12.14)
【出願人】(590004718)ジョンソン、マッセイ、パブリック、リミテッド、カンパニー (152)
【氏名又は名称原語表記】JOHNSON MATTHEY PUBLIC LIMITED COMPANY
【公開日】平成23年10月13日(2011.10.13)
【国際特許分類】
【出願日】平成23年5月27日(2011.5.27)
【分割の表示】特願2009−540878(P2009−540878)の分割
【原出願日】平成19年12月14日(2007.12.14)
【出願人】(590004718)ジョンソン、マッセイ、パブリック、リミテッド、カンパニー (152)
【氏名又は名称原語表記】JOHNSON MATTHEY PUBLIC LIMITED COMPANY
[ Back to top ]